
B型肝炎ウイルス表面抗原および界面活性剤の両方を含むワクチンの製造
【課題】非イオン性界面活性剤を含有する組み合わせワクチン用に、製造方法において界面活性剤を別の成分として添加する必要がない方法を提供する。
【解決手段】組み合わせワクチンにおいて使用するためのHBsAgを調製する際に、HBsAgを精製した後に非イオン性界面活性剤を添加することは知られている。しかし、HBsAgの精製後に界面活性剤を添加することは最適ではない。それは、製造中に別の処理ステップを必要とするからである。したがって本発明は、HBsAgの精製中に界面活性剤を使用する。
【解決手段】組み合わせワクチンにおいて使用するためのHBsAgを調製する際に、HBsAgを精製した後に非イオン性界面活性剤を添加することは知られている。しかし、HBsAgの精製後に界面活性剤を添加することは最適ではない。それは、製造中に別の処理ステップを必要とするからである。したがって本発明は、HBsAgの精製中に界面活性剤を使用する。
【発明の詳細な説明】
【技術分野】
【0001】
本明細書で引用するすべての文書は、その全容を参照によって組み込む。
【0002】
本発明は、2つ以上の病原体由来の混合型免疫原を含むワクチンであり、したがってそのワクチンの投与が2つ以上の病原体に対して対象を同時に免疫処置することができる、組み合わせワクチンの製造の分野にある。詳細には本発明は、組み合わせワクチンの製造における界面活性剤の使用に関する。
【背景技術】
【0003】
2種以上の病原性生物由来の抗原を単一用量中に含むワクチンは、「多価」または「混合」ワクチンとして知られている。ジフテリア、破傷風および百日咳に対する防御用の3価ワクチン(「DTP」ワクチン)ならびに麻疹、おたふく風邪および風疹に対する防御用の3価ワクチン(「MMR」ワクチン)を含めた、ヒトに使用するための様々な組み合わせワクチンが欧州および米国において承認されている。
【0004】
組み合わせワクチンには患者にとって受ける注射の回数が少なくなるという利点があり、特に小児予防接種に関して、これはコンプライアンスが高まるという臨床上の利点をもたらす(例えば、参考文献1の29章を参照)。しかし同時に、組み合わせワクチンは、抗原と他の成分の間の物理的および生化学的不適合性、免疫学的干渉、および安定性を含めた要因による製造上の難点を示す。
【0005】
ワクチンに非抗原成分を含有させることは必要であるが、困難を引き起こす可能性がある。界面活性剤は組み合わせワクチンにおいて特に問題である。それは、ある抗原は最適活性のために界面活性剤を必要とする可能性があるが、別の抗原は界面活性剤が存在すると負の影響を受ける可能性があるからである。さらに、界面活性剤が一般に安全であると認められていても、小児ワクチンに界面活性剤を含有させることはある患者群にとって問題である。
【0006】
ワクチン分野で特に興味深いのは、ポリオキシエチレンソルビタンエステル界面活性剤、特にポリソルベート20(「Tween20」またはポリオキシエチレンソルビタンモノラウレートとしても知られている)およびポリソルベート80(「Tween80」またはポリオキシエチレンソルビタンモノオレエートとしても知られている)種である。ポリソルベート20は1価HAVRIX(商標)A型肝炎ワクチンにおいて見られ、ポリソルベート80はTRIPEDIA(商標)およびINFANRIX(商標)系のワクチンなどの組み合わせワクチンにおいて見られる。これら2つの界面活性剤は、液状ロタウイルスワクチンを安定化させるためにも使用されている[2]。
【0007】
ポリソルベートはB型肝炎表面抗原(「HBsAg」)を含む組み合わせワクチンの製造においても使用されており、例えば参考文献3(特許文献1)および4(特許文献2)は、Tween20、Tween80またはTriton X−100などの非イオン性界面活性剤を添加することによってHBsAgのリン脂質成分への干渉が回避されている、4価D−T−P−HBsAgワクチンを作製するための方法を開示する。参考文献3(特許文献1)および4(特許文献2)の図2(本明細書の図1)中のデータから、界面活性剤はHBsAgの抗原性を維持するために必要とされるが、他の成分にはそれほど重要でないことが示される。試験された最高界面活性剤濃度はHBsAg20μg/mlに対して10μg/mlであり、この濃度はまた、最良の抗原性を示した。
【0008】
参考文献3(特許文献1)および4(特許文献2)の方法では、HBsAgを精製した後に非イオン性界面活性剤が添加される。しかし、HBsAgの精製後に界面活性剤を添加することは最適ではない。それは、製造中に別の処理ステップを必要とし、それにより処理時間が増大し、HBsAgが汚染されるリスクも高まるからである。組み合わせワクチンの作製時に汚染成分が使用されると、1価ワクチンの作製時より最終的な損失は大きく、例えば汚染HBsAg成分が清潔なD−T−P成分と混合されると、HBsAgのみでなくD−T−P−HBsAg混合物全体を廃棄しなければならない。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】国際公開第02/055105号パンフレット
【特許文献2】米国特許出願公開第2004/0048336号明細書
【発明の概要】
【発明が解決しようとする課題】
【0010】
したがって、非イオン性界面活性剤を含有する組み合わせワクチン用に、製造方法において界面活性剤を別の成分として添加する必要がない方法が依然として必要とされている。
【課題を解決するための手段】
【0011】
(要旨)
抗原を精製した後に非イオン性界面活性剤を抗原に添加する[3、4]のではなく、本発明は抗原の精製中に非イオン性界面活性剤を使用する。したがって、この界面活性剤は最終組み合わせワクチンにおいてその機能を果たすことができるが、汚染のリスク(したがって、調製後の組み合わせワクチン全体の損失のリスクも)は低下する。
【0012】
したがって本発明は、組み合わせワクチンを調製するための方法であって、ワクチンが(i)非イオン性界面活性剤、(ii)B型肝炎ウイルス(HBV)表面抗原、および(iii)少なくとも1つの非HBV病原体由来の抗原を含み、(i)組換え酵母細胞を非イオン性界面活性剤の存在下で破壊して精製HBsAg成分を得るステップを含む、酵母細胞からHBV表面抗原を精製すること、および(ii)精製HBsAg成分を非HBV病原体由来の少なくとも1つの他の抗原と合わせて組み合わせワクチンを得ることを含む方法を提供する。
【0013】
前に記載した汚染の難点を回避するために、この方法は、HBsAg精製後に非イオン性界面活性剤を別の成分として添加するステップを含まない。界面活性剤(または他の界面活性剤、イオン性または非イオン性)が、組み合わせワクチンを得るためにHBsAgと組み合わせる他の抗原成分中に存在する可能性はあるが、それ自体を別の成分として界面活性剤を添加することは回避される。したがって、界面活性剤は、別の成分として精製HBsAg成分に添加されず、抗原を組み合わせる際には添加されない。
【0014】
本発明は、組み合わせワクチンを調製するための方法であって、ワクチンが(i)非イオン性界面活性剤、(ii)B型肝炎ウイルス(HBV)表面抗原、および(iii)少なくとも1つの非HBV病原体由来の抗原を含み、精製HBV表面抗原を非HBV病原体由来の少なくとも1つの他の抗原と合わせて組み合わせワクチンを得るステップを含み、HBsAg発現組換え酵母細胞を非イオン性界面活性剤の存在下で破壊するプロセスによって精製HBV表面抗原が調製される方法も提供する。再度、界面活性剤の別個の添加は回避される。
【0015】
本発明は、(i)非イオン性界面活性剤、(ii)B型肝炎ウイルス(HBV)表面抗原、および(iii)少なくとも1つの非HBV病原体由来の抗原を含み、HBsAg発現組換え酵母細胞を非イオン性界面活性剤の存在下で破壊するプロセスによってHBV表面抗原が調製される、免疫原性組成物も提供する。再度、界面活性剤の別個の添加を回避しながらHBsAgを調製した。この生成物は、精製中に使用する非イオン性界面活性剤がHBsAg粒子中に保持される可能性があるので、異なる方法によってHBsAgを調製した生成物と区別することができる。
【発明を実施するための形態】
【0016】
非イオン性界面活性剤
本発明は、様々な非イオン性界面活性剤[5]、詳細にはワクチン製剤において見られる非イオン性界面活性剤を使用することができる。有機界面活性剤が好ましい。これらは典型的には、アルキレンオキシド(例えば、エチレンオキシド)と、高級アルコール、脂肪酸、アルキルフェノール、アルキルアミン、または少なくとも1つの活性水素原子を有する他の適切な化合物との反応生成物である。大部分の界面活性剤に関して、最も一般的なアルコール、アミンおよび酸はC8〜C18の範囲の炭素鎖長を有する。最も一般的なアルキルフェノールは、ノニルフェノールおよびオクチルフェノールである。ポリ(オキシエテン)残基を含む界面活性剤が特に好ましい。
【0017】
例えば、ポリオキシエチレンソルビタンエステル界面活性剤(一般にTweenと呼ばれる)、特にポリソルベート20およびポリソルベート80;DOWFAX(商標)の商品名で販売されているエチレンオキシド(EO)、プロピレンオキシド(PO)、および/またはブチレンオキシド(BO)のコポリマー、例えば直鎖状EO/POブロックコポリマー;反復エトキシ(オキシ−1,2−エタンジイル)基の数が変わり得るオクトキシノール、オクトキシノール−9(Triton X−100、またはt−オクチルフェノキシポリエトキシエタノール)が特に興味深い;(オクチルフェノキシ)ポリエトキシエタノール(IGEPAL CA−630/NP−40);ラウリル、セチル、ステアリルおよびオレイルアルコール(Brij界面活性剤として知られる)から誘導されるポリオキシエチレン脂肪族エーテル、例えばトリエチレングリコールモノラウリルエーテル(Brij30);およびソルビタンエステル(一般にSPANと呼ばれる)、例えばソルビタントリオレート(Span85)およびソルビタンモノラウレートだけには限られないが、これらを含めた界面活性剤と共に本発明を使用することができる。
【0018】
本発明は、ポリソルベート20と共に使用するのに特に適している。この界面活性剤は、ワクチン内を含めてヒトへの投与に関する立証済みの安全性プロファイルを有する。
【0019】
界面活性剤はその「HLB」(親水/親油平衡)によって分類することができる。本発明の好ましい界面活性剤は、少なくとも10、好ましくは少なくとも15、およびより好ましくは少なくとも16のHLBを有する。
【0020】
非イオン性界面活性剤は、本発明の組成物の成分である。患者への多用量の界面活性剤の投与を回避するために、組成物中の界面活性剤の濃度は30μg/mlを超えず、例えば25μg/ml以下、20μg/ml以下、15μg/ml以下、10μg/ml以下、5μg/ml以下などであることが好ましい。10μg/ml以下の濃度が好ましい。
【0021】
界面活性剤の濃度を特定するための代替として、組成物中の界面活性剤の量は、HBsAg各100μgに対して50μg未満(例えば40μg以下、30μg以下、25μg以下、20μg以下、15μg以下、10μg以下など)であることが好ましい。参考文献3および4から、界面活性剤:HBsAgの質量比が50%未満であることも示唆される。HBsAg100μg当たり25μg未満の界面活性剤が好ましい。
【0022】
以下でさらに詳細に言及するように、本発明の好ましい方法は、ジフテリアトキソイドおよび破傷風トキソイドを含む予め混合した成分を使用する。このD−T成分は非イオン性界面活性剤を実質的に含まず、および詳細にはポリソルベート20および80を含まないことが好ましい。同様に、予め混合したD−T−Pw成分は非イオン性界面活性剤、例えばポリソルベート20および80を含まない。
【0023】
B型肝炎ウイルス表面抗原
B型肝炎ウイルス(HBV)は、ウイルス肝炎を引き起こす既知の因子の1つである。HBVビリオンは外側タンパク質コートまたはカプシドに囲まれた内部コアからなり、ウイルスコアはウイルスDNAゲノムを含む。カプシドの主な成分は、典型的には約24kDaの分子量を有する226アミノ酸ポリペプチドである、HBV表面抗原、またはより一般的に「HBsAg」として知られるタンパク質である。HBsAgを含むすべての既存のB型肝炎ワクチンは、この抗原を通常なワクチンに投与すると、それはHBV感染に対して防御する抗HBsAg抗体の産生を刺激する。
【0024】
ワクチン製造用に、HBsAgは2つの方法で作製することができる。第1の方法は、HBV感染中に多量のHBsAgが肝臓中で合成され血流中に放出されるので、慢性B型肝炎キャリアの血漿から粒子形に抗原を精製することを含む。第2の方法は、組換えDNA法によってタンパク質を発現させることを含む。本発明の方法と共に使用するためのHBsAgは、酵母細胞中で組換えによって発現される。適切な酵母菌には、サッカロミセス属(出芽酵母(S.cerevisiae)など)またはハネンズラ属(Hanensula)(H.ポリモルファ(H.polymorpha)など)宿主がある。
【0025】
天然HBsAgと異なり(すなわち、血漿精製産物中と同様に)、酵母菌によって発現されるHBsAgは一般にグリコシル化されておらず、これは本発明と共に使用するためのHBsAgの最も好ましい形態である。酵母菌によって発現されるHBsAgは非常に免疫原性が高く、血液産物の汚染のリスクなしで調製することができる。
【0026】
HBsAgは一般に、リン脂質を含む脂質マトリクスを含む、実質的に球状の粒子の形(約20nmの平均径)であるはずである。酵母菌によって発現されるHBsAg粒子は、天然のHBVビリオン中に見られないホスファチジルイノシトールを含み得る。粒子は免疫系を刺激するために無毒量のLPSも含み得る[6]。
【0027】
HBsAgは、HBV亜型adw2に由来することが好ましい。
【0028】
HBsAgを精製するための多くの方法が当技術分野で知られている(例えば、参考文献7〜33を参照)。これらの方法は1価HBsAg調製物を作製する際に使用するために開示されるが、参考文献3および4中に開示された方法と異なり、これらはいずれも、特に組み合わせワクチンにおいて使用するためのHBsAgの精製に関するものではない。これらの方法および他の方法のいずれかを使用することができ、ただし、この方法は、組換え酵母細胞中での発現後の抗原を精製するのに適しており、この精製は、酵母細胞を非イオン性界面活性剤の存在下で破壊するステップを含む。
【0029】
HBsAgを精製するのに好ましい方法は、細胞破壊後の、限外濾過、サイズ排除クロマトグラフィー、アニオン交換クロマトグラフィー、超遠心分離、脱塩および滅菌濾過を含む。溶解物は(例えば、ポリエチレングリコールを使用して)細胞破壊後に沈殿させ、HBsAgを限外濾過用に溶液中に残しておいてよい。
【0030】
精製後HBsAgに透析を施すことができ(例えばシステインと)、これを使用して、HBsAg調製中に使用された可能性があるチメロサールなどの任意の水銀防腐剤を除去することができる[30、34]。
【0031】
HBsAgの量は典型的にはマイクログラムで表され、ワクチン用量当たりのHBsAgの典型的な量は10μgである。
【0032】
「S」配列以外に、表面抗原はプレ−S配列の全体または一部分、例えばプレ−S1および/またはプレ−S2配列の全体または一部分を含むことができる。
【0033】
非HBV抗原
本発明の免疫原性組成物は、少なくとも1つの非HBV病原体由来の少なくとも1つの防御抗原を含む。非HBV病原体はウイルス性および/または細菌性であってよい。
【0034】
典型的なウイルス性病原体には、ポリオウイルス、A型肝炎ウイルス、インフルエンザウイルス、麻疹ウイルス、おたふく風邪ウイルス、風疹ウイルスおよび水痘帯状疱疹ウイルスがあるが、これらだけには限られない。
【0035】
典型的な細菌性病原体には、ジフテリア菌(Corynebacterium diphtheriae);破傷風菌(Clostridium tetani);百日咳菌(Bordetella pertussis);b型および非分類系統を含めたインフルエンザ菌(Haemophilus influenzae);血清型A、B、C、W135および/またはYを含めた髄膜炎菌(Neisseria meningitidis);血清型4、6B、9V、14、18C、19F、および23Fを含めた肺炎連鎖球菌(Streptococcus pneumoniae);およびカタラリス菌(Moraxella catarrhalis)があるが、これらだけには限られない。
【0036】
ジフテリア菌はジフテリアを引き起こす。注射後に特異的抗トキシン抗体を誘導する能力を保持しながら、ジフテリア毒素を(例えば、ホルマリンまたはホルムアルデヒドを使用して)処理して毒性を除去することができる。これらのジフテリアトキソイドはジフテリアワクチンにおいて使用され、参考文献1の13章中により詳細に開示される。好ましいジフテリアトキソイドは、ホルムアルデヒド処理によって調製されるジフテリアトキソイドである。ジフテリアトキソイドは増殖培地(例えば、Fenton培地、またはLinggoudおよびFenton培地)中でジフテリア菌を増殖させることによって得ることができ、培地にウシ抽出物を補充することが可能であり、後にホルムアルデヒド処理、限外濾過および沈殿が可能である。トキソイド物質は、滅菌濾過および/または透析を含む方法によって次いで処理することができる。
【0037】
破傷風菌は破傷風を引き起こす。破傷風毒素を処理して防御トキソイドを与えることができる。これらのトキソイドは破傷風ワクチンにおいて使用され、参考文献1の27章中により詳細に開示される。好ましい破傷風トキソイドは、ホルムアルデヒド処理によって調製される破傷風トキソイドである。破傷風トキソイドは増殖培地(例えば、ウシカゼイン由来のLatham培地)中で破傷風菌を増殖させることによって得ることができ、後にホルムアルデヒド処理、限外濾過および沈殿が可能である。滅菌濾過および/または透析を含む方法によって、次いで物質を処理することができる。
【0038】
百日咳菌は百日咳を引き起こす。ワクチン中の百日咳抗原は、細胞性(完全細胞、不活性百日咳菌細胞の形;「wP」)あるいは無細胞(「aP」)のいずれかである。細胞性百日咳抗原の調製は充分に文書化されており[例えば、参考文献1の21章を参照]、例えば、それは培養第I期の百日咳菌の熱不活化によって得ることができる。無細胞抗原を使用する場合、1つ、2つまたは(好ましくは)3つの以下の抗原が含まれる:(1)解毒した百日咳毒素(百日咳トキソイド、すなわち「PT」);(2)繊維状ヘマグルチニン(「FHA」);(3)ペルタクチン(「69キロダルトンの外膜タンパク質」としても知られている)。この3つの抗原は、改変型Stainer−Scholte液状培地中で増殖させた百日咳菌培養物からの単離によって調製することが好ましい。PTおよびFHAは発酵ブロスから単離することができ(例えば、ヒドロキシアパタイトゲル上での吸着によって)、一方ペルタクチンは、熱処理および凝集反応(例えば、塩化バリウムを使用する)によって細胞からから抽出することができる。抗原は連続的なクロマトグラフィーおよび/または沈殿ステップにおいて精製することができる。PTおよびFHAは疎水性クロマトグラフィー、親和性クロマトグラフィーおよびサイズ排除クロマトグラフィーによって精製することができる。ペルタクチンはイオン交換クロマトグラフィー、疎水性クロマトグラフィーおよびサイズ排除クロマトグラフィーによって精製することができる。FHAおよびペルタクチンは、本発明による使用の前にホルムアルデヒドで処理することができる。ホルムアルデヒドおよび/またはグルタルアルデヒドを用いた処理によってPTを解毒することが好ましい。この化学的解毒手順に対する代替として、PTは突然変異誘発によって酵素活性が低下した突然変異PTであってよいが[35]、化学的処理による解毒が好ましい。
【0039】
b型インフルエンザ菌(「Hib」)は細菌性髄膜炎を引き起こす。Hibワクチンは典型的には被膜糖抗原に基づいており[例えば、参考文献1の14章]、その調製は充分に文書化されている[例えば、参考文献36〜45]。Hib糖は担体タンパク質と結合して、特に子供においてその免疫原性を増大させる。典型的な担体タンパク質は、破傷風トキソイド、ジフテリアトキソイド、ジフテリアトキソイドのCRM197誘導体、インフルエンザ菌タンパク質D、および血清型Bの髄膜炎菌由来の外膜タンパク質複合体である。破傷風トキソイドは、「PRP−T」と一般に呼ばれる生成物において使用される、好ましい担体である。臭化シアンを使用してHib被膜多糖を活性化させ、活性糖をアジピン酸リンカー((1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミドなど)、典型的には塩酸塩)と結合させ、次いでリンカー−糖物体と破傷風トキソイド担体タンパク質を反応させることによってPRP−Tを作製することができる。複合体の糖部分は、Hib細菌から調製した完全長のポリリボシルリビトールリン酸(PRP)、および/または完全長PRPの断片を含み得る。1:5(すなわち、過剰なタンパク質)と5:1(すなわち、過剰な糖)の間の糖:タンパク質比(w/w)、例えば1:2と5:1の間の比、および1:1.25と1:2.5の間の比を有する複合体を使用することができる。好ましいワクチンでは、しかし、糖と担体タンパク質の重量比は1:2.5と1:3.5の間である。破傷風トキソイドが抗原および担体タンパク質の両方として存在するワクチンでは、その時複合体中の糖と担体タンパク質の重量比は1:0.3と1:2の間である可能性がある[46]。Hib複合体の投与は0.15μg/ml以上、およびより好ましくは1μg/ml以上の抗PRP抗体濃度をもたらすことが好ましく、これらは標準的な応答閾値である。
【0040】
髄膜炎菌は細菌性髄膜炎を引き起こす。生物の被膜多糖に基づいて、A、B、C、H、I、K、L、29E、W135、X、YおよびZを含めたN.meningitidisの様々な血清型が同定されてきている。疾患と最も関係がある血清型は、A、B、C、W135およびYである。血清型A、C、W135およびYに対する現在のワクチンは被膜糖抗原に基づくものであるが、この手法は血清型Bには適しておらず、したがってタンパク質抗原および外膜小胞を代わりに使用する。被膜糖は担体タンパク質と結合して免疫原性を増大させる。典型的な担体タンパク質は、破傷風トキソイド、ジフテリアトキソイド、ジフテリアトキソイドのCRM197誘導体、およびインフルエンザ菌タンパク質Dである。複合体の糖部分は、髄膜炎菌から調製した完全長の糖、および/またはその断片を含み得る。血清型Cの糖は、OAc+またはOAc−系統のいずれかから調製することができる。血清型Aの糖に関しては、少なくとも50%(例えば少なくとも60%、70%、80%、90%、95%以上)のマンノースアミン残基が、C−3位置においてO−アセチル化されることが好ましい。1:10(すなわち、過剰なタンパク質)と10:1(すなわち、過剰な糖)の間の糖:タンパク質比(w/w)、例えば1:5と5:1の間、1:2.5と2.5:1の間の比、または1:1.25と1.25:1の間の比を有する髄膜炎菌複合体を使用することができる。複合体の投与によって、少なくとも4倍、および好ましくは少なくとも8倍の関連する血清型の血清細菌アッセイ(SBA)における力価の増大をもたらすことが好ましい。SBAにおける力価は、ベビーラビット補体またはヒト補体を使用して測定することができる[47]。
【0041】
肺炎連鎖球菌は細菌性髄膜炎を引き起こす。Hibおよび髄膜炎菌に関しては、既存のワクチンは被膜糖に基づく。肺炎連鎖球菌の2つ以上の血清型、および詳細には少なくとも血清型6B、14、19Fおよび23F由来の糖を含むことが好ましい。他の血清型は1、3、4、5、7F、9Vおよび18Cから選択されることが好ましい。例えば、23の異なる血清型に由来する多糖の混合物が広く使用されており、5〜11の異なる血清型に由来する多糖を含む複合体ワクチンも同様である[48]。例えば、PrevNar(商標)[49]は7つの血清型(4、6B、9V、14、18C、19F、および23F)に由来する複合体抗原を含む。糖は担体タンパク質と結合することが好ましい[例えば、参考文献50〜52]。典型的な担体タンパク質は、破傷風トキソイド、ジフテリアトキソイド、ジフテリアトキソイドのCRM197誘導体、およびインフルエンザ菌タンパク質Dである。PrevNar(商標)製品中の糖は、0.5ml用量当たり2μgの各糖(4μgの血清型6B)で、還元的アミノ化によってCRM197と個別に結合する。肺炎球菌に由来する糖抗原の使用に対する代替として、組成物は1つまたは複数のポリペプチド抗原を含むことができる。数系統の肺炎球菌のゲノム配列が利用可能であり[53、54]、これらは逆ワクチン法に供して[55〜58]、適切なポリペプチド抗原を同定することができる[59、60]。例えば組成物は、参考文献61中に定義されるように、以下の抗原:PhtA、PhtD、PhtB、PhtE、SpsA、LytB、LytC、LytA、Sp125、Sp101、Sp128、Sp130およびSp130の1つまたは複数を含むことができる。組成物は、2つ以上(例えば2、3、4、5、6、7、8、9、10、11、12、13または14個の)これらの抗原を含むことができる。いくつかの実施形態では、組成物は肺炎球菌に由来する糖とポリペプチド抗原の両方を含むことができる。これらは単純な混合物において使用することができ、あるいは肺炎球菌糖抗原は肺炎球菌タンパク質と結合させることが可能である。このような実施形態に適した担体タンパク質には、前述の肺炎球菌タンパク質抗原がある[61]。
