
BPBベースのカーゴ運搬システム
本発明は、(a)(i)鎖間(interstrand)非共有結合が形成されたパラレル(parallel)、アンチパラレル(antiparallel)、またはパラレル(parallel)とアンチパラレル(antiparallel)アミノ酸鎖を含む構造安定化部位(structure stabilizing region)と、(ii)前記構造安定化部位の両末端に結合されており、無作為的に選択されたそれぞれn及びm個のアミノ酸を含むターゲット結合部位I(target binding region I)及びターゲット結合部位II(target binding region II)と、を含む二座ペプチドバインダー(Bipodal Peptide Binder:BPB)と、(b)前記二座ペプチドバインダーに結合されたカーゴと、
を含むBPBベースのカーゴ運搬システム(Cargo Delivery System)に関し、本発明のBPBベースのカーゴ運搬システムは、BPBのターゲット結合性及び特異性に基づいて多用な物質を細胞表面または細胞内に運搬できる。二坐ペプチドバインダーは、非常に低い水準(例えば、nM水準)のKD値(解離定数)を示し、ターゲットに非常に高い親和度を示す。本発明のBPBベースのカーゴ運搬システムは、医薬、インビボ分子イメージング、インビトロ細胞イメージング及び薬物伝達用ターゲッティングをする際に利用でき、エスコート分子としても非常に有用に利用できる。
を含むBPBベースのカーゴ運搬システム(Cargo Delivery System)に関し、本発明のBPBベースのカーゴ運搬システムは、BPBのターゲット結合性及び特異性に基づいて多用な物質を細胞表面または細胞内に運搬できる。二坐ペプチドバインダーは、非常に低い水準(例えば、nM水準)のKD値(解離定数)を示し、ターゲットに非常に高い親和度を示す。本発明のBPBベースのカーゴ運搬システムは、医薬、インビボ分子イメージング、インビトロ細胞イメージング及び薬物伝達用ターゲッティングをする際に利用でき、エスコート分子としても非常に有用に利用できる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、BPBベースのカーゴ運搬システムに関する。
【背景技術】
【0002】
抗体は、B細胞が生産する血漿タンパク質の一種である免疫グロブリン蛋白質であって、外部から入った抗原の特定部位を特異的に認識して結合することにより、抗原を非活性化するか無力化させる。このような抗原−抗体反応の特異性と高度の親和度及び数千万種類の抗原を区別できる抗体の多様性を応用して、今日の診断剤と治療剤などを始めとした数多い種類の抗体製品が出現するようになった。現在FADでは、21個の単一クローン抗体を承認し、リツキシマブ(Rituximab)及びハーセプチン(Herceptin)のような抗体は、他の治療で全く反応を示さなかった患者の50%以上で効果を示し、実質的に様々な研究において、単一クローン抗体を利用してリンパ腫、大腸癌または乳癌などに成功的な臨床治療を示している。治療用抗体の全体市場規模は、2004年100億ドル規模から2010年には300億ドルに、年平均20%の成長率を示すと推定しており、その市場規模は、幾何級数的に増加すると推定されている。抗体を利用した新薬開発が活発になる理由は、薬品の開発期間が短く、投資費用が少なくて、副作用が容易に予測可能であるからである。また、抗体は生薬であるため、人体に影響がほとんどなく、体内における半減期が低分子量薬品に比べ圧倒的に長くて、患者に親和的である。このような有用性にもかかわらず、人間において単一クローン抗体は、外来抗原と認識され、酷いアレルギー反応または過敏反応を起こしたりもする。また、このような抗癌機能の単クローン抗体を臨床的に使用する場合、生産コストが高いため、治療剤としての値段が急激に上昇するという短所があり、抗体を培養する方法及び精製方法など、広範囲な分野の技術が各種知的所有権により保護されているため、高いライセンシング費を支払わなければならない。
【0003】
したがって、この問題を解決するために、米国を中心にヨーロッパ連合で、抗体代替蛋白質の開発が胎動期にある。抗体代替蛋白質は、抗体のように不変領域と可変領域を持てるように作った組み換え蛋白質であって、大きさが小さくて安定した蛋白質の一定部分を無作為配列のアミノ酸に変えてライブラリーを作り、これを標的物質に対してスクリーニングをして、高い親和力と良い特異性を有した物質を見つけることができる。例えば、抗体代替蛋白質の中、アビマー(avimer)とアフィボディ(affibody)は、標的物質に対してピコモル(picomole)程度の親和力を有した例示が報告されている。このような抗体代替蛋白質は、大きさが小さくて安定しているため、癌細胞に深く浸透可能であり、一般に免疫反応が少ないと報告されている。そして何よりも、広範囲な抗体特許問題から放れることができ、バクテリアから容易に大量精製できるため、生産コストが低くて、経済的に抗体より大きい長所を有する。現在開発された抗体代替蛋白質は40個があるが、この中、ベンチャー会社や多国的製薬会社で商用化を試みている抗体代替蛋白質は、フィブロネクチンタイプIIIドメイン、リポカリン、LDLR-Aドメイン、クリスタリン、プロテインA、アンキリンリピート(Ankyrin repeat)、BPTIという蛋白質を利用しており、ターゲットに対するピコモルで数ナノモル程度の高い親和力を有している。その中、アドネクチン(adnectin)、アビマー、クニッツ(Kunitz)ドメインは、現在FDA臨床実験が進行中である。
【0004】
本発明は、今までの蛋白質を利用した抗体代替蛋白質とは異なるペプチド基盤抗体代替蛋白質に焦点を合わせた。ペプチドは、抗体に比べ、適切な薬物動力学、大量生産性、低い毒性、抗原性抑制及び低い生産単価などにより、現在抗体治療剤に代わって多様に活用されている。治療用薬としてのペプチドの長所は、生産単価が低く、安全生及び反応性が高くて、特許ロイヤルティが相対的に低くて、望まない免疫システムに露出が少なく、ペプチド自体に対する抗体生産を抑制することができて、合成による変形が容易で正確であるということである。しかしながら、大部分のペプチドは、抗体に比べ、特定蛋白質ターゲットに対して低い親和力及び特異性を示すため、多様な応用分野に使用できないという短所がある。したがって、ペプチドの短所を克服できる新しいペプチド基盤抗体代替蛋白質の開発に対する要求が当業界に台頭されている。したがって、本発明者らは、生物学的ターゲット分子に高い親和性で特異的結合が可能なペプチド物質を開発するために鋭意研究した。これは、現在非常に多いターゲットに対して報告された、低い親和力を有したペプチドを利用して、速い時間内に高い親和性及び特異性を有した新薬候補を製造できる技術になると期待される。
【0005】
本明細書全体にかけて多数の特許文献及び論文が参照されて、その引用が表示されている。引用された特許文献及び論文の開示内容は、その全体が本明細書に参照として取り込まれ、本発明の属する技術分野の水準及び本発明の内容がより明確に説明される。
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明者らは、ターゲット結合性及び特異性に基づいて、多様な物質を細胞内または細胞表面に運搬できるシステムを開発するために鋭意研究した。その結果、比較的リジッド(rigid)なペプチド骨格を有する構造安定化部位の両末端に無作為的(random)にペプチドを結合させて、この二つのペプチドを共同にターゲット分子に結合させる場合は、大きく増加された結合能及び特異性を有する二座ペプチドバインダー(BPB)を得ることができ、このBPBに運搬対象のカーゴを結合させる場合は、BPBのターゲット結合性及び特異性に基づいて、多様な物質を細胞表面または細胞内に運搬できることを確認することにより、本発明を完成した。
【0007】
したがって、本発明の目的は、BPBベースのカーゴ運搬システムを提供することにある。
【0008】
本発明の他の目的及び利点は、発明の詳細な説明及び請求の範囲及び図面により、さらに明確にされる。
【課題を解決するための手段】
【0009】
本発明の一様態によると、本発明は、(a)(i)鎖間(interstrand)非共有結合が形成されたパラレル(parallel)、アンチパラレル(antiparallel)、またはパラレル(parallel)とアンチパラレル(antiparallel)アミノ酸鎖を含む構造安定化部位(structure stabilizing region)と、(ii)前記構造安定化部位の両末端に結合されており、無作為的に選択されたそれぞれn及びm個のアミノ酸を含むターゲット結合部位I(target binding region I)及びターゲット結合部位II(target binding region II)と、を含む二座ペプチドバインダー(Bipodal Peptide Binder:BPB)と、(b)前記二座ペプチドバインダーに結合されたカーゴと、を含むBPBベースのカーゴ運搬システム(Cargo Delivery System)を提供する。
【0010】
本発明の一様態によると、本発明は、(a)(i)鎖間(interstrand)非共有結合が形成されたパラレル(parallel)、アンチパラレル(antiparallel)、またはパラレル(parallel)とアンチパラレル(antiparallel)アミノ酸鎖を含む構造安定化部位(structure stabilizing region)と、(ii)前記構造安定化部位の両末端に結合されており、無作為的に選択されたそれぞれn及びm個のアミノ酸を含むターゲット結合部位I(target binding region I)及びターゲット結合部位II(target binding region II)と、を含む二座ペプチドバインダー(Bipodal Peptide Binder:BPB)と、(b)前記二座ペプチドバインダーに結合されたカーゴと、を含むBPBベースのカーゴ運搬システム(Cargo Delivery System)を、個体、組織または細胞に処理する段階を含むカーゴを運搬する方法を提供する。
【0011】
本発明者らは、ターゲット結合性及び特異性に基づいて、多様な物質を細胞内または細胞表面に運搬できるシステムを開発するために鋭意研究した。その結果、比較的リジッド(rigid)なペプチド骨格を有する構造安定化部位の両末端に無作為的(random)にペプチドを結合させて、この二つのペプチドを共同にターゲット分子に結合させる場合は、大きく増加された結合能及び特異性を有する二座ペプチドバインダー(BPB)を得ることができ、このBPBに運搬対象のカーゴを結合させる場合は、BPBのターゲット結合性及び特異性に基づいて、多様な物質を細胞表面または細胞内に運搬できることを確認した。
【0012】
本発明の基本的な戦略は、リジッドなペプチド骨格の両末端に、ターゲットに結合されるペプチドを連結することである。この場合、リジッドなペプチド骨格は、二座ペプチドバインダーの全体的な構造を安定化させる作用をして、ターゲット結合部位I及びターゲット結合部位IIがターゲット分子に結合されることを強化させる。
【0013】
本発明で利用可能な構造安定化部位は、パラレル、アンチパラレル、またはパラレルとアンチパラレルアミノ酸鎖を含み、鎖間(interstrand)水素結合、静電気的相互作用、疎水性相互作用、ファンデルワールス相互作用、パイ−パイ相互作用、陽イオン−パイ相互作用、またはこれらの組み合わせによる非共有結合が形成される蛋白質構造モチーフを含む。鎖間水素結合、静電気的相互作用、疎水性相互作用、ファンデルワールス相互作用、パイ−パイ相互作用、陽イオン−パイ相互作用、またはこれらの組み合わせにより形成される非共有結合は、構造安定化部位の堅固性(rigidity)に寄与する。
【0014】
本発明の好ましい具現例によると、構造安定化部位における鎖間(interstrand)非供給結合は、水素結合、疎水性相互作用、ファンデルワールス相互作用、パイ−パイ相互作用、またはこれらの組み合わせを含む。
【0015】
選択的に、構造化安定化部位に共有結合があり得る。例えば、構造化安定化部位に二硫化結合を形成させて、構造安定化部位の堅固性をさらに増加させることができる。このような共有結合による堅固性の増加は、二座ペプチドバインダーのターゲットに対する特異度及び親和度を考慮して付与する。
【0016】
本発明の好ましい具現例によると、構造安定化部位のアミノ酸鎖は、リンカーで連結されている。本明細書で鎖を言及しながら使用される用語‘リンカー’は、鎖と鎖を連結する物質を意味する。例えば、構造安定化部位としてβ−ヘアピンが利用される場合は、β−ヘアピンにあるターン配列がリンカーの役割をして、ロイシンジッパーが利用される場合は、ロイシンジッパーの二つのC-末端を連結する物質(例えば、ペプチドリンカー)がリンカーの役割をする。
【0017】
リンカーは、パラレル、アンチパラレル、またはパラレルとアンチパラレルアミノ酸鎖を連結する。例えば、パラレル方式で整列された少なくとも二つの鎖(好ましくは二つの鎖)、アンチパラレル方式で整列された少なくとも二つの鎖(好ましくは二つの鎖)、パラレル及びアンチパラレル方式で整列された少なくとも三つの鎖(好ましくは三つの鎖)をリンカーが連結するようになる。
【0018】
本発明の好ましい具現例によると、リンカーは、ターン配列またはペプチドリンカーである。
【0019】
本発明の好ましい具現例によると、前記ターン配列は、β−ターン、γ−ターン、α−ターン、π−ターンまたはω−loopである(Venkatachalam CM (1968), Biopolymers, 6, 1425-1436; Nemethy G and Printz MP. (1972), Macromolecules, 5, 755-758; Lewis PN et al., (1973), Biochim. Biophys. Acta, 303, 211-229; Toniolo C. (1980) CRC Crit. Rev. Biochem., 9, 1-44; Richardson JS. (1981), Adv. Protein Chem., 34, 167-339; Rose GD et al., (1985), Adv. Protein Chem., 37, 1-109; Milner-White EJ and Poet R. (1987), TIBS, 12, 189-192; Wilmot CM and Thornton JM. (1988), J. Mol. Biol., 203, 221-232; Milner-White EJ. (1990), J. Mol. Biol., 216, 385-397; Pavone V et al. (1996), Biopolymers, 38, 705-721; Rajashankar KR and Ramakumar S. (1996), Protein Sci., 5, 932-946)。最も好ましくは、本発明で利用されるターン配列は、β−ターンである。
【0020】
ターン配列としてβ−ターンが利用される場合、好ましくは、タイプI、タイプI’、タイプII、タイプII’、タイプIIIまたはタイプIII’ターン配列であり、より好ましくは、タイプI、タイプI’、タイプII、タイプII’ターン配列であり、さらに好ましくは、タイプI’、タイプII’ターン配列であって、最も好ましくは、タイプI’ターン配列である(B. L. Sibanda et al., J. Mol. Biol., 1989, 206, 4, 759-777; B. L. Sibanda et al., Methods Enzymol., 1991, 202, 59-82)。
【0021】
本発明の他の好ましい具現例によると、本発明でターン配列として利用できるものは、H. Jane Dyson et al., Eur. J. Biochem. 255:462-471(1998)に開示されており、前記文献は、本明細書に参照として取り込まれる。ターン配列として利用できるものは、次のアミノ酸配列を含む:X-Pro-Gly-Glu-Val; Ala-X-Gly-Glu-Val(Xは、20個のアミノ酸から選択される)。
【0022】
本発明の一具現例によって、構造安定化部位としてβ−シートまたはロイシンジッパーが利用される場合、パラレル方式で整列された二つの鎖またはアンチパラレル方式で整列された二つの鎖をペプチドリンカーが連結することが好ましい。
【0023】
ペプチドリンカーは、当業界に公知された如何なるものでも利用可能である。適したペプチドリンカーの配列は、下記のような要素を考慮して選択することができる:(a)柔軟な延長されたコンフォメーション(flexible extended conformation)に適用できる能力;(b)生物学的ターゲット物質と相互作用する二次構造を生成しない能力;及び(c)生物学的ターゲット分子と相互作用する疎水性残基または電荷を有する残基の不在。好ましいペプチドリンカーは、Gly、Asn及びSer残基を含む。Thr及びAlaのような他の中性アミノ酸もリンカー配列に含まれ得る。リンカーに適したアミノ酸配列は、Maratea et al., Gene 40:39-46(1985); Murphy et al., Proc. Natl. Acad Sci. USA 83:8258-8562(1986); 米国特許第4,935,233号、第4,751,180号及び第5,990,275号に開示されている。ペプチドリンカー配列は、1〜50アミノ酸残基で構成できる。
【0024】
本発明の好ましい具現例によると、構造安定化部位は、ヘアピン、β−ヘアピン、β−ターン、リンカーで連結されたβ−シートまたはリンカーで連結されたロイシンジッパーであり、より好ましくは、構造安定化部位は、β−ヘアピンまたはリンカーで連結されたβ−シートであって、最も好ましくは、β−ヘアピンである。
【0025】
本明細書で用語‘β−ヘアピン’は、二つのβ鎖を含む最も簡単な蛋白質モチーフを意味し、この二つのβ鎖は、互いにアンチパラレルな整列を示す。このβ−ヘアピンにおいて、二つのβ鎖は、一般にターン配列により連結される。
【0026】
好ましくは、β−ヘアピンに適用されるターン配列は、タイプI、タイプI’、タイプII、タイプII’、タイプIIIまたはタイプIII’ターン配列であり、より好ましくは、タイプI、タイプI’、タイプII、タイプII’ターン配列であり、さらに好ましくは、タイプI’、タイプII’ターン配列であって、最も好ましくは、タイプI’ターン配列である。また、X-Pro-Gly-Glu-Val;またはAla-X-Gly-Glu-Val(Xは、20個のアミノ酸から選択される)で表されるターン配列もβ−ヘアピンに利用できる。
【0027】
本発明の例示的な実施例によると、タイプIターン配列は、Asp-Asp-Ala-Thr-Lys-Thrであり、タイプI'ターン配列は、Glu-Asn-Gly-Lysであって、タイプIIターン配列は、X-Pro-Gly-Glu-Val;またはAla-X-Gly-Glu-Val(Xは、20個のアミノ酸から選択される)であり、タイプII'ターン配列は、Glu-Gly-Asn-LysまたはGlu-D-Pro-Asn-Lysである。
【0028】
β-ヘアピンコンフォメーションを有するペプチドは、当業界によく知られている。例えば、米国特許第6,914,123号及びAndrea G. Cochran et al., PNAS, 98(10):5578-5583)に開示されているトリプトファンジッパー、WO 2005/047503に開示されている鋳型-固定されたβ-ヘアピンミメティック、米国特許第5,807,979号に開示されているβ-ヘアピン変形体がよく知られている。その他にも、β-ヘアピンコンフォメーションを有するペプチドは、Smith & Regan (1995) Science 270:980-982; Chou & Fassman (1978) Annu. Rev. Biochem. 47:251-276; Kim & Berg (1993) Nature 362:267-270; Minor & Kim (1994) Nature 367:660-663; Minor & Kim (1993) Nature 371:264-267; Smith et al. Biochemistry (1994) 33:5510-5517; Searle et al. (1995) Nat. Struct. Biol. 2:999-1006; Haque & Gellman (1997) J. Am. Chem. Soc. 119:2303-2304; Blanco et al. (1993) J. Am. Chem. Soc. 115:5887-5888; de Alba et al. (1996) Fold. Des. 1: 133-144; de Alba et al. (1997) Protein Sci. 6:2548-2560; Ramirez-Alvarado et al. (1996) Nat. Struct. Biol. 3:604-612; Stanger & Gellman (1998) J. Am. Chem. Soc. 120:4236-4237; Maynard & Searle (1997) Chem. Commun. 1297-1298; Griffiths-Jones et al. (1998) Chem. Commun. 789-790; Maynard et al. (1998) J. Am. Chem. Soc. 120:1996-2007; 及びBlanco et al. (1994) Nat. Struct. Biol. 1:584-590に開示されており、前記文献は、本明細書に参照として取り込まれる。
【0029】
β-ヘアピンコンフォメーションを有するペプチドを構造安定化部位として利用する場合、最も好ましくは、トリプトファンジッパーを利用する。
【0030】
本発明の好ましい具現例によると、本発明で利用されるトリプトファンジッパーは、下記一般式Iで表される:
[一般式I]
X1-Trp(X2)X3-X4-X5(X'2)X6-X7
【0031】
X1は、SerまたはGly-Gluであり、X2及びX'2は、それぞれ独立してThr、His、Val、Ile、PheまたはTyrであり、X3は、TrpまたはTyrであり、X4は、タイプI、タイプI'、タイプII、タイプII'またはタイプIIIまたはタイプIII'ターン配列であり、X5は、TrpまたはPheであり、X6は、TrpまたはValであって、X7は、LysまたはThr-Gluである。
【0032】
より好ましくは、前記一般式Iにおいて、X1は、SerまたはGly-Gluであり、X2及びX'2は、それぞれ独立してThr、HisまたはValであり、X3は、TrpまたはTyrであり、X4は、タイプI、タイプI'、タイプIIまたはタイプII'ターン配列であり、X5は、TrpまたはPheであり、X6は、TrpまたはValであって、X7は、LysまたはThr-Gluである。
【0033】
さらに好ましくは、一般式Iにおいて、X1は、SerまたはGly-Gluであり、X2及びX'2は、それぞれ独立してThr、HisまたはValであり、X3は、Trpであり、X4は、タイプI、タイプI'、タイプIIまたはタイプII'ターン配列であり、X5は、Trpであり、X6は、Trpであって、X7は、LysまたはThr-Gluである。
【0034】
よりさらに好ましくは、一般式Iにおいて、X1は、Serであり、X2及びX'2は、Thrであり、X3は、Trpであり、X4は、タイプI'またはタイプII'ターン配列であり、X5は、Trpであり、X6は、Trpであって、X7は、Lysである。
【0035】
最も好ましくは、一般式Iにおいて、X1は、Serであり、X2及びX'2は、Thrであり、X3は、Trpであり、X4は、タイプI'ターン配列(ENGK)またはタイプII’ターン配列(EGNK)であり、X5は、Trpであり、X6は、Trpであって、X7は、Lysである。
【0036】
本発明に適したトリプトファンジッパーの例示的なアミノ酸配列は、配列番号1乃至3及び5乃至10に記載されている。
【0037】
本発明で構造安定化部位として利用可能なβ-ヘアピンペプチドは、蛋白質GのB1ドメイン由来のペプチド、即ち、GB1ペプチドである。
【0038】
本発明でGB1ペプチドが利用される場合、好ましくは、構造安定化部位は、下記一般式IIで表される:
[一般式II]
X1-Trp-X2-Tyr-X3-Phe-Thr-Val-X4
【0039】
X1は、Arg、Gly-GluまたはLys-Lysであり、X2は、GlnまたはThrであり、X3は、タイプI、タイプI'、タイプII、タイプII'またはタイプIIIまたはタイプIII'ターン配列であって、X4は、Gln、Thr-GluまたはGln-Gluである。
【0040】
より好ましくは、一般式IIの構造安定化部位は、下記一般式II’で表される:
[一般式II’]
X1-Trp-Thr-Tyr-X2-Phe-Thr-Val-X3
【0041】
X1は、Gly-GluまたはLys-Lysであり、X2は、タイプI、タイプI'、タイプII、タイプII'またはタイプIIIまたはタイプIII'ターン配列であり、X3は、Thr-GluまたはGln-Gluである。
【0042】
本発明に適したGB1β-ヘアピンの例示的なアミノ酸配列は、配列番号4及び14乃至15に記載されている。
【0043】
本発明で構造安定化部位として利用可能なβ-ヘアピンペプチドは、HPペプチドである。本発明でHPペプチドが利用される場合、好ましくは、構造安定化部位は、下記一般式IIIで表される:
[一般式III]
X1-X2-X3-Trp-X4-X5-Thr-X6-X7
【0044】
X1は、LysまたはLys-Lysであり、X2は、TrpまたはTyrであり、X3は、ValまたはThrであり、X4は、タイプI、タイプI'、タイプII、タイプII'またはタイプIIIまたはタイプIII'ターン配列であり、X5は、TrpまたはAlaであり、X6は、TrpまたはValであって、X7は、GluまたはGln-Gluである。
【0045】
本発明で構造安定化部位として利用可能なまた他のβ-ヘアピンペプチドは、下記一般式IVで表される:
[一般式IV]
X1-X2-X3-Trp-X4
【0046】
X1は、Lys-ThrまたはGlyであり、X2は、TrpまたはTyrであり、X3は、タイプI、タイプI'、タイプII、タイプII'またはタイプIIIまたはタイプIII'ターン配列であって、X4は、Thr-GluまたはGlyである。
【0047】
一般式III及びIVのβ-ヘアピンの例示的なアミノ酸配列は、配列番号11乃至12、15及び16乃至19に記載されている。
【0048】
本発明によると、構造安定化部位として、ヘアピン(α−ヘアピン、β−ヘアピン、γ−ヘアピン、p−ヘアピンなど)が利用できる。また、本発明によると、構造安定化部位としてβ−ターンが利用できる。
【0049】
本発明によると、構造安定化部位として、リンカーで連結されたβ-シートを利用することができる。β-シート構造において、パラレルまたはアンチパラレルな、好ましくは、アンチパラレルな二つのアミノ酸鎖が伸びた構造(extended form)からなっており、アミノ酸鎖間に水素結合が形成される。
【0050】
β-シート構造において、二つのアミノ酸鎖の隣接した二つの末端は、リンカーにより連結される。リンカーとしては、上述の多様なターン配列またはペプチドリンカーが利用できる。ターン配列がリンカーとして利用される場合、β-ターン配列が最も好ましい。
【0051】
本発明の他の変形例によると、構造安定化部位としてロイシンジッパーまたはリンカーで連結されたロイシンジッパーが利用できる。ロイシンジッパーは、パラレルな二つのα-鎖のダイマー化を引き起こす保存性ペプチドドメインであり、一般に、遺伝子発現に関与する蛋白質に発見されるダイマー化ドメインである("Leucine scissors". Glossary of Biochemistry and Molecular Biology (Revised). (1997). Ed. David M. Glick. London: Portland Press; Landschulz WH, et al. (1988) Science 240:1759-1764)。ロイシンジッパーは、一般にヘプタッド(heptad)反復配列を含み、ロイシン残基が4番目または5番目に位置している。例えば、本発明に利用できるロイシンジッパーは、LEALKEK, LKALEKE, LKKLVGE, LEDKVEE, LENEVARまたはLLSKNYHのアミノ酸配列を含む。本発明で利用されるロイシンジッパーの具体的な例は、配列番号39に記載されている。ロイシンジッパーのそれぞれの半分は、短いα-鎖からなっており、α-鎖間の直接的なロイシン接触がある。転写因子にあるロイシンジッパーは、一般に疎水性ロイシンジッパー部位及び塩基性部位(DNA分子の主グルーブと相互作用する部位)からなっている。本発明でロイシンジッパーが利用される場合は、塩基性部位は必ずしも必要なわけではない。ロイシンジッパー構造において、二つのアミノ酸鎖(即ち、二つのα-鎖)の隣接した二つの末端は、リンカーにより連結できる。リンカーとしては、上述の多様なターン配列またはペプチドリンカーが利用でき、好ましくは、ロイシンジッパーの構造に影響を及ぼさないペプチドリンカーが利用される。
【0052】
上述の構造安定化部位の両末端には、無作為アミノ酸配列が結合される。前記無作為アミノ酸配列がターゲット結合部位I及びターゲット結合部位IIを形成する。本発明の最も大きい特徴の一つは、構造安定化部位の両側末端にターゲット結合部位I及びターゲット結合部位IIを連結して、二座方式でペプチドバインダーを製作することである。ターゲット結合部位I及びターゲット結合部位IIは、互いに協同的に(cooperatively)ターゲットに結合することにより、ターゲットに対する親和度を大きく増加させる。
【0053】
ターゲット結合部位Iのアミノ酸数nは、特に制限されず、好ましくは2〜100の整数、より好ましくは2〜50の整数、さらに好ましくは、2〜20の整数、最も好ましくは、3〜10の整数である。
【0054】
ターゲット結合部位IIのアミノ酸数mは、特に制限されず、好ましくは2〜100の整数、より好ましくは2〜50の整数、さらに好ましくは、2〜20の整数、最も好ましくは、3〜10の整数である。
【0055】
ターゲット結合部位I及びターゲット結合部位IIには、それぞれ異なるまたは同一な数のアミノ酸残基が含まれる。ターゲット結合部位I及びターゲット結合部位IIには、それぞれ異なるまたは同一なアミノ酸配列が含まれて、好ましくは、それぞれ異なるアミノ酸配列が含まれる。
【0056】
ターゲット結合部位I及び/またはターゲット結合部位IIに含まれるアミノ酸配列は、線形のアミノ酸配列または環形のアミノ酸配列である。ターゲット結合部位のペプチド配列の安定性を増加させるために、ターゲット結合部位I及び/またはターゲット結合部位IIに含まれるアミノ酸配列の少なくとも一つのアミノ酸残基は、アセチル基、フルオレニルメトキシカルボニル基、ホルミル基、パルミトイル基、ミリスチル基、ステアリル基またはポリエチレングリコール(PEG)に変形できる。
【0057】
生物学的ターゲット分子に結合される本発明の二座ペプチドバインダーは、生体内生理学的反応の調節、生体内物質の検出、インビボ分子イメージング、インビトロ細胞イメージング及び薬物伝達用ターゲッティングをする際に利用でき、エスコート分子としても利用できる。
【0058】
本発明の好ましい具現例によると、構造安定化部位、ターゲット結合部位Iまたはターゲット結合部位II(より好ましくは、構造安定化部位、さらに好ましくは、構造安定化部位のリンカー)にカーゴが結合されている。前記カーゴの例は、請求項1において、前記カーゴは、化合物、化学薬物、バイオ薬物、無機粒子、ナノ粒子、タンパク質、ペプチド、核酸分子、脂質、炭水化物、リポソームまたは検出可能な信号を発生させるラベル分子を含むが、これらに限定されるものではない。
【0059】
前記検出可能な信号を発生させるラベルは、T1造影物質(例えば、Gdキレート化合物)、T2造影物質(例えば、超常磁性物質(例えば、マグネタイト、Fe3O4、γ-Fe2O3、マンガンフェライト、コバルトフェライト及びニッケルフェライト))、放射性同位元素(例えば、11C,15O, 13N, P32, S35, 44Sc, 45Ti, 118I, 136La, 198Tl, 200Tl, 205Bi及び206Bi)、蛍光物質(フルオレセイン(fluorescein)、フィコエリスリン(phycoerythrin)、ロダミン、リサミン(lissamine)、そしてCy3とCy5)、化学発光団、磁気粒子、マス標識または電子密集粒子を含むが、これらに制限されるものではない。
【0060】
前記化学薬物は、例えば、抗炎症剤、鎮痛剤、抗関節炎剤、鎮痙剤、抗鬱剤、抗精神病薬物、神経安定剤、抗不安剤、麻薬拮抗剤、抗パーキンソン疾患、コリン性アゴニスト、抗癌剤、抗血管新生抑制剤、免疫抑制剤、抗ウイルス剤、抗生剤、食欲抑制剤、鎮痛剤、抗コリン剤、抗ヒスタミン剤、抗片頭痛剤、ホルモン剤、冠状血管、脳血管または末梢血管拡張剤、避妊薬、抗血栓剤、利尿剤、抗高血圧剤、心血管疾患治療剤、美容成分(例えば、シワ改善剤、皮膚老化抑制剤及び皮膚美白剤)などを含むが、これらに限定されるものではない。
【0061】
前記バイオ薬物は、インシュリン、IGF-1(insulin-like growth factor 1)、成長ホルモン、エリスロポエチン、G-CSFs (granulocyte-colony stimulating factors)、GM-CSFs (granulocyte/macrophage-colony stimulating factors)、インターフェロンアルファ、インターフェロンベータ、インターフェロンガンマ、インターロイキン-1アルファ及びベータ、インターロイキン-3、インターロイキン-4、インターロイキン-6、インターロイキン-2、EGFs(epidermal growth factors)、カルシトニン(calcitonin)、ACTH(adrenocorticotropic hormone)、TNF(tumor necrosis factor)、アトビスバン(atobisban)、ブセレリン(buserelin)、セトロレリックス(cetrorelix)、デスロレリン(deslorelin)、デスモプレシン(desmopressin)、ジノルフィンA (dynorphin A) (1-13)、エルカトニン(elcatonin)、エレイドシン(eleidosin)、エプチフィバチド(eptifibatide)、GHRH-II(growth hormone releasing hormone-II)、ゴナドレリン(gonadorelin)、ゴセレリン(goserelin)、ヒストレリン(histrelin)、リュプロレリン(leuprorelin)、リプレシン(lypressin)、オクトレオチド(octreotide)、オキシトシン(oxytocin)、ピトレシン(pitressin)、セクレチン(secretin)、シンカリド(sincalide)、テルリプレシン(terlipressin)、チモペンチン(thymopentin)、チモシン(thymosine)α1、トリプトレリン(triptorelin)、ビバリルジン(bivalirudin)、カルベトシン(carbetocin)、シクロスポリン、エキセジン(exedine)、ランレオチド(lanreotide)、LHRH(luteinizing hormone-releasing hormone)、ナファレリン(nafarelin)、副甲状腺ホルモン、プラムリンチド(pramlintide)、T-20(enfuvirtide)、チマルファシン(thymalfasin)、ジコノチド、RNA、DNA、cDNA、アンチセンスオリゴヌクレオチド及びsiRNAなどがあるが、これらに限定されるものではない。
【0062】
ターゲット結合部位I及び/またはターゲット結合部位IIは、多様なターゲットに結合するアミノ酸配列を含むことができる。本発明の二座ペプチドバインダーによりターゲッティングできるものは、生化学物質、ペプチド、ポリペプチド、核酸、炭水化物、脂質、細胞及び組織のような生物学的ターゲット、化合物、金属または非金属物質を含み、好ましくは、生物学的ターゲットである。
【0063】
ターゲット結合部位が結合する生物学的ターゲットは、好ましくは、生化学物質、ペプチド、ポリペプチド、糖蛋白質、核酸、炭水化物、プロテオグリカン、脂質または糖脂質である。
【0064】
例えば、ターゲット結合部位が結合する生化学物質は、多様な生体内代謝産物(例えば、ATP、NADH、NADPH、炭水化物代謝産物、脂質代謝産物及びアミノ酸代謝産物)を含む。
【0065】
