
8−イソプラスタンの定量方法
【課題】精度よく8−イソプラスタンを定量できる方法を提供する。
【解決手段】本発明は、所定のキノキサリノン誘導体を8−イソプラスタンに対して過剰に用いて8−イソプラスタンを蛍光ラベル化するステップ(S101)と、蛍光ラベル化された8−イソプラスタンとキノキサリノン誘導体とを含む反応混合物を、スルホン酸又はスルホン塩が固定化された陽イオン交換担体に接触させて、ラベル化した8−イソプラスタンとキノキサリノン誘導体とを分離するステップ(S102)と、キノキサリノン誘導体と分離された、ラベル化した8−イソプラスタンを定量するステップ(S103)とを含む8−イソプラスタンの定量方法である。
【解決手段】本発明は、所定のキノキサリノン誘導体を8−イソプラスタンに対して過剰に用いて8−イソプラスタンを蛍光ラベル化するステップ(S101)と、蛍光ラベル化された8−イソプラスタンとキノキサリノン誘導体とを含む反応混合物を、スルホン酸又はスルホン塩が固定化された陽イオン交換担体に接触させて、ラベル化した8−イソプラスタンとキノキサリノン誘導体とを分離するステップ(S102)と、キノキサリノン誘導体と分離された、ラベル化した8−イソプラスタンを定量するステップ(S103)とを含む8−イソプラスタンの定量方法である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、8−イソプラスタンの定量方法に関する。
【背景技術】
【0002】
8−イソプラスタンは、細胞膜やリポ蛋白に含まれるリン脂質がフリーラジカルで酸化されることによって生じるプロスタグランジン様の化合物である。尿や血清などの生体試料中に含まれる8−イソプラスタンを測定することにより、生体内における酸化ストレスを非浸襲的に評価することができ、免疫測定により8−イソプラスタンを測定するためのキットも市販されている。
【0003】
特許文献1には、8−イソプラスタンの免疫測定に関する技術が記載されている。
【0004】
特許文献2には、LC−MS/MS法による8−イソプラスタンの分析に関する技術が記載されている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2004−157119号公報
【特許文献2】国際公開第2008/065895号パンフレット
【特許文献3】特開平3−291272号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかしながら、従来の8−イソプラスタンの分析方法では、定量するための精度が極めて低かった。そこで、発明者が鋭意検討したところ、特許文献3記載のキノキサリン誘導体で8−イソプラスタンを蛍光ラベル化することで、定量分析が可能になることが期待された。しかしながら、特許文献3の技術では、尿などの生体試料等に含まれる微量な8−イソプラスタンを収率よくラベル化し、定量分析の精度を向上させるには、未だ改善の余地があった。
【0007】
本発明は上記事情に鑑みてなされたものであり、その目的とするところは、精度よく8−イソプラスタンを定量できる技術を提供するものである。
【課題を解決するための手段】
【0008】
本発明によれば、
下記一般式(1)で表されるキノキサリノン誘導体を、8−イソプラスタンの物質量に対して過剰に用いて8−イソプラスタンを蛍光ラベル化するステップと、
蛍光ラベル化された8−イソプラスタンと、未反応のキノキサリノン誘導体とを含む反応混合物を、スルホン酸又はスルホン酸塩が固定化された陽イオン交換担体に接触させて、蛍光ラベル化された8−イソプラスタンと前記未反応のキノキサリノン誘導体とを分離するステップと、
前記未反応のキノキサリノン誘導体と分離された、蛍光ラベル化された8−イソプラスタンを定量するステップと、
を含む、8−イソプラスタンの定量方法が提供される。
【0009】
【化1】

【0010】
〔式(1)中、R1、R2及びR3は、炭素数1〜6のアルキル基であり、nは、0〜6の整数である。〕
【0011】
また、本発明によれば、上記の8−イソプラスタンの定量方法に用いられる、8−イソプラスタンを定量するためのキットが提供される。
【0012】
この発明によれば、キノキサリノン誘導体を用いて8−イソプラスタンを蛍光ラベル化し、ラベル化後に陽イオン交換担体を用いて蛍光ラベル化された8−イソプラスタンとキノキサリノン誘導体とを分離するため、微量の8−イソプラスタンを用いた場合であっても、過剰のキノキサリノン誘導体を用いて収率よくラベル化させ、かつ、過剰のキノキサリン誘導体を陽イオン交換担体に吸着させて除去することができる。したがって、蛍光ラベル化された8−イソプラスタンを定量することで、精度よく8−イソプラスタンを定量分析することが可能になる。
【発明の効果】
【0013】
本発明によれば、精度よく8−イソプラスタンを定量することができる。
【図面の簡単な説明】
【0014】
【図1】第1の実施形態に係る8−イソプラスタンの定量方法を示すフローチャートである。
【図2】第2の実施形態に係る8−イソプラスタンの定量方法を示すフローチャートである。
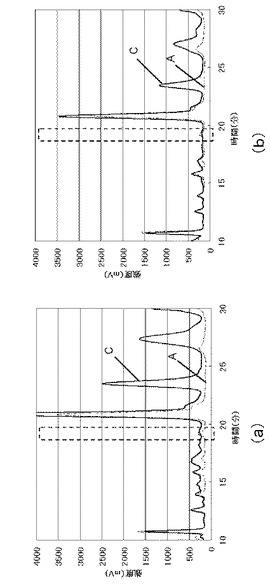
【図3】実施例1、2で得られた高速液体クロマトグラフィーのチャート図である。
【図4】実施例1、3で得られた高速液体クロマトグラフィーのチャート図である。
【発明を実施するための形態】
【0015】
以下、本発明の実施の形態について、図面を用いて説明する。尚、すべての図面において、同様な構成要素には同様の符号を付し、適宜説明を省略する。
【0016】
(第1の実施形態)
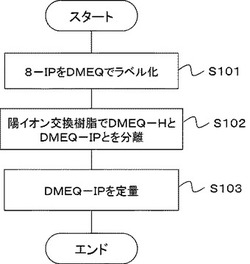
図1は、本実施の形態の8−イソプラスタン(8−IP)の定量方法を示すフローチャートである。本実施の形態は、キノキサリノン誘導体として1,2,3,4‐テトラヒドロ‐6,7‐ジメトキシ‐1‐メチル‐2(1H)‐オキソキノキサリン‐3‐プロピオン酸ヒドラジド(DMEQ−H、(上記式(1)において、R1、R2,R3がいずれもメチルであり、nが2である化合物))を、8−IPの物質量に対して過剰に用いて8−IPを蛍光ラベル化するステップ(S101)と、蛍光ラベル化された8−イソプラスタン(DMEQ−IP)と、未反応のDMEQ−Hとを含む反応混合物を、スルホン酸又はスルホン塩が固定化された陽イオン交換担体に接触させて、DMEQ−IPとDMEQ−Hとを分離するステップ(S102)と、未反応のDMEQ−Hと分離された、DMEQ−IPを定量するステップ(S103)とを含む。以下、各ステップについて詳細に説明する。
【0017】
[S101:8−IPを蛍光ラベル化するステップ]
ステップS101では、8−IPのカルボキシル基とDMEQのヒドラジド基とを反応させてアミド結合を形成し、DMEQ−IPを合成する。反応条件は特に制限されないが、溶媒中、塩基、及び、カルボン酸を活性化するための活性化剤存在下に反応させることが好ましい。溶媒としては、水、アルコール、非プロトン性極性溶媒を用いることが好ましく、ジメチルホルムアミドを用いると、より好ましい。塩基としては、脂肪族三級アミンや含窒素複素環化合物が好ましく、ピリジンがより好ましい。活性化剤としては、カルボジイミドが好ましく、ジシクロヘキシルカルボジイミド(DCC)、ジイソプロピルカルボジイミド(DIC)、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩(EDC)がより好ましく、EDCが特に好ましい。
【0018】
溶媒中で反応を行う場合、ラベル化させる8−IPの濃度は特に限定されないが、例えば、0.01〜0、5nmol/mLとすることができる。
【0019】
DMEQは、8−IPに対して過剰に用いることが好ましく、8−IPに対して、2×103倍モル以上用いることが好ましく、5×103倍モル以上用いることがより好ましく、1×105倍モル以上用いることが特に好ましい。上限は特にないが、2×105倍モル以下とすることができる。
【0020】
塩基(ピリジンなど)を用いる場合は、DMEQに対して、50〜1000倍モル用いることが好ましく、100〜500倍モル用いることがより好ましい。
【0021】
活性化剤(水溶性カルボジイミドなど)を用いる場合は、8−IPに対して過剰に用いることが好ましく、8−IPに対して、1×102〜5×105倍モル用いることが好ましく、2×103〜5×104倍モル用いることがより好ましい。
【0022】
反応温度は、室温(15〜30℃)以上であればよいが、反応効率を上げるため、50〜90℃加熱して行うことが好ましく、60〜80℃がより好ましい。反応時間は、制限されないが、例えば、10〜60分とすることができる。
【0023】
上記反応条件下における8−IPのラベル化収率は、50〜100%とすることができる。
【0024】
[S102:DMEQ−IPとDMEQ−Hとを分離するステップ]
ステップS102で用いる陽イオン交換担体は、スルホン酸又はスルホン酸塩が固定化されたものであればよいが、下記式(2)で表されるアルキルベンゼンスルホン酸又はその塩が固定化されたものが好ましい。
【0025】
【化2】
【0026】
〔式(2)中、mは、1〜6であり、B1は、樹脂基材又はシリカゲルである。〕
【0027】
また、式(2)で表される陽イオン交換担体としては、mが3のプロピルベンゼンスルホン酸又はその塩が固定化されたものがより好ましい。また、式(2)中、B1は、例えば、スチレンとジビニルベンゼンの共重合体等からなる樹脂基材又はシリカゲルであるが、シリカゲルがより好ましい。使用する陽イオン交換担体の量は、ステップS101において使用したDMEQに対して、スルホン酸又はスルホン酸塩の含量(eq)を1〜1000倍にすることが好ましい。
【0028】
DMEQ−IPとDMEQ−Hとを含む液体と、陽イオン交換担体との接触方法は特に限られず、バッチ法でもカラム法でもよいが、カラム法が好適である。陽イオン交換担体としてスルホン酸塩を用いる場合は、スルホン酸塩の対イオンをナトリウムイオンにしておくことが好ましい。水酸化ナトリウム水溶液や食塩水等を陽イオン交換担体に接触させることで、スルホン酸塩の対イオンをナトリウムイオンにすることができる。陽イオン交換担体は、充分な水及びアルコール、所定の緩衝液で平衡化しておくことが好ましい。緩衝液として、たとえばpH5.5〜8.5、好ましくは6.0〜8.0、より好ましくは6.5〜7.5に調節された緩衝液が挙げられる。本明細書において、緩衝液は、所定の酸を水に溶解したものであり、その種類は、pHによって適宜選択することができる。例えば、pH5.5〜5.6の範囲では酢酸緩衝液、pH5.5〜6.2の範囲ではクエン酸緩衝液、pH5.5〜7.0の範囲では、クエン酸−リン酸緩衝液、pH5.5〜8.5の範囲では、リン酸緩衝液、pH7.2〜8.5の範囲では、トリスリン酸緩衝液を用いることができる。また、この緩衝液は、0〜100体積%、好ましくは10〜80体積%の水溶性有機溶媒(特にエタノール)を含有することが好ましい。なお、本明細書において、pHは、25℃で測定したものをいう。
