
C型肝炎ウイルス感染の処置のための治療法
HCVに感染した患者におけるVX−222の薬物動態を改善する方法であって、該患者にVX−222およびVX−950を併用投与することを含む方法。HCVに感染した患者の処置方法であって、該患者にVX−222およびVX−950を投与することを含み、ここで、VX−222は、約20mgないし約400mg用量であり、VX−950は、約100mgないし約1,500mg用量である、方法。HCVに感染した患者の処置方法であって、治療的有効量のVX−222を投与することを含み、ここで、VX−222が、約20mgないし約2,000mg用量で1日1回投与される、方法。
【発明の詳細な説明】
【技術分野】
【0001】
関連出願
本出願は、2010年1月29日出願の米国仮特許出願番号第61/299,643号、2010年2月26日出願の同第61/308,506号、2010年3月1日出願の同第61/309,117号、および2010年4月15日出願の同第61/324,395号に優先権の利益を主張する。これらの出願の開示内容全体は、参照により本明細書に包含される。
【0002】
発明の技術分野
本発明は、C型肝炎ウイルス感染の処置法に関する。
【背景技術】
【0003】
発明の背景
C型肝炎ウイルス(“HCV”)による感染は、注目せざるを得ないヒトの医療問題である。HCVは、非A非B型肝炎のほとんどの症例の原因因子として認識されており、全世界で、概算で3%のヒト血清陽性率である(例えば、A. Alberti et al., “Natural History of Hepatitis C,” J. Hepatology, 31., (Suppl. 1), pp. 17−24 (1999)を参照)。米国だけでも400万名近くが感染している可能性がある(例えば、M.J. Alter et al., “The Epidemiology of Viral Hepatitis in the United States, Gastroenterol. Clin. North Am., 23, pp. 437−455 (1994); M. J. Alter “Hepatitis C Virus Infection in the United States,” J. Hepatology, 31., (Suppl. 1), pp. 88−91 (1999)を参照)。
【0004】
HCV感染者のうち、20−25%は、急性感染後にウイルスを除去することができるが、75−80%は、慢性感染症を発症する(例えば、preface, Frontiers in viral Hepatitis, Ed. RF Schinazi, J−P Sommadossi, and CM Rice, p. xi., Elsevier (2003)を参照のこと)。通常、これが肝炎の再発性および進行性の悪化を起こし、多くの場合、さらに重篤な病状、例えば、肝硬変および肝細胞癌を引き起こす(例えば、M.C. Kew, “Hepatitis C and Hepatocellular Carcinoma”, FEMS Microbiology Reviews, 14, pp. 211−220 (1994); I. Saito et. al., “Hepatitis C Virus Infection is Associated with the Development of Hepatocellular Carcinoma,” Proc. Natl. Acad. Sci. USA, 87, pp. 6547−6549 (1990)を参照)。残念なことに、慢性HCV感染症の進行を遅らせるのに広く有効な処置は存在しない。
【0005】
HCVゲノムは3010〜3033個のアミノ酸のポリタンパク質をコードする(例えば、Q.L. Choo, et. al., “Genetic Organization and Diversity of the Hepatitis C Virus.” Proc. Natl. Acad. Sci. USA, 88, pp. 2451−2455 (1991); N. Kato et al., “Molecular Cloning of the Human Hepatitis C Virus Genome From Japanese Patients with Non−A, Non−B Hepatitis,” Proc. Natl. Acad. Sci. USA, 87, pp. 9524−9528 (1990); A. Takamizawa et. al., “Structure and Organization of the Hepatitis C Virus Genome Isolated From Human Carriers,” J. Virol., 65, pp. 1105−1113 (1991)を参照)。HCVの非構造(NS)タンパク質は、ウイルス複製に必須の触媒機構を提供すると考えられている。NSタンパク質は、ポリタンパク質のタンパク質分解的切断に由来する(例えば、R. Bartenschlager et. al., “Nonstructural Protein 3 of the Hepatitis C Virus Encodes a Serine−Type Proteinase Required for Cleavage at the NS3/4 and NS4/5 Junctions,” J. Virol., 67, pp. 3835−3844 (1993); A. Grakoui et. al., “Characterization of the Hepatitis C Virus−Encoded Serine Proteinase: Determination of Proteinase−Dependent Polyprotein Cleavage Sites,” J. Virol., 67, pp. 2832−2843 (1993); A. Grakoui et. al., “Expression and Identification of Hepatitis C Virus Polyprotein Cleavage Products,” J. Virol., 67, pp. 1385−1395 (1993); L. Tomei et. al., “NS3 is a serine protease required for processing of hepatitis C virus polyprotein”, J. Virol., 67, pp. 4017−4026 (1993)を参照)。
【0006】
HCVのNSタンパク質3(NS3)には、大部分のウイルス酵素の合成を補助するセリンプロテアーゼ活性が含まれ、それ故に、後者はウイルス複製および感染力に必須であると見なされる。黄熱病ウイルスNS3プロテアーゼにおける変異は、ウイルス感染力を低減することが知られている(例えば、Chambers, T.J. et. al., “Evidence that the N−terminal Domain of Nonstructural Protein NS3 From Yellow Fever Virus is a Serine Protease Responsible for Site−Specific Cleavages in the Viral Polyprotein”, Proc. Natl. Acad. Sci. USA, 87, pp. 8898−8902 (1990)を参照)。NS3の最初の181個のアミノ酸(ウイルスポリタンパク質の残基1027〜1207)は、HCVポリタンパク質の4つ全ての下流部位を合成するNS3のセリンプロテアーゼドメインを含むことが示されている(例えば、C. Lin et al., “Hepatitis C Virus NS3 Serine Proteinase: Trans−Cleavage Requirements and Processing Kinetics”, J. Virol., 68, pp. 8147−8157 (1994)を参照)。
【0007】
HCV NS3 セリンプロテアーゼおよびそれと関係する補因子NS4Aは、全てのウイルス酵素プロセシングを補助し、故に、ウイルス複製に必須であると見なされる。このプロセシングはまた、ウイルス酵素プロセシングに関与する、ヒト免疫不全ウイルス アスパルチルプロテアーゼにより行われるものと同様のようである。ウイルスタンパク質プロセシングを阻害するHIVプロテアーゼ阻害剤は、ヒトにおける強力な抗ウイルス剤であり、ウイルス生活環のこの段階を阻止することは、結果として治療的有効剤であることを示す。結果として、HCV NS3 セリンプロテアーゼはまた、創薬の魅力的な標的である。
【0008】
現在、満足のいく抗HCV剤または処置剤は存在しない。最近まで、唯一の確立されたHCV疾患の治療法は、インターフェロンでの処置であった。HCV感染のために最初に承認された治療法は、標準的な(非ペグ化)インターフェロンαでの処置であった。しかし、インターフェロンは重い副作用を有し(例えば、M. A. Walker et al., “Hepatitis C Virus: An Overview of Current Approaches and Progress(C型肝炎ウイルス;最新の対処法と進歩の概説),” DDT, 4, pp. 518−29 (1999); D. Moradpour et al., “Current and Evolving Therapies for Hepatitis C,” Eur. J. Gastroenterol. Hepatol., 11, pp. 1199−1202 (1999); H. L. A. Janssen et al. “Suicide Associated with Alfa−Interferon Therapy for Chronic Viral Hepatitis,” J. Hepatol., 21, pp. 241−243 (1994); P.F. Renault et al., “Side Effects of Alpha Interferon” seminars in Liver Disease, 9, pp. 273−277. (1989)を参照)、長期間の寛解を誘導するのは、疾患の僅かな割合(〜25%)のみに限られる(例えば、O. Weiland, “Interferon Therapy in Chronic Hepatitis C Virus Infection”, FEMS Microbiol. Rev., 14, pp. 279−288 (1994)を参照)。処置レジメンへのリバビリンの追加は、わずかに治療応答率を増加させる。最近導入されたペグ化形態のインターフェロン(ペグ−イントロン(登録商標)およびペガシス(登録商標))とリバビリンとの併用もまた、寛解率の僅かながらの改善と副作用の部分的な軽減をもたらした。現在の標準的治療は、HCV遺伝子型のような予後因子、および治療への初期応答の発現に依拠しながら、24ないし48週継続する処置レジメンである。さらに、有効な抗HCVワクチンの見込みは、不明確なままである。
【0009】
従って、抗HCV療法および抗HCV化合物に適当な用量レジメンの必要性がある。
【0010】
HCVならびに他の疾患および障害は、肝臓損傷と関係がある。肝臓損傷を処置するための治療法および適当な用量レジメンの必要性がある。
【発明の概要】
【0011】
発明の概要
本発明は、一般的に、C型肝炎ウイルス(HCV)感染の処置法を提供する。本発明はまた、一般的に、C型肝炎ウイルス感染の臨床的続発症の予防法を提供する。
【0012】
一面において、本発明は、HCVに感染した患者におけるVX−222の薬物動態を改善する方法に関する。該方法は、患者に対して、VX−222およびVX−950を共投与することを含む。
【0013】
他の面において、本発明は、HCVに感染した患者の血漿中におけるVX−222への暴露を増大する方法に関する。該方法は、患者に対してVX−222およびVX−950を投与することを含む。
【0014】
他の面において、本発明は、HCVに感染した患者の処置法に関する。該方法は、患者にVX−222およびVX−950を投与することを含み、ここで、VX−222は、約20mgないし約400mgの量であり、VX−950は、約100mgないし約1500mgの量である。
【0015】
さらに他の面において、本発明は、治療的有効量のVX−222を投与することを含むHCVに感染した患者の処置法に関し、ここで、VX−222は、1日1回、約20mgないし約2000mgが投与される。
【0016】
さらに他の面において、本発明は、a)約20mgないし約400mgのVX−222;および、b) 約100mgないし約1500mgのVX−950を含む、薬学的に許容される組成物に関する。
【0017】
本発明はまた、HCVに感染した患者におけるVX−222のバイオアベイラビリティを増大するための医薬の製造における、VX−222およびVX−950の使用を提供する。
【0018】
本発明はまた、HCVに感染した患者の血漿中におけるVX−222のバイオアベイラビリティまたはVX−222への暴露を増大するための医薬の製造における、VX−222およびVX−950の使用を提供する。
【0019】
本発明はまた、HCVに感染した患者の処置のための医薬の製造におけるVX−222およびVX−950の使用を提供し、ここで、VX−222は、約20mgないし約400mgの量であり、VX−950は、約100mgないし約1500mgの量である。
【0020】
本発明はまた、HCVに感染した患者の処置のための医薬の製造におけるVX−222の使用を提供し、ここで、VX−222は、約20mgないし約2000mgの量で、または約50mgないし約2000mgの量で1日1回投与される。
【図面の簡単な説明】
【0021】
【図1】図1は、本発明の任意の態様の実験デザインを示すチャートである。
【図2】図2は、本発明の任意の態様の実験デザインを示すチャートである。
【図3】図3は、本発明の一態様の実験結果を示すチャートである。
【図4】図4は、本発明の一態様の実験結果を示すチャートである。
【図5】図5は、本発明の一態様の実験結果を示すチャートである。
【図6】図6は、本発明の一態様の実験結果を示すチャートである。
【図7】図7は、本発明の一態様の実験結果を示すチャートである。
【図8】図8は、本発明の一態様の実験結果を示すチャートである。
【図9】図9は、化合物1のプロドラッグおよび該プロドラッグの投与後にそれが変換される活性代謝物の血漿レベルを示すグラフである。
【発明を実施するための形態】
【0022】
本発明の詳細な説明
本発明は、VX−222を投与するための特定の用量および投与量レジメンに関する。本発明の目的に関して、VX−222は、化合物1ならびにその薬学的に許容される塩、溶媒和物およびプロドラッグ、また化合物1のプロドラッグの薬学的に許容される溶媒和物を含み、ここで、化合物1は、次の構造式:
【化1】

で示される。VX−222は、NS5Bポリメラーゼ阻害剤であり、WO2008/058393に記載されている。
【0023】
本発明はまた、VX−950を投与するための特定の用量および投与量レジメンに関する。VX−950は、7nmの定常状態結合定数(steady state binding constant)(ki*)を有する競合的な可逆性ペプチド模倣性NS3/4Aプロテアーゼ阻害剤である。例えば、WO02/018369を参照のこと。この目的に関して、VX−950は、化合物2ならびに化合物2の薬学的に許容される塩およびプロドラッグを含み、ここで、化合物2は、次の構造式:
【化2】
により示される。VX−950は、PCT公開番号WO02/018369、WO2006/050250およびWO2008144072に記載されている。VX−950の他の記載は、PCT公開番号WO07/098270およびWO08/106151に見出され得る。
【0024】
本明細書で用いる用語“薬学的に許容される塩”は、HCV感染の処置に対して安全かつ有効である塩を意味する。薬学的に許容される酸付加塩には、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、硝酸塩、硫酸塩、重硫酸塩、リン酸塩、過リン酸塩、イソニコチン酸塩、酢酸塩、および乳酸塩が含まれるが、これらに限定されない。種々のアミノ酸との薬学的に許容される塩もまた使用され得て、これらのアミノ酸塩の使用もまた、本発明の範囲内である。好適な塩基性塩には、アルミニウム塩、カルシウム塩、リチウム塩、マグネシウム塩、カリウム塩、ナトリウム塩、亜鉛塩およびジエタノールアミン塩が含まれるが、これらに限定されない。薬学的に許容される塩についての概要は、引用によりその内容全体を本明細書に包含させるberge et al., j. Pharm. Sci., 66, 1−19 (1977)を参照のこと。
【0025】
化合物1の薬学的に許容される塩の特定の例は、WO2008/058393に記載されており、例えば、アミノ酸(例えば、L−アルギニン、l−リシン)から誘導される塩類、アルカリ金属(例えば、ナトリウム、リチウム、カリウム)塩、アルカリ土類金属(例えば、カルシウム、マグネシウム)、アンモニウム、NR4+(式中、rは、C1−4アルキル)塩類、コリン塩およびトロメタミン塩類を含む適当な塩基から誘導される塩類である。一態様において、薬学的に許容される塩類は、ナトリウム塩である。別の態様において、薬学的に許容される塩類は、リチウム塩である。さらに別の態様において、薬学的に許容される塩は、カリウム塩である。さらに別の態様において、薬学的に許容される塩は、トロメタミン塩である。さらに別の態様において、薬学的に許容される塩は、l−アルギニン塩である。
【0026】
本明細書で用いる用語、化合物1の“薬学的に許容されるプロドラッグ”は、生理的条件下で、または化合物1の加溶媒分解により、またはHCV感染の処置においてその薬理学的効果を発揮する前に、化合物1の薬学的に許容される塩へ変換され得る化合物を意味する。本明細書で用いる用語、化合物2の“薬学的に許容されるプロドラッグ”は、生理的条件下で、または化合物2の加溶媒分解により、またはHCV感染の処置においてその薬理学的効果を発揮する前に、化合物2の薬学的に許容される塩へ変換され得る化合物を意味する。典型的に、プロドラッグは、改善された化学的安定性、改善された患者の受容性、改善されたバイオアベイラビリティ、延長された作用時間、改善された組織感受性、改善された剤形(例えば、増大した水溶性)、または低減された副作用(例えば、毒性)等の目的をもって製剤される。
【0027】
薬学的に許容されるプロドラッグは、当技術分野で公知の方法、例えば、Burger’s Medicinal Chemistry and Drug Chemistry, Vol. 1, 172−178 and 949−982, John Wiley & Sons (1995)に記載のような方法を用いて容易に製造され得る。Bertolini et al., J. Med. Chem., 40, 2011−2016 (1997); Shan et al., J. Pharm. Sci., 86(7), 765−767 (1997); Bagshawe, Drug Dev. Res., 34, 220−230 (1995); Bodor, Advances in Drug Res., 13, 224−331 (1984); Bundgaard, Design of Prodrugs, Elsevier Press (1985); および、Larsen, Design and Application of Prodrugs, Drug Design and Development (Krogsgaard−larsen et al., eds.), Harwood Academic Publishers (1991)も参照のこと。
【0028】
化合物1のプロドラッグの特定の例には、2010年6月28日出願のU.S.S.N. 61/359,164に記載のものが含まれる:
【化3】
【0029】
さらに、本明細書に記載の化合物が、異なる溶媒和物形態、例えば水和物形態で存在していてよく、さらに生物学的有効性を保持することは、当業者に理解され得る。かかる溶媒和物はまた、溶媒分子が、結晶化過程において化合物分子の結晶格子構造に挿入されるとき、形成され得る。本明細書で用いる用語、化合物1の“薬学的に許容される溶媒和物”は、溶媒分子(複数可)を含み、化合物1の生物学的有効性を保持する化合物1の薬学的に許容される溶媒和物形態を意味する。本明細書で用いる用語、化合物1のプロドラッグの“薬学的に許容される溶媒和物”は、溶媒分子(複数可)を含み、化合物1の生物学的有効性を保持する化合物1のプロドラッグの薬学的に許容される溶媒和物形態を意味する。
【0030】
1種以上の同位体に富む原子の存在でのみ化合物1および化合物2と異なる化合物は、本発明に包含される。例えば、水素を重水素またはトリチウムで置換したか、または炭素を13C−もしくは14C−に富む炭素により置換した以外は本発明化合物構造を有する化合物は、本発明の範囲内である。同位体に富む化合物2の任意の例は、WO2007/109080およびMaltais et al., J. of Medicinal Chemistry, “In Vitro and In Vivo isotope effects with Hepatitis c Protease Inhibitors: Enhanced Plasma Exposure of Deuterated Telaprevir versus Telaprevir in Rats” 2009;52(24):7993−8001に見出され得る。
【0031】
化合物1および化合物2は、それぞれ独立して、1個以上の不斉炭素原子を含んでいてよく、故に、ラセミ体およびラセミ混合物、単一のエナンチオマー、ジアステレオマー混合物および個々のジアステレオマーとして生じ得る。これらの化合物の全てのかかる同位体形態は、明示的に本発明に包含される。各立体中心炭素は、RまたはS配置であり得る。化合物2のn−プロピル側鎖でのD−およびL−異性体は、明示的に、本発明の範囲内に包含される。
【0032】
本明細書に記載の化合物が異なる多形形態で存在し得ることは、当業者に当然理解され得る。当技術分野で公知の通り、多形性は、2種以上の異なる結晶または“多形”体として結晶化する化合物の能力である。多形体は、固体状態の化合物分子が少なくとも2個の異なる配位または多形形態である化合物の固体結晶形である。何れかの化合物の多形形態は、同じ化学式または組成で示され、2個の異なる化合物の結晶構造として異なる化学物質である
【0033】
一面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1、またはその薬学的に許容される塩、溶媒和物もしくはプロドラッグ、または化合物1のプロドラッグの溶媒和物であり;VX−950は、化合物2、またはその薬学的に許容される塩もしくはプロドラッグである。
【0034】
別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1、またはその薬学的に許容される塩、溶媒和物もしくはプロドラッグ、または化合物1のプロドラッグの溶媒和物であり;VX−950は、化合物2またはその薬学的に許容される塩である。
【0035】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1、またはその薬学的に許容される塩、溶媒和物もしくはプロドラッグ、または化合物1のプロドラッグの溶媒和物であり;VX−950は化合物2である。
【0036】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1、またはその薬学的に許容される塩もしくは溶媒和物、もしくはプロドラッグであり;VX−950は、化合物2、またはその薬学的に許容される塩もしくはプロドラッグである。
【0037】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1、またはその薬学的に許容される塩もしくは溶媒和物、もしくはプロドラッグであり;VX−950は、化合物2またはその薬学的に許容される塩である。
【0038】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1、またはその薬学的に許容される塩もしくは溶媒和物、もしくはプロドラッグであり;VX−950は、化合物2である。
【0039】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1、またはその薬学的に許容される塩もしくは溶媒和物であり;VX−950は、化合物2、またはその薬学的に許容される塩もしくはプロドラッグである。
【0040】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1、またはその薬学的に許容される塩もしくは溶媒和物であり;VX−950は、化合物2またはその薬学的に許容される塩である。
【0041】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1、またはその薬学的に許容される塩もしくは溶媒和物であり;VX−950は化合物2である。
【0042】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1またはその薬学的に許容される塩であり;VX−950は、化合物2、またはその薬学的に許容される塩もしくはプロドラッグである。
【0043】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1またはその薬学的に許容される塩であり;VX−950は、化合物2またはその薬学的に許容される塩である。
【0044】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1またはその薬学的に許容される塩であり;VX−950は、化合物2である。
【0045】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は化合物1であり;VX−950は、化合物2、またはその薬学的に許容される塩もしくはプロドラッグである。
【0046】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は化合物1であり;VX−950は、化合物2またはその薬学的に許容される塩である。
【0047】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は化合物1であり;VX−950は化合物2である。
【0048】
一態様において、本発明は、HCVに感染した患者におけるVX−222の薬物動態を改善する方法に関する。該方法は、患者にVX−222およびVX−950を共投与することを含む。VX−222の改善された薬物動態には、患者の血漿、血液または肝臓におけるVX−222への暴露の増加が含まれる。別の態様において、本発明は、HCVに感染した患者の血漿におけるVX−222への暴露を増大する方法に関する。例えば、血漿におけるVX−222への暴露を、VX−222の、トラフ濃度(Ctrough)、平均血漿濃度(Cavg)、最大血漿濃度(Cmax)、またはAUC(曲線下面積)値により測定し得る。本明細書で用いる用語“トラフ濃度”(Ctrough)は、次の投与の直前の血漿中の薬物濃度、または2投与間の最小薬物濃度を意味する。本明細書で用いる用語“AUC”は、血漿(血清または血液)濃度−時間曲線の下部の面積を意味する。特定の態様において、VX−222への暴露は、AUC0−12(0ないし12時間)の値で示される。別の特定の態様において、VX−222への暴露は、AUC0−24(0ないし24時間)の値で示される。
【0049】
VX−950の共投与によるVX−222への暴露の増大は、VX−950なしで投与されるVX−222への暴露をVX−950と共投与されるVX−222への暴露と比較することにより決定され得る。1つの特定の態様において、VX−222への暴露は、VX−950なしで投与されるVX−222への暴露と比較して、約2ないし6倍増加する。1つの特定の態様において、VX−222への暴露は、VX−950なしで投与されるVX−222への暴露と比較して、約2ないし5倍増加する。1つの特定の態様において、VX−222への暴露は、VX−950なしで投与されるVX−222への暴露と比較して、約2または3倍増加する。いくつかの特定の態様において、該増大は、血漿におけるVX−222への暴露である。
【0050】
本発明において、各投与でのVX−950の量は、約100mgないし約1,500mg、約300mgないし約1,500mg、約500mgないし約1,500mg、約300mgないし約1,250mg、約450mg、約750mg、または約1,250mgであり得る。いくつかの特定の態様において、VX−950は、各投与にて約750mgである。いくつかの特定の態様において、VX−950は、各投与にて約1,125mgである。VX−950の量の好適な例は、WO2008/144072およびWO06/050250に記載され、その内容全体は、参照により本明細書に包含される。
【0051】
本発明において、各投与でのVX−222の量は、約20mgないし約2,000mg、約50mgないし約2,000mg、約100mgないし約1,500mg、約100mgないし約1,250mg、約100mgないし約1,000mg、約100mg、約250mg、約400mg、または約750mgであり得る。いくつかの特定の態様において、VX−222は、各投与にて約100mgである。いくつかの特定の態様において、VX−222は、各投与にて約250mgである。いくつかの特定の態様において、VX−222は、各投与にて約400mgである。いくつかの特定の態様において、VX−222は、各投与にて約750mgである。いくつかの特定の態様において、VX−222は、各投与にて約1,000mgである。いくつかの特定の態様において、VX−222は、各投与にて約1,500mgである。いくつかの特定の態様において、VX−222は、各投与にて約500mgである。いくつかの特定の態様において、VX−222は、各投与にて約1,125mgである。いくつかの特定の態様において、VX−222は、各投与にて約1,250mgである。
【0052】
さらに別の態様において、本発明は、HCVに感染した患者の処置方法であって、該患者にVX−222およびVX−950を投与することを含む方法に関する。特定の態様において、VX−222は、各投与にて約20mgないし約2,000mg、例えば約50mgないし約1,500mgであり、VX−950は、各投与にて約100mgないし約1,500mg、例えば約300mgないし約1,500mgである。別の特定の態様において、VX−222は、各投与にて約20mgないし約400mgの用量、例えば約50mgないし約400mgであり、VX−950は、各投与にて約100mgないし約1500mgである。さらに別の特定の態様において、VX−222は、各投与にて20mgと等しいか、または20mg以上ないし400mg未満の量である。さらに別の特定の態様において、VX−222は、各投与にて約20mgないし約300mgの量である。さらに別の特定の態様において、VX−222は、各投与にて約50mgないし約300mgの量である。さらに別の特定の態様において、VX−222は、各投与にて約100mgの量である。さらに別の特定の態様において、VX−222は、各投与にて約400mgの量である。さらに別の特定の態様において、VX−950は、各投与にて約300mgないし約1,500mgの量である。さらに別の特定の態様において、VX−950は、各投与にて約500mgないし約1,500mgの量である。さらに別の特定の態様において、VX−950は、各投与にて約750mgの量である。さらに別の特定の態様において、VX−950は、各投与にて約1,125mgの量である。
【0053】
さらに別の態様において、本発明は、HCVに感染した患者の処置方法であって、VX−222を投与すること(ここで、VX−222は、各投与にて、約20mgないし約2,000mgの量で投与される)を含む方法に関する。具体的には、VX−222の量は、各投与にて、約100mgないし約1,500mg、約100mgないし約1,250mg、約100mgないし約1,000mg、約100mg、約250mg、約400mg、約500mg、約750mg、約1000mg、約1125mg、または約1250mgであり得る。いくつかの態様において、VX−222は、1日1回投与される。特定の態様において、該方法は、1日1回、約50mgないし約2,000mgの量でVX−222を投与することを含む。いくつかの特定の態様において、VX−222の投与量は、約100mgを1日1回である。いくつかの特定の態様において、VX−222の投与量は、約250mgを1日1回である。いくつかの特定の態様において、VX−222の投与量は、約400mgを1日1回である。いくつかの特定の態様において、VX−222の投与量は、約500mgを1日1回である。いくつかの特定の態様において、VX−222の投与量は、約750mgを1日1回である。いくつかの特定の態様において、VX−222の投与量は、約1,000mgを1日1回である。いくつかの特定の態様において、VX−222の投与量は、約1,250mgを1日1回である。いくつかの特定の態様において、VX−222の投与量は、約1,125mgを1日1回である。いくつかの特定の態様において、VX−222の投与量は、約1,500mgを1日1回である。
【0054】
特定の態様において、本発明は、約20mgないし約2,000mg(または、約50mgないし約2,000mg、または上記の何れかの特定の投与量レジメン)のVX−222を1日1回投与し、さらにVX−222以外の1種以上のさらなるHCV薬を投与することによる、HCVに感染した患者の処置方法に関する。さらなるHCV薬の好適な例は、以下に詳細に記載され、VX−950、インターフェロンおよびリバビリンが含まれる。さらなる特定の態様において、VX−950が共投与される。VX−950の用量の典型例は、上記の通りである。別のさらなる特定の態様において、リバビリンの含有不含有に関わらず、インターフェロン(例えば、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bのようなペグ化インターフェロン)を共投与する。別のさらなる特定の態様において、VX−950;インターフェロン(例えば、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bのようなペグ化インターフェロン);および、リバビリンを共投与する。
【0055】
さらに別の態様において、本発明は、約20mgないし約2,000mgのVX−222;および約100mgないし約1,500mgのVX−950を含む薬学的に許容される組成物を提供する。所望により、薬学的に許容される担体もまた包含されてもよい。特定の態様において、本発明は、約20mgないし約1,500mg、または約50mgないし約1,500mgのVX−222を含む薬学的に許容される組成物を提供する。別の特定の態様において、これらの医薬組成物におけるVX−950の量は、約300mgないし約1500mg、約300mgないし約1250mg、約300mgないし約1,000mg、約300mgないし約750mg、または約375mgである。別の特定の態様において、これらの医薬組成物におけるVX−222の量は、50mgと等しいか、または50mg以上ないし400mg未満、約50mgないし約300mg、約50mg、約100mg、または約200mgである。これらの医薬組成物はそれぞれ、例えば、1日当たり、1回、2回または3回投与され得る。これらの組成物はそれぞれ、1種以上の投与形態(例えば、アンプル、カプセル、クリーム、乳剤、液剤、粒塊剤、ドロップ剤、注射剤、懸濁液、錠剤、粉剤)であり得る。これらの医薬組成物はそれぞれ、当業者により適当であると考えられる1種以上の経路(例えば、経口的、注入、注射、局所的または非経腸的)により投与され得て、投与形態によって変わる。
【0056】
一般的に、上記の本発明の方法において、VX−222およびVX−950は、それぞれ独立して、1日1回(QD)、1日2回(例えば、BID;q12h)、1日3回(例えば、TID;q8h)、または1日4回投与され得る。VX−222およびVX−950は、それぞれ独立して、食事の有無とは関係なく、投与され得る。
【0057】
いくつかの態様において、本発明の方法は、患者にVX−950を(a)各約450mg、8時間毎に1日3回;(b)各約750mg、8時間毎に1日3回;(c)各約1,125mg、12時間毎に1日2回;または、(d)各約1250mg、12時間毎に1日2回投与することを含む。
【0058】
いくつかの態様において、本発明の方法は、患者にVX−950を含む組成物の経口用量を投与することを含み、ここで、該用量は、患者に、投与後、VX−950の少なくとも約750ng/mlの平均血漿濃度(cavg)を提供する。いくつかの態様において、VX−950のcavgは、約1000ng/mlまたは約1250ng/mlである。いくつかの態様において、該用量は、本質的に約750mgのVX−950を含む。いくつかの態様において、cavgは、VX−950の投与後、3時間以内(例えば、2時間または1時間)で得られるか、または達成される。いくつかの態様において、VX−950のcavgは、約24時間以上(例えば、5週間または12週間)維持される。
【0059】
いくつかの態様において、本発明の方法は、患者にVX−950を投与することを含み、ここで、VX−950のトラフ血漿濃度は、24時間以上の間、最低でも約750、800、900または1000ng/mlで維持される。理論はともかく、約1500ng/ml以上のトラフ濃度は、本発明には必要ないと考えられる。従って、約750、800、900、1000ng/mlないし約1500ng/ml(特に、1000ないし約1500)のトラフ濃度は、本発明の範囲内である。
【0060】
理想的には、本発明の方法が、HCVに感染した患者の処置を伴うとき、該方法は、比較的迅速に、VX−950の治療的に有効な血漿濃度を達成し、次いで有効な治療応答が達成されるようなトラフ濃度を維持することを伴う。有効な治療応答は、好ましくは、a)持続的ウイルス応答を達成すること;および、b)少なくとも12週間(12週間以上)、血漿中において検出不可能なHCV RNAを達成すること、の一方または両方である。本明細書で用いる、HCV RNAについて“検出不可能”とは、HCV RNAが、現在市販されているアッセイにより決定される通り、好ましくはRoche COBAS TaqMan(商標) HCV/HPSアッセイにより決定される通り、10IU/ml未満で存在することを意味する。
【0061】
血漿濃度の比較的迅速な低下は、患者にある投与量を投与することにより得られ得る。一態様において、該投与量は、VX−950を約1250mgである。
【0062】
いくつかの態様において、本発明の方法は、VX−950を共投与することを含み、VX−950は、約750mgのVX−950(例えば、約375mgのVX−950の2つの錠剤)を含む投与形態であって、該投与形態を1日3回、例えば8時間毎(すなわち、q8h)に投与する。いくつかの態様において、本発明の方法は、VX−950を投与することを含み、VX−950は、約1125mgのVX−950(例えば、約375mgのVX−950の3つの錠剤)を含む投与形態であって、該投与形態を1日2回、例えば12時間毎(すなわち、q12h)に投与する。
【0063】
本発明において、VX−222および何れかのさらなるHCV薬(例えば、リバビリンの含有不含有に関わらず、VX−950;インターフェロン(例えば、ペグ化インターフェロン、例えばペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2b);または、VX−950、インターフェロン(例えば、ペグ化インターフェロン、例えばペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2b)、およびリバビリン)を、処置期間全体を通して独立して投与することができる。これらの態様において、VX−222処置期間およびさらなるHCV薬(複数可)の処置期間は同じである。
【0064】
あるいは、いくつかの態様において、VX−222およびさらなるHCV薬(例えば、リバビリンの含有不含有に関わらず、VX−950;インターフェロン(例えば、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bのようなペグ化インターフェロン);または、VX−950、インターフェロン(例えば、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bのようなペグ化インターフェロン)およびリバビリン)を、第一段階および第二段階の2つの段階に独立して投与することができる。VX−222および何れかのさらなるHCV薬はそれぞれ、第一段階もしくは第二段階のいずれか、または両方の段階で投与され得る。いくつかの態様において、VX−222を、第一段階でのみ投与し、インターフェロンを、第一段階および第二段階の両方で投与する。あるいは、いくつかの他の態様において、VX−222を第二段階でのみ投与し、インターフェロンを第一段階および第二段階の両方で投与する。いくつかの態様において、VX−222およびVX−950を共投与し、VX−222およびVX−950を第一段階のみで投与するか、または第二段階のみで投与する。いくつかの態様において、VX−222、VX−950、インターフェロン(例えば、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bのようなペグ化インターフェロン)、およびリバビリンを共投与し、VX−222およびVX−950を第一段階のみで投与し、インターフェロンおよびリバビリンを第一段階および第二段階の両方で投与する。
【0065】
第一段階および第二段階の期間の、好適な特定の例は、WO2008/144072に見出され得る。例えば、第一段階は、少なくとも約4週間、または約4週ないし約24週間(例えば、約4週間、約6週間、約8週間、約10週間、約12週間、約16週間、約20週間、約24週間など)の期間であってよく、第二段階は、少なくとも約12週間、例えば約12週間ないし約36週間の期間であり得る。ある態様において、第二段階は、約12週間である。他の態様において、第二段階は約24週間である。さらに他の態様において、第二段階は約36週間である。ある態様において、第一段階および第二段階の和は、約24週間ないし約48週間(例えば、約24週間、約36週間、または約48週間)である。いくつかの態様において、第一段階および第二段階は、同一期間であり得る。
【0066】
いくつかの態様において、本発明の方法は、VX−222およびインターフェロンを独立して、約4週間ないし約12週間(例えば、約4週間、約6週間、約8週間、または約12週間)の期間、約20週間ないし約28週間(例えば、約20週間、約24週間、または約28週間)の期間、または約8週間ないし約24週間(例えば、約8週間、約12週間、約16週間、または約24週間)の期間、共投与することを含む。これらの各態様の一面において、VX−222およびインターフェロンを独立して(第一段階)投与し、次いで約4週間ないし約36週間(例えば、約8週間ないし約36週間、約8週間ないし約24週間、約4週間ないし約24週間)の期間、インターフェロン(VX−222なし)を投与する(第二段階)。特定の例示的レジメンには、約24週間の全処置レジメン期間中、約16週間のインターフェロン(VX−222なし)の投与後に約8週間の間、VX−222およびインターフェロンを独立して投与すること;約24週間の全処置レジメン期間中、約12週間のインターフェロン(VX−222なし)の投与後に、約12週間の間、VX−222およびインターフェロンを独立して投与すること、が含まれる。かかるレジメンにおいて、所望により、全て(第一段階および第二段階の両方)、または各レジメンの一部(例えば、第一段階または第二段階のみ)の期間、リバビリンを投与してもよい。
