
A−βの排泄機構の機能低下によるアルツハイマー病の新規モデル非ヒト動物
【課題】
A-βが産生されることに加えて代謝/排泄が傷害され、特に孤発性のアルツハイマー病のモデルとして好適に利用できる、新規な非ヒト動物を提供することを目的とする。
【解決手段】
アミロイドペプチド前駆体タンパク質(APP)、プレセニリン1(PS1)、又はプレセニリン2(PS2)から選ばれる一以上をコードする遺伝子に変異を有する非ヒト動物に対し、αTTPをコードする遺伝子を欠失させ、A-βの代謝/排泄に障害を有するノックアウト非ヒト動物である。これは脳内での内因性のA-βの産生及び蓄積が起こって実際にADを発症するという、AD発症機構の初のモデル動物である。
A-βが産生されることに加えて代謝/排泄が傷害され、特に孤発性のアルツハイマー病のモデルとして好適に利用できる、新規な非ヒト動物を提供することを目的とする。
【解決手段】
アミロイドペプチド前駆体タンパク質(APP)、プレセニリン1(PS1)、又はプレセニリン2(PS2)から選ばれる一以上をコードする遺伝子に変異を有する非ヒト動物に対し、αTTPをコードする遺伝子を欠失させ、A-βの代謝/排泄に障害を有するノックアウト非ヒト動物である。これは脳内での内因性のA-βの産生及び蓄積が起こって実際にADを発症するという、AD発症機構の初のモデル動物である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アルツハイマー病のモデル動物として利用できる、αトコフェロール転移タンパク質(αTTP)遺伝子ノックアウト非ヒト動物に関する。
【背景技術】
【0002】
アルツハイマー病(AD)におけるアミロイドカスケード仮説の重要な分子の一つであるA-β 1-40、1-42は、アミロイドペプチド前駆体タンパク質(amyloid precursor protein;APP)から切り出されることにより産生されるペプチドである。アルツハイマー病では、このA-βが過剰となり老人斑として凝集すると考えられている。そして、脳組織内のA-βはネプリライシン(neprilysin;NEP)などにより分解されたり、血液脳関門(BBB)を介して脳から血液へと排泄されると考えられている。
【0003】
ところで、アルツハイマー病には、優性遺伝する家族性アルツハイマー病と、それ以外の孤発性アルツハイマー病の2種類が知られている。家族性ADでは、A-βの過剰産生が原因とされ、上述のAPPとプレセニリン1(Presenilin 1; PS1)、及びプレセニリン2 が原因遺伝子として特定されている。一方、アルツハイマー病の大部分(99%以上)を占める孤発性ADではA-βの代謝/排泄の障害が原因とされている。
【0004】
このようなアルツハイマー病の機序解明、及び治療薬開発のため、ADのモデル動物が従来作製されている。例えば、(特許文献1)には、スウェーデン型変異を含むヒトAPPポリペプチドをコードするDNAが、Thy-1プロモーターエレメントの転写制御下で発現される、ヒト以外のトランスジェニック動物が開示されている。
このトランスジェニック動物は、スウェーデン型変異APP遺伝子によってA-βが産生され、その結果、AD罹病患者で観察される認知障害を示すものである。
その他、APPをコードする遺伝子に変異を有するトランスジェニック動物としてはPDGF-APPマウス、Tg2576マウス、APP22/23マウス、NORβマウスが知られ、またPS1をコードする遺伝子に変異を有するトランスジェニック動物としてはPS-1M146L/Vマウス、PS-1A246Eマウス、PS-1L286V、HH136Rマウスが知られており、これらの組み合わせによるAPP×PS1ダブルトランスジェニックマウスなども知られている。
【0005】
しかし、上記のトランスジェニック動物は、A-βの過剰産生が原因といわれる家族性ADについてのモデル動物であり、アルツハイマー病の大部分を占める孤発性ADのモデルとしては不十分なものであった。
それゆえ、孤発性ADの機序解明のため、A-βが産生されることに加えて代謝/排泄が傷害されているADモデル動物の開発が望まれていた。
【0006】
【特許文献1】特表2000−515743号公報(請求項17)
【発明の開示】
【発明が解決しようとする課題】
【0007】
そこで本発明は、A-βが産生されることに加えて代謝/排泄が傷害され、特に孤発性のアルツハイマー病のモデルとして好適に利用できる、新規な非ヒト動物を提供することを目的とする。
【課題を解決するための手段】
【0008】
上記課題を解決するため、本発明者らは、αトコフェロール転移タンパク質(α-tocopherol transfer protein;αTTP)遺伝子を欠失させたαTTPノックアウト動物を作製し、このノックアウト動物とADとの関係について調べた。すなわち、αTTPノックアウト動物は、体内・脳内でのビタミンE欠乏のため、中枢神経系に慢性的な酸化ストレスがかかっており、この酸化ストレスがA-βの代謝/排泄に影響を及ぼすか否かについて検証を行った。
その結果、酸化ストレスはA-βの排泄障害を引き起こすことが明らかとなったため、この知見に基づき発明を完成した。
【0009】
すなわち、本発明は以下の(1)〜(7)から構成される。
(1) αトコフェロール転移タンパク質(αTTP)をコードする遺伝子を欠失させ、βアミロイドペプチド(A-β)の代謝/排泄に障害を有するノックアウト非ヒト動物。
(2) アミロイドペプチド前駆体タンパク質(Amyloid precursor protein; APP)、プレセニリン1(Presenilin 1; PS1)、又はプレセニリン2(Presenilin 2; PS2)から選ばれる一以上をコードする遺伝子に変異を有する非ヒト動物に対し、αTTPをコードする遺伝子を欠失させ、A-βの代謝/排泄に障害を有するノックアウト非ヒト動物。
(3) 上記(2)記載の非ヒト動物において、APPをコードする遺伝子の変異が、スウェーデン型変異(APP770についてLys670→Asn, Met671→Leu、APP751についてLys651→Asn, Met652→Leu、APP695についてLys695→Asn, Met596→Leu)であることを特徴とするノックアウト非ヒト動物。
(4) 上記(1)〜(3)のいずれか記載の非ヒト動物が、げっ歯類動物であることを特徴とするノックアウト非ヒト動物。
(5) 上記(4)記載のげっ歯類動物が、マウスであることを特徴とするノックアウト非ヒト動物。
(6) 上記(1)〜(5)のいずれか記載の非ヒト動物を用いたアルツハイマー病予防薬・治療薬のスクリーニング方法。
(7) 上記(6)記載のスクリーニング方法を用いて得られたアルツハイマー病予防薬・治療薬。
(8) 請求項1〜5のいずれか記載の非ヒト動物を用いた、PET、SPECT、もしくはMRIによる画像診断、又は髄液、血液、もしくは尿中の物質の定量によるアルツハイマー病の診断方法。
【発明の効果】
【0010】
家族性アルツハイマー病ではA-βの産生亢進によるA-β蓄積が重要な原因と考えられている。一方、患者の大多数を占める孤発性アルツハイマー病においてはA-β蓄積の原因としてその代謝あるいは排泄に低下があることが最近明らかになっている。しかし、A-βの排泄の異常が原因でAD症状をきたすモデル動物は未だ存在しなかった。
本発明のαTTPノックアウト動物は、中枢神経への慢性の酸化ストレスの結果、A-βの排泄が遅延するという表現型を持つ初めての動物モデルであり、したがって孤発性アルツハイマー病における、代謝/排泄の遅延の機序解明に有用である。
【0011】
特に、APP変異、PS1変異、又はPS2変異トランスジェニック動物と掛け合わせた場合には、AD同様に脳内のA-βが酸化ストレスによって排泄機構の機能低下を生じ、脳内での内因性のA-βの産生及び蓄積が起こって実際にADを発症するという、AD発症機構の初のモデル動物である。
したがって、この新規ノックアウト動物は、孤発性アルツハイマー病のドラックデスカバリー等の治療法の開発に現在唯一の動物モデルになり得ると考えられる。
【発明を実施するための最良の形態】
【0012】
以下、本発明について詳述する。
本発明のノックアウト非ヒト動物は、αトコフェロール転移タンパク質(αTTP)をコードする遺伝子を欠失させて作製される。ここで欠失とは、αTTPをコードする遺伝子の一部もしくは全部が破壊・欠損・置換・挿入等により不活性化され、野生型におけるαTTPを発現する機能を失った状態をいう。このαTTPノックアウト動物は、中枢神経への慢性的な酸化ストレスが加わることにより、後の実施例で述べるようにβアミロイドペプチド(A-β)の代謝/排泄に障害を有している。
【0013】
上記のαTTPノックアウト動物は、従来知られた手法を適宜利用して作製することができる。具体的には、クローニングされたαTTP遺伝子に、単純ヘルペスウイルスのチミジンキナーゼ遺伝子等のマーカー遺伝子を導入してターゲティングベクターを作製し、このターゲティングベクターをエレクトロポレーション法等によってES細胞に導入して培養した後、G418やガンシクロビア等の抗生物質により相同的組換えを起こしたES細胞を選択する。そして、組換えES細胞をマウス等の未分化胚芽細胞にマイクロインジェクションし、仮親の子宮に戻すことによってキメラ動物を得る。このキメラ動物を野生型の雌と交配させてαTTPヘテロ組換え動物(+/-)を作製し、そのαTTPヘテロ組換え動物の雄と雌を交配させることによって目的のαTTPノックアウト動物(-/-)を得ることができる。
【0014】
αTTPノックアウト動物の対象としては、マウスの他、ラット、ハムスター、モルモット、イヌ、ネコ、ウサギ、ウシ、ブタ、ヒツジ等の哺乳動物を用いることができる。その中でも、受精卵の培養、体外受精等の技術が進んでいる、マウス等のげっ歯類動物が特に好ましい。
【0015】
