
CD8α鎖に基づき免疫原性が減少した遺伝子治療ベクター
本発明は、発現ベクターおよびそのベクターでトランスフェクトされた標的細胞に対する宿主免疫反応を特異的に阻害するための組成物および方法を提供する。特に、CD8 α-鎖の使用に基づくベクター-関連抗原および標的-細胞関連抗原に対する宿主免疫反応の体液性および細胞性コンポーネントを特異的に阻害する方法を記載する。

【発明の詳細な説明】
【発明の詳細な説明】
【0001】
関連出願とのクロスレファレンス
この出願は、2003年3月19日出願の仮出願番号60/456,378の利益を主張する。
【0002】
本発明の分野
本発明は、一般的に遺伝子治療の分野に関係し、そしてより具体的には、遺伝子治療ベクターの免疫原性を低下させるための方法および組成物を提供する。
【0003】
発明の背景
遺伝子導入または遺伝子治療は後天性疾患および先天性疾患の治療のための見込みのある方法である。ますます多くの遺伝子(異常発現が生命を脅かすヒトの疾患と関連する遺伝子)が、クローニングされ、同定されている。ヒトにおいてそのようにクローニングされた遺伝子を発現する能力は、最終的に、多くの重要なヒトの疾患、現在の治療では不十分であるかまたは治療のない疾患の阻止および/または治癒を可能とする。例として、ヒト患者におけるコレステロール-調節遺伝子、HIV、または腫瘍抑制遺伝子の複製を選択的にブロックする遺伝子のインビボ発現は、心臓病、HIV、および癌、それぞれの治療を劇的に改善する。
【0004】
しかしながら、残念なことに、今日までに記載されている遺伝子治療プロトコールは、種々の問題により厄介であり、その中には、特に、ベクターからの遺伝子発現が短期間であることおよび同一ベクターを2回効果的に再投与できないことがあり、それらは、何れも、ベクターおよびその治療的ペイロードと関連する抗原に対する宿主免疫反応により生ずる。ウイルス性および/または治療遺伝子を組み込んだ組織は、CD8+ 細胞傷害性T細胞およびCD4+ ヘルパー T 細胞が仲介する宿主の細胞性免疫反応により最初に攻撃され、これは、ベクターからの遺伝子発現の持続を非常に制限する。更に、CD4+ T 細胞により仲介される宿主の体液性免疫反応は、更に、同一ベクターの再投与の成功を阻害することにより現在の遺伝子治療プロトコールの有効性を制限する。
【0005】
例えば、アデノウイルス性ベクターの最初の投与後、血清型-特異的抗体は、主要なウイルス性キャプシドタンパク質のエピトープ、すなわち、ペントン、ヘキソンおよびファイバーに対して生ずる。そのキャプシドタンパク質が、アデノウイルスそれ自身が細胞に結合しその後その細胞に感染する手段であるならば、次いでその抗体は、アデノウイルスの同一血清型により細胞の再感染を阻止するかまたは「中和」することができる。これは、遺伝子治療との関連で、その後に1以上の用量の外因性治療DNAを投与するために、アデノウイルスの異なる血清型を使用することを必要とする。加えて、治療およびウイルス性 遺伝子産物の両方が、標的細胞で発現し、それらは細胞性免疫反応に感受的となる。このため、それらは拒絶され、遺伝子治療の有益な効果が打ち消され、標的の器官または組織が破壊され得る。これら免疫が関連する障害の結果として、遺伝子治療プロトコールの進歩が妨げられている。
【0006】
