
AG013736を含んでなる剤形
本発明は、式(1)の化合物またはその医薬的に許容される塩、溶媒和物またはプロドラッグの剤形を提供する。さらに本発明は、該剤形を哺乳動物へ投与することによって、癌のような異常な細胞増殖を治療する方法を提供する。

【発明の詳細な説明】
【技術分野】
【0001】
本出願は、その開示がそのまま参照により本明細書に組み込まれる、米国仮特許出願第60/460,695号(2003年4月3日出願)および米国仮特許出願第60/491,771号(2003年7月31日出願)の利益を主張する。
【0002】
背景技術
本発明は、癌のような異常な細胞増殖の哺乳動物における治療に有用であるVEGFR阻害剤に関する。本発明はまた、異常な細胞増殖の哺乳動物、具体的にはヒトにおける治療にこうした化合物を使用する方法と、こうした化合物を含有する医薬組成物に関する。
【背景技術】
【0003】
式1:
【0004】
【化1】
【0005】
により表される化合物、6−[2−(メチルカルバモイル)フェニルスルファニル]−3−E−[2−(ピリジン−2−イル)エテニル]インダゾールは、結腸癌、黒色腫、乳癌および肺癌の異種移植モデルにおいて広汎な前臨床活性のある、VEGFR/PDGFRチロシンキナーゼの強力かつ選択的な阻害剤である(Hu−Lowe D,Heller D,Brekken J,Feeley R,Amundson K,Haines M,Troche G,Kim Y,Gonzalez D,Herrman M,Batugo M,Vekich S,Kania R,McTigue M,Gregory S,Bender S,Shalinsky D.,「VEGF/PDGF受容体チロシンキナーゼの低分子阻害剤、AG013736の薬理活性(Pharmacological Activities of AG013736,a Small Molecular Inhibitor of VEGF/PDGF Receptor Tyrosine Kinases)」Proc.Am.Assoc.Cancer Res.2002:抄録#5357)。ダイナミック対比増強MRI(dceMRI)を使用して評価した前臨床の腫瘍血管応答は、腫瘍増殖指標と対応することを示した(Wilmes LJ,Hylton NM,Wang D,Fleming LM,Gibbs J,Kim Y,Dillon R,Brasch RC,Park JW,Li K−L,Henry RG,Partridge SC,Shalinsky DR,Hu−Lowe D,McShane TM,およびPallavicini MG.,「新規VEGFR TK阻害剤、AG013736は、ヌードマウス中のヒトBT474乳癌異種移植片における腫瘍増殖および血管透過性を抑制する(AG013736,a Novel VEGFR TK Inhibitor,Suppresses Tumor Growth and Vascular Permeability in Human BT474 Breast Cancer Xenografts in Nude Mice)」Proc.Am.Assoc.Cancer Res.2003:抄録#3772)。
【発明の開示】
【発明が解決しようとする課題】
【0006】
発明の要約
本発明は、6−[2−(メチルカルバモイル)フェニルスルファニル]−3−E−[2−(ピリジン−2−イル)エテニル]インダゾールと体系的に命名可能である、式1:
【0007】
【化2】
【0008】
の化合物を使用する剤形と治療の方法を提供する。
1つの態様において、本発明は、哺乳動物への投与用の剤形を提供し、該剤形は、式1の化合物、その医薬的に許容される塩、溶媒和物、またはプロドラッグ、またはこれらの混合物を、哺乳動物への投与後に4500ng・時間/mL以下の式1の化合物またはその活性代謝産物の24時間AUC血漿値を提供するのに有効な量で含んでなる。24時間AUC血漿値は、本明細書の「詳細な説明」に記載のようにして決定可能である。
【0009】
この態様の具体的な側面において、24時間AUC血漿値の上限は、4000ng・時間/mL以下または3000ng・時間/mL以下または2500ng・時間/mL以下または2000ng・時間/mL以下または1500ng・時間/mL以下または1000ng・時間/mL以下または800ng・時間/mL以下または700ng・時間/mL以下である。好ましくは、そして列挙した上限のいずれとも組み合わせて、24時間AUC血漿値は、少なくとも10ng・時間/mLまたは少なくとも25ng・時間/mLまたは少なくとも50ng・時間/mLまたは少なくとも75ng・時間/mLまたは少なくとも100ng・時間/mLまたは少なくとも125ng・時間/mLである。24時間AUC血漿値の想定範囲には、列挙した下限のいずれかから列挙した上限のいずれかまでの範囲が含まれる。好ましい範囲の具体的な非限定例には、25〜4500ng・時間/mL、50〜2500ng・時間/mL、75〜1000ng・時間/mL、100〜800ng・時間/mL、および125〜700ng・時間/mLが含まれる。
【0010】
別の態様において、本発明は、上記に定義される式1の化合物、その医薬的に許容される塩、溶媒和物またはプロドラッグ、またはこれらの混合物を30mg以下の量で含んでなる剤形を提供する。化合物の全部または一部が塩、溶媒和物またはプロドラッグとして剤形にあるとき、その量は式1の化合物の同等量であり、それは分子量に基づいて当業者により容易に計算されると理解されたい。
【0011】
この態様の具体的な側面において、この量の上限は、20mg以下または15mg以下または12mg以下または10mg以下または8mg以下または7mg以下である。好ましくは、そして列挙した上限のいずれとも組み合わせて、この量は、少なくとも0.5mgまたは少なくとも1mgまたは少なくとも1.5mgまたは少なくとも2mgまたは少なくとも2.5mgまたは少なくとも3mgである。想定範囲には、列挙した下限のいずれかから列挙した上限のいずれかまでの範囲が含まれる。好ましい範囲の具体的な非限定例には、0.5〜30mg、1〜20mg、1.5〜15mg、2〜10mg、2.5〜8mg、および3〜7mgが含まれる。
【0012】
さらに本発明は、上記に定義されるような式1の化合物、その医薬的に許容される塩、溶媒和物またはプロドラッグ、またはこれらの混合物を、ヒトが含まれる哺乳動物への投与後に4500ng・時間/mL以下の式1の化合物またはその活性代謝産物の24時間AUC血漿値を提供するのに有効な量で哺乳動物へ投与することによって、異常な細胞増殖を哺乳動物において治療する方法を提供する。24時間AUC血漿値は、本明細書の「詳細な説明」に記載のようにして決定可能である。
【0013】
この態様の具体的な側面において、24時間AUC血漿値の上限は、4000ng・時間/mL以下または3000ng・時間/mL以下または2500ng・時間/mL以下または2000ng・時間/mL以下または1500ng・時間/mL以下または1000ng・時間/mL以下または800ng・時間/mL以下または700ng・時間/mL以下である。好ましくは、そして列挙した上限のいずれとも組み合わせて、24時間AUC血漿値は、少なくとも10ng・時間/mLまたは少なくとも25ng・時間/mLまたは少なくとも50ng・時間/mLまたは少なくとも75ng・時間/mLまたは少なくとも100ng・時間/mLまたは少なくとも125ng・時間/mLである。24時間AUC血漿値の想定範囲には、列挙した下限のいずれかから列挙した上限のいずれかまでの範囲が含まれる。好ましい範囲の具体的な非限定例には、25〜4500ng・時間/mL、50〜2500ng・時間/mL、75〜1000ng・時間/mL、100〜800ng・時間/mL、および125〜700ng・時間/mLが含まれる。
【0014】
さらに本発明は、上記に定義されるような式1の化合物、その医薬的に許容される塩、溶媒和物またはプロドラッグ、またはこれらの混合物を、投薬につき30mg以下の量でヒトが含まれる哺乳動物へ投与することによって、異常な細胞増殖を哺乳動物において治療する方法を提供する。化合物の全部または一部が塩、溶媒和物、またはプロドラッグとして剤形にあるとき、その量は式1の化合物の同等量であり、分子量に基づいて当業者により容易に計算されると理解されたい。
【0015】
この態様の具体的な側面において、この量の上限は、20mg以下または15mg以下または12mg以下または10mg以下または8mg以下または7mg以下である。好ましくは、そして列挙した上限のいずれとも組み合わせて、この量は、少なくとも0.5mgまたは少なくとも1mgまたは少なくとも1.5mgまたは少なくとも2mgまたは少なくとも2.5mgまたは少なくとも3mgである。想定範囲には、列挙した下限のいずれかから列挙した上限のいずれかまでの範囲が含まれる。好ましい範囲の具体的な非限定例には、0.5〜30mg、1〜20mg、1.5〜15mg、2〜10mg、2.5〜8mg、および3〜7mgが含まれる。
【0016】
本明細書に記載される本発明の方法のいずれの具体的な態様においても、異常な細胞増殖は癌であり、限定されないが、肺癌、骨癌、膵臓癌、皮膚癌(skin cancer)、頭頚部癌、皮膚(cutaneous)または眼内黒色腫、子宮癌、卵巣癌、直腸癌、肛門域の癌、胃癌、結腸癌、乳癌、子宮癌、卵管の癌、子宮内膜癌、子宮頚部癌、膣癌、外陰部の癌、ホジキン病、食道癌、小腸の癌、内分泌系の癌、甲状腺癌、副甲状腺癌、副腎の癌、柔組織の肉腫、尿道癌、陰茎癌、前立腺癌、慢性または急性白血病、リンパ球性リンパ腫、膀胱癌、腎臓または尿管の癌、腎細胞癌、腎盂癌(carcinoma of the renal pelvis)、中枢神経系(CNS)の新生物、原発性CNSリンパ腫、脊髄軸腫瘍、脳幹神経膠腫、下垂体腺腫、または1以上の前記癌の組合せが含まれる。前記方法の別の態様において、前記異常な細胞増殖は良性の増殖性疾患であり、限定されないが、乾癬、良性前立腺肥大症、または再狭窄が含まれる。
【0017】
別の態様において、本発明は、式1の化合物の療法的に許容される量を哺乳動物へ投与することによって、PDGFR BB仲介性の癌細胞遊走を哺乳動物において阻害する方法を提供する。
【0018】
別の態様において、本発明は、式1の化合物の療法的に許容される量を哺乳動物へ投与することによって、c−KIT活性を哺乳動物において阻害する方法を提供する。
本明細書に記載する本発明の方法のいずれかのさらに具体的な態様において、本方法は、抗腫瘍剤、抗血管新生剤、シグナル伝達阻害剤、および抗増殖剤より選択される1以上の物質の量を哺乳動物へ投与することをさらに含み、該量は、前記異常な細胞増殖を治療するのに全体として有効である。そうした物質には、PCT公開公報番号:WO00/38715、WO00/38716、WO00/38717、WO00/38718、WO00/38719、WO00/38730、WO00/38665、WO/0037107、およびWO00/38786に開示されるものが含まれ、この開示は、そのまま参照により本明細書に組み込まれる。
【0019】
抗腫瘍剤の例には、減数分裂阻害剤、例えば、ビンブラスチン、ビノレルビン、ビンデシン、およびビンクリスチンのような植物アルカロイド誘導体;コルキン、アロコキン、ハリコンドリン、N−ベンゾイルトリメチル−メチルエーテルコルヒチン酸、ドラスタチン10、マイスタンシン、リゾキシン、パクリタキセル(TaxolTM)、ドセタキセル(TaxotereTM)、2’−N−[3−(ジメチルアミノ)プロピル]グルタラメート(TaxolTM誘導体)のようなタキサン、チオコルヒチン、トリチルシステイン、テニポシド、メトトレキセート、アザチオプリン、フルオロウラシル、シトシンアラビノシド、2’,2’−ジフルオロデオキシシチジン(ゲンシタビン)、アドリアマイシン、およびミタマイシン;アルキル化剤、例えばシスプラチン、カルボプラチン、オキシプラチン、イプロプラチン、N−アセチル−DL−ザルコシル−L−ロイシンのエチルエステル(AsaleyまたはAsalex)、1,4−シクロヘキサジン−1,4−ジカルバミン酸、2,5−ビス(1−アジルジニル)−3,6−ジオキソ−、ジエチルエステル(ジアジクォン)、1,4−ビス(メタンスルホニルオキシ)ブタン(ビスルファンまたはロイコスルファン)、クロロゾトシン、クロメゾン、シアノモルホリノドキソルビシン、シクロジソン、ジアンヒドログラクチトール、フルオロドパン、ヘプスルファム、マイトマイシンC、ヒカンテオネマイトマイシンC、ミトゾラミド、1−(2−クロロエチル)−4−(3−クロロプロピル)−ピペラジン二塩酸塩、ピペラジンジオン、ピポブロマン、ポルフィロマイシン、スピロヒダントインマスタード、テロキシロン、テトラプラチン、チオテパ、トリエチレンミラミン、ウラシルナイトロジェンマスタード、ビス(3−メシルオキシプロピル)アミン塩酸塩、マイトマイシン、シクロヘキシル−クロロエチルニトロソ尿素、メチルシクロヘキシル−クロロエチルニトロソ尿素、1−(2−クロロエチル)−3−(2,6−ジオキソ−3−ピペリジニル)−1−ニトロソ尿素、ビス(2−クロロエチル)ニトロソ尿素のようなニトロソ尿素、プロカルバジン、ダカルバジン、メクロロエタミン、シクロホスファミド、イホスファミド、メルファラン、クロラムブシル、リン酸エストラムスチンナトリウム、ストルプトゾイン、およびテモゾラミドのようなナイトロジェンマスタード関連化合物;DNA代謝拮抗薬、例えば、5−フルオロウラシル、シトシンアラビノシド、ヒドロキシ尿素、2−[(3−ヒドロキシ−2−ピリノジビル)メチレン]−ヒドラジンカルボチオアミド、デオキシフルオロウリジン、5−ヒドロキシ−2−ホルミルピリジンチオセミカルバゾン、α−2’−デオキシ−6−チオグアノシン、グリシン酸アフィジコリン、5−アザデオキシシチジン、β−チオグアニンデオキシリボシド、シクロシチジン、グアナゾール、イノシングリコジアルデヒド、マクベシンII、ピラゾリミダゾール、クラドリビン、ペントスタチン、チアグアニン、メルカプトプリン、ブレオマイシン、2−シクロデオキシアデノシン、ラルチトレキセドおよびペメトレキセド二ナトリウムのようなチミジン酸シンターゼ阻害剤、クロファラビン、フロクスウリジン、およびフルダラビン;DNA/RNA代謝拮抗薬、例えば、L−アラノシン、5−アザシチジン、アシビシン、アミノプテリンとN−[2−クロロ−5−[[(2,4−ジアミノ−5−メチル−6−キナゾリニル)メチル]アミノ]ベンゾイル]−L−アスパラギン酸、N−[4−[[(2,4−ジアミノ−5−エチル−6−キナゾリニル)メチル]アミノ]ベンゾイル]−L−アスパラギン酸、N−[2−クロロ−4−[[(2,4−ジアミノプテリジニル)メチル]アミノ]ベンゾイル]−L−アスパラギン酸のようなその誘導体、可溶性ベーカーズ・アンチフォール(Baker’s antifol)、ジクロロアリルローソン、ブレキナー、フトラフール、ジヒドロ−5−アザシチジン、メトトレキセート、N−(ホスホノアセチル)−L−アスパラギン酸四ナトリウム塩、ピラゾフラン、トリメトレキセート、プリカマイシン、アクチノマイシンD、クリプトフィシン、およびクリプトフィシン−52のような類似体、または、例えば、N−(5−[N−(3,4−ジヒドロ−2−メチル−4−オキソキナゾリン−6−イルメチル)−N−メチルアミノ]−2−テノイル)−L−グルタミン酸のような、ヨーロッパ特許出願番号239362に開示される好ましい拮抗薬の1つ;増殖因子阻害剤;細胞周期阻害剤;挿入抗生物質、例えば、アドリアマイシンおよびブレオマイシン;タンパク質、例えば、インターフェロン;および、抗ホルモン剤、例えばNolvadexTM(タモキシフェン)のような抗エストロゲン、または例えば、CasodexTM(4’−シアノ−3−(4−フルオロフェニルスルホニル)−2−ヒドロキシ−2−メチル−3’−(トリフルオロメチル)プロピオンアニリド)のような抗アンドロゲンが含まれる。こうした併用治療は、治療の個別成分の同時、連続、または分離投薬により達成可能である。
が含まれる。
【0020】
抗血管新生剤には、MMP−2(マトリックス−メタロプロテイナーゼ2)阻害剤、MMP−9(マトリックス−メタロプロテイナーゼ9)阻害剤、およびCOX−II(シクロオキシゲナーゼII)阻害剤が含まれる。有用なCOX−II阻害剤の例には、CELEBREXTM(アレコキシブ)、バルデコキシブ、およびロフェコキシブが含まれる。有用なマトリックスメタロプロテイナーゼ阻害剤の例は、WO96/33172(1996年10月24日公開)、WO96/27583(1996年3月7日公開)、ヨーロッパ特許出願番号97304971.1(1997年7月8日出願)、ヨーロッパ特許出願番号99308617.2(1999年10月29日出願)、WO98/07697(1998年2月26日公開)、WO98/03516(1998年1月29日公開)、WO98/34918(1998年8月13日公開)、WO98/34915(1998年8月13日公開)、WO98/33768(1998年8月6日公開)、WO98/30566(1998年7月16日公開)、ヨーロッパ特許公開公報606,046(1994年7月13日公開)、ヨーロッパ特許公開公報931,788(1999年7月28日公開)、WO90/05719(1990年5月31日公開)、WO99/52910(1999年10月21日公開)、WO99/52889(1999年10月21日公開)、WO99/29667(1999年6月17日公開)、PCT国際特許出願番号PCT/IB98/01113(1998年7月21日出願)、ヨーロッパ特許出願番号99302232.1(1999年3月25日出願)、イギリス特許出願番号991296.1(1999年6月3日出願)、米国仮特許出願番号60/148,464(1999年8月12日出願)、米国特許第5,863,949号(1999年1月26日発行)、米国特許第5,861,510号(1999年1月19日発行)、およびヨーロッパ特許公開公報780,386(1997年6月25日公開)に記載され、これらはすべてそのまま参照により本明細書に組み込まれる。好ましいMMP−2およびMMP−9阻害剤は、MMP−1を阻害する活性をほとんどまたはまったく有さないものである。より好ましいのは、他のマトリックス−メタロプロテイナーゼ(即ち、MMP−1、MMP−3、MMP−4、MMP−5、MMP−6、MMP−7、MMP−8、MMP−10、MMP−11、MMP−12、およびMMP−13)に比較して、MMP−2および/またはMMP−9を選択的に阻害するものである。
【0021】
MMP阻害剤の例には、AG−3340、RO32−3555、RS13−0830、および以下のリストに列挙する化合物が含まれる:
3−[[4−(4−フルオロ−フェノキシ)−ベンゼンスルホニル]−(1−ヒドロキシカルバモイル−シクロペンチル)−アミノ]−プロピオン酸;
3−エクソ−3−[4−(4−フルオロ−フェノキシ)−ベンゼンスルホニルアミノ]−8−オキサ−ビシクロ[3.2.1]オクタン−3−カルボン酸ヒドロキシアミド;
(2R,3R)1−[4−(2−クロロ−4−フルオロ−ベンジルオキシ)−ベンゼンスルホニル]−3−ヒドロキシ−3−メチル−ピペリジン−2−カルボン酸ヒドロキシアミド;
4−[4−(4−フルオロ−フェノキシ)−ベンゼンスルホニルアミノ]−テトラヒドロ−ピラン−4−カルボン酸ヒドロキシアミド;
3−[[4−(4−フルオロ−フェノキシ)−ベンゼンスルホニル]−(1−ヒドロキシカルバモイル−シクロブチル)−アミノ]−プロピオン酸;
4−[4−(4−クロロ−フェノキシ)−ベンゼンスルホニルアミノ]−テトラヒドロ−ピラン−4−カルボン酸ヒドロキシアミド;
3−[4−(4−クロロ−フェノキシ)−ベンゼンスルホニルアミノ]−テトラヒドロ−ピラン−3−カルボン酸ヒドロキシアミド;
(2R,3R)1−[4−(4−フルオロ−2−メチル−ベンジルオキシ)−ベンゼンスルホニル]−3−ヒドロキシ−3−メチル−ピペリジン−2−カルボン酸ヒドロキシアミド;
3−[[4−(4−フルオロ−フェノキシ)−ベンゼンスルホニル]−(1−ヒドロキシカルバモイル−1−メチル−エチル)−アミノ]−プロピオン酸;
3−[[4−(4−フルオロ−フェノキシ)−ベンゼンスルホニル]−(4−ヒドロキシカルバモイル−テトラヒドロ−ピラン−4−イル)−アミノ]−プロピオン酸;
3−エクソ−3−[4−(4−クロロ−フェノキシ)−ベンゼンスルホニルアミノ]−8−オキサ−ビシクロ[3.2.1]オクタン−3−カルボン酸ヒドロキシアミド;
3−エンド−3−[4−(4−フルオロ−フェノキシ)−ベンゼンスルホニルアミノ]−8−オキサ−ビシクロ[3.2.1]オクタン−3−カルボン酸ヒドロキシアミド;および
3−[4−(4−フルオロ−フェノキシ)−ベンゼンスルホニルアミノ]−テトラヒドロ−フラン−3−カルボン酸ヒドロキシアミド;
および、前記化合物の医薬的に許容される塩、溶媒和物およびプロドラッグ。
【0022】
シグナル伝達阻害剤の例には、EGFR抗体、EGF抗体、およびEGFR阻害剤である分子のように、EGFR(上皮増殖因子受容体)応答を阻害することができる薬剤;VEGF(血管内皮増殖因子)阻害剤;および、erbB2受容体へ結合する有機分子または抗体のようなerbB2受容体阻害剤、例えばHERCEPTINTM(米国カリフォルニア州サウス・サンフランシスコのジェネンテク社)が含まれる。
【0023】
EGFR阻害剤は、例えば、WO95/19970(1995年7月27日公開)、WO98/14451(1998年4月9日公開)、WO98/02434(1998年1月22日公開)、および米国特許第5,747,498号(1998年5月5日発行)に記載されている。EGFR阻害剤には、限定されないが、モノクローナル抗体C225および抗EGFR22Mab(米国ニューヨーク州ニューヨークのイムクローン・システムズ社)、化合物ZD−1839(アストラゼネカ)、BIBX−1382(ベーリンガーインゲルハイム)、MDX−447(米国ニュージャージー州アナンデールのMedarex社)、およびOLX−103(米国ニュージャージー州ホワイトハウス・ステーションのメルク社)、VRCTC−310(Ventech Research)、およびEGF融合毒素(マサチューセッツ州ホプキントンのセラジェン社)が含まれる。