上記のαTTPノックアウト動物は、慢性の酸化ストレスによってA-βの代謝/排泄に障害を持つ初めての動物モデルであり、特に孤発性アルツハイマー病における、代謝/排泄の遅延の機序解明や治療法の開発に有用な動物モデルとして利用することができる。しかし、αTTPノックアウト動物では、脳内での内因性のA-βの産生がないためA-βは蓄積しない。これに対し本発明では、別の実施形態として、アミロイドペプチド前駆体タンパク質(APP)、プレセニリン1(PS1)、又はプレセニリン2(PS2)から選ばれる一以上をコードする遺伝子に変異を有する非ヒト動物に対し、αTTPをコードする遺伝子を欠失させることにより、A-βの産生を認めかつA-β排泄が傷害されている新規ノックアウト非ヒト動物を提供するものである。このノックアウト動物は、脳内にA-βが産生し、酸化ストレスによる排泄機構の機能低下が生じて、実際にA-βが蓄積しADを発症するという、最近有力なAD発症機構の初のモデル動物として利用することができる。
【0016】
上記のADモデル動物は、APP、PS1又はPS2の一以上に変異を有するトランスジェニック動物と、αTTPノックアウト動物とを掛け合わせることによって得ることができる。ここで、APP、PS1、又はPS2変異トランスジェニック動物は、既知の手法を用いて作製することができる。例えば変異APP遺伝子や変異PS1遺伝子、又は変異PS2遺伝子を受精卵の前核にマイクロインジェクションし、生き残ったものを仮親の卵管に移植する方法や、ES細胞に遺伝子を導入しておき、それを胚盤胞腔中に注入しキメラ胚を形成させ、それを仮親の子宮に移植する方法等を用いることができる。
【0017】
APPには、複数のイソ型(APP770、751、及び695)があり、それぞれにスウェーデン型変異、オランダ型変異、ロンドン型変異、フロリダ型、フランダース型等が知られているが、いずれも適用可能である。なお、スウェーデン型変異はAPP770についてLys670→Asn, Met671→Leu、APP751についてLys651→Asn, Met652→Leu、APP695についてLys695→Asn, Met596→Leuであり、オランダ型変異はAPP770についてAla692→Gly、APP751についてAla673→Gly、APP695についてAla617→Glyであり、ロンドン型変異はAPP770についてVal717→Ile、APP751についてVal698→Ile、APP695についてVal642→Ileである。また、家族性ADの疾患モデルであるSweden型変異APP770トランスジェニックマウス(Tg2576、Taconic社製)等が知られており、これをαTTPノックアウトマウスと掛け合わせるトランスジェニックマウスとして用いても良い。
【0018】
その他、APP、PS1、及びPS2には下記に示すような多数の変異が知られており、これらもαTTPノックアウト動物と掛け合わせる対象として用いることができる。
【0019】
<APP変異>
1.APP Glu665Asp (g.269483G>C) Not pathogenic
2.APP APPKM670/671NL; Swedish APP (g.[269498G>T;269499A>C])
3.APP Ala673Thr (g.269505G>A) Not pathogenic
4.APP His677Arg (g.269518A>G) Not pathogenic
5.APP Asp678Asn, Tottori APP (g.269520C>G)
6.APP Ala692Gly; Flemish APP (g.275267C>G)
7.APP Glu693Gln; Dutch APP (g.275269G>C)
8.APP Glu693Gly; Arctic APP (g.275270A>G)
9.APP Asp694Asn; Iowa APP (g.275272G>A)
10.APP Ala713Thr (g.[275329G>A;275337G>A]) (Pathogenic nature unclear)
11.APP Ala713Thr (g.275329G>A)
12.APP Ala713Val (g.275330C>T) Not pathogenic
13.APP Thr714Ala, Iranian APP (g.275332A>G)
14.APP Thr714Ile, Austrian APP (g.275333C>T)
15.APP Val715Met; French APP (g.275335G>A)
16.APP Val715Ala; German APP (g.275336T>C)
17.APP Ile716Val; Florida APP (g.275338A>G)
18.APP Ile716Thr (g.275339T>C)
19.APP Val717Ile; London APP (g.275341G>A)
20.APP Val717Leu (g.275341G>C)
21.APP Val717Phe (g.275341G>T)
22.APP Val717Gly (g.275342T>G)
23.APP Leu723Pro; Australian APP (g.275360T>C)
【0020】
<PS1変異>
1.PS1 Arg35Gln (g.22789G.A)
2.PS1 Ala79Val (g.22921C>T)
3.PS1 Val82Leu (g.22929G>C)
4.PS1 DeltaI83/M84 (g.22932-22937del)
5.PS1 Leu85Pro (g.22939T>C)
6.PS1 Val89Leu (g.22950G>T)
7.PS1 Cys92Ser (g.22960G>C)
8.PS1 Val94Met (g.22965G>A)
9.PS1 Val96Phe (g.22971G>T)
10.PS1 Phe105Leu (g.23000T>G)
11.PS1 Leu113Pro (g.23023T>C)
12.PS1 Intron4; InsTAC (g.23024delG)
13.PS1 Tyr115His (g.25546T>C)
14.PS1 Tyr115Asp (g.25546T>G)
15.PS1 Tyr115Cys (g.25547A>G)
16.PS1 Thr116Asn (g.25550C>A)
17.PS1 Thr116Ile (g.25550C>T)
18.PS1 Pro117Ser (g.25552C>T)
19.PS1 Pro117Arg (g.25553C>G)
20.PS1 Pro117Leu (g.25553C>T)
21.PS1 Glu120Lys (g.25561G>A)
22.PS1 Glu120Asp (g.25563A>C)
23.PS1 Glu120Asp (g.25563A>T)
24.PS1 Glu123Lys (g.25570G>A)
25.PS1 Asn135Asp (g.25606A>G)
26.PS1 Met139Val (g.25618A>G)
27.PS1 Met139Lys (g.25619T>A)
28.PS1 Met139Thr (g.25619T>C)
29.PS1 Met139Ile (g.25620G>A)
30.PS1 Ile143Phe (g.25630A>T)
31.PS1 Ile143Thr (g.25631T>C)
32.PS1 Ile143Met (g.25632T>G)
33.PS1 Met146Leu (g.25639A>C)
34.PS1 Met146Val (g.25639A>G)
35.PS1 Met146Leu (g.25639A>T)
36.PS1 Met146Ile (g.25641G>A)
37.PS1 Met146Ile (g.25641G>C)
38.PS1 Met146Ile (g.25641G>T)
39.PS1 Thr147Ile (g.25643C>T)
40.PS1 Leu153Val (g.25660C>G)
41.PS1 Tyr154Asn (g.25663T>A)
42.PS1 Tyr154Cys (g.25664A>G)
43.PS1 InsFI (g.25669_25670insTTATAT)
44.PS1 His163Tyr (g.38784C>T)
45.PS1 His163Arg (g.38785A>G)
46.PS1 Trp165Gly (g.38790T>G)
47.PS1 Trp165Cys (g.38792G>C)
48.PS1 Leu166Pro (g.38794T>C)
49.PS1 Leu166Arg (g.38794T>G)
50.PS1 DeltaI167 (g.38795_38797delTAT)
51.PS1 Ser169Pro (g.38802T>C)
52.PS1 Ser169Leu (g.38803C>T)
53.PS1 Leu171Pro (g.38809T>C)
54.PS1 Leu173Trp (g.38815T>G)
55.PS1 Leu174Met (g.38817C>A)
56.PS1 Phe175Ser (g.38821T>C) Not pathogenic
57.PS1 Phe177Leu (g.38826T>C)
58.PS1 Phe177Ser (g.38827T>C)
59.PS1 Ser178Pro (g.38829T>C)
60.PS1 Gly183Val (g.38845G>T)
61.PS1 Glu184Asp (g.44571A>C)
62.PS1 Gly206Ser (g.44635G>A)
63.PS1 Gly206Ala (g.44636G>C)
64.PS1 Gly206Val (g.44636G>T)
65.PS1 Gly209Arg (g.44644G>A)
66.PS1 Gly209Glu (g.44645G>A)
67.PS1 Gly209Val (g.44645G>T)
68.PS1 Ile213Leu (g.44656A>C)
69.PS1 Ile213Phe (g.44656A>T)
70.PS1 Ile213Thr (g.44657T>C)
71.PS1 Gly217Asp (g.44669G>A)
72.PS1 Leu219Phe (g.44674C>T)
73.PS1 Leu219Pro (g.44675T>C)
74.PS1 Gln222Arg (g.44684A>G)
75.PS1 Gln222His (g.44685G>C)
76.PS1 Leu226Arg (g.44696T>G)
77.PS1 Ile229Phe (g.44704A>T)
78.PS1 Ala231Thr (g.44710G>A)