従って、遺伝子治療発現ベクターおよびそのベクターによりトランスフェクトされた細胞に対する免疫反応を特異的に阻害する効果的な方法が当分野に非常に必要である。加えて、遺伝子治療ペイロードを投与しまたは導入するための改善された方法および組成物の必要がある。そのため、その遺伝子治療ベクターおよびその治療産物に対する細胞性および体液性免疫反応の両方を特異的に阻害し、それによって、そのベクターによりトランスフェクトされた細胞からの外因性遺伝子発現を増加させることが本発明の目的である。
【0007】
関連文献の要約
MHCクラスI-拘束(restricted)T 細胞(例えば、CD8+ CTL)の活性は、そのT 細胞レセプター複合体を通してシグナルを受け取るCTLがまたそのクラスI MHC 分子のα3 ドメインを通してシグナルを受け取るとき、抑制され得ることが知られている。これは、いわゆるvetoシグナルが、スティミュレーターまたは「veto」細胞により発現したCD8 分子により伝えられ得る。Sambhara and Miller, Science 252:1424-1427 (1991). 生じた免疫抑制は抗原-特異的でありかつMHC-拘束されており、それは、反応CTLによるveto細胞の一方向性認識から生ずるが、その逆は生じない。Rammensee et al., Eur. J. Immunol. 12:930-934 (1982); Fink et al., J. Exp. Med. 157:141-154 (1983); Rammensee et al., J. Immunol. 132:668-672 (1984). veto活性はCD8 α 鎖の存在と関連するため、それ故、CD8 α 鎖の発現のときにCD8の発現が消失し確認されるならばveto機能は喪失する。Hambor et al., J. Immunol. 145:1646-1652 (1990); Hambor et al., Intern. Immunol. 2:8856-8879 (1990); Kaplan et al., Proc. Natl. Acad. Sci. USA 86:8512-8515 (1989).
【0008】
多くのストラテジーが提唱され、望ましくない細胞傷害性T細胞反応を排除するこの抗原-特異的抑制経路が開発されている。1つのそのようなストラテジーは、CD8のveto活性を特異的な標的細胞に指示する第二リガンドに、CD8またはその機能ドメインを共有結合させるポリペプチド結合体(conjugate)の使用を含む。例えば、米国特許番号5,242,687、5,601,828および5,623,056参照。またあるいは、ハイブリッド抗体分子は、CD8 α 鎖の細胞外ドメインに結合するMHCクラスI 分子に対し特異性を有するモノクローナル抗体結合部位を有することが調べられている。Qi et al., J. Exp. Med. 183:1973-1980 (1996). しかし、その分子は、幾つかの欠点を有し、実際の臨床的有用性がまだ見い出されていない。
【0009】
更に最近では、WO 02/102852が、MHCクラスIに対する親和性が増加するように設計されたアミノ酸修飾を有する可溶性C8α 鎖変異体を用いるCTLの阻害を記載している。重要なことに、ここでは、提唱されたCD8α 組成物は、クラスI MHC 分子に特異的であり、それ故、CTLの反応のみを阻害すると予測されること、および更に他の免疫抑制剤との組合せが、細胞性および体液性免疫反応の他のエレメント、例えば、CD4+ T 細胞のようなMHCクラスII-拘束 T 細胞を含む状況で要求されることが教授されている。同文献 pp. 27-28.
