
C型肝炎ウイルス感染阻害剤及びそれを含有する医薬組成物
【課題】C型肝炎ウイルスの生活環における脱殻過程及び/又は感染後期過程でウイルス粒子の増殖を阻害する新規な作用機序のC型肝炎ウイルス感染阻害剤を提供し、C型肝炎を治療又は予防すること。
【解決手段】本発明は、以下の一般式(I)で表される化合物又はその薬学的に許容される塩を有効成分として含有する、C型肝炎ウイルス阻害剤を提供する。
【化1】

【解決手段】本発明は、以下の一般式(I)で表される化合物又はその薬学的に許容される塩を有効成分として含有する、C型肝炎ウイルス阻害剤を提供する。
【化1】
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、C型肝炎ウイルス感染阻害剤及びそれを含有する医薬組成物に関する。
【背景技術】
【0002】
C型肝炎ウイルス(Hepatitic C Virus;以下、特に断りのない限りHCVという。)は、フラビウイルス科ヘパシウイルス(Hepacivirus)属に分類されるRNAウイルスである。本ウイルスは、一本鎖RNA(+)ゲノムとエンベロープからなる構造を有する(非特許文献1)。
【0003】
同じ肝炎ウイルスであっても、DNAウイルスであるB型肝炎ウイルス(HBV)は、免疫能の未熟な新生児及び乳幼児期以外では、感染しても免疫機構により排除され、急性感染で終わる。一方、HCVは、未だ明らかではない原因により宿主の免疫機構を回避する。そのため免疫機構の発達した大人に感染した場合であっても、持続性感染に移行することが多い。
【0004】
HCV感染により引き起こされる肝炎(C型肝炎)は、発症後20数年の経過の中で肝硬変に至り、最終的には肝癌に至る可能性がある。肝癌は、手術で癌を摘出しても非癌部で引き続き起こる炎症により再発する患者の多いことが知られている。
【0005】
HCV感染者は、日本国内で約200万人、世界では約2億人存在すると推定されている。また、HCVに感染していることを自覚していない無症候性キャリアも多数存在している。現在、日本国内では、肝細胞癌による犠牲者は年間約3.5万人となっており、その8割はHCV感染によるものである(非特許文献2)。
【0006】
C型肝炎に対しては、現在、インターフェロンとリバビリン(Ribavirin;1‐β‐D−リボフラノシル‐1H‐1,2,4‐トリアゾール‐3−カルボキサミド)の併用療法が一般的である(非特許文献3)。しかしながら、この併用療法は、患者全体の約50%にしか有効ではなく、また、血球減少症や溶血性貧血等の重篤な副作用を起こすことが知られている(非特許文献4)。
【0007】
それゆえ、薬効の優れた新たなC型肝炎ウイルス感染阻害剤を創出することが求められており、そのためには、HCVがどのようにして宿主細胞に侵入し、複製増殖するのか、そして宿主細胞にどのような影響を与えるのかといったHCVの生活環の詳細を明らかにし、C型肝炎ウイルス感染阻害剤を探索する必要がある。
【0008】
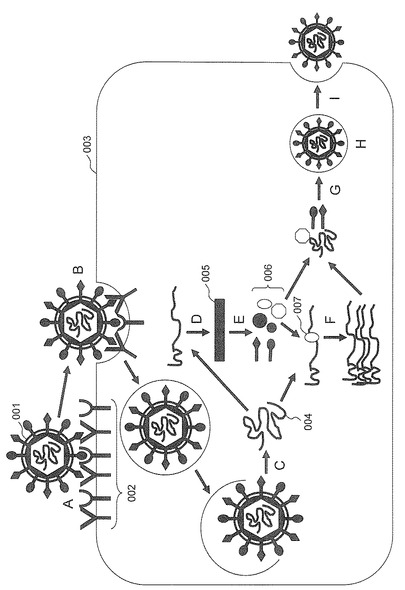
図1は、HCVの生活環を示している。HCV粒子(001)は、まず、宿主細胞(003)の細胞表面に存在するウイルス受容体(002)、具体的には、宿主細胞表面に存在するCD81(非特許文献5)及びClaudin-1(非特許文献6)等の受容体に吸着し(A:吸着過程)、エンドサイトーシスによって宿主細胞のエンドソーム内に取り込まれる(B:侵入過程)。次に、プラス鎖RNAであるウイルスゲノム(004)がウイルス粒子から宿主細胞質内に放出される(C:脱殻過程)。続いて、放出されたウイルスゲノムにコードされた前駆体タンパク質(005)をコードするmRNAが翻訳され(D:翻訳過程)、さらに生じた前駆体タンパク質がプロセッシング(E:プロセッシング過程)されることによって、ウイルス粒子を形成する構造タンパク質(Core、E1、E2)とウイルス粒子に含まれない非構造タンパク質(NS2、NS3、NS4A、NS4B、NS5A、NS5B)が生成される(006)。その後、ウイルスゲノムが、非構造タンパク質の一つであるRNA依存性RNAポリメラーゼ(NS5B)(007)によって複製される(F:複製過程)。複製されたウイルスゲノムは、構造タンパク質であるコアタンパク質(Core)及びエンベロープタンパク質(E1、E2)によってパッケージングされ、新たなウイルス粒子が形成される(G:パッケージング過程)。新たに形成されたウイルス粒子は、小胞体及びゴルジ体で成熟し(H:成熟過程)、最後に、宿主細胞膜を破り、細胞外に放出される(I:細胞外放出過程)(非特許文献7)。よって、上記HCVの生活環のいずれかの経路を阻害する化合物は、C型肝炎ウイルス感染阻害剤となり得る。
【0009】
1999年に、HCVを人工的に複製させる実験システムとしてサブゲノミックHCVレプリコンシステムが登場した(非特許文献8)。このシステムでは、HCVゲノムの複製に必須のNS3〜NS5B領域及びゲノムの両末端からなるHCVサブゲノムが細胞内で複製されるように構成されており、一細胞あたりのHCVサブゲノムのコピー数は数千というレベルに達する。それゆえ、当該評価系を用いて、HCV感染を阻害する化合物、すなわちC型肝炎ウイルス感染阻害剤を探索することが可能となった(非特許文献9)。
【0010】
上記サブゲノミックHCVレプリコンシステムから選択されたC型肝炎ウイルス感染阻害剤には、ウイルスタンパク質の翻訳後のプロセッシング過程(図1E)とHCV感染のウイルスゲノムの複製過程(図1F)とに作用するRNAポリメラーゼ、ヘリカーゼ、プロテアーゼ等のウイルス酵素を標的としたHCV酵素阻害剤が知られている(特許文献1、2)。しかしながら、ウイルス酵素をコードする遺伝子は、変異を生じ易いため、これらのHCV酵素阻害剤に対しては、耐性ウイルス株が容易に出現してしまう。それゆえ、これらHCV酵素阻害剤では薬効として不十分という問題がある(非特許文献10)。したがって、HCVの生活環において他の過程を阻害する新たなC型肝炎ウイルス感染阻害剤を見出すことが求められている。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】国際公開WO2008/098859
【特許文献2】特表2007-513971
【非特許文献】
【0012】
【非特許文献1】Rosenbergら、J. Mol. Biol.、 2001年、313巻、p.451-464
【非特許文献2】Saitoら、Proc. Natl, Acad. Sci. USA、1990年、87巻、p.6547-6549
【非特許文献3】HcHutchinsonら、N. Engl. J. Med.、1998年、339巻、p.1485-1492
【非特許文献4】Moriら、Biochem. Biophis. Res. Commun.、1992年、183巻、p.334-342
【非特許文献5】Bartoschら、J.Exp.Med.、2003年、197巻、p.633-642
【非特許文献6】Evans、Nature、2008年、446巻、p.801-805
【非特許文献7】Moradpourら、SWISS MED WKLY、2001年、131巻、p.291-298
【非特許文献8】Lomannら、Science、1999年、285巻、p.110-113
【非特許文献9】Bartenschlager、Nature Rev.Drug Discovery、2002年、1巻、p.911-916
【非特許文献10】Luら、Antimicro Agents Chemother.、2004年、48巻、p.2260-2266
【発明の概要】
【発明が解決しようとする課題】
【0013】
本発明は、HCVの生活環における脱殻過程又は感染後期過程でウイルス粒子の増殖を阻害する新規な作用機序のC型肝炎ウイルス感染阻害剤を提供し、C型肝炎を治療又は予防することを目的とする。
【課題を解決するための手段】
【0014】
上記目的を達成するため、本発明者らは、植物及び微生物由来の純天然物を含む化合物ライブラリーから、HCVの生活環における過程、特に脱殻過程(図1C)及び/又は感染後期過程(パッケージング過程(図1G)〜細胞外放出過程(図1I))における少なくとも一の過程を阻害できる化合物の単離を試みた。その結果、当該作用効果を有する化合物として、スピノシン誘導体が得られた。スピノシン誘導体は、マクロラクトン環及び二種のデオキシ糖を有する化合物である。この化合物は、主に放線菌の一種であるSaccharopolyspora spinosaの二次代謝物として得られる。特にスピノシンAとスピノシンDの混合物であるスピノサド(Spinosad)は、駆虫活性を有する農薬として知られている。例えば、ヒツジに付いたシラミの処置等に使用される他、ヒトに寄生するシラミ等の発生防除にも有用であることが報告されている(米国特許第6,063,771号)。このように、スピノシン誘導体の駆虫活性については、従来から多くの報告があったが、HCV感染の阻害活性については、これまで全く知られていなかった。本願発明は、当該新たな知見に基づいて完成されたものであり、すなわち、下記(1)〜(5)を提供する。
【0015】
(1) 以下の一般式(I)で示される化合物又はその薬学的に許容される塩を有効成分として含有する、C型肝炎ウイルス感染阻害剤。
【化1】
[式中、R1及びR2は、それぞれ独立して水素原子又は低級アルキル基を表し、R3は、水素原子又は以下の一般式(II)で示される基を表し、
【化2】
R4は、置換基で置換された若しくは置換されていない、低級アルキル基、低級シクロアルキル基又は低級シクロアルケニル基、を表し、R5〜R9は、それぞれ独立して水素原子又は低級アルキル基を表す。]
(2) R1及びR2は、それぞれ独立して水素原子、メチル基又はエチル基を表し、R3は、一般式(II)で示される基を表し、R4は、置換基で置換された若しくは置換されていないメチル基、エチル基、炭素数3〜8のシクロアルキル基又は炭素数3〜8のシクロアルケニル基を表し、R5〜R9は、それぞれ独立して水素原子又はメチル基を表す、上記(1)に記載のC型肝炎ウイルス感染阻害剤。
(3) 上記置換基は、ハロゲン、ヒドロキシル基、低級アルキル基、低級アルコキシ基又はシアノ基である、上記(1)又は(2)に記載のC型肝炎ウイルス感染阻害剤。
(4) C型肝炎ウイルスの生活環における、宿主細胞への吸着過程、侵入過程、前駆体タンパク質の翻訳過程及びプロセッシング過程並びにウイルスゲノムの複製過程、に対しては作用せず、脱殻過程及び/又はパッケージング過程から細胞外放出過程までの感染後期過程における少なくとも一の過程でウイルス粒子の増殖を阻害する、上記(1)〜(3)のいずれかに記載のC型肝炎ウイルス感染阻害剤。
(5) 上記(1)〜(4)のいずれかに記載のC型肝炎ウイルス阻害剤を有効成分として含有する、医薬組成物。
【0016】
なお、上記の一般式(I)で示される化合物は、突然変異によって出現する薬剤耐性のHCVに対してより効果的にその増殖を抑制することができるため、以下の化学式(Ia)及び/又は(Ib)で表される化合物であることが好ましい。
【0017】
【化3】
【0018】
【化4】
【発明の効果】
【0019】
本発明のC型肝炎ウイルス感染阻害剤によれば、突然変異によって出現する薬剤耐性のHCVに対しても効果的にその増殖を抑制することができ、C型肝炎の治療及び予防に対して有効な医薬を提供することができる。
【図面の簡単な説明】
【0020】
【図1】HCVの生活環を模式的に表した図である。
【図2】3種類の評価系(アッセイ)で得られる結果と阻害剤の関係を表した図である。
【図3】感染性完全長ゲノムHCVレプリコンを含む感染性HCV粒子作製用プラスミドベクターpHH/ZeoJFH1を模式的に表した図である。
【図4】C型肝炎ウイルス感染阻害剤のスクリーニング方法の概要を表した図である。
【図5】化合物1のHCVccアッセイの結果を表した図である。
【図6】サブゲノミックHCVレポーターレプリコン作製ベクターの構造を模式的に表した図である。遺伝子型1bを含むpSGR-Con1/Lucと遺伝子型p2aを含むpSGR-JFH/Lucとでは、図中網かけした領域がそれぞれで異なる。
【図7】化合物1の遺伝子型1bのサブゲノミックHCVレポーターレプリコンRNAを用いたレプリコンアッセイの結果を表した図である。Aは陽性対照であるIFN-αの結果を、またBは化合物1の結果を、それぞれ示す。
【図8】化合物1の遺伝子型2aのサブゲノミックHCVレポーターレプリコンRNAを用いたレプリコンアッセイの結果を表した図である。Aは陽性対照であるIFN-αの結果を、Bは化合物1の結果を、それぞれ示す。
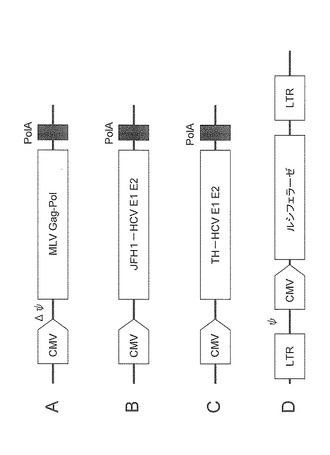
【図9】シュードウイルス作製用ベクター群(A〜D)を模式的に表した図である。
【図10】化合物1のHCVppアッセイの結果を表した図である。
【発明を実施するための形態】
【0021】
1.C型肝炎ウイルス感染阻害剤
本発明の第1の実施形態は、C型肝炎ウイルス感染阻害剤である。本発明のC型肝炎ウイルス感染阻害剤は、下記一般式(I)で表される化合物又はその薬学的に許容される塩を有効成分として含有することを特徴とする。
【0022】
「HCV」(C型肝炎ウイルス:Hepatitic C Virus)は、前述のように、フラビウイルス科ヘパシウイルス属に分類される一本鎖RNAウイルスであって、本発明の標的ウイルスである。
【0023】
「C型肝炎ウイルス感染阻害剤」とは、HCVの感染に必要な一以上の機能を阻害することによってHCV粒子の増殖を阻害又は抑制できる化合物をいう。本発明において「感染」とは、図1で示すHCVの生活環において、HCV粒子が宿主細胞の細胞表面に吸着し、宿主細胞内で増殖された後、細胞外に放出されるまでの過程(A〜I)をいう。つまり、C型肝炎ウイルス感染阻害剤は、吸着過程(A)、侵入過程(B)、脱穀過程(C)、翻訳過程(D)、プロセッシング過程(E)、複製過程(F)、パッケージング過程(G)、成熟過程(H)及び細胞外放出過程(I)のうち少なくとも一の過程に必要な機能を阻害する化合物である。
【0024】