【0067】
いくつかの態様において、本発明の方法は、VX−222およびVX−950を独立して、約4週間ないし約12週間(例えば、約4週間、約6週間、約8週間または約12週間)、約20週間ないし約28週間(例えば、約20週間、約24週間、または約28週間)、または約8週間ないし約24週間(例えば、約8週間、約12週間、約16週間または約24週間)の期間、共投与することを含む。これらの各態様の一面において、VX−222およびVX−950を独立して(第一段階)投与し、次いで約4週間ないし約36週間(例えば、約8週間ないし約36週間、約8週間ないし約24週間、約4週間ないし約24週間)の期間、インターフェロンおよびリバビリン(VX−222およびVX−950なし)を投与する(第二段階)。特定の例示的レジメンには、VX−222およびVX−950を独立して約8週間投与し、次いでインターフェロンおよびリバビリン(VX−222およびVX−950なし)を全体で約24週間の処置レジメンについて約16週間投与し;VX−222およびVX−950を独立して約12週間投与し、次いでインターフェロンおよびリバビリン(VX−222およびVX−950なし)を全体で約24週間の処置レジメンについて約12週間投与することが含まれる。かかるレジメンにおいて、VX−222およびVX−950の投与フェーズにインターフェロンおよびリバビリンの投与を供することも可能である。
【0068】
ある態様において、VX−222および所望によりVX−950またはインターフェロンを、約12週間未満の期間、独立して投与する。
ある態様において、VX−222および所望によりVX−950またはインターフェロンを、約8−12週間の期間、独立して投与する。
ある態様において、VX−222および所望によりVX−950またはインターフェロンを、約10週間の期間、独立して投与する。
ある態様において、VX−222および所望によりVX−950またはインターフェロンを、約10週間未満の期間、独立して投与する。
【0069】
ある態様において、VX−222および所望によりVX−950またはインターフェロンを、約2週間の期間、独立して投与する。
ある態様において、VX−222および所望によりVX−950またはインターフェロンを、約8週間未満(または約8週間)、約6週間未満(または約6週間)、または約4週間未満(または約4週間)の期間、独立して投与する。
ある態様において、VX−222およびVX−950を、約12週間(第一段階)の期間共投与し、次いで、所望によりインターフェロンおよびリバビリンを独立して約12週間(第二段階)投与してよい。
【0070】
ある態様において、VX−222、VX−950およびインターフェロンを約12週間共投与し、次いで、所望によりインターフェロンおよびリバビリンを独立して約12週間(第二段階)投与してよい。
ある態様において、VX−222、VX−950、インターフェロンおよびリバビリンを約12週間共投与し、次いで、所望によりインターフェロンおよびリバビリンを独立して約12週間(第二段階)投与してよい。
ある態様において、VX−222、インターフェロンおよびリバビリンを約12週間(第一段階)共投与し、次いで、所望によりインターフェロンおよびリバビリンを独立して約12週間(第二段階)投与してよい。
【0071】
ある態様において、VX−222およびVX−950を約12週間(第一段階)共投与し、次いで、所望によりインターフェロンおよびリバビリンを約12週間(第二段階)投与してよい。
ある態様において、VX−222、VX−950、インターフェロンおよびリバビリンを約12週間(第一段階)共投与し、次いで、所望によりインターフェロンおよびリバビリンを約12週間(第二段階)投与してよい。
ある態様において、VX−222、VX−950、インターフェロンおよびリバビリンを約12週間(第一段階)共投与し、次いで、所望によりインターフェロンおよびリバビリンを約36週間(第二段階)投与してよい。
【0072】
いくつかの態様において、上記の第一段階の何れかを、12週間未満の期間行い、第二段階を約12週間行うことができる。あるいは、第一段階を約12週間行い、第二段階を約24週間行うことができる。さらに他の面において、第一段階を約8週間行い、第二段階を約36週間行うことができる。さらに他の面において、第一段階を約4週間行い、第二段階を約36週間行うことができる。
【0073】
いくつかの態様において、上記の第一段階の何れかを、約8週間行い、第二段階を約16週間行うことができる。あるいは、第一段階を約8週間行い、第二段階を約40週間行うことができる。さらに他の面において、第一段階を約8週間行い、第二段階を約40週間行うことができる。
【0074】
いくつかの態様において、上記の第一段階および第二段階の何れかを、互いに入れ替えることができ、例えば、インターフェロン(所望によりリバビリンと共に)を第一段階に投与し、VX−222(所望によりVX−950と共に、またはVX−950、インターフェロンおよびリバビリンと共に)を第二段階に投与する。
【0075】
いくつかの態様において、上記の本発明の方法は、全処置の約12週間または約24週間の短期間の処置レジメンを評価する際に、治療応答ガイド基準(response−guided criteria)を用いる。これらの態様において、処置の第2週および第8週で検出不可能なHCV RNA(10IC/ml未満)を達成する患者は、全約24週間の処置中、12週に全ての処置を停止するか、またはpeg−IFN(ペグ化インターフェロン)およびRBV(リバビリン)治療をさらに12週間受けるように無作為化され;処置の第2週および第8週で検出不可能なHCV RNAを達成しない患者は、全24週間の処置中、peg−IFNおよびRBV(VX−222なし、またはVX−222およびVX−950なし)治療のさらに12週間を受ける。
【0076】
いくつかの態様において、上記の本発明の方法は、全処置が約12週間、約24週間または約36週間の短期処置レジメンの評価において、応答反応に基づく基準(response−guided criteria)を用いる。これらの態様において、処置の第2週および第8週で検出不可能なHCV RNA(10IC/ml未満)を達成する患者は、第12週で彼らに割り当てられた処置を終了し得る。第2週および第8週で検出不可能なHCV RNAを達成しない患者は、全24週間の処置についてさらに12週間のpeg−IFNおよびRBV(VX−222なし、またはVX−222およびVX−950なし)治療を受けるか、または全36週間の処置についてさらに24週間のpeg−IFNおよびRBV(VX−222なし、またはVX−222およびVX−950なし)治療を受ける。いくつかの特定の態様において、該方法は、VX−950およびVX−222(peg−IFNおよびRBVなし)を用い、例えば、1125mgのVX−950を1日2回および100mgまたは400mgのVX−222を1日2回等であり、処置の第2週および第8週で検出不可能なHCV RNA(10IC/ml未満)を達成する患者は、第12週で彼らの割り当てられた処置を終了してよく;第2週および第8週で検出不可能なHCV RNAを達成しない患者は、全36週間の処置についてさらに24週間のpeg−IFNおよびRBV(VX−222なし、またはVX−222およびVX−950なし)治療を受ける。いくつかの特定の態様において、該方法は、VX−950、VX−222、peg−IFNおよびRBV(例えば、1125mgのVX−950を1日2回、および100mgまたは400mgのVX−222を1日2回、180mcgのpeg−IFNを1週間に1回、ならびに800mg−1200mgのRBVを1日2回(例えば、体重75kg未満の患者に1000mg、または体重75kg以上の患者に1200mg))を受け、処置の第2週および第8週で検出不可能なHCV RNA(10IC/ml未満)を達成する患者は、第12週で彼らの割り当てられた処置を終了してよく;第2週および第8週で検出不可能なHCV RNAを達成しない患者は、全24週間の処置についてさらに12週間のpeg−IFNおよびRBV(VX−222なし、またはVX−222およびVX−950なし)治療を受ける。
【0077】
当業者には、本発明の方法が患者を予防的に処置するために用いられ、患者がC型肝炎ウイルスに感染したとき、該処置は、感染を処置し得ることが容易に理解され得る。故に、本発明の1つの態様は、患者におけるC型肝炎感染の処置法または予防法を提供する。
【0078】
C型肝炎に感染した患者の処置に加えて、本発明の方法は、C型肝炎に感染する可能性のある患者を予防するために用いられ得る。従って、本発明の1つの態様は、provides 患者におけるC型肝炎ウイルス感染の予防法であって、患者にVX−222と、要すれば何らかのさらなるHCV薬、例えば、上記の通り、VX−950;インターフェロン;インターフェロンおよびリバビリン;VX−950、インターフェロンおよびリバビリンを投与することを含む方法を提供する。
【0079】
本発明の方法はまた、免疫調節剤;抗ウイルス剤;HCVプロテアーゼの阻害剤(VX−222およびVX−950以外);HCV生活環の別の標的の阻害剤(NS3/4Aプロテアーゼ以外);内部リボソーム侵入の阻害剤、広域的ウイルス阻害剤;または、シトクロムP−450阻害剤;または、それらの組み合わせ、から選択されるさらなる薬剤を含む別の成分の投与を含み得る。さらなる薬剤はまた、ウイルスの細胞侵入の阻害剤からも選択される。
【0080】
従って、いくつかの態様において、さらなる薬剤は、別の抗ウイルス剤、好ましくは抗HCV剤(VX−222およびVX−950以外)である。そのような抗ウイルス剤には、α−、β−、およびγ−インターフェロンまたはチモシン、ペグ化誘導体化インターフェロン−α化合物、およびチモシンのような免疫調節剤;リバビリン、アマンタジン、およびテルビブジンのような他の抗ウイルス剤;C型肝炎プロテアーゼの他の阻害剤(NS2−NS3阻害剤およびNS3−NS4A阻害剤);ヘリカーゼ、ポリメラーゼ、およびメタロプロテアーゼ阻害剤を含む、HCV生活環における他の標的の阻害剤;内部リポソーム侵入の阻害剤;IMPDH阻害剤のような広域的ウイルス阻害剤(例えば、VX−497、VX−148、およびVX−944を含むが、それらに限定されない、米国特許第5,807,876号、同第6,498,178号、同第6,344,465号および同第6,054,472号ならびにPCT公報WO97/40028、WO98/40381およびWO00/56331に記載の化合物;ならびにミコフェノール酸およびその誘導体);または、上記のいずれかの組み合わせが含まれるが、それらに限定されない。
【0081】
本発明の化合物と併用され得る他の薬剤(例えば、非免疫調節または免疫調節化合物)には、引用により本明細書中に包含されるWO02/18369(例えば、273頁、9−22行目、および274頁、4行目から276頁、11行目、本開示は、引用により明確に本明細書中に包含される)に記載されるものが含まれるが、それらに限定されない。
【0082】
種々の公開された米国特許出願に記載されるさらに他の薬剤が包含される。これらの刊行物は、本発明の方法、特に肝炎の処置方法においてVX−950と併用され得る化合物および方法のさらなる教示を提供する。そのような方法および組成物は全て、本発明の方法および組成物と併用され得ると考えられる。簡単には、それらの刊行物の開示内容は、公開番号を引用することにより言及されるが、化合物の開示が、特に、引用により本明細書中に明確に包含されることが、特記されるべきである。そのような刊行物の例には、米国特許出願公開番号:US20040058982、US20050192212、US20050080005、US20050062522、US20050020503、US20040229818、US20040229817、US20040224900、US20040186125、US20040171626、US20040110747、US20040072788、US20040067901、US20030191067、US20030187018、US20030186895、US20030181363、US20020147160、US20040082574、US20050192212、US20050187192、US20050187165、US20050049220、およびUS20050222236が含まれる。
【0083】
さらなる他の薬剤には、Human Genome Sciencesから市販されるアルブフェロン(商標)(アルブミン−インターフェロンα);PEG−イントロン(登録商標)(ペグ化インターフェロンα−2b、Schering Corporation, Kenilworth, NJから市販される);イントロン−A(登録商標)(VIRAFERON(登録商標)、インターフェロンα−2b、Schering Corporation, Kenilworth, NJから市販される);リバビリン(1−β−D−リボフラノシル−1H−1,2,4−トリアゾール−3−カルボキサミド、ICN Pharmaceuticals, Inc., Costa Mesa, CAから市販される;the Merck Index, entry 8365, Twelfth Editionに記載される);REBETROL(登録商標)(Schering Corporation, Kenilworth, NJ);コーペガス(登録商標)(Hoffmann−La Roche, Nutley, NJ);ペガシス(登録商標)(ペグ化インターフェロンα−2a、Hoffmann−La Roche, Nutley, NJから市販される);ロフェロン(登録商標)(組換えインターフェロンα−2a、Hoffmann−La Roche, Nutley, NJから市販される);BEREFOR(登録商標)(インターフェロンα2、Boehringer Ingelheim Pharmaceutical, Inc., Ridgefield, CTから市販される);スミフェロン(登録商標)(スミフェロンのような、精製物と天然のαインターフェロンの混合物、住友製薬、日本から市販される);ウェルフェロン(登録商標)(インターフェロン α n1、Glaxo Wellcome Ltd., Great Britainから市販される);アルフェロン(登録商標)(Interferon Sciencesにより製造される天然αインターフェロンの混合物、およびPurdue Frederick Co., CTから市販される);α−インターフェロン;天然αインターフェロン 2a;天然αインターフェロン 2b;ペグ化αインターフェロン 2aまたは2b;コンセンサスαインターフェロン(Amgen, Inc., Newbury Park, CA);レベトロン(登録商標)(Schering Plough、インターフェロン−α 2B+リバビリン);ペグ化インターフェロンα(Reddy, K.R. et al. “Efficacy and Safety of Pegylated (40−kd) Interferon alpha−2a Compared with Interferon alpha−2a in Noncirrhotic Patients with Chronic Hepatitis C”(Hepatology, 33, pp. 433−438 (2001);コンセンサスインターフェロン(インファゲン(登録商標))(Kao, J.H., et al., “Efficacy of Consensus Interferon in the Treatment of Chronic Hepatitis” J. Gastroenterol. Hepatol. 15, pp. 1418−1423 (2000);リンパ芽球様または“天然”インターフェロン;インターフェロン tau(Clayette, P. et al., “IFN−tau, A New Interferon Type I with Antiretroviral activity” Pathol. Biol. (Paris) 47, pp. 553−559 (1999);インターロイキン−2(Davis, G.L. et al., “Future Options for the Management of Hepatitis C.” Seminars in Liver Disease, 19, pp. 103−112 (1999);インターロイキン−6(Davis et al. “Future Options for the Management of Hepatitis C.” Seminars in Liver Disease 19, pp. 103−112 (1999);インターロイキン−12(Davis, G.L. et al., “Future Options for the Management of Hepatitis C." Seminars in Liver Disease, 19, pp. 103−112 (1999);および、1型ヘルパーT細胞応答の発生を増強する化合物(Davis et al., “Future Options for the Management of Hepatitis C.” Seminars in Liver Disease, 19, pp. 103−112 (1999))が含まれるが、それらに限定されない。また、二本鎖RNA単独、またはトブラマイシンおよびイミキモド(3M Pharmaceuticals;Sauder, D.N. “Immunomodulatory and Pharmacologic Properties of Imiquimod” J. Am. Acad. Dermatol., 43 pp. S6−11 (2000))との組み合わせを含むが、これらに限定されない、細胞におけるインターフェロンの合成を刺激する化合物(Tazulakhova, E.B. et al., “Russian Experience in Screening, analysis, and Clinical Application of Novel Interferon Inducers” J. Interferon Cytokine Res., 21 pp. 65−73)も含まれる。また、WO02/18369、とりわけ272頁、15行目から273頁、8行目を参照のこと(この開示内容は、引用により本明細書中に明確に包含される)。
【0084】
シトクロムP450モノオキシゲナーゼ(“CYP”)阻害剤の好適な例には、リトナビル(WO94/14436)、ケトコナゾール、トロレアンドマイシン、4−メチルピラゾール、シクロスポリン、クロメチアゾール、シメチジン、イトラコナゾール、フルコナゾール、ミコナゾール、フルボキサミン、フルオキセチン、ネファゾドン、セルトラリン、インジナビル、ネルフィナビル、アンプレナビル、ホスアンプレナビル、サキナビル、ロピナビル、デラビルジン、エリスロマイシン、VX−944およびVX−497が含まれるが、これらに限定されない。好ましいCYP阻害剤には、リトナビル、ケトコナゾール、トロレアンドマイシン、4−メチルピラゾール、シクロスポリンおよびクロメチアゾールが含まれる。
【0085】
本発明の一態様は、CYP3A4の阻害剤を共投与する方法を提供する。
【0086】
本発明に用いられ得るインターフェロンの好適な例には、Human Genome Sciencesから市販されるアルブフェロン(商標)(アルブミン−インターフェロンα);PEG−イントロン(登録商標)(ペグ化インターフェロンα−2b、Schering Corporation, Kenilworth, NJから市販される);イントロン−A(登録商標)(VIRAFERON(登録商標)、インターフェロンα−2b、Schering Corporation, Kenilworth, NJから市販される);ペガシス(登録商標)(ペグ化インターフェロンα−2a、Hoffmann−La Roche, Nutley, NJから市販される);ロフェロン(登録商標)(組換えインターフェロンα−2a、Hoffmann−La Roche, Nutley, NJから市販される);BEREFOR(登録商標)(インターフェロンα2、Boehringer Ingelheim Pharmaceutical, Inc., Ridgefield, CTから市販される);スミフェロン(登録商標)(スミフェロンのような、精製物と天然のαインターフェロンの混合物、住友製薬、日本から市販される);ウェルフェロン(登録商標)(インターフェロン α n1、Glaxo Wellcome Ltd., Great Britainから市販される);アルフェロン(登録商標)(Interferon Sciencesにより製造される天然αインターフェロンの混合物、およびPurdue Frederick Co., CTから市販される);α−インターフェロン;天然αインターフェロン 2a;天然αインターフェロン 2b;ペグ化αインターフェロン 2aまたは2b;コンセンサスαインターフェロン(Amgen, Inc., Newbury Park, CA);レベトロン(登録商標)(Schering Plough、インターフェロン−α 2B+リバビリン);ペグ化インターフェロンα(Reddy, K.R. et al. “Efficacy and Safety of Pegylated (40−kd) Interferon alpha−2a Compared with Interferon alpha−2a in Noncirrhotic Patients with Chronic Hepatitis C”(Hepatology, 33, pp. 433−438 (2001);コンセンサスインターフェロン(インファゲン(登録商標))(Kao, J.H., et al., “Efficacy of Consensus Interferon in the Treatment of Chronic Hepatitis” J. Gastroenterol. Hepatol. 15, pp. 1418−1423 (2000);リンパ芽球様または“天然”インターフェロン;インターフェロン tau(Clayette, P. et al., “IFN−tau, A New Interferon Type I with Antiretroviral activity” Pathol. Biol. (Paris) 47, pp. 553−559 (1999);ならびに、インプランタブル・デュロス(登録商標)を介してオメガ・インターフェロンを提供するオメガ・デュロス(登録商標)(intarcia therapeutics, inc., mountain view, ca)が含まれる。
【0087】
いくつかの態様において、本発明の方法は、リバビリンの有無における、インターフェロンの共投与である。具体的には、インターフェロンは、ペグ化インターフェロン(peg−ifn)である。より具体的には、ペグ化インターフェロンは、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bのようなペグ化インターフェロンαである。
【0088】
一般的に、VX−222および何れかのさらなるHCV剤(VX−950、インターフェロンおよびリバビリンのような)は、独立して個別に、または共に投与され得る。一般に、VX−222およびVX−950は、独立して、経口、非経腸、舌下、吸入スプレーにより、局所、経直腸、経鼻、口腔、経膣、または埋め込みリザーバー(implanted reservoir)により投与され得る。インターフェロンは、通常、経口投与されないが、経口投与形態が開発されている。それにもかかわらず、本明細書中、本発明の方法または組み合わせを、何らかの特定の投与形態またはレジメンに限定すべきではない。当業者に認められる通り、インターフェロンの投与量は、一般的に、IUで特定される(例えば、約400万IUないし約1200万IU)。インターフェロンはまた、マイクログラム単位で投与され得る。例えば、Peg−イントロン(登録商標)の標準的用量は、約1.0−1.5μg/kg/週であり、ペガシス(登録商標)のそれは、180μg/週である。
【0089】
リバビリンは、通常、経口投与され、リバビリンの錠剤形態は、現在、市販されている。一般的基準では、リバビリン錠(例えば、約200mg錠剤)の1日投与量は、約800mgないし約1200mgである。例えば、リバビリン錠は、体重75kg未満の対象に約1000mg投与されるか、または体重75kg以上または75kgの対象に約1200mg投与される。それにもかかわらず、本明細書中、本発明の方法または組み合わせを、何らかの特定の投与形態またはレジメンに限定すべきではない。典型的に、リバビリンは、その商品ラベルに記載の投与量レジメンに従って投与され得る。
【0090】
ある態様において、VX−222およびVX−950(それを用いるとき)は、それぞれ独立して、経口的に、または静脈内に投与される。別のある態様において、VX−222およびVX−950(それを用いるとき)は、それぞれ独立して、経口投与される。
【0091】
いくつかの態様において、さらなる治療剤は、シトクロムp−450阻害剤である。CYP阻害剤について、1日当たり、体重1kg当たり、約0.001ないし約200mgの投与量が、一般的であり得る。より一般的には、1日当たり、体重1kg当たり、約0.1ないし約50mgまたは約1.1ないし約25mgの投与量であり得る。
【0092】
いくつかの態様において、さらなる治療剤はリトナビルである。リトナビルの具体的投与形態については、米国特許番号第6,037,157号およびそこに開示される文献:米国特許番号第5,484,801号、米国特許出願番号第08/402,690号、ならびにPCT公開番号WO95/07696およびWO95/09614を参照のこと。
【0093】
一般に、本発明において、1種以上の治療剤の“投与”または“共投与”(VX−950、インターフェロンおよびリバビリン、ならびにそれらの何れかの組み合わせを含む)は、各治療的活性剤を同じ投与形態で、または異なる投与形態で投与することを含む。異なる投与形態で投与するとき、治療的活性剤は、異なる時間に、同時に、または他の投与形態の投与中のいずれかの時間内に投与され得る。分割投与形態は、何れかの順序で投与され得る。すなわち、全ての投与形態は、他の投与形態の前に、共に、またはその後に投与され得る。
【0094】
一般に、種々の投与形態、製剤タイプおよび投与頻度、ならびにそれらの組み合わせが、本発明において用いられ得る。何れかの好適な投与形態および製剤タイプが、本発明において用いられ得る。
【0095】
いくつかの態様において、本発明の方法は、約100mgのVX−222(例えば、50mgのVX−222のカプセル2個)を含む投与形態でVX−222を投与することを含み、該投与形態は、1日1回、1日2回、例えば12時間毎(すなわち、q12h)に、または1日3回、例えば8時間毎(すなわち、q8h)に、投与され得る。いくつかの態様において、本発明の方法は、約400mgのVX−222(例えば、200mgのVX−222のカプセル2個)を含む投与形態でVX−222を投与することを含み、該投与形態は、1日1回、1日2回、例えば12時間毎(すなわち、q12h)に、または1日3回、例えば8時間毎(すなわち、q8h)に、投与され得る。いくつかの態様において、本発明の方法は、約750mgのVX−222(例えば、200mgのVX−222のカプセル3個および50mgのVX−222のカプセル3個)を含む投与形態でVX−222を投与することを含み、該投与形態は、1日1回、1日2回、例えば12時間毎(すなわち、q12h)に、または1日3回、例えば8時間毎(すなわち、q8h)に、投与され得る。いくつかの態様において、本発明の方法は、約1,500mgのVX−222(例えば、200mgのVX−222のカプセル7個および50mgのVX−222カプセル2個)を含む投与形態でVX−222を投与することを含み、該投与形態は、1日1回投与され得る。
【0096】
本発明の上記の方法のうち何れか1つの一面において、VX−950を含む組成物の経口用量が、それを必要とする患者に投与され、該用量は、患者に対して、投与後に少なくとも約750ng/mlのVX−950の平均血漿濃度(Cavg)を供する。いくつかの特定の態様において、VX−950のCavgは、約1000ng/mlまたは約1250ng/mlである。いくつかの特定の態様において、該用量は本質的に、750mgのVX−950を含む。いくつかの特定の態様において、Cavgは、VX−950の投与後3時間以内(例えば、2時間または1時間以内)に得られるか、または達せられる。いくつかの特定の態様において、VX−950のCavgは、約24時間(例えば、5週間または12週以上)維持される。他の面において、該経口用量は、患者に対して、24時間以上、約750ng/mlの最低トラフ濃度のVX−950血漿濃度を提供する。いくつかの特定の態様において、該投与形態は、24時間の間、血漿VX−950を、約800ng/ml(例えば、約900ng/mlまたは約1000ng/ml)の最低トラフ濃度に維持するために投与する。さらに別の面において、該経口用量で、治療的に有効な血漿濃度が得られ、あるトラフ濃度が維持され、ここで、VX−950の血漿トラフ濃度は、24時間の間、約750、800、900または1000ng/mlの最低レベルに維持される。ある具体的態様において、VX−950のトラフ濃度は、約750、800、900、1000ng/mlないし約1500ng/ml(例えば、1000ないし約1500)である。ある具体的態様において、VX−950のトラフ濃度は、約750、800、900、1000ng/mlないし約2500ng/ml(特に、1000ないし約2500)である。また、ヒトにVX−950を送達するため投与形態が提供され、ここで、該投与形態はVX−950を含み、該投与形態が24時間以内に少なくとも1回投与されるとき、VX−950の血漿トラフ濃度は、24時間の間、少なくとも約750ng/ml、800ng/ml、900ng/ml、または1000ng/mlないし約2500ng/ml(例えば、1000ng/mlないし約2500ng/ml、または1000ng/mlないし約1500ng/ml)に維持される。別の面において、該経口用量は、24時間の間、約30,000 hr*ng/mlないし約120,000 hr*ng/mlの範囲のVX−950の平均AUC(0−24時間)を患者に提供する。ある特定の態様において、VX−950のAUC(0−24時間)は、約50,000 hr*ng/mlないし約120,000 hr*ng/mlの範囲である。ある特定の態様において、VX−950のAUC(0−24時間)は、約60,000 hr*ng/mlないし約100,000 hr*ng/mlの範囲である。ある具体的な態様において、VX−950のAUC(0−24時間)は、約60,000 hr*ng/mlないし約90,000 hr*ng/mlの範囲である。WO2008/144072およびWO2005/25517に記載の他の具体的なVX−950の投与量レジメンもまた、本発明において用いられ得る。
【0097】
本発明の上記の方法のうち何れか1つの一面において、VX−222を含む組成物の経口用量が、それを必要とする患者に投与され、該用量は、患者に対して、投与後に少なくとも約750ng/mlのVX−222の平均最大血漿濃度(cmax)を与える。いくつかの特定の態様において、VX−222のcmaxは、少なくとも約1,000ng/mlである。いくつかの特定の態様において、VX−222のcmaxは、約750ng/mlないし約15,000ng/mlの範囲である。いくつかの特定の態様において、VX−222のcmaxは、約1,000ng/mlないし約15,000ng/mlの範囲である。いくつかの特定の態様において、VX−222のcmaxは、約3,000ng/mlないし約15,000ng/mlの範囲である。いくつかの特定の態様において、VX−222のcmaxは、約3,000ng/mlないし約12,000ng/mlの範囲である。他の面において、VX−222を含む組成物の経口用量は、それを必要とする患者に提供され、該用量は、患者に、24時間の間、約5,000 hr*ng/mlないし約150,000 hr*ng/mlの範囲のVX−222の平均AUC(0−24時間)を供する。いくつかの特定の態様において、VX−222のAUC(0−24時間)は、約5,000 hr*ng/mlないし約125,000 hr*ng/mlの範囲である。いくつかの特定の態様において、VX−222のAUC(0−24時間)は、約20,000 hr*ng/mlないし約100,000 hr*ng/mlの範囲である。いくつかの特定の態様において、VX−222のAUC(0−24時間)は、約20,000 hr*ng/mlないし約80,000 hr*ng/mlの範囲である。
【0098】
VX−222および何れかのさらなる薬剤は、分割投与形態で製剤され得る。あるいは、患者に投与される投与形態の数を減らすために、VX−222および何れかのさらなる薬剤は、任意の組み合わせで共に製剤され得る。何れかの別個の投与形態は、同時に、または異なる時間に投与され得る。投与形態は、その生物学的効果が有利になるように時間内に投与されるべきであることが理解されるべきである。
【0099】
例えば、本発明のVX−222およびVX−950の各量は、単一投与形態または2つ以上の投与形態で投与され得る。分割投与形態であるとき、各投与形態は、ほぼ同時に投与される。
【0100】
本発明において薬学的に許容される塩を治療的活性剤として用いるとき、それらの塩類は、典型的に、無機または有機酸およびそれらの塩基から誘導される。そのような酸性塩類には、酢酸塩、アジピン酸塩、アルギン酸塩、アスパラギン酸塩、安息香酸塩、ベンゼンスルホン酸塩、重硫酸塩、酪酸塩、クエン酸塩、ショウノウ酸塩(camphorate)、カンファースルホン酸塩(camphor sulfonate)、シクロペンタン−プロピオン酸塩、二グルコン酸塩、ドデシル硫酸塩、エタンスルホン酸塩、フマル酸塩、グルコヘプタン酸塩、グリセロリン酸塩、ヘミ硫酸塩、ヘプタン酸塩、ヘキサン酸塩、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、2−ヒドロキシエタンスルホン酸塩、乳酸塩、マレイン酸塩、メタンスルホン酸塩、2−ナフタレンスルホン酸塩、ニコチン酸塩、シュウ酸塩、パモ酸塩、ペクチン酸塩(pectinate)、過硫酸塩、3−フェニル−プロピオン酸塩、ピクリン酸塩、ピバリン酸塩、プロピオン酸塩、コハク酸塩、酒石酸塩、チオシアン酸塩、トシル酸塩およびウンデカン酸塩が含まれる。塩基性塩類には、アンモニウム塩、ナトリウムおよびカリウム塩のようなアルカリ金属塩、カルシウムおよびマグネシウム塩のようなアルカリ土類金属塩、ジシクロヘキシルアミン塩、N−メチル−D−グルカミンのような有機塩基との塩、ならびにアルギニン、リジンのようなアミノ酸との塩等が含まれる。
【0101】
また、塩基性窒素含有基は、塩化、臭化およびヨウ化メチル、エチル、プロピルおよびブチルのような低級ハロゲン化アルキル;硫化ジメチル、硫化ジエチル、硫化ジブチルおよび硫化ジアミルのような硫化ジアルキル;塩化、臭化およびヨウ化デシル、ラウリル、ミリスチルおよびステアリルのような長鎖ハロゲン化物;ベンジルおよびフェネチル臭化物のようなハロゲン化アラルキルなどのような物質によって四級化され得る。それにより、水または油に可溶性または分散性の生成物が得られる。
【0102】
本発明において、要すれば、治療剤(複数可)の修飾が可能であり、例えば、選択的生物学的特性を高めるのに適当な機能性を付加することにより修飾され得る。そのような修飾は、当技術分野で公知であり、所定の生物系(例えば、血液系、リンパ系、中枢神経系)への生物学的浸透を増加し、経口アベイラビリティーを増大し、注射による投与を可能にするための溶解性を増加し、代謝を変化させ、排泄速度を変化させることが含まれる。
【0103】
本発明のVX−222およびVX−950を含む1種以上の治療剤は、医薬組成物に包含され、該治療剤(複数可)は、単独で投与され得る。“医薬組成物”とは、本明細書に記載の治療剤を含む組成物を意味し、少なくとも1個の成分が、投与方法および投与形態の性質によって、薬学的に許容される担体、希釈剤、コーティング、アジュバント、賦形剤、または保存料のようなビークル、充填剤、崩壊剤、湿潤剤、乳化剤、乳化安定剤、懸濁化剤、等張剤、甘味剤、芳香剤、香味剤、着色剤、抗菌剤、抗細菌剤、他の治療剤、滑沢剤、吸収遅延または促進剤、ならびに分散剤を含む群から選択される。組成物は、錠剤、丸剤、顆粒、粉末、水性溶液または懸濁液、注射可能溶液、エリキシルまたはシロップの形態で存在し得る。
【0104】
懸濁化剤の例には、エトキシル化イソステアリルアルコール、ポリオキシエチレンソルビトールおよびソルビタンエステル、微結晶セルロース、アルミニウムメタヒドロキシド(aluminum metahydroxide)、ベントナイト、寒天−寒天およびトラガカント、ならびにこれらの物質の混合物が含まれる。微生物作用の予防のための抗菌剤および抗細菌剤の例には、パラベン、クロロブタノール、フェノール、ソルビン酸等が含まれる。等張剤の例には、糖類、塩化ナトリウム等が含まれる。吸収時間を延長するための吸収遅延剤の例には、モノステアリン酸アルミニウムおよびゼラチンが含まれる。吸収を増強するための吸収促進剤の例には、ジメチルスルホキシドおよび関連する類縁体が含まれる。担体、希釈剤、溶媒、ビークル、可溶化剤、乳化剤および乳化安定剤の例には、水、クロロホルム、スクロース、エタノール、イソプロピルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、テトラヒドロフルフリルアルコール、安息香酸ベンジル、ポリオール、プロピレングリコール、1,3−ブチレングリコール、グリセロール、ポリエチレングリコール、ジメチルホルムアミド、トゥイーン60、span@80、セトステアリルアルコール、ミリスチルアルコール、モノステアリン酸グリセロールおよびラウリル硫酸ナトリウム、ソルビタンの脂肪酸エステル、植物油(綿実油、落花生油、コーン油オリーブ油、ヒマシ油およびゴマ油)、およびオレイン酸エチルなどのような注射可能な有機エステル、またはこれらの物質の好適な混合物が含まれる。賦形剤の例には、ラクトース、乳糖、クエン酸ナトリウム、炭酸カルシウム、リン酸二カルシウムが含まれる。崩壊剤の例には、デンプン、アルギン酸およびある種のケイ酸塩複合体が含まれる。滑沢剤の例には、ステアリン酸マグネシウム、ラウリル硫酸ナトリウム、タルク、ならびに高分子量ポリエチレングリコールが含まれる。
【0105】
治療剤以外の医薬組成物中の物質の選択は、一般的に、溶解性のような治療剤の化学的特性、特定の投与方法および医薬業界で守られるべきによって決定される。例えば、ラクトース、クエン酸ナトリウム、炭酸カルシウム、リン酸二カルシウムのような賦形剤、デンプン、アルギン酸およびある種のケイ酸塩複合体のような崩壊剤、ならびにステアリン酸マグネシウム、ラウリル硫酸ナトリウムおよびタルクのような滑沢剤が、錠剤の製造に用いられ得る。
【0106】
医薬組成物は、錠剤、丸剤、顆粒、粉末、水性溶液または懸濁液、注射可能溶液、エリキシルまたはシロップのような種々の形態で存在し得る。
【0107】
“液体投与形態”は、患者に投与される治療剤の用量は、液体形態、例えば、薬学的許容されるエマルジョン、溶液、懸濁液、シロップおよびエリキシルを意味する。活性化合物に加えて、液体投与形態は、溶媒、可溶化剤および乳化剤のような当技術分野で常用される不活性希釈剤を包含し得る。
【0108】
固体組成物はまた、ラクトースまたはミルク糖のような賦形剤ならびに高分子量ポリエチレングリコールなどを用いて、軟および硬−充填ゼラチンカプセル中に充填剤として用いられ得る。
【0109】
水性懸濁液を用いるとき、それらは、懸濁を促進する乳化剤または薬剤を含み得る。
【0110】
エマルジョン医薬組成物の油相は、公知の方法に従い公知の成分から構成され得る。この相は単に乳化剤(別名エマルジェント(emulgent)として公知)を含み得るが、望ましくは、少なくとも1つの乳化剤と脂肪もしくは油との混合物、または脂肪および油の両方との混合物を含む。好ましくは、親水性乳化剤は安定剤として作用する親油性乳化剤と共に含まれる。また、好ましくは、油および脂肪の両方を含む。安定剤の有無にかかわらず、ともに乳化剤はいわゆる乳化蝋を構成し、該蝋は油および脂肪とともにクリーム製剤の油性分散相を形成する乳化軟膏基剤を構成する。
【0111】
要すれば、クリーム基剤の水相には、例えば、少なくとも30重量%の多価アルコール、すなわちプロピレングリコール、ブタン 1,3−ジオール、マンニトール、ソルビトール、グリセロールおよびポリエチレングリコール(PEG 400を含む)のような2個以上のヒドロキシル基を有するアルコールならびにそれらの混合物が含まれ得る。局所用製剤は望ましくは、皮膚または他の塗布領域を介する有効成分の吸収または浸透を増強する化合物を含み得る。
【0112】
製剤用の好適な油または脂肪の選択は、所望の美的(cosmetic)特性を達成することに基づく。故に、該クリームは、好ましくはチューブまたは他の容器からの漏れを避けるのに好適な濃度の、脂っこくなく、非着色で、かつ洗浄可能な製品であるべきである。
【0113】
直鎖または分枝鎖一または二塩基性アルキルエステル、例えばミリスチン酸ジイソプロピル、オレイン酸デシル、パルミチン酸イソプロピル、ステアリン酸ブチル、パルミチン酸2−エチルヘキシルまたはCrodamol CAPとして公知の分枝鎖エステルの混合物が使用され得る。これらは、必要な特性に応じて、単独でまたは組み合わせて使用され得る。あるいは、高融点脂質、例えば、白色軟質パラフィンおよび/または液体パラフィンまたは他の鉱油が使用され得る。
【0114】
一般的に、本明細書に記載の治療剤/医薬組成物は、好適な製剤形で、経口、吸入、直腸、経鼻、口腔(buccal)、舌下、経膣、経結腸、非経口(皮下、筋肉内、静脈内、皮内、鞘内、および硬膜外を含む)、嚢内および腹腔内を含む、局所または全身投与によりヒトおよび動物に投与され得る。好ましい経路は、例えばレシピエントの状態によって変わり得ることが認められ得る。
【0115】
“薬学的に許容される投与形態”は、本明細書に記載の治療剤の投与形態(VX−950を含む)を意味し、例えば、錠剤、粉末、エリキシル剤、シロップ、液体製剤(懸濁液、スプレー、吸入剤、錠剤、ロゼンジ、エマルジョン、溶液、顆粒、カプセルおよび坐薬を含む)、ならびにリポソーム製剤を含む注射用液体製剤が含まれる。一般に、製剤技術および製剤は、remington’s pharmaceutical sciences, mack publishing co., easton, pa, latest editionに見出され得る。
【0116】
“経口投与に好適な製剤”は、所定量の有効成分をそれぞれ含むカプセル剤、カシェ剤または錠剤として;粉剤または顆粒として;水性液体または非水性液体中の溶液または懸濁液として;または、油中水液体エマルジョンもしくは水中油液体エマルジョンのような分離した単位として提供され得る。有効成分はまた、ボーラス剤、舐剤またはペーストとして提供され得る。
【0117】
錠剤は、圧縮または成型により、必要に応じて1種以上の補助成分とともに、製造され得る。圧縮された錠剤は、好適な機械中で有効成分を自由流動形態(例えば、粉剤または顆粒)で、必要に応じてバインダー、潤滑剤、不活性希釈剤、防腐剤、表面活性剤または分散剤と混合し、圧縮することにより製造され得る。成型された錠剤は、好適な機械中で、不活性な液体希釈剤で湿らされた(moistened)粉末化された化合物の混合物を成型することにより製造され得る。錠剤は、必要に応じて、コーティングまたは割線を施され得、そして錠剤中の有効成分の遅いまたは制御された放出を提供するように処方され得る。