【0024】
VEGF阻害剤、例えばSU−5416およびSU−6668(米国カリフォルニア州サウス・サンフランシスコのスジェン社)も、式1の化合物と組合せまたは同時投与可能である。VEGF阻害剤は、例えば、WO99/24440(1999年5月20日公開)、PCT国際特許出願PCT/IB99/00797(1999年5月3日出願)、WO95/21613(1995年8月17日公開)、WO99/61422(1999年12月2日公開)、米国特許第5,834,504号(1998年11月10日発行)、WO98/50356(1998年11月12日公開)、米国特許第5,883,113号(1999年3月16日発行)、米国特許第5,886,020号(1999年3月23日発行)、米国特許第5,792,783号(1998年8月11日発行)、WO99/10349(1999年3月4日公開)、WO97/32856(1997年9月12日公開)、WO97/22596(1997年6月26日公開)、WO98/54093(1998年12月3日公開)、WO98/02438(1998年1月22日公開)、WO99/16755(1999年4月8日公開)、およびWO98/02437(1998年1月22日公開)に記載されて、これらはすべてそのまま参照により本明細書に組み込まれる。いくつかの具体的なVEGF阻害剤の他の例は、IM862(米国ワシントン州カークランドのCytran社);抗VEGFモノクローナル抗体、ベバシツマブ(bevacizumab)(カリフォルニア州サウス・サンフランシスコのジェネンテク社);並びに、Ribozyme(コロラド州ボルダー)およびカイロン(Chiron)(カリフォルニア州エメリヴィル)からの合成リボザイム、angiozymeTMである。
【0025】
GW−282974(グラクソ・ウェルカム社)とモノクローナル抗体のAR−209(米国テキサス州ウッドランズのAronex Pharmaceuticals社)および2B−1(カイロン)のようなErbB2受容体阻害剤は、式1の化合物と組み合わせて投与可能である。こうしたerbB2阻害剤には、WO98/02434(1998年1月22日公開)、WO99/35146(1999年7月15日公開)、WO99/35132(1999年7月15日公開)、WO98/02437(1998年1月22日公開)、WO97/13760(1997年4月17日公開)、WO95/19970(1995年7月27日公開)、米国特許第5,587,458号(1996年12月24日発行)、および米国特許第5,877,305号(1999年3月2日発行)に記載されるものが含まれ、このそれぞれは、そのまま参照により本明細書に組み込まれる。本発明において有用なerbB2受容体阻害剤は、米国仮特許出願番号60/117,341(1999年1月27日出願)および米国仮特許出願番号60/117,346(1999年1月27日出願)にも記載され、この両方ともそのまま参照により本明細書に組み込まれる。
【0026】
使用可能である他の抗増殖剤には、酵素ファルネシルタンパク質トランスフェラーゼの阻害剤と受容体チロシンキナーゼPDGFrの阻害剤が含まれ、以下の米国特許出願に開示されて特許請求される化合物:09/221946(1998年12月28日出願);09/454058(1999年12月2日出願);09/501163(2000年2月9日出願);09/539930(2000年3月31日出願);09/202796(1997年5月22日出願);09/384339(1999年8月26日出願);および09/383755(1999年8月26日出願)と、以下の米国仮特許出願に開示されて特許請求される化合物:60/168207(1999年11月30日出願);60/170119(1999年12月10日出願);60/177718(2000年1月21日出願);60/168217(1999年11月30日出願);および60/200834(2000年5月1日出願)が含まれる。上記の特許出願および仮特許出願のそれぞれは、そのまま参照により本明細書に組み込まれる。
【0027】
式1の化合物はまた、異常な細胞増殖または癌を治療するのに有用な他の薬剤と使用可能であり、限定されないが、CTLA4(細胞傷害性リンパ球抗原4)抗体のように抗腫瘍免疫応答を増強することが可能な薬剤と、CTLA4を阻止することが可能な他の薬剤;および、他のファルネシルタンパク質トランスフェラーゼ阻害剤のような抗増殖剤が含まれる。本発明において使用可能である具体的なCTLA4抗体には、そのまま参照により本明細書に組み込まれる米国仮特許出願60/113,647(1998年12月23日出願)に記載されるものが含まれる。
【0028】
別の態様において、本発明は、式1の化合物、またはその医薬的に許容される塩、溶媒和物またはプロドラッグと治療有効量のドセタキセルを含んでなる医薬組成物を提供する。
【0029】
別の態様において、本発明は、式1の化合物、またはその医薬的に許容される塩、溶媒和物またはプロドラッグと治療有効量のドセタキセルをヒトが含まれる哺乳動物へ投与することによって、異常な細胞増殖を該哺乳動物において治療する方法を提供する。式1の化合物とドセタキセルは、要望どおりに、別々に、または同じ組成物において投与可能であり、同じ投薬スケジュールでも、異なる投薬スケジュールでも投与可能である。
【課題を解決するための手段】
【0030】
定義
本明細書に使用する「異常な細胞増殖」は、他に述べなければ、正常な調節機序から独立している細胞増殖(例、接触阻害の消失)を意味する。これには:(1)突然変異したチロシンキナーゼを発現すること、または受容体チロシンキナーゼの過剰発現によって増殖する腫瘍細胞(腫瘍);(2)異常なチロシンキナーゼ活性化が起こる他の増殖性疾患の良性および悪性の細胞;および(4)受容体チロシンキナーゼによって増殖するあらゆる腫瘍の異常増殖が含まれる。
【0031】
本明細書に使用する用語「治療する」は、他に述べなければ、そうした用語が適用される障害または状態、またはそうした障害または状態の1以上の症状を逆転させること、緩和すること、その進行を阻害すること、または予防することを意味する。本明細書に使用する用語「治療」は、他に述べなければ、治療する行為を意味し、「治療する」は直前に定義されている。
【0032】
本明細書に使用する句「医薬的に許容される塩」には、他に述べなければ、化合物に存在し得る酸性または塩基性基の塩が含まれる。性質が塩基性である化合物は、様々な無機および有機酸と多種多様な塩を形成することが可能である。そうした塩基性化合物の医薬的に許容される酸付加塩を製造するために使用可能である酸は、無毒の酸付加塩、即ち、酢酸塩、ベンゼンスルホン酸塩、安息香酸塩、重炭酸塩、重硫酸塩、重酒石酸塩、ホウ酸塩、臭化物、エデト酸カルシウム、カムシラート、炭酸塩、塩化物、クラブラン酸塩、クエン酸塩、二塩酸塩、エデト酸塩、エジシレート、エストラート、エシレート、エチルコハク酸塩、フマル酸塩、グルセプタート、グルコン酸塩、グルタミン酸塩、グリコリルアルサニレート、ヘキシルレゾルシン酸塩、ヒドラバミン、臭化水素酸塩、塩酸塩、ヨウ化物、イソチオン酸塩、乳酸塩、ラクトビオン酸塩、ラウリル酸塩、リンゴ酸塩、マレイン酸塩、マンデル酸塩、メシレート、メチル硫酸塩、ムチン酸塩、ナプシラート、硝酸塩、オレイン酸塩、シュウ酸塩、パモ酸塩(エンボン酸塩)、パルミチン酸塩、パントテン酸塩、リン酸塩/二リン酸塩、ポリガラクツロン酸塩、サリチル酸塩、ステアリン酸塩、塩基性酢酸塩、コハク酸塩、タンニン酸塩、酒石酸塩、テオクラート、トシラート、トリエチオダイド、および吉草酸塩のような、薬理学的に許容されるアニオンを含有する塩を形成するものである。
【0033】
本明細書に使用する用語「プロドラッグ」は、他に述べなければ、投与に続き、何らかの化学的または生理学的プロセスを介して薬物をin vivoで放出する薬物前駆体である化合物を意味する(例えば、プロドラッグは、生理学的なpHへ置かれるとすぐに、所望される薬物形態へ変換される)。
【0034】
本発明にはまた、天然に通常見出される原子量または質量数と異なる原子量または質量数を有する原子に1以上の原子が置き換わっている事実を除けば、式1に引用する化合物と同一である、同位体標識化合物が含まれる。本発明の化合物へ取り込み可能である同位体の例には、2H、3H、13C、14C、15N、18O、17O、31P、32P、35S、18F、および36Clのように、それぞれ水素、炭素、窒素、酸素、リン、イオウ、フッ素および塩素の同位体が含まれる。上記の同位体および/または他の原子の他の同位体を含有する、本発明の化合物、そのプロドラッグ、前記化合物または前記プロドラッグのの医薬的に許容される塩および溶媒和物は、本発明の範囲内にある。本発明の特定の同位体標識化合物、例えば、3Hおよび14Cのような放射活性同位体が取り込まれているものは、薬物および/または基質の組織分布アッセイに有用である。トリチウム化、即ち3Hと炭素−14、即ち14Cの同位体は、その調製の容易さと検出可能性のために特に好ましい。さらに、重水素、即ち2Hのようなより重い同位体での置換は、より大きな代謝安定性より生じるある種の療法上の利点(例えば、in vivo半減期の増加または投与必要量の低下)をもたらすことができるので、ある状況においては好ましい場合がある。一般に、本発明の式1の同位体標識化合物とそのプロドラッグは、非標識化合物について記載した手順を行なうこと、容易に入手可能な同位体標識試薬を非同位体標識試薬に代用することによって製造可能である。
【0035】
発明の詳細な説明
式1の化合物は、そのまま参照により本明細書に組み込まれる、米国特許第6,531,491号および6,534,524号(それぞれ、2003年3月11日および2003年3月18日発行)に記載されるように製造可能である。ある種の出発材料は、当業者に馴染みの方法に従って製造可能であり、ある種の合成の変更は、当業者に馴染みの方法に従って行ってよい。
【0036】
式1の化合物は、様々な無機および有機酸と多種多様な異なる塩を形成することが可能である。そうした塩は、哺乳動物への投与について医薬的に許容されなければならないが、実際には、はじめに式1の化合物を反応混合物より医薬的に許容されない塩として単離してから、後者をアルカリ性の試薬での処理によって遊離塩基化合物へ戻し変換して、引き続き、後者の遊離塩基を医薬的に許容される酸付加塩へ変換することがしばしば望ましい。本発明の塩基性化合物の酸付加塩は、水系溶媒の媒体、またはメタノールまたはエタノールのような好適な有機溶液において実質的に同量の選択される鉱酸または有機酸で塩基性化合物を処理することによって容易に製造される。溶媒の慎重な蒸発と同時に、所望される固形の塩を容易に得る。所望される塩は、有機溶媒中の遊離塩基の溶液より、適正な鉱酸または有機酸を溶媒へ加えることによって沈殿させてもよい。
【0037】
式1の化合物の投与は、該化合物の作用部位への送達を可能にするどの方法によって実施してもよい。これらの方法には、経口ルート、十二指腸内ルート、非経口注射(静脈内、皮下、筋肉内、血管内、または注入が含まれる)、局所、および直腸の投与が含まれる。
【0038】
本化合物は、例えば、錠剤、カプセル剤、丸剤、散剤、持続放出製剤、溶液剤、懸濁液剤として経口投与に、無菌の溶液剤、懸濁液剤、または乳剤として非経口注射に、軟膏剤またはクリーム剤として局所投与に、または坐剤として直腸投与に適した形態で提供可能である。本化合物は、正確な投与量の単回投与に適した単位剤形であってよい。好ましくは、剤形には、慣用の医薬担体または賦形剤と有効成分としての式1の化合物が含まれる。さらに、剤形には、他の医療または医薬の薬剤、担体、アジュバント、等を含めてよい。
【0039】
例示の非経口投与型には、無菌の水溶液、例えば水性プロピレングリコールまたはデキストロース溶液中の溶液剤または懸濁液剤が含まれる。そうした剤形は、所望されるならば、好適に緩衝化してよい。
【0040】
好適な医薬担体には、不活性の希釈剤または充填剤、水、および様々な有機溶媒が含まれる。医薬組成物は、所望されるならば、芳香剤、結合剤、賦形剤、等のような追加成分を含有してよい。このように、経口投与では、クエン酸のような様々な賦形剤を含有する錠剤が、デンプン、アルギン酸、およびある種の複合ケイ酸塩のような様々な崩壊剤と、そしてショ糖、ゼラチン、およびアカシアのような結合剤と一緒に利用可能である。さらに、ステアリン酸マグネシウム、ラウリル硫酸ナトリウム、およびタルクのような滑沢剤が錠剤化の目的にしばしば有用である。同様の種類の固形組成物も、軟および硬充填のゼラチンカプセル剤に利用可能である。その好ましい材料には、ラクトースまたは乳糖と、高分子量ポリエチレングリコールが含まれる。水系の懸濁液剤またはエリキシル剤が経口投与に所望されるとき、そのなかの活性化合物は、様々な甘味剤または芳香剤、着色物質または色素と、そして所望されるならば、乳化剤または懸濁剤と、水、エタノール、ポリエチレングリコール、グリセリン、またはこれらの組合せのような希釈剤と一緒に組み合わせてよい。
【0041】
本発明の剤形の好ましい態様において、剤形は、経口剤形、より好ましくは、錠剤またはカプセル剤である。
本発明の方法の好ましい態様において、式1の化合物は、例えば、本明細書に記載するような経口剤形を使用することのように、経口で投与する。
【0042】
本方法には、望ましい投与方式を使用して、式1の化合物を投与することが含まれる。1つの具体的な態様において、本化合物は、1日につき1回(毎日、またはQD)、好ましくは1日につき2回(1日2回、またはBID)投与するが、より頻繁かまたは頻繁でない投与も本発明の範囲内にある。本化合物は、ヒトが含まれる哺乳動物へ、好ましくは絶食状態(投与の前および後の2時間以内は食物も飲み物もなし)で投与可能である。特に好ましい態様において、投与法はBID、絶食である。
【0043】
式1の化合物の具体的な量で様々な剤形を調製する方法は、当業者に知られているか、または明らかになろう。例えば、「レミントン製薬科学(Remington’s Pharmaceutical Sciences)」マック・パブリッシング・カンパニー、ペンシルヴェニア州イーストン、第15版(1975)を参照のこと。
【0044】
AUC血漿値は、式1の化合物またはその活性代謝産物の血漿濃度を、様々な時間間隔で液体クロマトグラフィータンデム質量分析法(LC−MS/MS)によるように直接測定して、時間曲線に対する血漿濃度下の面積を計算することによって定量可能である。AUCを計算するのに適した方法は、例えば、台形近似:
【0045】
【化3】
【0046】
[ここで、nは、データ点の数であり、tiおよびCiは、i番目のデータ点の時間および濃度(xおよびy値)である]を使用することによるように、当該技術分野でよく知られている。24時間AUC値は、投薬スケジュールに従って測定した血漿濃度を正規化することによって定量可能である。濃度標準液の調製用の復元溶液中の安定化剤として、酸性亜硫酸ナトリウムを加える。
【0047】
式1の化合物は、VEGF−R、FGF−R、CDK複合体、CHK1、CSF−R、および/またはLCKに関連したキナーゼ活性の変調および/または阻害に関して有利な特性を有する。
【0048】
以下の実施例に示すように、式1の化合物は、HUVECアポトーシスをin vitroで誘導すること、VEGF仲介性AktおよびeNOSリン酸化をHUVECにおいて阻害すること、HUVEC中のVEGFR−2リン酸化に対して化合物の投与中止後も存続する阻害効果を明示すること、およびマトリックスタンパク質のフィブロネクチンに対するPDGF BB誘発性の癌細胞遊走を阻害することが可能である。式1の化合物は、遊走および浸潤を阻害することによって、PDGFR推進性の腫瘍進行に抗する活性を有するかもしれない。
【0049】
式1の化合物はまた、TaxolTM、より好ましくはドセタキセルと組み合わせるとき、腫瘍増殖阻害においてより有効な活性を明示する。いずれかの薬剤単独よりも、同時療法で、より有意な腫瘍退縮を観察した。
【0050】
さらに本発明は、例えば哺乳動物組織において、式1の化合物を投与することによってプロテインキナーゼ活性を変調させるかまたは阻害する方法へ向けられる。本発明の化合物の、キナーゼの活性のようなプロテインキナーゼ活性のモジュレーターとしての活性は、in vivoおよび/またはin vitroアッセイが含まれる、当業者に利用可能な方法のいずれによっても測定可能である。活性測定に適したアッセイの例には、Parast C.ら,Biochemistry,37,16788−16801(1988);Jeffreyら,Nature,376,313−320(1995);WIPO国際特許公開公報番号WO97/34876;およびWIPO国際特許公開公報番号WO96/14843に記載されるものが含まれる。これらの特性は、例えば、以下の実施例に示す生物学的試験法の1以上を使用することによって評価可能である。
【実施例】
【0051】
以下に提供する実施例および製法は、本発明の剤形および方法をさらに例示して例証する。本発明の範囲は、以下の実施例の範囲により決して限定されないことを理解されたい。
【0052】
実施例1
式1の化合物について:(1)いくつかのスケジューリング:1日1回、週末投薬、休日および間欠性の投薬でのin vivo効力;(2)異種移植片モデルにおいてドセタキセルと組み合わせたときの効力;(3)内皮細胞におけるin vitro eNOSおよびAktリン酸化;(4)細胞培養およびin vivoでの酸化窒素および関連産物の濃度;および(5)全血液細胞中のc−Kitシグナルの、化合物の潜在バイオマーカーとしての使用を試験した。
【0053】
生物学的試験;酵素アッセイ
VEFG、FGF、その他のような増殖因子による細胞増殖の刺激は、そのそれぞれの受容体のチロシンキナーゼがそれぞれを自己リン酸化することの誘導に依存する。故に、これらの増殖因子によって誘導される細胞の増殖を妨げるプロテインキナーゼ阻害剤の能力は、受容体の自己リン酸化を妨げるその能力と直接的に相関する。化合物のプロテインキナーゼ阻害を測定するために、以下の構築体を設計した。
【0054】
アッセイ用のVEGF−R2構築体:この構築体は、チロシンキナーゼ活性を阻害する試験化合物の能力を決定する。キナーゼ挿入ドメインの68残基の中央50残基を欠く、ヒト血管内皮細胞増殖因子受容体2(VEGF−R2)のサイトゾルドメインの構築体(VEGF−R2Δ50)をバキュロウイルス/昆虫細胞系において発現させた。全長VEGF−R2の1356残基のうち、VEGF−R2Δ50は、残基806〜939および990〜1171と、キナーゼ挿入ドメイン内に、野生型VEGF−R2に対する1つの点突然変異(E990V)も含有する。この精製構築体の自己リン酸化を、5%グリセロールおよび5mM DTTを含有する100mM HEPES(pH7.5)中3mM ATPおよび40mM MgCl2の存在下、4μMの濃度の酵素の4℃で2時間のインキュベーションにより実施した。自己リン酸化の後で、この構築体は、野生型の自己リン酸化キナーゼドメイン構築体に本質的に同等な触媒活性を保有することが示された。Parastら,Biochemistry,37,16788−16801(1998)を参照のこと。
【0055】
アッセイ用のFGF−R1構築体:バキュロウイルスベクター発現系を使用して、ヒトFGF−R1の細胞内キナーゼドメインを、Mohammadiら,Mol.Cell.Biol.,16,977−989(1996)の残基番号付けシステムによる、内因性メチオニン残基456から出発してグルタミン酸766まで発現させた。さらに、この構築体はまた、以下の3つのアミノ酸置換を有する:L457V、C488A、およびC584S。
【0056】
アッセイ用のLCK構築体:昆虫細胞において、アミノ酸残基223から出発して残基509でのタンパク質の終わりまでのN−末端欠損体(以下の2つのアミノ酸置換がN末端にある:P233MおよびC224D)としてLCKチロシンキナーゼを発現させた。
【0057】
アッセイ用のCHK−1構築体:バキュロウイルス/昆虫細胞系を使用して、C末端Hisタグ付き全長ヒトCHK−1(FL−CHK−1)を発現させた。これは、476アミノ酸のヒトCHK−1のC末端に6つのヒスチジン残基(6xHis−タグ)を含有する。このタンパク質を慣用のクロマトグラフィー技術によって精製した。
【0058】
アッセイ用CDK2/サイクリンA構築体:バキュロウイルス発現ベクターで感染させた昆虫細胞より、公知の方法論(Rosenblattら,J.Mol.Biol.,230,1317−1319(1993))を使用して、CDK2を精製した。サイクリンAは、全長組換えサイクリンAを発現する大腸菌細胞より精製し、先端切断サイクリンA構築体は、限定したタンパク分解によって産生し、既報(Jeffreyら,Nature,376,313−320(1995))に記載のように精製した。
【0059】
アッセイ用CDK4/サイクリンD構築体:対応するバキュロウイルス発現ベクターで同時感染させた昆虫細胞より、従来の生化学のクロマトグラフィー技術を使用して、ヒトCDK4およびサイクリンD3の複合体、またはサイクリンD1とヒトCDK4およびグルタチオン−S−トランスフェラーゼ(GST−CDK4)の融合タンパク質の複合体を精製した。
【0060】
VEGF−R2アッセイ:共役分光測定(FLVK−P)アッセイ
