
B型肝炎ウイルス表面抗原に特異的に結合するヒト抗体

B型肝炎の予防や治療に効果的なHBVの表面抗原(HBsAg)に特異的に結合する抗体が提供される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、B型肝炎ウイルス表面抗原(Hepatitis B virus surface antigen、HBsAg)に特異的に結合する抗体に関するものである。
【背景技術】
【0002】
B型肝炎ウイルス(HBV)はヘパドナウイルス(Hepadnaviridae)ファミリーのメンバーであり、急性および慢性肝炎を引き起こす。世界の人口のうち、約3.5億人、特に韓国および中国では人口の5%〜8%が慢性HBV感染者であり、HBVは肝疾患や肝臓がんの主な原因である。HBVに対するワクチンの開発でB型肝炎を予防することが可能になったが、未だ多くの人々がHBV感染による慢性肝炎に苦しんでいる。HBV感染は、肝炎、肝硬変だけでなく、肝臓がんを誘発し、慢性感染症の患者における肝臓がんの発生頻度が健常者よりも300倍高い。WHO(世界保健機構)は、肝臓がんの約80%がHBV感染による慢性B型肝炎から始まると報告した。
【0003】
現在利用可能なB型肝炎の治療薬は、ラミブジン(lamivudine)とアデフォビル・ジピボキシル(adefovir dipivoxil)などのヌクレオシド類似体(nucleoside analogue)を含んでおり、これらはHBVポリメラーゼを阻害することが知られている。しかし、3年間投与後、75%の患者において耐性ウイルスが発生して治療効果を低減させるので、これらは垂直感染や肝移植後の感染症を予防するために、B型肝炎の抗体製剤と組み合わせて使用されている。
【0004】
現在使用されているB型肝炎の抗体製剤は、抗HBV抗体を持つヒト血液源から高度の精製法およびウイルス不活性化法を用いて製造されているが、高い抗HBV抗体を持つ高価なヒト血漿の利用可能性が低いだけではなく、ヒト血漿−由来のウイルスを不活性化するコストが高いので、上記の方法は、増加する需要に対処することができない。
【0005】
ケーラーとミルシュタイン(1975)によってモノクローナル抗体(monoclonal antibody;mAb)の製造技術が確立されてから、マウス由来のモノクローナル抗体は、診断用や一部の治療のために主に使われてきた。しかし、マウス抗体は、治療目的のために人体に適用する際のヒト抗マウス抗体(HAMA)を生成する可能性があるため、投与することができない。HAMAの問題を解決するために、キメリク抗体とヒト化抗体が開発されてきた。キメリク抗体は、抗体分子全体の〜30%を構成するマウスmAbの可変領域(Fvフラグメント)を含有する。これに対し、ヒト化抗体は、抗体分子全体の〜10%を構成するマウスmAbのCDR(相補性決定領域)を含有する。上記キメリクおよびヒト化抗体は、HAMA反応を大幅に減少させてはいるが、長期間および継続的な投与を要する慢性肝炎のような慢性疾患の治療にはヒト抗体を使用する方がよいと思われる。
【0006】
本願発明者らは、新規で改良された抗体を開発しようと努力しており、上記の抗体がHBV表面抗原に結合してHBVを不活性化するのに使用できることを確認した。
【発明の概要】
【発明が解決しようとする課題】
【0007】
したがって、本発明の目的は、B型肝炎ウイルスの表面抗原に特異的に結合する新規の抗体を提供することである。
【0008】
本発明の他の目的は、上記抗体の重鎖可変領域および軽鎖可変領域をそれぞれコードするDNAおよびそれを含む発現ベクターを提供することである。
【0009】
本発明のまた他の目的は、上記発現ベクターに形質転換された細胞株を提供することである。
【0010】
本発明のまた他の目的は、上記抗体を含むB型肝炎の予防または治療用薬学組成物を提供することである。
【課題を解決するための手段】
【0011】
本発明の一つの態様により、a)配列番号1のアミノ酸配列を持つ重鎖可変領域、b)配列番号2、3、および4のいずれか1つのアミノ酸配列を持つ軽鎖可変領域、c)重鎖定常領域、およびd)軽鎖定常領域を含んでいる、B型肝炎ウイルス表面抗原(HBsAg)に特異的に結合する抗体が提供される。
【0012】
本発明の他の態様により、配列番号1のアミノ酸配列を持つ重鎖可変領域をコードするDNAまたは配列番号2、3、および4のいずれか1つのアミノ酸配列を持つ軽鎖可変領域をコードするDNAおよびそれを含む発現ベクターが提供される。
【0013】
本発明の別の態様により、上記発現ベクターに形質転換された細胞株が提供される。
【0014】
本発明のまた別の態様により、上記抗体を含むB型肝炎の予防または治療用組成物が提供される。
【図面の簡単な説明】
【0015】
本発明の上記及びその他の目的と特徴は、添付の図面とともに考慮されるときに下記説明により明確になるもので、以下の通りである。
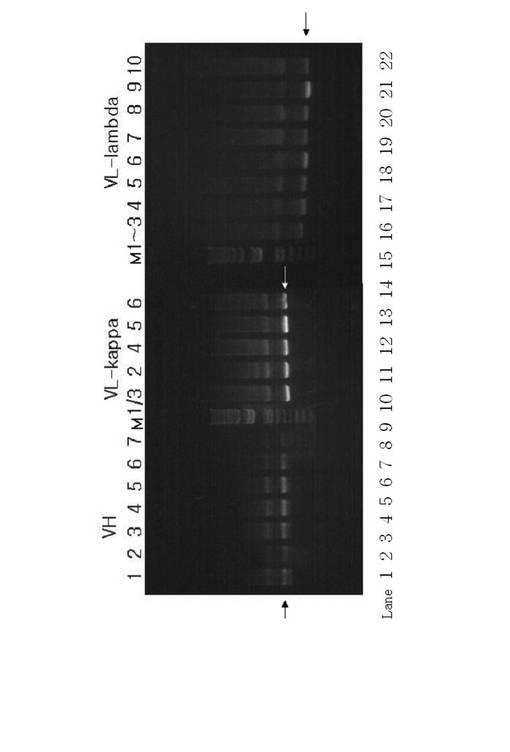
【図1】図1は、PCRによって合成された本発明の重鎖可変領域(VH)および軽鎖可変領域(VL)をそれぞれコードするDNAを示す電気泳動(1%アガロースゲル)の分析の写真結果である。
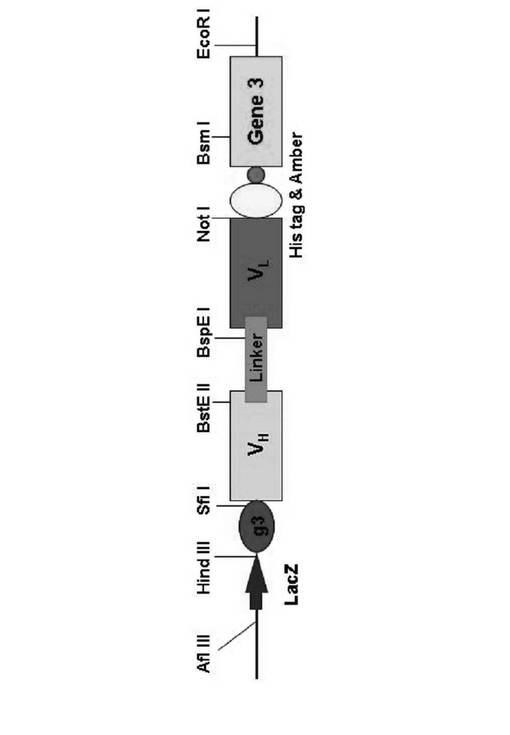
【図2】図2は、本発明の抗体の重鎖可変領域および軽鎖可変領域を含む、ファージディスプレイベクターpKS4Hの地図である。
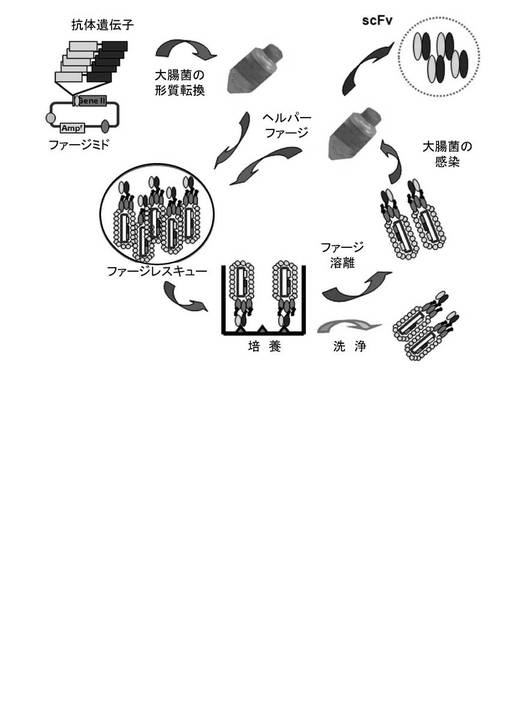
【図3】図3は、パンニング法を利用して、抗体ライブラリから抗体を選別する工程を示した図である。
【図4】図4は、抗体ライブラリから選別された本発明の抗体の一本鎖可変断片(ScFV)のアミノ酸配列である。
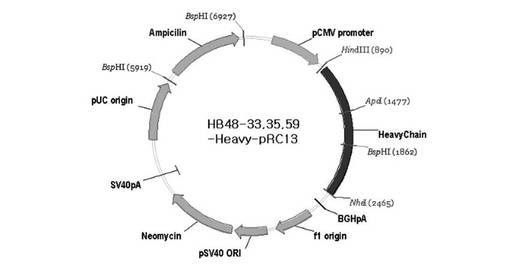
【図5】図5は、本発明のヒト抗体の重鎖を発現するための発現ベクター、HB48−33−Heavy−pRC13、HB48−35−Heavy−pRC13またはHB48−59−Heavy−pRC13の地図である。
【図6】図6は、本発明のヒト抗体の軽鎖を発現するための発現ベクター、HB48−33−Light−pKC12の地図である。
【図7】図7は、本発明のヒト抗体の軽鎖を発現するための発現ベクター、HB48−35−Light−pKC12の地図である。
【図8】図8は、本発明のヒト抗体の軽鎖を発現するための発現ベクター、HB48−59−Light−pKC12の地図である。
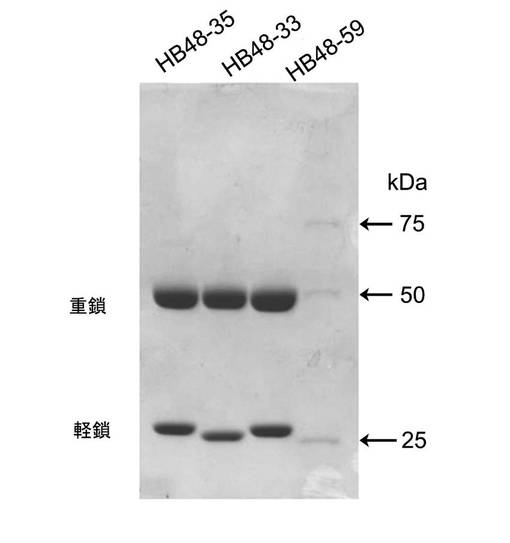
【図9】図9は、形質転換体から発現された重鎖および軽鎖のSDS−PAGE分析結果である。
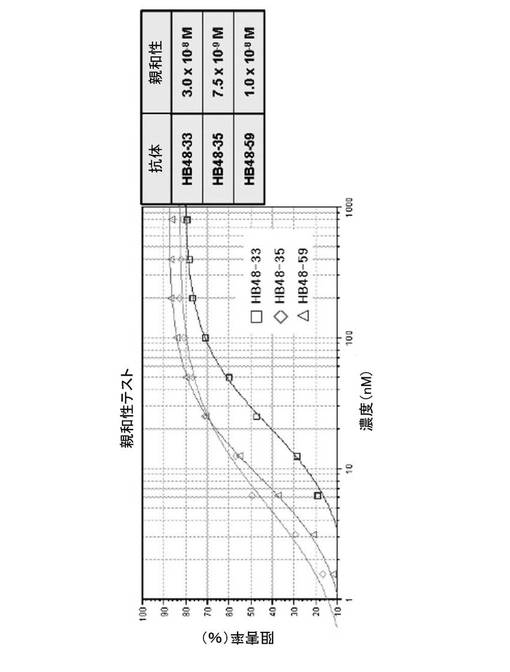
【図10】図10は、ヒト抗体(HB48−33、HB48−35およびHB48−59)のHBV表面抗原に対する相対的親和性である。
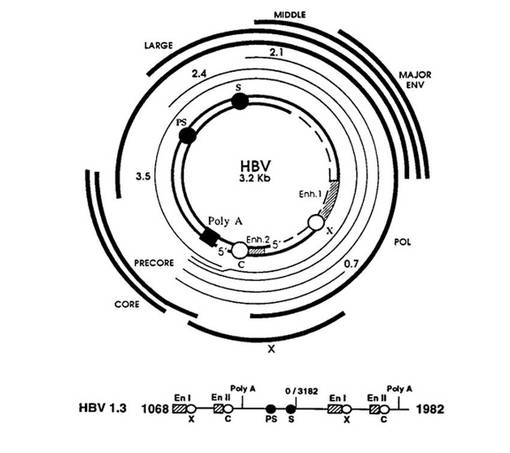
【図11】図11は、急性B型肝炎のマウスモデルの製造に使用された、pcDNA3.1にHBV DNA配列を挿入して製造されたベクターpHBV1.3−MBRIの地図である。
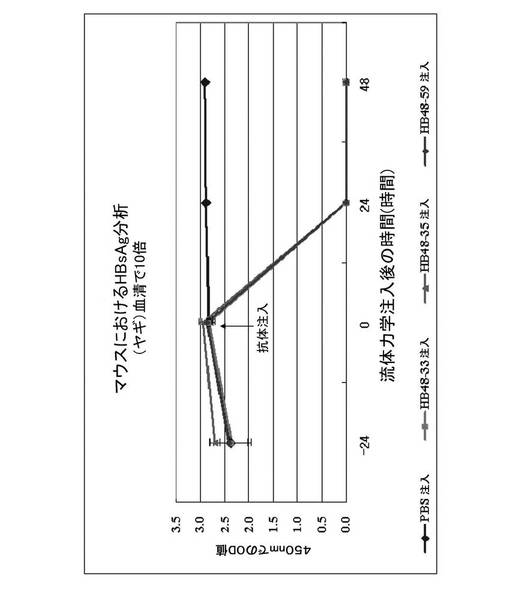
【図12】図12は、急性B型肝炎のマウスモデルを用いたin vivoの効果試験によって選別された本発明のヒト抗体HB48−33、HB48−35とHB48−59の中和能を示すグラフである。図11のプラスミドを注入して24時間後、HBV表面抗原(HBsAg)が発現したことを確認し、本発明の抗体の投与により、表面抗原が減少したことを確認した。表面抗原は、Genedia HBsAg ELISA 3.0(緑十字社 MS、韓国)で測定した。
【発明を実施するための形態】
【0016】
以下、本発明を詳細に説明する。
【0017】
本発明は、a)配列番号1のアミノ酸配列を持つ重鎖可変領域、b)配列番号2、3、および4のいずれか1つのアミノ酸配列を持つ軽鎖可変領域、c)重鎖定常領域、およびd)軽鎖定常領域を含んでいる、B型肝炎ウイルス表面抗原(HBsAg)に特異的に結合する抗体を提供する。本発明の抗体は、B型肝炎ウイルス表面抗原に特異的に結合してHBVを不活性化させることによってB型肝炎の予防または治療に効果を有することを特徴とする。
【0018】
上記HBV表面抗原に特異的に結合する抗体は、好ましくは、ファージディスプレイ法(Smith、Science、228、1315−1317、1985およびHoogenboom&Chames、Immunol Today、21、371−378、2000)を応用して選別することができる。上記のファージディスプレイ法では、フィラメント(filamentous)ファージ(例えば、M13、Fd、またはF1)の表面タンパク質をコードする遺伝子(gene III)が、目的の抗体をコードする遺伝子と融合され、表面に露出した融合抗体を持つ抗体−ファージ型のウイルス粒子を生成させる。その後、抗体の高い特異性および親和性、並びにファージの高い感染性を利用してバイオパンニング(biopanning)技術によってファージライブラリから目的とする抗体が選別できる(Burton&Barbas,Adv.Immunol.,57、191−280、1994、Winter et al.,Annu.Rev.Immunol.,12、433−455、1994、およびHoogenboom et al.,Immunotechnology、4、1−20、1998、 Kim et al.,Hybrid Hybridomics、21、 385−392、2002)。上記ファージディスプレイベクターは、pKS4H(韓国特許第0635370号を参照)またはpCANTAB5E、好ましくはpKS4Hである場合もある。
【0019】
本発明者らは、ファージライブラリから3つのヒト抗体(HB48−33、HB48−35およびHB48−59)を選別した後、HBV表面抗原に対する抗体の親和性と中和能を調べた(図10および11)。また、重鎖および軽鎖の可変領域のアミノ酸配列を分析した結果、すべての重鎖可変領域が配列番号1のアミノ酸配列を持ち、軽鎖可変領域がそれぞれ配列番号2,3、および4のアミノ酸配列を持つことを確認した。
【0020】
本発明の抗体の重鎖および軽鎖の不変領域はヒト抗体のものと同一であってもよい。
【0021】
