
6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメート
本発明は、有効成分6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートを含む固体医薬組成物に関する。本発明は又、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメート化合物の多形体にも関係する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、化合物6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートを有効成分として含む固体医薬組成物に関するものである。この発明は又、この医薬組成物の製法、その医薬、特にある種の癌の治療のための医薬(酵素ステロイドスルファターゼを標的とする化合物1)としての利用にも関係する。
【背景技術】
【0002】
化合物6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメート(以後、化合物1とも称する)を有効成分として含むこの発明による組成物は、安定な経口投与用医薬組成物であって、適当な生物学的利用能を提供するという利点を有する。
【0003】
下記の構造:
【化1】

の化合物1は、特許EP880514に記載されている。今日では、この化合物への注目は、L.W. Woo等(Chemistry & Biology, 2000, 7, 773-91)により記載されたようなそのスルファターゼ阻害活性及びこれが包含する治療用途のために増大している。ステロイド硫酸エステルの加水分解を担う酵素であるステロイドスルファターゼ(STS)の阻害は、例えば、ホルモン依存性乳癌を有する閉経後の患者に対する将来有望な新規な治療を代表する(Clin. Cancer Res. 2006; 12(5))。
【0004】
原則として、錠剤などの医薬組成物は、3通りの工程、湿式造粒法、乾式造粒法及び直接圧縮法によって製造することができる。
【0005】
化合物1は、G. Amidonにより提案された生物薬剤学的分類システム即ちBCS(G.L. Amidon等、「A theoretical basis for a biopharmaceutical drug classification: the correlation of in vitro drug dissolution and in vivo bioavailability」、Pharm. Res. 12 (1995) 413-420参照)のクラス2に属する。そうして、その吸収、及びそれ故、その生物学的利用能は、投与される医薬形態の溶解速度に強く依存する。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】欧州特許第880514号明細書
【非特許文献】
【0007】
【非特許文献1】L.W. Woo等、Chemistry & Biology, 2000, 7, 773-91
【非特許文献2】Clin. Cancer Res. 2006; 12(5)
【非特許文献3】G.L. Amidon等、Pharm. Res. 12 (1995) 413-420
【発明の概要】
【発明が解決しようとする課題】
【0008】
従来技術で公知の水相を用いる工程及び製剤、例えばクラス2の化合物の溶解度を増大させるために標準的様式において用いられる自己乳化性製剤(「自己乳化性薬物送達システム」)は、用いる賦形剤及び水相が化合物1と相容性でなく、製造段階において化学的劣化へと導くので、適当でない。
【0009】
化合物1の安定性及び低い溶解度の問題のために、経時的に十分に安定であって且つ十分な濃度の有効成分を含んでいる錠剤の製造を可能にする配合物を得ることは困難であった。その上、圧縮性の乏しい組成物は、乾式プレス中の破砕並びに余りに遅い溶解プロファイルを生じた。水の存在下で容易に加水分解されうる化合物1の不安定性のために、湿式造粒法を想定することは困難であった。
【課題を解決するための手段】
【0010】
予想外にも、化合物1の乾燥経口投与用形態を湿式造粒法により得ることを可能にする配合物が得られた。
湿式造粒法の場合には、成分を混合して、湿潤相中で結合剤を用いて造粒する。この結合剤は、湿潤相に溶解することができ又は造粒すべき粉末の混合物に取り込まれうる。次いでそれらの湿潤顆粒をふるいにかけ、乾燥させて、適宜粉砕した後に、錠剤に成形するために圧縮する。
【0011】
予想外にも、化合物1の新規な固体の医薬組成物が見出され、経口経路による投与が意図されてきており、これは、この有効成分に特異的な問題を解決すること及びそれを湿式造粒法により得ることを可能にする。
【0012】
この錠剤又はゼラチンカプセルの形態の経口投与用組成物は、固体形態の急速な溶解に安定であり、それ故、有効な生物学的利用能を伴う即時的放出を与える。
【0013】
以上より、本発明の主題は、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートを有効成分として含み且つ少なくとも一種類の製薬上許容しうる賦形剤を含む固体の医薬組成物である。
【0014】
本発明の主題は又、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートを有効成分として含み且つ少なくとも一種類の製薬上許容しうる賦形剤を含む、経口投与用の、好ましくは即時的に放出される経口投与用の固体の医薬組成物でもある。
【0015】
本発明の主題は又、経口経路による投与のための、化合物1を有効成分として含み且つ少なくとも一種類の崩壊剤並びに一種以上の水分に対する保護剤を含む固体の医薬組成物でもある。
【0016】
それ故、かかる薬剤は、有効成分を、加水分解に対して保護し、そうしてその劣化を防止する。好ましくは、化合物1は微粉化される。
【0017】
この劣化は、主として、この医薬組成物の製造中に起き、特に、湿式造粒法の段階において起きるということが認められた。予想外にも、当分野で結合剤及び希釈剤としての性質の故に用いられる賦形剤の添加が有効成分を保護する効果を与えるということが示された。
【0018】
本発明の主題は又、下記するような医薬組成物の製造方法でもある。従って、この発明の方法を、湿式造粒法を使用する錠剤の製造のための標準型の補助剤及び賦形剤を用いて実施することが可能である。
【0019】
「即時的放出を伴う経口投与」又は「即時的放出を伴う経口投与用形態」又は「即時的放出を伴う組成物」とは、この発明においては、化合物1用に開発された適当なイン・ビトロ溶解試験によって、45分以内に、好ましくは30分以内に有効成分の少なくとも80重量%のイン・ビトロでの溶解を与える投与又は経口投与用組成物を意味する。この試験は、パドル溶解装置を用いて、37℃で、100rpmで攪拌しながら、米国薬局方の基準に従って塩酸緩衝溶液(pH1.2)中で行なわれ、該装置は、0.1%界面活性剤、セチルトリメチルアンモニウムブロミドを含み、これは、有効成分の試験媒質中での十分な溶解度を得ることを可能にする。このアッセイは、紫外(UV)/可視光線の吸光度測定により、波長311nmで行なわれる。
【0020】
この発明により用いられる用語「滑剤(gliding agent)」又は「流動剤(flow agent)」とは、補助剤及び賦形剤をも意味し、ときには、潤滑剤及び顆粒の流動性及び流れを改善する薬剤と呼ばれる。
【0021】
この発明による用語「百分法配合」とは、組成物の全重量に対する、有効成分又は賦形剤の重量%での所与の割合を意味する。
【0022】
用語「結合剤」又は結着剤は、この発明によれば、生薬形態の構造及び結合力を維持するために用いられる補助剤又は賦形剤を示す。それらは、造粒段階における成分の顆粒形態での集合を可能にし、圧縮後の生薬形態の凝集を確実にする特性を有している。
【0023】
用語「崩壊剤」は、この発明によれば、液体環境、例えば水中又は胃液中での錠剤の崩壊を促進するために配合物に加えることのできる賦形剤又は補助剤を示す。
【発明の効果】
【0024】
本発明は、多くの利点を、特に、下記の組合せで示す:
・増大した安定性及び
・この医薬形態の即時的溶解(30分以内に少なくとも80%)にリンクした増大した生物学的利用能。
【図面の簡単な説明】
【0025】
【図1】化合物1の組成物の、時間及び組成物中の有効成分の微粉化の影響の関数としての比較用溶解曲線を示す図である。各曲線は、異なる微粉化の化合物1を含む固体組成物に対応している。
【図2】化合物1の種々の組成物の、時間の関数としての比較用溶解曲線を示す図である。各曲線は、異なる崩壊剤を含む固体組成物に対応している。
【図3】化合物1の2型のDMSO溶媒和化合物の推定されたX線回折ダイヤグラムを示す図である。
【図4】化合物1の2型のDMSO溶媒和化合物の実験によるX線粉末回折ダイヤグラムを示す図である。
【図5】化合物1の種類(variety)Iの推定されたX線粉末回折ダイヤグラムを示す図である。
【図6】化合物1の種類Iの実験によるX線粉末回折ダイヤグラムを示す図である。
【図7】化合物1の種類IのDSCサーモグラムを示す図である。
【図8】化合物1の種類IのIRスペクトルを示す図である。
【図9】化合物1の種類Iの固体のNMRスペクトルを示す図である。
【図10】化合物1の1型のDMSO溶媒和化合物の推定されたX線回折ダイヤグラムを示す図である。
【図11】化合物1の1型のDMSO溶媒和化合物の実験によるX線粉末回折ダイヤグラムを示す図である。
【図12】化合物1の3型のDMSO溶媒和化合物の推定されたX線回折ダイヤグラムを示す図である。
【図13】化合物1の3型のDMSO溶媒和化合物の実験によるX線粉末回折ダイヤグラムを示す図である。
【図14】化合物1の多形相(polymorphic form)IIIの推定されたX線回折ダイヤグラムを示す図である。
【図15】化合物1の多形相IIIの実験によるX線粉末回折ダイヤグラムを示す図である。
【図16】化合物1の多形相IIIのDSCサーモグラムを示す図である。
【図17】化合物1の多形相IIIのIRスペクトルを示す図である。
【図18】化合物1の多形相IIIの固体のNMRスペクトルを示す図である。
【図19】化合物1の1,4−ジオキサン半溶媒和化合物(hemisolvate)の推定されたX線回折ダイヤグラムを示す図である。
【図20】化合物1の1,4−ジオキサン半溶媒和化合物の実験によるX線粉末回折ダイヤグラムを示す図である。
【図21】化合物1の1,4−ジオキサン半溶媒和化合物のTG−DSCサーモグラムを示す図である。
【図22】化合物1の多形相IIの実験によるX線粉末回折ダイヤグラムを示す図である。
【図23】化合物1の多形相IIのDSCサーモグラムを示す図である。
【図24】化合物1の多形相IIのIRスペクトルを示す図である。
【図25】化合物1の多形相IIの固体のNMRスペクトルを示す図である。
【発明を実施するための形態】
【0026】
図1及び2は、賦形剤の効果並びに固体組成物のイン・ビトロでの振舞いを示している。
【0027】
好ましくは、この発明による錠剤の製造のために用いられる方法は、錠剤の形成段階の前に湿式造粒段階を通過する。
【0028】
事実、予想外にも、水相中での化合物1の低い安定性にもかかわらず、この錠剤は、湿式造粒段階を含む工程によって製造することができることが示された。この工程による錠剤の生産は、適切な賦形剤の存在下での化合物1の水相における安定化の恩恵を受ける。
【0029】
この発明の第一の特徴によれば、化合物1は、0.1〜20μmの粒子サイズを得るために、適当な処理により得られる。優先的に、化合物1は、1〜15μmの、一層優先的には、2〜10μmの粒子サイズを有する。尚一層優先的には、化合物1は、5μm±2μmのサイズを有する。
【0030】
この発明の医薬組成物の製造に適した製薬上許容しうる賦形剤は、例えば、マルトデキストリン、マンニトール、微結晶性セルロース、ラクトース、トウモロコシ澱粉、澱粉グリコール酸ナトリウム、クロスカルメロースナトリウム、部分的に架橋されたポリ(N−ビニル−2−ピロリドン)又はクロスポビドン、ポリビニルピロリドン、N−ビニル−2−ピロリドンと酢酸ビニルのコポリマー(又はコポビドン)例えばコリドンVA64共重合体、カルボキシメチルセルロース、アルファ化澱粉、メチルセルロース、ポリエチレングリコール、マクロゴール、ポリグリコール、ポリオキシエチレン、ピロリドン−2、コロイドシリカ、タルク、ステアリン酸マグネシウム、ナトリウムステアリルフマレート、ステアリン酸カルシウム、水素化植物油、ラウリル硫酸ナトリウム、リン酸カルシウム、糖類、デキストリン、澱粉、ゼラチン、セルロース、ワックスであってよく、又は水、有機溶媒例えばグリセロール又はグリコール並びにこれらの変化する割合の混合物(水を伴うか又は伴わない)であってもよい。これらの賦形剤は、有効成分に添加されるが、下記の機能を有している:
− 希釈剤、例えば、マンニトール、ラクトース又はラクトース一水和物、澱粉、炭酸カルシウム、微結晶性セルロース、又はマルトデキストリン;
− 崩壊剤、例えば、澱粉、クロスカルメロースナトリウム、澱粉グリコール酸ナトリウム、又はクロスポビドン;
− 結合剤、例えば、ポリビニルピロリドン、N−ビニル−2−ピロリドンと酢酸ビニルとのコポリマー(又はコポビドン)、カルボキシメチルセルロース、アルファ化澱粉、又はメチルセルロース;
− 流動剤又は滑剤、例えば、コロイドシリカ、又はタルク;
− 潤滑剤、例えば、ステアリン酸マグネシウム、ナトリウムステアリルフマレート、ステアリン酸カルシウム、ステアリン酸又は水素化植物油;
− 可溶化剤、例えば、マクロゴール(ポリエチレングリコール)、ポリグリコール、ポリオキシエチレングリコール、ポリジオール、ピロリドン−2、又はポリビニルピロリドン;
− 界面活性剤(又は表面活性剤)、例えば、ラウリル硫酸ナトリウム。
【0031】
本発明の主題は又、有効成分としての化合物1の経口経路による投与のための固体医薬組成物であって、化合物1及び少なくとも一の崩壊剤、一種以上の希釈剤及び一種以上の結合剤を含むことを特徴とする当該固体医薬組成物でもある。好ましくは、この化合物は、微粉化される。
【0032】
好ましくは、この発明による医薬組成物は、経口投与のための固体形態であり、ゼラチンカプセルである。好ましくは、この発明による医薬組成物は、経口投与のための固体形態であり、錠剤である。
【0033】
下記の組成物において、各種の賦形剤は、単独又は混合物であってよい。従って、「x%の結合剤」は、x%の単独の結合剤又は結合剤の混合物を意味する。
【0034】
この発明による組成物は、次の百分法の配合に従って、ゼラチンカプセル中に配合することができる:1〜30%の、好ましくは5〜20%の有効成分;40〜92%の、好ましくは65〜92%の、非常に優先的には75〜90%の希釈剤;0〜10%の結合剤、(好ましくは、0〜8%);0〜30%の崩壊剤(好ましくは、0〜20%);0〜5%の界面活性剤(好ましくは、0%);0〜5%の可溶化剤(好ましくは、0%);0.1〜3%の流動剤(好ましくは、0.9〜1.4%);0.5〜3%の潤滑剤(好ましくは、0.6〜2.8%)。
【0035】
優先的には、この医薬組成物は、上で規定したように、ゼラチンカプセルであり;一層優先的には、5〜30%の有効成分;40〜92%の希釈剤;0〜8%の結合剤(好ましくは、0〜5%);0〜30%の崩壊剤;0〜5%の界面活性剤;0〜5%の可溶化剤;0.1〜3%の流動剤;及び0.5〜3%の潤滑剤を含む百分法配合のゼラチンカプセルである。
【0036】
これらのゼラチンカプセルを配合するための好適な製薬上許容しうる賦形剤は、マンニトール、ラクトース、トウモロコシ澱粉、コロイドシリカ、ステアリン酸マグネシウム、及びラウリル硫酸ナトリウムであり、一層特には、マンニトール、ラクトース、コロイドシリカ、及びステアリン酸マグネシウムである。
【0037】
この発明による組成物は又、組成物の全重量に対して1〜30重量%の好ましくは5〜20重量%の、非常に優先的には8〜20重量%の有効成分;40〜92%の、好ましくは65〜92%の、非常に優先的には70〜85%の希釈剤;0.1〜20%の、好ましくは0.1〜10%の、非常に優先的には1〜5%の崩壊剤;0.1〜8%の、好ましくは2〜5%の結合剤;0.1〜3%の滑剤(好ましくは、0.5〜1.4%);0.2〜3%の潤滑剤(好ましくは、0.5〜2.8%)を含む百分法配合の錠剤(好ましくは、フィルムコート錠)中に配合することもできる。
【0038】
これらの錠剤の配合に好適な賦形剤は、マルトデキストリン、マンニトール、微結晶性セルロース、ラクトース又はラクトース一水和物、トウモロコシ澱粉、澱粉グリコール酸ナトリウム、クロスポビドン、ポリビニルピロリドン、コポビドン、カルボキシメチルセルロース、コロイドシリカ、ナトリウムステアリルフマレート、及びステアリン酸マグネシウムであり、一層特には、微結晶性セルロース、ラクトース、澱粉グリコール酸ナトリウム、コポビドン、コロイドシリカ、及びステアリン酸マグネシウムである。
【0039】
やはり好ましくは、上で規定した医薬組成物は、錠剤の全重量に対して、8〜20%の有効成分;70〜85%の希釈剤;1〜5%の崩壊剤;2〜5%の結合剤;0.5〜1.4%の流動剤;及び0.5〜2.8%の潤滑剤を含み、並びに、コート剤の全重量に対して約4.5〜5%、好ましくは4.8%の被覆溶液を含む百分法配合の錠剤(好ましくは、フィルムコート錠)である。
【0040】
好ましくは、本発明による医薬組成物は、次の賦形剤から選択される希釈剤を含む:マンニトール、ラクトース若しくはラクトース一水和物、澱粉、炭酸カルシウム、微結晶性セルロース、又はマルトデキストリン。
【0041】
好ましくは、本発明による医薬組成物は、次の賦形剤から選択される崩壊剤を含む:澱粉、クロスカルメロースナトリウム、澱粉グリコール酸ナトリウム、又はクロスポビドン。
【0042】
好ましくは、本発明による医薬組成物は、次の賦形剤から選択される結合剤を含む:ポリビニルピロリドン、N−ビニル−2−ピロリドンと酢酸ビニルとのコポリマー(コポビドン)、カルボキシメチルセルロース(CMC)、アルファ澱粉、又はメチルセルロース。
【0043】
好ましくは、本発明による医薬組成物は、次の賦形剤から選択される潤滑剤を含む:ステアリン酸マグネシウム、ナトリウムステアリルフマレート、ステアリン酸カルシウム又は水素化植物油。
【0044】
好ましくは、本発明による医薬組成物は、微結晶性セルロース(MCC)及び/若しくはコポビドン又は微結晶性セルロース(MCC)及び/若しくはカルボキシメチルセルロース(CMC)の何れかを含む。好ましくは、このCMCは、4〜6%のレベルである。非常に優先的には、本発明による医薬組成物は、微結晶性セルロース(MCC)を含む。非常に優先的には、本発明による医薬組成物は、コポビドンを含む。やはり非常に優先的には、本発明による医薬組成物は、微結晶性セルロース(MCC)及びコポビドンを含む。非常に優先的には、本発明による医薬組成物は、微結晶性セルロース(MCC)を含む。非常に優先的には、本発明による医薬組成物は、カルボキシメチルセルロース(CMC)を含む。非常に優先的には、本発明による医薬組成物は、微結晶性セルロース(MCC)及びカルボキシメチルセルロース(CMC)を含む。
【0045】
やはり好ましくは、選択される流動剤は、コロイドシリカ(即ち二酸化ケイ素のコロイド溶液)である。
【0046】
やはり好ましくは、選択される潤滑剤は、ステアリン酸マグネシウムである。これらの錠剤の配合物に賦形剤として加えられる崩壊剤は、溶解速度を増大させ、高い凝集力を有する錠剤を用いても、即時的溶解を達成することを可能にする。文献(例えば、J. Balasubramaniam, T. Bee, Pharmaceutical Technology Europe, Vol. 21, Number 9, 2009, p 44-49)によれば、最も有効な崩壊剤は、クロスポビドンA型及びB型と、その後のクロスカルメロースナトリウムである。
【0047】
好ましくは、本発明による錠剤の配合物において、用いられる崩壊剤は、澱粉グリコール酸ナトリウムであり、優先的には、1〜5%のレベルであり、非常に優先的には、3〜4%のレベルである。
【0048】
好ましくは、本発明による医薬組成物は、錠剤の全重量に対して、8〜20%の化合物1;20〜40%のラクトース及び25〜50%の微結晶性セルロース(希釈剤として使用);2〜8%のコポビドン(結合剤として使用);1〜5%の澱粉グリコール酸ナトリウム(崩壊剤として使用);0.2〜1.4%の流動剤;及び0.5〜2%の潤滑剤を含む錠剤であり、
非常に優先的には、錠剤の全重量に対して、8〜15%の化合物1;30〜40%のラクトース及び40〜50%の微結晶性セルロース(希釈剤として使用);2〜5%のコポビドン(結合剤として使用);3〜4.5%の澱粉グリコール酸ナトリウム(崩壊剤として使用);0.2〜1.4%の流動剤;及び0.5〜2%の潤滑剤を含む。
【0049】
やはり非常に優先的には、上記のように、この医薬組成物は、錠剤の全重量に対して、約10%の化合物1;36.5%のラクトース;45%の微結晶性セルロース;3%のコポビドン;4%の澱粉グリコール酸ナトリウム;0.5%のコロイドシリカ;1%のステアリン酸マグネシウムを含む錠剤である。用語「約」は、±0.5%を意味する。
【0050】
この発明の変形物によれば、この組成物は、コーティング又はフィルムコーティングを含むことができる。
【0051】
好ましくは、かかるコーティングは、有効成分の即時的放出に有意の影響を有さず、即ち、有効成分のイン・ビボでの放出速度論は、変化しない。
【0052】
やはり好ましくは、このコーティングは、有効成分の味をマスクするという利点を有し、固体組成物の錠剤の携帯での処理のための安全な取扱いを可能にする。
【0053】
この発明に記載された錠剤のために利用することのできるコーティング工程は、当業者に周知である。
【0054】
この錠剤がコーティングを含む場合には、後者は、好ましくは、ポリエチレングリコール、エチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルメチルセルロース(ヒプロメロース)により構成される群から選択されるポリマーを含むが、これらに限られない。特に、ポリエチレングリコール(マクロゴール)及び既に配合されたヒドロキシプロピルメチルセルロースを含むフィルムコーティング混合物を、Opadry(商標)IIホワイトの名称でColorcon社により市販されているもの(ラクトース一水和物、ヒプロメロース、二酸化チタン(E171)、トリアセチン)などの固体の経口投与用医薬形態の水性フィルムコーティングに利用することが可能である。Opadry(商標)IIホワイトは、水に可溶性であり、錠剤の形態で経口投与された医薬組成物の即時的崩壊を可能にする。
【0055】
好ましくは、本発明による固体医薬組成物は、錠剤の形態で与えられる。この発明は又、即時的溶解性の錠剤並びにコーティング又はフィルムでカバーされた錠剤にも関係する。
【0056】
医薬組成物は、例として記載されており、決して、この発明の範囲を制限するものではない。
【0057】
従って、この発明の主題は又、上記したようなこの発明の化合物の、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートからの製造方法でもあり、粒子サイズを減じる段階を含むことを特徴とする。
【0058】
優先的には、これらの粒子のサイズは、非プロトン性溶媒媒質中で、又は乾燥経路により、微粉状にすることにより低減される。
【0059】
やはり優先的には、これらの粒子のサイズは、非プロトン性有機溶媒を用いる湿式粉砕によって低減される。
【0060】
この発明は又、下記の段階を特徴とする、この発明の固体医薬組成物の製造方法にも関係する:
・ これらの成分をふるいにかけること;
・ コーティングを有しない錠剤の製造(湿式造粒法、混合及び圧縮);
・ コーティング溶液の製造。
【0061】
好ましくは、本発明による方法によって、湿式造粒段階において、有効成分及び予め混合した結合剤、希釈剤及び崩壊剤の全質量に対する水の質量は、約10〜30%である。
【0062】
好ましくは、この発明による方法によって、湿式造粒段階後に、これらの顆粒は、3%未満の残留水分が得られるまで乾燥される。
【0063】
この組成物は、この製造工程のすべての段階において、当業者に公知の慣用の装置によって製造される。
【0064】
本発明による配合物は、次の工程によって、製造することができる:第一の混合物を製造し、それは、有効成分により構成され、それは、先ず、結合剤、及び一種以上の希釈剤並びに崩壊剤を含む組成物に含まれるある種の賦形剤と同時にふるいにかけられる。
【0065】
このふるいかけ段階は、マニュアルシーブなどの適当なデバイスにて、ふるいを用いて行なわれる。
【0066】
これらのふるい分けした物質を、次いで、攪拌式造粒機に導入して、約5分間混合する。混合速度は、選択した装置による。
【0067】
次いで、この造粒のための水のマスを、造粒すべき固相に対するその割合が優先的に約10〜30%となるように調製する。造粒すべき固相とは、結合剤のマスに加えた有効成分及び予め混合された賦形剤の全マスを意味する。
【0068】
この造粒のための結合剤は、粉末形態で又は優先的には、造粒溶液を構成するために、あるかさの水に溶解されて混合物に添加される。
【0069】
この工程の出発時に得られる混合物を、次いで、好ましくは「高剪断力」型の攪拌造粒機にて、造粒溶液を用いる湿式経路によって造粒する。この造粒すべき溶液の添加後の造粒時間は、10分未満であり、優先的には、5分未満である。
【0070】
この最後の段階から生じる顆粒を、次いで、慣用の装置例えば流動層乾燥器で乾燥させることができる。好ましくは、これらの顆粒は、3%未満の残留水分が達成されるまで乾燥される。こうして乾燥された顆粒は、慣習的に錠剤の内相と呼ばれる部分を構成する。次いで、それらは、研摩によって調整される。
【0071】
予め得られた調整された顆粒及び、崩壊剤の一部分、粉末の流動性を改善することを可能にする流動剤、及び最後に潤滑剤により構成されるいわゆる外相により構成される第2の混合物を製造する。
【0072】
この外相の賦形剤は、先ず、マニュアルシーブなどの適当なデバイスにて、ふるいにかけられる。
【0073】
これらの顆粒、崩壊剤及び流動剤を、次いで、混合してから、潤滑剤を導入する。
【0074】
錠剤の形態の配合物の製造のために、最終生成混合物を、次いで、例えばロータリー式のプレスを用いて錠剤にすることができる。
【0075】
400mgの錠剤の硬さは、6〜12kパスカル(kPa)であってよい。100mgの錠剤の硬さは、4〜8kPaであってよい。
【0076】
これらの錠剤を、次いで、フィルムコーティングタービンにて、予め、好ましくは、純水に16%のOpadryII(商標)を添加することにより、用意した被覆用懸濁液をスプレーすることにより被覆することができる。
【0077】
フィルムコーティング中、製品の温度は、35〜55℃とする。
【0078】
好ましくは、乾燥後の錠剤の質量の増加は、フィルムコーティング前の錠剤の質量の4〜6%である。
【0079】
これらの錠剤は、パッケージに入れて、ブリスターパック又は瓶入りで保存することができる。
【0080】
この発明によれば、非被覆錠剤は、好ましくは、800mgの総重量を、好ましくは、400mgの総重量を超えない。
【0081】
この発明は、最終的には、前記のように、癌、優先的にはホルモン依存性の癌及びやはり優先的に乳癌、前立腺癌、子宮内膜癌又は卵巣癌を治療するための、この発明の医薬組成物の利用に関係する。
【0082】
本願は又、化合物6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメート(即ち、スルファミン酸6,7,8,9,10,11−ヘキサヒドロ−6−オキソベンゾ[b]シクロヘプタ[d]ピラン−3−イルエステル即ち化合物1)の多形体にも関係する。この発明は又、これらの多形相の製法、それらの有効成分としての利用、一層特にある種の癌の治療のための有効成分としての利用にも関係する。この発明は又、これらの多形相を有効成分として含む医薬組成物とも関係する。
