
CB2受容体を調節する化合物
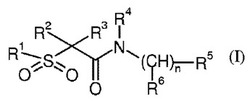
下記式(I)の化合物を開示する。本発明の化合物は、CB2受容体に結合し、かつCB2受容体の作動薬、拮抗薬又は逆作動薬であり、炎症の治療に有用である。作動薬である当該化合物は、さらに疼痛の治療に有用である。
【化1】

(I)
【化1】
(I)
【発明の詳細な説明】
【技術分野】
【0001】
(出願データ)
この出願は、2010年1月15日に出願された米国仮出願第61/295,201号の優先権を主張する。
(発明の背景)
1. 技術分野
本発明は、CB2受容体を調製する新規化合物及びそれらの薬物としての使用に関する。
【背景技術】
【0002】
2. 背景情報
カンナビノイドは、大麻(Cannabis sativa)(マリファナとしても知られる)に含まれている一群の約60の特徴的な化合物であり、最も代表的な分子はカンナビノール、カンナビジオール及びΔ9-テトラヒドロカンナビノール(THC)である。大麻の治療的利用は中国の古代王朝まで遡る可能性があり、食欲の欠如、嘔吐、痙攣、月経痛、痙縮からリウマチまで多岐にわたる種々の病気のための適用が含まれる。大麻使用の長い歴史がいくつかの医薬の開発につながった。例えば、THC及びその類似ナビロンに基づいているマリノール及びセサメット(Cesamet)は、それぞれ制吐薬及び食欲刺激薬として用いられる。臨床的利益にもかかわらず、大麻の治療的利用は、幻覚、中毒及び依存といったその精神活性作用のため制限されている。Mechoulam R, ed. Cannabinoids as Therapeutic Agents, Boca Raton, FL; CRC Press, 1986は、大麻の医薬的使用の概説を提供する。
【0003】
カンナビノイドの生理的作用は少なくとも2種のGタンパク質共役受容体CB1及びCB2によって媒介される。オートラジオグラフィー研究は、CB1受容体は中枢神経系内、特に大脳皮質、海馬、大脳基底核及び小脳内で主に発現されることを実証した。CB1受容体は、それほど多くはないが、生殖器系及び免疫系の末梢組織を含めた他の末梢組織内でも見られる。CB1受容体は、シナプス前ニューロンからの神経伝達物質の放出を調節し、かつ大麻の陶酔作用及び他の中枢神経系作用、例えばTHC誘導性リングカタレプシー(THC-induced ring-catalepsy)、運動性低下(hypomobility)、及び低体温等の多くを媒介すると考えられ、これらはCB1遺伝子欠失マウスには全く存在しないことが分かった(Zimmer et al., Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci U S A. (1999) 96:5780-5785.)。
CB2受容体はほとんど排他的に免疫系内で見られ、脾臓で最大密度である。免疫細胞内におけるCB2の発現レベルはCB1より約10〜100倍高いと見積もられている。免疫系内では、CB2は、B細胞、NK細胞、単細胞、ミクログリア細胞、好中球、T細胞、樹状細胞及びマスト細胞などの種々の細胞型内で見られ、CB2モジュレーターを介して種々多様の免疫機能を調節できることを示唆している(Klein et al., The cannabinoid system and immune system. J Leukoc Biol (2003) 74:.486-496)。このことは、CB2欠失マウスにはTHCの免疫調節作用が存在しないという知見によって支持される(Bicklet et al., Immunomodulation by cannabinoid is absent in mice deficient for the cannabinoid CB2 receptor. Eur J Pharmacol (2000) 396:141-149)。CB2選択性リガンドが開発され、種々の炎症環境内におけるそれらの作用について試験されている。例えば、炎症動物モデルにおいて、CB2選択性作動薬、逆作動薬及び拮抗薬が炎症の抑制に有効であることが示された(Hanus et al., HU-308: a specific agonist for CB(2), a peripheral cannabinoid receptor. Proc Natl Acad Sci U S A. (1999) 96:14228-14233, Ueda et al., Involvement of cannabinoid CB(2) receptor-mediated response and efficacy of cannabinoid CB(2) receptor inverse agonist, JTE-907, in cutaneous inflammation in mice. Eur J Pharmacol. (2005) 520:164-171 and Smith et al., The anti-inflammatory activities of cannabinoid receptor ligands in mouse peritonitis models Eur J Pharmacol. (2001) 432:107-119.)。さらに、CB2選択性作動薬は、多発性硬化症の動物モデルにおいて疾患の重症度及び痙縮性を抑制する(Baker et al., cannabinoids control spasticity and tremor in a multiple sclerosis model. Nature (2000) 404:84-87.Arevalo-Martin et al., Therapeutic action of cannabinoids in a murine model of multiple sclerosis J Neurosci. (2003) 23:2511-2516.)。まとめると、これらの結果は、炎症成分を有する医療状態の治療にCB2受容体モジュレーターを利用できるという概念を支持する。
【0004】
炎症に加えて、CB2作動薬は疼痛及び嘔吐を抑制することが分かっている。例えば、CB2選択性アゴニストは、熱又は他の刺激によって誘発される疼痛反応を鈍らせる(Malan et al., CB2 cannabinoid receptor-mediated peripheral antinociception. Pain. (2001) 93:239-45 and Nackley et al., Selective activation of cannabinoid CB(2) receptor suppresses spinal fos protein expression and pain behavior in a rat model of inflammation. Neuroscience (2003) 119:747-57.)。CB2活性化が神経障害性疼痛反応を抑制することも実証されている(Ibrahim et al., Activation of CB2 cannabinoid receptor by AM1241 inhibits experimental neuropathic pain: pain inhibition by receptor not present in the CNS. Proc Natl Acad Sci U S A. (2003) 100:10529-33.)。最後に、脳内でCB2を見つけていなかった初期データとは対照的に、最近の論文は、脳内のCB2の発現、脾臓内では約15%のレベルでの発現を実証した。この論文によってCB2活性化は内在性カンナビノイドの制吐作用に関与すると示されている(Van Sickle et al., Identification and functional characterization of brainstem cannabinoid CB2 receptor. Science. 2005 310:329-332.)。前述の結果は、CB2作動薬を炎症及び神経障害性疼痛並びに嘔吐の治療に使用できることを確証する。
WO2008014199及びWO2008039645は、そこで開示しているCB2受容体及びCB2作動活性を有するスルホン誘導体の治療的使用について考察している。
【発明の概要】
【発明が解決しようとする課題】
【0005】
(発明の概要)
本発明は、CB2受容体に結合してそれを調節する新規化合物を提供する。本発明は、治療量の本発明の化合物の投与を手段として炎症を治療するための方法及び医薬組成物をも提供する。最後に、本発明は、治療量の本発明の化合物の投与を手段として疼痛を治療するための方法及び医薬組成物を提供する。
【課題を解決するための手段】
【0006】
(発明の詳細な説明)
最も広い一般的実施形態1では、本発明は、下記式
【0007】
【化1】
(I)
【0008】
(式中、
R1は、C1-10アルキル、C3-10シクロアルキル、3〜10員飽和ヘテロ環式環、C1-5アルキル-ヘテロ環式環、5〜10員単環式若しくは二環式アリール環又はC1-5アルキル-アリール環であり、ここで、各R1は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C3-10シクロアルキル、C1-4アルコキシ、C1-4アルキルスルホニル、アシル、シアノ、ヒドロキシル及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
R2及びR3は、C1-4アルキル又は水素であり(但し、R2とR3が両方とも水素ではありえない);或いは
R2とR3が、それらが結合している炭素原子と一緒に3〜6員シクロアルキル環を形成し;
R4は、水素又はメチルであり;
R5は、C1-10アルキル、C3-10シクロアルキル、3-10員飽和ヘテロ環式環、5-10員単環式若しくは二環式アリール環、5〜10員単環式若しくは二環式ヘテロアリール環、-S-C1-5アルキル、-S-アリール、-S-CH2-アリール、-O-C1-5アルキル、-O-アリール、-O-CH2-アリール、-NH-C1-5アルキル、-NH-アリール、-NH-CH2-アリール又はアミノであり、ここで、各R5は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、アシル、-C(O)-ヘテロアリール、シアノ、ヒドロキシル及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒に4〜7員ヘテロ環式環(アリール、-C(O)-O-C1-5アルキル、-C(O)-ヘテロアリール、-C(O)-アリール、-SO2-ヘテロアリール及び-SO2-アリールから選択される置換基で置換されていてもよい)を形成していてもよく;
R6は、水素又はC1-4アルキルであり;
nは、0、1、2、3又は4であり;
ここで、式(I)又は前記いずれのR置換基上のいずれの炭素原子も、可能であれば、部分的又は完全にハロゲン化されていてもよい)
の化合物、
又はその医薬的に許容できる塩を提供する。
【0009】
別の実施形態2では、本発明は、上述した先行する一般的実施形態に従い、かつ式中、
R1が、C1-6アルキル、フェニル、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、-CH2-シクロプロピル、-CH2-シクロブチル、テトラヒドロフラニル、テトラヒドロピラニル、アゼチジニル、ピペリジニル;チオモルフォリニル、1,1-ジオキソ-1λ6-チオモルフォリニル、モルフォリニル、ピロリジニル、ピペラジニル、ベンジル、-CH2-テトラヒドロフラニル、-CH2-テトラヒドロピラニル、-CH2-CH2-テトラヒドロフラニル又は-CH2-CH2-テトラヒドロピラニルであり、ここで、各R1は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、C1-4アルキルスルホニル、アシル、シアノ、ヒドロキシル及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
R2及びR3が、独立にメチル、エチル、n-プロピル、イソプロピル、t-Bu、又は水素であり(但し、R2とR3が両方とも水素ではありえない);或いは
R2とR3が、それらが結合している炭素と一緒にシクロプロピル、シクロブチル、又はシクロペンチル環を形成し;
R4が、水素又はメチルであり;
R5が、C1-5アルキル、フェニル、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、テトラヒドロフラニル、テトラヒドロピラニル、アゼチジニル、ピペリジニル;チオモルフォリニル、1,1-ジオキソ-1λ6-チオモルフォリニル、モルフォリニル、ピロリジニル、ピペラジニル、インダニル、ベンゾオキサゾリル、ベンゾチアゾリル、ベンゾイミダゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、ピラゾリル、ピロリル、イミダゾリル、チエニル、チアジアゾリル、ピリジニル、ピリミジニル、ピリダジニル、ピラジニル、トリアジニル、キノリニル、ジヒドロ-2H-キノリニル、イソキノリニル、キナゾリニル、インダゾリル、インドリル、ベンゾフラニル、ベンゾピラニル、ベンゾジオキソリル、-S-C1-5アルキル、-S-フェニル、-S-CH2-フェニル、-O-C1-5アルキル、-O-フェニル、-O-CH2-フェニル、-NH-C1-5アルキル、-NH-フェニル、-NH-CH2-フェニル又はアミノであり、ここで、各R5は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、アシル、-C(O)-ヘテロアリール、シアノ、ヒドロキシル及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒に4〜7員ヘテロ環式環(0又は1個のさらなるヘテロ原子を含み、フェニル、-C(O)-O-C1-5アルキル、-C(O)-チエニル、-C(O)-フェニル、-SO2-ヘテロアリール及び-SO2-フェニルから選択される置換基で置換されていてもよい)を形成していてもよく;
R6が、水素又はC1-3アルキルである、
式(I)の化合物を提供する。
【0010】
別の実施形態3では、本発明は、先行する実施形態のいずれかに従い、かつ式中、
R1が、C1-6アルキル、フェニル、シクロペンチル、シクロヘキシル、テトラヒドロピラニル、ベンジル又は-CH2-テトラヒドロピラニルであり、ここで、各R1は、独立にC1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、C1-4アルキルスルホニル、及びハロゲンから選択される1〜3個の置換基で置換されていてもよい、
式(I)の化合物を提供する。
別の実施形態4では、本発明は、先行する実施形態のいずれかに従い、かつ式中、
R2及びR3が独立にメチルであり、或いは
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成している、
式(I)の化合物を提供する。
別の実施形態5では、本発明は、先行する実施形態のいずれかに従い、かつ式中、
R4が水素又はメチルであり;
R5が、C1-5アルキル、フェニル、シクロヘキシル、シクロヘプチル、モルフォリニル、インダニル、イソオキサゾリル、チエニル、ピリジニル、-S-C1-5アルキル、-S-フェニル、-S-CH2-フェニル、-O-C1-5アルキル、-O-フェニル、-NH-C1-5アルキル、-NH-フェニル又はアミノであり、ここで、各R5は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、アシル、及びハロゲンから選択される1〜3個の置換基で置換されていてもよい、
式(I)の化合物を提供する。
別の実施形態6では、本発明は、先行する実施形態1〜4のいずれかに従い、かつ式中、
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒に4〜7員ヘテロ環式環(0又は1個のさらなるヘテロ原子を含み、かつ、フェニル、-C(O)-O-C1-4アルキル、-C(O)-チエニル及び-SO2-フェニルから選択される置換基で置換されていてもよい)を形成していてもよい、
式(I)の化合物を提供する。
【0011】
別の実施形態7では、本発明は、実施形態2に従い、かつ式中、
R1が、C1-6アルキル、フェニル、シクロペンチル、シクロヘキシル、テトラヒドロピラニル、ベンジル又は-CH2-テトラヒドロピラニルであり、ここで、各R1は独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、C1-4アルキルスルホニル、及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
R2及びR3が独立にメチルであり、或いは
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成し;
R4が水素又はメチルであり;
R5が、C1-5アルキル、フェニル、シクロヘキシル、シクロヘプチル、モルフォリニル、インダニル、イソオキサゾリル、チエニル、ピリジニル、-S-C1-5アルキル、-S-フェニル、-S-CH2-フェニル、-O-C1-5アルキル、-O-フェニル、-NH-C1-5アルキル、-NH-フェニル又はアミノであり、ここで、各R5は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、アシル、及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒にピペリジニル又はピペラジニル(フェニル、-C(O)-O-C1-4アルキル、-C(O)-チエニル及び-SO2-フェニルから選択される置換基で置換されていてもよい)を形成していてもよく;
R6が水素又はメチルであり;
nが、0、1、2又は3である、
式(I)の化合物を提供する。
【0012】
別の実施形態8では、上記実施形態7に従い、かつ式中、
R1が、フェニル、シクロヘキシル又はベンジルであり、ここで、各R1は、独立に、トリフルオロメチル、及びクロロから選択される1〜2個の置換基で置換されていてもよく;
R2及びR3がメチルであり、或いは
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成し;
R4が水素であり;
R5が、C1-4アルキル、フェニル、シクロヘキシル、シクロヘプチル、チエニル、-S-C1-5アルキル、-S-フェニル、-S-CH2-フェニル、-O-C1-5アルキル、-O-フェニル又は-NH-フェニルであり、ここで、各R5は、独立に、C1-4アルキル、トリフルオロメチル、C1-4アルコキシ、フルオロ及びクロロから選択される1〜3個の置換基で置換されていてもよい、
式(I)の化合物を提供する。
別の実施形態9では、実施形態7に従い、かつ式中、
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒にピペリジニル又はピペラジニル(フェニル、-C(O)-O-C1-4アルキル及び-C(O)-チエニルから選択される置換基で置換されていてもよい)を形成していてもよい、
式(I)の化合物を提供する。
別の実施形態10では、実施形態8に従い、かつ式中、
R1がフェニル又はシクロヘキシルであり、それぞれトリフルオロメチル、及びクロロから選択される置換基で置換されていてもよく;
R2及びR3がメチルであり、或いは
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成し;
R4が水素であり;
R5が、C1-4アルキル、フェニル、シクロヘキシル、-S-C1-5アルキル、-S-フェニル又は-S-CH2-フェニルであり、ここで、各R5は、独立に、C1-4アルキル、トリフルオロメチル、メトキシ、フルオロ及びクロロから選択される1〜3個の置換基で置換されていてもよく;
nが0又は2である、
式(I)の化合物を提供する。
別の実施形態11では、実施形態9に従い、かつ式中、
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒に、フェニル基で置換されているピペラジニル環を形成している、
式(I)の化合物を提供する。
別の実施形態12では、上述した先行する実施形態のいずれかに従い、かつ式中、
R2及びR3がメチルである、
式(I)の化合物を提供する。
別の実施形態13では、先行する実施形態1〜12のいずれかに従い、かつ式中、
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成している、
式(I)の化合物を提供する。
別の実施形態では、下記式(IA)の化合物又はその医薬的に許容できる塩を提供する。
【0013】
【化2】
(IA)
【0014】
ここで、式(IA)の下記部分
【0015】
【化3】
【0016】
は、下表IのカラムA1〜A16から選択され、かつ式(IA)の下記部分
【0017】
【化4】
【0018】
は、下表IのカラムB1〜B48から選択される。
【0019】
表I
【0020】
別の実施形態では、本発明は、当技術分野で既知の一般的スキーム、例及び方法を考慮して作製できる下表IIの作製された化合物又はその医薬的に許容できる塩を提供する。
【0021】
表II
【発明を実施するための形態】
【0022】
この出願で上記開示化合物において、命名法が構造と矛盾する場合、化合物は構造によって定義されるものと解釈すべきである。
本発明は、式(I)の1種以上の化合物、又はその医薬的に許容できる誘導体を活性物質として含み、必要に応じて通常の賦形剤及び/又は担体と組み合わせた医薬製剤にも関する。
本発明の化合物は、それらの同位体標識形をも包含する。本発明の組合せの活性薬の同位体標識形は、前記活性薬の1つ以上の原子が、通常自然界に見られる前記原子の原子質量又は質量数と異なる原子質量又は質量数を有する原子と置き換わっているという事実を別にすれば、前記活性薬と同一である。商業的に容易に入手可能であり、かつよく確立された手順に従って本発明の組合せの活性薬に組み込める同位体の例としては、水素、炭素、窒素、酸素、リン、フッ素及び塩素の同位体、例えば、それぞれ、2H、3H、13C、14C、15N、18O、17O、31P、32P、35S、18F、及び36Clが挙げられる。本発明の組合せの1つ以上の上記同位体及び/又は他の原子の他の同位体を含有する活性薬、そのプロドラッグ、又はどちらかの医薬的に許容できる塩は、本発明の範囲内であると考えられる。
本発明は、ラセミ体及びラセミ混合物、単一のエナンチオマー、ジアステレオマー混合物及び個々のジアステレオマーとして存在し得る、1つ以上の不斉炭素原子を含有する上記いずれの化合物の使用をも包含する。異性体をエナンチオマー及びジアステレオマーと定義するものとする。これらの化合物の全ての該異性形が明白に本発明に包含される。各ステレオジェン炭素は、R若しくはS配置、又は組合せ配置であってよい。
式(I)の化合物が複数の互変異性形で存在することもある。本発明は、全ての該互変異性体を使用する方法を包含する。
【0023】
この明細書で使う全ての用語は、特に指定のない限り、当技術分野で知られているそれらの通常の意味に解釈するものとする。例えば、「C1-4アルコキシ」は、末端酸素を有するC1-4アルキルであり、例えばメトキシ、エトキシ、プロポキシ、ブトキシである。全てのアルキル、アルケニル及びアルキニル基は、構造的に可能であり、かつ特に指定のない限り、分岐又は不分岐であると解釈するものとする。他のさらに詳細な定義は以下のとおりである。
炭素環又はシクロアルキルは、3〜12個の炭素原子を含む炭化水素環を包含する。これらの炭素環は、芳香族又は非芳香族環系のどちらでもよい。非芳香族環系は、一不飽和又は多不飽和であってよい。好ましい炭素環としては、限定するものではないが、シクロプロピル、シクロブチル、シクロペンチル、シクロペンテニル、シクロヘキシル、シクロヘキセニル、シクロヘプタニル、シクロヘプテニル、フェニル、インダニル、インデニル、ベンゾシクロブタニル、ジヒドロナフチル、テトラヒドロナフチル、ナフチル、デカヒドロナフチル、ベンゾシクロヘプタニル及びベンゾシクロヘプテニルが挙げられる。シクロアルキルについての特定用語、例えばシクロブタニル及びシクロブチルは、互換的に使われるものとする。
用語「ヘテロ環」は、安定した非芳香族の3〜10員(好ましくは5又は6員)単環式又は非芳香族の8〜11員二環式ヘテロ環基を意味し、飽和又は不飽和であってよい。各ヘテロ環は、炭素原子と、窒素、酸素及びイオウから選択される1個以上、好ましくは1〜4個のヘテロ原子とから成る。ヘテロ環は、安定構造の生成をもたらす、環のいずれの原子によっても付着され得る。
用語「ヘテロアリール」は、N、O及びS等の1〜4個のヘテロ原子を含有する芳香族の5〜10員単環式又は8〜11員二環式環を意味すると解釈するものとする。
特に指定のない限り、ヘテロ環及びヘテロアリールとして、限定するものではないが、例えばベンゾオキサゾリル、ベンゾチアゾリル、ベンゾイミダゾリル、テトラヒドロピラニル、ジオキサニル、テトラヒドロフラニル、オキサゾリル、イソオキサゾリル、チアゾリル、ピラゾリル、ピロリル、イミダゾリル、チエニル、チアジアゾリル、トリアゾリル、チオモルフォリニル、1,1-ジオキソ-1λ6-チオモルフォリニル、モルフォリニル、ピリジニル、ピリミジニル、ピリダジニル、ピラジニル、トリアジニル、ピロリジニル、ピペリジニル、ピペラジニル、プリニル、キノリニル、ジヒドロ-2H-キノリニル、イソキノリニル、キナゾリニル、インダゾリル、チエノ[2,3-d]ピリミジニル、インドリル、イソインドリル、ベンゾフラニル、ベンゾピラニル及びベンゾジオキソリルが挙げられる。
本明細書では、用語「ヘテロ原子」は、炭素以外の原子、例えばO、N、S及びPを意味すると解釈するものとする。
【0024】
全てのアルキル基又は炭素鎖では、1個以上の炭素原子がヘテロ原子:O、S又はNと置き換わることがあり、Nが置換されない場合は、それはNHであると解釈するものとし、ヘテロ原子は分岐又は不分岐炭素鎖内の末端炭素原子又は内部炭素原子のどちらとも置き換わり得ることをも理解するものとする。このような基は、上述したように、オキソ等の基によって置換されて、限定するものではないが、アルコキシカルボニル、アシル、アミノ及びチオキソ等の定義をもたらし得る。
用語「アリール」は、本明細書では芳香族炭素環を意味すると解釈するものとする。
各アリール又はヘテロアリールは、特に指定のない限り、その部分的又は完全に水素化した誘導体を包含する。例えば、キノリニルにはデカヒドロキノリニル及びテトラヒドロキノリニルが含まれ、ナフチルには、その水素化誘導体、例えばテトラヒドロナフチルが含まれる。当業者には、本明細書に記載のアリール及びヘテロアリールの他の部分的又は完全に水素化した誘導体が明らかであろう。
本明細書では、「窒素」及び「イオウ」は、窒素及びイオウのいずれの酸化形並びにいずれの塩基性窒素の四級化形をも包含する。例えば、-S-C1-6アルキル基では、特に指定のない限り、これは-S(O)-C1-6アルキル及び-S(O)2-C1-6アルキルを包含すると解釈するものとする。
用語「ハロゲン」は、本明細書で使用する場合、臭素、塩素、フッ素又はヨウ素、好ましくはフッ素及び塩素を意味すると解釈するものとする。定義「部分的又は完全にハロゲン化」;「部分的又は完全にフッ素化」;「1個以上のハロゲン原子で置換された」は、例えば、1個以上の炭素原子に関するモノハロ、ジハロ又はトリハロ誘導体を包含する。アルキルについての非限定例は-CH2CHF2、-CF3等であろう。
【0025】
本発明の化合物は、当業者には明らかなように、「化学的に安定」であると考えられる化合物のみである。例えば、「ダングリング原子価」、又は「カルボアニオン」を有するであろう化合物は、ここに開示する発明方法によって予想される化合物ではない。
本発明は、式(I)の化合物の医薬的に許容できる誘導体を包含する。「医薬的に許容できる誘導体」とは、患者に投与すると、本発明に有用な化合物、又はその薬理学的に活性な代謝物又は薬理学的に活性な残基を(直接又は間接的に)もたらすことができるいずれの医薬的に許容できる塩若しくはエステル、又はいずれの他の化合物をも意味する。薬理学的に活性な代謝物は、酵素的又は化学的に代謝され得る本発明のいずれの化合物をも意味すると解釈するものとする。これには、例えば、式(I)のヒドロキシル化又は酸化誘導化合物が含まれる。
医薬的に許容できる塩には、医薬的に許容できる無機酸及び塩基並びに有機酸及び塩基から誘導される当該塩が含まれる。適切な酸の例としては、塩酸、臭化水素酸、硫酸、硝酸、過塩素酸、フマル酸、マレイン酸、リン酸、グリコール酸、乳酸、サリチル酸、コハク酸、トルエン-p-硫酸、酒石酸、酢酸、クエン酸、メタンスルホン酸、ギ酸、安息香酸、マロン酸、ナフタレン-2-硫酸及びベンゼンスルホン酸が挙げられる。他の酸、例えばシュウ酸は、それ自体医薬的に許容性ではないが、本化合物及びそれらの医薬的に許容できる酸付加塩を得るときの中間体として有用な塩の調製に利用し得る。適切な塩基から誘導される塩としては、アルカリ金属(例えば、ナトリウム)、アルカリ土類金属(例えば、マグネシウム)、アンモニウム及びN-(C1-C4アルキル)4+塩が挙げられる。
さらに、式(I)の化合物のプロドラッグの使用が本発明の範囲内である。プロドラッグには、簡単な化学変換が起こると、改変されて本発明の化合物を生成する当該化合物が含まれる。簡単な化学変換には、加水分解、酸化及び還元がある。詳細には、プロドラッグが患者に投与されると、プロドラッグは上記開示化合物に変換され、それによって所望の薬理作用を与え得る。