【0029】
カラム法の場合、コンディショニングされた陽イオン交換担体にDMEQ−IPと未反応のDMEQ−Hとを含む反応混合物を通液し、DMEQ−IP及びDMEQ−Hを陽イオン交換担体に保持させる。このとき、反応混合物は、エタノール等の水溶性有機溶媒、緩衝液又はこれらの混合液で反応混合物を希釈されていてもよい。
【0030】
その後、洗浄液によって、陽イオン交換担体に捕捉されていないアニオン物質や非イオン物質を洗い流す。洗浄液としては、水溶性有機溶媒と水とを主成分とするものが好ましく、水溶性有機溶媒としては、エタノール、アセトニトリル、メタノール等を用いることができ、その濃度は、洗浄液中、10〜100体積%、好ましくは、30〜100体積%とすることができる。洗浄液は、緩衝液を含んでいることが好ましく、緩衝液のpHは、例えば、5.5〜8.5、好ましくは6.0〜8.0、より好ましくは6.5〜7.5とすることができる。エタノールとリン酸緩衝液との混合液がより好ましく、洗浄液中、0〜100体積%、好ましくは30〜100体積%、より好ましくは50〜100体積%のエタノールを含有することがより好ましい。洗浄液の量は、0.1〜100mL、好ましくは0.1〜50mL、より好ましくは0.1〜20mLとすることができる。
【0031】
洗浄後、水溶性有機溶媒と水とを主成分とする溶出液を用いて、陽イオン交換担体に保持したDMEQ−IPを陽イオン交換担体から溶離する(S103)。溶出液は、エタノール、アセトニトリル、メタノール等の水溶性有機溶媒を含有することができ、その濃度は、溶出液中、10〜100体積%、好ましくは、30〜100体積%とすることができる。溶出液は、緩衝液を含むことが好ましく、緩衝液のpHは、例えば、5.5〜8.5、好ましくは6.0〜8.0、より好ましくは6.5〜7.5とする。また、エタノールとリン酸緩衝液との混合液がより好ましく、溶出液中、0〜100体積%、好ましくは30〜100体積%、より好ましくは50〜100体積%のエタノールを含有することがより好ましい。溶出液は、0.1〜10mLを通液させ、最初に通液された溶出液を約0.2〜0.3mLまで回収する。なお、溶出液量及び回収液量は、使用する担体の大きさ、サンプル量等に応じて、増減可能である。このようにして、DMEQ−IPを得る。DMEQ−Hは、陽イオン交換樹脂への吸着性が強いため、回収された液体中には、DMEQ−IPが含まれるが、DMEQ−Hを含んでいない。このようにして、DMEQ−IPとDMEQ−Hとを分離することができる。
【0032】
[S103:DMEQ−IPを定量するステップ]
ステップS102で得られたDMEQ−IPを用い、高速液体クロマトグラフィー(HPLC)法によりDMEQ−IPを定量する。S102において陽イオン交換樹脂から回収された回収液中の溶媒は、HPLCを行う前に適宜除去されてもよい。このHPLC法では、逆相カラムを用いた逆相カラムクロマトグラフィー法を用いることができる。また、カラムは、加熱することが好ましく、40〜60℃に加熱することが好ましい。移動相は、アルコールと緩衝液との混合液が好ましく、具体的には、メタノールと酢酸緩衝液との混合液が好ましい。移動相のpHは、弱酸性が好ましく、pH4〜6がより好ましい。アイソクラティックでも勾配をかけてもよいが、定量の精度を向上させるという観点から、アイソクラティックが好ましい。蛍光励起スペクトルは、最大367nm、蛍光発光スペクトルは、最大445nmが好ましい。DMEQ−IPの蛍光強度を測定することで、HPLC装置に注入したDMEQ−IPを定量することができる。
【0033】
DMEQ−IPの蛍光強度を定量の精度は、変動係数を20%以下にすることができる。また、1〜1000pmolの範囲であれば、ピークの高さとDMEQ−IPの量とが比例関係にあり、相関係数を0.99以上にすることができる。
【0034】
このようにして、求められたDMEQ−IPの量に基づきS101において使用した8−イソプラスタンの量を求めることができる。S103において得られたDMEQ−IPの物質量を、そのままラベル化に使用した8−IPの物質量としてもよいし、ラベル化収率を考慮して、使用した8−IPの物質量を算出してもよい。こうした本実施形態の8−IPの定量方法によれば、DMEQを用いて8−イソプラスタンを蛍光ラベル化し、ラベル化後に陽イオン交換担体を用いてDMEQ−IPとDMEQとを分離するため、100pmol以下、さらには、50pmol以下の微量の8−イソプラスタンを用いた場合であっても、過剰のDMEQを用いて収率よくラベル化させ、かつ、過剰のDMEQを陽イオン交換担体に吸着させて除去することができる。したがって、DMEQ−IPを定量することで、精度よく8−イソプラスタンを定量分析することが可能になる。
【0035】
上記本実施の形態に係る8−イソプラスタンを定量する方法に用いられる、8−イソプラスタンを定量するためのキットは、DMEQ−Hと、スルホン酸が固定化された陽イオン交換担体と、所定の洗浄液及び溶出液とを含むものであればよいが、DMEQ−Hは、ジメチルホルムアミド溶液であることが好ましく、陽イオン交換担体には、プロピルベンゼンスルホン酸又はその塩がシリカゲルに固定化された陽イオン交換担体であることが好ましく、洗浄液及び溶出液としては、エタノールとリン酸緩衝液との混合液であることが好ましい。この溶出液は、エタノールとリン酸緩衝液とを別々に包装し、ユーザにおいて調製する形態であってもよい。また、ピリジン等の塩基と、EDC等のカルボジイミド又はこれらの溶液を含んでいるとさらに好ましい。なお、こうしたキットには、本実施の形態に係る8−イソプラスタンの定量方法が記載された使用説明書が添付されている。
【0036】
(第2の実施形態)
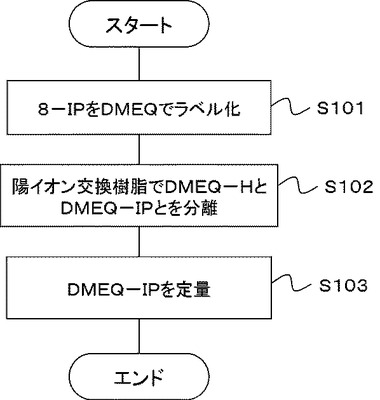
図2は、本実施の形態の8−イソプラスタン(8−IP)の定量方法の一部を示すフローチャートである。本実施の形態は、生体試料中の8−IPの定量方法である。本実施の形態では、生体試料中の8−IPを分離するステップ(S201)と、分離された8−IPを陰イオン交換担体を用いて精製するステップ(S202)とを含み、分離・精製された8−IPを用いて、第1の実施の形態で説明したステップS101、S102、S103を実行する。本実施の形態では、第1の実施の形態と異なる点のみを説明する。
【0037】
[S201:生体試料中の8−IPを分離するステップ]
S201において用意される生体試料としては、尿、血液、唾液等の体液が挙げられるが、以下、試料が尿である場合を例に説明する。採取する尿の量は、測定をより確実に行う観点から、たとえば0.5〜50mL、好適には1.0〜10mL、より好適には1.5〜5.0mLとする。採取された原尿そのものを使うことが可能である。また、尿を所定の緩衝液又は水で希釈して用いてもよい。このとき、他の添加剤、例えば、EDTA(エチレンジアミン四酢酸)等のキレート剤を加えても良い。尿は採取後直ちに濃縮されることが好ましいが、数時間から数日後であってもよい。
【0038】
S201において用いる逆相担体は、官能基として炭素数1〜30の直鎖の炭化水素基をもつ材料により構成されていることが好ましく、生体試料中の8−イソプラスタンをより効率的に処理する観点から、炭素数8〜22の直鎖の炭化水素基がより好ましく、炭素数10〜20の直鎖の炭化水素基が特に好ましい。逆相担体の量は、尿の重量に対して0.1〜10倍量とすることができる。
【0039】
逆相担体の材料として、具体的には、オクタデシルシリル(ODS)基を化学結合させたシリカゲルが挙げられる。このとき、オクタデシルシリル(ODS)基を化学結合させたシリカゲルにおけるシリル化剤の結合様式は、シリル化剤がシリカゲル中のシラノール基と1対1で結合している様式、つまりモノメリックな結合様式とするとよい。これにより、逆相系充填剤が過度に疎水性とならないようにすることができる。また、逆相担体は、さらに好適な疎水性を有するという観点から、担体表面の炭素(C)の含有量が、元素比で18%以下が好ましく、15%以下とするとより好ましい。
【0040】
また、逆相担体は、フロー式粒子像分析装置で測定される円相当径0.5〜10μmの粒子を粒子数として1〜20累積%含むことがより好ましい。また、さらに上記装置で測定される円相当径20〜100μmの粒子を粒子数として65〜99累積%含んでいてもよく、より好ましくは、78〜99累積%含んでいてもよい。こうすることで、分子量や性質等の性状が8−IPとわずかに異なる夾雑物が含まれている場合にも、これらを簡便で確実に分離することができる。この理由としては、粒子径が異なる粒子を特定の比率で配合することにより、大きな粒子の間に小さな粒子が入り込み、単位体積あたりの担体の表面積が最大となり、8−IPの保持力が向上することが考えられる。
【0041】
さらに、8−IPの分離を効率よく行う観点から、フロー式粒子像分析装置で測定される円相当径0.5〜10μmの粒子を粒子数として含む割合は、好ましくは4〜19累積%、さらに好ましくは4〜13累積%、特に好ましくは6〜13累積%である。また、上記装置で測定される円相当径20〜100μmの粒子を粒子数として含む割合は、好ましくは68〜95累積%、さらに好ましくは78〜93累積%、特に好ましくは85〜91累積%である。
【0042】
なお、フロー式粒子像分析装置は、測定対象の粒子を含む試料が流れるフローセルにストロボ光等の光を照射して、通過中の粒子の画像を取得し、画像解析により円相当径等の粒子形状を示すパラメータを算出する装置である。実際には隋円形などに崩れた粒子が存在するため、円相当径とは、実際に測定した粒子の粒子投影面積と同じ投影面積を持つ球を想定し、その球の直径と定義している。フロー式粒子像分析装置の具体例としては、シスメックス社製FPIA−3000が挙げられる。
【0043】
また、逆相担体には、沈降法で測定される粒径35〜60μmの粒子と、コールター法で測定される粒径10〜30μmの粒子との重量比が、80:20〜95:5の範囲にある粒子を含んでいても良い。このようにすれば、8−IPの回収率をさらに向上させることができる。粒径35〜60μmの粒子と粒径10〜30μmの粒子との重量比は、8−IPの回収率と分離時間とのバランスを向上させる観点から、好ましくは90:10とする。