【0042】
モラクセラカタラーリスは中耳炎および副鼻腔炎を引き起こし、時折咽頭炎の原因となる。参考文献62中に総説されるように、ワクチンは現在研究中である。
【0043】
HBVと同様に、HAVは肝炎を引き起こす。HAVワクチンは参考文献1の15章中に開示されている。好ましいHAV成分は不活性ウイルスに基づき、不活性化はホルマリン処理によって実施することができる。ウイルスはMRC−5細胞などのヒト胎児肺2倍体線維芽細胞において増殖し得る。好ましいHAV系統はHM175であるが、CR326Fを使用することもできる。ウイルス増殖を可能にする条件下で、細胞を増殖させることが可能である。細胞を溶解し、生成した懸濁液は限外濾過およびゲル透過クロマトグラフィーによって精製することができる。
【0044】
ポリオウイルスは灰白髄炎を引き起こす。経口ポリオウイルスワクチンを使用する代わりに、参考文献1の24章中により詳細に開示するように、本発明は不活化ポリオウイルスワクチン(IPV)を使用する。細胞培養中にポリオウイルスを増殖させることが可能であり、好ましい培養はサル腎臓に由来するVero細胞系を使用する。Vero細胞は、都合良くマイクロキャリア培養することができる。増殖後、限外濾過、ダイアフィルトレーション、およびクロマトグラフィーなどの技法を使用してビリオンを精製することができる。患者に投与する前に、ポリオウイルスは不活化しなければならず、これはホルムアルデヒドを用いた処理によって実施することができる。灰白髄炎は、ポリオウイルスの3つの型の1つによって引き起こされる可能性がある。3つの型は類似しており同一の症状を引き起こすが、これらは抗原的に非常に異なり、1つの型による感染は他の型による感染に対して防御しない。したがって、本発明では3つのポリオウイルス抗原:ポリオウイルス1型(例えば、Mahoney系統)、ポリオウイルス2型(例えば、MEF−1系統)、およびポリオウイルス3型(例えば、Saukett系統)を使用することが好ましい。これらのウイルスは個別に増殖、精製および不活化し、次いで組み合わせて本発明と共に使用するための多量の3価混合物を得ることが好ましい。IPVの量は典型的には「DU」単位(「D抗原単位」[63])で表す。
【0045】
麻疹、おたふく風邪および風疹ウイルスに対して防御するための抗原は、知られている1価および3価(「MMR」)ワクチンにおいて見られるように、典型的には生ウイルスである。麻疹ウイルスワクチンは、参考文献1の19章中により詳細に記載されている。おたふく風邪ウイルスワクチンは、参考文献1の20章中により詳細に記載されている。風疹ウイルスワクチンは、参考文献1の26章中により詳細に記載されている。典型的な麻疹ウイルス系統には、Moraten;Connaught;Schwarz;Edmonston−Zagreb;CAM−70;AIK−C;TD97;Leningrad−16;Shanghai−191などがある。SchwarzおよびMoraten系統が、米国および欧州で最も一般的に使用されている。典型的なおたふく風邪ウイルス系統には、Jeryl Lynn;RIT4385;Urabe;Hoshino;Rubini;Leningrad−3;Leningrad−Zagreb;Miyahara;Torii;NKM−46;S−12などがある。Jeryl Lynn、RIT4385、UrabeおよびLeningrad−Zagreb系統が、最も一般的な世界規模の系統である。典型的な風疹ウイルス系統には、RA27/3;Matsuba;TCRB19;Takahashi;Matsuura;TP−336などがある。RA27/3は、西欧諸国で使用されている最も一般的な系統である。
【0046】
水痘に対して防御するためのVZV抗原は典型的には、Oka系統のウイルスに基づく生ウイルスである。VZVワクチンは、参考文献1の28章中により詳細に記載されている。
【0047】
インフルエンザウイルス抗原は、参考文献1の17および18章中により詳細に記載されている。広義には、インフルエンザウイルスワクチンは生ウイルスまたは不活化ウイルスに基づいてよく、不活化ワクチンは完全ウイルス、「スプリット」ウイルスまたは精製表面抗原(ヘマグルチニンおよびノイラミニダーゼ含む)に基づいてよい。ワクチンを調製するために使用するウイルスは、卵または細胞培養物のいずれかで増殖させることが可能である。インフルエンザウイルス用のワクチン系統は季節ごとに変わる。現在の大流行間期では、ワクチンは典型的には2つのインフルエンザA系統(H1N1およびH3N2)および1つのインフルエンザB系統を含み、3価ワクチンが典型的である。本発明は、(特にインフルエンザAウイルスの)H2、H5、H7またはH9亜型系統などの流行系統(すなわち、ワクチンのレシピエントおよび一般的なヒト集団がそれに対する免疫がない系統)由来のウイルスも使用することができ、流行系統用のインフルエンザワクチンは1価であってよく、あるいは流行系統によって補充した通常の3価ワクチンに基づくものであってよい。インフルエンザウイルスはリアソータント系統であってよく、逆遺伝学的技法によって得ることができている。ウイルスは弱毒化することができる。ウイルスは温度感受性であってよい。ウイルスは低温適応性であってよい。
【0048】
ワクチンにおいて使用するためのこれらの病原体由来の抗原成分は、略称で一般に呼ばれる:「D」はジフテリアトキソイド、「T」は破傷風トキソイド、「P」は百日咳抗原、「Pa」は無細胞、「Pw」は細胞性、「Hib」はb型インフルエンザ菌被膜糖、「MenA」、「MenB」、「MenC」、「MenW」および「MenY」はそれぞれの髄膜炎菌の血清型、「IPV」は不活化ポリオウイルス、および「Spn」は肺炎球菌
。
【0049】
抗原成分をHBsAgと組み合わせて多価組成物を調製するとき、抗原は個別に加えることができ、あるいはHBsAgと組み合わせる前に抗原を予め混合することができる。DおよびT抗原を使用する場合、予め混合したD−T成分を使用することが好ましい。この2価成分は本発明の方法中で使用することができる、例えばそれをHBsAgと組み合わせて、3価D−T−HBV成分を作製することができる。代替として、D−T成分は他の非HBV抗原(例えば、無細胞百日咳抗原)と組み合わせることができ、したがってその成分はHBsAgなどと組み合わせることができる。D、TおよびPw抗原を使用する場合、予め混合したD−T−Pw成分を使用すること、したがって本発明の方法中にこの成分を使用することが好ましい。
【0050】
アジュバントを本発明の組成物中に含めるとき、アジュバントは様々な段階で加えることもできる。典型的には、本発明の方法中で使用する前に抗原をアジュバントと組み合わせているはずであるが(例えば、2価D−T混合物は本発明の方法中で使用する前にアルミニウム塩アジュバントに吸着しているはずであり、これはトキソイドを別々に調製し、その各々を水酸化アルミニウムアジュバントに別々に吸着させ、次いで2つの吸着トキソイドを(場合によっては他のアジュバントと)混合して本発明の方法中で使用するための物質を得ることによって都合良く実施することができる)、抗原を混合した後にアジュバントを加えること、またはアジュバントに抗原を加えることも可能である(例えば、水性アジュバントで始めるために、したがって抗原は個別あるいは予め混合して加える)。以下に記載するように、HBsAg成分は、非HBV抗原成分と組み合わせる前にリン酸アルミニウムアジュバントに吸着させることが好ましい。
【0051】
本発明の好ましい組成物は、HBsAg以外にD、TおよびP抗原(例えば、参考文献3および4)を少なくとも含む。特に好ましい組成物は以下の組合せである:
− HBsAg、D、T
− HBsAg、D、T、Pw
− HBsAg、D、T、Pw、Hib
− HBsAg、D、T、Pw、Hib、MenA、MenC
− HBsAg、D、T、Pw、Hib、MenA、MenC、MenW135
− HBsAg、D、T、Pw、Hib、MenA、MenC、MenY
− HBsAg、D、T、Pw、Hib、MenA、MenC、MenW135、MenY
− HBsAg、D、T、Pa
− HBsAg、D、T、Pa、Hib
− HBsAg、D、T、Pa、ポリオウイルス
− HBsAg、D、T、Pa、ポリオウイルス、Hib
− HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC
− HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC、MenA
− HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC、MenY
− HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC、MenW135
− HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC、MenA、MenW135、MenY
− HBsAg、Hib
− HBsAg、A型肝炎ウイルス。
【0052】
これらの組成物は列挙した抗原からなっていてよく、あるいは他の病原体由来の抗原をさらに含むことができる。したがって、これらは別々に、あるいは他のワクチンの成分として使用することができる。
【0053】
本発明のいくつかの実施形態では、組成物は5価D−T−Pa−HBV−IPVではない[30]。したがって組成物は、Pw成分および/または少なくとも1つの複合体を含むことができる。
【0054】
アジュバント
本発明の好ましい免疫原性組成物はアジュバントを含み、このアジュバントは1つまたは複数のアルミニウム塩、および特にリン酸アルミニウムアジュバントおよび/または水酸化アルミニウムアジュバントを含むことが好ましい。
【0055】
本発明の方法中で使用する抗原成分は、その方法中で使用する前にアルミニウムアジュバントを含む、すなわち抗原成分は、アジュバントと「予め混合されている」あるいはアジュバントに「予め吸着している」ことが好ましい。
【0056】
HBsAgおよびジフテリアトキソイドを含む組成物において、ジフテリアトキソイドを水酸化アルミニウムアジュバントに吸着させることが可能である。
【0057】
HBsAgおよび破傷風トキソイドを含む組成物において、破傷風トキソイドを水酸化アルミニウムアジュバントに吸着させることは可能であるが、これは必要ではない(例えば、全破傷風トキソイドの0〜10%の間の吸着を使用することができる)。
【0058】
HBsAgおよび完全細胞百日咳抗原を含む組成物において、wP抗原は水酸化アルミニウムアジュバントおよび/またはリン酸アルミニウムアジュバントと組み合わせることが好ましい。
【0059】
HBsAgおよび無細胞百日咳抗原を含む組成物において、百日咳抗原は1つまたは複数のアルミニウム塩アジュバントに吸着させることが可能であり、あるいは非吸着状態で加えることが可能である。
【0060】
ペルタクチンが組成物中に存在する場合、ペルタクチンは、本発明の方法中で使用する前に水酸化アルミニウムアジュバントに吸着することが好ましい。PTおよびFHAは、本発明の方法中で使用する前に、水酸化アルミニウムアジュバントまたはリン酸アルミニウムに吸着させることが可能である。好ましい実施形態では、本発明の方法中で使用する前にPT、FHAおよびペルタクチンを別々に、水酸化アルミニウムに予め吸着させる。
【0061】
HBsAgおよびHib抗原を含む組成物において、Hib複合体は非吸着状態であり得るが、それはリン酸アルミニウムアジュバントに吸着することが好ましい[64]。このような吸着は、D−T−Pw−Hib−HBsAg抗原を含むワクチンにおいて特に有用である。他の複合抗原(例えば、髄膜炎菌、肺炎球菌)を同様にアルミニウム塩(例えば、ホスフェート)に吸着させることが可能であり、あるいはこれらは非吸着状態であってよい[65]。
【0062】
IPV抗原は典型的には、本発明の方法中で使用する前に如何なるアジュバントにも吸着しないが、しかしIPV抗原は、他の成分に源を発するアルミニウムアジュバントに吸着した状態になり得る。
【0063】
参考文献66中に記載された方法を使用して、組成物中のHBsAgをリン酸アルミニウムに吸着させることが可能である。リン酸アルミニウムへの吸着はよく知られているENGERIX−B(商標)製品とは対照的であるが(この場合HBsAgは水酸化アルミニウムに吸着する)、HEPACCINE(商標)およびRECOMBIVAX(商標)製品と同じである。参考文献67中に言及されるように、リン酸アルミニウムは水酸化アルミニウムより良いHBsAgのアジュバントである可能性がある。(よく知られているENGERIX−B(商標)製品中と同様に)HBsAgは最終ワクチン中の水酸化アルミニウムアジュバントに吸着させることが可能であり、あるいは非吸着状態を保ち得るが、それは一般にリン酸アルミニウムアジュバントに吸着するはずである。さらにHBsAgは、本発明の方法中で使用する前にリン酸アルミニウムに予め吸着させることが好ましい。
【0064】
本発明の方法が、ジフテリアトキソイドと破傷風トキソイドがHBsAgとそれらを組み合わせる前に混合されている成分を使用する場合、このD−T混合物は、DおよびT抗原が吸着する水酸化アルミニウムアジュバントを含むことが好ましい。
【0065】
本発明の方法が、ジフテリアトキソイド、破傷風トキソイドおよび完全細胞百日咳抗原がHBsAgとそれらを組み合わせる前に混合されている成分を使用する場合、このD−T−Pw混合物は、DおよびT抗原が吸着する水酸化アルミニウムアジュバントと、リン酸アルミニウムアジュバントの両方を含むことが好ましい。
【0066】
現在使用されているアルミニウムアジュバントは典型的には、「水酸化アルミニウム」または「リン酸アルミニウム」アジュバントのいずれかと呼ばれている。しかし、いずれも存在する実際の化学化合物を正確に記載していないので、これらは便宜上の名称である(例えば、参考文献68の9章を参照)。本発明は、アジュバントとして一般に使用される「水酸化物」または「リン酸」塩のいずれかを使用することができる。
【0067】
「水酸化アルミニウム」として知られるアジュバントは典型的にはオキシ水酸化アルミニウム塩であり、これは通常少なくとも一部分が結晶である。式AlO(OH)によって表すことができるオキシ水酸化アルミニウムは、赤外線(IR)分光法によって、特に1070cm−1での吸着バンドおよび3090〜3100cm−1での強いピークの存在によって、水酸化アルミニウムAl(OH)3などの他のアルミニウム化合物と識別することができる(参考文献68の9章)。
【0068】
「リン酸アルミニウム」として知られるアジュバントは典型的には、少量のサルフェートもしばしば含むヒドロキシリン酸アルミニウムである。それらは沈殿によって得ることができ、沈殿中の反応条件および濃度は、塩中のホスフェートとヒドロキシルの置換度に影響を与える。ヒドロキシリン酸は一般に0.3と0.99の間のPO4/Alモル比を有する。ヒドロキシリン酸はヒドロキシル基の存在によって厳密なAlPO4と識別することができる。例えば、(例えば、200℃に加熱したときの)3164cm−1でのIRスペクトルバンドは、構造ヒドロキシルの存在を示す(参考文献68の9章)。
【0069】
リン酸アルミニウムアジュバントのPO4/Al3+モル比は、一般に0.3と1.2の間、好ましくは0.8と1.2の間、およびより好ましくは0.95±0.1であるはずである。リン酸アルミニウム、特にヒドロキシリン酸塩は一般に非晶質であるはずである。典型的なアジュバントは、0.6mgAl3+/mlで含まれる0.84と0.92の間のPO4/Alモル比を有する非晶質ヒドロキシリン酸アルミニウムである。リン酸アルミニウムは一般に微粒子であるはずである。粒子の典型的な直径は、任意の抗原吸着後に0.5〜20μm(例えば、約5〜10μm)の範囲である。
【0070】
リン酸アルミニウムのPZCはホスフェートとヒドロキシルの置換度と逆の関係にあり、沈殿によって塩を調製するために使用する反応物の反応条件および濃度に応じて、この置換度は変わる可能性がある。溶液中の遊離リン酸イオンの濃度を変えること(より多い量のリン酸=より酸性のPZC)、またはヒスチジンバッファーなどのバッファーを加えること(PZCをより塩基性にする)によってもPZCは変わる。本発明により使用するリン酸アルミニウムは、4.0と7.0の間のPZC、より好ましくは5.0と6.5の間、例えば約5.7のPZCを一般に有するはずである。
【0071】
本発明の組成物を調製するために使用するリン酸アルミニウム溶液は、バッファー(例えば、リン酸またはヒスチジンまたはトリスバッファー)を含み得るが、これは常に必要なわけではない。リン酸アルミニウム溶液は、滅菌済みであり発熱物質を含まないことが好ましい。リン酸アルミニウム溶液は、例えば1.0mMと20mMの間、好ましくは5mMと15mMの間、およびより好ましくは約10mMの濃度で存在する遊離水性リン酸イオンを含み得る。リン酸アルミニウム溶液は、塩化ナトリウムも含み得る。塩化ナトリウムの濃度は0.1〜100mg/ml(例えば、0.5〜50mg/ml、1〜20mg/ml、2〜10mg/ml)の範囲であることが好ましく、およびより好ましくは約3±1mg/mlである。NaClの存在は、抗原の吸着前のpHの正確な測定を容易にする。
【0072】
いくつかの実施形態では、本発明は、ポリソルベート80、Span85およびスクアレンの混合物を含む水中油型エマルジョンを含む組成物を除外することができる[69]。いくつかの実施形態では、本発明は、油、α−トコフェロールおよびポリソルベート80の混合物を含む組成物を除外することができる[70]。いくつかの実施形態では、本発明は、サポニンアジュバント(QS21など)および非イオン性界面活性剤(ポリソルベート40、60または80など)を含む組成物を除外することができる。いくつかの実施形態では、本発明は、サポニンアジュバント、代謝性油および非イオン性界面活性剤を含む組成物を除外することができる[71]。いくつかの実施形態では、本発明は、サポニンアジュバント、水中油型エマルジョンおよびステロールを含む組成物を除外することができる[72]。いくつかの実施形態では、本発明は、3d−MPL、QS21、トリグリセリドおよび水中油型エマルジョンを含む組成物を除外することができる[73]。
【0073】
精製HBsAgと他の抗原の組合せ
本発明の方法は、精製HBsAgを少なくとも1つの非HBV病原体の由来の少なくとも1つの抗原と合わせるステップを含む。
【0074】
抗原は個別に連続して組み合わせることができ、あるいは抗原を予め混合し、一緒に加えることができる。例えば、4価DTP−HBsAgワクチンは、容器へのHBsAg、D、TおよびP抗原の連続的な添加を含む方法によって、あるいはD、TおよびP抗原を予め混合し、次いでHBsAgとDTP混合物を組み合わせることによって作製することができる。
【0075】
抗原成分は任意の適切な順序で組み合わせることができる。
【0076】
非HBV病原体の由来の抗原は界面活性剤を含むことができ、これはHBsAg精製中に使用する非イオン性界面活性剤と同じであるか、あるいはそれと異なってよい。界面活性剤添加に関する多数の別ステップが回避されるので、他の抗原成分が界面活性剤を含む場合、本発明の方法は特に有用である。1つまたは複数の非HBV成分がポリソルベート80を含む方法が好ましい。
【0077】
ジフテリアトキソイドおよび破傷風トキソイドが本発明の組成物中に含まれる場合、それらはHBsAgと組み合わせる前に予め混合することが好ましい。したがって本発明の方法は、HBsAgを含む第1の成分を、D抗原とT抗原の両方を含む第2の成分と組み合わせることを含む。同様に、ジフテリアトキソイド、破傷風トキソイドおよび完全細胞百日咳抗原が組成物中に含まれる場合、それらは予め混合することが好ましく、したがっ
て本発明の方法は、HBsAgを含む第1の成分を、D、TおよびPw抗原を含む第2の成分と組み合わせることを含む。
【0078】
D−T混合物を使用する場合、本発明のワクチン中のジフテリアトキソイドと破傷風トキソイドの比は、通常(Lf単位で測定して)2:1と3:1の間、好ましくは2.4:1と2.6:1の間であり、2.5:1であることがより好ましい。
【0079】
アジュバントを本発明の組成物中に含めるとき、様々な段階でアジュバントを加えることも可能である。典型的には、抗原は、本発明の方法で使用する前にアジュバントと組み合わせるが(例えば、2価D−T混合物は、本発明の方法で使用する前にアルミニウム塩アジュバントに吸着する)、抗原を混合した後にアジュバントを加えること、あるいは抗原をアジュバントに加えることも可能である(例えば、水性アジュバントで始め、次いで抗原を個別にまたは予め混合して添加すること)。前に記載したように、非HBV抗原成分と組み合わせる前に、HBsAg成分をリン酸アルミニウムアジュバントに吸着させることが可能である。
【0080】
組み合わせワクチン
本発明の組成物は(a)抗原成分、および(b)非抗原成分を含むことができる。抗原成分は前に記載した抗原を含むか、あるいはそれらからなってよい。非抗原成分は、以下でさらに詳細に記載するように、担体、アジュバント、賦形剤、バッファーなどを含むことができる。これらの非抗原成分は様々な供給源を有し得る。例えばそれらは、製造中に使用する、あるいはこれらの成分と別に加えることができる抗原またはアジュバント物質の1つ中に存在する可能性がある。
【0081】
本発明の好ましい組成物は、1つまたは複数の医薬担体および/または賦形剤を含む。
【0082】
緊張性を調節するために、ナトリウム塩などの生理塩を含むことが好ましい。塩化ナトリウム(NaCl)が好ましく、これは1mg/mlと20mg/mlの間で存在してよい。
【0083】
組成物は一般に200mOsm/kgと400mOsm/kgの間、好ましくは240mOsm/kgと360mOsm/kgの間の浸透圧を有するはずであり、280mOsm/kg〜320mOsm/kgの範囲内に入ることがより好ましいはずである。浸透圧は予防接種によって引き起こされる痛みに対して影響がないことは以前に報告されているが[74]、この範囲の浸透圧を保つことはそれにもかかわらず好ましい。
【0084】
本発明の組成物は1種または複数種のバッファーを含むことができる。典型的なバッファーには、リン酸バッファー、トリスバッファー、ホウ酸バッファー、コハク酸バッファー、ヒスチジンバッファー、またはクエン酸バッファーがある。バッファーは典型的には5〜20mMの範囲で含まれるはずである。
【0085】
本発明の組成物のpHは一般に5.0と7.5の間、およびより典型的には最適な安定性のために5.0と6.0の間、あるいはジフテリアトキソイドおよび/または破傷風トキソイドが存在する場合、6.0と7.0の間であるはずである。したがって本発明の方法は、パッケージ前にバルクワクチンのpHを調節するステップを含むことができる。
【0086】
本発明の組成物は滅菌済みであることが好ましい。
【0087】
本発明の組成物は発熱物質を含まない、例えば用量当たり1EU未満(エンドトキシン単位、標準指標)、および好ましくは用量当たり0.1EU未満を含むことが好ましい。
【0088】
本発明の組成物はグルテンを含まないことが好ましい。
【0089】
HBsAgの吸着性のため、最終的なワクチン製品は、不透明な外見を有する懸濁液である可能性がある。この外見は、微生物による汚染が容易に目に見えないことを意味し、したがってワクチンは抗菌剤を含むことが好ましい。ワクチンを多用量容器中にパッケージするとき、これは特に重要である。封入するのに好ましい抗菌剤は、2−フェノキシエタノールおよびチメロサールである。しかし、本発明の方法中に水銀防腐剤(例えばチメロサール)は使用しないことが好ましい。したがって、方法中で使用する成分の1つからすべてが、水銀防腐剤を実質的に含まないでよい(特に、2価D−T成分;IPV成分;複合体成分)。しかし、本発明中で使用する前に成分(特にHBsAg)をこのような防腐剤で処理した場合、微量の存在が避けられない可能性がある。しかし安全性のために、最終組成物は約25ng/ml未満の水銀を含むことが好ましい。最終的なワクチン製品は、検出可能なチメロサールを含まないことがより好ましい。これは一般に、本発明の方法中のその添加前に抗原調製物から水銀防腐剤を除去すること、または組成物を生成するために使用する成分の調製中のチメロサールの使用を避けることによって実施されるはずである。
【0090】
2価D−T混合物を本発明の方法中で使用する場合、それはチメロサールを含まない状態でなければならない。いくつかの実施形態では、D−T混合物は2−フェノキシエタノールを含む可能性があるが、他の実施形態では、D−T混合物はチメロサールおよび2−フェノキシエタノールを含まない。3価D−T−Pw混合物を本発明の方法中に使用する場合、それは2−フェノキシエタノールを含まないでよいが、チメロサールを含む可能性がある。
【0091】
製造中、望ましい最終濃度を得るための成分の希釈は、通常WFI(注射用水)を用いて実施するはずである。
【0092】
Al3+の形態で表す本発明の組成物中のリン酸アルミニウムの濃度は5mg/ml未満、例えば4mg/ml以下、3mg/ml以下、2mg/ml以下、1mg/ml以下などであることが好ましい。
【0093】