【0010】
発明の概要
本発明は、CD8のような免疫修飾分子の標的化発現により仲介されるveto効果は、外因性遺伝的ペイロードを含む発現ベクターと関連する抗原に対する、およびトランスフェクトした標的細胞と関連する抗原に対する宿主免疫反応を効果的におよび特異的に阻害し得るという驚くべき発見に基づいている。本発明はまた、CD8αの標的化発現により仲介されるveto効果は、CD4+ T 細胞(MHCクラスII-拘束)およびCD8+ T 細胞(MHCクラスI-拘束)の反応を効果的におよび特異的に抑制し得るという更なる驚くべき発見、および該ベクター-関連抗原に対する宿主免疫反応の細胞性および体液性の両方のコンポーネントが阻害され得るという、得られた結論に基づいている。このため、本明細書で記載した方法および組成物を用いることにより、現在、ベクターからの遺伝子発現を制限し、遺伝子治療から最大の可能性を引き出すことを妨げるベクター-関連抗原に対する宿主免疫反応を阻害することにより遺伝子治療プロトコールは相乗的に促進され得る。
【0011】
従って、本発明は、発現ベクターおよびそのベクターでトランスフェクトされた標的細胞に対する宿主免疫反応を特異的に阻害するための組成物および方法を提供し、ここで、該ベクターは、veto効果を顕在化させることができる免疫修飾分子、好ましくは、CD8 ポリペプチド、より好ましくは、CD8 α-鎖、および最も好ましくは、CD8 α-鎖の細胞外および膜貫通ドメインの両方をコードする核酸配列を含む。対象の組成物および方法の性質、および上記CD8 α-鎖の従来技術の可溶性型の明かな不十分さを考慮すれば、CD8 α-鎖膜貫通ドメインまたは適当な代替の膜貫通領域の存在が重要であると思われる。
【0012】
1つの態様では、本発明は、発現ベクターに対する免疫反応を阻害するための方法であって、宿主標的細胞をインビボまたはエキソビボで、CD8 ポリペプチドのすべてまたは機能部分、好ましくは、CD8 α-鎖、および最も好ましくは、CD8 α-鎖の細胞外および膜貫通ドメインの両方をコードする発現ベクターと接触させること、ここで、該CD8 ポリペプチドは標的細胞の表面上で発現し、それにより、発現ベクターおよび標的細胞に対する免疫反応が特異的に阻害される、を含む該方法を提供する。組み換えベクターは、好ましくは、目的の治療タンパク質または分子をコードする1以上の更なる導入遺伝子を更にを含む。本明細書で記載および例示のように、免疫反応の体液性および細胞性コンポーネントの両方が、本発明の方法および組成物を使用することにより阻害される。
【0013】
更なる態様では、ベクター-関連抗原に対する宿主免疫反応の特異的阻害のための方法であって、CD8 ポリペプチドのすべてまたは機能部分、好ましくは、CD8 α-鎖、および最も好ましくは、CD8 α-鎖の細胞外および膜貫通ドメインの両方をコードする核酸を含む発現ベクターと宿主標的細胞をインビボまたはエキソビボで接触させること、ここで、CD8 ポリペプチドは標的細胞の表面上で発現し、それにより、ベクター-関連抗原に対する宿主免疫反応は特異的に阻害される、を含む該方法が提供される。
【0014】
更なる態様では、本発明は、宿主における治療導入遺伝子の発現を改善するのための方法であって、CD8 ポリペプチドのすべてまたは機能部分、好ましくは、CD8 α-鎖、および最も好ましくは、CD8 α-鎖の細胞外および膜貫通ドメインの両方をコードする核酸配列を含む発現ベクターを宿主に投与すること、ここで、CD8 ポリペプチドは宿主細胞の表面上で発現し、それにより、ベクター-関連抗原に対する宿主免疫反応が特異的に阻害される、を含む該方法を提供する。1つの実施態様では、治療導入遺伝子は、CD8 ポリペプチドと同一のベクターに含まれる。更に他の実施態様では、CD8 ポリペプチドおよび治療分子は別の発現ベクターによりコードされる。本明細書に記載のように、本発明の方法は、ベクター-関連抗原に対する宿主免疫反応の細胞性および体液性コンポーネントの両方を阻害し、それにより、宿主中の治療導入遺伝子を持続時間を延長させ、そしてその後の導入遺伝子発現のラウンドで発現ベクター再投与を可能とすることにより治療導入遺伝子の発現を改善する。