【0044】
生体試料は、予め水及びアルコールで十分にコンディショニングされた逆相担体と接触させることが好ましく、エタノール及び純水でコンディショニングされているとより好ましい。また、pH5.5〜8.5の緩衝液を用いてコンディショニングすることもできる。生体試料と逆相担体との接触方法は特に限られず、バッチ法でもカラム法でもよいが、効率よく8−イソプラスタンを濃縮できるという観点から、カラム法が好適である。ステップS201では、逆相担体と移動相との組み合わせとして、例えばオクタデシル(ODS)基を化学結合させたシリカゲルとリン酸緩衝液−エタノールの混合液との組み合わせが用いられる。
【0045】
その後、所定の緩衝液等を洗浄液として逆相担体に流し、逆相担体に捕捉されない物質を洗い流す。S202における洗浄液には、水溶性有機溶媒と水とを主成分とするものを用いることができる。水溶性有機溶媒には、エタノール、アセトニトリル、メタノール等を用いることができ、その濃度は、洗浄液中、10〜100体積%、好ましくは、30〜100体積%とすることができる。例えば、pHは、5.5〜8.5、好ましくは6.0〜8.0、より好ましくは6.5〜7.5に調節された緩衝液を用いることができる。中でも、エタノールとリン酸緩衝液との混合液が好ましく、洗浄液中、0〜70体積%、好ましくは0〜50体積%、より好ましくは0〜30体積%のエタノールを含有することがより好ましい。洗浄液の量は、1〜100mL、好ましくは1〜50mL、より好ましくは1〜20mLとすることができる。また、エタノールの含量を徐々に増加させるとより好ましく、0〜20体積%含有エタノール/リン酸緩衝液で洗浄した後、20〜40体積%含有エタノール/リン酸緩衝液で洗浄し、さらに、40〜60体積%含有エタノール/リン酸緩衝液で洗浄するとより好ましい。
【0046】
洗浄後、溶出液を用いて、逆相担体に保持した8−IPを逆相担体から溶離する。溶出液には、水溶性有機溶媒と水とを主成分とするものを用いることができる。水溶性有機溶媒には、エタノール、アセトニトリル、メタノール等を用いることができ、その濃度は、溶出液中、10〜100体積%、好ましくは、30〜100体積%とすることができる。溶出液は、pHが、例えば、5.5〜8.5、好ましくは6.0〜8.0、より好ましくは6.5〜7.5の緩衝液を含むことが好ましい。また、エタノールとリン酸緩衝液との混合液がより好ましく、溶出液中、10〜100体積%、好ましくは30〜100体積%、より好ましくは50〜100体積%のエタノールを含有することがより好ましい。溶出液は、0.5〜10mLを通液させ、0.7〜1.7mLにかけて溶出する約1mLを回収する。なお、溶出液量及び回収液量は使用する逆相担体の大きさ、サンプル量等に応じて、増減可能である。このようにして、生体試料から8−イソプラスタンを分離することができる。
【0047】
[S202:8−IPを精製するステップ]
ステップS202では、生体試料から分離した8−IPを、陰イオン交換担体を用いて精製する。陰イオン交換担体は、第一又は二級アミンが固定化された弱陰イオン交換担体、芳香族又は脂肪族第四級アンモニウムが固定化された陰イオン交換担体等が用いられるが、第四級アンモニウム塩が固定化された陰イオン交換担体が好ましい。第四級アンモニウム塩としては、式(3)で表されるテトラアルキルアンモニウム塩が好ましい。
【0048】
【化3】
【0049】
〔式(3)中、R4、R5、R6が炭素数1〜6のアルキル基であり、pが1〜5であり、B2が樹脂基材又はシリカゲルであり、X−が塩化物イオンである。〕
【0050】
式(3)で表される陰イオン交換担体としては、中でもR4、R5、R6がメチル基であり、pが3のトリメチルアンモニウムプロピル基が固定化されているとより好ましい。また、式(3)中、B2は、スチレンとジビニルベンゼンの共重合体等からなる樹脂基材又はシリカゲルであるが、シリカゲルがより好ましい。使用する陰イオン交換担体の量は、第四級アンモニウム塩の含量(eq)が、8−IPの物質量に対して、1〜10万倍にすることが好ましいが、例えば、尿中の8−IPを精製する場合、1mLの尿に対して、0.01〜1eqとなる量にすることができる。
【0051】
8−IPと陰イオン交換担体との接触方法は特に限られず、バッチ法でもカラム法でもよいが、カラム法が好適である。陰イオン交換担体として第四級アンモニウム塩を用いる場合、対イオンを塩化物イオンにしておくことが好ましい。塩酸や食塩水等を陰イオン交換担体に接触させることで、第四級アンモニウム塩の対イオンを塩化物イオンにすることができる。その後、充分な水及びアルコール、所定の緩衝液で平衡化しておく。緩衝液として、たとえばpH5.5〜8.5、好ましくは6.0〜8.0、より好ましくは6.5〜7.5に調節された緩衝液が挙げられる。また、この緩衝液は、0〜100体積%、好ましくは10〜80体積%の水溶性有機溶媒(特に、エタノール)を含有することが好ましい。
【0052】
カラム法の場合、コンディショニングされた陰イオン交換担体に液体試料を流し、8−IPを陰イオン交換担体に保持させた後、水溶性有機溶媒と水とを主成分とする洗浄液によって、陰イオン交換担体に捕捉されていないカチオン物質や非イオン物質を洗い流す。洗浄液として、水溶性有機溶媒と水とを主成分とするものを用いることができる。水溶性有機溶媒としては、エタノール、アセトニトリル、メタノール等を用いることができ、その濃度は、洗浄液中、10〜100体積%、好ましくは、30〜100体積%とすることができる。また、洗浄液は、緩衝液を含むことが好ましく、pHが、例えば、5.5〜8.5、好ましくは6.0〜8.0、より好ましくは6.5〜7.5の緩衝液を用いることができる。また、エタノールとリン酸緩衝液との混合液がより好ましく、洗浄液中、0〜100体積%、好ましくは30〜100体積%のエタノールを含有することがより好ましい。洗浄液の量は、1〜100mL、好ましくは1〜50mL、より好ましくは1〜20mLとすることができる。
【0053】
洗浄後、溶出液を用いて、陰イオン交換担体に保持した8−IPをイオン交換担体から溶離する。この溶出液は、水溶性有機溶媒と水とを主成分とするものが好ましい。水溶性有機溶媒は、エタノール、アセトニトリル、メタノール等を用いることができ、その濃度は、溶出液中、10〜100体積%、好ましくは、30〜100体積%とすることができる。溶出液は、pHが、例えば、5.5〜8.5、好ましくは6.0〜8.0、より好ましくは6.5〜7.5の緩衝液を含むことが好ましい。中でも、エタノールとリン酸緩衝液との混合液がより好ましく、溶出液中、0〜100体積%、好ましくは30〜100体積%、より好ましくは50〜100体積%のエタノールを含有することがより好ましい。溶出液は、0.1〜10mLを通液させ、1.5〜2.5mLにかけて溶出する約1mLを回収する。なお、溶出液量及び回収液量は、使用する担体の大きさ、サンプル量等に応じて、増減可能である。このようにして、8−IPの精製をすることができる。
【0054】
回収された液体中には、8−IPが濃縮されており、これを8−IPのラベル化(S101)に供することができる。なお、8−IPの分析前に回収液中の溶媒を、適宜除去してもよい。
【0055】
上記本実施の形態に係る8−IPの定量方法に用いられる、生体試料中に含まれる8−IPを定量するためのキットは、第一の実施形態で説明したものに加え、逆相担体と、水溶性有機溶媒と水とを主成分とする溶出液とをさらに備えるものとすることができる。この溶出液は、エタノールとリン酸緩衝液との混合液が好ましい。この溶出液もまた、エタノールとリン酸緩衝液とを別々に包装し、ユーザにおいて調製する形態であってもよい。また、第四級アンモニウム塩が固定化された陰イオン交換担体と、洗浄液と溶出液とをさらに含んでいてもよく、陰イオン交換担体として、トリメチルアンモニウムプロピル基がシリカゲルに固定化されたイオン交換担体、洗浄液及び溶出液として、エタノールとリン酸緩衝液との混合液が好ましい。この溶出液もまた、エタノールとリン酸緩衝液とを別々に包装し、ユーザにおいて調製する形態であってもよい。なお、こうしたキットには、本実施の形態に係る8−イソプラスタンを精製する方法が記載された使用説明書が添付されている。
【0056】
以上、図面を参照して本発明の実施形態について述べたが、これらは本発明の例示であり、上記以外の様々な構成を採用することもできる。
たとえば、上記の実施形態では、キノキサリノン誘導体として、DMEQを用いる例をあげて説明したが、式(1)で表されるキノキサリノン誘導体であれば、本発明の実施形態に記載された技術を用いることができる。例えば、1,2,3,4‐テトラヒドロ‐6,7‐ジメトキシ‐1‐メチル‐2(1H)‐オキソキノキサリン‐3‐カルボキシヒドラジド(式(1)において、R1、R2,R3がいずれもメチルであり、nが0である化合物)を用いることもできる。
【0057】
また、第2の実施形態では、生体試料から8−イソプラスタンを分離する方法と、分離した8−イソプラスタンを精製する方法とについて説明した。しかしながら、生体試料から分離した8−イソプラスタンを精製せずにステップS101〜S103を実行してもよい。また、化学的に合成した8−イソプラスタン、ステップS201以外の方法で生体試料から分離された8−イソプラスタンを用いてステップS202の8−イソプラスタンの精製を行い、ステップS101〜S103を実行してもよい。
【0058】
また、第2の実施形態では、尿中の8−イソプラスタンを精製する場合を例に説明したが、血清中の8−イソプラスタンを精製する場合は、8−イソプラスタンの正常値が尿中の100分の1程度であるため、定量前に、濃縮操作を行ってもよい。たとえば、血清サンプルを50℃のヒートブロックにセットし、窒素気流下で溶媒を除去するなど、100倍の濃縮操作を追加する。こうすることで、ステップS101〜103を実行し、蛍光ラベル化された8−イソプラスタンをHPLC分析するとき、血清中の8−イソプラスタンのピークを明瞭に観察することができる。
【実施例】
【0059】
実施例1