本発明の組成物中のHBsAgの濃度は60μg/ml未満、例えば55μg/ml以下、50μg/ml以下、45μg/ml以下、40μg/ml以下などであることが好ましい。約20μg/mlの濃度が典型的である。
【0094】
本発明の組成物中のジフテリアトキソイドの濃度は、典型的には少なくとも50IU/mlである。
【0095】
本発明の組成物中の破傷風トキソイドの濃度は、典型的には少なくとも100IU/mlである。
【0096】
本発明の組成物中のジフテリアトキソイドと破傷風トキソイドの比は、通常(Lf単位で測定して)2:1と3:1の間、好ましくは2.4:1と2.6:1の間であり、2.5:1であることがより好ましい。
【0097】
本発明の組成物中のwP抗原の量は、典型的には少なくとも8IU/mlである。
【0098】
本発明の組成物中の糖として測定したHib複合体の量は、典型的には10μg/mlと30μg/mlの間である。
【0099】
EU(Elisa単位)で測定したHAV抗原の量は、典型的には少なくとも600EU/mlである。
【0100】
IPV抗原の量は系統の血清型に依存する。1型ウイルスに関しては、組成物は約80DU/mlを典型的には含む。2型ウイルスに関しては、組成物は約16DU/mlを典型的には含む。3型ウイルスに関しては、組成物は約65DU/mlを典型的には含む。
【0101】
本発明の組成物中の糖として測定した髄膜炎菌複合体の量は、各血清型に関して典型的には5μg/mlと25μg/mlの間である。
【0102】
本発明の組成物中の糖として測定した肺炎球菌複合体の量は、各血清型に関して典型的には2μg/mlと20μg/mlの間である。
【0103】
本発明の組成物は、0.5mlの用量で患者に投与することが好ましい。0.5mlの用量という言及は、正規分布の分散、例えば0.5ml±0.05mlを含むことは理解されるはずである。
【0104】
本発明は、患者への投与用に次いで分配することができる、個々の用量にパッケージするのに適したバルク材料を提供することができる。前述の濃度は典型的には最終パッケージ用量における濃度であり、したがってバルクワクチンにおける濃度はさらに高い可能性がある(例えば、希釈によって最終濃度まで低下させるために)。
【0105】
本発明の組成物は、一般に水性形態であるはずである。
【0106】
個々の抗原成分由来の残留成分が、本発明の方法によって生成する最終ワクチン中に微量で存在する可能性もある。例えば、ホルムアルデヒドを使用してジフテリア、破傷風および百日咳のトキソイドを調製する場合、最終ワクチン製品は微量のホルムアルデヒド(例えば、10μg/ml未満、好ましくは5μg/ml未満)を保持する可能性がある。培地または安定剤をポリオウイルス調製中に使用することができた可能性があり(例えば、培地199)、これらは最終ワクチンまで存続する可能性がある。同様に、遊離アミノ酸(例えば、アラニン、アルギニン、アスパラギン酸、システインおよび/またはシスチン、グルタミン酸、グルタミン、グリシン、ヒスチジン、プロリンおよび/またはヒドロキシプロリン、イソロイシン、ロイシン、リシン、メチオニン、フェニルアラニン、セリン、スレオニン、トリプトファン、チロシンおよび/またはバリン)、ビタミン(例えば、コリン、アスコルビン酸など)、リン酸2ナトリウム、1カリウムリン酸塩、カルシウム、グルコース、硫酸アデニン、フェノールレッド、酢酸ナトリウム、塩化カリウムなどが、それぞれ100μg/ml以下、好ましくは10μg/ml未満で最終ワクチン中に保持される可能性がある。ネオマイシン(例えば、特にIPV成分由来の硫酸ネオマイシン)、ポリミキシンB(例えば、特にIPV成分由来の硫酸ポリミキシンB)などの、抗原調製物由来の他の成分も、用量当たりナノグラム未満の量で存在する可能性がある。抗原調製物に由来する最終ワクチンの他の考えられる成分は、抗原の完全ではない精製から生じる。少量の百日咳菌、ジフテリア菌、破傷風菌および出芽酵母のタンパク質および/またはゲノムDNAが、したがって存在する可能性がある。これらの残留成分を最少にするために、本発明の方法中で抗原を使用する前に、抗原調製物を処理してそれらを除去することが好ましい。
【0107】
IPV成分を使用する場合、それは一般にVero細胞において増殖しているはずである。最終ワクチンは好ましくは10ng/ml未満、好ましくは1ng/ml以下、例えば500pg/ml以下または50pg/ml以下のVero細胞DNA、例えば10ng/ml未満の50塩基対長以上であるVero細胞DNAを含む。
【0108】
本発明のパッケージ組成物
HBsAgとアジュバントを組み合わせた後、本発明の方法は容器に混合物の0.5mlサンプルを抽出およびパッケージするステップを含むことができる。多用量の状況に関しては、多用量を抽出し、1つの容器に一緒にパッケージするはずである。
【0109】
本発明の方法は、使用するために容器にワクチンをパッケージする他のステップを含むことができる。適切な容器にはバイアルおよび使い捨てシリンジがある(滅菌済みであるものが好ましい)。
【0110】
本発明の組成物をバイアルにパッケージする場合、これらはガラスまたはプラスチック材料でできていることが好ましい。組成物をバイアルに加える前に、バイアルは滅菌済みであることが好ましい。ラテックス感受性患者に関する問題を回避するために、ラテックスを含まないストッパーでバイアルを密封することが好ましい。バイアルは1回用量のワクチンを含むことができ、あるいはバイアルは、複数回用量、例えば10回用量を含むことができる(「多用量」バイアル)。多用量バイアルを使用する場合、バイアルの中身の汚染を回避することに注意を払って、厳密な無菌条件下で滅菌済みニードルおよびシリンジを用いて各用量を取り出さなければならない。好ましいバイアルは、無色のガラスでできている。
【0111】
バイアルは予め充填したシリンジをキャップに挿入することができ、シリンジの中身をバイアルに押し出すことができ(例えば、凍結乾燥物質をその中で還元するために)、バイアルの中身を除去してシリンジに戻すことができるように適合させたキャップ(例えばLuerロック)を有することができる。バイアルからのシリンジの除去後、次いでニードルを取り付けることができ、組成物は患者に投与することができる。キャップにアクセス可能である前にシールまたはカバーを除去する必要があるように、キャップはシールまたはカバーの内側に位置することが好ましい。
【0112】
組成物をシリンジにパッケージする場合、シリンジはそれに取り付けられたニードルを通常有していないはずであるが、構築および使用するための別のニードルをシリンジに供給することができる。安全なニードルが好ましい。1−インチ23−ゲージ、1−インチ25−ゲージおよび5/8−インチ25−ゲージのニードルが典型的である。記録をとるのを容易にするために中身のロット番号および有効期限をその上に印刷することができる剥離ラベルを、シリンジに与えることができる。シリンジ内のプランジャーは、吸引中にプランジャーが偶然に除去されるのを防ぐためのストッパーを有することが好ましい。シリンジはラテックスゴム製キャップおよび/またはプランジャーを有することができる。使い捨てシリンジは1回用量のワクチンを含む。シリンジは一般にニードルの取り付け前に先端を密封するための先端キャップを有するはずであり、先端キャップはブチルゴムでできていることが好ましい。シリンジとニードルを別々にパッケージする場合、ブチルゴム製シールドをニードルに取り付けることが好ましい。グレーのブチルゴムが好ましい。好ましいシリンジは、「Tip−Lok」(商標)の商品名で市場に出ているシリンジである。
【0113】
ガラス容器(例えば、シリンジまたはバイアル)を使用する場合、ソーダ石灰ガラスではなくホウ珪酸ガラスでできた容器を使用することがしたがって好ましい。
【0114】
組成物を容器にパッケージした後、分配用の箱の中、例えば段ボール箱の中に容器を次いで封入することができ、ワクチンの詳細、例えばその商品名、ワクチン中の抗原の一覧(例えば、「B型肝炎組換え体」など)、容器の表示(例えば、「使い捨ての予め充填したTip−Lokシリンジ」または「10×0.5mlのシリンジ1回用量バイアル」)、その用量(例えば、「それぞれ1回0.5mlの用量を含む」)、警告(例えば、「成人のみ使用」または「小児のみ使用」)、有効期限、指示(例えば、「(事前血液透析および血液透析を含めて)腎不全を有していた15歳以上の患者に関する、すべての知られている亜型によって引き起こされるB型肝炎ウイルス(HBV)感染に対する能動的免疫処置」など)、特許番号などがその箱にラベルされるはずである。それぞれの箱は、2つ以上のパッケージワクチン、例えば5または10のパッケージワクチン(特にバイアル用)を含むことができる。ワクチンがシリンジ中に含まれる場合、パッケージはシリンジの絵を示し得る。
【0115】
ワクチンは(例えば、同じ箱の中に)、ワクチンの詳細、例えば投与に関する指示、ワクチン中の抗原の詳細などを含む説明書と共に、1つにパッケージすることができる。指示は、警告、例えば予防接種後のアナフィラキシー反応の場合にアドレナリン溶液を容易に利用できる状態に保つなどの警告も含むことができる。
【0116】
パッケージワクチンは2℃と8℃の間で保存することが好ましい。それを凍結させてはならない。
【0117】
ワクチンは製造中に完全液状形態で与えることができ(すなわち、すべての抗原成分が水溶液または懸濁液中に存在する)、あるいはワクチンは、いくつかの成分が液状形態であり他の成分が凍結乾燥形態である形に調製することができる。したがって最終ワクチンは、2つの成分:(a)水性抗原を含む第1の成分と(b)凍結乾燥抗原を含む第2の成分を1つに混合することによって、使用時に即時調製することができる。2つの成分は別々の容器(例えば、バイアルおよび/またはシリンジ)中に存在することが好ましく、本発明は成分(a)および(b)を含むキットを提供する。この形式は複合体成分、特にHibおよび/または髄膜炎菌および/または肺炎球菌複合体を含むワクチンに特に有用である。それは、凍結乾燥形態でさらに安定する可能性があるからである。したがって複合体は、本発明と共にそれらを使用する前は凍結乾燥状態であってよい。他の成分を例えば安定剤として、凍結乾燥前に加えることもできる。含有するのに好ましい安定剤は、ラクトース、スクロースおよびマンニトール、ならびにこれらの混合物、例えばラクトース/スクロース混合物、スクロース/マンニトール混合物などである。したがって最終ワクチンは、ラクトースおよび/またはスクロースを含むことができる。スクロース/マンニトール混合物を使用することによって、乾燥プロセスをスピードアップすることが可能である。
【0118】
したがって本発明は、2容器の組み合わせワクチンを調製するための方法であって、
− 前に記載したように水性組み合わせワクチンを調製するが、ただし前記1つまたは複数の抗原が複合体被膜糖抗原を含まないステップ、
− 第1の容器(例えばシリンジ)中で前記組み合わせワクチンをパッケージするステップ、
− 複合体被膜糖抗原を凍結乾燥形に調製するステップ、
− 前記凍結乾燥抗原を第2の容器(例えばバイアル)中でパッケージするステップ、および
− 第1の容器と第2の容器をキットに1つにパッケージするステップを含む方法を提供する。
【0119】
その後、キットは医師に分配することができる。
【0120】
D、T、PおよびHBsAg成分は液状形態であることが好ましい。
【0121】
ワクチンの治療および投与法
本発明の組成物はヒト患者への投与に適しており、本発明は、本発明の組成物を患者に投与するステップを含む、患者における免疫応答を増大させる方法を提供する。
【0122】
本発明は、医薬において使用するための本発明の組成物も提供する。
【0123】
本発明は、患者に投与するための医薬品の製造における、(i)その精製法が非イオン性界面活性剤の存在下で酵母細胞を破壊することを含む組換え酵母細胞から精製したHBsAg、および(ii)1つまたは複数の非HBV抗原の使用も提供する。
【0124】
本発明の免疫原性組成物は、少なくともB型肝炎ウイルス感染の予防および/または治療において使用するためのワクチンであることが好ましい。本発明の組成物を与えた患者は、初回免疫処置後6週間で測定して、500mIU/ml以上の血清抗HBsAgGM力価を有することが好ましい。l2カ月後に測定して、力価が500mIU/ml以上であることがより好ましい。
【0125】
完全な有効性を有するために、子供用の典型的な初回免疫処置スケジュールは、2回以上の投与を施すことを含むことができる。例えば、投与は:0および6カ月(時間0は初回投与である)、0、1、2および6カ月;第0日、第21日、および次いで6カ月と12カ月の間に第3の投与;2、4および6カ月;3、4および5カ月;6、10および14週;または0、1、2、6および12カ月であってよい。
【0126】
例えば2歳の子供用に、追加抗原刺激投与として組成物を使用することもできる。
【0127】
本発明の組成物は、例えば腕または脚への筋肉内注射によって投与することができる。
【0128】
本発明によって生成されるワクチンは、Prevnar(商標)などの別の肺炎球菌複合体ワクチンと同時に患者に投与することができる。
【0129】
本発明の組成物がアルミニウム系アジュバントを含む場合、保存中に成分の沈殿が生じる可能性がある。したがって、組成物は患者に投与する前に攪拌しなければならない。攪拌した組成物は、白濁した懸濁液であるはずである。
【0130】
好ましいワクチン
本発明の特異的な多価免疫原性組成物は以下の組成物を含む:
・ HBsAg、D、T、PaおよびIPVを含む5価組成物。ワクチンは水性形態である。それは水酸化アルミニウムアジュバントとリン酸アルミニウムアジュバントの両方を含む。HBsAgはリン酸アルミニウムに吸着する。D、TおよびPaは水酸化アルミニウムに吸着する。1ml当たりの量:約50Lfのジフテリアトキソイド;約20Lfの破傷風トキソイド;約50μgのPT;約50μgのFHA;約16μgのペルタクチン;約20μgのHBsAg;約80DUの1型ポリオウイルス;約16DUの2型ポリオウイルス;約64DUの3型ポリオウイルス。用量:約0.5ml。予め充填したシリンジ中に存在してよい。
【0131】
・ HBsAg、D、T、PaおよびIPVを含む5価組成物。ワクチンは水性形態である。それは水酸化アルミニウムアジュバントとリン酸アルミニウムアジュバントの両方を含む。HBsAgはリン酸アルミニウムに吸着する。D、TおよびPaは水酸化アルミニウムに吸着する。1ml当たりの量:少なくとも60IUのジフテリアトキソイド;少なくとも80IUの破傷風トキソイド;約50μgのPT;約50μgのFHA;約16μgのペルタクチン;約20μgのHBsAg;約80DUの1型ポリオウイルス;約16DUの2型ポリオウイルス;約64DUの3型ポリオウイルス。用量:約0.5ml。予め充填したシリンジ中に存在してよい。
【0132】
・ HBsAg、D、TおよびPwを含む4価組成物。成分は水性形態である。それは水酸化アルミニウムアジュバントとリン酸アルミニウムアジュバントの両方を含む。HBsAgはリン酸アルミニウムに吸着する。DおよびTは水酸化アルミニウムに吸着する。組成物はチメロサールを含み、2−フェノキシエタノールは含まないことが好ましい。1ml当たりの量:少なくとも60IUのジフテリアトキソイド;少なくとも120IUの破傷風トキソイド;少なくとも8IUのPw、約20μgのHBsAg。用量:約0.5ml。
【0133】
・ HBsAg、D、T、PwおよびHib−T複合体を含む5価組成物。HBsAg、D、TおよびPw成分は水性形態であり、Hib−Tは凍結乾燥状態である。それは水酸化アルミニウムアジュバントとリン酸アルミニウムアジュバントの両方を含む。DおよびTは水酸化アルミニウムに吸着する。HBsAgおよびHib−Tはリン酸アルミニウムに吸着する。凍結乾燥Hib−Tはラクトースを含む。水性成分はチメロサールを含み得る。1ml当たりの量:少なくとも60IUのジフテリアトキソイド;少なくとも120IUの破傷風トキソイド(およびHib−T中の担体として5μgと25μgの間の破傷風トキソイド);少なくとも8IUのPw、約20μgのHBsAg;糖として測定して約5μgのHib−T。用量:約0.5ml。
【0134】
・ HBsAg、D、T、Pwならびに3つの複合体:Hib−T複合体、MenA複合体およびMenC複合体を含む7価組成物。HBsAg、D、TおよびPw成分は水性形態であり、3つの複合体は凍結乾燥状態である。それは水酸化アルミニウムアジュバントとリン酸アルミニウムアジュバントの両方を含む。DおよびTは水酸化アルミニウムに吸着する。HBsAgはリン酸アルミニウムに吸着する。凍結乾燥成分はラクトースおよび/またはスクロースを含み得る。水性成分はチメロサールを含み得る。考えられる1ml当たりの量:少なくとも60IUのジフテリアトキソイド;少なくとも120IUの破傷風トキソイド(およびHib−T中の担体として5μgと25μgの間の破傷風トキソイド);少なくとも8IUのPw、約20μgのHBsAg;糖として測定して約5μgの各複合体。用量:約0.5ml。
【0135】
これらの組成物はそれ自体ワクチンとして使用することができ、あるいは他のワクチンの成分として使用することができる。例えば本発明は、前に記載した5価HBsAg−D−T−Pa−IPV組成物、および凍結乾燥Hib−T複合体を含む6価組成物を提供する。凍結乾燥Hib−Tはアルミニウム塩に吸着しないことが好ましい。本発明は、前に記載した5価HBsAg−D−T−Pa−IPV組成物、および凍結乾燥Hib−TおよびMenC複合体を含む7価組成物も提供する。本発明は、前に記載した5価HBsAg−D−T−Pa−IPV組成物、および凍結乾燥Hib−T、MenCおよびMenY複合体を含む8価組成物も提供する。本発明は、前に記載した4価HBsAg−D−T−Pw組成物、および凍結乾燥Hib−T複合体を含む5価組成物も提供する。本発明は、前に記載した4価HBsAg−D−T−Pw組成物、およびHib−T複合体、MenA複合体およびMenC複合体の凍結乾燥混合物を含む7価組成物も提供する。最終ワクチンは、使用時に凍結乾燥物質を水性HBsAg含有物質で戻すことによって調製することができ、凍結乾燥成分と水性成分は、前に記載したようにキット内に1つにパッケージすることが好ましい。
【0136】
本発明の特異的な方法は以下のステップを含む方法を含む:
・ 本発明によるHBsAgの精製;HBsAgのリン酸アルミニウムアジュバントへの吸着;チメロサールを含まない水酸化アルミニウムアジュバントとの2価D−T混合物の入手;Pa成分用のPT、FHAおよびペルタクチンの入手;好ましくはアルミニウム塩アジュバントを含まない型1、2および3としてプールするIPV抗原の入手;最終的な5価混合を得るための任意の順序でのD−T、Pa、IPVおよびHBsAgの組合せ;場合によってはシリンジへのパッケージ。
【0137】
・ 本発明によるHBsAgの精製;HBsAgのリン酸アルミニウムアジュバントへの吸着;2−フェノキシエタノールを含まずチメロサールを含む水酸化アルミニウムアジュバントおよびリン酸アルミニウムアジュバントとの3価D−T−Pw混合物の入手;最終的な4価混合を得るためのD−T−PwとHBsAgの組合せ;場合によってはシリンジへのパッケージ;場合によっては凍結乾燥複合体成分、例えばHib−T;MenA、MenCと組み合わせたパッケージ。
【0138】
・ 本発明によるHBsAgの精製;HBsAgのリン酸アルミニウムアジュバントへの吸着;2−フェノキシエタノールを含まずチメロサールを含む水酸化アルミニウムアジュバントおよびリン酸アルミニウムアジュバントとの3価D−T−Pw混合物の入手;凍結乾燥Hib−T複合体の入手;水性4価成分を得るためのD−T−PwとHBsAgの組合せ;水性4価成分のガラスバイアルへのパッケージ;凍結乾燥Hib−Tおよび/またはMenCおよび/またはMenYのガラスバイアルへのパッケージ;5価組み合わせワクチンを得るための水戻し用の単一キット中に存在する2つのバイアルの組合せ。ガラスバイアルはI型ガラスであってよく、ブチルゴム製ストッパーを有する。
【0139】
・ 本発明によるHBsAgの精製;HBsAgのリン酸アルミニウムアジュバントへの吸着;2−フェノキシエタノールを含まずチメロサールを含む水酸化アルミニウムアジュバントおよびリン酸アルミニウムアジュバントとの3価D−T−Pw混合物の入手;好ましくはアルミニウム塩アジュバントを含まない型1、2および3としてプールするIPV抗原の入手;最終的な5価混合を得るための任意の順序でのD−T−Pa、IPVおよびHBsAgの組合せ;場合によってはシリンジへのパッケージ;場合によっては凍結乾燥複合体成分、例えばHib−T;MenA、MenCと組み合わせたパッケージ。
【0140】
他の成分、例えば塩化ナトリウム、アジュバント、防腐剤などを任意の段階で加えることができる。これらの方法を使用して、例えば前に記載したワクチンを調製することができる。
【0141】
異なる方法のステップは実質的に同時に実施することができ、あるいは別々に実施することができる。これらのステップは、同じ場所または異なる場所、異なる国においても実施することができ、例えばHBsAg精製は、Hib−T凍結乾燥と異なる場所で行うことができる。
【0142】
複合体の担体タンパク質
複合体糖抗原は、糖がそれに直接、あるいはリンカーを介して共有結合する担体タンパク質を含む。結合技法に関する一般的な情報は参考文献45において見ることができる。
【0143】
担体として使用するための様々なタンパク質が知られており、好ましい担体タンパク質は、ジフテリアトキソイドまたは破傷風トキソイドなどの細菌毒素またはトキソイドである。他の適切な担体タンパク質には、ジフテリア毒素のCRM197突然変異体[75〜77]、髄膜炎菌の外膜タンパク質[78]、合成ペプチド[79、80]、熱ショックタンパク質[81、82]、百日咳タンパク質[83、84]、サイトカイン[85]、リンホカイン[85]、ホルモン[85]、増殖因子[85]、様々な病原体由来の抗原に由来する多数のヒトCD4+T細胞エピトープを含む人工タンパク質[86]、例えばN19[87]、インフルエンザ菌由来のタンパク質D[88、89]、肺炎球菌表面タンパク質PspA[90]、ニューモリシン[91]、鉄摂取タンパク質[92]、C.ディフィシル(C.difficile)由来の毒素AまたはB[93]、B群連鎖球菌(S.agalactiae)タンパク質[94]などがあるが、これらだけには限られない。
【0144】
糖と担体の結合は、例えば担体タンパク質中のリシン残基、またはアルギニン残基の側鎖中の−NH2基を介した結合であることが好ましい。(例えば、システインの側鎖中の)−SH基との結合も考えられる。
【0145】
1:5(すなわち、過剰なタンパク質)と5:1(すなわち、過剰な糖)の間の糖:タンパク質比(w/w)を有する複合体が好ましい。
【0146】
組成物は少量の遊離担体を含むことができる。別の抗原として含まれる如何なる担体も無視すると、非結合担体は全体として組成物中の担体タンパク質の全量の5重量%を超えないことが好ましく、2重量%未満で存在することがより好ましい。
【0147】
例えば担体抑制のリスクを低下させるために、組成物中に2種類以上の担体タンパク質を含むことが考えられる。
【0148】
髄膜炎菌複合体に使用する担体タンパク質は、インフルエンザ菌由来のタンパク質Dであってよい。このタンパク質は参考文献95および96中に詳細に記載されており、複合体中の担体タンパク質としてのその使用は参考文献97中に記載されている。用語「タンパク質D」は、参考文献97中に開示された天然完全長タンパク質の断片、および完全長タンパク質Dまたはこれらの断片のいずれかを含む融合タンパク質(例えば、インフルエンザウイルスNS1タンパク質の断片とタンパク質Dの断片の融合体)も含む。これらの断片は、結合時にT非依存性糖抗原をT依存性抗原に変換する能力を保持しているはずである。典型的な断片は、タンパク質Dの少なくともN末端側1/3を含むはずである。このタンパク質は大腸菌中で好都合に発現される可能性があり[96]、この組換え物質は本発明と共に使用するのが好ましい[97]。
【0149】
一般事項
用語「comprising」は「including」ならびに「consisting」を包含し、例えば、Xを「含む(comprising)」組成物はXのみからなる可能性があるか、あるいは他の何かを含む、例えばX+Yである可能性がある。
【0150】
語句「実質的に」は「完全に」を除外するわけではない、例えば、Yを「実質的に含まない」組成物はYを完全に含まない可能性がある。必要な場合、語句「実質的に」は本発明の定義から除外することができる。
【0151】
数値xに関する用語「約」は、例えばx±10%を意味する。
【0152】
特に言及しない限り、2つ以上の成分を混合するステップを含む方法は、任意の特定の混合順を必要としない。したがって、成分は任意の順序で混合することができる。3つの成分が存在する場合、2成分を互いに組み合わせることができ、次いでその組合せを、第3の成分などと組み合わせることができる。
【0153】
ある抗原がアジュバントに「吸着する」として記載する場合、少なくとも50%(重量)、例えば50%、60%、70%、80%、90%、95%、98%あるいはそれより多くのその抗原が吸着することが好ましい。ジフテリアトキソイドと破傷風トキソイドは両方が完全に吸着する、すなわち上清中には何も検出できないことが好ましい。HBsAgの全体の吸着を使用することができる。
【0154】
