
CC−1065類似体の調製方法及び調製用化合物
【課題】CC−1065類似体の合成方法及び合成用化合物を提供すること。
【解決手段】上記の課題は、以下の化合物(I)又はその塩の合成方法により解決される。

(式中、R1及びR2は、各々独立して、H、アルキル、−C(O)OR’、−C(O)NR’R”又は保護基であり、R’及びR”は、独立して、H、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル及び非置換ヘテロシクロアルキルからなる群から選択され;R6は、H、置換若しくは非置換の低級アルキル、シアノ又はアルコキシであり;Xは、ハロゲンである)であって、保護基R1’及びR2’が、化合物(II)に付加され、
化合物(III)を形成し
(式中、R3は、アルキルである。)、化合物(III)のアミン窒素を含む5員環が生成される。
【解決手段】上記の課題は、以下の化合物(I)又はその塩の合成方法により解決される。
(式中、R1及びR2は、各々独立して、H、アルキル、−C(O)OR’、−C(O)NR’R”又は保護基であり、R’及びR”は、独立して、H、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル及び非置換ヘテロシクロアルキルからなる群から選択され;R6は、H、置換若しくは非置換の低級アルキル、シアノ又はアルコキシであり;Xは、ハロゲンである)であって、保護基R1’及びR2’が、化合物(II)に付加され、
化合物(III)を形成し
(式中、R3は、アルキルである。)、化合物(III)のアミン窒素を含む5員環が生成される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はCC−1065類似体の調製方法及び調製用化合物に関する。
関連出願の相互参照:本特許出願は、2005年10月26日に出願の米国仮特許出願第60/730,804号(全開示内容を参照によって本願明細書に援用する)の利益を主張するものである。
【背景技術】
【0002】
CC−1065は、強力なサイトトキシンであることが公知である。CC−1065は、アップジョン社(Upjohn Company)によって1981年にStreptomyces zelensisから初めて単離され(Hanka et al.,J.Antibiot.31:1211(1978);Martin et al,J.Antibiot.33:902(1980);Martin et al,J.Antibiot.34:1119(1981))、in vitro及び実験動物の両方において強力な抗腫瘍活性及び抗菌活性を有することが明らかにされた(Li et al.,Cancer Res.42:999(1982))。CC−1065は、5’−d(A/GNTTA)−3’及び5’−d(AAAAA)−3’の配列選択性により、副溝中で二本鎖B−DNAに結合し(Swenson et al,Cancer Res.42:2821(1982))、その分子中に存在するCPI左側部分によって3’−アデニンのN3位をアルキル化する(Hurley et al,Science 226:843(1984))。
【発明の開示】
【発明が解決しようとする課題】
【0003】
CC−1065は、その強力かつ広範な抗腫瘍活性にもかかわらず、それが実験動物の遅延死を引き起こすため、人間に対する使用を不可能にしている。
【0004】
CC−1065の多くのアナログ(類似体)及び誘導体が当該技術分野で公知である。その構造式、合成及び化合物の特性の多くが、これまで研究されている。例えば、Boger et al.,Angew.Chem.Int.Ed.Engl.35:1438(1996);及びBoger et al.,Chem.Rev.97:787(1997)を参照のこと。
【0005】
協和醗酵工業のグループは、多くのCC−1065誘導体を調製している。例えば、米国特許5,101,038号;同第5,641,780号;同第5,187,186号;同第5,070,092号;同第5,703,080号;同第5,070,092号;同第5,641,780号;同第5,101,038号;及び同第5,084,468号;並びにPCT出願公開第WO96/10405及び欧州特許出願公開第0537575A1号を参照のこと。
【0006】
また、アップジョン社はCC−1065の誘導体を多種にわたり調製している。例えば、米国特許第5,739,350号;同第4,978,757号、同第5,332,837号及び同第4,912,227号を参照のこと。
【課題を解決するための手段】
【0007】
ある実施態様は、化合物(I)又はその塩を合成する方法である。
【化1】
式中、R1及びR2は、各々独立して、H、アルキル、−C(O)OR’、−C(O)NR’R”又は保護基であり、R’及びR”は、独立して、H、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル及び非置換ヘテロシクロアルキルからなる群から選択され;R6は、H、置換若しくは非置換の低級アルキル、シアノ又はアルコキシであり;Xは、ハロゲンである。この方法では、保護基R1’及びR2’が、化合物(II)に付加され、
【化2】
化合物(III)を形成する。
【化3】
式中、R3は、H又はアルキルである。化合物(III)のアミン窒素を含む5員環が生成される。
【0008】
別の実施態様は、以下の式を有するCBI CC−1065類似体又はその医薬的に許容される塩を合成する方法である。
【化4】
式中、Xは、ハロであり;X1及びZは、各々独立して、O、S及びNR8から選択され、R8は、H、置換又は非置換のアルキル、置換又は非置換のヘテロアルキル及びアシルから選択されるメンバーであり;R4、R4,、R5及びR5,は、独立して、H、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル、非置換ヘテロシクロアルキル、ハロゲン、NO2、NR9R10、NC(O)R9、OC(O)NR9R10、OC(O)OR9、C(O)R9、SR9、OR9、CR9=NR10及びO(CH2)nNR11R11’からなる群から選択されるメンバーであり、R9及びR10は、独立して、H、置換若しくは非置換のアルキル、置換若しくは非置換のヘテロアルキル、置換若しくは非置換のアリール、置換若しくは非置換のヘテロアリール、置換若しくは非置換のヘテロシクロアルキル及び置換若しくは非置換のペプチジルから選択されるか、又はR9及びR10は、それらが結合する窒素原子と任意に結合して、任意に2つ以上のヘテロ原子を含む4〜6員環を有する置換若しくは非置換のヘテロシクロアルキル環系を形成し、R11及びR11’は、各々独立して、H又は低級アルキルであり;R6は、H、置換若しくは非置換の低級アルキル、シアノ又はアルコキシであり;R7は、H、置換アルキル、非置換アルキル、置換ヘテロアルキル、非置換ヘテロアルキル、ジホスフェート、トリホスフェート、アシル、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、R12、R13及びR14は、独立して、H、置換若しくは非置換のアルキル、置換若しくは非置換のヘテロアルキル及び置換若しくは非置換のアリールから選択されるメンバーであるか、又はR12及びR13は、それらが結合する窒素又は炭素原子と任意に結合して、任意に2つ以上のヘテロ原子を含む4〜6員環を有する置換若しくは非置換のヘテロシクロアルキル環系を形成する。
【0009】
上記方法は、保護基R1’及びR2’を化合物(II)に付加して
【化5】
化合物(III)を形成することを含む。
【化6】
式中、R3は、H又はアルキルである。化合物(III)のアミン窒素を含む5員環が生成される。結合部分が、化合物(III)に付加され、その結合部分は、以下を含む。
【化7】
【0010】
更に別の実施態様は、式(I)の化合物又はその医薬的に許容される塩である。
【化8】
式中、R1及びR2は、各々独立して、H、アルキル、−C(O)OR’、−C(O)NR’R’’又は保護基であり、R’及びR”は、独立してH、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル及び非置換ヘテロシクロアルキルからなる群から選択される。
【0011】
いかなる限定も包括もしない本発明の実施態様を図面を参照しつつ以下に記載する。図面においては、特に断りがない限り、種々の図面を通して付与された数字は部分部分を示す。図面と共に発明な詳細な説明を参照すれば本発明をさらに理解することができるであろう。
【発明を実施するための最良の形態】
【0012】
本願明細書において使用されるとき、「Boc」は、t−ブチルオキシカルボニルを指す。「CPI」は、シクロプロパピロロインドールを指す。「CBI」は、シクロプロパベンズインドールを指す。「Cbz」は、カルボベンゾキシである。「DCM」は、ジクロロメタンを指す。「DMF」は、N,N−ジメチルホルムアミドである。「FMOC」は、9−フルオレニルメチルオキシカルボニルを指す。「TEA」は、トリエチルアミンを指す。「THF」は、テトラヒドロフランを指す。「EDC」は、塩酸1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミドを指す。
【0013】
別途定義する場合を除き、本発明において使用する全ての技術用語及び科学用語は、本発明が属する分野における通常の技術を有する者によって一般に理解されるものと同じ意味で用いられる。本発明において使用する分類、並びに細胞培養、分子遺伝学、有機化学及び核酸化学における解析及び下記のハイブリダイゼーションの手順は、当該技術分野において良く知られ、一般に用いられているものを意味する。技術及び手順は、一般に、当該技術分野の従来法及び様々な一般的文献に従って実施される。本発明において使用する命名、及び分析における解析手順、並びに下記の有機合成は、当該技術分野において公知で、一般に用いられているものを意味する。化学合成及び化学分析においては、標準的技術又はその変法が用いられる。
【0014】
「治療薬」の用語は、治療上有効な量が存在する場合に、哺乳動物に所望の治療効果をもたらす化合物を意味する。腫瘍を処置するためには、治療薬が標的細胞に取り込まれるのが望ましい。
【0015】
「サイトトキシン」の用語は、癌細胞に対して毒性となる効果を有する治療薬を意味する。細胞に対する毒性とは、薬剤が細胞の成長を停止又は細胞を死滅させることを意味する。
【0016】
「プロドラッグ」及び「薬物コンジュゲート」の用語は、本願明細書において互換的に使用される。双方とも、コンジュゲートの形態をとる間は未だ細胞に対して比較的無害な化合物であるが、一定条件(例えば、標的細胞内又はその近傍に位置する酵素の存在)において選択的に分解され、薬理学的に活性な形態となるものを意味する。
【0017】
【化9】
なる符号は、結合部として使用されたとしても、また、結合に対して垂直に表示されたとしても、表示された部分が分子の残部や固体支持体、置換基等に付着する点を示す。
【0018】
「アルキル」の用語は、それ自体又は別の置換基の一部として、別途記載されない限り、直鎖状若しくは分枝状の鎖又は環状の炭化水素ラジカルあるいはそれらの組み合わせを意味し、それらは、完全に飽和していてもよいし、一不飽和又は多価不飽和であってもよく、示された炭素原子の数を有する(すなわちC1−C10は、1〜10個の炭素を意味する)二価及び多価のラジカルを含み得る。飽和炭化水素基の例として、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、t−ブチル、イソブチル、sec−ブチル、シクロヘキシル、(シクロヘキシル)メチル、シクロプロピルメチルなどの基、例えば、n−ペンチル、n−ヘキシル、n−ヘプチル、n−オクチル、等の相同体及び異性体が挙げられるが、これらに限定されない。不飽和アルキル基は、1以上の二重結合又は三重結合を有するものである。不飽和のアルキル基の例としては、ビニル、2−プロペニル、クロチル、2−イソペンテニル、2−(ブタジエニル)、2,4−ペンタジエニル、3−(1,4−ペンタジエニル)、エチニル、1−及び3−プロピニル、3−ブチニル並びにそれらの高い相同体及び異性体が挙げられるが、これらに限定されない。「アルキル」の用語は、別途に述べない限り、「ヘテロアルキル」など、以下に更に詳しく定義するアルキルの誘導体も含むものを意味する。アルキル基で炭化水素基に限られるものは、「ホモアルキル」と命名されている。
【0019】
「アルキレン」の用語は、それ自体又は別の置換基の一部として、アルカンから誘導される二価のラジカルを意味し、その例としては、−CH2CH2CH2CH2−が挙げられるが、これに限定されることはなく、更に「ヘテロアルキレン」と以下に記載される基が含まれる。望ましくは、アルキル(又はアルキレン)基は、1〜24の炭素原子を有するものであり、10以下の炭素原子を有するそれらの基が、本発明では望ましい。「低級アルキル」又は「低級アルキレン」は、短鎖のアルキル又はアルキレン基であり、概して8以下の炭素原子を有している。
【0020】
「ヘテロアルキル」の用語は、それ自体又は別の用語との組み合わせにおいて、別途記載されない限り、安定な直鎖状若しくは分枝状の鎖又は環状の炭化水素基あるいはそれらの組み合わせを意味し、多くの炭素原子並びにO、N、Si及びSからなる群から選択される少なくとも1つのヘテロ原子からなり、ここでその窒素、炭素及び硫黄原子は、任意に酸化されてもよく、また、窒素ヘテロ原子は、任意に四級化されていてもよい。ヘテロ原子(単数または複数)のO、N、S及びSiは、ヘテロアルキル基の内部の任意の位置、又はアルキル基が分子の残部に付着する位置に配置されうる。その例としては、−CH2−CH2−O−CH3、−CH2−CH2−NH−CH3、−CH2−CH2−N(CH3)−CH3、−CH2−S−CH2−CH3、−CH2−CH2、−S(O)−CH3、−CH2−CH2−S(O)2−CH3、−CH=CH−O−CH3、−Si(CH3)3、−CH2−CH=N−OCH3及び−CH=CH−N(CH3)−CH3が挙げられるが、これらに限定されない。例えば、−CH2−NH−OCH3及び−CH2−O−Si(CH3)3など、2つまでのヘテロ原子が連続してもよい。同様に、「ヘテロアルキレン」の用語は、それ自体又は別の置換基の一部として、ヘテロアルキルから誘導される二価のラジカルを意味し、その例としては、−CH2−CH2−S−CH2−CH2−及び−CH2−S−CH2−CH2−NH−CH2−が挙げられるが、これらに限定されない。ヘテロアルキレン基の場合、ヘテロ原子は鎖の末端の片方又は両方を占めることができる(例えば、アルキレンオキシ、アルキレンジオキシ、アルキレンアミノ、アルキレンジアミノ等)。「ヘテロアルキル」及び「ヘテロアルキレン」の用語には、ポリ(エチレングリコール)及びその誘導体(例えば、Shearwater Polymers Catalog、2001を参照されたい)が包含される。更にまた、アルキレン及びヘテロアルキレン置換基の場合、置換基の式が記載される方向によって、置換基の配向が規定されるものではない。例えば、式−C(O)2R’−は、−C(O)2R’−と−R5C(O)2−との両方を表す。
【0021】
「低級」の用語は、「アルキル」又は「ヘテロアルキル」の用語と組み合わせて、1〜6の炭素原子を有する部分をいう。
【0022】
「アルコキシ」、「アルキルアミノ」、「アルキルスルホニル」及び「アルキルチオ」(又はチオアルコキシ)の用語は、それらの従来の意味で使用され、それぞれ酸素原子、アミノ基、SO2基又は硫黄原子を介して分子の残部に付着したアルキル基を指す。「アリールスルホニル」の用語は、SO2基を介して分子の残部に付着したアリール基を指し、「スルフヒドリル」の用語は、SH基を指す。
【0023】
一般に、「アシル」置換基も、上に記載された群から選択される。本願明細書において使用されるとき、「アシル」置換基の用語は、本発明の化合物の多環式核に直接又は間接的に付着したカルボニル炭素に付着して、その原子価を満たす基を指す。
【0024】
「シクロアルキル」及び「ヘテロシクロアルキル」の用語は、それら自体、又は他の用語との組み合わせにおいて、別途に述べない限り、置換又は非置換「アルキル」、及び置換又は非置換「ヘテロアルキル」の環状体をそれぞれ表す。また、ヘテロシクロアルキルの場合、ヘテロ原子は、分子の残部にヘテロ環が付着する位置を占めることができる。シクロアルキルの例として、シクロペンチル、シクロヘキシル、1−シクロヘキセニル、3−シクロヘキセニル、シクロヘプチル等が挙げられるが、これらに限定されない。ヘテロシクロアルキルの例として、1−(1,2,5,6−テトラヒドロピリジル)、1−ピペリジニル、2−ピペリジニル、3−ピペリジニル、4−モルホリニル、3−モルホリニル、テトラヒドロフラン−2−イル、テトラヒドロフラン−3−イル、テトラヒドロチエン−2−イル、テトラヒドロチエン−3−イル、1−ピペラジニル、2−ピペラジニル基等が挙げられるが、これらに限定されない。環状構造のヘテロ原子及び炭素原子は、任意に酸化される。
【0025】
「ハロ」又は「ハロゲン」の用語は、それ自体又は別の置換基の一部として、別途記載されない限り、フッ素、塩素、臭素又はヨウ素原子を意味する。更に、「ハロアルキル」などの用語にはモノハロアルキル及びポリハロアルキルが含まれるものとする。例えば、「ハロ(C1−C4)アルキル」の用語は、トリフルオロメチル、2,2,2−トリフルオロエチル、4−クロロブチル、3−ブロモプロピルなどを含むことを意味するが、これらに限定されない。
【0026】
「アリール」の用語は、別途断りのない限り、置換又は非置換の、多価不飽和、芳香族及び炭化水素の置換基であり、単環又は多環(望ましくは1環から3環)状に融合若しくは共有結合するものを意味する。「ヘテロアリール」の用語は、N、O及びSから選択される1〜4個のヘテロ原子を含むアリール基(または環)を指し、ここで、その窒素、炭素及び硫黄原子は、任意に酸化され、窒素原子(単数又は複数)は、任意に四級化される。ヘテロアリール基は、ヘテロ原子を介して分子の残部に付着できる。アリール及びヘテロアリール基の例としては、フェニル、1−ナフチル、2−ナフチル、4−ビフェニル、1−ピロリル、2−ピロリル、3−ピロリル、3−ピラゾリル、2−イミダゾリル、4−イミダゾリル、ピラジニル、2−オキサゾリル、4−オキサゾリル、2−フェニル−4−オキサゾリル、5−オキサゾリル、3−イソキサゾリル、4−イソキサゾリル、5−イソキサゾリル、2−チアゾリル、4−チアゾリル、5−チアゾリル、2−フリル、3−フリル、2−チエニル、3−チエニル、2−ピリジル、3−ピリジル、4−ピリジル、2−ピリミジル、4−ピリミジル、5−ベンゾチアゾリル、プリニル、2−ベンズイミダゾリル、5−インドリル、1−イソキノリル、5−イソキノリル、2−キノキサリニル、5−キノキサリニル、3−キノリル、及び6−キノリルが挙げられるが、それに限定されない。上記アリール及びヘテロアリール環系の各々に対する置換基は、以下の許容される置換基の群から選択される。「アリール」及び「ヘテロアリール」は、アリール又はヘテロアリール系に、1つ以上の非芳香族の環系が縮合しているか、又は別の方法で結合している環系も包含する。
【0027】
略して、「アリール」の用語には、他の用語と組み合わせて使用する場合(例えば、アリールオキシ、アリールチオキシ、アリールアルキル)、上記定義のアリール及びヘテロアリール環の双方が含まれる。よって「アリールアルキル」の用語は、炭素原子(例えば、メチレン基)が例えば、酸素原子で置換されているアルキル基(例えば、フェノキシメチル、2−ピリジルオキシメチル、3−(1−ナフチルオキシ)プロピル等が挙げられる)を含め、アルキル基にアリール基が付着した基(例えば、ベンジル、フェネチル、ピリジルメチル等が挙げられる)を指す。
【0028】
上記の用語(例えば、「アルキル」、「ヘテロアルキル」、「アリール」及び「ヘテロアリール」)の各々には、示した基の置換及び非置換型の双方が含まれる。基の各々の型に望ましい置換基は、以下に示すとおりである。
【0029】
アルキル及びヘテロアルキルラジカル(アルキレン、アルケニル、ヘテロアルキレン、ヘテロアルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、シクロアルケニル及びヘテロシクロアルケニルと称されることが多い基を含む)に対する置換基は、一般に、それぞれ「アルキル置換基」及び「ヘテロアルキル置換基」と称され、それらは、0から(2m’+1)(m’は、かかるラジカルにおける炭素原子の総数である)までの範囲の数の、−OR’、=O、=NR’、=N−OR’、−NR’R”、−SR’、−ハロゲン、−SiR’R”R’”、−OC(O)R’、−C(O)R’、−CO2R’、−CONR’R”、−OC(O)NR’R”、−NR’’C(O)R’、−NR’−C(O)NR”R”’、−NR”C(O)2R’、−NR−C(NR’R”R’”)=NR””、−NR−C(NR’R”)=NR”’、−S(O)R’、−S(O)2R’、−S(O)2NR’R”、−NRSO2R’、−CN及び−NO2から選択される種々の基の1つ以上でありうるが、これらに限定されない。R’、R”、R’”及びR””は、各々好ましくは独立して、水素、置換若しくは非置換のヘテロアルキル、置換若しくは非置換のアリール、例えば、1〜3個のハロゲンで置換アリール、置換若しくは非置換のアルキル、アルコキシ又はチオアルコキシ基あるいはアリールアルキル基を指す。本発明の化合物が、1つより多いR基を含むとき、例えば、R’、R”、R’”及びR””基が1つより多く存在しているときのこれらの基の各々と同様に、R基の各々は、独立して選択される。R’及びR”が同じ窒素原子に付着する場合、それらは窒素原子と共に結合して5員環、6員環又は7員環を形成することができる。例えば、−NR’R”は、1−ピロリジニル及び4−モルホリニルを含むと意味されるが、これらに限定されない。置換基に関する上記の記載から、当業者は、「アルキル」の用語は、ハロアルキル(例えば、−CF3及び−CH2CF3)及びアシル(例えば、−C(O)CH3、−C(O)CF3、−C(O)CH2OCH3など)のような水素基以外の基に結合した炭素原子を含む基を含むことを意味すると理解するであろう。
【0030】
アルキルラジカルについて記載された置換基と同様に、アリール置換基及びヘテロアリール置換基は、一般に、それぞれ「アリール置換基」及び「ヘテロアリール置換基」を指し、また、それらは、多種多様なものであり、0から芳香族の環系における開放原子価の総数までの範囲の数で、例えば、ハロゲン、−OR’、=O’、=NR’、=N−OR’、−NR’R”、−SR’、−ハロゲン、−SiR’R”R”’、−OC(O)R’、−C(O)R’、−CO2R’、−CONR’R”、−OC(O)NR’R”、−NR”C(O)R’−NR’−C(O)NR”R’”、−NR”C(O)2R’、−NR−C(NR’R”)=NR”’、−S(O)R’、−S(O)2R’、−S(O)2NR’R”、−NRSO2R’、−CN及び−NO2、−R’、−N3、−CH(Ph)2、フルオロ(C1−C4)アルコキシ及びフルオロ(C1−C4)アルキルから選択され;R’、R”、R’”及びR””は、好ましくは独立して、水素、(C1−C8)アルキル及びヘテロアルキル、非置換アリール及びヘテロアリール、(非置換アリール)−(C1−C4)アルキル、並びに(非置換アリール)オキシ−(C1−C4)アルキルから選択される。本発明の化合物が、1つより多いR基を含むとき、例えば、R’、R”、R’”及びR””基が1つより多く存在しているときのこれらの基の各々と同様に、R基の各々は、独立して選択される。
【0031】
アリール又はヘテロアリール環の隣接した原子上のアリール置換基の2つは、式−T−C(O)−(CRR’)q−U−(式中、T及びUは、独立して、−NR−、−O−、−CRR’−又は単結合であり、qは、0から3の整数である)の置換基で任意に置換されてもよい。あるいは、アリール又はヘテロアリール環の隣接した原子上の置換基の2つは、式−A−(CH2)r−B−(式中、A及びBは、独立して、−CRR’−、−O−、−NR−、−S−、−S(O)−、−S(O)2−、−S(O)2NR’−又は単結合であり、rは、1から4の整数である)の置換基で任意に置換されてもよい。このようにして形成された新しい環の単結合の1つは、二重結合で任意に置換されてよい。あるいは、アリール又はヘテロアリール環の隣接した原子上の置換基の2つは、式−(CRR’)s−X−(CR”R’”)d−(式中、s及びdは、独立して0から3の整数であり、Xは、−O−、−NR’−、−S−、−S(O)−、−S(O)2−,又は−S(O)2NR’−である)の置換基で任意に置換されてもよい。置換基R、R’、R”及びR’”は、好ましくは独立して、水素又は置換若しくは非置換の(C1−C6)アルキルから選択される。
【0032】
本発明において、「ジホスフェート」の用語には、2つのリン酸基を含むリン酸のエステルが含まれるが、これらに限定されない。「トリホスフェート」の用語には、3つのリン酸基を含むリン酸のエステルが含まれるが、これらに限定されない。例えば、ジホスフェート又はトリホスフェートを有する特定の薬剤として、以下のものが挙げられる。
【化10】
本発明において、「ヘテロ原子」の用語には、酸素(O)、窒素(N)、イオウ(S)及びケイ素(Si)が含まれる。