後述する一般的合成方法(これも本発明の一部を構成する)を利用して式Iの化合物を作製することができる。
【0026】
(一般的合成方法)
本発明は、式(I)及び(IA)の化合物の作製方法をも提供する。全ての方法で、特に指定のない限り、下記式中のR、R1、R2、R3、R4、R5、R6及びnは、上述した本発明の式(I)中のR、R1、R2、R3、R4、R5、R6及びnの意味を有するものとする。
最適反応条件及び反応時間は、使用する特定の反応物質によって変化し得る。特に指定のない限り、当業者は、溶媒、温度、圧力、及び他の反応条件を容易に選択し得る。合成例セクションで具体的手順を提供する。典型的に、必要に応じて、薄層クロマトグラフィー(TLC)によって反応の進行をモニターすることができ、かつシリカゲル上クロマトグラフィー及び/又は再結晶によって中間体及び生成物を精製することができる。
以下の実施例は例示であり、当業者は認識しているように、過度の実験を行うことなく、個々の化合物に必要なように特定の試薬又は条件を変更できるであろう。下記方法で使用する出発原料及び中間体は、商業的に入手可能であり、或いは市販原料から当業者が容易に調製できる。WO2008098025、WO2008014199、WO2008039645、及びWO2009061652に開示されている合成方法を本発明の化合物の調製に利用してもよい。
下記スキーム1に示す方法で式(I)及び(IA)の化合物を合成することができる。
【0027】
【化5】
スキーム1
【0028】
スキーム1に示すように、式IIの酸を塩化チオニル又は塩化オキサリル等の試薬と反応させて酸塩化物を得て、適切な溶媒中、適切な塩基の存在下で式IIIのアミンと反応させて式(I)の化合物を得る。或いは、式IIの酸を標準的カップリング条件下で式IIIのアミンと結合して式(I)の化合物を得てもよい。これらの合成では技術上既知の標準的ペプチドカップリング反応(例えばM. Bodanszky, 1984, The Practice of Peptide Synthesis, Springer-Verlag参照)を利用してよい。適切なカップリング条件の例は、DMF等の適切な溶媒中のカルボン酸の溶液をEDC、HOBT、及び塩基、例えばジイソプロピルエチルアミンと処理した後、所望アミンと処理する。
技術上既知かつ下記実施例に示す方法による式(I)の初期生成物のさらなる改変を利用して本発明のさらなる化合物を調製することができる。
下記スキーム2に概要を示す方法で中間体酸IIを作製し得る。
【0029】
【化6】
スキーム2
【0030】
上述したように、適切な溶媒中、適切な塩基の存在下での式IVのチオールと式Vのブロモエチルエステルの反応が式VIチオエーテルをもたらす。式VIのチオエーテルを適切な酸化剤と反応させて、対応する式VIIのスルホンを得る。適切な溶媒中、水酸化リチウム等の適切な塩基の存在下における式VIIのスルホンのエステル基の加水分解が、対応する式IIの酸をもたらす。
下記スキーム3に概要を示す方法で中間体酸IIを作製してもよい。
【0031】
【化7】
スキーム3
【0032】
適切な溶媒中、式Vの出発ブロモエステルのチオ酢酸カリウム等の試薬との反応が式VIIIのチオ酢酸エステルをもたらす。適切な塩基の存在下でのチオ酢酸エステルVIIIと式IXの臭化物の反応が、対応する式VIのスルファニル酸エチルエステルをもたらす。スキーム2に示す工程順序によって式VIのスルファニル酸エチルエステルを式IIの中間体酸に変換し得る。
下記スキーム4に概要を示す方法によって中間体IIを作製し得る。
【0033】
【化8】
スキーム4
【0034】
スキーム4に示すように、適切な溶媒中、適切な塩基の存在下での式Xのアルコールとp-トルエンスルホニルクロリドの反応が式XIのスルホン酸エステルをもたらす。適切な溶媒中、式XIの化合物とチオ酢酸カリウムの反応が式XIIの化合物をもたらす。適切な溶媒中、適切な塩基の存在下での式XIIの中間体と式Vのブロモエステルの反応が式VIをもたらし、これをスキーム2に示す反応順序で式IIの所望中間体に変換することができる。
下記スキーム5に概要を示す方法で中間体IIを作製してもよい。
【0035】
【化9】
スキーム5
【0036】
スキーム5に示すように、文献に報告された手順を用いて式XIIIの塩化スルホニルを対応する式XIVのスルフィン酸ナトリウム塩に変換する。適切な溶媒中での式XIVのスルフィン酸ナトリウム塩と式Vのブロモエステルの反応が式VIIのスルホンエステルをもたらす。式VIIのスルホンエステルの加水分解が式IIの中間体酸をもたらす。
【0037】
(合成例)
下記実施例によって、本発明の化合物を作製可能な方法がさらに理解されるであろう。
酸方法A:
方法Aの酸をWO2008039645, Boehringer Ingelheim International GmbHに記載のように、或いはWO2008014199, Boehringer Ingelheim International GmbHに記載のように調製する。
2-シクロペンタンスルホニル-2-メチル-プロピオン酸の合成
【0038】
【化10】
【0039】
工程1:2-シクロペンチルスルファニル-2-メチル-プロピオン酸エチルエステルの合成
エタノール(50mL)中5g(48.7mmol)のシクロペンチルチオールの溶液に2.7g(48.75mmol)のKOHペレットを添加した後、9.5g(48.7mmol)のα-ブロモイソ酪酸エチルを加える。反応を加熱して2時間還流させてから室温に冷ます。固体(KBr)をろ過で分離し、エタノール(20mL)ですすぐ。ろ液を減圧下で濃縮し、残留物をDCM(50mL)に溶かす。有機層をNaHCO3飽和水溶液(50mL)で洗浄する。水性洗浄液をDCM(10mL)で逆抽出する。混ぜ合わせた有機物を食塩水で洗浄し、Na2SO4上で乾燥させる。ろ過及び減圧下での濃縮が8.1gの2-シクロペンチルスルファニル-2-メチル-プロピオン酸エチルエステルを与える。収率:77%、ES-MS:m/z 217 [M+H]
この手順に従い、下記変更に留意しながら以下のチオエーテルを合成する:合成例13〜14、17〜18及び21〜22を合成する場合の酸については、α-ブロモイソ酪酸の代わりにエチル-1-ブロモシクロブタンカルボキシラートを使用する。
【0040】
表1
*反応条件中にエチルエステルの加水分解が起こった
【0041】
工程2:2-シクロペンタンスルホニル-2-メチル-プロピオン酸エチルエステルの合成
1,4-ジオキサン/水(4/1,100mL)中6g(27.7mmol)の2-シクロペンチルスルファニル-2-メチル-プロピオン酸エチルエステルの溶液に数回に分けて51.2g(83mmol)の一過硫酸カリウム三重塩(OXONE(登録商標))を添加する。白色懸濁液を室温で3時間撹拌する。白色固体をろ過で分離し、1,4-ジオキサン(10mL)で洗浄する。ろ液を減圧下で濃縮して有機溶媒を除去する。結果として生じる水溶液をDCM(3×40mL)で抽出する。混ぜ合わせた有機抽出物をNaHCO3飽和水溶液、食塩水で洗浄し、Na2SO4上で乾燥させてろ過する。ろ液を減圧下で濃縮して5.4gの2-シクロペンタンスルホニル-2-メチル-プロピオン酸エチルエステルを得る。収率:78%、ES-MS:m/z 249 [M+H]
この手順に従って以下のスルホンを合成する。
表2
*酸化工程のため2-(4-クロロ-ベンジルスルファニル)-2-メチル-プロピオン酸を使用
【0042】
工程3:2-シクロペンタンスルホニル-2-メチル-プロピオン酸の合成
THF/水(4/1,60mL)中5.4g(21.7mmol)の2-シクロペンタンスルホニル-2-メチル-プロピオン酸エチルエステルの溶液に2.3g(56.6mmol)の水酸化リチウム一水和物をを添加する。反応を室温で18時間撹拌する。反応をさらに水(20mL)で希釈してからDCM(2×15mL)で洗浄する。塩基性水層を氷浴で冷却してから2M HCl水溶液で酸性にしてpHを2とする。酸性水層を2-プロパノール/クロロホルム(1/4,100mL)で抽出する。混ぜ合わせた有機抽出物を食塩水で洗浄し、Na2SO4上で乾燥させてろ過する。減圧下でろ液を濃縮して4.34gの2-シクロペンタンスルホニル-2-メチル-プロピオン酸を得る。収率:92%、ES-MS:m/z 221 [M+H]
この手順に従って以下の酸を合成する。
【0043】
表3
【0044】
酸方法B:
WO2008039645, Boehringer Ingelheim International GmbHに記載の方法を適応して方法Bの酸を調製する。
2-メチル-2-(テトラヒドロ-ピラン-4-イルメタンスルホニル)-プロピオン酸
【0045】
【化11】
【0046】
工程1:(テトラヒドロ-ピラン-4-イル)-メタノールの合成
窒素雰囲気下で、THF(200mL)中のLiAlH4溶液250mL(THF中2.3M溶液,0.575mol)にTHF(900mL)中130mL(0.974mol)のテトラヒドロ-ピラン-4-カルボン酸メチルエステルの溶液を滴下して添加する(注意:激しい発熱反応!)。氷浴で温度を40〜45℃に維持する。添加が完了したら、反応を室温で1.5時間撹拌する。反応を氷浴で冷まし、水(22mL)、15%NaOH水溶液(21mL)及び水(66mL)を添加してクエンチする。結果として生じる沈殿物をCelite(登録商標)でろ過して除去し、THF(300mL)ですすぐ。ろ液を減圧下で濃縮して102.5gの(テトラヒドロ-ピラン-4-イル)-メタノールを無色油として得る。収率:91%;1H NMR (400 MHz, クロロホルム-d) δ ppm 1.20 - 1.39 (2 H, m), 1.56 - 1.83 (3 H, m), 2.03 (1 H, br. s.), 3.29 - 3.52 (4 H, m), 3.89 - 4.05 (2 H, m)
【0047】
工程2:トルエン-4-スルホン酸テトラヒドロ-ピラン-4-イルメチルエステルの合成
下記文献参照の適応によって記載どおりに調製する:
Radziszewski, J.G. et al. J. Am. Chem. Soc. 1993, 115, 8401。
2-メチルテトラヒドロフラン(190mL)中97g(810mmol)の(テトラヒドロ-ピラン-4-イル)-メタノールの溶液に165mLの50%NaOH水溶液を加える。この撹拌溶液に2-メチルテトラヒドロフラン(280mL)中のp-トルエン-スルホニルクロリドの溶液(283g,1.46mol)を冷却しながら滴下して添加する。反応を30〜35℃で18時間撹拌する。懸濁液を氷水(280mL)とHCl水溶液(37%,203mL)の混合物中に注ぐ。メチルシクロヘキサン(1.4L)及びさらなる氷水(0.2L)の添加後、反応混合物を氷浴内で2時間撹拌する。結果として生じる結晶性沈殿物をろ過で単離し、メチルシクロヘキサン(0.5L)及び水(0.5L)で洗浄する。減圧下40℃で乾燥させて216gのトルエン-4-スルホン酸テトラヒドロ-ピラン-4-イルメチルエステルを白色結晶性固体として得る。収率:99%、ES-MS:m/z 271 [M+H]; 1H NMR (400 MHz, クロロホルム-d) δ ppm 1.19 - 1.35 (2 H, m), 1.54 - 1.63 (2 H, m), 1.85 - 2.02 (1 H, m), 2.45 (3 H, s), 3.28 - 3.39 (2 H, m), 3.86 (2H, d, J=6.60 Hz), 3.93 (2 H, dd, J=11.37, 4.52 Hz), 7.35 (2 H, d, J=9.29 Hz), 7.78 (2 H, d, J=8.31 Hz)
【0048】
工程3:チオ酢酸S-(テトラヒドロ-ピラン-4-イルメチル)エステルの合成
下記文献参照の適応によって記載どおりに調製する:
Watson, R.J. et al. Tetrahedron Lett. 2002, 43, 683-685。
メチルイソブチルケトン(1.6L)中224g(0.83mol)のトルエン-4-スルホン酸テトラヒドロ-ピラン-4-イルメチルエステルの溶液に189g(1.66mol)のチオ酢酸カリウムを加える。ベージュ色懸濁液を70℃で4.5時間撹拌する。反応混合物を室温に冷まして水(1.8L)を加える。有機層を10%K2CO3水溶液(1.8L)と水(1L)で洗浄する。有機層をcelite(登録商標)(20g)、活性炭(20g)及びNa2SO4(20g)を通してろ過し、ろ液を減圧下で濃縮する。残存油をメチルシクロヘキサン(200mL)及びn-ヘプタン(250mL)と共沸させて138gのチオ酢酸S-(テトラヒドロ-ピラン-4-イルメチル)エステルを黄橙色油として得る(注意:悪臭!)。収率:96%;ES-MS:m/z 175 [M+H]; 1H NMR (400 MHz, クロロホルム-d) δ ppm 1.23 - 1.40 (2 H, m), 1.59 - 1.78 (3 H, m), 2.33 (3 H, d, J=4.16 Hz), 2.82 (2 H, dd, J=6.24, 3.79 Hz), 3.27- 3.39 (2 H, m), 3.88 - 4.02 (2 H, m)
【0049】
工程4:2-メチル-2-(テトラヒドロ-ピラン-4-イルメタンスルホニル)-プロピオン酸エチルエステルの合成
トルエン(500mL)中90g(516mmol)のチオ酢酸S-(テトラヒドロ-ピラン-4-イルメチル)エステルの溶液を窒素雰囲気下にて氷浴内で冷却する。エタノール中ナトリウムエトキシドの溶液(21%,231mL)を添加して反応を50分間撹拌する。次に76mL(516mmol)のα-ブロモイソ酪酸エチルを加えて反応を1時間撹拌する。反応混合物に氷酢酸(8.9mL)と水(500mL)を加える。有機層を分けて水(500mL)で洗浄する。三口丸底フラスコに水(500mL)、oxone(登録商標)(477g,775mmol)及び硫酸水素テトラブチルアンモニウム(5g,15mmol)を入れて有機層を加える。二相性反応混合物を室温で2日間撹拌する。固体をろ過で除去し、ろ液の層を分ける。有機層を水(2×500mL)で洗浄する。減圧下で溶媒を除去し、さらにトルエンと共沸させて125gの2-メチル-2-(テトラヒドロ-ピラン-4-イルメタンスルホニル)-プロピオン酸エチルエステルを得る。収率:87%;ES-MS:m/z 279 [M+H];
1H NMR (250 MHz, クロロホルム-d) δppm 1.32 (3 H, t, J=7.16 Hz), 1.39 - 1.59 (2 H, m), 1.64 (6 H, s), 1.81 - 1.97 (2 H, m), 2.29 - 2.53 (1 H, m),3.15 (2 H, d, J=6.55 Hz), 3.45 (2 H, dd, J=1.83, 0.30 Hz), 3.88 - 4.03 (2 H, m), 4.26 (2 H, d, J=7.16 Hz)
【0050】
工程5:2-メチル-2-(テトラヒドロ-ピラン-4-イルメタンスルホニル)-プロピオン酸の合成
方法Aの工程3の適応によって記載どおりに調製する。
THF(450mL)中123g(0.44mol)の2-メチル-2-(テトラヒドロ-ピラン-4-イルメタンスルホニル)-プロピオン酸エチルエステルの溶液に663mLの2M水酸化ナトリウム水溶液(1.33mol)を加える。反応を室温で1時間撹拌する。反応混合物にTBME(1.25L)を加えて層を分ける。水層を氷浴内で冷却してから37%HCl水溶液(123mL)で酸性にする。結果として生じる沈殿物をろ過で単離し、水(200mL)で洗浄し、減圧下で50℃にて乾燥させて101gの2-メチル-2-(テトラヒドロ-ピラン-4-イルメタンスルホニル)-プロピオン酸を白色結晶性固体として得る。収率:91%;ES-MS:m/z 251 [M+H];1H NMR (400 MHz, DMSO-d6) δ ppm 1.31 - 1.45 (2 H, m), 1.49 (6 H, s), 1.70 - 1.79 (2 H, m), 2.13 - 2.28 (1 H, m), 3.24 (2 H, d, J=6.60 Hz), 3.28 - 3.38 (2 H, m), 3.76 - 3.85 (2 H, m), 13.65 (1 H, br. s.)
【0051】
酸方法C:
2-メチル-2-(4,4,4-トリフルオロ-ブタン-1-スルホニル)-プロピオン酸の合成
【0052】
【化12】
【0053】
工程1:2-アセチルスルファニル-2-メチル-プロピオン酸エチルエステルの合成
DMF(500mL)中α-ブロモイソ酪酸エチル(62g,0.32mol)の溶液に室温でチオ酢酸カリウム (72g,0.63mol)を加える。反応を16時間撹拌してから減圧下で濃縮する。残留物を2M塩酸水溶液(500mL)で希釈し、酢酸エチル(3×500mL)で抽出する。有機フラクションを混ぜ合わせて食塩水(300mL)で洗浄し、MgSO4上で乾燥させ、ろ過し、減圧下で濃縮する。ヘプタン/ジクロロメタンで溶出するシリカ上クロマトグラフィーで精製して44gの2-アセチルスルファニル-2-メチル-プロピオン酸エチルエステルを得る。収率:73%;m/z 191 [M+H];1H NMR (250 MHz, クロロホルム-d) δ ppm 1.18 - 1.30 (3 H, m), 1.57 (6 H, s), 2.27 (3 H, s), 4.19 (2 H, q, J=7.16 Hz)。
【0054】
工程1:2-メチル-2-(4,4,4-トリフルオロ-ブチルスルファニル)-プロピオン酸エチルエステルの合成
エタノール(1.2L,窒素下で1時間脱気した)中149g(785.4mmol)の2-アセチルスルファニル-2-メチル-プロピオン酸エチルエステルの溶液に169.7g(105mmol)のナトリウムメトキシドを添加した後、150g(785.4mmol)の1-ブロモ-4,4,4-トリフルオロブタンの溶液を加える。反応を85℃で3日間加熱する。減圧下で溶媒を除去する。残留物をDCM(1L)に溶かし、NaHCO3飽和水溶液で洗浄する(2×1L)。有機層をNa2SO4上で乾燥させ、ろ過し、ろ液を減圧下で濃縮して171gの2-メチル-2-(4,4,4-トリフルオロ-ブチルスルファニル)-プロピオン酸エチルエステルを褐色油として得る。収率:84%;ES-MS:m/z 259 [M+H];1H NMR (500 MHz, クロロホルム-d) δ ppm 1.29 (3 H, t, J=7.17 Hz), 1.51 (6 H, s), 1.76 - 1.86 (2 H, m), 2.12 - 2.27 (2 H, m), 2.69 (2 H, t, J=7.17 Hz), 4.18 (2 H, q, J=7.17 Hz)。
【0055】
工程2:2-メチル-2-(4,4,4-トリフルオロ-ブタン-1-スルホニル)-プロピオン酸エチルエステルの合成
1,4-ジオキサン/水(1/1,4L)中220g(851.7mmol)の2-メチル-2-(4,4,4-トリフルオロ-ブチルスルファニル)-プロピオン酸エチルエステルの溶液に1047g(1703.4mmol)のoxone(登録商標)を室温で0.5時間かけて少しずつ添加する。反応混合物を室温で18時間撹拌する。固体をろ過で除去して1,4-ジオキサン(0.5L)ですすぐ。ろ液を減圧下で濃縮して有機溶媒を除去する。水性残留物をDCM(2×1L)で抽出する。混ぜ合わせた有機抽出物をNaHCO3飽和水溶液(2L)で洗浄し、Na2SO4上で乾燥させてろ過する。ろ液を減圧下で濃縮して226gの2-メチル-2-(4,4,4-トリフルオロ-ブタン-1-スルホニル)-プロピオン酸エチルエステルを暗黄色油として得る。収率92%;ES-MS:m/z 291 [M+H];1H NMR (500 MHz, クロロホルム-d) δ ppm 1.32 (3 H, t, J=7.17 Hz), 1.66 (6 H, s), 2.20 (2 H, quin, J=7.59 Hz), 2.28 - 2.41 (2 H, m), 3.34 (2 H, t, J=7.48 Hz), 4.27 (2 H, q, J=7.17 Hz)。
【0056】
工程3:2-メチル-2-(4,4,4-トリフルオロ-ブタン-1-スルホニル)-プロピオン酸の合成
THF(3.4L)中170g(585.6mmol)の2-メチル-2-(4,4,4-トリフルオロ-ブタン-1-スルホニル)-プロピオン酸エチルエステルの溶液に225.4g(1756.8mmol)のカリウムトリメチルシラノラート(silanolate)を0.5時間かけて少しずつ添加する。反応を室温で18時間撹拌する。反応混合物を2M HCl水溶液(2L)で酸性にしてpHを2とし、DCM(2×2L)で抽出する。混ぜ合わせた有機抽出物を乾燥させて(Na2SO4)ろ過する。ろ液を減圧下で濃縮して143gの2-メチル-2-(4,4,4-トリフルオロ-ブタン-1-スルホニル)-プロピオン酸を黄色固体として得る。収率:93%;ES-MS:m/z 261 [M-H].1H NMR (500 MHz, クロロホルム-d) δ ppm 1.71 (6 H, s), 2.18 - 2.28 (2 H, m), 2.30 - 2.42 (2 H, m), 3.38 (2 H, t, J=7.48 Hz), 6.96 (1 H, br. s.)。
【0057】
酸方法D:
2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオン酸の合成
【0058】
【化13】
【0059】
工程1:テトラヒドロ-ピラン-4-オールの合成
THF(150mL)中75g(0.75mol)のテトラヒドロ-ピラン-4-オンの溶液にTHF(600mL)中28.4g(0.75mol)のLiAlH4の懸濁液を窒素雰囲気下で氷浴の助けを借りて30℃未満の温度に維持しながら添加する。次に反応を室温に戻して5時間撹拌する。NH4Cl飽和水溶液を起沸が止むまで添加して反応をクエンチする。結果として生じる沈殿物をCelite(登録商標)を通すろ過で除去し、THF(150mL)で洗浄する。ろ液を減圧下で濃縮して71.1gのテトラヒドロ-ピラン-4-オールを淡黄色油として得る。収率:92%、1H NMR (500 MHz, クロロホルム-d) δ ppm 1.54 (2 H, m), 1.81 - 1.92 (2 H, m), 2.11 (1 H, br. s.), 3.38 - 3.47 (2 H, m), 3.83 (1 H, tt, J=9.10, 4.38 Hz), 3.94 (2 H, dt, J=11.88, 4.15 Hz)。
【0060】
工程2:トルエン-4-スルホン酸テトラヒドロ-ピラン-4-イルエステルの合成
ピリジン(1.5L)中133g(1.31mol)のテトラヒドロ-ピラン-4-オールの溶液に373g(1.95mol)のp-トルエンスルホニルクロリドを10℃で何度かに分けて添加する。添加完了後、反応を室温に戻して18時間撹拌する。反応をHCl水溶液/氷の撹拌混合物上に注ぐ。結果として生じる沈殿物をろ過で単離してDCM(1L)に溶かす。有機層を1M HCl水溶液(1L)で洗浄した後、NaHCO3飽和水溶液(1L)で洗浄してからNa2SO4上で乾燥させる。ろ過及びろ液の減圧下での濃縮が300gのトルエン-4-スルホン酸テトラヒドロ-ピラン-4-イルエステルを橙色油として与える。収率:90%、ES-MS:m/z: 257 [M+H], 279 [M+Na]。
1H-NMR (250 MHz, クロロホルム-d) δ ppm 1.66 - 1.96 (4 H, m), 2.45 (3 H, s), 3.47 (2 H, ddd, J=11.76, 8.19, 3.50 Hz), 3.79 - 3.95 (2 H, m), 4.69 (1 H, tt, J=8.13, 4.13 Hz), 7.35 (2 H, d, J=8.07 Hz), 7.76 - 7.87 (2 H, m)
【0061】
工程3:チオ酢酸S-(テトラヒドロ-ピラン-4-イル)エステルの合成
DMF(3L)中300g(1.175mol)のトルエン-4-スルホン酸テトラヒドロ-ピラン-4-イルエステルの溶液に268g(2.35mol)のチオ酢酸カリウムを添加した後、触媒量のNaI(0.12g,10mol%)を室温で加える。添加完了後、反応を20時間50℃に加熱する。反応混合物をTBME(3L)と水(3L)に分配し、水層をTBME(2L)で抽出してからNaClで飽和させて再びTBME(2×2L)で抽出する。混ぜ合わせた有機抽出物をNa2SO4上で乾燥させ、ろ過し、減圧下で溶媒を除去して153gのチオ酢酸S-(テトラヒドロ-ピラン-4-イル)エステルを得る。収率:81%;ES-MS:m/z 161 [M+H];1H-NMR (250 MHz, クロロホルム-d) δ ppm 1.47 - 1.98 (4 H, m), 2.30 (3 H, s), 3.41 - 3.74 (3 H, m), 3.88 (2 H, dt, J=11.76, 3.86 Hz)
【0062】
工程4:2-メチル-2-(テトラヒドロ-ピラン-4-イルスルファニル)-プロピオン酸エチルエステルの合成
エタノール(3.5L)中153g(0.96mol)のチオ酢酸S-(テトラヒドロ-ピラン-4-イル)エステルの溶液を0.5時間にわたって窒素で脱気し、125g(2.23mol)のKOHを加える。次にEtOH(1L)中250mL(1.68mol)のα-ブロモイソ酪酸エチルの溶液を0.5時間かけて添加すると、その間に温度が40℃に上昇する。反応を窒素雰囲気下で室温にて18時間撹拌する。反応混合物をろ過し、固体をエタノール(0.5L)で洗浄し、ろ液を減圧下で濃縮する。粗製物質をシリカ上に乾式負荷(dryload)して乾式フラッシュカラムクロマトグラフィー(シリカ、溶出液:n-ヘプタン、2〜10%の酢酸エチル)で精製して158gの2-メチル-2-(テトラヒドロ-ピラン-4-イルスルファニル)-プロピオン酸エチルエステルを橙褐色油として得る。収率:71%;ES-MS:m/z 233 [M+H];1H-NMR (500 MHz, クロロホルム-d) δ ppm 1.28 (3 H, t, J=7.17 Hz), 1.52 (6 H, s), 1.56 - 1.67 (2 H, m), 1.85 (2 H, dt, J=13.35, 1.64 Hz), 3.04 (1 H, tt, J=10.60, 4.20 Hz), 3.40 - 3.49 (2 H, m), 3.88 (2 H, dt, J=11.75, 3.81 Hz), 4.14 - 4.20 (2 H, m)
【0063】
工程5:2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオン酸エチルエステルの合成
1,4-ジオキサン/水(4/1,1.6L)中158g(0.68mol)の2-メチル-2-(テトラヒドロ-ピラン-4-イルスルファニル)-プロピオン酸エチルエステルの溶液に835g(1.35mol)のoxone(登録商標)を50分かけて少しずつ添加する。反応混合物を室温で18時間撹拌する。固体をろ過で除去して1,4-ジオキサン(1L)で洗浄する。混ぜ合わせたろ液を減圧下で濃縮する。残留物を酢酸エチル(1.5L)に溶かして水(1L)で洗浄する。有機層をNa2SO4上で乾燥させ、ろ過し、減圧下で溶媒を除去して166gの2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオン酸エチルエステルを黄色油として得る。収率:92%、ES-MS:m/z 265 [M+H], 287 [M+Na];1H-NMR (250 MHz, クロロホルム-d) δ ppm 1.30 (3 H, t, J=7.08 Hz), 1.65 (6 H, s), 1.89 - 2.10 (4 H, m), 3.34 - 3.51 (2 H, m), 3.72 - 3.90 (1 H, m), 4.06 (2 H, dt, J=11.69, 3.60 Hz), 4.24 (2 H, q, J=7.16 Hz)
【0064】
工程6:2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオン酸の合成
THF/水(4/1,1.66L)中166g(0.63mol)の2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオン酸エチルエステルの溶液に50.5g(1.26mol)のNaOHペレットを20分かけて少しずつ加える。反応を室温で2.5日間撹拌する。減圧下で有機溶媒を除去し、水性残留物を水(2L)で希釈してDCM(2L)で洗浄する。水層を濃HClで酸性にしてpHを1〜2にしてからDCM(3×2L)で抽出する。酸性水溶液をさらにNaClで飽和させて再びDCM(6×2L)で抽出する。混ぜ合わせた有機抽出物を減圧下で濃縮して123gの2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオン酸を白色固体として得る。収率:83%、ES-MS:m/z 235 [M-H];1H-NMR (500 MHz, クロロホルム-d) δ ppm 1.71 (6 H, s), 1.94 - 2.12 (4 H, m), 3.47 (2 H, td, J=11.41, 2.98 Hz), 3.73 - 3.86 (1 H, m), 4.07 - 4.15 (2 H, m), 6.82 (1 H, br. s.)