ジフテリアトキソイドの量は国際単位(IU)で表すことができる。例えばNIBSCは、アンプル当たり160IUを含む、「吸着ジフテリアトキソイドの第3国際標準1999」[98、99]を与える。IUシステムに対する代替として、「Lf」単位(「凝集単位」または「境界凝集用量」)を、1国際単位のアンチトキシンと混合すると最適に凝集する混合物を生成する[100]トキソイドの量として定義する。例えばNIBSCは、アンプル当たり300LFを含む「ジフテリアトキソイド、プレイン」[101]を与え、アンプル当たり900LFを含む「凝集試験のジフテリアトキソイドの第1の国際参照試薬」[102]も与える。
【0155】
破傷風トキソイドの量は国際単位(IU)で表すことができる。例えばNIBSCは、アンプル当たり469IUを含む、「吸着破傷風トキソイドの第3国際標準2000」[103、104]を与える。IUシステムに対する代替として、「Lf」単位(「凝集単位」または「境界凝集用量」)を、1国際単位のアンチトキシンと混合すると最適に凝集する混合物を生成する[100]トキソイドの量として定義する。例えばNIBSCは、アンプル当たり1000LFを含む「凝集試験の破傷風トキソイドの第1の国際参照試薬」[105]を与える。
【0156】
wP抗原の量は国際単位(IU)で表すことができる。例えばNIBSCは、アンプル当たり46IUを含む「百日咳ワクチンの第3国際標準」[106]を与える。それぞれのアンプルは水溶液の2.0ml等分試料の凍結乾燥残留物を含み、この等分試料は8リットルのM/15SorensenのバッファーpH7.0で希釈した10リットルの細菌懸濁液(米国不透明度標準により180不透明度単位に等しい)を含んでいた。IUシステムに対する代替として、「OU」単位(「不透明度単位」)も使用される(例えば、4OUは約1IUであり得る)。
【0157】
複合体の量は一般に、担体の選択による変動を避けるために糖の質量によって示される(すなわち、全体としての複合体の用量(担体+糖)は述べられる用量より大きい)。
【0158】
動物(および特にウシ)の材料を細胞の培養において使用する場合、それらは感染性海綿状脳症(TSE)を含まない、および特にウシ海綿状脳症(BSE)を含まない供給源から入手しなければならない。
本発明はまた、以下の項目を提供する。
(項目1)
組み合わせワクチンを調製するためのプロセスであって、該ワクチンが(i)非イオン性界面活性剤、(ii)B型肝炎ウイルス(HBV)表面抗原、および(iii)少なくとも1つの非HBV病原体由来の抗原を含み、該プロセスが、(i)組換え酵母細胞を該非イオン性界面活性剤の存在下で破壊して精製HBsAg成分を得るステップを含む、該酵母細胞から該HBV表面抗原を精製すること、および(ii)該精製HBsAg成分を非HBV病原体由来の少なくとも1つの他の抗原と合わせて、該組み合わせワクチンを得ることを含む、プロセス。
(項目2)
組み合わせワクチンを調製するためのプロセスであって、該ワクチンが(i)非イオン性界面活性剤、(ii)B型肝炎ウイルス(HBV)表面抗原(HBsAg)、および(iii)少なくとも1つの非HBV病原体由来の抗原を含み、該プロセスが、精製HBsAgを非HBV病原体由来の少なくとも1つの他の抗原と合わせて、組み合わせワクチンを得るステップを含み、HBsAg発現組換え酵母細胞を該非イオン性界面活性剤の存在下で破壊するプロセスによって該HBsAgが調製される、プロセス。
(項目3)
(i)非イオン性界面活性剤、(ii)B型肝炎ウイルス(HBV)表面抗原、および(iii)少なくとも1つの非HBV病原体由来の抗原を含む免疫原性組成物であって、HBsAg発現組換え酵母細胞を該非イオン性界面活性剤の存在下で破壊するプロセスによって該HBV表面抗原が調製される、免疫原性組成物。
(項目4)
上記非イオン性界面活性剤がポリ(オキシエテン)残基を含む、項目1から3のいずれかに記載のプロセスまたは組成物。
(項目5)
上記非イオン性界面活性剤がポリオキシエチレンソルビタンエステルである、項目4に記載のプロセスまたは組成物。
(項目6)
上記非イオン性界面活性剤がポリソルベート20である、項目5に記載のプロセスまたは組成物。
(項目7)
上記非イオン性界面活性剤が30μg/ml以下で最終産物中に存在する、項目1から6のいずれかに記載のプロセスまたは組成物。
(項目8)
上記非イオン性界面活性剤がHBsAg各100μgに対して50μg以下で最終産物中に存在する、項目1から7のいずれかに記載のプロセスまたは組成物。
(項目9)
(i)上記少なくとも1つの非HBV病原体がジフテリア菌および破傷風菌を含み、(ii)これら2つの病原体由来の該抗原がジフテリアトキソイドおよび破傷風トキソイドであり、かつ(iii)該ジフテリアトキソイドと該破傷風トキソイドがポリソルベート20を実質的に含まない混合された形態で最初に存在する、項目1から8のいずれかに記載のプロセス。
(項目10)
上記HBV表面抗原がグリコシル化されていない、項目1から9のいずれかに記載のプロセスまたは組成物。
(項目11)
上記HBV表面抗原が、リン脂質を含む脂質マトリクスを含む粒子の形態である、項目1から10のいずれかに記載のプロセスまたは組成物。
(項目12)
上記HBV表面抗原がHBV亜型adw2に由来する、項目1から11のいずれかに記載のプロセスまたは組成物。
(項目13)
上記HBV表面抗原が用量当たり約10μgで最終組成物中に存在する、項目1から12のいずれかに記載のプロセスまたは組成物。
(項目14)
上記ワクチンがHib複合体、髄膜炎菌複合体、および/または肺炎球菌複合体を含む、項目1から13のいずれかに記載のプロセスまたは組成物。
(項目15)
上記最終組成物が3価HBsAg、D、T組成物;4価HBsAg、D、T、Pw組成物;5価HBsAg、D、T、Pw、Hib組成物;7価HBsAg、D、T、Pw、Hib、MenA、MenC組成物;8価HBsAg、D、T、Pw、Hib、MenA、MenC、MenW135組成物;8価HBsAg、D、T、Pw、Hib、MenA、MenC、MenY組成物;9価HBsAg、D、T、Pw、Hib、MenA、MenC、MenW135、MenY組成物;4価HBsAg、D、T、Pa組成物;5価HBsAg、D、T、Pa、Hib組成物;5価HBsAg、D、T、Pa、ポリオウイルス組成物;6価HBsAg、D、T、Pa、ポリオウイルス、Hib組成物;7価HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC組成物;8価HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC、MenA組成物;8価HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC、MenY組成物;8価HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC、MenW135組成物;10価HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC、MenA、MenW135、MenY組成物;2価HBsAg、Hib組成物;および2価HBsAg、A型肝炎ウイルス組成物から選択される、項目1から14のいずれかに記載のプロセスまたは組成物。
(項目16)
上記最終組成物がリン酸アルミニウムアジュバントおよび/または水酸化アルミニウムアジュバントを含む、項目1から15のいずれかに記載のプロセスまたは組成物。
(項目17)
上記最終組成物がリン酸アルミニウムアジュバントと水酸化アルミニウムアジュバントの両方を含む、項目1から16のいずれかに記載のプロセスまたは組成物。
(項目18)
上記最終組成物中のAl3+の濃度が5mg/ml以下である、項目16または17に記載のプロセスまたは組成物。
(項目19)
予め混合したD−T成分を加えることを含む、項目1から18のいずれかに記載のプロセス。
(項目20)
予め混合したD−T−Pw成分を加えることを含む、項目1から19のいずれかに記載のプロセス。
(項目21)
上記予め混合したD−T−Pw成分がリン酸アルミニウムアジュバントと水酸化アルミニウムアジュバントの両方を含む、項目20に記載のプロセス。
(項目22)
患者に投与するための薬剤の製造における、(i)組換え酵母細胞から精製したHBsAgであって、精製プロセスが非イオン性界面活性剤の存在下で該酵母細胞を破壊することを含む、HBsAg、および(ii)1つまたは複数の非HBV抗原の、使用。
(項目23)
5価組成物を調製するためのキットであって、該5価組成物がHBsAg、D、T、PwおよびHib−T複合体抗原を含み、該DおよびT抗原が水酸化アルミニウムアジュバントに吸着しており、該HBsAgおよびHib−Tがリン酸アルミニウムアジュバントに吸着しており、該HBsAg、D、TおよびPw成分が第1の容器中で水性形態であり、該Hib−Tが第2の容器中でラクトースと組み合わせた凍結乾燥形態であり、HBsAg発現組換え酵母細胞を非イオン性界面活性剤の存在下で破壊するプロセスによって、該HBsAgが調製される、キット。
(項目24)
7価組成物を調製するためのキットであって、該7価組成物がHBsAg、D、T、Pw、Hib−T複合体、MenA複合体およびMenC複合体を含み、該HBsAg、D、TおよびPw成分が第1の容器中で水性形態であり、該3つの複合体が第2の容器中で凍結乾燥形態であり、該第1の容器は水酸化アルミニウムアジュバントとリン酸アルミニウムアジュバントの両方も含み、該DおよびT抗原が水酸化アルミニウムに吸着しており、HBsAgがリン酸アルミニウムに吸着しており、HBsAg発現組換え酵母細胞を非イオン性界面活性剤の存在下で破壊するプロセスによって該HBsAgが調製される、キット。
(項目25)
上記第1の容器がチメロサールをさらに含む、項目24に記載のキット。
【図面の簡単な説明】
【0159】
【図1−1】図1は、参考文献3および4の図2のコピーであり、参考文献3および4は、吸着法に基づく抗原性および免疫をこの図が示すことを述べ、サンプル1は、過剰な水酸化アルミニウムゲルを各コンポーネントワクチンの組合せが終了した後に加えたサンプルであり、サンプル2は、同じ濃度の前記吸着剤を組合せが終了する前に事前に加えたサンプルである。図1Aおよび1Bは、それぞれサンプル1および2の相対的抗原性を示す。
【図1−2】図1は、参考文献3および4の図2のコピーであり、参考文献3および4は、吸着法に基づく抗原性および免疫をこの図が示すことを述べ、サンプル1は、過剰な水酸化アルミニウムゲルを各コンポーネントワクチンの組合せが終了した後に加えたサンプルであり、サンプル2は、同じ濃度の前記吸着剤を組合せが終了する前に事前に加えたサンプルである。図1Cおよび1Dは、それぞれサンプル1および2の各抗原に対する抗体形成の相対的レベルを示す。
【図2−1】図2は、組成物中のHBsAgの安定性に関するウエスタンブロットの結果を示す。図2A中、レーンは(1)Laemmliサンプルバッファー(LSB);(2)分子量マーカー;(3〜5)1μgの3つの別個の対照HBsAg調製物;(6〜8)3つの別個の5価ロットの上清;(9〜10)LSBである。図2Bおよび2C中、レーンは(1)分子量マーカー;(2〜4)1μgの3つの別個の対照HBsAg調製物;(5〜7)2〜8℃で2週間保存した、3つの別個の5価ロットの上清;(8〜10)36〜38℃で2週間保存した、3つの別個の5価ロットの上清である。図2D中、レーンは(1)分子量マーカー;(2)対照HBsAg;(3〜5)3つの別個の5価ロットの上清である。
【図2−2】図2は、組成物中のHBsAgの安定性に関するウエスタンブロットの結果を示す。図2A中、レーンは(1)Laemmliサンプルバッファー(LSB);(2)分子量マーカー;(3〜5)1μgの3つの別個の対照HBsAg調製物;(6〜8)3つの別個の5価ロットの上清;(9〜10)LSBである。図2Bおよび2C中、レーンは(1)分子量マーカー;(2〜4)1μgの3つの別個の対照HBsAg調製物;(5〜7)2〜8℃で2週間保存した、3つの別個の5価ロットの上清;(8〜10)36〜38℃で2週間保存した、3つの別個の5価ロットの上清である。図2D中、レーンは(1)分子量マーカー;(2)対照HBsAg;(3〜5)3つの別個の5価ロットの上清である。
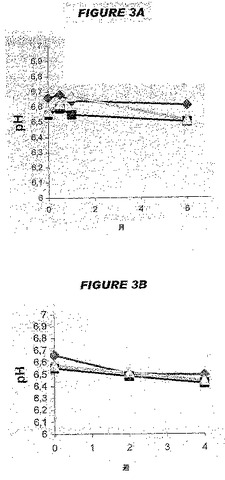
【図3】図3は、5価組成物の経時的なpHの変化を示す。
【図4】図4は、8価組成物中のHBsAgの安定性に関するウエスタンブロットの結果を示す。図4Aおよび4B中、レーン1は分子量マーカーを含み、レーン2は1μg/mlでHBsAg対照を含み、レーン3は8価組成物の上清を含む。図4B中、レーン4はLSBを含み、レーン5はレーン2と同じ対照を含み、レーン6はDOC/TCA抽出物を含む。
【実施例】
【0160】
(発明を実施するための様式)
HBsAg発現および精製
HBsAgをコードするH.ポリモルファ酵母菌宿主[107、108]を調製した。300リットルの発酵槽中で100リットルの培地を調製し、酵母菌を接種した。100グラム/リットルで細胞が存在するまで発酵を続けた。この段階で、宿主がメチロトローフ酵母菌であるので、メタノールを加え、発酵を停止させた。最終培養物の体積は160〜170リットルであった。遠心分離によって培養培地から細胞を採取した。
【0161】
精製後にHBsAgに非イオン性界面活性剤を添加する代わりに、参考文献3および4中に記載したように、タンパク質の精製中に界面活性剤を使用した。詳細には、採取した酵母細胞を0.1〜0.2%のポリソルベート20および10mMのEDTAを含むリン酸バッファーに懸濁させた。懸濁物を冷却し、高圧液体ホモジェナイザーを使用して破壊した。
【0162】
均質化した細胞懸濁液は、遠心分離によって次いで浄化した。上清にNaClを加えた後、ポリエチレングリコールを沈殿用に3〜5%の濃度までゆっくり加えた。溶液は5分間攪拌し、30分間放置し、10分間遠心分離にかけた。さらなるPEGを8〜10%まで上清にゆっくり加え、攪拌し2時間放置した。この溶液を10分間遠心分離にかけた。
【0163】
ヒュームドシリカを1〜2%の濃度で上清に加えた。溶液を12時間攪拌し、遠心分離にかけ、デオキシコール酸ナトリウムを含むホウ砂バッファー中で沈殿を完全に溶かした。水浴中で100〜120分間攪拌した後、溶液を遠心分離にかけ、上清を回収した。流速200mL/分でトリス/Clバッファーを用いて平衡状態にしたDEAEカラムに上清を充填し、NaCl溶液を含むトリス/Clバッファーで溶出した。塩化セシウムを1.2g/mLの濃度までDEAE溶出液に加え、溶液を40,000rpmで超遠心分離にかけた。脱塩したCsClプールをPBSで400μg/mlのタンパク質濃度まで希釈し、ホルマリンを0.025%の濃度まで加え、混合物は72時間放置した。最後に、残留ホルマリンはPBSを使用して除去した。
【0164】
このようにして、HBsAgはポリソルベート20を含めることによって精製したが、抗原を精製した後に加えたのではなく、組換え細胞を破壊する前に界面活性剤を添加した。
【0165】
完全液状多価ワクチン
酵母菌発現型HBsAg、ジフテリアトキソイド、破傷風トキソイドおよび完全細胞百日咳抗原を、アルミニウム塩アジュバントの懸濁液に加えた。混合物のpHを調節し、それがアルミニウムアジュバントに吸着した状態にならないようにHib−CRM197複合体を次いで加えた。この方法によって、以下の組成を有する5価ワクチンを得た:
【0166】
【表1】

他の実験では、血清型C、W135およびYの各々に由来する別個の髄膜炎菌CRM197複合体を、CRM−Hib成分の後に加えて8価ワクチンを得た:
【0167】
【表2】
ワクチンの安定性および有効性に関する重要なパラメータはHib複合体の加水分解率であり、臨床上の限界は25%の遊離糖である(参考文献109は、20%は臨床上の免疫原性に影響を与えなかったことを報告する)。このパラメータは、最少の分離および浄化でピコモルレベルにおいて非結合炭水化物の直接定量化を可能にするHPAEC−PADによって、5価ワクチンにおいて測定した。分析は遊離糖の量に焦点を当てた。
【0168】
遊離糖の量をアッセイし、全体の量の割合として表した。結果は以下の通りであった:
【0169】
【表3】
最大25%の遊離糖が臨床上許容される。すべての値がこの閾値未満であり、2〜8℃において4週間まで6.5%未満であった。熱ストレス条件下(36〜38℃で4週間)では、高レベルが見られたが、25%の値を依然大幅に下回り、最高はロット1の11.6%であった。多価Hibワクチンに関する初期の実験は、36〜38℃での1カ月の保存は、2〜8℃での2年の保存より多くのCRM−Hibの加水分解をもたらすことを示している。したがって許容される加水分解は、通常の保存条件下での少なくとも2年の時間規模を超えると予想することができる。
【0170】
遊離糖のHPAEC−PAD分析は、2〜8℃で6カ月後にも実施した。データは以下の通りであった:
【0171】
【表4】
このように、4週間と比較して6カ月では遊離糖の割合の非常にわずかな増大があるのみで、値は25%の値を依然大幅に下回る。したがってCRM197−Hibは、3つの配合物において非常に安定している。
【0172】
図3Aは、2〜8℃で保存した3つのロットに関する、6カ月間の5価ワクチンにおけるpHの変化を示す。図3Bは、36〜38℃で保存した3つのロットに関する4週間のpHの変化を示す。2〜8℃においてpHは6カ月間安定状態であったが、一方熱ストレス条件下では、2週間後に0.1pH単位のわずかな低下、および4週間後にさらにわずかな低下があった。そうではあったが、すべてのpH値は6.0〜7.0の許容範囲内に留まった。
【0173】
全3個の5価ロットの浸透圧は、312mOsm/Kgと315mOsm/Kgの間、つまり注射用ワクチンに関する240〜360mOsm/Kgの標的範囲内の中心にあった。
【0174】
抗原の効力および免疫原性の評価は、組み合わせワクチンの有効性を評価するために重要である。5価ワクチンにおけるジフテリア、破傷風および百日咳抗原の効力を評価し、CRM−HibとHBsAgの両方の免疫原性を試験した。ELISA分析を実施して、免疫処置後の特異的抗体のレベルを評価した。HBsAgの免疫原性の試験は、マウスモデルおよびHBVの効力に関して使用したスケジュールと異なる免疫処置スケジュールを使用して実施した。
【0175】
DTPの効力値は以下の通りであった:
【0176】
【表5】
これら3つの抗原のそれぞれに関して、効力試験の結果はいずれも許容下限を有意に上回り、これらの結果は、これら3つの抗原に関する優れた有効性を示す。
【0177】
HBsAgの免疫原性を評価するために、10CD1マウスの群に、第0日および第14日に皮下注射(0.5ml、生理食塩水に1:4に希釈)によって5価ワクチンを与えた。マウスは第21日に採血し、(a)「Enzygnost Anti−HBs II」試験(Dade Behring)または(b)「Ausab EIA」試験(Abbott)のいずれかを使用してELISAによってHBsAg特異的抗体を評価した。これらのELISA試験は、異なる形式およびHBsAgに対する異なる感度を有する。幾何学的平均力価は、したがって2試験間で比較可能ではない。しかし、それぞれの試験の範囲内では血清に関するGMT値が最適であった。結果は以下の通りであった:
【0178】
【表6】
この種のマウス免疫原性アッセイを実施して得たすべてのGMT値は、文献中で報告されている値より高い。応答率は約100%の最適レベルで両方の抗原に関して絶えず高い。
【0179】
Hibの免疫原性を評価するために、8CD1マウスの群に、第0、10および20日に皮下注射(0.5ml、生理食塩水に1:4に希釈)によって5価ワクチンを与えた。マウスは第34日に採血し、ELISAによってHib特異的抗体を評価した。結果は以下の通りであった:
【0180】
【表7】
HBsAgのアルミニウムアジュバントへの吸着はワクチンの免疫原性に関する重要な要因であり、このパラメータはイムノブロットによって測定した。従ったイムノブロット手順は、ほぼ以下の通りであった:1ml体積のワクチン上清をDOC/TCA沈殿させ、LSBを用いて変性させ、次いで12%アクリルアミドSDS−PAGEに充填し、1μgの各ロットのHBsAgを対照として充填し、ヤギ抗HBsAg抗体調製物は一次抗体(希釈1:1000)として使用し、抗ヤギPOD複合体(希釈1:2500)は二次抗体として使用した。
【0181】
5価ワクチンに関する結果を図2中に示す。図2Aのレーン6〜8は時間ゼロで組成物中に検出可能な可溶性HBsAgが存在しないことを示し、図2Bおよび2Cのレーン5〜10によって、2〜8℃または36〜38℃での保存の2週間および4週間後、これは依然として真実であることが確認される。3つの異なるロットにおいて、これらの様々な条件で約99%のHBsAgがアジュバントに依然として吸着した状態であった。
【0182】
他の安定性アッセイを、2〜8℃での保存の6カ月後に実施した(図2D)。3つのロットの各々に関して依然として約99%吸着した状態であった。
【0183】
図2中で使用した陽性対照は、1μgのHBsAgを含んでいた。Sペプチド(24kDa)に対応する1つのバンド、および凝集に特徴的なバンド(45kDaまで)が見られた。Pre−S2は見られなかった。
【0184】
8価ワクチンも、同様の方法でHBsAg吸着に関して試験した。1mlのサンプルを10分間3500rpmで遠心分離にかけた。上清は新たなチューブに除去し、DOC/TCAで沈殿させた。ペレットは200μlの抽出バッファーに再懸濁させ、5分間沸騰させ、10分間1300rpmで遠心分離にかけた。20μlのこの抽出物およびDOC/TCAで沈殿させた上清を、ウエスタンブロット用に12%SDS−PAGEに充填した。
【0185】
図4Aは時間ゼロでの8価ワクチンを示し、図4Bは2〜8℃での保存の8カ月後のワクチンを示す。図4Bのレーン3および6における任意の有意な染色の不在(対照レーン2および5中の1μgのHBsAgで見られるより確実に染色されていないこと)は、この保存期間中HBsAgの吸着は依然として安定状態であることを示す。
【0186】
液状/凍結乾燥多価ワクチン
3つの抗原成分を以下のように回収する:
− 3価D−T−Pw成分:水酸化アルミニウムアジュバントに吸着したジフテリアトキソイド、さらに水酸化アルミニウムアジュバントに吸着した破傷風トキソイド、および完全細胞百日咳抗原を含むD−T−Pw成分を調製した。D−T−Pw成分はリン酸アルミニウムアジュバントも含む。この成分はチメロサールを含むが、2−フェノキシエタノールは含まない。
【0187】
− HBsAg成分:HBsAgを組換え酵母菌から発現させ精製し、リン酸アルミニウム抗原に吸着させる[110]。
【0188】
− Hib複合体成分:Hib多糖をHib、系統20752から調製し、臭化シアンを用いた活性化およびアジピン酸ヒドラジドを用いた誘導体化後、約1:3の糖:担体重量比で、スペーサーをカルボジイミド系縮合によって破傷風トキソイドと共有結合させる。結合体はリン酸アルミニウムアジュバントに吸着させ[64]、次いでラクトースと共に凍結乾燥させる。
【0189】
D−T−Pw成分はHBsAg成分と混合させる。HBsAgのレベルは20μg/mlである。Dのレベルは60IU以上である。Tのレベルは120IU以上である。Pwのレベルは8IU以上である。白濁した懸濁液の形態である4価混合物は、ブチルゴム製ストッパーを有するガラスバイアルに水性形態でパッケージして、1用量(0.5ml)、2用量(1ml)または10用量(5ml)を含ませる。凍結乾燥Hib成分もバイアル中に置く。バイアル当たりの粉末の量は、バイアル当たり1用量、2用量または10用量での水戻し後、(糖として測定して)5μg/mlを得るように定量する。
【0190】
2つのバイアルは箱中に1つにパッケージする。箱は1バイアルの各成分(1用量または多用量バイアルのいずれか)または100バイアルの各成分のいずれかを含む。
【0191】
患者への投与用に、水性D−T−Pw−HBsAg物質をシリンジに取り出し、凍結乾燥複合体用バイアル中に導入する。凍結乾燥物質を再度活性化させた後、新しいニードルを介してそれをシリンジに再度取り出し、患者にすぐに投与することができる5価ワクチンを得る。様々な3用量の初回免疫処置スケジュール:2、4および6カ月;3、4、および5カ月;または6、10および14週を使用することができる。ワクチンは2年時の追加抗原刺激投与として使用することもできる。
【0192】
10用量のバイアルは、1バイアルで10患者を免疫処置するのに充分な物質を与える。水戻しは第1のシリンジを使用し、次いで1シリンジ当たり1用量で、新たなシリンジを用いて個々の用量を抽出する。他の(あまり好ましくない)アレンジメントでは、10用量を1シリンジに取り出し、ニードルは患者ごとに変える。
【0193】
本発明は単なる実施例によって記載しており、本発明の範囲および精神中に留まりながら変更形態を作製することができることは理解されるはずである。
【0194】
参考文献(これらの内容は、参考として本明細書に援用される)
【0195】
【化1】
【0196】
【化2】
【0197】
【化3】
【技術分野】
【0001】
本明細書で引用するすべての文書は、その全容を参照によって組み込む。
【0002】
本発明は、2つ以上の病原体由来の混合型免疫原を含むワクチンであり、したがってそのワクチンの投与が2つ以上の病原体に対して対象を同時に免疫処置することができる、組み合わせワクチンの製造の分野にある。詳細には本発明は、組み合わせワクチンの製造における界面活性剤の使用に関する。
