
CB−1アンタゴニスト/逆アゴニストの合成及び結晶体
本発明は結晶質3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリルと、その新規塩、溶媒和物、水和物及び多形体の製造方法に関する。

【発明の詳細な説明】
【技術分野】
【0001】
構造式I:
【0002】
【化1】
の化合物は既にUS2007/0123505、WO2007/062193及びWO2007/064566に開示されている。構造式Iの化合物とその新規多形体、溶媒和物、水和物及び塩はCB−1調節に関連する各種疾患の治療における中枢作用薬として有用な逆アゴニスト/アンタゴニストとして特徴付けられるCB1モジュレーターであり、このような疾患としては限定されないが、精神病、記憶障害、認知障害、アルツハイマー病、ハンチントン病、偏頭痛、神経障害、神経炎症障害(例えば多発性硬化症、ギラン・バレー症候群、並びにウイルス性脳炎、脳血管発作及び頭部外傷の炎症性後遺症)、不安障害、ストレス、癲癇、パーキンソン病、運動障害及び統合失調症が挙げられる。前記化合物は物質濫用障害の治療、肥満症又は摂食障害とそれに伴う合併症(例えば左心室肥大)の治療、並びに喘息、便秘、慢性腸偽閉塞及び肝硬変の治療にも有用である。
【0003】
本発明は、フッ素基を導入するためにHFの使用を必要とし、化合物1のジアステレオマーを分離するためにカラムクロマトグラフィーを必要とするリニア合成を使用して従来製造されていた強力なCB−1逆アゴニストである化合物Iの新規で効率的な合成方法について記載する。本発明の合成はコンバージェントであり、生成物の収率が高く、結晶質中間体が得られ、この点はクロマトグラフィーを使用しない単離精製に関する本発明の利点である。
【背景技術】
【0004】
ウィッティヒ反応とホーナー・ワズワース・エモンズ反応については、Bonadies,F.et al.,Tetrahedron Lett.1994,35,20,3383−3386;及びFukatsu K.et al.,J.Med.Chem.2002,45,4212−4221に記載されている。Tang,W.et al.,Chem.Rev.2003,103,3029にはエナンチオ選択的水素化用のキラルリンリガンドが記載されている。Kastrinsky,D.B.et al.J.Org.Chem.2004,69,2284;及びLewis,E.A.et al.,Tetrahedron Lett.2004,45,3059にはβ−カルボキシエステルの還元方法が記載されている。Weix,D.et al.,J.Am.Chem.Soc.127(4),1092−1093(2005);及びBolshan,Y.et al.,Org.Lett.7(8),1481−1484(2005)にはスルフィニルイミンへのアリールボロン酸のロジウム触媒付加が記載されている。Kitagawa,K.et al.,Angew.Chem.,Int.Ed.39(14),2481−2483(2000)にはn−BuLi/n−Bu2Mgを利用する金属−ハロゲン交換反応が記載されている。Hillier,M.C.;Chen,C−y.,J.Org.Chem.71,7885−7887(2006)にはビスアルキル化によるジオールからアゼチジンの製造が記載されている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】米国特許出願公開第2007/0123505号明細書
【特許文献2】国際公開第2007/062193号
【特許文献3】国際公開第2007/064566号
【非特許文献】
【0006】
【非特許文献1】Bonadies,F.et al.,Tetrahedron Lett.1994,35,20,3383−3386
【非特許文献2】Fukatsu K.et al.,J.Med.Chem.2002,45,4212−4221
【非特許文献3】Tang,W.et al.,Chem.Rev.2003,103,3029
【非特許文献4】Kastrinsky,D.B.et al.J.Org.Chem.2004,69,2284
【非特許文献5】Lewis,E.A.et al.,Tetrahedron Lett.2004,45,3059
【非特許文献6】Weix,D.et al.,J.Am.Chem.Soc.127(4),1092−1093(2005)
【非特許文献7】Bolshan,Y.et al.,Org.Lett.7(8),1481−1484(2005)
【非特許文献8】Kitagawa,K.et al.,Angew.Chem.,Int.Ed.39(14),2481−2483(2000)
【非特許文献9】Hillier,M.C.;Chen,C−y.,J.Org.Chem.71,7885−7887(2006)
【発明の概要】
【0007】
発明が解決しようとする課題及び課題を解決するための手段
本発明はベンズヒドリルアミンII及びシアノジオールIIIから構造式Iの3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリルを製造するための新規で効率的な方法を提供する。
【0008】
本発明は更に、1)化合物Iの遊離塩基無水多形体I;2)化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体I,タイプB;3)化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物多形体I,タイプA;並びに4)化合物IのHCl塩無水多形体A、B、C、D、E、F、G及びHと称する3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリルの11種類の同定された新規結晶体を提供する。これらの遊離塩基及び塩酸塩多形体の結晶体は新規であり、加工、取扱い及び投与し易さ等の化合物Iの医薬組成物の製造上の利点があると考えられる。特に、化合物Iの無水結晶質遊離塩基多形体Iは脂質溶解度;良好なpK暴露;化学的及び物理的安定性;純度;精製単離し易さ;並びに望ましい結晶寸法、結晶表面積及び結晶凝集回避による製剤化等の物理化学的性質の改善により、医薬製剤の製造に特に適している。化合物Iの新規HCl塩無水多形体Gは化合物Iの最も熱力学的に安定な結晶質HCl塩形態であるが、動力学的には多形体A、B、F及びHのほうが好ましい。
【0009】
本発明は更に、活性医薬成分として化合物Iの新規多形体及び塩を含有する医薬製剤と、CB−1関連疾患の治療におけるCB−1逆アゴニスト/アンタゴニストとしてのその使用方法にも関する。
【図面の簡単な説明】
【0010】
【図1】化合物Iの無水遊離塩基多形体IのX線回折(XRPD)パターンである。
【図2】化合物Iの無水遊離塩基多形体Iの熱重量分析(TGA)曲線である。
【図3】化合物Iの無水遊離塩基多形体Iの示差走査熱量(DSC)曲線である。
【図4】化合物Iの無水HCl塩多形体AのX線回折(XRPD)パターンである。
【図5】化合物Iの無水HCl塩多形体Aの示差走査熱量(DSC)曲線である。
【図6】化合物Iの無水HCl塩多形体BのX線回折(XRPD)パターンである。
【図7】化合物Iの無水HCl塩多形体Bの示差走査熱量(DSC)曲線である。
【図8】化合物Iの無水HCl塩多形体GのX線回折(XRPD)パターンである。
【図9】化合物Iの無水HCl塩多形体Gの示差走査熱量(DSC)曲線である。
【図10】化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物多形体IタイプAのX線回折(XRPD)パターンである。
【図11】化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物多形体IタイプAの示差走査熱量(DSC)曲線である。
【図12】化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物多形体IタイプAの熱重量分析(TGA)曲線である。
【図13】化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体IタイプBのX線回折(XRPD)パターンである。
【図14】化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体IタイプBの示差走査熱量(DSC)曲線である。
【図15】化合物IのHCl塩多形体CのX線回折(XRPD)パターンである。
【図16】化合物IのHCl塩多形体DのX線回折(XRPD)パターンである。
【図17】化合物IのHCl塩多形体Dの示差走査熱量(DSC)曲線である。
【図18】化合物IのHCl塩多形体Dの熱重量分析(TGA)曲線である。
【図19】化合物Iの無水HCl塩多形体EのX線回折(XRPD)パターンである。
【図20】化合物IのHCl塩多形体F水和物のX線回折(XRPD)パターンである。
【図21】化合物IのHCl塩多形体HのX線回折(XRPD)パターンである。
【発明を実施するための形態】
【0011】
本発明は構造式I:
【0012】
【化2】
の3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリルとその結晶質多形体、溶媒和物、水和物及び塩の製造方法を提供する。
【0013】
下記一般スキームに示すように、化合物IはベンズヒドリルアミンIIをシアノジオールIIIと反応させて保護オキサジアゾール化合物20を形成した後に保護基Pを開裂し、化合物Iを得ることにより製造することができる。
【0014】
【化3】
【0015】
化合物Iの遊離塩基には、無水遊離塩基多形体I、遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物多形体IタイプA及び遊離塩基トルエン/ヘプタン溶媒和物多形体IタイプBと称する3種類の公知結晶体ないし多形体がある。化合物Iの3種類の遊離塩基結晶体のX線粉末回折(XPRD)パターンを図1(多形体I)、図10(遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物多形体I,タイプA)及び図13(遊離塩基トルエン/ヘプタン溶媒和物多形体I,タイプB)に示す。図2の熱重量分析(TGA)曲線は化合物Iの無水遊離塩基多形体Iで10℃/分の昇温速度で窒素流下に得られ、室温から融点までに<0.1重量%の重量低下を示した。
【0016】
図3に示す化合物Iの無水遊離塩基多形体IのDSC曲線は外挿開始温度157.8℃、ピーク温度163.6℃及びそれに伴う熱量44.6J/gの単一吸熱を特徴とする。
【0017】
図10、11及び12は化合物Iの無水遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物多形体IタイプAのX線回折パターン、TGA曲線及びDSC曲線を示す。図13及び14は化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体IタイプBのX線回折パターンとDSC曲線を示す。
【0018】
化合物Iは更に以下に記載するように塩酸塩に変換することができる。図4及び5は化合物Iの塩酸塩形態AのX線回折パターンとDSC曲線を示す。図6及び7は化合物Iの塩酸塩形態BのX線回折パターンとDSC曲線を示す。図8及び9は化合物Iの塩酸塩形態GのX線回折パターンとDSC曲線を示す。図15は化合物Iの塩酸塩形態CのX線回折パターンを示す。図16、17及び18は化合物Iの塩酸塩形態DのX線回折パターン、TGA曲線及びDSC曲線を示す。図19は化合物Iの塩酸塩形態EのX線回折パターンを示す。図20は化合物Iの塩酸塩形態FのX線回折パターンを示す。最後に、図21は化合物Iの塩酸塩形態HのX線回折パターンを示す。
【0019】
本発明の1態様は式I:
【0020】
【化4】
の化合物又はその塩、水和物もしくは多形体の製造方法として、
A)化合物IIIのアルコール基を脱離基に変換した後にヒンダードアミン塩基で処理することにより式II:
【0021】
【化5】
(式中、Pは保護基である)の化合物又はその塩を式III:
【0022】
【化6】
の化合物とカップリングする段階と;
B)保護基Pを除去する段階を含む方法を提供する。
【0023】
本態様の1分類において、段階A及びBの保護基PはBoc及びCBZから選択される。
【0024】
本態様の別の分類において、段階Aの脱離基はトリフラート、トシラート、ノシラート及びメシラートから選択される。本態様の別の分類において、段階Aの脱離基はトリフラートであり、化合物IIIをトリフル酸無水物で処理し、ジトリフラート中間体を形成する。本態様の別の分類において、段階Aのヒンダードアミン塩基はジイソプロピルエチルアミン、トリエチルアミン、トリイソプロピルアミン及びジシクロヘキシルアミンから選択される。本態様の別の分類において、段階Aのヒンダードアミン塩基はジイソプロピルエチルアミンである。本態様の別の分類において、段階Aの反応はアセトニトリル中で実施される。
【0025】
本態様の別の分類において、保護基PはCBZである。本態様の別の分類において、保護基PはCBZであり、段階BのCBZ保護基は水素化により除去される。
【0026】
本態様の別の分類において、保護基PはBocである。本態様の別の分類において、保護基はBocであり、段階BのBoc保護基は酸を使用して除去される。本サブ分類の1サブ分類において、酸はHCl、H2SO4、H3PO4及びTFAから選択される。本サブ分類の別のサブ分類において、酸はHClである。本サブ分類の別のサブ分類において、酸はHClのイソプロパノール溶液である。本態様の別の分類において、段階Bは酢酸イソプロピル、イソプロパノール、塩化メチレン及びTHFから選択される溶媒中で実施される。本分類の1サブ分類において、段階Bの溶媒は酢酸イソプロピルである。
【0027】
本態様の1分類において、方法は更に式Iの化合物を単離する段階を含む。本分類の1サブ分類において、式Iの化合物はトルエン/ヘプタンから再結晶することにより単離される。
【0028】
本発明の別の態様は式I:
【0029】
【化7】
の化合物又はその塩、溶媒和物、水和物もしくは多形体の製造方法として、
式20:
【0030】
【化8】
の化合物の保護基Pを除去する段階を含む方法を提供する。
【0031】
本態様の1分類において、段階A及びBの保護基PはBoc及びCBZから選択される。
【0032】
本態様の別の分類において、保護基PはCBZである。本態様の別の分類において、保護基PはCBZであり、段階BのCBZ保護基は水素化により除去される。
【0033】
本態様の別の分類において、保護基PはBocである。本態様の別の分類において、保護基はBocであり、段階BのBoc保護基は酸を使用して除去される。本サブ分類の1サブ分類において、酸はHCl、H2SO4、H3PO4及びTFAから選択される。本サブ分類の別のサブ分類において、酸はHClである。本サブ分類の別のサブ分類において、酸はHClのイソプロパノール溶液である。本態様の別の分類において、段階Bは酢酸イソプロピル、イソプロパノール、塩化メチレン及びTHFから選択される溶媒中で実施される。本分類の1サブ分類において、段階Bの溶媒は酢酸イソプロピルである。
【0034】
本態様の1分類において、方法は更に式Iの化合物を単離する段階を含む。本分類の1サブ分類において、式Iの化合物はトルエン/ヘプタンから再結晶することにより単離される。
【0035】
本発明の別の態様は式I:
【0036】
【化9】
の化合物又はその塩、溶媒和物、水和物もしくは多形体の製造方法として、溶媒中で酸を使用して式20:
【0037】
【化10】
の化合物のBoc保護基を除去する段階を含む方法を提供する。
【0038】
本態様の1分類において、Boc保護基は酸を使用して除去される。本分類の1サブ分類において、酸はHCl、H2SO4、H3PO4及びTFAから選択される。本分類の別のサブ分類において、酸はHClである。本分類の別のサブ分類において、酸はHClのイソプロパノール溶液である。本態様の別の分類において、脱保護反応は酢酸イソプロピル、イソプロパノール、塩化メチレン及びTHFから選択される溶媒中で実施される。本分類の1サブ分類において、溶媒は酢酸イソプロピルである。
【0039】
本態様の1分類において、方法は更に式Iの化合物を単離する段階を含む。本分類の1サブ分類において、式Iの化合物はトルエン/ヘプタンから再結晶することにより単離される。
【0040】
本発明の別の態様は式20:
【0041】
【化11】
(式中、Pは保護基である)の化合物の製造方法として、化合物IIIのアルコール基を脱離基に変換した後にヒンダードアミン塩基で処理することにより式II:
【0042】
【化12】
(式中、Pは保護基である)の化合物又はその塩を式III:
【0043】
【化13】
の化合物とカップリングする段階を含む方法を提供する。
【0044】
本態様の1分類において、保護基PはBoc及びCBZから選択される。本態様の別の分類において、脱離基はトリフラート、トシラート、ノシラート及びメシラートから選択される。本態様の別の分類において、脱離基はトリフラートであり、化合物IIIをトリフル酸無水物で処理し、ジトリフラート中間体を形成する。本態様の別の分類において、ヒンダードアミン塩基はジイソプロピルエチルアミン、トリエチルアミン、トリイソプロピルアミン及びジシクロヘキシルアミンから選択される。本態様の別の分類において、ヒンダードアミン塩基はジイソプロピルエチルアミンである。本態様の別の分類において、反応はアセトニトリル中で実施される。
【0045】
本態様の別の分類において、方法は更に式20の化合物を単離する段階を含む。本分類の1サブ分類において、式20の化合物は酢酸イソプロピル、ジクロロメタン、アセトニトリル、ヘプタン又はその混合物から再結晶することにより単離される。本分類の別のサブ分類において、式20の化合物は酢酸イソプロピルとヘプタンから再結晶することにより単離される。
【0046】
本発明の別の態様は式II:
【0047】
【化14】
(式中、Pは保護基である)の化合物又はその塩の製造方法として、
(A)式1:
【0048】
【化15】
の化合物を塩基で処理した後にヒドラジンで処理することにより式3:
【0049】
【化16】
のヒドラジドを製造する段階と;
(B)式3のヒドラジドをカップリング剤で処理することにより式4:
【0050】
【化17】
のオキサジアゾールを形成する段階と;
(C)式4のオキサジアゾールをアルキルマグネシウム化合物で処理した後にアルキルリチウム化合物とDMFで処理することにより式5:
【0051】
【化18】
のアルデヒドを製造する段階と;
(D)式5のアルデヒドを触媒の存在下に(S)−tert−ブチルスルフィンアミドで処理することにより式6:
【0052】
【化19】
のN−tert−ブチルスルフィニルイミンを製造する段階と;
(E)式6のN−tert−ブチルスルフィニルイミンのオキサジアゾール窒素に保護基Pを付加することにより式7:
【0053】
【化20】
(式中、Pは保護基である)の保護オキサジアゾール化合物を形成する段階と;
(F)式7の化合物をロジウム触媒とリガンドの存在下にボロキシン10で処理することにより式8:
【0054】
【化21】
(式中、Pは保護基である)のN−tert−ブチルスルフィニルアミンを形成する段階と;
(G)式8の化合物のtert−ブチルスルホキシド基を開裂することにより式II:
【0055】
【化22】
(式中、Pは保護基である)の化合物を形成する段階を含む方法を提供する。
【0056】
本態様の1分類において、段階Aの塩基はDABCO、トリエチルアミン及びジイソプロピルエチルアミンから選択される。本分類の1サブ分類において、段階Aの塩基はDABCOである。本態様の別の分類において、段階Aの反応はメタノール中で実施される。本態様の別の分類において、段階Aの反応は50〜55℃で実施される。本態様の別の分類において、段階Aのヒドラジンは64%ヒドラジンである。本態様の別の分類において、段階Aの反応はメタノール中でDABCOで処理した後に64%ヒドラジンで処理することにより実施される。本態様の別の分類において、方法は更に式3のヒドラジドを単離する段階を含む。本分類の1サブ分類において、式3のヒドラジドは固体である。本態様の更に別の分類において、方法は更に段階Aの反応をワークアップする段階と、段階Bで式3のヒドラジドを溶液として使用する段階を含む。
【0057】
本態様の別の分類において、段階Bのカップリング剤はCDI、トリホスゲン及びホスゲンから選択される。本態様の別の分類において、段階Bのカップリング剤はCDIである。本態様の別の分類において、段階Bの反応は非プロトン性溶媒中で実施される。本分類の1サブ分類において、非プロトン性溶媒はTHF、トルエン及びエーテルである。本分類の別のサブ分類において、非プロトン性溶媒はTHFである。本態様の別の分類において、段階Bの反応は室温で実施される。本態様の別の分類において、段階Bの反応はTHF中でCDIを使用して実施される。本分類の1サブ分類において、反応は室温で実施される。本態様の別の分類において、方法は更に段階Bの式4のオキサジアゾールを単離する段階を含む。本分類の1サブ分類において、式4のオキサジアゾールは固体である。本態様の更に別の分類において、方法は更に段階Bの反応をワークアップする段階と、段階Cで式4のオキサジアゾールを溶液として使用する段階を含む。
【0058】
本態様の別の分類において、段階Cのアルキルマグネシウム化合物はジブチルマグネシウム、ジメチルマグネシウム、ジエチルマグネシウム及びジイソプロピルマグネシウムから選択される。本態様の別の分類において、段階Cのアルキルマグネシウム化合物はジ−n−ブチルマグネシウムである。本態様の別の分類において、段階Cのアルキルリチウム化合物はn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウム及びヘキシルリチウムから選択される。本態様の別の分類において、段階Cのアルキルリチウム化合物はn−ブチルリチウムである。本態様の別の分類において、段階Cの反応は非プロトン性溶媒中で実施される。本分類の1サブ分類において、段階Cの非プロトン性溶媒はTHF、トルエン、MTBE及びジエチルエーテルから選択される。本分類の別のサブ分類において、段階Cの非プロトン性溶媒はTHFである。本態様の別の分類において、段階Cの反応は約−20〜−78℃の温度で実施される。本分類の1サブ分類において、段階Cの反応は約−40〜−50℃の温度で実施される。本態様の別の分類において、段階Cの反応は約−20〜−78℃の温度にてTHF中でジ−n−ブチルマグネシウムとn−ブチルリチウムを使用して実施される。本分類の1サブ分類において、温度は約−40〜−50℃である。本態様の別の分類において、段階Cの反応は酸を使用してワークアップされる。本分類の1サブ分類において、段階Cの酸はHCl又はH2SO4である。本分類の1サブ分類において、段階Cの酸はHClである。本態様の別の分類において、方法は更に式5のアルデヒドを単離する段階を含む。本分類の1サブ分類において、式5のアルデヒドは固体である。本態様の更に別の分類において、方法は更に段階Cの反応をワークアップする段階と、段階Dで式5のアルデヒドを溶液として使用する段階を含む。
【0059】
本態様の別の分類において、段階Dの触媒はPPTS、KHSO4、BF3・エーテル錯体、Ti(OEt)4及びTiCl4/トリエチルアミンから選択される。本態様の別の分類において、段階Dの触媒はPPTSである。本態様の別の分類において、段階Dの反応はトルエン、塩化メチレン及びTHFから選択される溶媒中で実施される。本態様の別の分類において、段階Dの反応はトルエン中で実施される。本態様の別の分類において、段階Dの反応はトルエン中でPPTSを使用して実施される。本分類の1サブ分類において、段階Dの反応は約40℃で実施される。本態様の別の分類において、方法は更に段階Dの式6のN−tert−ブチルスルフィニルイミンを単離する段階を含む。本分類の1サブ分類において、式6のN−tert−ブチルスルフィニルイミンは固体である。本態様の更に別の分類において、方法は更に段階Dの反応をワークアップする段階と、段階Eで式6のN−tert−ブチルスルフィニルイミンを溶液として使用する段階を含む。
【0060】
本態様の1分類において、式7の化合物の保護基PはCBZ又はBoc基である。本態様の別の分類において、式7の化合物は保護基PがCBZであるN−CBZ保護オキサジアゾールである。本態様の別の分類において、式7の保護オキサジアゾール化合物は保護基PがBocであるN−Boc保護オキサジアゾールである。本分類の1サブ分類において、式7のN−Boc保護オキサジアゾールは式6のN−tert−ブチルスルフィニルイミンを塩基の存在下にboc酸無水物で処理することにより製造される。本サブ分類の1サブ分類において、塩基は第3級アミン塩基である。本サブ分類の別のサブ分類において、塩基はトリエチルアミンである。本分類の別のサブ分類において、N−Boc保護保護オキサジアゾールは非プロトン性溶媒中でトリエチルアミンの存在下に式6のN−tert−ブチルスルフィニルイミンをBoc酸無水物で処理することにより製造される。本サブ分類の1サブ分類において、非プロトン性溶媒はTHFである。本サブ分類の別のサブ分類において、段階Eの反応は約40℃で実施される。本態様の別の分類において、式7の化合物は式6のN−tert−ブチルスルフィニルイミンをTHF中でboc酸無水物とトリエチルアミンで処理することにより製造されるN−Boc保護オキサジアゾールである。本態様の別の分類において、方法は更に段階Eの式7の保護オキサジアゾール化合物を単離する段階を含む。本分類の1サブ分類において、式7の保護オキサジアゾール化合物は固体である。本態様の別の分類において、方法は更に段階Eの反応をワークアップする段階と、段階Fで式7の保護オキサジアゾール化合物を溶液として使用する段階を含む。
【0061】
本態様の別の分類において、段階Fの保護基PはCBZである。本態様の別の分類において、段階Fの保護基PはBocである。本態様の別の分類において、段階Fのロジウム触媒はRh(acac)(CH2CH2)2である。本態様の別の分類において、リガンドはホスフィンリガンドである。本分類の1サブ分類において、ホスフィンリガンドは1,2−ビス(ジフェニルホスフィノ)ベンゼンと1,2−ビス(ジフェニルホスフィノ)エタンから選択される。本分類の別のサブ分類において、ホスフィンリガンドは1,2−ビス(ジフェニルホスフィノ)ベンゼンである。本態様の別の分類において、溶媒はtert−アミルアルコール、tert−ブタノール、THF及びジオキサンから選択される。本分類の1サブ分類において、溶媒はtert−アミルアルコールである。本態様の別の分類において、段階Fの反応は約室温〜約45℃の温度で実施される。本態様の別の分類において、保護基PがBocである段階Fの反応はRh(acac)(CH2CH2)2と1,2−ビス(ジフェニルホスフィノ)ベンゼンの存在下で実施される。本分類の1サブ分類において、段階Fの反応はtert−アミルアルコール中で実施される。本分類の別のサブ分類において、段階Fの反応は約室温〜約45℃の温度で実施される。本態様の別の分類において、方法は更に段階Fの式8のN−tert−ブチルスルフィニルアミンを単離する段階を含む。本分類の1サブ分類において、式8のN−tert−ブチルスルフィニルアミンは固体である。本態様の別の分類において、方法は更に段階Fの反応をワークアップする段階と、段階Gで式8のN−tert−ブチルスルフィニルアミンを溶液として使用する段階を含む。
【0062】
本態様の別の分類において、段階Gの保護基PはCBZである。本態様の別の分類において、段階Gの保護基PはBocである。本態様の別の分類において、段階Gのtert−ブチルスルホキシド基は酸で開裂される。本態様の別の分類において、tert−ブチルスルホキシド基は塩酸、硫酸、リン酸及びトリフルオロ酢酸から構成される群から選択される酸で処理することにより開裂される。本態様の別の分類において、段階Gの開裂はハロゲン化溶媒中で実施される。本分類の1サブ分類において、ハロゲン化溶媒はジクロロメタン、クロロホルム及び四塩化炭素から選択される。本分類の別のサブ分類において、ハロゲン化溶媒はジクロロメタンである。本態様の別の分類において、段階Gの開裂は室温で実施される。本態様の別の分類において、段階Gの化合物8のtert−ブチルスルホキシド基は塩酸で処理することにより開裂される。本態様の別の分類において、方法は更に式IIの化合物を単離する段階を含む。本分類の1サブ分類において、式IIの化合物は固体である。本態様の別の分類において、方法は更に段階Gの反応をワークアップする段階と、カップリング反応で式IIaの化合物を溶液として使用し、化合物20を得る段階を含む。
【0063】
本発明の別の態様は式III:
【0064】
【化23】
の化合物又はその塩の製造方法として、
(A)式11:
【0065】
【化24】
の化合物をグリニャール試薬で処理した後に塩化イソブチリルで処理することにより式12:
【0066】
【化25】
の化合物を製造する段階と;
(B)ハロゲン化シリル又はシリルトリフラートの存在下にフッ素源と塩基で処理して式12の化合物をフッ素化することにより式13:
【0067】
【化26】
のフルオロケトン化合物を形成する段階と;
(C)式13の化合物を塩基の存在下にホスホノ酢酸トリメチルで処理することにより式14:
【0068】
【化27】
の化合物を製造する段階と;
(D)式14の化合物のエステルを加水分解することにより式15:
【0069】
【化28】
の化合物を製造する段階と;
(E)式15の化合物の二重結合を還元することにより式16:
【0070】
【化29】
の化合物を形成する段階と;
(F)式16の化合物のエステル化により式17:
【0071】
【化30】
(式中、R=C1−3アルキルである)の化合物を形成する段階と;
(G)式17の化合物のカルボキシル化により式18:
【0072】
【化31】
(式中、R=C1−3アルキルである)の化合物を形成する段階と;
(H)式18の化合物を還元することにより式19:
【0073】
【化32】
の化合物を形成する段階と;
(I)式19の化合物をシアノ化することにより式III
【0074】
【化33】
の化合物を形成する段階を含む方法を提供する。
【0075】
本態様の別の分類において、Rは−CH3である。本態様の別の分類において、RはCH2CH3である。本態様の更に別の分類において、Rは−CH2CH2CH3又は−CH(CH3)2である。
【0076】
本態様の別の分類において、段階Aのグリニャール試薬はイソプロピルマグネシウムクロリドである。本態様の別の分類において、段階Aの反応は1種以上の遷移金属ハロゲン化物塩触媒の存在下で実施される。本分類の1サブ分類において、遷移金属ハロゲン化物塩触媒はCuCl、ZnCl2及びCoCl2から選択される。本分類の別のサブ分類において、遷移金属ハロゲン化物塩触媒はCuClとZnCl2である。本態様の別の分類において、段階Aのグリニャール反応はエーテル溶媒中で実施される。本分類の1サブ分類において、エーテル溶媒はテトラヒドロフランである。本態様の別の分類において、段階Aの反応はCuClとZnCl2の存在下にテトラヒドロフラン中でイソプロピルマグネシウムクロリドを使用して実施される。本態様の別の分類において、方法は更に式12の化合物を単離する段階を含む。本態様の更に別の分類において、方法は更に段階Aの反応をワークアップする段階と、段階Bで式12の化合物をトルエン溶液として使用する段階を含む。
【0077】
本態様の別の分類において、段階Bのフッ素源はSelect−Fluor(登録商標)フッ素化剤である。本態様の別の分類において、段階Bの塩基はアルコキシド塩基又はナトリウムアミラートである。本分類の1サブ分類において、アルコキシド塩基はカリウムtert−ブトキシドである。本態様の別の分類において、段階Bの塩基はナトリウムアミラートである。本態様の別の分類において、段階Bのハロゲン化シリルとシリルトリフラートはterf−ブチルジメチルシリルクロリド、トリメチルシリルクロリド及びtert−ブチルジメチルシリルトリフラートから選択される。本分類の1サブ分類において、段階Bのハロゲン化シリルはtert−ブチルジメチルシリルクロリドである。本態様の別の分類において、段階Bのフッ素化源はSelect−Fluor(登録商標)フッ素化剤であり、塩基はナトリウムアミラートであり、ハロゲン化シリルはtert−ブチルジメチルシリルクロリドである。本態様の別の分類において、方法は更に式13の化合物を単離する段階を含む。本態様の更に別の分類において、方法は更に段階Bの反応をワークアップする段階と、段階Cで式13の化合物をトルエン溶液として使用する段階を含む。
【0078】
本態様の1分類において、段階Cの塩基は炭酸セシウム、炭酸カリウム、炭酸リチウム、カリウムtert−ブトキシド、水素化リチウム、水素化ナトリウム及びナトリウムアミラートから選択される。本分類の1サブ分類において、段階Cの塩基は炭酸カリウムである。本態様の別の分類では、段階Cのホスホノ酢酸トリメチルを式13の化合物に加える前に塩基で前処理する。本分類の1サブ分類において、段階Cの塩基は炭酸セシウム、炭酸カリウム、炭酸リチウム、カリウムtert−ブトキシド、水素化リチウム、水素化ナトリウム及びナトリウムアミラートから選択される。本サブ分類の1サブ分類において、段階Cの塩基は炭酸カリウムである。本態様の別の分類において、段階Cの反応は極性非プロトン性溶媒中で実施される。本分類の1サブ分類において、極性非プロトン性溶媒はジメチルホルムアミド、テトラヒドロフラン又はエーテルから選択される。本サブ分類の1サブ分類において、段階Cの溶媒はジメチルホルムアミドであるである。本態様の別の分類では、炭酸カリウムで前処理したホスホノ酢酸トリメチルと段階Cの式13の化合物を反応させる。本分類の1サブ分類において、段階Cの反応はジメチルホルムアミド中で実施される。本態様の別の分類において、方法は更に段階Cの式14の化合物を単離する段階を含む。本態様の更に別の分類において、方法は更に段階Cの反応をワークアップする段階と、段階Dで式14の化合物をトルエン中で使用する段階を含む。
【0079】
本態様の別の分類において、段階Dの加水分解は水酸化ナトリウム、水酸化リチウム又は水酸化カリウムを使用して実施される。本分類の1サブ分類において、段階Dの加水分解は水酸化ナトリウムを使用して実施される。本分類の別のサブ分類において、段階Dの加水分解は水性溶媒中で実施される。本サブ分類の1サブ分類において、水性溶媒はメタノール/水である。本サブ分類の別のサブ分類において、水性溶媒はメタノール/水/トルエンである。本態様の別の分類において、段階Dの加水分解はメタノール/水中で水酸化ナトリウムを使用して実施される。本態様の別の分類において、段階Dの加水分解はメタノール/水/トルエン中で水酸化ナトリウムを使用して実施される。本態様の別の分類において、方法は更に段階Dの式15の化合物を単離する段階を含む。
【0080】
本態様の別の分類において、段階Eの化合物15の還元は水素とルテニウム触媒の存在下の水素化である。本分類の1サブ分類において、ルテニウム触媒はアキシャルキラルリガンドをもつ。本サブ分類の1サブ分類において、アキシャルキラルリガンドはJosiphos(登録商標)型リガンド、Solphos(登録商標)型リガンド、CH3O−BIPHEP(登録商標)型リガンド、BINAP型リガンド又はSegphos(登録商標)型リガンドである。本サブ分類の別のサブ分類において、アキシャルキラルリガンドは(R)−Cl、CH3O−BIPHEP(登録商標)、(S)−Solphos(登録商標)、(R)−フリル−Solphos(登録商標)及びSL−J212−1である。本サブ分類の別のサブ分類において、Josiphos(登録商標)型アキシャルキラルリガンドはSL−J212−1である。本分類の別のサブ分類において、段階Eの水素化は加圧下に実施される。本サブ分類の1サブ分類において、段階Eの水素化は200psigで実施される。本分類の別のサブ分類において、段階Eの水素化は約40〜50℃で実施される。本態様の別の分類において、段階Eの還元はJosiphos(登録商標)型アキシャルキラルリガンドSL−J212−1をもつルテニウム触媒の存在下の水素化である。本分類の1サブ分類において、ルテニウム触媒は[(シメン)RuCl]2をSL−J212−1と反応させることにより製造される。本態様の別の分類において、方法は更に段階Eの式16の化合物を単離する段階を含む。本態様の別の分類において、方法は更に段階Eの式16の化合物を固体として単離する段階と、式16の化合物を再結晶させる段階を含む。本態様の別の分類において、方法は更に段階Eの反応をワークアップする段階と、段階Fで式16の化合物を溶液として使用する段階を含む。本態様の更に別の分類において、方法は更に段階Eの反応をワークアップする段階と、段階Fで式16の化合物をメタノール溶液として使用する段階を含む。
【0081】
本態様の別の分類において、段階Fのエステル化はアルコール溶媒中で酸塩化物の存在下に実施される。本分類の1サブ分類において、段階Fでエステル化により形成されるエステルはメチルエステルであり、エステル化はメタノール中で塩化アセチルの存在下に実施される。本サブ分類の1サブ分類において、エステル化は室温で実施される。本態様の別の分類において、方法は更に段階Fの式17の化合物を単離する段階を含む。
【0082】
