
C型慢性肝炎に対する治療効果を予測するためのプローブ
【課題】C型慢性肝炎の治療率に関与する一塩基多型(SNP)を検出するためのプローブ、ならびに、該プローブを用いたSNPの検出方法およびSNPを検出するためのキットの提供。
【解決手段】特定の塩基配列を有するプローブを用いる。
【解決手段】特定の塩基配列を有するプローブを用いる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、C型慢性肝炎の治療率に関与する一塩基多型(Single Nucleotide Polymorphism;SNP)を検出するためのプローブ、ならびに、該プローブを用いたSNPの検出方法およびSNPを検出するためのキットに関する。
【背景技術】
【0002】
C型肝炎ウイルス(Hepatitis C Virus;HCV)は感染者の70〜80%が慢性肝炎に移行すると言われている。さらに、慢性肝炎から肝硬変や肺がんへと移行する例も多数報告されている。
【0003】
C型肝炎の代表的な治療法としてペグインターフェロン+リバビリン(PEG−IFN/RBV)併用療法がある。しかし、この治療法の効果には個人差があることが知られてきた。
【0004】
PEG−IFN/RBV治療効果に個人差が生じる理由として、IL28B遺伝子周辺のSNPが関係あることが報告されている(非特許文献1、非特許文献2、特許文献1)。
【0005】
特許文献1にはSNP検査の実施例が記載されている。特許文献1ではシークエンス法やDigiTag2法という方法を用いている。シークエンス法は一般的に検体となるヒト遺伝子を核酸増幅し、その増幅産物の配列を同定するという方法で行う。しかし、この方法では核酸増幅産物のキャリーオーバーコンタミネーションによる誤診が発生するという懸念がある。また、検査開始から結果判明までに数時間から1日の期間が必要である。
【0006】
DigiTag2法は2本のプローブを用いて検出する方法である。プローブを用いる方法は核酸増幅と検出を同じ閉鎖系で行えばコンタミネーションを防ぐことができる。しかしDigiTag2法や蛍光共鳴エネルギー転移(FRET)を利用した方法では、検出には2本以上のプローブが必要であり、コストがかかるという欠点がある。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開2011−41526
【非特許文献】
【0008】
【非特許文献1】Ge D.et al.Nature 461,399−401,2009
【非特許文献2】Tanaka Y.et al.Nat Genet 41,1105−1109,2009
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明は上記従来のC型肝炎治療に関するSNP検査における問題点を解決しようとするものであり、その目的は該SNP検査を迅速、正確、かつ安価に行うための方法及び試薬を提供することにある。
【課題を解決するための手段】
【0010】
本発明者らは上記課題に鑑み鋭意検討した結果、C型肝炎の治療率に関与すると言われるSNPおよびその周辺の塩基配列に特異的に結合する1本の核酸プローブを用いることにより従来技術よりも正確に該SNPを検出できることを見出し、本発明を完成させるに至った。すなわち、本発明は以下のような構成からなる。
【0011】
[項1]
C型肝炎の治療率に関与すると言われるSNPを検出するためのプローブであって、配列番号1〜3のいずれかで示される塩基配列を有するプローブ。
[項2]
検出可能な標識物質により標識化された、項1に記載のプローブ。
[項3]
検出可能な標識物質が蛍光色素である、項2に記載のプローブ。
[項4]
末端のシトシンが標識化された、項2または3に記載のプローブ。
[項5]
C型肝炎の治療率に関与すると言われるSNPを検出する方法であって、以下(1)〜(3)の工程を全て含む方法。
(1)被検核酸と項1〜4のいずれかに記載のプローブとをハイブリダイズさせ複合体を形成させる工程
(2)工程(1)のあとに、連続的な温度上昇を行いながら、260nmの吸光度測定または前記プローブに付加した標識のシグナル強度を測定することにより、融解曲線分析を行う工程。
(3)工程(2)における、温度変化に伴うシグナル強度の変動から、前記被検核酸における変異の有無を決定する工程
[項6]
項5に記載の方法であって、以下(1)〜(3)の工程を全て含む方法。
(1)被検核酸と項1〜4のいずれかに記載のプローブとをハイブリダイズさせ複合体を形成させる工程
(2)工程(1)のあとに、連続的な温度上昇を行いながら、前記プローブを標識している蛍光色素の蛍光強度を測定することにより、融解曲線分析を行う工程。
(3)工程(2)における、温度変化に伴う蛍光強度の変動から、前記被検核酸における変異の有無を決定する工程
[項7]
工程(1)における被検核酸が、プライマー対を用いて鋳型核酸から増幅された物である、項5または6に記載の方法。
[項8]
核酸増幅に、100塩基/秒以上のDNA合成速度を有し、かつ、5’エキソヌクレアーゼ活性を持たないDNAポリメラーゼを用いる、項7に記載の方法。
[項9]
前記プライマー対が、下記(F)のオリゴヌクレオチドからなるフォワードプライマーおよび下記(R)のオリゴヌクレオチドからなるリバースプライマーを含む一対のプライマーである、項7または8に記載の方法。
(F)塩基配列が配列番号4〜6のいずれかで示されるオリゴヌクレオチド
(R)塩基配列が配列番号7〜9のいずれかで示されるオリゴヌクレオチド
[項10]
核酸増幅法によりC型肝炎の治療率に関与すると言われるSNPを増幅するためのプライマー対であって、下記(1)〜(3)のいずれかで構成されるプライマー対。
(1)配列番号4および配列番号7のそれぞれに示される塩基配列を有するプライマーからなるプライマー対
(2)配列番号5および配列番号8のそれぞれに示される塩基配列を有するプライマーからなるプライマー対
(3)配列番号6および配列番号9のそれぞれに示される塩基配列を有するプライマーからなるプライマー対
[項11]
項5に記載のC型肝炎の治療率に関与すると言われるSNPを検出する方法に用いるための変異検出キットであって、項1〜4のいずれかに記載のプローブを含む変異検出キット。
【発明の効果】
【0012】
本発明によれば、C型肝炎の治療率に関与すると言われるSNP検査を迅速、正確かつ簡便に行うことが可能になる。
【図面の簡単な説明】
【0013】
【図1】実施例1の結果を示す図である。
【図2】実施例2の結果を示す図である。
【図3】実施例3の結果を示す図である。
【図4】実施例4の結果を示す図である。
【発明を実施するための形態】
【0014】
本発明の実施形態の一つは、C型肝炎の治療率に関与すると言われるSNPを検出するためのプローブであって、配列番号1〜3のいずれかで示される塩基配列を有するプローブである。
【0015】
本発明の「C型肝炎の治療率に関与すると言われるSNP」とは、C型肝炎の治療,特にインターフェロン療法、中でもPEG−IFN/RBV併用療法の治療効果を予測できるSNPであって、より具体的には、rs8103142、rs8099917rs12979860で示される三つのSNPである。
【0016】
この三つのSNPはいずれもIL28B遺伝子またはその周辺に存在する。rs8103142およびその周辺の塩基配列は配列番号10で示される。rs8103142は配列番号10の389番目の塩基にあたり、配列番号10ではy(T/C)で示される。rs8099917およびその周辺の塩基配列は配列番号11で示される。rs8099917は配列番号11の301番目の塩基にあたり、配列番号11ではk(T/G)で示される。rs12979860及びその周辺の塩基配列は配列番号12で示される。rs12979860は配列番号12の301番目の塩基にあたり、配列番号12ではy(C/T)で示される。なお、上記SNPを表す番号(rs番号)や、配列表に記載の配列は、NCBIのデータベース(Bulid 35)に掲載されているヒトの遺伝子配列情報を参照している。
【0017】
本発明のプローブは、各被検核酸に対して1本ずつ特異的に結合するように設計されており、融解曲線分析に適している。
特許文献1に開示されている方法は、検出に2本以上のプローブを要し本発明とは全く異なる原理で被検核酸を検出する方法であるから、該方法に用いられているプローブを本発明に直接転用することは出来ないし、参考にすることも出来ない。
【0018】
配列番号1で示される本発明のプローブは、rs8103142を含む部分領域に相補的な配列で構成されており、なおかつrs8103142がマイナーアリルであるチミンの場合にアンチセンス鎖と完全な相補鎖となるように設計されていて、アンチセンス鎖におけるSNPを確認できる。
【0019】
配列番号2で示される本発明のプローブは、rs8099917を含む部分領域に相補的な配列で構成されており、なおかつrs8099917がマイナーアリルであるグアニンの場合にアンチセンス鎖と完全な相補鎖となるように設計されていて、アンチセンス鎖におけるSNPを確認できる。
【0020】
配列番号3で示される本発明のプローブは、rs12979860を含む部分領域に相補的な配列で構成されており、なおかつrs12979860がメジャーアリルであるグアニンの場合にアンチセンス鎖と完全な相補鎖となるように設計されていて、アンチセンス鎖におけるSNPを確認できる。
【0021】
本発明のプローブは、配列番号1から3のいずれかに示される核酸配列が好ましいが、rs8103142、rs8099917、rs12979860のいずれかのSNPを含む10塩基以上30塩基以下の核酸配列またはこれに相補的な核酸配列であれば、特に限定されない。
【0022】
本発明のプローブは、標識物質で標識化された標識化プローブであってもよい。好ましくは、蛍光色素によって標識された核酸プローブ、さらに好ましくは、末端が蛍光色素によって標識された核酸プローブを使用できる。
【0023】
本発明のプローブは、標識物質で標識化されたプローブであることが好ましい。前記標識物質は、制限されないが、蛍光色素(蛍光団)があげられる。前記標識化プローブの具体例として、例えば、前記蛍光色素で標識され、単独で蛍光を示し且つハイブリッド形成により蛍光が減少(例えば、消光)するプローブが好ましい。このような現象は、一般に、蛍光消光現象と呼ばれる。この蛍光消光現象を利用したプローブとしては、中でも、一般的にグアニン消光プローブとよばれるものが好ましい。このようなプローブは、いわゆるQProbe(登録商標)として知られている。グアニン消光プローブとは、例えば、オリゴヌクレオチドの3’末端もしくは5’末端の塩基がシトシンとなるように設計し、その末端の塩基シトシンが相補的な塩基グアニンに近づくと発光が弱くなる蛍光色素で前記末端を標識化したプローブである。本発明のプローブにおいては、例えば、蛍光消光現象を示す蛍光色素を、前記オリゴヌクレオチドの3’末端のシトシンに結合させてもよいし、前記オリゴヌクレオチドの5’末端をシトシンに設計し、これに結合させてもよい。
【0024】