【背景技術】
【0003】
2種以上の病原性生物由来の抗原を単一用量中に含むワクチンは、「多価」または「混合」ワクチンとして知られている。ジフテリア、破傷風および百日咳に対する防御用の3価ワクチン(「DTP」ワクチン)ならびに麻疹、おたふく風邪および風疹に対する防御用の3価ワクチン(「MMR」ワクチン)を含めた、ヒトに使用するための様々な組み合わせワクチンが欧州および米国において承認されている。
【0004】
組み合わせワクチンには患者にとって受ける注射の回数が少なくなるという利点があり、特に小児予防接種に関して、これはコンプライアンスが高まるという臨床上の利点をもたらす(例えば、参考文献1の29章を参照)。しかし同時に、組み合わせワクチンは、抗原と他の成分の間の物理的および生化学的不適合性、免疫学的干渉、および安定性を含めた要因による製造上の難点を示す。
【0005】
ワクチンに非抗原成分を含有させることは必要であるが、困難を引き起こす可能性がある。界面活性剤は組み合わせワクチンにおいて特に問題である。それは、ある抗原は最適活性のために界面活性剤を必要とする可能性があるが、別の抗原は界面活性剤が存在すると負の影響を受ける可能性があるからである。さらに、界面活性剤が一般に安全であると認められていても、小児ワクチンに界面活性剤を含有させることはある患者群にとって問題である。
【0006】
ワクチン分野で特に興味深いのは、ポリオキシエチレンソルビタンエステル界面活性剤、特にポリソルベート20(「Tween20」またはポリオキシエチレンソルビタンモノラウレートとしても知られている)およびポリソルベート80(「Tween80」またはポリオキシエチレンソルビタンモノオレエートとしても知られている)種である。ポリソルベート20は1価HAVRIX(商標)A型肝炎ワクチンにおいて見られ、ポリソルベート80はTRIPEDIA(商標)およびINFANRIX(商標)系のワクチンなどの組み合わせワクチンにおいて見られる。これら2つの界面活性剤は、液状ロタウイルスワクチンを安定化させるためにも使用されている[2]。
【0007】
ポリソルベートはB型肝炎表面抗原(「HBsAg」)を含む組み合わせワクチンの製造においても使用されており、例えば参考文献3(特許文献1)および4(特許文献2)は、Tween20、Tween80またはTriton X−100などの非イオン性界面活性剤を添加することによってHBsAgのリン脂質成分への干渉が回避されている、4価D−T−P−HBsAgワクチンを作製するための方法を開示する。参考文献3(特許文献1)および4(特許文献2)の図2(本明細書の図1)中のデータから、界面活性剤はHBsAgの抗原性を維持するために必要とされるが、他の成分にはそれほど重要でないことが示される。試験された最高界面活性剤濃度はHBsAg20μg/mlに対して10μg/mlであり、この濃度はまた、最良の抗原性を示した。
【0008】
参考文献3(特許文献1)および4(特許文献2)の方法では、HBsAgを精製した後に非イオン性界面活性剤が添加される。しかし、HBsAgの精製後に界面活性剤を添加することは最適ではない。それは、製造中に別の処理ステップを必要とし、それにより処理時間が増大し、HBsAgが汚染されるリスクも高まるからである。組み合わせワクチンの作製時に汚染成分が使用されると、1価ワクチンの作製時より最終的な損失は大きく、例えば汚染HBsAg成分が清潔なD−T−P成分と混合されると、HBsAgのみでなくD−T−P−HBsAg混合物全体を廃棄しなければならない。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】国際公開第02/055105号パンフレット
【特許文献2】米国特許出願公開第2004/0048336号明細書
【発明の概要】
【発明が解決しようとする課題】
【0010】
したがって、非イオン性界面活性剤を含有する組み合わせワクチン用に、製造方法において界面活性剤を別の成分として添加する必要がない方法が依然として必要とされている。
【課題を解決するための手段】
【0011】
(要旨)
抗原を精製した後に非イオン性界面活性剤を抗原に添加する[3、4]のではなく、本発明は抗原の精製中に非イオン性界面活性剤を使用する。したがって、この界面活性剤は最終組み合わせワクチンにおいてその機能を果たすことができるが、汚染のリスク(したがって、調製後の組み合わせワクチン全体の損失のリスクも)は低下する。
【0012】
したがって本発明は、組み合わせワクチンを調製するための方法であって、ワクチンが(i)非イオン性界面活性剤、(ii)B型肝炎ウイルス(HBV)表面抗原、および(iii)少なくとも1つの非HBV病原体由来の抗原を含み、(i)組換え酵母細胞を非イオン性界面活性剤の存在下で破壊して精製HBsAg成分を得るステップを含む、酵母細胞からHBV表面抗原を精製すること、および(ii)精製HBsAg成分を非HBV病原体由来の少なくとも1つの他の抗原と合わせて組み合わせワクチンを得ることを含む方法を提供する。
【0013】
前に記載した汚染の難点を回避するために、この方法は、HBsAg精製後に非イオン性界面活性剤を別の成分として添加するステップを含まない。界面活性剤(または他の界面活性剤、イオン性または非イオン性)が、組み合わせワクチンを得るためにHBsAgと組み合わせる他の抗原成分中に存在する可能性はあるが、それ自体を別の成分として界面活性剤を添加することは回避される。したがって、界面活性剤は、別の成分として精製HBsAg成分に添加されず、抗原を組み合わせる際には添加されない。
【0014】
本発明は、組み合わせワクチンを調製するための方法であって、ワクチンが(i)非イオン性界面活性剤、(ii)B型肝炎ウイルス(HBV)表面抗原、および(iii)少なくとも1つの非HBV病原体由来の抗原を含み、精製HBV表面抗原を非HBV病原体由来の少なくとも1つの他の抗原と合わせて組み合わせワクチンを得るステップを含み、HBsAg発現組換え酵母細胞を非イオン性界面活性剤の存在下で破壊するプロセスによって精製HBV表面抗原が調製される方法も提供する。再度、界面活性剤の別個の添加は回避される。
【0015】
本発明は、(i)非イオン性界面活性剤、(ii)B型肝炎ウイルス(HBV)表面抗原、および(iii)少なくとも1つの非HBV病原体由来の抗原を含み、HBsAg発現組換え酵母細胞を非イオン性界面活性剤の存在下で破壊するプロセスによってHBV表面抗原が調製される、免疫原性組成物も提供する。再度、界面活性剤の別個の添加を回避しながらHBsAgを調製した。この生成物は、精製中に使用する非イオン性界面活性剤がHBsAg粒子中に保持される可能性があるので、異なる方法によってHBsAgを調製した生成物と区別することができる。
【発明を実施するための形態】
【0016】
非イオン性界面活性剤
本発明は、様々な非イオン性界面活性剤[5]、詳細にはワクチン製剤において見られる非イオン性界面活性剤を使用することができる。有機界面活性剤が好ましい。これらは典型的には、アルキレンオキシド(例えば、エチレンオキシド)と、高級アルコール、脂肪酸、アルキルフェノール、アルキルアミン、または少なくとも1つの活性水素原子を有する他の適切な化合物との反応生成物である。大部分の界面活性剤に関して、最も一般的なアルコール、アミンおよび酸はC8〜C18の範囲の炭素鎖長を有する。最も一般的なアルキルフェノールは、ノニルフェノールおよびオクチルフェノールである。ポリ(オキシエテン)残基を含む界面活性剤が特に好ましい。
【0017】
例えば、ポリオキシエチレンソルビタンエステル界面活性剤(一般にTweenと呼ばれる)、特にポリソルベート20およびポリソルベート80;DOWFAX(商標)の商品名で販売されているエチレンオキシド(EO)、プロピレンオキシド(PO)、および/またはブチレンオキシド(BO)のコポリマー、例えば直鎖状EO/POブロックコポリマー;反復エトキシ(オキシ−1,2−エタンジイル)基の数が変わり得るオクトキシノール、オクトキシノール−9(Triton X−100、またはt−オクチルフェノキシポリエトキシエタノール)が特に興味深い;(オクチルフェノキシ)ポリエトキシエタノール(IGEPAL CA−630/NP−40);ラウリル、セチル、ステアリルおよびオレイルアルコール(Brij界面活性剤として知られる)から誘導されるポリオキシエチレン脂肪族エーテル、例えばトリエチレングリコールモノラウリルエーテル(Brij30);およびソルビタンエステル(一般にSPANと呼ばれる)、例えばソルビタントリオレート(Span85)およびソルビタンモノラウレートだけには限られないが、これらを含めた界面活性剤と共に本発明を使用することができる。
【0018】
本発明は、ポリソルベート20と共に使用するのに特に適している。この界面活性剤は、ワクチン内を含めてヒトへの投与に関する立証済みの安全性プロファイルを有する。
【0019】
界面活性剤はその「HLB」(親水/親油平衡)によって分類することができる。本発明の好ましい界面活性剤は、少なくとも10、好ましくは少なくとも15、およびより好ましくは少なくとも16のHLBを有する。
【0020】
非イオン性界面活性剤は、本発明の組成物の成分である。患者への多用量の界面活性剤の投与を回避するために、組成物中の界面活性剤の濃度は30μg/mlを超えず、例えば25μg/ml以下、20μg/ml以下、15μg/ml以下、10μg/ml以下、5μg/ml以下などであることが好ましい。10μg/ml以下の濃度が好ましい。
【0021】
界面活性剤の濃度を特定するための代替として、組成物中の界面活性剤の量は、HBsAg各100μgに対して50μg未満(例えば40μg以下、30μg以下、25μg以下、20μg以下、15μg以下、10μg以下など)であることが好ましい。参考文献3および4から、界面活性剤:HBsAgの質量比が50%未満であることも示唆される。HBsAg100μg当たり25μg未満の界面活性剤が好ましい。
【0022】
以下でさらに詳細に言及するように、本発明の好ましい方法は、ジフテリアトキソイドおよび破傷風トキソイドを含む予め混合した成分を使用する。このD−T成分は非イオン性界面活性剤を実質的に含まず、および詳細にはポリソルベート20および80を含まないことが好ましい。同様に、予め混合したD−T−Pw成分は非イオン性界面活性剤、例えばポリソルベート20および80を含まない。
【0023】
B型肝炎ウイルス表面抗原
B型肝炎ウイルス(HBV)は、ウイルス肝炎を引き起こす既知の因子の1つである。HBVビリオンは外側タンパク質コートまたはカプシドに囲まれた内部コアからなり、ウイルスコアはウイルスDNAゲノムを含む。カプシドの主な成分は、典型的には約24kDaの分子量を有する226アミノ酸ポリペプチドである、HBV表面抗原、またはより一般的に「HBsAg」として知られるタンパク質である。HBsAgを含むすべての既存のB型肝炎ワクチンは、この抗原を通常なワクチンに投与すると、それはHBV感染に対して防御する抗HBsAg抗体の産生を刺激する。
【0024】
ワクチン製造用に、HBsAgは2つの方法で作製することができる。第1の方法は、HBV感染中に多量のHBsAgが肝臓中で合成され血流中に放出されるので、慢性B型肝炎キャリアの血漿から粒子形に抗原を精製することを含む。第2の方法は、組換えDNA法によってタンパク質を発現させることを含む。本発明の方法と共に使用するためのHBsAgは、酵母細胞中で組換えによって発現される。適切な酵母菌には、サッカロミセス属(出芽酵母(S.cerevisiae)など)またはハネンズラ属(Hanensula)(H.ポリモルファ(H.polymorpha)など)宿主がある。
【0025】
天然HBsAgと異なり(すなわち、血漿精製産物中と同様に)、酵母菌によって発現されるHBsAgは一般にグリコシル化されておらず、これは本発明と共に使用するためのHBsAgの最も好ましい形態である。酵母菌によって発現されるHBsAgは非常に免疫原性が高く、血液産物の汚染のリスクなしで調製することができる。
【0026】
HBsAgは一般に、リン脂質を含む脂質マトリクスを含む、実質的に球状の粒子の形(約20nmの平均径)であるはずである。酵母菌によって発現されるHBsAg粒子は、天然のHBVビリオン中に見られないホスファチジルイノシトールを含み得る。粒子は免疫系を刺激するために無毒量のLPSも含み得る[6]。
【0027】
HBsAgは、HBV亜型adw2に由来することが好ましい。
【0028】
HBsAgを精製するための多くの方法が当技術分野で知られている(例えば、参考文献7〜33を参照)。これらの方法は1価HBsAg調製物を作製する際に使用するために開示されるが、参考文献3および4中に開示された方法と異なり、これらはいずれも、特に組み合わせワクチンにおいて使用するためのHBsAgの精製に関するものではない。これらの方法および他の方法のいずれかを使用することができ、ただし、この方法は、組換え酵母細胞中での発現後の抗原を精製するのに適しており、この精製は、酵母細胞を非イオン性界面活性剤の存在下で破壊するステップを含む。
【0029】
HBsAgを精製するのに好ましい方法は、細胞破壊後の、限外濾過、サイズ排除クロマトグラフィー、アニオン交換クロマトグラフィー、超遠心分離、脱塩および滅菌濾過を含む。溶解物は(例えば、ポリエチレングリコールを使用して)細胞破壊後に沈殿させ、HBsAgを限外濾過用に溶液中に残しておいてよい。
【0030】
精製後HBsAgに透析を施すことができ(例えばシステインと)、これを使用して、HBsAg調製中に使用された可能性があるチメロサールなどの任意の水銀防腐剤を除去することができる[30、34]。
【0031】
HBsAgの量は典型的にはマイクログラムで表され、ワクチン用量当たりのHBsAgの典型的な量は10μgである。
【0032】
「S」配列以外に、表面抗原はプレ−S配列の全体または一部分、例えばプレ−S1および/またはプレ−S2配列の全体または一部分を含むことができる。
【0033】
非HBV抗原
本発明の免疫原性組成物は、少なくとも1つの非HBV病原体由来の少なくとも1つの防御抗原を含む。非HBV病原体はウイルス性および/または細菌性であってよい。
【0034】
典型的なウイルス性病原体には、ポリオウイルス、A型肝炎ウイルス、インフルエンザウイルス、麻疹ウイルス、おたふく風邪ウイルス、風疹ウイルスおよび水痘帯状疱疹ウイルスがあるが、これらだけには限られない。
【0035】
典型的な細菌性病原体には、ジフテリア菌(Corynebacterium diphtheriae);破傷風菌(Clostridium tetani);百日咳菌(Bordetella pertussis);b型および非分類系統を含めたインフルエンザ菌(Haemophilus influenzae);血清型A、B、C、W135および/またはYを含めた髄膜炎菌(Neisseria meningitidis);血清型4、6B、9V、14、18C、19F、および23Fを含めた肺炎連鎖球菌(Streptococcus pneumoniae);およびカタラリス菌(Moraxella catarrhalis)があるが、これらだけには限られない。
【0036】
ジフテリア菌はジフテリアを引き起こす。注射後に特異的抗トキシン抗体を誘導する能力を保持しながら、ジフテリア毒素を(例えば、ホルマリンまたはホルムアルデヒドを使用して)処理して毒性を除去することができる。これらのジフテリアトキソイドはジフテリアワクチンにおいて使用され、参考文献1の13章中により詳細に開示される。好ましいジフテリアトキソイドは、ホルムアルデヒド処理によって調製されるジフテリアトキソイドである。ジフテリアトキソイドは増殖培地(例えば、Fenton培地、またはLinggoudおよびFenton培地)中でジフテリア菌を増殖させることによって得ることができ、培地にウシ抽出物を補充することが可能であり、後にホルムアルデヒド処理、限外濾過および沈殿が可能である。トキソイド物質は、滅菌濾過および/または透析を含む方法によって次いで処理することができる。
【0037】
破傷風菌は破傷風を引き起こす。破傷風毒素を処理して防御トキソイドを与えることができる。これらのトキソイドは破傷風ワクチンにおいて使用され、参考文献1の27章中により詳細に開示される。好ましい破傷風トキソイドは、ホルムアルデヒド処理によって調製される破傷風トキソイドである。破傷風トキソイドは増殖培地(例えば、ウシカゼイン由来のLatham培地)中で破傷風菌を増殖させることによって得ることができ、後にホルムアルデヒド処理、限外濾過および沈殿が可能である。滅菌濾過および/または透析を含む方法によって、次いで物質を処理することができる。
【0038】
百日咳菌は百日咳を引き起こす。ワクチン中の百日咳抗原は、細胞性(完全細胞、不活性百日咳菌細胞の形;「wP」)あるいは無細胞(「aP」)のいずれかである。細胞性百日咳抗原の調製は充分に文書化されており[例えば、参考文献1の21章を参照]、例えば、それは培養第I期の百日咳菌の熱不活化によって得ることができる。無細胞抗原を使用する場合、1つ、2つまたは(好ましくは)3つの以下の抗原が含まれる:(1)解毒した百日咳毒素(百日咳トキソイド、すなわち「PT」);(2)繊維状ヘマグルチニン(「FHA」);(3)ペルタクチン(「69キロダルトンの外膜タンパク質」としても知られている)。この3つの抗原は、改変型Stainer−Scholte液状培地中で増殖させた百日咳菌培養物からの単離によって調製することが好ましい。PTおよびFHAは発酵ブロスから単離することができ(例えば、ヒドロキシアパタイトゲル上での吸着によって)、一方ペルタクチンは、熱処理および凝集反応(例えば、塩化バリウムを使用する)によって細胞からから抽出することができる。抗原は連続的なクロマトグラフィーおよび/または沈殿ステップにおいて精製することができる。PTおよびFHAは疎水性クロマトグラフィー、親和性クロマトグラフィーおよびサイズ排除クロマトグラフィーによって精製することができる。ペルタクチンはイオン交換クロマトグラフィー、疎水性クロマトグラフィーおよびサイズ排除クロマトグラフィーによって精製することができる。FHAおよびペルタクチンは、本発明による使用の前にホルムアルデヒドで処理することができる。ホルムアルデヒドおよび/またはグルタルアルデヒドを用いた処理によってPTを解毒することが好ましい。この化学的解毒手順に対する代替として、PTは突然変異誘発によって酵素活性が低下した突然変異PTであってよいが[35]、化学的処理による解毒が好ましい。
【0039】
b型インフルエンザ菌(「Hib」)は細菌性髄膜炎を引き起こす。Hibワクチンは典型的には被膜糖抗原に基づいており[例えば、参考文献1の14章]、その調製は充分に文書化されている[例えば、参考文献36〜45]。Hib糖は担体タンパク質と結合して、特に子供においてその免疫原性を増大させる。典型的な担体タンパク質は、破傷風トキソイド、ジフテリアトキソイド、ジフテリアトキソイドのCRM197誘導体、インフルエンザ菌タンパク質D、および血清型Bの髄膜炎菌由来の外膜タンパク質複合体である。破傷風トキソイドは、「PRP−T」と一般に呼ばれる生成物において使用される、好ましい担体である。臭化シアンを使用してHib被膜多糖を活性化させ、活性糖をアジピン酸リンカー((1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミドなど)、典型的には塩酸塩)と結合させ、次いでリンカー−糖物体と破傷風トキソイド担体タンパク質を反応させることによってPRP−Tを作製することができる。複合体の糖部分は、Hib細菌から調製した完全長のポリリボシルリビトールリン酸(PRP)、および/または完全長PRPの断片を含み得る。1:5(すなわち、過剰なタンパク質)と5:1(すなわち、過剰な糖)の間の糖:タンパク質比(w/w)、例えば1:2と5:1の間の比、および1:1.25と1:2.5の間の比を有する複合体を使用することができる。好ましいワクチンでは、しかし、糖と担体タンパク質の重量比は1:2.5と1:3.5の間である。破傷風トキソイドが抗原および担体タンパク質の両方として存在するワクチンでは、その時複合体中の糖と担体タンパク質の重量比は1:0.3と1:2の間である可能性がある[46]。Hib複合体の投与は0.15μg/ml以上、およびより好ましくは1μg/ml以上の抗PRP抗体濃度をもたらすことが好ましく、これらは標準的な応答閾値である。
【0040】
髄膜炎菌は細菌性髄膜炎を引き起こす。生物の被膜多糖に基づいて、A、B、C、H、I、K、L、29E、W135、X、YおよびZを含めたN.meningitidisの様々な血清型が同定されてきている。疾患と最も関係がある血清型は、A、B、C、W135およびYである。血清型A、C、W135およびYに対する現在のワクチンは被膜糖抗原に基づくものであるが、この手法は血清型Bには適しておらず、したがってタンパク質抗原および外膜小胞を代わりに使用する。被膜糖は担体タンパク質と結合して免疫原性を増大させる。典型的な担体タンパク質は、破傷風トキソイド、ジフテリアトキソイド、ジフテリアトキソイドのCRM197誘導体、およびインフルエンザ菌タンパク質Dである。複合体の糖部分は、髄膜炎菌から調製した完全長の糖、および/またはその断片を含み得る。血清型Cの糖は、OAc+またはOAc−系統のいずれかから調製することができる。血清型Aの糖に関しては、少なくとも50%(例えば少なくとも60%、70%、80%、90%、95%以上)のマンノースアミン残基が、C−3位置においてO−アセチル化されることが好ましい。1:10(すなわち、過剰なタンパク質)と10:1(すなわち、過剰な糖)の間の糖:タンパク質比(w/w)、例えば1:5と5:1の間、1:2.5と2.5:1の間の比、または1:1.25と1.25:1の間の比を有する髄膜炎菌複合体を使用することができる。複合体の投与によって、少なくとも4倍、および好ましくは少なくとも8倍の関連する血清型の血清細菌アッセイ(SBA)における力価の増大をもたらすことが好ましい。SBAにおける力価は、ベビーラビット補体またはヒト補体を使用して測定することができる[47]。
【0041】
肺炎連鎖球菌は細菌性髄膜炎を引き起こす。Hibおよび髄膜炎菌に関しては、既存のワクチンは被膜糖に基づく。肺炎連鎖球菌の2つ以上の血清型、および詳細には少なくとも血清型6B、14、19Fおよび23F由来の糖を含むことが好ましい。他の血清型は1、3、4、5、7F、9Vおよび18Cから選択されることが好ましい。例えば、23の異なる血清型に由来する多糖の混合物が広く使用されており、5〜11の異なる血清型に由来する多糖を含む複合体ワクチンも同様である[48]。例えば、PrevNar(商標)[49]は7つの血清型(4、6B、9V、14、18C、19F、および23F)に由来する複合体抗原を含む。糖は担体タンパク質と結合することが好ましい[例えば、参考文献50〜52]。典型的な担体タンパク質は、破傷風トキソイド、ジフテリアトキソイド、ジフテリアトキソイドのCRM197誘導体、およびインフルエンザ菌タンパク質Dである。PrevNar(商標)製品中の糖は、0.5ml用量当たり2μgの各糖(4μgの血清型6B)で、還元的アミノ化によってCRM197と個別に結合する。肺炎球菌に由来する糖抗原の使用に対する代替として、組成物は1つまたは複数のポリペプチド抗原を含むことができる。数系統の肺炎球菌のゲノム配列が利用可能であり[53、54]、これらは逆ワクチン法に供して[55〜58]、適切なポリペプチド抗原を同定することができる[59、60]。例えば組成物は、参考文献61中に定義されるように、以下の抗原:PhtA、PhtD、PhtB、PhtE、SpsA、LytB、LytC、LytA、Sp125、Sp101、Sp128、Sp130およびSp130の1つまたは複数を含むことができる。組成物は、2つ以上(例えば2、3、4、5、6、7、8、9、10、11、12、13または14個の)これらの抗原を含むことができる。いくつかの実施形態では、組成物は肺炎球菌に由来する糖とポリペプチド抗原の両方を含むことができる。これらは単純な混合物において使用することができ、あるいは肺炎球菌糖抗原は肺炎球菌タンパク質と結合させることが可能である。このような実施形態に適した担体タンパク質には、前述の肺炎球菌タンパク質抗原がある[61]。
【0042】
モラクセラカタラーリスは中耳炎および副鼻腔炎を引き起こし、時折咽頭炎の原因となる。参考文献62中に総説されるように、ワクチンは現在研究中である。
【0043】
HBVと同様に、HAVは肝炎を引き起こす。HAVワクチンは参考文献1の15章中に開示されている。好ましいHAV成分は不活性ウイルスに基づき、不活性化はホルマリン処理によって実施することができる。ウイルスはMRC−5細胞などのヒト胎児肺2倍体線維芽細胞において増殖し得る。好ましいHAV系統はHM175であるが、CR326Fを使用することもできる。ウイルス増殖を可能にする条件下で、細胞を増殖させることが可能である。細胞を溶解し、生成した懸濁液は限外濾過およびゲル透過クロマトグラフィーによって精製することができる。
【0044】