ホスホリル転移に伴うADPのATPからの産生を、ホスホエノールピルビン酸塩(PEP)とピルビン酸キナーゼ(PK)および乳酸デヒドロゲナーゼ(LDH)を有する系を使用する、NADHの酸化へ共役した。Beckman DU650分光光度計を使用して、340nmでの吸光度(e340=6.22cm−1mM−1)の減少を追跡することによってNADHの酸化をモニタリングした。リン酸化VEGF−R2Δ50(以下の表においてFLVK−Pとして示す)のアッセイ条件は、以下の通りであった:200mM HEPES(pH7.5)中1mM PEP;250μM NADH;50ユニットのLDH/mL;20ユニットのPK/mL;5mM DTT;5.1mM ポリ(E4Y1);1mM ATP;および25mM MgCl2。非リン酸化VEGF−R2Δ50(表においてFLVKとして示す)のアッセイ条件は、以下の通りであった:200mM HEPES(pH7.5)中1mM PEP;250μM NADH;50ユニットのLDH/mL;20ユニットのPK/mL;5mM DTT;20mM ポリ(E4Y1);3mM ATP;および60mM MgCl2および2mM MnCl2。5〜40nMの酵素でアッセイを開始した。様々な濃度の試験化合物の存在下に酵素活性を測定することによって、Ki値を決定した。Enzyme KineticおよびKaleidagraphのソフトウェアを使用してデータを解析した。
【0061】
ELISAアッセイ:ビオチニル化ガストリンペプチド(1−17)を基質として使用して、ホスホガストリンの形成をモニタリングした。ストレプタビジン被覆96ウェルマイクロタイタープレートを使用してビオチニル化ホスホガストリンを固定化して、西洋ワサビペルオキシダーゼへ共役した抗ホスホチロシン抗体を使用する検出を続けた。2,2’−アジノ−ジ−[3−エチルベンザチアゾリンスルホネート(6)]ジアンモニウム塩(ABTS)を使用して、西洋ワサビペルオキシダーゼの活性をモニタリングした。典型的なアッセイ溶液は、200mM HEPES(pH7.5)中2μMビオチニル化ガストリンペプチド;5mM DTT;20μM ATP;26mM MgCl2;および2mM MnCl2を含有した。0.8nMのリン酸化VEGF−R2△50でアッセイを開始した。西洋ワサビペルオキシダーゼ活性は、ABTS,10mMを使用してアッセイした。酸(H2SO4)の添加により西洋ワサビペルオキシダーゼ反応を失活させ、405nmでの吸光度読取りを続けた。様々な濃度の試験化合物の存在下に酵素活性を測定することによって、Ki値を決定した。Enzyme KineticおよびKaleidagraphのソフトウェアを使用してデータを解析した。
【0062】
FGF−Rアッセイ:VEGF−R2についての上記の記載のように分光測定アッセイを行なったが、但し、濃度を以下のように変更した:FGF−R=50nM,ATP=2mM,およびポリ(E4Y1)=15mM。
【0063】
LCKアッセイ:VEGF−R2についての上記の記載のように分光測定アッセイを行なったが、但し、濃度を以下のように変更した:LCK=60nM,MgCl2=0mM,ポリ(E4Y1)=20mM。
【0064】
CHK−1アッセイ:合成基質ペプチドのSyntide−2(PLARTLSVAGLPGKK)へのホスホリル転移に伴うADPのATPからの産生を、ホスホエノールピルビン酸塩(PEP)を使用する、ピルビン酸キナーゼ(PK)および乳酸デヒドロゲナーゼ(LDH)の作用を介したNADHの酸化へ共役した。NADHの酸化は、HP8452分光光度計を使用して、340nmでの吸光度(ε340=6.22cm−1mM−1)の減少を追跡することによってモニタリングした。典型的な反応溶液は:50mM TRIS(pH7.5)中4mN PEP;0.15mM NADH;28ユニットのLDH/mL;16ユニットのPK/mL;3mM DTT;0.125mM Syntide−2;0.15mM ATP;25mM MgCl2;および400mL NaClを含有した。10nMのFL−CHK−1でアッセイを開始した。様々な濃度の試験化合物の存在下に初期酵素活性を測定することによって、Ki値を決定した。Enzyme KineticおよびKaleidagraphのソフトウェアを使用してデータを解析した。
【0065】
HUVEC増殖アッセイ:このアッセイは、ヒト臍静脈内皮細胞(「HUVEC」)の増殖因子刺激増殖を阻害する試験化合物の能力を決定する。HUVEC細胞(継代数3〜4、Clonetics社)をT75フラスコ中のEGM2培養基(Clonetics社)へ融かした。24時間後、このフラスコへ新鮮なEGM2培地を加えた。4〜5日後、細胞を別の培養基(10%胎仔ウシ血清(FBS)、60μg/mLの内皮細胞増殖サプリメント(ECGS)、および10μg/mL ヘパリンを補充したF12K培地)へ曝露した。その後の実験では、指数増殖期のHUVEC細胞を使用した。10,000〜12,000個のHUVEC細胞を96ウェル・ディッシュにおいて100μlの濃縮培養基(上記に記載)においてプレート培養した。この細胞を、この培地において24時間付着させた。次いで、培地を吸引により除去し、115μlの飢餓培地(F12K+1% FBS)を各ウェルへ加えた。18時間後、飢餓培地中1% DMSOに溶かした15μlの試験薬剤かまたはこの担体単独をそれぞれの処理ウェルへ加えた;最終DMSO濃度は0.1%であった。1時間後、飢餓培地中20μlの150ng/mL hrVEGF165を、未処理対照を含有するウェルを除くすべてのウェルへ加えた;最終VEGF濃度は20ng/mLであった。72時間後にMTT色素低下により細胞の増殖を定量したが、この時点で細胞をMTT(プロメガ社)へ4〜5時間曝露した。停止溶液(プロメガ社)の添加により色素低下を止め、570および630nmでの吸光度を96ウェル分光光度計プレートリーダーで決定した。
【0066】
癌細胞増殖(MV522)アッセイ:プロテインキナーゼ阻害剤が癌を治療するために血管新生を妨げる治療有用性を有するかどうかを決定するには、この阻害剤がキナーゼ受容体を発現しない細胞においては細胞増殖を非特異的に妨げないことを実証することが重要である。癌細胞を使用する増殖アッセイを実施することによってこのことを行う。癌細胞における細胞増殖を評価するためのプロトコールは、HUVEC細胞における評価について使用するものと同様である。2000個の肺癌細胞(MV522系、UCSDより獲得した)を増殖培地(2mMグルタミンおよび10% FBSを補充したRPMI1640培地)にまいた。試験薬剤および/または担体の添加に先立って、細胞を1日間付着させる。HUVECアッセイに使用するのと同じ試験薬剤で細胞を同時に処理する。試験薬剤への曝露の72時間後に、MTT色素低下アッセイによって細胞増殖を定量する。
【0067】
C−Kit効力決定:阻害剤によるc−Kitに抗する効力を決定するためにNCI−H526(ATCC)細胞を使用した。この細胞を亜集密まで増殖させて、飢餓培地において18時間インキュベートした。阻害剤を加え、細胞を2.3%アルブミンおよび1mM Na3VO4(シグマ)の存在下に37℃で45分間インキュベートした。この培養物へSCF、c−Kit増殖因子を50ng/mlの最終濃度で加えた。5分後、この細胞を冷PBSで2回濯ぎ、溶解緩衝液(50mM Tris,150mM NaCl,1mM PMSF,1% NP40,1mM Na3VO4およびプロテアーゼ阻害剤カクテル)で溶解させた。各溶解液より1mgの総タンパク質を使用し、4μg/ml CD117 ab−3(K45,Neomarkers)とともに4℃で一晩インキュベートして、免疫沈降を実施した。翌朝、この抗体複合体をプロテインAビーズへコンジュゲートした。リン酸化受容体用の抗ホスホチロシン抗体4G10(Upstate Biotechnology)、または抗c−Kit受容体抗体sc−1493(C−14,Santa Cruz)を1:1000で使用して、SDS PAGEとウェスタン・ブロット分析を実施した。化学発光試薬のECL Plusによってブロットを可視化した。ブロット中のシグナルの定量にphosphorimager(Storm 846,Molecular Dynamics)を使用した。
【0068】
動物および哺乳動物の全末梢血細胞中のc−kit陽性細胞集団の低下を式1の化合物の活性のバイオマーカーとして使用した。
ENOSおよびAktリン酸化測定:阻害剤によるeNOSおよびAktに抗する効力を決定するためにHUVEC(Clonetics)を使用した。この細胞を亜集密まで増殖させて、飢餓培地において18時間インキュベートした。阻害剤を加え、細胞を2.3%アルブミンおよび1mM Na3VO4(シグマ)の存在下に37℃で45分間インキュベートした。この培養基へVEGFを50ng/mlで加えた。5分後、この細胞を冷PBSで2回濯ぎ、溶解緩衝液(50mM Tris,150mM NaCl,1mM PMSF,1% NP40,1mM Na3VO4およびプロテアーゼ阻害剤カクテル)で溶解させた。30〜40μgの総タンパク質をウェスタン法によって分析した。eNOSおよびAktリン酸化は:Phospho−eNOS(Ser 1177)#9571またはPhospho−Akt(Ser 473)#9271の抗体(いずれもCell signalingより)を使用することによって評価した。タンパク質の検出は、NOS3(C−20)sc−654(Santa Cruz)またはAkt 抗体#9272(Cell signaling)を使用することによって達成した。いずれも、1:3000で使用する抗ウサギHRP連結二次抗体を必要とする。化学発光基質のSuper Signal West Dura(ピアス)によってブロットを可視化した。ブロット中のシグナルの定量にAlpha Imager 8800(Alpha Innotechより)を使用した。
【0069】
マウスPKアッセイ:薬物のマウスにおける薬物動態(例、吸収と消失)について、以下の実験を使用して解析した。試験化合物は、0.5% CMC担体中の懸濁液剤として、または30:70(PEG400:酸性化H2O)担体中の溶液剤として製剤化した。この懸濁液剤または溶液剤をC3H雌性マウス(n=4)へ経口(p.o.)または腹腔内(i.p.)投与した。0時間(投薬前)、投薬後0.5時間、1.0時間、2.0時間、および4.0時間の時点で眼窩出血により血液試料を採取した。各試料より、2500rpmで5分間の遠心分離によって血漿を入手した。この血漿より有機タンパク質沈殿法によって試験化合物を抽出した。各時点の出血につき、50μLの血漿を1.0mLのアセトニトリルと合わせ、2分間激しく振り混ぜてから、4000rpmで15分間スピンしてタンパク質を沈殿させ、試験化合物を抽出した。次いで、アセトニトリル上清(試験化合物を含有する抽出物)を新しい試験管へ注ぎ、N2ガスの蒸気下にホットプレート(25℃)で蒸発させた。乾燥した試験化合物の抽出物を含有するそれぞれの試験管へ125μLの移動相(60:40,0.025M NH4H2PO4+2.5mL/L TEA:アセトニトリル)を加えた。激しく撹拌することによって、試験化合物をこの移動相に再懸濁させ、4000rpmで5分間の遠心分離によりさらにタンパク質を除去した。UV検出付きヒューレット・パッカード1100シリーズHPLCでの試験化合物分析のために各試料をHPLCバイアルへ注いだ。各試料より95μLをPhenomenex−Prodigy逆相C−18,150x3.2mmカラムへ注入し、10分にわたる45〜50%アセトニトリル勾配ランで溶出させた。上記のやり方で血漿試料より抽出した試験化合物の既知濃度を使用して、標準曲線(ピーク面積x濃度μg/mL)との比較により、試験化合物の血漿濃度(μg/mL)を決定した。標準品と未知検体と一緒に、3群(n=4)の品質コントロール(0.25μg/mL、1.5μg/mL、および7.5μg/mL)を実施して、分析の一貫性を保証した。標準曲線はR2>0.99を有し、品質コントロールは、いずれもその予測値の10%範囲内にあった。定量した試験試料を、Kalidagraphソフトウェアを使用する可視ディスプレイのためにプロットし、WIN NONLINソフトウェアを使用して、その薬物動態パラメータを決定した。
【0070】
ヒト肝ミクロソーム(HLM)アッセイ:ヒト肝ミクロソームにおける化合物の代謝について、以下のようなLS−MS分析アッセイ手順によって測定した。はじめに、ヒト肝ミクロソーム(HLM)を融かし、冷100mMリン酸カリウム(KPO4)緩衝液で5mg/mLへ希釈した。適正量のKPO4緩衝液、NADPH再生溶液(B−NADP、グルコース−6−リン酸、グルコース−6−リン酸デヒドロゲナーゼ、およびMgCl2を含有する)、およびHLMを13x100mmのガラス試験管において37℃で10分間プレインキュベートした(試験化合物につき3つの試験管;同一3検体)。試験化合物(最終5μM)を各試験管へ加えて反応を開始させ、穏やかに振り混ぜて混合した後で、37℃でインキュベートした。t=0および2時間で、各インキュベーション試験管より250μLの試料を取り出し、1mLの氷冷アセトニトリル(+0.05μMレセルピン)を含有する分離用12x75mmガラス試験管へ移した。試料を4000rpmで20分間遠心分離して、タンパク質と塩を沈殿させた(ベックマン Allegra 6KR,S/N ALK98D06,#634)。上清を新しい12x75mmガラス試験管へ移し、Speed−Vac遠心分離真空エバポレーターにより蒸発させた。200μLの0.1%ギ酸/アセトニトリル(90/10)中で試料を復元し、激しく撹拌して溶かした。次いで、この試料を分離用ポリプロピレンミクロ遠心分離管へ移し、14000xgで10分間遠心分離した(Fisher Micro 14,S/N M0017580)。各時点(0および2時間)での各同一3検体(#1〜3)について、各試験化合物のアリコート試料を、以下に記載するLC−MS分析用の単回HPLCバイアルインサート(全部で6つの試料)へ合わせた。
【0071】
合わせた化合物試料を、ヒューレット・パッカード HP1100 ダイオードアレイHPLCとポジティブエレクトロスプレーSIRモードで作動するMicromass Quattro II三連四重極型質量分析計からなる(各試験化合物の分子イオンを特異的に走査するようにプログラムされている)、LC−MSシステムへ注入した。各試験化合物のピークを各時点で積分した。各化合物について、各時点でのピーク面積(n=3)を平均化して、この2時間での平均ピーク面積を0時間の時点での平均ピーク面積で割って、2時間で残存する試験化合物のパーセントを得た。
【0072】
in vitro HUVECアポトーシスアッセイ
ELISAによるアポトーシスの定量:細胞溶解液中の細胞質ヒストン会合DNA断片を定量するCell Death Detection Elisa PLUS(カタログ番号:1775425,ロッシュ・バイオケミカルズ、マンハイム、ドイツ)を使用して、HUVEC細胞のアポトーシスを測定した。製造業者の手引きに若干の変更を加えて、この手順を実施した。簡潔に言えば、飢餓状態のHUVEC細胞をVEGF(20ng/mL)の存在下に様々な濃度の化合物Aで処理した。この細胞のサイトゾル分画を様々な時点で採取し、マイクロタイタープレートへコートした一次抗ヒストンmAbとペルオキシダーゼへ結合した二次抗DNA mAbを用いるサンドイッチELISAにおける抗原の供給源として使用した。発色性ペルオキシダーゼ基質を加えて、分光光度計で405nmでの吸光度(基準波長:490nm)を測定することによって、アポトーシス細胞の数を定量した。
【0073】
TUNELによるアポトーシスの可視化:TdT仲介性dUTPニックエンド標識(TUNEL)技術を使用して、アポトーシス細胞のin situ検出を行った。簡潔に言えば、8ウェルLab−Tekチャンバ・スライドにおいて増殖させたHUVEC細胞をO/N飢餓させてから、様々な濃度の化合物Aで6時間処理した。次いで、製造業者の手引きに従って、この細胞を4%パラホルムアルデヒドに固定して、Triton X−100で透過性にして、ターミナルトランスフェラーゼとフルオレセイン−dUTP(Deadend Fluorometric TUNEL系、プロメガ、カタログ番号 G3250)が含まれるヌクレオチドの混合物において1時間インキュベートした。この細胞をヨウ化プロピジウム(PI)溶液で対比染色した。陽性染色されたフルオレセインおよびPI標識細胞を可視化して、蛍光顕微鏡によって写真撮影した。
【0074】
PDGF仲介性細胞遊走アッセイ:このアッセイには、U87MG細胞を使用した。6ウェルプレートを0.5ng/mL フィブロネクチンとともに一晩プレインキュベートする。翌日、U87MG細胞を各ウェルにおいてプレート培養して、集密まで増殖させる。この細胞を、0.1% FBSを含有する飢餓培地とともに一晩インキュベートした。ピペット先端を使用して約1cmのスクラッチをつくり、細胞を飢餓培地で洗浄した。次いで、プレートを0.5ng/mL フィブロネクチンとともに1時間インキュベートしてから、再び洗浄した。100ng/LI rhPDGF BBと化合物Aを飢餓培地中に含有する実験培地を導入した。0時間と15時間の間で細胞を写真撮影して、遊走を可視化した。
【0075】
細胞VEGFR−2および下流分子リン酸化アッセイ:HUVEC(Clonetics)を亜集密まで培養して、飢餓培地(F12K+0.1% FBS)において18時間インキュベートした。この細胞へ2.3%アルブミンおよび1mM Na3VO4(シグマ)の存在下に化合物Aを加えた。45分後、この培養物へVEGFを50ng/mLの最終濃度で加えた。5分後、この細胞を冷PBSで濯ぎ、溶解緩衝液(50mM Tris,150mM NaCl,1mM PMSF,1% NP40,1mM Na3VO4およびプロテアーゼ阻害剤カクテル)で溶解させた。溶解液由来の1ミリグラムの総タンパク質を、抗Flk−1 C−1158(Santa Cruz)を使用して免疫沈殿させた。この抗体複合体をプロテインAビーズへコンジュゲートして、SDS PAGE/ウェスタン分析を実施した。抗ホスホチロシン抗体4G10(Upstate Biotechnology)と抗Flk−1 C−20(Santa Cruz)によって、それぞれリン酸化VEGFR−2とそのタンパク質を検出した。eNOSおよびAktについては、細胞を上記と同じように処理した。全量30〜40μgのタンパク質を使用して、ウェスタン分析を実施した。eNOSおよびAktリン酸化は、Phospho−eNOS(Ser 1177,#9571)またはPhospho−Akt(Ser 473,#9271)の抗体(Cell signaling)を使用することによって探知した。タンパク質は、NOS3 C−20(sc−654,Santa Cruz)またはAkt 抗体#9272(Cell Signaling)を使用することによって評価した。HRP連結抗ウサギIgGを二次抗体として使用した。化学発光基質のSuper Signal West Dura(ピアス)によってすべてのブロットを可視化した。Alpha InnotechからのAlpha Imager 8800を使用して、シグナルを定量した。
【0076】
ウォッシュアウト実験:HUVEC細胞を上記に記載のように処理した。化合物A(10nM)と45分間のインキュベーションと、VEGF(50ng/ml)で5分間の刺激の後で、上清を除去し、洗浄し、VEGFおよびNa3VO4を含有する飢餓培地に置き換えた。この細胞を所望される長さの時間の間さらにインキュベートした後で、溶解させ、リン酸化および全体のVEGFR−2のために免疫沈殿およびウェスタンを使用して処理した(上記参照)。別の実験において、細胞を上記のように全体の時間の長さの間VEGFで処理し、所望される時点でのVEGFR−2リン酸化および全VEGFR−2を同様に評価した。ウォッシュアウトの間のシグナルをデンシトメトリーによって定量した。各実験からの最大刺激(5分)の強度を互いに正規化して、ホスホVEGFR−2の各時点での強度を2つの実験間で比較して、それにより、非処理であるがVEGF刺激した細胞に対して、VEGFR−2リン酸化の回復を決定した。
【0077】
腫瘍モデル:ヒトMV522(結腸癌)およびMDA−MB−231(乳癌)モデルでは、無胸腺マウス(n=8〜12)に5x106細胞/部位を移植(s.c.)した。マウスのルイス肺癌モデルでは、腫瘍断片(1〜2mm2)をB6D2F1マウスの右脇腹にトロカール移植した。投薬は、通常7日目(MV522)、または平均腫瘍サイズが150〜200mm3に達するとき(MDA−MB−231)に開始した。
【0078】
式1の化合物は、0.5% CMC/H2O中に製剤化して、PO、BID投与した。ドセタキセルは、7% EtOH/3% ポリソルベート/90% H2O中に製剤化して、週ごとに、静脈内で投薬した。通常、処理は3〜4週間続いた。腫瘍の幾何学的な長さおよび幅を、電子カリパスを使用して週3回測定した。腫瘍体積は、0.4x[長さx(幅)2]の積として算出した。データは、平均±SEMとして報告した。試験の最後に、腫瘍と組織を分析用に切除、秤量、および採取した。薬物濃度の分析用に血漿を採取した。
【0079】
結果を表1〜3に示す。
表1.化合物1の効力および選択性
【0080】
【表1】
【0081】
スクリーニングしたが、Ki計算の限度を超えた他の酵素は、cMet、LCK、c−Src、FAK、Pyk2、IRL、BTK、CDK1、CDK2、CDK4、PKA、PKC、PLK、およびChk1である。
【0082】
表2.MDA−MB−231ヒト乳癌モデルにおける化合物1およびドセタキセルの同時投与用の試験設計
【0083】
【表2】
【0084】
表3.ドセタキセルおよび化合物1の組合せ療法は、MDA−MB−231異種移植片モデルにおいてより大きな抗腫瘍活性をもたらした。
【0085】
【表3】
【0086】
上記の組合せ群は、完全および部分的な腫瘍退縮の増加した出現率を明示する。これらの薬剤を組み合わせるとき、腫瘍増殖率は、より大きな度合いまで低下した。この組合せ治療は、単剤単独と同じくらい十分に忍容された。
【0087】
実施例2