【0015】
更なる態様では、本発明は、免疫原性が減少した改善されたウイルス性発現ベクターを提供し、ここで、該発現ベクターは、本質的に、本明細書で開示したようなCD8 ポリペプチドをコードする核酸および目的の少なくとも1つの治療導入遺伝子をコードする核酸からなる非-ウイルス性核酸を含む。1つの実施態様では、治療導入遺伝子は免疫修飾分子以外のものである。好ましい実施態様では、CD8 ポリペプチドは、CD8 α-鎖のすべてまたは機能部分を含む。好ましくは、CD8 α-鎖の機能部分は、CD8 α-鎖の少なくとも細胞外ドメイン、およびより好ましくは、CD8 α-鎖の細胞外ドメインおよび膜貫通ドメインの両方を含む。一般的に、本明細書で提供する免疫修飾分子は、標的細胞のトランスフェクション後、標的細胞表面膜と結合し、例えば、該膜内に挿入されるか、またはそれと共有結合的または非共有結合的に結合される。
【0016】
本明細書での使用を意図する適当な発現ベクターは、組み換えおよび非-組み換えベクター、およびウイルス性(例えば、アデノウイルス性、レトロウイルス性、アデノ-随伴ウイルス性ベクターなど)および非-ウイルス性(例えば、細菌性プラスミド、ファージ、リポソームなど)ベクターを含む。ウイルスベクターが好ましく、アデノウイルス性ベクターが最も好ましい。
【0017】
多数の実施態様を開示しているが、本発明の他の実施態様が、本発明の例示となる実施態様を示し記載している、以下の詳細な説明から当業者には明らかとなるだろう。お解りのように、本発明は、種々の明らかな態様で修飾することができ、それらはすべて本発明の精神および範囲を逸脱することはない。従って、図面および詳細な説明は、事実上の例示であると見なされ、それらに限定すべきでない。
【0018】
図面の簡単な説明
図1は、様々な種由来のCD8 α-鎖 タンパク質および核酸配列を示す。示した配列の受け入れ番号もまた含まれている。
【0019】
図2A-Bは、野生型 CD8 α-鎖のアミノ酸および核酸配列を示し、ヒトおよびマウスのタンパク質の種々のドメインの区域(demarcation)が含まれている。
【0020】
図3は、C57BL/6 脾臓細胞で刺激されたBalb/c 脾臓細胞を示す。培養物に、マウス(A)またはヒト(B)由来の正常の繊維芽細胞 (黒丸)、培地(黒四角)、またはCD8を有する繊維芽細胞 (黒三角)が供された。培養物は回収され、C57BL/6-由来標的細胞に対するその溶菌能について試験した。
【0021】
図4は、対照繊維芽細胞(黒四角および黒三角)またはmCD8-トランスフェクトC57BL/6-(H-2b)由来(白丸および黒丸)繊維芽細胞を注射したBalb/c(H-2d)マウスを示す。2週間後、動物を屠殺し、脾臓細胞を回収し、C57BL/6(H-2b)(黒四角および白丸)またはCBA/J(H-2k)(黒丸および黒三角)脾臓細胞で刺激し、EL4 (H-2b)(黒四角および白丸)またはS.AKR (H-2k)(黒丸および黒三角)標的細胞に対する溶菌能について試験した。
【0022】
図5は、異型反応性(alloreactive) T 細胞(A) によるかまたは抗原-特異的 CTL(B)による溶菌に対する感受性について試験した標的細胞(黒三角)またはCD8-発現標的(黒四角)を示す。
【0023】
図6は、正常繊維芽細胞 (黒丸)およびmAdCD8 (A、黒三角)またはHAdCD8 (B、黒三角)で遺伝子導入された繊維芽細胞の存在下で設定したMLC(Balb/c 抗-C57B/6)を示す。繊維芽細胞は対照培養物(黒四角)には加えなかった。C57BL/6-由来標的に対するこれらの培養物の溶菌活性を培養期間の最後に測定した。