ポリオウイルスは灰白髄炎を引き起こす。経口ポリオウイルスワクチンを使用する代わりに、参考文献1の24章中により詳細に開示するように、本発明は不活化ポリオウイルスワクチン(IPV)を使用する。細胞培養中にポリオウイルスを増殖させることが可能であり、好ましい培養はサル腎臓に由来するVero細胞系を使用する。Vero細胞は、都合良くマイクロキャリア培養することができる。増殖後、限外濾過、ダイアフィルトレーション、およびクロマトグラフィーなどの技法を使用してビリオンを精製することができる。患者に投与する前に、ポリオウイルスは不活化しなければならず、これはホルムアルデヒドを用いた処理によって実施することができる。灰白髄炎は、ポリオウイルスの3つの型の1つによって引き起こされる可能性がある。3つの型は類似しており同一の症状を引き起こすが、これらは抗原的に非常に異なり、1つの型による感染は他の型による感染に対して防御しない。したがって、本発明では3つのポリオウイルス抗原:ポリオウイルス1型(例えば、Mahoney系統)、ポリオウイルス2型(例えば、MEF−1系統)、およびポリオウイルス3型(例えば、Saukett系統)を使用することが好ましい。これらのウイルスは個別に増殖、精製および不活化し、次いで組み合わせて本発明と共に使用するための多量の3価混合物を得ることが好ましい。IPVの量は典型的には「DU」単位(「D抗原単位」[63])で表す。
【0045】
麻疹、おたふく風邪および風疹ウイルスに対して防御するための抗原は、知られている1価および3価(「MMR」)ワクチンにおいて見られるように、典型的には生ウイルスである。麻疹ウイルスワクチンは、参考文献1の19章中により詳細に記載されている。おたふく風邪ウイルスワクチンは、参考文献1の20章中により詳細に記載されている。風疹ウイルスワクチンは、参考文献1の26章中により詳細に記載されている。典型的な麻疹ウイルス系統には、Moraten;Connaught;Schwarz;Edmonston−Zagreb;CAM−70;AIK−C;TD97;Leningrad−16;Shanghai−191などがある。SchwarzおよびMoraten系統が、米国および欧州で最も一般的に使用されている。典型的なおたふく風邪ウイルス系統には、Jeryl Lynn;RIT4385;Urabe;Hoshino;Rubini;Leningrad−3;Leningrad−Zagreb;Miyahara;Torii;NKM−46;S−12などがある。Jeryl Lynn、RIT4385、UrabeおよびLeningrad−Zagreb系統が、最も一般的な世界規模の系統である。典型的な風疹ウイルス系統には、RA27/3;Matsuba;TCRB19;Takahashi;Matsuura;TP−336などがある。RA27/3は、西欧諸国で使用されている最も一般的な系統である。
【0046】
水痘に対して防御するためのVZV抗原は典型的には、Oka系統のウイルスに基づく生ウイルスである。VZVワクチンは、参考文献1の28章中により詳細に記載されている。
【0047】
インフルエンザウイルス抗原は、参考文献1の17および18章中により詳細に記載されている。広義には、インフルエンザウイルスワクチンは生ウイルスまたは不活化ウイルスに基づいてよく、不活化ワクチンは完全ウイルス、「スプリット」ウイルスまたは精製表面抗原(ヘマグルチニンおよびノイラミニダーゼ含む)に基づいてよい。ワクチンを調製するために使用するウイルスは、卵または細胞培養物のいずれかで増殖させることが可能である。インフルエンザウイルス用のワクチン系統は季節ごとに変わる。現在の大流行間期では、ワクチンは典型的には2つのインフルエンザA系統(H1N1およびH3N2)および1つのインフルエンザB系統を含み、3価ワクチンが典型的である。本発明は、(特にインフルエンザAウイルスの)H2、H5、H7またはH9亜型系統などの流行系統(すなわち、ワクチンのレシピエントおよび一般的なヒト集団がそれに対する免疫がない系統)由来のウイルスも使用することができ、流行系統用のインフルエンザワクチンは1価であってよく、あるいは流行系統によって補充した通常の3価ワクチンに基づくものであってよい。インフルエンザウイルスはリアソータント系統であってよく、逆遺伝学的技法によって得ることができている。ウイルスは弱毒化することができる。ウイルスは温度感受性であってよい。ウイルスは低温適応性であってよい。
【0048】
ワクチンにおいて使用するためのこれらの病原体由来の抗原成分は、略称で一般に呼ばれる:「D」はジフテリアトキソイド、「T」は破傷風トキソイド、「P」は百日咳抗原、「Pa」は無細胞、「Pw」は細胞性、「Hib」はb型インフルエンザ菌被膜糖、「MenA」、「MenB」、「MenC」、「MenW」および「MenY」はそれぞれの髄膜炎菌の血清型、「IPV」は不活化ポリオウイルス、および「Spn」は肺炎球菌
。
【0049】
抗原成分をHBsAgと組み合わせて多価組成物を調製するとき、抗原は個別に加えることができ、あるいはHBsAgと組み合わせる前に抗原を予め混合することができる。DおよびT抗原を使用する場合、予め混合したD−T成分を使用することが好ましい。この2価成分は本発明の方法中で使用することができる、例えばそれをHBsAgと組み合わせて、3価D−T−HBV成分を作製することができる。代替として、D−T成分は他の非HBV抗原(例えば、無細胞百日咳抗原)と組み合わせることができ、したがってその成分はHBsAgなどと組み合わせることができる。D、TおよびPw抗原を使用する場合、予め混合したD−T−Pw成分を使用すること、したがって本発明の方法中にこの成分を使用することが好ましい。
【0050】
アジュバントを本発明の組成物中に含めるとき、アジュバントは様々な段階で加えることもできる。典型的には、本発明の方法中で使用する前に抗原をアジュバントと組み合わせているはずであるが(例えば、2価D−T混合物は本発明の方法中で使用する前にアルミニウム塩アジュバントに吸着しているはずであり、これはトキソイドを別々に調製し、その各々を水酸化アルミニウムアジュバントに別々に吸着させ、次いで2つの吸着トキソイドを(場合によっては他のアジュバントと)混合して本発明の方法中で使用するための物質を得ることによって都合良く実施することができる)、抗原を混合した後にアジュバントを加えること、またはアジュバントに抗原を加えることも可能である(例えば、水性アジュバントで始めるために、したがって抗原は個別あるいは予め混合して加える)。以下に記載するように、HBsAg成分は、非HBV抗原成分と組み合わせる前にリン酸アルミニウムアジュバントに吸着させることが好ましい。
【0051】
本発明の好ましい組成物は、HBsAg以外にD、TおよびP抗原(例えば、参考文献3および4)を少なくとも含む。特に好ましい組成物は以下の組合せである:
− HBsAg、D、T
− HBsAg、D、T、Pw
− HBsAg、D、T、Pw、Hib
− HBsAg、D、T、Pw、Hib、MenA、MenC
− HBsAg、D、T、Pw、Hib、MenA、MenC、MenW135
− HBsAg、D、T、Pw、Hib、MenA、MenC、MenY
− HBsAg、D、T、Pw、Hib、MenA、MenC、MenW135、MenY
− HBsAg、D、T、Pa
− HBsAg、D、T、Pa、Hib
− HBsAg、D、T、Pa、ポリオウイルス
− HBsAg、D、T、Pa、ポリオウイルス、Hib
− HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC
− HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC、MenA
− HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC、MenY
− HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC、MenW135
− HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC、MenA、MenW135、MenY
− HBsAg、Hib
− HBsAg、A型肝炎ウイルス。
【0052】
これらの組成物は列挙した抗原からなっていてよく、あるいは他の病原体由来の抗原をさらに含むことができる。したがって、これらは別々に、あるいは他のワクチンの成分として使用することができる。
【0053】
本発明のいくつかの実施形態では、組成物は5価D−T−Pa−HBV−IPVではない[30]。したがって組成物は、Pw成分および/または少なくとも1つの複合体を含むことができる。
【0054】
アジュバント
本発明の好ましい免疫原性組成物はアジュバントを含み、このアジュバントは1つまたは複数のアルミニウム塩、および特にリン酸アルミニウムアジュバントおよび/または水酸化アルミニウムアジュバントを含むことが好ましい。
【0055】
本発明の方法中で使用する抗原成分は、その方法中で使用する前にアルミニウムアジュバントを含む、すなわち抗原成分は、アジュバントと「予め混合されている」あるいはアジュバントに「予め吸着している」ことが好ましい。
【0056】
HBsAgおよびジフテリアトキソイドを含む組成物において、ジフテリアトキソイドを水酸化アルミニウムアジュバントに吸着させることが可能である。
【0057】
HBsAgおよび破傷風トキソイドを含む組成物において、破傷風トキソイドを水酸化アルミニウムアジュバントに吸着させることは可能であるが、これは必要ではない(例えば、全破傷風トキソイドの0〜10%の間の吸着を使用することができる)。
【0058】
HBsAgおよび完全細胞百日咳抗原を含む組成物において、wP抗原は水酸化アルミニウムアジュバントおよび/またはリン酸アルミニウムアジュバントと組み合わせることが好ましい。
【0059】
HBsAgおよび無細胞百日咳抗原を含む組成物において、百日咳抗原は1つまたは複数のアルミニウム塩アジュバントに吸着させることが可能であり、あるいは非吸着状態で加えることが可能である。
【0060】
ペルタクチンが組成物中に存在する場合、ペルタクチンは、本発明の方法中で使用する前に水酸化アルミニウムアジュバントに吸着することが好ましい。PTおよびFHAは、本発明の方法中で使用する前に、水酸化アルミニウムアジュバントまたはリン酸アルミニウムに吸着させることが可能である。好ましい実施形態では、本発明の方法中で使用する前にPT、FHAおよびペルタクチンを別々に、水酸化アルミニウムに予め吸着させる。
【0061】
HBsAgおよびHib抗原を含む組成物において、Hib複合体は非吸着状態であり得るが、それはリン酸アルミニウムアジュバントに吸着することが好ましい[64]。このような吸着は、D−T−Pw−Hib−HBsAg抗原を含むワクチンにおいて特に有用である。他の複合抗原(例えば、髄膜炎菌、肺炎球菌)を同様にアルミニウム塩(例えば、ホスフェート)に吸着させることが可能であり、あるいはこれらは非吸着状態であってよい[65]。
【0062】
IPV抗原は典型的には、本発明の方法中で使用する前に如何なるアジュバントにも吸着しないが、しかしIPV抗原は、他の成分に源を発するアルミニウムアジュバントに吸着した状態になり得る。
【0063】
参考文献66中に記載された方法を使用して、組成物中のHBsAgをリン酸アルミニウムに吸着させることが可能である。リン酸アルミニウムへの吸着はよく知られているENGERIX−B(商標)製品とは対照的であるが(この場合HBsAgは水酸化アルミニウムに吸着する)、HEPACCINE(商標)およびRECOMBIVAX(商標)製品と同じである。参考文献67中に言及されるように、リン酸アルミニウムは水酸化アルミニウムより良いHBsAgのアジュバントである可能性がある。(よく知られているENGERIX−B(商標)製品中と同様に)HBsAgは最終ワクチン中の水酸化アルミニウムアジュバントに吸着させることが可能であり、あるいは非吸着状態を保ち得るが、それは一般にリン酸アルミニウムアジュバントに吸着するはずである。さらにHBsAgは、本発明の方法中で使用する前にリン酸アルミニウムに予め吸着させることが好ましい。
【0064】
本発明の方法が、ジフテリアトキソイドと破傷風トキソイドがHBsAgとそれらを組み合わせる前に混合されている成分を使用する場合、このD−T混合物は、DおよびT抗原が吸着する水酸化アルミニウムアジュバントを含むことが好ましい。
【0065】
本発明の方法が、ジフテリアトキソイド、破傷風トキソイドおよび完全細胞百日咳抗原がHBsAgとそれらを組み合わせる前に混合されている成分を使用する場合、このD−T−Pw混合物は、DおよびT抗原が吸着する水酸化アルミニウムアジュバントと、リン酸アルミニウムアジュバントの両方を含むことが好ましい。
【0066】
現在使用されているアルミニウムアジュバントは典型的には、「水酸化アルミニウム」または「リン酸アルミニウム」アジュバントのいずれかと呼ばれている。しかし、いずれも存在する実際の化学化合物を正確に記載していないので、これらは便宜上の名称である(例えば、参考文献68の9章を参照)。本発明は、アジュバントとして一般に使用される「水酸化物」または「リン酸」塩のいずれかを使用することができる。
【0067】
「水酸化アルミニウム」として知られるアジュバントは典型的にはオキシ水酸化アルミニウム塩であり、これは通常少なくとも一部分が結晶である。式AlO(OH)によって表すことができるオキシ水酸化アルミニウムは、赤外線(IR)分光法によって、特に1070cm−1での吸着バンドおよび3090〜3100cm−1での強いピークの存在によって、水酸化アルミニウムAl(OH)3などの他のアルミニウム化合物と識別することができる(参考文献68の9章)。
【0068】
「リン酸アルミニウム」として知られるアジュバントは典型的には、少量のサルフェートもしばしば含むヒドロキシリン酸アルミニウムである。それらは沈殿によって得ることができ、沈殿中の反応条件および濃度は、塩中のホスフェートとヒドロキシルの置換度に影響を与える。ヒドロキシリン酸は一般に0.3と0.99の間のPO4/Alモル比を有する。ヒドロキシリン酸はヒドロキシル基の存在によって厳密なAlPO4と識別することができる。例えば、(例えば、200℃に加熱したときの)3164cm−1でのIRスペクトルバンドは、構造ヒドロキシルの存在を示す(参考文献68の9章)。
【0069】
リン酸アルミニウムアジュバントのPO4/Al3+モル比は、一般に0.3と1.2の間、好ましくは0.8と1.2の間、およびより好ましくは0.95±0.1であるはずである。リン酸アルミニウム、特にヒドロキシリン酸塩は一般に非晶質であるはずである。典型的なアジュバントは、0.6mgAl3+/mlで含まれる0.84と0.92の間のPO4/Alモル比を有する非晶質ヒドロキシリン酸アルミニウムである。リン酸アルミニウムは一般に微粒子であるはずである。粒子の典型的な直径は、任意の抗原吸着後に0.5〜20μm(例えば、約5〜10μm)の範囲である。
【0070】
リン酸アルミニウムのPZCはホスフェートとヒドロキシルの置換度と逆の関係にあり、沈殿によって塩を調製するために使用する反応物の反応条件および濃度に応じて、この置換度は変わる可能性がある。溶液中の遊離リン酸イオンの濃度を変えること(より多い量のリン酸=より酸性のPZC)、またはヒスチジンバッファーなどのバッファーを加えること(PZCをより塩基性にする)によってもPZCは変わる。本発明により使用するリン酸アルミニウムは、4.0と7.0の間のPZC、より好ましくは5.0と6.5の間、例えば約5.7のPZCを一般に有するはずである。
【0071】
本発明の組成物を調製するために使用するリン酸アルミニウム溶液は、バッファー(例えば、リン酸またはヒスチジンまたはトリスバッファー)を含み得るが、これは常に必要なわけではない。リン酸アルミニウム溶液は、滅菌済みであり発熱物質を含まないことが好ましい。リン酸アルミニウム溶液は、例えば1.0mMと20mMの間、好ましくは5mMと15mMの間、およびより好ましくは約10mMの濃度で存在する遊離水性リン酸イオンを含み得る。リン酸アルミニウム溶液は、塩化ナトリウムも含み得る。塩化ナトリウムの濃度は0.1〜100mg/ml(例えば、0.5〜50mg/ml、1〜20mg/ml、2〜10mg/ml)の範囲であることが好ましく、およびより好ましくは約3±1mg/mlである。NaClの存在は、抗原の吸着前のpHの正確な測定を容易にする。
【0072】
いくつかの実施形態では、本発明は、ポリソルベート80、Span85およびスクアレンの混合物を含む水中油型エマルジョンを含む組成物を除外することができる[69]。いくつかの実施形態では、本発明は、油、α−トコフェロールおよびポリソルベート80の混合物を含む組成物を除外することができる[70]。いくつかの実施形態では、本発明は、サポニンアジュバント(QS21など)および非イオン性界面活性剤(ポリソルベート40、60または80など)を含む組成物を除外することができる。いくつかの実施形態では、本発明は、サポニンアジュバント、代謝性油および非イオン性界面活性剤を含む組成物を除外することができる[71]。いくつかの実施形態では、本発明は、サポニンアジュバント、水中油型エマルジョンおよびステロールを含む組成物を除外することができる[72]。いくつかの実施形態では、本発明は、3d−MPL、QS21、トリグリセリドおよび水中油型エマルジョンを含む組成物を除外することができる[73]。
【0073】
精製HBsAgと他の抗原の組合せ
本発明の方法は、精製HBsAgを少なくとも1つの非HBV病原体の由来の少なくとも1つの抗原と合わせるステップを含む。
【0074】
抗原は個別に連続して組み合わせることができ、あるいは抗原を予め混合し、一緒に加えることができる。例えば、4価DTP−HBsAgワクチンは、容器へのHBsAg、D、TおよびP抗原の連続的な添加を含む方法によって、あるいはD、TおよびP抗原を予め混合し、次いでHBsAgとDTP混合物を組み合わせることによって作製することができる。
【0075】
抗原成分は任意の適切な順序で組み合わせることができる。
【0076】
非HBV病原体の由来の抗原は界面活性剤を含むことができ、これはHBsAg精製中に使用する非イオン性界面活性剤と同じであるか、あるいはそれと異なってよい。界面活性剤添加に関する多数の別ステップが回避されるので、他の抗原成分が界面活性剤を含む場合、本発明の方法は特に有用である。1つまたは複数の非HBV成分がポリソルベート80を含む方法が好ましい。
【0077】
ジフテリアトキソイドおよび破傷風トキソイドが本発明の組成物中に含まれる場合、それらはHBsAgと組み合わせる前に予め混合することが好ましい。したがって本発明の方法は、HBsAgを含む第1の成分を、D抗原とT抗原の両方を含む第2の成分と組み合わせることを含む。同様に、ジフテリアトキソイド、破傷風トキソイドおよび完全細胞百日咳抗原が組成物中に含まれる場合、それらは予め混合することが好ましく、したがっ
て本発明の方法は、HBsAgを含む第1の成分を、D、TおよびPw抗原を含む第2の成分と組み合わせることを含む。
【0078】
D−T混合物を使用する場合、本発明のワクチン中のジフテリアトキソイドと破傷風トキソイドの比は、通常(Lf単位で測定して)2:1と3:1の間、好ましくは2.4:1と2.6:1の間であり、2.5:1であることがより好ましい。
【0079】
アジュバントを本発明の組成物中に含めるとき、様々な段階でアジュバントを加えることも可能である。典型的には、抗原は、本発明の方法で使用する前にアジュバントと組み合わせるが(例えば、2価D−T混合物は、本発明の方法で使用する前にアルミニウム塩アジュバントに吸着する)、抗原を混合した後にアジュバントを加えること、あるいは抗原をアジュバントに加えることも可能である(例えば、水性アジュバントで始め、次いで抗原を個別にまたは予め混合して添加すること)。前に記載したように、非HBV抗原成分と組み合わせる前に、HBsAg成分をリン酸アルミニウムアジュバントに吸着させることが可能である。
【0080】
組み合わせワクチン
本発明の組成物は(a)抗原成分、および(b)非抗原成分を含むことができる。抗原成分は前に記載した抗原を含むか、あるいはそれらからなってよい。非抗原成分は、以下でさらに詳細に記載するように、担体、アジュバント、賦形剤、バッファーなどを含むことができる。これらの非抗原成分は様々な供給源を有し得る。例えばそれらは、製造中に使用する、あるいはこれらの成分と別に加えることができる抗原またはアジュバント物質の1つ中に存在する可能性がある。
【0081】
本発明の好ましい組成物は、1つまたは複数の医薬担体および/または賦形剤を含む。
【0082】
緊張性を調節するために、ナトリウム塩などの生理塩を含むことが好ましい。塩化ナトリウム(NaCl)が好ましく、これは1mg/mlと20mg/mlの間で存在してよい。
【0083】
組成物は一般に200mOsm/kgと400mOsm/kgの間、好ましくは240mOsm/kgと360mOsm/kgの間の浸透圧を有するはずであり、280mOsm/kg〜320mOsm/kgの範囲内に入ることがより好ましいはずである。浸透圧は予防接種によって引き起こされる痛みに対して影響がないことは以前に報告されているが[74]、この範囲の浸透圧を保つことはそれにもかかわらず好ましい。
【0084】
本発明の組成物は1種または複数種のバッファーを含むことができる。典型的なバッファーには、リン酸バッファー、トリスバッファー、ホウ酸バッファー、コハク酸バッファー、ヒスチジンバッファー、またはクエン酸バッファーがある。バッファーは典型的には5〜20mMの範囲で含まれるはずである。
【0085】
本発明の組成物のpHは一般に5.0と7.5の間、およびより典型的には最適な安定性のために5.0と6.0の間、あるいはジフテリアトキソイドおよび/または破傷風トキソイドが存在する場合、6.0と7.0の間であるはずである。したがって本発明の方法は、パッケージ前にバルクワクチンのpHを調節するステップを含むことができる。
【0086】
本発明の組成物は滅菌済みであることが好ましい。
【0087】
本発明の組成物は発熱物質を含まない、例えば用量当たり1EU未満(エンドトキシン単位、標準指標)、および好ましくは用量当たり0.1EU未満を含むことが好ましい。
【0088】
本発明の組成物はグルテンを含まないことが好ましい。
【0089】
HBsAgの吸着性のため、最終的なワクチン製品は、不透明な外見を有する懸濁液である可能性がある。この外見は、微生物による汚染が容易に目に見えないことを意味し、したがってワクチンは抗菌剤を含むことが好ましい。ワクチンを多用量容器中にパッケージするとき、これは特に重要である。封入するのに好ましい抗菌剤は、2−フェノキシエタノールおよびチメロサールである。しかし、本発明の方法中に水銀防腐剤(例えばチメロサール)は使用しないことが好ましい。したがって、方法中で使用する成分の1つからすべてが、水銀防腐剤を実質的に含まないでよい(特に、2価D−T成分;IPV成分;複合体成分)。しかし、本発明中で使用する前に成分(特にHBsAg)をこのような防腐剤で処理した場合、微量の存在が避けられない可能性がある。しかし安全性のために、最終組成物は約25ng/ml未満の水銀を含むことが好ましい。最終的なワクチン製品は、検出可能なチメロサールを含まないことがより好ましい。これは一般に、本発明の方法中のその添加前に抗原調製物から水銀防腐剤を除去すること、または組成物を生成するために使用する成分の調製中のチメロサールの使用を避けることによって実施されるはずである。
【0090】
2価D−T混合物を本発明の方法中で使用する場合、それはチメロサールを含まない状態でなければならない。いくつかの実施形態では、D−T混合物は2−フェノキシエタノールを含む可能性があるが、他の実施形態では、D−T混合物はチメロサールおよび2−フェノキシエタノールを含まない。3価D−T−Pw混合物を本発明の方法中に使用する場合、それは2−フェノキシエタノールを含まないでよいが、チメロサールを含む可能性がある。
【0091】
製造中、望ましい最終濃度を得るための成分の希釈は、通常WFI(注射用水)を用いて実施するはずである。
【0092】
Al3+の形態で表す本発明の組成物中のリン酸アルミニウムの濃度は5mg/ml未満、例えば4mg/ml以下、3mg/ml以下、2mg/ml以下、1mg/ml以下などであることが好ましい。
【0093】
本発明の組成物中のHBsAgの濃度は60μg/ml未満、例えば55μg/ml以下、50μg/ml以下、45μg/ml以下、40μg/ml以下などであることが好ましい。約20μg/mlの濃度が典型的である。
【0094】
本発明の組成物中のジフテリアトキソイドの濃度は、典型的には少なくとも50IU/mlである。
【0095】
本発明の組成物中の破傷風トキソイドの濃度は、典型的には少なくとも100IU/mlである。
【0096】
本発明の組成物中のジフテリアトキソイドと破傷風トキソイドの比は、通常(Lf単位で測定して)2:1と3:1の間、好ましくは2.4:1と2.6:1の間であり、2.5:1であることがより好ましい。
【0097】
本発明の組成物中のwP抗原の量は、典型的には少なくとも8IU/mlである。
【0098】
本発明の組成物中の糖として測定したHib複合体の量は、典型的には10μg/mlと30μg/mlの間である。
【0099】
EU(Elisa単位)で測定したHAV抗原の量は、典型的には少なくとも600EU/mlである。
【0100】
IPV抗原の量は系統の血清型に依存する。1型ウイルスに関しては、組成物は約80DU/mlを典型的には含む。2型ウイルスに関しては、組成物は約16DU/mlを典型的には含む。3型ウイルスに関しては、組成物は約65DU/mlを典型的には含む。
【0101】
本発明の組成物中の糖として測定した髄膜炎菌複合体の量は、各血清型に関して典型的には5μg/mlと25μg/mlの間である。