本発明は、配列番号1のアミノ酸配列を持つ抗体重鎖可変領域をコードするDNAを提供する。好ましくは、上記DNAは、配列番号1のアミノ酸配列をコードする配列番号5のヌクレオチド配列を持つポリヌクレオチドを含んでもよい。
【0022】
本発明は、配列番号2、3および4のいずれか1つのアミノ酸配列を持つ抗体の軽鎖可変領域をコードするDNAを提供する。好ましくは、上記DNAは、それぞれ配列番号2,3、および4のいずれか1つのアミノ酸配列をコードする配列番号6、7および8のいずれか1つのヌクレオチド配列を持つポリヌクレオチドを含んでもよい。
【0023】
本発明は、上記抗体の重鎖可変領域をコードするDNAを含んでいる、HBV表面抗原に特異的に結合する抗体の重鎖可変領域を発現するための発現ベクターを提供する。好ましくは、上記発現ベクターは、図5に地図で示した“HB48−33−Heavy−pRC13”、“HB48−35−Heavy−pRC13”または“HB48−59−Heavy−pRC13”であってもよい。
【0024】
具体的に、上記ベクターは、パンニング過程によって選別された抗体のVH断片(HB48VH)を適切なベクター、例えば、pRC13ベクター(寄託番号KCLRF−BP−00054、大韓民国特許第523732号を参照)に挿入して製造されることができる。
【0025】
本発明は、上記抗体の軽鎖可変領域をコードするDNAを含んでいる、HBV表面抗原に特異的に結合する抗体の軽鎖可変領域を発現するための発現ベクターを提供する。好ましくは、上記ベクターは、図6に地図で示した“HB48−33−Light−pKC13”、図7に地図で示した“HB48−35−Light−pKC13”または図8に地図で示した“HB48−59−Light−pKC13”であってよい。
【0026】
具体的に、上記ベクターは、パンニング過程によって選別された抗体のVL断片(HB48−33VL、HB48−35VLまたはHB48−59VL)を適切なベクター、例えば、pKC12ベクター(寄託番号KCLRF−BP−00054、大韓民国特許第523732号を参照)に挿入して製造することができる。
【0027】
本発明は、本発明の抗体の重鎖および軽鎖の可変領域を発現するための発現ベクターに形質転換された動物細胞株を提供する。上記動物細胞株は、CHO(Chinese hamster ovary)、HEK 293、またはNSO細胞株、好ましくはCHO(Chinese hamster ovary)細胞株でもよい。
【0028】
本発明に係る抗体は、上記重鎖および軽鎖の可変領域が互に結合することで製造することができる。
【0029】
抗原に対する本発明の抗体の親和性(affinity)は、例えば、競合ELISA(Kim et al.,Hybridoma、20、265−272、2001)によって測定することができる。図10に示すように、本発明のヒト抗体の中でHB48−35の親和性が最も高かったのに対し、HB48−59およびHB48−33の親和性は、HB48−35に比べてそれぞれ約1.3倍と4.0倍低かった。また、B型肝炎と類似の兆候を誘導するために、C57BL6マウス内にHBV DNAを流体力学注入(Hydrodynamic injection; Liu et al.,Gene Therapy、6、1258−1266、1999)して製造したHBVマウスモデルにおいて、HB48−33、HB48−35およびHB48−59抗体は、マウスの血液から、上記抗体によりHBVの表面抗原が基底レベルまで減少したHBVの中和能を示した(図12)。したがって、本発明の抗体は、HBVの表面抗原に結合してHBVを不活性化させることによって、B型肝炎を予防または治療するために使用することができる。
【0030】
上記の結果から本発明は、前記抗体を含むB型肝炎の予防または治療用薬学組成物を提供する。本発明の組成物は、通常の方法に応じて、薬学製剤に製造することができる。剤型の製造において、上記抗体は、好ましくは、担体と混合したり、担体で希釈されたり、容器の形態でもよい担体内に組み込まれる。担体が希釈剤として使用される場合、これは抗体に対する小胞(vesicle)、賦形剤または媒質(medium)として作用する固形、半固形または液状であってもよい。したがって、剤型は錠剤、丸剤、粉剤、サシェ(sachet)、カシェ(cachet)、エリクシール(elixir)、懸濁剤、乳剤、溶液剤、シロップ剤、エアロゾル、軟質または硬質ゼラチンカプセル剤、滅菌注射溶液、滅菌粉剤などの形を取ることができる。適切な担体、賦形剤および希釈剤の例として、ラクトース、デキストロース、スクロース、ソルビトール、マンニトール、カルシウムシリケート、セルロース、メチルセルロース、微晶質セルロース、ポリビニルピロリドン、水、メチルヒドロキシベンゾエイト、プロフィールヒドロキシベンゾエイト、タルク、ステアリン酸マグネシウムおよび鉱物油を挙げることができる。剤型は、充填剤、抗凝集剤、潤滑剤、湿潤剤、香料、乳化剤、防腐剤などをさらに含むことができる。本発明の抗体組成物は、上記抗体とともに、抗ウイルス剤として、インターフェロン、抗HBVモノクローナル抗体、抗HBVポリクローナル抗体、ヌクレオシド類似体、DNAポリメラーゼ阻害剤、siRNA製剤または治療用ワクチンをさらに含むことができる。
【0031】
HBV感染およびHBV関連の疾病は、本発明の組成物を、人間を含む哺乳動物に投与することで、予防または治療することができる。上記組成物内の抗体の投与量は、対象、疾病状態の重症度、投与速度、および医師の判断に左右され得る。有効成分として、上記抗体は、単一容量または分割用量で、一日に約0.001mg/kg体重〜10mg/kg体重、好ましくは0.005mg/kg体重〜1mg/kg体重範囲の有効量で経口以外の経路を介して哺乳動物に投与することができる。場合によっては、上記の投与量に比べて少ないかまたは多い量が投与量として適切であり、多量の投与量は、一日あたりに分割された少ない投与量で投与することができる。
【0032】
以下の実施例は、単に例示の目的で提供され、本発明の範囲を限定しようとするものではない。
【0033】
実施例1:RNAの分離
HBV表面抗原に特異的に結合する抗体を選別するために、ヒト抗体ライブラリを構築した。ヒト骨髄、ヒト胸腺、ヒト脾臓およびヒトB細胞から得られたRNAの混合物を使用した。ヒトB細胞RNAを除くすべてのRNAはClontech社(米国)から購入し、ヒトB細胞RNAは下記のように分離した。
【0034】
健康な成人から採血した50mLの血液を50mLのPBSと混合し希釈した。3mLのFicoll−Paque PLUS(GE Healthcare社、米国)を15mLチューブに入れ、ここに上記の希釈血液4mLを添加した。上記混合物を3,000rpmで遠心分離して白血球を分離した。上記分離した白血球2mLを6mLのPBSと混合し、100xgで10分間遠心分離した。上記の白血球100μLを1mLのトリゾール(登録商標)(Life Technology社、米国)と混合してRNAを分離した。
【0035】
一方、上記分離されたRNAを蒸留水で希釈した後、260nmで吸光度を測定し、その量を計算した(1.8μg/μL、フルスペクトル2000UV−VIS分光光度計、GE、米国)。詳細な手順は次のとおりである。
【0036】
1mLのトリゾールを100μLの白血球に添加してよく混ぜた後、常温(15℃〜30℃)で5分間放置した。そして、クロロホルム200μLを添加して15秒間よくかき混ぜてから、常温で3分間放置した。その後、上記混合物を2℃〜8℃、15分および15,000rpmの条件の下で遠心分離し、上清液を新しいチューブに移した。イソプロピルアルコール500μLを加えてよく混合した後、室温で10分間放置した。そして、上記混合物を2℃〜8℃および15,000rpmで5分間遠心分離して上清を除去した。これに75%エタノール1mLを添加し、上記混合物を2℃〜8℃、5分間、15,000rpmの条件下で遠心分離しエタノールを除去した後、RNAペレットを常温で5分間乾燥させた。これに蒸留水150μLを添加してRNAを溶解させた後、260nmにて上記懸濁液の吸光度を測定した。残りは−20℃で保管した。
【0037】
実施例2:抗体遺伝子の増幅
実施例1で分離したRNA1μgおよびpd(T)12〜18(0.5μg/μL)1μLを蒸留水と混合して最終体積を12.5μLに合わせた。上記混合物を70℃で2分間反応させ氷で冷却させた。その後、これに5X反応緩衝液、10mM dNTP混合物、組換えRNase阻害剤およびMMLV逆転写酵素(Clontech、米国)を添加して最終体積を20μLに合わせた後、42℃で1時間、95℃で5分間反応させてcDNAを合成した。LiquiMix QM Premix、Magenta(Neurotics Inc、韓国)、鋳型として4μLのcDNA、19μLの蒸留水並びにそれぞれscFv領域(single chain Fv)、重鎖可変領域、および軽鎖可変領域(kappaおよびlambda)のために考案された1μL(25pmol/μL)のプライマー[ScFv:配列番号9(Forward)、10(Reverse−κ)および11(Reverse−λ);重鎖可変領域:配列番号12(Forward−VH1)、13(Forward−VH2)、14(Forward−VH3)、15(Forward−VH4)、16(Forward−VH5)、17(Forward−VH6)、18(Forward−VH7)および19(Reverse);軽鎖可変領域κ鎖:配列番号20(Forward−VK1/3A)、21(Forward−VK1/3B)、22(Forward−VK2)、23(Forward−VK4)、24(Forward−VK5)、25(Forward−VK6)、26(Reverse−JK−A)、27(Reverse−JK−B)および28(Reverse−JK−C);軽鎖可変領域λ鎖:配列番号29(Forward−VL1〜3−A)、30(Forward−VL1〜3−B)、31(Forward−VL4)、32(Forward−VL5)、33(Forward−VL6)、34(Forward−VL7)、35(Forward−VL8)、36(Forward−VL9)、37(Forward−VL10)、38(Reverse−JL−A)および39(Reverse−JL−B)]を使用してPCR反応を行った。
【0038】
PCR反応は、95℃で5分、95℃で1分、55℃で2分、72℃で2分間を30回反応させた後、最後に72℃で15分間反応させて行われた。
