
A2A−アデノシンレセプターアゴニストおよびその多形体の調製方法
【課題】A2A−アデノシンレセプターアゴニストおよびその多形体の調製方法の提供。
【解決手段】本発明の開示は、A2A−アデノシンレセプターアゴニストの大スケールでの製造に適切な合成方法であり、またそれらの化合物の多形体、および特定の多形体を単離する方法に関する。より具体的な本発明の目的は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドおよびその多形体、好ましくはその一水和物の大スケールの調製のための便利な合成を提供することである。
【解決手段】本発明の開示は、A2A−アデノシンレセプターアゴニストの大スケールでの製造に適切な合成方法であり、またそれらの化合物の多形体、および特定の多形体を単離する方法に関する。より具体的な本発明の目的は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドおよびその多形体、好ましくはその一水和物の大スケールの調製のための便利な合成を提供することである。
【発明の詳細な説明】
【技術分野】
【0001】
本願は、米国仮特許出願第60/801,857号(2006年5月18日出願)および米国仮特許出願第60/765,114号(2006年2月3日出願)の優先権を主張し、これらの全部の開示は本明細書中で参考として援用する。
【0002】
(発明の分野)
本発明は、A2A−アデノシンレセプターアゴニストの大スケールでの調製のための方法に関し、またその化合物の多形体および特定の多形体を単離する工程の方法に関する。
【背景技術】
【0003】
(背景)
アデノシンは、天然に存在するヌクレオシドであり、A1、A2A、A2BおよびA3として公知のアデノシンレセプターのファミリーとの相互作用によりその生物学的効果を発揮し、それらレセプターはすべて、重要な生理学的プロセスを調整する。アデノシンの生物学的効果の1つは、冠動脈血管拡張薬として機能することであり;この結果はA2Aアデノシンレセプターとの相互作用により生ずる。このアデノシンの効果は、造影剤(imaging agent)(例えばタリウム201)の投与より前に冠動脈が拡張される場合、心臓の画像化に対する手助けとして有用であることが理解されており、従って生じる画像の観察によって、冠動脈疾患の存在または不在が決定され得る。このような技術の利点は、冠動脈疾患を有する患者に対して明らかに望ましくないトレッドミル上での運動によって、冠動脈血管拡張を引き起こす従来の方法を避けることである。
【0004】
しかし、アデノシンの投与はいくつかの不利な点を有する。アデノシンはヒトの体内で非常に短い半減期(10秒未満)を有し、またA1、A2A、A2BおよびA3レセプターアゴニストによる作用(agonism)に関するすべての効果を有する。従って、選択的なA2Aアデノシンレセプターアゴニストの使用は、冠動脈血管拡張を生ずるのによりすぐれた方法、特により長い半減期を有し、および副作用がほとんどないまたはまったくない方法を提供する。
【0005】
これらの所望の特性を有する化合物のクラスは、特許文献1に開示され、その全部の開示は本明細書中で参考として援用する。特に、特許文献1に開示される1つの化合物、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドは、高選択性A2A−アデノシンレセプターアゴニストとして示され、心臓の画像化において有用な冠動脈血管拡張薬として、臨床試験を現在行っている最中である。
【0006】
この化合物および類似の化合物において重要性が高まっていることに鑑みて、高収率および高純度で物質を大量に製造するための便利な方法を提供する、新しい合成方法を見つけることが望まれるようになった。この目的の化合物を開示する特許文献1は、この化合物を調製するためのいくつかの方法を提供する。しかしながら、これらの方法は小スケールでの合成に適しているが、この特許文献に開示されるすべての合成方法は保護基を利用しており、この方法は大スケールの合成に望ましくない。
【0007】
さらに、上記所望の生成物(つまり、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド)は、少なくとも3つの異なる結晶形状で存在することが可能であり、その中の最も安定なものは、一水和物である。この多形体は、相対湿度のストレス条件下で(under relative
humidity stress conditions)、その融点まで安定である。従って、新しい合成によって生成した最終生成物が安定な一水和物として得られることが望まれる。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】米国特許第6,403,567号明細書
【発明の概要】
【課題を解決するための手段】
【0009】
(発明の趣旨)
本発明の目的は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドおよびその多形体、好ましくはその一水和物の大スケールの調製のための便利な合成を提供することである。従って、第一の局面においては、本発明は式I:
【0010】
【化10】

の化合物の調製に関し、それは式(3):
【0011】
【化11】
の化合物をメチルアミンと接触させる工程を含む。
【0012】
第一の実施形態において、この反応はメチルアミンの水溶液中で、初期は約0〜5℃の温度で、続いて約50〜70℃に加温して実施される。あるいは、この反応は上記に記載のように、しかし封緘された圧力反応器中で実施される。
【0013】
第二の実施形態において、上記生成物は、この生成物を溶媒(例えば、ジメチルスルホキシド)に溶解し、精製水を加え、このようにして形成されるスラリーをろ過し、フィルターの内容物を水、続いてエタノールで洗浄し、そして残った固体を真空下で40℃を越えない温度で乾燥させることにより、純一水和物として単離される。
【0014】
第二の局面において、本発明は、式(3):
【0015】
【化12】
の化合物の調製に関し、それは式(2):
【0016】
【化13】
の化合物をエチル2−ホルミル−3−オキソプロピオネートと接触させる工程を含む。
【0017】
一実施形態において、この反応はエタノール中、約80℃の温度で、約1.1モル当量のエチル2−ホルミル−3−オキソプロピオネートを用いて、実施される。
【0018】
第三の局面において、本発明は、式(2):
【0019】
【化14】
の化合物の調製に関し、それは式(1):
【0020】
【化15】
の化合物をヒドラジンと接触させる工程を含む。
【0021】
上記に記載の合成は、所望の生成物の大スケールでの合成に適しており、この生成物は、最終生成物中に1種の少量の不純物が見られるが、良好な収率で提供される。この不純物は、式(2)のもとの中間体;つまり式:
【0022】
【化16】
の化合物であると示されている。この不純物は、結晶化により最終生成物より取り除かれ得るが、上記合成の利点のすべてを有するが、最終生成物中に式(2)の化合物を不純物として与えない代わりの合成を探求することが決定された。
【0023】
従って、第四の局面として、本発明は式(4):
【0024】
【化17】
の化合物をメチルアミンと接触させることにより、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドを合成する方法に関する。
【0025】
第一の実施形態において、この反応はメチルアミンの水溶液中で、初期は約0〜5℃の温度で、続いて約50〜70℃に加温して実施される。好ましくは、この反応は、封緘された圧力反応器中で実施される。
【0026】
第二の実施形態において、上記生成物は、この生成物を溶媒(例えば、ジメチルスルホキシド)に溶解し、精製水を加え、このようにして形成されるスラリーをろ過し、フィルターの内容物を水、続いてエタノールで洗浄し、そして残った固体を真空下で40℃を越えない温度で乾燥させることにより、純一水和物として単離される。
【0027】
第五の局面において、本発明は、式(4):
【0028】
【化18】
の化合物の合成に関し、それは式(2):
【0029】
【化19】
の化合物を過剰の、好ましくは2〜10倍過剰、より好ましくは5〜10倍過剰のエチル2−ホルミル−3−オキソプロピオネートと接触させる工程を含む。
【0030】
一実施形態において、この反応はエタノール中で、約80℃の温度で実施される。エチル2−ホルミル−3−オキソプロピオネートは5〜10倍過剰で存在する。
例えば、本願発明は以下の項目を提供する。
(項目1)
(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド:
【化1】
の大スケールでの調製のための方法であって、式(3)の化合物:
【化2】
をメチルアミンと反応させる工程を含む、方法。
(項目2)
項目1に記載の方法であって、前記式(3)の化合物をメチルアミン水溶液と、初期は約0〜5℃で、続いて約50〜70℃に加温して反応させる、方法。
(項目3)
前記反応が、封緘された圧力反応器で実施される、項目1に記載の方法。
(項目4)
項目1に記載の方法であって、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド最終生成物が、
(a)該生成物を溶媒に溶解する工程
(b)精製水を加える工程
(c)このようにして形成されたスラリーをろ過する工程
(d)フィルターの内容物を水、続いてエタノールで洗浄する工程、および
(e)残った固体を真空下で40℃を越えない温度で乾燥する工程
によって、純一水和物として単離される、方法。
(項目5)
項目4に記載の方法であって、前記工程(a)で使用される溶媒がジメチルスルホキシドである、方法。
(項目6)
式(3)の化合物:
【化3】
を調製する方法であって、式(2)の化合物:
【化4】
をエチル2−ホルミル−3−オキソプロピオネートと反応させる工程を含む、方法。
(項目7)
項目6に記載の方法であって、前記反応がエタノール中で実施される、方法。
(項目8)
項目7に記載の方法であって、前記反応が約80℃の温度で実施される、方法。
(項目9)
項目6に記載の方法であって、約1.1モル当量のエチル2−ホルミル−3−オキソプロピオネートが使用される、方法。
(項目10)
式(2)の化合物:
【化5】
を調製するための方法であって、式(1)の化合物:
【化6】