本発明のC型肝炎ウイルス感染阻害剤は、上記感染における脱殻過程(C)、及び/又は感染後期過程、すなわち、パッケージング過程(G)から細胞外放出過程(I)までの感染後期に関与する過程における少なくとも一の過程を阻害する作用を有することでHCVの宿主細胞への感染を阻害することができる。さらに、本発明のC型肝炎ウイルス感染阻害剤は、上記感染における、脱殻過程(C)、及び/又は感染後期過程の少なくとも一の過程を阻害する作用を有するが、吸着過程(A)、侵入過程(B)、翻訳過程(D)、プロセッシング過程(E)及び複製過程(F)に対しては阻害作用を示さない性質を有していてもよい。
【0025】
「宿主細胞」とは、HCVが感染する標的細胞をいう。具体的には、肝細胞又はリンパ球が該当する。宿主細胞の由来する生物種としては、ヒト、チンパンジー又はツパイが挙げられる。宿主細胞は、ヒトの初代培養肝細胞若しくは肝癌細胞株又はその両者を融合させた細胞、チンパンジーやツパイの初代肝細胞又はヒトリンパ球由来細胞株であってもよい。
【0026】
以下、本発明のC型肝炎ウイルス感染阻害剤について詳細に説明をする。
1−1.一般式(I)で表される化合物
本発明のC型肝炎ウイルス感染阻害剤は、有効成分として下記一般式(I)で示される化合物を少なくとも1種含有する。
【0027】
【化5】
[式中、R1及びR2は、それぞれ独立して水素原子、低級アルキル基を表し、R3は、水素原子又は一般式(II)で示される基を表し、
【化6】
R4は、置換基で置換された若しくは置換されていない、低級アルキル基、低級シクロアルキル基又は低級シクロアルケニル基を表し、R5〜R9は、それぞれ独立して水素原子又は低級アルキル基を表す。]
【0028】
「置換基で置換された若しくは置換されていない」とは、本発明の化合物がHCV感染阻害活性を有する限りにおいて、R4を構成する低級アルキル基、低級シクロアルキル基又は低級シクロアルケニル基が置換基で置換されてもよいし、又は置換されていなくてもよいことをいう。ここでいう「置換基」には、ハロゲン、ヒドロキシル基、低級アルキル基、低級アルケニル基、低級アルキニル基、低級アルコキシ基、アミノ基、低級アルキルアミノ基、アミド基、N-低級アルキルアミド基、シアノ基、ニトロ基、カルボキシル基、カルバモイル基、硫酸基又は亜硫酸基等が挙げられる。好ましくは、ハロゲン、ヒドロキシル基、低級アルキル基、低級アルコキシ基又はシアノ基である。上記置換基の数は、化合物あたり1個若しくは複数個、例えば、1〜4個、1〜3個、又は1若しくは2個である。
【0029】
「ハロゲン」は、フッ素原子(F)、塩素原子(Cl)、臭素原子(Br)又はヨウ素原子(I)である。
【0030】
ここで、「低級アルキル基」とは、炭素数1〜4個の直鎖状又は炭素数3〜4個の分枝状のアルキル基であり、例えば、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、tert−ブチル基等が挙げられる。本発明の一般式(I)で示される化合物の場合、R1、R2又はR4における低級アルキル基は、メチル基又はエチル基が、R5〜R9における低級アルキル基は、メチル基が特に好ましい。
【0031】
「低級シクロアルキル基」とは、炭素数3〜8個の環状アルキル基であり、例えば、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基、シクロヘプチル基、シクロオクチル基等が挙げられる。
【0032】
「低級シクロアルケニル基」とは、炭素数3〜8個の環状アルケニル基であり、例えば、シクロペンテニル基、シクロヘキセニル基等が挙げられる。
【0033】
「低級アルケニル基」とは、炭素数2〜8個、好ましくは2〜4個の直鎖状又は分岐状のアルケニル基である。具体的にはエテニル、プロペニル、ブテニル、ブタジエニル、ペンテニル、ペンタジエニル、ヘキセニル、ヘキサジエニル、ヘプテニル、ヘプタジエニル、オクテニル、オクタジエニル又はこれらの異性体等が含まれる。
【0034】
「低級アルキニル基」は、炭素数2〜8個、好ましくは2〜4個の直鎖状又は分岐状のアルキニル基である。具体的には、エチニル、プロピニル、ブチニル、ブタジイニル、ペンチニル、ペンタジイニル、ヘキシニル、ヘキサジイニル、ヘプチニル、ヘプタジイニル、オクチニル又はオクタジイニル等が含まれる。
【0035】
「低級アルコキシ基」とは、炭素数1〜4個の直鎖状又は炭素数3〜4個の分枝状のアルキル基であり、例えば、メトキシ基、エトキシ基、プロポキシ基、イソプロポキシ基、n−ブトキシ基、イソブトキシ基、tert−ブトキシ基等が挙げられる。
【0036】
「低級アルコキシ基」は、炭素数1〜6個、好ましくは1〜4個の直鎖状又は分岐状のアルコキシ基である。具体的にはメトキシ、エトキシ、n−プロポキシ、イソプロポキシ、n−ブトキシ、イソブトキシ、s−ブトキシ、t−ブトキシ、n−ペンチルオキシ、イソペンチルオキシ、n−ヘキシルオキシ又はイソヘキシルオキシ等が含まれる。
【0037】
本発明のC型肝炎ウイルス感染阻害剤は、一般式(I)で示される単一の化合物だけではなく、一般式(I)で示される二種類以上の化合物の混合物も包含する。例えば、本発明のC型肝炎ウイルス感染阻害剤は、一般式(I)において、R1が水素原子、R2及びR5〜R9がメチル基、R3が一般式(II)で示される基、R4が置換されていないエチル基、で表される以下の化学式(Ia)で示されるスピノシンA、及び、R1、R2及びR5〜R9がメチル基、R3が一般式(II)で示される基、R4が置換されていないエチル基、で表される以下の化学式(Ib)で示されるスピノシンD、をそれぞれ単独で有効成分として含有していてもよいし、スピノサドのようにスピノシンAとスピノシンDを組合せた混合物として含有していてもよい。
【0038】
【化7】
【0039】
【化8】
【0040】
1−2.薬学的に許容される塩
「薬学的に許容される塩」とは、一般式(I)で示される化合物の塩であって、これらの化合物上の特定の置換基(例えば、ヒドロキシル基)に基づいて、塩基又は酸を用いて調製された薬学的に非毒性の活性化合物の塩をいう。使用した塩基又は酸により塩基性付加塩と酸付加塩とに分類できる。
【0041】
「塩基性付加塩」としては、例えば、ナトリウム塩、カリウム塩等のアルカリ金属塩、カルシウム塩、マグネシウム塩等のアルカリ土類金属塩、トリメチルアミン塩、トリエチルアミン塩、ジシクロヘキシルアミン塩、エタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩、ブロカイン塩等の脂肪族アミン塩、N,N-ジベンジルエチレンジアミン等のアラルキルアミン塩、ピリジン塩、ピコリン塩、キノリン塩、イソキノリン塩等の複素環芳香族アミン塩、アルギニン塩、リジン塩等の塩基性アミノ酸塩、テトラメチルアンモニウム塩、テトラエチルアモニウム塩、ベンジルトリメチルアンモニウム塩、ベンジルトリエチルアンモニウム塩、ベンジルトリブチルアンモニウム塩、メチルトリオクチルアンモニウム塩、テトラブチルアンモニウム塩等の第4級アンモニウム塩、アンモニウム塩等が挙げられる。
【0042】
「酸付加塩」としては、例えば、塩酸塩、硫酸塩、硝酸塩、リン酸塩、炭酸塩、炭酸水素塩、過塩素酸塩等の無機酸塩、酢酸塩、プロピオン酸塩、乳酸塩、マレイン酸塩、フマル酸塩、酒石酸塩、リンゴ酸塩、クエン酸塩、アスコルビン酸塩等の有機酸塩、メタンスルホン酸塩、イセチオン酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩等のスルホン酸塩、アスパラギン酸塩、グルタミン酸塩等の酸性アミノ酸等が挙げられる。
【0043】
一般式(I)で示される化合物又はその薬学的に許容される塩は、本発明のC型肝炎ウイルス感染阻害剤又は後述する医薬組成物中でプロドラッグの形態で存在していてもよい。ここでいう「プロドラッグ」は、生理学的条件下で容易に化学変化を受け、その結果として上記C型肝炎ウイルス感染阻害剤又はその活性形態を提供する化合物である。例えば、投与前は、一般式(I)で示される化合物とは異なる構造の化合物としてC型肝炎ウイルス感染阻害剤又は医薬組成物中に存在し、投与後、例えば、消化管内で消化酵素の作用によって一般式(I)で示される化合物又はその活性型に変換されるものをいう。あるいは、ex vivo環境で化学的又は生化学的方法によって一般式(I)で示される化合物又はその活性型に変換されるものも含み得る。
【0044】
プロドラッグの形態は、例えば、一般式(I)で示される化合物又はその薬学的に許容される塩を経口投与しても生体内で直接利用ができない場合や、利用困難な場合に特に有効である。また、プロドラッグは、一般式(I)で示される化合物又はその薬学的に許容される塩よりもC型肝炎ウイルス感染阻害剤又は医薬組成物での溶解度が高い場合が多く、その点においても有用である。
【0045】
本発明のC型肝炎ウイルス感染阻害剤は、一般式(I)で示される化合物又はその薬学的に許容される塩を薬学的に許容可能な溶媒に溶解して利用することができる。
【0046】
薬学的に許容可能な溶媒としては、例えば、水、エタノール、プロピレングリコール、エトキシ化イソステアリルアルコール、ポリオキシ化イソステアリルアルコール、ポリオキシエチレンソルビタン脂肪酸エステル類等が挙げられる。これらは、殺菌されていることが望ましく、必要に応じて血液と等張に調整されていることが好ましい。
【0047】
2.医薬組成物
本発明の第2の実施形態は、医薬組成物である。本発明の医薬組成物は、C型肝炎の治療又は予防するため医薬であって、本発明の上記実施形態1のC型肝炎ウイルス感染阻害剤を有効成分として含有することを特徴とする。
【0048】
本明細書において「治療」とは、C型肝炎及び/又はそれに伴う症状を緩和又は除去することをいい、「予防」とはC型肝炎に罹患しないようにすることをいう。
【0049】
2−1.薬学的に許容可能な担体
本発明の医薬組成物は、薬学的に許容可能な担体を含むことができる。「薬学的に許容可能な担体」とは、製剤技術分野において通常使用し得る添加剤をいう。例えば、賦形剤、結合剤、崩壊剤、充填剤、乳化剤、流動添加調節剤、滑沢剤等が挙げられる。
【0050】
賦形剤としては、例えば、単糖、二糖類、シクロデキストリン、多糖類等の糖(より具体的には、例えば、グルコース、スクロース、ラクトース、ラフィノース、マンニトール、ソルビトール、イノシトール、デキストリン、マルトデキストリン、デンプン、セルロース等)、金属塩(例えば、塩化ナトリウム、リン酸ナトリウム、リン酸カルシウム、硫酸カルシウム、硫酸マグネシウム、炭酸カルシウム等)、クエン酸、酒石酸、グリシン、ポリエチレングリコール(PEG)、プルロニック、カオリン、ケイ酸、それらの組み合わせ等が挙げられる。
【0051】
結合剤としては、例えば、トウモロコシ、コムギ、コメ、ジャガイモのデンプンを用いたデンプン糊、単シロップ、グルコース液、ゼラチン、トラガカント、メチルセルロース、ヒドロキシプロピルメチルセルロース、カルボキシメチルセルロースナトリウム、セラック、ポリビニルピロリドン等が挙げられる。
【0052】
崩壊剤としては、例えば、上記デンプンや、乳糖、カルボキシメチルデンプン、架橋ポリビニルピロリドン、アガー、ラミナラン末、炭酸水素ナトリウム、炭酸カルシウム、アルギン酸、アルギン酸ナトリウム、ポリオキシエチレンソルビタン脂肪酸エステル、ラウリル硫酸ナトリウム、ステアリン酸モノグリセリドや、それらの塩等が挙げられる。
【0053】
充填剤としては、例えば、上記糖、リン酸カルシウム(例えば、リン酸三カルシウム又はリン酸水素カルシウム等)等が挙げられる。
【0054】
乳化剤としては、例えば、ソルビタン脂肪酸エステル、グリセリン脂肪酸エステル、ショ糖脂肪酸エステル、プロピレングリコール脂肪酸エステル等が挙げられる。
【0055】
流動添加調節剤及び滑沢剤としては、例えば、ケイ酸塩、タルク、ステアリン酸塩、ポリエチレングリコール等が挙げられる。
【0056】
このような担体は、主として医薬組成物の剤形形成を容易にし、また剤形及び薬剤効果を維持するために用いられるものであり、必要に応じて適当な種類を適宜組み合わせて使用すればよい。本発明の医薬組成物は、上記の添加剤の他、必要に応じて矯味矯臭剤、溶解補助剤(可溶化剤)、懸濁剤、希釈剤、界面活性剤、安定剤、吸収促進剤(例えば、第4級アンモニウム塩類、ラウリル硫酸ナトリウム等)、増量剤、付湿剤、保湿剤(例えば、グリセリン、澱粉等)、吸着剤(例えば、澱粉、乳糖、カオリン、ベントナイト、コロイド状ケイ酸等)、崩壊抑制剤(例えば、白糖、ステアリン、カカオバター、水素添加油等)、コーティング剤、着色剤、保存剤、抗酸化剤、香料、風味剤、甘味剤、緩衝剤等を含むこともできる。
【0057】
2−2.医薬組成物の剤形
本発明の医薬組成物の剤形は、投与方法及び/又は処方条件によって異なる。投与方法については後述するが、通常は経口投与又は非経口投与に大別することができる。
【0058】
経口投与に適した剤形としては、例えば、固形剤(錠剤、丸剤、舌下剤、カプセル剤、ドロップ剤、トローチ剤を含む)、顆粒剤、粉剤、散剤、液剤等を挙げることができる。さらに固形剤は、必要に応じ、当該分野で公知の剤皮を施した剤形、例えば、糖衣錠、ゼラチン被包錠、腸溶錠、フィルムコーティング錠、二重錠、多層錠とすることができる。
【0059】
非経口投与は、さらに組織内投与、局所投与、経直腸的投与に細分でき、それぞれの投与方法に適した剤形にすることができる。組織内投与に適した剤形としては、例えば、液剤である注射剤が挙げられる。局所投与に適した剤形としては、例えば、液剤(塗布剤、点眼剤、点鼻剤、吸引剤を含む)、懸濁剤(乳剤、クリーム剤を含む)、粉剤(点鼻剤、吸引剤を含む)、ペースト剤、ゲル剤、軟膏剤、硬膏剤等を挙げることができる。経直腸的投与に適した剤形としては、例えば、坐剤等を挙げることができる。
【0060】
上記各剤形の形状、大きさについては、いずれもそれぞれの剤形において当該分野で公知の剤形の範囲内にあればよく、特に限定はしない。
【0061】
2−3.医薬組成物の製造