【0039】
上記増幅された抗体DNAを1.2%アガロースゲルにおいて電気泳動によって確認し(図1参照)、キアゲンキット(Qiagen社、ドイツ)を用いて精製した。図1に示すように、重鎖および軽鎖(kappaおよびlambda)の可変領域に対応する1,350bpのDNAバンドが得られた。図1においてMは、サイズマーカー、VHは重鎖可変領域(レーン1:重鎖可変領域タイプI、レーン2:重鎖可変領域タイプII、レーン3:重鎖可変領域タイプIII、レーン4:重鎖可変領域タイプIV;レーン5:重鎖可変領域タイプV、レーン6:重鎖可変領域タイプVI、およびレーン7:重鎖可変領域タイプVII)、およびVLは、軽鎖可変領域(レーン9:軽鎖可変領域1/3κ、レーン10:軽鎖可変領域2κ、レーン11:軽鎖可変領域4κ、レーン12:軽鎖可変領域5κ、およびレーン13:軽鎖可変領域6κ、レーン15:軽鎖可変領域1λ〜3λ、レーン16:軽鎖可変領域4λ、レーン17:軽鎖可変領域5λ、レーン18:軽鎖可変領域6λ、レーン19:軽鎖可変領域7λ、レーン20:軽鎖可変領域8λ、レーン21:軽鎖可変領域9λおよびレーン22:軽鎖可変領域10λ)を指す。
【0040】
実施例3:抗体DNAの制限酵素切断
実施例2で準備したVHおよびVLをそれぞれ制限酵素SfiI/BspEIおよびBspEI/NotIで切断し、上記切断した断片を1.2%のアガロースゲル電気泳動で分離した後、キアゲンキットを用いて精製した。
【0041】
実施例4:抗体DNAのライゲーションおよびライブラリの製造
ファージディスプレイベクターpKS4H(緑十字社、韓国、韓国特許第635370号参照)を制限酵素SfiI/BspEIを使用して切断し、1.2%アガロースゲル電気泳動を利用して分離した後、キアゲンキットを利用して精製した。40μgのpKS4Hを実施例3で用意された10μgのVHと混合し、これにT4 DNAリガーゼ(New England BioLabs、米国)を添加した後、25℃で一晩中反応させた。上記リガーゼ反応混合物を、キアゲンキットを用いて精製し、大腸菌XL1−blue(Stratagene社、米国)に電気穿孔法(electroporation)により形質転換させた。上記形質転換体を、100mLの培地で一晩培養し、プラスミドを分離した。上記のプラスミドを“pKS4H−VH−△VL”と命名した。
【0042】
上記プラスミドpKS4H−VH−△VLを制限酵素BspEI/NotIで切断し、上記記載のように精製した。その後、40μgのpKS4H−VH−△VLプラスミドを実施例3で用意された10μgのVL PCR DNAおよびT4 DNAリガーゼ(New England BioLabs、米国)と混合し、25℃で一晩反応させた。上記リガーゼ反応混合物を、キアゲンキットを用いて精製し、大腸菌XL1−blueに電気穿孔法により形質転換させた。上記形質転換体をカルベニシリン(carbenicillin)およびテトラサイクリン(tetracyclin)を含有する100mLの培地において37℃で2時間培養した。そして、上記培地にM13ヘルパーファージ(Stratagene社、米国)を接種し、16時間培養してエンベルグ等(Engberg et al.,Mol.Biotechnol.,6、287−310、1996)で報告されたように、ファージライブラリを作製した。一方、上記大腸菌からプラスミドを分離して、“pKS4H−VH−VL”と命名した。上記プラスミドの地図を図2に示している。
【0043】
実施例5:HBV表面抗原に結合する抗体の選別
HBV表面抗原に結合する抗体をパンニング法(Engberg et al.,Mol.Biotechnol.,6、287−310、1996およびKim et al.,Gene、241、19−25、2000; Kim et al., Hybrid Hybridomics、21、385−392、2002)を応用して選別した。具体的に、HBVの表面抗原(緑十字社、韓国)をPBSバッファーで希釈し、それぞれのイムノチューブ(immunotube、NUNC社、デンマーク)を、上記希釈された抗原でコーティングした。そして、上記のコーティングされたイムノチューブに実施例4で製造したファージライブラリを添加し、37℃で2時間培養した。HBVに結合するファージを、0.1Mグリシンバッファー(pH2.0)を使用して溶出させた。以降、上記ファージで大腸菌XL1−blue細胞を感染させヘルパーファージ(Stratagene社、米国)を添加した。上記大腸菌(E.coli)を一晩培養し、これにPEG溶液(20%PEG8000および15%NaCl)を添加した。そして、沈殿したファージを回収して(ファージレスキュー)、上記ファージを再度HBVの表面抗原がコーティングされたイムノチューブと反応させて、上記過程を4回繰り返した。上記過程からHBVの表面抗原に結合する3つのヒト抗体を選別し、これをHB48−33、HB48−35およびHB48−59と命名した。ファージディスプレイライブラリを用いてヒト抗体を選別する過程を図3に示した。
【0044】
4回のパンニングから得られた各コロニーを公知の方法(Kim et al.、Gene、241、19−25、2000)によって2mLの培地で成長させ、IPTG(isopropyl bD−thiogalactopyranoside)を処理して抗体発現を誘導した。抗体の発現をHBVの表面抗原がコーティングされた96−ウェルプレートを使用してELISA(Enzyme−Linked Immunosorbent Assay)により測定した。
【0045】
実施例6:選別された抗体の配列分析
実施例5で選別されたヒト抗体HB48−33、HB48−35とHB48−59を分泌するコロニーを50μg/mLのカルベニシリンを含有する10 mLのLB培地で一晩培養した後、キアゲンプラスミドミニキット(Qiagen、Valencia、CA、米国)を使用して、プラスミドを分離した。上記のプラスミドをSfiI/NotIで切断した後、抗体の断片の挿入を確認するためにアガロースゲルで電気泳動を行った。プラスミドに挿入されたscFvのDNA配列を分析した。scFvのヌクレオチド配列をGenotech社(大田、韓国)にてシーケンスプライマである配列番号40のp033で分析した。
【0046】
ヒト抗体HB48−33、HB48−35およびHB48−59のscFvのDNA配列を、Webベースのプログラム(www.expasy.org:DNA to Protein translate tool)を使用して、アミノ酸に翻訳し、上記翻訳アミノ酸配列を図4に示した。図4に示すように、ヒト抗体HB48−33、HB48−35およびHB48−59は、同一のアミノ酸配列で示される重鎖可変領域および異なるアミノ酸配列で示される軽鎖可変領域を持っていた。
【0047】
実施例7:発現ベクターの製造
上記の抗体断片を完全な免疫グロブリンに転換させるために、抗体発現ベクターpRC13およびpKC12を使用した(韓国特許第523732号参照、寄託番号KCLRF−BP−00054)。
【0048】
重鎖発現ベクターpRC13は、抗体のVH断片がHindIIIおよびApaI部位内に簡単に挿入できるベクターである。図5に例示のように、本発明では、ヒト抗体HB48−33、HB48−35またはHB48−59の重鎖可変領域をコードするDNAを配列番号41(Forward)および42(Reverse)のプライマーを使用して、95℃で1分、55℃で2分、72℃で2分の条件下で増幅させた後、HindIII/ApaIで切断して、同じ制限酵素で切断されたpRC13に挿入した。上記組換えベクターを、“HB48−33−Heavy−pRC13”、“HB48−35−Heavy−pRC13”または“HB48−59−Heavy−pRC13”と命名した(図5参照)。
【0049】
一方、軽鎖発現ベクターpKC12ベクターは抗体のVL断片がNheIおよびApaI部位内に簡単に挿入できるベクターである。図6、図7および図8に示したように、ヒト抗体HB48−35とHB48−59とのk軽鎖可変領域をコードするDNAを配列番号43(Forward)および44(Reverse)のプライマーを使用して、95℃で1分、55℃で2分、72℃で2分の条件下で増幅させ、ヒト抗体HB48−33のλ軽鎖可変領域をコードするDNAを配列番号43(Forward)および45(Reverse)のプライマーを使用して95℃で1分、55℃で2分、72℃で2分の条件下で増幅させた。上記増幅されたDNAをNheI/ApaIで切断し、同じ制限酵素で切断されたPKC12内に挿入した。上記組換えベクターを、それぞれ“HB48−33−Light−pKC12”、“HB48−35−Light−pKC12”および“HB48−59−Light−pKC12”と命名した(図6、図7および図8を参照)。
【0050】
実施例8:抗体分泌動物細胞株の製造
形質感染24時間前に、2×105個のCHO(chinese hamster ovary)細胞を10%のFBS(Life Technologies社、米国)を含有するα−MEM培地(Life Technologies社、米国)が入ったT−25フラスコ(NUNC社、デンマーク)で、培養密度(confluency)が50%に到達する直前まで、5%CO2の存在下で、37℃のインキュベーターで培養した。その後、30μgのリポペクチン(Life Technologies社、米国)を1.5mLのOpti−MEM(Life Technologies社、米国)に入れて常温で90分間放置した。同時に、8μgのHB48−33−Heavy−pRC13および7μgのHB48−33−Light−pKC12、8μgのHB48−35−Heavy−pRC13および7μgのHB48−35−Light−pKC12、8μgのHB48−59−Heavy−pRC13および7μgのHB48−59−Light−pKC12をそれぞれ混合し、1.5mLのOpti−MEMに入れてから、常温で90分間放置した。90分後、リポペクチンを含有する培地をHB48−33−Heavy−pRC13&HB48−33−Light−pKC12、HB48−35−Heavy−pRC13&HB48−35−Light−pKC12、およびHB48−59−Heavy−pRC13&HB48−59−Light−pKC12を含有する培地と混合した後、15分間室温で反応させた。反応させる間に、上記細胞から培地を除去し、細胞をPBSで3回洗浄した。上記洗浄された細胞に上記の反応混合物を入れて6時間培養した。