79.PS1 Ala231Val (g.44711C>T)
80.PS1 Met233Leu (g.44716A>C)
81.PS1 Met233Val (g.44716A>G)
82.PS1 Met233Thr (g.44717T>C)
83.PS1 Leu235Val (g.44722C>G)
84.PS1 Leu235Pro (g.44723T>C)
85.PS1 Phe237Ile (g.44728T>A)
86.PS1 Phe237Leu (g.44728T>C)
87.PS1 Ala246Glu (g.44756C>A)
88.PS1 Leu250Val (g.44767T>G)
89.PS1 Leu250Ser (g .44768T>C)
90.PS1 Tyr256Ser (g.44786A>C)
91.PS1 Ala260Val (g.49964C>T)
92.PS1 Val261Phe (g.49966G>T)
93.PS1 Leu262Phe (g.49971G>C)
94.PS1 Cys263Arg (g.49972T>C)
95.PS1 Cys263Phe (g.49973G>T)
96.PS1 Pro264Leu (g.49976C>T)
97.PS1 Pro267Ser (g.49984C>T)
98.PS1 Pro267Leu (g.49985C>T)
99.PS1 Arg269Gly (g.49990C>G)
100.PS1 Arg269His (g.49991G>A)
101.PS1 Leu271Val (g.49996C>G)
102.PS1 Val272Ala (g.50000T>C)
103.PS1 Glu273Ala (g.50003A>C)
104.PS1 Thr274Arg (g.50006C>G)
105.PS1 Arg278Lys (g.50018G>A)
106.PS1 Arg278Thr (g.50018G>C)
107.PS1 Arg278Ile (g.50018G>T)
108.PS1 Glu280Ala (g.50024A>C)
109.PS1 Glu280Gly (g.50024A>G)
110.PS1 Leu282Val (g.50029C>G)
111.PS1 Leu282Arg (g.50030T>G)
112.PS1 Pro284Leu (g.50036C>T)
113.PS1 Ala285Val (g.50039C>T)
114.PS1 Leu286Val (g.50041C>G)
115.PS1 Delta9 (g.56305-62162del)
116.PS1 Delta9Finn (g.56681-61235del)
117.PS1 Delta9 (g.58304G>A)
118.PS1 Delta9 (g.58304G>T)
119.PS1 Glu318Gly (g.58389A>G) Not pathogenic
120.PS1 InsR (g.63786_63787insTCG)
121.PS1 Thr354Ile (g.63792C>T)
122.PS1 Arg358Gln (g.63804G>A)
123.PS1 Ser365Tyr (g.63825C>A)
124.PS1 Arg377Met (g.69044G>T)
125.PS1 Gly378Glu (g.69047G>A)
126.PS1 Gly378Val (g.69047G>T)
127.PS1 Gly384Ala (g.69065G>C)
128.PS1 Ser390Ile (g.69083G>T)
129.PS1 Leu392Val (g.69088C>G)
130.PS1 Leu392Pro (g.69089T>C)
131.PS1 Gly394Val (g.69095G>T)
132.PS1 Asn405Ser (g.69128A>G)
133.PS1 Ala409Thr (g.69139G>A)
134.PS1 Cys410Tyr (g.69143G>A)
135.PS1 Leu418Phe (g.71058G>T)
136.PS1 Leu424Arg (g.71075T>G)
137.PS1 Ala426Pro (g.71080G>C)
138.PS1 Ala431Glu (g.71096C>A)
139.PS1 Ala431Val (g.71096C>T)
140.PS1 Ala434Cys (g.[71104G>T;71105C>G])
141.PS1 Leu435Phe (g.71107C>T)
142.PS1 Pro436Ser (g.71110C>T)
143.PS1 Pro436Gln (g.71111C>A)
144.PS1 Ile439Val (g.71119A>G)
【0021】
<PS2変異>
1.PS2 Arg62His ( g.1839G>A)
2.PS2 Thr122Pro (g.3636A>C)
3.PS2 Ser130Leu (g.3661C>T)
4.PS2 Asn141Ile (g.3694A>T)
5.PS2 Val148Ile (g.3714G>A)
6.PS2 Gln228Leu (g.7039A>T)
7.PS2 Met239Val (g.7071A>G)
8.PS2 Met239Ile (g.7073G>A)
9.PS2 Pro334Arg (g.9867C>G) Not pathogenic
10.PS2 Thr430Met (g.13615C>T)
11.PS2 Asp439Ala (g.13642A>C)
【0022】
上記のAPP、PS1、又はPS 2変異トランスジェニック動物とαTTPノックアウト動物とを掛け合わせることにより目的のADモデル動物を得ることができる。掛け合わせる方法としては、例えば雄(雌)のAPP変異トランスジェニック動物と雌(雄)のαTTPノックアウト動物とを繰り返し交配させることによって適宜行うことができる。
【0023】
以上のような、掛け合わせたADモデル動物は、アルツハイマー病、特に孤発性ADの治療薬のスクリーニングに利用することができる。すなわち、治療薬の候補物質をモデル動物に投与し、特に脳レベルでの種々の生化学的又は組織学的変化を観察したり、投与前後における動物の行動解析等によって、ペプチドやタンパク質分子、抗体、キメラ分子、アンチセンスDNA・RNA、リボザイム、siRNA、低分子化合物等から構成される適切な治療薬を特定することができる。
【0024】
また、上記ADモデル動物は、PET、SPECT、もしくはMRIによる画像診断、又は髄液、血液、もしくは尿中の物質の定量によるアルツハイマー病の診断方法の開発にも利用することができる。
【実施例】
【0025】
以下、実施例を示して本発明をさらに詳細に説明する。
(実施例1)
<αTTPノックアウトマウスの作製>
まず、エクソン1を含む8.8kベースのαTTPゲノム断端からαTTPターゲットベクターを作製した。続いて、ホスホグリセリン酸塩キナーゼ-ネオカセット断端をエクソン1に挿入し、単純ヘルペスウイルスチミジンキナーゼをエクソン2の下流に入れた。このコンストラクトをAB2.2-Prime ES細胞にエレクトロポレートし、G148ガンシクロビル耐性のクローンをPCRでスクリーニングした後、対立遺伝子が分裂したES細胞をC57BL/6Jの未分化胚芽細胞に注射した。これによって得られたオスのキメラマウスをC57BL/6メスマウスと交配し、αTTPヘテロマウスを得た。そして、このαTTPヘテロマウスを交配することによりαTTPノックアウトマウスを作製した。
ここで得られたαTTPノックアウトマウスはC57BL/6Jと129/SvEvBrdを半分ずつ持ち合わせた遺伝背景をもつが、このマウスを10世代以上C57BL/6Jと交配し、C57BL/6Jでバッククロスを行った。
【0026】
<実験方法>
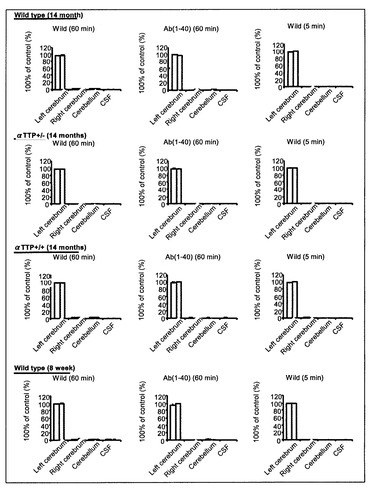
αTTP(-/-)、(+/-)ならびに野生型(WT)の片側前頭葉(S2)にラジオアイソトープでラベルしたA-β([125I]A-β 1-40(Perkin Elmer))を0.02μCiの濃度でインジェクションした。60分後もしくは5分後にsacrificeし、インジェクション側大脳半球、対側大脳半球、両側小脳半球、脳脊髄液(5μl)を採取した。それぞれの組織の放射活性を測定し、インジェクションした内の残存量を測定した。
【0027】
<結果>
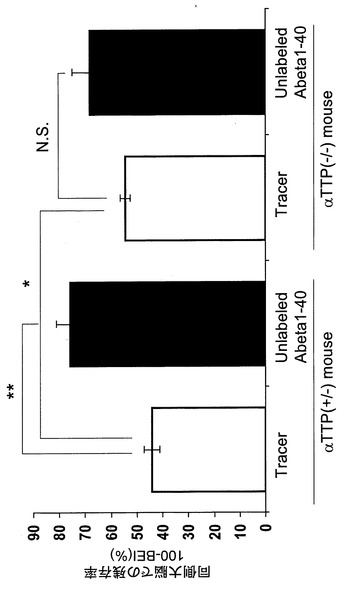
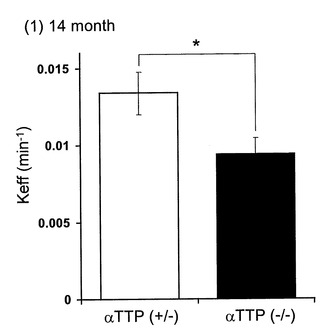
αTTP(+/-)マウスにおいて[125I]A-β 1-40脳内投与60分後における脳内残存率は44.2%であり、20μM A-β 1-40の共存下においてその値は76.0%まで上昇した(図1)。また、5分と60分の脳内残存率から求めた[125I]A-β 1-40の排出速度定数(Keff)は0.0134±0.00138min-1であった(図2)。αTTP(-/-)マウスにおいて、[125I]A-β 1-40脳内投与60分後における脳内残存率は54.5%であり、20μM A-β 1-40の共存下においてその値は68.2%まで上昇した(図1)。また、5分と60分の脳内残存率から求めた[125I]A-β 1-40の排出速度定数(Keff)は0.0094±0.00104min-1であった(図2)。またαTTP(+/-)マウス及びαTTP(-/-)マウスとも、対側大脳、小脳、脳脊髄液から有意な放射活性は得られなかった。
αTTP(-/-)マウスにおける[125I]A-β 1-40脳内投与後60分における脳内残存率はαTTP(+/-)マウスより有意に上昇しKeffは有意に減少したことから、ビタミンE欠乏による酸化ストレスによりA-β 1-40の分解または転送が抑制され、A-βのクリアランスが低下し、その結果、脳内のA-β濃度が上昇しアルツハイマー病を発症する可能性が示唆された。
【0028】
(実施例2)
<APP変異トランスジェニックマウス(Tg2576)の作製>
ハムスタープリオン蛋白オープンリーディングフレーム(ORF)を、若年性家族性アルツハイマー病の原因であるスウェーデン型変異のヒトAPP遺伝子(Lys670→Asn,Met671→Leu)のORFで置換した後、プリオン蛋白コスミッドベクターに挿入した。これによりヒトAPPをC57B6/SJL F2マウスに過剰発現させ、Tg2576を作製した。