前記蛍光色素は、制限されないが、例えば、フルオレセイン、リン光体、ローダミン、ポリメチン色素誘導体等があげられ、市販の蛍光色素としては、例えば、BODIPY FL(商標名、モレキュラープローブ社製)、FluorePrime(商品名、アマシャムファルマシア社製)、Fluoredite(商品名、ミリポア社製)、FAM(ABI社製)、Cy3およびCy5(アマシャムファルマシア社製)、TAMRA(モレキュラープローブ社製)等があげられる。プローブの検出条件は、特に制限されず、使用する蛍光色素により適宜決定できるが、例えば、Pacific Blueは、検出波長450〜480nm、TAMRAは、検出波長585〜700nm、BODIPY FLは、検出波長515〜555nmで検出できる。このようなプローブを使用すれば、シグナルの変動により、ハイブリダイズと解離とを容易に確認することができる。
【0025】
本発明のプローブは、例えば、3’末端にリン酸基が付加されてもよい。後述するように、変異の有無を検出する被検核酸(標的核酸)は、PCR等の核酸増幅法によって調製することができ、この際、本発明のプローブを核酸増幅反応の反応系に共存させることができる。このような場合、プローブの3’末端にリン酸基を付加させておけば、プローブ自体が核酸増幅反応によって伸長することを十分に防止できる。また、3’末端に前述のような標識物質を付加することによっても、同様の効果が得られる。
【0026】
本発明のプローブは、前述のように、C型肝炎治療に関するSNPの検出に使用することができる。この変異の検出方法は、何ら制限されず、被検核酸と前記プローブとのハイブリダイズを利用する方法であればよい。
【0027】
本発明の実施形態の一つは、C型肝炎の治療率に関与すると言われるSNPを検出する方法であって、以下(1)〜(3)の工程を全て含むことを特徴とする方法である。
C型肝炎の治療率に関与すると言われるSNPを検出する方法であって、以下(1)〜(3)の工程を全て含む方法。
(1)被検核酸と項1〜4のいずれかに記載のプローブとをハイブリダイズさせ複合体を形成させる工程
(2)工程(1)のあとに、連続的な温度上昇を行いながら、260nmの吸光度測定または前記プローブに付加した標識のシグナル強度を測定することにより、融解曲線分析を行う工程。
(3)工程(2)における、温度変化に伴うシグナル強度の変動から、前記被検核酸における変異の有無を決定する工程
【0028】
本発明のSNP検出方法によれば、配列番号1〜3のいずれかのプローブを使用することでそれぞれrs8103142、rs8099917、rs12979860のSNPを検出できる。
【0029】
本発明における被検核酸は、例えば、一本鎖でもよいし、二本鎖でもよい。二本鎖の場合は、例えば、被検核酸とプローブとをハイブリダイズさせてハイブリッド体を形成するために、加熱により前記二本鎖を一本鎖に解離させる工程を含むことが好ましい。
【0030】
前記被検核酸の種類としては、特に制限されないが、例えば、DNAや、トータルRNA、mRNA等のRNA等があげられる。また、前記被検核酸は、例えば、生体試料等の試料に含まれる核酸があげられる。前記試料中の核酸は、例えば、生体試料中に元来含まれている核酸でもよいが、例えば、変異検出の精度を向上できることから、前記生体試料中の核酸を鋳型として核酸増幅法により増幅させた増幅産物等があげられる。具体例としては、例えば、前記生体試料に元来含まれているDNAを鋳型として、核酸増幅法により増幅させた増幅産物や、前記生体試料に元来含まれているRNAから逆転写反応(RT−PCR)により生成させたcDNAを鋳型として、核酸増幅法により増幅させた増幅産物があげられる。これらの増幅産物を、本発明における被検核酸としてもよい。前記増幅産物の長さは、特に制限されないが、例えば、50〜1000塩基であり、好ましくは80〜300塩基である。
【0031】
前記生体試料としては、特に制限されないが、例えば、白血球細胞等の血球試料、全血試料、口腔拭い液、咽頭拭い液等があげられる。なお、本発明において、試料の採取方法、DNAやRNA等の核酸の調製方法等は、制限されず、従来公知の方法が採用できる。
【0032】
本発明の方法で用いる融解曲線分析について説明する。例えば、二本鎖DNAを含む溶液を加熱していくと、260nmにおける吸光度が上昇する。これは、二本鎖DNAにおける両鎖間の水素結合が加熱によってほどけ、一本鎖DNAに解離(DNAの融解)することが原因である。そして、全ての二本鎖DNAが解離して一本鎖DNAになると、その吸光度は、加熱開始時の吸光度(二本鎖DNAのみの吸光度)の約1.5倍程度を示し、これによって融解が完了したと判断できる。
【0033】
本発明において、融解曲線分析を行うための温度変化に伴うシグナル変動の測定は、前述のような原理から、260nmの吸光度測定により行うこともできるが、本発明のプローブに付加した標識のシグナルを測定することが好ましい。このため、本発明のプローブとして、前述の標識化プローブを使用することが好ましい。
前記標識化プローブとしては、例えば、単独でシグナルを示し且つハイブリッド形成によりシグナルを示さない標識化プローブ、または、単独でシグナルを示さず且つハイブリッド形成によりシグナルを示す標識化プローブがあげられる。前者のプローブであれば、検出対象配列とハイブリッド(二本鎖)を形成している際にはシグナルを示さず、加熱によりプローブが遊離するとシグナルを示す。また、後者のプローブであれば、検出対象配列とハイブリッド(二本鎖)を形成することによってシグナルを示し、加熱によりプローブが遊離するとシグナルが減少(消失)する。したがって、この標識によるシグナルをシグナル特有の条件(吸光度等)で検出することによって、前記260nmの吸光度測定と同様に、融解の進行を把握することができる。標識化プローブにおける標識物質は、例えば、前述のとおりである。
好ましい標識物質は、蛍光色素である。標識物質に蛍光色素を用いた場合、上記方法において、前記プローブに付加した蛍光色素のシグナル強度を測定することにより、融解曲線分析を行い、この工程における、温度変化に伴うシグナル強度の変動から、前記被検核酸における変異の有無を決定することができる。
【0034】
前述のように、被検核酸が、鋳型核酸から核酸増幅法により増幅させた増幅産物である場合、前記工程(1)は、フォワードプライマーおよびリバースプライマーのプライマー対を用いて鋳型核酸から核酸を増幅する工程を含んでも良い。言い換えれば、前記工程(1)における被検核酸は、フォワードプライマーおよびリバースプライマーのプライマー対を用いて鋳型核酸から増幅された物であっても良い。
【0035】
前記プライマー対としては、IL28B遺伝子および/またはIL28B遺伝子周辺領域の本発明のプローブがハイブリダイズ可能な領域が特異的に増幅されるものであれば、特に制限されない。前記増幅領域は、例えば、前記プローブがハイブリダイズする領域のみでもよいし、前記ハイブリダイズ領域を含む領域であってもよい。
【0036】
具体的には、例えば、下記(F)のオリゴヌクレオチドからなるフォワードプライマーおよび下記(R)のオリゴヌクレオチドからなるリバースプライマーを含む一対のプライマーが例示できる。
(F)塩基配列が配列番号4〜6のいずれかで示されるオリゴヌクレオチド
(R)塩基配列が配列番号7〜9のいずれかで示されるオリゴヌクレオチド
【0037】
前記核酸増幅工程に用いられる具体的な核酸増幅方法としては特に限定されず、適宜公知の方法を用いることができる。例えば、PCR(Polymerase Chain Reaction)法、NASBA(Nucleic acid sequence based amplification)法、TMA(Transcription−mediated amplification)法、SDA(Strand Displacement Amplification)法等があげられるが、PCR法を用いることが好ましい。なお、これらの各方法において、増幅反応の条件は特に制限されず、従来公知の方法により行うことができる。
【0038】
核酸増幅にPCR法を用いる場合、DNAポリメラーゼには、α型DNAポリメラーゼを用いることが好ましい。その理由を以下に説明する。
【0039】
本発明プローブが含まれる反応系でIL28B遺伝子および/またはIL28B遺伝子周辺領域を増幅する場合、核酸増幅工程中に該核酸プローブが試料のIL28B遺伝子および/またはIL28B遺伝子周辺領域またはそれらの増幅産物と結合しうる。核酸増幅工程中にIL28B遺伝子および/またはIL28B遺伝子周辺領域と結合した該核酸プローブは、核酸プライマーとDNAポリメラーゼによる核酸増幅反応を阻害する。
【0040】
Taq DNA PolymeraseなどPolI型のDNAポリメラーゼは5’→ 3’エキソヌクレアーゼ活性を持つことが知られている。この活性のため、核酸増幅反応中に鋳型となるIL28B遺伝子および/またはIL28B遺伝子周辺領域と結合した核酸がある場合、該結合核酸はエキソヌクレアーゼ活性によって分解されてしまう。このため、反応系中の該核酸プローブが減少し核酸検出工程に問題が生じる可能性がある。従って、PolI型DNAポリメラーゼを用いて本発明を実施することは好ましくない。
【0041】
他方、KOD DNA Polymerase(超好熱始原菌Thermococcus kodakaraensis KOD1由来)などα型のDNAポリメラーゼは5’→ 3’エキソヌクレアーゼ活性は持たず、3’→ 5’エキソヌクレアーゼ活性を持つ。従って、α型DNAポリメラーゼを用いれば上記問題を解決できるのみならず、3’→ 5’エキソヌクレアーゼ活性により核酸増幅工程において高い正確性が発揮される。
【0042】
通常、α型DNAポリメラーゼは3’→ 5’エキソヌクレアーゼ活性のため、核酸増幅速度はPolI型酵素と比較して低い傾向がある。しかし、KOD DNA Polymeraseはα型DNAポリメラーゼでありながらDNA合成活性が高く100塩基/秒以上のDNA合成速度を有し伸長効率が優れている。従って、本発明の実施にはα型DNAポリメラーゼの中でも、KOD DNA Polymerase(東洋紡績製、商標)を用いることが好ましい。
【0043】
さらに、α型DNAポリメラーゼを変異させて100塩基/秒以上のデオキシリボ核酸合成速度を達成させた変異型、あるいは、野生型および/または変異型の組み合わせにより当該性能を達成させたDNAポリメラーゼ組成物も、本発明の実施に適したDNAポリメラーゼとして用いることができる。
例えば、上記KOD DNA Polymerase以外に100塩基/秒以上のデオキシリボ核酸合成速度を有するDNAポリメラーゼとして、「KOD FX(東洋紡績製、登録商標)」、「KOD −Plus−(東洋紡績製、商標)」、「KOD Dash(東洋紡績製、登録商標)」、PrimeSTAR HS DNAポリメラーゼ(タカラバイオ製、登録商標)なども利用できる。
【0044】
本発明において、DNA合成活性とは鋳型DNAにアニールされたオリゴヌクレオチドまたはポリヌクレオチドの3’−ヒドロキシル基にデオキシリボヌクレオシド5’−トリホスフェートのα−ホスフェートを共有結合せしめることにより、デオキシリボ核酸にデオキシリボヌクレオシド5’−モノホスフェートを鋳型依存的に導入する反応を触媒する活性をいう。
【0045】