本発明のC型肝炎ウイルス感染阻害剤を含有する医薬組成物を製剤化するには、原則として当該分野で公知の方法を利用することができる。例えば、Remington’s Pharmaceutical Sciences(Merck Publishing Co.,Easton,Pa.)に記載の方法を用いればよい。
【0062】
医薬組成物における上記のC型肝炎ウイルス感染阻害剤の含有量は、使用するC型肝炎ウイルス感染阻害剤の種類及び/又はその有効量、医薬組成物の剤形(形態、大きさを含む)、並びに添加する担体の種類によって異なり、それぞれの条件において適宜選択される。
【0063】
本明細書で使用する場合、「有効量」とは、上記一般式(I)で示される化合物又はその薬学的に許容される塩が有効成分としての機能を発揮する上で必要な量、すなわち、本発明ではC型肝炎ウイルス感染阻害剤がHCVの宿主細胞への感染を阻害する上で必要な量であって、かつそれを投与する患者に対して有害な副作用をほとんど又は全く付与しない量をいう。この有効量は、患者の情報及び投与経路等の様々な条件によって変化し得る。
【0064】
「患者の情報」とは、病気の進行度や重症度、全身の健康状態、年齢、体重、性別、食生活、薬剤感受性、併用医薬の有無及び治療に対する耐性等を含む。上記のC型肝炎ウイルス感染阻害剤及び医薬組成物の最終的な投与量及び有効量は、個々の患者の情報等に応じて、最終的には医師の判断によって決定される。
【0065】
医薬組成物における本発明のC型肝炎ウイルス感染阻害剤の含有量の一例としては、本発明の医薬組成物を上記固形剤として調製する場合であれば、1錠重量に対して通常0.1重量%〜30重量%、好ましくは0.5重量%〜20重量%、より好ましくは1重量%〜10重量%のC型肝炎ウイルス感染阻害剤を含有していればよい。具体的には、例えば、1錠200mgの錠剤を製造する場合、一般式(I)で示される化合物を1錠あたり通常200μg〜60mg含んでいればよい。これ以外には、薬学的に許容可能な担体として、上記添加剤を当該分野で公知の含有率で含むことができる。
【0066】
固形剤の場合、C型肝炎ウイルス感染阻害剤の有効量は、剤数によって調整することが可能であるため、必ずしも1錠中に有効量を含有させる必要はなく、一投与単位中に、すなわち、複数の錠剤等を服用する場合、1回の服用単位中に、総計でC型肝炎ウイルス感染阻害剤が有効量含有されていればよい。
【0067】
また、医薬組成物における本発明のC型肝炎ウイルス感染阻害剤の含有量の他の例として、本発明の医薬組成物を注射剤として調製する場合には、薬学的に許容可能な溶媒、好ましくは血液と等張にした希釈剤に溶解した上記HCV阻害剤を用いて当該分野で慣用されている方法により製造することができる。一投与単位の注射剤中に含有される上記HCV阻害剤量は、通常0.1%(w/v)〜30%(w/v)、好ましくは0.1%(w/v)〜20%(w/v)、より好ましくは0.1%(w/v)〜30%(w/v)である。具体的には、例えば、1回2mLの注射剤を製造する場合、一般式(I)で示される化合物を通常2μg〜600μg含むことができる。
【0068】
注射剤には、等張性の溶液を調製するのに充分な量の食塩、グルコース、グリセリン、通常の溶解補助剤、緩衝剤、pH調整剤、無痛化剤等も配合することができる。これら溶液、乳剤及び懸濁剤は、一般式(I)で示される化合物又はその薬学的に許容される塩の所定量を、水性や油性の非毒性の溶媒及び希釈剤に溶解又は懸濁し、さらに必要に応じて等張化剤等を加え、滅菌することにより調製することができる。
【0069】
本発明の医薬組成物を局所剤として調製する場合には、一般式(I)で示される化合物又はその薬学的に許容される塩の所定量を、外用製剤の目的に適合する香料、着色料、充填剤、界面活性剤、保湿剤、皮膚軟化剤、ゲル化剤、担体、保存剤、安定剤等のうちの一種以上と組み合わせることにより調製することができる。
【0070】
本発明の医薬組成物を坐剤として調製する場合には、基材としてこの分野で従来公知のものを広く使用することができる。このような基材としては、例えばパルミチン酸ミリスチルエステル等の高級エステル類、高級アルコール類、ポリエチレングリコール、カカオ脂、ゼラチン、半合成グリセリドや、これらの混合物などの低融点基材を挙げることができる。坐剤は、一般式(I)で示される化合物又はその薬学的に許容される塩の所定量を上記基材に混入し、成型することにより調製することができる。
【0071】
3.C型肝炎ウイルス感染阻害剤及びその医薬組成物の投与方法
本発明のC型肝炎ウイルス感染阻害剤及びその医薬組成物の投与方法は、当該分野で周知の投与単位形態で投与することができる。投与単位形態には、例えば、経口投与、組織内投与(例えば、皮下投与、筋肉内投与、静脈内投与等)、局所投与(例えば、経皮投与等)又は経直腸的投与等が挙げられる。上記のC型肝炎ウイルス感染阻害剤及びその医薬組成物は、これらの投与単位形態のいずれを使用してもよい。また、C型肝炎ウイルス感染阻害剤及びその医薬組成物は、使用する投与単位形態に適した剤形で投与することが好ましい。
例えば、経口投与であれば、固形、粉末又は液剤で投与することが好ましい。
【0072】
組織内投与の場合、皮下投与、筋肉内投与又は静脈内投与等のいずれの方法も使用できる。本発明の目的が一般式(I)で表される化合物又はその薬学的に許容される塩の作用効果によりHCVの宿主細胞への感染を阻害し、それによりC型肝炎を治療又は予防すること、また、HCVが体液を介して伝播されること、さらに、主たる宿主細胞である肝細胞が存在する肝臓が無数の毛細血管を有することを鑑みれば、血流を介した投与が好ましい。それゆえ、このような血流を介した投与において好ましい投与方法は、注射である。
【0073】
注射の場合、注入部位は特に限定しない。例えば、静脈内、動脈内、肝臓内、筋肉内、関節内、骨髄内、髄腔内、心室内、経皮、皮下、皮内、腹腔内、鼻腔内、腸内、舌下等が挙げられる。好ましくは、静脈内注射、動脈内注射等の血管内への注射である。血流を介して本発明の医薬組成物を直ちに全身に行き渡らせることが可能であり、また侵襲性が比較的低く、患者に与える負担が小さいからである。あるいは、肝臓内又は肝門脈に注射してもよい。これは、本発明のC型肝炎ウイルス感染阻害剤及びその医薬組成物をHCVが局在する部位に直接的に作用させることができるからである。
【0074】
本発明のC型肝炎ウイルス感染阻害剤及びその医薬組成物を投与する場合、前述のように、一投与単位中にHCV阻害活性が発揮され得る有効量が含有されていることが好ましい。
【0075】
具体的な投与量の一例として、例えば、C型肝炎発症初期であって、他の医薬の併用を必要としないヒト成人男子(体重60kg)に経口投与する場合、一日当たりのC型肝炎ウイルス感染阻害剤の有効量は、通常1〜2000mg、好ましくは1〜1000mg、より好ましくは1〜500mgの範囲である。患者の状態又は投与経路等に応じて、上記範囲未満又は上記範囲を超える用量を投与することもできる。HCV感染阻害効果を得る上で、上記のC型肝炎ウイルス感染阻害剤及びその医薬組成物の大量投与が必要な場合、患者に対する負担軽減のために数回に分割して投与することもできる。
【0076】
本発明のC型肝炎ウイルス感染阻害剤及びその医薬組成物は、以下の実施例で示すように、HCVの宿主細胞への感染、特にHCVの宿主細胞への感染における、HCVの脱殻過程及びパッケージングから細胞外放出までの感染後期過程の少なくとも一の過程を阻害する。HCVの脱殻過程及び/又は感染後期過程の少なくとも一の過程を阻害するC型肝炎ウイルス感染阻害剤は、これまでに知られておらず、また、このような薬剤は、現在臨床試験が進行中であるHCV酵素阻害剤と比較すると、薬剤耐性ウイルス株が出現しにくいことが期待される。したがって、これらをC型肝炎の治療又は予防に有効な新規の医薬品として利用できる。
【実施例】
【0077】
以下、実施例を挙げて本発明について具体的に説明するが、本発明は以下の実施例によって限定されるものではない。
【0078】
<実施例1:C型肝炎ウイルス感染阻害剤の探索>
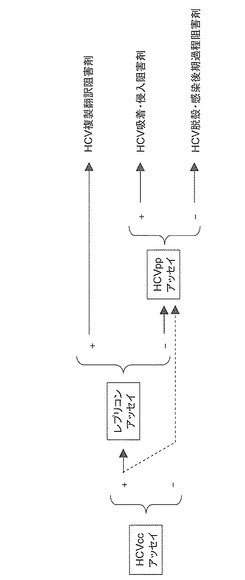
HCVの生活環における経路を阻害する新たなC型肝炎ウイルス感染阻害剤を探索するために、HCVの生活環(図1)を利用した3種類の評価方法によるスクリーニングを行なった。すなわち、第一の方法は、HCVの感染経路(図1A〜Iのいずれか一以上)を阻害する化合物(C型肝炎ウイルス感染阻害剤)をスクリーニングする評価方法(図2:HCVccアッセイ)である。第二の方法は、HCVの生活環のうちHCVゲノムの翻訳過程から複製過程(図1D〜F)を阻害する化合物(HCV複製翻訳阻害剤)をスクリーニングする評価方法(図2:レプリコンアッセイ)である。そして、第三の方法は、HCVの生活環のうちHCVの細胞への吸着過程(図1A)から侵入過程(図1B)を阻害する化合物(HCV吸着・侵入阻害剤)をスクリーニングするための評価方法(図2:HCVppアッセイ)である。
【0079】
ここで、図2に示すように、これらの3つのスクリーニング方法で、第一の方法(HCVccアッセイ)で効果があり(図2の「+」に相当)、第二及び第三の方法(それぞれレプリコンアッセイ及びHCVppアッセイ)で効果が得られない(図2の「−」に相当)化合物は、HCVの宿主細胞での脱殻過程(図1C)及び/又はパッケージング(図1G)から細胞外放出(図1I)までの感染後期過程の少なくとも一の過程を阻害する化合物(HCV脱穀・感染後期過程阻害剤)といえる(図2)。
【0080】
以下、各評価方法によるスクリーニングとその結果について説明をする。
1.HCVccアッセイによるスクリーニング
(1)感染性HCVの調製
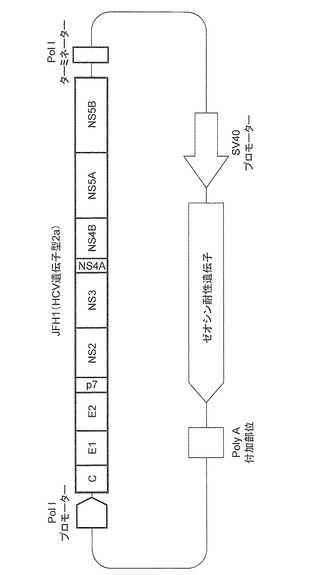
国際公開WO07/037428 /及びJ.Virol.(2010)84、5824−35(Masaki,T.et al.)に記載の方法に従い、図3に示すRNA PolIプロモーター/ターミネーター系を利用したJFH1株(遺伝子型2a)のHCV cDNAを含むプラスミドベクター(pHH/ZeoJFH1)を構築した。pHH/ZeoJFH1は、感染性完全長ゲノムHCVレプリコン、すなわち、コアタンパク質(c)、2つのエンベロープタンパク質(E1、E2)及び複製に必要な非構造タンパク質(NS2、NS3、NS4A、NS4B、NS5A、NS5B)を少なくとも含み、コアタンパク質(c)をコードする配列の上流にPolIプロモーター配列を、またNS5Bタンパク質をコードする配列の下流にPolIターミネーター配列を配置させてなる。
【0081】
なお、PolIプロモーター配列は、ヒト肝癌由来Huh7細胞においてHCVが自律複製能を発揮し得るように配置されるものであればよく、特に限定されない。また、ターミネーター配列は、必要に応じて除くこともできる。さらに、コアタンパク質(c)をコードする配列の上流に一のIRES(リボソーム内部侵入部位:internal ribosomal entry site)配列を含んでもよく、加えて一の選択マーカー遺伝子やリポーター遺伝子を含んでいてもよい。なお、pHH/ZeoJFH1は、選択マーカー遺伝子として、図3に示すようにゼオシン耐性遺伝子を包含している。
【0082】
感染性HCVを得るために、上記プラスミドベクターpHH/ZeoJFH1をヒト肝癌由来のHuh7.5.1細胞に導入した。得られた細胞株(Huh7.5.1/JFH1zeoR 細胞)を、5%CO2下で37℃にて150cm2のフラスコあたり30〜50mLの細胞培養用培地(10%FCS、10mM Hepes緩衝液、1mM MEMピルビン酸ナトリウム、1×MEM非必須アミノ酸溶液、100単位/mLペニシリン及び100μg/mLストレプトマイシンを含有するダルベッコ変法イーグル培地:DMEM)を用いて6日間培養し、得られた培養液上清を感染性HCVストック液とした。なお、培養期間は、3〜10日間、好ましくは4〜7日間、より好ましくは5〜6日間とすることができる。この培養液上清中のHCVコアタンパク質の数(fmol/mL)を「HCV量」とした。HCV量の測定にはオーソHCV抗原IRMAテストを用いた(Aoyagi et al.、J.Clin.Microbiol.、(1999)37、1802−1808)。
【0083】
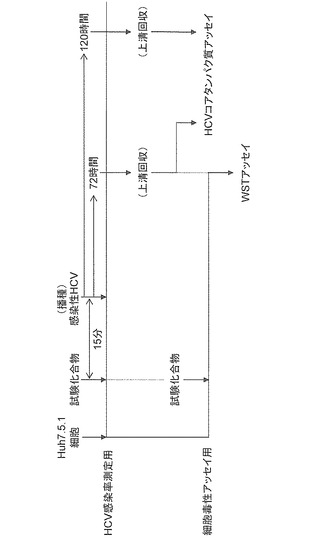
(2)C型肝炎ウイルス感染阻害剤のスクリーニング
C型肝炎ウイルス感染阻害剤のスクリーニング方法の概要は、図4に示す通りである。まず、HCV感染率を測定するために、ヒト肝癌由来Huh7.5.1細胞に様々な試験化合物を加え、その15分後に上記(1)で調製した感染性HCVを添加して培養した。次に、感染性HCV添加後72時間及び120時間を経た培養液上清中のHCV量を測定した。また、試験化合物の細胞毒性アッセイ用として、試験化合物のみで処理したHuh7.5.1細胞の生存率(survival rate)(%)を測定した。その結果、生存率が約80%以上で、かつHCV量から算出されるHCV感染率(%)が約50%以下となる物質を第一候補化合物として選出した。続いて、得られた第一候補化合物のうち選択性(selective index:Si)が10以上の化合物をC型肝炎ウイルス感染阻害剤とした。ここで、「選択性」とは、CC50/EC50の値をいう。本式中、CC50は細胞が50%傷害を受ける薬剤の濃度であり、EC50値はウイルスの増殖を50%抑制する薬剤の濃度である。
【0084】
以下、C型肝炎ウイルス感染阻害剤の検出方法を詳述する。
(i)HCV感染率の算出
まず、Huh7.5.1細胞(継代数が43回目までのものを使用)を上記細胞培養用培地に懸濁し、1×104個/100μL/ウェルとなるように96ウェルのマイクロプレートに播種した。次に、5μM及び0.5μMの試験化合物を、上記マイクロプレートの細胞を培養した各ウェルに加えた。試験化合物には800種類の化合物(並木商事より購入)を用いた。また、陰性対照サンプルとして試験化合物を添加しないウェル(候補阻害剤なし)、陽性対照としてインターフェロンα(IFN-α)を5U/mL及び0.5U/mLを加えたウェルを同時に調製した。続いて、試験化合物の添加15分後に、感染性HCVを0.2fmol/ウェルで播種し、培養した。播種72時間後と120時間後に培養液から50μLを回収し、培養液中のHCV量を測定した。陰性対照サンプル中のHCV量を100%としたときの各試験化合物添加サンプル中のHCV量をHCV感染率(%)として算出した。