6時間後、上記反応混合物を除去し、α−MEM培地を入れて48時間培養した。そして、上記細胞をトリプシン(Life Technologies社、米国)で処理してフラスコから外した後、α−MEM培地で希釈し、96−ウェルプレート(NUNC社、デンマーク)に継代した。このとき、α−MEM培地はリボヌクレオシドとデオキシリボヌクレオシドを含有していないのに対し、10%の透析されたFBS(Life Technologies社、米国)および550μg/mLのG418(Sigma社、米国)を含有する。2日ごとに上記培地を新しい培地に交換した。ELISA分析のために、コロニーを形成した培養上清を回収し、選別された細胞を12−ウェルプレートに移した。細胞が12−ウェルプレートでよく育てば、細胞を6−ウェルプレートに移して、細胞が6−ウェルプレートでもよく育つと、これにメトトレキサート(MTX、中外製薬、韓国)を処理した。MTXの初期濃度は20nMであり、細胞の適応に応じて、80nM、320nMおよび1μMで増加した。1μMの濃度で生存して高い量の抗体分泌を持つ細胞株を選別した。上記選別された細胞株を、スピナーフラスコと無血清培地とを使用して65rpm、5%CO2および37℃のインキュベーターで大量に培養した。100mLの無血清培地が入った250mLフラスコで108細胞を培養した。細胞数が初期濃度の2倍になると、1,000rpmで5分間遠心分離して細胞を回収した。上記回収した細胞を再び200mLの培地が入った500mLフラスコで培養した。細胞数が初期濃度の2倍になると、1,000rpmで5分間遠心分離して細胞を回収し、1Lの培地が入った3Lスピナーフラスコに移した。ブチル酸ナトリウム(Aldrich社、米国)を最終濃度2mMになるように添加し、上記細胞を5日間培養した後、培地から上清を回収した。スピナーフラスコから回収した上清から、Protein A−アガロースカラム(Amersham Pharmacia Biotech社、米国)を使用して抗体を精製し、SDS−PAGEにより分析した。
【0051】
図9に示すように、約50kDaの重鎖と25kDaの軽鎖とのバンドが観察され、これは抗体が正確に合成されたことを示す。
【0052】
実施例9:抗体の親和性の測定
実施例8で得られた抗体のHBV表面抗原に対する親和性を競合ELISA法(Kim et al.,Hybridoma、20、265−272、2001)により決定し、上記の結果を図10に示した。簡略な手順は次のとおりである。
【0053】
(1)適正抗体濃度の決定
A.プレートの準備
PBS中で2μg/mLで希釈された、100μLのHBV表面抗原(緑十字社、韓国)をELISAプレートの各ウェルに入れ、4℃で一晩培養した。プレートの各ウェルをPBSTで1回洗浄し、各ウェルに300μLの1%BSA−PBS溶液を加えた後、室温で1時間培養した。
【0054】
B.1次反応
100μLのそれぞれ精製された抗体(0.5μg/mL)をプレートの各ウェルに入れ、室温で2時間反応させた後、PBSTで4回洗浄した。
【0055】
C.2次反応
1%BSA−PBSで1:5,000に希釈された100μLのヤギ抗ヒトIgG(Fab specific)−パーオキシダーゼ接合体(Sigma)を各ウェルに添加し、室温で1時間培養した後、PBSTで4回洗浄した。
【0056】
D.基質反応
100μLのTMB(3,3’、5,5’−テトラメチルベンジジン(Tetramethylbenzidine)、マイクロウェル パーオキシダーゼ サブストレート システム(KPL、MD、米国))を各ウェルに添加した後、405nmでO.D値を測定した。抗体の適正濃度を半分−最大結合値(half−maximum binding)を示す抗体濃度として決定した。
【0057】
(2)競合ELISA
A.プレートの準備
PBS中で2μg/mLで希釈された、100μLのHBV表面抗原(緑十字社、韓国)をELISAプレートの各ウェルに入れ、4℃で一晩培養した。各ウェルをPBSTで1回洗浄し、各ウェルに300μLの1%BSA−PBS溶液を加えた後、室温で1時間培養した。
【0058】
B.1次反応
2μgのHBV表面抗原を2の倍数で連続希釈し、プレートの各ウェルに10μLの上記希釈されたHBsAgを添加した。それから、(1)で決定された適正濃度に希釈した90μLの抗体を各ウェルに添加し、室温で2時間反応させた後、PBSTで4回洗浄した。
【0059】
C.2次反応
ヤギ抗−ヒトIgG(Fab specific)−パーオキシダーゼ接合体(Sigma)1%BSA−PBS溶液で5,000倍に希釈し、各ウェルに100μLずつ添加し、室温で1時間反応させた後、PBSTで4回洗浄した。
【0060】
1%BSA−PBSで1:5,000に希釈された、100μLのヤギ抗−ヒトIgG(Fab specific)−パーオキシダーゼ接合体(Sigma)を各ウェルに添加し、室温で1時間培養した後、PBSTで4回洗浄した。
【0061】
D.基質反応
TMB(3,3’、5,5’−テトラメチルベンジジン(Tetramethylbenzidine)、マイクロウェルパーオキシダーゼサブストレートシステム(KPL、MD、米国))溶液を100μLずつ各ウェルに添加した後、405nmでO.D値を測定した。最大結合値(競合する上皮成長因子受容体がない状態のELISA O.D値)の50%を阻害するHBV表面抗原の濃度を親和性(Kd)で算定した。
【0062】
100μLのTMB(3,3’、5,5’−テトラメチルベンジジン(Tetramethylbenzidine)、マイクロウェルパーオキシダーゼサブストレートシステム(KPL、MD、米国))を各ウェルに添加した後、405nmでO.D値を測定した。最大結合値(競合するEGFRが存在しないO.D値)の50%を阻害するHBsAGの濃度をKdに決定した。
【0063】
図10に示すように、HB48−35の親和性が、本発明のヒト抗体の中で最も高く、これに対してHB48−59とHB48−33の親和性は、HB48−35に比べてそれぞれ約1.3倍および4.0倍低かった。
【0064】
実施例10:急性B型肝炎のマウスモデルを用いたin vivoの効果テスト
急性B型肝炎の類似兆候を示すように流体力学注入法(Hydrodynamic injection)によってHBV DNAを注入して製造したC57BL6マウスモデルにおいて、HB48−33、HB48−35およびHB48−59のHBsAgに対する中和能を比較した。
【0065】
4週齢の約20gの体重の20匹のC57BL6マウスをチャールズリバー研究所(Charles Liver Laboratory、MA、USA)から購入して、表1に示すように5匹ずつ4群に分けた。
【0066】
【表1】

【0067】
HBV DNAをpcDNA3.1(Invitrogen、米国)内に挿入して製造した20μgのベクターpHBV−MBRI(Shin et al.、Virus Research119、146−153、2006;図11を参照)をすべてのマウスにマウスの尾静脈を介して体重の9.5%の体積で、0.3mL/minの速度で注射して急性B型肝炎を誘発した。24時間後、表1に列挙された試験物質0.2mLをマウスの尾静脈を介して注射した。注射前(0時間)、注射後24時間および注射後48時間で血清を分離した。上記の分離された血清をヤギ血清(goat serum)で10倍希釈した後、Genedia HBsAg ELISA3.0(緑十字社 MS、韓国)を使用して、HBsAgの血中濃度を測定し、上記の結果を図12に示した。図12に示すように、PBSを静脈注射した対照群において、48時間HBsAgの血中濃度がピークに保持された。これに対し、0.1mg HB48−33(実験群I)、0.1mg HB48−35(実験群II)および0.1mg HB48−59(実験群III)で処理された郡で、24時間〜48時間の間HBsAgの血中濃度が検出されなかった。したがって、本発明の抗体は、HBVの表面抗原を中和させるのに非常に有効であることが確認された。
【0068】
本発明は、上記特定の実施例と関連して説明されたにもかかわらず、本技術分野の熟練者により、本発明の様々な変形および変化が行えることと認識されるべきであり、これはまた添付の請求項によって定義されるような本発明の範囲に属する。
【技術分野】
【0001】
本発明は、B型肝炎ウイルス表面抗原(Hepatitis B virus surface antigen、HBsAg)に特異的に結合する抗体に関するものである。
【背景技術】
【0002】
B型肝炎ウイルス(HBV)はヘパドナウイルス(Hepadnaviridae)ファミリーのメンバーであり、急性および慢性肝炎を引き起こす。世界の人口のうち、約3.5億人、特に韓国および中国では人口の5%〜8%が慢性HBV感染者であり、HBVは肝疾患や肝臓がんの主な原因である。HBVに対するワクチンの開発でB型肝炎を予防することが可能になったが、未だ多くの人々がHBV感染による慢性肝炎に苦しんでいる。HBV感染は、肝炎、肝硬変だけでなく、肝臓がんを誘発し、慢性感染症の患者における肝臓がんの発生頻度が健常者よりも300倍高い。WHO(世界保健機構)は、肝臓がんの約80%がHBV感染による慢性B型肝炎から始まると報告した。
【0003】
現在利用可能なB型肝炎の治療薬は、ラミブジン(lamivudine)とアデフォビル・ジピボキシル(adefovir dipivoxil)などのヌクレオシド類似体(nucleoside analogue)を含んでおり、これらはHBVポリメラーゼを阻害することが知られている。しかし、3年間投与後、75%の患者において耐性ウイルスが発生して治療効果を低減させるので、これらは垂直感染や肝移植後の感染症を予防するために、B型肝炎の抗体製剤と組み合わせて使用されている。
【0004】
現在使用されているB型肝炎の抗体製剤は、抗HBV抗体を持つヒト血液源から高度の精製法およびウイルス不活性化法を用いて製造されているが、高い抗HBV抗体を持つ高価なヒト血漿の利用可能性が低いだけではなく、ヒト血漿−由来のウイルスを不活性化するコストが高いので、上記の方法は、増加する需要に対処することができない。
【0005】