その活性測定法は、酵素活性が高い場合には、保存緩衝液でサンプルを希釈して測定を行う。本発明では、下記A液25μl、B液およびC液各5μlおよび滅菌水10μlをエッペンドルフチューブに加えて攪拌混合した後、上記酵素液5μlを加えて75℃で10分間反応する。その後、氷冷し、E液50μl、D液100μlを加えて、攪拌後、さらに10分間氷冷する。この液をガラスフィルター(ワットマンGF/Cフィルター)で濾過し、D液及びエタノールで充分洗浄し、フィルターの放射活性を液体シンチレーションカウンター(パッカード社製)で計測し、鋳型DNAへのヌクレオチドの取り込みを測定する。酵素活性の1単位はこの条件下で30分あたり10nモルのヌクレオチドを酸不溶性画分に取り込む酵素量とする。
A: 40mM Tris−HCl(pH7.5)
16mM 塩化マグネシウム
15mM ジチオスレイトール
100μg/ml BSA
B: 2μg/μl 活性化仔牛胸腺DNA
C: 1.5mM dNTP(250cpm/pmol〔 3H〕dTTP)
D: 20% トリクロロ酢酸(2mMピロリン酸ナトリウム)
E: 1μg/μl キャリアーDNA
【0046】
本発明において、3’−5’エキソヌクレアーゼ活性とは、DNAの3’末端領域を切除し、5’−モノヌクレオチドを遊離する活性をいう。
その活性測定法は、50μlの反応液(120mM Tris−HCl(pH8.8 at 25℃), 10mM KCl, 6mM 硫酸アンモニウム,1mM MgCl2, 0.1% Triton X−100, 0.001% BSA,5 μg トリチウムラベルされた大腸菌DNA)を1.5mlのエッペンチューブに分注し、DNAポリメラーゼを加える。75℃で10分間反応させた後、氷冷によって反応を停止し、次にキャリアーとして、0.1%のBSAを50μl加え、さらに10%のトリクロロ酢酸、2%ピロリン酸ナトリウム溶液を100μl加え混合する。氷上で15分放置した後、12,000回転で10分間遠心し沈殿を分離する。上清100μlの放射活性を液体シンチレーションカウンター(パッカード社製)で計測し、酸可溶性画分に遊離したヌクレオチド量を測定する。
【0047】
本発明において、DNAポリメラーゼのDNA合成速度とは単位時間当たりのDNAの合成数をいう。DNA合成速度は、市販品であれば該市販品の説明書等に記載されている値、説明書等に記載がない場合は文献等に記載されている値を指すものとする。これらの方法で特定できない場合は、以下の方法で求めることができる。
【0048】
DNA合成速度の測定法
DNAポリメラーゼの反応液(20mM Tris−HCl(pH7.5),8mM塩化マグネシウム、7.5mM ジチオスレイトール、100 μg/ml BSA、0.1mM dNTP、0.2μCi [α−32P]dCTP)を、プライマーをアニーリングさせたM13mp181本鎖DNAと75℃で反応させる。反応停止は等量の反応停止液(50mM 水酸化ナトリウム、10mM EDTA、5% フィコール、0.05% ブロモフェノールブルー)を加えることにより行う。上記反応にて合成されたDNAをアルカリアガロースゲル電気泳動にて分画した後、ゲルを乾燥させオートラジオグラフィーを行う。DNAサイズマーカーとしてはラベルしたλ/HindIIIを用いる。このマーカーのバンドを指標として合成されたDNAのサイズを測定することによって、DNA合成速度を求める。
【0049】
次に、本発明の変異検出方法について、本発明のプローブとして、蛍光色素で標識化された標識化プローブを使用する例をあげて説明する。なお、本発明の変異検出方法は、以下の説明によって何ら制限されることはない。
【0050】
まず、例えば全血からゲノムDNAを単離する。単離は、例えば、市販のゲノムDNA単離キットを用いて行うことができる。
次に、単離したゲノムDNAを含む試料に、標識化プローブとして、例えば、QProbeを添加する。
【0051】
前記プローブの添加のタイミングは、特に制限されず、例えば、後述する増幅反応前、増幅反応途中および増幅反応後のいずれに、増幅反応の反応系に添加してもよい。
中でも、増幅反応と、前記工程(2)とを連続的に行うことができるため、増幅反応前に添加することが好ましい。このように核酸増幅反応の前に前記プローブを添加する場合は、例えば、前述のように、その3’末端に、蛍光色素を付加したり、リン酸基を付加することが好ましい。
【0052】
前記プローブは、単離したゲノムDNAを含む液体試料に添加してもよいし、溶媒中でゲノムDNAと混合してもよい。前記溶媒としては、特に制限されず、例えば、Tris−HCl等の緩衝液、KCl、MgCl2、MgSO4、グリセロール等を含む溶媒、PCR反応液等、従来公知のものがあげられる。
【0053】
続いて、単離したゲノムDNAを鋳型として、PCR等の核酸増幅法によって、検出目的の塩基部位を含む配列を増幅させる。なお、PCR等の条件は、特に制限されず、従来公知の方法により行うことができる。
【0054】
PCRのプライマーの配列は、検出目的の塩基部位を含む配列を増幅できるものであれば特に制限されず、前述のように、目的の配列に応じて、従来公知の方法により適宜設計できる。プライマーの長さは、特に制限されず、一般的な長さ(例えば、10〜40mer)に設定できる。
【0055】
本発明の検出方法に使用するプライマーセットとしては、例えば、前述の本発明のプライマーセットがあげられる。なお、本発明のプライマーセットの用途は、本発明の検出方法には制限されず、また、本発明の検出方法に使用するプライマーセットも、本発明のプライマーセットには限定されない。
【0056】
次に、得られたPCR増幅産物の解離、および、解離により得られた一本鎖DNAと前記標識化プローブとのハイブリダイズを行う。これは、例えば、前記反応液の温度変化によって行うことができる。
【0057】
前記解離工程における加熱温度は、前記増幅産物が解離できる温度であれば特に制限されないが、例えば、85〜98℃である。加熱時間も特に制限されないが、通常、1秒〜10分であり、好ましくは1秒〜5分である。
【0058】
また、解離した一本鎖DNAと前記標識化プローブとのハイブリダイズは、例えば、前記解離工程の後、前記解離工程における加熱温度を降下させることによって行うことができる。温度条件としては、例えば、35〜50℃である。
【0059】
ハイブリダイズ工程の反応系(反応系)における各組成の体積や濃度は、特に制限されない。具体例としては、前記反応系において、DNAの濃度は、例えば、0.01〜1μmol/Lであり、好ましくは0.1〜0.5μmol/L、前記標識化プローブの濃度は、例えば、前記DNAに対する添加割合を満たす範囲が好ましく、例えば、0.001〜10μmol/Lであり、好ましくは0.001〜1μmol/Lである。
【0060】
そして、前記反応液の温度を変化させ、前記増幅産物と前記標識化プローブとのハイブリッド形成体の融解状態を示すシグナル値を測定する。具体的には、例えば、前記反応液(前記一本鎖DNAと前記標識化プローブとのハイブリッド形成体)を加熱し、温度上昇に伴うシグナル値の変動を測定する。前述のように、末端のC塩基が標識化されたプローブ(グアニン消光プローブ)を使用した場合、一本鎖DNAとハイブリダイズした状態では、蛍光が減少(または消光)し、解離した状態では、蛍光を発する。したがって、例えば、蛍光が減少(または消光)しているハイブリッド形成体を徐々に加熱し、温度上昇に伴う蛍光強度の増加を測定すればよい。
【0061】
蛍光強度の変動を測定する際の温度範囲は、特に制限されないが、例えば、開始温度が室温〜85℃であり、好ましくは25〜70℃であり、終了温度は、例えば、40〜105℃である。また、温度の上昇速度は、特に制限されないが、例えば、0.05〜20℃/秒であり、好ましくは0.08〜5℃/秒である。
【0062】
目的の塩基部位における遺伝子型(野生型、変異型など)の決定は、例えば、ハイブリッド形成時におけるシグナル変動を測定することによって行いうる。すなわち、前記プローブを含む反応液の温度を降下させてハイブリッド形成体を形成する際に、前記温度降下に伴うシグナル変動を測定する。
【0063】
具体例として、単独でシグナルを示し且つハイブリッド形成によりシグナルを示さない標識化プローブ(例えば、グアニン消光プローブ)を使用した場合、一本鎖DNAとプローブとが解離している状態では蛍光を発しているが、温度の降下によりハイブリッドを形成すると、前記蛍光が減少(または消光)する。したがって、例えば、前記反応液の温度を徐々に降下させて、温度下降に伴う蛍光強度の減少を測定すればよい。他方、単独でシグナルを示さず且つハイブリッド形成によりシグナルを示す標識化プローブを使用した場合、一本鎖DNAとプローブとが解離している状態では蛍光を発していないが、温度の降下によりハイブリッドを形成すると、蛍光を発するようになる。したがって、例えば、前記反応液の温度を徐々に降下させて、温度下降に伴う蛍光強度の増加を測定すればよい。
【0064】
また、本発明においては、目的の塩基部位における遺伝子型の決定のために、前記シグナルの変動を解析してTm(melting temperature)値として決定してもよい。
【実施例】
【0065】
以下に実施例を示して本発明を具体的に説明するが、本発明は実施例に限定されるものではない。
【0066】
〔実施例1:精製したヒトゲノムからのrs8103142の検出〕
(1)試料の調製
既にrs8103142がメジャーホモ(T/T)であることが判明している精製されたヒトゲノム(以下HM)、rs8103142がヘテロ(T/C)であることが判明している精製されたヒトゲノム(以下HH)、そしてrs8103142がマイナーホモ(C/C)であることが判明している精製されたヒトゲノム(以下Hm)を試料とし、陰性コントロール(NC)として10mMのTris−HCl(pH7.5)を試料とした。
(2)核酸増幅および融解曲線分析
上記ヒトゲノム試料および陰性コントロールにそれぞれ下記試薬を添加して、下記条件によりrs8103142を検出した。核酸増幅および融解曲線分析には東洋紡績社製GENECUBE(登録商標)を使用した。
【0067】
試薬
以下の試薬を含む13.2μl溶液を調製した。
核酸プライマー(配列番号4) 300nM
核酸プライマー(配列番号7) 1500nM
核酸プローブ(配列番号1、3’末端をBODIPY−FL標識) 300nM
×10緩衝液 1.3μl
dNTP 0.2mM
MgSO4 4mM
BSA 1.3μg
DMSO 0.98μL
KOD plus DNA polymerase(東洋紡績製) 0.4U
試料 4μl
【0068】
核酸増幅および融解曲線分析
94℃・2分
(以上1サイクル)
97℃・1秒
60℃・3秒
63℃・5秒
(以上50サイクル)
94℃・30秒
39℃・30秒
39℃〜75℃(0.09℃/秒で温度上昇)
【0069】
結果
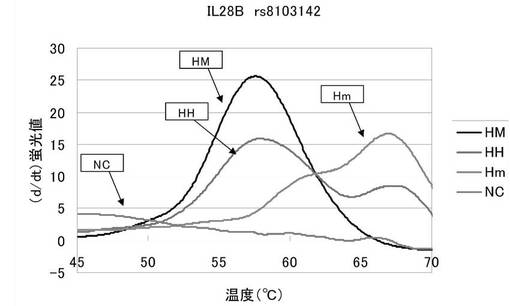