【0065】
方法E:
2-(4-メタンスルホニル-ベンゼンスルホニル)-2-メチル-プロピオン酸の合成
【0066】
【化14】
【0067】
工程1:4-メタンスルホニル-ベンゼンスルフィン酸ナトリウムの合成
下記参考文献の適応によって記載どおりに調製した:Faucher, A.-M. et al. J. Med. Chem. 2004, 47, 19-21; Binsiti, C. Eur. J. Med. Chem. Chim. Ther. 2001, 36, 809-828.
Field, L.; Clark, R.D. Org. Synth. 1958, 38, 62-64。
水(5.0mL)中0.57gのNaHCO3(7.1mmol)と1.1gのNa2SO3(8.8mmol)の溶液に1.0g(3.9mmol)の4-メタンスルホニル-ベンゼン塩化スルホニルを加えた。反応を80℃で3時間加熱した。減圧下で溶媒を除去した。ろ液を減圧下で濃縮して4-メタンスルホニル-ベンゼンスルフィン酸ナトリウムを得た。
【0068】
工程2:2-(4-メタンスルホニル-ベンゼンスルホニル)-2-メチル-プロピオン酸エチルエステルの合成
下記参考文献の適応によって記載どおりに調製した:Faucher, A.-M. et al. J. Med. Chem. 2004, 47, 19-21; Binsiti, C. Eur. J. Med. Chem. Chim. Ther. 2001, 36, 809-828;
Field, L.; Clark, R.D. Org. Synth. 1958, 38, 62-64; Troeger; Uhde;, J. Prakt. Chem. 1899, 59, 320-349。
粗製4-メタンスルホニル-ベンゼンスルフィン酸ナトリウム(3.9mmol,バリエーションAで合成した)をDMF(20mL)に懸濁させた。ピリジン(0.6mL)とα-ブロモイソ酪酸エチル(0.7mL)を添加した。反応を窒素下で室温にて18時間撹拌した。反応混合物を減圧下で濃縮してからDCM(20mL)と水(20mL)で溶解させた。層を分けて有機層をNa2SO4上で乾燥させた。ろ過、減圧下での濃縮後、カラムクロマトグラフィー(シリカ,溶出系:DCM,0〜10%の酢酸エチル)により0.67gの2-(4-メタンスルホニル-ベンゼンスルホニル)-2-メチル-プロピオン酸エチルエステルを得た(収率51%)。1H NMR (400 MHz, CDCl3) ppm 1.25 (3H, t, J=20 Hz), 1.64 (6H, s), 3.11 (3H, s), 4.16 (2H, q, J=18 Hz), 8.07 (2H, d, J=22 Hz), 8.13 (2H, d, J=21 Hz)
【0069】
工程3:2-(4-メタンスルホニル-ベンゼンスルホニル)-2-メチル-プロピオン酸の合成
THF/水(4/1,10mL)中0.67g(2.07mmol)の2-(4-メタンスルホニル-ベンゼンスルホニル)-2-メチル-プロピオン酸エチルエステルの溶液に100mg(4.14mmol)の水酸化リチウム一水和物を加えた。反応を室温で18時間撹拌した。反応をさらに水(20mL)で希釈してからDCM(2×15mL)で洗浄した。塩基性水層を氷浴で冷却し、1M HCl水溶液で酸性にしてpHを2とした。酸性水層をイソプロパノール/クロロホルム(1/1,3×20mL)で抽出した。混ぜ合わせた有機抽出物を食塩水で洗浄し、Na2SO4上で乾燥させ、ろ過した。減圧下でろ液を濃縮して461mgの2-(4-メタンスルホニル-ベンゼンスルホニル)-2-メチル-プロピオン酸を得た(収率73%)。1H NMR (400 MHz, DMSO-d6) ppm 1.51 (6H, s), 3.35 (3H, s), 8.08 (2H, d, J=22 Hz), 8.20 (2H, d, J=21 Hz), 13.64 (1H, s)
【0070】
アミド方法A:
2-(4-クロロ-ベンゼンスルホニル)-N-シクロヘキシル-2-メチル-プロピオンアミド(実施例39,表4)の合成
【0071】
【化15】
【0072】
対応酸塩化物としての26mg(0.1mmol)の2-(4-クロロ-ベンゼンスルホニル)-2-メチル-プロピオン酸の活性化を塩化チオニル(0.5mL)と50℃で2時間処理することにより達成した。反応を室温に冷まして過剰の塩化チオニルを減圧下で除去した。
シクロヘキシルアミン(15mg,0.15mmol)とN,N,N-トリエチルアミン(41.8μLs,0.3mmol)を無水DCM(1.0mL)に溶かす。酸塩化物(28mg,0.1mmol)をDCM(0.5mL)に溶かして前記アミン溶液に加える。反応をオービタルシェーカー上に16時間置く。反応を濃縮する。粗生成物を10%H2O/DMSO(1mL)に溶かす。分取HPLCで精製して2-(4-クロロ-ベンゼンスルホニル)-N-シクロヘキシル-2-メチル-プロピオンアミド(19mgs,0.056mmol)を得る。収率:56%;ES-MS:m/z 344.3 [M+H]
この手順に従って表4のアミド方法Aの化合物を作製する。
【0073】
アミド方法B:
2-(4-クロロ-ベンゼンスルホニル)-2-メチル-N-(2-フェニルアミノ-エチル)-プロピオンアミド(実施例19,表4)の合成
【0074】
【化16】
【0075】
対応する酸塩化物としての100mg(0.38mmol)の2-(4-クロロ-ベンゼンスルホニル)-2-メチル-プロピオン酸の活性化を塩化チオニル(2mL)と80℃で2時間処理することにより達成する。反応を室温に冷まして過剰の塩化チオニルを減圧下で除去する。粗製酸塩化物をDCM(1mL)に溶かし、DCM(1mL)中N-フェニルエチレンジアミン(52mg,0.38mmol)とN,N-ジイソプロピルエチルアミン(66μL,0.38mmol)の溶液に滴下して加える。反応を室温で16時間撹拌する。反応混合物をNaHCO3飽和水溶液で洗浄する。有機層を分け、乾燥させ(Na2SO4)、ろ過してろ液を減圧下で濃縮する。残留物をカラムクロマトグラフィー(シリカ,溶出液:DCM,酢酸エチル)で精製して110mgの実施例19を得る。収率:76%;ES-MS:m/z 381 [M+H]
この手順に従って表4のアミド方法Bの化合物を作製する。
【0076】
アミド方法C:
2-メチル-N-(2-フェノキシ-エチル)-2-(4-トリフルオロメチル-ベンゼンスルホニル)-プロピオンアミド(実施例3,表4)の合成
【0077】
【化17】
【0078】
DCM(4mL)中80mg(0.27mmol)の2-メチル-2-(4-トリフルオロメチル-ベンゼンスルホニル)-プロピオン酸の溶液にN,N-ジイソプロピルエチルアミン(56μL,0.328mmol)、PS-DCC(506mg,PolymerLabs,負荷1.6mmol/g)及びDMAP(cat.)を添加する。オービタルシェーカー上で反応を室温にて16時間振盪させる。残留物をろ過で分離してDCM(5mL)で洗浄する。ろ液をNaHCO3飽和水溶液で洗浄する。有機層を分け、乾燥させ(Na2SO4)、ろ過し、ろ液を減圧下で濃縮する。残留物をカラムクロマトグラフィー(シリカ,溶出液:DCM,酢酸エチル)で精製して17mgの2-メチル-N-(2-フェノキシ-エチル)-2-(4-トリフルオロメチル-ベンゼンスルホニル)-プロピオンアミドを得る。収率:15%;ES-MS:m/z 416 [M+H]。
この手順に従って表4のアミド方法Cの化合物を作製する。
【0079】
アミド方法D:
N-(3-tert-ブチル-イソオキサゾール-5-イルメチル)-2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオンアミド(実施例70,表4)の合成
【0080】
【化18】
【0081】
工程1:3-tert-ブチル-イソオキサゾール-5-カルボン酸アミドの合成
対応する酸塩化物としての420mg(2.48mmol)の3-tert-ブチル-イソオキサゾール-5-カルボン酸の活性化を塩化オキサリル(0.25mL,2.98mmol)及びDMF(1滴)と室温で18時間処理することにより達成する。反応を減圧下で濃縮する。粗製酸塩化物をDCM(1mL)に溶かし、アンモニア水溶液(30wt%,10mL,)に0℃で滴下して添加する。反応を室温で16時間撹拌する。層を分けて水相をDCM(3×20mL)で抽出する。混ぜ合わせた有機抽出物を乾燥させ(MgSO4)、ろ過し、ろ液を減圧下で濃縮して334mgの3-tert-ブチル-イソオキサゾール-5-カルボン酸アミドを得る。収率:77%;ES-MS:m/z 169 [M+H];1H NMR (500 MHz, クロロホルム-d) δ ppm 1.36 (9 H, s), 5.75 (1 H, br. s.), 6.46 (1 H, br. s.), 6.89 (1 H, s)
【0082】
工程2:(3-tert-ブチル-1,2-オキサゾール-5-イル)メタンアミンの合成
下記参考文献の適応によって(3-tert-ブチル-1,2-オキサゾール-5-イル)メタンアミンを合成する:Dannhardt, G.; Kiefer, W.; Lambrecht, G.; Laufer, S.; Mutschler, E.; Schweiger, J.; Striegel, HG. Eur. J. Med. Chem. 1995, 30, 839-850。
無水THF(10mL)中220mg(1.27mmol)の3-tert-ブチル-イソオキサゾール-5-カルボン酸アミドの溶液に2.54mL(5.08mmol,THF中2M溶液)のボラン-メチルスルフィド複合体を窒素雰囲気下で室温にて加える。反応を加熱して3時間還流させる。さらなる分量のボラン-メチルスルフィド複合体(1.3mL)を加えて反応を4時間加熱する。メタノールを添加して反応混合物をクエンチし、室温で16時間静置する。混合物を減圧下で濃縮し、1M HCl水溶液(8mL)を加える。混合物を加熱して1時間還流させてから0℃に冷却し、6M NaOH水溶液及び固体K2CO3を添加して中和する。混合物をジエチルエーテル(5×10mL)で抽出し、混ぜ合わせた有機抽出物を乾燥させ(MgSO4)、ろ過し、ろ液を減圧下で濃縮する。残留物をカラムクロマトグラフィー(シリカ,溶出液:DCM,0〜10%のメタノール)で精製して84mgの(3-tert-ブチル-1,2-オキサゾール-5-イル)メタンアミンを得る。収率:34%;ES-MS:m/z 155 [M+H];1H NMR (250 MHz, クロロホルム-d) δ ppm 1.33 (9 H, s), 1.84 (2 H+ H2O, br. s.), 3.96 (2 H, s), 6.07 (1 H, s)
【0083】
工程3:N-(3-tert-ブチル-イソオキサゾール-5-イルメチル)-2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオンアミドの合成
対応する酸塩化物としての51mg(0.22mmol)の2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオン酸の活性化をDCM(2mL)中の塩化オキサリル(0.04mL)及びDMF(1滴)と室温にて1時間処理することにより達成する。反応を減圧下で濃縮する。粗製酸塩化物をDCM(1mL)に溶かし、DCM(2mL)中の(3-tert-ブチル-1,2-オキサゾール-5-イル)メタンアミン(42mg,80%,0.22mmol)とN,N-ジイソプロピルエチルアミン(114μL,0.66mmol)の溶液に滴下して添加する。反応を減圧下で濃縮する。残留物をカラムクロマトグラフィー(シリカ,溶出液:ヘプタン,0〜50%の酢酸エチル→DCM,20%の酢酸エチル)で2回、次に分取HPLC(中和法)で精製して36mgのN-(3-tert-ブチル-イソオキサゾール-5-イルメチル)-2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオンアミドを得る。収率:44%;ES-MS:m/z 373 [M+H]。
この手順に従って表4のアミド方法Dの化合物を作製する。
【0084】
表4
【0085】
(生物学的特性の評価)
後述するアッセイを利用して式Iの化合物の生物学的特性を評価する。
A. ヒトCB1及びCB2受容体結合:
実験方法:
CB2膜を購入する。CB2膜はヒトCB2受容体cDNAを安定的に形質移入したHEK293 EBNA細胞から作製される(Perkin Elmer Life and Analytical Sciences)。ヒトCB1受容体及びGα16 cDNAを安定的に同時形質移入したHEK細胞からCB1膜を単離する。膜調製物を50mMトリス、pH 7.5、2.5mM EDTA、5mM MgCl2、0.8%の脂肪酸フリーウシ血清アルブミンを含有するアッセイ緩衝液内で室温にて4時間シンチレーションビーズ(Ysi-Poly-L-lysine SPA beads, GE Healthcare)に結合させる。アッセイ緩衝液で洗浄することによって未結合膜を除去する。膜-ビーズ混合物を96ウェルプレートに15ugの膜/ウェル(CB2)又は2.5ug/ウェル(CB1)及び1mgのSPAビーズ/ウェルの量で添加する。1×10-5M〜1×10-10Mの範囲の用量反応濃度(0.25% DMSO、最終)で化合物を膜-ビーズ混合物に添加する。1.5nM(CB2)又は2.5nM(CB1)の最終濃度で3H-CP55940(Perkin Elmer Life and Analytical Sciences)を添加して競合反応を惹起する。反応を室温で18時間インキュベートし、TopCount NXTプレートリーダーで解読する。1.25uMのWin 55212(Sigma)の非存在下及び存在下で全体的及び非特異的結合を決定する。XLFit 4.1 4パラメーターロジスティックモデルを用いて、受容体への放射活性標識リガンドの特異的結合を50%阻害する化合物の濃度として各化合物のIC50値を計算する。チェン・プルソフの等式(Cheng-Prusoff equation)を用いてIC50値を阻害定数(Ki)値に換算する。
【0086】
B. cAMP合成のCB2R媒介調節:
以下の実験方法に従って本発明の化合物をそれらのCB2作動又は逆アゴニスト活性について評価する。上記結合アッセイによってCB2に結合することが分かっているが、このアッセイでcAMP合成のCB2R媒介調節を示すことが示されない化合物は、CB2拮抗薬であると推定される。
実験方法:
ヒトCB2Rを発現しているCHO細胞(Euroscreen)を384ウェルプレートに5000細胞/ウェルの密度で蒔いて37℃で一晩インキュベートする。培地の除去後、1mM IBMX、0.25%BSA及び10uMフォルスコリン(Forskolin)を含有する刺激緩衝液で希釈した試験化合物で細胞を処理する。このアッセイを37℃で30分間インキュベートする。細胞を溶解させ、DiscoverX-XS cAMPキットを用い、製造業者のプロトコルに従ってcAMP濃度を測定する。この環境では、アゴニストはcAMPのフォルスコリン誘発産生を減らすが、逆アゴニストはcAMPのフォルスコリン誘発産生をさらに増やすであろう。アゴニストのEC50を次のように計算する。1uMのCP55940によって阻害されたcAMPのレベルと比較してフォルスコリンによって産生されたcAMPの最大量を100%と定義する。フォルスコリン刺激cAMP合成の50%を阻害する濃度として各試験化合物のEC50値を決定する。4パラメーターロジスティックモデル(XLfit 4.0のModel 205)を用いてデータを解析する。
【0087】
C. cAMP合成のCB1R媒介調節:
以下の実験方法に従って本発明の化合物をそれらのCB1アゴニスト又は逆アゴニスト活性について評価する。上記結合アッセイによってCB1に結合することが分かっているが、このアッセイでcAMP合成のCB1R媒介調節を示すことが分からない化合物は、CB1アゴニストであると推定される。
実験方法:
ヒトCB1Rを発現しているCHO細胞(Euroscreen)を384ウェルプレートに5000細胞/ウェルの密度で蒔いて37℃で一晩インキュベートする。培地の除去後、1mM IBMX、0.25%BSA及び10uMフォルスコリンを含有する刺激緩衝液で希釈した試験化合物で細胞を処理する。このアッセイを37℃で30分間インキュベートする。細胞を溶解させ、DiscoverX-XS cAMPキットを用い、製造業者のプロトコルに従ってcAMP濃度を測定する。この環境では、アゴニストはcAMPのフォルスコリン誘発産生を減らすが、逆アゴニストはcAMPのフォルスコリン誘発産生をさらに増やすであろう。作動薬のEC50を次のように計算する。1uMのCP55940によって阻害されたcAMPのレベルと比較してフォルスコリンによって産生されたcAMPの最大量を100%と定義する。フォルスコリン刺激cAMP合成の50%を阻害する濃度として各試験化合物のEC50値を決定する。4パラメーターロジスティックモデル(XLfit 4.0のModel 205)を用いてデータを解析する。
【0088】
(アゴニスト活性を有する化合物)
上記アッセイの使用を通じて、化合物がアゴニスト活性を示し、ひいては疼痛の治療並びに炎症の治療に特に良く適していることが分かる。本発明の好ましい化合物はCB2の活性範囲(<500nM)を有するであろう。
(治療的使用)
上記アッセイによって実証できるように、本発明の化合物はCB2受容体機能を調節するのに有用である。この事実の理由で、これらの化合物は、CB2受容体機能によって媒介されるか又はCB2受容体機能の調節から利益を得るであろう疾患状態及び状況を治療する際に治療的使用を有するであろう。
本発明の化合物はCB2受容体機能を調節するので、それらは非常に有用な抗炎症活性及び免疫抑制活性を有し、かつ疾患状態及び状況の治療用薬物、特に後述する医薬組成物の形態の薬物として患者に使用することができる。
前述したように、CB2アゴニストである当該化合物は、疼痛の治療のためにも利用可能である。
【0089】
本発明のアゴニスト、拮抗薬及び逆アゴニスト化合物は、炎症プロセスを伴う以下の疾患状態又は適応症の治療用薬物として患者に使用可能である。
(i)肺疾患:例えば、喘息、気管支炎、アレルギー性鼻炎、肺気腫、成人呼吸窮迫症候群(ARDS)、ハト愛好家病、農夫肺、慢性閉塞性肺疾患(COPD)、喘息(例えばアレルギー性喘息(アトピー性又は非アトピー性)のみならず運動誘発気管支収縮、職業性喘息、喘息のウイルス増悪又は細菌増悪、他の非アレルギー性喘息及び「喘鳴する乳幼児症候群(wheezy-infant syndrome)」など)、塵肺症(例えばアルミニウム肺症、炭粉症、石綿肺症、石粉症(chalicosis)、睫毛脱落症(ptilosis)、鉄沈着症、珪肺症、タバコ肺症及び綿肺症など);
(ii)リウマチ性疾患又は自己免疫疾患又は筋骨格疾患:全ての形態のリウマチ性疾患、特に関節リウマチ、急性リウマチ熱、及びリウマチ性多発筋痛症;反応性関節炎;リウマチ性軟組織疾患;他の起源の炎症性軟組織疾患;変性関節疾患(関節症)の関節炎症状;腱炎、滑液包炎、変形性関節症、外傷性関節炎;いずれもの起源の膠原病、例えば、全身性エリテマトーデス、強皮症、多発性筋炎、皮膚筋炎、シェーグレン症候群、スチル病、フェルティ症候群;並びに骨粗しょう症及び他の骨吸収疾患;
(iii)アレルギー性疾患:全ての形態のアレルギー反応、例えば、血管神経性浮腫、枯草熱、昆虫刺咬症、薬物、血液誘導体、造影剤などに対するアレルギー反応、アナフィラキシーショック(即時型過敏反応)、蕁麻疹、血管神経性浮腫、及び接触皮膚炎;
(iv)血管疾患:結節性汎動脈炎(panarteritis nodosa)、結節性多発動脈炎、結節性動脈周囲炎、側頭動脈炎(arteritis temporalis)、ウェゲナー肉芽腫症(Wegner granulomatosis)、巨細胞性動脈炎、アテローム性動脈硬化症、再灌流傷害及び結節性紅斑;
(v)皮膚疾患:例えば皮膚炎、乾癬;日焼け、やけど、湿疹;
(vi)腎疾患:例えばネフローゼ症候群;及び全てのタイプの腎炎、例えば、糸球体腎炎;膵炎(pancreatits);
(vii)肝疾患:例えば急性肝細胞崩壊;種々起源の急性肝炎、例えばウイルス、毒物、薬物誘発性肝炎;及び慢性進行性及び/又は慢性間欠性肝炎;
(viii)胃腸疾患:例えば炎症性腸疾患、過敏性腸症候群、限局性腸炎(クローン病)、潰瘍性大腸炎;胃炎;アフタ性潰瘍、セリアック病、限局性回腸炎、胃食道逆流症;
(ix)神経保護:例えば、脳卒中後の神経変性;心停止;肺バイパス;外傷性脳損傷;脊髄損傷などの治療における神経保護;
(x)眼病:アレルギー性角膜炎、ぶどう膜炎、又は虹彩炎;結膜炎;眼瞼炎;視神経炎(neuritis nervi optici);脈絡膜炎;緑内障及び交感神経性眼炎;
(xi)耳鼻咽喉(ENT)科の疾患:例えば耳鳴;アレルギー性鼻炎又は枯草熱;外耳炎;接触性湿疹、感染などに起因する疾患;及び中耳炎;
(xii)神経疾患:例えば脳浮腫、特に腫瘍関連脳浮腫;多発性硬化症;急性脳脊髄炎;髄膜炎;急性脊髄損傷;心的外傷;認知症、特に変性認知症(老人性認知症、アルツハイマー病;パーキンソン病及びクロイツフェルト・ヤコブ病;ハンチントン舞踏病、ピック病;運動ニューロン疾患など)、血管性認知症(多発梗塞性認知症など)並びに頭蓋内占拠性病変に関連する認知症;感染及び関連状態(HIV感染など);ギラン・バレー症候群;重症筋無力症、脳卒中;及び種々の形態の発作、例えば、点頭痙攣;
(xiii)血液疾患:後天性溶血性貧血;再生不良性貧血、及び特発性血小板減少症;
(xiv)腫瘍疾患:急性リンパ性白血病;ホジキン病、悪性リンパ腫;リンパ肉芽腫症;リンパ肉腫;固形悪性腫瘍;広範な転移;
(xv)内分泌疾患:内分泌性眼疾患;内分泌性眼窩症(orbitopathia);甲状腺クリーゼ;ドケルヴァン甲状腺炎(Thyroiditis de Quervain);橋本甲状腺炎;バセドー病;肉芽腫性甲状腺炎;リンパ腫性甲状腺腫(struma lymphomatosa);及びグレーブス病;I型糖尿病(インスリン依存性糖尿病);
(xvi)臓器及び組織移植並びに移植片対宿主病;
(xvii)重篤状態のショック、例えば、敗血性ショック、アナフラキシーショック、及び全身性炎症反応症候群(SIRS);
(xviii)急性疼痛、例えば歯痛、周術期、術後の疼痛、外傷性疼痛、筋肉痛、やけどした皮膚、日焼けの疼痛、三叉神経痛、日焼け;胃腸管又は子宮の痙攣、疝痛;
(xix)内臓痛、例え慢性骨盤痛、膵炎、消化性潰瘍、間質性膀胱炎、腎疝痛、狭心症、月経痛(dysmenorrhoea)、月経、婦人科痛、過敏性腸症候群(IBS)、非潰瘍性胃腸障害、非心臓性胸痛、心筋虚血と関連する疼痛;
(xx)神経因性疼痛、例えば腰背部痛、非ヘルペス性神経痛、ヘルペス後神経痛、糖尿病性神経障害、神経損傷、後天性免疫不全症候群(AIDS)関連神経因性疼痛、頭部外傷、有痛性外傷性単神経障害、毒物及び化学療法誘発疼痛、幻肢痛、有痛性多発神経障害、視床痛症候群、脳卒中後痛、中枢神経系損傷、術後痛、断端痛、反復運動痛、乳房切除術後症候群、多発性硬化症、歯根裂離(root avulsions)、開胸術後症候群によって誘発される疼痛、痛覚過敏及びアロディニア関連神経痛;
(xxi)骨関節炎、関節リウマチ、リウマチ性疾患、腱鞘炎、痛風、外陰部痛、筋筋膜痛(筋肉損傷、線維筋痛症)、腱炎、骨関節炎、若年性関節炎、脊椎炎、痛風関節炎、乾癬性関節炎、筋骨格痛、線維筋痛症、捻挫及び筋挫傷などの障害によって誘発されるか又は関連する炎症性/侵害受容性疼痛、交感神経的に維持される疼痛、筋炎、片頭痛、歯痛、インフルエンザ及び他のウイルス感染(例えば感冒、リウマチ熱、全身性エリテマトーデス)と関連する疼痛;
(xxii)腫瘍、例えばリンパ性白血病;ホジキン病、悪性リンパ腫;リンパ肉芽腫症;リンパ肉腫;固形悪性腫瘍;広範な転移に誘発されるか又は関連する癌性疼痛;
(xxiii)頭痛、例えば群発頭痛、前兆がある場合及び前兆がない場合の片頭痛、緊張型頭痛、様々な起源の頭痛、頭痛障害(予防的及び急性使用を含め);
(xxiv)種々の他の疾患状態又は状況、例えば、経皮的冠動脈形成術後の再狭窄、急性及び慢性疼痛、アテローム性動脈硬化症、再灌流傷害、うっ血性心不全、心筋梗塞、熱傷、外傷続発性多臓器損傷、壊死性腸炎並びに血液透析、白血球フェレーシス、及び顆粒球輸血と関連する症候群、サルコイドーシス、歯肉炎、発熱;瘤(bums)、捻挫又は骨折を伴う外傷に起因する浮腫、脳浮腫及び血管浮腫、糖尿病、例えば糖尿病性脈管障害、糖尿病性神経障害、糖尿病性網膜症、膵島炎に伴う毛細血管抵抗後又は糖尿病性症状(例えば高血糖、多尿、蛋白尿及び亜硝酸塩増加及びカリクレイン尿中排泄)。
【0090】
他の適応症も挙げられる:てんかん、敗血性ショック(例えば抗血液量減少及び/又は抗血圧降下薬として)、癌、敗血症、骨粗しょう症、良性前立腺肥大及び過活動膀胱、そう痒症、白斑症、一般的胃腸障害、呼吸器、尿生殖器、胃腸又は血管領域における内臓運動性の障害、創傷、やけど、組織損傷及び術後熱、そう痒関連症候群。
ヒトの治療に有用であることに加えて、これらの化合物はペット、珍しい動物並びに哺乳類、げっ歯類などを含めた家畜の治療にも有用である。
上記疾患及び状態の治療のため、治療的に有効な用量は、一般的に本発明の化合物の投薬につき約0.01mg〜約100mg/kg(体重);好ましくは投薬につき約0.1mg〜約20mg/kg(体重)であろう。例えば、70kgの人に投与するためには、用量範囲は、本発明の化合物の投薬につき約0.7mg〜約7000mg、好ましくは投薬につき約7.0mg〜約1400mgである。最適な投与レベル及びパターンを決定するためには、ある程度の日常的用量の最適化が必要であろう。活性成分を1日1〜6回投与してよい。
【0091】
(一般的投与及び医薬組成物)
医薬品として使用する場合、典型的に医薬組成物の形態で本発明の化合物を投与する。該医薬組成物は、医薬品分野で周知の手順を利用して調製可能であり、少なくとも1種の本発明の化合物を含む。本発明の化合物を単独で投与してもよく、或いは本発明の化合物の安定性を向上させ、特定の実施形態では本化合物を含む医薬組成物の投与を容易にし、溶解及び分散を高め、阻害活性を高め、補助治療をもたらす等のアジュバントと併用してよい。本発明の化合物を単独で使用してよく、或いは本発明の他の活性物質と共に使用してよく、必要に応じて他の薬理学的に活性な物質と共に使用してもよい。一般に、本発明の化合物は、治療的又は医薬的に有効な量で投与されるが、診断又は他の目的のためにはより少ない量で投与し得る。