式1の化合物、6−[2−(メチルカルバモイル)フェニルスルファニル]−3−E−[2−(ピリジン−2−イル)エテニル]インダゾールを固形腫瘍の患者へ様々な用量で投与した。式1の化合物を経口の錠剤形において、BIDまたはQDのスケジュールで使用して、30名(男性13名、女性17名)の患者を治療した。サイクルは、それぞれ28日であった。具体的な腫瘍の診断は、乳房(11)、甲状腺(5)、腎細胞(5)、肺(4)および、その他(5)であった。液体クロマトグラフィータンデム質量分析法(LC−MS/MS)によって、薬物動態データを測定した。このサイクルの15日目に、投与後0.5時間、1時間、2時間、4時間、8時間、および12時間の時点で血液試料を採取した。
【0088】
薬物動態結果(15日目の平均値)を表4に示す。患者は、他に述べなければ、絶食ではなかった。括弧内の数字は、百分率として表される変動係数である。この表において、Cmaxは、観測される式1の化合物の最高血漿濃度であり、AUC(0−24)は、24時間のAUC血漿濃度であり、T1/2は、濃度対時間のプロットより決定される半減期である。見出し項目の「PKの患者数」は、薬物動態データを入手した患者の数を示す。
【0089】
表4
【0090】
【表4】
【0091】
さらに、第一コホートの患者(n=6)は、10mg QD〜30mg BIDに及ぶ個別化された用量を受けた(PK示さず)。絶食状態では、摂食状態に比べて、血漿曝露がより高く(約49%)、患者内の変動は抑えられた。現時点での最大耐薬量(MTD)は、絶食状態で5mg BIDであると決定した。MTDより高い用量での投薬制限毒性(DLT)は、高血圧(HTN)、発作、肝機能検査値の上昇、膵炎、無呼吸、および胃炎であった。さらに、応答した2名のNSCLC患者は、致命的な喀血を起こし、1名は3週後にこの化合物での治療を中止した。非投薬制限性のタンパク尿も観察された。MTD以下の用量では、1名の患者で、グレード2の胃炎へDLTが限定された。7/14名の患者で非投薬制限性のHTNが観察され、慣用の降圧用医薬品により管理した。RECIST判定基準による2つの永続性のある一部応答が観察され(腎細胞と上顎洞の腺嚢腫において)、この十分に前処置した集団のうち5名の患者で、安定した疾患が4ヶ月以上(4〜13+ヶ月の範囲)続いた。dceMRIを使用して、21名の患者で予備分析を実施して、式1の化合物により誘発される血管系への効果をベースラインと2、28、および56日目で測定した。平均Ktransの百分率変化(P.S.Tofts,G.Brix,D.L.Buckley,J.L.Evelhoch,E.Henderson,M.V.Knopp,H.B.W.Larsson,T.Lee,N.A.Mayr,G.J.M.Parker,R.E.Port,J.TaylorおよびR.M.Weisskoff,「分散可能トレーサーのダイナミック対比増強T1重み付けMRIからの動態パラメータの評価:量および記号の標準化」(Estimating Kinetic Parameters Dynamic Contrast−Enhanced T1−Weighted MRI of a Diffusable Tracer:Standardized Quantities and Symbols)Journal of Magnetic Resonance Imaging,10:223−232(1999)とコントラスト強度X時間曲線下の初期面積(IAUC)をそれぞれの指標腫瘍(n=1〜4/患者)につき計算した。腫瘍血管系の応答は、ベースライン変数値が2日目までに50%以上減少するものと定義した。腫瘍血管系応答の急激な(2日目)の減少(KtransおよびIAUCの50%以上の減少)が18名の評価可能な患者の6名で観察され、11/18名で、KtransおよびIAUCの両方の40%以上の減少が明示された。スキャンに伴う技術課題により、3/21の画像セットが評価可能でなかった。本実施例は、式1の化合物が、臨床応答と急性の腫瘍血管系変化により顕現されるように、きわめて有効な薬剤であることを示す。
【0092】
実施例3
30mg遊離塩基/kg用量の式1の[14C]標識化合物をインタクトまたは胆管カニューレ挿入ビーグル犬へ経口投与した後で、広汎な代謝を観察した。生物変換経路には、酸素化(モノまたはジ)、グルクロニド化、グルコシル化、および酸素化に続く硫酸化またはグルコシル化が含まれた。図1は、同定された代謝産物を示す。血漿中では、M12(N−オキシド)が検出可能な唯一の代謝産物である。尿中では、M5(デピリジニルカルボン酸)が主要な代謝産物である。主要な胆汁代謝産物には、M8(硫酸塩)とM12が含まれる。主要な糞中代謝産物M1の化学構造は、不明のままである。
【0093】
本化合物の単回経口投薬に続く、[14C]由来放射活性のビーグル犬における排泄パターンは、雄と雌で同様であり、放射活性は、主に糞より排泄された。インタクトな雌の糞中80.9%と尿中7.0%の回収率と比較して、インタクトな雄の平均回収率は、糞中85.5%と尿中5.3%であった。胆管カニューレ挿入した雄イヌは、比較的少ない分量の放射活性を胆汁に排泄し(8.3%の回収率)、さらなる放射活性は、糞(52.7%)と尿(11.3%)に回収された。胆管カニューレを挿入したイヌからの尿および胆汁の放射活性を合わせると、投与した放射活性のほぼ20%が胃腸吸収を受けたことを示唆する。すべての試料の全体平均回収率は、インタクトな雄と雌についてそれぞれ92.4%と92.6%、胆管カニューレ挿入した雄で89.6%であった。放射HPLC検出器(β−RAM)とインライン共役したHPCLと、エレクトロスプレー(ESI)および大気圧化学イオン化(APCI)供給源のMS検出をポジティブまたはネガティブ形式で使用して、すべての代謝産物のプロファイリングおよび構造解明を実施した。
【0094】
実施例4
式1の化合物は、CD−1マウスにおいて、[14C]標識化合物の単回経口投与に続き、広汎な代謝を受ける。低い百分率の未変化薬物が尿および糞中に回収され、多様な第I相および第II相の代謝産物が観察された。生物変換経路には、酸素化(モノまたはジ)、グルクロニド化、グルコシル化、および酸素化に続くグルクロニド化またはグルコシル化のいずれかが含まれた。同定された代謝産物を図2に示す。血漿中では、未変化薬物とM12(N−オキシド)が2つの主要な成分を代表した。M7(グルクロニド)は、尿と糞の両方で最も顕著な代謝産物を代表した。
【0095】
50mg遊離塩基/kg用量の[14C]AG−013736を雄性CD−1マウスへ単回経口投与後、[14C]由来放射活性の大部分が糞中で回収された。投薬後48時間での放射活性の平均(n=2)回収率(用量の%)は、糞中65.8%と尿中12.7%であった。排泄物中の放射活性の消失速度は速やかで、ほぼ72%の用量が投薬後24時間以内に回収された。LC−RAM−MS法を使用して、代謝産物の放射活性プロファイリングと構造特性決定を実施した。
【0096】
図1および2に示す代謝産物に加えて、他の既知の代謝産物には、式1aに示す活性des−メチル代謝産物が含まれる。
【0097】
【化4】
【0098】
実施例5
免疫組織化学を使用して、腫瘍微小血管密度(MVD)を測定することによって、血管新生を評価した。凍結した腫瘍切片を血管表面マーカーのCD−31用に染色し、組織切片の数領域中の血管の量をマニュアルで定量した。種々の試験は、式1の化合物の2〜3週のPO BID投与が、対照腫瘍と比較して、処理した腫瘍中の血管の数を70%低下させることを示した。処理後の微小血管密度の減少は、LLC、MV522、およびM24metが含まれる、使用したすべての腫瘍モデルで観察された。LLC腫瘍モデルでは、浸透Alzetポンプを介して連続的に送達するとき、式1の化合物は、有意な増殖阻害をもたらした。3回の試験からのデータは、LLCモデルにおけるこのクラスの薬剤により達成可能である最大の腫瘍増殖阻害が78%であることを示した。55±17ng/mL(N=3)ほどの低さの血漿濃度では、90%の最大増殖阻害が達成された。この濃度を生物学的活性濃度(BAC)と名付けた。50%最大増殖阻害は、28±11ng/mL(N=3)の血漿濃度と関連した。この濃度を最小有効濃度(MEC)と名付けた。1つの試験群で、本化合物の連続注入により産生される70%のMGIは、574ng・時間/mLのAUC(0−24)と関連したが、同じ試験において、PO BID投薬後の720ng・時間/mLのAUC(0−24)は、40%の最大増殖阻害(MGI)を生じた。これらの結果は、このモデルにおいて見られる抗腫瘍効力がトラフ(谷)濃度により推進されること、そしてマウスでは、最大抗腫瘍効力をもたらすのに、本化合物の連続した低濃度が十分であり得ることを示唆する。
【0099】
式1の化合物は、ヒト乳癌異種移植片モデル、MDA−MB−231において単剤として有効であった。このモデルにおける式1の化合物とドセタキセルの組合せを用いた効力試験に備えて、ナイーブなヌードマウスで予備試験を行って、PKおよび耐性に対する潜在的な薬物−薬物相互作用の効果を決定した。15または30mg/kgのドセタキセル、週1回3週間IV投与の後で、対照と比べた体重の減少(それぞれ、7%と11%)をドセタキセル処理動物で確認した。ドセタキセル単独で処理した動物と、ドセタキセルと式1の化合物(30mg/kg/日、16日間;PO)の組合せを与えたものとの間では、体重の差異を認めなかった。ドセタキセル投与は、式1の化合物のAUCに影響を及ぼさなかったが、AG−013736のCmax値は、式1の化合物単独と比べて、組合せ群において有意に低下した。
【0100】
選択した組織(肝臓、腎臓、心臓、脾臓、胃、小腸および大腸、卵巣、胸骨、関節)の組織学的検査は、この試験において単剤としての式1の化合物で処理したマウスにおける標的臓器効果を明らかにしなかった。ドセタキセル処理マウスに認められた変化には、卵胞の壊死とごく軽症〜軽症の骨髄低細胞性が含まれた。式1の化合物とドセタキセルの組合せ治療は、卵巣に対するドセタキセルの効果を悪化させなかったが、式1の化合物/ドセタキセル組合せを与えた動物において骨髄低細胞性の強度増加(ごく軽症〜中等症)を認めた。さらに、組合せで処理した動物においては、低細胞性の強度増加の二次効果と見られる、骨髄出血が観察された。
【0101】
式1の化合物とドセタキセルをMDA−MB−231腫瘍モデルにおける効力評価のために組み合わせた。式1の化合物単独(25、5、および1mg/kg,PO,BID,3週間投与)は、用量依存性の腫瘍増殖阻害をもたらした。ドセタキセル単独(IV、週ごと)20および10mg/kgも有効であった(しかし、2mg/kgは無効であった)。式1の化合物とドセタキセルの間には、有益な療法上の相互作用があるように見られた。この利益は、これらの薬剤を高用量および中用量の両方で組み合わせるときにより明らかであった。高用量および中用量の組合せアームでの部分退縮(腫瘍サイズの16%〜97%の低下)および完全応答の出現率は、個々の薬剤単独の同じ用量での群の出現率よりずっと大きかった。この試験の限られた群と比較的短い時間フレームのために、上記のことは決定的な知見ではない。式1の化合物は、すべての用量で十分に耐えられた。高用量の組合せ群(25mg/kgの化合物1と20mg/kgのドセタキセル)では、他のすべての群と比べて、化学療法剤の第三回目の投薬後に、平均体重の3%〜7%の低下があった。薬物動態分析は、式1の化合物のAUC値がドセタキセルの存在下で影響を受けないこと、しかしCmax値は、式1の化合物単独と比較して、組合せ群で有意に低下することを明示した。
【0102】
ドセタキセルとの組合せにおける式1の化合物の抗腫瘍効果をLLCモデルにおいて検討した。LLCモデルは、ドセタキセルに対してきわめて抵抗性である。その細胞傷害剤では、報告されているMTD(30mg/kg,週ごとに投薬、iv)で腫瘍増殖遅延(TGD)がほとんど見られなかった(TGD=3.2日)。すべてのマウスが大きな原発腫瘍により実験の28日以内に安楽死した。対照的に、式1の化合物単剤は、用量依存性で統計学的に有意なTGDをもたらした(10mg/kgで13.4日、30mg/kgで15.4日、PO,BID)。しかしながら、この薬剤は、遅延させただけで、肺への転移を止めることはなかった。低用量組合せ群(TGD=15.2日)ではなく、高用量組合せ群のTGD(20.4日)は、単剤単独のいずれよりも統計学的に異なっていた(それぞれ、P=0.0079およびP=0.254)。高用量組合せ群ではより多くの動物(3/10)が客観的なエンドポイントに達したが、低用量組合せ群では達しなかった。結論として、式1の化合物とドセタキセルの高用量組合せ療法は、いずれの単独療法単独よりも大きい原発腫瘍増殖および転移の遅延をもたらす可能性があるが、完全な治癒をもたらすわけではない。
【0103】
MV522腫瘍モデルを使用する1つの試験は、式1の化合物の1日1回(QD)60mg/kg PO投薬が30mg/kg PO,BIDと同様の腫瘍増殖阻害をもたらす(p=0.154)ことを明示した。さらに、抗腫瘍効果は、30mg/kgで連続5日間PO,BID投薬後に2日間休薬するとき、同じ用量濃度を使用して毎日PO,BID投薬することと比べて低下するようには見えなかった(p=0.223)。これらの結果は、この非臨床腫瘍モデルにおいて、式1の化合物をQDで投与しても、またはある種の臨時投薬スケジュールで投与しても、有意な抗腫瘍効果を達成することを期待してよいことを示唆する。
【0104】
MV522異種移植片モデルにおいて抗腫瘍効果をもたらすのに必要とされる受容体阻害の時間量と式1の化合物の濃度について検討した。この結果は、PO投薬(QDまたはBID)では、50%以上の抗腫瘍効果のためには、ほぼ24時間毎日EC50(5ng/mL)より高く曝露することが必要であることを示した。90%の腫瘍増殖阻害を達成するためには、40〜60ng/mL以上の血漿濃度に毎日少なくとも4時間曝露することが必要であった。上記の閾値を超えた曝露は、さらなる効果を保証しなかった。BIDまたはQD群のいずれでも、同様の体重損失があり、いずれも5%未満であった。従って、適正な用量と曝露時間があれば、QD処方は、BID処方と同じくらい有効であるかもしれない。
【0105】
Alzetポンプを介した連続曝露は、規則的な定期投薬と比べて、式1の化合物によるより大きな抗腫瘍効果をもたらした。このポンプによる10mg/mLでの送達は、30ng/mLの一定した平均全身曝露をもたらし、腫瘍の静止状態を生じた。対照的に、予測EC90に優る式1の化合物の血漿濃度を生じる、飽和量(PO,BID)では、腫瘍増殖遅延をもたらすことができただけである。従って、式1の化合物の連続全身曝露は、腫瘍を治療するのに、1日2回の経口投薬処方より有効であると思われた。
【0106】
間欠性の投薬処方を使用する式1の化合物の抗腫瘍効果も試験した。治療群は、以下の通りであった:担体の連日投薬、担体の間欠投薬、30mg/kg(BID)の連日投薬、および30mg/kgの間欠投薬。間欠投薬のスケジュールは、以下の通りであった:サイクル1(12〜18日目に投薬、19〜28日目に休薬)、およびサイクル2(29〜36日目に投薬、37〜44日目に休薬)。平均腫瘍サイズが250mm3になったときに投薬を開始し;いずれにもAG−013736(PO,BID)を与えた。全体的に見て、間欠および連日のBID投薬の間には有意差があり、連続した連日投薬処方は、増殖遅延をもたらすのにより有効であった。間欠投薬された薬物群では、投薬を中止して3〜4日以内に腫瘍が通常の増殖速度を再び獲得した。しかしながら、サイクル2投薬の2日以内には、腫瘍増殖阻害が再開した。予測されるように、どの群でも退縮は見られなかった。
【0107】
具体的で好ましい態様を参照にして本発明を例示したが、当業者は、定型的な実験と本発明の実践により、種々の変更および改良がなし得ることを理解されよう。従って、本発明は、上記の記載によって限定されず、付帯の特許請求項とその同等物により規定されるものとする。
【図面の簡単な説明】
【0108】
【図1】図1は、14C標識化合物の単回経口投薬に続く、イヌにおいて同定される式1の化合物の代謝産物を示す。
【図2】図2は、14C標識化合物の単回経口投薬に続く、マウスにおいて同定される式1の化合物の代謝産物を示す。
【技術分野】
【0001】
本出願は、その開示がそのまま参照により本明細書に組み込まれる、米国仮特許出願第60/460,695号(2003年4月3日出願)および米国仮特許出願第60/491,771号(2003年7月31日出願)の利益を主張する。
【0002】
背景技術
本発明は、癌のような異常な細胞増殖の哺乳動物における治療に有用であるVEGFR阻害剤に関する。本発明はまた、異常な細胞増殖の哺乳動物、具体的にはヒトにおける治療にこうした化合物を使用する方法と、こうした化合物を含有する医薬組成物に関する。
【背景技術】
【0003】
式1:
【0004】
【化1】
【0005】
により表される化合物、6−[2−(メチルカルバモイル)フェニルスルファニル]−3−E−[2−(ピリジン−2−イル)エテニル]インダゾールは、結腸癌、黒色腫、乳癌および肺癌の異種移植モデルにおいて広汎な前臨床活性のある、VEGFR/PDGFRチロシンキナーゼの強力かつ選択的な阻害剤である(Hu−Lowe D,Heller D,Brekken J,Feeley R,Amundson K,Haines M,Troche G,Kim Y,Gonzalez D,Herrman M,Batugo M,Vekich S,Kania R,McTigue M,Gregory S,Bender S,Shalinsky D.,「VEGF/PDGF受容体チロシンキナーゼの低分子阻害剤、AG013736の薬理活性(Pharmacological Activities of AG013736,a Small Molecular Inhibitor of VEGF/PDGF Receptor Tyrosine Kinases)」Proc.Am.Assoc.Cancer Res.2002:抄録#5357)。ダイナミック対比増強MRI(dceMRI)を使用して評価した前臨床の腫瘍血管応答は、腫瘍増殖指標と対応することを示した(Wilmes LJ,Hylton NM,Wang D,Fleming LM,Gibbs J,Kim Y,Dillon R,Brasch RC,Park JW,Li K−L,Henry RG,Partridge SC,Shalinsky DR,Hu−Lowe D,McShane TM,およびPallavicini MG.,「新規VEGFR TK阻害剤、AG013736は、ヌードマウス中のヒトBT474乳癌異種移植片における腫瘍増殖および血管透過性を抑制する(AG013736,a Novel VEGFR TK Inhibitor,Suppresses Tumor Growth and Vascular Permeability in Human BT474 Breast Cancer Xenografts in Nude Mice)」Proc.Am.Assoc.Cancer Res.2003:抄録#3772)。
【発明の開示】
【発明が解決しようとする課題】
【0006】
発明の要約
本発明は、6−[2−(メチルカルバモイル)フェニルスルファニル]−3−E−[2−(ピリジン−2−イル)エテニル]インダゾールと体系的に命名可能である、式1:
【0007】
【化2】
【0008】
の化合物を使用する剤形と治療の方法を提供する。
1つの態様において、本発明は、哺乳動物への投与用の剤形を提供し、該剤形は、式1の化合物、その医薬的に許容される塩、溶媒和物、またはプロドラッグ、またはこれらの混合物を、哺乳動物への投与後に4500ng・時間/mL以下の式1の化合物またはその活性代謝産物の24時間AUC血漿値を提供するのに有効な量で含んでなる。24時間AUC血漿値は、本明細書の「詳細な説明」に記載のようにして決定可能である。
【0009】
この態様の具体的な側面において、24時間AUC血漿値の上限は、4000ng・時間/mL以下または3000ng・時間/mL以下または2500ng・時間/mL以下または2000ng・時間/mL以下または1500ng・時間/mL以下または1000ng・時間/mL以下または800ng・時間/mL以下または700ng・時間/mL以下である。好ましくは、そして列挙した上限のいずれとも組み合わせて、24時間AUC血漿値は、少なくとも10ng・時間/mLまたは少なくとも25ng・時間/mLまたは少なくとも50ng・時間/mLまたは少なくとも75ng・時間/mLまたは少なくとも100ng・時間/mLまたは少なくとも125ng・時間/mLである。24時間AUC血漿値の想定範囲には、列挙した下限のいずれかから列挙した上限のいずれかまでの範囲が含まれる。好ましい範囲の具体的な非限定例には、25〜4500ng・時間/mL、50〜2500ng・時間/mL、75〜1000ng・時間/mL、100〜800ng・時間/mL、および125〜700ng・時間/mLが含まれる。
【0010】
別の態様において、本発明は、上記に定義される式1の化合物、その医薬的に許容される塩、溶媒和物またはプロドラッグ、またはこれらの混合物を30mg以下の量で含んでなる剤形を提供する。化合物の全部または一部が塩、溶媒和物またはプロドラッグとして剤形にあるとき、その量は式1の化合物の同等量であり、それは分子量に基づいて当業者により容易に計算されると理解されたい。
【0011】
この態様の具体的な側面において、この量の上限は、20mg以下または15mg以下または12mg以下または10mg以下または8mg以下または7mg以下である。好ましくは、そして列挙した上限のいずれとも組み合わせて、この量は、少なくとも0.5mgまたは少なくとも1mgまたは少なくとも1.5mgまたは少なくとも2mgまたは少なくとも2.5mgまたは少なくとも3mgである。想定範囲には、列挙した下限のいずれかから列挙した上限のいずれかまでの範囲が含まれる。好ましい範囲の具体的な非限定例には、0.5〜30mg、1〜20mg、1.5〜15mg、2〜10mg、2.5〜8mg、および3〜7mgが含まれる。