8−イソプラスタンの標準品(Cayman社製)2ngを8%(v/v)エタノール含有10mMリン酸緩衝液(pH7.0)0.2mLに溶解して10ng/mL(30pmol/mL)8−イソプラスタン溶液を調製した。これを150μLとり、75μLの試薬I(8−イソプラスタンに対して5×104倍モル)と、60μLの試薬II(DMEQ−Hに対して500倍モル)と、15μLの試薬III(8−イソプラスタンに対して2.5×105倍モル)とに混合し、70℃で20分間反応させた。その後、10分間流水で冷却した。Varian製のSCX(イオン交換容量:0.8meq/g、充てん剤量0.25g、カラムサイズ4mL、カラム形状注射筒型、ポリプロピレン)をエタノール及び純水により順にコンディショニングして準備し、冷却した反応液を導入した。8%(v/v)エタノール含有10mMリン酸緩衝液(pH7.0)0.2mLで洗浄した後、8%(v/v)エタノール含有10mMリン酸緩衝液(pH7.0)0.2mLを通液して得られるフラクションを8−イソプラスタンの分析サンプルとして回収した。回収した分析サンプルは、そのままHPLCに注入した。
<試薬>
・試薬I:10mM 1,2,3,4‐テトラヒドロ‐6,7‐ジメトキシ‐1‐メチル‐2(1H)‐オキソキノキサリン‐3‐プロピオン酸ヒドラジド(DMEQ−H)/ジメチルホルムアミド
・試薬II:10%(v/v)ピリジン/20mM塩酸含有エタノール
・試薬III:0.5M 1−エチル−3−(3−ジメチルアミノプロピル)カルボジミド塩酸塩/精製水
<HPLC条件>
流速 1.2mL/分
カラムオーブン 50℃
分析カラム Divelosil ODS MG5(250mm、φ4.6mm)
注入量 10μL
蛍光検出器 Ex:367nm、Em:445nm
移動相 メタノール:25mM 酢酸/酢酸ナトリウム(pH4.5)=55:45(アイソクラティック)
【0060】
実施例2
逆相担体として、YMC社製ODS−AQの粒径50μm及び20μmを、50μm:20μm=90:10の重量比で、均質になるように混合して、逆相カラム充填材を調製した。粒径50μmは沈降法、粒径20μmはコールター法により測定した。調製した逆相カラム充填材は、フロー式粒子像分析装置(シスメックス社製FPIA−3000)で測定される円相当径0.5〜10μmの粒子径を粒子数として10累積%含み、上記装置で測定される円相当径20〜100μmの粒子を粒子数として90累積%含んでいた。この充填材を800mg充填した逆相カラムを準備し、エタノール、水の順で通液し、コンディショニングを行った。この逆相カラムに、1.4mLの尿(産業技術総合研究所ボランティア(ヒト)、産業技術総合研究所倫理審査委員会承認番号15000−A−20081215−001)と、0.6mLの80mMリン酸緩衝液(pH7.0、4mM EDTA(エチレンジアミン四酢酸)含有)とを混ぜ合わせた合計2mLのサンプルを導入した。そして、洗浄液として、下記(i)、(ii)、(iii)を用い、順に洗浄した。
(i)2%(v/v)エタノール含有10mMリン酸緩衝液(pH7.0):6.0mL
(ii)30%(v/v)エタノール含有10mMリン酸緩衝液(pH7.0):4.0mL
(iii)50%(v/v)エタノール含有10mMリン酸緩衝液(pH7.0):0.8mL
その後、50%(v/v)エタノール含有10mMリン酸緩衝液(pH7.0)0.7mLを通液して得られるフラクションを8−イソプラスタンを含有するフラクションとして回収した。ついで、イオン交換担体として、Varian製のSAX(イオン交換容量:0.8meq/g、充てん剤量0.5g、カラムサイズ3mL、カラム形状注射筒型、ポリプロピレン)を準備し、逆相担体から回収した8−イソプラスタンを含有するフラクションをコンディショニングしたSAXに導入した。SAXのコンディショニンングは、エタノール及び純水を順に通液して行った。8−イソプラスタンが導入されたSAXは、0.4mLの50%(v/v)エタノール含有10mMリン酸緩衝液(pH7.0)で洗浄した後、50%(v/v)エタノール含有10mMリン酸緩衝液(pH7.0)0.8mLを通液して得られるフラクションを8−イソプラスタンを含有するフラクションとして回収した。このフラクションを150μLとり、75μLの試薬I、60μLの試薬II、15μLの試薬IIIを加えて、実施例1と同様に、SCXによる処理を経てHPLC分析を行った。
【0061】
実施例3
実施例2において、異なる検体から得られた尿サンプルを用いた以外は、同様にして行った。
【0062】
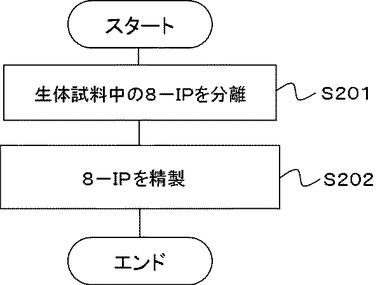
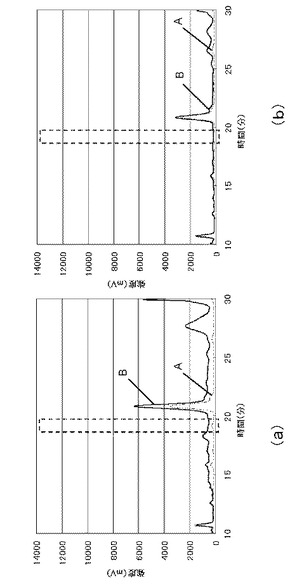
実施例1、2で得られたHPLCチャートを図3に示す。図3中、A(破線)が実施例1、B(実線)が実施例2である。実施例2は2回行い、図3(a)のBは、1回目に行った実施例1のチャート図を示し、図3(b)のBは、2回目に行った実施例1のチャート図を示した。また、実施例1、3で得られたHPLCチャートを図4に示す。A(破線)が実施例1であり、C(実線)が実施例3である。実施例3も2回行い、図4(a)のCは、1回目に行った実施例3のチャート図を示し、図4(b)のCは、2回目に行った実施例3のチャート図を示した。図3、4中、8−イソプラスタンの保持時間は約19分である。なお、実施例1のサンプルは、実施例2又は3のサンプルと同時に注入し、保持時間約19分のピークが重なることを確認した。
【0063】
実施例2について、尿の代わりに、8−イソプラスタン溶液を1、2、5、10、20、50ng/mLにふって調製したものを用い、それぞれについて同様な操作を行って検量線を作成した。なお、8−イソプラスタン溶液は、8−イソプラスタンの標準品(Cayman社製)を8%(v/v)エタノール含有10mMリン酸緩衝液に溶解して調製した。また、10、50ng/mLの8−イソプラスタン溶液については、それぞれn=10の平均値を用い、1、2、5、20ng/mLの8−イソプラスタン溶液については、それぞれn=5の平均値を用いて検量線を作成した。変動係数は、2.8〜8.5%であった。また、検量線の相関係数は、R2=0.9984であった。
なお、実施例1について、8−イソプラスタン溶液の調製を2、4、10、20、40、100ng/mLにして行っても、同様な検量線が得られることも確認した。
実施例2、3の分析結果、及び、得られた検量線から、この結果から、実施例2で使用した尿に含まれる8−イソプラスタンの量は、15pmol/mlであり、実施例3で使用した尿に含まれる8−イソプラスタンの量は、11pmol/mlであることが求められた。
【技術分野】
【0001】
本発明は、8−イソプラスタンの定量方法に関する。
【背景技術】
【0002】
8−イソプラスタンは、細胞膜やリポ蛋白に含まれるリン脂質がフリーラジカルで酸化されることによって生じるプロスタグランジン様の化合物である。尿や血清などの生体試料中に含まれる8−イソプラスタンを測定することにより、生体内における酸化ストレスを非浸襲的に評価することができ、免疫測定により8−イソプラスタンを測定するためのキットも市販されている。
【0003】
特許文献1には、8−イソプラスタンの免疫測定に関する技術が記載されている。
【0004】
特許文献2には、LC−MS/MS法による8−イソプラスタンの分析に関する技術が記載されている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2004−157119号公報
【特許文献2】国際公開第2008/065895号パンフレット
【特許文献3】特開平3−291272号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかしながら、従来の8−イソプラスタンの分析方法では、定量するための精度が極めて低かった。そこで、発明者が鋭意検討したところ、特許文献3記載のキノキサリン誘導体で8−イソプラスタンを蛍光ラベル化することで、定量分析が可能になることが期待された。しかしながら、特許文献3の技術では、尿などの生体試料等に含まれる微量な8−イソプラスタンを収率よくラベル化し、定量分析の精度を向上させるには、未だ改善の余地があった。
【0007】
本発明は上記事情に鑑みてなされたものであり、その目的とするところは、精度よく8−イソプラスタンを定量できる技術を提供するものである。
【課題を解決するための手段】
【0008】
本発明によれば、
下記一般式(1)で表されるキノキサリノン誘導体を、8−イソプラスタンの物質量に対して過剰に用いて8−イソプラスタンを蛍光ラベル化するステップと、
蛍光ラベル化された8−イソプラスタンと、未反応のキノキサリノン誘導体とを含む反応混合物を、スルホン酸又はスルホン酸塩が固定化された陽イオン交換担体に接触させて、蛍光ラベル化された8−イソプラスタンと前記未反応のキノキサリノン誘導体とを分離するステップと、
前記未反応のキノキサリノン誘導体と分離された、蛍光ラベル化された8−イソプラスタンを定量するステップと、
を含む、8−イソプラスタンの定量方法が提供される。
【0009】
【化1】
【0010】
〔式(1)中、R1、R2及びR3は、炭素数1〜6のアルキル基であり、nは、0〜6の整数である。〕
【0011】
また、本発明によれば、上記の8−イソプラスタンの定量方法に用いられる、8−イソプラスタンを定量するためのキットが提供される。
【0012】
この発明によれば、キノキサリノン誘導体を用いて8−イソプラスタンを蛍光ラベル化し、ラベル化後に陽イオン交換担体を用いて蛍光ラベル化された8−イソプラスタンとキノキサリノン誘導体とを分離するため、微量の8−イソプラスタンを用いた場合であっても、過剰のキノキサリノン誘導体を用いて収率よくラベル化させ、かつ、過剰のキノキサリン誘導体を陽イオン交換担体に吸着させて除去することができる。したがって、蛍光ラベル化された8−イソプラスタンを定量することで、精度よく8−イソプラスタンを定量分析することが可能になる。
【発明の効果】
【0013】
本発明によれば、精度よく8−イソプラスタンを定量することができる。
【図面の簡単な説明】
【0014】
【図1】第1の実施形態に係る8−イソプラスタンの定量方法を示すフローチャートである。
【図2】第2の実施形態に係る8−イソプラスタンの定量方法を示すフローチャートである。
【図3】実施例1、2で得られた高速液体クロマトグラフィーのチャート図である。
【図4】実施例1、3で得られた高速液体クロマトグラフィーのチャート図である。
【発明を実施するための形態】
【0015】
以下、本発明の実施の形態について、図面を用いて説明する。尚、すべての図面において、同様な構成要素には同様の符号を付し、適宜説明を省略する。
【0016】
(第1の実施形態)