純粋形態又は適切な医薬組成物での本発明の化合物の投与は、医薬組成物の投与の一般に認められたいずれの様式を利用しても行える。従って、投与は、例えば、経口、頬側(例えば、舌下)、経鼻、非経口、局所、経皮、経膣、又は経直腸的に、固体、半固体、凍結乾燥散剤、又は液体剤形、例えば、錠剤、座剤、丸剤、軟弾性及び硬ゼラチンカプセル剤、散剤、溶液、懸濁液、又はエアロゾル等で、好ましくは正確な薬用量の簡単な投与に適した単位剤形であってよい。医薬組成物は、一般的に通常の医薬担体又は賦形剤と、活性物質として本発明の化合物とを含み、かつそれに加えて、他の薬用剤、医薬、担体、アジュバント、希釈剤、ビヒクル、又はその組合せを含んでよい。このような医薬的に許容できる賦形剤、担体、又は添加剤並びに種々の様式又は投与のための医薬組成物の製造方法は当業者に周知である。技術水準は、例えば、Remington: The Science and Practice of Pharmacy, 20th Edition, A. Gennaro (ed.), Lippincott Williams & Wilkins, 2000; Handbook of Pharmaceutical Additives, Michael & Irene Ash (eds.), Gower, 1995; Handbook of Pharmaceutical Excipients, A.H. Kibbe (ed.), American Pharmaceutical Ass’n, 2000; H.C. Ansel and N.G. Popovish, Pharmaceutical Dosage Forms and Drug Delivery Systems, 5th ed., Lea and Febiger, 1990によって証明される。なお、技術水準をよりよく示すため、これらの文献の内容全体を参照によってここに援用する。
【0092】
当業者が予想するように、特定の医薬製剤で利用する本発明の化合物の形態(例えば塩)は、その製剤が有効であるために必要とされる適切な物理的特性(例えば、水溶性)を有するように選択されるであろう。
頬側(舌下)投与に適した医薬組成物には、香味づけ基剤、普通はスクロースと、アカシア又はトラガカントに本発明の化合物を含むロゼンジ剤、及びゼラチンとグリセリン又はスクロースとアカシアのような不活性基剤に本発明の化合物を含むトローチ剤(pastille)がある。
非経口投与に適した医薬組成物は、本発明の化合物の無菌水性製剤を含む。これらの製剤は好ましくは静脈内投与されるが、皮下、筋肉内、又は皮内注射経由で投与を達成することもできる。注射用医薬製剤は一般的に注射用無菌食塩水、リン酸緩衝食塩水、油性懸濁液、又は技術上周知の他の注射用担体を基礎とし、通常は滅菌され、かつ血液と等張の状態にされる。従って、注射用医薬製剤を無毒の非経口的に許容できる希釈剤又は溶媒中の滅菌注射用溶液又は懸濁液として提供し得る。このような希釈剤又は溶媒としては、1,3-ブタンジオール、水、リンゲル液、等張塩化ナトリウム溶液、固定油、例えば合成モノグリセリド若しくはジグリセリド、脂肪酸、例えばオレイン酸などが挙げられる。既知技術に従い、適切な分散剤又は硬化剤及び懸濁化剤を用いて該注射用医薬製剤を処方する。注射用組成物は通常0.1〜5%w/wの本発明の化合物を含むであろう。
【0093】
化合物の経口投与用固体剤形には、カプセル剤、錠剤、丸剤、散剤、顆粒剤がある。該経口投与のためには、本発明の化合物を含む医薬的に許容できる組成物は、例えば、医薬品グレードのマンニトール、ラクトース、デンプン、アルファ化デンプン、ステアリン酸マグネシウム、ナトリウムサッカリン、タルカム、セルロースエーテル誘導体、グルコース、ゼラチン、スクロース、シトラート、没食子酸プロピル等のような通常用いられるいずれかの賦形剤の組み入れによって形成される。このような固体医薬製剤には、技術上周知なように、いくつかの機構によって胃腸管への薬物の遅延性又は持続性送達をもたらすための製剤が含まれる。このような機構としては、限定するものではないが、小腸のpHを変えることに基づく剤形からのpH感受性放出、錠剤又はカプセル剤の緩徐浸食、製剤の物理的性質に基づく胃内での保持、腸管の粘膜内層への剤形の生体接着、又は剤形からの活性薬物の酵素的放出などが挙げられる。
化合物の経口投与用の液体剤形としては、エマルション、マイクロエマルション、溶液、懸濁液、シロップ、及びエリキシル剤が挙げられ、必要に応じて、例えば、水、食塩水、デキストロース水溶液、グリセロール、エタノール等の担体中の医薬アジュバントを含む。これらの組成物は、湿潤剤、乳化剤、懸濁化剤、甘味料、調味料、及び香料などのさらなるアジュバントを含むこともできる。
【0094】
化合物の局所剤形には、軟膏、ペースト、クリーム、ローション、ゲル、散剤、溶液、スプレー、吸入剤、眼軟膏又は点耳薬、含浸包帯剤及びエアロゾルがあり、適切な通常の添加剤、例えば保存剤、薬物の浸透を助けるための溶媒並びに軟膏及びクリームには皮膚軟化剤を含有し得る。局所適用は1日1回でも複数回でもよく、通常の医学的考慮によって決まる。さらに、本発明に好ましい化合物を適切な鼻腔内ビヒクルの局所使用によって鼻腔内形態で投与することができる。製剤はクリーム又は軟膏基剤、またローションではエタノール又はオレイルアルコール等の適合性の通常の担体を含んでもよい。このような担体は、製剤の約1%から約98%まで存在してよいが、さらに一般的にはそれらは製剤の約80%までを形成するであろう。
経皮投与も可能である。長期間レシピエントの表皮と密接に接触し続けるのに適合した個別パッチとして、経皮投与に適した医薬組成物を提供することができる。経皮送達システムの形態で投与されるため、当然に、薬用量の投与は、投与計画全体を通じて間欠的ではなく連続的であろう。該パッチは、接着剤に溶解及び/又は分散させ、或いはポリマーに分散させた必要に応じて緩衝化した水溶液に適切に本発明の化合物を含む。活性化合物の適切な濃度は、約1%〜35%、好ましくは約3%〜15%である。
【0095】
吸入による投与のためには、本発明の化合物は通常、噴霧ガスが不要なポンプスプレー装置から或いは適切な噴霧剤、例えば、ジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、テトラフルオロエタン、ヘプタフルオロプロパン、二酸化炭素、若しくは他の適切なガスを使用する加圧パック又は噴霧器からエアロゾルスプレーの形態で送達される。いずれの場合も、定量を送達するためのバルブを設け、結果としての定量吸入器(metered dose inhaler)(MDI)を用いて本発明の化合物を再現性のある制御されたやり方で投与することによって、エアロゾルスプレー投薬単位を決定し得る。このような吸入器、噴霧器、又はアトマイザー装置は技術上周知であり、例えば、PCT国際出願公開第WO 97/12687号(特にその図6は市販のRESPIMAT(登録商標)噴霧器の基本である);第WO 94/07607号;第WO 97/12683号;及び第WO 97/20590号がある。この結果、上記文献は参照され、それぞれ全体が参照によって本明細書に援用される。
直腸投与は、化合物を低融点の水溶性又は不溶性固体、例えば脂肪、ココアバター、グリセリンゼラチン、水素化植物油、種々の分子量のポリエチレングリコールの混合物、又はポリエチレングリコールの脂肪酸エステル等と混合する単位用量座剤を利用して達成し得る。活性化合物は普通は少量成分であり、多くの場合、約0.05〜10質量%であり、残りが基剤成分である。
【0096】
全ての上記医薬組成物において、本発明の化合物は許容可能な担体又は賦形剤と処方される。当然に、使用する担体又は賦形剤は、組成物の他の成分と適合性であるという意味で許容性でなければならず、かつ患者にとって有害であってはならない。担体又は賦形剤は固体若しくは液体、又は両者であってよく、好ましくは単位用量組成物、例えば、0.05質量%〜95質量%の活性化合物を含有し得る錠剤として、本発明の化合物と処方される。このような担体又は賦形剤としては、不活性な充填剤若しくは希釈剤、結合剤、潤沢剤、崩壊剤、溶解遅延剤、再吸収促進剤、吸収剤、及び着色料が挙げられる。適切な結合剤としては、デンプン、ゼラチン、天然糖、例えばグルコース又はβ-ラクトース、コーン甘味料、天然及び合成ガム、例えばアカシア、トラガカント又はアルギン酸ナトリウム、カルボキシメチルセルロース、ポリエチレングリコール、蝋などが挙げられる。潤沢剤としては、オレイン酸ナトリウム、ステアリン酸ナトリウム、ステアリン酸マグネシウム、安息香酸ナトリウム、酢酸ナトリウム、塩化ナトリウム等が挙げられる。崩壊剤としては、デンプン、メチルセルロース、寒天、ベントナイト、キサンタンガム等が挙げられる。
医薬的に許容できる担体及び賦形剤は、前述した全ての添加剤などを包含する。
【技術分野】
【0001】
(出願データ)
この出願は、2010年1月15日に出願された米国仮出願第61/295,201号の優先権を主張する。
(発明の背景)
1. 技術分野
本発明は、CB2受容体を調製する新規化合物及びそれらの薬物としての使用に関する。
【背景技術】
【0002】
2. 背景情報
カンナビノイドは、大麻(Cannabis sativa)(マリファナとしても知られる)に含まれている一群の約60の特徴的な化合物であり、最も代表的な分子はカンナビノール、カンナビジオール及びΔ9-テトラヒドロカンナビノール(THC)である。大麻の治療的利用は中国の古代王朝まで遡る可能性があり、食欲の欠如、嘔吐、痙攣、月経痛、痙縮からリウマチまで多岐にわたる種々の病気のための適用が含まれる。大麻使用の長い歴史がいくつかの医薬の開発につながった。例えば、THC及びその類似ナビロンに基づいているマリノール及びセサメット(Cesamet)は、それぞれ制吐薬及び食欲刺激薬として用いられる。臨床的利益にもかかわらず、大麻の治療的利用は、幻覚、中毒及び依存といったその精神活性作用のため制限されている。Mechoulam R, ed. Cannabinoids as Therapeutic Agents, Boca Raton, FL; CRC Press, 1986は、大麻の医薬的使用の概説を提供する。
【0003】
カンナビノイドの生理的作用は少なくとも2種のGタンパク質共役受容体CB1及びCB2によって媒介される。オートラジオグラフィー研究は、CB1受容体は中枢神経系内、特に大脳皮質、海馬、大脳基底核及び小脳内で主に発現されることを実証した。CB1受容体は、それほど多くはないが、生殖器系及び免疫系の末梢組織を含めた他の末梢組織内でも見られる。CB1受容体は、シナプス前ニューロンからの神経伝達物質の放出を調節し、かつ大麻の陶酔作用及び他の中枢神経系作用、例えばTHC誘導性リングカタレプシー(THC-induced ring-catalepsy)、運動性低下(hypomobility)、及び低体温等の多くを媒介すると考えられ、これらはCB1遺伝子欠失マウスには全く存在しないことが分かった(Zimmer et al., Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci U S A. (1999) 96:5780-5785.)。
CB2受容体はほとんど排他的に免疫系内で見られ、脾臓で最大密度である。免疫細胞内におけるCB2の発現レベルはCB1より約10〜100倍高いと見積もられている。免疫系内では、CB2は、B細胞、NK細胞、単細胞、ミクログリア細胞、好中球、T細胞、樹状細胞及びマスト細胞などの種々の細胞型内で見られ、CB2モジュレーターを介して種々多様の免疫機能を調節できることを示唆している(Klein et al., The cannabinoid system and immune system. J Leukoc Biol (2003) 74:.486-496)。このことは、CB2欠失マウスにはTHCの免疫調節作用が存在しないという知見によって支持される(Bicklet et al., Immunomodulation by cannabinoid is absent in mice deficient for the cannabinoid CB2 receptor. Eur J Pharmacol (2000) 396:141-149)。CB2選択性リガンドが開発され、種々の炎症環境内におけるそれらの作用について試験されている。例えば、炎症動物モデルにおいて、CB2選択性作動薬、逆作動薬及び拮抗薬が炎症の抑制に有効であることが示された(Hanus et al., HU-308: a specific agonist for CB(2), a peripheral cannabinoid receptor. Proc Natl Acad Sci U S A. (1999) 96:14228-14233, Ueda et al., Involvement of cannabinoid CB(2) receptor-mediated response and efficacy of cannabinoid CB(2) receptor inverse agonist, JTE-907, in cutaneous inflammation in mice. Eur J Pharmacol. (2005) 520:164-171 and Smith et al., The anti-inflammatory activities of cannabinoid receptor ligands in mouse peritonitis models Eur J Pharmacol. (2001) 432:107-119.)。さらに、CB2選択性作動薬は、多発性硬化症の動物モデルにおいて疾患の重症度及び痙縮性を抑制する(Baker et al., cannabinoids control spasticity and tremor in a multiple sclerosis model. Nature (2000) 404:84-87.Arevalo-Martin et al., Therapeutic action of cannabinoids in a murine model of multiple sclerosis J Neurosci. (2003) 23:2511-2516.)。まとめると、これらの結果は、炎症成分を有する医療状態の治療にCB2受容体モジュレーターを利用できるという概念を支持する。
【0004】
炎症に加えて、CB2作動薬は疼痛及び嘔吐を抑制することが分かっている。例えば、CB2選択性アゴニストは、熱又は他の刺激によって誘発される疼痛反応を鈍らせる(Malan et al., CB2 cannabinoid receptor-mediated peripheral antinociception. Pain. (2001) 93:239-45 and Nackley et al., Selective activation of cannabinoid CB(2) receptor suppresses spinal fos protein expression and pain behavior in a rat model of inflammation. Neuroscience (2003) 119:747-57.)。CB2活性化が神経障害性疼痛反応を抑制することも実証されている(Ibrahim et al., Activation of CB2 cannabinoid receptor by AM1241 inhibits experimental neuropathic pain: pain inhibition by receptor not present in the CNS. Proc Natl Acad Sci U S A. (2003) 100:10529-33.)。最後に、脳内でCB2を見つけていなかった初期データとは対照的に、最近の論文は、脳内のCB2の発現、脾臓内では約15%のレベルでの発現を実証した。この論文によってCB2活性化は内在性カンナビノイドの制吐作用に関与すると示されている(Van Sickle et al., Identification and functional characterization of brainstem cannabinoid CB2 receptor. Science. 2005 310:329-332.)。前述の結果は、CB2作動薬を炎症及び神経障害性疼痛並びに嘔吐の治療に使用できることを確証する。
WO2008014199及びWO2008039645は、そこで開示しているCB2受容体及びCB2作動活性を有するスルホン誘導体の治療的使用について考察している。
【発明の概要】
【発明が解決しようとする課題】
【0005】
(発明の概要)
本発明は、CB2受容体に結合してそれを調節する新規化合物を提供する。本発明は、治療量の本発明の化合物の投与を手段として炎症を治療するための方法及び医薬組成物をも提供する。最後に、本発明は、治療量の本発明の化合物の投与を手段として疼痛を治療するための方法及び医薬組成物を提供する。
【課題を解決するための手段】
【0006】
(発明の詳細な説明)
最も広い一般的実施形態1では、本発明は、下記式
【0007】
【化1】
(I)
【0008】
(式中、
R1は、C1-10アルキル、C3-10シクロアルキル、3〜10員飽和ヘテロ環式環、C1-5アルキル-ヘテロ環式環、5〜10員単環式若しくは二環式アリール環又はC1-5アルキル-アリール環であり、ここで、各R1は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C3-10シクロアルキル、C1-4アルコキシ、C1-4アルキルスルホニル、アシル、シアノ、ヒドロキシル及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
R2及びR3は、C1-4アルキル又は水素であり(但し、R2とR3が両方とも水素ではありえない);或いは
R2とR3が、それらが結合している炭素原子と一緒に3〜6員シクロアルキル環を形成し;
R4は、水素又はメチルであり;
R5は、C1-10アルキル、C3-10シクロアルキル、3-10員飽和ヘテロ環式環、5-10員単環式若しくは二環式アリール環、5〜10員単環式若しくは二環式ヘテロアリール環、-S-C1-5アルキル、-S-アリール、-S-CH2-アリール、-O-C1-5アルキル、-O-アリール、-O-CH2-アリール、-NH-C1-5アルキル、-NH-アリール、-NH-CH2-アリール又はアミノであり、ここで、各R5は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、アシル、-C(O)-ヘテロアリール、シアノ、ヒドロキシル及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒に4〜7員ヘテロ環式環(アリール、-C(O)-O-C1-5アルキル、-C(O)-ヘテロアリール、-C(O)-アリール、-SO2-ヘテロアリール及び-SO2-アリールから選択される置換基で置換されていてもよい)を形成していてもよく;
R6は、水素又はC1-4アルキルであり;
nは、0、1、2、3又は4であり;
ここで、式(I)又は前記いずれのR置換基上のいずれの炭素原子も、可能であれば、部分的又は完全にハロゲン化されていてもよい)
の化合物、
又はその医薬的に許容できる塩を提供する。
【0009】
別の実施形態2では、本発明は、上述した先行する一般的実施形態に従い、かつ式中、
R1が、C1-6アルキル、フェニル、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、-CH2-シクロプロピル、-CH2-シクロブチル、テトラヒドロフラニル、テトラヒドロピラニル、アゼチジニル、ピペリジニル;チオモルフォリニル、1,1-ジオキソ-1λ6-チオモルフォリニル、モルフォリニル、ピロリジニル、ピペラジニル、ベンジル、-CH2-テトラヒドロフラニル、-CH2-テトラヒドロピラニル、-CH2-CH2-テトラヒドロフラニル又は-CH2-CH2-テトラヒドロピラニルであり、ここで、各R1は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、C1-4アルキルスルホニル、アシル、シアノ、ヒドロキシル及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
R2及びR3が、独立にメチル、エチル、n-プロピル、イソプロピル、t-Bu、又は水素であり(但し、R2とR3が両方とも水素ではありえない);或いは
R2とR3が、それらが結合している炭素と一緒にシクロプロピル、シクロブチル、又はシクロペンチル環を形成し;
R4が、水素又はメチルであり;
R5が、C1-5アルキル、フェニル、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、テトラヒドロフラニル、テトラヒドロピラニル、アゼチジニル、ピペリジニル;チオモルフォリニル、1,1-ジオキソ-1λ6-チオモルフォリニル、モルフォリニル、ピロリジニル、ピペラジニル、インダニル、ベンゾオキサゾリル、ベンゾチアゾリル、ベンゾイミダゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、ピラゾリル、ピロリル、イミダゾリル、チエニル、チアジアゾリル、ピリジニル、ピリミジニル、ピリダジニル、ピラジニル、トリアジニル、キノリニル、ジヒドロ-2H-キノリニル、イソキノリニル、キナゾリニル、インダゾリル、インドリル、ベンゾフラニル、ベンゾピラニル、ベンゾジオキソリル、-S-C1-5アルキル、-S-フェニル、-S-CH2-フェニル、-O-C1-5アルキル、-O-フェニル、-O-CH2-フェニル、-NH-C1-5アルキル、-NH-フェニル、-NH-CH2-フェニル又はアミノであり、ここで、各R5は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、アシル、-C(O)-ヘテロアリール、シアノ、ヒドロキシル及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒に4〜7員ヘテロ環式環(0又は1個のさらなるヘテロ原子を含み、フェニル、-C(O)-O-C1-5アルキル、-C(O)-チエニル、-C(O)-フェニル、-SO2-ヘテロアリール及び-SO2-フェニルから選択される置換基で置換されていてもよい)を形成していてもよく;
R6が、水素又はC1-3アルキルである、
式(I)の化合物を提供する。
【0010】
別の実施形態3では、本発明は、先行する実施形態のいずれかに従い、かつ式中、
R1が、C1-6アルキル、フェニル、シクロペンチル、シクロヘキシル、テトラヒドロピラニル、ベンジル又は-CH2-テトラヒドロピラニルであり、ここで、各R1は、独立にC1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、C1-4アルキルスルホニル、及びハロゲンから選択される1〜3個の置換基で置換されていてもよい、
式(I)の化合物を提供する。
別の実施形態4では、本発明は、先行する実施形態のいずれかに従い、かつ式中、
R2及びR3が独立にメチルであり、或いは
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成している、
式(I)の化合物を提供する。
別の実施形態5では、本発明は、先行する実施形態のいずれかに従い、かつ式中、
R4が水素又はメチルであり;
R5が、C1-5アルキル、フェニル、シクロヘキシル、シクロヘプチル、モルフォリニル、インダニル、イソオキサゾリル、チエニル、ピリジニル、-S-C1-5アルキル、-S-フェニル、-S-CH2-フェニル、-O-C1-5アルキル、-O-フェニル、-NH-C1-5アルキル、-NH-フェニル又はアミノであり、ここで、各R5は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、アシル、及びハロゲンから選択される1〜3個の置換基で置換されていてもよい、
式(I)の化合物を提供する。
別の実施形態6では、本発明は、先行する実施形態1〜4のいずれかに従い、かつ式中、
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒に4〜7員ヘテロ環式環(0又は1個のさらなるヘテロ原子を含み、かつ、フェニル、-C(O)-O-C1-4アルキル、-C(O)-チエニル及び-SO2-フェニルから選択される置換基で置換されていてもよい)を形成していてもよい、
式(I)の化合物を提供する。
【0011】
別の実施形態7では、本発明は、実施形態2に従い、かつ式中、
R1が、C1-6アルキル、フェニル、シクロペンチル、シクロヘキシル、テトラヒドロピラニル、ベンジル又は-CH2-テトラヒドロピラニルであり、ここで、各R1は独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、C1-4アルキルスルホニル、及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
R2及びR3が独立にメチルであり、或いは
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成し;
R4が水素又はメチルであり;
R5が、C1-5アルキル、フェニル、シクロヘキシル、シクロヘプチル、モルフォリニル、インダニル、イソオキサゾリル、チエニル、ピリジニル、-S-C1-5アルキル、-S-フェニル、-S-CH2-フェニル、-O-C1-5アルキル、-O-フェニル、-NH-C1-5アルキル、-NH-フェニル又はアミノであり、ここで、各R5は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、アシル、及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒にピペリジニル又はピペラジニル(フェニル、-C(O)-O-C1-4アルキル、-C(O)-チエニル及び-SO2-フェニルから選択される置換基で置換されていてもよい)を形成していてもよく;
R6が水素又はメチルであり;
nが、0、1、2又は3である、
式(I)の化合物を提供する。
【0012】
別の実施形態8では、上記実施形態7に従い、かつ式中、
R1が、フェニル、シクロヘキシル又はベンジルであり、ここで、各R1は、独立に、トリフルオロメチル、及びクロロから選択される1〜2個の置換基で置換されていてもよく;
R2及びR3がメチルであり、或いは
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成し;
R4が水素であり;
R5が、C1-4アルキル、フェニル、シクロヘキシル、シクロヘプチル、チエニル、-S-C1-5アルキル、-S-フェニル、-S-CH2-フェニル、-O-C1-5アルキル、-O-フェニル又は-NH-フェニルであり、ここで、各R5は、独立に、C1-4アルキル、トリフルオロメチル、C1-4アルコキシ、フルオロ及びクロロから選択される1〜3個の置換基で置換されていてもよい、
式(I)の化合物を提供する。
別の実施形態9では、実施形態7に従い、かつ式中、