【0118】
直腸投与用の固体組成物は、公知の方法で製剤される坐薬および少なくとも1個の本発明の化合物を含む。
【0119】
要すれば、より有効な分散性に関して、本明細書に記載の治療剤は、マイクロカプセル化されるか、または生体適合性の、生分解可能なポリマーマトリクス(例えば、ポリ(d,l−ラクチド コ−グリコチド))、リポソームおよびミクロスフェアのような遅延放出また標的送達システムと結合され得て、いわゆる皮下または筋肉内デポー技術により皮下または筋肉内に注射されて、2週間またはそれ以上の間、化合物をゆっくり放出し続ける。治療剤は、例えば、細菌保持フィルターを介して濾過されるか、または使用直前に滅菌水、または他の滅菌注射可能媒体に溶解され得る、滅菌固体組成物形態に滅菌剤を挿入されて、滅菌され得る。
【0120】
“経鼻または吸入投与に適する製剤”は、患者に経鼻的に、または吸入により投与されるのに適する形態の製剤を意味する。該製剤は、例えば1ないし500ミクロンの範囲の粒子サイズ(30ミクロン、35ミクロンなどのような5ミクロン増加の20ないし500ミクロンの範囲の粒子サイズを含む)を有する粉末形態の担体を含み得る。担体が、例えば、経鼻スプレーまたは点鼻薬として投与するための液体である好適な製剤は、有効成分の水性または油性溶液を含む。エアロゾル投与に適する製剤は、常套法に従って製造され得て、他の治療剤と共に送達され得る。吸入治療剤は、定量吸入器により容易に投与される。
【0121】
“経口投与に好適な製剤”は、患者に経口的に投与されるのに好適な形態の製剤を意味する。該製剤は、一定量の有効成分をそれぞれ含むカプセル、カシェ剤または錠剤のような;粉末もしくは顆粒として;水性液体もしくは非水性液体中、溶液もしくは懸濁液のような;または、油中水液体エマルジョンもしくは水中油液体エマルジョンのような分離した単位として存在し得る。治療剤はまた、ボーラス剤、舐剤またはペーストとしても存在し得る。
【0122】
“非経腸投与に好適な製剤”は、患者に非経腸的投与されるのに好適な形態の製剤を意味する。該製剤は、滅菌され、エマルジョン、懸濁液、水性および非水性注射溶液を含み、それらは、懸濁化剤および濃化剤および抗酸化剤、緩衝剤、バクテリオスタットならびに製剤を意図されるレシピエントの血液と等張にし、pHを適切に調節する溶質を含む。
【0123】
“経直腸または経膣投与に好適な製剤”は、患者に経直腸または経膣投与するのに適する形態の製剤を意味する。該製剤は、好ましくは、本発明の化合物と好適な非刺激性の賦形剤または担体、例えばカカオバター、ポリエチレングリコールまたは坐薬ワックス(こでは、常温では固体であるが、体温では液体であり、故に直腸または膣腔で融解し、有効成分を放出する)を混合することにより製造され得る坐薬の形態である。
【0124】
“全身投与に好適な製剤”は、患者に全身的に投与されるのに適する形態の製剤を意味する。該製剤は、好ましくは筋層内、静脈内、腹腔内および皮下を含む注射により投与される。注射に関して、本発明の化合物は、液体溶液、好ましくは、ハンク溶液またはリンゲル溶液のような生理的に許容可能な緩衝液に製剤される。さらに、該化合物は、固体形態で製剤され得て、使用直前に再溶解されるか、または懸濁され得る。凍結乾燥形態もまた包含される、全身投与はまた、経粘膜または経皮手段によっても可能であるか、または化合物は経口投与され得る。経粘膜または経皮投与に関して、透過されるバリアに適する浸透剤が製剤に用いられる。かかる浸透剤は一般的に、当技術分野で公知であり、例えば、経粘膜投与のための胆汁酸塩およびフシジン酸誘導体を含む。さらに、洗浄剤が浸透を容易にするために用いられ得る。経粘膜投与には、経鼻スプレー、または例えば坐薬が使用され得る。経口投与に関して、該化合物は、カプセル、錠剤およびトニック剤のような常套の経口投与形態に製剤される。
【0125】
“局所投与に好適な製剤”は、患者に局所的に投与されるのに適する形態の製剤を意味する。該製剤は、局所用軟膏、軟膏(salve)、粉剤、スプレーおよび吸入剤、ゲル(水またはアルコール基剤)、クリームとして、当技術分野で通常知られている形態で存在し得るか、または経皮バリアを介して化合物の制御放出を可能にし得る、パッチにおける適用のためのマトリックスベース中に挿入され得る。軟膏形態に製剤されるとき、有効成分は、パラフィン性または水性の混合可能な軟膏基剤と共に使用され得る。あるいは、有効成分は、油中水クリーム基剤と共にクリームに製剤され得る。眼への局所投与に適する製剤には、有効成分が溶解されるか、または好適な担体、とりわけ有効成分用の水性溶媒中に懸濁される点眼剤が含まれる。口への局所投与に適する製剤には、風味基剤、通常スクロースおよびアカシアまたはトラガカント中に有効成分を含むロゼンジ;ゼラチンおよびグリセリン、またはスクロースおよびアカシアのような不活性基剤中に有効成分を含むトローチ;ならびに、好適な液体担体中に有効成分を含むマウスウォッシュが含まれる。
【0126】
“固体投与形態”は、本明細書に記載の治療剤の投与形態が固体形態、例えばカプセル剤、錠剤、丸剤、粉剤、糖衣錠または顆粒であることを意味する。かかる固体投与形態において、本発明の化合物は、クエン酸ナトリウムまたはリン酸二カルシウムのような少なくとも1種の不活性な慣習的な賦形剤(または担体)、または(a)充填剤もしくは増量剤、例えば、デンプン、ラクトース、スクロース、グルコース、マンニトールおよびケイ酸、(b)結合剤、例えば、カルボキシメチルセルロース、アルギネート類(alignate)、ゼラチン、ポリビニルピロリドン、スクロースおよびアカシア、(c)湿潤剤、例えば、グリセロール、(d)崩壊剤、例えば、寒天−寒天、炭酸カルシウム、ジャガイモまたはタピオカデンプン、アルギン酸、任意のケイ酸塩および炭酸ナトリウムの混合物、(e)溶液抑制剤、例えばパラフィン、(f)吸収促進剤、例えば、第四級アンモニウム化合物、(g)湿潤剤、例えば、セチルアルコールおよびモノステアリン酸グリセロール、(h)吸着剤、例えば、カオリンおよびベントナイト、(i)潤滑剤、例えば、タルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体ポリエチレングリコール、ラウリル硫酸ナトリウム、(j)乳白剤、(k)緩衝剤、および腸管のある部分で本発明の化合物を遅延放出する薬剤と混合される。
【0127】
担体および/または賦形剤と結合されて単一投与形態を製造し得る有効な治療剤の量は、処置される宿主および特定の投与方法によって変わり得る。典型的な製剤は、約5%ないし約95%の有効治療剤(w/w)を含み得る。好ましくは、かかる製剤は、治療剤を約20%ないし約80%含む。
【0128】
製剤は、薬剤学分野で周知の何れかの方法により単位投与形態で製造され得る。そのような方法には、有効成分を1個以上の補助成分を構成する担体と合わせる工程が含まれる。一般的に、該製剤は、有効成分を液体担体または微細に分割した固体担体またはその両方と合わせて、その後、必要であれば、製品を成形して、均一かつ密に結合して製造される。
【0129】
製剤は、単一用量または複数用量の容器、例えば弾性ストッパーを有する密閉アンプルおよびバイアル中に存在し得て、使用直前に滅菌液体担体、例えば注射用水の添加のみを要する凍結乾燥状態で貯蔵され得る。即時調製の注射溶液および懸濁液は、上記の種類の滅菌粉末、顆粒および錠剤から製造され得る。
【0130】
本明細書に記載の医薬組成物および用量製剤は、好ましくはインビボでの使用のためのものである。それにも関わらず、何れかの目的のための医薬組成物および用量製剤の使用に限定されることを意図しない。例えば、本明細書に記載の医薬組成物で予め処理された生物学的物質もまた、本発明に使用され得る。かかる生物学的物質には、血液およびその成分、例えば血漿、血小板、血液細胞の亜集団など;腎臓、肝臓、心臓、肺などのような臓器;精子および卵子;骨髄およびその成分、ならびに食塩水、デキストロースなどのような患者に注入される他の液体が含まれるが、これらに限定されない。
【0131】
何れかの特定の患者に対する特定の投与量および処置レジメンが、用いる特定の化合物の活性、年齢、体重、一般的健康状態、性別、食事、投与時間、排泄速度、混合薬、担当医の判断、および処置すべき特定の疾患の重症度、これまでの治療歴、共存疾患または併用薬、ベースラインのウイルス量、人種、疾患の期間、肝機能の状態および肝線維症/肝硬変の程度、ならびに治療目標(移植によるウイルス循環の排除またはウイルスの根絶)を含む、様々な因子によって変わり得ることも理解されるべきである。有効成分の量は、特定の記載した化合物、ならびに組成物中のさらなる抗ウイルス剤の存在または不存在、および性質によっても変わり得る。
【0132】
本発明の処置レジメンおよび用量形態に従い、VX−950およびインターフェロンの共投与は、サンプルまたは患者における、ウイルスの生活環に必要なNS3/4Aセリンプロテアーゼをコードする(または、本発明の実施に有効な量の)ウイルスを有効に減少させる。従って、本発明はまた、ウイルスの生活環に必要なNS3/4Aセリンプロテアーゼをコードするウイルスにより特徴付けられるウイルスに感染した患者の、上記のVX−950およびインターフェロン(および、所望により1種以上のさらなる治療剤)の投与による処置方法を提供する。
【0133】
本発明において、本発明に用いられる各活性治療剤は、独立して、食事の有無で患者に投与され得る。いくつかの態様において、VX−222および/または何れかのさらなるHCV薬は独立して、食事と共に投与される。本明細書で用いる用語“食事と組み合わせて”は、有効治療剤(複数可)が食事の食べ終わり約90分以内、例えば、食事後の約90分以内および食事前の約90分以内に投与されることを意味する。いくつかの態様において、有効治療剤(複数可)は、食事の食べ終わり前約30分まで、または食べ終わり後30分までに投与される。必要ではないが、全ての種類の食品(高脂肪および低脂肪食)が摂取され得て、高脂肪食は、より低脂肪食と比較して、改善された吸収を提供し得る。本明細書で用いる“高脂肪”は、約30%以上のカロリーが脂肪により提供される食事を意味する。任意の態様において、食事は、少なくとも約50カロリーを有する。他の任意の態様において、食事は、少なくとも約100カロリーである。さらに他の任意の態様において、食事は、少なくとも約50−100カロリーないし約3,000カロリーまで、約2,000カロリーまで、または約1,000カロリーまでである。さらに他の任意の態様において、食事には、脂肪からの、その総カロリーの少なくとも約30%が含まれる。
【0134】
一般に、本発明において、有効治療応答が達成されるように、処置はHCVウイルス感染を完全に根絶するか、またはその重症度を低減させ得る。有効治療応答は、例えば、a)継続的なウイルス応答を達成している;およびb)血漿中、検出不可能なHCV RNAを少なくとも約12週間(約12週間またはそれ以上)達成しているの、一方または両方であり得る。用語“検出不可能な”は、上記に定義の通りである。
【0135】
他の態様において、本発明の方法は、投与後の患者のHCV RNAレベルが、処置前よりも少なくとも約2log10(例えば、少なくとも約4log10)低下するように、HCVに感染した患者を処置する。
【0136】
いくつかの態様において、ウイルス血漿濃度の比較的迅速な低下は、患者に負荷用量の投与を行うことにより得られ得る。一態様において、該負荷用量は、約1250mgのVX−950である。
【0137】
いくつかの態様において、本発明の方法は、4週目のRVRおよび12週目の検出不可能な状態を達成できる。
【0138】
一般に、本発明において、“患者”には、哺乳動物、特にヒトが含まれる。
【0139】
ある態様において、本発明の方法は、遺伝子型1型C型肝炎ウイルスに感染した患者の処置を提供する。遺伝子型1型HCV感染は、アメリカ合衆国において、処置の最も困難なHCV株であり、最も蔓延している株である。
【0140】
有利には、両HCV未処置の患者および以前に処置された患者は、本発明の方法に有益である。誤解を避けるために、本発明の方法で処置され得る患者には、HCV処置が、試されなかった患者か、または、非応答、リバウンド、再発および進行した患者を含む、処置に失敗した患者が含まれる。任意の態様において、本発明の方法は、HCV未処置の患者を処置する。本明細書で用いる“HCV未処置”患者は、アメリカ食品医薬品局(FDA)または他のアメリカの機関もしくはFDAと同等の国際的機関によって承認されたか、または承認申請中の薬剤で以前にHCV処置を行なわれていないことを意味する。
【0141】
本発明の方法は、長期または短期療法として用いられ得る。本発明の方法が、患者を予防的に処置するのに使用され、該患者がC型肝炎ウイルスに感染した患者となるとき、該方法は、その後、感染を処置し得ることは、当業者に容易に明らかであり得る。故に、本発明の一態様は、患者におけるC型肝炎感染の処置または予防のための方法を提供する。
【0142】
患者の血漿中のVX−222およびVX−950濃度を決定するためのアッセイは、当技術分野で公知の方法により行った。例えば, Wasley, A. Et al., Semin. Liver Dis., 20: 1−16, 2000; Alter, H.J. et al., Semin. Liver dis., 20: 17−35, 2000; Brown, R.S. Jr. et al., Liver Transpl., 9: S10−S13, 2003; Defrancesco, R. et al., Nature, 436(7053): 953−960, 2005; Bowen, D.G. et al., J. Hepatol., 42: 408−417, 2005; Hoofnagle, J.H., Hepatology, 36: S21−S29, 2002, Brown, R.S. Jr. et al., Nature, 436 (7053): 973−978, 2005; および、Chisari, F.V., Nature, 436(7053): 930−932, 2005を参照のこと。
【0143】
本発明に関係する投与を、長期または短期療法として用い得る。単一投与形態を製造するために担体物質と併用され得る有効成分の量は、処置される宿主および特定の投与方法によって変わり得る。典型的に製剤は、約5%ないし約95%の有効化合物(w/w)を含み得る。好ましくは、かかる製剤は、約20%ないし約80%の有効化合物を含み得る。
【0144】
患者の状態の改善後は、必要ならば、本発明の化合物、組成物または組み合わせの維持量が投与され得る。次いで、投与の量あるいは頻度、または両方を、その症状の関数として、症状が所望のレベルに軽減され、処置を終えるべきときに、改善された状態が維持されるレベルに減らし得る。しかしながら、患者は、何らかの疾患症状の再発により長期的に断続的な処置を必要とし得る。
【0145】
何れかの特定の患者に対する特定の投与量および処置レジメンが、用いる特定の化合物の活性、年齢、体重、一般的健康状態、性別、食事、投与時間、排泄速度、混合薬、担当医の判断、および処置すべき特定の疾患の重症度、これまでの治療歴、共存疾患または併用薬、ベースラインのウイルス量、人種、疾患の期間、肝機能の状態および肝線維症/肝硬変の程度、ならびに治療目標(移植によるウイルス循環の排除またはウイルスの根絶)を含む、様々な因子によって変わり得ることも理解されるべきである。有効成分の量は、特定の記載した化合物、ならびに組成物中のさらなる抗ウイルス剤の存在または不存在、および性質によっても変わり得る。
【0146】
他の態様によれば、本発明は、本発明の薬学的に許容される組成物を該患者に投与することにより、ウイルスの生活環に必要なNS3/4Aセリンプロテアーゼをコードするウイルスにより特徴付けられるウイルスに感染した患者の処置方法を提供する。好ましくは、本発明の方法は、HCV感染を有する患者の処置に用いられる。かかる処置は、ウイルス感染を完全に根絶するか、またはその重症度を低減し得る。好ましくは、該患者は哺乳動物である。より好ましくは、該患者はヒトである。
【0147】
本明細書に記載の投与量は、好ましくはインビボでの使用用である。それにもかかわらず、これは目的に応じてこれらの量の、例えばVX−222またはVX−950の使用に限定することを意図していない。さらに別の態様において、本発明は、生物学的物質と、本発明の化合物を含む薬学的に許容される組成物を合わせる段階を含む、患者に投与することを目的として該生物学的物質を前処理する方法を提供する。かかる生物学的物質には、血液および血漿、血小板、血液細胞の亜集団などのその成分;腎臓、肝臓、心臓、肺などの臓器;精子および卵子;骨髄およびその成分、ならびに食塩水、デキストロースなどのような患者に注入される他の液体が含まれるが、それらに限定されない。
【0148】
本発明はまた、VX−222、VX−950、および薬学的に許容される担体、アジュバント、またはビヒクルを組み合わせる工程を含む、VX−222、VX−950、および薬学的に許容される担体、アジュバント、またはビヒクルを含む組成物を製造するための方法を提供する(ここで、組成物中のVX−222およびVX−950の投与量は、本発明の何れかの態様に従う。)。本発明の別の態様は、該組成物が、本明細書に記載の1個以上のさらなる薬剤を含む方法を提供する。
【0149】
医薬組成物はまた、単一パッケージ、通常ブリスターパッケージに一連の処置全体を含む“患者パック(patient pack)”で患者に処方され得る。患者パックには、従来の処方薬には通常添付されていない添付文書が含まれていて患者がいつでも利用できる点で、薬剤師が、大量供給物(bulk supply)から患者への薬剤分配を行なう従来の処方よりも有利である。添付文書の包含は、患者に医者の指示の順守を改善させることが証明されている。
【0150】
患者が本発明の正しい使用をするように指示する添付文書を包含する単一の患者パック、または各製剤の患者パックを用いる本発明の併用投与は、本発明の望ましいさらなる特徴であることが理解され得る。
【0151】
さらなる一面によれば、本発明は、VX−222(本発明の投与量)および本発明の併用使用の指示書を含む添付情報を含むパッケージである。全ての組成物、投与形態、治療レジメンまたは本発明の他の態様は、医薬パッケージ中に存在し得る。本発明の別の態様において、該医薬パッケージは、本明細書に記載の1個以上のさらなる薬剤をさらに含む。さらなる薬剤または複数の薬剤は、同一のパッケージまたは異なるパッケージで提供され得る。
【0152】
本発明の他の一面は、単一または複数の各医薬成分の製剤;貯蔵中および投与前に製剤(複数可)を保存する容器;ならびに、HCV感染の処置または予防のための有効な方法で薬剤投与を行うための指示書を含む、HCV感染の処置またはHCV感染の予防における使用のための(または、本発明の他の方法における使用のための)患者用のパッケージ化されたキットを含む。
【0153】
従って、本発明は、VX−222(および、所望によりさらなる薬剤)の用量の同時または連続投与のためのキットを提供する。典型的に、そのようなキットは、例えば、薬学的に許容される担体中(および、単一または複数の製剤中)、各化合物の組成物および任意のさらなる薬剤(複数可)、ならびに同時または連続投与のための指示書を含み得る。
【0154】
別の態様において、パッケージキットは、自己投与のための1個以上の投与形態;貯蔵中および使用前に該投与形態を保存するための、好ましくは密閉された容器;および、薬剤投与を行うための患者への指示書を包含して提供される。該指示書は、一般的に、添付文書、ラベル、および/またはキットの他の成分の指示が記載され、投与形態または複数の投与形態が、本明細書中に記載されている。各投与形態は、一枚の金属箔−プラスチック薄板で形成される個々の空間(cell)または半球形(bubble)中に、他から分離されたそれぞれの投与形態を包含させるか、または投与形態は、プラスチック容器のような単一の容器中に包含され得る。本発明のキットはまた、典型的には、個々のキット成分をパッケージングするための手段、すなわち、投与形態、容器手段、および使用のための指示書を含み得る。そのようなパッケージング手段は、段ボール箱または紙箱、プラスチック容器または金属箔容器などの形態であり得る。
【0155】
本発明のキットは、何れかの組成物、投与形態、治療レジメンまたは医薬パッケージのような、本発明のいずれかの一面を具現化し得る。
【0156】
本発明のパッケージおよびキットは、所望により、複数の組成物または投与形態を含む。従って、1個の組成物または2個以上の組成物を含むパッケージおよびキットは、本発明の範囲内に包含され得る。
【0157】
ある例示的態様は、以下に説明され、記載されているが、本発明の化合物が、当業者に一般的に利用可能な適当な出発物質を用いて、一般に上記の方法に従い製造され得ることは、当然のことである。
【0158】
VX−222は、一般的に、当業者に周知の方法で製造され得る(例えば、WO2002/100851およびWO2008/058393を参照のこと)。当技術分野で周知の全ての好適な製剤が、本発明において使用され得る。例えば、WO2002/100851およびWO2008/058393に記載の製剤が、本発明で使用され得る。本発明で使用され得る1つの特定の例には、VX−222の遊離酸形; Avicel PH 101;ラクトース一水和物;Poloxamer 188;ラウリル硫酸ナトリウム;プロビドンK29/32; Avicel PH 102;ラクトース一水和物;クロスカルメロースナトリウム;ステアリン酸マグネシウムが含まれる。本発明で用いられ得る特定の製剤は、実施例5に例示される。
【0159】
本発明の一態様は、VX−222の遊離酸形(構造式(I)で示される化合物); Avicel PH 101;ラクトース一水和物; Poloxamer (例えば、Polxamer 188);ラウリル硫酸ナトリウム;プロビドン K29/32; Avicel PH 102;ラクトース一水和物;クロスカルメロースナトリウム;ならびに、ステアリン酸マグネシウムを含むVX−222の製剤である。特定の態様において、製剤は、約45−60重量%のVX−222の遊離酸形;約5−20重量%のAvicel PH 101;約10−20重量%のラクトース一水和物;約1−10重量%のPoloxamer(例えば、Polxamer 188);約1−5重量%のラウリル硫酸ナトリウム;約1−10重量%のProvidone (例えば、プロビドンK29/32);約1−10重量%のAvicel PH 102;約1−10重量%のラクトース一水和物;約1−10重量%のクロスカルメロースナトリウム;ならびに、約0.1−5重量%のステアリン酸マグネシウムを含む。実施例5に記載の製剤はまた、本発明に包含される。
【0160】
VX−950は、一般的に、当業者に周知の方法で製造され得る(例えば、WO02/18369を参照のこと)。当技術分野で周知の全ての好適な製剤が、本発明において使用され得る。例えば、WO2005/123075、WO2007/109604、WO2007/109605およびWO2008/080167に記載の製剤が本発明で使用され得る。本発明で使用され得る特定の製剤は、実施例4に例示される。他の具体的な例は、以下を含む:
【表1】
【表2】
【表3】
【表4】
ここで、HPMC(ヒドロキシプロピルメチルセルロース 60SH 50cP (Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)(Hypromellose Acetate Succinate, HG grade, Shin−Etsu Chemical Co.)、HPC(ヒドロキシプロピルセルロース)、PVP(ポリビニルピロリドン)およびSLS(ラウリル硫酸ナトリウム)は、WO2005/123075に記載されている。任意の態様において、上記の固体分散体は、1%HPMC、0.002%シメチコン溶液(1重量%のHPMC、0.002重量%のシメチコンおよび99重量%の水)に懸濁され得る。さらなる例には、1:1 VX950:PVPK30、1重量%のSLS(Refreshed Tox.);Niro−49重量% HPMCAS/1重量% SLS/1重量% SDBS/49%VX−950;40.5重量% PVP−VA/10重量% ETPGS/49.5重量% VX−950;40.5重量% HPMC/10重量% ETPGS/49.5重量% VX−950;49重量% VX950、49重量% HPMCAS、1重量% SLS、1重量% SDBS;および、49重量% VX950、16重量% HPPh、33重量% HPC、1重量% SLS、□重量% SDBS(ここで、PVPK30(ポリビニルピロリドンK30)、SDBS(ドデシルベンゼンスルホン酸ナトリウム)、HPMCAS(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、ビタミンETPGS、PVP(ポリビニルピロリドン)およびSLS(ラウリル硫酸ナトリウム)、ならびにこれらの製剤の製造の詳細は、WO2005/123075に見出され得る)を含む。さらなる例には、WO2007/109604に記載のものが含まれる:
【0161】
55重量% VX−950、24.4重量% HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、19.6重量% HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量% ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
55重量% VX−950、14.7重量% HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、29.3重量% HPMC−60SH(ヒドロキシプロピルメチルセルロース60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
60重量% VX−950、24.4重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、14.6重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量% ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
65重量%VX−950、17重量% HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、17重量% HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
70重量% VX−950、9.7重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、19.3重量% HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP (Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量% ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
【0162】
60重量% VX−950、39重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
49.5重量% VX−950、24.5重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、24.5重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
83重量% VX−950、8重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、8重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
49.5重量%VX−950、24.5重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、24.5重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
70重量%VX−950、14.5重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、14.5重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
【0163】
65重量%VX−950、14.6重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、19.4重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
65重量%VX−950、9.7重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、24.3重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
60重量%VX−950、19.5重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、19.5重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
60重量%VX−950、14.6重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、24.4重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
【0164】
70重量%VX−950、9.7重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、19.3重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
49.5重量%VX−950、24.5重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、24.5重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
83重量%VX−950、8重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、8重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
49.5重量%VX−950、49.5重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
【0165】
83重量%VX−950、16重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
82.44重量%VX−950、15.89重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、および1.67重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
49.5重量%VX−950、24.75重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、24.75重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
60重量%VX−950、24.6重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード),14.4重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
【0166】
60重量%VX−950、39重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;ならびに、
49.5重量%VX−950、49.5重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体。
【0167】
これらの固体分散体の製造の詳細は、WO2007/109604に記載されている。さらなる具体例には、WO2007/109604に記載のVX−950のスプレー乾燥分散体を含む錠剤製剤が含まれる:
【表5】
【0168】
さらなる具体例には、WO2008/080167に記載の錠剤製剤が含まれる:
【表6】
【表7】
【0169】
【表8】
【表9】
【表10】
【0170】
全ての引用文献は、引用により本明細書中に包含される。
【0171】
本発明をより十分に理解するために、以下の製造例および実験例を記載する。これらの実施例は、説明のみを目的とし、いかなる点でも本発明の範囲を限定するものと解釈してはならない。
【実施例】
【0172】
実施例
以下の実施例において、VX−222は化合物1を、VX−950は化合物2を意味する。
実施例1:HCV レプリコン細胞アッセイプロトコール
C型肝炎ウイルス(HCV)レプリコンを含む細胞を、10%ウシ胎仔血清(FBS)、0.25mg/mLのG418、適当な添加剤を含むDMEM(媒体A)中で維持した。
【0173】
1日目に、レプリコン細胞単層を、トリプシン:EDTA混合物で処理し、除去し、その後、媒体Aを最終濃度100,000細胞/mLまで希釈した。10,000細胞/100μLを、96ウェル組織培養プレートの各ウェル中に播種し、37℃にて、組織培養インキュベーター中で一晩培養した。
【0174】
2日目、化合物(100%DMSO中)を、2%FBS、0.5%DMSO、適当な添加剤を含むDMEM(媒体B)中に連続的に希釈した。DMSOの最終濃度を、一連の希釈を通して0.5%に維持した。
【0175】
レプリコン細胞単層上の媒体Aを除去し、その後、種々の濃度の化合物を含む媒体Bを添加した。化合物を含まない媒体Bを、対照として他のウェルに添加した。
【0176】
細胞を、37℃の組織培養インキュベーター中、媒体B中化合物または0.5%DMSOを、48時間インキュベートした。48時間のインキュベーションの最後に、媒体Bを除去し、レプリコン細胞単層をPBSで一度洗浄し、RNA抽出前に−80℃で貯蔵した。
【0177】
処理したレプリコン細胞単層を含む培養プレートを解凍し、一定量のウシウイルス性下痢ウイルス(BVDV)のような別のRNAウイルスを、各ウェルにて細胞に添加した。RNA抽出試薬(RNeasyキットの試薬のような)を、RNA分解を避けるために細胞に直ちに添加した。全RNAを、抽出効率および一貫性を改善するための改良を伴う、製造業者の指示書に従って抽出した。最後に、HCVレプリコンRNAを含む全細胞RNAを溶出させ、さらなる処理まで−80℃で貯蔵した。
【0178】
TaqmanリアルタイムRT−PCR定量アッセイを、特定のプライマーおよびプローブの2個のセットを用いて行った。1つは、HCVについて、もう1つは、BVDVについて行った。処理したHCVレプリコン細胞由来の全RNA抽出物を、同じPCRウェル中に、HCVおよびBVDV RNAの両方の定量のために、PCR反応に追加した。実験的失敗に印をつけ、各ウェル中のBVDV RNAレベルを基に却下した。各ウェル中のHCV RNAレベルを、同じPCRプレートで実行する標準曲線に従い計算した。VX−950処理によるHCV RNAレベルの阻害または減少の割合を、DMSOを用いて計算し、VX−950不含有対照を0%阻害とした。IC50(HCV RNAレベルの50%阻害が見られる濃度)を、いずれかのVX−950濃度の滴定曲線から計算した。
【0179】
VX−950は、レプリコンアッセイにおいて顕著な活性を有することが示された。VX−950は、240ng/mLのIC50および476ng/mLのIC90を有した。
【0180】
実施例2:HCV Ki アッセイプロトコール
5AB基質および生成物の単離のためのHPLCマイクロボア法
基質:
NH2−Glu−Asp−Val−Val−(α)Abu−Cys−Ser−Met−Ser−Tyr−COOH(配列番号1)
【0181】
20mM 5AB(または、各自の選択濃度)の原液を、DMSOおよび0.2M DTTで作製した。これを−20℃にてアリコートで貯蔵した。
緩衝液:50mM HEPES、pH7.8;20%グリセロール;100mM NaCl。
【0182】
総アッセイ容量は、100μlであった:
【表11】
【0183】
緩衝液、KK4A、DTT、およびtNS3を合わせ;96ウェルプレートの各ウェルに各78μLずつ分配した。これを、30℃で約5分ないし10分間インキュベートした。2.5μLの適当な濃度の試験化合物を、DMSO中に溶解し(DMSOのみを対照とする)、各ウェルに添加した。これを、室温で15分間インキュベートした。20μLの250μM 5AB基質の添加により反応を開始させた(25μM濃度は、5ABのKm値と等しいか、わずかに低い)。得られた混合物を30℃で20分間インキュベートし、その後、25μLの10%TFAを添加して反応を終了させ、そして分析のために混合物をHPLCバイアルに移した(120μLアリコート)。SMSY産物を、以下の方法により、基質およびKK4Aから単離した:
【0184】
マイクロボア単離法
装置:Agilent 1100
Degasser G1322A
Binary pump G1312A
Autosampler G1313A
Column thermostated chamber G1316A
Diode array detector G1315A
カラム:
Phenomenex Jupiter;5マイクロン C18;300オングストローム;150×2mm;P/O 00F−4053−B0
カラムサーモスタット:40℃
注入量:100μL
溶媒A=HPLC等級純水+0.1%TFA
溶媒B=HPLC等級アセトニトリル+0.1%TFA
【0185】
【表12】
【0186】
実施例3
VX−950を、無作為化、二重盲検、プラセボ−対照の単回用量漸増試験において試験した。25名の健康な男性ボランティアが登録された。各対象は、VX−950の複数の単回用量を少なくとも7日間、3種の用量のVX−950を増加用量レベルで投与され、ならびに1用量のプラセボを投与された。
【0187】
25mgないし1250mgの用量を評価した。2倍ずつ増量しながら、より低用量範囲で攻撃的であり、より高用量範囲で保守的になるようにフィボナッチ数を改変した、用量漸増スキームを用いた。
【0188】
VX−950は、全ての用量レベルで良好な耐容性であるという結果を示した。重大な有害事象は試験中には報告されなかった。そして、用量レベルの増加により有害事象が増大することはなかった。
【0189】
薬物動態分析を、統計学的モーメント手法を用いて行った。薬物動態分析は、VX−950が、3時間の平均tmaxで吸収されたことを示した。2%未満のVX−950は尿中で変化なく除かれ、薬剤が、主に代謝経路から排泄されることを示した。
【0190】
実施例4
経口投与製剤を下記の通りに製造した。VX−950およびpovidone K29/32を塩化メチレン中に溶解し、次いでラウリル硫酸ナトリウムを添加し、溶液中に分散させて、均一な懸濁液を形成した。この懸濁液を、90℃の入口温度および56℃の出口温度を用いてスプレー乾燥させて、生成物をサイクロンから集めた。スプレー乾燥分散体を、75℃にて8時間、流動床乾燥させた。得られた粉末を、ガラスバイアル中で予め測定し、投与直前に、対象への投与のために水(30mL)に懸濁した。投与に関して、各バイアルを、全量が90mLである水の3分割量で洗浄した。
【表13】
【0191】
実施例5
VX−222(本明細書中、化合物1)の2つの異なる経口投与形態を以下の通りに製造した。
【表14】
【表15】
【0192】
VX−222(本明細書中、化合物1)のフォームAの任意の特性は以下の通りである:
【表16】
【0193】
XRPDパターンを、密封チューブ源およびHi−Star 領域検出器を備えるBruker D8 Discover 回折計(Asset Tag V012842)(Bruker AXS, Madison, WI)を、室温にて、反射モードで用いることにより得ることができる。X線発生器を、40kVおよび35mAの電圧で操作した。該粉末サンプルを、アルミニウムホルダー上に置いた。2個のフレームを、フレーム当たり120秒の露光時間で記録した。データを0.02o 2θの刻み幅の4o−40o 2θの範囲で統合し、1つの連続したパターンにまとめた。
【0194】
VX−222のフォームAを、以下に記載の工程に従い製造し得る:
−10gのVX−222(WO2008/058393に記載の通りに製造した化合物1)を反応器に入れる。
−20gのメタノールを加え、60℃まで加熱して溶解させる。
−10℃まで冷却し、固体が形成するまで待つ。
−固体を濾過する。
−20gのアセトンを25℃で添加する。
−1時間撹拌する。
−固体を濾過する。
−75℃で12時間乾燥させる。
【0195】
実施例6:実験VX−222−002からの薬物動態データ:VX−222およびVX−950の併用処置
VX−222について実施例5に記載の製剤を用いた。実験において、20名の健康な対象をコホート1またはコホート2(1コホート当たり10名の対象)に登録した。コホート1およびコホート2の対象は、実験の3つの期間の全て、処置期間1、処置期間2および処置期間3を完了した。処置期間1において、対象は、VX−222またはVX−222プラセボを食事と共に10日間投与された。処置期間2において、対象は、VX−950またはVX−950プラセボを食事と共に10日間投与された。処置期間3において、対象は、同時に、VX−222およびVX−950の両方またはVX−222プラセボおよびVX−950プラセボの両方を食事と共に10日間投与された。VX−222を処置期間1に投与し、VX−950を処置期間2に投与するために、処置期間1および処置期間2を7日間のウォッシュアウト期間で分けた。コホート1の対象に、処置期間1および3中、400mgのVX−222またはVX−222プラセボを12時間毎(q12h)に投与した。コホート2の対象に、処置期間1および3中、1,000mgのVX−222またはVX−222プラセボをq12hに投与した。コホート1およびコホート2の対象に、処置期間2および3中、1125mgのVX−950またはVX−950プラセボをq12hに投与した。この実験の実験デザインを図1に示す:
【表17】
【0196】
予備安全解析報告は、処置期間3において、重症化または重篤な有害事象(SAE)は報告されなかったことを示した。主な報告された有害事象は、軽度のものであり、予期しない有害事象の出現または傾向はなかった。下痢、食欲減退、掻痒、鼻血および鼻づまりを含むいくつかの有害事象は、処置期間1および処置期間3と比較して、処置期間3においてより多く見られた。
【0197】
薬物動態学的評価を以下の通りに行った:
VCH−222血漿:
−期間1
1日目:0(投与前)、投与後0.5、1、1.5、2、4、6、8および12時間
3日目、5日目、7日目、8日目および9日目:0(投与前)
10日目:0(投与前)、投与後0.5、1、1.5、2、4、6、8、12、24、48および72時間
−期間3
31日目および33日目:0(投与前)
37日目:0(投与前)、投与後0.5、1、1.5、2、4、6、8、12および24時間
VX−950(および代謝物)血漿:
−期間2
22日目、24日目および26日目:0(投与前)
27日目:0(投与前)、投与後0.5、1、1.5、2、4、6、8および12時間
