
6,7−不飽和−7−カルバモイルモルヒナン誘導体の結晶およびその製造方法
【課題】6,7−不飽和−7−カルバモイルモルヒナン誘導体を医薬品として工業的に製造するための化合物、及び該化合物からモルヒナン誘導体とする製造方法の提供。
【解決手段】式(IID)で示される化合物、および転位反応により式(IA)で示される化合物の製造方法。

【解決手段】式(IID)で示される化合物、および転位反応により式(IA)で示される化合物の製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、モルヒナン誘導体の結晶およびその製造方法に関する。より詳しくは、6,7−不飽和−7−カルバモイルモルヒナン誘導体、その酸付加塩および/またはそれらの溶媒和物の結晶、およびそれらの製造方法に関する。
【背景技術】
【0002】
薬物デリバリーにおいては、有用な優れた化学的および/または物理的特性を有する結晶形態が望まれている。
特許文献1には、下式:
【化1】
で表される6,7−不飽和−7−カルバモイルモルヒナン誘導体が嘔吐および/または便秘の治療および/または予防剤として有用であることが記載されている。またその実施例には、以下の化合物(I−284):
【化2】
が遊離塩の形態で開示されているが、その酸付加塩および/または溶媒和物は、具体的に開示されていない。また、その結晶についても全く記載はない。
また、6,7−不飽和−7−カルバモイルモルヒナン誘導体の製造方法としては、以下の式:
【化3】
で示されるように、7位カルボキシル誘導体から、対応する7位カルバモイル体を合成する製造方法が開示されているに過ぎない。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】国際公開第2006/126637号パンフレット
【特許文献2】国際公開第2001/002375号パンフレット
【非特許文献】
【0004】
【非特許文献1】Chemical Communications, 1998, vol.23, 2575-2576
【非特許文献2】Synthesis, 1989, vol.2, 131-132
【発明の概要】
【発明が解決しようとする課題】
【0005】
医薬活性成分は、それぞれの固体形態によって、実質的に異なる物理的特性を有し得る。このような物理的特性の違いは、例えば医薬活性成分の製造方法もしくは投与方法、又は医薬活性成分を含む医薬組成物に影響を与え得る。
6,7−不飽和−7−カルバモイルモルヒナン誘導体は既に開示されているものの、医薬品として使用または医薬品として工業的に製造するために、好適な塩および/または安定な結晶形ならびにより好ましい製造方法の確立が望まれている。
【課題を解決するための手段】
【0006】
本発明者等は、鋭意検討した結果、下式(IA):
【化4】
で示される化合物、その酸付加塩、および/またはそれらの溶媒和物から安定な結晶が得られることを見出して以下の発明を完成した。
本発明者等は、さらに、下式(II):
【化5】
(式中、R1は水素原子または水酸基の保護基、R2は置換基を有していてもよい低級アルキル、R3は置換基を有していてもよい低級アルキル、置換基を有していてもよいシクロアルキル、置換基を有していてもよいアリール、または置換基を有していてもよいヘテロアリール)
で表されるカルバメート誘導体を塩基存在下で反応させ、必要により保護基R1を脱保護することで、下式(I):
【化6】
(式中、R2およびR3は上記と同意義)
の化合物が得られることを見出し、6,7−不飽和−7−カルバモイルモルヒナン誘導体の新たな製造方法に係る発明を完成させた。
【0007】
本発明は以下の通りである。
(1)下式(IA):
【化7】
で示される化合物のp−トルエンスルホン酸塩、酢酸塩若しくは塩酸塩、または当該化合物若しくは当該酸付加塩の溶媒和物。
【0008】
(2)下式(IA):
【化8】
で示される化合物のp−トルエンスルホン酸塩の結晶、または当該酸付加塩の溶媒和物の結晶。
【0009】
(3)粉末X線回折スペクトルにおいて、回折角度(2θ):7.8°±0.2°、10.6°±0.2°、15.6°±0.2°、17.8°±0.2°および21.5°±0.2にピークを有する、(2)に記載のp−トルエンスルホン酸塩の結晶。
(4)粉末X線回折スペクトルにおいて、回折角度(2θ):7.8°±0.2°、10.6°±0.2°、15.6°±0.2°、17.8°±0.2°、18.6°±0.2°、20.4°±0.2°、21.5°±0.2°、21.9°±0.2°、23.6°±0.2°および25.5°±0.2°にピークを有する、(2)に記載のp−トルエンスルホン酸塩の結晶。
(5)図1に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、(2)に記載のp−トルエンスルホン酸塩の結晶。
【0010】
(6)粉末X線回折スペクトルにおいて、回折角度(2θ):12.9°±0.2°、17.6°±0.2°、22.4°±0.2°、25.4°±0.2°および28.7°±0.2°にピークを有する、(2)に記載のp−トルエンスルホン酸塩水和物のI形結晶。
(7)粉末X線回折スペクトルにおいて、回折角度(2θ):6.6°±0.2°、8.9°±0.2°、11.4°±0.2°、12.9°±0.2°、14.0°±0.2°、15.0°±0.2°、17.6°±0.2°、18.2°±0.2°、22.4°±0.2°、25.4°±0.2°および28.7°±0.2°にピークを有する、(2)に記載のp−トルエンスルホン酸塩水和物のI形結晶。
(8)図2に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、(2)に記載のp−トルエンスルホン酸塩水和物のI形結晶。
【0011】
(9)粉末X線回折スペクトルにおいて、回折角度(2θ):8.8°±0.2°、17.5°±0.2°、21.9°±0.2°、23.7°±0.2°および26.1°±0.2°にピークを有する、(2)に記載のp−トルエンスルホン酸塩水和物のII形結晶。
(10)粉末X線回折スペクトルにおいて、回折角度(2θ):7.1°±0.2°、8.8°±0.2°、17.5°±0.2°、19.2°±0.2°、19.7°±0.2°、21.2°±0.2°、21.9°±0.2°、23.7°±0.2°、24.5°±0.2°および26.1°±0.2°にピークを有する、(2)に記載のp−トルエンスルホン酸塩水和物のII形結晶。
(11)図3に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、(2)に記載のp−トルエンスルホン酸塩水和物のII形結晶。
【0012】
(12)式(IA):
【化9】
で示される化合物の酢酸塩の結晶、または当該酸付加塩の溶媒和物の結晶。
(13)粉末X線回折スペクトルにおいて、回折角度(2θ):5.6°±0.2°、10.3°±0.2°、12.0°±0.2°、14.6°±0.2°および26.0°±0.2°にピークを有する、(12)に記載の酢酸塩の結晶。
(14)粉末X線回折スペクトルにおいて、回折角度(2θ):5.6°±0.2°、8.3±0.2°、9.1±0.2°、10.3°±0.2°、12.0°±0.2°、13.5±0.2°、14.6°±0.2°、16.3±0.2°および26.0°±0.2°にピークを有する、(12)に記載の酢酸塩の結晶。
(15)図4に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、(12)に記載の酢酸塩の結晶。
【0013】
(16)式(IA):
【化10】
で示される化合物の塩酸塩の結晶、または当該酸付加塩の溶媒和物の結晶。
(17)粉末X線回折スペクトルにおいて、回折角度(2θ):8.5°±0.2°、12.7°±0.2°、15.6°±0.2°、17.3°±0.2°および23.9°±0.2°にピークを有する、(16)に記載の塩酸塩の結晶。
(18)粉末X線回折スペクトルにおいて、回折角度(2θ):8.5°±0.2°、10.8°±0.2°、11.3°±0.2°、12.7°±0.2°、13.9°±0.2°、15.6°±0.2°、17.3°±0.2°、19.2°±0.2°、20.1°±0.2°および23.9°±0.2°にピークを有する、(16)に記載の塩酸塩の結晶。
(19)図5に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、(16)に記載の塩酸塩の結晶。
【0014】
(20)式(IA):
【化11】
で示される化合物の結晶、またはその溶媒和物の結晶。
(21)粉末X線回折スペクトルにおいて、回折角度(2θ):13.5°±0.2°、21.6°±0.2°、22.1°±0.2°、23.4°±0.2°および26.7°±0.2°にピークを有する、(20)に記載の式(IA)で示される化合物の結晶。
(22)粉末X線回折スペクトルにおいて、回折角度(2θ):6.8°±0.2°、11.7°±0.2°、13.5°±0.2°、15.6°±0.2°、16.7°±0.2°、21.6°±0.2°、22.1°±0.2°、23.4°±0.2°、26.7°±0.2°および30.1°±0.2°にピークを有する、(20)に記載の式(IA)で示される化合物の結晶。
(23)図7に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、(20)に記載の式(IA)で示される化合物の結晶。
【0015】
(24)粉末X線回折スペクトルにおいて、回折角度(2θ):11.0°±0.2°、16.5°±0.2°、20.5°±0.2°、21.8°±0.2°および22.6°±0.2°にピークを有する、(20)に記載のエタノール和物の結晶。
(25)粉末X線回折スペクトルにおいて、回折角度(2θ):6.9°±0.2°、11.0°±0.2°、12.9°±0.2°、13.4°±0.2°、16.5°±0.2°、20.5°±0.2°、21.3°±0.2°、21.8°±0.2°、22.6°±0.2°および25.1°±0.2°にピークを有する、(20)に記載のエタノール和物の結晶。
(26)図6に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、(20)に記載のエタノール和物の結晶。
(27)上記(2)〜(26)のいずれかに記載の結晶を含む医薬組成物。
(27A)上記(2)〜(26)のいずれかに記載の結晶を含有することを特徴とするオピオイド受容体拮抗剤。
(27B)上記(2)〜(26)のいずれかに記載の結晶を含有することを特徴とする、嘔気、嘔吐および/または便秘の治療および/または予防剤。
(27C)上記(2)〜(26)のいずれかに記載の結晶を含有することを特徴とする、オピオイド受容体アゴニスト作用を有する化合物により誘発される副作用の軽減および/または予防剤。
(27D)副作用が嘔気、嘔吐および/または便秘である、上記(27C)記載の治療および/または予防剤。
(27E)オピオイド受容体アゴニスト作用を有する化合物がモルヒネ、オキシコドン、ハイドロコドン、トラマドール、またはそれらの製薬上許容される塩またはそれらの溶媒和物である、上記(27C)または(27D)記載の治療および/または予防剤。
(27F)嘔気、嘔吐および/または便秘の治療および/または予防のための医薬を製造するための、上記(2)〜(26)のいずれかに記載の結晶の使用。
(27G)オピオイド受容体アゴニスト作用を有する化合物により誘発される副作用の軽減および/または予防のための医薬を製造するための、上記(2)〜(26)のいずれかに記載の結晶の使用。
(27H)上記(2)〜(26)のいずれかに記載の結晶を含む医薬組成物を投与することを特徴とする、嘔気、嘔吐および/または便秘の治療および/または予防方法。
(27I)上記(2)〜(26)のいずれかに記載の結晶を含む医薬組成物を投与することを特徴とする、オピオイド受容体アゴニスト作用を有する化合物により誘発される副作用の軽減および/または予防方法。
(27J)嘔気、嘔吐および/または便秘の治療および/または予防をするための、上記(2)〜(26)のいずれかに記載の結晶を含む医薬組成物。
(27K)オピオイド受容体アゴニスト作用を有する化合物により誘発される副作用の軽減および/または予防をするための、上記(2)〜(26)のいずれかに記載の結晶を含む医薬組成物。
(27L)オピオイド受容体アゴニスト作用を有する化合物と、当該化合物投与により誘発される副作用の軽減および/または予防のために効果的な量の上記(2)〜(26)のいずれかに記載の結晶とを組み合わせてなる鎮痛剤。
(27M)オピオイド受容体アゴニスト作用を有する化合物と、当該化合物投与により誘発される嘔気、嘔吐および/または便秘の治療および/または予防のために効果的な量の上記(2)〜(26)のいずれかに記載の結晶とを組み合わせてなる鎮痛剤。
(27N)オピオイド受容体アゴニスト作用を有する化合物がモルヒネ、オキシコドン、ハイドロコドン、トラマドール、またはそれらの製薬上許容される塩またはそれらの溶媒和物である、上記(27L)または(27M)記載の鎮痛剤。
【0016】
(28)式(IA):
【化12】
で示される化合物に酸を添加し、必要に応じて溶媒中から結晶化することを特徴とする、(2)〜(19)のいずれかに記載の、式(IA)で示される化合物の酸付加塩の結晶、または当該酸付加塩の溶媒和物の結晶の製造方法。
(29)式(IID):
【化13】
(式中、R1は水素原子または水酸基の保護基)
で示される化合物を塩基で処理し、必要であればR1を脱保護した後、p−トルエンスルホン酸を添加し、必要に応じて溶媒中から結晶化することを特徴とする、(2)に記載の結晶の製造方法。
(30)式(IIE):
【化14】
(式中、R1aは水素原子または塩基で脱保護可能な水酸基の保護基)
で示される化合物を塩基で処理し、次いでp−トルエンスルホン酸を添加し、必要に応じて溶媒中から結晶化することを特徴とする、(29)の製造方法。
(31)式(III):
【化15】
(式中、R1bは水酸基の保護基;R2は置換基を有していてもよい低級アルキル)で示される化合物を、酸の存在下または非存在下、式:R3−N=C=O(式中、R3は置換基を有していてもよい低級アルキル、置換基を有していてもよいシクロアルキル、置換もしくは非置換アリール、または置換もしくは非置換ヘテロアリール)で示される化合物または式:R3−NH−C(=O)−X(式中、R3は前記と同意義、Xは脱離基)で示される化合物と反応させることを特徴とする、式(II):
【化16】
(式中、R1b、R2およびR3は、前記と同意義)で示される化合物の製造方法。
(32)式(IV):
【化17】
(式中、R2は(31)と同意義)で示される化合物の水酸基を保護することにより、当該式(III):
【化18】
(式中、R1bおよびR2は(31)と同意義)
で示される化合物を得ることを特徴とする、(31)に記載の製造方法。
(33)式(IV):
【化19】
(式中、R2は(31)と同意義)
で示される化合物の水酸基を保護し、式(III):
【化20】
(式中、R1bおよびR2は(31)と同意義)
で示される化合物を得る工程、および式(III)で示される化合物を、酸の存在下または非存在下、式:R3−N=C=O(式中、R3は(31)と同意義)で示される化合物または式:R3−NH−C(=O)−X(式中、R3は前記と同意義、Xは脱離基)で示される化合物と反応させる工程を連続して行う、(32)記載の製造方法。
ここで、「工程を連続して行う」とは、前工程の反応により生成した化合物を単離することなく、次工程を行うことを包含する。例えば、ワンポットで2つの工程を行うことが挙げられる。
(34)酸の存在下で行う、(31)〜(33)のいずれかに記載の製造方法。
(35)酸がルイス酸である、(34)記載の製造方法。
(36)ルイス酸触媒が、CuCl、CuCl2、CuBr、CuI、CuBr、CuSO4、Cu、Zn(OAc)2、ZnBr2またはZnCl2である、(35)に記載の製造方法。
(37)式(III)で示される化合物に対して、約0.00005〜約1.0当量の酸の存在下で反応させることを特徴とする、(31)〜(36)のいずれかに記載の製造方法。
(38)R1bが塩基で脱保護可能な水酸基の保護基である、(31)〜(37)のいずれかに記載の製造方法。
(39)式(IIA):
【化21】
(式中、R2およびR3は(31)と同意義)で示される化合物を、塩基で処理することを特徴とする、式(I):
【化22】
(式中、R2およびR3は、前記と同意義)で示される化合物の製造方法。
(40)式(IIC):
【化23】
(式中、R1cは塩基で脱保護可能な水酸基の保護基;R2およびR3は(31)と同意義)
で示される化合物を、塩基で処理することを特徴とする、式(I):
【化24】
(式中、R2およびR3は、前記と同意義)で示される化合物の製造方法。
(41)式(IIB):
【化25】
(式中、R1bは水酸基の保護基;R2およびR3は(31)と同意義)
で示される化合物を、塩基で処理することを特徴とする、式(IB):
【化26】
(式中、R1dは塩基では脱保護されない水酸基の保護基または水素原子;R2およびR3は、前記と同意義)
で示される化合物の製造方法。
(42)塩基が無機塩基である(39)〜(41)のいずれかに記載の製造方法。
(43)塩基が水酸化カリウム、水酸化ナトリウム、水酸化リチウムまたは水酸化セシウムである(39)〜(41)のいずれかに記載の製造方法。
(44)反応温度が30℃〜100℃である(39)〜(43)のいずれかに記載の製造方法。
(45)式(VIIIa):
【化27】
(式中、R5は低級アルキル)で示される化合物を、ルイス酸および塩基の存在下で反応させることを特徴とする、式(IX):
【化28】
で示される化合物の製造方法。
(46)式(X):
【化29】
(式中、R5は低級アルキル)
で示される化合物と、式(XI):
【化30】
で示される化合物を反応させ、当該式(VIIIa)で示される化合物を得ることを特徴とする、(45)記載の製造方法。
(47)ルイス酸がAlCl3またはTiCl4である(45)または(46)に記載の製造方法。
(48)式(IIIA):
【化31】
(式中、R1cは、塩基で脱保護可能な水酸基の保護基;R2は(31)と同意義)
で示される化合物を、ルイス酸触媒の存在下または非存在下、式:R3−N=C=O(式中、R3は(31)と同意義)で示される化合物または式:R3−NH−C(=O)−X(式中、R3は前記と同意義、Xは脱離基)で示される化合物と反応させ、式(IIC):
【化32】
(式中、R1c、R2およびR3は、前記と同意義)
で示される化合物を得る工程、
上記式(IIC)で示される化合物を、塩基で処理し、式(I):
【化33】
(式中、R2およびR3は、前記と同意義)
で示される化合物を得る工程、および
上記式(I)で示される化合物に酸を添加して酸付加塩とする工程を包含する、式(I)で示される化合物の酸付加塩の製造方法。
酸を添加して酸付加塩とした後、必要に応じて該反応液を冷却してもよい。
ここで、式(IIC)で示される化合物を得る工程において、一つの態様として、ルイス酸触媒存在下、式(IIIA)で示される化合物と式:R3−N=C=O(式中、R3は上記と同意義)で示される化合物を反応させる工程が挙げられる。一つの態様として、ルイス酸触媒非存在下、式(IIIA)で示される化合物と式:R3−N=C=O(式中、R3は上記と同意義)で示される化合物を反応させる工程が挙げられる。別の一つの態様として、ルイス酸触媒非存在下、式(IIIA)で示される化合物と式:R3−NH−C(=O)−X(式中、R3およびXは上記と同意義)で示される化合物を反応させる工程が挙げられる。
(49)式(I)で示される化合物の酸付加塩が、p−トルエンスルホン酸塩、酢酸塩または塩酸塩、またはそれらの溶媒和物である、(48)の製造方法。
(50)p−トルエンスルホン酸塩、酢酸塩または塩酸塩、またはそれらの溶媒和物が結晶である、(49)の製造方法。
(51)式(IID):
【化34】
(式中、R1は水素原子または水酸基の保護基)
で示される化合物。
(52)式(VII):
【化35】
[式中、−R6は、−N=C=Oまたは−NH−C(=O)−X(式中、Xは脱離基)で表される基]で示される化合物。
上記式(II)で示される化合物、式(IIA)で示される化合物、式(IIB)で示される化合物、式(IIC)で示される化合物、式(IID)で示される化合物および式(IIE)で示される化合物において、モルヒナン骨格の7位側鎖である「−O−C(=O)−NH−」基の「−NH−」の水素原子は、アミノ基の保護基に置き換わってもよい。
【0017】
本明細書中、「ハロゲン」とはフッ素、塩素、臭素およびヨウ素を包含する。「ハロゲノ低級アルキル」、「ハロゲノ低級アルコキシ」、「ハロゲノ低級アルキルチオ」のハロゲン部分も同様である。
「低級アルキル」とは、炭素数1〜10、好ましくは炭素数1〜6、さらに好ましくは炭素数1〜3の直鎖または分枝状のアルキルを包含し、例えばメチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチル、n−ペンチル、イソペンチル、ネオペンチル、ヘキシル、イソヘキシル、n−へプチル、イソヘプチル、n−オクチル、イソオクチル、n−ノニルおよびn−デシル等が挙げられる。好ましくはメチル、エチル、イソプロピル、n−ブチル、sec−ブチル、tert−ブチル、1-エチルプロピル等である。
【0018】
「置換基を有していてもよい低級アルキル」の置換基としてはハロゲン、ヒドロキシ、低級アルコキシ、ハロゲノ低級アルコキシ、ヒドロキシ低級アルコキシ、低級アルキルチオ、低級アルキルアミノ、アシルアミノ、アシル、アシルオキシ、シアノ、カルボキシ、低級アルコキシカルボニル、カルバモイル、低級アルキルカルバモイル、シアノカルバモイル、低級アルキルスルホニルカルバモイル、アリールスルホニルカルバモイル、スルファモイル、低級アルキルスルファモイル、低級アルキルスルホニル、置換基群αから選択される1以上の基で置換されていてもよいシクロアルキル(ここで置換基群αとはハロゲン、ヒドロキシ、低級アルキル、ハロゲノ低級アルキル、ヒドロキシ低級アルキル、低級アルコキシ低級アルキル、カルボキシ低級アルキル、低級アルコキシカルボニル低級アルキル、アミノ低級アルキル、低級アルキルアミノ低級アルキル、アシルアミノ低級アルキル、シアノ低級アルキル、低級アルコキシ、ハロゲノ低級アルコキシ、ヒドロキシ低級アルコキシ、低級アルキルチオ、ハロゲノ低級アルキルチオ、アシル、アシルオキシ、アミノ、低級アルキルアミノ、アシルアミノ、シアノ、カルボキシ、低級アルコキシカルボニル、カルバモイル、低級アルキルカルバモイル、アリールカルバモイル、シアノカルバモイル、低級アルキルスルホニルカルバモイル、スルファモイル、低級アルキルスルファモイル、低級アルキルスルホニル、低級アルキレンジオキシで置換されていてもよいアリールおよびヘテロ環式基である)、置換基群αから選択される1以上の基で置換されていてもよいシクロアルケニル、置換基群αから選択される1以上の基で置換されていてもよいアリール、置換基群αから選択される1以上の基で置換されていてもよいアリールオキシ、置換基群αから選択される1以上の基で置換されていてもよいアリールチオ、置換基群αから選択される1以上の基で置換されていてもよいヘテロ環式基、置換基群αから選択される1以上の基で置換されていてもよいヘテロ環オキシ等が挙げられる。
【0019】
「ハロゲノ低級アルキル」、「ヒドロキシ低級アルキル」、「アミノ低級アルキル」、「アシルアミノ低級アルキル」、「アシルオキシ低級アルキル」、「シクロアルキル低級アルキル」、「低級アルコキシ」、「ハロゲノ低級アルコキシ」、「ヒドロキシ低級アルコキシ」、「低級アルコキシ低級アルキル」、「低級アルコキシカルボニル」、「カルボキシ低級アルキル」、「低級アルコキシカルボニル低級アルキル」、「低級アルキルチオ」、「ハロゲノ低級アルキルチオ」、「低級アルキルアミノ」、「低級アルキルアミノ低級アルキル」、「低級アルキルカルバモイル」、「低級アルキルスルファモイル」、「低級アルキルスルホニル」、「アリール低級アルキル」、「トリ低級アルキルシリル」、「低級アルキルジアリールシリル」、「トリアリール低級アルキルシリル」、「低級アルコキシ低級アルコキシ低級アルキル」、「低級アルキルチオ低級アルキル」、「アリール低級アルコキシ低級アルキル」、「低級アルキルスルホニル」、「低級アルキルスルホニルカルバモイル」、「低級アルキルカルボニル」、「シアノ低級アルキル」、「低級アルコキシカルボニルアミノ」、「低級アルキレンジオキシ」、「ヘテロ環低級アルキル」の低級アルキル部分は上記「低級アルキル」と同様である。
「置換基を有していてもよい低級アルコキシ」、「置換基を有していてもよい低級アルキルチオ」、「置換基を有していてもよい低級アルキルスルホニル」の置換基は上記「置換基を有していてもよい低級アルキル」の置換基と同様である。
【0020】
「低級アルケニル」とは、任意の位置に1以上の二重結合を有する炭素数2〜10、好ましくは炭素数2〜8、さらに好ましくは炭素数3〜6の直鎖または分枝状のアルケニルを包含する。具体的にはビニル、アリル、プロペニル、イソプロペニル、ブテニル、イソブテニル、プレニル、ブタジエニル、ペンテニル、イソペンテニル、ペンタジエニル、ヘキセニル、イソヘキセニル、ヘキサジエニル、ヘプテニル、オクテニル、ノネニルおよびデセニル等を包含する。
「置換基を有していてもよい低級アルケニル」の置換基は上記「置換基を有していてもよい低級アルキル」と同様である。
「低級アルキニル」とは、任意の位置に1以上の三重結合を有する炭素数2〜10、好ましくは炭素数2〜8、さらに好ましくは炭素数3〜6の直鎖または分枝状のアルキニルを包含する。具体的には、エチニル、プロピニル、ブチニル、ペンチニル、ヘキシニル、ヘプチニル、オクチニル、ノニニル、デシニル等を包含する。これらはさらに任意の位置に二重結合を有していてもよい。
【0021】
「置換基を有していてもよい低級アルキニル」の置換基は上記「置換基を有していてもよい低級アルキル」の置換基と同様である。
「置換基を有していてもよいアミノ」の置換基としては、置換基群αから選択される1以上の基で置換されていてもよい低級アルキル、置換基群αから選択される1以上の基で置換されていてもよいシクロアルキル、置換基群αから選択される1以上の基で置換されていてもよいアシル、置換基群αから選択される1以上の基で置換されていてもよいアミノ、置換基群αから選択される1以上の基で置換されていてもよいアリール、スルファモイル、置換基群αから選択される1以上の基で置換されていてもよい低級アルキルスルファモイル、置換基群αから選択される1以上の基で置換されていてもよいアリールスルファモイル、置換基群αから選択される1以上の基で置換されていてもよい低級アルキルスルホニル、置換基群αから選択される1以上の基で置換されていてもよいアリールスルホニル、置換基群αから選択される1以上の基で置換されていてもよいアリールアミノ、置換基群αから選択される1以上の基で置換されていてもよいヘテロ環式基等が挙げられる。
【0022】
「置換基を有していてもよいカルバモイル」の置換基は上記「置換基を有していてもよいアミノ」の置換基と同様である。
「シクロアルキル」とは炭素数3〜10、好ましくは炭素数3〜8、より好ましくは炭素数4〜8の炭素環式基であり、例えばシクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、シクロノニルおよびシクロデシル等を包含する。これらはさらに任意の位置で後述の「アリール」または後述の「ヘテロ環式基」と縮合していてもよい。
「シクロアルキル低級アルキル」、「シクロアルキルカルボニル」のシクロアルキル部分は上記「シクロアルキル」と同様である。
【0023】
「置換基を有していてもよいシクロアルキル」の置換基は上述の置換基群αから選択される1以上の基が挙げられる。置換基は任意の位置に置換することができ、シクロアルキルの結合手を有する炭素原子に置換してもよい。
「シクロアルケニル」とは、上記シクロアルキルの環中の任意の位置に1以上の二重結合を有しているものを包含し、具体的にはシクロプロペニル、シクロブテニル、シクロペンテニル、シクロヘキセニル、シクロへプテニル、シクロオクテニルおよびシクロヘキサジエニル等が挙げられる。
「シクロアルケニルカルボニル」のシクロアルケニル部分は上記「シクロアルケニル」と同様である。
【0024】
「置換基を有していてもよいシクロアルケニル」の置換基は上記「置換基を有していてもよいシクロアルキル」と同様である。
「アリール」とは、フェニル、ナフチル、アントリルおよびフェナントリル等を包含し、特にフェニルが好ましい。
「アリールオキシ」、「アリールチオ」、「アリール低級アルキル」、「低級アルキルジアリールシリル」、「トリアリール低級アルキルシリル」、「アリール低級アルキルオキシ低級アルキル」、「アリールスルホニル」、「アリールスルファモイル」、「アリールアミノ」、「アリールカルバモイル」、「アリールスルホニルカルバモイル」のアリール部分も上記「アリール」と同様である。
【0025】
「置換基を有していてもよいアリール」、「置換基を有していてもよいフェニル」、「置換基を有していてもよいアリールスルホニル」の置換基としては、上述の置換基群α、置換基群αから選択される1以上の基で置換されたフェニル、置換基群αから選択される1以上の基で置換されたフェノキシ、または低級アルキレンジオキシ等が挙げられる。
「ヘテロ環式基」とは、O、SおよびNから任意に選択されるヘテロ原子を環内に1以上有するヘテロ環式基を包含し、具体的にはピロリル、イミダゾリル、ピラゾリル、ピリジル、ピリダジニル、ピリミジニル、ピラジニル、トリアゾリル、トリアジニル、テトラゾリル、イソオキサゾリル、オキサゾリル、オキサジアゾリル、イソチアゾリル、チアゾリル、チアジアゾリル、フリルおよびチエニル等の5〜6員のヘテロアリール;インドリル、イソインドリル、インダゾリル、インドリジニル、インドリニル、イソインドリニル、キノリル、イソキノリル、シンノリニル、フタラジニル、キナゾリニル、ナフチリジニル、キノキサリニル、プリニル、プテリジニル、ベンゾピラニル、ベンズイミダゾリル、ベンズイソオキサゾリル、ベンズオキサゾリル、ベンズオキサジアゾリル、ベンゾイソチアゾリル、ベンゾチアゾリル、ベンゾチアジアゾリル、ベンゾフリル、イソベンゾフリル、ベンゾチエニル、ベンゾトリアゾリル、イミダゾピリジル、トリアゾロピリジル、イミダゾチアゾリル、ピラジノピリダジニル、キナゾリニル、キノリル、イソキノリル、ナフチリジニル、ジヒドロピリジル、テトラヒドロキノリル、テトラヒドロベンゾチエニル等の2環の縮合ヘテロ環式基;カルバゾリル、アクリジニル、キサンテニル、フェノチアジニル、フェノキサチイニル、フェノキサジニル、ジベンゾフリル等の3環の縮合ヘテロ環式基;ジオキサニル、チイラニル、チオラニル、チエタニル、オキシラニル、オキセタニル、オキサチオラニル、アゼチジニル、チアニル、ピロリジニル、ピロリニル、イミダゾリジニル、イミダゾリニル、ピラゾリジニル、ピラゾリニル、ピペリジル、ピペラジニル、モルホリニル、モルホリノ、チオモルホリニル、チオモルホリノ、ジヒドロピリジル、ジヒドロフリル、テトラヒドロフリル、テトラヒドロピラニル、テトラヒドロチアゾリル、テトラヒドロイソチアゾリル等の非芳香族ヘテロ環式基を包含する。好ましくは5〜6員のヘテロアリールまたは非芳香族ヘテロ環式基である。