本態様の別の分類において、段階Gのカルボキシル化は式17の化合物をエーテル又は極性溶媒中で塩基で処理した後にCO2を加えることにより実施される。本分類の1サブ分類において、塩基はリチウムヘキサメチルジシラジド、ナトリウムヘキサメチルジシラジド、カリウムヘキサメチルジシラジドム及びLDAから選択される。本分類の別のサブ分類において、エーテル又は極性溶媒はTHF、THF、MTBE、DME、トルエン及びDMPUの1種以上から選択される。本分類の別のサブ分類では、式17の化合物をTHF又はトルエン中でリチウムヘキサメチルジシラジドで処理した後にCO2を加えることによりカルボキシル化反応を実施した。本分類の別のサブ分類では、式17の化合物をTHF又はトルエンとDMPU中でリチウムヘキサメチルジシラジドで処理した後にCO2を加えることによりカルボキシル化反応を実施した。本分類の別のサブ分類では、式17の化合物をDME/トルエンとDMPU中でリチウムヘキサメチルジシラジドで処理した後にCO2を加えることによりカルボキシル化反応を実施した。本態様の別の分類において、方法は更に段階Gの式18の化合物を単離する段階を含む。
【0083】
本態様の1分類では、還元剤の存在下で段階Hの還元を実施した。本分類の1サブ分類において、還元剤は水素化ホウ素ナトリウム、水素化ホウ素ナトリウム/I2、水素化ホウ素ナトリウム/Br2、水素化ホウ素ナトリウムBF3/テトラヒドロフラン錯体、BF3/エーテル錯体、ボラン/テトラヒドロフラン錯体及びボラン/ジメチルスルフィド錯体である。本分類の別のサブ分類において、還元剤は水素化ホウ素ナトリウム/Br2である。本分類の別のサブ分類において、段階Hの還元はトルエン、DME、THF、DME/トルエン及びジクロロメタンの1種以上から選択される溶媒中で実施される。本分類の別のサブ分類において、段階Hの還元はDME及びトルエンから選択される溶媒中で実施される。本態様の別の分類において、方法は更に段階Hの式19の化合物を単離する段階を含む。
【0084】
本態様の1分類において、段階Iのシアノ化は亜鉛、臭素、Zn(CN)2及びパラジウム触媒の存在下で実施される。本分類の1サブ分類において、パラジウム触媒は二座又は単座パラジウム触媒である。本分類の別のサブ分類において、パラジウム触媒はパラジウムホスフィン触媒である。本サブ分類の1サブ分類において、パラジウム触媒はパラジウムテトラキストリフェニルホスフィンである。本分類の別のサブ分類において、パラジウム触媒はPd(dppf)2である。本分類の別のサブ分類において、段階Iのシアノ化はDMF中で実施される。本分類の別のサブ分類において、段階Iのシアノ化はDMF中で80℃にて実施される。
【0085】
本態様の別の分類において、方法は更に段階Iの式IIIの化合物を単離する段階を含む。
【0086】
本発明の別の態様はCB−1調節に関連する疾患の予防又は治療方法として、前記疾患の予防又は治療を必要とする対象に治療有効量の化合物Iの多形体、水和物又は塩を投与する段階を含む方法を提供する。
【0087】
本発明の別の態様はCB−1調節に関連する疾患の治療、抑制又は予防を必要とする対象における前記治療、抑制又は予防に有用な医薬の製造用としての治療有効量の化合物Iの多形体、溶媒和物、水和物又は塩の使用を提供する。
【0088】
本発明の別の態様は肥満症、摂食障害又は肥満関連障害の予防又は治療方法として、前記予防又は治療を必要とする対象に治療有効量の化合物Iの多形体、水和物又は塩を投与する段階を含む方法を提供する。
【0089】
本発明の別の態様は肥満症、摂食障害又は肥満関連障害の治療、抑制又は予防を必要とする対象における前記治療、抑制又は予防に有用な医薬の製造用としての治療有効量の化合物Iの多形体、溶媒和物、水和物又は塩の使用を提供する。
【0090】
「3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル」なる用語は固体状の3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリルのみならず、昇温により3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリルに変換可能なそのガラス体、凍結乾燥物及び混合物等の任意非晶質又は部分結晶質固体状の3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリルも包含する。
【0091】
多形体とは化学組成が同一でありながら結晶構造の異なる化合物である。多形性とは同一の化学物質が異なる結晶構造として存在する性質である。構造式Iの化合物3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリルとそのHCl塩は結晶化条件の入念な制御により各々形成することが可能な少なくとも11種類の多形体ないし結晶体として存在することが判明した。
【0092】
「水和物」なる用語は化合物Iの完全水和物、複合水和物及び部分水和物の全水和物を包含するものとし、限定されないが、一水和物、半水和物及びビス水和物が挙げられる。
【0093】
「溶媒和物」なる用語は化合物Iの結晶構造内に溶媒分子を含む化合物形態、又は化合物Iと結合もしくは会合した溶媒分子を含む化合物形態を包含するものとし、溶媒としては限定されないが、トルエン、ヘプタン、酢酸イソプロピル、酢酸エチル、メチルシクロヘキサン及び水が挙げられる。
【0094】
「非晶質」なる用語は長距離分子秩序をもたない固体形態を意味する。
【0095】
構造式Iの3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリルは結晶質塩酸塩を形成することが判明した。
【0096】
式Iの化合物の他の塩とは、医薬的に許容可能な通常の塩を意味し、例えば化合物がカルボキシル基をもつ場合にはカルボキシル基の塩基付加塩を意味し、化合物がアミノ又は塩基性複素環基をもつ場合にはアミノ又は塩基性複素環の酸付加塩を意味し、他の化合物についても同様である。「医薬的に許容可能な塩」なる用語は無機塩基又は酸と有機塩基又は酸を含む医薬的に許容可能な非毒性塩基又は酸から製造される塩を意味する。塩基付加塩としては、アルカリ金属(限定されないが、例えばナトリウム、カリウム);アルカリ土類金属(限定されないが、例えばカルシウム、マグネシウム);アンモニウム又は有機アミン(限定されないが、例えばトリメチルアミン、トリエチルアミン、ジシクロヘキシルアミン、エタノールアミン、ジエタノールアミン、トリエタノールアミン、プロカイン、N,N’−ジベンジルエチレンジアミン)等との塩が挙げられる。酸付加塩としては、無機酸(限定されないが、例えば塩酸、硫酸、硝酸、リン酸、過塩素酸)、有機酸(限定されないが、例えば酢酸、蓚酸、マレイン酸、フマル酸、酒石酸、クエン酸、アスコルビン酸、トリフルオロ酢酸、酢酸)、スルホン酸(限定されないが、例えばメタンスルホン酸、イセチオン酸、ベンゼンスルホン酸、p−トルエンスルホン酸、p−トルエンスルホン酸一水和物、p−トルエンスルホン酸水和物、樟脳スルホン酸)等との塩が挙げられる。
【0097】
本発明の1態様では、3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)をその遊離塩基、塩、水和物又は多形体として含有する医薬組成物を提供する。本態様の1分類において、化合物Iは実質的に純粋な形態である。本態様の別の分類において、化合物Iは結晶質である。本態様の別の分類において、化合物Iは結晶質無水遊離塩基である。本態様の別の分類において、化合物Iは結晶質遊離塩基溶媒和物である。本態様の別の分類において、化合物Iは結晶質無水塩である。本態様の別の分類において、化合物Iは結晶質塩水和物である。本態様の別の分類において、化合物Iは結晶質無水HCl塩である。本態様の別の分類において、化合物Iは結晶質HCl塩水和物である。本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物Iの遊離塩基形態Iとして含有する。本分類の1サブ分類において、化合物Iの遊離塩基形態Iは実質的に純粋な形態である。本分類の別のサブ分類において、化合物Iの遊離塩基形態Iは結晶質である。本分類の別のサブ分類において、化合物Iの遊離塩基形態Iは無水である。本分類の別のサブ分類において、化合物Iの遊離塩基形態Iは無水結晶質である。
【0098】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物Iの遊離塩基トルエン/ヘプタン溶媒和物形態I,タイプBとして含有する。本分類の1サブ分類において、化合物Iの遊離塩基トルエン/ヘプタン溶媒和物形態I,タイプBは実質的に純粋な形態である。本分類の別のサブ分類において、化合物Iの遊離塩基トルエン/ヘプタン溶媒和物形態I,タイプBは結晶質である。本分類の別のサブ分類において、化合物Iの遊離塩基トルエン/ヘプタン溶媒和物形態I,タイプBは無水である。本分類の別のサブ分類において、化合物Iの遊離塩基トルエン/ヘプタン溶媒和物形態I,タイプBは無水結晶質である。
【0099】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物形態I,タイプAとして含有する。本分類の1サブ分類において、化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物形態I,タイプAは実質的に純粋な形態である。本分類の別のサブ分類において、化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物形態I,タイプAは結晶質である。本分類の別のサブ分類において、化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物形態I,タイプAは無水である。本分類の別のサブ分類において、化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物形態I,タイプAは無水結晶質である。
【0100】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物IのHCl塩形態Aとして含有する。本分類の1サブ分類において、化合物IのHCl塩形態Aは実質的に純粋な形態である。本分類の別のサブ分類において、化合物IのHCl塩形態Aは結晶質である。本分類の別のサブ分類において、化合物IのHCl塩形態Aは無水である。本分類の別のサブ分類において、化合物IのHCl塩形態Aは無水結晶質である。
【0101】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物IのHCl塩形態Bとして含有する。本分類の1サブ分類において、化合物IのHCl塩形態Bは実質的に純粋な形態である。本分類の別のサブ分類において、化合物IのHCl塩形態Bは結晶質である。本分類の別のサブ分類において、化合物IのHCl塩形態Bは無水である。本分類の別のサブ分類において、化合物IのHCl塩形態Bは無水結晶質である。
【0102】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物IのHCl塩形態Cとして含有する。本分類の1サブ分類において、化合物IのHCl塩形態Cは実質的に純粋な形態である。本分類の別のサブ分類において、化合物IのHCl塩形態Cは結晶質である。本分類の別のサブ分類において、化合物IのHCl塩形態Cは無水である。本分類の別のサブ分類において、化合物IのHCl塩形態Cは無水結晶質である。
【0103】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物IのHCl塩形態Dとして含有する。本分類の1サブ分類において、化合物IのHCl塩形態Dは実質的に純粋な形態である。本分類の別のサブ分類において、化合物IのHCl塩形態Dは結晶質である。本分類の別のサブ分類において、化合物IのHCl塩形態Dは無水である。本分類の別のサブ分類において、化合物IのHCl塩形態Dは無水結晶質である。
【0104】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物IのHCl塩形態Eとして含有する。本分類の1サブ分類において、化合物IのHCl塩形態Eは実質的に純粋な形態である。本分類の別のサブ分類において、化合物IのHCl塩形態Eは結晶質である。本分類の別のサブ分類において、化合物IのHCl塩形態Eは無水である。本分類の別のサブ分類において、化合物IのHCl塩形態Eは無水結晶質である。
【0105】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物IのHCl塩形態Fとして含有する。本分類の1サブ分類において、化合物IのHCl塩形態Fは実質的に純粋な形態である。本分類の別のサブ分類において、化合物IのHCl塩形態Fは結晶質である。本分類の別のサブ分類において、化合物IのHCl塩形態Fは水和物である。本分類の別のサブ分類において、化合物IのHCl塩形態Fは結晶質水和物である。
【0106】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物IのHCl塩形態Gとして含有する。本分類の1サブ分類において、化合物IのHCl塩形態Gは実質的に純粋な形態である。本分類の別のサブ分類において、化合物IのHCl塩形態Gは結晶質である。本分類の別のサブ分類において、化合物IのHCl塩形態Gは無水である。本分類の別のサブ分類において、化合物IのHCl塩形態Gは無水結晶質である。
【0107】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物IのHCl塩形態Hとして含有する。本分類の1サブ分類において、化合物IのHCl塩形態Hは実質的に純粋な形態である。本分類の別のサブ分類において、化合物IのHCl塩形態Hは結晶質である。本分類の別のサブ分類において、化合物IのHCl塩形態Hは無水である。本分類の別のサブ分類において、化合物IのHCl塩形態Hは無水結晶質である。
【0108】
本発明の方法における化合物は置換方式に応じて光学異性体、ジアステレオマー及び幾何異性体等の立体異性体又は互変異性体を含む。本発明は本発明の組成物に含まれる化合物のこのような全異性体とその混合物を包含するものとする。上記化合物の全水和物、溶媒和物及び多形結晶体とその使用(本発明の方法におけるその使用を含む)が本発明の範囲に含まれる。
【0109】
ニューロキニン−1(NK−1)受容体アンタゴニストを本発明の化合物と有利に併用することができる。本発明で使用されるNK−1受容体アンタゴニストは文献に詳細に記載されている。本発明で使用される特定ニューロキニン−1受容体アンタゴニストとしては、(±)−(2R3R,2S3S)−N−{[2−シクロプロポキシ−5−(トリフルオロメトキシ)フェニル]メチル}−2−フェニルピペリジン−3−アミン;2−(R)−(1−(R)−(3,5−ビス(トリフルオロメチル)フェニル)エトキシ)−3−(S)−(4−フルオロフェニル)−4−(3−(5−オキソ−1H,4H−1,2,4−トリアゾロ)メチル)モルホリン;アプレピタント;CJ17493;GW597599;GW679769;R673;RO67319;R1124;R1204;SSR146977;SSR240600;T−2328;及びT2763;又はその医薬的に許容可能な塩が挙げられる。式I、II又はIIIの化合物と併用することができる他の抗肥満薬の例は“Patent focus on new anti−obesity agents,”Exp.Opin.Ther.Patents,10:819−831(2000);“Novel anti−obesity drugs,”Exp.Opin.Invest.Drugs,9:1317−1326(2000);及び“Recent advances in feeding suppressing agents:potential therapeutic strategy for the treatment of obesity,Exp.Opin.Ther.Patents,11:1677−1692(2001)に開示されている。肥満におけるニューロペプチドYの役割はExp.Opin.Invest.Drugs,9:1327−1346(2000)に記載されている。カンナビノイド受容体リガンドはExp.Opin.Invest.Drugs,9:1553−1571(2000)に記載されている。
【0110】
本発明の別の側面は化合物Iの多形体、水和物又は塩と、医薬的に許容可能なキャリヤーを含有する医薬組成物を提供する。本発明の医薬組成物は活性成分として式Iの化合物又はその医薬的に許容可能な塩を含有しており、更に医薬的に許容可能なキャリヤーと場合により他の治療成分を加えてもよい。
【0111】
組成物としては経口、直腸、局所、非経口(例えば皮下、筋肉内及び静脈内)、眼球内(眼科)、肺(鼻腔又は口腔吸入)又は鼻腔内投与に適した組成物が挙げられるが、任意の所与症例に最適な経路は治療する病態の種類及び重篤度と活性成分の種類により異なる。組成物は単位用量形態が適切であり、製薬分野で周知の方法のいずれかにより製造することができる。
【0112】
実際の使用では、従来の医薬配合技術に従って化合物Iの多形体、水和物及び塩を活性成分として医薬キャリヤーと混和することができる。キャリヤーは例えば経口又は非経口(例えば静脈内)等の投与に所望される製剤形態に応じて多様な形態を取ることができる。経口剤形用組成物を製造する際には、通常の医薬媒体のいずれかを利用することができ、例えば懸濁液剤、エリキシル剤及び溶液剤等の経口液体製剤の場合には例えば水、グリコール、油類、アルコール類、フレーバー剤、防腐剤、着色剤等が挙げられ、例えば散剤、ハード及びソフトカプセル剤並びに錠剤等の経口固体製剤の場合には澱粉、糖類、微結晶セルロース、希釈剤、顆粒化剤、滑沢剤、結合剤、崩壊剤等のキャリヤーが挙げられ、固体経口製剤のほうが液体製剤よりも好ましい。
【0113】
投与し易いという理由から、錠剤とカプセル剤が最も有利な経口用量単位形態であり、その場合には当然のことながら固体医薬キャリヤーを使用する。所望により、標準水性又は非水性技術により錠剤にコーティングしてもよい。このような組成物及び製剤は少なくとも0.1%の活性化合物を含有すべきである。これらの組成物中の活性化合物の百分率は当然のことながら変動させることができ、単位重量の約2%〜約60%が適切であると思われる。このような治療薬として有用な組成物中の活性化合物の量は有効用量が得られるような量とする。活性化合物は鼻腔内投与することもできる(例えば液体滴剤又はスプレー)。
【0114】
錠剤、ピル、カプセル剤等にはトラガカントガム、アラビアガム、コーンスターチ又はゼラチン等の結合剤;リン酸2カルシウム等の賦形剤;コーンスターチ、ジャガイモ澱粉、アルギン酸等の崩壊剤;ステアリン酸マグネシウム等の滑沢剤;及びスクロース、ラクトース又はサッカリン等の甘味剤も添加してもよい。用量単位形態がカプセルの場合には、上記型の材料に加え、脂肪油等の液体キャリヤーを加えることができる。
【0115】
コーティングとして又は用量単位の物理的形状を変更するために他の各種材料を加えてもよい。例えば、錠剤にはシェラック、糖又は両者をコーティングすることができる。シロップ又はエリキシル剤には活性成分に加え、甘味剤としてスクロース、防腐剤としてメチル及びプロピルパラベン、色素並びにチェリー又はオレンジフレーバー等のフレーバー剤を加えることができる。
【0116】
化合物Iの多形体、水和物及び塩は非経口投与することもできる。ヒドロキシプロピルセルロース等の界面活性剤と適切に混合した水中でこれらの活性化合物の溶液剤又は懸濁液剤を製造することができる。グリセロール、液体ポリエチレングリコール及びその油中混液で分散液剤を製造することもできる。通常の保存及び使用条件下で、これらの製剤には微生物の増殖を防ぐために防腐剤を添加することができる。
【0117】
注射用に適した剤形としては、滅菌水溶液又は水分散液と、滅菌注射用溶液又は分散液の即席調製用滅菌粉末が挙げられる。いずれの場合も、剤形は無菌でなければならず、注射し易い程度まで流動性でなければならない。製造及び保存条件下で安定でなければならず、細菌や真菌等の微生物の汚染作用から保護する必要がある。キャリヤーは例えば水、エタノール、ポリオール(例えばグリセロール、プロピレングリコール及び液体ポリエチレングリコール)、適切なその混液及び植物油を含有する溶媒又は分散媒とすることができる。
【0118】
本発明は肥満症及び肥満関連障害の治療及び/又は予防を必要とする対象における肥満症及び肥満関連障害の治療及び/又は予防方法として、前記治療を必要とする対象に治療有効量の化合物Iの水和物、塩又は多形体を投与する段階を含む方法を提供する。本発明は更に精神病、記憶障害、認知障害、アルツハイマー病、偏頭痛、神経障害、神経炎症障害(例えば多発性硬化症、ギラン・バレー症候群、並びにウイルス性脳炎、脳血管発作及び頭部外傷の炎症性後遺症)、不安障害、ストレス、癲癇、パーキンソン病、運動障害及び統合失調症等のCB−1調節障害の予防及び/又は治療用医薬の製造用としての化合物Iの水和物、塩及び多形体の使用を提供する。前記化合物は物質濫用障害の治療、肥満症又は摂食障害、肥満関連障害とそれに伴う合併症(例えば左心室肥大)の治療、並びに喘息、便秘、慢性腸偽閉塞及び肝硬変の治療にも有用である。
【0119】
本発明における肥満関連障害は肥満に付随、誘発又は起因する。肥満関連障害の例としては再狭窄、アテローム性動脈硬化症、動脈硬化症、過食症及び多食症、高血圧、糖尿病、血漿インスリン濃度及びインスリン抵抗性上昇、脂質異常症、高脂血症、子宮体癌、乳癌、前立腺癌、結腸癌、変形性関節症、閉塞型睡眠時無呼吸、胆石症、胆石、心疾患、心拍リズム異常及び不整脈、心筋梗塞、鬱血性心不全、冠動脈性心疾患、突然死、脳卒中、多嚢胞性卵巣疾患、頭蓋咽頭腫、プラダー・ウィリー症候群、フローリッヒ症候群、GH欠損対象、正常変異型低身長、ターナー症候群及び合計除脂肪体重の百分率としての代謝活性低下又は安静時エネルギー消費量低下を示す他の病態(例えば急性リンパ芽球性白血病、代謝症候群、インスリン抵抗性症候群の小児)、生殖ホルモン異常、性機能及び生殖機能不全(例えば受精障害、不妊症、男性性腺機能低下症及び女性多毛症)、妊婦肥満に伴う胎児異常、胃腸運動障害(例えば肥満関連胃食道逆流症)、呼吸器障害(例えば肥満低換気症候群(ピックウィック症候群)、息切れ)、心血管障害、炎症(例えば脈管構造の全身性炎症)、動脈硬化症、高コレステロール血症、高尿酸血症、腰痛、胆嚢疾患、通風、腎臓癌、知覚麻痺の危険の増加、左心室肥大、アルツハイマー病が挙げられる。
【0120】
(肥満症及び肥満関連障害の)「治療」とは肥満対象の体重を低下又は維持するための本発明の化合物又は併用剤の投与を意味する。(肥満症及び肥満関連障害の)「予防」とは肥満の危険のある対象の体重を低下又は維持するための本発明の化合物又は併用剤の投与を意味する。
【0121】
本明細書で使用する「対象」なる用語は治療、観察又は実験の対象とした動物、好ましくは哺乳動物、最も好ましくはヒトを意味する。「〜を必要とする対象」なる用語は研究者、獣医、医師又は他の臨床医の判断により治療又は予防を必要とする対象を意味する。1態様において、治療を必要とする対象は肥満症の哺乳動物である。別の態様において、治療を必要とする対象は1種以上の肥満関連併存症を伴う肥満症のヒトである。別の態様において、治療を必要とする対象は肥満関連併存症を伴わない肥満症のヒトである。本明細書で使用する「治療有効量」なる用語は治療する障害の症状の緩和を含めて研究者、獣医、医師又は他の臨床医により求められる生物学的又は医学的応答を組織、系、対象又はヒトに誘発する組成物中の活性化合物又は薬剤の量を意味する。
【0122】
化合物Iの塩、水和物又は多形体の予防又は治療用量値は当然のことながら治療する病態の重篤度の性質と、組成物中の特定化合物及びその投与経路により異なる。また、個々の患者の年齢、体重及び応答によっても異なる。一般に、肥満症又は肥満関連障害の治療には、対象の体重当たり約0.0001mg/kg〜約100mg/kg、好ましくは約0.001mg/kg〜約100mg/kg、より好ましくは約0.001mg/kg〜約10mg/kgの1日用量の化合物Iの塩、水和物又は多形体を1日1回もしくは2〜6回に分けて又は徐放形態で投与する。他方、場合によってはこれらの範囲外の用量を使用することが必要な場合もある。本発明の化合物は化合物毎に固有の用量範囲でヒトに投与することができる。経口投与では、治療する対象の症状に用量を合わせて各活性成分0.01mg〜1,000mg、好ましくは0.01、0.05、0.1、0.2、0.5、1.0、1.5、2.0、2.5、3.0、3.5、4.0、4.5、5.0、5.5、6.0、7.5、10、15、20、25、30、40、50、60、75、80、100、125、150、175、200、225、250、500、750、850及び1,000mgを含有する錠剤として組成物を提供することが好ましい。この投与レジメンは最適治療応答が得られるように調節することができる。
【0123】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)とその塩、水和物及び溶媒和物の結晶体のX線粉末回折パターンを作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した。
【0124】
N2流下に昇温速度10℃/分でTA Instruments DSC−2910示差走査熱量計を使用してDSCデータを取得した。TA Instruments DSC 2910又は同等機器。2〜6mgサンプルを開放アルミニウムパンに配量する。このパンを次にクリンプし、熱量計セルのサンプル位置に置く。閉鎖パンでサンプルを加熱する。空のパンを参照位置に置く。熱量計セルを閉じ、セルに窒素流を通す。サンプルを昇温速度10℃/分で約250℃の温度まで昇温するように昇温プログラムを設定する。昇温プログラムを開始する。プログラムが完了したら、システムソフトウェアに同梱のDSC分析プログラムを使用してデータを分析する。吸熱を観察する温度範囲の上下の基線温度点の間で融解吸熱を積分する。報告するデータは開始温度、ピーク温度及びエンタルピーである。
【0125】
Perkin Elmer TGA−7熱重量分析器を使用してTGAデータを取得した。5〜20mgサンプルを白金パンに配量する。炉を上昇させ、窒素流をサンプル上に流す。窒素流下に昇温速度10℃/分で約250℃の温度までサンプルを昇温するように昇温プログラムを設定する。昇温プログラムを開始する。プログラムが完了したら、システムソフトウェアに同梱の分析プログラムのΔY関数を使用してデータを分析する。昇温プログラムの開始からサンプルの融解/分解までサンプルによる百分率重量低下を計算する。
【0126】
下記スキーム及び実施例において、各種試薬記号及び略語は以下の意味である:acacはアセチルアセトナートであり;aqは水溶液であり;t−AmOHはtert−アミルアルコールであり;BINAPは2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチルであり;BuLi又はn−BuLiはブチルリチウムであり;Bu2Mgはジブチルマグネシウムであり;Bocはtert−ブトキシカルボニルであり;Boc酸無水物はtert−ブトキシカルボニル酸無水物であり;CBZはカルボベンジルオキシであり;CDIは1,1’−カルボニルジイミダゾールであり;DABCOは1,4−ジアザビシクロ[2.2.2]オクタンであり;DMEはエチレングリコールジメチルエーテルであり;DMFはジメチルホルムアミドであり;DMPUは1,3−ジメチル−3,4,5,6−テトラヒドロ−2(1H)ピリミジノンであり;dppbenzは1,2−ビス(ジフェニルホスフィノ)ベンゼンであり;dppeはジフェニルホスフィノエテンであり;dppf=(フェニル)2PC5H4FeC5H4P(フェニル)2であり、gはグラムであり;hは時間であり;GMPは製造管理及び品質管理規則であり;HClは塩酸であり;IPAはイソプロピルアルコールであり;IPAc又はiPAcは酢酸イソプロピルであり;i−ProHはイソプロパノールであり;KFはカール・フィッシャーであり;kgはキログラムであり;Lはリットルであり;LCAPは液体クロマトグラフィー分析純度であり;LDAはリチウムジイソプロピルアミドであり;LiHMDSはリチウムヘキサメチルジシラジドであり;Meはメチルであり;MeOHはメタノールであり;minは分であり;mLはミリリットルであり;molはモルであり;mmolはミリモルであり;MTBEはtert−ブチルメチルエーテルであり;Nはノルマルであり;PPTSはp−トルエンスルホン酸ピリジニウムであり;rtは室温であり;SL−J212−1は(R)−1−[(S)−2−ジ−2−フリルホスフィノ)フェロセニル]エチルジ−tert.−ブチルホスフィンであり,TBSClはtert−ブチルジメチルシリルクロリドてあり;TFAはトリフルオロ酢酸であり、THFはテトラヒドロフランである。
【0127】
新規方法を利用する代表的な実験手順を下記スキーム及び実施例に詳細に記載する。以下のスキーム及び実施例は本発明を例証するために記載するものであり、本発明の範囲を限定するとみなすべきではない。
【0128】
本発明は強力なCB−1逆アゴニスト化合物Iの効率的な合成方法について記載する。スキーム1に示すように、市販の3−ブロモベンゾイルクロリド1から完全に官能基付与したキラルベンズヒドリルアミンIIを合成する。DABCOとヒドラジンで処理することにより3−ブロモベンゾイルクロリド1をヒドラジド3に変換した。ヒドラジド3をCDIで処理してオキサジアゾール4を得、金属−ハロゲン交換反応によりアルデヒド5に変換した。PPTSと(S)−スルフィンアミドで処理することによりアルデヒド5をエルマンのイミン化合物6に変換した。次に化合物6のオキサジアゾール窒素をBoc基で保護してN−Bocイミン7を得た後、高度に立体選択的なRh触媒付加によりアリールボロキシン10をN−Bocイミン7に付加し、スルフィンアミド8を得た。保護オキサジアゾールの存在下でスルフィンアミド8を選択的に脱保護し、ベンズヒドリルアミンIIを得た。
【0129】
スキーム2はシアノジオールIII中間体の合成を示す。シアノジオールIIIは1,3−ジブロモフルオロベンゼン11から開始し、イソプロピルマグネシウムクロリドによるグリニャール形成後にCuCl/ZnCl2触媒により塩化イソブチリルに付加し、ケトン12を得た。ケトン12のシリルエノールエーテルの形成後、Select−Fluor(登録商標)フッ素化剤でin situフッ素化し、フルオロケトン13を得た。フルオロケトン13を炭酸カリウムとホスホノ酢酸トリメチルで処理し、ホーナー・ワズワース・エモンズ付加物であるα,β−不飽和エステル14を得た後、この化合物をNaOHでin situ加水分解し、α,β−不飽和酸15を得た。α,β−不飽和酸15のロジウム触媒不斉水素化により飽和酸16を得、in situエステル化後に飽和エステル17を得た。飽和エステル17のカルボキシル化によりβ−カルボキシエステル18を得、DME中で水素化ホウ素ナトリウム−臭素により還元することによりブロモジオール19に還元した。最後に、ブロモジオール19のパラジウム触媒シアノ化により、必要なシアノジオールIIIを得た。
【0130】
スキーム3は中間体20のアゼチジン環を導入するために使用するシアノジオールIIIとベンズヒドリルアミンIIの高度にコンバージェントなカップリングを示す。最後に、中間体20からN−Boc保護基を除去することにより化合物Iの合成を完了する。この経路における全中間体は結晶質であり、単離精製に関する本発明の利点である。
【0131】
【化34】
【実施例1】
【0132】
tert−ブチル−5−{3−[(S)−アミノ(4−クロロフェニル)メチル]フェニル]−2−オキソ−1,3,4−オキサジアゾール−3(2H)カルボキシレート(化合物II)の製造
ステップA:ヒドラジド3の製造。100L丸底フラスコに窒素下でDABCO(2.81kg,25.06mol)とMeOH(35L)を加えた。3−ブロモベンゾイルクロリド1(5.0kg,22.78mol)を20〜25℃で30分間かけて加え、氷水浴を使用して温度を制御した。混合物を室温で10〜20分間撹拌した。ヒドラジン(64%,8.8L,182mol)を20分間かけて加えた後、反応混合物を50〜55℃まで3時間加熱した。水(35L)を加え、室温で1時間かけてバッチを結晶化させた。得られたスラリーを室温で1〜2時間撹拌し、濾過した。湿潤ケーキを水洗し(3×15L)、室温で減圧/N2スイープ下に乾燥し、ヒドラジド3を白色固体として得た。ヒドラジド3のHPLC保持時間=6.75分(5ミクロン,4.6×250mmのWaters Symmetry C−18カラム);20℃,215nmでUV検出;勾配流速1.0mL/分;A=水+0.1%H3PO4;B=アセトニトリル;勾配溶出:0分:95%A/5%B;5分:55%A/45%B;10分:25%A/75%B;13分:10%A/90%B;及び26分:10%A/90%B。1H NMR(DMSO−d6):δ9.88(1H,s),7.98(1H,m),7.81(1H,m),7.70(1H,m),7.41(1H,m),4.52(2H,s)。
【0133】
ステップB:オキサジアゾール4の製造。75L丸底フラスコに窒素下で3−ブロモ安息香酸ヒドラジド3(3.5kg,16.3mol)とTHF(35L)を加えた。スラリーを室温で5〜10分間撹拌した。次に20〜25℃で10〜20分間かけてCDI(3.17kg,19.5mol)を加え、氷水浴を使用して温度を制御した。反応混合物は徐々に透明溶液となり、溶液を室温で2〜3時間撹拌した。IPAc(35L)と水(35L)を溶液に加えた。層分離し、水層をIPAc(15L)で抽出した。有機層を合わせて水(2×35L)、次いでブライン(20L)で洗浄し、濃縮した。濃縮中にスラリーが形成された。スラリーにヘプタン(2×10L)を流し、最終容量を30Lに調整した。得られたスラリー混合物を室温で1〜2時間撹拌後、濾過した。得られた湿潤ケーキをヘプタン(2×10L)で洗浄し、室温で減圧/N2スイープ下に乾燥し、オキサジアゾール4を白色固体として得た。オキサジアゾール4のHPLC保持時間=11.20分(5ミクロン,4.6×250mmのWaters Symmetry C−18カラム);20℃,215nmでUV検出;勾配流速1.0mL/分;A=水+0.1%H3PO4;B=アセトニトリル;勾配溶出:0分:95%A/5%B;5分:55%A/45%B;10分:25%A/75%B;13分:10%A/90%B;及び26分:10%A/90%B。1H NMR(400MHz,dmso−d6)δ12.65(1H,s),7.85(1H,s),7.72(2H,m),7.42(1H,t,J=7.8Hz)。
【0134】
ステップC:アルデヒド5の製造。75L丸底フラスコに窒素下でオキサジアゾール4(3.3kg,13.7mol)とTHF(33L)を加えた。溶液を−50℃まで冷却し、Bu2Mg(ヘプタン中1N,2N,10.3L,10.3mol)を−40〜−45℃で30〜50分間かけて加えた。ドライアイスアセトン浴を使用して温度を制御した。得られた不均質混合物を同一温度で1時間撹拌した。次にn−BuLi(ヘキサン中1.6M,10.3L,16.6mol)を−40〜−45℃で30分間かけて加えた。ドライアイスアセトン浴を使用して温度を制御した。スラリーを−40〜−45℃で2〜3時間撹拌した後、DMF(3.2L,41.1mol)を−40℃で1時間かけて加えた。反応混合物を0〜10℃まで昇温し、同一温度で3〜5時間撹拌した。バッチを0℃まで冷却し、バッチ温度を10℃未満に維持しながら2N HCl(10L)を20分間かけて加えることにより反応混合物をクエンチした。バッチを160Lエクストラクターに移し、EtOAc(33L)と2N HCl(23L)を加えた。得られた層を分離し、水層をEtOAc(10L)で抽出した。有機層を合わせて水(33L)、次いでブライン(20L)で洗浄し、濃縮し、ヘプタン(10L)を流した。スラリーを1:2 EtOAc/ヘプタン(12L)中で室温にて2時間撹拌し、濾過した。湿潤ケーキをヘプタン(2×8L)で洗浄し、室温で減圧/N2スイープ下に乾燥し、アルデヒド5を白色固体として得た。アルデヒド5のHPLC保持時間=8.73分(5ミクロン,4.6×250mmのWaters Symmetry C−18カラム);20℃,215nmでUV検出;勾配流速1.0mL/分;A=水+0.1%H3PO4;B=アセトニトリル;勾配溶出:0分:95%A/5%B;5分:55%A/45%B;10分:25%A/75%B;13分:10%A/90%B;及び26分:10%A/90%B。1H NMR(400MHz,dmso−d6)δ12.65(1H,s),10.04(1H,s),8.28(1H,s),8.11(2H,m),7.79(1H,m)。
【0135】
ステップD:N−tert−ブタンスルフィニルイミン6の製造。75L丸底フラスコに窒素下でアルデヒド5(2.1kg,11.0mol)と、(S)−tert−ブタンスルフィンアミド(1.46kg,12.1mol)と、PPTS(1.38kg,5.5mol)と、トルエン(20L)を加えた。得られたスラリーを40℃まで20時間加熱した。バッチを25℃まで冷却し、得られた固体を濾過した。湿潤ケーキをトルエン(3×15L)、水(3×15L)、次いでヘプタン(3×12L)で洗浄した。湿潤ケーキを室温で減圧/N2スイープ下に乾燥後、エルマンイミン6を白色固体として単離した。エルマンイミン6のHPLC保持時間=10.64分(5ミクロン,4.6×250mmのWaters Symmetry C−18カラム);20℃,215nmでUV検出;勾配流速1.0mL/分;A=水+0.1%H3PO4;B=アセトニトリル;勾配溶出:0分:95%A/5%B;5分:55%A/45%B;10分:25%A/75%B;13分:10%A/90%B;及び26分:10%A/90%B。1H NMR(400MHz,dmso−d6)δ12.63(1H,br s),8.62(1H,s),8.27(1H,s),8.09(1H,d,J=7.8Hz),7.95(1H,d,J=7.8Hz),7.69(1H,t,J=7.8Hz),1.20(9H,s)。
