
9E−15−(2−ピロリジン−1−イル−エトキシ)−7,12,25−トリオキサ−19,21,24−トリアザ−テトラシクロ[18.3.1.1(2,5).1(14,18)]ヘキサコサ−1(24),2,4,9,14,16,18(26),20,22−ノナエンのクエン酸塩
本発明は、向上した特性を有することがことが見出された9E−15−(2−ピロリジン−1−イル−エトキシ)−7,12,25−トリオキサ−19,21,24−トリアザ−テトラシクロ[18.3.1.1(2,5).1(14,18)]ヘキサコサ−1(24),2,4,9,14,16,18(26),20,22−ノナエン(化合物I)の特定の塩に関する。本発明は、特に、この化合物のクエン酸塩に関する。本発明は、また、そのクエン酸塩を含有する医薬組成物及び特定の病状の処置におけるそのクエン酸塩の使用方法にも関する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、9E−15−(2−ピロリジン−1−イル−エトキシ)−7,12,25−トリオキサ−19,21,24−トリアザ−テトラシクロ[18.3.1.1(2,5).1(14,18)]ヘキサコサ−1(24),2,4,9,14,16,18(26),20,22−ノナエンのクエン酸塩に関する。さらに、本発明は、そのクエン酸塩を含有する医薬組成物及び特定の病状の処置におけるその塩の使用方法に関する。
【背景技術】
【0002】
9E−15−(2−ピロリジン−1−イル−エトキシ)−7,12,25−トリオキサ−19,21,24−トリアザ−テトラシクロ[18.3.1.1(2,5).1(14,18)]ヘキサコサ−1(24),2,4,9,14,16,18(26),20,22−ノナエン(化合物I)の化合物は、PCT/SG2006/000352に最初に記載され、多くの病状の処置のための薬学的に活性な薬剤としての大きな将来性を示した。この化合物が示す活性プロファイルに基づいて、本化合物の医薬開発が進められている。
【0003】
【化1】
【0004】
大量生産及び最終的には商業的使用に適した薬剤の開発において、対象者に対する薬剤活性の受容レベルは考慮されるべき重要な変数のひとつに過ぎない。例えば、医薬組成物の製剤においては、薬学的に活性な物質が、商業的製造プロセスにおいて確実に再生産でき、かつ、その薬学的に活性な物質が、それが曝される諸条件に耐えうる頑丈な形態であることが必須である。
【0005】
製造の観点から、薬学的に活性な物質の商業的製造プロセスは、同一の製造条件が用いられた場合は、同一の物質を生産するというものであることが重要である。加えて、薬学的に活性な物質は、固体形態で存在し、製造条件にわずかな変更があった場合に、それが、生産される薬学的に活性な物質の固体形態に大きな変化をもたらさないことが望ましい。例えば、製造プロセスが、確実に同一の結晶特性を有する物質を生産すること、そしてまた、そのプロセスが、同一レベルの水和状態を有する物質を生産することが重要である。
【0006】
加えて、薬学的に活性な物質は、分解、吸湿、さらにその固体形態に生じるその後の変化に対して安定であることが重要である。これは、薬学的に有効な成分の医薬製剤への組み込みを容易にする上で重要である。薬学的に活性な物質が、時間の経過とともに水を吸収するという意味において吸湿性(「粘着性」)であるなら、同一の投薬量を提供するために添加されるその薬学的に活性な物質の量が、水和の程度に依存して大幅に変化するであろうことから、その薬学的に活性な物質を薬剤に信頼性をもって配合することはほとんど不可能である。さらに、水和又は固体形態(「多形性」)におけるばらつきは、溶解性又は溶解速度といった物理化学的特性における変化をもたらす可能性があり、このことは、ひいては患者における一貫性のない経口吸収という結果を招きうる。
【0007】
したがって、薬学的に活性な薬剤の化学的安定性、固体状態の安定性及び「保存可能期間」は、極めて重要な要因である。理想的な状況では、薬学的に活性な薬剤及びそれを含有するあらゆる組成物は、活性、含水量、溶解度特性、固体形態等の有効成分の物理化学的特性に有意な変化を示すことなく、相当な期間にわたって有効に保存されうるものであるべきである。
【0008】
9E−15−(2−ピロリジン−1−イル−エトキシ)−7,12,25−トリオキサ−19,21,24−トリアザ−テトラシクロ[18.3.1.1(2,5).1(14,18)]ヘキサコサ−1(24),2,4,9,14,16,18(26),20,22−ノナエンに関して、最初の研究は、塩酸塩に関して実施され、多形が優勢であり、その化合物は、製造条件によって、複数の結晶形をとることが見出された。加えて、製造条件が一定だった場合ですら、バッチごとに多形の比率が異なることが観察された。これらバッチ間の一貫性の欠如が、商業的観点から、塩酸塩をあまり望ましくないものとした。
【0009】
したがって、上に示した課題の一つ以上を克服するか又は改善する9E−15−(2−ピロリジン−1−イル−エトキシ)−7,12,25−トリオキサ−19,21,24−トリアザ−テトラシクロ[18.3.1.1(2,5).1(14,18)]ヘキサコサ−1(24),2,4,9,14,16,18(26),20,22−ノナエンの塩類を開発することが望ましい。
【発明の概要】
【0010】
本発明は、9E−15−(2−ピロリジン−1−イル−エトキシ)−7,12,25−トリオキサ−19,21,24−トリアザ−テトラシクロ[18.3.1.1(2,5).1(14,18)]ヘキサコサ1(24),2,4,9,14,16,18(26),20,22−ノナエンのクエン酸塩を提供する。
【0011】
いくつかの実施態様において、塩は、結晶性である。
【0012】
いくつかの実施態様において、塩は、1:1のクエン酸塩である。いくつかの実施態様において、クエン酸塩は、X線回折において、2シータスケールで、22.4o±0.5oでピークを示す。
【0013】
いくつかの実施態様において、クエン酸塩はまた、X線回折において、2シータスケールで、10.0°±0.5°、15.6°±0.5°及び17.2°±0.5°でピークを示す。
【0014】
いくつかの実施態様において、クエン酸塩は、X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、21.1°±0.5°及び22.4°±0.5°からなる群より選択される、少なくとも4つのピークを示す。
【0015】
いくつかの実施態様において、クエン酸塩は、X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、21.1°±0.5°及び22.4°±0.5°からなる群より選択される、少なくとも6つのピークを示す。
【0016】
いくつかの実施態様において、クエン酸塩は、X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、21.1°±0.5°及び22.4°±0.5°でピークを示す。
【0017】
いくつかの実施態様において、クエン酸塩は、X線回折において、2シータスケールで、11.1°±0.5°、18.1°±0.5°、21.8°±0.5°、23.2°±0.5°及び27.6°±0.5°でピークを示す。
【0018】
いくつかの実施態様において、クエン酸塩は、X線回折において、2シータスケールで、7.0°±0.5°、14.0°±0.5°、19.0°±0.5°、19.8°±0.5°、23.6°±0.5°、24.3°±0.5°、25.2°±0.5°、25.7°±0.5°、26.1°±0.5°、26.5°±0.5°及び32.1°±0.5°でピークを示す。
【0019】
また、本発明は、上記のとおりの塩を含む医薬組成物を提供する。
【0020】
もう一つの実施態様では、本発明は、本発明の治療有効量の塩を、それを必要とする患者に投与することを含む、増殖性障害を治療又は予防する方法を提供する。いくつかの実施態様において、増殖性障害は、癌である。
【0021】
もう1つの実施態様において、本発明は、増殖性障害の処置における本発明の塩の使用を提供する。いくつかの実施態様において、増殖性障害は、癌である。
【0022】
もう一つの実施態様において、本発明は、増殖性障害の処置のための薬剤の製造における本発明の塩の使用を提供する。いくつかの実施態様において、増殖性障害は、癌である。
【図面の簡単な説明】
【0023】
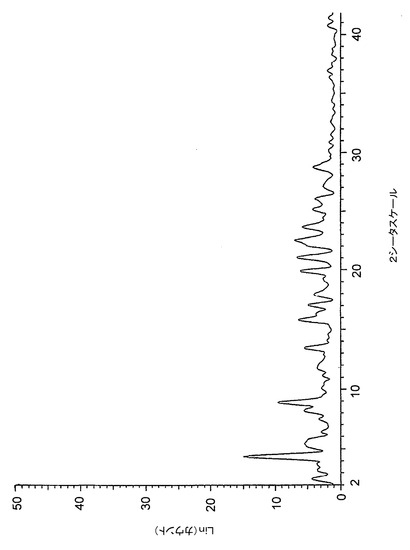
【図1】図1は、バッチ1(THF中で調製したHCl塩)に関する高分解能粉末X線回折(XRPD)ディフラクトグラムを示す。
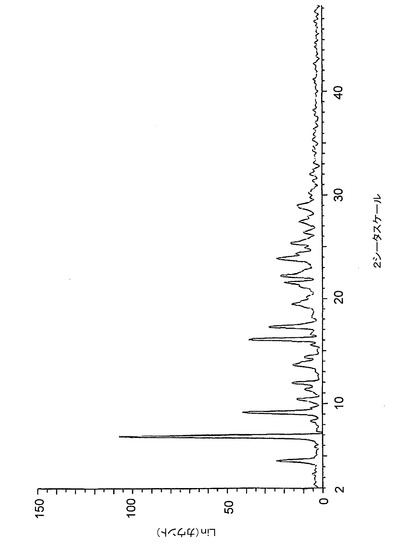
【図2】図2は、バッチ2(MeCN中で調製したHCl塩)に関する高分解能粉末X線回折(XRPD)ディフラクトグラムを示す。
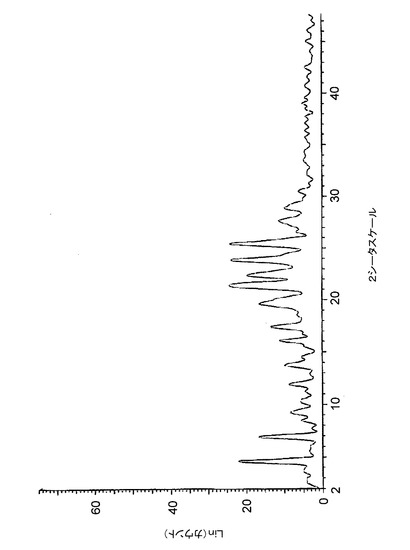
【図3】図3は、バッチ3(アセトン中で調製したHCl塩)に関する高分解能粉末X線回折(XRPD)ディフラクトグラムを示す。
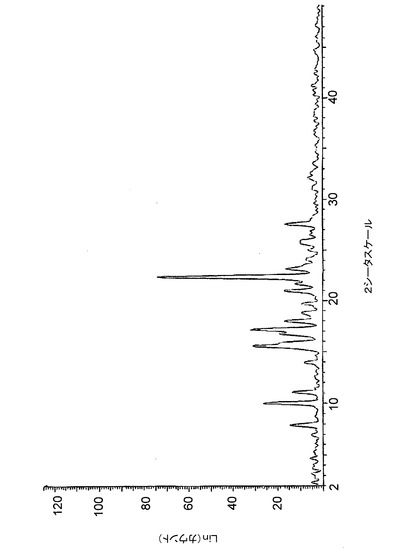
【図4】図4は、バッチ4(THF中で調製したクエン酸塩)に関する高分解能粉末X線回折(XRPD)ディフラクトグラムを示す。
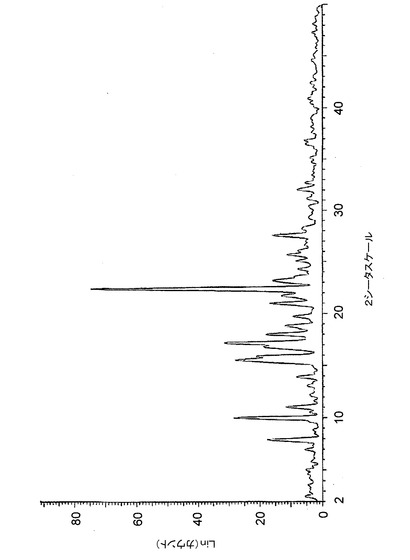
【図5】図5は、バッチ5(MeCN中で調製したクエン酸塩)に関する高分解能粉末X線回折(XRPD)ディフラクトグラムを示す。