ターゲット結合部位が結合する例示的なペプチドまたはポリペプチドは、酵素、リガンド、受容体、バイオマーカー、ホルモン、転写因子、成長因子、免疫グロブリン、信号伝達蛋白質、結合蛋白質、イオンチャンネル、抗原、付着蛋白質、構造蛋白質、調節蛋白質、毒素蛋白質、サイトカイン及び血液凝固因子を含むが、これらに限定されるものではない。より詳細には、二座ペプチドバインダーのターゲットは、フィブロネクチン extra domain B(ED-B), VEGF(vascular endothelial growth factor), VEGFR(vascular endothelial growth factor receptor), VCAM1(vascular cell adhesion molecule-1), nAchR(Nicotinic acetylcholine receptor), HSA(Human serum albumin), MyD88, EGFR(Epidermal Growth Factor Receptor), HER2/neu, CD20, CD33, CD52, EpCAM (Epithelial Cell Adhesion Molecule), TNF-α(Tumor Necrosis Factor-α), IgE (Immunoglobulin E), CD11A (α-chain of lymphocyte function-associated antigen 1), CD3, CD25, Glycoprotein IIb/IIIa, インテグリン, AFP(Alpha-fetoprotein), β2M (Beta2-microglobulin), BTA(Bladder Tumor Antigens), NMP22, Cancer Antigen 125, Cancer Antigen 15-3, カルシトニン, Carcinoembryonic Antigen, Chromogranin A,エストロゲン受容体、プロゲステロン受容体, Human Chorionic Gonadotropin, Neuron-Specific Enolase, PSA(Prostate-Specific Antigen), PAP(Prostatic Acid Phosphatase)及びThyroglobulinを含むが、これらに限定されるものではない。
【0066】
ターゲット結合部位が結合する例示的な核酸分子は、gDNA, mRNA, cDNA, rRNA(ribosomal RNA), rDNA(ribosomal DNA)及びtRNAを含むが、これらに限定されるものではない。ターゲット結合部位が結合する例示的な炭水化物は、生体内炭水化物であって、単糖類、二糖類、三糖類及び多糖類を含むが、これらに限定されるものではない。ターゲット結合部位が結合する例示的な脂質は、脂肪酸、トリアシルグリセロール、スフィンゴ脂質、ガングリオシド及びコレステロールを含むが、これらに限定されるものではない。
【0067】
本発明の二座ペプチドバインダーは、細胞表面に露出した生体分子(例えば、蛋白質)に結合することもできるが、細胞内生体分子(例えば、蛋白質)にも結合し、生体分子の活性を調節することができる。
【0068】
二座ペプチドバインダーが細胞内蛋白質をターゲッティングする場合、好ましくは、二座ペプチドバインダーは、追加的に細胞膜透過ペプチド(CPP)を含む。
【0069】
前記CPPは、当業界に公知された多様なCPPを含み、例えば、HIV-1 Tat蛋白質、オリゴアルギニン、ANTPペプチド、HSV VP22転写調節蛋白質、vFGF由来のMTSペプチド、Penetratin、Transportan、Pep-1ペプチド、Pep-7ペプチド、Buforin II、MAP(Model Amphiphatic Peptide)、k-FGF、Ku 70、pVEC、SynB1またはHN-1を含むが、これらに限定されるものではない。前記CPPを二座ペプチドに結合させる方法は、多様な方法があって、例えば、二座ペプチドの構造安定化部位にあるループ部分のリジン残基とCPPを共有結合させる。
【0070】
細胞内には、生理活性に重要な役割をするたくさんのターゲット蛋白質があり、CPPが結合された二座ペプチドバインダーは、細胞内に流入されて、このターゲット蛋白質に結合して活性を調節(例えば、抑制)する。
【0071】
上述のように、本発明の二座ペプチドバインダーは、典型的に‘N-ターゲット結合部位I−構造安定化部位の一つの鎖−リンカー−構造安定化部位の他の鎖−ターゲット結合部位II−C’のコンストラクトを有する。
【0072】
本発明の好ましい具現例によると、本発明の二座ペプチドバインダーにおいて、ターゲット結合部位Iと構造安定化部位の一鎖との間及び/または構造安定化部位の他の鎖−ターゲット結合部位IIの間には、ターゲット結合部位と構造安定化部位間の相互構造的影響を遮断する構造影響抑制部位(structure influence inhibiting region)を含む。回転部位には、ペプチド分子でΦとΨの回転が比較的自由なアミノ酸が位置する。好ましくは、ΦとΨの回転が比較的自由なアミノ酸は、グリシン、アラニン及びセリンである。構造影響抑制部位には、1〜10個、好ましくは、1〜8個、より好ましくは、1〜3個のアミノ酸残基が位置することができる。
【0073】
上述のコンストラクトを有する本発明の二座ペプチドバインダーのライブラリーは、当業界に公知された多様な方法で得られる。このライブラリーにおいて、二座ペプチドバインダーは無作為配列を有するようになり、これは、ターゲット結合部位I及び/またはターゲット結合部位IIのどの位置でも配列選好度(sequence preference)がないか、または指定(または固定)されたアミノ酸残基がないことを意味する。
【0074】
例えば、二座ペプチドバインダーのライブラリーは、固状支持体(例えば、ポリスチレンまたはポリアクリルアミド樹脂)上で行われるスプリット合成方法(Lam et al. (1991) Nature 354:82; WO 92/00091)によって構築できる。
【0075】
本発明の好ましい具現例によると、二座ペプチドバインダーのライブラリーは、細胞表面展示(cell surface display)方式(例えば、ファージディスプレイ、バクテリアディスプレイまたはイーストディスプレイ)で構築される。好ましくは、二座ペプチドバインダーのライブラリーは、プラスミド、バクテリオファージ、ファージミド、イースト、バクテリア、mRNAまたはリボゾームを基にするディスプレイ方法により製作できる。
【0076】
ファージディスプレイは、ファージの表面上のコート蛋白質に融合された蛋白質形態で多様なポリペプチドをディスプレイする技術である(Scott, J. K. and Smith, G. P. (1990) Science 249: 386; Sambrook, J. et al., Molecular Cloning. A Laboratory Manual, 3rd ed. ColdSpring HarborPress(2001); Clackson and Lowman, Phage Display, Oxford UniversityPress(2004))。フィラメント性ファージ(例えば、M13)の遺伝子IIIまたは遺伝子VIIIに発現しようとする遺伝子を融合させて、無作為ペプチドをディスプレイする。
【0077】
ファージディスプレイには、ファージミドが利用できる。ファージミドは、バクテリアの複製原点(例えば、ColE1)及びバクテリオファージのintergenic部位の一コピーを有するプラスミドベクターである。このファージミドにクローニングされたDNA断片は、プラスミドのように増殖される。
【0078】
二座ペプチドバインダーのライブラリーをファージディスプレイ方式で構築する場合、本発明の好ましい具現例は、下記の段階を含む:(i)ファージコート蛋白質(例えば、M13のようなフィラメント性ファージの遺伝子IIIまたは遺伝子VIIIコート蛋白質)をコーディングする遺伝子と二座ペプチドバインダーをコーディングする遺伝子が融合された融合遺伝子;及び前記融合遺伝子に作動的に結合された転写調節配列(例えば、lacプロモーター)を含む発現ベクターのライブラリーを製作する段階;(ii)前記発現ベクターライブラリーを適した宿主に導入させる段階;(iii)前記宿主細胞を培養し、組み換えファージまたはファージミドウイルスパーティクルを形成させて、融合蛋白質が表面にディスプレイされるようにする段階;(iv)生物学的ターゲット分子と前記ウイルスパーティクルを接触させて、パーティクルをターゲット分子に結合させる段階;及び(v)ターゲット分子に結合しなかったパーティクルを分離する段階。
【0079】
ファージディスプレイを利用してペプチドライブラリーを構築して、これらのライブラリーをスクリーニングする方法は、米国特許第5,723,286号、第5,432,018号、第5,580,717号、第5,427,908号、第5,498,530号、第5,770,434号、第5,734,018号、第5,698,426号、第5,763,192号、及び第5,723,323号に開示されている。
【0080】
二座ペプチドバインダーの遺伝子を含む発現ベクターを製作する方法は、当業界に公知された方法によって行うことができる。例えば、公知のファージミドまたはファージベクター(例えば、pIGT2, fUSE5, fAFF1, fd-CAT1, m663, fdtetDOG, pHEN1, pComb3, pComb8, pCANTAB 5E (Pharmacia), LamdaSurfZap, pIF4, PM48, PM52, PM54, fdH及びp8V5)に二座ペプチドバインダーの遺伝子を挿入させて発現ベクターを製作することができる。
【0081】
大部分のファージディスプレイ方法がフィラメント性ファージを利用して行われるが、λファージディスプレイ(WO 95/34683;米国特許第5,627,024号)、T4ファージディスプレイ(Ren et al. (1998) Gene 215:439; Zhu (1997) CAN 33:534)及びT7ファージディスプレイ(米国特許第5,766,905号)も、二座ペプチドバインダーのライブラリーを構築する際に利用できる。
【0082】
ベクターライブラリーを適した宿主細胞に導入させる方法は、多様な形質転換方法によって行うことができ、最も好ましくは、電気穿孔(electroporation)方法によって行われる(参照:米国特許第5,186,800号, 第5,422,272号, 第5,750,373号)。本発明に適する宿主は、細胞は、E. coliのようなグラム陰性バクテリア細胞であり、適したE. coli宿主は、JM101, E. coli K12 strain 294, E. coli strain W3110及びE. coli XL-1Blue(Stratagene)を含むが、これらに限定されるものではない。宿主細胞は、形質転換前にコンピテンス細胞として用意することが好ましい(Sambrook, J. et al., Molecular Cloning. A Laboratory Manual, 3rd ed. Cold Spring HarborPress(2001))。形質転換された細胞の選別は、一般に抗生剤(例えば、テトラサイクリン及びアンピシリン)を含む培地で培養して行われる。選別された形質転換細胞をヘルパーファージの存在下で追加的に培養し、組み換えファージまたはファージミドウイルスパーティクルを生成させる。前記ヘルパーファージとして適したものは、Exヘルパーファージ、M13-KO7、M13-VCS及びR408を含むが、これらに限定されるものではない。
【0083】
生物学的ターゲット分子と結合するウイルスパーティクルの選別は、通常的にバイオパニング過程を通じて行うことができる(Sambrook, J. et al., Molecular Cloning. A Laboratory Manual, 3rd ed. Cold SpringHarbor Press(2001); Clackson and Lowman, Phage Display, Oxford University Press(2004))。
【0084】
本発明の二座ペプチドバインダーの具体的な例は、配列番号20〜38及び40〜41に記載されている。
【0085】
本発明のまた他の様態によると、本発明は、上述の二座ペプチドバインダーをコーディングする核酸分子を提供する。
【0086】
本発明の他の様態によると、本発明は、二座ペプチドバインダーをコーディングする核酸分子を含む二座ペプチドバインダーの発現用ベクターを提供する。
【0087】
本発明の他の様態によると、本発明は、二座ペプチドバインダーの発現用ベクターを含む形質転換体を提供する。
【0088】
本明細書において、用語‘核酸分子’は、DNA(gDNA及びcDNA)、そしてRNA分子を包括的に含む意味を有して、核酸分子において基本構成単位であるヌクレオチドは、天然のヌクレオチドだけではなく、糖または塩基部位が変形された類似体(analogue)も含む(Scheit,Nucleotide Analogs,John Wiley,New York(1980);Uhlman及びPeyman, Chemical Reviews, 90:543-584(1990))。
【0089】
本発明の好ましい具現例によると、本発明のベクターは、二座ペプチドバインダーをコーディングする核酸分子の他に、前記核酸分子に、転写を進行させることのできる強力なプロモーター(例えば、tacプロモーター、lacプロモーター、lacUV5プロモーター、lppプロモーター、pLλプロモーター、pRλプロモーター、rac5プロモーター、ampプロモーター、recAプロモーター、SP6プロモーター、trpプロモーター及びT7プロモーターなど)、解読の開始のためのリボゾーム結合座及び転写/解読終結配列を含む。
【0090】
本発明の好ましい具現例によると、本発明のベクターは、二座ペプチドバインダーをコーディングする核酸分子の5’-方向側にシグナル配列(例えば、pelB)を追加的に含むことができる。また、本発明の好ましい具現例によると、本発明のベクターは、二座ペプチドバインダーがファージの表面によく発現されたのかを確認するためのタギング配列(例えば、myc tag)を追加的に含む。
【0091】
本発明の好ましい具現例によると、本発明のベクターは、ファージコート蛋白質、好ましくは、M13のようなフィラメント性ファージの遺伝子IIIまたは遺伝子VIIIコート蛋白質をコーディングする遺伝子を含む。本発明の好ましい具現例によると、本発明のベクターは、バクテリアの複製原点(例えば、ColE1)及び/またはバクテリオファージの複製原点を含む。一方、本発明のベクターは、選択標識として、当業界で通常的に利用される抗生剤耐性遺伝子を含むことができるが、例えば、アンピシリン、ゲンタマイシン、カベニシリン、クロラムフェニコール、ストレプトマイシン、カナマイシン、ゲネチシン、ネオマイシン及びテトラサイクリンに対する耐性遺伝子を含むことができる。
【0092】
本発明の形質転換体は、好ましくは、E.coliのようなグラム陰性バクテリア細胞であり、適するE.coli宿主は、JM101, E. coli K12 strain 294, E. coli strain W3110及びE. coli XL-1Blue(Stratagene)を含むが、これらに限定されるものではない。本発明のベクターを宿主細胞内に運搬する方法は、CaCl2方法(Cohen, S.N. et al., Proc. Natl. Acac. Sci. USA, 9:2110-2114(1973))、ハナハン方法(Cohen, S.N. et al., Proc. Natl. Acac. Sci. USA, 9:2110-2114(1973);及びHanahan, D., J. Mol. Biol., 166:557-580(1983))及び電気穿孔方法(米国特許第5,186,800号、第5,422,272号、第5,750,373号)などにより行うことができる。
【0093】
本発明の二座ペプチドバインダーは、非常に低い水準(例えば、nM水準)のKD値(解離常数)を示し、生物学的ターゲット分子に非常に高い親和度を示すペプチドを提供する。下記実施例に記載のように、モノポダル方式で製作されたバインダーと比較し、二座ペプチドバインダーは、約102〜105倍(好ましくは、約103〜104倍)高い親和度を示す。
【0094】
本発明の二座ペプチドバインダーは、医薬としての用途を有するだけではなく、生体内物質の検出、インビボ分子イメージング、インビトロ細胞イメージング及び薬物伝達用ターゲッティングをする際に利用でき、エスコート分子としても利用できる。
【発明の効果】
【0095】
本発明の特徴及び利点を要約すると、以下のようである:
(i)本発明は、BPBベースのカーゴ運搬システムを提供する。
(ii)本発明の二座ペプチドバインダーにおいて、構造安定性部位の両末端に結合されている離隔された(distal)二つのターゲット結合部位は、互いに協同的に(cooperatively)、シナージック(synergetically)にターゲットに結合する。
(iii)このため、本発明の二座ペプチドバインダーは、非常に低い水準(例えば、nM水準)のKD値(解離定数)を示し、ターゲットに非常に高い親和度を示す。
(iv)本発明のBPBベースのカーゴ運搬システムは、BPBのターゲット結合性及び特異性に基づいて、多様な物質を細胞表面または細胞内に運搬することができる。
【図面の簡単な説明】
【0096】
【図1a】構造安定化部位としてβ−ヘアピン(hairpin)を含む二座ペプチドバインダー(bipodal-peptide binder)の模式図を示す。
【図1b】構造安定化部位としてリンカーで連結されたβ−シートを含む二座ペプチドバインダー(bipodal-peptide binder)の模式図を示す。
【図1c】構造安定化部位としてリンカーで連結されたロイシンジッパーを含む二座ペプチドバインダー(bipodal-peptide binder)の模式図を示す。
【図1d】構造安定化部位としてリンカーで連結されたロイシン-リッチモチーフ(leucine-rich motif)を含む二座ペプチドバインダー(bipodal-peptide binder)の模式図を示す。
【図2】二座ペプチドバインダーライブラリーをクローニングするための戦略を示す。pIGT2ファージミドベクターマップにおいて、pelBシグナル配列、myc tagは、目的遺伝子がファージの表面によく発現されたのかを確認するためのタギング配列である。プロモーターとしてlacプロモーターが利用された。
【図3】フィブロネクチンED-Bバイオパニング過程の間、インプットファージに対するED-B、ストレプトアビジン及びBSAのバイオパニング結果を示す。
【図4】フィブロネクチンED-Bバイオパニング過程の間、二座ペプチドバインダーライブラリーのバイオパニングの3次段階で回収した60個の組み換えファージのED-B及びBSAに対するELISA結果である。
【図5a】フィブロネクチンED-B蛋白質に結合する特定二座ペプチドバインダーの親和力を測定した結果である。
【図5b】VEGFに結合する特定二座ペプチドバインダーの親和力を測定した結果である。
【図5c】VCAM1に結合する特定二座ペプチドバインダーの親和力を測定した結果である。
【図5d】nAchR(Nicotinic acetylcholine receptor)に結合する特定二座ペプチドバインダーの親和力を測定した結果である。
【図5e】HSA(Human Serum Albumin)に結合する特定二座ペプチドバインダーの親和力を測定した結果である。
【図6a】フィブロネクチンED-Bに対して特異性検査をするために、二座ペプチドバインダーを有している組み換えファージを、種々の蛋白質に対してELISAを行って吸光度を測定した結果である。左側バーからストレプトアビジン、ED-B、アセチルコリン、α1、BSA、VCAM、TNF-α、トロンビン、ミオグロブリン、リソザイム及びビスファチンに対する結果である。
【図6b】VEGFに対して特異性検査をするために、二座ペプチドバインダーを有している組み換えファージを、種々の蛋白質に対してELISAを行って吸光度を測定した結果である。
【図6c】VCAM1に対して特異性検査をするために、二座ペプチドバインダーを有している組み換えファージを、種々の蛋白質に対してELISAを行って吸光度を測定した結果である。
【図6d】nAchR断片ペプチドに対して特異性検査をするために、二座ペプチドバインダーを有している組み換えファージを、種々の蛋白質に対してELISAを行って吸光度を測定した結果である。
【図6e】HSAに対して特異性検査をするために、二座ペプチドバインダーを有している組み換えファージを、種々の蛋白質に対してELISAを行って吸光度を測定した結果である。
【図6f】MyD88に対して特異性検査をするために、二座ペプチドバインダーを有している組み換えファージを、種々の蛋白質に対してELISAを行って吸光度を測定した結果である。
【図7】二座ペプチドバインダーの共同作用効果の証明のための親和力を測定した結果である。
【図8】二座ペプチドバインダーにおいて、構造安定化部位をトリプトファンジッパーの代わりに種々のβ-ヘアピンモチーフに変えて、二座ペプチドバインダーの親和力を測定した結果である。
【図9】二座ペプチドバインダーにおいて、構造安定化部位をトリプトファンジッパーの代わりにロイシンジッパーに変えて、二座ペプチドバインダーの親和力を測定した結果である。
【図10】癌バイオマーカーであるフィブロネクチンED-Bに特異的な二座ペプチドバインダーの癌標的化結果である。時間が経過するにつれて、癌に二座ペプチドバインダーが蓄積されることが分かる。それぞれの臓器を分離して蛍光を測定した時も、癌に相当部分蓄積されることが分かる。
【図11a】GST-BPB fusion proteinの精製を示す。
【図11b】GST-BPBのフィブロネクチン EDBに対するaffinity測定結果である。
【図12a】TNFα−BPBの精製を示す。
【図12b】TNFα−BPBのフィブロネクチン EDBに対するaffinity測定結果である。
【図12c】TNFα−BPBのcytotoxicitiy測定結果である。
【図13a】BPBcss-LS内に9R/siRNAのencapsulation efficiencyを分析した結果である。Green boxは、9R/siRNA複合体を含むliposomal fractionを示し、Red boxは、free unencapsulated siRNA fractionを示す。
【図13b】EDB over-expressing cell lineにおいて、BPBCSS−LSのuptakeを分析した結果である。Negativeは、未処理細胞を示し、LSは、リポソームのみを処理した細胞、CSS-LSは、BPBCSS-LSで処理した細胞を示す。
【図13c】EDB non-expressing cell lineにおいて、BPBCSS−LSのuptakeを分析した結果である。Negativeは、未処理細胞を示し、LSは、リポソームのみを処理した細胞、CSS-LSは、BPBCSS-LSで処理した細胞を示す。
【図13d】MCF-7 cellにVEGF-C siRNA knockdown efficiencyを分析した結果である。
【図14a】DSPE-PEG2000-MalとBPB(SSS) peptideのconjugationをMALDIを利用して分析した結果である。
【図14b】一週間SPION-BPBのHydrodynamic size変化を観察したELS分析結果である。
【図14c】(a)DSPE-PEG2000coated SPION及び(b)BPB conjugated DSPE-PEG2000coated SPIONのTEMイメージである。
【図14d】DSPE-PEG2000 coated SPION(DSPE-SPION)及びBPB conjugated DSPE-PEG2000 coated SPION(SSS-SPION)のT2-weighted MR phantom study結果である。
【図14e】DSPE-PEG2000 coated SPION及びBPB conjugated DSPE-PEG2000 coated SPIONを処理した細胞の(a)MRIイメージと(b)T2信号を分析した結果である。
【図14f】DSPE-PEG2000coated SPIONとBPB conjugated DSPE-PEG2000coated SPIONを処理した細胞のconfocal顕微鏡イメージである。
【図14g】DSPE-PEG2000 coated SPIONとBPB conjugated DSPE-PEG2000 coated SPIONのMTT assay結果である。
【図14h】DSPE-PEG2000coated SPION及びBPB conjugated DSPE-PEG2000coated SPIONの脳癌動物モデルにおけるMRIイメージである。
【図15a】Gold Nanoparticle-BPBのTEM imageである。
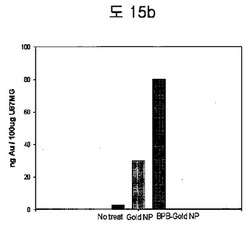
【図15b】U87MG細胞を利用してBPB conjugated金ナノ粒子を同一な濃度で入れた後、ICP-MSを利用して、細胞内のAuの量の差を測定した結果である。
【図15c】U87MG細胞に(a)GNPまたは(b)BPB-GNPを処理した後、Silver enhancementした顕微鏡写真である。
【発明を実施するための形態】
【0097】
以下、実施例を通じて本発明をさらに詳細に説明するが、これら実施例は、本発明をより具体的に説明するためのものであって、本発明の範囲がこれら実施例に限定されないことは、本発明の属する技術分野で通常の知識を有する者にとっては自明なことであろう。
【実施例】
【0098】
実験材料及び実験方法
実施例1:ライブラリーの製作
二座ペプチドバインダー遺伝子の製作及びファージミドベクターへの挿入
二つのオリゴヌクレオチドBeta-F1(5'-TTCTATGCGGCCCAGCTGGCC (NNK)6GGATCTTGGACATGGGAAAACGGAAAA-3')及びBeta-B1 (5'-AACAGTTTCTGCGGCCGCTCCTCC TCC(MNN)6TCCCTTCCATGTCCATTTTCCGTT-3')(Nは、A, T, GまたはC; Kは、GまたはT; Mは、CまたはA)を合成した。二重鎖を作るために、Beta-F1 4μM, Beta-B1 4μM, 2.5mM dNTP混合液4μl、ExTaq DNA重合酵素1μl(Takara, Seoul, Korea)及び10×PCRバッファ5μlを混合して、総50μlになるように蒸留水を添加した混合液を25個作った。この混合液をPCR反応(94℃で5分、60サイクル: 30℃で30秒、72℃で30秒及び72℃で7分間)をして、二重鎖を作った後、PCR精製キット(GeneAll, Seoul, Korea)を利用して精製し、二座ペプチドバインダー遺伝子を得た。二座ペプチドバインダーに挿入させる遺伝子をpIGT2ファージミドベクター(Ig therapy, Chuncheon, Korea)に連結するために、インサート遺伝子とpIGT2ファージミドベクターに制限酵素を処理した。約11μgのインサートDNAをSfiI(New England Biolabs(NEB, Ipswich)及びNotI(NEB, Ipswich)でそれぞれ4時間ずつ反応した後、PCR精製キットを利用して精製した。また、約40μgのpIGT2ファージミドベクターをSfiI及びNotIでそれぞれ4時間ずつ反応した後、CIAP(Calf Intestinal Alkaline Phosphatase)(NEB, Ipswich)を入れて1時間反応した後、PCR精製キットを利用して精製した。これらをUV-可視光線分光器(Ultrospec 2100pro, Amersham Bioscience)で定量し、2.9μgのインサート遺伝子を、T4 DNAリガーゼ(Bioneer, Daejeon, Korea)を利用してpIGT2ファージミドベクター12μgと18℃で15時間連結した後、エタノールで沈殿させてTEバッファ100μlでDNAを溶解させた。
【0099】
コンピテンス細胞の用意
E.coli XL1-BLUE細胞(American Type Culture Collection, Manassas, USA)をLB寒天プレートに線状塗抹した。寒天プレート培地で育った群落を5mlのLB培地に接種した後、37℃で200rpmの速度で混合しながら一日間培養した。培養された10mlの細胞を2lのLB培地に接種して、同じ方式で600nmの波長で吸光度が0.3-0.4になるまで培養した。培養されたフラスコを30分間氷に放置した後、4℃で4,000×gで20分間遠心分離し、沈殿した細胞を除いた上澄み液を全て除去し、1lの冷却された滅菌蒸留水で懸濁させた。これを再び同じ方法で遠心分離して、上澄み液を除去した後、1lの冷却された滅菌蒸留水で再懸濁させて、同じ方式で10%グリセロール溶液40mlで洗浄を繰り返して遠心分離した後、最後に10%グリセロール溶液4mlで懸濁させた後、200μlずつ分株して液体窒素に冷凍させた後、-80℃に保管した。
【0100】
電気穿孔法
ファージミドベクター12μgと二座ペプチドバインダーにインサートDNA 2.9μgを連結反応させた100μlを25個に分株して、電気穿孔を行った。コンピテンス細胞を氷の上で溶かして、200mlのコンピテンス細胞を連結反応した溶液4μlと混合した後、冷却して用意した0.2cmのキュベットに入れた後、1分間氷の上に放置した。電気穿孔機(BioRad, Hercules, CA)を200Ωで25μF及び2.5kVの条件にプログラムして、用意したキュベットの水気を除去して電気穿孔機に位置させた後、パルスを与えた(時間常数は、4.5〜5msec)。その後、直に37℃に用意した20mMのグルコースの含まれた1mlのLB液体培地に入れて、得られた25mlの細胞を100ml試験管に移した。一時間37℃で200rpmの速度で混合しながら培養した後、ライブラリーの数を測定するために、10μlを稀釈してアンピシリン寒天培地に塗抹した。残りの細胞を、1lのLBに20mMグルコース及び50μg/mlのアンピシリンを入れて、30℃で一日間培養した。4℃で4,000×gで20分間遠心分離し、沈殿した細胞を除いた上澄み液を全て除去し、40mlのLBで再懸濁させた後、グリセロールを最終濃度20%以上入れて、-80℃に保管した。
【0101】
ライブラリーで組み換えファージの生産とPEG沈殿
-80℃に貯蔵された二座ペプチドバインダーライブラリーで組み換えファージを生産した。500mlフラスコに100mlのLB液体培地にアンピシリン(50μg/ml)及び20mMのグルコースを入れた後、-80℃に保管されたライブラリー1mlを追加して、一時間37℃で150rpmの速度で混合しながら培養した。これに1×1011pfuのExヘルパーファージ(Ig therapy, Chuncheon, Korea)を入れて、再び一時間同じ条件で培養した。1,000×gで10分間遠心分離して上澄み液を除去し、これにアンピシリン(50μg/ml)及びカナマイシン(25μg/ml)が含まれたLB液体培地100mlを入れて、一日間培養し、組み換えファージを生産した。培養液を3,000×gで10分間遠心分離し、得られた上澄み液100mlにPEG/NaCl 25mlを混合して、氷に1時間放置させた後、4℃で20分間10,000×gで遠心分離して、上澄み液は注意して除去し、2mlのPBS(pH 7.4)でペレットを再懸濁させた。
【0102】
実施例2:蛋白質の準備
実施例に利用するフィブロネクチン(フィブロネクチン) ED-B, VEGF(vascular endothelial growth factor), VCAM1(vascular cell adhesion molecule-1), nAchR(Nicotinic acetylcholine receptor), HSA(Human serum albumin)及びMyD88を下記のように用意した。
【0103】
フィブロネクチンED-B遺伝子の製作及び発現ベクターへの挿入
韓国生命工学研究院から部分的なヒトのフィブロネクチンED-B(ID=KU017225)遺伝子を供給してもらった。プライマーEDB_F1(5'-TTCATAACATATGCCAGAGGTGCCCCAA-3')及びEDB_B1(5'-ATTGGATCCTTACGTTTGTTGTGTCAGTGTAGTAGGGGCACTCTCGCCGCCATTAATGAGAGTGATAACGCTGATATCATAGTCAATGCCCGGCTCCAGCCCTGTG-3')を合成して、EDB-F1 20 pmol, EDB-B1 20 pmol, 2.5 mM dNTP混合液4μl, ExTaq DNA重合酵素1μl(10 U)及び10×PCRバッファ5μlを混合して、50μlになるように蒸留水を添加した混合液を製造した。この混合液をPCR反応(94℃で5分, 30サイクル: 55℃で30秒, 72℃で1分及び94℃で30秒)をしてEDBインサートにした後、PCR精製キットを利用して精製した。EDBインサート遺伝子をpET28bベクター(Novagen)に連結するために、EDBインサート遺伝子及びpET28bベクターに制限酵素を処理した。約2μgのインサートDNAをBamHI(NEB, Ipswich)及びNdeI(NEB, Ipswich)で4時間ずつ反応した後、PCR精製キットを利用して精製した。また、約2μgのpIGT2ファージミドベクターをBamHI及びNdeIで3時間ずつ反応した後、CIAPを入れて1時間反応した後、PCR精製キットを利用して精製した。これらをベクターとインサートが約1:3のモル比率になるように添加して、T4 DNAリガーゼ(Bioneer, Daejeon, Korea)を利用して18℃で10時間連結させて、XL-1コンピテント細胞に形質転換をした後、カナマイシンの含まれた寒天培地に塗抹した。寒天プレート培地で育った群落を5mlのLB培地に接種した後、37℃で200rpmの速度で混合しながら一日間培養した後、プラスミドフラップキット(GeneAll, Seoul, Korea)を利用してプラスミドを精製し、シーケンシングをして、クローニングが成功したかどうかを確認した。
【0104】
VEGF121遺伝子の製作及び発現ベクターへの挿入
サイトカイン銀行(Jeonju, Korea)から部分的なヒトのVEGF(ID=G157)遺伝子を供給してもらった。プライマーVEGF_F1(5'-ATAGAATTCGCACCCATGGCAGAA-3')及びVEGF_B1(5'-ATTAAGCTTTCACCGCCTCGGCTTGTCACAATTTTCTTGTCTTGC-3')を合成して、VEGF-F1 20 pmol, VEGF-B1 20 pmol, 2.5 mM dNTP混合液4μl、ExTaq DNA重合酵素1μl(10 U)及び10×PCRバッファ5μlを混合して、50μlになるように蒸留水を添加した混合液を製造した。この混合液をPCR反応(94℃で5分, 30サイクル: 55℃で30秒, 72℃で1分及び94℃で30秒)をしてVEGFインサートにした後、PCR精製キットを利用して精製した。VEGFインサート遺伝子をpET32aベクター(Novagen)に連結するために、VEGFインサート遺伝子及びpET32aベクターに制限酵素を処理した。約2μgのインサートDNAをEcoRI(NEB, Ipswich)及びHindIII(NEB, Ipswich)で4時間ずつ反応した後、PCR精製キットを利用して精製した。これらをベクターとインサートが約1:3のモル比率になるように添加して、T4 DNAリガーゼ(Bioneer, Daejeon, Korea)を利用して18℃で10時間連結させて、XL-1コンピテント細胞に形質転換をした後、アンピシリンの含まれた寒天培地に塗抹した。寒天プレート培地で育った群落を5mlのLB培地に接種した後、37℃で200rpmの速度で混合しながら一日間培養した後、プラスミドフラップキット(GeneAll, Seoul, Korea)を利用してプラスミドを精製し、シーケンシングをして、クローニングが成功したかどうかを確認した。
【0105】
VCAM1遺伝子の製作及び発現ベクターへの挿入
韓国生命工学研究院からヒトのVCAM1遺伝子を供給してもらった。VCAM1インサート遺伝子をpET32aベクターに連結するために、VCAM1インサート遺伝子及びpET32aベクターに制限酵素を処理した。これらをベクターとインサートが約1:3のモル比率になるように添加して、T4 DNAリガーゼ(Bioneer, Daejeon, Korea)を利用して18℃で10時間連結させて、XL-1コンピテント細胞に形質転換をした後、アンピシリンの含まれた寒天培地に塗抹した。寒天プレート培地で育った群落を5mlのLB培地に接種した後、37℃で200rpmの速度で混合しながら一日間培養した後、プラスミドフラップキット(GeneAll, Seoul, Korea)を利用してプラスミドを精製し、シーケンシングをして、クローニングが成功したかどうかを確認した。
【0106】
フィブロネクチンED-Bの発現及び精製