【0033】
「R」の符号は、置換又は非置換アルキル、置換又は非置換ヘテロアルキル、置換又は非置換アリール、置換又は非置換ヘテロアリール、及び置換又は非置換ヘテロシクリル基より選択される置換基を表す一般略記である。
【0034】
「保護基」の用語に関しては、当業者は、選択された反応条件のセットによる干渉から特定の官能基をどのようにして保護するかを理解するであろう。例えば、有用な保護基については、Greene et al.,PROTECTIVE GROUPS IN ORGANIC SYNTHESIS,John Wiley & Sons,New York、1991を参照のこと。適当な保護基の例としては、BOC、FMOC、2−トリメチルシリルエトキシカルボニル、アリルオキシカルボニル、4−メチル−1−ピペラジンカルボニル、1−メチル−1−(4−ビフェニリル)エトキシカルボニル、ジフェニルオキシカルボニル、ベンジル、t−ブチル、テトラヒドロピラン、トリメチルシリル、t−ブチルジメチルシリル、トリイソプロピルシリル、t−ブチルジフェニルシリル、2,2,2−トリクロロエチルオキシカルボニル、ジイソプロピルメチルオキシカルボニル、ビニルオキシカルボニル、メトキシベンジルオキシカルボニル、ニトロベニズル(nitrobenyzl)オキシカルボニル、シクロヘキシルオキシカルボニル、シクロペンチルオキシカルボニル、ベンジルオキシカルボニル、ホルミル、アセチル、トリハロアセチル、ベンゾイル、ニトロフェニルアセチル、2−ニトロベンゼンスルホニル、フタルイミド及びジチアスクシノイルが挙げられるが、これらに限定されない。
【0035】
「医薬的に許容される担体」の用語は、本発明において、化学物質の運搬又は輸送に関与する、液体若しくは固体充填剤、希釈剤、賦形剤、溶媒、又は封入材料などの、医薬的に許容される材料、組成物又は溶剤を意味する。医薬的に許容される担体には医薬的に許容される塩が含まれ、ここで「医薬的に許容される塩」の用語には、本願明細書に記載の化合物に見られる特定の置換基に応じ、比較的無毒の酸又は塩基を用いて調製される活性化合物の塩が含まれる。本発明の化合物が比較的酸性の官能基を含む場合、塩基付加塩は、このような化合物の中性型を充分量の所望の塩基と、そのままか又は望ましい不活性溶媒中で接触させることによって得ることができる。医薬的に許容される塩基付加塩の例として、ナトリウム、カリウム、カルシウム、アンモニウム、有機アミノ、若しくはマグネシウム塩、又は類似の塩が挙げられる。本発明の化合物が比較的塩基性の官能基を含む場合、酸付加塩は、このような化合物の中性型を充分量の所望の酸と、そのままか又は望ましい不活性溶媒中で接触させることによって得ることができる。医薬的に許容される酸付加塩の例として、塩化水素酸、塩化臭素酸、硝酸、炭酸、一水素炭酸、リン酸、一水素リン酸、二水素リン酸、硫酸、一水素硫酸、ヨウ化水素酸、又はリン酸等のような無機酸に由来するものや、酢酸、プロピオン酸、イソ酪酸、マレイン酸、マロン酸、安息香酸、コハク酸、スベリン酸、フマル酸、乳酸、マンデル酸、フタル酸、ベンゼンスルホン酸、p−トリルスルホン酸、クエン酸、酒石酸、メタンスルホン酸等のような比較的無毒の有機酸に由来する塩が挙げられる。更に挙げられるのは、アルギン酸塩などの、アミノ酸の塩等、及びグルクロン酸又はガラクツロン酸のような有機酸の塩等である(例えば、Bergeら、「Pharmaceutical Salts」、Journal of Pharmaceutical Science、1977、66、1−19を参照)。本発明の特異的な所定の化合物は、その化合物を塩基又は酸付加塩のいずれかに変換させる、塩基性及び酸性官能基の双方を含む。
【0036】
天然型化合物は、望ましくは、上記の塩を塩基又は酸に接触させ、従来法にて中性構造の化合物を単離することによって得られる。化合物の原型は、極性溶媒中での溶解性など特定の物理的特性ではそれらの様々な塩と異なるが、その他の点では、それらの塩は本発明の目的とする化合物の中性構造と同等である。
【0037】
塩の型に加えて、本発明はプロドラッグ型の化合物を提供する。本願明細書に記載の化合物のプロドラッグとは、生理的条件下で化学的変化を容易に起こして本発明の化合物を提供する化合物である。加えて、プロドラッグは、ex vivoの環境において化学的又は生化学的方法によって本発明の化合物に変換されうる。例えば、プロドラッグは、望ましい酵素又は化学試薬と共に経皮リザーバーパッチに添加した場合、本発明の化合物に徐々に変換されうる。
【0038】
本発明の特定の化合物は、非溶媒和物や、水和物を含めた溶媒和物の形態にて存在しうる。一般的に、溶媒和物は非溶媒和物と同等であり、本発明の範囲に包含されるものである。本発明の特定の化合物は、多結晶又は非晶形にて存在することもある。一般的に、すべての物理的な型が本発明により企図される用途において同等であり、本発明の範囲に含まれる。
【0039】
本発明の特定の化合物は、不斉炭素原子(光学的中心)又は二重結合を有しており、ラセミ体、ジアステレオマー、幾何異性体、及び個々の異性体が、本発明の範囲に包含される。
【0040】
本発明の化合物はまた、かかる化合物を構成する1つ以上の原子において、非天然の割合で原子同位体も含みうる。例えば、化合物は、例えばトリチウム(3H)、ヨウ素−125(125I)又は炭素−14(14C)などの放射性同位体で放射標識されてもよい。本発明の化合物の同位体のすべてが、放射性の有無に関わらず、本発明の範囲に包含される。
【0041】
「ポリペプチド」、「ペプチド」及び「タンパク質」の用語は、本発明において、アミノ酸のポリマーを言うのに互換的に使用される。これらの用語は、天然アミノ酸ポリマー及び非天然アミノ酸ポリマーに適用するのと同様に、1つ以上のアミノ酸残基が、それに対応する天然アミノ酸の人工的な化学模倣物であるアミノ酸ポリマーにも適用する。またこれらの用語は、「抗体」の用語も包含する。
【0042】
「アミノ酸」の用語は、天然及び合成アミノ酸や、天然アミノ酸と同様に機能するアミノ酸類似体及びアミノ酸模倣物をいう。天然アミノ酸とは、遺伝暗号によってコードされるものや、事後的に修飾されるそれらのアミノ酸、例えば、ヒドロキシプロリン、γ−カルボキシグルタメート、及びO−ホスホセリンをいう。アミノ酸類似体は、天然アミノ酸と同じ基本化学構造、すなわち、水素、カルボキシル基、アミノ基及びR基に結合したα炭素を有する化合物、例えば、ホモセリン、ノルロイシン、メチオニンスルホキシド、メチオニンメチルスルホニウムを指す。このようなアミン酸類似体は、修飾されたR基(例えば、ノルロイシン)又は修飾されたペプチド骨格を有するが、天然アミノ酸と同じ基本化学構造を保持している。特に使用しても良い1つのアミノ酸はシトルリンであって、これはアルギニンの前駆体であり、肝臓における尿素の形成に関与する。アミノ酸模倣物は、アミノ酸の一般化学構造とは異なる構造を有するが、天然アミノ酸と同様に機能する化学的化合物をいう。「非天然アミノ酸」の用語は、上記の20の天然アミノ酸の「D」型立体構造を有するものを表す。非天然アミノ酸の用語には、天然アミノ酸の相同体、及び天然アミノ酸の合成的に修飾された型とが含まれることが更に理解される。合成的に修飾された型には、上限2つの炭素原子にまで短縮又は延長されたアルキレン鎖を有するアミノ酸、任意に置換アリール基を含むアミノ酸、及びハロゲン化基、望ましくはハロゲン化アルキル及びアリール基を含むアミノ酸が含まれるが、これらに限定されない。本発明のリンカー又はコンジュゲートに付着すると、アミノ酸は、アミノ酸のカルボン酸基がケト(C(O))基に置換されている「アミノ酸側鎖」の形態となる。従って、例えば、アラニン側鎖は、−C(O)−CH(NH2)−CH3などである。
【0043】
アミノ酸及びペプチドは、保護基によって保護されてもよい。保護基は、アミノ酸又はペプチドのN−末端を望ましくない反応から保護する原子又は化学的部分であり、薬剤−リガンドコンジュゲートの合成の際に使用することができる。これは、合成中はN−末端に付着した状態が望ましく、薬剤コンジュゲートの合成の完了後に、その除去を選択的に行う化学的条件又はその他の条件によって除去するのがよい。N−末端保護に望ましい保護基は、ペプチド化学の技術において公知である。保護基の例として、水素、D−アミノ酸、及びカルボベンゾキシ(Cbz)クロライドが挙げられるが、これらに限定されない。
【0044】
「抗体」の用語は、本発明においては全抗体、及び抗原結合断片のいずれか(すなわち、「抗原結合部分」)、又はその一本鎖を含む。「抗体」とは、ジスルフィド結合により相互連結する、少なくとも2本の重(H)鎖と2本の軽(L)鎖とを含む糖タンパク質、又はその抗原結合部分をいう。各重鎖は、重鎖可変領域(VH)及び重鎖定常領域を含む。重鎖定常領域は、3つのドメイン、CH1、CH2及びCH3を含み、ミュー、デルタ、ガンマ、アルファ又はイプシロンアイソタイプでありうる。各軽鎖は、軽鎖可変領域(VL)及び軽鎖定常領域を含む。軽鎖定常領域は、1つのドメインCLを含み、それは、カッパー又はラムダアイソタイプでありうる。VH及びVL領域は、フレームワーク領域(FR)と称される、より保存された領域が介在する、相補性決定領域(CDR)と称される超可変性の領域に更に細分されうる。VH及びVLの各々は、3つのCDR及び4つのFRを含み、アミノ末端からカルボキシ末端に向かって以下の順序で配置される:FR1、CDR1、FR2、CDR2、FR3、CDR3、FR4。重鎖と軽鎖の可変領域は、抗原と相互作用する結合ドメインを含んでいる。抗体の定常領域は、宿主組織、又は、免疫系の様々な細胞(例えば、エフェクター細胞)及び古典補体系の第一成分(Clq)を含む因子への、免疫グロブリンの結合を媒介しうる。
【0045】
抗体の「抗体断片」又は「抗原結合部分」(又は単に「抗体部分」)の用語は、本発明において、抗原への特異的な結合能を保持する、抗体の1以上の断片をいう。抗体の抗原結合機能は、全長の抗体の断片によって行えることが示されている。「抗体断片」又は抗体の「抗原結合部分」の用語内に包含される結合断片の例としては、(i)VL、VH、CL及びCH1ドメインからなる一価断片であるFab断片;(ii)ヒンジ領域におけるジスルフィド架橋によって連結された2つのFab断片を含む二価断片であるF(ab’)2断片;(iii)VH及びCH1ドメインからなるFd断片;(iv)抗体の単腕のVL及びVHドメインからなるFv断片、(v)VHドメインからなるdAb断片(Ward et al.,(1989)Nature 341 :544−546);及び(vi)単離された相補性決定領域(CDR)が挙げられる。更に、Fv断片の2つのドメインであるVL及びVHは、別々の遺伝子によってコードされるが、それらをVL領域とVH領域とが対を形成して一価分子を形成する一本鎖タンパク質(一本鎖Fv(scFv)として公知である;例えば、Bird et al.(1988)Science 242:423−426;及びHuston et al.(1988)Proc.Natl.Acad.Sci.USA 85:5879−5883を参照のこと)として作製することができる合成リンカーによって、組換え法を用いてそれらを連結することができる。このような一本鎖抗体もまた、抗体の「抗原結合部分」の用語に包含されることが意図される。これらの抗体断片は、当業者に知られた従来の技術を用いて得られ、それら断片は通常の抗体と同様に、その用途に基づきスクリーニングされる。
【0046】
「モノクローナル抗体」の用語は、本発明において、単一の分子組成の抗体分子の調製物をいう。モノクローナル抗体組成物は、特定のエピトープに対する単一の結合特異性及び親和性を呈する。
【0047】
「固体支持体」は、本願明細書において使用されるとき、選択された溶媒系に実質的に不溶性である材料、又はその支持体が溶解性である選択された溶媒系から容易に分離することができる(例えば、沈殿によって)材料を指す。本発明を実施するのに有用な固体支持体は、選択された種がその固体支持体に結合させられるように活性化されているか、又は活性化することのできる基を含むことができる。固体支持体はまた、例えば、チップ、ウエハー又はウェルなどといった基材で、その上に個々の、又は1以上の本発明の化合物が結合されるものでもよい。
【0048】
本発明の化合物は、単一の異性体(例えば、エナンチオマー、シス−トランス異性体、ジアステレオマー)として、又は異性体の混合物として調製される。望ましい実施形態において、化合物は、実質的に単一の異性体として調製される。実質的に異性体として純粋な化合物を調製する方法は、当該技術分野において知られている。例えば、エナンチオマーとして濃縮された混合物、及び純粋なエナンチオマー化合物は、エナンチオマーとして純粋なエナンチオマーの合成中間体を、キラル中心での立体化学を不変のままにするか又はその完全な反転を引き起こす反応と組み合わせて使用することにより、調製することができる。あるいは、最終産物又はその合成経路での中間体は、単一の立体異性体に分割することができる。特定の立体中心を反転するか又は不変のままにするための技術、及び立体異性体の混合物を分離するための技術は、当該技術分野においてよく知られており、特定の状況に対して方法を選択及び充当することは、当業者が充分になしうる範囲にある。一般に、Furniss et al.(eds.),VOGEL’S ENCYCLOPEDIA OF PRACTICAL ORGANIC CHEMISTRY 5TH ED.,Longman Scientific and Technical Ltd.,Essex,1991,pp.809−816;及びHeller,Acc.Chem.Res.23:128(1990)を参照のこと。
【0049】
CC−1065の細胞傷害性類似体は、CC−1065のシクロプロパピロロインドール(CPI)部分の代わりにアルキル化部分としてシクロプロパベンズインドール(CBI)部分を用いて形成することができる。一例として、CC−1065 CBI類似体としては、以下の式を有する化合物(またはその医薬的に許容される塩)が挙げられるがこれに限定されない。
【化11】
式中、Xは、ハロである。好ましくは、Xは、Cl又はBrであり、より好ましくは、Xは、Brである。
【0050】
X1及びZは、各々独立して、O、S及びNR8から選択され、R8は、H、置換又は非置換のアルキル、置換又は非置換のヘテロアルキル及びアシルから選択されるメンバーである。
【0051】
R4、R4,、R5及びR5,は、独立して、H、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル、非置換ヘテロシクロアルキル、ハロゲン、NO2、NR9R10、NC(O)R9、OC(O)NR9R10、OC(O)OR9、C(O)R9、SR9、OR9、CR9=NR10及びO(CH2)nNR11R11’からなる群から選択されるメンバーであり、R9及びR10は、独立して、H、置換又は非置換のアルキル、置換又は非置換のヘテロアルキル、置換又は非置換のアリール、置換又は非置換のヘテロアリール、置換又は非置換のヘテロシクロアルキル及び置換又は非置換のペプチジルから選択されるか、又はR9及びR10は、それらが結合する窒素原子と任意に結合して、任意に2つ以上のヘテロ原子を含む4〜6員環を有する置換又は非置換のヘテロシクロアルキル環系を形成し、R11及びR11’は、各々独立して、H又は低級アルキルである。
【0052】
R6は、H、置換若しくは非置換の低級アルキル、シアノ又はアルコキシである。好ましくは、R6は、メチル、シアノ又はHである。より好ましくは、R6は、Hである。
【0053】
R7は、H、置換アルキル、非置換アルキル、置換ヘテロアルキル、非置換ヘテロアルキル、ジホスフェート、トリホスフェート、アシル、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、R12、R13及びR14は、独立して、H、置換若しくは非置換のアルキル、置換若しくは非置換のヘテロアルキル及び置換若しくは非置換のアリールから選択されるメンバーであるか、又はR12及びR13は、それらが結合する窒素若しくは炭素原子と任意に結合して、任意に2つ以上のヘテロ原子を含む4〜6員環を有する置換若しくは非置換のヘテロシクロアルキル環系を形成する。
【0054】
CBI CC−1065類似体の例は、共同所有の(co−owned)米国特許出願第10/160,972号;同第10/161,233号;同第10/161,234号、同第11/134,685号及び同第11/134,826号(これらのすべてが参照によって本願明細書に援用される)に記載されている。これらの参考文献はまた、これらの化合物についての合成及び使用の例も記載している。これらの化合物は、治療薬(例えば、薬物)及びプロドラッグとして使用することができる。少なくとも幾つかの実施態様では、CBI CC−1065類似体は、細胞傷害性CBI CC−1065類似体を癌腫細胞などの所望の標的細胞に選択的に送達する医薬組成物において使用するために標的化剤(例えば、抗体、受容体、ペプチド、レクチン、糖、核酸又はそれらの組み合わせ)とコンジュゲートすることができる。
【0055】
これらの化合物によって標的化されうる前癌状態の代表的な例としては、化生、過形成、異形成、結腸直腸ポリープ、光線性角化症(actinic ketatosis)、光線口唇炎、ヒトパピローマウイルス症、白斑症、扁平苔癬(lychen planus)及びボーエン病が挙げられるが、これらに限定されない。
【0056】
これらの化合物によって標的化されうる癌又は腫瘍の代表的な例としては、肺癌、結腸癌、前立腺癌、リンパ腫、メラノーマ、乳癌、卵巣癌、精巣癌、CNS癌、腎癌、腎臓癌、膵癌、胃癌、口腔癌、鼻腔癌、子宮頸癌及び白血病が挙げられるがこれらに限定されない。薬物で処置されるべき腫瘍組織にその薬物を標的化するように特定の標的化剤を選択することができる(すなわち、腫瘍特異的抗原に特異的な標的化剤が選択される)ことは、当業者には容易に明らかであろう。そのような標的化剤の例は、当該技術分野で周知であり、それらの例としては、乳癌を処置するための抗Her2、リンパ腫を処置するための抗CD20、前立腺癌を処置するための抗PSMA、及び非ホジキンリンパ腫を含むリンパ腫を処置するための抗CD30が挙げられるがこれらに限定されない。
【0057】
これらの化合物は、細胞を死滅させる方法を提供する。この方法は、細胞に本発明の化合物の当該細胞を死滅させるのに充分な量を投与することを含む。望ましい実施形態において、化合物は、その細胞を担持している患者に投与される。更なる望ましい実施形態において、投与は、その細胞(例えば、細胞は腫瘍細胞でありうる)を含む腫瘍の成長を遅延又は停止させるのに有用である。成長を遅延させるための投与について、細胞の成長速度は、投与前の成長速度の少なくとも10%は低くなるのが望ましい。更には、成長速度は少なくとも20%、30%、40%、50%、60%、70%、80%、90%遅延され、又は完全に停止するのが望ましい。
【0058】
医薬組成物は、治療上有効な量、すなわち、その意図された目的を達成するのに有効な量の活性成分が含まれている組成物を含む。特定の用途に有効な実際の量はとりわけ、処置すべき状態に依存するものである。有効量の決定は、特に本願明細書の詳細な開示に鑑み、当業者が充分なしうる。
【0059】
本願明細書に記載のいずれの化合物についても、治療上有効な量を最初に細胞培養アッセイにより調べることができる。標的血漿濃度は、細胞成長又は分裂を阻害することができる活性化合物(単数又は複数)の濃度となる。望ましい実施形態において、細胞活性は少なくとも25%阻害される。又は、少なくとも約50%、75%、更には90%以上の細胞活性の阻害を誘発できる活性化合物(単数又は複数)の標的血漿濃度が、現在のところ望ましい。患者における細胞活性の阻害の割合をモニターし、達成される血漿薬剤濃度の適切さを評価でき、所望の阻害割合が得られるように増減して薬用量を調整することができる。
【0060】
当該技術分野において公知であるように、ヒトで使用するのに治療上有効な量は動物モデルから決定することもできる。例えば、ヒト用の投薬量は、動物で有効であると見出されている循環濃度を得るように製剤化できる。ヒトでの薬用量は、細胞阻害をモニターし、上記のように薬用量を増減して調整することができる。
【0061】
治療上有効な投薬量は、類似の薬理学的活性を呈することが知られている化合物に対するヒトのデータから決定することもできる。適用される薬用量は、相対的な生物学的利用能、及び投与化合物の既知化合物と比較した作用強度に基づいて調整することができる。
【0062】
上記の方法、及び当該技術分野において公知である他の方法に基づき、ヒトで最高の効能を達成する薬用量を調整することは、当業者が充分なしうる範囲内にある。
【0063】
局所投与の場合、投与化合物の全身循環濃度は、特別に重要ではない。このような場合、意図した結果を得るのに有効な局部領域での濃度となるように、化合物を投与する。
【0064】
異常な細胞性増殖に関わる疾病の予防及び/又は処置における使用のため、約0.001μM〜20μMの投与化合物の循環濃度が好ましく、更に約0.01μM〜5μMが望ましい。
【0065】
本願明細書に記載の化合物の経口投与に対する患者の薬用量は、望ましくは約1mg/日〜約10,000mg/日、より望ましくは約10mg/日〜約1,000mg/日、最も望ましくは約50mg/日〜約500mg/日の範囲にある。患者の体重に換算すると、典型的な薬用量は、約0.01〜約150mg/kg/日、更に望ましくは約0.1〜約15mg/kg/日、最も望ましくは約1〜約10mg/kg/日の範囲、例えば、5mg/kg/日、又は3mg/kg/日である。
【0066】
少なくとも幾つかの実施態様では、腫瘍成長を遅延又は阻害する患者の薬用量は、1□mol/kg/日以下でありうる。例えば、患者薬用量は、薬物又は薬物コンジュゲート(例えば、抗体−薬物コンジュゲート)の0.9、0.6、0.5、0.45、0.3、0.2、0.15又は0.1□mol/kg/日以下(薬物のモルに対して)でありうる。好ましくは、薬剤又は薬剤コンジュゲートを少なくとも5日間にわたり連日投与することにより、腫瘍の成長を抑止する。少なくとも幾つかの実施態様では、腫瘍はSCIDマウスにおけるヒト型腫瘍である。例として、SCIDマウスは、CB17.SCIDマウスであってもよい(Taconic、Germantown、NYから市販)。
【0067】
投与のその他の態様については、用量及び間隔を、処置すべき特定の臨床適応症に対して有効な投与化合物の血漿レベルをもたらすように、個々に調整することができる。例えば、一実施形態において、本発明による化合物は、1日に何度も、比較的高濃度で投与される。あるいは、本発明の化合物を最低有効量投与し、より頻度の少ない投与計画を用いることがより望ましい。これにより、個々の疾病の重症度に見合う治療計画が提供されることになる。
【0068】
本願明細書における開示を応用して、実質的な毒性を引き起こさず、しかも特定の患者によって示される臨床的症状を処置するのに完全に有効な、治療法を計画することができる。この計画には、化合物の作用強度、相対的生物学的利用能、患者の体重、望まない副作用の存在及び重度、望ましい投与の形態及び選択薬剤の毒性プロファイルなどの因子を考慮することにより、活性化合物を注意深く選択することが含まれる。
【0069】
一般に、CBl部分は、以下の式を有する。
【化12】
式中、置換基は、酸素及び窒素原子に付着することができ、Xは、ハロであり、R6は、H、置換若しくは非置換の低級アルキル、シアノ又はアルコキシである。好ましくは、R6は、H、メチル又はシアノである。より好ましくは、R6は、Hである。更に、Xは、好ましくはCl又はBrであり、より好ましくは、Xは、Brである。一般に、結合部分は、CBI部分のアミン置換基に付着されうる。適当な結合部分の例としては、以下が挙げられるが、これに限定されない。
【化13】
式中、X1、Z、R4、R4,、R5及びR5,は、上で定義したとおりである。
【0070】
適当な結合部分の例は、米国特許出願第10/160,972号;同第10/161,233号;同第10/161,234号、同第11/134,685号及び同第11/134,826号;並びに米国特許第6,534,660号(参照によって本願明細書に援用される)に例示及び記載されている。この式中の適当な結合部分はまた、以下のような複数の縮合環を有する結合部分も含む。
【化14】
式中、Z’は、独立してO、S及びNR8から選択され、R8は、H、置換又は非置換のアルキル、置換又は非置換のヘテロアルキル及びアシルから選択されるメンバーである。
【0071】
R4,,、R4,,,、R5,,及びR5,,,は、独立して、H、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル、非置換ヘテロシクロアルキル、ハロゲン、NO2、NR9R10、NC(O)R9、OC(O)NR9R10、OC(O)OR9、C(O)R9、SR9、OR9、CR9=NR10及びO(CH2)nNR11R11’からなる群から選択されるメンバーであり、R9及びR10は、独立して、H、置換若しくは非置換のアルキル、置換若しくは非置換のヘテロアルキル、置換若しくは非置換のアリール、置換若しくは非置換のヘテロアリール、置換若しくは非置換のヘテロシクロアルキル及び置換若しくは非置換のペプチジルから選択されるか、又はR9及びR10は、それらが結合する窒素原子と任意に結合して、任意に2つ以上のヘテロ原子を含む4〜6員環を有する置換若しくは非置換のヘテロシクロアルキル環系を形成し、R11及びR11’は、各々独立して、H又は低級アルキルである。R15は、H、置換又は非置換のアルキルでありうるか、又はR15及びR4’又はR5’は、結合して、環(例えば、5員環又は6員環)を形成しうる。