【0083】
Wooにより簡潔に記載されて、以後種類Iと呼ばれる形態は、特徴的な赤外吸収バンドを生成する。出願人は、構造の完全な特性決定のために適当な大きさと品質の単結晶を得た。
【0084】
従来技術において得られた結晶は、良好な生物学的利用能を与えず、それらの調製物は、産業的規模に移行することができない。
【0085】
その上、出願人は、化合物1の結晶を、産業的過程により得た。顕微鏡観察は、結晶の進行性の不透明化を約140〜145℃で観察することを可能にする。この現象は、不可逆的であり:結晶は、ゆっくりした又は急速な冷却後に、その最初の外観を回復しない。DSCにおいては、固−固転移に対応する吸熱現象が、140℃から観察される。第2の吸熱のピークは、未変換の種類Iの溶融に対応する170℃で観察される。第3の吸熱のピークは、180℃で観察される。この第3のピークは、固−固転移中に生成された他の多形相の溶融に対応する。連続的に160℃まで加熱してから18〜25℃にまで冷却されたこの化合物に対して行なわれたX線粉末回析により、化合物1の種類Iのこれらの特性に加えて、幾つかのピークが観察され、これらのピークは、加熱中に生成した他の多形相に対応している。
【0086】
従って、産業的条件下で得られた結晶は、DSCにより及び/又は160℃への加熱後のX線粉末回折により検出された不均質性を示す。この不均質性は、化合物の加熱中のグレーン成長及び他の結晶形の成長を与え、これは、加熱中の生成物を不安定にする。
【0087】
不均質性とは、ここでは、変色又はポリタイプを含む結晶中又は結晶上の欠陥を意味し、これは、慣用の光学顕微鏡又は偏光装置を備えた顕微鏡によって及び/又は示差エンタルピー分析及び/又はX線粉末回折により及び/又は共焦点ラマン顕微鏡観察によって検出することができる。従って、結晶中のかかる不均質性の存在は、有効成分の保存中に、又は錠剤、ゼラチンカプセル、クリーム、又は他の生薬形態の製造工程中に、多くの問題へと導く。例えば、相対湿度ストレスは、医薬有効成分に対して許容しえない挙動へと導きうる。その上、Y. Mnyukh Fundamentals of solid-state phase transitions, ferromagnetism and ferroelectricity (2001)により示されたように、これらの欠陥は、固−固転換をある機構(グレーン成長)によってプレコード化(pre-code)しうる。
【0088】
現在、この分子の臨床的開発のためには、ある結晶形を選択し、産業的条件下でのその生産を達成することが必要であり、鍵となる因子は、その熱安定性及び生物学的利用能である。
【0089】
それ故、出願人が解決することを提案する技術的課題は、化合物1の、産業的規模で用いうる条件下で安定で且つ生物学的利用能のある形態である。
【0090】
当業者には、機械的処理による粒子サイズの低減を目的とした処理は、結晶化相に、欠陥、残留ストレス、多形転移、部分的又は全体的非晶質化(状況によっては、化学分解を伴う)を生じうることは公知である(Jet-milling; from a particle perspective predicting particle fracture based on mechanical material properties Onno M. de Vegt; PhD thesis University of Groningen; 19 October 2007; Garnier, S.; Petit, S.J Mallet, F.; Petit, M.-N.; Lemarchand, D.; Coste, S.; Lefebvre, J.; Coquerel, G., Influence of aging, grinding and preheating on the thermal behaviour of α-lactose monohydrate. Int. J. Pharm. 2008, 361, 131-140)。
【0091】
化合物の、一つより多くの結晶形で存在する能力は、用語「多形性」により規定され、その異なる結晶形は、「多形種類(polymorphic varieties)」又は「多形体(他形相)」として知られる。多形性は、固体状態の有効成分の多くの特性に影響を与えうる。一の物質の種々の多形性は、例えばそれらの溶解度に直接影響しうるそれらの物性により、互いに、相当に異なることがありうる。多形性は、多くの有機化合物について示されている。
【0092】
非常に予想外なことに、出願人は、今や、粒子サイズの低減を目的とする産業的条件下で得られる化合物1の適当な処理が、有効成分の配合物と適合する粒子サイズを有する化合物1の種類Iを生成することができ、同時に、結晶中の異成分の濃度を付随的に低減させ、そうして、化合物1の安定で生物学的利用能のある形態を産業的規模で用いることのできる条件下で与えることを見出した。実験部に記載したDSC及びX線粉末回折分析は、こうして得られた種類Iが増大した安定性を有することを示すことを可能にする。
【0093】
従って、この発明の主題である低粒子サイズの種類Iは、種々の分析法例えば固体のX線粉末回折、示差走査熱分析(DSC)、ラマン分光法、赤外線分光法又はNMRによって特性決定することができる。
【0094】
この発明の主題は、化合物1の結晶DMSOの2型の溶媒和化合物でもあり、これは、X線粉末回折を利用して、角度(°2シータ)で表される特徴的なピークを示す(±0.1°2シータ):7.6;18.7;24.2;29.9。
【0095】
非常に予想外なことに、出願人は、今や、グレーン成長及び他の結晶の成長を与える異成分を有する産業的条件下で得られたI型の結晶が、化合物の加熱中に、高温で安定なIII型を形成することを見出した。本発明の主題は、この化合物1の新規なIII型である。このIII型は、145℃より高温で、熱力学的に安定であるという利点を有している。
【0096】
この発明のIII型の化合物1に対する幾つかの合成経路があり、その一つは、II型と呼ばれる化合物1の他の新規な結晶形の中間体を経由するものである。
【0097】
化合物1の種類Iは、X線粉末回折により特性決定することができ、回折ダイヤグラムは、特に、角度(°2シータ)で表される特徴的ピークを有する(約±0.1°2シータ):7.5;10.9;13.1;17.7;19.0。
【0098】
これらの±0.1°2シータの誤差を伴う角度(°2シータ)は、Braggの法則による反射角、即ち、試料へのX線の入射角を表す。
【0099】
粒子サイズの低減前の化合物1の種類Iは、単結晶のX線分析によって特性決定することができる。
【0100】
低減された粒子サイズの化合物1の種類Iは、X線粉末回折ダイヤグラムによって特性決定することができる(図6)。
【0101】
化合物1の種類Iは、上記のように、DSCサーモグラムによって特性決定することもできる(図7)。
【0102】
化合物1の種類Iは、上記のように、赤外線スペクトルによって特性決定することもできる(図8)。
【0103】
化合物1の種類Iは、上記のように、固体のNMRスペクトルによって特性決定することもできる(図9)。
【0104】
それ故、出願人は、適当な処理により得られた化合物1の種類Iを、0.1〜20μmの粒子サイズを得るために提供する。
【0105】
本発明は、多くの利点を、特に、増大した安定性及び生物学的利用能に有する。
【0106】
それ故、本発明の主題は、0.1〜20μmの粒子サイズの6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートの多形性化合物であり、これは、X線粉末回折により、角度(°2シータ)で表される特徴的なピークを示す(±0.1°2シータ):7.5;10.9;13.1;17.7;19.0。
【0107】
優先的に、この発明の化合物は、1〜15μmの粒子サイズを有する。
【0108】
一層優先的には、この発明の化合物は、3〜7μmの粒子サイズを有する。
【0109】
尚一層優先的には、この発明による化合物は、5μm±1μmのサイズを有する。
【0110】
好ましくは、上で規定した化合物は、X線粉末回折により、角度(°2シータ)で表される特徴的なピークを示す(±0.1°2シータ):7.5;10.9;13.1;15.0;15.8;17.0;17.7;19.0;22.5。
【0111】
やはり優先的に、上で規定した化合物は、160℃まで加熱してから18〜25℃に戻した後に行なわれるX線粉末回折により、角度(°2シータ)で表される次の特徴的ピークを示す(±0.1°2シータ):7.5;10.9;13.1;17.7;19.0(160℃への加熱中に生成された型に対応する更なるピークを含まない)。
【0112】
非常に優先的には、上で規定した化合物は、5℃/分の温度勾配を有するDSCを用いて、吸熱性溶融ピークを170℃±5℃に示し且つ吸熱性ピークを180℃±2℃に示し、180℃でのピークは、170℃での溶融中に交換されたエンタルピーの最大で10%を表すということが理解される。
【0113】
やはり非常に優先的には、上で規定した化合物は、DSCを用いて、140〜155℃で吸熱事象を示さない。
【0114】
尚一層優先的には、上で規定した化合物は、赤外線分光法を用いて、cm-1で表される特徴的なピークを示し(±5cm-1):3310;3167;3059;891;798;733;679;455;尚一層優先的には、次の特徴的なピークを示した(±5cm-1):3310;3167;3059;2928;2858;1690;1605;1454;1385;1261;1188;1126;941;891;853;798;733;679;598;544;455。
【0115】
一変形によれば、この発明は、上で医薬として規定した化合物に関係する。
【0116】
この発明による化合物は、様々な医薬組成物に配合することができる。固体形態の場合には、それは、例えば、粉末、顆粒、錠剤、ゼラチンカプセル、リポソーム又は坐薬であってよい。液体形態の場合には、それは、例えば、溶液、乳濁液、懸濁液又はシロップであってよい。この発明の化合物の投与は、例えば、局所経路、経口経路により、筋肉注射、皮下注射により、そして特に、ゼラチンカプセル、錠剤、パッチ又はクリームの形態で行なうことができる。
【0117】
適当な支持体又は賦形剤は、例えば、マルトデキストリン、マンニトール、微結晶性セルロース、ラクトース、トウモロコシ澱粉、澱粉グリコール酸ナトリウム、クロスカルメロースナトリウム、ポリビニル−ポリピロリドン(又はクロスポビドン)、ポリビニルピロリドン、カルボキシメチルセルロース、アルファ澱粉、メチルセルロース、ポリエチレングリコール、マクロゴール、ポリグリコール、ポリオキシエチレン、ポリジオール、ピロリドン−2、コロイドシリカ、タルク、ステアリン酸マグネシウム、ナトリウムステアリルフマレート、ステアリン酸カルシウム、水素化植物油、ラウリル硫酸ナトリウム、リン酸カルシウム、糖類、デキストリン、澱粉、ゼラチン、セルロース、ワックスであってよく、又は水、有機溶媒例えばグリセロール又はグリコール類、それらの割合を変化させた混合物でもよい(水を伴っても伴わなくてもよい)。有効成分に添加されるこれらの支持体は、下記するように役立つ:
− 希釈剤、例えば、マンニトール、ラクトース、澱粉、炭酸カルシウム、微結晶性セルロース、又はマルトデキストリン;
− 崩壊剤、例えば、澱粉、ナトリウムクロスカルメロース、澱粉グリコール酸ナトリウム、又はクロスポビドン;
− 結合剤、例えば、ポリビニルピロリドン、カルボキシメチルセルロース、アルファ澱粉、又はメチルセルロース;
− 流動剤、例えば、コロイドシリカ、又はタルク;
− 潤滑剤、例えば、ステアリン酸マグネシウム、ナトリウムステアリルフマレート、ステアリン酸カルシウム又は水素化植物油;
− 可溶化剤、例えば、ポリエチレングリコール、マクロゴール、ポリグリコール、ポリオキシエチレン、ポリジオール、ピロリドン−2、又はポリビニルピロリドン;
− 界面活性剤(又は、表面活性剤)、例えば、ラウリル硫酸ナトリウム。
【0118】
この発明の組成物は、ゼラチンカプセル中に、次のように配合することができる:5〜30%の有効成分(優先的には、6〜13%);40〜92%の希釈剤(優先的には、65〜92%;非常に優先的には85〜90%);0〜30%の崩壊剤(優先的には、0〜22%;非常に優先的には0%);0〜5%の界面活性剤(優先的には、0%);0〜5%の可溶化剤(優先的には、0%);0.1〜3%の流動剤(優先的には、0.9〜1.4%);0.5〜3%の潤滑剤(優先的には、0.6〜2.8%)。
【0119】
これらのゼラチンカプセルを配合するための好適な賦形剤は、マンニトール、ラクトース、トウモロコシ澱粉、ステアリン酸マグネシウム、及びラウリル硫酸ナトリウムであり、一層特には、マンニトール、ラクトース、コロイドシリカ、及びステアリン酸マグネシウムである。
【0120】
この発明による組成物は又、5〜30%の有効成分(優先的には、7〜15%、非常に優先的には10〜15%);40〜92%の希釈剤(優先的には、34〜89%、非常に優先的には70〜85%);0〜40%の崩壊剤(優先的には、0〜20%、非常に優先的には、3〜5%);0〜8%の結合剤(優先的には、2〜5%);0.1〜3%の流動剤(0.5〜1.4%);0.5〜3%の潤滑剤(優先的には、0.5〜2.8%)を含む錠剤中に配合することもできる。
【0121】
これらの錠剤を配合するのに適した賦形剤は、マルトデキストリン、マンニトール、微結晶性セルロース、ラクトース、トウモロコシ澱粉、澱粉グリコール酸ナトリウム、クロスポビドン、ポリビニルピロリドン、カルボキシメチルセルロース、コロイドシリカ、マグネシウムステアリルフマレート、及びステアリン酸マグネシウムであり、一層特には、微結晶性セルロース、ラクトース、澱粉グリコール酸ナトリウム、ポリビニルピロリドン、コロイドシリカ、及びステアリン酸マグネシウムである。
【0122】
変形物によれば、この発明は、有効成分として、0.1〜20μmの粒子サイズの6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートを、少なくとも一種の製薬上許容しうる支持体と共に含む医薬組成物に関係し;優先的には、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートは、1〜15μmの粒子サイズを有し;一層優先的には、3〜7μmのの粒子サイズを有し、更に優先的には、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートは、5μm±1μmのサイズを有する。
【0123】
優先的には、この医薬組成物は、上記のように、有効成分として、前に規定したように、種類Iの化合物1を含む。
【0124】
非常に優先的には、この医薬組成物は、上で規定したように、ゼラチンカプセルであり;一層優先的には、5〜30%の有効成分;40〜92%の希釈剤;0〜30%の崩壊剤;0〜5%の界面活性剤;0〜5%の可溶化剤;0.1〜3%の流動剤;及び0.5〜3%の潤滑剤を含むゼラチンカプセルでもある。
【0125】
やはり非常に優先的には、この医薬組成物は、上で規定したように、錠剤であり、一層優先的には、5〜30%の有効成分;40〜92%の希釈剤;0〜40%の崩壊剤;0〜8%の結合剤;0.1〜3%の流動剤;及び0.5〜3%の潤滑剤を含む錠剤でもある。
【0126】
化合物1の合成経路は、特許EP880514に記載されたように又はL.W. Woo等、Chemistry & Biology, 2000, 7, 773-91によって、従来技術において構想されている。
【0127】
出願人は、今や、この化合物が2つの化学段階にて合成されうることを見出した。その第一段階は、強酸(例えば、硫酸、トリフルオロ酢酸又はメタンスルホン酸)中での、2−カルベトキシシクロヘプタノンのレゾルシノールとの縮合よりなり、中間体3−ヒドロキシ−8,9,10,11−テトラヒドロシクロヘプタ[c]クロメン−6(7H)−オンが、アルコール/水混合物を用いる(例えば、エタノールを加えてから水を加える)沈澱により単離される。第二段階においては、スルホニルイソシアネートクロリドが、トルエン溶液中で、蟻酸の作用により、変換されて、スルファモイルクロリドとなり、次いで、溶剤例えばDMA中に溶解された前記の中間体と縮合される。この反応媒質を水で処理し、有機溶媒(好ましくは、2−メチルテトラヒドロフラン:MeTHF)により抽出し、次いで、粗化合物1が、貧溶媒(antisolvent)(好ましくは、メチルシクロヘキサン)を添加することにより、有機相から沈澱される。最後に、純粋な化合物1が、アセトン又は酢酸エチルへの高温での溶解及び貧溶媒(好ましくは、メチルシクロヘキサン)添加による沈澱による粗生成物の再結晶化によって得られる。
【0128】
結果として、この発明の主題は又、下記の段階を含むことを特徴とする、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートの製造方法でもある:
− 2−カルベトキシシクロヘプタノンの、強酸中でのレゾルシノールとの縮合、
− こうして得られた3−ヒドロキシ−8,9,10,11−テトラヒドロシクロヘプタ[c]クロメン−6(7H)−オンの、アルコール/水混合物を用いる沈澱による単離、
− 3−ヒドロキシ−8,9,10,11−テトラヒドロシクロヘプタ[c]クロメン−6(7H)−オンの、非プロトン性溶媒中でのクロリドスルファモイルとの縮合、
− 得られた粗生成物の高温アセトン又は酢酸エチル中への溶解及び貧溶媒例えばメチルシクロヘキサンの添加による再結晶。
【0129】
この発明の化合物は、前に記載のように、化合物1から、粒子サイズの低減を目的とした処理によって、種類Iの形態で得られる。
【0130】
従って、この発明の主題は又、上記のこの発明の化合物の、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートからの、粒子サイズの低減の段階を含むことを特徴とする製造方法でもある。
【0131】
優先的には、これらの粒子のサイズは、微粉状にすることにより低減される。
【0132】
やはり優先的には、これらの粒子のサイズは、湿式研削により、有機非プロトン性溶媒を用いて低減される。
【0133】
本発明の主題は又、X線粉末回折を用いて、角度(°2シータ)で表される特徴的なピーク(±0.1°2シータ):8.6;11.3;28.6を示す、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートの多形性化合物でもある(III型の化合物)。好ましくは、この化合物のIII型は、角度(°2シータ)で表される特徴的なピークを示す(±0.1°2シータ):8.6;11.3;12.0;16.6;20.9;23.0;28.6。
【0134】
この発明の一層特別の主題は、上で規定したIII型の化合物であり、それは、単結晶X線回折を利用して、下記のセルパラメーターを示し:
【表1】
下記の換算(reduced)座標(×104)及び同等の等方性の運動パラメーター(Å2×103)を示し:
【表2】
下記の水素原子の座標(×104)及び同等の等方性転置パラメーター(Å2×103)(U(eq)は、直交テンソルUijの値の3分の1に等しい)を示し:
【表3】
下記の格子面間隔を示す:
【表4】
【0135】
優先的には、この発明の主題は、上で規定したような、5℃/分でのDSCを用いて、180℃±2℃の吸熱性溶融ピークを示す化合物(IIII型)である。
【0136】
好ましくは、この発明による化合物(III型)は、上で規定したように、赤外線分光分析を用いて、次のcm-1で表される特徴的なピークを示す(±5cm-1):3406;3217;1678;1011;563;
【0137】
そして、非常に優先的には、次のcm-1で表される特徴的なピークを示す(±5cm-1):3406;3217;3082;2924;1678;1385;1269;1134;1011;934;845;601;563;536。
【0138】
変形物によれば、この発明の主題は、医薬として、上で規定したように、III型の化合物である。
【0139】
この発明の主題は又、有効成分として、上で規定したようなIII型の化合物を、少なくとも一の製薬上許容しうる支持体と共に含む医薬組成物でもある。
【0140】
やはり、この発明の主題は、化合物1の1型の結晶DMSO溶媒和化合物でもあり、これは、X線粉末回折を用いて、角度(°2シータ)で表される特徴的なピークを示す(±0.1°2シータ):6.6;10.9;13.1;14.3;15.7;16.7;17.4;18.3;19.6;20.8;21.9;22.6;23.0;24.7;24.9;25.2;25.5;25.8;26.6;26.9;27.2;28.3。
【0141】
やはり、この発明の主題は、化合物1の3型の結晶DMSO溶媒和化合物でもあり、これは、X線粉末回折を用いて、角度(°2シータ)で表される特徴的なピークを示す(±0.1°2シータ):9.8;13.9;16.0;17.7;19.1;22.1。
【0142】
更に、この発明の主題は又、化合物1の結晶1,4−ジオキサン半溶媒和化合物でもあり、これは、X線粉末回折を用いて、角度(°2シータ)で表される特徴的なピークを示す(±0.1°2シータ):9.8;10.0;10.8;13.6;13.9;14.1;15.9;16.1;18.0;18.3;19.0;19.6;20.0;20.1;20.2;20.6;21.4;21.7;21.8;22.0;22.2;22.3;23.4;23.9;24.3;24.4;24.9;25.3;25.4;26.2;27.0;27.1;27.3;27.5;28.2;28.4;28.5;28.7;29.1;29.4。
【0143】
その上さらに、この発明の主題は、化合物1の多形化合物、II型化合物でもあり、これは、X線粉末回折を用いて、角度(°2シータ)で表される特徴的なピークを示し(±0.1°2シータ):9.4;10.7;12.8;18.2;18.9;19.7;20.4;23.2;優先的には、角度(°2シータ)で表される特徴的なピークを示す(±0.1°2シータ):9.4;10.7;12.2;12.8;15.1;18.2;18.9;19.7;20.4;21.1;22.0;23.2。
【0144】
優先的には、このII型の化合物は、5℃/分でのDSCを用いて、165℃±5℃の吸熱性溶融ピークを示し;一層優先的には、それは、赤外線分光法を用いて、特徴的なピークを示し(cm-1〜±5cm-1で表示):3356;3321;3186;1504;872;787;尚一層優先的には、特徴的なピーク(cm-1〜±5cm-1で表示):3356;3321;3186;3078;2932;2851;1693;1609;1504;1462;1377;1265;1192;1123;937;872;837;787;594を示す。
【0145】
この発明による化合物は、上記のように、化合物1から、更なる段階により得られ、それは、下記のものであってよい:
− 水中の1型のDMSO溶媒和化合物の脱溶媒、
− 水中の3型のDMSO溶媒和化合物の脱溶媒、
− エタノール中での噴霧化、
− アセトン中での噴霧化、
− 還流下でのクメン中での再糊状化、
− 熱処理と、その後の噴霧化と、その後の第二の熱処理、又は
− II型の熱処理。
【0146】
こうして、変形物によれば、この発明の主題は、上で規定したようなIII型の化合物の、化合物1から出発する製造方法であって、下記の方法の一つによる方法である:
− a)上で規定したような1型の結晶DMSO溶媒和化合物へと導くための攪拌及びDMSOからの沈澱(この化合物は、次いで、水に溶解される);
− b)上で規定したような3型の結晶DMSO溶媒和化合物へと導くための攪拌及びDMSOからの沈澱(この化合物は、次いで、水に溶解される);
− c)エタノール中での噴霧化;
− d)アセトン中での噴霧化;
− e)還流下でのクメン中での再糊状化;
− f)155〜165℃の温度での、10〜20分間の熱処理と、その後の噴霧化と、その後の155〜165℃で10〜20分間の第二の熱処理、
− g)上で規定したようなII型の化合物の処理、そして:
− 1,4−ジオキサン中での化合物1の噴霧化により、
− 又は、上で規定したような結晶DMSO半溶媒和化合物へ導くための化合物1の攪拌及び1,4−ジオキサンからの沈澱により得られ、この化合物は、次いで、不活性気流中で5℃/分で20〜80℃に加熱することにより脱溶媒される、
この処理は、155〜165℃で6〜10分間の熱処理よりなる。
【0147】
優先的には、この発明による方法は、上で規定したような1型の結晶DMSO溶媒和化合物の脱溶媒を、水中で完了する。
【0148】
優先的には、この発明による方法は、上で規定したような3型の結晶DMSO溶媒和化合物の脱溶媒を、水中で完了する。
【0149】
やはり優先的には、この発明による方法は、噴霧化を、エタノール中で完了する。
【0150】
やはり優先的には、この発明による方法は、噴霧化を、アセトン中で完了する。
【0151】
非常に優先的には、この発明による方法は、再糊状化を、還流下でクメン中で完了する。
【0152】
尚一層優先的には、この発明による方法は、155〜165℃で10〜20分間の熱処理と、その後の微粉化と、その後の155〜165℃で10〜20分間の第二の熱処理を完了し;
そして優先的には、これらの熱処理は、160℃±1℃で15分間行なわれる。
【0153】
やはり尚一層優先的には、この発明による方法は、上で規定したように、II型の化合物の熱処理を、155〜165℃で6〜10分間で完了し;
そして優先的には、この熱処理は、160℃±1℃で7.5分間行なわれる。
【0154】
やはり好ましくは、このII型の化合物は、上で規定したように、1,4−ジオキサン中での化合物1の噴霧化により得られ;又はII型の化合物は、上で規定したように、結晶1,4−ジオキサン半溶媒和化合物の脱溶媒により得られる。
【0155】
変形物によれば、この発明の主題は、上で規定したような1型の結晶DMSO溶媒和化合物の製造方法であって、化合物1から出発して、攪拌及びDMSOからの沈澱による、当該製造方法である。
【0156】
変形物によれば、この発明の主題は、上で規定したような3型の結晶DMSO溶媒和化合物の製造方法であって、化合物1から出発して、攪拌及びDMSOからの沈澱による、当該製造方法である。
【0157】
変形物によれば、この発明の主題は、上で規定したような結晶1,4−ジオキサン半溶媒和化合物の製造方法であって、化合物1から出発して、攪拌及び1,4−ジオキサンからの沈澱による、当該製造方法である。
【0158】
上記のII型について、それは、2つの合成経路によって得られうる:
− 直接に、1,4−ジオキサン中での噴霧化による
− 予め1,4−ジオキサンからの沈澱により得られた化合物1の1−4−ジオキサン半溶媒和化合物の脱溶媒による。
【0159】
やはり、他の変形によれば、この発明の主題は、上記のII型の化合物の製造方法であって、化合物1から出発し、下記の方法の一つによる、当該製造方法である:
− 1,4−ジオキサン中での噴霧化;
− 上記の結晶1,4−ジオキサン半溶媒和化合物を導くための、攪拌及び1,4−ジオキサンからの沈澱、該化合物は、次いで、不活性気流中での20〜80℃で5℃/分の加熱による脱溶媒に付される。
【0160】
好ましくは、上記のII型の化合物の製造方法は、上記の結晶1,4−ジオキサン半溶媒和化合物の脱溶媒を経る。
【0161】
やはり好ましくは、上記のII型の化合物の製造方法は、1,4−ジオキサン中での噴霧化を経る。
【0162】
或は、この発明の主題は、上で規定した、種類Iの又はIII型の化合物の、癌;優先的にホルモン依存性の癌、やはり優先的には乳癌、前立腺癌、子宮内膜癌又は卵巣癌から選択される癌を治療することを意図した医薬を製造するための利用である。
【0163】
後述の実験部は、上記の手順を説明するために与えられるものであり、この発明の範囲を限定するものと考えるべきではない。
【0164】
別途規定しない限り、本願で用いられる技術的及び科学的用語は、当業者が通常理解するのと同じ意味を有する。その上、すべての特許(又は特許出願)並びに他の参照を、参考として援用する。
【実施例】
【0165】
実験部:
1.この発明による組成物
実施例1a:
錠剤の形態であって、化合物1を有効成分として含む組成物を下記の表1に示した。かかる組成物は、5kgのバッチ及び40mgの投与量につき、下記のダイヤグラムに従って、湿式造粒法により製造することができる。
【0166】
【表5】
【0167】
【表6】
【0168】
40mgの錠剤に塗布されたコーティングは、Opadry(商標)IIホワイト(Colorcon)よりなり;これは、即時放出型錠剤に用いるための市販されているコーティングの混合物である。このコーティングの主な目的は、薬物の悪い味をマスクすることである。それは又、強い有効成分を含む錠剤の包装作業中の取扱いに関連したリスクをも低減させる。
【0169】
実施例1b:
ゼラチンカプセルの形態であって、化合物1を有効成分として含む組成物を下記の表2に示した。
【0170】
【表7】
【0171】
2.崩壊剤の溶解プロファイルに対する効果
すべての賦形剤を、欧州薬局方集成に従って、分析する。
【0172】
崩壊剤の効果は、下記の第三節に記載した組成物2と17の、澱粉グリコール酸ナトリウムなしでの、30分での解散価値(dissolution values)の比較により見えてくる。
【0173】
崩壊剤の添加の、溶解プロファイルに対する効果について、種々の試験が行なわれてきており、錠剤の崩壊時間が研究された。澱粉グリコール酸ナトリウムA型(Explotab(商標))とアルファ化トウモロコシ澱粉(Starch 1500 (商標))の2種の崩壊剤の効果を研究するために、4つの組成の錠剤を生成した。
【0174】
【表8】
【0175】
これらの得られた結果は、崩壊剤の添加が錠剤の溶解プロファイルを加速して、参照用カプセルに近いものとしていることを示している(図2参照)。
【0176】
3.湿式造粒工程中の有効成分に対する結合剤の保護効果
結合剤及び/又は希釈剤(微結晶性セルロース、コポビドン、カルボキシメチルセルロース)の保護効果を示す研究が、最終的な不純物のレベル並びにこれらの配合物の製造中の挙動の比較により行なわれた。湿式造粒法により得られた種々の固体組成物を、Waters社製の2487 UV 検出器を有するAlliance 2695システムにて、下記の表(表4)に記載された条件にしたがってHPLC(高性能液体クロマトグラフィー)により分析した。
【0177】
【表9】
【0178】
【表10】
【0179】
4.ブリスターパック内における6カ月及び12カ月での、貯蔵条件の関数としての、5mg錠剤及び40mg錠剤の安定性
ある錠剤形態(実施例1aの百分法配合)は、一次包装後にブリスターパック内で安定であった。
【0180】
この目的は、この形態の、6カ月及び12カ月の期間にわたる安定性をチェックすることである。
【0181】
推奨される通常の貯蔵条件は、25℃及び60%相対湿度(25℃/60%RH)であり、これらの条件下で生成されたデータを、それぞれ、5mg錠剤及び40mg錠剤について、表6及び7に示してある。
【0182】
更に、他の貯蔵条件(例えば、40℃/75%RH)も又、6カ月安定性期間の最後まで調べて、生成されたデータを、それぞれ、5mg錠剤及び40mg錠剤について、表8及び9に示した。
【0183】
【表11】
【0184】
表6は、複雑なアッセイを規定しており、不純物、及び5mgの有効成分を含み、ブリスターパックに包装されて、25℃/60%RHで貯蔵された錠剤の安定性研究からの溶解試験結果を与えるものである。
【0185】
【表12】
【0186】
表7は、複雑なアッセイを規定し、不純物、及び40mgの有効成分を含み、ブリスターパックに包装されて、25℃/60%RHで貯蔵された錠剤の安定性研究からの溶解試験結果を与えるものである。
【0187】
【表13】
【0188】
表8は、複雑なアッセイを規定しており、不純物、及び5mgの有効成分を含み、ブリスターパックに包装されて、種々の貯蔵条件下で貯蔵された錠剤の安定性研究からの溶解試験結果を与えるものである。
【0189】
【表14】
【0190】
表9において、アッセイ、不純物及び、40mgの有効成分を含みブリスターパックに包装されて種々の貯蔵条件化で貯蔵された錠剤の安定性研究の溶解の試験が一緒に分類されている。
【0191】
25℃/60%RHでの12カ月の貯蔵の後、このアッセイ及び溶解について、何らの有意の変化は認められなかった。
【0192】
95%の信頼区間での回帰線により得られる結果の統計的分析は、少なくとも3年間の製品品質規格との一致を約束する。
【0193】
40℃の温度及び75%相対湿度(40℃/75%RH)での促進された安定性は、製品品質規格と一致して結果を示す。
【0194】
最も厳しい条件下での製品の安定性は、25℃/60%RHで貯蔵された錠剤につき行なわれた計画において信頼を強化する。
【0195】
5.化合物1種類Iの製造
5.1 化合物1の合成
実施例5: 第一段階は、メタンスルホン酸中での、2−カルベトキシシクロヘプタノンのレゾルシノールとの縮合からなり、この反応は、4時間にわたって、25℃に達する。こうして形成された中間体の3−ヒドロキシ−8,9,10,11−テトラヒドロシクロヘプタ[c]クロメン−6(7H)−オンは、エタノールを添加してから水を添加することにより沈澱し、それは、濾過により単離され、真空下で60℃で乾燥されて、78%の収率が得られる。第二段階において、スルホニルイソシアネートクロリドが、トルエン溶液中で、蟻酸の作用によって、スルファモイルクロリドに転化され、次いで、N,N−ジメチルアセタミド(DMA)中に溶解された前記中間体と縮合される。この反応媒質を、水で処理して、2−メチルテトラヒドロフラン(2−MeTHF)で抽出する。次いで、有機相にメチルシクロヘキサンを加えることにより得られた沈澱の濾過により、粗化合物1が得られる。最後に、純水な化合物1が、熱いアセトンに溶解させてメチルシクロヘキサンを加えることによる沈澱による、粗生成物の再結晶により得られる(貧溶媒メチルシクロヘキサンの添加は、収率の増大を主な目的としている)。
【0196】
化合物1は、こうして、収率65%で得られ、以下に供せられる。
【0197】
こうして得られた実施例1を、粒子サイズの低減を目的とした適当な処理例えば微粉化(ポイント5.3未満)又は湿式粉砕(ポイント5未満)にかけることができる。
【0198】
5.2 水中の化合物1の2型のDMSO溶媒和化合物の脱溶媒
実施例5a:化合物1の2型のDMSO溶媒和化合物
1mLのDMSO(沸点=189℃)を丸薬容器に注ぎ、次いで、1gの化合物1(実施例1)を加える。その溶液を、マグネチックスターラーバー及びマグネチックスターラーを用いて攪拌する。固体を溶液中に急速に通す。1gの化合物1を、再び、マグネチックスターラーで攪拌しながら加える。固体が部分的に溶解してから、ケーキングが認められる。このケーキングの試料を、まだ湿っている間に、X線粉末回折により分析する。この分析は、以下で与えるが、これが化合物1の2型のDMSO溶媒和化合物であることを示す(実施例5a)。
【0199】
実施例5b:
この化合物1の2型のDMSO溶媒和化合物(実施例5b)を冷水に浸し、周囲温度で5分間にわたって攪拌し続ける。その懸濁液を、次いで、濾過して乾燥させる。
【0200】
こうして、化合物1が、50%の収率で得られ、以下に供せられる。
【0201】
こうして得られた実施例5bを、粒子サイズの低減を目的とする適当な処理例えば微粉化又は湿式粉砕にかけることができる。
【0202】
5.3 微粉化
微粉化は、エアジェット超微粉砕機を利用して、18〜25℃の温度で行なわれる。使用する圧搾空気の特性は、以下の通りである:
− CO:<5 ppm、
− CO2:<500 ppm、
− 炭水化物:<0.5 mg.m-3、
− 1立方フィート(0.3048m)当りの粒子数>0.5μ:<10000、
− 最大性能:13バール(13×106ダイン/cm2)で、2300 cfm(立方フィート(0.3048m)/分)、
− 最大運転作業圧:13バール(106ダイン/cm2)。
【0203】
実施例5c:粒子サイズ3μmを有する化合物1の種類I
249gの化合物1(実施例1)を、エアジェット超微粉砕機を利用して微粉化する。この微粉化のパラメーターは、80psi(0.0703kg/cm2)のベンチュリ圧、110psi(0.0703kg/cm2)の超微粉砕機圧及び12kg/時の供給速度である。
【0204】
実施例3−1は、こうして、97%の収率で得られる。
【0205】
実施例5d:粒子サイズ5μmを有する化合物1の種類I
16.5kgの化合物1(実施例5)を、圧搾エアジェット超微粉砕機を用いて微粉化する。微粉化パラメーターは、80psi(0.0703kg/cm2)のベンチュリ圧、32psi(0.0703kg/cm2)の超微粉砕機圧及び14.4kg/時の供給速度である。
【0206】
実施例5dは、こうして、98%の収率で得られて、以下に供される。
【0207】
実施例5e:粒子サイズ9μmを有する化合物1の種類I
150gの化合物1(実施例51)を、圧搾エアジェット超微粉砕機を用いて微粉化する。微粉化パラメーターは、50psi(0.0703kg/cm2)のベンチュリ圧、60psi(0.0703kg/cm2)の超微粉砕機圧及び15kg/時の供給速度である。
【0208】
実施例5eは、こうして、98%の収率で得られる。
【0209】
実施例5f:粒子サイズ15μmを有する化合物1の種類I
39gの化合物1(実施例5)を、圧搾エアジェット超微粉砕機を用いて微粉化する。微粉化パラメーターは、30psi(0.0703kg/cm2)のベンチュリ圧、10psi(0.0703kg/cm2)の超微粉砕機圧及び15分間に40gの供給速度である。
【0210】
実施例5fは、こうして、74%の収率で得られる。
【0211】
5.4 湿式粉砕
実施例5g:0.69gの化合物1(実施例5)を、0.068gのメチルシクロヘキサンと共に、Fritsch型遊星モデルP4ボールミル中で粉砕する。
【0212】
湿式粉砕のパラメーターは、直径10mmの7個のめのうボール、100−100rpmの粉砕トルクΩ(ディスクの回転速度)−ω(フラスコの回転速度)、72回の「10分間の粉砕−5分間の休止」シーケンスに対応する全18時間中での有効な12時間、14.1のボールの質量/溶質質量比;22℃の温度及び9%の化合物1の質量/メチルシクロヘキサン質量比である。
【0213】
実施例3は、こうして、収率99%で得られて、以下に供される。
【0214】
6.得られた結晶の説明
6.1 用いた装置
6.1.1 X線粉末及び単結晶回折
ジーメンスD5005回折計、シンチレーション検出器
・ 波長:1.54056 Cu,電圧40KV、強度40mA
・ 測定レンジ:3°−30°2シータ
・ 間隔:0.04°2シータ
・ 間隔の持続期間:4秒
・ 固定スロット:1.6mm
・ Kβフィルター(Ni)
・ 内部参照なし
・ データ処理のためのEVAソフトウェア(v12.0)
【0215】
Smart Apex Bruker回折計、2次元検出器
・ 結晶マトリクスのパラメーター及び配向の決定のためのSMARTソフトウェア
・ データ積分及び処理のためのSAINTソフトウェア
・ 空間群の決定及び構造分解能のためのWinGXソフトウェア
【0216】
6.1.2 DSC
・ Netzsch DSC 204F1
・ アルミニウムるつぼ及び穴をあけたふた
・ 大気:ヘリウム
・ 初期温度:20℃
・ 最終温度:200℃
・ 温度勾配:5℃/分
【0217】
6.1.3 IR
・ KBrペレット
・ Bruker IFS28型分光計
・ スペクトル範囲:400〜4000cm-1
【0218】
6.1.4 固体のNMR
・ Bruker Avance 500 MHz分光計
・ MAS(マジック角回転)4mmプローブ
・ Bruker XwinNMRソフトウェア
・ VACP(可変振幅直交偏波)及びMAS(11kHz)及びプロトン分離(スピナル64、65kHz)
・ 外部参照:アダマンタン
【0219】
6.1.5 粒子サイズの分布
・ Malvern Mastersizer Sレーザー粒度計
・ 試料のサイズ:30〜50mg
・ 分散媒:0.50%(m/v)Nonidet(水中)
・ 相対的インデックスとして:粒子RI=1.55;虚のRI=1.00を用いるMie理論
・ 超音波処理時間:60秒
・ 超音波処理エネルギー:50〜60Hz
・ 再循環時間:30秒/2000rpm
・ あいまい化:15〜25%
【0220】
6.2. 実施例の特性表示
6.2.1 5及び5bの実施例
単結晶を、エタノール中の飽和溶液の4℃で2日間にわたる、ゆっくりとした蒸発によって単離することは、可能となっている。これらの単結晶は、これらの単結晶は、、化合物1の種類Iの完全な構造を、X線回折により明らかにすることを可能にした。
【0221】
X線による単結晶の結晶回折:化合物1種類Iの結晶構造は、解明されており、下記のセルパラメーターに示してある:
【表15】
【0222】
化合物1の種類Iの換算座標(×104)及び同等の等方性位置パラメーター(Å2×103)は、次の通りである:
【表16】
【0223】
化合物1の種類Iの換算座標(×104)は、次の通りである:
【表17】
【0224】
化合物1の種類Iの格子面間隔は、次の通りである:
【表18】
【0225】
6.2.2 実施例5a
単結晶を、周囲温度で、DMSOの飽和溶液からのゆっくりとした蒸発によって単離することは可能であった。これらの単結晶は、化合物1の2型のDMSO溶媒和化合物の完全な構造をX線回折によって解明することを可能にした。
【0226】
単結晶X線回折:化合物1の2型のDMSO溶媒和化合物の結晶構造を解明し、セルパラメーターを下記に示した:
【表19】
【0227】
この化合物1の2型のDMSO溶媒和化合物のセル中の原子の原子座標(×104)及び同等の等方性転置パラメーター(Å2×103)(U(eq)は、直交テンソルUijの値の3分の1に等しい)は、下記の通りである:
【表20】
【0228】
化合物1の2型のDMSO溶媒和化合物の水素原子の座標(×104)及び同等の等方性転置パラメーター(Å2×103)(U(eq)は、直交テンソルUijの値の3分の1に等しい)は、下記の通りである:
【表21】
【0229】
化合物1の2型のDMSO溶媒和化合物の格子面間隔は、下記の通りである:
【表22】
【0230】
化合物1の2型のDMSO溶媒和化合物のX線粉末回折ダイヤグラムは、ある角度(°2シータ)で表される次の特徴的ピークを示す(約±0.1°2シータ):(図3及び4)。7.6;18.7;24.2;29.9。
【0231】
6.2.3 実施例5c − 5g
6.2.3.1 X線粉末回折(図5及び6)
微粉化後又は湿式粉砕後の化合物1の種類IのX線粉末回折ダイヤグラムは、次の角度(°2シータ)で表される特徴的なピークを示す(約±0.1°2シータ):7.5;10.9;11.7;13.1;14.6;15.0;15.8;17.0;17.3;17.5;17.7;17.9;19.0;19.9;20.0;21.4;21.8;22.5;23.5;23.9;24.7;24.9;25.5;25.9;26.2;26.4;26.6;27.2;27.4;27.6;27.8;28.2;28.8;29.0;29.4;29.8。
【0232】
微粉化又は湿式粉砕後に160℃に加熱した後の化合物1の種類IのX線粉末回折ダイヤグラムは、次の角度(°2シータ)で表される特徴的なピークを示し(約±0.1°2シータ):7.5;10.9;11.7;13.1;14.6;15.0;15.8;17.0;17.3;17.5;17.7;17.9;19.0;19.9;20.0;21.4;21.8;22.5;23.5;23.9;24.7;24.9;25.5;25.9;26.2;26.4;26.6;27.2;27.4;27.6;27.8;28.2;28.8;29.0;29.4;29.8、160℃までの加熱の際に生成された新規な形態に対応する更なるピークはない。
【0233】
6.2.3.2 DSC(図7)
DSCを利用して、微粉化又は湿式粉砕後の化合物1の種類Iの、5℃/分で得られたサーモグラムは、種類Iの溶融ピーク(170℃±5℃で開始)及び、適宜、分析中(180℃±2℃で開始)に生成される新規な多形の小さい溶融ピークを含み、それは、種類Iの溶融ピーク中に交換されるエンタルピーの10%未満を表し、140〜155℃では如何なる吸熱事象(又は、0.5J/g未満)をも示さない。
【0234】
6.2.3.3 IR(図8)
微粉化又は湿式粉砕後の化合物1の種類IのIRスペクトルは、次の特徴的なピークを示す(ほぼ±5cm-1で、cm-1で表示):3310;3167;3059;2928;2858;1690;1605;1454;1385;1261;1188;1126;941;891;853;798;733;679;598;544;455。
【0235】
6.2.3.4 固体のNMR(図9)
微粉化又は湿式粉砕後の化合物1の種類Iの、固体のNMRにより得られるスペクトルは、次の特徴的なピークを示す(ほぼ±0.2ppmまでのppmで表示)163.1;156.1;153.8;153.1;130.4;127.6;107.6;35;25。
【0236】
6.2.3.5 粒子サイズ
実施例2−2による微粉化後の化合物1の種類Iの粒子サイズ分布は、次の通りである:D10(%)=1.2μm;D50(%)=5.2μm及びD90(%)=11.0μm。
【0237】
実施例3による湿式粉砕後の化合物1の種類Iの粒子サイズ分布は、次の通りである:D10(%)=0.6μm;D50(%)=3.1μm及びD90(%)=13.7μm。
【0238】
7.配合物
7.1 ゼラチンカプセル形態の配合物
この発明による化合物1の種類Iは、5〜30%の有効成分(好ましくは、6〜13%)、40〜92%の希釈剤(好ましくは、65〜92%、非常に好ましくは85〜90%)、0〜30%の崩壊剤(好ましくは、0〜22%、非常に好ましくは、0%)、0〜5%の界面活性剤(好ましくは、0%)、0〜5%の可溶化剤(好ましくは、0%)、0.1〜3%の流動剤(好ましくは、0.9〜1.4%)、0.5〜3%の潤滑剤(好ましくは、0.6〜2.8%)を含むゼラチンカプセルに配合することができる。
【0239】
これらのゼラチンカプセルを配合するための好適な賦形剤は、マンニトール、ラクトース、トウモロコシ澱粉、コロイドシリカ、ステアリン酸マグネシウム、及びラウリル硫酸ナトリウムであり、一層特には、マンニトール、ラクトース、コロイドシリカ、及びステアリン酸マグネシウムである。
【0240】
下記のゼラチンカプセルを、粉末を、当業者に公知の標準的技術によって混合することにより作成した。
【表23】
M=マンニトール; L=ラクトース; CS=トウモロコシ澱粉; SLS=ラウリル硫酸ナトリウム; PEG=ポリエチレングリコール; CS=コロイドシリカ; MS=ステアリン酸マグネシウム
【0241】
実施例7j:ゼラチンカプセル7jの実施例は、実施例5(40mgの化合物1を260mgのラクトースと混合して、ゼラチンカプセルに入れる)の化合物1から製造される。
【0242】
実施例7k:ゼラチンカプセル7kの実施例は、有効成分として実施例5dの代わりに実施例5cを用いる配合7gにより製造される。
【0243】
実施例7l:ゼラチンカプセル7lの実施例は、7.2%の実施例5d、31.9%のマンニトール、59.4%のラクトース及び1.5%のコロイドシリカを用いて製造される。
【0244】
実施例7m:ゼラチンカプセル7mの実施例は、配合7gにより、実施例5dを有効成分として用いて製造される。
【0245】
実施例7n:ゼラチンカプセル4nの実施例は、配合4gにより、実施例5eを実施例5dの代わりに有効成分として用いて製造される。
【0246】
実施例7o:ゼラチンカプセル4oの実施例は、有効成分として、実施例5f(40mgの実施例5fを、260mgのラクトースと混合して、ゼラチンカプセル内に入れる)を用いて製造される。
【0247】
7.2 錠剤の形態の配合物
この発明による化合物1の種類Iは、以下を含む錠剤に配合することができる:5〜30%の有効成分(好ましくは、7〜20%、非常に好ましくは、10〜15%)、40〜92%の希釈剤(好ましくは、34〜89%、非常に好ましくは、70〜85%)、0〜40%の崩壊剤(好ましくは、0〜20%、非常に好ましくは、3〜5%)、0〜8%の結合剤(好ましくは、2〜5%)、0.1〜3%の流動剤(好ましくは、0.5〜1.4%)、0.5〜3%の潤滑剤(好ましくは、0.5〜2.8%)。
【0248】
これらのゼラチンカプセルの配合のための好適な賦形剤は、マルトデキストリン、マンニトール、微結晶性セルロース、ラクトース、トウモロコシ澱粉、澱粉グリコール酸ナトリウム、クロスポビドン、ポリビニルピロリドン、カルボキシメチルセルロース、コロイドシリカ、フマル酸ステアリルマグネシウム、及びステアリン酸マグネシウム、一層特には、微結晶性セルロース、ラクトース、澱粉グリコール酸ナトリウム、ポリビニルピロリドン、コロイドシリカ、及びステアリン酸マグネシウムである。
【0249】
下記の錠剤は、当業者に公知の標準的技術により、湿式造粒法によって製造したものである。
【表24】
【0250】
8.物理化学的及び生物学的特性
8.1 溶解速度論
溶解速度論(時間の関数として溶解した化合物1のパーセンテージとして表示)は、当業者に公知の標準的技術によって測定され、下記の表に与えてある。
【表25】
【0251】
上記の溶解結果は、理論強度のパーセンテージとして表されている。ゼラチンカプセル4j〜4nの理論強度は、有効成分の40mgである。これらの実施例は、実験的なものであり、有効成分の重量測定時の僅かな過多はありうることであり、最終溶解点における100%より大きいパーセンテージを説明する。
【0252】
8.2 生物学的利用能
化合物1の比較用の生物学的利用能を、イヌにおいて、2つのゼラチンカプセルの経口経路による単一投与後に研究した。投与後、1;1.5;2;4;6;9;12;24;30及び48時間で、血液試料を採取する。曲線下領域(AUC)は、これらのイヌからの血漿試料において測定される薬物速度論的パラメーターの一つである。得られた結果を下記の表に与える。
【表26】
【0253】
8.3 化学的安定性
安定性の研究を100℃で、化合物1のI型と特許EP880514に記載された化合物1との間で行なった。化合物1の化学的安定性は、異なるスコアのHPLCを用いて研究される。
【0254】
HPLC法の操作条件は、次の通りである:
・ カラム:Interchim UP3HDO-15XS, 150×4.6mm、
・ 溶離剤A: −水 2500
−トリフルオロ酢酸 0.5
・ 溶離剤B: −アセトニトリル
・ 勾配:
【表27】
・ 検出:205nm、
・ 注入:20マイクロリットル、
・ 温度:40℃、
・ 注入した溶液:0.5mg.mL-1(アセトニトリル)
【表28】
【0255】
9.III型の化合物1の製造
本願に記載したように化合物1のIII型は、化合物1(実施例5)の合成の段階と、その後の更なる段階により得られ、それは、下記であってよい:
− 水中での1型のDMSO溶媒和化合物の脱溶媒:実施例9a、
− 水中での3型のDMSO溶媒和化合物の脱溶媒:実施例9b、
− エタノール中での微粒化:実施例9c、
− アセトン中での微粒化:実施例9d、
− クメン中での、還流下での再糊状化:実施例9e、
− 熱処理と、その後の微粒化と、その後の第二の熱処理:実施例9f、
− II型の熱処理:実施例9g。
【0256】
9.1 1型のDMSO溶媒和化合物の水中での脱溶媒
実施例9aa:化合物1の1型のDMSO溶媒和化合物
1mLのDMSO(沸点=189℃)を、丸薬容器に注ぎ、次いで、1gの化合物1(実施例5)を加える。この溶液を、マグネチックスターラーバー及びマグネチックスターラーを用いて攪拌する。この固体は、溶液中へと急速に消え去る。1gの化合物1を、再び、やはりマグネチックスターラーで攪拌しながら加える。この固体は、部分的に溶解し、その後、ケーキングが認められる。このケーキングの試料を、未だ湿気があるうちに、X線粉末回折により分析する。この分析は、以下に与えるが、これが化合物1の1型のDMSO溶媒和化合物(実施例9aa)であることを示す。
【0257】
実施例9a:
この化合物1の2型のDMSO溶媒和化合物(実施例9aa)を、冷水に浸し、攪拌下に、周囲温度で5分間置く。その懸濁液を、次いで、濾過して乾燥する。
こうして、III型の化合物1は、収率50%で得られ、以下に供せられる。
【0258】
9.2 3型のDMSO溶媒和化合物の水中での脱溶媒
実施例9ba:化合物1の3型のDMSO溶媒和化合物
1mLのDMSO(沸点=189℃)を、丸薬容器に注ぎ、次いで、1gの化合物1(実施例1)を加える。この溶液を、マグネチックスターラーバー及びマグネチックスターラーを用いて攪拌する。固体は、急速に、溶液中に消え去る。この溶液を、マグネチックスターラーによる攪拌下に24時間放置する。次いで、ケーキングが認められる。ケーキングの試料を、未だ湿気のあるうちに、X線粉末回折により分析する。この分析は、以下で与えるが、これが化合物1の3型のDMSO溶媒和化合物(実施例9ba)であることを示す。
【0259】
実施例9b:
化合物1の3型のDMSO溶媒和化合物(実施例3ba)を、冷水に浸し、攪拌しながら周囲温度で5分間放置する。次いで、上清を濾過して乾燥させる。
こうして、III型の化合物1が、収率50%で、得られ、以下に供せられる。
【0260】
9.3 エタノール中での微粒化
実施例9c:
得られた生成物は、Buchi 190 スプレードライヤーを用いて生成される。2gの化合物1(実施例5)を、200mLのエタノール(沸点=78℃)に溶解させる。乾燥空気入口温度は、3バール(3×106ダイン/cm2)の圧力で、90℃に調節される。出口温度は、微粒化中、38℃で測定される。出口での流量は、700Nl/時に調節される。570mgの乾燥粉末が回収され、X線粉末回折により分析される。この分析は、これが、化合物1のIII型であり、それが、構造的に純粋であることを、解明された単結晶構造から計算されたダイヤグラムとの比較により示す。
こうして、III型の化合物1は、収率29%で得られ、以下に供せられる。
【0261】
9.4 アセトン中での微粒化
実施例9d:
得られた生成物は、不活性ループを含むBuchi 290 スプレードライヤーを用いて生成される。2gの化合物1(実施例1)を100mLのアセトン(沸点=56℃)に溶解させる。乾燥空気入口温度を、70℃に調節する。出口温度は、微粒化中、50℃にて測定する。出口流量は、7600Nl/時に調節する。1gの乾燥粉末を、回収してX線粉末回折により分析する。この分析は、これが、化合物1のIII型であり、それが、構造的に純粋であることを、解明された単結晶構造から計算されたダイヤグラムとの比較により示す。
こうして、III型の化合物1が、収率50%で得られ、以下に供せられる。
【0262】
9.5 還流下でのクメン中での再糊状化
実施例9e:
10gの化合物1(実施例5)を、100mLのクメン(沸点=152℃)中に懸濁させる。この混合物を、攪拌下に、還流させ、次いで、周囲温度まで冷却する。回収されて乾燥された粉末の分析は、それが、化合物1のIII型であることを示す。
こうして、III型の化合物1が、収率99%で得られ、以下に供せられる。
【0263】
9.6 熱処理と、その後の微粉化と、その後の第二の熱処理
実施例9f:
150gの化合物1を、通風式オーブン中で、160℃で15分間加熱する。そうして加熱した生成物を、次いで、圧搾エアジェット超微粉砕機を用いて微粉化する。微粉化パラメーターは、第一の関門としての、80psi(5.624kg/cm2)のベンチュリ圧、120psi(8.436kg/cm2)の超微粉砕機の圧力及び1.2kg/時の供給速度及び第二の関門としての、50psi(3.515kg/cm2)のベンチュリ圧、50psi(3.515kg/cm2)の超微粉砕機の圧力及び1.2kg/時の供給速度である。微粉化の収率は、69%である。こうして得られた15gの生成物を、160℃で15分間、通風式オーブン中で加熱する。
こうして、III型の化合物1が、収率69%で得られ、以下に供せられる。
【0264】
9.7 II型の熱処理
9.7.1 II型の獲得
化合物1のII型は、2つの合成経路によって得ることができる:
− 予め1,4−ジオキサンからの沈澱により得られた化合物1の1,4−ジオキサン半溶媒和化合物の脱溶媒;
− 1,4−ジオキサンにおける直接的微粉化。
【0265】
実施例9h:化合物1の1,4−ジオキサン半溶媒和化合物
1,4−ジオキサン中の化合物1(実施例5)の溶液を、周囲温度で調製し、攪拌下に、24時間置いた。攪拌中、有意の沈澱が認められた。次いで、その固体を濾過して、X線粉末回折により分析した。この段階の最後で得られた生成物は、化合物1の1,4−ジオキサン半溶媒和化合物である。
こうして、化合物1の1,4−ジオキサン半溶媒和化合物が、収率80%で得られ、以下に供せられる。
【0266】
実施例9i:II型の化合物1
第一の合成経路に従って、化合物1(実施例9h)の1,4−ジオキサン半溶媒和化合物を、不活性ガス流中で、20℃から80℃まで、5℃/分で加熱し、脱溶媒により、化合物1のII型を生成する(X線回折パターンにより特性決定)。
こうして、II型の化合物1が、収率99%で得られ、以下に供せられる。
【0267】
第二の合成経路に従って、II型の化合物1を次のように得ることができる:1gの化合物1(実施例5)を100mLの1,4−ジオキサン(沸点=101℃)に溶解させた。入口温度:無塵の乾燥空気を、3バール(3×106ダイン/cm2)の圧力で、130℃に調節した。出口温度を、微粉化中、88℃で測定した。出口流量を、700Nl/時に調節した。この段階の最後に得られた生成物は、II型である。
こうして、II型の化合物1が、収率50%で得られ、以下に供せられる。
【0268】
9.7.2 III型の獲得
実施例9g:
II型の化合物1(実施例9i)の回収された乾燥粉末を、160℃のオーブン中に、6〜10分間、好ましくは7.5分間置いた。この処理の後に回収され、X線粉末回折により分析された粉末は、化合物1のIII型であり、これは、解明された単結晶構造から得られる計算されたダイヤグラムとの比較により構造的に純粋である。
こうして、III型の化合物1が、収率99%で得られ、以下に供せられる。
【0269】
10.得られた固相の特性決定
10.1 使用する機器
10.1.1 X線粉末回折及び単結晶回折
シーメンスD5005回折計、シンチレーション検出器
− 波長:1.54056 Cu、電圧 40 KV、電流の強さ 40 mA
− 測定レンジ:3° - 30°2シータ
− 0.04°2シータ
− 間隔:0.04°2シータ
− 間隔の持続時間:4秒
− 固定スロット:1.6mm
− Kβフィルター(Ni)
− 内部標準なし
− データ処理用EVAソフトウェア(v12.0)
【0270】
Smart Apex Bruker回折計、二次元検出器
− 結晶マトリクスのパラメーター及び配向の決定のためのSMARTソフトウェア
− データの積分及び処理のためのSAINTソフトウェア
− 空間群の決定及び構造解明のためのWinGXソフトウェア
【0271】
10.1.2 DSC
− NetzschDSC204F1
− アルミニウム製るつぼ及び穴の開いた蓋
− 大気:ヘリウム
− 初期温度:20℃
− 最終温度:200℃
− 温度勾配:5℃/分
【0272】
10.1.3 TG−DSC
− NetzschSTA449C
− アルミニウム製るつぼ及び穴の開いた蓋
− 大気:ヘリウム
− 初期温度:25℃
− 最終温度:200℃
− 温度勾配:5℃/分
【0273】
10.1.4 IR
− KBrペレット
− Bruker IFS28型分光計
− スペクトル範囲:400〜4000cm-1
【0274】
10.1.5 固体のNMR
− Bruker Avance 500MHz分光計
− MAS(Magic Angle Spinning) 4mmプローブ
− Bruker XwinNMRソフトウェア
− VACP(Variable Amplitude Cross-Polarization)及びMAS(11kHz)及びプロトン分離(スピナル64、65kHz)
− 外部標準:アダマンタン
【0275】
10.2 これらの実施例の特性決定
10.2.1 化合物1のI型
化合物1のI型のX線粉末回折ダイヤグラムは、角度(°2シータ)で表される次の特徴的なピークを示す(約±0.1°2シータ):7.5;10.9;11.7;13.1;14.6;15.0;15.8;17.0;17.3;17.5;17.7;17.9;19.0;19.9;20.0;21.4;21.8;22.5;23.5;23.9;24.7;24.9;25.5;25.9;26.2;26.4;26.6;27.2;27.4;27.6;27.8;28.2;28.8;29.0;29.4;29.8。
【0276】
10.2.2 化合物1の1型のDMSO溶媒和化合物
単結晶を、DMSOの飽和溶液を周囲温度でゆっくり蒸発させることによって単離することは可能であった。これらの単結晶は、化合物1の1型のDMSO溶媒和化合物の完全な構造をX線回折により解明することを可能にした。
【0277】
単結晶のX線回折:化合物1の1型のDMSO溶媒和化合物の結晶構造は、下記のセルパラメーターを示す。
【表29】
【0278】
セル中の原子(×104)の原子座標及び同等の等方性置換パラメーター(Å2×103)(U(eq)は、直交テンソルUijの値の3分の1に等しい)は、下記の通りである:
【表30】
【0279】
これらの水素原子(×104)の座標及び同等の等方性置換パラメーター(Å2×103)(U(eq)は、直交テンソルUijの値の3分の1に等しい)は、下記の通りである:
【表31】
【0280】
DMSO溶媒和化合物1型の格子面間隔は、下記の通りである:
【表32】
【0281】
化合物1の1型のDMSO溶媒和化合物のX線粉末回折ダイヤグラムは、角度(°2シータ)で表される下記の特徴的ピークを示す(約±0.1°2シータ):6.6;10.9;13.1;14.3;15.7;16.7;17.4;18.3;19.6;20.8;21.9;22.6;23.0;24.7;24.9;25.2;25.5;25.8;26.6;26.9;27.2;28.3(図1及び2)。
【0282】
2.2.3 化合物1の3型のDMSO溶媒和物化合物
単結晶を、DMSOの飽和溶液を周囲温度でゆっくりと蒸発させることにより単離することが可能であった。これらの単結晶は、化合物1の3型のDMSO溶媒和化合物を、X線回折により、完全な構造を解明することを可能にした。
【0283】
単結晶のX線回折:化合物1の3型のDMSO溶媒和化合物の結晶構造を、解明して、下記のセルパラメーターを示す:
【表33】
【0284】
これらの水素原子(×104)の座標及び同等の等方性置換パラメーター(Å2×103)(U(eq)は、直交テンソルUijの値の3分の1に等しい)は、下記の通りである:
【表34】
【0285】
これらの水素原子(×104)の座標及び同等の等方性置換パラメーター(Å2×103)(U(eq)は、直交テンソルUijの値の3分の1に等しい)は、下記の通りである:
【表35】
【0286】
DMSO溶媒和化合物3型の格子面間隔は、次の通りである:
【表36】
【0287】
化合物1の3型のDMSO溶媒和化合物のX線粉末回折ダイヤグラムは、角度(°2シータ)で表される下記の特徴的ピークを示す(約±0.1°2シータ):(図3及び4)。9.7;13.9;16.0;17.8;19.1;22.1。
【0288】
2.2.4 III型の単結晶のX線回折
アセトン/n−ヘプタン混合物(50% v/v)中の化合物1の飽和溶液を周囲温度でゆっくり蒸発させることにより、X線回折により化合物1のIII型の完全な構造を解明することを可能にした単結晶を単離することが可能であった。
【0289】
単結晶X線回折:III型の化合物1の結晶構造を解明して、下記のセルパラメーターを示す。
【表37】
【0290】
III型の化合物1の換算座標(×104)及び同等の等方性位置パラメーター(Å2×103)は、次の通りである:
【表38】
【0291】
これらの水素原子の座標(×104)及び同等の等方性転置パラメーター(Å2×103)(U(eq)は、直交テンソルUijの値の3分の1に等しい)は、下記の通りである:
【表39】
【0292】
格子面間隔は、下記の通りである:
【表40】
【0293】
10.2.5 実施例9a〜9g:III型の化合物1
この固体のX線粉末回折、単結晶X線回折、DSC、IR及びNMRによる分析は、実施例9aが実施例9b、実施例9c、実施例9d、実施例9e、実施例9f及び実施例9gと等しいことを示している。
【0294】
10.2.5.1 X線粉末回折
化合物1のIII型のX線粉末回折は、角度(°2シータ)で表される次の特徴的ピークを示す(約±0.1°2シータ):8.6;11.3;12.0;16.7;17.4;17.9;20.9;22.6;23.0;23.4;23.8;25.7;28.6(図5及び6)。
【0295】
10.2.5.2 DSC
DSCにおいて、化合物1のIII型のサーモグラムは、III型の溶融ピーク(180℃±2℃で開始)を含む(図7)。
【0296】
10.2.5.3 IR
化合物1のIII型のIRスペクトルは、cm-1で現される特徴的ピークを示す(約±5cm-1):3406;3217;3082;2924;1678;1385;1269;1134;1011;934;845;601;563;536(図8)。
【0297】
10.2.5.4 固体のNMR
この固体のNMRにより得られたIII型のスペクトルは、次のppmで表される特徴的ピークを示す(約±0.2ppm):162.9;156.4;151.3;126.1;124.7;118.8;117.1;112.9;35;20(図9)。
【0298】
10.2.6 実施例9h:化合物1の1,4−ジオキサン半溶媒和化合物
10.2.6.1 単結晶のX線回折
1,4−ジオキサン中の化合物1の飽和溶液を周囲温度でゆっくりと蒸発させることにより、化合物1の1,4−ジオキサン半溶媒和化合物の完全な構造をX線回折によって解明することを可能にする単結晶を単離することが可能であった。
【0299】
化合物1の1,4−ジオキサン半溶媒和化合物の結晶構造は、下記のセルパラメーターを示す:
【表41】
【0300】
化合物1の1,4−ジオキサン半溶媒和化合物の換算座標(×104)及び同等の等方性運動パラメーター(Å2×103)は、下記の通りである:
【表42】
【0301】
化合物1の1,4−ジオキサン半溶媒和化合物の水素の座標(×104)及び同等の等方性転置パラメーター(Å2×103)は、下記の通りである:
【表43】
【0302】
1,4−ジオキサン半溶媒和化合物の格子面間隔は、下記の通りである:
【表44】
【0303】
10.2.6.2 X線粉末回折
化合物1の1,4−ジオキサン半溶媒和化合物のX線粉末回折ダイヤグラムは、角度(°2シータ)で表される次の特徴的ピークを示す(約±0.1°2シータ):9.8;10.0;10.8;13.6;13.9;14.1;15.9;16.1;18.0;18.3;19.0;19.6;20.0;20.1;20.2;20.6;21.4;21.7;21.8;22.0;22.2;22.3;23.4;23.9;24.3;24.4;24.9;25.3;25.4;26.2;27.0;27.1;27.3;27.5;28.2;28.4;28.5;28.7;29.1;29.4(図10及び11)。
【0304】
10.2.6.2 TG−DSC
TG−DSCを用いて、化合物1の1,4−ジオキサン半溶媒和化合物のサーモグラムは、この溶媒和化合物が、化合物1のII型を生成するために、75℃から、1,4−ジオキサンを放出し始めていることを示している(図21)。
【0305】
10.2.7 実施例9i:II型の化合物1
10.2.7.1 X線粉末回折
化合物1のII型のX線粉末回折ダイヤグラムは、角度(°2シータ)で表される次の特徴的ピークを示す(約±0.1°2シータ):9.4;10.7;12.2;12.8;14.3;15.1;15.9;16.9;18.2;18.9;19.4;19.7;20.4;21.1;21.4;21.8;22.0;22.7;23.0;23.2;23.7;23.9;24.4;24.8;25.5;26.8;27.1;27.4;28.1;28.3;29.5(図12及び13)。
【0306】
10.2.7.2 DSC
DSCを用いて、化合物1のII型のサーモグラムは、吸熱現象を含み、これは、II型の165℃±5℃での準安定性溶融と、その後の他の型への再結晶化と、それに続く別の溶融に対応している(図23)。
【0307】
10.2.7.3 IR
化合物1のII型のIRスペクトルは、cm-1で表される特徴的ピークを示す(約±5-1):3356;3321;3186;3078;2932;2851;1693;1609;1504;1462;1377;1265;1192;1123;937;872;837;787;594(図14及び15)。
【0308】
10.2.7.4 固体のNMR
この固体のNMRにより得られたII型のスペクトルは、次のppmで表される特徴的ピークを示す(約±0.2ppm):163.0;154.5;153.5;151.6;128.6;118.5;111.4;108.2;35;25(図16)。
【0309】
11. 物理化学的及び生物学的特性
11.1 転化実験
出願人は、化合物1の種々の多形相間での転化実験を行なっており、これは、III型が高温(T>145℃)で安定な型であることを示している。この有意の特性は、極微量の溶媒を真空下での加熱により除去することを可能にする(Polymorphism in the Pharmaceutical Industry, Wiley 2006, Hilfiker編; Wiley, ISBN: 978-3-527-31146-0)。周囲温度に戻すことにより、化合物1のIII型は、不変のままであり、これも又、製薬産業における主たる利点を構成する。
【0310】
11.2 化学的性
安定性の研究を、III型の化合物1と特許EP880514に記載された化合物1との間で、150℃で行なった。この化合物1の化学的安定性は、HPLCを用いて行なった。
【0311】
HPLC法の操作条件は、下記の通りである:
・カラム:Interchim UP3HDO-15XS, 150 × 4.6 mm、
・ 溶離液A: −水 .................. 2500
−トリフルオロ酢酸 ... 0.5
・溶離液B: −アセトニトリル ..........
・ 勾配:
【表45】
・検出:205nm、
・注入:20マイクロリットル、
・ 温度:40℃、
・注入される溶液:0.5mg.mL-1(アセトニトリル)
【表46】
【技術分野】
【0001】
本発明は、化合物6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートを有効成分として含む固体医薬組成物に関するものである。この発明は又、この医薬組成物の製法、その医薬、特にある種の癌の治療のための医薬(酵素ステロイドスルファターゼを標的とする化合物1)としての利用にも関係する。
【背景技術】
【0002】
化合物6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメート(以後、化合物1とも称する)を有効成分として含むこの発明による組成物は、安定な経口投与用医薬組成物であって、適当な生物学的利用能を提供するという利点を有する。
【0003】
下記の構造:
【化1】
の化合物1は、特許EP880514に記載されている。今日では、この化合物への注目は、L.W. Woo等(Chemistry & Biology, 2000, 7, 773-91)により記載されたようなそのスルファターゼ阻害活性及びこれが包含する治療用途のために増大している。ステロイド硫酸エステルの加水分解を担う酵素であるステロイドスルファターゼ(STS)の阻害は、例えば、ホルモン依存性乳癌を有する閉経後の患者に対する将来有望な新規な治療を代表する(Clin. Cancer Res. 2006; 12(5))。
【0004】
原則として、錠剤などの医薬組成物は、3通りの工程、湿式造粒法、乾式造粒法及び直接圧縮法によって製造することができる。
【0005】
化合物1は、G. Amidonにより提案された生物薬剤学的分類システム即ちBCS(G.L. Amidon等、「A theoretical basis for a biopharmaceutical drug classification: the correlation of in vitro drug dissolution and in vivo bioavailability」、Pharm. Res. 12 (1995) 413-420参照)のクラス2に属する。そうして、その吸収、及びそれ故、その生物学的利用能は、投与される医薬形態の溶解速度に強く依存する。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】欧州特許第880514号明細書
【非特許文献】
【0007】
【非特許文献1】L.W. Woo等、Chemistry & Biology, 2000, 7, 773-91
【非特許文献2】Clin. Cancer Res. 2006; 12(5)
【非特許文献3】G.L. Amidon等、Pharm. Res. 12 (1995) 413-420
【発明の概要】
【発明が解決しようとする課題】
【0008】
従来技術で公知の水相を用いる工程及び製剤、例えばクラス2の化合物の溶解度を増大させるために標準的様式において用いられる自己乳化性製剤(「自己乳化性薬物送達システム」)は、用いる賦形剤及び水相が化合物1と相容性でなく、製造段階において化学的劣化へと導くので、適当でない。
【0009】
化合物1の安定性及び低い溶解度の問題のために、経時的に十分に安定であって且つ十分な濃度の有効成分を含んでいる錠剤の製造を可能にする配合物を得ることは困難であった。その上、圧縮性の乏しい組成物は、乾式プレス中の破砕並びに余りに遅い溶解プロファイルを生じた。水の存在下で容易に加水分解されうる化合物1の不安定性のために、湿式造粒法を想定することは困難であった。
【課題を解決するための手段】
【0010】
予想外にも、化合物1の乾燥経口投与用形態を湿式造粒法により得ることを可能にする配合物が得られた。
湿式造粒法の場合には、成分を混合して、湿潤相中で結合剤を用いて造粒する。この結合剤は、湿潤相に溶解することができ又は造粒すべき粉末の混合物に取り込まれうる。次いでそれらの湿潤顆粒をふるいにかけ、乾燥させて、適宜粉砕した後に、錠剤に成形するために圧縮する。
【0011】
予想外にも、化合物1の新規な固体の医薬組成物が見出され、経口経路による投与が意図されてきており、これは、この有効成分に特異的な問題を解決すること及びそれを湿式造粒法により得ることを可能にする。
【0012】
この錠剤又はゼラチンカプセルの形態の経口投与用組成物は、固体形態の急速な溶解に安定であり、それ故、有効な生物学的利用能を伴う即時的放出を与える。
【0013】
以上より、本発明の主題は、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートを有効成分として含み且つ少なくとも一種類の製薬上許容しうる賦形剤を含む固体の医薬組成物である。
【0014】
本発明の主題は又、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートを有効成分として含み且つ少なくとも一種類の製薬上許容しうる賦形剤を含む、経口投与用の、好ましくは即時的に放出される経口投与用の固体の医薬組成物でもある。
【0015】
本発明の主題は又、経口経路による投与のための、化合物1を有効成分として含み且つ少なくとも一種類の崩壊剤並びに一種以上の水分に対する保護剤を含む固体の医薬組成物でもある。
【0016】
それ故、かかる薬剤は、有効成分を、加水分解に対して保護し、そうしてその劣化を防止する。好ましくは、化合物1は微粉化される。
【0017】
この劣化は、主として、この医薬組成物の製造中に起き、特に、湿式造粒法の段階において起きるということが認められた。予想外にも、当分野で結合剤及び希釈剤としての性質の故に用いられる賦形剤の添加が有効成分を保護する効果を与えるということが示された。
【0018】
本発明の主題は又、下記するような医薬組成物の製造方法でもある。従って、この発明の方法を、湿式造粒法を使用する錠剤の製造のための標準型の補助剤及び賦形剤を用いて実施することが可能である。
【0019】
「即時的放出を伴う経口投与」又は「即時的放出を伴う経口投与用形態」又は「即時的放出を伴う組成物」とは、この発明においては、化合物1用に開発された適当なイン・ビトロ溶解試験によって、45分以内に、好ましくは30分以内に有効成分の少なくとも80重量%のイン・ビトロでの溶解を与える投与又は経口投与用組成物を意味する。この試験は、パドル溶解装置を用いて、37℃で、100rpmで攪拌しながら、米国薬局方の基準に従って塩酸緩衝溶液(pH1.2)中で行なわれ、該装置は、0.1%界面活性剤、セチルトリメチルアンモニウムブロミドを含み、これは、有効成分の試験媒質中での十分な溶解度を得ることを可能にする。このアッセイは、紫外(UV)/可視光線の吸光度測定により、波長311nmで行なわれる。
【0020】
この発明により用いられる用語「滑剤(gliding agent)」又は「流動剤(flow agent)」とは、補助剤及び賦形剤をも意味し、ときには、潤滑剤及び顆粒の流動性及び流れを改善する薬剤と呼ばれる。
【0021】
この発明による用語「百分法配合」とは、組成物の全重量に対する、有効成分又は賦形剤の重量%での所与の割合を意味する。
【0022】
用語「結合剤」又は結着剤は、この発明によれば、生薬形態の構造及び結合力を維持するために用いられる補助剤又は賦形剤を示す。それらは、造粒段階における成分の顆粒形態での集合を可能にし、圧縮後の生薬形態の凝集を確実にする特性を有している。
【0023】
用語「崩壊剤」は、この発明によれば、液体環境、例えば水中又は胃液中での錠剤の崩壊を促進するために配合物に加えることのできる賦形剤又は補助剤を示す。
【発明の効果】
【0024】
本発明は、多くの利点を、特に、下記の組合せで示す:
・増大した安定性及び
・この医薬形態の即時的溶解(30分以内に少なくとも80%)にリンクした増大した生物学的利用能。
【図面の簡単な説明】
【0025】
【図1】化合物1の組成物の、時間及び組成物中の有効成分の微粉化の影響の関数としての比較用溶解曲線を示す図である。各曲線は、異なる微粉化の化合物1を含む固体組成物に対応している。
【図2】化合物1の種々の組成物の、時間の関数としての比較用溶解曲線を示す図である。各曲線は、異なる崩壊剤を含む固体組成物に対応している。
【図3】化合物1の2型のDMSO溶媒和化合物の推定されたX線回折ダイヤグラムを示す図である。
【図4】化合物1の2型のDMSO溶媒和化合物の実験によるX線粉末回折ダイヤグラムを示す図である。
【図5】化合物1の種類(variety)Iの推定されたX線粉末回折ダイヤグラムを示す図である。
【図6】化合物1の種類Iの実験によるX線粉末回折ダイヤグラムを示す図である。
【図7】化合物1の種類IのDSCサーモグラムを示す図である。
【図8】化合物1の種類IのIRスペクトルを示す図である。
【図9】化合物1の種類Iの固体のNMRスペクトルを示す図である。
【図10】化合物1の1型のDMSO溶媒和化合物の推定されたX線回折ダイヤグラムを示す図である。
【図11】化合物1の1型のDMSO溶媒和化合物の実験によるX線粉末回折ダイヤグラムを示す図である。
【図12】化合物1の3型のDMSO溶媒和化合物の推定されたX線回折ダイヤグラムを示す図である。
【図13】化合物1の3型のDMSO溶媒和化合物の実験によるX線粉末回折ダイヤグラムを示す図である。
【図14】化合物1の多形相(polymorphic form)IIIの推定されたX線回折ダイヤグラムを示す図である。
【図15】化合物1の多形相IIIの実験によるX線粉末回折ダイヤグラムを示す図である。
【図16】化合物1の多形相IIIのDSCサーモグラムを示す図である。
【図17】化合物1の多形相IIIのIRスペクトルを示す図である。
【図18】化合物1の多形相IIIの固体のNMRスペクトルを示す図である。
【図19】化合物1の1,4−ジオキサン半溶媒和化合物(hemisolvate)の推定されたX線回折ダイヤグラムを示す図である。
【図20】化合物1の1,4−ジオキサン半溶媒和化合物の実験によるX線粉末回折ダイヤグラムを示す図である。
【図21】化合物1の1,4−ジオキサン半溶媒和化合物のTG−DSCサーモグラムを示す図である。
【図22】化合物1の多形相IIの実験によるX線粉末回折ダイヤグラムを示す図である。
【図23】化合物1の多形相IIのDSCサーモグラムを示す図である。
【図24】化合物1の多形相IIのIRスペクトルを示す図である。
【図25】化合物1の多形相IIの固体のNMRスペクトルを示す図である。
【発明を実施するための形態】
【0026】
図1及び2は、賦形剤の効果並びに固体組成物のイン・ビトロでの振舞いを示している。
【0027】
好ましくは、この発明による錠剤の製造のために用いられる方法は、錠剤の形成段階の前に湿式造粒段階を通過する。
【0028】
事実、予想外にも、水相中での化合物1の低い安定性にもかかわらず、この錠剤は、湿式造粒段階を含む工程によって製造することができることが示された。この工程による錠剤の生産は、適切な賦形剤の存在下での化合物1の水相における安定化の恩恵を受ける。
【0029】
この発明の第一の特徴によれば、化合物1は、0.1〜20μmの粒子サイズを得るために、適当な処理により得られる。優先的に、化合物1は、1〜15μmの、一層優先的には、2〜10μmの粒子サイズを有する。尚一層優先的には、化合物1は、5μm±2μmのサイズを有する。
【0030】
この発明の医薬組成物の製造に適した製薬上許容しうる賦形剤は、例えば、マルトデキストリン、マンニトール、微結晶性セルロース、ラクトース、トウモロコシ澱粉、澱粉グリコール酸ナトリウム、クロスカルメロースナトリウム、部分的に架橋されたポリ(N−ビニル−2−ピロリドン)又はクロスポビドン、ポリビニルピロリドン、N−ビニル−2−ピロリドンと酢酸ビニルのコポリマー(又はコポビドン)例えばコリドンVA64共重合体、カルボキシメチルセルロース、アルファ化澱粉、メチルセルロース、ポリエチレングリコール、マクロゴール、ポリグリコール、ポリオキシエチレン、ピロリドン−2、コロイドシリカ、タルク、ステアリン酸マグネシウム、ナトリウムステアリルフマレート、ステアリン酸カルシウム、水素化植物油、ラウリル硫酸ナトリウム、リン酸カルシウム、糖類、デキストリン、澱粉、ゼラチン、セルロース、ワックスであってよく、又は水、有機溶媒例えばグリセロール又はグリコール並びにこれらの変化する割合の混合物(水を伴うか又は伴わない)であってもよい。これらの賦形剤は、有効成分に添加されるが、下記の機能を有している:
− 希釈剤、例えば、マンニトール、ラクトース又はラクトース一水和物、澱粉、炭酸カルシウム、微結晶性セルロース、又はマルトデキストリン;
− 崩壊剤、例えば、澱粉、クロスカルメロースナトリウム、澱粉グリコール酸ナトリウム、又はクロスポビドン;
− 結合剤、例えば、ポリビニルピロリドン、N−ビニル−2−ピロリドンと酢酸ビニルとのコポリマー(又はコポビドン)、カルボキシメチルセルロース、アルファ化澱粉、又はメチルセルロース;
− 流動剤又は滑剤、例えば、コロイドシリカ、又はタルク;
− 潤滑剤、例えば、ステアリン酸マグネシウム、ナトリウムステアリルフマレート、ステアリン酸カルシウム、ステアリン酸又は水素化植物油;
− 可溶化剤、例えば、マクロゴール(ポリエチレングリコール)、ポリグリコール、ポリオキシエチレングリコール、ポリジオール、ピロリドン−2、又はポリビニルピロリドン;
− 界面活性剤(又は表面活性剤)、例えば、ラウリル硫酸ナトリウム。
【0031】
本発明の主題は又、有効成分としての化合物1の経口経路による投与のための固体医薬組成物であって、化合物1及び少なくとも一の崩壊剤、一種以上の希釈剤及び一種以上の結合剤を含むことを特徴とする当該固体医薬組成物でもある。好ましくは、この化合物は、微粉化される。
【0032】
好ましくは、この発明による医薬組成物は、経口投与のための固体形態であり、ゼラチンカプセルである。好ましくは、この発明による医薬組成物は、経口投与のための固体形態であり、錠剤である。