フィブロネクチンED-BをクローニングしたpET28bベクターをBL21細胞に形質転換した後、カナマイシンの含まれた寒天培地に塗抹した。寒天プレート培地で育った群落をカナマイシン(25μg/ml)の含まれた5mlのLB培地に接種した後、37℃で200rpmの速度で混合しながら一日間培養した後、カナマイシン(25μg/ml)の含まれた50mlのLB培地に移して3時間培養した。培養したE. coliをカナマイシン(25μg/ml)の含まれた2lのLBに接種して、OD=0.6-0.8まで培養した。その後、1mMイソプロピル-β-D-チオガラクトピラノシド(IPTG)を入れて、37℃で200rpmの速度で混合しながら8時間培養した。4,000×gで20分間遠心分離して、沈殿された細胞を除いた上澄み液を全て除去し、ライシスバッファ(50mMリン酸ナトリウム(pH 8.0), 300mM NaCl及び5mMイミダゾール)で懸濁させた。-80℃に一日間保管した後、ソニケーターを利用してE. coliを溶解させた後、15,000×gで1時間遠心分離して、上澄み液をNi-NTA親和性レジン(Elpisbio, Daejeon, Korea)に結合させる。ライシスバッファでレジンを洗浄した後、イリュージョンバッファ(50mMリン酸ナトリウム(pH 8.0), 300mM NaCl及び300mMイミダゾール)を利用して、N-末端His-tag ED-B蛋白質を溶出させて獲得した。このように獲得した蛋白質をSuperdex75カラム(GE Healthcare, United Kingdom)及びPBS(pH 7.4)バッファを利用して、ゲルろ過法(gel filtration)で純度の高いED-B蛋白質を得た。バイオパニングのためにED-B蛋白質にビオチンを連結した。6mgのスルホ-NHS-SS-ビオチン(PIERCE, Illinois, USA)及び1.5mgのED-B蛋白質を常温で0.1Mホウ酸ナトリウム(pH 9.0)下で2時間反応し、反応しなかったスルホ-NHS-SS-ビオチンを除去するために、蛋白質をSuperdex75カラム及びPBS(pH 7.4)バッファを利用してゲルろ過法(gel filtration)でビオチン-EDB蛋白質を精製した。
【0107】
VEGF121とVCAM1-Trxの発現及び精製
VEGF121とVCAM1をクローニングしたpET32aベクターをそれぞれAD494細胞に形質転換した後、アンピシリンの含まれた寒天培地に塗抹した。寒天プレート培地で育った群落をアンピシリン(25μg/ml)の含まれた5mlのLB培地に接種した後、37℃で200rpmの速度で混合しながら一日間培養した後、アンピシリン(25μg/ml)の含まれた50mlのLB培地に移して3時間培養した。培養したE. coliをアンピシリン(25μg/ml)の含まれた2lのLBに接種して、OD=0.6-0.8まで培養した。その後、1mMイソプロピル-β-D-チオガラクトピラノシド(IPTG)を入れて、37℃で200rpmの速度で混合しながら8時間培養した。4,000×gで20分間遠心分離して、沈殿された細胞を除いた上澄み液を全て除去し、ライシスバッファ(50mMリン酸ナトリウム(pH 8.0), 300mM NaCl及び5mMイミダゾール)で懸濁させた。-80℃に一日間保管した後、ソニケーターを利用してE. coliを溶解させた後、15,000×gで1時間遠心分離して、上澄み液をNi-NTA親和性レジン(Elpisbio, Daejeon, Korea)に結合させる。ライシスバッファでレジンを洗浄した後、イリュージョンバッファ(50mMリン酸ナトリウム(pH 8.0), 300mM NaCl及び300mMイミダゾール)を利用して、Trx-VEGF121蛋白質を溶出させて獲得した。このように獲得した蛋白質をSuperdex75カラム(GE Healthcare, United Kingdom)及びPBS(pH 7.4)バッファを利用して、ゲルろ過法(gel filtration)で純度の高いVEGF-TrxとVCAM1-Trx蛋白質を得た。純粋なVEGFを得るために、トロンビンでVEGFとTrx間を切断し、VEGF121を得た。
【0108】
一方、HSAは、Genetex会社(Irvine)から購入して利用した。nAchR(Nicotinic acetylcholine receptor)の断片ペプチドであるbiotin-SGEWVIKEARGWKHWVFYSCCPTTPYLDITYH(32mer)は、ANYGEN(Korea, Kwangju)で合成した。Human MyD88は、Santa Cruz Biotechnology(sc-4540WB)(California)から購入した。
【0109】
実施例3:バイオパニング方法
BiotinフィブロネクチンED-B蛋白質とBiotin-nAchRペプチドのバイオパニング方法
2mlのストレプトアビジン(10μg/ml)を96ウェルELISAプレート(Corning)の40個のウェルに50μlずつ入れた後、4℃で一晩中放置し、翌日20個のウェルにのみ0.1%のPBST(tween-20)で3回洗浄した後、ビオチンED-BとビオチンnAchR(10μg/ml)をそれぞれ入れて、常温で1時間放置した。その後、40個のウェルの全てを0.1%PBSTで3回洗浄して、PBSで稀釈した2%BSAを使用して常温で2時間ブロッキングした後、溶液を全て捨てて、0.1%PBSTで3回洗浄した。これに二座ペプチドバインダー組み換えファージ含有溶液800μl及び10%BSA 200μlを混合し、ストレプトアビジン及びBSAに結合するファージを除去するために、ストレプトアビジンにBSAコーティングした20個のウェルに入れて1時間27℃に放置した。上澄み液を回収してED-BとnAchRが結合した20個のウェルに移して、27℃で45分間放置した。20個のウェルの溶液を全て除去して、0.5%PBSTで15回洗浄した後、0.2Mグリシン/HCl(pH2.2)1mlを各ウェル当たり50μlずつ入れて20分間ファージを溶離させて、1mlをチューブに集めて、これに2M Tris-base(pH 9.0)150μlを入れて溶液を中和させた。各バイオパニング毎にインプットファージ及びアウトプットファージの数を測定するために、OD=0.7のXL-1 BLUE細胞に混ぜて、アンピシリンの含まれた寒天培地に塗抹した。パニングを繰り返すために10mlのE. coli XL1-BLUE細胞と混ぜて、37℃で1時間200rpmの速度で混合しながら培養した。アンピシリン(50μg/ml)及び20mMグルコースを混合した後、2×1010 pfuのExヘルパーファージを添加して、37℃で1時間200rpmの速度で混合しながら培養した。培養液を1,000×gで10分間遠心分離した後、上澄み液は全て除去し、沈殿された細胞をアンピシリン(50μg/ml)及びカナマイシン(25μg/ml)の含まれた40ml LB液体培地で再懸濁して、30℃で200 rpmの速度で混合しながら一日間培養した。培養液を4,000×g、20分間及び4℃の条件で遠心分離した。上澄み液に8mlの5x PEG/NaCl[20% PEG(w/v)及び15% NaCl(w/v)]を添加した後、4℃で1時間静置させた。遠心分離後、PEG溶液を完全に除去して、1mlのPBS溶液でファージペプチドペレットを溶かした後、2次バイオパニングに使用した。各パニング段階毎に上記と同一な方法を使用したが、洗浄過程は、段階別にそれぞれ25回及び35回(0.5% PBST)増加させた。
【0110】
VEGFとVCAM1-TrxとHuman serum albumin(HSA), MyD88のバイオパニング方法
VEGFとVCAM1-trxとHSAとMyD 88(5μg/ml)を96ウェルELISAプレート(Corning)の10個のウェルに50μlずつ入れた後、4℃で一晩中放置し、翌日2%BSAを使用して常温で2時間ブロッキングした後、溶液を全て捨てて、0.1%PBSTで3回洗浄した。これに二座ペプチドバインダー組み換えファージ含有溶液800μl及び10%BSA 200μlを混合し、VEGFとVCAM1-TrxとHSAが結合した10個のウェルに移して常温で1時間放置した。10個のウェルの溶液を全て除去して、0.1%PBSTで10回洗浄した後、0.2Mグリシン/HCl(pH2.2)1mlを各ウェル当たり50μlずつ入れて20分間ファージを溶離させて、1mlをチューブに集め、これに2M Tris-base(pH 9.0)150μlを入れて溶液を中和させた。各バイオパニング毎にインプットファージ及びアウトプットファージの数を測定するために、OD=0.7のXL-1 BLUE細胞に混ぜて、アンピシリンの含まれた寒天培地に塗抹した。パニングを繰り返すために10mlのE. coli XL1-BLUE細胞と混ぜて、37℃で1時間200rpmの速度で混合しながら培養した。アンピシリン(50μg/ml)及び20mMグルコースを混合した後、2×1010 pfuのExヘルパーファージを添加して、37℃で1時間200rpmの速度で混合しながら培養した。培養液を1,000×gで10分間遠心分離した後、上澄み液は除去し、沈殿された細胞をアンピシリン(50μg/ml)及びカナマイシン(25μg/ml)の含まれた40ml LB液体培地で再懸濁して、30℃で200 rpmの速度で混合しながら一日間培養した。培養液を4,000×g、20分間及び4℃の条件で遠心分離した。上澄み液に8mlの5x PEG/NaCl[20% PEG(w/v)及び15% NaCl(w/v)]を添加した後、4℃で1時間静置させた。遠心分離後、PEG溶液を完全に除去して、1mlのPBS溶液でファージペプチドペレットを溶かした後、2次バイオパニングに使用した。各パニング段階毎に上記と同一な方法を使用したが、洗浄過程は、段階別にそれぞれ20回及び30回(0.1% PBST)増加させた。
【0111】
実施例4:フィブロネクチンED-BのインプットファージのELISA
二座ペプチドバインダーライブラリーの各インプットファージのELISAを、ストレプトアビジン、BSA及びED-Bに対して行った。96ウェルELISAプレートに10μg/mlストレプトアビジンを各ウェル当たり50μlずつ18個のウェルに入れて、10μg/ml BSAを各ウェル当たり50μlずつ9個のウェルに入れて、4℃で一晩中放置した。翌日、ストレプトアビジンのある18個のウェルの中、9個のウェルのみを0.1% PBST(tween-20)で3回洗浄し、ビオチンED-B(10μg/ml)を入れて常温で1時間放置した。その後、全てのウェルを0.1% PBSTで3回洗浄し、PBSで稀釈した2% BSAを使用して常温で2時間ブロッキングした後、溶液を全て捨てて0.1% PBSTで3回洗浄した。これに二座ペプチドバインダー組み換えファージである一番目、二番目及び三番目のインプットファージ800μl及び10% BSA 200μlを混合して、100μlずつED-B, ストレプトアビジン及びBSAウェルにそれぞれ3ウェルずつ分株して、27℃で1時間30分間静置した。0.1% PBST溶液で10回洗浄した後、HRP-コンジュゲート抗-M13抗体(GE Healthcare)を1:1,000に稀釈して、27℃で1時間反応した。0.1% PBSTで5回洗浄した後、ペロキシダーゼの基質であるテトラメチルベンジジン(TMB)(BD Science)溶液100μlを分株して、発色反応を誘導した後、100μlの1 M HClを添加して反応を中止した。その後、450nmで吸光度を測定した。
【0112】
実施例5:フィブロネクチンED-B, VEGF, VCAM1, nAchR, HSA, MyD88蛋白質に特異的なファージペプチド検索(ファージELISA)
アウトプットファージ/インプットファージ比率が最も高いバイオパニング段階で回収したファージをXL1-BLUE細胞に感染させた後、プラークがプレート当たり100-200個程度になるように塗抹した。滅菌されたチップを利用し、プラーク60個を2mlのLB-アンピシリン(50μg/ml)培養液に接種した後、37℃で5時間振とう培養し、OD=0.8-1で5×109pfuのExヘルパーファージを添加して、37℃で1時間200 rpmの速度で混合しながら培養した。培養液を1,000×gで10分間遠心分離した後、上澄み液は全て除去し、沈殿された細胞をアンピシリン(50μg/ml)及びカナマイシン(25μg/ml)の含まれた1ml LB液体培地で再懸濁して、30℃で200 rpmの速度で混合しながら一日間培養した。培養液を10,000×g, 20分間及び4℃の条件で遠心分離して上澄み液を回収した後、2%の脱脂牛乳を入れて、これをファージペプチド検索に使用した。96ウェルELISAプレートに5μg/mlのフィブロネクチン ED-B, VEGF, VCAM1, Nicotinic acetylcholine receptor(nAchR), Human serum albumin, MyD88を各ウェル当たり50μlずつ30個のウェルに入れて、また、10μg/mlのBSAを各ウェル当たり50μlずつ30個のウェルに入れて4℃で一日間放置した。翌日、全てのウェルを0.1% PBSTで3回洗浄し、PBSで稀釈した2%脱脂牛乳を使用して常温で2時間ブロッキングした後、溶液を全て捨てて0.1% PBSTで3回洗浄した。各クローン別に増幅されたファージペプチド溶液100μlずつを全てのウェルに分株して、27℃で1時間30分間静置した。0.1% PBST溶液で5回洗浄した後、HRP-コンジュゲート抗-M13抗体(GE Healthcare)を1:1,000に稀釈して、27℃で1時間反応した。0.1% PBSTで5回洗浄した後、TMB溶液100μlを分株して発色反応を誘導した後、100μlの1 M HClを添加して反応を中止した。450 nmで吸光度を測定し、BSAに比べて吸光度の高いクーロンを選択した。これらのファージをXL1細胞に感染させた後、プラークがプレート当たり100-200個程度になるように塗抹した。滅菌されたチップを利用し、プラークを4mlのLB-アンピシリン(50μg/ml)培養液に接種した後、37℃で一日間振とう培養して、プラスミドフラップキットを利用してプラスミドを精製し、シーケンシングを依頼した(Genotech, Daejeon, Korea)。シーケンシングプライマーは、ベクターシーケンスである5'-GATTACGCCAAGCTTTGGAGC-3'を使用した。
【0113】
実施例6:フィブロネクチンED-B, VEGF, nAchRバインディングアッセイ
DNAシーケンシングで重複して出たED-B, VEGF, nAchRに特異的な二座ペプチドバインダーペプチドを合成(Anygen,韓国)した。親和度の測定は、BIAcore X(Biacore AB, Uppsala, Sweden)を利用して行った。ED-BとnAchRは、ストレプトアビジンSAチップ(Biacore)にビオチン-EDBを2,000 RUだけ流して固定させた。VEGFは、EDC/NHSを利用してCM5チップ(Biacore)に固定した。ランニングバッファとしてはPBS(pH 7.4)を使用して、フローは、分当たり30μl流しながら、多様な濃度で動力学を測定した後、BIAevaluationソフトウェア(Biacore AB, Uppsala, Sweden)で親和度を計算した。
【0114】
実施例7:癌バイオマーカーであるフィブロネクチンED-Bに特異的な二座ペプチドバインダーの癌標的化
癌にたくさん分布するフィブロネクチンED-Bをターゲットする二座ペプチドバインダー(ペプチド2)にCy5.5-NHS蛍光ダイ(flurorescence dye)(Amersham Pharmacia, Piscataway)を50 mMホウ酸ナトリウム緩衝液(pH 9.7)で12時間常温反応した。反応後にSephadex G25(Pharmacia Biotech, Uppsala, Sweden)でCy5.5と二座ペプチドバインダー-Cy5.5を分離した。Balb/cヌードマウスにHuman U87MG(ATCC) 2 x 106 cellを皮下に入れて癌を10日間育てた後、0.5nmolの二座ペプチドバインダー-Cy5.5を静脈注射で入れて、IVIS(Caliper Life Sience, Hopkinton)で蛍光を測定した。この実験は、癌バイオマーカーであるフィブロネクチンED-Bに特異的な二座ペプチドバインダーが、インビボ動物において癌に蓄積されることを証明する結果であって、実際、癌診断剤としての応用性を示す(図11)。
【0115】
実施例8:細胞内に存在するMyD88に特異的な二座ペプチドバインダーの活性抑制実験
MyD88は、細胞内に存在する蛋白質であるため、二座ペプチドバインダーのloopのリジン(lysine)残基を利用し、細胞透過ペプチド(cell penetrating peptide)である9個のアルギニン(arginine)(Anygen, 韓国)を、EDC/NHS(Sigma)を利用して二座ペプチドバインダーに共有結合させて、細胞透過を可能にした。MyD88の活性が活性化されるとMMP-13の量が増加するため、MMP-13の量を測定すれば、MyD88の活性が抑制されるかどうか判別できる。MyD88の活性を活性化させるIL-1beta(10 ng/ml)(R&D systems, Minneapolis MN)を軟骨細胞に処理した。その後、MyD88に特異的な二座ペプチドバインダー(表3fのペプチド1)を軟骨細胞に10μM処理して、12時間後にmRNAを分離した後、MMP-13とGAPDHに対してRT-PCRを進行した。また、軟骨細胞を破壊して細胞内蛋白質を得た後、Anti-MMP 13抗体(Abcam, ab3208, Cambridge)とセミドライtransfer機械(Amersham Bioscience, Piscataway)を利用しウェスタンブロッティングを行って、MMP-13の量を測定した。
【0116】
実験結果
実施例9:二座ペプチドバインダーライブラリーの製作
二座ペプチドバインダーの構造安定化部位としては、安定したβ-ヘアピンモチーフを使用した。特に、トリプトファン-トリプトファンアミノ酸の相互作用によりβ-ヘアピンモチーフ構造を安定にできるトリプトファンジッパー(Andrea et al., Proc. Natl. Acad. Sci. 98:5578-5583(2001))を利用した。骨格であるトリプトファンジッパーのN-及びC-末端部分にそれぞれ6個のアミノ酸を無作為に配列することにより、二つの部分に可変的部位を生成した(図1a)。これを二座ペプチドバインダーと命名し、両側の可変的部位を有しているため、抗原に共同作用で付くことができ、高い親和力及び特異性を有することができる。また、二座ペプチドバインダーの構造安定化部位は、図1b乃至図1eのように様々に構成できる。
【0117】
合成した二つの無作為配列オリゴヌクレオチドをPCR反応を通じて二重鎖にした後、制限酵素のSfiI及びNotIで切断した後、pIGT2ファージミドベクターにクローニングして、8×108以上のライブラリーを構築した(図2)。
【0118】
実施例10:バイオパニング結果
二座ペプチドバインダーライブラリーをフィブロネクチンED-B, VEGF, VCAM1, nAchR, HSA蛋白質に対して3〜5回にかけてバイオパニングを行って、各パニング段階で回収したファージペプチドのアウトプットファージ/インプットファージの比率を決定した(表1a)。
【0119】
【表1a】

【0120】
【表1b】
【0121】
【表1c】
【0122】
【表1d】
【0123】
【表1e】
【0124】
【表1f】
【0125】
実施例11:フィブロネクチンED-BのインプットファージELISA結果
二座ペプチドバインダーのライブラリーの各インプットファージをED-B、ストレプトアビジン及びBSAに対してELISAを行った。一番目のインプットファージの反応性は、ED-B、ストレプトアビジン及びBSAのいずれも吸光度がほぼ等しいが、二番目のインプットファージからED-B吸光度が、ストレプトアビジンに比べ5.1倍、BSAに比べ3.4倍高かった。最後に三番目のインプットファージでは、ED-B吸光度が、ストレプトアビジンに比べ22倍、BSAに比べ15倍高い反応性を示し、ED-Bに対して成功的にバイオパニングがなされることが分かる(図3及び表2)。
【0126】
【表2】
【0127】
実施例12:フィブロネクチンED-B, VEGF, VCAM1, nAchR, HSA及びMyD88に対して特異的なファージペプチド検索(ファージELISA)及びシーケンシング
各ライブラリーのパニング段階の中、アウトプット/インプットの比率が最も高い段階で回収したファージをプラーク形態として確保した。各プラークから60個のファージを増幅させた後、BSAに対してELISAを行った(図4)。BSAに比べて吸光度の高いクローンを選択し、DNAシーケンシングを依頼した。これから重複したそれぞれの蛋白質に特異的なペプチドシーケンスを得た(表3)。
【0128】
【表3a】
【0129】
【表3b】
【0130】
【表3c】
【0131】
【表3d】
【0132】
【表3e】
【0133】
【表3f】
【0134】
実施例13:フィブロネクチンED-B, VEGF, VCAM1, nAchR及びHSAの親和度の測定
フィブロネクチンED-B, VEGF, VCAM1, nAchR及びHSAに対する前記ペプチドを合成し、SPR Biacore system(Biacore AB, Uppsala, Sweden)を利用して親和度を測定した。フィブロネクチンED-Bに対する親和度を測定した結果、ペプチド1は、620nMを示して、ペプチド2は、75nMを示し、ペプチド3は、2.5μMを示した(図5a)。VEGFに対する親和度を測定した結果、ペプチド1は、60nM、ペプチド2は、326nMを示した(図5b)。VCAM1の断片ペプチドに対する親和度を測定した結果、ペプチド1は、318nMを示した(図5c)。nAchRの断片ペプチドに対する親和度を測定した結果、ペプチド1は、73nMを示した(図5d)。HSAの断片ペプチドに対する親和度を測定した結果、ペプチド1は、115nMを示した(図5e)。
【0135】
実施例14:フィブロネクチンED-B, VEGF, VCAM1, nAchR, HSA及びMyD88に対する特異性分析
それぞれの蛋白質に特異的に結合する組み換えファージを、種々の蛋白質に対して特異性検査をELISAを利用して行った。96ウェルELISAプレートにそれぞれの蛋白質5μg/mlを50μlずつウェルに入れて、翌日0.1% PBST(tween-20)で3回洗浄し、2%BSAを使用して常温で2時間ブロッキングした後、溶液を全て捨てて0.1% PBSTで3回洗浄した。これに本発明のペプチドを有する組み換えファージを2%BSAとよく混合し、100μlずつ10個の蛋白質が結合されているウェルに分株し、27℃で2時間静置した。0.1% PBST溶液で5回洗浄した後、HRP-コンジュゲーション抗-M13抗体(GE Healthcare)を1:1,000に稀釈して、27℃で1時間反応した。0.1% PBSTで5回洗浄した後、TMB溶液100μlを分株して発色反応を誘導した後、100μlの1 M HClを添加して反応を中止した後、450 nmで吸光度を測定した。図6aから分かるように、二座ペプチドバインダーで見つけたED-Bに特異的な表3aのペプチド2は、他の蛋白質の吸光度と比較すると、30倍以上の差を示し、これは、ペプチド2シーケンスがED-Bに対して特異的であることを示す結果である。図6b〜6fから分かるように、表3b〜3fのそれぞれのペプチド1に対する特異性分析結果、これらのそれぞれのペプチドは、VEGF, VCAM1, nAchR, HSA及びMyD88に対して特異性を有するということが分かる。
【0136】
実施例15:SPR(Surface Plasmon Resonance)の共同作用効果の確認
二座ペプチドバインダーの抗原に対する共同作用効果を証明するために、親和力が最もよい表3aのED-Bに対するペプチド2のN-及びC-末端の一側部位のみをそれぞれ除去したペプチドを合成し、親和力を測定した。N-末端部分は、592μMを有して、C-末端部分は、12.8μMを示した(図7)。二座ペプチドバインダーにおいて二座(bipodal)を有していることから表す共同作用効果は、43nMの親和力であることを証明した(図5a)。
【0137】
実施例16:他のβ-ヘアピンに対するバインディングアッセイ
トリプトファンジッパーの外に、異なるβ-ヘアピン骨格であるGB1m3及びHP7に、ED-Bに特異的に結合するペプチド2のN-末端シーケンス(HCSSAV)とC-末端シーケンス(IIRLEQ)を有するようにペプチドを合成した(Anygen, 韓国)。即ち、トリプトファンジッパーを含む二座ペプチドバインダーのシーケンスは、HCSSAVGSWTWENGKWTWKGIIRLEQであり、GB1m3を含む二座ペプチドバインダーは、HCSSAVGKKWTYNPATGKFTVQEGIIRLEQであって、HP7を含む二座ペプチドバインダーは、HCSSAVGKTWNPATGKWTEGIIRLEQである。各ペプチドの親和度は、BIAcore X(Biacore AB, Uppsala, Sweden)を使用して測定した。ストレプトアビジンSAチップ(Biacore AB, Uppsala, Sweden)にビオチン-EDBを2,000 RUだけ流して固定した。ランニングバッファとしてはPBS(pH 7.4)を使用して、フローは、分当たり30μl流しながら、多様な濃度で動力学を測定した後、BIAevaluationで親和度を計算した。 親和度を測定した結果、GB1m3が70nM、HP7が84nMであって、トリプトファンジッパー(43nM)と等しい親和力を有することを確認した(図8)。これは、全ての安定したβ-ヘアピンモチーフが構造安定化部位として機能することを証明する結果である。
【0138】
実施例17:構造安定化部位としてロイシンジッパーを含む二座ペプチドバインダーに対するバインディングアッセイ
β-ヘアピン骨格の代わりに、構造安定化部位としてロイシンジッパーに、ED-Bに特異的に結合するペプチド2のN-末端シーケンス(HCSSAV)とC-末端シーケンス(IIRLEQ)を有するようにCSSPIQGGSMKQLEDKVEELLSKNYHLENEVARLKKLVGER及びIIRLEQGGSMKQLEDKVEELLSKNYHLENEVARLKKLVGERペプチドを合成した(Anygen, 韓国)。二つのペプチドをダイマーにした後、BIAcore X(Biacore AB, Uppsala, Sweden)を使用して親和度を測定した。親和度を測定した結果、ロイシンジッパーは、5μMの親和度を示して、これは、トリプトファンジッパー(43nM)の親和度に劣る親和度ではあるが、ロイシンジッパーも二座ペプチドバインダーの構造安定化部位として機能することができることが分かる(図9)。
【0139】
実施例18:癌バイオマーカーであるフィブロネクチンED-Bに特異的な二座ペプチドバインダーの癌標的化
癌にたくさん分布するフィブロネクチンED-Bをターゲッティングする二座ペプチドバインダーにCy5.5蛍光ダイを付着した後、Human U87MG癌が存在するマウスに静脈注射で投与して、IVIS機会を利用し、二座ペプチドバインダーが癌にターゲットするかどうか蛍光を確認した(図10)。実験結果、癌バイオマーカーであるフィブロネクチンED-Bに特異的な二座ペプチドバインダーは、癌組織に蓄積されることが観察されて、これは、本発明の二座ペプチドバインダーがインビボイメージングに利用できることを示す結果である。
【0140】
実施例19:GST-BPB fusion protein for フィブロネクチンEDB
1.GST-BPB遺伝子の製作
Glutathione S-transferase(GST)遺伝子の含まれたpGEX4T-1ベクター(GE Healthcare)にフィブロネクチン EDBを認識するBPB遺伝子シーケンスをクローニングした。二つのオリゴヌクレオチドGST-F1(5’-ACCGGATCCCATTGTTCTAGT-3’)とGST-B1(5'-ATTCTCGAGTTACGCTCCTCCTCC-3')を利用して、EDBタンパク質に付くファージから得たphagemid vectorをtemplate(実施例1及び3)でPCRして、フィブロネクチンEDBに付くBPBペプチド遺伝子を得た。このBPBペプチドのペプチド配列は、HCSSAVGSWTWENGKWTWKGIIRLEQである。GST-F1 20pmol, GST-B1 20pmol, 鋳型選別したphagemid template 1ul, 2.5mM dNTP mixture 4μl, Ex Taq DNA polymerase 1μl (10 U), 10 x PCR buffer 5μlを混ぜて、総50μlになるように蒸留水を追加した混合液を作った。この混合液をPCR反応(94℃で5分, 30cycle: 55℃で30秒と72℃で1分, 94℃で30秒)して、BPB Insertにした後、PCR purification kit(GeneAll, Seoul, Korea)を利用して精製した。BPB Insert遺伝子をpGEX4T-1 ベクターに連結するために、BPB Insert遺伝子とpGEX4T-1ベクターに制限酵素処理を行った。約2μgのInsert DNAをBamHI (NEB)とXhoI (NEB)で4時間ずつ反応させた後、PCR purification kitを利用して精製した。これらをmolar ratio vector:Insert=1:3でT4 DNA ligase(Bioneer)を利用して18℃、10時間連結した後、E. coli XL-1 competent cell(Stratagene)にtransformationをした後、Ampicillin agar培地に塗抹した。寒天平板培地で育った集落を5mlのLB培地に接種した後、37℃で200rpmの速度で混ぜながら、一日中培養した後、plasmid preparation kit(GeneAll, Seoul, Korea)を利用してplasmidを精製し、シーケンシングをして、クローニングが成功したかどうかを確認した。
【0141】
2. GST-BPB発現及び精製
GST-BPBをクローニングしたベクターをE. coli BL21にtransformationした後、2L LBにアンピシリン(25ug/ml)を入れて、OD=0.6-0.8まで育てた。その後、1mM isopropyl-β-D-thiogalactopyranoside(IPTG)を入れて、37℃で200rpmの速度で混ぜながら、8時間育てた。4,000gで20分間遠心分離して、沈んだ細胞を除いた上澄み液を全て除去して、lysis buffer (50 mM sodium phosphate (pH 8.0), 300mM NaClと5mM imidazole)で再浮遊させた。-80℃で一日間保管した後、Sonicatorを利用してE. coli溶解させた後、15,000gで1時間遠心分離して、上澄み液をGST affinity resin(Peptron)に結合させた。PBS bufferでresinを洗浄した後、Elution buffer(20mM GSH in 10mM Tris-HCl, pH 8)を利用して、GST-BPBタンパク質を分離して収集した。このように集めたタンパク質をSuperdex75 column(Amersham)とPBS(pH7.4) bufferを利用してgel filtrationをして、純度高いGST-BPBタンパク質を収集した。
【0142】
3. GST-BPBのフィブロネクチン EDB binding実験
精製されたGST-BPBを、フィブロネクチンEDBにbindingするかをSPR機械を利用して確認した。Affinity測定は、BIAcore Xを利用して行った。Streptavidin SA chip(Biacore)にbiotin-EDBを2000RUだけ流して固定させた。Running bufferとしてはPBS(pH7.4)を使用して、流速は30ul/minとして流しながら、様々な濃度でkineticsを測定した後、BIAevaluationでaffinityを計算した。
【0143】
4.実験結果
(1) GST-BPBの製作
フィブロネクチンED-Bを特異的に認識するBPBをGST fusion vectorにクローニングした後、GST-BPBを大腸菌で発現して精製した(図11a)。
【0144】
(2) GST-BPBの親和度測定
GST-BPBをSPR biacore systemを利用してフィブロネクチンフィブロネクチン EDBに対するaffinityを測定した。Affinityを測定した結果、284nMのaffinityを有して、GSTがあるにもかかわらず、十分フィブロネクチンEDBを認識できることを示した(図11b)。
【0145】
実施例20:TNFα-BPB fusion protein
1.TNFα-BPB遺伝子の製作
pET28b vector(Novagen)にhuman TNFα(tumor necrosis factor α)とフィブロネクチンEDBを認識するBPB遺伝子シーケンスをクローニングした。二つのオリゴヌクレオチドTNF-F1(AATAAAACATATG TCTCGAACCCCGA)とTNF-B1(ATGGATCCCAGGGCAATGATC)を利用して、human TNFαがクローニングされているベクターGh75(Cytokine Bank, Korea)でTNFα遺伝子を増幅して、これをpET28bにクローニングした。そして二つのオリゴヌクレオチドBPB-F1(AAT GAATTC TCTTCCTCATCGGGTTCTTCCTCATCGGGTTGTAGTTCTCCTATTC)とBPB-B1(AAT AAGCTT TCA TTGCTCCAACCTAAT)を利用し、EDBタンパク質に付くファージで得たphagemid vector(実施例1及び3)をtemplateでPCRして、フィブロネクチンEDBに付くBPBペプチド遺伝子を得た。このBPBペプチドのペプチド配列は、CSSPIQGSWTWENGKWTWKGIIRLEQである。
【0146】
前記BPB遺伝子を、TNFαの入ったpET28bベクターにクローニングした。plasmid preparation kitを利用してplasmidを精製し、シーケンシングをして、クローニングが成功したかどうかを確認した。
【0147】
2. TNFα-BPB発現及び精製
TNFα-BPBをクローニングしたpET28bベクターをBL21 cellにtransformationした後、kanamycine agar培地に塗抹した。2L LBにカナマイシン(25ug/ml)を入れて、OD=0.6-0.8まで育てた後、1mM isopropyl-β-D-thiogalactopyranoside(IPTG)を入れて、37℃で200rpmの速度で混ぜながら、8時間育てた。4,000gで20分間遠心分離して、沈んだ細胞を除いた上澄み液を全て除去して、lysis buffer (50 mM sodium phosphate (pH 8.0), 300mM NaClと5mM imidazole)で再浮遊させた。-80℃で一日間保管した後、Sonicatorを利用してE. coli溶解させた後、15,000gで1時間遠心分離して、上澄み液をNi-NTA affinity resin(ELPIS biotech.)に結合させた。Lysis bufferでresinを洗浄した後、Elution buffer(50mM sodium phosphate (pH 8.0), 300 mM NaCl and 300 mM imidazole)を利用して、TNFα-BPBタンパク質を分離して収集した。このように集めたタンパク質をSuperdex75 columnとPBS(pH7.4) bufferを利用してgel filtrationをして、純度高いTNFα-BPBタンパク質を収集した。
【0148】
3. TNFα-BPBのフィブロネクチン EDB binding実験
精製されたTNFα-BPBを、フィブロネクチンEDBにbindingするかをSPR機械を利用して確認した。Affinity測定は、BIAcore Xを利用して行った。Streptavidin SA chip(Biacore)にbiotin-EDBを2000RUだけ流して固定させた。Running bufferとしてはPBS(pH7.4)を使用して、流速は30ul/minとして流しながら、様々な濃度でkineticsを測定した後、BIAevaluationでaffinityを計算した。
【0149】
4. TNFα-BPBのcytotoxicity実験
L-M mouse fibroblastsを利用して、TNFα-BPBとTNFαのcytotoxicitiyをMTT分析で測定した。まずL-M mouse fibroblastを96well microplateに5000個を敷いた後、18時間後にTNFαとTNFα-BPBを濃度別に入れて、30時間後にMTTを行って、cytotoxicitiyを測定した。
【0150】
5.実験結果
(1)TNFα-BPB製作
フィブロネクチンED-Bを特異的に認識するBPBをTNFαにクローニングした後、 TNFα-BPBを大腸菌で発現して精製した(図12a)。
【0151】
(2)TNFα-BPBの親和度測定
TNFα-BPBを、SPR biacore systemを利用してフィブロネクチン EDBに対するaffinityを測定した。Affinityを測定した結果、80nMのaffinityを有して、TNFαがあるにもかかわらず、十分フィブロネクチンEDBを認識できることを示した(図12b).