【0136】
ステップE:N−Bocイミン7の製造。75L丸底フラスコに窒素下でN−tertブタンスルフィニルイミン6(3.1kg,9.83mol)と、THF(39L)と、Et3N(2.5kg,24.7mol)と、Boc酸無水物(3.2kg,14.66mol)を加えた。得られた混合物を40℃に5時間、室温に8〜10時間加熱した。混合物を濃縮し、IPAc(25L)、次いでヘプタン(10L)を流した。濃縮中に固体が形成され、その後、混合物容量を約22Lに調整した。スラリーを室温で1〜2時間撹拌し、濾過した。得られた湿潤ケーキを1:4 IPAc/ヘプタン(15L)、1:10 IPAc/ヘプタン(15L)及びヘプタン(15L)で洗浄した。湿潤ケーキを室温で減圧/N2スイープ下に乾燥後、N−Bocイミン生成物7を白色固体して単離した。N−Bocイミン7のHPLC保持時間=14.00分(5ミクロン,4.6×250mmのWaters Symmetry C−18カラム);20℃,215nmでUV検出;勾配流速1.0mL/分;A=水+0.1%H3PO4;B=アセトニトリル;勾配溶出:0分:95%A/5%B;5分:55%A/45%B;10分:25%A/75%B;13分:10%A/90%B;及び26分:10%A/90%B。1H NMR(400MHz,CDCl3)δ8.64(1H,s),8.43(1H,s),8.09(1H,d,J=7.8Hz),8.02(1H,d,J=7.8Hz),7.63(1H,t,J=7.8Hz),1.69(9H,s),1.30(9H,s)。
【0137】
ステップF:ボロキシン10の製造。オーバーヘッドスターラー、蒸留装置(バッチ型濃縮器)を取付けた50L丸底フラスコに窒素下で4−クロロフェニルボロン酸9(3.6kg,23mol)とトルエン(30L)を加えた。得られたスラリーを沸騰するまで(95〜110℃)加熱し、共沸蒸留により水を除去した。容器内に一定容量を維持するために蒸留中にトルエン(合計約30〜35L)を補充した。溶媒約30Lの蒸留後、バッチを室温まで冷却し、0.5〜1時間撹拌した。生成物を濾過し、トルエン(2×10L)、次いでヘプタン(1×10L)で洗浄し、室温で減圧/N2スイープ下に乾燥し、ボロキシン10を白色固体として得た。
【0138】
ステップG:スルフィンアミド8の製造。75L丸底フラスコに窒素下でN−Bocイミン7(3.1kg,7.88mol)と、ボロキシン10(1.64kg,3.94mol)と、t−AmOH(62L)を加えた。反応混合物を45℃まで加熱し、1,2−ビス(ジフェニルホスフィノ)ベンゼン(93g,0.208mol)を加えた。得られた混合物に45℃で20分間窒素ガスをスパージ後、Rh(acac)(CH2CH2)2(45g,0.174mol)を加えた。反応混合物に同一温度で更に10分間窒素ガスをスパージした後、45℃に3〜5時間加熱した。バッチを室温まで冷却し、EtOAc(25L)で希釈した。EtOAc(20L)と0.5M Na2CO3水溶液(22L)を加えた160Lエクストラクターに得られた低粘度スラリーを移した。10分間室温で激しく混合後、層分離した。上部の有機層を3%ブライン(3×20L)、次いでブライン(20L)で洗浄した。有機溶液を25−30Lまで濃縮し、ヘプタン(20L)を流した。濃縮中に、生成物が沈殿した後、バッチ容量をヘプタンで約60Lに調整した。得られたスラリーを室温で1〜2時間撹拌後、濾過した。得られた湿潤ケーキを1:4 EtOAc/ヘプタン(2×20L)とヘプタン(30L)で洗浄した。次に湿潤ケーキを室温で減圧/N2スイープ下に乾燥し、スルフィンアミド8を薄黄色綿毛状固体として単離した。スルフィンアミド8のHPLC保持時間=15.15分(Phenomenex Synergi,4ミクロン,Hydro−RP 80A,250×4.6mm);20℃,215nmでUV検出;勾配流速1.0mL/分;A=水+0.1%H3PO4;B=アセトニトリル;勾配溶出:0分:95%A/5%B;5分:55%A/45%B;10分:25%A/75%B;13分:10%A/90%B;及び26分:10%A/90%B。1H NMR(400MHz,CDCl3)δ7.83(1H,s),7.74(1H,d,J=7.7Hz),7.55(1H,d,J=7.7Hz),7.39(1H,t,J=7.7Hz),7.26(4H,m),5.59(1H,s),4.04(1H,s),1.60(9H,s),1.21(9H,s)。
【0139】
ステップH:ベンズヒドリルアミンIIの製造。熱電対、窒素流導入管及びオーバーヘッドスターラーを取付けた100L容器にスルフィンアミド8(3.0kg,5.93mol)とジクロロメタン(15L)を加えた。溶液にHClのi−PrOH溶液(0.62Mを10Lと0.65Mを6.8L,10.08mol)を加えた。室温で3時間エージング後に反応は完了した。反応混合物を水(30L,発熱)でクエンチした後、飽和NaHCO3水溶液(15L)をゆっくりと加え、CO2発生を抑えた。混合物を室温で30分間撹拌した。次に反応混合物を100Lエクストラクターに移し、ジクロロメタン(6L)でリンスした。層分離した。有機層を1ミクロンWhatmanインラインフィルターで濾過しながら75L容器に注入した。溶液を濃縮し、内部温度〜45℃で更にi−PrOH(6L)を加えて共沸蒸留し、粘稠スラリーを得た後、ジクロロメタン18Lを加えて〜8Lまで濃縮することにより溶媒をジクロロメタンに交換した。次に混合物を35℃まで加熱し、全固形分を完全に溶解し、貧溶媒としてのi−PrOH(24L)を滴下漏斗によりゆっくりとバッチに加えた。i−PrOHの添加後、バッチを周囲温度まで一晩ゆっくりと冷却した。次に温度を2時間0〜5℃まで下げた。次に固形分を濾過し、得られたケーキスラリーをi−PrOH(2×7.5L)とn−ヘプタン(3×9L)で洗浄した。週末にかけて、底から窒素スイープと真空引きしながらケーキをフィルターポットで乾燥し、ベンズヒドリルアミンIIを得た。ベンズヒドリルアミンIIのHPLC保持時間=8.95(Zorbax RX C8,5.0ミクロン,4.6mm×250mm,P.N.:880967);温度:25℃,210nmでUV検出,カラム流速は1.0ml/分とし、溶媒Aはアセトニトリルとし、溶媒Bは0.1%H3PO4緩衝H2Oとする;勾配溶出:0分:10%A/90%B;1分:90%A/10%B;及び15分:98%A/2%B。1H(CDCl3)δ7.99(t,J=1.6Hz,1H),7.80(dt,J=7.6,1.6Hz,1H),7.57(dt,J=7.6,1.6Hz,1H),7.44(t,J=7.6Hz,1H),7.33(m,2H),7.27(m,2H),5.26(s,1H),1.77(brs,2H),1.66(s,9H)。1H NMR(400MHz,CDCl3)δ8.00(1H,s),7.80(1H,d,J=7.7Hz),7.08(1H,d,J=7.7Hz),7.44(1H,t,J=7.7Hz),7.30(4H,m),5.26(1H,br s),1.60(2H,br s),1.68(9H,s)。
【0140】
【化35】
【実施例2】
【0141】
3−フルオロ−5−{(1S)−2−フルオロ−1−[2−ヒドロキシ−1−(ヒドロキシメチル)エチル]−2−メチルプロピル}ベンゾニトリル(化合物III)の製造
ステップA:イソプロピルケトン12の合成。熱電対と5L滴下漏斗を取付けた50L丸底フラスコに窒素雰囲気下でZnCl2(32.7g,0.24mol)とCuCl(23.3g,0.24mol)を加えた。THF(12L)を加え、N2をスラリーに5分間バブリングすることにより溶液を脱気した。その後、2,3−ジブロモフルオロベンゼン11(6.0kg,24mol)を一度に加えた。反応混合物を10℃まで冷却し、温度を15〜20℃に維持しながらイソプロピルマグネシウムクロリド(2M,14L,28mol)を1時間20分間かけて滴下した。100L円筒形容器に塩化イソブチリル(3.3kg,31mol)とTHF(6L)を加えた。混合物を6℃まで冷却後、温度を10℃〜18℃に維持しながら、予め調製しておいたアリールグリニャール溶液を70分間かけてポンプ導入した。添加の完了後、反応混合物を20分間エージングさせた後、変換についてアッセイした。変換の完了後、反応混合物をトルエン(11L)と1Nクエン酸(10L)で希釈した。2相混合物を10分間撹拌後、層分離した。次に有機層を1Nクエン酸(10L)で処理し、更に10分間撹拌した。層分離し、有機層を1N K2HPO4(2×11L)と水(11L)で順次洗浄した。次に同一サイズの第2のバッチを処理しながら、湿潤トルエンバッチを36時間保持した。次にイソプロピルケトン12を含む湿潤トルエンバッチを合わせ、約2〜3容量及びKF<200ppmまで濃縮し、次段階で直接使用した。イソプロピルケトン12のHPLC保持時間=8.1分(25cm Zorbax SB C−18でMeCN/0.1 H3PO4を1mL/分で使用);勾配溶出:70%MeCN 5分後、10分で90%MeCNまで;210nm;5分間保持。1H NMR(400MHz,CDCl3)δ7.85(1H,br.s,Ar−H),7.56(1H,dd,8.9Hz,1.0Hz,Ar−H),7.43(1H,dd,8Hz,1.0Hz,Ar−H),3.43(1H,sep.6.9Hz,C−H),1.21(6H,d,6.9Hz,(CH3)2CH)。
【0142】
ステップB:フルオロケトン13の合成。熱電対を取付けた100L丸底フラスコにN2雰囲気下でナトリウムアミラート(3.0kg,27.3mol)とDMF(11L)を順次加えた。塩基のほぼ全部が溶解するまでスラリーを30分間エージングさせた。次に溶液を10℃まで冷却し、イソプロピルケトンを制御下に添加することにより温度を15〜20℃に維持しながら、イソプロピルケトン12の〜50重量%溶液(10.8kg,50重量%,5.4kg,22mol)を55分間かけて加えた。添加の完了後、溶液を30分間エージングさせ、10℃まで冷却し、TBSClの制御下の添加により温度を20〜28℃に維持しながら、TBSCl(4.3kg,28.5mol)で30分間処理した。反応混合物を30分間エージングさせた後、反応温度を28〜35℃に維持しながらSelect−Fluor(登録商標)(8.5kg,24mol,Air Products)を1.5時間かけて加え、スラリーを1時間エージングさせた。完全に変換したら、水(18L)を加え、次いでトルエン(12L)を加えた。得られた2相混合物を100L円筒形容器に移し、10分間エージングさせた。層分離し、有機相を更に水洗(9L)した。同一サイズの第2のバッチを処理しながら、フルオロケトン13を含有する湿潤トルエン溶液を36時間保持した。次にフルオロケトン13を含有するトルエンバッチを合わせ、KF<200ppm及び約50重量%まで濃縮した。得られたフルオロケトン13のトルエン溶液を次段階で直接使用した。フルオロケトン13のHPLC保持時間=8.5分(25cm Zorbax SB C−18でMeCN/0.1 H3PO4を1mL/分で使用);勾配溶出:70%MeCN 5分後、10分で90%MeCNまで、210nmで5分間保持。IRvmax/cm−13083(C−H),2989(C−H),2941(C−H),1696(C=O),1577(C=C);1H NMR(500MHz,CDCl3)δ8.02(1H,s,ArH),7.74(1H,br d,JHF=9.1Hz,ArH),7.65(1H,dt,JHF=7.66Hz,JH,H=2.0Hz,ArH),1.69(6H,d,JH,F=21.7Hz,2×CH3);13C NMR(125MHz,CDCl3)δ198.2(dd,J=27.1Hz,1.8Hz,C=O),162.2(d,J=252.3Hz,ArCF),136.9(dd,J=6.8,4.3Hz,ArCCO),128.9(dd,J=8.6,3.1Hz,ArCH),123.5(d,J=24.6Hz,ArCH),122.8(dd,J=9.2,1.2Hz,ArCBr),115.8(dd,J=22.8,9.2Hz,ArCH),99.9(d,J=180.3Hz,C(CH3)2),25.5(d,J=24.0Hz,2×CH3);19F NMR(470MHz,CDCl3)δ−109.96,Ar−F),−144.68(C−F)。
【0143】
ステップC:α,β−不飽和エステル14の合成。熱電対を取付けた50L丸底フラスコにN2雰囲気下でDMF(10L)と、炭酸カリウム(5.3kg,38.4mol)と、ホスホノ酢酸トリメチル(5.3kg,29.1mol)を順次加えた。スラリーを70℃まで加熱し、フルオロケトン13のトルエン溶液(50重量%溶液9.9kg,4.96kg,18.8mol)で46分間処理した。反応混合物を80〜82℃まで加熱し、この温度で10時間エージングさせた。次に反応混合物を室温まで冷却し、一晩10時間エージングさせた。フルオロケトン13が完全に消費されたら、2Nクエン酸(20L)とトルエン(10L)を加えた100L円筒形容器にスラリーを加えて逆クエンチした。層を10分間混合後、分離した。α,β−不飽和エステル14を含むトルエン有機相を水洗(10L)し、次段階で使用した。
【0144】
ステップD:α.β−不飽和酸15の合成。熱電対を取付けた50L丸底フラスコにN2雰囲気下でステップCのα,β−不飽和エステル14を含有するトルエン溶液を加えた。次に、メタノール(17L)と5N NaOH(8L)を加えた。混合物を45℃まで加熱し、3.5時間迅速に撹拌した。次に、反応混合物を20℃まで冷却し、ヘプタン(15L)と水(15L)を加えた100L円筒形容器に移した。得られた2相混合物を10分間迅速に撹拌し、相分離した。水相をヘプタン(10L)で洗浄し、層分離した。水層を50L丸底フラスコに加え、pH=5になるまで5N HClで酸性化した。次に、α,β−不飽和酸15シード(60g)を加え、5N HClの滴下により溶液をpH2まで1時間かけてゆっくりと酸性化した。固体α,β−不飽和酸15を濾取し、1:1メタノール/水(15L)で洗浄し、ケーキからN2を36時間吸引することにより乾燥した。同一規模の第2のバッチを処理し、α,β−不飽和酸15をオフホワイト固体として得た。α,β−不飽和酸15のHPLC保持時間=13.3分(25cm Zorbax SB C−18でMeCN/0.1 H3PO4を1mL/分で使用);勾配溶出:10%MeCN−50%MeCN 0〜5分後、5〜20分間で50→90%MeCN,210nmで更に5分間90%MeCN保持。IRvmax/cm−1(膜)3087(O−H),2986(C−H),2940(C−H),1703(C=O),1645(C=C);1H NMR(500MHz,CDCl3)δ7.25(1H,dt,JH,F=8.3Hz,JH,H=1.9Hz,Ar−H),7.06(1H,s,Ar−H),6.81(1H,br.dt,JH,F=8.7Hz,JH,H=1.6Hz,Ar−H),6.27(1H,s,C=CH),1.5(6H,d,JH,F=21.9Hz);13C NMR(125MHz,CDCl3)δ170.0(C=O),162.0(d,J=251.0Hz,ArC−F),159.7(dd,J=19.1Hz,J=1.8Hz,C−CO2H),139.4(dd,J=8.6Hz,J=3.1,ArCC),126.7(d,J=3.1Hz,ArCH),122.1(d,J=3.1Hz,ArCBr),118.6(d,J=24Hz,ArCH),117.0(d,=13.5Hz,C=CCO2H),114.3(d,J=22.8Hz,ArCH),95.3(d,J=179.0Hz,C−F),26.4(d,J=25.2Hz,2xCH3);19F NMR(470MHz,CDCl3)δ−110.4(Ar−F),−138.09(C−F)。
【0145】
ステップE:α,β−飽和酸16の合成。α,β−不飽和酸15(2.2kg,7.2mol)を4Lビーカーに配量し、ポリ容器に移した。次にビーカーをMeOH(2L)でリンスし、溶液をポリ容器に移した。次に、トリエチルアミン(252mL,1.8mol)を加え、漏斗を残りのMeOH(7L)でリンスした。激しく混合後、バッチを5ガロン容器に移した後、容器を脱気した。次に触媒を次のように調製した:[Ru(シメン)Cl]2前駆体(1.7g,0.0055mol,0.076mol%)と(R)−1−[(S)−2−ジ−2−フリルホスフィノ)フェロセニル]エチルジ−tert−ブチルホスフィン(ないしSL−J212−1)リガンド(3.1g,0.006mol,0.083mol%)をN2雰囲気下で100mL丸底フラスコに加えた。次に、固形分を1:3 CH2Cl2/MeOH(21mL)に溶解し、磁気撹拌棒を使用して1時間撹拌した。1時間エージング後、MeOH 20mLを使用して触媒溶液をステンレス鋼レザバーに移した。更にMeOH 40mLをリンス用にステンレス鋼レザバーに移した。レザバーを密閉後、溶液を200psig下に50℃にてH2で10時間処理し、92%eeのα,β−飽和酸16のメタノール溶液を得、次段階で使用した。
【0146】
ステップF:α,β−飽和エステル17の製造。100L丸底フラスコにα,β−飽和酸16(4.50kg,14.6mol)のメタノール溶液を加えた。溶液を−15℃まで冷却後、ドライアイス/アセトン浴で温度を−15℃〜−5℃に維持しながら塩化アセチル(2.2kg,2.0L,28.6mol)を30分間かけて滴下した。次に反応混合物を室温まで昇温し、HPLCにより完了したと判断されるまで3時間エージングさせた。溶液を100L円筒形容器に移した。トルエン(20L)を加えた後、7%NaCl水溶液(18L)を加えた。得られた2相溶液を30℃まで加熱し、10分間撹拌した。次に相を分離し、α,β−飽和エステル17を含む有機相をK2HPO4溶液(10L)で洗浄した。α,β−飽和酸16の第2の4.5kgバッチでこのプロセスを反復した。α,β−飽和エステル17を含有する湿潤トルエンバッチを合わせ、約2容量まで濃縮した。α,β−飽和エステル17のHPLC保持時間=7.5分(ACE−111−15030カラムで勾配溶出:0.1% H3PO4水溶液/CH3CN 60%→20%0〜6分後、20%→5%6〜8分を使用);38℃,220nm。1H NMR(400MHz,CDCl3)δ7.22(1H,br.s,Ar−H),7.15(1H,dt,6.1,1.9Hz,Ar−H),6.94(1H,br,d,9.5Hz,Ar−H),3.58(3H,s,OCH3),3.28(1H,ddd,20.6,10.2,4.5Hz,CH),2.88(1H,dd,16.2,4.5,CH2),2.73(1H,dd,16.2,10.2Hz,CH2),1.38(3H,d,21.4Hz,CH3),1.23(3H,d,21.6Hz,CH3)。
【0147】
ステップG:β−カルボキシエステル18の合成。75L丸底フラスコにα,β−飽和エステル17のトルエン溶液(3〜4容量,合計18L)を加えた後、DMPU(1.7kg,13.3mol)を加えた。溶液を−43℃まで冷却後、ドライアイス/アセトン浴で発熱を抑えながら、LiHMDS(12.2L,1.3M,15.9mol)のTHF溶液を1時間かけてゆっくりと加えた。完了後、反応混合物を−66℃まで冷却し、反応温度を<−58〜−60℃に維持しながら乾燥CO2ガスを溶液にゆっくりと1時間バブリングした。反応混合物を−66℃まで再冷却し、水性混合物の温度を0〜15℃に維持しながら水に逆クエンチした。有機相を分離した(水相pH=10〜11)。次にトルエン(18L)を水層に加えた後、pHが<3になるまでH3PO4(85重量%,3.5L)をゆっくりと加えた。水相を分離し、有機相を2.5N HCl(11kg)で2回洗浄した。上記手順を使用して更に4.1kgのα,β−飽和エステル17を処理した。次にβ−カルボキシエステル18を含有するトルエン抽出相を合わせて約3〜4容量まで濃縮した。β−カルボキシエステル18のHPLC保持時間=12.0分(4.6*150mm,直径5μmのZorbax Eclipse XD8−C8カラムでH2O中CH3CN/0.1%H3PO4を使用);1ml/分,5μl注入,210nm,勾配溶出:t=0分,90%H2O/CH3CN;t=15分,10%H2O/CH3CN;及びt=20分,10%H2O/CH3CN。
【0148】
ステップH:ブロモジオール19の合成。100L丸底フラスコにN2下でDME(23.9kg)を加えた後、NaBH4(2.2kg,59.2mol.)を加えた。得られたスラリーを−20℃まで冷却後、温度を−10℃〜−20℃に維持しながら滴下漏斗で2.5時間かけて臭素(1.4L,26.5mol)を加えた。臭素の添加後、溶液を2時間エージングさせた後、10℃まで昇温した。次に添加中に反応温度を≦35℃に維持するように氷/水浴で冷却しながらβ−カルボキシエステル18のDME/トルエン溶液(10.6kg溶液,推定値4.4kg,12.0mol)を臭素溶液に2時間かけてゆっくりと加えた。次に反応混合物を19時間室温でエージングさせた後、HPLC分析はβ−カルボキシエステルが完全に消費されたことを示した。発熱を抑えるように冷却しながら白色不均質混合物をトルエン(4L)と2N K2CO3(38.6kg)に5℃で逆クエンチした。更にトルエン(7L)を使用してガラス容器をリンスした。トルエン溶液を合わせて35℃まで2時間昇温後、室温まで放冷させ、15時間エージングさせた。層分離し(水溶液pH=11〜12)、有機層を0.5N HCl(9.4kg;水溶液pH=0〜1)で洗浄し、次いで2N K2CO3(22kg,5分間エージング;水溶液pH=13)と2N K2CO3(9.1kg,5分間エージング,水溶液pH=13〜14)で洗浄した。次に層分離し、ブロモジオール19を含有する湿潤DME/トルエン層を得た。4.4kgのβ−カルボキシエステル18の第2のバッチを同様に処理した。ブロモジオール19を含有するDME/トルエン層を合わせ、約2容量まで蒸留させた。ブロモジオール19のHPLC保持時間=4.36分(ACE−111−1503,勾配溶出:60%−5% 0.1%H3PO4,220nm)。1H NMR(400MHz,CDCl3)δ7.26−7.24(1H,m,Ar−H),7.17−7.14(1H,m,Ar−H),6.91(1H,dt,9.7,1.7Hz,Ar−H),3.89(1H,dd,11.0,6.0Hz,CH),3.81−3.69(2H,m,2xCH),3.29(1H,dd,11.0,7.3Hz,CH),3.26−3.20(2H,m,CH,OH),2.99(1H,dd,22.3,8.3,CH),2.32−2.25(1H,m,OH),1.37(3H,d,21.8Hz,CH3),1.31(3H,d,22.3Hz,CH3)。
【0149】
ステップI:シアノジオールIIIの合成。粗ブロモジオール19(5kg,15.5mol)のトルエン(〜20重量%)溶液をDMF(2.5容量,約12.5L)に溶媒交換し、HPLCにより<5LCAPと判断されるまでトルエンを還元した。Zn粉末(615g,9.4mol,<10ミクロン,Aldrich)とDMF(3容量,約15L)のスラリーに室温でN2下に臭素(90mL,1.75mol)をゆっくりと加えた。混合物を室温で20分間撹拌すると、この間にオレンジ色から無色に変色した。次に、Zn(CN)2(1.1kg,9.4mol)と、ブロモジオール19の溶液(DMF2.5容量に溶解,DMF0.5容量でリンス)と、PPh3(485g,1.85mol)と、Pd(OAc)2(103g,0.46mol)を順次加え、表面下窒素流を使用して得られた混合物を30分間脱気した。次に反応混合物を窒素雰囲気下で80℃まで昇温した後、3時間エージングさせた後、室温まで冷却し、一晩エージングさせた。混合物を30%NH4OH(0.84容量)で処理し、1時間エージングさせた。得られたスラリーをSolka Floc(登録商標)で濾過し、濾過層をIPAc(85L)で洗浄した。10%NH4OH(60L)を加えたエクストラクターに濾液を移した。有機溶液を5%NaCl(30L)と水(30L)で1回洗浄した。IPAcが<1mol%(GCによるアッセイ)に低下するまで〜40℃にて一定容量で有機層をトルエン(〜9L/kgシアノジオール)に溶媒交換した。ヘプタン(5L/kgシアノジオール)を〜45℃で加え、得られたスラリーを室温まで冷却後、0℃まで冷却した。スラリーを濾過し、30:70ヘプタン:トルエン(15L)で洗浄し、シアノジオールIIIを白色固体(98LCAP,95%ee)として得た。シアノジオールIIIのHPLC保持時間=8.9分(4.6*250mm,直径5μmのWaters Symmetry C18);溶離液:H2O中CH3CN/0.1%H3PO4;1ml/分,5μl注入,210nm,勾配溶出:t=0分,65%H2O/CH3CN;t=4分,65%H2O/CH3CN;t=25分,20%H2O/CH3CN;t=26分,0%H2O/CH3CN;及びt=30分 0%H2O/CH3CN。αD=+26.0°;IRcm−(膜)3395(O−H),2912(C−H,2233(CN),1593,1439;1H NMR(400MHz,CDCl3)δ7.37(1H,t,J=1.4Hz,Ar−H),7.31−7.25(2H,m,Ar−H),3.98(1H,dd,J=11.2,5.3Hz),3.84(1H,dd,J=11.0,3.2Hz),3.76(1H,ddd,J=11.2,4.7,1.0Hz),3.28(1H,dd,J=11.0,6.7Hz),3.20(1H,dd,J=23.2,8.1Hz),1.37(3H,d,J=21.7Hz),1.36(3H,d,J=22.2Hz);13C NMR(125MHz,CDCl3)δ163.3,160.8,144.0,143.9,143.85,129.4,122.0,121.7,118.1,117.8,117.59,117.56,113.85,113.75,99.0,97.31,64.52,64.47,63.78,63.76,52.13,52.11,51.90,51.89,43.01,43.00,28.13,27.89,24.80,24.55;19F NMR(470MHz,CDCl3)δ−109.6(Ar−F),−137.5(C−F);m/z 269.1227;HRMS 269.1235。
【0150】
【化36】
【実施例3】
【0151】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)無水遊離塩基多形体Iの製造
ステップA:アゼチジン20の製造。熱電対、滴下漏斗及びオーバーヘッドスターラーを取付けた75L容器にシアノジオールIII(0.94kg,3.48mol)とアセトニトリル(14L)を加え、アセトン/ドライアイスを使用して内容物を−30℃まで冷却した。溶液にトリフル酸無水物(1.29L,7.66mol)をゆっくりと加えた後、ジイソプロピルエチルアミン(1.52L,2.5eq)をゆっくりと加えた。どちらの添加も内部温度を−20℃未満に維持しながら実施した。−30℃で1時間後にビストリフラートを形成する反応が完了したことがHPLCにより確認された。更にジイソプロピルエチルアミン(1.52L,2.5eq)をゆっくりと加えた後、内部温度を−20℃未満に維持しながら、ベンズヒドリルアミンII(1.4kg,3.48mol)をジクロロメタン(5.6L)溶液として滴下漏斗によりゆっくりと加えた。次に滴下漏斗をジクロロメタン(0.7L)でリンスした。冷却浴を除去し、反応混合物を室温まで一晩昇温した。次に反応混合物を濃縮し、IPAc 28Lを使用して溶媒をIPAcに交換し、最終容量〜10.5Lまで濃縮した。得られたスラリーをヘプタン(5.6L)で希釈し、IPAc/ヘプタンの〜3:2混合物を得た。室温で一晩エージング後、スラリーを濾過し、ジイソプロピルエチルアミントリフル酸塩を除去し、得られた塩ケーキをIPAc/ヘプタンの3:2混液(3×4.9L)で洗浄した。濾液を〜15Lまで濃縮し、Nuchar Aquaguard Powder(登録商標)(0.56kg)で処理した。混合物を1時間かけて50℃まで加熱した後、室温まで一晩冷却した。得られたスラリーをSolka Floc(登録商標)パッド(1.4kg)で濾過し、ケーキをIPAc(5×2.8L)で洗浄した。濾液を50Lエクストラクターに移し、GMP水(1×7L)で洗浄した。1ミクロンインラインフィルターを通して有機層を50L容器に移した。濾液を〜10Lまで濃縮後、IPAc 42Lを使用して溶媒をIPAcに交換し、〜6.4Lまで濃縮した。溶液を室温まで一晩冷却した。ヘプタン(8.4L)を1時間かけてゆっくりと加えた後、シード(7g)を加えた。1時間室温でエージング後、更にヘプタン(8.4L)を2時間かけてゆっくりと加えた後、室温で1時間エージングさせた。室温で1時間かけてヘプタン(4.2L)を加え、スラリーを室温で一晩エージングさせた。こうして2種類の結晶(微結晶と顆粒状結晶)を得た。濾過前に温度を2時間で0〜5℃まで下げた。スラリーを濾過し、得られたケーキスラリーをIPAc/ヘプタン1:5混液(1×2.8L)、次いでヘプタン(2×2.8L)で洗浄した。底から窒素スイープと真空引きしながらケーキをフィルターポットで一晩乾燥した。得られたアゼチジン20は純度92wt%及び97.8LCAPで得られた。アゼチジン20のHPLC保持時間=11.23(Zorbax RX C8,Analytical 5.0ミクロン,4.6mm×250mm,P.N.:880967−901);温度:25℃;210nmでUV検出;カラム流速は1.0ml/分とし;溶媒Aはアセトニトリルとし;溶媒Bは0.1%H3PO4緩衝H2Oとする,勾配溶出:0分:10%溶媒A,90%溶媒B;10分:90%溶媒A,10%溶媒B;15分:98%溶媒A,2%溶媒B。1H(CDCl3)δ7.92(t,J=1.6Hz,1H),7.72(dt,J=7.6,1.6Hz,1H),7.53(d,J=7.6Hz,1H),7.37(d,J=7.6Hz,1H),7.34(m,2H),7.28−7.25(om,3H),7.21(ddd,J=7.6,2.4,,1.4Hz,1H),7.16(m,1H),4.27(s,1H),3.61(t,J=6.4Hz,1H),3.16(m,1H)3.05(t,J=6.4Hz,1H),2.91(dd,J=19.1,11.0Hz,1H),2.85(t,J=7.6Hz,1H),2.27(t,J=7.6Hz,1H),1.66(s,9H),1.27(d,J=21.3Hz,3H),1.19(d,J=21.5Hz,3H)。19F(CDCl3)δ−110.0,−144.3。
【0152】
ステップB:化合物Iの製造。熱電対、滴下漏斗及びオーバーヘッドスターラーを取付けた50L容器にアゼチジン20(1.85アッセイkg,2.91mol)と、IPAc(5.6L)と、HClのIPA溶液(7.4L,4.55M)を温和な吸熱(〜6℃)下に加えた。HPLCによると、室温で一晩エージング後に反応は完了した。飽和NaHCO3(30.0L)を加えた100Lエクストラクターに温和な発熱(〜5℃)下にバッチを移すことにより反応混合物をクエンチした。更に飽和NaHCO3(5.0L,合計35.0L)をゆっくりと加え、pHを〜7に調整した。更にIPAc(13.0L)を加えた後、層分離し、有機層をGMP水(1×9.3L)で洗浄した。1ミクロンインラインフィルターを通して有機層を50L容器に移した。有機層を〜10Lまで濃縮し、IPAc 18Lを使用して溶媒をIPAcに交換し、〜18Lまで濃縮した。次に混合物をNuchar Aquaguard Powder(登録商標)(0.45kg)で処理し、50℃まで1時間加熱後、室温まで一晩冷却した。得られたスラリーをSolka Floc(登録商標)パッド(1.85kg)で濾過し、ケーキをIPAc(4×7.0L)で洗浄した。1ミクロンインラインフィルターを通して有機層を50L容器に移し、〜6Lまで濃縮し、トルエン28Lを使用して溶媒をトルエンに交換した。得られた溶液を〜9.0Lまで濃縮し、45℃までゆっくりと加熱し、内部温度を40〜45℃に維持しながらヘプタン(1.9L)で1時間かけてゆっくりと処理した。バッチにシード添加し(7g)、1時間45℃でエージングさせた。更にトルエン(1.0L)を加えた後、反応温度を40〜45℃に維持しながら、ヘプタン(1.9L)を45℃で2時間かけてゆっくりと加えた。45℃で1時間エージング後、反応混合物を室温まで一晩冷却した。スラリーを濾過し、ケーキスラリーをトルエン/ヘプタンの3:2混液(1×6.0L)、次いでヘプタン(2×6.0L)で洗浄した。次に底から窒素スイープと真空引きしながらケーキをフィルターポットで一晩乾燥し、純度96.8wt%及び98.6LCAPで化合物Iを得た。化合物IのHPLC保持時間=9.54(Zorbax RX C8,Analytical 5.0ミクロン,4.6mm×250mm,P.N.:880967−901);温度:25℃;210nmでUV検出;カラム流速:1.0ml/分;溶媒A:アセトニトリル;溶媒B:0.1%H3PO4緩衝H2O,勾配溶出:0分:10%溶媒A,90%溶媒B;10分:90%溶媒A,10%溶媒B;15分:98%溶媒A,2%溶媒B。1H(CDCl3)δ10.85(brs,1H),7.99(t,J=1.6Hz,1H),7.67(dt,J=7.6,1.6Hz,1H),7.46(d,J=7.6Hz,1H),7.39−7.35(om,3H),7.30−7.28(om,3H),7.24−7.18(om,2H),4.35(s,1H),3.70(t,J=6.8Hz,1H),3.33(m,1H),3.1 1(td,J=7.6,1.6Hz,1H),3.01−2.93(om,2H),2.40(t,J=8.0Hz,1H),1.27(d,J=21.3Hz,3H),1.22(d,J=21.7Hz,3H)。19F(CDCl3)δ−109.7,−142.7。
【実施例4】
【0153】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)遊離塩基トルエン/ヘプタン溶媒和物多形体I,タイプBの製造
化合物I 3.36kgをトルエン20Lに溶解し、1ミクロンインラインフィルターで濾過した。スラリーを53℃まで加熱後、45℃まで冷却した。次に、10重量%シードスラリー(化合物I 101gを6:4トルエン/ヘプタンに媒体で粉砕)1.01kgを混合物に加えた後、6:4トルエン/ヘプタン(779g)を加えた。得られたスラリーを1時間エージングさせた。較正済みEncynova(登録商標)配量ポンプを使用してヘプタン(10.1L)を4時間40分かけて38mL/分の速度で加えた。得られたスラリーを室温まで冷却し、一晩エージングさせた。混合物を濾過し、6:4トルエン/ヘプタン(14L)で洗浄した。次に得られたケーキをヘプタン28Lで洗浄し、化合物Iを結晶質遊離塩基トルエン/ヘプタン溶媒和物形態I,タイプBとして得た。
【0154】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体I,タイプBのX線粉末回折スペクトル(図13)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(2θ),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0155】
【表1】
【0156】
化合物Iの無水遊離塩基トルエン/ヘプタン溶媒和物多形体I,タイプBは表1に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体I,タイプBは角度θ値4.7°により同定することができる。化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体I,タイプBは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)4.7°;
b)4.7°及び7.1°;
c)4.7°、7.1°及び8.4°;
d)4.7°、7.1°、8.4°及び8.7°;
e)4.7°、7.1°、8.4°、8.7°及び9.5°;
f)4.7°、7.1°、8.4°、8.7°、9.5°及び11.6°;
g)4.7°、7.1°、8.4°、8.7°、9.5°、11.6°及び14.3°;
h)4.7°、7.1°、8.4°、8.7°、9.5°、11.6°、14.3°及び15.4°;
i)4.7°、7.1°、8.4°、8.7°、9.5°、11.6°、14.3°、15.4°及び17.3°。
【0157】
化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体I、タイプBはCu線を使用して得られたX線粉末回折パターンからd間隔18.800、12.450、10.526、10.163、9.309、7.628、6.194、5.754及び5.126Åの1個以上の反射により同定することもできる。
【0158】
Perkin Elmer TGA−7機器で窒素流下に昇温速度10℃/分で化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体I,タイプBの熱重量(TG)分析曲線(図14)を得た。
【実施例5】
【0159】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)無水遊離塩基多形体Iの製造
実施例4に記載したように製造したヘプタン/トルエン溶媒和物を乾燥することにより化合物Iの無水遊離塩基形態Iを得た。フィルターポットを一晩乾燥後、白色固体をトレーに移し、窒素スイープ下に25℃/75Torrで減圧乾燥し、化合物Iを98.62LCAP及び>99%eeの無水遊離塩基多形体Iとして得た。