【0033】
下記の組成物において、各種の賦形剤は、単独又は混合物であってよい。従って、「x%の結合剤」は、x%の単独の結合剤又は結合剤の混合物を意味する。
【0034】
この発明による組成物は、次の百分法の配合に従って、ゼラチンカプセル中に配合することができる:1〜30%の、好ましくは5〜20%の有効成分;40〜92%の、好ましくは65〜92%の、非常に優先的には75〜90%の希釈剤;0〜10%の結合剤、(好ましくは、0〜8%);0〜30%の崩壊剤(好ましくは、0〜20%);0〜5%の界面活性剤(好ましくは、0%);0〜5%の可溶化剤(好ましくは、0%);0.1〜3%の流動剤(好ましくは、0.9〜1.4%);0.5〜3%の潤滑剤(好ましくは、0.6〜2.8%)。
【0035】
優先的には、この医薬組成物は、上で規定したように、ゼラチンカプセルであり;一層優先的には、5〜30%の有効成分;40〜92%の希釈剤;0〜8%の結合剤(好ましくは、0〜5%);0〜30%の崩壊剤;0〜5%の界面活性剤;0〜5%の可溶化剤;0.1〜3%の流動剤;及び0.5〜3%の潤滑剤を含む百分法配合のゼラチンカプセルである。
【0036】
これらのゼラチンカプセルを配合するための好適な製薬上許容しうる賦形剤は、マンニトール、ラクトース、トウモロコシ澱粉、コロイドシリカ、ステアリン酸マグネシウム、及びラウリル硫酸ナトリウムであり、一層特には、マンニトール、ラクトース、コロイドシリカ、及びステアリン酸マグネシウムである。
【0037】
この発明による組成物は又、組成物の全重量に対して1〜30重量%の好ましくは5〜20重量%の、非常に優先的には8〜20重量%の有効成分;40〜92%の、好ましくは65〜92%の、非常に優先的には70〜85%の希釈剤;0.1〜20%の、好ましくは0.1〜10%の、非常に優先的には1〜5%の崩壊剤;0.1〜8%の、好ましくは2〜5%の結合剤;0.1〜3%の滑剤(好ましくは、0.5〜1.4%);0.2〜3%の潤滑剤(好ましくは、0.5〜2.8%)を含む百分法配合の錠剤(好ましくは、フィルムコート錠)中に配合することもできる。
【0038】
これらの錠剤の配合に好適な賦形剤は、マルトデキストリン、マンニトール、微結晶性セルロース、ラクトース又はラクトース一水和物、トウモロコシ澱粉、澱粉グリコール酸ナトリウム、クロスポビドン、ポリビニルピロリドン、コポビドン、カルボキシメチルセルロース、コロイドシリカ、ナトリウムステアリルフマレート、及びステアリン酸マグネシウムであり、一層特には、微結晶性セルロース、ラクトース、澱粉グリコール酸ナトリウム、コポビドン、コロイドシリカ、及びステアリン酸マグネシウムである。
【0039】
やはり好ましくは、上で規定した医薬組成物は、錠剤の全重量に対して、8〜20%の有効成分;70〜85%の希釈剤;1〜5%の崩壊剤;2〜5%の結合剤;0.5〜1.4%の流動剤;及び0.5〜2.8%の潤滑剤を含み、並びに、コート剤の全重量に対して約4.5〜5%、好ましくは4.8%の被覆溶液を含む百分法配合の錠剤(好ましくは、フィルムコート錠)である。
【0040】
好ましくは、本発明による医薬組成物は、次の賦形剤から選択される希釈剤を含む:マンニトール、ラクトース若しくはラクトース一水和物、澱粉、炭酸カルシウム、微結晶性セルロース、又はマルトデキストリン。
【0041】
好ましくは、本発明による医薬組成物は、次の賦形剤から選択される崩壊剤を含む:澱粉、クロスカルメロースナトリウム、澱粉グリコール酸ナトリウム、又はクロスポビドン。
【0042】
好ましくは、本発明による医薬組成物は、次の賦形剤から選択される結合剤を含む:ポリビニルピロリドン、N−ビニル−2−ピロリドンと酢酸ビニルとのコポリマー(コポビドン)、カルボキシメチルセルロース(CMC)、アルファ澱粉、又はメチルセルロース。
【0043】
好ましくは、本発明による医薬組成物は、次の賦形剤から選択される潤滑剤を含む:ステアリン酸マグネシウム、ナトリウムステアリルフマレート、ステアリン酸カルシウム又は水素化植物油。
【0044】
好ましくは、本発明による医薬組成物は、微結晶性セルロース(MCC)及び/若しくはコポビドン又は微結晶性セルロース(MCC)及び/若しくはカルボキシメチルセルロース(CMC)の何れかを含む。好ましくは、このCMCは、4〜6%のレベルである。非常に優先的には、本発明による医薬組成物は、微結晶性セルロース(MCC)を含む。非常に優先的には、本発明による医薬組成物は、コポビドンを含む。やはり非常に優先的には、本発明による医薬組成物は、微結晶性セルロース(MCC)及びコポビドンを含む。非常に優先的には、本発明による医薬組成物は、微結晶性セルロース(MCC)を含む。非常に優先的には、本発明による医薬組成物は、カルボキシメチルセルロース(CMC)を含む。非常に優先的には、本発明による医薬組成物は、微結晶性セルロース(MCC)及びカルボキシメチルセルロース(CMC)を含む。
【0045】
やはり好ましくは、選択される流動剤は、コロイドシリカ(即ち二酸化ケイ素のコロイド溶液)である。
【0046】
やはり好ましくは、選択される潤滑剤は、ステアリン酸マグネシウムである。これらの錠剤の配合物に賦形剤として加えられる崩壊剤は、溶解速度を増大させ、高い凝集力を有する錠剤を用いても、即時的溶解を達成することを可能にする。文献(例えば、J. Balasubramaniam, T. Bee, Pharmaceutical Technology Europe, Vol. 21, Number 9, 2009, p 44-49)によれば、最も有効な崩壊剤は、クロスポビドンA型及びB型と、その後のクロスカルメロースナトリウムである。
【0047】
好ましくは、本発明による錠剤の配合物において、用いられる崩壊剤は、澱粉グリコール酸ナトリウムであり、優先的には、1〜5%のレベルであり、非常に優先的には、3〜4%のレベルである。
【0048】
好ましくは、本発明による医薬組成物は、錠剤の全重量に対して、8〜20%の化合物1;20〜40%のラクトース及び25〜50%の微結晶性セルロース(希釈剤として使用);2〜8%のコポビドン(結合剤として使用);1〜5%の澱粉グリコール酸ナトリウム(崩壊剤として使用);0.2〜1.4%の流動剤;及び0.5〜2%の潤滑剤を含む錠剤であり、
非常に優先的には、錠剤の全重量に対して、8〜15%の化合物1;30〜40%のラクトース及び40〜50%の微結晶性セルロース(希釈剤として使用);2〜5%のコポビドン(結合剤として使用);3〜4.5%の澱粉グリコール酸ナトリウム(崩壊剤として使用);0.2〜1.4%の流動剤;及び0.5〜2%の潤滑剤を含む。
【0049】
やはり非常に優先的には、上記のように、この医薬組成物は、錠剤の全重量に対して、約10%の化合物1;36.5%のラクトース;45%の微結晶性セルロース;3%のコポビドン;4%の澱粉グリコール酸ナトリウム;0.5%のコロイドシリカ;1%のステアリン酸マグネシウムを含む錠剤である。用語「約」は、±0.5%を意味する。
【0050】
この発明の変形物によれば、この組成物は、コーティング又はフィルムコーティングを含むことができる。
【0051】
好ましくは、かかるコーティングは、有効成分の即時的放出に有意の影響を有さず、即ち、有効成分のイン・ビボでの放出速度論は、変化しない。
【0052】
やはり好ましくは、このコーティングは、有効成分の味をマスクするという利点を有し、固体組成物の錠剤の携帯での処理のための安全な取扱いを可能にする。
【0053】
この発明に記載された錠剤のために利用することのできるコーティング工程は、当業者に周知である。
【0054】
この錠剤がコーティングを含む場合には、後者は、好ましくは、ポリエチレングリコール、エチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルメチルセルロース(ヒプロメロース)により構成される群から選択されるポリマーを含むが、これらに限られない。特に、ポリエチレングリコール(マクロゴール)及び既に配合されたヒドロキシプロピルメチルセルロースを含むフィルムコーティング混合物を、Opadry(商標)IIホワイトの名称でColorcon社により市販されているもの(ラクトース一水和物、ヒプロメロース、二酸化チタン(E171)、トリアセチン)などの固体の経口投与用医薬形態の水性フィルムコーティングに利用することが可能である。Opadry(商標)IIホワイトは、水に可溶性であり、錠剤の形態で経口投与された医薬組成物の即時的崩壊を可能にする。
【0055】
好ましくは、本発明による固体医薬組成物は、錠剤の形態で与えられる。この発明は又、即時的溶解性の錠剤並びにコーティング又はフィルムでカバーされた錠剤にも関係する。
【0056】
医薬組成物は、例として記載されており、決して、この発明の範囲を制限するものではない。
【0057】
従って、この発明の主題は又、上記したようなこの発明の化合物の、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートからの製造方法でもあり、粒子サイズを減じる段階を含むことを特徴とする。
【0058】
優先的には、これらの粒子のサイズは、非プロトン性溶媒媒質中で、又は乾燥経路により、微粉状にすることにより低減される。
【0059】
やはり優先的には、これらの粒子のサイズは、非プロトン性有機溶媒を用いる湿式粉砕によって低減される。
【0060】
この発明は又、下記の段階を特徴とする、この発明の固体医薬組成物の製造方法にも関係する:
・ これらの成分をふるいにかけること;
・ コーティングを有しない錠剤の製造(湿式造粒法、混合及び圧縮);
・ コーティング溶液の製造。
【0061】
好ましくは、本発明による方法によって、湿式造粒段階において、有効成分及び予め混合した結合剤、希釈剤及び崩壊剤の全質量に対する水の質量は、約10〜30%である。
【0062】
好ましくは、この発明による方法によって、湿式造粒段階後に、これらの顆粒は、3%未満の残留水分が得られるまで乾燥される。
【0063】
この組成物は、この製造工程のすべての段階において、当業者に公知の慣用の装置によって製造される。
【0064】
本発明による配合物は、次の工程によって、製造することができる:第一の混合物を製造し、それは、有効成分により構成され、それは、先ず、結合剤、及び一種以上の希釈剤並びに崩壊剤を含む組成物に含まれるある種の賦形剤と同時にふるいにかけられる。
【0065】
このふるいかけ段階は、マニュアルシーブなどの適当なデバイスにて、ふるいを用いて行なわれる。
【0066】
これらのふるい分けした物質を、次いで、攪拌式造粒機に導入して、約5分間混合する。混合速度は、選択した装置による。
【0067】
次いで、この造粒のための水のマスを、造粒すべき固相に対するその割合が優先的に約10〜30%となるように調製する。造粒すべき固相とは、結合剤のマスに加えた有効成分及び予め混合された賦形剤の全マスを意味する。
【0068】
この造粒のための結合剤は、粉末形態で又は優先的には、造粒溶液を構成するために、あるかさの水に溶解されて混合物に添加される。
【0069】
この工程の出発時に得られる混合物を、次いで、好ましくは「高剪断力」型の攪拌造粒機にて、造粒溶液を用いる湿式経路によって造粒する。この造粒すべき溶液の添加後の造粒時間は、10分未満であり、優先的には、5分未満である。
【0070】
この最後の段階から生じる顆粒を、次いで、慣用の装置例えば流動層乾燥器で乾燥させることができる。好ましくは、これらの顆粒は、3%未満の残留水分が達成されるまで乾燥される。こうして乾燥された顆粒は、慣習的に錠剤の内相と呼ばれる部分を構成する。次いで、それらは、研摩によって調整される。
【0071】
予め得られた調整された顆粒及び、崩壊剤の一部分、粉末の流動性を改善することを可能にする流動剤、及び最後に潤滑剤により構成されるいわゆる外相により構成される第2の混合物を製造する。
【0072】
この外相の賦形剤は、先ず、マニュアルシーブなどの適当なデバイスにて、ふるいにかけられる。
【0073】
これらの顆粒、崩壊剤及び流動剤を、次いで、混合してから、潤滑剤を導入する。
【0074】
錠剤の形態の配合物の製造のために、最終生成混合物を、次いで、例えばロータリー式のプレスを用いて錠剤にすることができる。
【0075】
400mgの錠剤の硬さは、6〜12kパスカル(kPa)であってよい。100mgの錠剤の硬さは、4〜8kPaであってよい。
【0076】
これらの錠剤を、次いで、フィルムコーティングタービンにて、予め、好ましくは、純水に16%のOpadryII(商標)を添加することにより、用意した被覆用懸濁液をスプレーすることにより被覆することができる。
【0077】
フィルムコーティング中、製品の温度は、35〜55℃とする。
【0078】
好ましくは、乾燥後の錠剤の質量の増加は、フィルムコーティング前の錠剤の質量の4〜6%である。
【0079】
これらの錠剤は、パッケージに入れて、ブリスターパック又は瓶入りで保存することができる。
【0080】
この発明によれば、非被覆錠剤は、好ましくは、800mgの総重量を、好ましくは、400mgの総重量を超えない。
【0081】
この発明は、最終的には、前記のように、癌、優先的にはホルモン依存性の癌及びやはり優先的に乳癌、前立腺癌、子宮内膜癌又は卵巣癌を治療するための、この発明の医薬組成物の利用に関係する。
【0082】
本願は又、化合物6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメート(即ち、スルファミン酸6,7,8,9,10,11−ヘキサヒドロ−6−オキソベンゾ[b]シクロヘプタ[d]ピラン−3−イルエステル即ち化合物1)の多形体にも関係する。この発明は又、これらの多形相の製法、それらの有効成分としての利用、一層特にある種の癌の治療のための有効成分としての利用にも関係する。この発明は又、これらの多形相を有効成分として含む医薬組成物とも関係する。
【0083】
Wooにより簡潔に記載されて、以後種類Iと呼ばれる形態は、特徴的な赤外吸収バンドを生成する。出願人は、構造の完全な特性決定のために適当な大きさと品質の単結晶を得た。
【0084】
従来技術において得られた結晶は、良好な生物学的利用能を与えず、それらの調製物は、産業的規模に移行することができない。
【0085】
その上、出願人は、化合物1の結晶を、産業的過程により得た。顕微鏡観察は、結晶の進行性の不透明化を約140〜145℃で観察することを可能にする。この現象は、不可逆的であり:結晶は、ゆっくりした又は急速な冷却後に、その最初の外観を回復しない。DSCにおいては、固−固転移に対応する吸熱現象が、140℃から観察される。第2の吸熱のピークは、未変換の種類Iの溶融に対応する170℃で観察される。第3の吸熱のピークは、180℃で観察される。この第3のピークは、固−固転移中に生成された他の多形相の溶融に対応する。連続的に160℃まで加熱してから18〜25℃にまで冷却されたこの化合物に対して行なわれたX線粉末回析により、化合物1の種類Iのこれらの特性に加えて、幾つかのピークが観察され、これらのピークは、加熱中に生成した他の多形相に対応している。
【0086】
従って、産業的条件下で得られた結晶は、DSCにより及び/又は160℃への加熱後のX線粉末回折により検出された不均質性を示す。この不均質性は、化合物の加熱中のグレーン成長及び他の結晶形の成長を与え、これは、加熱中の生成物を不安定にする。
【0087】
不均質性とは、ここでは、変色又はポリタイプを含む結晶中又は結晶上の欠陥を意味し、これは、慣用の光学顕微鏡又は偏光装置を備えた顕微鏡によって及び/又は示差エンタルピー分析及び/又はX線粉末回折により及び/又は共焦点ラマン顕微鏡観察によって検出することができる。従って、結晶中のかかる不均質性の存在は、有効成分の保存中に、又は錠剤、ゼラチンカプセル、クリーム、又は他の生薬形態の製造工程中に、多くの問題へと導く。例えば、相対湿度ストレスは、医薬有効成分に対して許容しえない挙動へと導きうる。その上、Y. Mnyukh Fundamentals of solid-state phase transitions, ferromagnetism and ferroelectricity (2001)により示されたように、これらの欠陥は、固−固転換をある機構(グレーン成長)によってプレコード化(pre-code)しうる。
【0088】
現在、この分子の臨床的開発のためには、ある結晶形を選択し、産業的条件下でのその生産を達成することが必要であり、鍵となる因子は、その熱安定性及び生物学的利用能である。
【0089】
それ故、出願人が解決することを提案する技術的課題は、化合物1の、産業的規模で用いうる条件下で安定で且つ生物学的利用能のある形態である。
【0090】
当業者には、機械的処理による粒子サイズの低減を目的とした処理は、結晶化相に、欠陥、残留ストレス、多形転移、部分的又は全体的非晶質化(状況によっては、化学分解を伴う)を生じうることは公知である(Jet-milling; from a particle perspective predicting particle fracture based on mechanical material properties Onno M. de Vegt; PhD thesis University of Groningen; 19 October 2007; Garnier, S.; Petit, S.J Mallet, F.; Petit, M.-N.; Lemarchand, D.; Coste, S.; Lefebvre, J.; Coquerel, G., Influence of aging, grinding and preheating on the thermal behaviour of α-lactose monohydrate. Int. J. Pharm. 2008, 361, 131-140)。
【0091】
化合物の、一つより多くの結晶形で存在する能力は、用語「多形性」により規定され、その異なる結晶形は、「多形種類(polymorphic varieties)」又は「多形体(他形相)」として知られる。多形性は、固体状態の有効成分の多くの特性に影響を与えうる。一の物質の種々の多形性は、例えばそれらの溶解度に直接影響しうるそれらの物性により、互いに、相当に異なることがありうる。多形性は、多くの有機化合物について示されている。
【0092】
非常に予想外なことに、出願人は、今や、粒子サイズの低減を目的とする産業的条件下で得られる化合物1の適当な処理が、有効成分の配合物と適合する粒子サイズを有する化合物1の種類Iを生成することができ、同時に、結晶中の異成分の濃度を付随的に低減させ、そうして、化合物1の安定で生物学的利用能のある形態を産業的規模で用いることのできる条件下で与えることを見出した。実験部に記載したDSC及びX線粉末回折分析は、こうして得られた種類Iが増大した安定性を有することを示すことを可能にする。
【0093】
従って、この発明の主題である低粒子サイズの種類Iは、種々の分析法例えば固体のX線粉末回折、示差走査熱分析(DSC)、ラマン分光法、赤外線分光法又はNMRによって特性決定することができる。
【0094】
この発明の主題は、化合物1の結晶DMSOの2型の溶媒和化合物でもあり、これは、X線粉末回折を利用して、角度(°2シータ)で表される特徴的なピークを示す(±0.1°2シータ):7.6;18.7;24.2;29.9。
【0095】
非常に予想外なことに、出願人は、今や、グレーン成長及び他の結晶の成長を与える異成分を有する産業的条件下で得られたI型の結晶が、化合物の加熱中に、高温で安定なIII型を形成することを見出した。本発明の主題は、この化合物1の新規なIII型である。このIII型は、145℃より高温で、熱力学的に安定であるという利点を有している。
【0096】
この発明のIII型の化合物1に対する幾つかの合成経路があり、その一つは、II型と呼ばれる化合物1の他の新規な結晶形の中間体を経由するものである。
【0097】
化合物1の種類Iは、X線粉末回折により特性決定することができ、回折ダイヤグラムは、特に、角度(°2シータ)で表される特徴的ピークを有する(約±0.1°2シータ):7.5;10.9;13.1;17.7;19.0。
【0098】
これらの±0.1°2シータの誤差を伴う角度(°2シータ)は、Braggの法則による反射角、即ち、試料へのX線の入射角を表す。
【0099】
粒子サイズの低減前の化合物1の種類Iは、単結晶のX線分析によって特性決定することができる。
【0100】
低減された粒子サイズの化合物1の種類Iは、X線粉末回折ダイヤグラムによって特性決定することができる(図6)。
【0101】
化合物1の種類Iは、上記のように、DSCサーモグラムによって特性決定することもできる(図7)。
【0102】
化合物1の種類Iは、上記のように、赤外線スペクトルによって特性決定することもできる(図8)。
【0103】
化合物1の種類Iは、上記のように、固体のNMRスペクトルによって特性決定することもできる(図9)。
【0104】
それ故、出願人は、適当な処理により得られた化合物1の種類Iを、0.1〜20μmの粒子サイズを得るために提供する。
【0105】
本発明は、多くの利点を、特に、増大した安定性及び生物学的利用能に有する。
【0106】
それ故、本発明の主題は、0.1〜20μmの粒子サイズの6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートの多形性化合物であり、これは、X線粉末回折により、角度(°2シータ)で表される特徴的なピークを示す(±0.1°2シータ):7.5;10.9;13.1;17.7;19.0。
【0107】
優先的に、この発明の化合物は、1〜15μmの粒子サイズを有する。
【0108】
一層優先的には、この発明の化合物は、3〜7μmの粒子サイズを有する。
【0109】
尚一層優先的には、この発明による化合物は、5μm±1μmのサイズを有する。
【0110】
好ましくは、上で規定した化合物は、X線粉末回折により、角度(°2シータ)で表される特徴的なピークを示す(±0.1°2シータ):7.5;10.9;13.1;15.0;15.8;17.0;17.7;19.0;22.5。
【0111】
やはり優先的に、上で規定した化合物は、160℃まで加熱してから18〜25℃に戻した後に行なわれるX線粉末回折により、角度(°2シータ)で表される次の特徴的ピークを示す(±0.1°2シータ):7.5;10.9;13.1;17.7;19.0(160℃への加熱中に生成された型に対応する更なるピークを含まない)。
【0112】
非常に優先的には、上で規定した化合物は、5℃/分の温度勾配を有するDSCを用いて、吸熱性溶融ピークを170℃±5℃に示し且つ吸熱性ピークを180℃±2℃に示し、180℃でのピークは、170℃での溶融中に交換されたエンタルピーの最大で10%を表すということが理解される。
【0113】
やはり非常に優先的には、上で規定した化合物は、DSCを用いて、140〜155℃で吸熱事象を示さない。
【0114】
尚一層優先的には、上で規定した化合物は、赤外線分光法を用いて、cm-1で表される特徴的なピークを示し(±5cm-1):3310;3167;3059;891;798;733;679;455;尚一層優先的には、次の特徴的なピークを示した(±5cm-1):3310;3167;3059;2928;2858;1690;1605;1454;1385;1261;1188;1126;941;891;853;798;733;679;598;544;455。
【0115】
一変形によれば、この発明は、上で医薬として規定した化合物に関係する。
【0116】
この発明による化合物は、様々な医薬組成物に配合することができる。固体形態の場合には、それは、例えば、粉末、顆粒、錠剤、ゼラチンカプセル、リポソーム又は坐薬であってよい。液体形態の場合には、それは、例えば、溶液、乳濁液、懸濁液又はシロップであってよい。この発明の化合物の投与は、例えば、局所経路、経口経路により、筋肉注射、皮下注射により、そして特に、ゼラチンカプセル、錠剤、パッチ又はクリームの形態で行なうことができる。
【0117】
適当な支持体又は賦形剤は、例えば、マルトデキストリン、マンニトール、微結晶性セルロース、ラクトース、トウモロコシ澱粉、澱粉グリコール酸ナトリウム、クロスカルメロースナトリウム、ポリビニル−ポリピロリドン(又はクロスポビドン)、ポリビニルピロリドン、カルボキシメチルセルロース、アルファ澱粉、メチルセルロース、ポリエチレングリコール、マクロゴール、ポリグリコール、ポリオキシエチレン、ポリジオール、ピロリドン−2、コロイドシリカ、タルク、ステアリン酸マグネシウム、ナトリウムステアリルフマレート、ステアリン酸カルシウム、水素化植物油、ラウリル硫酸ナトリウム、リン酸カルシウム、糖類、デキストリン、澱粉、ゼラチン、セルロース、ワックスであってよく、又は水、有機溶媒例えばグリセロール又はグリコール類、それらの割合を変化させた混合物でもよい(水を伴っても伴わなくてもよい)。有効成分に添加されるこれらの支持体は、下記するように役立つ:
− 希釈剤、例えば、マンニトール、ラクトース、澱粉、炭酸カルシウム、微結晶性セルロース、又はマルトデキストリン;
− 崩壊剤、例えば、澱粉、ナトリウムクロスカルメロース、澱粉グリコール酸ナトリウム、又はクロスポビドン;
− 結合剤、例えば、ポリビニルピロリドン、カルボキシメチルセルロース、アルファ澱粉、又はメチルセルロース;
− 流動剤、例えば、コロイドシリカ、又はタルク;
− 潤滑剤、例えば、ステアリン酸マグネシウム、ナトリウムステアリルフマレート、ステアリン酸カルシウム又は水素化植物油;
− 可溶化剤、例えば、ポリエチレングリコール、マクロゴール、ポリグリコール、ポリオキシエチレン、ポリジオール、ピロリドン−2、又はポリビニルピロリドン;
− 界面活性剤(又は、表面活性剤)、例えば、ラウリル硫酸ナトリウム。
【0118】
この発明の組成物は、ゼラチンカプセル中に、次のように配合することができる:5〜30%の有効成分(優先的には、6〜13%);40〜92%の希釈剤(優先的には、65〜92%;非常に優先的には85〜90%);0〜30%の崩壊剤(優先的には、0〜22%;非常に優先的には0%);0〜5%の界面活性剤(優先的には、0%);0〜5%の可溶化剤(優先的には、0%);0.1〜3%の流動剤(優先的には、0.9〜1.4%);0.5〜3%の潤滑剤(優先的には、0.6〜2.8%)。
【0119】
これらのゼラチンカプセルを配合するための好適な賦形剤は、マンニトール、ラクトース、トウモロコシ澱粉、ステアリン酸マグネシウム、及びラウリル硫酸ナトリウムであり、一層特には、マンニトール、ラクトース、コロイドシリカ、及びステアリン酸マグネシウムである。
【0120】
この発明による組成物は又、5〜30%の有効成分(優先的には、7〜15%、非常に優先的には10〜15%);40〜92%の希釈剤(優先的には、34〜89%、非常に優先的には70〜85%);0〜40%の崩壊剤(優先的には、0〜20%、非常に優先的には、3〜5%);0〜8%の結合剤(優先的には、2〜5%);0.1〜3%の流動剤(0.5〜1.4%);0.5〜3%の潤滑剤(優先的には、0.5〜2.8%)を含む錠剤中に配合することもできる。
【0121】
これらの錠剤を配合するのに適した賦形剤は、マルトデキストリン、マンニトール、微結晶性セルロース、ラクトース、トウモロコシ澱粉、澱粉グリコール酸ナトリウム、クロスポビドン、ポリビニルピロリドン、カルボキシメチルセルロース、コロイドシリカ、マグネシウムステアリルフマレート、及びステアリン酸マグネシウムであり、一層特には、微結晶性セルロース、ラクトース、澱粉グリコール酸ナトリウム、ポリビニルピロリドン、コロイドシリカ、及びステアリン酸マグネシウムである。
【0122】
変形物によれば、この発明は、有効成分として、0.1〜20μmの粒子サイズの6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートを、少なくとも一種の製薬上許容しうる支持体と共に含む医薬組成物に関係し;優先的には、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートは、1〜15μmの粒子サイズを有し;一層優先的には、3〜7μmのの粒子サイズを有し、更に優先的には、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートは、5μm±1μmのサイズを有する。
