
CD56陽性T細胞増強方法
【課題】ドナーから採取した血液に包含されるT細胞及び単球を用いて、簡便かつ効率的にCD56+T細胞を増強することのできる方法を開発し、提供することである。また、本発明の他の目的は、前記増強方法によって得られるCD56+T細胞強化型血液製剤を比較的安価で提供することである。
【解決手段】T細胞をCD56陽性樹状細胞及びサイトカインと混合培養することによってCD56陽性T細胞を増強させる。この際、CD56陽性CD8陽性細胞傷害性T細胞をより増強される場合には、疾患抗原ペプチドを、またCD56陽性γδT細胞をより増強させるにはビスホスホネート誘導体又はその塩若しくはその水和物でCD56陽性樹状細胞を感作させる。
【解決手段】T細胞をCD56陽性樹状細胞及びサイトカインと混合培養することによってCD56陽性T細胞を増強させる。この際、CD56陽性CD8陽性細胞傷害性T細胞をより増強される場合には、疾患抗原ペプチドを、またCD56陽性γδT細胞をより増強させるにはビスホスホネート誘導体又はその塩若しくはその水和物でCD56陽性樹状細胞を感作させる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、CD56陽性T細胞を増強させる方法、その方法によって得られるCD56陽性T細胞を含むCD56陽性T細胞強化型血液製剤、及びCD56陽性T細胞増強剤に関する。
【背景技術】
【0002】
悪性新生物である癌は、1981年以降日本人における死因の第1位となっており、全死亡原因の約3割を占めている。医学の進歩により、癌の治癒率、生存率は、著しく改善されているが、現在もなお難治性の疾患であることに変わりはない。
【0003】
癌の治療方法は、手術により腫瘍部を切除する外科手術療法、抗ガン剤を用いた化学療法、及び腫瘍に放射線を照射する放射線療法を標準治療法とするが、近年では、免疫細胞を利用した免疫療法が新たな治療法として注目されている(非特許文献1)。
【0004】
免疫療法とは、体内の免疫系を強化することによって癌やウイルス感染症等の治療を行う方法である。特に、癌に対しては、外科療法、化学療法、放射線療法と並ぶ新たな治療法と注目されており、様々な方法が開発されている。例えば、サイトカイン療法、ワクチン療法、BRM(生物応答調整剤:Biological Response Modifier)療法、細胞免疫療法等が挙げられる。
【0005】
サイトカイン療法とは、T細胞やNK細胞等のリンパ球を増殖若しくは活性化させる作用を有するサイトカインを生体内に直接投与することによって、癌細胞やウイルス感染細胞を殺傷する治療法である。例えば、インターロイキン2(IL-2)やインターフェロンの投与によるサイトカイン療法等が該当する(非引用文献2)。しかし、この療法は、臨床結果では期待ほどの効果が得られず、また、臓器機能不全や体液貯留(IL-2投与の場合)、感冒症状若しくは精神障害(インターフェロン投与の場合)等の重篤な副作用を生じるという問題があった。
【0006】
ワクチン療法とは、癌細胞特異的抗原ペプチド等を直接人体に接種し、その抗原ペプチドに対する免疫系を活性化させる治療法である(非特許文献3)。当該治療法は、有効例がいくつか報告されているが、HLAクラスIを発現していない腫瘍等に対しては効果がない等の問題があった。
【0007】
BRM療法とは、腫瘍細胞等に対する患者の生物学的応答性を修飾する物質による治療法である(非特許文献4)。BRMとしては、PSKやベスタチン、OK432等が知られている。この治療方法は、一部の癌等では有効性が認められているが、本来外科療法や化学療法のように免疫能が低下する他の治療法と併用して用いることで効果が得られる補助的療法という側面が強い。また、必ずしも免疫力が強化されるとは限らず、単独での抗癌効果等は弱いという問題があった。
【0008】
細胞免疫療法は、患者から採取した免疫細胞を生体外で増殖、活性化等の処理を行った後に再びその患者の体内に戻すことによって当該患者の免疫力を高める治療法である(非特許文献5)。この細胞免疫療法は、生体外で処理する免疫細胞の種類によって、活性化リンパ球療法と樹状細胞(以下、しばしば「DC」(Dendritic Cell)と表わす)療法に、分類される。
【0009】
活性化リンパ球療法は、生体外でT細胞に対して活性・増殖処理を行う活性化T細胞療法、とNK細胞に対して活性・増殖処理を行う活性化NK細胞療法とに、さらに分類される。
【0010】
活性化T細胞療法は、例えば、LAK(Lymphokine Activated Killer cells;リンフォカイン活性化キラー細胞)療法や、TIL(腫瘍組織浸潤リンパ球)療法、細胞傷害性リンパ球(Cytotoxic T Lymphocyte;以下、本明細書ではしばしば「CTL」と表わす)療法等が該当する。
【0011】
DC療法とは、疾患抗原として、例えば、癌抗原ペプチド又は感染症抗原ペプチドを、直接又は細胞内でプロセッシングした後、そのペプチドの一部をMHC(Major Histocompatibility antigen complex;主要組織適合抗原複合体)に提示し、DCを用いて、体内で抗原を特異的に攻撃するCTLを誘導することで治療を行う方法である。このDC療法に用いられるDCの調製には、大きく分けて2つ方法が知られている。
【0012】
1つ目の方法は、末梢血から末梢血単核球(Peripheral Blood Mononuclear Cells:以下「PBMCs」とする)分画を分離し、次いで細胞付着法にて付着細胞分画を単球として分離して、又はマグネットビーズを用いたMagnetic Cell Sorting(以下、「MACS」と記す)法(Miltenyi Biotec)にてCD14陽性(以下、「CD14+」と表わす。本明細書においては、以下、しばしば「陽性」を肩付の「+」で、また「陰性」を肩付の「−」で、表わす)分画を単球として分離して、これら単球成分にGM-CSFとIL-4を添加培養することによって未熟樹状細胞を調製した後、さらにTNF-a、PGE2、IL-1β若しくはIL-6等の成熟用サイトカイン又は樹状細胞上のTLR(Toll like receptor)と結合するリガンドであるOK432やCpG等を反応させて成熟樹状細胞を調製する方法である。
【0013】
もう一つの方法は、比重遠心分離法によってサイトカインを用いることなく末梢血よりDC前駆細胞を分離した後、数日間培養することによりDCを分離する方法である。
【0014】
上記いずれの方法で調製された場合にも、DCとしての特徴を示すHLA-DR、 CD80、及びCD86は陽性であるが、CD14とCD56は陰性である。ところが、このようなDC(CD14−CD56−DC)を用いた場合、in vitro又はin vivoのいずれにおいてもCTLの十分な誘導を引き起こすことができず、その結果、DC療法の治療効果が低いという問題があった。
【0015】
この問題を解決するため、DCに疾患抗原ペプチドと共にビスホスホネートを共感作させることでCTL誘導効率を増強する方法が報告されている(非特許文献6)。しかし、このCTL誘導効果はin vitroでの結果のみであり、in vivoにおいては確認されていない。
【0016】
それ故、より高いCTL誘導能を有するDCとそれを用いたCTL増強方法の開発が急務であった。
【先行技術文献】
【特許文献】
【0017】
【特許文献1】WO2007/029689
【非特許文献】
【0018】
【非特許文献1】Milani V, et al., 2009, J trans Res, 7(50):1-18.
【非特許文献2】Rosenberg SA, et al., 1985, J Exp Med., 161:1169-88.
【非特許文献3】Bendandi, M. et al., 1999, Nature Med,5:1171-1177.
【非特許文献4】Fisher M,et al., 2002, Anticancer Res.,22:1737-54.
【非特許文献5】Takayama Y et al., 2000, Lancet,356:802-807.
【非特許文献6】J of Leukocyte Biology, 2008, 83: 742-754
【発明の概要】
【発明が解決しようとする課題】
【0019】
本発明の目的は、ドナーから採取した血液に包含されるT細胞及び単球を用いて、簡便かつ効率的にCD56+T細胞を増強することのできる方法を開発し、提供することである。
【0020】
また、本発明の他の目的は、前記増強方法によって得られるCD56+T細胞強化型血液製剤を比較的安価で提供することである。
【課題を解決するための手段】
【0021】
前記課題を解決するために、本発明者らは鋭意研究を重ねた結果、CD14+CD56+DC(以下、しばしば「CD56+DC」と略記する)がCD14−CD56−DCと比較して非常に高いCTLの誘導能を有することを見出した。この知見に基づいて、T細胞とCD56+DC及びIL-2を混合培養するとCD56+T細胞を効率的に増強することができる。
【0022】
さらに、疾患抗原ペプチドでCD56+DCを感作することで、CD56+T細胞のうち疾患抗原特異的CD56+CD8+CTLがより増強されること、又は疾患抗原ペプチドに代えてビスホスホネート誘導体、又はその塩若しくはその水和物でCD56+DCを感作することで、CD56+T細胞のうちCD56+γδT細胞がより増強されることを見出した。
【0023】
CD14+CD56+DCは、末梢血中の単球をIFN-αとGM-CSFで3日間培養することで調製できること、この方法で得られたCD14+CD56+DCはTRAILを発現しており、それを介して直接K562細胞株を殺傷し、自己以外の(allogeneic)のT細胞を刺激する活性が高いことはPapewalisらによって既に報告されている(Papewalis C. et al., J. Immunology, 2008, 180:1462-1470)。しかし、CD14+CD56+DCがCD14−CD56−DCと比較して、CD56+T細胞の増強活性が高いことは検討されておらず、これまで知られていなかった。本発明は、これらの知見に基づくものであり、以下を提供する。
【0024】
(1)T細胞をCD56+DC及びサイトカインと混合培養する工程を含むCD56+T細胞の増強方法。
(2)前記サイトカインがインターロイキン2である、(1)に記載の増強方法。
(3)疾患抗原ペプチドをさらに含む、(1)又は(2)に記載の増強方法。
(4)前記疾患が腫瘍又は感染症である、(3)に記載の増殖方法。
(5)前記混合培養時における培養液中のインターロイキン2の終濃度が1U/mL〜200U/mLである、(3)又は(4)に記載の増強方法。
【0025】
(6)ビスホスホネート誘導体又はその塩若しくはその水和物をさらに含む、(3)〜(5)のいずれかに記載の増強方法。
(7)前記ビスホスホネート誘導体がゾレドロン酸、パミドロン酸、アレンドロン酸、リセドロン酸、イパンドロン酸、インカドロン酸、エチドロン酸及びそれらの組み合わせからなる群から選択される、(6)に記載の増強方法。
(8)前記CD56+T細胞が疾患抗原特異的CD56+CD8+CTLである、(3)〜(7)のいずれかに記載の増強方法。
(9)ビスホスホネート誘導体又はその塩若しくはその水和物をさらに含む、(1)に記載の増強方法。
(10)前記ビスホスホネート誘導体がゾレドロン酸、パミドロン酸、アレンドロン酸、リセドロン酸、イパンドロン酸、インカドロン酸、エチドロン酸及びそれらの組み合わせからなる群から選択される、(9)に記載の増強方法。
【0026】
(11)前記混合培養時における培養液中のインターロイキン2の終濃度が300U/mL〜3000U/mLである、(9)又は(10)に記載の増強方法。
(12)前記CD56+T細胞がCD56+γδ型T細胞である、(9)〜(11)のいずれかに記載の増強方法。
(13)(1)〜(12)のいずれかに記載の増強方法によって増強されたCD56+T細胞を含むCD56+T細胞強化型血液製剤。
(14)CD56+DC及びサイトカインを含むCD56+T細胞増強剤。
(15)前記サイトカインがインターロイキン2である、(14)に記載のCD56+T細胞増強剤。
【0027】
(16)ビスホスホネート誘導体又はその塩若しくはその水和物をさらに含む、(15)に記載のCD56+T細胞増強剤。
(17)前記ビスホスホネート誘導体がゾレドロン酸、パミドロン酸、アレンドロン酸、リセドロン酸、イパンドロン酸、インカドロン酸、エチドロン酸及びそれらの組み合わせからなる群から選択される、(16)に記載のCD56+T細胞増強剤。
(18)前記CD56+T細胞がCD56+γδ型T細胞である、(16)又は(17)に記載のCD56+T細胞増強剤。
(19)疾患抗原ペプチドをさらに含む、(15)〜(17)のいずれかに記載のCD56+T細胞増強剤。
(20)前記CD56+T細胞が疾患抗原特異的CD56+CD8+CTLである、(19)に記載のCD56+T細胞増強剤。
【発明の効果】
【0028】
本発明のCD56+T細胞増強方法によれば、血液ドナー及びレシピエントである患者に対する侵襲性が低く、かつ被検体から採取した血液中のT細胞を用いて、簡便かつ効率的にCD56+T細胞、特に疾患抗原特異的CD56+CD8+CTL又はCD56+γδT細胞を増強することができる。
【0029】
本発明のCD56+T細胞強化型血液製剤によれば、CD56+T細胞が増強された血液製剤を比較的安価で提供することができる。
【0030】
本発明のCD56+T細胞増強剤によれば、T細胞の培養液への添加によって容易にCD56+T細胞を増強することができる。
【図面の簡単な説明】
【0031】
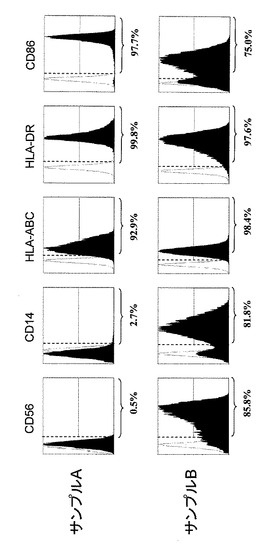
【図1】実施例におけるサンプルA及びサンプルBで得られたDCの表面抗原を各表面抗原特異的抗体で検出したフローサイトメトリー(FACS)の結果(黒色ヒストグラム)を示す。各ヒストグラムにおいて示す%は、全DCにおいて各表面抗原を発現している細胞(各ヒストグラムにおいて縦波線よりも右領域の細胞で示される)の割合を示す。また、白色ヒストグラムは、各表面抗原抗体と同一の標識子で標識されたIgG1抗体(ネガテイブコントロール)のヒストグラムである。
【図2】表2に示したドナーαにおける各サンプルのサイトグラムを示す。各サイトグラムにおいて、横軸は、FITC標識した抗CD8抗体(FITC-CD8)の、縦軸は、PE標識したMART-1改変型A27L特異的テトラマー(PE-M1t)の蛍光強度をそれぞれ対数スケールで示している。また、前記各種蛍光の強度に基づいてサイトグラムを4つの区画(X1〜X4)に分画した。X1にはCD8−A27L+T細胞が、X2にはCD8+A27L+T細胞(疾患抗原特異的CD8+CTLに相当)が、X3にはCD8−A27L−T細胞が、そしてX4にはCD8+A27L−T細胞が、それぞれ分布する。表2に示す%は、測定した細胞(a〜cは、全T細胞;d〜fは、全CD8+T細胞)においてX2区画に含まれる細胞の割合(%)を表す。また、培養0日目のa、dは、反応細胞のみを、培養12日目のb、eは、反応細胞にCD56−DC及びMART-1改変型A27Lを加えて、そして培養12日目のc、fは、反応細胞にCD56+DC及びMART-1改変型A27Lを加えて、培養したものである。
【図3】A:表2ドナーαにおいて疾患抗原A27Lで感作されたCD56+DCで誘導されたCD8+T細胞中のCD56+T細胞(CD56+CD8+T細胞)の割合をPC5標識した抗CD56抗体で検出したフローサイトメトリーの結果(黒色ヒストグラム)を示す。B:図2において疾患抗原A27Lで感作されたCD56+DCで誘導されたA27L抗原特異的CD8+CTL中のCD56+T細胞(A27L抗原特異的CD56+CD8+CTL)の割合をPC5標識した抗CD56抗体で検出したフローサイトメトリーの結果(黒色ヒストグラム)を示す。(Y軸は、細胞数を示す。)各ヒストグラムにおいて示す%は、CD56抗原を発現している細胞(各ヒストグラムにおいて縦波線よりも右領域の細胞で示される)の割合を示す。A、Bのいずれも白色ヒストグラムは、各表面抗原抗体と同一の標識子で標識されたIgG1抗体(ネガテイブコントロール)のヒストグラムである。
【図4】表2に示したドナーαにおける各サンプルのサイトグラムを示す。本図の基本的な説明は、図2に準ずる。ただし、本図においてはa、dは、反応細胞のみを、b、eは、反応細胞にCD56−DC、A27L及びゾメタを加えて、そしてc、fは、反応細胞にCD56+DC、A27L及びゾメタを加えて、培養したものである。
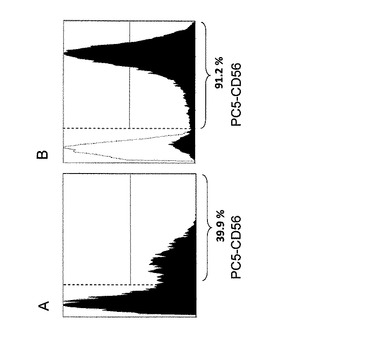
【図5】ドナーαにおいてゾメタで感作されたCD56−DCにより誘導されたγδT細胞中のCD56+γδT細胞の割合(A)とゾメタで感作されたCD56+DCにより誘導されたγδT細胞中のCD56+γδT細胞の割合(B)を、PC5標識した抗CD56抗体で検出したフローサイトメトリーの結果(黒色ヒストグラム)で示す。各ヒストグラムにおいて下記に示す%は、CD56抗原を発現している細胞(各ヒストグラムにおいて縦波線よりも右領域の細胞で示される)の数比を示す。また、白色ヒストグラムは、各表面抗原抗体と同一の標識子で標識されたIgG1抗体(ネガテイブコントロール)のヒストグラムである。
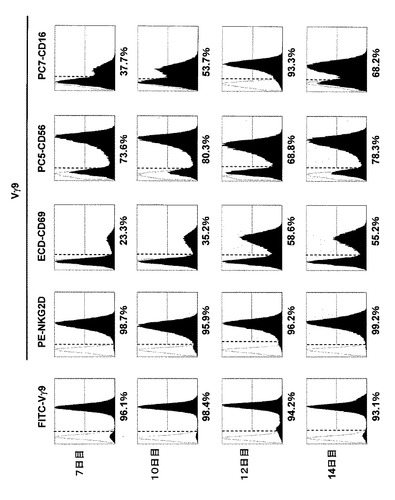
【図6】癌患者由来の末梢血から調製されたCD56+DCと反応細胞を用いて、本発明の方法に基づいてゾメタで感作したものを培養期間(7、10、12、14日)ごとにFITC標識したVγ9抗体を用いたフローサイトメトリーで検出したときの結果(黒色ヒストグラム)を示す。各ヒストグラムにおいて縦波線よりも右領域の細胞が、Vγ9+細胞(γδT細胞)であり、ヒストグラム下に示す%は、測定した全T細胞中のVγ9+細胞の数比を示す。
【0032】
また、測定されたVγ9+細胞において、γδT細胞の表現抗原である、NKG2D、CD69、CD56及びCD16の発現をそれぞれPE標識抗NKG2D抗体、ECD標識抗CD69抗体、PC5標識抗CD56抗体及びPC7標識抗CD16抗体を用いたフローサイトメトリーで検出したときの結果(黒色ヒストグラム)を示す。各ヒストグラムにおいて縦波線よりも右領域の細胞が、それぞれの抗体の陽性細胞であり、ヒストグラム下に示す%は、測定した全Vγ9+細胞中のそれぞれの抗原陽性細胞の数比を示す。
【発明を実施するための形態】
【0033】
1.CD56陽性T細胞増強方法
本発明の第1の実施形態は、CD56陽性T細胞(CD56+T細胞)増強方法である。
本明細書において「CD56+T細胞(の)増強」とは、CD56+T細胞を誘導し、増殖させ、かつ活性化することをいう。例えば、T細胞の細胞傷害機能が強化されること、すなわちCTLが増加すること、及び/又はT細胞表面の活性及び/又は増殖に関するレセプターの発現が増強されること等が含まれる。
【0034】
本発明のCD56+T細胞増強方法は、混合培養工程を含む。また、必要に応じて、その混合培養工程内において、さらに選択ステップを含むことができる。以下、本工程及び選択ステップの構成について詳説する。なお、本実施形態において、各操作は、原則として滅菌処理済の試薬、培地、器具等を使用し、培養はクリーンルーム内のクリーンベンチ等の無菌的環境下で行うことを前提とする。これは、雑菌等のコンタミネーションを防止するためである。
【0035】
1−1.混合培養工程
「混合培養工程」とは、T細胞をCD56+DC及びサイトカインと混合培養する工程である。
1−1−1.T細胞とその調製
「T細胞」とは、骨髄中の造血幹細胞に由来し、原則として胸腺内で分化・成熟したリンパ球の1種をいう。細胞表面にαβ型レセプターを発現するαβT細胞とγδ型レセプターを発現するγδT細胞が知られるが、本明細書においてはいずれも包含する。また、ここでいうαβT細胞は、獲得型免疫応答の調整に関与するヘルパーT細胞とインターフェロンγ(INF-γ)産生作用と細胞傷害活性を示すNKT細胞のいずれも含む。さらに、γδT細胞は、細胞傷害活性を示すキラーT細胞を含む。本工程で使用するT細胞は、本実施形態のCD56+T細胞増強方法の反応細胞として供される。