−期間3
31日目および33日目:0(投与前)
37日目:0(投与前)、投与後0.5、1、1.5、2、4、6、8および12時間
VCH−222 尿中:
−10日目および37日目:投与後0ないし4、4ないし8、8ないし12および12ないし24(10日目のみ)時間。
【0198】
以下の表1は、この実験の予備的薬物動態学的(pk)結果を提供する。表に示す通り、VX−222血漿暴露は、増加した。
【表18】
【0199】
実施例7.パートAにおける実験VX−222−102からの薬物動態データ:VX−222処置
VX−222について実施例5に記載の製剤を用いた。実験102のパートAにおいて、対象をVX−222またはプラセボ 6:2(VX−222:プラセボ)の割当比で無作為化し、コホート1、コホート2、コホート3またはコホート4とした。コホート1、コホート2およびコホート3に登録された対象は、250mg、500mgまたは750mgのVX−222またはプラセボを1日2回(BID)、それぞれ3日間投与された。コホート4に登録された対象は、1,500mgのVX−222またはプラセボを1日1回(QD)、3日間投与された。標準的ケア処置について、医師により適当と判断されたとき、peg−ifn−アルファ−2aおよびRBVが48週間までの一部にて投与の最後に対象に供された。
【0200】
A.予備的結果
予備的安全性解析報告:遺伝子型1型慢性C型肝炎に感染した対象は、VX−222またはプラセボを250mg(コホート1)、500mg(コホート2)、または750mg(コホート3)(BID)で3日間、多数回投与された。予備的安全性解析報告は、重症化または重篤な有害事象は報告されなかったことを示した。主な報告された有害事象は、軽度のものであり、予期しない有害事象の出現または傾向はなかった。
【0201】
予備的薬物動態学的(PK)分析:コホート1、コホート2およびコホート3からの予備的pkパラメーターのまとめを、表2に示す。
【表19】
【0202】
予備的HCV RNA分析:コホート1、コホート2およびコホート3からの予備的HCV RNA分析のまとめを表3に提供する。コホート1、コホート2およびコホート3における遺伝子型1型HCVに感染した対象についての4日目の平均log HCV RNA減少は、それぞれ3.1、3.4および3.2であった。1,500mgのVX−222を1日1回投与された、コホート4における遺伝子型1型HCVに感染した対象についての4日目の平均log HCV RNA減少は、3.6であった。
【表20】
【0203】
B.さらなる結果
実験VX−222−102のさらなる結果を図2−8に示す:
250mgのVX−222 BID、500mgのVX−222 BID、750mgのVX−222 BID、および1,500mgのVX−222 QDを投与された各用量群における6名の患者を含む、32名の慢性遺伝子型1型HCVに感染した未処置患者をこの臨床試験に登録した。4種の用量群にて2名の患者がプラセボを投与され、全部で8名の患者がプラセボ投与された。試験のパートAを、アメリカ合衆国、カナダおよびアルゼンチンの10施設で行った。試験に登録された患者のうち、24名の患者が遺伝子型1a型HCV感染を有し、8名の患者が遺伝子型1b型HCV感染を有した。試験に登録された6名の患者は、アフリカ系アメリカ人であり、25名が白人であり、1名がインド人/アラスカ系であった。
【0204】
試験デザインaおよび人種
多施設、無作為化、二重盲検、プラセボ対照、漸増用量試験
−−HCV遺伝子型1型に感染した患者
・肝硬変の発現なし
・ALT値<5xULM
・スクリーニングで血漿 HCV RNAレベル≧5log10 IU/mL
−−4群の比較実験(VX−222:プラセボ 6:2無作為化)
−−この漸増用量試験に登録された順
・250mg BID、3日間
・500mg BID、3日間
・750mg BID、3日間
・1500mg QD、3日間。
【0205】
ベースライン特性を図3にまとめた。HCV RNA変化を図4−6に示す。図7は、VX−222薬物動態を示す。図7に示す通り、Tmaxには投与後2−6時間で達し、VX−222暴露は、およそ用量比例的に増大した。図8は、3日目におけるVX−222薬物動態のまとめを示す。
【0206】
ウイルス動態結果
VX−222での処置は、4種のVX−222用量群で血漿HCV RNAの3 log10以上の平均減少という結果となった。さらに、漸増用量応答が、500mg、750mgおよび1,500mg用量群で非常に似た結果で4種の用量群で観察された。250mg BID、500mg BIDおよび750mg BIDのVX−222を投与した3日後に達成された平均HCV RNA減少は、それぞれ3.1 log10(範囲:2.0ないし4.2)、3.4 log10(範囲:3.2ないし3.6)、および3.2 log10(範囲:2.3ないし3.8)であった。1,500mg QDのVX−222を投与した3日後に達成されたは、3.4 log10(範囲:3.1ないし3.9)であった。プラセボを受容する患者において、特記すべきHCV RNAの減少は観察されなかった。同様のウイルス減少が、遺伝子型1aおよび1b型に感染した患者で観察された。
【0207】
本試験のパートAのこれらの結果は、VX−222の750mg BIDで、予め3日間処置された5名の患者のウイルス薬物動態実験からの発見と一致する。
【0208】
安全性および耐容性結果
この試験のパートAで集められた安全性および耐容性の情報は伏せられたままであり、故に、本発明で供される安全性の情報には、プラセボまたはVX−222の投与後の患者についての収集データが含まれる。プラセボまたはVX−222は、4種の用量群の全てで良好な耐容性であって、重症化または重篤な有害事象は報告されず、処置の中止も生じなかった。プラセボまたはVX−222の投与後に報告された全ての有害事象は、軽度または中等症であった。用量群あたり少なくとも2名の患者に生じた最も多く報告された有害事象は、下痢、頭痛、悪心、無力症および発熱であった。
【0209】
実施例8.パートCにおける実験VX−222−102からの薬物動態データ:VX−222での処置
実験101のパートCにおいて、VX−222を、未処置の慢性C型肝炎を有する対象に750mg b.i.d.の複数用量で3日間投与した。さらに、実験102のパートAにおいて、VX−222を、未処置の慢性C型肝炎を有する対象に250mgないし750mg BID複数用量および1500mgを1日1回、3日間投与した。
【0210】
実験101(最終データ)および実験102(予備データ)にて評価されたVX−222のPKパラメーターを以下に概説する:
・AUCの蓄積指数は1.90倍であり、Cmaxのそれは、1.75倍であった。
・反復測定分析は、安定状態が処置の3日以内に達成されたことを示唆した。
・VX−222暴露は、平均Cmax、AUCτおよびCτにおける増加により示される通り、用量増加に伴い増加した。VX−222暴露は、250ないし750mg b.i.dの範囲の用量でおよそ用量に比例して増加した。
・VX−222吸収はゆっくりであり、2ないし6時間の範囲の安定状態での平均tmaxであった。
・蓄積指数は、BIDレジメンに関して約2倍であった。QDレジメンに関して、3日目の1500mg VX−222への暴露は、1日目に観察されたものと同様であった。
・平均t1/2は、全ての用量で変化しなかった。
・VX−222 t1/2は約5時間であった。
・投与間隔の最後でのVX−222濃度は、全ての対象についてインビトロでのIC90(319ng/mL)より高かった。
・一般に、対象におけるVX−222暴露は、健康な対象と比較して未処置HCV対象にて約2倍高かった。
【0211】
パートCにおけるVX−222臨床試験効果
実験101のパートCの主目的は、遺伝子型1型慢性C型肝炎感染を有する未処置対象におけるVX−222の薬理作用を評価することであった。750mg b.i.d.で連続3日間投与したVX−222の最終的な薬力学的パラメーターを以下に概説する:
・平均未変換ベースライン(1日目)HCV血漿RNAレベルは、4962600IU/mL(log10=6.4927)であった。
・2ないし4日目のlog10投与前HCV血漿RNAを用いて測定したベースラインからの平均最大減少は、−3.6784であった。投与前RNAレベルとlog10HCV RNAにおける減少のの振幅との相関は、比較的低かった。
これらの結果は健康な対象と一致し、VX−222とVX−950の共投与がVX−222単独投与よりも2倍高かった。
【0212】
実施例9:薬物併用アッセイ:VX−950およびVX−222
材料および方法
細胞
レプリコン細胞株Huh−7、Huh−7肝細胞癌細胞株由来のET細胞を、Ralf bartenschlager 博士(Bartenschlager, R. Innovation: Hepatitis C virus replicons: potential role for drug development. Nat. Rev. Drug Discov. 2002, 1, 911−916. Krieger, N.; Lohmann, V.; Bartenschlager, R. Enhancement of hepatitis C virus RNA replication by cell culture−adaptive mutations. J. Virol. 2001, 75, 4614−4624. Lohmann, V.; Korner, F.; Koch, J.−O.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999, 285, 110−113.) (Reblikon GmbH, Gau−Odernheim, Germany)から入手した。該Huh−7 ET細胞株は、ネオマイシン遺伝子に加えて、ホタルルシフェラーゼ遺伝子に融合されたコピーを担持する、高度に細胞培養に適合したレプリコンi389Luc−UBI−neo/NS3−3’/5.1コンストラクト(HCV NS3およびNS5遺伝子内の3つの適応変異の存在によっても特徴付けられる)を含む(Vrolijk, J.M.; Kaul, A.; Hansen, B.E.; Lohmann, V., Haagmans, B.L.; Schalm, S.W.; Bartenschlager, R. A replicon−based bioassay for the measurement of interferons in patients with chronic hepatitis C. Journal of Virol. Methods. 2003, 110, 201−209)。この細胞株は、ルシフェラーゼ活性を測定することにより、HCV RNA複製および形質転換の測定が可能である。ルシフェラーゼ活性は、これらの細胞におけるレプリコンRNAレベルに密接に従うことが既報である(j. Virol. 2001, 75, 4614−4624, journal of virol. Methods. 2003, 110, 201−209)。DMEM(wisent inc., st−bruno, qc, canada)からなる細胞培養に用いられる培地に、supplemented with 最終濃度、10%ウシ胎仔血清と1%ペニシリン/ストレプトマイシン、1%グルタミン、1%ピルビン酸ナトリウム、1%非必須アミノ酸および180μg/mlのジェネテシン(G418)(invitrogen, burlington, on, canada)を補った。細胞を、37℃、5%CO2雰囲気下でインキュベートし、サブコンフルエンスで週2回継代した。
【0213】
レプリコン細胞株Huh−7、Huh−7肝細胞癌細胞株由来の9−13細胞を、Ralf bartenschlager 博士(reblikon gmbh, gau−odernheim, germany)から入手した。Huh−7、9−13細胞株は、HCVサブゲノム レプリコンpfk i377/ns3−3’/wtを含み(Koutsoudakis, G.; Kaul, A.; Steinmann, E.; Kallis, S.; Lohmann, V.; Pietschmann, T.; Bartenschlager, R. Characterization of the Early Steps of Hepatitis C Virus Infection by Using Luciferase Reporter Viruses. J Virol. 2006, 80, 5308−5320)、リアルタイムPCRアッセイに用いられる。定量的リアルタイムPCRアッセイ(taqman)は、基本的に、レポーター色素およびクエンチャー色素がそれに結合される蛍光オリゴヌクレオチドプローブの使用と組み合わせた標準的PCRである。PCR中に、該プローブは、プライマー部位の上流と下流の間の標的にアニーリングする。各伸張反応中、プローブは、Taq DNA ポリメラーゼの5’ヌクレアーゼ活性により切断される。これは、クエンチャー色素からレポーター色素を分離させ、レポーター色素の蛍光強度を増大させる。蛍光強度は、標的DNAの存在量に比例する。
【0214】
薬物併用アッセイ
ルシフェラーゼアッセイ(MacSynergy分析を用いる4日目の実験)。Huh−7、ETレプリコン細胞を、乳白色の96ウェル細胞培養マイクロタイタープレート中、100μl培地中にサブコンフルエント密度(3x103 細胞/ウェル)で播種した。アッセイに用いる細胞培養培地は、G418とフェノールレッドを含まないこと以外は、上記と同様である。チオフェンと種々の濃度の異なる抗HCV薬との組み合わせを含むストック溶液のマトリックスを深い96ウェルプレートに製造した。3−4時間、37℃でインキュベート後、該マトリックスストック溶液プレートからの化合物(100μl)を、最終容量200μlを細胞に添加し、ここで、ある化合物は水平方向に増量させ、ある化合物は垂直方向に増量される。少なくとも4個の細胞プレートが各薬物−薬物相互作用試験に用いられ、各併用が少なくとも2回行われる。次いで、細胞はさらに、37℃、5%CO2雰囲気下で4日間インキュベートされる。その後、培養培地を除去し、細胞を95μlのルシフェラーゼ緩衝液(緩衝洗浄液中のルシフェリン基質)の添加により細胞溶解させた。細胞溶解物を少なくとも10分間室温でインキュベートし、直接照明から保護する。プレートを、照度計(Wallac Microbeta Trilux, Perkin Elmer(商標), MA, USA)を用いてルシフェラーゼ数を読み出す。
【0215】
化合物の併用が相加的、相乗的または拮抗的であるかどうかを決定するために、チオフェンと他の各薬物との4日間の処置の薬物併用効果を、MacSynergy II(商標) program (Prichard, M.N.; Prichard, L.E.; Shipman, C. Strategic design and three−dimensional analysis of antiviral drug combinations. Antimicrob. Agents Chemother. 1993, 37:540−545. Prichard, M.N. and Shipman, C. A three−dimensional model to analyze drug−drug interactions. Antiviral Res. 1990, 14:181−205)により計算する。この方法は、統計的確率に基づくBlissの独立ヌル・モデルを用いて薬物の組合せを試験し、2つの薬物が複製を阻害するのに独立して作用すると仮定する。この方法を用いて、理論的相加相互作用が、単独で作用する個々の薬物の用量応答曲線から計算される。その後、予測された相加効果は、用量応答曲線における相違を明らかにするため、実験的に決定された効果から差し引かれる。相互作用が相加的であるとき、結果として得られる曲線は0%差の水平面で現される。平面よりも高いピークは全て、予想を上回る効果(相乗効果)を示す。反対に、平面より低いピークは、期待された効果未満(拮抗作用)を示す。実験的用量応答曲線周辺の信頼区間は、統計学的にデータを評価するのに用いられピークの面積は、もたらされる相乗効果または拮抗作用を定量するために計算される。
【0216】
単独で試験されるとき、全ての薬物の阻害作用の50%阻害濃度(IC50)もまた、1化合物当たり7ないし9種の濃度を用いて用量応答曲線から決定される。曲線は、非線形回帰分析法を用いるデータ点に適合され、IC50は、GraphPad Prism software, version 2.0 (GraphPad Software Inc., San Diego, CA, USA)を用いて得られる曲線から補間される:
≦25uM2%:相乗作用ほとんどなし
25uM2%−50uM2%:わずかな相乗作用
50uM2%−100uM2%:中程度の相乗作用
≧100uM2%:強い相乗作用。
【0217】
リアルタイムPCRアッセイ(HCVレプリコンウイルスRNA排除およびリバウンド実験)。
レプリコン細胞株Huh−7、9−13細胞を、12ウェル培養ディッシュ中、1ml容量で、各ウェル当たり3x104細胞密度で播種する。該アッセイに用いる細胞培養培地は、10%ウシ胎仔血清、1%ペニシリン/ストレプトマイシン、1%グルタミン、1%ピルビン酸ナトリウムおよび1%非必須アミノ酸を補ったDMEMである。3ないし4時間インキュベート後、化合物を、最終容量2mlで種々の濃度で添加する。その後、細胞を37℃、5%CO2雰囲気下でさらに14日間インキュベートする。細胞を3ないし4日毎に剥がし、培地および阻害剤を補充し、細胞のサンプルをリアルタイムPCRによりRNA定量のために集める。14日間のインキュベート後、細胞を剥がし、抗ウイルス化合物を含まない新鮮な培地中に播く。18日目に、細胞を剥がし、0.25mg/mlのG418抗生物質を含む新鮮な選択培地に播く。培養をG418の存在下で42日目まで行い、細胞を3ないし4日毎に剥がし、該細胞サンプルを、リアルタイムPCRによりRNA定量を行う。全RNA(細胞およびウイルス起源)を、製造業者のプロトコールに従いQiagen RNeasy reagent (Qiagen Inc., Mississauga, ON, Canada, kit: 74106)を用いて抽出し、MMLV RT酵素を用いてcDNA合成を行う。この工程は、PCRのリアルタイム検出のためのABI PRISM(登録商標) 7700 Sequence Detection System (Applied Biosystems, Foster City, CA, USA)で適当なオリゴヌクレオチド、TaqmanプローブおよびDNA taq ポリメラーゼを用いてPCR反応により行われる。18S RNAレベルが、各ウェルにおける総RNA量の標準化に用いられる。
【0218】
単独で試験されるとき、全ての薬剤の阻害効果のための50%および90%阻害濃度(IC50およびIC90)もまた、1化合物当たりデュプリケートで6種の濃度を用いる用量応答曲線から決定される。曲線は、非線形回帰分析法を用いるデータ点に適合され、IC50およびIC90は、GraphPad Prism software, version 2.0 (GraphPad Software Inc., San Diego, CA, USA)を用いて得られる曲線から補間される。
【0219】
チオフェン化合物とウイルスプロテアーゼ阻害剤の併用実験
選択されたチオフェン化合物の併用実験では、上記の材料および方法を用いた。試験のために選択したHCV NS3 プロテアーゼ阻害剤はVX−950である。Macsynergy(商標)ソフトウェアを用いるこれらの化合物の相乗効果の結果を、表4に記載する。
【表21】
【0220】
実施例10:化合物1の任意のプロドラッグの合成
本明細書で用いる用語“RT(分)”は、化合物と関連するLCMS保持時間(分)を意味する。ある特定の化合物のNMRおよび質量分析データを、表5にまとめる。
化合物Aの製造
【化4】
5−(3,3−ジメチルブト−1−イニル)−3−[(トランス−4−ヒドロキシシクロヘキシル)−(4−トランス メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸(化合物(1)、300mg、0.67mmol)を、ジクロロメタン(DCM、15mL)に溶解した。これに、(2S)−2−(tert−ブトキシカルボニルアミノ)−3−メチル−ブタン酸 Boc−L−バリン(176mg、0.81mmol)、N,N−ジメチルピリジン−4−アミン(DMAP、8.22mg、0.067mmol)、トリエチルアミン(Et3N、136mg、187μL、1.35mmol)、および3−(エチルイミノメチレンアミノ)−N,N−ジメチル−プロパン−1−アミン塩酸塩(EDC、129mg、0.67mmol)を添加した。反応物を一晩撹拌した。次いで、反応混合物を濃縮し、酢酸エチル(EtOAc)で希釈し、水で洗浄し、合わせた有機相を塩水で洗浄し、硫酸ナトリウムで乾燥させた。濾過し、濃縮して黄色油状物を得て、それをカラムクロマトグラフィーにより精製した。次いで、得られた生成物を、ジオキサン中、4N HCl(15mL)で処理して、所望の化合物AをHCl塩として得た(100mg、26%):MS:m/z(obs.):545.4[M+H]+;保持時間:3.45分;1H NMR (300 MHz, MeOH) δ 7.04 (s, 1H), 4.75−4.58 (m, 1H), 4.39 (dt, J=14.5, 9.4 Hz, 1H), 3.85 (d, J=4.4 Hz, 1H), 3.80−3.68 (m, 1H), 3.61−3.51 (m, 1H), 2.24 (dt, J=14.0, 6.9 Hz, 1H), 2.01 (dd, J=15.2, 7.3 Hz, 6H), 1.60 (dd, J=28.5, 14.8 Hz, 9H), 1.34 (s, 9H), 1.18−0.99 (m, 3H), 0.81 (d, J=6.5 Hz, 3H), 0.66 (dd, J=25.3, 12.9 Hz, 1H)。
【0221】
化合物Bの製造
【化5】
5−(3,3−ジメチルブト−1−イニル)−3−[(トランス 4−ヒドロキシシクロヘキシル)−(トランス 4−メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸(化合物(1)、100mg、0.12mmol)を、ジクロロメタン(DCM、10.0mL)に溶解し、0℃まで冷却した。テトラゾール(4.0mg、0.058mmol)を添加し、次いでN−(ジ−tert−ブトキシホスファニル)−N−エチル−エタナミン(288mg、322μL、1.16mmol)を添加した。反応物を室温で一晩撹拌し、次いで−78℃まで冷却した。3−クロロベンゼンカルボペルオキソ酸(3−Chlorobenzenecarboperoxoic acid)(MCPBA)(99.7mg、0.58mmol)を添加し、反応物を2時間撹拌し、次いでNa2SO3水溶液でクエンチした。混合物を酢酸エチルで抽出し、抽出物を水で洗浄した。有機相を集め、無色油状物を得て、それをISCOシリカゲルクロマトグラフィーにより精製して、次工程に直接用いた。生成物にCH2Cl2(5mL)および2,2,2−トリフルオロ酢酸(TFA)(5mL)を添加した。反応物を2時間撹拌し、次いで濃縮し、生成物BをHPLCにより精製した:MS:m/z(obs.):526.39[M+H]+;保持時間:6.51分;1H NMR (300 MHz, d6−DMSO) δ 7.18 (s, 1H), 4.29 (t, J=11.8 Hz, 1H), 3.83 (s, 1H), 2.53 (d, J=8.2 Hz, 3H), 1.84 (s, 2H), 1.75−1.33 (m, 7H), 1.30 (s, 9H), 1.27−1.09 (m, 3H), 0.90 (d, J=12.9 Hz, 2H), 0.76 (d, J=6.5 Hz, 2H), 0.70−0.47 (m, 2H); 31P NMR (121.5 MHz, d6−DMSO) δ −2.01 (s)。
【0222】
化合物Cの製造
【化6】
CH2Cl2(15mL)中、5−(3,3−ジメチルブト−1−イニル)−3−[(4−トランス−ヒドロキシシクロヘキシル)−(4−トランス−メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸(化合物(1)、75mg、0.17mmol)およびN−Boc−グリシン(44.2mg、0.25mmol)の溶液に、3−(エチルイミノメチレンアミノ)−N,N−ジメチル−プロパン−1−アミン塩酸塩(EDC)(32.2mg、0.17mmol)、N,N−ジメチルピリジン−4−アミン(DMAP)(10.3mg、0.084mmol)およびEt3N(34mg、0.33mmol)を添加した。反応混合物を環境温度で一晩撹拌し、次いで反応混合物を蒸発させ、ISCOシリカゲルクロマトグラフィーにより精製して、化合物(b4)、[O−(N−t−ブトキシカルボニル)−グリシル]−5−(3,3−ジメチルブト−1−イニル)−3−[(4−トランス−ヒドロキシシクロヘキシル)−(4−トランス−メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸を得た:MS:m/z(obs.):603.17[M+H]+;保持時間:2.31分。
【0223】
【化7】
[O−(N−t−ブトキシカルボニル)−グリシル]−5−(3,3−ジメチルブト−1−イニル)−3−[(4−トランス−ヒドロキシシクロヘキシル)−(4−トランス−メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸(化合物(b4)、40mg、0.066mmol)を、ジオキサン中、4N HCl(1mL)で処理し、室温で一晩撹拌した。次いで、反応混合物を濃縮し、HPLCにより精製して、化合物Cを得た(11mg):MS:m/z(obs.):503.35[M+H]+;保持時間:2.24分。
【0224】
化合物Dの製造
【化8】
化合物(a5)、[O−(N−t−ブトキシカルボニル)−D−イソロイシル]−5−(3,3−ジメチルブト−1−イニル)−3−[(4−トランス−ヒドロキシシクロヘキシル)−(4−トランス−メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸(Boc−D−イソロイシンから上記の化合物1および4に記載の通りに製造)を、ジオキサン中、4N HCl(10mL)で処理し、室温で一晩撹拌した。次いで、反応混合物を濃縮し、HPLCにより精製して化合物Dを得た:MS:m/z(obs.):559.4[M+H]+;保持時間:2.39分。
【0225】
化合物Eの製造
【化9】
化合物(a6)、[O−(N−t−ブトキシカルボニル)−D−バリニル]−5−(3,3−ジメチルブト−1−イニル)−3−[(4−トランス−ヒドロキシシクロヘキシル)−(4−トランス−メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸(30mg)(Boc−D−バリンから上記の化合物1および4に記載の通りに製造)を、ジオキサン中、4N HCl(10mL)で処理し、室温で一晩撹拌した。次いで、反応混合物を濃縮し、HPLCにより精製して化合物Eを得た:MS:m/z(obs.):545.39[M+H]+;保持時間:2.35分。
【0226】
化合物Fの製造
【化10】
化合物(a8)、(O−(N−t−ブトキシカルボニル)−L−イソロイシル)−5−(3,3−ジメチルブト−1−イニル)−3−[(4−トランス−ヒドロキシシクロヘキシル)−(4−トランス−メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸(Boc−L−イソロイシンから上記化合物AおよびCに記載の通りに製造)(35mg)を、ジオキサン中、4N HCl(10mL)で処理し、室温で一晩撹拌した。次いで、反応混合物を濃縮し、HPLCにより精製して、化合物8を得た:MS:m/z(obs.):559.47[M+H]+;保持時間:3.2分。
【0227】
化合物Gの製造
【化11】
化合物(a9)、(O−(N−t−ブトキシカルボニル)−L−アラニル)−5−(3,3−ジメチルブト−1−イニル)−3−[(4−トランス−ヒドロキシシクロヘキシル)−(4−トランス−メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸(Boc−L−アラニンから上記化合物AおよびCに記載の通りに製造)(25mg)を、ジオキサン中、4N HClで処理し、室温で一晩撹拌した。次いで、反応混合物を濃縮し、HPLCにより精製して化合物Gを得た:MS:m/z(obs.):517.43[M+H]+;保持時間:2.99分。
【0228】
化合物Hの製造
【化12】
化合物(a10)、(O−(N−t−ブトキシカルボニル)−D−アラニル)−5−(3,3−ジメチルブト−1−イニル)−3−[(4−トランス−ヒドロキシシクロヘキシル)−(4−トランス−メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸(Boc−D−アラニンから上記化合物1および4に記載の通りに製造)(35mg、0.058mmol)を、ジオキサン中、4N HCl(10mL)で処理し、室温で一晩撹拌した。次いで、反応混合物を濃縮し、HPLCにより精製して化合物Hを得た:MS:m/z(obs.):517.43[M+H]+;保持時間:3.0分。
【0229】
【表22】
【0230】
実施例10:化合物1のプロドラッグのPKパラメーター
PKパラメーターが決定されるべきプロドラッグは、0.5%mc/0.5%トゥイーン 80/99%水中の溶液として製剤され得て、3mg/kg用量でラットに強制経口される。実験の前日にラットを計測する。ラットの血漿を、Instech自動血液サンプリング装置を用いて、投与前、および投与後15分、30分、1時間、2時間、3時間、4時間、6時間、8時間、12時間および24時間にサンプル採取する。血液をK2−EDTAを含むチューブに集め、110μlの血漿を分析のために抽出する。ラットは自由に餌を摂り、標準的なIACUCおよびSOPプロトコールが適用される。血漿サンプルおよび用量サンプルは、プロドラッグ化合物および活性代謝産物の両方に関してLC/MS/MSを用いて分析される。各対象に関する両分析物のPKパラメーターを、プロドラッグの定量した用量を用いて計算する。
【0231】
化合物H(図9にて“化合物10”と称される、化合物1のプロドラッグ)のPKパラメーターを、上記の通りに測定し、図9に記載した。図9に示す通り、化合物HのO−アラニル基は、インビボで、OH活性代謝産物に変換される。
【技術分野】
【0001】
関連出願
本出願は、2010年1月29日出願の米国仮特許出願番号第61/299,643号、2010年2月26日出願の同第61/308,506号、2010年3月1日出願の同第61/309,117号、および2010年4月15日出願の同第61/324,395号に優先権の利益を主張する。これらの出願の開示内容全体は、参照により本明細書に包含される。
【0002】
発明の技術分野
本発明は、C型肝炎ウイルス感染の処置法に関する。
【背景技術】
【0003】
発明の背景
C型肝炎ウイルス(“HCV”)による感染は、注目せざるを得ないヒトの医療問題である。HCVは、非A非B型肝炎のほとんどの症例の原因因子として認識されており、全世界で、概算で3%のヒト血清陽性率である(例えば、A. Alberti et al., “Natural History of Hepatitis C,” J. Hepatology, 31., (Suppl. 1), pp. 17−24 (1999)を参照)。米国だけでも400万名近くが感染している可能性がある(例えば、M.J. Alter et al., “The Epidemiology of Viral Hepatitis in the United States, Gastroenterol. Clin. North Am., 23, pp. 437−455 (1994); M. J. Alter “Hepatitis C Virus Infection in the United States,” J. Hepatology, 31., (Suppl. 1), pp. 88−91 (1999)を参照)。
【0004】
HCV感染者のうち、20−25%は、急性感染後にウイルスを除去することができるが、75−80%は、慢性感染症を発症する(例えば、preface, Frontiers in viral Hepatitis, Ed. RF Schinazi, J−P Sommadossi, and CM Rice, p. xi., Elsevier (2003)を参照のこと)。通常、これが肝炎の再発性および進行性の悪化を起こし、多くの場合、さらに重篤な病状、例えば、肝硬変および肝細胞癌を引き起こす(例えば、M.C. Kew, “Hepatitis C and Hepatocellular Carcinoma”, FEMS Microbiology Reviews, 14, pp. 211−220 (1994); I. Saito et. al., “Hepatitis C Virus Infection is Associated with the Development of Hepatocellular Carcinoma,” Proc. Natl. Acad. Sci. USA, 87, pp. 6547−6549 (1990)を参照)。残念なことに、慢性HCV感染症の進行を遅らせるのに広く有効な処置は存在しない。
【0005】
HCVゲノムは3010〜3033個のアミノ酸のポリタンパク質をコードする(例えば、Q.L. Choo, et. al., “Genetic Organization and Diversity of the Hepatitis C Virus.” Proc. Natl. Acad. Sci. USA, 88, pp. 2451−2455 (1991); N. Kato et al., “Molecular Cloning of the Human Hepatitis C Virus Genome From Japanese Patients with Non−A, Non−B Hepatitis,” Proc. Natl. Acad. Sci. USA, 87, pp. 9524−9528 (1990); A. Takamizawa et. al., “Structure and Organization of the Hepatitis C Virus Genome Isolated From Human Carriers,” J. Virol., 65, pp. 1105−1113 (1991)を参照)。HCVの非構造(NS)タンパク質は、ウイルス複製に必須の触媒機構を提供すると考えられている。NSタンパク質は、ポリタンパク質のタンパク質分解的切断に由来する(例えば、R. Bartenschlager et. al., “Nonstructural Protein 3 of the Hepatitis C Virus Encodes a Serine−Type Proteinase Required for Cleavage at the NS3/4 and NS4/5 Junctions,” J. Virol., 67, pp. 3835−3844 (1993); A. Grakoui et. al., “Characterization of the Hepatitis C Virus−Encoded Serine Proteinase: Determination of Proteinase−Dependent Polyprotein Cleavage Sites,” J. Virol., 67, pp. 2832−2843 (1993); A. Grakoui et. al., “Expression and Identification of Hepatitis C Virus Polyprotein Cleavage Products,” J. Virol., 67, pp. 1385−1395 (1993); L. Tomei et. al., “NS3 is a serine protease required for processing of hepatitis C virus polyprotein”, J. Virol., 67, pp. 4017−4026 (1993)を参照)。
【0006】
HCVのNSタンパク質3(NS3)には、大部分のウイルス酵素の合成を補助するセリンプロテアーゼ活性が含まれ、それ故に、後者はウイルス複製および感染力に必須であると見なされる。黄熱病ウイルスNS3プロテアーゼにおける変異は、ウイルス感染力を低減することが知られている(例えば、Chambers, T.J. et. al., “Evidence that the N−terminal Domain of Nonstructural Protein NS3 From Yellow Fever Virus is a Serine Protease Responsible for Site−Specific Cleavages in the Viral Polyprotein”, Proc. Natl. Acad. Sci. USA, 87, pp. 8898−8902 (1990)を参照)。NS3の最初の181個のアミノ酸(ウイルスポリタンパク質の残基1027〜1207)は、HCVポリタンパク質の4つ全ての下流部位を合成するNS3のセリンプロテアーゼドメインを含むことが示されている(例えば、C. Lin et al., “Hepatitis C Virus NS3 Serine Proteinase: Trans−Cleavage Requirements and Processing Kinetics”, J. Virol., 68, pp. 8147−8157 (1994)を参照)。
【0007】
HCV NS3 セリンプロテアーゼおよびそれと関係する補因子NS4Aは、全てのウイルス酵素プロセシングを補助し、故に、ウイルス複製に必須であると見なされる。このプロセシングはまた、ウイルス酵素プロセシングに関与する、ヒト免疫不全ウイルス アスパルチルプロテアーゼにより行われるものと同様のようである。ウイルスタンパク質プロセシングを阻害するHIVプロテアーゼ阻害剤は、ヒトにおける強力な抗ウイルス剤であり、ウイルス生活環のこの段階を阻止することは、結果として治療的有効剤であることを示す。結果として、HCV NS3 セリンプロテアーゼはまた、創薬の魅力的な標的である。
【0008】
現在、満足のいく抗HCV剤または処置剤は存在しない。最近まで、唯一の確立されたHCV疾患の治療法は、インターフェロンでの処置であった。HCV感染のために最初に承認された治療法は、標準的な(非ペグ化)インターフェロンαでの処置であった。しかし、インターフェロンは重い副作用を有し(例えば、M. A. Walker et al., “Hepatitis C Virus: An Overview of Current Approaches and Progress(C型肝炎ウイルス;最新の対処法と進歩の概説),” DDT, 4, pp. 518−29 (1999); D. Moradpour et al., “Current and Evolving Therapies for Hepatitis C,” Eur. J. Gastroenterol. Hepatol., 11, pp. 1199−1202 (1999); H. L. A. Janssen et al. “Suicide Associated with Alfa−Interferon Therapy for Chronic Viral Hepatitis,” J. Hepatol., 21, pp. 241−243 (1994); P.F. Renault et al., “Side Effects of Alpha Interferon” seminars in Liver Disease, 9, pp. 273−277. (1989)を参照)、長期間の寛解を誘導するのは、疾患の僅かな割合(〜25%)のみに限られる(例えば、O. Weiland, “Interferon Therapy in Chronic Hepatitis C Virus Infection”, FEMS Microbiol. Rev., 14, pp. 279−288 (1994)を参照)。処置レジメンへのリバビリンの追加は、わずかに治療応答率を増加させる。最近導入されたペグ化形態のインターフェロン(ペグ−イントロン(登録商標)およびペガシス(登録商標))とリバビリンとの併用もまた、寛解率の僅かながらの改善と副作用の部分的な軽減をもたらした。現在の標準的治療は、HCV遺伝子型のような予後因子、および治療への初期応答の発現に依拠しながら、24ないし48週継続する処置レジメンである。さらに、有効な抗HCVワクチンの見込みは、不明確なままである。
【0009】
従って、抗HCV療法および抗HCV化合物に適当な用量レジメンの必要性がある。
【0010】
HCVならびに他の疾患および障害は、肝臓損傷と関係がある。肝臓損傷を処置するための治療法および適当な用量レジメンの必要性がある。
【発明の概要】
【0011】
発明の概要
本発明は、一般的に、C型肝炎ウイルス(HCV)感染の処置法を提供する。本発明はまた、一般的に、C型肝炎ウイルス感染の臨床的続発症の予防法を提供する。
【0012】
一面において、本発明は、HCVに感染した患者におけるVX−222の薬物動態を改善する方法に関する。該方法は、患者に対して、VX−222およびVX−950を共投与することを含む。
【0013】
他の面において、本発明は、HCVに感染した患者の血漿中におけるVX−222への暴露を増大する方法に関する。該方法は、患者に対してVX−222およびVX−950を投与することを含む。
【0014】
他の面において、本発明は、HCVに感染した患者の処置法に関する。該方法は、患者にVX−222およびVX−950を投与することを含み、ここで、VX−222は、約20mgないし約400mgの量であり、VX−950は、約100mgないし約1500mgの量である。
【0015】
さらに他の面において、本発明は、治療的有効量のVX−222を投与することを含むHCVに感染した患者の処置法に関し、ここで、VX−222は、1日1回、約20mgないし約2000mgが投与される。
【0016】
さらに他の面において、本発明は、a)約20mgないし約400mgのVX−222;および、b) 約100mgないし約1500mgのVX−950を含む、薬学的に許容される組成物に関する。
【0017】
本発明はまた、HCVに感染した患者におけるVX−222のバイオアベイラビリティを増大するための医薬の製造における、VX−222およびVX−950の使用を提供する。
【0018】
本発明はまた、HCVに感染した患者の血漿中におけるVX−222のバイオアベイラビリティまたはVX−222への暴露を増大するための医薬の製造における、VX−222およびVX−950の使用を提供する。
【0019】
本発明はまた、HCVに感染した患者の処置のための医薬の製造におけるVX−222およびVX−950の使用を提供し、ここで、VX−222は、約20mgないし約400mgの量であり、VX−950は、約100mgないし約1500mgの量である。
【0020】
本発明はまた、HCVに感染した患者の処置のための医薬の製造におけるVX−222の使用を提供し、ここで、VX−222は、約20mgないし約2000mgの量で、または約50mgないし約2000mgの量で1日1回投与される。
【図面の簡単な説明】
【0021】
【図1】図1は、本発明の任意の態様の実験デザインを示すチャートである。