をヒドラジンと反応させる工程を含む、方法。
(項目11)
(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドを合成する方法であって、式(4)の化合物:
【化7】
をメチルアミンに接触させることにより合成する、方法。
(項目12)
項目11に記載の方法であって、前記式(4)の化合物をメチルアミン水溶液と、初期は約0〜5℃で、続いて約50〜70℃に加温して反応させる、方法。
(項目13)
前記反応が、封緘された圧力反応器で実施される、項目11に記載の方法。
(項目14)
項目11に記載の方法であって、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド最終生成物が、
(a)項目11に記載の生成物を溶媒に溶解する工程
(b)精製水を加える工程
(c)このようにして形成されたスラリーをろ過する工程
(d)フィルターの内容物を水、続いてエタノールで洗浄する工程、および
(e)残った固体を真空下で40℃を越えない温度で乾燥する工程
によって、純一水和物として単離される、方法。
(項目15)
項目14に記載の方法であって、前記工程(a)で使用される溶媒がジメチルスルホキシドである、方法。
(項目16)
式(4)の化合物:
【化8】
を合成する方法であって、式(2)の化合物:
【化9】
を過剰のエチル2−ホルミル−3−オキソプロピオネートと接触させる工程を含む、方法。
(項目17)
項目16に記載の方法であって、前記反応がエタノール中で実施される、方法。
(項目18)
項目17に記載の方法であって、前記反応が約80℃の温度で実施される、方法。
(項目19)
項目16に記載の方法であって、約2〜10倍のモル過剰のエチル2−ホルミル−3−オキソプロピオネートが使用される、方法。
(項目20)
項目19に記載の方法であって、約5〜10倍のモル過剰のエチル2−ホルミル−3−オキソプロピオネートが使用される、方法。
【図面の簡単な説明】
【0031】
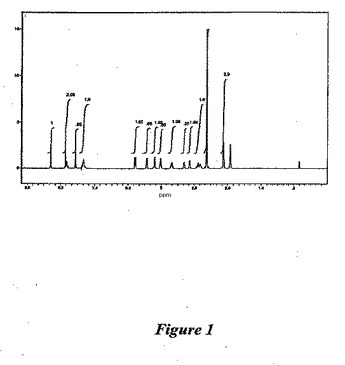
【図1】図1は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド一水和物(形状A)の1H NMRスペクトルである。
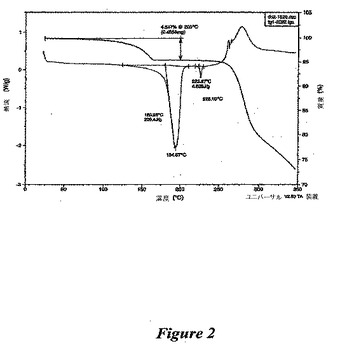
【図2】図2は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド一水和物の熱的分析である。
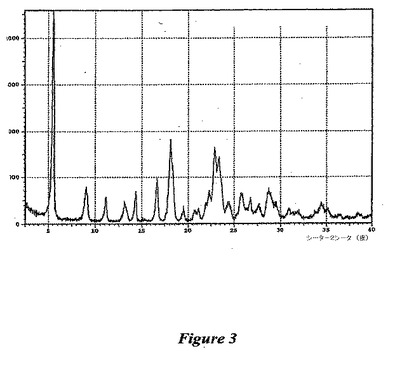
【図3】図3は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド一水和物のX線回折パターンである。
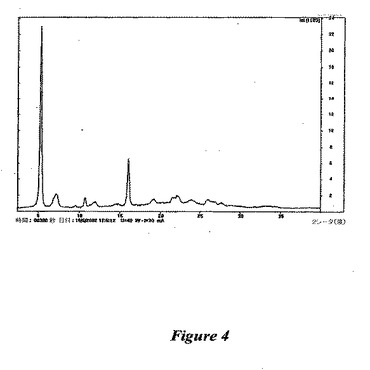
【図4】図4は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドの形状BのX線回折パターンである。
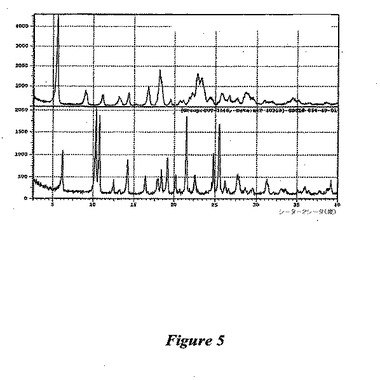
【図5】図5は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドの形状Cの、形状Aと比較したX線回折パターンである。
【発明を実施するための形態】
【0032】
(定義および一般的なパラメーター)
本明細書で使用される場合、以下の単語および句は一般的に、それらが使用される文脈が別の方法で示す程度までを除いて、以下に記載の意味を有することが意味される。
【0033】
「任意の」または「必要に応じて」は、続いて記載される事象または状況が起こっても起こらなくてもよいことを意味し、そしてその記載が、その事象または状況が起こる場合および、その事象または状況が起こらない場合を包含することを意味する。
【0034】
用語「治療上の有効量」は、以下に定義されるような処置が必要な哺乳動物に投与される場合に、その処置をもたらすのに充分な式Iの化合物の量、に言及する。この治療上の有効量は、処置される被験体および疾患状態、被験体の体重および年齢、疾患状態の重大さ、投与の様式などに依存して変わり、そして当業者により容易に決定され得る。
【0035】
用語「処置」または「処置すること」は、哺乳動物における任意の疾患の処置を意味し、以下:
(i)上記疾患を予防すること、すなわち上記疾患の臨床的症状を発症させないようにすること;
(ii)上記疾患を抑制すること、すなわち臨床的症状の進行を止めること;
および/または
(iii)上記疾患を軽減すること、すなわち臨床的症状の後退を引き起こすこと;
が挙げられる。
【0036】
本明細書で使用される場合、「薬学的に受容可能なキャリアー」としては、任意のおよびすべての溶媒、分散媒体、コーティング、抗菌剤および抗真菌剤、等張性遅延剤(isotonic delaying agent)および吸収遅延剤(absorption delaying agent)などが挙げられる。薬学的に活性な物質のためのこのような媒体および薬剤の使用は、当該分野で周知である。任意の従来の媒体または薬剤が活性成分と不適合である範囲を除き、治療的組成物中でのその使用が企図される。補助的な活性成分もまた、上記組成物中に組み込まれ得る。
【0037】
用語「多形体」は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドのアモルファスおよび溶媒和物を含むことが意図される。
【0038】
この化合物は少なくとも3つの異なる結晶形状(本明細書中で、形状A、形状B、形状Cと言及される)およびアモルファス生成物として存在することが可能である、ということがわかった。
【0039】
形状A:この多形体は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドをプロトン性溶媒(例えば、エタノールまたはエタノール/水混合物)または極性溶媒(例えば、ジメチルスルホキシド/水)より結晶化することにより生成され得る。形状Aは一水和物であることが示されており、および周囲温度で種々の多形体の中で最も安定である。形状Aは相対湿度のストレス条件下で、その融点まで安定である。
【0040】
形状B:この多形体は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドのトリフルオロエタノール溶液を、周囲温度で減圧下でエバポレーションすることによって生成される。この結晶のX線解析は、任意の他の多形体と明確に異なった(図4を参照のこと)が、上記X線解析が乱れた幅広いピークを与え、そして上記多形体がさまざまな量の水を含むため、その構造の決定は困難であった。この多形体の調製を信頼性をもって再現することは困難であることが分かった。
【0041】
形状C:この多形体は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドをアセトニトリル中に60℃で長い時間をかけてスラリー化することにより生成される。この結晶のX線解析は、任意の他の多形体と明確に異なった(図5を参照のこと)。多形体Cは種々の水和物であることが示され、これらは昇温すると、不安的な形状に脱溶媒和する。
【0042】
アモルファス物質:この多形体は形状Aの多形体を200℃までの温度に加熱することにより生成される。この多形体は空気中の水分の存在下で不安定であり、種々の水和物を形成する。
【0043】
(形状A、B、Cおよびアモルファス物質の解析のための技術)
X線粉末回折
X線粉末回折(XRPD)解析は、Shimadzu XRD−6000 X線粉末回折器で、Cu Kα照射を用いて実施した。上記装置は精密な焦点のX線チューブを備え、そしてこのチューブの電圧および電流をそれぞれ40kVおよび40mAにセットした。発散スリットおよび散乱スリットを1’’mmにセットし、そして受光スリットを0.15mmにセットした。回折照射はNaIシンチレーション検出器により検出した。2.5〜40°の2θより3°/分(0.4秒/0.02°ステップ)で、シータ−2シータ連続スキャンを使用した。ケイ素基準を装置配列のチェックに使用した。データを集め、XRD−6000v.4.1ソフトウェアを用いて解析した。
【0044】
X線粉末回折(XRPD)解析はまた、CPS(屈曲位置感度(Curved Position Sensitive))検出器を備えたInel XRG−3000回折器を用いて、120°の28範囲において実施した。装置の較正はケイ素基準を用いて実施した。このチューブの電圧および電流をそれぞれ40kVおよび30mAにセットした。モノクロメータースリットは5mm×80μmにセットした。サンプルを、シリコンインサートを備えたアルミニウムサンプルホルダー内またはガラスXRPD−品質(quiality)キャピラリー内に置いた。それぞれのキャピラリーを、データ捕捉の間にこのキャピラリーがスピンするのが可能なようにモーターを備えるゴニオメーターの上部に据え付けた。実時間処理データをCu−Kα照射を用いて、0.03°の2θの分解能で収集した。代表的に、データは300秒の時間を越えて収集した。2.5〜40°の2θの範囲内のデータ点のみを、プロットされたXPRDパターン内に表示した。
【0045】
(熱的解析)
熱重量(TG)分析をTA装置2050熱重量分析器またはTA装置2950熱重量分析器にて実施した。較正基準は、ニッケルおよびAlumelTMであった。サンプルをアルミニウムサンプルパン内に置き、TGファーネス中に挿入し、ならびに厳密に重量を測定した。このサンプルを窒素中で10℃/分の速度で300℃か350℃のいずれかまで加熱した。他の方法で述べられなければ、サンプルの重量は分析前にTGAファーネス内で25℃で平衡化した。
【0046】
示差走査熱量測定(DSC)分析をTA装置示差走査熱量測定器2920にて実施した。厳密に重量を測定されたサンプルを、圧力を解放させるための小さい穴を備える曲がったパンか密封して封緘されたパンのいずれかの中に置いた。それぞれのサンプルを、窒素下で10℃/分の速度で300℃か350℃のいずれかまで加熱した。インジウム金属を較正基準として使用した。温度は最大転移点で記録した。
【0047】
(赤外分光法)