ケーラーとミルシュタイン(1975)によってモノクローナル抗体(monoclonal antibody;mAb)の製造技術が確立されてから、マウス由来のモノクローナル抗体は、診断用や一部の治療のために主に使われてきた。しかし、マウス抗体は、治療目的のために人体に適用する際のヒト抗マウス抗体(HAMA)を生成する可能性があるため、投与することができない。HAMAの問題を解決するために、キメリク抗体とヒト化抗体が開発されてきた。キメリク抗体は、抗体分子全体の〜30%を構成するマウスmAbの可変領域(Fvフラグメント)を含有する。これに対し、ヒト化抗体は、抗体分子全体の〜10%を構成するマウスmAbのCDR(相補性決定領域)を含有する。上記キメリクおよびヒト化抗体は、HAMA反応を大幅に減少させてはいるが、長期間および継続的な投与を要する慢性肝炎のような慢性疾患の治療にはヒト抗体を使用する方がよいと思われる。
【0006】
本願発明者らは、新規で改良された抗体を開発しようと努力しており、上記の抗体がHBV表面抗原に結合してHBVを不活性化するのに使用できることを確認した。
【発明の概要】
【発明が解決しようとする課題】
【0007】
したがって、本発明の目的は、B型肝炎ウイルスの表面抗原に特異的に結合する新規の抗体を提供することである。
【0008】
本発明の他の目的は、上記抗体の重鎖可変領域および軽鎖可変領域をそれぞれコードするDNAおよびそれを含む発現ベクターを提供することである。
【0009】
本発明のまた他の目的は、上記発現ベクターに形質転換された細胞株を提供することである。
【0010】
本発明のまた他の目的は、上記抗体を含むB型肝炎の予防または治療用薬学組成物を提供することである。
【課題を解決するための手段】
【0011】
本発明の一つの態様により、a)配列番号1のアミノ酸配列を持つ重鎖可変領域、b)配列番号2、3、および4のいずれか1つのアミノ酸配列を持つ軽鎖可変領域、c)重鎖定常領域、およびd)軽鎖定常領域を含んでいる、B型肝炎ウイルス表面抗原(HBsAg)に特異的に結合する抗体が提供される。
【0012】
本発明の他の態様により、配列番号1のアミノ酸配列を持つ重鎖可変領域をコードするDNAまたは配列番号2、3、および4のいずれか1つのアミノ酸配列を持つ軽鎖可変領域をコードするDNAおよびそれを含む発現ベクターが提供される。
【0013】
本発明の別の態様により、上記発現ベクターに形質転換された細胞株が提供される。
【0014】
本発明のまた別の態様により、上記抗体を含むB型肝炎の予防または治療用組成物が提供される。
【図面の簡単な説明】
【0015】
本発明の上記及びその他の目的と特徴は、添付の図面とともに考慮されるときに下記説明により明確になるもので、以下の通りである。
【図1】図1は、PCRによって合成された本発明の重鎖可変領域(VH)および軽鎖可変領域(VL)をそれぞれコードするDNAを示す電気泳動(1%アガロースゲル)の分析の写真結果である。
【図2】図2は、本発明の抗体の重鎖可変領域および軽鎖可変領域を含む、ファージディスプレイベクターpKS4Hの地図である。
【図3】図3は、パンニング法を利用して、抗体ライブラリから抗体を選別する工程を示した図である。
【図4】図4は、抗体ライブラリから選別された本発明の抗体の一本鎖可変断片(ScFV)のアミノ酸配列である。
【図5】図5は、本発明のヒト抗体の重鎖を発現するための発現ベクター、HB48−33−Heavy−pRC13、HB48−35−Heavy−pRC13またはHB48−59−Heavy−pRC13の地図である。
【図6】図6は、本発明のヒト抗体の軽鎖を発現するための発現ベクター、HB48−33−Light−pKC12の地図である。
【図7】図7は、本発明のヒト抗体の軽鎖を発現するための発現ベクター、HB48−35−Light−pKC12の地図である。
【図8】図8は、本発明のヒト抗体の軽鎖を発現するための発現ベクター、HB48−59−Light−pKC12の地図である。
【図9】図9は、形質転換体から発現された重鎖および軽鎖のSDS−PAGE分析結果である。
【図10】図10は、ヒト抗体(HB48−33、HB48−35およびHB48−59)のHBV表面抗原に対する相対的親和性である。
【図11】図11は、急性B型肝炎のマウスモデルの製造に使用された、pcDNA3.1にHBV DNA配列を挿入して製造されたベクターpHBV1.3−MBRIの地図である。
【図12】図12は、急性B型肝炎のマウスモデルを用いたin vivoの効果試験によって選別された本発明のヒト抗体HB48−33、HB48−35とHB48−59の中和能を示すグラフである。図11のプラスミドを注入して24時間後、HBV表面抗原(HBsAg)が発現したことを確認し、本発明の抗体の投与により、表面抗原が減少したことを確認した。表面抗原は、Genedia HBsAg ELISA 3.0(緑十字社 MS、韓国)で測定した。
【発明を実施するための形態】
【0016】
以下、本発明を詳細に説明する。
【0017】
本発明は、a)配列番号1のアミノ酸配列を持つ重鎖可変領域、b)配列番号2、3、および4のいずれか1つのアミノ酸配列を持つ軽鎖可変領域、c)重鎖定常領域、およびd)軽鎖定常領域を含んでいる、B型肝炎ウイルス表面抗原(HBsAg)に特異的に結合する抗体を提供する。本発明の抗体は、B型肝炎ウイルス表面抗原に特異的に結合してHBVを不活性化させることによってB型肝炎の予防または治療に効果を有することを特徴とする。
【0018】
上記HBV表面抗原に特異的に結合する抗体は、好ましくは、ファージディスプレイ法(Smith、Science、228、1315−1317、1985およびHoogenboom&Chames、Immunol Today、21、371−378、2000)を応用して選別することができる。上記のファージディスプレイ法では、フィラメント(filamentous)ファージ(例えば、M13、Fd、またはF1)の表面タンパク質をコードする遺伝子(gene III)が、目的の抗体をコードする遺伝子と融合され、表面に露出した融合抗体を持つ抗体−ファージ型のウイルス粒子を生成させる。その後、抗体の高い特異性および親和性、並びにファージの高い感染性を利用してバイオパンニング(biopanning)技術によってファージライブラリから目的とする抗体が選別できる(Burton&Barbas,Adv.Immunol.,57、191−280、1994、Winter et al.,Annu.Rev.Immunol.,12、433−455、1994、およびHoogenboom et al.,Immunotechnology、4、1−20、1998、 Kim et al.,Hybrid Hybridomics、21、 385−392、2002)。上記ファージディスプレイベクターは、pKS4H(韓国特許第0635370号を参照)またはpCANTAB5E、好ましくはpKS4Hである場合もある。
【0019】
本発明者らは、ファージライブラリから3つのヒト抗体(HB48−33、HB48−35およびHB48−59)を選別した後、HBV表面抗原に対する抗体の親和性と中和能を調べた(図10および11)。また、重鎖および軽鎖の可変領域のアミノ酸配列を分析した結果、すべての重鎖可変領域が配列番号1のアミノ酸配列を持ち、軽鎖可変領域がそれぞれ配列番号2,3、および4のアミノ酸配列を持つことを確認した。
【0020】
本発明の抗体の重鎖および軽鎖の不変領域はヒト抗体のものと同一であってもよい。
【0021】
本発明は、配列番号1のアミノ酸配列を持つ抗体重鎖可変領域をコードするDNAを提供する。好ましくは、上記DNAは、配列番号1のアミノ酸配列をコードする配列番号5のヌクレオチド配列を持つポリヌクレオチドを含んでもよい。
【0022】
本発明は、配列番号2、3および4のいずれか1つのアミノ酸配列を持つ抗体の軽鎖可変領域をコードするDNAを提供する。好ましくは、上記DNAは、それぞれ配列番号2,3、および4のいずれか1つのアミノ酸配列をコードする配列番号6、7および8のいずれか1つのヌクレオチド配列を持つポリヌクレオチドを含んでもよい。
【0023】
本発明は、上記抗体の重鎖可変領域をコードするDNAを含んでいる、HBV表面抗原に特異的に結合する抗体の重鎖可変領域を発現するための発現ベクターを提供する。好ましくは、上記発現ベクターは、図5に地図で示した“HB48−33−Heavy−pRC13”、“HB48−35−Heavy−pRC13”または“HB48−59−Heavy−pRC13”であってもよい。
【0024】
具体的に、上記ベクターは、パンニング過程によって選別された抗体のVH断片(HB48VH)を適切なベクター、例えば、pRC13ベクター(寄託番号KCLRF−BP−00054、大韓民国特許第523732号を参照)に挿入して製造されることができる。
【0025】
本発明は、上記抗体の軽鎖可変領域をコードするDNAを含んでいる、HBV表面抗原に特異的に結合する抗体の軽鎖可変領域を発現するための発現ベクターを提供する。好ましくは、上記ベクターは、図6に地図で示した“HB48−33−Light−pKC13”、図7に地図で示した“HB48−35−Light−pKC13”または図8に地図で示した“HB48−59−Light−pKC13”であってよい。
【0026】
具体的に、上記ベクターは、パンニング過程によって選別された抗体のVL断片(HB48−33VL、HB48−35VLまたはHB48−59VL)を適切なベクター、例えば、pKC12ベクター(寄託番号KCLRF−BP−00054、大韓民国特許第523732号を参照)に挿入して製造することができる。
【0027】
本発明は、本発明の抗体の重鎖および軽鎖の可変領域を発現するための発現ベクターに形質転換された動物細胞株を提供する。上記動物細胞株は、CHO(Chinese hamster ovary)、HEK 293、またはNSO細胞株、好ましくはCHO(Chinese hamster ovary)細胞株でもよい。
【0028】
本発明に係る抗体は、上記重鎖および軽鎖の可変領域が互に結合することで製造することができる。
【0029】
抗原に対する本発明の抗体の親和性(affinity)は、例えば、競合ELISA(Kim et al.,Hybridoma、20、265−272、2001)によって測定することができる。図10に示すように、本発明のヒト抗体の中でHB48−35の親和性が最も高かったのに対し、HB48−59およびHB48−33の親和性は、HB48−35に比べてそれぞれ約1.3倍と4.0倍低かった。また、B型肝炎と類似の兆候を誘導するために、C57BL6マウス内にHBV DNAを流体力学注入(Hydrodynamic injection; Liu et al.,Gene Therapy、6、1258−1266、1999)して製造したHBVマウスモデルにおいて、HB48−33、HB48−35およびHB48−59抗体は、マウスの血液から、上記抗体によりHBVの表面抗原が基底レベルまで減少したHBVの中和能を示した(図12)。したがって、本発明の抗体は、HBVの表面抗原に結合してHBVを不活性化させることによって、B型肝炎を予防または治療するために使用することができる。