図1は、本実施の形態の8−イソプラスタン(8−IP)の定量方法を示すフローチャートである。本実施の形態は、キノキサリノン誘導体として1,2,3,4‐テトラヒドロ‐6,7‐ジメトキシ‐1‐メチル‐2(1H)‐オキソキノキサリン‐3‐プロピオン酸ヒドラジド(DMEQ−H、(上記式(1)において、R1、R2,R3がいずれもメチルであり、nが2である化合物))を、8−IPの物質量に対して過剰に用いて8−IPを蛍光ラベル化するステップ(S101)と、蛍光ラベル化された8−イソプラスタン(DMEQ−IP)と、未反応のDMEQ−Hとを含む反応混合物を、スルホン酸又はスルホン塩が固定化された陽イオン交換担体に接触させて、DMEQ−IPとDMEQ−Hとを分離するステップ(S102)と、未反応のDMEQ−Hと分離された、DMEQ−IPを定量するステップ(S103)とを含む。以下、各ステップについて詳細に説明する。
【0017】
[S101:8−IPを蛍光ラベル化するステップ]
ステップS101では、8−IPのカルボキシル基とDMEQのヒドラジド基とを反応させてアミド結合を形成し、DMEQ−IPを合成する。反応条件は特に制限されないが、溶媒中、塩基、及び、カルボン酸を活性化するための活性化剤存在下に反応させることが好ましい。溶媒としては、水、アルコール、非プロトン性極性溶媒を用いることが好ましく、ジメチルホルムアミドを用いると、より好ましい。塩基としては、脂肪族三級アミンや含窒素複素環化合物が好ましく、ピリジンがより好ましい。活性化剤としては、カルボジイミドが好ましく、ジシクロヘキシルカルボジイミド(DCC)、ジイソプロピルカルボジイミド(DIC)、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩(EDC)がより好ましく、EDCが特に好ましい。
【0018】
溶媒中で反応を行う場合、ラベル化させる8−IPの濃度は特に限定されないが、例えば、0.01〜0、5nmol/mLとすることができる。
【0019】
DMEQは、8−IPに対して過剰に用いることが好ましく、8−IPに対して、2×103倍モル以上用いることが好ましく、5×103倍モル以上用いることがより好ましく、1×105倍モル以上用いることが特に好ましい。上限は特にないが、2×105倍モル以下とすることができる。
【0020】
塩基(ピリジンなど)を用いる場合は、DMEQに対して、50〜1000倍モル用いることが好ましく、100〜500倍モル用いることがより好ましい。
【0021】
活性化剤(水溶性カルボジイミドなど)を用いる場合は、8−IPに対して過剰に用いることが好ましく、8−IPに対して、1×102〜5×105倍モル用いることが好ましく、2×103〜5×104倍モル用いることがより好ましい。
【0022】
反応温度は、室温(15〜30℃)以上であればよいが、反応効率を上げるため、50〜90℃加熱して行うことが好ましく、60〜80℃がより好ましい。反応時間は、制限されないが、例えば、10〜60分とすることができる。
【0023】
上記反応条件下における8−IPのラベル化収率は、50〜100%とすることができる。
【0024】
[S102:DMEQ−IPとDMEQ−Hとを分離するステップ]
ステップS102で用いる陽イオン交換担体は、スルホン酸又はスルホン酸塩が固定化されたものであればよいが、下記式(2)で表されるアルキルベンゼンスルホン酸又はその塩が固定化されたものが好ましい。
【0025】
【化2】
【0026】
〔式(2)中、mは、1〜6であり、B1は、樹脂基材又はシリカゲルである。〕
【0027】
また、式(2)で表される陽イオン交換担体としては、mが3のプロピルベンゼンスルホン酸又はその塩が固定化されたものがより好ましい。また、式(2)中、B1は、例えば、スチレンとジビニルベンゼンの共重合体等からなる樹脂基材又はシリカゲルであるが、シリカゲルがより好ましい。使用する陽イオン交換担体の量は、ステップS101において使用したDMEQに対して、スルホン酸又はスルホン酸塩の含量(eq)を1〜1000倍にすることが好ましい。
【0028】
DMEQ−IPとDMEQ−Hとを含む液体と、陽イオン交換担体との接触方法は特に限られず、バッチ法でもカラム法でもよいが、カラム法が好適である。陽イオン交換担体としてスルホン酸塩を用いる場合は、スルホン酸塩の対イオンをナトリウムイオンにしておくことが好ましい。水酸化ナトリウム水溶液や食塩水等を陽イオン交換担体に接触させることで、スルホン酸塩の対イオンをナトリウムイオンにすることができる。陽イオン交換担体は、充分な水及びアルコール、所定の緩衝液で平衡化しておくことが好ましい。緩衝液として、たとえばpH5.5〜8.5、好ましくは6.0〜8.0、より好ましくは6.5〜7.5に調節された緩衝液が挙げられる。本明細書において、緩衝液は、所定の酸を水に溶解したものであり、その種類は、pHによって適宜選択することができる。例えば、pH5.5〜5.6の範囲では酢酸緩衝液、pH5.5〜6.2の範囲ではクエン酸緩衝液、pH5.5〜7.0の範囲では、クエン酸−リン酸緩衝液、pH5.5〜8.5の範囲では、リン酸緩衝液、pH7.2〜8.5の範囲では、トリスリン酸緩衝液を用いることができる。また、この緩衝液は、0〜100体積%、好ましくは10〜80体積%の水溶性有機溶媒(特にエタノール)を含有することが好ましい。なお、本明細書において、pHは、25℃で測定したものをいう。
【0029】
カラム法の場合、コンディショニングされた陽イオン交換担体にDMEQ−IPと未反応のDMEQ−Hとを含む反応混合物を通液し、DMEQ−IP及びDMEQ−Hを陽イオン交換担体に保持させる。このとき、反応混合物は、エタノール等の水溶性有機溶媒、緩衝液又はこれらの混合液で反応混合物を希釈されていてもよい。
【0030】
その後、洗浄液によって、陽イオン交換担体に捕捉されていないアニオン物質や非イオン物質を洗い流す。洗浄液としては、水溶性有機溶媒と水とを主成分とするものが好ましく、水溶性有機溶媒としては、エタノール、アセトニトリル、メタノール等を用いることができ、その濃度は、洗浄液中、10〜100体積%、好ましくは、30〜100体積%とすることができる。洗浄液は、緩衝液を含んでいることが好ましく、緩衝液のpHは、例えば、5.5〜8.5、好ましくは6.0〜8.0、より好ましくは6.5〜7.5とすることができる。エタノールとリン酸緩衝液との混合液がより好ましく、洗浄液中、0〜100体積%、好ましくは30〜100体積%、より好ましくは50〜100体積%のエタノールを含有することがより好ましい。洗浄液の量は、0.1〜100mL、好ましくは0.1〜50mL、より好ましくは0.1〜20mLとすることができる。
【0031】
洗浄後、水溶性有機溶媒と水とを主成分とする溶出液を用いて、陽イオン交換担体に保持したDMEQ−IPを陽イオン交換担体から溶離する(S103)。溶出液は、エタノール、アセトニトリル、メタノール等の水溶性有機溶媒を含有することができ、その濃度は、溶出液中、10〜100体積%、好ましくは、30〜100体積%とすることができる。溶出液は、緩衝液を含むことが好ましく、緩衝液のpHは、例えば、5.5〜8.5、好ましくは6.0〜8.0、より好ましくは6.5〜7.5とする。また、エタノールとリン酸緩衝液との混合液がより好ましく、溶出液中、0〜100体積%、好ましくは30〜100体積%、より好ましくは50〜100体積%のエタノールを含有することがより好ましい。溶出液は、0.1〜10mLを通液させ、最初に通液された溶出液を約0.2〜0.3mLまで回収する。なお、溶出液量及び回収液量は、使用する担体の大きさ、サンプル量等に応じて、増減可能である。このようにして、DMEQ−IPを得る。DMEQ−Hは、陽イオン交換樹脂への吸着性が強いため、回収された液体中には、DMEQ−IPが含まれるが、DMEQ−Hを含んでいない。このようにして、DMEQ−IPとDMEQ−Hとを分離することができる。
【0032】
[S103:DMEQ−IPを定量するステップ]
ステップS102で得られたDMEQ−IPを用い、高速液体クロマトグラフィー(HPLC)法によりDMEQ−IPを定量する。S102において陽イオン交換樹脂から回収された回収液中の溶媒は、HPLCを行う前に適宜除去されてもよい。このHPLC法では、逆相カラムを用いた逆相カラムクロマトグラフィー法を用いることができる。また、カラムは、加熱することが好ましく、40〜60℃に加熱することが好ましい。移動相は、アルコールと緩衝液との混合液が好ましく、具体的には、メタノールと酢酸緩衝液との混合液が好ましい。移動相のpHは、弱酸性が好ましく、pH4〜6がより好ましい。アイソクラティックでも勾配をかけてもよいが、定量の精度を向上させるという観点から、アイソクラティックが好ましい。蛍光励起スペクトルは、最大367nm、蛍光発光スペクトルは、最大445nmが好ましい。DMEQ−IPの蛍光強度を測定することで、HPLC装置に注入したDMEQ−IPを定量することができる。
【0033】
DMEQ−IPの蛍光強度を定量の精度は、変動係数を20%以下にすることができる。また、1〜1000pmolの範囲であれば、ピークの高さとDMEQ−IPの量とが比例関係にあり、相関係数を0.99以上にすることができる。
【0034】
このようにして、求められたDMEQ−IPの量に基づきS101において使用した8−イソプラスタンの量を求めることができる。S103において得られたDMEQ−IPの物質量を、そのままラベル化に使用した8−IPの物質量としてもよいし、ラベル化収率を考慮して、使用した8−IPの物質量を算出してもよい。こうした本実施形態の8−IPの定量方法によれば、DMEQを用いて8−イソプラスタンを蛍光ラベル化し、ラベル化後に陽イオン交換担体を用いてDMEQ−IPとDMEQとを分離するため、100pmol以下、さらには、50pmol以下の微量の8−イソプラスタンを用いた場合であっても、過剰のDMEQを用いて収率よくラベル化させ、かつ、過剰のDMEQを陽イオン交換担体に吸着させて除去することができる。したがって、DMEQ−IPを定量することで、精度よく8−イソプラスタンを定量分析することが可能になる。
【0035】
上記本実施の形態に係る8−イソプラスタンを定量する方法に用いられる、8−イソプラスタンを定量するためのキットは、DMEQ−Hと、スルホン酸が固定化された陽イオン交換担体と、所定の洗浄液及び溶出液とを含むものであればよいが、DMEQ−Hは、ジメチルホルムアミド溶液であることが好ましく、陽イオン交換担体には、プロピルベンゼンスルホン酸又はその塩がシリカゲルに固定化された陽イオン交換担体であることが好ましく、洗浄液及び溶出液としては、エタノールとリン酸緩衝液との混合液であることが好ましい。この溶出液は、エタノールとリン酸緩衝液とを別々に包装し、ユーザにおいて調製する形態であってもよい。また、ピリジン等の塩基と、EDC等のカルボジイミド又はこれらの溶液を含んでいるとさらに好ましい。なお、こうしたキットには、本実施の形態に係る8−イソプラスタンの定量方法が記載された使用説明書が添付されている。