赤外線スペクトルは、Ever−Glo中赤外線/遠赤外線供給源、拡大範囲臭化カリウムビームスプリッター、および重水素化トリグリシンスルフェート(DTGS)検出器を備えたMagna860(登録商標)フーリエ変換赤外(FT−IR)分光器(Nicolet Instrument Corp.)により捕捉した。他の方法で述べられなければ、Spectra−Tech,Inc.の拡散反射率付属品(diffuse reflectance accessory)(CollectorTM)をサンプリングに使用した。それぞれのスペクトルは、4cm−1のスペクトル分解能で、256共付加(co−add)スキャンを示す。化合物のサンプル調製は、サンプルをマイクロカップ内に置く工程、および物質をつや消しガラススライドで平らにする工程よりなった。バックグラウンドのデータセットは、適切な位置の調整ミラー(alignment mirror)を用いて得た。スペクトルは、バックグラウンドの単一ビームデータセットに対するサンプルの単一ビームデータセットの比を示す。装置の波長較正はポリスチレンを用いて実施した。
【0048】
(NMR分光法)
液相1H NMRスペクトルは周囲温度で、Brukerモデル AM−250分光計で、5.87T(ラーモア回転数:1H=250MHz)で捕捉した。時間−領域データを、7.5psのパルス幅および1.6834秒の捕捉時間を使用して、5000Hzのスペクトルウィンドウ(window)に対して捕捉した。全部で16,384データポイントを収集した。5秒の緩和遅延時間を過渡状態間に使用した。それぞれのデータセットは、代表的に128共平均(coaveraged)過渡状態よりなった。スペクトルはGRAMS 132 A1ソフトウェア、バージョン6.00を利用して処理した。自由誘導減衰(FID)は、フーリエ変換より前に、ゼロフィリングして(zero−filled)、データポイントの数を4倍し、0.61Hzの指数関数(line−broadening factor)で指数的に積算した。1Hスペクトルは内部標準として加えられたテトラメチルシラン(0ppm)を内部測定基準とした。
【0049】
あるいは、NMR解析を実施例4に記載のように実施した。
【0050】
(水分吸着/脱着解析)
水分吸着/脱着解析データを、VTI SGA−100 気化吸着解析器で収集した。吸着データおよび脱着データを、5%〜95%の範囲の相対湿度(RH)にわたり、10%RH間隔で、窒素パージ下で収集した。塩化ナトリウム(NaCl)およびポリビニルピロリドン(PVP)を較正基準として使用した。解析に使用する平衡基準は、5分以内で0.0100%未満の重量変化であり、重量基準が合わない場合は180分の最大平衡時間を有した。プロットされるデータは、初期の湿度含有量については調整していない。
【0051】
(命名法)
化合物(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドの構造は、以下のとおりである:
【0052】
【化20】
(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドの合成)
((1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドの大スケールの合成のための一つの方法が、反応スキームIに示される。
【0053】
【化21】
工程1−式(2)の調製
式(2)の化合物は、溶媒なしでのヒドラジン一水和物との反応より式(1)の化合物より調製される。この反応は約40℃+/−5℃の温度で実施される。反応が完了すると、式(2)の生成物は、式(2)の化合物が限られた溶解性を有するプロトン性溶媒(例えば、エタノールまたはイソプロパノール)中で撹拌されることにより単離される。この混合物は約1〜5時間撹拌され、続いてろ過させる。固体は、水で撹拌し、ろ過し、および水で、続いてイソプロパノールで洗浄することにより不純物を取り除かれ、ならびに減圧下で乾燥される。この固体は精製なしで次の工程に利用される。
【0054】
工程2−式(3)の調製
次に式(2)の化合物は、約1〜1.2モル当量のエチル2−ホルミル−3−オキソプロピオネートとの反応により式(3)の化合物へと変換される。この反応はプロトン性溶媒中、好ましくは、エタノール中で、ほぼ還流温度で、約2〜4時間かけて実施される。約0℃に冷却した後、固体はろ過され、冷エタノールで洗浄され、減圧下で乾燥される。式(3)の生成物は精製なしで次の工程に利用される。
【0055】
工程3−最終生成物の調製
最終生成物は、メチルアミン、好ましくは水性のメチルアミンとの反応により式(3)の化合物より調製される。この反応は約室温で、約4時間かけて実施される。式Iの生成物は、従来の手段(例えば、ろ過し、固体を冷エタノールで洗浄し、ならびに減圧下で乾燥する)で単離される。
【0056】
(出発原料の調製)
(4S,2R,3R,5R)−2−(6−アミノ−2−クロロプリン−9−イル)−5−(ヒドロキシメチル)オキソラン−3,4−ジオールは、工程1の出発原料として使用される。この化合物は市販されている。
【0057】
エチル2−ホルミル−3−オキソプロパノエートは、工程2の出発原料として使用される。これは市販されており、また反応スキームIIで示すように作製され得る。
【0058】
【化22】
エチル3,3−ジエトキシプロピオネートは強塩基、好ましくは水素化ナトリウムの存在下でギ酸エチルエステルと反応させる。この反応は約0〜5℃で、約24時間かけて実施される。生成物は従来の手段(例えば、水を添加し、従来の溶媒(例えばt−ブチルメチルエーテル)で不純物を抽出し、水相を例えば塩酸で酸性化し、次にジクロロメタンのような溶媒で抽出し、および減圧下でこの乾燥させた抽出物よりこの溶媒を除く)により単離される。
【0059】
(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドの大スケールでの合成に好ましい方法は、反応スキームIIIに示される。
【0060】
【化23】
工程1−式(2)の調製
式(2)の化合物は、溶媒なしでのヒドラジン一水和物との反応より式(1)の化合物より調製される。この反応は約45〜55℃+/−5℃の温度で実施される。反応が完了すると、式(2)の生成物は、式(2)の化合物が限られた溶解性を有するプロトン性溶媒(例えば、エタノールまたはイソプロパノール)中で撹拌されることにより単離される。この混合物は約1〜5時間撹拌され、続いてろ過される。固体は、水で撹拌し、ろ過し、および水で、続いてエタノールまたはイソプロパノールで洗浄することにより不純物を取り除かれ、ならびに減圧下で乾燥される。この固体は精製なしで次の工程に利用される。
【0061】
工程2−式(4)の調製
式(2)の化合物は次に過剰の、例えば2〜10倍過剰、より好ましくは約5〜10倍過剰のエチル2−ホルミル−3−オキソプロピオネートとの反応により式(4)の化合物に変換される。この反応は、プロトン性溶媒中、例えばエタノール中で、ほぼ還流温度で、約2〜4時間かけて実施される。約0℃に冷却後、固体はろ過され、冷エタノールで洗浄され、および減圧下で乾燥されて、そして式(4)の生成物は精製なしで次の工程に利用される。
【0062】
式(4)の化合物は、(2E)アルケン誘導体として記載される。なぜなら、(2E)アルケン誘導体がこの反応で形成される主要異性体であるからである。しかし、かなりの量の(2Z)アルケン誘導体もこの反応で形成し得ることは注記されるべきであり;それは:
【0063】
【化24】
であり、エチル(2Z)−3−({9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)−オキソラン−2−イル]−2−[4−(エトキシカルボニル)ピラゾリル]プリン−6−イル}アミノ)−2−ホルミルプロペ−2−ノエートと命名される。
【0064】
従って、式(4)の化合物は(2E)アルケン誘導体のみとして示されるが、用語「式(4)の化合物」は、この式(4)の化合物が単一の(2E)異性体である場合と、および生成物の大部分が(2E)異性体でありかつ(2Z)異性体の小量部分もまた存在する場合との両方を包含することが意図される。工程3に記載されるメチルアミンとの反応による式(4)の化合物の最終生成物への変換は、同様の様式で進行し、ここで式(4)の化合物は(2E)異性体として存在するか、または(2E)異性体と(2Z)異性体との混合物として存在するかのどちらかである。
【0065】
工程3−最終生成物の調製
最終生成物は、メチルアミン、好ましくは水性のメチルアミンとの反応により式(4)の化合物より調製される。この反応は初期は約0〜5℃で約8時間かけて、好ましくは圧力反応器内で、続いて、50〜60℃の温度に約1時間かけて昇温し、15〜30分その温度を維持することにより実施される。この生成物は、従来の手段、例えば、0〜5℃まで冷却し、約1時間真空を維持して、メチルアミンを除く工程で単離される。真空を解除し、そして残った内容物は0〜5℃に少なくとも30分保持され、続いてろ過される。このようにして得られた固体は次に水で、続いてエタノールで洗浄され、減圧下で乾燥される。
【0066】
このプロセスは、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドをその一水和物として提供する。この多形体は、ジメチルスルホキシドに溶解し、任意の固体不純物を溶液よりろ過し、ならびに水を加えることによって溶液から一水和物を沈殿させることにより、さらに精製され得る。
【実施例】
【0067】
(実施例1)
(エチル−2−ホルミル−3−オキソプロピオネートの調製)
【0068】
【化25】
磁気撹拌バー、熱電対、デジタル温度計、ガス吹き込み管およびガス排出管および滴下ろうとを備えた三つ口または四つ口の丸底フラスコにアルゴンを充填した。テトラヒドロフラン中のエチル3,3−ジメトキシプロピオネート(64.5g)を滴下ろうとに充填した。水素化ナトリウム(60%分散物の21.2g)を反応フラスコに充填し、続いてテトラヒドロフランを充填した。フラスコの内容物を氷浴で0〜5℃に冷却し、そしてギ酸エチルエステル(257g)を加えた。この混合物を0〜5℃に冷却し、そしてフラスコ内の温度を5℃未満に維持しながら、滴下ろうとの内容物を滴下した。氷浴を取り除き、そして内容物を周囲温度まで戻した。エチル3,3−ジメトキシプロピオネートの消費をTLC分析にてモニタリングした。この反応を氷水(10.6容量)を加えてクエンチし、メチルt−ブチルエーテル(各5.4容量)にて3回抽出し、そして有機層を破棄した。水相を濃塩酸でpHが1〜1.5になるまで酸性化した。酸性の水層をジクロロメタンで3回抽出し、合わせた有機層を硫酸ナトリウムで乾燥した。溶媒を減圧下で除き、そして真空でこの残渣を蒸留してエチル2−ホルミル−3−オキソプロピオネートを27.92g、70%収率で準備した。