【図2】図2は、本発明の任意の態様の実験デザインを示すチャートである。
【図3】図3は、本発明の一態様の実験結果を示すチャートである。
【図4】図4は、本発明の一態様の実験結果を示すチャートである。
【図5】図5は、本発明の一態様の実験結果を示すチャートである。
【図6】図6は、本発明の一態様の実験結果を示すチャートである。
【図7】図7は、本発明の一態様の実験結果を示すチャートである。
【図8】図8は、本発明の一態様の実験結果を示すチャートである。
【図9】図9は、化合物1のプロドラッグおよび該プロドラッグの投与後にそれが変換される活性代謝物の血漿レベルを示すグラフである。
【発明を実施するための形態】
【0022】
本発明の詳細な説明
本発明は、VX−222を投与するための特定の用量および投与量レジメンに関する。本発明の目的に関して、VX−222は、化合物1ならびにその薬学的に許容される塩、溶媒和物およびプロドラッグ、また化合物1のプロドラッグの薬学的に許容される溶媒和物を含み、ここで、化合物1は、次の構造式:
【化1】
で示される。VX−222は、NS5Bポリメラーゼ阻害剤であり、WO2008/058393に記載されている。
【0023】
本発明はまた、VX−950を投与するための特定の用量および投与量レジメンに関する。VX−950は、7nmの定常状態結合定数(steady state binding constant)(ki*)を有する競合的な可逆性ペプチド模倣性NS3/4Aプロテアーゼ阻害剤である。例えば、WO02/018369を参照のこと。この目的に関して、VX−950は、化合物2ならびに化合物2の薬学的に許容される塩およびプロドラッグを含み、ここで、化合物2は、次の構造式:
【化2】
により示される。VX−950は、PCT公開番号WO02/018369、WO2006/050250およびWO2008144072に記載されている。VX−950の他の記載は、PCT公開番号WO07/098270およびWO08/106151に見出され得る。
【0024】
本明細書で用いる用語“薬学的に許容される塩”は、HCV感染の処置に対して安全かつ有効である塩を意味する。薬学的に許容される酸付加塩には、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、硝酸塩、硫酸塩、重硫酸塩、リン酸塩、過リン酸塩、イソニコチン酸塩、酢酸塩、および乳酸塩が含まれるが、これらに限定されない。種々のアミノ酸との薬学的に許容される塩もまた使用され得て、これらのアミノ酸塩の使用もまた、本発明の範囲内である。好適な塩基性塩には、アルミニウム塩、カルシウム塩、リチウム塩、マグネシウム塩、カリウム塩、ナトリウム塩、亜鉛塩およびジエタノールアミン塩が含まれるが、これらに限定されない。薬学的に許容される塩についての概要は、引用によりその内容全体を本明細書に包含させるberge et al., j. Pharm. Sci., 66, 1−19 (1977)を参照のこと。
【0025】
化合物1の薬学的に許容される塩の特定の例は、WO2008/058393に記載されており、例えば、アミノ酸(例えば、L−アルギニン、l−リシン)から誘導される塩類、アルカリ金属(例えば、ナトリウム、リチウム、カリウム)塩、アルカリ土類金属(例えば、カルシウム、マグネシウム)、アンモニウム、NR4+(式中、rは、C1−4アルキル)塩類、コリン塩およびトロメタミン塩類を含む適当な塩基から誘導される塩類である。一態様において、薬学的に許容される塩類は、ナトリウム塩である。別の態様において、薬学的に許容される塩類は、リチウム塩である。さらに別の態様において、薬学的に許容される塩は、カリウム塩である。さらに別の態様において、薬学的に許容される塩は、トロメタミン塩である。さらに別の態様において、薬学的に許容される塩は、l−アルギニン塩である。
【0026】
本明細書で用いる用語、化合物1の“薬学的に許容されるプロドラッグ”は、生理的条件下で、または化合物1の加溶媒分解により、またはHCV感染の処置においてその薬理学的効果を発揮する前に、化合物1の薬学的に許容される塩へ変換され得る化合物を意味する。本明細書で用いる用語、化合物2の“薬学的に許容されるプロドラッグ”は、生理的条件下で、または化合物2の加溶媒分解により、またはHCV感染の処置においてその薬理学的効果を発揮する前に、化合物2の薬学的に許容される塩へ変換され得る化合物を意味する。典型的に、プロドラッグは、改善された化学的安定性、改善された患者の受容性、改善されたバイオアベイラビリティ、延長された作用時間、改善された組織感受性、改善された剤形(例えば、増大した水溶性)、または低減された副作用(例えば、毒性)等の目的をもって製剤される。
【0027】
薬学的に許容されるプロドラッグは、当技術分野で公知の方法、例えば、Burger’s Medicinal Chemistry and Drug Chemistry, Vol. 1, 172−178 and 949−982, John Wiley & Sons (1995)に記載のような方法を用いて容易に製造され得る。Bertolini et al., J. Med. Chem., 40, 2011−2016 (1997); Shan et al., J. Pharm. Sci., 86(7), 765−767 (1997); Bagshawe, Drug Dev. Res., 34, 220−230 (1995); Bodor, Advances in Drug Res., 13, 224−331 (1984); Bundgaard, Design of Prodrugs, Elsevier Press (1985); および、Larsen, Design and Application of Prodrugs, Drug Design and Development (Krogsgaard−larsen et al., eds.), Harwood Academic Publishers (1991)も参照のこと。
【0028】
化合物1のプロドラッグの特定の例には、2010年6月28日出願のU.S.S.N. 61/359,164に記載のものが含まれる:
【化3】
【0029】
さらに、本明細書に記載の化合物が、異なる溶媒和物形態、例えば水和物形態で存在していてよく、さらに生物学的有効性を保持することは、当業者に理解され得る。かかる溶媒和物はまた、溶媒分子が、結晶化過程において化合物分子の結晶格子構造に挿入されるとき、形成され得る。本明細書で用いる用語、化合物1の“薬学的に許容される溶媒和物”は、溶媒分子(複数可)を含み、化合物1の生物学的有効性を保持する化合物1の薬学的に許容される溶媒和物形態を意味する。本明細書で用いる用語、化合物1のプロドラッグの“薬学的に許容される溶媒和物”は、溶媒分子(複数可)を含み、化合物1の生物学的有効性を保持する化合物1のプロドラッグの薬学的に許容される溶媒和物形態を意味する。
【0030】
1種以上の同位体に富む原子の存在でのみ化合物1および化合物2と異なる化合物は、本発明に包含される。例えば、水素を重水素またはトリチウムで置換したか、または炭素を13C−もしくは14C−に富む炭素により置換した以外は本発明化合物構造を有する化合物は、本発明の範囲内である。同位体に富む化合物2の任意の例は、WO2007/109080およびMaltais et al., J. of Medicinal Chemistry, “In Vitro and In Vivo isotope effects with Hepatitis c Protease Inhibitors: Enhanced Plasma Exposure of Deuterated Telaprevir versus Telaprevir in Rats” 2009;52(24):7993−8001に見出され得る。
【0031】
化合物1および化合物2は、それぞれ独立して、1個以上の不斉炭素原子を含んでいてよく、故に、ラセミ体およびラセミ混合物、単一のエナンチオマー、ジアステレオマー混合物および個々のジアステレオマーとして生じ得る。これらの化合物の全てのかかる同位体形態は、明示的に本発明に包含される。各立体中心炭素は、RまたはS配置であり得る。化合物2のn−プロピル側鎖でのD−およびL−異性体は、明示的に、本発明の範囲内に包含される。
【0032】
本明細書に記載の化合物が異なる多形形態で存在し得ることは、当業者に当然理解され得る。当技術分野で公知の通り、多形性は、2種以上の異なる結晶または“多形”体として結晶化する化合物の能力である。多形体は、固体状態の化合物分子が少なくとも2個の異なる配位または多形形態である化合物の固体結晶形である。何れかの化合物の多形形態は、同じ化学式または組成で示され、2個の異なる化合物の結晶構造として異なる化学物質である
【0033】
一面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1、またはその薬学的に許容される塩、溶媒和物もしくはプロドラッグ、または化合物1のプロドラッグの溶媒和物であり;VX−950は、化合物2、またはその薬学的に許容される塩もしくはプロドラッグである。
【0034】
別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1、またはその薬学的に許容される塩、溶媒和物もしくはプロドラッグ、または化合物1のプロドラッグの溶媒和物であり;VX−950は、化合物2またはその薬学的に許容される塩である。
【0035】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1、またはその薬学的に許容される塩、溶媒和物もしくはプロドラッグ、または化合物1のプロドラッグの溶媒和物であり;VX−950は化合物2である。
【0036】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1、またはその薬学的に許容される塩もしくは溶媒和物、もしくはプロドラッグであり;VX−950は、化合物2、またはその薬学的に許容される塩もしくはプロドラッグである。
【0037】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1、またはその薬学的に許容される塩もしくは溶媒和物、もしくはプロドラッグであり;VX−950は、化合物2またはその薬学的に許容される塩である。
【0038】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1、またはその薬学的に許容される塩もしくは溶媒和物、もしくはプロドラッグであり;VX−950は、化合物2である。
【0039】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1、またはその薬学的に許容される塩もしくは溶媒和物であり;VX−950は、化合物2、またはその薬学的に許容される塩もしくはプロドラッグである。
【0040】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1、またはその薬学的に許容される塩もしくは溶媒和物であり;VX−950は、化合物2またはその薬学的に許容される塩である。
【0041】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1、またはその薬学的に許容される塩もしくは溶媒和物であり;VX−950は化合物2である。
【0042】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1またはその薬学的に許容される塩であり;VX−950は、化合物2、またはその薬学的に許容される塩もしくはプロドラッグである。
【0043】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1またはその薬学的に許容される塩であり;VX−950は、化合物2またはその薬学的に許容される塩である。
【0044】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は、化合物1またはその薬学的に許容される塩であり;VX−950は、化合物2である。
【0045】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は化合物1であり;VX−950は、化合物2、またはその薬学的に許容される塩もしくはプロドラッグである。
【0046】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は化合物1であり;VX−950は、化合物2またはその薬学的に許容される塩である。
【0047】
さらに別の面において、以下に記載の本発明の態様のいずれか1つに関して、VX−222は化合物1であり;VX−950は化合物2である。
【0048】
一態様において、本発明は、HCVに感染した患者におけるVX−222の薬物動態を改善する方法に関する。該方法は、患者にVX−222およびVX−950を共投与することを含む。VX−222の改善された薬物動態には、患者の血漿、血液または肝臓におけるVX−222への暴露の増加が含まれる。別の態様において、本発明は、HCVに感染した患者の血漿におけるVX−222への暴露を増大する方法に関する。例えば、血漿におけるVX−222への暴露を、VX−222の、トラフ濃度(Ctrough)、平均血漿濃度(Cavg)、最大血漿濃度(Cmax)、またはAUC(曲線下面積)値により測定し得る。本明細書で用いる用語“トラフ濃度”(Ctrough)は、次の投与の直前の血漿中の薬物濃度、または2投与間の最小薬物濃度を意味する。本明細書で用いる用語“AUC”は、血漿(血清または血液)濃度−時間曲線の下部の面積を意味する。特定の態様において、VX−222への暴露は、AUC0−12(0ないし12時間)の値で示される。別の特定の態様において、VX−222への暴露は、AUC0−24(0ないし24時間)の値で示される。
【0049】
VX−950の共投与によるVX−222への暴露の増大は、VX−950なしで投与されるVX−222への暴露をVX−950と共投与されるVX−222への暴露と比較することにより決定され得る。1つの特定の態様において、VX−222への暴露は、VX−950なしで投与されるVX−222への暴露と比較して、約2ないし6倍増加する。1つの特定の態様において、VX−222への暴露は、VX−950なしで投与されるVX−222への暴露と比較して、約2ないし5倍増加する。1つの特定の態様において、VX−222への暴露は、VX−950なしで投与されるVX−222への暴露と比較して、約2または3倍増加する。いくつかの特定の態様において、該増大は、血漿におけるVX−222への暴露である。
【0050】
本発明において、各投与でのVX−950の量は、約100mgないし約1,500mg、約300mgないし約1,500mg、約500mgないし約1,500mg、約300mgないし約1,250mg、約450mg、約750mg、または約1,250mgであり得る。いくつかの特定の態様において、VX−950は、各投与にて約750mgである。いくつかの特定の態様において、VX−950は、各投与にて約1,125mgである。VX−950の量の好適な例は、WO2008/144072およびWO06/050250に記載され、その内容全体は、参照により本明細書に包含される。
【0051】
本発明において、各投与でのVX−222の量は、約20mgないし約2,000mg、約50mgないし約2,000mg、約100mgないし約1,500mg、約100mgないし約1,250mg、約100mgないし約1,000mg、約100mg、約250mg、約400mg、または約750mgであり得る。いくつかの特定の態様において、VX−222は、各投与にて約100mgである。いくつかの特定の態様において、VX−222は、各投与にて約250mgである。いくつかの特定の態様において、VX−222は、各投与にて約400mgである。いくつかの特定の態様において、VX−222は、各投与にて約750mgである。いくつかの特定の態様において、VX−222は、各投与にて約1,000mgである。いくつかの特定の態様において、VX−222は、各投与にて約1,500mgである。いくつかの特定の態様において、VX−222は、各投与にて約500mgである。いくつかの特定の態様において、VX−222は、各投与にて約1,125mgである。いくつかの特定の態様において、VX−222は、各投与にて約1,250mgである。
【0052】
さらに別の態様において、本発明は、HCVに感染した患者の処置方法であって、該患者にVX−222およびVX−950を投与することを含む方法に関する。特定の態様において、VX−222は、各投与にて約20mgないし約2,000mg、例えば約50mgないし約1,500mgであり、VX−950は、各投与にて約100mgないし約1,500mg、例えば約300mgないし約1,500mgである。別の特定の態様において、VX−222は、各投与にて約20mgないし約400mgの用量、例えば約50mgないし約400mgであり、VX−950は、各投与にて約100mgないし約1500mgである。さらに別の特定の態様において、VX−222は、各投与にて20mgと等しいか、または20mg以上ないし400mg未満の量である。さらに別の特定の態様において、VX−222は、各投与にて約20mgないし約300mgの量である。さらに別の特定の態様において、VX−222は、各投与にて約50mgないし約300mgの量である。さらに別の特定の態様において、VX−222は、各投与にて約100mgの量である。さらに別の特定の態様において、VX−222は、各投与にて約400mgの量である。さらに別の特定の態様において、VX−950は、各投与にて約300mgないし約1,500mgの量である。さらに別の特定の態様において、VX−950は、各投与にて約500mgないし約1,500mgの量である。さらに別の特定の態様において、VX−950は、各投与にて約750mgの量である。さらに別の特定の態様において、VX−950は、各投与にて約1,125mgの量である。
【0053】
さらに別の態様において、本発明は、HCVに感染した患者の処置方法であって、VX−222を投与すること(ここで、VX−222は、各投与にて、約20mgないし約2,000mgの量で投与される)を含む方法に関する。具体的には、VX−222の量は、各投与にて、約100mgないし約1,500mg、約100mgないし約1,250mg、約100mgないし約1,000mg、約100mg、約250mg、約400mg、約500mg、約750mg、約1000mg、約1125mg、または約1250mgであり得る。いくつかの態様において、VX−222は、1日1回投与される。特定の態様において、該方法は、1日1回、約50mgないし約2,000mgの量でVX−222を投与することを含む。いくつかの特定の態様において、VX−222の投与量は、約100mgを1日1回である。いくつかの特定の態様において、VX−222の投与量は、約250mgを1日1回である。いくつかの特定の態様において、VX−222の投与量は、約400mgを1日1回である。いくつかの特定の態様において、VX−222の投与量は、約500mgを1日1回である。いくつかの特定の態様において、VX−222の投与量は、約750mgを1日1回である。いくつかの特定の態様において、VX−222の投与量は、約1,000mgを1日1回である。いくつかの特定の態様において、VX−222の投与量は、約1,250mgを1日1回である。いくつかの特定の態様において、VX−222の投与量は、約1,125mgを1日1回である。いくつかの特定の態様において、VX−222の投与量は、約1,500mgを1日1回である。
【0054】
特定の態様において、本発明は、約20mgないし約2,000mg(または、約50mgないし約2,000mg、または上記の何れかの特定の投与量レジメン)のVX−222を1日1回投与し、さらにVX−222以外の1種以上のさらなるHCV薬を投与することによる、HCVに感染した患者の処置方法に関する。さらなるHCV薬の好適な例は、以下に詳細に記載され、VX−950、インターフェロンおよびリバビリンが含まれる。さらなる特定の態様において、VX−950が共投与される。VX−950の用量の典型例は、上記の通りである。別のさらなる特定の態様において、リバビリンの含有不含有に関わらず、インターフェロン(例えば、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bのようなペグ化インターフェロン)を共投与する。別のさらなる特定の態様において、VX−950;インターフェロン(例えば、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bのようなペグ化インターフェロン);および、リバビリンを共投与する。
【0055】
さらに別の態様において、本発明は、約20mgないし約2,000mgのVX−222;および約100mgないし約1,500mgのVX−950を含む薬学的に許容される組成物を提供する。所望により、薬学的に許容される担体もまた包含されてもよい。特定の態様において、本発明は、約20mgないし約1,500mg、または約50mgないし約1,500mgのVX−222を含む薬学的に許容される組成物を提供する。別の特定の態様において、これらの医薬組成物におけるVX−950の量は、約300mgないし約1500mg、約300mgないし約1250mg、約300mgないし約1,000mg、約300mgないし約750mg、または約375mgである。別の特定の態様において、これらの医薬組成物におけるVX−222の量は、50mgと等しいか、または50mg以上ないし400mg未満、約50mgないし約300mg、約50mg、約100mg、または約200mgである。これらの医薬組成物はそれぞれ、例えば、1日当たり、1回、2回または3回投与され得る。これらの組成物はそれぞれ、1種以上の投与形態(例えば、アンプル、カプセル、クリーム、乳剤、液剤、粒塊剤、ドロップ剤、注射剤、懸濁液、錠剤、粉剤)であり得る。これらの医薬組成物はそれぞれ、当業者により適当であると考えられる1種以上の経路(例えば、経口的、注入、注射、局所的または非経腸的)により投与され得て、投与形態によって変わる。
【0056】
一般的に、上記の本発明の方法において、VX−222およびVX−950は、それぞれ独立して、1日1回(QD)、1日2回(例えば、BID;q12h)、1日3回(例えば、TID;q8h)、または1日4回投与され得る。VX−222およびVX−950は、それぞれ独立して、食事の有無とは関係なく、投与され得る。
【0057】
いくつかの態様において、本発明の方法は、患者にVX−950を(a)各約450mg、8時間毎に1日3回;(b)各約750mg、8時間毎に1日3回;(c)各約1,125mg、12時間毎に1日2回;または、(d)各約1250mg、12時間毎に1日2回投与することを含む。
【0058】
いくつかの態様において、本発明の方法は、患者にVX−950を含む組成物の経口用量を投与することを含み、ここで、該用量は、患者に、投与後、VX−950の少なくとも約750ng/mlの平均血漿濃度(cavg)を提供する。いくつかの態様において、VX−950のcavgは、約1000ng/mlまたは約1250ng/mlである。いくつかの態様において、該用量は、本質的に約750mgのVX−950を含む。いくつかの態様において、cavgは、VX−950の投与後、3時間以内(例えば、2時間または1時間)で得られるか、または達成される。いくつかの態様において、VX−950のcavgは、約24時間以上(例えば、5週間または12週間)維持される。
【0059】
いくつかの態様において、本発明の方法は、患者にVX−950を投与することを含み、ここで、VX−950のトラフ血漿濃度は、24時間以上の間、最低でも約750、800、900または1000ng/mlで維持される。理論はともかく、約1500ng/ml以上のトラフ濃度は、本発明には必要ないと考えられる。従って、約750、800、900、1000ng/mlないし約1500ng/ml(特に、1000ないし約1500)のトラフ濃度は、本発明の範囲内である。
【0060】
理想的には、本発明の方法が、HCVに感染した患者の処置を伴うとき、該方法は、比較的迅速に、VX−950の治療的に有効な血漿濃度を達成し、次いで有効な治療応答が達成されるようなトラフ濃度を維持することを伴う。有効な治療応答は、好ましくは、a)持続的ウイルス応答を達成すること;および、b)少なくとも12週間(12週間以上)、血漿中において検出不可能なHCV RNAを達成すること、の一方または両方である。本明細書で用いる、HCV RNAについて“検出不可能”とは、HCV RNAが、現在市販されているアッセイにより決定される通り、好ましくはRoche COBAS TaqMan(商標) HCV/HPSアッセイにより決定される通り、10IU/ml未満で存在することを意味する。
【0061】
血漿濃度の比較的迅速な低下は、患者にある投与量を投与することにより得られ得る。一態様において、該投与量は、VX−950を約1250mgである。
【0062】
いくつかの態様において、本発明の方法は、VX−950を共投与することを含み、VX−950は、約750mgのVX−950(例えば、約375mgのVX−950の2つの錠剤)を含む投与形態であって、該投与形態を1日3回、例えば8時間毎(すなわち、q8h)に投与する。いくつかの態様において、本発明の方法は、VX−950を投与することを含み、VX−950は、約1125mgのVX−950(例えば、約375mgのVX−950の3つの錠剤)を含む投与形態であって、該投与形態を1日2回、例えば12時間毎(すなわち、q12h)に投与する。
【0063】
本発明において、VX−222および何れかのさらなるHCV薬(例えば、リバビリンの含有不含有に関わらず、VX−950;インターフェロン(例えば、ペグ化インターフェロン、例えばペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2b);または、VX−950、インターフェロン(例えば、ペグ化インターフェロン、例えばペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2b)、およびリバビリン)を、処置期間全体を通して独立して投与することができる。これらの態様において、VX−222処置期間およびさらなるHCV薬(複数可)の処置期間は同じである。
【0064】
あるいは、いくつかの態様において、VX−222およびさらなるHCV薬(例えば、リバビリンの含有不含有に関わらず、VX−950;インターフェロン(例えば、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bのようなペグ化インターフェロン);または、VX−950、インターフェロン(例えば、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bのようなペグ化インターフェロン)およびリバビリン)を、第一段階および第二段階の2つの段階に独立して投与することができる。VX−222および何れかのさらなるHCV薬はそれぞれ、第一段階もしくは第二段階のいずれか、または両方の段階で投与され得る。いくつかの態様において、VX−222を、第一段階でのみ投与し、インターフェロンを、第一段階および第二段階の両方で投与する。あるいは、いくつかの他の態様において、VX−222を第二段階でのみ投与し、インターフェロンを第一段階および第二段階の両方で投与する。いくつかの態様において、VX−222およびVX−950を共投与し、VX−222およびVX−950を第一段階のみで投与するか、または第二段階のみで投与する。いくつかの態様において、VX−222、VX−950、インターフェロン(例えば、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bのようなペグ化インターフェロン)、およびリバビリンを共投与し、VX−222およびVX−950を第一段階のみで投与し、インターフェロンおよびリバビリンを第一段階および第二段階の両方で投与する。
【0065】
第一段階および第二段階の期間の、好適な特定の例は、WO2008/144072に見出され得る。例えば、第一段階は、少なくとも約4週間、または約4週ないし約24週間(例えば、約4週間、約6週間、約8週間、約10週間、約12週間、約16週間、約20週間、約24週間など)の期間であってよく、第二段階は、少なくとも約12週間、例えば約12週間ないし約36週間の期間であり得る。ある態様において、第二段階は、約12週間である。他の態様において、第二段階は約24週間である。さらに他の態様において、第二段階は約36週間である。ある態様において、第一段階および第二段階の和は、約24週間ないし約48週間(例えば、約24週間、約36週間、または約48週間)である。いくつかの態様において、第一段階および第二段階は、同一期間であり得る。
【0066】
いくつかの態様において、本発明の方法は、VX−222およびインターフェロンを独立して、約4週間ないし約12週間(例えば、約4週間、約6週間、約8週間、または約12週間)の期間、約20週間ないし約28週間(例えば、約20週間、約24週間、または約28週間)の期間、または約8週間ないし約24週間(例えば、約8週間、約12週間、約16週間、または約24週間)の期間、共投与することを含む。これらの各態様の一面において、VX−222およびインターフェロンを独立して(第一段階)投与し、次いで約4週間ないし約36週間(例えば、約8週間ないし約36週間、約8週間ないし約24週間、約4週間ないし約24週間)の期間、インターフェロン(VX−222なし)を投与する(第二段階)。特定の例示的レジメンには、約24週間の全処置レジメン期間中、約16週間のインターフェロン(VX−222なし)の投与後に約8週間の間、VX−222およびインターフェロンを独立して投与すること;約24週間の全処置レジメン期間中、約12週間のインターフェロン(VX−222なし)の投与後に、約12週間の間、VX−222およびインターフェロンを独立して投与すること、が含まれる。かかるレジメンにおいて、所望により、全て(第一段階および第二段階の両方)、または各レジメンの一部(例えば、第一段階または第二段階のみ)の期間、リバビリンを投与してもよい。
【0067】
いくつかの態様において、本発明の方法は、VX−222およびVX−950を独立して、約4週間ないし約12週間(例えば、約4週間、約6週間、約8週間または約12週間)、約20週間ないし約28週間(例えば、約20週間、約24週間、または約28週間)、または約8週間ないし約24週間(例えば、約8週間、約12週間、約16週間または約24週間)の期間、共投与することを含む。これらの各態様の一面において、VX−222およびVX−950を独立して(第一段階)投与し、次いで約4週間ないし約36週間(例えば、約8週間ないし約36週間、約8週間ないし約24週間、約4週間ないし約24週間)の期間、インターフェロンおよびリバビリン(VX−222およびVX−950なし)を投与する(第二段階)。特定の例示的レジメンには、VX−222およびVX−950を独立して約8週間投与し、次いでインターフェロンおよびリバビリン(VX−222およびVX−950なし)を全体で約24週間の処置レジメンについて約16週間投与し;VX−222およびVX−950を独立して約12週間投与し、次いでインターフェロンおよびリバビリン(VX−222およびVX−950なし)を全体で約24週間の処置レジメンについて約12週間投与することが含まれる。かかるレジメンにおいて、VX−222およびVX−950の投与フェーズにインターフェロンおよびリバビリンの投与を供することも可能である。
【0068】
ある態様において、VX−222および所望によりVX−950またはインターフェロンを、約12週間未満の期間、独立して投与する。
ある態様において、VX−222および所望によりVX−950またはインターフェロンを、約8−12週間の期間、独立して投与する。
ある態様において、VX−222および所望によりVX−950またはインターフェロンを、約10週間の期間、独立して投与する。
ある態様において、VX−222および所望によりVX−950またはインターフェロンを、約10週間未満の期間、独立して投与する。
【0069】
ある態様において、VX−222および所望によりVX−950またはインターフェロンを、約2週間の期間、独立して投与する。
ある態様において、VX−222および所望によりVX−950またはインターフェロンを、約8週間未満(または約8週間)、約6週間未満(または約6週間)、または約4週間未満(または約4週間)の期間、独立して投与する。
ある態様において、VX−222およびVX−950を、約12週間(第一段階)の期間共投与し、次いで、所望によりインターフェロンおよびリバビリンを独立して約12週間(第二段階)投与してよい。
【0070】
ある態様において、VX−222、VX−950およびインターフェロンを約12週間共投与し、次いで、所望によりインターフェロンおよびリバビリンを独立して約12週間(第二段階)投与してよい。
ある態様において、VX−222、VX−950、インターフェロンおよびリバビリンを約12週間共投与し、次いで、所望によりインターフェロンおよびリバビリンを独立して約12週間(第二段階)投与してよい。
ある態様において、VX−222、インターフェロンおよびリバビリンを約12週間(第一段階)共投与し、次いで、所望によりインターフェロンおよびリバビリンを独立して約12週間(第二段階)投与してよい。
【0071】
ある態様において、VX−222およびVX−950を約12週間(第一段階)共投与し、次いで、所望によりインターフェロンおよびリバビリンを約12週間(第二段階)投与してよい。
ある態様において、VX−222、VX−950、インターフェロンおよびリバビリンを約12週間(第一段階)共投与し、次いで、所望によりインターフェロンおよびリバビリンを約12週間(第二段階)投与してよい。
ある態様において、VX−222、VX−950、インターフェロンおよびリバビリンを約12週間(第一段階)共投与し、次いで、所望によりインターフェロンおよびリバビリンを約36週間(第二段階)投与してよい。
【0072】
いくつかの態様において、上記の第一段階の何れかを、12週間未満の期間行い、第二段階を約12週間行うことができる。あるいは、第一段階を約12週間行い、第二段階を約24週間行うことができる。さらに他の面において、第一段階を約8週間行い、第二段階を約36週間行うことができる。さらに他の面において、第一段階を約4週間行い、第二段階を約36週間行うことができる。
【0073】
いくつかの態様において、上記の第一段階の何れかを、約8週間行い、第二段階を約16週間行うことができる。あるいは、第一段階を約8週間行い、第二段階を約40週間行うことができる。さらに他の面において、第一段階を約8週間行い、第二段階を約40週間行うことができる。
【0074】
いくつかの態様において、上記の第一段階および第二段階の何れかを、互いに入れ替えることができ、例えば、インターフェロン(所望によりリバビリンと共に)を第一段階に投与し、VX−222(所望によりVX−950と共に、またはVX−950、インターフェロンおよびリバビリンと共に)を第二段階に投与する。
【0075】
いくつかの態様において、上記の本発明の方法は、全処置の約12週間または約24週間の短期間の処置レジメンを評価する際に、治療応答ガイド基準(response−guided criteria)を用いる。これらの態様において、処置の第2週および第8週で検出不可能なHCV RNA(10IC/ml未満)を達成する患者は、全約24週間の処置中、12週に全ての処置を停止するか、またはpeg−IFN(ペグ化インターフェロン)およびRBV(リバビリン)治療をさらに12週間受けるように無作為化され;処置の第2週および第8週で検出不可能なHCV RNAを達成しない患者は、全24週間の処置中、peg−IFNおよびRBV(VX−222なし、またはVX−222およびVX−950なし)治療のさらに12週間を受ける。
【0076】
いくつかの態様において、上記の本発明の方法は、全処置が約12週間、約24週間または約36週間の短期処置レジメンの評価において、応答反応に基づく基準(response−guided criteria)を用いる。これらの態様において、処置の第2週および第8週で検出不可能なHCV RNA(10IC/ml未満)を達成する患者は、第12週で彼らに割り当てられた処置を終了し得る。第2週および第8週で検出不可能なHCV RNAを達成しない患者は、全24週間の処置についてさらに12週間のpeg−IFNおよびRBV(VX−222なし、またはVX−222およびVX−950なし)治療を受けるか、または全36週間の処置についてさらに24週間のpeg−IFNおよびRBV(VX−222なし、またはVX−222およびVX−950なし)治療を受ける。いくつかの特定の態様において、該方法は、VX−950およびVX−222(peg−IFNおよびRBVなし)を用い、例えば、1125mgのVX−950を1日2回および100mgまたは400mgのVX−222を1日2回等であり、処置の第2週および第8週で検出不可能なHCV RNA(10IC/ml未満)を達成する患者は、第12週で彼らの割り当てられた処置を終了してよく;第2週および第8週で検出不可能なHCV RNAを達成しない患者は、全36週間の処置についてさらに24週間のpeg−IFNおよびRBV(VX−222なし、またはVX−222およびVX−950なし)治療を受ける。いくつかの特定の態様において、該方法は、VX−950、VX−222、peg−IFNおよびRBV(例えば、1125mgのVX−950を1日2回、および100mgまたは400mgのVX−222を1日2回、180mcgのpeg−IFNを1週間に1回、ならびに800mg−1200mgのRBVを1日2回(例えば、体重75kg未満の患者に1000mg、または体重75kg以上の患者に1200mg))を受け、処置の第2週および第8週で検出不可能なHCV RNA(10IC/ml未満)を達成する患者は、第12週で彼らの割り当てられた処置を終了してよく;第2週および第8週で検出不可能なHCV RNAを達成しない患者は、全24週間の処置についてさらに12週間のpeg−IFNおよびRBV(VX−222なし、またはVX−222およびVX−950なし)治療を受ける。
【0077】
当業者には、本発明の方法が患者を予防的に処置するために用いられ、患者がC型肝炎ウイルスに感染したとき、該処置は、感染を処置し得ることが容易に理解され得る。故に、本発明の1つの態様は、患者におけるC型肝炎感染の処置法または予防法を提供する。
【0078】
C型肝炎に感染した患者の処置に加えて、本発明の方法は、C型肝炎に感染する可能性のある患者を予防するために用いられ得る。従って、本発明の1つの態様は、provides 患者におけるC型肝炎ウイルス感染の予防法であって、患者にVX−222と、要すれば何らかのさらなるHCV薬、例えば、上記の通り、VX−950;インターフェロン;インターフェロンおよびリバビリン;VX−950、インターフェロンおよびリバビリンを投与することを含む方法を提供する。
【0079】
本発明の方法はまた、免疫調節剤;抗ウイルス剤;HCVプロテアーゼの阻害剤(VX−222およびVX−950以外);HCV生活環の別の標的の阻害剤(NS3/4Aプロテアーゼ以外);内部リボソーム侵入の阻害剤、広域的ウイルス阻害剤;または、シトクロムP−450阻害剤;または、それらの組み合わせ、から選択されるさらなる薬剤を含む別の成分の投与を含み得る。さらなる薬剤はまた、ウイルスの細胞侵入の阻害剤からも選択される。
【0080】
従って、いくつかの態様において、さらなる薬剤は、別の抗ウイルス剤、好ましくは抗HCV剤(VX−222およびVX−950以外)である。そのような抗ウイルス剤には、α−、β−、およびγ−インターフェロンまたはチモシン、ペグ化誘導体化インターフェロン−α化合物、およびチモシンのような免疫調節剤;リバビリン、アマンタジン、およびテルビブジンのような他の抗ウイルス剤;C型肝炎プロテアーゼの他の阻害剤(NS2−NS3阻害剤およびNS3−NS4A阻害剤);ヘリカーゼ、ポリメラーゼ、およびメタロプロテアーゼ阻害剤を含む、HCV生活環における他の標的の阻害剤;内部リポソーム侵入の阻害剤;IMPDH阻害剤のような広域的ウイルス阻害剤(例えば、VX−497、VX−148、およびVX−944を含むが、それらに限定されない、米国特許第5,807,876号、同第6,498,178号、同第6,344,465号および同第6,054,472号ならびにPCT公報WO97/40028、WO98/40381およびWO00/56331に記載の化合物;ならびにミコフェノール酸およびその誘導体);または、上記のいずれかの組み合わせが含まれるが、それらに限定されない。
【0081】
本発明の化合物と併用され得る他の薬剤(例えば、非免疫調節または免疫調節化合物)には、引用により本明細書中に包含されるWO02/18369(例えば、273頁、9−22行目、および274頁、4行目から276頁、11行目、本開示は、引用により明確に本明細書中に包含される)に記載されるものが含まれるが、それらに限定されない。
【0082】
種々の公開された米国特許出願に記載されるさらに他の薬剤が包含される。これらの刊行物は、本発明の方法、特に肝炎の処置方法においてVX−950と併用され得る化合物および方法のさらなる教示を提供する。そのような方法および組成物は全て、本発明の方法および組成物と併用され得ると考えられる。簡単には、それらの刊行物の開示内容は、公開番号を引用することにより言及されるが、化合物の開示が、特に、引用により本明細書中に明確に包含されることが、特記されるべきである。そのような刊行物の例には、米国特許出願公開番号:US20040058982、US20050192212、US20050080005、US20050062522、US20050020503、US20040229818、US20040229817、US20040224900、US20040186125、US20040171626、US20040110747、US20040072788、US20040067901、US20030191067、US20030187018、US20030186895、US20030181363、US20020147160、US20040082574、US20050192212、US20050187192、US20050187165、US20050049220、およびUS20050222236が含まれる。
【0083】