【0012】
さらに本発明は、上記に定義されるような式1の化合物、その医薬的に許容される塩、溶媒和物またはプロドラッグ、またはこれらの混合物を、ヒトが含まれる哺乳動物への投与後に4500ng・時間/mL以下の式1の化合物またはその活性代謝産物の24時間AUC血漿値を提供するのに有効な量で哺乳動物へ投与することによって、異常な細胞増殖を哺乳動物において治療する方法を提供する。24時間AUC血漿値は、本明細書の「詳細な説明」に記載のようにして決定可能である。
【0013】
この態様の具体的な側面において、24時間AUC血漿値の上限は、4000ng・時間/mL以下または3000ng・時間/mL以下または2500ng・時間/mL以下または2000ng・時間/mL以下または1500ng・時間/mL以下または1000ng・時間/mL以下または800ng・時間/mL以下または700ng・時間/mL以下である。好ましくは、そして列挙した上限のいずれとも組み合わせて、24時間AUC血漿値は、少なくとも10ng・時間/mLまたは少なくとも25ng・時間/mLまたは少なくとも50ng・時間/mLまたは少なくとも75ng・時間/mLまたは少なくとも100ng・時間/mLまたは少なくとも125ng・時間/mLである。24時間AUC血漿値の想定範囲には、列挙した下限のいずれかから列挙した上限のいずれかまでの範囲が含まれる。好ましい範囲の具体的な非限定例には、25〜4500ng・時間/mL、50〜2500ng・時間/mL、75〜1000ng・時間/mL、100〜800ng・時間/mL、および125〜700ng・時間/mLが含まれる。
【0014】
さらに本発明は、上記に定義されるような式1の化合物、その医薬的に許容される塩、溶媒和物またはプロドラッグ、またはこれらの混合物を、投薬につき30mg以下の量でヒトが含まれる哺乳動物へ投与することによって、異常な細胞増殖を哺乳動物において治療する方法を提供する。化合物の全部または一部が塩、溶媒和物、またはプロドラッグとして剤形にあるとき、その量は式1の化合物の同等量であり、分子量に基づいて当業者により容易に計算されると理解されたい。
【0015】
この態様の具体的な側面において、この量の上限は、20mg以下または15mg以下または12mg以下または10mg以下または8mg以下または7mg以下である。好ましくは、そして列挙した上限のいずれとも組み合わせて、この量は、少なくとも0.5mgまたは少なくとも1mgまたは少なくとも1.5mgまたは少なくとも2mgまたは少なくとも2.5mgまたは少なくとも3mgである。想定範囲には、列挙した下限のいずれかから列挙した上限のいずれかまでの範囲が含まれる。好ましい範囲の具体的な非限定例には、0.5〜30mg、1〜20mg、1.5〜15mg、2〜10mg、2.5〜8mg、および3〜7mgが含まれる。
【0016】
本明細書に記載される本発明の方法のいずれの具体的な態様においても、異常な細胞増殖は癌であり、限定されないが、肺癌、骨癌、膵臓癌、皮膚癌(skin cancer)、頭頚部癌、皮膚(cutaneous)または眼内黒色腫、子宮癌、卵巣癌、直腸癌、肛門域の癌、胃癌、結腸癌、乳癌、子宮癌、卵管の癌、子宮内膜癌、子宮頚部癌、膣癌、外陰部の癌、ホジキン病、食道癌、小腸の癌、内分泌系の癌、甲状腺癌、副甲状腺癌、副腎の癌、柔組織の肉腫、尿道癌、陰茎癌、前立腺癌、慢性または急性白血病、リンパ球性リンパ腫、膀胱癌、腎臓または尿管の癌、腎細胞癌、腎盂癌(carcinoma of the renal pelvis)、中枢神経系(CNS)の新生物、原発性CNSリンパ腫、脊髄軸腫瘍、脳幹神経膠腫、下垂体腺腫、または1以上の前記癌の組合せが含まれる。前記方法の別の態様において、前記異常な細胞増殖は良性の増殖性疾患であり、限定されないが、乾癬、良性前立腺肥大症、または再狭窄が含まれる。
【0017】
別の態様において、本発明は、式1の化合物の療法的に許容される量を哺乳動物へ投与することによって、PDGFR BB仲介性の癌細胞遊走を哺乳動物において阻害する方法を提供する。
【0018】
別の態様において、本発明は、式1の化合物の療法的に許容される量を哺乳動物へ投与することによって、c−KIT活性を哺乳動物において阻害する方法を提供する。
本明細書に記載する本発明の方法のいずれかのさらに具体的な態様において、本方法は、抗腫瘍剤、抗血管新生剤、シグナル伝達阻害剤、および抗増殖剤より選択される1以上の物質の量を哺乳動物へ投与することをさらに含み、該量は、前記異常な細胞増殖を治療するのに全体として有効である。そうした物質には、PCT公開公報番号:WO00/38715、WO00/38716、WO00/38717、WO00/38718、WO00/38719、WO00/38730、WO00/38665、WO/0037107、およびWO00/38786に開示されるものが含まれ、この開示は、そのまま参照により本明細書に組み込まれる。
【0019】
抗腫瘍剤の例には、減数分裂阻害剤、例えば、ビンブラスチン、ビノレルビン、ビンデシン、およびビンクリスチンのような植物アルカロイド誘導体;コルキン、アロコキン、ハリコンドリン、N−ベンゾイルトリメチル−メチルエーテルコルヒチン酸、ドラスタチン10、マイスタンシン、リゾキシン、パクリタキセル(TaxolTM)、ドセタキセル(TaxotereTM)、2’−N−[3−(ジメチルアミノ)プロピル]グルタラメート(TaxolTM誘導体)のようなタキサン、チオコルヒチン、トリチルシステイン、テニポシド、メトトレキセート、アザチオプリン、フルオロウラシル、シトシンアラビノシド、2’,2’−ジフルオロデオキシシチジン(ゲンシタビン)、アドリアマイシン、およびミタマイシン;アルキル化剤、例えばシスプラチン、カルボプラチン、オキシプラチン、イプロプラチン、N−アセチル−DL−ザルコシル−L−ロイシンのエチルエステル(AsaleyまたはAsalex)、1,4−シクロヘキサジン−1,4−ジカルバミン酸、2,5−ビス(1−アジルジニル)−3,6−ジオキソ−、ジエチルエステル(ジアジクォン)、1,4−ビス(メタンスルホニルオキシ)ブタン(ビスルファンまたはロイコスルファン)、クロロゾトシン、クロメゾン、シアノモルホリノドキソルビシン、シクロジソン、ジアンヒドログラクチトール、フルオロドパン、ヘプスルファム、マイトマイシンC、ヒカンテオネマイトマイシンC、ミトゾラミド、1−(2−クロロエチル)−4−(3−クロロプロピル)−ピペラジン二塩酸塩、ピペラジンジオン、ピポブロマン、ポルフィロマイシン、スピロヒダントインマスタード、テロキシロン、テトラプラチン、チオテパ、トリエチレンミラミン、ウラシルナイトロジェンマスタード、ビス(3−メシルオキシプロピル)アミン塩酸塩、マイトマイシン、シクロヘキシル−クロロエチルニトロソ尿素、メチルシクロヘキシル−クロロエチルニトロソ尿素、1−(2−クロロエチル)−3−(2,6−ジオキソ−3−ピペリジニル)−1−ニトロソ尿素、ビス(2−クロロエチル)ニトロソ尿素のようなニトロソ尿素、プロカルバジン、ダカルバジン、メクロロエタミン、シクロホスファミド、イホスファミド、メルファラン、クロラムブシル、リン酸エストラムスチンナトリウム、ストルプトゾイン、およびテモゾラミドのようなナイトロジェンマスタード関連化合物;DNA代謝拮抗薬、例えば、5−フルオロウラシル、シトシンアラビノシド、ヒドロキシ尿素、2−[(3−ヒドロキシ−2−ピリノジビル)メチレン]−ヒドラジンカルボチオアミド、デオキシフルオロウリジン、5−ヒドロキシ−2−ホルミルピリジンチオセミカルバゾン、α−2’−デオキシ−6−チオグアノシン、グリシン酸アフィジコリン、5−アザデオキシシチジン、β−チオグアニンデオキシリボシド、シクロシチジン、グアナゾール、イノシングリコジアルデヒド、マクベシンII、ピラゾリミダゾール、クラドリビン、ペントスタチン、チアグアニン、メルカプトプリン、ブレオマイシン、2−シクロデオキシアデノシン、ラルチトレキセドおよびペメトレキセド二ナトリウムのようなチミジン酸シンターゼ阻害剤、クロファラビン、フロクスウリジン、およびフルダラビン;DNA/RNA代謝拮抗薬、例えば、L−アラノシン、5−アザシチジン、アシビシン、アミノプテリンとN−[2−クロロ−5−[[(2,4−ジアミノ−5−メチル−6−キナゾリニル)メチル]アミノ]ベンゾイル]−L−アスパラギン酸、N−[4−[[(2,4−ジアミノ−5−エチル−6−キナゾリニル)メチル]アミノ]ベンゾイル]−L−アスパラギン酸、N−[2−クロロ−4−[[(2,4−ジアミノプテリジニル)メチル]アミノ]ベンゾイル]−L−アスパラギン酸のようなその誘導体、可溶性ベーカーズ・アンチフォール(Baker’s antifol)、ジクロロアリルローソン、ブレキナー、フトラフール、ジヒドロ−5−アザシチジン、メトトレキセート、N−(ホスホノアセチル)−L−アスパラギン酸四ナトリウム塩、ピラゾフラン、トリメトレキセート、プリカマイシン、アクチノマイシンD、クリプトフィシン、およびクリプトフィシン−52のような類似体、または、例えば、N−(5−[N−(3,4−ジヒドロ−2−メチル−4−オキソキナゾリン−6−イルメチル)−N−メチルアミノ]−2−テノイル)−L−グルタミン酸のような、ヨーロッパ特許出願番号239362に開示される好ましい拮抗薬の1つ;増殖因子阻害剤;細胞周期阻害剤;挿入抗生物質、例えば、アドリアマイシンおよびブレオマイシン;タンパク質、例えば、インターフェロン;および、抗ホルモン剤、例えばNolvadexTM(タモキシフェン)のような抗エストロゲン、または例えば、CasodexTM(4’−シアノ−3−(4−フルオロフェニルスルホニル)−2−ヒドロキシ−2−メチル−3’−(トリフルオロメチル)プロピオンアニリド)のような抗アンドロゲンが含まれる。こうした併用治療は、治療の個別成分の同時、連続、または分離投薬により達成可能である。
が含まれる。
【0020】
抗血管新生剤には、MMP−2(マトリックス−メタロプロテイナーゼ2)阻害剤、MMP−9(マトリックス−メタロプロテイナーゼ9)阻害剤、およびCOX−II(シクロオキシゲナーゼII)阻害剤が含まれる。有用なCOX−II阻害剤の例には、CELEBREXTM(アレコキシブ)、バルデコキシブ、およびロフェコキシブが含まれる。有用なマトリックスメタロプロテイナーゼ阻害剤の例は、WO96/33172(1996年10月24日公開)、WO96/27583(1996年3月7日公開)、ヨーロッパ特許出願番号97304971.1(1997年7月8日出願)、ヨーロッパ特許出願番号99308617.2(1999年10月29日出願)、WO98/07697(1998年2月26日公開)、WO98/03516(1998年1月29日公開)、WO98/34918(1998年8月13日公開)、WO98/34915(1998年8月13日公開)、WO98/33768(1998年8月6日公開)、WO98/30566(1998年7月16日公開)、ヨーロッパ特許公開公報606,046(1994年7月13日公開)、ヨーロッパ特許公開公報931,788(1999年7月28日公開)、WO90/05719(1990年5月31日公開)、WO99/52910(1999年10月21日公開)、WO99/52889(1999年10月21日公開)、WO99/29667(1999年6月17日公開)、PCT国際特許出願番号PCT/IB98/01113(1998年7月21日出願)、ヨーロッパ特許出願番号99302232.1(1999年3月25日出願)、イギリス特許出願番号991296.1(1999年6月3日出願)、米国仮特許出願番号60/148,464(1999年8月12日出願)、米国特許第5,863,949号(1999年1月26日発行)、米国特許第5,861,510号(1999年1月19日発行)、およびヨーロッパ特許公開公報780,386(1997年6月25日公開)に記載され、これらはすべてそのまま参照により本明細書に組み込まれる。好ましいMMP−2およびMMP−9阻害剤は、MMP−1を阻害する活性をほとんどまたはまったく有さないものである。より好ましいのは、他のマトリックス−メタロプロテイナーゼ(即ち、MMP−1、MMP−3、MMP−4、MMP−5、MMP−6、MMP−7、MMP−8、MMP−10、MMP−11、MMP−12、およびMMP−13)に比較して、MMP−2および/またはMMP−9を選択的に阻害するものである。
【0021】
MMP阻害剤の例には、AG−3340、RO32−3555、RS13−0830、および以下のリストに列挙する化合物が含まれる:
3−[[4−(4−フルオロ−フェノキシ)−ベンゼンスルホニル]−(1−ヒドロキシカルバモイル−シクロペンチル)−アミノ]−プロピオン酸;
3−エクソ−3−[4−(4−フルオロ−フェノキシ)−ベンゼンスルホニルアミノ]−8−オキサ−ビシクロ[3.2.1]オクタン−3−カルボン酸ヒドロキシアミド;
(2R,3R)1−[4−(2−クロロ−4−フルオロ−ベンジルオキシ)−ベンゼンスルホニル]−3−ヒドロキシ−3−メチル−ピペリジン−2−カルボン酸ヒドロキシアミド;
4−[4−(4−フルオロ−フェノキシ)−ベンゼンスルホニルアミノ]−テトラヒドロ−ピラン−4−カルボン酸ヒドロキシアミド;
3−[[4−(4−フルオロ−フェノキシ)−ベンゼンスルホニル]−(1−ヒドロキシカルバモイル−シクロブチル)−アミノ]−プロピオン酸;
4−[4−(4−クロロ−フェノキシ)−ベンゼンスルホニルアミノ]−テトラヒドロ−ピラン−4−カルボン酸ヒドロキシアミド;
3−[4−(4−クロロ−フェノキシ)−ベンゼンスルホニルアミノ]−テトラヒドロ−ピラン−3−カルボン酸ヒドロキシアミド;
(2R,3R)1−[4−(4−フルオロ−2−メチル−ベンジルオキシ)−ベンゼンスルホニル]−3−ヒドロキシ−3−メチル−ピペリジン−2−カルボン酸ヒドロキシアミド;
3−[[4−(4−フルオロ−フェノキシ)−ベンゼンスルホニル]−(1−ヒドロキシカルバモイル−1−メチル−エチル)−アミノ]−プロピオン酸;
3−[[4−(4−フルオロ−フェノキシ)−ベンゼンスルホニル]−(4−ヒドロキシカルバモイル−テトラヒドロ−ピラン−4−イル)−アミノ]−プロピオン酸;
3−エクソ−3−[4−(4−クロロ−フェノキシ)−ベンゼンスルホニルアミノ]−8−オキサ−ビシクロ[3.2.1]オクタン−3−カルボン酸ヒドロキシアミド;
3−エンド−3−[4−(4−フルオロ−フェノキシ)−ベンゼンスルホニルアミノ]−8−オキサ−ビシクロ[3.2.1]オクタン−3−カルボン酸ヒドロキシアミド;および
3−[4−(4−フルオロ−フェノキシ)−ベンゼンスルホニルアミノ]−テトラヒドロ−フラン−3−カルボン酸ヒドロキシアミド;
および、前記化合物の医薬的に許容される塩、溶媒和物およびプロドラッグ。
【0022】
シグナル伝達阻害剤の例には、EGFR抗体、EGF抗体、およびEGFR阻害剤である分子のように、EGFR(上皮増殖因子受容体)応答を阻害することができる薬剤;VEGF(血管内皮増殖因子)阻害剤;および、erbB2受容体へ結合する有機分子または抗体のようなerbB2受容体阻害剤、例えばHERCEPTINTM(米国カリフォルニア州サウス・サンフランシスコのジェネンテク社)が含まれる。
【0023】
EGFR阻害剤は、例えば、WO95/19970(1995年7月27日公開)、WO98/14451(1998年4月9日公開)、WO98/02434(1998年1月22日公開)、および米国特許第5,747,498号(1998年5月5日発行)に記載されている。EGFR阻害剤には、限定されないが、モノクローナル抗体C225および抗EGFR22Mab(米国ニューヨーク州ニューヨークのイムクローン・システムズ社)、化合物ZD−1839(アストラゼネカ)、BIBX−1382(ベーリンガーインゲルハイム)、MDX−447(米国ニュージャージー州アナンデールのMedarex社)、およびOLX−103(米国ニュージャージー州ホワイトハウス・ステーションのメルク社)、VRCTC−310(Ventech Research)、およびEGF融合毒素(マサチューセッツ州ホプキントンのセラジェン社)が含まれる。
【0024】
VEGF阻害剤、例えばSU−5416およびSU−6668(米国カリフォルニア州サウス・サンフランシスコのスジェン社)も、式1の化合物と組合せまたは同時投与可能である。VEGF阻害剤は、例えば、WO99/24440(1999年5月20日公開)、PCT国際特許出願PCT/IB99/00797(1999年5月3日出願)、WO95/21613(1995年8月17日公開)、WO99/61422(1999年12月2日公開)、米国特許第5,834,504号(1998年11月10日発行)、WO98/50356(1998年11月12日公開)、米国特許第5,883,113号(1999年3月16日発行)、米国特許第5,886,020号(1999年3月23日発行)、米国特許第5,792,783号(1998年8月11日発行)、WO99/10349(1999年3月4日公開)、WO97/32856(1997年9月12日公開)、WO97/22596(1997年6月26日公開)、WO98/54093(1998年12月3日公開)、WO98/02438(1998年1月22日公開)、WO99/16755(1999年4月8日公開)、およびWO98/02437(1998年1月22日公開)に記載されて、これらはすべてそのまま参照により本明細書に組み込まれる。いくつかの具体的なVEGF阻害剤の他の例は、IM862(米国ワシントン州カークランドのCytran社);抗VEGFモノクローナル抗体、ベバシツマブ(bevacizumab)(カリフォルニア州サウス・サンフランシスコのジェネンテク社);並びに、Ribozyme(コロラド州ボルダー)およびカイロン(Chiron)(カリフォルニア州エメリヴィル)からの合成リボザイム、angiozymeTMである。
【0025】
GW−282974(グラクソ・ウェルカム社)とモノクローナル抗体のAR−209(米国テキサス州ウッドランズのAronex Pharmaceuticals社)および2B−1(カイロン)のようなErbB2受容体阻害剤は、式1の化合物と組み合わせて投与可能である。こうしたerbB2阻害剤には、WO98/02434(1998年1月22日公開)、WO99/35146(1999年7月15日公開)、WO99/35132(1999年7月15日公開)、WO98/02437(1998年1月22日公開)、WO97/13760(1997年4月17日公開)、WO95/19970(1995年7月27日公開)、米国特許第5,587,458号(1996年12月24日発行)、および米国特許第5,877,305号(1999年3月2日発行)に記載されるものが含まれ、このそれぞれは、そのまま参照により本明細書に組み込まれる。本発明において有用なerbB2受容体阻害剤は、米国仮特許出願番号60/117,341(1999年1月27日出願)および米国仮特許出願番号60/117,346(1999年1月27日出願)にも記載され、この両方ともそのまま参照により本明細書に組み込まれる。
【0026】
使用可能である他の抗増殖剤には、酵素ファルネシルタンパク質トランスフェラーゼの阻害剤と受容体チロシンキナーゼPDGFrの阻害剤が含まれ、以下の米国特許出願に開示されて特許請求される化合物:09/221946(1998年12月28日出願);09/454058(1999年12月2日出願);09/501163(2000年2月9日出願);09/539930(2000年3月31日出願);09/202796(1997年5月22日出願);09/384339(1999年8月26日出願);および09/383755(1999年8月26日出願)と、以下の米国仮特許出願に開示されて特許請求される化合物:60/168207(1999年11月30日出願);60/170119(1999年12月10日出願);60/177718(2000年1月21日出願);60/168217(1999年11月30日出願);および60/200834(2000年5月1日出願)が含まれる。上記の特許出願および仮特許出願のそれぞれは、そのまま参照により本明細書に組み込まれる。
【0027】
式1の化合物はまた、異常な細胞増殖または癌を治療するのに有用な他の薬剤と使用可能であり、限定されないが、CTLA4(細胞傷害性リンパ球抗原4)抗体のように抗腫瘍免疫応答を増強することが可能な薬剤と、CTLA4を阻止することが可能な他の薬剤;および、他のファルネシルタンパク質トランスフェラーゼ阻害剤のような抗増殖剤が含まれる。本発明において使用可能である具体的なCTLA4抗体には、そのまま参照により本明細書に組み込まれる米国仮特許出願60/113,647(1998年12月23日出願)に記載されるものが含まれる。
【0028】
別の態様において、本発明は、式1の化合物、またはその医薬的に許容される塩、溶媒和物またはプロドラッグと治療有効量のドセタキセルを含んでなる医薬組成物を提供する。
【0029】
別の態様において、本発明は、式1の化合物、またはその医薬的に許容される塩、溶媒和物またはプロドラッグと治療有効量のドセタキセルをヒトが含まれる哺乳動物へ投与することによって、異常な細胞増殖を該哺乳動物において治療する方法を提供する。式1の化合物とドセタキセルは、要望どおりに、別々に、または同じ組成物において投与可能であり、同じ投薬スケジュールでも、異なる投薬スケジュールでも投与可能である。
【課題を解決するための手段】
【0030】
定義
本明細書に使用する「異常な細胞増殖」は、他に述べなければ、正常な調節機序から独立している細胞増殖(例、接触阻害の消失)を意味する。これには:(1)突然変異したチロシンキナーゼを発現すること、または受容体チロシンキナーゼの過剰発現によって増殖する腫瘍細胞(腫瘍);(2)異常なチロシンキナーゼ活性化が起こる他の増殖性疾患の良性および悪性の細胞;および(4)受容体チロシンキナーゼによって増殖するあらゆる腫瘍の異常増殖が含まれる。
【0031】