【0072】
CC−1065 CBI類似体の形成において有用な1つの中間体化合物は、式(I)を有する。
【化15】
式中、R1及びR2は、各々独立して、H、アルキル、−C(O)OR’、−C(O)NR’R”又は保護基であり、R’及びR”は、独立して、H、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル及び非置換ヘテロシクロアルキルからなる群から選択され、Xは、ハロゲンである。
【0073】
化合物(I)のための1つの従来の合成方法において、出発物質は、1,3−ジヒドロキシナフタレンであり、それを加圧チャンバー(例えば、ボンベ)内でアンモニアと反応させることにより、ナフタレンの3位におけるヒドロキシ基をアミンで置換する。この合成方法の例は、米国特許第6,534,660号及びD.L.Boger et al.,J.Org.Chem.57,2873−2876(1992)(これら両方が参照によって援用される)に見出されうる。このアミノ化反応の後、ヒドロキシ部分及びアミン部分の両方に保護基を付加する。
【0074】
このアミノ化反応は、小規模において許容できる収率でありうるが、通常、実質的に1気圧を超える圧力下(約1.01×105Pa)及び一般に少なくとも1.5気圧の圧力下(1.52×105Pa)において行う加圧反応を含むためにボンベを使用するので、この反応をスケールアップすることは困難でありうる。この合成方法は、スケールアップすると実質的に収率が低下することが見出されている。
【0075】
従来の方法とは対照的に、出発物質を、図1及び2の合成スキームに例示されるように化合物(II)とすることができる。
【化16】
式中、R3は、H又はアルキルである。好ましくは、R3は、C1−5アルキルであり、より好ましくは、メチルである。例えば、4−メトキシ−2−ナフチルアミンは、Aldrich Chemical Company,Inc.,Milwaukee,WIから市販されている。R3がアルキルであり、保護基R1’をヒドロキシ部分及びアミン部分の両方に付加して化合物(III)を形成する場合、その化合物の加水分解が比較的容易である。
【化17】
幾つかの実施態様では、図1及び2に例示されるように、アミン及びヒドロキシ官能基に対して様々な保護基をもたらすことが望ましい。したがって、最初の保護基であるR1’を、これらの部分の1つから(例えば、ヒドロキシ部分から)除去して、第2の異なる保護基であるR2’で置換することにより、化合物(IV)をもたらすことができる。
【化18】
化合物(IV)の1つの特定の例は、以下の式を有する。
【化19】
一方の保護基をそのままにしておきながら、他方の保護基を選択的に除去することができる場合、ヒドロキシル置換基とアミン置換基とで異なる保護基を有することにより、その後の反応ステップを容易にすることができる。あるいは、異なる保護基を最初にヒドロキシル及びアミン置換基に付加することもできる。
【0076】
スキーム1及び2(図1及び2)には、化合物(IV)から化合物(I)を形成する際の残存ステップの一実施態様が例示されている。これらのステップは、例えば、アミン基の窒素を用いて環を形成することを含みうる。これは、例えば、窒素に隣接したアリール環のアルキル化の後の閉環ステップによって達成されうる。一実施態様において、N−ヨードスクシンイミドによる化合物(IV)のヨウ素化により化合物が生成され、その後、それを1,3−ジブロモプロペン又は1,3−ジクロロプロペンを用いてアルキル化することができる。その後、2,2’−アゾビスイソブチロニトリル(AIBN)の存在下で水素化トリブチルスズを用いて閉環を行うことができ、それにより、ラセミのCBI誘導体である化合物(V)が得られる。
【化20】
所望であれば、保護基を除去してCBIを形成することができる。これらの反応ステップにおいて様々な反応物及び触媒を使用することができることが理解されるだろう。例は、Boger,Chemical Reviews,97,787−828(1997,)(参照によって本願明細書に援用される)に見出されうる。
【0077】
クロマトグラフィ法の使用を含む公知のエナンチオマーの分離技術を用いて、ラセミのCBI誘導体を分離することができる。特に有用な1つの技術は、キラルカラムを使用した高圧液体クロマトグラフィ(HPLC)である。例えば、HPLC Chiralcelカラム及びヘキサン/イソプロパノール(99:1)溶出剤を用いてそのようなエナンチオマーの分離を行うことにより、化合物(I)が得られる。
【0078】
化合物(I)の特定の例としては、以下が挙げられるが、これらに限定されない。
【化21】
【0079】
化合物(I)を使用して、例えば、共同所有の米国特許出願第10/160,972号;同第10/161,233号;同第10/161,234号、同第11/134,685号及び同第11/134,826号(これらのすべてが参照によって本願明細書に援用される)に記載されているようなCBI CC−1065類似体を形成することができる。例えば、アミン置換基を脱保護し、脱保護されたアミンと結合部分を含む化合物とを反応させることによって、化合物(I)に結合部分を付加することができる。酸素を脱保護し、それを適切な反応物(単数又は複数)と反応させることによって、さらなる置換基をCBI化合物の酸素原子に付加することができる。
【実施例】
【0080】
当業者は、さらなる詳述なしに、前述の記載を使用して本発明を最大限に利用することができると考えられる。それゆえ、以下の好ましい特定の実施態様は、単なる例示にすぎず、いかなる様式でも本開示の残りの部分を限定すると解釈されるべきでない。前述及び以下の実施例において、すべての温度を摂氏温度で、未修正で示し、すべての部及びパーセントは、別途記載されない限り、それぞれ質量部及び質量パーセントである。
【0081】
実施例1−スキーム1(図1)
N−(tert−ブチルオキシカルボニル)−4−O−(tert−ブチルオキシカルボニル)−2−ナフチルアミン(2)の合成
氷酢酸(9.6mL)及び臭化水素酸水溶液(16mL,48%)中の4−メトキシ−2−ナフチルアミン(230mg,1.33mmol)の溶液をN2下で4時間還流した。少量のサンプル(0.1mL)を酢酸エチル(0.5mL)で希釈し、次いで、水(0.5mL)及びTEA(0.1mL)を加えた。有機層のTLC(20:1DCM/メタノール)では、出発物質は検出されなくなり、それより低い新しいスポットが検出された(Rf=0.1)。溶媒を減圧下で除去し、生成物を真空下で乾燥することにより、中間体である4−ヒドロキシ−2−ナフチルアミンが得られ、それをいかなる精製もすることなく次の工程に使用した。ジオキサン(10mL)中の4−ヒドロキシ−2−ナフチルアミンの溶液に、TEA(1mL)及びジ−tert−ブチルジカーボネート(1.149g,5.27mmol)を加えた。その反応混合物をN2下で4時間還流した。TLC(4:1ヘキサン/酢酸エチル)では、出発物質は検出されなくなり、それより高い新しいスポットが検出された(Rf=0.55)。その反応混合物を酢酸エチル(50mL)で希釈し、水で洗浄した。その水層を酢酸エチル(2×50mL)で抽出し、有機層を併せ、ブラインで洗浄した。その有機物を無水Na2SO4で乾燥し、濾過し、減圧下で濃縮することにより、N−(tert−ブチルオキシカルボニル)−4−O−(tert−ブチルオキシカルボニル)−2−ナフチルアミン(2,80%収率)を油状物として得た。
【0082】
化合物3の合成:
アセトン(10mL)中の化合物2の溶液に、NaOH水溶液(10mL,1M)を加えた。その反応混合物を室温で一晩撹拌した。TLC(4:1ヘキサン/酢酸エチル)では、出発物質は検出されなくなり、それより低い新しいスポットが検出された。その反応混合物を酢酸エチル(50mL)で抽出し、水で洗浄した。水層を酢酸エチル(2×50mL)で抽出し、有機層をブラインで洗浄し、無水Na2SO4で乾燥し、濃縮した。その残渣を、ヘキサン溶液中の10−20%酢酸エチルを用いた10gのシリカゲルカラムにおいて精製することにより、化合物3(181mg,53%)を油状物として得た。
【0083】
化合物4の合成:
窒素雰囲気下の無水DMF(50ml)中の化合物3(5g,19.3mmol)の溶液を、臭化ベンジル(4g,23.1mol)、炭酸カリウム(3.7g,27mol)及びヨウ化テトラブチルアンモニウム(70mg,0.01mmol)で処理した。その反応混合物を室温で8時間撹拌した。その反応混合物を減圧下で濃縮した。クロマトグラフィを用いた分離(4×10cm SiO2,10−20%EtOAc−ヘキサン勾配溶出)によって、純粋な化合物4(5.48g,83%)をクリーム色の粉末として得た。1H NMR(CDCl3,400MHz,ppm)8.22(d,1H J=8.1Hz,C5−H),7.68(d,1H,J=8.2Hz,C8−H),7.3−7.5(m,8H,C1−H,C6−H,C7−H,CH2C6H5),7.06(d,1H,J=1.1Hz,C3−H),6.62(br s,1H,NH),5.23(S,2H,OCH2(C6H5),1.55(s,9H,OC(CH3)3).
【0084】
化合物5の合成:
スターラーバー及びゴム隔壁を備えた1000ml丸底フラスコにて化合物4(13g,0.0372mol)及びTHF(300ml)を混合した。透明の黄色溶液を、ドライアイス浴を用いて窒素雰囲気下において−20Cに冷却した。p−トルエンスルホン酸(0.10g,0.0005mol)をその反応物に加え、その溶液を10分間撹拌した。N−ヨードスクシンイミド(10g,0.0446mol)をTHF(50ml)に溶解し、カニューレを用いてその反応物に加えた(約1時間)。その溶液を氷浴中で2時間撹拌したところ、茶色がかった色を呈した。次いで、その溶液を氷浴から取り出し、窒素下で1.5時間室温に温めた。TLC(2:1ヘキサン/DCM)では、出発物質は検出されなくなり、それより高い新しいスポットが検出された。その反応物を飽和ΝaHCO3(200ml)でクエンチし、白色固体が形成された。その溶液を10分間撹拌した後、その反応物にEtOAc(200ml)及び水(100ml)を加えた。水層をEtOAc(2×100ml)で抽出し、有機層を併せ、ブライン(100ml)で抽出した。その有機層をMgSO4で乾燥し、濾過し、真空下で濃縮することにより、暗赤茶色の固体が得られた。その固体を、溶離剤として2:1ヘキサン/DCMを用いるカラムクロマトグラフィで精製することにより、化合物5(14g,79%)を茶色固体として得た。
【0085】
化合物6の合成:
スターラーバー及び窒素導入口を備えた500ml丸底フラスコにて、化合物5(22.5g,0.0473mol)及び無水DMF(250ml)を混合した。その黄色−橙色溶液を、窒素雰囲気下において氷/塩浴を用いて0℃に冷却した。その反応物にNaH(60%,5.6g,0.146mol)を一度に加えた。その溶液は濁り、気体を発生した。その反応物を氷浴中で15分間撹拌し、次いで、その氷浴を取り除き、その溶液を更に15分間撹拌して、その反応物にシス/トランス−1,3−ジブロモプロペン(14ml,0.14mol)を、注射器を用いて少しずつ加えた。その反応物を窒素下で室温にて1時間撹拌したところ、その反応物は、濁った茶色を呈した。温度を40℃に上げた。その反応物を室温に冷却した。TLC(4:1ヘキサン/EtOAc)では、出発物質は検出されなくなり、それより低い新しいスポットが検出された。その反応物を水(500ml)でクエンチした。その水層をEtOAc(4×100ml)で抽出し、有機層をブラインで洗浄した(2×75ml)。その有機層をMgSO4で乾燥し、濾過し、真空下で濃縮することより、茶色油状物が得られた。その生成物を、溶離剤として1:1DCM/ヘキサンを用いるカラムクロマトグラフィで精製することにより、化合物6(25g,89%)を茶色油状物として得た。
【0086】
化合物7の合成:
スターラーバー、温度プローブ、還流冷却器及び窒素導入口を備えた1000ml三首丸底フラスコにて、化合物6(25g,0.0421mol)、トルエン(500ml)、2,2’−アゾビス(2−メチルプロピオニトリル)(0.15g,0.0009mol)及び水素化トリブチルスズ(3.4ml,0.0126mol)を混合した[注射器を用いて]。その溶液に15分間窒素を泡立たせながら通し、次いで、その反応物を窒素下で80℃に加熱した。15分間80℃に加熱した後、その反応物に注射器を用いて水素化トリブチルスズ(3.4ml,0.0126mol)を加えた。更に15分間加熱した後、その反応物に注射器を用いて水素化トリブチルスズ(3.4ml,0.0126mol)を加えた。更に15分後、その反応物に注射器を用いて水素化トリブチルスズ(3.4ml,0.0126mol)を加えた。加えた水素化トリブチルスズの総量は、13.6ml,0.0505molであった。その反応物を30分間80℃に加熱し、次いで、室温に冷却した。TLC(10%EtOAc/ヘキサン)では、出発物質及びそれより高い新しいスポットが検出された。その溶液を真空下で濃縮することにより、黄色固体が得られた。その固体を、溶離剤として1:1ジクロロメタン(dicloromethane)/ヘキサンを用いるカラムクロマトグラフィで精製することにより、黄色固体が得られた。その固体を、ヘキサンから再結晶化する(200ml、30分間45Cにし、冷蔵庫にて2時間冷却し、濾過により回収し、真空下で乾燥させる)ことにより、化合物7(11.70g,59%収率)を淡黄色固体として得た。NMR(1H,CDCl3,400MHz):□1.61(9H,s,C−(CH3)3);3.30(1H,t,J=26Hz,CH−CH2−N);3.82(1H,d,J=26Hz,Br−CH2−CH);4.04(1H,d,J=19Hz,Br−CH2−CH)4.14(1H,t,J=26Hz,CH−CH2−N);4.21(1H,m,CH2−CH−CH2);5.26(2H,s,O−CH2−C6H5);7.3−7.55(8H,m,O−CH2−C6H5,C10H5);7.63(1H,d,J=21Hz,C10H5);8.3(1H,d,J=21Hz,C10H5)
【0087】
化合物7の分割:
ラセミの化合物7をDCM(50mg,1ml)に溶解した。次いで、その溶液をヘキサン(9ml)で希釈した。次いで、その溶液をChiralcel OD prepカラム(10ミクロン,20×250mm)に充填し、ヘキサン/イソプロパノール(99:1,15ml/分)を用いて分離した。第1のエナンチオマー(7a)は、10〜15分に溶出し、第2のエナンチオマー(7b)は、17.5〜25分に溶出する。分析カラム(Chiralcel OD,0.46×25cm,20マイクロメートル)は、7aについては7.71分及び7bについては12.9分の保持時間を与える(99:1ヘキサン/IPA,1ml/分,15分間の流入)。NMR(1H,CDCl3,400MHz):□1.61(9H,s,C−(CH3)3);3.30(1H,t,J=26Hz,CH−CH2−N);3.82(1H,d,J=26Hz,Br−CH2−CH);4.04(1H,d,J=19Hz,Br−CH2−CH)4.14(1H,t,J=26Hz,CH−CH2−N);4.21(1H,m,CH2−CH−CH2);5.26(2H,s,O−CH2−C6H5);7.3−7.55(8H,m,O−CH2−C6H5,C10H5);7.63(1H,d,J=21Hz,C10H5);8.3(1H,d,J=21Hz,C10H5)
【0088】
実施例2−スキーム2(図2)
合成方法の手順は、化合物5の合成を通じて実施例1において上に記載したとおりである。化合物8の合成スターラーバー及び窒素導入口を備えた250ml丸底フラスコにて、化合物5(8.4g,0.0177mol)及び無水DMF(125ml)を混合した。その黄色−橙色溶液を、氷/塩浴を用いて窒素雰囲気下において0℃に冷却した。その反応物にNaH(60%,2.22g,0.0554mol)を一度に加えた。その溶液は濁り、気体が発生した。その反応物を氷浴中で15分間撹拌し、次いで、氷浴を取り除き、その溶液を更に15分間撹拌した。その反応物にシス/トランス−1,3−ジクロロプロペン(5.3ml,0.0571mol)を、注射器を用いて少しずつ加えた。その反応物を窒素下で室温にて3時間撹拌したところ、濁った茶色を呈した。TLC(4:1ヘキサン/EtOAc)では、出発物質は検出されなくなり、それより低い新しいスポットが検出された。その反応物を水(250ml)でクエンチした。水層をEtOAc(3×100ml)で抽出し、有機層をブラインで洗浄した(2×50ml)。その有機層をMgSO4で乾燥し、濾過し、真空下で濃縮することより、茶色油状物が得られた。その生成物を、溶離剤として1:1DCM/ヘキサンを用いるカラムクロマトグラフィで精製することにより、(E/Z)−tert−ブチル4−(ベンジルオキシ)−1−ヨードナフタレン−2−イル(3−クロロアリル)カルバメート(8)(9g,93%)を黄色油状物として得た。
【0089】
化合物9の合成:
スターラーバー、温度プローブ、還流冷却器及び窒素導入口を備えた500ml三首丸底フラスコにて、化合物8(9g,0.0164mol)、トルエン(200ml)、2,2’−アゾビス(2−メチルプロピオニトリル)(0.15g,0.0009mol)及び水素化トリブチルスズ(1.5ml,0.0056mol)を混合した[注射器を用いて]。その溶液に15分間窒素を泡立たせながら通し、次いで、その反応物を窒素下で80℃に加熱した。15分間80℃に加熱した後、その反応物に注射器を用いて水素化トリブチルスズ(1.5ml,0.0056mol)を加えた。更に15分間加熱した後、その反応物に注射器を用いて水素化トリブチルスズ(1.5ml,0.0056mol)を加えた。更に15分後、その反応物に注射器を用いて水素化トリブチルスズ(1.0ml,0.0037mol)を加えた。加えた水素化トリブチルスズの総量は、5.5ml,0.0204molであった。その反応物を30分間80℃に加熱し、次いで、室温に冷却した。TLC(10%EtOAc/ヘキサン)では、出発物質は検出されなくなり、それより高い新しいスポットが検出された。その溶液を真空下で濃縮することにより、黄色油状物が得られた。その油状物を、溶離剤として100%ヘキサン、5%EtOAc/ヘキサン、10%EtOAc/ヘキサンを用いるカラムクロマトグラフィで精製することにより、淡黄色固体が得られた。その固体を、ヘキサンから再結晶化する(100ml、30分間45Cにし、冷蔵庫にて2時間冷却し、濾過により回収し、真空下で乾燥させる)ことにより、化合物9を(4.16g,60%収率)白色固体として得た。
【0090】
化合物9の分割:
ラセミの化合物9をDCM(50mg,1ml)に溶解した。次いで、その溶液をヘキサン(9ml)で希釈した。次いで、その溶液をChiralcel OD prepカラム(10ミクロン,20×250mm)に充填し、ヘキサン/イソプロパノール(99:1,15ml/分)を用いて分離した。第1のエナンチオマー(9a)は、11.5〜15分に溶出し、第2のエナンチオマー(9b)は、17.5〜25分に溶出する。分析カラム(Chiralcel OD,0.46×25cm,20ミクロン)は、9aについては6.5分及び9bについては10.6分の保持時間を与える(99:1ヘキサン/IPA,1ml/分,15分間の流入)。NMR(1H,CDCl3,400MHz):d 1.61(9H,s,C−(CH3)3);3.44(1H,t,J=25Hz,CH−CH2−N);3.9−4.0(2H,m,Cl−CH2−CH);4.12(1H,t,J=26Hz,CH−CH2−N);4.25(1H,m,CH2−CH−CH2);5.26(2H,s,O−CH2−C6H5);7.2−7.5(8H,m,O−CH2−C6H5,C10H5);7.63(1H,d,J=21Hz,C10H5);8.3(1H,d,J=21Hz,C10H5)
【0091】
実施例3−スキーム3(図3)
合成方法の手順は、化合物3の合成を通じて実施例1において上に記載したとおりである。
【0092】
化合物10の合成:
無水DCM(20ml)中のtert−ブチル−4−ヒドロキシナフタレン−2−イルカルバメート(3)(500mg,2.89mmol)、4−メチル−1−ピペラジンカルボニルクロリド塩酸塩(858mg,4.34mmol)、無水ピリジン(4.98ml,57.8mmol)及びアリルアルコール(4.98ml,73.2mmol)の溶液を室温で一晩撹拌した。TLC(9:1DCM/MeOH)では、出発物質は検出されなくなり、それより一層低いスポットが検出された。その反応混合物を水でクエンチした。水層をEtOAc(3×50ml)で抽出し、有機層をブラインで洗浄した(2×50ml)。その有機層をNa2SO4で乾燥し、濾過し、真空下で濃縮することにより、茶色油状物が得られた。その粗生成物を、DCM中の1−5%メタノールを用いるカラムクロマトグラフィで精製することにより、10(602mg,82%)を黄色固体として得た。1HNMR(DMSO−d6)δ9.68(s,1H),7.91(s,1H),7.80(d,1H),7.69(s,1H),7.47(t,1H),7.42(s,1H),7.39(t,1H),3.78(s,2H),3.42(s,2H),2.44(s,2H),2.39(s,2H),2.21(s,3H),1.50(s,9H)
【0093】
化合物11の合成:
無水THF(5ml)中の化合物10(82mg,0.21mmol)、p−トルエンスルホン酸(10mg,0.05mmol)及びN−ヨードスクシンイミド(96mg,0.43mmol)の溶液を室温で一晩撹拌した。TLC(9:1DCM/MeOH)では、少量の出発物質及びそれより高いスポットが検出された。その反応混合物を飽和ΝaHCO3(10ml)でクエンチした。室温で10分間撹拌した後、その反応混合物をEtOAc(3×20ml)で抽出し、その有機層をブラインで洗浄した(2×20ml)。その有機層をNa2SO4で乾燥し、濾過し、真空下で濃縮することにより、茶色油状物が得られた。その粗生成物を、DCM中の1−5%メタノールを用いるカラムクロマトグラフィで精製することにより、11(52mg,48%)を黄色油状物として得た。1HNMR(DMSO−d6)δ8.81(s,1H),8.14(d,1H),7.79(d,1H),7.65(t,1H),7.58(t,1H),7.45(t,1H),3.78(s,2H),3.42(s,2H),2.44(s,2H),2.39(s,2H),2.21(s,3H),1.50(s,9H)
【0094】
化合物12の合成:
無水DMF(5ml)中の化合物11(102mg,0.2mmol)の溶液を氷浴中で冷却した。その反応物に、水素化ナトリウム(鉱油中60%,32mg,0.8mmol)を加えた。その反応混合物を0℃で15分間及び室温で15分間撹拌した。その反応物にシス/トランス−1,3−ジクロロプレペン(Dichloroprepene)(83.36μl,0.9mmol)を加えた。その反応混合物を室温で1時間撹拌した。TLC(9:1DCM/MeOH)では、出発物質は検出されなかった。その反応混合物を水でクエンチした。その水層をEtOAc(3×10ml)で抽出し、その有機層をブラインで洗浄した(2×10ml)。その有機層をNa2SO4で乾燥し、濾過し、真空下で濃縮することにより、茶色油状物が得られた。その粗生成物を、DCM中の1−5%メタノールを用いるカラムクロマトグラフィで精製することにより、12(82mg,70%)を黄色固体として得た。1HNMR(DMSO−d6)δ8.20(d,1H),7.82(d,1H),7.62(m,2H),7.38(d,1H),6.38(m,1H),6.18(m,1H),3.98−4.46(dd,2H),3.78(s,2H),3.42(s,2H),2.44(s,2H),2.39(s,2H),2.21(s,3H),1.50(s,9H)
【0095】
化合物13の合成:
無水トルエン(3ml)中の12(82mg,0.14mmol)の溶液に、乾燥窒素を15分間泡立たせた。その反応物に水素化トリブチルスズ(47.1μl,0.18mmol)及び2,2’−アゾビスイソブチロニトリル(10mg,0.06mmol)を加えた。その反応混合物を窒素下で15分間80℃に加熱した。TLC(9:1DCM/MeOH)では、新しい青いスポットが検出され、出発物質は検出されなかった。その反応混合物を真空下で濃縮することにより、黄色油状物が得られた。その粗生成物を、DCM中の1−5%メタノールを用いるカラムクロマトグラフィで精製することにより、13(52mg,82%)を白色固体として得た。1HNMR(DMSO−d6)δ7.92(d,1H),7.83(m,1H),7.78(d,1H),7.58(t,1H),7.42(t,1H),4.20(m,2H),4.04(m,2H),3.92(m,1H),3.78(s,2H),3.42(s,2H),2.44(s,2H),2.39(s,2H),2.21(s,3H),1.50(s,9H).