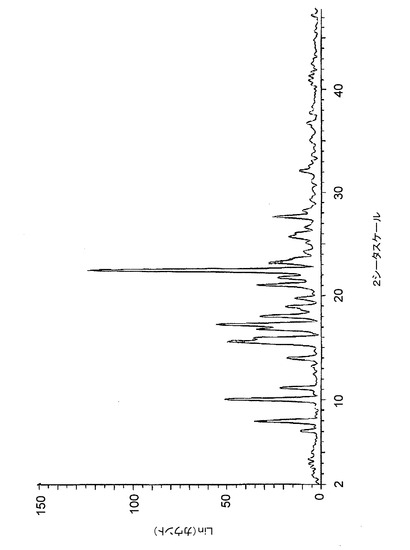
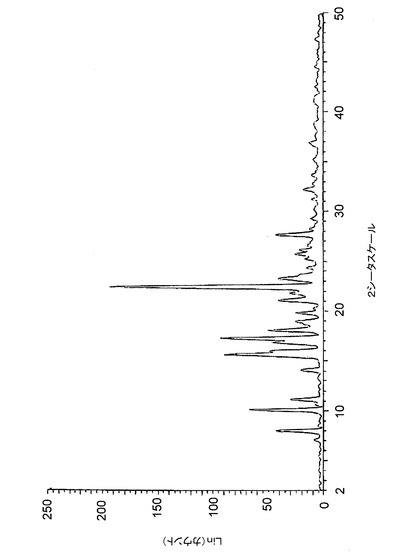
【図6】図6は、バッチ6(アセトン中で調製したクエン酸塩)に関する高分解能粉末X線回折(XRPD)ディフラクトグラムを示す。
【図7】図7は、バッチ7(アセトン中で調製したクエン酸塩(20gスケール))に関する高分解能粉末X線回折(XRPD)ディフラクトグラムを示す。
【図8】図8は、バッチ8(アセトン中で調製したクエン酸塩(20gスケール))に関する高分解能粉末X線回折(XRPD)ディフラクトグラムを示す。
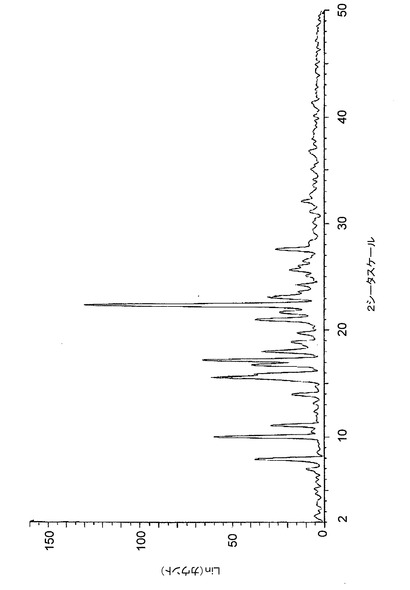
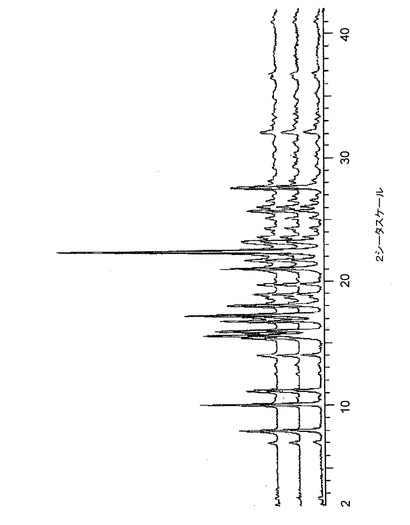
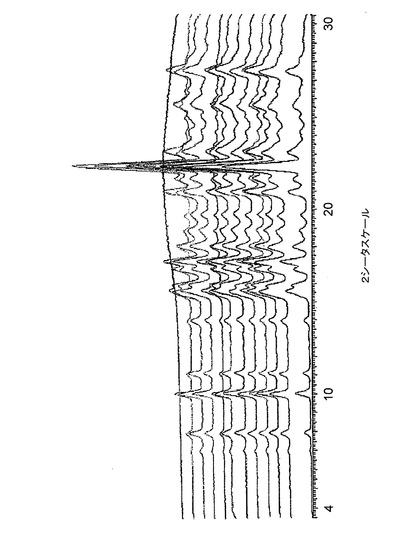
【図9】図9は、バッチ4〜6に関する高分解能粉末X線回折(XRPD)ディフラクトグラムを示す。
【図10】図10は、バッチ4〜6に関する低分解能粉末X線回折ディフラクトグラムを示す。
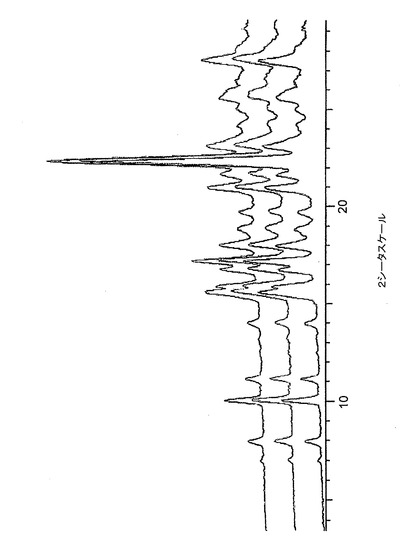
【図11】図11は、バッチ4に関する、高分解能及び低分解能粉末X線回折ディフラクトグラムのオーバーレイを示す。
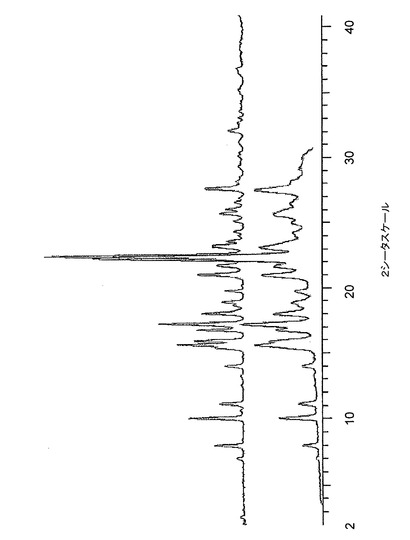
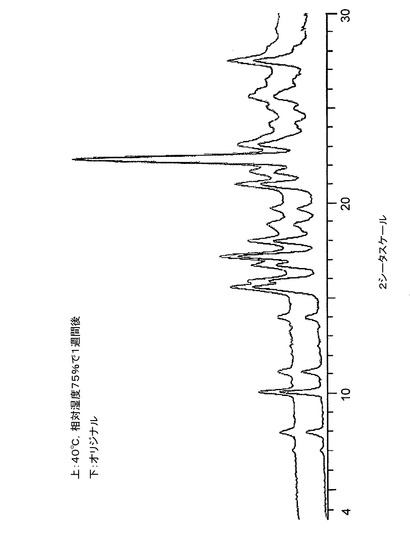
【図12】図12は、40℃及び相対湿度75%で1週間保存前後の、バッチ4に関する粉末X線回折トレースである。
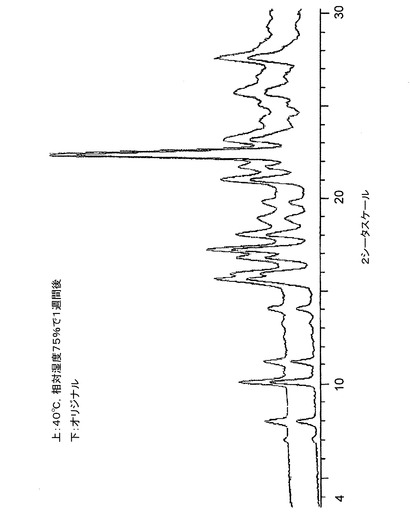
【図13】図13は、40℃及び相対湿度75%で1週間保存前後の、バッチ5に関する粉末X線回折トレースである。
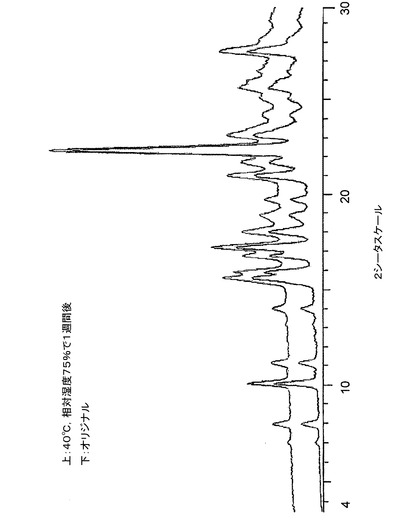
【図14】図14は、40℃及び相対湿度75%で1週間保存前後の、バッチ6に関する粉末X線回折トレースである。
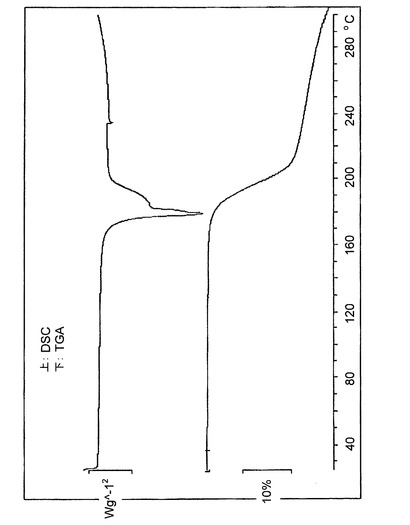
【図15】図15は、バッチ4に関する示差走査熱量測定(DSC、上)及び熱重量分析(TGA)のデータを示す。
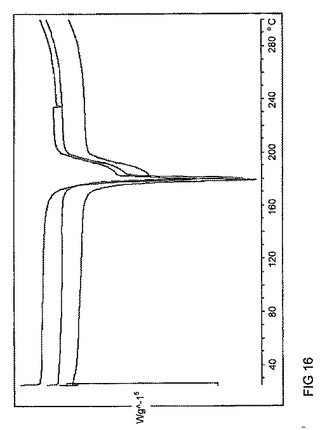
【図16】図16は、バッチ4〜6に関するDSCトレースのオーバーレイを示す。
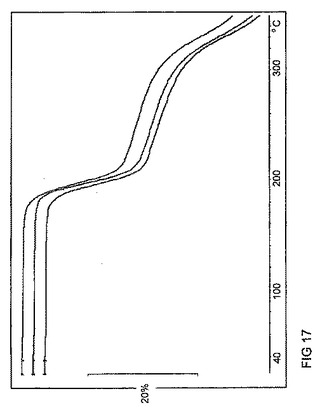
【図17】図17は、バッチ4〜6に関するTGAトレースのオーバーレイを示す。
【図18】図18は、バッチ4に関する重量蒸気収着の運動エネルギーのプロットを示す。
【図19】図19は、バッチ4に関する重量蒸気収着の等温線のプロットを示す。
【図20】図20は、バッチ4に関する重量蒸気収着の実験前後の、粉末X線回折トレースを示す。
【図21】図21は、溶解性スクリーンからのサンプルの粉末X線回折トレースを示す。
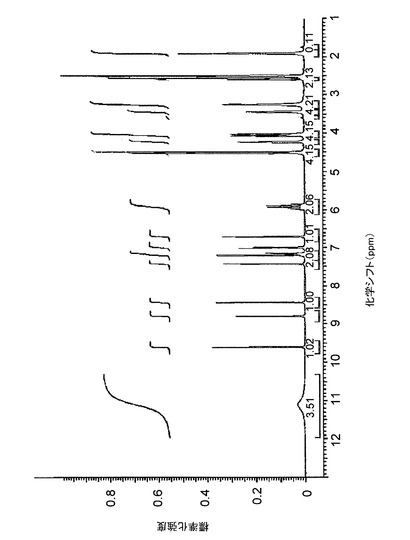
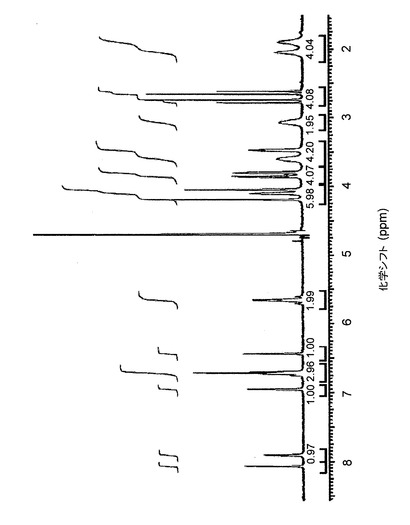
【図22】図22は、d6−DMSO中のバッチ4に関する1H NMRスペクトルを示す。
【図23】図23は、D2O中のバッチ4に関する1H NMRスペクトルを示す。
【0024】
発明の詳細な説明
上述のように、今や、9E−15−(2−ピロリジン−1−イル−エトキシ)−7,12,25−トリオキサ−19,21,24−トリアザ−テトラシクロ[18.3.1.1(2,5).1(14,18)]ヘキサコサ−1(24),2,4,9,14,16,18(26),20,22−ノナエンの特定の塩類が、単一の頑丈な多形体として存在することがわかった。特に、本出願人は、この化合物のクエン酸塩が、単一の多形体として存在することを見出した。
【0025】
クエン酸の構造は、従来技術に熟練した読み手に明らかであると考えられるが、あらゆる不確実性を回避するために、その構造を以下に示す。
【化2】
【0026】
本願明細書に記載した塩酸塩及びクエン酸塩に関する比較研究は、表1に記載するバッチで実施した。
【0027】
【表1】
【0028】
化合物1に関する最初の研究は、塩酸塩を対象とした。以下にまとめるように、最初に調製された塩酸塩は、一貫性のない固体状態を生じ、粉末X線回折(XRPD)データにおいて著しいばらつきを示すことが分かった。
【0029】
塩酸塩としての化合物1は、バッチ1(THF中で調製)、バッチ2(アセトニトリル中で調製)及びバッチ3(アセトン中で調製)を結晶性材料として与える3つの異なる溶媒において調製された。図1、2及び3は、XRPDディフラクトグラムにおける、これらのバッチ間での著しいばらつきを示し、これらのHCl塩の結晶構造には、異なる溶媒中、同様の条件下で調製した場合でも、全般的な相違があることを示している。
【0030】
上記のように、塩酸塩に許容し得ないばらつきが見られる結果、これに代わる頑丈な固体状態が必要とされた。更なる発見努力により、クエン酸塩が、そのような頑丈な固体状態の一つであると確認された。
【0031】
9E−15−(2−ピロリジン−1−イル−エトキシ)−7,12,25−トリオキサ−19,21,24−トリアザ−テトラシクロ[18.3.1.1(2,5).1(14,18)]ヘキサコサ−1(24),2,4,9,14,16,18(26),20,22−ノナエンクエン酸塩の5つのバッチを特徴づけした。分析の結果を、以下の実施例で示す。
【0032】
粉末X線回折(XRPD)を用いて化合物1のクエン酸塩を特徴づけた。本発明のクエン酸塩に関する有意なX線回折ピーク(高分解能条件下で収集した)のリストを表2中に示す。
【0033】
【表2】
【0034】
表から分かるように、本クエン酸塩は、X線回折において、2シータスケールで、22.4°±0.5°でピークを示すものとして特徴づけることができる。
【0035】
また、本クエン酸塩は、X線回折において、2シータスケールで、10.0°±0.5°、15.6°±0.5°及び17.2°±0.5°でピークを示すものとして特徴づけることもできる。
【0036】
いくつかの実施態様において、本クエン酸塩は、さらに、X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、21.1°±0.5°及び22.4°±0.5°からなる群より選択される、少なくとも4つのピークを示すものとして特徴づけることもできる。
【0037】
いくつかの実施態様において、本クエン酸塩は、さらに、X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、21.1°±0.5°及び22.4°±0.5°からなる群より選択される、少なくとも6つのピークを示すものとして特徴づけることもできる。
【0038】
いくつかの実施態様において、本クエン酸塩は、さらに、X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、21.1°±0.5°及び22.4°±0.5°でピークを示すものとして特徴づけることもできる。
【0039】
いくつかの実施態様において、また、本クエン酸塩は、X線回折において、2シータスケールで、11.1°±0.5°、18.1°±0.5°、21.8°±0.5°、23.2°±0.5°及び27.6°±0.5°でピークを示す。
【0040】
いくつかの実施態様において、本クエン酸塩は、さらに、X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、11.1°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、18.1°±0.5°、21.8°±0.5°、21.1°±0.5°、22.4°±0.5°、23.2°±0.5°及び27.6°±0.5°でピークを示すものとして特徴づけることもできる。
【0041】
上記のピークは、特徴的なピークであるが、本クエン酸塩は、また、X線回折において、2シータスケールで、7.0°±0.5°、14.0°±0.5°、19.0°±0.5°、19.8°±0.5°、23.6°±0.5°、24.3°±0.5°、25.2°±0.5°、25.7°±0.5°、26.1°±0.5°、26.5°±0.5°及び32.1°±0.5°でピークを示す場合もある。
【0042】
当業者は理解されることであろうが、回折の相対的強度は、多くの要因(例えば試料調製の方法及び使用する計測器の型)に応じて変化しうる。加えて、特定の事例においては、上記ピークのいくつかは、検出可能でなくてもよい。
【0043】
本発明の塩は、化合物1の遊離塩基と適切な溶媒中のクエン酸とを反応させ、その反応混合物から、得られた塩を、結晶化、沈殿又は蒸発後に回収することにより製造することができる。
【0044】
塩を形成する反応は、遊離塩基が適切な溶解度を有する、任意の非干渉性溶媒又は溶媒混合物において実施することができる。この種の適切な溶媒の例は、アセトニトリル、テトラヒドロフラン及びアセトンを含む。この方法は、通常、20℃を超すような高い温度で、遊離塩基を適切な溶媒に溶解させることを含む。ある実施態様(例えばアセトン)において、遊離塩基は、約56℃の温度で溶媒中に溶解される。ある実施態様(例えばアセトニトリル)において、遊離塩基は、約82℃の温度で溶媒中に溶解される。
【0045】