【0036】
本工程で使用するT細胞の調製方法は、特に限定はしない。当該分野で公知の方法、例えば、PMBCsからMACS法によってCD3+細胞として分離されたT細胞を用いることができる(Langman R.S., et al., 2007, Blood, 109:1113-1122)。その他にも、例えば、次項の「(2)CD56+DCとその調製」において、血液からCD14+細胞を単離した後の細胞(CD14−細胞)を利用することができる。具体的には、CD14-細胞をAIM-V培地(インビトロジェン)等に懸濁すればよい。また、必要に応じて0.5〜20%の牛血清(BSA)、牛胎児血清(FBS)、ヒト血清、ヒト血漿等を培地に添加することができる。CD14−細胞は、凍結保存したものを解凍、洗浄後懸濁したものを用いてもよい。この方法で得られるCD14−細胞集団は、その多くがT細胞である。B細胞も僅かに包含するものの、それ自体は、本発明の方法では増強されないことから本工程で使用するT細胞に混在していても問題はない。
【0037】
1−1−2.CD56陽性樹状細胞とその調製
本明細書において「CD56陽性樹状細胞(CD56+DC)」とは、細胞表面にCD56抗原を発現する樹状細胞である。
【0038】
本工程で使用するCD56+DCの調製方法は、特に限定はしない。例えば、当該分野で公知の方法、例えば、血液から直接単離する方法を用いてもよい。以下、CD56+DCの調製方法を、具体例を挙げて説明するが、本発明において使用するCD56+DCの調製方法は、この方法に限られない。
【0039】
(1)血液の採取
CD56+DCの調製は、まず、DCの前駆細胞を血液から取得することから始める。本発明において「血液」とは、DC及びDC前駆細胞を含む血液成分をいう。例えば、全血(末梢血を含む)、臍帯血、骨髄液やその成分の一部、例えば、単核球等が該当する。好ましくは単核球である。中でも末梢血から得られる末梢血単核球(Peripheral Blood Mononuclear Cells:以下「PBMCs」とする)は、時期を選ばずに容易に採取できる上にドナーへの侵襲性が低いため特に好ましい。
【0040】
前記「血液」を提供するドナーは、原則として生きている哺乳動物である。哺乳動物の種類は問わないが、好ましくはヒトである。対象とするドナーは、本発明の製造方法で得られるCD56+T細胞を投与する動物と同一の種類であることが望ましい。例えば、本発明の製造方法で得られるCD56+T細胞をヒトに投与する場合、ドナーはヒトであることが好ましい。また、細胞免疫療法を前提とした場合、ドナーはCD56+T細胞を投与するレシピエント自身となる。自己の血液由来の血液製剤であれば、投与後の拒絶反応の可能性を限りなく排除することができるからである。細胞免疫療法を前提とする場合には、レシピエントでもあるドナーが健常体である必要はない。例えば、癌やウイルス感染症に罹患した患者であってもよい。血液は、ドナーから採取されたものにヘパリンやクエン酸等を添加して抗凝固処理を施した後に、一旦冷蔵若しくは冷凍で保存した保存血液等でもよい。このような冷凍又は冷蔵血液を用いる場合、解凍及び加温処理は、公知の方法に基づいて行えばよい。例えば、凍結保存されたPBMCsにRPMI-1640培地を添加して解凍後、37℃、5%CO2下で3時間インキュベートする方法が挙げられる。
【0041】
ドナーからの採血方法は、公知の方法に従えばよい。例えば、末梢血であれば末梢部の静脈等に注射をして採取すればよく、骨髄液であれば骨髄穿刺(マルク)によって採取すればよく、臍帯血であれば分娩後胎盤の娩出前の臍帯に針を刺して採取すればよい。以下、末梢血の採取について、一例を挙げて具体的に説明をする。
【0042】
末梢全血は、生体の末梢部血管、例えば、静脈又は動脈に注射針を刺して真空採血管、採血バッグ等による公知の全血採取法によって採取することができる。採取する容量は、ドナーの負担にならず、かつ所定のCD56+T細胞量を製造する上で必要な量である。例えば、大人一人に対して一回に投与するCD56+T細胞を製造する場合、通常20mL〜50mLあればよい。ただし、癌患者のように血中のPBMCs数が低下している場合や多量のPBMCsを必要とする場合には、成分採血(アフェレーシス)により、PBMCsだけを必要量選択的に採取することもできる。採取後、血液が凝固しないように、例えば、シリンジ内部等をヘパリン等の血液凝固阻止剤若しくは血液凝固阻害剤で予めコーティングしておくか、又は採取した血液にヘパリンやクエン酸等を添加しておくことが好ましい。また、末梢全血から血漿を分離し、血球成分を使用してもよい。血漿は、例えば、末梢全血を遠心管に移して、2000rpm〜4000rpmで5〜20分間遠心し、その上清を得ればよい。それを56℃にて30分間程度加温して非働化し、2000rpm〜4000rpmで5〜20分間遠心して、血小板など沈殿物を除去後、細胞培養の栄養原として使用することもできる。
【0043】
(2)単球の分離
次に、末梢全血からPBMCsを分離する。PBMCsの分離方法は、赤血球から有核細胞を分離するあらゆる方法を利用することができる。例えば、フィコール・ハイパック(Ficoll−Hypaque)やフィコール・コンレイ(Ficoll−Conray)を比重液とした密度勾配遠心法を用いて、末梢全血又は血漿分離後の血球成分から得る方法が挙げられる。これらの比重液は、市販の分離液等を利用すると便利である。例えば、Ficoll−Paque PLUS(Amersham社)やLYMPHOPREP(AXIS−SHIELD社)等が利用できる。PBMCsの分離方法については、キット添付のプロトコールに従えばよい。
【0044】
分離後のPBMCsは、比重液を除去するために生理食塩水、PBS(-)や培養細胞用の培地で数回洗浄する。ここで培養細胞用の培地としては、例えば、血清を含まないPBS(-)、AIM-V培地、RPMI1640培地又は培養に用いる無血清培地等を用いればよい。前記PBS(-)若しくは培地で洗浄後、回収したPBMCsの数を血球計算板でカウントしておくことが好ましい。健康なヒト成人の場合、通常、20mL〜60mLの末梢全血から2×107個〜6×107個以上のPBMCsを回収することができる。
【0045】
(3)CD14+細胞の分離
続いて、回収したPBMCsから、DCの前駆細胞である単球のマーカーCD14を発現しているCD14+細胞(単球細胞)を分離する。分離方法は、CD14+細胞を分離可能な公知のあらゆる方法を利用することができる。例えば、抗CD14抗体標識されたマグネットビーズを用いたMagnetic Cell Sorting(以下、「MACS」と記す)を利用して単球を単離、回収する方法は、簡単でかつ単球細胞の回収率が高いため好ましい。MACSの詳細な方法については、Miltenyi Biotec, Auburn社の分離プロトコールを参照すればよい。他の方法として、回収したPBMCsを培養フラスコ等の培養容器に移し,34℃〜38℃、好ましくは37℃で、2%〜10%、好ましくは5%のCO2濃度条件下で1時間以上培養した後、容器壁面に付着した細胞をDC前駆細胞として用いる方法等を利用することもできる。
【0046】
(4)CD56+DCの誘導
得られたDC前駆細胞(CD14+DC)をCD56+DC(CD14+CD56+DC)に誘導する。誘導方法は、前記誘導を達成できる方法であれば、特に限定はしない。例えば、Papewalis C et al., J. Immunology, 2008, 180:1462-1470に記載の方法を用いることができる。この方法では、DCをGM-CSF及びIFN-αと共に34℃〜38℃、好ましくは37℃で、2%〜10%、好ましくは5%のCO2濃度条件下で、RPMI-V培地、AIM-V培地等を用いて3〜7日間培養することで達成できる。また、培養液中に、必要に応じて、0.5〜20%のBSA、FBS、ヒト血清、ヒト血漿等を培地に添加することができる。誘導後のCD56+DCは、表面抗原に基づく抗体(抗CD56抗体)を用いたMACS等によって分離することによってその純度を高めることができる
【0047】
1−1−3.サイトカイン
本明細書において「サイトカイン」とは、細胞間の情報伝達を担う多種多様なタンパク質性のホルモンであって、免疫系においては、前述のようにT細胞やNK細胞等のリンパ球を増殖若しくは活性化させる作用を有する。例えば、インターロイキン(Interleukin)やINF、TNF、MCP等が挙げられる。本発明のCD56+T細胞増強方法に適当なサイトカインとしては、例えば、IL-2、IL-12、IL-15、IL-18、IL-21が挙げられる。これらの一以上のサイトカインであれば、いずれのサイトカインであってもよいが、特に好ましいサイトカインは、IL-2である。サイトカインは、各メーカー(例えば、Chiron、Smitomo等)から市販されているものを利用すればよい。細胞免疫療法に用いるのであれば、医療用グレードの抗体を使用することが好ましい。
【0048】
1−1−4.混合培養方法
本工程では、混合培養によりT細胞にCD56+DC及びサイトカインを接触させて、CD56+T細胞を増強することを特徴とする。
【0049】
混合培養方法の具体的な例として、まず、上記で調製したT細胞を、例えば、培地で細胞密度1〜2×106個/mLとなるように調製する。ここで用いる培地は、前述した細胞培養用の適当な培地に、非働化処理をしたBSA、FBS、ヒト血清若しくは血漿を容量比(V/V)で5〜20%程度添加したものを用いればよい。細胞免疫療法に用いるためにCD56+T細胞を増強する場合には、無血清培地に自己血漿を添加したものを用いることが望ましい。自己血漿は、血液採取工程後に得られる血液から調製すればよい。例えば、採取した末梢全血を室温(10℃〜30℃:以下同じ。)にて3000rpmで10分間程度遠心して得た上清を自己血漿とすることができる。また、培地には必要に応じて、ストレプトマイシン、ペニシリン、カナマイシン、ゲンタマイシン等の抗生物質を添加してもよい。
【0050】
次に、T細胞を含む培地に上記で調製したCD56+DCとサイトカインを添加する。CD56+DCは、上記培地に細胞密度1×105〜2×105個/mLとなるように添加する。また、サイトカインは、上記培地に終濃度が0.1〜5000U/mL、好ましくは1〜3000U/mLとなるように添加する。添加後、培地を十分に混合し、34℃〜38℃、好ましくは37℃で、2%〜10%、好ましくは5%のCO2濃度条件下で5〜30日間、好ましくは7〜28日間、より好ましくは7〜14日間培養する。その間、必要に応じて、培地交換を行ってもよい。
【0051】
1−2.選択ステップ
前記混合培養工程は、その工程内に選択ステップを含むことができる。
本明細書において「選択ステップ」とは、本発明のCD56+T細胞増強方法において、特定のCD56+T細胞を他のCD56+T細胞よりも強く増強させたい場合に、必要に応じて採用し得るステップである。例えば、疾患抗原特異的CD56+CD8+CTL増強ステップ、又はCD56+γδT細胞増強ステップが挙げられる。
以下、それぞれのステップの構成について、具体的に説明をする。
【0052】
1−2−1.疾患抗原特異的CD56+CD8+CTL増強ステップ
「疾患抗原特異的CD56+CD8+CTL増強ステップ」とは、本発明のCD56+T細胞増強方法において、疾患抗原特異的なCD56+CD8+CTLをより強く増強させるためのステップである。本ステップは、疾患抗原ペプチド感作手段、及び/又はビスホスホネート補助感作手段を含む。
【0053】
なお、本ステップを選択する場合、前記混合培養時における培養液中のサイトカイン、好ましくはIL-2の終濃度は、1〜200U/mL、好ましくは10〜150U/mL、より好ましくは10〜50U/mLとなるようにしておく。
【0054】
(1)疾患抗原ペプチド感作手段
「疾患抗原ペプチド感作手段」とは、前記混合培養工程に疾患抗原ペプチドを加えて、CD56+DCをその疾患抗原ペプチドで感作する手段である。疾患抗原特異的CD56+CD8+CTL増強ステップにおいて本手段を行うことによって、疾患抗原特異的CD56+CD8+CTLを、選択的に増強することができる。
【0055】
本明細書において「感作」とは、CD56+DCを所定の物質と反応させることをいう。好ましくは前記所定の物質をCD56+DCの表面上に直接的に又は間接的に提示させることである。前記物質は、例えば、疾患抗原ペプチド及び/又は後述するビスホスホネート誘導体又はその塩若しくはその水和物が挙げられる。
【0056】
本明細書において「抗原ペプチド」とは、抗原となり得るペプチドであって、原則として一以上のエピトープを含む。ペプチドの長さは特に限定はしないが、前述のように少なくとも一のエピトープを含む必要があることから、下限は、通常は5アミノ酸、好ましくは6アミノ酸、より好ましくは7アミノ酸、さらに好ましくは8アミノ酸あればよい。また、上限は、タンパク質の全長である。通常は、前記下限以上500アミノ酸以下、300アミノ酸以下、200アミノ酸以下、100アミノ酸以下、80アミノ酸以下、60アミノ酸以下、50アミノ酸以下、40アミノ酸以下、30アミノ酸以下、又は20アミノ酸以下のペプチドが好ましく利用される。
【0057】
また、本明細書において「疾患抗原ペプチド」とは、疾患原因となる標的タンパク質又はその断片由来の抗原ペプチドをいう。ここでいう「疾患」は、宿主の身体や精神に機能的障害又は器質的障害がもたらされた結果、その宿主の身体的又は精神的健康が損なわれた状態、すなわち病気の状態をいう。例えば、腫瘍又は感染症を含む。腫瘍は、良性腫瘍及び悪性腫瘍(悪性新生物、癌;以下「癌」として示す)を含む。癌の種類は問わない。例えば、上皮性腫瘍や肉腫、白血病、骨髄腫等が該当する。より具体的には、脳腫瘍、網膜芽腫、基底細胞癌、悪性黒色腫、舌癌、食道癌、胃癌、大腸癌、肺癌、白血病、リンパ腫、乳癌、子宮頸癌、子宮体癌、卵巣癌、前立腺癌、精巣腫瘍、膀胱癌、腎臓癌、肝臓癌、膵臓癌及び線維肉腫等が挙げられる。感染症は、例えば、ウイルス感染症、細菌感染症及び寄生虫感染症を含む。ここでいう「ウイルス感染症」は、ウイルス感染による疾患全般を指すが、特に治癒が難治の慢性ウイルス感染症や急性ウイルス感染症の予防が該当する。当該難治な慢性ウイルス感染症としては、例えば、AIDSを引き起こすHIV感染症、ウイルス性慢性肝炎、子宮頸癌を引き起こすヒトパピローマウイルス感染症が挙げられる。また、急性ウイルス感染症としてはインフルエンザ等のウイルス性呼吸器感染症や免疫不全状態での急性ウイルス感染症が挙げられる。ウイルスは、DNAウイルス又はRNAウイルスを問わない。また、また、「細菌感染症」とは、真正細菌(グラム陽性菌及びグラム陰性菌を含む)又は真菌(糸状菌、酵母等及び担子菌を含む)による感染症である。例えば、カンジダ感染症、ブラストミセス症、ヒストプラスマ症等が挙げられる。さらに、ここでいう「寄生虫感染症」は、原虫又は蠕虫による疾患全般を指す。例えば、マラリア、リューシュマニア症、フィラリア症、エキノコックス症、日本住血吸虫症等が挙げられる。
【0058】
本手段で使用する疾患ペプチド抗原は、標的とする疾患に対する抗原ペプチドを使用することが好ましい。例えば、標的とする疾患がメラノーマ(悪性黒色腫)であれば、メラノーマに特異的な抗原ペプチドを使用すればよい。疾患ペプチド抗原は、単一種であってもよいし、二以上の、好ましくは同一の疾患に対する、異なる疾患ペプチド抗原を用いてもよい。
【0059】
疾患抗原ペプチドは、天然のタンパク質又はその断片由来であってもよいし、それらの配列に基づいた合成ペプチドであってもよい。合成ペプチドの使用は、疾患原因となる標的タンパク質を得るために被検体から血液や組織等を採取する必要がないため、被検体に対する侵襲性が極めて低いことから好ましく利用できる。特に各癌抗原ペプチドは、表1に示す公知の抗原を、メーカーから購入又は公知のアミノ酸配列に基づいて合成したものを利用してもよい。各癌種に対する疾患抗原ペプチドの具体例として、下記表1に記載の抗原ペプチドが挙げられる。表1に記載の抗原ペプチドは、そのアミノ酸配列が公知であり、化学合成によって、また市販品の購入によって容易に入手可能である。
【0060】
【表1】

【0061】
本手段は、前記混合培養工程において、CD56+DCを疾患抗原ペプチドで感作可能な限り、いかなる態様でも行うことができる。例えば、前記混合培養工程において、T細胞、CD56+DC、サイトカインと共に疾患抗原ペプチドを培地に加えて、混合培養することによって、CD56+DCを疾患抗原ペプチドで感作させてもよい。また、CD56+DCを疾患抗原ペプチドと共に1時間〜24時間培養して予備感作させた後に、T細胞及びサイトカインを含む培地にそれらを混合して、引き続き培養しても構わない。さらに、T細胞、CD56+DC及びサイトカインを混合し、1時間〜24時間培養した後に、疾患抗原ペプチドを追加してもよい。
【0062】
疾患抗原ペプチドの濃度は、通常DCを疾患抗原ペプチドにより感作する濃度であればよく、特に限定はされない。例えば、Cance Research,1999,Vol.59,2167-2173、The Journal of Immunology,1995,Vol.154,2257-2265、又は、The Journal of Immunology,1994,153,996-1003に記載されているように、培養液における終濃度が0.01〜20μg/mL、好ましくは0.1〜2μg/mLであればよい。
【0063】
(2)ビスホスホネート補助感作手段
本発明において「ビスホスホネート補助感作手段」とは、CD56+DCをビスホスホネート誘導体又はその塩若しくはその水和物(以下、本明細書では「ビスホスホネート誘導体等」とする)で感作する手段である。本手段は、疾患抗原特異的CD56+CD8+CTL増強ステップにおいて、前記疾患抗原ペプチド感作手段に加えて、必要に応じて追加される補助的手段である。したがって、本手段を選択した場合、CD56+DCは、ビスホスホネート誘導体等及び疾患抗原ペプチドで共感作されることとなる。本発明において、「共感作」とは、同時、連続的、又は断続的に2以上の物質で感作することをいう。本手段を行うことにより、疾患抗原ペプチド感作手段のみの場合よりも、さらに疾患抗原特異的CD56+CD8+CTLを増強することができる。
【0064】
本明細書において「ビスホスホネート誘導体」とは、下記一般式1で示される化合物をいう。
【0065】
【化1】
【0066】
上記式中、R1は、水素原子(H)又は低級アルキル基を表し、R2とR3は、それぞれ独立して、水素原子、ハロゲン、ヒドロキシル基、アミノ基、チオール基、置換された若しくは置換されていないアリール基、置換された若しくは置換されていないアルキル基、低級アルキルアミノ基、アルアルキル基、シクロアルキル基又は複素環式基を表し、又はR2とR3は、それらを含む環状構造の一部を形成し、該環状構造を形成する置換基は、R2とR3において、それぞれ独立して、ハロゲン、低級アルキル基、ヒドロキシル基、チオール基、アミノ基、アルコキシ基、アリール基、アリールチオ基、アリールオキシ基、アルキルチオ基、シクロアルキル基又は複素環式基に由来する。
【0067】
ビスホスホネート誘導体の具体例としては、ゾレドロン酸、パミドロン酸、アレンドロン酸、リセドロン酸、イパンドロン酸、インカドロン酸、エチドロン酸が挙げられる。本発明においては、一以上のビスホスホネート誘導体又はその塩若しくはその水和物をNK細胞増殖刺激因子として添加することができる。本発明において、特に好ましいビスホスホネート誘導体は、ゾレドロン酸又はNK細胞の増殖/活性化を有するゾレドロン酸誘導体又はその塩若しくはその水和物である。
【0068】