【0029】
<αTTPノックアウトマウスとTg2576との掛け合わせ>
オスαTTP KOマウスとメスTg2576、ならびにメスαTTP KOマウスとオスTg2576をSPF環境の下それぞれ自然交配し、2種類の遺伝子型のF1産仔すなわち(1)[αTTP(+/-), APP(+)]と(2)[αTTP(+/-), APP(-)]を得た。
これを再度、オス[αTTP(+/-), APP(+)]とメス[αTTP(+/-), APP(-)]、ならびにメス[αTTP(+/-), APP(+)]とオス[αTTP(+/-), APP(-)]でそれぞれ自然交配し、4種類の遺伝子型のF2産仔すなわち、(1)[αTTP(-/-), APP(-)]、(2)[αTTP(-/-), APP(+)]、(3)[αTTP(+/+), APP(-)]と(4)[αTTP(+/+), APP(+)]を得た。ここで、(2)[αTTP(-/-), APP(+)]が掛け合わせマウスに相当する。
また、この4群は親であるF1メスの出産直後よりF1に対する餌のα-トコフェロールを排除し、遺伝子型が決まり次第αTTP(+/+)の遺伝子型をもつ個体は通常餌(α-トコフェロール36mg/kg)に変更した。αTTP(-/-)の遺伝子型をもつ個体は一定の時期よりα-トコフェロール750mg/kg含む餌を与え始める群とα-トコフェロールを全く含まない餌を継続する群を作製した。
【0030】
<実験方法>
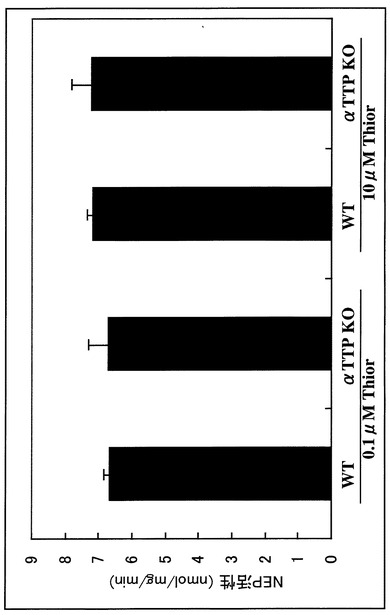
上記の掛け合わせマウスについて、モリスの水迷路(Morris water maze;MWM)をはじめとした行動解析によりその認知・記憶障害を検証するとともに、脳の病理学的検討(免疫染色、銀染色など)、生化学的検索(ELISA、ウエスタンブロッティング)によりA-βの蓄積を定量した。さらに、その消失速度の低下がネプリライシン(NEP)で代表されるA-β分解酵素の活性低下によるものか、A-βの脳からの排泄障害によるものかを検討するため、αTTP KOでNEP活性を定量した。
【0031】
<結果>
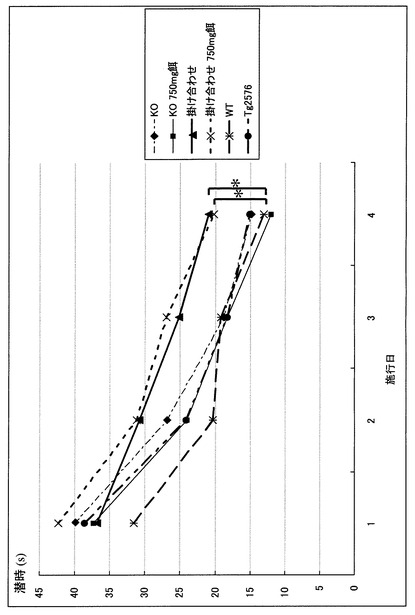
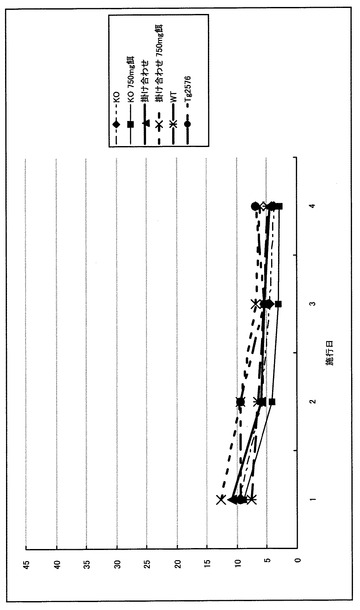
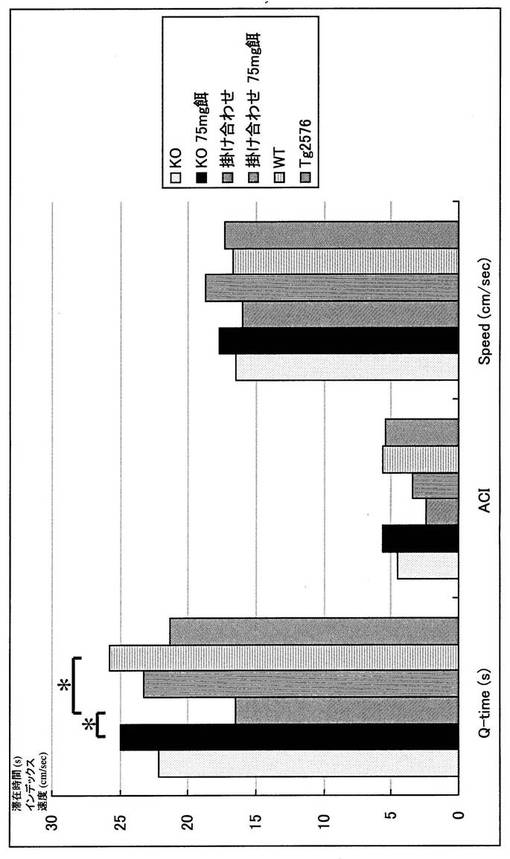
MWMでは統計学的な有意差をもって新規マウスの記憶・認知能力の低下を認めた。すなわち、Tg2576が野生型(WT)と差を認めえぬ条件下での水面下プラットフォーム(Hidden platform)においても、新規マウスは反復測定分散分析(repeated measure ANOVA)で有意に潜時の延長を認めた(P<0.05)(図3)。また、プローブトライアルにおいても同様に、1要因分散分析(one-factor ANOVA)にてターゲット4分割(target quadrant)における滞在時間の短縮を認めた(P<0.05)(図4)。このタスクにおける十分な運動能力・動機づけの確認のため水面上プラットフォーム(Visible platform)を行ったが、ここでは各群での有意差は認めずMWMが適正な条件下で行われていることが確認された(図5)。さらに、この新規マウスにおいても大脳に老人斑が出現することを銀染色にて確認し(図6)、かつA-β 1-40、1-42もELISAで確認した(図7、8)。この掛け合わせマウスでのA-β蓄積増加と痴呆症状の憎悪は、掛け合わせたαTTPノックアウトマウスにて証明された中枢神経での慢性酸化ストレスによるA-βの排泄障害と考えられる。
なぜなら、αTTP KOでは[125I]A-β1-40脳内投与による分解・排泄障害を認めるが(図9〜11)、この脳内からの消失障害にA-βを分解する代表的酵素であるネプリライシン(NEP)には異常を認めなかったからである(図12)。これにより、酸化ストレスはA-βの排泄障害を引き起こしていることが示された。
以上より、この新規マウスは、脳内で産生されたA-βが、酸化ストレスにより排泄されずADを発症する、孤発性ADモデルマウスと言うことができる。
【産業上の利用可能性】
【0032】
本発明に係るノックアウト動物は、中枢神経への慢性の酸化ストレスの結果、A-βの排泄が遅延するという表現型を持ち、また、APP、PS1、又はPS2変異トランスジェニック動物と掛け合わせた場合には脳内での内因性のA-βの産生及び蓄積が起こって実際にADを発症する。したがって、このノックアウト動物は、特に孤発性アルツハイマー病におけるA-βの代謝/排泄遅延の機序解明や、アルツハイマー病予防薬・治療薬、及び診断法の開発に利用することができる。
【図面の簡単な説明】
【0033】
【図1】[125I]A-β 1-40注入後60分での残存率を示すグラフである。
【図2】[125I]A-β 1-40の消失速度を示すグラフである。
【図3】モリス型水迷路hidden platform testの結果を示すグラフである。 Day1からDay4にかけプラットフォーム到達潜時が全群において徐々に短くなっており、学習能力を認める。野生型(WT)に対してTg2576やαTTP KOの潜時短縮はほぼ同程度であるが、掛け合わせの群は有意差をもって潜時短縮が傷害されている。※:P<0.05(repeated measure ANOVA)
【図4】モリス型水迷路probe testの結果を示すグラフである。 Day5にプラットフォームを取り除き60秒間のprobe testを施行した。Q-time(ターゲットクアドラントを泳いでいた秒数)はWTに対して掛け合わせの群は有意差をもって短縮しており、ACIも同様の傾向を認めた。なお、Speed(個体の泳いだ速さ)は各群で有意差はなく、運動能力の差は認めなかった。※:P<0.05(one-factor ANOVA)ACI:Annuls crossing index=[プラットフォームがあるはずの場所を横切った回数]-[その他3箇所の地点を横切った回数]/3
【図5】モリス型水迷路visible platform testの結果を示すグラフである。 Day6からDay9にかけて行ったvisible platform testではプラットフォーム到達潜時に全群とも有意差を認めず、動機づけは適正に行われ運動能力にも差がないことが確認された。
【図6】大脳皮質老人斑の染色(Campbell-Switzer法による銀染色)の結果を示す図である。 10ヶ月齢メスの掛け合わせマウスならびにTg2576での老人斑の染色を示している。両者ともに老人斑の出現を認めている。
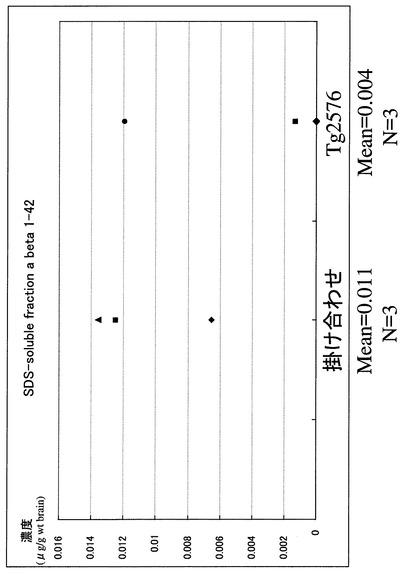
【図7】SDS可溶性分画でのA-β 1-40の定量(ELISA)の結果を示す図である。 8ヶ月齢メスの掛け合わせマウスならびにTg2576でのA-β 1-40の定量結果である。両者ともにA-β 1-40の蓄積が認められる。
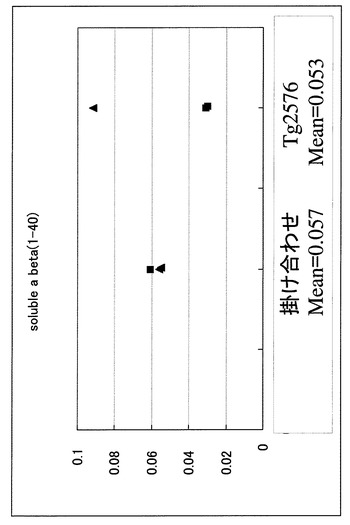
【図8】SDS可溶性分画でのA-β 1-42の定量(ELISA)の結果を示す図である。 8ヶ月齢メスの掛け合わせマウスならびにTg2576でのA-β 1-42の定量結果である。両者ともにA-β 1-42の蓄積が認められる。
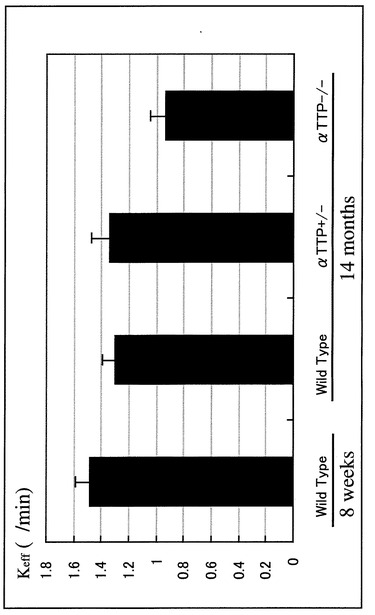
【図9】[125I]A-β 1-40のBBB排出速度定数を示すグラフである。 ラジオアイソトープで標識した[125I]A-β 1-40を左大脳S2領域に投与し、注入5分後及び60分後の[125I]A-β 1-40の残存量を測定し、BBB排出速度定数(Keff)を計算した。8週齢WTに対し14ヶ月齢WTは定数が12.2%減少し、14ヶ月齢WTに対しαTTP KOはさらに27.7%の減少を認めた。
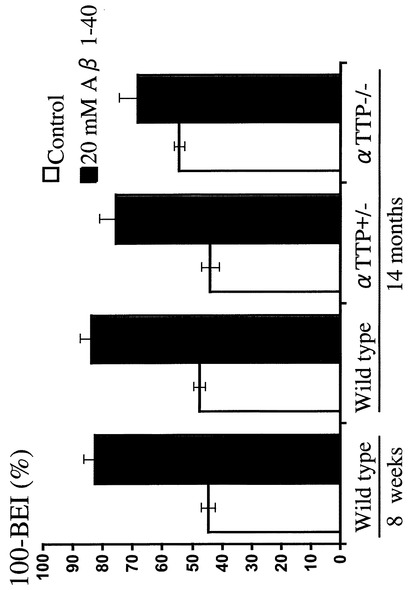
【図10】非標識A-β 1-40同時注入後脳内残存率を示すグラフである。 大量の非標識体A-β 1-40を同時に注入し60分後の[125I]A-β 1-40を測定することにより、飽和性の有無を検討した。8週及び14ヶ月齢WT、αTTP(+/-)マウスにおいて投与後60分における100-BEI(%)値は対照群に比べて顕著に増加し、αTTP KOでは増加傾向が見られた。以上より排泄輸送は飽和性を有していると考えられた。
【図11】組織中の[125I]A-β 1-40濃度を示すグラフである。 この実験において、各群の反投与側大脳、小脳及びCSFからは有意な[125I]A-β 1-40は検出されなかった。
【図12】大脳ネプリライシン活性測定の結果を示すグラフである。 大脳ホモジェネートのNEP依存性中性エンドペプチダーゼ活性を蛍光分析により測定した。NEPの特異的阻害剤であるThiorphanと反応させた大脳ホモジェネートでの測定値と比較することにより、ホモジェネート中のネプリライシン活性を定量した。NEPの活性は10μM Thiorphan、0.1μM Thiorphanとも遺伝子型による差は認めなかった。
【技術分野】
【0001】
本発明は、アルツハイマー病のモデル動物として利用できる、αトコフェロール転移タンパク質(αTTP)遺伝子ノックアウト非ヒト動物に関する。
【背景技術】
【0002】
アルツハイマー病(AD)におけるアミロイドカスケード仮説の重要な分子の一つであるA-β 1-40、1-42は、アミロイドペプチド前駆体タンパク質(amyloid precursor protein;APP)から切り出されることにより産生されるペプチドである。アルツハイマー病では、このA-βが過剰となり老人斑として凝集すると考えられている。そして、脳組織内のA-βはネプリライシン(neprilysin;NEP)などにより分解されたり、血液脳関門(BBB)を介して脳から血液へと排泄されると考えられている。
【0003】