一度遊離塩基が適切な溶媒に溶解したら、本方法は、次に、適切な量の酸を添加することを含む。酸は、通常、適切な溶媒(通常、遊離塩基を溶解に用いるのと同じ溶媒)中の溶液として加えられる。使用する酸の量は、通常、化学量論的に等量又は化学量論的にわずかに過剰量ではあるが、酸の量は変化させ得る。酸の添加に続いて、次に、本方法は、通常、添加温度で反応混合物を1時間撹拌し、その後、結晶化を容易にするために、反応混合物を反応温度未満の温度に冷却することを含む。一度所望のレベルの結晶形成が生じたら、その結晶を、濾過により分離し、従来技術において通常の手段を用いて乾燥させることができる。
【0046】
本発明のもう一つの実施態様は、増殖性障害の処置における本発明の塩の使用を提供する。この種の化合物の使用に関する配合及び方法論、並びにそれによって治療され得る障害は、PCT/SG2006/000352にて開示されているとおりである。
【0047】
本発明を、ここで、以下の非限定的な実施例によって記載する。塩酸塩を、比較例用に上記のように調製し、同様の方法で分析した。
【0048】
実施例1−溶媒としてのTHF中のHCl塩(バッチ1)の形成:
【0049】
化合物1の遊離塩基(0.200g、0.432mmoles、1当量)を、THF15mLに加えた。溶液を、溶解の完了が観察されるまで加熱還流し、1時間保持した。次に、1N HCl(0.518mL、0.518mmoles、1.2当量)を、還流条件下で徐々に加えた。混合物を、更に15分間還流し、次に冷却した。段階的な冷却によって、結晶化が観察された。結晶を、室温で12時間撹拌し、真空下で濾過した。生成物を、真空下で乾燥させて、165mgを得た。
【0050】
実施例2−溶媒としてのCH3CN中のHCl塩(バッチ2)の形成:
【0051】
化合物1(0.300g、0.648mmoles、1当量)の遊離塩基を、CH3CN70mLに加えた。溶液を、溶解の完了が観察されるまで加熱還流し、1時間保持した。次に1N HCl(0.778mL、0.778mmoles、1.2当量)を、還流条件下で徐々に加えた。混合物を、更に15分間還流し、次に冷却した。段階的な冷却によって、結晶化が観察された。結晶を、12時間室温で撹拌し、真空下で濾過した。生成物を、真空下で乾燥させて、190mgを得た。
【0052】
実施例3−溶媒としてのアセトン中のHCl塩(バッチ3)の形成:
【0053】
化合物1(0.200g、0.432mmoles、1当量)の遊離塩基を、アセトン50mLに加えた。溶液を、溶解の完了が観察されるまで加熱還流し、1時間保持した。次に1N HCl(0.518mL、0.518mmoles、1.2当量)を、還流条件下で徐々に加えた。混合物をさらに15分間還流し、次に冷却した。段階的な冷却によって、結晶化が観察された。結晶を、室温で12時間撹拌し、真空下で濾過した。生成物を、真空下で乾燥させて、180mgを得た。
【0054】
実施例4−溶媒としてのTHF中のクエン酸塩(バッチ4)の形成:
【0055】
化合物1(0.300g、0.648mmoles、1当量)の遊離塩基を、THF 12mLに加えた。溶液を、溶解の完了が観察されるまで加熱還流し、1時間保持した。次にTHF12mLに溶解したクエン酸(0.149g、0.778mmoles、1.2当量)の溶液を、還流条件下で徐々に加えた。混合物を、更に15分間還流し、次に冷却した。段階的な冷却によって、結晶化が観察された。結晶を室温で12時間撹拌し、減圧下で濾過した。生成物を、真空下で乾燥させて、250mgを得た。
【0056】
実施例5−溶媒としてのCH3CN中のクエン酸塩(バッチ5)の形成:
【0057】
化合物1(0.200g、0.432mmoles、1当量)の遊離塩基を、CH3CN 45mLに加えた。溶液を、溶解の完了が観察されるまで加熱還流し、1時間保持した。次にCH3CN 12mLに溶解したクエン酸(0.099g、0.518mmoles、1.2当量)の溶液を、還流条件下で徐々に加えた。混合物を、更に15分間還流し、次に冷却した。段階的な冷却によって、結晶化が観察された。結晶を、室温で12時間撹拌し、真空下で濾過した。生成物を、真空下で乾燥させて、220mgを得た。
【0058】
実施例6−溶媒としてのアセトン中のクエン酸塩(バッチ6)の形成:
【0059】
化合物1(0.200g、0.432mmoles、1当量)の遊離塩基をアセトン50mLに加えた。溶液を、溶解の完了が観察されるまで加熱還流し、1時間保持した。次に、アセトン20mLに溶解したクエン酸(0.099g、0.518mmoles、1.2当量)の溶液を、還流条件下で徐々に加えた。混合物を、更に15分間還流し、次に冷却した。段階的な冷却によって、結晶化が観察された。結晶を、室温で12時間撹拌し、真空下で濾過した。生成物を、真空下で乾燥させて、198mgを得た。
【0060】
実施例7−粉末X線回折研究
【0061】
条件1a(高分解能)
【0062】
粉末X線回折(XRPD)パターンを、CuK放射(1.54A)(40kV、ステップ・サイズ0.03°及びステップ時間0.5秒での30連続走査モードは、θ〜θmAであった)を用いたαSIEMENS D5000回折計で収集した。使用したθの範囲は、有効な22°〜50°を与えるサンプル−検出器距離であった。サンプル分析時間(X線ビームに曝される時間)は、13分33秒であった。データ収集に用いたソフトウェアは、DIFFRACplus-D5000 #1であり、データは、Diffrac Plus -D5000 #1を用いて分析して提示した。
【0063】
周囲条件下でテストするサンプルは、粉砕せずに調製した粉体を用いて、平板試料として調製した。サンプルの約100〜200mgを、ガラススライドに軽く押し付けて平坦な面を得た。
【0064】
条件1b(高分解能)
【0065】
粉末X線回折(XRPD)パターンを、Cu Kα放射(40kV、40mA)、自動化XYZステージ、オートサンプルポジショニング用レーザー・ビデオ顕微鏡及びHiStar二次元領域検出器を用いて、Bruker AXS C2 GADDS回折計で収集した。X線光学系は、0.3mmのピンホールコリメーターに接続した単一のGoebel多層ミラーからなる。ビーム拡散(すなわちサンプルに対するX線ビームの有効サイズ)は、約4mmであった。3.2°〜29.7°の有効な2θ範囲を与えるサンプル−検出器距離20cmでθ−θ連続走査モードを使用した。通常、サンプルは、120秒間X線束に曝される。データ収集に使用したソフトウェアは、WNT 4.1.16用のGADDSであり、データは、Diffrac Plus EVA v 9.0.0.2 又はv 13.0.0.2で分析して提示した。周囲条件下でテストするサンプルは、粉砕せずに調製された粉体を用いて、平板試料として調製した。サンプルの約1〜2mgを、ガラススライドに軽く押し付けて平坦な面を得た。
【0066】
条件2(低分解能)
【0067】
粉末X線回折パターンは、また、Cu Kα放射(40kV、40mA)、θ−2θゴニオメーター及びV4発散及び受光スリット、Geモノクロメーター及びLynxeye検出器を用いて、Bruker D8 回折計で収集した。計測器は、認証されたコランダム標準(NIST1976)を使用して、性能点検される。データ収集に使用したソフトウェアは Diffrac Plus XRD Commander v 2.5.0であり、データは、Diffrac Plus EVA v 11.0.0.2 又はv 13.0.0.2を用いて分析して提示した。サンプルは、入手した粉体そのままを使用した平板試料として、周囲条件下でテストした。研磨したゼロバックグラウンド(510)シリコンウエハーに切り込んだ空洞に、サンプル約15mgを緩やかに詰めた。分析の間、試料をそれ自体の面内で回転させた。データ収集の詳細は、以下の通りである:
○角度範囲:2〜42°2θ
○ステップサイズ:0.05°2θ
○収集時間:0.5秒/ステップ
【0068】
高分解能XRPDトレース(条件1a)を、サンプルの各々に対して得た。そして、図4〜8に示された結果は、クエン酸塩の5つのサンプルがすべて同じ結晶相のものであることを示している。また、バッチ4〜6に関するデータは、条件1b下でも収集され、図9に示すオーバレイは、これらのパターンが極めて類似しており、このことは、これらがすべて、同一の結晶質相のものであることを示している。
【0069】
低分解能XRPDトレース(条件2)も、Bruker GADDS回折計を用いて収集したので、多形性スクリーン分析用の標準パターンが得られた。バッチ4〜6のトレースのオーバレイを図10に示し、高分解能及び低分解能トレースの比較を図11に示す。
【0070】
低分解能XRPDトレースの収集のために調製したサンプルを、温度40度及び相対湿度75%に維持される容器に入れた。1週間後、相変化について調べるために、サンプルを、低分解能XRPD(条件2)で再分析した。結果を、最初のXRPDトレースと比較して、図12〜図14に示す。相変化が発生しなかったこと、そして、本発明のクエン酸塩はこれらの条件下で少なくとも1週間は安定であることがわかった。
【0071】
実施例8−核磁気共鳴(NMR)研究
【0072】
1H NMRスペクトルを、オートサンプラーを備え、DRX400コンソールによって制御されるBruker 400MHz測定器で収集した。自動化した実験は、標準Bruker搭載実験装置を用いて、Topspin v 1.3(パッチレベル8)で動作するICON-NMR v 4.0.4(build 1)を用いることで用意した。サンプルは、d6−DMSO又はD2O中で調製した。オフライン分析は、ACD SpecManager v 9.09(build 7703)を用いて行なった。