IR:vmax(cm−1)(膜)。1H NMR(400MHz,CDCl3)δ11.5−11.8(1H,bs,NH),8.07(1H,s,ArH),7.66(1H,d,J=7.7Hz,ArH),7.47(1H,d,J=7.74,ArH)7.39−1.21(8H,m,ArH),4.44(1H,s,ArArCHN),3.75(1H,dd,J=6.8,6.7Hz,NCHH),3.43−3.37(1H,m,NCH2CH),3.16(1H,dd,J=6.8,6.7Hz,NCHH),3.06−2.99(2H,m,NCHH,ArCHCH),2.49(1H,dd,J=8.2,8.1Hz,NCHH),1.26(3H,d,J=21.7Hz,CH3CF),1.22(3H,d,J=21.7Hz,CH3CF)。13C NMR(100MHz,CDCl3)δ163.3,160.8,155.7,154.9,143.4,143.3,143.3,141.6,138.9,133.7,131.1,129.5,129.1,128.8,125.3,125.0,124.5,121.5,121.3,118.1,117.8,117.6,117.6,113.7,113.6,97.2,95.5,77.6,61.1,59.4,59.2,58.2,30.3,25.9,25.7,25.3,25.1。19F NMR(376MHz,CDCl3)δ−109.7(Ar−F),−142.3(C−F)。
【0160】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物Iの無水遊離塩基多形体IのX線粉末回折スペクトル(図1)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(2θ),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0161】
【表2】
【0162】
化合物Iの無水遊離塩基多形体Iは表2に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物Iの無水遊離塩基多形体Iは角度θ値5.2°により同定することができる。化合物Iの無水遊離塩基多形体Iは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)5.2°;
b)5.2°及び15.4°;
c)5.2°、15.4°及び9.3°;
d)5.2°、15.4°、9.3°及び17.4°;
e)5.2°、15.4°、9.3°、17.4°及び7.0°;
f)5.2°、15.4°、9.3°、17.4°、7.0°及び15.7°;
g)5.2°、15.4°、9.3°、17.4°、7.0°、15.7°及び22.5°;
h)5.2°、15.4°、9.3°、17.4°、7.0°、15.7°、22.5°及び11.8°;
i)5.2°、15.4°、9.3°、17.4°、7.0°、15.7°、22.5°、11.8°及び16.4°。
【0163】
化合物Iの無水遊離塩基多形体IはCu線を使用して得られたX線粉末回折パターンからd間隔16.994、5.754、9.509、5.096、12.627、5.644、3.951、7.499及び5.405Åの1個以上の反射により同定することもできる。
【0164】
Perkin Elmer TGA−7機器で窒素流下に昇温速度10℃/分で化合物Iの無水遊離塩基多形体Iの熱重量(TG)分析曲線(図2)を得た。減量は融点までで<0.1%であった。TA Instruments DSC−2910示差走査熱量計でN2流下に昇温速度10℃/分で化合物Iの無水遊離塩基多形体IのDSC曲線(図3)を得た。サンプルを閉鎖パンで加熱した。開始温度157.8C、ピーク温度163.6C及びそれに伴うエンタルピー44.6J/gの単一吸熱。
【実施例6】
【0165】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)酢酸イソプロピル/メチルシクロヘキサン溶媒和物遊離塩基多形体IタイプAの製造
化合物Iの遊離塩基300mgを酢酸イソプロピル0.25mLに溶解した。混合物を70℃まで加熱した後、メチルシクロヘキサン0.25mLを加えた。混合物を40℃まで加熱後、化合物Iの遊離塩基をシード添加した。固形分がゆっくりと沈殿し始めた。溶液を室温まで冷却し、30分間撹拌した。次に酢酸イソプロピル0.1mLとメチルシクロヘキサン0.5mLを加えた。得られた固形分を濾過し、化合物Iのメチルシクロヘキサン/酢酸イソプロピル溶媒和物遊離塩基多形体Iを得た。
【0166】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物Iの酢酸イソプロピル/メチルシクロヘキサン溶媒和物遊離塩基多形体I,タイプAのX線粉末回折スペクトル(図10)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(2θ),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0167】
【表3】
【0168】
化合物Iの酢酸イソプロピル/メチルシクロヘキサン溶媒和物遊離塩基多形体I,タイプAは表3に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物Iの酢酸イソプロピル/メチルシクロヘキサン溶媒和物遊離塩基多形体I,タイプAは角度θ値4.6°により同定することができる。化合物Iの酢酸イソプロピル/メチルシクロヘキサン溶媒和物遊離塩基多形体I,タイプAは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)4.6°;
b)4.6°及び7.5°;
c)4.6°、7.5°及び9.1°;
d)4.6°、7.5°、9.1°及び9.7°;
e)4.6°、7.5°、9.1°、9.7°及び14.5°;
f)4.6°、7.5°、9.1°、9.7°、14.5°及び15.1°;
g)4.6°、7.5°、9.1°、9.7°、14.5°、15.1°及び16.8°;
h)4.6°、7.5°、9.1°、9.7°、14.5°、15.1°、16.8°及び17.8°;
i)4.6°、7.5°、9.1°、9.7°、14.5°、15.1°、16.8°、17.8°及び18.2°。
【0169】
化合物Iの酢酸イソプロピル/メチルシクロヘキサン溶媒和物遊離塩基多形体I、タイプAはCu線を使用して得られたX線粉末回折パターンからd間隔19.209、11.787、9.718、9.118、6.109、5.867、5.277、4.983及び4.874Åの1個以上の反射により同定することもできる。Perkin Elmer TGA−7機器で窒素流下に昇温速度10℃/分で化合物Iの酢酸イソプロピル/メチルシクロヘキサン溶媒和物遊離塩基多形体IタイプAの熱重量(TG)分析曲線(図12)を得た。TA Instruments DSC−2910示差走査熱量計でN2流下に昇温速度10℃/分で化合物Iの酢酸イソプロピル/メチルシクロヘキサン溶媒和物遊離塩基多形体IタイプAのDSC曲線(図11)を得た。
【実施例7】
【0170】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)無水HCl塩多形体Aの製造
化合物Iの非晶質遊離塩基0.5gをアセトニトリル2.5mLに溶解した。次に1.0M HCl/Et2O 0.93mLを滴下すると、透明溶液が得られた。約2分後に、白色固体が溶液から晶析した。混合物を更に10分間撹拌後、濾過した。得られた固形分を減圧窒素スイープ下にフリットで乾燥し、化合物Iの無水HCl塩多形体Aを得た。
【0171】
あるいは、a)エタノールから化合物IのHCl塩を晶析させ、乾燥する方法;及びb)実施例14の化合物IのHCl塩多形体Hを乾燥する方法により、化合物Iの無水HCl塩多形体Aを形成することもできる。
【0172】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物Iの無水HCl塩多形体AのX線粉末回折スペクトル(図4)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(2θ),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0173】
【表4】
【0174】
化合物Iの無水HCl塩多形体Aは表4に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物Iの無水HCl塩多形体Aは角度θ値13.0°により同定することができる。化合物Iの無水HCl塩形態Aは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)13.0°;
b)13.0°及び19.3°;
c)13.0°、19.3°及び15.8°;
d)13.0°、19.3°、15.8°及び16.1°;
e)13.0°、19.3°、15.8°、16.1°及び17.3°;
f)13.0°、19.3°、15.8°、16.1°、17.3°及び13.6°;
g)13.0°、19.3°、15.8°、16.1°、17.3°、13.6°及び9.3°;
h)13.0°、19.3°、15.8°、16.1°、17.3°、13.6°、9.3°及び16.6°;
i)13.0°、19.3°、15.8°、16.1°、17.3°、13.6°、9.3°、16.6°及び9.6°。
【0175】
化合物Iの無水HCl塩多形体AはCu線を使用して得られたX線粉末回折パターンからd間隔6.810、4.599、9.509、5.609、5.505、5.126、9.213、6.511及び5.340Åの1個以上の反射により同定することもできる。
【0176】
化合物Iの無水HCl塩多形体Aは示差走査熱量分析(DSC)によっても特徴付けられる。TA Instruments DSC−2910示差走査熱量計でN2流下に昇温速度10℃/分で化合物Iの無水HCl塩多形体AのDSC曲線(図5)を得た。サンプルを閉鎖パンで加熱した。開始温度213.3Cで単一吸熱が観察される。
【実施例8】
【0177】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)無水HCl塩多形体Bの製造
酢酸イソプロピル35mLを化合物Iの非晶質遊離塩基3.5gに加えた。次に、HClのIPA溶液(5〜6M)1.44mLを加えた。反応混合物を85〜90℃まで加熱した後、圧力管に密封した。150℃に設定した加熱マントルで反応混合物を一晩加熱した。翌朝、反応混合物を冷却し、濾過し、化合物IのHCl塩多形体Bを得た。
【0178】
あるいは、a)実施例9の化合物IのHCl塩多形体Cを酢酸イソプロピルでスラリー化し、乾燥する方法;b)実施例10の化合物IのHCl塩多形体Dを化合物IのHCl塩多形体Bシードと共にエタノールでスラリー化し、乾燥する方法;c)実施例7の化合物IのHCl塩多形体Aを酢酸イソプロピルでスラリー化し、乾燥する方法;及びd)215℃を上回る温度まで化合物IのHCl塩多形体Aを加熱する方法により、化合物Iの無水HCl塩多形体Bを形成することもできる。
【0179】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物Iの無水HCl塩多形体BのX線粉末回折スペクトル(図6)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(2θ),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0180】
【表5】
【0181】
化合物Iの無水HCl塩多形体Bは表5に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物Iの無水HCl塩多形体Bは角度θ値17.9°により同定することができる。化合物Iの無水HCl塩多形体Bは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)17.9°;
b)17.9°及び15.3°;
c)17.9°、15.3°及び19.4°;
d)17.9°、15.3°、19.4°及び24.9°;
e)17.9°、15.3°、19.4°、24.9°及び6.1°;
f)17.9°、15.3°、19.4°、24.9°、6.1°及び8.1°;
g)17.9°、15.3°、19.4°、24.9°、6.1°、8.1°及び
21.5°;
h)17.9°、15.3°、19.4°、24.9°、6.1°、8.1°、21.5°及び14.2°;
i)17.9°、15.3°、19.4°、24.9°、6.1°、8.1°、21.5°、14.2°及び10.9°。
【0182】
化合物Iの無水HCl塩多形体BはCu線を使用して得られたX線粉末回折パターンからd間隔4.955、14.488、5.791、10.915、8.117、6.237、4.575、4.133及び3.576Åの1個以上の反射により同定することもできる。
【0183】
化合物Iの無水HCl塩多形体Bは示差走査熱量分析(DSC)によっても特徴付けられる。TA Instruments DSC−2910示差走査熱量計でN2流下に昇温速度10℃/分で化合物Iの無水HCl塩多形体BのDSC曲線(図7)を得た。サンプルを閉鎖パンで加熱した。開始温度249.9Cで単一吸熱が観察される。
【実施例9】
【0184】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)HCl塩多形体Cの製造
化合物Iの非晶質遊離塩基0.5gをイソプロパノール2.5mLに溶解した。次に5〜6N HCl/イソプロピルアルコール1.1モル当量を滴下した。30分間撹拌後、得られた固形分を濾過した。固形分を減圧窒素スイープ下にフリットで乾燥し、化合物IのHCl塩多形体Cを得た。
【0185】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物IのHCl塩多形体CのX線粉末回折スペクトル(図15)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(20),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0186】
【表6】
【0187】
化合物IのHCl塩多形体Cは表6に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物IのHCl塩多形体Cは角度θ値6.5°により同定することができる。化合物IのHCl塩形態Cは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
j)6.5°;
k)6.5°及び8.2°;
1)6.5°、8.2°及び9.1°;
m)6.5°、8.2°、9.1°及び10°;
n)6.5°、8.2°、9.1°、10°及び13.8°;
o)6.5°、8.2°、9.1°、10°、13.8°及び14.2°;
p)6.5°、8.2°、9.1°、10°、13.8°、14.2°及び14.9°;
q)6.5°、8.2°、9.1°、10°、13.8°、14.2°、14.9°及び15.8°;
r)6.5°、8.2°、9.1°、10°、13.8°、14.2°、14.9°、15.8°及び18.1°。
【0188】
化合物IのHCl塩多形体CはCu線を使用して得られたX線粉末回折パターンからd間隔13.598、10.782、9.718、8.845、6.417、6.237、5.945、5.609及び4.901Åの1個以上の反射により同定することもできる。
【実施例10】
【0189】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物T)HCl塩多形体Dの製造
結晶質HCl塩多形体F(水和物)を35℃の真空炉で乾燥することにより化合物IのHCl塩多形体Dを製造した。
【0190】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物IのHCl塩多形体DのX線粉末回折スペクトル(図16)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(2θ),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0191】
【表7】
【0192】
化合物IのHCl塩多形体Dは表7に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物IのHCl塩多形体Dは角度θ値7.2°により同定することができる。化合物IのHCl塩形態Dは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)7.2°;
b)7.2°及び12.3°;
c)7.2°、12.3°及び6.2°;
d)7.2°、12.3°、6.2°及び11.1°;
e)7.2°、12.3°、6.2°、11.1°及び13.8°;
f)7.2°、12.3°、6.2°、11.1°、13.8°及び18°;
g)7.2°、12.3°、6.2°、11.1°、13.8°、18°及び8.8°;
h)7.2°、12.3°、6.2°、11.1°、13.8°、18°、8.8°及び22°;
i)7.2°、12.3°、6.2°、11.1°、13.8°、18°、8.8°、22°及び24.2°。
【0193】
化合物IのHCl塩多形体DはCu線を使用して得られたX線粉末回折パターンからd間隔12.277、7.196、14.255、7.971、6.417、4.928、10.048、4.040及び3.678Åの1個以上の反射により同定することもできる。
【0194】
Perkin Elmer TGA−7機器で窒素流下に昇温速度10℃/分で化合物IのHCl塩多形体Dの熱重量(TG)分析曲線(図18)を得た。TA Instruments DSC−2910示差走査熱量計でN2流下に昇温速度10℃/分で化合物IのHCl塩多形体DのDSC曲線(図17)を得た。
【実施例11】
【0195】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)無水HCl塩多形体Eの製造
HCl塩多形体Dを温度>185℃まで乾燥することにより無水HCl塩多形体Eを製造した。
【0196】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物Iの無水HCl塩多形体EのX線粉末回折スペクトル(図19)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(2θ),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0197】
【表8】
【0198】
化合物Iの無水HCl塩多形体Eは表8に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物Iの無水HCl塩多形体Eは角度θ値7.4°により同定することができる。化合物Iの無水HCl塩形態Eは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)7.4°;
b)7.4°及び8.2°;
c)7.4°、8.2°及び8.9°;
d)7.4°、8.2°、8.9°及び10.2°;
e)7.4°、8.2°、8.9°、10.2°及び12.8°;
f)7.4°、8.2°、8.9°、10.2°、12.8°及び14.3°;
g)7.4°、8.2°、8.9°、10.2°、12.8°、14.3°及び14.8°;
h)7.4°、8.2°、8.9°、10.2°、12.8°、14.3°、14.8°及び15.4°;
i)7.4°、8.2°、8.9°、10.2°、12.8°、14.3°、14.8°、15.4°及び19.5°。
【0199】
化合物Iの無水HCl塩多形体EはCu線を使用して得られたX線粉末回折パターンからd間隔11.946、10.782、9.936、8.672、6.916、6.194、5.985、5.754及び4.552Åの1個以上の反射により同定することもできる。
【実施例12】
【0200】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物1)HCl塩多形体F水和物の製造
化合物Iの遊離塩基2.5gをエタノール10mLに溶解した。次に溶液0.685gを容器に加えた後、エタノール2.5mLを加えた。次に0.411M HCl水溶液0.75mLを容器に加えた。1%シードを加え、スラリーを1時間エージングさせた。次に遊離塩基溶液の残りの11.3mLと1.4M HCl水溶液3.1mLを8時間かけて加えた。次にスラリーを濾過し、1:1エタノール/水で洗浄し、水和物HCl塩多形体Fを得た。
【0201】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物IのHCl塩多形体F水和物のX線粉末回折スペクトル(図20)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(2θ),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0202】
【表9】
【0203】
化合物IのHCl塩多形体F水和物は表9に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物IのHCl塩多形体F水和物は角度θ値6.1°により同定することができる。化合物IのHCl塩形態F水和物は以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)6.1°;
b)6.1°及び6.4°;
c)6.1°、6.4°及び7.3°;
d)6.1°、6.4°、7.3°及び11°;
e)6.1°、6.4°、7.3°、11°及び11.5°;
f)6.1°、6.4°、7.3°、11°、11.5°及び12°;
g)6.1°、6.4°、7.3°、11°、11.5°、12°及び16.2°;
h)6.1°、6.4°、7.3°、11°、11.5°、12°、16.2°及び18°;
i)6.1°、6.4°、7.3°、11°、11.5°、12°、16.2°、18°及び19°。
【0204】
化合物IのHCl塩多形体F水和物はCu線を使用して得られたX線粉末回折パターンからd間隔14.488、13.810、12.109、8.043、7.694、7.375、5.471、4.928及び4.671Åの1個以上の反射により同定することもできる。
【実施例13】
【0205】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)無水HCl塩多形体Gの製造
化合物Iの結晶質遊離塩基2.0gをエタノール3.7mLに溶解した。次に1.25M HClエタノール溶液0.05mLを加えた後、多形体Gシード80mgを加えた。次に0.2mL/時の速度で1.25M HClエタノール溶液3.2mLをゆっくりと加えた。得られたスラリーを濾過し、エタノール6mLで洗浄し、減圧窒素スイープ下にフリットで乾燥し、化合物Iの無水HCl塩多形体Gを得た。
【0206】
あるいは、a)実施例10の化合物IのHCl塩多形体Dを化合物IのHCl塩多形体Gと共にスラリー化する方法;b)実施例8の化合物IのHCl塩多形体Bを化合物IのHCl塩多形体Gシードと共にエタノールでスラリー化する方法;c)実施例8の化合物IのHCl塩多形体Bを25%水/エタノールでスラリー化する方法;及びd)実施例7の化合物IのHCl塩多形体Aを化合物IのHCl塩多形体Gと共にエタノールでスラリー化する方法により、化合物Iの無水HCl塩多形体Gを形成することもできる。
【0207】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物Iの無水HCl塩多形体GのX線粉末回折スペクトル(図8)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(2θ),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0208】
【表10】
【0209】
化合物Iの無水HCl塩多形体Gは表10に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物Iの無水HCl塩多形体Gは角度θ値8.2°により同定することができる。化合物Iの無水HCl塩多形体Gは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)8.2°;
b)8.2°及び20.8°;
c)8.2°、20.8°及び16.8°;
d)8.2°、20.8°、16.8°及び21.6°;
e)8.2°、20.8°、16.8°、21.6°及び12.6°;
f)8.2°、20.8°、16.8°、21.6°、12.6°及び10.4°;
g)8.2°、20.8°、16.8°、21.6°、12.6°、10.4°及び12.1°;
h)8.2°、20.8°、16.8°、21.6°、12.6°、10.4°、12.1°及び14.1°;
i)8.2°、20.8°、16.8°、21.6°、12.6°、10.4°、12.1°、14.1°及び13.3°。
【0210】
化合物Iの無水HCl塩多形体GはCu線を使用して得られたX線粉末回折パターンからd間隔10.782、4.270、8.506、7.025、7.314、6.657、6.281、5.277及び4.114Åの1個以上の反射により同定することもできる。
【0211】
化合物Iの無水HCl塩多形体Gは示差走査熱量分析(DSC)によっても特徴付けられる。TA Instruments DSC−2910示差走査熱量計でN2流下に昇温速度10℃/分で化合物Iの無水HCl塩多形体GのDSC曲線(図9)を得た。サンプルを閉鎖パンで加熱した。開始温度267.3Cで単一吸熱が観察される。
【実施例14】
【0212】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)HCl塩多形体Hの製造
化合物Iの結晶質遊離塩基2.0gをエタノール3.7mLに溶解した。次に1.25M HClエタノール溶液0.05mLを加えた後に多形体Gシード5mgを加えた。次に1.25M HClエタノール溶液3.2mLを0.2mL/時の速度でゆっくりと加えた。得られたスラリーを濾過し、エタノール6mLで洗浄し、乾燥しなかった。
【0213】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物IのHCl塩多形体HのX線粉末回折スペクトル(図21)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(20),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0214】
【表11】
【0215】
化合物IのHCl塩多形体Hは表11に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物IのHCl塩多形体Hは角度θ値9.1°により同定することができる。化合物IのHCl塩形態Hは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)9.1°;
b)9.1°及び9.4°;
c)9.1°、9.4°及び12.8°;
d)9.1°、9.4°、12.8°及び13.4°;
e)9.1°、9.4°、12.8°、13.4°及び14.3°;
f)9.1°、9.4°、12.8°、13.4°、14.3°及び14.9°;
g)9.1°、9.4°、12.8°、13.4°、14.3°、14.9°及び15.3°;
h)9.1°、9.4°、12.8°、13.4°、14.3°、14.9°、15.3°及び16.9°;
i)9.1°、9.4°、12.8°、13.4°、14.3°、14.9°、15.3°、16.9°及び18.2°。
【0216】
化合物IのHCl塩多形体CはCu線を使用して得られたX線粉末回折パターンからd間隔9.718、9.408、6.916、6.607、6.194、5.945、5.791、5.246及び4.874Åの1個以上の反射により同定することもできる。
【技術分野】
【0001】
構造式I:
【0002】
【化1】
の化合物は既にUS2007/0123505、WO2007/062193及びWO2007/064566に開示されている。構造式Iの化合物とその新規多形体、溶媒和物、水和物及び塩はCB−1調節に関連する各種疾患の治療における中枢作用薬として有用な逆アゴニスト/アンタゴニストとして特徴付けられるCB1モジュレーターであり、このような疾患としては限定されないが、精神病、記憶障害、認知障害、アルツハイマー病、ハンチントン病、偏頭痛、神経障害、神経炎症障害(例えば多発性硬化症、ギラン・バレー症候群、並びにウイルス性脳炎、脳血管発作及び頭部外傷の炎症性後遺症)、不安障害、ストレス、癲癇、パーキンソン病、運動障害及び統合失調症が挙げられる。前記化合物は物質濫用障害の治療、肥満症又は摂食障害とそれに伴う合併症(例えば左心室肥大)の治療、並びに喘息、便秘、慢性腸偽閉塞及び肝硬変の治療にも有用である。
【0003】
本発明は、フッ素基を導入するためにHFの使用を必要とし、化合物1のジアステレオマーを分離するためにカラムクロマトグラフィーを必要とするリニア合成を使用して従来製造されていた強力なCB−1逆アゴニストである化合物Iの新規で効率的な合成方法について記載する。本発明の合成はコンバージェントであり、生成物の収率が高く、結晶質中間体が得られ、この点はクロマトグラフィーを使用しない単離精製に関する本発明の利点である。
【背景技術】
【0004】
ウィッティヒ反応とホーナー・ワズワース・エモンズ反応については、Bonadies,F.et al.,Tetrahedron Lett.1994,35,20,3383−3386;及びFukatsu K.et al.,J.Med.Chem.2002,45,4212−4221に記載されている。Tang,W.et al.,Chem.Rev.2003,103,3029にはエナンチオ選択的水素化用のキラルリンリガンドが記載されている。Kastrinsky,D.B.et al.J.Org.Chem.2004,69,2284;及びLewis,E.A.et al.,Tetrahedron Lett.2004,45,3059にはβ−カルボキシエステルの還元方法が記載されている。Weix,D.et al.,J.Am.Chem.Soc.127(4),1092−1093(2005);及びBolshan,Y.et al.,Org.Lett.7(8),1481−1484(2005)にはスルフィニルイミンへのアリールボロン酸のロジウム触媒付加が記載されている。Kitagawa,K.et al.,Angew.Chem.,Int.Ed.39(14),2481−2483(2000)にはn−BuLi/n−Bu2Mgを利用する金属−ハロゲン交換反応が記載されている。Hillier,M.C.;Chen,C−y.,J.Org.Chem.71,7885−7887(2006)にはビスアルキル化によるジオールからアゼチジンの製造が記載されている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】米国特許出願公開第2007/0123505号明細書
【特許文献2】国際公開第2007/062193号
【特許文献3】国際公開第2007/064566号
【非特許文献】
【0006】
【非特許文献1】Bonadies,F.et al.,Tetrahedron Lett.1994,35,20,3383−3386
【非特許文献2】Fukatsu K.et al.,J.Med.Chem.2002,45,4212−4221
【非特許文献3】Tang,W.et al.,Chem.Rev.2003,103,3029
【非特許文献4】Kastrinsky,D.B.et al.J.Org.Chem.2004,69,2284
【非特許文献5】Lewis,E.A.et al.,Tetrahedron Lett.2004,45,3059
【非特許文献6】Weix,D.et al.,J.Am.Chem.Soc.127(4),1092−1093(2005)
【非特許文献7】Bolshan,Y.et al.,Org.Lett.7(8),1481−1484(2005)
【非特許文献8】Kitagawa,K.et al.,Angew.Chem.,Int.Ed.39(14),2481−2483(2000)
【非特許文献9】Hillier,M.C.;Chen,C−y.,J.Org.Chem.71,7885−7887(2006)
【発明の概要】
【0007】
発明が解決しようとする課題及び課題を解決するための手段
本発明はベンズヒドリルアミンII及びシアノジオールIIIから構造式Iの3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリルを製造するための新規で効率的な方法を提供する。
【0008】
本発明は更に、1)化合物Iの遊離塩基無水多形体I;2)化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体I,タイプB;3)化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物多形体I,タイプA;並びに4)化合物IのHCl塩無水多形体A、B、C、D、E、F、G及びHと称する3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリルの11種類の同定された新規結晶体を提供する。これらの遊離塩基及び塩酸塩多形体の結晶体は新規であり、加工、取扱い及び投与し易さ等の化合物Iの医薬組成物の製造上の利点があると考えられる。特に、化合物Iの無水結晶質遊離塩基多形体Iは脂質溶解度;良好なpK暴露;化学的及び物理的安定性;純度;精製単離し易さ;並びに望ましい結晶寸法、結晶表面積及び結晶凝集回避による製剤化等の物理化学的性質の改善により、医薬製剤の製造に特に適している。化合物Iの新規HCl塩無水多形体Gは化合物Iの最も熱力学的に安定な結晶質HCl塩形態であるが、動力学的には多形体A、B、F及びHのほうが好ましい。
【0009】
本発明は更に、活性医薬成分として化合物Iの新規多形体及び塩を含有する医薬製剤と、CB−1関連疾患の治療におけるCB−1逆アゴニスト/アンタゴニストとしてのその使用方法にも関する。
【図面の簡単な説明】
【0010】
【図1】化合物Iの無水遊離塩基多形体IのX線回折(XRPD)パターンである。
【図2】化合物Iの無水遊離塩基多形体Iの熱重量分析(TGA)曲線である。
【図3】化合物Iの無水遊離塩基多形体Iの示差走査熱量(DSC)曲線である。
【図4】化合物Iの無水HCl塩多形体AのX線回折(XRPD)パターンである。
【図5】化合物Iの無水HCl塩多形体Aの示差走査熱量(DSC)曲線である。
【図6】化合物Iの無水HCl塩多形体BのX線回折(XRPD)パターンである。
【図7】化合物Iの無水HCl塩多形体Bの示差走査熱量(DSC)曲線である。
【図8】化合物Iの無水HCl塩多形体GのX線回折(XRPD)パターンである。
【図9】化合物Iの無水HCl塩多形体Gの示差走査熱量(DSC)曲線である。
【図10】化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物多形体IタイプAのX線回折(XRPD)パターンである。
【図11】化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物多形体IタイプAの示差走査熱量(DSC)曲線である。
【図12】化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物多形体IタイプAの熱重量分析(TGA)曲線である。
【図13】化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体IタイプBのX線回折(XRPD)パターンである。
【図14】化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体IタイプBの示差走査熱量(DSC)曲線である。
【図15】化合物IのHCl塩多形体CのX線回折(XRPD)パターンである。
【図16】化合物IのHCl塩多形体DのX線回折(XRPD)パターンである。
【図17】化合物IのHCl塩多形体Dの示差走査熱量(DSC)曲線である。
【図18】化合物IのHCl塩多形体Dの熱重量分析(TGA)曲線である。
【図19】化合物Iの無水HCl塩多形体EのX線回折(XRPD)パターンである。
【図20】化合物IのHCl塩多形体F水和物のX線回折(XRPD)パターンである。
【図21】化合物IのHCl塩多形体HのX線回折(XRPD)パターンである。
【発明を実施するための形態】
【0011】
本発明は構造式I:
【0012】
【化2】
の3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリルとその結晶質多形体、溶媒和物、水和物及び塩の製造方法を提供する。
【0013】
下記一般スキームに示すように、化合物IはベンズヒドリルアミンIIをシアノジオールIIIと反応させて保護オキサジアゾール化合物20を形成した後に保護基Pを開裂し、化合物Iを得ることにより製造することができる。
【0014】
【化3】
【0015】