【0085】
(ii)Huh7.5.1細胞生存率の算出
試験化合物で処理したHuh7.5.1細胞生存率は、Premix WST細胞増殖アッセイキット(タカラバイオ)を用いて試験化合物の細胞毒性アッセイ法(WSTアッセイ)により測定した。本キットは、テトラゾリウム塩WST-1の生成したホルマザンの吸光度を直接測定する方法に基づくものである。まず、Huh7.5.1細胞(継代数が43回目までのものを使用)を上記細胞培養用培地に懸濁し、1×104個/100μL/ウェルとなるように96ウェルのマイクロプレートに播種した。次に、5μMの上記800種類の試験化合物を上記各ウェルに加えた。続いて、試験化合物を添加した48時間後(HCV感染率測定用の実験における感染性HCV添加の72時間後に相当する)に各ウェルにPremix WST-1溶液を10μL加え、引き続き4時間培養して、EIAリーダーにて420nm〜480nmの波長で吸光度を測定し、細胞生存率を算出した。
【0086】
(3)結果
スクリーニングの結果、化合物1がC型肝炎ウイルス感染阻害剤の有効成分となる第一候補化合物として得られた(表1)。化合物1は、スピノシンA:スピノシンD=7:3の混合物である。
【0087】
【表1】
【0088】
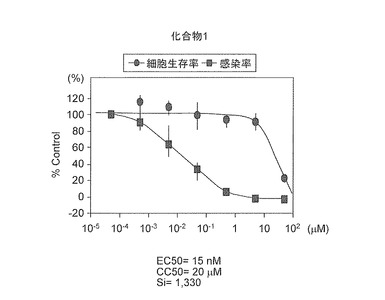
次に、第一候補化合物である化合物1を50μMから0.00005μMまで10倍段階希釈し、各濃度の感染阻害効果及び細胞毒性を評価した。結果を図5に示す。化合物1は、EC50=15nM及びCC50=20μMであったことからSi=1330であった。したがって、化合物1は、新規C型肝炎ウイルス感染阻害剤として同定された。
【0089】
2.HCVゲノムの複製過程を阻害する化合物(HCV複製翻訳阻害剤)としての評価(レプリコンアッセイ)
新規HCV阻害剤として単離された化合物1がHCVゲノムの複製過程を阻害する化合物であるか否かを評価した。評価には、サブゲノミックHCVレポーターレプリコンRNAを複製する細胞を用いた。本明細書で「サブゲノミックHCVレポーターレプリコン」とは、サブゲノミックHCVレプリコン中にレポーター遺伝子を含むものをいう。後述するように、本実施例における評価系では、レポーター遺伝子としてルシフェラーゼ遺伝子を用いている。それゆえ、本レプリコンRNAを導入した細胞が該サブゲノミックHCVレポーターレプリコンを複製しているか否かを確認するには、レポーターレプリコンRNAから翻訳されるルシフェラーゼの活性を測定するか、又はHCVタンパク質をウエスタンブロット等で検出すればよい。本実施例では、上記選択性(Si)が10以上の化合物をHCV複製翻訳阻害剤とした。
【0090】
サブゲノミックHCVレポーターレプリコンRNAを複製する細胞に試験化合物であるHCV阻害剤を接触させ、ルシフェラーゼの活性又はHCVタンパク質の発現レベルを、該HCV阻害剤を接触させない時のそれらと比較することで、得られたHCV阻害剤がHCVの複製翻訳を阻害する化合物であるか否かを評価することができる。
【0091】
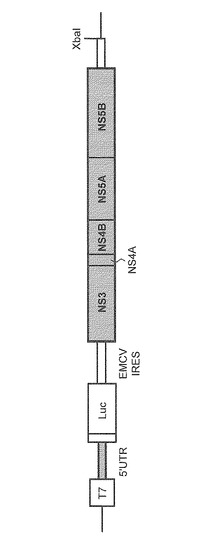
(1)サブゲノミックHCVレポーターレプリコンRNAの合成:
まず、図6に示す構造を有する遺伝子型1bのpSGR-Con1/Luc(Murayama et al.、J.Virol.2007、81:8030-8040)及び遺伝子型2aの pSGR−JFH/Luc(Kato et al.、J.Clin.Microbiol.、2005;43(11):5679-5684)プラスミドを制限酵素XbaIで開裂し、Mung Bean Nuclease(タカラバイオ)で平滑末端とした。なお、pSGR-Con1/LucとpSGR−JFH/Lucとでは、図6において網掛けした領域の塩基配列がそれぞれ異なる。
【0092】
得られた直鎖状DNAを鋳型として、MEGAscript(Ambion社)を用いてサブゲノミックHCVレポーターレプリコンRNAをin vitro合成した。
【0093】
(2)サブゲノミックHCVレポーターレプリコンRNA複製細胞の調製
次に、Huh7.5.1細胞(継代数が43回目までのものを使用)を細胞培養用培地に4×105個/mLとなるように懸濁し、25cm2培養フラスコに10mL播種した。その後、培養フラスコを5%CO2下で37℃にて培養した。24時間後、培養したHuh7.5.1細胞に上記(1)で合成したサブゲノミックHCVレポーターレプリコンRNAをリポフェクション法で導入した。なお、細胞導入にはエレクトロポレーション法を用いてもよい。翌日、培養フラスコから細胞を剥離し、5×104個/mLになるように培地で調製した後、96ウェルのマイクロプレートの各ウェルに90μLずつ播種し(約4500個/ウェル)、5%CO2下で37℃にて約20時間培養した。その後、化合物1及び陽性対照としてインターフェロンα(IFN-α)の溶液を10μL加え、引き続き同条件下で培養した(最終濃度:3倍段階希釈で、化合物1は、5μM〜0.02μM、IFN-αは、100U/mL〜0.14U/mL)。化合物1を添加してから72時間後、細胞をPBSで2回洗浄した。測定キットに添付の細胞溶解バッファ(Promega社)で溶解し、ルシフェラーゼの活性を測定した。ルシフェラーゼの測定にはPromega社のSteady-Glo抽出測定キットを使用した。
【0094】
また、化合物1で処理したHuh7.5.1細胞生存率は、Premix WST細胞増殖アッセイキット(タカラバイオ)を用いて測定した。
【0095】
(3)結果
図7に遺伝子型1bのサブゲノミックHCVレポーターレプリコンRNAの、また図8に遺伝子型2aのサブゲノミックHCVレポーターレプリコンRNAの結果を示す。
【0096】
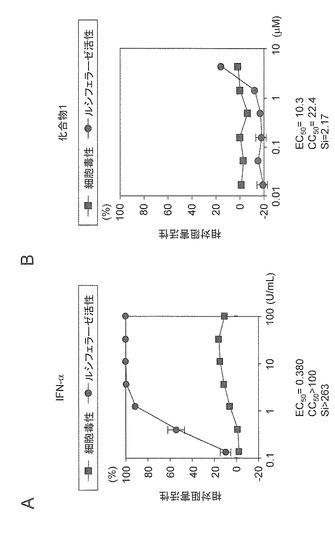
遺伝子型1bのサブゲノミックHCVレポーターレプリコンRNAの複製阻害活性評価系においてIFN-αは、EC50=0.38U/mL及びCC50>100U/mLであったことからSiは>263であった(図7A)。一方、化合物1は、EC50=10.3μM及びCC50=22.4μMであったことからSi=2.17であった(図7B)。
【0097】
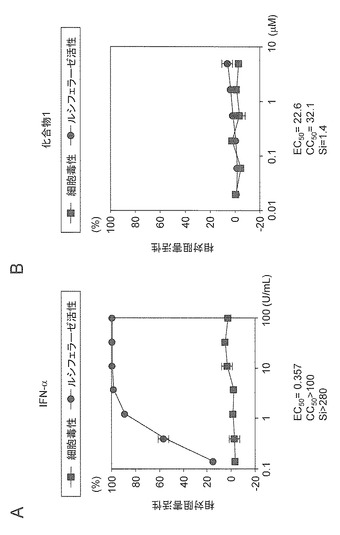
遺伝子型2aのサブゲノミックHCVレポーターレプリコンRNAの複製阻害活性評価系においてIFN-αは、EC50=0.357U/mL及びCC50>100U/mLであったことからSiは>280であった(図8A)。一方、化合物1は、EC50=22.6μM及びCC50=32.1μMであったことからSi=1.4であった(図8B)。
【0098】
これらの結果は、化合物1がHCV阻害剤ではあるがHCVの複製翻訳過程を阻害しないことを示している。したがって、化合物1は、HCV生活環のうち、複製翻訳過程に作用しないこと、すなわちHCV複製翻訳阻害剤ではないことが判明した。
【0099】
3.HCVppアッセイによるスクリーニング
続いて、化合物1が、HCVの吸着過程から侵入過程を阻害するか否かをHCVppアッセイにより評価した。HCVのエンベロープタンパク質を感染性レトロウイルスに提示させたシュードウイルスは、HCV受容体の1つと考えられているCD81を介して宿主細胞内に侵入することが報告されている(Bartosch、B.et al.、J.Exp.Med.、197:633-642、2003)。したがって、このシュードウイルスを用いてHCV吸着・侵入阻害剤であるか否かを評価した。
【0100】
(1)シュードウイルスHCVppの作製
HCVppを作製するためのベクターを図9に示す。マウスレトロウイルスのgag及びpolタンパク質をコードする発現ベクターとして、マウス白血病ウイルス(MLV)Gag−Pol発現ベクターAを用いた。また、HCVのエンベロープタンパク質であるE1、E2タンパク質をコードする発現ベクターとして、HCVエンベロープタンパク質発現ベクターB、及びCを用いた。ベクターB中のJFH1-HCVE1E2における「JFH1」は、HCVの遺伝子型2aのJFH1株由来のエンベロープタンパク質を有する発現ベクターであることを意味し、同様にベクターC中のTH-HCVE1E2における「TH」は、HCVの遺伝子型1bのTH株由来のエンベロープタンパク質を有する発現ベクターであることを意味する。さらに、ルシフェラーゼタンパク質をコードし、かつレトロウイルスパッケージングサイトを持つ発現ベクターとしてルシフェラーゼ発現ベクターDを用いた。
【0101】
発現ベクターA、B及びD、又は発現ベクターA、C及びDを293T細胞にリポフェクション法で共導入することにより、ルシフェラーゼがHCVのE1及びE2タンパク質の外皮(エンベロープ)で包れたシュードウイルス HCVpp(JFH1)又はHCVpp(TH)が産生される。このHCVppをHCV吸着・侵入阻害剤の評価に利用した。
【0102】
(2)HCVppを用いたHCVの吸着過程から侵入過程に対する阻害効果の評価
Huh7.5.1細胞を1×104個/ウェルとなるように96ウェルのマイクロプレートに播種し、37℃の5%CO2インキュベーターにて一夜培養した。
【0103】
次に96ウェルのマイクロプレートに培養したHuh7.5.1細胞から培養液を除去し、化合物1を図10に示す最終濃度(3〜300μM)の1.5倍となるように細胞培養液で希釈調製し、この溶液をウェル当たり100μL加え、2時間培養した後、調製したHCVppを各ウェルに50μL加えた。その後、37℃の5%CO2インキュベーターにて3時間培養した。培養後、上清を除去し、PBSで洗浄後、新鮮な培地100μLを加え、引き続き同条件下で培養した。67時間後(化合物を添加して72時間後)、細胞をPBSで2回洗浄し、60μLの測定キットに添付の細胞溶解バッファ(Promega社)を加え、ピペッティングにより細胞を溶解した。細胞溶解液中のルシフェラーゼ活性をPromega社のSteady-Glo抽出測定キットを用いて測定した。化合物無添加のサンプル(コントロール)におけるルシフェラーゼ活性を100%としたときの、化合物1を添加したルシフェラーゼの活性(%)を算出した。HCVppには、JFH1株のエンベロープタンパク質を持つHCVpp(JFH1)と遺伝子型1bのTH株のエンベロープタンパク質を持つHCVpp(TH)を用いた。化合物1は、3、10、30、100及び300μMの濃度で評価した。
【0104】
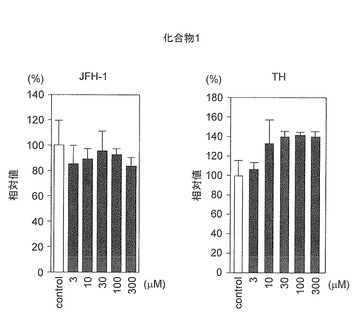
(3)結果:
図10に結果を示す。化合物1は、3〜300μMの濃度範囲においてHCVppの吸着過程及び侵入過程を阻害しなかった。上記レプリコンアッセイ及び当該HCVppアッセイの結果から、化合物1は、HCVの宿主細胞内での脱殻過程及び/又は感染後期過程、すなわちパッケージング過程から細胞外放出過程までの感染後期過程の少なくとも一の過程を阻害する化合物であることが判明した。
【0105】
4.CD81の発現に及ぼす効果の評価
新規HCV阻害剤として単離された化合物1が、HCVの受容体の一つであるCD81(Bartosch、B.et al.、J.Exp.Med.、197:633-642、2003)の発現を抑制し、その結果、HCVの感染が阻害される可能性について検討した。
【0106】
(1)フローサイトメトリーによるHuh7細胞におけるCD81の発現解析
Huh7細胞で発現し、HCV受容体の一つとして考えられているCD81の発現が化合物1により影響を受けるか否かについてフローサイトメトリーを用いて解析した。
【0107】
Huh7細胞を1.6×105個/ウェルとなるように6穴プレートに播種し、37℃の5%CO2インキュベーターにて培養した。48時間後、Huh7細胞から培養液を除去し、10μMの濃度となるように化合物1をHuh7細胞に加え、37℃の5%CO2インキュベーターにて2時間培養した。培養後、上清を除去し、PBSで洗浄後、新鮮な培地3mLを加え、引き続き同条件下で培養した。24時間後に細胞を回収して、FITC標識抗CD81抗体にて染色し、フローサイトメトリーにて解析した。
【0108】
(2)結果
化合物1は、10μMの濃度でHuh7細胞におけるCD81の発現に影響を及ぼさなかった。以上の結果から、化合物1はHCVの受容体の一つであるCD81の発現を抑制して感染を阻害するのではないことが判明した。
【技術分野】
【0001】
本発明は、C型肝炎ウイルス感染阻害剤及びそれを含有する医薬組成物に関する。
【背景技術】
【0002】
C型肝炎ウイルス(Hepatitic C Virus;以下、特に断りのない限りHCVという。)は、フラビウイルス科ヘパシウイルス(Hepacivirus)属に分類されるRNAウイルスである。本ウイルスは、一本鎖RNA(+)ゲノムとエンベロープからなる構造を有する(非特許文献1)。
【0003】
同じ肝炎ウイルスであっても、DNAウイルスであるB型肝炎ウイルス(HBV)は、免疫能の未熟な新生児及び乳幼児期以外では、感染しても免疫機構により排除され、急性感染で終わる。一方、HCVは、未だ明らかではない原因により宿主の免疫機構を回避する。そのため免疫機構の発達した大人に感染した場合であっても、持続性感染に移行することが多い。
【0004】
HCV感染により引き起こされる肝炎(C型肝炎)は、発症後20数年の経過の中で肝硬変に至り、最終的には肝癌に至る可能性がある。肝癌は、手術で癌を摘出しても非癌部で引き続き起こる炎症により再発する患者の多いことが知られている。
【0005】
HCV感染者は、日本国内で約200万人、世界では約2億人存在すると推定されている。また、HCVに感染していることを自覚していない無症候性キャリアも多数存在している。現在、日本国内では、肝細胞癌による犠牲者は年間約3.5万人となっており、その8割はHCV感染によるものである(非特許文献2)。