【0073】
1H NMRは、3つのサンプルすべてが同一化合物であることを示している。クエン酸塩の化学量論の決定は、対イオンのシグナルの積分によって行なった。しかし、これらのシグナルは、(バッチ4、図22の)スペクトルにおいては、DMSOシグナルの下に現れるので、クエン酸のシグナルの積分は実施できなかった。図23は、D2O中のバッチ4の1H−NMRを示す。この溶媒において、クエン酸のシグナルの積分は、化学量論が、予想どおり1:1であることを示した。
【0074】
実施例9−示差走査熱量測定(DSC)及び熱重量分析(TGA)
【0075】
示差走査熱量測定(DSC)データは、34ポジション・オートサンプラーを備えたMettler DSC 823eで収集した。計測器は、認定インジウムを使用して、エネルギー及び温度較正を行った。典型的には、0.5〜1.5mgの各サンプルを、ピンホール付きアルミニウムパンで、10℃/分で25℃から350℃に加熱した。50mL/分での窒素パージを、サンプル全体に維持した。計測器制御及びデータ分析ソフトウェアは、STARe v 9.10であった。
【0076】
熱重量分析(TGA)データを、34ポジション・オートサンプラーを備えたMettler TGA/SDTA 851eで収集した。計測器は、認定インジウムを使用して、温度較正を行った。典型的には、5〜30mgの各サンプルを、予め重量測定したアルミニウムるつぼに入れて10℃/分で周囲温度から350℃に加熱した。50mL/分での窒素パージを、サンプル全体に維持した。計測器制御及びデータ分析ソフトウェアは、STARe v 9.10であった。
【0077】
バッチ4(図15)に関するDSCトレースは、176℃で著しい熱イベントがあることを示している。TGAでは、対応して約20%の重量損失が見られる(図15)。この重量損失は、DSC吸熱の複雑な形状と共に、>176℃で全体量の減衰が生じていることを示している。理論に束縛されることは望まないが、このことは、塩の解離及びその後のクエン酸の分解を示している可能性がある。
【0078】
バッチ5及び6は、同様のDSC及びTGAトレース(図16及び17は、それぞれDSC及びTGAデータのオーバレイを示す)を示した。
【0079】
実施例10−重量蒸気収着(GVS)
【0080】
SMS Analysis Suiteソフトウェアで制御するSMS DVS Intrinsic moisture sorption analyserを用いて、収着等温線を得た。サンプル温度を、計測器の制御により25℃に維持した。湿度は、総流量200mL/分で、乾燥及び湿潤窒素の流れを混合することによって制御した。相対湿度は、サンプル近辺に位置した較正済Rotronicプローブ(1.0〜100%RHのダイナミックレンジ)で測定した。%RHの関数としてのサンプル中の重量変化(質量緩和)は、微量天秤(精度±0.005mg)で常にモニターした。典型的には5〜20mgのサンプルを、周囲条件下、風袋計量したメッシュ・ステンレス・スチールバスケットに入れた。サンプルを40%RH及び25℃(典型的な室温条件)にて充填し、取り出した。水分収着等温線解析は、以下に概説するように行った(2走査で1サイクルを完結)。標準等温線解析を、25℃で、10%RH間隔で、0.5〜90%RH範囲にわたって実施した。
【0081】
【表3】
【0082】
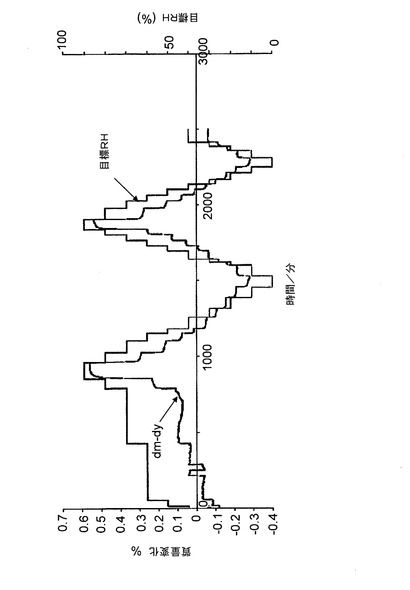
本クエン酸塩の吸湿性を、バッチ4に関する重量蒸気収着実験を行うことによって調べた。環境の湿度を2つの完全なサイクルを通して変化させながら、サンプル約20mgを、25℃に保った。図18に示す動的プロットは、バッチ4のサンプルが、各%RHステップで平衡重量に達することを示している。サンプルは、実験の初期において、平衡に達するのにより長くかかっている。これは、残留溶媒の置換による可能性がある。
【0083】
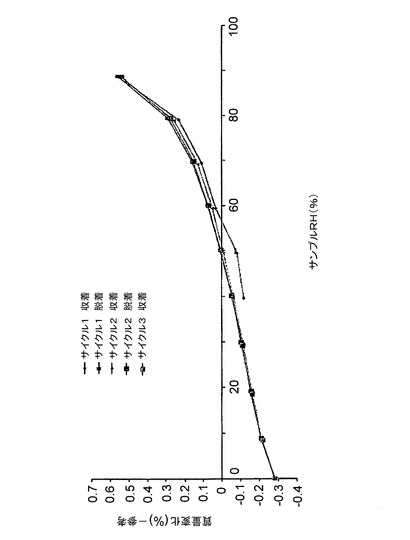
図19に示す等温線プロットは、サンプルが、40%RH〜90%RHの間で、<0.6%の水を吸収することを示している。最大重量差(0%RH〜90%RH)は、1% w/w未満であり、このことは、本クエン酸塩が、吸湿性ではないことを示している。加えて、本クエン酸塩が水和形態で存在することに関する証拠はない。
【0084】
【表4】
【0085】
GVS実験終了時、サンプルを回収し、XRPDで分析し、あらゆる全体的な相変化を調べた。結果(図20)は、全体的な相変化はないことを示している。
【0086】
実施例11−高速液クロマトグラフィ(HPLC)による化学純度の決定
【0087】
ダイオードアレイ検出器を備え、ChemStation software v B.02.01-SR1を用いたAgilent HP1100シリーズシステムで、純度分析を行った。使用したパラメーターを表5にまとめた。
【0088】
【表5】
【0089】
本クエン酸塩のバッチ4〜6化学純度は、このHPLC手順を用いて決定した。数値結果を表6に示す。
【0090】
【表6】
【0091】
表から分かるように、各サンプルの測定された純度は、98.1%を超えている。
【0092】
実施例12−溶解性及び多形性評価
【0093】
調査した各溶媒について、化合物1の約8mgを、8mLのスクリューキャップ付きガラスバイアルに量り入れた。溶媒は、10倍容量のアリコートを加え、混合物を超音波処理し、(ホットエアガンで)温めて溶解を促した。100倍容量の溶媒を加えても、溶解が成し遂げられない場合は、更に100倍容量のアリコートを加えた。各実験及び観察の詳細(表7)は、完全に溶解が成し遂げられたのは、水だけにおいてであることを示している。
【0094】
【表7】
【0095】
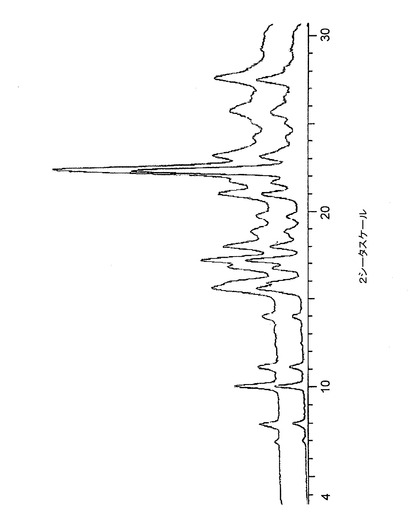
次に、バイアルを、湿度チャンバーに置き、25℃/50℃(8時間サイクル)で24時間のサイクルに付した。この時間の終わりに、サンプルを検査し、次に緩く蓋をして、溶媒が蒸発するにまかせた。乾燥させたこれらのサンプルを、次に石英アレイに移し、一方、溶媒がまだ残っているものは、シンター上に減圧下で濾過した。サンプルをXRPDによって分析し、それらの結晶状態及び形態を評価した。XRPD分析(図21)の結果は、(水を唯一の例外として)すべてのサンプルが形態1であったことを示している。水溶液から得られたサンプルは、非晶質(図21、一番上のトレースは、明確なピークがないことを示している)であった。
【0096】
有機溶媒中の化合物1クエン酸塩の溶解性は、極めて限られていることが証明された。黄色の結晶と接触した溶媒が着色しなかったことは、溶解度が極めてわずかであったことを示している。有機溶媒スクリーニング・サンプルからの結晶残渣は、すべて形態1であった。本クエン酸塩は、100mg/分レベルで水に溶解することがわかった。溶液を蒸発させて回収した固体は、非晶質であることがわかった。溶解度スクリーンは、いかなる溶媒和化合物の存在も、又はクエン酸塩の多形をも示さなかった。
【0097】
実施例1〜12の結果を以下の表にまとめる。
【0098】
【表8】
【0099】
本発明に記載されている特定の実施態様の詳細は、制限として解釈すべきものではない。本発明の本質及び範囲を逸脱しない範囲で様々な等価物及び変更がなされてよく、そして、かかる等価な実施態様は、本発明の一部であると理解される。
【技術分野】
【0001】
本発明は、9E−15−(2−ピロリジン−1−イル−エトキシ)−7,12,25−トリオキサ−19,21,24−トリアザ−テトラシクロ[18.3.1.1(2,5).1(14,18)]ヘキサコサ−1(24),2,4,9,14,16,18(26),20,22−ノナエンのクエン酸塩に関する。さらに、本発明は、そのクエン酸塩を含有する医薬組成物及び特定の病状の処置におけるその塩の使用方法に関する。
【背景技術】
【0002】
9E−15−(2−ピロリジン−1−イル−エトキシ)−7,12,25−トリオキサ−19,21,24−トリアザ−テトラシクロ[18.3.1.1(2,5).1(14,18)]ヘキサコサ−1(24),2,4,9,14,16,18(26),20,22−ノナエン(化合物I)の化合物は、PCT/SG2006/000352に最初に記載され、多くの病状の処置のための薬学的に活性な薬剤としての大きな将来性を示した。この化合物が示す活性プロファイルに基づいて、本化合物の医薬開発が進められている。
【0003】
【化1】
【0004】
大量生産及び最終的には商業的使用に適した薬剤の開発において、対象者に対する薬剤活性の受容レベルは考慮されるべき重要な変数のひとつに過ぎない。例えば、医薬組成物の製剤においては、薬学的に活性な物質が、商業的製造プロセスにおいて確実に再生産でき、かつ、その薬学的に活性な物質が、それが曝される諸条件に耐えうる頑丈な形態であることが必須である。
【0005】
製造の観点から、薬学的に活性な物質の商業的製造プロセスは、同一の製造条件が用いられた場合は、同一の物質を生産するというものであることが重要である。加えて、薬学的に活性な物質は、固体形態で存在し、製造条件にわずかな変更があった場合に、それが、生産される薬学的に活性な物質の固体形態に大きな変化をもたらさないことが望ましい。例えば、製造プロセスが、確実に同一の結晶特性を有する物質を生産すること、そしてまた、そのプロセスが、同一レベルの水和状態を有する物質を生産することが重要である。