さらなる他の薬剤には、Human Genome Sciencesから市販されるアルブフェロン(商標)(アルブミン−インターフェロンα);PEG−イントロン(登録商標)(ペグ化インターフェロンα−2b、Schering Corporation, Kenilworth, NJから市販される);イントロン−A(登録商標)(VIRAFERON(登録商標)、インターフェロンα−2b、Schering Corporation, Kenilworth, NJから市販される);リバビリン(1−β−D−リボフラノシル−1H−1,2,4−トリアゾール−3−カルボキサミド、ICN Pharmaceuticals, Inc., Costa Mesa, CAから市販される;the Merck Index, entry 8365, Twelfth Editionに記載される);REBETROL(登録商標)(Schering Corporation, Kenilworth, NJ);コーペガス(登録商標)(Hoffmann−La Roche, Nutley, NJ);ペガシス(登録商標)(ペグ化インターフェロンα−2a、Hoffmann−La Roche, Nutley, NJから市販される);ロフェロン(登録商標)(組換えインターフェロンα−2a、Hoffmann−La Roche, Nutley, NJから市販される);BEREFOR(登録商標)(インターフェロンα2、Boehringer Ingelheim Pharmaceutical, Inc., Ridgefield, CTから市販される);スミフェロン(登録商標)(スミフェロンのような、精製物と天然のαインターフェロンの混合物、住友製薬、日本から市販される);ウェルフェロン(登録商標)(インターフェロン α n1、Glaxo Wellcome Ltd., Great Britainから市販される);アルフェロン(登録商標)(Interferon Sciencesにより製造される天然αインターフェロンの混合物、およびPurdue Frederick Co., CTから市販される);α−インターフェロン;天然αインターフェロン 2a;天然αインターフェロン 2b;ペグ化αインターフェロン 2aまたは2b;コンセンサスαインターフェロン(Amgen, Inc., Newbury Park, CA);レベトロン(登録商標)(Schering Plough、インターフェロン−α 2B+リバビリン);ペグ化インターフェロンα(Reddy, K.R. et al. “Efficacy and Safety of Pegylated (40−kd) Interferon alpha−2a Compared with Interferon alpha−2a in Noncirrhotic Patients with Chronic Hepatitis C”(Hepatology, 33, pp. 433−438 (2001);コンセンサスインターフェロン(インファゲン(登録商標))(Kao, J.H., et al., “Efficacy of Consensus Interferon in the Treatment of Chronic Hepatitis” J. Gastroenterol. Hepatol. 15, pp. 1418−1423 (2000);リンパ芽球様または“天然”インターフェロン;インターフェロン tau(Clayette, P. et al., “IFN−tau, A New Interferon Type I with Antiretroviral activity” Pathol. Biol. (Paris) 47, pp. 553−559 (1999);インターロイキン−2(Davis, G.L. et al., “Future Options for the Management of Hepatitis C.” Seminars in Liver Disease, 19, pp. 103−112 (1999);インターロイキン−6(Davis et al. “Future Options for the Management of Hepatitis C.” Seminars in Liver Disease 19, pp. 103−112 (1999);インターロイキン−12(Davis, G.L. et al., “Future Options for the Management of Hepatitis C." Seminars in Liver Disease, 19, pp. 103−112 (1999);および、1型ヘルパーT細胞応答の発生を増強する化合物(Davis et al., “Future Options for the Management of Hepatitis C.” Seminars in Liver Disease, 19, pp. 103−112 (1999))が含まれるが、それらに限定されない。また、二本鎖RNA単独、またはトブラマイシンおよびイミキモド(3M Pharmaceuticals;Sauder, D.N. “Immunomodulatory and Pharmacologic Properties of Imiquimod” J. Am. Acad. Dermatol., 43 pp. S6−11 (2000))との組み合わせを含むが、これらに限定されない、細胞におけるインターフェロンの合成を刺激する化合物(Tazulakhova, E.B. et al., “Russian Experience in Screening, analysis, and Clinical Application of Novel Interferon Inducers” J. Interferon Cytokine Res., 21 pp. 65−73)も含まれる。また、WO02/18369、とりわけ272頁、15行目から273頁、8行目を参照のこと(この開示内容は、引用により本明細書中に明確に包含される)。
【0084】
シトクロムP450モノオキシゲナーゼ(“CYP”)阻害剤の好適な例には、リトナビル(WO94/14436)、ケトコナゾール、トロレアンドマイシン、4−メチルピラゾール、シクロスポリン、クロメチアゾール、シメチジン、イトラコナゾール、フルコナゾール、ミコナゾール、フルボキサミン、フルオキセチン、ネファゾドン、セルトラリン、インジナビル、ネルフィナビル、アンプレナビル、ホスアンプレナビル、サキナビル、ロピナビル、デラビルジン、エリスロマイシン、VX−944およびVX−497が含まれるが、これらに限定されない。好ましいCYP阻害剤には、リトナビル、ケトコナゾール、トロレアンドマイシン、4−メチルピラゾール、シクロスポリンおよびクロメチアゾールが含まれる。
【0085】
本発明の一態様は、CYP3A4の阻害剤を共投与する方法を提供する。
【0086】
本発明に用いられ得るインターフェロンの好適な例には、Human Genome Sciencesから市販されるアルブフェロン(商標)(アルブミン−インターフェロンα);PEG−イントロン(登録商標)(ペグ化インターフェロンα−2b、Schering Corporation, Kenilworth, NJから市販される);イントロン−A(登録商標)(VIRAFERON(登録商標)、インターフェロンα−2b、Schering Corporation, Kenilworth, NJから市販される);ペガシス(登録商標)(ペグ化インターフェロンα−2a、Hoffmann−La Roche, Nutley, NJから市販される);ロフェロン(登録商標)(組換えインターフェロンα−2a、Hoffmann−La Roche, Nutley, NJから市販される);BEREFOR(登録商標)(インターフェロンα2、Boehringer Ingelheim Pharmaceutical, Inc., Ridgefield, CTから市販される);スミフェロン(登録商標)(スミフェロンのような、精製物と天然のαインターフェロンの混合物、住友製薬、日本から市販される);ウェルフェロン(登録商標)(インターフェロン α n1、Glaxo Wellcome Ltd., Great Britainから市販される);アルフェロン(登録商標)(Interferon Sciencesにより製造される天然αインターフェロンの混合物、およびPurdue Frederick Co., CTから市販される);α−インターフェロン;天然αインターフェロン 2a;天然αインターフェロン 2b;ペグ化αインターフェロン 2aまたは2b;コンセンサスαインターフェロン(Amgen, Inc., Newbury Park, CA);レベトロン(登録商標)(Schering Plough、インターフェロン−α 2B+リバビリン);ペグ化インターフェロンα(Reddy, K.R. et al. “Efficacy and Safety of Pegylated (40−kd) Interferon alpha−2a Compared with Interferon alpha−2a in Noncirrhotic Patients with Chronic Hepatitis C”(Hepatology, 33, pp. 433−438 (2001);コンセンサスインターフェロン(インファゲン(登録商標))(Kao, J.H., et al., “Efficacy of Consensus Interferon in the Treatment of Chronic Hepatitis” J. Gastroenterol. Hepatol. 15, pp. 1418−1423 (2000);リンパ芽球様または“天然”インターフェロン;インターフェロン tau(Clayette, P. et al., “IFN−tau, A New Interferon Type I with Antiretroviral activity” Pathol. Biol. (Paris) 47, pp. 553−559 (1999);ならびに、インプランタブル・デュロス(登録商標)を介してオメガ・インターフェロンを提供するオメガ・デュロス(登録商標)(intarcia therapeutics, inc., mountain view, ca)が含まれる。
【0087】
いくつかの態様において、本発明の方法は、リバビリンの有無における、インターフェロンの共投与である。具体的には、インターフェロンは、ペグ化インターフェロン(peg−ifn)である。より具体的には、ペグ化インターフェロンは、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bのようなペグ化インターフェロンαである。
【0088】
一般的に、VX−222および何れかのさらなるHCV剤(VX−950、インターフェロンおよびリバビリンのような)は、独立して個別に、または共に投与され得る。一般に、VX−222およびVX−950は、独立して、経口、非経腸、舌下、吸入スプレーにより、局所、経直腸、経鼻、口腔、経膣、または埋め込みリザーバー(implanted reservoir)により投与され得る。インターフェロンは、通常、経口投与されないが、経口投与形態が開発されている。それにもかかわらず、本明細書中、本発明の方法または組み合わせを、何らかの特定の投与形態またはレジメンに限定すべきではない。当業者に認められる通り、インターフェロンの投与量は、一般的に、IUで特定される(例えば、約400万IUないし約1200万IU)。インターフェロンはまた、マイクログラム単位で投与され得る。例えば、Peg−イントロン(登録商標)の標準的用量は、約1.0−1.5μg/kg/週であり、ペガシス(登録商標)のそれは、180μg/週である。
【0089】
リバビリンは、通常、経口投与され、リバビリンの錠剤形態は、現在、市販されている。一般的基準では、リバビリン錠(例えば、約200mg錠剤)の1日投与量は、約800mgないし約1200mgである。例えば、リバビリン錠は、体重75kg未満の対象に約1000mg投与されるか、または体重75kg以上または75kgの対象に約1200mg投与される。それにもかかわらず、本明細書中、本発明の方法または組み合わせを、何らかの特定の投与形態またはレジメンに限定すべきではない。典型的に、リバビリンは、その商品ラベルに記載の投与量レジメンに従って投与され得る。
【0090】
ある態様において、VX−222およびVX−950(それを用いるとき)は、それぞれ独立して、経口的に、または静脈内に投与される。別のある態様において、VX−222およびVX−950(それを用いるとき)は、それぞれ独立して、経口投与される。
【0091】
いくつかの態様において、さらなる治療剤は、シトクロムp−450阻害剤である。CYP阻害剤について、1日当たり、体重1kg当たり、約0.001ないし約200mgの投与量が、一般的であり得る。より一般的には、1日当たり、体重1kg当たり、約0.1ないし約50mgまたは約1.1ないし約25mgの投与量であり得る。
【0092】
いくつかの態様において、さらなる治療剤はリトナビルである。リトナビルの具体的投与形態については、米国特許番号第6,037,157号およびそこに開示される文献:米国特許番号第5,484,801号、米国特許出願番号第08/402,690号、ならびにPCT公開番号WO95/07696およびWO95/09614を参照のこと。
【0093】
一般に、本発明において、1種以上の治療剤の“投与”または“共投与”(VX−950、インターフェロンおよびリバビリン、ならびにそれらの何れかの組み合わせを含む)は、各治療的活性剤を同じ投与形態で、または異なる投与形態で投与することを含む。異なる投与形態で投与するとき、治療的活性剤は、異なる時間に、同時に、または他の投与形態の投与中のいずれかの時間内に投与され得る。分割投与形態は、何れかの順序で投与され得る。すなわち、全ての投与形態は、他の投与形態の前に、共に、またはその後に投与され得る。
【0094】
一般に、種々の投与形態、製剤タイプおよび投与頻度、ならびにそれらの組み合わせが、本発明において用いられ得る。何れかの好適な投与形態および製剤タイプが、本発明において用いられ得る。
【0095】
いくつかの態様において、本発明の方法は、約100mgのVX−222(例えば、50mgのVX−222のカプセル2個)を含む投与形態でVX−222を投与することを含み、該投与形態は、1日1回、1日2回、例えば12時間毎(すなわち、q12h)に、または1日3回、例えば8時間毎(すなわち、q8h)に、投与され得る。いくつかの態様において、本発明の方法は、約400mgのVX−222(例えば、200mgのVX−222のカプセル2個)を含む投与形態でVX−222を投与することを含み、該投与形態は、1日1回、1日2回、例えば12時間毎(すなわち、q12h)に、または1日3回、例えば8時間毎(すなわち、q8h)に、投与され得る。いくつかの態様において、本発明の方法は、約750mgのVX−222(例えば、200mgのVX−222のカプセル3個および50mgのVX−222のカプセル3個)を含む投与形態でVX−222を投与することを含み、該投与形態は、1日1回、1日2回、例えば12時間毎(すなわち、q12h)に、または1日3回、例えば8時間毎(すなわち、q8h)に、投与され得る。いくつかの態様において、本発明の方法は、約1,500mgのVX−222(例えば、200mgのVX−222のカプセル7個および50mgのVX−222カプセル2個)を含む投与形態でVX−222を投与することを含み、該投与形態は、1日1回投与され得る。
【0096】
本発明の上記の方法のうち何れか1つの一面において、VX−950を含む組成物の経口用量が、それを必要とする患者に投与され、該用量は、患者に対して、投与後に少なくとも約750ng/mlのVX−950の平均血漿濃度(Cavg)を供する。いくつかの特定の態様において、VX−950のCavgは、約1000ng/mlまたは約1250ng/mlである。いくつかの特定の態様において、該用量は本質的に、750mgのVX−950を含む。いくつかの特定の態様において、Cavgは、VX−950の投与後3時間以内(例えば、2時間または1時間以内)に得られるか、または達せられる。いくつかの特定の態様において、VX−950のCavgは、約24時間(例えば、5週間または12週以上)維持される。他の面において、該経口用量は、患者に対して、24時間以上、約750ng/mlの最低トラフ濃度のVX−950血漿濃度を提供する。いくつかの特定の態様において、該投与形態は、24時間の間、血漿VX−950を、約800ng/ml(例えば、約900ng/mlまたは約1000ng/ml)の最低トラフ濃度に維持するために投与する。さらに別の面において、該経口用量で、治療的に有効な血漿濃度が得られ、あるトラフ濃度が維持され、ここで、VX−950の血漿トラフ濃度は、24時間の間、約750、800、900または1000ng/mlの最低レベルに維持される。ある具体的態様において、VX−950のトラフ濃度は、約750、800、900、1000ng/mlないし約1500ng/ml(例えば、1000ないし約1500)である。ある具体的態様において、VX−950のトラフ濃度は、約750、800、900、1000ng/mlないし約2500ng/ml(特に、1000ないし約2500)である。また、ヒトにVX−950を送達するため投与形態が提供され、ここで、該投与形態はVX−950を含み、該投与形態が24時間以内に少なくとも1回投与されるとき、VX−950の血漿トラフ濃度は、24時間の間、少なくとも約750ng/ml、800ng/ml、900ng/ml、または1000ng/mlないし約2500ng/ml(例えば、1000ng/mlないし約2500ng/ml、または1000ng/mlないし約1500ng/ml)に維持される。別の面において、該経口用量は、24時間の間、約30,000 hr*ng/mlないし約120,000 hr*ng/mlの範囲のVX−950の平均AUC(0−24時間)を患者に提供する。ある特定の態様において、VX−950のAUC(0−24時間)は、約50,000 hr*ng/mlないし約120,000 hr*ng/mlの範囲である。ある特定の態様において、VX−950のAUC(0−24時間)は、約60,000 hr*ng/mlないし約100,000 hr*ng/mlの範囲である。ある具体的な態様において、VX−950のAUC(0−24時間)は、約60,000 hr*ng/mlないし約90,000 hr*ng/mlの範囲である。WO2008/144072およびWO2005/25517に記載の他の具体的なVX−950の投与量レジメンもまた、本発明において用いられ得る。
【0097】
本発明の上記の方法のうち何れか1つの一面において、VX−222を含む組成物の経口用量が、それを必要とする患者に投与され、該用量は、患者に対して、投与後に少なくとも約750ng/mlのVX−222の平均最大血漿濃度(cmax)を与える。いくつかの特定の態様において、VX−222のcmaxは、少なくとも約1,000ng/mlである。いくつかの特定の態様において、VX−222のcmaxは、約750ng/mlないし約15,000ng/mlの範囲である。いくつかの特定の態様において、VX−222のcmaxは、約1,000ng/mlないし約15,000ng/mlの範囲である。いくつかの特定の態様において、VX−222のcmaxは、約3,000ng/mlないし約15,000ng/mlの範囲である。いくつかの特定の態様において、VX−222のcmaxは、約3,000ng/mlないし約12,000ng/mlの範囲である。他の面において、VX−222を含む組成物の経口用量は、それを必要とする患者に提供され、該用量は、患者に、24時間の間、約5,000 hr*ng/mlないし約150,000 hr*ng/mlの範囲のVX−222の平均AUC(0−24時間)を供する。いくつかの特定の態様において、VX−222のAUC(0−24時間)は、約5,000 hr*ng/mlないし約125,000 hr*ng/mlの範囲である。いくつかの特定の態様において、VX−222のAUC(0−24時間)は、約20,000 hr*ng/mlないし約100,000 hr*ng/mlの範囲である。いくつかの特定の態様において、VX−222のAUC(0−24時間)は、約20,000 hr*ng/mlないし約80,000 hr*ng/mlの範囲である。
【0098】
VX−222および何れかのさらなる薬剤は、分割投与形態で製剤され得る。あるいは、患者に投与される投与形態の数を減らすために、VX−222および何れかのさらなる薬剤は、任意の組み合わせで共に製剤され得る。何れかの別個の投与形態は、同時に、または異なる時間に投与され得る。投与形態は、その生物学的効果が有利になるように時間内に投与されるべきであることが理解されるべきである。
【0099】
例えば、本発明のVX−222およびVX−950の各量は、単一投与形態または2つ以上の投与形態で投与され得る。分割投与形態であるとき、各投与形態は、ほぼ同時に投与される。
【0100】
本発明において薬学的に許容される塩を治療的活性剤として用いるとき、それらの塩類は、典型的に、無機または有機酸およびそれらの塩基から誘導される。そのような酸性塩類には、酢酸塩、アジピン酸塩、アルギン酸塩、アスパラギン酸塩、安息香酸塩、ベンゼンスルホン酸塩、重硫酸塩、酪酸塩、クエン酸塩、ショウノウ酸塩(camphorate)、カンファースルホン酸塩(camphor sulfonate)、シクロペンタン−プロピオン酸塩、二グルコン酸塩、ドデシル硫酸塩、エタンスルホン酸塩、フマル酸塩、グルコヘプタン酸塩、グリセロリン酸塩、ヘミ硫酸塩、ヘプタン酸塩、ヘキサン酸塩、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、2−ヒドロキシエタンスルホン酸塩、乳酸塩、マレイン酸塩、メタンスルホン酸塩、2−ナフタレンスルホン酸塩、ニコチン酸塩、シュウ酸塩、パモ酸塩、ペクチン酸塩(pectinate)、過硫酸塩、3−フェニル−プロピオン酸塩、ピクリン酸塩、ピバリン酸塩、プロピオン酸塩、コハク酸塩、酒石酸塩、チオシアン酸塩、トシル酸塩およびウンデカン酸塩が含まれる。塩基性塩類には、アンモニウム塩、ナトリウムおよびカリウム塩のようなアルカリ金属塩、カルシウムおよびマグネシウム塩のようなアルカリ土類金属塩、ジシクロヘキシルアミン塩、N−メチル−D−グルカミンのような有機塩基との塩、ならびにアルギニン、リジンのようなアミノ酸との塩等が含まれる。
【0101】
また、塩基性窒素含有基は、塩化、臭化およびヨウ化メチル、エチル、プロピルおよびブチルのような低級ハロゲン化アルキル;硫化ジメチル、硫化ジエチル、硫化ジブチルおよび硫化ジアミルのような硫化ジアルキル;塩化、臭化およびヨウ化デシル、ラウリル、ミリスチルおよびステアリルのような長鎖ハロゲン化物;ベンジルおよびフェネチル臭化物のようなハロゲン化アラルキルなどのような物質によって四級化され得る。それにより、水または油に可溶性または分散性の生成物が得られる。
【0102】
本発明において、要すれば、治療剤(複数可)の修飾が可能であり、例えば、選択的生物学的特性を高めるのに適当な機能性を付加することにより修飾され得る。そのような修飾は、当技術分野で公知であり、所定の生物系(例えば、血液系、リンパ系、中枢神経系)への生物学的浸透を増加し、経口アベイラビリティーを増大し、注射による投与を可能にするための溶解性を増加し、代謝を変化させ、排泄速度を変化させることが含まれる。
【0103】
本発明のVX−222およびVX−950を含む1種以上の治療剤は、医薬組成物に包含され、該治療剤(複数可)は、単独で投与され得る。“医薬組成物”とは、本明細書に記載の治療剤を含む組成物を意味し、少なくとも1個の成分が、投与方法および投与形態の性質によって、薬学的に許容される担体、希釈剤、コーティング、アジュバント、賦形剤、または保存料のようなビークル、充填剤、崩壊剤、湿潤剤、乳化剤、乳化安定剤、懸濁化剤、等張剤、甘味剤、芳香剤、香味剤、着色剤、抗菌剤、抗細菌剤、他の治療剤、滑沢剤、吸収遅延または促進剤、ならびに分散剤を含む群から選択される。組成物は、錠剤、丸剤、顆粒、粉末、水性溶液または懸濁液、注射可能溶液、エリキシルまたはシロップの形態で存在し得る。
【0104】
懸濁化剤の例には、エトキシル化イソステアリルアルコール、ポリオキシエチレンソルビトールおよびソルビタンエステル、微結晶セルロース、アルミニウムメタヒドロキシド(aluminum metahydroxide)、ベントナイト、寒天−寒天およびトラガカント、ならびにこれらの物質の混合物が含まれる。微生物作用の予防のための抗菌剤および抗細菌剤の例には、パラベン、クロロブタノール、フェノール、ソルビン酸等が含まれる。等張剤の例には、糖類、塩化ナトリウム等が含まれる。吸収時間を延長するための吸収遅延剤の例には、モノステアリン酸アルミニウムおよびゼラチンが含まれる。吸収を増強するための吸収促進剤の例には、ジメチルスルホキシドおよび関連する類縁体が含まれる。担体、希釈剤、溶媒、ビークル、可溶化剤、乳化剤および乳化安定剤の例には、水、クロロホルム、スクロース、エタノール、イソプロピルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、テトラヒドロフルフリルアルコール、安息香酸ベンジル、ポリオール、プロピレングリコール、1,3−ブチレングリコール、グリセロール、ポリエチレングリコール、ジメチルホルムアミド、トゥイーン60、span@80、セトステアリルアルコール、ミリスチルアルコール、モノステアリン酸グリセロールおよびラウリル硫酸ナトリウム、ソルビタンの脂肪酸エステル、植物油(綿実油、落花生油、コーン油オリーブ油、ヒマシ油およびゴマ油)、およびオレイン酸エチルなどのような注射可能な有機エステル、またはこれらの物質の好適な混合物が含まれる。賦形剤の例には、ラクトース、乳糖、クエン酸ナトリウム、炭酸カルシウム、リン酸二カルシウムが含まれる。崩壊剤の例には、デンプン、アルギン酸およびある種のケイ酸塩複合体が含まれる。滑沢剤の例には、ステアリン酸マグネシウム、ラウリル硫酸ナトリウム、タルク、ならびに高分子量ポリエチレングリコールが含まれる。
【0105】
治療剤以外の医薬組成物中の物質の選択は、一般的に、溶解性のような治療剤の化学的特性、特定の投与方法および医薬業界で守られるべきによって決定される。例えば、ラクトース、クエン酸ナトリウム、炭酸カルシウム、リン酸二カルシウムのような賦形剤、デンプン、アルギン酸およびある種のケイ酸塩複合体のような崩壊剤、ならびにステアリン酸マグネシウム、ラウリル硫酸ナトリウムおよびタルクのような滑沢剤が、錠剤の製造に用いられ得る。
【0106】
医薬組成物は、錠剤、丸剤、顆粒、粉末、水性溶液または懸濁液、注射可能溶液、エリキシルまたはシロップのような種々の形態で存在し得る。
【0107】
“液体投与形態”は、患者に投与される治療剤の用量は、液体形態、例えば、薬学的許容されるエマルジョン、溶液、懸濁液、シロップおよびエリキシルを意味する。活性化合物に加えて、液体投与形態は、溶媒、可溶化剤および乳化剤のような当技術分野で常用される不活性希釈剤を包含し得る。
【0108】
固体組成物はまた、ラクトースまたはミルク糖のような賦形剤ならびに高分子量ポリエチレングリコールなどを用いて、軟および硬−充填ゼラチンカプセル中に充填剤として用いられ得る。
【0109】
水性懸濁液を用いるとき、それらは、懸濁を促進する乳化剤または薬剤を含み得る。
【0110】
エマルジョン医薬組成物の油相は、公知の方法に従い公知の成分から構成され得る。この相は単に乳化剤(別名エマルジェント(emulgent)として公知)を含み得るが、望ましくは、少なくとも1つの乳化剤と脂肪もしくは油との混合物、または脂肪および油の両方との混合物を含む。好ましくは、親水性乳化剤は安定剤として作用する親油性乳化剤と共に含まれる。また、好ましくは、油および脂肪の両方を含む。安定剤の有無にかかわらず、ともに乳化剤はいわゆる乳化蝋を構成し、該蝋は油および脂肪とともにクリーム製剤の油性分散相を形成する乳化軟膏基剤を構成する。
【0111】
要すれば、クリーム基剤の水相には、例えば、少なくとも30重量%の多価アルコール、すなわちプロピレングリコール、ブタン 1,3−ジオール、マンニトール、ソルビトール、グリセロールおよびポリエチレングリコール(PEG 400を含む)のような2個以上のヒドロキシル基を有するアルコールならびにそれらの混合物が含まれ得る。局所用製剤は望ましくは、皮膚または他の塗布領域を介する有効成分の吸収または浸透を増強する化合物を含み得る。
【0112】
製剤用の好適な油または脂肪の選択は、所望の美的(cosmetic)特性を達成することに基づく。故に、該クリームは、好ましくはチューブまたは他の容器からの漏れを避けるのに好適な濃度の、脂っこくなく、非着色で、かつ洗浄可能な製品であるべきである。
【0113】
直鎖または分枝鎖一または二塩基性アルキルエステル、例えばミリスチン酸ジイソプロピル、オレイン酸デシル、パルミチン酸イソプロピル、ステアリン酸ブチル、パルミチン酸2−エチルヘキシルまたはCrodamol CAPとして公知の分枝鎖エステルの混合物が使用され得る。これらは、必要な特性に応じて、単独でまたは組み合わせて使用され得る。あるいは、高融点脂質、例えば、白色軟質パラフィンおよび/または液体パラフィンまたは他の鉱油が使用され得る。
【0114】
一般的に、本明細書に記載の治療剤/医薬組成物は、好適な製剤形で、経口、吸入、直腸、経鼻、口腔(buccal)、舌下、経膣、経結腸、非経口(皮下、筋肉内、静脈内、皮内、鞘内、および硬膜外を含む)、嚢内および腹腔内を含む、局所または全身投与によりヒトおよび動物に投与され得る。好ましい経路は、例えばレシピエントの状態によって変わり得ることが認められ得る。
【0115】
“薬学的に許容される投与形態”は、本明細書に記載の治療剤の投与形態(VX−950を含む)を意味し、例えば、錠剤、粉末、エリキシル剤、シロップ、液体製剤(懸濁液、スプレー、吸入剤、錠剤、ロゼンジ、エマルジョン、溶液、顆粒、カプセルおよび坐薬を含む)、ならびにリポソーム製剤を含む注射用液体製剤が含まれる。一般に、製剤技術および製剤は、remington’s pharmaceutical sciences, mack publishing co., easton, pa, latest editionに見出され得る。
【0116】
“経口投与に好適な製剤”は、所定量の有効成分をそれぞれ含むカプセル剤、カシェ剤または錠剤として;粉剤または顆粒として;水性液体または非水性液体中の溶液または懸濁液として;または、油中水液体エマルジョンもしくは水中油液体エマルジョンのような分離した単位として提供され得る。有効成分はまた、ボーラス剤、舐剤またはペーストとして提供され得る。
【0117】
錠剤は、圧縮または成型により、必要に応じて1種以上の補助成分とともに、製造され得る。圧縮された錠剤は、好適な機械中で有効成分を自由流動形態(例えば、粉剤または顆粒)で、必要に応じてバインダー、潤滑剤、不活性希釈剤、防腐剤、表面活性剤または分散剤と混合し、圧縮することにより製造され得る。成型された錠剤は、好適な機械中で、不活性な液体希釈剤で湿らされた(moistened)粉末化された化合物の混合物を成型することにより製造され得る。錠剤は、必要に応じて、コーティングまたは割線を施され得、そして錠剤中の有効成分の遅いまたは制御された放出を提供するように処方され得る。
【0118】
直腸投与用の固体組成物は、公知の方法で製剤される坐薬および少なくとも1個の本発明の化合物を含む。
【0119】
要すれば、より有効な分散性に関して、本明細書に記載の治療剤は、マイクロカプセル化されるか、または生体適合性の、生分解可能なポリマーマトリクス(例えば、ポリ(d,l−ラクチド コ−グリコチド))、リポソームおよびミクロスフェアのような遅延放出また標的送達システムと結合され得て、いわゆる皮下または筋肉内デポー技術により皮下または筋肉内に注射されて、2週間またはそれ以上の間、化合物をゆっくり放出し続ける。治療剤は、例えば、細菌保持フィルターを介して濾過されるか、または使用直前に滅菌水、または他の滅菌注射可能媒体に溶解され得る、滅菌固体組成物形態に滅菌剤を挿入されて、滅菌され得る。
【0120】
“経鼻または吸入投与に適する製剤”は、患者に経鼻的に、または吸入により投与されるのに適する形態の製剤を意味する。該製剤は、例えば1ないし500ミクロンの範囲の粒子サイズ(30ミクロン、35ミクロンなどのような5ミクロン増加の20ないし500ミクロンの範囲の粒子サイズを含む)を有する粉末形態の担体を含み得る。担体が、例えば、経鼻スプレーまたは点鼻薬として投与するための液体である好適な製剤は、有効成分の水性または油性溶液を含む。エアロゾル投与に適する製剤は、常套法に従って製造され得て、他の治療剤と共に送達され得る。吸入治療剤は、定量吸入器により容易に投与される。
【0121】
“経口投与に好適な製剤”は、患者に経口的に投与されるのに好適な形態の製剤を意味する。該製剤は、一定量の有効成分をそれぞれ含むカプセル、カシェ剤または錠剤のような;粉末もしくは顆粒として;水性液体もしくは非水性液体中、溶液もしくは懸濁液のような;または、油中水液体エマルジョンもしくは水中油液体エマルジョンのような分離した単位として存在し得る。治療剤はまた、ボーラス剤、舐剤またはペーストとしても存在し得る。
【0122】
“非経腸投与に好適な製剤”は、患者に非経腸的投与されるのに好適な形態の製剤を意味する。該製剤は、滅菌され、エマルジョン、懸濁液、水性および非水性注射溶液を含み、それらは、懸濁化剤および濃化剤および抗酸化剤、緩衝剤、バクテリオスタットならびに製剤を意図されるレシピエントの血液と等張にし、pHを適切に調節する溶質を含む。
【0123】
“経直腸または経膣投与に好適な製剤”は、患者に経直腸または経膣投与するのに適する形態の製剤を意味する。該製剤は、好ましくは、本発明の化合物と好適な非刺激性の賦形剤または担体、例えばカカオバター、ポリエチレングリコールまたは坐薬ワックス(こでは、常温では固体であるが、体温では液体であり、故に直腸または膣腔で融解し、有効成分を放出する)を混合することにより製造され得る坐薬の形態である。
【0124】
“全身投与に好適な製剤”は、患者に全身的に投与されるのに適する形態の製剤を意味する。該製剤は、好ましくは筋層内、静脈内、腹腔内および皮下を含む注射により投与される。注射に関して、本発明の化合物は、液体溶液、好ましくは、ハンク溶液またはリンゲル溶液のような生理的に許容可能な緩衝液に製剤される。さらに、該化合物は、固体形態で製剤され得て、使用直前に再溶解されるか、または懸濁され得る。凍結乾燥形態もまた包含される、全身投与はまた、経粘膜または経皮手段によっても可能であるか、または化合物は経口投与され得る。経粘膜または経皮投与に関して、透過されるバリアに適する浸透剤が製剤に用いられる。かかる浸透剤は一般的に、当技術分野で公知であり、例えば、経粘膜投与のための胆汁酸塩およびフシジン酸誘導体を含む。さらに、洗浄剤が浸透を容易にするために用いられ得る。経粘膜投与には、経鼻スプレー、または例えば坐薬が使用され得る。経口投与に関して、該化合物は、カプセル、錠剤およびトニック剤のような常套の経口投与形態に製剤される。
【0125】
“局所投与に好適な製剤”は、患者に局所的に投与されるのに適する形態の製剤を意味する。該製剤は、局所用軟膏、軟膏(salve)、粉剤、スプレーおよび吸入剤、ゲル(水またはアルコール基剤)、クリームとして、当技術分野で通常知られている形態で存在し得るか、または経皮バリアを介して化合物の制御放出を可能にし得る、パッチにおける適用のためのマトリックスベース中に挿入され得る。軟膏形態に製剤されるとき、有効成分は、パラフィン性または水性の混合可能な軟膏基剤と共に使用され得る。あるいは、有効成分は、油中水クリーム基剤と共にクリームに製剤され得る。眼への局所投与に適する製剤には、有効成分が溶解されるか、または好適な担体、とりわけ有効成分用の水性溶媒中に懸濁される点眼剤が含まれる。口への局所投与に適する製剤には、風味基剤、通常スクロースおよびアカシアまたはトラガカント中に有効成分を含むロゼンジ;ゼラチンおよびグリセリン、またはスクロースおよびアカシアのような不活性基剤中に有効成分を含むトローチ;ならびに、好適な液体担体中に有効成分を含むマウスウォッシュが含まれる。
【0126】
“固体投与形態”は、本明細書に記載の治療剤の投与形態が固体形態、例えばカプセル剤、錠剤、丸剤、粉剤、糖衣錠または顆粒であることを意味する。かかる固体投与形態において、本発明の化合物は、クエン酸ナトリウムまたはリン酸二カルシウムのような少なくとも1種の不活性な慣習的な賦形剤(または担体)、または(a)充填剤もしくは増量剤、例えば、デンプン、ラクトース、スクロース、グルコース、マンニトールおよびケイ酸、(b)結合剤、例えば、カルボキシメチルセルロース、アルギネート類(alignate)、ゼラチン、ポリビニルピロリドン、スクロースおよびアカシア、(c)湿潤剤、例えば、グリセロール、(d)崩壊剤、例えば、寒天−寒天、炭酸カルシウム、ジャガイモまたはタピオカデンプン、アルギン酸、任意のケイ酸塩および炭酸ナトリウムの混合物、(e)溶液抑制剤、例えばパラフィン、(f)吸収促進剤、例えば、第四級アンモニウム化合物、(g)湿潤剤、例えば、セチルアルコールおよびモノステアリン酸グリセロール、(h)吸着剤、例えば、カオリンおよびベントナイト、(i)潤滑剤、例えば、タルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体ポリエチレングリコール、ラウリル硫酸ナトリウム、(j)乳白剤、(k)緩衝剤、および腸管のある部分で本発明の化合物を遅延放出する薬剤と混合される。
【0127】
担体および/または賦形剤と結合されて単一投与形態を製造し得る有効な治療剤の量は、処置される宿主および特定の投与方法によって変わり得る。典型的な製剤は、約5%ないし約95%の有効治療剤(w/w)を含み得る。好ましくは、かかる製剤は、治療剤を約20%ないし約80%含む。
【0128】
製剤は、薬剤学分野で周知の何れかの方法により単位投与形態で製造され得る。そのような方法には、有効成分を1個以上の補助成分を構成する担体と合わせる工程が含まれる。一般的に、該製剤は、有効成分を液体担体または微細に分割した固体担体またはその両方と合わせて、その後、必要であれば、製品を成形して、均一かつ密に結合して製造される。
【0129】
製剤は、単一用量または複数用量の容器、例えば弾性ストッパーを有する密閉アンプルおよびバイアル中に存在し得て、使用直前に滅菌液体担体、例えば注射用水の添加のみを要する凍結乾燥状態で貯蔵され得る。即時調製の注射溶液および懸濁液は、上記の種類の滅菌粉末、顆粒および錠剤から製造され得る。
【0130】
本明細書に記載の医薬組成物および用量製剤は、好ましくはインビボでの使用のためのものである。それにも関わらず、何れかの目的のための医薬組成物および用量製剤の使用に限定されることを意図しない。例えば、本明細書に記載の医薬組成物で予め処理された生物学的物質もまた、本発明に使用され得る。かかる生物学的物質には、血液およびその成分、例えば血漿、血小板、血液細胞の亜集団など;腎臓、肝臓、心臓、肺などのような臓器;精子および卵子;骨髄およびその成分、ならびに食塩水、デキストロースなどのような患者に注入される他の液体が含まれるが、これらに限定されない。
【0131】
何れかの特定の患者に対する特定の投与量および処置レジメンが、用いる特定の化合物の活性、年齢、体重、一般的健康状態、性別、食事、投与時間、排泄速度、混合薬、担当医の判断、および処置すべき特定の疾患の重症度、これまでの治療歴、共存疾患または併用薬、ベースラインのウイルス量、人種、疾患の期間、肝機能の状態および肝線維症/肝硬変の程度、ならびに治療目標(移植によるウイルス循環の排除またはウイルスの根絶)を含む、様々な因子によって変わり得ることも理解されるべきである。有効成分の量は、特定の記載した化合物、ならびに組成物中のさらなる抗ウイルス剤の存在または不存在、および性質によっても変わり得る。
【0132】
本発明の処置レジメンおよび用量形態に従い、VX−950およびインターフェロンの共投与は、サンプルまたは患者における、ウイルスの生活環に必要なNS3/4Aセリンプロテアーゼをコードする(または、本発明の実施に有効な量の)ウイルスを有効に減少させる。従って、本発明はまた、ウイルスの生活環に必要なNS3/4Aセリンプロテアーゼをコードするウイルスにより特徴付けられるウイルスに感染した患者の、上記のVX−950およびインターフェロン(および、所望により1種以上のさらなる治療剤)の投与による処置方法を提供する。
【0133】
本発明において、本発明に用いられる各活性治療剤は、独立して、食事の有無で患者に投与され得る。いくつかの態様において、VX−222および/または何れかのさらなるHCV薬は独立して、食事と共に投与される。本明細書で用いる用語“食事と組み合わせて”は、有効治療剤(複数可)が食事の食べ終わり約90分以内、例えば、食事後の約90分以内および食事前の約90分以内に投与されることを意味する。いくつかの態様において、有効治療剤(複数可)は、食事の食べ終わり前約30分まで、または食べ終わり後30分までに投与される。必要ではないが、全ての種類の食品(高脂肪および低脂肪食)が摂取され得て、高脂肪食は、より低脂肪食と比較して、改善された吸収を提供し得る。本明細書で用いる“高脂肪”は、約30%以上のカロリーが脂肪により提供される食事を意味する。任意の態様において、食事は、少なくとも約50カロリーを有する。他の任意の態様において、食事は、少なくとも約100カロリーである。さらに他の任意の態様において、食事は、少なくとも約50−100カロリーないし約3,000カロリーまで、約2,000カロリーまで、または約1,000カロリーまでである。さらに他の任意の態様において、食事には、脂肪からの、その総カロリーの少なくとも約30%が含まれる。
【0134】
一般に、本発明において、有効治療応答が達成されるように、処置はHCVウイルス感染を完全に根絶するか、またはその重症度を低減させ得る。有効治療応答は、例えば、a)継続的なウイルス応答を達成している;およびb)血漿中、検出不可能なHCV RNAを少なくとも約12週間(約12週間またはそれ以上)達成しているの、一方または両方であり得る。用語“検出不可能な”は、上記に定義の通りである。
【0135】
他の態様において、本発明の方法は、投与後の患者のHCV RNAレベルが、処置前よりも少なくとも約2log10(例えば、少なくとも約4log10)低下するように、HCVに感染した患者を処置する。
【0136】
いくつかの態様において、ウイルス血漿濃度の比較的迅速な低下は、患者に負荷用量の投与を行うことにより得られ得る。一態様において、該負荷用量は、約1250mgのVX−950である。
【0137】
いくつかの態様において、本発明の方法は、4週目のRVRおよび12週目の検出不可能な状態を達成できる。
【0138】
一般に、本発明において、“患者”には、哺乳動物、特にヒトが含まれる。
【0139】
ある態様において、本発明の方法は、遺伝子型1型C型肝炎ウイルスに感染した患者の処置を提供する。遺伝子型1型HCV感染は、アメリカ合衆国において、処置の最も困難なHCV株であり、最も蔓延している株である。
【0140】
有利には、両HCV未処置の患者および以前に処置された患者は、本発明の方法に有益である。誤解を避けるために、本発明の方法で処置され得る患者には、HCV処置が、試されなかった患者か、または、非応答、リバウンド、再発および進行した患者を含む、処置に失敗した患者が含まれる。任意の態様において、本発明の方法は、HCV未処置の患者を処置する。本明細書で用いる“HCV未処置”患者は、アメリカ食品医薬品局(FDA)または他のアメリカの機関もしくはFDAと同等の国際的機関によって承認されたか、または承認申請中の薬剤で以前にHCV処置を行なわれていないことを意味する。
【0141】
本発明の方法は、長期または短期療法として用いられ得る。本発明の方法が、患者を予防的に処置するのに使用され、該患者がC型肝炎ウイルスに感染した患者となるとき、該方法は、その後、感染を処置し得ることは、当業者に容易に明らかであり得る。故に、本発明の一態様は、患者におけるC型肝炎感染の処置または予防のための方法を提供する。
【0142】
患者の血漿中のVX−222およびVX−950濃度を決定するためのアッセイは、当技術分野で公知の方法により行った。例えば, Wasley, A. Et al., Semin. Liver Dis., 20: 1−16, 2000; Alter, H.J. et al., Semin. Liver dis., 20: 17−35, 2000; Brown, R.S. Jr. et al., Liver Transpl., 9: S10−S13, 2003; Defrancesco, R. et al., Nature, 436(7053): 953−960, 2005; Bowen, D.G. et al., J. Hepatol., 42: 408−417, 2005; Hoofnagle, J.H., Hepatology, 36: S21−S29, 2002, Brown, R.S. Jr. et al., Nature, 436 (7053): 973−978, 2005; および、Chisari, F.V., Nature, 436(7053): 930−932, 2005を参照のこと。