化合物Iの遊離塩基には、無水遊離塩基多形体I、遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物多形体IタイプA及び遊離塩基トルエン/ヘプタン溶媒和物多形体IタイプBと称する3種類の公知結晶体ないし多形体がある。化合物Iの3種類の遊離塩基結晶体のX線粉末回折(XPRD)パターンを図1(多形体I)、図10(遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物多形体I,タイプA)及び図13(遊離塩基トルエン/ヘプタン溶媒和物多形体I,タイプB)に示す。図2の熱重量分析(TGA)曲線は化合物Iの無水遊離塩基多形体Iで10℃/分の昇温速度で窒素流下に得られ、室温から融点までに<0.1重量%の重量低下を示した。
【0016】
図3に示す化合物Iの無水遊離塩基多形体IのDSC曲線は外挿開始温度157.8℃、ピーク温度163.6℃及びそれに伴う熱量44.6J/gの単一吸熱を特徴とする。
【0017】
図10、11及び12は化合物Iの無水遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物多形体IタイプAのX線回折パターン、TGA曲線及びDSC曲線を示す。図13及び14は化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体IタイプBのX線回折パターンとDSC曲線を示す。
【0018】
化合物Iは更に以下に記載するように塩酸塩に変換することができる。図4及び5は化合物Iの塩酸塩形態AのX線回折パターンとDSC曲線を示す。図6及び7は化合物Iの塩酸塩形態BのX線回折パターンとDSC曲線を示す。図8及び9は化合物Iの塩酸塩形態GのX線回折パターンとDSC曲線を示す。図15は化合物Iの塩酸塩形態CのX線回折パターンを示す。図16、17及び18は化合物Iの塩酸塩形態DのX線回折パターン、TGA曲線及びDSC曲線を示す。図19は化合物Iの塩酸塩形態EのX線回折パターンを示す。図20は化合物Iの塩酸塩形態FのX線回折パターンを示す。最後に、図21は化合物Iの塩酸塩形態HのX線回折パターンを示す。
【0019】
本発明の1態様は式I:
【0020】
【化4】
の化合物又はその塩、水和物もしくは多形体の製造方法として、
A)化合物IIIのアルコール基を脱離基に変換した後にヒンダードアミン塩基で処理することにより式II:
【0021】
【化5】
(式中、Pは保護基である)の化合物又はその塩を式III:
【0022】
【化6】
の化合物とカップリングする段階と;
B)保護基Pを除去する段階を含む方法を提供する。
【0023】
本態様の1分類において、段階A及びBの保護基PはBoc及びCBZから選択される。
【0024】
本態様の別の分類において、段階Aの脱離基はトリフラート、トシラート、ノシラート及びメシラートから選択される。本態様の別の分類において、段階Aの脱離基はトリフラートであり、化合物IIIをトリフル酸無水物で処理し、ジトリフラート中間体を形成する。本態様の別の分類において、段階Aのヒンダードアミン塩基はジイソプロピルエチルアミン、トリエチルアミン、トリイソプロピルアミン及びジシクロヘキシルアミンから選択される。本態様の別の分類において、段階Aのヒンダードアミン塩基はジイソプロピルエチルアミンである。本態様の別の分類において、段階Aの反応はアセトニトリル中で実施される。
【0025】
本態様の別の分類において、保護基PはCBZである。本態様の別の分類において、保護基PはCBZであり、段階BのCBZ保護基は水素化により除去される。
【0026】
本態様の別の分類において、保護基PはBocである。本態様の別の分類において、保護基はBocであり、段階BのBoc保護基は酸を使用して除去される。本サブ分類の1サブ分類において、酸はHCl、H2SO4、H3PO4及びTFAから選択される。本サブ分類の別のサブ分類において、酸はHClである。本サブ分類の別のサブ分類において、酸はHClのイソプロパノール溶液である。本態様の別の分類において、段階Bは酢酸イソプロピル、イソプロパノール、塩化メチレン及びTHFから選択される溶媒中で実施される。本分類の1サブ分類において、段階Bの溶媒は酢酸イソプロピルである。
【0027】
本態様の1分類において、方法は更に式Iの化合物を単離する段階を含む。本分類の1サブ分類において、式Iの化合物はトルエン/ヘプタンから再結晶することにより単離される。
【0028】
本発明の別の態様は式I:
【0029】
【化7】
の化合物又はその塩、溶媒和物、水和物もしくは多形体の製造方法として、
式20:
【0030】
【化8】
の化合物の保護基Pを除去する段階を含む方法を提供する。
【0031】
本態様の1分類において、段階A及びBの保護基PはBoc及びCBZから選択される。
【0032】
本態様の別の分類において、保護基PはCBZである。本態様の別の分類において、保護基PはCBZであり、段階BのCBZ保護基は水素化により除去される。
【0033】
本態様の別の分類において、保護基PはBocである。本態様の別の分類において、保護基はBocであり、段階BのBoc保護基は酸を使用して除去される。本サブ分類の1サブ分類において、酸はHCl、H2SO4、H3PO4及びTFAから選択される。本サブ分類の別のサブ分類において、酸はHClである。本サブ分類の別のサブ分類において、酸はHClのイソプロパノール溶液である。本態様の別の分類において、段階Bは酢酸イソプロピル、イソプロパノール、塩化メチレン及びTHFから選択される溶媒中で実施される。本分類の1サブ分類において、段階Bの溶媒は酢酸イソプロピルである。
【0034】
本態様の1分類において、方法は更に式Iの化合物を単離する段階を含む。本分類の1サブ分類において、式Iの化合物はトルエン/ヘプタンから再結晶することにより単離される。
【0035】
本発明の別の態様は式I:
【0036】
【化9】
の化合物又はその塩、溶媒和物、水和物もしくは多形体の製造方法として、溶媒中で酸を使用して式20:
【0037】
【化10】
の化合物のBoc保護基を除去する段階を含む方法を提供する。
【0038】
本態様の1分類において、Boc保護基は酸を使用して除去される。本分類の1サブ分類において、酸はHCl、H2SO4、H3PO4及びTFAから選択される。本分類の別のサブ分類において、酸はHClである。本分類の別のサブ分類において、酸はHClのイソプロパノール溶液である。本態様の別の分類において、脱保護反応は酢酸イソプロピル、イソプロパノール、塩化メチレン及びTHFから選択される溶媒中で実施される。本分類の1サブ分類において、溶媒は酢酸イソプロピルである。
【0039】
本態様の1分類において、方法は更に式Iの化合物を単離する段階を含む。本分類の1サブ分類において、式Iの化合物はトルエン/ヘプタンから再結晶することにより単離される。
【0040】
本発明の別の態様は式20:
【0041】
【化11】
(式中、Pは保護基である)の化合物の製造方法として、化合物IIIのアルコール基を脱離基に変換した後にヒンダードアミン塩基で処理することにより式II:
【0042】
【化12】
(式中、Pは保護基である)の化合物又はその塩を式III:
【0043】
【化13】
の化合物とカップリングする段階を含む方法を提供する。
【0044】
本態様の1分類において、保護基PはBoc及びCBZから選択される。本態様の別の分類において、脱離基はトリフラート、トシラート、ノシラート及びメシラートから選択される。本態様の別の分類において、脱離基はトリフラートであり、化合物IIIをトリフル酸無水物で処理し、ジトリフラート中間体を形成する。本態様の別の分類において、ヒンダードアミン塩基はジイソプロピルエチルアミン、トリエチルアミン、トリイソプロピルアミン及びジシクロヘキシルアミンから選択される。本態様の別の分類において、ヒンダードアミン塩基はジイソプロピルエチルアミンである。本態様の別の分類において、反応はアセトニトリル中で実施される。
【0045】
本態様の別の分類において、方法は更に式20の化合物を単離する段階を含む。本分類の1サブ分類において、式20の化合物は酢酸イソプロピル、ジクロロメタン、アセトニトリル、ヘプタン又はその混合物から再結晶することにより単離される。本分類の別のサブ分類において、式20の化合物は酢酸イソプロピルとヘプタンから再結晶することにより単離される。
【0046】
本発明の別の態様は式II:
【0047】
【化14】
(式中、Pは保護基である)の化合物又はその塩の製造方法として、
(A)式1:
【0048】
【化15】
の化合物を塩基で処理した後にヒドラジンで処理することにより式3:
【0049】
【化16】
のヒドラジドを製造する段階と;
(B)式3のヒドラジドをカップリング剤で処理することにより式4:
【0050】
【化17】
のオキサジアゾールを形成する段階と;
(C)式4のオキサジアゾールをアルキルマグネシウム化合物で処理した後にアルキルリチウム化合物とDMFで処理することにより式5:
【0051】
【化18】
のアルデヒドを製造する段階と;
(D)式5のアルデヒドを触媒の存在下に(S)−tert−ブチルスルフィンアミドで処理することにより式6:
【0052】
【化19】
のN−tert−ブチルスルフィニルイミンを製造する段階と;
(E)式6のN−tert−ブチルスルフィニルイミンのオキサジアゾール窒素に保護基Pを付加することにより式7:
【0053】
【化20】
(式中、Pは保護基である)の保護オキサジアゾール化合物を形成する段階と;
(F)式7の化合物をロジウム触媒とリガンドの存在下にボロキシン10で処理することにより式8:
【0054】
【化21】
(式中、Pは保護基である)のN−tert−ブチルスルフィニルアミンを形成する段階と;
(G)式8の化合物のtert−ブチルスルホキシド基を開裂することにより式II:
【0055】
【化22】
(式中、Pは保護基である)の化合物を形成する段階を含む方法を提供する。
【0056】
本態様の1分類において、段階Aの塩基はDABCO、トリエチルアミン及びジイソプロピルエチルアミンから選択される。本分類の1サブ分類において、段階Aの塩基はDABCOである。本態様の別の分類において、段階Aの反応はメタノール中で実施される。本態様の別の分類において、段階Aの反応は50〜55℃で実施される。本態様の別の分類において、段階Aのヒドラジンは64%ヒドラジンである。本態様の別の分類において、段階Aの反応はメタノール中でDABCOで処理した後に64%ヒドラジンで処理することにより実施される。本態様の別の分類において、方法は更に式3のヒドラジドを単離する段階を含む。本分類の1サブ分類において、式3のヒドラジドは固体である。本態様の更に別の分類において、方法は更に段階Aの反応をワークアップする段階と、段階Bで式3のヒドラジドを溶液として使用する段階を含む。
【0057】
本態様の別の分類において、段階Bのカップリング剤はCDI、トリホスゲン及びホスゲンから選択される。本態様の別の分類において、段階Bのカップリング剤はCDIである。本態様の別の分類において、段階Bの反応は非プロトン性溶媒中で実施される。本分類の1サブ分類において、非プロトン性溶媒はTHF、トルエン及びエーテルである。本分類の別のサブ分類において、非プロトン性溶媒はTHFである。本態様の別の分類において、段階Bの反応は室温で実施される。本態様の別の分類において、段階Bの反応はTHF中でCDIを使用して実施される。本分類の1サブ分類において、反応は室温で実施される。本態様の別の分類において、方法は更に段階Bの式4のオキサジアゾールを単離する段階を含む。本分類の1サブ分類において、式4のオキサジアゾールは固体である。本態様の更に別の分類において、方法は更に段階Bの反応をワークアップする段階と、段階Cで式4のオキサジアゾールを溶液として使用する段階を含む。
【0058】
本態様の別の分類において、段階Cのアルキルマグネシウム化合物はジブチルマグネシウム、ジメチルマグネシウム、ジエチルマグネシウム及びジイソプロピルマグネシウムから選択される。本態様の別の分類において、段階Cのアルキルマグネシウム化合物はジ−n−ブチルマグネシウムである。本態様の別の分類において、段階Cのアルキルリチウム化合物はn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウム及びヘキシルリチウムから選択される。本態様の別の分類において、段階Cのアルキルリチウム化合物はn−ブチルリチウムである。本態様の別の分類において、段階Cの反応は非プロトン性溶媒中で実施される。本分類の1サブ分類において、段階Cの非プロトン性溶媒はTHF、トルエン、MTBE及びジエチルエーテルから選択される。本分類の別のサブ分類において、段階Cの非プロトン性溶媒はTHFである。本態様の別の分類において、段階Cの反応は約−20〜−78℃の温度で実施される。本分類の1サブ分類において、段階Cの反応は約−40〜−50℃の温度で実施される。本態様の別の分類において、段階Cの反応は約−20〜−78℃の温度にてTHF中でジ−n−ブチルマグネシウムとn−ブチルリチウムを使用して実施される。本分類の1サブ分類において、温度は約−40〜−50℃である。本態様の別の分類において、段階Cの反応は酸を使用してワークアップされる。本分類の1サブ分類において、段階Cの酸はHCl又はH2SO4である。本分類の1サブ分類において、段階Cの酸はHClである。本態様の別の分類において、方法は更に式5のアルデヒドを単離する段階を含む。本分類の1サブ分類において、式5のアルデヒドは固体である。本態様の更に別の分類において、方法は更に段階Cの反応をワークアップする段階と、段階Dで式5のアルデヒドを溶液として使用する段階を含む。
【0059】
本態様の別の分類において、段階Dの触媒はPPTS、KHSO4、BF3・エーテル錯体、Ti(OEt)4及びTiCl4/トリエチルアミンから選択される。本態様の別の分類において、段階Dの触媒はPPTSである。本態様の別の分類において、段階Dの反応はトルエン、塩化メチレン及びTHFから選択される溶媒中で実施される。本態様の別の分類において、段階Dの反応はトルエン中で実施される。本態様の別の分類において、段階Dの反応はトルエン中でPPTSを使用して実施される。本分類の1サブ分類において、段階Dの反応は約40℃で実施される。本態様の別の分類において、方法は更に段階Dの式6のN−tert−ブチルスルフィニルイミンを単離する段階を含む。本分類の1サブ分類において、式6のN−tert−ブチルスルフィニルイミンは固体である。本態様の更に別の分類において、方法は更に段階Dの反応をワークアップする段階と、段階Eで式6のN−tert−ブチルスルフィニルイミンを溶液として使用する段階を含む。
【0060】
本態様の1分類において、式7の化合物の保護基PはCBZ又はBoc基である。本態様の別の分類において、式7の化合物は保護基PがCBZであるN−CBZ保護オキサジアゾールである。本態様の別の分類において、式7の保護オキサジアゾール化合物は保護基PがBocであるN−Boc保護オキサジアゾールである。本分類の1サブ分類において、式7のN−Boc保護オキサジアゾールは式6のN−tert−ブチルスルフィニルイミンを塩基の存在下にboc酸無水物で処理することにより製造される。本サブ分類の1サブ分類において、塩基は第3級アミン塩基である。本サブ分類の別のサブ分類において、塩基はトリエチルアミンである。本分類の別のサブ分類において、N−Boc保護保護オキサジアゾールは非プロトン性溶媒中でトリエチルアミンの存在下に式6のN−tert−ブチルスルフィニルイミンをBoc酸無水物で処理することにより製造される。本サブ分類の1サブ分類において、非プロトン性溶媒はTHFである。本サブ分類の別のサブ分類において、段階Eの反応は約40℃で実施される。本態様の別の分類において、式7の化合物は式6のN−tert−ブチルスルフィニルイミンをTHF中でboc酸無水物とトリエチルアミンで処理することにより製造されるN−Boc保護オキサジアゾールである。本態様の別の分類において、方法は更に段階Eの式7の保護オキサジアゾール化合物を単離する段階を含む。本分類の1サブ分類において、式7の保護オキサジアゾール化合物は固体である。本態様の別の分類において、方法は更に段階Eの反応をワークアップする段階と、段階Fで式7の保護オキサジアゾール化合物を溶液として使用する段階を含む。
【0061】
本態様の別の分類において、段階Fの保護基PはCBZである。本態様の別の分類において、段階Fの保護基PはBocである。本態様の別の分類において、段階Fのロジウム触媒はRh(acac)(CH2CH2)2である。本態様の別の分類において、リガンドはホスフィンリガンドである。本分類の1サブ分類において、ホスフィンリガンドは1,2−ビス(ジフェニルホスフィノ)ベンゼンと1,2−ビス(ジフェニルホスフィノ)エタンから選択される。本分類の別のサブ分類において、ホスフィンリガンドは1,2−ビス(ジフェニルホスフィノ)ベンゼンである。本態様の別の分類において、溶媒はtert−アミルアルコール、tert−ブタノール、THF及びジオキサンから選択される。本分類の1サブ分類において、溶媒はtert−アミルアルコールである。本態様の別の分類において、段階Fの反応は約室温〜約45℃の温度で実施される。本態様の別の分類において、保護基PがBocである段階Fの反応はRh(acac)(CH2CH2)2と1,2−ビス(ジフェニルホスフィノ)ベンゼンの存在下で実施される。本分類の1サブ分類において、段階Fの反応はtert−アミルアルコール中で実施される。本分類の別のサブ分類において、段階Fの反応は約室温〜約45℃の温度で実施される。本態様の別の分類において、方法は更に段階Fの式8のN−tert−ブチルスルフィニルアミンを単離する段階を含む。本分類の1サブ分類において、式8のN−tert−ブチルスルフィニルアミンは固体である。本態様の別の分類において、方法は更に段階Fの反応をワークアップする段階と、段階Gで式8のN−tert−ブチルスルフィニルアミンを溶液として使用する段階を含む。
【0062】
本態様の別の分類において、段階Gの保護基PはCBZである。本態様の別の分類において、段階Gの保護基PはBocである。本態様の別の分類において、段階Gのtert−ブチルスルホキシド基は酸で開裂される。本態様の別の分類において、tert−ブチルスルホキシド基は塩酸、硫酸、リン酸及びトリフルオロ酢酸から構成される群から選択される酸で処理することにより開裂される。本態様の別の分類において、段階Gの開裂はハロゲン化溶媒中で実施される。本分類の1サブ分類において、ハロゲン化溶媒はジクロロメタン、クロロホルム及び四塩化炭素から選択される。本分類の別のサブ分類において、ハロゲン化溶媒はジクロロメタンである。本態様の別の分類において、段階Gの開裂は室温で実施される。本態様の別の分類において、段階Gの化合物8のtert−ブチルスルホキシド基は塩酸で処理することにより開裂される。本態様の別の分類において、方法は更に式IIの化合物を単離する段階を含む。本分類の1サブ分類において、式IIの化合物は固体である。本態様の別の分類において、方法は更に段階Gの反応をワークアップする段階と、カップリング反応で式IIaの化合物を溶液として使用し、化合物20を得る段階を含む。
【0063】
本発明の別の態様は式III:
【0064】
【化23】
の化合物又はその塩の製造方法として、
(A)式11:
【0065】
【化24】
の化合物をグリニャール試薬で処理した後に塩化イソブチリルで処理することにより式12:
【0066】
【化25】
の化合物を製造する段階と;
(B)ハロゲン化シリル又はシリルトリフラートの存在下にフッ素源と塩基で処理して式12の化合物をフッ素化することにより式13:
【0067】
【化26】
のフルオロケトン化合物を形成する段階と;
(C)式13の化合物を塩基の存在下にホスホノ酢酸トリメチルで処理することにより式14:
【0068】
【化27】
の化合物を製造する段階と;
(D)式14の化合物のエステルを加水分解することにより式15:
【0069】
【化28】
の化合物を製造する段階と;
(E)式15の化合物の二重結合を還元することにより式16:
【0070】
【化29】
の化合物を形成する段階と;
(F)式16の化合物のエステル化により式17:
【0071】
【化30】
(式中、R=C1−3アルキルである)の化合物を形成する段階と;
(G)式17の化合物のカルボキシル化により式18:
【0072】
【化31】
(式中、R=C1−3アルキルである)の化合物を形成する段階と;
(H)式18の化合物を還元することにより式19:
【0073】
【化32】
の化合物を形成する段階と;
(I)式19の化合物をシアノ化することにより式III
【0074】
【化33】
の化合物を形成する段階を含む方法を提供する。
【0075】
本態様の別の分類において、Rは−CH3である。本態様の別の分類において、RはCH2CH3である。本態様の更に別の分類において、Rは−CH2CH2CH3又は−CH(CH3)2である。
【0076】
本態様の別の分類において、段階Aのグリニャール試薬はイソプロピルマグネシウムクロリドである。本態様の別の分類において、段階Aの反応は1種以上の遷移金属ハロゲン化物塩触媒の存在下で実施される。本分類の1サブ分類において、遷移金属ハロゲン化物塩触媒はCuCl、ZnCl2及びCoCl2から選択される。本分類の別のサブ分類において、遷移金属ハロゲン化物塩触媒はCuClとZnCl2である。本態様の別の分類において、段階Aのグリニャール反応はエーテル溶媒中で実施される。本分類の1サブ分類において、エーテル溶媒はテトラヒドロフランである。本態様の別の分類において、段階Aの反応はCuClとZnCl2の存在下にテトラヒドロフラン中でイソプロピルマグネシウムクロリドを使用して実施される。本態様の別の分類において、方法は更に式12の化合物を単離する段階を含む。本態様の更に別の分類において、方法は更に段階Aの反応をワークアップする段階と、段階Bで式12の化合物をトルエン溶液として使用する段階を含む。
【0077】
本態様の別の分類において、段階Bのフッ素源はSelect−Fluor(登録商標)フッ素化剤である。本態様の別の分類において、段階Bの塩基はアルコキシド塩基又はナトリウムアミラートである。本分類の1サブ分類において、アルコキシド塩基はカリウムtert−ブトキシドである。本態様の別の分類において、段階Bの塩基はナトリウムアミラートである。本態様の別の分類において、段階Bのハロゲン化シリルとシリルトリフラートはterf−ブチルジメチルシリルクロリド、トリメチルシリルクロリド及びtert−ブチルジメチルシリルトリフラートから選択される。本分類の1サブ分類において、段階Bのハロゲン化シリルはtert−ブチルジメチルシリルクロリドである。本態様の別の分類において、段階Bのフッ素化源はSelect−Fluor(登録商標)フッ素化剤であり、塩基はナトリウムアミラートであり、ハロゲン化シリルはtert−ブチルジメチルシリルクロリドである。本態様の別の分類において、方法は更に式13の化合物を単離する段階を含む。本態様の更に別の分類において、方法は更に段階Bの反応をワークアップする段階と、段階Cで式13の化合物をトルエン溶液として使用する段階を含む。
【0078】
本態様の1分類において、段階Cの塩基は炭酸セシウム、炭酸カリウム、炭酸リチウム、カリウムtert−ブトキシド、水素化リチウム、水素化ナトリウム及びナトリウムアミラートから選択される。本分類の1サブ分類において、段階Cの塩基は炭酸カリウムである。本態様の別の分類では、段階Cのホスホノ酢酸トリメチルを式13の化合物に加える前に塩基で前処理する。本分類の1サブ分類において、段階Cの塩基は炭酸セシウム、炭酸カリウム、炭酸リチウム、カリウムtert−ブトキシド、水素化リチウム、水素化ナトリウム及びナトリウムアミラートから選択される。本サブ分類の1サブ分類において、段階Cの塩基は炭酸カリウムである。本態様の別の分類において、段階Cの反応は極性非プロトン性溶媒中で実施される。本分類の1サブ分類において、極性非プロトン性溶媒はジメチルホルムアミド、テトラヒドロフラン又はエーテルから選択される。本サブ分類の1サブ分類において、段階Cの溶媒はジメチルホルムアミドであるである。本態様の別の分類では、炭酸カリウムで前処理したホスホノ酢酸トリメチルと段階Cの式13の化合物を反応させる。本分類の1サブ分類において、段階Cの反応はジメチルホルムアミド中で実施される。本態様の別の分類において、方法は更に段階Cの式14の化合物を単離する段階を含む。本態様の更に別の分類において、方法は更に段階Cの反応をワークアップする段階と、段階Dで式14の化合物をトルエン中で使用する段階を含む。
【0079】
本態様の別の分類において、段階Dの加水分解は水酸化ナトリウム、水酸化リチウム又は水酸化カリウムを使用して実施される。本分類の1サブ分類において、段階Dの加水分解は水酸化ナトリウムを使用して実施される。本分類の別のサブ分類において、段階Dの加水分解は水性溶媒中で実施される。本サブ分類の1サブ分類において、水性溶媒はメタノール/水である。本サブ分類の別のサブ分類において、水性溶媒はメタノール/水/トルエンである。本態様の別の分類において、段階Dの加水分解はメタノール/水中で水酸化ナトリウムを使用して実施される。本態様の別の分類において、段階Dの加水分解はメタノール/水/トルエン中で水酸化ナトリウムを使用して実施される。本態様の別の分類において、方法は更に段階Dの式15の化合物を単離する段階を含む。
【0080】
本態様の別の分類において、段階Eの化合物15の還元は水素とルテニウム触媒の存在下の水素化である。本分類の1サブ分類において、ルテニウム触媒はアキシャルキラルリガンドをもつ。本サブ分類の1サブ分類において、アキシャルキラルリガンドはJosiphos(登録商標)型リガンド、Solphos(登録商標)型リガンド、CH3O−BIPHEP(登録商標)型リガンド、BINAP型リガンド又はSegphos(登録商標)型リガンドである。本サブ分類の別のサブ分類において、アキシャルキラルリガンドは(R)−Cl、CH3O−BIPHEP(登録商標)、(S)−Solphos(登録商標)、(R)−フリル−Solphos(登録商標)及びSL−J212−1である。本サブ分類の別のサブ分類において、Josiphos(登録商標)型アキシャルキラルリガンドはSL−J212−1である。本分類の別のサブ分類において、段階Eの水素化は加圧下に実施される。本サブ分類の1サブ分類において、段階Eの水素化は200psigで実施される。本分類の別のサブ分類において、段階Eの水素化は約40〜50℃で実施される。本態様の別の分類において、段階Eの還元はJosiphos(登録商標)型アキシャルキラルリガンドSL−J212−1をもつルテニウム触媒の存在下の水素化である。本分類の1サブ分類において、ルテニウム触媒は[(シメン)RuCl]2をSL−J212−1と反応させることにより製造される。本態様の別の分類において、方法は更に段階Eの式16の化合物を単離する段階を含む。本態様の別の分類において、方法は更に段階Eの式16の化合物を固体として単離する段階と、式16の化合物を再結晶させる段階を含む。本態様の別の分類において、方法は更に段階Eの反応をワークアップする段階と、段階Fで式16の化合物を溶液として使用する段階を含む。本態様の更に別の分類において、方法は更に段階Eの反応をワークアップする段階と、段階Fで式16の化合物をメタノール溶液として使用する段階を含む。
【0081】
本態様の別の分類において、段階Fのエステル化はアルコール溶媒中で酸塩化物の存在下に実施される。本分類の1サブ分類において、段階Fでエステル化により形成されるエステルはメチルエステルであり、エステル化はメタノール中で塩化アセチルの存在下に実施される。本サブ分類の1サブ分類において、エステル化は室温で実施される。本態様の別の分類において、方法は更に段階Fの式17の化合物を単離する段階を含む。
【0082】
本態様の別の分類において、段階Gのカルボキシル化は式17の化合物をエーテル又は極性溶媒中で塩基で処理した後にCO2を加えることにより実施される。本分類の1サブ分類において、塩基はリチウムヘキサメチルジシラジド、ナトリウムヘキサメチルジシラジド、カリウムヘキサメチルジシラジドム及びLDAから選択される。本分類の別のサブ分類において、エーテル又は極性溶媒はTHF、THF、MTBE、DME、トルエン及びDMPUの1種以上から選択される。本分類の別のサブ分類では、式17の化合物をTHF又はトルエン中でリチウムヘキサメチルジシラジドで処理した後にCO2を加えることによりカルボキシル化反応を実施した。本分類の別のサブ分類では、式17の化合物をTHF又はトルエンとDMPU中でリチウムヘキサメチルジシラジドで処理した後にCO2を加えることによりカルボキシル化反応を実施した。本分類の別のサブ分類では、式17の化合物をDME/トルエンとDMPU中でリチウムヘキサメチルジシラジドで処理した後にCO2を加えることによりカルボキシル化反応を実施した。本態様の別の分類において、方法は更に段階Gの式18の化合物を単離する段階を含む。
【0083】
本態様の1分類では、還元剤の存在下で段階Hの還元を実施した。本分類の1サブ分類において、還元剤は水素化ホウ素ナトリウム、水素化ホウ素ナトリウム/I2、水素化ホウ素ナトリウム/Br2、水素化ホウ素ナトリウムBF3/テトラヒドロフラン錯体、BF3/エーテル錯体、ボラン/テトラヒドロフラン錯体及びボラン/ジメチルスルフィド錯体である。本分類の別のサブ分類において、還元剤は水素化ホウ素ナトリウム/Br2である。本分類の別のサブ分類において、段階Hの還元はトルエン、DME、THF、DME/トルエン及びジクロロメタンの1種以上から選択される溶媒中で実施される。本分類の別のサブ分類において、段階Hの還元はDME及びトルエンから選択される溶媒中で実施される。本態様の別の分類において、方法は更に段階Hの式19の化合物を単離する段階を含む。
【0084】
本態様の1分類において、段階Iのシアノ化は亜鉛、臭素、Zn(CN)2及びパラジウム触媒の存在下で実施される。本分類の1サブ分類において、パラジウム触媒は二座又は単座パラジウム触媒である。本分類の別のサブ分類において、パラジウム触媒はパラジウムホスフィン触媒である。本サブ分類の1サブ分類において、パラジウム触媒はパラジウムテトラキストリフェニルホスフィンである。本分類の別のサブ分類において、パラジウム触媒はPd(dppf)2である。本分類の別のサブ分類において、段階Iのシアノ化はDMF中で実施される。本分類の別のサブ分類において、段階Iのシアノ化はDMF中で80℃にて実施される。
【0085】
本態様の別の分類において、方法は更に段階Iの式IIIの化合物を単離する段階を含む。
【0086】
本発明の別の態様はCB−1調節に関連する疾患の予防又は治療方法として、前記疾患の予防又は治療を必要とする対象に治療有効量の化合物Iの多形体、水和物又は塩を投与する段階を含む方法を提供する。
【0087】
本発明の別の態様はCB−1調節に関連する疾患の治療、抑制又は予防を必要とする対象における前記治療、抑制又は予防に有用な医薬の製造用としての治療有効量の化合物Iの多形体、溶媒和物、水和物又は塩の使用を提供する。
【0088】
本発明の別の態様は肥満症、摂食障害又は肥満関連障害の予防又は治療方法として、前記予防又は治療を必要とする対象に治療有効量の化合物Iの多形体、水和物又は塩を投与する段階を含む方法を提供する。
【0089】
本発明の別の態様は肥満症、摂食障害又は肥満関連障害の治療、抑制又は予防を必要とする対象における前記治療、抑制又は予防に有用な医薬の製造用としての治療有効量の化合物Iの多形体、溶媒和物、水和物又は塩の使用を提供する。
【0090】
「3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル」なる用語は固体状の3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリルのみならず、昇温により3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリルに変換可能なそのガラス体、凍結乾燥物及び混合物等の任意非晶質又は部分結晶質固体状の3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリルも包含する。
【0091】
多形体とは化学組成が同一でありながら結晶構造の異なる化合物である。多形性とは同一の化学物質が異なる結晶構造として存在する性質である。構造式Iの化合物3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリルとそのHCl塩は結晶化条件の入念な制御により各々形成することが可能な少なくとも11種類の多形体ないし結晶体として存在することが判明した。
【0092】
「水和物」なる用語は化合物Iの完全水和物、複合水和物及び部分水和物の全水和物を包含するものとし、限定されないが、一水和物、半水和物及びビス水和物が挙げられる。
【0093】
「溶媒和物」なる用語は化合物Iの結晶構造内に溶媒分子を含む化合物形態、又は化合物Iと結合もしくは会合した溶媒分子を含む化合物形態を包含するものとし、溶媒としては限定されないが、トルエン、ヘプタン、酢酸イソプロピル、酢酸エチル、メチルシクロヘキサン及び水が挙げられる。
【0094】
「非晶質」なる用語は長距離分子秩序をもたない固体形態を意味する。
【0095】
構造式Iの3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリルは結晶質塩酸塩を形成することが判明した。
【0096】
式Iの化合物の他の塩とは、医薬的に許容可能な通常の塩を意味し、例えば化合物がカルボキシル基をもつ場合にはカルボキシル基の塩基付加塩を意味し、化合物がアミノ又は塩基性複素環基をもつ場合にはアミノ又は塩基性複素環の酸付加塩を意味し、他の化合物についても同様である。「医薬的に許容可能な塩」なる用語は無機塩基又は酸と有機塩基又は酸を含む医薬的に許容可能な非毒性塩基又は酸から製造される塩を意味する。塩基付加塩としては、アルカリ金属(限定されないが、例えばナトリウム、カリウム);アルカリ土類金属(限定されないが、例えばカルシウム、マグネシウム);アンモニウム又は有機アミン(限定されないが、例えばトリメチルアミン、トリエチルアミン、ジシクロヘキシルアミン、エタノールアミン、ジエタノールアミン、トリエタノールアミン、プロカイン、N,N’−ジベンジルエチレンジアミン)等との塩が挙げられる。酸付加塩としては、無機酸(限定されないが、例えば塩酸、硫酸、硝酸、リン酸、過塩素酸)、有機酸(限定されないが、例えば酢酸、蓚酸、マレイン酸、フマル酸、酒石酸、クエン酸、アスコルビン酸、トリフルオロ酢酸、酢酸)、スルホン酸(限定されないが、例えばメタンスルホン酸、イセチオン酸、ベンゼンスルホン酸、p−トルエンスルホン酸、p−トルエンスルホン酸一水和物、p−トルエンスルホン酸水和物、樟脳スルホン酸)等との塩が挙げられる。
【0097】
本発明の1態様では、3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)をその遊離塩基、塩、水和物又は多形体として含有する医薬組成物を提供する。本態様の1分類において、化合物Iは実質的に純粋な形態である。本態様の別の分類において、化合物Iは結晶質である。本態様の別の分類において、化合物Iは結晶質無水遊離塩基である。本態様の別の分類において、化合物Iは結晶質遊離塩基溶媒和物である。本態様の別の分類において、化合物Iは結晶質無水塩である。本態様の別の分類において、化合物Iは結晶質塩水和物である。本態様の別の分類において、化合物Iは結晶質無水HCl塩である。本態様の別の分類において、化合物Iは結晶質HCl塩水和物である。本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物Iの遊離塩基形態Iとして含有する。本分類の1サブ分類において、化合物Iの遊離塩基形態Iは実質的に純粋な形態である。本分類の別のサブ分類において、化合物Iの遊離塩基形態Iは結晶質である。本分類の別のサブ分類において、化合物Iの遊離塩基形態Iは無水である。本分類の別のサブ分類において、化合物Iの遊離塩基形態Iは無水結晶質である。
【0098】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物Iの遊離塩基トルエン/ヘプタン溶媒和物形態I,タイプBとして含有する。本分類の1サブ分類において、化合物Iの遊離塩基トルエン/ヘプタン溶媒和物形態I,タイプBは実質的に純粋な形態である。本分類の別のサブ分類において、化合物Iの遊離塩基トルエン/ヘプタン溶媒和物形態I,タイプBは結晶質である。本分類の別のサブ分類において、化合物Iの遊離塩基トルエン/ヘプタン溶媒和物形態I,タイプBは無水である。本分類の別のサブ分類において、化合物Iの遊離塩基トルエン/ヘプタン溶媒和物形態I,タイプBは無水結晶質である。
【0099】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物形態I,タイプAとして含有する。本分類の1サブ分類において、化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物形態I,タイプAは実質的に純粋な形態である。本分類の別のサブ分類において、化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物形態I,タイプAは結晶質である。本分類の別のサブ分類において、化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物形態I,タイプAは無水である。本分類の別のサブ分類において、化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物形態I,タイプAは無水結晶質である。