【0006】
加えて、薬学的に活性な物質は、分解、吸湿、さらにその固体形態に生じるその後の変化に対して安定であることが重要である。これは、薬学的に有効な成分の医薬製剤への組み込みを容易にする上で重要である。薬学的に活性な物質が、時間の経過とともに水を吸収するという意味において吸湿性(「粘着性」)であるなら、同一の投薬量を提供するために添加されるその薬学的に活性な物質の量が、水和の程度に依存して大幅に変化するであろうことから、その薬学的に活性な物質を薬剤に信頼性をもって配合することはほとんど不可能である。さらに、水和又は固体形態(「多形性」)におけるばらつきは、溶解性又は溶解速度といった物理化学的特性における変化をもたらす可能性があり、このことは、ひいては患者における一貫性のない経口吸収という結果を招きうる。
【0007】
したがって、薬学的に活性な薬剤の化学的安定性、固体状態の安定性及び「保存可能期間」は、極めて重要な要因である。理想的な状況では、薬学的に活性な薬剤及びそれを含有するあらゆる組成物は、活性、含水量、溶解度特性、固体形態等の有効成分の物理化学的特性に有意な変化を示すことなく、相当な期間にわたって有効に保存されうるものであるべきである。
【0008】
9E−15−(2−ピロリジン−1−イル−エトキシ)−7,12,25−トリオキサ−19,21,24−トリアザ−テトラシクロ[18.3.1.1(2,5).1(14,18)]ヘキサコサ−1(24),2,4,9,14,16,18(26),20,22−ノナエンに関して、最初の研究は、塩酸塩に関して実施され、多形が優勢であり、その化合物は、製造条件によって、複数の結晶形をとることが見出された。加えて、製造条件が一定だった場合ですら、バッチごとに多形の比率が異なることが観察された。これらバッチ間の一貫性の欠如が、商業的観点から、塩酸塩をあまり望ましくないものとした。
【0009】
したがって、上に示した課題の一つ以上を克服するか又は改善する9E−15−(2−ピロリジン−1−イル−エトキシ)−7,12,25−トリオキサ−19,21,24−トリアザ−テトラシクロ[18.3.1.1(2,5).1(14,18)]ヘキサコサ−1(24),2,4,9,14,16,18(26),20,22−ノナエンの塩類を開発することが望ましい。
【発明の概要】
【0010】
本発明は、9E−15−(2−ピロリジン−1−イル−エトキシ)−7,12,25−トリオキサ−19,21,24−トリアザ−テトラシクロ[18.3.1.1(2,5).1(14,18)]ヘキサコサ1(24),2,4,9,14,16,18(26),20,22−ノナエンのクエン酸塩を提供する。
【0011】
いくつかの実施態様において、塩は、結晶性である。
【0012】
いくつかの実施態様において、塩は、1:1のクエン酸塩である。いくつかの実施態様において、クエン酸塩は、X線回折において、2シータスケールで、22.4o±0.5oでピークを示す。
【0013】
いくつかの実施態様において、クエン酸塩はまた、X線回折において、2シータスケールで、10.0°±0.5°、15.6°±0.5°及び17.2°±0.5°でピークを示す。
【0014】
いくつかの実施態様において、クエン酸塩は、X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、21.1°±0.5°及び22.4°±0.5°からなる群より選択される、少なくとも4つのピークを示す。
【0015】
いくつかの実施態様において、クエン酸塩は、X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、21.1°±0.5°及び22.4°±0.5°からなる群より選択される、少なくとも6つのピークを示す。
【0016】
いくつかの実施態様において、クエン酸塩は、X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、21.1°±0.5°及び22.4°±0.5°でピークを示す。
【0017】
いくつかの実施態様において、クエン酸塩は、X線回折において、2シータスケールで、11.1°±0.5°、18.1°±0.5°、21.8°±0.5°、23.2°±0.5°及び27.6°±0.5°でピークを示す。
【0018】
いくつかの実施態様において、クエン酸塩は、X線回折において、2シータスケールで、7.0°±0.5°、14.0°±0.5°、19.0°±0.5°、19.8°±0.5°、23.6°±0.5°、24.3°±0.5°、25.2°±0.5°、25.7°±0.5°、26.1°±0.5°、26.5°±0.5°及び32.1°±0.5°でピークを示す。
【0019】
また、本発明は、上記のとおりの塩を含む医薬組成物を提供する。
【0020】
もう一つの実施態様では、本発明は、本発明の治療有効量の塩を、それを必要とする患者に投与することを含む、増殖性障害を治療又は予防する方法を提供する。いくつかの実施態様において、増殖性障害は、癌である。
【0021】
もう1つの実施態様において、本発明は、増殖性障害の処置における本発明の塩の使用を提供する。いくつかの実施態様において、増殖性障害は、癌である。
【0022】
もう一つの実施態様において、本発明は、増殖性障害の処置のための薬剤の製造における本発明の塩の使用を提供する。いくつかの実施態様において、増殖性障害は、癌である。
【図面の簡単な説明】
【0023】
【図1】図1は、バッチ1(THF中で調製したHCl塩)に関する高分解能粉末X線回折(XRPD)ディフラクトグラムを示す。
【図2】図2は、バッチ2(MeCN中で調製したHCl塩)に関する高分解能粉末X線回折(XRPD)ディフラクトグラムを示す。
【図3】図3は、バッチ3(アセトン中で調製したHCl塩)に関する高分解能粉末X線回折(XRPD)ディフラクトグラムを示す。
【図4】図4は、バッチ4(THF中で調製したクエン酸塩)に関する高分解能粉末X線回折(XRPD)ディフラクトグラムを示す。
【図5】図5は、バッチ5(MeCN中で調製したクエン酸塩)に関する高分解能粉末X線回折(XRPD)ディフラクトグラムを示す。
【図6】図6は、バッチ6(アセトン中で調製したクエン酸塩)に関する高分解能粉末X線回折(XRPD)ディフラクトグラムを示す。
【図7】図7は、バッチ7(アセトン中で調製したクエン酸塩(20gスケール))に関する高分解能粉末X線回折(XRPD)ディフラクトグラムを示す。
【図8】図8は、バッチ8(アセトン中で調製したクエン酸塩(20gスケール))に関する高分解能粉末X線回折(XRPD)ディフラクトグラムを示す。
【図9】図9は、バッチ4〜6に関する高分解能粉末X線回折(XRPD)ディフラクトグラムを示す。
【図10】図10は、バッチ4〜6に関する低分解能粉末X線回折ディフラクトグラムを示す。
【図11】図11は、バッチ4に関する、高分解能及び低分解能粉末X線回折ディフラクトグラムのオーバーレイを示す。
【図12】図12は、40℃及び相対湿度75%で1週間保存前後の、バッチ4に関する粉末X線回折トレースである。
【図13】図13は、40℃及び相対湿度75%で1週間保存前後の、バッチ5に関する粉末X線回折トレースである。
【図14】図14は、40℃及び相対湿度75%で1週間保存前後の、バッチ6に関する粉末X線回折トレースである。
【図15】図15は、バッチ4に関する示差走査熱量測定(DSC、上)及び熱重量分析(TGA)のデータを示す。
【図16】図16は、バッチ4〜6に関するDSCトレースのオーバーレイを示す。
【図17】図17は、バッチ4〜6に関するTGAトレースのオーバーレイを示す。
【図18】図18は、バッチ4に関する重量蒸気収着の運動エネルギーのプロットを示す。
【図19】図19は、バッチ4に関する重量蒸気収着の等温線のプロットを示す。
【図20】図20は、バッチ4に関する重量蒸気収着の実験前後の、粉末X線回折トレースを示す。
【図21】図21は、溶解性スクリーンからのサンプルの粉末X線回折トレースを示す。
【図22】図22は、d6−DMSO中のバッチ4に関する1H NMRスペクトルを示す。
【図23】図23は、D2O中のバッチ4に関する1H NMRスペクトルを示す。
【0024】
発明の詳細な説明
上述のように、今や、9E−15−(2−ピロリジン−1−イル−エトキシ)−7,12,25−トリオキサ−19,21,24−トリアザ−テトラシクロ[18.3.1.1(2,5).1(14,18)]ヘキサコサ−1(24),2,4,9,14,16,18(26),20,22−ノナエンの特定の塩類が、単一の頑丈な多形体として存在することがわかった。特に、本出願人は、この化合物のクエン酸塩が、単一の多形体として存在することを見出した。
【0025】
クエン酸の構造は、従来技術に熟練した読み手に明らかであると考えられるが、あらゆる不確実性を回避するために、その構造を以下に示す。
【化2】
【0026】
本願明細書に記載した塩酸塩及びクエン酸塩に関する比較研究は、表1に記載するバッチで実施した。
【0027】
【表1】
【0028】
化合物1に関する最初の研究は、塩酸塩を対象とした。以下にまとめるように、最初に調製された塩酸塩は、一貫性のない固体状態を生じ、粉末X線回折(XRPD)データにおいて著しいばらつきを示すことが分かった。
【0029】
塩酸塩としての化合物1は、バッチ1(THF中で調製)、バッチ2(アセトニトリル中で調製)及びバッチ3(アセトン中で調製)を結晶性材料として与える3つの異なる溶媒において調製された。図1、2及び3は、XRPDディフラクトグラムにおける、これらのバッチ間での著しいばらつきを示し、これらのHCl塩の結晶構造には、異なる溶媒中、同様の条件下で調製した場合でも、全般的な相違があることを示している。