【0026】
「ヘテロ環オキシ」、「ヘテロ環低級アルキル」のヘテロ環部分も上記「ヘテロ環式基」と同様である。
「置換基を有していてもよいヘテロ環式基」および「置換基を有してもよいヘテロ環オキシ」の置換基としては上述の置換基群αおよびオキソからなる群から選択される1以上の基が挙げられる。置換基は任意の位置に置換することができ、ヘテロ環式基の結合手を有する炭素原子または窒素原子に置換してもよい。
【0027】
「アシル」とは炭素数1〜10、好ましくは炭素数1〜6、さらに好ましくは炭素数1〜4の直鎖または分枝の鎖状脂肪族アシル、炭素数4〜9、好ましくは炭素数4〜7の環状脂肪族アシル、アロイルおよびヘテロ環カルボニルを包含する。ここで「鎖状脂肪族」とは上記「低級アルキル」、上記「低級アルケニル」、および上記「低級アルキニル」を包含する。「環状脂肪族」とは上記「シクロアルキル」および上記「シクロアルケニル」を包含する。ヘテロ環カルボニルのヘテロ環部分も上記「ヘテロ環式基」と同様である。アシルの具体例としては、ホルミル、アセチル、プロピオニル、ブチリル、イソブチリル、バレリル、ピバロイル、ヘキサノイル、アクリロイル、プロピオロイル、メタクリロイル、クロトノイル、シクロプロピルカルボニル、シクロヘキシルカルボニル、シクロオクチルカルボニル、ベンゾイル、ピリジンカルボニル、ピペリジンカルボニル、ピペラジンカルボニル、モルホリノカルボニル等を包含する。
「アシルオキシ」、「アシルアミノ」、「アシルアミノ低級アルキル」および「アシルオキシ低級アルキル」のアシル部分は上記「アシル」と同様である。
「置換基を有していてもよいアシル」または「置換基を有していてもよいアシルオキシ」の置換基は、「アシル」が鎖状脂肪族アシルの場合、上記「置換基を有していてもよい低級アルキル」の置換基と同様であり、「アシル」が環状脂肪族アシル、アロイルおよびヘテロ環カルボニルの場合、上述の置換基群αから選択される1以上の基が挙げられる。
【0028】
「溶媒和物」とは、例えば有機溶媒(エタノール、2−プロパノール、酢酸メチル、酢酸エチル、酢酸n−プロピル、1、2−ジメトキシエタン、メチルイソブチルケトン、アセトニトリル等)との溶媒和物、水和物等を包含する。水和物を形成する時は、任意の数の水分子と配位していてもよい。
「水酸基の保護基」とは、ベンジル基、p−メトキシフェニルベンジル基、アセチル基、ホルミル基、ベンゾイル基、クロロアセチル基、ピバロイル基、メチルカーボネート基、イソブチルカーボネート基、ベンジルカーボネート基、ビニルカーボネート基、フェニルカーバメート基、メシル基、トシル基、トリメチルシリル基、トリエチルシリル基、t−ブチルジメチルシリル基、メトキシメチル基、ベンジルオキシメチル基、メトキシエトキシメチル基、2-(トリメチルシリル)エトキシメチル基、プロペニル基、フェナシル基、テトラヒドロピラニル基等が挙げられる。
「塩基で脱保護可能な水酸基の保護基」とは、アセチル基、ホルミル基、ベンゾイル基、クロロアセチル基、ピバロイル基、メチルカーボネート基、イソブチルカーボネート基、ベンジルカーボネート基、ビニルカーボネート基、フェニルカーバメート基、メシル基、トシル基等が挙げられる。一つの態様として、アセチル基、ホルミル基、ベンゾイル基、クロロアセチル基、ピバロイル基が挙げられる。別の態様として、アセチル基が挙げられる。
「塩基では脱保護されない水酸基の保護基」とは、ベンジル基、p−メトキシフェニルベンジル基、トリメチルシリル基、トリエチルシリル基、t−ブチルジメチルシリル基、メトキシメチル基、ベンジルオキシメチル基、メトキシエトキシメチル基、2-(トリメチルシリル)エトキシメチル基、プロペニル基、フェナシル基、テトラヒドロピラニル基等が挙げられる。
「脱離基」とは、置換基を有していてもよいフェノキシ(例えば、フェノキシ、p−ニトロフェノキシ、o−ニトロフェノキシが挙げられる)、ヘテロ環式基(例えば、1−イミダゾリル、1−ピラゾリルが挙げられる)、置換基を有してもよいヘテロ環オキシ(例えば,ピリジルオキシが挙げられる)等が挙げられる。
「式(I)で示される化合物の酸付加塩」および「式(IA)で表される酸付加塩」の「酸付加塩」とは、無機酸(例えば、塩酸、硫酸、硝酸、炭酸、臭化水素酸、リン酸、ヨウ化水素酸等)、および有機酸(例えば、ギ酸、酢酸、プロピオン酸、トリフルオロ酢酸、クエン酸、乳酸、酒石酸、シュウ酸、マレイン酸、フマル酸、マンデル酸、グルタル酸、リンゴ酸、安息香酸、フタル酸、アスコルビン酸、ベンゼンスルホン酸、p−トルエンスルホン酸、メタンスルホン酸、エタンスルホン酸等)との塩が挙げられる。例えば、「式(I)で示される化合物の酸付加塩」および「式(IA)で表される酸付加塩」の酸付加塩としては、p−トルエンスルホン酸塩、酢酸塩および塩酸塩が挙げられる。
「アミノ基の保護基」とは、t−ブチルジメチルシリル基、t−ブトキシカルボニル基、アリル基、9-フルオレニルメチルオキシカルボニル基、ベンジル基、p−メトキシベンジル基、メトキシメチル基、ベンジルオキシメチル基、ベンズヒドリル基およびトリチル基が挙げられる。
【発明の効果】
【0029】
本発明によって、6,7−不飽和−7−カルバモイルモルヒナン誘導体、その酸付加塩および/またはそれらの溶媒和物の結晶体が提供される。当該結晶体は、安定性が良好であり医薬品製造用原体として用いることができる。
また新規な製造方法は、製造工程の短縮化、収率の向上等に貢献することができる。
【図面の簡単な説明】
【0030】
【図1】本発明に係る化合物(IA)のp−トルエンスルホン酸塩の結晶体(非溶媒和物)の粉末X線回折パターンを示す。
【図2】本発明に係る化合物(IA)のp−トルエンスルホン酸塩水和物の結晶体(I形)の粉末X線回折パターンを示す。
【図3】本発明に係る化合物(IA)のp−トルエンスルホン酸塩水和物の結晶体(II形)の粉末X線回折パターンを示す。
【図4】本発明に係る化合物(IA)の酢酸塩の結晶体の粉末X線回折パターンを示す。
【図5】本発明に係る化合物(IA)の塩酸塩の結晶体の粉末X線回折パターンを示す。
【図6】本発明に係る化合物(IA)のエタノール和物の結晶体の粉末X線回折パターンを示す。
【図7】本発明に係る化合物(IA)の遊離体の結晶体の粉末X線回折パターンを示す。
【図8】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・酢酸メチル和物の結晶体の粉末X線回折パターンを示す。
【図9】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・(酢酸エチルおよび2−プロパノール)和物の結晶体の粉末X線回折パターンを示す。
【図10】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・(酢酸n‐プロピルおよび2−プロパノール)和物の結晶体の粉末X線回折パターンを示す。
【図11】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・アセトニトリル和物の結晶体の粉末X線回折パターンを示す。
【図12】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・1、2−ジメトキシエタン和物の結晶体の粉末X線回折パターンを示す。
【図13】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・メチルイソブチルケトン和物の結晶体の粉末X線回折パターンを示す。
【図14】本発明に係る化合物(IA)のp−トルエンスルホン酸塩の結晶体(非溶媒和物)のTG/DTA分析結果を示す。
【図15】本発明に係る化合物(IA)のp−トルエンスルホン酸塩水和物の結晶体(I形)のTG/DTA分析結果を示す。
【図16】本発明に係る化合物(IA)のp−トルエンスルホン酸塩水和物の結晶体(II形)のTG/DTA分析結果を示す。
【図17】本発明に係る化合物(IA)の酢酸塩のTG/DTA分析結果を示す。
【図18】本発明に係る化合物(IA)の塩酸塩のTG/DTA分析結果を示す。
【図19】本発明に係る化合物(IA)のエタノール和物のTG/DTA分析結果を示す。
【図20】本発明に係る化合物(IA)の遊離体のTG/DTA分析結果を示す。
【図21】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・酢酸メチル和物のTG/DTA分析結果を示す。
【図22】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・(酢酸エチルおよび2−プロパノール)和物のTG/DTA分析結果を示す。
【図23】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・(酢酸n‐プロピルおよび2−プロパノール)和物のTG/DTA分析結果を示す。
【図24】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・アセトニトリル和物のTG/DTA分析結果を示す。
【図25】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・1、2−ジメトキシエタン和物のTG/DTA分析結果を示す。
【図26】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・メチルイソブチルケトン和物のTG/DTA分析結果を示す。
【発明を実施するための形態】
【0031】
本発明の結晶体は、式(I)で示される化合物の酸付加塩または該酸付加塩の溶媒和物として得られる。ここで用いられる酸としては、p−トルエンスルホン酸、酢酸または塩酸等が例示される。中でもp−トルエンスルホン酸の結晶体は、吸湿性がなく、安定性も優れていると考えられる。溶媒和物を形成する際の溶媒としては、水、エタノール、2−プロパノール、酢酸メチル、酢酸エチル、酢酸n‐プロピル、1、2−ジメトキシエタン、メチルイソブチルケトン、アセトニトリル等が例示できる。
酸付加塩の結晶は、式(I)で示される化合物の溶液に、通常1.0〜10.0当量の酸を、0℃〜室温で、若しくは溶媒の沸点以下に加温して添加した後、必要に応じて溶液を冷却および/または濃縮して晶析させる。
溶媒和物の結晶の調製は、式(I)で示される化合物の酸付加塩を、少なくとも溶媒和させる溶媒を含む可溶性溶媒に、室温で、または溶媒の沸点以下に加温して溶解し、溶媒和させたい溶媒を加え、0℃〜室温で数時間〜1日、撹拌または静置することにより行う。晶析した溶媒和物は、濾過または遠心分離等の通常の分離手段で溶媒から分離し、洗浄、乾燥等の通常の精製手段により単離することができる。
式(I)で示される化合物の溶媒和物もまた、本発明の結晶体に包含される。溶媒としては水、エタノール等が例示される。式(I)で示される化合物の溶媒和物の場合も、上記酸付加塩の溶媒和物と同様にして調製することができる。
本発明の結晶体の具体例としては、化合物(IA)の場合、p−トルエンスルホン酸塩(非溶媒和物)、p−トルエンスルホン酸塩・水和物、p−トルエンスルホン酸塩・酢酸メチル和物、p−トルエンスルホン酸塩・(酢酸エチル・2−プロパノール)和物、p−トルエンスルホン酸塩・(酢酸n‐プロピル・2−プロパノール)和物、p−トルエンスルホン酸塩・アセトニトリル和物、p−トルエンスルホン酸塩・1、2−ジメトキシエタン和物、p−トルエンスルホン酸塩・メチルイソブチルケトン和物、塩酸塩、酢酸塩、遊離体のエタノール和物等が例示できる。
例えば、式(IA)で示される化合物のp−トルエンスルホン酸塩(非溶媒和物)は、以下のようにして得られる。即ち、化合物(IA)を含む有機層に2−プロパノールと酢酸n−プロピルを加えて濃縮し、50〜70℃で1〜10.0当量のp−トルエンスルホン酸を2−プロパノールに溶解して滴下し晶析する。得られる未乾固体を、メタノールと酢酸n−プロピルに再び加温溶解し、不溶物をろ過した後、減圧濃縮して晶析させ、得られた結晶を50〜70℃で2〜5時間減圧下で乾燥すると目的のp−トルエンスルホン酸塩(非溶媒和物)を得ることができる。
【0032】
以下に本発明の結晶体を特定する方法につき説明する。
特に言及がなければ、本明細書中および特許請求の範囲記載の数値は、おおよその値である。数値の変動は、装置キャリブレーション、装置エラー、物質の純度、結晶サイズ、サンプルサイズ、その他の因子に起因する。
【0033】
本明細書中で用いる「結晶」とは、秩序だった長い範囲の分子構造を有する物質を意味する。結晶形態の結晶化度は、例えば、粉末X線回折、水分吸着、示差、熱量分析、溶液比色、溶解特性を含めた多くの技術によって測定することができる。
【0034】
一般に結晶性有機化合物は、3次元空間に周期的に配列された多数の原子よりなる。構造周期性は、通例、ほとんどの分光学的プローブ(例えば、X線回折、赤外スペクトル、ラマンスペクトルおよび固体NMR)によって明確に区別可能な物理的特性を発現する。
中でも粉末X線回折(XRPD)は、固体の結晶性を測定するための最も感度の良い分析法のうちの1つである。X線が結晶に照射されると、結晶格子面で反射し、互いに干渉しあい、ブラッグ則よって予測される条件を満たす方向の回折線のみ強度が増大し、それ以外は打ち消しあって観測されない。一方、非晶質固体については広範囲の秩序だった回折線は認められない。非晶質固体は、通常、反復する結晶格子の広い範囲の秩序が不存在であるため、ハローパターンと呼ばれるブロードなXRPDパターンを示す。
【0035】
本出願で開示する6,7−不飽和−7−カルバモイルモルヒナン誘導体、その酸付加塩および/またはそれらの溶媒和物の結晶形態は、好ましくは、区別可能な粉末X線回折プロフィールを有する。例えば、化合物(IA)のp−トルエンスルホン酸塩の場合、好ましくは、特徴的な回折ピークの存在によって各結晶体を特定し、他の結晶体と区別することができる。本明細書中で用いる特徴的な回折ピークは、観察された回折パターンから選択されるピークである。好ましくは、特徴的なピークは、回折パターンにおける約20本、より好ましくは約10本、最も好ましくは約5本から選択される。
【0036】
一般に、粉末X線回折における回折角度(2θ)は±0.2°の範囲内で誤差が生じ得るので、上記の回折角度の値は±0.2°程度の範囲内の数値も含むものとして理解される必要がある。したがって、粉末X線回折におけるピークの回折角度が完全に一致する結晶だけでなく、ピークの回折角度が±0.2°程度の誤差で一致する結晶も本発明に含まれる。
【0037】
一般に、後述の表及び図において表示されるピークの相対強度は、多くの因子、例えばX線ビームに対する結晶の配向効果、分析される物質の純度又はサンプルの結晶化度によって変動し得ることが知られている。また、ピーク位置についても、サンプル高の変動に基づいてシフトし得る。さらに、異なる波長を使用して測定するとブラッグ式(nλ=2dsinθ)に従って異なるシフトが得られるが、このような別の波長の使用により得られる別のXRPDパターンも、本発明の範囲に含まれる。
【0038】
本発明に係る化合物(IA)のp−トルエンスルホン酸塩の結晶体(非溶媒和物)、p−トルエンスルホン酸塩水和物(I形)およびp−トルエンスルホン酸塩水和物(II形)は図1〜3のような粉末X線回折パターンを示す。各結晶体は少なくとも表1に示されるような特徴的ピークを示す。
【表1】
【0039】
本発明の結晶体は熱分析の手法によっても特定することができる。
ここでTG/DTA(示差熱熱重量同時測定)は、熱分析の主要な測定方法のひとつであって、原子・分子の集合体としての物質の重量および熱的性質を測定する方法である。
TG/DTAは医薬活性成分の温度または時間に係る重量および熱量の変化を測定する方法であり、得られたデータを温度または時間に対してプロットすることにより、TG(熱重量)およびDTA(示差熱)曲線が得られる。TG/DTA曲線より、医薬活性成分の分解、脱水、酸化、還元、昇華、蒸発に関する重量および熱量変化の情報を得ることができる。
TG/DTAにおいて、「融点」とは、オンセット温度をいう。
TG/DTAについて、観察される温度、重量変化は、温度変化速度ならびに用いる試料調製技法および特定の装置に依存し得ることが知られている。結晶の同一性の認定においては、全体的なパターンが重要であり、測定条件によって多少変化し得る。
【0040】
本発明に係る化合物(IA)のp−トルエンスルホン酸塩の結晶体、p−トルエンスルホン酸塩水和物(I形)、p−トルエンスルホン酸塩水和物(II形)、酢酸塩および塩酸塩のTG/DTA分析結果は図14〜18に表される。
【0041】
本発明の式(IA)で示される化合物、その酸付加塩、および/またはそれらの溶媒和物の結晶は、オピオイド受容体(特にオピオイドδ、μ受容体)拮抗作用を有している。従って、オピオイド受容体アゴニスト作用を有する化合物に誘発される嘔気・嘔吐・便秘の他、急性消化不良、急性アルコール中毒、食中毒、感冒、胃潰瘍、十二指腸潰瘍、胃がん、腸閉塞、虫垂炎、腹膜炎、胆石症、肝炎、肝臓炎、脳炎、髄膜炎、脳圧亢進、頭部外傷、乗り物酔い、つわり、化学療法による副作用、放射線療法による副作用、抗癌剤等による副作用、消化管の圧迫・狭窄や手術後の腸管癒着などの原因により起こる消化管通過障害、脳腫瘍・脳出血・髄膜炎・脳への放射線照射などによる脳圧上昇等の原因により起こる嘔気、嘔吐の治療および/または予防、腸閉塞、十二指腸潰瘍または虫垂炎等を原因とする急性便秘、神経障害、低栄養、全身衰弱、ビタミン欠乏症、貧血、感受性低下または機械的刺激不足等を原因とする弛緩性便秘、ストレス等を原因とするけいれん性便秘の治療および/または予防に対して有効である。
【0042】
本発明の式(IA)で示される化合物、その酸付加塩、および/またはそれらの溶媒和物の結晶は、脳移行性が低いため、疼痛を伴う疾患(例えば癌性疼痛(骨転移、神経圧迫、頭蓋内圧亢進、軟部組織浸潤、便秘または筋の攣縮による痛み、内臓、筋・筋膜、腰または肩関節周囲の痛み、術後の慢性的な痛み)、AIDS等)の患者に対して投与されるオピオイド受容体アゴニスト作用を有する化合物の鎮痛作用をほとんど阻害することなく、オピオイド受容体アゴニストにより誘発される嘔気、嘔吐、便秘等の副作用に対して高い軽減効果を示す。また、本発明の結晶はオピオイド受容体に対して純粋なアンタゴニスト活性を有し、hERGチャンネル阻害作用が弱く、心毒性の懸念がない、等の安全性の面における利点も有している。さらに、本発明の結晶は高い経口吸収性、ヒト血漿中における高い安定性、高いバイオアベイラビリティー等の体内動態における有利な特徴をも有しており、医薬品として非常に有効である。
【0043】
オピオイド受容体アゴニスト作用を有する化合物により誘発される嘔気、嘔吐、便秘に対して本発明結晶またはその医薬組成物を投与する場合、その投与はオピオイド受容体アゴニスト作用を有する化合物の投与前、投与後または同時投与のいずれであってもよい。これらの2種の薬物の投与間隔は特に限定されるものではない。例えば、本発明結晶またはその結晶を含む医薬組成物を、オピオイド受容体アゴニスト作用を有する化合物の投与後に投与する場合、オピオイド受容体アゴニストの投与直後〜約3日以内、投与直後〜約1日以内であれば、より有効に作用する。また、オピオイド受容体アゴニスト投与前に、本発明にかかる結晶またはその結晶を含む医薬組成物を投与する場合、オピオイド受容体アゴニスト投与直前〜約1日前、直前〜約12時間前であれば、より有効に作用する。
本発明結晶またはその結晶を含む医薬組成物を嘔気、嘔吐および/または便秘治療剤および/または予防剤として投与する際には、他の嘔気、嘔吐および/または便秘治療剤および/または予防剤と併用してもよい。例えば、塩酸オンダンセトロン、副腎皮質ステロイド(メチルプレドニゾロン、プレドニゾロン、デキサメタゾン等)、プロクロルペラジン、ハロペリドール、チミペロン、ペルフェナジン、メトクロプラミド、ドンペリドン、スコポラミン、塩酸クロルプロマジン、ドロペリドール、刺激性緩下薬(センノシド、ピコスルファートナトリウムなど)、浸透圧性緩下薬(ラクツロース)や塩類緩下薬(酸化マグネシウムなど)等との併用が可能である。
また、本発明結晶またはその結晶を含む医薬組成物は、オピオイド受容体アゴニスト作用を有する化合物および/または他の嘔気、嘔吐および/または便秘治療剤および/または予防剤、ならびに必要に応じて各種医薬用添加剤を配合した合剤とすることも可能である。
【0044】
本発明の結晶はそれ自体でヒト患者に投与することができるか、または、前記した結晶を適当な担体または賦形剤と混合した医薬組成物にて投与することができる。薬物の処方および投与のための技術は、「Remington's Pharmacological Sciences」Mack Publishing Co.、Easton、PA.最新版に見出すことができる。
【0045】
適当な投与経路は、限定されるものではないが、経口、直腸、経粘膜または腸投与あるいは筋肉内、皮下、脊髄内、鞘内、直接的心室内、静脈内、硝子体内、腹腔内、鼻腔内、眼内、注射を含むことができる。好ましい投与経路は経口および非経口である。
本発明の医薬組成物は、当該分野でよく知られた製法、例えば、慣用的な混合、溶解、顆粒化、糖衣−作成、粉末化、乳化、カプセル化、包括、凍結乾燥プロセスによって製造することができる。
【0046】
本発明で用いられる医薬組成物は、本発明の結晶の、医薬的に使用することができる製剤への製造を容易とする賦形剤および補助剤を含めた1以上の製薬学上許容される担体を用いて公知の方法で処方することができる。適切な処方は、選択された投与経路に依存する。
【0047】
注射による投与を行う場合は、本発明の結晶を溶解した水性溶液、好ましくは、生理学上適合する、リンゲル液または生理食塩水のような緩衝液に溶解して投与することができる。経粘膜投与を行う場合は、浸透させるべきバリアーに適した浸透剤を用いて投与することができる。該浸透剤は一般に当該分野で知られているものを用いることができる。
【0048】
経口投与を行う場合は、本発明の結晶と、当該分野でよく知られた医薬上許容される担体とを合わせることによって投与することができる。該担体により、本発明の結晶を、患者による経口摂取のために、錠剤、丸剤、ロゼンジ、糖衣錠、カプセル剤、液剤、ゲル、シロップ、スラリー、懸濁剤として投与することができる。経口用途の医薬組成物は、固体賦形剤を用い、所望であれば他の適切な補助剤を添加した後、得られた混合物を粉砕し、顆粒の混合物を処理して錠剤または糖衣錠コアを得ることによって作成することができる。有用な賦形剤は、特に、ラクトース、スクロース、マンニトール、またはソルビトールを含めた糖などの充填剤、例えば、とうもろこし澱粉、小麦澱粉、米澱粉およびじゃがいも澱粉などのセルロース調製物およびゼラチン、トラガカントガム、メチルセルロース、ヒドロキシプロピルメチルセルロースおよび/またはカルボキシメチルセルロースナトリウムなどである。必要であれば、寒天、アルギン酸などの崩壊剤を添加することができる。アルギン酸ナトリウムなどの塩を用いることもできる。
【0049】
経口用途の医薬組成物は、ゼラチンで作成されたプッシュフィットカプセルならびにゼラチンおよびグリセロールまたはソルビトールなどの可塑剤で作成された密封カプセル剤を含む。プッシュフィットカプセルはラクトースなどの充填剤、澱粉などのバインダーおよび/またはタルクまたはステアリン酸マグネシウムなどの滑沢剤および所望により、安定化剤と混合した本発明結晶を含むことができる。ソフトカプセル剤として用いる場合は、本発明の結晶を、脂肪油、流動パラフィンまたは液状ポリエチレングリコールなどの適当な液体に溶解または懸濁させて用いることができる。安定化剤をこれらの処方に加えることもできる。
【0050】
医薬組成物は、適当な固体またはゲル相の担体または賦形剤を含むこともできる。そのような担体または賦形剤は、例えば、炭酸カルシウム、リン酸カルシウム、種々の糖、澱粉、セルロース誘導体、ゼラチン、ポリエチレングリコールなどのポリマーなどが挙げられる。
【0051】
本発明の結晶またはその結晶を含む医薬組成物の治療上有効な薬効成分量は、最初に細胞培養アッセイから見積もることができる。次いで、細胞培養で決定されたIC50(すなわち、PK活性の最大の半分の阻害を達成する本発明の結晶またはその医薬組成物の濃度)を含む循環濃度範囲を達成するように、動物モデルで用いるために投与量を多く処方することができる。次いで、そのような情報を用いて、ヒトにおける有用な量をより正確に決定することができる。
【0052】
本発明の結晶またはその結晶を含む医薬組成物の毒性および治療効果は、例えば、本発明の主題の結晶またはその医薬組成物についてのIC50を決定することによって、細胞培養または実験動物において、標準的な医薬手法によって測定することができる。これらの細胞培養アッセイおよび動物実験から得られたデータは、ヒトで用いる投与量の範囲を処方するために用いることができる。投与量は、使用する投与形態および利用する投与経路に応じて変化させることができる。正確な処方投与経路および投与量は、患者の状態を考慮して個々の医師が選択することができる(例えば、Finglら、1975、in“The Pharmacological Basis of Therapeutics”、Ch.1p.l参照)。
また、投与量は、疾患の状態、投与ルート、患者の年齢、または体重によっても異なるが、成人に経口で投与する場合、通常0.1μg〜1g/日であり、好ましくは0.01〜200mg/日であり、非経口投与の場合には通常1μg〜10g/日であり、好ましくは0.1mg〜10mg/日である。
【0053】
次に本発明による化合物(I)の製造方法を説明する。
本発明による化合物(I)の製造方法をスキーム1に示した。
【化36】
(式中、R1bは水酸基の保護基;R2は置換基を有していてもよいアルキル;R3は置換基を有していてもよいアルキル、置換基を有していてもよいシクロアルキル、置換もしくは非置換アリール、または置換もしくは非置換ヘテロアリール;R7は置換基を有してもよいフェニル基をそれぞれ表す。)
本発明によれば、カルバメート誘導体(II)に塩基を添加し、室温〜溶媒の沸点以下の温度で1〜10時間反応させることにより化合物(I)を得ることができる。塩基としては水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化セシウム等の無機塩基が好ましく、カルバメート誘導体(II)に対して1〜10当量のアルカリを水溶液として添加することが好ましい。カルバメート誘導体は親水性溶媒、例えばメタノール、エタノール、2−プロパノール、DMSO等の溶媒に溶解し、上記アルカリ水溶液を添加することで好適に反応させることができる。
カルバメート誘導体(II)の水酸基保護基R1bは特に限定されないが、例えばアセチル基等の塩基で脱保護される保護基を用いると化合物(I)を直接得ることができる。R1bが塩基では脱保護されない保護基の場合、上記の塩基処理の前または処理後に適宜保護基を脱保護すればよい。
カルバメート誘導体(II)は化合物(III)にイソシアネート体(V)を反応させることにより得られる。反応は、化合物(III)の溶液に、化合物(III)に対して0.5〜5当量、好ましくは、1.0〜1.2当量のイソシアネート体(V)溶液を加え、室温〜溶媒の沸点以下の温度で1〜10時間反応させる。ここで、例えば、CuCl2のようなルイス酸触媒を0.00005〜1当量、好ましくは0.0001〜0.1当量、さらに好ましくは0.0001〜0.01当量添加することが好ましい。反応溶媒は特に制限はないが酢酸エチル、アセトニトリル、アセトン、トルエン等が使用できる。
ここで用いるイソシアネート体(V)は、下記スキームに従い、
【化37】
例えば、その前駆体であるカルバミン酸エステル(VIII)(式中、R5は低級アルキル)を、ルイス酸および塩基の存在下で反応させることにより得られる。
イソシアネート体に代えて、カルバミン酸活性エステル(VI)(式中、R7は置換基を有していてもよいフェニル基)を用いてカルバメート体(II)を調製することもできる。この活性エステルは例えばアミノ体R3−NH2に相当するフェノールのクロロ蟻酸エステルを反応させて得ることができる。
【化38】
ここでR7としては、OR7が脱離基Xとして機能するものが好ましく、具体的にはフェニル基、p−ニトロフェニル基、p−クロロフェニル基等が例示される。
【実施例】
【0054】
本発明を以下の実施例によりさらに詳しく説明する。これらは本発明を限定するものではない。数値(例えば、量、温度など)に関して正確性を保証する努力をしているが、いくらかの誤差および偏差は考慮されるべきである。特に示さなければ、%は成分の重量%および組成物の全重量の重量%である。圧力は大気圧かまたはそれに近い圧力である。本明細書で使用する他の略語は以下のように定義される:gはグラム、Lはリットル、mgはミリグラム、mLはミリリットル、Bocはt−ブトキシカルボニル基、Acはアセチル基、Meはメチル基、Etはエチル基、Prはプロピル基を表す。
(粉末X線回折パターンの測定)
各実施例で得られた結晶の粉末X線回折測定は、日本薬局方の一般試験法に記載された粉末X線回折測定法に従い、以下の測定条件で行った。
(装置)
Bruker社製D−8Discover
(操作方法)
試料について、以下の条件で測定を行った。
測定法:反射法
光源の種類:Cu管球
使用波長:CuKα線
管電流:40mA
管電圧:40Kv
試料プレート:ガラス
測定範囲:3°―40°
【0055】
(TG/DTAデータの測定)
また、各実施例で得られた各結晶約5mgを量り、アルミニウムパンにつめ、開放系にて測定した。測定条件は以下のとおりである。
(測定条件)
装置:SEIKO社製TG/DTA6300
測定温度範囲:25℃−300℃
昇温速度:10℃/分
【0056】
[実施例1−1]
化合物(IA)のp−トルエンスルホン酸塩[化合物(9)]の製造
【化39】
【0057】
工程1 化合物(3)の合成
t−ブトキシカルボニルアミノイソ酪酸(1)(25.0g、123mmol)の酢酸n−プロピル(150ml)溶液に、0℃でジイソプロピルエチルアミン(17.5g、135.4mmol)を加えた。同温下、混合液にクロロギ酸イソブチル(17.6g、128.9mmol)を滴下し、1時間攪拌した。同反応液にベンズアミドオキシム(2)(17.6g、129.3mmol)の酢酸n−プロピル(100ml)溶液を加え、0℃で1時間攪拌後、95℃で5時間攪拌した。酸水溶液を加え分離後、有機層を水と炭酸水素ナトリウムで洗浄し、減圧濃縮した。同反応液に塩酸を加え2.5時間攪拌し、析出した結晶を濾取し、洗浄、乾燥後、化合物(3)(27.34g、92.7%)を得た。
1H NMR(300MHz, DMSO‐d6) δ 1.80 (6H, s), 7.59-7.64 (3H, m), 8.01-8.05 (2H, m), 9.26(3H, br).