ゾレドロン酸(商品名ゾメタ(登録商標)、ノバルティスファーマ社)は、骨吸収抑制活性を有するビスホスホネートで、悪性腫瘍による高カルシウム血症又は多発性骨髄腫による骨病変及び固形癌骨転移による骨病変に対する治療薬として知られている。また、その化学構造の中に窒素含有ビスホスホネート(N-BPs:Nitrogen containing-BisphosPhonate)を包含するため、細胞内でFarnesyl PyrophosPhate (FPP)の合成を抑制して、その結果、前駆体であるIsoPentenyl Pyrophosphate(IPP)が蓄積する。それによって、その生体の免疫反応を活性化できることが報告されている(van Beek E, et al., 1999, Biochem Biophys Res Commun, 264:108-11; Gober HJ, et al., 2003, J Exp Med, 197:163-8.)。
【0069】
「その塩」とは、前記ビスホスホネート誘導体、好ましくはゾレドロン酸の塩基性付加塩をいう。塩基性付加塩としては、例えば、ナトリウム塩若しくはカリウム塩のようなアルカリ金属塩、カルシウム塩若しくはマグネシウム塩のようなアルカリ土類金属塩、トリメチルアミン塩、トリエチルアミン塩、ジシクロヘキシルアミン塩、エタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩若しくはブロカイン塩のような脂肪族アミン塩、N,N-ジベンジルエチレンジアミンのようなアラルキルアミン塩、ピリジン塩、ピコリン塩、キノリン塩若しくはイソキノリン塩のような複素環芳香族アミン塩、アルギニン塩若しくはリジン塩のような塩基性アミノ酸塩、アンモニウム塩又はテトラメチルアンモニウム塩、テトラエチルアモニウム塩、ベンジルトリメチルアンモニウム塩、ベンジルトリエチルアンモニウム塩、ベンジルトリブチルアンモニウム塩、メチルトリオクチルアンモニウム塩若しくはテトラブチルアンモニウム塩のような第4級アンモニウム塩が挙げられる。
【0070】
本手段は、前記混合培養工程において、CD56+DCがビスホスホネート誘導体等で感作される限り、いかなる態様でも行うことができる。また、結果として、CD56+DCがそれらに共感作されれば足りることから、CD56+DCを疾患抗原ペプチドとビスホスホネート誘導体等で感作する順序は問わない。例えば、本ステップにおいて、本手段を疾患抗原ペプチド手段と同時に、手段前に、又は手段後に行うことができる。具体的には、前記混合培養工程において、T細胞、CD56+DC、サイトカイン、疾患抗原ペプチドと共にビスホスホネート誘導体等を培地に加えて、混合培養することによって、CD56+DCを共感作する態様であってもよい。また、CD56+DCを疾患抗原ペプチド及びビスホスホネート誘導体等と共に12時間〜24時間培養して予め共感作した後に、T細胞及びサイトカインを含む培地にそれらを混合して、引き続き培養しても構わない。あるいは、CD56+DCを疾患抗原ペプチドと共に1時間〜24時間培養した後、さらにビスホスホネート誘導体等を加えて12時〜48時間培養し、その後にT細胞及びサイトカインを含む培地にそれらを混合する方法であってもよいし、逆に、CD56+DCをビスホスホネート誘導体等と共に12時〜48時間培養した後、さらに疾患抗原ペプチドを加えて1時間〜24時間培養し、その後にT細胞及びサイトカインを含む培地にそれらを混合する方法であってもよい。また、疾患抗原ペプチド感作手段後に行う場合には、疾患抗原ペプチド感作手段後の培養液にビスホスホネート誘導体等を添加すればよい。
【0071】
ビスホスホネート誘導体等の濃度は、通常細胞をビスホスホネート誘導体等により感作する濃度であればよく、特に限定はされないが、例えば、The Journal of Immunology,2001,Vol.166,5508-5514、又は、Blood,2001,Vol.98,No.5,1616-1618]に記載されているように培養液における終濃度が0.001μM〜20μMであればよい。好ましくは0.001μM〜5μMである。
【0072】
1−2−2.CD56+γδT細胞増強ステップ
「CD56+γδT細胞増強ステップ」とは、本発明のCD56+T細胞増強方法において、CD56+γδT細胞をより強く増強させるためのステップである。本ステップは、ビスホスホネート感作手段を含む。本ステップでは、前記「疾患抗原ペプチド感作手段」に相当する、CD56+DCを疾患ペプチドで感作する手段を含まない点において、疾患抗原特異的CD56+CD8+CTL増強ステップとは異なる。
【0073】
なお、本ステップを選択する場合、前記混合培養時における培養液中のサイトカイン、好ましくはIL-2の終濃度は、300〜3000U/mL、好ましくは400〜200U/mL、より好ましくは500〜1000U/mLとなるようにしておく。
【0074】
(1)ビスホスホネート感作手段
「ビスホスホネート感作手段」とは、前記CD56+DCをビスホスホネート誘導体等で感作する手段である。基本的な操作手段は、前記「ビスホスホネート誘導体補助感作手段」と同じでよい。CD56+γδT細胞増強ステップにおいて、本手段を行うことによって、CD56+γδT細胞がより強く増強される。CD56+γδT細胞は、CD56−γδT細胞と比較して細胞傷害活性と相関するCD107a分子の発現が高く、腫瘍細胞株に対して高い傷害活性を有することが知られている(Alan A. et al., Clin Cancer Res, 2008, Vol. 14:4232-4240)。
【0075】
本手段で使用するビスホスホネート誘導体等は、前記「ビスホスホネート誘導体補助感作手段」で詳説したことから、ここでの説明は省略する。
【0076】
本手段は、前記混合培養工程において、CD56+DCをビスホスホネート誘導体等で感作可能な限り、いかなる態様でも行うことができる。例えば、前記混合培養工程において、T細胞、CD56+DC、サイトカインと共にビスホスホネート誘導体等を培地に加えて、混合培養することによって、CD56+DCをビスホスホネート誘導体等で感作させることができる。また、CD56+DCをビスホスホネート誘導体等と共に12時間〜24時間培養して予備感作させた後に、T細胞及びサイトカインを含む培地にそれらを混合して、引き続き培養しても構わない。さらに、T細胞、CD56+DC及びサイトカインを混合し、12時間〜24時間培養した後に、ビスホスホネート誘導体等を追加してもよい。
【0077】
ビスホスホネート誘導体等の濃度は、通常細胞をビスホスホネート誘導体等により感作する濃度であればよく、特に限定はされないが、例えば、The Journal of Immunology,2001,Vol.166,5508-5514、又は、Blood,2001,Vol.98,No.5,1616-1618]に記載されているように培養液における終濃度が0.001μM〜20μMであればよい。好ましくは5μM〜10μMである。
【0078】
1−3.効果
本発明によれば、ドナー及びレシピエントに対する侵襲性が低く、かつ少量の血液中に含まれるT細胞から簡便かつ効率的にCD56+T細胞、特に疾患抗原特異的CD56+CD8+CTL又はCD56+γδT細胞を増強することができる。具体例を挙げると、40mLの末梢血を採取して、本発明の方法を用いて14日間培養することによって約1×109個のCD56+γδT細胞を調製できる。ここで、健常な大人の血中T細胞数は、1500〜4500細胞/mLであり、γδT細胞は、そのうちの約5%、すなわち75〜225細胞/mLであることが知られている。つまり、γδT細胞は、健常人の全血液量(約5L)中にも3.75×105〜11.25×105細胞しか含まれていない。ところが、本発明の方法を用いることで、僅か40mLの末梢血から、健常人の全血液の約2600〜7800倍にも及ぶγδT細胞数が増強することができる。
【0079】
2.CD56+T細胞強化型血液製剤
本発明の第2の実施形態は、CD56+T細胞強化型血液製剤である。本発明のCD56+T細胞強化型血液製剤は、前記第1実施形態に記載のCD56+T細胞の増強方法によって増強されたCD56+T細胞を主成分として含むことを特徴とする。
【0080】
本発明において「強化」とは、細胞を増殖及び活性化させること、又はさせたことを意味する。
【0081】
CD56+T細胞強化型血液製剤において培養で用いた培地や当該培地に添加したサイトカイン、疾患抗原ペプチド、ビスホスホネート誘導体等の添加物は不要である。したがって、CD56+T細胞強化型血液製剤を使用するに当たっては、前記培養液から培地や前記添加物を可能な限り除去し、増強されたCD56+T細胞が主成分となるように調製しておくことが好ましい。ただし、本実施形態のCD56+T細胞強化型血液製剤を調製するにあたり第1実施形態で用いたCD56+DCは、必ずしも除去する必要はない。CD56+DCもまた、免疫療法において有効成分となり得るからである。
【0082】
第1実施形態のCD56+T細胞増強方法実施後の培養液から培地や添加物の除去する方法の一具体例として、まず、増強されたCD56+T細胞を含む培養液を滅菌済み遠心チューブに移し、1200rpmにて8分間ほど室温にて遠心し、添加物を含む上清の培地を除去する。CD56+T細胞やCD56+DC(以下、「CD56+T細胞等」とする)は、沈殿物として回収できる。回収されたCD56+T細胞等は、2回以上PBS(-)で洗浄することが好ましい。洗浄後のCD56+T細胞等は、血球計算板を用いて細胞数をカウントし、10mL〜200mLの乳酸リンゲル液又は生理食塩水で調整する。こうして、本実施形態のCD56+T細胞強化型血液製剤を調製することができる。当該製剤からCD56+DCを除去したい場合には、例えば、抗CD14抗体を用いたMACSを利用してCD56+DCのみを分離除去すればよい。調製後、必要に応じて当該血液製剤にサイトカイン等を添加することも可能である。
【0083】
本実施形態の血液製剤は、含有するCD56+T細胞の70%以上がCTLであることが望ましい。CTL率、すなわちCD56+T細胞の活性化率は、K562白血病細胞株を用いた細胞傷害活性又は活性化マーカーの発現によって判断できる。活性化マーカーは、CD69等の公知のマーカーが利用できる。また、それらの検出には、それぞれのマーカーに対する抗体を用いればよい。
【0084】
本実施形態のCD56+T細胞強化型血液製剤は、製造後直ちに使用することも、また0℃〜8℃の温度下で所定の期間、又は保存液等を加えて超低温下(約-80℃)若しくは液体窒素中で数年に渡る長期間保存することも可能である。前記保存液としては、市販のリンパ球保存液を用いると便利である。例えば、バンバンカー(日本ジェネティックス社)、ケーエムバンカーII(コスモバイオ社)等が利用できる。
【0085】
本実施形態のCD56+T細胞強化型血液製剤は、1×107〜1×108個/mLの増強されたCD56+T細胞を包含する。それ故、この血液製剤の投与によって被検体のCD56+T細胞数を速やかに増大させることができる。したがって、CD56+T細胞強化型血液製剤の投与により、腫瘍等の疾患を有する被検体の免疫応答を増強し、その疾患の進行の抑制、治癒が可能となる。
【0086】
本実施形態のCD56+T細胞強化型血液製剤によれば、増強されたCD56+T細胞を多数含む血液製剤を凍結保存できることから、必要時に必要量を被検体に投与することができる。
【0087】
3.CD56+T細胞増強剤
本発明の第3の実施形態は、CD56+T細胞増強剤である。本発明のCD56+T細胞増強剤は、T細胞の培養液に添加して混合培養することによって、CD56+T細胞を増強することができる。
【0088】
3−1.構成
3−1−1.必須成分
本発明のCD56+T細胞増強剤は、CD56+DC及びサイトカインを必須の成分として含有する。CD56+T細胞増強剤に含まれるCD56+DCは、前記第1実施形態の「1−1−2.CD56+DCとその調製」の項に記載した方法で調製したものを利用できる。また、サイトカインは、IL-2、IL-12、IL-15、IL-18又はIL-21のうちいずれか一以上が含まれていればよい。特に好ましいのは、IL-2である。
【0089】
CD56+DCとサイトカインは、CD56+T細胞増強剤において、必ずしも混合状態である必要はない。すなわち、CD56+T細胞増強剤とは、各成分を同時に又は別個にT細胞の培養液に添加することによって、CD56+T細胞を増強し得る薬剤である。
【0090】
3−1−2.選択成分
本明細書において、「選択成分」とは、本発明のCD56+T細胞増強剤に必要に応じて添加することのできる成分である。本発明のCD56+T細胞増強剤は、前記必須成分にさらなる選択成分を加えることで、疾患抗原特異的CD56+CD8+CTLやCD56+γδT細胞のような特定のCD56+T細胞をより効率的に増強することができる。選択成分としては、例えば、疾患抗原ペプチドビスホスホネート誘導体等が挙げられる。疾患抗原ペプチド及びビスホスホネート誘導体等については、いずれも前記第1実施形態で詳説していることから、ここではその説明を省略する。
【0091】
以下、疾患抗原特異的CD56+CD8+CTL増強剤、及びCD56+γδT細胞増強剤について、具体的に説明をする。
(1)疾患抗原特異的CD56+CD8+CTL増強剤
「CD56+CD8+CTL増強剤」とは、前記必須成分に加えて、疾患抗原特異的CD56+CD8+CTLを増強するための選択成分を含むCD56+T細胞増強剤である。疾患抗原特異的CD56+CD8+CTL増強剤は、選択成分として疾患抗原ペプチドを含有する。ここで使用する疾患抗原ペプチドは、原則として標的とする疾患に特有の疾患抗原ペプチドである。例えば、標的とする疾患が、癌であり、かつその癌がメラノーマである場合、本発明の疾患抗原特異的CD56+CD8+CTL増強剤に含まれる疾患抗原ペプチドは、メラノーマに対する抗原ペプチド、好ましくはメラノーマ特異的な抗原ペプチドとなる。例えば、表1に記載のCDC27、CDK4、CTAG1、CTAG2、gp100、MART-1、MUM-1、NAP1L1、NFE2L2等が挙げられる。なお、疾患抗原特異的CD56+CD8+CTL増強剤においては、包含するサイトカイン、好ましくはIL-2がT細胞の培養液に添加後に終濃度で1〜200U/mL、好ましくは10〜150U/mL、より好ましくは50〜100U/mLとなるように調整しておく。
【0092】
疾患抗原特異的CD56+CD8+CTL増強剤は、選択成分として疾患抗原ペプチドに加えて、ビスホスホネート誘導体等をさらに含んでいてもよい。本発明の疾患抗原特異的CD56+CD8+CTL増強剤で使用するビスホスホネート誘導体等は、前記第1実施形態の「ビスホスホネート誘導体補助感作手段」で詳説したことから、ここでの説明は省略する。なお、疾患抗原特異的CD56+CD8+CTL増強剤においては、包含するビスホスホネート誘導体等がT細胞の培養液に添加後に終濃度で0.001μM〜20μMであればよい。好ましくは0.001μM〜5μMとなるように調整しておく。
【0093】
(2)CD56+γδT細胞増強剤
「CD56+T細胞増強剤」とは、前記必須成分に加えて、CD56+γδT細胞を増強するための選択成分を含むCD56+T細胞増強剤である。CD56+γδT細胞増強剤は、選択成分としてビスホスホネート誘導体等を含有する。本発明のCD56+γδTで使用するビスホスホネート誘導体等は、前記第1実施形態の「ビスホスホネート誘導体補助感作手段」で詳説したことから、ここでの説明は省略する。なお、CD56+γδT細胞増強剤においては、包含するビスホスホネート誘導体等がT細胞の培養液に添加後に終濃度で0.001μM〜20μMであればよい。好ましくは5μM〜10μMとなるように調整しておく。
【0094】
疾患抗原特異的CD56+CD8+CTL増強剤とCD56+γδT細胞増強剤との違いは、前者が疾患抗原ペプチドを必要構成成分として包含するのに対して、後者は包含しない点、前者がビスホスホネート誘導等を補助成分として必要に応じて適宜包含すればよいのに対して、後者は必要構成成分として包含する点、及び単位量あたりのサイトカインの量が異なる点である。
【0095】
3−2.剤形
本発明のCD56+T細胞増強剤の剤形は特に問わない。適当なバッファに溶解した液体状態、粉末状態、粉末を適当な賦形剤等を添加し錠剤化したものとすることができる。また、必須成分又は選択成分を問わず、各成分が最終的にT細胞に作用できればよいことから、本発明のCD56+T細胞増強剤は、各成分が必ずしも統合されている必要はなく、個別に分離された状態であってもよい。それ故、成分ごとに剤形が異なっていてもよい。例えば、CD56+DCは培地に懸濁された状態であり、サイトカインは粉末状態でもよい。このように、成分が分離された状態の場合には、各成分を同時に又は別個にT細胞の培養液に添加することによって、CD56+T細胞を増強し得る。
【0096】
4.疾患を治療する細胞免疫療法
本発明の第4の実施形態は、第2実施形態のCD56+T細胞強化型血液製剤を生体に投与する細胞免疫療法である。
【0097】
4−1.構成
本実施形態でいう「細胞免疫療法」とは、前記第2実施形態のCD56+T細胞強化型血液製剤を被検体に投与することで、被検体における血中の単位体積あたりのCD56+T細胞、すなわち免疫力を有するT細胞の絶対数を通常血液の平均値よりも増大させて、その被検体における、癌、ウイルス感染症、細菌感染症又は寄生虫感染症に対する細胞傷害活性を増強して、疾患の症状を軽減、改善、又は治療する方法である。特に、本実施形態の細胞免疫療法は、血液ドナーとレシピエントが同一である細胞免疫療法を前提としたものである事が好ましい。これは、拒絶反応の危険性がほとんどないからである。
【0098】
「免疫力を有するT細胞」とは、免疫系における機能が強化されたT細胞を意味する。例えば、細胞傷害活性が活性化されたCD56+CD8+CTL、キラーT細胞、CD56+γδT細胞、NKT細胞が該当する。また、ここでいう「単位体積あたりの〜通常血液の平均値」とは、健常な個体の血液で一般に観察される免疫力を有するT細胞数の単位体積あたりの平均値を意味する。
【0099】
4−2.方法
本実施形態の細胞免疫療法におけるCD56+T細胞強化型血液製剤の投与方法について、細胞免疫療法を行う場合を例として以下で説明をする。当該投与方法は、実施形態1のCD56+T細胞強化型血液製剤を投与する点を除けば、従来の細胞免疫療法で知られる方法と基本的に同様である。したがって、投与方法に関しては公知の細胞免疫療法における投与方法に準じて行えばよい。例えば、前記第1実施形態に記載のCD56+T細胞増強方法によって患者から採取した血液から増強されたCD56+T細胞を主成分として含む第2実施形態のCD56+T細胞強化型血液製剤を、採取後約2週間後にその患者の体内に静脈注射又は点滴等を用いて投与する方法等が挙げられる。
【0100】
本実施形態におけるCD56+T細胞強化型血液製剤の1回あたりの投与量は、一般的な大人へ投与する場合には細胞数にして5×108個〜1×1010個のCD56+T細胞を含む容量であればよい。実際の投与では、投与する者の年齢、性別、体重、疾患の状態、体力等を勘案して適宜調節することが好ましい。
【0101】
本実施形態における細胞免疫療法の一例として、前記投与方法を1サイクルとして、約2週間間隔で1クール(6サイクル)以上投与を継続する方法が挙げられる。細胞免疫療法でない場合も、患者以外のドナー由来の血液で製造されたCD56+T細胞強化型血液製剤を投与する点を除いては、同様の方法で当該細胞免疫療法を行えばよい。
【0102】
4−3.効果
本実施形態の細胞免疫療法によれば、従来の免疫療法、特に細胞免疫療法と比較して、癌等の疾患に対する治癒に高い有効性を有する。また、従来の細胞免疫療法と基本的な操作技術等は同様であることから、細胞免疫療法の技術を有する者であれば特段の技術習得をすることなく実施できる。
【実施例】
【0103】
以下の実施例をもって本発明を具体的に説明する。なお、以下の実施例は、単に本発明を例示するのみであり、本発明はこれらの実施例によって何ら限定されるものではない。また、本実施例で使用された温度、量、時間等の数値に関して、実験上の多少の誤差及び偏差は斟酌される。
【0104】
<実施例1:CD56+T細胞の増強>
1.DCの調製
(方法)
(1)DCの分化誘導