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒にピペリジニル又はピペラジニル(フェニル、-C(O)-O-C1-4アルキル及び-C(O)-チエニルから選択される置換基で置換されていてもよい)を形成していてもよい、
式(I)の化合物を提供する。
別の実施形態10では、実施形態8に従い、かつ式中、
R1がフェニル又はシクロヘキシルであり、それぞれトリフルオロメチル、及びクロロから選択される置換基で置換されていてもよく;
R2及びR3がメチルであり、或いは
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成し;
R4が水素であり;
R5が、C1-4アルキル、フェニル、シクロヘキシル、-S-C1-5アルキル、-S-フェニル又は-S-CH2-フェニルであり、ここで、各R5は、独立に、C1-4アルキル、トリフルオロメチル、メトキシ、フルオロ及びクロロから選択される1〜3個の置換基で置換されていてもよく;
nが0又は2である、
式(I)の化合物を提供する。
別の実施形態11では、実施形態9に従い、かつ式中、
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒に、フェニル基で置換されているピペラジニル環を形成している、
式(I)の化合物を提供する。
別の実施形態12では、上述した先行する実施形態のいずれかに従い、かつ式中、
R2及びR3がメチルである、
式(I)の化合物を提供する。
別の実施形態13では、先行する実施形態1〜12のいずれかに従い、かつ式中、
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成している、
式(I)の化合物を提供する。
別の実施形態では、下記式(IA)の化合物又はその医薬的に許容できる塩を提供する。
【0013】
【化2】
(IA)
【0014】
ここで、式(IA)の下記部分
【0015】
【化3】
【0016】
は、下表IのカラムA1〜A16から選択され、かつ式(IA)の下記部分
【0017】
【化4】
【0018】
は、下表IのカラムB1〜B48から選択される。
【0019】
表I
【0020】
別の実施形態では、本発明は、当技術分野で既知の一般的スキーム、例及び方法を考慮して作製できる下表IIの作製された化合物又はその医薬的に許容できる塩を提供する。
【0021】
表II
【発明を実施するための形態】
【0022】
この出願で上記開示化合物において、命名法が構造と矛盾する場合、化合物は構造によって定義されるものと解釈すべきである。
本発明は、式(I)の1種以上の化合物、又はその医薬的に許容できる誘導体を活性物質として含み、必要に応じて通常の賦形剤及び/又は担体と組み合わせた医薬製剤にも関する。
本発明の化合物は、それらの同位体標識形をも包含する。本発明の組合せの活性薬の同位体標識形は、前記活性薬の1つ以上の原子が、通常自然界に見られる前記原子の原子質量又は質量数と異なる原子質量又は質量数を有する原子と置き換わっているという事実を別にすれば、前記活性薬と同一である。商業的に容易に入手可能であり、かつよく確立された手順に従って本発明の組合せの活性薬に組み込める同位体の例としては、水素、炭素、窒素、酸素、リン、フッ素及び塩素の同位体、例えば、それぞれ、2H、3H、13C、14C、15N、18O、17O、31P、32P、35S、18F、及び36Clが挙げられる。本発明の組合せの1つ以上の上記同位体及び/又は他の原子の他の同位体を含有する活性薬、そのプロドラッグ、又はどちらかの医薬的に許容できる塩は、本発明の範囲内であると考えられる。
本発明は、ラセミ体及びラセミ混合物、単一のエナンチオマー、ジアステレオマー混合物及び個々のジアステレオマーとして存在し得る、1つ以上の不斉炭素原子を含有する上記いずれの化合物の使用をも包含する。異性体をエナンチオマー及びジアステレオマーと定義するものとする。これらの化合物の全ての該異性形が明白に本発明に包含される。各ステレオジェン炭素は、R若しくはS配置、又は組合せ配置であってよい。
式(I)の化合物が複数の互変異性形で存在することもある。本発明は、全ての該互変異性体を使用する方法を包含する。
【0023】
この明細書で使う全ての用語は、特に指定のない限り、当技術分野で知られているそれらの通常の意味に解釈するものとする。例えば、「C1-4アルコキシ」は、末端酸素を有するC1-4アルキルであり、例えばメトキシ、エトキシ、プロポキシ、ブトキシである。全てのアルキル、アルケニル及びアルキニル基は、構造的に可能であり、かつ特に指定のない限り、分岐又は不分岐であると解釈するものとする。他のさらに詳細な定義は以下のとおりである。
炭素環又はシクロアルキルは、3〜12個の炭素原子を含む炭化水素環を包含する。これらの炭素環は、芳香族又は非芳香族環系のどちらでもよい。非芳香族環系は、一不飽和又は多不飽和であってよい。好ましい炭素環としては、限定するものではないが、シクロプロピル、シクロブチル、シクロペンチル、シクロペンテニル、シクロヘキシル、シクロヘキセニル、シクロヘプタニル、シクロヘプテニル、フェニル、インダニル、インデニル、ベンゾシクロブタニル、ジヒドロナフチル、テトラヒドロナフチル、ナフチル、デカヒドロナフチル、ベンゾシクロヘプタニル及びベンゾシクロヘプテニルが挙げられる。シクロアルキルについての特定用語、例えばシクロブタニル及びシクロブチルは、互換的に使われるものとする。
用語「ヘテロ環」は、安定した非芳香族の3〜10員(好ましくは5又は6員)単環式又は非芳香族の8〜11員二環式ヘテロ環基を意味し、飽和又は不飽和であってよい。各ヘテロ環は、炭素原子と、窒素、酸素及びイオウから選択される1個以上、好ましくは1〜4個のヘテロ原子とから成る。ヘテロ環は、安定構造の生成をもたらす、環のいずれの原子によっても付着され得る。
用語「ヘテロアリール」は、N、O及びS等の1〜4個のヘテロ原子を含有する芳香族の5〜10員単環式又は8〜11員二環式環を意味すると解釈するものとする。
特に指定のない限り、ヘテロ環及びヘテロアリールとして、限定するものではないが、例えばベンゾオキサゾリル、ベンゾチアゾリル、ベンゾイミダゾリル、テトラヒドロピラニル、ジオキサニル、テトラヒドロフラニル、オキサゾリル、イソオキサゾリル、チアゾリル、ピラゾリル、ピロリル、イミダゾリル、チエニル、チアジアゾリル、トリアゾリル、チオモルフォリニル、1,1-ジオキソ-1λ6-チオモルフォリニル、モルフォリニル、ピリジニル、ピリミジニル、ピリダジニル、ピラジニル、トリアジニル、ピロリジニル、ピペリジニル、ピペラジニル、プリニル、キノリニル、ジヒドロ-2H-キノリニル、イソキノリニル、キナゾリニル、インダゾリル、チエノ[2,3-d]ピリミジニル、インドリル、イソインドリル、ベンゾフラニル、ベンゾピラニル及びベンゾジオキソリルが挙げられる。
本明細書では、用語「ヘテロ原子」は、炭素以外の原子、例えばO、N、S及びPを意味すると解釈するものとする。
【0024】
全てのアルキル基又は炭素鎖では、1個以上の炭素原子がヘテロ原子:O、S又はNと置き換わることがあり、Nが置換されない場合は、それはNHであると解釈するものとし、ヘテロ原子は分岐又は不分岐炭素鎖内の末端炭素原子又は内部炭素原子のどちらとも置き換わり得ることをも理解するものとする。このような基は、上述したように、オキソ等の基によって置換されて、限定するものではないが、アルコキシカルボニル、アシル、アミノ及びチオキソ等の定義をもたらし得る。
用語「アリール」は、本明細書では芳香族炭素環を意味すると解釈するものとする。
各アリール又はヘテロアリールは、特に指定のない限り、その部分的又は完全に水素化した誘導体を包含する。例えば、キノリニルにはデカヒドロキノリニル及びテトラヒドロキノリニルが含まれ、ナフチルには、その水素化誘導体、例えばテトラヒドロナフチルが含まれる。当業者には、本明細書に記載のアリール及びヘテロアリールの他の部分的又は完全に水素化した誘導体が明らかであろう。
本明細書では、「窒素」及び「イオウ」は、窒素及びイオウのいずれの酸化形並びにいずれの塩基性窒素の四級化形をも包含する。例えば、-S-C1-6アルキル基では、特に指定のない限り、これは-S(O)-C1-6アルキル及び-S(O)2-C1-6アルキルを包含すると解釈するものとする。
用語「ハロゲン」は、本明細書で使用する場合、臭素、塩素、フッ素又はヨウ素、好ましくはフッ素及び塩素を意味すると解釈するものとする。定義「部分的又は完全にハロゲン化」;「部分的又は完全にフッ素化」;「1個以上のハロゲン原子で置換された」は、例えば、1個以上の炭素原子に関するモノハロ、ジハロ又はトリハロ誘導体を包含する。アルキルについての非限定例は-CH2CHF2、-CF3等であろう。
【0025】
本発明の化合物は、当業者には明らかなように、「化学的に安定」であると考えられる化合物のみである。例えば、「ダングリング原子価」、又は「カルボアニオン」を有するであろう化合物は、ここに開示する発明方法によって予想される化合物ではない。
本発明は、式(I)の化合物の医薬的に許容できる誘導体を包含する。「医薬的に許容できる誘導体」とは、患者に投与すると、本発明に有用な化合物、又はその薬理学的に活性な代謝物又は薬理学的に活性な残基を(直接又は間接的に)もたらすことができるいずれの医薬的に許容できる塩若しくはエステル、又はいずれの他の化合物をも意味する。薬理学的に活性な代謝物は、酵素的又は化学的に代謝され得る本発明のいずれの化合物をも意味すると解釈するものとする。これには、例えば、式(I)のヒドロキシル化又は酸化誘導化合物が含まれる。
医薬的に許容できる塩には、医薬的に許容できる無機酸及び塩基並びに有機酸及び塩基から誘導される当該塩が含まれる。適切な酸の例としては、塩酸、臭化水素酸、硫酸、硝酸、過塩素酸、フマル酸、マレイン酸、リン酸、グリコール酸、乳酸、サリチル酸、コハク酸、トルエン-p-硫酸、酒石酸、酢酸、クエン酸、メタンスルホン酸、ギ酸、安息香酸、マロン酸、ナフタレン-2-硫酸及びベンゼンスルホン酸が挙げられる。他の酸、例えばシュウ酸は、それ自体医薬的に許容性ではないが、本化合物及びそれらの医薬的に許容できる酸付加塩を得るときの中間体として有用な塩の調製に利用し得る。適切な塩基から誘導される塩としては、アルカリ金属(例えば、ナトリウム)、アルカリ土類金属(例えば、マグネシウム)、アンモニウム及びN-(C1-C4アルキル)4+塩が挙げられる。
さらに、式(I)の化合物のプロドラッグの使用が本発明の範囲内である。プロドラッグには、簡単な化学変換が起こると、改変されて本発明の化合物を生成する当該化合物が含まれる。簡単な化学変換には、加水分解、酸化及び還元がある。詳細には、プロドラッグが患者に投与されると、プロドラッグは上記開示化合物に変換され、それによって所望の薬理作用を与え得る。
後述する一般的合成方法(これも本発明の一部を構成する)を利用して式Iの化合物を作製することができる。
【0026】
(一般的合成方法)
本発明は、式(I)及び(IA)の化合物の作製方法をも提供する。全ての方法で、特に指定のない限り、下記式中のR、R1、R2、R3、R4、R5、R6及びnは、上述した本発明の式(I)中のR、R1、R2、R3、R4、R5、R6及びnの意味を有するものとする。
最適反応条件及び反応時間は、使用する特定の反応物質によって変化し得る。特に指定のない限り、当業者は、溶媒、温度、圧力、及び他の反応条件を容易に選択し得る。合成例セクションで具体的手順を提供する。典型的に、必要に応じて、薄層クロマトグラフィー(TLC)によって反応の進行をモニターすることができ、かつシリカゲル上クロマトグラフィー及び/又は再結晶によって中間体及び生成物を精製することができる。
以下の実施例は例示であり、当業者は認識しているように、過度の実験を行うことなく、個々の化合物に必要なように特定の試薬又は条件を変更できるであろう。下記方法で使用する出発原料及び中間体は、商業的に入手可能であり、或いは市販原料から当業者が容易に調製できる。WO2008098025、WO2008014199、WO2008039645、及びWO2009061652に開示されている合成方法を本発明の化合物の調製に利用してもよい。
下記スキーム1に示す方法で式(I)及び(IA)の化合物を合成することができる。
【0027】
【化5】
スキーム1
【0028】
スキーム1に示すように、式IIの酸を塩化チオニル又は塩化オキサリル等の試薬と反応させて酸塩化物を得て、適切な溶媒中、適切な塩基の存在下で式IIIのアミンと反応させて式(I)の化合物を得る。或いは、式IIの酸を標準的カップリング条件下で式IIIのアミンと結合して式(I)の化合物を得てもよい。これらの合成では技術上既知の標準的ペプチドカップリング反応(例えばM. Bodanszky, 1984, The Practice of Peptide Synthesis, Springer-Verlag参照)を利用してよい。適切なカップリング条件の例は、DMF等の適切な溶媒中のカルボン酸の溶液をEDC、HOBT、及び塩基、例えばジイソプロピルエチルアミンと処理した後、所望アミンと処理する。
技術上既知かつ下記実施例に示す方法による式(I)の初期生成物のさらなる改変を利用して本発明のさらなる化合物を調製することができる。
下記スキーム2に概要を示す方法で中間体酸IIを作製し得る。
【0029】
【化6】
スキーム2
【0030】
上述したように、適切な溶媒中、適切な塩基の存在下での式IVのチオールと式Vのブロモエチルエステルの反応が式VIチオエーテルをもたらす。式VIのチオエーテルを適切な酸化剤と反応させて、対応する式VIIのスルホンを得る。適切な溶媒中、水酸化リチウム等の適切な塩基の存在下における式VIIのスルホンのエステル基の加水分解が、対応する式IIの酸をもたらす。
下記スキーム3に概要を示す方法で中間体酸IIを作製してもよい。
【0031】
【化7】
スキーム3
【0032】
適切な溶媒中、式Vの出発ブロモエステルのチオ酢酸カリウム等の試薬との反応が式VIIIのチオ酢酸エステルをもたらす。適切な塩基の存在下でのチオ酢酸エステルVIIIと式IXの臭化物の反応が、対応する式VIのスルファニル酸エチルエステルをもたらす。スキーム2に示す工程順序によって式VIのスルファニル酸エチルエステルを式IIの中間体酸に変換し得る。
下記スキーム4に概要を示す方法によって中間体IIを作製し得る。
【0033】
【化8】
スキーム4
【0034】
スキーム4に示すように、適切な溶媒中、適切な塩基の存在下での式Xのアルコールとp-トルエンスルホニルクロリドの反応が式XIのスルホン酸エステルをもたらす。適切な溶媒中、式XIの化合物とチオ酢酸カリウムの反応が式XIIの化合物をもたらす。適切な溶媒中、適切な塩基の存在下での式XIIの中間体と式Vのブロモエステルの反応が式VIをもたらし、これをスキーム2に示す反応順序で式IIの所望中間体に変換することができる。
下記スキーム5に概要を示す方法で中間体IIを作製してもよい。
【0035】
【化9】
スキーム5
【0036】
スキーム5に示すように、文献に報告された手順を用いて式XIIIの塩化スルホニルを対応する式XIVのスルフィン酸ナトリウム塩に変換する。適切な溶媒中での式XIVのスルフィン酸ナトリウム塩と式Vのブロモエステルの反応が式VIIのスルホンエステルをもたらす。式VIIのスルホンエステルの加水分解が式IIの中間体酸をもたらす。
【0037】
(合成例)
下記実施例によって、本発明の化合物を作製可能な方法がさらに理解されるであろう。
酸方法A:
方法Aの酸をWO2008039645, Boehringer Ingelheim International GmbHに記載のように、或いはWO2008014199, Boehringer Ingelheim International GmbHに記載のように調製する。
2-シクロペンタンスルホニル-2-メチル-プロピオン酸の合成
【0038】
【化10】
【0039】
工程1:2-シクロペンチルスルファニル-2-メチル-プロピオン酸エチルエステルの合成
エタノール(50mL)中5g(48.7mmol)のシクロペンチルチオールの溶液に2.7g(48.75mmol)のKOHペレットを添加した後、9.5g(48.7mmol)のα-ブロモイソ酪酸エチルを加える。反応を加熱して2時間還流させてから室温に冷ます。固体(KBr)をろ過で分離し、エタノール(20mL)ですすぐ。ろ液を減圧下で濃縮し、残留物をDCM(50mL)に溶かす。有機層をNaHCO3飽和水溶液(50mL)で洗浄する。水性洗浄液をDCM(10mL)で逆抽出する。混ぜ合わせた有機物を食塩水で洗浄し、Na2SO4上で乾燥させる。ろ過及び減圧下での濃縮が8.1gの2-シクロペンチルスルファニル-2-メチル-プロピオン酸エチルエステルを与える。収率:77%、ES-MS:m/z 217 [M+H]
この手順に従い、下記変更に留意しながら以下のチオエーテルを合成する:合成例13〜14、17〜18及び21〜22を合成する場合の酸については、α-ブロモイソ酪酸の代わりにエチル-1-ブロモシクロブタンカルボキシラートを使用する。
【0040】
表1
*反応条件中にエチルエステルの加水分解が起こった
【0041】
工程2:2-シクロペンタンスルホニル-2-メチル-プロピオン酸エチルエステルの合成
1,4-ジオキサン/水(4/1,100mL)中6g(27.7mmol)の2-シクロペンチルスルファニル-2-メチル-プロピオン酸エチルエステルの溶液に数回に分けて51.2g(83mmol)の一過硫酸カリウム三重塩(OXONE(登録商標))を添加する。白色懸濁液を室温で3時間撹拌する。白色固体をろ過で分離し、1,4-ジオキサン(10mL)で洗浄する。ろ液を減圧下で濃縮して有機溶媒を除去する。結果として生じる水溶液をDCM(3×40mL)で抽出する。混ぜ合わせた有機抽出物をNaHCO3飽和水溶液、食塩水で洗浄し、Na2SO4上で乾燥させてろ過する。ろ液を減圧下で濃縮して5.4gの2-シクロペンタンスルホニル-2-メチル-プロピオン酸エチルエステルを得る。収率:78%、ES-MS:m/z 249 [M+H]
この手順に従って以下のスルホンを合成する。
表2
*酸化工程のため2-(4-クロロ-ベンジルスルファニル)-2-メチル-プロピオン酸を使用
【0042】
工程3:2-シクロペンタンスルホニル-2-メチル-プロピオン酸の合成
THF/水(4/1,60mL)中5.4g(21.7mmol)の2-シクロペンタンスルホニル-2-メチル-プロピオン酸エチルエステルの溶液に2.3g(56.6mmol)の水酸化リチウム一水和物をを添加する。反応を室温で18時間撹拌する。反応をさらに水(20mL)で希釈してからDCM(2×15mL)で洗浄する。塩基性水層を氷浴で冷却してから2M HCl水溶液で酸性にしてpHを2とする。酸性水層を2-プロパノール/クロロホルム(1/4,100mL)で抽出する。混ぜ合わせた有機抽出物を食塩水で洗浄し、Na2SO4上で乾燥させてろ過する。減圧下でろ液を濃縮して4.34gの2-シクロペンタンスルホニル-2-メチル-プロピオン酸を得る。収率:92%、ES-MS:m/z 221 [M+H]
この手順に従って以下の酸を合成する。
【0043】
表3
【0044】
酸方法B:
WO2008039645, Boehringer Ingelheim International GmbHに記載の方法を適応して方法Bの酸を調製する。
2-メチル-2-(テトラヒドロ-ピラン-4-イルメタンスルホニル)-プロピオン酸
【0045】
【化11】
【0046】
工程1:(テトラヒドロ-ピラン-4-イル)-メタノールの合成
窒素雰囲気下で、THF(200mL)中のLiAlH4溶液250mL(THF中2.3M溶液,0.575mol)にTHF(900mL)中130mL(0.974mol)のテトラヒドロ-ピラン-4-カルボン酸メチルエステルの溶液を滴下して添加する(注意:激しい発熱反応!)。氷浴で温度を40〜45℃に維持する。添加が完了したら、反応を室温で1.5時間撹拌する。反応を氷浴で冷まし、水(22mL)、15%NaOH水溶液(21mL)及び水(66mL)を添加してクエンチする。結果として生じる沈殿物をCelite(登録商標)でろ過して除去し、THF(300mL)ですすぐ。ろ液を減圧下で濃縮して102.5gの(テトラヒドロ-ピラン-4-イル)-メタノールを無色油として得る。収率:91%;1H NMR (400 MHz, クロロホルム-d) δ ppm 1.20 - 1.39 (2 H, m), 1.56 - 1.83 (3 H, m), 2.03 (1 H, br. s.), 3.29 - 3.52 (4 H, m), 3.89 - 4.05 (2 H, m)
【0047】
工程2:トルエン-4-スルホン酸テトラヒドロ-ピラン-4-イルメチルエステルの合成
下記文献参照の適応によって記載どおりに調製する:
Radziszewski, J.G. et al. J. Am. Chem. Soc. 1993, 115, 8401。
2-メチルテトラヒドロフラン(190mL)中97g(810mmol)の(テトラヒドロ-ピラン-4-イル)-メタノールの溶液に165mLの50%NaOH水溶液を加える。この撹拌溶液に2-メチルテトラヒドロフラン(280mL)中のp-トルエン-スルホニルクロリドの溶液(283g,1.46mol)を冷却しながら滴下して添加する。反応を30〜35℃で18時間撹拌する。懸濁液を氷水(280mL)とHCl水溶液(37%,203mL)の混合物中に注ぐ。メチルシクロヘキサン(1.4L)及びさらなる氷水(0.2L)の添加後、反応混合物を氷浴内で2時間撹拌する。結果として生じる結晶性沈殿物をろ過で単離し、メチルシクロヘキサン(0.5L)及び水(0.5L)で洗浄する。減圧下40℃で乾燥させて216gのトルエン-4-スルホン酸テトラヒドロ-ピラン-4-イルメチルエステルを白色結晶性固体として得る。収率:99%、ES-MS:m/z 271 [M+H]; 1H NMR (400 MHz, クロロホルム-d) δ ppm 1.19 - 1.35 (2 H, m), 1.54 - 1.63 (2 H, m), 1.85 - 2.02 (1 H, m), 2.45 (3 H, s), 3.28 - 3.39 (2 H, m), 3.86 (2H, d, J=6.60 Hz), 3.93 (2 H, dd, J=11.37, 4.52 Hz), 7.35 (2 H, d, J=9.29 Hz), 7.78 (2 H, d, J=8.31 Hz)
【0048】
工程3:チオ酢酸S-(テトラヒドロ-ピラン-4-イルメチル)エステルの合成
下記文献参照の適応によって記載どおりに調製する:
Watson, R.J. et al. Tetrahedron Lett. 2002, 43, 683-685。
メチルイソブチルケトン(1.6L)中224g(0.83mol)のトルエン-4-スルホン酸テトラヒドロ-ピラン-4-イルメチルエステルの溶液に189g(1.66mol)のチオ酢酸カリウムを加える。ベージュ色懸濁液を70℃で4.5時間撹拌する。反応混合物を室温に冷まして水(1.8L)を加える。有機層を10%K2CO3水溶液(1.8L)と水(1L)で洗浄する。有機層をcelite(登録商標)(20g)、活性炭(20g)及びNa2SO4(20g)を通してろ過し、ろ液を減圧下で濃縮する。残存油をメチルシクロヘキサン(200mL)及びn-ヘプタン(250mL)と共沸させて138gのチオ酢酸S-(テトラヒドロ-ピラン-4-イルメチル)エステルを黄橙色油として得る(注意:悪臭!)。収率:96%;ES-MS:m/z 175 [M+H]; 1H NMR (400 MHz, クロロホルム-d) δ ppm 1.23 - 1.40 (2 H, m), 1.59 - 1.78 (3 H, m), 2.33 (3 H, d, J=4.16 Hz), 2.82 (2 H, dd, J=6.24, 3.79 Hz), 3.27- 3.39 (2 H, m), 3.88 - 4.02 (2 H, m)
【0049】
工程4:2-メチル-2-(テトラヒドロ-ピラン-4-イルメタンスルホニル)-プロピオン酸エチルエステルの合成
トルエン(500mL)中90g(516mmol)のチオ酢酸S-(テトラヒドロ-ピラン-4-イルメチル)エステルの溶液を窒素雰囲気下にて氷浴内で冷却する。エタノール中ナトリウムエトキシドの溶液(21%,231mL)を添加して反応を50分間撹拌する。次に76mL(516mmol)のα-ブロモイソ酪酸エチルを加えて反応を1時間撹拌する。反応混合物に氷酢酸(8.9mL)と水(500mL)を加える。有機層を分けて水(500mL)で洗浄する。三口丸底フラスコに水(500mL)、oxone(登録商標)(477g,775mmol)及び硫酸水素テトラブチルアンモニウム(5g,15mmol)を入れて有機層を加える。二相性反応混合物を室温で2日間撹拌する。固体をろ過で除去し、ろ液の層を分ける。有機層を水(2×500mL)で洗浄する。減圧下で溶媒を除去し、さらにトルエンと共沸させて125gの2-メチル-2-(テトラヒドロ-ピラン-4-イルメタンスルホニル)-プロピオン酸エチルエステルを得る。収率:87%;ES-MS:m/z 279 [M+H];
1H NMR (250 MHz, クロロホルム-d) δppm 1.32 (3 H, t, J=7.16 Hz), 1.39 - 1.59 (2 H, m), 1.64 (6 H, s), 1.81 - 1.97 (2 H, m), 2.29 - 2.53 (1 H, m),3.15 (2 H, d, J=6.55 Hz), 3.45 (2 H, dd, J=1.83, 0.30 Hz), 3.88 - 4.03 (2 H, m), 4.26 (2 H, d, J=7.16 Hz)
【0050】
工程5:2-メチル-2-(テトラヒドロ-ピラン-4-イルメタンスルホニル)-プロピオン酸の合成
方法Aの工程3の適応によって記載どおりに調製する。
THF(450mL)中123g(0.44mol)の2-メチル-2-(テトラヒドロ-ピラン-4-イルメタンスルホニル)-プロピオン酸エチルエステルの溶液に663mLの2M水酸化ナトリウム水溶液(1.33mol)を加える。反応を室温で1時間撹拌する。反応混合物にTBME(1.25L)を加えて層を分ける。水層を氷浴内で冷却してから37%HCl水溶液(123mL)で酸性にする。結果として生じる沈殿物をろ過で単離し、水(200mL)で洗浄し、減圧下で50℃にて乾燥させて101gの2-メチル-2-(テトラヒドロ-ピラン-4-イルメタンスルホニル)-プロピオン酸を白色結晶性固体として得る。収率:91%;ES-MS:m/z 251 [M+H];1H NMR (400 MHz, DMSO-d6) δ ppm 1.31 - 1.45 (2 H, m), 1.49 (6 H, s), 1.70 - 1.79 (2 H, m), 2.13 - 2.28 (1 H, m), 3.24 (2 H, d, J=6.60 Hz), 3.28 - 3.38 (2 H, m), 3.76 - 3.85 (2 H, m), 13.65 (1 H, br. s.)