【0152】
(3)TNFα-BPBのcytotoxicitiy測定
L-M mouse fibroblast cellでTNFnとTNFn-BPBのcytotoxicityをMTTで測定した。TNFα-BPBがTNFαより5倍程度cytotoxicityが高い(図12c)。
【0153】
実施例21:リポソーム-BPB
1. Conjugation of BPBcssto Mal-PEG2000-DSPE
Mal-PEG2000-DSPEを利用し、BPBEDB(CSSPIQGSWTWENGK(Cys, lysine残基にcysを連結)WTWKGIIRLEQ)のシステイン残基にコンジュゲーションさせた。簡単には、Mal-PEG2000-DSPEの1.1 fold molar excessをchloroform/DMSO(1:1 v/v)でBPBEDBと混合した。12時間常温で攪拌して、反応を進行した。
【0154】
2. Encapsulation efficiency of 9R/siRNA in BPBcss-LS
9R peptide及びVEGF-C siRNA(sense 5' CAG AUG GAU UCC AUG ACA dTdT, anti-sense 5' AUG UCA UGG AAU CCA UGU G dTdT)をHEPES-buffered 5% glucose (pH 7.4)で希釈して、最終容量250μlになるようにして、室温で10分間インキュベーションした。前記溶液を混合してボルテキシングして、500μl 9R/siRNA complexes at a +/- charge ratio of 1:6を得た。同時にBPB-DSPEが内包されたリポソームを作るためのPOPC/Chol/POPG/PEG2000-DSPE/BPB-DSPE (4:3:3:1:0.5)mixtureと対照群リポソームのためのPOPC/Chol/POPG/PEG2000-DSPE (4:3:3:1)mixtureを利用してlipid filmを作る。これに500ul Hepes buffer glucose 5% (HBG 5%)を入れた後、lipid filmに500ul 9R/siRNA complexを入れた。この混合物を簡単にソニケーションした後、hand-held extruder (Avanti Polar Lipid, AL, USA)を利用して二つのpolycarbonate membranes (100 nm pore size)のスタックを通じて11回押出した。9R/siRNA-loaded BPB-リポソームs(BPBcss-LS)と対照群である9R/siRNA-loaded-リポソームsをsize-exclusion chromatography (CL-4B column)で精製した。Sepharose CL4B column eluentの分画をOliGreen (Invitrogen)で分析してsiRNA含量を分析した。
【0155】
3. 大きさ分布及びzeta-potentials
9R/siRNAがローディングされたBPBcss-LSをHBS(Hepes Buffer Saline)で希釈し、最適のscattering intensityを得た。Hydrodynamic diameter及びzeta potentialを、ELS 8000装置(Otsuka Electronics Korea, Seoul, South Korea)を利用してelectrophoretic light scattering方法で測定した。
【0156】
4. Uptake of BPBcss-LS in EDB positive and EDB negative cell line
細胞をcover glass coated with 2% gelatin上で一晩中培養した。24時間後、細胞に200ugのBPB-リポソーム(0.3% rhodomaime含み)と対照群の200ugのリポソーム(0.3% rhodamine)をそれぞれ処理した。37℃で1時間処理後、細胞を3回洗浄し、4%パラホルムアルデヒドparaformaldehydeで固定化した後、共焦点顕微鏡で観察した。
【0157】
5. siRNA transfection efficiency in MCF-7 cells in vitro
BPBcss-LSにencapsulatされたsiRNAのMCF-7 cell内へのtransfection効率を評価するために、MCF-7 cellを50nM VEGF-C siRNA in lipofectamine(positive control), 50 nM BPBcss-LS及び100 nM BPBcss-LSで形質転換させた。4時間経過後、細胞を24時間さらにインキュベーションして、次いでRNA分離、cDNA合成及び実時間RT-PCRを行った。
【0158】
6.実験結果
(1) BPB-リポソーム大きさ及びゼータ電位
BPBcss-LSの測定された大きさ及びゼータ電位は、下記表のようである。
【0159】
【表4】
【0160】
(2) BPB-リポソームにsiRNA内包
図13aから分かるように、BPB-リポソーム(CSS-LS)にsiRNAを内包したものとfree siRNAを除去するために、size exculsion CL-4Bカラムを利用して分離した。大きさの大きいsiRNA/BPB-リポソームが先に出て、その後free siRNAが出ることを観察することができる。この方法でsiRNAの内包された、精製されたBPB-リポソームを得ることができる。
【0161】
(3) BPB-リポソームの特異性
ターゲットタンパク質であるEDBが過発現されるU87MG, MCF-7, MCF-7/ADR細胞株を利用して、BPB-リポソームがよく認識するかを確認した。0.3%ロダミンの含まれたBPB-リポソームと対照群のリポソームを三つの細胞に一時間処理した後、洗浄して、顕微鏡で観察した。図13bから分かるように、BPB-リポソーム(CSS-LS)は、全ての細胞によく付着することを確認した。しかし、対照群であるBPBのないリポソーム(LS)は、EDB過発現細胞株に付着しなかった。これは、BPB-リポソームが EDBを細胞内で特異的に認識できることを示す結果である。
【0162】
また、EDB negative cellであるPC3とLnCapを利用し、上記と同様な方法でBPB-リポソームと対照群のリポソームを処理した時、両方ともそれぞれの細胞に付着しないことを図13cから確認できる。これは、ターゲットタンパク質であるEDBがないと、BPB-リポソームであっても付着しないことを示し、BPB-リポソームがEDBに非常に特異的であることを示す。
【0163】
(4) MCF-7細胞にBPB-LS/siRNA内包効能テスト
図13dから分かるように、VEGF-C siRNAを内包したBPB-LSが細胞内でVEGF-CのmRNAを抑制できるかをMCF-7細胞で確認した。50nMと100nM VEGF-C siRNAを内包したBPB-LSを細胞に処理した時、一般によく使用されるリポフェクタミンとほぼ等しくVEGF-C mRNAを抑制できることを示した。
【0164】
実施例22:Oleic SPION-BPB
1. Oleic SPION-BPBの製造
500ug/100ul(CHCl3) DSPE-PEG2000-Mal(Avanti Polar Lipids, Alabaster, AL, USA)(1,2-Distearoyl-sn-Glycero-3-Phosphoethanolamine-N-[Maleimide(Polyethylene Glycol)2000])とBPB(SSS) peptide(SSSPIQGSWTWENGK(Cys)WTWKGIIRLEQ) 250ug/100ul(DMSO)をCHCl3/DMSO co-solvent下で一晩中反応した。反応後、MALDI-TOF-MS(SHIMADZU Axima-CFR,SHIMADZE, Japen)を利用して反応を確認した。DSPE-PEG2000polymerとBPB conjugated DSPE-PEG2000 及びRhodamine-DSPEを(5:94:1)の比率でD.W.に入れた後、Oleic SPION(superparamagnetic iron oxide nanoparticle, 熱分解法で合成)をHexaneに溶かした後、上記のpolymer solutionに入れて、probeタイプのsonicatorを利用してsonicationした後、stirring方法とcentrifuge、そしてmagnetを利用して精製した。そして、BPBのないグループも上記の同様な方法で合成した。
【0165】
2.合成後、Size測定ELSとTEMを利用してBPB conjugatedされたDSPE-PEG2000 コーティングOleic SPION確認
electrophoretic light scattering(ELS 8000 instrument of Otsuka Electronics, Otsuka Electronics Korea, Seoul, South Korea)を利用してHydrodynamic sizeを測定し、BPB conjugate有無のサイズ変化を測定して、また、一定時間D.W.における安定性を確認し、transmission electron microscopy(Philips TECNAI F20 instrument (Philips ElectronicInstruments Corp., Mahwah, NJ))を利用してナノ粒子のaggregation程度を確認した。
【0166】
3. T2-weighted MRファントム研究を通じて合成したBPB conjugatedされたDSPE-PEG2000コーティング Oleic SPIONがMR造影剤として使用可能であるかの確認
BPB conjugated DSPE-PEG2000 coated oleic SPIONとDSPE-PEG2000 coated oleic SPIONをそれぞれ濃度別にe-tubeに入れて3T MRI scanner (Magnetom Avanto, Siemens, Germany)を利用してT2 Timeを測定し、r2値を計算した。
【0167】
4.細胞実験を利用してactive targeting確認
6 well plateにU87MG細胞を37℃ CO2 インキュベーターで培養密度90-100%に培養後、同じ濃度のBPB conjugated DSGPE-PEG2000 coated SPIONとDSGPE-PEG2000coated SPIONを培地に混ぜて30分間培養した後、細胞を集めて、3T MRI scanner (Magnetom Avanto, Siemens, Germany)を通じて信号強度の差を確認した。また、confocal microscope (Olympus, FV1000)を利用し、細胞におけるロダミン信号の差を確認した。
【0168】
5.細胞毒性実験
96ウェルプレートにU87MG細胞を37℃ CO2 インキュベーターで培養密度50%に培養後、同じ濃度のBPB conjugated DSPE-PEG2000 coated oleic SPIONとDSPE-PEG2000coated oleic SPIONを培地に混ぜて12時間培養した後、MTT assayを通じて細胞毒性有無を確認した。
【0169】
6.脳癌動物モデルを利用したactive targeting MR実験
Balb/c nude(female) 6 weeks miceの後ろ右足の上部にU87MG細胞5 x 106 個の細胞を移植し、2週間程度育てた後、癌の大きさが約100-150mm3 になった時、BPB conjugated DSPE-PEG2000coated oleic SPIONとDSPE-PEG2000 coated oleic SPIONを20mg Fe/kgずつ尻尾静脈に投与後、3T MRIを利用して癌部位の信号強度を時間別にモニターした。
【0170】
7. 実験結果
(1) SPION-BPBの製作
DSPE-PEG2000-MalとBPB(SSS)ペプチドのコンジュゲーションを、MALDIを利用して確認した(図14a)。
【0171】
DSPEを利用してSPIONを合成する場合、Hydrodynamic size(水溶液状態でsize測定)を測定すると、約30nm程度のsizeが出て、BPB(SSS peptide)をコンジュゲートしたDSPEを使用する場合、sizeが約20nm程度増加した。
【0172】
【表5】
【0173】
一週間ELSを利用してHydrodynamic size変化を観察したが、一週間ナノ粒子の大きさに大きい変化がなく、安定した状態であることを確認した(図14b)。TEMイメージを通じて、ナノ粒子(SPION)にBPBがコンジュゲートされた後にもよく分散されることを確認した。また、中心コアのサイズが約12nm程度になることを確認した(図14c)。
【0174】
(2) T2-weighted MRファントム研究を通じて、BPB conjugated DSPE-PEG2000 コーティングOleic SPIONがMR造影剤として使用可能であるかの確認
DSPE-PEG2000 coated oleic SPION及びBPB conjugated DSPE-PEG2000 coated oleic SPIONのいずれも、r2値は120 mM-1S-1であって、MRI T2造影剤として使用可能であることを確認した(図14d)。
【0175】
(3)細胞実験を利用して active targeting確認
細胞実験でBPBのあるグループとないグループ間のMRイメージ及び信号強度の差を確認した(図14e)。BPB conjugated DSPE-PEG2000 coated SPIONが細胞にさらに多くuptakeされて、MRイメージでさらに黒く表れて、またMR信号比較(ROI信号比較)においても、DSPE-PEG2000 coated SPIONよりさらに低い値を示した。
【0176】
Confocal顕微鏡を利用して、DSPE-PEG2000 coated SPIONとBPB conjugated DSPE-PEG2000 coated SPIONのロダミン信号の差を確認した(図14f)。BPBコンジュゲーションされたグループから細胞のロダミン信号がさらに強く表れた。
【0177】
(4)細胞毒性実験
MTT実験を通じて、DSPE-PEG2000 coated SPIONとBPB conjugated DSPE-PEG2000 coated SPIONの細胞毒性実験を行った結果、両グループとも低い細胞毒性を示した(図14g)。
【0178】
(5)脳癌動物モデルを利用したactive targeting MR実験
脳癌動物モデルにおいて、BPB conjugated DSPE-PEG2000coated SPIONグループがinjection 1時間後、癌部位が黒く表れることを確認した(図14h)。それに対し、BPB conjugateされなかったDSPE-PEG2000 coated SPIONグループでは、injectionの前と後において大きい変化が現れなかった。
【0179】
実施例23:Gold NPs-BPB
1. Pegylated Gold nanoparticleの製造
金イオンを、還元剤を使用し、約5nm大きさを有する金ナノ粒子を合成した。5 nm金ナノ粒子が生体内における安定性、即ちRES(reticuloendothelial system)などへのuptakeを減らして、目標細胞への伝達効率を増加させるために、金ナノ粒子表面を生体的合成高分子のPEGでコーティングが必要である。したがって、PEGにチオールグループを導入するために、ジチオールグループとカルボキシルグループが含まれたlipoic acidと反応させた。Lipoic acidにPEGを結合するために、まず、lipoic acidのカルボキシルグループをDCC(dicyclohexylcarbodiimide)とNHS (N-hydroxylsucciniimide)で活性化させた後、100ugのNH2-PEG-OCH3と反応させることによりlipoic-PEGを合成した。金ナノ粒子にPEGがコーティングされることを、1M NaClを添加してaggregationされるかを測定して、PEGが完璧にコーティングされることを測定した。
【0180】
2. Gold nanoparticle-BPBコンジュゲーション
合成されたlipoic-PEG-maleimdeとチオールグループが露出されたBPB(CSSpペプチド, CSSPIQGSWTWENGK(cys)WTWKGIIRLEQ)をCHCl3/DMSO(3:1) co-solvent下で一晩中反応した後、lipoic-PEG-maleimde-BPB利用してgold nanoparticleをコーティングした。
【0181】
3. Gold nanoparticle-BPBの癌細胞認識実験
BPB-金ナノ粒子がフィブロネクチンED-Bを認識するかどうか、フィブロネクチン ED-BがoverexpressionされるU87MG glioblastomaを利用して、癌細胞認識を確認した。Human U87MG glioblastoma cellを90-100% confluencyでcover slipに育てた後、BPB-金ナノ粒子を1時間37℃でcellとインキュベーションした。その後、PBSで3回洗浄した後、細胞をトリプシンで分離した後、細胞を集めてInductively coupled plasma mass spectrometry (ICP-MS)を利用し、細胞内Auの量を測定した。
【0182】
4. 金ナノ粒子に両性イオン(zwitterions)コーティング
金ナノ粒子は、既存の造影剤が有している短所(短い映像時間、腎臓毒性)を克服することができるが、金ナノ粒子も同様に大きさなどの問題によって、体外に排出されない問題点を有している。このような問題点を克服するために、PEGの代わりに、PEGのような役割をしながらもナノ粒子のhydrodynamicサイズを大きく増加させない両性イオンを金ナノ粒子にコーティングさせることにより、最終金ナノ粒子のhydrodynamic大きさを8nm以下に減らして、体外に排出できるようにナノ粒子をデザインした。また、本研究陳で使用した両性イオンは、金ナノ粒子と結合できる部分として、モノチオール(mono-thiol)グループではなく、環状ジチオール(cyclic dithiol)グループを導入することにより、血液内で金ナノ粒子と両性イオン間の安定性(stability)を増加させた。両性イオンの構造は、下記のようである。
【化1】
【0183】
次に、金ナノ粒子が血清(serum)と1M NaCl下で安定するために必要な両性イオンと金ナノ粒子間の最適の比率を、金ナノ粒子の表面プラズモン(surface Plasmon resonance)最大ピーク(peak)の変化を観察しながら測定した。まず、金ナノ粒子と両性イオン間の多様な比率(1:3000, 1:5000, 1:7000, 1:10000, 1:12000, 1:15000)を混ぜて、12時間反応後、遠心分離機を利用して分離した。10%血清と1M NaClを添加して、24時間が経過した後、表面プラズモン共鳴最大ピークをUV-Vis spectrumを利用して測定した。
【0184】
5. 金ナノ粒子にBPBを付着するためのBPB変形
金ナノ粒子とBPB間の結合力、即ち、血液内での安定性を増加させるために、BPBでも同様に環状ジチオール(cyclic dithiol)グループを導入した。即ち、lipoic acidをDCC(N,N`-Dicyclohexylcarbodiimide)/NHS(N-hydroxl succinimides)とTHF(tetrahydrofuran)で72時間反応し、lipoic-NHS形態を作って、トルエンによる再結晶方法で精製した。精製したlipoic-NHSは、N-末端基がアセチル化(acetylation)されたBPBのリジン(lysine)残基のアミン(NH2)グループとDMSO(Dimethyl sulfoxide)で2時間反応し、BPBに環状ジチオールグループを導入することができた。
【0185】
6.実験結果
(1) Gold Nanoparticle-BPBの製作
金ナノ粒子は、現在広く使用されているヨードベースのCT造影剤の短所である腎臓毒性、短い映像化時間などを克服できるだけではなく、肝癌造影が可能な生体適合性高分子でコーティングした金ナノ粒子を次世代CT造影剤として開発することができる。ここでは、癌標的BPBを付着することにより、CTで癌をイメージングできる金ナノ粒子を開発した。5nmの金ナノ粒子にフィブロネクチンEDBを認識するBPBをコンジュゲーションした。TEMイメージを通じて、ナノ粒子が、BPBがコンジュゲートされた後にもよく分散されているかを確認した(図15a)。
【0186】
(2) Gold Nanoparticle-BPBの癌細胞標的実験
U87MG細胞を利用して、BPB conjugated金ナノ粒子を同一な濃度として入れた後、ICP-MSを利用して細胞内のAuの量の差を測定した。BPBのあるグループが、BPBのないグループより細胞内Auの量が多いことが確認された。したがって、BPB conjugated金ナノ粒子の方がEDBによる細胞表面吸着と細胞内伝達にさらに優れていることを確認した(図15b)。また、BPBがコンジュゲーションされなかった金ナノ粒子よりBPBのコンジュゲーションされた金ナノ粒子の方が選択的にU87MG細胞に吸着することを、silver enhancement方法を利用して顕微鏡観察によっても確認することができた(図15c)。
【0187】
(3)金ナノ粒子の両性イオンコーティング
BPBを金ナノ粒子に付着した時、選択的に癌細胞に吸着されることを確認した後、金ナノ粒子が生体内に注入された時、血液内で安定するように、金ナノ粒子表面を両性イオンでコーティングした。金ナノ粒子と両性イオン間の比率が1:10000以上である時、金ナノ粒子の表面プラズモン共鳴最大ピークが、血清及び1M NaClを添加して24時間が経っても変化がないことから、血清及び1M NaClの環境においても金ナノ粒子が安定したことを確認することができた。
【0188】
(4)金ナノ粒子にBPBを付着するためのBPB変形
BPBに環状ジチオールグループを導入したLipoic-BPBをHPLCで分離し、MALDI-TOFを通じて合成物を確認することができた。
【0189】
実施例24:Drug-BPB conjugates
1. Docetaxel(DTX)-NHSの製造及び確認
DTXのヒドロキシル基をsuccinic anhydrideと常温で12時間反応し、DTX succinic acidを製造した。BPBのlysineが有しているfree amineにDTX succinic acidをコンジュゲーションさせるために、まずアミンに反応するNHSグループをDTX succinic acidに結合させた。反応条件は、DTX succinic acidをMC(methylene choloride)溶液に溶かして、EDC/NHSを加えて常温で12時間反応した。この際、モル比は、DTX succinic acide : EDC : NHS = 1 : 1.2 : 1.5として行った。反応後、移動相溶媒をEA(ethyl acetate)としてTLC(thin liquid chromatography)分析を行った。
【0190】
2. DTX-NHSとBPBコンジュゲーション
DTX-NHSをコンジュゲーション反応直前にDMFに溶かして、BPBを追加して溶かした。完全に溶けない場合、DMSOを少しずつ添加した。これにBPBのfree amine形成を助けるbase DIEA(di-isoethyl amine)を添加して、常温で12時間反応した。DIEAは、BPBの2当量以上入れた。BPB配列は、N-terminal acetylated BPBであって、配列は、SSSPIQGSWTWENGKWTWRGIIRLEQである。
【0191】
3. DTX-BPBの精製及び分析
DTX-NHSとBPBコンジュゲーション反応液を真空乾燥機で乾燥させた後、50% ACNに溶かしてHPLCで分析し、DTX-BPBコンジュゲートに該当するHPLCピークサンプルを、質量分析(MALD TOF)を行ってDTX-BPBコンジュゲート形成を確認した。
【0192】
以上、本発明の望ましい具現例を詳細に記述したが、当業界の通常の知識を有する者にとっては、このような具体的な記述はただ望ましい具現例に過ぎなく、これに本発明の範囲が限定されないことは明らかである。従って、本発明の実質的な範囲は、添付の請求項とその等価物により定義されると言える。
【技術分野】
【0001】
本発明は、BPBベースのカーゴ運搬システムに関する。
【背景技術】
【0002】
抗体は、B細胞が生産する血漿タンパク質の一種である免疫グロブリン蛋白質であって、外部から入った抗原の特定部位を特異的に認識して結合することにより、抗原を非活性化するか無力化させる。このような抗原−抗体反応の特異性と高度の親和度及び数千万種類の抗原を区別できる抗体の多様性を応用して、今日の診断剤と治療剤などを始めとした数多い種類の抗体製品が出現するようになった。現在FADでは、21個の単一クローン抗体を承認し、リツキシマブ(Rituximab)及びハーセプチン(Herceptin)のような抗体は、他の治療で全く反応を示さなかった患者の50%以上で効果を示し、実質的に様々な研究において、単一クローン抗体を利用してリンパ腫、大腸癌または乳癌などに成功的な臨床治療を示している。治療用抗体の全体市場規模は、2004年100億ドル規模から2010年には300億ドルに、年平均20%の成長率を示すと推定しており、その市場規模は、幾何級数的に増加すると推定されている。抗体を利用した新薬開発が活発になる理由は、薬品の開発期間が短く、投資費用が少なくて、副作用が容易に予測可能であるからである。また、抗体は生薬であるため、人体に影響がほとんどなく、体内における半減期が低分子量薬品に比べ圧倒的に長くて、患者に親和的である。このような有用性にもかかわらず、人間において単一クローン抗体は、外来抗原と認識され、酷いアレルギー反応または過敏反応を起こしたりもする。また、このような抗癌機能の単クローン抗体を臨床的に使用する場合、生産コストが高いため、治療剤としての値段が急激に上昇するという短所があり、抗体を培養する方法及び精製方法など、広範囲な分野の技術が各種知的所有権により保護されているため、高いライセンシング費を支払わなければならない。
【0003】
したがって、この問題を解決するために、米国を中心にヨーロッパ連合で、抗体代替蛋白質の開発が胎動期にある。抗体代替蛋白質は、抗体のように不変領域と可変領域を持てるように作った組み換え蛋白質であって、大きさが小さくて安定した蛋白質の一定部分を無作為配列のアミノ酸に変えてライブラリーを作り、これを標的物質に対してスクリーニングをして、高い親和力と良い特異性を有した物質を見つけることができる。例えば、抗体代替蛋白質の中、アビマー(avimer)とアフィボディ(affibody)は、標的物質に対してピコモル(picomole)程度の親和力を有した例示が報告されている。このような抗体代替蛋白質は、大きさが小さくて安定しているため、癌細胞に深く浸透可能であり、一般に免疫反応が少ないと報告されている。そして何よりも、広範囲な抗体特許問題から放れることができ、バクテリアから容易に大量精製できるため、生産コストが低くて、経済的に抗体より大きい長所を有する。現在開発された抗体代替蛋白質は40個があるが、この中、ベンチャー会社や多国的製薬会社で商用化を試みている抗体代替蛋白質は、フィブロネクチンタイプIIIドメイン、リポカリン、LDLR-Aドメイン、クリスタリン、プロテインA、アンキリンリピート(Ankyrin repeat)、BPTIという蛋白質を利用しており、ターゲットに対するピコモルで数ナノモル程度の高い親和力を有している。その中、アドネクチン(adnectin)、アビマー、クニッツ(Kunitz)ドメインは、現在FDA臨床実験が進行中である。
【0004】
本発明は、今までの蛋白質を利用した抗体代替蛋白質とは異なるペプチド基盤抗体代替蛋白質に焦点を合わせた。ペプチドは、抗体に比べ、適切な薬物動力学、大量生産性、低い毒性、抗原性抑制及び低い生産単価などにより、現在抗体治療剤に代わって多様に活用されている。治療用薬としてのペプチドの長所は、生産単価が低く、安全生及び反応性が高くて、特許ロイヤルティが相対的に低くて、望まない免疫システムに露出が少なく、ペプチド自体に対する抗体生産を抑制することができて、合成による変形が容易で正確であるということである。しかしながら、大部分のペプチドは、抗体に比べ、特定蛋白質ターゲットに対して低い親和力及び特異性を示すため、多様な応用分野に使用できないという短所がある。したがって、ペプチドの短所を克服できる新しいペプチド基盤抗体代替蛋白質の開発に対する要求が当業界に台頭されている。したがって、本発明者らは、生物学的ターゲット分子に高い親和性で特異的結合が可能なペプチド物質を開発するために鋭意研究した。これは、現在非常に多いターゲットに対して報告された、低い親和力を有したペプチドを利用して、速い時間内に高い親和性及び特異性を有した新薬候補を製造できる技術になると期待される。
【0005】
本明細書全体にかけて多数の特許文献及び論文が参照されて、その引用が表示されている。引用された特許文献及び論文の開示内容は、その全体が本明細書に参照として取り込まれ、本発明の属する技術分野の水準及び本発明の内容がより明確に説明される。
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明者らは、ターゲット結合性及び特異性に基づいて、多様な物質を細胞内または細胞表面に運搬できるシステムを開発するために鋭意研究した。その結果、比較的リジッド(rigid)なペプチド骨格を有する構造安定化部位の両末端に無作為的(random)にペプチドを結合させて、この二つのペプチドを共同にターゲット分子に結合させる場合は、大きく増加された結合能及び特異性を有する二座ペプチドバインダー(BPB)を得ることができ、このBPBに運搬対象のカーゴを結合させる場合は、BPBのターゲット結合性及び特異性に基づいて、多様な物質を細胞表面または細胞内に運搬できることを確認することにより、本発明を完成した。
【0007】
したがって、本発明の目的は、BPBベースのカーゴ運搬システムを提供することにある。
【0008】
本発明の他の目的及び利点は、発明の詳細な説明及び請求の範囲及び図面により、さらに明確にされる。
【課題を解決するための手段】
【0009】
本発明の一様態によると、本発明は、(a)(i)鎖間(interstrand)非共有結合が形成されたパラレル(parallel)、アンチパラレル(antiparallel)、またはパラレル(parallel)とアンチパラレル(antiparallel)アミノ酸鎖を含む構造安定化部位(structure stabilizing region)と、(ii)前記構造安定化部位の両末端に結合されており、無作為的に選択されたそれぞれn及びm個のアミノ酸を含むターゲット結合部位I(target binding region I)及びターゲット結合部位II(target binding region II)と、を含む二座ペプチドバインダー(Bipodal Peptide Binder:BPB)と、(b)前記二座ペプチドバインダーに結合されたカーゴと、を含むBPBベースのカーゴ運搬システム(Cargo Delivery System)を提供する。
【0010】
本発明の一様態によると、本発明は、(a)(i)鎖間(interstrand)非共有結合が形成されたパラレル(parallel)、アンチパラレル(antiparallel)、またはパラレル(parallel)とアンチパラレル(antiparallel)アミノ酸鎖を含む構造安定化部位(structure stabilizing region)と、(ii)前記構造安定化部位の両末端に結合されており、無作為的に選択されたそれぞれn及びm個のアミノ酸を含むターゲット結合部位I(target binding region I)及びターゲット結合部位II(target binding region II)と、を含む二座ペプチドバインダー(Bipodal Peptide Binder:BPB)と、(b)前記二座ペプチドバインダーに結合されたカーゴと、を含むBPBベースのカーゴ運搬システム(Cargo Delivery System)を、個体、組織または細胞に処理する段階を含むカーゴを運搬する方法を提供する。
【0011】
本発明者らは、ターゲット結合性及び特異性に基づいて、多様な物質を細胞内または細胞表面に運搬できるシステムを開発するために鋭意研究した。その結果、比較的リジッド(rigid)なペプチド骨格を有する構造安定化部位の両末端に無作為的(random)にペプチドを結合させて、この二つのペプチドを共同にターゲット分子に結合させる場合は、大きく増加された結合能及び特異性を有する二座ペプチドバインダー(BPB)を得ることができ、このBPBに運搬対象のカーゴを結合させる場合は、BPBのターゲット結合性及び特異性に基づいて、多様な物質を細胞表面または細胞内に運搬できることを確認した。
【0012】
本発明の基本的な戦略は、リジッドなペプチド骨格の両末端に、ターゲットに結合されるペプチドを連結することである。この場合、リジッドなペプチド骨格は、二座ペプチドバインダーの全体的な構造を安定化させる作用をして、ターゲット結合部位I及びターゲット結合部位IIがターゲット分子に結合されることを強化させる。
【0013】
本発明で利用可能な構造安定化部位は、パラレル、アンチパラレル、またはパラレルとアンチパラレルアミノ酸鎖を含み、鎖間(interstrand)水素結合、静電気的相互作用、疎水性相互作用、ファンデルワールス相互作用、パイ−パイ相互作用、陽イオン−パイ相互作用、またはこれらの組み合わせによる非共有結合が形成される蛋白質構造モチーフを含む。鎖間水素結合、静電気的相互作用、疎水性相互作用、ファンデルワールス相互作用、パイ−パイ相互作用、陽イオン−パイ相互作用、またはこれらの組み合わせにより形成される非共有結合は、構造安定化部位の堅固性(rigidity)に寄与する。
【0014】
本発明の好ましい具現例によると、構造安定化部位における鎖間(interstrand)非供給結合は、水素結合、疎水性相互作用、ファンデルワールス相互作用、パイ−パイ相互作用、またはこれらの組み合わせを含む。
【0015】
選択的に、構造化安定化部位に共有結合があり得る。例えば、構造化安定化部位に二硫化結合を形成させて、構造安定化部位の堅固性をさらに増加させることができる。このような共有結合による堅固性の増加は、二座ペプチドバインダーのターゲットに対する特異度及び親和度を考慮して付与する。
【0016】
本発明の好ましい具現例によると、構造安定化部位のアミノ酸鎖は、リンカーで連結されている。本明細書で鎖を言及しながら使用される用語‘リンカー’は、鎖と鎖を連結する物質を意味する。例えば、構造安定化部位としてβ−ヘアピンが利用される場合は、β−ヘアピンにあるターン配列がリンカーの役割をして、ロイシンジッパーが利用される場合は、ロイシンジッパーの二つのC-末端を連結する物質(例えば、ペプチドリンカー)がリンカーの役割をする。
【0017】