健常ドナーから末梢血を50mL採血した。次に、血液分離比重液((Lymphoprep:AXIS-SHIELD、Norway)を用いて2400rpmで20分間遠心分離後、単核細胞成分分画を回収した。回収した分画をAIM-V medium(インビトロジェン)40mLに懸濁した後、50mLフラスコへ20mLずつ2分して、1〜2時間37℃でインキュベーションした。その後、それぞれにフラスコ内部を軽くピペッティングして非付着細胞成分を回収した。回収された非付着細胞成分は、主にT細胞集団であり、後述する「2.疾患抗原特異的CD56+CD8+CTLの調製(1)」、「3.疾患抗原特異的CD56+CD8+CTLの調製(2)」及び「4.CD56+γδT細胞の調製」における、反応細胞として使用した。
【0105】
また、純度の高いDCを得るために、前記非付着細胞成分回収後の各フラスコに、さらに新たに培地を加えて内壁を強くピぺッテイングし、残存する非付着細胞成分を可能な限り除去した。付着細胞成分からのDCの調製にはFBS(Nichirei Biosciences Inc.)又は5〜10%のAB血清(DSファーマバイオメデイカルKK)を含むAIM-Vmedium(インビトロジェン)を用いた。
【0106】
続いて、一方のフラスコ(サンプルA)にGM-CSF(Primmune Inc)とIL-4(Primmune Inc)を、最終濃度がそれぞれ1000U/mLと500U/mLなるように添加して、37℃、5% CO2濃度下において5日培養して未熟DCを調製した。さらに、TNFα (Wako Pure Chemical Industries, LTD)を最終濃度で10ng/mLとなるように加えて、引き続き37℃、5% CO2濃度下において2日間培養した。
【0107】
他方のフラスコ(サンプルB)にはGM-CSF(上記)とIFN-α(Astellas)を最終濃度が1000U/mLになるように添加して、37℃、5% CO2濃度下において3日間培養した。
【0108】
(2)DCの状態の確認
得られたDCの表面抗原の検出は、フローサイトメーター(Epics XL-MCL; Beckman Coulter )を用いて行った。まず、測定する細胞を1×103cells/μLとなるようにPBSに懸濁した溶液100μLに目的の抗体を添加し、遮光の状態で4℃、15分間染色した。抗体は、いずれも標識子で標識されたPC5-抗CD56抗体、PC7-抗CD14抗体、PE-抗HLA-ABC抗体、FITC-抗HLA-DR抗体、及びPE-抗CD86抗体を用いた(全てBeckman Coulter)。ネガテイブコントロールとして、それぞれの抗体と同一の標識子で標識されたIgG1抗体(Beckman)を用いた。細胞は、染色後、PBSで洗浄して測定した。
【0109】
(結果)
結果を図1に示す。サンプルAの細胞は、CD56陽性細胞率が0.5%であるのに対して、サンプルBの細胞は、85.8%であった。この結果から、サンプルAの細胞は、CD14−CD56−DC(以下、単に「CD56−DC」とする)、またサンプルBの細胞は、CD14+CD56+DC(以下、単に「CD56+DC」とする)であることが確認された。
【0110】
その他の表面抗原である、CD14、MHC分子(HLA-ABC、HLA-DR)及び共刺激分子(CD86)についても、CD56-DCとCD56+DCでは同程度の発現を示した。
【0111】
2.疾患抗原特異的CD56+CD8+CTLの調製(1)
(方法)
ドナーα及びβにおいて前記「1.DCの調製」で調製したサンプルA由来のCD56−DC又はサンプルB由来のCD56+DCのそれぞれ2×105個/mLを、同じく「1.DCの調整」で調製した反応細胞(主にT細胞からなる)2×106個(反応細胞:DC=10:1)と共に24ウェルプレート(SUMILON)に入れた。各ウェルの混合培養液中には、疾患抗原ペプチドとして2μg/mLのヒト主要組織適合抗原(HLA)A*0201拘束性のメラノーマ特異的抗原ペプチドMART-1改変型A27L (ELAGIGILTV:配列番号1)(SCURUM)をこの混合培養溶液中に最終濃度で50U/mLのIL-2(Chiron)を加え、37℃、5%CO2条件下で7日間培養した。培養を通して5〜10%のFBS又はAB血清を含むAIM-V培地を用いた。
【0112】
DCと反応細胞の混合培養開始から7日以降に適時、FITC標識した抗CD8抗体(Beckman Coulter)又はPE標識したMART-1改変型A27L特異的テトラマー(PE-M1t)(MBL)を用いてフローサイトメーターで全T細胞中、又は全CD8+T細胞中に占めるA27L特異的CD8+CTL(疾患抗原特異的CD8+CTL)の割合を測定した。
【0113】
さらに、PC5標識した抗CD56抗体(Beckman Coulter)を用いて、フローサイトメーターで全CD8+T細胞中、又は全A27L特異的CD8+CTL中に占めるA27L特異的CD56+CD8+CTLの割合を測定した。
【0114】
(結果)
A27L特異的CD8+CTLの割合を表2及び図2に、また、A27L特異的CD56+CD8+CTLの割合を図3に示す。図2は、表2で示すドナーαのサイトグラムである。
【0115】
【表2】
【0116】
表中「+」及び「−」は、それぞれ培養液中の存在の有無を示す。「CD56+」は、「CD56+DCを、「CD56−」は、CD56−DCを、それぞれ示す。ドナーα及びβにおいて、反応細胞のみが「+」のサンプルは、前記「1.DCの調整」で調製した反応細胞のみのサンプルであり、培養前コントロールとして用いた。
【0117】
表2から、ドナーα及びβ共にCD56+DCを用いたときが、DC56−DCを用いたときよりも、全細胞中、又は全CD8+細胞中におけるA27L抗原特異的CTLの存在率が高かった。すなわち、CD56+DCを用いた場合の方がより高いA27L抗原特異的CTL誘導能を有することが示された。
【0118】
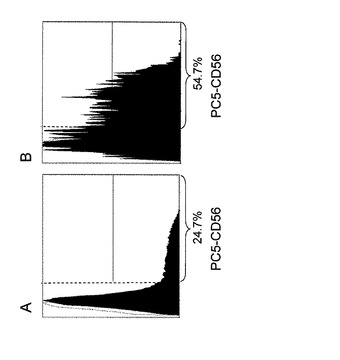
また、図3から、CD56+DCで刺激して増殖してくるCD8+T細胞全体の中でCD56+T細胞の割合(図中縦破線より右領域)は24.7%(図3A)であるのに対して、A27L抗原特異的CD8+CTL全体(Mart1 tetramer gate)の中では54.7%であった(図3B)。このように全CD8+T細胞の中でCD56を共発現しているCD56+CD8+T細胞の割合に比べて、疾患抗原特異的CD8+CTLの中でCD56を共発現している疾患抗原特異的CD56+CD8+CTLの割合が高いという事実は、CD56の発現が疾患抗原特異的CTLの傷害活性と相関していることを示唆している。この事実は、本発明者らが見出した新規知見である。また、疾患抗原特異的CD56+CD8+CTLは、細胞傷害に関与する細胞内パーフォリンやグランザイムを多く含んでおり、高い細胞傷害活性を有していることが確認されているCD8T細胞のサブセットである(J. Immunology 2000, 164:1148-1152)。したがって、本発明の増強方法によって得られる疾患抗原特異的CD56+CD8+CTLを被検体に投与すれば、その被検体の体内における疾患抗原特異的CD56+CD8+CTLを優位に増強できることから、細胞免疫療法において治療効果の高い有用な免疫細胞を提供できる。
【0119】
3.疾患抗原特異的CD56+CD8+CTLの調製(2)
(方法)
基本操作は、前記「疾患抗原特異的CD56+CD8+CTLの調製(1)」と同じである。ただし、ここでは、CD56−DC又はCD56+DC、及び反応細胞とIL-2(Chiron)に加えて、アミノビスホスホネート剤であるゾメタ(商品名、Novartis)を、最終濃度が0.01μMとなるように添加した。
【0120】
DCと反応細胞の混合培養開始から12日以降に適時、FITC標識した抗CD8抗体(BeckmanCoulter)又はPE標識したMART-1改変型A27L特異的テトラマー(MBL)を用いてフローサイトメーターで全細胞中、又は全CD8+T細胞中に占めるA27L特異的CD8+CTL(疾患抗原特異的CD8+CTL)の割合を測定した。また、混合培養液中にゾメタを添加した場合はγδT細胞の増殖が予測されるので、上記MART-1改変型A27L特異的テトラマーの測定に加えて増殖してくる全細胞中のVγ9γδT細胞の割合を、FITC標識化抗Vγ9抗体(Beckman Coulter)を用いて同様にフローサイトメーターで測定した。
【0121】
(結果)
結果を表3及び図4に示す。
【0122】
【表3】
【0123】
表中の説明は、表2と同じである。ゾメタ及びA27Lペプチドで感作したDCをT細胞と共にIL-2存在下で培養することにより誘導されるA27L抗原特異的CD8+CTLの全細胞中に占める割合は、ドナーα及びβ共にCD56+DCを用いたときが、DC56−DCを用いたときよりも、全細胞中、又は全CD8+細胞中におけるA27L抗原特異的CTLの存在率が高かった。また、コントロールに対する増殖倍率(表中、鍵カッコで示す)に関して、表2の結果と比較すると、ゾメタを加えたときの方がさらに高かった。すなわち、CD56+DCを疾患抗原ペプチドとゾメタで共感作させると、疾患抗原特異的CTL誘導がより増強されるがことが示された。
【0124】
4.CD56+γδT細胞の調製
(方法)
前記「1.DCの調整」で調製したドナーα由来のサンプルA由来のCD56−DC又はサンプルB由来のCD56+DCのそれぞれ2×105個/mLを、同じく「1.DCの調整」で調製した反応細胞(主にT細胞からなる)2×106個(反応細胞:DC=10:1)と共に24ウェルプレート(SUMILON)に入れた。各ウェルの混合培養液中には、ゾメタ(Novartis)及びIL-2(Chiron)を、最終濃度がそれぞれ5μM及び1000U/mLとなるように加え、37℃、5%CO2条件下で14日間培養した。培養を通して5〜10%のFBS又はAB血清を含むAIM-V培地を用いた。
【0125】
培養後、FITC標識した抗Vγ9抗体(Beckman Coulter)とPC5標識した抗CD56抗体(Beckman Coulter)を用いて、フローサイトメーターでVγ9陽性細胞にgateをかけて、得られたVγ9+細胞を100%としてCD56+細胞全γδT細胞中に占めるCD56+T細胞の割合を測定した。
【0126】
(結果)
結果を表4及び図5に示す。
【0127】
【表4】
【0128】
全γδT細胞中のCD56+γδT細胞率は、CD56-DCを用いた場合が39.9%であったのに対して(表3、図5A)、CD56+DCを用いた場合は91.2%であった(表4、図5B)。この結果は、CD56+DCがCD56−DCよりもCD56+γδT細胞をより強く増強できることを示している。
【0129】
<実施例2:CD56+γδT細胞上の表面抗原の確認>
前記実施例1の「4.CD56+γδT細胞の調製」の結果から、健常ドナーの末梢血から調製したCD56+DCと反応細胞を培養すると、CD56−DCを用いたときよりも、より効率的にCD56+γδT細胞を増殖できることが確認された。そこで、癌患者由来の末梢血でも同様の効果が得られるかを検証した。
【0130】
(方法)
肺癌癌患者(ステージIV)由来の末梢血から、上記実施例1の「1.DCの調製」及び「4.CD56+γδT細胞の調製」に記載の方法と同様の方法でγδT細胞の調製を行った。培養によって増殖した全T細胞中のγδT細胞の割合、及びその増殖したγδT細胞において発現している表面抗原NKG2D、CD69、CD56及びCD16のそれぞれの発現を、培養時間を追って測定した。NKG2Dは、活性型のNKレセプターを示し、この抗原の発現が高いと細胞傷害活性が高くなる。CD69は、細胞が活性化に伴い発現する抗原で、IFN-aの産生と相関しており、CD69の発現の増加と共に抗腫瘍活性も高まる。またCD16は、Fcγレセプターを示し、この抗原の発現が高いと、抗体依存性細胞傷害活性が高まる。なお、培養前のPBMCs中のγδTの割合は8.0%であり、CD56+γδTの割合は4.1%であった。
【0131】
増殖してくる全T細胞中のγδT細胞の割合は、γδT細胞のマーカー分子であるVγ9をFITC標識した抗Vγ9抗体(Beckman Coulter)を用いて測定した。また、Vγ9+細胞(γδT細胞)上の表面分子NKG2D、CD69、CD56及びCD16抗原を、それぞれPE標識抗NKG2D抗体、ECD標識抗CD69抗体、PC5標識抗CD56抗体、及びPC7標識抗CD16抗体(いずれもBeckman Coulter)を用いて測定した。前記測定された全γδT細胞に対する 、各抗原を発現している細胞の割合を算出した。
【0132】
(結果)
結果を図6に示す。図6で示すように、全T細胞中のVγ9+細胞の割合は、培養7〜14日の期間で、いずれも90%以上高いの割合を示した。また、Vγ9+細胞中のCD16+細胞(CD16+Vγ9+細胞)の割合は、12日目で93.3%を示し、CD56+DCと反応細胞を混合培養することによって、癌患者由来の血液であってもNK様の細胞表面を有するVγ9細胞を増強できることが確認できた。
【0133】
抗体療法との併用療法を考えるときNK様の細胞表面を有するγδT細胞を増殖させることはその細胞の細胞傷害性を増強し、免疫力を強化する上で重要である。本発明の方法によれば、癌患者由来の末梢血であっても、わずか50mL採血(包含されるVγ9+細胞数は、2.3×106)から14日間の培養でVγ9+細胞数は、元の血液の500倍以上に相当する1.2×109個に達する。免疫細胞療法において、通常、その効果が期待できるγδT細胞数は、109オーダーであるが、本発明のCD56+T細胞増強方法によれば、そのオーダーのγδT細胞を簡便に調製することが可能となる。
【技術分野】
【0001】
本発明は、CD56陽性T細胞を増強させる方法、その方法によって得られるCD56陽性T細胞を含むCD56陽性T細胞強化型血液製剤、及びCD56陽性T細胞増強剤に関する。
【背景技術】
【0002】
悪性新生物である癌は、1981年以降日本人における死因の第1位となっており、全死亡原因の約3割を占めている。医学の進歩により、癌の治癒率、生存率は、著しく改善されているが、現在もなお難治性の疾患であることに変わりはない。
【0003】
癌の治療方法は、手術により腫瘍部を切除する外科手術療法、抗ガン剤を用いた化学療法、及び腫瘍に放射線を照射する放射線療法を標準治療法とするが、近年では、免疫細胞を利用した免疫療法が新たな治療法として注目されている(非特許文献1)。
【0004】
免疫療法とは、体内の免疫系を強化することによって癌やウイルス感染症等の治療を行う方法である。特に、癌に対しては、外科療法、化学療法、放射線療法と並ぶ新たな治療法と注目されており、様々な方法が開発されている。例えば、サイトカイン療法、ワクチン療法、BRM(生物応答調整剤:Biological Response Modifier)療法、細胞免疫療法等が挙げられる。
【0005】
サイトカイン療法とは、T細胞やNK細胞等のリンパ球を増殖若しくは活性化させる作用を有するサイトカインを生体内に直接投与することによって、癌細胞やウイルス感染細胞を殺傷する治療法である。例えば、インターロイキン2(IL-2)やインターフェロンの投与によるサイトカイン療法等が該当する(非引用文献2)。しかし、この療法は、臨床結果では期待ほどの効果が得られず、また、臓器機能不全や体液貯留(IL-2投与の場合)、感冒症状若しくは精神障害(インターフェロン投与の場合)等の重篤な副作用を生じるという問題があった。
【0006】
ワクチン療法とは、癌細胞特異的抗原ペプチド等を直接人体に接種し、その抗原ペプチドに対する免疫系を活性化させる治療法である(非特許文献3)。当該治療法は、有効例がいくつか報告されているが、HLAクラスIを発現していない腫瘍等に対しては効果がない等の問題があった。
【0007】
BRM療法とは、腫瘍細胞等に対する患者の生物学的応答性を修飾する物質による治療法である(非特許文献4)。BRMとしては、PSKやベスタチン、OK432等が知られている。この治療方法は、一部の癌等では有効性が認められているが、本来外科療法や化学療法のように免疫能が低下する他の治療法と併用して用いることで効果が得られる補助的療法という側面が強い。また、必ずしも免疫力が強化されるとは限らず、単独での抗癌効果等は弱いという問題があった。
【0008】
細胞免疫療法は、患者から採取した免疫細胞を生体外で増殖、活性化等の処理を行った後に再びその患者の体内に戻すことによって当該患者の免疫力を高める治療法である(非特許文献5)。この細胞免疫療法は、生体外で処理する免疫細胞の種類によって、活性化リンパ球療法と樹状細胞(以下、しばしば「DC」(Dendritic Cell)と表わす)療法に、分類される。
【0009】
活性化リンパ球療法は、生体外でT細胞に対して活性・増殖処理を行う活性化T細胞療法、とNK細胞に対して活性・増殖処理を行う活性化NK細胞療法とに、さらに分類される。
【0010】
活性化T細胞療法は、例えば、LAK(Lymphokine Activated Killer cells;リンフォカイン活性化キラー細胞)療法や、TIL(腫瘍組織浸潤リンパ球)療法、細胞傷害性リンパ球(Cytotoxic T Lymphocyte;以下、本明細書ではしばしば「CTL」と表わす)療法等が該当する。
【0011】
DC療法とは、疾患抗原として、例えば、癌抗原ペプチド又は感染症抗原ペプチドを、直接又は細胞内でプロセッシングした後、そのペプチドの一部をMHC(Major Histocompatibility antigen complex;主要組織適合抗原複合体)に提示し、DCを用いて、体内で抗原を特異的に攻撃するCTLを誘導することで治療を行う方法である。このDC療法に用いられるDCの調製には、大きく分けて2つ方法が知られている。
【0012】
1つ目の方法は、末梢血から末梢血単核球(Peripheral Blood Mononuclear Cells:以下「PBMCs」とする)分画を分離し、次いで細胞付着法にて付着細胞分画を単球として分離して、又はマグネットビーズを用いたMagnetic Cell Sorting(以下、「MACS」と記す)法(Miltenyi Biotec)にてCD14陽性(以下、「CD14+」と表わす。本明細書においては、以下、しばしば「陽性」を肩付の「+」で、また「陰性」を肩付の「−」で、表わす)分画を単球として分離して、これら単球成分にGM-CSFとIL-4を添加培養することによって未熟樹状細胞を調製した後、さらにTNF-a、PGE2、IL-1β若しくはIL-6等の成熟用サイトカイン又は樹状細胞上のTLR(Toll like receptor)と結合するリガンドであるOK432やCpG等を反応させて成熟樹状細胞を調製する方法である。
【0013】
もう一つの方法は、比重遠心分離法によってサイトカインを用いることなく末梢血よりDC前駆細胞を分離した後、数日間培養することによりDCを分離する方法である。
【0014】
上記いずれの方法で調製された場合にも、DCとしての特徴を示すHLA-DR、 CD80、及びCD86は陽性であるが、CD14とCD56は陰性である。ところが、このようなDC(CD14−CD56−DC)を用いた場合、in vitro又はin vivoのいずれにおいてもCTLの十分な誘導を引き起こすことができず、その結果、DC療法の治療効果が低いという問題があった。
【0015】
この問題を解決するため、DCに疾患抗原ペプチドと共にビスホスホネートを共感作させることでCTL誘導効率を増強する方法が報告されている(非特許文献6)。しかし、このCTL誘導効果はin vitroでの結果のみであり、in vivoにおいては確認されていない。
【0016】
それ故、より高いCTL誘導能を有するDCとそれを用いたCTL増強方法の開発が急務であった。
【先行技術文献】
【特許文献】
【0017】
【特許文献1】WO2007/029689
【非特許文献】
【0018】
【非特許文献1】Milani V, et al., 2009, J trans Res, 7(50):1-18.