【0006】
C型肝炎に対しては、現在、インターフェロンとリバビリン(Ribavirin;1‐β‐D−リボフラノシル‐1H‐1,2,4‐トリアゾール‐3−カルボキサミド)の併用療法が一般的である(非特許文献3)。しかしながら、この併用療法は、患者全体の約50%にしか有効ではなく、また、血球減少症や溶血性貧血等の重篤な副作用を起こすことが知られている(非特許文献4)。
【0007】
それゆえ、薬効の優れた新たなC型肝炎ウイルス感染阻害剤を創出することが求められており、そのためには、HCVがどのようにして宿主細胞に侵入し、複製増殖するのか、そして宿主細胞にどのような影響を与えるのかといったHCVの生活環の詳細を明らかにし、C型肝炎ウイルス感染阻害剤を探索する必要がある。
【0008】
図1は、HCVの生活環を示している。HCV粒子(001)は、まず、宿主細胞(003)の細胞表面に存在するウイルス受容体(002)、具体的には、宿主細胞表面に存在するCD81(非特許文献5)及びClaudin-1(非特許文献6)等の受容体に吸着し(A:吸着過程)、エンドサイトーシスによって宿主細胞のエンドソーム内に取り込まれる(B:侵入過程)。次に、プラス鎖RNAであるウイルスゲノム(004)がウイルス粒子から宿主細胞質内に放出される(C:脱殻過程)。続いて、放出されたウイルスゲノムにコードされた前駆体タンパク質(005)をコードするmRNAが翻訳され(D:翻訳過程)、さらに生じた前駆体タンパク質がプロセッシング(E:プロセッシング過程)されることによって、ウイルス粒子を形成する構造タンパク質(Core、E1、E2)とウイルス粒子に含まれない非構造タンパク質(NS2、NS3、NS4A、NS4B、NS5A、NS5B)が生成される(006)。その後、ウイルスゲノムが、非構造タンパク質の一つであるRNA依存性RNAポリメラーゼ(NS5B)(007)によって複製される(F:複製過程)。複製されたウイルスゲノムは、構造タンパク質であるコアタンパク質(Core)及びエンベロープタンパク質(E1、E2)によってパッケージングされ、新たなウイルス粒子が形成される(G:パッケージング過程)。新たに形成されたウイルス粒子は、小胞体及びゴルジ体で成熟し(H:成熟過程)、最後に、宿主細胞膜を破り、細胞外に放出される(I:細胞外放出過程)(非特許文献7)。よって、上記HCVの生活環のいずれかの経路を阻害する化合物は、C型肝炎ウイルス感染阻害剤となり得る。
【0009】
1999年に、HCVを人工的に複製させる実験システムとしてサブゲノミックHCVレプリコンシステムが登場した(非特許文献8)。このシステムでは、HCVゲノムの複製に必須のNS3〜NS5B領域及びゲノムの両末端からなるHCVサブゲノムが細胞内で複製されるように構成されており、一細胞あたりのHCVサブゲノムのコピー数は数千というレベルに達する。それゆえ、当該評価系を用いて、HCV感染を阻害する化合物、すなわちC型肝炎ウイルス感染阻害剤を探索することが可能となった(非特許文献9)。
【0010】
上記サブゲノミックHCVレプリコンシステムから選択されたC型肝炎ウイルス感染阻害剤には、ウイルスタンパク質の翻訳後のプロセッシング過程(図1E)とHCV感染のウイルスゲノムの複製過程(図1F)とに作用するRNAポリメラーゼ、ヘリカーゼ、プロテアーゼ等のウイルス酵素を標的としたHCV酵素阻害剤が知られている(特許文献1、2)。しかしながら、ウイルス酵素をコードする遺伝子は、変異を生じ易いため、これらのHCV酵素阻害剤に対しては、耐性ウイルス株が容易に出現してしまう。それゆえ、これらHCV酵素阻害剤では薬効として不十分という問題がある(非特許文献10)。したがって、HCVの生活環において他の過程を阻害する新たなC型肝炎ウイルス感染阻害剤を見出すことが求められている。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】国際公開WO2008/098859
【特許文献2】特表2007-513971
【非特許文献】
【0012】
【非特許文献1】Rosenbergら、J. Mol. Biol.、 2001年、313巻、p.451-464
【非特許文献2】Saitoら、Proc. Natl, Acad. Sci. USA、1990年、87巻、p.6547-6549
【非特許文献3】HcHutchinsonら、N. Engl. J. Med.、1998年、339巻、p.1485-1492
【非特許文献4】Moriら、Biochem. Biophis. Res. Commun.、1992年、183巻、p.334-342
【非特許文献5】Bartoschら、J.Exp.Med.、2003年、197巻、p.633-642
【非特許文献6】Evans、Nature、2008年、446巻、p.801-805
【非特許文献7】Moradpourら、SWISS MED WKLY、2001年、131巻、p.291-298
【非特許文献8】Lomannら、Science、1999年、285巻、p.110-113
【非特許文献9】Bartenschlager、Nature Rev.Drug Discovery、2002年、1巻、p.911-916
【非特許文献10】Luら、Antimicro Agents Chemother.、2004年、48巻、p.2260-2266
【発明の概要】
【発明が解決しようとする課題】
【0013】
本発明は、HCVの生活環における脱殻過程又は感染後期過程でウイルス粒子の増殖を阻害する新規な作用機序のC型肝炎ウイルス感染阻害剤を提供し、C型肝炎を治療又は予防することを目的とする。
【課題を解決するための手段】
【0014】
上記目的を達成するため、本発明者らは、植物及び微生物由来の純天然物を含む化合物ライブラリーから、HCVの生活環における過程、特に脱殻過程(図1C)及び/又は感染後期過程(パッケージング過程(図1G)〜細胞外放出過程(図1I))における少なくとも一の過程を阻害できる化合物の単離を試みた。その結果、当該作用効果を有する化合物として、スピノシン誘導体が得られた。スピノシン誘導体は、マクロラクトン環及び二種のデオキシ糖を有する化合物である。この化合物は、主に放線菌の一種であるSaccharopolyspora spinosaの二次代謝物として得られる。特にスピノシンAとスピノシンDの混合物であるスピノサド(Spinosad)は、駆虫活性を有する農薬として知られている。例えば、ヒツジに付いたシラミの処置等に使用される他、ヒトに寄生するシラミ等の発生防除にも有用であることが報告されている(米国特許第6,063,771号)。このように、スピノシン誘導体の駆虫活性については、従来から多くの報告があったが、HCV感染の阻害活性については、これまで全く知られていなかった。本願発明は、当該新たな知見に基づいて完成されたものであり、すなわち、下記(1)〜(5)を提供する。
【0015】
(1) 以下の一般式(I)で示される化合物又はその薬学的に許容される塩を有効成分として含有する、C型肝炎ウイルス感染阻害剤。
【化1】
[式中、R1及びR2は、それぞれ独立して水素原子又は低級アルキル基を表し、R3は、水素原子又は以下の一般式(II)で示される基を表し、
【化2】
R4は、置換基で置換された若しくは置換されていない、低級アルキル基、低級シクロアルキル基又は低級シクロアルケニル基、を表し、R5〜R9は、それぞれ独立して水素原子又は低級アルキル基を表す。]
(2) R1及びR2は、それぞれ独立して水素原子、メチル基又はエチル基を表し、R3は、一般式(II)で示される基を表し、R4は、置換基で置換された若しくは置換されていないメチル基、エチル基、炭素数3〜8のシクロアルキル基又は炭素数3〜8のシクロアルケニル基を表し、R5〜R9は、それぞれ独立して水素原子又はメチル基を表す、上記(1)に記載のC型肝炎ウイルス感染阻害剤。
(3) 上記置換基は、ハロゲン、ヒドロキシル基、低級アルキル基、低級アルコキシ基又はシアノ基である、上記(1)又は(2)に記載のC型肝炎ウイルス感染阻害剤。
(4) C型肝炎ウイルスの生活環における、宿主細胞への吸着過程、侵入過程、前駆体タンパク質の翻訳過程及びプロセッシング過程並びにウイルスゲノムの複製過程、に対しては作用せず、脱殻過程及び/又はパッケージング過程から細胞外放出過程までの感染後期過程における少なくとも一の過程でウイルス粒子の増殖を阻害する、上記(1)〜(3)のいずれかに記載のC型肝炎ウイルス感染阻害剤。
(5) 上記(1)〜(4)のいずれかに記載のC型肝炎ウイルス阻害剤を有効成分として含有する、医薬組成物。
【0016】
なお、上記の一般式(I)で示される化合物は、突然変異によって出現する薬剤耐性のHCVに対してより効果的にその増殖を抑制することができるため、以下の化学式(Ia)及び/又は(Ib)で表される化合物であることが好ましい。
【0017】
【化3】
【0018】
【化4】
【発明の効果】
【0019】
本発明のC型肝炎ウイルス感染阻害剤によれば、突然変異によって出現する薬剤耐性のHCVに対しても効果的にその増殖を抑制することができ、C型肝炎の治療及び予防に対して有効な医薬を提供することができる。
【図面の簡単な説明】
【0020】
【図1】HCVの生活環を模式的に表した図である。
【図2】3種類の評価系(アッセイ)で得られる結果と阻害剤の関係を表した図である。
【図3】感染性完全長ゲノムHCVレプリコンを含む感染性HCV粒子作製用プラスミドベクターpHH/ZeoJFH1を模式的に表した図である。
【図4】C型肝炎ウイルス感染阻害剤のスクリーニング方法の概要を表した図である。
【図5】化合物1のHCVccアッセイの結果を表した図である。
【図6】サブゲノミックHCVレポーターレプリコン作製ベクターの構造を模式的に表した図である。遺伝子型1bを含むpSGR-Con1/Lucと遺伝子型p2aを含むpSGR-JFH/Lucとでは、図中網かけした領域がそれぞれで異なる。
【図7】化合物1の遺伝子型1bのサブゲノミックHCVレポーターレプリコンRNAを用いたレプリコンアッセイの結果を表した図である。Aは陽性対照であるIFN-αの結果を、またBは化合物1の結果を、それぞれ示す。
【図8】化合物1の遺伝子型2aのサブゲノミックHCVレポーターレプリコンRNAを用いたレプリコンアッセイの結果を表した図である。Aは陽性対照であるIFN-αの結果を、Bは化合物1の結果を、それぞれ示す。
【図9】シュードウイルス作製用ベクター群(A〜D)を模式的に表した図である。
【図10】化合物1のHCVppアッセイの結果を表した図である。
【発明を実施するための形態】
【0021】
1.C型肝炎ウイルス感染阻害剤
本発明の第1の実施形態は、C型肝炎ウイルス感染阻害剤である。本発明のC型肝炎ウイルス感染阻害剤は、下記一般式(I)で表される化合物又はその薬学的に許容される塩を有効成分として含有することを特徴とする。
【0022】
「HCV」(C型肝炎ウイルス:Hepatitic C Virus)は、前述のように、フラビウイルス科ヘパシウイルス属に分類される一本鎖RNAウイルスであって、本発明の標的ウイルスである。
【0023】
「C型肝炎ウイルス感染阻害剤」とは、HCVの感染に必要な一以上の機能を阻害することによってHCV粒子の増殖を阻害又は抑制できる化合物をいう。本発明において「感染」とは、図1で示すHCVの生活環において、HCV粒子が宿主細胞の細胞表面に吸着し、宿主細胞内で増殖された後、細胞外に放出されるまでの過程(A〜I)をいう。つまり、C型肝炎ウイルス感染阻害剤は、吸着過程(A)、侵入過程(B)、脱穀過程(C)、翻訳過程(D)、プロセッシング過程(E)、複製過程(F)、パッケージング過程(G)、成熟過程(H)及び細胞外放出過程(I)のうち少なくとも一の過程に必要な機能を阻害する化合物である。
【0024】
本発明のC型肝炎ウイルス感染阻害剤は、上記感染における脱殻過程(C)、及び/又は感染後期過程、すなわち、パッケージング過程(G)から細胞外放出過程(I)までの感染後期に関与する過程における少なくとも一の過程を阻害する作用を有することでHCVの宿主細胞への感染を阻害することができる。さらに、本発明のC型肝炎ウイルス感染阻害剤は、上記感染における、脱殻過程(C)、及び/又は感染後期過程の少なくとも一の過程を阻害する作用を有するが、吸着過程(A)、侵入過程(B)、翻訳過程(D)、プロセッシング過程(E)及び複製過程(F)に対しては阻害作用を示さない性質を有していてもよい。
【0025】
「宿主細胞」とは、HCVが感染する標的細胞をいう。具体的には、肝細胞又はリンパ球が該当する。宿主細胞の由来する生物種としては、ヒト、チンパンジー又はツパイが挙げられる。宿主細胞は、ヒトの初代培養肝細胞若しくは肝癌細胞株又はその両者を融合させた細胞、チンパンジーやツパイの初代肝細胞又はヒトリンパ球由来細胞株であってもよい。
【0026】
以下、本発明のC型肝炎ウイルス感染阻害剤について詳細に説明をする。
1−1.一般式(I)で表される化合物
本発明のC型肝炎ウイルス感染阻害剤は、有効成分として下記一般式(I)で示される化合物を少なくとも1種含有する。
【0027】
【化5】
[式中、R1及びR2は、それぞれ独立して水素原子、低級アルキル基を表し、R3は、水素原子又は一般式(II)で示される基を表し、
【化6】
R4は、置換基で置換された若しくは置換されていない、低級アルキル基、低級シクロアルキル基又は低級シクロアルケニル基を表し、R5〜R9は、それぞれ独立して水素原子又は低級アルキル基を表す。]
【0028】
「置換基で置換された若しくは置換されていない」とは、本発明の化合物がHCV感染阻害活性を有する限りにおいて、R4を構成する低級アルキル基、低級シクロアルキル基又は低級シクロアルケニル基が置換基で置換されてもよいし、又は置換されていなくてもよいことをいう。ここでいう「置換基」には、ハロゲン、ヒドロキシル基、低級アルキル基、低級アルケニル基、低級アルキニル基、低級アルコキシ基、アミノ基、低級アルキルアミノ基、アミド基、N-低級アルキルアミド基、シアノ基、ニトロ基、カルボキシル基、カルバモイル基、硫酸基又は亜硫酸基等が挙げられる。好ましくは、ハロゲン、ヒドロキシル基、低級アルキル基、低級アルコキシ基又はシアノ基である。上記置換基の数は、化合物あたり1個若しくは複数個、例えば、1〜4個、1〜3個、又は1若しくは2個である。