【0024】
図7は、アデノウイルス性veto転移ベクター、mAdCD8による免疫化を示す。C57BL/6 マウスは上記のベクターで感染させた。10日後、脾臓細胞を回収し、Adβgal ウイルスの存在下、培養した。芽細胞の数を示す。
【0025】
図8 は、mAdCD8による陰性免疫化(negative immunization)を示す(A)。C57BL/6 マウスはAdβgalまたはmAdCD8により静脈注射で一度免疫化された。(A)のように処置した動物は、5日後、Adβgalで再免疫化した(B)。最後の注射の7日後、動物は屠殺し、その脾臓細胞はAdβgalの存在下、培養した。培養の5日後、細胞は、Adβgal-感染性同系標的細胞の溶菌能について試験した。
【0026】
図9は、示したように形質導入した、1x106 (または全くなし)スティミュレーター細胞と共にインキュベートした3x106 C7Bl/6 脾臓細胞を示す。4日後、培養物を、免疫蛍光によりCD4+ T リンパ芽球の存在について分析した。
【0027】
図10A-Dは、種々のウイルスコンストラクトによる感染後のマウスおよびヒト CD8 α-鎖の表面発現を示す。A. 感染細胞: Mc57T 繊維芽細胞; パネル 1: 偽-感染; パネル 2: hAdCD8による感染. B. 感染細胞: MC57T 繊維芽細胞; パネル 1: 偽感染; パネル 2: mAdCD8による感染. C. 感染細胞: Balbc 非選択骨髄細胞; パネル 1: lacZ アデノウイルス性ベクター (AdLacZ)による感染; パネル l2: mAdCD8による感染. D. 感染細胞: MC57T 繊維芽細胞; パネル 1: 偽-感染; パネル 2: pAAV-mCD8による感染; パネル 3: pAAV-hCD8による感染.
【0028】
図11は、MLC (Balb/c 抗-C57BL/6)を、形質導入後、MLCに加えられる前、0 または 5 時間培養されたこれらの繊維芽細胞の存在下、設定したものを示す。培養の最後に、リンパ芽球の数を蛍光活性化細胞アナライザーで測定した。
【0029】
図12は、veto転移ベクターによるインビトロ阻害を示す。BALB/c 抗-C57BL/6混合リンパ球培養 (MLC)は、非感染またはmAdCD8-感染MC57繊維芽細胞 (H-2b)の非存在下または存在下で行った(X)。CTL反応は、EL4 (H-2b)標的細胞で測定した。
【0030】
図13は、AdLacZ または mAdCD8で免疫化されたBalb/c マウスを示す。それらの脾臓細胞は、AdLacZの存在下で培養し、AdLacZ-感染性同系P815標的細胞に対する特異的溶菌活性について試験した。
【0031】
図14は、AdLacZ (黒四角)またはmAdCD8 (黒三角)で免疫化した(A) C57BL/6 動物を示す。同系AdLacZ EL4 標的細胞に対する脾臓細胞の溶菌活性を試験した。(B)その動物は、AdLacz-感染化EL4 標的に対する溶菌活性を試験する前にAdLacZで再免疫化した。
【0032】
図15は、ヘモグロビン βのmRNA 配列を示す。
【0033】
図16は、GATA結合タンパク質のmRNA 配列を示す。
【0034】
図17は、d-アミノエブリネート(aminoevulinate)シンターゼのmRNA 配列を示す。
【0035】
図18は、グルコース-6-ホスフェート-デヒドロゲナーゼのmRNA 配列を示す。
【0036】
図19は、オルニチンカルバモイルトランスフェラーゼのmRNA 配列を示す。
【0037】
図20は、α-L-イズロニダーゼのmRNA 配列を示す。
【0038】
図21は、β-グルコシダーゼのmRNA 配列を示す。
【0039】
図22は、α-ガラクトシダーゼのmRNA 配列を示す。
【0040】
詳細な説明