図1は、融解曲線における温度上昇にともなう蛍光強度の変化を、グラフの横軸を温度、縦軸を蛍光シグナルの微分値として解析した結果を表した図である。HMは57〜58℃にピークが現れており、Hmは67℃付近でピークが現れている。そしてHHはその両方の温度でピークが現れている。本実施例で用いた配列番号1のプローブは末端がBODIPY−FL標識されている。このプローブは相補的な配列の核酸と結合している間は蛍光が消光しており、逆にプローブが単独で遊離している間は蛍光が発光しているという特徴を有する。配列番号1のプローブはマイナーホモのrs8103142を含む核酸と最も強く結合し、メジャーホモのrs8103142を含む核酸とは一塩基のミスマッチが生じているため、より弱く結合する。従って、図1の57〜58℃でのピークは被検核酸中のrs8103142がメジャーアリルであることを示しており、67℃付近のピークは被検核酸中のrs8103142がマイナーアリルであることを示している。そしてその両方のピークを発した被検核酸はメジャーアリルとマイナーアリルの両方を有するヘテロであることを示している。
以上より、本発明プローブを用いることでrs8103142のアリルを容易に識別できることが示された。
【0070】
〔実施例2:精製したヒトゲノムからのrs8099917の検出〕
(1)試料の調製
既にrs8099917がメジャーホモ(T/T)であることが判明している精製されたヒトゲノム(以下HM)、rs8099917がヘテロ(T/G)であることが判明している精製されたヒトゲノム(以下HH)、そしてrs8099917がマイナーホモ(G/G)であることが判明している精製されたヒトゲノム(以下Hm)を試料とし、陰性コントロール(NC)として10mMのTris−HCl(pH7.5)を試料とした。
(2)核酸増幅および融解曲線分析
上記ヒトゲノム試料および陰性コントロールにそれぞれ下記試薬を添加して、下記条件によりrs8099917を検出した。核酸増幅および融解曲線分析には東洋紡績社製GENECUBE(登録商標)を使用した。
【0071】
試薬
以下の試薬を含む13.2μl溶液を調製した。
核酸プライマー(配列番号5) 500nM
核酸プライマー(配列番号8) 2500nM
核酸プローブ(配列番号2、3’末端をBODIPY−FL標識) 250nM
×10緩衝液 1.3μl
dNTP 0.2mM
MgSO4 4mM
BSA 1.3μg
DMSO 0.98μL
KOD plus DNA polymerase(東洋紡績製) 0.4U
試料 4μl
【0072】
核酸増幅および融解曲線分析
実施例1と同じ。
【0073】
結果
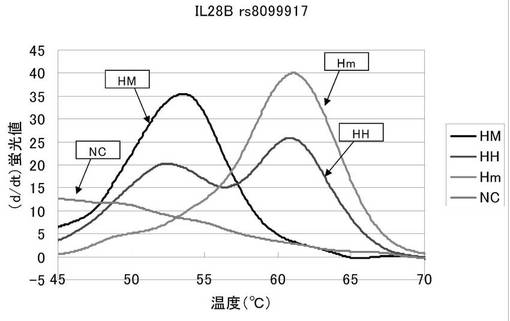
図2は、融解曲線における温度上昇にともなう蛍光強度の変化を、グラフの横軸を温度、縦軸を蛍光シグナルの微分値として解析した結果を表した図である。HMは54℃付近にピークが現れており、Hmは61℃付近でピークが現れている。そしてHHはその両方の温度でピークが現れている。本実施例で用いた配列番号2のプローブは末端がBODIPY−FL標識されている。このプローブは相補的な配列の核酸と結合している間は蛍光が消光しており、逆にプローブが単独で遊離している間は蛍光が発光しているという特徴を有する。配列番号2のプローブはマイナーホモのrs8099917を含む核酸と最も強く結合し、メジャーホモのrs8103142を含む核酸とは一塩基のミスマッチが生じているため、より弱く結合する。従って、図2の54℃付近でのピークは被検核酸中のrs8099917がメジャーアリルであることを示しており、61℃付近のピークは被検核酸中のrs8099917がマイナーアリルであることを示している。そしてその両方のピークを発した被検核酸はメジャーアリルとマイナーアリルの両方を有するヘテロであることを示している。
以上より、本発明プローブを用いることでrs8099917のアリルを容易に識別できることが示された。
【0074】
〔実施例3:精製したヒトゲノムからのrs12979860の検出〕
(1)試料の調製
既にrs12979860がメジャーホモ(C/C)であることが判明している精製されたヒトゲノム(以下HM)、rs12979860がヘテロ(C/T)であることが判明している精製されたヒトゲノム(以下HH)、そしてrs12979860がマイナーホモ(T/T)であることが判明している精製されたヒトゲノム(以下Hm)を試料とし、陰性コントロール(NC)として10mMのTris−HCl(pH7.5)を試料とした。
(2)核酸増幅および融解曲線分析
上記ヒトゲノム試料および陰性コントロールにそれぞれ下記試薬を添加して、下記条件によりrs12979860を検出した。核酸増幅および融解曲線分析には東洋紡績社製GENECUBE(登録商標)を使用した。
【0075】
試薬
以下の試薬を含む13.2μl溶液を調製した。
核酸プライマー(配列番号6) 300nM
核酸プライマー(配列番号9) 1500nM
核酸プローブ(配列番号3、3’末端をBODIPY−FL標識) 300nM
×10緩衝液 1.3μl
dNTP 0.2mM
MgSO4 4mM
BSA 1.3μg
DMSO 0.98μL
KOD plus DNA polymerase(東洋紡績製) 0.4U
試料 4μl
【0076】
核酸増幅および融解曲線分析
実施例1と同じ。
【0077】
結果
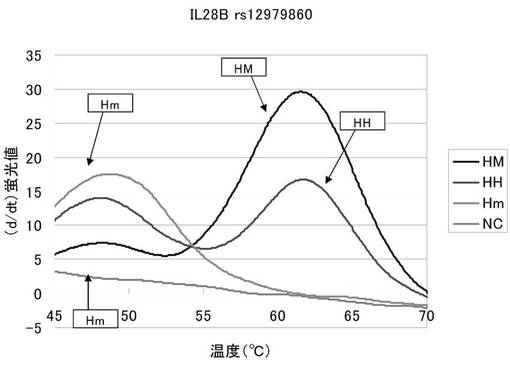
図3は、融解曲線における温度上昇にともなう蛍光強度の変化を、グラフの横軸を温度、縦軸を蛍光シグナルの微分値として解析した結果を表した図である。HMは62℃付近にピークが現れており、Hmは48℃付近でピークが現れている。そしてHHはその両方の温度でピークが現れている。本実施例で用いた配列番号3のプローブは末端がBODIPY−FL標識されている。このプローブは相補的な配列の核酸と結合している間は蛍光が消光しており、逆にプローブが単独で遊離している間は蛍光が発光しているという特徴を有する。配列番号3のプローブはメジャーホモのrs12979860を含む核酸と最も強く結合し、マイナーホモのrs12979860を含む核酸とは一塩基のミスマッチが生じているため、より弱く結合する。従って、図3の48℃付近でのピークは被検核酸中のrs12979860がマイナーアリルであることを示しており、62℃付近のピークは被検核酸中のrs12979860がメジャーアリルであることを示している。そしてその両方のピークを発した被検核酸はメジャーアリルとマイナーアリルの両方を有するヘテロであることを示している。
以上より、本発明プローブを用いることでrs12979860のアリルを容易に識別できることが示された。
【0078】
〔実施例4:全血希釈液からのrs8099917の検出〕
(1)試料の調製
全血10μlに10mMTris−HCl(pH7.5)を90μl加えて、希釈倍率10倍の全血希釈液を調製した。この全血希釈液を試料とした。検出系は10μlであり、試料は1μlを供するため、検出系での全血の終濃度は1%となる。
(2)核酸増幅および融解曲線分析
上記全血希釈液に下記試薬を添加して、下記条件によりrs8099917を検出した。核酸増幅および融解曲線分析には東洋紡績社製GENECUBE(登録商標)を使用した。
【0079】
試薬
以下の試薬を含む10μl溶液を調製した。
核酸プライマー(配列番号5) 500nM
核酸プライマー(配列番号8) 2500nM
核酸プローブ(配列番号2、3’末端をBODIPY−FL標識) 250nM
×10緩衝液 1μl
dNTP 0.2mM
MgSO4 4mM
BSA 1μg
DMSO 0.75μL
KOD plus DNA polymerase(東洋紡績製) 0.3U
試料 1μl
【0080】
核酸増幅および融解曲線分析
実施例1と同じ。
【0081】
結果
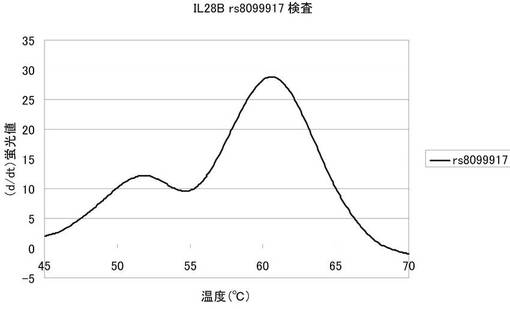
図5は、融解曲線における温度上昇にともなう蛍光強度の変化を、グラフの横軸を温度、縦軸を蛍光シグナルの微分値として解析した結果を表した図である。図5から明らかなように、本発明プローブを用いることによって全血の希釈液からでもrs8099917を検出することができる。
【産業上の利用可能性】
【0082】
本発明をC型肝炎治療に関するSNP検査およびC型肝炎治療に関するSNP検査試薬開発に適用することで、迅速、確実かつ簡便な慢性骨髄増殖性疾患関連遺伝子の変異検出を行うことができる。
【技術分野】
【0001】
本発明は、C型慢性肝炎の治療率に関与する一塩基多型(Single Nucleotide Polymorphism;SNP)を検出するためのプローブ、ならびに、該プローブを用いたSNPの検出方法およびSNPを検出するためのキットに関する。
【背景技術】
【0002】
C型肝炎ウイルス(Hepatitis C Virus;HCV)は感染者の70〜80%が慢性肝炎に移行すると言われている。さらに、慢性肝炎から肝硬変や肺がんへと移行する例も多数報告されている。
【0003】
C型肝炎の代表的な治療法としてペグインターフェロン+リバビリン(PEG−IFN/RBV)併用療法がある。しかし、この治療法の効果には個人差があることが知られてきた。
【0004】
PEG−IFN/RBV治療効果に個人差が生じる理由として、IL28B遺伝子周辺のSNPが関係あることが報告されている(非特許文献1、非特許文献2、特許文献1)。
【0005】
特許文献1にはSNP検査の実施例が記載されている。特許文献1ではシークエンス法やDigiTag2法という方法を用いている。シークエンス法は一般的に検体となるヒト遺伝子を核酸増幅し、その増幅産物の配列を同定するという方法で行う。しかし、この方法では核酸増幅産物のキャリーオーバーコンタミネーションによる誤診が発生するという懸念がある。また、検査開始から結果判明までに数時間から1日の期間が必要である。
【0006】
DigiTag2法は2本のプローブを用いて検出する方法である。プローブを用いる方法は核酸増幅と検出を同じ閉鎖系で行えばコンタミネーションを防ぐことができる。しかしDigiTag2法や蛍光共鳴エネルギー転移(FRET)を利用した方法では、検出には2本以上のプローブが必要であり、コストがかかるという欠点がある。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開2011−41526
【非特許文献】
【0008】
【非特許文献1】Ge D.et al.Nature 461,399−401,2009
【非特許文献2】Tanaka Y.et al.Nat Genet 41,1105−1109,2009
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明は上記従来のC型肝炎治療に関するSNP検査における問題点を解決しようとするものであり、その目的は該SNP検査を迅速、正確、かつ安価に行うための方法及び試薬を提供することにある。
【課題を解決するための手段】
【0010】
本発明者らは上記課題に鑑み鋭意検討した結果、C型肝炎の治療率に関与すると言われるSNPおよびその周辺の塩基配列に特異的に結合する1本の核酸プローブを用いることにより従来技術よりも正確に該SNPを検出できることを見出し、本発明を完成させるに至った。すなわち、本発明は以下のような構成からなる。
【0011】
[項1]