ところで、アルツハイマー病には、優性遺伝する家族性アルツハイマー病と、それ以外の孤発性アルツハイマー病の2種類が知られている。家族性ADでは、A-βの過剰産生が原因とされ、上述のAPPとプレセニリン1(Presenilin 1; PS1)、及びプレセニリン2 が原因遺伝子として特定されている。一方、アルツハイマー病の大部分(99%以上)を占める孤発性ADではA-βの代謝/排泄の障害が原因とされている。
【0004】
このようなアルツハイマー病の機序解明、及び治療薬開発のため、ADのモデル動物が従来作製されている。例えば、(特許文献1)には、スウェーデン型変異を含むヒトAPPポリペプチドをコードするDNAが、Thy-1プロモーターエレメントの転写制御下で発現される、ヒト以外のトランスジェニック動物が開示されている。
このトランスジェニック動物は、スウェーデン型変異APP遺伝子によってA-βが産生され、その結果、AD罹病患者で観察される認知障害を示すものである。
その他、APPをコードする遺伝子に変異を有するトランスジェニック動物としてはPDGF-APPマウス、Tg2576マウス、APP22/23マウス、NORβマウスが知られ、またPS1をコードする遺伝子に変異を有するトランスジェニック動物としてはPS-1M146L/Vマウス、PS-1A246Eマウス、PS-1L286V、HH136Rマウスが知られており、これらの組み合わせによるAPP×PS1ダブルトランスジェニックマウスなども知られている。
【0005】
しかし、上記のトランスジェニック動物は、A-βの過剰産生が原因といわれる家族性ADについてのモデル動物であり、アルツハイマー病の大部分を占める孤発性ADのモデルとしては不十分なものであった。
それゆえ、孤発性ADの機序解明のため、A-βが産生されることに加えて代謝/排泄が傷害されているADモデル動物の開発が望まれていた。
【0006】
【特許文献1】特表2000−515743号公報(請求項17)
【発明の開示】
【発明が解決しようとする課題】
【0007】
そこで本発明は、A-βが産生されることに加えて代謝/排泄が傷害され、特に孤発性のアルツハイマー病のモデルとして好適に利用できる、新規な非ヒト動物を提供することを目的とする。
【課題を解決するための手段】
【0008】
上記課題を解決するため、本発明者らは、αトコフェロール転移タンパク質(α-tocopherol transfer protein;αTTP)遺伝子を欠失させたαTTPノックアウト動物を作製し、このノックアウト動物とADとの関係について調べた。すなわち、αTTPノックアウト動物は、体内・脳内でのビタミンE欠乏のため、中枢神経系に慢性的な酸化ストレスがかかっており、この酸化ストレスがA-βの代謝/排泄に影響を及ぼすか否かについて検証を行った。
その結果、酸化ストレスはA-βの排泄障害を引き起こすことが明らかとなったため、この知見に基づき発明を完成した。
【0009】
すなわち、本発明は以下の(1)〜(7)から構成される。
(1) αトコフェロール転移タンパク質(αTTP)をコードする遺伝子を欠失させ、βアミロイドペプチド(A-β)の代謝/排泄に障害を有するノックアウト非ヒト動物。
(2) アミロイドペプチド前駆体タンパク質(Amyloid precursor protein; APP)、プレセニリン1(Presenilin 1; PS1)、又はプレセニリン2(Presenilin 2; PS2)から選ばれる一以上をコードする遺伝子に変異を有する非ヒト動物に対し、αTTPをコードする遺伝子を欠失させ、A-βの代謝/排泄に障害を有するノックアウト非ヒト動物。
(3) 上記(2)記載の非ヒト動物において、APPをコードする遺伝子の変異が、スウェーデン型変異(APP770についてLys670→Asn, Met671→Leu、APP751についてLys651→Asn, Met652→Leu、APP695についてLys695→Asn, Met596→Leu)であることを特徴とするノックアウト非ヒト動物。
(4) 上記(1)〜(3)のいずれか記載の非ヒト動物が、げっ歯類動物であることを特徴とするノックアウト非ヒト動物。
(5) 上記(4)記載のげっ歯類動物が、マウスであることを特徴とするノックアウト非ヒト動物。
(6) 上記(1)〜(5)のいずれか記載の非ヒト動物を用いたアルツハイマー病予防薬・治療薬のスクリーニング方法。
(7) 上記(6)記載のスクリーニング方法を用いて得られたアルツハイマー病予防薬・治療薬。
(8) 請求項1〜5のいずれか記載の非ヒト動物を用いた、PET、SPECT、もしくはMRIによる画像診断、又は髄液、血液、もしくは尿中の物質の定量によるアルツハイマー病の診断方法。
【発明の効果】
【0010】
家族性アルツハイマー病ではA-βの産生亢進によるA-β蓄積が重要な原因と考えられている。一方、患者の大多数を占める孤発性アルツハイマー病においてはA-β蓄積の原因としてその代謝あるいは排泄に低下があることが最近明らかになっている。しかし、A-βの排泄の異常が原因でAD症状をきたすモデル動物は未だ存在しなかった。
本発明のαTTPノックアウト動物は、中枢神経への慢性の酸化ストレスの結果、A-βの排泄が遅延するという表現型を持つ初めての動物モデルであり、したがって孤発性アルツハイマー病における、代謝/排泄の遅延の機序解明に有用である。
【0011】
特に、APP変異、PS1変異、又はPS2変異トランスジェニック動物と掛け合わせた場合には、AD同様に脳内のA-βが酸化ストレスによって排泄機構の機能低下を生じ、脳内での内因性のA-βの産生及び蓄積が起こって実際にADを発症するという、AD発症機構の初のモデル動物である。
したがって、この新規ノックアウト動物は、孤発性アルツハイマー病のドラックデスカバリー等の治療法の開発に現在唯一の動物モデルになり得ると考えられる。
【発明を実施するための最良の形態】
【0012】
以下、本発明について詳述する。
本発明のノックアウト非ヒト動物は、αトコフェロール転移タンパク質(αTTP)をコードする遺伝子を欠失させて作製される。ここで欠失とは、αTTPをコードする遺伝子の一部もしくは全部が破壊・欠損・置換・挿入等により不活性化され、野生型におけるαTTPを発現する機能を失った状態をいう。このαTTPノックアウト動物は、中枢神経への慢性的な酸化ストレスが加わることにより、後の実施例で述べるようにβアミロイドペプチド(A-β)の代謝/排泄に障害を有している。
【0013】
上記のαTTPノックアウト動物は、従来知られた手法を適宜利用して作製することができる。具体的には、クローニングされたαTTP遺伝子に、単純ヘルペスウイルスのチミジンキナーゼ遺伝子等のマーカー遺伝子を導入してターゲティングベクターを作製し、このターゲティングベクターをエレクトロポレーション法等によってES細胞に導入して培養した後、G418やガンシクロビア等の抗生物質により相同的組換えを起こしたES細胞を選択する。そして、組換えES細胞をマウス等の未分化胚芽細胞にマイクロインジェクションし、仮親の子宮に戻すことによってキメラ動物を得る。このキメラ動物を野生型の雌と交配させてαTTPヘテロ組換え動物(+/-)を作製し、そのαTTPヘテロ組換え動物の雄と雌を交配させることによって目的のαTTPノックアウト動物(-/-)を得ることができる。
【0014】
αTTPノックアウト動物の対象としては、マウスの他、ラット、ハムスター、モルモット、イヌ、ネコ、ウサギ、ウシ、ブタ、ヒツジ等の哺乳動物を用いることができる。その中でも、受精卵の培養、体外受精等の技術が進んでいる、マウス等のげっ歯類動物が特に好ましい。
【0015】
上記のαTTPノックアウト動物は、慢性の酸化ストレスによってA-βの代謝/排泄に障害を持つ初めての動物モデルであり、特に孤発性アルツハイマー病における、代謝/排泄の遅延の機序解明や治療法の開発に有用な動物モデルとして利用することができる。しかし、αTTPノックアウト動物では、脳内での内因性のA-βの産生がないためA-βは蓄積しない。これに対し本発明では、別の実施形態として、アミロイドペプチド前駆体タンパク質(APP)、プレセニリン1(PS1)、又はプレセニリン2(PS2)から選ばれる一以上をコードする遺伝子に変異を有する非ヒト動物に対し、αTTPをコードする遺伝子を欠失させることにより、A-βの産生を認めかつA-β排泄が傷害されている新規ノックアウト非ヒト動物を提供するものである。このノックアウト動物は、脳内にA-βが産生し、酸化ストレスによる排泄機構の機能低下が生じて、実際にA-βが蓄積しADを発症するという、最近有力なAD発症機構の初のモデル動物として利用することができる。
【0016】
上記のADモデル動物は、APP、PS1又はPS2の一以上に変異を有するトランスジェニック動物と、αTTPノックアウト動物とを掛け合わせることによって得ることができる。ここで、APP、PS1、又はPS2変異トランスジェニック動物は、既知の手法を用いて作製することができる。例えば変異APP遺伝子や変異PS1遺伝子、又は変異PS2遺伝子を受精卵の前核にマイクロインジェクションし、生き残ったものを仮親の卵管に移植する方法や、ES細胞に遺伝子を導入しておき、それを胚盤胞腔中に注入しキメラ胚を形成させ、それを仮親の子宮に移植する方法等を用いることができる。
【0017】
APPには、複数のイソ型(APP770、751、及び695)があり、それぞれにスウェーデン型変異、オランダ型変異、ロンドン型変異、フロリダ型、フランダース型等が知られているが、いずれも適用可能である。なお、スウェーデン型変異はAPP770についてLys670→Asn, Met671→Leu、APP751についてLys651→Asn, Met652→Leu、APP695についてLys695→Asn, Met596→Leuであり、オランダ型変異はAPP770についてAla692→Gly、APP751についてAla673→Gly、APP695についてAla617→Glyであり、ロンドン型変異はAPP770についてVal717→Ile、APP751についてVal698→Ile、APP695についてVal642→Ileである。また、家族性ADの疾患モデルであるSweden型変異APP770トランスジェニックマウス(Tg2576、Taconic社製)等が知られており、これをαTTPノックアウトマウスと掛け合わせるトランスジェニックマウスとして用いても良い。
【0018】