【0143】
本発明に関係する投与を、長期または短期療法として用い得る。単一投与形態を製造するために担体物質と併用され得る有効成分の量は、処置される宿主および特定の投与方法によって変わり得る。典型的に製剤は、約5%ないし約95%の有効化合物(w/w)を含み得る。好ましくは、かかる製剤は、約20%ないし約80%の有効化合物を含み得る。
【0144】
患者の状態の改善後は、必要ならば、本発明の化合物、組成物または組み合わせの維持量が投与され得る。次いで、投与の量あるいは頻度、または両方を、その症状の関数として、症状が所望のレベルに軽減され、処置を終えるべきときに、改善された状態が維持されるレベルに減らし得る。しかしながら、患者は、何らかの疾患症状の再発により長期的に断続的な処置を必要とし得る。
【0145】
何れかの特定の患者に対する特定の投与量および処置レジメンが、用いる特定の化合物の活性、年齢、体重、一般的健康状態、性別、食事、投与時間、排泄速度、混合薬、担当医の判断、および処置すべき特定の疾患の重症度、これまでの治療歴、共存疾患または併用薬、ベースラインのウイルス量、人種、疾患の期間、肝機能の状態および肝線維症/肝硬変の程度、ならびに治療目標(移植によるウイルス循環の排除またはウイルスの根絶)を含む、様々な因子によって変わり得ることも理解されるべきである。有効成分の量は、特定の記載した化合物、ならびに組成物中のさらなる抗ウイルス剤の存在または不存在、および性質によっても変わり得る。
【0146】
他の態様によれば、本発明は、本発明の薬学的に許容される組成物を該患者に投与することにより、ウイルスの生活環に必要なNS3/4Aセリンプロテアーゼをコードするウイルスにより特徴付けられるウイルスに感染した患者の処置方法を提供する。好ましくは、本発明の方法は、HCV感染を有する患者の処置に用いられる。かかる処置は、ウイルス感染を完全に根絶するか、またはその重症度を低減し得る。好ましくは、該患者は哺乳動物である。より好ましくは、該患者はヒトである。
【0147】
本明細書に記載の投与量は、好ましくはインビボでの使用用である。それにもかかわらず、これは目的に応じてこれらの量の、例えばVX−222またはVX−950の使用に限定することを意図していない。さらに別の態様において、本発明は、生物学的物質と、本発明の化合物を含む薬学的に許容される組成物を合わせる段階を含む、患者に投与することを目的として該生物学的物質を前処理する方法を提供する。かかる生物学的物質には、血液および血漿、血小板、血液細胞の亜集団などのその成分;腎臓、肝臓、心臓、肺などの臓器;精子および卵子;骨髄およびその成分、ならびに食塩水、デキストロースなどのような患者に注入される他の液体が含まれるが、それらに限定されない。
【0148】
本発明はまた、VX−222、VX−950、および薬学的に許容される担体、アジュバント、またはビヒクルを組み合わせる工程を含む、VX−222、VX−950、および薬学的に許容される担体、アジュバント、またはビヒクルを含む組成物を製造するための方法を提供する(ここで、組成物中のVX−222およびVX−950の投与量は、本発明の何れかの態様に従う。)。本発明の別の態様は、該組成物が、本明細書に記載の1個以上のさらなる薬剤を含む方法を提供する。
【0149】
医薬組成物はまた、単一パッケージ、通常ブリスターパッケージに一連の処置全体を含む“患者パック(patient pack)”で患者に処方され得る。患者パックには、従来の処方薬には通常添付されていない添付文書が含まれていて患者がいつでも利用できる点で、薬剤師が、大量供給物(bulk supply)から患者への薬剤分配を行なう従来の処方よりも有利である。添付文書の包含は、患者に医者の指示の順守を改善させることが証明されている。
【0150】
患者が本発明の正しい使用をするように指示する添付文書を包含する単一の患者パック、または各製剤の患者パックを用いる本発明の併用投与は、本発明の望ましいさらなる特徴であることが理解され得る。
【0151】
さらなる一面によれば、本発明は、VX−222(本発明の投与量)および本発明の併用使用の指示書を含む添付情報を含むパッケージである。全ての組成物、投与形態、治療レジメンまたは本発明の他の態様は、医薬パッケージ中に存在し得る。本発明の別の態様において、該医薬パッケージは、本明細書に記載の1個以上のさらなる薬剤をさらに含む。さらなる薬剤または複数の薬剤は、同一のパッケージまたは異なるパッケージで提供され得る。
【0152】
本発明の他の一面は、単一または複数の各医薬成分の製剤;貯蔵中および投与前に製剤(複数可)を保存する容器;ならびに、HCV感染の処置または予防のための有効な方法で薬剤投与を行うための指示書を含む、HCV感染の処置またはHCV感染の予防における使用のための(または、本発明の他の方法における使用のための)患者用のパッケージ化されたキットを含む。
【0153】
従って、本発明は、VX−222(および、所望によりさらなる薬剤)の用量の同時または連続投与のためのキットを提供する。典型的に、そのようなキットは、例えば、薬学的に許容される担体中(および、単一または複数の製剤中)、各化合物の組成物および任意のさらなる薬剤(複数可)、ならびに同時または連続投与のための指示書を含み得る。
【0154】
別の態様において、パッケージキットは、自己投与のための1個以上の投与形態;貯蔵中および使用前に該投与形態を保存するための、好ましくは密閉された容器;および、薬剤投与を行うための患者への指示書を包含して提供される。該指示書は、一般的に、添付文書、ラベル、および/またはキットの他の成分の指示が記載され、投与形態または複数の投与形態が、本明細書中に記載されている。各投与形態は、一枚の金属箔−プラスチック薄板で形成される個々の空間(cell)または半球形(bubble)中に、他から分離されたそれぞれの投与形態を包含させるか、または投与形態は、プラスチック容器のような単一の容器中に包含され得る。本発明のキットはまた、典型的には、個々のキット成分をパッケージングするための手段、すなわち、投与形態、容器手段、および使用のための指示書を含み得る。そのようなパッケージング手段は、段ボール箱または紙箱、プラスチック容器または金属箔容器などの形態であり得る。
【0155】
本発明のキットは、何れかの組成物、投与形態、治療レジメンまたは医薬パッケージのような、本発明のいずれかの一面を具現化し得る。
【0156】
本発明のパッケージおよびキットは、所望により、複数の組成物または投与形態を含む。従って、1個の組成物または2個以上の組成物を含むパッケージおよびキットは、本発明の範囲内に包含され得る。
【0157】
ある例示的態様は、以下に説明され、記載されているが、本発明の化合物が、当業者に一般的に利用可能な適当な出発物質を用いて、一般に上記の方法に従い製造され得ることは、当然のことである。
【0158】
VX−222は、一般的に、当業者に周知の方法で製造され得る(例えば、WO2002/100851およびWO2008/058393を参照のこと)。当技術分野で周知の全ての好適な製剤が、本発明において使用され得る。例えば、WO2002/100851およびWO2008/058393に記載の製剤が、本発明で使用され得る。本発明で使用され得る1つの特定の例には、VX−222の遊離酸形; Avicel PH 101;ラクトース一水和物;Poloxamer 188;ラウリル硫酸ナトリウム;プロビドンK29/32; Avicel PH 102;ラクトース一水和物;クロスカルメロースナトリウム;ステアリン酸マグネシウムが含まれる。本発明で用いられ得る特定の製剤は、実施例5に例示される。
【0159】
本発明の一態様は、VX−222の遊離酸形(構造式(I)で示される化合物); Avicel PH 101;ラクトース一水和物; Poloxamer (例えば、Polxamer 188);ラウリル硫酸ナトリウム;プロビドン K29/32; Avicel PH 102;ラクトース一水和物;クロスカルメロースナトリウム;ならびに、ステアリン酸マグネシウムを含むVX−222の製剤である。特定の態様において、製剤は、約45−60重量%のVX−222の遊離酸形;約5−20重量%のAvicel PH 101;約10−20重量%のラクトース一水和物;約1−10重量%のPoloxamer(例えば、Polxamer 188);約1−5重量%のラウリル硫酸ナトリウム;約1−10重量%のProvidone (例えば、プロビドンK29/32);約1−10重量%のAvicel PH 102;約1−10重量%のラクトース一水和物;約1−10重量%のクロスカルメロースナトリウム;ならびに、約0.1−5重量%のステアリン酸マグネシウムを含む。実施例5に記載の製剤はまた、本発明に包含される。
【0160】
VX−950は、一般的に、当業者に周知の方法で製造され得る(例えば、WO02/18369を参照のこと)。当技術分野で周知の全ての好適な製剤が、本発明において使用され得る。例えば、WO2005/123075、WO2007/109604、WO2007/109605およびWO2008/080167に記載の製剤が本発明で使用され得る。本発明で使用され得る特定の製剤は、実施例4に例示される。他の具体的な例は、以下を含む:
【表1】
【表2】
【表3】
【表4】
ここで、HPMC(ヒドロキシプロピルメチルセルロース 60SH 50cP (Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)(Hypromellose Acetate Succinate, HG grade, Shin−Etsu Chemical Co.)、HPC(ヒドロキシプロピルセルロース)、PVP(ポリビニルピロリドン)およびSLS(ラウリル硫酸ナトリウム)は、WO2005/123075に記載されている。任意の態様において、上記の固体分散体は、1%HPMC、0.002%シメチコン溶液(1重量%のHPMC、0.002重量%のシメチコンおよび99重量%の水)に懸濁され得る。さらなる例には、1:1 VX950:PVPK30、1重量%のSLS(Refreshed Tox.);Niro−49重量% HPMCAS/1重量% SLS/1重量% SDBS/49%VX−950;40.5重量% PVP−VA/10重量% ETPGS/49.5重量% VX−950;40.5重量% HPMC/10重量% ETPGS/49.5重量% VX−950;49重量% VX950、49重量% HPMCAS、1重量% SLS、1重量% SDBS;および、49重量% VX950、16重量% HPPh、33重量% HPC、1重量% SLS、□重量% SDBS(ここで、PVPK30(ポリビニルピロリドンK30)、SDBS(ドデシルベンゼンスルホン酸ナトリウム)、HPMCAS(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、ビタミンETPGS、PVP(ポリビニルピロリドン)およびSLS(ラウリル硫酸ナトリウム)、ならびにこれらの製剤の製造の詳細は、WO2005/123075に見出され得る)を含む。さらなる例には、WO2007/109604に記載のものが含まれる:
【0161】
55重量% VX−950、24.4重量% HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、19.6重量% HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量% ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
55重量% VX−950、14.7重量% HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、29.3重量% HPMC−60SH(ヒドロキシプロピルメチルセルロース60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
60重量% VX−950、24.4重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、14.6重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量% ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
65重量%VX−950、17重量% HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、17重量% HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
70重量% VX−950、9.7重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、19.3重量% HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP (Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量% ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
【0162】
60重量% VX−950、39重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
49.5重量% VX−950、24.5重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、24.5重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
83重量% VX−950、8重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、8重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
49.5重量%VX−950、24.5重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、24.5重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
70重量%VX−950、14.5重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、14.5重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
【0163】
65重量%VX−950、14.6重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、19.4重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
65重量%VX−950、9.7重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、24.3重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
60重量%VX−950、19.5重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、19.5重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
60重量%VX−950、14.6重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、24.4重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
【0164】
70重量%VX−950、9.7重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、19.3重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
49.5重量%VX−950、24.5重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、24.5重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
83重量%VX−950、8重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、8重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
49.5重量%VX−950、49.5重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
【0165】
83重量%VX−950、16重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
82.44重量%VX−950、15.89重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、および1.67重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
49.5重量%VX−950、24.75重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、24.75重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
60重量%VX−950、24.6重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード),14.4重量%HPMC−60SH(ヒドロキシプロピルメチルセルロース 60SH 50cP(Biddle SawyerまたはShin−Etsu Metolose, HPMC60SH50)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;
【0166】
60重量%VX−950、39重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体;ならびに、
49.5重量%VX−950、49.5重量%HPMCAS−HG(ヒドロキシプロピルメチルセルロースアセテートサクシネート)、JPE(Biddle SawyerまたはShin−Etsu HPMCAS−HG グレード)、および1重量%ラウリル硫酸ナトリウム(SLS)を含む固体分散体。
【0167】
これらの固体分散体の製造の詳細は、WO2007/109604に記載されている。さらなる具体例には、WO2007/109604に記載のVX−950のスプレー乾燥分散体を含む錠剤製剤が含まれる:
【表5】
【0168】
さらなる具体例には、WO2008/080167に記載の錠剤製剤が含まれる:
【表6】
【表7】
【0169】
【表8】
【表9】
【表10】
【0170】
全ての引用文献は、引用により本明細書中に包含される。
【0171】
本発明をより十分に理解するために、以下の製造例および実験例を記載する。これらの実施例は、説明のみを目的とし、いかなる点でも本発明の範囲を限定するものと解釈してはならない。
【実施例】
【0172】
実施例
以下の実施例において、VX−222は化合物1を、VX−950は化合物2を意味する。
実施例1:HCV レプリコン細胞アッセイプロトコール
C型肝炎ウイルス(HCV)レプリコンを含む細胞を、10%ウシ胎仔血清(FBS)、0.25mg/mLのG418、適当な添加剤を含むDMEM(媒体A)中で維持した。
【0173】
1日目に、レプリコン細胞単層を、トリプシン:EDTA混合物で処理し、除去し、その後、媒体Aを最終濃度100,000細胞/mLまで希釈した。10,000細胞/100μLを、96ウェル組織培養プレートの各ウェル中に播種し、37℃にて、組織培養インキュベーター中で一晩培養した。
【0174】
2日目、化合物(100%DMSO中)を、2%FBS、0.5%DMSO、適当な添加剤を含むDMEM(媒体B)中に連続的に希釈した。DMSOの最終濃度を、一連の希釈を通して0.5%に維持した。
【0175】
レプリコン細胞単層上の媒体Aを除去し、その後、種々の濃度の化合物を含む媒体Bを添加した。化合物を含まない媒体Bを、対照として他のウェルに添加した。
【0176】
細胞を、37℃の組織培養インキュベーター中、媒体B中化合物または0.5%DMSOを、48時間インキュベートした。48時間のインキュベーションの最後に、媒体Bを除去し、レプリコン細胞単層をPBSで一度洗浄し、RNA抽出前に−80℃で貯蔵した。
【0177】
処理したレプリコン細胞単層を含む培養プレートを解凍し、一定量のウシウイルス性下痢ウイルス(BVDV)のような別のRNAウイルスを、各ウェルにて細胞に添加した。RNA抽出試薬(RNeasyキットの試薬のような)を、RNA分解を避けるために細胞に直ちに添加した。全RNAを、抽出効率および一貫性を改善するための改良を伴う、製造業者の指示書に従って抽出した。最後に、HCVレプリコンRNAを含む全細胞RNAを溶出させ、さらなる処理まで−80℃で貯蔵した。
【0178】
TaqmanリアルタイムRT−PCR定量アッセイを、特定のプライマーおよびプローブの2個のセットを用いて行った。1つは、HCVについて、もう1つは、BVDVについて行った。処理したHCVレプリコン細胞由来の全RNA抽出物を、同じPCRウェル中に、HCVおよびBVDV RNAの両方の定量のために、PCR反応に追加した。実験的失敗に印をつけ、各ウェル中のBVDV RNAレベルを基に却下した。各ウェル中のHCV RNAレベルを、同じPCRプレートで実行する標準曲線に従い計算した。VX−950処理によるHCV RNAレベルの阻害または減少の割合を、DMSOを用いて計算し、VX−950不含有対照を0%阻害とした。IC50(HCV RNAレベルの50%阻害が見られる濃度)を、いずれかのVX−950濃度の滴定曲線から計算した。
【0179】
VX−950は、レプリコンアッセイにおいて顕著な活性を有することが示された。VX−950は、240ng/mLのIC50および476ng/mLのIC90を有した。
【0180】
実施例2:HCV Ki アッセイプロトコール
5AB基質および生成物の単離のためのHPLCマイクロボア法
基質:
NH2−Glu−Asp−Val−Val−(α)Abu−Cys−Ser−Met−Ser−Tyr−COOH(配列番号1)
【0181】
20mM 5AB(または、各自の選択濃度)の原液を、DMSOおよび0.2M DTTで作製した。これを−20℃にてアリコートで貯蔵した。
緩衝液:50mM HEPES、pH7.8;20%グリセロール;100mM NaCl。
【0182】
総アッセイ容量は、100μlであった:
【表11】
【0183】
緩衝液、KK4A、DTT、およびtNS3を合わせ;96ウェルプレートの各ウェルに各78μLずつ分配した。これを、30℃で約5分ないし10分間インキュベートした。2.5μLの適当な濃度の試験化合物を、DMSO中に溶解し(DMSOのみを対照とする)、各ウェルに添加した。これを、室温で15分間インキュベートした。20μLの250μM 5AB基質の添加により反応を開始させた(25μM濃度は、5ABのKm値と等しいか、わずかに低い)。得られた混合物を30℃で20分間インキュベートし、その後、25μLの10%TFAを添加して反応を終了させ、そして分析のために混合物をHPLCバイアルに移した(120μLアリコート)。SMSY産物を、以下の方法により、基質およびKK4Aから単離した:
【0184】
マイクロボア単離法
装置:Agilent 1100
Degasser G1322A
Binary pump G1312A
Autosampler G1313A
Column thermostated chamber G1316A
Diode array detector G1315A
カラム:
Phenomenex Jupiter;5マイクロン C18;300オングストローム;150×2mm;P/O 00F−4053−B0
カラムサーモスタット:40℃
注入量:100μL
溶媒A=HPLC等級純水+0.1%TFA
溶媒B=HPLC等級アセトニトリル+0.1%TFA
【0185】
【表12】
【0186】
実施例3
VX−950を、無作為化、二重盲検、プラセボ−対照の単回用量漸増試験において試験した。25名の健康な男性ボランティアが登録された。各対象は、VX−950の複数の単回用量を少なくとも7日間、3種の用量のVX−950を増加用量レベルで投与され、ならびに1用量のプラセボを投与された。
【0187】
25mgないし1250mgの用量を評価した。2倍ずつ増量しながら、より低用量範囲で攻撃的であり、より高用量範囲で保守的になるようにフィボナッチ数を改変した、用量漸増スキームを用いた。
【0188】
VX−950は、全ての用量レベルで良好な耐容性であるという結果を示した。重大な有害事象は試験中には報告されなかった。そして、用量レベルの増加により有害事象が増大することはなかった。
【0189】
薬物動態分析を、統計学的モーメント手法を用いて行った。薬物動態分析は、VX−950が、3時間の平均tmaxで吸収されたことを示した。2%未満のVX−950は尿中で変化なく除かれ、薬剤が、主に代謝経路から排泄されることを示した。
【0190】
実施例4
経口投与製剤を下記の通りに製造した。VX−950およびpovidone K29/32を塩化メチレン中に溶解し、次いでラウリル硫酸ナトリウムを添加し、溶液中に分散させて、均一な懸濁液を形成した。この懸濁液を、90℃の入口温度および56℃の出口温度を用いてスプレー乾燥させて、生成物をサイクロンから集めた。スプレー乾燥分散体を、75℃にて8時間、流動床乾燥させた。得られた粉末を、ガラスバイアル中で予め測定し、投与直前に、対象への投与のために水(30mL)に懸濁した。投与に関して、各バイアルを、全量が90mLである水の3分割量で洗浄した。
【表13】
【0191】
実施例5
VX−222(本明細書中、化合物1)の2つの異なる経口投与形態を以下の通りに製造した。
【表14】
【表15】
【0192】
VX−222(本明細書中、化合物1)のフォームAの任意の特性は以下の通りである:
【表16】
【0193】
XRPDパターンを、密封チューブ源およびHi−Star 領域検出器を備えるBruker D8 Discover 回折計(Asset Tag V012842)(Bruker AXS, Madison, WI)を、室温にて、反射モードで用いることにより得ることができる。X線発生器を、40kVおよび35mAの電圧で操作した。該粉末サンプルを、アルミニウムホルダー上に置いた。2個のフレームを、フレーム当たり120秒の露光時間で記録した。データを0.02o 2θの刻み幅の4o−40o 2θの範囲で統合し、1つの連続したパターンにまとめた。
【0194】
VX−222のフォームAを、以下に記載の工程に従い製造し得る:
−10gのVX−222(WO2008/058393に記載の通りに製造した化合物1)を反応器に入れる。
−20gのメタノールを加え、60℃まで加熱して溶解させる。
−10℃まで冷却し、固体が形成するまで待つ。
−固体を濾過する。
−20gのアセトンを25℃で添加する。
−1時間撹拌する。
−固体を濾過する。
−75℃で12時間乾燥させる。
【0195】
実施例6:実験VX−222−002からの薬物動態データ:VX−222およびVX−950の併用処置
VX−222について実施例5に記載の製剤を用いた。実験において、20名の健康な対象をコホート1またはコホート2(1コホート当たり10名の対象)に登録した。コホート1およびコホート2の対象は、実験の3つの期間の全て、処置期間1、処置期間2および処置期間3を完了した。処置期間1において、対象は、VX−222またはVX−222プラセボを食事と共に10日間投与された。処置期間2において、対象は、VX−950またはVX−950プラセボを食事と共に10日間投与された。処置期間3において、対象は、同時に、VX−222およびVX−950の両方またはVX−222プラセボおよびVX−950プラセボの両方を食事と共に10日間投与された。VX−222を処置期間1に投与し、VX−950を処置期間2に投与するために、処置期間1および処置期間2を7日間のウォッシュアウト期間で分けた。コホート1の対象に、処置期間1および3中、400mgのVX−222またはVX−222プラセボを12時間毎(q12h)に投与した。コホート2の対象に、処置期間1および3中、1,000mgのVX−222またはVX−222プラセボをq12hに投与した。コホート1およびコホート2の対象に、処置期間2および3中、1125mgのVX−950またはVX−950プラセボをq12hに投与した。この実験の実験デザインを図1に示す:
【表17】
【0196】
予備安全解析報告は、処置期間3において、重症化または重篤な有害事象(SAE)は報告されなかったことを示した。主な報告された有害事象は、軽度のものであり、予期しない有害事象の出現または傾向はなかった。下痢、食欲減退、掻痒、鼻血および鼻づまりを含むいくつかの有害事象は、処置期間1および処置期間3と比較して、処置期間3においてより多く見られた。
【0197】
薬物動態学的評価を以下の通りに行った:
VCH−222血漿:
−期間1
1日目:0(投与前)、投与後0.5、1、1.5、2、4、6、8および12時間
3日目、5日目、7日目、8日目および9日目:0(投与前)
10日目:0(投与前)、投与後0.5、1、1.5、2、4、6、8、12、24、48および72時間
−期間3
31日目および33日目:0(投与前)
37日目:0(投与前)、投与後0.5、1、1.5、2、4、6、8、12および24時間
VX−950(および代謝物)血漿:
−期間2
22日目、24日目および26日目:0(投与前)
27日目:0(投与前)、投与後0.5、1、1.5、2、4、6、8および12時間
−期間3
31日目および33日目:0(投与前)
37日目:0(投与前)、投与後0.5、1、1.5、2、4、6、8および12時間
VCH−222 尿中:
−10日目および37日目:投与後0ないし4、4ないし8、8ないし12および12ないし24(10日目のみ)時間。
【0198】
以下の表1は、この実験の予備的薬物動態学的(pk)結果を提供する。表に示す通り、VX−222血漿暴露は、増加した。
【表18】
【0199】
実施例7.パートAにおける実験VX−222−102からの薬物動態データ:VX−222処置
VX−222について実施例5に記載の製剤を用いた。実験102のパートAにおいて、対象をVX−222またはプラセボ 6:2(VX−222:プラセボ)の割当比で無作為化し、コホート1、コホート2、コホート3またはコホート4とした。コホート1、コホート2およびコホート3に登録された対象は、250mg、500mgまたは750mgのVX−222またはプラセボを1日2回(BID)、それぞれ3日間投与された。コホート4に登録された対象は、1,500mgのVX−222またはプラセボを1日1回(QD)、3日間投与された。標準的ケア処置について、医師により適当と判断されたとき、peg−ifn−アルファ−2aおよびRBVが48週間までの一部にて投与の最後に対象に供された。
【0200】
A.予備的結果
予備的安全性解析報告:遺伝子型1型慢性C型肝炎に感染した対象は、VX−222またはプラセボを250mg(コホート1)、500mg(コホート2)、または750mg(コホート3)(BID)で3日間、多数回投与された。予備的安全性解析報告は、重症化または重篤な有害事象は報告されなかったことを示した。主な報告された有害事象は、軽度のものであり、予期しない有害事象の出現または傾向はなかった。
【0201】
予備的薬物動態学的(PK)分析:コホート1、コホート2およびコホート3からの予備的pkパラメーターのまとめを、表2に示す。
【表19】
【0202】
予備的HCV RNA分析:コホート1、コホート2およびコホート3からの予備的HCV RNA分析のまとめを表3に提供する。コホート1、コホート2およびコホート3における遺伝子型1型HCVに感染した対象についての4日目の平均log HCV RNA減少は、それぞれ3.1、3.4および3.2であった。1,500mgのVX−222を1日1回投与された、コホート4における遺伝子型1型HCVに感染した対象についての4日目の平均log HCV RNA減少は、3.6であった。
【表20】
【0203】
B.さらなる結果
実験VX−222−102のさらなる結果を図2−8に示す:
250mgのVX−222 BID、500mgのVX−222 BID、750mgのVX−222 BID、および1,500mgのVX−222 QDを投与された各用量群における6名の患者を含む、32名の慢性遺伝子型1型HCVに感染した未処置患者をこの臨床試験に登録した。4種の用量群にて2名の患者がプラセボを投与され、全部で8名の患者がプラセボ投与された。試験のパートAを、アメリカ合衆国、カナダおよびアルゼンチンの10施設で行った。試験に登録された患者のうち、24名の患者が遺伝子型1a型HCV感染を有し、8名の患者が遺伝子型1b型HCV感染を有した。試験に登録された6名の患者は、アフリカ系アメリカ人であり、25名が白人であり、1名がインド人/アラスカ系であった。
【0204】
試験デザインaおよび人種
多施設、無作為化、二重盲検、プラセボ対照、漸増用量試験
−−HCV遺伝子型1型に感染した患者
・肝硬変の発現なし
・ALT値<5xULM
・スクリーニングで血漿 HCV RNAレベル≧5log10 IU/mL
−−4群の比較実験(VX−222:プラセボ 6:2無作為化)
−−この漸増用量試験に登録された順
・250mg BID、3日間
・500mg BID、3日間
・750mg BID、3日間
・1500mg QD、3日間。
【0205】
ベースライン特性を図3にまとめた。HCV RNA変化を図4−6に示す。図7は、VX−222薬物動態を示す。図7に示す通り、Tmaxには投与後2−6時間で達し、VX−222暴露は、およそ用量比例的に増大した。図8は、3日目におけるVX−222薬物動態のまとめを示す。
【0206】
ウイルス動態結果
VX−222での処置は、4種のVX−222用量群で血漿HCV RNAの3 log10以上の平均減少という結果となった。さらに、漸増用量応答が、500mg、750mgおよび1,500mg用量群で非常に似た結果で4種の用量群で観察された。250mg BID、500mg BIDおよび750mg BIDのVX−222を投与した3日後に達成された平均HCV RNA減少は、それぞれ3.1 log10(範囲:2.0ないし4.2)、3.4 log10(範囲:3.2ないし3.6)、および3.2 log10(範囲:2.3ないし3.8)であった。1,500mg QDのVX−222を投与した3日後に達成されたは、3.4 log10(範囲:3.1ないし3.9)であった。プラセボを受容する患者において、特記すべきHCV RNAの減少は観察されなかった。同様のウイルス減少が、遺伝子型1aおよび1b型に感染した患者で観察された。
【0207】
本試験のパートAのこれらの結果は、VX−222の750mg BIDで、予め3日間処置された5名の患者のウイルス薬物動態実験からの発見と一致する。
【0208】
安全性および耐容性結果
この試験のパートAで集められた安全性および耐容性の情報は伏せられたままであり、故に、本発明で供される安全性の情報には、プラセボまたはVX−222の投与後の患者についての収集データが含まれる。プラセボまたはVX−222は、4種の用量群の全てで良好な耐容性であって、重症化または重篤な有害事象は報告されず、処置の中止も生じなかった。プラセボまたはVX−222の投与後に報告された全ての有害事象は、軽度または中等症であった。用量群あたり少なくとも2名の患者に生じた最も多く報告された有害事象は、下痢、頭痛、悪心、無力症および発熱であった。
【0209】
実施例8.パートCにおける実験VX−222−102からの薬物動態データ:VX−222での処置
実験101のパートCにおいて、VX−222を、未処置の慢性C型肝炎を有する対象に750mg b.i.d.の複数用量で3日間投与した。さらに、実験102のパートAにおいて、VX−222を、未処置の慢性C型肝炎を有する対象に250mgないし750mg BID複数用量および1500mgを1日1回、3日間投与した。
【0210】
実験101(最終データ)および実験102(予備データ)にて評価されたVX−222のPKパラメーターを以下に概説する:
・AUCの蓄積指数は1.90倍であり、Cmaxのそれは、1.75倍であった。
・反復測定分析は、安定状態が処置の3日以内に達成されたことを示唆した。
・VX−222暴露は、平均Cmax、AUCτおよびCτにおける増加により示される通り、用量増加に伴い増加した。VX−222暴露は、250ないし750mg b.i.dの範囲の用量でおよそ用量に比例して増加した。
・VX−222吸収はゆっくりであり、2ないし6時間の範囲の安定状態での平均tmaxであった。
・蓄積指数は、BIDレジメンに関して約2倍であった。QDレジメンに関して、3日目の1500mg VX−222への暴露は、1日目に観察されたものと同様であった。
・平均t1/2は、全ての用量で変化しなかった。
・VX−222 t1/2は約5時間であった。
・投与間隔の最後でのVX−222濃度は、全ての対象についてインビトロでのIC90(319ng/mL)より高かった。
・一般に、対象におけるVX−222暴露は、健康な対象と比較して未処置HCV対象にて約2倍高かった。
【0211】
パートCにおけるVX−222臨床試験効果
実験101のパートCの主目的は、遺伝子型1型慢性C型肝炎感染を有する未処置対象におけるVX−222の薬理作用を評価することであった。750mg b.i.d.で連続3日間投与したVX−222の最終的な薬力学的パラメーターを以下に概説する:
・平均未変換ベースライン(1日目)HCV血漿RNAレベルは、4962600IU/mL(log10=6.4927)であった。
・2ないし4日目のlog10投与前HCV血漿RNAを用いて測定したベースラインからの平均最大減少は、−3.6784であった。投与前RNAレベルとlog10HCV RNAにおける減少のの振幅との相関は、比較的低かった。
これらの結果は健康な対象と一致し、VX−222とVX−950の共投与がVX−222単独投与よりも2倍高かった。
【0212】
実施例9:薬物併用アッセイ:VX−950およびVX−222
材料および方法
細胞
レプリコン細胞株Huh−7、Huh−7肝細胞癌細胞株由来のET細胞を、Ralf bartenschlager 博士(Bartenschlager, R. Innovation: Hepatitis C virus replicons: potential role for drug development. Nat. Rev. Drug Discov. 2002, 1, 911−916. Krieger, N.; Lohmann, V.; Bartenschlager, R. Enhancement of hepatitis C virus RNA replication by cell culture−adaptive mutations. J. Virol. 2001, 75, 4614−4624. Lohmann, V.; Korner, F.; Koch, J.−O.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999, 285, 110−113.) (Reblikon GmbH, Gau−Odernheim, Germany)から入手した。該Huh−7 ET細胞株は、ネオマイシン遺伝子に加えて、ホタルルシフェラーゼ遺伝子に融合されたコピーを担持する、高度に細胞培養に適合したレプリコンi389Luc−UBI−neo/NS3−3’/5.1コンストラクト(HCV NS3およびNS5遺伝子内の3つの適応変異の存在によっても特徴付けられる)を含む(Vrolijk, J.M.; Kaul, A.; Hansen, B.E.; Lohmann, V., Haagmans, B.L.; Schalm, S.W.; Bartenschlager, R. A replicon−based bioassay for the measurement of interferons in patients with chronic hepatitis C. Journal of Virol. Methods. 2003, 110, 201−209)。この細胞株は、ルシフェラーゼ活性を測定することにより、HCV RNA複製および形質転換の測定が可能である。ルシフェラーゼ活性は、これらの細胞におけるレプリコンRNAレベルに密接に従うことが既報である(j. Virol. 2001, 75, 4614−4624, journal of virol. Methods. 2003, 110, 201−209)。DMEM(wisent inc., st−bruno, qc, canada)からなる細胞培養に用いられる培地に、supplemented with 最終濃度、10%ウシ胎仔血清と1%ペニシリン/ストレプトマイシン、1%グルタミン、1%ピルビン酸ナトリウム、1%非必須アミノ酸および180μg/mlのジェネテシン(G418)(invitrogen, burlington, on, canada)を補った。細胞を、37℃、5%CO2雰囲気下でインキュベートし、サブコンフルエンスで週2回継代した。
【0213】
レプリコン細胞株Huh−7、Huh−7肝細胞癌細胞株由来の9−13細胞を、Ralf bartenschlager 博士(reblikon gmbh, gau−odernheim, germany)から入手した。Huh−7、9−13細胞株は、HCVサブゲノム レプリコンpfk i377/ns3−3’/wtを含み(Koutsoudakis, G.; Kaul, A.; Steinmann, E.; Kallis, S.; Lohmann, V.; Pietschmann, T.; Bartenschlager, R. Characterization of the Early Steps of Hepatitis C Virus Infection by Using Luciferase Reporter Viruses. J Virol. 2006, 80, 5308−5320)、リアルタイムPCRアッセイに用いられる。定量的リアルタイムPCRアッセイ(taqman)は、基本的に、レポーター色素およびクエンチャー色素がそれに結合される蛍光オリゴヌクレオチドプローブの使用と組み合わせた標準的PCRである。PCR中に、該プローブは、プライマー部位の上流と下流の間の標的にアニーリングする。各伸張反応中、プローブは、Taq DNA ポリメラーゼの5’ヌクレアーゼ活性により切断される。これは、クエンチャー色素からレポーター色素を分離させ、レポーター色素の蛍光強度を増大させる。蛍光強度は、標的DNAの存在量に比例する。
【0214】
薬物併用アッセイ