【非特許文献2】Rosenberg SA, et al., 1985, J Exp Med., 161:1169-88.
【非特許文献3】Bendandi, M. et al., 1999, Nature Med,5:1171-1177.
【非特許文献4】Fisher M,et al., 2002, Anticancer Res.,22:1737-54.
【非特許文献5】Takayama Y et al., 2000, Lancet,356:802-807.
【非特許文献6】J of Leukocyte Biology, 2008, 83: 742-754
【発明の概要】
【発明が解決しようとする課題】
【0019】
本発明の目的は、ドナーから採取した血液に包含されるT細胞及び単球を用いて、簡便かつ効率的にCD56+T細胞を増強することのできる方法を開発し、提供することである。
【0020】
また、本発明の他の目的は、前記増強方法によって得られるCD56+T細胞強化型血液製剤を比較的安価で提供することである。
【課題を解決するための手段】
【0021】
前記課題を解決するために、本発明者らは鋭意研究を重ねた結果、CD14+CD56+DC(以下、しばしば「CD56+DC」と略記する)がCD14−CD56−DCと比較して非常に高いCTLの誘導能を有することを見出した。この知見に基づいて、T細胞とCD56+DC及びIL-2を混合培養するとCD56+T細胞を効率的に増強することができる。
【0022】
さらに、疾患抗原ペプチドでCD56+DCを感作することで、CD56+T細胞のうち疾患抗原特異的CD56+CD8+CTLがより増強されること、又は疾患抗原ペプチドに代えてビスホスホネート誘導体、又はその塩若しくはその水和物でCD56+DCを感作することで、CD56+T細胞のうちCD56+γδT細胞がより増強されることを見出した。
【0023】
CD14+CD56+DCは、末梢血中の単球をIFN-αとGM-CSFで3日間培養することで調製できること、この方法で得られたCD14+CD56+DCはTRAILを発現しており、それを介して直接K562細胞株を殺傷し、自己以外の(allogeneic)のT細胞を刺激する活性が高いことはPapewalisらによって既に報告されている(Papewalis C. et al., J. Immunology, 2008, 180:1462-1470)。しかし、CD14+CD56+DCがCD14−CD56−DCと比較して、CD56+T細胞の増強活性が高いことは検討されておらず、これまで知られていなかった。本発明は、これらの知見に基づくものであり、以下を提供する。
【0024】
(1)T細胞をCD56+DC及びサイトカインと混合培養する工程を含むCD56+T細胞の増強方法。
(2)前記サイトカインがインターロイキン2である、(1)に記載の増強方法。
(3)疾患抗原ペプチドをさらに含む、(1)又は(2)に記載の増強方法。
(4)前記疾患が腫瘍又は感染症である、(3)に記載の増殖方法。
(5)前記混合培養時における培養液中のインターロイキン2の終濃度が1U/mL〜200U/mLである、(3)又は(4)に記載の増強方法。
【0025】
(6)ビスホスホネート誘導体又はその塩若しくはその水和物をさらに含む、(3)〜(5)のいずれかに記載の増強方法。
(7)前記ビスホスホネート誘導体がゾレドロン酸、パミドロン酸、アレンドロン酸、リセドロン酸、イパンドロン酸、インカドロン酸、エチドロン酸及びそれらの組み合わせからなる群から選択される、(6)に記載の増強方法。
(8)前記CD56+T細胞が疾患抗原特異的CD56+CD8+CTLである、(3)〜(7)のいずれかに記載の増強方法。
(9)ビスホスホネート誘導体又はその塩若しくはその水和物をさらに含む、(1)に記載の増強方法。
(10)前記ビスホスホネート誘導体がゾレドロン酸、パミドロン酸、アレンドロン酸、リセドロン酸、イパンドロン酸、インカドロン酸、エチドロン酸及びそれらの組み合わせからなる群から選択される、(9)に記載の増強方法。
【0026】
(11)前記混合培養時における培養液中のインターロイキン2の終濃度が300U/mL〜3000U/mLである、(9)又は(10)に記載の増強方法。
(12)前記CD56+T細胞がCD56+γδ型T細胞である、(9)〜(11)のいずれかに記載の増強方法。
(13)(1)〜(12)のいずれかに記載の増強方法によって増強されたCD56+T細胞を含むCD56+T細胞強化型血液製剤。
(14)CD56+DC及びサイトカインを含むCD56+T細胞増強剤。
(15)前記サイトカインがインターロイキン2である、(14)に記載のCD56+T細胞増強剤。
【0027】
(16)ビスホスホネート誘導体又はその塩若しくはその水和物をさらに含む、(15)に記載のCD56+T細胞増強剤。
(17)前記ビスホスホネート誘導体がゾレドロン酸、パミドロン酸、アレンドロン酸、リセドロン酸、イパンドロン酸、インカドロン酸、エチドロン酸及びそれらの組み合わせからなる群から選択される、(16)に記載のCD56+T細胞増強剤。
(18)前記CD56+T細胞がCD56+γδ型T細胞である、(16)又は(17)に記載のCD56+T細胞増強剤。
(19)疾患抗原ペプチドをさらに含む、(15)〜(17)のいずれかに記載のCD56+T細胞増強剤。
(20)前記CD56+T細胞が疾患抗原特異的CD56+CD8+CTLである、(19)に記載のCD56+T細胞増強剤。
【発明の効果】
【0028】
本発明のCD56+T細胞増強方法によれば、血液ドナー及びレシピエントである患者に対する侵襲性が低く、かつ被検体から採取した血液中のT細胞を用いて、簡便かつ効率的にCD56+T細胞、特に疾患抗原特異的CD56+CD8+CTL又はCD56+γδT細胞を増強することができる。
【0029】
本発明のCD56+T細胞強化型血液製剤によれば、CD56+T細胞が増強された血液製剤を比較的安価で提供することができる。
【0030】
本発明のCD56+T細胞増強剤によれば、T細胞の培養液への添加によって容易にCD56+T細胞を増強することができる。
【図面の簡単な説明】
【0031】
【図1】実施例におけるサンプルA及びサンプルBで得られたDCの表面抗原を各表面抗原特異的抗体で検出したフローサイトメトリー(FACS)の結果(黒色ヒストグラム)を示す。各ヒストグラムにおいて示す%は、全DCにおいて各表面抗原を発現している細胞(各ヒストグラムにおいて縦波線よりも右領域の細胞で示される)の割合を示す。また、白色ヒストグラムは、各表面抗原抗体と同一の標識子で標識されたIgG1抗体(ネガテイブコントロール)のヒストグラムである。
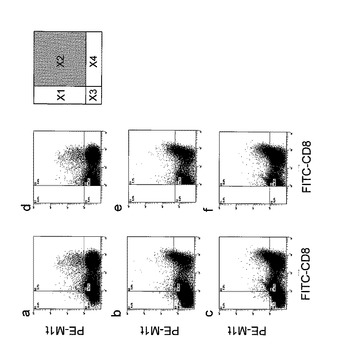
【図2】表2に示したドナーαにおける各サンプルのサイトグラムを示す。各サイトグラムにおいて、横軸は、FITC標識した抗CD8抗体(FITC-CD8)の、縦軸は、PE標識したMART-1改変型A27L特異的テトラマー(PE-M1t)の蛍光強度をそれぞれ対数スケールで示している。また、前記各種蛍光の強度に基づいてサイトグラムを4つの区画(X1〜X4)に分画した。X1にはCD8−A27L+T細胞が、X2にはCD8+A27L+T細胞(疾患抗原特異的CD8+CTLに相当)が、X3にはCD8−A27L−T細胞が、そしてX4にはCD8+A27L−T細胞が、それぞれ分布する。表2に示す%は、測定した細胞(a〜cは、全T細胞;d〜fは、全CD8+T細胞)においてX2区画に含まれる細胞の割合(%)を表す。また、培養0日目のa、dは、反応細胞のみを、培養12日目のb、eは、反応細胞にCD56−DC及びMART-1改変型A27Lを加えて、そして培養12日目のc、fは、反応細胞にCD56+DC及びMART-1改変型A27Lを加えて、培養したものである。
【図3】A:表2ドナーαにおいて疾患抗原A27Lで感作されたCD56+DCで誘導されたCD8+T細胞中のCD56+T細胞(CD56+CD8+T細胞)の割合をPC5標識した抗CD56抗体で検出したフローサイトメトリーの結果(黒色ヒストグラム)を示す。B:図2において疾患抗原A27Lで感作されたCD56+DCで誘導されたA27L抗原特異的CD8+CTL中のCD56+T細胞(A27L抗原特異的CD56+CD8+CTL)の割合をPC5標識した抗CD56抗体で検出したフローサイトメトリーの結果(黒色ヒストグラム)を示す。(Y軸は、細胞数を示す。)各ヒストグラムにおいて示す%は、CD56抗原を発現している細胞(各ヒストグラムにおいて縦波線よりも右領域の細胞で示される)の割合を示す。A、Bのいずれも白色ヒストグラムは、各表面抗原抗体と同一の標識子で標識されたIgG1抗体(ネガテイブコントロール)のヒストグラムである。
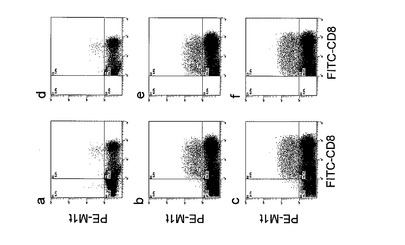
【図4】表2に示したドナーαにおける各サンプルのサイトグラムを示す。本図の基本的な説明は、図2に準ずる。ただし、本図においてはa、dは、反応細胞のみを、b、eは、反応細胞にCD56−DC、A27L及びゾメタを加えて、そしてc、fは、反応細胞にCD56+DC、A27L及びゾメタを加えて、培養したものである。
【図5】ドナーαにおいてゾメタで感作されたCD56−DCにより誘導されたγδT細胞中のCD56+γδT細胞の割合(A)とゾメタで感作されたCD56+DCにより誘導されたγδT細胞中のCD56+γδT細胞の割合(B)を、PC5標識した抗CD56抗体で検出したフローサイトメトリーの結果(黒色ヒストグラム)で示す。各ヒストグラムにおいて下記に示す%は、CD56抗原を発現している細胞(各ヒストグラムにおいて縦波線よりも右領域の細胞で示される)の数比を示す。また、白色ヒストグラムは、各表面抗原抗体と同一の標識子で標識されたIgG1抗体(ネガテイブコントロール)のヒストグラムである。
【図6】癌患者由来の末梢血から調製されたCD56+DCと反応細胞を用いて、本発明の方法に基づいてゾメタで感作したものを培養期間(7、10、12、14日)ごとにFITC標識したVγ9抗体を用いたフローサイトメトリーで検出したときの結果(黒色ヒストグラム)を示す。各ヒストグラムにおいて縦波線よりも右領域の細胞が、Vγ9+細胞(γδT細胞)であり、ヒストグラム下に示す%は、測定した全T細胞中のVγ9+細胞の数比を示す。
【0032】
また、測定されたVγ9+細胞において、γδT細胞の表現抗原である、NKG2D、CD69、CD56及びCD16の発現をそれぞれPE標識抗NKG2D抗体、ECD標識抗CD69抗体、PC5標識抗CD56抗体及びPC7標識抗CD16抗体を用いたフローサイトメトリーで検出したときの結果(黒色ヒストグラム)を示す。各ヒストグラムにおいて縦波線よりも右領域の細胞が、それぞれの抗体の陽性細胞であり、ヒストグラム下に示す%は、測定した全Vγ9+細胞中のそれぞれの抗原陽性細胞の数比を示す。
【発明を実施するための形態】
【0033】
1.CD56陽性T細胞増強方法
本発明の第1の実施形態は、CD56陽性T細胞(CD56+T細胞)増強方法である。
本明細書において「CD56+T細胞(の)増強」とは、CD56+T細胞を誘導し、増殖させ、かつ活性化することをいう。例えば、T細胞の細胞傷害機能が強化されること、すなわちCTLが増加すること、及び/又はT細胞表面の活性及び/又は増殖に関するレセプターの発現が増強されること等が含まれる。
【0034】
本発明のCD56+T細胞増強方法は、混合培養工程を含む。また、必要に応じて、その混合培養工程内において、さらに選択ステップを含むことができる。以下、本工程及び選択ステップの構成について詳説する。なお、本実施形態において、各操作は、原則として滅菌処理済の試薬、培地、器具等を使用し、培養はクリーンルーム内のクリーンベンチ等の無菌的環境下で行うことを前提とする。これは、雑菌等のコンタミネーションを防止するためである。
【0035】
1−1.混合培養工程
「混合培養工程」とは、T細胞をCD56+DC及びサイトカインと混合培養する工程である。
1−1−1.T細胞とその調製
「T細胞」とは、骨髄中の造血幹細胞に由来し、原則として胸腺内で分化・成熟したリンパ球の1種をいう。細胞表面にαβ型レセプターを発現するαβT細胞とγδ型レセプターを発現するγδT細胞が知られるが、本明細書においてはいずれも包含する。また、ここでいうαβT細胞は、獲得型免疫応答の調整に関与するヘルパーT細胞とインターフェロンγ(INF-γ)産生作用と細胞傷害活性を示すNKT細胞のいずれも含む。さらに、γδT細胞は、細胞傷害活性を示すキラーT細胞を含む。本工程で使用するT細胞は、本実施形態のCD56+T細胞増強方法の反応細胞として供される。
【0036】
本工程で使用するT細胞の調製方法は、特に限定はしない。当該分野で公知の方法、例えば、PMBCsからMACS法によってCD3+細胞として分離されたT細胞を用いることができる(Langman R.S., et al., 2007, Blood, 109:1113-1122)。その他にも、例えば、次項の「(2)CD56+DCとその調製」において、血液からCD14+細胞を単離した後の細胞(CD14−細胞)を利用することができる。具体的には、CD14-細胞をAIM-V培地(インビトロジェン)等に懸濁すればよい。また、必要に応じて0.5〜20%の牛血清(BSA)、牛胎児血清(FBS)、ヒト血清、ヒト血漿等を培地に添加することができる。CD14−細胞は、凍結保存したものを解凍、洗浄後懸濁したものを用いてもよい。この方法で得られるCD14−細胞集団は、その多くがT細胞である。B細胞も僅かに包含するものの、それ自体は、本発明の方法では増強されないことから本工程で使用するT細胞に混在していても問題はない。
【0037】
1−1−2.CD56陽性樹状細胞とその調製
本明細書において「CD56陽性樹状細胞(CD56+DC)」とは、細胞表面にCD56抗原を発現する樹状細胞である。
【0038】
本工程で使用するCD56+DCの調製方法は、特に限定はしない。例えば、当該分野で公知の方法、例えば、血液から直接単離する方法を用いてもよい。以下、CD56+DCの調製方法を、具体例を挙げて説明するが、本発明において使用するCD56+DCの調製方法は、この方法に限られない。
【0039】
(1)血液の採取
CD56+DCの調製は、まず、DCの前駆細胞を血液から取得することから始める。本発明において「血液」とは、DC及びDC前駆細胞を含む血液成分をいう。例えば、全血(末梢血を含む)、臍帯血、骨髄液やその成分の一部、例えば、単核球等が該当する。好ましくは単核球である。中でも末梢血から得られる末梢血単核球(Peripheral Blood Mononuclear Cells:以下「PBMCs」とする)は、時期を選ばずに容易に採取できる上にドナーへの侵襲性が低いため特に好ましい。
【0040】
前記「血液」を提供するドナーは、原則として生きている哺乳動物である。哺乳動物の種類は問わないが、好ましくはヒトである。対象とするドナーは、本発明の製造方法で得られるCD56+T細胞を投与する動物と同一の種類であることが望ましい。例えば、本発明の製造方法で得られるCD56+T細胞をヒトに投与する場合、ドナーはヒトであることが好ましい。また、細胞免疫療法を前提とした場合、ドナーはCD56+T細胞を投与するレシピエント自身となる。自己の血液由来の血液製剤であれば、投与後の拒絶反応の可能性を限りなく排除することができるからである。細胞免疫療法を前提とする場合には、レシピエントでもあるドナーが健常体である必要はない。例えば、癌やウイルス感染症に罹患した患者であってもよい。血液は、ドナーから採取されたものにヘパリンやクエン酸等を添加して抗凝固処理を施した後に、一旦冷蔵若しくは冷凍で保存した保存血液等でもよい。このような冷凍又は冷蔵血液を用いる場合、解凍及び加温処理は、公知の方法に基づいて行えばよい。例えば、凍結保存されたPBMCsにRPMI-1640培地を添加して解凍後、37℃、5%CO2下で3時間インキュベートする方法が挙げられる。
【0041】
ドナーからの採血方法は、公知の方法に従えばよい。例えば、末梢血であれば末梢部の静脈等に注射をして採取すればよく、骨髄液であれば骨髄穿刺(マルク)によって採取すればよく、臍帯血であれば分娩後胎盤の娩出前の臍帯に針を刺して採取すればよい。以下、末梢血の採取について、一例を挙げて具体的に説明をする。
【0042】
末梢全血は、生体の末梢部血管、例えば、静脈又は動脈に注射針を刺して真空採血管、採血バッグ等による公知の全血採取法によって採取することができる。採取する容量は、ドナーの負担にならず、かつ所定のCD56+T細胞量を製造する上で必要な量である。例えば、大人一人に対して一回に投与するCD56+T細胞を製造する場合、通常20mL〜50mLあればよい。ただし、癌患者のように血中のPBMCs数が低下している場合や多量のPBMCsを必要とする場合には、成分採血(アフェレーシス)により、PBMCsだけを必要量選択的に採取することもできる。採取後、血液が凝固しないように、例えば、シリンジ内部等をヘパリン等の血液凝固阻止剤若しくは血液凝固阻害剤で予めコーティングしておくか、又は採取した血液にヘパリンやクエン酸等を添加しておくことが好ましい。また、末梢全血から血漿を分離し、血球成分を使用してもよい。血漿は、例えば、末梢全血を遠心管に移して、2000rpm〜4000rpmで5〜20分間遠心し、その上清を得ればよい。それを56℃にて30分間程度加温して非働化し、2000rpm〜4000rpmで5〜20分間遠心して、血小板など沈殿物を除去後、細胞培養の栄養原として使用することもできる。
【0043】
(2)単球の分離
次に、末梢全血からPBMCsを分離する。PBMCsの分離方法は、赤血球から有核細胞を分離するあらゆる方法を利用することができる。例えば、フィコール・ハイパック(Ficoll−Hypaque)やフィコール・コンレイ(Ficoll−Conray)を比重液とした密度勾配遠心法を用いて、末梢全血又は血漿分離後の血球成分から得る方法が挙げられる。これらの比重液は、市販の分離液等を利用すると便利である。例えば、Ficoll−Paque PLUS(Amersham社)やLYMPHOPREP(AXIS−SHIELD社)等が利用できる。PBMCsの分離方法については、キット添付のプロトコールに従えばよい。
【0044】