【0058】
工程2 化合物(4)の合成
化合物(3)(19.0g、79.2mmol)のトルエン152ml懸濁液に、25℃でアルカリ水を加え攪拌した。50℃で、クロロギ酸メチル(8.3g、88.0mmol)を加え1時間攪拌後、有機層を分離し、塩酸水、炭酸水素ナトリウム水および水で順次洗浄を行い、減圧濃縮した。この反応液に1.0mol/l三塩化ホウ素のトルエン溶液(7.3ml)を加え、50℃でトリエチルアミンを滴下し、2時間攪拌後、反応液を濃縮して化合物(4)の溶液を得た。
【0059】
工程3 化合物(6)の合成
市販のナルトレキソン塩酸塩(5)(20.0g、52.9mmol)の酢酸エチル160ml溶液にトリエチルアミン(11.3g、111.7mmol)と無水酢酸(5.7g、55.8mmol)を加えた後、40℃で2時間攪拌した。反応液を冷却し、水洗後、減圧濃縮し、化合物(6)溶液を得た。
1H-NMR (300 MHz, DMSO‐d6) δ 0.14 (2H, d, J=4.8 Hz), 0.49 (2H, d, J=7.8 Hz), 0.88 (1H, m), 1.29 (1H, d, J=9.9 Hz),1.46 (1H, td, J=14.1, 3.3 Hz), 1.79(1H, dt, J=12.0, 3.3 Hz), 1.90-2.00(1H, m), 2.11(1H, dt, J=14.4, 3.3 Hz), 2.26 (3H, s), 2.30-2.46 (3H, m), 2.52-2.72(2H, m), 2.92(1H, td, J=14.1, 4.8 Hz), 3.07(1H, d, J=18.9 Hz), 3.17(1H, d, J=5.7 Hz), 4.91(1H, s), 5.18(1H, s), 6.71(1H, d, J=8.1 Hz), 6.83(1H, d, J=8.1 Hz)
【0060】
工程4 化合物(7)の合成
化合物(6)溶液に、化合物(4)の反応液と酢酸エチルを加えた後、この混合液に塩化銅(II)水溶液を加え、25℃で4時間攪拌した。反応液にヘプタンを加えて晶析し、ろ過、洗浄後、乾燥して化合物(7)(89.2%)を得た。
1H-NMR (300 MHz, DMSO‐d6) δ 0.20-0.40 (2H, m), 0.60-0.90 (1H, m), 1.20-1.50 (2H, m), 1.67 (3H, s), 1.74 (3H, s), 1.90-2.10 (2H, m), 2.10-2.20 (2H, m), 2.26 (3H, s), 2.30-2.55 (2H, m), 2.58-2.80 (4H, m), 3.03(2H, m), 4.31(1H, s), 4.81(1H, s), 6.71(1H, d, J=8.1 Hz), 6.85(1H, d, J=7.8 Hz), 7.50-7.70(3H, m), 7.92-8.01(2H, m), 8.11(1H, s).
【0061】
工程5 化合物(8)の合成
化合物(7)(5.5g、9.0mmol)の2−プロパノール22mlの懸濁液に、水酸化カリウム水溶液を滴下し、80℃で5時間攪拌した。反応液をトルエン洗浄した後、pH7.0〜8.0に調整し、酢酸n-プロピルで抽出した。有機層を水洗後、2-プロパノールと酢酸n-プロピルを加え濃縮後、60℃でp−トルエンスルホン酸2-プロパノール溶液(1.5g,8.1mmol)を滴下し、晶析した。冷却後、析出した固体をろ過し、未乾固体(8)[p−トルエンスルホン酸塩・(酢酸n−プロピルおよび2−プロパノール)和物]を得た。
未乾固体(8)[p−トルエンスルホン酸塩・(酢酸n−プロピルおよび2−プロパノール)和物]の粉末X線回折およびTG/DTA分析の結果を下記参考例3に示す。
【0062】
工程6 化合物(9)の合成
未乾固体(8)に、メタノールと酢酸n−プロピルを加え、加温溶解し、ろ過、洗浄後、減圧濃縮した。析出物を濾取後、洗浄し、得られた粗生成物を60℃で3時間減圧乾燥し、化合物(9)の結晶(非溶媒和物:66.3%)を得た。
1H-NMR (300MHz,DMSO-d6) δ 13.37 (1H, s), 9.44 (1H, s), 8.95 (1H, br s), 8.12 (1H, s), 7.99-7.96 (2H, m), 7.60-7.53 (3H, m), 7.49-7.45 (2H, m), 7.11 (2H, d, J=8.4Hz), 6.69 (2H, ABq.), 6.56 (1H, s), 4.94 (1H, s), 3.95 (1H, d, J=5.1Hz), 3.50-3.25 (2H, m), 3.07 (2H, br d, J=12Hz), 3.00-2.90 (1H, m), 2.75-2.60 (1H, m), 2.60-2.40 (2H, m), 2.29 (3H, s), 2.10 (1H, d, J=14.7Hz), 1.70 (6H, s), 1.75-1.60 (1H, m), 1.15-0.95 (1H, m), 0.80-0.55 (2H, m), 0.55-0.35 (2H, m).
粉末X線回折の結果を図1および表2に示す。
【表2】
粉末X線回折スペクトルにおいて、回折角度(2θ):7.8°±0.2°、10.6°±0.2°、15.6°±0.2°、17.8°±0.2°、18.6°±0.2°、20.4°±0.2°、21.5°±0.2°、21.9°±0.2°、23.6°±0.2°および25.5°±0.2°にピークが認められた。
TG/DTA分析結果を図14に示す。
【0063】
[実施例1−2]
式(IA)で示される化合物のp−トルエンスルホン酸塩[化合物(9)]の製造(別法A)
【化40】
【0064】
工程1 化合物(11)の合成
メトキシカルボニル‐2‐メチルアラニン(10)(5.00g、31.0mmol)とアセトニトリル25mlの懸濁液に、0℃でCDI(カルボニルジイミダゾール、5.28g、31.1mmol)とアセトニトリル5mlを加えて1.5時間攪拌した。同温でベンズアミドオキシム(2)(4.65g、34.2mmol)とアセトニトリル20mlを加え2時間攪拌した。同反応液に、化合物(10)に対して0.10当量の炭酸カリウム(0.43g)を水15mlに溶解した炭酸カリウム水溶液全量を加え、溶媒の沸点以下の温度で1〜5時間反応させた。減圧濃縮後、水を加えて析出した粗生成物を濾取、洗浄した。未乾結晶を乾燥し化合物(11)[7.43g、収率91.7%]を得た。
化合物(11):
1H-NMR(300MHz, CDCl3) δ1.81(6H, s), 3.65 (3H, s), 5.46(1H, s), 7.49-7.50 (3H, m), 8.05-8.08 (2H, m).
【0065】
工程2 化合物(4)の合成
化合物(11)(15.12g、57.41mmol)と三塩化アルミニウム(9.19g、68.89mmol)のトルエン溶液に、50℃でトリエチルアミン(7.55g、74.63mmol)を滴下し、同温度で2.5時間攪拌した。有機層を分離し、濃縮後、化合物(4)を反応液として得た。
1H-NMR(300MHz, CDCl3) δ1.84(6H, s), 7.31-7.55 (3H, m), 8.05-8.13 (2H, m).
13C-NMR(75MHz, CDCl3) δ29.85, 55.71, 126.16, 127,44, 128.78, 131.35, 168.23, 180.88.
IR (cm-1)1446, 1478, 1570, 1638, 2256, 2986, 3337.
【0066】
工程3−6 化合物(9)の合成
実施例1−1記載の同じ工程で化合物(5)から化合物(9)(非溶媒和物)を合成した。
【0067】
[実施例1−3]
式(IA)で示される化合物のp−トルエンスルホン酸塩[化合物(9)]の製造(別法B)
【化41】
【0068】
工程1
実施例1−1記載の工程1と同様の方法で合成した化合物(3)(2.03g、10.0mmol)をアセトニトリル20mlに溶解し、氷冷下、ピリジン(0.89ml、11.0mmol)及びクロロ蟻酸4−ニトロフェニルエステル(2.22g、11.0mmol)を加え、室温で1.5時間攪拌した。反応液を、2mol/L塩酸と氷水の中に注ぎ、酢酸エチルで2回抽出した。抽出液を食塩水で洗浄後、無水硫酸ナトリウム上で乾燥し、ろ過、濃縮した。得られた単黄色油状物4.88gに、ヘキサン約20mlを加え、氷冷下、固化させた。得られた固体を濾取し、ヘキサンで洗浄することにより、目的とする化合物(16)(3.74g)を白色固体として得た。
1H-NMR (CDCl3) δ 8.24 (2H, d, J=9.3Hz), 8.09 (2H, m), 7.53-7.45 (3H, m), 7.33 (2H, br d, J=8.7Hz), 5.99 (1H, br s), 1.92 (6H, s).
【0069】
工程2
実施例1−1記載の工程3と同様の方法で合成した化合物(6)(3.28g、8.56mmol)及び化合物(16)(3.79g、10.3mmol)をアセトニトリル10mlに溶解し、22時間還流した。反応液を室温に戻し、氷水の中に注ぎ、酢酸エチルで2回抽出した。抽出液を0.1mol/L水酸化ナトリウム水溶液で2回、食塩水で1回洗浄後、無水硫酸マグネシウム上で乾燥し、ろ過、濃縮した。得られた化合物(7)の非晶形固体(5.46g)は、精製せず、次の反応に使用した。
1H-NMR (DMSO-d6) δ 8.0-7.9 (2H, m), 7.6-7.5 (3H, m), 6.9-6.7 (2H, Abq.), 4.32 (1H, s), 3.2-1.2 (12H, m), 2.26 (3H, s), 1.71 (1H, d, J=21.6Hz), 1.61 (6H, s), 0.95-0.65 (1H, m), 0.55-0.2 (2H, m), 0.2-0.5 (2H, m).
【0070】
工程3
化合物(7)(500mg)をジメチルスルホキシド2mlに溶解し、2mol/L水酸化カリウム水溶液2mlを加え、80℃で6時間加熱攪拌した。反応液を室温に戻し、2mol/L塩酸で中和し、酢酸エチルで2回抽出した。抽出液を0.1mol/L水酸化ナトリウム水溶液、食塩水で順次洗浄後、無水硫酸ナトリウム上で乾燥し、ろ過、濃縮した。得られた淡黄色の非晶形固体412mgをメタノール2mlに溶解し、p−トルエンスルホン酸水和物165mgを加え、30分間静置した。その後、アセトニトリル20mlを加えて、5℃で一晩静置した。析出物を濾取し、減圧下乾燥することにより、目的とするp−トルエンスルホン酸塩(9)(非溶媒和物:282mg)を結晶として得た[化合物(6)から48%の収率]。
1H-NMR (DMSO-d6) δ 13.37 (1H, s), 9.44 (1H, s), 8.95 (1H, br s), 8.12 (1H, s), 7.99-7.96 (2H, m), 7.60-7.53 (3H, m), 7.49-7.45 (2H, m), 7.11 (2H, d, J=8.4Hz), 6.69 (2H, ABq.), 6.56 (1H, s), 4.94 (1H, s), 3.95 (1H, d, J=5.1Hz), 3.50-3.25 (2H, m), 3.07 (2H, br d, J=12Hz), 3.00-2.90 (1H, m), 2.75-2.60 (1H, m), 2.60-2.40 (2H, m), 2.29 (3H, s), 2.10 (1H, d, J=14.7Hz), 1.70 (6H, s), 1.75-1.60 (1H, m), 1.15-0.95 (1H, m), 0.80-0.55 (2H, m), 0.55-0.35 (2H, m).
【0071】
[実施例1−4]
式(IA)で示される化合物のp−トルエンスルホン酸塩[化合物(9)]の製造(別法C)
【化42】
【0072】
工程1 化合物(4)の合成
実施例1−2記載の工程1と同様の方法で合成した化合物(11)(0.5g、1.91mmol)にトルエンを加えた後、TiCl4(2.30mmol)を加え50℃に加熱した。この反応液にトリエチルアミン(2.30mmol)を加え、同温で2時間攪拌し、化合物(4)を反応液として得た。
工程2−7 化合物(9)の合成
実施例1−1記載の同じ工程で化合物(5)から化合物(9)(非溶媒和物)を合成した。
【0073】
[実施例2]
化合物(20)の合成
【化43】
【0074】
工程1
化合物(17)(4.51g、36mmol)をアセトニトリル45mlに溶解し、氷冷下、ピリジン(3.20ml、39.6mmol)及びクロロ蟻酸フェニルエステル(5.00ml、39.6mmol)を加えた。ジメチルホルムアミド9ml及びアセトニトリル30mlを加えて、室温で45分間攪拌した。反応液から析出物を濾取し、冷メタノール、水で洗浄後、減圧乾燥することにより、目的とする化合物(18)(7.02g)を白色固体として得た。
1H-NMR (CDCl3) δ 8.61 (1H, s), 7.43 (2H, t, J=7.8Hz), 7.41 (1H, s), 7.29 (1H, t, J=7.8Hz), 7.21 (2H, d, J=7.8Hz), 3.97 (3H, s).
【0075】
工程2
実施例1−1記載の工程3と同様の方法で合成した化合物(6)(1.92g、5.00mmol)及び化合物18(1.84g、7.50mmol)をジメチルホルムアミド10mlに溶解し、120℃で4時間加熱攪拌した。反応液を室温に戻し、アセトニトリル50mlを加え、析出物を濾去した。濾液を60℃で減圧濃縮し、ジメチルホルムアミドを留去した。残渣にアセトニトリル100mlを加え、氷冷下、30分間攪拌した。析出した結晶を濾取し、冷アセトニトリルで洗浄、減圧乾燥し、目的とする化合物(19)の第一晶(1.66g)を得た。さらに、母液を濃縮した後、残渣にジエチルエーテルを加え、室温で攪拌することにより、第二晶(306mg)、第三晶(71mg)を得た。
1H-NMR (DMSO-d6) δ 10.58 (1H, br s), 8.52 (1H, s), 7.19 (1H, s), 6.83 (2H, Abq.), 4.78 (1H, s), 4.44 (1H, d, J=5.4Hz), 3.90 (3H, s), 3.12 (1H, d, J=18.6Hz), 2.9-2.55 (4H, m), 2.35 (1H, dd, J=6.3Hz, 12.6Hz), 2.27 (3H, s), 2.25-2.12 (3H, m), 2.1-1.9 (1H, m), 1.62-1.48 (1H, m), 1.28-1.20 (1H, m), 0.75-0.62 (1H, m), 0.35 (2H, d, J=7.5Hz), 0.1-0.5 (2H, m).
【0076】
工程3
化合物(19)(2.02mg、3.78mmol)をメタノール9.5mlに溶解し、2mol/L水酸化カリウム水溶液9.5mlを加え、60℃で2.5時間加熱攪拌した。反応液を室温に戻し、氷冷下、2mol/L塩酸で中和した後、メタノールを留去した。析出した粗結晶を濾取した後、酢酸エチルとメタノールの混液(1:1)から再結晶し、目的とする化合物(20)(1.44g)を結晶として得た(収率77%)。
1H-NMR (DMSO-d6) δ 14.2 (1H, br s), 9.19 (1H, s), 8.8 (1H, br s), 8.32 (1H, s), 7.49 (1H, s), 6.56 (2H, ABq.), 6.1 (1H, br s), 4.53 (1H, br s), 3.82 (3H, s), 3.5-2.3 (9H, m), 1.82 (1H, d, J=15.6Hz), 1.53 (1H, br d, J=13.5Hz), 1.15-0.95 (1H, m), 0.75-0.5 (2H, m), 0.5-0.3 (2H, m).