【0036】
(第2の実施形態)
図2は、本実施の形態の8−イソプラスタン(8−IP)の定量方法の一部を示すフローチャートである。本実施の形態は、生体試料中の8−IPの定量方法である。本実施の形態では、生体試料中の8−IPを分離するステップ(S201)と、分離された8−IPを陰イオン交換担体を用いて精製するステップ(S202)とを含み、分離・精製された8−IPを用いて、第1の実施の形態で説明したステップS101、S102、S103を実行する。本実施の形態では、第1の実施の形態と異なる点のみを説明する。
【0037】
[S201:生体試料中の8−IPを分離するステップ]
S201において用意される生体試料としては、尿、血液、唾液等の体液が挙げられるが、以下、試料が尿である場合を例に説明する。採取する尿の量は、測定をより確実に行う観点から、たとえば0.5〜50mL、好適には1.0〜10mL、より好適には1.5〜5.0mLとする。採取された原尿そのものを使うことが可能である。また、尿を所定の緩衝液又は水で希釈して用いてもよい。このとき、他の添加剤、例えば、EDTA(エチレンジアミン四酢酸)等のキレート剤を加えても良い。尿は採取後直ちに濃縮されることが好ましいが、数時間から数日後であってもよい。
【0038】
S201において用いる逆相担体は、官能基として炭素数1〜30の直鎖の炭化水素基をもつ材料により構成されていることが好ましく、生体試料中の8−イソプラスタンをより効率的に処理する観点から、炭素数8〜22の直鎖の炭化水素基がより好ましく、炭素数10〜20の直鎖の炭化水素基が特に好ましい。逆相担体の量は、尿の重量に対して0.1〜10倍量とすることができる。
【0039】
逆相担体の材料として、具体的には、オクタデシルシリル(ODS)基を化学結合させたシリカゲルが挙げられる。このとき、オクタデシルシリル(ODS)基を化学結合させたシリカゲルにおけるシリル化剤の結合様式は、シリル化剤がシリカゲル中のシラノール基と1対1で結合している様式、つまりモノメリックな結合様式とするとよい。これにより、逆相系充填剤が過度に疎水性とならないようにすることができる。また、逆相担体は、さらに好適な疎水性を有するという観点から、担体表面の炭素(C)の含有量が、元素比で18%以下が好ましく、15%以下とするとより好ましい。
【0040】
また、逆相担体は、フロー式粒子像分析装置で測定される円相当径0.5〜10μmの粒子を粒子数として1〜20累積%含むことがより好ましい。また、さらに上記装置で測定される円相当径20〜100μmの粒子を粒子数として65〜99累積%含んでいてもよく、より好ましくは、78〜99累積%含んでいてもよい。こうすることで、分子量や性質等の性状が8−IPとわずかに異なる夾雑物が含まれている場合にも、これらを簡便で確実に分離することができる。この理由としては、粒子径が異なる粒子を特定の比率で配合することにより、大きな粒子の間に小さな粒子が入り込み、単位体積あたりの担体の表面積が最大となり、8−IPの保持力が向上することが考えられる。
【0041】
さらに、8−IPの分離を効率よく行う観点から、フロー式粒子像分析装置で測定される円相当径0.5〜10μmの粒子を粒子数として含む割合は、好ましくは4〜19累積%、さらに好ましくは4〜13累積%、特に好ましくは6〜13累積%である。また、上記装置で測定される円相当径20〜100μmの粒子を粒子数として含む割合は、好ましくは68〜95累積%、さらに好ましくは78〜93累積%、特に好ましくは85〜91累積%である。
【0042】
なお、フロー式粒子像分析装置は、測定対象の粒子を含む試料が流れるフローセルにストロボ光等の光を照射して、通過中の粒子の画像を取得し、画像解析により円相当径等の粒子形状を示すパラメータを算出する装置である。実際には隋円形などに崩れた粒子が存在するため、円相当径とは、実際に測定した粒子の粒子投影面積と同じ投影面積を持つ球を想定し、その球の直径と定義している。フロー式粒子像分析装置の具体例としては、シスメックス社製FPIA−3000が挙げられる。
【0043】
また、逆相担体には、沈降法で測定される粒径35〜60μmの粒子と、コールター法で測定される粒径10〜30μmの粒子との重量比が、80:20〜95:5の範囲にある粒子を含んでいても良い。このようにすれば、8−IPの回収率をさらに向上させることができる。粒径35〜60μmの粒子と粒径10〜30μmの粒子との重量比は、8−IPの回収率と分離時間とのバランスを向上させる観点から、好ましくは90:10とする。
【0044】
生体試料は、予め水及びアルコールで十分にコンディショニングされた逆相担体と接触させることが好ましく、エタノール及び純水でコンディショニングされているとより好ましい。また、pH5.5〜8.5の緩衝液を用いてコンディショニングすることもできる。生体試料と逆相担体との接触方法は特に限られず、バッチ法でもカラム法でもよいが、効率よく8−イソプラスタンを濃縮できるという観点から、カラム法が好適である。ステップS201では、逆相担体と移動相との組み合わせとして、例えばオクタデシル(ODS)基を化学結合させたシリカゲルとリン酸緩衝液−エタノールの混合液との組み合わせが用いられる。
【0045】
その後、所定の緩衝液等を洗浄液として逆相担体に流し、逆相担体に捕捉されない物質を洗い流す。S202における洗浄液には、水溶性有機溶媒と水とを主成分とするものを用いることができる。水溶性有機溶媒には、エタノール、アセトニトリル、メタノール等を用いることができ、その濃度は、洗浄液中、10〜100体積%、好ましくは、30〜100体積%とすることができる。例えば、pHは、5.5〜8.5、好ましくは6.0〜8.0、より好ましくは6.5〜7.5に調節された緩衝液を用いることができる。中でも、エタノールとリン酸緩衝液との混合液が好ましく、洗浄液中、0〜70体積%、好ましくは0〜50体積%、より好ましくは0〜30体積%のエタノールを含有することがより好ましい。洗浄液の量は、1〜100mL、好ましくは1〜50mL、より好ましくは1〜20mLとすることができる。また、エタノールの含量を徐々に増加させるとより好ましく、0〜20体積%含有エタノール/リン酸緩衝液で洗浄した後、20〜40体積%含有エタノール/リン酸緩衝液で洗浄し、さらに、40〜60体積%含有エタノール/リン酸緩衝液で洗浄するとより好ましい。
【0046】
洗浄後、溶出液を用いて、逆相担体に保持した8−IPを逆相担体から溶離する。溶出液には、水溶性有機溶媒と水とを主成分とするものを用いることができる。水溶性有機溶媒には、エタノール、アセトニトリル、メタノール等を用いることができ、その濃度は、溶出液中、10〜100体積%、好ましくは、30〜100体積%とすることができる。溶出液は、pHが、例えば、5.5〜8.5、好ましくは6.0〜8.0、より好ましくは6.5〜7.5の緩衝液を含むことが好ましい。また、エタノールとリン酸緩衝液との混合液がより好ましく、溶出液中、10〜100体積%、好ましくは30〜100体積%、より好ましくは50〜100体積%のエタノールを含有することがより好ましい。溶出液は、0.5〜10mLを通液させ、0.7〜1.7mLにかけて溶出する約1mLを回収する。なお、溶出液量及び回収液量は使用する逆相担体の大きさ、サンプル量等に応じて、増減可能である。このようにして、生体試料から8−イソプラスタンを分離することができる。
【0047】
[S202:8−IPを精製するステップ]
ステップS202では、生体試料から分離した8−IPを、陰イオン交換担体を用いて精製する。陰イオン交換担体は、第一又は二級アミンが固定化された弱陰イオン交換担体、芳香族又は脂肪族第四級アンモニウムが固定化された陰イオン交換担体等が用いられるが、第四級アンモニウム塩が固定化された陰イオン交換担体が好ましい。第四級アンモニウム塩としては、式(3)で表されるテトラアルキルアンモニウム塩が好ましい。
【0048】
【化3】
【0049】
〔式(3)中、R4、R5、R6が炭素数1〜6のアルキル基であり、pが1〜5であり、B2が樹脂基材又はシリカゲルであり、X−が塩化物イオンである。〕
【0050】
式(3)で表される陰イオン交換担体としては、中でもR4、R5、R6がメチル基であり、pが3のトリメチルアンモニウムプロピル基が固定化されているとより好ましい。また、式(3)中、B2は、スチレンとジビニルベンゼンの共重合体等からなる樹脂基材又はシリカゲルであるが、シリカゲルがより好ましい。使用する陰イオン交換担体の量は、第四級アンモニウム塩の含量(eq)が、8−IPの物質量に対して、1〜10万倍にすることが好ましいが、例えば、尿中の8−IPを精製する場合、1mLの尿に対して、0.01〜1eqとなる量にすることができる。
【0051】
8−IPと陰イオン交換担体との接触方法は特に限られず、バッチ法でもカラム法でもよいが、カラム法が好適である。陰イオン交換担体として第四級アンモニウム塩を用いる場合、対イオンを塩化物イオンにしておくことが好ましい。塩酸や食塩水等を陰イオン交換担体に接触させることで、第四級アンモニウム塩の対イオンを塩化物イオンにすることができる。その後、充分な水及びアルコール、所定の緩衝液で平衡化しておく。緩衝液として、たとえばpH5.5〜8.5、好ましくは6.0〜8.0、より好ましくは6.5〜7.5に調節された緩衝液が挙げられる。また、この緩衝液は、0〜100体積%、好ましくは10〜80体積%の水溶性有機溶媒(特に、エタノール)を含有することが好ましい。
【0052】
カラム法の場合、コンディショニングされた陰イオン交換担体に液体試料を流し、8−IPを陰イオン交換担体に保持させた後、水溶性有機溶媒と水とを主成分とする洗浄液によって、陰イオン交換担体に捕捉されていないカチオン物質や非イオン物質を洗い流す。洗浄液として、水溶性有機溶媒と水とを主成分とするものを用いることができる。水溶性有機溶媒としては、エタノール、アセトニトリル、メタノール等を用いることができ、その濃度は、洗浄液中、10〜100体積%、好ましくは、30〜100体積%とすることができる。また、洗浄液は、緩衝液を含むことが好ましく、pHが、例えば、5.5〜8.5、好ましくは6.0〜8.0、より好ましくは6.5〜7.5の緩衝液を用いることができる。また、エタノールとリン酸緩衝液との混合液がより好ましく、洗浄液中、0〜100体積%、好ましくは30〜100体積%のエタノールを含有することがより好ましい。洗浄液の量は、1〜100mL、好ましくは1〜50mL、より好ましくは1〜20mLとすることができる。