【0069】
(実施例2)
(A.2−ヒドラジノアデノシン(2)の調製)
【0070】
【化26】
メカニカルスターラー、ガス吹き込み管、ガス排出管および熱電対を備えたフラスコをアルゴンで洗い流した。2−クロロアデノシンヘミ水和物(hemihydrate)(53.1g)を加え、続いてヒドラジン一水和物(134g)を加えた。この混合物を40〜45℃で2時間加熱しながら撹拌した。この反応の進行をTLC分析にて追跡した。反応が完了すると、熱源を取り除き、エタノール(800ml)を加えた。この混合物を室温で2時間撹拌し、次に沈殿物をろ過により収集した。ろ過ケークをエタノールで洗浄し、減圧下で30分乾燥した。この固体を、メカニカルスターラーを備えたきれいなフラスコに移し、そして水(300ml)を加えた。この懸濁液を室温で18時間撹拌し、そして固体をろ過して単離した。このろ過ケークを氷水(300ml)で洗浄し、続いて氷冷エタノール(300ml)で洗浄した。固体を減圧下で乾燥し、2−ヒドラジノアデノシン(41.38g、81.4%収率、99.3%純度)を準備した。
【0071】
(B.2−ヒドラジノアデノシン(2)の調製の代替法)
ヒドラジン水和物(258g、250ml)を含む反応容器を40〜50℃に加熱した。加熱した混合物に、2−クロロアデノシンヘミ水和物(100g)を温度を45〜55℃の間に維持しながら一部ずつ加えた。温度をこの温度で2時間維持し、次に脱イオン水(500ml)を温度を45〜55℃の間に維持しながら30分の時間をかけて加えた。この混合物を次に0〜5℃に3時間かけて冷却し、続いてこの温度でさらに30分撹拌した。この固体を次にろ過し、冷脱イオン水(200ml、2〜5℃)で、続いてエタノール(400ml)で洗浄した。この固体を12時間真空乾燥し、2−ヒドラジノアデノシンを準備した。
【0072】
(実施例3)
(エチル1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル)ピラゾール−4−カルボキシレート(3)の調製)
【0073】
【化27】
エチル2−ホルミル−3−オキソプロピオネート(23.93g、0.17mol)を、メカニカルスターラー、ガス吹き込み管、ガス排出管および還流冷却管を備えたフラスコ内に据えた。2−プロパノールをこのフラスコに加え、続いて2−ヒドラジノアデノシン(44.45g、0.15mol)を加えた。この混合物を2〜4時間撹拌しながら加熱して還流させ、次に反応の進行をTLC分析した。反応が完了したことを判断して、熱源を取り除き、この混合物を室温まで冷却した。懸濁液を撹拌しながら氷浴で1.5〜2時間冷却した。固体を真空ろ過で単離し、氷冷2−プロパノールで洗浄した。生成物のエチル1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル)ピラゾール−4−カルボキシレートを減圧下で、一定の重量まで乾燥した。収量54.29g、純度(HPLCによる)96.6%。
【0074】
(実施例4)
((1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドの調製)
【0075】
【化28】
エチル1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル)ピラゾール−4−カルボキシレート(46.4g)とメチルアミン(40%水中、600ml)との混合物を周囲温度で約4時間撹拌し、次に反応の進行をHPLC分析した。過剰のメチルアミンの大部分を減圧下で除去し、残った混合物を0℃で2時間冷却した。固体物質をろ過し、氷冷200プルーフエタノールで洗浄し、および減圧下で乾燥して、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドを一水和物として準備した(36.6g、純度99.6%)。
【0076】
この物質の構造は1H NMRにて確認した(図1および以下の参照のこと)。熱的分析(図2を参照のこと)は、1分子の水の存在と一致する結果を提供した。X線粉末回折パターンを得た(図3)。
【0077】
【化29】
1H NMRおよび13C NMRスペクトルを以下の様式で得た。上記の得られた物質の2つのサンプルの重量を測定し、d6−DMSOに溶解した。このサンプルを1Hスペクトルには5.3mg使用し、13Cスペクトルには20.8mg使用した。すべてのスペクトルは、周囲温度で、JEOL Eclipse+ 400 分光計において、1Hは400MHz、13Cは100MHzで操作することにより得た。
【0078】
【化30】
((1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド一水和物の精製)
ジメチルスルホキシド(300ml)中の(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド一水和物(100g)の溶液を、0.6〜0.8ミクロンプレフィルターおよび0.2ミクロンフィルターを通してろ過し、任意の固体不純物を取り除いた。次にこのろ液に1時間かけて攪拌しながら脱イオン水(1リットル)をゆっくり加え、そしてこの生成したスラリーを続いて1時間以上撹拌した。固体をろ過し、脱イオン水(2×1リットル)で洗浄し、ならびに1時間以上真空乾燥した。次に乾燥した生成物を、脱イオン水(1.5リットル)により、2時間以上かけて再度スラリー化し、ろ過し、ならびに脱イオン水(1リットル)、続いて純エタノール(750ml)で洗浄した。精製された生成物を40℃以下の温度で、12時間以上かけて真空乾燥し、任意の2−ヒドラジンアデノシン不純物のない、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド一水和物を準備した。
【0079】
(実施例5)
(エチル(2E)−3−({9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−2−[4−(エトキシカルボニル)ピラゾリル]プリン−6−イル}アミノ)−2−ホルミルプロプ−2−エノエートの調製)
【0080】
【化31】
2−ヒドラジノアデノシン(100g、0.34mol)とエチル2−ホルミル−3−オキソプロピオネート(242g、1.7mol)と純エタノールとの混合物を反応器に充填し、そしてこの混合物を2時間加熱して還流させた。反応が完了したことを判断して、熱源を取り除き、この混合物を3時間かけて徐々に5〜10℃に冷却した。この温度でこのスラリーを30分撹拌し、そして混合物をろ過した。固体物質を冷純エタノール(5〜10℃)で洗浄し、続いて40℃を超えない温度で真空乾燥し、エチル(2E)−3−({9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−2−[4−(エトキシカルボニル)ピラゾリル]プリン−6−イル}アミノ)−2−ホルミルプロプ−2−エノエートを準備した。
【0081】
元素分析は、以下の結果を与えた:C,48.75%;H,4.86%;N,18.05%;O,27.57.理論値:C,49.72%;H,4.74%;N,18.45%;O,27.09.この分析は、所望の生成物のヘミ水和物に対する実験的誤差限界に一
致する(C,48.89%;H,4.81%;N,18.1%;O,28.12)。
【0082】
1H NMRおよび13C NMRスペクトルは、以下の様式で得た。20.2mgの式(4)の化合物を約0.75mlのDMSO−d6に溶解し、そしてスペクトルを、周囲温度で、JEOL ECX−400 NMR分光計において、1Hは400MHz、13Cは100MHzで操作することにより得た。ケミカルシフトは、DMSO溶媒において1Hで2.50ppmおよび13Cで39.5ppmを参照した。
【0083】
(結果)
1Hケミカルシフトおよび13Cケミカルシフトは表1に記載される。約60/30の比の2つの異性体が1Hスペクトルと13Cスペクトルとの両方より観測された。表中に、主および副とラベル化した。
【0084】
【化32】
式(4)の化合物は以下の2つの異性体の混合物であることを確認した:
【0085】
【化33】
(実施例6)
(化合物(4)からの(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドの調製)
【0086】
【化34】
40%メチルアミン水溶液(1300ml)を圧力反応器に据え、0〜5℃に冷却し、そして実施例5の生成物(エチル(2E)−3−({9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−2−[4−(エトキシカルボニル)ピラゾリル]プリン−6−イル}アミノ)−2−ホルミルプロプ−2−エノエート)(100g)を加えた。この混合物を0〜5℃で少なくとも8時間撹拌し、反応の完了をモニタリングした。反応が完了すると、この混合物を温めて、50〜60℃の間の温度で1時間維持し、続いて1時間かけて30℃未満まで冷却した。温度が30℃未満になると、この混合物を100〜150mmHgの圧力で脱ガスし、温度を0〜5℃に低下させた。この混合物を、100〜150mmHgの圧力を維持しながら、0〜5℃で少なくとも1時間撹拌した。次に温度を0〜5℃に維持しながら、30分以上かけてこの真空状態を停止し、窒素に置換した。固体生成物を次にろ過し、水(3×500ml)、続いて純エタノール(625ml)で洗浄した。生成物を40℃を越えさせない温度で真空乾燥し、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドを一水和物として準備した。
【0087】
1H NMRおよび13C NMRスペクトルを以下の様式で得た。上記の得られた物質の2つのサンプルの重量を測定し、d6−DMSOに溶解した。サンプルを1Hスペクトルには5.3mg使用し、13Cスペクトルには20.8mg使用した。すべてのスペクトルは、周囲温度で、JEOL Eclipse+ 400 分光計において、1Hは400MHz、13Cは100MHzで操作することにより得た。
【0088】
【化35】
元素分析は、以下の結果を与えた:C,43.96%;H,4.94%;N,27.94.理論値:C,44.12%;H,4.94%;N,27.44%;O,27.09.