分離後のPBMCsは、比重液を除去するために生理食塩水、PBS(-)や培養細胞用の培地で数回洗浄する。ここで培養細胞用の培地としては、例えば、血清を含まないPBS(-)、AIM-V培地、RPMI1640培地又は培養に用いる無血清培地等を用いればよい。前記PBS(-)若しくは培地で洗浄後、回収したPBMCsの数を血球計算板でカウントしておくことが好ましい。健康なヒト成人の場合、通常、20mL〜60mLの末梢全血から2×107個〜6×107個以上のPBMCsを回収することができる。
【0045】
(3)CD14+細胞の分離
続いて、回収したPBMCsから、DCの前駆細胞である単球のマーカーCD14を発現しているCD14+細胞(単球細胞)を分離する。分離方法は、CD14+細胞を分離可能な公知のあらゆる方法を利用することができる。例えば、抗CD14抗体標識されたマグネットビーズを用いたMagnetic Cell Sorting(以下、「MACS」と記す)を利用して単球を単離、回収する方法は、簡単でかつ単球細胞の回収率が高いため好ましい。MACSの詳細な方法については、Miltenyi Biotec, Auburn社の分離プロトコールを参照すればよい。他の方法として、回収したPBMCsを培養フラスコ等の培養容器に移し,34℃〜38℃、好ましくは37℃で、2%〜10%、好ましくは5%のCO2濃度条件下で1時間以上培養した後、容器壁面に付着した細胞をDC前駆細胞として用いる方法等を利用することもできる。
【0046】
(4)CD56+DCの誘導
得られたDC前駆細胞(CD14+DC)をCD56+DC(CD14+CD56+DC)に誘導する。誘導方法は、前記誘導を達成できる方法であれば、特に限定はしない。例えば、Papewalis C et al., J. Immunology, 2008, 180:1462-1470に記載の方法を用いることができる。この方法では、DCをGM-CSF及びIFN-αと共に34℃〜38℃、好ましくは37℃で、2%〜10%、好ましくは5%のCO2濃度条件下で、RPMI-V培地、AIM-V培地等を用いて3〜7日間培養することで達成できる。また、培養液中に、必要に応じて、0.5〜20%のBSA、FBS、ヒト血清、ヒト血漿等を培地に添加することができる。誘導後のCD56+DCは、表面抗原に基づく抗体(抗CD56抗体)を用いたMACS等によって分離することによってその純度を高めることができる
【0047】
1−1−3.サイトカイン
本明細書において「サイトカイン」とは、細胞間の情報伝達を担う多種多様なタンパク質性のホルモンであって、免疫系においては、前述のようにT細胞やNK細胞等のリンパ球を増殖若しくは活性化させる作用を有する。例えば、インターロイキン(Interleukin)やINF、TNF、MCP等が挙げられる。本発明のCD56+T細胞増強方法に適当なサイトカインとしては、例えば、IL-2、IL-12、IL-15、IL-18、IL-21が挙げられる。これらの一以上のサイトカインであれば、いずれのサイトカインであってもよいが、特に好ましいサイトカインは、IL-2である。サイトカインは、各メーカー(例えば、Chiron、Smitomo等)から市販されているものを利用すればよい。細胞免疫療法に用いるのであれば、医療用グレードの抗体を使用することが好ましい。
【0048】
1−1−4.混合培養方法
本工程では、混合培養によりT細胞にCD56+DC及びサイトカインを接触させて、CD56+T細胞を増強することを特徴とする。
【0049】
混合培養方法の具体的な例として、まず、上記で調製したT細胞を、例えば、培地で細胞密度1〜2×106個/mLとなるように調製する。ここで用いる培地は、前述した細胞培養用の適当な培地に、非働化処理をしたBSA、FBS、ヒト血清若しくは血漿を容量比(V/V)で5〜20%程度添加したものを用いればよい。細胞免疫療法に用いるためにCD56+T細胞を増強する場合には、無血清培地に自己血漿を添加したものを用いることが望ましい。自己血漿は、血液採取工程後に得られる血液から調製すればよい。例えば、採取した末梢全血を室温(10℃〜30℃:以下同じ。)にて3000rpmで10分間程度遠心して得た上清を自己血漿とすることができる。また、培地には必要に応じて、ストレプトマイシン、ペニシリン、カナマイシン、ゲンタマイシン等の抗生物質を添加してもよい。
【0050】
次に、T細胞を含む培地に上記で調製したCD56+DCとサイトカインを添加する。CD56+DCは、上記培地に細胞密度1×105〜2×105個/mLとなるように添加する。また、サイトカインは、上記培地に終濃度が0.1〜5000U/mL、好ましくは1〜3000U/mLとなるように添加する。添加後、培地を十分に混合し、34℃〜38℃、好ましくは37℃で、2%〜10%、好ましくは5%のCO2濃度条件下で5〜30日間、好ましくは7〜28日間、より好ましくは7〜14日間培養する。その間、必要に応じて、培地交換を行ってもよい。
【0051】
1−2.選択ステップ
前記混合培養工程は、その工程内に選択ステップを含むことができる。
本明細書において「選択ステップ」とは、本発明のCD56+T細胞増強方法において、特定のCD56+T細胞を他のCD56+T細胞よりも強く増強させたい場合に、必要に応じて採用し得るステップである。例えば、疾患抗原特異的CD56+CD8+CTL増強ステップ、又はCD56+γδT細胞増強ステップが挙げられる。
以下、それぞれのステップの構成について、具体的に説明をする。
【0052】
1−2−1.疾患抗原特異的CD56+CD8+CTL増強ステップ
「疾患抗原特異的CD56+CD8+CTL増強ステップ」とは、本発明のCD56+T細胞増強方法において、疾患抗原特異的なCD56+CD8+CTLをより強く増強させるためのステップである。本ステップは、疾患抗原ペプチド感作手段、及び/又はビスホスホネート補助感作手段を含む。
【0053】
なお、本ステップを選択する場合、前記混合培養時における培養液中のサイトカイン、好ましくはIL-2の終濃度は、1〜200U/mL、好ましくは10〜150U/mL、より好ましくは10〜50U/mLとなるようにしておく。
【0054】
(1)疾患抗原ペプチド感作手段
「疾患抗原ペプチド感作手段」とは、前記混合培養工程に疾患抗原ペプチドを加えて、CD56+DCをその疾患抗原ペプチドで感作する手段である。疾患抗原特異的CD56+CD8+CTL増強ステップにおいて本手段を行うことによって、疾患抗原特異的CD56+CD8+CTLを、選択的に増強することができる。
【0055】
本明細書において「感作」とは、CD56+DCを所定の物質と反応させることをいう。好ましくは前記所定の物質をCD56+DCの表面上に直接的に又は間接的に提示させることである。前記物質は、例えば、疾患抗原ペプチド及び/又は後述するビスホスホネート誘導体又はその塩若しくはその水和物が挙げられる。
【0056】
本明細書において「抗原ペプチド」とは、抗原となり得るペプチドであって、原則として一以上のエピトープを含む。ペプチドの長さは特に限定はしないが、前述のように少なくとも一のエピトープを含む必要があることから、下限は、通常は5アミノ酸、好ましくは6アミノ酸、より好ましくは7アミノ酸、さらに好ましくは8アミノ酸あればよい。また、上限は、タンパク質の全長である。通常は、前記下限以上500アミノ酸以下、300アミノ酸以下、200アミノ酸以下、100アミノ酸以下、80アミノ酸以下、60アミノ酸以下、50アミノ酸以下、40アミノ酸以下、30アミノ酸以下、又は20アミノ酸以下のペプチドが好ましく利用される。
【0057】
また、本明細書において「疾患抗原ペプチド」とは、疾患原因となる標的タンパク質又はその断片由来の抗原ペプチドをいう。ここでいう「疾患」は、宿主の身体や精神に機能的障害又は器質的障害がもたらされた結果、その宿主の身体的又は精神的健康が損なわれた状態、すなわち病気の状態をいう。例えば、腫瘍又は感染症を含む。腫瘍は、良性腫瘍及び悪性腫瘍(悪性新生物、癌;以下「癌」として示す)を含む。癌の種類は問わない。例えば、上皮性腫瘍や肉腫、白血病、骨髄腫等が該当する。より具体的には、脳腫瘍、網膜芽腫、基底細胞癌、悪性黒色腫、舌癌、食道癌、胃癌、大腸癌、肺癌、白血病、リンパ腫、乳癌、子宮頸癌、子宮体癌、卵巣癌、前立腺癌、精巣腫瘍、膀胱癌、腎臓癌、肝臓癌、膵臓癌及び線維肉腫等が挙げられる。感染症は、例えば、ウイルス感染症、細菌感染症及び寄生虫感染症を含む。ここでいう「ウイルス感染症」は、ウイルス感染による疾患全般を指すが、特に治癒が難治の慢性ウイルス感染症や急性ウイルス感染症の予防が該当する。当該難治な慢性ウイルス感染症としては、例えば、AIDSを引き起こすHIV感染症、ウイルス性慢性肝炎、子宮頸癌を引き起こすヒトパピローマウイルス感染症が挙げられる。また、急性ウイルス感染症としてはインフルエンザ等のウイルス性呼吸器感染症や免疫不全状態での急性ウイルス感染症が挙げられる。ウイルスは、DNAウイルス又はRNAウイルスを問わない。また、また、「細菌感染症」とは、真正細菌(グラム陽性菌及びグラム陰性菌を含む)又は真菌(糸状菌、酵母等及び担子菌を含む)による感染症である。例えば、カンジダ感染症、ブラストミセス症、ヒストプラスマ症等が挙げられる。さらに、ここでいう「寄生虫感染症」は、原虫又は蠕虫による疾患全般を指す。例えば、マラリア、リューシュマニア症、フィラリア症、エキノコックス症、日本住血吸虫症等が挙げられる。
【0058】
本手段で使用する疾患ペプチド抗原は、標的とする疾患に対する抗原ペプチドを使用することが好ましい。例えば、標的とする疾患がメラノーマ(悪性黒色腫)であれば、メラノーマに特異的な抗原ペプチドを使用すればよい。疾患ペプチド抗原は、単一種であってもよいし、二以上の、好ましくは同一の疾患に対する、異なる疾患ペプチド抗原を用いてもよい。
【0059】
疾患抗原ペプチドは、天然のタンパク質又はその断片由来であってもよいし、それらの配列に基づいた合成ペプチドであってもよい。合成ペプチドの使用は、疾患原因となる標的タンパク質を得るために被検体から血液や組織等を採取する必要がないため、被検体に対する侵襲性が極めて低いことから好ましく利用できる。特に各癌抗原ペプチドは、表1に示す公知の抗原を、メーカーから購入又は公知のアミノ酸配列に基づいて合成したものを利用してもよい。各癌種に対する疾患抗原ペプチドの具体例として、下記表1に記載の抗原ペプチドが挙げられる。表1に記載の抗原ペプチドは、そのアミノ酸配列が公知であり、化学合成によって、また市販品の購入によって容易に入手可能である。
【0060】
【表1】
【0061】
本手段は、前記混合培養工程において、CD56+DCを疾患抗原ペプチドで感作可能な限り、いかなる態様でも行うことができる。例えば、前記混合培養工程において、T細胞、CD56+DC、サイトカインと共に疾患抗原ペプチドを培地に加えて、混合培養することによって、CD56+DCを疾患抗原ペプチドで感作させてもよい。また、CD56+DCを疾患抗原ペプチドと共に1時間〜24時間培養して予備感作させた後に、T細胞及びサイトカインを含む培地にそれらを混合して、引き続き培養しても構わない。さらに、T細胞、CD56+DC及びサイトカインを混合し、1時間〜24時間培養した後に、疾患抗原ペプチドを追加してもよい。
【0062】
疾患抗原ペプチドの濃度は、通常DCを疾患抗原ペプチドにより感作する濃度であればよく、特に限定はされない。例えば、Cance Research,1999,Vol.59,2167-2173、The Journal of Immunology,1995,Vol.154,2257-2265、又は、The Journal of Immunology,1994,153,996-1003に記載されているように、培養液における終濃度が0.01〜20μg/mL、好ましくは0.1〜2μg/mLであればよい。
【0063】
(2)ビスホスホネート補助感作手段
本発明において「ビスホスホネート補助感作手段」とは、CD56+DCをビスホスホネート誘導体又はその塩若しくはその水和物(以下、本明細書では「ビスホスホネート誘導体等」とする)で感作する手段である。本手段は、疾患抗原特異的CD56+CD8+CTL増強ステップにおいて、前記疾患抗原ペプチド感作手段に加えて、必要に応じて追加される補助的手段である。したがって、本手段を選択した場合、CD56+DCは、ビスホスホネート誘導体等及び疾患抗原ペプチドで共感作されることとなる。本発明において、「共感作」とは、同時、連続的、又は断続的に2以上の物質で感作することをいう。本手段を行うことにより、疾患抗原ペプチド感作手段のみの場合よりも、さらに疾患抗原特異的CD56+CD8+CTLを増強することができる。
【0064】
本明細書において「ビスホスホネート誘導体」とは、下記一般式1で示される化合物をいう。
【0065】
【化1】
【0066】
上記式中、R1は、水素原子(H)又は低級アルキル基を表し、R2とR3は、それぞれ独立して、水素原子、ハロゲン、ヒドロキシル基、アミノ基、チオール基、置換された若しくは置換されていないアリール基、置換された若しくは置換されていないアルキル基、低級アルキルアミノ基、アルアルキル基、シクロアルキル基又は複素環式基を表し、又はR2とR3は、それらを含む環状構造の一部を形成し、該環状構造を形成する置換基は、R2とR3において、それぞれ独立して、ハロゲン、低級アルキル基、ヒドロキシル基、チオール基、アミノ基、アルコキシ基、アリール基、アリールチオ基、アリールオキシ基、アルキルチオ基、シクロアルキル基又は複素環式基に由来する。
【0067】
ビスホスホネート誘導体の具体例としては、ゾレドロン酸、パミドロン酸、アレンドロン酸、リセドロン酸、イパンドロン酸、インカドロン酸、エチドロン酸が挙げられる。本発明においては、一以上のビスホスホネート誘導体又はその塩若しくはその水和物をNK細胞増殖刺激因子として添加することができる。本発明において、特に好ましいビスホスホネート誘導体は、ゾレドロン酸又はNK細胞の増殖/活性化を有するゾレドロン酸誘導体又はその塩若しくはその水和物である。
【0068】
ゾレドロン酸(商品名ゾメタ(登録商標)、ノバルティスファーマ社)は、骨吸収抑制活性を有するビスホスホネートで、悪性腫瘍による高カルシウム血症又は多発性骨髄腫による骨病変及び固形癌骨転移による骨病変に対する治療薬として知られている。また、その化学構造の中に窒素含有ビスホスホネート(N-BPs:Nitrogen containing-BisphosPhonate)を包含するため、細胞内でFarnesyl PyrophosPhate (FPP)の合成を抑制して、その結果、前駆体であるIsoPentenyl Pyrophosphate(IPP)が蓄積する。それによって、その生体の免疫反応を活性化できることが報告されている(van Beek E, et al., 1999, Biochem Biophys Res Commun, 264:108-11; Gober HJ, et al., 2003, J Exp Med, 197:163-8.)。
【0069】
「その塩」とは、前記ビスホスホネート誘導体、好ましくはゾレドロン酸の塩基性付加塩をいう。塩基性付加塩としては、例えば、ナトリウム塩若しくはカリウム塩のようなアルカリ金属塩、カルシウム塩若しくはマグネシウム塩のようなアルカリ土類金属塩、トリメチルアミン塩、トリエチルアミン塩、ジシクロヘキシルアミン塩、エタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩若しくはブロカイン塩のような脂肪族アミン塩、N,N-ジベンジルエチレンジアミンのようなアラルキルアミン塩、ピリジン塩、ピコリン塩、キノリン塩若しくはイソキノリン塩のような複素環芳香族アミン塩、アルギニン塩若しくはリジン塩のような塩基性アミノ酸塩、アンモニウム塩又はテトラメチルアンモニウム塩、テトラエチルアモニウム塩、ベンジルトリメチルアンモニウム塩、ベンジルトリエチルアンモニウム塩、ベンジルトリブチルアンモニウム塩、メチルトリオクチルアンモニウム塩若しくはテトラブチルアンモニウム塩のような第4級アンモニウム塩が挙げられる。
【0070】
本手段は、前記混合培養工程において、CD56+DCがビスホスホネート誘導体等で感作される限り、いかなる態様でも行うことができる。また、結果として、CD56+DCがそれらに共感作されれば足りることから、CD56+DCを疾患抗原ペプチドとビスホスホネート誘導体等で感作する順序は問わない。例えば、本ステップにおいて、本手段を疾患抗原ペプチド手段と同時に、手段前に、又は手段後に行うことができる。具体的には、前記混合培養工程において、T細胞、CD56+DC、サイトカイン、疾患抗原ペプチドと共にビスホスホネート誘導体等を培地に加えて、混合培養することによって、CD56+DCを共感作する態様であってもよい。また、CD56+DCを疾患抗原ペプチド及びビスホスホネート誘導体等と共に12時間〜24時間培養して予め共感作した後に、T細胞及びサイトカインを含む培地にそれらを混合して、引き続き培養しても構わない。あるいは、CD56+DCを疾患抗原ペプチドと共に1時間〜24時間培養した後、さらにビスホスホネート誘導体等を加えて12時〜48時間培養し、その後にT細胞及びサイトカインを含む培地にそれらを混合する方法であってもよいし、逆に、CD56+DCをビスホスホネート誘導体等と共に12時〜48時間培養した後、さらに疾患抗原ペプチドを加えて1時間〜24時間培養し、その後にT細胞及びサイトカインを含む培地にそれらを混合する方法であってもよい。また、疾患抗原ペプチド感作手段後に行う場合には、疾患抗原ペプチド感作手段後の培養液にビスホスホネート誘導体等を添加すればよい。
【0071】
ビスホスホネート誘導体等の濃度は、通常細胞をビスホスホネート誘導体等により感作する濃度であればよく、特に限定はされないが、例えば、The Journal of Immunology,2001,Vol.166,5508-5514、又は、Blood,2001,Vol.98,No.5,1616-1618]に記載されているように培養液における終濃度が0.001μM〜20μMであればよい。好ましくは0.001μM〜5μMである。
【0072】
1−2−2.CD56+γδT細胞増強ステップ