【0096】
化合物13の分割:
ラセミの化合物13をメタノールに溶解した。次いで、その溶液をCHIRALPAK AD prepカラム(20ミクロン,20×250mm)に充填し、メタノール(15ml/分)を用いて分離した。第1のエナンチオマー(13a)は、5.1分から溶出し、第2のエナンチオマー(13b)は、7.1分から溶出する。1HNMR(DMSO−d6)δ7.92(d,1H),7.83(m,1H),7.78(d,1H),7.58(t,1H),7.42(t,1H),4.20(m,2H),4.04(m,2H),3.92(m,1H),3.78(s,2H),3.42(s,2H),2.44(s,2H),2.39(s,2H),2.21(s,3H),1.50(s,9H)
【0097】
実施例4−CBI CC−1065類似体の合成
【化22】
【0098】
化合物(14)の合成:
MeOH/CH2Cl2(1/2,10ml)中の9b(100mg,0.24mmol)及び10%Pd−C(35mg)の溶液を真空中で40秒間脱気した。得られた混合物を水素雰囲気下に置き、25℃で7時間撹拌した。その反応混合物をセライトで濾過した(CH2Cl2洗浄)。溶媒を真空中で除去した。EtOAc/Hex(2/8)を用いて溶出する、シリカゲルによるクロマトグラフィから14(77mg,98%)が得られた。1NMR DMSO−d6)δ10.36(s,1H),8.04(d,1H,J=8.2Hz),7.72(d,1H,J=8.2Hz),7.61(br s,1H),7.45(t,1H,J=8.4Hz),7.261(t,1H,J=8.4Hz),4.06(m,4H),3.73(m,1H),1.52(s,9H)
【0099】
化合物(16)の合成:
4M HCl−EtOAc(5ml)中の14(35mg,0.1mmol)の溶液をAr下、25℃にて30分間撹拌した。溶媒を真空中で除去した。その残渣に、5−アセチルインドン−2−カルボン酸(24.4mg,0.12mmol)を加えた。DMF(3ml)中のEDC(22.9mg,0.12mmol)の溶液を加え、その反応混合物を25℃で5時間撹拌した。溶媒を除去した。その粗生成物を、CH2Cl2中の10%MeOHを用いて溶出する、シリカゲルによるクロマトグラフィに供することにより、16(40.7mg,93%)が得られた。1HNMR DMSO−d6)δ12.13(s,1H),10.47(s,1H),8.45(s,1H),8.10(d,1H,J=8.4Hz),7.96(br s,1H),7.85(d,2H,J=8.4Hz),7.54(d,1H,J=8.4Hz),7.51(t,1H,J=8.2Hz),7.36(t,1H,J=7.6),7.35(s,1H),4.81(t,1H,11.2Hz),4.54(dd,1H,8.8Hz),4.23(m,1H),4.01(dd,1H,J=10.2Hz),3.86(dd,1H,J=10.7Hz),2.61(s,3H)
【0100】
化合物(17)の合成:
4−メチル−1−ピペラジンカルボニルクロリド塩酸塩(19.9mg,0.1mmol)を、乾燥塩化メチレン(4ml)中の3%アリルアルコール中の16(20mg,0.05mmol)及び無水ピリジン(25μml,0.3mmol)の溶液に加え、その混合物を16時間撹拌した。その粗生成物のシリカゲルによる精製により、17(23.6mg,91%)が得られた。1NMR DMSO−d6)δ12.03(s,1H),8.41(s,1H),8.21(s,1H),8.01(d,1H,J=8.4Hz),7.88(d,1H,J=8.4Hz),7.82(dd,1H,J=8.4Hz),7.58(t,1H,J=8.1Hz),7.51(d,1H,J=8.4Hz),7.46(t,1H,J=7.6Hz),7.37(s,1H),4.86(t,1H,J=10.8Hz),4.57(dd,1H,J=10.8Hz),4.38(m,1H),4.06(dd,1H,J=10.8Hz),3.86(dd,1H,J=11Hz),3.41(br,4H),3.29(br,4H),2.82(s,3H),2.57(s,3H)
【0101】
化合物(19)の合成:
乾燥塩化メチレン(1ml)中の5%酢酸中の17(13mg,24μmol)及びリンカー18(16.9mg,31μmol)の溶液を25℃で30分間撹拌した。溶媒を真空中で完全に除去し、HPLC(SymmetryPrep C18,7μm,19×150mmカラム)で精製することにより、19(18.5mg,81%)が得られた。MS:C48H57ClN8O11に対する算出値(M+H)m/z 958.38,実測値958.10
【0102】
実施例5:増殖アッセイ
確立された3H−チミジン増殖アッセイを用いて、本発明の細胞傷害性化合物の生物学的活性をアッセイすることができる。これは、外来性の放射標識された3H−チミジンの取り込みを測定することによってDNA合成を評価するので、細胞増殖を定量化するための簡便な方法である。このアッセイは再現性が高く、多数の化合物に適応できる。
【0103】
アッセイでは、最初に前骨髄性白血病細胞(HL−60)を、熱不活性ウシ胎仔血清(FCS)を10%含有するRPMI培地で培養した。実験の第1日目に、細胞を回収し、洗浄し、10%FCSを含有するRPMI中に0.5×106細胞/mlの濃度に再懸濁する。100μlの細胞懸濁液を96ウェルプレートに加える。ドキソルビシン(ポジティブコントロール)又は試験化合物の系列希釈(3倍濃縮系列)を調製し、ウェル当たり化合物100μlで添加した。最終的に、1ウェルあたり10μlの100μCi/mlの3H−チミジンを添加し、そのプレートを24時間インキュベートする。プレートから、96ウェルハーヴェスタ(Packard Instruments社)を使用して回収し、Packard Top Count counterで計測した。Prismソフトウェアを使用して、薬物のモル濃度の関数として4パラメータによるロジスティック曲線を3H−チミジン取り込みに当てはめ、IC50値を決定した。
【0104】
CBI CC−1065類似体(例えば、実施例4の化合物19)は、一般に、上記アッセイにおいて、約1pMから約100nM、好ましくは約10pMから約10nMというIC50値を有する。
【0105】
実施例6:抗体への薬物分子のコンジュゲーション
この実施例では、本発明の薬物分子(他の基(例えば、スペーサー、反応性の官能基など)を任意に含む)を、標的化剤としての抗体とコンジュゲートするための反応条件及び方法論について記載する。その条件及び方法論は、例示することだけを意図しており、非限定的なものである。薬物分子を抗体とコンジュゲートするための他のアプローチは、当該技術分野で公知である。
【0106】
本願明細書中に記載されるコンジュゲーション方法は、抗体のリシンと2−イミノチオランとの反応の後、薬物−リンカー分子と活性マレイミド基との反応による、抗体への遊離チオール基の導入に基づくものである。最初に、コンジュゲートされる抗体の緩衝液を、50mM NaCl、2mM DTPA,pH8.0を含有する0.1Mリン酸緩衝液pH8.0に交換し、5〜10mg/mlに濃縮する。その抗体に2−イミノチオランを加えることによりチオール化を達成する。加える2−イミノチオランの量は、予備実験において決定し、抗体によって変化するものである。抗体に加える2−イミノチオランの量を徐々に増やし、その後、その抗体を室温で1時間インキュベートするという力価測定である予備実験において、その抗体を、Sephadex G−25カラムを用いて50mM HEPES緩衝液pH6.0中に脱塩し、導入されたチオール基の数を、ジチオジピリジン(DTDP)との反応によって迅速に決定する。チオール基とDTDPとの反応により、チオピリジンが遊離し、それを324nmにおいてモニターする。0.5〜1.0mg/mlというタンパク質濃度のサンプルを使用することができる。280nmでの吸光度を用いて、サンプル中のタンパク質の濃度を正確に決定し、次いで、各サンプルのアリコート(0.9ml)を0.1mlのDTDP(エタノール中5mM原液)と室温にて10分間インキュベートする。緩衝液のみ+DTDPのブランクサンプルもまた一緒にインキュベートする。10分後、324nmにおける吸光度を測定し、19800M−1のチオピリジンに対する吸光係数を用いて、存在するチオールの数を定量する。
【0107】
通常、1抗体あたり3つのチオール基というチオール化レベルが望ましい。例えばこれは、1つの特定の抗体を用いて、モル濃度が15倍過剰の2−イミノチオランを加え、その後、室温で1時間インキュベートすることによって達成することができる。それゆえ、コンジュゲートされる抗体を、所望のモル比率で2−イミノチオランとともにインキュベートし、次いで、コンジュゲーション緩衝液(5mMグリシン、3%グリセロール及び2mM DTPAを含有する50mM HEPES緩衝液pH6.0)中に脱塩する。導入されたチオールの数を上に記載したように定量する間は、このチオール化された材料を氷上で維持する。
【0108】
導入されたチオールの数を検証した後、活性マレイミド基を含む薬物分子(例えば、実施例3の化合物15)を、1チオールあたり3倍過剰のモル濃度で加えた。
【0109】
コンジュゲーション反応は、最終濃度が5%のエチレングリコールジメチルエーテル(または適当な代替の溶媒)を含むコンジュゲーション緩衝液中で行う。通常、薬物原液を90%エチレングリコールジメチルエーテル、10%ジメチルスルホキシドに溶解する。抗体に加える場合、その原液を、最終濃度を5%とするのに十分なエチレングリコールジメチルエーテルを有するチオール化抗体に直接加えるか、又は、最終濃度が10%のエチレングリコールジメチルエーテルを含むコンジュゲーション緩衝液に予め希釈した後、等容積のチオール化抗体を加える。コンジュゲーション反応物を、室温で2時間、混合しながらインキュベートする。インキュベート後、その反応混合物を14000RPMで15分間遠心し、精製をすぐに行わない場合はpHを7.2に調整することができる。コンジュゲートの精製は、多数の方法を用いるクロマトグラフィによって達成することができる。コンジュゲートは、5mMグリシン、50mM NaCl及び3%グリセロールを含有する50mM HEPES緩衝液pH7.2で予め平衡化されたSephacryl S200カラムにおけるサイズ排除クロマトグラフィを用いて精製することができる。クロマトグラフィは、28cm/hという線流速で行うことができる。コンジュゲートを含む画分を回収し、プールし、濃縮することができる。あるいは、精製は、イオン交換クロマトグラフィによって達成することができる。条件は、抗体によって異なり、各場合において最適化する必要がある。例えば、抗体−薬物コンジュゲート反応混合物を、50mM HEPES、5mMグリシン、3%グリセロール,pH6.0で予め平衡化されたSP−Sepharoseカラムに適用することができる。その抗体コンジュゲートを、平衡化緩衝液中の0〜1M NaClの勾配を用いて溶出することができる。コンジュゲートを含む画分をプールすることができ、pHを7.2に調節することができ、必要であれば、そのサンプルを濃縮することができる。
【0110】
本願明細書中で引用されたすべての特許出願、特許及び刊行物の全開示内容は、参照によって本願明細書に援用される。
【0111】
前述の実施例において使用された反応物及び/又は使用条件の代わりに一般的又は特に記載された反応物及び/又は本発明の使用条件を用いることによって同様に首尾よく前述の実施例を繰り返すことができる。
【0112】
前述の記載から、当業者は、本発明の本質的な特徴を容易に確認することができ、また、本発明の技術範囲から逸脱することなく、本発明を様々な用途及び条件に適合するように本発明の様々な変更及び改変を行うことができる。
【図面の簡単な説明】
【0113】
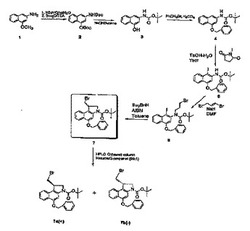
【図1】CBI CC−1065類似体の調整法の一態様を示す合成経路図である。
【図2】CBI CC−1065類似体の調整法の別の態様を示す合成経路図である。
【図3】CBI CC−1065類似体の調整法の第三の態様を示す合成経路図である。
【技術分野】
【0001】
本発明はCC−1065類似体の調製方法及び調製用化合物に関する。
関連出願の相互参照:本特許出願は、2005年10月26日に出願の米国仮特許出願第60/730,804号(全開示内容を参照によって本願明細書に援用する)の利益を主張するものである。
【背景技術】
【0002】
CC−1065は、強力なサイトトキシンであることが公知である。CC−1065は、アップジョン社(Upjohn Company)によって1981年にStreptomyces zelensisから初めて単離され(Hanka et al.,J.Antibiot.31:1211(1978);Martin et al,J.Antibiot.33:902(1980);Martin et al,J.Antibiot.34:1119(1981))、in vitro及び実験動物の両方において強力な抗腫瘍活性及び抗菌活性を有することが明らかにされた(Li et al.,Cancer Res.42:999(1982))。CC−1065は、5’−d(A/GNTTA)−3’及び5’−d(AAAAA)−3’の配列選択性により、副溝中で二本鎖B−DNAに結合し(Swenson et al,Cancer Res.42:2821(1982))、その分子中に存在するCPI左側部分によって3’−アデニンのN3位をアルキル化する(Hurley et al,Science 226:843(1984))。
【発明の開示】
【発明が解決しようとする課題】
【0003】
CC−1065は、その強力かつ広範な抗腫瘍活性にもかかわらず、それが実験動物の遅延死を引き起こすため、人間に対する使用を不可能にしている。
【0004】
CC−1065の多くのアナログ(類似体)及び誘導体が当該技術分野で公知である。その構造式、合成及び化合物の特性の多くが、これまで研究されている。例えば、Boger et al.,Angew.Chem.Int.Ed.Engl.35:1438(1996);及びBoger et al.,Chem.Rev.97:787(1997)を参照のこと。
【0005】
協和醗酵工業のグループは、多くのCC−1065誘導体を調製している。例えば、米国特許5,101,038号;同第5,641,780号;同第5,187,186号;同第5,070,092号;同第5,703,080号;同第5,070,092号;同第5,641,780号;同第5,101,038号;及び同第5,084,468号;並びにPCT出願公開第WO96/10405及び欧州特許出願公開第0537575A1号を参照のこと。
【0006】
また、アップジョン社はCC−1065の誘導体を多種にわたり調製している。例えば、米国特許第5,739,350号;同第4,978,757号、同第5,332,837号及び同第4,912,227号を参照のこと。
【課題を解決するための手段】
【0007】
ある実施態様は、化合物(I)又はその塩を合成する方法である。
【化1】
式中、R1及びR2は、各々独立して、H、アルキル、−C(O)OR’、−C(O)NR’R”又は保護基であり、R’及びR”は、独立して、H、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル及び非置換ヘテロシクロアルキルからなる群から選択され;R6は、H、置換若しくは非置換の低級アルキル、シアノ又はアルコキシであり;Xは、ハロゲンである。この方法では、保護基R1’及びR2’が、化合物(II)に付加され、
【化2】
化合物(III)を形成する。
【化3】
式中、R3は、H又はアルキルである。化合物(III)のアミン窒素を含む5員環が生成される。
【0008】
別の実施態様は、以下の式を有するCBI CC−1065類似体又はその医薬的に許容される塩を合成する方法である。
【化4】
式中、Xは、ハロであり;X1及びZは、各々独立して、O、S及びNR8から選択され、R8は、H、置換又は非置換のアルキル、置換又は非置換のヘテロアルキル及びアシルから選択されるメンバーであり;R4、R4,、R5及びR5,は、独立して、H、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル、非置換ヘテロシクロアルキル、ハロゲン、NO2、NR9R10、NC(O)R9、OC(O)NR9R10、OC(O)OR9、C(O)R9、SR9、OR9、CR9=NR10及びO(CH2)nNR11R11’からなる群から選択されるメンバーであり、R9及びR10は、独立して、H、置換若しくは非置換のアルキル、置換若しくは非置換のヘテロアルキル、置換若しくは非置換のアリール、置換若しくは非置換のヘテロアリール、置換若しくは非置換のヘテロシクロアルキル及び置換若しくは非置換のペプチジルから選択されるか、又はR9及びR10は、それらが結合する窒素原子と任意に結合して、任意に2つ以上のヘテロ原子を含む4〜6員環を有する置換若しくは非置換のヘテロシクロアルキル環系を形成し、R11及びR11’は、各々独立して、H又は低級アルキルであり;R6は、H、置換若しくは非置換の低級アルキル、シアノ又はアルコキシであり;R7は、H、置換アルキル、非置換アルキル、置換ヘテロアルキル、非置換ヘテロアルキル、ジホスフェート、トリホスフェート、アシル、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、R12、R13及びR14は、独立して、H、置換若しくは非置換のアルキル、置換若しくは非置換のヘテロアルキル及び置換若しくは非置換のアリールから選択されるメンバーであるか、又はR12及びR13は、それらが結合する窒素又は炭素原子と任意に結合して、任意に2つ以上のヘテロ原子を含む4〜6員環を有する置換若しくは非置換のヘテロシクロアルキル環系を形成する。
【0009】
上記方法は、保護基R1’及びR2’を化合物(II)に付加して
【化5】
化合物(III)を形成することを含む。
【化6】
式中、R3は、H又はアルキルである。化合物(III)のアミン窒素を含む5員環が生成される。結合部分が、化合物(III)に付加され、その結合部分は、以下を含む。
【化7】
【0010】
更に別の実施態様は、式(I)の化合物又はその医薬的に許容される塩である。
【化8】
式中、R1及びR2は、各々独立して、H、アルキル、−C(O)OR’、−C(O)NR’R’’又は保護基であり、R’及びR”は、独立してH、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル及び非置換ヘテロシクロアルキルからなる群から選択される。
【0011】
いかなる限定も包括もしない本発明の実施態様を図面を参照しつつ以下に記載する。図面においては、特に断りがない限り、種々の図面を通して付与された数字は部分部分を示す。図面と共に発明な詳細な説明を参照すれば本発明をさらに理解することができるであろう。
【発明を実施するための最良の形態】
【0012】
本願明細書において使用されるとき、「Boc」は、t−ブチルオキシカルボニルを指す。「CPI」は、シクロプロパピロロインドールを指す。「CBI」は、シクロプロパベンズインドールを指す。「Cbz」は、カルボベンゾキシである。「DCM」は、ジクロロメタンを指す。「DMF」は、N,N−ジメチルホルムアミドである。「FMOC」は、9−フルオレニルメチルオキシカルボニルを指す。「TEA」は、トリエチルアミンを指す。「THF」は、テトラヒドロフランを指す。「EDC」は、塩酸1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミドを指す。
【0013】
別途定義する場合を除き、本発明において使用する全ての技術用語及び科学用語は、本発明が属する分野における通常の技術を有する者によって一般に理解されるものと同じ意味で用いられる。本発明において使用する分類、並びに細胞培養、分子遺伝学、有機化学及び核酸化学における解析及び下記のハイブリダイゼーションの手順は、当該技術分野において良く知られ、一般に用いられているものを意味する。技術及び手順は、一般に、当該技術分野の従来法及び様々な一般的文献に従って実施される。本発明において使用する命名、及び分析における解析手順、並びに下記の有機合成は、当該技術分野において公知で、一般に用いられているものを意味する。化学合成及び化学分析においては、標準的技術又はその変法が用いられる。
【0014】
「治療薬」の用語は、治療上有効な量が存在する場合に、哺乳動物に所望の治療効果をもたらす化合物を意味する。腫瘍を処置するためには、治療薬が標的細胞に取り込まれるのが望ましい。
【0015】
「サイトトキシン」の用語は、癌細胞に対して毒性となる効果を有する治療薬を意味する。細胞に対する毒性とは、薬剤が細胞の成長を停止又は細胞を死滅させることを意味する。
【0016】
「プロドラッグ」及び「薬物コンジュゲート」の用語は、本願明細書において互換的に使用される。双方とも、コンジュゲートの形態をとる間は未だ細胞に対して比較的無害な化合物であるが、一定条件(例えば、標的細胞内又はその近傍に位置する酵素の存在)において選択的に分解され、薬理学的に活性な形態となるものを意味する。
【0017】
【化9】
なる符号は、結合部として使用されたとしても、また、結合に対して垂直に表示されたとしても、表示された部分が分子の残部や固体支持体、置換基等に付着する点を示す。
【0018】
「アルキル」の用語は、それ自体又は別の置換基の一部として、別途記載されない限り、直鎖状若しくは分枝状の鎖又は環状の炭化水素ラジカルあるいはそれらの組み合わせを意味し、それらは、完全に飽和していてもよいし、一不飽和又は多価不飽和であってもよく、示された炭素原子の数を有する(すなわちC1−C10は、1〜10個の炭素を意味する)二価及び多価のラジカルを含み得る。飽和炭化水素基の例として、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、t−ブチル、イソブチル、sec−ブチル、シクロヘキシル、(シクロヘキシル)メチル、シクロプロピルメチルなどの基、例えば、n−ペンチル、n−ヘキシル、n−ヘプチル、n−オクチル、等の相同体及び異性体が挙げられるが、これらに限定されない。不飽和アルキル基は、1以上の二重結合又は三重結合を有するものである。不飽和のアルキル基の例としては、ビニル、2−プロペニル、クロチル、2−イソペンテニル、2−(ブタジエニル)、2,4−ペンタジエニル、3−(1,4−ペンタジエニル)、エチニル、1−及び3−プロピニル、3−ブチニル並びにそれらの高い相同体及び異性体が挙げられるが、これらに限定されない。「アルキル」の用語は、別途に述べない限り、「ヘテロアルキル」など、以下に更に詳しく定義するアルキルの誘導体も含むものを意味する。アルキル基で炭化水素基に限られるものは、「ホモアルキル」と命名されている。
【0019】
「アルキレン」の用語は、それ自体又は別の置換基の一部として、アルカンから誘導される二価のラジカルを意味し、その例としては、−CH2CH2CH2CH2−が挙げられるが、これに限定されることはなく、更に「ヘテロアルキレン」と以下に記載される基が含まれる。望ましくは、アルキル(又はアルキレン)基は、1〜24の炭素原子を有するものであり、10以下の炭素原子を有するそれらの基が、本発明では望ましい。「低級アルキル」又は「低級アルキレン」は、短鎖のアルキル又はアルキレン基であり、概して8以下の炭素原子を有している。
【0020】