【0077】
[実施例3−1]
化合物(7)の合成(その1)
【化44】
【0078】
実施例1−1記載の工程3と同様の方法で合成した化合物(6)(ナルトレキソン塩酸塩5.00g相当)の酢酸エチル溶液に、酢酸(0.1当量)加えた後50℃に昇温し、実施例1−1記載の工程1および工程2と同じ方法で合成した化合物(4)の反応液を加え、同温で6.5時間攪拌した。反応液にヘプタンを加え、析出固体を濾取し、洗浄後、乾燥して化合物(7)(6.92g、85.3%)を得た。
【0079】
[実施例3−2]
化合物(7)の合成(その2)
【化45】
実施例1−1記載の工程3と同様の方法で合成した化合物(6)の酢酸エチル溶液に、化合物(4)(1.5当量)とトルエンを加え、70℃で8時間攪拌し、化合物(7)を得た。
【0080】
[実施例4]
化合物(IA)のp−トルエンスルホン酸塩水和物結晶(I形)の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)5.00gに2−プロパノール25mL−水2.5mL混液を加え加温溶解した。アセトニトリル50mLを加え室温攪拌を4時間行った。析出晶を濾別し、85℃で減圧乾燥を4時間行い、4.68gの結晶を得た。
元素分析:
計算値:C, 60.28; H, 5.94; N, 7.21; S, 4.13(1.9H2Oとしての計算値)
実測値:C, 60.5; H, 6.17; N, 7.21; S, 3.83
粉末X線回折の結果を図2および表3に示す。
【表3】
粉末X線回折スペクトルにおいて、回折角度(2θ):6.6°±0.2°、8.9°±0.2°、11.4°±0.2°、12.9°±0.2°、14.0°±0.2°、15.0°±0.2°、17.6°±0.2°、18.2°±0.2°、22.4°±0.2°、25.4°±0.2°および28.7°±0.2°にピークが認められた。
TG/DTA分析結果を図15に示す。この結果から、重量減少率を算出すると、2.42%であった。従って、1水相当の水を含むことがわかる。
以上の測定結果から、本I形結晶は、1〜2水相当の水を含む結晶であると考えられる。
【0081】
[実施例5]
化合物(IA)のp−トルエンスルホン酸塩水和物結晶(II形)の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)5.00gにテトラヒドロフラン12.5mLを加え溶解させた。酢酸n−プロピル50mLを加え、室温攪拌を4時間行った。析出晶を濾別し、85℃で減圧乾燥を4時間行い、4.77gの結晶を得た。
元素分析:
計算値:C, 61.56; H, 5.83; N, 7.36; S, 4.21(1.0H2Oとしての計算値)
実測値:C, 61.68; H, 5.78; N, 7.39; S, 4.07
粉末X線回折の結果を図3および表4に示す。
【表4】
粉末X線回折スペクトルにおいて、回折角度(2θ):7.1°±0.2°、8.8°±0.2°、17.5°±0.2°、19.2°±0.2°、19.7°±0.2°、21.2°±0.2°、21.9°±0.2°、23.7°±0.2°、24.5°±0.2°および26.1°±0.2°にピークが認められた。
TG/DTA分析結果を図16に示す。この結果から、重量減少率を算出すると、1.42%であった。従って、0.5水相当の水を含むことがわかる。
以上の測定結果から、本II形結晶は、0.5〜1水相当の水を含む結晶であると考えられる。
【0082】
[実施例6]
化合物(IA)の酢酸塩の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)20.00gに酢酸エチル100mL、炭酸ナトリウム3.18g(p−トルエンスルホン酸塩に対し1.1等量)を溶かした水50mLを加え、分液操作を行った。酢酸エチル層を1%炭酸ナトリウム水溶液50mL、飽和食塩水50mLで洗浄し、各水層を酢酸エチル50mLで逆抽出した。酢酸エチル層を硫酸ナトリウムで脱水し、約30gまで濃縮した。濃縮液にアセトニトリル200mL、酢酸4.6mL(p−トルエンスルホン酸塩に対し3等量)を加え、室温攪拌し、結晶の析出が認められてから、室温下1日静置した。析出晶を濾別し、16.17gの結晶を得た。
粉末X線回折の結果を図4および表5に示す。
【表5】
粉末X線回折スペクトルにおいて、回折角度(2θ):5.6°±0.2°、8.3±0.2°、9.1±0.2°、10.3°±0.2°、12.0°±0.2°、13.5±0.2°、14.6°±0.2°、16.3±0.2°および26.0°±0.2°にピークが認められた。
TG/DTA分析結果を図17に示す。
【0083】
[実施例7]
化合物(IA)の塩酸塩の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)20.00gに酢酸エチル100mL、炭酸ナトリウム3.18g(p−トルエンスルホン酸塩に対し1.1等量)を溶かした水50mLを加え、分液操作を行った。酢酸エチル層を水50mLで2回洗浄し、各水層を酢酸エチル50mLで逆抽出した。酢酸エチル層を硫酸ナトリウムで脱水し、アセトニトリル200mL、4mol/L塩酸−酢酸エチル10mL(p−トルエンスルホン酸塩に対し1.5等量)を加え、約50gまで濃縮した。濃縮液にアセトニトリル200mLを加え、室温攪拌を1時間行った。析出晶を濾別し、10.01gの結晶を得た。
粉末X線回折の結果を図5および表6に示す。
【表6】
粉末X線回折スペクトルにおいて、回折角度(2θ):8.5°±0.2°、10.8°±0.2°、11.3°±0.2°、12.7°±0.2°、13.9°±0.2°、15.6°±0.2°、17.3°±0.2°、19.2°±0.2°、20.1°±0.2°および23.9°±0.2°にピークが認められた。
TG/DTA分析結果を図18に示す。
【0084】
[実施例8]
化合物(IA)のエタノール和物の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)20.00gに酢酸エチル100mL、炭酸ナトリウム3.18g(p−トルエンスルホン酸塩に対し1.1等量)を溶かした水50mLを加え、分液操作を行った。酢酸エチル層を1%炭酸ナトリウム水溶液50mL、飽和食塩水50mLで洗浄し、各水層を酢酸エチル50mLで逆抽出を行った。酢酸エチル層を硫酸ナトリウムで脱水し、約30gまで濃縮した。濃縮液にエタノール100mLを加え、溶液が濁る寸前までイソプロピルエーテルを加えた。室温攪拌し、結晶の析出が認められてから、室温下で1日静置した。析出晶を濾別し、9.57gの結晶を得た。
粉末X線回折の結果を図6および表7に示す。
【表7】
粉末X線回折スペクトルにおいて、回折角度(2θ):6.9°±0.2°、11.0°±0.2°、12.9°±0.2°、13.4°±0.2°、16.5°±0.2°、20.5°±0.2°、21.3°±0.2°、21.8°±0.2°、22.6°±0.2°および25.1°±0.2°にピークが認められた。
TG/DTA分析結果を図19に示す。
【0085】
[実施例9]
化合物(IA)の遊離体の製造
実施例8記載のエタノール和物を120℃で10時間減圧乾燥し、核磁気共鳴スペクトルでエタノールが残留していないことを確認した。
粉末X線回折の結果を図7および表8に示す。
【表8】
粉末X線回折スペクトルにおいて、回折角度(2θ):6.8°±0.2°、11.7°±0.2°、13.5°±0.2°、15.6°±0.2°、16.7°±0.2°、21.6°±0.2°、22.1°±0.2°、23.4°±0.2°、26.7°±0.2°および30.1°±0.2°にピークが認められた。
TG/DTA分析結果を図20に示す。
【0086】
[実施例10]
化合物(IA)のp−トルエンスルホン酸塩・酢酸メチル和物の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)1.00gに2−プロパノール5mL−水0.5mL混液を加え、加温溶解させた。酢酸メチル20mLを加え、室温攪拌を4時間行った。析出晶を濾別し、0.98gの結晶を得た。
粉末X線回折の結果を図8および表9に示す。
【表9】
粉末X線回折スペクトルにおいて、回折角度(2θ):17.5°±0.2°、21.8°±0.2°、23.2°±0.2°、25.1°±0.2°および30.6°±0.2°にピークが認められた。
TG/DTA分析結果を図21に示す。
【0087】
[実施例11]
化合物(IA)のp−トルエンスルホン酸塩・(酢酸エチルおよび2−プロパノール)和物の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)1.00gに2−プロパノール5mL−水0.5mL混液を加え、加温溶解させた。酢酸エチル20mLを加え、室温攪拌を4時間行った。析出晶を濾別し、0.96gの結晶を得た。
粉末X線回折の結果を図9および表10に示す。
【表10】
粉末X線回折スペクトルにおいて、回折角度(2θ):6.6°±0.2°、7.8°±0.2°、17.0°±0.2°、17.8°±0.2°、18.5°±0.2°、21.9°±0.2°および23.6°±0.2°にピークが認められた。
TG/DTA分析結果を図22に示す。
【0088】
[実施例12]
化合物(IA)のp−トルエンスルホン酸塩・(酢酸n−プロピルおよび2−プロパノール)和物の製造
上記実施例1−1の工程5で得た未乾固体(8)の粉末X線回折の結果を図10および表11に示す。
【表11】
粉末X線回折スペクトルにおいて、回折角度(2θ):6.5°±0.2°、8.3°±0.2°、15.5°±0.2°、16.8°±0.2°および18.3°±0.2°にピークが認められた。
TG/DTA分析結果を図23に示す。
【0089】
[実施例13]
化合物(IA)のp−トルエンスルホン酸塩・アセトニトリル和物の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)1.00gに2−プロパノール5mL−水0.5mL混液を加え、加温溶解させた。アセトニトリル15mLを加え、室温攪拌を4時間行った。析出晶を濾別し、1.02gの結晶を得た。
粉末X線回折の結果を図11および表12に示す。
【表12】
粉末X線回折スペクトルにおいて、回折角度(2θ):11.3°±0.2°、17.6°±0.2°、21.7°±0.2°、22.3°±0.2°および28.5°±0.2°にピークが認められた。
TG/DTA分析結果を図24に示す。
【0090】
[実施例14]
化合物(IA)のp−トルエンスルホン酸塩・1、2−ジメトキシエタン和物の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)1.00gにテトラヒドロフラン5mLを加え加温溶解させた。1、2−ジメトキシエタン20mLを加え、室温攪拌を4時間行った。析出晶を濾別し、1.05gの結晶を得た。
粉末X線回折の結果を図12および表13に示す。
【表13】
粉末X線回折スペクトルにおいて、回折角度(2θ):8.5°±0.2°、13.0°±0.2°、14.0°±0.2°、14.9°±0.2°、17.3°±0.2°、20.0°±0.2°、21.9°±0.2°、22.7°±0.2°、25.6°±0.2°および27.2°±0.2°にピークが認められた。
TG/DTA分析結果を図25に示す。
【0091】
[実施例15]
化合物(IA)のp−トルエンスルホン酸塩・メチルイソブチルケトン和物の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)1.00gにテトラヒドロフラン5mLを加え加温溶解させた。メチルイソブチルケトン20mLを加え、室温攪拌を4時間行った。析出晶を濾別し、1.02gの結晶を得た。
粉末X線回折の結果を図13および表14に示す。
【表14】
粉末X線回折スペクトルにおいて、回折角度(2θ):5.9°±0.2°、7.9°±0.2°、13.5°±0.2°、15.2°±0.2°、23.2°±0.2°、23.7°±0.2°、24.0°±0.2°、25.0°±0.2°、27.0°±0.2°および28.5°±0.2°にピークが認められた。
TG/DTA分析結果を図26に示す。
【0092】
試験例1
結晶の固体安定性試験
結晶約10mgをポリエチレン製栓付2mL硝子容器に正確に秤取した。該硝子容器を閉栓後、パラフイルムを巻き、40℃または60℃で、2週間または4週間保存した。40℃で保存したサンプルを40℃密栓保存品といい、60℃で保存したサンプルを60℃密栓保存品という。該硝子容器を開栓した状態で、40℃相対湿度89%または60℃で、2週間または4週間保存した。40℃相対湿度89%で保存したサンプルを40℃相対湿度89%保存品といい、60℃で保存したサンプルを60℃開栓保存品という。−40℃密栓保存品を標準品として、下記条件下のHPLC法で絶対検量線法により含量を測定した。
式(IA)のp−トルエンスルホン酸塩(非溶媒和物)の結晶の外観変化の観察結果および結晶の残存率を表16に示した。
式(IA)のp−トルエンスルホン酸塩水和物結晶(I形)の外観変化の観察結果および結晶の残存率を表17に示した。
式(IA)のp−トルエンスルホン酸塩水和物結晶(II形)の外観変化の観察結果および結晶の残存率を表18に示した。
ここで、外観変化がなかったものを(−)、わずかに外観変化が認められたものを(±)とした。
式(IA)のp−トルエンスルホン酸塩(非溶媒和物)の結晶、式(IA)のp−トルエンスルホン酸塩水和物結晶(I形)および式(IA)のp−トルエンスルホン酸塩水和物結晶(II形)の外観変化はなく、いずれの結晶の残存率も低下することなく、安定であることを確認した。
HPLC条件
カラム:CAPCELL PAK C18 AQ (3μm 3.0x150mm)
カラム温度:50℃
UV検出波長:231nm
移動相:[A]10mmol/L ギ酸アンモニウムおよび10mmol/L 塩化マグネシウムの混合液、[B]アセトニトリルを表15のようにグラジエントした。
流速:0.6mL/分
【表15】
【表16】
【表17】
【表18】
【0093】
試験例2
結晶の吸湿性確認試験
式(IA)のp−トルエンスルホン酸塩(非溶媒和物)の結晶約10mgを、水蒸気吸脱着測定装置用試料容器に量り取り、25℃において乾燥窒素雰囲気下で乾燥させた。乾燥後、相対湿度を0%から95%の範囲において5%間隔で連続的に変化させ、試料の水蒸気吸脱着量を水蒸気吸脱着測定装置DVS Advantage(surface measurement systems社製)で測定した。
結果を表19に示す。25℃における相対湿度0%から95%の範囲での式(IA)のp−トルエンスルホン酸塩(非溶媒和物)の結晶の最大吸湿量は1.2%未満であり、該結晶は、ほとんど吸湿性を示さなかった。
【表19】
【0094】
製剤例1
以下の成分を含有する顆粒剤を製造する。
【表20】
上記の「式(IA)で示される化合物」は、式(IA)で示される化合物の遊離体、その酸付加塩および/またはそれらの溶媒和物の結晶を包含する。
式(IA)で表わされる化合物と乳糖を60メッシュのふるいに通す。コーンスターチを120メッシュのふるいに通す。これらをV型混合機にて混合する。混合末にHPC-L(低粘度ヒドロキシプロピルセルロース)水溶液を添加し、練合、造粒(押し出し造粒 孔径0.5〜1mm)、乾燥工程する。得られた乾燥顆粒を振動ふるい(12/60メッシュ)で櫛過し顆粒剤を得る。
【0095】
製剤例2
以下の成分を含有するカプセル充填用顆粒剤を製造する。
【表21】
上記の「式(IA)で示される化合物」は、式(IA)で示される化合物の遊離体、その酸付加塩および/またはそれらの溶媒和物の結晶を包含する。
式(IA)で表わされる化合物、乳糖を60メッシュのふるいに通す。コーンスターチを120メッシュのふるいに通す。これらを混合し、混合末にHPC-L溶液を添加して練合、造粒、乾燥する。得られた乾燥顆粒を整粒後、その150mgを4号硬ゼラチンカプセルに充填する。
【0096】
製剤例3
以下の成分を含有する錠剤を製造する。
【表22】
上記の「式(IA)で示される化合物」は、式(IA)で示される化合物の遊離体、その酸付加塩および/またはそれらの溶媒和物の結晶を包含する。
式(IA)で表わされる化合物、乳糖、微結晶セルロース、CMC-Na(カルボキシメチルセルロース ナトリウム塩)を60メッシュのふるいに通し、混合する。混合末にステアリン酸マグネシウム混合し、製錠用混合末を得る。本混合末を直打し、150mgの錠剤を得る。
【0097】
製剤例4
以下の成分を加温混合後、滅菌して注射剤とする。
【表23】
上記の「式(IA)で示される化合物」は、式(IA)で示される化合物の遊離体、その酸付加塩および/またはそれらの溶媒和物の結晶を包含する。
【産業上の利用可能性】
【0098】
本発明は、医薬品製造用原体として有用な6,7−不飽和−7−カルバモイルモルヒナン誘導体、その酸付加塩、それらの溶媒和物、またはそれらの安定な結晶体を提供する。
また本発明は、オピオイド受容体アゴニスト作用を有する化合物に誘発される嘔気、嘔吐および/または便秘の治療および/または予防剤として有用な、6,7−不飽和−7−カルバモイルモルヒナン誘導体、その酸付加塩、それらの溶媒和物、またはそれらの結晶の新たな製造方法を提供するものである。
【技術分野】
【0001】
本発明は、モルヒナン誘導体の結晶およびその製造方法に関する。より詳しくは、6,7−不飽和−7−カルバモイルモルヒナン誘導体、その酸付加塩および/またはそれらの溶媒和物の結晶、およびそれらの製造方法に関する。
【背景技術】
【0002】
薬物デリバリーにおいては、有用な優れた化学的および/または物理的特性を有する結晶形態が望まれている。
特許文献1には、下式:
【化1】
で表される6,7−不飽和−7−カルバモイルモルヒナン誘導体が嘔吐および/または便秘の治療および/または予防剤として有用であることが記載されている。またその実施例には、以下の化合物(I−284):
【化2】
が遊離塩の形態で開示されているが、その酸付加塩および/または溶媒和物は、具体的に開示されていない。また、その結晶についても全く記載はない。
また、6,7−不飽和−7−カルバモイルモルヒナン誘導体の製造方法としては、以下の式:
【化3】
で示されるように、7位カルボキシル誘導体から、対応する7位カルバモイル体を合成する製造方法が開示されているに過ぎない。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】国際公開第2006/126637号パンフレット
【特許文献2】国際公開第2001/002375号パンフレット
【非特許文献】
【0004】
【非特許文献1】Chemical Communications, 1998, vol.23, 2575-2576
【非特許文献2】Synthesis, 1989, vol.2, 131-132
【発明の概要】
【発明が解決しようとする課題】
【0005】
医薬活性成分は、それぞれの固体形態によって、実質的に異なる物理的特性を有し得る。このような物理的特性の違いは、例えば医薬活性成分の製造方法もしくは投与方法、又は医薬活性成分を含む医薬組成物に影響を与え得る。
6,7−不飽和−7−カルバモイルモルヒナン誘導体は既に開示されているものの、医薬品として使用または医薬品として工業的に製造するために、好適な塩および/または安定な結晶形ならびにより好ましい製造方法の確立が望まれている。
【課題を解決するための手段】
【0006】
本発明者等は、鋭意検討した結果、下式(IA):
【化4】
で示される化合物、その酸付加塩、および/またはそれらの溶媒和物から安定な結晶が得られることを見出して以下の発明を完成した。
本発明者等は、さらに、下式(II):
【化5】
(式中、R1は水素原子または水酸基の保護基、R2は置換基を有していてもよい低級アルキル、R3は置換基を有していてもよい低級アルキル、置換基を有していてもよいシクロアルキル、置換基を有していてもよいアリール、または置換基を有していてもよいヘテロアリール)
で表されるカルバメート誘導体を塩基存在下で反応させ、必要により保護基R1を脱保護することで、下式(I):
【化6】
(式中、R2およびR3は上記と同意義)
の化合物が得られることを見出し、6,7−不飽和−7−カルバモイルモルヒナン誘導体の新たな製造方法に係る発明を完成させた。
【0007】
本発明は以下の通りである。
(1)下式(IA):
【化7】
で示される化合物のp−トルエンスルホン酸塩、酢酸塩若しくは塩酸塩、または当該化合物若しくは当該酸付加塩の溶媒和物。
【0008】
(2)下式(IA):
【化8】
で示される化合物のp−トルエンスルホン酸塩の結晶、または当該酸付加塩の溶媒和物の結晶。
【0009】
(3)粉末X線回折スペクトルにおいて、回折角度(2θ):7.8°±0.2°、10.6°±0.2°、15.6°±0.2°、17.8°±0.2°および21.5°±0.2にピークを有する、(2)に記載のp−トルエンスルホン酸塩の結晶。
(4)粉末X線回折スペクトルにおいて、回折角度(2θ):7.8°±0.2°、10.6°±0.2°、15.6°±0.2°、17.8°±0.2°、18.6°±0.2°、20.4°±0.2°、21.5°±0.2°、21.9°±0.2°、23.6°±0.2°および25.5°±0.2°にピークを有する、(2)に記載のp−トルエンスルホン酸塩の結晶。
(5)図1に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、(2)に記載のp−トルエンスルホン酸塩の結晶。
【0010】
(6)粉末X線回折スペクトルにおいて、回折角度(2θ):12.9°±0.2°、17.6°±0.2°、22.4°±0.2°、25.4°±0.2°および28.7°±0.2°にピークを有する、(2)に記載のp−トルエンスルホン酸塩水和物のI形結晶。
(7)粉末X線回折スペクトルにおいて、回折角度(2θ):6.6°±0.2°、8.9°±0.2°、11.4°±0.2°、12.9°±0.2°、14.0°±0.2°、15.0°±0.2°、17.6°±0.2°、18.2°±0.2°、22.4°±0.2°、25.4°±0.2°および28.7°±0.2°にピークを有する、(2)に記載のp−トルエンスルホン酸塩水和物のI形結晶。
(8)図2に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、(2)に記載のp−トルエンスルホン酸塩水和物のI形結晶。
【0011】
(9)粉末X線回折スペクトルにおいて、回折角度(2θ):8.8°±0.2°、17.5°±0.2°、21.9°±0.2°、23.7°±0.2°および26.1°±0.2°にピークを有する、(2)に記載のp−トルエンスルホン酸塩水和物のII形結晶。
(10)粉末X線回折スペクトルにおいて、回折角度(2θ):7.1°±0.2°、8.8°±0.2°、17.5°±0.2°、19.2°±0.2°、19.7°±0.2°、21.2°±0.2°、21.9°±0.2°、23.7°±0.2°、24.5°±0.2°および26.1°±0.2°にピークを有する、(2)に記載のp−トルエンスルホン酸塩水和物のII形結晶。
(11)図3に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、(2)に記載のp−トルエンスルホン酸塩水和物のII形結晶。
【0012】
(12)式(IA):
【化9】
で示される化合物の酢酸塩の結晶、または当該酸付加塩の溶媒和物の結晶。
(13)粉末X線回折スペクトルにおいて、回折角度(2θ):5.6°±0.2°、10.3°±0.2°、12.0°±0.2°、14.6°±0.2°および26.0°±0.2°にピークを有する、(12)に記載の酢酸塩の結晶。
(14)粉末X線回折スペクトルにおいて、回折角度(2θ):5.6°±0.2°、8.3±0.2°、9.1±0.2°、10.3°±0.2°、12.0°±0.2°、13.5±0.2°、14.6°±0.2°、16.3±0.2°および26.0°±0.2°にピークを有する、(12)に記載の酢酸塩の結晶。
(15)図4に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、(12)に記載の酢酸塩の結晶。
【0013】
(16)式(IA):
【化10】
で示される化合物の塩酸塩の結晶、または当該酸付加塩の溶媒和物の結晶。
(17)粉末X線回折スペクトルにおいて、回折角度(2θ):8.5°±0.2°、12.7°±0.2°、15.6°±0.2°、17.3°±0.2°および23.9°±0.2°にピークを有する、(16)に記載の塩酸塩の結晶。
(18)粉末X線回折スペクトルにおいて、回折角度(2θ):8.5°±0.2°、10.8°±0.2°、11.3°±0.2°、12.7°±0.2°、13.9°±0.2°、15.6°±0.2°、17.3°±0.2°、19.2°±0.2°、20.1°±0.2°および23.9°±0.2°にピークを有する、(16)に記載の塩酸塩の結晶。
(19)図5に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、(16)に記載の塩酸塩の結晶。
【0014】
(20)式(IA):
【化11】
で示される化合物の結晶、またはその溶媒和物の結晶。
(21)粉末X線回折スペクトルにおいて、回折角度(2θ):13.5°±0.2°、21.6°±0.2°、22.1°±0.2°、23.4°±0.2°および26.7°±0.2°にピークを有する、(20)に記載の式(IA)で示される化合物の結晶。
(22)粉末X線回折スペクトルにおいて、回折角度(2θ):6.8°±0.2°、11.7°±0.2°、13.5°±0.2°、15.6°±0.2°、16.7°±0.2°、21.6°±0.2°、22.1°±0.2°、23.4°±0.2°、26.7°±0.2°および30.1°±0.2°にピークを有する、(20)に記載の式(IA)で示される化合物の結晶。
(23)図7に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、(20)に記載の式(IA)で示される化合物の結晶。
【0015】
(24)粉末X線回折スペクトルにおいて、回折角度(2θ):11.0°±0.2°、16.5°±0.2°、20.5°±0.2°、21.8°±0.2°および22.6°±0.2°にピークを有する、(20)に記載のエタノール和物の結晶。
(25)粉末X線回折スペクトルにおいて、回折角度(2θ):6.9°±0.2°、11.0°±0.2°、12.9°±0.2°、13.4°±0.2°、16.5°±0.2°、20.5°±0.2°、21.3°±0.2°、21.8°±0.2°、22.6°±0.2°および25.1°±0.2°にピークを有する、(20)に記載のエタノール和物の結晶。
(26)図6に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、(20)に記載のエタノール和物の結晶。
(27)上記(2)〜(26)のいずれかに記載の結晶を含む医薬組成物。
(27A)上記(2)〜(26)のいずれかに記載の結晶を含有することを特徴とするオピオイド受容体拮抗剤。
(27B)上記(2)〜(26)のいずれかに記載の結晶を含有することを特徴とする、嘔気、嘔吐および/または便秘の治療および/または予防剤。
(27C)上記(2)〜(26)のいずれかに記載の結晶を含有することを特徴とする、オピオイド受容体アゴニスト作用を有する化合物により誘発される副作用の軽減および/または予防剤。
(27D)副作用が嘔気、嘔吐および/または便秘である、上記(27C)記載の治療および/または予防剤。
(27E)オピオイド受容体アゴニスト作用を有する化合物がモルヒネ、オキシコドン、ハイドロコドン、トラマドール、またはそれらの製薬上許容される塩またはそれらの溶媒和物である、上記(27C)または(27D)記載の治療および/または予防剤。
(27F)嘔気、嘔吐および/または便秘の治療および/または予防のための医薬を製造するための、上記(2)〜(26)のいずれかに記載の結晶の使用。
(27G)オピオイド受容体アゴニスト作用を有する化合物により誘発される副作用の軽減および/または予防のための医薬を製造するための、上記(2)〜(26)のいずれかに記載の結晶の使用。
(27H)上記(2)〜(26)のいずれかに記載の結晶を含む医薬組成物を投与することを特徴とする、嘔気、嘔吐および/または便秘の治療および/または予防方法。