【0029】
「ハロゲン」は、フッ素原子(F)、塩素原子(Cl)、臭素原子(Br)又はヨウ素原子(I)である。
【0030】
ここで、「低級アルキル基」とは、炭素数1〜4個の直鎖状又は炭素数3〜4個の分枝状のアルキル基であり、例えば、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、tert−ブチル基等が挙げられる。本発明の一般式(I)で示される化合物の場合、R1、R2又はR4における低級アルキル基は、メチル基又はエチル基が、R5〜R9における低級アルキル基は、メチル基が特に好ましい。
【0031】
「低級シクロアルキル基」とは、炭素数3〜8個の環状アルキル基であり、例えば、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基、シクロヘプチル基、シクロオクチル基等が挙げられる。
【0032】
「低級シクロアルケニル基」とは、炭素数3〜8個の環状アルケニル基であり、例えば、シクロペンテニル基、シクロヘキセニル基等が挙げられる。
【0033】
「低級アルケニル基」とは、炭素数2〜8個、好ましくは2〜4個の直鎖状又は分岐状のアルケニル基である。具体的にはエテニル、プロペニル、ブテニル、ブタジエニル、ペンテニル、ペンタジエニル、ヘキセニル、ヘキサジエニル、ヘプテニル、ヘプタジエニル、オクテニル、オクタジエニル又はこれらの異性体等が含まれる。
【0034】
「低級アルキニル基」は、炭素数2〜8個、好ましくは2〜4個の直鎖状又は分岐状のアルキニル基である。具体的には、エチニル、プロピニル、ブチニル、ブタジイニル、ペンチニル、ペンタジイニル、ヘキシニル、ヘキサジイニル、ヘプチニル、ヘプタジイニル、オクチニル又はオクタジイニル等が含まれる。
【0035】
「低級アルコキシ基」とは、炭素数1〜4個の直鎖状又は炭素数3〜4個の分枝状のアルキル基であり、例えば、メトキシ基、エトキシ基、プロポキシ基、イソプロポキシ基、n−ブトキシ基、イソブトキシ基、tert−ブトキシ基等が挙げられる。
【0036】
「低級アルコキシ基」は、炭素数1〜6個、好ましくは1〜4個の直鎖状又は分岐状のアルコキシ基である。具体的にはメトキシ、エトキシ、n−プロポキシ、イソプロポキシ、n−ブトキシ、イソブトキシ、s−ブトキシ、t−ブトキシ、n−ペンチルオキシ、イソペンチルオキシ、n−ヘキシルオキシ又はイソヘキシルオキシ等が含まれる。
【0037】
本発明のC型肝炎ウイルス感染阻害剤は、一般式(I)で示される単一の化合物だけではなく、一般式(I)で示される二種類以上の化合物の混合物も包含する。例えば、本発明のC型肝炎ウイルス感染阻害剤は、一般式(I)において、R1が水素原子、R2及びR5〜R9がメチル基、R3が一般式(II)で示される基、R4が置換されていないエチル基、で表される以下の化学式(Ia)で示されるスピノシンA、及び、R1、R2及びR5〜R9がメチル基、R3が一般式(II)で示される基、R4が置換されていないエチル基、で表される以下の化学式(Ib)で示されるスピノシンD、をそれぞれ単独で有効成分として含有していてもよいし、スピノサドのようにスピノシンAとスピノシンDを組合せた混合物として含有していてもよい。
【0038】
【化7】
【0039】
【化8】
【0040】
1−2.薬学的に許容される塩
「薬学的に許容される塩」とは、一般式(I)で示される化合物の塩であって、これらの化合物上の特定の置換基(例えば、ヒドロキシル基)に基づいて、塩基又は酸を用いて調製された薬学的に非毒性の活性化合物の塩をいう。使用した塩基又は酸により塩基性付加塩と酸付加塩とに分類できる。
【0041】
「塩基性付加塩」としては、例えば、ナトリウム塩、カリウム塩等のアルカリ金属塩、カルシウム塩、マグネシウム塩等のアルカリ土類金属塩、トリメチルアミン塩、トリエチルアミン塩、ジシクロヘキシルアミン塩、エタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩、ブロカイン塩等の脂肪族アミン塩、N,N-ジベンジルエチレンジアミン等のアラルキルアミン塩、ピリジン塩、ピコリン塩、キノリン塩、イソキノリン塩等の複素環芳香族アミン塩、アルギニン塩、リジン塩等の塩基性アミノ酸塩、テトラメチルアンモニウム塩、テトラエチルアモニウム塩、ベンジルトリメチルアンモニウム塩、ベンジルトリエチルアンモニウム塩、ベンジルトリブチルアンモニウム塩、メチルトリオクチルアンモニウム塩、テトラブチルアンモニウム塩等の第4級アンモニウム塩、アンモニウム塩等が挙げられる。
【0042】
「酸付加塩」としては、例えば、塩酸塩、硫酸塩、硝酸塩、リン酸塩、炭酸塩、炭酸水素塩、過塩素酸塩等の無機酸塩、酢酸塩、プロピオン酸塩、乳酸塩、マレイン酸塩、フマル酸塩、酒石酸塩、リンゴ酸塩、クエン酸塩、アスコルビン酸塩等の有機酸塩、メタンスルホン酸塩、イセチオン酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩等のスルホン酸塩、アスパラギン酸塩、グルタミン酸塩等の酸性アミノ酸等が挙げられる。
【0043】
一般式(I)で示される化合物又はその薬学的に許容される塩は、本発明のC型肝炎ウイルス感染阻害剤又は後述する医薬組成物中でプロドラッグの形態で存在していてもよい。ここでいう「プロドラッグ」は、生理学的条件下で容易に化学変化を受け、その結果として上記C型肝炎ウイルス感染阻害剤又はその活性形態を提供する化合物である。例えば、投与前は、一般式(I)で示される化合物とは異なる構造の化合物としてC型肝炎ウイルス感染阻害剤又は医薬組成物中に存在し、投与後、例えば、消化管内で消化酵素の作用によって一般式(I)で示される化合物又はその活性型に変換されるものをいう。あるいは、ex vivo環境で化学的又は生化学的方法によって一般式(I)で示される化合物又はその活性型に変換されるものも含み得る。
【0044】
プロドラッグの形態は、例えば、一般式(I)で示される化合物又はその薬学的に許容される塩を経口投与しても生体内で直接利用ができない場合や、利用困難な場合に特に有効である。また、プロドラッグは、一般式(I)で示される化合物又はその薬学的に許容される塩よりもC型肝炎ウイルス感染阻害剤又は医薬組成物での溶解度が高い場合が多く、その点においても有用である。
【0045】
本発明のC型肝炎ウイルス感染阻害剤は、一般式(I)で示される化合物又はその薬学的に許容される塩を薬学的に許容可能な溶媒に溶解して利用することができる。
【0046】
薬学的に許容可能な溶媒としては、例えば、水、エタノール、プロピレングリコール、エトキシ化イソステアリルアルコール、ポリオキシ化イソステアリルアルコール、ポリオキシエチレンソルビタン脂肪酸エステル類等が挙げられる。これらは、殺菌されていることが望ましく、必要に応じて血液と等張に調整されていることが好ましい。
【0047】
2.医薬組成物
本発明の第2の実施形態は、医薬組成物である。本発明の医薬組成物は、C型肝炎の治療又は予防するため医薬であって、本発明の上記実施形態1のC型肝炎ウイルス感染阻害剤を有効成分として含有することを特徴とする。
【0048】
本明細書において「治療」とは、C型肝炎及び/又はそれに伴う症状を緩和又は除去することをいい、「予防」とはC型肝炎に罹患しないようにすることをいう。
【0049】
2−1.薬学的に許容可能な担体
本発明の医薬組成物は、薬学的に許容可能な担体を含むことができる。「薬学的に許容可能な担体」とは、製剤技術分野において通常使用し得る添加剤をいう。例えば、賦形剤、結合剤、崩壊剤、充填剤、乳化剤、流動添加調節剤、滑沢剤等が挙げられる。
【0050】
賦形剤としては、例えば、単糖、二糖類、シクロデキストリン、多糖類等の糖(より具体的には、例えば、グルコース、スクロース、ラクトース、ラフィノース、マンニトール、ソルビトール、イノシトール、デキストリン、マルトデキストリン、デンプン、セルロース等)、金属塩(例えば、塩化ナトリウム、リン酸ナトリウム、リン酸カルシウム、硫酸カルシウム、硫酸マグネシウム、炭酸カルシウム等)、クエン酸、酒石酸、グリシン、ポリエチレングリコール(PEG)、プルロニック、カオリン、ケイ酸、それらの組み合わせ等が挙げられる。
【0051】
結合剤としては、例えば、トウモロコシ、コムギ、コメ、ジャガイモのデンプンを用いたデンプン糊、単シロップ、グルコース液、ゼラチン、トラガカント、メチルセルロース、ヒドロキシプロピルメチルセルロース、カルボキシメチルセルロースナトリウム、セラック、ポリビニルピロリドン等が挙げられる。
【0052】
崩壊剤としては、例えば、上記デンプンや、乳糖、カルボキシメチルデンプン、架橋ポリビニルピロリドン、アガー、ラミナラン末、炭酸水素ナトリウム、炭酸カルシウム、アルギン酸、アルギン酸ナトリウム、ポリオキシエチレンソルビタン脂肪酸エステル、ラウリル硫酸ナトリウム、ステアリン酸モノグリセリドや、それらの塩等が挙げられる。
【0053】
充填剤としては、例えば、上記糖、リン酸カルシウム(例えば、リン酸三カルシウム又はリン酸水素カルシウム等)等が挙げられる。
【0054】
乳化剤としては、例えば、ソルビタン脂肪酸エステル、グリセリン脂肪酸エステル、ショ糖脂肪酸エステル、プロピレングリコール脂肪酸エステル等が挙げられる。
【0055】
流動添加調節剤及び滑沢剤としては、例えば、ケイ酸塩、タルク、ステアリン酸塩、ポリエチレングリコール等が挙げられる。
【0056】
このような担体は、主として医薬組成物の剤形形成を容易にし、また剤形及び薬剤効果を維持するために用いられるものであり、必要に応じて適当な種類を適宜組み合わせて使用すればよい。本発明の医薬組成物は、上記の添加剤の他、必要に応じて矯味矯臭剤、溶解補助剤(可溶化剤)、懸濁剤、希釈剤、界面活性剤、安定剤、吸収促進剤(例えば、第4級アンモニウム塩類、ラウリル硫酸ナトリウム等)、増量剤、付湿剤、保湿剤(例えば、グリセリン、澱粉等)、吸着剤(例えば、澱粉、乳糖、カオリン、ベントナイト、コロイド状ケイ酸等)、崩壊抑制剤(例えば、白糖、ステアリン、カカオバター、水素添加油等)、コーティング剤、着色剤、保存剤、抗酸化剤、香料、風味剤、甘味剤、緩衝剤等を含むこともできる。
【0057】
2−2.医薬組成物の剤形
本発明の医薬組成物の剤形は、投与方法及び/又は処方条件によって異なる。投与方法については後述するが、通常は経口投与又は非経口投与に大別することができる。
【0058】
経口投与に適した剤形としては、例えば、固形剤(錠剤、丸剤、舌下剤、カプセル剤、ドロップ剤、トローチ剤を含む)、顆粒剤、粉剤、散剤、液剤等を挙げることができる。さらに固形剤は、必要に応じ、当該分野で公知の剤皮を施した剤形、例えば、糖衣錠、ゼラチン被包錠、腸溶錠、フィルムコーティング錠、二重錠、多層錠とすることができる。
【0059】
非経口投与は、さらに組織内投与、局所投与、経直腸的投与に細分でき、それぞれの投与方法に適した剤形にすることができる。組織内投与に適した剤形としては、例えば、液剤である注射剤が挙げられる。局所投与に適した剤形としては、例えば、液剤(塗布剤、点眼剤、点鼻剤、吸引剤を含む)、懸濁剤(乳剤、クリーム剤を含む)、粉剤(点鼻剤、吸引剤を含む)、ペースト剤、ゲル剤、軟膏剤、硬膏剤等を挙げることができる。経直腸的投与に適した剤形としては、例えば、坐剤等を挙げることができる。
【0060】
上記各剤形の形状、大きさについては、いずれもそれぞれの剤形において当該分野で公知の剤形の範囲内にあればよく、特に限定はしない。
【0061】
2−3.医薬組成物の製造
本発明のC型肝炎ウイルス感染阻害剤を含有する医薬組成物を製剤化するには、原則として当該分野で公知の方法を利用することができる。例えば、Remington’s Pharmaceutical Sciences(Merck Publishing Co.,Easton,Pa.)に記載の方法を用いればよい。
【0062】
医薬組成物における上記のC型肝炎ウイルス感染阻害剤の含有量は、使用するC型肝炎ウイルス感染阻害剤の種類及び/又はその有効量、医薬組成物の剤形(形態、大きさを含む)、並びに添加する担体の種類によって異なり、それぞれの条件において適宜選択される。
【0063】
本明細書で使用する場合、「有効量」とは、上記一般式(I)で示される化合物又はその薬学的に許容される塩が有効成分としての機能を発揮する上で必要な量、すなわち、本発明ではC型肝炎ウイルス感染阻害剤がHCVの宿主細胞への感染を阻害する上で必要な量であって、かつそれを投与する患者に対して有害な副作用をほとんど又は全く付与しない量をいう。この有効量は、患者の情報及び投与経路等の様々な条件によって変化し得る。
【0064】
「患者の情報」とは、病気の進行度や重症度、全身の健康状態、年齢、体重、性別、食生活、薬剤感受性、併用医薬の有無及び治療に対する耐性等を含む。上記のC型肝炎ウイルス感染阻害剤及び医薬組成物の最終的な投与量及び有効量は、個々の患者の情報等に応じて、最終的には医師の判断によって決定される。
【0065】
医薬組成物における本発明のC型肝炎ウイルス感染阻害剤の含有量の一例としては、本発明の医薬組成物を上記固形剤として調製する場合であれば、1錠重量に対して通常0.1重量%〜30重量%、好ましくは0.5重量%〜20重量%、より好ましくは1重量%〜10重量%のC型肝炎ウイルス感染阻害剤を含有していればよい。具体的には、例えば、1錠200mgの錠剤を製造する場合、一般式(I)で示される化合物を1錠あたり通常200μg〜60mg含んでいればよい。これ以外には、薬学的に許容可能な担体として、上記添加剤を当該分野で公知の含有率で含むことができる。
【0066】
固形剤の場合、C型肝炎ウイルス感染阻害剤の有効量は、剤数によって調整することが可能であるため、必ずしも1錠中に有効量を含有させる必要はなく、一投与単位中に、すなわち、複数の錠剤等を服用する場合、1回の服用単位中に、総計でC型肝炎ウイルス感染阻害剤が有効量含有されていればよい。
【0067】