本明細書に使用する用語「治療する」は、他に述べなければ、そうした用語が適用される障害または状態、またはそうした障害または状態の1以上の症状を逆転させること、緩和すること、その進行を阻害すること、または予防することを意味する。本明細書に使用する用語「治療」は、他に述べなければ、治療する行為を意味し、「治療する」は直前に定義されている。
【0032】
本明細書に使用する句「医薬的に許容される塩」には、他に述べなければ、化合物に存在し得る酸性または塩基性基の塩が含まれる。性質が塩基性である化合物は、様々な無機および有機酸と多種多様な塩を形成することが可能である。そうした塩基性化合物の医薬的に許容される酸付加塩を製造するために使用可能である酸は、無毒の酸付加塩、即ち、酢酸塩、ベンゼンスルホン酸塩、安息香酸塩、重炭酸塩、重硫酸塩、重酒石酸塩、ホウ酸塩、臭化物、エデト酸カルシウム、カムシラート、炭酸塩、塩化物、クラブラン酸塩、クエン酸塩、二塩酸塩、エデト酸塩、エジシレート、エストラート、エシレート、エチルコハク酸塩、フマル酸塩、グルセプタート、グルコン酸塩、グルタミン酸塩、グリコリルアルサニレート、ヘキシルレゾルシン酸塩、ヒドラバミン、臭化水素酸塩、塩酸塩、ヨウ化物、イソチオン酸塩、乳酸塩、ラクトビオン酸塩、ラウリル酸塩、リンゴ酸塩、マレイン酸塩、マンデル酸塩、メシレート、メチル硫酸塩、ムチン酸塩、ナプシラート、硝酸塩、オレイン酸塩、シュウ酸塩、パモ酸塩(エンボン酸塩)、パルミチン酸塩、パントテン酸塩、リン酸塩/二リン酸塩、ポリガラクツロン酸塩、サリチル酸塩、ステアリン酸塩、塩基性酢酸塩、コハク酸塩、タンニン酸塩、酒石酸塩、テオクラート、トシラート、トリエチオダイド、および吉草酸塩のような、薬理学的に許容されるアニオンを含有する塩を形成するものである。
【0033】
本明細書に使用する用語「プロドラッグ」は、他に述べなければ、投与に続き、何らかの化学的または生理学的プロセスを介して薬物をin vivoで放出する薬物前駆体である化合物を意味する(例えば、プロドラッグは、生理学的なpHへ置かれるとすぐに、所望される薬物形態へ変換される)。
【0034】
本発明にはまた、天然に通常見出される原子量または質量数と異なる原子量または質量数を有する原子に1以上の原子が置き換わっている事実を除けば、式1に引用する化合物と同一である、同位体標識化合物が含まれる。本発明の化合物へ取り込み可能である同位体の例には、2H、3H、13C、14C、15N、18O、17O、31P、32P、35S、18F、および36Clのように、それぞれ水素、炭素、窒素、酸素、リン、イオウ、フッ素および塩素の同位体が含まれる。上記の同位体および/または他の原子の他の同位体を含有する、本発明の化合物、そのプロドラッグ、前記化合物または前記プロドラッグのの医薬的に許容される塩および溶媒和物は、本発明の範囲内にある。本発明の特定の同位体標識化合物、例えば、3Hおよび14Cのような放射活性同位体が取り込まれているものは、薬物および/または基質の組織分布アッセイに有用である。トリチウム化、即ち3Hと炭素−14、即ち14Cの同位体は、その調製の容易さと検出可能性のために特に好ましい。さらに、重水素、即ち2Hのようなより重い同位体での置換は、より大きな代謝安定性より生じるある種の療法上の利点(例えば、in vivo半減期の増加または投与必要量の低下)をもたらすことができるので、ある状況においては好ましい場合がある。一般に、本発明の式1の同位体標識化合物とそのプロドラッグは、非標識化合物について記載した手順を行なうこと、容易に入手可能な同位体標識試薬を非同位体標識試薬に代用することによって製造可能である。
【0035】
発明の詳細な説明
式1の化合物は、そのまま参照により本明細書に組み込まれる、米国特許第6,531,491号および6,534,524号(それぞれ、2003年3月11日および2003年3月18日発行)に記載されるように製造可能である。ある種の出発材料は、当業者に馴染みの方法に従って製造可能であり、ある種の合成の変更は、当業者に馴染みの方法に従って行ってよい。
【0036】
式1の化合物は、様々な無機および有機酸と多種多様な異なる塩を形成することが可能である。そうした塩は、哺乳動物への投与について医薬的に許容されなければならないが、実際には、はじめに式1の化合物を反応混合物より医薬的に許容されない塩として単離してから、後者をアルカリ性の試薬での処理によって遊離塩基化合物へ戻し変換して、引き続き、後者の遊離塩基を医薬的に許容される酸付加塩へ変換することがしばしば望ましい。本発明の塩基性化合物の酸付加塩は、水系溶媒の媒体、またはメタノールまたはエタノールのような好適な有機溶液において実質的に同量の選択される鉱酸または有機酸で塩基性化合物を処理することによって容易に製造される。溶媒の慎重な蒸発と同時に、所望される固形の塩を容易に得る。所望される塩は、有機溶媒中の遊離塩基の溶液より、適正な鉱酸または有機酸を溶媒へ加えることによって沈殿させてもよい。
【0037】
式1の化合物の投与は、該化合物の作用部位への送達を可能にするどの方法によって実施してもよい。これらの方法には、経口ルート、十二指腸内ルート、非経口注射(静脈内、皮下、筋肉内、血管内、または注入が含まれる)、局所、および直腸の投与が含まれる。
【0038】
本化合物は、例えば、錠剤、カプセル剤、丸剤、散剤、持続放出製剤、溶液剤、懸濁液剤として経口投与に、無菌の溶液剤、懸濁液剤、または乳剤として非経口注射に、軟膏剤またはクリーム剤として局所投与に、または坐剤として直腸投与に適した形態で提供可能である。本化合物は、正確な投与量の単回投与に適した単位剤形であってよい。好ましくは、剤形には、慣用の医薬担体または賦形剤と有効成分としての式1の化合物が含まれる。さらに、剤形には、他の医療または医薬の薬剤、担体、アジュバント、等を含めてよい。
【0039】
例示の非経口投与型には、無菌の水溶液、例えば水性プロピレングリコールまたはデキストロース溶液中の溶液剤または懸濁液剤が含まれる。そうした剤形は、所望されるならば、好適に緩衝化してよい。
【0040】
好適な医薬担体には、不活性の希釈剤または充填剤、水、および様々な有機溶媒が含まれる。医薬組成物は、所望されるならば、芳香剤、結合剤、賦形剤、等のような追加成分を含有してよい。このように、経口投与では、クエン酸のような様々な賦形剤を含有する錠剤が、デンプン、アルギン酸、およびある種の複合ケイ酸塩のような様々な崩壊剤と、そしてショ糖、ゼラチン、およびアカシアのような結合剤と一緒に利用可能である。さらに、ステアリン酸マグネシウム、ラウリル硫酸ナトリウム、およびタルクのような滑沢剤が錠剤化の目的にしばしば有用である。同様の種類の固形組成物も、軟および硬充填のゼラチンカプセル剤に利用可能である。その好ましい材料には、ラクトースまたは乳糖と、高分子量ポリエチレングリコールが含まれる。水系の懸濁液剤またはエリキシル剤が経口投与に所望されるとき、そのなかの活性化合物は、様々な甘味剤または芳香剤、着色物質または色素と、そして所望されるならば、乳化剤または懸濁剤と、水、エタノール、ポリエチレングリコール、グリセリン、またはこれらの組合せのような希釈剤と一緒に組み合わせてよい。
【0041】
本発明の剤形の好ましい態様において、剤形は、経口剤形、より好ましくは、錠剤またはカプセル剤である。
本発明の方法の好ましい態様において、式1の化合物は、例えば、本明細書に記載するような経口剤形を使用することのように、経口で投与する。
【0042】
本方法には、望ましい投与方式を使用して、式1の化合物を投与することが含まれる。1つの具体的な態様において、本化合物は、1日につき1回(毎日、またはQD)、好ましくは1日につき2回(1日2回、またはBID)投与するが、より頻繁かまたは頻繁でない投与も本発明の範囲内にある。本化合物は、ヒトが含まれる哺乳動物へ、好ましくは絶食状態(投与の前および後の2時間以内は食物も飲み物もなし)で投与可能である。特に好ましい態様において、投与法はBID、絶食である。
【0043】
式1の化合物の具体的な量で様々な剤形を調製する方法は、当業者に知られているか、または明らかになろう。例えば、「レミントン製薬科学(Remington’s Pharmaceutical Sciences)」マック・パブリッシング・カンパニー、ペンシルヴェニア州イーストン、第15版(1975)を参照のこと。
【0044】
AUC血漿値は、式1の化合物またはその活性代謝産物の血漿濃度を、様々な時間間隔で液体クロマトグラフィータンデム質量分析法(LC−MS/MS)によるように直接測定して、時間曲線に対する血漿濃度下の面積を計算することによって定量可能である。AUCを計算するのに適した方法は、例えば、台形近似:
【0045】
【化3】
【0046】
[ここで、nは、データ点の数であり、tiおよびCiは、i番目のデータ点の時間および濃度(xおよびy値)である]を使用することによるように、当該技術分野でよく知られている。24時間AUC値は、投薬スケジュールに従って測定した血漿濃度を正規化することによって定量可能である。濃度標準液の調製用の復元溶液中の安定化剤として、酸性亜硫酸ナトリウムを加える。
【0047】
式1の化合物は、VEGF−R、FGF−R、CDK複合体、CHK1、CSF−R、および/またはLCKに関連したキナーゼ活性の変調および/または阻害に関して有利な特性を有する。
【0048】
以下の実施例に示すように、式1の化合物は、HUVECアポトーシスをin vitroで誘導すること、VEGF仲介性AktおよびeNOSリン酸化をHUVECにおいて阻害すること、HUVEC中のVEGFR−2リン酸化に対して化合物の投与中止後も存続する阻害効果を明示すること、およびマトリックスタンパク質のフィブロネクチンに対するPDGF BB誘発性の癌細胞遊走を阻害することが可能である。式1の化合物は、遊走および浸潤を阻害することによって、PDGFR推進性の腫瘍進行に抗する活性を有するかもしれない。
【0049】
式1の化合物はまた、TaxolTM、より好ましくはドセタキセルと組み合わせるとき、腫瘍増殖阻害においてより有効な活性を明示する。いずれかの薬剤単独よりも、同時療法で、より有意な腫瘍退縮を観察した。
【0050】
さらに本発明は、例えば哺乳動物組織において、式1の化合物を投与することによってプロテインキナーゼ活性を変調させるかまたは阻害する方法へ向けられる。本発明の化合物の、キナーゼの活性のようなプロテインキナーゼ活性のモジュレーターとしての活性は、in vivoおよび/またはin vitroアッセイが含まれる、当業者に利用可能な方法のいずれによっても測定可能である。活性測定に適したアッセイの例には、Parast C.ら,Biochemistry,37,16788−16801(1988);Jeffreyら,Nature,376,313−320(1995);WIPO国際特許公開公報番号WO97/34876;およびWIPO国際特許公開公報番号WO96/14843に記載されるものが含まれる。これらの特性は、例えば、以下の実施例に示す生物学的試験法の1以上を使用することによって評価可能である。
【実施例】
【0051】
以下に提供する実施例および製法は、本発明の剤形および方法をさらに例示して例証する。本発明の範囲は、以下の実施例の範囲により決して限定されないことを理解されたい。
【0052】
実施例1
式1の化合物について:(1)いくつかのスケジューリング:1日1回、週末投薬、休日および間欠性の投薬でのin vivo効力;(2)異種移植片モデルにおいてドセタキセルと組み合わせたときの効力;(3)内皮細胞におけるin vitro eNOSおよびAktリン酸化;(4)細胞培養およびin vivoでの酸化窒素および関連産物の濃度;および(5)全血液細胞中のc−Kitシグナルの、化合物の潜在バイオマーカーとしての使用を試験した。
【0053】
生物学的試験;酵素アッセイ
VEFG、FGF、その他のような増殖因子による細胞増殖の刺激は、そのそれぞれの受容体のチロシンキナーゼがそれぞれを自己リン酸化することの誘導に依存する。故に、これらの増殖因子によって誘導される細胞の増殖を妨げるプロテインキナーゼ阻害剤の能力は、受容体の自己リン酸化を妨げるその能力と直接的に相関する。化合物のプロテインキナーゼ阻害を測定するために、以下の構築体を設計した。
【0054】
アッセイ用のVEGF−R2構築体:この構築体は、チロシンキナーゼ活性を阻害する試験化合物の能力を決定する。キナーゼ挿入ドメインの68残基の中央50残基を欠く、ヒト血管内皮細胞増殖因子受容体2(VEGF−R2)のサイトゾルドメインの構築体(VEGF−R2Δ50)をバキュロウイルス/昆虫細胞系において発現させた。全長VEGF−R2の1356残基のうち、VEGF−R2Δ50は、残基806〜939および990〜1171と、キナーゼ挿入ドメイン内に、野生型VEGF−R2に対する1つの点突然変異(E990V)も含有する。この精製構築体の自己リン酸化を、5%グリセロールおよび5mM DTTを含有する100mM HEPES(pH7.5)中3mM ATPおよび40mM MgCl2の存在下、4μMの濃度の酵素の4℃で2時間のインキュベーションにより実施した。自己リン酸化の後で、この構築体は、野生型の自己リン酸化キナーゼドメイン構築体に本質的に同等な触媒活性を保有することが示された。Parastら,Biochemistry,37,16788−16801(1998)を参照のこと。
【0055】
アッセイ用のFGF−R1構築体:バキュロウイルスベクター発現系を使用して、ヒトFGF−R1の細胞内キナーゼドメインを、Mohammadiら,Mol.Cell.Biol.,16,977−989(1996)の残基番号付けシステムによる、内因性メチオニン残基456から出発してグルタミン酸766まで発現させた。さらに、この構築体はまた、以下の3つのアミノ酸置換を有する:L457V、C488A、およびC584S。
【0056】
アッセイ用のLCK構築体:昆虫細胞において、アミノ酸残基223から出発して残基509でのタンパク質の終わりまでのN−末端欠損体(以下の2つのアミノ酸置換がN末端にある:P233MおよびC224D)としてLCKチロシンキナーゼを発現させた。
【0057】
アッセイ用のCHK−1構築体:バキュロウイルス/昆虫細胞系を使用して、C末端Hisタグ付き全長ヒトCHK−1(FL−CHK−1)を発現させた。これは、476アミノ酸のヒトCHK−1のC末端に6つのヒスチジン残基(6xHis−タグ)を含有する。このタンパク質を慣用のクロマトグラフィー技術によって精製した。
【0058】
アッセイ用CDK2/サイクリンA構築体:バキュロウイルス発現ベクターで感染させた昆虫細胞より、公知の方法論(Rosenblattら,J.Mol.Biol.,230,1317−1319(1993))を使用して、CDK2を精製した。サイクリンAは、全長組換えサイクリンAを発現する大腸菌細胞より精製し、先端切断サイクリンA構築体は、限定したタンパク分解によって産生し、既報(Jeffreyら,Nature,376,313−320(1995))に記載のように精製した。
【0059】
アッセイ用CDK4/サイクリンD構築体:対応するバキュロウイルス発現ベクターで同時感染させた昆虫細胞より、従来の生化学のクロマトグラフィー技術を使用して、ヒトCDK4およびサイクリンD3の複合体、またはサイクリンD1とヒトCDK4およびグルタチオン−S−トランスフェラーゼ(GST−CDK4)の融合タンパク質の複合体を精製した。
【0060】
VEGF−R2アッセイ:共役分光測定(FLVK−P)アッセイ
ホスホリル転移に伴うADPのATPからの産生を、ホスホエノールピルビン酸塩(PEP)とピルビン酸キナーゼ(PK)および乳酸デヒドロゲナーゼ(LDH)を有する系を使用する、NADHの酸化へ共役した。Beckman DU650分光光度計を使用して、340nmでの吸光度(e340=6.22cm−1mM−1)の減少を追跡することによってNADHの酸化をモニタリングした。リン酸化VEGF−R2Δ50(以下の表においてFLVK−Pとして示す)のアッセイ条件は、以下の通りであった:200mM HEPES(pH7.5)中1mM PEP;250μM NADH;50ユニットのLDH/mL;20ユニットのPK/mL;5mM DTT;5.1mM ポリ(E4Y1);1mM ATP;および25mM MgCl2。非リン酸化VEGF−R2Δ50(表においてFLVKとして示す)のアッセイ条件は、以下の通りであった:200mM HEPES(pH7.5)中1mM PEP;250μM NADH;50ユニットのLDH/mL;20ユニットのPK/mL;5mM DTT;20mM ポリ(E4Y1);3mM ATP;および60mM MgCl2および2mM MnCl2。5〜40nMの酵素でアッセイを開始した。様々な濃度の試験化合物の存在下に酵素活性を測定することによって、Ki値を決定した。Enzyme KineticおよびKaleidagraphのソフトウェアを使用してデータを解析した。
【0061】
ELISAアッセイ:ビオチニル化ガストリンペプチド(1−17)を基質として使用して、ホスホガストリンの形成をモニタリングした。ストレプタビジン被覆96ウェルマイクロタイタープレートを使用してビオチニル化ホスホガストリンを固定化して、西洋ワサビペルオキシダーゼへ共役した抗ホスホチロシン抗体を使用する検出を続けた。2,2’−アジノ−ジ−[3−エチルベンザチアゾリンスルホネート(6)]ジアンモニウム塩(ABTS)を使用して、西洋ワサビペルオキシダーゼの活性をモニタリングした。典型的なアッセイ溶液は、200mM HEPES(pH7.5)中2μMビオチニル化ガストリンペプチド;5mM DTT;20μM ATP;26mM MgCl2;および2mM MnCl2を含有した。0.8nMのリン酸化VEGF−R2△50でアッセイを開始した。西洋ワサビペルオキシダーゼ活性は、ABTS,10mMを使用してアッセイした。酸(H2SO4)の添加により西洋ワサビペルオキシダーゼ反応を失活させ、405nmでの吸光度読取りを続けた。様々な濃度の試験化合物の存在下に酵素活性を測定することによって、Ki値を決定した。Enzyme KineticおよびKaleidagraphのソフトウェアを使用してデータを解析した。
【0062】
FGF−Rアッセイ:VEGF−R2についての上記の記載のように分光測定アッセイを行なったが、但し、濃度を以下のように変更した:FGF−R=50nM,ATP=2mM,およびポリ(E4Y1)=15mM。
【0063】
LCKアッセイ:VEGF−R2についての上記の記載のように分光測定アッセイを行なったが、但し、濃度を以下のように変更した:LCK=60nM,MgCl2=0mM,ポリ(E4Y1)=20mM。
【0064】
CHK−1アッセイ:合成基質ペプチドのSyntide−2(PLARTLSVAGLPGKK)へのホスホリル転移に伴うADPのATPからの産生を、ホスホエノールピルビン酸塩(PEP)を使用する、ピルビン酸キナーゼ(PK)および乳酸デヒドロゲナーゼ(LDH)の作用を介したNADHの酸化へ共役した。NADHの酸化は、HP8452分光光度計を使用して、340nmでの吸光度(ε340=6.22cm−1mM−1)の減少を追跡することによってモニタリングした。典型的な反応溶液は:50mM TRIS(pH7.5)中4mN PEP;0.15mM NADH;28ユニットのLDH/mL;16ユニットのPK/mL;3mM DTT;0.125mM Syntide−2;0.15mM ATP;25mM MgCl2;および400mL NaClを含有した。10nMのFL−CHK−1でアッセイを開始した。様々な濃度の試験化合物の存在下に初期酵素活性を測定することによって、Ki値を決定した。Enzyme KineticおよびKaleidagraphのソフトウェアを使用してデータを解析した。
【0065】
HUVEC増殖アッセイ:このアッセイは、ヒト臍静脈内皮細胞(「HUVEC」)の増殖因子刺激増殖を阻害する試験化合物の能力を決定する。HUVEC細胞(継代数3〜4、Clonetics社)をT75フラスコ中のEGM2培養基(Clonetics社)へ融かした。24時間後、このフラスコへ新鮮なEGM2培地を加えた。4〜5日後、細胞を別の培養基(10%胎仔ウシ血清(FBS)、60μg/mLの内皮細胞増殖サプリメント(ECGS)、および10μg/mL ヘパリンを補充したF12K培地)へ曝露した。その後の実験では、指数増殖期のHUVEC細胞を使用した。10,000〜12,000個のHUVEC細胞を96ウェル・ディッシュにおいて100μlの濃縮培養基(上記に記載)においてプレート培養した。この細胞を、この培地において24時間付着させた。次いで、培地を吸引により除去し、115μlの飢餓培地(F12K+1% FBS)を各ウェルへ加えた。18時間後、飢餓培地中1% DMSOに溶かした15μlの試験薬剤かまたはこの担体単独をそれぞれの処理ウェルへ加えた;最終DMSO濃度は0.1%であった。1時間後、飢餓培地中20μlの150ng/mL hrVEGF165を、未処理対照を含有するウェルを除くすべてのウェルへ加えた;最終VEGF濃度は20ng/mLであった。72時間後にMTT色素低下により細胞の増殖を定量したが、この時点で細胞をMTT(プロメガ社)へ4〜5時間曝露した。停止溶液(プロメガ社)の添加により色素低下を止め、570および630nmでの吸光度を96ウェル分光光度計プレートリーダーで決定した。
【0066】
癌細胞増殖(MV522)アッセイ:プロテインキナーゼ阻害剤が癌を治療するために血管新生を妨げる治療有用性を有するかどうかを決定するには、この阻害剤がキナーゼ受容体を発現しない細胞においては細胞増殖を非特異的に妨げないことを実証することが重要である。癌細胞を使用する増殖アッセイを実施することによってこのことを行う。癌細胞における細胞増殖を評価するためのプロトコールは、HUVEC細胞における評価について使用するものと同様である。2000個の肺癌細胞(MV522系、UCSDより獲得した)を増殖培地(2mMグルタミンおよび10% FBSを補充したRPMI1640培地)にまいた。試験薬剤および/または担体の添加に先立って、細胞を1日間付着させる。HUVECアッセイに使用するのと同じ試験薬剤で細胞を同時に処理する。試験薬剤への曝露の72時間後に、MTT色素低下アッセイによって細胞増殖を定量する。