(27I)上記(2)〜(26)のいずれかに記載の結晶を含む医薬組成物を投与することを特徴とする、オピオイド受容体アゴニスト作用を有する化合物により誘発される副作用の軽減および/または予防方法。
(27J)嘔気、嘔吐および/または便秘の治療および/または予防をするための、上記(2)〜(26)のいずれかに記載の結晶を含む医薬組成物。
(27K)オピオイド受容体アゴニスト作用を有する化合物により誘発される副作用の軽減および/または予防をするための、上記(2)〜(26)のいずれかに記載の結晶を含む医薬組成物。
(27L)オピオイド受容体アゴニスト作用を有する化合物と、当該化合物投与により誘発される副作用の軽減および/または予防のために効果的な量の上記(2)〜(26)のいずれかに記載の結晶とを組み合わせてなる鎮痛剤。
(27M)オピオイド受容体アゴニスト作用を有する化合物と、当該化合物投与により誘発される嘔気、嘔吐および/または便秘の治療および/または予防のために効果的な量の上記(2)〜(26)のいずれかに記載の結晶とを組み合わせてなる鎮痛剤。
(27N)オピオイド受容体アゴニスト作用を有する化合物がモルヒネ、オキシコドン、ハイドロコドン、トラマドール、またはそれらの製薬上許容される塩またはそれらの溶媒和物である、上記(27L)または(27M)記載の鎮痛剤。
【0016】
(28)式(IA):
【化12】
で示される化合物に酸を添加し、必要に応じて溶媒中から結晶化することを特徴とする、(2)〜(19)のいずれかに記載の、式(IA)で示される化合物の酸付加塩の結晶、または当該酸付加塩の溶媒和物の結晶の製造方法。
(29)式(IID):
【化13】
(式中、R1は水素原子または水酸基の保護基)
で示される化合物を塩基で処理し、必要であればR1を脱保護した後、p−トルエンスルホン酸を添加し、必要に応じて溶媒中から結晶化することを特徴とする、(2)に記載の結晶の製造方法。
(30)式(IIE):
【化14】
(式中、R1aは水素原子または塩基で脱保護可能な水酸基の保護基)
で示される化合物を塩基で処理し、次いでp−トルエンスルホン酸を添加し、必要に応じて溶媒中から結晶化することを特徴とする、(29)の製造方法。
(31)式(III):
【化15】
(式中、R1bは水酸基の保護基;R2は置換基を有していてもよい低級アルキル)で示される化合物を、酸の存在下または非存在下、式:R3−N=C=O(式中、R3は置換基を有していてもよい低級アルキル、置換基を有していてもよいシクロアルキル、置換もしくは非置換アリール、または置換もしくは非置換ヘテロアリール)で示される化合物または式:R3−NH−C(=O)−X(式中、R3は前記と同意義、Xは脱離基)で示される化合物と反応させることを特徴とする、式(II):
【化16】
(式中、R1b、R2およびR3は、前記と同意義)で示される化合物の製造方法。
(32)式(IV):
【化17】
(式中、R2は(31)と同意義)で示される化合物の水酸基を保護することにより、当該式(III):
【化18】
(式中、R1bおよびR2は(31)と同意義)
で示される化合物を得ることを特徴とする、(31)に記載の製造方法。
(33)式(IV):
【化19】
(式中、R2は(31)と同意義)
で示される化合物の水酸基を保護し、式(III):
【化20】
(式中、R1bおよびR2は(31)と同意義)
で示される化合物を得る工程、および式(III)で示される化合物を、酸の存在下または非存在下、式:R3−N=C=O(式中、R3は(31)と同意義)で示される化合物または式:R3−NH−C(=O)−X(式中、R3は前記と同意義、Xは脱離基)で示される化合物と反応させる工程を連続して行う、(32)記載の製造方法。
ここで、「工程を連続して行う」とは、前工程の反応により生成した化合物を単離することなく、次工程を行うことを包含する。例えば、ワンポットで2つの工程を行うことが挙げられる。
(34)酸の存在下で行う、(31)〜(33)のいずれかに記載の製造方法。
(35)酸がルイス酸である、(34)記載の製造方法。
(36)ルイス酸触媒が、CuCl、CuCl2、CuBr、CuI、CuBr、CuSO4、Cu、Zn(OAc)2、ZnBr2またはZnCl2である、(35)に記載の製造方法。
(37)式(III)で示される化合物に対して、約0.00005〜約1.0当量の酸の存在下で反応させることを特徴とする、(31)〜(36)のいずれかに記載の製造方法。
(38)R1bが塩基で脱保護可能な水酸基の保護基である、(31)〜(37)のいずれかに記載の製造方法。
(39)式(IIA):
【化21】
(式中、R2およびR3は(31)と同意義)で示される化合物を、塩基で処理することを特徴とする、式(I):
【化22】
(式中、R2およびR3は、前記と同意義)で示される化合物の製造方法。
(40)式(IIC):
【化23】
(式中、R1cは塩基で脱保護可能な水酸基の保護基;R2およびR3は(31)と同意義)
で示される化合物を、塩基で処理することを特徴とする、式(I):
【化24】
(式中、R2およびR3は、前記と同意義)で示される化合物の製造方法。
(41)式(IIB):
【化25】
(式中、R1bは水酸基の保護基;R2およびR3は(31)と同意義)
で示される化合物を、塩基で処理することを特徴とする、式(IB):
【化26】
(式中、R1dは塩基では脱保護されない水酸基の保護基または水素原子;R2およびR3は、前記と同意義)
で示される化合物の製造方法。
(42)塩基が無機塩基である(39)〜(41)のいずれかに記載の製造方法。
(43)塩基が水酸化カリウム、水酸化ナトリウム、水酸化リチウムまたは水酸化セシウムである(39)〜(41)のいずれかに記載の製造方法。
(44)反応温度が30℃〜100℃である(39)〜(43)のいずれかに記載の製造方法。
(45)式(VIIIa):
【化27】
(式中、R5は低級アルキル)で示される化合物を、ルイス酸および塩基の存在下で反応させることを特徴とする、式(IX):
【化28】
で示される化合物の製造方法。
(46)式(X):
【化29】
(式中、R5は低級アルキル)
で示される化合物と、式(XI):
【化30】
で示される化合物を反応させ、当該式(VIIIa)で示される化合物を得ることを特徴とする、(45)記載の製造方法。
(47)ルイス酸がAlCl3またはTiCl4である(45)または(46)に記載の製造方法。
(48)式(IIIA):
【化31】
(式中、R1cは、塩基で脱保護可能な水酸基の保護基;R2は(31)と同意義)
で示される化合物を、ルイス酸触媒の存在下または非存在下、式:R3−N=C=O(式中、R3は(31)と同意義)で示される化合物または式:R3−NH−C(=O)−X(式中、R3は前記と同意義、Xは脱離基)で示される化合物と反応させ、式(IIC):
【化32】
(式中、R1c、R2およびR3は、前記と同意義)
で示される化合物を得る工程、
上記式(IIC)で示される化合物を、塩基で処理し、式(I):
【化33】
(式中、R2およびR3は、前記と同意義)
で示される化合物を得る工程、および
上記式(I)で示される化合物に酸を添加して酸付加塩とする工程を包含する、式(I)で示される化合物の酸付加塩の製造方法。
酸を添加して酸付加塩とした後、必要に応じて該反応液を冷却してもよい。
ここで、式(IIC)で示される化合物を得る工程において、一つの態様として、ルイス酸触媒存在下、式(IIIA)で示される化合物と式:R3−N=C=O(式中、R3は上記と同意義)で示される化合物を反応させる工程が挙げられる。一つの態様として、ルイス酸触媒非存在下、式(IIIA)で示される化合物と式:R3−N=C=O(式中、R3は上記と同意義)で示される化合物を反応させる工程が挙げられる。別の一つの態様として、ルイス酸触媒非存在下、式(IIIA)で示される化合物と式:R3−NH−C(=O)−X(式中、R3およびXは上記と同意義)で示される化合物を反応させる工程が挙げられる。
(49)式(I)で示される化合物の酸付加塩が、p−トルエンスルホン酸塩、酢酸塩または塩酸塩、またはそれらの溶媒和物である、(48)の製造方法。
(50)p−トルエンスルホン酸塩、酢酸塩または塩酸塩、またはそれらの溶媒和物が結晶である、(49)の製造方法。
(51)式(IID):
【化34】
(式中、R1は水素原子または水酸基の保護基)
で示される化合物。
(52)式(VII):
【化35】
[式中、−R6は、−N=C=Oまたは−NH−C(=O)−X(式中、Xは脱離基)で表される基]で示される化合物。
上記式(II)で示される化合物、式(IIA)で示される化合物、式(IIB)で示される化合物、式(IIC)で示される化合物、式(IID)で示される化合物および式(IIE)で示される化合物において、モルヒナン骨格の7位側鎖である「−O−C(=O)−NH−」基の「−NH−」の水素原子は、アミノ基の保護基に置き換わってもよい。
【0017】
本明細書中、「ハロゲン」とはフッ素、塩素、臭素およびヨウ素を包含する。「ハロゲノ低級アルキル」、「ハロゲノ低級アルコキシ」、「ハロゲノ低級アルキルチオ」のハロゲン部分も同様である。
「低級アルキル」とは、炭素数1〜10、好ましくは炭素数1〜6、さらに好ましくは炭素数1〜3の直鎖または分枝状のアルキルを包含し、例えばメチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチル、n−ペンチル、イソペンチル、ネオペンチル、ヘキシル、イソヘキシル、n−へプチル、イソヘプチル、n−オクチル、イソオクチル、n−ノニルおよびn−デシル等が挙げられる。好ましくはメチル、エチル、イソプロピル、n−ブチル、sec−ブチル、tert−ブチル、1-エチルプロピル等である。
【0018】
「置換基を有していてもよい低級アルキル」の置換基としてはハロゲン、ヒドロキシ、低級アルコキシ、ハロゲノ低級アルコキシ、ヒドロキシ低級アルコキシ、低級アルキルチオ、低級アルキルアミノ、アシルアミノ、アシル、アシルオキシ、シアノ、カルボキシ、低級アルコキシカルボニル、カルバモイル、低級アルキルカルバモイル、シアノカルバモイル、低級アルキルスルホニルカルバモイル、アリールスルホニルカルバモイル、スルファモイル、低級アルキルスルファモイル、低級アルキルスルホニル、置換基群αから選択される1以上の基で置換されていてもよいシクロアルキル(ここで置換基群αとはハロゲン、ヒドロキシ、低級アルキル、ハロゲノ低級アルキル、ヒドロキシ低級アルキル、低級アルコキシ低級アルキル、カルボキシ低級アルキル、低級アルコキシカルボニル低級アルキル、アミノ低級アルキル、低級アルキルアミノ低級アルキル、アシルアミノ低級アルキル、シアノ低級アルキル、低級アルコキシ、ハロゲノ低級アルコキシ、ヒドロキシ低級アルコキシ、低級アルキルチオ、ハロゲノ低級アルキルチオ、アシル、アシルオキシ、アミノ、低級アルキルアミノ、アシルアミノ、シアノ、カルボキシ、低級アルコキシカルボニル、カルバモイル、低級アルキルカルバモイル、アリールカルバモイル、シアノカルバモイル、低級アルキルスルホニルカルバモイル、スルファモイル、低級アルキルスルファモイル、低級アルキルスルホニル、低級アルキレンジオキシで置換されていてもよいアリールおよびヘテロ環式基である)、置換基群αから選択される1以上の基で置換されていてもよいシクロアルケニル、置換基群αから選択される1以上の基で置換されていてもよいアリール、置換基群αから選択される1以上の基で置換されていてもよいアリールオキシ、置換基群αから選択される1以上の基で置換されていてもよいアリールチオ、置換基群αから選択される1以上の基で置換されていてもよいヘテロ環式基、置換基群αから選択される1以上の基で置換されていてもよいヘテロ環オキシ等が挙げられる。
【0019】
「ハロゲノ低級アルキル」、「ヒドロキシ低級アルキル」、「アミノ低級アルキル」、「アシルアミノ低級アルキル」、「アシルオキシ低級アルキル」、「シクロアルキル低級アルキル」、「低級アルコキシ」、「ハロゲノ低級アルコキシ」、「ヒドロキシ低級アルコキシ」、「低級アルコキシ低級アルキル」、「低級アルコキシカルボニル」、「カルボキシ低級アルキル」、「低級アルコキシカルボニル低級アルキル」、「低級アルキルチオ」、「ハロゲノ低級アルキルチオ」、「低級アルキルアミノ」、「低級アルキルアミノ低級アルキル」、「低級アルキルカルバモイル」、「低級アルキルスルファモイル」、「低級アルキルスルホニル」、「アリール低級アルキル」、「トリ低級アルキルシリル」、「低級アルキルジアリールシリル」、「トリアリール低級アルキルシリル」、「低級アルコキシ低級アルコキシ低級アルキル」、「低級アルキルチオ低級アルキル」、「アリール低級アルコキシ低級アルキル」、「低級アルキルスルホニル」、「低級アルキルスルホニルカルバモイル」、「低級アルキルカルボニル」、「シアノ低級アルキル」、「低級アルコキシカルボニルアミノ」、「低級アルキレンジオキシ」、「ヘテロ環低級アルキル」の低級アルキル部分は上記「低級アルキル」と同様である。
「置換基を有していてもよい低級アルコキシ」、「置換基を有していてもよい低級アルキルチオ」、「置換基を有していてもよい低級アルキルスルホニル」の置換基は上記「置換基を有していてもよい低級アルキル」の置換基と同様である。
【0020】
「低級アルケニル」とは、任意の位置に1以上の二重結合を有する炭素数2〜10、好ましくは炭素数2〜8、さらに好ましくは炭素数3〜6の直鎖または分枝状のアルケニルを包含する。具体的にはビニル、アリル、プロペニル、イソプロペニル、ブテニル、イソブテニル、プレニル、ブタジエニル、ペンテニル、イソペンテニル、ペンタジエニル、ヘキセニル、イソヘキセニル、ヘキサジエニル、ヘプテニル、オクテニル、ノネニルおよびデセニル等を包含する。
「置換基を有していてもよい低級アルケニル」の置換基は上記「置換基を有していてもよい低級アルキル」と同様である。
「低級アルキニル」とは、任意の位置に1以上の三重結合を有する炭素数2〜10、好ましくは炭素数2〜8、さらに好ましくは炭素数3〜6の直鎖または分枝状のアルキニルを包含する。具体的には、エチニル、プロピニル、ブチニル、ペンチニル、ヘキシニル、ヘプチニル、オクチニル、ノニニル、デシニル等を包含する。これらはさらに任意の位置に二重結合を有していてもよい。
【0021】
「置換基を有していてもよい低級アルキニル」の置換基は上記「置換基を有していてもよい低級アルキル」の置換基と同様である。
「置換基を有していてもよいアミノ」の置換基としては、置換基群αから選択される1以上の基で置換されていてもよい低級アルキル、置換基群αから選択される1以上の基で置換されていてもよいシクロアルキル、置換基群αから選択される1以上の基で置換されていてもよいアシル、置換基群αから選択される1以上の基で置換されていてもよいアミノ、置換基群αから選択される1以上の基で置換されていてもよいアリール、スルファモイル、置換基群αから選択される1以上の基で置換されていてもよい低級アルキルスルファモイル、置換基群αから選択される1以上の基で置換されていてもよいアリールスルファモイル、置換基群αから選択される1以上の基で置換されていてもよい低級アルキルスルホニル、置換基群αから選択される1以上の基で置換されていてもよいアリールスルホニル、置換基群αから選択される1以上の基で置換されていてもよいアリールアミノ、置換基群αから選択される1以上の基で置換されていてもよいヘテロ環式基等が挙げられる。
【0022】
「置換基を有していてもよいカルバモイル」の置換基は上記「置換基を有していてもよいアミノ」の置換基と同様である。
「シクロアルキル」とは炭素数3〜10、好ましくは炭素数3〜8、より好ましくは炭素数4〜8の炭素環式基であり、例えばシクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、シクロノニルおよびシクロデシル等を包含する。これらはさらに任意の位置で後述の「アリール」または後述の「ヘテロ環式基」と縮合していてもよい。
「シクロアルキル低級アルキル」、「シクロアルキルカルボニル」のシクロアルキル部分は上記「シクロアルキル」と同様である。
【0023】
「置換基を有していてもよいシクロアルキル」の置換基は上述の置換基群αから選択される1以上の基が挙げられる。置換基は任意の位置に置換することができ、シクロアルキルの結合手を有する炭素原子に置換してもよい。
「シクロアルケニル」とは、上記シクロアルキルの環中の任意の位置に1以上の二重結合を有しているものを包含し、具体的にはシクロプロペニル、シクロブテニル、シクロペンテニル、シクロヘキセニル、シクロへプテニル、シクロオクテニルおよびシクロヘキサジエニル等が挙げられる。
「シクロアルケニルカルボニル」のシクロアルケニル部分は上記「シクロアルケニル」と同様である。
【0024】
「置換基を有していてもよいシクロアルケニル」の置換基は上記「置換基を有していてもよいシクロアルキル」と同様である。
「アリール」とは、フェニル、ナフチル、アントリルおよびフェナントリル等を包含し、特にフェニルが好ましい。
「アリールオキシ」、「アリールチオ」、「アリール低級アルキル」、「低級アルキルジアリールシリル」、「トリアリール低級アルキルシリル」、「アリール低級アルキルオキシ低級アルキル」、「アリールスルホニル」、「アリールスルファモイル」、「アリールアミノ」、「アリールカルバモイル」、「アリールスルホニルカルバモイル」のアリール部分も上記「アリール」と同様である。
【0025】
「置換基を有していてもよいアリール」、「置換基を有していてもよいフェニル」、「置換基を有していてもよいアリールスルホニル」の置換基としては、上述の置換基群α、置換基群αから選択される1以上の基で置換されたフェニル、置換基群αから選択される1以上の基で置換されたフェノキシ、または低級アルキレンジオキシ等が挙げられる。
「ヘテロ環式基」とは、O、SおよびNから任意に選択されるヘテロ原子を環内に1以上有するヘテロ環式基を包含し、具体的にはピロリル、イミダゾリル、ピラゾリル、ピリジル、ピリダジニル、ピリミジニル、ピラジニル、トリアゾリル、トリアジニル、テトラゾリル、イソオキサゾリル、オキサゾリル、オキサジアゾリル、イソチアゾリル、チアゾリル、チアジアゾリル、フリルおよびチエニル等の5〜6員のヘテロアリール;インドリル、イソインドリル、インダゾリル、インドリジニル、インドリニル、イソインドリニル、キノリル、イソキノリル、シンノリニル、フタラジニル、キナゾリニル、ナフチリジニル、キノキサリニル、プリニル、プテリジニル、ベンゾピラニル、ベンズイミダゾリル、ベンズイソオキサゾリル、ベンズオキサゾリル、ベンズオキサジアゾリル、ベンゾイソチアゾリル、ベンゾチアゾリル、ベンゾチアジアゾリル、ベンゾフリル、イソベンゾフリル、ベンゾチエニル、ベンゾトリアゾリル、イミダゾピリジル、トリアゾロピリジル、イミダゾチアゾリル、ピラジノピリダジニル、キナゾリニル、キノリル、イソキノリル、ナフチリジニル、ジヒドロピリジル、テトラヒドロキノリル、テトラヒドロベンゾチエニル等の2環の縮合ヘテロ環式基;カルバゾリル、アクリジニル、キサンテニル、フェノチアジニル、フェノキサチイニル、フェノキサジニル、ジベンゾフリル等の3環の縮合ヘテロ環式基;ジオキサニル、チイラニル、チオラニル、チエタニル、オキシラニル、オキセタニル、オキサチオラニル、アゼチジニル、チアニル、ピロリジニル、ピロリニル、イミダゾリジニル、イミダゾリニル、ピラゾリジニル、ピラゾリニル、ピペリジル、ピペラジニル、モルホリニル、モルホリノ、チオモルホリニル、チオモルホリノ、ジヒドロピリジル、ジヒドロフリル、テトラヒドロフリル、テトラヒドロピラニル、テトラヒドロチアゾリル、テトラヒドロイソチアゾリル等の非芳香族ヘテロ環式基を包含する。好ましくは5〜6員のヘテロアリールまたは非芳香族ヘテロ環式基である。
【0026】
「ヘテロ環オキシ」、「ヘテロ環低級アルキル」のヘテロ環部分も上記「ヘテロ環式基」と同様である。
「置換基を有していてもよいヘテロ環式基」および「置換基を有してもよいヘテロ環オキシ」の置換基としては上述の置換基群αおよびオキソからなる群から選択される1以上の基が挙げられる。置換基は任意の位置に置換することができ、ヘテロ環式基の結合手を有する炭素原子または窒素原子に置換してもよい。
【0027】
「アシル」とは炭素数1〜10、好ましくは炭素数1〜6、さらに好ましくは炭素数1〜4の直鎖または分枝の鎖状脂肪族アシル、炭素数4〜9、好ましくは炭素数4〜7の環状脂肪族アシル、アロイルおよびヘテロ環カルボニルを包含する。ここで「鎖状脂肪族」とは上記「低級アルキル」、上記「低級アルケニル」、および上記「低級アルキニル」を包含する。「環状脂肪族」とは上記「シクロアルキル」および上記「シクロアルケニル」を包含する。ヘテロ環カルボニルのヘテロ環部分も上記「ヘテロ環式基」と同様である。アシルの具体例としては、ホルミル、アセチル、プロピオニル、ブチリル、イソブチリル、バレリル、ピバロイル、ヘキサノイル、アクリロイル、プロピオロイル、メタクリロイル、クロトノイル、シクロプロピルカルボニル、シクロヘキシルカルボニル、シクロオクチルカルボニル、ベンゾイル、ピリジンカルボニル、ピペリジンカルボニル、ピペラジンカルボニル、モルホリノカルボニル等を包含する。
「アシルオキシ」、「アシルアミノ」、「アシルアミノ低級アルキル」および「アシルオキシ低級アルキル」のアシル部分は上記「アシル」と同様である。
「置換基を有していてもよいアシル」または「置換基を有していてもよいアシルオキシ」の置換基は、「アシル」が鎖状脂肪族アシルの場合、上記「置換基を有していてもよい低級アルキル」の置換基と同様であり、「アシル」が環状脂肪族アシル、アロイルおよびヘテロ環カルボニルの場合、上述の置換基群αから選択される1以上の基が挙げられる。
【0028】
「溶媒和物」とは、例えば有機溶媒(エタノール、2−プロパノール、酢酸メチル、酢酸エチル、酢酸n−プロピル、1、2−ジメトキシエタン、メチルイソブチルケトン、アセトニトリル等)との溶媒和物、水和物等を包含する。水和物を形成する時は、任意の数の水分子と配位していてもよい。
「水酸基の保護基」とは、ベンジル基、p−メトキシフェニルベンジル基、アセチル基、ホルミル基、ベンゾイル基、クロロアセチル基、ピバロイル基、メチルカーボネート基、イソブチルカーボネート基、ベンジルカーボネート基、ビニルカーボネート基、フェニルカーバメート基、メシル基、トシル基、トリメチルシリル基、トリエチルシリル基、t−ブチルジメチルシリル基、メトキシメチル基、ベンジルオキシメチル基、メトキシエトキシメチル基、2-(トリメチルシリル)エトキシメチル基、プロペニル基、フェナシル基、テトラヒドロピラニル基等が挙げられる。
「塩基で脱保護可能な水酸基の保護基」とは、アセチル基、ホルミル基、ベンゾイル基、クロロアセチル基、ピバロイル基、メチルカーボネート基、イソブチルカーボネート基、ベンジルカーボネート基、ビニルカーボネート基、フェニルカーバメート基、メシル基、トシル基等が挙げられる。一つの態様として、アセチル基、ホルミル基、ベンゾイル基、クロロアセチル基、ピバロイル基が挙げられる。別の態様として、アセチル基が挙げられる。
「塩基では脱保護されない水酸基の保護基」とは、ベンジル基、p−メトキシフェニルベンジル基、トリメチルシリル基、トリエチルシリル基、t−ブチルジメチルシリル基、メトキシメチル基、ベンジルオキシメチル基、メトキシエトキシメチル基、2-(トリメチルシリル)エトキシメチル基、プロペニル基、フェナシル基、テトラヒドロピラニル基等が挙げられる。
「脱離基」とは、置換基を有していてもよいフェノキシ(例えば、フェノキシ、p−ニトロフェノキシ、o−ニトロフェノキシが挙げられる)、ヘテロ環式基(例えば、1−イミダゾリル、1−ピラゾリルが挙げられる)、置換基を有してもよいヘテロ環オキシ(例えば,ピリジルオキシが挙げられる)等が挙げられる。
「式(I)で示される化合物の酸付加塩」および「式(IA)で表される酸付加塩」の「酸付加塩」とは、無機酸(例えば、塩酸、硫酸、硝酸、炭酸、臭化水素酸、リン酸、ヨウ化水素酸等)、および有機酸(例えば、ギ酸、酢酸、プロピオン酸、トリフルオロ酢酸、クエン酸、乳酸、酒石酸、シュウ酸、マレイン酸、フマル酸、マンデル酸、グルタル酸、リンゴ酸、安息香酸、フタル酸、アスコルビン酸、ベンゼンスルホン酸、p−トルエンスルホン酸、メタンスルホン酸、エタンスルホン酸等)との塩が挙げられる。例えば、「式(I)で示される化合物の酸付加塩」および「式(IA)で表される酸付加塩」の酸付加塩としては、p−トルエンスルホン酸塩、酢酸塩および塩酸塩が挙げられる。
「アミノ基の保護基」とは、t−ブチルジメチルシリル基、t−ブトキシカルボニル基、アリル基、9-フルオレニルメチルオキシカルボニル基、ベンジル基、p−メトキシベンジル基、メトキシメチル基、ベンジルオキシメチル基、ベンズヒドリル基およびトリチル基が挙げられる。
【発明の効果】
【0029】
本発明によって、6,7−不飽和−7−カルバモイルモルヒナン誘導体、その酸付加塩および/またはそれらの溶媒和物の結晶体が提供される。当該結晶体は、安定性が良好であり医薬品製造用原体として用いることができる。
また新規な製造方法は、製造工程の短縮化、収率の向上等に貢献することができる。
【図面の簡単な説明】
【0030】
【図1】本発明に係る化合物(IA)のp−トルエンスルホン酸塩の結晶体(非溶媒和物)の粉末X線回折パターンを示す。
【図2】本発明に係る化合物(IA)のp−トルエンスルホン酸塩水和物の結晶体(I形)の粉末X線回折パターンを示す。
【図3】本発明に係る化合物(IA)のp−トルエンスルホン酸塩水和物の結晶体(II形)の粉末X線回折パターンを示す。
【図4】本発明に係る化合物(IA)の酢酸塩の結晶体の粉末X線回折パターンを示す。
【図5】本発明に係る化合物(IA)の塩酸塩の結晶体の粉末X線回折パターンを示す。
【図6】本発明に係る化合物(IA)のエタノール和物の結晶体の粉末X線回折パターンを示す。
【図7】本発明に係る化合物(IA)の遊離体の結晶体の粉末X線回折パターンを示す。
【図8】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・酢酸メチル和物の結晶体の粉末X線回折パターンを示す。
【図9】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・(酢酸エチルおよび2−プロパノール)和物の結晶体の粉末X線回折パターンを示す。
【図10】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・(酢酸n‐プロピルおよび2−プロパノール)和物の結晶体の粉末X線回折パターンを示す。
【図11】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・アセトニトリル和物の結晶体の粉末X線回折パターンを示す。
【図12】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・1、2−ジメトキシエタン和物の結晶体の粉末X線回折パターンを示す。
【図13】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・メチルイソブチルケトン和物の結晶体の粉末X線回折パターンを示す。
【図14】本発明に係る化合物(IA)のp−トルエンスルホン酸塩の結晶体(非溶媒和物)のTG/DTA分析結果を示す。
【図15】本発明に係る化合物(IA)のp−トルエンスルホン酸塩水和物の結晶体(I形)のTG/DTA分析結果を示す。
【図16】本発明に係る化合物(IA)のp−トルエンスルホン酸塩水和物の結晶体(II形)のTG/DTA分析結果を示す。
【図17】本発明に係る化合物(IA)の酢酸塩のTG/DTA分析結果を示す。
【図18】本発明に係る化合物(IA)の塩酸塩のTG/DTA分析結果を示す。
【図19】本発明に係る化合物(IA)のエタノール和物のTG/DTA分析結果を示す。
【図20】本発明に係る化合物(IA)の遊離体のTG/DTA分析結果を示す。
【図21】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・酢酸メチル和物のTG/DTA分析結果を示す。
【図22】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・(酢酸エチルおよび2−プロパノール)和物のTG/DTA分析結果を示す。
【図23】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・(酢酸n‐プロピルおよび2−プロパノール)和物のTG/DTA分析結果を示す。