【0053】
洗浄後、溶出液を用いて、陰イオン交換担体に保持した8−IPをイオン交換担体から溶離する。この溶出液は、水溶性有機溶媒と水とを主成分とするものが好ましい。水溶性有機溶媒は、エタノール、アセトニトリル、メタノール等を用いることができ、その濃度は、溶出液中、10〜100体積%、好ましくは、30〜100体積%とすることができる。溶出液は、pHが、例えば、5.5〜8.5、好ましくは6.0〜8.0、より好ましくは6.5〜7.5の緩衝液を含むことが好ましい。中でも、エタノールとリン酸緩衝液との混合液がより好ましく、溶出液中、0〜100体積%、好ましくは30〜100体積%、より好ましくは50〜100体積%のエタノールを含有することがより好ましい。溶出液は、0.1〜10mLを通液させ、1.5〜2.5mLにかけて溶出する約1mLを回収する。なお、溶出液量及び回収液量は、使用する担体の大きさ、サンプル量等に応じて、増減可能である。このようにして、8−IPの精製をすることができる。
【0054】
回収された液体中には、8−IPが濃縮されており、これを8−IPのラベル化(S101)に供することができる。なお、8−IPの分析前に回収液中の溶媒を、適宜除去してもよい。
【0055】
上記本実施の形態に係る8−IPの定量方法に用いられる、生体試料中に含まれる8−IPを定量するためのキットは、第一の実施形態で説明したものに加え、逆相担体と、水溶性有機溶媒と水とを主成分とする溶出液とをさらに備えるものとすることができる。この溶出液は、エタノールとリン酸緩衝液との混合液が好ましい。この溶出液もまた、エタノールとリン酸緩衝液とを別々に包装し、ユーザにおいて調製する形態であってもよい。また、第四級アンモニウム塩が固定化された陰イオン交換担体と、洗浄液と溶出液とをさらに含んでいてもよく、陰イオン交換担体として、トリメチルアンモニウムプロピル基がシリカゲルに固定化されたイオン交換担体、洗浄液及び溶出液として、エタノールとリン酸緩衝液との混合液が好ましい。この溶出液もまた、エタノールとリン酸緩衝液とを別々に包装し、ユーザにおいて調製する形態であってもよい。なお、こうしたキットには、本実施の形態に係る8−イソプラスタンを精製する方法が記載された使用説明書が添付されている。
【0056】
以上、図面を参照して本発明の実施形態について述べたが、これらは本発明の例示であり、上記以外の様々な構成を採用することもできる。
たとえば、上記の実施形態では、キノキサリノン誘導体として、DMEQを用いる例をあげて説明したが、式(1)で表されるキノキサリノン誘導体であれば、本発明の実施形態に記載された技術を用いることができる。例えば、1,2,3,4‐テトラヒドロ‐6,7‐ジメトキシ‐1‐メチル‐2(1H)‐オキソキノキサリン‐3‐カルボキシヒドラジド(式(1)において、R1、R2,R3がいずれもメチルであり、nが0である化合物)を用いることもできる。
【0057】
また、第2の実施形態では、生体試料から8−イソプラスタンを分離する方法と、分離した8−イソプラスタンを精製する方法とについて説明した。しかしながら、生体試料から分離した8−イソプラスタンを精製せずにステップS101〜S103を実行してもよい。また、化学的に合成した8−イソプラスタン、ステップS201以外の方法で生体試料から分離された8−イソプラスタンを用いてステップS202の8−イソプラスタンの精製を行い、ステップS101〜S103を実行してもよい。
【0058】
また、第2の実施形態では、尿中の8−イソプラスタンを精製する場合を例に説明したが、血清中の8−イソプラスタンを精製する場合は、8−イソプラスタンの正常値が尿中の100分の1程度であるため、定量前に、濃縮操作を行ってもよい。たとえば、血清サンプルを50℃のヒートブロックにセットし、窒素気流下で溶媒を除去するなど、100倍の濃縮操作を追加する。こうすることで、ステップS101〜103を実行し、蛍光ラベル化された8−イソプラスタンをHPLC分析するとき、血清中の8−イソプラスタンのピークを明瞭に観察することができる。
【実施例】
【0059】
実施例1
8−イソプラスタンの標準品(Cayman社製)2ngを8%(v/v)エタノール含有10mMリン酸緩衝液(pH7.0)0.2mLに溶解して10ng/mL(30pmol/mL)8−イソプラスタン溶液を調製した。これを150μLとり、75μLの試薬I(8−イソプラスタンに対して5×104倍モル)と、60μLの試薬II(DMEQ−Hに対して500倍モル)と、15μLの試薬III(8−イソプラスタンに対して2.5×105倍モル)とに混合し、70℃で20分間反応させた。その後、10分間流水で冷却した。Varian製のSCX(イオン交換容量:0.8meq/g、充てん剤量0.25g、カラムサイズ4mL、カラム形状注射筒型、ポリプロピレン)をエタノール及び純水により順にコンディショニングして準備し、冷却した反応液を導入した。8%(v/v)エタノール含有10mMリン酸緩衝液(pH7.0)0.2mLで洗浄した後、8%(v/v)エタノール含有10mMリン酸緩衝液(pH7.0)0.2mLを通液して得られるフラクションを8−イソプラスタンの分析サンプルとして回収した。回収した分析サンプルは、そのままHPLCに注入した。
<試薬>
・試薬I:10mM 1,2,3,4‐テトラヒドロ‐6,7‐ジメトキシ‐1‐メチル‐2(1H)‐オキソキノキサリン‐3‐プロピオン酸ヒドラジド(DMEQ−H)/ジメチルホルムアミド
・試薬II:10%(v/v)ピリジン/20mM塩酸含有エタノール
・試薬III:0.5M 1−エチル−3−(3−ジメチルアミノプロピル)カルボジミド塩酸塩/精製水
<HPLC条件>
流速 1.2mL/分
カラムオーブン 50℃
分析カラム Divelosil ODS MG5(250mm、φ4.6mm)
注入量 10μL
蛍光検出器 Ex:367nm、Em:445nm
移動相 メタノール:25mM 酢酸/酢酸ナトリウム(pH4.5)=55:45(アイソクラティック)
【0060】
実施例2
逆相担体として、YMC社製ODS−AQの粒径50μm及び20μmを、50μm:20μm=90:10の重量比で、均質になるように混合して、逆相カラム充填材を調製した。粒径50μmは沈降法、粒径20μmはコールター法により測定した。調製した逆相カラム充填材は、フロー式粒子像分析装置(シスメックス社製FPIA−3000)で測定される円相当径0.5〜10μmの粒子径を粒子数として10累積%含み、上記装置で測定される円相当径20〜100μmの粒子を粒子数として90累積%含んでいた。この充填材を800mg充填した逆相カラムを準備し、エタノール、水の順で通液し、コンディショニングを行った。この逆相カラムに、1.4mLの尿(産業技術総合研究所ボランティア(ヒト)、産業技術総合研究所倫理審査委員会承認番号15000−A−20081215−001)と、0.6mLの80mMリン酸緩衝液(pH7.0、4mM EDTA(エチレンジアミン四酢酸)含有)とを混ぜ合わせた合計2mLのサンプルを導入した。そして、洗浄液として、下記(i)、(ii)、(iii)を用い、順に洗浄した。
(i)2%(v/v)エタノール含有10mMリン酸緩衝液(pH7.0):6.0mL
(ii)30%(v/v)エタノール含有10mMリン酸緩衝液(pH7.0):4.0mL
(iii)50%(v/v)エタノール含有10mMリン酸緩衝液(pH7.0):0.8mL
その後、50%(v/v)エタノール含有10mMリン酸緩衝液(pH7.0)0.7mLを通液して得られるフラクションを8−イソプラスタンを含有するフラクションとして回収した。ついで、イオン交換担体として、Varian製のSAX(イオン交換容量:0.8meq/g、充てん剤量0.5g、カラムサイズ3mL、カラム形状注射筒型、ポリプロピレン)を準備し、逆相担体から回収した8−イソプラスタンを含有するフラクションをコンディショニングしたSAXに導入した。SAXのコンディショニンングは、エタノール及び純水を順に通液して行った。8−イソプラスタンが導入されたSAXは、0.4mLの50%(v/v)エタノール含有10mMリン酸緩衝液(pH7.0)で洗浄した後、50%(v/v)エタノール含有10mMリン酸緩衝液(pH7.0)0.8mLを通液して得られるフラクションを8−イソプラスタンを含有するフラクションとして回収した。このフラクションを150μLとり、75μLの試薬I、60μLの試薬II、15μLの試薬IIIを加えて、実施例1と同様に、SCXによる処理を経てHPLC分析を行った。
【0061】
実施例3
実施例2において、異なる検体から得られた尿サンプルを用いた以外は、同様にして行った。
【0062】
実施例1、2で得られたHPLCチャートを図3に示す。図3中、A(破線)が実施例1、B(実線)が実施例2である。実施例2は2回行い、図3(a)のBは、1回目に行った実施例1のチャート図を示し、図3(b)のBは、2回目に行った実施例1のチャート図を示した。また、実施例1、3で得られたHPLCチャートを図4に示す。A(破線)が実施例1であり、C(実線)が実施例3である。実施例3も2回行い、図4(a)のCは、1回目に行った実施例3のチャート図を示し、図4(b)のCは、2回目に行った実施例3のチャート図を示した。図3、4中、8−イソプラスタンの保持時間は約19分である。なお、実施例1のサンプルは、実施例2又は3のサンプルと同時に注入し、保持時間約19分のピークが重なることを確認した。
【0063】
実施例2について、尿の代わりに、8−イソプラスタン溶液を1、2、5、10、20、50ng/mLにふって調製したものを用い、それぞれについて同様な操作を行って検量線を作成した。なお、8−イソプラスタン溶液は、8−イソプラスタンの標準品(Cayman社製)を8%(v/v)エタノール含有10mMリン酸緩衝液に溶解して調製した。また、10、50ng/mLの8−イソプラスタン溶液については、それぞれn=10の平均値を用い、1、2、5、20ng/mLの8−イソプラスタン溶液については、それぞれn=5の平均値を用いて検量線を作成した。変動係数は、2.8〜8.5%であった。また、検量線の相関係数は、R2=0.9984であった。
なお、実施例1について、8−イソプラスタン溶液の調製を2、4、10、20、40、100ng/mLにして行っても、同様な検量線が得られることも確認した。