【0067】
C−Kit効力決定:阻害剤によるc−Kitに抗する効力を決定するためにNCI−H526(ATCC)細胞を使用した。この細胞を亜集密まで増殖させて、飢餓培地において18時間インキュベートした。阻害剤を加え、細胞を2.3%アルブミンおよび1mM Na3VO4(シグマ)の存在下に37℃で45分間インキュベートした。この培養物へSCF、c−Kit増殖因子を50ng/mlの最終濃度で加えた。5分後、この細胞を冷PBSで2回濯ぎ、溶解緩衝液(50mM Tris,150mM NaCl,1mM PMSF,1% NP40,1mM Na3VO4およびプロテアーゼ阻害剤カクテル)で溶解させた。各溶解液より1mgの総タンパク質を使用し、4μg/ml CD117 ab−3(K45,Neomarkers)とともに4℃で一晩インキュベートして、免疫沈降を実施した。翌朝、この抗体複合体をプロテインAビーズへコンジュゲートした。リン酸化受容体用の抗ホスホチロシン抗体4G10(Upstate Biotechnology)、または抗c−Kit受容体抗体sc−1493(C−14,Santa Cruz)を1:1000で使用して、SDS PAGEとウェスタン・ブロット分析を実施した。化学発光試薬のECL Plusによってブロットを可視化した。ブロット中のシグナルの定量にphosphorimager(Storm 846,Molecular Dynamics)を使用した。
【0068】
動物および哺乳動物の全末梢血細胞中のc−kit陽性細胞集団の低下を式1の化合物の活性のバイオマーカーとして使用した。
ENOSおよびAktリン酸化測定:阻害剤によるeNOSおよびAktに抗する効力を決定するためにHUVEC(Clonetics)を使用した。この細胞を亜集密まで増殖させて、飢餓培地において18時間インキュベートした。阻害剤を加え、細胞を2.3%アルブミンおよび1mM Na3VO4(シグマ)の存在下に37℃で45分間インキュベートした。この培養基へVEGFを50ng/mlで加えた。5分後、この細胞を冷PBSで2回濯ぎ、溶解緩衝液(50mM Tris,150mM NaCl,1mM PMSF,1% NP40,1mM Na3VO4およびプロテアーゼ阻害剤カクテル)で溶解させた。30〜40μgの総タンパク質をウェスタン法によって分析した。eNOSおよびAktリン酸化は:Phospho−eNOS(Ser 1177)#9571またはPhospho−Akt(Ser 473)#9271の抗体(いずれもCell signalingより)を使用することによって評価した。タンパク質の検出は、NOS3(C−20)sc−654(Santa Cruz)またはAkt 抗体#9272(Cell signaling)を使用することによって達成した。いずれも、1:3000で使用する抗ウサギHRP連結二次抗体を必要とする。化学発光基質のSuper Signal West Dura(ピアス)によってブロットを可視化した。ブロット中のシグナルの定量にAlpha Imager 8800(Alpha Innotechより)を使用した。
【0069】
マウスPKアッセイ:薬物のマウスにおける薬物動態(例、吸収と消失)について、以下の実験を使用して解析した。試験化合物は、0.5% CMC担体中の懸濁液剤として、または30:70(PEG400:酸性化H2O)担体中の溶液剤として製剤化した。この懸濁液剤または溶液剤をC3H雌性マウス(n=4)へ経口(p.o.)または腹腔内(i.p.)投与した。0時間(投薬前)、投薬後0.5時間、1.0時間、2.0時間、および4.0時間の時点で眼窩出血により血液試料を採取した。各試料より、2500rpmで5分間の遠心分離によって血漿を入手した。この血漿より有機タンパク質沈殿法によって試験化合物を抽出した。各時点の出血につき、50μLの血漿を1.0mLのアセトニトリルと合わせ、2分間激しく振り混ぜてから、4000rpmで15分間スピンしてタンパク質を沈殿させ、試験化合物を抽出した。次いで、アセトニトリル上清(試験化合物を含有する抽出物)を新しい試験管へ注ぎ、N2ガスの蒸気下にホットプレート(25℃)で蒸発させた。乾燥した試験化合物の抽出物を含有するそれぞれの試験管へ125μLの移動相(60:40,0.025M NH4H2PO4+2.5mL/L TEA:アセトニトリル)を加えた。激しく撹拌することによって、試験化合物をこの移動相に再懸濁させ、4000rpmで5分間の遠心分離によりさらにタンパク質を除去した。UV検出付きヒューレット・パッカード1100シリーズHPLCでの試験化合物分析のために各試料をHPLCバイアルへ注いだ。各試料より95μLをPhenomenex−Prodigy逆相C−18,150x3.2mmカラムへ注入し、10分にわたる45〜50%アセトニトリル勾配ランで溶出させた。上記のやり方で血漿試料より抽出した試験化合物の既知濃度を使用して、標準曲線(ピーク面積x濃度μg/mL)との比較により、試験化合物の血漿濃度(μg/mL)を決定した。標準品と未知検体と一緒に、3群(n=4)の品質コントロール(0.25μg/mL、1.5μg/mL、および7.5μg/mL)を実施して、分析の一貫性を保証した。標準曲線はR2>0.99を有し、品質コントロールは、いずれもその予測値の10%範囲内にあった。定量した試験試料を、Kalidagraphソフトウェアを使用する可視ディスプレイのためにプロットし、WIN NONLINソフトウェアを使用して、その薬物動態パラメータを決定した。
【0070】
ヒト肝ミクロソーム(HLM)アッセイ:ヒト肝ミクロソームにおける化合物の代謝について、以下のようなLS−MS分析アッセイ手順によって測定した。はじめに、ヒト肝ミクロソーム(HLM)を融かし、冷100mMリン酸カリウム(KPO4)緩衝液で5mg/mLへ希釈した。適正量のKPO4緩衝液、NADPH再生溶液(B−NADP、グルコース−6−リン酸、グルコース−6−リン酸デヒドロゲナーゼ、およびMgCl2を含有する)、およびHLMを13x100mmのガラス試験管において37℃で10分間プレインキュベートした(試験化合物につき3つの試験管;同一3検体)。試験化合物(最終5μM)を各試験管へ加えて反応を開始させ、穏やかに振り混ぜて混合した後で、37℃でインキュベートした。t=0および2時間で、各インキュベーション試験管より250μLの試料を取り出し、1mLの氷冷アセトニトリル(+0.05μMレセルピン)を含有する分離用12x75mmガラス試験管へ移した。試料を4000rpmで20分間遠心分離して、タンパク質と塩を沈殿させた(ベックマン Allegra 6KR,S/N ALK98D06,#634)。上清を新しい12x75mmガラス試験管へ移し、Speed−Vac遠心分離真空エバポレーターにより蒸発させた。200μLの0.1%ギ酸/アセトニトリル(90/10)中で試料を復元し、激しく撹拌して溶かした。次いで、この試料を分離用ポリプロピレンミクロ遠心分離管へ移し、14000xgで10分間遠心分離した(Fisher Micro 14,S/N M0017580)。各時点(0および2時間)での各同一3検体(#1〜3)について、各試験化合物のアリコート試料を、以下に記載するLC−MS分析用の単回HPLCバイアルインサート(全部で6つの試料)へ合わせた。
【0071】
合わせた化合物試料を、ヒューレット・パッカード HP1100 ダイオードアレイHPLCとポジティブエレクトロスプレーSIRモードで作動するMicromass Quattro II三連四重極型質量分析計からなる(各試験化合物の分子イオンを特異的に走査するようにプログラムされている)、LC−MSシステムへ注入した。各試験化合物のピークを各時点で積分した。各化合物について、各時点でのピーク面積(n=3)を平均化して、この2時間での平均ピーク面積を0時間の時点での平均ピーク面積で割って、2時間で残存する試験化合物のパーセントを得た。
【0072】
in vitro HUVECアポトーシスアッセイ
ELISAによるアポトーシスの定量:細胞溶解液中の細胞質ヒストン会合DNA断片を定量するCell Death Detection Elisa PLUS(カタログ番号:1775425,ロッシュ・バイオケミカルズ、マンハイム、ドイツ)を使用して、HUVEC細胞のアポトーシスを測定した。製造業者の手引きに若干の変更を加えて、この手順を実施した。簡潔に言えば、飢餓状態のHUVEC細胞をVEGF(20ng/mL)の存在下に様々な濃度の化合物Aで処理した。この細胞のサイトゾル分画を様々な時点で採取し、マイクロタイタープレートへコートした一次抗ヒストンmAbとペルオキシダーゼへ結合した二次抗DNA mAbを用いるサンドイッチELISAにおける抗原の供給源として使用した。発色性ペルオキシダーゼ基質を加えて、分光光度計で405nmでの吸光度(基準波長:490nm)を測定することによって、アポトーシス細胞の数を定量した。
【0073】
TUNELによるアポトーシスの可視化:TdT仲介性dUTPニックエンド標識(TUNEL)技術を使用して、アポトーシス細胞のin situ検出を行った。簡潔に言えば、8ウェルLab−Tekチャンバ・スライドにおいて増殖させたHUVEC細胞をO/N飢餓させてから、様々な濃度の化合物Aで6時間処理した。次いで、製造業者の手引きに従って、この細胞を4%パラホルムアルデヒドに固定して、Triton X−100で透過性にして、ターミナルトランスフェラーゼとフルオレセイン−dUTP(Deadend Fluorometric TUNEL系、プロメガ、カタログ番号 G3250)が含まれるヌクレオチドの混合物において1時間インキュベートした。この細胞をヨウ化プロピジウム(PI)溶液で対比染色した。陽性染色されたフルオレセインおよびPI標識細胞を可視化して、蛍光顕微鏡によって写真撮影した。
【0074】
PDGF仲介性細胞遊走アッセイ:このアッセイには、U87MG細胞を使用した。6ウェルプレートを0.5ng/mL フィブロネクチンとともに一晩プレインキュベートする。翌日、U87MG細胞を各ウェルにおいてプレート培養して、集密まで増殖させる。この細胞を、0.1% FBSを含有する飢餓培地とともに一晩インキュベートした。ピペット先端を使用して約1cmのスクラッチをつくり、細胞を飢餓培地で洗浄した。次いで、プレートを0.5ng/mL フィブロネクチンとともに1時間インキュベートしてから、再び洗浄した。100ng/LI rhPDGF BBと化合物Aを飢餓培地中に含有する実験培地を導入した。0時間と15時間の間で細胞を写真撮影して、遊走を可視化した。
【0075】
細胞VEGFR−2および下流分子リン酸化アッセイ:HUVEC(Clonetics)を亜集密まで培養して、飢餓培地(F12K+0.1% FBS)において18時間インキュベートした。この細胞へ2.3%アルブミンおよび1mM Na3VO4(シグマ)の存在下に化合物Aを加えた。45分後、この培養物へVEGFを50ng/mLの最終濃度で加えた。5分後、この細胞を冷PBSで濯ぎ、溶解緩衝液(50mM Tris,150mM NaCl,1mM PMSF,1% NP40,1mM Na3VO4およびプロテアーゼ阻害剤カクテル)で溶解させた。溶解液由来の1ミリグラムの総タンパク質を、抗Flk−1 C−1158(Santa Cruz)を使用して免疫沈殿させた。この抗体複合体をプロテインAビーズへコンジュゲートして、SDS PAGE/ウェスタン分析を実施した。抗ホスホチロシン抗体4G10(Upstate Biotechnology)と抗Flk−1 C−20(Santa Cruz)によって、それぞれリン酸化VEGFR−2とそのタンパク質を検出した。eNOSおよびAktについては、細胞を上記と同じように処理した。全量30〜40μgのタンパク質を使用して、ウェスタン分析を実施した。eNOSおよびAktリン酸化は、Phospho−eNOS(Ser 1177,#9571)またはPhospho−Akt(Ser 473,#9271)の抗体(Cell signaling)を使用することによって探知した。タンパク質は、NOS3 C−20(sc−654,Santa Cruz)またはAkt 抗体#9272(Cell Signaling)を使用することによって評価した。HRP連結抗ウサギIgGを二次抗体として使用した。化学発光基質のSuper Signal West Dura(ピアス)によってすべてのブロットを可視化した。Alpha InnotechからのAlpha Imager 8800を使用して、シグナルを定量した。
【0076】
ウォッシュアウト実験:HUVEC細胞を上記に記載のように処理した。化合物A(10nM)と45分間のインキュベーションと、VEGF(50ng/ml)で5分間の刺激の後で、上清を除去し、洗浄し、VEGFおよびNa3VO4を含有する飢餓培地に置き換えた。この細胞を所望される長さの時間の間さらにインキュベートした後で、溶解させ、リン酸化および全体のVEGFR−2のために免疫沈殿およびウェスタンを使用して処理した(上記参照)。別の実験において、細胞を上記のように全体の時間の長さの間VEGFで処理し、所望される時点でのVEGFR−2リン酸化および全VEGFR−2を同様に評価した。ウォッシュアウトの間のシグナルをデンシトメトリーによって定量した。各実験からの最大刺激(5分)の強度を互いに正規化して、ホスホVEGFR−2の各時点での強度を2つの実験間で比較して、それにより、非処理であるがVEGF刺激した細胞に対して、VEGFR−2リン酸化の回復を決定した。
【0077】
腫瘍モデル:ヒトMV522(結腸癌)およびMDA−MB−231(乳癌)モデルでは、無胸腺マウス(n=8〜12)に5x106細胞/部位を移植(s.c.)した。マウスのルイス肺癌モデルでは、腫瘍断片(1〜2mm2)をB6D2F1マウスの右脇腹にトロカール移植した。投薬は、通常7日目(MV522)、または平均腫瘍サイズが150〜200mm3に達するとき(MDA−MB−231)に開始した。
【0078】
式1の化合物は、0.5% CMC/H2O中に製剤化して、PO、BID投与した。ドセタキセルは、7% EtOH/3% ポリソルベート/90% H2O中に製剤化して、週ごとに、静脈内で投薬した。通常、処理は3〜4週間続いた。腫瘍の幾何学的な長さおよび幅を、電子カリパスを使用して週3回測定した。腫瘍体積は、0.4x[長さx(幅)2]の積として算出した。データは、平均±SEMとして報告した。試験の最後に、腫瘍と組織を分析用に切除、秤量、および採取した。薬物濃度の分析用に血漿を採取した。
【0079】
結果を表1〜3に示す。
表1.化合物1の効力および選択性
【0080】
【表1】
【0081】
スクリーニングしたが、Ki計算の限度を超えた他の酵素は、cMet、LCK、c−Src、FAK、Pyk2、IRL、BTK、CDK1、CDK2、CDK4、PKA、PKC、PLK、およびChk1である。
【0082】
表2.MDA−MB−231ヒト乳癌モデルにおける化合物1およびドセタキセルの同時投与用の試験設計
【0083】
【表2】
【0084】
表3.ドセタキセルおよび化合物1の組合せ療法は、MDA−MB−231異種移植片モデルにおいてより大きな抗腫瘍活性をもたらした。
【0085】
【表3】
【0086】
上記の組合せ群は、完全および部分的な腫瘍退縮の増加した出現率を明示する。これらの薬剤を組み合わせるとき、腫瘍増殖率は、より大きな度合いまで低下した。この組合せ治療は、単剤単独と同じくらい十分に忍容された。
【0087】
実施例2
式1の化合物、6−[2−(メチルカルバモイル)フェニルスルファニル]−3−E−[2−(ピリジン−2−イル)エテニル]インダゾールを固形腫瘍の患者へ様々な用量で投与した。式1の化合物を経口の錠剤形において、BIDまたはQDのスケジュールで使用して、30名(男性13名、女性17名)の患者を治療した。サイクルは、それぞれ28日であった。具体的な腫瘍の診断は、乳房(11)、甲状腺(5)、腎細胞(5)、肺(4)および、その他(5)であった。液体クロマトグラフィータンデム質量分析法(LC−MS/MS)によって、薬物動態データを測定した。このサイクルの15日目に、投与後0.5時間、1時間、2時間、4時間、8時間、および12時間の時点で血液試料を採取した。
【0088】
薬物動態結果(15日目の平均値)を表4に示す。患者は、他に述べなければ、絶食ではなかった。括弧内の数字は、百分率として表される変動係数である。この表において、Cmaxは、観測される式1の化合物の最高血漿濃度であり、AUC(0−24)は、24時間のAUC血漿濃度であり、T1/2は、濃度対時間のプロットより決定される半減期である。見出し項目の「PKの患者数」は、薬物動態データを入手した患者の数を示す。
【0089】
表4
【0090】
【表4】
【0091】
さらに、第一コホートの患者(n=6)は、10mg QD〜30mg BIDに及ぶ個別化された用量を受けた(PK示さず)。絶食状態では、摂食状態に比べて、血漿曝露がより高く(約49%)、患者内の変動は抑えられた。現時点での最大耐薬量(MTD)は、絶食状態で5mg BIDであると決定した。MTDより高い用量での投薬制限毒性(DLT)は、高血圧(HTN)、発作、肝機能検査値の上昇、膵炎、無呼吸、および胃炎であった。さらに、応答した2名のNSCLC患者は、致命的な喀血を起こし、1名は3週後にこの化合物での治療を中止した。非投薬制限性のタンパク尿も観察された。MTD以下の用量では、1名の患者で、グレード2の胃炎へDLTが限定された。7/14名の患者で非投薬制限性のHTNが観察され、慣用の降圧用医薬品により管理した。RECIST判定基準による2つの永続性のある一部応答が観察され(腎細胞と上顎洞の腺嚢腫において)、この十分に前処置した集団のうち5名の患者で、安定した疾患が4ヶ月以上(4〜13+ヶ月の範囲)続いた。dceMRIを使用して、21名の患者で予備分析を実施して、式1の化合物により誘発される血管系への効果をベースラインと2、28、および56日目で測定した。平均Ktransの百分率変化(P.S.Tofts,G.Brix,D.L.Buckley,J.L.Evelhoch,E.Henderson,M.V.Knopp,H.B.W.Larsson,T.Lee,N.A.Mayr,G.J.M.Parker,R.E.Port,J.TaylorおよびR.M.Weisskoff,「分散可能トレーサーのダイナミック対比増強T1重み付けMRIからの動態パラメータの評価:量および記号の標準化」(Estimating Kinetic Parameters Dynamic Contrast−Enhanced T1−Weighted MRI of a Diffusable Tracer:Standardized Quantities and Symbols)Journal of Magnetic Resonance Imaging,10:223−232(1999)とコントラスト強度X時間曲線下の初期面積(IAUC)をそれぞれの指標腫瘍(n=1〜4/患者)につき計算した。腫瘍血管系の応答は、ベースライン変数値が2日目までに50%以上減少するものと定義した。腫瘍血管系応答の急激な(2日目)の減少(KtransおよびIAUCの50%以上の減少)が18名の評価可能な患者の6名で観察され、11/18名で、KtransおよびIAUCの両方の40%以上の減少が明示された。スキャンに伴う技術課題により、3/21の画像セットが評価可能でなかった。本実施例は、式1の化合物が、臨床応答と急性の腫瘍血管系変化により顕現されるように、きわめて有効な薬剤であることを示す。
【0092】
実施例3
30mg遊離塩基/kg用量の式1の[14C]標識化合物をインタクトまたは胆管カニューレ挿入ビーグル犬へ経口投与した後で、広汎な代謝を観察した。生物変換経路には、酸素化(モノまたはジ)、グルクロニド化、グルコシル化、および酸素化に続く硫酸化またはグルコシル化が含まれた。図1は、同定された代謝産物を示す。血漿中では、M12(N−オキシド)が検出可能な唯一の代謝産物である。尿中では、M5(デピリジニルカルボン酸)が主要な代謝産物である。主要な胆汁代謝産物には、M8(硫酸塩)とM12が含まれる。主要な糞中代謝産物M1の化学構造は、不明のままである。
【0093】
本化合物の単回経口投薬に続く、[14C]由来放射活性のビーグル犬における排泄パターンは、雄と雌で同様であり、放射活性は、主に糞より排泄された。インタクトな雌の糞中80.9%と尿中7.0%の回収率と比較して、インタクトな雄の平均回収率は、糞中85.5%と尿中5.3%であった。胆管カニューレ挿入した雄イヌは、比較的少ない分量の放射活性を胆汁に排泄し(8.3%の回収率)、さらなる放射活性は、糞(52.7%)と尿(11.3%)に回収された。胆管カニューレを挿入したイヌからの尿および胆汁の放射活性を合わせると、投与した放射活性のほぼ20%が胃腸吸収を受けたことを示唆する。すべての試料の全体平均回収率は、インタクトな雄と雌についてそれぞれ92.4%と92.6%、胆管カニューレ挿入した雄で89.6%であった。放射HPLC検出器(β−RAM)とインライン共役したHPCLと、エレクトロスプレー(ESI)および大気圧化学イオン化(APCI)供給源のMS検出をポジティブまたはネガティブ形式で使用して、すべての代謝産物のプロファイリングおよび構造解明を実施した。
【0094】
実施例4
式1の化合物は、CD−1マウスにおいて、[14C]標識化合物の単回経口投与に続き、広汎な代謝を受ける。低い百分率の未変化薬物が尿および糞中に回収され、多様な第I相および第II相の代謝産物が観察された。生物変換経路には、酸素化(モノまたはジ)、グルクロニド化、グルコシル化、および酸素化に続くグルクロニド化またはグルコシル化のいずれかが含まれた。同定された代謝産物を図2に示す。血漿中では、未変化薬物とM12(N−オキシド)が2つの主要な成分を代表した。M7(グルクロニド)は、尿と糞の両方で最も顕著な代謝産物を代表した。