【0100】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物IのHCl塩形態Aとして含有する。本分類の1サブ分類において、化合物IのHCl塩形態Aは実質的に純粋な形態である。本分類の別のサブ分類において、化合物IのHCl塩形態Aは結晶質である。本分類の別のサブ分類において、化合物IのHCl塩形態Aは無水である。本分類の別のサブ分類において、化合物IのHCl塩形態Aは無水結晶質である。
【0101】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物IのHCl塩形態Bとして含有する。本分類の1サブ分類において、化合物IのHCl塩形態Bは実質的に純粋な形態である。本分類の別のサブ分類において、化合物IのHCl塩形態Bは結晶質である。本分類の別のサブ分類において、化合物IのHCl塩形態Bは無水である。本分類の別のサブ分類において、化合物IのHCl塩形態Bは無水結晶質である。
【0102】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物IのHCl塩形態Cとして含有する。本分類の1サブ分類において、化合物IのHCl塩形態Cは実質的に純粋な形態である。本分類の別のサブ分類において、化合物IのHCl塩形態Cは結晶質である。本分類の別のサブ分類において、化合物IのHCl塩形態Cは無水である。本分類の別のサブ分類において、化合物IのHCl塩形態Cは無水結晶質である。
【0103】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物IのHCl塩形態Dとして含有する。本分類の1サブ分類において、化合物IのHCl塩形態Dは実質的に純粋な形態である。本分類の別のサブ分類において、化合物IのHCl塩形態Dは結晶質である。本分類の別のサブ分類において、化合物IのHCl塩形態Dは無水である。本分類の別のサブ分類において、化合物IのHCl塩形態Dは無水結晶質である。
【0104】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物IのHCl塩形態Eとして含有する。本分類の1サブ分類において、化合物IのHCl塩形態Eは実質的に純粋な形態である。本分類の別のサブ分類において、化合物IのHCl塩形態Eは結晶質である。本分類の別のサブ分類において、化合物IのHCl塩形態Eは無水である。本分類の別のサブ分類において、化合物IのHCl塩形態Eは無水結晶質である。
【0105】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物IのHCl塩形態Fとして含有する。本分類の1サブ分類において、化合物IのHCl塩形態Fは実質的に純粋な形態である。本分類の別のサブ分類において、化合物IのHCl塩形態Fは結晶質である。本分類の別のサブ分類において、化合物IのHCl塩形態Fは水和物である。本分類の別のサブ分類において、化合物IのHCl塩形態Fは結晶質水和物である。
【0106】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物IのHCl塩形態Gとして含有する。本分類の1サブ分類において、化合物IのHCl塩形態Gは実質的に純粋な形態である。本分類の別のサブ分類において、化合物IのHCl塩形態Gは結晶質である。本分類の別のサブ分類において、化合物IのHCl塩形態Gは無水である。本分類の別のサブ分類において、化合物IのHCl塩形態Gは無水結晶質である。
【0107】
本態様の別の分類において、組成物は3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)を化合物IのHCl塩形態Hとして含有する。本分類の1サブ分類において、化合物IのHCl塩形態Hは実質的に純粋な形態である。本分類の別のサブ分類において、化合物IのHCl塩形態Hは結晶質である。本分類の別のサブ分類において、化合物IのHCl塩形態Hは無水である。本分類の別のサブ分類において、化合物IのHCl塩形態Hは無水結晶質である。
【0108】
本発明の方法における化合物は置換方式に応じて光学異性体、ジアステレオマー及び幾何異性体等の立体異性体又は互変異性体を含む。本発明は本発明の組成物に含まれる化合物のこのような全異性体とその混合物を包含するものとする。上記化合物の全水和物、溶媒和物及び多形結晶体とその使用(本発明の方法におけるその使用を含む)が本発明の範囲に含まれる。
【0109】
ニューロキニン−1(NK−1)受容体アンタゴニストを本発明の化合物と有利に併用することができる。本発明で使用されるNK−1受容体アンタゴニストは文献に詳細に記載されている。本発明で使用される特定ニューロキニン−1受容体アンタゴニストとしては、(±)−(2R3R,2S3S)−N−{[2−シクロプロポキシ−5−(トリフルオロメトキシ)フェニル]メチル}−2−フェニルピペリジン−3−アミン;2−(R)−(1−(R)−(3,5−ビス(トリフルオロメチル)フェニル)エトキシ)−3−(S)−(4−フルオロフェニル)−4−(3−(5−オキソ−1H,4H−1,2,4−トリアゾロ)メチル)モルホリン;アプレピタント;CJ17493;GW597599;GW679769;R673;RO67319;R1124;R1204;SSR146977;SSR240600;T−2328;及びT2763;又はその医薬的に許容可能な塩が挙げられる。式I、II又はIIIの化合物と併用することができる他の抗肥満薬の例は“Patent focus on new anti−obesity agents,”Exp.Opin.Ther.Patents,10:819−831(2000);“Novel anti−obesity drugs,”Exp.Opin.Invest.Drugs,9:1317−1326(2000);及び“Recent advances in feeding suppressing agents:potential therapeutic strategy for the treatment of obesity,Exp.Opin.Ther.Patents,11:1677−1692(2001)に開示されている。肥満におけるニューロペプチドYの役割はExp.Opin.Invest.Drugs,9:1327−1346(2000)に記載されている。カンナビノイド受容体リガンドはExp.Opin.Invest.Drugs,9:1553−1571(2000)に記載されている。
【0110】
本発明の別の側面は化合物Iの多形体、水和物又は塩と、医薬的に許容可能なキャリヤーを含有する医薬組成物を提供する。本発明の医薬組成物は活性成分として式Iの化合物又はその医薬的に許容可能な塩を含有しており、更に医薬的に許容可能なキャリヤーと場合により他の治療成分を加えてもよい。
【0111】
組成物としては経口、直腸、局所、非経口(例えば皮下、筋肉内及び静脈内)、眼球内(眼科)、肺(鼻腔又は口腔吸入)又は鼻腔内投与に適した組成物が挙げられるが、任意の所与症例に最適な経路は治療する病態の種類及び重篤度と活性成分の種類により異なる。組成物は単位用量形態が適切であり、製薬分野で周知の方法のいずれかにより製造することができる。
【0112】
実際の使用では、従来の医薬配合技術に従って化合物Iの多形体、水和物及び塩を活性成分として医薬キャリヤーと混和することができる。キャリヤーは例えば経口又は非経口(例えば静脈内)等の投与に所望される製剤形態に応じて多様な形態を取ることができる。経口剤形用組成物を製造する際には、通常の医薬媒体のいずれかを利用することができ、例えば懸濁液剤、エリキシル剤及び溶液剤等の経口液体製剤の場合には例えば水、グリコール、油類、アルコール類、フレーバー剤、防腐剤、着色剤等が挙げられ、例えば散剤、ハード及びソフトカプセル剤並びに錠剤等の経口固体製剤の場合には澱粉、糖類、微結晶セルロース、希釈剤、顆粒化剤、滑沢剤、結合剤、崩壊剤等のキャリヤーが挙げられ、固体経口製剤のほうが液体製剤よりも好ましい。
【0113】
投与し易いという理由から、錠剤とカプセル剤が最も有利な経口用量単位形態であり、その場合には当然のことながら固体医薬キャリヤーを使用する。所望により、標準水性又は非水性技術により錠剤にコーティングしてもよい。このような組成物及び製剤は少なくとも0.1%の活性化合物を含有すべきである。これらの組成物中の活性化合物の百分率は当然のことながら変動させることができ、単位重量の約2%〜約60%が適切であると思われる。このような治療薬として有用な組成物中の活性化合物の量は有効用量が得られるような量とする。活性化合物は鼻腔内投与することもできる(例えば液体滴剤又はスプレー)。
【0114】
錠剤、ピル、カプセル剤等にはトラガカントガム、アラビアガム、コーンスターチ又はゼラチン等の結合剤;リン酸2カルシウム等の賦形剤;コーンスターチ、ジャガイモ澱粉、アルギン酸等の崩壊剤;ステアリン酸マグネシウム等の滑沢剤;及びスクロース、ラクトース又はサッカリン等の甘味剤も添加してもよい。用量単位形態がカプセルの場合には、上記型の材料に加え、脂肪油等の液体キャリヤーを加えることができる。
【0115】
コーティングとして又は用量単位の物理的形状を変更するために他の各種材料を加えてもよい。例えば、錠剤にはシェラック、糖又は両者をコーティングすることができる。シロップ又はエリキシル剤には活性成分に加え、甘味剤としてスクロース、防腐剤としてメチル及びプロピルパラベン、色素並びにチェリー又はオレンジフレーバー等のフレーバー剤を加えることができる。
【0116】
化合物Iの多形体、水和物及び塩は非経口投与することもできる。ヒドロキシプロピルセルロース等の界面活性剤と適切に混合した水中でこれらの活性化合物の溶液剤又は懸濁液剤を製造することができる。グリセロール、液体ポリエチレングリコール及びその油中混液で分散液剤を製造することもできる。通常の保存及び使用条件下で、これらの製剤には微生物の増殖を防ぐために防腐剤を添加することができる。
【0117】
注射用に適した剤形としては、滅菌水溶液又は水分散液と、滅菌注射用溶液又は分散液の即席調製用滅菌粉末が挙げられる。いずれの場合も、剤形は無菌でなければならず、注射し易い程度まで流動性でなければならない。製造及び保存条件下で安定でなければならず、細菌や真菌等の微生物の汚染作用から保護する必要がある。キャリヤーは例えば水、エタノール、ポリオール(例えばグリセロール、プロピレングリコール及び液体ポリエチレングリコール)、適切なその混液及び植物油を含有する溶媒又は分散媒とすることができる。
【0118】
本発明は肥満症及び肥満関連障害の治療及び/又は予防を必要とする対象における肥満症及び肥満関連障害の治療及び/又は予防方法として、前記治療を必要とする対象に治療有効量の化合物Iの水和物、塩又は多形体を投与する段階を含む方法を提供する。本発明は更に精神病、記憶障害、認知障害、アルツハイマー病、偏頭痛、神経障害、神経炎症障害(例えば多発性硬化症、ギラン・バレー症候群、並びにウイルス性脳炎、脳血管発作及び頭部外傷の炎症性後遺症)、不安障害、ストレス、癲癇、パーキンソン病、運動障害及び統合失調症等のCB−1調節障害の予防及び/又は治療用医薬の製造用としての化合物Iの水和物、塩及び多形体の使用を提供する。前記化合物は物質濫用障害の治療、肥満症又は摂食障害、肥満関連障害とそれに伴う合併症(例えば左心室肥大)の治療、並びに喘息、便秘、慢性腸偽閉塞及び肝硬変の治療にも有用である。
【0119】
本発明における肥満関連障害は肥満に付随、誘発又は起因する。肥満関連障害の例としては再狭窄、アテローム性動脈硬化症、動脈硬化症、過食症及び多食症、高血圧、糖尿病、血漿インスリン濃度及びインスリン抵抗性上昇、脂質異常症、高脂血症、子宮体癌、乳癌、前立腺癌、結腸癌、変形性関節症、閉塞型睡眠時無呼吸、胆石症、胆石、心疾患、心拍リズム異常及び不整脈、心筋梗塞、鬱血性心不全、冠動脈性心疾患、突然死、脳卒中、多嚢胞性卵巣疾患、頭蓋咽頭腫、プラダー・ウィリー症候群、フローリッヒ症候群、GH欠損対象、正常変異型低身長、ターナー症候群及び合計除脂肪体重の百分率としての代謝活性低下又は安静時エネルギー消費量低下を示す他の病態(例えば急性リンパ芽球性白血病、代謝症候群、インスリン抵抗性症候群の小児)、生殖ホルモン異常、性機能及び生殖機能不全(例えば受精障害、不妊症、男性性腺機能低下症及び女性多毛症)、妊婦肥満に伴う胎児異常、胃腸運動障害(例えば肥満関連胃食道逆流症)、呼吸器障害(例えば肥満低換気症候群(ピックウィック症候群)、息切れ)、心血管障害、炎症(例えば脈管構造の全身性炎症)、動脈硬化症、高コレステロール血症、高尿酸血症、腰痛、胆嚢疾患、通風、腎臓癌、知覚麻痺の危険の増加、左心室肥大、アルツハイマー病が挙げられる。
【0120】
(肥満症及び肥満関連障害の)「治療」とは肥満対象の体重を低下又は維持するための本発明の化合物又は併用剤の投与を意味する。(肥満症及び肥満関連障害の)「予防」とは肥満の危険のある対象の体重を低下又は維持するための本発明の化合物又は併用剤の投与を意味する。
【0121】
本明細書で使用する「対象」なる用語は治療、観察又は実験の対象とした動物、好ましくは哺乳動物、最も好ましくはヒトを意味する。「〜を必要とする対象」なる用語は研究者、獣医、医師又は他の臨床医の判断により治療又は予防を必要とする対象を意味する。1態様において、治療を必要とする対象は肥満症の哺乳動物である。別の態様において、治療を必要とする対象は1種以上の肥満関連併存症を伴う肥満症のヒトである。別の態様において、治療を必要とする対象は肥満関連併存症を伴わない肥満症のヒトである。本明細書で使用する「治療有効量」なる用語は治療する障害の症状の緩和を含めて研究者、獣医、医師又は他の臨床医により求められる生物学的又は医学的応答を組織、系、対象又はヒトに誘発する組成物中の活性化合物又は薬剤の量を意味する。
【0122】
化合物Iの塩、水和物又は多形体の予防又は治療用量値は当然のことながら治療する病態の重篤度の性質と、組成物中の特定化合物及びその投与経路により異なる。また、個々の患者の年齢、体重及び応答によっても異なる。一般に、肥満症又は肥満関連障害の治療には、対象の体重当たり約0.0001mg/kg〜約100mg/kg、好ましくは約0.001mg/kg〜約100mg/kg、より好ましくは約0.001mg/kg〜約10mg/kgの1日用量の化合物Iの塩、水和物又は多形体を1日1回もしくは2〜6回に分けて又は徐放形態で投与する。他方、場合によってはこれらの範囲外の用量を使用することが必要な場合もある。本発明の化合物は化合物毎に固有の用量範囲でヒトに投与することができる。経口投与では、治療する対象の症状に用量を合わせて各活性成分0.01mg〜1,000mg、好ましくは0.01、0.05、0.1、0.2、0.5、1.0、1.5、2.0、2.5、3.0、3.5、4.0、4.5、5.0、5.5、6.0、7.5、10、15、20、25、30、40、50、60、75、80、100、125、150、175、200、225、250、500、750、850及び1,000mgを含有する錠剤として組成物を提供することが好ましい。この投与レジメンは最適治療応答が得られるように調節することができる。
【0123】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)とその塩、水和物及び溶媒和物の結晶体のX線粉末回折パターンを作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した。
【0124】
N2流下に昇温速度10℃/分でTA Instruments DSC−2910示差走査熱量計を使用してDSCデータを取得した。TA Instruments DSC 2910又は同等機器。2〜6mgサンプルを開放アルミニウムパンに配量する。このパンを次にクリンプし、熱量計セルのサンプル位置に置く。閉鎖パンでサンプルを加熱する。空のパンを参照位置に置く。熱量計セルを閉じ、セルに窒素流を通す。サンプルを昇温速度10℃/分で約250℃の温度まで昇温するように昇温プログラムを設定する。昇温プログラムを開始する。プログラムが完了したら、システムソフトウェアに同梱のDSC分析プログラムを使用してデータを分析する。吸熱を観察する温度範囲の上下の基線温度点の間で融解吸熱を積分する。報告するデータは開始温度、ピーク温度及びエンタルピーである。
【0125】
Perkin Elmer TGA−7熱重量分析器を使用してTGAデータを取得した。5〜20mgサンプルを白金パンに配量する。炉を上昇させ、窒素流をサンプル上に流す。窒素流下に昇温速度10℃/分で約250℃の温度までサンプルを昇温するように昇温プログラムを設定する。昇温プログラムを開始する。プログラムが完了したら、システムソフトウェアに同梱の分析プログラムのΔY関数を使用してデータを分析する。昇温プログラムの開始からサンプルの融解/分解までサンプルによる百分率重量低下を計算する。
【0126】
下記スキーム及び実施例において、各種試薬記号及び略語は以下の意味である:acacはアセチルアセトナートであり;aqは水溶液であり;t−AmOHはtert−アミルアルコールであり;BINAPは2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチルであり;BuLi又はn−BuLiはブチルリチウムであり;Bu2Mgはジブチルマグネシウムであり;Bocはtert−ブトキシカルボニルであり;Boc酸無水物はtert−ブトキシカルボニル酸無水物であり;CBZはカルボベンジルオキシであり;CDIは1,1’−カルボニルジイミダゾールであり;DABCOは1,4−ジアザビシクロ[2.2.2]オクタンであり;DMEはエチレングリコールジメチルエーテルであり;DMFはジメチルホルムアミドであり;DMPUは1,3−ジメチル−3,4,5,6−テトラヒドロ−2(1H)ピリミジノンであり;dppbenzは1,2−ビス(ジフェニルホスフィノ)ベンゼンであり;dppeはジフェニルホスフィノエテンであり;dppf=(フェニル)2PC5H4FeC5H4P(フェニル)2であり、gはグラムであり;hは時間であり;GMPは製造管理及び品質管理規則であり;HClは塩酸であり;IPAはイソプロピルアルコールであり;IPAc又はiPAcは酢酸イソプロピルであり;i−ProHはイソプロパノールであり;KFはカール・フィッシャーであり;kgはキログラムであり;Lはリットルであり;LCAPは液体クロマトグラフィー分析純度であり;LDAはリチウムジイソプロピルアミドであり;LiHMDSはリチウムヘキサメチルジシラジドであり;Meはメチルであり;MeOHはメタノールであり;minは分であり;mLはミリリットルであり;molはモルであり;mmolはミリモルであり;MTBEはtert−ブチルメチルエーテルであり;Nはノルマルであり;PPTSはp−トルエンスルホン酸ピリジニウムであり;rtは室温であり;SL−J212−1は(R)−1−[(S)−2−ジ−2−フリルホスフィノ)フェロセニル]エチルジ−tert.−ブチルホスフィンであり,TBSClはtert−ブチルジメチルシリルクロリドてあり;TFAはトリフルオロ酢酸であり、THFはテトラヒドロフランである。
【0127】
新規方法を利用する代表的な実験手順を下記スキーム及び実施例に詳細に記載する。以下のスキーム及び実施例は本発明を例証するために記載するものであり、本発明の範囲を限定するとみなすべきではない。
【0128】
本発明は強力なCB−1逆アゴニスト化合物Iの効率的な合成方法について記載する。スキーム1に示すように、市販の3−ブロモベンゾイルクロリド1から完全に官能基付与したキラルベンズヒドリルアミンIIを合成する。DABCOとヒドラジンで処理することにより3−ブロモベンゾイルクロリド1をヒドラジド3に変換した。ヒドラジド3をCDIで処理してオキサジアゾール4を得、金属−ハロゲン交換反応によりアルデヒド5に変換した。PPTSと(S)−スルフィンアミドで処理することによりアルデヒド5をエルマンのイミン化合物6に変換した。次に化合物6のオキサジアゾール窒素をBoc基で保護してN−Bocイミン7を得た後、高度に立体選択的なRh触媒付加によりアリールボロキシン10をN−Bocイミン7に付加し、スルフィンアミド8を得た。保護オキサジアゾールの存在下でスルフィンアミド8を選択的に脱保護し、ベンズヒドリルアミンIIを得た。
【0129】
スキーム2はシアノジオールIII中間体の合成を示す。シアノジオールIIIは1,3−ジブロモフルオロベンゼン11から開始し、イソプロピルマグネシウムクロリドによるグリニャール形成後にCuCl/ZnCl2触媒により塩化イソブチリルに付加し、ケトン12を得た。ケトン12のシリルエノールエーテルの形成後、Select−Fluor(登録商標)フッ素化剤でin situフッ素化し、フルオロケトン13を得た。フルオロケトン13を炭酸カリウムとホスホノ酢酸トリメチルで処理し、ホーナー・ワズワース・エモンズ付加物であるα,β−不飽和エステル14を得た後、この化合物をNaOHでin situ加水分解し、α,β−不飽和酸15を得た。α,β−不飽和酸15のロジウム触媒不斉水素化により飽和酸16を得、in situエステル化後に飽和エステル17を得た。飽和エステル17のカルボキシル化によりβ−カルボキシエステル18を得、DME中で水素化ホウ素ナトリウム−臭素により還元することによりブロモジオール19に還元した。最後に、ブロモジオール19のパラジウム触媒シアノ化により、必要なシアノジオールIIIを得た。
【0130】
スキーム3は中間体20のアゼチジン環を導入するために使用するシアノジオールIIIとベンズヒドリルアミンIIの高度にコンバージェントなカップリングを示す。最後に、中間体20からN−Boc保護基を除去することにより化合物Iの合成を完了する。この経路における全中間体は結晶質であり、単離精製に関する本発明の利点である。
【0131】
【化34】
【実施例1】
【0132】
tert−ブチル−5−{3−[(S)−アミノ(4−クロロフェニル)メチル]フェニル]−2−オキソ−1,3,4−オキサジアゾール−3(2H)カルボキシレート(化合物II)の製造
ステップA:ヒドラジド3の製造。100L丸底フラスコに窒素下でDABCO(2.81kg,25.06mol)とMeOH(35L)を加えた。3−ブロモベンゾイルクロリド1(5.0kg,22.78mol)を20〜25℃で30分間かけて加え、氷水浴を使用して温度を制御した。混合物を室温で10〜20分間撹拌した。ヒドラジン(64%,8.8L,182mol)を20分間かけて加えた後、反応混合物を50〜55℃まで3時間加熱した。水(35L)を加え、室温で1時間かけてバッチを結晶化させた。得られたスラリーを室温で1〜2時間撹拌し、濾過した。湿潤ケーキを水洗し(3×15L)、室温で減圧/N2スイープ下に乾燥し、ヒドラジド3を白色固体として得た。ヒドラジド3のHPLC保持時間=6.75分(5ミクロン,4.6×250mmのWaters Symmetry C−18カラム);20℃,215nmでUV検出;勾配流速1.0mL/分;A=水+0.1%H3PO4;B=アセトニトリル;勾配溶出:0分:95%A/5%B;5分:55%A/45%B;10分:25%A/75%B;13分:10%A/90%B;及び26分:10%A/90%B。1H NMR(DMSO−d6):δ9.88(1H,s),7.98(1H,m),7.81(1H,m),7.70(1H,m),7.41(1H,m),4.52(2H,s)。
【0133】
ステップB:オキサジアゾール4の製造。75L丸底フラスコに窒素下で3−ブロモ安息香酸ヒドラジド3(3.5kg,16.3mol)とTHF(35L)を加えた。スラリーを室温で5〜10分間撹拌した。次に20〜25℃で10〜20分間かけてCDI(3.17kg,19.5mol)を加え、氷水浴を使用して温度を制御した。反応混合物は徐々に透明溶液となり、溶液を室温で2〜3時間撹拌した。IPAc(35L)と水(35L)を溶液に加えた。層分離し、水層をIPAc(15L)で抽出した。有機層を合わせて水(2×35L)、次いでブライン(20L)で洗浄し、濃縮した。濃縮中にスラリーが形成された。スラリーにヘプタン(2×10L)を流し、最終容量を30Lに調整した。得られたスラリー混合物を室温で1〜2時間撹拌後、濾過した。得られた湿潤ケーキをヘプタン(2×10L)で洗浄し、室温で減圧/N2スイープ下に乾燥し、オキサジアゾール4を白色固体として得た。オキサジアゾール4のHPLC保持時間=11.20分(5ミクロン,4.6×250mmのWaters Symmetry C−18カラム);20℃,215nmでUV検出;勾配流速1.0mL/分;A=水+0.1%H3PO4;B=アセトニトリル;勾配溶出:0分:95%A/5%B;5分:55%A/45%B;10分:25%A/75%B;13分:10%A/90%B;及び26分:10%A/90%B。1H NMR(400MHz,dmso−d6)δ12.65(1H,s),7.85(1H,s),7.72(2H,m),7.42(1H,t,J=7.8Hz)。
【0134】
ステップC:アルデヒド5の製造。75L丸底フラスコに窒素下でオキサジアゾール4(3.3kg,13.7mol)とTHF(33L)を加えた。溶液を−50℃まで冷却し、Bu2Mg(ヘプタン中1N,2N,10.3L,10.3mol)を−40〜−45℃で30〜50分間かけて加えた。ドライアイスアセトン浴を使用して温度を制御した。得られた不均質混合物を同一温度で1時間撹拌した。次にn−BuLi(ヘキサン中1.6M,10.3L,16.6mol)を−40〜−45℃で30分間かけて加えた。ドライアイスアセトン浴を使用して温度を制御した。スラリーを−40〜−45℃で2〜3時間撹拌した後、DMF(3.2L,41.1mol)を−40℃で1時間かけて加えた。反応混合物を0〜10℃まで昇温し、同一温度で3〜5時間撹拌した。バッチを0℃まで冷却し、バッチ温度を10℃未満に維持しながら2N HCl(10L)を20分間かけて加えることにより反応混合物をクエンチした。バッチを160Lエクストラクターに移し、EtOAc(33L)と2N HCl(23L)を加えた。得られた層を分離し、水層をEtOAc(10L)で抽出した。有機層を合わせて水(33L)、次いでブライン(20L)で洗浄し、濃縮し、ヘプタン(10L)を流した。スラリーを1:2 EtOAc/ヘプタン(12L)中で室温にて2時間撹拌し、濾過した。湿潤ケーキをヘプタン(2×8L)で洗浄し、室温で減圧/N2スイープ下に乾燥し、アルデヒド5を白色固体として得た。アルデヒド5のHPLC保持時間=8.73分(5ミクロン,4.6×250mmのWaters Symmetry C−18カラム);20℃,215nmでUV検出;勾配流速1.0mL/分;A=水+0.1%H3PO4;B=アセトニトリル;勾配溶出:0分:95%A/5%B;5分:55%A/45%B;10分:25%A/75%B;13分:10%A/90%B;及び26分:10%A/90%B。1H NMR(400MHz,dmso−d6)δ12.65(1H,s),10.04(1H,s),8.28(1H,s),8.11(2H,m),7.79(1H,m)。
【0135】
ステップD:N−tert−ブタンスルフィニルイミン6の製造。75L丸底フラスコに窒素下でアルデヒド5(2.1kg,11.0mol)と、(S)−tert−ブタンスルフィンアミド(1.46kg,12.1mol)と、PPTS(1.38kg,5.5mol)と、トルエン(20L)を加えた。得られたスラリーを40℃まで20時間加熱した。バッチを25℃まで冷却し、得られた固体を濾過した。湿潤ケーキをトルエン(3×15L)、水(3×15L)、次いでヘプタン(3×12L)で洗浄した。湿潤ケーキを室温で減圧/N2スイープ下に乾燥後、エルマンイミン6を白色固体として単離した。エルマンイミン6のHPLC保持時間=10.64分(5ミクロン,4.6×250mmのWaters Symmetry C−18カラム);20℃,215nmでUV検出;勾配流速1.0mL/分;A=水+0.1%H3PO4;B=アセトニトリル;勾配溶出:0分:95%A/5%B;5分:55%A/45%B;10分:25%A/75%B;13分:10%A/90%B;及び26分:10%A/90%B。1H NMR(400MHz,dmso−d6)δ12.63(1H,br s),8.62(1H,s),8.27(1H,s),8.09(1H,d,J=7.8Hz),7.95(1H,d,J=7.8Hz),7.69(1H,t,J=7.8Hz),1.20(9H,s)。
【0136】
ステップE:N−Bocイミン7の製造。75L丸底フラスコに窒素下でN−tertブタンスルフィニルイミン6(3.1kg,9.83mol)と、THF(39L)と、Et3N(2.5kg,24.7mol)と、Boc酸無水物(3.2kg,14.66mol)を加えた。得られた混合物を40℃に5時間、室温に8〜10時間加熱した。混合物を濃縮し、IPAc(25L)、次いでヘプタン(10L)を流した。濃縮中に固体が形成され、その後、混合物容量を約22Lに調整した。スラリーを室温で1〜2時間撹拌し、濾過した。得られた湿潤ケーキを1:4 IPAc/ヘプタン(15L)、1:10 IPAc/ヘプタン(15L)及びヘプタン(15L)で洗浄した。湿潤ケーキを室温で減圧/N2スイープ下に乾燥後、N−Bocイミン生成物7を白色固体して単離した。N−Bocイミン7のHPLC保持時間=14.00分(5ミクロン,4.6×250mmのWaters Symmetry C−18カラム);20℃,215nmでUV検出;勾配流速1.0mL/分;A=水+0.1%H3PO4;B=アセトニトリル;勾配溶出:0分:95%A/5%B;5分:55%A/45%B;10分:25%A/75%B;13分:10%A/90%B;及び26分:10%A/90%B。1H NMR(400MHz,CDCl3)δ8.64(1H,s),8.43(1H,s),8.09(1H,d,J=7.8Hz),8.02(1H,d,J=7.8Hz),7.63(1H,t,J=7.8Hz),1.69(9H,s),1.30(9H,s)。
【0137】
ステップF:ボロキシン10の製造。オーバーヘッドスターラー、蒸留装置(バッチ型濃縮器)を取付けた50L丸底フラスコに窒素下で4−クロロフェニルボロン酸9(3.6kg,23mol)とトルエン(30L)を加えた。得られたスラリーを沸騰するまで(95〜110℃)加熱し、共沸蒸留により水を除去した。容器内に一定容量を維持するために蒸留中にトルエン(合計約30〜35L)を補充した。溶媒約30Lの蒸留後、バッチを室温まで冷却し、0.5〜1時間撹拌した。生成物を濾過し、トルエン(2×10L)、次いでヘプタン(1×10L)で洗浄し、室温で減圧/N2スイープ下に乾燥し、ボロキシン10を白色固体として得た。
【0138】
ステップG:スルフィンアミド8の製造。75L丸底フラスコに窒素下でN−Bocイミン7(3.1kg,7.88mol)と、ボロキシン10(1.64kg,3.94mol)と、t−AmOH(62L)を加えた。反応混合物を45℃まで加熱し、1,2−ビス(ジフェニルホスフィノ)ベンゼン(93g,0.208mol)を加えた。得られた混合物に45℃で20分間窒素ガスをスパージ後、Rh(acac)(CH2CH2)2(45g,0.174mol)を加えた。反応混合物に同一温度で更に10分間窒素ガスをスパージした後、45℃に3〜5時間加熱した。バッチを室温まで冷却し、EtOAc(25L)で希釈した。EtOAc(20L)と0.5M Na2CO3水溶液(22L)を加えた160Lエクストラクターに得られた低粘度スラリーを移した。10分間室温で激しく混合後、層分離した。上部の有機層を3%ブライン(3×20L)、次いでブライン(20L)で洗浄した。有機溶液を25−30Lまで濃縮し、ヘプタン(20L)を流した。濃縮中に、生成物が沈殿した後、バッチ容量をヘプタンで約60Lに調整した。得られたスラリーを室温で1〜2時間撹拌後、濾過した。得られた湿潤ケーキを1:4 EtOAc/ヘプタン(2×20L)とヘプタン(30L)で洗浄した。次に湿潤ケーキを室温で減圧/N2スイープ下に乾燥し、スルフィンアミド8を薄黄色綿毛状固体として単離した。スルフィンアミド8のHPLC保持時間=15.15分(Phenomenex Synergi,4ミクロン,Hydro−RP 80A,250×4.6mm);20℃,215nmでUV検出;勾配流速1.0mL/分;A=水+0.1%H3PO4;B=アセトニトリル;勾配溶出:0分:95%A/5%B;5分:55%A/45%B;10分:25%A/75%B;13分:10%A/90%B;及び26分:10%A/90%B。1H NMR(400MHz,CDCl3)δ7.83(1H,s),7.74(1H,d,J=7.7Hz),7.55(1H,d,J=7.7Hz),7.39(1H,t,J=7.7Hz),7.26(4H,m),5.59(1H,s),4.04(1H,s),1.60(9H,s),1.21(9H,s)。
【0139】
ステップH:ベンズヒドリルアミンIIの製造。熱電対、窒素流導入管及びオーバーヘッドスターラーを取付けた100L容器にスルフィンアミド8(3.0kg,5.93mol)とジクロロメタン(15L)を加えた。溶液にHClのi−PrOH溶液(0.62Mを10Lと0.65Mを6.8L,10.08mol)を加えた。室温で3時間エージング後に反応は完了した。反応混合物を水(30L,発熱)でクエンチした後、飽和NaHCO3水溶液(15L)をゆっくりと加え、CO2発生を抑えた。混合物を室温で30分間撹拌した。次に反応混合物を100Lエクストラクターに移し、ジクロロメタン(6L)でリンスした。層分離した。有機層を1ミクロンWhatmanインラインフィルターで濾過しながら75L容器に注入した。溶液を濃縮し、内部温度〜45℃で更にi−PrOH(6L)を加えて共沸蒸留し、粘稠スラリーを得た後、ジクロロメタン18Lを加えて〜8Lまで濃縮することにより溶媒をジクロロメタンに交換した。次に混合物を35℃まで加熱し、全固形分を完全に溶解し、貧溶媒としてのi−PrOH(24L)を滴下漏斗によりゆっくりとバッチに加えた。i−PrOHの添加後、バッチを周囲温度まで一晩ゆっくりと冷却した。次に温度を2時間0〜5℃まで下げた。次に固形分を濾過し、得られたケーキスラリーをi−PrOH(2×7.5L)とn−ヘプタン(3×9L)で洗浄した。週末にかけて、底から窒素スイープと真空引きしながらケーキをフィルターポットで乾燥し、ベンズヒドリルアミンIIを得た。ベンズヒドリルアミンIIのHPLC保持時間=8.95(Zorbax RX C8,5.0ミクロン,4.6mm×250mm,P.N.:880967);温度:25℃,210nmでUV検出,カラム流速は1.0ml/分とし、溶媒Aはアセトニトリルとし、溶媒Bは0.1%H3PO4緩衝H2Oとする;勾配溶出:0分:10%A/90%B;1分:90%A/10%B;及び15分:98%A/2%B。1H(CDCl3)δ7.99(t,J=1.6Hz,1H),7.80(dt,J=7.6,1.6Hz,1H),7.57(dt,J=7.6,1.6Hz,1H),7.44(t,J=7.6Hz,1H),7.33(m,2H),7.27(m,2H),5.26(s,1H),1.77(brs,2H),1.66(s,9H)。1H NMR(400MHz,CDCl3)δ8.00(1H,s),7.80(1H,d,J=7.7Hz),7.08(1H,d,J=7.7Hz),7.44(1H,t,J=7.7Hz),7.30(4H,m),5.26(1H,br s),1.60(2H,br s),1.68(9H,s)。
【0140】
【化35】
【実施例2】
【0141】
3−フルオロ−5−{(1S)−2−フルオロ−1−[2−ヒドロキシ−1−(ヒドロキシメチル)エチル]−2−メチルプロピル}ベンゾニトリル(化合物III)の製造
ステップA:イソプロピルケトン12の合成。熱電対と5L滴下漏斗を取付けた50L丸底フラスコに窒素雰囲気下でZnCl2(32.7g,0.24mol)とCuCl(23.3g,0.24mol)を加えた。THF(12L)を加え、N2をスラリーに5分間バブリングすることにより溶液を脱気した。その後、2,3−ジブロモフルオロベンゼン11(6.0kg,24mol)を一度に加えた。反応混合物を10℃まで冷却し、温度を15〜20℃に維持しながらイソプロピルマグネシウムクロリド(2M,14L,28mol)を1時間20分間かけて滴下した。100L円筒形容器に塩化イソブチリル(3.3kg,31mol)とTHF(6L)を加えた。混合物を6℃まで冷却後、温度を10℃〜18℃に維持しながら、予め調製しておいたアリールグリニャール溶液を70分間かけてポンプ導入した。添加の完了後、反応混合物を20分間エージングさせた後、変換についてアッセイした。変換の完了後、反応混合物をトルエン(11L)と1Nクエン酸(10L)で希釈した。2相混合物を10分間撹拌後、層分離した。次に有機層を1Nクエン酸(10L)で処理し、更に10分間撹拌した。層分離し、有機層を1N K2HPO4(2×11L)と水(11L)で順次洗浄した。次に同一サイズの第2のバッチを処理しながら、湿潤トルエンバッチを36時間保持した。次にイソプロピルケトン12を含む湿潤トルエンバッチを合わせ、約2〜3容量及びKF<200ppmまで濃縮し、次段階で直接使用した。イソプロピルケトン12のHPLC保持時間=8.1分(25cm Zorbax SB C−18でMeCN/0.1 H3PO4を1mL/分で使用);勾配溶出:70%MeCN 5分後、10分で90%MeCNまで;210nm;5分間保持。1H NMR(400MHz,CDCl3)δ7.85(1H,br.s,Ar−H),7.56(1H,dd,8.9Hz,1.0Hz,Ar−H),7.43(1H,dd,8Hz,1.0Hz,Ar−H),3.43(1H,sep.6.9Hz,C−H),1.21(6H,d,6.9Hz,(CH3)2CH)。
【0142】