【0051】
酸方法C:
2-メチル-2-(4,4,4-トリフルオロ-ブタン-1-スルホニル)-プロピオン酸の合成
【0052】
【化12】
【0053】
工程1:2-アセチルスルファニル-2-メチル-プロピオン酸エチルエステルの合成
DMF(500mL)中α-ブロモイソ酪酸エチル(62g,0.32mol)の溶液に室温でチオ酢酸カリウム (72g,0.63mol)を加える。反応を16時間撹拌してから減圧下で濃縮する。残留物を2M塩酸水溶液(500mL)で希釈し、酢酸エチル(3×500mL)で抽出する。有機フラクションを混ぜ合わせて食塩水(300mL)で洗浄し、MgSO4上で乾燥させ、ろ過し、減圧下で濃縮する。ヘプタン/ジクロロメタンで溶出するシリカ上クロマトグラフィーで精製して44gの2-アセチルスルファニル-2-メチル-プロピオン酸エチルエステルを得る。収率:73%;m/z 191 [M+H];1H NMR (250 MHz, クロロホルム-d) δ ppm 1.18 - 1.30 (3 H, m), 1.57 (6 H, s), 2.27 (3 H, s), 4.19 (2 H, q, J=7.16 Hz)。
【0054】
工程1:2-メチル-2-(4,4,4-トリフルオロ-ブチルスルファニル)-プロピオン酸エチルエステルの合成
エタノール(1.2L,窒素下で1時間脱気した)中149g(785.4mmol)の2-アセチルスルファニル-2-メチル-プロピオン酸エチルエステルの溶液に169.7g(105mmol)のナトリウムメトキシドを添加した後、150g(785.4mmol)の1-ブロモ-4,4,4-トリフルオロブタンの溶液を加える。反応を85℃で3日間加熱する。減圧下で溶媒を除去する。残留物をDCM(1L)に溶かし、NaHCO3飽和水溶液で洗浄する(2×1L)。有機層をNa2SO4上で乾燥させ、ろ過し、ろ液を減圧下で濃縮して171gの2-メチル-2-(4,4,4-トリフルオロ-ブチルスルファニル)-プロピオン酸エチルエステルを褐色油として得る。収率:84%;ES-MS:m/z 259 [M+H];1H NMR (500 MHz, クロロホルム-d) δ ppm 1.29 (3 H, t, J=7.17 Hz), 1.51 (6 H, s), 1.76 - 1.86 (2 H, m), 2.12 - 2.27 (2 H, m), 2.69 (2 H, t, J=7.17 Hz), 4.18 (2 H, q, J=7.17 Hz)。
【0055】
工程2:2-メチル-2-(4,4,4-トリフルオロ-ブタン-1-スルホニル)-プロピオン酸エチルエステルの合成
1,4-ジオキサン/水(1/1,4L)中220g(851.7mmol)の2-メチル-2-(4,4,4-トリフルオロ-ブチルスルファニル)-プロピオン酸エチルエステルの溶液に1047g(1703.4mmol)のoxone(登録商標)を室温で0.5時間かけて少しずつ添加する。反応混合物を室温で18時間撹拌する。固体をろ過で除去して1,4-ジオキサン(0.5L)ですすぐ。ろ液を減圧下で濃縮して有機溶媒を除去する。水性残留物をDCM(2×1L)で抽出する。混ぜ合わせた有機抽出物をNaHCO3飽和水溶液(2L)で洗浄し、Na2SO4上で乾燥させてろ過する。ろ液を減圧下で濃縮して226gの2-メチル-2-(4,4,4-トリフルオロ-ブタン-1-スルホニル)-プロピオン酸エチルエステルを暗黄色油として得る。収率92%;ES-MS:m/z 291 [M+H];1H NMR (500 MHz, クロロホルム-d) δ ppm 1.32 (3 H, t, J=7.17 Hz), 1.66 (6 H, s), 2.20 (2 H, quin, J=7.59 Hz), 2.28 - 2.41 (2 H, m), 3.34 (2 H, t, J=7.48 Hz), 4.27 (2 H, q, J=7.17 Hz)。
【0056】
工程3:2-メチル-2-(4,4,4-トリフルオロ-ブタン-1-スルホニル)-プロピオン酸の合成
THF(3.4L)中170g(585.6mmol)の2-メチル-2-(4,4,4-トリフルオロ-ブタン-1-スルホニル)-プロピオン酸エチルエステルの溶液に225.4g(1756.8mmol)のカリウムトリメチルシラノラート(silanolate)を0.5時間かけて少しずつ添加する。反応を室温で18時間撹拌する。反応混合物を2M HCl水溶液(2L)で酸性にしてpHを2とし、DCM(2×2L)で抽出する。混ぜ合わせた有機抽出物を乾燥させて(Na2SO4)ろ過する。ろ液を減圧下で濃縮して143gの2-メチル-2-(4,4,4-トリフルオロ-ブタン-1-スルホニル)-プロピオン酸を黄色固体として得る。収率:93%;ES-MS:m/z 261 [M-H].1H NMR (500 MHz, クロロホルム-d) δ ppm 1.71 (6 H, s), 2.18 - 2.28 (2 H, m), 2.30 - 2.42 (2 H, m), 3.38 (2 H, t, J=7.48 Hz), 6.96 (1 H, br. s.)。
【0057】
酸方法D:
2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオン酸の合成
【0058】
【化13】
【0059】
工程1:テトラヒドロ-ピラン-4-オールの合成
THF(150mL)中75g(0.75mol)のテトラヒドロ-ピラン-4-オンの溶液にTHF(600mL)中28.4g(0.75mol)のLiAlH4の懸濁液を窒素雰囲気下で氷浴の助けを借りて30℃未満の温度に維持しながら添加する。次に反応を室温に戻して5時間撹拌する。NH4Cl飽和水溶液を起沸が止むまで添加して反応をクエンチする。結果として生じる沈殿物をCelite(登録商標)を通すろ過で除去し、THF(150mL)で洗浄する。ろ液を減圧下で濃縮して71.1gのテトラヒドロ-ピラン-4-オールを淡黄色油として得る。収率:92%、1H NMR (500 MHz, クロロホルム-d) δ ppm 1.54 (2 H, m), 1.81 - 1.92 (2 H, m), 2.11 (1 H, br. s.), 3.38 - 3.47 (2 H, m), 3.83 (1 H, tt, J=9.10, 4.38 Hz), 3.94 (2 H, dt, J=11.88, 4.15 Hz)。
【0060】
工程2:トルエン-4-スルホン酸テトラヒドロ-ピラン-4-イルエステルの合成
ピリジン(1.5L)中133g(1.31mol)のテトラヒドロ-ピラン-4-オールの溶液に373g(1.95mol)のp-トルエンスルホニルクロリドを10℃で何度かに分けて添加する。添加完了後、反応を室温に戻して18時間撹拌する。反応をHCl水溶液/氷の撹拌混合物上に注ぐ。結果として生じる沈殿物をろ過で単離してDCM(1L)に溶かす。有機層を1M HCl水溶液(1L)で洗浄した後、NaHCO3飽和水溶液(1L)で洗浄してからNa2SO4上で乾燥させる。ろ過及びろ液の減圧下での濃縮が300gのトルエン-4-スルホン酸テトラヒドロ-ピラン-4-イルエステルを橙色油として与える。収率:90%、ES-MS:m/z: 257 [M+H], 279 [M+Na]。
1H-NMR (250 MHz, クロロホルム-d) δ ppm 1.66 - 1.96 (4 H, m), 2.45 (3 H, s), 3.47 (2 H, ddd, J=11.76, 8.19, 3.50 Hz), 3.79 - 3.95 (2 H, m), 4.69 (1 H, tt, J=8.13, 4.13 Hz), 7.35 (2 H, d, J=8.07 Hz), 7.76 - 7.87 (2 H, m)
【0061】
工程3:チオ酢酸S-(テトラヒドロ-ピラン-4-イル)エステルの合成
DMF(3L)中300g(1.175mol)のトルエン-4-スルホン酸テトラヒドロ-ピラン-4-イルエステルの溶液に268g(2.35mol)のチオ酢酸カリウムを添加した後、触媒量のNaI(0.12g,10mol%)を室温で加える。添加完了後、反応を20時間50℃に加熱する。反応混合物をTBME(3L)と水(3L)に分配し、水層をTBME(2L)で抽出してからNaClで飽和させて再びTBME(2×2L)で抽出する。混ぜ合わせた有機抽出物をNa2SO4上で乾燥させ、ろ過し、減圧下で溶媒を除去して153gのチオ酢酸S-(テトラヒドロ-ピラン-4-イル)エステルを得る。収率:81%;ES-MS:m/z 161 [M+H];1H-NMR (250 MHz, クロロホルム-d) δ ppm 1.47 - 1.98 (4 H, m), 2.30 (3 H, s), 3.41 - 3.74 (3 H, m), 3.88 (2 H, dt, J=11.76, 3.86 Hz)
【0062】
工程4:2-メチル-2-(テトラヒドロ-ピラン-4-イルスルファニル)-プロピオン酸エチルエステルの合成
エタノール(3.5L)中153g(0.96mol)のチオ酢酸S-(テトラヒドロ-ピラン-4-イル)エステルの溶液を0.5時間にわたって窒素で脱気し、125g(2.23mol)のKOHを加える。次にEtOH(1L)中250mL(1.68mol)のα-ブロモイソ酪酸エチルの溶液を0.5時間かけて添加すると、その間に温度が40℃に上昇する。反応を窒素雰囲気下で室温にて18時間撹拌する。反応混合物をろ過し、固体をエタノール(0.5L)で洗浄し、ろ液を減圧下で濃縮する。粗製物質をシリカ上に乾式負荷(dryload)して乾式フラッシュカラムクロマトグラフィー(シリカ、溶出液:n-ヘプタン、2〜10%の酢酸エチル)で精製して158gの2-メチル-2-(テトラヒドロ-ピラン-4-イルスルファニル)-プロピオン酸エチルエステルを橙褐色油として得る。収率:71%;ES-MS:m/z 233 [M+H];1H-NMR (500 MHz, クロロホルム-d) δ ppm 1.28 (3 H, t, J=7.17 Hz), 1.52 (6 H, s), 1.56 - 1.67 (2 H, m), 1.85 (2 H, dt, J=13.35, 1.64 Hz), 3.04 (1 H, tt, J=10.60, 4.20 Hz), 3.40 - 3.49 (2 H, m), 3.88 (2 H, dt, J=11.75, 3.81 Hz), 4.14 - 4.20 (2 H, m)
【0063】
工程5:2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオン酸エチルエステルの合成
1,4-ジオキサン/水(4/1,1.6L)中158g(0.68mol)の2-メチル-2-(テトラヒドロ-ピラン-4-イルスルファニル)-プロピオン酸エチルエステルの溶液に835g(1.35mol)のoxone(登録商標)を50分かけて少しずつ添加する。反応混合物を室温で18時間撹拌する。固体をろ過で除去して1,4-ジオキサン(1L)で洗浄する。混ぜ合わせたろ液を減圧下で濃縮する。残留物を酢酸エチル(1.5L)に溶かして水(1L)で洗浄する。有機層をNa2SO4上で乾燥させ、ろ過し、減圧下で溶媒を除去して166gの2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオン酸エチルエステルを黄色油として得る。収率:92%、ES-MS:m/z 265 [M+H], 287 [M+Na];1H-NMR (250 MHz, クロロホルム-d) δ ppm 1.30 (3 H, t, J=7.08 Hz), 1.65 (6 H, s), 1.89 - 2.10 (4 H, m), 3.34 - 3.51 (2 H, m), 3.72 - 3.90 (1 H, m), 4.06 (2 H, dt, J=11.69, 3.60 Hz), 4.24 (2 H, q, J=7.16 Hz)
【0064】
工程6:2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオン酸の合成
THF/水(4/1,1.66L)中166g(0.63mol)の2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオン酸エチルエステルの溶液に50.5g(1.26mol)のNaOHペレットを20分かけて少しずつ加える。反応を室温で2.5日間撹拌する。減圧下で有機溶媒を除去し、水性残留物を水(2L)で希釈してDCM(2L)で洗浄する。水層を濃HClで酸性にしてpHを1〜2にしてからDCM(3×2L)で抽出する。酸性水溶液をさらにNaClで飽和させて再びDCM(6×2L)で抽出する。混ぜ合わせた有機抽出物を減圧下で濃縮して123gの2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオン酸を白色固体として得る。収率:83%、ES-MS:m/z 235 [M-H];1H-NMR (500 MHz, クロロホルム-d) δ ppm 1.71 (6 H, s), 1.94 - 2.12 (4 H, m), 3.47 (2 H, td, J=11.41, 2.98 Hz), 3.73 - 3.86 (1 H, m), 4.07 - 4.15 (2 H, m), 6.82 (1 H, br. s.)
【0065】
方法E:
2-(4-メタンスルホニル-ベンゼンスルホニル)-2-メチル-プロピオン酸の合成
【0066】
【化14】
【0067】
工程1:4-メタンスルホニル-ベンゼンスルフィン酸ナトリウムの合成
下記参考文献の適応によって記載どおりに調製した:Faucher, A.-M. et al. J. Med. Chem. 2004, 47, 19-21; Binsiti, C. Eur. J. Med. Chem. Chim. Ther. 2001, 36, 809-828.