【0095】
50mg遊離塩基/kg用量の[14C]AG−013736を雄性CD−1マウスへ単回経口投与後、[14C]由来放射活性の大部分が糞中で回収された。投薬後48時間での放射活性の平均(n=2)回収率(用量の%)は、糞中65.8%と尿中12.7%であった。排泄物中の放射活性の消失速度は速やかで、ほぼ72%の用量が投薬後24時間以内に回収された。LC−RAM−MS法を使用して、代謝産物の放射活性プロファイリングと構造特性決定を実施した。
【0096】
図1および2に示す代謝産物に加えて、他の既知の代謝産物には、式1aに示す活性des−メチル代謝産物が含まれる。
【0097】
【化4】
【0098】
実施例5
免疫組織化学を使用して、腫瘍微小血管密度(MVD)を測定することによって、血管新生を評価した。凍結した腫瘍切片を血管表面マーカーのCD−31用に染色し、組織切片の数領域中の血管の量をマニュアルで定量した。種々の試験は、式1の化合物の2〜3週のPO BID投与が、対照腫瘍と比較して、処理した腫瘍中の血管の数を70%低下させることを示した。処理後の微小血管密度の減少は、LLC、MV522、およびM24metが含まれる、使用したすべての腫瘍モデルで観察された。LLC腫瘍モデルでは、浸透Alzetポンプを介して連続的に送達するとき、式1の化合物は、有意な増殖阻害をもたらした。3回の試験からのデータは、LLCモデルにおけるこのクラスの薬剤により達成可能である最大の腫瘍増殖阻害が78%であることを示した。55±17ng/mL(N=3)ほどの低さの血漿濃度では、90%の最大増殖阻害が達成された。この濃度を生物学的活性濃度(BAC)と名付けた。50%最大増殖阻害は、28±11ng/mL(N=3)の血漿濃度と関連した。この濃度を最小有効濃度(MEC)と名付けた。1つの試験群で、本化合物の連続注入により産生される70%のMGIは、574ng・時間/mLのAUC(0−24)と関連したが、同じ試験において、PO BID投薬後の720ng・時間/mLのAUC(0−24)は、40%の最大増殖阻害(MGI)を生じた。これらの結果は、このモデルにおいて見られる抗腫瘍効力がトラフ(谷)濃度により推進されること、そしてマウスでは、最大抗腫瘍効力をもたらすのに、本化合物の連続した低濃度が十分であり得ることを示唆する。
【0099】
式1の化合物は、ヒト乳癌異種移植片モデル、MDA−MB−231において単剤として有効であった。このモデルにおける式1の化合物とドセタキセルの組合せを用いた効力試験に備えて、ナイーブなヌードマウスで予備試験を行って、PKおよび耐性に対する潜在的な薬物−薬物相互作用の効果を決定した。15または30mg/kgのドセタキセル、週1回3週間IV投与の後で、対照と比べた体重の減少(それぞれ、7%と11%)をドセタキセル処理動物で確認した。ドセタキセル単独で処理した動物と、ドセタキセルと式1の化合物(30mg/kg/日、16日間;PO)の組合せを与えたものとの間では、体重の差異を認めなかった。ドセタキセル投与は、式1の化合物のAUCに影響を及ぼさなかったが、AG−013736のCmax値は、式1の化合物単独と比べて、組合せ群において有意に低下した。
【0100】
選択した組織(肝臓、腎臓、心臓、脾臓、胃、小腸および大腸、卵巣、胸骨、関節)の組織学的検査は、この試験において単剤としての式1の化合物で処理したマウスにおける標的臓器効果を明らかにしなかった。ドセタキセル処理マウスに認められた変化には、卵胞の壊死とごく軽症〜軽症の骨髄低細胞性が含まれた。式1の化合物とドセタキセルの組合せ治療は、卵巣に対するドセタキセルの効果を悪化させなかったが、式1の化合物/ドセタキセル組合せを与えた動物において骨髄低細胞性の強度増加(ごく軽症〜中等症)を認めた。さらに、組合せで処理した動物においては、低細胞性の強度増加の二次効果と見られる、骨髄出血が観察された。
【0101】
式1の化合物とドセタキセルをMDA−MB−231腫瘍モデルにおける効力評価のために組み合わせた。式1の化合物単独(25、5、および1mg/kg,PO,BID,3週間投与)は、用量依存性の腫瘍増殖阻害をもたらした。ドセタキセル単独(IV、週ごと)20および10mg/kgも有効であった(しかし、2mg/kgは無効であった)。式1の化合物とドセタキセルの間には、有益な療法上の相互作用があるように見られた。この利益は、これらの薬剤を高用量および中用量の両方で組み合わせるときにより明らかであった。高用量および中用量の組合せアームでの部分退縮(腫瘍サイズの16%〜97%の低下)および完全応答の出現率は、個々の薬剤単独の同じ用量での群の出現率よりずっと大きかった。この試験の限られた群と比較的短い時間フレームのために、上記のことは決定的な知見ではない。式1の化合物は、すべての用量で十分に耐えられた。高用量の組合せ群(25mg/kgの化合物1と20mg/kgのドセタキセル)では、他のすべての群と比べて、化学療法剤の第三回目の投薬後に、平均体重の3%〜7%の低下があった。薬物動態分析は、式1の化合物のAUC値がドセタキセルの存在下で影響を受けないこと、しかしCmax値は、式1の化合物単独と比較して、組合せ群で有意に低下することを明示した。
【0102】
ドセタキセルとの組合せにおける式1の化合物の抗腫瘍効果をLLCモデルにおいて検討した。LLCモデルは、ドセタキセルに対してきわめて抵抗性である。その細胞傷害剤では、報告されているMTD(30mg/kg,週ごとに投薬、iv)で腫瘍増殖遅延(TGD)がほとんど見られなかった(TGD=3.2日)。すべてのマウスが大きな原発腫瘍により実験の28日以内に安楽死した。対照的に、式1の化合物単剤は、用量依存性で統計学的に有意なTGDをもたらした(10mg/kgで13.4日、30mg/kgで15.4日、PO,BID)。しかしながら、この薬剤は、遅延させただけで、肺への転移を止めることはなかった。低用量組合せ群(TGD=15.2日)ではなく、高用量組合せ群のTGD(20.4日)は、単剤単独のいずれよりも統計学的に異なっていた(それぞれ、P=0.0079およびP=0.254)。高用量組合せ群ではより多くの動物(3/10)が客観的なエンドポイントに達したが、低用量組合せ群では達しなかった。結論として、式1の化合物とドセタキセルの高用量組合せ療法は、いずれの単独療法単独よりも大きい原発腫瘍増殖および転移の遅延をもたらす可能性があるが、完全な治癒をもたらすわけではない。
【0103】
MV522腫瘍モデルを使用する1つの試験は、式1の化合物の1日1回(QD)60mg/kg PO投薬が30mg/kg PO,BIDと同様の腫瘍増殖阻害をもたらす(p=0.154)ことを明示した。さらに、抗腫瘍効果は、30mg/kgで連続5日間PO,BID投薬後に2日間休薬するとき、同じ用量濃度を使用して毎日PO,BID投薬することと比べて低下するようには見えなかった(p=0.223)。これらの結果は、この非臨床腫瘍モデルにおいて、式1の化合物をQDで投与しても、またはある種の臨時投薬スケジュールで投与しても、有意な抗腫瘍効果を達成することを期待してよいことを示唆する。
【0104】
MV522異種移植片モデルにおいて抗腫瘍効果をもたらすのに必要とされる受容体阻害の時間量と式1の化合物の濃度について検討した。この結果は、PO投薬(QDまたはBID)では、50%以上の抗腫瘍効果のためには、ほぼ24時間毎日EC50(5ng/mL)より高く曝露することが必要であることを示した。90%の腫瘍増殖阻害を達成するためには、40〜60ng/mL以上の血漿濃度に毎日少なくとも4時間曝露することが必要であった。上記の閾値を超えた曝露は、さらなる効果を保証しなかった。BIDまたはQD群のいずれでも、同様の体重損失があり、いずれも5%未満であった。従って、適正な用量と曝露時間があれば、QD処方は、BID処方と同じくらい有効であるかもしれない。
【0105】
Alzetポンプを介した連続曝露は、規則的な定期投薬と比べて、式1の化合物によるより大きな抗腫瘍効果をもたらした。このポンプによる10mg/mLでの送達は、30ng/mLの一定した平均全身曝露をもたらし、腫瘍の静止状態を生じた。対照的に、予測EC90に優る式1の化合物の血漿濃度を生じる、飽和量(PO,BID)では、腫瘍増殖遅延をもたらすことができただけである。従って、式1の化合物の連続全身曝露は、腫瘍を治療するのに、1日2回の経口投薬処方より有効であると思われた。
【0106】
間欠性の投薬処方を使用する式1の化合物の抗腫瘍効果も試験した。治療群は、以下の通りであった:担体の連日投薬、担体の間欠投薬、30mg/kg(BID)の連日投薬、および30mg/kgの間欠投薬。間欠投薬のスケジュールは、以下の通りであった:サイクル1(12〜18日目に投薬、19〜28日目に休薬)、およびサイクル2(29〜36日目に投薬、37〜44日目に休薬)。平均腫瘍サイズが250mm3になったときに投薬を開始し;いずれにもAG−013736(PO,BID)を与えた。全体的に見て、間欠および連日のBID投薬の間には有意差があり、連続した連日投薬処方は、増殖遅延をもたらすのにより有効であった。間欠投薬された薬物群では、投薬を中止して3〜4日以内に腫瘍が通常の増殖速度を再び獲得した。しかしながら、サイクル2投薬の2日以内には、腫瘍増殖阻害が再開した。予測されるように、どの群でも退縮は見られなかった。
【0107】
具体的で好ましい態様を参照にして本発明を例示したが、当業者は、定型的な実験と本発明の実践により、種々の変更および改良がなし得ることを理解されよう。従って、本発明は、上記の記載によって限定されず、付帯の特許請求項とその同等物により規定されるものとする。
【図面の簡単な説明】
【0108】
【図1】図1は、14C標識化合物の単回経口投薬に続く、イヌにおいて同定される式1の化合物の代謝産物を示す。
【図2】図2は、14C標識化合物の単回経口投薬に続く、マウスにおいて同定される式1の化合物の代謝産物を示す。
【特許請求の範囲】
【請求項1】
哺乳動物への投与用の剤形であって、式1:
【化1】
の化合物、その医薬的に許容される塩または溶媒和物、またはこれらの混合物を、哺乳動物への投与後に25〜4500ng・時間/mLの式1の化合物またはその活性代謝産物の24時間AUC血漿値を提供するのに有効な量で含んでなる、前記剤形。
【請求項2】
24時間AUC血漿値が100〜800ng・時間/mLである、請求項1の剤形。
【請求項3】
経口剤形である、請求項1の剤形。
【請求項4】
式1:
【化2】
の化合物、その医薬的に許容される塩または溶媒和物、またはこれらの混合物を0.5〜30mgの量で含んでなる剤形。
【請求項5】
量が2〜10mgである、請求項4の剤形。
【請求項6】
経口剤形である、請求項4の剤形。
【請求項7】
異常な細胞増殖を哺乳動物において治療する方法であって、請求項1〜6のいずれかの剤形を哺乳動物へ投与することを含んでなる、前記方法。
【請求項8】
剤形を経口で投与する、請求項7の方法。
【請求項9】
剤形を1日につき少なくとも1回の投与頻度で投与する、請求項7または8の方法。
【請求項10】
剤形を1日につき少なくとも2回の投与頻度で投与する、請求項7または8の方法。
【請求項11】
哺乳動物が、投与の工程前少なくとも2時間、投与の工程後少なくとも2時間、または投与の工程の前後ともに少なくとも2時間絶食する、請求項7〜10のいずれかの方法。
【請求項12】
異常な細胞増殖が癌である、請求項7〜11のいずれかの方法。
【請求項13】
癌が、肺癌、骨癌、膵臓癌、皮膚癌(skin cancer)、頭頚部癌、皮膚(cutaneous)または眼内黒色腫、子宮癌、卵巣癌、直腸癌、肛門域の癌、胃癌、結腸癌、乳癌、卵管の癌、子宮内膜癌、子宮頚部癌、膣癌、外陰部の癌、ホジキン病、食道癌、小腸の癌、内分泌系の癌、甲状腺癌、副甲状腺癌、副腎の癌、柔組織の肉腫、尿道癌、陰茎癌、前立腺癌、慢性または急性白血病、リンパ球性リンパ腫、膀胱癌、腎臓または尿管の癌、腎細胞癌、腎盂癌(carcinoma of the renal pelvis)、中枢神経系(CNS)の新生物、原発性CNSリンパ腫、脊髄軸腫瘍、脳幹神経膠腫、下垂体腺腫、およびこれらの組合せより選択される、請求項12の方法。
【請求項14】
有糸分裂阻害剤、アルキル化剤、代謝拮抗薬、挿入抗生物質、増殖因子阻害剤、細胞周期阻害剤、酵素、トポイソメラーゼ阻害剤、生物学的応答調節剤、抗体、細胞傷害剤、抗ホルモン剤、抗アンドロゲン剤、およびこれらの混合物からなる群より選択される抗腫瘍剤を同時投与することをさらに含む、請求項7〜13のいずれかの方法。
【請求項15】
抗腫瘍剤がドセタキセルである、請求項14の方法。
【特許請求の範囲】
【請求項1】
哺乳動物への投与用の剤形であって、式1:
【化1】
の化合物、その医薬的に許容される塩または溶媒和物、またはこれらの混合物を、哺乳動物への投与後に25〜4500ng・時間/mLの式1の化合物またはその活性代謝産物の24時間AUC血漿値を提供するのに有効な量で含んでなる、前記剤形。
【請求項2】
24時間AUC血漿値が100〜800ng・時間/mLである、請求項1の剤形。
【請求項3】
経口剤形である、請求項1の剤形。
【請求項4】
式1:
【化2】
の化合物、その医薬的に許容される塩または溶媒和物、またはこれらの混合物を0.5〜30mgの量で含んでなる剤形。
【請求項5】
量が2〜10mgである、請求項4の剤形。
【請求項6】
経口剤形である、請求項4の剤形。
【請求項7】
異常な細胞増殖を哺乳動物において治療するための、請求項1〜6のいずれかの剤形。
【請求項8】
剤形を経口で投与する、請求項7の剤形。
【請求項9】
剤形を1日につき少なくとも1回の投与頻度で投与する、請求項7または8の剤形。
【請求項10】
剤形を1日につき少なくとも2回の投与頻度で投与する、請求項7または8の剤形。
【請求項11】
哺乳動物が、投与の工程前少なくとも2時間、投与の工程後少なくとも2時間、または投与の工程の前後ともに少なくとも2時間絶食する、請求項7〜10のいずれかの剤形。
【請求項12】
異常な細胞増殖が癌である、請求項7〜11のいずれかの剤形。
【請求項13】
癌が、肺癌、骨癌、膵臓癌、皮膚癌(skin cancer)、頭頚部癌、皮膚(cutaneous)または眼内黒色腫、子宮癌、卵巣癌、直腸癌、肛門域の癌、胃癌、結腸癌、乳癌、卵管の癌、子宮内膜癌、子宮頚部癌、膣癌、外陰部の癌、ホジキン病、食道癌、小腸の癌、内分泌系の癌、甲状腺癌、副甲状腺癌、副腎の癌、柔組織の肉腫、尿道癌、陰茎癌、前立腺癌、慢性または急性白血病、リンパ球性リンパ腫、膀胱癌、腎臓または尿管の癌、腎細胞癌、腎盂癌(carcinoma of the renal pelvis)、中枢神経系(CNS)の新生物、原発性CNSリンパ腫、脊髄軸腫瘍、脳幹神経膠腫、下垂体腺腫、およびこれらの組合せより選択される、請求項12の剤形。
【請求項14】
有糸分裂阻害剤、アルキル化剤、代謝拮抗薬、挿入抗生物質、増殖因子阻害剤、細胞周期阻害剤、酵素、トポイソメラーゼ阻害剤、生物学的応答調節剤、抗体、細胞傷害剤、抗ホルモン剤、抗アンドロゲン剤、およびこれらの混合物からなる群より選択される抗腫瘍剤を同時投与することを特徴とする、請求項7〜13のいずれかの剤形。
【請求項15】
抗腫瘍剤がドセタキセルである、請求項14の剤形。
【請求項1】
哺乳動物への投与用の剤形であって、式1:
【化1】
の化合物、その医薬的に許容される塩または溶媒和物、またはこれらの混合物を、哺乳動物への投与後に25〜4500ng・時間/mLの式1の化合物またはその活性代謝産物の24時間AUC血漿値を提供するのに有効な量で含んでなる、前記剤形。
【請求項2】
24時間AUC血漿値が100〜800ng・時間/mLである、請求項1の剤形。
【請求項3】
経口剤形である、請求項1の剤形。
【請求項4】
式1:
【化2】
の化合物、その医薬的に許容される塩または溶媒和物、またはこれらの混合物を0.5〜30mgの量で含んでなる剤形。
【請求項5】
量が2〜10mgである、請求項4の剤形。
【請求項6】
経口剤形である、請求項4の剤形。
【請求項7】
異常な細胞増殖を哺乳動物において治療する方法であって、請求項1〜6のいずれかの剤形を哺乳動物へ投与することを含んでなる、前記方法。
【請求項8】
剤形を経口で投与する、請求項7の方法。
【請求項9】
剤形を1日につき少なくとも1回の投与頻度で投与する、請求項7または8の方法。
【請求項10】
剤形を1日につき少なくとも2回の投与頻度で投与する、請求項7または8の方法。
【請求項11】
哺乳動物が、投与の工程前少なくとも2時間、投与の工程後少なくとも2時間、または投与の工程の前後ともに少なくとも2時間絶食する、請求項7〜10のいずれかの方法。
【請求項12】
異常な細胞増殖が癌である、請求項7〜11のいずれかの方法。
【請求項13】
癌が、肺癌、骨癌、膵臓癌、皮膚癌(skin cancer)、頭頚部癌、皮膚(cutaneous)または眼内黒色腫、子宮癌、卵巣癌、直腸癌、肛門域の癌、胃癌、結腸癌、乳癌、卵管の癌、子宮内膜癌、子宮頚部癌、膣癌、外陰部の癌、ホジキン病、食道癌、小腸の癌、内分泌系の癌、甲状腺癌、副甲状腺癌、副腎の癌、柔組織の肉腫、尿道癌、陰茎癌、前立腺癌、慢性または急性白血病、リンパ球性リンパ腫、膀胱癌、腎臓または尿管の癌、腎細胞癌、腎盂癌(carcinoma of the renal pelvis)、中枢神経系(CNS)の新生物、原発性CNSリンパ腫、脊髄軸腫瘍、脳幹神経膠腫、下垂体腺腫、およびこれらの組合せより選択される、請求項12の方法。
【請求項14】
有糸分裂阻害剤、アルキル化剤、代謝拮抗薬、挿入抗生物質、増殖因子阻害剤、細胞周期阻害剤、酵素、トポイソメラーゼ阻害剤、生物学的応答調節剤、抗体、細胞傷害剤、抗ホルモン剤、抗アンドロゲン剤、およびこれらの混合物からなる群より選択される抗腫瘍剤を同時投与することをさらに含む、請求項7〜13のいずれかの方法。
【請求項15】
抗腫瘍剤がドセタキセルである、請求項14の方法。
【特許請求の範囲】
【請求項1】
哺乳動物への投与用の剤形であって、式1:
【化1】
の化合物、その医薬的に許容される塩または溶媒和物、またはこれらの混合物を、哺乳動物への投与後に25〜4500ng・時間/mLの式1の化合物またはその活性代謝産物の24時間AUC血漿値を提供するのに有効な量で含んでなる、前記剤形。
【請求項2】
24時間AUC血漿値が100〜800ng・時間/mLである、請求項1の剤形。
【請求項3】
経口剤形である、請求項1の剤形。
【請求項4】
式1:
【化2】
の化合物、その医薬的に許容される塩または溶媒和物、またはこれらの混合物を0.5〜30mgの量で含んでなる剤形。
【請求項5】
量が2〜10mgである、請求項4の剤形。
【請求項6】
経口剤形である、請求項4の剤形。
【請求項7】
異常な細胞増殖を哺乳動物において治療するための、請求項1〜6のいずれかの剤形。
【請求項8】
剤形を経口で投与する、請求項7の剤形。
【請求項9】
剤形を1日につき少なくとも1回の投与頻度で投与する、請求項7または8の剤形。
【請求項10】
剤形を1日につき少なくとも2回の投与頻度で投与する、請求項7または8の剤形。
【請求項11】
哺乳動物が、投与の工程前少なくとも2時間、投与の工程後少なくとも2時間、または投与の工程の前後ともに少なくとも2時間絶食する、請求項7〜10のいずれかの剤形。
【請求項12】
異常な細胞増殖が癌である、請求項7〜11のいずれかの剤形。
【請求項13】
癌が、肺癌、骨癌、膵臓癌、皮膚癌(skin cancer)、頭頚部癌、皮膚(cutaneous)または眼内黒色腫、子宮癌、卵巣癌、直腸癌、肛門域の癌、胃癌、結腸癌、乳癌、卵管の癌、子宮内膜癌、子宮頚部癌、膣癌、外陰部の癌、ホジキン病、食道癌、小腸の癌、内分泌系の癌、甲状腺癌、副甲状腺癌、副腎の癌、柔組織の肉腫、尿道癌、陰茎癌、前立腺癌、慢性または急性白血病、リンパ球性リンパ腫、膀胱癌、腎臓または尿管の癌、腎細胞癌、腎盂癌(carcinoma of the renal pelvis)、中枢神経系(CNS)の新生物、原発性CNSリンパ腫、脊髄軸腫瘍、脳幹神経膠腫、下垂体腺腫、およびこれらの組合せより選択される、請求項12の剤形。
【請求項14】
有糸分裂阻害剤、アルキル化剤、代謝拮抗薬、挿入抗生物質、増殖因子阻害剤、細胞周期阻害剤、酵素、トポイソメラーゼ阻害剤、生物学的応答調節剤、抗体、細胞傷害剤、抗ホルモン剤、抗アンドロゲン剤、およびこれらの混合物からなる群より選択される抗腫瘍剤を同時投与することを特徴とする、請求項7〜13のいずれかの剤形。
【請求項15】
抗腫瘍剤がドセタキセルである、請求項14の剤形。
【図1】

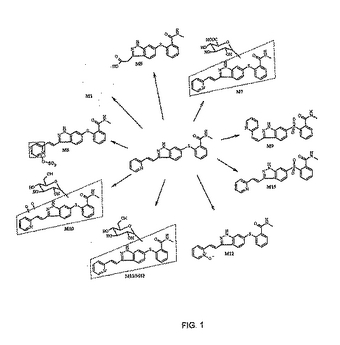
【図2】
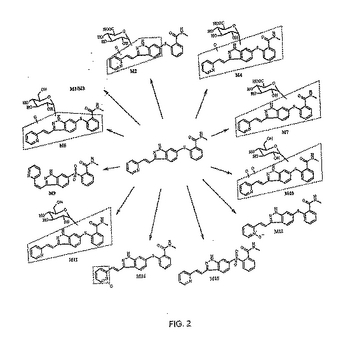
【図2】
【公表番号】特表2006−522087(P2006−522087A)
【公表日】平成18年9月28日(2006.9.28)
【国際特許分類】
【出願番号】特願2006−506378(P2006−506378)
【出願日】平成16年3月17日(2004.3.17)
【国際出願番号】PCT/IB2004/000867
【国際公開番号】WO2004/087152
【国際公開日】平成16年10月14日(2004.10.14)
【出願人】(593141953)ファイザー・インク (302)
【Fターム(参考)】
【公表日】平成18年9月28日(2006.9.28)
【国際特許分類】
【出願日】平成16年3月17日(2004.3.17)
【国際出願番号】PCT/IB2004/000867
【国際公開番号】WO2004/087152
【国際公開日】平成16年10月14日(2004.10.14)
【出願人】(593141953)ファイザー・インク (302)
【Fターム(参考)】
[ Back to top ]