この分析は、一水和物に対する実験的誤差限界に一致する。
【技術分野】
【0001】
本願は、米国仮特許出願第60/801,857号(2006年5月18日出願)および米国仮特許出願第60/765,114号(2006年2月3日出願)の優先権を主張し、これらの全部の開示は本明細書中で参考として援用する。
【0002】
(発明の分野)
本発明は、A2A−アデノシンレセプターアゴニストの大スケールでの調製のための方法に関し、またその化合物の多形体および特定の多形体を単離する工程の方法に関する。
【背景技術】
【0003】
(背景)
アデノシンは、天然に存在するヌクレオシドであり、A1、A2A、A2BおよびA3として公知のアデノシンレセプターのファミリーとの相互作用によりその生物学的効果を発揮し、それらレセプターはすべて、重要な生理学的プロセスを調整する。アデノシンの生物学的効果の1つは、冠動脈血管拡張薬として機能することであり;この結果はA2Aアデノシンレセプターとの相互作用により生ずる。このアデノシンの効果は、造影剤(imaging agent)(例えばタリウム201)の投与より前に冠動脈が拡張される場合、心臓の画像化に対する手助けとして有用であることが理解されており、従って生じる画像の観察によって、冠動脈疾患の存在または不在が決定され得る。このような技術の利点は、冠動脈疾患を有する患者に対して明らかに望ましくないトレッドミル上での運動によって、冠動脈血管拡張を引き起こす従来の方法を避けることである。
【0004】
しかし、アデノシンの投与はいくつかの不利な点を有する。アデノシンはヒトの体内で非常に短い半減期(10秒未満)を有し、またA1、A2A、A2BおよびA3レセプターアゴニストによる作用(agonism)に関するすべての効果を有する。従って、選択的なA2Aアデノシンレセプターアゴニストの使用は、冠動脈血管拡張を生ずるのによりすぐれた方法、特により長い半減期を有し、および副作用がほとんどないまたはまったくない方法を提供する。
【0005】
これらの所望の特性を有する化合物のクラスは、特許文献1に開示され、その全部の開示は本明細書中で参考として援用する。特に、特許文献1に開示される1つの化合物、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドは、高選択性A2A−アデノシンレセプターアゴニストとして示され、心臓の画像化において有用な冠動脈血管拡張薬として、臨床試験を現在行っている最中である。
【0006】
この化合物および類似の化合物において重要性が高まっていることに鑑みて、高収率および高純度で物質を大量に製造するための便利な方法を提供する、新しい合成方法を見つけることが望まれるようになった。この目的の化合物を開示する特許文献1は、この化合物を調製するためのいくつかの方法を提供する。しかしながら、これらの方法は小スケールでの合成に適しているが、この特許文献に開示されるすべての合成方法は保護基を利用しており、この方法は大スケールの合成に望ましくない。
【0007】
さらに、上記所望の生成物(つまり、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド)は、少なくとも3つの異なる結晶形状で存在することが可能であり、その中の最も安定なものは、一水和物である。この多形体は、相対湿度のストレス条件下で(under relative
humidity stress conditions)、その融点まで安定である。従って、新しい合成によって生成した最終生成物が安定な一水和物として得られることが望まれる。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】米国特許第6,403,567号明細書
【発明の概要】
【課題を解決するための手段】
【0009】
(発明の趣旨)
本発明の目的は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドおよびその多形体、好ましくはその一水和物の大スケールの調製のための便利な合成を提供することである。従って、第一の局面においては、本発明は式I:
【0010】
【化10】
の化合物の調製に関し、それは式(3):
【0011】
【化11】
の化合物をメチルアミンと接触させる工程を含む。
【0012】
第一の実施形態において、この反応はメチルアミンの水溶液中で、初期は約0〜5℃の温度で、続いて約50〜70℃に加温して実施される。あるいは、この反応は上記に記載のように、しかし封緘された圧力反応器中で実施される。
【0013】
第二の実施形態において、上記生成物は、この生成物を溶媒(例えば、ジメチルスルホキシド)に溶解し、精製水を加え、このようにして形成されるスラリーをろ過し、フィルターの内容物を水、続いてエタノールで洗浄し、そして残った固体を真空下で40℃を越えない温度で乾燥させることにより、純一水和物として単離される。
【0014】
第二の局面において、本発明は、式(3):
【0015】
【化12】
の化合物の調製に関し、それは式(2):
【0016】
【化13】
の化合物をエチル2−ホルミル−3−オキソプロピオネートと接触させる工程を含む。
【0017】
一実施形態において、この反応はエタノール中、約80℃の温度で、約1.1モル当量のエチル2−ホルミル−3−オキソプロピオネートを用いて、実施される。
【0018】
第三の局面において、本発明は、式(2):
【0019】
【化14】
の化合物の調製に関し、それは式(1):
【0020】
【化15】
の化合物をヒドラジンと接触させる工程を含む。
【0021】
上記に記載の合成は、所望の生成物の大スケールでの合成に適しており、この生成物は、最終生成物中に1種の少量の不純物が見られるが、良好な収率で提供される。この不純物は、式(2)のもとの中間体;つまり式:
【0022】
【化16】
の化合物であると示されている。この不純物は、結晶化により最終生成物より取り除かれ得るが、上記合成の利点のすべてを有するが、最終生成物中に式(2)の化合物を不純物として与えない代わりの合成を探求することが決定された。
【0023】
従って、第四の局面として、本発明は式(4):
【0024】
【化17】
の化合物をメチルアミンと接触させることにより、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドを合成する方法に関する。
【0025】
第一の実施形態において、この反応はメチルアミンの水溶液中で、初期は約0〜5℃の温度で、続いて約50〜70℃に加温して実施される。好ましくは、この反応は、封緘された圧力反応器中で実施される。
【0026】
第二の実施形態において、上記生成物は、この生成物を溶媒(例えば、ジメチルスルホキシド)に溶解し、精製水を加え、このようにして形成されるスラリーをろ過し、フィルターの内容物を水、続いてエタノールで洗浄し、そして残った固体を真空下で40℃を越えない温度で乾燥させることにより、純一水和物として単離される。
【0027】
第五の局面において、本発明は、式(4):
【0028】
【化18】
の化合物の合成に関し、それは式(2):
【0029】
【化19】
の化合物を過剰の、好ましくは2〜10倍過剰、より好ましくは5〜10倍過剰のエチル2−ホルミル−3−オキソプロピオネートと接触させる工程を含む。
【0030】
一実施形態において、この反応はエタノール中で、約80℃の温度で実施される。エチル2−ホルミル−3−オキソプロピオネートは5〜10倍過剰で存在する。
例えば、本願発明は以下の項目を提供する。
(項目1)
(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド:
【化1】
の大スケールでの調製のための方法であって、式(3)の化合物:
【化2】
をメチルアミンと反応させる工程を含む、方法。
(項目2)
項目1に記載の方法であって、前記式(3)の化合物をメチルアミン水溶液と、初期は約0〜5℃で、続いて約50〜70℃に加温して反応させる、方法。
(項目3)
前記反応が、封緘された圧力反応器で実施される、項目1に記載の方法。
(項目4)
項目1に記載の方法であって、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド最終生成物が、
(a)該生成物を溶媒に溶解する工程
(b)精製水を加える工程
(c)このようにして形成されたスラリーをろ過する工程
(d)フィルターの内容物を水、続いてエタノールで洗浄する工程、および
(e)残った固体を真空下で40℃を越えない温度で乾燥する工程
によって、純一水和物として単離される、方法。
(項目5)
項目4に記載の方法であって、前記工程(a)で使用される溶媒がジメチルスルホキシドである、方法。
(項目6)
式(3)の化合物:
【化3】
を調製する方法であって、式(2)の化合物:
【化4】
をエチル2−ホルミル−3−オキソプロピオネートと反応させる工程を含む、方法。
(項目7)
項目6に記載の方法であって、前記反応がエタノール中で実施される、方法。
(項目8)
項目7に記載の方法であって、前記反応が約80℃の温度で実施される、方法。
(項目9)
項目6に記載の方法であって、約1.1モル当量のエチル2−ホルミル−3−オキソプロピオネートが使用される、方法。
(項目10)
式(2)の化合物:
【化5】
を調製するための方法であって、式(1)の化合物:
【化6】
をヒドラジンと反応させる工程を含む、方法。
(項目11)
(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドを合成する方法であって、式(4)の化合物:
【化7】
をメチルアミンに接触させることにより合成する、方法。
(項目12)
項目11に記載の方法であって、前記式(4)の化合物をメチルアミン水溶液と、初期は約0〜5℃で、続いて約50〜70℃に加温して反応させる、方法。
(項目13)
前記反応が、封緘された圧力反応器で実施される、項目11に記載の方法。
(項目14)
項目11に記載の方法であって、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド最終生成物が、
(a)項目11に記載の生成物を溶媒に溶解する工程
(b)精製水を加える工程
(c)このようにして形成されたスラリーをろ過する工程
(d)フィルターの内容物を水、続いてエタノールで洗浄する工程、および
(e)残った固体を真空下で40℃を越えない温度で乾燥する工程
によって、純一水和物として単離される、方法。
(項目15)