C型肝炎の治療率に関与すると言われるSNPを検出するためのプローブであって、配列番号1〜3のいずれかで示される塩基配列を有するプローブ。
[項2]
検出可能な標識物質により標識化された、項1に記載のプローブ。
[項3]
検出可能な標識物質が蛍光色素である、項2に記載のプローブ。
[項4]
末端のシトシンが標識化された、項2または3に記載のプローブ。
[項5]
C型肝炎の治療率に関与すると言われるSNPを検出する方法であって、以下(1)〜(3)の工程を全て含む方法。
(1)被検核酸と項1〜4のいずれかに記載のプローブとをハイブリダイズさせ複合体を形成させる工程
(2)工程(1)のあとに、連続的な温度上昇を行いながら、260nmの吸光度測定または前記プローブに付加した標識のシグナル強度を測定することにより、融解曲線分析を行う工程。
(3)工程(2)における、温度変化に伴うシグナル強度の変動から、前記被検核酸における変異の有無を決定する工程
[項6]
項5に記載の方法であって、以下(1)〜(3)の工程を全て含む方法。
(1)被検核酸と項1〜4のいずれかに記載のプローブとをハイブリダイズさせ複合体を形成させる工程
(2)工程(1)のあとに、連続的な温度上昇を行いながら、前記プローブを標識している蛍光色素の蛍光強度を測定することにより、融解曲線分析を行う工程。
(3)工程(2)における、温度変化に伴う蛍光強度の変動から、前記被検核酸における変異の有無を決定する工程
[項7]
工程(1)における被検核酸が、プライマー対を用いて鋳型核酸から増幅された物である、項5または6に記載の方法。
[項8]
核酸増幅に、100塩基/秒以上のDNA合成速度を有し、かつ、5’エキソヌクレアーゼ活性を持たないDNAポリメラーゼを用いる、項7に記載の方法。
[項9]
前記プライマー対が、下記(F)のオリゴヌクレオチドからなるフォワードプライマーおよび下記(R)のオリゴヌクレオチドからなるリバースプライマーを含む一対のプライマーである、項7または8に記載の方法。
(F)塩基配列が配列番号4〜6のいずれかで示されるオリゴヌクレオチド
(R)塩基配列が配列番号7〜9のいずれかで示されるオリゴヌクレオチド
[項10]
核酸増幅法によりC型肝炎の治療率に関与すると言われるSNPを増幅するためのプライマー対であって、下記(1)〜(3)のいずれかで構成されるプライマー対。
(1)配列番号4および配列番号7のそれぞれに示される塩基配列を有するプライマーからなるプライマー対
(2)配列番号5および配列番号8のそれぞれに示される塩基配列を有するプライマーからなるプライマー対
(3)配列番号6および配列番号9のそれぞれに示される塩基配列を有するプライマーからなるプライマー対
[項11]
項5に記載のC型肝炎の治療率に関与すると言われるSNPを検出する方法に用いるための変異検出キットであって、項1〜4のいずれかに記載のプローブを含む変異検出キット。
【発明の効果】
【0012】
本発明によれば、C型肝炎の治療率に関与すると言われるSNP検査を迅速、正確かつ簡便に行うことが可能になる。
【図面の簡単な説明】
【0013】
【図1】実施例1の結果を示す図である。
【図2】実施例2の結果を示す図である。
【図3】実施例3の結果を示す図である。
【図4】実施例4の結果を示す図である。
【発明を実施するための形態】
【0014】
本発明の実施形態の一つは、C型肝炎の治療率に関与すると言われるSNPを検出するためのプローブであって、配列番号1〜3のいずれかで示される塩基配列を有するプローブである。
【0015】
本発明の「C型肝炎の治療率に関与すると言われるSNP」とは、C型肝炎の治療,特にインターフェロン療法、中でもPEG−IFN/RBV併用療法の治療効果を予測できるSNPであって、より具体的には、rs8103142、rs8099917rs12979860で示される三つのSNPである。
【0016】
この三つのSNPはいずれもIL28B遺伝子またはその周辺に存在する。rs8103142およびその周辺の塩基配列は配列番号10で示される。rs8103142は配列番号10の389番目の塩基にあたり、配列番号10ではy(T/C)で示される。rs8099917およびその周辺の塩基配列は配列番号11で示される。rs8099917は配列番号11の301番目の塩基にあたり、配列番号11ではk(T/G)で示される。rs12979860及びその周辺の塩基配列は配列番号12で示される。rs12979860は配列番号12の301番目の塩基にあたり、配列番号12ではy(C/T)で示される。なお、上記SNPを表す番号(rs番号)や、配列表に記載の配列は、NCBIのデータベース(Bulid 35)に掲載されているヒトの遺伝子配列情報を参照している。
【0017】
本発明のプローブは、各被検核酸に対して1本ずつ特異的に結合するように設計されており、融解曲線分析に適している。
特許文献1に開示されている方法は、検出に2本以上のプローブを要し本発明とは全く異なる原理で被検核酸を検出する方法であるから、該方法に用いられているプローブを本発明に直接転用することは出来ないし、参考にすることも出来ない。
【0018】
配列番号1で示される本発明のプローブは、rs8103142を含む部分領域に相補的な配列で構成されており、なおかつrs8103142がマイナーアリルであるチミンの場合にアンチセンス鎖と完全な相補鎖となるように設計されていて、アンチセンス鎖におけるSNPを確認できる。
【0019】
配列番号2で示される本発明のプローブは、rs8099917を含む部分領域に相補的な配列で構成されており、なおかつrs8099917がマイナーアリルであるグアニンの場合にアンチセンス鎖と完全な相補鎖となるように設計されていて、アンチセンス鎖におけるSNPを確認できる。
【0020】
配列番号3で示される本発明のプローブは、rs12979860を含む部分領域に相補的な配列で構成されており、なおかつrs12979860がメジャーアリルであるグアニンの場合にアンチセンス鎖と完全な相補鎖となるように設計されていて、アンチセンス鎖におけるSNPを確認できる。
【0021】
本発明のプローブは、配列番号1から3のいずれかに示される核酸配列が好ましいが、rs8103142、rs8099917、rs12979860のいずれかのSNPを含む10塩基以上30塩基以下の核酸配列またはこれに相補的な核酸配列であれば、特に限定されない。
【0022】
本発明のプローブは、標識物質で標識化された標識化プローブであってもよい。好ましくは、蛍光色素によって標識された核酸プローブ、さらに好ましくは、末端が蛍光色素によって標識された核酸プローブを使用できる。
【0023】
本発明のプローブは、標識物質で標識化されたプローブであることが好ましい。前記標識物質は、制限されないが、蛍光色素(蛍光団)があげられる。前記標識化プローブの具体例として、例えば、前記蛍光色素で標識され、単独で蛍光を示し且つハイブリッド形成により蛍光が減少(例えば、消光)するプローブが好ましい。このような現象は、一般に、蛍光消光現象と呼ばれる。この蛍光消光現象を利用したプローブとしては、中でも、一般的にグアニン消光プローブとよばれるものが好ましい。このようなプローブは、いわゆるQProbe(登録商標)として知られている。グアニン消光プローブとは、例えば、オリゴヌクレオチドの3’末端もしくは5’末端の塩基がシトシンとなるように設計し、その末端の塩基シトシンが相補的な塩基グアニンに近づくと発光が弱くなる蛍光色素で前記末端を標識化したプローブである。本発明のプローブにおいては、例えば、蛍光消光現象を示す蛍光色素を、前記オリゴヌクレオチドの3’末端のシトシンに結合させてもよいし、前記オリゴヌクレオチドの5’末端をシトシンに設計し、これに結合させてもよい。
【0024】
前記蛍光色素は、制限されないが、例えば、フルオレセイン、リン光体、ローダミン、ポリメチン色素誘導体等があげられ、市販の蛍光色素としては、例えば、BODIPY FL(商標名、モレキュラープローブ社製)、FluorePrime(商品名、アマシャムファルマシア社製)、Fluoredite(商品名、ミリポア社製)、FAM(ABI社製)、Cy3およびCy5(アマシャムファルマシア社製)、TAMRA(モレキュラープローブ社製)等があげられる。プローブの検出条件は、特に制限されず、使用する蛍光色素により適宜決定できるが、例えば、Pacific Blueは、検出波長450〜480nm、TAMRAは、検出波長585〜700nm、BODIPY FLは、検出波長515〜555nmで検出できる。このようなプローブを使用すれば、シグナルの変動により、ハイブリダイズと解離とを容易に確認することができる。
【0025】
本発明のプローブは、例えば、3’末端にリン酸基が付加されてもよい。後述するように、変異の有無を検出する被検核酸(標的核酸)は、PCR等の核酸増幅法によって調製することができ、この際、本発明のプローブを核酸増幅反応の反応系に共存させることができる。このような場合、プローブの3’末端にリン酸基を付加させておけば、プローブ自体が核酸増幅反応によって伸長することを十分に防止できる。また、3’末端に前述のような標識物質を付加することによっても、同様の効果が得られる。
【0026】
本発明のプローブは、前述のように、C型肝炎治療に関するSNPの検出に使用することができる。この変異の検出方法は、何ら制限されず、被検核酸と前記プローブとのハイブリダイズを利用する方法であればよい。
【0027】
本発明の実施形態の一つは、C型肝炎の治療率に関与すると言われるSNPを検出する方法であって、以下(1)〜(3)の工程を全て含むことを特徴とする方法である。
C型肝炎の治療率に関与すると言われるSNPを検出する方法であって、以下(1)〜(3)の工程を全て含む方法。
(1)被検核酸と項1〜4のいずれかに記載のプローブとをハイブリダイズさせ複合体を形成させる工程
(2)工程(1)のあとに、連続的な温度上昇を行いながら、260nmの吸光度測定または前記プローブに付加した標識のシグナル強度を測定することにより、融解曲線分析を行う工程。
(3)工程(2)における、温度変化に伴うシグナル強度の変動から、前記被検核酸における変異の有無を決定する工程
【0028】
本発明のSNP検出方法によれば、配列番号1〜3のいずれかのプローブを使用することでそれぞれrs8103142、rs8099917、rs12979860のSNPを検出できる。
【0029】
本発明における被検核酸は、例えば、一本鎖でもよいし、二本鎖でもよい。二本鎖の場合は、例えば、被検核酸とプローブとをハイブリダイズさせてハイブリッド体を形成するために、加熱により前記二本鎖を一本鎖に解離させる工程を含むことが好ましい。
【0030】
前記被検核酸の種類としては、特に制限されないが、例えば、DNAや、トータルRNA、mRNA等のRNA等があげられる。また、前記被検核酸は、例えば、生体試料等の試料に含まれる核酸があげられる。前記試料中の核酸は、例えば、生体試料中に元来含まれている核酸でもよいが、例えば、変異検出の精度を向上できることから、前記生体試料中の核酸を鋳型として核酸増幅法により増幅させた増幅産物等があげられる。具体例としては、例えば、前記生体試料に元来含まれているDNAを鋳型として、核酸増幅法により増幅させた増幅産物や、前記生体試料に元来含まれているRNAから逆転写反応(RT−PCR)により生成させたcDNAを鋳型として、核酸増幅法により増幅させた増幅産物があげられる。これらの増幅産物を、本発明における被検核酸としてもよい。前記増幅産物の長さは、特に制限されないが、例えば、50〜1000塩基であり、好ましくは80〜300塩基である。