実施例2、3の分析結果、及び、得られた検量線から、この結果から、実施例2で使用した尿に含まれる8−イソプラスタンの量は、15pmol/mlであり、実施例3で使用した尿に含まれる8−イソプラスタンの量は、11pmol/mlであることが求められた。
【特許請求の範囲】
【請求項1】
下記一般式(1):
【化1】
〔式(1)中、R1、R2及びR3は、炭素数1〜6のアルキル基であり、nは、0〜6の整数である。〕
で表されるキノキサリノン誘導体を、8−イソプラスタンの物質量に対して過剰に用いて8−イソプラスタンを蛍光ラベル化するステップと、
蛍光ラベル化された8−イソプラスタンと、未反応のキノキサリノン誘導体とを含む反応混合物を、スルホン酸又はスルホン酸塩が固定化された陽イオン交換担体に接触させて、蛍光ラベル化された8−イソプラスタンと前記未反応のキノキサリノン誘導体とを分離するステップと、
前記未反応のキノキサリノン誘導体と分離された、蛍光ラベル化された8−イソプラスタンを定量するステップと、
を含む、8−イソプラスタンの定量方法。
【請求項2】
前記キノキサリノン誘導体が1,2,3,4‐テトラヒドロ‐6,7‐ジメトキシ‐1‐メチル‐2(1H)‐オキソキノキサリン‐3‐プロピオン酸ヒドラジドである、請求項1に記載の8−イソプラスタンの定量方法。
【請求項3】
8−イソプラスタンを蛍光ラベル化する前記ステップにおいて、8−イソプラスタンに対して5×103倍モル以上の前記キノキサリノン誘導体を用いて、8−イソプラスタンを蛍光ラベル化する、請求項1又は2に記載の8−イソプラスタンの定量方法。
【請求項4】
8−イソプラスタンを蛍光ラベル化する前記ステップにおいて、50〜90℃に加熱して前記キノキサリノン誘導体と8−イソプラスタンとを反応させることにより蛍光ラベル化する、請求項1乃至3いずれか1項に記載の8−イソプラスタンの定量方法。
【請求項5】
蛍光ラベル化された8−イソプラスタンを定量する前記ステップにおいて、逆相カラムを用いたアイソクラティックによる高速液体クロマトグラフィー法を用いて蛍光ラベル化された8−イソプラスタンを分析する、請求項1乃至4いずれか1項に記載の8−イソプラスタンの定量方法。
【請求項6】
前記陽イオン交換担体が、下記式(2)で表されるアルキルベンゼンスルホン酸又はその塩が固定化されたものである、請求項1乃至5いずれか1項に記載の8−イソプラスタンの定量方法。
【化2】
〔式(2)中、mは、1〜6であり、B1は、樹脂基材又はシリカゲルである。〕
【請求項7】
生体試料中の8−イソプラスタンを分離するステップをさらに含み、分離された8−イソプラスタンを用いて、蛍光ラベル化する前記ステップを実行する、請求項1乃至6いずれか1項に記載の8−イソプラスタンの定量方法。
【請求項8】
前記生体試料中の8−イソプラスタンを分離する前記ステップにおいて、前記生体試料と逆相担体とを接触させて、前記生体試料から8−イソプラスタンを分離する、請求項7に記載の8−イソプラスタンの定量方法。
【請求項9】
前記逆相担体が、官能基として炭素数1〜30の直鎖の炭化水素基を有し、担体表面の炭素含有量が元素比で18%以下である材料により構成されている、請求項8に記載の8−イソプラスタンの定量方法。
【請求項10】
前記逆相担体が、フロー式粒子像分析装置で測定される円相当径0.5〜10μmの粒子を粒子数として1〜20累積%含む、請求項9に記載の8−イソプラスタンの定量方法。
【請求項11】
前記逆相担体が、さらに、フロー式粒子像分析装置で測定される円相当径20〜100μmの粒子を粒子数として65〜99累積%含む、請求項10に記載の8−イソプラスタンの定量方法。
【請求項12】
前記逆相担体の材料が、オクタデシルシリル基を有するシリカゲルである、請求項8乃至11いずれか1項に記載の8−イソプラスタンの定量方法。
【請求項13】
前記逆相担体が、沈降法で測定される粒径35μm以上60μm以下の粒子とコールター法で測定される粒径10μm以上30μm以下の粒子とを含み、
粒径35μm以上60μm以下の前記粒子と、粒径10μm以上30μm以下の前記粒子との重量比が、80:20〜95:5の範囲である、請求項8乃至13いずれか1項に記載の8−イソプラスタンの定量方法。
【請求項14】
前記生体試料が尿、唾液又は血液である、請求項7乃至13いずれか1項に記載の8−イソプラスタンの定量方法。
【請求項15】
8−イソプラスタンを陰イオン交換担体を用いて精製するステップをさらに含み、精製された8−イソプラスタンを用いて、蛍光ラベル化する前記ステップを実行する、請求項1乃至14いずれか1項に記載の8−イソプラスタンの定量方法。
【請求項16】
前記陰イオン交換担体が、第四級アンモニウム塩が固定化された陰イオン交換担体である、請求項15に記載の8−イソプラスタンの定量方法。
【請求項17】
請求項1乃至16いずれか1項に記載の8−イソプラスタンの定量方法に用いられる、8−イソプラスタンを定量するためのキット。
【請求項1】
下記一般式(1):
【化1】
〔式(1)中、R1、R2及びR3は、炭素数1〜6のアルキル基であり、nは、0〜6の整数である。〕
で表されるキノキサリノン誘導体を、8−イソプラスタンの物質量に対して過剰に用いて8−イソプラスタンを蛍光ラベル化するステップと、
蛍光ラベル化された8−イソプラスタンと、未反応のキノキサリノン誘導体とを含む反応混合物を、スルホン酸又はスルホン酸塩が固定化された陽イオン交換担体に接触させて、蛍光ラベル化された8−イソプラスタンと前記未反応のキノキサリノン誘導体とを分離するステップと、
前記未反応のキノキサリノン誘導体と分離された、蛍光ラベル化された8−イソプラスタンを定量するステップと、
を含む、8−イソプラスタンの定量方法。
【請求項2】
前記キノキサリノン誘導体が1,2,3,4‐テトラヒドロ‐6,7‐ジメトキシ‐1‐メチル‐2(1H)‐オキソキノキサリン‐3‐プロピオン酸ヒドラジドである、請求項1に記載の8−イソプラスタンの定量方法。
【請求項3】
8−イソプラスタンを蛍光ラベル化する前記ステップにおいて、8−イソプラスタンに対して5×103倍モル以上の前記キノキサリノン誘導体を用いて、8−イソプラスタンを蛍光ラベル化する、請求項1又は2に記載の8−イソプラスタンの定量方法。
【請求項4】
8−イソプラスタンを蛍光ラベル化する前記ステップにおいて、50〜90℃に加熱して前記キノキサリノン誘導体と8−イソプラスタンとを反応させることにより蛍光ラベル化する、請求項1乃至3いずれか1項に記載の8−イソプラスタンの定量方法。
【請求項5】
蛍光ラベル化された8−イソプラスタンを定量する前記ステップにおいて、逆相カラムを用いたアイソクラティックによる高速液体クロマトグラフィー法を用いて蛍光ラベル化された8−イソプラスタンを分析する、請求項1乃至4いずれか1項に記載の8−イソプラスタンの定量方法。
【請求項6】
前記陽イオン交換担体が、下記式(2)で表されるアルキルベンゼンスルホン酸又はその塩が固定化されたものである、請求項1乃至5いずれか1項に記載の8−イソプラスタンの定量方法。
【化2】
〔式(2)中、mは、1〜6であり、B1は、樹脂基材又はシリカゲルである。〕
【請求項7】
生体試料中の8−イソプラスタンを分離するステップをさらに含み、分離された8−イソプラスタンを用いて、蛍光ラベル化する前記ステップを実行する、請求項1乃至6いずれか1項に記載の8−イソプラスタンの定量方法。
【請求項8】
前記生体試料中の8−イソプラスタンを分離する前記ステップにおいて、前記生体試料と逆相担体とを接触させて、前記生体試料から8−イソプラスタンを分離する、請求項7に記載の8−イソプラスタンの定量方法。
【請求項9】
前記逆相担体が、官能基として炭素数1〜30の直鎖の炭化水素基を有し、担体表面の炭素含有量が元素比で18%以下である材料により構成されている、請求項8に記載の8−イソプラスタンの定量方法。
【請求項10】
前記逆相担体が、フロー式粒子像分析装置で測定される円相当径0.5〜10μmの粒子を粒子数として1〜20累積%含む、請求項9に記載の8−イソプラスタンの定量方法。
【請求項11】
前記逆相担体が、さらに、フロー式粒子像分析装置で測定される円相当径20〜100μmの粒子を粒子数として65〜99累積%含む、請求項10に記載の8−イソプラスタンの定量方法。
【請求項12】
前記逆相担体の材料が、オクタデシルシリル基を有するシリカゲルである、請求項8乃至11いずれか1項に記載の8−イソプラスタンの定量方法。
【請求項13】
前記逆相担体が、沈降法で測定される粒径35μm以上60μm以下の粒子とコールター法で測定される粒径10μm以上30μm以下の粒子とを含み、
粒径35μm以上60μm以下の前記粒子と、粒径10μm以上30μm以下の前記粒子との重量比が、80:20〜95:5の範囲である、請求項8乃至13いずれか1項に記載の8−イソプラスタンの定量方法。
【請求項14】
前記生体試料が尿、唾液又は血液である、請求項7乃至13いずれか1項に記載の8−イソプラスタンの定量方法。
【請求項15】
8−イソプラスタンを陰イオン交換担体を用いて精製するステップをさらに含み、精製された8−イソプラスタンを用いて、蛍光ラベル化する前記ステップを実行する、請求項1乃至14いずれか1項に記載の8−イソプラスタンの定量方法。
【請求項16】
前記陰イオン交換担体が、第四級アンモニウム塩が固定化された陰イオン交換担体である、請求項15に記載の8−イソプラスタンの定量方法。
【請求項17】
請求項1乃至16いずれか1項に記載の8−イソプラスタンの定量方法に用いられる、8−イソプラスタンを定量するためのキット。
【図1】

【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公開番号】特開2012−194178(P2012−194178A)
【公開日】平成24年10月11日(2012.10.11)
【国際特許分類】
【出願番号】特願2012−13213(P2012−13213)
【出願日】平成24年1月25日(2012.1.25)
【出願人】(000133179)株式会社タニタ (303)
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【公開日】平成24年10月11日(2012.10.11)
【国際特許分類】
【出願日】平成24年1月25日(2012.1.25)
【出願人】(000133179)株式会社タニタ (303)
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
[ Back to top ]