【0123】
優先的には、この医薬組成物は、上記のように、有効成分として、前に規定したように、種類Iの化合物1を含む。
【0124】
非常に優先的には、この医薬組成物は、上で規定したように、ゼラチンカプセルであり;一層優先的には、5〜30%の有効成分;40〜92%の希釈剤;0〜30%の崩壊剤;0〜5%の界面活性剤;0〜5%の可溶化剤;0.1〜3%の流動剤;及び0.5〜3%の潤滑剤を含むゼラチンカプセルでもある。
【0125】
やはり非常に優先的には、この医薬組成物は、上で規定したように、錠剤であり、一層優先的には、5〜30%の有効成分;40〜92%の希釈剤;0〜40%の崩壊剤;0〜8%の結合剤;0.1〜3%の流動剤;及び0.5〜3%の潤滑剤を含む錠剤でもある。
【0126】
化合物1の合成経路は、特許EP880514に記載されたように又はL.W. Woo等、Chemistry & Biology, 2000, 7, 773-91によって、従来技術において構想されている。
【0127】
出願人は、今や、この化合物が2つの化学段階にて合成されうることを見出した。その第一段階は、強酸(例えば、硫酸、トリフルオロ酢酸又はメタンスルホン酸)中での、2−カルベトキシシクロヘプタノンのレゾルシノールとの縮合よりなり、中間体3−ヒドロキシ−8,9,10,11−テトラヒドロシクロヘプタ[c]クロメン−6(7H)−オンが、アルコール/水混合物を用いる(例えば、エタノールを加えてから水を加える)沈澱により単離される。第二段階においては、スルホニルイソシアネートクロリドが、トルエン溶液中で、蟻酸の作用により、変換されて、スルファモイルクロリドとなり、次いで、溶剤例えばDMA中に溶解された前記の中間体と縮合される。この反応媒質を水で処理し、有機溶媒(好ましくは、2−メチルテトラヒドロフラン:MeTHF)により抽出し、次いで、粗化合物1が、貧溶媒(antisolvent)(好ましくは、メチルシクロヘキサン)を添加することにより、有機相から沈澱される。最後に、純粋な化合物1が、アセトン又は酢酸エチルへの高温での溶解及び貧溶媒(好ましくは、メチルシクロヘキサン)添加による沈澱による粗生成物の再結晶化によって得られる。
【0128】
結果として、この発明の主題は又、下記の段階を含むことを特徴とする、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートの製造方法でもある:
− 2−カルベトキシシクロヘプタノンの、強酸中でのレゾルシノールとの縮合、
− こうして得られた3−ヒドロキシ−8,9,10,11−テトラヒドロシクロヘプタ[c]クロメン−6(7H)−オンの、アルコール/水混合物を用いる沈澱による単離、
− 3−ヒドロキシ−8,9,10,11−テトラヒドロシクロヘプタ[c]クロメン−6(7H)−オンの、非プロトン性溶媒中でのクロリドスルファモイルとの縮合、
− 得られた粗生成物の高温アセトン又は酢酸エチル中への溶解及び貧溶媒例えばメチルシクロヘキサンの添加による再結晶。
【0129】
この発明の化合物は、前に記載のように、化合物1から、粒子サイズの低減を目的とした処理によって、種類Iの形態で得られる。
【0130】
従って、この発明の主題は又、上記のこの発明の化合物の、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートからの、粒子サイズの低減の段階を含むことを特徴とする製造方法でもある。
【0131】
優先的には、これらの粒子のサイズは、微粉状にすることにより低減される。
【0132】
やはり優先的には、これらの粒子のサイズは、湿式研削により、有機非プロトン性溶媒を用いて低減される。
【0133】
本発明の主題は又、X線粉末回折を用いて、角度(°2シータ)で表される特徴的なピーク(±0.1°2シータ):8.6;11.3;28.6を示す、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートの多形性化合物でもある(III型の化合物)。好ましくは、この化合物のIII型は、角度(°2シータ)で表される特徴的なピークを示す(±0.1°2シータ):8.6;11.3;12.0;16.6;20.9;23.0;28.6。
【0134】
この発明の一層特別の主題は、上で規定したIII型の化合物であり、それは、単結晶X線回折を利用して、下記のセルパラメーターを示し:
【表1】
下記の換算(reduced)座標(×104)及び同等の等方性の運動パラメーター(Å2×103)を示し:
【表2】
下記の水素原子の座標(×104)及び同等の等方性転置パラメーター(Å2×103)(U(eq)は、直交テンソルUijの値の3分の1に等しい)を示し:
【表3】
下記の格子面間隔を示す:
【表4】
【0135】
優先的には、この発明の主題は、上で規定したような、5℃/分でのDSCを用いて、180℃±2℃の吸熱性溶融ピークを示す化合物(IIII型)である。
【0136】
好ましくは、この発明による化合物(III型)は、上で規定したように、赤外線分光分析を用いて、次のcm-1で表される特徴的なピークを示す(±5cm-1):3406;3217;1678;1011;563;
【0137】
そして、非常に優先的には、次のcm-1で表される特徴的なピークを示す(±5cm-1):3406;3217;3082;2924;1678;1385;1269;1134;1011;934;845;601;563;536。
【0138】
変形物によれば、この発明の主題は、医薬として、上で規定したように、III型の化合物である。
【0139】
この発明の主題は又、有効成分として、上で規定したようなIII型の化合物を、少なくとも一の製薬上許容しうる支持体と共に含む医薬組成物でもある。
【0140】
やはり、この発明の主題は、化合物1の1型の結晶DMSO溶媒和化合物でもあり、これは、X線粉末回折を用いて、角度(°2シータ)で表される特徴的なピークを示す(±0.1°2シータ):6.6;10.9;13.1;14.3;15.7;16.7;17.4;18.3;19.6;20.8;21.9;22.6;23.0;24.7;24.9;25.2;25.5;25.8;26.6;26.9;27.2;28.3。
【0141】
やはり、この発明の主題は、化合物1の3型の結晶DMSO溶媒和化合物でもあり、これは、X線粉末回折を用いて、角度(°2シータ)で表される特徴的なピークを示す(±0.1°2シータ):9.8;13.9;16.0;17.7;19.1;22.1。
【0142】
更に、この発明の主題は又、化合物1の結晶1,4−ジオキサン半溶媒和化合物でもあり、これは、X線粉末回折を用いて、角度(°2シータ)で表される特徴的なピークを示す(±0.1°2シータ):9.8;10.0;10.8;13.6;13.9;14.1;15.9;16.1;18.0;18.3;19.0;19.6;20.0;20.1;20.2;20.6;21.4;21.7;21.8;22.0;22.2;22.3;23.4;23.9;24.3;24.4;24.9;25.3;25.4;26.2;27.0;27.1;27.3;27.5;28.2;28.4;28.5;28.7;29.1;29.4。
【0143】
その上さらに、この発明の主題は、化合物1の多形化合物、II型化合物でもあり、これは、X線粉末回折を用いて、角度(°2シータ)で表される特徴的なピークを示し(±0.1°2シータ):9.4;10.7;12.8;18.2;18.9;19.7;20.4;23.2;優先的には、角度(°2シータ)で表される特徴的なピークを示す(±0.1°2シータ):9.4;10.7;12.2;12.8;15.1;18.2;18.9;19.7;20.4;21.1;22.0;23.2。
【0144】
優先的には、このII型の化合物は、5℃/分でのDSCを用いて、165℃±5℃の吸熱性溶融ピークを示し;一層優先的には、それは、赤外線分光法を用いて、特徴的なピークを示し(cm-1〜±5cm-1で表示):3356;3321;3186;1504;872;787;尚一層優先的には、特徴的なピーク(cm-1〜±5cm-1で表示):3356;3321;3186;3078;2932;2851;1693;1609;1504;1462;1377;1265;1192;1123;937;872;837;787;594を示す。
【0145】
この発明による化合物は、上記のように、化合物1から、更なる段階により得られ、それは、下記のものであってよい:
− 水中の1型のDMSO溶媒和化合物の脱溶媒、
− 水中の3型のDMSO溶媒和化合物の脱溶媒、
− エタノール中での噴霧化、
− アセトン中での噴霧化、
− 還流下でのクメン中での再糊状化、
− 熱処理と、その後の噴霧化と、その後の第二の熱処理、又は
− II型の熱処理。
【0146】
こうして、変形物によれば、この発明の主題は、上で規定したようなIII型の化合物の、化合物1から出発する製造方法であって、下記の方法の一つによる方法である:
− a)上で規定したような1型の結晶DMSO溶媒和化合物へと導くための攪拌及びDMSOからの沈澱(この化合物は、次いで、水に溶解される);
− b)上で規定したような3型の結晶DMSO溶媒和化合物へと導くための攪拌及びDMSOからの沈澱(この化合物は、次いで、水に溶解される);
− c)エタノール中での噴霧化;
− d)アセトン中での噴霧化;
− e)還流下でのクメン中での再糊状化;
− f)155〜165℃の温度での、10〜20分間の熱処理と、その後の噴霧化と、その後の155〜165℃で10〜20分間の第二の熱処理、
− g)上で規定したようなII型の化合物の処理、そして:
− 1,4−ジオキサン中での化合物1の噴霧化により、
− 又は、上で規定したような結晶DMSO半溶媒和化合物へ導くための化合物1の攪拌及び1,4−ジオキサンからの沈澱により得られ、この化合物は、次いで、不活性気流中で5℃/分で20〜80℃に加熱することにより脱溶媒される、
この処理は、155〜165℃で6〜10分間の熱処理よりなる。
【0147】
優先的には、この発明による方法は、上で規定したような1型の結晶DMSO溶媒和化合物の脱溶媒を、水中で完了する。
【0148】
優先的には、この発明による方法は、上で規定したような3型の結晶DMSO溶媒和化合物の脱溶媒を、水中で完了する。
【0149】
やはり優先的には、この発明による方法は、噴霧化を、エタノール中で完了する。
【0150】
やはり優先的には、この発明による方法は、噴霧化を、アセトン中で完了する。
【0151】
非常に優先的には、この発明による方法は、再糊状化を、還流下でクメン中で完了する。
【0152】
尚一層優先的には、この発明による方法は、155〜165℃で10〜20分間の熱処理と、その後の微粉化と、その後の155〜165℃で10〜20分間の第二の熱処理を完了し;
そして優先的には、これらの熱処理は、160℃±1℃で15分間行なわれる。
【0153】
やはり尚一層優先的には、この発明による方法は、上で規定したように、II型の化合物の熱処理を、155〜165℃で6〜10分間で完了し;
そして優先的には、この熱処理は、160℃±1℃で7.5分間行なわれる。
【0154】
やはり好ましくは、このII型の化合物は、上で規定したように、1,4−ジオキサン中での化合物1の噴霧化により得られ;又はII型の化合物は、上で規定したように、結晶1,4−ジオキサン半溶媒和化合物の脱溶媒により得られる。
【0155】
変形物によれば、この発明の主題は、上で規定したような1型の結晶DMSO溶媒和化合物の製造方法であって、化合物1から出発して、攪拌及びDMSOからの沈澱による、当該製造方法である。
【0156】
変形物によれば、この発明の主題は、上で規定したような3型の結晶DMSO溶媒和化合物の製造方法であって、化合物1から出発して、攪拌及びDMSOからの沈澱による、当該製造方法である。
【0157】
変形物によれば、この発明の主題は、上で規定したような結晶1,4−ジオキサン半溶媒和化合物の製造方法であって、化合物1から出発して、攪拌及び1,4−ジオキサンからの沈澱による、当該製造方法である。
【0158】
上記のII型について、それは、2つの合成経路によって得られうる:
− 直接に、1,4−ジオキサン中での噴霧化による
− 予め1,4−ジオキサンからの沈澱により得られた化合物1の1−4−ジオキサン半溶媒和化合物の脱溶媒による。
【0159】
やはり、他の変形によれば、この発明の主題は、上記のII型の化合物の製造方法であって、化合物1から出発し、下記の方法の一つによる、当該製造方法である:
− 1,4−ジオキサン中での噴霧化;
− 上記の結晶1,4−ジオキサン半溶媒和化合物を導くための、攪拌及び1,4−ジオキサンからの沈澱、該化合物は、次いで、不活性気流中での20〜80℃で5℃/分の加熱による脱溶媒に付される。
【0160】
好ましくは、上記のII型の化合物の製造方法は、上記の結晶1,4−ジオキサン半溶媒和化合物の脱溶媒を経る。
【0161】
やはり好ましくは、上記のII型の化合物の製造方法は、1,4−ジオキサン中での噴霧化を経る。
【0162】
或は、この発明の主題は、上で規定した、種類Iの又はIII型の化合物の、癌;優先的にホルモン依存性の癌、やはり優先的には乳癌、前立腺癌、子宮内膜癌又は卵巣癌から選択される癌を治療することを意図した医薬を製造するための利用である。
【0163】
後述の実験部は、上記の手順を説明するために与えられるものであり、この発明の範囲を限定するものと考えるべきではない。
【0164】
別途規定しない限り、本願で用いられる技術的及び科学的用語は、当業者が通常理解するのと同じ意味を有する。その上、すべての特許(又は特許出願)並びに他の参照を、参考として援用する。
【実施例】
【0165】
実験部:
1.この発明による組成物
実施例1a:
錠剤の形態であって、化合物1を有効成分として含む組成物を下記の表1に示した。かかる組成物は、5kgのバッチ及び40mgの投与量につき、下記のダイヤグラムに従って、湿式造粒法により製造することができる。
【0166】
【表5】
【0167】
【表6】
【0168】
40mgの錠剤に塗布されたコーティングは、Opadry(商標)IIホワイト(Colorcon)よりなり;これは、即時放出型錠剤に用いるための市販されているコーティングの混合物である。このコーティングの主な目的は、薬物の悪い味をマスクすることである。それは又、強い有効成分を含む錠剤の包装作業中の取扱いに関連したリスクをも低減させる。
【0169】
実施例1b:
ゼラチンカプセルの形態であって、化合物1を有効成分として含む組成物を下記の表2に示した。
【0170】
【表7】
【0171】
2.崩壊剤の溶解プロファイルに対する効果
すべての賦形剤を、欧州薬局方集成に従って、分析する。
【0172】
崩壊剤の効果は、下記の第三節に記載した組成物2と17の、澱粉グリコール酸ナトリウムなしでの、30分での解散価値(dissolution values)の比較により見えてくる。
【0173】
崩壊剤の添加の、溶解プロファイルに対する効果について、種々の試験が行なわれてきており、錠剤の崩壊時間が研究された。澱粉グリコール酸ナトリウムA型(Explotab(商標))とアルファ化トウモロコシ澱粉(Starch 1500 (商標))の2種の崩壊剤の効果を研究するために、4つの組成の錠剤を生成した。
【0174】
【表8】
【0175】
これらの得られた結果は、崩壊剤の添加が錠剤の溶解プロファイルを加速して、参照用カプセルに近いものとしていることを示している(図2参照)。
【0176】
3.湿式造粒工程中の有効成分に対する結合剤の保護効果
結合剤及び/又は希釈剤(微結晶性セルロース、コポビドン、カルボキシメチルセルロース)の保護効果を示す研究が、最終的な不純物のレベル並びにこれらの配合物の製造中の挙動の比較により行なわれた。湿式造粒法により得られた種々の固体組成物を、Waters社製の2487 UV 検出器を有するAlliance 2695システムにて、下記の表(表4)に記載された条件にしたがってHPLC(高性能液体クロマトグラフィー)により分析した。
【0177】
【表9】
【0178】
【表10】
【0179】
4.ブリスターパック内における6カ月及び12カ月での、貯蔵条件の関数としての、5mg錠剤及び40mg錠剤の安定性
ある錠剤形態(実施例1aの百分法配合)は、一次包装後にブリスターパック内で安定であった。
【0180】
この目的は、この形態の、6カ月及び12カ月の期間にわたる安定性をチェックすることである。
【0181】
推奨される通常の貯蔵条件は、25℃及び60%相対湿度(25℃/60%RH)であり、これらの条件下で生成されたデータを、それぞれ、5mg錠剤及び40mg錠剤について、表6及び7に示してある。
【0182】
更に、他の貯蔵条件(例えば、40℃/75%RH)も又、6カ月安定性期間の最後まで調べて、生成されたデータを、それぞれ、5mg錠剤及び40mg錠剤について、表8及び9に示した。
【0183】
【表11】
【0184】
表6は、複雑なアッセイを規定しており、不純物、及び5mgの有効成分を含み、ブリスターパックに包装されて、25℃/60%RHで貯蔵された錠剤の安定性研究からの溶解試験結果を与えるものである。
【0185】
【表12】
【0186】
表7は、複雑なアッセイを規定し、不純物、及び40mgの有効成分を含み、ブリスターパックに包装されて、25℃/60%RHで貯蔵された錠剤の安定性研究からの溶解試験結果を与えるものである。
【0187】
【表13】
【0188】
表8は、複雑なアッセイを規定しており、不純物、及び5mgの有効成分を含み、ブリスターパックに包装されて、種々の貯蔵条件下で貯蔵された錠剤の安定性研究からの溶解試験結果を与えるものである。
【0189】
【表14】
【0190】
表9において、アッセイ、不純物及び、40mgの有効成分を含みブリスターパックに包装されて種々の貯蔵条件化で貯蔵された錠剤の安定性研究の溶解の試験が一緒に分類されている。
【0191】
25℃/60%RHでの12カ月の貯蔵の後、このアッセイ及び溶解について、何らの有意の変化は認められなかった。
【0192】
95%の信頼区間での回帰線により得られる結果の統計的分析は、少なくとも3年間の製品品質規格との一致を約束する。
【0193】
40℃の温度及び75%相対湿度(40℃/75%RH)での促進された安定性は、製品品質規格と一致して結果を示す。
【0194】
最も厳しい条件下での製品の安定性は、25℃/60%RHで貯蔵された錠剤につき行なわれた計画において信頼を強化する。
【0195】
5.化合物1種類Iの製造
5.1 化合物1の合成
実施例5: 第一段階は、メタンスルホン酸中での、2−カルベトキシシクロヘプタノンのレゾルシノールとの縮合からなり、この反応は、4時間にわたって、25℃に達する。こうして形成された中間体の3−ヒドロキシ−8,9,10,11−テトラヒドロシクロヘプタ[c]クロメン−6(7H)−オンは、エタノールを添加してから水を添加することにより沈澱し、それは、濾過により単離され、真空下で60℃で乾燥されて、78%の収率が得られる。第二段階において、スルホニルイソシアネートクロリドが、トルエン溶液中で、蟻酸の作用によって、スルファモイルクロリドに転化され、次いで、N,N−ジメチルアセタミド(DMA)中に溶解された前記中間体と縮合される。この反応媒質を、水で処理して、2−メチルテトラヒドロフラン(2−MeTHF)で抽出する。次いで、有機相にメチルシクロヘキサンを加えることにより得られた沈澱の濾過により、粗化合物1が得られる。最後に、純水な化合物1が、熱いアセトンに溶解させてメチルシクロヘキサンを加えることによる沈澱による、粗生成物の再結晶により得られる(貧溶媒メチルシクロヘキサンの添加は、収率の増大を主な目的としている)。
【0196】
化合物1は、こうして、収率65%で得られ、以下に供せられる。
【0197】
こうして得られた実施例1を、粒子サイズの低減を目的とした適当な処理例えば微粉化(ポイント5.3未満)又は湿式粉砕(ポイント5未満)にかけることができる。
【0198】
5.2 水中の化合物1の2型のDMSO溶媒和化合物の脱溶媒
実施例5a:化合物1の2型のDMSO溶媒和化合物
1mLのDMSO(沸点=189℃)を丸薬容器に注ぎ、次いで、1gの化合物1(実施例1)を加える。その溶液を、マグネチックスターラーバー及びマグネチックスターラーを用いて攪拌する。固体を溶液中に急速に通す。1gの化合物1を、再び、マグネチックスターラーで攪拌しながら加える。固体が部分的に溶解してから、ケーキングが認められる。このケーキングの試料を、まだ湿っている間に、X線粉末回折により分析する。この分析は、以下で与えるが、これが化合物1の2型のDMSO溶媒和化合物であることを示す(実施例5a)。
【0199】
実施例5b:
この化合物1の2型のDMSO溶媒和化合物(実施例5b)を冷水に浸し、周囲温度で5分間にわたって攪拌し続ける。その懸濁液を、次いで、濾過して乾燥させる。
【0200】
こうして、化合物1が、50%の収率で得られ、以下に供せられる。
【0201】
こうして得られた実施例5bを、粒子サイズの低減を目的とする適当な処理例えば微粉化又は湿式粉砕にかけることができる。
【0202】
5.3 微粉化
微粉化は、エアジェット超微粉砕機を利用して、18〜25℃の温度で行なわれる。使用する圧搾空気の特性は、以下の通りである:
− CO:<5 ppm、
− CO2:<500 ppm、
− 炭水化物:<0.5 mg.m-3、
− 1立方フィート(0.3048m)当りの粒子数>0.5μ:<10000、
− 最大性能:13バール(13×106ダイン/cm2)で、2300 cfm(立方フィート(0.3048m)/分)、
− 最大運転作業圧:13バール(106ダイン/cm2)。
【0203】
実施例5c:粒子サイズ3μmを有する化合物1の種類I
249gの化合物1(実施例1)を、エアジェット超微粉砕機を利用して微粉化する。この微粉化のパラメーターは、80psi(0.0703kg/cm2)のベンチュリ圧、110psi(0.0703kg/cm2)の超微粉砕機圧及び12kg/時の供給速度である。
【0204】
実施例3−1は、こうして、97%の収率で得られる。
【0205】
実施例5d:粒子サイズ5μmを有する化合物1の種類I
16.5kgの化合物1(実施例5)を、圧搾エアジェット超微粉砕機を用いて微粉化する。微粉化パラメーターは、80psi(0.0703kg/cm2)のベンチュリ圧、32psi(0.0703kg/cm2)の超微粉砕機圧及び14.4kg/時の供給速度である。
【0206】
実施例5dは、こうして、98%の収率で得られて、以下に供される。
【0207】
実施例5e:粒子サイズ9μmを有する化合物1の種類I
150gの化合物1(実施例51)を、圧搾エアジェット超微粉砕機を用いて微粉化する。微粉化パラメーターは、50psi(0.0703kg/cm2)のベンチュリ圧、60psi(0.0703kg/cm2)の超微粉砕機圧及び15kg/時の供給速度である。
【0208】
実施例5eは、こうして、98%の収率で得られる。
【0209】
実施例5f:粒子サイズ15μmを有する化合物1の種類I
39gの化合物1(実施例5)を、圧搾エアジェット超微粉砕機を用いて微粉化する。微粉化パラメーターは、30psi(0.0703kg/cm2)のベンチュリ圧、10psi(0.0703kg/cm2)の超微粉砕機圧及び15分間に40gの供給速度である。
【0210】
実施例5fは、こうして、74%の収率で得られる。
【0211】
5.4 湿式粉砕
実施例5g:0.69gの化合物1(実施例5)を、0.068gのメチルシクロヘキサンと共に、Fritsch型遊星モデルP4ボールミル中で粉砕する。
【0212】
湿式粉砕のパラメーターは、直径10mmの7個のめのうボール、100−100rpmの粉砕トルクΩ(ディスクの回転速度)−ω(フラスコの回転速度)、72回の「10分間の粉砕−5分間の休止」シーケンスに対応する全18時間中での有効な12時間、14.1のボールの質量/溶質質量比;22℃の温度及び9%の化合物1の質量/メチルシクロヘキサン質量比である。
【0213】
実施例3は、こうして、収率99%で得られて、以下に供される。
【0214】
6.得られた結晶の説明
6.1 用いた装置
6.1.1 X線粉末及び単結晶回折
ジーメンスD5005回折計、シンチレーション検出器
・ 波長:1.54056 Cu,電圧40KV、強度40mA
・ 測定レンジ:3°−30°2シータ
・ 間隔:0.04°2シータ
・ 間隔の持続期間:4秒
・ 固定スロット:1.6mm
・ Kβフィルター(Ni)
・ 内部参照なし
・ データ処理のためのEVAソフトウェア(v12.0)
【0215】
Smart Apex Bruker回折計、2次元検出器
・ 結晶マトリクスのパラメーター及び配向の決定のためのSMARTソフトウェア
・ データ積分及び処理のためのSAINTソフトウェア
・ 空間群の決定及び構造分解能のためのWinGXソフトウェア
【0216】
6.1.2 DSC
・ Netzsch DSC 204F1
・ アルミニウムるつぼ及び穴をあけたふた
・ 大気:ヘリウム
・ 初期温度:20℃
・ 最終温度:200℃
・ 温度勾配:5℃/分
【0217】
6.1.3 IR
・ KBrペレット
・ Bruker IFS28型分光計
・ スペクトル範囲:400〜4000cm-1
【0218】
6.1.4 固体のNMR
・ Bruker Avance 500 MHz分光計
・ MAS(マジック角回転)4mmプローブ
・ Bruker XwinNMRソフトウェア
・ VACP(可変振幅直交偏波)及びMAS(11kHz)及びプロトン分離(スピナル64、65kHz)
・ 外部参照:アダマンタン
【0219】
6.1.5 粒子サイズの分布
・ Malvern Mastersizer Sレーザー粒度計
・ 試料のサイズ:30〜50mg
・ 分散媒:0.50%(m/v)Nonidet(水中)
・ 相対的インデックスとして:粒子RI=1.55;虚のRI=1.00を用いるMie理論
・ 超音波処理時間:60秒
・ 超音波処理エネルギー:50〜60Hz
・ 再循環時間:30秒/2000rpm
・ あいまい化:15〜25%
【0220】
6.2. 実施例の特性表示
6.2.1 5及び5bの実施例
単結晶を、エタノール中の飽和溶液の4℃で2日間にわたる、ゆっくりとした蒸発によって単離することは、可能となっている。これらの単結晶は、これらの単結晶は、、化合物1の種類Iの完全な構造を、X線回折により明らかにすることを可能にした。
【0221】
X線による単結晶の結晶回折:化合物1種類Iの結晶構造は、解明されており、下記のセルパラメーターに示してある:
【表15】
【0222】
化合物1の種類Iの換算座標(×104)及び同等の等方性位置パラメーター(Å2×103)は、次の通りである:
【表16】
【0223】
化合物1の種類Iの換算座標(×104)は、次の通りである:
【表17】
【0224】
化合物1の種類Iの格子面間隔は、次の通りである:
【表18】
【0225】
6.2.2 実施例5a
単結晶を、周囲温度で、DMSOの飽和溶液からのゆっくりとした蒸発によって単離することは可能であった。これらの単結晶は、化合物1の2型のDMSO溶媒和化合物の完全な構造をX線回折によって解明することを可能にした。
【0226】
単結晶X線回折:化合物1の2型のDMSO溶媒和化合物の結晶構造を解明し、セルパラメーターを下記に示した:
【表19】
【0227】
この化合物1の2型のDMSO溶媒和化合物のセル中の原子の原子座標(×104)及び同等の等方性転置パラメーター(Å2×103)(U(eq)は、直交テンソルUijの値の3分の1に等しい)は、下記の通りである:
【表20】
【0228】
化合物1の2型のDMSO溶媒和化合物の水素原子の座標(×104)及び同等の等方性転置パラメーター(Å2×103)(U(eq)は、直交テンソルUijの値の3分の1に等しい)は、下記の通りである:
【表21】
【0229】
化合物1の2型のDMSO溶媒和化合物の格子面間隔は、下記の通りである:
【表22】
【0230】