【0031】
前記生体試料としては、特に制限されないが、例えば、白血球細胞等の血球試料、全血試料、口腔拭い液、咽頭拭い液等があげられる。なお、本発明において、試料の採取方法、DNAやRNA等の核酸の調製方法等は、制限されず、従来公知の方法が採用できる。
【0032】
本発明の方法で用いる融解曲線分析について説明する。例えば、二本鎖DNAを含む溶液を加熱していくと、260nmにおける吸光度が上昇する。これは、二本鎖DNAにおける両鎖間の水素結合が加熱によってほどけ、一本鎖DNAに解離(DNAの融解)することが原因である。そして、全ての二本鎖DNAが解離して一本鎖DNAになると、その吸光度は、加熱開始時の吸光度(二本鎖DNAのみの吸光度)の約1.5倍程度を示し、これによって融解が完了したと判断できる。
【0033】
本発明において、融解曲線分析を行うための温度変化に伴うシグナル変動の測定は、前述のような原理から、260nmの吸光度測定により行うこともできるが、本発明のプローブに付加した標識のシグナルを測定することが好ましい。このため、本発明のプローブとして、前述の標識化プローブを使用することが好ましい。
前記標識化プローブとしては、例えば、単独でシグナルを示し且つハイブリッド形成によりシグナルを示さない標識化プローブ、または、単独でシグナルを示さず且つハイブリッド形成によりシグナルを示す標識化プローブがあげられる。前者のプローブであれば、検出対象配列とハイブリッド(二本鎖)を形成している際にはシグナルを示さず、加熱によりプローブが遊離するとシグナルを示す。また、後者のプローブであれば、検出対象配列とハイブリッド(二本鎖)を形成することによってシグナルを示し、加熱によりプローブが遊離するとシグナルが減少(消失)する。したがって、この標識によるシグナルをシグナル特有の条件(吸光度等)で検出することによって、前記260nmの吸光度測定と同様に、融解の進行を把握することができる。標識化プローブにおける標識物質は、例えば、前述のとおりである。
好ましい標識物質は、蛍光色素である。標識物質に蛍光色素を用いた場合、上記方法において、前記プローブに付加した蛍光色素のシグナル強度を測定することにより、融解曲線分析を行い、この工程における、温度変化に伴うシグナル強度の変動から、前記被検核酸における変異の有無を決定することができる。
【0034】
前述のように、被検核酸が、鋳型核酸から核酸増幅法により増幅させた増幅産物である場合、前記工程(1)は、フォワードプライマーおよびリバースプライマーのプライマー対を用いて鋳型核酸から核酸を増幅する工程を含んでも良い。言い換えれば、前記工程(1)における被検核酸は、フォワードプライマーおよびリバースプライマーのプライマー対を用いて鋳型核酸から増幅された物であっても良い。
【0035】
前記プライマー対としては、IL28B遺伝子および/またはIL28B遺伝子周辺領域の本発明のプローブがハイブリダイズ可能な領域が特異的に増幅されるものであれば、特に制限されない。前記増幅領域は、例えば、前記プローブがハイブリダイズする領域のみでもよいし、前記ハイブリダイズ領域を含む領域であってもよい。
【0036】
具体的には、例えば、下記(F)のオリゴヌクレオチドからなるフォワードプライマーおよび下記(R)のオリゴヌクレオチドからなるリバースプライマーを含む一対のプライマーが例示できる。
(F)塩基配列が配列番号4〜6のいずれかで示されるオリゴヌクレオチド
(R)塩基配列が配列番号7〜9のいずれかで示されるオリゴヌクレオチド
【0037】
前記核酸増幅工程に用いられる具体的な核酸増幅方法としては特に限定されず、適宜公知の方法を用いることができる。例えば、PCR(Polymerase Chain Reaction)法、NASBA(Nucleic acid sequence based amplification)法、TMA(Transcription−mediated amplification)法、SDA(Strand Displacement Amplification)法等があげられるが、PCR法を用いることが好ましい。なお、これらの各方法において、増幅反応の条件は特に制限されず、従来公知の方法により行うことができる。
【0038】
核酸増幅にPCR法を用いる場合、DNAポリメラーゼには、α型DNAポリメラーゼを用いることが好ましい。その理由を以下に説明する。
【0039】
本発明プローブが含まれる反応系でIL28B遺伝子および/またはIL28B遺伝子周辺領域を増幅する場合、核酸増幅工程中に該核酸プローブが試料のIL28B遺伝子および/またはIL28B遺伝子周辺領域またはそれらの増幅産物と結合しうる。核酸増幅工程中にIL28B遺伝子および/またはIL28B遺伝子周辺領域と結合した該核酸プローブは、核酸プライマーとDNAポリメラーゼによる核酸増幅反応を阻害する。
【0040】
Taq DNA PolymeraseなどPolI型のDNAポリメラーゼは5’→ 3’エキソヌクレアーゼ活性を持つことが知られている。この活性のため、核酸増幅反応中に鋳型となるIL28B遺伝子および/またはIL28B遺伝子周辺領域と結合した核酸がある場合、該結合核酸はエキソヌクレアーゼ活性によって分解されてしまう。このため、反応系中の該核酸プローブが減少し核酸検出工程に問題が生じる可能性がある。従って、PolI型DNAポリメラーゼを用いて本発明を実施することは好ましくない。
【0041】
他方、KOD DNA Polymerase(超好熱始原菌Thermococcus kodakaraensis KOD1由来)などα型のDNAポリメラーゼは5’→ 3’エキソヌクレアーゼ活性は持たず、3’→ 5’エキソヌクレアーゼ活性を持つ。従って、α型DNAポリメラーゼを用いれば上記問題を解決できるのみならず、3’→ 5’エキソヌクレアーゼ活性により核酸増幅工程において高い正確性が発揮される。
【0042】
通常、α型DNAポリメラーゼは3’→ 5’エキソヌクレアーゼ活性のため、核酸増幅速度はPolI型酵素と比較して低い傾向がある。しかし、KOD DNA Polymeraseはα型DNAポリメラーゼでありながらDNA合成活性が高く100塩基/秒以上のDNA合成速度を有し伸長効率が優れている。従って、本発明の実施にはα型DNAポリメラーゼの中でも、KOD DNA Polymerase(東洋紡績製、商標)を用いることが好ましい。
【0043】
さらに、α型DNAポリメラーゼを変異させて100塩基/秒以上のデオキシリボ核酸合成速度を達成させた変異型、あるいは、野生型および/または変異型の組み合わせにより当該性能を達成させたDNAポリメラーゼ組成物も、本発明の実施に適したDNAポリメラーゼとして用いることができる。
例えば、上記KOD DNA Polymerase以外に100塩基/秒以上のデオキシリボ核酸合成速度を有するDNAポリメラーゼとして、「KOD FX(東洋紡績製、登録商標)」、「KOD −Plus−(東洋紡績製、商標)」、「KOD Dash(東洋紡績製、登録商標)」、PrimeSTAR HS DNAポリメラーゼ(タカラバイオ製、登録商標)なども利用できる。
【0044】
本発明において、DNA合成活性とは鋳型DNAにアニールされたオリゴヌクレオチドまたはポリヌクレオチドの3’−ヒドロキシル基にデオキシリボヌクレオシド5’−トリホスフェートのα−ホスフェートを共有結合せしめることにより、デオキシリボ核酸にデオキシリボヌクレオシド5’−モノホスフェートを鋳型依存的に導入する反応を触媒する活性をいう。
【0045】
その活性測定法は、酵素活性が高い場合には、保存緩衝液でサンプルを希釈して測定を行う。本発明では、下記A液25μl、B液およびC液各5μlおよび滅菌水10μlをエッペンドルフチューブに加えて攪拌混合した後、上記酵素液5μlを加えて75℃で10分間反応する。その後、氷冷し、E液50μl、D液100μlを加えて、攪拌後、さらに10分間氷冷する。この液をガラスフィルター(ワットマンGF/Cフィルター)で濾過し、D液及びエタノールで充分洗浄し、フィルターの放射活性を液体シンチレーションカウンター(パッカード社製)で計測し、鋳型DNAへのヌクレオチドの取り込みを測定する。酵素活性の1単位はこの条件下で30分あたり10nモルのヌクレオチドを酸不溶性画分に取り込む酵素量とする。
A: 40mM Tris−HCl(pH7.5)
16mM 塩化マグネシウム
15mM ジチオスレイトール
100μg/ml BSA
B: 2μg/μl 活性化仔牛胸腺DNA
C: 1.5mM dNTP(250cpm/pmol〔 3H〕dTTP)
D: 20% トリクロロ酢酸(2mMピロリン酸ナトリウム)
E: 1μg/μl キャリアーDNA
【0046】
本発明において、3’−5’エキソヌクレアーゼ活性とは、DNAの3’末端領域を切除し、5’−モノヌクレオチドを遊離する活性をいう。
その活性測定法は、50μlの反応液(120mM Tris−HCl(pH8.8 at 25℃), 10mM KCl, 6mM 硫酸アンモニウム,1mM MgCl2, 0.1% Triton X−100, 0.001% BSA,5 μg トリチウムラベルされた大腸菌DNA)を1.5mlのエッペンチューブに分注し、DNAポリメラーゼを加える。75℃で10分間反応させた後、氷冷によって反応を停止し、次にキャリアーとして、0.1%のBSAを50μl加え、さらに10%のトリクロロ酢酸、2%ピロリン酸ナトリウム溶液を100μl加え混合する。氷上で15分放置した後、12,000回転で10分間遠心し沈殿を分離する。上清100μlの放射活性を液体シンチレーションカウンター(パッカード社製)で計測し、酸可溶性画分に遊離したヌクレオチド量を測定する。
【0047】
本発明において、DNAポリメラーゼのDNA合成速度とは単位時間当たりのDNAの合成数をいう。DNA合成速度は、市販品であれば該市販品の説明書等に記載されている値、説明書等に記載がない場合は文献等に記載されている値を指すものとする。これらの方法で特定できない場合は、以下の方法で求めることができる。
【0048】
DNA合成速度の測定法
DNAポリメラーゼの反応液(20mM Tris−HCl(pH7.5),8mM塩化マグネシウム、7.5mM ジチオスレイトール、100 μg/ml BSA、0.1mM dNTP、0.2μCi [α−32P]dCTP)を、プライマーをアニーリングさせたM13mp181本鎖DNAと75℃で反応させる。反応停止は等量の反応停止液(50mM 水酸化ナトリウム、10mM EDTA、5% フィコール、0.05% ブロモフェノールブルー)を加えることにより行う。上記反応にて合成されたDNAをアルカリアガロースゲル電気泳動にて分画した後、ゲルを乾燥させオートラジオグラフィーを行う。DNAサイズマーカーとしてはラベルしたλ/HindIIIを用いる。このマーカーのバンドを指標として合成されたDNAのサイズを測定することによって、DNA合成速度を求める。
【0049】