【0102】
本発明の組成物中の糖として測定した肺炎球菌複合体の量は、各血清型に関して典型的には2μg/mlと20μg/mlの間である。
【0103】
本発明の組成物は、0.5mlの用量で患者に投与することが好ましい。0.5mlの用量という言及は、正規分布の分散、例えば0.5ml±0.05mlを含むことは理解されるはずである。
【0104】
本発明は、患者への投与用に次いで分配することができる、個々の用量にパッケージするのに適したバルク材料を提供することができる。前述の濃度は典型的には最終パッケージ用量における濃度であり、したがってバルクワクチンにおける濃度はさらに高い可能性がある(例えば、希釈によって最終濃度まで低下させるために)。
【0105】
本発明の組成物は、一般に水性形態であるはずである。
【0106】
個々の抗原成分由来の残留成分が、本発明の方法によって生成する最終ワクチン中に微量で存在する可能性もある。例えば、ホルムアルデヒドを使用してジフテリア、破傷風および百日咳のトキソイドを調製する場合、最終ワクチン製品は微量のホルムアルデヒド(例えば、10μg/ml未満、好ましくは5μg/ml未満)を保持する可能性がある。培地または安定剤をポリオウイルス調製中に使用することができた可能性があり(例えば、培地199)、これらは最終ワクチンまで存続する可能性がある。同様に、遊離アミノ酸(例えば、アラニン、アルギニン、アスパラギン酸、システインおよび/またはシスチン、グルタミン酸、グルタミン、グリシン、ヒスチジン、プロリンおよび/またはヒドロキシプロリン、イソロイシン、ロイシン、リシン、メチオニン、フェニルアラニン、セリン、スレオニン、トリプトファン、チロシンおよび/またはバリン)、ビタミン(例えば、コリン、アスコルビン酸など)、リン酸2ナトリウム、1カリウムリン酸塩、カルシウム、グルコース、硫酸アデニン、フェノールレッド、酢酸ナトリウム、塩化カリウムなどが、それぞれ100μg/ml以下、好ましくは10μg/ml未満で最終ワクチン中に保持される可能性がある。ネオマイシン(例えば、特にIPV成分由来の硫酸ネオマイシン)、ポリミキシンB(例えば、特にIPV成分由来の硫酸ポリミキシンB)などの、抗原調製物由来の他の成分も、用量当たりナノグラム未満の量で存在する可能性がある。抗原調製物に由来する最終ワクチンの他の考えられる成分は、抗原の完全ではない精製から生じる。少量の百日咳菌、ジフテリア菌、破傷風菌および出芽酵母のタンパク質および/またはゲノムDNAが、したがって存在する可能性がある。これらの残留成分を最少にするために、本発明の方法中で抗原を使用する前に、抗原調製物を処理してそれらを除去することが好ましい。
【0107】
IPV成分を使用する場合、それは一般にVero細胞において増殖しているはずである。最終ワクチンは好ましくは10ng/ml未満、好ましくは1ng/ml以下、例えば500pg/ml以下または50pg/ml以下のVero細胞DNA、例えば10ng/ml未満の50塩基対長以上であるVero細胞DNAを含む。
【0108】
本発明のパッケージ組成物
HBsAgとアジュバントを組み合わせた後、本発明の方法は容器に混合物の0.5mlサンプルを抽出およびパッケージするステップを含むことができる。多用量の状況に関しては、多用量を抽出し、1つの容器に一緒にパッケージするはずである。
【0109】
本発明の方法は、使用するために容器にワクチンをパッケージする他のステップを含むことができる。適切な容器にはバイアルおよび使い捨てシリンジがある(滅菌済みであるものが好ましい)。
【0110】
本発明の組成物をバイアルにパッケージする場合、これらはガラスまたはプラスチック材料でできていることが好ましい。組成物をバイアルに加える前に、バイアルは滅菌済みであることが好ましい。ラテックス感受性患者に関する問題を回避するために、ラテックスを含まないストッパーでバイアルを密封することが好ましい。バイアルは1回用量のワクチンを含むことができ、あるいはバイアルは、複数回用量、例えば10回用量を含むことができる(「多用量」バイアル)。多用量バイアルを使用する場合、バイアルの中身の汚染を回避することに注意を払って、厳密な無菌条件下で滅菌済みニードルおよびシリンジを用いて各用量を取り出さなければならない。好ましいバイアルは、無色のガラスでできている。
【0111】
バイアルは予め充填したシリンジをキャップに挿入することができ、シリンジの中身をバイアルに押し出すことができ(例えば、凍結乾燥物質をその中で還元するために)、バイアルの中身を除去してシリンジに戻すことができるように適合させたキャップ(例えばLuerロック)を有することができる。バイアルからのシリンジの除去後、次いでニードルを取り付けることができ、組成物は患者に投与することができる。キャップにアクセス可能である前にシールまたはカバーを除去する必要があるように、キャップはシールまたはカバーの内側に位置することが好ましい。
【0112】
組成物をシリンジにパッケージする場合、シリンジはそれに取り付けられたニードルを通常有していないはずであるが、構築および使用するための別のニードルをシリンジに供給することができる。安全なニードルが好ましい。1−インチ23−ゲージ、1−インチ25−ゲージおよび5/8−インチ25−ゲージのニードルが典型的である。記録をとるのを容易にするために中身のロット番号および有効期限をその上に印刷することができる剥離ラベルを、シリンジに与えることができる。シリンジ内のプランジャーは、吸引中にプランジャーが偶然に除去されるのを防ぐためのストッパーを有することが好ましい。シリンジはラテックスゴム製キャップおよび/またはプランジャーを有することができる。使い捨てシリンジは1回用量のワクチンを含む。シリンジは一般にニードルの取り付け前に先端を密封するための先端キャップを有するはずであり、先端キャップはブチルゴムでできていることが好ましい。シリンジとニードルを別々にパッケージする場合、ブチルゴム製シールドをニードルに取り付けることが好ましい。グレーのブチルゴムが好ましい。好ましいシリンジは、「Tip−Lok」(商標)の商品名で市場に出ているシリンジである。
【0113】
ガラス容器(例えば、シリンジまたはバイアル)を使用する場合、ソーダ石灰ガラスではなくホウ珪酸ガラスでできた容器を使用することがしたがって好ましい。
【0114】
組成物を容器にパッケージした後、分配用の箱の中、例えば段ボール箱の中に容器を次いで封入することができ、ワクチンの詳細、例えばその商品名、ワクチン中の抗原の一覧(例えば、「B型肝炎組換え体」など)、容器の表示(例えば、「使い捨ての予め充填したTip−Lokシリンジ」または「10×0.5mlのシリンジ1回用量バイアル」)、その用量(例えば、「それぞれ1回0.5mlの用量を含む」)、警告(例えば、「成人のみ使用」または「小児のみ使用」)、有効期限、指示(例えば、「(事前血液透析および血液透析を含めて)腎不全を有していた15歳以上の患者に関する、すべての知られている亜型によって引き起こされるB型肝炎ウイルス(HBV)感染に対する能動的免疫処置」など)、特許番号などがその箱にラベルされるはずである。それぞれの箱は、2つ以上のパッケージワクチン、例えば5または10のパッケージワクチン(特にバイアル用)を含むことができる。ワクチンがシリンジ中に含まれる場合、パッケージはシリンジの絵を示し得る。
【0115】
ワクチンは(例えば、同じ箱の中に)、ワクチンの詳細、例えば投与に関する指示、ワクチン中の抗原の詳細などを含む説明書と共に、1つにパッケージすることができる。指示は、警告、例えば予防接種後のアナフィラキシー反応の場合にアドレナリン溶液を容易に利用できる状態に保つなどの警告も含むことができる。
【0116】
パッケージワクチンは2℃と8℃の間で保存することが好ましい。それを凍結させてはならない。
【0117】
ワクチンは製造中に完全液状形態で与えることができ(すなわち、すべての抗原成分が水溶液または懸濁液中に存在する)、あるいはワクチンは、いくつかの成分が液状形態であり他の成分が凍結乾燥形態である形に調製することができる。したがって最終ワクチンは、2つの成分:(a)水性抗原を含む第1の成分と(b)凍結乾燥抗原を含む第2の成分を1つに混合することによって、使用時に即時調製することができる。2つの成分は別々の容器(例えば、バイアルおよび/またはシリンジ)中に存在することが好ましく、本発明は成分(a)および(b)を含むキットを提供する。この形式は複合体成分、特にHibおよび/または髄膜炎菌および/または肺炎球菌複合体を含むワクチンに特に有用である。それは、凍結乾燥形態でさらに安定する可能性があるからである。したがって複合体は、本発明と共にそれらを使用する前は凍結乾燥状態であってよい。他の成分を例えば安定剤として、凍結乾燥前に加えることもできる。含有するのに好ましい安定剤は、ラクトース、スクロースおよびマンニトール、ならびにこれらの混合物、例えばラクトース/スクロース混合物、スクロース/マンニトール混合物などである。したがって最終ワクチンは、ラクトースおよび/またはスクロースを含むことができる。スクロース/マンニトール混合物を使用することによって、乾燥プロセスをスピードアップすることが可能である。
【0118】
したがって本発明は、2容器の組み合わせワクチンを調製するための方法であって、
− 前に記載したように水性組み合わせワクチンを調製するが、ただし前記1つまたは複数の抗原が複合体被膜糖抗原を含まないステップ、
− 第1の容器(例えばシリンジ)中で前記組み合わせワクチンをパッケージするステップ、
− 複合体被膜糖抗原を凍結乾燥形に調製するステップ、
− 前記凍結乾燥抗原を第2の容器(例えばバイアル)中でパッケージするステップ、および
− 第1の容器と第2の容器をキットに1つにパッケージするステップを含む方法を提供する。
【0119】
その後、キットは医師に分配することができる。
【0120】
D、T、PおよびHBsAg成分は液状形態であることが好ましい。
【0121】
ワクチンの治療および投与法
本発明の組成物はヒト患者への投与に適しており、本発明は、本発明の組成物を患者に投与するステップを含む、患者における免疫応答を増大させる方法を提供する。
【0122】
本発明は、医薬において使用するための本発明の組成物も提供する。
【0123】
本発明は、患者に投与するための医薬品の製造における、(i)その精製法が非イオン性界面活性剤の存在下で酵母細胞を破壊することを含む組換え酵母細胞から精製したHBsAg、および(ii)1つまたは複数の非HBV抗原の使用も提供する。
【0124】
本発明の免疫原性組成物は、少なくともB型肝炎ウイルス感染の予防および/または治療において使用するためのワクチンであることが好ましい。本発明の組成物を与えた患者は、初回免疫処置後6週間で測定して、500mIU/ml以上の血清抗HBsAgGM力価を有することが好ましい。l2カ月後に測定して、力価が500mIU/ml以上であることがより好ましい。
【0125】
完全な有効性を有するために、子供用の典型的な初回免疫処置スケジュールは、2回以上の投与を施すことを含むことができる。例えば、投与は:0および6カ月(時間0は初回投与である)、0、1、2および6カ月;第0日、第21日、および次いで6カ月と12カ月の間に第3の投与;2、4および6カ月;3、4および5カ月;6、10および14週;または0、1、2、6および12カ月であってよい。
【0126】
例えば2歳の子供用に、追加抗原刺激投与として組成物を使用することもできる。
【0127】
本発明の組成物は、例えば腕または脚への筋肉内注射によって投与することができる。
【0128】
本発明によって生成されるワクチンは、Prevnar(商標)などの別の肺炎球菌複合体ワクチンと同時に患者に投与することができる。
【0129】
本発明の組成物がアルミニウム系アジュバントを含む場合、保存中に成分の沈殿が生じる可能性がある。したがって、組成物は患者に投与する前に攪拌しなければならない。攪拌した組成物は、白濁した懸濁液であるはずである。
【0130】
好ましいワクチン
本発明の特異的な多価免疫原性組成物は以下の組成物を含む:
・ HBsAg、D、T、PaおよびIPVを含む5価組成物。ワクチンは水性形態である。それは水酸化アルミニウムアジュバントとリン酸アルミニウムアジュバントの両方を含む。HBsAgはリン酸アルミニウムに吸着する。D、TおよびPaは水酸化アルミニウムに吸着する。1ml当たりの量:約50Lfのジフテリアトキソイド;約20Lfの破傷風トキソイド;約50μgのPT;約50μgのFHA;約16μgのペルタクチン;約20μgのHBsAg;約80DUの1型ポリオウイルス;約16DUの2型ポリオウイルス;約64DUの3型ポリオウイルス。用量:約0.5ml。予め充填したシリンジ中に存在してよい。
【0131】
・ HBsAg、D、T、PaおよびIPVを含む5価組成物。ワクチンは水性形態である。それは水酸化アルミニウムアジュバントとリン酸アルミニウムアジュバントの両方を含む。HBsAgはリン酸アルミニウムに吸着する。D、TおよびPaは水酸化アルミニウムに吸着する。1ml当たりの量:少なくとも60IUのジフテリアトキソイド;少なくとも80IUの破傷風トキソイド;約50μgのPT;約50μgのFHA;約16μgのペルタクチン;約20μgのHBsAg;約80DUの1型ポリオウイルス;約16DUの2型ポリオウイルス;約64DUの3型ポリオウイルス。用量:約0.5ml。予め充填したシリンジ中に存在してよい。
【0132】
・ HBsAg、D、TおよびPwを含む4価組成物。成分は水性形態である。それは水酸化アルミニウムアジュバントとリン酸アルミニウムアジュバントの両方を含む。HBsAgはリン酸アルミニウムに吸着する。DおよびTは水酸化アルミニウムに吸着する。組成物はチメロサールを含み、2−フェノキシエタノールは含まないことが好ましい。1ml当たりの量:少なくとも60IUのジフテリアトキソイド;少なくとも120IUの破傷風トキソイド;少なくとも8IUのPw、約20μgのHBsAg。用量:約0.5ml。
【0133】
・ HBsAg、D、T、PwおよびHib−T複合体を含む5価組成物。HBsAg、D、TおよびPw成分は水性形態であり、Hib−Tは凍結乾燥状態である。それは水酸化アルミニウムアジュバントとリン酸アルミニウムアジュバントの両方を含む。DおよびTは水酸化アルミニウムに吸着する。HBsAgおよびHib−Tはリン酸アルミニウムに吸着する。凍結乾燥Hib−Tはラクトースを含む。水性成分はチメロサールを含み得る。1ml当たりの量:少なくとも60IUのジフテリアトキソイド;少なくとも120IUの破傷風トキソイド(およびHib−T中の担体として5μgと25μgの間の破傷風トキソイド);少なくとも8IUのPw、約20μgのHBsAg;糖として測定して約5μgのHib−T。用量:約0.5ml。
【0134】
・ HBsAg、D、T、Pwならびに3つの複合体:Hib−T複合体、MenA複合体およびMenC複合体を含む7価組成物。HBsAg、D、TおよびPw成分は水性形態であり、3つの複合体は凍結乾燥状態である。それは水酸化アルミニウムアジュバントとリン酸アルミニウムアジュバントの両方を含む。DおよびTは水酸化アルミニウムに吸着する。HBsAgはリン酸アルミニウムに吸着する。凍結乾燥成分はラクトースおよび/またはスクロースを含み得る。水性成分はチメロサールを含み得る。考えられる1ml当たりの量:少なくとも60IUのジフテリアトキソイド;少なくとも120IUの破傷風トキソイド(およびHib−T中の担体として5μgと25μgの間の破傷風トキソイド);少なくとも8IUのPw、約20μgのHBsAg;糖として測定して約5μgの各複合体。用量:約0.5ml。
【0135】
これらの組成物はそれ自体ワクチンとして使用することができ、あるいは他のワクチンの成分として使用することができる。例えば本発明は、前に記載した5価HBsAg−D−T−Pa−IPV組成物、および凍結乾燥Hib−T複合体を含む6価組成物を提供する。凍結乾燥Hib−Tはアルミニウム塩に吸着しないことが好ましい。本発明は、前に記載した5価HBsAg−D−T−Pa−IPV組成物、および凍結乾燥Hib−TおよびMenC複合体を含む7価組成物も提供する。本発明は、前に記載した5価HBsAg−D−T−Pa−IPV組成物、および凍結乾燥Hib−T、MenCおよびMenY複合体を含む8価組成物も提供する。本発明は、前に記載した4価HBsAg−D−T−Pw組成物、および凍結乾燥Hib−T複合体を含む5価組成物も提供する。本発明は、前に記載した4価HBsAg−D−T−Pw組成物、およびHib−T複合体、MenA複合体およびMenC複合体の凍結乾燥混合物を含む7価組成物も提供する。最終ワクチンは、使用時に凍結乾燥物質を水性HBsAg含有物質で戻すことによって調製することができ、凍結乾燥成分と水性成分は、前に記載したようにキット内に1つにパッケージすることが好ましい。
【0136】
本発明の特異的な方法は以下のステップを含む方法を含む:
・ 本発明によるHBsAgの精製;HBsAgのリン酸アルミニウムアジュバントへの吸着;チメロサールを含まない水酸化アルミニウムアジュバントとの2価D−T混合物の入手;Pa成分用のPT、FHAおよびペルタクチンの入手;好ましくはアルミニウム塩アジュバントを含まない型1、2および3としてプールするIPV抗原の入手;最終的な5価混合を得るための任意の順序でのD−T、Pa、IPVおよびHBsAgの組合せ;場合によってはシリンジへのパッケージ。
【0137】
・ 本発明によるHBsAgの精製;HBsAgのリン酸アルミニウムアジュバントへの吸着;2−フェノキシエタノールを含まずチメロサールを含む水酸化アルミニウムアジュバントおよびリン酸アルミニウムアジュバントとの3価D−T−Pw混合物の入手;最終的な4価混合を得るためのD−T−PwとHBsAgの組合せ;場合によってはシリンジへのパッケージ;場合によっては凍結乾燥複合体成分、例えばHib−T;MenA、MenCと組み合わせたパッケージ。
【0138】
・ 本発明によるHBsAgの精製;HBsAgのリン酸アルミニウムアジュバントへの吸着;2−フェノキシエタノールを含まずチメロサールを含む水酸化アルミニウムアジュバントおよびリン酸アルミニウムアジュバントとの3価D−T−Pw混合物の入手;凍結乾燥Hib−T複合体の入手;水性4価成分を得るためのD−T−PwとHBsAgの組合せ;水性4価成分のガラスバイアルへのパッケージ;凍結乾燥Hib−Tおよび/またはMenCおよび/またはMenYのガラスバイアルへのパッケージ;5価組み合わせワクチンを得るための水戻し用の単一キット中に存在する2つのバイアルの組合せ。ガラスバイアルはI型ガラスであってよく、ブチルゴム製ストッパーを有する。
【0139】
・ 本発明によるHBsAgの精製;HBsAgのリン酸アルミニウムアジュバントへの吸着;2−フェノキシエタノールを含まずチメロサールを含む水酸化アルミニウムアジュバントおよびリン酸アルミニウムアジュバントとの3価D−T−Pw混合物の入手;好ましくはアルミニウム塩アジュバントを含まない型1、2および3としてプールするIPV抗原の入手;最終的な5価混合を得るための任意の順序でのD−T−Pa、IPVおよびHBsAgの組合せ;場合によってはシリンジへのパッケージ;場合によっては凍結乾燥複合体成分、例えばHib−T;MenA、MenCと組み合わせたパッケージ。
【0140】
他の成分、例えば塩化ナトリウム、アジュバント、防腐剤などを任意の段階で加えることができる。これらの方法を使用して、例えば前に記載したワクチンを調製することができる。
【0141】
異なる方法のステップは実質的に同時に実施することができ、あるいは別々に実施することができる。これらのステップは、同じ場所または異なる場所、異なる国においても実施することができ、例えばHBsAg精製は、Hib−T凍結乾燥と異なる場所で行うことができる。
【0142】
複合体の担体タンパク質
複合体糖抗原は、糖がそれに直接、あるいはリンカーを介して共有結合する担体タンパク質を含む。結合技法に関する一般的な情報は参考文献45において見ることができる。
【0143】
担体として使用するための様々なタンパク質が知られており、好ましい担体タンパク質は、ジフテリアトキソイドまたは破傷風トキソイドなどの細菌毒素またはトキソイドである。他の適切な担体タンパク質には、ジフテリア毒素のCRM197突然変異体[75〜77]、髄膜炎菌の外膜タンパク質[78]、合成ペプチド[79、80]、熱ショックタンパク質[81、82]、百日咳タンパク質[83、84]、サイトカイン[85]、リンホカイン[85]、ホルモン[85]、増殖因子[85]、様々な病原体由来の抗原に由来する多数のヒトCD4+T細胞エピトープを含む人工タンパク質[86]、例えばN19[87]、インフルエンザ菌由来のタンパク質D[88、89]、肺炎球菌表面タンパク質PspA[90]、ニューモリシン[91]、鉄摂取タンパク質[92]、C.ディフィシル(C.difficile)由来の毒素AまたはB[93]、B群連鎖球菌(S.agalactiae)タンパク質[94]などがあるが、これらだけには限られない。
【0144】
糖と担体の結合は、例えば担体タンパク質中のリシン残基、またはアルギニン残基の側鎖中の−NH2基を介した結合であることが好ましい。(例えば、システインの側鎖中の)−SH基との結合も考えられる。
【0145】
1:5(すなわち、過剰なタンパク質)と5:1(すなわち、過剰な糖)の間の糖:タンパク質比(w/w)を有する複合体が好ましい。
【0146】
組成物は少量の遊離担体を含むことができる。別の抗原として含まれる如何なる担体も無視すると、非結合担体は全体として組成物中の担体タンパク質の全量の5重量%を超えないことが好ましく、2重量%未満で存在することがより好ましい。
【0147】
例えば担体抑制のリスクを低下させるために、組成物中に2種類以上の担体タンパク質を含むことが考えられる。
【0148】
髄膜炎菌複合体に使用する担体タンパク質は、インフルエンザ菌由来のタンパク質Dであってよい。このタンパク質は参考文献95および96中に詳細に記載されており、複合体中の担体タンパク質としてのその使用は参考文献97中に記載されている。用語「タンパク質D」は、参考文献97中に開示された天然完全長タンパク質の断片、および完全長タンパク質Dまたはこれらの断片のいずれかを含む融合タンパク質(例えば、インフルエンザウイルスNS1タンパク質の断片とタンパク質Dの断片の融合体)も含む。これらの断片は、結合時にT非依存性糖抗原をT依存性抗原に変換する能力を保持しているはずである。典型的な断片は、タンパク質Dの少なくともN末端側1/3を含むはずである。このタンパク質は大腸菌中で好都合に発現される可能性があり[96]、この組換え物質は本発明と共に使用するのが好ましい[97]。
【0149】
一般事項
用語「comprising」は「including」ならびに「consisting」を包含し、例えば、Xを「含む(comprising)」組成物はXのみからなる可能性があるか、あるいは他の何かを含む、例えばX+Yである可能性がある。
【0150】
語句「実質的に」は「完全に」を除外するわけではない、例えば、Yを「実質的に含まない」組成物はYを完全に含まない可能性がある。必要な場合、語句「実質的に」は本発明の定義から除外することができる。
【0151】
数値xに関する用語「約」は、例えばx±10%を意味する。
【0152】
特に言及しない限り、2つ以上の成分を混合するステップを含む方法は、任意の特定の混合順を必要としない。したがって、成分は任意の順序で混合することができる。3つの成分が存在する場合、2成分を互いに組み合わせることができ、次いでその組合せを、第3の成分などと組み合わせることができる。
【0153】
ある抗原がアジュバントに「吸着する」として記載する場合、少なくとも50%(重量)、例えば50%、60%、70%、80%、90%、95%、98%あるいはそれより多くのその抗原が吸着することが好ましい。ジフテリアトキソイドと破傷風トキソイドは両方が完全に吸着する、すなわち上清中には何も検出できないことが好ましい。HBsAgの全体の吸着を使用することができる。
【0154】
ジフテリアトキソイドの量は国際単位(IU)で表すことができる。例えばNIBSCは、アンプル当たり160IUを含む、「吸着ジフテリアトキソイドの第3国際標準1999」[98、99]を与える。IUシステムに対する代替として、「Lf」単位(「凝集単位」または「境界凝集用量」)を、1国際単位のアンチトキシンと混合すると最適に凝集する混合物を生成する[100]トキソイドの量として定義する。例えばNIBSCは、アンプル当たり300LFを含む「ジフテリアトキソイド、プレイン」[101]を与え、アンプル当たり900LFを含む「凝集試験のジフテリアトキソイドの第1の国際参照試薬」[102]も与える。
【0155】
破傷風トキソイドの量は国際単位(IU)で表すことができる。例えばNIBSCは、アンプル当たり469IUを含む、「吸着破傷風トキソイドの第3国際標準2000」[103、104]を与える。IUシステムに対する代替として、「Lf」単位(「凝集単位」または「境界凝集用量」)を、1国際単位のアンチトキシンと混合すると最適に凝集する混合物を生成する[100]トキソイドの量として定義する。例えばNIBSCは、アンプル当たり1000LFを含む「凝集試験の破傷風トキソイドの第1の国際参照試薬」[105]を与える。
【0156】