リンカーは、パラレル、アンチパラレル、またはパラレルとアンチパラレルアミノ酸鎖を連結する。例えば、パラレル方式で整列された少なくとも二つの鎖(好ましくは二つの鎖)、アンチパラレル方式で整列された少なくとも二つの鎖(好ましくは二つの鎖)、パラレル及びアンチパラレル方式で整列された少なくとも三つの鎖(好ましくは三つの鎖)をリンカーが連結するようになる。
【0018】
本発明の好ましい具現例によると、リンカーは、ターン配列またはペプチドリンカーである。
【0019】
本発明の好ましい具現例によると、前記ターン配列は、β−ターン、γ−ターン、α−ターン、π−ターンまたはω−loopである(Venkatachalam CM (1968), Biopolymers, 6, 1425-1436; Nemethy G and Printz MP. (1972), Macromolecules, 5, 755-758; Lewis PN et al., (1973), Biochim. Biophys. Acta, 303, 211-229; Toniolo C. (1980) CRC Crit. Rev. Biochem., 9, 1-44; Richardson JS. (1981), Adv. Protein Chem., 34, 167-339; Rose GD et al., (1985), Adv. Protein Chem., 37, 1-109; Milner-White EJ and Poet R. (1987), TIBS, 12, 189-192; Wilmot CM and Thornton JM. (1988), J. Mol. Biol., 203, 221-232; Milner-White EJ. (1990), J. Mol. Biol., 216, 385-397; Pavone V et al. (1996), Biopolymers, 38, 705-721; Rajashankar KR and Ramakumar S. (1996), Protein Sci., 5, 932-946)。最も好ましくは、本発明で利用されるターン配列は、β−ターンである。
【0020】
ターン配列としてβ−ターンが利用される場合、好ましくは、タイプI、タイプI’、タイプII、タイプII’、タイプIIIまたはタイプIII’ターン配列であり、より好ましくは、タイプI、タイプI’、タイプII、タイプII’ターン配列であり、さらに好ましくは、タイプI’、タイプII’ターン配列であって、最も好ましくは、タイプI’ターン配列である(B. L. Sibanda et al., J. Mol. Biol., 1989, 206, 4, 759-777; B. L. Sibanda et al., Methods Enzymol., 1991, 202, 59-82)。
【0021】
本発明の他の好ましい具現例によると、本発明でターン配列として利用できるものは、H. Jane Dyson et al., Eur. J. Biochem. 255:462-471(1998)に開示されており、前記文献は、本明細書に参照として取り込まれる。ターン配列として利用できるものは、次のアミノ酸配列を含む:X-Pro-Gly-Glu-Val; Ala-X-Gly-Glu-Val(Xは、20個のアミノ酸から選択される)。
【0022】
本発明の一具現例によって、構造安定化部位としてβ−シートまたはロイシンジッパーが利用される場合、パラレル方式で整列された二つの鎖またはアンチパラレル方式で整列された二つの鎖をペプチドリンカーが連結することが好ましい。
【0023】
ペプチドリンカーは、当業界に公知された如何なるものでも利用可能である。適したペプチドリンカーの配列は、下記のような要素を考慮して選択することができる:(a)柔軟な延長されたコンフォメーション(flexible extended conformation)に適用できる能力;(b)生物学的ターゲット物質と相互作用する二次構造を生成しない能力;及び(c)生物学的ターゲット分子と相互作用する疎水性残基または電荷を有する残基の不在。好ましいペプチドリンカーは、Gly、Asn及びSer残基を含む。Thr及びAlaのような他の中性アミノ酸もリンカー配列に含まれ得る。リンカーに適したアミノ酸配列は、Maratea et al., Gene 40:39-46(1985); Murphy et al., Proc. Natl. Acad Sci. USA 83:8258-8562(1986); 米国特許第4,935,233号、第4,751,180号及び第5,990,275号に開示されている。ペプチドリンカー配列は、1〜50アミノ酸残基で構成できる。
【0024】
本発明の好ましい具現例によると、構造安定化部位は、ヘアピン、β−ヘアピン、β−ターン、リンカーで連結されたβ−シートまたはリンカーで連結されたロイシンジッパーであり、より好ましくは、構造安定化部位は、β−ヘアピンまたはリンカーで連結されたβ−シートであって、最も好ましくは、β−ヘアピンである。
【0025】
本明細書で用語‘β−ヘアピン’は、二つのβ鎖を含む最も簡単な蛋白質モチーフを意味し、この二つのβ鎖は、互いにアンチパラレルな整列を示す。このβ−ヘアピンにおいて、二つのβ鎖は、一般にターン配列により連結される。
【0026】
好ましくは、β−ヘアピンに適用されるターン配列は、タイプI、タイプI’、タイプII、タイプII’、タイプIIIまたはタイプIII’ターン配列であり、より好ましくは、タイプI、タイプI’、タイプII、タイプII’ターン配列であり、さらに好ましくは、タイプI’、タイプII’ターン配列であって、最も好ましくは、タイプI’ターン配列である。また、X-Pro-Gly-Glu-Val;またはAla-X-Gly-Glu-Val(Xは、20個のアミノ酸から選択される)で表されるターン配列もβ−ヘアピンに利用できる。
【0027】
本発明の例示的な実施例によると、タイプIターン配列は、Asp-Asp-Ala-Thr-Lys-Thrであり、タイプI'ターン配列は、Glu-Asn-Gly-Lysであって、タイプIIターン配列は、X-Pro-Gly-Glu-Val;またはAla-X-Gly-Glu-Val(Xは、20個のアミノ酸から選択される)であり、タイプII'ターン配列は、Glu-Gly-Asn-LysまたはGlu-D-Pro-Asn-Lysである。
【0028】
β-ヘアピンコンフォメーションを有するペプチドは、当業界によく知られている。例えば、米国特許第6,914,123号及びAndrea G. Cochran et al., PNAS, 98(10):5578-5583)に開示されているトリプトファンジッパー、WO 2005/047503に開示されている鋳型-固定されたβ-ヘアピンミメティック、米国特許第5,807,979号に開示されているβ-ヘアピン変形体がよく知られている。その他にも、β-ヘアピンコンフォメーションを有するペプチドは、Smith & Regan (1995) Science 270:980-982; Chou & Fassman (1978) Annu. Rev. Biochem. 47:251-276; Kim & Berg (1993) Nature 362:267-270; Minor & Kim (1994) Nature 367:660-663; Minor & Kim (1993) Nature 371:264-267; Smith et al. Biochemistry (1994) 33:5510-5517; Searle et al. (1995) Nat. Struct. Biol. 2:999-1006; Haque & Gellman (1997) J. Am. Chem. Soc. 119:2303-2304; Blanco et al. (1993) J. Am. Chem. Soc. 115:5887-5888; de Alba et al. (1996) Fold. Des. 1: 133-144; de Alba et al. (1997) Protein Sci. 6:2548-2560; Ramirez-Alvarado et al. (1996) Nat. Struct. Biol. 3:604-612; Stanger & Gellman (1998) J. Am. Chem. Soc. 120:4236-4237; Maynard & Searle (1997) Chem. Commun. 1297-1298; Griffiths-Jones et al. (1998) Chem. Commun. 789-790; Maynard et al. (1998) J. Am. Chem. Soc. 120:1996-2007; 及びBlanco et al. (1994) Nat. Struct. Biol. 1:584-590に開示されており、前記文献は、本明細書に参照として取り込まれる。
【0029】
β-ヘアピンコンフォメーションを有するペプチドを構造安定化部位として利用する場合、最も好ましくは、トリプトファンジッパーを利用する。
【0030】
本発明の好ましい具現例によると、本発明で利用されるトリプトファンジッパーは、下記一般式Iで表される:
[一般式I]
X1-Trp(X2)X3-X4-X5(X'2)X6-X7
【0031】
X1は、SerまたはGly-Gluであり、X2及びX'2は、それぞれ独立してThr、His、Val、Ile、PheまたはTyrであり、X3は、TrpまたはTyrであり、X4は、タイプI、タイプI'、タイプII、タイプII'またはタイプIIIまたはタイプIII'ターン配列であり、X5は、TrpまたはPheであり、X6は、TrpまたはValであって、X7は、LysまたはThr-Gluである。
【0032】
より好ましくは、前記一般式Iにおいて、X1は、SerまたはGly-Gluであり、X2及びX'2は、それぞれ独立してThr、HisまたはValであり、X3は、TrpまたはTyrであり、X4は、タイプI、タイプI'、タイプIIまたはタイプII'ターン配列であり、X5は、TrpまたはPheであり、X6は、TrpまたはValであって、X7は、LysまたはThr-Gluである。
【0033】
さらに好ましくは、一般式Iにおいて、X1は、SerまたはGly-Gluであり、X2及びX'2は、それぞれ独立してThr、HisまたはValであり、X3は、Trpであり、X4は、タイプI、タイプI'、タイプIIまたはタイプII'ターン配列であり、X5は、Trpであり、X6は、Trpであって、X7は、LysまたはThr-Gluである。
【0034】
よりさらに好ましくは、一般式Iにおいて、X1は、Serであり、X2及びX'2は、Thrであり、X3は、Trpであり、X4は、タイプI'またはタイプII'ターン配列であり、X5は、Trpであり、X6は、Trpであって、X7は、Lysである。
【0035】
最も好ましくは、一般式Iにおいて、X1は、Serであり、X2及びX'2は、Thrであり、X3は、Trpであり、X4は、タイプI'ターン配列(ENGK)またはタイプII’ターン配列(EGNK)であり、X5は、Trpであり、X6は、Trpであって、X7は、Lysである。
【0036】
本発明に適したトリプトファンジッパーの例示的なアミノ酸配列は、配列番号1乃至3及び5乃至10に記載されている。
【0037】
本発明で構造安定化部位として利用可能なβ-ヘアピンペプチドは、蛋白質GのB1ドメイン由来のペプチド、即ち、GB1ペプチドである。
【0038】
本発明でGB1ペプチドが利用される場合、好ましくは、構造安定化部位は、下記一般式IIで表される:
[一般式II]
X1-Trp-X2-Tyr-X3-Phe-Thr-Val-X4
【0039】
X1は、Arg、Gly-GluまたはLys-Lysであり、X2は、GlnまたはThrであり、X3は、タイプI、タイプI'、タイプII、タイプII'またはタイプIIIまたはタイプIII'ターン配列であって、X4は、Gln、Thr-GluまたはGln-Gluである。
【0040】
より好ましくは、一般式IIの構造安定化部位は、下記一般式II’で表される:
[一般式II’]
X1-Trp-Thr-Tyr-X2-Phe-Thr-Val-X3
【0041】
X1は、Gly-GluまたはLys-Lysであり、X2は、タイプI、タイプI'、タイプII、タイプII'またはタイプIIIまたはタイプIII'ターン配列であり、X3は、Thr-GluまたはGln-Gluである。
【0042】
本発明に適したGB1β-ヘアピンの例示的なアミノ酸配列は、配列番号4及び14乃至15に記載されている。
【0043】
本発明で構造安定化部位として利用可能なβ-ヘアピンペプチドは、HPペプチドである。本発明でHPペプチドが利用される場合、好ましくは、構造安定化部位は、下記一般式IIIで表される:
[一般式III]
X1-X2-X3-Trp-X4-X5-Thr-X6-X7
【0044】
X1は、LysまたはLys-Lysであり、X2は、TrpまたはTyrであり、X3は、ValまたはThrであり、X4は、タイプI、タイプI'、タイプII、タイプII'またはタイプIIIまたはタイプIII'ターン配列であり、X5は、TrpまたはAlaであり、X6は、TrpまたはValであって、X7は、GluまたはGln-Gluである。
【0045】
本発明で構造安定化部位として利用可能なまた他のβ-ヘアピンペプチドは、下記一般式IVで表される:
[一般式IV]
X1-X2-X3-Trp-X4
【0046】
X1は、Lys-ThrまたはGlyであり、X2は、TrpまたはTyrであり、X3は、タイプI、タイプI'、タイプII、タイプII'またはタイプIIIまたはタイプIII'ターン配列であって、X4は、Thr-GluまたはGlyである。
【0047】
一般式III及びIVのβ-ヘアピンの例示的なアミノ酸配列は、配列番号11乃至12、15及び16乃至19に記載されている。
【0048】
本発明によると、構造安定化部位として、ヘアピン(α−ヘアピン、β−ヘアピン、γ−ヘアピン、p−ヘアピンなど)が利用できる。また、本発明によると、構造安定化部位としてβ−ターンが利用できる。
【0049】
本発明によると、構造安定化部位として、リンカーで連結されたβ-シートを利用することができる。β-シート構造において、パラレルまたはアンチパラレルな、好ましくは、アンチパラレルな二つのアミノ酸鎖が伸びた構造(extended form)からなっており、アミノ酸鎖間に水素結合が形成される。
【0050】
β-シート構造において、二つのアミノ酸鎖の隣接した二つの末端は、リンカーにより連結される。リンカーとしては、上述の多様なターン配列またはペプチドリンカーが利用できる。ターン配列がリンカーとして利用される場合、β-ターン配列が最も好ましい。
【0051】
本発明の他の変形例によると、構造安定化部位としてロイシンジッパーまたはリンカーで連結されたロイシンジッパーが利用できる。ロイシンジッパーは、パラレルな二つのα-鎖のダイマー化を引き起こす保存性ペプチドドメインであり、一般に、遺伝子発現に関与する蛋白質に発見されるダイマー化ドメインである("Leucine scissors". Glossary of Biochemistry and Molecular Biology (Revised). (1997). Ed. David M. Glick. London: Portland Press; Landschulz WH, et al. (1988) Science 240:1759-1764)。ロイシンジッパーは、一般にヘプタッド(heptad)反復配列を含み、ロイシン残基が4番目または5番目に位置している。例えば、本発明に利用できるロイシンジッパーは、LEALKEK, LKALEKE, LKKLVGE, LEDKVEE, LENEVARまたはLLSKNYHのアミノ酸配列を含む。本発明で利用されるロイシンジッパーの具体的な例は、配列番号39に記載されている。ロイシンジッパーのそれぞれの半分は、短いα-鎖からなっており、α-鎖間の直接的なロイシン接触がある。転写因子にあるロイシンジッパーは、一般に疎水性ロイシンジッパー部位及び塩基性部位(DNA分子の主グルーブと相互作用する部位)からなっている。本発明でロイシンジッパーが利用される場合は、塩基性部位は必ずしも必要なわけではない。ロイシンジッパー構造において、二つのアミノ酸鎖(即ち、二つのα-鎖)の隣接した二つの末端は、リンカーにより連結できる。リンカーとしては、上述の多様なターン配列またはペプチドリンカーが利用でき、好ましくは、ロイシンジッパーの構造に影響を及ぼさないペプチドリンカーが利用される。
【0052】
上述の構造安定化部位の両末端には、無作為アミノ酸配列が結合される。前記無作為アミノ酸配列がターゲット結合部位I及びターゲット結合部位IIを形成する。本発明の最も大きい特徴の一つは、構造安定化部位の両側末端にターゲット結合部位I及びターゲット結合部位IIを連結して、二座方式でペプチドバインダーを製作することである。ターゲット結合部位I及びターゲット結合部位IIは、互いに協同的に(cooperatively)ターゲットに結合することにより、ターゲットに対する親和度を大きく増加させる。
【0053】
ターゲット結合部位Iのアミノ酸数nは、特に制限されず、好ましくは2〜100の整数、より好ましくは2〜50の整数、さらに好ましくは、2〜20の整数、最も好ましくは、3〜10の整数である。
【0054】
ターゲット結合部位IIのアミノ酸数mは、特に制限されず、好ましくは2〜100の整数、より好ましくは2〜50の整数、さらに好ましくは、2〜20の整数、最も好ましくは、3〜10の整数である。
【0055】
ターゲット結合部位I及びターゲット結合部位IIには、それぞれ異なるまたは同一な数のアミノ酸残基が含まれる。ターゲット結合部位I及びターゲット結合部位IIには、それぞれ異なるまたは同一なアミノ酸配列が含まれて、好ましくは、それぞれ異なるアミノ酸配列が含まれる。
【0056】
ターゲット結合部位I及び/またはターゲット結合部位IIに含まれるアミノ酸配列は、線形のアミノ酸配列または環形のアミノ酸配列である。ターゲット結合部位のペプチド配列の安定性を増加させるために、ターゲット結合部位I及び/またはターゲット結合部位IIに含まれるアミノ酸配列の少なくとも一つのアミノ酸残基は、アセチル基、フルオレニルメトキシカルボニル基、ホルミル基、パルミトイル基、ミリスチル基、ステアリル基またはポリエチレングリコール(PEG)に変形できる。
【0057】
生物学的ターゲット分子に結合される本発明の二座ペプチドバインダーは、生体内生理学的反応の調節、生体内物質の検出、インビボ分子イメージング、インビトロ細胞イメージング及び薬物伝達用ターゲッティングをする際に利用でき、エスコート分子としても利用できる。
【0058】
本発明の好ましい具現例によると、構造安定化部位、ターゲット結合部位Iまたはターゲット結合部位II(より好ましくは、構造安定化部位、さらに好ましくは、構造安定化部位のリンカー)にカーゴが結合されている。前記カーゴの例は、請求項1において、前記カーゴは、化合物、化学薬物、バイオ薬物、無機粒子、ナノ粒子、タンパク質、ペプチド、核酸分子、脂質、炭水化物、リポソームまたは検出可能な信号を発生させるラベル分子を含むが、これらに限定されるものではない。
【0059】
前記検出可能な信号を発生させるラベルは、T1造影物質(例えば、Gdキレート化合物)、T2造影物質(例えば、超常磁性物質(例えば、マグネタイト、Fe3O4、γ-Fe2O3、マンガンフェライト、コバルトフェライト及びニッケルフェライト))、放射性同位元素(例えば、11C,15O, 13N, P32, S35, 44Sc, 45Ti, 118I, 136La, 198Tl, 200Tl, 205Bi及び206Bi)、蛍光物質(フルオレセイン(fluorescein)、フィコエリスリン(phycoerythrin)、ロダミン、リサミン(lissamine)、そしてCy3とCy5)、化学発光団、磁気粒子、マス標識または電子密集粒子を含むが、これらに制限されるものではない。
【0060】
前記化学薬物は、例えば、抗炎症剤、鎮痛剤、抗関節炎剤、鎮痙剤、抗鬱剤、抗精神病薬物、神経安定剤、抗不安剤、麻薬拮抗剤、抗パーキンソン疾患、コリン性アゴニスト、抗癌剤、抗血管新生抑制剤、免疫抑制剤、抗ウイルス剤、抗生剤、食欲抑制剤、鎮痛剤、抗コリン剤、抗ヒスタミン剤、抗片頭痛剤、ホルモン剤、冠状血管、脳血管または末梢血管拡張剤、避妊薬、抗血栓剤、利尿剤、抗高血圧剤、心血管疾患治療剤、美容成分(例えば、シワ改善剤、皮膚老化抑制剤及び皮膚美白剤)などを含むが、これらに限定されるものではない。
【0061】
前記バイオ薬物は、インシュリン、IGF-1(insulin-like growth factor 1)、成長ホルモン、エリスロポエチン、G-CSFs (granulocyte-colony stimulating factors)、GM-CSFs (granulocyte/macrophage-colony stimulating factors)、インターフェロンアルファ、インターフェロンベータ、インターフェロンガンマ、インターロイキン-1アルファ及びベータ、インターロイキン-3、インターロイキン-4、インターロイキン-6、インターロイキン-2、EGFs(epidermal growth factors)、カルシトニン(calcitonin)、ACTH(adrenocorticotropic hormone)、TNF(tumor necrosis factor)、アトビスバン(atobisban)、ブセレリン(buserelin)、セトロレリックス(cetrorelix)、デスロレリン(deslorelin)、デスモプレシン(desmopressin)、ジノルフィンA (dynorphin A) (1-13)、エルカトニン(elcatonin)、エレイドシン(eleidosin)、エプチフィバチド(eptifibatide)、GHRH-II(growth hormone releasing hormone-II)、ゴナドレリン(gonadorelin)、ゴセレリン(goserelin)、ヒストレリン(histrelin)、リュプロレリン(leuprorelin)、リプレシン(lypressin)、オクトレオチド(octreotide)、オキシトシン(oxytocin)、ピトレシン(pitressin)、セクレチン(secretin)、シンカリド(sincalide)、テルリプレシン(terlipressin)、チモペンチン(thymopentin)、チモシン(thymosine)α1、トリプトレリン(triptorelin)、ビバリルジン(bivalirudin)、カルベトシン(carbetocin)、シクロスポリン、エキセジン(exedine)、ランレオチド(lanreotide)、LHRH(luteinizing hormone-releasing hormone)、ナファレリン(nafarelin)、副甲状腺ホルモン、プラムリンチド(pramlintide)、T-20(enfuvirtide)、チマルファシン(thymalfasin)、ジコノチド、RNA、DNA、cDNA、アンチセンスオリゴヌクレオチド及びsiRNAなどがあるが、これらに限定されるものではない。
【0062】
ターゲット結合部位I及び/またはターゲット結合部位IIは、多様なターゲットに結合するアミノ酸配列を含むことができる。本発明の二座ペプチドバインダーによりターゲッティングできるものは、生化学物質、ペプチド、ポリペプチド、核酸、炭水化物、脂質、細胞及び組織のような生物学的ターゲット、化合物、金属または非金属物質を含み、好ましくは、生物学的ターゲットである。
【0063】
ターゲット結合部位が結合する生物学的ターゲットは、好ましくは、生化学物質、ペプチド、ポリペプチド、糖蛋白質、核酸、炭水化物、プロテオグリカン、脂質または糖脂質である。
【0064】
例えば、ターゲット結合部位が結合する生化学物質は、多様な生体内代謝産物(例えば、ATP、NADH、NADPH、炭水化物代謝産物、脂質代謝産物及びアミノ酸代謝産物)を含む。
【0065】
ターゲット結合部位が結合する例示的なペプチドまたはポリペプチドは、酵素、リガンド、受容体、バイオマーカー、ホルモン、転写因子、成長因子、免疫グロブリン、信号伝達蛋白質、結合蛋白質、イオンチャンネル、抗原、付着蛋白質、構造蛋白質、調節蛋白質、毒素蛋白質、サイトカイン及び血液凝固因子を含むが、これらに限定されるものではない。より詳細には、二座ペプチドバインダーのターゲットは、フィブロネクチン extra domain B(ED-B), VEGF(vascular endothelial growth factor), VEGFR(vascular endothelial growth factor receptor), VCAM1(vascular cell adhesion molecule-1), nAchR(Nicotinic acetylcholine receptor), HSA(Human serum albumin), MyD88, EGFR(Epidermal Growth Factor Receptor), HER2/neu, CD20, CD33, CD52, EpCAM (Epithelial Cell Adhesion Molecule), TNF-α(Tumor Necrosis Factor-α), IgE (Immunoglobulin E), CD11A (α-chain of lymphocyte function-associated antigen 1), CD3, CD25, Glycoprotein IIb/IIIa, インテグリン, AFP(Alpha-fetoprotein), β2M (Beta2-microglobulin), BTA(Bladder Tumor Antigens), NMP22, Cancer Antigen 125, Cancer Antigen 15-3, カルシトニン, Carcinoembryonic Antigen, Chromogranin A,エストロゲン受容体、プロゲステロン受容体, Human Chorionic Gonadotropin, Neuron-Specific Enolase, PSA(Prostate-Specific Antigen), PAP(Prostatic Acid Phosphatase)及びThyroglobulinを含むが、これらに限定されるものではない。
【0066】
ターゲット結合部位が結合する例示的な核酸分子は、gDNA, mRNA, cDNA, rRNA(ribosomal RNA), rDNA(ribosomal DNA)及びtRNAを含むが、これらに限定されるものではない。ターゲット結合部位が結合する例示的な炭水化物は、生体内炭水化物であって、単糖類、二糖類、三糖類及び多糖類を含むが、これらに限定されるものではない。ターゲット結合部位が結合する例示的な脂質は、脂肪酸、トリアシルグリセロール、スフィンゴ脂質、ガングリオシド及びコレステロールを含むが、これらに限定されるものではない。
【0067】
本発明の二座ペプチドバインダーは、細胞表面に露出した生体分子(例えば、蛋白質)に結合することもできるが、細胞内生体分子(例えば、蛋白質)にも結合し、生体分子の活性を調節することができる。
【0068】
二座ペプチドバインダーが細胞内蛋白質をターゲッティングする場合、好ましくは、二座ペプチドバインダーは、追加的に細胞膜透過ペプチド(CPP)を含む。
【0069】
前記CPPは、当業界に公知された多様なCPPを含み、例えば、HIV-1 Tat蛋白質、オリゴアルギニン、ANTPペプチド、HSV VP22転写調節蛋白質、vFGF由来のMTSペプチド、Penetratin、Transportan、Pep-1ペプチド、Pep-7ペプチド、Buforin II、MAP(Model Amphiphatic Peptide)、k-FGF、Ku 70、pVEC、SynB1またはHN-1を含むが、これらに限定されるものではない。前記CPPを二座ペプチドに結合させる方法は、多様な方法があって、例えば、二座ペプチドの構造安定化部位にあるループ部分のリジン残基とCPPを共有結合させる。
【0070】
細胞内には、生理活性に重要な役割をするたくさんのターゲット蛋白質があり、CPPが結合された二座ペプチドバインダーは、細胞内に流入されて、このターゲット蛋白質に結合して活性を調節(例えば、抑制)する。
【0071】
上述のように、本発明の二座ペプチドバインダーは、典型的に‘N-ターゲット結合部位I−構造安定化部位の一つの鎖−リンカー−構造安定化部位の他の鎖−ターゲット結合部位II−C’のコンストラクトを有する。
【0072】
本発明の好ましい具現例によると、本発明の二座ペプチドバインダーにおいて、ターゲット結合部位Iと構造安定化部位の一鎖との間及び/または構造安定化部位の他の鎖−ターゲット結合部位IIの間には、ターゲット結合部位と構造安定化部位間の相互構造的影響を遮断する構造影響抑制部位(structure influence inhibiting region)を含む。回転部位には、ペプチド分子でΦとΨの回転が比較的自由なアミノ酸が位置する。好ましくは、ΦとΨの回転が比較的自由なアミノ酸は、グリシン、アラニン及びセリンである。構造影響抑制部位には、1〜10個、好ましくは、1〜8個、より好ましくは、1〜3個のアミノ酸残基が位置することができる。
【0073】
上述のコンストラクトを有する本発明の二座ペプチドバインダーのライブラリーは、当業界に公知された多様な方法で得られる。このライブラリーにおいて、二座ペプチドバインダーは無作為配列を有するようになり、これは、ターゲット結合部位I及び/またはターゲット結合部位IIのどの位置でも配列選好度(sequence preference)がないか、または指定(または固定)されたアミノ酸残基がないことを意味する。
【0074】
例えば、二座ペプチドバインダーのライブラリーは、固状支持体(例えば、ポリスチレンまたはポリアクリルアミド樹脂)上で行われるスプリット合成方法(Lam et al. (1991) Nature 354:82; WO 92/00091)によって構築できる。
【0075】
本発明の好ましい具現例によると、二座ペプチドバインダーのライブラリーは、細胞表面展示(cell surface display)方式(例えば、ファージディスプレイ、バクテリアディスプレイまたはイーストディスプレイ)で構築される。好ましくは、二座ペプチドバインダーのライブラリーは、プラスミド、バクテリオファージ、ファージミド、イースト、バクテリア、mRNAまたはリボゾームを基にするディスプレイ方法により製作できる。
【0076】
ファージディスプレイは、ファージの表面上のコート蛋白質に融合された蛋白質形態で多様なポリペプチドをディスプレイする技術である(Scott, J. K. and Smith, G. P. (1990) Science 249: 386; Sambrook, J. et al., Molecular Cloning. A Laboratory Manual, 3rd ed. ColdSpring HarborPress(2001); Clackson and Lowman, Phage Display, Oxford UniversityPress(2004))。フィラメント性ファージ(例えば、M13)の遺伝子IIIまたは遺伝子VIIIに発現しようとする遺伝子を融合させて、無作為ペプチドをディスプレイする。
【0077】
ファージディスプレイには、ファージミドが利用できる。ファージミドは、バクテリアの複製原点(例えば、ColE1)及びバクテリオファージのintergenic部位の一コピーを有するプラスミドベクターである。このファージミドにクローニングされたDNA断片は、プラスミドのように増殖される。
【0078】
二座ペプチドバインダーのライブラリーをファージディスプレイ方式で構築する場合、本発明の好ましい具現例は、下記の段階を含む:(i)ファージコート蛋白質(例えば、M13のようなフィラメント性ファージの遺伝子IIIまたは遺伝子VIIIコート蛋白質)をコーディングする遺伝子と二座ペプチドバインダーをコーディングする遺伝子が融合された融合遺伝子;及び前記融合遺伝子に作動的に結合された転写調節配列(例えば、lacプロモーター)を含む発現ベクターのライブラリーを製作する段階;(ii)前記発現ベクターライブラリーを適した宿主に導入させる段階;(iii)前記宿主細胞を培養し、組み換えファージまたはファージミドウイルスパーティクルを形成させて、融合蛋白質が表面にディスプレイされるようにする段階;(iv)生物学的ターゲット分子と前記ウイルスパーティクルを接触させて、パーティクルをターゲット分子に結合させる段階;及び(v)ターゲット分子に結合しなかったパーティクルを分離する段階。
【0079】
ファージディスプレイを利用してペプチドライブラリーを構築して、これらのライブラリーをスクリーニングする方法は、米国特許第5,723,286号、第5,432,018号、第5,580,717号、第5,427,908号、第5,498,530号、第5,770,434号、第5,734,018号、第5,698,426号、第5,763,192号、及び第5,723,323号に開示されている。
【0080】
二座ペプチドバインダーの遺伝子を含む発現ベクターを製作する方法は、当業界に公知された方法によって行うことができる。例えば、公知のファージミドまたはファージベクター(例えば、pIGT2, fUSE5, fAFF1, fd-CAT1, m663, fdtetDOG, pHEN1, pComb3, pComb8, pCANTAB 5E (Pharmacia), LamdaSurfZap, pIF4, PM48, PM52, PM54, fdH及びp8V5)に二座ペプチドバインダーの遺伝子を挿入させて発現ベクターを製作することができる。
【0081】
大部分のファージディスプレイ方法がフィラメント性ファージを利用して行われるが、λファージディスプレイ(WO 95/34683;米国特許第5,627,024号)、T4ファージディスプレイ(Ren et al. (1998) Gene 215:439; Zhu (1997) CAN 33:534)及びT7ファージディスプレイ(米国特許第5,766,905号)も、二座ペプチドバインダーのライブラリーを構築する際に利用できる。
【0082】
ベクターライブラリーを適した宿主細胞に導入させる方法は、多様な形質転換方法によって行うことができ、最も好ましくは、電気穿孔(electroporation)方法によって行われる(参照:米国特許第5,186,800号, 第5,422,272号, 第5,750,373号)。本発明に適する宿主は、細胞は、E. coliのようなグラム陰性バクテリア細胞であり、適したE. coli宿主は、JM101, E. coli K12 strain 294, E. coli strain W3110及びE. coli XL-1Blue(Stratagene)を含むが、これらに限定されるものではない。宿主細胞は、形質転換前にコンピテンス細胞として用意することが好ましい(Sambrook, J. et al., Molecular Cloning. A Laboratory Manual, 3rd ed. Cold Spring HarborPress(2001))。形質転換された細胞の選別は、一般に抗生剤(例えば、テトラサイクリン及びアンピシリン)を含む培地で培養して行われる。選別された形質転換細胞をヘルパーファージの存在下で追加的に培養し、組み換えファージまたはファージミドウイルスパーティクルを生成させる。前記ヘルパーファージとして適したものは、Exヘルパーファージ、M13-KO7、M13-VCS及びR408を含むが、これらに限定されるものではない。