【0030】
上記の結果から本発明は、前記抗体を含むB型肝炎の予防または治療用薬学組成物を提供する。本発明の組成物は、通常の方法に応じて、薬学製剤に製造することができる。剤型の製造において、上記抗体は、好ましくは、担体と混合したり、担体で希釈されたり、容器の形態でもよい担体内に組み込まれる。担体が希釈剤として使用される場合、これは抗体に対する小胞(vesicle)、賦形剤または媒質(medium)として作用する固形、半固形または液状であってもよい。したがって、剤型は錠剤、丸剤、粉剤、サシェ(sachet)、カシェ(cachet)、エリクシール(elixir)、懸濁剤、乳剤、溶液剤、シロップ剤、エアロゾル、軟質または硬質ゼラチンカプセル剤、滅菌注射溶液、滅菌粉剤などの形を取ることができる。適切な担体、賦形剤および希釈剤の例として、ラクトース、デキストロース、スクロース、ソルビトール、マンニトール、カルシウムシリケート、セルロース、メチルセルロース、微晶質セルロース、ポリビニルピロリドン、水、メチルヒドロキシベンゾエイト、プロフィールヒドロキシベンゾエイト、タルク、ステアリン酸マグネシウムおよび鉱物油を挙げることができる。剤型は、充填剤、抗凝集剤、潤滑剤、湿潤剤、香料、乳化剤、防腐剤などをさらに含むことができる。本発明の抗体組成物は、上記抗体とともに、抗ウイルス剤として、インターフェロン、抗HBVモノクローナル抗体、抗HBVポリクローナル抗体、ヌクレオシド類似体、DNAポリメラーゼ阻害剤、siRNA製剤または治療用ワクチンをさらに含むことができる。
【0031】
HBV感染およびHBV関連の疾病は、本発明の組成物を、人間を含む哺乳動物に投与することで、予防または治療することができる。上記組成物内の抗体の投与量は、対象、疾病状態の重症度、投与速度、および医師の判断に左右され得る。有効成分として、上記抗体は、単一容量または分割用量で、一日に約0.001mg/kg体重〜10mg/kg体重、好ましくは0.005mg/kg体重〜1mg/kg体重範囲の有効量で経口以外の経路を介して哺乳動物に投与することができる。場合によっては、上記の投与量に比べて少ないかまたは多い量が投与量として適切であり、多量の投与量は、一日あたりに分割された少ない投与量で投与することができる。
【0032】
以下の実施例は、単に例示の目的で提供され、本発明の範囲を限定しようとするものではない。
【0033】
実施例1:RNAの分離
HBV表面抗原に特異的に結合する抗体を選別するために、ヒト抗体ライブラリを構築した。ヒト骨髄、ヒト胸腺、ヒト脾臓およびヒトB細胞から得られたRNAの混合物を使用した。ヒトB細胞RNAを除くすべてのRNAはClontech社(米国)から購入し、ヒトB細胞RNAは下記のように分離した。
【0034】
健康な成人から採血した50mLの血液を50mLのPBSと混合し希釈した。3mLのFicoll−Paque PLUS(GE Healthcare社、米国)を15mLチューブに入れ、ここに上記の希釈血液4mLを添加した。上記混合物を3,000rpmで遠心分離して白血球を分離した。上記分離した白血球2mLを6mLのPBSと混合し、100xgで10分間遠心分離した。上記の白血球100μLを1mLのトリゾール(登録商標)(Life Technology社、米国)と混合してRNAを分離した。
【0035】
一方、上記分離されたRNAを蒸留水で希釈した後、260nmで吸光度を測定し、その量を計算した(1.8μg/μL、フルスペクトル2000UV−VIS分光光度計、GE、米国)。詳細な手順は次のとおりである。
【0036】
1mLのトリゾールを100μLの白血球に添加してよく混ぜた後、常温(15℃〜30℃)で5分間放置した。そして、クロロホルム200μLを添加して15秒間よくかき混ぜてから、常温で3分間放置した。その後、上記混合物を2℃〜8℃、15分および15,000rpmの条件の下で遠心分離し、上清液を新しいチューブに移した。イソプロピルアルコール500μLを加えてよく混合した後、室温で10分間放置した。そして、上記混合物を2℃〜8℃および15,000rpmで5分間遠心分離して上清を除去した。これに75%エタノール1mLを添加し、上記混合物を2℃〜8℃、5分間、15,000rpmの条件下で遠心分離しエタノールを除去した後、RNAペレットを常温で5分間乾燥させた。これに蒸留水150μLを添加してRNAを溶解させた後、260nmにて上記懸濁液の吸光度を測定した。残りは−20℃で保管した。
【0037】
実施例2:抗体遺伝子の増幅
実施例1で分離したRNA1μgおよびpd(T)12〜18(0.5μg/μL)1μLを蒸留水と混合して最終体積を12.5μLに合わせた。上記混合物を70℃で2分間反応させ氷で冷却させた。その後、これに5X反応緩衝液、10mM dNTP混合物、組換えRNase阻害剤およびMMLV逆転写酵素(Clontech、米国)を添加して最終体積を20μLに合わせた後、42℃で1時間、95℃で5分間反応させてcDNAを合成した。LiquiMix QM Premix、Magenta(Neurotics Inc、韓国)、鋳型として4μLのcDNA、19μLの蒸留水並びにそれぞれscFv領域(single chain Fv)、重鎖可変領域、および軽鎖可変領域(kappaおよびlambda)のために考案された1μL(25pmol/μL)のプライマー[ScFv:配列番号9(Forward)、10(Reverse−κ)および11(Reverse−λ);重鎖可変領域:配列番号12(Forward−VH1)、13(Forward−VH2)、14(Forward−VH3)、15(Forward−VH4)、16(Forward−VH5)、17(Forward−VH6)、18(Forward−VH7)および19(Reverse);軽鎖可変領域κ鎖:配列番号20(Forward−VK1/3A)、21(Forward−VK1/3B)、22(Forward−VK2)、23(Forward−VK4)、24(Forward−VK5)、25(Forward−VK6)、26(Reverse−JK−A)、27(Reverse−JK−B)および28(Reverse−JK−C);軽鎖可変領域λ鎖:配列番号29(Forward−VL1〜3−A)、30(Forward−VL1〜3−B)、31(Forward−VL4)、32(Forward−VL5)、33(Forward−VL6)、34(Forward−VL7)、35(Forward−VL8)、36(Forward−VL9)、37(Forward−VL10)、38(Reverse−JL−A)および39(Reverse−JL−B)]を使用してPCR反応を行った。
【0038】
PCR反応は、95℃で5分、95℃で1分、55℃で2分、72℃で2分間を30回反応させた後、最後に72℃で15分間反応させて行われた。
【0039】
上記増幅された抗体DNAを1.2%アガロースゲルにおいて電気泳動によって確認し(図1参照)、キアゲンキット(Qiagen社、ドイツ)を用いて精製した。図1に示すように、重鎖および軽鎖(kappaおよびlambda)の可変領域に対応する1,350bpのDNAバンドが得られた。図1においてMは、サイズマーカー、VHは重鎖可変領域(レーン1:重鎖可変領域タイプI、レーン2:重鎖可変領域タイプII、レーン3:重鎖可変領域タイプIII、レーン4:重鎖可変領域タイプIV;レーン5:重鎖可変領域タイプV、レーン6:重鎖可変領域タイプVI、およびレーン7:重鎖可変領域タイプVII)、およびVLは、軽鎖可変領域(レーン9:軽鎖可変領域1/3κ、レーン10:軽鎖可変領域2κ、レーン11:軽鎖可変領域4κ、レーン12:軽鎖可変領域5κ、およびレーン13:軽鎖可変領域6κ、レーン15:軽鎖可変領域1λ〜3λ、レーン16:軽鎖可変領域4λ、レーン17:軽鎖可変領域5λ、レーン18:軽鎖可変領域6λ、レーン19:軽鎖可変領域7λ、レーン20:軽鎖可変領域8λ、レーン21:軽鎖可変領域9λおよびレーン22:軽鎖可変領域10λ)を指す。
【0040】
実施例3:抗体DNAの制限酵素切断
実施例2で準備したVHおよびVLをそれぞれ制限酵素SfiI/BspEIおよびBspEI/NotIで切断し、上記切断した断片を1.2%のアガロースゲル電気泳動で分離した後、キアゲンキットを用いて精製した。
【0041】
実施例4:抗体DNAのライゲーションおよびライブラリの製造
ファージディスプレイベクターpKS4H(緑十字社、韓国、韓国特許第635370号参照)を制限酵素SfiI/BspEIを使用して切断し、1.2%アガロースゲル電気泳動を利用して分離した後、キアゲンキットを利用して精製した。40μgのpKS4Hを実施例3で用意された10μgのVHと混合し、これにT4 DNAリガーゼ(New England BioLabs、米国)を添加した後、25℃で一晩中反応させた。上記リガーゼ反応混合物を、キアゲンキットを用いて精製し、大腸菌XL1−blue(Stratagene社、米国)に電気穿孔法(electroporation)により形質転換させた。上記形質転換体を、100mLの培地で一晩培養し、プラスミドを分離した。上記のプラスミドを“pKS4H−VH−△VL”と命名した。
【0042】
上記プラスミドpKS4H−VH−△VLを制限酵素BspEI/NotIで切断し、上記記載のように精製した。その後、40μgのpKS4H−VH−△VLプラスミドを実施例3で用意された10μgのVL PCR DNAおよびT4 DNAリガーゼ(New England BioLabs、米国)と混合し、25℃で一晩反応させた。上記リガーゼ反応混合物を、キアゲンキットを用いて精製し、大腸菌XL1−blueに電気穿孔法により形質転換させた。上記形質転換体をカルベニシリン(carbenicillin)およびテトラサイクリン(tetracyclin)を含有する100mLの培地において37℃で2時間培養した。そして、上記培地にM13ヘルパーファージ(Stratagene社、米国)を接種し、16時間培養してエンベルグ等(Engberg et al.,Mol.Biotechnol.,6、287−310、1996)で報告されたように、ファージライブラリを作製した。一方、上記大腸菌からプラスミドを分離して、“pKS4H−VH−VL”と命名した。上記プラスミドの地図を図2に示している。