【図24】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・アセトニトリル和物のTG/DTA分析結果を示す。
【図25】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・1、2−ジメトキシエタン和物のTG/DTA分析結果を示す。
【図26】本発明に係る化合物(IA)のp−トルエンスルホン酸塩・メチルイソブチルケトン和物のTG/DTA分析結果を示す。
【発明を実施するための形態】
【0031】
本発明の結晶体は、式(I)で示される化合物の酸付加塩または該酸付加塩の溶媒和物として得られる。ここで用いられる酸としては、p−トルエンスルホン酸、酢酸または塩酸等が例示される。中でもp−トルエンスルホン酸の結晶体は、吸湿性がなく、安定性も優れていると考えられる。溶媒和物を形成する際の溶媒としては、水、エタノール、2−プロパノール、酢酸メチル、酢酸エチル、酢酸n‐プロピル、1、2−ジメトキシエタン、メチルイソブチルケトン、アセトニトリル等が例示できる。
酸付加塩の結晶は、式(I)で示される化合物の溶液に、通常1.0〜10.0当量の酸を、0℃〜室温で、若しくは溶媒の沸点以下に加温して添加した後、必要に応じて溶液を冷却および/または濃縮して晶析させる。
溶媒和物の結晶の調製は、式(I)で示される化合物の酸付加塩を、少なくとも溶媒和させる溶媒を含む可溶性溶媒に、室温で、または溶媒の沸点以下に加温して溶解し、溶媒和させたい溶媒を加え、0℃〜室温で数時間〜1日、撹拌または静置することにより行う。晶析した溶媒和物は、濾過または遠心分離等の通常の分離手段で溶媒から分離し、洗浄、乾燥等の通常の精製手段により単離することができる。
式(I)で示される化合物の溶媒和物もまた、本発明の結晶体に包含される。溶媒としては水、エタノール等が例示される。式(I)で示される化合物の溶媒和物の場合も、上記酸付加塩の溶媒和物と同様にして調製することができる。
本発明の結晶体の具体例としては、化合物(IA)の場合、p−トルエンスルホン酸塩(非溶媒和物)、p−トルエンスルホン酸塩・水和物、p−トルエンスルホン酸塩・酢酸メチル和物、p−トルエンスルホン酸塩・(酢酸エチル・2−プロパノール)和物、p−トルエンスルホン酸塩・(酢酸n‐プロピル・2−プロパノール)和物、p−トルエンスルホン酸塩・アセトニトリル和物、p−トルエンスルホン酸塩・1、2−ジメトキシエタン和物、p−トルエンスルホン酸塩・メチルイソブチルケトン和物、塩酸塩、酢酸塩、遊離体のエタノール和物等が例示できる。
例えば、式(IA)で示される化合物のp−トルエンスルホン酸塩(非溶媒和物)は、以下のようにして得られる。即ち、化合物(IA)を含む有機層に2−プロパノールと酢酸n−プロピルを加えて濃縮し、50〜70℃で1〜10.0当量のp−トルエンスルホン酸を2−プロパノールに溶解して滴下し晶析する。得られる未乾固体を、メタノールと酢酸n−プロピルに再び加温溶解し、不溶物をろ過した後、減圧濃縮して晶析させ、得られた結晶を50〜70℃で2〜5時間減圧下で乾燥すると目的のp−トルエンスルホン酸塩(非溶媒和物)を得ることができる。
【0032】
以下に本発明の結晶体を特定する方法につき説明する。
特に言及がなければ、本明細書中および特許請求の範囲記載の数値は、おおよその値である。数値の変動は、装置キャリブレーション、装置エラー、物質の純度、結晶サイズ、サンプルサイズ、その他の因子に起因する。
【0033】
本明細書中で用いる「結晶」とは、秩序だった長い範囲の分子構造を有する物質を意味する。結晶形態の結晶化度は、例えば、粉末X線回折、水分吸着、示差、熱量分析、溶液比色、溶解特性を含めた多くの技術によって測定することができる。
【0034】
一般に結晶性有機化合物は、3次元空間に周期的に配列された多数の原子よりなる。構造周期性は、通例、ほとんどの分光学的プローブ(例えば、X線回折、赤外スペクトル、ラマンスペクトルおよび固体NMR)によって明確に区別可能な物理的特性を発現する。
中でも粉末X線回折(XRPD)は、固体の結晶性を測定するための最も感度の良い分析法のうちの1つである。X線が結晶に照射されると、結晶格子面で反射し、互いに干渉しあい、ブラッグ則よって予測される条件を満たす方向の回折線のみ強度が増大し、それ以外は打ち消しあって観測されない。一方、非晶質固体については広範囲の秩序だった回折線は認められない。非晶質固体は、通常、反復する結晶格子の広い範囲の秩序が不存在であるため、ハローパターンと呼ばれるブロードなXRPDパターンを示す。
【0035】
本出願で開示する6,7−不飽和−7−カルバモイルモルヒナン誘導体、その酸付加塩および/またはそれらの溶媒和物の結晶形態は、好ましくは、区別可能な粉末X線回折プロフィールを有する。例えば、化合物(IA)のp−トルエンスルホン酸塩の場合、好ましくは、特徴的な回折ピークの存在によって各結晶体を特定し、他の結晶体と区別することができる。本明細書中で用いる特徴的な回折ピークは、観察された回折パターンから選択されるピークである。好ましくは、特徴的なピークは、回折パターンにおける約20本、より好ましくは約10本、最も好ましくは約5本から選択される。
【0036】
一般に、粉末X線回折における回折角度(2θ)は±0.2°の範囲内で誤差が生じ得るので、上記の回折角度の値は±0.2°程度の範囲内の数値も含むものとして理解される必要がある。したがって、粉末X線回折におけるピークの回折角度が完全に一致する結晶だけでなく、ピークの回折角度が±0.2°程度の誤差で一致する結晶も本発明に含まれる。
【0037】
一般に、後述の表及び図において表示されるピークの相対強度は、多くの因子、例えばX線ビームに対する結晶の配向効果、分析される物質の純度又はサンプルの結晶化度によって変動し得ることが知られている。また、ピーク位置についても、サンプル高の変動に基づいてシフトし得る。さらに、異なる波長を使用して測定するとブラッグ式(nλ=2dsinθ)に従って異なるシフトが得られるが、このような別の波長の使用により得られる別のXRPDパターンも、本発明の範囲に含まれる。
【0038】
本発明に係る化合物(IA)のp−トルエンスルホン酸塩の結晶体(非溶媒和物)、p−トルエンスルホン酸塩水和物(I形)およびp−トルエンスルホン酸塩水和物(II形)は図1〜3のような粉末X線回折パターンを示す。各結晶体は少なくとも表1に示されるような特徴的ピークを示す。
【表1】
【0039】
本発明の結晶体は熱分析の手法によっても特定することができる。
ここでTG/DTA(示差熱熱重量同時測定)は、熱分析の主要な測定方法のひとつであって、原子・分子の集合体としての物質の重量および熱的性質を測定する方法である。
TG/DTAは医薬活性成分の温度または時間に係る重量および熱量の変化を測定する方法であり、得られたデータを温度または時間に対してプロットすることにより、TG(熱重量)およびDTA(示差熱)曲線が得られる。TG/DTA曲線より、医薬活性成分の分解、脱水、酸化、還元、昇華、蒸発に関する重量および熱量変化の情報を得ることができる。
TG/DTAにおいて、「融点」とは、オンセット温度をいう。
TG/DTAについて、観察される温度、重量変化は、温度変化速度ならびに用いる試料調製技法および特定の装置に依存し得ることが知られている。結晶の同一性の認定においては、全体的なパターンが重要であり、測定条件によって多少変化し得る。
【0040】
本発明に係る化合物(IA)のp−トルエンスルホン酸塩の結晶体、p−トルエンスルホン酸塩水和物(I形)、p−トルエンスルホン酸塩水和物(II形)、酢酸塩および塩酸塩のTG/DTA分析結果は図14〜18に表される。
【0041】
本発明の式(IA)で示される化合物、その酸付加塩、および/またはそれらの溶媒和物の結晶は、オピオイド受容体(特にオピオイドδ、μ受容体)拮抗作用を有している。従って、オピオイド受容体アゴニスト作用を有する化合物に誘発される嘔気・嘔吐・便秘の他、急性消化不良、急性アルコール中毒、食中毒、感冒、胃潰瘍、十二指腸潰瘍、胃がん、腸閉塞、虫垂炎、腹膜炎、胆石症、肝炎、肝臓炎、脳炎、髄膜炎、脳圧亢進、頭部外傷、乗り物酔い、つわり、化学療法による副作用、放射線療法による副作用、抗癌剤等による副作用、消化管の圧迫・狭窄や手術後の腸管癒着などの原因により起こる消化管通過障害、脳腫瘍・脳出血・髄膜炎・脳への放射線照射などによる脳圧上昇等の原因により起こる嘔気、嘔吐の治療および/または予防、腸閉塞、十二指腸潰瘍または虫垂炎等を原因とする急性便秘、神経障害、低栄養、全身衰弱、ビタミン欠乏症、貧血、感受性低下または機械的刺激不足等を原因とする弛緩性便秘、ストレス等を原因とするけいれん性便秘の治療および/または予防に対して有効である。
【0042】
本発明の式(IA)で示される化合物、その酸付加塩、および/またはそれらの溶媒和物の結晶は、脳移行性が低いため、疼痛を伴う疾患(例えば癌性疼痛(骨転移、神経圧迫、頭蓋内圧亢進、軟部組織浸潤、便秘または筋の攣縮による痛み、内臓、筋・筋膜、腰または肩関節周囲の痛み、術後の慢性的な痛み)、AIDS等)の患者に対して投与されるオピオイド受容体アゴニスト作用を有する化合物の鎮痛作用をほとんど阻害することなく、オピオイド受容体アゴニストにより誘発される嘔気、嘔吐、便秘等の副作用に対して高い軽減効果を示す。また、本発明の結晶はオピオイド受容体に対して純粋なアンタゴニスト活性を有し、hERGチャンネル阻害作用が弱く、心毒性の懸念がない、等の安全性の面における利点も有している。さらに、本発明の結晶は高い経口吸収性、ヒト血漿中における高い安定性、高いバイオアベイラビリティー等の体内動態における有利な特徴をも有しており、医薬品として非常に有効である。
【0043】
オピオイド受容体アゴニスト作用を有する化合物により誘発される嘔気、嘔吐、便秘に対して本発明結晶またはその医薬組成物を投与する場合、その投与はオピオイド受容体アゴニスト作用を有する化合物の投与前、投与後または同時投与のいずれであってもよい。これらの2種の薬物の投与間隔は特に限定されるものではない。例えば、本発明結晶またはその結晶を含む医薬組成物を、オピオイド受容体アゴニスト作用を有する化合物の投与後に投与する場合、オピオイド受容体アゴニストの投与直後〜約3日以内、投与直後〜約1日以内であれば、より有効に作用する。また、オピオイド受容体アゴニスト投与前に、本発明にかかる結晶またはその結晶を含む医薬組成物を投与する場合、オピオイド受容体アゴニスト投与直前〜約1日前、直前〜約12時間前であれば、より有効に作用する。
本発明結晶またはその結晶を含む医薬組成物を嘔気、嘔吐および/または便秘治療剤および/または予防剤として投与する際には、他の嘔気、嘔吐および/または便秘治療剤および/または予防剤と併用してもよい。例えば、塩酸オンダンセトロン、副腎皮質ステロイド(メチルプレドニゾロン、プレドニゾロン、デキサメタゾン等)、プロクロルペラジン、ハロペリドール、チミペロン、ペルフェナジン、メトクロプラミド、ドンペリドン、スコポラミン、塩酸クロルプロマジン、ドロペリドール、刺激性緩下薬(センノシド、ピコスルファートナトリウムなど)、浸透圧性緩下薬(ラクツロース)や塩類緩下薬(酸化マグネシウムなど)等との併用が可能である。
また、本発明結晶またはその結晶を含む医薬組成物は、オピオイド受容体アゴニスト作用を有する化合物および/または他の嘔気、嘔吐および/または便秘治療剤および/または予防剤、ならびに必要に応じて各種医薬用添加剤を配合した合剤とすることも可能である。
【0044】
本発明の結晶はそれ自体でヒト患者に投与することができるか、または、前記した結晶を適当な担体または賦形剤と混合した医薬組成物にて投与することができる。薬物の処方および投与のための技術は、「Remington's Pharmacological Sciences」Mack Publishing Co.、Easton、PA.最新版に見出すことができる。
【0045】
適当な投与経路は、限定されるものではないが、経口、直腸、経粘膜または腸投与あるいは筋肉内、皮下、脊髄内、鞘内、直接的心室内、静脈内、硝子体内、腹腔内、鼻腔内、眼内、注射を含むことができる。好ましい投与経路は経口および非経口である。
本発明の医薬組成物は、当該分野でよく知られた製法、例えば、慣用的な混合、溶解、顆粒化、糖衣−作成、粉末化、乳化、カプセル化、包括、凍結乾燥プロセスによって製造することができる。
【0046】
本発明で用いられる医薬組成物は、本発明の結晶の、医薬的に使用することができる製剤への製造を容易とする賦形剤および補助剤を含めた1以上の製薬学上許容される担体を用いて公知の方法で処方することができる。適切な処方は、選択された投与経路に依存する。
【0047】
注射による投与を行う場合は、本発明の結晶を溶解した水性溶液、好ましくは、生理学上適合する、リンゲル液または生理食塩水のような緩衝液に溶解して投与することができる。経粘膜投与を行う場合は、浸透させるべきバリアーに適した浸透剤を用いて投与することができる。該浸透剤は一般に当該分野で知られているものを用いることができる。
【0048】
経口投与を行う場合は、本発明の結晶と、当該分野でよく知られた医薬上許容される担体とを合わせることによって投与することができる。該担体により、本発明の結晶を、患者による経口摂取のために、錠剤、丸剤、ロゼンジ、糖衣錠、カプセル剤、液剤、ゲル、シロップ、スラリー、懸濁剤として投与することができる。経口用途の医薬組成物は、固体賦形剤を用い、所望であれば他の適切な補助剤を添加した後、得られた混合物を粉砕し、顆粒の混合物を処理して錠剤または糖衣錠コアを得ることによって作成することができる。有用な賦形剤は、特に、ラクトース、スクロース、マンニトール、またはソルビトールを含めた糖などの充填剤、例えば、とうもろこし澱粉、小麦澱粉、米澱粉およびじゃがいも澱粉などのセルロース調製物およびゼラチン、トラガカントガム、メチルセルロース、ヒドロキシプロピルメチルセルロースおよび/またはカルボキシメチルセルロースナトリウムなどである。必要であれば、寒天、アルギン酸などの崩壊剤を添加することができる。アルギン酸ナトリウムなどの塩を用いることもできる。
【0049】
経口用途の医薬組成物は、ゼラチンで作成されたプッシュフィットカプセルならびにゼラチンおよびグリセロールまたはソルビトールなどの可塑剤で作成された密封カプセル剤を含む。プッシュフィットカプセルはラクトースなどの充填剤、澱粉などのバインダーおよび/またはタルクまたはステアリン酸マグネシウムなどの滑沢剤および所望により、安定化剤と混合した本発明結晶を含むことができる。ソフトカプセル剤として用いる場合は、本発明の結晶を、脂肪油、流動パラフィンまたは液状ポリエチレングリコールなどの適当な液体に溶解または懸濁させて用いることができる。安定化剤をこれらの処方に加えることもできる。
【0050】
医薬組成物は、適当な固体またはゲル相の担体または賦形剤を含むこともできる。そのような担体または賦形剤は、例えば、炭酸カルシウム、リン酸カルシウム、種々の糖、澱粉、セルロース誘導体、ゼラチン、ポリエチレングリコールなどのポリマーなどが挙げられる。
【0051】
本発明の結晶またはその結晶を含む医薬組成物の治療上有効な薬効成分量は、最初に細胞培養アッセイから見積もることができる。次いで、細胞培養で決定されたIC50(すなわち、PK活性の最大の半分の阻害を達成する本発明の結晶またはその医薬組成物の濃度)を含む循環濃度範囲を達成するように、動物モデルで用いるために投与量を多く処方することができる。次いで、そのような情報を用いて、ヒトにおける有用な量をより正確に決定することができる。
【0052】
本発明の結晶またはその結晶を含む医薬組成物の毒性および治療効果は、例えば、本発明の主題の結晶またはその医薬組成物についてのIC50を決定することによって、細胞培養または実験動物において、標準的な医薬手法によって測定することができる。これらの細胞培養アッセイおよび動物実験から得られたデータは、ヒトで用いる投与量の範囲を処方するために用いることができる。投与量は、使用する投与形態および利用する投与経路に応じて変化させることができる。正確な処方投与経路および投与量は、患者の状態を考慮して個々の医師が選択することができる(例えば、Finglら、1975、in“The Pharmacological Basis of Therapeutics”、Ch.1p.l参照)。
また、投与量は、疾患の状態、投与ルート、患者の年齢、または体重によっても異なるが、成人に経口で投与する場合、通常0.1μg〜1g/日であり、好ましくは0.01〜200mg/日であり、非経口投与の場合には通常1μg〜10g/日であり、好ましくは0.1mg〜10mg/日である。
【0053】
次に本発明による化合物(I)の製造方法を説明する。
本発明による化合物(I)の製造方法をスキーム1に示した。
【化36】
(式中、R1bは水酸基の保護基;R2は置換基を有していてもよいアルキル;R3は置換基を有していてもよいアルキル、置換基を有していてもよいシクロアルキル、置換もしくは非置換アリール、または置換もしくは非置換ヘテロアリール;R7は置換基を有してもよいフェニル基をそれぞれ表す。)
本発明によれば、カルバメート誘導体(II)に塩基を添加し、室温〜溶媒の沸点以下の温度で1〜10時間反応させることにより化合物(I)を得ることができる。塩基としては水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化セシウム等の無機塩基が好ましく、カルバメート誘導体(II)に対して1〜10当量のアルカリを水溶液として添加することが好ましい。カルバメート誘導体は親水性溶媒、例えばメタノール、エタノール、2−プロパノール、DMSO等の溶媒に溶解し、上記アルカリ水溶液を添加することで好適に反応させることができる。
カルバメート誘導体(II)の水酸基保護基R1bは特に限定されないが、例えばアセチル基等の塩基で脱保護される保護基を用いると化合物(I)を直接得ることができる。R1bが塩基では脱保護されない保護基の場合、上記の塩基処理の前または処理後に適宜保護基を脱保護すればよい。
カルバメート誘導体(II)は化合物(III)にイソシアネート体(V)を反応させることにより得られる。反応は、化合物(III)の溶液に、化合物(III)に対して0.5〜5当量、好ましくは、1.0〜1.2当量のイソシアネート体(V)溶液を加え、室温〜溶媒の沸点以下の温度で1〜10時間反応させる。ここで、例えば、CuCl2のようなルイス酸触媒を0.00005〜1当量、好ましくは0.0001〜0.1当量、さらに好ましくは0.0001〜0.01当量添加することが好ましい。反応溶媒は特に制限はないが酢酸エチル、アセトニトリル、アセトン、トルエン等が使用できる。
ここで用いるイソシアネート体(V)は、下記スキームに従い、
【化37】
例えば、その前駆体であるカルバミン酸エステル(VIII)(式中、R5は低級アルキル)を、ルイス酸および塩基の存在下で反応させることにより得られる。
イソシアネート体に代えて、カルバミン酸活性エステル(VI)(式中、R7は置換基を有していてもよいフェニル基)を用いてカルバメート体(II)を調製することもできる。この活性エステルは例えばアミノ体R3−NH2に相当するフェノールのクロロ蟻酸エステルを反応させて得ることができる。
【化38】
ここでR7としては、OR7が脱離基Xとして機能するものが好ましく、具体的にはフェニル基、p−ニトロフェニル基、p−クロロフェニル基等が例示される。
【実施例】
【0054】
本発明を以下の実施例によりさらに詳しく説明する。これらは本発明を限定するものではない。数値(例えば、量、温度など)に関して正確性を保証する努力をしているが、いくらかの誤差および偏差は考慮されるべきである。特に示さなければ、%は成分の重量%および組成物の全重量の重量%である。圧力は大気圧かまたはそれに近い圧力である。本明細書で使用する他の略語は以下のように定義される:gはグラム、Lはリットル、mgはミリグラム、mLはミリリットル、Bocはt−ブトキシカルボニル基、Acはアセチル基、Meはメチル基、Etはエチル基、Prはプロピル基を表す。
(粉末X線回折パターンの測定)
各実施例で得られた結晶の粉末X線回折測定は、日本薬局方の一般試験法に記載された粉末X線回折測定法に従い、以下の測定条件で行った。
(装置)
Bruker社製D−8Discover
(操作方法)
試料について、以下の条件で測定を行った。
測定法:反射法
光源の種類:Cu管球
使用波長:CuKα線
管電流:40mA
管電圧:40Kv
試料プレート:ガラス
測定範囲:3°―40°
【0055】
(TG/DTAデータの測定)
また、各実施例で得られた各結晶約5mgを量り、アルミニウムパンにつめ、開放系にて測定した。測定条件は以下のとおりである。
(測定条件)
装置:SEIKO社製TG/DTA6300
測定温度範囲:25℃−300℃
昇温速度:10℃/分
【0056】
[実施例1−1]
化合物(IA)のp−トルエンスルホン酸塩[化合物(9)]の製造
【化39】
【0057】
工程1 化合物(3)の合成
t−ブトキシカルボニルアミノイソ酪酸(1)(25.0g、123mmol)の酢酸n−プロピル(150ml)溶液に、0℃でジイソプロピルエチルアミン(17.5g、135.4mmol)を加えた。同温下、混合液にクロロギ酸イソブチル(17.6g、128.9mmol)を滴下し、1時間攪拌した。同反応液にベンズアミドオキシム(2)(17.6g、129.3mmol)の酢酸n−プロピル(100ml)溶液を加え、0℃で1時間攪拌後、95℃で5時間攪拌した。酸水溶液を加え分離後、有機層を水と炭酸水素ナトリウムで洗浄し、減圧濃縮した。同反応液に塩酸を加え2.5時間攪拌し、析出した結晶を濾取し、洗浄、乾燥後、化合物(3)(27.34g、92.7%)を得た。
1H NMR(300MHz, DMSO‐d6) δ 1.80 (6H, s), 7.59-7.64 (3H, m), 8.01-8.05 (2H, m), 9.26(3H, br).
【0058】
工程2 化合物(4)の合成
化合物(3)(19.0g、79.2mmol)のトルエン152ml懸濁液に、25℃でアルカリ水を加え攪拌した。50℃で、クロロギ酸メチル(8.3g、88.0mmol)を加え1時間攪拌後、有機層を分離し、塩酸水、炭酸水素ナトリウム水および水で順次洗浄を行い、減圧濃縮した。この反応液に1.0mol/l三塩化ホウ素のトルエン溶液(7.3ml)を加え、50℃でトリエチルアミンを滴下し、2時間攪拌後、反応液を濃縮して化合物(4)の溶液を得た。
【0059】
工程3 化合物(6)の合成
市販のナルトレキソン塩酸塩(5)(20.0g、52.9mmol)の酢酸エチル160ml溶液にトリエチルアミン(11.3g、111.7mmol)と無水酢酸(5.7g、55.8mmol)を加えた後、40℃で2時間攪拌した。反応液を冷却し、水洗後、減圧濃縮し、化合物(6)溶液を得た。
1H-NMR (300 MHz, DMSO‐d6) δ 0.14 (2H, d, J=4.8 Hz), 0.49 (2H, d, J=7.8 Hz), 0.88 (1H, m), 1.29 (1H, d, J=9.9 Hz),1.46 (1H, td, J=14.1, 3.3 Hz), 1.79(1H, dt, J=12.0, 3.3 Hz), 1.90-2.00(1H, m), 2.11(1H, dt, J=14.4, 3.3 Hz), 2.26 (3H, s), 2.30-2.46 (3H, m), 2.52-2.72(2H, m), 2.92(1H, td, J=14.1, 4.8 Hz), 3.07(1H, d, J=18.9 Hz), 3.17(1H, d, J=5.7 Hz), 4.91(1H, s), 5.18(1H, s), 6.71(1H, d, J=8.1 Hz), 6.83(1H, d, J=8.1 Hz)
【0060】
工程4 化合物(7)の合成
化合物(6)溶液に、化合物(4)の反応液と酢酸エチルを加えた後、この混合液に塩化銅(II)水溶液を加え、25℃で4時間攪拌した。反応液にヘプタンを加えて晶析し、ろ過、洗浄後、乾燥して化合物(7)(89.2%)を得た。
1H-NMR (300 MHz, DMSO‐d6) δ 0.20-0.40 (2H, m), 0.60-0.90 (1H, m), 1.20-1.50 (2H, m), 1.67 (3H, s), 1.74 (3H, s), 1.90-2.10 (2H, m), 2.10-2.20 (2H, m), 2.26 (3H, s), 2.30-2.55 (2H, m), 2.58-2.80 (4H, m), 3.03(2H, m), 4.31(1H, s), 4.81(1H, s), 6.71(1H, d, J=8.1 Hz), 6.85(1H, d, J=7.8 Hz), 7.50-7.70(3H, m), 7.92-8.01(2H, m), 8.11(1H, s).
【0061】
工程5 化合物(8)の合成
化合物(7)(5.5g、9.0mmol)の2−プロパノール22mlの懸濁液に、水酸化カリウム水溶液を滴下し、80℃で5時間攪拌した。反応液をトルエン洗浄した後、pH7.0〜8.0に調整し、酢酸n-プロピルで抽出した。有機層を水洗後、2-プロパノールと酢酸n-プロピルを加え濃縮後、60℃でp−トルエンスルホン酸2-プロパノール溶液(1.5g,8.1mmol)を滴下し、晶析した。冷却後、析出した固体をろ過し、未乾固体(8)[p−トルエンスルホン酸塩・(酢酸n−プロピルおよび2−プロパノール)和物]を得た。
未乾固体(8)[p−トルエンスルホン酸塩・(酢酸n−プロピルおよび2−プロパノール)和物]の粉末X線回折およびTG/DTA分析の結果を下記参考例3に示す。
【0062】
工程6 化合物(9)の合成
未乾固体(8)に、メタノールと酢酸n−プロピルを加え、加温溶解し、ろ過、洗浄後、減圧濃縮した。析出物を濾取後、洗浄し、得られた粗生成物を60℃で3時間減圧乾燥し、化合物(9)の結晶(非溶媒和物:66.3%)を得た。
1H-NMR (300MHz,DMSO-d6) δ 13.37 (1H, s), 9.44 (1H, s), 8.95 (1H, br s), 8.12 (1H, s), 7.99-7.96 (2H, m), 7.60-7.53 (3H, m), 7.49-7.45 (2H, m), 7.11 (2H, d, J=8.4Hz), 6.69 (2H, ABq.), 6.56 (1H, s), 4.94 (1H, s), 3.95 (1H, d, J=5.1Hz), 3.50-3.25 (2H, m), 3.07 (2H, br d, J=12Hz), 3.00-2.90 (1H, m), 2.75-2.60 (1H, m), 2.60-2.40 (2H, m), 2.29 (3H, s), 2.10 (1H, d, J=14.7Hz), 1.70 (6H, s), 1.75-1.60 (1H, m), 1.15-0.95 (1H, m), 0.80-0.55 (2H, m), 0.55-0.35 (2H, m).
粉末X線回折の結果を図1および表2に示す。
【表2】
粉末X線回折スペクトルにおいて、回折角度(2θ):7.8°±0.2°、10.6°±0.2°、15.6°±0.2°、17.8°±0.2°、18.6°±0.2°、20.4°±0.2°、21.5°±0.2°、21.9°±0.2°、23.6°±0.2°および25.5°±0.2°にピークが認められた。
TG/DTA分析結果を図14に示す。
【0063】
[実施例1−2]
式(IA)で示される化合物のp−トルエンスルホン酸塩[化合物(9)]の製造(別法A)
【化40】
【0064】
工程1 化合物(11)の合成
メトキシカルボニル‐2‐メチルアラニン(10)(5.00g、31.0mmol)とアセトニトリル25mlの懸濁液に、0℃でCDI(カルボニルジイミダゾール、5.28g、31.1mmol)とアセトニトリル5mlを加えて1.5時間攪拌した。同温でベンズアミドオキシム(2)(4.65g、34.2mmol)とアセトニトリル20mlを加え2時間攪拌した。同反応液に、化合物(10)に対して0.10当量の炭酸カリウム(0.43g)を水15mlに溶解した炭酸カリウム水溶液全量を加え、溶媒の沸点以下の温度で1〜5時間反応させた。減圧濃縮後、水を加えて析出した粗生成物を濾取、洗浄した。未乾結晶を乾燥し化合物(11)[7.43g、収率91.7%]を得た。
化合物(11):
1H-NMR(300MHz, CDCl3) δ1.81(6H, s), 3.65 (3H, s), 5.46(1H, s), 7.49-7.50 (3H, m), 8.05-8.08 (2H, m).