ステップB:フルオロケトン13の合成。熱電対を取付けた100L丸底フラスコにN2雰囲気下でナトリウムアミラート(3.0kg,27.3mol)とDMF(11L)を順次加えた。塩基のほぼ全部が溶解するまでスラリーを30分間エージングさせた。次に溶液を10℃まで冷却し、イソプロピルケトンを制御下に添加することにより温度を15〜20℃に維持しながら、イソプロピルケトン12の〜50重量%溶液(10.8kg,50重量%,5.4kg,22mol)を55分間かけて加えた。添加の完了後、溶液を30分間エージングさせ、10℃まで冷却し、TBSClの制御下の添加により温度を20〜28℃に維持しながら、TBSCl(4.3kg,28.5mol)で30分間処理した。反応混合物を30分間エージングさせた後、反応温度を28〜35℃に維持しながらSelect−Fluor(登録商標)(8.5kg,24mol,Air Products)を1.5時間かけて加え、スラリーを1時間エージングさせた。完全に変換したら、水(18L)を加え、次いでトルエン(12L)を加えた。得られた2相混合物を100L円筒形容器に移し、10分間エージングさせた。層分離し、有機相を更に水洗(9L)した。同一サイズの第2のバッチを処理しながら、フルオロケトン13を含有する湿潤トルエン溶液を36時間保持した。次にフルオロケトン13を含有するトルエンバッチを合わせ、KF<200ppm及び約50重量%まで濃縮した。得られたフルオロケトン13のトルエン溶液を次段階で直接使用した。フルオロケトン13のHPLC保持時間=8.5分(25cm Zorbax SB C−18でMeCN/0.1 H3PO4を1mL/分で使用);勾配溶出:70%MeCN 5分後、10分で90%MeCNまで、210nmで5分間保持。IRvmax/cm−13083(C−H),2989(C−H),2941(C−H),1696(C=O),1577(C=C);1H NMR(500MHz,CDCl3)δ8.02(1H,s,ArH),7.74(1H,br d,JHF=9.1Hz,ArH),7.65(1H,dt,JHF=7.66Hz,JH,H=2.0Hz,ArH),1.69(6H,d,JH,F=21.7Hz,2×CH3);13C NMR(125MHz,CDCl3)δ198.2(dd,J=27.1Hz,1.8Hz,C=O),162.2(d,J=252.3Hz,ArCF),136.9(dd,J=6.8,4.3Hz,ArCCO),128.9(dd,J=8.6,3.1Hz,ArCH),123.5(d,J=24.6Hz,ArCH),122.8(dd,J=9.2,1.2Hz,ArCBr),115.8(dd,J=22.8,9.2Hz,ArCH),99.9(d,J=180.3Hz,C(CH3)2),25.5(d,J=24.0Hz,2×CH3);19F NMR(470MHz,CDCl3)δ−109.96,Ar−F),−144.68(C−F)。
【0143】
ステップC:α,β−不飽和エステル14の合成。熱電対を取付けた50L丸底フラスコにN2雰囲気下でDMF(10L)と、炭酸カリウム(5.3kg,38.4mol)と、ホスホノ酢酸トリメチル(5.3kg,29.1mol)を順次加えた。スラリーを70℃まで加熱し、フルオロケトン13のトルエン溶液(50重量%溶液9.9kg,4.96kg,18.8mol)で46分間処理した。反応混合物を80〜82℃まで加熱し、この温度で10時間エージングさせた。次に反応混合物を室温まで冷却し、一晩10時間エージングさせた。フルオロケトン13が完全に消費されたら、2Nクエン酸(20L)とトルエン(10L)を加えた100L円筒形容器にスラリーを加えて逆クエンチした。層を10分間混合後、分離した。α,β−不飽和エステル14を含むトルエン有機相を水洗(10L)し、次段階で使用した。
【0144】
ステップD:α.β−不飽和酸15の合成。熱電対を取付けた50L丸底フラスコにN2雰囲気下でステップCのα,β−不飽和エステル14を含有するトルエン溶液を加えた。次に、メタノール(17L)と5N NaOH(8L)を加えた。混合物を45℃まで加熱し、3.5時間迅速に撹拌した。次に、反応混合物を20℃まで冷却し、ヘプタン(15L)と水(15L)を加えた100L円筒形容器に移した。得られた2相混合物を10分間迅速に撹拌し、相分離した。水相をヘプタン(10L)で洗浄し、層分離した。水層を50L丸底フラスコに加え、pH=5になるまで5N HClで酸性化した。次に、α,β−不飽和酸15シード(60g)を加え、5N HClの滴下により溶液をpH2まで1時間かけてゆっくりと酸性化した。固体α,β−不飽和酸15を濾取し、1:1メタノール/水(15L)で洗浄し、ケーキからN2を36時間吸引することにより乾燥した。同一規模の第2のバッチを処理し、α,β−不飽和酸15をオフホワイト固体として得た。α,β−不飽和酸15のHPLC保持時間=13.3分(25cm Zorbax SB C−18でMeCN/0.1 H3PO4を1mL/分で使用);勾配溶出:10%MeCN−50%MeCN 0〜5分後、5〜20分間で50→90%MeCN,210nmで更に5分間90%MeCN保持。IRvmax/cm−1(膜)3087(O−H),2986(C−H),2940(C−H),1703(C=O),1645(C=C);1H NMR(500MHz,CDCl3)δ7.25(1H,dt,JH,F=8.3Hz,JH,H=1.9Hz,Ar−H),7.06(1H,s,Ar−H),6.81(1H,br.dt,JH,F=8.7Hz,JH,H=1.6Hz,Ar−H),6.27(1H,s,C=CH),1.5(6H,d,JH,F=21.9Hz);13C NMR(125MHz,CDCl3)δ170.0(C=O),162.0(d,J=251.0Hz,ArC−F),159.7(dd,J=19.1Hz,J=1.8Hz,C−CO2H),139.4(dd,J=8.6Hz,J=3.1,ArCC),126.7(d,J=3.1Hz,ArCH),122.1(d,J=3.1Hz,ArCBr),118.6(d,J=24Hz,ArCH),117.0(d,=13.5Hz,C=CCO2H),114.3(d,J=22.8Hz,ArCH),95.3(d,J=179.0Hz,C−F),26.4(d,J=25.2Hz,2xCH3);19F NMR(470MHz,CDCl3)δ−110.4(Ar−F),−138.09(C−F)。
【0145】
ステップE:α,β−飽和酸16の合成。α,β−不飽和酸15(2.2kg,7.2mol)を4Lビーカーに配量し、ポリ容器に移した。次にビーカーをMeOH(2L)でリンスし、溶液をポリ容器に移した。次に、トリエチルアミン(252mL,1.8mol)を加え、漏斗を残りのMeOH(7L)でリンスした。激しく混合後、バッチを5ガロン容器に移した後、容器を脱気した。次に触媒を次のように調製した:[Ru(シメン)Cl]2前駆体(1.7g,0.0055mol,0.076mol%)と(R)−1−[(S)−2−ジ−2−フリルホスフィノ)フェロセニル]エチルジ−tert−ブチルホスフィン(ないしSL−J212−1)リガンド(3.1g,0.006mol,0.083mol%)をN2雰囲気下で100mL丸底フラスコに加えた。次に、固形分を1:3 CH2Cl2/MeOH(21mL)に溶解し、磁気撹拌棒を使用して1時間撹拌した。1時間エージング後、MeOH 20mLを使用して触媒溶液をステンレス鋼レザバーに移した。更にMeOH 40mLをリンス用にステンレス鋼レザバーに移した。レザバーを密閉後、溶液を200psig下に50℃にてH2で10時間処理し、92%eeのα,β−飽和酸16のメタノール溶液を得、次段階で使用した。
【0146】
ステップF:α,β−飽和エステル17の製造。100L丸底フラスコにα,β−飽和酸16(4.50kg,14.6mol)のメタノール溶液を加えた。溶液を−15℃まで冷却後、ドライアイス/アセトン浴で温度を−15℃〜−5℃に維持しながら塩化アセチル(2.2kg,2.0L,28.6mol)を30分間かけて滴下した。次に反応混合物を室温まで昇温し、HPLCにより完了したと判断されるまで3時間エージングさせた。溶液を100L円筒形容器に移した。トルエン(20L)を加えた後、7%NaCl水溶液(18L)を加えた。得られた2相溶液を30℃まで加熱し、10分間撹拌した。次に相を分離し、α,β−飽和エステル17を含む有機相をK2HPO4溶液(10L)で洗浄した。α,β−飽和酸16の第2の4.5kgバッチでこのプロセスを反復した。α,β−飽和エステル17を含有する湿潤トルエンバッチを合わせ、約2容量まで濃縮した。α,β−飽和エステル17のHPLC保持時間=7.5分(ACE−111−15030カラムで勾配溶出:0.1% H3PO4水溶液/CH3CN 60%→20%0〜6分後、20%→5%6〜8分を使用);38℃,220nm。1H NMR(400MHz,CDCl3)δ7.22(1H,br.s,Ar−H),7.15(1H,dt,6.1,1.9Hz,Ar−H),6.94(1H,br,d,9.5Hz,Ar−H),3.58(3H,s,OCH3),3.28(1H,ddd,20.6,10.2,4.5Hz,CH),2.88(1H,dd,16.2,4.5,CH2),2.73(1H,dd,16.2,10.2Hz,CH2),1.38(3H,d,21.4Hz,CH3),1.23(3H,d,21.6Hz,CH3)。
【0147】
ステップG:β−カルボキシエステル18の合成。75L丸底フラスコにα,β−飽和エステル17のトルエン溶液(3〜4容量,合計18L)を加えた後、DMPU(1.7kg,13.3mol)を加えた。溶液を−43℃まで冷却後、ドライアイス/アセトン浴で発熱を抑えながら、LiHMDS(12.2L,1.3M,15.9mol)のTHF溶液を1時間かけてゆっくりと加えた。完了後、反応混合物を−66℃まで冷却し、反応温度を<−58〜−60℃に維持しながら乾燥CO2ガスを溶液にゆっくりと1時間バブリングした。反応混合物を−66℃まで再冷却し、水性混合物の温度を0〜15℃に維持しながら水に逆クエンチした。有機相を分離した(水相pH=10〜11)。次にトルエン(18L)を水層に加えた後、pHが<3になるまでH3PO4(85重量%,3.5L)をゆっくりと加えた。水相を分離し、有機相を2.5N HCl(11kg)で2回洗浄した。上記手順を使用して更に4.1kgのα,β−飽和エステル17を処理した。次にβ−カルボキシエステル18を含有するトルエン抽出相を合わせて約3〜4容量まで濃縮した。β−カルボキシエステル18のHPLC保持時間=12.0分(4.6*150mm,直径5μmのZorbax Eclipse XD8−C8カラムでH2O中CH3CN/0.1%H3PO4を使用);1ml/分,5μl注入,210nm,勾配溶出:t=0分,90%H2O/CH3CN;t=15分,10%H2O/CH3CN;及びt=20分,10%H2O/CH3CN。
【0148】
ステップH:ブロモジオール19の合成。100L丸底フラスコにN2下でDME(23.9kg)を加えた後、NaBH4(2.2kg,59.2mol.)を加えた。得られたスラリーを−20℃まで冷却後、温度を−10℃〜−20℃に維持しながら滴下漏斗で2.5時間かけて臭素(1.4L,26.5mol)を加えた。臭素の添加後、溶液を2時間エージングさせた後、10℃まで昇温した。次に添加中に反応温度を≦35℃に維持するように氷/水浴で冷却しながらβ−カルボキシエステル18のDME/トルエン溶液(10.6kg溶液,推定値4.4kg,12.0mol)を臭素溶液に2時間かけてゆっくりと加えた。次に反応混合物を19時間室温でエージングさせた後、HPLC分析はβ−カルボキシエステルが完全に消費されたことを示した。発熱を抑えるように冷却しながら白色不均質混合物をトルエン(4L)と2N K2CO3(38.6kg)に5℃で逆クエンチした。更にトルエン(7L)を使用してガラス容器をリンスした。トルエン溶液を合わせて35℃まで2時間昇温後、室温まで放冷させ、15時間エージングさせた。層分離し(水溶液pH=11〜12)、有機層を0.5N HCl(9.4kg;水溶液pH=0〜1)で洗浄し、次いで2N K2CO3(22kg,5分間エージング;水溶液pH=13)と2N K2CO3(9.1kg,5分間エージング,水溶液pH=13〜14)で洗浄した。次に層分離し、ブロモジオール19を含有する湿潤DME/トルエン層を得た。4.4kgのβ−カルボキシエステル18の第2のバッチを同様に処理した。ブロモジオール19を含有するDME/トルエン層を合わせ、約2容量まで蒸留させた。ブロモジオール19のHPLC保持時間=4.36分(ACE−111−1503,勾配溶出:60%−5% 0.1%H3PO4,220nm)。1H NMR(400MHz,CDCl3)δ7.26−7.24(1H,m,Ar−H),7.17−7.14(1H,m,Ar−H),6.91(1H,dt,9.7,1.7Hz,Ar−H),3.89(1H,dd,11.0,6.0Hz,CH),3.81−3.69(2H,m,2xCH),3.29(1H,dd,11.0,7.3Hz,CH),3.26−3.20(2H,m,CH,OH),2.99(1H,dd,22.3,8.3,CH),2.32−2.25(1H,m,OH),1.37(3H,d,21.8Hz,CH3),1.31(3H,d,22.3Hz,CH3)。
【0149】
ステップI:シアノジオールIIIの合成。粗ブロモジオール19(5kg,15.5mol)のトルエン(〜20重量%)溶液をDMF(2.5容量,約12.5L)に溶媒交換し、HPLCにより<5LCAPと判断されるまでトルエンを還元した。Zn粉末(615g,9.4mol,<10ミクロン,Aldrich)とDMF(3容量,約15L)のスラリーに室温でN2下に臭素(90mL,1.75mol)をゆっくりと加えた。混合物を室温で20分間撹拌すると、この間にオレンジ色から無色に変色した。次に、Zn(CN)2(1.1kg,9.4mol)と、ブロモジオール19の溶液(DMF2.5容量に溶解,DMF0.5容量でリンス)と、PPh3(485g,1.85mol)と、Pd(OAc)2(103g,0.46mol)を順次加え、表面下窒素流を使用して得られた混合物を30分間脱気した。次に反応混合物を窒素雰囲気下で80℃まで昇温した後、3時間エージングさせた後、室温まで冷却し、一晩エージングさせた。混合物を30%NH4OH(0.84容量)で処理し、1時間エージングさせた。得られたスラリーをSolka Floc(登録商標)で濾過し、濾過層をIPAc(85L)で洗浄した。10%NH4OH(60L)を加えたエクストラクターに濾液を移した。有機溶液を5%NaCl(30L)と水(30L)で1回洗浄した。IPAcが<1mol%(GCによるアッセイ)に低下するまで〜40℃にて一定容量で有機層をトルエン(〜9L/kgシアノジオール)に溶媒交換した。ヘプタン(5L/kgシアノジオール)を〜45℃で加え、得られたスラリーを室温まで冷却後、0℃まで冷却した。スラリーを濾過し、30:70ヘプタン:トルエン(15L)で洗浄し、シアノジオールIIIを白色固体(98LCAP,95%ee)として得た。シアノジオールIIIのHPLC保持時間=8.9分(4.6*250mm,直径5μmのWaters Symmetry C18);溶離液:H2O中CH3CN/0.1%H3PO4;1ml/分,5μl注入,210nm,勾配溶出:t=0分,65%H2O/CH3CN;t=4分,65%H2O/CH3CN;t=25分,20%H2O/CH3CN;t=26分,0%H2O/CH3CN;及びt=30分 0%H2O/CH3CN。αD=+26.0°;IRcm−(膜)3395(O−H),2912(C−H,2233(CN),1593,1439;1H NMR(400MHz,CDCl3)δ7.37(1H,t,J=1.4Hz,Ar−H),7.31−7.25(2H,m,Ar−H),3.98(1H,dd,J=11.2,5.3Hz),3.84(1H,dd,J=11.0,3.2Hz),3.76(1H,ddd,J=11.2,4.7,1.0Hz),3.28(1H,dd,J=11.0,6.7Hz),3.20(1H,dd,J=23.2,8.1Hz),1.37(3H,d,J=21.7Hz),1.36(3H,d,J=22.2Hz);13C NMR(125MHz,CDCl3)δ163.3,160.8,144.0,143.9,143.85,129.4,122.0,121.7,118.1,117.8,117.59,117.56,113.85,113.75,99.0,97.31,64.52,64.47,63.78,63.76,52.13,52.11,51.90,51.89,43.01,43.00,28.13,27.89,24.80,24.55;19F NMR(470MHz,CDCl3)δ−109.6(Ar−F),−137.5(C−F);m/z 269.1227;HRMS 269.1235。
【0150】
【化36】
【実施例3】
【0151】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)無水遊離塩基多形体Iの製造
ステップA:アゼチジン20の製造。熱電対、滴下漏斗及びオーバーヘッドスターラーを取付けた75L容器にシアノジオールIII(0.94kg,3.48mol)とアセトニトリル(14L)を加え、アセトン/ドライアイスを使用して内容物を−30℃まで冷却した。溶液にトリフル酸無水物(1.29L,7.66mol)をゆっくりと加えた後、ジイソプロピルエチルアミン(1.52L,2.5eq)をゆっくりと加えた。どちらの添加も内部温度を−20℃未満に維持しながら実施した。−30℃で1時間後にビストリフラートを形成する反応が完了したことがHPLCにより確認された。更にジイソプロピルエチルアミン(1.52L,2.5eq)をゆっくりと加えた後、内部温度を−20℃未満に維持しながら、ベンズヒドリルアミンII(1.4kg,3.48mol)をジクロロメタン(5.6L)溶液として滴下漏斗によりゆっくりと加えた。次に滴下漏斗をジクロロメタン(0.7L)でリンスした。冷却浴を除去し、反応混合物を室温まで一晩昇温した。次に反応混合物を濃縮し、IPAc 28Lを使用して溶媒をIPAcに交換し、最終容量〜10.5Lまで濃縮した。得られたスラリーをヘプタン(5.6L)で希釈し、IPAc/ヘプタンの〜3:2混合物を得た。室温で一晩エージング後、スラリーを濾過し、ジイソプロピルエチルアミントリフル酸塩を除去し、得られた塩ケーキをIPAc/ヘプタンの3:2混液(3×4.9L)で洗浄した。濾液を〜15Lまで濃縮し、Nuchar Aquaguard Powder(登録商標)(0.56kg)で処理した。混合物を1時間かけて50℃まで加熱した後、室温まで一晩冷却した。得られたスラリーをSolka Floc(登録商標)パッド(1.4kg)で濾過し、ケーキをIPAc(5×2.8L)で洗浄した。濾液を50Lエクストラクターに移し、GMP水(1×7L)で洗浄した。1ミクロンインラインフィルターを通して有機層を50L容器に移した。濾液を〜10Lまで濃縮後、IPAc 42Lを使用して溶媒をIPAcに交換し、〜6.4Lまで濃縮した。溶液を室温まで一晩冷却した。ヘプタン(8.4L)を1時間かけてゆっくりと加えた後、シード(7g)を加えた。1時間室温でエージング後、更にヘプタン(8.4L)を2時間かけてゆっくりと加えた後、室温で1時間エージングさせた。室温で1時間かけてヘプタン(4.2L)を加え、スラリーを室温で一晩エージングさせた。こうして2種類の結晶(微結晶と顆粒状結晶)を得た。濾過前に温度を2時間で0〜5℃まで下げた。スラリーを濾過し、得られたケーキスラリーをIPAc/ヘプタン1:5混液(1×2.8L)、次いでヘプタン(2×2.8L)で洗浄した。底から窒素スイープと真空引きしながらケーキをフィルターポットで一晩乾燥した。得られたアゼチジン20は純度92wt%及び97.8LCAPで得られた。アゼチジン20のHPLC保持時間=11.23(Zorbax RX C8,Analytical 5.0ミクロン,4.6mm×250mm,P.N.:880967−901);温度:25℃;210nmでUV検出;カラム流速は1.0ml/分とし;溶媒Aはアセトニトリルとし;溶媒Bは0.1%H3PO4緩衝H2Oとする,勾配溶出:0分:10%溶媒A,90%溶媒B;10分:90%溶媒A,10%溶媒B;15分:98%溶媒A,2%溶媒B。1H(CDCl3)δ7.92(t,J=1.6Hz,1H),7.72(dt,J=7.6,1.6Hz,1H),7.53(d,J=7.6Hz,1H),7.37(d,J=7.6Hz,1H),7.34(m,2H),7.28−7.25(om,3H),7.21(ddd,J=7.6,2.4,,1.4Hz,1H),7.16(m,1H),4.27(s,1H),3.61(t,J=6.4Hz,1H),3.16(m,1H)3.05(t,J=6.4Hz,1H),2.91(dd,J=19.1,11.0Hz,1H),2.85(t,J=7.6Hz,1H),2.27(t,J=7.6Hz,1H),1.66(s,9H),1.27(d,J=21.3Hz,3H),1.19(d,J=21.5Hz,3H)。19F(CDCl3)δ−110.0,−144.3。
【0152】
ステップB:化合物Iの製造。熱電対、滴下漏斗及びオーバーヘッドスターラーを取付けた50L容器にアゼチジン20(1.85アッセイkg,2.91mol)と、IPAc(5.6L)と、HClのIPA溶液(7.4L,4.55M)を温和な吸熱(〜6℃)下に加えた。HPLCによると、室温で一晩エージング後に反応は完了した。飽和NaHCO3(30.0L)を加えた100Lエクストラクターに温和な発熱(〜5℃)下にバッチを移すことにより反応混合物をクエンチした。更に飽和NaHCO3(5.0L,合計35.0L)をゆっくりと加え、pHを〜7に調整した。更にIPAc(13.0L)を加えた後、層分離し、有機層をGMP水(1×9.3L)で洗浄した。1ミクロンインラインフィルターを通して有機層を50L容器に移した。有機層を〜10Lまで濃縮し、IPAc 18Lを使用して溶媒をIPAcに交換し、〜18Lまで濃縮した。次に混合物をNuchar Aquaguard Powder(登録商標)(0.45kg)で処理し、50℃まで1時間加熱後、室温まで一晩冷却した。得られたスラリーをSolka Floc(登録商標)パッド(1.85kg)で濾過し、ケーキをIPAc(4×7.0L)で洗浄した。1ミクロンインラインフィルターを通して有機層を50L容器に移し、〜6Lまで濃縮し、トルエン28Lを使用して溶媒をトルエンに交換した。得られた溶液を〜9.0Lまで濃縮し、45℃までゆっくりと加熱し、内部温度を40〜45℃に維持しながらヘプタン(1.9L)で1時間かけてゆっくりと処理した。バッチにシード添加し(7g)、1時間45℃でエージングさせた。更にトルエン(1.0L)を加えた後、反応温度を40〜45℃に維持しながら、ヘプタン(1.9L)を45℃で2時間かけてゆっくりと加えた。45℃で1時間エージング後、反応混合物を室温まで一晩冷却した。スラリーを濾過し、ケーキスラリーをトルエン/ヘプタンの3:2混液(1×6.0L)、次いでヘプタン(2×6.0L)で洗浄した。次に底から窒素スイープと真空引きしながらケーキをフィルターポットで一晩乾燥し、純度96.8wt%及び98.6LCAPで化合物Iを得た。化合物IのHPLC保持時間=9.54(Zorbax RX C8,Analytical 5.0ミクロン,4.6mm×250mm,P.N.:880967−901);温度:25℃;210nmでUV検出;カラム流速:1.0ml/分;溶媒A:アセトニトリル;溶媒B:0.1%H3PO4緩衝H2O,勾配溶出:0分:10%溶媒A,90%溶媒B;10分:90%溶媒A,10%溶媒B;15分:98%溶媒A,2%溶媒B。1H(CDCl3)δ10.85(brs,1H),7.99(t,J=1.6Hz,1H),7.67(dt,J=7.6,1.6Hz,1H),7.46(d,J=7.6Hz,1H),7.39−7.35(om,3H),7.30−7.28(om,3H),7.24−7.18(om,2H),4.35(s,1H),3.70(t,J=6.8Hz,1H),3.33(m,1H),3.1 1(td,J=7.6,1.6Hz,1H),3.01−2.93(om,2H),2.40(t,J=8.0Hz,1H),1.27(d,J=21.3Hz,3H),1.22(d,J=21.7Hz,3H)。19F(CDCl3)δ−109.7,−142.7。
【実施例4】
【0153】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)遊離塩基トルエン/ヘプタン溶媒和物多形体I,タイプBの製造
化合物I 3.36kgをトルエン20Lに溶解し、1ミクロンインラインフィルターで濾過した。スラリーを53℃まで加熱後、45℃まで冷却した。次に、10重量%シードスラリー(化合物I 101gを6:4トルエン/ヘプタンに媒体で粉砕)1.01kgを混合物に加えた後、6:4トルエン/ヘプタン(779g)を加えた。得られたスラリーを1時間エージングさせた。較正済みEncynova(登録商標)配量ポンプを使用してヘプタン(10.1L)を4時間40分かけて38mL/分の速度で加えた。得られたスラリーを室温まで冷却し、一晩エージングさせた。混合物を濾過し、6:4トルエン/ヘプタン(14L)で洗浄した。次に得られたケーキをヘプタン28Lで洗浄し、化合物Iを結晶質遊離塩基トルエン/ヘプタン溶媒和物形態I,タイプBとして得た。
【0154】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体I,タイプBのX線粉末回折スペクトル(図13)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(2θ),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0155】
【表1】
【0156】
化合物Iの無水遊離塩基トルエン/ヘプタン溶媒和物多形体I,タイプBは表1に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体I,タイプBは角度θ値4.7°により同定することができる。化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体I,タイプBは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)4.7°;
b)4.7°及び7.1°;
c)4.7°、7.1°及び8.4°;
d)4.7°、7.1°、8.4°及び8.7°;
e)4.7°、7.1°、8.4°、8.7°及び9.5°;
f)4.7°、7.1°、8.4°、8.7°、9.5°及び11.6°;
g)4.7°、7.1°、8.4°、8.7°、9.5°、11.6°及び14.3°;
h)4.7°、7.1°、8.4°、8.7°、9.5°、11.6°、14.3°及び15.4°;
i)4.7°、7.1°、8.4°、8.7°、9.5°、11.6°、14.3°、15.4°及び17.3°。
【0157】
化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体I、タイプBはCu線を使用して得られたX線粉末回折パターンからd間隔18.800、12.450、10.526、10.163、9.309、7.628、6.194、5.754及び5.126Åの1個以上の反射により同定することもできる。
【0158】
Perkin Elmer TGA−7機器で窒素流下に昇温速度10℃/分で化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体I,タイプBの熱重量(TG)分析曲線(図14)を得た。
【実施例5】
【0159】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)無水遊離塩基多形体Iの製造
実施例4に記載したように製造したヘプタン/トルエン溶媒和物を乾燥することにより化合物Iの無水遊離塩基形態Iを得た。フィルターポットを一晩乾燥後、白色固体をトレーに移し、窒素スイープ下に25℃/75Torrで減圧乾燥し、化合物Iを98.62LCAP及び>99%eeの無水遊離塩基多形体Iとして得た。
IR:vmax(cm−1)(膜)。1H NMR(400MHz,CDCl3)δ11.5−11.8(1H,bs,NH),8.07(1H,s,ArH),7.66(1H,d,J=7.7Hz,ArH),7.47(1H,d,J=7.74,ArH)7.39−1.21(8H,m,ArH),4.44(1H,s,ArArCHN),3.75(1H,dd,J=6.8,6.7Hz,NCHH),3.43−3.37(1H,m,NCH2CH),3.16(1H,dd,J=6.8,6.7Hz,NCHH),3.06−2.99(2H,m,NCHH,ArCHCH),2.49(1H,dd,J=8.2,8.1Hz,NCHH),1.26(3H,d,J=21.7Hz,CH3CF),1.22(3H,d,J=21.7Hz,CH3CF)。13C NMR(100MHz,CDCl3)δ163.3,160.8,155.7,154.9,143.4,143.3,143.3,141.6,138.9,133.7,131.1,129.5,129.1,128.8,125.3,125.0,124.5,121.5,121.3,118.1,117.8,117.6,117.6,113.7,113.6,97.2,95.5,77.6,61.1,59.4,59.2,58.2,30.3,25.9,25.7,25.3,25.1。19F NMR(376MHz,CDCl3)δ−109.7(Ar−F),−142.3(C−F)。
【0160】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物Iの無水遊離塩基多形体IのX線粉末回折スペクトル(図1)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(2θ),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0161】
【表2】
【0162】
化合物Iの無水遊離塩基多形体Iは表2に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物Iの無水遊離塩基多形体Iは角度θ値5.2°により同定することができる。化合物Iの無水遊離塩基多形体Iは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)5.2°;
b)5.2°及び15.4°;
c)5.2°、15.4°及び9.3°;
d)5.2°、15.4°、9.3°及び17.4°;
e)5.2°、15.4°、9.3°、17.4°及び7.0°;
f)5.2°、15.4°、9.3°、17.4°、7.0°及び15.7°;
g)5.2°、15.4°、9.3°、17.4°、7.0°、15.7°及び22.5°;
h)5.2°、15.4°、9.3°、17.4°、7.0°、15.7°、22.5°及び11.8°;
i)5.2°、15.4°、9.3°、17.4°、7.0°、15.7°、22.5°、11.8°及び16.4°。
【0163】
化合物Iの無水遊離塩基多形体IはCu線を使用して得られたX線粉末回折パターンからd間隔16.994、5.754、9.509、5.096、12.627、5.644、3.951、7.499及び5.405Åの1個以上の反射により同定することもできる。
【0164】
Perkin Elmer TGA−7機器で窒素流下に昇温速度10℃/分で化合物Iの無水遊離塩基多形体Iの熱重量(TG)分析曲線(図2)を得た。減量は融点までで<0.1%であった。TA Instruments DSC−2910示差走査熱量計でN2流下に昇温速度10℃/分で化合物Iの無水遊離塩基多形体IのDSC曲線(図3)を得た。サンプルを閉鎖パンで加熱した。開始温度157.8C、ピーク温度163.6C及びそれに伴うエンタルピー44.6J/gの単一吸熱。
【実施例6】
【0165】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)酢酸イソプロピル/メチルシクロヘキサン溶媒和物遊離塩基多形体IタイプAの製造
化合物Iの遊離塩基300mgを酢酸イソプロピル0.25mLに溶解した。混合物を70℃まで加熱した後、メチルシクロヘキサン0.25mLを加えた。混合物を40℃まで加熱後、化合物Iの遊離塩基をシード添加した。固形分がゆっくりと沈殿し始めた。溶液を室温まで冷却し、30分間撹拌した。次に酢酸イソプロピル0.1mLとメチルシクロヘキサン0.5mLを加えた。得られた固形分を濾過し、化合物Iのメチルシクロヘキサン/酢酸イソプロピル溶媒和物遊離塩基多形体Iを得た。
【0166】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物Iの酢酸イソプロピル/メチルシクロヘキサン溶媒和物遊離塩基多形体I,タイプAのX線粉末回折スペクトル(図10)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(2θ),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0167】
【表3】
【0168】
化合物Iの酢酸イソプロピル/メチルシクロヘキサン溶媒和物遊離塩基多形体I,タイプAは表3に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物Iの酢酸イソプロピル/メチルシクロヘキサン溶媒和物遊離塩基多形体I,タイプAは角度θ値4.6°により同定することができる。化合物Iの酢酸イソプロピル/メチルシクロヘキサン溶媒和物遊離塩基多形体I,タイプAは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)4.6°;
b)4.6°及び7.5°;
c)4.6°、7.5°及び9.1°;
d)4.6°、7.5°、9.1°及び9.7°;
e)4.6°、7.5°、9.1°、9.7°及び14.5°;
f)4.6°、7.5°、9.1°、9.7°、14.5°及び15.1°;
g)4.6°、7.5°、9.1°、9.7°、14.5°、15.1°及び16.8°;
h)4.6°、7.5°、9.1°、9.7°、14.5°、15.1°、16.8°及び17.8°;
i)4.6°、7.5°、9.1°、9.7°、14.5°、15.1°、16.8°、17.8°及び18.2°。
【0169】
化合物Iの酢酸イソプロピル/メチルシクロヘキサン溶媒和物遊離塩基多形体I、タイプAはCu線を使用して得られたX線粉末回折パターンからd間隔19.209、11.787、9.718、9.118、6.109、5.867、5.277、4.983及び4.874Åの1個以上の反射により同定することもできる。Perkin Elmer TGA−7機器で窒素流下に昇温速度10℃/分で化合物Iの酢酸イソプロピル/メチルシクロヘキサン溶媒和物遊離塩基多形体IタイプAの熱重量(TG)分析曲線(図12)を得た。TA Instruments DSC−2910示差走査熱量計でN2流下に昇温速度10℃/分で化合物Iの酢酸イソプロピル/メチルシクロヘキサン溶媒和物遊離塩基多形体IタイプAのDSC曲線(図11)を得た。
【実施例7】
【0170】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)無水HCl塩多形体Aの製造
化合物Iの非晶質遊離塩基0.5gをアセトニトリル2.5mLに溶解した。次に1.0M HCl/Et2O 0.93mLを滴下すると、透明溶液が得られた。約2分後に、白色固体が溶液から晶析した。混合物を更に10分間撹拌後、濾過した。得られた固形分を減圧窒素スイープ下にフリットで乾燥し、化合物Iの無水HCl塩多形体Aを得た。
【0171】
あるいは、a)エタノールから化合物IのHCl塩を晶析させ、乾燥する方法;及びb)実施例14の化合物IのHCl塩多形体Hを乾燥する方法により、化合物Iの無水HCl塩多形体Aを形成することもできる。
【0172】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物Iの無水HCl塩多形体AのX線粉末回折スペクトル(図4)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(2θ),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0173】
【表4】
【0174】
化合物Iの無水HCl塩多形体Aは表4に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物Iの無水HCl塩多形体Aは角度θ値13.0°により同定することができる。化合物Iの無水HCl塩形態Aは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)13.0°;
b)13.0°及び19.3°;
c)13.0°、19.3°及び15.8°;
d)13.0°、19.3°、15.8°及び16.1°;
e)13.0°、19.3°、15.8°、16.1°及び17.3°;
f)13.0°、19.3°、15.8°、16.1°、17.3°及び13.6°;
g)13.0°、19.3°、15.8°、16.1°、17.3°、13.6°及び9.3°;
h)13.0°、19.3°、15.8°、16.1°、17.3°、13.6°、9.3°及び16.6°;
i)13.0°、19.3°、15.8°、16.1°、17.3°、13.6°、9.3°、16.6°及び9.6°。
【0175】
化合物Iの無水HCl塩多形体AはCu線を使用して得られたX線粉末回折パターンからd間隔6.810、4.599、9.509、5.609、5.505、5.126、9.213、6.511及び5.340Åの1個以上の反射により同定することもできる。
【0176】