項目14に記載の方法であって、前記工程(a)で使用される溶媒がジメチルスルホキシドである、方法。
(項目16)
式(4)の化合物:
【化8】
を合成する方法であって、式(2)の化合物:
【化9】
を過剰のエチル2−ホルミル−3−オキソプロピオネートと接触させる工程を含む、方法。
(項目17)
項目16に記載の方法であって、前記反応がエタノール中で実施される、方法。
(項目18)
項目17に記載の方法であって、前記反応が約80℃の温度で実施される、方法。
(項目19)
項目16に記載の方法であって、約2〜10倍のモル過剰のエチル2−ホルミル−3−オキソプロピオネートが使用される、方法。
(項目20)
項目19に記載の方法であって、約5〜10倍のモル過剰のエチル2−ホルミル−3−オキソプロピオネートが使用される、方法。
【図面の簡単な説明】
【0031】
【図1】図1は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド一水和物(形状A)の1H NMRスペクトルである。
【図2】図2は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド一水和物の熱的分析である。
【図3】図3は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド一水和物のX線回折パターンである。
【図4】図4は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドの形状BのX線回折パターンである。
【図5】図5は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドの形状Cの、形状Aと比較したX線回折パターンである。
【発明を実施するための形態】
【0032】
(定義および一般的なパラメーター)
本明細書で使用される場合、以下の単語および句は一般的に、それらが使用される文脈が別の方法で示す程度までを除いて、以下に記載の意味を有することが意味される。
【0033】
「任意の」または「必要に応じて」は、続いて記載される事象または状況が起こっても起こらなくてもよいことを意味し、そしてその記載が、その事象または状況が起こる場合および、その事象または状況が起こらない場合を包含することを意味する。
【0034】
用語「治療上の有効量」は、以下に定義されるような処置が必要な哺乳動物に投与される場合に、その処置をもたらすのに充分な式Iの化合物の量、に言及する。この治療上の有効量は、処置される被験体および疾患状態、被験体の体重および年齢、疾患状態の重大さ、投与の様式などに依存して変わり、そして当業者により容易に決定され得る。
【0035】
用語「処置」または「処置すること」は、哺乳動物における任意の疾患の処置を意味し、以下:
(i)上記疾患を予防すること、すなわち上記疾患の臨床的症状を発症させないようにすること;
(ii)上記疾患を抑制すること、すなわち臨床的症状の進行を止めること;
および/または
(iii)上記疾患を軽減すること、すなわち臨床的症状の後退を引き起こすこと;
が挙げられる。
【0036】
本明細書で使用される場合、「薬学的に受容可能なキャリアー」としては、任意のおよびすべての溶媒、分散媒体、コーティング、抗菌剤および抗真菌剤、等張性遅延剤(isotonic delaying agent)および吸収遅延剤(absorption delaying agent)などが挙げられる。薬学的に活性な物質のためのこのような媒体および薬剤の使用は、当該分野で周知である。任意の従来の媒体または薬剤が活性成分と不適合である範囲を除き、治療的組成物中でのその使用が企図される。補助的な活性成分もまた、上記組成物中に組み込まれ得る。
【0037】
用語「多形体」は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドのアモルファスおよび溶媒和物を含むことが意図される。
【0038】
この化合物は少なくとも3つの異なる結晶形状(本明細書中で、形状A、形状B、形状Cと言及される)およびアモルファス生成物として存在することが可能である、ということがわかった。
【0039】
形状A:この多形体は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドをプロトン性溶媒(例えば、エタノールまたはエタノール/水混合物)または極性溶媒(例えば、ジメチルスルホキシド/水)より結晶化することにより生成され得る。形状Aは一水和物であることが示されており、および周囲温度で種々の多形体の中で最も安定である。形状Aは相対湿度のストレス条件下で、その融点まで安定である。
【0040】
形状B:この多形体は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドのトリフルオロエタノール溶液を、周囲温度で減圧下でエバポレーションすることによって生成される。この結晶のX線解析は、任意の他の多形体と明確に異なった(図4を参照のこと)が、上記X線解析が乱れた幅広いピークを与え、そして上記多形体がさまざまな量の水を含むため、その構造の決定は困難であった。この多形体の調製を信頼性をもって再現することは困難であることが分かった。
【0041】
形状C:この多形体は、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドをアセトニトリル中に60℃で長い時間をかけてスラリー化することにより生成される。この結晶のX線解析は、任意の他の多形体と明確に異なった(図5を参照のこと)。多形体Cは種々の水和物であることが示され、これらは昇温すると、不安的な形状に脱溶媒和する。
【0042】
アモルファス物質:この多形体は形状Aの多形体を200℃までの温度に加熱することにより生成される。この多形体は空気中の水分の存在下で不安定であり、種々の水和物を形成する。
【0043】
(形状A、B、Cおよびアモルファス物質の解析のための技術)
X線粉末回折
X線粉末回折(XRPD)解析は、Shimadzu XRD−6000 X線粉末回折器で、Cu Kα照射を用いて実施した。上記装置は精密な焦点のX線チューブを備え、そしてこのチューブの電圧および電流をそれぞれ40kVおよび40mAにセットした。発散スリットおよび散乱スリットを1’’mmにセットし、そして受光スリットを0.15mmにセットした。回折照射はNaIシンチレーション検出器により検出した。2.5〜40°の2θより3°/分(0.4秒/0.02°ステップ)で、シータ−2シータ連続スキャンを使用した。ケイ素基準を装置配列のチェックに使用した。データを集め、XRD−6000v.4.1ソフトウェアを用いて解析した。
【0044】
X線粉末回折(XRPD)解析はまた、CPS(屈曲位置感度(Curved Position Sensitive))検出器を備えたInel XRG−3000回折器を用いて、120°の28範囲において実施した。装置の較正はケイ素基準を用いて実施した。このチューブの電圧および電流をそれぞれ40kVおよび30mAにセットした。モノクロメータースリットは5mm×80μmにセットした。サンプルを、シリコンインサートを備えたアルミニウムサンプルホルダー内またはガラスXRPD−品質(quiality)キャピラリー内に置いた。それぞれのキャピラリーを、データ捕捉の間にこのキャピラリーがスピンするのが可能なようにモーターを備えるゴニオメーターの上部に据え付けた。実時間処理データをCu−Kα照射を用いて、0.03°の2θの分解能で収集した。代表的に、データは300秒の時間を越えて収集した。2.5〜40°の2θの範囲内のデータ点のみを、プロットされたXPRDパターン内に表示した。
【0045】
(熱的解析)
熱重量(TG)分析をTA装置2050熱重量分析器またはTA装置2950熱重量分析器にて実施した。較正基準は、ニッケルおよびAlumelTMであった。サンプルをアルミニウムサンプルパン内に置き、TGファーネス中に挿入し、ならびに厳密に重量を測定した。このサンプルを窒素中で10℃/分の速度で300℃か350℃のいずれかまで加熱した。他の方法で述べられなければ、サンプルの重量は分析前にTGAファーネス内で25℃で平衡化した。
【0046】
示差走査熱量測定(DSC)分析をTA装置示差走査熱量測定器2920にて実施した。厳密に重量を測定されたサンプルを、圧力を解放させるための小さい穴を備える曲がったパンか密封して封緘されたパンのいずれかの中に置いた。それぞれのサンプルを、窒素下で10℃/分の速度で300℃か350℃のいずれかまで加熱した。インジウム金属を較正基準として使用した。温度は最大転移点で記録した。
【0047】
(赤外分光法)
赤外線スペクトルは、Ever−Glo中赤外線/遠赤外線供給源、拡大範囲臭化カリウムビームスプリッター、および重水素化トリグリシンスルフェート(DTGS)検出器を備えたMagna860(登録商標)フーリエ変換赤外(FT−IR)分光器(Nicolet Instrument Corp.)により捕捉した。他の方法で述べられなければ、Spectra−Tech,Inc.の拡散反射率付属品(diffuse reflectance accessory)(CollectorTM)をサンプリングに使用した。それぞれのスペクトルは、4cm−1のスペクトル分解能で、256共付加(co−add)スキャンを示す。化合物のサンプル調製は、サンプルをマイクロカップ内に置く工程、および物質をつや消しガラススライドで平らにする工程よりなった。バックグラウンドのデータセットは、適切な位置の調整ミラー(alignment mirror)を用いて得た。スペクトルは、バックグラウンドの単一ビームデータセットに対するサンプルの単一ビームデータセットの比を示す。装置の波長較正はポリスチレンを用いて実施した。
【0048】
(NMR分光法)
液相1H NMRスペクトルは周囲温度で、Brukerモデル AM−250分光計で、5.87T(ラーモア回転数:1H=250MHz)で捕捉した。時間−領域データを、7.5psのパルス幅および1.6834秒の捕捉時間を使用して、5000Hzのスペクトルウィンドウ(window)に対して捕捉した。全部で16,384データポイントを収集した。5秒の緩和遅延時間を過渡状態間に使用した。それぞれのデータセットは、代表的に128共平均(coaveraged)過渡状態よりなった。スペクトルはGRAMS 132 A1ソフトウェア、バージョン6.00を利用して処理した。自由誘導減衰(FID)は、フーリエ変換より前に、ゼロフィリングして(zero−filled)、データポイントの数を4倍し、0.61Hzの指数関数(line−broadening factor)で指数的に積算した。1Hスペクトルは内部標準として加えられたテトラメチルシラン(0ppm)を内部測定基準とした。