【0065】
工程2 化合物(4)の合成
化合物(11)(15.12g、57.41mmol)と三塩化アルミニウム(9.19g、68.89mmol)のトルエン溶液に、50℃でトリエチルアミン(7.55g、74.63mmol)を滴下し、同温度で2.5時間攪拌した。有機層を分離し、濃縮後、化合物(4)を反応液として得た。
1H-NMR(300MHz, CDCl3) δ1.84(6H, s), 7.31-7.55 (3H, m), 8.05-8.13 (2H, m).
13C-NMR(75MHz, CDCl3) δ29.85, 55.71, 126.16, 127,44, 128.78, 131.35, 168.23, 180.88.
IR (cm-1)1446, 1478, 1570, 1638, 2256, 2986, 3337.
【0066】
工程3−6 化合物(9)の合成
実施例1−1記載の同じ工程で化合物(5)から化合物(9)(非溶媒和物)を合成した。
【0067】
[実施例1−3]
式(IA)で示される化合物のp−トルエンスルホン酸塩[化合物(9)]の製造(別法B)
【化41】
【0068】
工程1
実施例1−1記載の工程1と同様の方法で合成した化合物(3)(2.03g、10.0mmol)をアセトニトリル20mlに溶解し、氷冷下、ピリジン(0.89ml、11.0mmol)及びクロロ蟻酸4−ニトロフェニルエステル(2.22g、11.0mmol)を加え、室温で1.5時間攪拌した。反応液を、2mol/L塩酸と氷水の中に注ぎ、酢酸エチルで2回抽出した。抽出液を食塩水で洗浄後、無水硫酸ナトリウム上で乾燥し、ろ過、濃縮した。得られた単黄色油状物4.88gに、ヘキサン約20mlを加え、氷冷下、固化させた。得られた固体を濾取し、ヘキサンで洗浄することにより、目的とする化合物(16)(3.74g)を白色固体として得た。
1H-NMR (CDCl3) δ 8.24 (2H, d, J=9.3Hz), 8.09 (2H, m), 7.53-7.45 (3H, m), 7.33 (2H, br d, J=8.7Hz), 5.99 (1H, br s), 1.92 (6H, s).
【0069】
工程2
実施例1−1記載の工程3と同様の方法で合成した化合物(6)(3.28g、8.56mmol)及び化合物(16)(3.79g、10.3mmol)をアセトニトリル10mlに溶解し、22時間還流した。反応液を室温に戻し、氷水の中に注ぎ、酢酸エチルで2回抽出した。抽出液を0.1mol/L水酸化ナトリウム水溶液で2回、食塩水で1回洗浄後、無水硫酸マグネシウム上で乾燥し、ろ過、濃縮した。得られた化合物(7)の非晶形固体(5.46g)は、精製せず、次の反応に使用した。
1H-NMR (DMSO-d6) δ 8.0-7.9 (2H, m), 7.6-7.5 (3H, m), 6.9-6.7 (2H, Abq.), 4.32 (1H, s), 3.2-1.2 (12H, m), 2.26 (3H, s), 1.71 (1H, d, J=21.6Hz), 1.61 (6H, s), 0.95-0.65 (1H, m), 0.55-0.2 (2H, m), 0.2-0.5 (2H, m).
【0070】
工程3
化合物(7)(500mg)をジメチルスルホキシド2mlに溶解し、2mol/L水酸化カリウム水溶液2mlを加え、80℃で6時間加熱攪拌した。反応液を室温に戻し、2mol/L塩酸で中和し、酢酸エチルで2回抽出した。抽出液を0.1mol/L水酸化ナトリウム水溶液、食塩水で順次洗浄後、無水硫酸ナトリウム上で乾燥し、ろ過、濃縮した。得られた淡黄色の非晶形固体412mgをメタノール2mlに溶解し、p−トルエンスルホン酸水和物165mgを加え、30分間静置した。その後、アセトニトリル20mlを加えて、5℃で一晩静置した。析出物を濾取し、減圧下乾燥することにより、目的とするp−トルエンスルホン酸塩(9)(非溶媒和物:282mg)を結晶として得た[化合物(6)から48%の収率]。
1H-NMR (DMSO-d6) δ 13.37 (1H, s), 9.44 (1H, s), 8.95 (1H, br s), 8.12 (1H, s), 7.99-7.96 (2H, m), 7.60-7.53 (3H, m), 7.49-7.45 (2H, m), 7.11 (2H, d, J=8.4Hz), 6.69 (2H, ABq.), 6.56 (1H, s), 4.94 (1H, s), 3.95 (1H, d, J=5.1Hz), 3.50-3.25 (2H, m), 3.07 (2H, br d, J=12Hz), 3.00-2.90 (1H, m), 2.75-2.60 (1H, m), 2.60-2.40 (2H, m), 2.29 (3H, s), 2.10 (1H, d, J=14.7Hz), 1.70 (6H, s), 1.75-1.60 (1H, m), 1.15-0.95 (1H, m), 0.80-0.55 (2H, m), 0.55-0.35 (2H, m).
【0071】
[実施例1−4]
式(IA)で示される化合物のp−トルエンスルホン酸塩[化合物(9)]の製造(別法C)
【化42】
【0072】
工程1 化合物(4)の合成
実施例1−2記載の工程1と同様の方法で合成した化合物(11)(0.5g、1.91mmol)にトルエンを加えた後、TiCl4(2.30mmol)を加え50℃に加熱した。この反応液にトリエチルアミン(2.30mmol)を加え、同温で2時間攪拌し、化合物(4)を反応液として得た。
工程2−7 化合物(9)の合成
実施例1−1記載の同じ工程で化合物(5)から化合物(9)(非溶媒和物)を合成した。
【0073】
[実施例2]
化合物(20)の合成
【化43】
【0074】
工程1
化合物(17)(4.51g、36mmol)をアセトニトリル45mlに溶解し、氷冷下、ピリジン(3.20ml、39.6mmol)及びクロロ蟻酸フェニルエステル(5.00ml、39.6mmol)を加えた。ジメチルホルムアミド9ml及びアセトニトリル30mlを加えて、室温で45分間攪拌した。反応液から析出物を濾取し、冷メタノール、水で洗浄後、減圧乾燥することにより、目的とする化合物(18)(7.02g)を白色固体として得た。
1H-NMR (CDCl3) δ 8.61 (1H, s), 7.43 (2H, t, J=7.8Hz), 7.41 (1H, s), 7.29 (1H, t, J=7.8Hz), 7.21 (2H, d, J=7.8Hz), 3.97 (3H, s).
【0075】
工程2
実施例1−1記載の工程3と同様の方法で合成した化合物(6)(1.92g、5.00mmol)及び化合物18(1.84g、7.50mmol)をジメチルホルムアミド10mlに溶解し、120℃で4時間加熱攪拌した。反応液を室温に戻し、アセトニトリル50mlを加え、析出物を濾去した。濾液を60℃で減圧濃縮し、ジメチルホルムアミドを留去した。残渣にアセトニトリル100mlを加え、氷冷下、30分間攪拌した。析出した結晶を濾取し、冷アセトニトリルで洗浄、減圧乾燥し、目的とする化合物(19)の第一晶(1.66g)を得た。さらに、母液を濃縮した後、残渣にジエチルエーテルを加え、室温で攪拌することにより、第二晶(306mg)、第三晶(71mg)を得た。
1H-NMR (DMSO-d6) δ 10.58 (1H, br s), 8.52 (1H, s), 7.19 (1H, s), 6.83 (2H, Abq.), 4.78 (1H, s), 4.44 (1H, d, J=5.4Hz), 3.90 (3H, s), 3.12 (1H, d, J=18.6Hz), 2.9-2.55 (4H, m), 2.35 (1H, dd, J=6.3Hz, 12.6Hz), 2.27 (3H, s), 2.25-2.12 (3H, m), 2.1-1.9 (1H, m), 1.62-1.48 (1H, m), 1.28-1.20 (1H, m), 0.75-0.62 (1H, m), 0.35 (2H, d, J=7.5Hz), 0.1-0.5 (2H, m).
【0076】
工程3
化合物(19)(2.02mg、3.78mmol)をメタノール9.5mlに溶解し、2mol/L水酸化カリウム水溶液9.5mlを加え、60℃で2.5時間加熱攪拌した。反応液を室温に戻し、氷冷下、2mol/L塩酸で中和した後、メタノールを留去した。析出した粗結晶を濾取した後、酢酸エチルとメタノールの混液(1:1)から再結晶し、目的とする化合物(20)(1.44g)を結晶として得た(収率77%)。
1H-NMR (DMSO-d6) δ 14.2 (1H, br s), 9.19 (1H, s), 8.8 (1H, br s), 8.32 (1H, s), 7.49 (1H, s), 6.56 (2H, ABq.), 6.1 (1H, br s), 4.53 (1H, br s), 3.82 (3H, s), 3.5-2.3 (9H, m), 1.82 (1H, d, J=15.6Hz), 1.53 (1H, br d, J=13.5Hz), 1.15-0.95 (1H, m), 0.75-0.5 (2H, m), 0.5-0.3 (2H, m).
【0077】
[実施例3−1]
化合物(7)の合成(その1)
【化44】
【0078】
実施例1−1記載の工程3と同様の方法で合成した化合物(6)(ナルトレキソン塩酸塩5.00g相当)の酢酸エチル溶液に、酢酸(0.1当量)加えた後50℃に昇温し、実施例1−1記載の工程1および工程2と同じ方法で合成した化合物(4)の反応液を加え、同温で6.5時間攪拌した。反応液にヘプタンを加え、析出固体を濾取し、洗浄後、乾燥して化合物(7)(6.92g、85.3%)を得た。
【0079】
[実施例3−2]
化合物(7)の合成(その2)
【化45】
実施例1−1記載の工程3と同様の方法で合成した化合物(6)の酢酸エチル溶液に、化合物(4)(1.5当量)とトルエンを加え、70℃で8時間攪拌し、化合物(7)を得た。
【0080】
[実施例4]
化合物(IA)のp−トルエンスルホン酸塩水和物結晶(I形)の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)5.00gに2−プロパノール25mL−水2.5mL混液を加え加温溶解した。アセトニトリル50mLを加え室温攪拌を4時間行った。析出晶を濾別し、85℃で減圧乾燥を4時間行い、4.68gの結晶を得た。
元素分析:
計算値:C, 60.28; H, 5.94; N, 7.21; S, 4.13(1.9H2Oとしての計算値)
実測値:C, 60.5; H, 6.17; N, 7.21; S, 3.83
粉末X線回折の結果を図2および表3に示す。
【表3】
粉末X線回折スペクトルにおいて、回折角度(2θ):6.6°±0.2°、8.9°±0.2°、11.4°±0.2°、12.9°±0.2°、14.0°±0.2°、15.0°±0.2°、17.6°±0.2°、18.2°±0.2°、22.4°±0.2°、25.4°±0.2°および28.7°±0.2°にピークが認められた。
TG/DTA分析結果を図15に示す。この結果から、重量減少率を算出すると、2.42%であった。従って、1水相当の水を含むことがわかる。
以上の測定結果から、本I形結晶は、1〜2水相当の水を含む結晶であると考えられる。
【0081】
[実施例5]
化合物(IA)のp−トルエンスルホン酸塩水和物結晶(II形)の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)5.00gにテトラヒドロフラン12.5mLを加え溶解させた。酢酸n−プロピル50mLを加え、室温攪拌を4時間行った。析出晶を濾別し、85℃で減圧乾燥を4時間行い、4.77gの結晶を得た。
元素分析:
計算値:C, 61.56; H, 5.83; N, 7.36; S, 4.21(1.0H2Oとしての計算値)
実測値:C, 61.68; H, 5.78; N, 7.39; S, 4.07
粉末X線回折の結果を図3および表4に示す。
【表4】
粉末X線回折スペクトルにおいて、回折角度(2θ):7.1°±0.2°、8.8°±0.2°、17.5°±0.2°、19.2°±0.2°、19.7°±0.2°、21.2°±0.2°、21.9°±0.2°、23.7°±0.2°、24.5°±0.2°および26.1°±0.2°にピークが認められた。
TG/DTA分析結果を図16に示す。この結果から、重量減少率を算出すると、1.42%であった。従って、0.5水相当の水を含むことがわかる。
以上の測定結果から、本II形結晶は、0.5〜1水相当の水を含む結晶であると考えられる。
【0082】
[実施例6]
化合物(IA)の酢酸塩の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)20.00gに酢酸エチル100mL、炭酸ナトリウム3.18g(p−トルエンスルホン酸塩に対し1.1等量)を溶かした水50mLを加え、分液操作を行った。酢酸エチル層を1%炭酸ナトリウム水溶液50mL、飽和食塩水50mLで洗浄し、各水層を酢酸エチル50mLで逆抽出した。酢酸エチル層を硫酸ナトリウムで脱水し、約30gまで濃縮した。濃縮液にアセトニトリル200mL、酢酸4.6mL(p−トルエンスルホン酸塩に対し3等量)を加え、室温攪拌し、結晶の析出が認められてから、室温下1日静置した。析出晶を濾別し、16.17gの結晶を得た。
粉末X線回折の結果を図4および表5に示す。
【表5】
粉末X線回折スペクトルにおいて、回折角度(2θ):5.6°±0.2°、8.3±0.2°、9.1±0.2°、10.3°±0.2°、12.0°±0.2°、13.5±0.2°、14.6°±0.2°、16.3±0.2°および26.0°±0.2°にピークが認められた。
TG/DTA分析結果を図17に示す。
【0083】
[実施例7]
化合物(IA)の塩酸塩の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)20.00gに酢酸エチル100mL、炭酸ナトリウム3.18g(p−トルエンスルホン酸塩に対し1.1等量)を溶かした水50mLを加え、分液操作を行った。酢酸エチル層を水50mLで2回洗浄し、各水層を酢酸エチル50mLで逆抽出した。酢酸エチル層を硫酸ナトリウムで脱水し、アセトニトリル200mL、4mol/L塩酸−酢酸エチル10mL(p−トルエンスルホン酸塩に対し1.5等量)を加え、約50gまで濃縮した。濃縮液にアセトニトリル200mLを加え、室温攪拌を1時間行った。析出晶を濾別し、10.01gの結晶を得た。
粉末X線回折の結果を図5および表6に示す。
【表6】
粉末X線回折スペクトルにおいて、回折角度(2θ):8.5°±0.2°、10.8°±0.2°、11.3°±0.2°、12.7°±0.2°、13.9°±0.2°、15.6°±0.2°、17.3°±0.2°、19.2°±0.2°、20.1°±0.2°および23.9°±0.2°にピークが認められた。
TG/DTA分析結果を図18に示す。
【0084】
[実施例8]
化合物(IA)のエタノール和物の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)20.00gに酢酸エチル100mL、炭酸ナトリウム3.18g(p−トルエンスルホン酸塩に対し1.1等量)を溶かした水50mLを加え、分液操作を行った。酢酸エチル層を1%炭酸ナトリウム水溶液50mL、飽和食塩水50mLで洗浄し、各水層を酢酸エチル50mLで逆抽出を行った。酢酸エチル層を硫酸ナトリウムで脱水し、約30gまで濃縮した。濃縮液にエタノール100mLを加え、溶液が濁る寸前までイソプロピルエーテルを加えた。室温攪拌し、結晶の析出が認められてから、室温下で1日静置した。析出晶を濾別し、9.57gの結晶を得た。
粉末X線回折の結果を図6および表7に示す。
【表7】
粉末X線回折スペクトルにおいて、回折角度(2θ):6.9°±0.2°、11.0°±0.2°、12.9°±0.2°、13.4°±0.2°、16.5°±0.2°、20.5°±0.2°、21.3°±0.2°、21.8°±0.2°、22.6°±0.2°および25.1°±0.2°にピークが認められた。
TG/DTA分析結果を図19に示す。
【0085】
[実施例9]
化合物(IA)の遊離体の製造
実施例8記載のエタノール和物を120℃で10時間減圧乾燥し、核磁気共鳴スペクトルでエタノールが残留していないことを確認した。
粉末X線回折の結果を図7および表8に示す。
【表8】
粉末X線回折スペクトルにおいて、回折角度(2θ):6.8°±0.2°、11.7°±0.2°、13.5°±0.2°、15.6°±0.2°、16.7°±0.2°、21.6°±0.2°、22.1°±0.2°、23.4°±0.2°、26.7°±0.2°および30.1°±0.2°にピークが認められた。
TG/DTA分析結果を図20に示す。
【0086】
[実施例10]
化合物(IA)のp−トルエンスルホン酸塩・酢酸メチル和物の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)1.00gに2−プロパノール5mL−水0.5mL混液を加え、加温溶解させた。酢酸メチル20mLを加え、室温攪拌を4時間行った。析出晶を濾別し、0.98gの結晶を得た。
粉末X線回折の結果を図8および表9に示す。
【表9】
粉末X線回折スペクトルにおいて、回折角度(2θ):17.5°±0.2°、21.8°±0.2°、23.2°±0.2°、25.1°±0.2°および30.6°±0.2°にピークが認められた。
TG/DTA分析結果を図21に示す。
【0087】
[実施例11]
化合物(IA)のp−トルエンスルホン酸塩・(酢酸エチルおよび2−プロパノール)和物の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)1.00gに2−プロパノール5mL−水0.5mL混液を加え、加温溶解させた。酢酸エチル20mLを加え、室温攪拌を4時間行った。析出晶を濾別し、0.96gの結晶を得た。
粉末X線回折の結果を図9および表10に示す。
【表10】
粉末X線回折スペクトルにおいて、回折角度(2θ):6.6°±0.2°、7.8°±0.2°、17.0°±0.2°、17.8°±0.2°、18.5°±0.2°、21.9°±0.2°および23.6°±0.2°にピークが認められた。
TG/DTA分析結果を図22に示す。
【0088】
[実施例12]
化合物(IA)のp−トルエンスルホン酸塩・(酢酸n−プロピルおよび2−プロパノール)和物の製造
上記実施例1−1の工程5で得た未乾固体(8)の粉末X線回折の結果を図10および表11に示す。
【表11】
粉末X線回折スペクトルにおいて、回折角度(2θ):6.5°±0.2°、8.3°±0.2°、15.5°±0.2°、16.8°±0.2°および18.3°±0.2°にピークが認められた。
TG/DTA分析結果を図23に示す。
【0089】
[実施例13]
化合物(IA)のp−トルエンスルホン酸塩・アセトニトリル和物の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)1.00gに2−プロパノール5mL−水0.5mL混液を加え、加温溶解させた。アセトニトリル15mLを加え、室温攪拌を4時間行った。析出晶を濾別し、1.02gの結晶を得た。
粉末X線回折の結果を図11および表12に示す。
【表12】
粉末X線回折スペクトルにおいて、回折角度(2θ):11.3°±0.2°、17.6°±0.2°、21.7°±0.2°、22.3°±0.2°および28.5°±0.2°にピークが認められた。
TG/DTA分析結果を図24に示す。
【0090】
[実施例14]
化合物(IA)のp−トルエンスルホン酸塩・1、2−ジメトキシエタン和物の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)1.00gにテトラヒドロフラン5mLを加え加温溶解させた。1、2−ジメトキシエタン20mLを加え、室温攪拌を4時間行った。析出晶を濾別し、1.05gの結晶を得た。
粉末X線回折の結果を図12および表13に示す。
【表13】
粉末X線回折スペクトルにおいて、回折角度(2θ):8.5°±0.2°、13.0°±0.2°、14.0°±0.2°、14.9°±0.2°、17.3°±0.2°、20.0°±0.2°、21.9°±0.2°、22.7°±0.2°、25.6°±0.2°および27.2°±0.2°にピークが認められた。