化合物Iの無水HCl塩多形体Aは示差走査熱量分析(DSC)によっても特徴付けられる。TA Instruments DSC−2910示差走査熱量計でN2流下に昇温速度10℃/分で化合物Iの無水HCl塩多形体AのDSC曲線(図5)を得た。サンプルを閉鎖パンで加熱した。開始温度213.3Cで単一吸熱が観察される。
【実施例8】
【0177】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)無水HCl塩多形体Bの製造
酢酸イソプロピル35mLを化合物Iの非晶質遊離塩基3.5gに加えた。次に、HClのIPA溶液(5〜6M)1.44mLを加えた。反応混合物を85〜90℃まで加熱した後、圧力管に密封した。150℃に設定した加熱マントルで反応混合物を一晩加熱した。翌朝、反応混合物を冷却し、濾過し、化合物IのHCl塩多形体Bを得た。
【0178】
あるいは、a)実施例9の化合物IのHCl塩多形体Cを酢酸イソプロピルでスラリー化し、乾燥する方法;b)実施例10の化合物IのHCl塩多形体Dを化合物IのHCl塩多形体Bシードと共にエタノールでスラリー化し、乾燥する方法;c)実施例7の化合物IのHCl塩多形体Aを酢酸イソプロピルでスラリー化し、乾燥する方法;及びd)215℃を上回る温度まで化合物IのHCl塩多形体Aを加熱する方法により、化合物Iの無水HCl塩多形体Bを形成することもできる。
【0179】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物Iの無水HCl塩多形体BのX線粉末回折スペクトル(図6)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(2θ),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0180】
【表5】
【0181】
化合物Iの無水HCl塩多形体Bは表5に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物Iの無水HCl塩多形体Bは角度θ値17.9°により同定することができる。化合物Iの無水HCl塩多形体Bは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)17.9°;
b)17.9°及び15.3°;
c)17.9°、15.3°及び19.4°;
d)17.9°、15.3°、19.4°及び24.9°;
e)17.9°、15.3°、19.4°、24.9°及び6.1°;
f)17.9°、15.3°、19.4°、24.9°、6.1°及び8.1°;
g)17.9°、15.3°、19.4°、24.9°、6.1°、8.1°及び
21.5°;
h)17.9°、15.3°、19.4°、24.9°、6.1°、8.1°、21.5°及び14.2°;
i)17.9°、15.3°、19.4°、24.9°、6.1°、8.1°、21.5°、14.2°及び10.9°。
【0182】
化合物Iの無水HCl塩多形体BはCu線を使用して得られたX線粉末回折パターンからd間隔4.955、14.488、5.791、10.915、8.117、6.237、4.575、4.133及び3.576Åの1個以上の反射により同定することもできる。
【0183】
化合物Iの無水HCl塩多形体Bは示差走査熱量分析(DSC)によっても特徴付けられる。TA Instruments DSC−2910示差走査熱量計でN2流下に昇温速度10℃/分で化合物Iの無水HCl塩多形体BのDSC曲線(図7)を得た。サンプルを閉鎖パンで加熱した。開始温度249.9Cで単一吸熱が観察される。
【実施例9】
【0184】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)HCl塩多形体Cの製造
化合物Iの非晶質遊離塩基0.5gをイソプロパノール2.5mLに溶解した。次に5〜6N HCl/イソプロピルアルコール1.1モル当量を滴下した。30分間撹拌後、得られた固形分を濾過した。固形分を減圧窒素スイープ下にフリットで乾燥し、化合物IのHCl塩多形体Cを得た。
【0185】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物IのHCl塩多形体CのX線粉末回折スペクトル(図15)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(20),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0186】
【表6】
【0187】
化合物IのHCl塩多形体Cは表6に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物IのHCl塩多形体Cは角度θ値6.5°により同定することができる。化合物IのHCl塩形態Cは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
j)6.5°;
k)6.5°及び8.2°;
1)6.5°、8.2°及び9.1°;
m)6.5°、8.2°、9.1°及び10°;
n)6.5°、8.2°、9.1°、10°及び13.8°;
o)6.5°、8.2°、9.1°、10°、13.8°及び14.2°;
p)6.5°、8.2°、9.1°、10°、13.8°、14.2°及び14.9°;
q)6.5°、8.2°、9.1°、10°、13.8°、14.2°、14.9°及び15.8°;
r)6.5°、8.2°、9.1°、10°、13.8°、14.2°、14.9°、15.8°及び18.1°。
【0188】
化合物IのHCl塩多形体CはCu線を使用して得られたX線粉末回折パターンからd間隔13.598、10.782、9.718、8.845、6.417、6.237、5.945、5.609及び4.901Åの1個以上の反射により同定することもできる。
【実施例10】
【0189】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物T)HCl塩多形体Dの製造
結晶質HCl塩多形体F(水和物)を35℃の真空炉で乾燥することにより化合物IのHCl塩多形体Dを製造した。
【0190】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物IのHCl塩多形体DのX線粉末回折スペクトル(図16)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(2θ),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0191】
【表7】
【0192】
化合物IのHCl塩多形体Dは表7に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物IのHCl塩多形体Dは角度θ値7.2°により同定することができる。化合物IのHCl塩形態Dは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)7.2°;
b)7.2°及び12.3°;
c)7.2°、12.3°及び6.2°;
d)7.2°、12.3°、6.2°及び11.1°;
e)7.2°、12.3°、6.2°、11.1°及び13.8°;
f)7.2°、12.3°、6.2°、11.1°、13.8°及び18°;
g)7.2°、12.3°、6.2°、11.1°、13.8°、18°及び8.8°;
h)7.2°、12.3°、6.2°、11.1°、13.8°、18°、8.8°及び22°;
i)7.2°、12.3°、6.2°、11.1°、13.8°、18°、8.8°、22°及び24.2°。
【0193】
化合物IのHCl塩多形体DはCu線を使用して得られたX線粉末回折パターンからd間隔12.277、7.196、14.255、7.971、6.417、4.928、10.048、4.040及び3.678Åの1個以上の反射により同定することもできる。
【0194】
Perkin Elmer TGA−7機器で窒素流下に昇温速度10℃/分で化合物IのHCl塩多形体Dの熱重量(TG)分析曲線(図18)を得た。TA Instruments DSC−2910示差走査熱量計でN2流下に昇温速度10℃/分で化合物IのHCl塩多形体DのDSC曲線(図17)を得た。
【実施例11】
【0195】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)無水HCl塩多形体Eの製造
HCl塩多形体Dを温度>185℃まで乾燥することにより無水HCl塩多形体Eを製造した。
【0196】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物Iの無水HCl塩多形体EのX線粉末回折スペクトル(図19)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(2θ),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0197】
【表8】
【0198】
化合物Iの無水HCl塩多形体Eは表8に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物Iの無水HCl塩多形体Eは角度θ値7.4°により同定することができる。化合物Iの無水HCl塩形態Eは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)7.4°;
b)7.4°及び8.2°;
c)7.4°、8.2°及び8.9°;
d)7.4°、8.2°、8.9°及び10.2°;
e)7.4°、8.2°、8.9°、10.2°及び12.8°;
f)7.4°、8.2°、8.9°、10.2°、12.8°及び14.3°;
g)7.4°、8.2°、8.9°、10.2°、12.8°、14.3°及び14.8°;
h)7.4°、8.2°、8.9°、10.2°、12.8°、14.3°、14.8°及び15.4°;
i)7.4°、8.2°、8.9°、10.2°、12.8°、14.3°、14.8°、15.4°及び19.5°。
【0199】
化合物Iの無水HCl塩多形体EはCu線を使用して得られたX線粉末回折パターンからd間隔11.946、10.782、9.936、8.672、6.916、6.194、5.985、5.754及び4.552Åの1個以上の反射により同定することもできる。
【実施例12】
【0200】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物1)HCl塩多形体F水和物の製造
化合物Iの遊離塩基2.5gをエタノール10mLに溶解した。次に溶液0.685gを容器に加えた後、エタノール2.5mLを加えた。次に0.411M HCl水溶液0.75mLを容器に加えた。1%シードを加え、スラリーを1時間エージングさせた。次に遊離塩基溶液の残りの11.3mLと1.4M HCl水溶液3.1mLを8時間かけて加えた。次にスラリーを濾過し、1:1エタノール/水で洗浄し、水和物HCl塩多形体Fを得た。
【0201】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物IのHCl塩多形体F水和物のX線粉末回折スペクトル(図20)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(2θ),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0202】
【表9】
【0203】
化合物IのHCl塩多形体F水和物は表9に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物IのHCl塩多形体F水和物は角度θ値6.1°により同定することができる。化合物IのHCl塩形態F水和物は以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)6.1°;
b)6.1°及び6.4°;
c)6.1°、6.4°及び7.3°;
d)6.1°、6.4°、7.3°及び11°;
e)6.1°、6.4°、7.3°、11°及び11.5°;
f)6.1°、6.4°、7.3°、11°、11.5°及び12°;
g)6.1°、6.4°、7.3°、11°、11.5°、12°及び16.2°;
h)6.1°、6.4°、7.3°、11°、11.5°、12°、16.2°及び18°;
i)6.1°、6.4°、7.3°、11°、11.5°、12°、16.2°、18°及び19°。
【0204】
化合物IのHCl塩多形体F水和物はCu線を使用して得られたX線粉末回折パターンからd間隔14.488、13.810、12.109、8.043、7.694、7.375、5.471、4.928及び4.671Åの1個以上の反射により同定することもできる。
【実施例13】
【0205】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)無水HCl塩多形体Gの製造
化合物Iの結晶質遊離塩基2.0gをエタノール3.7mLに溶解した。次に1.25M HClエタノール溶液0.05mLを加えた後、多形体Gシード80mgを加えた。次に0.2mL/時の速度で1.25M HClエタノール溶液3.2mLをゆっくりと加えた。得られたスラリーを濾過し、エタノール6mLで洗浄し、減圧窒素スイープ下にフリットで乾燥し、化合物Iの無水HCl塩多形体Gを得た。
【0206】
あるいは、a)実施例10の化合物IのHCl塩多形体Dを化合物IのHCl塩多形体Gと共にスラリー化する方法;b)実施例8の化合物IのHCl塩多形体Bを化合物IのHCl塩多形体Gシードと共にエタノールでスラリー化する方法;c)実施例8の化合物IのHCl塩多形体Bを25%水/エタノールでスラリー化する方法;及びd)実施例7の化合物IのHCl塩多形体Aを化合物IのHCl塩多形体Gと共にエタノールでスラリー化する方法により、化合物Iの無水HCl塩多形体Gを形成することもできる。
【0207】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物Iの無水HCl塩多形体GのX線粉末回折スペクトル(図8)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(2θ),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0208】
【表10】
【0209】
化合物Iの無水HCl塩多形体Gは表10に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物Iの無水HCl塩多形体Gは角度θ値8.2°により同定することができる。化合物Iの無水HCl塩多形体Gは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)8.2°;
b)8.2°及び20.8°;
c)8.2°、20.8°及び16.8°;
d)8.2°、20.8°、16.8°及び21.6°;
e)8.2°、20.8°、16.8°、21.6°及び12.6°;
f)8.2°、20.8°、16.8°、21.6°、12.6°及び10.4°;
g)8.2°、20.8°、16.8°、21.6°、12.6°、10.4°及び12.1°;
h)8.2°、20.8°、16.8°、21.6°、12.6°、10.4°、12.1°及び14.1°;
i)8.2°、20.8°、16.8°、21.6°、12.6°、10.4°、12.1°、14.1°及び13.3°。
【0210】
化合物Iの無水HCl塩多形体GはCu線を使用して得られたX線粉末回折パターンからd間隔10.782、4.270、8.506、7.025、7.314、6.657、6.281、5.277及び4.114Åの1個以上の反射により同定することもできる。
【0211】
化合物Iの無水HCl塩多形体Gは示差走査熱量分析(DSC)によっても特徴付けられる。TA Instruments DSC−2910示差走査熱量計でN2流下に昇温速度10℃/分で化合物Iの無水HCl塩多形体GのDSC曲線(図9)を得た。サンプルを閉鎖パンで加熱した。開始温度267.3Cで単一吸熱が観察される。
【実施例14】
【0212】
3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル(化合物I)HCl塩多形体Hの製造
化合物Iの結晶質遊離塩基2.0gをエタノール3.7mLに溶解した。次に1.25M HClエタノール溶液0.05mLを加えた後に多形体Gシード5mgを加えた。次に1.25M HClエタノール溶液3.2mLを0.2mL/時の速度でゆっくりと加えた。得られたスラリーを濾過し、エタノール6mLで洗浄し、乾燥しなかった。
【0213】
Philips Analytical X’Pert PRO X線回折システムでPW3040/60コンソールを使用して化合物IのHCl塩多形体HのX線粉末回折スペクトル(図21)を作成した。PW3373/00セラミックCu LFF X線管Kα線を線源として使用した。X線粉末回折スペクトルを周囲温度で記録した(CuKα線,2°〜40°(20),0.0167°刻み,1刻み当たり5.08秒)。波長1.54178ÅのCuKαをd間隔計算に使用した。
【0214】
【表11】
【0215】
化合物IのHCl塩多形体Hは表11に示す角度2θ値の完全な集合を特徴とするが、このような同定に全値は必要ない。化合物IのHCl塩多形体Hは角度θ値9.1°により同定することができる。化合物IのHCl塩形態Hは以下の角度θ値のいずれか1個、又は以下の角度θ値集合のいずれか1個により同定することができる:
a)9.1°;
b)9.1°及び9.4°;
c)9.1°、9.4°及び12.8°;
d)9.1°、9.4°、12.8°及び13.4°;
e)9.1°、9.4°、12.8°、13.4°及び14.3°;
f)9.1°、9.4°、12.8°、13.4°、14.3°及び14.9°;
g)9.1°、9.4°、12.8°、13.4°、14.3°、14.9°及び15.3°;
h)9.1°、9.4°、12.8°、13.4°、14.3°、14.9°、15.3°及び16.9°;
i)9.1°、9.4°、12.8°、13.4°、14.3°、14.9°、15.3°、16.9°及び18.2°。
【0216】
化合物IのHCl塩多形体CはCu線を使用して得られたX線粉末回折パターンからd間隔9.718、9.408、6.916、6.607、6.194、5.945、5.791、5.246及び4.874Åの1個以上の反射により同定することもできる。
【特許請求の範囲】
【請求項1】
式I:
【化1】
の化合物又はその塩、溶媒和物、水和物もしくは多形体の製造方法であって、
(A)式20:
【化2】
の化合物の保護基Pを除去する段階と;
(B)得られた生成物を単離する段階を含む前記方法。
【請求項2】
保護基PがCBZであり、水素化によりCBZ保護基を除去する請求項1に記載の方法。
【請求項3】
保護基PがBocであり、酸処理によりBoc保護基を除去する請求項1に記載の方法。
【請求項4】
更にトルエン/ヘプタンから結晶化させることにより式Iの化合物を単離する段階を含む請求項1に記載の方法。
【請求項5】
式Iの化合物の塩が塩酸塩である請求項1に記載の方法。
【請求項6】
式Iの単離化合物の多形体が、
(1)図1のX線粉末回折パターンを特徴とする化合物Iの無水遊離塩基多形体I;
(2)図13のX線粉末回折パターンを特徴とする化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体IタイプB;
(4)図10のX線粉末回折パターンを特徴とする化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物多形体IタイプA;
(5)図4のX線粉末回折パターンを特徴とする化合物Iの無水HCl塩多形体A;
(6)図6のX線粉末回折パターンを特徴とする化合物Iの無水HCl塩多形体B;
(7)図15のX線粉末回折パターンを特徴とする化合物IのHCl塩多形体C;
(8)図16のX線粉末回折パターンを特徴とする化合物IのHCl塩多形体D;
(9)図19のX線粉末回折パターンを特徴とする化合物Iの無水HCl塩多形体E;
(10)図20のX線粉末回折パターンを特徴とする化合物IのHCl塩多形体F水和物;
(11)図8のX線粉末回折パターンを特徴とする化合物Iの無水HCl塩多形体G;及び
(12)図21のX線粉末回折パターンを特徴とする化合物IのHCl塩多形体H
から構成される群から選択される請求項1に記載の方法。
【請求項7】
化合物I:
【化3】
の無水遊離塩基多形体Iである化合物。
【請求項8】
図1のX線粉末回折パターンを特徴とする請求項7に記載の化合物Iの無水遊離塩基多形体I。
【請求項9】
Cu線を使用して得られるX線粉末回折パターンが5.2°〜28.5°の角度2θ値を含む請求項7に記載の化合物。
【請求項10】
Cu線を使用して得られるX線粉末回折パターンが5.2°の角度2θ値を含む請求項7に記載の化合物。
【請求項11】
Cu線を使用して得られるX線粉末回折パターンが5.2°及び7.0°の角度2θ値を含む請求項7に記載の化合物。
【請求項12】
Cu線を使用して得られるX線粉末回折パターンが5.2°及び7.0°の角度2θ値と、9.3°,11.8°、15.4°、15.7°、16.4°、17.4°及び22.5°から構成される群から選択される少なくとも1個の角度θ値を含む請求項7に記載の化合物。
【請求項13】
Cu線を使用して得られるX線粉末回折パターンがd間隔16.99Åの反射を特徴とする請求項7に記載の化合物。
【請求項14】
Cu線を使用して得られるX線粉末回折パターンがd間隔16.99Åの反射および、12.63Å、9.51Å、7.5Å、5.75Å、5.64Å、5.40Å、5.09Å及び3.95Åから構成される群から選択されるd間隔の少なくとも1個の反射を特徴とする請求項7に記載の化合物。
【請求項15】
示差走査熱量分析ピーク融点温度が約163.57℃である請求項7に記載の化合物。
【請求項16】
治療有効量の請求項7に記載の化合物Iの無水遊離塩基多形体Iおよび、医薬的に許容可能なキャリヤーを含有する医薬的組成物。
【請求項17】
肥満症、糖尿病、アルツハイマー病又は肥満関連障害の治療を必要とする対象における肥満症、糖尿病、アルツハイマー病又は肥満関連障害の治療方法であって、治療有効量の請求項7に記載の化合物Iの無水遊離塩基多形体Iを前記対象に投与する段階を含む前記方法。
【請求項18】
肥満症、糖尿病、アルツハイマー病又は肥満関連障害の治療又は予防を必要とする対象における肥満症、糖尿病、アルツハイマー病又は肥満関連障害の治療又は予防に有用な医薬の製造用としての請求項7に記載の化合物Iの無水遊離塩基多形体Iの使用。
【請求項19】
式II:
【化4】
(式中、PはBoc又はCBZである)の化合物又はその塩、水和物もしくは溶媒和物。
【請求項20】
式III:
【化5】
の化合物又はその塩、水和物もしくは溶媒和物。
【請求項21】
更に式20:
【化6】
(式中Pは保護基である)の化合物を製造する段階を含み、
前記段階が、化合物IIIのアルコール基を脱離基に変換した後にヒンダードアミン塩基で処理した後に、式II:
【化7】
(式中、Pは保護基である)の化合物又はその塩を式III:
【化8】
の化合物とカップリングする段階を含む請求項1に記載の方法。
【請求項22】
脱離基がトリフラートであり、化合物IIIをトリフル酸無水物で処理してジトリフラート中間体を形成し、ヒンダードアミン塩基がジイソプロピルエチルアミンである請求項21に記載の方法。
【請求項23】
更に式II:
【化9】
(式中、Pは保護基である)の化合物又はその塩を製造する段階を含み、前記段階が、
(A)式1:
【化10】
の化合物を塩基で処理した後にヒドラジンで処理することにより式3:
【化11】
のヒドラジドを製造する段階と;
(B)式3のヒドラジドをカップリング剤で処理することにより式4:
【化12】
のオキサジアゾールを形成する段階と;
(C)式4のオキサジアゾールをアルキルマグネシウム化合物で処理した後にアルキルリチウム化合物とDMFで処理することにより式5:
【化13】
のアルデヒドを製造する段階と;
(D)式5のアルデヒドを触媒の存在下に(S)−tert−ブチルスルフィンアミドで処理することにより式6:
【化14】
のN−tert−ブチルスルフィニルイミンを製造する段階と;
(E)式6のN−tert−ブチルスルフィニルイミンのオキサジアゾール窒素に保護基Pを付加することにより式7:
【化15】
(式中、Pは保護基である)の保護オキサジアゾール化合物を形成する段階と;
(F)式7の保護オキサジアゾール化合物をロジウム触媒とリガンドの存在下にボロキシン10で処理することにより式8:
【化16】
(式中、Pは保護基である)のN−tert−ブチルスルフィニルアミンを形成する段階と;
(G)式8のN−tert−ブチルスルフィニルアミンのtert−ブチルスルホキシド基を開裂することにより式II:
【化17】
(式中、Pは保護基である)の化合物を形成する段階を含む請求項21に記載の方法。
【請求項24】
更に式III:
【化18】
の化合物又はその塩を製造する段階を含み、前記段階が、
(A)式11:
【化19】
の化合物をグリニャール試薬で処理した後に塩化イソブチリルで処理することにより式12:
【化20】
の化合物を製造する段階と;
(B)ハロゲン化シリル又はシリルトリフラートの存在下にフッ素源と塩基で処理して式12の化合物をフッ素化することにより式13:
【化21】
のフルオロケトン化合物を形成する段階と;
(C)式13の化合物を塩基の存在下にホスホノ酢酸トリメチルで処理することにより式14:
【化22】
の化合物を製造する段階と;
(D)式14の化合物のエステルを加水分解することにより式15:
【化23】
の化合物を製造する段階と;
(E)式15の化合物の二重結合を還元することにより式16:
【化24】
の化合物を形成する段階と;
(F)式16の化合物のエステル化により式17:
【化25】
(式中、R=C1−3アルキルである)の化合物を形成する段階と;
(G)式17の化合物のカルボキシル化により式18:
【化26】
(式中、R=C1−3アルキルである)の化合物を形成する段階と;
(H)式18の化合物を還元することにより式19:
【化27】
の化合物を形成する段階と;
(I)式19の化合物をシアノ化することにより式III
【化28】
の化合物を形成する段階を含む請求項21に記載の方法。
【請求項1】
式I:
【化1】
の化合物又はその塩、溶媒和物、水和物もしくは多形体の製造方法であって、
(A)式20:
【化2】
の化合物の保護基Pを除去する段階と;
(B)得られた生成物を単離する段階を含む前記方法。
【請求項2】
保護基PがCBZであり、水素化によりCBZ保護基を除去する請求項1に記載の方法。
【請求項3】
保護基PがBocであり、酸処理によりBoc保護基を除去する請求項1に記載の方法。
【請求項4】
更にトルエン/ヘプタンから結晶化させることにより式Iの化合物を単離する段階を含む請求項1に記載の方法。
【請求項5】
式Iの化合物の塩が塩酸塩である請求項1に記載の方法。
【請求項6】
式Iの単離化合物の多形体が、
(1)図1のX線粉末回折パターンを特徴とする化合物Iの無水遊離塩基多形体I;
(2)図13のX線粉末回折パターンを特徴とする化合物Iの遊離塩基トルエン/ヘプタン溶媒和物多形体IタイプB;
(4)図10のX線粉末回折パターンを特徴とする化合物Iの遊離塩基酢酸イソプロピル/メチルシクロヘキサン溶媒和物多形体IタイプA;
(5)図4のX線粉末回折パターンを特徴とする化合物Iの無水HCl塩多形体A;
(6)図6のX線粉末回折パターンを特徴とする化合物Iの無水HCl塩多形体B;
(7)図15のX線粉末回折パターンを特徴とする化合物IのHCl塩多形体C;
(8)図16のX線粉末回折パターンを特徴とする化合物IのHCl塩多形体D;
(9)図19のX線粉末回折パターンを特徴とする化合物Iの無水HCl塩多形体E;
(10)図20のX線粉末回折パターンを特徴とする化合物IのHCl塩多形体F水和物;
(11)図8のX線粉末回折パターンを特徴とする化合物Iの無水HCl塩多形体G;及び
(12)図21のX線粉末回折パターンを特徴とする化合物IのHCl塩多形体H
から構成される群から選択される請求項1に記載の方法。
【請求項7】
化合物I:
【化3】
の無水遊離塩基多形体Iである化合物。
【請求項8】
図1のX線粉末回折パターンを特徴とする請求項7に記載の化合物Iの無水遊離塩基多形体I。
【請求項9】
Cu線を使用して得られるX線粉末回折パターンが5.2°〜28.5°の角度2θ値を含む請求項7に記載の化合物。
【請求項10】
Cu線を使用して得られるX線粉末回折パターンが5.2°の角度2θ値を含む請求項7に記載の化合物。
【請求項11】
Cu線を使用して得られるX線粉末回折パターンが5.2°及び7.0°の角度2θ値を含む請求項7に記載の化合物。
【請求項12】
Cu線を使用して得られるX線粉末回折パターンが5.2°及び7.0°の角度2θ値と、9.3°,11.8°、15.4°、15.7°、16.4°、17.4°及び22.5°から構成される群から選択される少なくとも1個の角度θ値を含む請求項7に記載の化合物。
【請求項13】
Cu線を使用して得られるX線粉末回折パターンがd間隔16.99Åの反射を特徴とする請求項7に記載の化合物。
【請求項14】
Cu線を使用して得られるX線粉末回折パターンがd間隔16.99Åの反射および、12.63Å、9.51Å、7.5Å、5.75Å、5.64Å、5.40Å、5.09Å及び3.95Åから構成される群から選択されるd間隔の少なくとも1個の反射を特徴とする請求項7に記載の化合物。
【請求項15】
示差走査熱量分析ピーク融点温度が約163.57℃である請求項7に記載の化合物。
【請求項16】
治療有効量の請求項7に記載の化合物Iの無水遊離塩基多形体Iおよび、医薬的に許容可能なキャリヤーを含有する医薬的組成物。
【請求項17】
肥満症、糖尿病、アルツハイマー病又は肥満関連障害の治療を必要とする対象における肥満症、糖尿病、アルツハイマー病又は肥満関連障害の治療方法であって、治療有効量の請求項7に記載の化合物Iの無水遊離塩基多形体Iを前記対象に投与する段階を含む前記方法。
【請求項18】
肥満症、糖尿病、アルツハイマー病又は肥満関連障害の治療又は予防を必要とする対象における肥満症、糖尿病、アルツハイマー病又は肥満関連障害の治療又は予防に有用な医薬の製造用としての請求項7に記載の化合物Iの無水遊離塩基多形体Iの使用。
【請求項19】
式II:
【化4】
(式中、PはBoc又はCBZである)の化合物又はその塩、水和物もしくは溶媒和物。
【請求項20】
式III:
【化5】
の化合物又はその塩、水和物もしくは溶媒和物。
【請求項21】
更に式20:
【化6】
(式中Pは保護基である)の化合物を製造する段階を含み、
前記段階が、化合物IIIのアルコール基を脱離基に変換した後にヒンダードアミン塩基で処理した後に、式II:
【化7】
(式中、Pは保護基である)の化合物又はその塩を式III:
【化8】
の化合物とカップリングする段階を含む請求項1に記載の方法。
【請求項22】
脱離基がトリフラートであり、化合物IIIをトリフル酸無水物で処理してジトリフラート中間体を形成し、ヒンダードアミン塩基がジイソプロピルエチルアミンである請求項21に記載の方法。
【請求項23】
更に式II:
【化9】
(式中、Pは保護基である)の化合物又はその塩を製造する段階を含み、前記段階が、
(A)式1:
【化10】
の化合物を塩基で処理した後にヒドラジンで処理することにより式3:
【化11】
のヒドラジドを製造する段階と;
(B)式3のヒドラジドをカップリング剤で処理することにより式4:
【化12】
のオキサジアゾールを形成する段階と;
(C)式4のオキサジアゾールをアルキルマグネシウム化合物で処理した後にアルキルリチウム化合物とDMFで処理することにより式5:
【化13】
のアルデヒドを製造する段階と;
(D)式5のアルデヒドを触媒の存在下に(S)−tert−ブチルスルフィンアミドで処理することにより式6:
【化14】
のN−tert−ブチルスルフィニルイミンを製造する段階と;
(E)式6のN−tert−ブチルスルフィニルイミンのオキサジアゾール窒素に保護基Pを付加することにより式7:
【化15】
(式中、Pは保護基である)の保護オキサジアゾール化合物を形成する段階と;
(F)式7の保護オキサジアゾール化合物をロジウム触媒とリガンドの存在下にボロキシン10で処理することにより式8:
【化16】
(式中、Pは保護基である)のN−tert−ブチルスルフィニルアミンを形成する段階と;
(G)式8のN−tert−ブチルスルフィニルアミンのtert−ブチルスルホキシド基を開裂することにより式II:
【化17】
(式中、Pは保護基である)の化合物を形成する段階を含む請求項21に記載の方法。
【請求項24】
更に式III:
【化18】
の化合物又はその塩を製造する段階を含み、前記段階が、
(A)式11:
【化19】
の化合物をグリニャール試薬で処理した後に塩化イソブチリルで処理することにより式12:
【化20】
の化合物を製造する段階と;
(B)ハロゲン化シリル又はシリルトリフラートの存在下にフッ素源と塩基で処理して式12の化合物をフッ素化することにより式13:
【化21】
のフルオロケトン化合物を形成する段階と;
(C)式13の化合物を塩基の存在下にホスホノ酢酸トリメチルで処理することにより式14:
【化22】
の化合物を製造する段階と;
(D)式14の化合物のエステルを加水分解することにより式15:
【化23】
の化合物を製造する段階と;
(E)式15の化合物の二重結合を還元することにより式16:
【化24】
の化合物を形成する段階と;
(F)式16の化合物のエステル化により式17:
【化25】
(式中、R=C1−3アルキルである)の化合物を形成する段階と;
(G)式17の化合物のカルボキシル化により式18:
【化26】
(式中、R=C1−3アルキルである)の化合物を形成する段階と;
(H)式18の化合物を還元することにより式19:
【化27】
の化合物を形成する段階と;
(I)式19の化合物をシアノ化することにより式III
【化28】
の化合物を形成する段階を含む請求項21に記載の方法。
【図1】

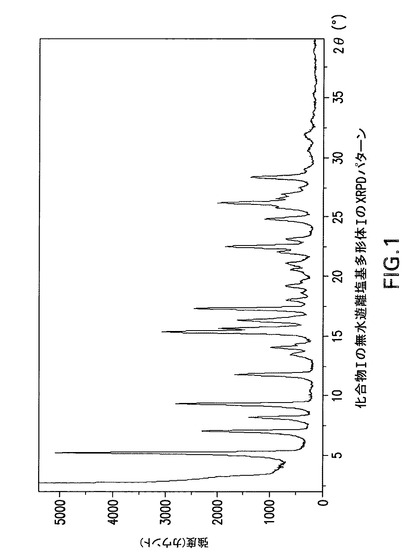
【図2】
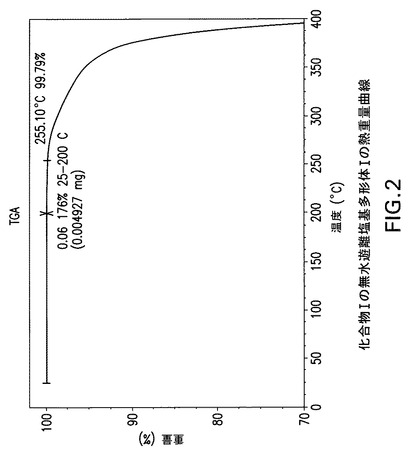
【図3】
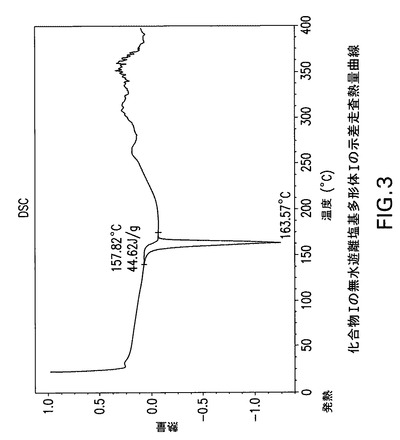
【図4】
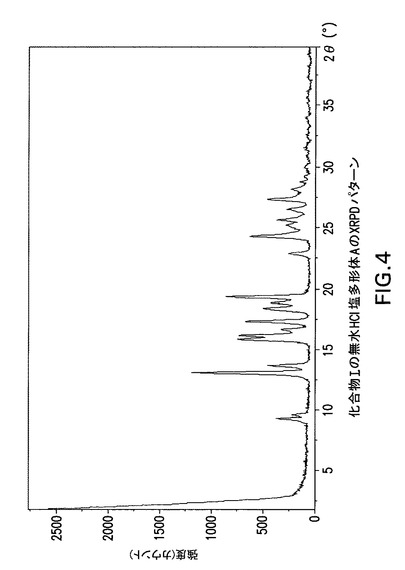
【図5】
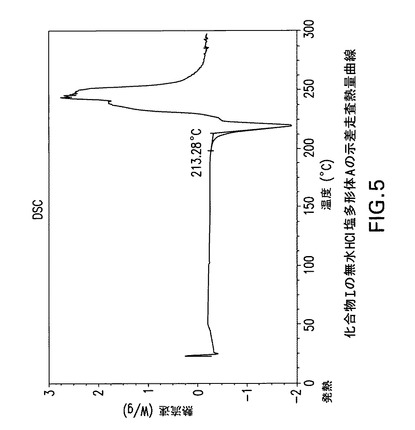
【図6】
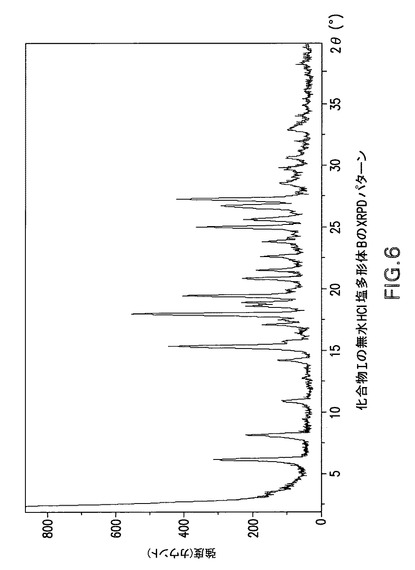
【図7】
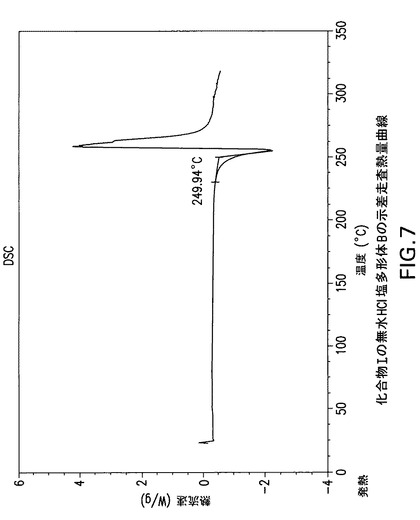
【図8】
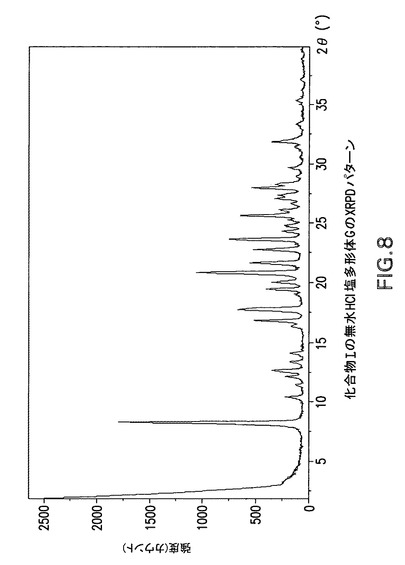
【図9】
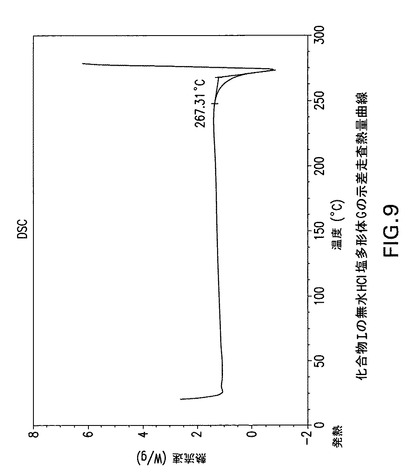
【図10】
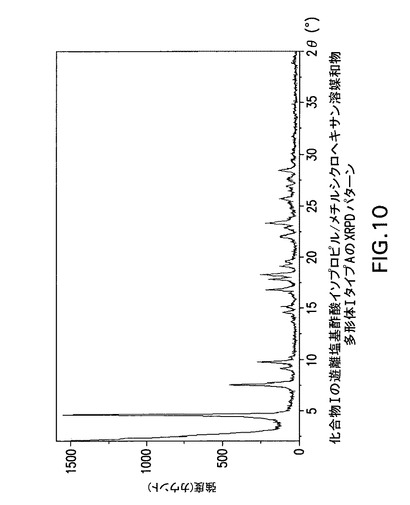
【図11】
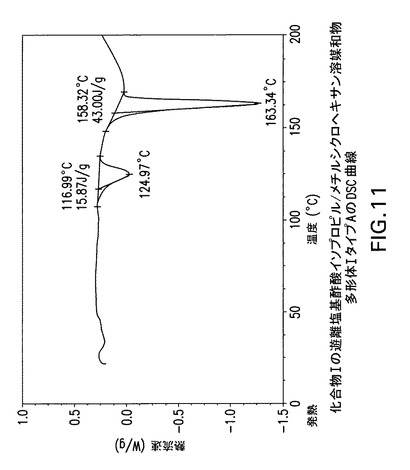
【図12】
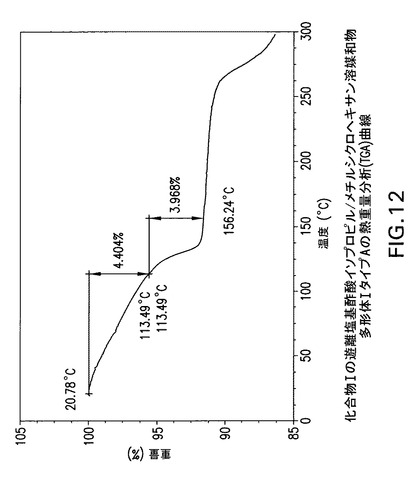
【図13】
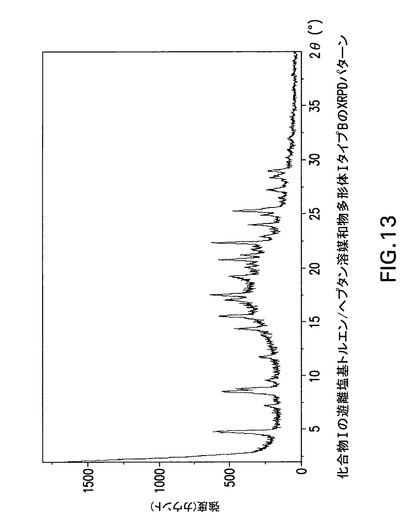
【図14】
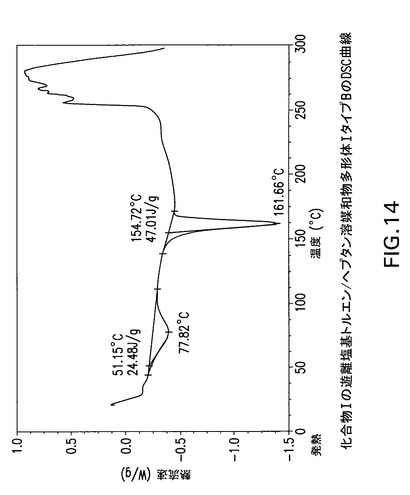
【図15】
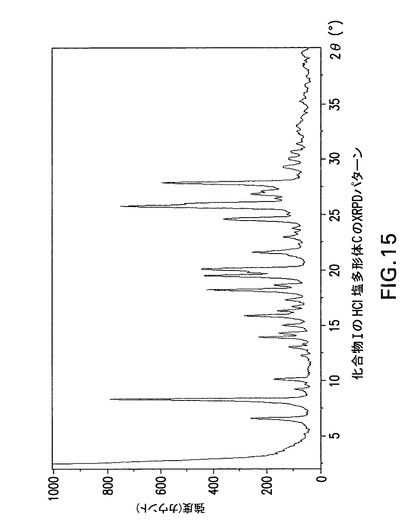
【図16】
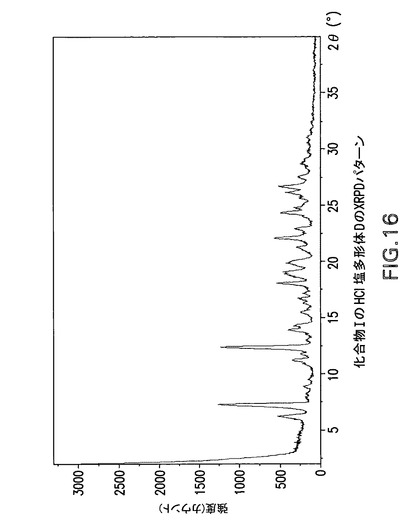
【図17】
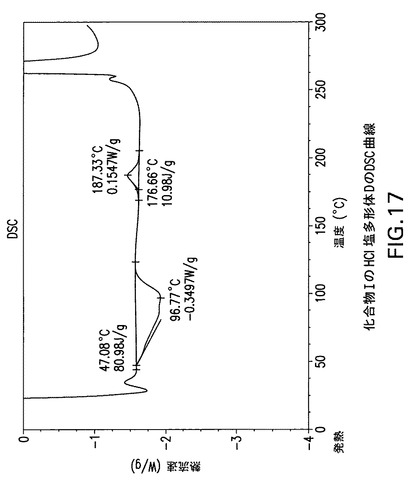
【図18】
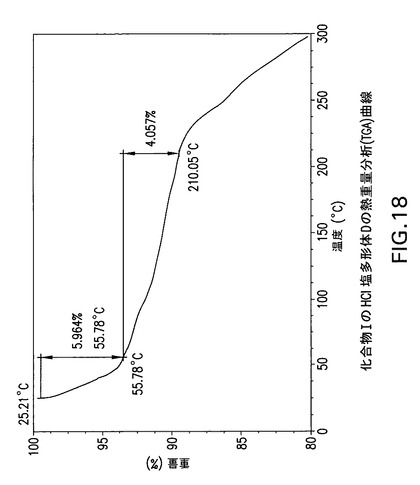
【図19】
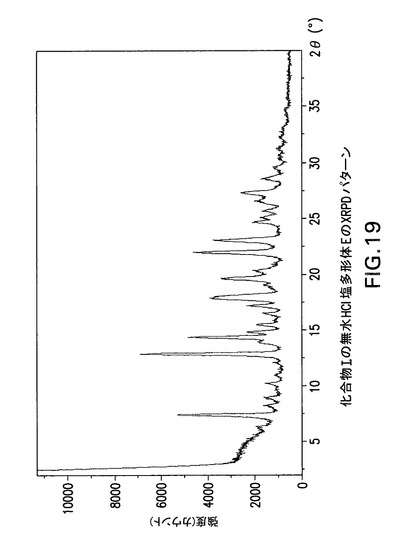
【図20】
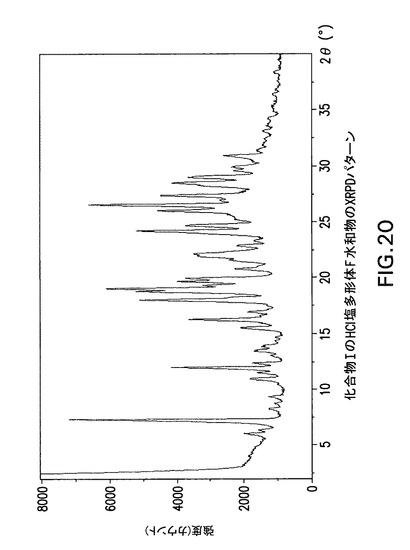
【図21】
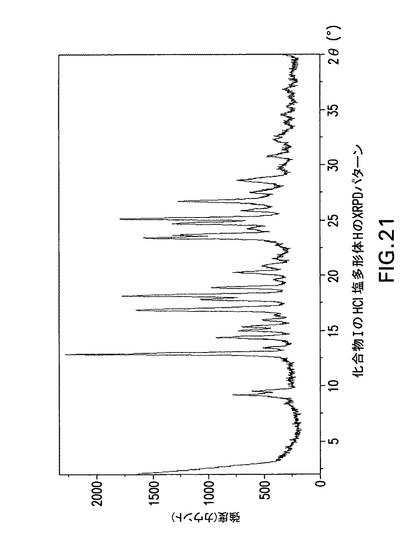
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【公表番号】特表2011−502119(P2011−502119A)
【公表日】平成23年1月20日(2011.1.20)
【国際特許分類】
【出願番号】特願2010−531013(P2010−531013)
【出願日】平成20年10月20日(2008.10.20)
【国際出願番号】PCT/US2008/011927
【国際公開番号】WO2009/054923
【国際公開日】平成21年4月30日(2009.4.30)
【出願人】(390023526)メルク・シャープ・エンド・ドーム・コーポレイション (924)
【Fターム(参考)】
【公表日】平成23年1月20日(2011.1.20)
【国際特許分類】
【出願日】平成20年10月20日(2008.10.20)
【国際出願番号】PCT/US2008/011927
【国際公開番号】WO2009/054923
【国際公開日】平成21年4月30日(2009.4.30)
【出願人】(390023526)メルク・シャープ・エンド・ドーム・コーポレイション (924)
【Fターム(参考)】
[ Back to top ]