wP抗原の量は国際単位(IU)で表すことができる。例えばNIBSCは、アンプル当たり46IUを含む「百日咳ワクチンの第3国際標準」[106]を与える。それぞれのアンプルは水溶液の2.0ml等分試料の凍結乾燥残留物を含み、この等分試料は8リットルのM/15SorensenのバッファーpH7.0で希釈した10リットルの細菌懸濁液(米国不透明度標準により180不透明度単位に等しい)を含んでいた。IUシステムに対する代替として、「OU」単位(「不透明度単位」)も使用される(例えば、4OUは約1IUであり得る)。
【0157】
複合体の量は一般に、担体の選択による変動を避けるために糖の質量によって示される(すなわち、全体としての複合体の用量(担体+糖)は述べられる用量より大きい)。
【0158】
動物(および特にウシ)の材料を細胞の培養において使用する場合、それらは感染性海綿状脳症(TSE)を含まない、および特にウシ海綿状脳症(BSE)を含まない供給源から入手しなければならない。
本発明はまた、以下の項目を提供する。
(項目1)
組み合わせワクチンを調製するためのプロセスであって、該ワクチンが(i)非イオン性界面活性剤、(ii)B型肝炎ウイルス(HBV)表面抗原、および(iii)少なくとも1つの非HBV病原体由来の抗原を含み、該プロセスが、(i)組換え酵母細胞を該非イオン性界面活性剤の存在下で破壊して精製HBsAg成分を得るステップを含む、該酵母細胞から該HBV表面抗原を精製すること、および(ii)該精製HBsAg成分を非HBV病原体由来の少なくとも1つの他の抗原と合わせて、該組み合わせワクチンを得ることを含む、プロセス。
(項目2)
組み合わせワクチンを調製するためのプロセスであって、該ワクチンが(i)非イオン性界面活性剤、(ii)B型肝炎ウイルス(HBV)表面抗原(HBsAg)、および(iii)少なくとも1つの非HBV病原体由来の抗原を含み、該プロセスが、精製HBsAgを非HBV病原体由来の少なくとも1つの他の抗原と合わせて、組み合わせワクチンを得るステップを含み、HBsAg発現組換え酵母細胞を該非イオン性界面活性剤の存在下で破壊するプロセスによって該HBsAgが調製される、プロセス。
(項目3)
(i)非イオン性界面活性剤、(ii)B型肝炎ウイルス(HBV)表面抗原、および(iii)少なくとも1つの非HBV病原体由来の抗原を含む免疫原性組成物であって、HBsAg発現組換え酵母細胞を該非イオン性界面活性剤の存在下で破壊するプロセスによって該HBV表面抗原が調製される、免疫原性組成物。
(項目4)
上記非イオン性界面活性剤がポリ(オキシエテン)残基を含む、項目1から3のいずれかに記載のプロセスまたは組成物。
(項目5)
上記非イオン性界面活性剤がポリオキシエチレンソルビタンエステルである、項目4に記載のプロセスまたは組成物。
(項目6)
上記非イオン性界面活性剤がポリソルベート20である、項目5に記載のプロセスまたは組成物。
(項目7)
上記非イオン性界面活性剤が30μg/ml以下で最終産物中に存在する、項目1から6のいずれかに記載のプロセスまたは組成物。
(項目8)
上記非イオン性界面活性剤がHBsAg各100μgに対して50μg以下で最終産物中に存在する、項目1から7のいずれかに記載のプロセスまたは組成物。
(項目9)
(i)上記少なくとも1つの非HBV病原体がジフテリア菌および破傷風菌を含み、(ii)これら2つの病原体由来の該抗原がジフテリアトキソイドおよび破傷風トキソイドであり、かつ(iii)該ジフテリアトキソイドと該破傷風トキソイドがポリソルベート20を実質的に含まない混合された形態で最初に存在する、項目1から8のいずれかに記載のプロセス。
(項目10)
上記HBV表面抗原がグリコシル化されていない、項目1から9のいずれかに記載のプロセスまたは組成物。
(項目11)
上記HBV表面抗原が、リン脂質を含む脂質マトリクスを含む粒子の形態である、項目1から10のいずれかに記載のプロセスまたは組成物。
(項目12)
上記HBV表面抗原がHBV亜型adw2に由来する、項目1から11のいずれかに記載のプロセスまたは組成物。
(項目13)
上記HBV表面抗原が用量当たり約10μgで最終組成物中に存在する、項目1から12のいずれかに記載のプロセスまたは組成物。
(項目14)
上記ワクチンがHib複合体、髄膜炎菌複合体、および/または肺炎球菌複合体を含む、項目1から13のいずれかに記載のプロセスまたは組成物。
(項目15)
上記最終組成物が3価HBsAg、D、T組成物;4価HBsAg、D、T、Pw組成物;5価HBsAg、D、T、Pw、Hib組成物;7価HBsAg、D、T、Pw、Hib、MenA、MenC組成物;8価HBsAg、D、T、Pw、Hib、MenA、MenC、MenW135組成物;8価HBsAg、D、T、Pw、Hib、MenA、MenC、MenY組成物;9価HBsAg、D、T、Pw、Hib、MenA、MenC、MenW135、MenY組成物;4価HBsAg、D、T、Pa組成物;5価HBsAg、D、T、Pa、Hib組成物;5価HBsAg、D、T、Pa、ポリオウイルス組成物;6価HBsAg、D、T、Pa、ポリオウイルス、Hib組成物;7価HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC組成物;8価HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC、MenA組成物;8価HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC、MenY組成物;8価HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC、MenW135組成物;10価HBsAg、D、T、Pa、ポリオウイルス、Hib、MenC、MenA、MenW135、MenY組成物;2価HBsAg、Hib組成物;および2価HBsAg、A型肝炎ウイルス組成物から選択される、項目1から14のいずれかに記載のプロセスまたは組成物。
(項目16)
上記最終組成物がリン酸アルミニウムアジュバントおよび/または水酸化アルミニウムアジュバントを含む、項目1から15のいずれかに記載のプロセスまたは組成物。
(項目17)
上記最終組成物がリン酸アルミニウムアジュバントと水酸化アルミニウムアジュバントの両方を含む、項目1から16のいずれかに記載のプロセスまたは組成物。
(項目18)
上記最終組成物中のAl3+の濃度が5mg/ml以下である、項目16または17に記載のプロセスまたは組成物。
(項目19)
予め混合したD−T成分を加えることを含む、項目1から18のいずれかに記載のプロセス。
(項目20)
予め混合したD−T−Pw成分を加えることを含む、項目1から19のいずれかに記載のプロセス。
(項目21)
上記予め混合したD−T−Pw成分がリン酸アルミニウムアジュバントと水酸化アルミニウムアジュバントの両方を含む、項目20に記載のプロセス。
(項目22)
患者に投与するための薬剤の製造における、(i)組換え酵母細胞から精製したHBsAgであって、精製プロセスが非イオン性界面活性剤の存在下で該酵母細胞を破壊することを含む、HBsAg、および(ii)1つまたは複数の非HBV抗原の、使用。
(項目23)
5価組成物を調製するためのキットであって、該5価組成物がHBsAg、D、T、PwおよびHib−T複合体抗原を含み、該DおよびT抗原が水酸化アルミニウムアジュバントに吸着しており、該HBsAgおよびHib−Tがリン酸アルミニウムアジュバントに吸着しており、該HBsAg、D、TおよびPw成分が第1の容器中で水性形態であり、該Hib−Tが第2の容器中でラクトースと組み合わせた凍結乾燥形態であり、HBsAg発現組換え酵母細胞を非イオン性界面活性剤の存在下で破壊するプロセスによって、該HBsAgが調製される、キット。
(項目24)
7価組成物を調製するためのキットであって、該7価組成物がHBsAg、D、T、Pw、Hib−T複合体、MenA複合体およびMenC複合体を含み、該HBsAg、D、TおよびPw成分が第1の容器中で水性形態であり、該3つの複合体が第2の容器中で凍結乾燥形態であり、該第1の容器は水酸化アルミニウムアジュバントとリン酸アルミニウムアジュバントの両方も含み、該DおよびT抗原が水酸化アルミニウムに吸着しており、HBsAgがリン酸アルミニウムに吸着しており、HBsAg発現組換え酵母細胞を非イオン性界面活性剤の存在下で破壊するプロセスによって該HBsAgが調製される、キット。
(項目25)
上記第1の容器がチメロサールをさらに含む、項目24に記載のキット。
【図面の簡単な説明】
【0159】
【図1−1】図1は、参考文献3および4の図2のコピーであり、参考文献3および4は、吸着法に基づく抗原性および免疫をこの図が示すことを述べ、サンプル1は、過剰な水酸化アルミニウムゲルを各コンポーネントワクチンの組合せが終了した後に加えたサンプルであり、サンプル2は、同じ濃度の前記吸着剤を組合せが終了する前に事前に加えたサンプルである。図1Aおよび1Bは、それぞれサンプル1および2の相対的抗原性を示す。
【図1−2】図1は、参考文献3および4の図2のコピーであり、参考文献3および4は、吸着法に基づく抗原性および免疫をこの図が示すことを述べ、サンプル1は、過剰な水酸化アルミニウムゲルを各コンポーネントワクチンの組合せが終了した後に加えたサンプルであり、サンプル2は、同じ濃度の前記吸着剤を組合せが終了する前に事前に加えたサンプルである。図1Cおよび1Dは、それぞれサンプル1および2の各抗原に対する抗体形成の相対的レベルを示す。
【図2−1】図2は、組成物中のHBsAgの安定性に関するウエスタンブロットの結果を示す。図2A中、レーンは(1)Laemmliサンプルバッファー(LSB);(2)分子量マーカー;(3〜5)1μgの3つの別個の対照HBsAg調製物;(6〜8)3つの別個の5価ロットの上清;(9〜10)LSBである。図2Bおよび2C中、レーンは(1)分子量マーカー;(2〜4)1μgの3つの別個の対照HBsAg調製物;(5〜7)2〜8℃で2週間保存した、3つの別個の5価ロットの上清;(8〜10)36〜38℃で2週間保存した、3つの別個の5価ロットの上清である。図2D中、レーンは(1)分子量マーカー;(2)対照HBsAg;(3〜5)3つの別個の5価ロットの上清である。
【図2−2】図2は、組成物中のHBsAgの安定性に関するウエスタンブロットの結果を示す。図2A中、レーンは(1)Laemmliサンプルバッファー(LSB);(2)分子量マーカー;(3〜5)1μgの3つの別個の対照HBsAg調製物;(6〜8)3つの別個の5価ロットの上清;(9〜10)LSBである。図2Bおよび2C中、レーンは(1)分子量マーカー;(2〜4)1μgの3つの別個の対照HBsAg調製物;(5〜7)2〜8℃で2週間保存した、3つの別個の5価ロットの上清;(8〜10)36〜38℃で2週間保存した、3つの別個の5価ロットの上清である。図2D中、レーンは(1)分子量マーカー;(2)対照HBsAg;(3〜5)3つの別個の5価ロットの上清である。
【図3】図3は、5価組成物の経時的なpHの変化を示す。
【図4】図4は、8価組成物中のHBsAgの安定性に関するウエスタンブロットの結果を示す。図4Aおよび4B中、レーン1は分子量マーカーを含み、レーン2は1μg/mlでHBsAg対照を含み、レーン3は8価組成物の上清を含む。図4B中、レーン4はLSBを含み、レーン5はレーン2と同じ対照を含み、レーン6はDOC/TCA抽出物を含む。
【実施例】
【0160】
(発明を実施するための様式)
HBsAg発現および精製
HBsAgをコードするH.ポリモルファ酵母菌宿主[107、108]を調製した。300リットルの発酵槽中で100リットルの培地を調製し、酵母菌を接種した。100グラム/リットルで細胞が存在するまで発酵を続けた。この段階で、宿主がメチロトローフ酵母菌であるので、メタノールを加え、発酵を停止させた。最終培養物の体積は160〜170リットルであった。遠心分離によって培養培地から細胞を採取した。
【0161】
精製後にHBsAgに非イオン性界面活性剤を添加する代わりに、参考文献3および4中に記載したように、タンパク質の精製中に界面活性剤を使用した。詳細には、採取した酵母細胞を0.1〜0.2%のポリソルベート20および10mMのEDTAを含むリン酸バッファーに懸濁させた。懸濁物を冷却し、高圧液体ホモジェナイザーを使用して破壊した。
【0162】
均質化した細胞懸濁液は、遠心分離によって次いで浄化した。上清にNaClを加えた後、ポリエチレングリコールを沈殿用に3〜5%の濃度までゆっくり加えた。溶液は5分間攪拌し、30分間放置し、10分間遠心分離にかけた。さらなるPEGを8〜10%まで上清にゆっくり加え、攪拌し2時間放置した。この溶液を10分間遠心分離にかけた。
【0163】
ヒュームドシリカを1〜2%の濃度で上清に加えた。溶液を12時間攪拌し、遠心分離にかけ、デオキシコール酸ナトリウムを含むホウ砂バッファー中で沈殿を完全に溶かした。水浴中で100〜120分間攪拌した後、溶液を遠心分離にかけ、上清を回収した。流速200mL/分でトリス/Clバッファーを用いて平衡状態にしたDEAEカラムに上清を充填し、NaCl溶液を含むトリス/Clバッファーで溶出した。塩化セシウムを1.2g/mLの濃度までDEAE溶出液に加え、溶液を40,000rpmで超遠心分離にかけた。脱塩したCsClプールをPBSで400μg/mlのタンパク質濃度まで希釈し、ホルマリンを0.025%の濃度まで加え、混合物は72時間放置した。最後に、残留ホルマリンはPBSを使用して除去した。
【0164】
このようにして、HBsAgはポリソルベート20を含めることによって精製したが、抗原を精製した後に加えたのではなく、組換え細胞を破壊する前に界面活性剤を添加した。
【0165】
完全液状多価ワクチン
酵母菌発現型HBsAg、ジフテリアトキソイド、破傷風トキソイドおよび完全細胞百日咳抗原を、アルミニウム塩アジュバントの懸濁液に加えた。混合物のpHを調節し、それがアルミニウムアジュバントに吸着した状態にならないようにHib−CRM197複合体を次いで加えた。この方法によって、以下の組成を有する5価ワクチンを得た:
【0166】
【表1】
他の実験では、血清型C、W135およびYの各々に由来する別個の髄膜炎菌CRM197複合体を、CRM−Hib成分の後に加えて8価ワクチンを得た:
【0167】
【表2】
ワクチンの安定性および有効性に関する重要なパラメータはHib複合体の加水分解率であり、臨床上の限界は25%の遊離糖である(参考文献109は、20%は臨床上の免疫原性に影響を与えなかったことを報告する)。このパラメータは、最少の分離および浄化でピコモルレベルにおいて非結合炭水化物の直接定量化を可能にするHPAEC−PADによって、5価ワクチンにおいて測定した。分析は遊離糖の量に焦点を当てた。
【0168】
遊離糖の量をアッセイし、全体の量の割合として表した。結果は以下の通りであった:
【0169】
【表3】
最大25%の遊離糖が臨床上許容される。すべての値がこの閾値未満であり、2〜8℃において4週間まで6.5%未満であった。熱ストレス条件下(36〜38℃で4週間)では、高レベルが見られたが、25%の値を依然大幅に下回り、最高はロット1の11.6%であった。多価Hibワクチンに関する初期の実験は、36〜38℃での1カ月の保存は、2〜8℃での2年の保存より多くのCRM−Hibの加水分解をもたらすことを示している。したがって許容される加水分解は、通常の保存条件下での少なくとも2年の時間規模を超えると予想することができる。
【0170】
遊離糖のHPAEC−PAD分析は、2〜8℃で6カ月後にも実施した。データは以下の通りであった:
【0171】
【表4】
このように、4週間と比較して6カ月では遊離糖の割合の非常にわずかな増大があるのみで、値は25%の値を依然大幅に下回る。したがってCRM197−Hibは、3つの配合物において非常に安定している。
【0172】
図3Aは、2〜8℃で保存した3つのロットに関する、6カ月間の5価ワクチンにおけるpHの変化を示す。図3Bは、36〜38℃で保存した3つのロットに関する4週間のpHの変化を示す。2〜8℃においてpHは6カ月間安定状態であったが、一方熱ストレス条件下では、2週間後に0.1pH単位のわずかな低下、および4週間後にさらにわずかな低下があった。そうではあったが、すべてのpH値は6.0〜7.0の許容範囲内に留まった。
【0173】
全3個の5価ロットの浸透圧は、312mOsm/Kgと315mOsm/Kgの間、つまり注射用ワクチンに関する240〜360mOsm/Kgの標的範囲内の中心にあった。
【0174】
抗原の効力および免疫原性の評価は、組み合わせワクチンの有効性を評価するために重要である。5価ワクチンにおけるジフテリア、破傷風および百日咳抗原の効力を評価し、CRM−HibとHBsAgの両方の免疫原性を試験した。ELISA分析を実施して、免疫処置後の特異的抗体のレベルを評価した。HBsAgの免疫原性の試験は、マウスモデルおよびHBVの効力に関して使用したスケジュールと異なる免疫処置スケジュールを使用して実施した。
【0175】
DTPの効力値は以下の通りであった:
【0176】
【表5】
これら3つの抗原のそれぞれに関して、効力試験の結果はいずれも許容下限を有意に上回り、これらの結果は、これら3つの抗原に関する優れた有効性を示す。
【0177】
HBsAgの免疫原性を評価するために、10CD1マウスの群に、第0日および第14日に皮下注射(0.5ml、生理食塩水に1:4に希釈)によって5価ワクチンを与えた。マウスは第21日に採血し、(a)「Enzygnost Anti−HBs II」試験(Dade Behring)または(b)「Ausab EIA」試験(Abbott)のいずれかを使用してELISAによってHBsAg特異的抗体を評価した。これらのELISA試験は、異なる形式およびHBsAgに対する異なる感度を有する。幾何学的平均力価は、したがって2試験間で比較可能ではない。しかし、それぞれの試験の範囲内では血清に関するGMT値が最適であった。結果は以下の通りであった:
【0178】
【表6】
この種のマウス免疫原性アッセイを実施して得たすべてのGMT値は、文献中で報告されている値より高い。応答率は約100%の最適レベルで両方の抗原に関して絶えず高い。
【0179】
Hibの免疫原性を評価するために、8CD1マウスの群に、第0、10および20日に皮下注射(0.5ml、生理食塩水に1:4に希釈)によって5価ワクチンを与えた。マウスは第34日に採血し、ELISAによってHib特異的抗体を評価した。結果は以下の通りであった:
【0180】
【表7】
HBsAgのアルミニウムアジュバントへの吸着はワクチンの免疫原性に関する重要な要因であり、このパラメータはイムノブロットによって測定した。従ったイムノブロット手順は、ほぼ以下の通りであった:1ml体積のワクチン上清をDOC/TCA沈殿させ、LSBを用いて変性させ、次いで12%アクリルアミドSDS−PAGEに充填し、1μgの各ロットのHBsAgを対照として充填し、ヤギ抗HBsAg抗体調製物は一次抗体(希釈1:1000)として使用し、抗ヤギPOD複合体(希釈1:2500)は二次抗体として使用した。
【0181】
5価ワクチンに関する結果を図2中に示す。図2Aのレーン6〜8は時間ゼロで組成物中に検出可能な可溶性HBsAgが存在しないことを示し、図2Bおよび2Cのレーン5〜10によって、2〜8℃または36〜38℃での保存の2週間および4週間後、これは依然として真実であることが確認される。3つの異なるロットにおいて、これらの様々な条件で約99%のHBsAgがアジュバントに依然として吸着した状態であった。
【0182】
他の安定性アッセイを、2〜8℃での保存の6カ月後に実施した(図2D)。3つのロットの各々に関して依然として約99%吸着した状態であった。
【0183】
図2中で使用した陽性対照は、1μgのHBsAgを含んでいた。Sペプチド(24kDa)に対応する1つのバンド、および凝集に特徴的なバンド(45kDaまで)が見られた。Pre−S2は見られなかった。
【0184】
8価ワクチンも、同様の方法でHBsAg吸着に関して試験した。1mlのサンプルを10分間3500rpmで遠心分離にかけた。上清は新たなチューブに除去し、DOC/TCAで沈殿させた。ペレットは200μlの抽出バッファーに再懸濁させ、5分間沸騰させ、10分間1300rpmで遠心分離にかけた。20μlのこの抽出物およびDOC/TCAで沈殿させた上清を、ウエスタンブロット用に12%SDS−PAGEに充填した。
【0185】
図4Aは時間ゼロでの8価ワクチンを示し、図4Bは2〜8℃での保存の8カ月後のワクチンを示す。図4Bのレーン3および6における任意の有意な染色の不在(対照レーン2および5中の1μgのHBsAgで見られるより確実に染色されていないこと)は、この保存期間中HBsAgの吸着は依然として安定状態であることを示す。
【0186】
液状/凍結乾燥多価ワクチン
3つの抗原成分を以下のように回収する:
− 3価D−T−Pw成分:水酸化アルミニウムアジュバントに吸着したジフテリアトキソイド、さらに水酸化アルミニウムアジュバントに吸着した破傷風トキソイド、および完全細胞百日咳抗原を含むD−T−Pw成分を調製した。D−T−Pw成分はリン酸アルミニウムアジュバントも含む。この成分はチメロサールを含むが、2−フェノキシエタノールは含まない。
【0187】
− HBsAg成分:HBsAgを組換え酵母菌から発現させ精製し、リン酸アルミニウム抗原に吸着させる[110]。
【0188】
− Hib複合体成分:Hib多糖をHib、系統20752から調製し、臭化シアンを用いた活性化およびアジピン酸ヒドラジドを用いた誘導体化後、約1:3の糖:担体重量比で、スペーサーをカルボジイミド系縮合によって破傷風トキソイドと共有結合させる。結合体はリン酸アルミニウムアジュバントに吸着させ[64]、次いでラクトースと共に凍結乾燥させる。
【0189】
D−T−Pw成分はHBsAg成分と混合させる。HBsAgのレベルは20μg/mlである。Dのレベルは60IU以上である。Tのレベルは120IU以上である。Pwのレベルは8IU以上である。白濁した懸濁液の形態である4価混合物は、ブチルゴム製ストッパーを有するガラスバイアルに水性形態でパッケージして、1用量(0.5ml)、2用量(1ml)または10用量(5ml)を含ませる。凍結乾燥Hib成分もバイアル中に置く。バイアル当たりの粉末の量は、バイアル当たり1用量、2用量または10用量での水戻し後、(糖として測定して)5μg/mlを得るように定量する。
【0190】
2つのバイアルは箱中に1つにパッケージする。箱は1バイアルの各成分(1用量または多用量バイアルのいずれか)または100バイアルの各成分のいずれかを含む。
【0191】
患者への投与用に、水性D−T−Pw−HBsAg物質をシリンジに取り出し、凍結乾燥複合体用バイアル中に導入する。凍結乾燥物質を再度活性化させた後、新しいニードルを介してそれをシリンジに再度取り出し、患者にすぐに投与することができる5価ワクチンを得る。様々な3用量の初回免疫処置スケジュール:2、4および6カ月;3、4、および5カ月;または6、10および14週を使用することができる。ワクチンは2年時の追加抗原刺激投与として使用することもできる。
【0192】
10用量のバイアルは、1バイアルで10患者を免疫処置するのに充分な物質を与える。水戻しは第1のシリンジを使用し、次いで1シリンジ当たり1用量で、新たなシリンジを用いて個々の用量を抽出する。他の(あまり好ましくない)アレンジメントでは、10用量を1シリンジに取り出し、ニードルは患者ごとに変える。
【0193】
本発明は単なる実施例によって記載しており、本発明の範囲および精神中に留まりながら変更形態を作製することができることは理解されるはずである。
【0194】
参考文献(これらの内容は、参考として本明細書に援用される)
【0195】
【化1】
【0196】
【化2】
【0197】
【化3】
【特許請求の範囲】
【請求項1】
本明細書の一部に記載の発明。
【請求項1】
本明細書の一部に記載の発明。
【図1−1】

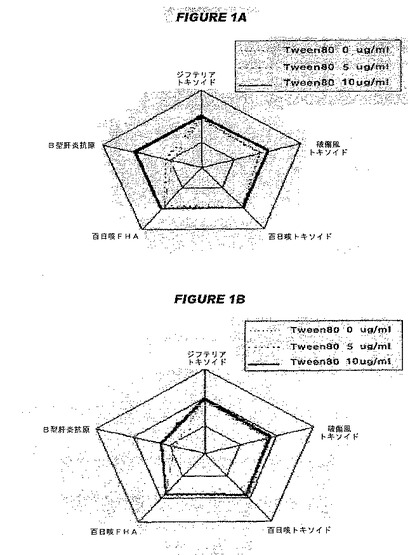
【図1−2】
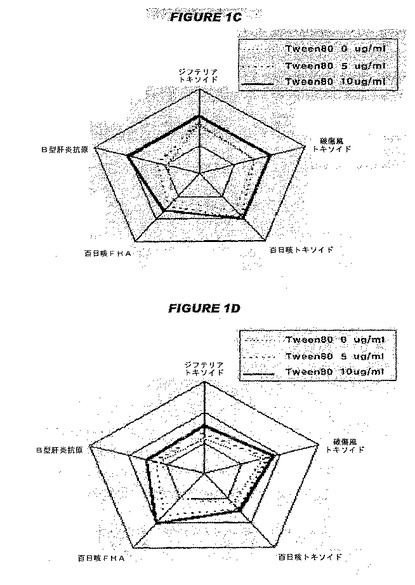
【図2−1】
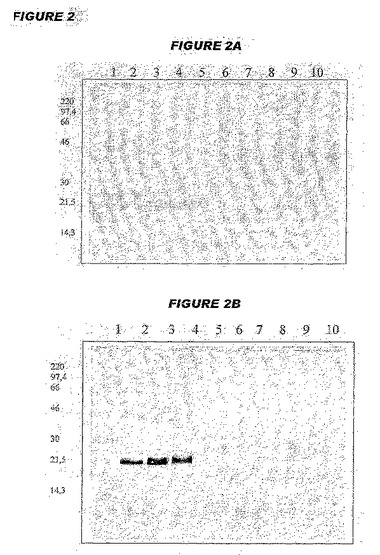
【図2−2】

【図3】
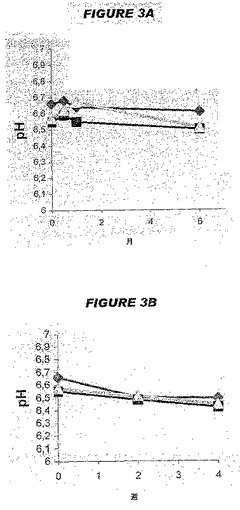
【図4】

【図1−2】
【図2−1】
【図2−2】
【図3】
【図4】
【公開番号】特開2013−56940(P2013−56940A)
【公開日】平成25年3月28日(2013.3.28)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−279058(P2012−279058)
【出願日】平成24年12月21日(2012.12.21)
【分割の表示】特願2008−539536(P2008−539536)の分割
【原出願日】平成18年11月7日(2006.11.7)
【出願人】(507238285)ノバルティス ヴァクシンズ アンド ダイアグノスティクス エスアールエル (35)
【Fターム(参考)】
【公開日】平成25年3月28日(2013.3.28)
【国際特許分類】
【出願番号】特願2012−279058(P2012−279058)
【出願日】平成24年12月21日(2012.12.21)
【分割の表示】特願2008−539536(P2008−539536)の分割
【原出願日】平成18年11月7日(2006.11.7)
【出願人】(507238285)ノバルティス ヴァクシンズ アンド ダイアグノスティクス エスアールエル (35)
【Fターム(参考)】
[ Back to top ]