TG/DTA分析結果を図25に示す。
【0091】
[実施例15]
化合物(IA)のp−トルエンスルホン酸塩・メチルイソブチルケトン和物の製造
上記実施例1−1に従って合成したp−トルエンスルホン酸塩(非溶媒和物)1.00gにテトラヒドロフラン5mLを加え加温溶解させた。メチルイソブチルケトン20mLを加え、室温攪拌を4時間行った。析出晶を濾別し、1.02gの結晶を得た。
粉末X線回折の結果を図13および表14に示す。
【表14】
粉末X線回折スペクトルにおいて、回折角度(2θ):5.9°±0.2°、7.9°±0.2°、13.5°±0.2°、15.2°±0.2°、23.2°±0.2°、23.7°±0.2°、24.0°±0.2°、25.0°±0.2°、27.0°±0.2°および28.5°±0.2°にピークが認められた。
TG/DTA分析結果を図26に示す。
【0092】
試験例1
結晶の固体安定性試験
結晶約10mgをポリエチレン製栓付2mL硝子容器に正確に秤取した。該硝子容器を閉栓後、パラフイルムを巻き、40℃または60℃で、2週間または4週間保存した。40℃で保存したサンプルを40℃密栓保存品といい、60℃で保存したサンプルを60℃密栓保存品という。該硝子容器を開栓した状態で、40℃相対湿度89%または60℃で、2週間または4週間保存した。40℃相対湿度89%で保存したサンプルを40℃相対湿度89%保存品といい、60℃で保存したサンプルを60℃開栓保存品という。−40℃密栓保存品を標準品として、下記条件下のHPLC法で絶対検量線法により含量を測定した。
式(IA)のp−トルエンスルホン酸塩(非溶媒和物)の結晶の外観変化の観察結果および結晶の残存率を表16に示した。
式(IA)のp−トルエンスルホン酸塩水和物結晶(I形)の外観変化の観察結果および結晶の残存率を表17に示した。
式(IA)のp−トルエンスルホン酸塩水和物結晶(II形)の外観変化の観察結果および結晶の残存率を表18に示した。
ここで、外観変化がなかったものを(−)、わずかに外観変化が認められたものを(±)とした。
式(IA)のp−トルエンスルホン酸塩(非溶媒和物)の結晶、式(IA)のp−トルエンスルホン酸塩水和物結晶(I形)および式(IA)のp−トルエンスルホン酸塩水和物結晶(II形)の外観変化はなく、いずれの結晶の残存率も低下することなく、安定であることを確認した。
HPLC条件
カラム:CAPCELL PAK C18 AQ (3μm 3.0x150mm)
カラム温度:50℃
UV検出波長:231nm
移動相:[A]10mmol/L ギ酸アンモニウムおよび10mmol/L 塩化マグネシウムの混合液、[B]アセトニトリルを表15のようにグラジエントした。
流速:0.6mL/分
【表15】
【表16】
【表17】
【表18】
【0093】
試験例2
結晶の吸湿性確認試験
式(IA)のp−トルエンスルホン酸塩(非溶媒和物)の結晶約10mgを、水蒸気吸脱着測定装置用試料容器に量り取り、25℃において乾燥窒素雰囲気下で乾燥させた。乾燥後、相対湿度を0%から95%の範囲において5%間隔で連続的に変化させ、試料の水蒸気吸脱着量を水蒸気吸脱着測定装置DVS Advantage(surface measurement systems社製)で測定した。
結果を表19に示す。25℃における相対湿度0%から95%の範囲での式(IA)のp−トルエンスルホン酸塩(非溶媒和物)の結晶の最大吸湿量は1.2%未満であり、該結晶は、ほとんど吸湿性を示さなかった。
【表19】
【0094】
製剤例1
以下の成分を含有する顆粒剤を製造する。
【表20】
上記の「式(IA)で示される化合物」は、式(IA)で示される化合物の遊離体、その酸付加塩および/またはそれらの溶媒和物の結晶を包含する。
式(IA)で表わされる化合物と乳糖を60メッシュのふるいに通す。コーンスターチを120メッシュのふるいに通す。これらをV型混合機にて混合する。混合末にHPC-L(低粘度ヒドロキシプロピルセルロース)水溶液を添加し、練合、造粒(押し出し造粒 孔径0.5〜1mm)、乾燥工程する。得られた乾燥顆粒を振動ふるい(12/60メッシュ)で櫛過し顆粒剤を得る。
【0095】
製剤例2
以下の成分を含有するカプセル充填用顆粒剤を製造する。
【表21】
上記の「式(IA)で示される化合物」は、式(IA)で示される化合物の遊離体、その酸付加塩および/またはそれらの溶媒和物の結晶を包含する。
式(IA)で表わされる化合物、乳糖を60メッシュのふるいに通す。コーンスターチを120メッシュのふるいに通す。これらを混合し、混合末にHPC-L溶液を添加して練合、造粒、乾燥する。得られた乾燥顆粒を整粒後、その150mgを4号硬ゼラチンカプセルに充填する。
【0096】
製剤例3
以下の成分を含有する錠剤を製造する。
【表22】
上記の「式(IA)で示される化合物」は、式(IA)で示される化合物の遊離体、その酸付加塩および/またはそれらの溶媒和物の結晶を包含する。
式(IA)で表わされる化合物、乳糖、微結晶セルロース、CMC-Na(カルボキシメチルセルロース ナトリウム塩)を60メッシュのふるいに通し、混合する。混合末にステアリン酸マグネシウム混合し、製錠用混合末を得る。本混合末を直打し、150mgの錠剤を得る。
【0097】
製剤例4
以下の成分を加温混合後、滅菌して注射剤とする。
【表23】
上記の「式(IA)で示される化合物」は、式(IA)で示される化合物の遊離体、その酸付加塩および/またはそれらの溶媒和物の結晶を包含する。
【産業上の利用可能性】
【0098】
本発明は、医薬品製造用原体として有用な6,7−不飽和−7−カルバモイルモルヒナン誘導体、その酸付加塩、それらの溶媒和物、またはそれらの安定な結晶体を提供する。
また本発明は、オピオイド受容体アゴニスト作用を有する化合物に誘発される嘔気、嘔吐および/または便秘の治療および/または予防剤として有用な、6,7−不飽和−7−カルバモイルモルヒナン誘導体、その酸付加塩、それらの溶媒和物、またはそれらの結晶の新たな製造方法を提供するものである。
【特許請求の範囲】
【請求項1】
式(III):
【化1】
(式中、R1bは水酸基の保護基;R2は置換基を有していてもよい低級アルキル)で示される化合物を、酸の存在下または非存在下、式:R3−N=C=O(式中、R3は置換基を有していてもよい低級アルキル、置換基を有していてもよいシクロアルキル、置換もしくは非置換アリール、または置換もしくは非置換ヘテロアリール)で示される化合物または式:R3−NH−C(=O)−X(式中、R3は前記と同意義、Xは脱離基)で示される化合物と反応させることを特徴とする、式(II):
【化2】
(式中、R1b、R2およびR3は、前記と同意義)で示される化合物の製造方法。
【請求項2】
式(IV):
【化3】
(式中、R2は請求項1と同意義)で示される化合物の水酸基を保護することにより、当該式(III):
【化4】
(式中、R1bおよびR2は請求項1と同意義)で示される化合物を得ることを特徴とする、請求項1記載の製造方法。
【請求項3】
式(IV):
【化5】
(式中、R2は請求項1と同意義)で示される化合物の水酸基を保護し、式(III):
【化6】
(式中、R1bおよびR2は請求項1と同意義)で示される化合物を得る工程、および式(III)で示される化合物を、酸の存在下または非存在下、式:R3−N=C=O(式中、R3は請求項3と同意義)で示される化合物または式:R3−NH−C(=O)−X(式中、R3は前記と同意義、Xは脱離基)で示される化合物と反応させる工程を連続して行う、請求項2記載の製造方法。
【請求項4】
酸の存在下で行う、請求項1〜3のいずれかに記載の製造方法。
【請求項5】
酸がルイス酸である、請求項4記載の製造方法。
【請求項6】
ルイス酸触媒が、CuCl、CuCl2、CuBr、CuI、CuBr、CuSO4、Cu、Zn(OAc)2、ZnBr2またはZnCl2である、請求項5に記載の製造方法。
【請求項7】
式(III)で示される化合物に対して、約0.00005〜約1.0当量の酸の存在下で反応させることを特徴とする、請求項1〜6のいずれかに記載の製造方法。
【請求項8】
R1bが塩基で脱保護可能な水酸基の保護基である、請求項1〜7のいずれかに記載の製造方法。
【請求項9】
式(IIA):
【化7】
(式中、R2およびR3は請求項1と同意義)で示される化合物を、塩基で処理することを特徴とする、式(I):
【化8】
(式中、R2およびR3は、前記と同意義)で示される化合物の製造方法。
【請求項10】
式(IIC):
【化9】
(式中、R1cは塩基で脱保護可能な水酸基の保護基;R2およびR3は請求項1と同意義)
で示される化合物を、塩基で処理することを特徴とする、式(I):
【化10】
(式中、R2およびR3は、前記と同意義)で示される化合物の製造方法。
【請求項11】
式(IIB):
【化11】
(式中、R1bは水酸基の保護基;R2およびR3は請求項1と同意義)で示される化合物を、塩基で処理することを特徴とする、式(IB):
【化12】
(式中、R1dは塩基では脱保護されない水酸基の保護基または水素原子;R2およびR3は、前記と同意義)で示される化合物の製造方法。
【請求項12】
塩基が無機塩基である請求項9〜11のいずれかに記載の製造方法。
【請求項13】
塩基が水酸化カリウム、水酸化ナトリウム、水酸化リチウムまたは水酸化セシウムである請求項9〜11のいずれかに記載の製造方法。
【請求項14】
反応温度が約30℃〜約100℃である請求項9〜13のいずれかに記載の製造方法。
【請求項15】
式(VIIIa):
【化13】
(式中、R5は低級アルキル)で示される化合物を、ルイス酸および塩基の存在下で反応させることを特徴とする、式(IX):
【化14】
で示される化合物の製造方法。
【請求項16】
式(X):
【化15】
(式中、R5は低級アルキル)で示される化合物と、式(XI):
【化16】
で示される化合物を反応させ、当該式(VIIIa)で示される化合物を得ることを特徴とする、請求項15記載の製造方法。
【請求項17】
ルイス酸がAlCl3またはTiCl4である請求項15または16に記載の製造方法。
【請求項18】
式(IIIA):
【化17】
(式中、R1cは、塩基で脱保護可能な水酸基の保護基;R2は請求項1と同意義)で示される化合物を、ルイス酸触媒の存在下または非存在下、式:R3−N=C=O(式中、R3は請求項1と同意義)で示される化合物または式:R3−NH−C(=O)−X(式中、R3は前記と同意義、Xは脱離基)で示される化合物と反応させ、式(IIC):
【化18】
(式中、R1c、R2およびR3は、前記と同意義)で示される化合物を得る工程、
上記式(IIC)で示される化合物を、塩基で処理し、式(I):
【化19】
(式中、R2およびR3は、前記と同意義)で示される化合物を得る工程、および
上記式(I)で示される化合物に酸を添加して酸付加塩とする工程を包含する、式(I)で示される化合物の酸付加塩の製造方法。
【請求項19】
式(I)で示される化合物の酸付加塩が、p−トルエンスルホン酸塩、酢酸塩または塩酸塩、またはそれらの溶媒和物である、請求項18の製造方法。
【請求項20】
p−トルエンスルホン酸塩、酢酸塩または塩酸塩、またはそれらの溶媒和物が結晶である、請求項19の製造方法。
【請求項21】
式(IID):
【化20】
(式中、R1は水素原子または水酸基の保護基)で示される化合物。
【請求項1】
式(III):
【化1】
(式中、R1bは水酸基の保護基;R2は置換基を有していてもよい低級アルキル)で示される化合物を、酸の存在下または非存在下、式:R3−N=C=O(式中、R3は置換基を有していてもよい低級アルキル、置換基を有していてもよいシクロアルキル、置換もしくは非置換アリール、または置換もしくは非置換ヘテロアリール)で示される化合物または式:R3−NH−C(=O)−X(式中、R3は前記と同意義、Xは脱離基)で示される化合物と反応させることを特徴とする、式(II):
【化2】
(式中、R1b、R2およびR3は、前記と同意義)で示される化合物の製造方法。
【請求項2】
式(IV):
【化3】
(式中、R2は請求項1と同意義)で示される化合物の水酸基を保護することにより、当該式(III):
【化4】
(式中、R1bおよびR2は請求項1と同意義)で示される化合物を得ることを特徴とする、請求項1記載の製造方法。
【請求項3】
式(IV):
【化5】
(式中、R2は請求項1と同意義)で示される化合物の水酸基を保護し、式(III):
【化6】
(式中、R1bおよびR2は請求項1と同意義)で示される化合物を得る工程、および式(III)で示される化合物を、酸の存在下または非存在下、式:R3−N=C=O(式中、R3は請求項3と同意義)で示される化合物または式:R3−NH−C(=O)−X(式中、R3は前記と同意義、Xは脱離基)で示される化合物と反応させる工程を連続して行う、請求項2記載の製造方法。
【請求項4】
酸の存在下で行う、請求項1〜3のいずれかに記載の製造方法。
【請求項5】
酸がルイス酸である、請求項4記載の製造方法。
【請求項6】
ルイス酸触媒が、CuCl、CuCl2、CuBr、CuI、CuBr、CuSO4、Cu、Zn(OAc)2、ZnBr2またはZnCl2である、請求項5に記載の製造方法。
【請求項7】
式(III)で示される化合物に対して、約0.00005〜約1.0当量の酸の存在下で反応させることを特徴とする、請求項1〜6のいずれかに記載の製造方法。
【請求項8】
R1bが塩基で脱保護可能な水酸基の保護基である、請求項1〜7のいずれかに記載の製造方法。
【請求項9】
式(IIA):
【化7】
(式中、R2およびR3は請求項1と同意義)で示される化合物を、塩基で処理することを特徴とする、式(I):
【化8】
(式中、R2およびR3は、前記と同意義)で示される化合物の製造方法。
【請求項10】
式(IIC):
【化9】
(式中、R1cは塩基で脱保護可能な水酸基の保護基;R2およびR3は請求項1と同意義)
で示される化合物を、塩基で処理することを特徴とする、式(I):
【化10】
(式中、R2およびR3は、前記と同意義)で示される化合物の製造方法。
【請求項11】
式(IIB):
【化11】
(式中、R1bは水酸基の保護基;R2およびR3は請求項1と同意義)で示される化合物を、塩基で処理することを特徴とする、式(IB):
【化12】
(式中、R1dは塩基では脱保護されない水酸基の保護基または水素原子;R2およびR3は、前記と同意義)で示される化合物の製造方法。
【請求項12】
塩基が無機塩基である請求項9〜11のいずれかに記載の製造方法。
【請求項13】
塩基が水酸化カリウム、水酸化ナトリウム、水酸化リチウムまたは水酸化セシウムである請求項9〜11のいずれかに記載の製造方法。
【請求項14】
反応温度が約30℃〜約100℃である請求項9〜13のいずれかに記載の製造方法。
【請求項15】
式(VIIIa):
【化13】
(式中、R5は低級アルキル)で示される化合物を、ルイス酸および塩基の存在下で反応させることを特徴とする、式(IX):
【化14】
で示される化合物の製造方法。
【請求項16】
式(X):
【化15】
(式中、R5は低級アルキル)で示される化合物と、式(XI):
【化16】
で示される化合物を反応させ、当該式(VIIIa)で示される化合物を得ることを特徴とする、請求項15記載の製造方法。
【請求項17】
ルイス酸がAlCl3またはTiCl4である請求項15または16に記載の製造方法。
【請求項18】
式(IIIA):
【化17】
(式中、R1cは、塩基で脱保護可能な水酸基の保護基;R2は請求項1と同意義)で示される化合物を、ルイス酸触媒の存在下または非存在下、式:R3−N=C=O(式中、R3は請求項1と同意義)で示される化合物または式:R3−NH−C(=O)−X(式中、R3は前記と同意義、Xは脱離基)で示される化合物と反応させ、式(IIC):
【化18】
(式中、R1c、R2およびR3は、前記と同意義)で示される化合物を得る工程、
上記式(IIC)で示される化合物を、塩基で処理し、式(I):
【化19】
(式中、R2およびR3は、前記と同意義)で示される化合物を得る工程、および
上記式(I)で示される化合物に酸を添加して酸付加塩とする工程を包含する、式(I)で示される化合物の酸付加塩の製造方法。
【請求項19】
式(I)で示される化合物の酸付加塩が、p−トルエンスルホン酸塩、酢酸塩または塩酸塩、またはそれらの溶媒和物である、請求項18の製造方法。
【請求項20】
p−トルエンスルホン酸塩、酢酸塩または塩酸塩、またはそれらの溶媒和物が結晶である、請求項19の製造方法。
【請求項21】
式(IID):
【化20】
(式中、R1は水素原子または水酸基の保護基)で示される化合物。
【図1】

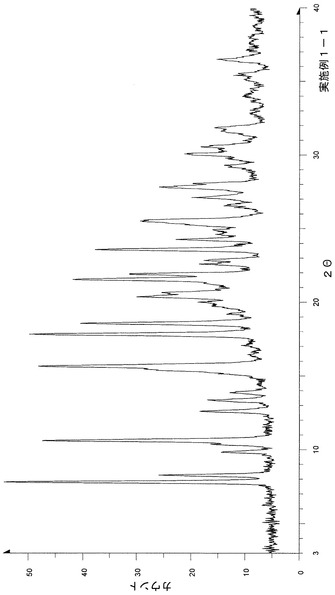
【図2】
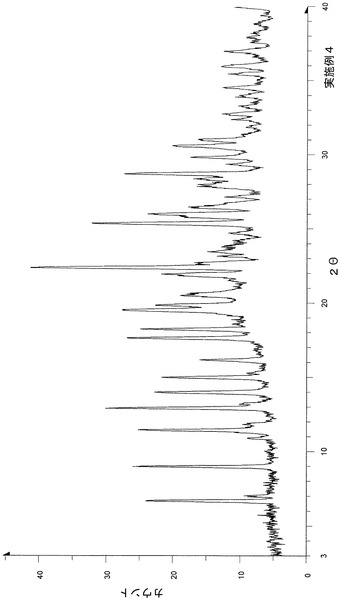
【図3】
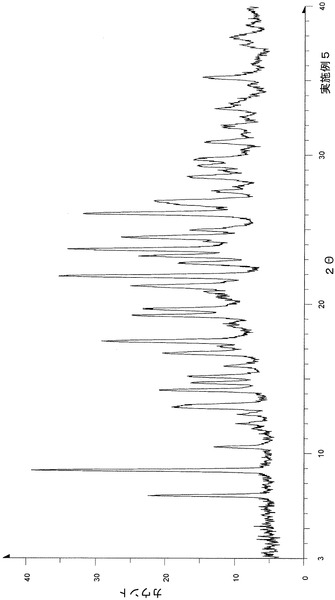
【図4】
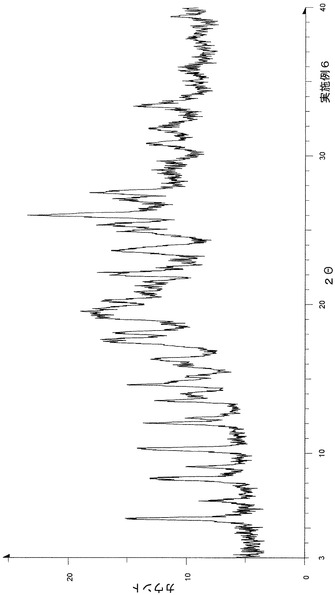
【図5】
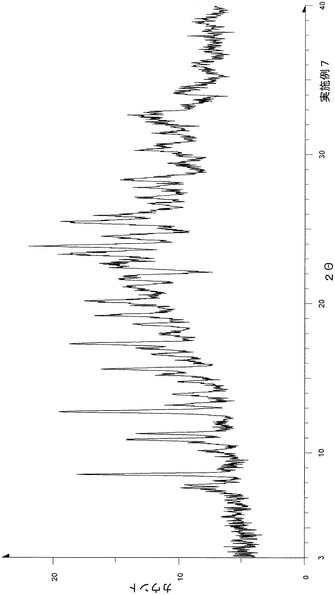
【図6】
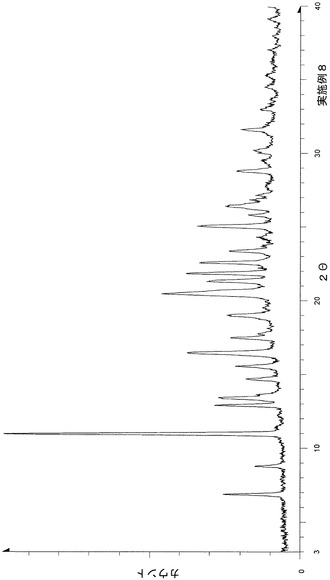
【図7】
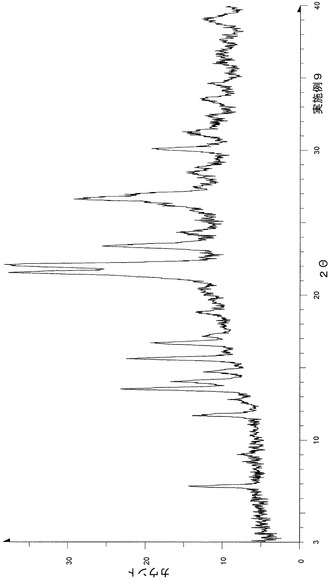
【図8】
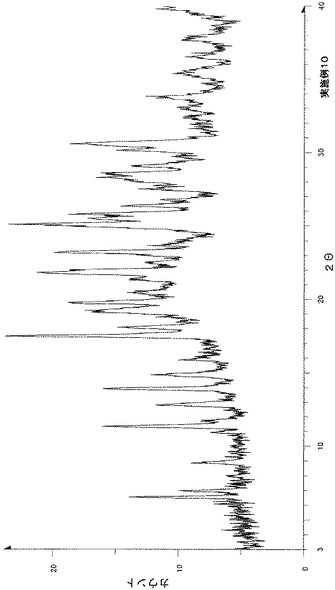
【図9】
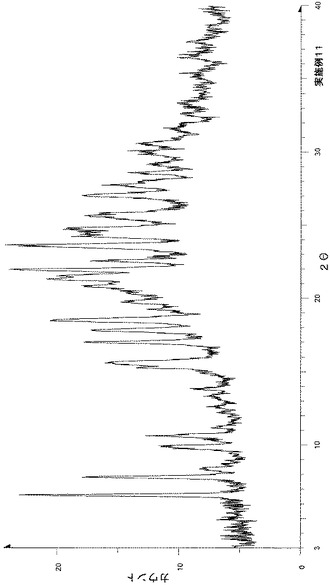
【図10】
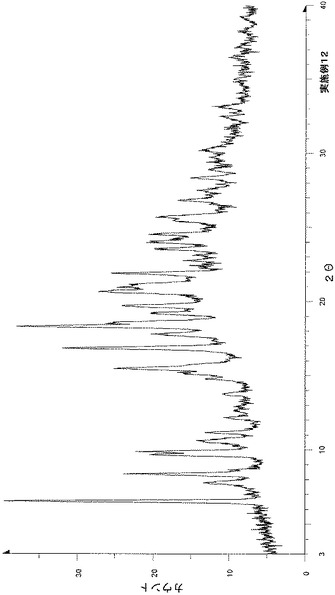
【図11】
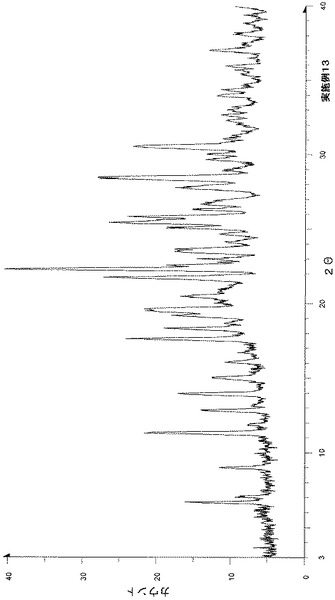
【図12】
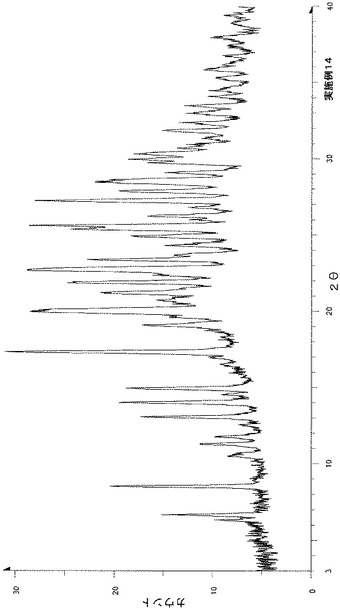
【図13】
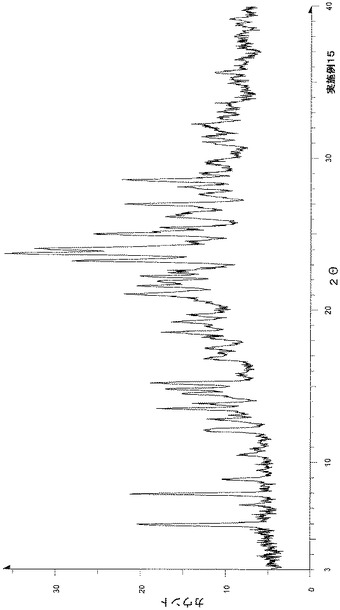
【図14】
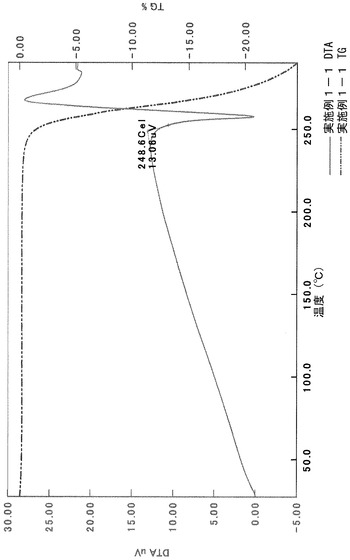
【図15】
【図16】
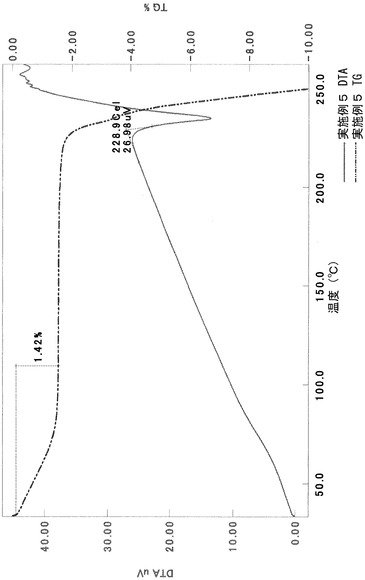
【図17】
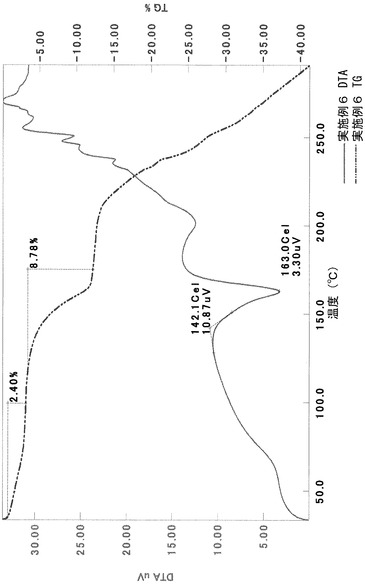
【図18】
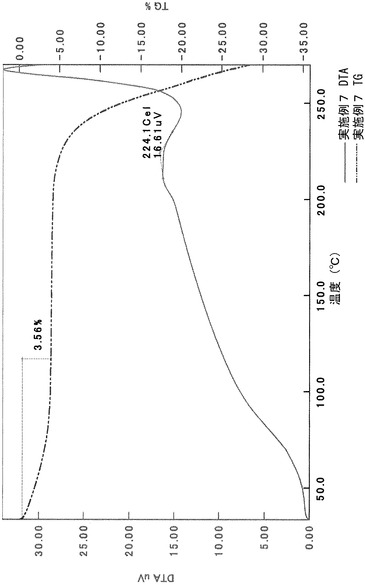
【図19】
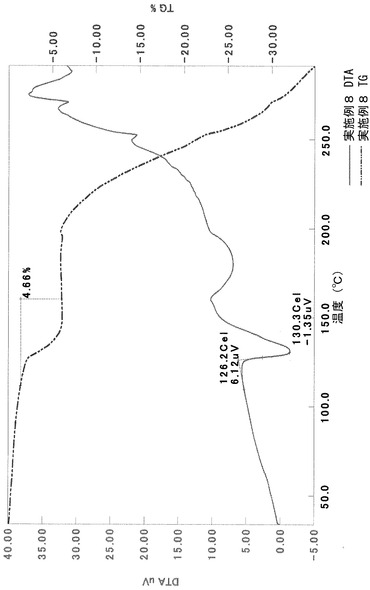
【図20】
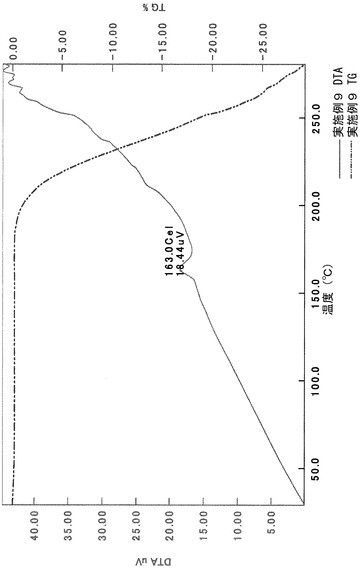
【図21】
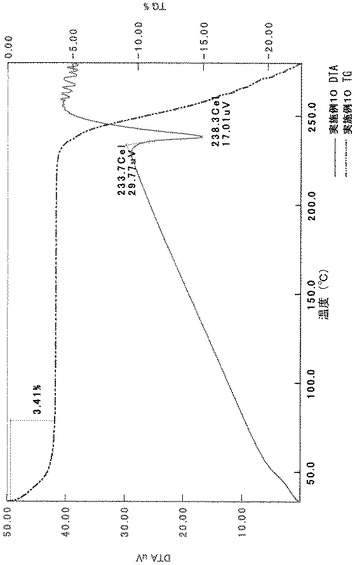
【図22】
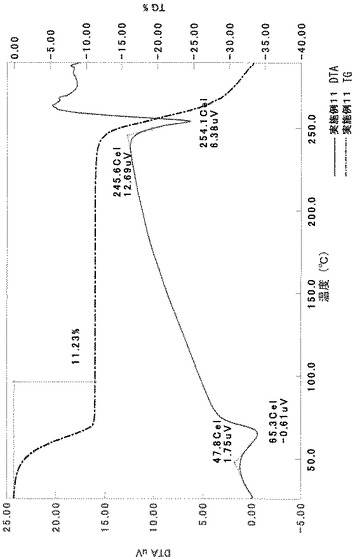
【図23】
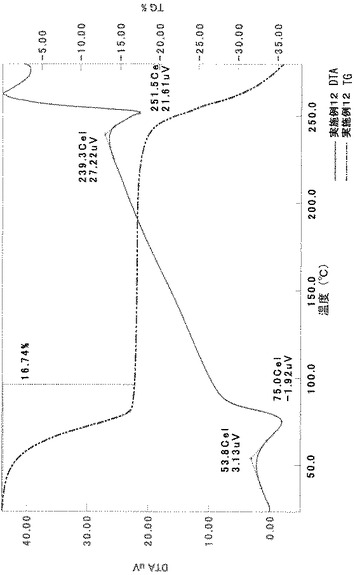
【図24】
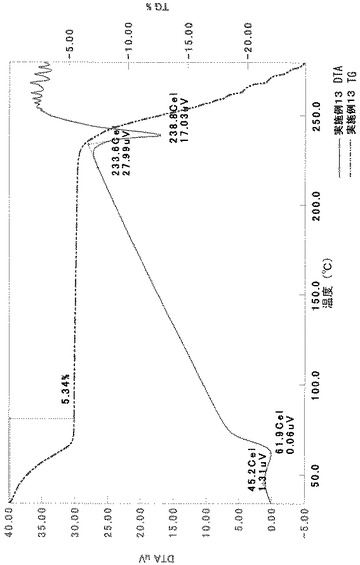
【図25】
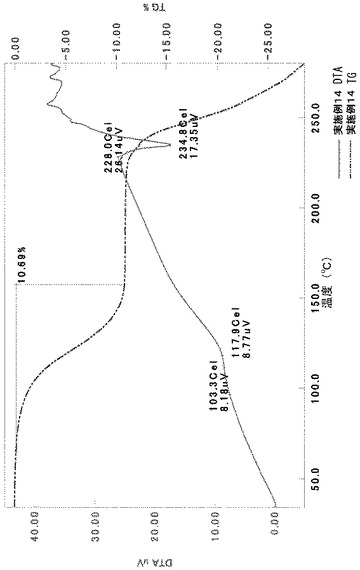
【図26】
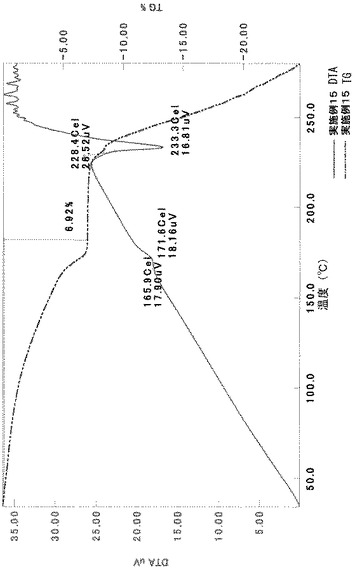
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【公開番号】特開2012−162527(P2012−162527A)
【公開日】平成24年8月30日(2012.8.30)
【国際特許分類】
【出願番号】特願2012−49520(P2012−49520)
【出願日】平成24年3月6日(2012.3.6)
【分割の表示】特願2012−512162(P2012−512162)の分割
【原出願日】平成23年11月11日(2011.11.11)
【出願人】(000001926)塩野義製薬株式会社 (229)
【Fターム(参考)】
【公開日】平成24年8月30日(2012.8.30)
【国際特許分類】
【出願日】平成24年3月6日(2012.3.6)
【分割の表示】特願2012−512162(P2012−512162)の分割
【原出願日】平成23年11月11日(2011.11.11)
【出願人】(000001926)塩野義製薬株式会社 (229)
【Fターム(参考)】
[ Back to top ]