「CD56+γδT細胞増強ステップ」とは、本発明のCD56+T細胞増強方法において、CD56+γδT細胞をより強く増強させるためのステップである。本ステップは、ビスホスホネート感作手段を含む。本ステップでは、前記「疾患抗原ペプチド感作手段」に相当する、CD56+DCを疾患ペプチドで感作する手段を含まない点において、疾患抗原特異的CD56+CD8+CTL増強ステップとは異なる。
【0073】
なお、本ステップを選択する場合、前記混合培養時における培養液中のサイトカイン、好ましくはIL-2の終濃度は、300〜3000U/mL、好ましくは400〜200U/mL、より好ましくは500〜1000U/mLとなるようにしておく。
【0074】
(1)ビスホスホネート感作手段
「ビスホスホネート感作手段」とは、前記CD56+DCをビスホスホネート誘導体等で感作する手段である。基本的な操作手段は、前記「ビスホスホネート誘導体補助感作手段」と同じでよい。CD56+γδT細胞増強ステップにおいて、本手段を行うことによって、CD56+γδT細胞がより強く増強される。CD56+γδT細胞は、CD56−γδT細胞と比較して細胞傷害活性と相関するCD107a分子の発現が高く、腫瘍細胞株に対して高い傷害活性を有することが知られている(Alan A. et al., Clin Cancer Res, 2008, Vol. 14:4232-4240)。
【0075】
本手段で使用するビスホスホネート誘導体等は、前記「ビスホスホネート誘導体補助感作手段」で詳説したことから、ここでの説明は省略する。
【0076】
本手段は、前記混合培養工程において、CD56+DCをビスホスホネート誘導体等で感作可能な限り、いかなる態様でも行うことができる。例えば、前記混合培養工程において、T細胞、CD56+DC、サイトカインと共にビスホスホネート誘導体等を培地に加えて、混合培養することによって、CD56+DCをビスホスホネート誘導体等で感作させることができる。また、CD56+DCをビスホスホネート誘導体等と共に12時間〜24時間培養して予備感作させた後に、T細胞及びサイトカインを含む培地にそれらを混合して、引き続き培養しても構わない。さらに、T細胞、CD56+DC及びサイトカインを混合し、12時間〜24時間培養した後に、ビスホスホネート誘導体等を追加してもよい。
【0077】
ビスホスホネート誘導体等の濃度は、通常細胞をビスホスホネート誘導体等により感作する濃度であればよく、特に限定はされないが、例えば、The Journal of Immunology,2001,Vol.166,5508-5514、又は、Blood,2001,Vol.98,No.5,1616-1618]に記載されているように培養液における終濃度が0.001μM〜20μMであればよい。好ましくは5μM〜10μMである。
【0078】
1−3.効果
本発明によれば、ドナー及びレシピエントに対する侵襲性が低く、かつ少量の血液中に含まれるT細胞から簡便かつ効率的にCD56+T細胞、特に疾患抗原特異的CD56+CD8+CTL又はCD56+γδT細胞を増強することができる。具体例を挙げると、40mLの末梢血を採取して、本発明の方法を用いて14日間培養することによって約1×109個のCD56+γδT細胞を調製できる。ここで、健常な大人の血中T細胞数は、1500〜4500細胞/mLであり、γδT細胞は、そのうちの約5%、すなわち75〜225細胞/mLであることが知られている。つまり、γδT細胞は、健常人の全血液量(約5L)中にも3.75×105〜11.25×105細胞しか含まれていない。ところが、本発明の方法を用いることで、僅か40mLの末梢血から、健常人の全血液の約2600〜7800倍にも及ぶγδT細胞数が増強することができる。
【0079】
2.CD56+T細胞強化型血液製剤
本発明の第2の実施形態は、CD56+T細胞強化型血液製剤である。本発明のCD56+T細胞強化型血液製剤は、前記第1実施形態に記載のCD56+T細胞の増強方法によって増強されたCD56+T細胞を主成分として含むことを特徴とする。
【0080】
本発明において「強化」とは、細胞を増殖及び活性化させること、又はさせたことを意味する。
【0081】
CD56+T細胞強化型血液製剤において培養で用いた培地や当該培地に添加したサイトカイン、疾患抗原ペプチド、ビスホスホネート誘導体等の添加物は不要である。したがって、CD56+T細胞強化型血液製剤を使用するに当たっては、前記培養液から培地や前記添加物を可能な限り除去し、増強されたCD56+T細胞が主成分となるように調製しておくことが好ましい。ただし、本実施形態のCD56+T細胞強化型血液製剤を調製するにあたり第1実施形態で用いたCD56+DCは、必ずしも除去する必要はない。CD56+DCもまた、免疫療法において有効成分となり得るからである。
【0082】
第1実施形態のCD56+T細胞増強方法実施後の培養液から培地や添加物の除去する方法の一具体例として、まず、増強されたCD56+T細胞を含む培養液を滅菌済み遠心チューブに移し、1200rpmにて8分間ほど室温にて遠心し、添加物を含む上清の培地を除去する。CD56+T細胞やCD56+DC(以下、「CD56+T細胞等」とする)は、沈殿物として回収できる。回収されたCD56+T細胞等は、2回以上PBS(-)で洗浄することが好ましい。洗浄後のCD56+T細胞等は、血球計算板を用いて細胞数をカウントし、10mL〜200mLの乳酸リンゲル液又は生理食塩水で調整する。こうして、本実施形態のCD56+T細胞強化型血液製剤を調製することができる。当該製剤からCD56+DCを除去したい場合には、例えば、抗CD14抗体を用いたMACSを利用してCD56+DCのみを分離除去すればよい。調製後、必要に応じて当該血液製剤にサイトカイン等を添加することも可能である。
【0083】
本実施形態の血液製剤は、含有するCD56+T細胞の70%以上がCTLであることが望ましい。CTL率、すなわちCD56+T細胞の活性化率は、K562白血病細胞株を用いた細胞傷害活性又は活性化マーカーの発現によって判断できる。活性化マーカーは、CD69等の公知のマーカーが利用できる。また、それらの検出には、それぞれのマーカーに対する抗体を用いればよい。
【0084】
本実施形態のCD56+T細胞強化型血液製剤は、製造後直ちに使用することも、また0℃〜8℃の温度下で所定の期間、又は保存液等を加えて超低温下(約-80℃)若しくは液体窒素中で数年に渡る長期間保存することも可能である。前記保存液としては、市販のリンパ球保存液を用いると便利である。例えば、バンバンカー(日本ジェネティックス社)、ケーエムバンカーII(コスモバイオ社)等が利用できる。
【0085】
本実施形態のCD56+T細胞強化型血液製剤は、1×107〜1×108個/mLの増強されたCD56+T細胞を包含する。それ故、この血液製剤の投与によって被検体のCD56+T細胞数を速やかに増大させることができる。したがって、CD56+T細胞強化型血液製剤の投与により、腫瘍等の疾患を有する被検体の免疫応答を増強し、その疾患の進行の抑制、治癒が可能となる。
【0086】
本実施形態のCD56+T細胞強化型血液製剤によれば、増強されたCD56+T細胞を多数含む血液製剤を凍結保存できることから、必要時に必要量を被検体に投与することができる。
【0087】
3.CD56+T細胞増強剤
本発明の第3の実施形態は、CD56+T細胞増強剤である。本発明のCD56+T細胞増強剤は、T細胞の培養液に添加して混合培養することによって、CD56+T細胞を増強することができる。
【0088】
3−1.構成
3−1−1.必須成分
本発明のCD56+T細胞増強剤は、CD56+DC及びサイトカインを必須の成分として含有する。CD56+T細胞増強剤に含まれるCD56+DCは、前記第1実施形態の「1−1−2.CD56+DCとその調製」の項に記載した方法で調製したものを利用できる。また、サイトカインは、IL-2、IL-12、IL-15、IL-18又はIL-21のうちいずれか一以上が含まれていればよい。特に好ましいのは、IL-2である。
【0089】
CD56+DCとサイトカインは、CD56+T細胞増強剤において、必ずしも混合状態である必要はない。すなわち、CD56+T細胞増強剤とは、各成分を同時に又は別個にT細胞の培養液に添加することによって、CD56+T細胞を増強し得る薬剤である。
【0090】
3−1−2.選択成分
本明細書において、「選択成分」とは、本発明のCD56+T細胞増強剤に必要に応じて添加することのできる成分である。本発明のCD56+T細胞増強剤は、前記必須成分にさらなる選択成分を加えることで、疾患抗原特異的CD56+CD8+CTLやCD56+γδT細胞のような特定のCD56+T細胞をより効率的に増強することができる。選択成分としては、例えば、疾患抗原ペプチドビスホスホネート誘導体等が挙げられる。疾患抗原ペプチド及びビスホスホネート誘導体等については、いずれも前記第1実施形態で詳説していることから、ここではその説明を省略する。
【0091】
以下、疾患抗原特異的CD56+CD8+CTL増強剤、及びCD56+γδT細胞増強剤について、具体的に説明をする。
(1)疾患抗原特異的CD56+CD8+CTL増強剤
「CD56+CD8+CTL増強剤」とは、前記必須成分に加えて、疾患抗原特異的CD56+CD8+CTLを増強するための選択成分を含むCD56+T細胞増強剤である。疾患抗原特異的CD56+CD8+CTL増強剤は、選択成分として疾患抗原ペプチドを含有する。ここで使用する疾患抗原ペプチドは、原則として標的とする疾患に特有の疾患抗原ペプチドである。例えば、標的とする疾患が、癌であり、かつその癌がメラノーマである場合、本発明の疾患抗原特異的CD56+CD8+CTL増強剤に含まれる疾患抗原ペプチドは、メラノーマに対する抗原ペプチド、好ましくはメラノーマ特異的な抗原ペプチドとなる。例えば、表1に記載のCDC27、CDK4、CTAG1、CTAG2、gp100、MART-1、MUM-1、NAP1L1、NFE2L2等が挙げられる。なお、疾患抗原特異的CD56+CD8+CTL増強剤においては、包含するサイトカイン、好ましくはIL-2がT細胞の培養液に添加後に終濃度で1〜200U/mL、好ましくは10〜150U/mL、より好ましくは50〜100U/mLとなるように調整しておく。
【0092】
疾患抗原特異的CD56+CD8+CTL増強剤は、選択成分として疾患抗原ペプチドに加えて、ビスホスホネート誘導体等をさらに含んでいてもよい。本発明の疾患抗原特異的CD56+CD8+CTL増強剤で使用するビスホスホネート誘導体等は、前記第1実施形態の「ビスホスホネート誘導体補助感作手段」で詳説したことから、ここでの説明は省略する。なお、疾患抗原特異的CD56+CD8+CTL増強剤においては、包含するビスホスホネート誘導体等がT細胞の培養液に添加後に終濃度で0.001μM〜20μMであればよい。好ましくは0.001μM〜5μMとなるように調整しておく。
【0093】
(2)CD56+γδT細胞増強剤
「CD56+T細胞増強剤」とは、前記必須成分に加えて、CD56+γδT細胞を増強するための選択成分を含むCD56+T細胞増強剤である。CD56+γδT細胞増強剤は、選択成分としてビスホスホネート誘導体等を含有する。本発明のCD56+γδTで使用するビスホスホネート誘導体等は、前記第1実施形態の「ビスホスホネート誘導体補助感作手段」で詳説したことから、ここでの説明は省略する。なお、CD56+γδT細胞増強剤においては、包含するビスホスホネート誘導体等がT細胞の培養液に添加後に終濃度で0.001μM〜20μMであればよい。好ましくは5μM〜10μMとなるように調整しておく。
【0094】
疾患抗原特異的CD56+CD8+CTL増強剤とCD56+γδT細胞増強剤との違いは、前者が疾患抗原ペプチドを必要構成成分として包含するのに対して、後者は包含しない点、前者がビスホスホネート誘導等を補助成分として必要に応じて適宜包含すればよいのに対して、後者は必要構成成分として包含する点、及び単位量あたりのサイトカインの量が異なる点である。
【0095】
3−2.剤形
本発明のCD56+T細胞増強剤の剤形は特に問わない。適当なバッファに溶解した液体状態、粉末状態、粉末を適当な賦形剤等を添加し錠剤化したものとすることができる。また、必須成分又は選択成分を問わず、各成分が最終的にT細胞に作用できればよいことから、本発明のCD56+T細胞増強剤は、各成分が必ずしも統合されている必要はなく、個別に分離された状態であってもよい。それ故、成分ごとに剤形が異なっていてもよい。例えば、CD56+DCは培地に懸濁された状態であり、サイトカインは粉末状態でもよい。このように、成分が分離された状態の場合には、各成分を同時に又は別個にT細胞の培養液に添加することによって、CD56+T細胞を増強し得る。
【0096】
4.疾患を治療する細胞免疫療法
本発明の第4の実施形態は、第2実施形態のCD56+T細胞強化型血液製剤を生体に投与する細胞免疫療法である。
【0097】
4−1.構成
本実施形態でいう「細胞免疫療法」とは、前記第2実施形態のCD56+T細胞強化型血液製剤を被検体に投与することで、被検体における血中の単位体積あたりのCD56+T細胞、すなわち免疫力を有するT細胞の絶対数を通常血液の平均値よりも増大させて、その被検体における、癌、ウイルス感染症、細菌感染症又は寄生虫感染症に対する細胞傷害活性を増強して、疾患の症状を軽減、改善、又は治療する方法である。特に、本実施形態の細胞免疫療法は、血液ドナーとレシピエントが同一である細胞免疫療法を前提としたものである事が好ましい。これは、拒絶反応の危険性がほとんどないからである。
【0098】
「免疫力を有するT細胞」とは、免疫系における機能が強化されたT細胞を意味する。例えば、細胞傷害活性が活性化されたCD56+CD8+CTL、キラーT細胞、CD56+γδT細胞、NKT細胞が該当する。また、ここでいう「単位体積あたりの〜通常血液の平均値」とは、健常な個体の血液で一般に観察される免疫力を有するT細胞数の単位体積あたりの平均値を意味する。
【0099】
4−2.方法
本実施形態の細胞免疫療法におけるCD56+T細胞強化型血液製剤の投与方法について、細胞免疫療法を行う場合を例として以下で説明をする。当該投与方法は、実施形態1のCD56+T細胞強化型血液製剤を投与する点を除けば、従来の細胞免疫療法で知られる方法と基本的に同様である。したがって、投与方法に関しては公知の細胞免疫療法における投与方法に準じて行えばよい。例えば、前記第1実施形態に記載のCD56+T細胞増強方法によって患者から採取した血液から増強されたCD56+T細胞を主成分として含む第2実施形態のCD56+T細胞強化型血液製剤を、採取後約2週間後にその患者の体内に静脈注射又は点滴等を用いて投与する方法等が挙げられる。
【0100】
本実施形態におけるCD56+T細胞強化型血液製剤の1回あたりの投与量は、一般的な大人へ投与する場合には細胞数にして5×108個〜1×1010個のCD56+T細胞を含む容量であればよい。実際の投与では、投与する者の年齢、性別、体重、疾患の状態、体力等を勘案して適宜調節することが好ましい。
【0101】
本実施形態における細胞免疫療法の一例として、前記投与方法を1サイクルとして、約2週間間隔で1クール(6サイクル)以上投与を継続する方法が挙げられる。細胞免疫療法でない場合も、患者以外のドナー由来の血液で製造されたCD56+T細胞強化型血液製剤を投与する点を除いては、同様の方法で当該細胞免疫療法を行えばよい。
【0102】
4−3.効果
本実施形態の細胞免疫療法によれば、従来の免疫療法、特に細胞免疫療法と比較して、癌等の疾患に対する治癒に高い有効性を有する。また、従来の細胞免疫療法と基本的な操作技術等は同様であることから、細胞免疫療法の技術を有する者であれば特段の技術習得をすることなく実施できる。
【実施例】
【0103】
以下の実施例をもって本発明を具体的に説明する。なお、以下の実施例は、単に本発明を例示するのみであり、本発明はこれらの実施例によって何ら限定されるものではない。また、本実施例で使用された温度、量、時間等の数値に関して、実験上の多少の誤差及び偏差は斟酌される。
【0104】
<実施例1:CD56+T細胞の増強>
1.DCの調製
(方法)
(1)DCの分化誘導
健常ドナーから末梢血を50mL採血した。次に、血液分離比重液((Lymphoprep:AXIS-SHIELD、Norway)を用いて2400rpmで20分間遠心分離後、単核細胞成分分画を回収した。回収した分画をAIM-V medium(インビトロジェン)40mLに懸濁した後、50mLフラスコへ20mLずつ2分して、1〜2時間37℃でインキュベーションした。その後、それぞれにフラスコ内部を軽くピペッティングして非付着細胞成分を回収した。回収された非付着細胞成分は、主にT細胞集団であり、後述する「2.疾患抗原特異的CD56+CD8+CTLの調製(1)」、「3.疾患抗原特異的CD56+CD8+CTLの調製(2)」及び「4.CD56+γδT細胞の調製」における、反応細胞として使用した。
【0105】
また、純度の高いDCを得るために、前記非付着細胞成分回収後の各フラスコに、さらに新たに培地を加えて内壁を強くピぺッテイングし、残存する非付着細胞成分を可能な限り除去した。付着細胞成分からのDCの調製にはFBS(Nichirei Biosciences Inc.)又は5〜10%のAB血清(DSファーマバイオメデイカルKK)を含むAIM-Vmedium(インビトロジェン)を用いた。
【0106】
続いて、一方のフラスコ(サンプルA)にGM-CSF(Primmune Inc)とIL-4(Primmune Inc)を、最終濃度がそれぞれ1000U/mLと500U/mLなるように添加して、37℃、5% CO2濃度下において5日培養して未熟DCを調製した。さらに、TNFα (Wako Pure Chemical Industries, LTD)を最終濃度で10ng/mLとなるように加えて、引き続き37℃、5% CO2濃度下において2日間培養した。
【0107】
他方のフラスコ(サンプルB)にはGM-CSF(上記)とIFN-α(Astellas)を最終濃度が1000U/mLになるように添加して、37℃、5% CO2濃度下において3日間培養した。
【0108】
(2)DCの状態の確認