次に、本発明の変異検出方法について、本発明のプローブとして、蛍光色素で標識化された標識化プローブを使用する例をあげて説明する。なお、本発明の変異検出方法は、以下の説明によって何ら制限されることはない。
【0050】
まず、例えば全血からゲノムDNAを単離する。単離は、例えば、市販のゲノムDNA単離キットを用いて行うことができる。
次に、単離したゲノムDNAを含む試料に、標識化プローブとして、例えば、QProbeを添加する。
【0051】
前記プローブの添加のタイミングは、特に制限されず、例えば、後述する増幅反応前、増幅反応途中および増幅反応後のいずれに、増幅反応の反応系に添加してもよい。
中でも、増幅反応と、前記工程(2)とを連続的に行うことができるため、増幅反応前に添加することが好ましい。このように核酸増幅反応の前に前記プローブを添加する場合は、例えば、前述のように、その3’末端に、蛍光色素を付加したり、リン酸基を付加することが好ましい。
【0052】
前記プローブは、単離したゲノムDNAを含む液体試料に添加してもよいし、溶媒中でゲノムDNAと混合してもよい。前記溶媒としては、特に制限されず、例えば、Tris−HCl等の緩衝液、KCl、MgCl2、MgSO4、グリセロール等を含む溶媒、PCR反応液等、従来公知のものがあげられる。
【0053】
続いて、単離したゲノムDNAを鋳型として、PCR等の核酸増幅法によって、検出目的の塩基部位を含む配列を増幅させる。なお、PCR等の条件は、特に制限されず、従来公知の方法により行うことができる。
【0054】
PCRのプライマーの配列は、検出目的の塩基部位を含む配列を増幅できるものであれば特に制限されず、前述のように、目的の配列に応じて、従来公知の方法により適宜設計できる。プライマーの長さは、特に制限されず、一般的な長さ(例えば、10〜40mer)に設定できる。
【0055】
本発明の検出方法に使用するプライマーセットとしては、例えば、前述の本発明のプライマーセットがあげられる。なお、本発明のプライマーセットの用途は、本発明の検出方法には制限されず、また、本発明の検出方法に使用するプライマーセットも、本発明のプライマーセットには限定されない。
【0056】
次に、得られたPCR増幅産物の解離、および、解離により得られた一本鎖DNAと前記標識化プローブとのハイブリダイズを行う。これは、例えば、前記反応液の温度変化によって行うことができる。
【0057】
前記解離工程における加熱温度は、前記増幅産物が解離できる温度であれば特に制限されないが、例えば、85〜98℃である。加熱時間も特に制限されないが、通常、1秒〜10分であり、好ましくは1秒〜5分である。
【0058】
また、解離した一本鎖DNAと前記標識化プローブとのハイブリダイズは、例えば、前記解離工程の後、前記解離工程における加熱温度を降下させることによって行うことができる。温度条件としては、例えば、35〜50℃である。
【0059】
ハイブリダイズ工程の反応系(反応系)における各組成の体積や濃度は、特に制限されない。具体例としては、前記反応系において、DNAの濃度は、例えば、0.01〜1μmol/Lであり、好ましくは0.1〜0.5μmol/L、前記標識化プローブの濃度は、例えば、前記DNAに対する添加割合を満たす範囲が好ましく、例えば、0.001〜10μmol/Lであり、好ましくは0.001〜1μmol/Lである。
【0060】
そして、前記反応液の温度を変化させ、前記増幅産物と前記標識化プローブとのハイブリッド形成体の融解状態を示すシグナル値を測定する。具体的には、例えば、前記反応液(前記一本鎖DNAと前記標識化プローブとのハイブリッド形成体)を加熱し、温度上昇に伴うシグナル値の変動を測定する。前述のように、末端のC塩基が標識化されたプローブ(グアニン消光プローブ)を使用した場合、一本鎖DNAとハイブリダイズした状態では、蛍光が減少(または消光)し、解離した状態では、蛍光を発する。したがって、例えば、蛍光が減少(または消光)しているハイブリッド形成体を徐々に加熱し、温度上昇に伴う蛍光強度の増加を測定すればよい。
【0061】
蛍光強度の変動を測定する際の温度範囲は、特に制限されないが、例えば、開始温度が室温〜85℃であり、好ましくは25〜70℃であり、終了温度は、例えば、40〜105℃である。また、温度の上昇速度は、特に制限されないが、例えば、0.05〜20℃/秒であり、好ましくは0.08〜5℃/秒である。
【0062】
目的の塩基部位における遺伝子型(野生型、変異型など)の決定は、例えば、ハイブリッド形成時におけるシグナル変動を測定することによって行いうる。すなわち、前記プローブを含む反応液の温度を降下させてハイブリッド形成体を形成する際に、前記温度降下に伴うシグナル変動を測定する。
【0063】
具体例として、単独でシグナルを示し且つハイブリッド形成によりシグナルを示さない標識化プローブ(例えば、グアニン消光プローブ)を使用した場合、一本鎖DNAとプローブとが解離している状態では蛍光を発しているが、温度の降下によりハイブリッドを形成すると、前記蛍光が減少(または消光)する。したがって、例えば、前記反応液の温度を徐々に降下させて、温度下降に伴う蛍光強度の減少を測定すればよい。他方、単独でシグナルを示さず且つハイブリッド形成によりシグナルを示す標識化プローブを使用した場合、一本鎖DNAとプローブとが解離している状態では蛍光を発していないが、温度の降下によりハイブリッドを形成すると、蛍光を発するようになる。したがって、例えば、前記反応液の温度を徐々に降下させて、温度下降に伴う蛍光強度の増加を測定すればよい。
【0064】
また、本発明においては、目的の塩基部位における遺伝子型の決定のために、前記シグナルの変動を解析してTm(melting temperature)値として決定してもよい。
【実施例】
【0065】
以下に実施例を示して本発明を具体的に説明するが、本発明は実施例に限定されるものではない。
【0066】
〔実施例1:精製したヒトゲノムからのrs8103142の検出〕
(1)試料の調製
既にrs8103142がメジャーホモ(T/T)であることが判明している精製されたヒトゲノム(以下HM)、rs8103142がヘテロ(T/C)であることが判明している精製されたヒトゲノム(以下HH)、そしてrs8103142がマイナーホモ(C/C)であることが判明している精製されたヒトゲノム(以下Hm)を試料とし、陰性コントロール(NC)として10mMのTris−HCl(pH7.5)を試料とした。
(2)核酸増幅および融解曲線分析
上記ヒトゲノム試料および陰性コントロールにそれぞれ下記試薬を添加して、下記条件によりrs8103142を検出した。核酸増幅および融解曲線分析には東洋紡績社製GENECUBE(登録商標)を使用した。
【0067】
試薬
以下の試薬を含む13.2μl溶液を調製した。
核酸プライマー(配列番号4) 300nM
核酸プライマー(配列番号7) 1500nM
核酸プローブ(配列番号1、3’末端をBODIPY−FL標識) 300nM
×10緩衝液 1.3μl
dNTP 0.2mM
MgSO4 4mM
BSA 1.3μg
DMSO 0.98μL
KOD plus DNA polymerase(東洋紡績製) 0.4U
試料 4μl
【0068】
核酸増幅および融解曲線分析
94℃・2分
(以上1サイクル)
97℃・1秒
60℃・3秒
63℃・5秒
(以上50サイクル)
94℃・30秒
39℃・30秒
39℃〜75℃(0.09℃/秒で温度上昇)
【0069】
結果
図1は、融解曲線における温度上昇にともなう蛍光強度の変化を、グラフの横軸を温度、縦軸を蛍光シグナルの微分値として解析した結果を表した図である。HMは57〜58℃にピークが現れており、Hmは67℃付近でピークが現れている。そしてHHはその両方の温度でピークが現れている。本実施例で用いた配列番号1のプローブは末端がBODIPY−FL標識されている。このプローブは相補的な配列の核酸と結合している間は蛍光が消光しており、逆にプローブが単独で遊離している間は蛍光が発光しているという特徴を有する。配列番号1のプローブはマイナーホモのrs8103142を含む核酸と最も強く結合し、メジャーホモのrs8103142を含む核酸とは一塩基のミスマッチが生じているため、より弱く結合する。従って、図1の57〜58℃でのピークは被検核酸中のrs8103142がメジャーアリルであることを示しており、67℃付近のピークは被検核酸中のrs8103142がマイナーアリルであることを示している。そしてその両方のピークを発した被検核酸はメジャーアリルとマイナーアリルの両方を有するヘテロであることを示している。
以上より、本発明プローブを用いることでrs8103142のアリルを容易に識別できることが示された。
【0070】
〔実施例2:精製したヒトゲノムからのrs8099917の検出〕
(1)試料の調製
既にrs8099917がメジャーホモ(T/T)であることが判明している精製されたヒトゲノム(以下HM)、rs8099917がヘテロ(T/G)であることが判明している精製されたヒトゲノム(以下HH)、そしてrs8099917がマイナーホモ(G/G)であることが判明している精製されたヒトゲノム(以下Hm)を試料とし、陰性コントロール(NC)として10mMのTris−HCl(pH7.5)を試料とした。
(2)核酸増幅および融解曲線分析
上記ヒトゲノム試料および陰性コントロールにそれぞれ下記試薬を添加して、下記条件によりrs8099917を検出した。核酸増幅および融解曲線分析には東洋紡績社製GENECUBE(登録商標)を使用した。
【0071】
試薬
以下の試薬を含む13.2μl溶液を調製した。
核酸プライマー(配列番号5) 500nM
核酸プライマー(配列番号8) 2500nM
核酸プローブ(配列番号2、3’末端をBODIPY−FL標識) 250nM
×10緩衝液 1.3μl
dNTP 0.2mM
MgSO4 4mM
BSA 1.3μg
DMSO 0.98μL
KOD plus DNA polymerase(東洋紡績製) 0.4U
試料 4μl
【0072】
核酸増幅および融解曲線分析
実施例1と同じ。
【0073】
結果
図2は、融解曲線における温度上昇にともなう蛍光強度の変化を、グラフの横軸を温度、縦軸を蛍光シグナルの微分値として解析した結果を表した図である。HMは54℃付近にピークが現れており、Hmは61℃付近でピークが現れている。そしてHHはその両方の温度でピークが現れている。本実施例で用いた配列番号2のプローブは末端がBODIPY−FL標識されている。このプローブは相補的な配列の核酸と結合している間は蛍光が消光しており、逆にプローブが単独で遊離している間は蛍光が発光しているという特徴を有する。配列番号2のプローブはマイナーホモのrs8099917を含む核酸と最も強く結合し、メジャーホモのrs8103142を含む核酸とは一塩基のミスマッチが生じているため、より弱く結合する。従って、図2の54℃付近でのピークは被検核酸中のrs8099917がメジャーアリルであることを示しており、61℃付近のピークは被検核酸中のrs8099917がマイナーアリルであることを示している。そしてその両方のピークを発した被検核酸はメジャーアリルとマイナーアリルの両方を有するヘテロであることを示している。