【0030】
上記のように、塩酸塩に許容し得ないばらつきが見られる結果、これに代わる頑丈な固体状態が必要とされた。更なる発見努力により、クエン酸塩が、そのような頑丈な固体状態の一つであると確認された。
【0031】
9E−15−(2−ピロリジン−1−イル−エトキシ)−7,12,25−トリオキサ−19,21,24−トリアザ−テトラシクロ[18.3.1.1(2,5).1(14,18)]ヘキサコサ−1(24),2,4,9,14,16,18(26),20,22−ノナエンクエン酸塩の5つのバッチを特徴づけした。分析の結果を、以下の実施例で示す。
【0032】
粉末X線回折(XRPD)を用いて化合物1のクエン酸塩を特徴づけた。本発明のクエン酸塩に関する有意なX線回折ピーク(高分解能条件下で収集した)のリストを表2中に示す。
【0033】
【表2】
【0034】
表から分かるように、本クエン酸塩は、X線回折において、2シータスケールで、22.4°±0.5°でピークを示すものとして特徴づけることができる。
【0035】
また、本クエン酸塩は、X線回折において、2シータスケールで、10.0°±0.5°、15.6°±0.5°及び17.2°±0.5°でピークを示すものとして特徴づけることもできる。
【0036】
いくつかの実施態様において、本クエン酸塩は、さらに、X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、21.1°±0.5°及び22.4°±0.5°からなる群より選択される、少なくとも4つのピークを示すものとして特徴づけることもできる。
【0037】
いくつかの実施態様において、本クエン酸塩は、さらに、X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、21.1°±0.5°及び22.4°±0.5°からなる群より選択される、少なくとも6つのピークを示すものとして特徴づけることもできる。
【0038】
いくつかの実施態様において、本クエン酸塩は、さらに、X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、21.1°±0.5°及び22.4°±0.5°でピークを示すものとして特徴づけることもできる。
【0039】
いくつかの実施態様において、また、本クエン酸塩は、X線回折において、2シータスケールで、11.1°±0.5°、18.1°±0.5°、21.8°±0.5°、23.2°±0.5°及び27.6°±0.5°でピークを示す。
【0040】
いくつかの実施態様において、本クエン酸塩は、さらに、X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、11.1°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、18.1°±0.5°、21.8°±0.5°、21.1°±0.5°、22.4°±0.5°、23.2°±0.5°及び27.6°±0.5°でピークを示すものとして特徴づけることもできる。
【0041】
上記のピークは、特徴的なピークであるが、本クエン酸塩は、また、X線回折において、2シータスケールで、7.0°±0.5°、14.0°±0.5°、19.0°±0.5°、19.8°±0.5°、23.6°±0.5°、24.3°±0.5°、25.2°±0.5°、25.7°±0.5°、26.1°±0.5°、26.5°±0.5°及び32.1°±0.5°でピークを示す場合もある。
【0042】
当業者は理解されることであろうが、回折の相対的強度は、多くの要因(例えば試料調製の方法及び使用する計測器の型)に応じて変化しうる。加えて、特定の事例においては、上記ピークのいくつかは、検出可能でなくてもよい。
【0043】
本発明の塩は、化合物1の遊離塩基と適切な溶媒中のクエン酸とを反応させ、その反応混合物から、得られた塩を、結晶化、沈殿又は蒸発後に回収することにより製造することができる。
【0044】
塩を形成する反応は、遊離塩基が適切な溶解度を有する、任意の非干渉性溶媒又は溶媒混合物において実施することができる。この種の適切な溶媒の例は、アセトニトリル、テトラヒドロフラン及びアセトンを含む。この方法は、通常、20℃を超すような高い温度で、遊離塩基を適切な溶媒に溶解させることを含む。ある実施態様(例えばアセトン)において、遊離塩基は、約56℃の温度で溶媒中に溶解される。ある実施態様(例えばアセトニトリル)において、遊離塩基は、約82℃の温度で溶媒中に溶解される。
【0045】
一度遊離塩基が適切な溶媒に溶解したら、本方法は、次に、適切な量の酸を添加することを含む。酸は、通常、適切な溶媒(通常、遊離塩基を溶解に用いるのと同じ溶媒)中の溶液として加えられる。使用する酸の量は、通常、化学量論的に等量又は化学量論的にわずかに過剰量ではあるが、酸の量は変化させ得る。酸の添加に続いて、次に、本方法は、通常、添加温度で反応混合物を1時間撹拌し、その後、結晶化を容易にするために、反応混合物を反応温度未満の温度に冷却することを含む。一度所望のレベルの結晶形成が生じたら、その結晶を、濾過により分離し、従来技術において通常の手段を用いて乾燥させることができる。
【0046】
本発明のもう一つの実施態様は、増殖性障害の処置における本発明の塩の使用を提供する。この種の化合物の使用に関する配合及び方法論、並びにそれによって治療され得る障害は、PCT/SG2006/000352にて開示されているとおりである。
【0047】
本発明を、ここで、以下の非限定的な実施例によって記載する。塩酸塩を、比較例用に上記のように調製し、同様の方法で分析した。
【0048】
実施例1−溶媒としてのTHF中のHCl塩(バッチ1)の形成:
【0049】
化合物1の遊離塩基(0.200g、0.432mmoles、1当量)を、THF15mLに加えた。溶液を、溶解の完了が観察されるまで加熱還流し、1時間保持した。次に、1N HCl(0.518mL、0.518mmoles、1.2当量)を、還流条件下で徐々に加えた。混合物を、更に15分間還流し、次に冷却した。段階的な冷却によって、結晶化が観察された。結晶を、室温で12時間撹拌し、真空下で濾過した。生成物を、真空下で乾燥させて、165mgを得た。
【0050】
実施例2−溶媒としてのCH3CN中のHCl塩(バッチ2)の形成:
【0051】
化合物1(0.300g、0.648mmoles、1当量)の遊離塩基を、CH3CN70mLに加えた。溶液を、溶解の完了が観察されるまで加熱還流し、1時間保持した。次に1N HCl(0.778mL、0.778mmoles、1.2当量)を、還流条件下で徐々に加えた。混合物を、更に15分間還流し、次に冷却した。段階的な冷却によって、結晶化が観察された。結晶を、12時間室温で撹拌し、真空下で濾過した。生成物を、真空下で乾燥させて、190mgを得た。
【0052】
実施例3−溶媒としてのアセトン中のHCl塩(バッチ3)の形成:
【0053】
化合物1(0.200g、0.432mmoles、1当量)の遊離塩基を、アセトン50mLに加えた。溶液を、溶解の完了が観察されるまで加熱還流し、1時間保持した。次に1N HCl(0.518mL、0.518mmoles、1.2当量)を、還流条件下で徐々に加えた。混合物をさらに15分間還流し、次に冷却した。段階的な冷却によって、結晶化が観察された。結晶を、室温で12時間撹拌し、真空下で濾過した。生成物を、真空下で乾燥させて、180mgを得た。
【0054】
実施例4−溶媒としてのTHF中のクエン酸塩(バッチ4)の形成:
【0055】
化合物1(0.300g、0.648mmoles、1当量)の遊離塩基を、THF 12mLに加えた。溶液を、溶解の完了が観察されるまで加熱還流し、1時間保持した。次にTHF12mLに溶解したクエン酸(0.149g、0.778mmoles、1.2当量)の溶液を、還流条件下で徐々に加えた。混合物を、更に15分間還流し、次に冷却した。段階的な冷却によって、結晶化が観察された。結晶を室温で12時間撹拌し、減圧下で濾過した。生成物を、真空下で乾燥させて、250mgを得た。
【0056】
実施例5−溶媒としてのCH3CN中のクエン酸塩(バッチ5)の形成:
【0057】
化合物1(0.200g、0.432mmoles、1当量)の遊離塩基を、CH3CN 45mLに加えた。溶液を、溶解の完了が観察されるまで加熱還流し、1時間保持した。次にCH3CN 12mLに溶解したクエン酸(0.099g、0.518mmoles、1.2当量)の溶液を、還流条件下で徐々に加えた。混合物を、更に15分間還流し、次に冷却した。段階的な冷却によって、結晶化が観察された。結晶を、室温で12時間撹拌し、真空下で濾過した。生成物を、真空下で乾燥させて、220mgを得た。
【0058】
実施例6−溶媒としてのアセトン中のクエン酸塩(バッチ6)の形成:
【0059】
化合物1(0.200g、0.432mmoles、1当量)の遊離塩基をアセトン50mLに加えた。溶液を、溶解の完了が観察されるまで加熱還流し、1時間保持した。次に、アセトン20mLに溶解したクエン酸(0.099g、0.518mmoles、1.2当量)の溶液を、還流条件下で徐々に加えた。混合物を、更に15分間還流し、次に冷却した。段階的な冷却によって、結晶化が観察された。結晶を、室温で12時間撹拌し、真空下で濾過した。生成物を、真空下で乾燥させて、198mgを得た。