「ヘテロアルキル」の用語は、それ自体又は別の用語との組み合わせにおいて、別途記載されない限り、安定な直鎖状若しくは分枝状の鎖又は環状の炭化水素基あるいはそれらの組み合わせを意味し、多くの炭素原子並びにO、N、Si及びSからなる群から選択される少なくとも1つのヘテロ原子からなり、ここでその窒素、炭素及び硫黄原子は、任意に酸化されてもよく、また、窒素ヘテロ原子は、任意に四級化されていてもよい。ヘテロ原子(単数または複数)のO、N、S及びSiは、ヘテロアルキル基の内部の任意の位置、又はアルキル基が分子の残部に付着する位置に配置されうる。その例としては、−CH2−CH2−O−CH3、−CH2−CH2−NH−CH3、−CH2−CH2−N(CH3)−CH3、−CH2−S−CH2−CH3、−CH2−CH2、−S(O)−CH3、−CH2−CH2−S(O)2−CH3、−CH=CH−O−CH3、−Si(CH3)3、−CH2−CH=N−OCH3及び−CH=CH−N(CH3)−CH3が挙げられるが、これらに限定されない。例えば、−CH2−NH−OCH3及び−CH2−O−Si(CH3)3など、2つまでのヘテロ原子が連続してもよい。同様に、「ヘテロアルキレン」の用語は、それ自体又は別の置換基の一部として、ヘテロアルキルから誘導される二価のラジカルを意味し、その例としては、−CH2−CH2−S−CH2−CH2−及び−CH2−S−CH2−CH2−NH−CH2−が挙げられるが、これらに限定されない。ヘテロアルキレン基の場合、ヘテロ原子は鎖の末端の片方又は両方を占めることができる(例えば、アルキレンオキシ、アルキレンジオキシ、アルキレンアミノ、アルキレンジアミノ等)。「ヘテロアルキル」及び「ヘテロアルキレン」の用語には、ポリ(エチレングリコール)及びその誘導体(例えば、Shearwater Polymers Catalog、2001を参照されたい)が包含される。更にまた、アルキレン及びヘテロアルキレン置換基の場合、置換基の式が記載される方向によって、置換基の配向が規定されるものではない。例えば、式−C(O)2R’−は、−C(O)2R’−と−R5C(O)2−との両方を表す。
【0021】
「低級」の用語は、「アルキル」又は「ヘテロアルキル」の用語と組み合わせて、1〜6の炭素原子を有する部分をいう。
【0022】
「アルコキシ」、「アルキルアミノ」、「アルキルスルホニル」及び「アルキルチオ」(又はチオアルコキシ)の用語は、それらの従来の意味で使用され、それぞれ酸素原子、アミノ基、SO2基又は硫黄原子を介して分子の残部に付着したアルキル基を指す。「アリールスルホニル」の用語は、SO2基を介して分子の残部に付着したアリール基を指し、「スルフヒドリル」の用語は、SH基を指す。
【0023】
一般に、「アシル」置換基も、上に記載された群から選択される。本願明細書において使用されるとき、「アシル」置換基の用語は、本発明の化合物の多環式核に直接又は間接的に付着したカルボニル炭素に付着して、その原子価を満たす基を指す。
【0024】
「シクロアルキル」及び「ヘテロシクロアルキル」の用語は、それら自体、又は他の用語との組み合わせにおいて、別途に述べない限り、置換又は非置換「アルキル」、及び置換又は非置換「ヘテロアルキル」の環状体をそれぞれ表す。また、ヘテロシクロアルキルの場合、ヘテロ原子は、分子の残部にヘテロ環が付着する位置を占めることができる。シクロアルキルの例として、シクロペンチル、シクロヘキシル、1−シクロヘキセニル、3−シクロヘキセニル、シクロヘプチル等が挙げられるが、これらに限定されない。ヘテロシクロアルキルの例として、1−(1,2,5,6−テトラヒドロピリジル)、1−ピペリジニル、2−ピペリジニル、3−ピペリジニル、4−モルホリニル、3−モルホリニル、テトラヒドロフラン−2−イル、テトラヒドロフラン−3−イル、テトラヒドロチエン−2−イル、テトラヒドロチエン−3−イル、1−ピペラジニル、2−ピペラジニル基等が挙げられるが、これらに限定されない。環状構造のヘテロ原子及び炭素原子は、任意に酸化される。
【0025】
「ハロ」又は「ハロゲン」の用語は、それ自体又は別の置換基の一部として、別途記載されない限り、フッ素、塩素、臭素又はヨウ素原子を意味する。更に、「ハロアルキル」などの用語にはモノハロアルキル及びポリハロアルキルが含まれるものとする。例えば、「ハロ(C1−C4)アルキル」の用語は、トリフルオロメチル、2,2,2−トリフルオロエチル、4−クロロブチル、3−ブロモプロピルなどを含むことを意味するが、これらに限定されない。
【0026】
「アリール」の用語は、別途断りのない限り、置換又は非置換の、多価不飽和、芳香族及び炭化水素の置換基であり、単環又は多環(望ましくは1環から3環)状に融合若しくは共有結合するものを意味する。「ヘテロアリール」の用語は、N、O及びSから選択される1〜4個のヘテロ原子を含むアリール基(または環)を指し、ここで、その窒素、炭素及び硫黄原子は、任意に酸化され、窒素原子(単数又は複数)は、任意に四級化される。ヘテロアリール基は、ヘテロ原子を介して分子の残部に付着できる。アリール及びヘテロアリール基の例としては、フェニル、1−ナフチル、2−ナフチル、4−ビフェニル、1−ピロリル、2−ピロリル、3−ピロリル、3−ピラゾリル、2−イミダゾリル、4−イミダゾリル、ピラジニル、2−オキサゾリル、4−オキサゾリル、2−フェニル−4−オキサゾリル、5−オキサゾリル、3−イソキサゾリル、4−イソキサゾリル、5−イソキサゾリル、2−チアゾリル、4−チアゾリル、5−チアゾリル、2−フリル、3−フリル、2−チエニル、3−チエニル、2−ピリジル、3−ピリジル、4−ピリジル、2−ピリミジル、4−ピリミジル、5−ベンゾチアゾリル、プリニル、2−ベンズイミダゾリル、5−インドリル、1−イソキノリル、5−イソキノリル、2−キノキサリニル、5−キノキサリニル、3−キノリル、及び6−キノリルが挙げられるが、それに限定されない。上記アリール及びヘテロアリール環系の各々に対する置換基は、以下の許容される置換基の群から選択される。「アリール」及び「ヘテロアリール」は、アリール又はヘテロアリール系に、1つ以上の非芳香族の環系が縮合しているか、又は別の方法で結合している環系も包含する。
【0027】
略して、「アリール」の用語には、他の用語と組み合わせて使用する場合(例えば、アリールオキシ、アリールチオキシ、アリールアルキル)、上記定義のアリール及びヘテロアリール環の双方が含まれる。よって「アリールアルキル」の用語は、炭素原子(例えば、メチレン基)が例えば、酸素原子で置換されているアルキル基(例えば、フェノキシメチル、2−ピリジルオキシメチル、3−(1−ナフチルオキシ)プロピル等が挙げられる)を含め、アルキル基にアリール基が付着した基(例えば、ベンジル、フェネチル、ピリジルメチル等が挙げられる)を指す。
【0028】
上記の用語(例えば、「アルキル」、「ヘテロアルキル」、「アリール」及び「ヘテロアリール」)の各々には、示した基の置換及び非置換型の双方が含まれる。基の各々の型に望ましい置換基は、以下に示すとおりである。
【0029】
アルキル及びヘテロアルキルラジカル(アルキレン、アルケニル、ヘテロアルキレン、ヘテロアルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、シクロアルケニル及びヘテロシクロアルケニルと称されることが多い基を含む)に対する置換基は、一般に、それぞれ「アルキル置換基」及び「ヘテロアルキル置換基」と称され、それらは、0から(2m’+1)(m’は、かかるラジカルにおける炭素原子の総数である)までの範囲の数の、−OR’、=O、=NR’、=N−OR’、−NR’R”、−SR’、−ハロゲン、−SiR’R”R’”、−OC(O)R’、−C(O)R’、−CO2R’、−CONR’R”、−OC(O)NR’R”、−NR’’C(O)R’、−NR’−C(O)NR”R”’、−NR”C(O)2R’、−NR−C(NR’R”R’”)=NR””、−NR−C(NR’R”)=NR”’、−S(O)R’、−S(O)2R’、−S(O)2NR’R”、−NRSO2R’、−CN及び−NO2から選択される種々の基の1つ以上でありうるが、これらに限定されない。R’、R”、R’”及びR””は、各々好ましくは独立して、水素、置換若しくは非置換のヘテロアルキル、置換若しくは非置換のアリール、例えば、1〜3個のハロゲンで置換アリール、置換若しくは非置換のアルキル、アルコキシ又はチオアルコキシ基あるいはアリールアルキル基を指す。本発明の化合物が、1つより多いR基を含むとき、例えば、R’、R”、R’”及びR””基が1つより多く存在しているときのこれらの基の各々と同様に、R基の各々は、独立して選択される。R’及びR”が同じ窒素原子に付着する場合、それらは窒素原子と共に結合して5員環、6員環又は7員環を形成することができる。例えば、−NR’R”は、1−ピロリジニル及び4−モルホリニルを含むと意味されるが、これらに限定されない。置換基に関する上記の記載から、当業者は、「アルキル」の用語は、ハロアルキル(例えば、−CF3及び−CH2CF3)及びアシル(例えば、−C(O)CH3、−C(O)CF3、−C(O)CH2OCH3など)のような水素基以外の基に結合した炭素原子を含む基を含むことを意味すると理解するであろう。
【0030】
アルキルラジカルについて記載された置換基と同様に、アリール置換基及びヘテロアリール置換基は、一般に、それぞれ「アリール置換基」及び「ヘテロアリール置換基」を指し、また、それらは、多種多様なものであり、0から芳香族の環系における開放原子価の総数までの範囲の数で、例えば、ハロゲン、−OR’、=O’、=NR’、=N−OR’、−NR’R”、−SR’、−ハロゲン、−SiR’R”R”’、−OC(O)R’、−C(O)R’、−CO2R’、−CONR’R”、−OC(O)NR’R”、−NR”C(O)R’−NR’−C(O)NR”R’”、−NR”C(O)2R’、−NR−C(NR’R”)=NR”’、−S(O)R’、−S(O)2R’、−S(O)2NR’R”、−NRSO2R’、−CN及び−NO2、−R’、−N3、−CH(Ph)2、フルオロ(C1−C4)アルコキシ及びフルオロ(C1−C4)アルキルから選択され;R’、R”、R’”及びR””は、好ましくは独立して、水素、(C1−C8)アルキル及びヘテロアルキル、非置換アリール及びヘテロアリール、(非置換アリール)−(C1−C4)アルキル、並びに(非置換アリール)オキシ−(C1−C4)アルキルから選択される。本発明の化合物が、1つより多いR基を含むとき、例えば、R’、R”、R’”及びR””基が1つより多く存在しているときのこれらの基の各々と同様に、R基の各々は、独立して選択される。
【0031】
アリール又はヘテロアリール環の隣接した原子上のアリール置換基の2つは、式−T−C(O)−(CRR’)q−U−(式中、T及びUは、独立して、−NR−、−O−、−CRR’−又は単結合であり、qは、0から3の整数である)の置換基で任意に置換されてもよい。あるいは、アリール又はヘテロアリール環の隣接した原子上の置換基の2つは、式−A−(CH2)r−B−(式中、A及びBは、独立して、−CRR’−、−O−、−NR−、−S−、−S(O)−、−S(O)2−、−S(O)2NR’−又は単結合であり、rは、1から4の整数である)の置換基で任意に置換されてもよい。このようにして形成された新しい環の単結合の1つは、二重結合で任意に置換されてよい。あるいは、アリール又はヘテロアリール環の隣接した原子上の置換基の2つは、式−(CRR’)s−X−(CR”R’”)d−(式中、s及びdは、独立して0から3の整数であり、Xは、−O−、−NR’−、−S−、−S(O)−、−S(O)2−,又は−S(O)2NR’−である)の置換基で任意に置換されてもよい。置換基R、R’、R”及びR’”は、好ましくは独立して、水素又は置換若しくは非置換の(C1−C6)アルキルから選択される。
【0032】
本発明において、「ジホスフェート」の用語には、2つのリン酸基を含むリン酸のエステルが含まれるが、これらに限定されない。「トリホスフェート」の用語には、3つのリン酸基を含むリン酸のエステルが含まれるが、これらに限定されない。例えば、ジホスフェート又はトリホスフェートを有する特定の薬剤として、以下のものが挙げられる。
【化10】
本発明において、「ヘテロ原子」の用語には、酸素(O)、窒素(N)、イオウ(S)及びケイ素(Si)が含まれる。
【0033】
「R」の符号は、置換又は非置換アルキル、置換又は非置換ヘテロアルキル、置換又は非置換アリール、置換又は非置換ヘテロアリール、及び置換又は非置換ヘテロシクリル基より選択される置換基を表す一般略記である。
【0034】
「保護基」の用語に関しては、当業者は、選択された反応条件のセットによる干渉から特定の官能基をどのようにして保護するかを理解するであろう。例えば、有用な保護基については、Greene et al.,PROTECTIVE GROUPS IN ORGANIC SYNTHESIS,John Wiley & Sons,New York、1991を参照のこと。適当な保護基の例としては、BOC、FMOC、2−トリメチルシリルエトキシカルボニル、アリルオキシカルボニル、4−メチル−1−ピペラジンカルボニル、1−メチル−1−(4−ビフェニリル)エトキシカルボニル、ジフェニルオキシカルボニル、ベンジル、t−ブチル、テトラヒドロピラン、トリメチルシリル、t−ブチルジメチルシリル、トリイソプロピルシリル、t−ブチルジフェニルシリル、2,2,2−トリクロロエチルオキシカルボニル、ジイソプロピルメチルオキシカルボニル、ビニルオキシカルボニル、メトキシベンジルオキシカルボニル、ニトロベニズル(nitrobenyzl)オキシカルボニル、シクロヘキシルオキシカルボニル、シクロペンチルオキシカルボニル、ベンジルオキシカルボニル、ホルミル、アセチル、トリハロアセチル、ベンゾイル、ニトロフェニルアセチル、2−ニトロベンゼンスルホニル、フタルイミド及びジチアスクシノイルが挙げられるが、これらに限定されない。
【0035】
「医薬的に許容される担体」の用語は、本発明において、化学物質の運搬又は輸送に関与する、液体若しくは固体充填剤、希釈剤、賦形剤、溶媒、又は封入材料などの、医薬的に許容される材料、組成物又は溶剤を意味する。医薬的に許容される担体には医薬的に許容される塩が含まれ、ここで「医薬的に許容される塩」の用語には、本願明細書に記載の化合物に見られる特定の置換基に応じ、比較的無毒の酸又は塩基を用いて調製される活性化合物の塩が含まれる。本発明の化合物が比較的酸性の官能基を含む場合、塩基付加塩は、このような化合物の中性型を充分量の所望の塩基と、そのままか又は望ましい不活性溶媒中で接触させることによって得ることができる。医薬的に許容される塩基付加塩の例として、ナトリウム、カリウム、カルシウム、アンモニウム、有機アミノ、若しくはマグネシウム塩、又は類似の塩が挙げられる。本発明の化合物が比較的塩基性の官能基を含む場合、酸付加塩は、このような化合物の中性型を充分量の所望の酸と、そのままか又は望ましい不活性溶媒中で接触させることによって得ることができる。医薬的に許容される酸付加塩の例として、塩化水素酸、塩化臭素酸、硝酸、炭酸、一水素炭酸、リン酸、一水素リン酸、二水素リン酸、硫酸、一水素硫酸、ヨウ化水素酸、又はリン酸等のような無機酸に由来するものや、酢酸、プロピオン酸、イソ酪酸、マレイン酸、マロン酸、安息香酸、コハク酸、スベリン酸、フマル酸、乳酸、マンデル酸、フタル酸、ベンゼンスルホン酸、p−トリルスルホン酸、クエン酸、酒石酸、メタンスルホン酸等のような比較的無毒の有機酸に由来する塩が挙げられる。更に挙げられるのは、アルギン酸塩などの、アミノ酸の塩等、及びグルクロン酸又はガラクツロン酸のような有機酸の塩等である(例えば、Bergeら、「Pharmaceutical Salts」、Journal of Pharmaceutical Science、1977、66、1−19を参照)。本発明の特異的な所定の化合物は、その化合物を塩基又は酸付加塩のいずれかに変換させる、塩基性及び酸性官能基の双方を含む。
【0036】
天然型化合物は、望ましくは、上記の塩を塩基又は酸に接触させ、従来法にて中性構造の化合物を単離することによって得られる。化合物の原型は、極性溶媒中での溶解性など特定の物理的特性ではそれらの様々な塩と異なるが、その他の点では、それらの塩は本発明の目的とする化合物の中性構造と同等である。
【0037】
塩の型に加えて、本発明はプロドラッグ型の化合物を提供する。本願明細書に記載の化合物のプロドラッグとは、生理的条件下で化学的変化を容易に起こして本発明の化合物を提供する化合物である。加えて、プロドラッグは、ex vivoの環境において化学的又は生化学的方法によって本発明の化合物に変換されうる。例えば、プロドラッグは、望ましい酵素又は化学試薬と共に経皮リザーバーパッチに添加した場合、本発明の化合物に徐々に変換されうる。
【0038】
本発明の特定の化合物は、非溶媒和物や、水和物を含めた溶媒和物の形態にて存在しうる。一般的に、溶媒和物は非溶媒和物と同等であり、本発明の範囲に包含されるものである。本発明の特定の化合物は、多結晶又は非晶形にて存在することもある。一般的に、すべての物理的な型が本発明により企図される用途において同等であり、本発明の範囲に含まれる。
【0039】
本発明の特定の化合物は、不斉炭素原子(光学的中心)又は二重結合を有しており、ラセミ体、ジアステレオマー、幾何異性体、及び個々の異性体が、本発明の範囲に包含される。
【0040】
本発明の化合物はまた、かかる化合物を構成する1つ以上の原子において、非天然の割合で原子同位体も含みうる。例えば、化合物は、例えばトリチウム(3H)、ヨウ素−125(125I)又は炭素−14(14C)などの放射性同位体で放射標識されてもよい。本発明の化合物の同位体のすべてが、放射性の有無に関わらず、本発明の範囲に包含される。
【0041】
「ポリペプチド」、「ペプチド」及び「タンパク質」の用語は、本発明において、アミノ酸のポリマーを言うのに互換的に使用される。これらの用語は、天然アミノ酸ポリマー及び非天然アミノ酸ポリマーに適用するのと同様に、1つ以上のアミノ酸残基が、それに対応する天然アミノ酸の人工的な化学模倣物であるアミノ酸ポリマーにも適用する。またこれらの用語は、「抗体」の用語も包含する。
【0042】
「アミノ酸」の用語は、天然及び合成アミノ酸や、天然アミノ酸と同様に機能するアミノ酸類似体及びアミノ酸模倣物をいう。天然アミノ酸とは、遺伝暗号によってコードされるものや、事後的に修飾されるそれらのアミノ酸、例えば、ヒドロキシプロリン、γ−カルボキシグルタメート、及びO−ホスホセリンをいう。アミノ酸類似体は、天然アミノ酸と同じ基本化学構造、すなわち、水素、カルボキシル基、アミノ基及びR基に結合したα炭素を有する化合物、例えば、ホモセリン、ノルロイシン、メチオニンスルホキシド、メチオニンメチルスルホニウムを指す。このようなアミン酸類似体は、修飾されたR基(例えば、ノルロイシン)又は修飾されたペプチド骨格を有するが、天然アミノ酸と同じ基本化学構造を保持している。特に使用しても良い1つのアミノ酸はシトルリンであって、これはアルギニンの前駆体であり、肝臓における尿素の形成に関与する。アミノ酸模倣物は、アミノ酸の一般化学構造とは異なる構造を有するが、天然アミノ酸と同様に機能する化学的化合物をいう。「非天然アミノ酸」の用語は、上記の20の天然アミノ酸の「D」型立体構造を有するものを表す。非天然アミノ酸の用語には、天然アミノ酸の相同体、及び天然アミノ酸の合成的に修飾された型とが含まれることが更に理解される。合成的に修飾された型には、上限2つの炭素原子にまで短縮又は延長されたアルキレン鎖を有するアミノ酸、任意に置換アリール基を含むアミノ酸、及びハロゲン化基、望ましくはハロゲン化アルキル及びアリール基を含むアミノ酸が含まれるが、これらに限定されない。本発明のリンカー又はコンジュゲートに付着すると、アミノ酸は、アミノ酸のカルボン酸基がケト(C(O))基に置換されている「アミノ酸側鎖」の形態となる。従って、例えば、アラニン側鎖は、−C(O)−CH(NH2)−CH3などである。
【0043】
アミノ酸及びペプチドは、保護基によって保護されてもよい。保護基は、アミノ酸又はペプチドのN−末端を望ましくない反応から保護する原子又は化学的部分であり、薬剤−リガンドコンジュゲートの合成の際に使用することができる。これは、合成中はN−末端に付着した状態が望ましく、薬剤コンジュゲートの合成の完了後に、その除去を選択的に行う化学的条件又はその他の条件によって除去するのがよい。N−末端保護に望ましい保護基は、ペプチド化学の技術において公知である。保護基の例として、水素、D−アミノ酸、及びカルボベンゾキシ(Cbz)クロライドが挙げられるが、これらに限定されない。
【0044】
「抗体」の用語は、本発明においては全抗体、及び抗原結合断片のいずれか(すなわち、「抗原結合部分」)、又はその一本鎖を含む。「抗体」とは、ジスルフィド結合により相互連結する、少なくとも2本の重(H)鎖と2本の軽(L)鎖とを含む糖タンパク質、又はその抗原結合部分をいう。各重鎖は、重鎖可変領域(VH)及び重鎖定常領域を含む。重鎖定常領域は、3つのドメイン、CH1、CH2及びCH3を含み、ミュー、デルタ、ガンマ、アルファ又はイプシロンアイソタイプでありうる。各軽鎖は、軽鎖可変領域(VL)及び軽鎖定常領域を含む。軽鎖定常領域は、1つのドメインCLを含み、それは、カッパー又はラムダアイソタイプでありうる。VH及びVL領域は、フレームワーク領域(FR)と称される、より保存された領域が介在する、相補性決定領域(CDR)と称される超可変性の領域に更に細分されうる。VH及びVLの各々は、3つのCDR及び4つのFRを含み、アミノ末端からカルボキシ末端に向かって以下の順序で配置される:FR1、CDR1、FR2、CDR2、FR3、CDR3、FR4。重鎖と軽鎖の可変領域は、抗原と相互作用する結合ドメインを含んでいる。抗体の定常領域は、宿主組織、又は、免疫系の様々な細胞(例えば、エフェクター細胞)及び古典補体系の第一成分(Clq)を含む因子への、免疫グロブリンの結合を媒介しうる。
【0045】
抗体の「抗体断片」又は「抗原結合部分」(又は単に「抗体部分」)の用語は、本発明において、抗原への特異的な結合能を保持する、抗体の1以上の断片をいう。抗体の抗原結合機能は、全長の抗体の断片によって行えることが示されている。「抗体断片」又は抗体の「抗原結合部分」の用語内に包含される結合断片の例としては、(i)VL、VH、CL及びCH1ドメインからなる一価断片であるFab断片;(ii)ヒンジ領域におけるジスルフィド架橋によって連結された2つのFab断片を含む二価断片であるF(ab’)2断片;(iii)VH及びCH1ドメインからなるFd断片;(iv)抗体の単腕のVL及びVHドメインからなるFv断片、(v)VHドメインからなるdAb断片(Ward et al.,(1989)Nature 341 :544−546);及び(vi)単離された相補性決定領域(CDR)が挙げられる。更に、Fv断片の2つのドメインであるVL及びVHは、別々の遺伝子によってコードされるが、それらをVL領域とVH領域とが対を形成して一価分子を形成する一本鎖タンパク質(一本鎖Fv(scFv)として公知である;例えば、Bird et al.(1988)Science 242:423−426;及びHuston et al.(1988)Proc.Natl.Acad.Sci.USA 85:5879−5883を参照のこと)として作製することができる合成リンカーによって、組換え法を用いてそれらを連結することができる。このような一本鎖抗体もまた、抗体の「抗原結合部分」の用語に包含されることが意図される。これらの抗体断片は、当業者に知られた従来の技術を用いて得られ、それら断片は通常の抗体と同様に、その用途に基づきスクリーニングされる。