【0049】
あるいは、NMR解析を実施例4に記載のように実施した。
【0050】
(水分吸着/脱着解析)
水分吸着/脱着解析データを、VTI SGA−100 気化吸着解析器で収集した。吸着データおよび脱着データを、5%〜95%の範囲の相対湿度(RH)にわたり、10%RH間隔で、窒素パージ下で収集した。塩化ナトリウム(NaCl)およびポリビニルピロリドン(PVP)を較正基準として使用した。解析に使用する平衡基準は、5分以内で0.0100%未満の重量変化であり、重量基準が合わない場合は180分の最大平衡時間を有した。プロットされるデータは、初期の湿度含有量については調整していない。
【0051】
(命名法)
化合物(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドの構造は、以下のとおりである:
【0052】
【化20】
(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドの合成)
((1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドの大スケールの合成のための一つの方法が、反応スキームIに示される。
【0053】
【化21】
工程1−式(2)の調製
式(2)の化合物は、溶媒なしでのヒドラジン一水和物との反応より式(1)の化合物より調製される。この反応は約40℃+/−5℃の温度で実施される。反応が完了すると、式(2)の生成物は、式(2)の化合物が限られた溶解性を有するプロトン性溶媒(例えば、エタノールまたはイソプロパノール)中で撹拌されることにより単離される。この混合物は約1〜5時間撹拌され、続いてろ過させる。固体は、水で撹拌し、ろ過し、および水で、続いてイソプロパノールで洗浄することにより不純物を取り除かれ、ならびに減圧下で乾燥される。この固体は精製なしで次の工程に利用される。
【0054】
工程2−式(3)の調製
次に式(2)の化合物は、約1〜1.2モル当量のエチル2−ホルミル−3−オキソプロピオネートとの反応により式(3)の化合物へと変換される。この反応はプロトン性溶媒中、好ましくは、エタノール中で、ほぼ還流温度で、約2〜4時間かけて実施される。約0℃に冷却した後、固体はろ過され、冷エタノールで洗浄され、減圧下で乾燥される。式(3)の生成物は精製なしで次の工程に利用される。
【0055】
工程3−最終生成物の調製
最終生成物は、メチルアミン、好ましくは水性のメチルアミンとの反応により式(3)の化合物より調製される。この反応は約室温で、約4時間かけて実施される。式Iの生成物は、従来の手段(例えば、ろ過し、固体を冷エタノールで洗浄し、ならびに減圧下で乾燥する)で単離される。
【0056】
(出発原料の調製)
(4S,2R,3R,5R)−2−(6−アミノ−2−クロロプリン−9−イル)−5−(ヒドロキシメチル)オキソラン−3,4−ジオールは、工程1の出発原料として使用される。この化合物は市販されている。
【0057】
エチル2−ホルミル−3−オキソプロパノエートは、工程2の出発原料として使用される。これは市販されており、また反応スキームIIで示すように作製され得る。
【0058】
【化22】
エチル3,3−ジエトキシプロピオネートは強塩基、好ましくは水素化ナトリウムの存在下でギ酸エチルエステルと反応させる。この反応は約0〜5℃で、約24時間かけて実施される。生成物は従来の手段(例えば、水を添加し、従来の溶媒(例えばt−ブチルメチルエーテル)で不純物を抽出し、水相を例えば塩酸で酸性化し、次にジクロロメタンのような溶媒で抽出し、および減圧下でこの乾燥させた抽出物よりこの溶媒を除く)により単離される。
【0059】
(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドの大スケールでの合成に好ましい方法は、反応スキームIIIに示される。
【0060】
【化23】
工程1−式(2)の調製
式(2)の化合物は、溶媒なしでのヒドラジン一水和物との反応より式(1)の化合物より調製される。この反応は約45〜55℃+/−5℃の温度で実施される。反応が完了すると、式(2)の生成物は、式(2)の化合物が限られた溶解性を有するプロトン性溶媒(例えば、エタノールまたはイソプロパノール)中で撹拌されることにより単離される。この混合物は約1〜5時間撹拌され、続いてろ過される。固体は、水で撹拌し、ろ過し、および水で、続いてエタノールまたはイソプロパノールで洗浄することにより不純物を取り除かれ、ならびに減圧下で乾燥される。この固体は精製なしで次の工程に利用される。
【0061】
工程2−式(4)の調製
式(2)の化合物は次に過剰の、例えば2〜10倍過剰、より好ましくは約5〜10倍過剰のエチル2−ホルミル−3−オキソプロピオネートとの反応により式(4)の化合物に変換される。この反応は、プロトン性溶媒中、例えばエタノール中で、ほぼ還流温度で、約2〜4時間かけて実施される。約0℃に冷却後、固体はろ過され、冷エタノールで洗浄され、および減圧下で乾燥されて、そして式(4)の生成物は精製なしで次の工程に利用される。
【0062】
式(4)の化合物は、(2E)アルケン誘導体として記載される。なぜなら、(2E)アルケン誘導体がこの反応で形成される主要異性体であるからである。しかし、かなりの量の(2Z)アルケン誘導体もこの反応で形成し得ることは注記されるべきであり;それは:
【0063】
【化24】
であり、エチル(2Z)−3−({9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)−オキソラン−2−イル]−2−[4−(エトキシカルボニル)ピラゾリル]プリン−6−イル}アミノ)−2−ホルミルプロペ−2−ノエートと命名される。
【0064】
従って、式(4)の化合物は(2E)アルケン誘導体のみとして示されるが、用語「式(4)の化合物」は、この式(4)の化合物が単一の(2E)異性体である場合と、および生成物の大部分が(2E)異性体でありかつ(2Z)異性体の小量部分もまた存在する場合との両方を包含することが意図される。工程3に記載されるメチルアミンとの反応による式(4)の化合物の最終生成物への変換は、同様の様式で進行し、ここで式(4)の化合物は(2E)異性体として存在するか、または(2E)異性体と(2Z)異性体との混合物として存在するかのどちらかである。
【0065】
工程3−最終生成物の調製
最終生成物は、メチルアミン、好ましくは水性のメチルアミンとの反応により式(4)の化合物より調製される。この反応は初期は約0〜5℃で約8時間かけて、好ましくは圧力反応器内で、続いて、50〜60℃の温度に約1時間かけて昇温し、15〜30分その温度を維持することにより実施される。この生成物は、従来の手段、例えば、0〜5℃まで冷却し、約1時間真空を維持して、メチルアミンを除く工程で単離される。真空を解除し、そして残った内容物は0〜5℃に少なくとも30分保持され、続いてろ過される。このようにして得られた固体は次に水で、続いてエタノールで洗浄され、減圧下で乾燥される。
【0066】
このプロセスは、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドをその一水和物として提供する。この多形体は、ジメチルスルホキシドに溶解し、任意の固体不純物を溶液よりろ過し、ならびに水を加えることによって溶液から一水和物を沈殿させることにより、さらに精製され得る。
【実施例】
【0067】
(実施例1)
(エチル−2−ホルミル−3−オキソプロピオネートの調製)
【0068】
【化25】
磁気撹拌バー、熱電対、デジタル温度計、ガス吹き込み管およびガス排出管および滴下ろうとを備えた三つ口または四つ口の丸底フラスコにアルゴンを充填した。テトラヒドロフラン中のエチル3,3−ジメトキシプロピオネート(64.5g)を滴下ろうとに充填した。水素化ナトリウム(60%分散物の21.2g)を反応フラスコに充填し、続いてテトラヒドロフランを充填した。フラスコの内容物を氷浴で0〜5℃に冷却し、そしてギ酸エチルエステル(257g)を加えた。この混合物を0〜5℃に冷却し、そしてフラスコ内の温度を5℃未満に維持しながら、滴下ろうとの内容物を滴下した。氷浴を取り除き、そして内容物を周囲温度まで戻した。エチル3,3−ジメトキシプロピオネートの消費をTLC分析にてモニタリングした。この反応を氷水(10.6容量)を加えてクエンチし、メチルt−ブチルエーテル(各5.4容量)にて3回抽出し、そして有機層を破棄した。水相を濃塩酸でpHが1〜1.5になるまで酸性化した。酸性の水層をジクロロメタンで3回抽出し、合わせた有機層を硫酸ナトリウムで乾燥した。溶媒を減圧下で除き、そして真空でこの残渣を蒸留してエチル2−ホルミル−3−オキソプロピオネートを27.92g、70%収率で準備した。
【0069】
(実施例2)
(A.2−ヒドラジノアデノシン(2)の調製)
【0070】
【化26】
メカニカルスターラー、ガス吹き込み管、ガス排出管および熱電対を備えたフラスコをアルゴンで洗い流した。2−クロロアデノシンヘミ水和物(hemihydrate)(53.1g)を加え、続いてヒドラジン一水和物(134g)を加えた。この混合物を40〜45℃で2時間加熱しながら撹拌した。この反応の進行をTLC分析にて追跡した。反応が完了すると、熱源を取り除き、エタノール(800ml)を加えた。この混合物を室温で2時間撹拌し、次に沈殿物をろ過により収集した。ろ過ケークをエタノールで洗浄し、減圧下で30分乾燥した。この固体を、メカニカルスターラーを備えたきれいなフラスコに移し、そして水(300ml)を加えた。この懸濁液を室温で18時間撹拌し、そして固体をろ過して単離した。このろ過ケークを氷水(300ml)で洗浄し、続いて氷冷エタノール(300ml)で洗浄した。固体を減圧下で乾燥し、2−ヒドラジノアデノシン(41.38g、81.4%収率、99.3%純度)を準備した。
【0071】
(B.2−ヒドラジノアデノシン(2)の調製の代替法)