【0083】
生物学的ターゲット分子と結合するウイルスパーティクルの選別は、通常的にバイオパニング過程を通じて行うことができる(Sambrook, J. et al., Molecular Cloning. A Laboratory Manual, 3rd ed. Cold SpringHarbor Press(2001); Clackson and Lowman, Phage Display, Oxford University Press(2004))。
【0084】
本発明の二座ペプチドバインダーの具体的な例は、配列番号20〜38及び40〜41に記載されている。
【0085】
本発明のまた他の様態によると、本発明は、上述の二座ペプチドバインダーをコーディングする核酸分子を提供する。
【0086】
本発明の他の様態によると、本発明は、二座ペプチドバインダーをコーディングする核酸分子を含む二座ペプチドバインダーの発現用ベクターを提供する。
【0087】
本発明の他の様態によると、本発明は、二座ペプチドバインダーの発現用ベクターを含む形質転換体を提供する。
【0088】
本明細書において、用語‘核酸分子’は、DNA(gDNA及びcDNA)、そしてRNA分子を包括的に含む意味を有して、核酸分子において基本構成単位であるヌクレオチドは、天然のヌクレオチドだけではなく、糖または塩基部位が変形された類似体(analogue)も含む(Scheit,Nucleotide Analogs,John Wiley,New York(1980);Uhlman及びPeyman, Chemical Reviews, 90:543-584(1990))。
【0089】
本発明の好ましい具現例によると、本発明のベクターは、二座ペプチドバインダーをコーディングする核酸分子の他に、前記核酸分子に、転写を進行させることのできる強力なプロモーター(例えば、tacプロモーター、lacプロモーター、lacUV5プロモーター、lppプロモーター、pLλプロモーター、pRλプロモーター、rac5プロモーター、ampプロモーター、recAプロモーター、SP6プロモーター、trpプロモーター及びT7プロモーターなど)、解読の開始のためのリボゾーム結合座及び転写/解読終結配列を含む。
【0090】
本発明の好ましい具現例によると、本発明のベクターは、二座ペプチドバインダーをコーディングする核酸分子の5’-方向側にシグナル配列(例えば、pelB)を追加的に含むことができる。また、本発明の好ましい具現例によると、本発明のベクターは、二座ペプチドバインダーがファージの表面によく発現されたのかを確認するためのタギング配列(例えば、myc tag)を追加的に含む。
【0091】
本発明の好ましい具現例によると、本発明のベクターは、ファージコート蛋白質、好ましくは、M13のようなフィラメント性ファージの遺伝子IIIまたは遺伝子VIIIコート蛋白質をコーディングする遺伝子を含む。本発明の好ましい具現例によると、本発明のベクターは、バクテリアの複製原点(例えば、ColE1)及び/またはバクテリオファージの複製原点を含む。一方、本発明のベクターは、選択標識として、当業界で通常的に利用される抗生剤耐性遺伝子を含むことができるが、例えば、アンピシリン、ゲンタマイシン、カベニシリン、クロラムフェニコール、ストレプトマイシン、カナマイシン、ゲネチシン、ネオマイシン及びテトラサイクリンに対する耐性遺伝子を含むことができる。
【0092】
本発明の形質転換体は、好ましくは、E.coliのようなグラム陰性バクテリア細胞であり、適するE.coli宿主は、JM101, E. coli K12 strain 294, E. coli strain W3110及びE. coli XL-1Blue(Stratagene)を含むが、これらに限定されるものではない。本発明のベクターを宿主細胞内に運搬する方法は、CaCl2方法(Cohen, S.N. et al., Proc. Natl. Acac. Sci. USA, 9:2110-2114(1973))、ハナハン方法(Cohen, S.N. et al., Proc. Natl. Acac. Sci. USA, 9:2110-2114(1973);及びHanahan, D., J. Mol. Biol., 166:557-580(1983))及び電気穿孔方法(米国特許第5,186,800号、第5,422,272号、第5,750,373号)などにより行うことができる。
【0093】
本発明の二座ペプチドバインダーは、非常に低い水準(例えば、nM水準)のKD値(解離常数)を示し、生物学的ターゲット分子に非常に高い親和度を示すペプチドを提供する。下記実施例に記載のように、モノポダル方式で製作されたバインダーと比較し、二座ペプチドバインダーは、約102〜105倍(好ましくは、約103〜104倍)高い親和度を示す。
【0094】
本発明の二座ペプチドバインダーは、医薬としての用途を有するだけではなく、生体内物質の検出、インビボ分子イメージング、インビトロ細胞イメージング及び薬物伝達用ターゲッティングをする際に利用でき、エスコート分子としても利用できる。
【発明の効果】
【0095】
本発明の特徴及び利点を要約すると、以下のようである:
(i)本発明は、BPBベースのカーゴ運搬システムを提供する。
(ii)本発明の二座ペプチドバインダーにおいて、構造安定性部位の両末端に結合されている離隔された(distal)二つのターゲット結合部位は、互いに協同的に(cooperatively)、シナージック(synergetically)にターゲットに結合する。
(iii)このため、本発明の二座ペプチドバインダーは、非常に低い水準(例えば、nM水準)のKD値(解離定数)を示し、ターゲットに非常に高い親和度を示す。
(iv)本発明のBPBベースのカーゴ運搬システムは、BPBのターゲット結合性及び特異性に基づいて、多様な物質を細胞表面または細胞内に運搬することができる。
【図面の簡単な説明】
【0096】
【図1a】構造安定化部位としてβ−ヘアピン(hairpin)を含む二座ペプチドバインダー(bipodal-peptide binder)の模式図を示す。
【図1b】構造安定化部位としてリンカーで連結されたβ−シートを含む二座ペプチドバインダー(bipodal-peptide binder)の模式図を示す。
【図1c】構造安定化部位としてリンカーで連結されたロイシンジッパーを含む二座ペプチドバインダー(bipodal-peptide binder)の模式図を示す。
【図1d】構造安定化部位としてリンカーで連結されたロイシン-リッチモチーフ(leucine-rich motif)を含む二座ペプチドバインダー(bipodal-peptide binder)の模式図を示す。
【図2】二座ペプチドバインダーライブラリーをクローニングするための戦略を示す。pIGT2ファージミドベクターマップにおいて、pelBシグナル配列、myc tagは、目的遺伝子がファージの表面によく発現されたのかを確認するためのタギング配列である。プロモーターとしてlacプロモーターが利用された。
【図3】フィブロネクチンED-Bバイオパニング過程の間、インプットファージに対するED-B、ストレプトアビジン及びBSAのバイオパニング結果を示す。
【図4】フィブロネクチンED-Bバイオパニング過程の間、二座ペプチドバインダーライブラリーのバイオパニングの3次段階で回収した60個の組み換えファージのED-B及びBSAに対するELISA結果である。
【図5a】フィブロネクチンED-B蛋白質に結合する特定二座ペプチドバインダーの親和力を測定した結果である。
【図5b】VEGFに結合する特定二座ペプチドバインダーの親和力を測定した結果である。
【図5c】VCAM1に結合する特定二座ペプチドバインダーの親和力を測定した結果である。
【図5d】nAchR(Nicotinic acetylcholine receptor)に結合する特定二座ペプチドバインダーの親和力を測定した結果である。
【図5e】HSA(Human Serum Albumin)に結合する特定二座ペプチドバインダーの親和力を測定した結果である。
【図6a】フィブロネクチンED-Bに対して特異性検査をするために、二座ペプチドバインダーを有している組み換えファージを、種々の蛋白質に対してELISAを行って吸光度を測定した結果である。左側バーからストレプトアビジン、ED-B、アセチルコリン、α1、BSA、VCAM、TNF-α、トロンビン、ミオグロブリン、リソザイム及びビスファチンに対する結果である。
【図6b】VEGFに対して特異性検査をするために、二座ペプチドバインダーを有している組み換えファージを、種々の蛋白質に対してELISAを行って吸光度を測定した結果である。
【図6c】VCAM1に対して特異性検査をするために、二座ペプチドバインダーを有している組み換えファージを、種々の蛋白質に対してELISAを行って吸光度を測定した結果である。
【図6d】nAchR断片ペプチドに対して特異性検査をするために、二座ペプチドバインダーを有している組み換えファージを、種々の蛋白質に対してELISAを行って吸光度を測定した結果である。
【図6e】HSAに対して特異性検査をするために、二座ペプチドバインダーを有している組み換えファージを、種々の蛋白質に対してELISAを行って吸光度を測定した結果である。
【図6f】MyD88に対して特異性検査をするために、二座ペプチドバインダーを有している組み換えファージを、種々の蛋白質に対してELISAを行って吸光度を測定した結果である。
【図7】二座ペプチドバインダーの共同作用効果の証明のための親和力を測定した結果である。
【図8】二座ペプチドバインダーにおいて、構造安定化部位をトリプトファンジッパーの代わりに種々のβ-ヘアピンモチーフに変えて、二座ペプチドバインダーの親和力を測定した結果である。
【図9】二座ペプチドバインダーにおいて、構造安定化部位をトリプトファンジッパーの代わりにロイシンジッパーに変えて、二座ペプチドバインダーの親和力を測定した結果である。
【図10】癌バイオマーカーであるフィブロネクチンED-Bに特異的な二座ペプチドバインダーの癌標的化結果である。時間が経過するにつれて、癌に二座ペプチドバインダーが蓄積されることが分かる。それぞれの臓器を分離して蛍光を測定した時も、癌に相当部分蓄積されることが分かる。
【図11a】GST-BPB fusion proteinの精製を示す。
【図11b】GST-BPBのフィブロネクチン EDBに対するaffinity測定結果である。
【図12a】TNFα−BPBの精製を示す。
【図12b】TNFα−BPBのフィブロネクチン EDBに対するaffinity測定結果である。
【図12c】TNFα−BPBのcytotoxicitiy測定結果である。
【図13a】BPBcss-LS内に9R/siRNAのencapsulation efficiencyを分析した結果である。Green boxは、9R/siRNA複合体を含むliposomal fractionを示し、Red boxは、free unencapsulated siRNA fractionを示す。
【図13b】EDB over-expressing cell lineにおいて、BPBCSS−LSのuptakeを分析した結果である。Negativeは、未処理細胞を示し、LSは、リポソームのみを処理した細胞、CSS-LSは、BPBCSS-LSで処理した細胞を示す。
【図13c】EDB non-expressing cell lineにおいて、BPBCSS−LSのuptakeを分析した結果である。Negativeは、未処理細胞を示し、LSは、リポソームのみを処理した細胞、CSS-LSは、BPBCSS-LSで処理した細胞を示す。
【図13d】MCF-7 cellにVEGF-C siRNA knockdown efficiencyを分析した結果である。
【図14a】DSPE-PEG2000-MalとBPB(SSS) peptideのconjugationをMALDIを利用して分析した結果である。
【図14b】一週間SPION-BPBのHydrodynamic size変化を観察したELS分析結果である。
【図14c】(a)DSPE-PEG2000coated SPION及び(b)BPB conjugated DSPE-PEG2000coated SPIONのTEMイメージである。
【図14d】DSPE-PEG2000 coated SPION(DSPE-SPION)及びBPB conjugated DSPE-PEG2000 coated SPION(SSS-SPION)のT2-weighted MR phantom study結果である。
【図14e】DSPE-PEG2000 coated SPION及びBPB conjugated DSPE-PEG2000 coated SPIONを処理した細胞の(a)MRIイメージと(b)T2信号を分析した結果である。
【図14f】DSPE-PEG2000coated SPIONとBPB conjugated DSPE-PEG2000coated SPIONを処理した細胞のconfocal顕微鏡イメージである。
【図14g】DSPE-PEG2000 coated SPIONとBPB conjugated DSPE-PEG2000 coated SPIONのMTT assay結果である。
【図14h】DSPE-PEG2000coated SPION及びBPB conjugated DSPE-PEG2000coated SPIONの脳癌動物モデルにおけるMRIイメージである。
【図15a】Gold Nanoparticle-BPBのTEM imageである。
【図15b】U87MG細胞を利用してBPB conjugated金ナノ粒子を同一な濃度で入れた後、ICP-MSを利用して、細胞内のAuの量の差を測定した結果である。
【図15c】U87MG細胞に(a)GNPまたは(b)BPB-GNPを処理した後、Silver enhancementした顕微鏡写真である。
【発明を実施するための形態】
【0097】
以下、実施例を通じて本発明をさらに詳細に説明するが、これら実施例は、本発明をより具体的に説明するためのものであって、本発明の範囲がこれら実施例に限定されないことは、本発明の属する技術分野で通常の知識を有する者にとっては自明なことであろう。
【実施例】
【0098】
実験材料及び実験方法
実施例1:ライブラリーの製作
二座ペプチドバインダー遺伝子の製作及びファージミドベクターへの挿入
二つのオリゴヌクレオチドBeta-F1(5'-TTCTATGCGGCCCAGCTGGCC (NNK)6GGATCTTGGACATGGGAAAACGGAAAA-3')及びBeta-B1 (5'-AACAGTTTCTGCGGCCGCTCCTCC TCC(MNN)6TCCCTTCCATGTCCATTTTCCGTT-3')(Nは、A, T, GまたはC; Kは、GまたはT; Mは、CまたはA)を合成した。二重鎖を作るために、Beta-F1 4μM, Beta-B1 4μM, 2.5mM dNTP混合液4μl、ExTaq DNA重合酵素1μl(Takara, Seoul, Korea)及び10×PCRバッファ5μlを混合して、総50μlになるように蒸留水を添加した混合液を25個作った。この混合液をPCR反応(94℃で5分、60サイクル: 30℃で30秒、72℃で30秒及び72℃で7分間)をして、二重鎖を作った後、PCR精製キット(GeneAll, Seoul, Korea)を利用して精製し、二座ペプチドバインダー遺伝子を得た。二座ペプチドバインダーに挿入させる遺伝子をpIGT2ファージミドベクター(Ig therapy, Chuncheon, Korea)に連結するために、インサート遺伝子とpIGT2ファージミドベクターに制限酵素を処理した。約11μgのインサートDNAをSfiI(New England Biolabs(NEB, Ipswich)及びNotI(NEB, Ipswich)でそれぞれ4時間ずつ反応した後、PCR精製キットを利用して精製した。また、約40μgのpIGT2ファージミドベクターをSfiI及びNotIでそれぞれ4時間ずつ反応した後、CIAP(Calf Intestinal Alkaline Phosphatase)(NEB, Ipswich)を入れて1時間反応した後、PCR精製キットを利用して精製した。これらをUV-可視光線分光器(Ultrospec 2100pro, Amersham Bioscience)で定量し、2.9μgのインサート遺伝子を、T4 DNAリガーゼ(Bioneer, Daejeon, Korea)を利用してpIGT2ファージミドベクター12μgと18℃で15時間連結した後、エタノールで沈殿させてTEバッファ100μlでDNAを溶解させた。
【0099】
コンピテンス細胞の用意
E.coli XL1-BLUE細胞(American Type Culture Collection, Manassas, USA)をLB寒天プレートに線状塗抹した。寒天プレート培地で育った群落を5mlのLB培地に接種した後、37℃で200rpmの速度で混合しながら一日間培養した。培養された10mlの細胞を2lのLB培地に接種して、同じ方式で600nmの波長で吸光度が0.3-0.4になるまで培養した。培養されたフラスコを30分間氷に放置した後、4℃で4,000×gで20分間遠心分離し、沈殿した細胞を除いた上澄み液を全て除去し、1lの冷却された滅菌蒸留水で懸濁させた。これを再び同じ方法で遠心分離して、上澄み液を除去した後、1lの冷却された滅菌蒸留水で再懸濁させて、同じ方式で10%グリセロール溶液40mlで洗浄を繰り返して遠心分離した後、最後に10%グリセロール溶液4mlで懸濁させた後、200μlずつ分株して液体窒素に冷凍させた後、-80℃に保管した。
【0100】
電気穿孔法
ファージミドベクター12μgと二座ペプチドバインダーにインサートDNA 2.9μgを連結反応させた100μlを25個に分株して、電気穿孔を行った。コンピテンス細胞を氷の上で溶かして、200mlのコンピテンス細胞を連結反応した溶液4μlと混合した後、冷却して用意した0.2cmのキュベットに入れた後、1分間氷の上に放置した。電気穿孔機(BioRad, Hercules, CA)を200Ωで25μF及び2.5kVの条件にプログラムして、用意したキュベットの水気を除去して電気穿孔機に位置させた後、パルスを与えた(時間常数は、4.5〜5msec)。その後、直に37℃に用意した20mMのグルコースの含まれた1mlのLB液体培地に入れて、得られた25mlの細胞を100ml試験管に移した。一時間37℃で200rpmの速度で混合しながら培養した後、ライブラリーの数を測定するために、10μlを稀釈してアンピシリン寒天培地に塗抹した。残りの細胞を、1lのLBに20mMグルコース及び50μg/mlのアンピシリンを入れて、30℃で一日間培養した。4℃で4,000×gで20分間遠心分離し、沈殿した細胞を除いた上澄み液を全て除去し、40mlのLBで再懸濁させた後、グリセロールを最終濃度20%以上入れて、-80℃に保管した。
【0101】
ライブラリーで組み換えファージの生産とPEG沈殿
-80℃に貯蔵された二座ペプチドバインダーライブラリーで組み換えファージを生産した。500mlフラスコに100mlのLB液体培地にアンピシリン(50μg/ml)及び20mMのグルコースを入れた後、-80℃に保管されたライブラリー1mlを追加して、一時間37℃で150rpmの速度で混合しながら培養した。これに1×1011pfuのExヘルパーファージ(Ig therapy, Chuncheon, Korea)を入れて、再び一時間同じ条件で培養した。1,000×gで10分間遠心分離して上澄み液を除去し、これにアンピシリン(50μg/ml)及びカナマイシン(25μg/ml)が含まれたLB液体培地100mlを入れて、一日間培養し、組み換えファージを生産した。培養液を3,000×gで10分間遠心分離し、得られた上澄み液100mlにPEG/NaCl 25mlを混合して、氷に1時間放置させた後、4℃で20分間10,000×gで遠心分離して、上澄み液は注意して除去し、2mlのPBS(pH 7.4)でペレットを再懸濁させた。
【0102】
実施例2:蛋白質の準備
実施例に利用するフィブロネクチン(フィブロネクチン) ED-B, VEGF(vascular endothelial growth factor), VCAM1(vascular cell adhesion molecule-1), nAchR(Nicotinic acetylcholine receptor), HSA(Human serum albumin)及びMyD88を下記のように用意した。
【0103】
フィブロネクチンED-B遺伝子の製作及び発現ベクターへの挿入
韓国生命工学研究院から部分的なヒトのフィブロネクチンED-B(ID=KU017225)遺伝子を供給してもらった。プライマーEDB_F1(5'-TTCATAACATATGCCAGAGGTGCCCCAA-3')及びEDB_B1(5'-ATTGGATCCTTACGTTTGTTGTGTCAGTGTAGTAGGGGCACTCTCGCCGCCATTAATGAGAGTGATAACGCTGATATCATAGTCAATGCCCGGCTCCAGCCCTGTG-3')を合成して、EDB-F1 20 pmol, EDB-B1 20 pmol, 2.5 mM dNTP混合液4μl, ExTaq DNA重合酵素1μl(10 U)及び10×PCRバッファ5μlを混合して、50μlになるように蒸留水を添加した混合液を製造した。この混合液をPCR反応(94℃で5分, 30サイクル: 55℃で30秒, 72℃で1分及び94℃で30秒)をしてEDBインサートにした後、PCR精製キットを利用して精製した。EDBインサート遺伝子をpET28bベクター(Novagen)に連結するために、EDBインサート遺伝子及びpET28bベクターに制限酵素を処理した。約2μgのインサートDNAをBamHI(NEB, Ipswich)及びNdeI(NEB, Ipswich)で4時間ずつ反応した後、PCR精製キットを利用して精製した。また、約2μgのpIGT2ファージミドベクターをBamHI及びNdeIで3時間ずつ反応した後、CIAPを入れて1時間反応した後、PCR精製キットを利用して精製した。これらをベクターとインサートが約1:3のモル比率になるように添加して、T4 DNAリガーゼ(Bioneer, Daejeon, Korea)を利用して18℃で10時間連結させて、XL-1コンピテント細胞に形質転換をした後、カナマイシンの含まれた寒天培地に塗抹した。寒天プレート培地で育った群落を5mlのLB培地に接種した後、37℃で200rpmの速度で混合しながら一日間培養した後、プラスミドフラップキット(GeneAll, Seoul, Korea)を利用してプラスミドを精製し、シーケンシングをして、クローニングが成功したかどうかを確認した。
【0104】
VEGF121遺伝子の製作及び発現ベクターへの挿入
サイトカイン銀行(Jeonju, Korea)から部分的なヒトのVEGF(ID=G157)遺伝子を供給してもらった。プライマーVEGF_F1(5'-ATAGAATTCGCACCCATGGCAGAA-3')及びVEGF_B1(5'-ATTAAGCTTTCACCGCCTCGGCTTGTCACAATTTTCTTGTCTTGC-3')を合成して、VEGF-F1 20 pmol, VEGF-B1 20 pmol, 2.5 mM dNTP混合液4μl、ExTaq DNA重合酵素1μl(10 U)及び10×PCRバッファ5μlを混合して、50μlになるように蒸留水を添加した混合液を製造した。この混合液をPCR反応(94℃で5分, 30サイクル: 55℃で30秒, 72℃で1分及び94℃で30秒)をしてVEGFインサートにした後、PCR精製キットを利用して精製した。VEGFインサート遺伝子をpET32aベクター(Novagen)に連結するために、VEGFインサート遺伝子及びpET32aベクターに制限酵素を処理した。約2μgのインサートDNAをEcoRI(NEB, Ipswich)及びHindIII(NEB, Ipswich)で4時間ずつ反応した後、PCR精製キットを利用して精製した。これらをベクターとインサートが約1:3のモル比率になるように添加して、T4 DNAリガーゼ(Bioneer, Daejeon, Korea)を利用して18℃で10時間連結させて、XL-1コンピテント細胞に形質転換をした後、アンピシリンの含まれた寒天培地に塗抹した。寒天プレート培地で育った群落を5mlのLB培地に接種した後、37℃で200rpmの速度で混合しながら一日間培養した後、プラスミドフラップキット(GeneAll, Seoul, Korea)を利用してプラスミドを精製し、シーケンシングをして、クローニングが成功したかどうかを確認した。
【0105】
VCAM1遺伝子の製作及び発現ベクターへの挿入
韓国生命工学研究院からヒトのVCAM1遺伝子を供給してもらった。VCAM1インサート遺伝子をpET32aベクターに連結するために、VCAM1インサート遺伝子及びpET32aベクターに制限酵素を処理した。これらをベクターとインサートが約1:3のモル比率になるように添加して、T4 DNAリガーゼ(Bioneer, Daejeon, Korea)を利用して18℃で10時間連結させて、XL-1コンピテント細胞に形質転換をした後、アンピシリンの含まれた寒天培地に塗抹した。寒天プレート培地で育った群落を5mlのLB培地に接種した後、37℃で200rpmの速度で混合しながら一日間培養した後、プラスミドフラップキット(GeneAll, Seoul, Korea)を利用してプラスミドを精製し、シーケンシングをして、クローニングが成功したかどうかを確認した。
【0106】
フィブロネクチンED-Bの発現及び精製
フィブロネクチンED-BをクローニングしたpET28bベクターをBL21細胞に形質転換した後、カナマイシンの含まれた寒天培地に塗抹した。寒天プレート培地で育った群落をカナマイシン(25μg/ml)の含まれた5mlのLB培地に接種した後、37℃で200rpmの速度で混合しながら一日間培養した後、カナマイシン(25μg/ml)の含まれた50mlのLB培地に移して3時間培養した。培養したE. coliをカナマイシン(25μg/ml)の含まれた2lのLBに接種して、OD=0.6-0.8まで培養した。その後、1mMイソプロピル-β-D-チオガラクトピラノシド(IPTG)を入れて、37℃で200rpmの速度で混合しながら8時間培養した。4,000×gで20分間遠心分離して、沈殿された細胞を除いた上澄み液を全て除去し、ライシスバッファ(50mMリン酸ナトリウム(pH 8.0), 300mM NaCl及び5mMイミダゾール)で懸濁させた。-80℃に一日間保管した後、ソニケーターを利用してE. coliを溶解させた後、15,000×gで1時間遠心分離して、上澄み液をNi-NTA親和性レジン(Elpisbio, Daejeon, Korea)に結合させる。ライシスバッファでレジンを洗浄した後、イリュージョンバッファ(50mMリン酸ナトリウム(pH 8.0), 300mM NaCl及び300mMイミダゾール)を利用して、N-末端His-tag ED-B蛋白質を溶出させて獲得した。このように獲得した蛋白質をSuperdex75カラム(GE Healthcare, United Kingdom)及びPBS(pH 7.4)バッファを利用して、ゲルろ過法(gel filtration)で純度の高いED-B蛋白質を得た。バイオパニングのためにED-B蛋白質にビオチンを連結した。6mgのスルホ-NHS-SS-ビオチン(PIERCE, Illinois, USA)及び1.5mgのED-B蛋白質を常温で0.1Mホウ酸ナトリウム(pH 9.0)下で2時間反応し、反応しなかったスルホ-NHS-SS-ビオチンを除去するために、蛋白質をSuperdex75カラム及びPBS(pH 7.4)バッファを利用してゲルろ過法(gel filtration)でビオチン-EDB蛋白質を精製した。
【0107】
VEGF121とVCAM1-Trxの発現及び精製
VEGF121とVCAM1をクローニングしたpET32aベクターをそれぞれAD494細胞に形質転換した後、アンピシリンの含まれた寒天培地に塗抹した。寒天プレート培地で育った群落をアンピシリン(25μg/ml)の含まれた5mlのLB培地に接種した後、37℃で200rpmの速度で混合しながら一日間培養した後、アンピシリン(25μg/ml)の含まれた50mlのLB培地に移して3時間培養した。培養したE. coliをアンピシリン(25μg/ml)の含まれた2lのLBに接種して、OD=0.6-0.8まで培養した。その後、1mMイソプロピル-β-D-チオガラクトピラノシド(IPTG)を入れて、37℃で200rpmの速度で混合しながら8時間培養した。4,000×gで20分間遠心分離して、沈殿された細胞を除いた上澄み液を全て除去し、ライシスバッファ(50mMリン酸ナトリウム(pH 8.0), 300mM NaCl及び5mMイミダゾール)で懸濁させた。-80℃に一日間保管した後、ソニケーターを利用してE. coliを溶解させた後、15,000×gで1時間遠心分離して、上澄み液をNi-NTA親和性レジン(Elpisbio, Daejeon, Korea)に結合させる。ライシスバッファでレジンを洗浄した後、イリュージョンバッファ(50mMリン酸ナトリウム(pH 8.0), 300mM NaCl及び300mMイミダゾール)を利用して、Trx-VEGF121蛋白質を溶出させて獲得した。このように獲得した蛋白質をSuperdex75カラム(GE Healthcare, United Kingdom)及びPBS(pH 7.4)バッファを利用して、ゲルろ過法(gel filtration)で純度の高いVEGF-TrxとVCAM1-Trx蛋白質を得た。純粋なVEGFを得るために、トロンビンでVEGFとTrx間を切断し、VEGF121を得た。
【0108】
一方、HSAは、Genetex会社(Irvine)から購入して利用した。nAchR(Nicotinic acetylcholine receptor)の断片ペプチドであるbiotin-SGEWVIKEARGWKHWVFYSCCPTTPYLDITYH(32mer)は、ANYGEN(Korea, Kwangju)で合成した。Human MyD88は、Santa Cruz Biotechnology(sc-4540WB)(California)から購入した。
【0109】
実施例3:バイオパニング方法
BiotinフィブロネクチンED-B蛋白質とBiotin-nAchRペプチドのバイオパニング方法
2mlのストレプトアビジン(10μg/ml)を96ウェルELISAプレート(Corning)の40個のウェルに50μlずつ入れた後、4℃で一晩中放置し、翌日20個のウェルにのみ0.1%のPBST(tween-20)で3回洗浄した後、ビオチンED-BとビオチンnAchR(10μg/ml)をそれぞれ入れて、常温で1時間放置した。その後、40個のウェルの全てを0.1%PBSTで3回洗浄して、PBSで稀釈した2%BSAを使用して常温で2時間ブロッキングした後、溶液を全て捨てて、0.1%PBSTで3回洗浄した。これに二座ペプチドバインダー組み換えファージ含有溶液800μl及び10%BSA 200μlを混合し、ストレプトアビジン及びBSAに結合するファージを除去するために、ストレプトアビジンにBSAコーティングした20個のウェルに入れて1時間27℃に放置した。上澄み液を回収してED-BとnAchRが結合した20個のウェルに移して、27℃で45分間放置した。20個のウェルの溶液を全て除去して、0.5%PBSTで15回洗浄した後、0.2Mグリシン/HCl(pH2.2)1mlを各ウェル当たり50μlずつ入れて20分間ファージを溶離させて、1mlをチューブに集めて、これに2M Tris-base(pH 9.0)150μlを入れて溶液を中和させた。各バイオパニング毎にインプットファージ及びアウトプットファージの数を測定するために、OD=0.7のXL-1 BLUE細胞に混ぜて、アンピシリンの含まれた寒天培地に塗抹した。パニングを繰り返すために10mlのE. coli XL1-BLUE細胞と混ぜて、37℃で1時間200rpmの速度で混合しながら培養した。アンピシリン(50μg/ml)及び20mMグルコースを混合した後、2×1010 pfuのExヘルパーファージを添加して、37℃で1時間200rpmの速度で混合しながら培養した。培養液を1,000×gで10分間遠心分離した後、上澄み液は全て除去し、沈殿された細胞をアンピシリン(50μg/ml)及びカナマイシン(25μg/ml)の含まれた40ml LB液体培地で再懸濁して、30℃で200 rpmの速度で混合しながら一日間培養した。培養液を4,000×g、20分間及び4℃の条件で遠心分離した。上澄み液に8mlの5x PEG/NaCl[20% PEG(w/v)及び15% NaCl(w/v)]を添加した後、4℃で1時間静置させた。遠心分離後、PEG溶液を完全に除去して、1mlのPBS溶液でファージペプチドペレットを溶かした後、2次バイオパニングに使用した。各パニング段階毎に上記と同一な方法を使用したが、洗浄過程は、段階別にそれぞれ25回及び35回(0.5% PBST)増加させた。
【0110】
VEGFとVCAM1-TrxとHuman serum albumin(HSA), MyD88のバイオパニング方法
VEGFとVCAM1-trxとHSAとMyD 88(5μg/ml)を96ウェルELISAプレート(Corning)の10個のウェルに50μlずつ入れた後、4℃で一晩中放置し、翌日2%BSAを使用して常温で2時間ブロッキングした後、溶液を全て捨てて、0.1%PBSTで3回洗浄した。これに二座ペプチドバインダー組み換えファージ含有溶液800μl及び10%BSA 200μlを混合し、VEGFとVCAM1-TrxとHSAが結合した10個のウェルに移して常温で1時間放置した。10個のウェルの溶液を全て除去して、0.1%PBSTで10回洗浄した後、0.2Mグリシン/HCl(pH2.2)1mlを各ウェル当たり50μlずつ入れて20分間ファージを溶離させて、1mlをチューブに集め、これに2M Tris-base(pH 9.0)150μlを入れて溶液を中和させた。各バイオパニング毎にインプットファージ及びアウトプットファージの数を測定するために、OD=0.7のXL-1 BLUE細胞に混ぜて、アンピシリンの含まれた寒天培地に塗抹した。パニングを繰り返すために10mlのE. coli XL1-BLUE細胞と混ぜて、37℃で1時間200rpmの速度で混合しながら培養した。アンピシリン(50μg/ml)及び20mMグルコースを混合した後、2×1010 pfuのExヘルパーファージを添加して、37℃で1時間200rpmの速度で混合しながら培養した。培養液を1,000×gで10分間遠心分離した後、上澄み液は除去し、沈殿された細胞をアンピシリン(50μg/ml)及びカナマイシン(25μg/ml)の含まれた40ml LB液体培地で再懸濁して、30℃で200 rpmの速度で混合しながら一日間培養した。培養液を4,000×g、20分間及び4℃の条件で遠心分離した。上澄み液に8mlの5x PEG/NaCl[20% PEG(w/v)及び15% NaCl(w/v)]を添加した後、4℃で1時間静置させた。遠心分離後、PEG溶液を完全に除去して、1mlのPBS溶液でファージペプチドペレットを溶かした後、2次バイオパニングに使用した。各パニング段階毎に上記と同一な方法を使用したが、洗浄過程は、段階別にそれぞれ20回及び30回(0.1% PBST)増加させた。