その他、APP、PS1、及びPS2には下記に示すような多数の変異が知られており、これらもαTTPノックアウト動物と掛け合わせる対象として用いることができる。
【0019】
<APP変異>
1.APP Glu665Asp (g.269483G>C) Not pathogenic
2.APP APPKM670/671NL; Swedish APP (g.[269498G>T;269499A>C])
3.APP Ala673Thr (g.269505G>A) Not pathogenic
4.APP His677Arg (g.269518A>G) Not pathogenic
5.APP Asp678Asn, Tottori APP (g.269520C>G)
6.APP Ala692Gly; Flemish APP (g.275267C>G)
7.APP Glu693Gln; Dutch APP (g.275269G>C)
8.APP Glu693Gly; Arctic APP (g.275270A>G)
9.APP Asp694Asn; Iowa APP (g.275272G>A)
10.APP Ala713Thr (g.[275329G>A;275337G>A]) (Pathogenic nature unclear)
11.APP Ala713Thr (g.275329G>A)
12.APP Ala713Val (g.275330C>T) Not pathogenic
13.APP Thr714Ala, Iranian APP (g.275332A>G)
14.APP Thr714Ile, Austrian APP (g.275333C>T)
15.APP Val715Met; French APP (g.275335G>A)
16.APP Val715Ala; German APP (g.275336T>C)
17.APP Ile716Val; Florida APP (g.275338A>G)
18.APP Ile716Thr (g.275339T>C)
19.APP Val717Ile; London APP (g.275341G>A)
20.APP Val717Leu (g.275341G>C)
21.APP Val717Phe (g.275341G>T)
22.APP Val717Gly (g.275342T>G)
23.APP Leu723Pro; Australian APP (g.275360T>C)
【0020】
<PS1変異>
1.PS1 Arg35Gln (g.22789G.A)
2.PS1 Ala79Val (g.22921C>T)
3.PS1 Val82Leu (g.22929G>C)
4.PS1 DeltaI83/M84 (g.22932-22937del)
5.PS1 Leu85Pro (g.22939T>C)
6.PS1 Val89Leu (g.22950G>T)
7.PS1 Cys92Ser (g.22960G>C)
8.PS1 Val94Met (g.22965G>A)
9.PS1 Val96Phe (g.22971G>T)
10.PS1 Phe105Leu (g.23000T>G)
11.PS1 Leu113Pro (g.23023T>C)
12.PS1 Intron4; InsTAC (g.23024delG)
13.PS1 Tyr115His (g.25546T>C)
14.PS1 Tyr115Asp (g.25546T>G)
15.PS1 Tyr115Cys (g.25547A>G)
16.PS1 Thr116Asn (g.25550C>A)
17.PS1 Thr116Ile (g.25550C>T)
18.PS1 Pro117Ser (g.25552C>T)
19.PS1 Pro117Arg (g.25553C>G)
20.PS1 Pro117Leu (g.25553C>T)
21.PS1 Glu120Lys (g.25561G>A)
22.PS1 Glu120Asp (g.25563A>C)
23.PS1 Glu120Asp (g.25563A>T)
24.PS1 Glu123Lys (g.25570G>A)
25.PS1 Asn135Asp (g.25606A>G)
26.PS1 Met139Val (g.25618A>G)
27.PS1 Met139Lys (g.25619T>A)
28.PS1 Met139Thr (g.25619T>C)
29.PS1 Met139Ile (g.25620G>A)
30.PS1 Ile143Phe (g.25630A>T)
31.PS1 Ile143Thr (g.25631T>C)
32.PS1 Ile143Met (g.25632T>G)
33.PS1 Met146Leu (g.25639A>C)
34.PS1 Met146Val (g.25639A>G)
35.PS1 Met146Leu (g.25639A>T)
36.PS1 Met146Ile (g.25641G>A)
37.PS1 Met146Ile (g.25641G>C)
38.PS1 Met146Ile (g.25641G>T)
39.PS1 Thr147Ile (g.25643C>T)
40.PS1 Leu153Val (g.25660C>G)
41.PS1 Tyr154Asn (g.25663T>A)
42.PS1 Tyr154Cys (g.25664A>G)
43.PS1 InsFI (g.25669_25670insTTATAT)
44.PS1 His163Tyr (g.38784C>T)
45.PS1 His163Arg (g.38785A>G)
46.PS1 Trp165Gly (g.38790T>G)
47.PS1 Trp165Cys (g.38792G>C)
48.PS1 Leu166Pro (g.38794T>C)
49.PS1 Leu166Arg (g.38794T>G)
50.PS1 DeltaI167 (g.38795_38797delTAT)
51.PS1 Ser169Pro (g.38802T>C)
52.PS1 Ser169Leu (g.38803C>T)
53.PS1 Leu171Pro (g.38809T>C)
54.PS1 Leu173Trp (g.38815T>G)
55.PS1 Leu174Met (g.38817C>A)
56.PS1 Phe175Ser (g.38821T>C) Not pathogenic
57.PS1 Phe177Leu (g.38826T>C)
58.PS1 Phe177Ser (g.38827T>C)
59.PS1 Ser178Pro (g.38829T>C)
60.PS1 Gly183Val (g.38845G>T)
61.PS1 Glu184Asp (g.44571A>C)
62.PS1 Gly206Ser (g.44635G>A)
63.PS1 Gly206Ala (g.44636G>C)
64.PS1 Gly206Val (g.44636G>T)
65.PS1 Gly209Arg (g.44644G>A)
66.PS1 Gly209Glu (g.44645G>A)
67.PS1 Gly209Val (g.44645G>T)
68.PS1 Ile213Leu (g.44656A>C)
69.PS1 Ile213Phe (g.44656A>T)
70.PS1 Ile213Thr (g.44657T>C)
71.PS1 Gly217Asp (g.44669G>A)
72.PS1 Leu219Phe (g.44674C>T)
73.PS1 Leu219Pro (g.44675T>C)
74.PS1 Gln222Arg (g.44684A>G)
75.PS1 Gln222His (g.44685G>C)
76.PS1 Leu226Arg (g.44696T>G)
77.PS1 Ile229Phe (g.44704A>T)
78.PS1 Ala231Thr (g.44710G>A)
79.PS1 Ala231Val (g.44711C>T)
80.PS1 Met233Leu (g.44716A>C)
81.PS1 Met233Val (g.44716A>G)
82.PS1 Met233Thr (g.44717T>C)
83.PS1 Leu235Val (g.44722C>G)
84.PS1 Leu235Pro (g.44723T>C)
85.PS1 Phe237Ile (g.44728T>A)
86.PS1 Phe237Leu (g.44728T>C)
87.PS1 Ala246Glu (g.44756C>A)
88.PS1 Leu250Val (g.44767T>G)
89.PS1 Leu250Ser (g .44768T>C)
90.PS1 Tyr256Ser (g.44786A>C)
91.PS1 Ala260Val (g.49964C>T)
92.PS1 Val261Phe (g.49966G>T)
93.PS1 Leu262Phe (g.49971G>C)
94.PS1 Cys263Arg (g.49972T>C)
95.PS1 Cys263Phe (g.49973G>T)
96.PS1 Pro264Leu (g.49976C>T)
97.PS1 Pro267Ser (g.49984C>T)
98.PS1 Pro267Leu (g.49985C>T)
99.PS1 Arg269Gly (g.49990C>G)
100.PS1 Arg269His (g.49991G>A)
101.PS1 Leu271Val (g.49996C>G)
102.PS1 Val272Ala (g.50000T>C)
103.PS1 Glu273Ala (g.50003A>C)
104.PS1 Thr274Arg (g.50006C>G)
105.PS1 Arg278Lys (g.50018G>A)
106.PS1 Arg278Thr (g.50018G>C)
107.PS1 Arg278Ile (g.50018G>T)
108.PS1 Glu280Ala (g.50024A>C)
109.PS1 Glu280Gly (g.50024A>G)
110.PS1 Leu282Val (g.50029C>G)
111.PS1 Leu282Arg (g.50030T>G)
112.PS1 Pro284Leu (g.50036C>T)
113.PS1 Ala285Val (g.50039C>T)