【0046】
「モノクローナル抗体」の用語は、本発明において、単一の分子組成の抗体分子の調製物をいう。モノクローナル抗体組成物は、特定のエピトープに対する単一の結合特異性及び親和性を呈する。
【0047】
「固体支持体」は、本願明細書において使用されるとき、選択された溶媒系に実質的に不溶性である材料、又はその支持体が溶解性である選択された溶媒系から容易に分離することができる(例えば、沈殿によって)材料を指す。本発明を実施するのに有用な固体支持体は、選択された種がその固体支持体に結合させられるように活性化されているか、又は活性化することのできる基を含むことができる。固体支持体はまた、例えば、チップ、ウエハー又はウェルなどといった基材で、その上に個々の、又は1以上の本発明の化合物が結合されるものでもよい。
【0048】
本発明の化合物は、単一の異性体(例えば、エナンチオマー、シス−トランス異性体、ジアステレオマー)として、又は異性体の混合物として調製される。望ましい実施形態において、化合物は、実質的に単一の異性体として調製される。実質的に異性体として純粋な化合物を調製する方法は、当該技術分野において知られている。例えば、エナンチオマーとして濃縮された混合物、及び純粋なエナンチオマー化合物は、エナンチオマーとして純粋なエナンチオマーの合成中間体を、キラル中心での立体化学を不変のままにするか又はその完全な反転を引き起こす反応と組み合わせて使用することにより、調製することができる。あるいは、最終産物又はその合成経路での中間体は、単一の立体異性体に分割することができる。特定の立体中心を反転するか又は不変のままにするための技術、及び立体異性体の混合物を分離するための技術は、当該技術分野においてよく知られており、特定の状況に対して方法を選択及び充当することは、当業者が充分になしうる範囲にある。一般に、Furniss et al.(eds.),VOGEL’S ENCYCLOPEDIA OF PRACTICAL ORGANIC CHEMISTRY 5TH ED.,Longman Scientific and Technical Ltd.,Essex,1991,pp.809−816;及びHeller,Acc.Chem.Res.23:128(1990)を参照のこと。
【0049】
CC−1065の細胞傷害性類似体は、CC−1065のシクロプロパピロロインドール(CPI)部分の代わりにアルキル化部分としてシクロプロパベンズインドール(CBI)部分を用いて形成することができる。一例として、CC−1065 CBI類似体としては、以下の式を有する化合物(またはその医薬的に許容される塩)が挙げられるがこれに限定されない。
【化11】
式中、Xは、ハロである。好ましくは、Xは、Cl又はBrであり、より好ましくは、Xは、Brである。
【0050】
X1及びZは、各々独立して、O、S及びNR8から選択され、R8は、H、置換又は非置換のアルキル、置換又は非置換のヘテロアルキル及びアシルから選択されるメンバーである。
【0051】
R4、R4,、R5及びR5,は、独立して、H、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル、非置換ヘテロシクロアルキル、ハロゲン、NO2、NR9R10、NC(O)R9、OC(O)NR9R10、OC(O)OR9、C(O)R9、SR9、OR9、CR9=NR10及びO(CH2)nNR11R11’からなる群から選択されるメンバーであり、R9及びR10は、独立して、H、置換又は非置換のアルキル、置換又は非置換のヘテロアルキル、置換又は非置換のアリール、置換又は非置換のヘテロアリール、置換又は非置換のヘテロシクロアルキル及び置換又は非置換のペプチジルから選択されるか、又はR9及びR10は、それらが結合する窒素原子と任意に結合して、任意に2つ以上のヘテロ原子を含む4〜6員環を有する置換又は非置換のヘテロシクロアルキル環系を形成し、R11及びR11’は、各々独立して、H又は低級アルキルである。
【0052】
R6は、H、置換若しくは非置換の低級アルキル、シアノ又はアルコキシである。好ましくは、R6は、メチル、シアノ又はHである。より好ましくは、R6は、Hである。
【0053】
R7は、H、置換アルキル、非置換アルキル、置換ヘテロアルキル、非置換ヘテロアルキル、ジホスフェート、トリホスフェート、アシル、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、R12、R13及びR14は、独立して、H、置換若しくは非置換のアルキル、置換若しくは非置換のヘテロアルキル及び置換若しくは非置換のアリールから選択されるメンバーであるか、又はR12及びR13は、それらが結合する窒素若しくは炭素原子と任意に結合して、任意に2つ以上のヘテロ原子を含む4〜6員環を有する置換若しくは非置換のヘテロシクロアルキル環系を形成する。
【0054】
CBI CC−1065類似体の例は、共同所有の(co−owned)米国特許出願第10/160,972号;同第10/161,233号;同第10/161,234号、同第11/134,685号及び同第11/134,826号(これらのすべてが参照によって本願明細書に援用される)に記載されている。これらの参考文献はまた、これらの化合物についての合成及び使用の例も記載している。これらの化合物は、治療薬(例えば、薬物)及びプロドラッグとして使用することができる。少なくとも幾つかの実施態様では、CBI CC−1065類似体は、細胞傷害性CBI CC−1065類似体を癌腫細胞などの所望の標的細胞に選択的に送達する医薬組成物において使用するために標的化剤(例えば、抗体、受容体、ペプチド、レクチン、糖、核酸又はそれらの組み合わせ)とコンジュゲートすることができる。
【0055】
これらの化合物によって標的化されうる前癌状態の代表的な例としては、化生、過形成、異形成、結腸直腸ポリープ、光線性角化症(actinic ketatosis)、光線口唇炎、ヒトパピローマウイルス症、白斑症、扁平苔癬(lychen planus)及びボーエン病が挙げられるが、これらに限定されない。
【0056】
これらの化合物によって標的化されうる癌又は腫瘍の代表的な例としては、肺癌、結腸癌、前立腺癌、リンパ腫、メラノーマ、乳癌、卵巣癌、精巣癌、CNS癌、腎癌、腎臓癌、膵癌、胃癌、口腔癌、鼻腔癌、子宮頸癌及び白血病が挙げられるがこれらに限定されない。薬物で処置されるべき腫瘍組織にその薬物を標的化するように特定の標的化剤を選択することができる(すなわち、腫瘍特異的抗原に特異的な標的化剤が選択される)ことは、当業者には容易に明らかであろう。そのような標的化剤の例は、当該技術分野で周知であり、それらの例としては、乳癌を処置するための抗Her2、リンパ腫を処置するための抗CD20、前立腺癌を処置するための抗PSMA、及び非ホジキンリンパ腫を含むリンパ腫を処置するための抗CD30が挙げられるがこれらに限定されない。
【0057】
これらの化合物は、細胞を死滅させる方法を提供する。この方法は、細胞に本発明の化合物の当該細胞を死滅させるのに充分な量を投与することを含む。望ましい実施形態において、化合物は、その細胞を担持している患者に投与される。更なる望ましい実施形態において、投与は、その細胞(例えば、細胞は腫瘍細胞でありうる)を含む腫瘍の成長を遅延又は停止させるのに有用である。成長を遅延させるための投与について、細胞の成長速度は、投与前の成長速度の少なくとも10%は低くなるのが望ましい。更には、成長速度は少なくとも20%、30%、40%、50%、60%、70%、80%、90%遅延され、又は完全に停止するのが望ましい。
【0058】
医薬組成物は、治療上有効な量、すなわち、その意図された目的を達成するのに有効な量の活性成分が含まれている組成物を含む。特定の用途に有効な実際の量はとりわけ、処置すべき状態に依存するものである。有効量の決定は、特に本願明細書の詳細な開示に鑑み、当業者が充分なしうる。
【0059】
本願明細書に記載のいずれの化合物についても、治療上有効な量を最初に細胞培養アッセイにより調べることができる。標的血漿濃度は、細胞成長又は分裂を阻害することができる活性化合物(単数又は複数)の濃度となる。望ましい実施形態において、細胞活性は少なくとも25%阻害される。又は、少なくとも約50%、75%、更には90%以上の細胞活性の阻害を誘発できる活性化合物(単数又は複数)の標的血漿濃度が、現在のところ望ましい。患者における細胞活性の阻害の割合をモニターし、達成される血漿薬剤濃度の適切さを評価でき、所望の阻害割合が得られるように増減して薬用量を調整することができる。
【0060】
当該技術分野において公知であるように、ヒトで使用するのに治療上有効な量は動物モデルから決定することもできる。例えば、ヒト用の投薬量は、動物で有効であると見出されている循環濃度を得るように製剤化できる。ヒトでの薬用量は、細胞阻害をモニターし、上記のように薬用量を増減して調整することができる。
【0061】
治療上有効な投薬量は、類似の薬理学的活性を呈することが知られている化合物に対するヒトのデータから決定することもできる。適用される薬用量は、相対的な生物学的利用能、及び投与化合物の既知化合物と比較した作用強度に基づいて調整することができる。
【0062】
上記の方法、及び当該技術分野において公知である他の方法に基づき、ヒトで最高の効能を達成する薬用量を調整することは、当業者が充分なしうる範囲内にある。
【0063】
局所投与の場合、投与化合物の全身循環濃度は、特別に重要ではない。このような場合、意図した結果を得るのに有効な局部領域での濃度となるように、化合物を投与する。
【0064】
異常な細胞性増殖に関わる疾病の予防及び/又は処置における使用のため、約0.001μM〜20μMの投与化合物の循環濃度が好ましく、更に約0.01μM〜5μMが望ましい。
【0065】
本願明細書に記載の化合物の経口投与に対する患者の薬用量は、望ましくは約1mg/日〜約10,000mg/日、より望ましくは約10mg/日〜約1,000mg/日、最も望ましくは約50mg/日〜約500mg/日の範囲にある。患者の体重に換算すると、典型的な薬用量は、約0.01〜約150mg/kg/日、更に望ましくは約0.1〜約15mg/kg/日、最も望ましくは約1〜約10mg/kg/日の範囲、例えば、5mg/kg/日、又は3mg/kg/日である。
【0066】
少なくとも幾つかの実施態様では、腫瘍成長を遅延又は阻害する患者の薬用量は、1□mol/kg/日以下でありうる。例えば、患者薬用量は、薬物又は薬物コンジュゲート(例えば、抗体−薬物コンジュゲート)の0.9、0.6、0.5、0.45、0.3、0.2、0.15又は0.1□mol/kg/日以下(薬物のモルに対して)でありうる。好ましくは、薬剤又は薬剤コンジュゲートを少なくとも5日間にわたり連日投与することにより、腫瘍の成長を抑止する。少なくとも幾つかの実施態様では、腫瘍はSCIDマウスにおけるヒト型腫瘍である。例として、SCIDマウスは、CB17.SCIDマウスであってもよい(Taconic、Germantown、NYから市販)。
【0067】
投与のその他の態様については、用量及び間隔を、処置すべき特定の臨床適応症に対して有効な投与化合物の血漿レベルをもたらすように、個々に調整することができる。例えば、一実施形態において、本発明による化合物は、1日に何度も、比較的高濃度で投与される。あるいは、本発明の化合物を最低有効量投与し、より頻度の少ない投与計画を用いることがより望ましい。これにより、個々の疾病の重症度に見合う治療計画が提供されることになる。
【0068】
本願明細書における開示を応用して、実質的な毒性を引き起こさず、しかも特定の患者によって示される臨床的症状を処置するのに完全に有効な、治療法を計画することができる。この計画には、化合物の作用強度、相対的生物学的利用能、患者の体重、望まない副作用の存在及び重度、望ましい投与の形態及び選択薬剤の毒性プロファイルなどの因子を考慮することにより、活性化合物を注意深く選択することが含まれる。
【0069】
一般に、CBl部分は、以下の式を有する。
【化12】
式中、置換基は、酸素及び窒素原子に付着することができ、Xは、ハロであり、R6は、H、置換若しくは非置換の低級アルキル、シアノ又はアルコキシである。好ましくは、R6は、H、メチル又はシアノである。より好ましくは、R6は、Hである。更に、Xは、好ましくはCl又はBrであり、より好ましくは、Xは、Brである。一般に、結合部分は、CBI部分のアミン置換基に付着されうる。適当な結合部分の例としては、以下が挙げられるが、これに限定されない。
【化13】
式中、X1、Z、R4、R4,、R5及びR5,は、上で定義したとおりである。
【0070】
適当な結合部分の例は、米国特許出願第10/160,972号;同第10/161,233号;同第10/161,234号、同第11/134,685号及び同第11/134,826号;並びに米国特許第6,534,660号(参照によって本願明細書に援用される)に例示及び記載されている。この式中の適当な結合部分はまた、以下のような複数の縮合環を有する結合部分も含む。
【化14】
式中、Z’は、独立してO、S及びNR8から選択され、R8は、H、置換又は非置換のアルキル、置換又は非置換のヘテロアルキル及びアシルから選択されるメンバーである。
【0071】
R4,,、R4,,,、R5,,及びR5,,,は、独立して、H、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル、非置換ヘテロシクロアルキル、ハロゲン、NO2、NR9R10、NC(O)R9、OC(O)NR9R10、OC(O)OR9、C(O)R9、SR9、OR9、CR9=NR10及びO(CH2)nNR11R11’からなる群から選択されるメンバーであり、R9及びR10は、独立して、H、置換若しくは非置換のアルキル、置換若しくは非置換のヘテロアルキル、置換若しくは非置換のアリール、置換若しくは非置換のヘテロアリール、置換若しくは非置換のヘテロシクロアルキル及び置換若しくは非置換のペプチジルから選択されるか、又はR9及びR10は、それらが結合する窒素原子と任意に結合して、任意に2つ以上のヘテロ原子を含む4〜6員環を有する置換若しくは非置換のヘテロシクロアルキル環系を形成し、R11及びR11’は、各々独立して、H又は低級アルキルである。R15は、H、置換又は非置換のアルキルでありうるか、又はR15及びR4’又はR5’は、結合して、環(例えば、5員環又は6員環)を形成しうる。
【0072】
CC−1065 CBI類似体の形成において有用な1つの中間体化合物は、式(I)を有する。
【化15】
式中、R1及びR2は、各々独立して、H、アルキル、−C(O)OR’、−C(O)NR’R”又は保護基であり、R’及びR”は、独立して、H、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル及び非置換ヘテロシクロアルキルからなる群から選択され、Xは、ハロゲンである。
【0073】
化合物(I)のための1つの従来の合成方法において、出発物質は、1,3−ジヒドロキシナフタレンであり、それを加圧チャンバー(例えば、ボンベ)内でアンモニアと反応させることにより、ナフタレンの3位におけるヒドロキシ基をアミンで置換する。この合成方法の例は、米国特許第6,534,660号及びD.L.Boger et al.,J.Org.Chem.57,2873−2876(1992)(これら両方が参照によって援用される)に見出されうる。このアミノ化反応の後、ヒドロキシ部分及びアミン部分の両方に保護基を付加する。
【0074】
このアミノ化反応は、小規模において許容できる収率でありうるが、通常、実質的に1気圧を超える圧力下(約1.01×105Pa)及び一般に少なくとも1.5気圧の圧力下(1.52×105Pa)において行う加圧反応を含むためにボンベを使用するので、この反応をスケールアップすることは困難でありうる。この合成方法は、スケールアップすると実質的に収率が低下することが見出されている。
【0075】
従来の方法とは対照的に、出発物質を、図1及び2の合成スキームに例示されるように化合物(II)とすることができる。
【化16】
式中、R3は、H又はアルキルである。好ましくは、R3は、C1−5アルキルであり、より好ましくは、メチルである。例えば、4−メトキシ−2−ナフチルアミンは、Aldrich Chemical Company,Inc.,Milwaukee,WIから市販されている。R3がアルキルであり、保護基R1’をヒドロキシ部分及びアミン部分の両方に付加して化合物(III)を形成する場合、その化合物の加水分解が比較的容易である。
【化17】
幾つかの実施態様では、図1及び2に例示されるように、アミン及びヒドロキシ官能基に対して様々な保護基をもたらすことが望ましい。したがって、最初の保護基であるR1’を、これらの部分の1つから(例えば、ヒドロキシ部分から)除去して、第2の異なる保護基であるR2’で置換することにより、化合物(IV)をもたらすことができる。
【化18】
化合物(IV)の1つの特定の例は、以下の式を有する。
【化19】
一方の保護基をそのままにしておきながら、他方の保護基を選択的に除去することができる場合、ヒドロキシル置換基とアミン置換基とで異なる保護基を有することにより、その後の反応ステップを容易にすることができる。あるいは、異なる保護基を最初にヒドロキシル及びアミン置換基に付加することもできる。
【0076】
スキーム1及び2(図1及び2)には、化合物(IV)から化合物(I)を形成する際の残存ステップの一実施態様が例示されている。これらのステップは、例えば、アミン基の窒素を用いて環を形成することを含みうる。これは、例えば、窒素に隣接したアリール環のアルキル化の後の閉環ステップによって達成されうる。一実施態様において、N−ヨードスクシンイミドによる化合物(IV)のヨウ素化により化合物が生成され、その後、それを1,3−ジブロモプロペン又は1,3−ジクロロプロペンを用いてアルキル化することができる。その後、2,2’−アゾビスイソブチロニトリル(AIBN)の存在下で水素化トリブチルスズを用いて閉環を行うことができ、それにより、ラセミのCBI誘導体である化合物(V)が得られる。
【化20】
所望であれば、保護基を除去してCBIを形成することができる。これらの反応ステップにおいて様々な反応物及び触媒を使用することができることが理解されるだろう。例は、Boger,Chemical Reviews,97,787−828(1997,)(参照によって本願明細書に援用される)に見出されうる。
【0077】
クロマトグラフィ法の使用を含む公知のエナンチオマーの分離技術を用いて、ラセミのCBI誘導体を分離することができる。特に有用な1つの技術は、キラルカラムを使用した高圧液体クロマトグラフィ(HPLC)である。例えば、HPLC Chiralcelカラム及びヘキサン/イソプロパノール(99:1)溶出剤を用いてそのようなエナンチオマーの分離を行うことにより、化合物(I)が得られる。
【0078】
化合物(I)の特定の例としては、以下が挙げられるが、これらに限定されない。
【化21】
【0079】
化合物(I)を使用して、例えば、共同所有の米国特許出願第10/160,972号;同第10/161,233号;同第10/161,234号、同第11/134,685号及び同第11/134,826号(これらのすべてが参照によって本願明細書に援用される)に記載されているようなCBI CC−1065類似体を形成することができる。例えば、アミン置換基を脱保護し、脱保護されたアミンと結合部分を含む化合物とを反応させることによって、化合物(I)に結合部分を付加することができる。酸素を脱保護し、それを適切な反応物(単数又は複数)と反応させることによって、さらなる置換基をCBI化合物の酸素原子に付加することができる。
【実施例】
【0080】
当業者は、さらなる詳述なしに、前述の記載を使用して本発明を最大限に利用することができると考えられる。それゆえ、以下の好ましい特定の実施態様は、単なる例示にすぎず、いかなる様式でも本開示の残りの部分を限定すると解釈されるべきでない。前述及び以下の実施例において、すべての温度を摂氏温度で、未修正で示し、すべての部及びパーセントは、別途記載されない限り、それぞれ質量部及び質量パーセントである。
【0081】
実施例1−スキーム1(図1)
N−(tert−ブチルオキシカルボニル)−4−O−(tert−ブチルオキシカルボニル)−2−ナフチルアミン(2)の合成
氷酢酸(9.6mL)及び臭化水素酸水溶液(16mL,48%)中の4−メトキシ−2−ナフチルアミン(230mg,1.33mmol)の溶液をN2下で4時間還流した。少量のサンプル(0.1mL)を酢酸エチル(0.5mL)で希釈し、次いで、水(0.5mL)及びTEA(0.1mL)を加えた。有機層のTLC(20:1DCM/メタノール)では、出発物質は検出されなくなり、それより低い新しいスポットが検出された(Rf=0.1)。溶媒を減圧下で除去し、生成物を真空下で乾燥することにより、中間体である4−ヒドロキシ−2−ナフチルアミンが得られ、それをいかなる精製もすることなく次の工程に使用した。ジオキサン(10mL)中の4−ヒドロキシ−2−ナフチルアミンの溶液に、TEA(1mL)及びジ−tert−ブチルジカーボネート(1.149g,5.27mmol)を加えた。その反応混合物をN2下で4時間還流した。TLC(4:1ヘキサン/酢酸エチル)では、出発物質は検出されなくなり、それより高い新しいスポットが検出された(Rf=0.55)。その反応混合物を酢酸エチル(50mL)で希釈し、水で洗浄した。その水層を酢酸エチル(2×50mL)で抽出し、有機層を併せ、ブラインで洗浄した。その有機物を無水Na2SO4で乾燥し、濾過し、減圧下で濃縮することにより、N−(tert−ブチルオキシカルボニル)−4−O−(tert−ブチルオキシカルボニル)−2−ナフチルアミン(2,80%収率)を油状物として得た。
【0082】
化合物3の合成:
アセトン(10mL)中の化合物2の溶液に、NaOH水溶液(10mL,1M)を加えた。その反応混合物を室温で一晩撹拌した。TLC(4:1ヘキサン/酢酸エチル)では、出発物質は検出されなくなり、それより低い新しいスポットが検出された。その反応混合物を酢酸エチル(50mL)で抽出し、水で洗浄した。水層を酢酸エチル(2×50mL)で抽出し、有機層をブラインで洗浄し、無水Na2SO4で乾燥し、濃縮した。その残渣を、ヘキサン溶液中の10−20%酢酸エチルを用いた10gのシリカゲルカラムにおいて精製することにより、化合物3(181mg,53%)を油状物として得た。
【0083】
化合物4の合成:
窒素雰囲気下の無水DMF(50ml)中の化合物3(5g,19.3mmol)の溶液を、臭化ベンジル(4g,23.1mol)、炭酸カリウム(3.7g,27mol)及びヨウ化テトラブチルアンモニウム(70mg,0.01mmol)で処理した。その反応混合物を室温で8時間撹拌した。その反応混合物を減圧下で濃縮した。クロマトグラフィを用いた分離(4×10cm SiO2,10−20%EtOAc−ヘキサン勾配溶出)によって、純粋な化合物4(5.48g,83%)をクリーム色の粉末として得た。1H NMR(CDCl3,400MHz,ppm)8.22(d,1H J=8.1Hz,C5−H),7.68(d,1H,J=8.2Hz,C8−H),7.3−7.5(m,8H,C1−H,C6−H,C7−H,CH2C6H5),7.06(d,1H,J=1.1Hz,C3−H),6.62(br s,1H,NH),5.23(S,2H,OCH2(C6H5),1.55(s,9H,OC(CH3)3).