また、医薬組成物における本発明のC型肝炎ウイルス感染阻害剤の含有量の他の例として、本発明の医薬組成物を注射剤として調製する場合には、薬学的に許容可能な溶媒、好ましくは血液と等張にした希釈剤に溶解した上記HCV阻害剤を用いて当該分野で慣用されている方法により製造することができる。一投与単位の注射剤中に含有される上記HCV阻害剤量は、通常0.1%(w/v)〜30%(w/v)、好ましくは0.1%(w/v)〜20%(w/v)、より好ましくは0.1%(w/v)〜30%(w/v)である。具体的には、例えば、1回2mLの注射剤を製造する場合、一般式(I)で示される化合物を通常2μg〜600μg含むことができる。
【0068】
注射剤には、等張性の溶液を調製するのに充分な量の食塩、グルコース、グリセリン、通常の溶解補助剤、緩衝剤、pH調整剤、無痛化剤等も配合することができる。これら溶液、乳剤及び懸濁剤は、一般式(I)で示される化合物又はその薬学的に許容される塩の所定量を、水性や油性の非毒性の溶媒及び希釈剤に溶解又は懸濁し、さらに必要に応じて等張化剤等を加え、滅菌することにより調製することができる。
【0069】
本発明の医薬組成物を局所剤として調製する場合には、一般式(I)で示される化合物又はその薬学的に許容される塩の所定量を、外用製剤の目的に適合する香料、着色料、充填剤、界面活性剤、保湿剤、皮膚軟化剤、ゲル化剤、担体、保存剤、安定剤等のうちの一種以上と組み合わせることにより調製することができる。
【0070】
本発明の医薬組成物を坐剤として調製する場合には、基材としてこの分野で従来公知のものを広く使用することができる。このような基材としては、例えばパルミチン酸ミリスチルエステル等の高級エステル類、高級アルコール類、ポリエチレングリコール、カカオ脂、ゼラチン、半合成グリセリドや、これらの混合物などの低融点基材を挙げることができる。坐剤は、一般式(I)で示される化合物又はその薬学的に許容される塩の所定量を上記基材に混入し、成型することにより調製することができる。
【0071】
3.C型肝炎ウイルス感染阻害剤及びその医薬組成物の投与方法
本発明のC型肝炎ウイルス感染阻害剤及びその医薬組成物の投与方法は、当該分野で周知の投与単位形態で投与することができる。投与単位形態には、例えば、経口投与、組織内投与(例えば、皮下投与、筋肉内投与、静脈内投与等)、局所投与(例えば、経皮投与等)又は経直腸的投与等が挙げられる。上記のC型肝炎ウイルス感染阻害剤及びその医薬組成物は、これらの投与単位形態のいずれを使用してもよい。また、C型肝炎ウイルス感染阻害剤及びその医薬組成物は、使用する投与単位形態に適した剤形で投与することが好ましい。
例えば、経口投与であれば、固形、粉末又は液剤で投与することが好ましい。
【0072】
組織内投与の場合、皮下投与、筋肉内投与又は静脈内投与等のいずれの方法も使用できる。本発明の目的が一般式(I)で表される化合物又はその薬学的に許容される塩の作用効果によりHCVの宿主細胞への感染を阻害し、それによりC型肝炎を治療又は予防すること、また、HCVが体液を介して伝播されること、さらに、主たる宿主細胞である肝細胞が存在する肝臓が無数の毛細血管を有することを鑑みれば、血流を介した投与が好ましい。それゆえ、このような血流を介した投与において好ましい投与方法は、注射である。
【0073】
注射の場合、注入部位は特に限定しない。例えば、静脈内、動脈内、肝臓内、筋肉内、関節内、骨髄内、髄腔内、心室内、経皮、皮下、皮内、腹腔内、鼻腔内、腸内、舌下等が挙げられる。好ましくは、静脈内注射、動脈内注射等の血管内への注射である。血流を介して本発明の医薬組成物を直ちに全身に行き渡らせることが可能であり、また侵襲性が比較的低く、患者に与える負担が小さいからである。あるいは、肝臓内又は肝門脈に注射してもよい。これは、本発明のC型肝炎ウイルス感染阻害剤及びその医薬組成物をHCVが局在する部位に直接的に作用させることができるからである。
【0074】
本発明のC型肝炎ウイルス感染阻害剤及びその医薬組成物を投与する場合、前述のように、一投与単位中にHCV阻害活性が発揮され得る有効量が含有されていることが好ましい。
【0075】
具体的な投与量の一例として、例えば、C型肝炎発症初期であって、他の医薬の併用を必要としないヒト成人男子(体重60kg)に経口投与する場合、一日当たりのC型肝炎ウイルス感染阻害剤の有効量は、通常1〜2000mg、好ましくは1〜1000mg、より好ましくは1〜500mgの範囲である。患者の状態又は投与経路等に応じて、上記範囲未満又は上記範囲を超える用量を投与することもできる。HCV感染阻害効果を得る上で、上記のC型肝炎ウイルス感染阻害剤及びその医薬組成物の大量投与が必要な場合、患者に対する負担軽減のために数回に分割して投与することもできる。
【0076】
本発明のC型肝炎ウイルス感染阻害剤及びその医薬組成物は、以下の実施例で示すように、HCVの宿主細胞への感染、特にHCVの宿主細胞への感染における、HCVの脱殻過程及びパッケージングから細胞外放出までの感染後期過程の少なくとも一の過程を阻害する。HCVの脱殻過程及び/又は感染後期過程の少なくとも一の過程を阻害するC型肝炎ウイルス感染阻害剤は、これまでに知られておらず、また、このような薬剤は、現在臨床試験が進行中であるHCV酵素阻害剤と比較すると、薬剤耐性ウイルス株が出現しにくいことが期待される。したがって、これらをC型肝炎の治療又は予防に有効な新規の医薬品として利用できる。
【実施例】
【0077】
以下、実施例を挙げて本発明について具体的に説明するが、本発明は以下の実施例によって限定されるものではない。
【0078】
<実施例1:C型肝炎ウイルス感染阻害剤の探索>
HCVの生活環における経路を阻害する新たなC型肝炎ウイルス感染阻害剤を探索するために、HCVの生活環(図1)を利用した3種類の評価方法によるスクリーニングを行なった。すなわち、第一の方法は、HCVの感染経路(図1A〜Iのいずれか一以上)を阻害する化合物(C型肝炎ウイルス感染阻害剤)をスクリーニングする評価方法(図2:HCVccアッセイ)である。第二の方法は、HCVの生活環のうちHCVゲノムの翻訳過程から複製過程(図1D〜F)を阻害する化合物(HCV複製翻訳阻害剤)をスクリーニングする評価方法(図2:レプリコンアッセイ)である。そして、第三の方法は、HCVの生活環のうちHCVの細胞への吸着過程(図1A)から侵入過程(図1B)を阻害する化合物(HCV吸着・侵入阻害剤)をスクリーニングするための評価方法(図2:HCVppアッセイ)である。
【0079】
ここで、図2に示すように、これらの3つのスクリーニング方法で、第一の方法(HCVccアッセイ)で効果があり(図2の「+」に相当)、第二及び第三の方法(それぞれレプリコンアッセイ及びHCVppアッセイ)で効果が得られない(図2の「−」に相当)化合物は、HCVの宿主細胞での脱殻過程(図1C)及び/又はパッケージング(図1G)から細胞外放出(図1I)までの感染後期過程の少なくとも一の過程を阻害する化合物(HCV脱穀・感染後期過程阻害剤)といえる(図2)。
【0080】
以下、各評価方法によるスクリーニングとその結果について説明をする。
1.HCVccアッセイによるスクリーニング
(1)感染性HCVの調製
国際公開WO07/037428 /及びJ.Virol.(2010)84、5824−35(Masaki,T.et al.)に記載の方法に従い、図3に示すRNA PolIプロモーター/ターミネーター系を利用したJFH1株(遺伝子型2a)のHCV cDNAを含むプラスミドベクター(pHH/ZeoJFH1)を構築した。pHH/ZeoJFH1は、感染性完全長ゲノムHCVレプリコン、すなわち、コアタンパク質(c)、2つのエンベロープタンパク質(E1、E2)及び複製に必要な非構造タンパク質(NS2、NS3、NS4A、NS4B、NS5A、NS5B)を少なくとも含み、コアタンパク質(c)をコードする配列の上流にPolIプロモーター配列を、またNS5Bタンパク質をコードする配列の下流にPolIターミネーター配列を配置させてなる。
【0081】
なお、PolIプロモーター配列は、ヒト肝癌由来Huh7細胞においてHCVが自律複製能を発揮し得るように配置されるものであればよく、特に限定されない。また、ターミネーター配列は、必要に応じて除くこともできる。さらに、コアタンパク質(c)をコードする配列の上流に一のIRES(リボソーム内部侵入部位:internal ribosomal entry site)配列を含んでもよく、加えて一の選択マーカー遺伝子やリポーター遺伝子を含んでいてもよい。なお、pHH/ZeoJFH1は、選択マーカー遺伝子として、図3に示すようにゼオシン耐性遺伝子を包含している。
【0082】
感染性HCVを得るために、上記プラスミドベクターpHH/ZeoJFH1をヒト肝癌由来のHuh7.5.1細胞に導入した。得られた細胞株(Huh7.5.1/JFH1zeoR 細胞)を、5%CO2下で37℃にて150cm2のフラスコあたり30〜50mLの細胞培養用培地(10%FCS、10mM Hepes緩衝液、1mM MEMピルビン酸ナトリウム、1×MEM非必須アミノ酸溶液、100単位/mLペニシリン及び100μg/mLストレプトマイシンを含有するダルベッコ変法イーグル培地:DMEM)を用いて6日間培養し、得られた培養液上清を感染性HCVストック液とした。なお、培養期間は、3〜10日間、好ましくは4〜7日間、より好ましくは5〜6日間とすることができる。この培養液上清中のHCVコアタンパク質の数(fmol/mL)を「HCV量」とした。HCV量の測定にはオーソHCV抗原IRMAテストを用いた(Aoyagi et al.、J.Clin.Microbiol.、(1999)37、1802−1808)。
【0083】
(2)C型肝炎ウイルス感染阻害剤のスクリーニング
C型肝炎ウイルス感染阻害剤のスクリーニング方法の概要は、図4に示す通りである。まず、HCV感染率を測定するために、ヒト肝癌由来Huh7.5.1細胞に様々な試験化合物を加え、その15分後に上記(1)で調製した感染性HCVを添加して培養した。次に、感染性HCV添加後72時間及び120時間を経た培養液上清中のHCV量を測定した。また、試験化合物の細胞毒性アッセイ用として、試験化合物のみで処理したHuh7.5.1細胞の生存率(survival rate)(%)を測定した。その結果、生存率が約80%以上で、かつHCV量から算出されるHCV感染率(%)が約50%以下となる物質を第一候補化合物として選出した。続いて、得られた第一候補化合物のうち選択性(selective index:Si)が10以上の化合物をC型肝炎ウイルス感染阻害剤とした。ここで、「選択性」とは、CC50/EC50の値をいう。本式中、CC50は細胞が50%傷害を受ける薬剤の濃度であり、EC50値はウイルスの増殖を50%抑制する薬剤の濃度である。
【0084】
以下、C型肝炎ウイルス感染阻害剤の検出方法を詳述する。
(i)HCV感染率の算出
まず、Huh7.5.1細胞(継代数が43回目までのものを使用)を上記細胞培養用培地に懸濁し、1×104個/100μL/ウェルとなるように96ウェルのマイクロプレートに播種した。次に、5μM及び0.5μMの試験化合物を、上記マイクロプレートの細胞を培養した各ウェルに加えた。試験化合物には800種類の化合物(並木商事より購入)を用いた。また、陰性対照サンプルとして試験化合物を添加しないウェル(候補阻害剤なし)、陽性対照としてインターフェロンα(IFN-α)を5U/mL及び0.5U/mLを加えたウェルを同時に調製した。続いて、試験化合物の添加15分後に、感染性HCVを0.2fmol/ウェルで播種し、培養した。播種72時間後と120時間後に培養液から50μLを回収し、培養液中のHCV量を測定した。陰性対照サンプル中のHCV量を100%としたときの各試験化合物添加サンプル中のHCV量をHCV感染率(%)として算出した。
【0085】
(ii)Huh7.5.1細胞生存率の算出
試験化合物で処理したHuh7.5.1細胞生存率は、Premix WST細胞増殖アッセイキット(タカラバイオ)を用いて試験化合物の細胞毒性アッセイ法(WSTアッセイ)により測定した。本キットは、テトラゾリウム塩WST-1の生成したホルマザンの吸光度を直接測定する方法に基づくものである。まず、Huh7.5.1細胞(継代数が43回目までのものを使用)を上記細胞培養用培地に懸濁し、1×104個/100μL/ウェルとなるように96ウェルのマイクロプレートに播種した。次に、5μMの上記800種類の試験化合物を上記各ウェルに加えた。続いて、試験化合物を添加した48時間後(HCV感染率測定用の実験における感染性HCV添加の72時間後に相当する)に各ウェルにPremix WST-1溶液を10μL加え、引き続き4時間培養して、EIAリーダーにて420nm〜480nmの波長で吸光度を測定し、細胞生存率を算出した。
【0086】
(3)結果
スクリーニングの結果、化合物1がC型肝炎ウイルス感染阻害剤の有効成分となる第一候補化合物として得られた(表1)。化合物1は、スピノシンA:スピノシンD=7:3の混合物である。
【0087】
【表1】
【0088】
次に、第一候補化合物である化合物1を50μMから0.00005μMまで10倍段階希釈し、各濃度の感染阻害効果及び細胞毒性を評価した。結果を図5に示す。化合物1は、EC50=15nM及びCC50=20μMであったことからSi=1330であった。したがって、化合物1は、新規C型肝炎ウイルス感染阻害剤として同定された。
【0089】
2.HCVゲノムの複製過程を阻害する化合物(HCV複製翻訳阻害剤)としての評価(レプリコンアッセイ)
新規HCV阻害剤として単離された化合物1がHCVゲノムの複製過程を阻害する化合物であるか否かを評価した。評価には、サブゲノミックHCVレポーターレプリコンRNAを複製する細胞を用いた。本明細書で「サブゲノミックHCVレポーターレプリコン」とは、サブゲノミックHCVレプリコン中にレポーター遺伝子を含むものをいう。後述するように、本実施例における評価系では、レポーター遺伝子としてルシフェラーゼ遺伝子を用いている。それゆえ、本レプリコンRNAを導入した細胞が該サブゲノミックHCVレポーターレプリコンを複製しているか否かを確認するには、レポーターレプリコンRNAから翻訳されるルシフェラーゼの活性を測定するか、又はHCVタンパク質をウエスタンブロット等で検出すればよい。本実施例では、上記選択性(Si)が10以上の化合物をHCV複製翻訳阻害剤とした。