ヒドラジン水和物(258g、250ml)を含む反応容器を40〜50℃に加熱した。加熱した混合物に、2−クロロアデノシンヘミ水和物(100g)を温度を45〜55℃の間に維持しながら一部ずつ加えた。温度をこの温度で2時間維持し、次に脱イオン水(500ml)を温度を45〜55℃の間に維持しながら30分の時間をかけて加えた。この混合物を次に0〜5℃に3時間かけて冷却し、続いてこの温度でさらに30分撹拌した。この固体を次にろ過し、冷脱イオン水(200ml、2〜5℃)で、続いてエタノール(400ml)で洗浄した。この固体を12時間真空乾燥し、2−ヒドラジノアデノシンを準備した。
【0072】
(実施例3)
(エチル1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル)ピラゾール−4−カルボキシレート(3)の調製)
【0073】
【化27】
エチル2−ホルミル−3−オキソプロピオネート(23.93g、0.17mol)を、メカニカルスターラー、ガス吹き込み管、ガス排出管および還流冷却管を備えたフラスコ内に据えた。2−プロパノールをこのフラスコに加え、続いて2−ヒドラジノアデノシン(44.45g、0.15mol)を加えた。この混合物を2〜4時間撹拌しながら加熱して還流させ、次に反応の進行をTLC分析した。反応が完了したことを判断して、熱源を取り除き、この混合物を室温まで冷却した。懸濁液を撹拌しながら氷浴で1.5〜2時間冷却した。固体を真空ろ過で単離し、氷冷2−プロパノールで洗浄した。生成物のエチル1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル)ピラゾール−4−カルボキシレートを減圧下で、一定の重量まで乾燥した。収量54.29g、純度(HPLCによる)96.6%。
【0074】
(実施例4)
((1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドの調製)
【0075】
【化28】
エチル1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル)ピラゾール−4−カルボキシレート(46.4g)とメチルアミン(40%水中、600ml)との混合物を周囲温度で約4時間撹拌し、次に反応の進行をHPLC分析した。過剰のメチルアミンの大部分を減圧下で除去し、残った混合物を0℃で2時間冷却した。固体物質をろ過し、氷冷200プルーフエタノールで洗浄し、および減圧下で乾燥して、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドを一水和物として準備した(36.6g、純度99.6%)。
【0076】
この物質の構造は1H NMRにて確認した(図1および以下の参照のこと)。熱的分析(図2を参照のこと)は、1分子の水の存在と一致する結果を提供した。X線粉末回折パターンを得た(図3)。
【0077】
【化29】
1H NMRおよび13C NMRスペクトルを以下の様式で得た。上記の得られた物質の2つのサンプルの重量を測定し、d6−DMSOに溶解した。このサンプルを1Hスペクトルには5.3mg使用し、13Cスペクトルには20.8mg使用した。すべてのスペクトルは、周囲温度で、JEOL Eclipse+ 400 分光計において、1Hは400MHz、13Cは100MHzで操作することにより得た。
【0078】
【化30】
((1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド一水和物の精製)
ジメチルスルホキシド(300ml)中の(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド一水和物(100g)の溶液を、0.6〜0.8ミクロンプレフィルターおよび0.2ミクロンフィルターを通してろ過し、任意の固体不純物を取り除いた。次にこのろ液に1時間かけて攪拌しながら脱イオン水(1リットル)をゆっくり加え、そしてこの生成したスラリーを続いて1時間以上撹拌した。固体をろ過し、脱イオン水(2×1リットル)で洗浄し、ならびに1時間以上真空乾燥した。次に乾燥した生成物を、脱イオン水(1.5リットル)により、2時間以上かけて再度スラリー化し、ろ過し、ならびに脱イオン水(1リットル)、続いて純エタノール(750ml)で洗浄した。精製された生成物を40℃以下の温度で、12時間以上かけて真空乾燥し、任意の2−ヒドラジンアデノシン不純物のない、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミド一水和物を準備した。
【0079】
(実施例5)
(エチル(2E)−3−({9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−2−[4−(エトキシカルボニル)ピラゾリル]プリン−6−イル}アミノ)−2−ホルミルプロプ−2−エノエートの調製)
【0080】
【化31】
2−ヒドラジノアデノシン(100g、0.34mol)とエチル2−ホルミル−3−オキソプロピオネート(242g、1.7mol)と純エタノールとの混合物を反応器に充填し、そしてこの混合物を2時間加熱して還流させた。反応が完了したことを判断して、熱源を取り除き、この混合物を3時間かけて徐々に5〜10℃に冷却した。この温度でこのスラリーを30分撹拌し、そして混合物をろ過した。固体物質を冷純エタノール(5〜10℃)で洗浄し、続いて40℃を超えない温度で真空乾燥し、エチル(2E)−3−({9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−2−[4−(エトキシカルボニル)ピラゾリル]プリン−6−イル}アミノ)−2−ホルミルプロプ−2−エノエートを準備した。
【0081】
元素分析は、以下の結果を与えた:C,48.75%;H,4.86%;N,18.05%;O,27.57.理論値:C,49.72%;H,4.74%;N,18.45%;O,27.09.この分析は、所望の生成物のヘミ水和物に対する実験的誤差限界に一
致する(C,48.89%;H,4.81%;N,18.1%;O,28.12)。
【0082】
1H NMRおよび13C NMRスペクトルは、以下の様式で得た。20.2mgの式(4)の化合物を約0.75mlのDMSO−d6に溶解し、そしてスペクトルを、周囲温度で、JEOL ECX−400 NMR分光計において、1Hは400MHz、13Cは100MHzで操作することにより得た。ケミカルシフトは、DMSO溶媒において1Hで2.50ppmおよび13Cで39.5ppmを参照した。
【0083】
(結果)
1Hケミカルシフトおよび13Cケミカルシフトは表1に記載される。約60/30の比の2つの異性体が1Hスペクトルと13Cスペクトルとの両方より観測された。表中に、主および副とラベル化した。
【0084】
【化32】
式(4)の化合物は以下の2つの異性体の混合物であることを確認した:
【0085】
【化33】
(実施例6)
(化合物(4)からの(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドの調製)
【0086】
【化34】
40%メチルアミン水溶液(1300ml)を圧力反応器に据え、0〜5℃に冷却し、そして実施例5の生成物(エチル(2E)−3−({9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−2−[4−(エトキシカルボニル)ピラゾリル]プリン−6−イル}アミノ)−2−ホルミルプロプ−2−エノエート)(100g)を加えた。この混合物を0〜5℃で少なくとも8時間撹拌し、反応の完了をモニタリングした。反応が完了すると、この混合物を温めて、50〜60℃の間の温度で1時間維持し、続いて1時間かけて30℃未満まで冷却した。温度が30℃未満になると、この混合物を100〜150mmHgの圧力で脱ガスし、温度を0〜5℃に低下させた。この混合物を、100〜150mmHgの圧力を維持しながら、0〜5℃で少なくとも1時間撹拌した。次に温度を0〜5℃に維持しながら、30分以上かけてこの真空状態を停止し、窒素に置換した。固体生成物を次にろ過し、水(3×500ml)、続いて純エタノール(625ml)で洗浄した。生成物を40℃を越えさせない温度で真空乾燥し、(1−{9−[(4S,2R,3R,5R)−3,4−ジヒドロキシ−5−(ヒドロキシメチル)オキソラン−2−イル]−6−アミノプリン−2−イル}ピラゾール−4−イル)−N−メチルカルボキサミドを一水和物として準備した。
【0087】
1H NMRおよび13C NMRスペクトルを以下の様式で得た。上記の得られた物質の2つのサンプルの重量を測定し、d6−DMSOに溶解した。サンプルを1Hスペクトルには5.3mg使用し、13Cスペクトルには20.8mg使用した。すべてのスペクトルは、周囲温度で、JEOL Eclipse+ 400 分光計において、1Hは400MHz、13Cは100MHzで操作することにより得た。
【0088】
【化35】
元素分析は、以下の結果を与えた:C,43.96%;H,4.94%;N,27.94.理論値:C,44.12%;H,4.94%;N,27.44%;O,27.09.
この分析は、一水和物に対する実験的誤差限界に一致する。
【特許請求の範囲】
【請求項1】
本願明細書に記載された発明。
【請求項1】
本願明細書に記載された発明。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2013−14620(P2013−14620A)
【公開日】平成25年1月24日(2013.1.24)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−226343(P2012−226343)
【出願日】平成24年10月11日(2012.10.11)
【分割の表示】特願2008−553396(P2008−553396)の分割
【原出願日】平成19年2月2日(2007.2.2)
【出願人】(504003226)ギリアード・パロ・アルト・インコーポレイテッド (62)
【氏名又は名称原語表記】Gilead Palo Alto,Inc.
【Fターム(参考)】
【公開日】平成25年1月24日(2013.1.24)
【国際特許分類】
【出願番号】特願2012−226343(P2012−226343)
【出願日】平成24年10月11日(2012.10.11)
【分割の表示】特願2008−553396(P2008−553396)の分割
【原出願日】平成19年2月2日(2007.2.2)
【出願人】(504003226)ギリアード・パロ・アルト・インコーポレイテッド (62)
【氏名又は名称原語表記】Gilead Palo Alto,Inc.
【Fターム(参考)】
[ Back to top ]