【0060】
実施例7−粉末X線回折研究
【0061】
条件1a(高分解能)
【0062】
粉末X線回折(XRPD)パターンを、CuK放射(1.54A)(40kV、ステップ・サイズ0.03°及びステップ時間0.5秒での30連続走査モードは、θ〜θmAであった)を用いたαSIEMENS D5000回折計で収集した。使用したθの範囲は、有効な22°〜50°を与えるサンプル−検出器距離であった。サンプル分析時間(X線ビームに曝される時間)は、13分33秒であった。データ収集に用いたソフトウェアは、DIFFRACplus-D5000 #1であり、データは、Diffrac Plus -D5000 #1を用いて分析して提示した。
【0063】
周囲条件下でテストするサンプルは、粉砕せずに調製した粉体を用いて、平板試料として調製した。サンプルの約100〜200mgを、ガラススライドに軽く押し付けて平坦な面を得た。
【0064】
条件1b(高分解能)
【0065】
粉末X線回折(XRPD)パターンを、Cu Kα放射(40kV、40mA)、自動化XYZステージ、オートサンプルポジショニング用レーザー・ビデオ顕微鏡及びHiStar二次元領域検出器を用いて、Bruker AXS C2 GADDS回折計で収集した。X線光学系は、0.3mmのピンホールコリメーターに接続した単一のGoebel多層ミラーからなる。ビーム拡散(すなわちサンプルに対するX線ビームの有効サイズ)は、約4mmであった。3.2°〜29.7°の有効な2θ範囲を与えるサンプル−検出器距離20cmでθ−θ連続走査モードを使用した。通常、サンプルは、120秒間X線束に曝される。データ収集に使用したソフトウェアは、WNT 4.1.16用のGADDSであり、データは、Diffrac Plus EVA v 9.0.0.2 又はv 13.0.0.2で分析して提示した。周囲条件下でテストするサンプルは、粉砕せずに調製された粉体を用いて、平板試料として調製した。サンプルの約1〜2mgを、ガラススライドに軽く押し付けて平坦な面を得た。
【0066】
条件2(低分解能)
【0067】
粉末X線回折パターンは、また、Cu Kα放射(40kV、40mA)、θ−2θゴニオメーター及びV4発散及び受光スリット、Geモノクロメーター及びLynxeye検出器を用いて、Bruker D8 回折計で収集した。計測器は、認証されたコランダム標準(NIST1976)を使用して、性能点検される。データ収集に使用したソフトウェアは Diffrac Plus XRD Commander v 2.5.0であり、データは、Diffrac Plus EVA v 11.0.0.2 又はv 13.0.0.2を用いて分析して提示した。サンプルは、入手した粉体そのままを使用した平板試料として、周囲条件下でテストした。研磨したゼロバックグラウンド(510)シリコンウエハーに切り込んだ空洞に、サンプル約15mgを緩やかに詰めた。分析の間、試料をそれ自体の面内で回転させた。データ収集の詳細は、以下の通りである:
○角度範囲:2〜42°2θ
○ステップサイズ:0.05°2θ
○収集時間:0.5秒/ステップ
【0068】
高分解能XRPDトレース(条件1a)を、サンプルの各々に対して得た。そして、図4〜8に示された結果は、クエン酸塩の5つのサンプルがすべて同じ結晶相のものであることを示している。また、バッチ4〜6に関するデータは、条件1b下でも収集され、図9に示すオーバレイは、これらのパターンが極めて類似しており、このことは、これらがすべて、同一の結晶質相のものであることを示している。
【0069】
低分解能XRPDトレース(条件2)も、Bruker GADDS回折計を用いて収集したので、多形性スクリーン分析用の標準パターンが得られた。バッチ4〜6のトレースのオーバレイを図10に示し、高分解能及び低分解能トレースの比較を図11に示す。
【0070】
低分解能XRPDトレースの収集のために調製したサンプルを、温度40度及び相対湿度75%に維持される容器に入れた。1週間後、相変化について調べるために、サンプルを、低分解能XRPD(条件2)で再分析した。結果を、最初のXRPDトレースと比較して、図12〜図14に示す。相変化が発生しなかったこと、そして、本発明のクエン酸塩はこれらの条件下で少なくとも1週間は安定であることがわかった。
【0071】
実施例8−核磁気共鳴(NMR)研究
【0072】
1H NMRスペクトルを、オートサンプラーを備え、DRX400コンソールによって制御されるBruker 400MHz測定器で収集した。自動化した実験は、標準Bruker搭載実験装置を用いて、Topspin v 1.3(パッチレベル8)で動作するICON-NMR v 4.0.4(build 1)を用いることで用意した。サンプルは、d6−DMSO又はD2O中で調製した。オフライン分析は、ACD SpecManager v 9.09(build 7703)を用いて行なった。
【0073】
1H NMRは、3つのサンプルすべてが同一化合物であることを示している。クエン酸塩の化学量論の決定は、対イオンのシグナルの積分によって行なった。しかし、これらのシグナルは、(バッチ4、図22の)スペクトルにおいては、DMSOシグナルの下に現れるので、クエン酸のシグナルの積分は実施できなかった。図23は、D2O中のバッチ4の1H−NMRを示す。この溶媒において、クエン酸のシグナルの積分は、化学量論が、予想どおり1:1であることを示した。
【0074】
実施例9−示差走査熱量測定(DSC)及び熱重量分析(TGA)
【0075】
示差走査熱量測定(DSC)データは、34ポジション・オートサンプラーを備えたMettler DSC 823eで収集した。計測器は、認定インジウムを使用して、エネルギー及び温度較正を行った。典型的には、0.5〜1.5mgの各サンプルを、ピンホール付きアルミニウムパンで、10℃/分で25℃から350℃に加熱した。50mL/分での窒素パージを、サンプル全体に維持した。計測器制御及びデータ分析ソフトウェアは、STARe v 9.10であった。
【0076】
熱重量分析(TGA)データを、34ポジション・オートサンプラーを備えたMettler TGA/SDTA 851eで収集した。計測器は、認定インジウムを使用して、温度較正を行った。典型的には、5〜30mgの各サンプルを、予め重量測定したアルミニウムるつぼに入れて10℃/分で周囲温度から350℃に加熱した。50mL/分での窒素パージを、サンプル全体に維持した。計測器制御及びデータ分析ソフトウェアは、STARe v 9.10であった。
【0077】
バッチ4(図15)に関するDSCトレースは、176℃で著しい熱イベントがあることを示している。TGAでは、対応して約20%の重量損失が見られる(図15)。この重量損失は、DSC吸熱の複雑な形状と共に、>176℃で全体量の減衰が生じていることを示している。理論に束縛されることは望まないが、このことは、塩の解離及びその後のクエン酸の分解を示している可能性がある。
【0078】
バッチ5及び6は、同様のDSC及びTGAトレース(図16及び17は、それぞれDSC及びTGAデータのオーバレイを示す)を示した。
【0079】
実施例10−重量蒸気収着(GVS)
【0080】
SMS Analysis Suiteソフトウェアで制御するSMS DVS Intrinsic moisture sorption analyserを用いて、収着等温線を得た。サンプル温度を、計測器の制御により25℃に維持した。湿度は、総流量200mL/分で、乾燥及び湿潤窒素の流れを混合することによって制御した。相対湿度は、サンプル近辺に位置した較正済Rotronicプローブ(1.0〜100%RHのダイナミックレンジ)で測定した。%RHの関数としてのサンプル中の重量変化(質量緩和)は、微量天秤(精度±0.005mg)で常にモニターした。典型的には5〜20mgのサンプルを、周囲条件下、風袋計量したメッシュ・ステンレス・スチールバスケットに入れた。サンプルを40%RH及び25℃(典型的な室温条件)にて充填し、取り出した。水分収着等温線解析は、以下に概説するように行った(2走査で1サイクルを完結)。標準等温線解析を、25℃で、10%RH間隔で、0.5〜90%RH範囲にわたって実施した。
【0081】
【表3】
【0082】
本クエン酸塩の吸湿性を、バッチ4に関する重量蒸気収着実験を行うことによって調べた。環境の湿度を2つの完全なサイクルを通して変化させながら、サンプル約20mgを、25℃に保った。図18に示す動的プロットは、バッチ4のサンプルが、各%RHステップで平衡重量に達することを示している。サンプルは、実験の初期において、平衡に達するのにより長くかかっている。これは、残留溶媒の置換による可能性がある。
【0083】
図19に示す等温線プロットは、サンプルが、40%RH〜90%RHの間で、<0.6%の水を吸収することを示している。最大重量差(0%RH〜90%RH)は、1% w/w未満であり、このことは、本クエン酸塩が、吸湿性ではないことを示している。加えて、本クエン酸塩が水和形態で存在することに関する証拠はない。
【0084】
【表4】
【0085】
GVS実験終了時、サンプルを回収し、XRPDで分析し、あらゆる全体的な相変化を調べた。結果(図20)は、全体的な相変化はないことを示している。
【0086】
実施例11−高速液クロマトグラフィ(HPLC)による化学純度の決定
【0087】
ダイオードアレイ検出器を備え、ChemStation software v B.02.01-SR1を用いたAgilent HP1100シリーズシステムで、純度分析を行った。使用したパラメーターを表5にまとめた。