【0111】
実施例4:フィブロネクチンED-BのインプットファージのELISA
二座ペプチドバインダーライブラリーの各インプットファージのELISAを、ストレプトアビジン、BSA及びED-Bに対して行った。96ウェルELISAプレートに10μg/mlストレプトアビジンを各ウェル当たり50μlずつ18個のウェルに入れて、10μg/ml BSAを各ウェル当たり50μlずつ9個のウェルに入れて、4℃で一晩中放置した。翌日、ストレプトアビジンのある18個のウェルの中、9個のウェルのみを0.1% PBST(tween-20)で3回洗浄し、ビオチンED-B(10μg/ml)を入れて常温で1時間放置した。その後、全てのウェルを0.1% PBSTで3回洗浄し、PBSで稀釈した2% BSAを使用して常温で2時間ブロッキングした後、溶液を全て捨てて0.1% PBSTで3回洗浄した。これに二座ペプチドバインダー組み換えファージである一番目、二番目及び三番目のインプットファージ800μl及び10% BSA 200μlを混合して、100μlずつED-B, ストレプトアビジン及びBSAウェルにそれぞれ3ウェルずつ分株して、27℃で1時間30分間静置した。0.1% PBST溶液で10回洗浄した後、HRP-コンジュゲート抗-M13抗体(GE Healthcare)を1:1,000に稀釈して、27℃で1時間反応した。0.1% PBSTで5回洗浄した後、ペロキシダーゼの基質であるテトラメチルベンジジン(TMB)(BD Science)溶液100μlを分株して、発色反応を誘導した後、100μlの1 M HClを添加して反応を中止した。その後、450nmで吸光度を測定した。
【0112】
実施例5:フィブロネクチンED-B, VEGF, VCAM1, nAchR, HSA, MyD88蛋白質に特異的なファージペプチド検索(ファージELISA)
アウトプットファージ/インプットファージ比率が最も高いバイオパニング段階で回収したファージをXL1-BLUE細胞に感染させた後、プラークがプレート当たり100-200個程度になるように塗抹した。滅菌されたチップを利用し、プラーク60個を2mlのLB-アンピシリン(50μg/ml)培養液に接種した後、37℃で5時間振とう培養し、OD=0.8-1で5×109pfuのExヘルパーファージを添加して、37℃で1時間200 rpmの速度で混合しながら培養した。培養液を1,000×gで10分間遠心分離した後、上澄み液は全て除去し、沈殿された細胞をアンピシリン(50μg/ml)及びカナマイシン(25μg/ml)の含まれた1ml LB液体培地で再懸濁して、30℃で200 rpmの速度で混合しながら一日間培養した。培養液を10,000×g, 20分間及び4℃の条件で遠心分離して上澄み液を回収した後、2%の脱脂牛乳を入れて、これをファージペプチド検索に使用した。96ウェルELISAプレートに5μg/mlのフィブロネクチン ED-B, VEGF, VCAM1, Nicotinic acetylcholine receptor(nAchR), Human serum albumin, MyD88を各ウェル当たり50μlずつ30個のウェルに入れて、また、10μg/mlのBSAを各ウェル当たり50μlずつ30個のウェルに入れて4℃で一日間放置した。翌日、全てのウェルを0.1% PBSTで3回洗浄し、PBSで稀釈した2%脱脂牛乳を使用して常温で2時間ブロッキングした後、溶液を全て捨てて0.1% PBSTで3回洗浄した。各クローン別に増幅されたファージペプチド溶液100μlずつを全てのウェルに分株して、27℃で1時間30分間静置した。0.1% PBST溶液で5回洗浄した後、HRP-コンジュゲート抗-M13抗体(GE Healthcare)を1:1,000に稀釈して、27℃で1時間反応した。0.1% PBSTで5回洗浄した後、TMB溶液100μlを分株して発色反応を誘導した後、100μlの1 M HClを添加して反応を中止した。450 nmで吸光度を測定し、BSAに比べて吸光度の高いクーロンを選択した。これらのファージをXL1細胞に感染させた後、プラークがプレート当たり100-200個程度になるように塗抹した。滅菌されたチップを利用し、プラークを4mlのLB-アンピシリン(50μg/ml)培養液に接種した後、37℃で一日間振とう培養して、プラスミドフラップキットを利用してプラスミドを精製し、シーケンシングを依頼した(Genotech, Daejeon, Korea)。シーケンシングプライマーは、ベクターシーケンスである5'-GATTACGCCAAGCTTTGGAGC-3'を使用した。
【0113】
実施例6:フィブロネクチンED-B, VEGF, nAchRバインディングアッセイ
DNAシーケンシングで重複して出たED-B, VEGF, nAchRに特異的な二座ペプチドバインダーペプチドを合成(Anygen,韓国)した。親和度の測定は、BIAcore X(Biacore AB, Uppsala, Sweden)を利用して行った。ED-BとnAchRは、ストレプトアビジンSAチップ(Biacore)にビオチン-EDBを2,000 RUだけ流して固定させた。VEGFは、EDC/NHSを利用してCM5チップ(Biacore)に固定した。ランニングバッファとしてはPBS(pH 7.4)を使用して、フローは、分当たり30μl流しながら、多様な濃度で動力学を測定した後、BIAevaluationソフトウェア(Biacore AB, Uppsala, Sweden)で親和度を計算した。
【0114】
実施例7:癌バイオマーカーであるフィブロネクチンED-Bに特異的な二座ペプチドバインダーの癌標的化
癌にたくさん分布するフィブロネクチンED-Bをターゲットする二座ペプチドバインダー(ペプチド2)にCy5.5-NHS蛍光ダイ(flurorescence dye)(Amersham Pharmacia, Piscataway)を50 mMホウ酸ナトリウム緩衝液(pH 9.7)で12時間常温反応した。反応後にSephadex G25(Pharmacia Biotech, Uppsala, Sweden)でCy5.5と二座ペプチドバインダー-Cy5.5を分離した。Balb/cヌードマウスにHuman U87MG(ATCC) 2 x 106 cellを皮下に入れて癌を10日間育てた後、0.5nmolの二座ペプチドバインダー-Cy5.5を静脈注射で入れて、IVIS(Caliper Life Sience, Hopkinton)で蛍光を測定した。この実験は、癌バイオマーカーであるフィブロネクチンED-Bに特異的な二座ペプチドバインダーが、インビボ動物において癌に蓄積されることを証明する結果であって、実際、癌診断剤としての応用性を示す(図11)。
【0115】
実施例8:細胞内に存在するMyD88に特異的な二座ペプチドバインダーの活性抑制実験
MyD88は、細胞内に存在する蛋白質であるため、二座ペプチドバインダーのloopのリジン(lysine)残基を利用し、細胞透過ペプチド(cell penetrating peptide)である9個のアルギニン(arginine)(Anygen, 韓国)を、EDC/NHS(Sigma)を利用して二座ペプチドバインダーに共有結合させて、細胞透過を可能にした。MyD88の活性が活性化されるとMMP-13の量が増加するため、MMP-13の量を測定すれば、MyD88の活性が抑制されるかどうか判別できる。MyD88の活性を活性化させるIL-1beta(10 ng/ml)(R&D systems, Minneapolis MN)を軟骨細胞に処理した。その後、MyD88に特異的な二座ペプチドバインダー(表3fのペプチド1)を軟骨細胞に10μM処理して、12時間後にmRNAを分離した後、MMP-13とGAPDHに対してRT-PCRを進行した。また、軟骨細胞を破壊して細胞内蛋白質を得た後、Anti-MMP 13抗体(Abcam, ab3208, Cambridge)とセミドライtransfer機械(Amersham Bioscience, Piscataway)を利用しウェスタンブロッティングを行って、MMP-13の量を測定した。
【0116】
実験結果
実施例9:二座ペプチドバインダーライブラリーの製作
二座ペプチドバインダーの構造安定化部位としては、安定したβ-ヘアピンモチーフを使用した。特に、トリプトファン-トリプトファンアミノ酸の相互作用によりβ-ヘアピンモチーフ構造を安定にできるトリプトファンジッパー(Andrea et al., Proc. Natl. Acad. Sci. 98:5578-5583(2001))を利用した。骨格であるトリプトファンジッパーのN-及びC-末端部分にそれぞれ6個のアミノ酸を無作為に配列することにより、二つの部分に可変的部位を生成した(図1a)。これを二座ペプチドバインダーと命名し、両側の可変的部位を有しているため、抗原に共同作用で付くことができ、高い親和力及び特異性を有することができる。また、二座ペプチドバインダーの構造安定化部位は、図1b乃至図1eのように様々に構成できる。
【0117】
合成した二つの無作為配列オリゴヌクレオチドをPCR反応を通じて二重鎖にした後、制限酵素のSfiI及びNotIで切断した後、pIGT2ファージミドベクターにクローニングして、8×108以上のライブラリーを構築した(図2)。
【0118】
実施例10:バイオパニング結果
二座ペプチドバインダーライブラリーをフィブロネクチンED-B, VEGF, VCAM1, nAchR, HSA蛋白質に対して3〜5回にかけてバイオパニングを行って、各パニング段階で回収したファージペプチドのアウトプットファージ/インプットファージの比率を決定した(表1a)。
【0119】
【表1a】
【0120】
【表1b】
【0121】
【表1c】
【0122】
【表1d】
【0123】
【表1e】
【0124】
【表1f】
【0125】
実施例11:フィブロネクチンED-BのインプットファージELISA結果
二座ペプチドバインダーのライブラリーの各インプットファージをED-B、ストレプトアビジン及びBSAに対してELISAを行った。一番目のインプットファージの反応性は、ED-B、ストレプトアビジン及びBSAのいずれも吸光度がほぼ等しいが、二番目のインプットファージからED-B吸光度が、ストレプトアビジンに比べ5.1倍、BSAに比べ3.4倍高かった。最後に三番目のインプットファージでは、ED-B吸光度が、ストレプトアビジンに比べ22倍、BSAに比べ15倍高い反応性を示し、ED-Bに対して成功的にバイオパニングがなされることが分かる(図3及び表2)。
【0126】
【表2】
【0127】
実施例12:フィブロネクチンED-B, VEGF, VCAM1, nAchR, HSA及びMyD88に対して特異的なファージペプチド検索(ファージELISA)及びシーケンシング
各ライブラリーのパニング段階の中、アウトプット/インプットの比率が最も高い段階で回収したファージをプラーク形態として確保した。各プラークから60個のファージを増幅させた後、BSAに対してELISAを行った(図4)。BSAに比べて吸光度の高いクローンを選択し、DNAシーケンシングを依頼した。これから重複したそれぞれの蛋白質に特異的なペプチドシーケンスを得た(表3)。
【0128】
【表3a】
【0129】
【表3b】
【0130】
【表3c】
【0131】
【表3d】
【0132】
【表3e】
【0133】
【表3f】
【0134】
実施例13:フィブロネクチンED-B, VEGF, VCAM1, nAchR及びHSAの親和度の測定
フィブロネクチンED-B, VEGF, VCAM1, nAchR及びHSAに対する前記ペプチドを合成し、SPR Biacore system(Biacore AB, Uppsala, Sweden)を利用して親和度を測定した。フィブロネクチンED-Bに対する親和度を測定した結果、ペプチド1は、620nMを示して、ペプチド2は、75nMを示し、ペプチド3は、2.5μMを示した(図5a)。VEGFに対する親和度を測定した結果、ペプチド1は、60nM、ペプチド2は、326nMを示した(図5b)。VCAM1の断片ペプチドに対する親和度を測定した結果、ペプチド1は、318nMを示した(図5c)。nAchRの断片ペプチドに対する親和度を測定した結果、ペプチド1は、73nMを示した(図5d)。HSAの断片ペプチドに対する親和度を測定した結果、ペプチド1は、115nMを示した(図5e)。
【0135】
実施例14:フィブロネクチンED-B, VEGF, VCAM1, nAchR, HSA及びMyD88に対する特異性分析
それぞれの蛋白質に特異的に結合する組み換えファージを、種々の蛋白質に対して特異性検査をELISAを利用して行った。96ウェルELISAプレートにそれぞれの蛋白質5μg/mlを50μlずつウェルに入れて、翌日0.1% PBST(tween-20)で3回洗浄し、2%BSAを使用して常温で2時間ブロッキングした後、溶液を全て捨てて0.1% PBSTで3回洗浄した。これに本発明のペプチドを有する組み換えファージを2%BSAとよく混合し、100μlずつ10個の蛋白質が結合されているウェルに分株し、27℃で2時間静置した。0.1% PBST溶液で5回洗浄した後、HRP-コンジュゲーション抗-M13抗体(GE Healthcare)を1:1,000に稀釈して、27℃で1時間反応した。0.1% PBSTで5回洗浄した後、TMB溶液100μlを分株して発色反応を誘導した後、100μlの1 M HClを添加して反応を中止した後、450 nmで吸光度を測定した。図6aから分かるように、二座ペプチドバインダーで見つけたED-Bに特異的な表3aのペプチド2は、他の蛋白質の吸光度と比較すると、30倍以上の差を示し、これは、ペプチド2シーケンスがED-Bに対して特異的であることを示す結果である。図6b〜6fから分かるように、表3b〜3fのそれぞれのペプチド1に対する特異性分析結果、これらのそれぞれのペプチドは、VEGF, VCAM1, nAchR, HSA及びMyD88に対して特異性を有するということが分かる。
【0136】
実施例15:SPR(Surface Plasmon Resonance)の共同作用効果の確認
二座ペプチドバインダーの抗原に対する共同作用効果を証明するために、親和力が最もよい表3aのED-Bに対するペプチド2のN-及びC-末端の一側部位のみをそれぞれ除去したペプチドを合成し、親和力を測定した。N-末端部分は、592μMを有して、C-末端部分は、12.8μMを示した(図7)。二座ペプチドバインダーにおいて二座(bipodal)を有していることから表す共同作用効果は、43nMの親和力であることを証明した(図5a)。
【0137】
実施例16:他のβ-ヘアピンに対するバインディングアッセイ
トリプトファンジッパーの外に、異なるβ-ヘアピン骨格であるGB1m3及びHP7に、ED-Bに特異的に結合するペプチド2のN-末端シーケンス(HCSSAV)とC-末端シーケンス(IIRLEQ)を有するようにペプチドを合成した(Anygen, 韓国)。即ち、トリプトファンジッパーを含む二座ペプチドバインダーのシーケンスは、HCSSAVGSWTWENGKWTWKGIIRLEQであり、GB1m3を含む二座ペプチドバインダーは、HCSSAVGKKWTYNPATGKFTVQEGIIRLEQであって、HP7を含む二座ペプチドバインダーは、HCSSAVGKTWNPATGKWTEGIIRLEQである。各ペプチドの親和度は、BIAcore X(Biacore AB, Uppsala, Sweden)を使用して測定した。ストレプトアビジンSAチップ(Biacore AB, Uppsala, Sweden)にビオチン-EDBを2,000 RUだけ流して固定した。ランニングバッファとしてはPBS(pH 7.4)を使用して、フローは、分当たり30μl流しながら、多様な濃度で動力学を測定した後、BIAevaluationで親和度を計算した。 親和度を測定した結果、GB1m3が70nM、HP7が84nMであって、トリプトファンジッパー(43nM)と等しい親和力を有することを確認した(図8)。これは、全ての安定したβ-ヘアピンモチーフが構造安定化部位として機能することを証明する結果である。
【0138】
実施例17:構造安定化部位としてロイシンジッパーを含む二座ペプチドバインダーに対するバインディングアッセイ
β-ヘアピン骨格の代わりに、構造安定化部位としてロイシンジッパーに、ED-Bに特異的に結合するペプチド2のN-末端シーケンス(HCSSAV)とC-末端シーケンス(IIRLEQ)を有するようにCSSPIQGGSMKQLEDKVEELLSKNYHLENEVARLKKLVGER及びIIRLEQGGSMKQLEDKVEELLSKNYHLENEVARLKKLVGERペプチドを合成した(Anygen, 韓国)。二つのペプチドをダイマーにした後、BIAcore X(Biacore AB, Uppsala, Sweden)を使用して親和度を測定した。親和度を測定した結果、ロイシンジッパーは、5μMの親和度を示して、これは、トリプトファンジッパー(43nM)の親和度に劣る親和度ではあるが、ロイシンジッパーも二座ペプチドバインダーの構造安定化部位として機能することができることが分かる(図9)。
【0139】
実施例18:癌バイオマーカーであるフィブロネクチンED-Bに特異的な二座ペプチドバインダーの癌標的化
癌にたくさん分布するフィブロネクチンED-Bをターゲッティングする二座ペプチドバインダーにCy5.5蛍光ダイを付着した後、Human U87MG癌が存在するマウスに静脈注射で投与して、IVIS機会を利用し、二座ペプチドバインダーが癌にターゲットするかどうか蛍光を確認した(図10)。実験結果、癌バイオマーカーであるフィブロネクチンED-Bに特異的な二座ペプチドバインダーは、癌組織に蓄積されることが観察されて、これは、本発明の二座ペプチドバインダーがインビボイメージングに利用できることを示す結果である。
【0140】
実施例19:GST-BPB fusion protein for フィブロネクチンEDB
1.GST-BPB遺伝子の製作
Glutathione S-transferase(GST)遺伝子の含まれたpGEX4T-1ベクター(GE Healthcare)にフィブロネクチン EDBを認識するBPB遺伝子シーケンスをクローニングした。二つのオリゴヌクレオチドGST-F1(5’-ACCGGATCCCATTGTTCTAGT-3’)とGST-B1(5'-ATTCTCGAGTTACGCTCCTCCTCC-3')を利用して、EDBタンパク質に付くファージから得たphagemid vectorをtemplate(実施例1及び3)でPCRして、フィブロネクチンEDBに付くBPBペプチド遺伝子を得た。このBPBペプチドのペプチド配列は、HCSSAVGSWTWENGKWTWKGIIRLEQである。GST-F1 20pmol, GST-B1 20pmol, 鋳型選別したphagemid template 1ul, 2.5mM dNTP mixture 4μl, Ex Taq DNA polymerase 1μl (10 U), 10 x PCR buffer 5μlを混ぜて、総50μlになるように蒸留水を追加した混合液を作った。この混合液をPCR反応(94℃で5分, 30cycle: 55℃で30秒と72℃で1分, 94℃で30秒)して、BPB Insertにした後、PCR purification kit(GeneAll, Seoul, Korea)を利用して精製した。BPB Insert遺伝子をpGEX4T-1 ベクターに連結するために、BPB Insert遺伝子とpGEX4T-1ベクターに制限酵素処理を行った。約2μgのInsert DNAをBamHI (NEB)とXhoI (NEB)で4時間ずつ反応させた後、PCR purification kitを利用して精製した。これらをmolar ratio vector:Insert=1:3でT4 DNA ligase(Bioneer)を利用して18℃、10時間連結した後、E. coli XL-1 competent cell(Stratagene)にtransformationをした後、Ampicillin agar培地に塗抹した。寒天平板培地で育った集落を5mlのLB培地に接種した後、37℃で200rpmの速度で混ぜながら、一日中培養した後、plasmid preparation kit(GeneAll, Seoul, Korea)を利用してplasmidを精製し、シーケンシングをして、クローニングが成功したかどうかを確認した。
【0141】
2. GST-BPB発現及び精製
GST-BPBをクローニングしたベクターをE. coli BL21にtransformationした後、2L LBにアンピシリン(25ug/ml)を入れて、OD=0.6-0.8まで育てた。その後、1mM isopropyl-β-D-thiogalactopyranoside(IPTG)を入れて、37℃で200rpmの速度で混ぜながら、8時間育てた。4,000gで20分間遠心分離して、沈んだ細胞を除いた上澄み液を全て除去して、lysis buffer (50 mM sodium phosphate (pH 8.0), 300mM NaClと5mM imidazole)で再浮遊させた。-80℃で一日間保管した後、Sonicatorを利用してE. coli溶解させた後、15,000gで1時間遠心分離して、上澄み液をGST affinity resin(Peptron)に結合させた。PBS bufferでresinを洗浄した後、Elution buffer(20mM GSH in 10mM Tris-HCl, pH 8)を利用して、GST-BPBタンパク質を分離して収集した。このように集めたタンパク質をSuperdex75 column(Amersham)とPBS(pH7.4) bufferを利用してgel filtrationをして、純度高いGST-BPBタンパク質を収集した。
【0142】
3. GST-BPBのフィブロネクチン EDB binding実験
精製されたGST-BPBを、フィブロネクチンEDBにbindingするかをSPR機械を利用して確認した。Affinity測定は、BIAcore Xを利用して行った。Streptavidin SA chip(Biacore)にbiotin-EDBを2000RUだけ流して固定させた。Running bufferとしてはPBS(pH7.4)を使用して、流速は30ul/minとして流しながら、様々な濃度でkineticsを測定した後、BIAevaluationでaffinityを計算した。
【0143】
4.実験結果
(1) GST-BPBの製作
フィブロネクチンED-Bを特異的に認識するBPBをGST fusion vectorにクローニングした後、GST-BPBを大腸菌で発現して精製した(図11a)。
【0144】
(2) GST-BPBの親和度測定
GST-BPBをSPR biacore systemを利用してフィブロネクチンフィブロネクチン EDBに対するaffinityを測定した。Affinityを測定した結果、284nMのaffinityを有して、GSTがあるにもかかわらず、十分フィブロネクチンEDBを認識できることを示した(図11b)。
【0145】
実施例20:TNFα-BPB fusion protein
1.TNFα-BPB遺伝子の製作
pET28b vector(Novagen)にhuman TNFα(tumor necrosis factor α)とフィブロネクチンEDBを認識するBPB遺伝子シーケンスをクローニングした。二つのオリゴヌクレオチドTNF-F1(AATAAAACATATG TCTCGAACCCCGA)とTNF-B1(ATGGATCCCAGGGCAATGATC)を利用して、human TNFαがクローニングされているベクターGh75(Cytokine Bank, Korea)でTNFα遺伝子を増幅して、これをpET28bにクローニングした。そして二つのオリゴヌクレオチドBPB-F1(AAT GAATTC TCTTCCTCATCGGGTTCTTCCTCATCGGGTTGTAGTTCTCCTATTC)とBPB-B1(AAT AAGCTT TCA TTGCTCCAACCTAAT)を利用し、EDBタンパク質に付くファージで得たphagemid vector(実施例1及び3)をtemplateでPCRして、フィブロネクチンEDBに付くBPBペプチド遺伝子を得た。このBPBペプチドのペプチド配列は、CSSPIQGSWTWENGKWTWKGIIRLEQである。
【0146】
前記BPB遺伝子を、TNFαの入ったpET28bベクターにクローニングした。plasmid preparation kitを利用してplasmidを精製し、シーケンシングをして、クローニングが成功したかどうかを確認した。
【0147】
2. TNFα-BPB発現及び精製
TNFα-BPBをクローニングしたpET28bベクターをBL21 cellにtransformationした後、kanamycine agar培地に塗抹した。2L LBにカナマイシン(25ug/ml)を入れて、OD=0.6-0.8まで育てた後、1mM isopropyl-β-D-thiogalactopyranoside(IPTG)を入れて、37℃で200rpmの速度で混ぜながら、8時間育てた。4,000gで20分間遠心分離して、沈んだ細胞を除いた上澄み液を全て除去して、lysis buffer (50 mM sodium phosphate (pH 8.0), 300mM NaClと5mM imidazole)で再浮遊させた。-80℃で一日間保管した後、Sonicatorを利用してE. coli溶解させた後、15,000gで1時間遠心分離して、上澄み液をNi-NTA affinity resin(ELPIS biotech.)に結合させた。Lysis bufferでresinを洗浄した後、Elution buffer(50mM sodium phosphate (pH 8.0), 300 mM NaCl and 300 mM imidazole)を利用して、TNFα-BPBタンパク質を分離して収集した。このように集めたタンパク質をSuperdex75 columnとPBS(pH7.4) bufferを利用してgel filtrationをして、純度高いTNFα-BPBタンパク質を収集した。
【0148】
3. TNFα-BPBのフィブロネクチン EDB binding実験
精製されたTNFα-BPBを、フィブロネクチンEDBにbindingするかをSPR機械を利用して確認した。Affinity測定は、BIAcore Xを利用して行った。Streptavidin SA chip(Biacore)にbiotin-EDBを2000RUだけ流して固定させた。Running bufferとしてはPBS(pH7.4)を使用して、流速は30ul/minとして流しながら、様々な濃度でkineticsを測定した後、BIAevaluationでaffinityを計算した。
【0149】
4. TNFα-BPBのcytotoxicity実験
L-M mouse fibroblastsを利用して、TNFα-BPBとTNFαのcytotoxicitiyをMTT分析で測定した。まずL-M mouse fibroblastを96well microplateに5000個を敷いた後、18時間後にTNFαとTNFα-BPBを濃度別に入れて、30時間後にMTTを行って、cytotoxicitiyを測定した。
【0150】
5.実験結果
(1)TNFα-BPB製作
フィブロネクチンED-Bを特異的に認識するBPBをTNFαにクローニングした後、 TNFα-BPBを大腸菌で発現して精製した(図12a)。
【0151】
(2)TNFα-BPBの親和度測定
TNFα-BPBを、SPR biacore systemを利用してフィブロネクチン EDBに対するaffinityを測定した。Affinityを測定した結果、80nMのaffinityを有して、TNFαがあるにもかかわらず、十分フィブロネクチンEDBを認識できることを示した(図12b).
【0152】
(3)TNFα-BPBのcytotoxicitiy測定
L-M mouse fibroblast cellでTNFnとTNFn-BPBのcytotoxicityをMTTで測定した。TNFα-BPBがTNFαより5倍程度cytotoxicityが高い(図12c)。
【0153】
実施例21:リポソーム-BPB
1. Conjugation of BPBcssto Mal-PEG2000-DSPE
Mal-PEG2000-DSPEを利用し、BPBEDB(CSSPIQGSWTWENGK(Cys, lysine残基にcysを連結)WTWKGIIRLEQ)のシステイン残基にコンジュゲーションさせた。簡単には、Mal-PEG2000-DSPEの1.1 fold molar excessをchloroform/DMSO(1:1 v/v)でBPBEDBと混合した。12時間常温で攪拌して、反応を進行した。
【0154】
2. Encapsulation efficiency of 9R/siRNA in BPBcss-LS
9R peptide及びVEGF-C siRNA(sense 5' CAG AUG GAU UCC AUG ACA dTdT, anti-sense 5' AUG UCA UGG AAU CCA UGU G dTdT)をHEPES-buffered 5% glucose (pH 7.4)で希釈して、最終容量250μlになるようにして、室温で10分間インキュベーションした。前記溶液を混合してボルテキシングして、500μl 9R/siRNA complexes at a +/- charge ratio of 1:6を得た。同時にBPB-DSPEが内包されたリポソームを作るためのPOPC/Chol/POPG/PEG2000-DSPE/BPB-DSPE (4:3:3:1:0.5)mixtureと対照群リポソームのためのPOPC/Chol/POPG/PEG2000-DSPE (4:3:3:1)mixtureを利用してlipid filmを作る。これに500ul Hepes buffer glucose 5% (HBG 5%)を入れた後、lipid filmに500ul 9R/siRNA complexを入れた。この混合物を簡単にソニケーションした後、hand-held extruder (Avanti Polar Lipid, AL, USA)を利用して二つのpolycarbonate membranes (100 nm pore size)のスタックを通じて11回押出した。9R/siRNA-loaded BPB-リポソームs(BPBcss-LS)と対照群である9R/siRNA-loaded-リポソームsをsize-exclusion chromatography (CL-4B column)で精製した。Sepharose CL4B column eluentの分画をOliGreen (Invitrogen)で分析してsiRNA含量を分析した。
【0155】
3. 大きさ分布及びzeta-potentials
9R/siRNAがローディングされたBPBcss-LSをHBS(Hepes Buffer Saline)で希釈し、最適のscattering intensityを得た。Hydrodynamic diameter及びzeta potentialを、ELS 8000装置(Otsuka Electronics Korea, Seoul, South Korea)を利用してelectrophoretic light scattering方法で測定した。
【0156】
4. Uptake of BPBcss-LS in EDB positive and EDB negative cell line
細胞をcover glass coated with 2% gelatin上で一晩中培養した。24時間後、細胞に200ugのBPB-リポソーム(0.3% rhodomaime含み)と対照群の200ugのリポソーム(0.3% rhodamine)をそれぞれ処理した。37℃で1時間処理後、細胞を3回洗浄し、4%パラホルムアルデヒドparaformaldehydeで固定化した後、共焦点顕微鏡で観察した。
【0157】
5. siRNA transfection efficiency in MCF-7 cells in vitro
BPBcss-LSにencapsulatされたsiRNAのMCF-7 cell内へのtransfection効率を評価するために、MCF-7 cellを50nM VEGF-C siRNA in lipofectamine(positive control), 50 nM BPBcss-LS及び100 nM BPBcss-LSで形質転換させた。4時間経過後、細胞を24時間さらにインキュベーションして、次いでRNA分離、cDNA合成及び実時間RT-PCRを行った。
【0158】
6.実験結果
(1) BPB-リポソーム大きさ及びゼータ電位
BPBcss-LSの測定された大きさ及びゼータ電位は、下記表のようである。
【0159】
【表4】
【0160】
(2) BPB-リポソームにsiRNA内包
図13aから分かるように、BPB-リポソーム(CSS-LS)にsiRNAを内包したものとfree siRNAを除去するために、size exculsion CL-4Bカラムを利用して分離した。大きさの大きいsiRNA/BPB-リポソームが先に出て、その後free siRNAが出ることを観察することができる。この方法でsiRNAの内包された、精製されたBPB-リポソームを得ることができる。
【0161】
(3) BPB-リポソームの特異性
ターゲットタンパク質であるEDBが過発現されるU87MG, MCF-7, MCF-7/ADR細胞株を利用して、BPB-リポソームがよく認識するかを確認した。0.3%ロダミンの含まれたBPB-リポソームと対照群のリポソームを三つの細胞に一時間処理した後、洗浄して、顕微鏡で観察した。図13bから分かるように、BPB-リポソーム(CSS-LS)は、全ての細胞によく付着することを確認した。しかし、対照群であるBPBのないリポソーム(LS)は、EDB過発現細胞株に付着しなかった。これは、BPB-リポソームが EDBを細胞内で特異的に認識できることを示す結果である。
【0162】
また、EDB negative cellであるPC3とLnCapを利用し、上記と同様な方法でBPB-リポソームと対照群のリポソームを処理した時、両方ともそれぞれの細胞に付着しないことを図13cから確認できる。これは、ターゲットタンパク質であるEDBがないと、BPB-リポソームであっても付着しないことを示し、BPB-リポソームがEDBに非常に特異的であることを示す。
【0163】
(4) MCF-7細胞にBPB-LS/siRNA内包効能テスト
図13dから分かるように、VEGF-C siRNAを内包したBPB-LSが細胞内でVEGF-CのmRNAを抑制できるかをMCF-7細胞で確認した。50nMと100nM VEGF-C siRNAを内包したBPB-LSを細胞に処理した時、一般によく使用されるリポフェクタミンとほぼ等しくVEGF-C mRNAを抑制できることを示した。
【0164】