【0043】
実施例5:HBV表面抗原に結合する抗体の選別
HBV表面抗原に結合する抗体をパンニング法(Engberg et al.,Mol.Biotechnol.,6、287−310、1996およびKim et al.,Gene、241、19−25、2000; Kim et al., Hybrid Hybridomics、21、385−392、2002)を応用して選別した。具体的に、HBVの表面抗原(緑十字社、韓国)をPBSバッファーで希釈し、それぞれのイムノチューブ(immunotube、NUNC社、デンマーク)を、上記希釈された抗原でコーティングした。そして、上記のコーティングされたイムノチューブに実施例4で製造したファージライブラリを添加し、37℃で2時間培養した。HBVに結合するファージを、0.1Mグリシンバッファー(pH2.0)を使用して溶出させた。以降、上記ファージで大腸菌XL1−blue細胞を感染させヘルパーファージ(Stratagene社、米国)を添加した。上記大腸菌(E.coli)を一晩培養し、これにPEG溶液(20%PEG8000および15%NaCl)を添加した。そして、沈殿したファージを回収して(ファージレスキュー)、上記ファージを再度HBVの表面抗原がコーティングされたイムノチューブと反応させて、上記過程を4回繰り返した。上記過程からHBVの表面抗原に結合する3つのヒト抗体を選別し、これをHB48−33、HB48−35およびHB48−59と命名した。ファージディスプレイライブラリを用いてヒト抗体を選別する過程を図3に示した。
【0044】
4回のパンニングから得られた各コロニーを公知の方法(Kim et al.、Gene、241、19−25、2000)によって2mLの培地で成長させ、IPTG(isopropyl bD−thiogalactopyranoside)を処理して抗体発現を誘導した。抗体の発現をHBVの表面抗原がコーティングされた96−ウェルプレートを使用してELISA(Enzyme−Linked Immunosorbent Assay)により測定した。
【0045】
実施例6:選別された抗体の配列分析
実施例5で選別されたヒト抗体HB48−33、HB48−35とHB48−59を分泌するコロニーを50μg/mLのカルベニシリンを含有する10 mLのLB培地で一晩培養した後、キアゲンプラスミドミニキット(Qiagen、Valencia、CA、米国)を使用して、プラスミドを分離した。上記のプラスミドをSfiI/NotIで切断した後、抗体の断片の挿入を確認するためにアガロースゲルで電気泳動を行った。プラスミドに挿入されたscFvのDNA配列を分析した。scFvのヌクレオチド配列をGenotech社(大田、韓国)にてシーケンスプライマである配列番号40のp033で分析した。
【0046】
ヒト抗体HB48−33、HB48−35およびHB48−59のscFvのDNA配列を、Webベースのプログラム(www.expasy.org:DNA to Protein translate tool)を使用して、アミノ酸に翻訳し、上記翻訳アミノ酸配列を図4に示した。図4に示すように、ヒト抗体HB48−33、HB48−35およびHB48−59は、同一のアミノ酸配列で示される重鎖可変領域および異なるアミノ酸配列で示される軽鎖可変領域を持っていた。
【0047】
実施例7:発現ベクターの製造
上記の抗体断片を完全な免疫グロブリンに転換させるために、抗体発現ベクターpRC13およびpKC12を使用した(韓国特許第523732号参照、寄託番号KCLRF−BP−00054)。
【0048】
重鎖発現ベクターpRC13は、抗体のVH断片がHindIIIおよびApaI部位内に簡単に挿入できるベクターである。図5に例示のように、本発明では、ヒト抗体HB48−33、HB48−35またはHB48−59の重鎖可変領域をコードするDNAを配列番号41(Forward)および42(Reverse)のプライマーを使用して、95℃で1分、55℃で2分、72℃で2分の条件下で増幅させた後、HindIII/ApaIで切断して、同じ制限酵素で切断されたpRC13に挿入した。上記組換えベクターを、“HB48−33−Heavy−pRC13”、“HB48−35−Heavy−pRC13”または“HB48−59−Heavy−pRC13”と命名した(図5参照)。
【0049】
一方、軽鎖発現ベクターpKC12ベクターは抗体のVL断片がNheIおよびApaI部位内に簡単に挿入できるベクターである。図6、図7および図8に示したように、ヒト抗体HB48−35とHB48−59とのk軽鎖可変領域をコードするDNAを配列番号43(Forward)および44(Reverse)のプライマーを使用して、95℃で1分、55℃で2分、72℃で2分の条件下で増幅させ、ヒト抗体HB48−33のλ軽鎖可変領域をコードするDNAを配列番号43(Forward)および45(Reverse)のプライマーを使用して95℃で1分、55℃で2分、72℃で2分の条件下で増幅させた。上記増幅されたDNAをNheI/ApaIで切断し、同じ制限酵素で切断されたPKC12内に挿入した。上記組換えベクターを、それぞれ“HB48−33−Light−pKC12”、“HB48−35−Light−pKC12”および“HB48−59−Light−pKC12”と命名した(図6、図7および図8を参照)。
【0050】
実施例8:抗体分泌動物細胞株の製造
形質感染24時間前に、2×105個のCHO(chinese hamster ovary)細胞を10%のFBS(Life Technologies社、米国)を含有するα−MEM培地(Life Technologies社、米国)が入ったT−25フラスコ(NUNC社、デンマーク)で、培養密度(confluency)が50%に到達する直前まで、5%CO2の存在下で、37℃のインキュベーターで培養した。その後、30μgのリポペクチン(Life Technologies社、米国)を1.5mLのOpti−MEM(Life Technologies社、米国)に入れて常温で90分間放置した。同時に、8μgのHB48−33−Heavy−pRC13および7μgのHB48−33−Light−pKC12、8μgのHB48−35−Heavy−pRC13および7μgのHB48−35−Light−pKC12、8μgのHB48−59−Heavy−pRC13および7μgのHB48−59−Light−pKC12をそれぞれ混合し、1.5mLのOpti−MEMに入れてから、常温で90分間放置した。90分後、リポペクチンを含有する培地をHB48−33−Heavy−pRC13&HB48−33−Light−pKC12、HB48−35−Heavy−pRC13&HB48−35−Light−pKC12、およびHB48−59−Heavy−pRC13&HB48−59−Light−pKC12を含有する培地と混合した後、15分間室温で反応させた。反応させる間に、上記細胞から培地を除去し、細胞をPBSで3回洗浄した。上記洗浄された細胞に上記の反応混合物を入れて6時間培養した。6時間後、上記反応混合物を除去し、α−MEM培地を入れて48時間培養した。そして、上記細胞をトリプシン(Life Technologies社、米国)で処理してフラスコから外した後、α−MEM培地で希釈し、96−ウェルプレート(NUNC社、デンマーク)に継代した。このとき、α−MEM培地はリボヌクレオシドとデオキシリボヌクレオシドを含有していないのに対し、10%の透析されたFBS(Life Technologies社、米国)および550μg/mLのG418(Sigma社、米国)を含有する。2日ごとに上記培地を新しい培地に交換した。ELISA分析のために、コロニーを形成した培養上清を回収し、選別された細胞を12−ウェルプレートに移した。細胞が12−ウェルプレートでよく育てば、細胞を6−ウェルプレートに移して、細胞が6−ウェルプレートでもよく育つと、これにメトトレキサート(MTX、中外製薬、韓国)を処理した。MTXの初期濃度は20nMであり、細胞の適応に応じて、80nM、320nMおよび1μMで増加した。1μMの濃度で生存して高い量の抗体分泌を持つ細胞株を選別した。上記選別された細胞株を、スピナーフラスコと無血清培地とを使用して65rpm、5%CO2および37℃のインキュベーターで大量に培養した。100mLの無血清培地が入った250mLフラスコで108細胞を培養した。細胞数が初期濃度の2倍になると、1,000rpmで5分間遠心分離して細胞を回収した。上記回収した細胞を再び200mLの培地が入った500mLフラスコで培養した。細胞数が初期濃度の2倍になると、1,000rpmで5分間遠心分離して細胞を回収し、1Lの培地が入った3Lスピナーフラスコに移した。ブチル酸ナトリウム(Aldrich社、米国)を最終濃度2mMになるように添加し、上記細胞を5日間培養した後、培地から上清を回収した。スピナーフラスコから回収した上清から、Protein A−アガロースカラム(Amersham Pharmacia Biotech社、米国)を使用して抗体を精製し、SDS−PAGEにより分析した。
【0051】
図9に示すように、約50kDaの重鎖と25kDaの軽鎖とのバンドが観察され、これは抗体が正確に合成されたことを示す。
【0052】
実施例9:抗体の親和性の測定
実施例8で得られた抗体のHBV表面抗原に対する親和性を競合ELISA法(Kim et al.,Hybridoma、20、265−272、2001)により決定し、上記の結果を図10に示した。簡略な手順は次のとおりである。
【0053】
(1)適正抗体濃度の決定
A.プレートの準備
PBS中で2μg/mLで希釈された、100μLのHBV表面抗原(緑十字社、韓国)をELISAプレートの各ウェルに入れ、4℃で一晩培養した。プレートの各ウェルをPBSTで1回洗浄し、各ウェルに300μLの1%BSA−PBS溶液を加えた後、室温で1時間培養した。
【0054】
B.1次反応
100μLのそれぞれ精製された抗体(0.5μg/mL)をプレートの各ウェルに入れ、室温で2時間反応させた後、PBSTで4回洗浄した。
【0055】
C.2次反応