化合物1の2型のDMSO溶媒和化合物のX線粉末回折ダイヤグラムは、ある角度(°2シータ)で表される次の特徴的ピークを示す(約±0.1°2シータ):(図3及び4)。7.6;18.7;24.2;29.9。
【0231】
6.2.3 実施例5c − 5g
6.2.3.1 X線粉末回折(図5及び6)
微粉化後又は湿式粉砕後の化合物1の種類IのX線粉末回折ダイヤグラムは、次の角度(°2シータ)で表される特徴的なピークを示す(約±0.1°2シータ):7.5;10.9;11.7;13.1;14.6;15.0;15.8;17.0;17.3;17.5;17.7;17.9;19.0;19.9;20.0;21.4;21.8;22.5;23.5;23.9;24.7;24.9;25.5;25.9;26.2;26.4;26.6;27.2;27.4;27.6;27.8;28.2;28.8;29.0;29.4;29.8。
【0232】
微粉化又は湿式粉砕後に160℃に加熱した後の化合物1の種類IのX線粉末回折ダイヤグラムは、次の角度(°2シータ)で表される特徴的なピークを示し(約±0.1°2シータ):7.5;10.9;11.7;13.1;14.6;15.0;15.8;17.0;17.3;17.5;17.7;17.9;19.0;19.9;20.0;21.4;21.8;22.5;23.5;23.9;24.7;24.9;25.5;25.9;26.2;26.4;26.6;27.2;27.4;27.6;27.8;28.2;28.8;29.0;29.4;29.8、160℃までの加熱の際に生成された新規な形態に対応する更なるピークはない。
【0233】
6.2.3.2 DSC(図7)
DSCを利用して、微粉化又は湿式粉砕後の化合物1の種類Iの、5℃/分で得られたサーモグラムは、種類Iの溶融ピーク(170℃±5℃で開始)及び、適宜、分析中(180℃±2℃で開始)に生成される新規な多形の小さい溶融ピークを含み、それは、種類Iの溶融ピーク中に交換されるエンタルピーの10%未満を表し、140〜155℃では如何なる吸熱事象(又は、0.5J/g未満)をも示さない。
【0234】
6.2.3.3 IR(図8)
微粉化又は湿式粉砕後の化合物1の種類IのIRスペクトルは、次の特徴的なピークを示す(ほぼ±5cm-1で、cm-1で表示):3310;3167;3059;2928;2858;1690;1605;1454;1385;1261;1188;1126;941;891;853;798;733;679;598;544;455。
【0235】
6.2.3.4 固体のNMR(図9)
微粉化又は湿式粉砕後の化合物1の種類Iの、固体のNMRにより得られるスペクトルは、次の特徴的なピークを示す(ほぼ±0.2ppmまでのppmで表示)163.1;156.1;153.8;153.1;130.4;127.6;107.6;35;25。
【0236】
6.2.3.5 粒子サイズ
実施例2−2による微粉化後の化合物1の種類Iの粒子サイズ分布は、次の通りである:D10(%)=1.2μm;D50(%)=5.2μm及びD90(%)=11.0μm。
【0237】
実施例3による湿式粉砕後の化合物1の種類Iの粒子サイズ分布は、次の通りである:D10(%)=0.6μm;D50(%)=3.1μm及びD90(%)=13.7μm。
【0238】
7.配合物
7.1 ゼラチンカプセル形態の配合物
この発明による化合物1の種類Iは、5〜30%の有効成分(好ましくは、6〜13%)、40〜92%の希釈剤(好ましくは、65〜92%、非常に好ましくは85〜90%)、0〜30%の崩壊剤(好ましくは、0〜22%、非常に好ましくは、0%)、0〜5%の界面活性剤(好ましくは、0%)、0〜5%の可溶化剤(好ましくは、0%)、0.1〜3%の流動剤(好ましくは、0.9〜1.4%)、0.5〜3%の潤滑剤(好ましくは、0.6〜2.8%)を含むゼラチンカプセルに配合することができる。
【0239】
これらのゼラチンカプセルを配合するための好適な賦形剤は、マンニトール、ラクトース、トウモロコシ澱粉、コロイドシリカ、ステアリン酸マグネシウム、及びラウリル硫酸ナトリウムであり、一層特には、マンニトール、ラクトース、コロイドシリカ、及びステアリン酸マグネシウムである。
【0240】
下記のゼラチンカプセルを、粉末を、当業者に公知の標準的技術によって混合することにより作成した。
【表23】
M=マンニトール; L=ラクトース; CS=トウモロコシ澱粉; SLS=ラウリル硫酸ナトリウム; PEG=ポリエチレングリコール; CS=コロイドシリカ; MS=ステアリン酸マグネシウム
【0241】
実施例7j:ゼラチンカプセル7jの実施例は、実施例5(40mgの化合物1を260mgのラクトースと混合して、ゼラチンカプセルに入れる)の化合物1から製造される。
【0242】
実施例7k:ゼラチンカプセル7kの実施例は、有効成分として実施例5dの代わりに実施例5cを用いる配合7gにより製造される。
【0243】
実施例7l:ゼラチンカプセル7lの実施例は、7.2%の実施例5d、31.9%のマンニトール、59.4%のラクトース及び1.5%のコロイドシリカを用いて製造される。
【0244】
実施例7m:ゼラチンカプセル7mの実施例は、配合7gにより、実施例5dを有効成分として用いて製造される。
【0245】
実施例7n:ゼラチンカプセル4nの実施例は、配合4gにより、実施例5eを実施例5dの代わりに有効成分として用いて製造される。
【0246】
実施例7o:ゼラチンカプセル4oの実施例は、有効成分として、実施例5f(40mgの実施例5fを、260mgのラクトースと混合して、ゼラチンカプセル内に入れる)を用いて製造される。
【0247】
7.2 錠剤の形態の配合物
この発明による化合物1の種類Iは、以下を含む錠剤に配合することができる:5〜30%の有効成分(好ましくは、7〜20%、非常に好ましくは、10〜15%)、40〜92%の希釈剤(好ましくは、34〜89%、非常に好ましくは、70〜85%)、0〜40%の崩壊剤(好ましくは、0〜20%、非常に好ましくは、3〜5%)、0〜8%の結合剤(好ましくは、2〜5%)、0.1〜3%の流動剤(好ましくは、0.5〜1.4%)、0.5〜3%の潤滑剤(好ましくは、0.5〜2.8%)。
【0248】
これらのゼラチンカプセルの配合のための好適な賦形剤は、マルトデキストリン、マンニトール、微結晶性セルロース、ラクトース、トウモロコシ澱粉、澱粉グリコール酸ナトリウム、クロスポビドン、ポリビニルピロリドン、カルボキシメチルセルロース、コロイドシリカ、フマル酸ステアリルマグネシウム、及びステアリン酸マグネシウム、一層特には、微結晶性セルロース、ラクトース、澱粉グリコール酸ナトリウム、ポリビニルピロリドン、コロイドシリカ、及びステアリン酸マグネシウムである。
【0249】
下記の錠剤は、当業者に公知の標準的技術により、湿式造粒法によって製造したものである。
【表24】
【0250】
8.物理化学的及び生物学的特性
8.1 溶解速度論
溶解速度論(時間の関数として溶解した化合物1のパーセンテージとして表示)は、当業者に公知の標準的技術によって測定され、下記の表に与えてある。
【表25】
【0251】
上記の溶解結果は、理論強度のパーセンテージとして表されている。ゼラチンカプセル4j〜4nの理論強度は、有効成分の40mgである。これらの実施例は、実験的なものであり、有効成分の重量測定時の僅かな過多はありうることであり、最終溶解点における100%より大きいパーセンテージを説明する。
【0252】
8.2 生物学的利用能
化合物1の比較用の生物学的利用能を、イヌにおいて、2つのゼラチンカプセルの経口経路による単一投与後に研究した。投与後、1;1.5;2;4;6;9;12;24;30及び48時間で、血液試料を採取する。曲線下領域(AUC)は、これらのイヌからの血漿試料において測定される薬物速度論的パラメーターの一つである。得られた結果を下記の表に与える。
【表26】
【0253】
8.3 化学的安定性
安定性の研究を100℃で、化合物1のI型と特許EP880514に記載された化合物1との間で行なった。化合物1の化学的安定性は、異なるスコアのHPLCを用いて研究される。
【0254】
HPLC法の操作条件は、次の通りである:
・ カラム:Interchim UP3HDO-15XS, 150×4.6mm、
・ 溶離剤A: −水 2500
−トリフルオロ酢酸 0.5
・ 溶離剤B: −アセトニトリル
・ 勾配:
【表27】
・ 検出:205nm、
・ 注入:20マイクロリットル、
・ 温度:40℃、
・ 注入した溶液:0.5mg.mL-1(アセトニトリル)
【表28】
【0255】
9.III型の化合物1の製造
本願に記載したように化合物1のIII型は、化合物1(実施例5)の合成の段階と、その後の更なる段階により得られ、それは、下記であってよい:
− 水中での1型のDMSO溶媒和化合物の脱溶媒:実施例9a、
− 水中での3型のDMSO溶媒和化合物の脱溶媒:実施例9b、
− エタノール中での微粒化:実施例9c、
− アセトン中での微粒化:実施例9d、
− クメン中での、還流下での再糊状化:実施例9e、
− 熱処理と、その後の微粒化と、その後の第二の熱処理:実施例9f、
− II型の熱処理:実施例9g。
【0256】
9.1 1型のDMSO溶媒和化合物の水中での脱溶媒
実施例9aa:化合物1の1型のDMSO溶媒和化合物
1mLのDMSO(沸点=189℃)を、丸薬容器に注ぎ、次いで、1gの化合物1(実施例5)を加える。この溶液を、マグネチックスターラーバー及びマグネチックスターラーを用いて攪拌する。この固体は、溶液中へと急速に消え去る。1gの化合物1を、再び、やはりマグネチックスターラーで攪拌しながら加える。この固体は、部分的に溶解し、その後、ケーキングが認められる。このケーキングの試料を、未だ湿気があるうちに、X線粉末回折により分析する。この分析は、以下に与えるが、これが化合物1の1型のDMSO溶媒和化合物(実施例9aa)であることを示す。
【0257】
実施例9a:
この化合物1の2型のDMSO溶媒和化合物(実施例9aa)を、冷水に浸し、攪拌下に、周囲温度で5分間置く。その懸濁液を、次いで、濾過して乾燥する。
こうして、III型の化合物1は、収率50%で得られ、以下に供せられる。
【0258】
9.2 3型のDMSO溶媒和化合物の水中での脱溶媒
実施例9ba:化合物1の3型のDMSO溶媒和化合物
1mLのDMSO(沸点=189℃)を、丸薬容器に注ぎ、次いで、1gの化合物1(実施例1)を加える。この溶液を、マグネチックスターラーバー及びマグネチックスターラーを用いて攪拌する。固体は、急速に、溶液中に消え去る。この溶液を、マグネチックスターラーによる攪拌下に24時間放置する。次いで、ケーキングが認められる。ケーキングの試料を、未だ湿気のあるうちに、X線粉末回折により分析する。この分析は、以下で与えるが、これが化合物1の3型のDMSO溶媒和化合物(実施例9ba)であることを示す。
【0259】
実施例9b:
化合物1の3型のDMSO溶媒和化合物(実施例3ba)を、冷水に浸し、攪拌しながら周囲温度で5分間放置する。次いで、上清を濾過して乾燥させる。
こうして、III型の化合物1が、収率50%で、得られ、以下に供せられる。
【0260】
9.3 エタノール中での微粒化
実施例9c:
得られた生成物は、Buchi 190 スプレードライヤーを用いて生成される。2gの化合物1(実施例5)を、200mLのエタノール(沸点=78℃)に溶解させる。乾燥空気入口温度は、3バール(3×106ダイン/cm2)の圧力で、90℃に調節される。出口温度は、微粒化中、38℃で測定される。出口での流量は、700Nl/時に調節される。570mgの乾燥粉末が回収され、X線粉末回折により分析される。この分析は、これが、化合物1のIII型であり、それが、構造的に純粋であることを、解明された単結晶構造から計算されたダイヤグラムとの比較により示す。
こうして、III型の化合物1は、収率29%で得られ、以下に供せられる。
【0261】
9.4 アセトン中での微粒化
実施例9d:
得られた生成物は、不活性ループを含むBuchi 290 スプレードライヤーを用いて生成される。2gの化合物1(実施例1)を100mLのアセトン(沸点=56℃)に溶解させる。乾燥空気入口温度を、70℃に調節する。出口温度は、微粒化中、50℃にて測定する。出口流量は、7600Nl/時に調節する。1gの乾燥粉末を、回収してX線粉末回折により分析する。この分析は、これが、化合物1のIII型であり、それが、構造的に純粋であることを、解明された単結晶構造から計算されたダイヤグラムとの比較により示す。
こうして、III型の化合物1が、収率50%で得られ、以下に供せられる。
【0262】
9.5 還流下でのクメン中での再糊状化
実施例9e:
10gの化合物1(実施例5)を、100mLのクメン(沸点=152℃)中に懸濁させる。この混合物を、攪拌下に、還流させ、次いで、周囲温度まで冷却する。回収されて乾燥された粉末の分析は、それが、化合物1のIII型であることを示す。
こうして、III型の化合物1が、収率99%で得られ、以下に供せられる。
【0263】
9.6 熱処理と、その後の微粉化と、その後の第二の熱処理
実施例9f:
150gの化合物1を、通風式オーブン中で、160℃で15分間加熱する。そうして加熱した生成物を、次いで、圧搾エアジェット超微粉砕機を用いて微粉化する。微粉化パラメーターは、第一の関門としての、80psi(5.624kg/cm2)のベンチュリ圧、120psi(8.436kg/cm2)の超微粉砕機の圧力及び1.2kg/時の供給速度及び第二の関門としての、50psi(3.515kg/cm2)のベンチュリ圧、50psi(3.515kg/cm2)の超微粉砕機の圧力及び1.2kg/時の供給速度である。微粉化の収率は、69%である。こうして得られた15gの生成物を、160℃で15分間、通風式オーブン中で加熱する。
こうして、III型の化合物1が、収率69%で得られ、以下に供せられる。
【0264】
9.7 II型の熱処理
9.7.1 II型の獲得
化合物1のII型は、2つの合成経路によって得ることができる:
− 予め1,4−ジオキサンからの沈澱により得られた化合物1の1,4−ジオキサン半溶媒和化合物の脱溶媒;
− 1,4−ジオキサンにおける直接的微粉化。
【0265】
実施例9h:化合物1の1,4−ジオキサン半溶媒和化合物
1,4−ジオキサン中の化合物1(実施例5)の溶液を、周囲温度で調製し、攪拌下に、24時間置いた。攪拌中、有意の沈澱が認められた。次いで、その固体を濾過して、X線粉末回折により分析した。この段階の最後で得られた生成物は、化合物1の1,4−ジオキサン半溶媒和化合物である。
こうして、化合物1の1,4−ジオキサン半溶媒和化合物が、収率80%で得られ、以下に供せられる。
【0266】
実施例9i:II型の化合物1
第一の合成経路に従って、化合物1(実施例9h)の1,4−ジオキサン半溶媒和化合物を、不活性ガス流中で、20℃から80℃まで、5℃/分で加熱し、脱溶媒により、化合物1のII型を生成する(X線回折パターンにより特性決定)。
こうして、II型の化合物1が、収率99%で得られ、以下に供せられる。
【0267】
第二の合成経路に従って、II型の化合物1を次のように得ることができる:1gの化合物1(実施例5)を100mLの1,4−ジオキサン(沸点=101℃)に溶解させた。入口温度:無塵の乾燥空気を、3バール(3×106ダイン/cm2)の圧力で、130℃に調節した。出口温度を、微粉化中、88℃で測定した。出口流量を、700Nl/時に調節した。この段階の最後に得られた生成物は、II型である。
こうして、II型の化合物1が、収率50%で得られ、以下に供せられる。
【0268】
9.7.2 III型の獲得
実施例9g:
II型の化合物1(実施例9i)の回収された乾燥粉末を、160℃のオーブン中に、6〜10分間、好ましくは7.5分間置いた。この処理の後に回収され、X線粉末回折により分析された粉末は、化合物1のIII型であり、これは、解明された単結晶構造から得られる計算されたダイヤグラムとの比較により構造的に純粋である。
こうして、III型の化合物1が、収率99%で得られ、以下に供せられる。
【0269】
10.得られた固相の特性決定
10.1 使用する機器
10.1.1 X線粉末回折及び単結晶回折
シーメンスD5005回折計、シンチレーション検出器
− 波長:1.54056 Cu、電圧 40 KV、電流の強さ 40 mA
− 測定レンジ:3° - 30°2シータ
− 0.04°2シータ
− 間隔:0.04°2シータ
− 間隔の持続時間:4秒
− 固定スロット:1.6mm
− Kβフィルター(Ni)
− 内部標準なし
− データ処理用EVAソフトウェア(v12.0)
【0270】
Smart Apex Bruker回折計、二次元検出器
− 結晶マトリクスのパラメーター及び配向の決定のためのSMARTソフトウェア
− データの積分及び処理のためのSAINTソフトウェア
− 空間群の決定及び構造解明のためのWinGXソフトウェア
【0271】
10.1.2 DSC
− NetzschDSC204F1
− アルミニウム製るつぼ及び穴の開いた蓋
− 大気:ヘリウム
− 初期温度:20℃
− 最終温度:200℃
− 温度勾配:5℃/分
【0272】
10.1.3 TG−DSC
− NetzschSTA449C
− アルミニウム製るつぼ及び穴の開いた蓋
− 大気:ヘリウム
− 初期温度:25℃
− 最終温度:200℃
− 温度勾配:5℃/分
【0273】
10.1.4 IR
− KBrペレット
− Bruker IFS28型分光計
− スペクトル範囲:400〜4000cm-1
【0274】
10.1.5 固体のNMR
− Bruker Avance 500MHz分光計
− MAS(Magic Angle Spinning) 4mmプローブ
− Bruker XwinNMRソフトウェア
− VACP(Variable Amplitude Cross-Polarization)及びMAS(11kHz)及びプロトン分離(スピナル64、65kHz)
− 外部標準:アダマンタン
【0275】
10.2 これらの実施例の特性決定
10.2.1 化合物1のI型
化合物1のI型のX線粉末回折ダイヤグラムは、角度(°2シータ)で表される次の特徴的なピークを示す(約±0.1°2シータ):7.5;10.9;11.7;13.1;14.6;15.0;15.8;17.0;17.3;17.5;17.7;17.9;19.0;19.9;20.0;21.4;21.8;22.5;23.5;23.9;24.7;24.9;25.5;25.9;26.2;26.4;26.6;27.2;27.4;27.6;27.8;28.2;28.8;29.0;29.4;29.8。
【0276】
10.2.2 化合物1の1型のDMSO溶媒和化合物
単結晶を、DMSOの飽和溶液を周囲温度でゆっくり蒸発させることによって単離することは可能であった。これらの単結晶は、化合物1の1型のDMSO溶媒和化合物の完全な構造をX線回折により解明することを可能にした。
【0277】
単結晶のX線回折:化合物1の1型のDMSO溶媒和化合物の結晶構造は、下記のセルパラメーターを示す。
【表29】
【0278】
セル中の原子(×104)の原子座標及び同等の等方性置換パラメーター(Å2×103)(U(eq)は、直交テンソルUijの値の3分の1に等しい)は、下記の通りである:
【表30】
【0279】
これらの水素原子(×104)の座標及び同等の等方性置換パラメーター(Å2×103)(U(eq)は、直交テンソルUijの値の3分の1に等しい)は、下記の通りである:
【表31】
【0280】
DMSO溶媒和化合物1型の格子面間隔は、下記の通りである:
【表32】
【0281】
化合物1の1型のDMSO溶媒和化合物のX線粉末回折ダイヤグラムは、角度(°2シータ)で表される下記の特徴的ピークを示す(約±0.1°2シータ):6.6;10.9;13.1;14.3;15.7;16.7;17.4;18.3;19.6;20.8;21.9;22.6;23.0;24.7;24.9;25.2;25.5;25.8;26.6;26.9;27.2;28.3(図1及び2)。
【0282】
2.2.3 化合物1の3型のDMSO溶媒和物化合物
単結晶を、DMSOの飽和溶液を周囲温度でゆっくりと蒸発させることにより単離することが可能であった。これらの単結晶は、化合物1の3型のDMSO溶媒和化合物を、X線回折により、完全な構造を解明することを可能にした。
【0283】
単結晶のX線回折:化合物1の3型のDMSO溶媒和化合物の結晶構造を、解明して、下記のセルパラメーターを示す:
【表33】
【0284】
これらの水素原子(×104)の座標及び同等の等方性置換パラメーター(Å2×103)(U(eq)は、直交テンソルUijの値の3分の1に等しい)は、下記の通りである:
【表34】
【0285】
これらの水素原子(×104)の座標及び同等の等方性置換パラメーター(Å2×103)(U(eq)は、直交テンソルUijの値の3分の1に等しい)は、下記の通りである:
【表35】
【0286】
DMSO溶媒和化合物3型の格子面間隔は、次の通りである:
【表36】
【0287】
化合物1の3型のDMSO溶媒和化合物のX線粉末回折ダイヤグラムは、角度(°2シータ)で表される下記の特徴的ピークを示す(約±0.1°2シータ):(図3及び4)。9.7;13.9;16.0;17.8;19.1;22.1。
【0288】
2.2.4 III型の単結晶のX線回折
アセトン/n−ヘプタン混合物(50% v/v)中の化合物1の飽和溶液を周囲温度でゆっくり蒸発させることにより、X線回折により化合物1のIII型の完全な構造を解明することを可能にした単結晶を単離することが可能であった。
【0289】
単結晶X線回折:III型の化合物1の結晶構造を解明して、下記のセルパラメーターを示す。
【表37】
【0290】
III型の化合物1の換算座標(×104)及び同等の等方性位置パラメーター(Å2×103)は、次の通りである:
【表38】
【0291】
これらの水素原子の座標(×104)及び同等の等方性転置パラメーター(Å2×103)(U(eq)は、直交テンソルUijの値の3分の1に等しい)は、下記の通りである:
【表39】
【0292】
格子面間隔は、下記の通りである:
【表40】
【0293】
10.2.5 実施例9a〜9g:III型の化合物1
この固体のX線粉末回折、単結晶X線回折、DSC、IR及びNMRによる分析は、実施例9aが実施例9b、実施例9c、実施例9d、実施例9e、実施例9f及び実施例9gと等しいことを示している。
【0294】
10.2.5.1 X線粉末回折
化合物1のIII型のX線粉末回折は、角度(°2シータ)で表される次の特徴的ピークを示す(約±0.1°2シータ):8.6;11.3;12.0;16.7;17.4;17.9;20.9;22.6;23.0;23.4;23.8;25.7;28.6(図5及び6)。
【0295】
10.2.5.2 DSC
DSCにおいて、化合物1のIII型のサーモグラムは、III型の溶融ピーク(180℃±2℃で開始)を含む(図7)。
【0296】
10.2.5.3 IR
化合物1のIII型のIRスペクトルは、cm-1で現される特徴的ピークを示す(約±5cm-1):3406;3217;3082;2924;1678;1385;1269;1134;1011;934;845;601;563;536(図8)。
【0297】
10.2.5.4 固体のNMR
この固体のNMRにより得られたIII型のスペクトルは、次のppmで表される特徴的ピークを示す(約±0.2ppm):162.9;156.4;151.3;126.1;124.7;118.8;117.1;112.9;35;20(図9)。
【0298】
10.2.6 実施例9h:化合物1の1,4−ジオキサン半溶媒和化合物
10.2.6.1 単結晶のX線回折
1,4−ジオキサン中の化合物1の飽和溶液を周囲温度でゆっくりと蒸発させることにより、化合物1の1,4−ジオキサン半溶媒和化合物の完全な構造をX線回折によって解明することを可能にする単結晶を単離することが可能であった。
【0299】
化合物1の1,4−ジオキサン半溶媒和化合物の結晶構造は、下記のセルパラメーターを示す:
【表41】
【0300】
化合物1の1,4−ジオキサン半溶媒和化合物の換算座標(×104)及び同等の等方性運動パラメーター(Å2×103)は、下記の通りである:
【表42】
【0301】
化合物1の1,4−ジオキサン半溶媒和化合物の水素の座標(×104)及び同等の等方性転置パラメーター(Å2×103)は、下記の通りである:
【表43】
【0302】
1,4−ジオキサン半溶媒和化合物の格子面間隔は、下記の通りである:
【表44】
【0303】
10.2.6.2 X線粉末回折
化合物1の1,4−ジオキサン半溶媒和化合物のX線粉末回折ダイヤグラムは、角度(°2シータ)で表される次の特徴的ピークを示す(約±0.1°2シータ):9.8;10.0;10.8;13.6;13.9;14.1;15.9;16.1;18.0;18.3;19.0;19.6;20.0;20.1;20.2;20.6;21.4;21.7;21.8;22.0;22.2;22.3;23.4;23.9;24.3;24.4;24.9;25.3;25.4;26.2;27.0;27.1;27.3;27.5;28.2;28.4;28.5;28.7;29.1;29.4(図10及び11)。
【0304】
10.2.6.2 TG−DSC
TG−DSCを用いて、化合物1の1,4−ジオキサン半溶媒和化合物のサーモグラムは、この溶媒和化合物が、化合物1のII型を生成するために、75℃から、1,4−ジオキサンを放出し始めていることを示している(図21)。
【0305】
10.2.7 実施例9i:II型の化合物1
10.2.7.1 X線粉末回折
化合物1のII型のX線粉末回折ダイヤグラムは、角度(°2シータ)で表される次の特徴的ピークを示す(約±0.1°2シータ):9.4;10.7;12.2;12.8;14.3;15.1;15.9;16.9;18.2;18.9;19.4;19.7;20.4;21.1;21.4;21.8;22.0;22.7;23.0;23.2;23.7;23.9;24.4;24.8;25.5;26.8;27.1;27.4;28.1;28.3;29.5(図12及び13)。
【0306】
10.2.7.2 DSC
DSCを用いて、化合物1のII型のサーモグラムは、吸熱現象を含み、これは、II型の165℃±5℃での準安定性溶融と、その後の他の型への再結晶化と、それに続く別の溶融に対応している(図23)。
【0307】
10.2.7.3 IR
化合物1のII型のIRスペクトルは、cm-1で表される特徴的ピークを示す(約±5-1):3356;3321;3186;3078;2932;2851;1693;1609;1504;1462;1377;1265;1192;1123;937;872;837;787;594(図14及び15)。
【0308】
10.2.7.4 固体のNMR
この固体のNMRにより得られたII型のスペクトルは、次のppmで表される特徴的ピークを示す(約±0.2ppm):163.0;154.5;153.5;151.6;128.6;118.5;111.4;108.2;35;25(図16)。
【0309】
11. 物理化学的及び生物学的特性
11.1 転化実験
出願人は、化合物1の種々の多形相間での転化実験を行なっており、これは、III型が高温(T>145℃)で安定な型であることを示している。この有意の特性は、極微量の溶媒を真空下での加熱により除去することを可能にする(Polymorphism in the Pharmaceutical Industry, Wiley 2006, Hilfiker編; Wiley, ISBN: 978-3-527-31146-0)。周囲温度に戻すことにより、化合物1のIII型は、不変のままであり、これも又、製薬産業における主たる利点を構成する。
【0310】
11.2 化学的性
安定性の研究を、III型の化合物1と特許EP880514に記載された化合物1との間で、150℃で行なった。この化合物1の化学的安定性は、HPLCを用いて行なった。
【0311】
HPLC法の操作条件は、下記の通りである:
・カラム:Interchim UP3HDO-15XS, 150 × 4.6 mm、
・ 溶離液A: −水 .................. 2500
−トリフルオロ酢酸 ... 0.5
・溶離液B: −アセトニトリル ..........