以上より、本発明プローブを用いることでrs8099917のアリルを容易に識別できることが示された。
【0074】
〔実施例3:精製したヒトゲノムからのrs12979860の検出〕
(1)試料の調製
既にrs12979860がメジャーホモ(C/C)であることが判明している精製されたヒトゲノム(以下HM)、rs12979860がヘテロ(C/T)であることが判明している精製されたヒトゲノム(以下HH)、そしてrs12979860がマイナーホモ(T/T)であることが判明している精製されたヒトゲノム(以下Hm)を試料とし、陰性コントロール(NC)として10mMのTris−HCl(pH7.5)を試料とした。
(2)核酸増幅および融解曲線分析
上記ヒトゲノム試料および陰性コントロールにそれぞれ下記試薬を添加して、下記条件によりrs12979860を検出した。核酸増幅および融解曲線分析には東洋紡績社製GENECUBE(登録商標)を使用した。
【0075】
試薬
以下の試薬を含む13.2μl溶液を調製した。
核酸プライマー(配列番号6) 300nM
核酸プライマー(配列番号9) 1500nM
核酸プローブ(配列番号3、3’末端をBODIPY−FL標識) 300nM
×10緩衝液 1.3μl
dNTP 0.2mM
MgSO4 4mM
BSA 1.3μg
DMSO 0.98μL
KOD plus DNA polymerase(東洋紡績製) 0.4U
試料 4μl
【0076】
核酸増幅および融解曲線分析
実施例1と同じ。
【0077】
結果
図3は、融解曲線における温度上昇にともなう蛍光強度の変化を、グラフの横軸を温度、縦軸を蛍光シグナルの微分値として解析した結果を表した図である。HMは62℃付近にピークが現れており、Hmは48℃付近でピークが現れている。そしてHHはその両方の温度でピークが現れている。本実施例で用いた配列番号3のプローブは末端がBODIPY−FL標識されている。このプローブは相補的な配列の核酸と結合している間は蛍光が消光しており、逆にプローブが単独で遊離している間は蛍光が発光しているという特徴を有する。配列番号3のプローブはメジャーホモのrs12979860を含む核酸と最も強く結合し、マイナーホモのrs12979860を含む核酸とは一塩基のミスマッチが生じているため、より弱く結合する。従って、図3の48℃付近でのピークは被検核酸中のrs12979860がマイナーアリルであることを示しており、62℃付近のピークは被検核酸中のrs12979860がメジャーアリルであることを示している。そしてその両方のピークを発した被検核酸はメジャーアリルとマイナーアリルの両方を有するヘテロであることを示している。
以上より、本発明プローブを用いることでrs12979860のアリルを容易に識別できることが示された。
【0078】
〔実施例4:全血希釈液からのrs8099917の検出〕
(1)試料の調製
全血10μlに10mMTris−HCl(pH7.5)を90μl加えて、希釈倍率10倍の全血希釈液を調製した。この全血希釈液を試料とした。検出系は10μlであり、試料は1μlを供するため、検出系での全血の終濃度は1%となる。
(2)核酸増幅および融解曲線分析
上記全血希釈液に下記試薬を添加して、下記条件によりrs8099917を検出した。核酸増幅および融解曲線分析には東洋紡績社製GENECUBE(登録商標)を使用した。
【0079】
試薬
以下の試薬を含む10μl溶液を調製した。
核酸プライマー(配列番号5) 500nM
核酸プライマー(配列番号8) 2500nM
核酸プローブ(配列番号2、3’末端をBODIPY−FL標識) 250nM
×10緩衝液 1μl
dNTP 0.2mM
MgSO4 4mM
BSA 1μg
DMSO 0.75μL
KOD plus DNA polymerase(東洋紡績製) 0.3U
試料 1μl
【0080】
核酸増幅および融解曲線分析
実施例1と同じ。
【0081】
結果
図5は、融解曲線における温度上昇にともなう蛍光強度の変化を、グラフの横軸を温度、縦軸を蛍光シグナルの微分値として解析した結果を表した図である。図5から明らかなように、本発明プローブを用いることによって全血の希釈液からでもrs8099917を検出することができる。
【産業上の利用可能性】
【0082】
本発明をC型肝炎治療に関するSNP検査およびC型肝炎治療に関するSNP検査試薬開発に適用することで、迅速、確実かつ簡便な慢性骨髄増殖性疾患関連遺伝子の変異検出を行うことができる。
【特許請求の範囲】
【請求項1】
C型肝炎の治療率に関与すると言われるSNPを検出するためのプローブであって、配列番号1〜3のいずれかで示される塩基配列を有するプローブ。
【請求項2】
検出可能な標識物質により標識化された、請求項1に記載のプローブ。
【請求項3】
検出可能な標識物質が蛍光色素である、請求項2に記載のプローブ。
【請求項4】
末端のシトシンが標識化された、請求項2または3に記載のプローブ。
【請求項5】
C型肝炎の治療率に関与すると言われるSNPを検出する方法であって、以下(1)〜(3)の工程を全て含む方法。
(1)被検核酸と項1〜4のいずれかに記載のプローブとをハイブリダイズさせ複合体を形成させる工程
(2)工程(1)のあとに、連続的な温度上昇を行いながら、260nmの吸光度測定または前記プローブに付加した標識のシグナル強度を測定することにより、融解曲線分析を行う工程。
(3)工程(2)における、温度変化に伴うシグナル強度の変動から、前記被検核酸における変異の有無を決定する工程
【請求項6】
請求項5に記載の方法であって、以下(1)〜(3)の工程を全て含む方法。
(1)被検核酸と項1〜4のいずれかに記載のプローブとをハイブリダイズさせ複合体を形成させる工程
(2)工程(1)のあとに、連続的な温度上昇を行いながら、前記プローブを標識している蛍光色素の蛍光強度を測定することにより、融解曲線分析を行う工程。
(3)工程(2)における、温度変化に伴う蛍光強度の変動から、前記被検核酸における変異の有無を決定する工程
【請求項7】
工程(1)における被検核酸が、プライマー対を用いて鋳型核酸から増幅された物である、請求項5または6に記載の方法。
【請求項8】
核酸増幅に、100塩基/秒以上のDNA合成速度を有し、かつ、5’エキソヌクレアーゼ活性を持たないDNAポリメラーゼを用いる、請求項7に記載の方法。
【請求項9】
前記プライマー対が、下記(F)のオリゴヌクレオチドからなるフォワードプライマーおよび下記(R)のオリゴヌクレオチドからなるリバースプライマーを含む一対のプライマーである、請求項7または8に記載の方法。
(F)塩基配列が配列番号4〜6のいずれかで示されるオリゴヌクレオチド
(R)塩基配列が配列番号7〜9のいずれかで示されるオリゴヌクレオチド
【請求項10】
核酸増幅法によりC型肝炎の治療率に関与すると言われるSNPを増幅するためのプライマー対であって、下記(1)〜(3)のいずれかで構成されるプライマー対。
(1)配列番号4および配列番号7のそれぞれに示される塩基配列を有するプライマーからなるプライマー対
(2)配列番号5および配列番号8のそれぞれに示される塩基配列を有するプライマーからなるプライマー対
(3)配列番号6および配列番号9のそれぞれに示される塩基配列を有するプライマーからなるプライマー対
【請求項11】
請求項5に記載のC型肝炎の治療率に関与すると言われるSNPを検出する方法に用いるための変異検出キットであって、項1〜4のいずれかに記載のプローブを含む変異検出キット。
【請求項1】
C型肝炎の治療率に関与すると言われるSNPを検出するためのプローブであって、配列番号1〜3のいずれかで示される塩基配列を有するプローブ。
【請求項2】
検出可能な標識物質により標識化された、請求項1に記載のプローブ。
【請求項3】
検出可能な標識物質が蛍光色素である、請求項2に記載のプローブ。
【請求項4】
末端のシトシンが標識化された、請求項2または3に記載のプローブ。
【請求項5】
C型肝炎の治療率に関与すると言われるSNPを検出する方法であって、以下(1)〜(3)の工程を全て含む方法。
(1)被検核酸と項1〜4のいずれかに記載のプローブとをハイブリダイズさせ複合体を形成させる工程
(2)工程(1)のあとに、連続的な温度上昇を行いながら、260nmの吸光度測定または前記プローブに付加した標識のシグナル強度を測定することにより、融解曲線分析を行う工程。
(3)工程(2)における、温度変化に伴うシグナル強度の変動から、前記被検核酸における変異の有無を決定する工程
【請求項6】
請求項5に記載の方法であって、以下(1)〜(3)の工程を全て含む方法。
(1)被検核酸と項1〜4のいずれかに記載のプローブとをハイブリダイズさせ複合体を形成させる工程
(2)工程(1)のあとに、連続的な温度上昇を行いながら、前記プローブを標識している蛍光色素の蛍光強度を測定することにより、融解曲線分析を行う工程。
(3)工程(2)における、温度変化に伴う蛍光強度の変動から、前記被検核酸における変異の有無を決定する工程
【請求項7】
工程(1)における被検核酸が、プライマー対を用いて鋳型核酸から増幅された物である、請求項5または6に記載の方法。
【請求項8】
核酸増幅に、100塩基/秒以上のDNA合成速度を有し、かつ、5’エキソヌクレアーゼ活性を持たないDNAポリメラーゼを用いる、請求項7に記載の方法。
【請求項9】
前記プライマー対が、下記(F)のオリゴヌクレオチドからなるフォワードプライマーおよび下記(R)のオリゴヌクレオチドからなるリバースプライマーを含む一対のプライマーである、請求項7または8に記載の方法。
(F)塩基配列が配列番号4〜6のいずれかで示されるオリゴヌクレオチド
(R)塩基配列が配列番号7〜9のいずれかで示されるオリゴヌクレオチド
【請求項10】
核酸増幅法によりC型肝炎の治療率に関与すると言われるSNPを増幅するためのプライマー対であって、下記(1)〜(3)のいずれかで構成されるプライマー対。
(1)配列番号4および配列番号7のそれぞれに示される塩基配列を有するプライマーからなるプライマー対
(2)配列番号5および配列番号8のそれぞれに示される塩基配列を有するプライマーからなるプライマー対
(3)配列番号6および配列番号9のそれぞれに示される塩基配列を有するプライマーからなるプライマー対
【請求項11】
請求項5に記載のC型肝炎の治療率に関与すると言われるSNPを検出する方法に用いるための変異検出キットであって、項1〜4のいずれかに記載のプローブを含む変異検出キット。
【図1】


【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公開番号】特開2013−70639(P2013−70639A)
【公開日】平成25年4月22日(2013.4.22)
【国際特許分類】
【出願番号】特願2011−210612(P2011−210612)
【出願日】平成23年9月27日(2011.9.27)
【出願人】(000003160)東洋紡株式会社 (3,622)
【Fターム(参考)】
【公開日】平成25年4月22日(2013.4.22)
【国際特許分類】
【出願日】平成23年9月27日(2011.9.27)
【出願人】(000003160)東洋紡株式会社 (3,622)
【Fターム(参考)】
[ Back to top ]