1%BSA−PBSで1:5,000に希釈された100μLのヤギ抗ヒトIgG(Fab specific)−パーオキシダーゼ接合体(Sigma)を各ウェルに添加し、室温で1時間培養した後、PBSTで4回洗浄した。
【0056】
D.基質反応
100μLのTMB(3,3’、5,5’−テトラメチルベンジジン(Tetramethylbenzidine)、マイクロウェル パーオキシダーゼ サブストレート システム(KPL、MD、米国))を各ウェルに添加した後、405nmでO.D値を測定した。抗体の適正濃度を半分−最大結合値(half−maximum binding)を示す抗体濃度として決定した。
【0057】
(2)競合ELISA
A.プレートの準備
PBS中で2μg/mLで希釈された、100μLのHBV表面抗原(緑十字社、韓国)をELISAプレートの各ウェルに入れ、4℃で一晩培養した。各ウェルをPBSTで1回洗浄し、各ウェルに300μLの1%BSA−PBS溶液を加えた後、室温で1時間培養した。
【0058】
B.1次反応
2μgのHBV表面抗原を2の倍数で連続希釈し、プレートの各ウェルに10μLの上記希釈されたHBsAgを添加した。それから、(1)で決定された適正濃度に希釈した90μLの抗体を各ウェルに添加し、室温で2時間反応させた後、PBSTで4回洗浄した。
【0059】
C.2次反応
ヤギ抗−ヒトIgG(Fab specific)−パーオキシダーゼ接合体(Sigma)1%BSA−PBS溶液で5,000倍に希釈し、各ウェルに100μLずつ添加し、室温で1時間反応させた後、PBSTで4回洗浄した。
【0060】
1%BSA−PBSで1:5,000に希釈された、100μLのヤギ抗−ヒトIgG(Fab specific)−パーオキシダーゼ接合体(Sigma)を各ウェルに添加し、室温で1時間培養した後、PBSTで4回洗浄した。
【0061】
D.基質反応
TMB(3,3’、5,5’−テトラメチルベンジジン(Tetramethylbenzidine)、マイクロウェルパーオキシダーゼサブストレートシステム(KPL、MD、米国))溶液を100μLずつ各ウェルに添加した後、405nmでO.D値を測定した。最大結合値(競合する上皮成長因子受容体がない状態のELISA O.D値)の50%を阻害するHBV表面抗原の濃度を親和性(Kd)で算定した。
【0062】
100μLのTMB(3,3’、5,5’−テトラメチルベンジジン(Tetramethylbenzidine)、マイクロウェルパーオキシダーゼサブストレートシステム(KPL、MD、米国))を各ウェルに添加した後、405nmでO.D値を測定した。最大結合値(競合するEGFRが存在しないO.D値)の50%を阻害するHBsAGの濃度をKdに決定した。
【0063】
図10に示すように、HB48−35の親和性が、本発明のヒト抗体の中で最も高く、これに対してHB48−59とHB48−33の親和性は、HB48−35に比べてそれぞれ約1.3倍および4.0倍低かった。
【0064】
実施例10:急性B型肝炎のマウスモデルを用いたin vivoの効果テスト
急性B型肝炎の類似兆候を示すように流体力学注入法(Hydrodynamic injection)によってHBV DNAを注入して製造したC57BL6マウスモデルにおいて、HB48−33、HB48−35およびHB48−59のHBsAgに対する中和能を比較した。
【0065】
4週齢の約20gの体重の20匹のC57BL6マウスをチャールズリバー研究所(Charles Liver Laboratory、MA、USA)から購入して、表1に示すように5匹ずつ4群に分けた。
【0066】
【表1】
【0067】
HBV DNAをpcDNA3.1(Invitrogen、米国)内に挿入して製造した20μgのベクターpHBV−MBRI(Shin et al.、Virus Research119、146−153、2006;図11を参照)をすべてのマウスにマウスの尾静脈を介して体重の9.5%の体積で、0.3mL/minの速度で注射して急性B型肝炎を誘発した。24時間後、表1に列挙された試験物質0.2mLをマウスの尾静脈を介して注射した。注射前(0時間)、注射後24時間および注射後48時間で血清を分離した。上記の分離された血清をヤギ血清(goat serum)で10倍希釈した後、Genedia HBsAg ELISA3.0(緑十字社 MS、韓国)を使用して、HBsAgの血中濃度を測定し、上記の結果を図12に示した。図12に示すように、PBSを静脈注射した対照群において、48時間HBsAgの血中濃度がピークに保持された。これに対し、0.1mg HB48−33(実験群I)、0.1mg HB48−35(実験群II)および0.1mg HB48−59(実験群III)で処理された郡で、24時間〜48時間の間HBsAgの血中濃度が検出されなかった。したがって、本発明の抗体は、HBVの表面抗原を中和させるのに非常に有効であることが確認された。
【0068】
本発明は、上記特定の実施例と関連して説明されたにもかかわらず、本技術分野の熟練者により、本発明の様々な変形および変化が行えることと認識されるべきであり、これはまた添付の請求項によって定義されるような本発明の範囲に属する。
【特許請求の範囲】
【請求項1】
a)配列番号1のアミノ酸配列を持つ重鎖可変領域と、
b)配列番号2、3、および4のいずれかのアミノ酸配列を持つ軽鎖可変領域と、
c)重鎖定常領域と、
d)軽鎖定常領域とを含む、B型肝炎ウイルス表面抗原(HBsAg)に特異的に結合するヒト抗体。
【請求項2】
配列番号1のアミノ酸配列を持つ抗体重鎖可変領域をコードするDNA。
【請求項3】
上記DNAが配列番号5のヌクレオチド配列を持つポリヌクレオチドを含む、請求項2に記載のDNA。
【請求項4】
配列番号2、3、および4のいずれか1つのアミノ酸配列を持つ抗体軽鎖可変領域をコードするDNA。
【請求項5】
上記DNAが配列番号6、7、および8のいずれか1つのヌクレオチド配列を持つポリヌクレオチドを含む、請求項4に記載のDNA。
【請求項6】
請求項2のDNAを含む、HBV表面抗原に特異的に結合する抗体の重鎖可変領域を発現するための発現ベクター。
【請求項7】
上記ベクターは、図5に地図で示す、HB48−33−Heavy−pRC13、HB48−35−Heavy−pRC13、またはHB48−59−Heavy−pRC13である、請求項6に記載の発現ベクター。
【請求項8】
請求項4のDNAを含む、HBV表面抗原に特異的に結合する抗体の軽鎖可変領域を発現するための発現ベクター。
【請求項9】
上記ベクターは、図6に地図で示すHB48−33−Light−pKC13、図7に地図で示すHB48−35−Light−pKC13、または図8に地図で示すHB48−59−Light−pKC13である、請求項8に記載の発現ベクター。
【請求項10】
請求項6の発現ベクター及び請求項8の発現ベクターに形質転換された動物細胞株。
【請求項11】
上記動物細胞株が、CHO(Chinese hamster ovary)、HEK 293、またはNSO細胞株である、請求項10に記載の動物細胞株。
【請求項12】
請求項1の抗体を含むB型肝炎の予防または治療用薬学組成物。
【請求項1】
a)配列番号1のアミノ酸配列を持つ重鎖可変領域と、
b)配列番号2、3、および4のいずれかのアミノ酸配列を持つ軽鎖可変領域と、
c)重鎖定常領域と、
d)軽鎖定常領域とを含む、B型肝炎ウイルス表面抗原(HBsAg)に特異的に結合するヒト抗体。
【請求項2】
配列番号1のアミノ酸配列を持つ抗体重鎖可変領域をコードするDNA。
【請求項3】
上記DNAが配列番号5のヌクレオチド配列を持つポリヌクレオチドを含む、請求項2に記載のDNA。
【請求項4】
配列番号2、3、および4のいずれか1つのアミノ酸配列を持つ抗体軽鎖可変領域をコードするDNA。
【請求項5】
上記DNAが配列番号6、7、および8のいずれか1つのヌクレオチド配列を持つポリヌクレオチドを含む、請求項4に記載のDNA。
【請求項6】
請求項2のDNAを含む、HBV表面抗原に特異的に結合する抗体の重鎖可変領域を発現するための発現ベクター。
【請求項7】
上記ベクターは、図5に地図で示す、HB48−33−Heavy−pRC13、HB48−35−Heavy−pRC13、またはHB48−59−Heavy−pRC13である、請求項6に記載の発現ベクター。
【請求項8】
請求項4のDNAを含む、HBV表面抗原に特異的に結合する抗体の軽鎖可変領域を発現するための発現ベクター。
【請求項9】
上記ベクターは、図6に地図で示すHB48−33−Light−pKC13、図7に地図で示すHB48−35−Light−pKC13、または図8に地図で示すHB48−59−Light−pKC13である、請求項8に記載の発現ベクター。
【請求項10】
請求項6の発現ベクター及び請求項8の発現ベクターに形質転換された動物細胞株。
【請求項11】
上記動物細胞株が、CHO(Chinese hamster ovary)、HEK 293、またはNSO細胞株である、請求項10に記載の動物細胞株。
【請求項12】
請求項1の抗体を含むB型肝炎の予防または治療用薬学組成物。
【図1】

【図2】
【図3】
【図4】

【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公表番号】特表2013−515477(P2013−515477A)
【公表日】平成25年5月9日(2013.5.9)
【国際特許分類】
【出願番号】特願2012−545832(P2012−545832)
【出願日】平成22年7月8日(2010.7.8)
【国際出願番号】PCT/KR2010/004445
【国際公開番号】WO2011/078456
【国際公開日】平成23年6月30日(2011.6.30)
【出願人】(512165396)グリーン クロス コーポレーション (1)
【氏名又は名称原語表記】GREEN CROSS CORPORATION
【Fターム(参考)】
【公表日】平成25年5月9日(2013.5.9)
【国際特許分類】
【出願日】平成22年7月8日(2010.7.8)
【国際出願番号】PCT/KR2010/004445
【国際公開番号】WO2011/078456
【国際公開日】平成23年6月30日(2011.6.30)
【出願人】(512165396)グリーン クロス コーポレーション (1)
【氏名又は名称原語表記】GREEN CROSS CORPORATION
【Fターム(参考)】
[ Back to top ]