Field, L.; Clark, R.D. Org. Synth. 1958, 38, 62-64。
水(5.0mL)中0.57gのNaHCO3(7.1mmol)と1.1gのNa2SO3(8.8mmol)の溶液に1.0g(3.9mmol)の4-メタンスルホニル-ベンゼン塩化スルホニルを加えた。反応を80℃で3時間加熱した。減圧下で溶媒を除去した。ろ液を減圧下で濃縮して4-メタンスルホニル-ベンゼンスルフィン酸ナトリウムを得た。
【0068】
工程2:2-(4-メタンスルホニル-ベンゼンスルホニル)-2-メチル-プロピオン酸エチルエステルの合成
下記参考文献の適応によって記載どおりに調製した:Faucher, A.-M. et al. J. Med. Chem. 2004, 47, 19-21; Binsiti, C. Eur. J. Med. Chem. Chim. Ther. 2001, 36, 809-828;
Field, L.; Clark, R.D. Org. Synth. 1958, 38, 62-64; Troeger; Uhde;, J. Prakt. Chem. 1899, 59, 320-349。
粗製4-メタンスルホニル-ベンゼンスルフィン酸ナトリウム(3.9mmol,バリエーションAで合成した)をDMF(20mL)に懸濁させた。ピリジン(0.6mL)とα-ブロモイソ酪酸エチル(0.7mL)を添加した。反応を窒素下で室温にて18時間撹拌した。反応混合物を減圧下で濃縮してからDCM(20mL)と水(20mL)で溶解させた。層を分けて有機層をNa2SO4上で乾燥させた。ろ過、減圧下での濃縮後、カラムクロマトグラフィー(シリカ,溶出系:DCM,0〜10%の酢酸エチル)により0.67gの2-(4-メタンスルホニル-ベンゼンスルホニル)-2-メチル-プロピオン酸エチルエステルを得た(収率51%)。1H NMR (400 MHz, CDCl3) ppm 1.25 (3H, t, J=20 Hz), 1.64 (6H, s), 3.11 (3H, s), 4.16 (2H, q, J=18 Hz), 8.07 (2H, d, J=22 Hz), 8.13 (2H, d, J=21 Hz)
【0069】
工程3:2-(4-メタンスルホニル-ベンゼンスルホニル)-2-メチル-プロピオン酸の合成
THF/水(4/1,10mL)中0.67g(2.07mmol)の2-(4-メタンスルホニル-ベンゼンスルホニル)-2-メチル-プロピオン酸エチルエステルの溶液に100mg(4.14mmol)の水酸化リチウム一水和物を加えた。反応を室温で18時間撹拌した。反応をさらに水(20mL)で希釈してからDCM(2×15mL)で洗浄した。塩基性水層を氷浴で冷却し、1M HCl水溶液で酸性にしてpHを2とした。酸性水層をイソプロパノール/クロロホルム(1/1,3×20mL)で抽出した。混ぜ合わせた有機抽出物を食塩水で洗浄し、Na2SO4上で乾燥させ、ろ過した。減圧下でろ液を濃縮して461mgの2-(4-メタンスルホニル-ベンゼンスルホニル)-2-メチル-プロピオン酸を得た(収率73%)。1H NMR (400 MHz, DMSO-d6) ppm 1.51 (6H, s), 3.35 (3H, s), 8.08 (2H, d, J=22 Hz), 8.20 (2H, d, J=21 Hz), 13.64 (1H, s)
【0070】
アミド方法A:
2-(4-クロロ-ベンゼンスルホニル)-N-シクロヘキシル-2-メチル-プロピオンアミド(実施例39,表4)の合成
【0071】
【化15】
【0072】
対応酸塩化物としての26mg(0.1mmol)の2-(4-クロロ-ベンゼンスルホニル)-2-メチル-プロピオン酸の活性化を塩化チオニル(0.5mL)と50℃で2時間処理することにより達成した。反応を室温に冷まして過剰の塩化チオニルを減圧下で除去した。
シクロヘキシルアミン(15mg,0.15mmol)とN,N,N-トリエチルアミン(41.8μLs,0.3mmol)を無水DCM(1.0mL)に溶かす。酸塩化物(28mg,0.1mmol)をDCM(0.5mL)に溶かして前記アミン溶液に加える。反応をオービタルシェーカー上に16時間置く。反応を濃縮する。粗生成物を10%H2O/DMSO(1mL)に溶かす。分取HPLCで精製して2-(4-クロロ-ベンゼンスルホニル)-N-シクロヘキシル-2-メチル-プロピオンアミド(19mgs,0.056mmol)を得る。収率:56%;ES-MS:m/z 344.3 [M+H]
この手順に従って表4のアミド方法Aの化合物を作製する。
【0073】
アミド方法B:
2-(4-クロロ-ベンゼンスルホニル)-2-メチル-N-(2-フェニルアミノ-エチル)-プロピオンアミド(実施例19,表4)の合成
【0074】
【化16】
【0075】
対応する酸塩化物としての100mg(0.38mmol)の2-(4-クロロ-ベンゼンスルホニル)-2-メチル-プロピオン酸の活性化を塩化チオニル(2mL)と80℃で2時間処理することにより達成する。反応を室温に冷まして過剰の塩化チオニルを減圧下で除去する。粗製酸塩化物をDCM(1mL)に溶かし、DCM(1mL)中N-フェニルエチレンジアミン(52mg,0.38mmol)とN,N-ジイソプロピルエチルアミン(66μL,0.38mmol)の溶液に滴下して加える。反応を室温で16時間撹拌する。反応混合物をNaHCO3飽和水溶液で洗浄する。有機層を分け、乾燥させ(Na2SO4)、ろ過してろ液を減圧下で濃縮する。残留物をカラムクロマトグラフィー(シリカ,溶出液:DCM,酢酸エチル)で精製して110mgの実施例19を得る。収率:76%;ES-MS:m/z 381 [M+H]
この手順に従って表4のアミド方法Bの化合物を作製する。
【0076】
アミド方法C:
2-メチル-N-(2-フェノキシ-エチル)-2-(4-トリフルオロメチル-ベンゼンスルホニル)-プロピオンアミド(実施例3,表4)の合成
【0077】
【化17】
【0078】
DCM(4mL)中80mg(0.27mmol)の2-メチル-2-(4-トリフルオロメチル-ベンゼンスルホニル)-プロピオン酸の溶液にN,N-ジイソプロピルエチルアミン(56μL,0.328mmol)、PS-DCC(506mg,PolymerLabs,負荷1.6mmol/g)及びDMAP(cat.)を添加する。オービタルシェーカー上で反応を室温にて16時間振盪させる。残留物をろ過で分離してDCM(5mL)で洗浄する。ろ液をNaHCO3飽和水溶液で洗浄する。有機層を分け、乾燥させ(Na2SO4)、ろ過し、ろ液を減圧下で濃縮する。残留物をカラムクロマトグラフィー(シリカ,溶出液:DCM,酢酸エチル)で精製して17mgの2-メチル-N-(2-フェノキシ-エチル)-2-(4-トリフルオロメチル-ベンゼンスルホニル)-プロピオンアミドを得る。収率:15%;ES-MS:m/z 416 [M+H]。
この手順に従って表4のアミド方法Cの化合物を作製する。
【0079】
アミド方法D:
N-(3-tert-ブチル-イソオキサゾール-5-イルメチル)-2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオンアミド(実施例70,表4)の合成
【0080】
【化18】
【0081】
工程1:3-tert-ブチル-イソオキサゾール-5-カルボン酸アミドの合成
対応する酸塩化物としての420mg(2.48mmol)の3-tert-ブチル-イソオキサゾール-5-カルボン酸の活性化を塩化オキサリル(0.25mL,2.98mmol)及びDMF(1滴)と室温で18時間処理することにより達成する。反応を減圧下で濃縮する。粗製酸塩化物をDCM(1mL)に溶かし、アンモニア水溶液(30wt%,10mL,)に0℃で滴下して添加する。反応を室温で16時間撹拌する。層を分けて水相をDCM(3×20mL)で抽出する。混ぜ合わせた有機抽出物を乾燥させ(MgSO4)、ろ過し、ろ液を減圧下で濃縮して334mgの3-tert-ブチル-イソオキサゾール-5-カルボン酸アミドを得る。収率:77%;ES-MS:m/z 169 [M+H];1H NMR (500 MHz, クロロホルム-d) δ ppm 1.36 (9 H, s), 5.75 (1 H, br. s.), 6.46 (1 H, br. s.), 6.89 (1 H, s)
【0082】
工程2:(3-tert-ブチル-1,2-オキサゾール-5-イル)メタンアミンの合成
下記参考文献の適応によって(3-tert-ブチル-1,2-オキサゾール-5-イル)メタンアミンを合成する:Dannhardt, G.; Kiefer, W.; Lambrecht, G.; Laufer, S.; Mutschler, E.; Schweiger, J.; Striegel, HG. Eur. J. Med. Chem. 1995, 30, 839-850。
無水THF(10mL)中220mg(1.27mmol)の3-tert-ブチル-イソオキサゾール-5-カルボン酸アミドの溶液に2.54mL(5.08mmol,THF中2M溶液)のボラン-メチルスルフィド複合体を窒素雰囲気下で室温にて加える。反応を加熱して3時間還流させる。さらなる分量のボラン-メチルスルフィド複合体(1.3mL)を加えて反応を4時間加熱する。メタノールを添加して反応混合物をクエンチし、室温で16時間静置する。混合物を減圧下で濃縮し、1M HCl水溶液(8mL)を加える。混合物を加熱して1時間還流させてから0℃に冷却し、6M NaOH水溶液及び固体K2CO3を添加して中和する。混合物をジエチルエーテル(5×10mL)で抽出し、混ぜ合わせた有機抽出物を乾燥させ(MgSO4)、ろ過し、ろ液を減圧下で濃縮する。残留物をカラムクロマトグラフィー(シリカ,溶出液:DCM,0〜10%のメタノール)で精製して84mgの(3-tert-ブチル-1,2-オキサゾール-5-イル)メタンアミンを得る。収率:34%;ES-MS:m/z 155 [M+H];1H NMR (250 MHz, クロロホルム-d) δ ppm 1.33 (9 H, s), 1.84 (2 H+ H2O, br. s.), 3.96 (2 H, s), 6.07 (1 H, s)
【0083】
工程3:N-(3-tert-ブチル-イソオキサゾール-5-イルメチル)-2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオンアミドの合成
対応する酸塩化物としての51mg(0.22mmol)の2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオン酸の活性化をDCM(2mL)中の塩化オキサリル(0.04mL)及びDMF(1滴)と室温にて1時間処理することにより達成する。反応を減圧下で濃縮する。粗製酸塩化物をDCM(1mL)に溶かし、DCM(2mL)中の(3-tert-ブチル-1,2-オキサゾール-5-イル)メタンアミン(42mg,80%,0.22mmol)とN,N-ジイソプロピルエチルアミン(114μL,0.66mmol)の溶液に滴下して添加する。反応を減圧下で濃縮する。残留物をカラムクロマトグラフィー(シリカ,溶出液:ヘプタン,0〜50%の酢酸エチル→DCM,20%の酢酸エチル)で2回、次に分取HPLC(中和法)で精製して36mgのN-(3-tert-ブチル-イソオキサゾール-5-イルメチル)-2-メチル-2-(テトラヒドロ-ピラン-4-スルホニル)-プロピオンアミドを得る。収率:44%;ES-MS:m/z 373 [M+H]。
この手順に従って表4のアミド方法Dの化合物を作製する。
【0084】
表4
【0085】
(生物学的特性の評価)
後述するアッセイを利用して式Iの化合物の生物学的特性を評価する。
A. ヒトCB1及びCB2受容体結合:
実験方法:
CB2膜を購入する。CB2膜はヒトCB2受容体cDNAを安定的に形質移入したHEK293 EBNA細胞から作製される(Perkin Elmer Life and Analytical Sciences)。ヒトCB1受容体及びGα16 cDNAを安定的に同時形質移入したHEK細胞からCB1膜を単離する。膜調製物を50mMトリス、pH 7.5、2.5mM EDTA、5mM MgCl2、0.8%の脂肪酸フリーウシ血清アルブミンを含有するアッセイ緩衝液内で室温にて4時間シンチレーションビーズ(Ysi-Poly-L-lysine SPA beads, GE Healthcare)に結合させる。アッセイ緩衝液で洗浄することによって未結合膜を除去する。膜-ビーズ混合物を96ウェルプレートに15ugの膜/ウェル(CB2)又は2.5ug/ウェル(CB1)及び1mgのSPAビーズ/ウェルの量で添加する。1×10-5M〜1×10-10Mの範囲の用量反応濃度(0.25% DMSO、最終)で化合物を膜-ビーズ混合物に添加する。1.5nM(CB2)又は2.5nM(CB1)の最終濃度で3H-CP55940(Perkin Elmer Life and Analytical Sciences)を添加して競合反応を惹起する。反応を室温で18時間インキュベートし、TopCount NXTプレートリーダーで解読する。1.25uMのWin 55212(Sigma)の非存在下及び存在下で全体的及び非特異的結合を決定する。XLFit 4.1 4パラメーターロジスティックモデルを用いて、受容体への放射活性標識リガンドの特異的結合を50%阻害する化合物の濃度として各化合物のIC50値を計算する。チェン・プルソフの等式(Cheng-Prusoff equation)を用いてIC50値を阻害定数(Ki)値に換算する。
【0086】
B. cAMP合成のCB2R媒介調節:
以下の実験方法に従って本発明の化合物をそれらのCB2作動又は逆アゴニスト活性について評価する。上記結合アッセイによってCB2に結合することが分かっているが、このアッセイでcAMP合成のCB2R媒介調節を示すことが示されない化合物は、CB2拮抗薬であると推定される。
実験方法:
ヒトCB2Rを発現しているCHO細胞(Euroscreen)を384ウェルプレートに5000細胞/ウェルの密度で蒔いて37℃で一晩インキュベートする。培地の除去後、1mM IBMX、0.25%BSA及び10uMフォルスコリン(Forskolin)を含有する刺激緩衝液で希釈した試験化合物で細胞を処理する。このアッセイを37℃で30分間インキュベートする。細胞を溶解させ、DiscoverX-XS cAMPキットを用い、製造業者のプロトコルに従ってcAMP濃度を測定する。この環境では、アゴニストはcAMPのフォルスコリン誘発産生を減らすが、逆アゴニストはcAMPのフォルスコリン誘発産生をさらに増やすであろう。アゴニストのEC50を次のように計算する。1uMのCP55940によって阻害されたcAMPのレベルと比較してフォルスコリンによって産生されたcAMPの最大量を100%と定義する。フォルスコリン刺激cAMP合成の50%を阻害する濃度として各試験化合物のEC50値を決定する。4パラメーターロジスティックモデル(XLfit 4.0のModel 205)を用いてデータを解析する。
【0087】
C. cAMP合成のCB1R媒介調節:
以下の実験方法に従って本発明の化合物をそれらのCB1アゴニスト又は逆アゴニスト活性について評価する。上記結合アッセイによってCB1に結合することが分かっているが、このアッセイでcAMP合成のCB1R媒介調節を示すことが分からない化合物は、CB1アゴニストであると推定される。
実験方法:
ヒトCB1Rを発現しているCHO細胞(Euroscreen)を384ウェルプレートに5000細胞/ウェルの密度で蒔いて37℃で一晩インキュベートする。培地の除去後、1mM IBMX、0.25%BSA及び10uMフォルスコリンを含有する刺激緩衝液で希釈した試験化合物で細胞を処理する。このアッセイを37℃で30分間インキュベートする。細胞を溶解させ、DiscoverX-XS cAMPキットを用い、製造業者のプロトコルに従ってcAMP濃度を測定する。この環境では、アゴニストはcAMPのフォルスコリン誘発産生を減らすが、逆アゴニストはcAMPのフォルスコリン誘発産生をさらに増やすであろう。作動薬のEC50を次のように計算する。1uMのCP55940によって阻害されたcAMPのレベルと比較してフォルスコリンによって産生されたcAMPの最大量を100%と定義する。フォルスコリン刺激cAMP合成の50%を阻害する濃度として各試験化合物のEC50値を決定する。4パラメーターロジスティックモデル(XLfit 4.0のModel 205)を用いてデータを解析する。
【0088】
(アゴニスト活性を有する化合物)
上記アッセイの使用を通じて、化合物がアゴニスト活性を示し、ひいては疼痛の治療並びに炎症の治療に特に良く適していることが分かる。本発明の好ましい化合物はCB2の活性範囲(<500nM)を有するであろう。
(治療的使用)
上記アッセイによって実証できるように、本発明の化合物はCB2受容体機能を調節するのに有用である。この事実の理由で、これらの化合物は、CB2受容体機能によって媒介されるか又はCB2受容体機能の調節から利益を得るであろう疾患状態及び状況を治療する際に治療的使用を有するであろう。
本発明の化合物はCB2受容体機能を調節するので、それらは非常に有用な抗炎症活性及び免疫抑制活性を有し、かつ疾患状態及び状況の治療用薬物、特に後述する医薬組成物の形態の薬物として患者に使用することができる。
前述したように、CB2アゴニストである当該化合物は、疼痛の治療のためにも利用可能である。
【0089】
本発明のアゴニスト、拮抗薬及び逆アゴニスト化合物は、炎症プロセスを伴う以下の疾患状態又は適応症の治療用薬物として患者に使用可能である。
(i)肺疾患:例えば、喘息、気管支炎、アレルギー性鼻炎、肺気腫、成人呼吸窮迫症候群(ARDS)、ハト愛好家病、農夫肺、慢性閉塞性肺疾患(COPD)、喘息(例えばアレルギー性喘息(アトピー性又は非アトピー性)のみならず運動誘発気管支収縮、職業性喘息、喘息のウイルス増悪又は細菌増悪、他の非アレルギー性喘息及び「喘鳴する乳幼児症候群(wheezy-infant syndrome)」など)、塵肺症(例えばアルミニウム肺症、炭粉症、石綿肺症、石粉症(chalicosis)、睫毛脱落症(ptilosis)、鉄沈着症、珪肺症、タバコ肺症及び綿肺症など);
(ii)リウマチ性疾患又は自己免疫疾患又は筋骨格疾患:全ての形態のリウマチ性疾患、特に関節リウマチ、急性リウマチ熱、及びリウマチ性多発筋痛症;反応性関節炎;リウマチ性軟組織疾患;他の起源の炎症性軟組織疾患;変性関節疾患(関節症)の関節炎症状;腱炎、滑液包炎、変形性関節症、外傷性関節炎;いずれもの起源の膠原病、例えば、全身性エリテマトーデス、強皮症、多発性筋炎、皮膚筋炎、シェーグレン症候群、スチル病、フェルティ症候群;並びに骨粗しょう症及び他の骨吸収疾患;
(iii)アレルギー性疾患:全ての形態のアレルギー反応、例えば、血管神経性浮腫、枯草熱、昆虫刺咬症、薬物、血液誘導体、造影剤などに対するアレルギー反応、アナフィラキシーショック(即時型過敏反応)、蕁麻疹、血管神経性浮腫、及び接触皮膚炎;
(iv)血管疾患:結節性汎動脈炎(panarteritis nodosa)、結節性多発動脈炎、結節性動脈周囲炎、側頭動脈炎(arteritis temporalis)、ウェゲナー肉芽腫症(Wegner granulomatosis)、巨細胞性動脈炎、アテローム性動脈硬化症、再灌流傷害及び結節性紅斑;
(v)皮膚疾患:例えば皮膚炎、乾癬;日焼け、やけど、湿疹;
(vi)腎疾患:例えばネフローゼ症候群;及び全てのタイプの腎炎、例えば、糸球体腎炎;膵炎(pancreatits);
(vii)肝疾患:例えば急性肝細胞崩壊;種々起源の急性肝炎、例えばウイルス、毒物、薬物誘発性肝炎;及び慢性進行性及び/又は慢性間欠性肝炎;
(viii)胃腸疾患:例えば炎症性腸疾患、過敏性腸症候群、限局性腸炎(クローン病)、潰瘍性大腸炎;胃炎;アフタ性潰瘍、セリアック病、限局性回腸炎、胃食道逆流症;
(ix)神経保護:例えば、脳卒中後の神経変性;心停止;肺バイパス;外傷性脳損傷;脊髄損傷などの治療における神経保護;
(x)眼病:アレルギー性角膜炎、ぶどう膜炎、又は虹彩炎;結膜炎;眼瞼炎;視神経炎(neuritis nervi optici);脈絡膜炎;緑内障及び交感神経性眼炎;
(xi)耳鼻咽喉(ENT)科の疾患:例えば耳鳴;アレルギー性鼻炎又は枯草熱;外耳炎;接触性湿疹、感染などに起因する疾患;及び中耳炎;
(xii)神経疾患:例えば脳浮腫、特に腫瘍関連脳浮腫;多発性硬化症;急性脳脊髄炎;髄膜炎;急性脊髄損傷;心的外傷;認知症、特に変性認知症(老人性認知症、アルツハイマー病;パーキンソン病及びクロイツフェルト・ヤコブ病;ハンチントン舞踏病、ピック病;運動ニューロン疾患など)、血管性認知症(多発梗塞性認知症など)並びに頭蓋内占拠性病変に関連する認知症;感染及び関連状態(HIV感染など);ギラン・バレー症候群;重症筋無力症、脳卒中;及び種々の形態の発作、例えば、点頭痙攣;
(xiii)血液疾患:後天性溶血性貧血;再生不良性貧血、及び特発性血小板減少症;
(xiv)腫瘍疾患:急性リンパ性白血病;ホジキン病、悪性リンパ腫;リンパ肉芽腫症;リンパ肉腫;固形悪性腫瘍;広範な転移;
(xv)内分泌疾患:内分泌性眼疾患;内分泌性眼窩症(orbitopathia);甲状腺クリーゼ;ドケルヴァン甲状腺炎(Thyroiditis de Quervain);橋本甲状腺炎;バセドー病;肉芽腫性甲状腺炎;リンパ腫性甲状腺腫(struma lymphomatosa);及びグレーブス病;I型糖尿病(インスリン依存性糖尿病);
(xvi)臓器及び組織移植並びに移植片対宿主病;
(xvii)重篤状態のショック、例えば、敗血性ショック、アナフラキシーショック、及び全身性炎症反応症候群(SIRS);
(xviii)急性疼痛、例えば歯痛、周術期、術後の疼痛、外傷性疼痛、筋肉痛、やけどした皮膚、日焼けの疼痛、三叉神経痛、日焼け;胃腸管又は子宮の痙攣、疝痛;
(xix)内臓痛、例え慢性骨盤痛、膵炎、消化性潰瘍、間質性膀胱炎、腎疝痛、狭心症、月経痛(dysmenorrhoea)、月経、婦人科痛、過敏性腸症候群(IBS)、非潰瘍性胃腸障害、非心臓性胸痛、心筋虚血と関連する疼痛;
(xx)神経因性疼痛、例えば腰背部痛、非ヘルペス性神経痛、ヘルペス後神経痛、糖尿病性神経障害、神経損傷、後天性免疫不全症候群(AIDS)関連神経因性疼痛、頭部外傷、有痛性外傷性単神経障害、毒物及び化学療法誘発疼痛、幻肢痛、有痛性多発神経障害、視床痛症候群、脳卒中後痛、中枢神経系損傷、術後痛、断端痛、反復運動痛、乳房切除術後症候群、多発性硬化症、歯根裂離(root avulsions)、開胸術後症候群によって誘発される疼痛、痛覚過敏及びアロディニア関連神経痛;
(xxi)骨関節炎、関節リウマチ、リウマチ性疾患、腱鞘炎、痛風、外陰部痛、筋筋膜痛(筋肉損傷、線維筋痛症)、腱炎、骨関節炎、若年性関節炎、脊椎炎、痛風関節炎、乾癬性関節炎、筋骨格痛、線維筋痛症、捻挫及び筋挫傷などの障害によって誘発されるか又は関連する炎症性/侵害受容性疼痛、交感神経的に維持される疼痛、筋炎、片頭痛、歯痛、インフルエンザ及び他のウイルス感染(例えば感冒、リウマチ熱、全身性エリテマトーデス)と関連する疼痛;
(xxii)腫瘍、例えばリンパ性白血病;ホジキン病、悪性リンパ腫;リンパ肉芽腫症;リンパ肉腫;固形悪性腫瘍;広範な転移に誘発されるか又は関連する癌性疼痛;
(xxiii)頭痛、例えば群発頭痛、前兆がある場合及び前兆がない場合の片頭痛、緊張型頭痛、様々な起源の頭痛、頭痛障害(予防的及び急性使用を含め);
(xxiv)種々の他の疾患状態又は状況、例えば、経皮的冠動脈形成術後の再狭窄、急性及び慢性疼痛、アテローム性動脈硬化症、再灌流傷害、うっ血性心不全、心筋梗塞、熱傷、外傷続発性多臓器損傷、壊死性腸炎並びに血液透析、白血球フェレーシス、及び顆粒球輸血と関連する症候群、サルコイドーシス、歯肉炎、発熱;瘤(bums)、捻挫又は骨折を伴う外傷に起因する浮腫、脳浮腫及び血管浮腫、糖尿病、例えば糖尿病性脈管障害、糖尿病性神経障害、糖尿病性網膜症、膵島炎に伴う毛細血管抵抗後又は糖尿病性症状(例えば高血糖、多尿、蛋白尿及び亜硝酸塩増加及びカリクレイン尿中排泄)。
【0090】
他の適応症も挙げられる:てんかん、敗血性ショック(例えば抗血液量減少及び/又は抗血圧降下薬として)、癌、敗血症、骨粗しょう症、良性前立腺肥大及び過活動膀胱、そう痒症、白斑症、一般的胃腸障害、呼吸器、尿生殖器、胃腸又は血管領域における内臓運動性の障害、創傷、やけど、組織損傷及び術後熱、そう痒関連症候群。
ヒトの治療に有用であることに加えて、これらの化合物はペット、珍しい動物並びに哺乳類、げっ歯類などを含めた家畜の治療にも有用である。
上記疾患及び状態の治療のため、治療的に有効な用量は、一般的に本発明の化合物の投薬につき約0.01mg〜約100mg/kg(体重);好ましくは投薬につき約0.1mg〜約20mg/kg(体重)であろう。例えば、70kgの人に投与するためには、用量範囲は、本発明の化合物の投薬につき約0.7mg〜約7000mg、好ましくは投薬につき約7.0mg〜約1400mgである。最適な投与レベル及びパターンを決定するためには、ある程度の日常的用量の最適化が必要であろう。活性成分を1日1〜6回投与してよい。
【0091】
(一般的投与及び医薬組成物)
医薬品として使用する場合、典型的に医薬組成物の形態で本発明の化合物を投与する。該医薬組成物は、医薬品分野で周知の手順を利用して調製可能であり、少なくとも1種の本発明の化合物を含む。本発明の化合物を単独で投与してもよく、或いは本発明の化合物の安定性を向上させ、特定の実施形態では本化合物を含む医薬組成物の投与を容易にし、溶解及び分散を高め、阻害活性を高め、補助治療をもたらす等のアジュバントと併用してよい。本発明の化合物を単独で使用してよく、或いは本発明の他の活性物質と共に使用してよく、必要に応じて他の薬理学的に活性な物質と共に使用してもよい。一般に、本発明の化合物は、治療的又は医薬的に有効な量で投与されるが、診断又は他の目的のためにはより少ない量で投与し得る。
純粋形態又は適切な医薬組成物での本発明の化合物の投与は、医薬組成物の投与の一般に認められたいずれの様式を利用しても行える。従って、投与は、例えば、経口、頬側(例えば、舌下)、経鼻、非経口、局所、経皮、経膣、又は経直腸的に、固体、半固体、凍結乾燥散剤、又は液体剤形、例えば、錠剤、座剤、丸剤、軟弾性及び硬ゼラチンカプセル剤、散剤、溶液、懸濁液、又はエアロゾル等で、好ましくは正確な薬用量の簡単な投与に適した単位剤形であってよい。医薬組成物は、一般的に通常の医薬担体又は賦形剤と、活性物質として本発明の化合物とを含み、かつそれに加えて、他の薬用剤、医薬、担体、アジュバント、希釈剤、ビヒクル、又はその組合せを含んでよい。このような医薬的に許容できる賦形剤、担体、又は添加剤並びに種々の様式又は投与のための医薬組成物の製造方法は当業者に周知である。技術水準は、例えば、Remington: The Science and Practice of Pharmacy, 20th Edition, A. Gennaro (ed.), Lippincott Williams & Wilkins, 2000; Handbook of Pharmaceutical Additives, Michael & Irene Ash (eds.), Gower, 1995; Handbook of Pharmaceutical Excipients, A.H. Kibbe (ed.), American Pharmaceutical Ass’n, 2000; H.C. Ansel and N.G. Popovish, Pharmaceutical Dosage Forms and Drug Delivery Systems, 5th ed., Lea and Febiger, 1990によって証明される。なお、技術水準をよりよく示すため、これらの文献の内容全体を参照によってここに援用する。
【0092】
当業者が予想するように、特定の医薬製剤で利用する本発明の化合物の形態(例えば塩)は、その製剤が有効であるために必要とされる適切な物理的特性(例えば、水溶性)を有するように選択されるであろう。
頬側(舌下)投与に適した医薬組成物には、香味づけ基剤、普通はスクロースと、アカシア又はトラガカントに本発明の化合物を含むロゼンジ剤、及びゼラチンとグリセリン又はスクロースとアカシアのような不活性基剤に本発明の化合物を含むトローチ剤(pastille)がある。
非経口投与に適した医薬組成物は、本発明の化合物の無菌水性製剤を含む。これらの製剤は好ましくは静脈内投与されるが、皮下、筋肉内、又は皮内注射経由で投与を達成することもできる。注射用医薬製剤は一般的に注射用無菌食塩水、リン酸緩衝食塩水、油性懸濁液、又は技術上周知の他の注射用担体を基礎とし、通常は滅菌され、かつ血液と等張の状態にされる。従って、注射用医薬製剤を無毒の非経口的に許容できる希釈剤又は溶媒中の滅菌注射用溶液又は懸濁液として提供し得る。このような希釈剤又は溶媒としては、1,3-ブタンジオール、水、リンゲル液、等張塩化ナトリウム溶液、固定油、例えば合成モノグリセリド若しくはジグリセリド、脂肪酸、例えばオレイン酸などが挙げられる。既知技術に従い、適切な分散剤又は硬化剤及び懸濁化剤を用いて該注射用医薬製剤を処方する。注射用組成物は通常0.1〜5%w/wの本発明の化合物を含むであろう。
【0093】
化合物の経口投与用固体剤形には、カプセル剤、錠剤、丸剤、散剤、顆粒剤がある。該経口投与のためには、本発明の化合物を含む医薬的に許容できる組成物は、例えば、医薬品グレードのマンニトール、ラクトース、デンプン、アルファ化デンプン、ステアリン酸マグネシウム、ナトリウムサッカリン、タルカム、セルロースエーテル誘導体、グルコース、ゼラチン、スクロース、シトラート、没食子酸プロピル等のような通常用いられるいずれかの賦形剤の組み入れによって形成される。このような固体医薬製剤には、技術上周知なように、いくつかの機構によって胃腸管への薬物の遅延性又は持続性送達をもたらすための製剤が含まれる。このような機構としては、限定するものではないが、小腸のpHを変えることに基づく剤形からのpH感受性放出、錠剤又はカプセル剤の緩徐浸食、製剤の物理的性質に基づく胃内での保持、腸管の粘膜内層への剤形の生体接着、又は剤形からの活性薬物の酵素的放出などが挙げられる。
化合物の経口投与用の液体剤形としては、エマルション、マイクロエマルション、溶液、懸濁液、シロップ、及びエリキシル剤が挙げられ、必要に応じて、例えば、水、食塩水、デキストロース水溶液、グリセロール、エタノール等の担体中の医薬アジュバントを含む。これらの組成物は、湿潤剤、乳化剤、懸濁化剤、甘味料、調味料、及び香料などのさらなるアジュバントを含むこともできる。