【0088】
【表5】
【0089】
本クエン酸塩のバッチ4〜6化学純度は、このHPLC手順を用いて決定した。数値結果を表6に示す。
【0090】
【表6】
【0091】
表から分かるように、各サンプルの測定された純度は、98.1%を超えている。
【0092】
実施例12−溶解性及び多形性評価
【0093】
調査した各溶媒について、化合物1の約8mgを、8mLのスクリューキャップ付きガラスバイアルに量り入れた。溶媒は、10倍容量のアリコートを加え、混合物を超音波処理し、(ホットエアガンで)温めて溶解を促した。100倍容量の溶媒を加えても、溶解が成し遂げられない場合は、更に100倍容量のアリコートを加えた。各実験及び観察の詳細(表7)は、完全に溶解が成し遂げられたのは、水だけにおいてであることを示している。
【0094】
【表7】
【0095】
次に、バイアルを、湿度チャンバーに置き、25℃/50℃(8時間サイクル)で24時間のサイクルに付した。この時間の終わりに、サンプルを検査し、次に緩く蓋をして、溶媒が蒸発するにまかせた。乾燥させたこれらのサンプルを、次に石英アレイに移し、一方、溶媒がまだ残っているものは、シンター上に減圧下で濾過した。サンプルをXRPDによって分析し、それらの結晶状態及び形態を評価した。XRPD分析(図21)の結果は、(水を唯一の例外として)すべてのサンプルが形態1であったことを示している。水溶液から得られたサンプルは、非晶質(図21、一番上のトレースは、明確なピークがないことを示している)であった。
【0096】
有機溶媒中の化合物1クエン酸塩の溶解性は、極めて限られていることが証明された。黄色の結晶と接触した溶媒が着色しなかったことは、溶解度が極めてわずかであったことを示している。有機溶媒スクリーニング・サンプルからの結晶残渣は、すべて形態1であった。本クエン酸塩は、100mg/分レベルで水に溶解することがわかった。溶液を蒸発させて回収した固体は、非晶質であることがわかった。溶解度スクリーンは、いかなる溶媒和化合物の存在も、又はクエン酸塩の多形をも示さなかった。
【0097】
実施例1〜12の結果を以下の表にまとめる。
【0098】
【表8】
【0099】
本発明に記載されている特定の実施態様の詳細は、制限として解釈すべきものではない。本発明の本質及び範囲を逸脱しない範囲で様々な等価物及び変更がなされてよく、そして、かかる等価な実施態様は、本発明の一部であると理解される。
【特許請求の範囲】
【請求項1】
9E−15−(2−ピロリジン−1−イル−エトキシ)−7,12,25−トリオキサ−19,21,24−トリアザ−テトラシクロ[18.3.1.1(2,5).1(14,18)]ヘキサコサ−1(24),2,4,9,14,16,18(26),20,22−ノナエンのクエン酸塩。
【請求項2】
塩が結晶である、請求項1記載の塩。
【請求項3】
塩が1:1塩である、請求項1又は2記載の塩。
【請求項4】
X線回折において、2シータスケールで、22.4o±0.5oでピークを示す、請求項1〜3のいずれか一項記載の塩。
【請求項5】
X線回折において、2シータスケールで、10.0°±0.5°、15.6°±0.5°及び17.2°±0.5°でもピークを示す請求項4記載の塩。
【請求項6】
X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、21.1°±0.5°及び22.4°±0.5°からなる群より選択される、少なくとも4つのピークを示す、請求項1〜5のいずれか一項記載の塩。
【請求項7】
X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、21.1°±0.5°及び22.4°±0.5°からなる群より選択される、少なくとも6つのピークを示す、請求項5記載の塩。
【請求項8】
X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、21.1°±0.5°及び22.4°±0.5°でピークを示す、請求項6記載の塩。
【請求項9】
X線回折において、2シータスケールで、11.1°±0.5°、18.1°±0.5°、21.8°±0.5°、23.2°±0.5°及び27.6°±0.5°でもピークを示す、請求項8記載の塩。
【請求項10】
X線回折において、2シータスケールで、7.0°±0.5°、14.0°±0.5°、19.0°±0.5°、19.8°±0.5°、23.6°±0.5°、24.3°±0.5°、25.2°±0.5°、25.7°±0.5°、26.1°±0.5°、26.5°±0.5°及び32.1°±0.5°でもピークを示す、請求項9記載の塩。
【請求項11】
請求項1〜10のいずれか一項記載の塩を含む、医薬組成物。
【請求項12】
請求項1〜10のいずれか一項記載の治療有効量の塩を、それを必要とする患者に投与することを含む、増殖性障害を治療又は予防する方法。
【請求項13】
増殖性障害が癌である、請求項12記載の方法。
【請求項14】
増殖性障害の処置における、請求項1〜10のいずれか一項記載の塩の使用。
【請求項15】
増殖性障害が癌である、請求項14記載の使用。
【請求項16】
増殖性障害の処置のための薬剤の製造における、請求項1〜10のいずれか一項記載の塩の使用。
【請求項17】
増殖性障害が癌である、請求項16記載の使用。
【請求項1】
9E−15−(2−ピロリジン−1−イル−エトキシ)−7,12,25−トリオキサ−19,21,24−トリアザ−テトラシクロ[18.3.1.1(2,5).1(14,18)]ヘキサコサ−1(24),2,4,9,14,16,18(26),20,22−ノナエンのクエン酸塩。
【請求項2】
塩が結晶である、請求項1記載の塩。
【請求項3】
塩が1:1塩である、請求項1又は2記載の塩。
【請求項4】
X線回折において、2シータスケールで、22.4o±0.5oでピークを示す、請求項1〜3のいずれか一項記載の塩。
【請求項5】
X線回折において、2シータスケールで、10.0°±0.5°、15.6°±0.5°及び17.2°±0.5°でもピークを示す請求項4記載の塩。
【請求項6】
X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、21.1°±0.5°及び22.4°±0.5°からなる群より選択される、少なくとも4つのピークを示す、請求項1〜5のいずれか一項記載の塩。
【請求項7】
X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、21.1°±0.5°及び22.4°±0.5°からなる群より選択される、少なくとも6つのピークを示す、請求項5記載の塩。
【請求項8】
X線回折において、2シータスケールで、7.9°±0.5°、10.0°±0.5°、15.6°±0.5°、15.9°±0.5°、16.8°±0.5°、17.2°±0.5°、21.1°±0.5°及び22.4°±0.5°でピークを示す、請求項6記載の塩。
【請求項9】
X線回折において、2シータスケールで、11.1°±0.5°、18.1°±0.5°、21.8°±0.5°、23.2°±0.5°及び27.6°±0.5°でもピークを示す、請求項8記載の塩。
【請求項10】
X線回折において、2シータスケールで、7.0°±0.5°、14.0°±0.5°、19.0°±0.5°、19.8°±0.5°、23.6°±0.5°、24.3°±0.5°、25.2°±0.5°、25.7°±0.5°、26.1°±0.5°、26.5°±0.5°及び32.1°±0.5°でもピークを示す、請求項9記載の塩。
【請求項11】
請求項1〜10のいずれか一項記載の塩を含む、医薬組成物。
【請求項12】
請求項1〜10のいずれか一項記載の治療有効量の塩を、それを必要とする患者に投与することを含む、増殖性障害を治療又は予防する方法。
【請求項13】
増殖性障害が癌である、請求項12記載の方法。
【請求項14】
増殖性障害の処置における、請求項1〜10のいずれか一項記載の塩の使用。
【請求項15】
増殖性障害が癌である、請求項14記載の使用。
【請求項16】
増殖性障害の処置のための薬剤の製造における、請求項1〜10のいずれか一項記載の塩の使用。
【請求項17】
増殖性障害が癌である、請求項16記載の使用。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【公表番号】特表2012−533539(P2012−533539A)
【公表日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願番号】特願2012−520571(P2012−520571)
【出願日】平成22年7月14日(2010.7.14)
【国際出願番号】PCT/SG2010/000265
【国際公開番号】WO2011/008172
【国際公開日】平成23年1月20日(2011.1.20)
【出願人】(506096349)エス*バイオ プライベート リミティッド (10)
【Fターム(参考)】
【公表日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願日】平成22年7月14日(2010.7.14)
【国際出願番号】PCT/SG2010/000265
【国際公開番号】WO2011/008172
【国際公開日】平成23年1月20日(2011.1.20)
【出願人】(506096349)エス*バイオ プライベート リミティッド (10)
【Fターム(参考)】
[ Back to top ]