得られたDCの表面抗原の検出は、フローサイトメーター(Epics XL-MCL; Beckman Coulter )を用いて行った。まず、測定する細胞を1×103cells/μLとなるようにPBSに懸濁した溶液100μLに目的の抗体を添加し、遮光の状態で4℃、15分間染色した。抗体は、いずれも標識子で標識されたPC5-抗CD56抗体、PC7-抗CD14抗体、PE-抗HLA-ABC抗体、FITC-抗HLA-DR抗体、及びPE-抗CD86抗体を用いた(全てBeckman Coulter)。ネガテイブコントロールとして、それぞれの抗体と同一の標識子で標識されたIgG1抗体(Beckman)を用いた。細胞は、染色後、PBSで洗浄して測定した。
【0109】
(結果)
結果を図1に示す。サンプルAの細胞は、CD56陽性細胞率が0.5%であるのに対して、サンプルBの細胞は、85.8%であった。この結果から、サンプルAの細胞は、CD14−CD56−DC(以下、単に「CD56−DC」とする)、またサンプルBの細胞は、CD14+CD56+DC(以下、単に「CD56+DC」とする)であることが確認された。
【0110】
その他の表面抗原である、CD14、MHC分子(HLA-ABC、HLA-DR)及び共刺激分子(CD86)についても、CD56-DCとCD56+DCでは同程度の発現を示した。
【0111】
2.疾患抗原特異的CD56+CD8+CTLの調製(1)
(方法)
ドナーα及びβにおいて前記「1.DCの調製」で調製したサンプルA由来のCD56−DC又はサンプルB由来のCD56+DCのそれぞれ2×105個/mLを、同じく「1.DCの調整」で調製した反応細胞(主にT細胞からなる)2×106個(反応細胞:DC=10:1)と共に24ウェルプレート(SUMILON)に入れた。各ウェルの混合培養液中には、疾患抗原ペプチドとして2μg/mLのヒト主要組織適合抗原(HLA)A*0201拘束性のメラノーマ特異的抗原ペプチドMART-1改変型A27L (ELAGIGILTV:配列番号1)(SCURUM)をこの混合培養溶液中に最終濃度で50U/mLのIL-2(Chiron)を加え、37℃、5%CO2条件下で7日間培養した。培養を通して5〜10%のFBS又はAB血清を含むAIM-V培地を用いた。
【0112】
DCと反応細胞の混合培養開始から7日以降に適時、FITC標識した抗CD8抗体(Beckman Coulter)又はPE標識したMART-1改変型A27L特異的テトラマー(PE-M1t)(MBL)を用いてフローサイトメーターで全T細胞中、又は全CD8+T細胞中に占めるA27L特異的CD8+CTL(疾患抗原特異的CD8+CTL)の割合を測定した。
【0113】
さらに、PC5標識した抗CD56抗体(Beckman Coulter)を用いて、フローサイトメーターで全CD8+T細胞中、又は全A27L特異的CD8+CTL中に占めるA27L特異的CD56+CD8+CTLの割合を測定した。
【0114】
(結果)
A27L特異的CD8+CTLの割合を表2及び図2に、また、A27L特異的CD56+CD8+CTLの割合を図3に示す。図2は、表2で示すドナーαのサイトグラムである。
【0115】
【表2】
【0116】
表中「+」及び「−」は、それぞれ培養液中の存在の有無を示す。「CD56+」は、「CD56+DCを、「CD56−」は、CD56−DCを、それぞれ示す。ドナーα及びβにおいて、反応細胞のみが「+」のサンプルは、前記「1.DCの調整」で調製した反応細胞のみのサンプルであり、培養前コントロールとして用いた。
【0117】
表2から、ドナーα及びβ共にCD56+DCを用いたときが、DC56−DCを用いたときよりも、全細胞中、又は全CD8+細胞中におけるA27L抗原特異的CTLの存在率が高かった。すなわち、CD56+DCを用いた場合の方がより高いA27L抗原特異的CTL誘導能を有することが示された。
【0118】
また、図3から、CD56+DCで刺激して増殖してくるCD8+T細胞全体の中でCD56+T細胞の割合(図中縦破線より右領域)は24.7%(図3A)であるのに対して、A27L抗原特異的CD8+CTL全体(Mart1 tetramer gate)の中では54.7%であった(図3B)。このように全CD8+T細胞の中でCD56を共発現しているCD56+CD8+T細胞の割合に比べて、疾患抗原特異的CD8+CTLの中でCD56を共発現している疾患抗原特異的CD56+CD8+CTLの割合が高いという事実は、CD56の発現が疾患抗原特異的CTLの傷害活性と相関していることを示唆している。この事実は、本発明者らが見出した新規知見である。また、疾患抗原特異的CD56+CD8+CTLは、細胞傷害に関与する細胞内パーフォリンやグランザイムを多く含んでおり、高い細胞傷害活性を有していることが確認されているCD8T細胞のサブセットである(J. Immunology 2000, 164:1148-1152)。したがって、本発明の増強方法によって得られる疾患抗原特異的CD56+CD8+CTLを被検体に投与すれば、その被検体の体内における疾患抗原特異的CD56+CD8+CTLを優位に増強できることから、細胞免疫療法において治療効果の高い有用な免疫細胞を提供できる。
【0119】
3.疾患抗原特異的CD56+CD8+CTLの調製(2)
(方法)
基本操作は、前記「疾患抗原特異的CD56+CD8+CTLの調製(1)」と同じである。ただし、ここでは、CD56−DC又はCD56+DC、及び反応細胞とIL-2(Chiron)に加えて、アミノビスホスホネート剤であるゾメタ(商品名、Novartis)を、最終濃度が0.01μMとなるように添加した。
【0120】
DCと反応細胞の混合培養開始から12日以降に適時、FITC標識した抗CD8抗体(BeckmanCoulter)又はPE標識したMART-1改変型A27L特異的テトラマー(MBL)を用いてフローサイトメーターで全細胞中、又は全CD8+T細胞中に占めるA27L特異的CD8+CTL(疾患抗原特異的CD8+CTL)の割合を測定した。また、混合培養液中にゾメタを添加した場合はγδT細胞の増殖が予測されるので、上記MART-1改変型A27L特異的テトラマーの測定に加えて増殖してくる全細胞中のVγ9γδT細胞の割合を、FITC標識化抗Vγ9抗体(Beckman Coulter)を用いて同様にフローサイトメーターで測定した。
【0121】
(結果)
結果を表3及び図4に示す。
【0122】
【表3】
【0123】
表中の説明は、表2と同じである。ゾメタ及びA27Lペプチドで感作したDCをT細胞と共にIL-2存在下で培養することにより誘導されるA27L抗原特異的CD8+CTLの全細胞中に占める割合は、ドナーα及びβ共にCD56+DCを用いたときが、DC56−DCを用いたときよりも、全細胞中、又は全CD8+細胞中におけるA27L抗原特異的CTLの存在率が高かった。また、コントロールに対する増殖倍率(表中、鍵カッコで示す)に関して、表2の結果と比較すると、ゾメタを加えたときの方がさらに高かった。すなわち、CD56+DCを疾患抗原ペプチドとゾメタで共感作させると、疾患抗原特異的CTL誘導がより増強されるがことが示された。
【0124】
4.CD56+γδT細胞の調製
(方法)
前記「1.DCの調整」で調製したドナーα由来のサンプルA由来のCD56−DC又はサンプルB由来のCD56+DCのそれぞれ2×105個/mLを、同じく「1.DCの調整」で調製した反応細胞(主にT細胞からなる)2×106個(反応細胞:DC=10:1)と共に24ウェルプレート(SUMILON)に入れた。各ウェルの混合培養液中には、ゾメタ(Novartis)及びIL-2(Chiron)を、最終濃度がそれぞれ5μM及び1000U/mLとなるように加え、37℃、5%CO2条件下で14日間培養した。培養を通して5〜10%のFBS又はAB血清を含むAIM-V培地を用いた。
【0125】
培養後、FITC標識した抗Vγ9抗体(Beckman Coulter)とPC5標識した抗CD56抗体(Beckman Coulter)を用いて、フローサイトメーターでVγ9陽性細胞にgateをかけて、得られたVγ9+細胞を100%としてCD56+細胞全γδT細胞中に占めるCD56+T細胞の割合を測定した。
【0126】
(結果)
結果を表4及び図5に示す。
【0127】
【表4】
【0128】
全γδT細胞中のCD56+γδT細胞率は、CD56-DCを用いた場合が39.9%であったのに対して(表3、図5A)、CD56+DCを用いた場合は91.2%であった(表4、図5B)。この結果は、CD56+DCがCD56−DCよりもCD56+γδT細胞をより強く増強できることを示している。
【0129】
<実施例2:CD56+γδT細胞上の表面抗原の確認>
前記実施例1の「4.CD56+γδT細胞の調製」の結果から、健常ドナーの末梢血から調製したCD56+DCと反応細胞を培養すると、CD56−DCを用いたときよりも、より効率的にCD56+γδT細胞を増殖できることが確認された。そこで、癌患者由来の末梢血でも同様の効果が得られるかを検証した。
【0130】
(方法)
肺癌癌患者(ステージIV)由来の末梢血から、上記実施例1の「1.DCの調製」及び「4.CD56+γδT細胞の調製」に記載の方法と同様の方法でγδT細胞の調製を行った。培養によって増殖した全T細胞中のγδT細胞の割合、及びその増殖したγδT細胞において発現している表面抗原NKG2D、CD69、CD56及びCD16のそれぞれの発現を、培養時間を追って測定した。NKG2Dは、活性型のNKレセプターを示し、この抗原の発現が高いと細胞傷害活性が高くなる。CD69は、細胞が活性化に伴い発現する抗原で、IFN-aの産生と相関しており、CD69の発現の増加と共に抗腫瘍活性も高まる。またCD16は、Fcγレセプターを示し、この抗原の発現が高いと、抗体依存性細胞傷害活性が高まる。なお、培養前のPBMCs中のγδTの割合は8.0%であり、CD56+γδTの割合は4.1%であった。
【0131】
増殖してくる全T細胞中のγδT細胞の割合は、γδT細胞のマーカー分子であるVγ9をFITC標識した抗Vγ9抗体(Beckman Coulter)を用いて測定した。また、Vγ9+細胞(γδT細胞)上の表面分子NKG2D、CD69、CD56及びCD16抗原を、それぞれPE標識抗NKG2D抗体、ECD標識抗CD69抗体、PC5標識抗CD56抗体、及びPC7標識抗CD16抗体(いずれもBeckman Coulter)を用いて測定した。前記測定された全γδT細胞に対する 、各抗原を発現している細胞の割合を算出した。
【0132】
(結果)
結果を図6に示す。図6で示すように、全T細胞中のVγ9+細胞の割合は、培養7〜14日の期間で、いずれも90%以上高いの割合を示した。また、Vγ9+細胞中のCD16+細胞(CD16+Vγ9+細胞)の割合は、12日目で93.3%を示し、CD56+DCと反応細胞を混合培養することによって、癌患者由来の血液であってもNK様の細胞表面を有するVγ9細胞を増強できることが確認できた。
【0133】
抗体療法との併用療法を考えるときNK様の細胞表面を有するγδT細胞を増殖させることはその細胞の細胞傷害性を増強し、免疫力を強化する上で重要である。本発明の方法によれば、癌患者由来の末梢血であっても、わずか50mL採血(包含されるVγ9+細胞数は、2.3×106)から14日間の培養でVγ9+細胞数は、元の血液の500倍以上に相当する1.2×109個に達する。免疫細胞療法において、通常、その効果が期待できるγδT細胞数は、109オーダーであるが、本発明のCD56+T細胞増強方法によれば、そのオーダーのγδT細胞を簡便に調製することが可能となる。
【特許請求の範囲】
【請求項1】
T細胞をCD56陽性樹状細胞及びサイトカインと混合培養する工程を含むCD56陽性T細胞の増強方法。
【請求項2】
前記サイトカインがインターロイキン2である、請求項1に記載の増強方法。
【請求項3】
疾患抗原ペプチドをさらに含む、請求項1又は2に記載の増強方法。
【請求項4】
前記疾患が腫瘍又は感染症である、請求項3に記載の増殖方法。
【請求項5】
前記混合培養時における培養液中のインターロイキン2の終濃度が1U/mL〜200U/mLである、請求項3又は4に記載の増強方法。
【請求項6】
ビスホスホネート誘導体又はその塩若しくはその水和物をさらに含む、請求項3〜5のいずれか一項に記載の増強方法。
【請求項7】
前記ビスホスホネート誘導体がゾレドロン酸、パミドロン酸、アレンドロン酸、リセドロン酸、イパンドロン酸、インカドロン酸、エチドロン酸及びそれらの組み合わせからなる群から選択される、請求項6に記載の増強方法。
【請求項8】
前記CD56陽性T細胞が疾患抗原特異的CD56陽性CD8陽性細胞傷害性T細胞である、請求3〜7のいずれか一項に記載の増強方法。
【請求項9】
ビスホスホネート誘導体又はその塩若しくはその水和物をさらに含む、請求項1に記載の増強方法。
【請求項10】
前記ビスホスホネート誘導体がゾレドロン酸、パミドロン酸、アレンドロン酸、リセドロン酸、イパンドロン酸、インカドロン酸、エチドロン酸及びそれらの組み合わせからなる群から選択される、請求項9に記載の増強方法。
【請求項11】
前記混合培養時における培養液中のインターロイキン2の終濃度が300U/mL〜3000U/mLである、請求項9又は10に記載の増強方法。
【請求項12】
前記CD56陽性T細胞がCD56陽性γδ型T細胞である、請求項9〜11のいずれか一項に記載の増強方法。
【請求項13】
請求項1〜12のいずれか一項に記載の増強方法によって増強されたCD56陽性T細胞を含むCD56陽性T細胞強化型血液製剤。
【請求項14】
CD56陽性樹状細胞及びサイトカインを含むCD56陽性T細胞増強剤。
【請求項15】
前記サイトカインがインターロイキン2である、請求項14に記載のCD56陽性T細胞増強剤。
【請求項16】
ビスホスホネート誘導体又はその塩若しくはその水和物をさらに含む、請求項15に記載のCD56陽性T細胞増強剤。
【請求項17】
前記ビスホスホネート誘導体がゾレドロン酸、パミドロン酸、アレンドロン酸、リセドロン酸、イパンドロン酸、インカドロン酸、エチドロン酸及びそれらの組み合わせからなる群から選択される、請求項16に記載のCD56陽性T細胞増強剤。
【請求項18】
前記CD56陽性T細胞がCD56陽性γδ型T細胞である、請求項16又は17に記載のCD56陽性T細胞増強剤。
【請求項19】
疾患抗原ペプチドをさらに含む、請求項15〜17のいずれか一項に記載のCD56陽性T細胞増強剤。
【請求項20】
前記CD56陽性T細胞が疾患抗原特異的CD56陽性CD8陽性CTL細胞である、請求19に記載のCD56陽性T細胞増強剤。
【請求項1】
T細胞をCD56陽性樹状細胞及びサイトカインと混合培養する工程を含むCD56陽性T細胞の増強方法。
【請求項2】
前記サイトカインがインターロイキン2である、請求項1に記載の増強方法。
【請求項3】
疾患抗原ペプチドをさらに含む、請求項1又は2に記載の増強方法。
【請求項4】
前記疾患が腫瘍又は感染症である、請求項3に記載の増殖方法。
【請求項5】
前記混合培養時における培養液中のインターロイキン2の終濃度が1U/mL〜200U/mLである、請求項3又は4に記載の増強方法。
【請求項6】
ビスホスホネート誘導体又はその塩若しくはその水和物をさらに含む、請求項3〜5のいずれか一項に記載の増強方法。
【請求項7】
前記ビスホスホネート誘導体がゾレドロン酸、パミドロン酸、アレンドロン酸、リセドロン酸、イパンドロン酸、インカドロン酸、エチドロン酸及びそれらの組み合わせからなる群から選択される、請求項6に記載の増強方法。
【請求項8】
前記CD56陽性T細胞が疾患抗原特異的CD56陽性CD8陽性細胞傷害性T細胞である、請求3〜7のいずれか一項に記載の増強方法。
【請求項9】
ビスホスホネート誘導体又はその塩若しくはその水和物をさらに含む、請求項1に記載の増強方法。
【請求項10】
前記ビスホスホネート誘導体がゾレドロン酸、パミドロン酸、アレンドロン酸、リセドロン酸、イパンドロン酸、インカドロン酸、エチドロン酸及びそれらの組み合わせからなる群から選択される、請求項9に記載の増強方法。
【請求項11】
前記混合培養時における培養液中のインターロイキン2の終濃度が300U/mL〜3000U/mLである、請求項9又は10に記載の増強方法。
【請求項12】
前記CD56陽性T細胞がCD56陽性γδ型T細胞である、請求項9〜11のいずれか一項に記載の増強方法。
【請求項13】
請求項1〜12のいずれか一項に記載の増強方法によって増強されたCD56陽性T細胞を含むCD56陽性T細胞強化型血液製剤。
【請求項14】
CD56陽性樹状細胞及びサイトカインを含むCD56陽性T細胞増強剤。
【請求項15】
前記サイトカインがインターロイキン2である、請求項14に記載のCD56陽性T細胞増強剤。
【請求項16】
ビスホスホネート誘導体又はその塩若しくはその水和物をさらに含む、請求項15に記載のCD56陽性T細胞増強剤。
【請求項17】
前記ビスホスホネート誘導体がゾレドロン酸、パミドロン酸、アレンドロン酸、リセドロン酸、イパンドロン酸、インカドロン酸、エチドロン酸及びそれらの組み合わせからなる群から選択される、請求項16に記載のCD56陽性T細胞増強剤。
【請求項18】
前記CD56陽性T細胞がCD56陽性γδ型T細胞である、請求項16又は17に記載のCD56陽性T細胞増強剤。
【請求項19】
疾患抗原ペプチドをさらに含む、請求項15〜17のいずれか一項に記載のCD56陽性T細胞増強剤。
【請求項20】
前記CD56陽性T細胞が疾患抗原特異的CD56陽性CD8陽性CTL細胞である、請求19に記載のCD56陽性T細胞増強剤。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2013−81428(P2013−81428A)
【公開日】平成25年5月9日(2013.5.9)
【国際特許分類】
【出願番号】特願2011−224048(P2011−224048)
【出願日】平成23年10月11日(2011.10.11)
【出願人】(510035912)株式会社日本バイオセラピー研究所 (2)
【Fターム(参考)】
【公開日】平成25年5月9日(2013.5.9)
【国際特許分類】
【出願日】平成23年10月11日(2011.10.11)
【出願人】(510035912)株式会社日本バイオセラピー研究所 (2)
【Fターム(参考)】
[ Back to top ]