【0090】
サブゲノミックHCVレポーターレプリコンRNAを複製する細胞に試験化合物であるHCV阻害剤を接触させ、ルシフェラーゼの活性又はHCVタンパク質の発現レベルを、該HCV阻害剤を接触させない時のそれらと比較することで、得られたHCV阻害剤がHCVの複製翻訳を阻害する化合物であるか否かを評価することができる。
【0091】
(1)サブゲノミックHCVレポーターレプリコンRNAの合成:
まず、図6に示す構造を有する遺伝子型1bのpSGR-Con1/Luc(Murayama et al.、J.Virol.2007、81:8030-8040)及び遺伝子型2aの pSGR−JFH/Luc(Kato et al.、J.Clin.Microbiol.、2005;43(11):5679-5684)プラスミドを制限酵素XbaIで開裂し、Mung Bean Nuclease(タカラバイオ)で平滑末端とした。なお、pSGR-Con1/LucとpSGR−JFH/Lucとでは、図6において網掛けした領域の塩基配列がそれぞれ異なる。
【0092】
得られた直鎖状DNAを鋳型として、MEGAscript(Ambion社)を用いてサブゲノミックHCVレポーターレプリコンRNAをin vitro合成した。
【0093】
(2)サブゲノミックHCVレポーターレプリコンRNA複製細胞の調製
次に、Huh7.5.1細胞(継代数が43回目までのものを使用)を細胞培養用培地に4×105個/mLとなるように懸濁し、25cm2培養フラスコに10mL播種した。その後、培養フラスコを5%CO2下で37℃にて培養した。24時間後、培養したHuh7.5.1細胞に上記(1)で合成したサブゲノミックHCVレポーターレプリコンRNAをリポフェクション法で導入した。なお、細胞導入にはエレクトロポレーション法を用いてもよい。翌日、培養フラスコから細胞を剥離し、5×104個/mLになるように培地で調製した後、96ウェルのマイクロプレートの各ウェルに90μLずつ播種し(約4500個/ウェル)、5%CO2下で37℃にて約20時間培養した。その後、化合物1及び陽性対照としてインターフェロンα(IFN-α)の溶液を10μL加え、引き続き同条件下で培養した(最終濃度:3倍段階希釈で、化合物1は、5μM〜0.02μM、IFN-αは、100U/mL〜0.14U/mL)。化合物1を添加してから72時間後、細胞をPBSで2回洗浄した。測定キットに添付の細胞溶解バッファ(Promega社)で溶解し、ルシフェラーゼの活性を測定した。ルシフェラーゼの測定にはPromega社のSteady-Glo抽出測定キットを使用した。
【0094】
また、化合物1で処理したHuh7.5.1細胞生存率は、Premix WST細胞増殖アッセイキット(タカラバイオ)を用いて測定した。
【0095】
(3)結果
図7に遺伝子型1bのサブゲノミックHCVレポーターレプリコンRNAの、また図8に遺伝子型2aのサブゲノミックHCVレポーターレプリコンRNAの結果を示す。
【0096】
遺伝子型1bのサブゲノミックHCVレポーターレプリコンRNAの複製阻害活性評価系においてIFN-αは、EC50=0.38U/mL及びCC50>100U/mLであったことからSiは>263であった(図7A)。一方、化合物1は、EC50=10.3μM及びCC50=22.4μMであったことからSi=2.17であった(図7B)。
【0097】
遺伝子型2aのサブゲノミックHCVレポーターレプリコンRNAの複製阻害活性評価系においてIFN-αは、EC50=0.357U/mL及びCC50>100U/mLであったことからSiは>280であった(図8A)。一方、化合物1は、EC50=22.6μM及びCC50=32.1μMであったことからSi=1.4であった(図8B)。
【0098】
これらの結果は、化合物1がHCV阻害剤ではあるがHCVの複製翻訳過程を阻害しないことを示している。したがって、化合物1は、HCV生活環のうち、複製翻訳過程に作用しないこと、すなわちHCV複製翻訳阻害剤ではないことが判明した。
【0099】
3.HCVppアッセイによるスクリーニング
続いて、化合物1が、HCVの吸着過程から侵入過程を阻害するか否かをHCVppアッセイにより評価した。HCVのエンベロープタンパク質を感染性レトロウイルスに提示させたシュードウイルスは、HCV受容体の1つと考えられているCD81を介して宿主細胞内に侵入することが報告されている(Bartosch、B.et al.、J.Exp.Med.、197:633-642、2003)。したがって、このシュードウイルスを用いてHCV吸着・侵入阻害剤であるか否かを評価した。
【0100】
(1)シュードウイルスHCVppの作製
HCVppを作製するためのベクターを図9に示す。マウスレトロウイルスのgag及びpolタンパク質をコードする発現ベクターとして、マウス白血病ウイルス(MLV)Gag−Pol発現ベクターAを用いた。また、HCVのエンベロープタンパク質であるE1、E2タンパク質をコードする発現ベクターとして、HCVエンベロープタンパク質発現ベクターB、及びCを用いた。ベクターB中のJFH1-HCVE1E2における「JFH1」は、HCVの遺伝子型2aのJFH1株由来のエンベロープタンパク質を有する発現ベクターであることを意味し、同様にベクターC中のTH-HCVE1E2における「TH」は、HCVの遺伝子型1bのTH株由来のエンベロープタンパク質を有する発現ベクターであることを意味する。さらに、ルシフェラーゼタンパク質をコードし、かつレトロウイルスパッケージングサイトを持つ発現ベクターとしてルシフェラーゼ発現ベクターDを用いた。
【0101】
発現ベクターA、B及びD、又は発現ベクターA、C及びDを293T細胞にリポフェクション法で共導入することにより、ルシフェラーゼがHCVのE1及びE2タンパク質の外皮(エンベロープ)で包れたシュードウイルス HCVpp(JFH1)又はHCVpp(TH)が産生される。このHCVppをHCV吸着・侵入阻害剤の評価に利用した。
【0102】
(2)HCVppを用いたHCVの吸着過程から侵入過程に対する阻害効果の評価
Huh7.5.1細胞を1×104個/ウェルとなるように96ウェルのマイクロプレートに播種し、37℃の5%CO2インキュベーターにて一夜培養した。
【0103】
次に96ウェルのマイクロプレートに培養したHuh7.5.1細胞から培養液を除去し、化合物1を図10に示す最終濃度(3〜300μM)の1.5倍となるように細胞培養液で希釈調製し、この溶液をウェル当たり100μL加え、2時間培養した後、調製したHCVppを各ウェルに50μL加えた。その後、37℃の5%CO2インキュベーターにて3時間培養した。培養後、上清を除去し、PBSで洗浄後、新鮮な培地100μLを加え、引き続き同条件下で培養した。67時間後(化合物を添加して72時間後)、細胞をPBSで2回洗浄し、60μLの測定キットに添付の細胞溶解バッファ(Promega社)を加え、ピペッティングにより細胞を溶解した。細胞溶解液中のルシフェラーゼ活性をPromega社のSteady-Glo抽出測定キットを用いて測定した。化合物無添加のサンプル(コントロール)におけるルシフェラーゼ活性を100%としたときの、化合物1を添加したルシフェラーゼの活性(%)を算出した。HCVppには、JFH1株のエンベロープタンパク質を持つHCVpp(JFH1)と遺伝子型1bのTH株のエンベロープタンパク質を持つHCVpp(TH)を用いた。化合物1は、3、10、30、100及び300μMの濃度で評価した。
【0104】
(3)結果:
図10に結果を示す。化合物1は、3〜300μMの濃度範囲においてHCVppの吸着過程及び侵入過程を阻害しなかった。上記レプリコンアッセイ及び当該HCVppアッセイの結果から、化合物1は、HCVの宿主細胞内での脱殻過程及び/又は感染後期過程、すなわちパッケージング過程から細胞外放出過程までの感染後期過程の少なくとも一の過程を阻害する化合物であることが判明した。
【0105】
4.CD81の発現に及ぼす効果の評価
新規HCV阻害剤として単離された化合物1が、HCVの受容体の一つであるCD81(Bartosch、B.et al.、J.Exp.Med.、197:633-642、2003)の発現を抑制し、その結果、HCVの感染が阻害される可能性について検討した。
【0106】
(1)フローサイトメトリーによるHuh7細胞におけるCD81の発現解析
Huh7細胞で発現し、HCV受容体の一つとして考えられているCD81の発現が化合物1により影響を受けるか否かについてフローサイトメトリーを用いて解析した。
【0107】
Huh7細胞を1.6×105個/ウェルとなるように6穴プレートに播種し、37℃の5%CO2インキュベーターにて培養した。48時間後、Huh7細胞から培養液を除去し、10μMの濃度となるように化合物1をHuh7細胞に加え、37℃の5%CO2インキュベーターにて2時間培養した。培養後、上清を除去し、PBSで洗浄後、新鮮な培地3mLを加え、引き続き同条件下で培養した。24時間後に細胞を回収して、FITC標識抗CD81抗体にて染色し、フローサイトメトリーにて解析した。
【0108】
(2)結果
化合物1は、10μMの濃度でHuh7細胞におけるCD81の発現に影響を及ぼさなかった。以上の結果から、化合物1はHCVの受容体の一つであるCD81の発現を抑制して感染を阻害するのではないことが判明した。
【特許請求の範囲】
【請求項1】
以下の一般式(I)で示される化合物又はその薬学的に許容される塩を有効成分として含有する、C型肝炎ウイルス感染阻害剤。
【化1】
[式中、R1及びR2は、それぞれ独立して水素原子又は低級アルキル基を表し、R3は、水素原子又は以下の一般式(II)で示される基を表し、
【化2】
R4は、置換基で置換された若しくは置換されていない、低級アルキル基、低級シクロアルキル基又は低級シクロアルケニル基、を表し、R5〜R9は、それぞれ独立して水素原子又は低級アルキル基を表す。]
【請求項2】
R1及びR2は、それぞれ独立して水素原子、メチル基又はエチル基を表し、
R3は、一般式(II)で示される基を表し、
R4は、置換基で置換された若しくは置換されていないメチル基、エチル基、炭素数3〜8のシクロアルキル基又は炭素数3〜8のシクロアルケニル基を表し、
R5〜R9は、それぞれ独立して水素原子又はメチル基を表す、
請求項1記載のC型肝炎ウイルス感染阻害剤。
【請求項3】
前記置換基は、ハロゲン、ヒドロキシル基、低級アルキル基、低級アルコキシ基又はシアノ基である、請求項1又は2記載のC型肝炎ウイルス感染阻害剤。
【請求項4】
C型肝炎ウイルスの生活環における、宿主細胞への吸着過程、侵入過程、前駆体タンパク質の翻訳過程及びプロセッシング過程並びにウイルスゲノムの複製過程、に対しては作用せず、脱殻過程及び/又はパッケージング過程から細胞外放出過程までの感染後期過程における少なくとも一の過程でウイルス粒子の増殖を阻害する、請求項1〜3のいずれか一項記載のC型肝炎ウイルス感染阻害剤。
【請求項5】
請求項1〜4のいずれか一項記載のC型肝炎ウイルス阻害剤を有効成分として含有する、医薬組成物。
【請求項1】
以下の一般式(I)で示される化合物又はその薬学的に許容される塩を有効成分として含有する、C型肝炎ウイルス感染阻害剤。
【化1】
[式中、R1及びR2は、それぞれ独立して水素原子又は低級アルキル基を表し、R3は、水素原子又は以下の一般式(II)で示される基を表し、
【化2】
R4は、置換基で置換された若しくは置換されていない、低級アルキル基、低級シクロアルキル基又は低級シクロアルケニル基、を表し、R5〜R9は、それぞれ独立して水素原子又は低級アルキル基を表す。]
【請求項2】
R1及びR2は、それぞれ独立して水素原子、メチル基又はエチル基を表し、
R3は、一般式(II)で示される基を表し、
R4は、置換基で置換された若しくは置換されていないメチル基、エチル基、炭素数3〜8のシクロアルキル基又は炭素数3〜8のシクロアルケニル基を表し、
R5〜R9は、それぞれ独立して水素原子又はメチル基を表す、
請求項1記載のC型肝炎ウイルス感染阻害剤。
【請求項3】
前記置換基は、ハロゲン、ヒドロキシル基、低級アルキル基、低級アルコキシ基又はシアノ基である、請求項1又は2記載のC型肝炎ウイルス感染阻害剤。
【請求項4】
C型肝炎ウイルスの生活環における、宿主細胞への吸着過程、侵入過程、前駆体タンパク質の翻訳過程及びプロセッシング過程並びにウイルスゲノムの複製過程、に対しては作用せず、脱殻過程及び/又はパッケージング過程から細胞外放出過程までの感染後期過程における少なくとも一の過程でウイルス粒子の増殖を阻害する、請求項1〜3のいずれか一項記載のC型肝炎ウイルス感染阻害剤。
【請求項5】
請求項1〜4のいずれか一項記載のC型肝炎ウイルス阻害剤を有効成分として含有する、医薬組成物。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公開番号】特開2012−31098(P2012−31098A)
【公開日】平成24年2月16日(2012.2.16)
【国際特許分類】
【出願番号】特願2010−171906(P2010−171906)
【出願日】平成22年7月30日(2010.7.30)
【出願人】(000003159)東レ株式会社 (7,677)
【出願人】(591222245)国立感染症研究所長 (48)
【出願人】(591063394)財団法人 東京都医学総合研究所 (69)
【Fターム(参考)】
【公開日】平成24年2月16日(2012.2.16)
【国際特許分類】
【出願日】平成22年7月30日(2010.7.30)
【出願人】(000003159)東レ株式会社 (7,677)
【出願人】(591222245)国立感染症研究所長 (48)
【出願人】(591063394)財団法人 東京都医学総合研究所 (69)
【Fターム(参考)】
[ Back to top ]