【0094】
化合物の局所剤形には、軟膏、ペースト、クリーム、ローション、ゲル、散剤、溶液、スプレー、吸入剤、眼軟膏又は点耳薬、含浸包帯剤及びエアロゾルがあり、適切な通常の添加剤、例えば保存剤、薬物の浸透を助けるための溶媒並びに軟膏及びクリームには皮膚軟化剤を含有し得る。局所適用は1日1回でも複数回でもよく、通常の医学的考慮によって決まる。さらに、本発明に好ましい化合物を適切な鼻腔内ビヒクルの局所使用によって鼻腔内形態で投与することができる。製剤はクリーム又は軟膏基剤、またローションではエタノール又はオレイルアルコール等の適合性の通常の担体を含んでもよい。このような担体は、製剤の約1%から約98%まで存在してよいが、さらに一般的にはそれらは製剤の約80%までを形成するであろう。
経皮投与も可能である。長期間レシピエントの表皮と密接に接触し続けるのに適合した個別パッチとして、経皮投与に適した医薬組成物を提供することができる。経皮送達システムの形態で投与されるため、当然に、薬用量の投与は、投与計画全体を通じて間欠的ではなく連続的であろう。該パッチは、接着剤に溶解及び/又は分散させ、或いはポリマーに分散させた必要に応じて緩衝化した水溶液に適切に本発明の化合物を含む。活性化合物の適切な濃度は、約1%〜35%、好ましくは約3%〜15%である。
【0095】
吸入による投与のためには、本発明の化合物は通常、噴霧ガスが不要なポンプスプレー装置から或いは適切な噴霧剤、例えば、ジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、テトラフルオロエタン、ヘプタフルオロプロパン、二酸化炭素、若しくは他の適切なガスを使用する加圧パック又は噴霧器からエアロゾルスプレーの形態で送達される。いずれの場合も、定量を送達するためのバルブを設け、結果としての定量吸入器(metered dose inhaler)(MDI)を用いて本発明の化合物を再現性のある制御されたやり方で投与することによって、エアロゾルスプレー投薬単位を決定し得る。このような吸入器、噴霧器、又はアトマイザー装置は技術上周知であり、例えば、PCT国際出願公開第WO 97/12687号(特にその図6は市販のRESPIMAT(登録商標)噴霧器の基本である);第WO 94/07607号;第WO 97/12683号;及び第WO 97/20590号がある。この結果、上記文献は参照され、それぞれ全体が参照によって本明細書に援用される。
直腸投与は、化合物を低融点の水溶性又は不溶性固体、例えば脂肪、ココアバター、グリセリンゼラチン、水素化植物油、種々の分子量のポリエチレングリコールの混合物、又はポリエチレングリコールの脂肪酸エステル等と混合する単位用量座剤を利用して達成し得る。活性化合物は普通は少量成分であり、多くの場合、約0.05〜10質量%であり、残りが基剤成分である。
【0096】
全ての上記医薬組成物において、本発明の化合物は許容可能な担体又は賦形剤と処方される。当然に、使用する担体又は賦形剤は、組成物の他の成分と適合性であるという意味で許容性でなければならず、かつ患者にとって有害であってはならない。担体又は賦形剤は固体若しくは液体、又は両者であってよく、好ましくは単位用量組成物、例えば、0.05質量%〜95質量%の活性化合物を含有し得る錠剤として、本発明の化合物と処方される。このような担体又は賦形剤としては、不活性な充填剤若しくは希釈剤、結合剤、潤沢剤、崩壊剤、溶解遅延剤、再吸収促進剤、吸収剤、及び着色料が挙げられる。適切な結合剤としては、デンプン、ゼラチン、天然糖、例えばグルコース又はβ-ラクトース、コーン甘味料、天然及び合成ガム、例えばアカシア、トラガカント又はアルギン酸ナトリウム、カルボキシメチルセルロース、ポリエチレングリコール、蝋などが挙げられる。潤沢剤としては、オレイン酸ナトリウム、ステアリン酸ナトリウム、ステアリン酸マグネシウム、安息香酸ナトリウム、酢酸ナトリウム、塩化ナトリウム等が挙げられる。崩壊剤としては、デンプン、メチルセルロース、寒天、ベントナイト、キサンタンガム等が挙げられる。
医薬的に許容できる担体及び賦形剤は、前述した全ての添加剤などを包含する。
【特許請求の範囲】
【請求項1】
下記式(I)
【化1】
(I)
(式中、
R1は、C1-10アルキル、C3-10シクロアルキル、3〜10員飽和ヘテロ環式環、C1-5アルキル-ヘテロ環式環、5〜10員単環式若しくは二環式アリール環又はC1-5アルキル-アリール環であり、ここで、各R1は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C3-10シクロアルキル、C1-4アルコキシ、C1-4アルキルスルホニル、アシル、シアノ、ヒドロキシル及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
R2及びR3は、C1-4アルキル又は水素であり(但し、R2とR3が両方とも水素ではありえない);或いは
R2とR3が、それらが結合している炭素原子と一緒に3〜6員シクロアルキル環を形成し;
R4は、水素又はメチルであり;
R5は、C1-10アルキル、C3-10シクロアルキル、3〜10員飽和ヘテロ環式環、5〜10員単環式若しくは二環式アリール環、5〜10員単環式若しくは二環式ヘテロアリール環、-S-C1-5アルキル、-S-アリール、-S-CH2-アリール、-O-C1-5アルキル、-O-アリール、-O-CH2-アリール、-NH-C1-5アルキル、-NH-アリール、-NH-CH2-アリール又はアミノであり、ここで、各R5は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、アシル、-C(O)-ヘテロアリール、シアノ、ヒドロキシル及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒に4〜7員ヘテロ環式環(アリール、-C(O)-O-C1-5アルキル、-C(O)-ヘテロアリール、-C(O)-アリール、-SO2-ヘテロアリール及び-SO2-アリールから選択される置換基で置換されていてもよい)を形成していてもよく;
R6は、水素又はC1-4アルキルであり;
nは、0、1、2、3又は4であり;
ここで、式(I)又は前記いずれのR置換基上のいずれの炭素原子も、可能であれば部分的又は完全にハロゲン化されていてもよい)
の化合物、
又はその医薬的に許容できる塩。
【請求項2】
式中、
R1が、C1-6アルキル、フェニル、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、-CH2-シクロプロピル、-CH2-シクロブチル、テトラヒドロフラニル、テトラヒドロピラニル、アゼチジニル、ピペリジニル;チオモルフォリニル、1,1-ジオキソ-1λ6-チオモルフォリニル、モルフォリニル、ピロリジニル、ピペラジニル、ベンジル、-CH2-テトラヒドロフラニル、-CH2-テトラヒドロピラニル、-CH2-CH2-テトラヒドロフラニル又は-CH2-CH2-テトラヒドロピラニルであり、ここで、各R1は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、C1-4アルキルスルホニル、アシル、シアノ、ヒドロキシル及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
R2及びR3が、独立にメチル、エチル、n-プロピル、イソプロピル、t-Bu、又は水素であり(但し、R2とR3が両方とも水素ではありえない);或いは
R2とR3が、それらが結合している炭素と一緒にシクロプロピル、シクロブチル、又はシクロペンチル環を形成し;
R4が、水素又はメチルであり;
R5が、C1-5アルキル、フェニル、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、テトラヒドロフラニル、テトラヒドロピラニル、アゼチジニル、ピペリジニル;チオモルフォリニル、1,1-ジオキソ-1λ6-チオモルフォリニル、モルフォリニル、ピロリジニル、ピペラジニル、インダニル、ベンゾオキサゾリル、ベンゾチアゾリル、ベンゾイミダゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、ピラゾリル、ピロリル、イミダゾリル、チエニル、チアジアゾリル、ピリジニル、ピリミジニル、ピリダジニル、ピラジニル、トリアジニル、キノリニル、ジヒドロ-2H-キノリニル、イソキノリニル、キナゾリニル、インダゾリル、インドリル、ベンゾフラニル、ベンゾピラニル、ベンゾジオキソリル、-S-C1-5アルキル、-S-フェニル、-S-CH2-フェニル、-O-C1-5アルキル、-O-フェニル、-O-CH2-フェニル、-NH-C1-5アルキル、-NH-フェニル、-NH-CH2-フェニル又はアミノであり、ここで、各R5は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、アシル、-C(O)-ヘテロアリール、シアノ、ヒドロキシル及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒に4〜7員ヘテロ環式環(0又は1個のさらなるヘテロ原子を含み、かつフェニル、-C(O)-O-C1-5アルキル、-C(O)-チエニル、-C(O)-フェニル、-SO2-ヘテロアリール及び-SO2-フェニルから選択される置換基で置換されていてもよい)を形成していてもよく;
R6が、水素又はC1-3アルキルである、
請求項1に記載の化合物。
【請求項3】
式中、
R1が、C1-6アルキル、フェニル、シクロペンチル、シクロヘキシル、テトラヒドロピラニル、ベンジル又は-CH2-テトラヒドロピラニルであり、ここで、各R1は、独立にC1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、C1-4アルキルスルホニル、及びハロゲンから選択される1〜3個の置換基で置換されていてもよい、
請求項2に記載の化合物。
【請求項4】
式中、
R2及びR3が独立にメチルであり、或いは
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成している、
請求項3に記載の化合物。
【請求項5】
式中、
R4が水素又はメチルであり;
R5が、C1-5アルキル、フェニル、シクロヘキシル、シクロヘプチル、モルフォリニル、インダニル、イソオキサゾリル、チエニル、ピリジニル、-S-C1-5アルキル、-S-フェニル、-S-CH2-フェニル、-O-C1-5アルキル、-O-フェニル、-NH-C1-5アルキル、-NH-フェニル又はアミノであり、ここで、各R5は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、アシル、及びハロゲンから選択される1〜3個の置換基で置換されていてもよい、
請求項4に記載の化合物。
【請求項6】
式中、
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒に4〜7員ヘテロ環式環(0又は1個のさらなるヘテロ原子を含み、かつフェニル、-C(O)-O-C1-4アルキル、-C(O)-チエニル及び-SO2-フェニルから選択される置換基で置換されていてもよい)を形成していてもよい、
請求項5に記載の化合物。
【請求項7】
式中、
R1が、C1-6アルキル、フェニル、シクロペンチル、シクロヘキシル、テトラヒドロピラニル、ベンジル又は-CH2-テトラヒドロピラニルであり、ここで、各R1は、独立にC1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、C1-4アルキルスルホニル、及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
R2及びR3が独立にメチルであり、或いは
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成し;
R4が水素又はメチルであり;
R5が、C1-5アルキル、フェニル、シクロヘキシル、シクロヘプチル、モルフォリニル、インダニル、イソオキサゾリル、チエニル、ピリジニル、-S-C1-5アルキル、-S-フェニル、-S-CH2-フェニル、-O-C1-5アルキル、-O-フェニル、-NH-C1-5アルキル、-NH-フェニル又はアミノであり、ここで、各R5は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、アシル、及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒にピペリジニル又はピペラジニル(フェニル、-C(O)-O-C1-4アルキル、-C(O)-チエニル及び-SO2-フェニルから選択される置換基で置換されていてもよい)を形成していてもよく;
R6が水素又はメチルであり;
nが、0、1、2又は3である、
請求項2に記載の化合物。
【請求項8】
式中、
R1が、フェニル、シクロヘキシル又はベンジルであり、ここで、各R1は、独立に、トリフルオロメチル、及びクロロから選択される1〜2個の置換基で置換されていてもよく;
R2及びR3がメチルであり、或いは
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成し;
R4が水素であり;
R5が、C1-4アルキル、フェニル、シクロヘキシル、シクロヘプチル、チエニル、-S-C1-5アルキル、-S-フェニル、-S-CH2-フェニル、-O-C1-5アルキル、-O-フェニル又は-NH-フェニルであり、ここで、各R5は、独立に、C1-4アルキル、トリフルオロメチル、C1-4アルコキシ、フルオロ及びクロロから選択される1〜3個の置換基で置換されていてもよい、
請求項7に記載の化合物。
【請求項9】
式中、
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒にピペリジニル又はピペラジニル(フェニル、-C(O)-O-C1-4アルキル及び-C(O)-チエニルから選択される置換基で置換されていてもよい)を形成していてもよい、
請求項7に記載の化合物。
【請求項10】
式中、
R1がフェニル又はシクロヘキシルであり、それぞれ、トリフルオロメチル、及びクロロから選択される置換基で置換されていてもよく;
R2及びR3がメチルであり、或いは
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成し;
R4が水素であり;
R5が、C1-4アルキル、フェニル、シクロヘキシル、-S-C1-5アルキル、-S-フェニル又は-S-CH2-フェニルであり、ここで、各R5は、独立に、C1-4アルキル、トリフルオロメチル、メトキシ、フルオロ及びクロロから選択される1〜3個の置換基で置換されていてもよく;
nが0又は2である、
請求項8に記載の化合物。
【請求項11】
式中、
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒に、フェニル基で置換されているピペラジニル環を形成している、
請求項9に記載の化合物。
【請求項12】
式中、
R2及びR3がメチルである、
請求項1に記載の化合物。
【請求項13】
式中、
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成している、
請求項1に記載の化合物。
【請求項14】
下記式(IA)の化合物又はその医薬的に許容できる塩。
【化2】
(IA)
(ここで、式(IA)の下記部分
【化3】
は、下表IのA1〜A16から選択され、かつ式(IA)の下記部分
【化4】
は、下表IのB1〜B48から選択される。)
表I
【請求項15】
下記化合物から選択される化合物又はその医薬的に許容できる塩。
【請求項16】
治療的に有効な量の請求項1に記載の化合物と、1種以上の医薬的に許容できる担体及び/又はアジュバントとを含んでなる医薬組成物。
【請求項17】
治療的に有効な量の請求項1に記載の化合物を投与することを含んでなる疼痛の治療方法。
【請求項18】
肺疾患、リウマチ性疾患、自己免疫疾患、筋骨格疾患、アレルギー性疾患、アレルギー反応、血管疾患、皮膚疾患、腎疾患、肝疾患、胃腸疾患、神経変性、眼病、耳鼻咽喉疾患、神経疾患、血液疾患、腫瘍、内分泌疾患、臓器及び組織移植並びに移植片対宿主病、重篤状態のショック、急性疼痛、内臓痛、胃腸管又は子宮の痙攣、疝痛、神経因性疼痛、炎症性及び侵害受容性疼痛、癌性疼痛、頭痛、再狭窄、アテローム性動脈硬化症、再灌流傷害、うっ血性心不全、心筋梗塞、熱傷、外傷続発性多臓器損傷、壊死性腸炎並びに血液透析、白血球フェレーシス、及び顆粒球輸血と関連する症候群、サルコイドーシス、歯肉炎及び発熱から選択される疾患又は状態の治療方法であって、治療的に有効な量の請求項1に記載の化合物を投与することを含んでなる方法。
【請求項1】
下記式(I)
【化1】
(I)
(式中、
R1は、C1-10アルキル、C3-10シクロアルキル、3〜10員飽和ヘテロ環式環、C1-5アルキル-ヘテロ環式環、5〜10員単環式若しくは二環式アリール環又はC1-5アルキル-アリール環であり、ここで、各R1は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C3-10シクロアルキル、C1-4アルコキシ、C1-4アルキルスルホニル、アシル、シアノ、ヒドロキシル及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
R2及びR3は、C1-4アルキル又は水素であり(但し、R2とR3が両方とも水素ではありえない);或いは
R2とR3が、それらが結合している炭素原子と一緒に3〜6員シクロアルキル環を形成し;
R4は、水素又はメチルであり;
R5は、C1-10アルキル、C3-10シクロアルキル、3〜10員飽和ヘテロ環式環、5〜10員単環式若しくは二環式アリール環、5〜10員単環式若しくは二環式ヘテロアリール環、-S-C1-5アルキル、-S-アリール、-S-CH2-アリール、-O-C1-5アルキル、-O-アリール、-O-CH2-アリール、-NH-C1-5アルキル、-NH-アリール、-NH-CH2-アリール又はアミノであり、ここで、各R5は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、アシル、-C(O)-ヘテロアリール、シアノ、ヒドロキシル及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒に4〜7員ヘテロ環式環(アリール、-C(O)-O-C1-5アルキル、-C(O)-ヘテロアリール、-C(O)-アリール、-SO2-ヘテロアリール及び-SO2-アリールから選択される置換基で置換されていてもよい)を形成していてもよく;
R6は、水素又はC1-4アルキルであり;
nは、0、1、2、3又は4であり;
ここで、式(I)又は前記いずれのR置換基上のいずれの炭素原子も、可能であれば部分的又は完全にハロゲン化されていてもよい)
の化合物、
又はその医薬的に許容できる塩。
【請求項2】
式中、
R1が、C1-6アルキル、フェニル、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、-CH2-シクロプロピル、-CH2-シクロブチル、テトラヒドロフラニル、テトラヒドロピラニル、アゼチジニル、ピペリジニル;チオモルフォリニル、1,1-ジオキソ-1λ6-チオモルフォリニル、モルフォリニル、ピロリジニル、ピペラジニル、ベンジル、-CH2-テトラヒドロフラニル、-CH2-テトラヒドロピラニル、-CH2-CH2-テトラヒドロフラニル又は-CH2-CH2-テトラヒドロピラニルであり、ここで、各R1は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、C1-4アルキルスルホニル、アシル、シアノ、ヒドロキシル及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
R2及びR3が、独立にメチル、エチル、n-プロピル、イソプロピル、t-Bu、又は水素であり(但し、R2とR3が両方とも水素ではありえない);或いは
R2とR3が、それらが結合している炭素と一緒にシクロプロピル、シクロブチル、又はシクロペンチル環を形成し;
R4が、水素又はメチルであり;
R5が、C1-5アルキル、フェニル、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、テトラヒドロフラニル、テトラヒドロピラニル、アゼチジニル、ピペリジニル;チオモルフォリニル、1,1-ジオキソ-1λ6-チオモルフォリニル、モルフォリニル、ピロリジニル、ピペラジニル、インダニル、ベンゾオキサゾリル、ベンゾチアゾリル、ベンゾイミダゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、ピラゾリル、ピロリル、イミダゾリル、チエニル、チアジアゾリル、ピリジニル、ピリミジニル、ピリダジニル、ピラジニル、トリアジニル、キノリニル、ジヒドロ-2H-キノリニル、イソキノリニル、キナゾリニル、インダゾリル、インドリル、ベンゾフラニル、ベンゾピラニル、ベンゾジオキソリル、-S-C1-5アルキル、-S-フェニル、-S-CH2-フェニル、-O-C1-5アルキル、-O-フェニル、-O-CH2-フェニル、-NH-C1-5アルキル、-NH-フェニル、-NH-CH2-フェニル又はアミノであり、ここで、各R5は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、アシル、-C(O)-ヘテロアリール、シアノ、ヒドロキシル及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒に4〜7員ヘテロ環式環(0又は1個のさらなるヘテロ原子を含み、かつフェニル、-C(O)-O-C1-5アルキル、-C(O)-チエニル、-C(O)-フェニル、-SO2-ヘテロアリール及び-SO2-フェニルから選択される置換基で置換されていてもよい)を形成していてもよく;
R6が、水素又はC1-3アルキルである、
請求項1に記載の化合物。
【請求項3】
式中、
R1が、C1-6アルキル、フェニル、シクロペンチル、シクロヘキシル、テトラヒドロピラニル、ベンジル又は-CH2-テトラヒドロピラニルであり、ここで、各R1は、独立にC1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、C1-4アルキルスルホニル、及びハロゲンから選択される1〜3個の置換基で置換されていてもよい、
請求項2に記載の化合物。
【請求項4】
式中、
R2及びR3が独立にメチルであり、或いは
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成している、
請求項3に記載の化合物。
【請求項5】
式中、
R4が水素又はメチルであり;
R5が、C1-5アルキル、フェニル、シクロヘキシル、シクロヘプチル、モルフォリニル、インダニル、イソオキサゾリル、チエニル、ピリジニル、-S-C1-5アルキル、-S-フェニル、-S-CH2-フェニル、-O-C1-5アルキル、-O-フェニル、-NH-C1-5アルキル、-NH-フェニル又はアミノであり、ここで、各R5は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、アシル、及びハロゲンから選択される1〜3個の置換基で置換されていてもよい、
請求項4に記載の化合物。
【請求項6】
式中、
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒に4〜7員ヘテロ環式環(0又は1個のさらなるヘテロ原子を含み、かつフェニル、-C(O)-O-C1-4アルキル、-C(O)-チエニル及び-SO2-フェニルから選択される置換基で置換されていてもよい)を形成していてもよい、
請求項5に記載の化合物。
【請求項7】
式中、
R1が、C1-6アルキル、フェニル、シクロペンチル、シクロヘキシル、テトラヒドロピラニル、ベンジル又は-CH2-テトラヒドロピラニルであり、ここで、各R1は、独立にC1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、C1-4アルキルスルホニル、及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
R2及びR3が独立にメチルであり、或いは
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成し;
R4が水素又はメチルであり;
R5が、C1-5アルキル、フェニル、シクロヘキシル、シクロヘプチル、モルフォリニル、インダニル、イソオキサゾリル、チエニル、ピリジニル、-S-C1-5アルキル、-S-フェニル、-S-CH2-フェニル、-O-C1-5アルキル、-O-フェニル、-NH-C1-5アルキル、-NH-フェニル又はアミノであり、ここで、各R5は、独立に、C1-4アルキル基(1〜3個のハロゲンで置換されていてもよい)、C1-4アルコキシ、アシル、及びハロゲンから選択される1〜3個の置換基で置換されていてもよく;
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒にピペリジニル又はピペラジニル(フェニル、-C(O)-O-C1-4アルキル、-C(O)-チエニル及び-SO2-フェニルから選択される置換基で置換されていてもよい)を形成していてもよく;
R6が水素又はメチルであり;
nが、0、1、2又は3である、
請求項2に記載の化合物。
【請求項8】
式中、
R1が、フェニル、シクロヘキシル又はベンジルであり、ここで、各R1は、独立に、トリフルオロメチル、及びクロロから選択される1〜2個の置換基で置換されていてもよく;
R2及びR3がメチルであり、或いは
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成し;
R4が水素であり;
R5が、C1-4アルキル、フェニル、シクロヘキシル、シクロヘプチル、チエニル、-S-C1-5アルキル、-S-フェニル、-S-CH2-フェニル、-O-C1-5アルキル、-O-フェニル又は-NH-フェニルであり、ここで、各R5は、独立に、C1-4アルキル、トリフルオロメチル、C1-4アルコキシ、フルオロ及びクロロから選択される1〜3個の置換基で置換されていてもよい、
請求項7に記載の化合物。
【請求項9】
式中、
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒にピペリジニル又はピペラジニル(フェニル、-C(O)-O-C1-4アルキル及び-C(O)-チエニルから選択される置換基で置換されていてもよい)を形成していてもよい、
請求項7に記載の化合物。
【請求項10】
式中、
R1がフェニル又はシクロヘキシルであり、それぞれ、トリフルオロメチル、及びクロロから選択される置換基で置換されていてもよく;
R2及びR3がメチルであり、或いは
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成し;
R4が水素であり;
R5が、C1-4アルキル、フェニル、シクロヘキシル、-S-C1-5アルキル、-S-フェニル又は-S-CH2-フェニルであり、ここで、各R5は、独立に、C1-4アルキル、トリフルオロメチル、メトキシ、フルオロ及びクロロから選択される1〜3個の置換基で置換されていてもよく;
nが0又は2である、
請求項8に記載の化合物。
【請求項11】
式中、
n=0の場合、R4とR5が、それらが結合している窒素原子と一緒に、フェニル基で置換されているピペラジニル環を形成している、
請求項9に記載の化合物。
【請求項12】
式中、
R2及びR3がメチルである、
請求項1に記載の化合物。
【請求項13】
式中、
R2とR3が、それらが結合している炭素と一緒にシクロブチル環を形成している、
請求項1に記載の化合物。
【請求項14】
下記式(IA)の化合物又はその医薬的に許容できる塩。
【化2】
(IA)
(ここで、式(IA)の下記部分
【化3】
は、下表IのA1〜A16から選択され、かつ式(IA)の下記部分
【化4】
は、下表IのB1〜B48から選択される。)
表I
【請求項15】
下記化合物から選択される化合物又はその医薬的に許容できる塩。
【請求項16】
治療的に有効な量の請求項1に記載の化合物と、1種以上の医薬的に許容できる担体及び/又はアジュバントとを含んでなる医薬組成物。
【請求項17】
治療的に有効な量の請求項1に記載の化合物を投与することを含んでなる疼痛の治療方法。
【請求項18】
肺疾患、リウマチ性疾患、自己免疫疾患、筋骨格疾患、アレルギー性疾患、アレルギー反応、血管疾患、皮膚疾患、腎疾患、肝疾患、胃腸疾患、神経変性、眼病、耳鼻咽喉疾患、神経疾患、血液疾患、腫瘍、内分泌疾患、臓器及び組織移植並びに移植片対宿主病、重篤状態のショック、急性疼痛、内臓痛、胃腸管又は子宮の痙攣、疝痛、神経因性疼痛、炎症性及び侵害受容性疼痛、癌性疼痛、頭痛、再狭窄、アテローム性動脈硬化症、再灌流傷害、うっ血性心不全、心筋梗塞、熱傷、外傷続発性多臓器損傷、壊死性腸炎並びに血液透析、白血球フェレーシス、及び顆粒球輸血と関連する症候群、サルコイドーシス、歯肉炎及び発熱から選択される疾患又は状態の治療方法であって、治療的に有効な量の請求項1に記載の化合物を投与することを含んでなる方法。
【公表番号】特表2013−517271(P2013−517271A)
【公表日】平成25年5月16日(2013.5.16)
【国際特許分類】
【出願番号】特願2012−548997(P2012−548997)
【出願日】平成23年1月11日(2011.1.11)
【国際出願番号】PCT/US2011/020767
【国際公開番号】WO2011/088015
【国際公開日】平成23年7月21日(2011.7.21)
【出願人】(503385923)ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング (976)
【Fターム(参考)】
【公表日】平成25年5月16日(2013.5.16)
【国際特許分類】
【出願日】平成23年1月11日(2011.1.11)
【国際出願番号】PCT/US2011/020767
【国際公開番号】WO2011/088015
【国際公開日】平成23年7月21日(2011.7.21)
【出願人】(503385923)ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング (976)
【Fターム(参考)】
[ Back to top ]