ルシフェラーゼアッセイ(MacSynergy分析を用いる4日目の実験)。Huh−7、ETレプリコン細胞を、乳白色の96ウェル細胞培養マイクロタイタープレート中、100μl培地中にサブコンフルエント密度(3x103 細胞/ウェル)で播種した。アッセイに用いる細胞培養培地は、G418とフェノールレッドを含まないこと以外は、上記と同様である。チオフェンと種々の濃度の異なる抗HCV薬との組み合わせを含むストック溶液のマトリックスを深い96ウェルプレートに製造した。3−4時間、37℃でインキュベート後、該マトリックスストック溶液プレートからの化合物(100μl)を、最終容量200μlを細胞に添加し、ここで、ある化合物は水平方向に増量させ、ある化合物は垂直方向に増量される。少なくとも4個の細胞プレートが各薬物−薬物相互作用試験に用いられ、各併用が少なくとも2回行われる。次いで、細胞はさらに、37℃、5%CO2雰囲気下で4日間インキュベートされる。その後、培養培地を除去し、細胞を95μlのルシフェラーゼ緩衝液(緩衝洗浄液中のルシフェリン基質)の添加により細胞溶解させた。細胞溶解物を少なくとも10分間室温でインキュベートし、直接照明から保護する。プレートを、照度計(Wallac Microbeta Trilux, Perkin Elmer(商標), MA, USA)を用いてルシフェラーゼ数を読み出す。
【0215】
化合物の併用が相加的、相乗的または拮抗的であるかどうかを決定するために、チオフェンと他の各薬物との4日間の処置の薬物併用効果を、MacSynergy II(商標) program (Prichard, M.N.; Prichard, L.E.; Shipman, C. Strategic design and three−dimensional analysis of antiviral drug combinations. Antimicrob. Agents Chemother. 1993, 37:540−545. Prichard, M.N. and Shipman, C. A three−dimensional model to analyze drug−drug interactions. Antiviral Res. 1990, 14:181−205)により計算する。この方法は、統計的確率に基づくBlissの独立ヌル・モデルを用いて薬物の組合せを試験し、2つの薬物が複製を阻害するのに独立して作用すると仮定する。この方法を用いて、理論的相加相互作用が、単独で作用する個々の薬物の用量応答曲線から計算される。その後、予測された相加効果は、用量応答曲線における相違を明らかにするため、実験的に決定された効果から差し引かれる。相互作用が相加的であるとき、結果として得られる曲線は0%差の水平面で現される。平面よりも高いピークは全て、予想を上回る効果(相乗効果)を示す。反対に、平面より低いピークは、期待された効果未満(拮抗作用)を示す。実験的用量応答曲線周辺の信頼区間は、統計学的にデータを評価するのに用いられピークの面積は、もたらされる相乗効果または拮抗作用を定量するために計算される。
【0216】
単独で試験されるとき、全ての薬物の阻害作用の50%阻害濃度(IC50)もまた、1化合物当たり7ないし9種の濃度を用いて用量応答曲線から決定される。曲線は、非線形回帰分析法を用いるデータ点に適合され、IC50は、GraphPad Prism software, version 2.0 (GraphPad Software Inc., San Diego, CA, USA)を用いて得られる曲線から補間される:
≦25uM2%:相乗作用ほとんどなし
25uM2%−50uM2%:わずかな相乗作用
50uM2%−100uM2%:中程度の相乗作用
≧100uM2%:強い相乗作用。
【0217】
リアルタイムPCRアッセイ(HCVレプリコンウイルスRNA排除およびリバウンド実験)。
レプリコン細胞株Huh−7、9−13細胞を、12ウェル培養ディッシュ中、1ml容量で、各ウェル当たり3x104細胞密度で播種する。該アッセイに用いる細胞培養培地は、10%ウシ胎仔血清、1%ペニシリン/ストレプトマイシン、1%グルタミン、1%ピルビン酸ナトリウムおよび1%非必須アミノ酸を補ったDMEMである。3ないし4時間インキュベート後、化合物を、最終容量2mlで種々の濃度で添加する。その後、細胞を37℃、5%CO2雰囲気下でさらに14日間インキュベートする。細胞を3ないし4日毎に剥がし、培地および阻害剤を補充し、細胞のサンプルをリアルタイムPCRによりRNA定量のために集める。14日間のインキュベート後、細胞を剥がし、抗ウイルス化合物を含まない新鮮な培地中に播く。18日目に、細胞を剥がし、0.25mg/mlのG418抗生物質を含む新鮮な選択培地に播く。培養をG418の存在下で42日目まで行い、細胞を3ないし4日毎に剥がし、該細胞サンプルを、リアルタイムPCRによりRNA定量を行う。全RNA(細胞およびウイルス起源)を、製造業者のプロトコールに従いQiagen RNeasy reagent (Qiagen Inc., Mississauga, ON, Canada, kit: 74106)を用いて抽出し、MMLV RT酵素を用いてcDNA合成を行う。この工程は、PCRのリアルタイム検出のためのABI PRISM(登録商標) 7700 Sequence Detection System (Applied Biosystems, Foster City, CA, USA)で適当なオリゴヌクレオチド、TaqmanプローブおよびDNA taq ポリメラーゼを用いてPCR反応により行われる。18S RNAレベルが、各ウェルにおける総RNA量の標準化に用いられる。
【0218】
単独で試験されるとき、全ての薬剤の阻害効果のための50%および90%阻害濃度(IC50およびIC90)もまた、1化合物当たりデュプリケートで6種の濃度を用いる用量応答曲線から決定される。曲線は、非線形回帰分析法を用いるデータ点に適合され、IC50およびIC90は、GraphPad Prism software, version 2.0 (GraphPad Software Inc., San Diego, CA, USA)を用いて得られる曲線から補間される。
【0219】
チオフェン化合物とウイルスプロテアーゼ阻害剤の併用実験
選択されたチオフェン化合物の併用実験では、上記の材料および方法を用いた。試験のために選択したHCV NS3 プロテアーゼ阻害剤はVX−950である。Macsynergy(商標)ソフトウェアを用いるこれらの化合物の相乗効果の結果を、表4に記載する。
【表21】
【0220】
実施例10:化合物1の任意のプロドラッグの合成
本明細書で用いる用語“RT(分)”は、化合物と関連するLCMS保持時間(分)を意味する。ある特定の化合物のNMRおよび質量分析データを、表5にまとめる。
化合物Aの製造
【化4】
5−(3,3−ジメチルブト−1−イニル)−3−[(トランス−4−ヒドロキシシクロヘキシル)−(4−トランス メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸(化合物(1)、300mg、0.67mmol)を、ジクロロメタン(DCM、15mL)に溶解した。これに、(2S)−2−(tert−ブトキシカルボニルアミノ)−3−メチル−ブタン酸 Boc−L−バリン(176mg、0.81mmol)、N,N−ジメチルピリジン−4−アミン(DMAP、8.22mg、0.067mmol)、トリエチルアミン(Et3N、136mg、187μL、1.35mmol)、および3−(エチルイミノメチレンアミノ)−N,N−ジメチル−プロパン−1−アミン塩酸塩(EDC、129mg、0.67mmol)を添加した。反応物を一晩撹拌した。次いで、反応混合物を濃縮し、酢酸エチル(EtOAc)で希釈し、水で洗浄し、合わせた有機相を塩水で洗浄し、硫酸ナトリウムで乾燥させた。濾過し、濃縮して黄色油状物を得て、それをカラムクロマトグラフィーにより精製した。次いで、得られた生成物を、ジオキサン中、4N HCl(15mL)で処理して、所望の化合物AをHCl塩として得た(100mg、26%):MS:m/z(obs.):545.4[M+H]+;保持時間:3.45分;1H NMR (300 MHz, MeOH) δ 7.04 (s, 1H), 4.75−4.58 (m, 1H), 4.39 (dt, J=14.5, 9.4 Hz, 1H), 3.85 (d, J=4.4 Hz, 1H), 3.80−3.68 (m, 1H), 3.61−3.51 (m, 1H), 2.24 (dt, J=14.0, 6.9 Hz, 1H), 2.01 (dd, J=15.2, 7.3 Hz, 6H), 1.60 (dd, J=28.5, 14.8 Hz, 9H), 1.34 (s, 9H), 1.18−0.99 (m, 3H), 0.81 (d, J=6.5 Hz, 3H), 0.66 (dd, J=25.3, 12.9 Hz, 1H)。
【0221】
化合物Bの製造
【化5】
5−(3,3−ジメチルブト−1−イニル)−3−[(トランス 4−ヒドロキシシクロヘキシル)−(トランス 4−メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸(化合物(1)、100mg、0.12mmol)を、ジクロロメタン(DCM、10.0mL)に溶解し、0℃まで冷却した。テトラゾール(4.0mg、0.058mmol)を添加し、次いでN−(ジ−tert−ブトキシホスファニル)−N−エチル−エタナミン(288mg、322μL、1.16mmol)を添加した。反応物を室温で一晩撹拌し、次いで−78℃まで冷却した。3−クロロベンゼンカルボペルオキソ酸(3−Chlorobenzenecarboperoxoic acid)(MCPBA)(99.7mg、0.58mmol)を添加し、反応物を2時間撹拌し、次いでNa2SO3水溶液でクエンチした。混合物を酢酸エチルで抽出し、抽出物を水で洗浄した。有機相を集め、無色油状物を得て、それをISCOシリカゲルクロマトグラフィーにより精製して、次工程に直接用いた。生成物にCH2Cl2(5mL)および2,2,2−トリフルオロ酢酸(TFA)(5mL)を添加した。反応物を2時間撹拌し、次いで濃縮し、生成物BをHPLCにより精製した:MS:m/z(obs.):526.39[M+H]+;保持時間:6.51分;1H NMR (300 MHz, d6−DMSO) δ 7.18 (s, 1H), 4.29 (t, J=11.8 Hz, 1H), 3.83 (s, 1H), 2.53 (d, J=8.2 Hz, 3H), 1.84 (s, 2H), 1.75−1.33 (m, 7H), 1.30 (s, 9H), 1.27−1.09 (m, 3H), 0.90 (d, J=12.9 Hz, 2H), 0.76 (d, J=6.5 Hz, 2H), 0.70−0.47 (m, 2H); 31P NMR (121.5 MHz, d6−DMSO) δ −2.01 (s)。
【0222】
化合物Cの製造
【化6】
CH2Cl2(15mL)中、5−(3,3−ジメチルブト−1−イニル)−3−[(4−トランス−ヒドロキシシクロヘキシル)−(4−トランス−メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸(化合物(1)、75mg、0.17mmol)およびN−Boc−グリシン(44.2mg、0.25mmol)の溶液に、3−(エチルイミノメチレンアミノ)−N,N−ジメチル−プロパン−1−アミン塩酸塩(EDC)(32.2mg、0.17mmol)、N,N−ジメチルピリジン−4−アミン(DMAP)(10.3mg、0.084mmol)およびEt3N(34mg、0.33mmol)を添加した。反応混合物を環境温度で一晩撹拌し、次いで反応混合物を蒸発させ、ISCOシリカゲルクロマトグラフィーにより精製して、化合物(b4)、[O−(N−t−ブトキシカルボニル)−グリシル]−5−(3,3−ジメチルブト−1−イニル)−3−[(4−トランス−ヒドロキシシクロヘキシル)−(4−トランス−メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸を得た:MS:m/z(obs.):603.17[M+H]+;保持時間:2.31分。
【0223】
【化7】
[O−(N−t−ブトキシカルボニル)−グリシル]−5−(3,3−ジメチルブト−1−イニル)−3−[(4−トランス−ヒドロキシシクロヘキシル)−(4−トランス−メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸(化合物(b4)、40mg、0.066mmol)を、ジオキサン中、4N HCl(1mL)で処理し、室温で一晩撹拌した。次いで、反応混合物を濃縮し、HPLCにより精製して、化合物Cを得た(11mg):MS:m/z(obs.):503.35[M+H]+;保持時間:2.24分。
【0224】
化合物Dの製造
【化8】
化合物(a5)、[O−(N−t−ブトキシカルボニル)−D−イソロイシル]−5−(3,3−ジメチルブト−1−イニル)−3−[(4−トランス−ヒドロキシシクロヘキシル)−(4−トランス−メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸(Boc−D−イソロイシンから上記の化合物1および4に記載の通りに製造)を、ジオキサン中、4N HCl(10mL)で処理し、室温で一晩撹拌した。次いで、反応混合物を濃縮し、HPLCにより精製して化合物Dを得た:MS:m/z(obs.):559.4[M+H]+;保持時間:2.39分。
【0225】
化合物Eの製造
【化9】
化合物(a6)、[O−(N−t−ブトキシカルボニル)−D−バリニル]−5−(3,3−ジメチルブト−1−イニル)−3−[(4−トランス−ヒドロキシシクロヘキシル)−(4−トランス−メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸(30mg)(Boc−D−バリンから上記の化合物1および4に記載の通りに製造)を、ジオキサン中、4N HCl(10mL)で処理し、室温で一晩撹拌した。次いで、反応混合物を濃縮し、HPLCにより精製して化合物Eを得た:MS:m/z(obs.):545.39[M+H]+;保持時間:2.35分。
【0226】
化合物Fの製造
【化10】
化合物(a8)、(O−(N−t−ブトキシカルボニル)−L−イソロイシル)−5−(3,3−ジメチルブト−1−イニル)−3−[(4−トランス−ヒドロキシシクロヘキシル)−(4−トランス−メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸(Boc−L−イソロイシンから上記化合物AおよびCに記載の通りに製造)(35mg)を、ジオキサン中、4N HCl(10mL)で処理し、室温で一晩撹拌した。次いで、反応混合物を濃縮し、HPLCにより精製して、化合物8を得た:MS:m/z(obs.):559.47[M+H]+;保持時間:3.2分。
【0227】
化合物Gの製造
【化11】
化合物(a9)、(O−(N−t−ブトキシカルボニル)−L−アラニル)−5−(3,3−ジメチルブト−1−イニル)−3−[(4−トランス−ヒドロキシシクロヘキシル)−(4−トランス−メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸(Boc−L−アラニンから上記化合物AおよびCに記載の通りに製造)(25mg)を、ジオキサン中、4N HClで処理し、室温で一晩撹拌した。次いで、反応混合物を濃縮し、HPLCにより精製して化合物Gを得た:MS:m/z(obs.):517.43[M+H]+;保持時間:2.99分。
【0228】
化合物Hの製造
【化12】
化合物(a10)、(O−(N−t−ブトキシカルボニル)−D−アラニル)−5−(3,3−ジメチルブト−1−イニル)−3−[(4−トランス−ヒドロキシシクロヘキシル)−(4−トランス−メチルシクロヘキサンカルボニル)アミノ]チオフェン−2−カルボン酸(Boc−D−アラニンから上記化合物1および4に記載の通りに製造)(35mg、0.058mmol)を、ジオキサン中、4N HCl(10mL)で処理し、室温で一晩撹拌した。次いで、反応混合物を濃縮し、HPLCにより精製して化合物Hを得た:MS:m/z(obs.):517.43[M+H]+;保持時間:3.0分。
【0229】
【表22】
【0230】
実施例10:化合物1のプロドラッグのPKパラメーター
PKパラメーターが決定されるべきプロドラッグは、0.5%mc/0.5%トゥイーン 80/99%水中の溶液として製剤され得て、3mg/kg用量でラットに強制経口される。実験の前日にラットを計測する。ラットの血漿を、Instech自動血液サンプリング装置を用いて、投与前、および投与後15分、30分、1時間、2時間、3時間、4時間、6時間、8時間、12時間および24時間にサンプル採取する。血液をK2−EDTAを含むチューブに集め、110μlの血漿を分析のために抽出する。ラットは自由に餌を摂り、標準的なIACUCおよびSOPプロトコールが適用される。血漿サンプルおよび用量サンプルは、プロドラッグ化合物および活性代謝産物の両方に関してLC/MS/MSを用いて分析される。各対象に関する両分析物のPKパラメーターを、プロドラッグの定量した用量を用いて計算する。
【0231】
化合物H(図9にて“化合物10”と称される、化合物1のプロドラッグ)のPKパラメーターを、上記の通りに測定し、図9に記載した。図9に示す通り、化合物HのO−アラニル基は、インビボで、OH活性代謝産物に変換される。
【特許請求の範囲】
【請求項1】
HCVに感染した患者においてVX−222の薬物動態を改善する方法であって、該患者にVX−222およびVX−950を共投与することを含む、方法。
【請求項2】
患者の血漿、血液または肝臓におけるVX−222への暴露が改善される、請求項1に記載の方法。
【請求項3】
HCVに感染した患者の血漿中におけるVX−222への暴露を増大する方法であって、該患者にVX−222およびVX−950を投与することを含む、方法。
【請求項4】
患者の血漿中、VX−222への暴露が、VX−950を投与しないときのVX−222への血漿暴露と比較して2−6倍増大する、請求項1−3のいずれか一項に記載の方法。
【請求項5】
患者の血漿中、VX−222への暴露が、VX−950を投与しないときのVX−222への血漿暴露と比較して2−4倍増大する、請求項4に記載の方法。
【請求項6】
VX−222のC(trough)レベルが増大する、請求項1−5のいずれか1項に記載の方法。
【請求項7】
VX−222のC(max)値が増大する、請求項1−5のいずれか1項に記載の方法。
【請求項8】
VX−222のAUC値が増大する、請求項1−5のいずれか1項に記載の方法。
【請求項9】
VX−222が、各投与にて約20mgないし約2,000mg投与される、請求項1−8のいずれか1項に記載の方法。
【請求項10】
VX−222が、各投与にて約50mgないし約2,000mg投与される、請求項9に記載の方法。
【請求項11】
VX−222が、各投与にて約100mgないし約1,500mg投与される、請求項10に記載の方法。
【請求項12】
VX−222が、各投与にて約100mg投与される、請求項11に記載の方法。
【請求項13】
VX−222が、各投与にて約400mg投与される、請求項11に記載の方法。
【請求項14】
VX−222が、各投与にて約250mg投与される、請求項11に記載の方法。
【請求項15】
VX−222が、各投与にて約500mg投与される、請求項11に記載の方法。
【請求項16】
VX−222が、各投与にて約750mg投与される、請求項11に記載の方法。
【請求項17】
VX−222が1日1回投与される、請求項1−16のいずれか一項に記載の方法。
【請求項18】
VX−222が1日2回投与される、請求項1−16のいずれか一項に記載の方法。
【請求項19】
VX−222が、各投与にて約1,500mg投与される、請求項11に記載の方法。
【請求項20】
VX−222が1日1回投与される、請求項19に記載の方法。
【請求項21】
VX−950が、各投与にて約100mgないし約1,500mg投与される、請求項1−20のいずれか一項に記載の方法。
【請求項22】
VX−950が、各投与にて約500mgないし約1,500mg投与される、請求項21に記載の方法。
【請求項23】
VX−950が、各投与にて約750mg投与される、請求項21に記載の方法。
【請求項24】
VX−950が、約750mgを1日3回投与される、請求項23に記載の方法。
【請求項25】
VX−950が、各投与にて約1,125mg投与される、請求項22に記載の方法。
【請求項26】
VX−950が、約1,125mgを1日2回投与される、請求項25に記載の方法。
【請求項27】
VX−950およびVX−222以外の1種以上のさらなるHCV薬を患者に投与することをさらに含む、請求項1−26のいずれか一項に記載の方法。
【請求項28】
インターフェロンを共投与する、請求項27に記載の方法。
【請求項29】
インターフェロンがペグ化インターフェロンである、請求項28に記載の方法。
【請求項30】
インターフェロンがペグ化インターフェロンαである、請求項28に記載の方法。
【請求項31】
ペグ化インターフェロンαが、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bである、請求項30に記載の方法。
【請求項32】
リバビリンを共投与する、請求項27または28に記載の方法。
【請求項33】
約8週間ないし約24週間の期間に、VX−950およびVX−222を共投与する、請求項1−32のいずれか一項に記載の方法。
【請求項34】
VX−950およびVX−222を約12週間共投与する、請求項33に記載の方法。
【請求項35】
ペグ化インターフェロンαおよびリバビリンを投与する、請求項27に記載の方法。
【請求項36】
ペグ化インターフェロンαが、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bである、請求項35に記載の方法。
【請求項37】
VX−950およびVX−222を約12週間共投与する、請求項35または36に記載の方法。
【請求項38】
ペグ化インターフェロンおよびリバビリンを約12週間共投与する、請求項37に記載の方法。
【請求項39】
ペグ化インターフェロンおよびリバビリンを約24週間共投与する、請求項37に記載の方法。
【請求項40】
HCVに感染した患者の処置方法であって、該患者にVX−222およびVX−950を投与することを含み、ここで、VX−222は、各投与にて約20mgないし約400mg投与され、VX−950は、各投与にて約100mgないし約1500mg投与される、方法。
【請求項41】
VX−222が、各投与にて20mgまたは20mgより多く、かつ400mg未満の量で投与される、請求項40に記載の方法。
【請求項42】
VX−222が、各投与にて約20mgないし約300mg投与される、請求項40に記載の方法。
【請求項43】
VX−950が、各投与にて約300mgないし約1500mg投与される、請求項40に記載の方法。
【請求項44】
VX−950が、約750mgを1日3回投与される、請求項40に記載の方法。
【請求項45】
VX−950が、約1125mgを1日2回投与される、請求項40に記載の方法。
【請求項46】
VX−222が、各投与にて約100mg投与される、請求項40−45のいずれか一項に記載の方法。
【請求項47】
VX−222が、各投与にて約400mg投与される、請求項40−45のいずれか一項に記載の方法。
【請求項48】
VX−222が1日1回投与される、請求項40−47のいずれか一項に記載の方法。
【請求項49】
VX−222が1日2回投与される、請求項40−47のいずれか一項に記載の方法。
【請求項50】
患者にVX−950およびVX−222以外の1種以上のさらなるHCV薬を投与することをさらに含む、請求項40−49のいずれか一項に記載の方法。
【請求項51】
インターフェロンを共投与する、請求項50に記載の方法。
【請求項52】
インターフェロンがペグ化インターフェロンである、請求項51に記載の方法。
【請求項53】
インターフェロンがペグ化インターフェロンαである、請求項52に記載の方法。
【請求項54】
ペグ化インターフェロンαが、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bである、請求項52に記載の方法。
【請求項55】
リバビリンを共投与する、請求項50または51に記載の方法。
【請求項56】
約8週ないし約24週間の期間に、VX−950およびVX−222を共投与する、請求項40−55のいずれか一項に記載の方法。
【請求項57】
VX−950およびVX−222を、約12週間共投与する、請求項56に記載の方法。
【請求項58】
ペグ化インターフェロンαおよびリバビリンを共投与する、請求項50に記載の方法。
【請求項59】
ペグ化インターフェロンαが、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bである、請求項58に記載の方法。
【請求項60】
VX−950およびVX−222を約12週間共投与する、請求項58または59に記載の方法。
【請求項61】
ペグ化インターフェロンおよびリバビリンを約12週間共投与する、請求項60に記載の方法。
【請求項62】
ペグ化インターフェロンおよびリバビリンを24週間共投与する、請求項61に記載の方法。
【請求項63】
HCVに感染した患者の処置方法であって、治療的有効量のVX−222を投与することを含み、ここで、VX−222は、約20mgないし約2,000mgの量で1日1回投与される、方法。
【請求項64】
VX−222が、約100mgないし約1,500mgの量で1日1回投与される、請求項63に記載の方法。
【請求項65】
VX−222が、約1,500mgの量で1日1回投与される、請求項64に記載の方法。
【請求項66】
VX−222が、約750mgの量で1日1回投与される、請求項64に記載の方法。
【請求項67】
VX−222が、約500mgの量で1日1回投与される、請求項64に記載の方法。
【請求項68】
VX−222が、約400mgの量で1日1回投与される、請求項64に記載の方法。
【請求項69】
VX−222が、約250mgの量で1日1回投与される、請求項64に記載の方法。
【請求項70】
VX−222が、約100mgの量で1日1回投与される、請求項64に記載の方法。
【請求項71】
患者にVX−222以外の1種以上のさらなるHCV薬を投与することをさらに含む、請求項63−70のいずれか一項に記載の方法。
【請求項72】
VX−950を共投与する、請求項71に記載の方法。
【請求項73】
VX−950が、各投与にて約500mgないし約1,500mg投与される、請求項72に記載の方法。
【請求項74】
VX−950が、750mgを1日3回投与される、請求項72に記載の方法。
【請求項75】
VX−950が、1125mgを1日2回投与される、請求項73に記載の方法。
【請求項76】
VX−950およびVX−222が、約8週間ないし約24週間の期間投与される、請求項72−75のいずれか一項に記載の方法。
【請求項77】
インターフェロンが共投与される、請求項76に記載の方法。
【請求項78】
インターフェロンがペグ化インターフェロンである、請求項77に記載の方法。
【請求項79】
インターフェロンがペグ化インターフェロンαである、請求項78に記載の方法。
【請求項80】
ペグ化インターフェロンαが、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bである、請求項79に記載の方法。
【請求項81】
リバビリンが共投与される、請求項71−77のいずれか一項に記載の方法。
【請求項82】
ペグ化インターフェロンαおよびリバビリンが共投与される、請求項71−76のいずれか一項に記載の方法。
【請求項83】
ペグ化インターフェロンαが、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bである、請求項82に記載の方法。
【請求項84】
VX−950;ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2b;および、リバビリンが共投与される、請求項82に記載の方法。
【請求項85】
VX−950およびVX−222を約12週間共投与する、請求項84に記載の方法。
【請求項86】
ペグ化インターフェロンおよびリバビリンを約12週間共投与する、請求項84に記載の方法。
【請求項87】
ペグ化インターフェロンおよびリバビリンを約24週間共投与する、請求項84に記載の方法。
【請求項88】
a)約20mgないし約400mg量のVX−222;および
b)約100mgないし約1,500mg量のVX−950
を含む、薬学的に許容される組成物。
【請求項89】
VX−222が、50mgまたは50mgより多く、かつ400mg未満の量である、請求項88に記載の組成物。
【請求項90】
VX−222が約100mgないし約400mgの量であり、VX−950が約300mgないし約750mgの量である、請求項88に記載の組成物。
【請求項91】
VX−950が約375mgの量である、請求項88−90のいずれか一項に記載の組成物。
【請求項92】
VX−222が約50mgの量である、請求項88−90のいずれか一項に記載の組成物。
【請求項93】
VX−222が約200mgの量である、請求項88−90のいずれか一項に記載の組成物。
【請求項1】
HCVに感染した患者においてVX−222の薬物動態を改善する方法であって、該患者にVX−222およびVX−950を共投与することを含む、方法。
【請求項2】
患者の血漿、血液または肝臓におけるVX−222への暴露が改善される、請求項1に記載の方法。
【請求項3】
HCVに感染した患者の血漿中におけるVX−222への暴露を増大する方法であって、該患者にVX−222およびVX−950を投与することを含む、方法。
【請求項4】
患者の血漿中、VX−222への暴露が、VX−950を投与しないときのVX−222への血漿暴露と比較して2−6倍増大する、請求項1−3のいずれか一項に記載の方法。
【請求項5】
患者の血漿中、VX−222への暴露が、VX−950を投与しないときのVX−222への血漿暴露と比較して2−4倍増大する、請求項4に記載の方法。
【請求項6】
VX−222のC(trough)レベルが増大する、請求項1−5のいずれか1項に記載の方法。
【請求項7】
VX−222のC(max)値が増大する、請求項1−5のいずれか1項に記載の方法。
【請求項8】
VX−222のAUC値が増大する、請求項1−5のいずれか1項に記載の方法。
【請求項9】
VX−222が、各投与にて約20mgないし約2,000mg投与される、請求項1−8のいずれか1項に記載の方法。
【請求項10】
VX−222が、各投与にて約50mgないし約2,000mg投与される、請求項9に記載の方法。
【請求項11】
VX−222が、各投与にて約100mgないし約1,500mg投与される、請求項10に記載の方法。
【請求項12】
VX−222が、各投与にて約100mg投与される、請求項11に記載の方法。
【請求項13】
VX−222が、各投与にて約400mg投与される、請求項11に記載の方法。
【請求項14】
VX−222が、各投与にて約250mg投与される、請求項11に記載の方法。
【請求項15】
VX−222が、各投与にて約500mg投与される、請求項11に記載の方法。
【請求項16】
VX−222が、各投与にて約750mg投与される、請求項11に記載の方法。
【請求項17】
VX−222が1日1回投与される、請求項1−16のいずれか一項に記載の方法。
【請求項18】
VX−222が1日2回投与される、請求項1−16のいずれか一項に記載の方法。
【請求項19】
VX−222が、各投与にて約1,500mg投与される、請求項11に記載の方法。
【請求項20】
VX−222が1日1回投与される、請求項19に記載の方法。
【請求項21】
VX−950が、各投与にて約100mgないし約1,500mg投与される、請求項1−20のいずれか一項に記載の方法。
【請求項22】
VX−950が、各投与にて約500mgないし約1,500mg投与される、請求項21に記載の方法。
【請求項23】
VX−950が、各投与にて約750mg投与される、請求項21に記載の方法。
【請求項24】
VX−950が、約750mgを1日3回投与される、請求項23に記載の方法。
【請求項25】
VX−950が、各投与にて約1,125mg投与される、請求項22に記載の方法。
【請求項26】
VX−950が、約1,125mgを1日2回投与される、請求項25に記載の方法。
【請求項27】
VX−950およびVX−222以外の1種以上のさらなるHCV薬を患者に投与することをさらに含む、請求項1−26のいずれか一項に記載の方法。
【請求項28】
インターフェロンを共投与する、請求項27に記載の方法。
【請求項29】
インターフェロンがペグ化インターフェロンである、請求項28に記載の方法。
【請求項30】
インターフェロンがペグ化インターフェロンαである、請求項28に記載の方法。
【請求項31】
ペグ化インターフェロンαが、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bである、請求項30に記載の方法。
【請求項32】
リバビリンを共投与する、請求項27または28に記載の方法。
【請求項33】
約8週間ないし約24週間の期間に、VX−950およびVX−222を共投与する、請求項1−32のいずれか一項に記載の方法。
【請求項34】
VX−950およびVX−222を約12週間共投与する、請求項33に記載の方法。
【請求項35】
ペグ化インターフェロンαおよびリバビリンを投与する、請求項27に記載の方法。
【請求項36】
ペグ化インターフェロンαが、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bである、請求項35に記載の方法。
【請求項37】
VX−950およびVX−222を約12週間共投与する、請求項35または36に記載の方法。
【請求項38】
ペグ化インターフェロンおよびリバビリンを約12週間共投与する、請求項37に記載の方法。
【請求項39】
ペグ化インターフェロンおよびリバビリンを約24週間共投与する、請求項37に記載の方法。
【請求項40】
HCVに感染した患者の処置方法であって、該患者にVX−222およびVX−950を投与することを含み、ここで、VX−222は、各投与にて約20mgないし約400mg投与され、VX−950は、各投与にて約100mgないし約1500mg投与される、方法。
【請求項41】
VX−222が、各投与にて20mgまたは20mgより多く、かつ400mg未満の量で投与される、請求項40に記載の方法。
【請求項42】
VX−222が、各投与にて約20mgないし約300mg投与される、請求項40に記載の方法。
【請求項43】
VX−950が、各投与にて約300mgないし約1500mg投与される、請求項40に記載の方法。
【請求項44】
VX−950が、約750mgを1日3回投与される、請求項40に記載の方法。
【請求項45】
VX−950が、約1125mgを1日2回投与される、請求項40に記載の方法。
【請求項46】
VX−222が、各投与にて約100mg投与される、請求項40−45のいずれか一項に記載の方法。
【請求項47】
VX−222が、各投与にて約400mg投与される、請求項40−45のいずれか一項に記載の方法。
【請求項48】
VX−222が1日1回投与される、請求項40−47のいずれか一項に記載の方法。
【請求項49】
VX−222が1日2回投与される、請求項40−47のいずれか一項に記載の方法。
【請求項50】
患者にVX−950およびVX−222以外の1種以上のさらなるHCV薬を投与することをさらに含む、請求項40−49のいずれか一項に記載の方法。
【請求項51】
インターフェロンを共投与する、請求項50に記載の方法。
【請求項52】
インターフェロンがペグ化インターフェロンである、請求項51に記載の方法。
【請求項53】
インターフェロンがペグ化インターフェロンαである、請求項52に記載の方法。
【請求項54】
ペグ化インターフェロンαが、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bである、請求項52に記載の方法。
【請求項55】
リバビリンを共投与する、請求項50または51に記載の方法。
【請求項56】
約8週ないし約24週間の期間に、VX−950およびVX−222を共投与する、請求項40−55のいずれか一項に記載の方法。
【請求項57】
VX−950およびVX−222を、約12週間共投与する、請求項56に記載の方法。
【請求項58】
ペグ化インターフェロンαおよびリバビリンを共投与する、請求項50に記載の方法。
【請求項59】
ペグ化インターフェロンαが、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bである、請求項58に記載の方法。
【請求項60】
VX−950およびVX−222を約12週間共投与する、請求項58または59に記載の方法。
【請求項61】
ペグ化インターフェロンおよびリバビリンを約12週間共投与する、請求項60に記載の方法。
【請求項62】
ペグ化インターフェロンおよびリバビリンを24週間共投与する、請求項61に記載の方法。
【請求項63】
HCVに感染した患者の処置方法であって、治療的有効量のVX−222を投与することを含み、ここで、VX−222は、約20mgないし約2,000mgの量で1日1回投与される、方法。
【請求項64】
VX−222が、約100mgないし約1,500mgの量で1日1回投与される、請求項63に記載の方法。
【請求項65】
VX−222が、約1,500mgの量で1日1回投与される、請求項64に記載の方法。
【請求項66】
VX−222が、約750mgの量で1日1回投与される、請求項64に記載の方法。
【請求項67】
VX−222が、約500mgの量で1日1回投与される、請求項64に記載の方法。
【請求項68】
VX−222が、約400mgの量で1日1回投与される、請求項64に記載の方法。
【請求項69】
VX−222が、約250mgの量で1日1回投与される、請求項64に記載の方法。
【請求項70】
VX−222が、約100mgの量で1日1回投与される、請求項64に記載の方法。
【請求項71】
患者にVX−222以外の1種以上のさらなるHCV薬を投与することをさらに含む、請求項63−70のいずれか一項に記載の方法。
【請求項72】
VX−950を共投与する、請求項71に記載の方法。
【請求項73】
VX−950が、各投与にて約500mgないし約1,500mg投与される、請求項72に記載の方法。
【請求項74】
VX−950が、750mgを1日3回投与される、請求項72に記載の方法。
【請求項75】
VX−950が、1125mgを1日2回投与される、請求項73に記載の方法。
【請求項76】
VX−950およびVX−222が、約8週間ないし約24週間の期間投与される、請求項72−75のいずれか一項に記載の方法。
【請求項77】
インターフェロンが共投与される、請求項76に記載の方法。
【請求項78】
インターフェロンがペグ化インターフェロンである、請求項77に記載の方法。
【請求項79】
インターフェロンがペグ化インターフェロンαである、請求項78に記載の方法。
【請求項80】
ペグ化インターフェロンαが、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bである、請求項79に記載の方法。
【請求項81】
リバビリンが共投与される、請求項71−77のいずれか一項に記載の方法。
【請求項82】
ペグ化インターフェロンαおよびリバビリンが共投与される、請求項71−76のいずれか一項に記載の方法。
【請求項83】
ペグ化インターフェロンαが、ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2bである、請求項82に記載の方法。
【請求項84】
VX−950;ペグ化インターフェロン α−2aまたはペグ化インターフェロン α−2b;および、リバビリンが共投与される、請求項82に記載の方法。
【請求項85】
VX−950およびVX−222を約12週間共投与する、請求項84に記載の方法。
【請求項86】
ペグ化インターフェロンおよびリバビリンを約12週間共投与する、請求項84に記載の方法。
【請求項87】
ペグ化インターフェロンおよびリバビリンを約24週間共投与する、請求項84に記載の方法。
【請求項88】
a)約20mgないし約400mg量のVX−222;および
b)約100mgないし約1,500mg量のVX−950
を含む、薬学的に許容される組成物。
【請求項89】
VX−222が、50mgまたは50mgより多く、かつ400mg未満の量である、請求項88に記載の組成物。
【請求項90】
VX−222が約100mgないし約400mgの量であり、VX−950が約300mgないし約750mgの量である、請求項88に記載の組成物。
【請求項91】
VX−950が約375mgの量である、請求項88−90のいずれか一項に記載の組成物。
【請求項92】
VX−222が約50mgの量である、請求項88−90のいずれか一項に記載の組成物。
【請求項93】
VX−222が約200mgの量である、請求項88−90のいずれか一項に記載の組成物。
【図1】

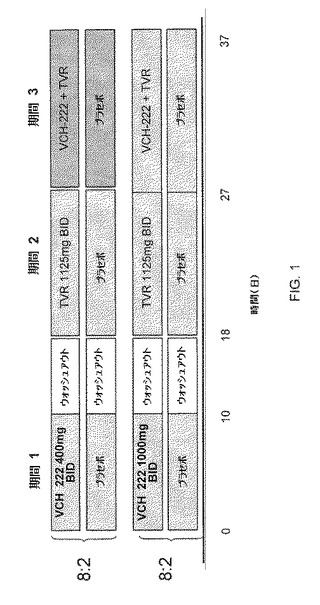
【図2】
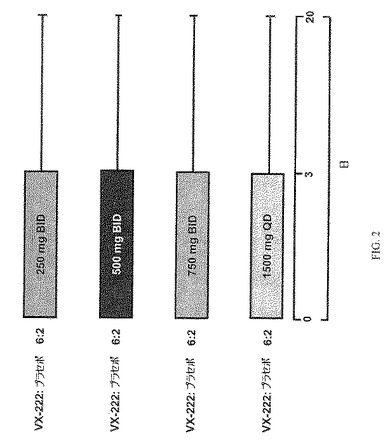
【図3】
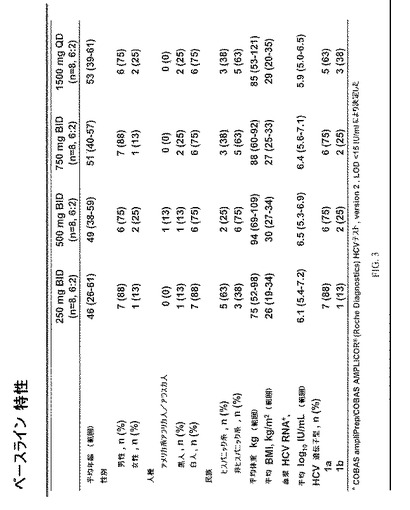
【図4】
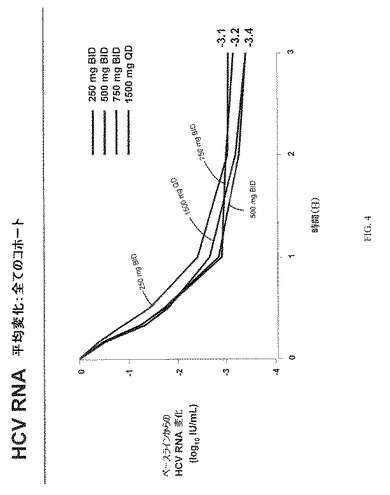
【図5】
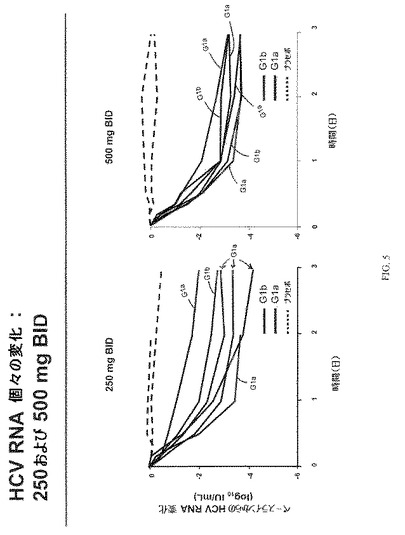
【図6】
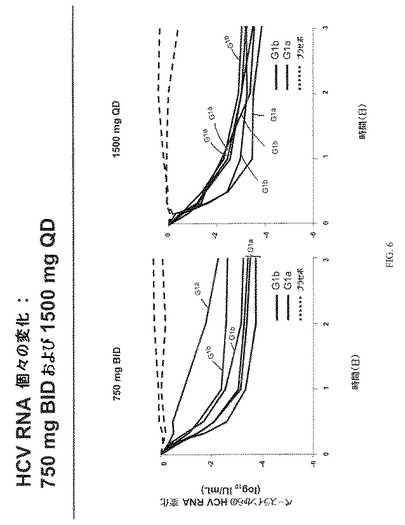
【図7】
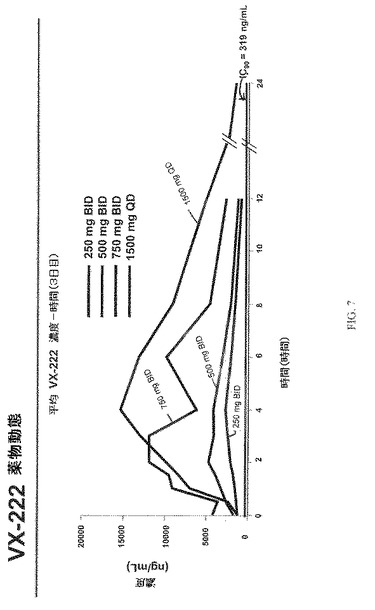
【図8】

【図9】
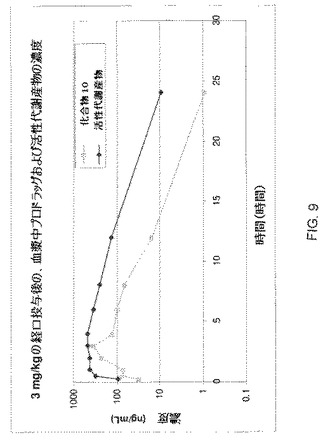
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公表番号】特表2013−518124(P2013−518124A)
【公表日】平成25年5月20日(2013.5.20)
【国際特許分類】
【出願番号】特願2012−551302(P2012−551302)
【出願日】平成23年1月28日(2011.1.28)
【国際出願番号】PCT/US2011/022854
【国際公開番号】WO2011/094489
【国際公開日】平成23年8月4日(2011.8.4)
【出願人】(598032106)バーテックス ファーマシューティカルズ インコーポレイテッド (414)
【氏名又は名称原語表記】VERTEX PHARMACEUTICALS INCORPORATED
【住所又は居所原語表記】130 Waverly Street, Camridge, Massachusetts 02139−4242, U.S.A.
【Fターム(参考)】
【公表日】平成25年5月20日(2013.5.20)
【国際特許分類】
【出願日】平成23年1月28日(2011.1.28)
【国際出願番号】PCT/US2011/022854
【国際公開番号】WO2011/094489
【国際公開日】平成23年8月4日(2011.8.4)
【出願人】(598032106)バーテックス ファーマシューティカルズ インコーポレイテッド (414)
【氏名又は名称原語表記】VERTEX PHARMACEUTICALS INCORPORATED
【住所又は居所原語表記】130 Waverly Street, Camridge, Massachusetts 02139−4242, U.S.A.
【Fターム(参考)】
[ Back to top ]