実施例22:Oleic SPION-BPB
1. Oleic SPION-BPBの製造
500ug/100ul(CHCl3) DSPE-PEG2000-Mal(Avanti Polar Lipids, Alabaster, AL, USA)(1,2-Distearoyl-sn-Glycero-3-Phosphoethanolamine-N-[Maleimide(Polyethylene Glycol)2000])とBPB(SSS) peptide(SSSPIQGSWTWENGK(Cys)WTWKGIIRLEQ) 250ug/100ul(DMSO)をCHCl3/DMSO co-solvent下で一晩中反応した。反応後、MALDI-TOF-MS(SHIMADZU Axima-CFR,SHIMADZE, Japen)を利用して反応を確認した。DSPE-PEG2000polymerとBPB conjugated DSPE-PEG2000 及びRhodamine-DSPEを(5:94:1)の比率でD.W.に入れた後、Oleic SPION(superparamagnetic iron oxide nanoparticle, 熱分解法で合成)をHexaneに溶かした後、上記のpolymer solutionに入れて、probeタイプのsonicatorを利用してsonicationした後、stirring方法とcentrifuge、そしてmagnetを利用して精製した。そして、BPBのないグループも上記の同様な方法で合成した。
【0165】
2.合成後、Size測定ELSとTEMを利用してBPB conjugatedされたDSPE-PEG2000 コーティングOleic SPION確認
electrophoretic light scattering(ELS 8000 instrument of Otsuka Electronics, Otsuka Electronics Korea, Seoul, South Korea)を利用してHydrodynamic sizeを測定し、BPB conjugate有無のサイズ変化を測定して、また、一定時間D.W.における安定性を確認し、transmission electron microscopy(Philips TECNAI F20 instrument (Philips ElectronicInstruments Corp., Mahwah, NJ))を利用してナノ粒子のaggregation程度を確認した。
【0166】
3. T2-weighted MRファントム研究を通じて合成したBPB conjugatedされたDSPE-PEG2000コーティング Oleic SPIONがMR造影剤として使用可能であるかの確認
BPB conjugated DSPE-PEG2000 coated oleic SPIONとDSPE-PEG2000 coated oleic SPIONをそれぞれ濃度別にe-tubeに入れて3T MRI scanner (Magnetom Avanto, Siemens, Germany)を利用してT2 Timeを測定し、r2値を計算した。
【0167】
4.細胞実験を利用してactive targeting確認
6 well plateにU87MG細胞を37℃ CO2 インキュベーターで培養密度90-100%に培養後、同じ濃度のBPB conjugated DSGPE-PEG2000 coated SPIONとDSGPE-PEG2000coated SPIONを培地に混ぜて30分間培養した後、細胞を集めて、3T MRI scanner (Magnetom Avanto, Siemens, Germany)を通じて信号強度の差を確認した。また、confocal microscope (Olympus, FV1000)を利用し、細胞におけるロダミン信号の差を確認した。
【0168】
5.細胞毒性実験
96ウェルプレートにU87MG細胞を37℃ CO2 インキュベーターで培養密度50%に培養後、同じ濃度のBPB conjugated DSPE-PEG2000 coated oleic SPIONとDSPE-PEG2000coated oleic SPIONを培地に混ぜて12時間培養した後、MTT assayを通じて細胞毒性有無を確認した。
【0169】
6.脳癌動物モデルを利用したactive targeting MR実験
Balb/c nude(female) 6 weeks miceの後ろ右足の上部にU87MG細胞5 x 106 個の細胞を移植し、2週間程度育てた後、癌の大きさが約100-150mm3 になった時、BPB conjugated DSPE-PEG2000coated oleic SPIONとDSPE-PEG2000 coated oleic SPIONを20mg Fe/kgずつ尻尾静脈に投与後、3T MRIを利用して癌部位の信号強度を時間別にモニターした。
【0170】
7. 実験結果
(1) SPION-BPBの製作
DSPE-PEG2000-MalとBPB(SSS)ペプチドのコンジュゲーションを、MALDIを利用して確認した(図14a)。
【0171】
DSPEを利用してSPIONを合成する場合、Hydrodynamic size(水溶液状態でsize測定)を測定すると、約30nm程度のsizeが出て、BPB(SSS peptide)をコンジュゲートしたDSPEを使用する場合、sizeが約20nm程度増加した。
【0172】
【表5】
【0173】
一週間ELSを利用してHydrodynamic size変化を観察したが、一週間ナノ粒子の大きさに大きい変化がなく、安定した状態であることを確認した(図14b)。TEMイメージを通じて、ナノ粒子(SPION)にBPBがコンジュゲートされた後にもよく分散されることを確認した。また、中心コアのサイズが約12nm程度になることを確認した(図14c)。
【0174】
(2) T2-weighted MRファントム研究を通じて、BPB conjugated DSPE-PEG2000 コーティングOleic SPIONがMR造影剤として使用可能であるかの確認
DSPE-PEG2000 coated oleic SPION及びBPB conjugated DSPE-PEG2000 coated oleic SPIONのいずれも、r2値は120 mM-1S-1であって、MRI T2造影剤として使用可能であることを確認した(図14d)。
【0175】
(3)細胞実験を利用して active targeting確認
細胞実験でBPBのあるグループとないグループ間のMRイメージ及び信号強度の差を確認した(図14e)。BPB conjugated DSPE-PEG2000 coated SPIONが細胞にさらに多くuptakeされて、MRイメージでさらに黒く表れて、またMR信号比較(ROI信号比較)においても、DSPE-PEG2000 coated SPIONよりさらに低い値を示した。
【0176】
Confocal顕微鏡を利用して、DSPE-PEG2000 coated SPIONとBPB conjugated DSPE-PEG2000 coated SPIONのロダミン信号の差を確認した(図14f)。BPBコンジュゲーションされたグループから細胞のロダミン信号がさらに強く表れた。
【0177】
(4)細胞毒性実験
MTT実験を通じて、DSPE-PEG2000 coated SPIONとBPB conjugated DSPE-PEG2000 coated SPIONの細胞毒性実験を行った結果、両グループとも低い細胞毒性を示した(図14g)。
【0178】
(5)脳癌動物モデルを利用したactive targeting MR実験
脳癌動物モデルにおいて、BPB conjugated DSPE-PEG2000coated SPIONグループがinjection 1時間後、癌部位が黒く表れることを確認した(図14h)。それに対し、BPB conjugateされなかったDSPE-PEG2000 coated SPIONグループでは、injectionの前と後において大きい変化が現れなかった。
【0179】
実施例23:Gold NPs-BPB
1. Pegylated Gold nanoparticleの製造
金イオンを、還元剤を使用し、約5nm大きさを有する金ナノ粒子を合成した。5 nm金ナノ粒子が生体内における安定性、即ちRES(reticuloendothelial system)などへのuptakeを減らして、目標細胞への伝達効率を増加させるために、金ナノ粒子表面を生体的合成高分子のPEGでコーティングが必要である。したがって、PEGにチオールグループを導入するために、ジチオールグループとカルボキシルグループが含まれたlipoic acidと反応させた。Lipoic acidにPEGを結合するために、まず、lipoic acidのカルボキシルグループをDCC(dicyclohexylcarbodiimide)とNHS (N-hydroxylsucciniimide)で活性化させた後、100ugのNH2-PEG-OCH3と反応させることによりlipoic-PEGを合成した。金ナノ粒子にPEGがコーティングされることを、1M NaClを添加してaggregationされるかを測定して、PEGが完璧にコーティングされることを測定した。
【0180】
2. Gold nanoparticle-BPBコンジュゲーション
合成されたlipoic-PEG-maleimdeとチオールグループが露出されたBPB(CSSpペプチド, CSSPIQGSWTWENGK(cys)WTWKGIIRLEQ)をCHCl3/DMSO(3:1) co-solvent下で一晩中反応した後、lipoic-PEG-maleimde-BPB利用してgold nanoparticleをコーティングした。
【0181】
3. Gold nanoparticle-BPBの癌細胞認識実験
BPB-金ナノ粒子がフィブロネクチンED-Bを認識するかどうか、フィブロネクチン ED-BがoverexpressionされるU87MG glioblastomaを利用して、癌細胞認識を確認した。Human U87MG glioblastoma cellを90-100% confluencyでcover slipに育てた後、BPB-金ナノ粒子を1時間37℃でcellとインキュベーションした。その後、PBSで3回洗浄した後、細胞をトリプシンで分離した後、細胞を集めてInductively coupled plasma mass spectrometry (ICP-MS)を利用し、細胞内Auの量を測定した。
【0182】
4. 金ナノ粒子に両性イオン(zwitterions)コーティング
金ナノ粒子は、既存の造影剤が有している短所(短い映像時間、腎臓毒性)を克服することができるが、金ナノ粒子も同様に大きさなどの問題によって、体外に排出されない問題点を有している。このような問題点を克服するために、PEGの代わりに、PEGのような役割をしながらもナノ粒子のhydrodynamicサイズを大きく増加させない両性イオンを金ナノ粒子にコーティングさせることにより、最終金ナノ粒子のhydrodynamic大きさを8nm以下に減らして、体外に排出できるようにナノ粒子をデザインした。また、本研究陳で使用した両性イオンは、金ナノ粒子と結合できる部分として、モノチオール(mono-thiol)グループではなく、環状ジチオール(cyclic dithiol)グループを導入することにより、血液内で金ナノ粒子と両性イオン間の安定性(stability)を増加させた。両性イオンの構造は、下記のようである。
【化1】
【0183】
次に、金ナノ粒子が血清(serum)と1M NaCl下で安定するために必要な両性イオンと金ナノ粒子間の最適の比率を、金ナノ粒子の表面プラズモン(surface Plasmon resonance)最大ピーク(peak)の変化を観察しながら測定した。まず、金ナノ粒子と両性イオン間の多様な比率(1:3000, 1:5000, 1:7000, 1:10000, 1:12000, 1:15000)を混ぜて、12時間反応後、遠心分離機を利用して分離した。10%血清と1M NaClを添加して、24時間が経過した後、表面プラズモン共鳴最大ピークをUV-Vis spectrumを利用して測定した。
【0184】
5. 金ナノ粒子にBPBを付着するためのBPB変形
金ナノ粒子とBPB間の結合力、即ち、血液内での安定性を増加させるために、BPBでも同様に環状ジチオール(cyclic dithiol)グループを導入した。即ち、lipoic acidをDCC(N,N`-Dicyclohexylcarbodiimide)/NHS(N-hydroxl succinimides)とTHF(tetrahydrofuran)で72時間反応し、lipoic-NHS形態を作って、トルエンによる再結晶方法で精製した。精製したlipoic-NHSは、N-末端基がアセチル化(acetylation)されたBPBのリジン(lysine)残基のアミン(NH2)グループとDMSO(Dimethyl sulfoxide)で2時間反応し、BPBに環状ジチオールグループを導入することができた。
【0185】
6.実験結果
(1) Gold Nanoparticle-BPBの製作
金ナノ粒子は、現在広く使用されているヨードベースのCT造影剤の短所である腎臓毒性、短い映像化時間などを克服できるだけではなく、肝癌造影が可能な生体適合性高分子でコーティングした金ナノ粒子を次世代CT造影剤として開発することができる。ここでは、癌標的BPBを付着することにより、CTで癌をイメージングできる金ナノ粒子を開発した。5nmの金ナノ粒子にフィブロネクチンEDBを認識するBPBをコンジュゲーションした。TEMイメージを通じて、ナノ粒子が、BPBがコンジュゲートされた後にもよく分散されているかを確認した(図15a)。
【0186】
(2) Gold Nanoparticle-BPBの癌細胞標的実験
U87MG細胞を利用して、BPB conjugated金ナノ粒子を同一な濃度として入れた後、ICP-MSを利用して細胞内のAuの量の差を測定した。BPBのあるグループが、BPBのないグループより細胞内Auの量が多いことが確認された。したがって、BPB conjugated金ナノ粒子の方がEDBによる細胞表面吸着と細胞内伝達にさらに優れていることを確認した(図15b)。また、BPBがコンジュゲーションされなかった金ナノ粒子よりBPBのコンジュゲーションされた金ナノ粒子の方が選択的にU87MG細胞に吸着することを、silver enhancement方法を利用して顕微鏡観察によっても確認することができた(図15c)。
【0187】
(3)金ナノ粒子の両性イオンコーティング
BPBを金ナノ粒子に付着した時、選択的に癌細胞に吸着されることを確認した後、金ナノ粒子が生体内に注入された時、血液内で安定するように、金ナノ粒子表面を両性イオンでコーティングした。金ナノ粒子と両性イオン間の比率が1:10000以上である時、金ナノ粒子の表面プラズモン共鳴最大ピークが、血清及び1M NaClを添加して24時間が経っても変化がないことから、血清及び1M NaClの環境においても金ナノ粒子が安定したことを確認することができた。
【0188】
(4)金ナノ粒子にBPBを付着するためのBPB変形
BPBに環状ジチオールグループを導入したLipoic-BPBをHPLCで分離し、MALDI-TOFを通じて合成物を確認することができた。
【0189】
実施例24:Drug-BPB conjugates
1. Docetaxel(DTX)-NHSの製造及び確認
DTXのヒドロキシル基をsuccinic anhydrideと常温で12時間反応し、DTX succinic acidを製造した。BPBのlysineが有しているfree amineにDTX succinic acidをコンジュゲーションさせるために、まずアミンに反応するNHSグループをDTX succinic acidに結合させた。反応条件は、DTX succinic acidをMC(methylene choloride)溶液に溶かして、EDC/NHSを加えて常温で12時間反応した。この際、モル比は、DTX succinic acide : EDC : NHS = 1 : 1.2 : 1.5として行った。反応後、移動相溶媒をEA(ethyl acetate)としてTLC(thin liquid chromatography)分析を行った。
【0190】
2. DTX-NHSとBPBコンジュゲーション
DTX-NHSをコンジュゲーション反応直前にDMFに溶かして、BPBを追加して溶かした。完全に溶けない場合、DMSOを少しずつ添加した。これにBPBのfree amine形成を助けるbase DIEA(di-isoethyl amine)を添加して、常温で12時間反応した。DIEAは、BPBの2当量以上入れた。BPB配列は、N-terminal acetylated BPBであって、配列は、SSSPIQGSWTWENGKWTWRGIIRLEQである。
【0191】
3. DTX-BPBの精製及び分析
DTX-NHSとBPBコンジュゲーション反応液を真空乾燥機で乾燥させた後、50% ACNに溶かしてHPLCで分析し、DTX-BPBコンジュゲートに該当するHPLCピークサンプルを、質量分析(MALD TOF)を行ってDTX-BPBコンジュゲート形成を確認した。
【0192】
以上、本発明の望ましい具現例を詳細に記述したが、当業界の通常の知識を有する者にとっては、このような具体的な記述はただ望ましい具現例に過ぎなく、これに本発明の範囲が限定されないことは明らかである。従って、本発明の実質的な範囲は、添付の請求項とその等価物により定義されると言える。
【特許請求の範囲】
【請求項1】
(a)(i)鎖間(interstrand)非共有結合が形成されたパラレル(parallel)、アンチパラレル(antiparallel)、またはパラレル(parallel)とアンチパラレル(antiparallel)アミノ酸鎖を含む構造安定化部位(structure stabilizing region)と、(ii)前記構造安定化部位の両末端に結合されており、無作為的に選択されたそれぞれn及びm個のアミノ酸を含むターゲット結合部位I(target binding region I)及びターゲット結合部位II(target binding region II)と、を含む二座ペプチドバインダー(Bipodal Peptide Binder:BPB)と、
(b)前記二座ペプチドバインダーに結合されたカーゴと、
を含む、BPBベースのカーゴ運搬システム(Cargo Delivery System)。
【請求項2】
前記構造安定化部位に形成された鎖間非共有結合は、水素結合、静電気的相互作用、疎水性相互作用、ファンデルワールス相互作用、パイ−パイ相互作用、陽イオン−パイ相互作用、またはこれらの組み合わせであることを特徴とする、請求項1に記載の運搬システム。
【請求項3】
前記構造安定化部位のアミノ酸鎖は、リンカーで連結されたことを特徴とする、請求項1に記載の運搬システム。
【請求項4】
前記構造安定化部位は、β−ヘアピン、リンカーで連結されたβ−シート、ロイシンジッパーまたはリンカーで連結されたロイシンジッパー、ロイシンリッチモチーフであることを特徴とする、請求項1に記載の運搬システム。
【請求項5】
前記構造安定化部位は、β−ヘアピンであることを特徴とする、請求項1に記載の運搬システム。
【請求項6】
前記β−ヘアピンは、配列番号1乃至19からなる群から選択されることを特徴とする、請求項5に記載の運搬システム。
【請求項7】
前記β−ヘアピンは、配列番号2、14、または18から選択されることを特徴とする、請求項6に記載の運搬システム。
【請求項8】
前記β−ヘアピンは、下記一般式Iで表されることを特徴とする、請求項5に記載の運搬システム。
[一般式I]
X1-Trp(X2)X3-X4-X5(X'2)X6-X7
X1は、SerまたはGly-Gluであり、X2及びX'2は、それぞれ独立してThr、His、Val、Ile、PheまたはTyrであり、X3は、TrpまたはTyrであり、X4は、タイプI、タイプI'、タイプII、タイプII'またはタイプIIIまたはタイプIII'ターン配列であり、X5は、TrpまたはPheであり、X6は、TrpまたはValであって、X7は、LysまたはThr-Gluである。
【請求項9】
前記β−ヘアピンは、下記一般式IIで表されることを特徴とする、請求項5に記載の運搬システム。
[一般式II]
X1-Trp-X2-Tyr-X3-Phe-Thr-Val-X4
X1は、Arg、Gly-GluまたはLys-Lysであり、X2は、GlnまたはThrであり、X3は、タイプI、タイプI'、タイプII、タイプII'またはタイプIIIまたはタイプIII'ターン配列であって、X4は、Gln、Thr-GluまたはGln-Gluである。
【請求項10】
前記β−ヘアピンは、下記一般式IIIで表されることを特徴とする、請求項5に記載の運搬システム。
[一般式III]
X1-X2-X3-Trp-X4-X5-Thr-X6-X7
X1は、LysまたはLys-Lysであり、X2は、TrpまたはTyrであり、X3は、ValまたはThrであり、X4は、タイプI、タイプI'、タイプII、タイプII'またはタイプIIIまたはタイプIII'ターン配列であり、X5は、TrpまたはAlaであり、X6は、TrpまたはValであって、X7は、GluまたはGln-Gluである。
【請求項11】
前記β−ヘアピンは、下記一般式IVで表されることを特徴とする、請求項5に記載の運搬システム。
[一般式IV]
X1-X2-X3-Trp-X4
X1は、Lys-ThrまたはGlyであり、X2は、TrpまたはTyrであり、X3は、タイプI、タイプI'、タイプII、タイプII'またはタイプIIIまたはタイプIII'ターン配列であって、X4は、Thr-GluまたはGlyである。
【請求項12】
前記β−ヘアピンは、前記一般式Iにおいて、X1は、SerまたはGly-Gluであり、X2及びX'2は、それぞれ独立してThr、HisまたはValであり、X3は、TrpまたはTyrであり、X4は、タイプI、タイプI'、タイプIIまたはタイプII'配列であり、X5は、TrpまたはPheであり、X6は、TrpまたはValであって、X7は、LysまたはThr-Gluであることを特徴とする、請求項8に記載の運搬システム。
【請求項13】
前記ターゲット結合部位Iのアミノ酸の数nは、2〜20であることを特徴とする、請求項1に記載の運搬システム。
【請求項14】
前記ターゲット結合部位IIのアミノ酸の数mは、2〜20であることを特徴とする、請求項1に記載の運搬システム。
【請求項15】
前記ターゲット結合部位I及びターゲット結合部位IIは、ターゲットに共同に作用して結合することを特徴とする、請求項1に記載の運搬システム。
【請求項16】
前記構造安定化部位、ターゲット結合部位Iまたはターゲット結合部位IIに、他の追加的な機能性分子が結合されていることを特徴とする、請求項1に記載の運搬システム。
【請求項17】
前記機能性分子は、細胞膜透過ペプチド(CPP)であることを特徴とする、請求項16に記載の運搬システム。
【請求項18】
前記ターゲットは、生化学物質、ペプチド、ポリペプチド、核酸、炭水化物、脂質、細胞、組織、化合物、金属または非金属物質であることを特徴とする、請求項1に記載の運搬システム。
【請求項19】
前記カーゴは、化合物、化学薬物、バイオ薬物、無機粒子、ナノ粒子、タンパク質、ペプチド、核酸分子、脂質、炭水化物、リポソームまたは検出可能な信号を発生させるラベル分子であることを特徴とする、請求項1に記載の運搬システム。
【請求項1】
(a)(i)鎖間(interstrand)非共有結合が形成されたパラレル(parallel)、アンチパラレル(antiparallel)、またはパラレル(parallel)とアンチパラレル(antiparallel)アミノ酸鎖を含む構造安定化部位(structure stabilizing region)と、(ii)前記構造安定化部位の両末端に結合されており、無作為的に選択されたそれぞれn及びm個のアミノ酸を含むターゲット結合部位I(target binding region I)及びターゲット結合部位II(target binding region II)と、を含む二座ペプチドバインダー(Bipodal Peptide Binder:BPB)と、
(b)前記二座ペプチドバインダーに結合されたカーゴと、
を含む、BPBベースのカーゴ運搬システム(Cargo Delivery System)。
【請求項2】
前記構造安定化部位に形成された鎖間非共有結合は、水素結合、静電気的相互作用、疎水性相互作用、ファンデルワールス相互作用、パイ−パイ相互作用、陽イオン−パイ相互作用、またはこれらの組み合わせであることを特徴とする、請求項1に記載の運搬システム。
【請求項3】
前記構造安定化部位のアミノ酸鎖は、リンカーで連結されたことを特徴とする、請求項1に記載の運搬システム。
【請求項4】
前記構造安定化部位は、β−ヘアピン、リンカーで連結されたβ−シート、ロイシンジッパーまたはリンカーで連結されたロイシンジッパー、ロイシンリッチモチーフであることを特徴とする、請求項1に記載の運搬システム。
【請求項5】
前記構造安定化部位は、β−ヘアピンであることを特徴とする、請求項1に記載の運搬システム。
【請求項6】
前記β−ヘアピンは、配列番号1乃至19からなる群から選択されることを特徴とする、請求項5に記載の運搬システム。
【請求項7】
前記β−ヘアピンは、配列番号2、14、または18から選択されることを特徴とする、請求項6に記載の運搬システム。
【請求項8】
前記β−ヘアピンは、下記一般式Iで表されることを特徴とする、請求項5に記載の運搬システム。
[一般式I]
X1-Trp(X2)X3-X4-X5(X'2)X6-X7
X1は、SerまたはGly-Gluであり、X2及びX'2は、それぞれ独立してThr、His、Val、Ile、PheまたはTyrであり、X3は、TrpまたはTyrであり、X4は、タイプI、タイプI'、タイプII、タイプII'またはタイプIIIまたはタイプIII'ターン配列であり、X5は、TrpまたはPheであり、X6は、TrpまたはValであって、X7は、LysまたはThr-Gluである。
【請求項9】
前記β−ヘアピンは、下記一般式IIで表されることを特徴とする、請求項5に記載の運搬システム。
[一般式II]
X1-Trp-X2-Tyr-X3-Phe-Thr-Val-X4
X1は、Arg、Gly-GluまたはLys-Lysであり、X2は、GlnまたはThrであり、X3は、タイプI、タイプI'、タイプII、タイプII'またはタイプIIIまたはタイプIII'ターン配列であって、X4は、Gln、Thr-GluまたはGln-Gluである。
【請求項10】
前記β−ヘアピンは、下記一般式IIIで表されることを特徴とする、請求項5に記載の運搬システム。
[一般式III]
X1-X2-X3-Trp-X4-X5-Thr-X6-X7
X1は、LysまたはLys-Lysであり、X2は、TrpまたはTyrであり、X3は、ValまたはThrであり、X4は、タイプI、タイプI'、タイプII、タイプII'またはタイプIIIまたはタイプIII'ターン配列であり、X5は、TrpまたはAlaであり、X6は、TrpまたはValであって、X7は、GluまたはGln-Gluである。
【請求項11】
前記β−ヘアピンは、下記一般式IVで表されることを特徴とする、請求項5に記載の運搬システム。
[一般式IV]
X1-X2-X3-Trp-X4
X1は、Lys-ThrまたはGlyであり、X2は、TrpまたはTyrであり、X3は、タイプI、タイプI'、タイプII、タイプII'またはタイプIIIまたはタイプIII'ターン配列であって、X4は、Thr-GluまたはGlyである。
【請求項12】
前記β−ヘアピンは、前記一般式Iにおいて、X1は、SerまたはGly-Gluであり、X2及びX'2は、それぞれ独立してThr、HisまたはValであり、X3は、TrpまたはTyrであり、X4は、タイプI、タイプI'、タイプIIまたはタイプII'配列であり、X5は、TrpまたはPheであり、X6は、TrpまたはValであって、X7は、LysまたはThr-Gluであることを特徴とする、請求項8に記載の運搬システム。
【請求項13】
前記ターゲット結合部位Iのアミノ酸の数nは、2〜20であることを特徴とする、請求項1に記載の運搬システム。
【請求項14】
前記ターゲット結合部位IIのアミノ酸の数mは、2〜20であることを特徴とする、請求項1に記載の運搬システム。
【請求項15】
前記ターゲット結合部位I及びターゲット結合部位IIは、ターゲットに共同に作用して結合することを特徴とする、請求項1に記載の運搬システム。
【請求項16】
前記構造安定化部位、ターゲット結合部位Iまたはターゲット結合部位IIに、他の追加的な機能性分子が結合されていることを特徴とする、請求項1に記載の運搬システム。
【請求項17】
前記機能性分子は、細胞膜透過ペプチド(CPP)であることを特徴とする、請求項16に記載の運搬システム。
【請求項18】
前記ターゲットは、生化学物質、ペプチド、ポリペプチド、核酸、炭水化物、脂質、細胞、組織、化合物、金属または非金属物質であることを特徴とする、請求項1に記載の運搬システム。
【請求項19】
前記カーゴは、化合物、化学薬物、バイオ薬物、無機粒子、ナノ粒子、タンパク質、ペプチド、核酸分子、脂質、炭水化物、リポソームまたは検出可能な信号を発生させるラベル分子であることを特徴とする、請求項1に記載の運搬システム。
【図1b】

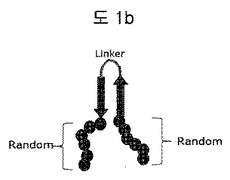
【図1c】
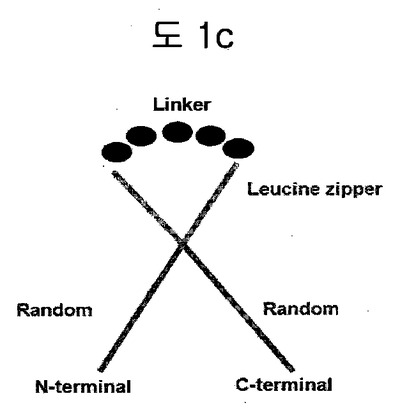
【図1d】
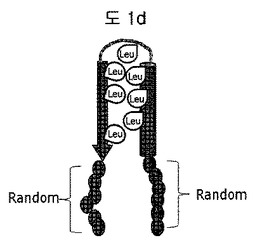
【図2】
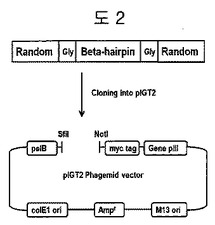
【図5a】
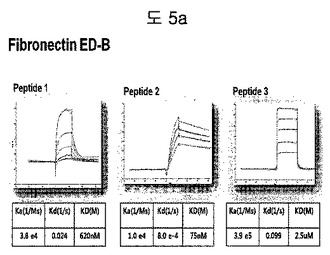
【図5b】
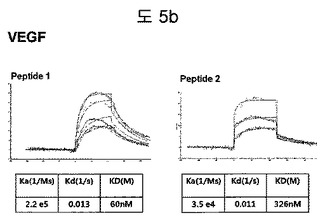
【図5c】
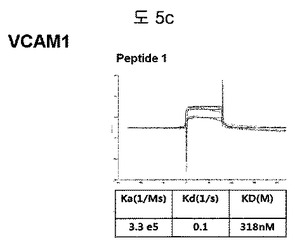
【図5d】
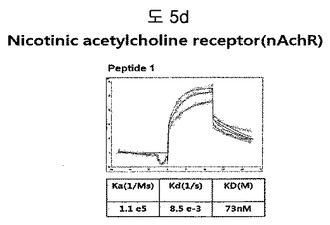
【図5e】
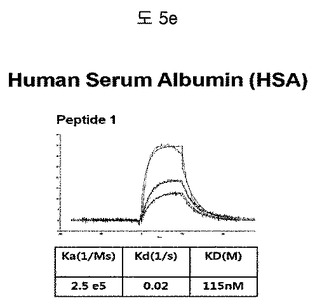
【図6f】
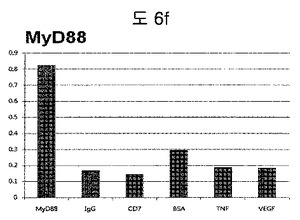
【図7】
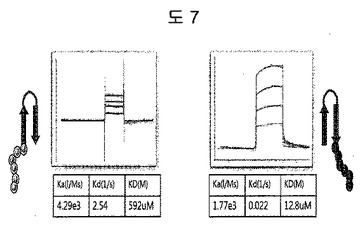
【図8】
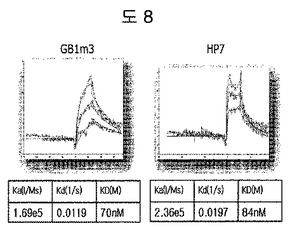
【図9】
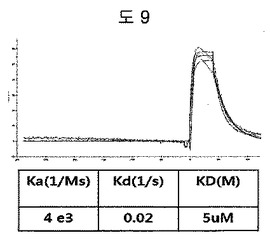
【図10】
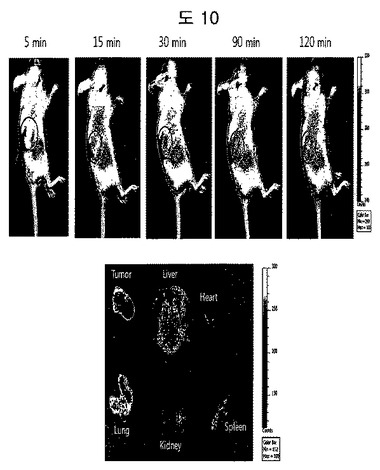
【図11a】
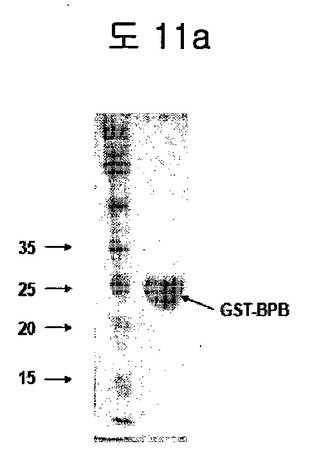
【図11b】
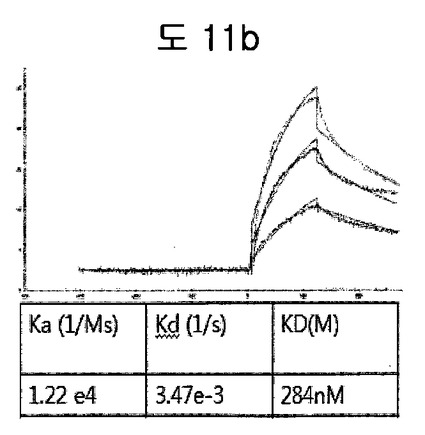
【図12a】
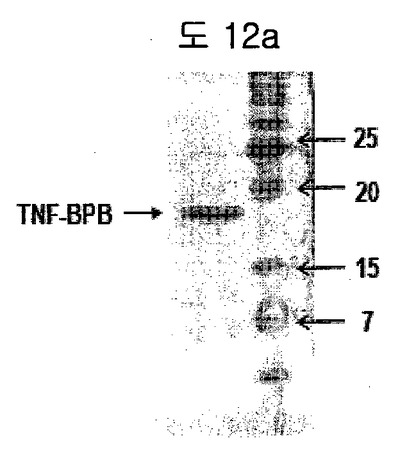
【図12b】
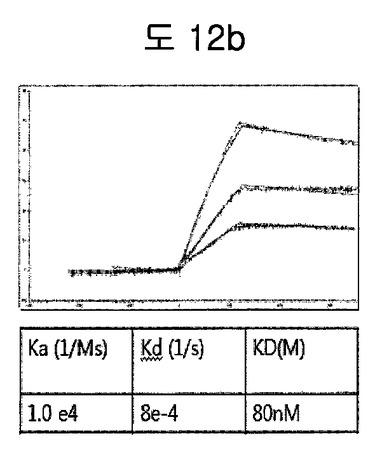
【図12c】
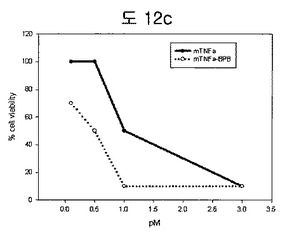
【図13a】
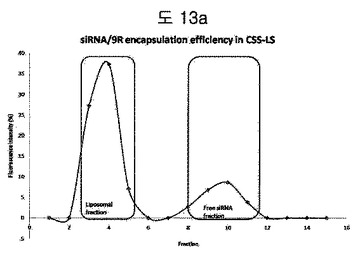
【図13b】
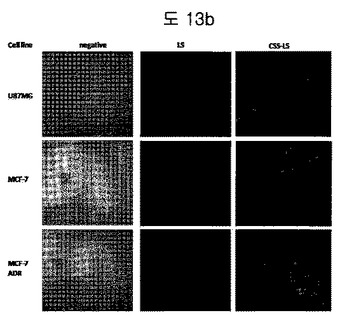
【図13c】

【図13d】
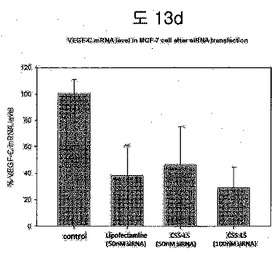
【図14a】
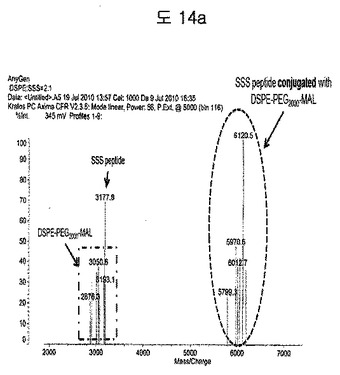
【図14b】
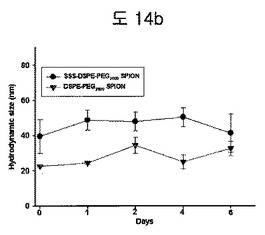
【図14c】
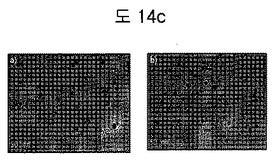
【図14d】
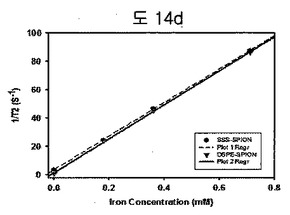
【図14e】
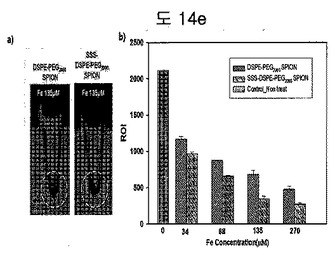
【図14f】
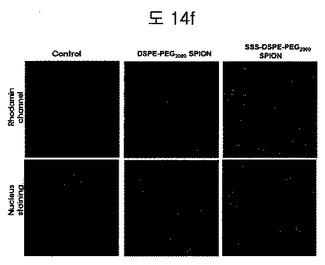
【図14g】
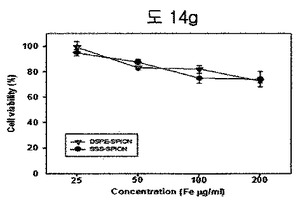
【図14h】
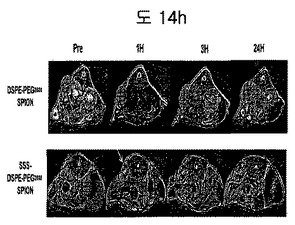
【図15a】

【図15b】
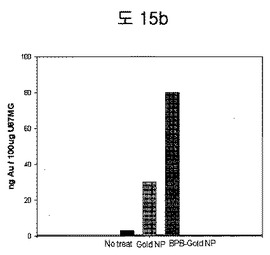
【図15c】

【図1a】
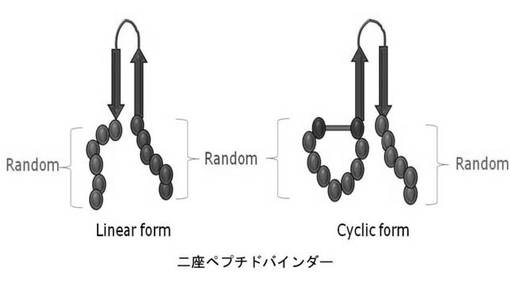
【図3】
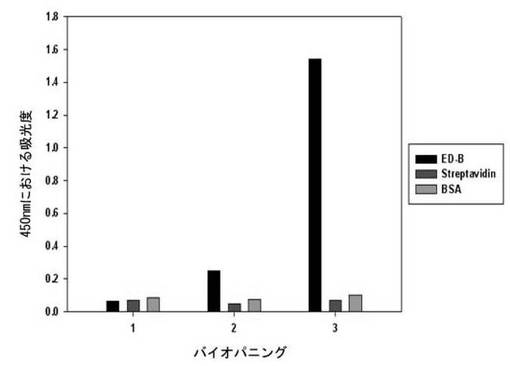
【図4】
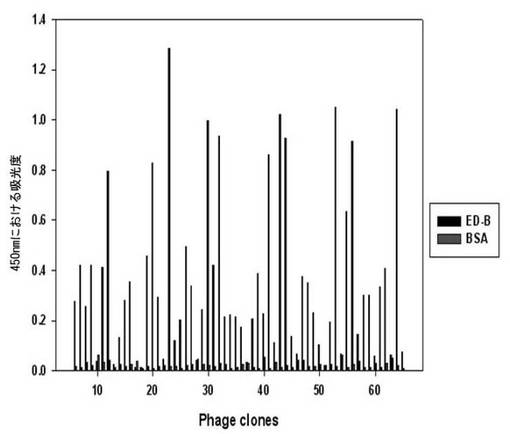
【図6a】
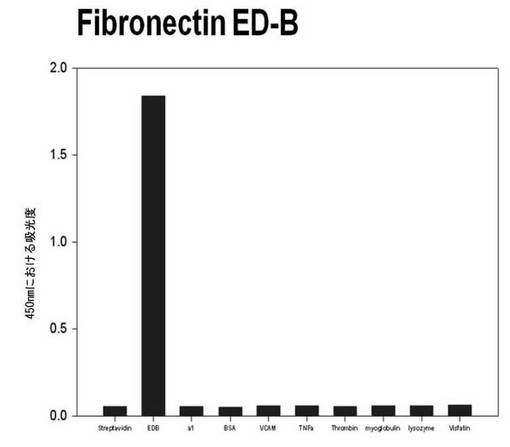
【図6b】
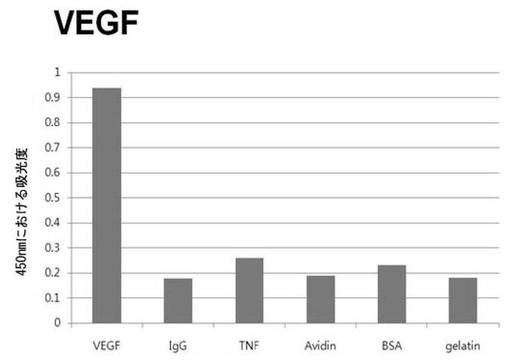
【図6c】
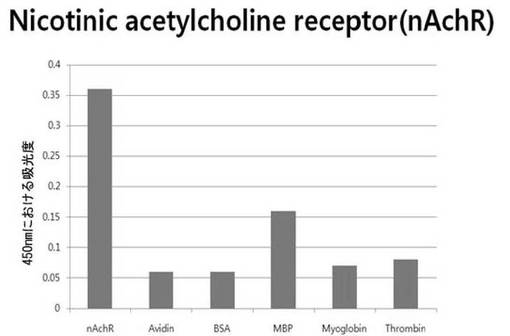
【図6d】
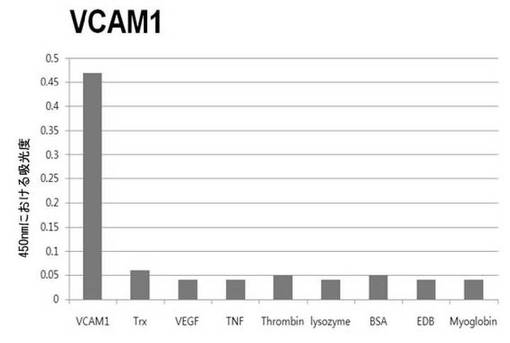
【図6e】
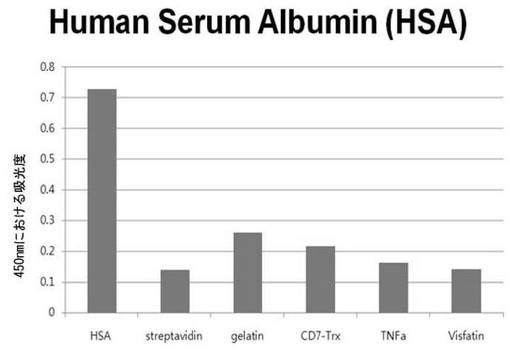
【図1c】
【図1d】
【図2】
【図5a】
【図5b】
【図5c】
【図5d】
【図5e】
【図6f】
【図7】
【図8】
【図9】
【図10】
【図11a】
【図11b】
【図12a】
【図12b】
【図12c】
【図13a】
【図13b】
【図13c】
【図13d】
【図14a】
【図14b】
【図14c】
【図14d】
【図14e】
【図14f】
【図14g】
【図14h】
【図15a】
【図15b】
【図15c】
【図1a】
【図3】
【図4】
【図6a】
【図6b】
【図6c】
【図6d】
【図6e】
【公表番号】特表2013−513601(P2013−513601A)
【公表日】平成25年4月22日(2013.4.22)
【国際特許分類】
【出願番号】特願2012−543020(P2012−543020)
【出願日】平成22年12月3日(2010.12.3)
【国際出願番号】PCT/KR2010/008645
【国際公開番号】WO2011/071279
【国際公開日】平成23年6月16日(2011.6.16)
【出願人】(509143332)グワンジュ・インスティテュート・オブ・サイエンス・アンド・テクノロジー (4)
【Fターム(参考)】
【公表日】平成25年4月22日(2013.4.22)
【国際特許分類】
【出願日】平成22年12月3日(2010.12.3)
【国際出願番号】PCT/KR2010/008645
【国際公開番号】WO2011/071279
【国際公開日】平成23年6月16日(2011.6.16)
【出願人】(509143332)グワンジュ・インスティテュート・オブ・サイエンス・アンド・テクノロジー (4)
【Fターム(参考)】
[ Back to top ]