114.PS1 Leu286Val (g.50041C>G)
115.PS1 Delta9 (g.56305-62162del)
116.PS1 Delta9Finn (g.56681-61235del)
117.PS1 Delta9 (g.58304G>A)
118.PS1 Delta9 (g.58304G>T)
119.PS1 Glu318Gly (g.58389A>G) Not pathogenic
120.PS1 InsR (g.63786_63787insTCG)
121.PS1 Thr354Ile (g.63792C>T)
122.PS1 Arg358Gln (g.63804G>A)
123.PS1 Ser365Tyr (g.63825C>A)
124.PS1 Arg377Met (g.69044G>T)
125.PS1 Gly378Glu (g.69047G>A)
126.PS1 Gly378Val (g.69047G>T)
127.PS1 Gly384Ala (g.69065G>C)
128.PS1 Ser390Ile (g.69083G>T)
129.PS1 Leu392Val (g.69088C>G)
130.PS1 Leu392Pro (g.69089T>C)
131.PS1 Gly394Val (g.69095G>T)
132.PS1 Asn405Ser (g.69128A>G)
133.PS1 Ala409Thr (g.69139G>A)
134.PS1 Cys410Tyr (g.69143G>A)
135.PS1 Leu418Phe (g.71058G>T)
136.PS1 Leu424Arg (g.71075T>G)
137.PS1 Ala426Pro (g.71080G>C)
138.PS1 Ala431Glu (g.71096C>A)
139.PS1 Ala431Val (g.71096C>T)
140.PS1 Ala434Cys (g.[71104G>T;71105C>G])
141.PS1 Leu435Phe (g.71107C>T)
142.PS1 Pro436Ser (g.71110C>T)
143.PS1 Pro436Gln (g.71111C>A)
144.PS1 Ile439Val (g.71119A>G)
【0021】
<PS2変異>
1.PS2 Arg62His ( g.1839G>A)
2.PS2 Thr122Pro (g.3636A>C)
3.PS2 Ser130Leu (g.3661C>T)
4.PS2 Asn141Ile (g.3694A>T)
5.PS2 Val148Ile (g.3714G>A)
6.PS2 Gln228Leu (g.7039A>T)
7.PS2 Met239Val (g.7071A>G)
8.PS2 Met239Ile (g.7073G>A)
9.PS2 Pro334Arg (g.9867C>G) Not pathogenic
10.PS2 Thr430Met (g.13615C>T)
11.PS2 Asp439Ala (g.13642A>C)
【0022】
上記のAPP、PS1、又はPS 2変異トランスジェニック動物とαTTPノックアウト動物とを掛け合わせることにより目的のADモデル動物を得ることができる。掛け合わせる方法としては、例えば雄(雌)のAPP変異トランスジェニック動物と雌(雄)のαTTPノックアウト動物とを繰り返し交配させることによって適宜行うことができる。
【0023】
以上のような、掛け合わせたADモデル動物は、アルツハイマー病、特に孤発性ADの治療薬のスクリーニングに利用することができる。すなわち、治療薬の候補物質をモデル動物に投与し、特に脳レベルでの種々の生化学的又は組織学的変化を観察したり、投与前後における動物の行動解析等によって、ペプチドやタンパク質分子、抗体、キメラ分子、アンチセンスDNA・RNA、リボザイム、siRNA、低分子化合物等から構成される適切な治療薬を特定することができる。
【0024】
また、上記ADモデル動物は、PET、SPECT、もしくはMRIによる画像診断、又は髄液、血液、もしくは尿中の物質の定量によるアルツハイマー病の診断方法の開発にも利用することができる。
【実施例】
【0025】
以下、実施例を示して本発明をさらに詳細に説明する。
(実施例1)
<αTTPノックアウトマウスの作製>
まず、エクソン1を含む8.8kベースのαTTPゲノム断端からαTTPターゲットベクターを作製した。続いて、ホスホグリセリン酸塩キナーゼ-ネオカセット断端をエクソン1に挿入し、単純ヘルペスウイルスチミジンキナーゼをエクソン2の下流に入れた。このコンストラクトをAB2.2-Prime ES細胞にエレクトロポレートし、G148ガンシクロビル耐性のクローンをPCRでスクリーニングした後、対立遺伝子が分裂したES細胞をC57BL/6Jの未分化胚芽細胞に注射した。これによって得られたオスのキメラマウスをC57BL/6メスマウスと交配し、αTTPヘテロマウスを得た。そして、このαTTPヘテロマウスを交配することによりαTTPノックアウトマウスを作製した。
ここで得られたαTTPノックアウトマウスはC57BL/6Jと129/SvEvBrdを半分ずつ持ち合わせた遺伝背景をもつが、このマウスを10世代以上C57BL/6Jと交配し、C57BL/6Jでバッククロスを行った。
【0026】
<実験方法>
αTTP(-/-)、(+/-)ならびに野生型(WT)の片側前頭葉(S2)にラジオアイソトープでラベルしたA-β([125I]A-β 1-40(Perkin Elmer))を0.02μCiの濃度でインジェクションした。60分後もしくは5分後にsacrificeし、インジェクション側大脳半球、対側大脳半球、両側小脳半球、脳脊髄液(5μl)を採取した。それぞれの組織の放射活性を測定し、インジェクションした内の残存量を測定した。
【0027】
<結果>
αTTP(+/-)マウスにおいて[125I]A-β 1-40脳内投与60分後における脳内残存率は44.2%であり、20μM A-β 1-40の共存下においてその値は76.0%まで上昇した(図1)。また、5分と60分の脳内残存率から求めた[125I]A-β 1-40の排出速度定数(Keff)は0.0134±0.00138min-1であった(図2)。αTTP(-/-)マウスにおいて、[125I]A-β 1-40脳内投与60分後における脳内残存率は54.5%であり、20μM A-β 1-40の共存下においてその値は68.2%まで上昇した(図1)。また、5分と60分の脳内残存率から求めた[125I]A-β 1-40の排出速度定数(Keff)は0.0094±0.00104min-1であった(図2)。またαTTP(+/-)マウス及びαTTP(-/-)マウスとも、対側大脳、小脳、脳脊髄液から有意な放射活性は得られなかった。
αTTP(-/-)マウスにおける[125I]A-β 1-40脳内投与後60分における脳内残存率はαTTP(+/-)マウスより有意に上昇しKeffは有意に減少したことから、ビタミンE欠乏による酸化ストレスによりA-β 1-40の分解または転送が抑制され、A-βのクリアランスが低下し、その結果、脳内のA-β濃度が上昇しアルツハイマー病を発症する可能性が示唆された。
【0028】
(実施例2)
<APP変異トランスジェニックマウス(Tg2576)の作製>
ハムスタープリオン蛋白オープンリーディングフレーム(ORF)を、若年性家族性アルツハイマー病の原因であるスウェーデン型変異のヒトAPP遺伝子(Lys670→Asn,Met671→Leu)のORFで置換した後、プリオン蛋白コスミッドベクターに挿入した。これによりヒトAPPをC57B6/SJL F2マウスに過剰発現させ、Tg2576を作製した。
【0029】
<αTTPノックアウトマウスとTg2576との掛け合わせ>
オスαTTP KOマウスとメスTg2576、ならびにメスαTTP KOマウスとオスTg2576をSPF環境の下それぞれ自然交配し、2種類の遺伝子型のF1産仔すなわち(1)[αTTP(+/-), APP(+)]と(2)[αTTP(+/-), APP(-)]を得た。
これを再度、オス[αTTP(+/-), APP(+)]とメス[αTTP(+/-), APP(-)]、ならびにメス[αTTP(+/-), APP(+)]とオス[αTTP(+/-), APP(-)]でそれぞれ自然交配し、4種類の遺伝子型のF2産仔すなわち、(1)[αTTP(-/-), APP(-)]、(2)[αTTP(-/-), APP(+)]、(3)[αTTP(+/+), APP(-)]と(4)[αTTP(+/+), APP(+)]を得た。ここで、(2)[αTTP(-/-), APP(+)]が掛け合わせマウスに相当する。
また、この4群は親であるF1メスの出産直後よりF1に対する餌のα-トコフェロールを排除し、遺伝子型が決まり次第αTTP(+/+)の遺伝子型をもつ個体は通常餌(α-トコフェロール36mg/kg)に変更した。αTTP(-/-)の遺伝子型をもつ個体は一定の時期よりα-トコフェロール750mg/kg含む餌を与え始める群とα-トコフェロールを全く含まない餌を継続する群を作製した。
【0030】
<実験方法>
上記の掛け合わせマウスについて、モリスの水迷路(Morris water maze;MWM)をはじめとした行動解析によりその認知・記憶障害を検証するとともに、脳の病理学的検討(免疫染色、銀染色など)、生化学的検索(ELISA、ウエスタンブロッティング)によりA-βの蓄積を定量した。さらに、その消失速度の低下がネプリライシン(NEP)で代表されるA-β分解酵素の活性低下によるものか、A-βの脳からの排泄障害によるものかを検討するため、αTTP KOでNEP活性を定量した。
【0031】
<結果>