【0084】
化合物5の合成:
スターラーバー及びゴム隔壁を備えた1000ml丸底フラスコにて化合物4(13g,0.0372mol)及びTHF(300ml)を混合した。透明の黄色溶液を、ドライアイス浴を用いて窒素雰囲気下において−20Cに冷却した。p−トルエンスルホン酸(0.10g,0.0005mol)をその反応物に加え、その溶液を10分間撹拌した。N−ヨードスクシンイミド(10g,0.0446mol)をTHF(50ml)に溶解し、カニューレを用いてその反応物に加えた(約1時間)。その溶液を氷浴中で2時間撹拌したところ、茶色がかった色を呈した。次いで、その溶液を氷浴から取り出し、窒素下で1.5時間室温に温めた。TLC(2:1ヘキサン/DCM)では、出発物質は検出されなくなり、それより高い新しいスポットが検出された。その反応物を飽和ΝaHCO3(200ml)でクエンチし、白色固体が形成された。その溶液を10分間撹拌した後、その反応物にEtOAc(200ml)及び水(100ml)を加えた。水層をEtOAc(2×100ml)で抽出し、有機層を併せ、ブライン(100ml)で抽出した。その有機層をMgSO4で乾燥し、濾過し、真空下で濃縮することにより、暗赤茶色の固体が得られた。その固体を、溶離剤として2:1ヘキサン/DCMを用いるカラムクロマトグラフィで精製することにより、化合物5(14g,79%)を茶色固体として得た。
【0085】
化合物6の合成:
スターラーバー及び窒素導入口を備えた500ml丸底フラスコにて、化合物5(22.5g,0.0473mol)及び無水DMF(250ml)を混合した。その黄色−橙色溶液を、窒素雰囲気下において氷/塩浴を用いて0℃に冷却した。その反応物にNaH(60%,5.6g,0.146mol)を一度に加えた。その溶液は濁り、気体を発生した。その反応物を氷浴中で15分間撹拌し、次いで、その氷浴を取り除き、その溶液を更に15分間撹拌して、その反応物にシス/トランス−1,3−ジブロモプロペン(14ml,0.14mol)を、注射器を用いて少しずつ加えた。その反応物を窒素下で室温にて1時間撹拌したところ、その反応物は、濁った茶色を呈した。温度を40℃に上げた。その反応物を室温に冷却した。TLC(4:1ヘキサン/EtOAc)では、出発物質は検出されなくなり、それより低い新しいスポットが検出された。その反応物を水(500ml)でクエンチした。その水層をEtOAc(4×100ml)で抽出し、有機層をブラインで洗浄した(2×75ml)。その有機層をMgSO4で乾燥し、濾過し、真空下で濃縮することより、茶色油状物が得られた。その生成物を、溶離剤として1:1DCM/ヘキサンを用いるカラムクロマトグラフィで精製することにより、化合物6(25g,89%)を茶色油状物として得た。
【0086】
化合物7の合成:
スターラーバー、温度プローブ、還流冷却器及び窒素導入口を備えた1000ml三首丸底フラスコにて、化合物6(25g,0.0421mol)、トルエン(500ml)、2,2’−アゾビス(2−メチルプロピオニトリル)(0.15g,0.0009mol)及び水素化トリブチルスズ(3.4ml,0.0126mol)を混合した[注射器を用いて]。その溶液に15分間窒素を泡立たせながら通し、次いで、その反応物を窒素下で80℃に加熱した。15分間80℃に加熱した後、その反応物に注射器を用いて水素化トリブチルスズ(3.4ml,0.0126mol)を加えた。更に15分間加熱した後、その反応物に注射器を用いて水素化トリブチルスズ(3.4ml,0.0126mol)を加えた。更に15分後、その反応物に注射器を用いて水素化トリブチルスズ(3.4ml,0.0126mol)を加えた。加えた水素化トリブチルスズの総量は、13.6ml,0.0505molであった。その反応物を30分間80℃に加熱し、次いで、室温に冷却した。TLC(10%EtOAc/ヘキサン)では、出発物質及びそれより高い新しいスポットが検出された。その溶液を真空下で濃縮することにより、黄色固体が得られた。その固体を、溶離剤として1:1ジクロロメタン(dicloromethane)/ヘキサンを用いるカラムクロマトグラフィで精製することにより、黄色固体が得られた。その固体を、ヘキサンから再結晶化する(200ml、30分間45Cにし、冷蔵庫にて2時間冷却し、濾過により回収し、真空下で乾燥させる)ことにより、化合物7(11.70g,59%収率)を淡黄色固体として得た。NMR(1H,CDCl3,400MHz):□1.61(9H,s,C−(CH3)3);3.30(1H,t,J=26Hz,CH−CH2−N);3.82(1H,d,J=26Hz,Br−CH2−CH);4.04(1H,d,J=19Hz,Br−CH2−CH)4.14(1H,t,J=26Hz,CH−CH2−N);4.21(1H,m,CH2−CH−CH2);5.26(2H,s,O−CH2−C6H5);7.3−7.55(8H,m,O−CH2−C6H5,C10H5);7.63(1H,d,J=21Hz,C10H5);8.3(1H,d,J=21Hz,C10H5)
【0087】
化合物7の分割:
ラセミの化合物7をDCM(50mg,1ml)に溶解した。次いで、その溶液をヘキサン(9ml)で希釈した。次いで、その溶液をChiralcel OD prepカラム(10ミクロン,20×250mm)に充填し、ヘキサン/イソプロパノール(99:1,15ml/分)を用いて分離した。第1のエナンチオマー(7a)は、10〜15分に溶出し、第2のエナンチオマー(7b)は、17.5〜25分に溶出する。分析カラム(Chiralcel OD,0.46×25cm,20マイクロメートル)は、7aについては7.71分及び7bについては12.9分の保持時間を与える(99:1ヘキサン/IPA,1ml/分,15分間の流入)。NMR(1H,CDCl3,400MHz):□1.61(9H,s,C−(CH3)3);3.30(1H,t,J=26Hz,CH−CH2−N);3.82(1H,d,J=26Hz,Br−CH2−CH);4.04(1H,d,J=19Hz,Br−CH2−CH)4.14(1H,t,J=26Hz,CH−CH2−N);4.21(1H,m,CH2−CH−CH2);5.26(2H,s,O−CH2−C6H5);7.3−7.55(8H,m,O−CH2−C6H5,C10H5);7.63(1H,d,J=21Hz,C10H5);8.3(1H,d,J=21Hz,C10H5)
【0088】
実施例2−スキーム2(図2)
合成方法の手順は、化合物5の合成を通じて実施例1において上に記載したとおりである。化合物8の合成スターラーバー及び窒素導入口を備えた250ml丸底フラスコにて、化合物5(8.4g,0.0177mol)及び無水DMF(125ml)を混合した。その黄色−橙色溶液を、氷/塩浴を用いて窒素雰囲気下において0℃に冷却した。その反応物にNaH(60%,2.22g,0.0554mol)を一度に加えた。その溶液は濁り、気体が発生した。その反応物を氷浴中で15分間撹拌し、次いで、氷浴を取り除き、その溶液を更に15分間撹拌した。その反応物にシス/トランス−1,3−ジクロロプロペン(5.3ml,0.0571mol)を、注射器を用いて少しずつ加えた。その反応物を窒素下で室温にて3時間撹拌したところ、濁った茶色を呈した。TLC(4:1ヘキサン/EtOAc)では、出発物質は検出されなくなり、それより低い新しいスポットが検出された。その反応物を水(250ml)でクエンチした。水層をEtOAc(3×100ml)で抽出し、有機層をブラインで洗浄した(2×50ml)。その有機層をMgSO4で乾燥し、濾過し、真空下で濃縮することより、茶色油状物が得られた。その生成物を、溶離剤として1:1DCM/ヘキサンを用いるカラムクロマトグラフィで精製することにより、(E/Z)−tert−ブチル4−(ベンジルオキシ)−1−ヨードナフタレン−2−イル(3−クロロアリル)カルバメート(8)(9g,93%)を黄色油状物として得た。
【0089】
化合物9の合成:
スターラーバー、温度プローブ、還流冷却器及び窒素導入口を備えた500ml三首丸底フラスコにて、化合物8(9g,0.0164mol)、トルエン(200ml)、2,2’−アゾビス(2−メチルプロピオニトリル)(0.15g,0.0009mol)及び水素化トリブチルスズ(1.5ml,0.0056mol)を混合した[注射器を用いて]。その溶液に15分間窒素を泡立たせながら通し、次いで、その反応物を窒素下で80℃に加熱した。15分間80℃に加熱した後、その反応物に注射器を用いて水素化トリブチルスズ(1.5ml,0.0056mol)を加えた。更に15分間加熱した後、その反応物に注射器を用いて水素化トリブチルスズ(1.5ml,0.0056mol)を加えた。更に15分後、その反応物に注射器を用いて水素化トリブチルスズ(1.0ml,0.0037mol)を加えた。加えた水素化トリブチルスズの総量は、5.5ml,0.0204molであった。その反応物を30分間80℃に加熱し、次いで、室温に冷却した。TLC(10%EtOAc/ヘキサン)では、出発物質は検出されなくなり、それより高い新しいスポットが検出された。その溶液を真空下で濃縮することにより、黄色油状物が得られた。その油状物を、溶離剤として100%ヘキサン、5%EtOAc/ヘキサン、10%EtOAc/ヘキサンを用いるカラムクロマトグラフィで精製することにより、淡黄色固体が得られた。その固体を、ヘキサンから再結晶化する(100ml、30分間45Cにし、冷蔵庫にて2時間冷却し、濾過により回収し、真空下で乾燥させる)ことにより、化合物9を(4.16g,60%収率)白色固体として得た。
【0090】
化合物9の分割:
ラセミの化合物9をDCM(50mg,1ml)に溶解した。次いで、その溶液をヘキサン(9ml)で希釈した。次いで、その溶液をChiralcel OD prepカラム(10ミクロン,20×250mm)に充填し、ヘキサン/イソプロパノール(99:1,15ml/分)を用いて分離した。第1のエナンチオマー(9a)は、11.5〜15分に溶出し、第2のエナンチオマー(9b)は、17.5〜25分に溶出する。分析カラム(Chiralcel OD,0.46×25cm,20ミクロン)は、9aについては6.5分及び9bについては10.6分の保持時間を与える(99:1ヘキサン/IPA,1ml/分,15分間の流入)。NMR(1H,CDCl3,400MHz):d 1.61(9H,s,C−(CH3)3);3.44(1H,t,J=25Hz,CH−CH2−N);3.9−4.0(2H,m,Cl−CH2−CH);4.12(1H,t,J=26Hz,CH−CH2−N);4.25(1H,m,CH2−CH−CH2);5.26(2H,s,O−CH2−C6H5);7.2−7.5(8H,m,O−CH2−C6H5,C10H5);7.63(1H,d,J=21Hz,C10H5);8.3(1H,d,J=21Hz,C10H5)
【0091】
実施例3−スキーム3(図3)
合成方法の手順は、化合物3の合成を通じて実施例1において上に記載したとおりである。
【0092】
化合物10の合成:
無水DCM(20ml)中のtert−ブチル−4−ヒドロキシナフタレン−2−イルカルバメート(3)(500mg,2.89mmol)、4−メチル−1−ピペラジンカルボニルクロリド塩酸塩(858mg,4.34mmol)、無水ピリジン(4.98ml,57.8mmol)及びアリルアルコール(4.98ml,73.2mmol)の溶液を室温で一晩撹拌した。TLC(9:1DCM/MeOH)では、出発物質は検出されなくなり、それより一層低いスポットが検出された。その反応混合物を水でクエンチした。水層をEtOAc(3×50ml)で抽出し、有機層をブラインで洗浄した(2×50ml)。その有機層をNa2SO4で乾燥し、濾過し、真空下で濃縮することにより、茶色油状物が得られた。その粗生成物を、DCM中の1−5%メタノールを用いるカラムクロマトグラフィで精製することにより、10(602mg,82%)を黄色固体として得た。1HNMR(DMSO−d6)δ9.68(s,1H),7.91(s,1H),7.80(d,1H),7.69(s,1H),7.47(t,1H),7.42(s,1H),7.39(t,1H),3.78(s,2H),3.42(s,2H),2.44(s,2H),2.39(s,2H),2.21(s,3H),1.50(s,9H)
【0093】
化合物11の合成:
無水THF(5ml)中の化合物10(82mg,0.21mmol)、p−トルエンスルホン酸(10mg,0.05mmol)及びN−ヨードスクシンイミド(96mg,0.43mmol)の溶液を室温で一晩撹拌した。TLC(9:1DCM/MeOH)では、少量の出発物質及びそれより高いスポットが検出された。その反応混合物を飽和ΝaHCO3(10ml)でクエンチした。室温で10分間撹拌した後、その反応混合物をEtOAc(3×20ml)で抽出し、その有機層をブラインで洗浄した(2×20ml)。その有機層をNa2SO4で乾燥し、濾過し、真空下で濃縮することにより、茶色油状物が得られた。その粗生成物を、DCM中の1−5%メタノールを用いるカラムクロマトグラフィで精製することにより、11(52mg,48%)を黄色油状物として得た。1HNMR(DMSO−d6)δ8.81(s,1H),8.14(d,1H),7.79(d,1H),7.65(t,1H),7.58(t,1H),7.45(t,1H),3.78(s,2H),3.42(s,2H),2.44(s,2H),2.39(s,2H),2.21(s,3H),1.50(s,9H)
【0094】
化合物12の合成:
無水DMF(5ml)中の化合物11(102mg,0.2mmol)の溶液を氷浴中で冷却した。その反応物に、水素化ナトリウム(鉱油中60%,32mg,0.8mmol)を加えた。その反応混合物を0℃で15分間及び室温で15分間撹拌した。その反応物にシス/トランス−1,3−ジクロロプレペン(Dichloroprepene)(83.36μl,0.9mmol)を加えた。その反応混合物を室温で1時間撹拌した。TLC(9:1DCM/MeOH)では、出発物質は検出されなかった。その反応混合物を水でクエンチした。その水層をEtOAc(3×10ml)で抽出し、その有機層をブラインで洗浄した(2×10ml)。その有機層をNa2SO4で乾燥し、濾過し、真空下で濃縮することにより、茶色油状物が得られた。その粗生成物を、DCM中の1−5%メタノールを用いるカラムクロマトグラフィで精製することにより、12(82mg,70%)を黄色固体として得た。1HNMR(DMSO−d6)δ8.20(d,1H),7.82(d,1H),7.62(m,2H),7.38(d,1H),6.38(m,1H),6.18(m,1H),3.98−4.46(dd,2H),3.78(s,2H),3.42(s,2H),2.44(s,2H),2.39(s,2H),2.21(s,3H),1.50(s,9H)
【0095】
化合物13の合成:
無水トルエン(3ml)中の12(82mg,0.14mmol)の溶液に、乾燥窒素を15分間泡立たせた。その反応物に水素化トリブチルスズ(47.1μl,0.18mmol)及び2,2’−アゾビスイソブチロニトリル(10mg,0.06mmol)を加えた。その反応混合物を窒素下で15分間80℃に加熱した。TLC(9:1DCM/MeOH)では、新しい青いスポットが検出され、出発物質は検出されなかった。その反応混合物を真空下で濃縮することにより、黄色油状物が得られた。その粗生成物を、DCM中の1−5%メタノールを用いるカラムクロマトグラフィで精製することにより、13(52mg,82%)を白色固体として得た。1HNMR(DMSO−d6)δ7.92(d,1H),7.83(m,1H),7.78(d,1H),7.58(t,1H),7.42(t,1H),4.20(m,2H),4.04(m,2H),3.92(m,1H),3.78(s,2H),3.42(s,2H),2.44(s,2H),2.39(s,2H),2.21(s,3H),1.50(s,9H).