・ 勾配:
【表45】
・検出:205nm、
・注入:20マイクロリットル、
・ 温度:40℃、
・注入される溶液:0.5mg.mL-1(アセトニトリル)
【表46】
【特許請求の範囲】
【請求項1】
6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートを有効成分として含み且つ少なくとも一種類の製薬上許容しうる賦形剤を含む固体の医薬組成物。
【請求項2】
経口投与のための固体形態がゼラチンカプセルである、請求項1に記載の医薬組成物。
【請求項3】
固体形態が錠剤である、請求項1に記載の医薬組成物。
【請求項4】
錠剤が、組成物の全重量に対して、1〜30%の有効成分;40〜92%の希釈剤;0.1〜20%の崩壊剤;0.1〜8%の結合剤;0.1〜3%の滑剤;及び0.5〜3%の潤滑剤を含み、並びに被覆錠剤の全重量に対して約4.8%の被覆溶液を成分として含む、請求項3に記載の医薬組成物。
【請求項5】
希釈剤が、次の賦形剤から選択される、請求項4に記載の医薬組成物:マンニトール、ラクトース若しくはラクトース一水和物、澱粉、炭酸カルシウム、微結晶性セルロース、又はマルトデキストリン。
【請求項6】
崩壊剤が、次の賦形剤から選択される、請求項4に記載の医薬組成物:澱粉、クロスカルメロースナトリウム、澱粉グリコール酸ナトリウム、又はクロスポビドン。
【請求項7】
結合剤が、次の賦形剤から選択される、請求項4に記載の医薬組成物:ポリビニルピロリドン、N−ビニル−2−ピロリドンと酢酸ビニルとのコポリマー(即ち、コポビドン)、カルボキシメチルセルロース(CMC)、アルファ澱粉、又はメチルセルロース。
【請求項8】
潤滑剤が、次の賦形剤から選択される、請求項4に記載の医薬組成物:ステアリン酸マグネシウム、ナトリウムステアリルフマレート、ステアリン酸カルシウム又は水素化植物油。
【請求項9】
微結晶性セルロース(MCC)及び/若しくはコポビドン又は微結晶性セルロース(MCC)及び/若しくはカルボキシメチルセルロース(CMC)の何れかを含む、請求項1及び2〜8に記載の医薬組成物。
【請求項10】
錠剤の形態の、錠剤の全重量に対して、8〜20%の有効成分;20〜40%のラクトース及び25〜50%の微結晶性セルロース(希釈剤として使用);2〜8%のコポビドン(結合剤として使用);1〜5%の澱粉グリコール酸ナトリウム(崩壊剤として使用);0.2〜1.4%の流動剤;及び0.5〜2%の潤滑剤を含む、請求項1及び3〜9の一つに記載の医薬組成物。
【請求項11】
6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートが、1〜20μmの粒子サイズを有する、請求項1〜10いずれか一つに記載の医薬組成物。
【請求項12】
6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートが、1〜15μmの粒子サイズを有する、請求項11に記載の医薬組成物。
【請求項13】
即時的放出のためであることを特徴とする、請求項1〜12いずれか一つに記載の医薬組成物。
【請求項14】
請求項1〜13いずれか一つに記載の医薬組成物であって、非被覆錠剤は、800mgの総重量を、好ましくは400mgの総重量を超えない当該医薬組成物。
【請求項15】
6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートから出発する請求項1〜14いずれか一つに記載の固体医薬組成物の製造方法であって、有効成分の粒子サイズを減じる段階を含むことを特徴とする当該製造方法。
【請求項16】
粒子サイズを微粉化により減じる、請求項15に記載の方法。
【請求項17】
下記の段階を含む、請求項1〜14いずれか一つに記載の固体医薬組成物の製造方法であって、混合物が、粉末に加えられた場合にその流動性を改善することを可能にする物質、滑剤等を含むことができる当該製造方法:
・ これらの成分をふるいにかけること
・ これらの成分の湿式造粒とその後の混合及び圧縮
・ コーティング溶液の製造。
【請求項18】
滑剤が、二酸化ケイ素のコロイド溶液である、請求項17に記載の方法。
【請求項19】
湿式造粒段階において、有効成分及び予め混合した結合剤、希釈剤及び崩壊剤の全質量に対する水の質量が、約10〜30%である、請求項17に記載の方法。
【請求項20】
湿式造粒段階後に、これらの顆粒が、3%未満の残留水分が得られるまで乾燥される、請求項17に記載の方法。
【請求項21】
0.1〜20μmの粒子サイズの6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートの多形性化合物であって、X線粉末回折を用いて、角度(°2シータ)で表される特徴的ピーク(±0.1°2シータ):7.5;10.9;13.1;17.7;19.0を示す当該多形性化合物。
【請求項22】
粒子サイズが1〜15μmである、請求項21に記載の化合物。
【請求項23】
粒子サイズが3〜7μmである、請求項21に記載の化合物。
【請求項24】
5μm±1μmのサイズを有する、請求項21に記載の化合物。
【請求項25】
X線粉末回折により、角度(°2シータ)で表される次の特徴的ピークを示す、請求項21〜24の一つに記載の化合物(±0.1°2シータ):7.5;10.9;13.1;15.0;15.8;17.0;17.7;19.0;22.5。
【請求項26】
160℃まで加熱してから18〜25℃の温度に戻した後に行なわれるX線粉末回折により、角度(°2シータ)で表される次の特徴的ピークを示す、請求項21〜25の一つに記載の化合物(±0.1°2シータ):7.5;10.9;13.1;17.7;19.0(160℃への加熱中に生成された型に対応する更なるピークを含まない)。
【請求項27】
5℃/分の温度勾配のDSCを用いて、吸熱性溶融ピークを170℃±5℃に示し且つ吸熱性ピークを180℃±2℃に示し、180℃でのピークは、170℃での溶融中に交換されたエンタルピーの最大で10%を表すと理解される、請求項21〜25の一つに記載の化合物。
【請求項28】
DSCを用いて、140〜155℃で吸熱事象を示さない、請求項1〜7の一つに記載の化合物。
【請求項29】
赤外線分光法を用いて、cm-1で表される次の特徴的なピークを示す、請求項1〜8の一つに記載の化合物(±5cm-1):3310;3167;3059;891;798;733;679;455。
【請求項30】
X線粉末回折を用いて、角度(°2シータ)で表される次の特徴的なピーク(±0.1°2シータ):8.6;11.3;28.6を示す、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートの多形性化合物。
【請求項31】
X線粉末回折を用いて、角度(°2シータ)で表される次の特徴的なピークを示す、請求項30に記載の化合物(±0.1°2シータ):8.6;11.3;12.0;16.6;20.9;23.0;28.6。
【請求項32】
単結晶X線回折を利用して、下記のセルパラメーターを示し:
【表1】
下記の換算座標(×104)及び同等の等方性の運動パラメーター(Å2×103)を示す、請求項30又は31の一つに記載の化合物:
【表2】
【請求項33】
5℃/分までのDSCを用いて、180℃±2℃の吸熱性溶融ピークを示す、請求項30〜32いずれか一つに記載の化合物。
【請求項34】
赤外線分光分析を用いて、cm-1で表される次の特徴的なピークを示す(±5cm-1):3406;3217;1678;1011;563、請求項30〜33の一つに記載の化合物。
【請求項35】
赤外線分光分析を用いて、cm-1で表される次の特徴的なピークを示す(±5cm-1):3406;3217;3082;2924;1678;1385;1269;1134;1011;934;845;601;563;536、請求項34に記載の化合物。
【請求項1】
6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートを有効成分として含み且つ少なくとも一種類の製薬上許容しうる賦形剤を含む固体の医薬組成物。
【請求項2】
経口投与のための固体形態がゼラチンカプセルである、請求項1に記載の医薬組成物。
【請求項3】
固体形態が錠剤である、請求項1に記載の医薬組成物。
【請求項4】
錠剤が、組成物の全重量に対して、1〜30%の有効成分;40〜92%の希釈剤;0.1〜20%の崩壊剤;0.1〜8%の結合剤;0.1〜3%の滑剤;及び0.5〜3%の潤滑剤を含み、並びに被覆錠剤の全重量に対して約4.8%の被覆溶液を成分として含む、請求項3に記載の医薬組成物。
【請求項5】
希釈剤が、次の賦形剤から選択される、請求項4に記載の医薬組成物:マンニトール、ラクトース若しくはラクトース一水和物、澱粉、炭酸カルシウム、微結晶性セルロース、又はマルトデキストリン。
【請求項6】
崩壊剤が、次の賦形剤から選択される、請求項4に記載の医薬組成物:澱粉、クロスカルメロースナトリウム、澱粉グリコール酸ナトリウム、又はクロスポビドン。
【請求項7】
結合剤が、次の賦形剤から選択される、請求項4に記載の医薬組成物:ポリビニルピロリドン、N−ビニル−2−ピロリドンと酢酸ビニルとのコポリマー(即ち、コポビドン)、カルボキシメチルセルロース(CMC)、アルファ澱粉、又はメチルセルロース。
【請求項8】
潤滑剤が、次の賦形剤から選択される、請求項4に記載の医薬組成物:ステアリン酸マグネシウム、ナトリウムステアリルフマレート、ステアリン酸カルシウム又は水素化植物油。
【請求項9】
微結晶性セルロース(MCC)及び/若しくはコポビドン又は微結晶性セルロース(MCC)及び/若しくはカルボキシメチルセルロース(CMC)の何れかを含む、請求項1及び2〜8に記載の医薬組成物。
【請求項10】
錠剤の形態の、錠剤の全重量に対して、8〜20%の有効成分;20〜40%のラクトース及び25〜50%の微結晶性セルロース(希釈剤として使用);2〜8%のコポビドン(結合剤として使用);1〜5%の澱粉グリコール酸ナトリウム(崩壊剤として使用);0.2〜1.4%の流動剤;及び0.5〜2%の潤滑剤を含む、請求項1及び3〜9の一つに記載の医薬組成物。
【請求項11】
6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートが、1〜20μmの粒子サイズを有する、請求項1〜10いずれか一つに記載の医薬組成物。
【請求項12】
6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートが、1〜15μmの粒子サイズを有する、請求項11に記載の医薬組成物。
【請求項13】
即時的放出のためであることを特徴とする、請求項1〜12いずれか一つに記載の医薬組成物。
【請求項14】
請求項1〜13いずれか一つに記載の医薬組成物であって、非被覆錠剤は、800mgの総重量を、好ましくは400mgの総重量を超えない当該医薬組成物。
【請求項15】
6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートから出発する請求項1〜14いずれか一つに記載の固体医薬組成物の製造方法であって、有効成分の粒子サイズを減じる段階を含むことを特徴とする当該製造方法。
【請求項16】
粒子サイズを微粉化により減じる、請求項15に記載の方法。
【請求項17】
下記の段階を含む、請求項1〜14いずれか一つに記載の固体医薬組成物の製造方法であって、混合物が、粉末に加えられた場合にその流動性を改善することを可能にする物質、滑剤等を含むことができる当該製造方法:
・ これらの成分をふるいにかけること
・ これらの成分の湿式造粒とその後の混合及び圧縮
・ コーティング溶液の製造。
【請求項18】
滑剤が、二酸化ケイ素のコロイド溶液である、請求項17に記載の方法。
【請求項19】
湿式造粒段階において、有効成分及び予め混合した結合剤、希釈剤及び崩壊剤の全質量に対する水の質量が、約10〜30%である、請求項17に記載の方法。
【請求項20】
湿式造粒段階後に、これらの顆粒が、3%未満の残留水分が得られるまで乾燥される、請求項17に記載の方法。
【請求項21】
0.1〜20μmの粒子サイズの6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートの多形性化合物であって、X線粉末回折を用いて、角度(°2シータ)で表される特徴的ピーク(±0.1°2シータ):7.5;10.9;13.1;17.7;19.0を示す当該多形性化合物。
【請求項22】
粒子サイズが1〜15μmである、請求項21に記載の化合物。
【請求項23】
粒子サイズが3〜7μmである、請求項21に記載の化合物。
【請求項24】
5μm±1μmのサイズを有する、請求項21に記載の化合物。
【請求項25】
X線粉末回折により、角度(°2シータ)で表される次の特徴的ピークを示す、請求項21〜24の一つに記載の化合物(±0.1°2シータ):7.5;10.9;13.1;15.0;15.8;17.0;17.7;19.0;22.5。
【請求項26】
160℃まで加熱してから18〜25℃の温度に戻した後に行なわれるX線粉末回折により、角度(°2シータ)で表される次の特徴的ピークを示す、請求項21〜25の一つに記載の化合物(±0.1°2シータ):7.5;10.9;13.1;17.7;19.0(160℃への加熱中に生成された型に対応する更なるピークを含まない)。
【請求項27】
5℃/分の温度勾配のDSCを用いて、吸熱性溶融ピークを170℃±5℃に示し且つ吸熱性ピークを180℃±2℃に示し、180℃でのピークは、170℃での溶融中に交換されたエンタルピーの最大で10%を表すと理解される、請求項21〜25の一つに記載の化合物。
【請求項28】
DSCを用いて、140〜155℃で吸熱事象を示さない、請求項1〜7の一つに記載の化合物。
【請求項29】
赤外線分光法を用いて、cm-1で表される次の特徴的なピークを示す、請求項1〜8の一つに記載の化合物(±5cm-1):3310;3167;3059;891;798;733;679;455。
【請求項30】
X線粉末回折を用いて、角度(°2シータ)で表される次の特徴的なピーク(±0.1°2シータ):8.6;11.3;28.6を示す、6−オキソ−6,7,8,9,10,11−ヘキサヒドロシクロヘプタ[c]クロメン−3−イルスルファメートの多形性化合物。
【請求項31】
X線粉末回折を用いて、角度(°2シータ)で表される次の特徴的なピークを示す、請求項30に記載の化合物(±0.1°2シータ):8.6;11.3;12.0;16.6;20.9;23.0;28.6。
【請求項32】
単結晶X線回折を利用して、下記のセルパラメーターを示し:
【表1】
下記の換算座標(×104)及び同等の等方性の運動パラメーター(Å2×103)を示す、請求項30又は31の一つに記載の化合物:
【表2】
【請求項33】
5℃/分までのDSCを用いて、180℃±2℃の吸熱性溶融ピークを示す、請求項30〜32いずれか一つに記載の化合物。
【請求項34】
赤外線分光分析を用いて、cm-1で表される次の特徴的なピークを示す(±5cm-1):3406;3217;1678;1011;563、請求項30〜33の一つに記載の化合物。
【請求項35】
赤外線分光分析を用いて、cm-1で表される次の特徴的なピークを示す(±5cm-1):3406;3217;3082;2924;1678;1385;1269;1134;1011;934;845;601;563;536、請求項34に記載の化合物。
【図1】

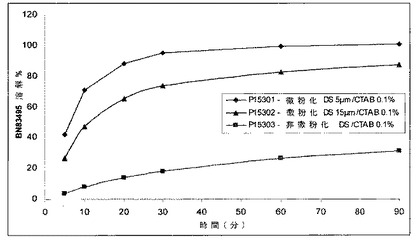
【図2】
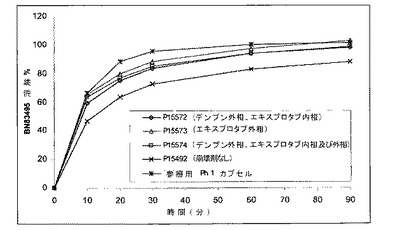
【図3】
【図4】
【図5】
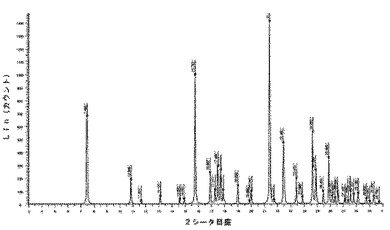
【図6】
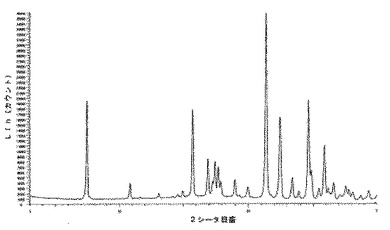
【図7】
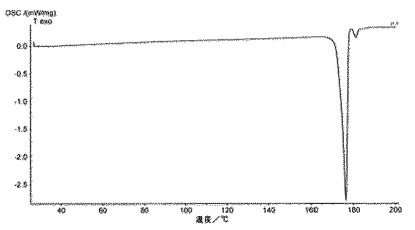
【図8】
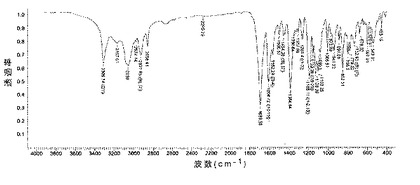
【図9】
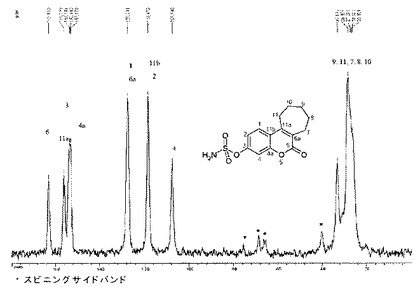
【図10】
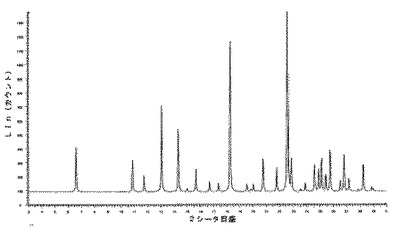
【図11】
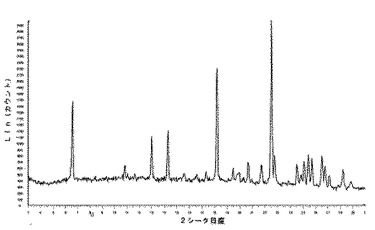
【図12】
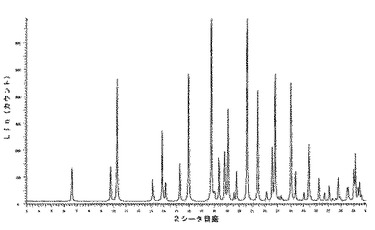
【図13】
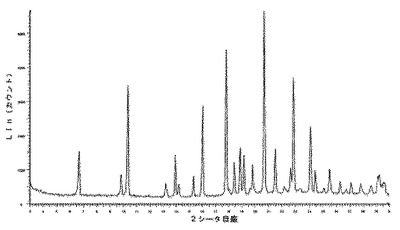
【図14】
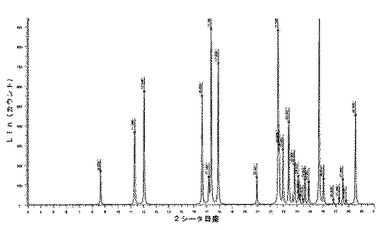
【図15】
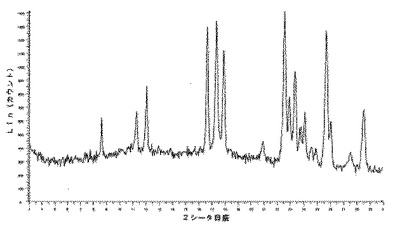
【図16】
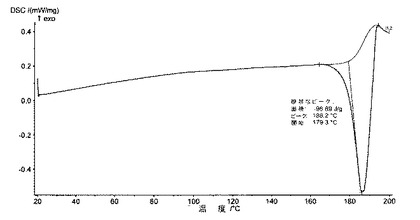
【図17】
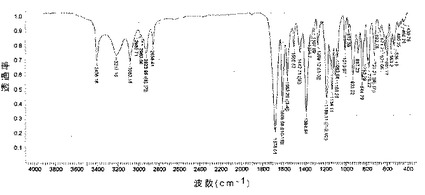
【図18】
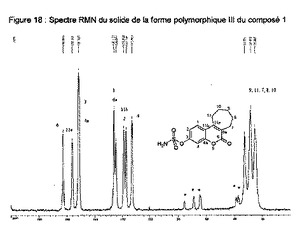
【図19】
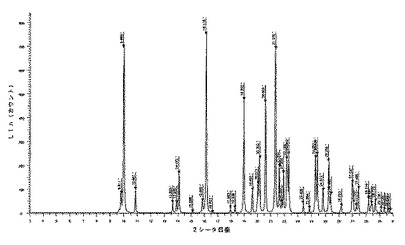
【図20】
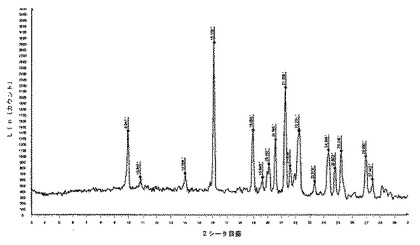
【図21】
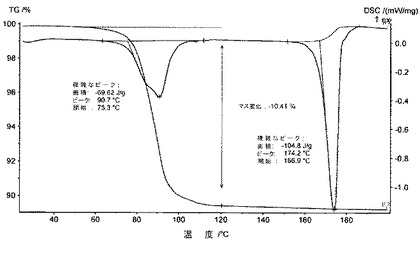
【図22】
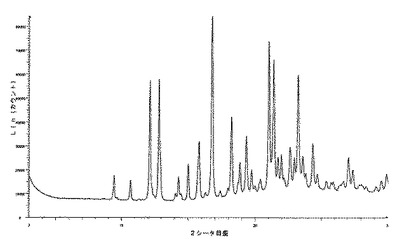
【図23】
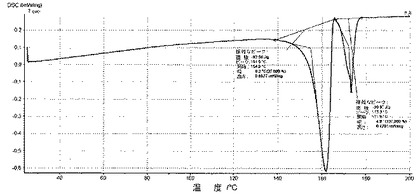
【図24】
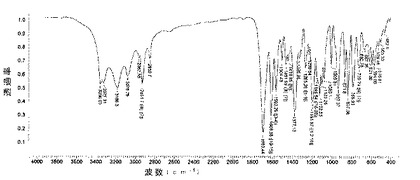
【図25】
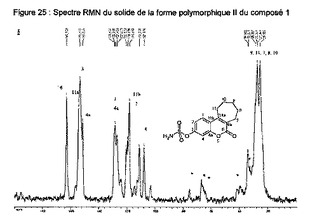
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【公表番号】特表2012−517988(P2012−517988A)
【公表日】平成24年8月9日(2012.8.9)
【国際特許分類】
【出願番号】特願2011−549626(P2011−549626)
【出願日】平成22年2月12日(2010.2.12)
【国際出願番号】PCT/FR2010/000117
【国際公開番号】WO2010/092260
【国際公開日】平成22年8月19日(2010.8.19)
【出願人】(509120469)イプセン ファルマ ソシエテ パール アクシオン サンプリフィエ (51)
【氏名又は名称原語表記】IPSEN PHARMA S.A.S.
【住所又は居所原語表記】65 Quai Georges Gorse,F−92100 Boulogne Billancourt FRANCE
【Fターム(参考)】
【公表日】平成24年8月9日(2012.8.9)
【国際特許分類】
【出願日】平成22年2月12日(2010.2.12)
【国際出願番号】PCT/FR2010/000117
【国際公開番号】WO2010/092260
【国際公開日】平成22年8月19日(2010.8.19)
【出願人】(509120469)イプセン ファルマ ソシエテ パール アクシオン サンプリフィエ (51)
【氏名又は名称原語表記】IPSEN PHARMA S.A.S.
【住所又は居所原語表記】65 Quai Georges Gorse,F−92100 Boulogne Billancourt FRANCE
【Fターム(参考)】
[ Back to top ]