MWMでは統計学的な有意差をもって新規マウスの記憶・認知能力の低下を認めた。すなわち、Tg2576が野生型(WT)と差を認めえぬ条件下での水面下プラットフォーム(Hidden platform)においても、新規マウスは反復測定分散分析(repeated measure ANOVA)で有意に潜時の延長を認めた(P<0.05)(図3)。また、プローブトライアルにおいても同様に、1要因分散分析(one-factor ANOVA)にてターゲット4分割(target quadrant)における滞在時間の短縮を認めた(P<0.05)(図4)。このタスクにおける十分な運動能力・動機づけの確認のため水面上プラットフォーム(Visible platform)を行ったが、ここでは各群での有意差は認めずMWMが適正な条件下で行われていることが確認された(図5)。さらに、この新規マウスにおいても大脳に老人斑が出現することを銀染色にて確認し(図6)、かつA-β 1-40、1-42もELISAで確認した(図7、8)。この掛け合わせマウスでのA-β蓄積増加と痴呆症状の憎悪は、掛け合わせたαTTPノックアウトマウスにて証明された中枢神経での慢性酸化ストレスによるA-βの排泄障害と考えられる。
なぜなら、αTTP KOでは[125I]A-β1-40脳内投与による分解・排泄障害を認めるが(図9〜11)、この脳内からの消失障害にA-βを分解する代表的酵素であるネプリライシン(NEP)には異常を認めなかったからである(図12)。これにより、酸化ストレスはA-βの排泄障害を引き起こしていることが示された。
以上より、この新規マウスは、脳内で産生されたA-βが、酸化ストレスにより排泄されずADを発症する、孤発性ADモデルマウスと言うことができる。
【産業上の利用可能性】
【0032】
本発明に係るノックアウト動物は、中枢神経への慢性の酸化ストレスの結果、A-βの排泄が遅延するという表現型を持ち、また、APP、PS1、又はPS2変異トランスジェニック動物と掛け合わせた場合には脳内での内因性のA-βの産生及び蓄積が起こって実際にADを発症する。したがって、このノックアウト動物は、特に孤発性アルツハイマー病におけるA-βの代謝/排泄遅延の機序解明や、アルツハイマー病予防薬・治療薬、及び診断法の開発に利用することができる。
【図面の簡単な説明】
【0033】
【図1】[125I]A-β 1-40注入後60分での残存率を示すグラフである。
【図2】[125I]A-β 1-40の消失速度を示すグラフである。
【図3】モリス型水迷路hidden platform testの結果を示すグラフである。 Day1からDay4にかけプラットフォーム到達潜時が全群において徐々に短くなっており、学習能力を認める。野生型(WT)に対してTg2576やαTTP KOの潜時短縮はほぼ同程度であるが、掛け合わせの群は有意差をもって潜時短縮が傷害されている。※:P<0.05(repeated measure ANOVA)
【図4】モリス型水迷路probe testの結果を示すグラフである。 Day5にプラットフォームを取り除き60秒間のprobe testを施行した。Q-time(ターゲットクアドラントを泳いでいた秒数)はWTに対して掛け合わせの群は有意差をもって短縮しており、ACIも同様の傾向を認めた。なお、Speed(個体の泳いだ速さ)は各群で有意差はなく、運動能力の差は認めなかった。※:P<0.05(one-factor ANOVA)ACI:Annuls crossing index=[プラットフォームがあるはずの場所を横切った回数]-[その他3箇所の地点を横切った回数]/3
【図5】モリス型水迷路visible platform testの結果を示すグラフである。 Day6からDay9にかけて行ったvisible platform testではプラットフォーム到達潜時に全群とも有意差を認めず、動機づけは適正に行われ運動能力にも差がないことが確認された。
【図6】大脳皮質老人斑の染色(Campbell-Switzer法による銀染色)の結果を示す図である。 10ヶ月齢メスの掛け合わせマウスならびにTg2576での老人斑の染色を示している。両者ともに老人斑の出現を認めている。
【図7】SDS可溶性分画でのA-β 1-40の定量(ELISA)の結果を示す図である。 8ヶ月齢メスの掛け合わせマウスならびにTg2576でのA-β 1-40の定量結果である。両者ともにA-β 1-40の蓄積が認められる。
【図8】SDS可溶性分画でのA-β 1-42の定量(ELISA)の結果を示す図である。 8ヶ月齢メスの掛け合わせマウスならびにTg2576でのA-β 1-42の定量結果である。両者ともにA-β 1-42の蓄積が認められる。
【図9】[125I]A-β 1-40のBBB排出速度定数を示すグラフである。 ラジオアイソトープで標識した[125I]A-β 1-40を左大脳S2領域に投与し、注入5分後及び60分後の[125I]A-β 1-40の残存量を測定し、BBB排出速度定数(Keff)を計算した。8週齢WTに対し14ヶ月齢WTは定数が12.2%減少し、14ヶ月齢WTに対しαTTP KOはさらに27.7%の減少を認めた。
【図10】非標識A-β 1-40同時注入後脳内残存率を示すグラフである。 大量の非標識体A-β 1-40を同時に注入し60分後の[125I]A-β 1-40を測定することにより、飽和性の有無を検討した。8週及び14ヶ月齢WT、αTTP(+/-)マウスにおいて投与後60分における100-BEI(%)値は対照群に比べて顕著に増加し、αTTP KOでは増加傾向が見られた。以上より排泄輸送は飽和性を有していると考えられた。
【図11】組織中の[125I]A-β 1-40濃度を示すグラフである。 この実験において、各群の反投与側大脳、小脳及びCSFからは有意な[125I]A-β 1-40は検出されなかった。
【図12】大脳ネプリライシン活性測定の結果を示すグラフである。 大脳ホモジェネートのNEP依存性中性エンドペプチダーゼ活性を蛍光分析により測定した。NEPの特異的阻害剤であるThiorphanと反応させた大脳ホモジェネートでの測定値と比較することにより、ホモジェネート中のネプリライシン活性を定量した。NEPの活性は10μM Thiorphan、0.1μM Thiorphanとも遺伝子型による差は認めなかった。
【特許請求の範囲】
【請求項1】
αトコフェロール転移タンパク質(αTTP)をコードする遺伝子を欠失させ、βアミロイドペプチド(A-β)の代謝/排泄に障害を有するノックアウト非ヒト動物。
【請求項2】
アミロイドペプチド前駆体タンパク質(Amyloid precursor protein; APP)、プレセニリン1(Presenilin 1; PS1)、又はプレセニリン2(Presenilin 2; PS2)から選ばれる一以上をコードする遺伝子に変異を有する非ヒト動物に対し、αTTPをコードする遺伝子を欠失させ、A-βの代謝/排泄に障害を有するノックアウト非ヒト動物。
【請求項3】
請求項2記載の非ヒト動物において、APPをコードする遺伝子の変異が、スウェーデン型変異(APP770についてLys670→Asn, Met671→Leu、APP751についてLys651→Asn, Met652→Leu、APP695についてLys695→Asn, Met596→Leu)であることを特徴とするノックアウト非ヒト動物。
【請求項4】
請求項1〜3のいずれか記載の非ヒト動物が、げっ歯類動物であることを特徴とするノックアウト非ヒト動物。
【請求項5】
請求項4記載のげっ歯類動物が、マウスであることを特徴とするノックアウト非ヒト動物。
【請求項6】
請求項1〜5のいずれか記載の非ヒト動物を用いたアルツハイマー病予防薬・治療薬のスクリーニング方法。
【請求項7】
請求項6記載のスクリーニング方法を用いて得られたアルツハイマー病予防薬・治療薬。
【請求項8】
請求項1〜5のいずれか記載の非ヒト動物を用いた、PET、SPECT、もしくはMRIによる画像診断、又は髄液、血液、もしくは尿中の物質の定量によるアルツハイマー病の診断方法。
【請求項1】
αトコフェロール転移タンパク質(αTTP)をコードする遺伝子を欠失させ、βアミロイドペプチド(A-β)の代謝/排泄に障害を有するノックアウト非ヒト動物。
【請求項2】
アミロイドペプチド前駆体タンパク質(Amyloid precursor protein; APP)、プレセニリン1(Presenilin 1; PS1)、又はプレセニリン2(Presenilin 2; PS2)から選ばれる一以上をコードする遺伝子に変異を有する非ヒト動物に対し、αTTPをコードする遺伝子を欠失させ、A-βの代謝/排泄に障害を有するノックアウト非ヒト動物。
【請求項3】
請求項2記載の非ヒト動物において、APPをコードする遺伝子の変異が、スウェーデン型変異(APP770についてLys670→Asn, Met671→Leu、APP751についてLys651→Asn, Met652→Leu、APP695についてLys695→Asn, Met596→Leu)であることを特徴とするノックアウト非ヒト動物。
【請求項4】
請求項1〜3のいずれか記載の非ヒト動物が、げっ歯類動物であることを特徴とするノックアウト非ヒト動物。
【請求項5】
請求項4記載のげっ歯類動物が、マウスであることを特徴とするノックアウト非ヒト動物。
【請求項6】
請求項1〜5のいずれか記載の非ヒト動物を用いたアルツハイマー病予防薬・治療薬のスクリーニング方法。
【請求項7】
請求項6記載のスクリーニング方法を用いて得られたアルツハイマー病予防薬・治療薬。
【請求項8】
請求項1〜5のいずれか記載の非ヒト動物を用いた、PET、SPECT、もしくはMRIによる画像診断、又は髄液、血液、もしくは尿中の物質の定量によるアルツハイマー病の診断方法。
【図1】


【図2】
【図3】
【図5】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図4】
【図6】

【図2】
【図3】
【図5】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図4】
【図6】
【公開番号】特開2008−178297(P2008−178297A)
【公開日】平成20年8月7日(2008.8.7)
【国際特許分類】
【出願番号】特願2005−119052(P2005−119052)
【出願日】平成17年4月15日(2005.4.15)
【出願人】(504179255)国立大学法人 東京医科歯科大学 (228)
【Fターム(参考)】
【公開日】平成20年8月7日(2008.8.7)
【国際特許分類】
【出願日】平成17年4月15日(2005.4.15)
【出願人】(504179255)国立大学法人 東京医科歯科大学 (228)
【Fターム(参考)】
[ Back to top ]