【0096】
化合物13の分割:
ラセミの化合物13をメタノールに溶解した。次いで、その溶液をCHIRALPAK AD prepカラム(20ミクロン,20×250mm)に充填し、メタノール(15ml/分)を用いて分離した。第1のエナンチオマー(13a)は、5.1分から溶出し、第2のエナンチオマー(13b)は、7.1分から溶出する。1HNMR(DMSO−d6)δ7.92(d,1H),7.83(m,1H),7.78(d,1H),7.58(t,1H),7.42(t,1H),4.20(m,2H),4.04(m,2H),3.92(m,1H),3.78(s,2H),3.42(s,2H),2.44(s,2H),2.39(s,2H),2.21(s,3H),1.50(s,9H)
【0097】
実施例4−CBI CC−1065類似体の合成
【化22】
【0098】
化合物(14)の合成:
MeOH/CH2Cl2(1/2,10ml)中の9b(100mg,0.24mmol)及び10%Pd−C(35mg)の溶液を真空中で40秒間脱気した。得られた混合物を水素雰囲気下に置き、25℃で7時間撹拌した。その反応混合物をセライトで濾過した(CH2Cl2洗浄)。溶媒を真空中で除去した。EtOAc/Hex(2/8)を用いて溶出する、シリカゲルによるクロマトグラフィから14(77mg,98%)が得られた。1NMR DMSO−d6)δ10.36(s,1H),8.04(d,1H,J=8.2Hz),7.72(d,1H,J=8.2Hz),7.61(br s,1H),7.45(t,1H,J=8.4Hz),7.261(t,1H,J=8.4Hz),4.06(m,4H),3.73(m,1H),1.52(s,9H)
【0099】
化合物(16)の合成:
4M HCl−EtOAc(5ml)中の14(35mg,0.1mmol)の溶液をAr下、25℃にて30分間撹拌した。溶媒を真空中で除去した。その残渣に、5−アセチルインドン−2−カルボン酸(24.4mg,0.12mmol)を加えた。DMF(3ml)中のEDC(22.9mg,0.12mmol)の溶液を加え、その反応混合物を25℃で5時間撹拌した。溶媒を除去した。その粗生成物を、CH2Cl2中の10%MeOHを用いて溶出する、シリカゲルによるクロマトグラフィに供することにより、16(40.7mg,93%)が得られた。1HNMR DMSO−d6)δ12.13(s,1H),10.47(s,1H),8.45(s,1H),8.10(d,1H,J=8.4Hz),7.96(br s,1H),7.85(d,2H,J=8.4Hz),7.54(d,1H,J=8.4Hz),7.51(t,1H,J=8.2Hz),7.36(t,1H,J=7.6),7.35(s,1H),4.81(t,1H,11.2Hz),4.54(dd,1H,8.8Hz),4.23(m,1H),4.01(dd,1H,J=10.2Hz),3.86(dd,1H,J=10.7Hz),2.61(s,3H)
【0100】
化合物(17)の合成:
4−メチル−1−ピペラジンカルボニルクロリド塩酸塩(19.9mg,0.1mmol)を、乾燥塩化メチレン(4ml)中の3%アリルアルコール中の16(20mg,0.05mmol)及び無水ピリジン(25μml,0.3mmol)の溶液に加え、その混合物を16時間撹拌した。その粗生成物のシリカゲルによる精製により、17(23.6mg,91%)が得られた。1NMR DMSO−d6)δ12.03(s,1H),8.41(s,1H),8.21(s,1H),8.01(d,1H,J=8.4Hz),7.88(d,1H,J=8.4Hz),7.82(dd,1H,J=8.4Hz),7.58(t,1H,J=8.1Hz),7.51(d,1H,J=8.4Hz),7.46(t,1H,J=7.6Hz),7.37(s,1H),4.86(t,1H,J=10.8Hz),4.57(dd,1H,J=10.8Hz),4.38(m,1H),4.06(dd,1H,J=10.8Hz),3.86(dd,1H,J=11Hz),3.41(br,4H),3.29(br,4H),2.82(s,3H),2.57(s,3H)
【0101】
化合物(19)の合成:
乾燥塩化メチレン(1ml)中の5%酢酸中の17(13mg,24μmol)及びリンカー18(16.9mg,31μmol)の溶液を25℃で30分間撹拌した。溶媒を真空中で完全に除去し、HPLC(SymmetryPrep C18,7μm,19×150mmカラム)で精製することにより、19(18.5mg,81%)が得られた。MS:C48H57ClN8O11に対する算出値(M+H)m/z 958.38,実測値958.10
【0102】
実施例5:増殖アッセイ
確立された3H−チミジン増殖アッセイを用いて、本発明の細胞傷害性化合物の生物学的活性をアッセイすることができる。これは、外来性の放射標識された3H−チミジンの取り込みを測定することによってDNA合成を評価するので、細胞増殖を定量化するための簡便な方法である。このアッセイは再現性が高く、多数の化合物に適応できる。
【0103】
アッセイでは、最初に前骨髄性白血病細胞(HL−60)を、熱不活性ウシ胎仔血清(FCS)を10%含有するRPMI培地で培養した。実験の第1日目に、細胞を回収し、洗浄し、10%FCSを含有するRPMI中に0.5×106細胞/mlの濃度に再懸濁する。100μlの細胞懸濁液を96ウェルプレートに加える。ドキソルビシン(ポジティブコントロール)又は試験化合物の系列希釈(3倍濃縮系列)を調製し、ウェル当たり化合物100μlで添加した。最終的に、1ウェルあたり10μlの100μCi/mlの3H−チミジンを添加し、そのプレートを24時間インキュベートする。プレートから、96ウェルハーヴェスタ(Packard Instruments社)を使用して回収し、Packard Top Count counterで計測した。Prismソフトウェアを使用して、薬物のモル濃度の関数として4パラメータによるロジスティック曲線を3H−チミジン取り込みに当てはめ、IC50値を決定した。
【0104】
CBI CC−1065類似体(例えば、実施例4の化合物19)は、一般に、上記アッセイにおいて、約1pMから約100nM、好ましくは約10pMから約10nMというIC50値を有する。
【0105】
実施例6:抗体への薬物分子のコンジュゲーション
この実施例では、本発明の薬物分子(他の基(例えば、スペーサー、反応性の官能基など)を任意に含む)を、標的化剤としての抗体とコンジュゲートするための反応条件及び方法論について記載する。その条件及び方法論は、例示することだけを意図しており、非限定的なものである。薬物分子を抗体とコンジュゲートするための他のアプローチは、当該技術分野で公知である。
【0106】
本願明細書中に記載されるコンジュゲーション方法は、抗体のリシンと2−イミノチオランとの反応の後、薬物−リンカー分子と活性マレイミド基との反応による、抗体への遊離チオール基の導入に基づくものである。最初に、コンジュゲートされる抗体の緩衝液を、50mM NaCl、2mM DTPA,pH8.0を含有する0.1Mリン酸緩衝液pH8.0に交換し、5〜10mg/mlに濃縮する。その抗体に2−イミノチオランを加えることによりチオール化を達成する。加える2−イミノチオランの量は、予備実験において決定し、抗体によって変化するものである。抗体に加える2−イミノチオランの量を徐々に増やし、その後、その抗体を室温で1時間インキュベートするという力価測定である予備実験において、その抗体を、Sephadex G−25カラムを用いて50mM HEPES緩衝液pH6.0中に脱塩し、導入されたチオール基の数を、ジチオジピリジン(DTDP)との反応によって迅速に決定する。チオール基とDTDPとの反応により、チオピリジンが遊離し、それを324nmにおいてモニターする。0.5〜1.0mg/mlというタンパク質濃度のサンプルを使用することができる。280nmでの吸光度を用いて、サンプル中のタンパク質の濃度を正確に決定し、次いで、各サンプルのアリコート(0.9ml)を0.1mlのDTDP(エタノール中5mM原液)と室温にて10分間インキュベートする。緩衝液のみ+DTDPのブランクサンプルもまた一緒にインキュベートする。10分後、324nmにおける吸光度を測定し、19800M−1のチオピリジンに対する吸光係数を用いて、存在するチオールの数を定量する。
【0107】
通常、1抗体あたり3つのチオール基というチオール化レベルが望ましい。例えばこれは、1つの特定の抗体を用いて、モル濃度が15倍過剰の2−イミノチオランを加え、その後、室温で1時間インキュベートすることによって達成することができる。それゆえ、コンジュゲートされる抗体を、所望のモル比率で2−イミノチオランとともにインキュベートし、次いで、コンジュゲーション緩衝液(5mMグリシン、3%グリセロール及び2mM DTPAを含有する50mM HEPES緩衝液pH6.0)中に脱塩する。導入されたチオールの数を上に記載したように定量する間は、このチオール化された材料を氷上で維持する。
【0108】
導入されたチオールの数を検証した後、活性マレイミド基を含む薬物分子(例えば、実施例3の化合物15)を、1チオールあたり3倍過剰のモル濃度で加えた。
【0109】
コンジュゲーション反応は、最終濃度が5%のエチレングリコールジメチルエーテル(または適当な代替の溶媒)を含むコンジュゲーション緩衝液中で行う。通常、薬物原液を90%エチレングリコールジメチルエーテル、10%ジメチルスルホキシドに溶解する。抗体に加える場合、その原液を、最終濃度を5%とするのに十分なエチレングリコールジメチルエーテルを有するチオール化抗体に直接加えるか、又は、最終濃度が10%のエチレングリコールジメチルエーテルを含むコンジュゲーション緩衝液に予め希釈した後、等容積のチオール化抗体を加える。コンジュゲーション反応物を、室温で2時間、混合しながらインキュベートする。インキュベート後、その反応混合物を14000RPMで15分間遠心し、精製をすぐに行わない場合はpHを7.2に調整することができる。コンジュゲートの精製は、多数の方法を用いるクロマトグラフィによって達成することができる。コンジュゲートは、5mMグリシン、50mM NaCl及び3%グリセロールを含有する50mM HEPES緩衝液pH7.2で予め平衡化されたSephacryl S200カラムにおけるサイズ排除クロマトグラフィを用いて精製することができる。クロマトグラフィは、28cm/hという線流速で行うことができる。コンジュゲートを含む画分を回収し、プールし、濃縮することができる。あるいは、精製は、イオン交換クロマトグラフィによって達成することができる。条件は、抗体によって異なり、各場合において最適化する必要がある。例えば、抗体−薬物コンジュゲート反応混合物を、50mM HEPES、5mMグリシン、3%グリセロール,pH6.0で予め平衡化されたSP−Sepharoseカラムに適用することができる。その抗体コンジュゲートを、平衡化緩衝液中の0〜1M NaClの勾配を用いて溶出することができる。コンジュゲートを含む画分をプールすることができ、pHを7.2に調節することができ、必要であれば、そのサンプルを濃縮することができる。
【0110】
本願明細書中で引用されたすべての特許出願、特許及び刊行物の全開示内容は、参照によって本願明細書に援用される。
【0111】
前述の実施例において使用された反応物及び/又は使用条件の代わりに一般的又は特に記載された反応物及び/又は本発明の使用条件を用いることによって同様に首尾よく前述の実施例を繰り返すことができる。
【0112】
前述の記載から、当業者は、本発明の本質的な特徴を容易に確認することができ、また、本発明の技術範囲から逸脱することなく、本発明を様々な用途及び条件に適合するように本発明の様々な変更及び改変を行うことができる。
【図面の簡単な説明】
【0113】
【図1】CBI CC−1065類似体の調整法の一態様を示す合成経路図である。
【図2】CBI CC−1065類似体の調整法の別の態様を示す合成経路図である。
【図3】CBI CC−1065類似体の調整法の第三の態様を示す合成経路図である。
【特許請求の範囲】
【請求項1】
化合物(I)又はその塩の合成方法
【化1】
(式中、R1及びR2は、各々独立して、H、アルキル、−C(O)OR’、−C(O)NR’R”又は保護基であり、R’及びR”は、独立して、H、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル及び非置換ヘテロシクロアルキルからなる群から選択され;R6は、H、置換若しくは非置換の低級アルキル、シアノ又はアルコキシであり;Xは、ハロゲンである)であって、保護基R1’及びR2’が、化合物(II)に付加され、
【化2】
化合物(III)を形成し
【化3】
(式中、R3は、アルキルである。)、化合物(III)のアミン窒素を含む5員環が生成される、前記合成方法。
【請求項2】
R3がメチルである、請求項1に記載の方法。
【請求項3】
XがCl又はBrである、請求項1に記載の方法。
【請求項4】
XがBrである、請求項3に記載の方法。
【請求項5】
R1’及びR2’が異なる保護基である、請求項1に記載の方法。
【請求項6】
R1’及びR2’の付加がR1’を化合物(II)に付加して化合物(III’)を形成し、
【化4】
ヒドロキシ置換基の保護基R1’を保護基R2’で置換する、請求項5に記載の方法。
【請求項7】
R1’がtert−ブチルオキシカルボニルである、請求項5に記載の方法。
【請求項8】
R2’が−CH2Phである、請求項7に記載の方法。
【請求項9】
R1’及びR2’が同一である、請求項1に記載の方法。
【請求項10】
さらに、5員環形成後にR1’及びR2’が水素により置換される、請求項1に記載の方法。
【請求項11】
R1’及びR1が同一であり、R2’及びR2が同一である、請求項1に記載の方法。
【請求項12】
前記5員環の形成が化合物(III)のアミン置換基に隣接する炭素のヨウ素化と、その後に続く1,3−ジハロプロペンを用いたアルキル化とを含んでなる、請求項1に記載の方法。
【請求項13】
以下の式を有するCBI CC−1065類似体又はその医薬的に許容される塩の合成方法であって、
【化5】
(式中、Xは、ハロであり;X1及びZは、各々独立して、O、S及びNR8から選択され、R8は、H、置換又は非置換のアルキル、置換又は非置換のヘテロアルキル及びアシルから選択されるメンバーであり;R4、R4,、R5及びR5,は、独立して、H、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル、非置換ヘテロシクロアルキル、ハロゲン、NO2、NR9R10、NC(O)R9、OC(O)NR9R10、OC(O)OR9、C(O)R9、SR9、OR9、CR9=NR10及びO(CH2)nNR11R11’からなる群から選択されるメンバーであり、R9及びR10は、独立して、H、置換若しくは非置換のアルキル、置換若しくは非置換のヘテロアルキル、置換若しくは非置換のアリール、置換若しくは非置換のヘテロアリール、置換若しくは非置換のヘテロシクロアルキル及び置換若しくは非置換のペプチジルから選択されるか、又はR9及びR10は、それらが結合する窒素原子と任意に結合して、任意に2つ以上のヘテロ原子を含む4〜6員環を有する置換若しくは非置換のヘテロシクロアルキル環系を形成し、R11及びR11’は、各々独立して、H又は低級アルキルであり;R6は、H、置換若しくは非置換の低級アルキル、シアノ又はアルコキシであり;R7は、H、置換アルキル、非置換アルキル、置換ヘテロアルキル、非置換ヘテロアルキル、ジホスフェート、トリホスフェート、アシル、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、R12、R13及びR14は、独立して、H、置換若しくは非置換のアルキル、置換若しくは非置換のヘテロアルキル及び置換若しくは非置換のアリールから選択されるメンバーであるか、又はR12及びR13は、それらが結合する窒素又は炭素原子と任意に結合して、任意に2つ以上のヘテロ原子を含む4〜6員環を有する置換若しくは非置換のヘテロシクロアルキル環系を形成する。)であって、保護基R1’及びR2’を化合物(II)に付加して
【化6】
化合物(III)を形成し
【化7】
(式中、R3はアルキルである。)、そして、化合物(III)のアミン窒素を含む5員環を形成し、以下を含む
【化8】
結合部分を、化合物(III)に付加することを含んでなる、前記方法。
【請求項14】
前記5員環の形成が化合物(III)のアミン置換基に隣接する炭素のヨウ素化と、その後に続く5員環形成前の1,3−ジハロプロペンを用いたアルキル化とを含んでなる、請求項13に記載の方法。
【請求項15】
前記結合部分の付加が保護基R1’の脱離を含んでなる、請求項13に記載の方法。
【請求項16】
前記結合部分の付加が、前記結合部分のアミン置換基への付加を更に含んでなる、請求項15に記載の方法。
【請求項17】
R6がHである、請求項13に記載の方法。
【請求項18】
式(I)の化合物又はその医薬的に許容される塩
【化9】
(式中、R1及びR2は、各々独立して、H、アルキル、−C(O)OR’、−C(O)NR’R”又は保護基であり、R’及びR”は、独立してH、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル及び非置換ヘテロシクロアルキルからなる群から選択され、R6は、H、置換若しくは非置換の低級アルキル、シアノ又はアルコキシである。)。
【請求項19】
R1及びR2が異なる保護基である、請求項18記載の化合物。
【請求項20】
以下の化合物である、請求項18記載の化合物。
【化10】
【請求項1】
化合物(I)又はその塩の合成方法
【化1】
(式中、R1及びR2は、各々独立して、H、アルキル、−C(O)OR’、−C(O)NR’R”又は保護基であり、R’及びR”は、独立して、H、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル及び非置換ヘテロシクロアルキルからなる群から選択され;R6は、H、置換若しくは非置換の低級アルキル、シアノ又はアルコキシであり;Xは、ハロゲンである)であって、保護基R1’及びR2’が、化合物(II)に付加され、
【化2】
化合物(III)を形成し
【化3】
(式中、R3は、アルキルである。)、化合物(III)のアミン窒素を含む5員環が生成される、前記合成方法。
【請求項2】
R3がメチルである、請求項1に記載の方法。
【請求項3】
XがCl又はBrである、請求項1に記載の方法。
【請求項4】
XがBrである、請求項3に記載の方法。
【請求項5】
R1’及びR2’が異なる保護基である、請求項1に記載の方法。
【請求項6】
R1’及びR2’の付加がR1’を化合物(II)に付加して化合物(III’)を形成し、
【化4】
ヒドロキシ置換基の保護基R1’を保護基R2’で置換する、請求項5に記載の方法。
【請求項7】
R1’がtert−ブチルオキシカルボニルである、請求項5に記載の方法。
【請求項8】
R2’が−CH2Phである、請求項7に記載の方法。
【請求項9】
R1’及びR2’が同一である、請求項1に記載の方法。
【請求項10】
さらに、5員環形成後にR1’及びR2’が水素により置換される、請求項1に記載の方法。
【請求項11】
R1’及びR1が同一であり、R2’及びR2が同一である、請求項1に記載の方法。
【請求項12】
前記5員環の形成が化合物(III)のアミン置換基に隣接する炭素のヨウ素化と、その後に続く1,3−ジハロプロペンを用いたアルキル化とを含んでなる、請求項1に記載の方法。
【請求項13】
以下の式を有するCBI CC−1065類似体又はその医薬的に許容される塩の合成方法であって、
【化5】
(式中、Xは、ハロであり;X1及びZは、各々独立して、O、S及びNR8から選択され、R8は、H、置換又は非置換のアルキル、置換又は非置換のヘテロアルキル及びアシルから選択されるメンバーであり;R4、R4,、R5及びR5,は、独立して、H、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル、非置換ヘテロシクロアルキル、ハロゲン、NO2、NR9R10、NC(O)R9、OC(O)NR9R10、OC(O)OR9、C(O)R9、SR9、OR9、CR9=NR10及びO(CH2)nNR11R11’からなる群から選択されるメンバーであり、R9及びR10は、独立して、H、置換若しくは非置換のアルキル、置換若しくは非置換のヘテロアルキル、置換若しくは非置換のアリール、置換若しくは非置換のヘテロアリール、置換若しくは非置換のヘテロシクロアルキル及び置換若しくは非置換のペプチジルから選択されるか、又はR9及びR10は、それらが結合する窒素原子と任意に結合して、任意に2つ以上のヘテロ原子を含む4〜6員環を有する置換若しくは非置換のヘテロシクロアルキル環系を形成し、R11及びR11’は、各々独立して、H又は低級アルキルであり;R6は、H、置換若しくは非置換の低級アルキル、シアノ又はアルコキシであり;R7は、H、置換アルキル、非置換アルキル、置換ヘテロアルキル、非置換ヘテロアルキル、ジホスフェート、トリホスフェート、アシル、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、R12、R13及びR14は、独立して、H、置換若しくは非置換のアルキル、置換若しくは非置換のヘテロアルキル及び置換若しくは非置換のアリールから選択されるメンバーであるか、又はR12及びR13は、それらが結合する窒素又は炭素原子と任意に結合して、任意に2つ以上のヘテロ原子を含む4〜6員環を有する置換若しくは非置換のヘテロシクロアルキル環系を形成する。)であって、保護基R1’及びR2’を化合物(II)に付加して
【化6】
化合物(III)を形成し
【化7】
(式中、R3はアルキルである。)、そして、化合物(III)のアミン窒素を含む5員環を形成し、以下を含む
【化8】
結合部分を、化合物(III)に付加することを含んでなる、前記方法。
【請求項14】
前記5員環の形成が化合物(III)のアミン置換基に隣接する炭素のヨウ素化と、その後に続く5員環形成前の1,3−ジハロプロペンを用いたアルキル化とを含んでなる、請求項13に記載の方法。
【請求項15】
前記結合部分の付加が保護基R1’の脱離を含んでなる、請求項13に記載の方法。
【請求項16】
前記結合部分の付加が、前記結合部分のアミン置換基への付加を更に含んでなる、請求項15に記載の方法。
【請求項17】
R6がHである、請求項13に記載の方法。
【請求項18】
式(I)の化合物又はその医薬的に許容される塩
【化9】
(式中、R1及びR2は、各々独立して、H、アルキル、−C(O)OR’、−C(O)NR’R”又は保護基であり、R’及びR”は、独立してH、置換アルキル、非置換アルキル、置換アリール、非置換アリール、置換ヘテロアリール、非置換ヘテロアリール、置換ヘテロシクロアルキル及び非置換ヘテロシクロアルキルからなる群から選択され、R6は、H、置換若しくは非置換の低級アルキル、シアノ又はアルコキシである。)。
【請求項19】
R1及びR2が異なる保護基である、請求項18記載の化合物。
【請求項20】
以下の化合物である、請求項18記載の化合物。
【化10】
【図1】

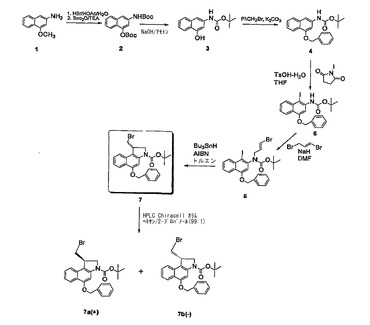
【図2】

【図3】
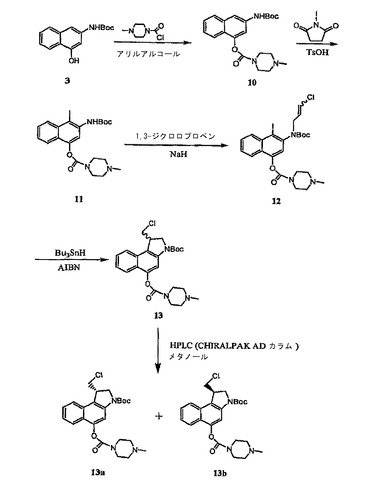
【図2】
【図3】
【公表番号】特表2009−513676(P2009−513676A)
【公表日】平成21年4月2日(2009.4.2)
【国際特許分類】
【出願番号】特願2008−538132(P2008−538132)
【出願日】平成18年10月18日(2006.10.18)
【国際出願番号】PCT/US2006/060050
【国際公開番号】WO2007/051081
【国際公開日】平成19年5月3日(2007.5.3)
【出願人】(504378238)メダレックス インコーポレイテッド (20)
【Fターム(参考)】
【公表日】平成21年4月2日(2009.4.2)
【国際特許分類】
【出願日】平成18年10月18日(2006.10.18)
【国際出願番号】PCT/US2006/060050
【国際公開番号】WO2007/051081
【国際公開日】平成19年5月3日(2007.5.3)
【出願人】(504378238)メダレックス インコーポレイテッド (20)
【Fターム(参考)】
[ Back to top ]