発現ベクターと関連するタンパク質に対する宿主免疫反応は、遺伝子治療技術の開発に厄介であり、ここで、該反応の細胞性コンポーネントは、ベクター内に含まれる遺伝子の発現を厳格に制限し、該反応の体液性コンポーネントは、免疫能のある動物における同一ベクターの再投与を面倒にする。本発明の成功は、発現ベクターでトランスフェクトされた標的細胞におけるCD8のような免疫修飾分子の発現は、CD4+ T 細胞およびCD8+ T 細胞の両方の反応を抑制し、それにより、ベクター-関連抗原に対する宿主免疫反応の体液性および細胞性コンポーネントの両方を効果的におよび特異的に阻害するという驚くべき発見に起因する。
【0041】
このため、本明細書で記載する組成物および方法は、宿主細胞における発現ベクターの持続を向上させ、それにより、ベクター内に含まれる治療導入遺伝子の発現を改善させ、そして、同一ベクター(例えば、同一血清型の組み換えアデノウイルス性ベクター)による宿主細胞への再投与を成功させ得ることにより、インビボおよびエキソビボ遺伝子治療プロトコールを劇的に改善することができる。1つの実施態様では、免疫修飾分子、好ましくは、CD8 ポリペプチド、より好ましくは、CD8 α鎖、および最も好ましくは、CD8 α-鎖の細胞外ドメインおよび膜貫通ドメインの両方をコードする核酸、および目的の1以上の治療分子をコードする核酸配列を含む発現ベクターが提供される。更なる他の実施態様では、別々の発現ベクターを提供し、その1つはCD8 ポリペプチドをコードし、そしてその1つは、宿主に対する共-投与のための望ましい治療分子をコードする。
【0042】
本発明はまた、発現ベクター、特に、アデノウイルス性ベクター、アデノ-随伴ウイルス性ベクター、ヘルペスウイルス性ベクターまたはレトロウイルス性ベクターのような組み換えベクターに対する免疫反応を阻害するのための方法であって、標的細胞を、例えばインビボおよびエキソビボ遺伝子治療との関連で免疫修飾分子および目的の1以上の治療分子をコードする発現ベクターと接触させることを含む該方法を提供する。本明細書で記載および例示のように、本発明により達成される宿主免疫反応の抗原-特異的阻害は、細胞における発現ベクターのより持続的な存在、およびベクター内に含まれる治療導入遺伝子の付随的に改善した発現を可能とし、ならびに遺伝子治療を続けるための同一ベクターの再投与の成功が可能となる。
【0043】
従って、本発明は、遺伝子治療のための組成物および方法を提供し、ここで、遺伝子治療送達ビヒクルと関連する抗原に対する細胞性および体液性免疫反応は、完全になくなるか減少する。一般的に、本発明は、発現ベクター、遺伝子治療ベクター、遺伝子治療ベクターで感染させた標的細胞または標的細胞の子孫に対する細胞性および/または体液性免疫反応の両方を低下させるか減らすための方法および組成物を目的とする。
【0044】
“インビボ遺伝子治療”および “インビトロ遺伝子治療”は、エキソビボの応用を含む “遺伝子治療”の分野の当業者にとって通常既知のことおよび該当業者が引用することに関する、過去、現在および将来のすべての変更および修飾を含むことを意図する。
【0045】
“発現ベクター”は、標的細胞への核酸の送達のための任意のビヒクルを意味する。発現ベクターは、一般的に、ウイルス性ベクターおよび非-ウイルス性ベクターに分けることができる。ウイルス性ベクターは、以下に限定されないが、アデノウイルス性ベクター、アデノ-随伴ベクター、レトロウイルス性ベクター、レンチウイルス性ベクターなどを意味する。非-ウイルス性ベクターは、プラスミドベクター、裸のDNA、種々の担体と結合するかまたはリポソームもしくは他の脂質調製物と関連する裸のDNAを意味する。一般的に、発現ベクターは組み換え体であるが、幾つかの実施態様では、例えば、リポソームまたは細胞切除、例えば、遺伝子銃技術を用いるとき、それらは、組み換え体ではない。本明細書で使用のための好ましい組み換えベクターは、プラスミドベクター、およびアデノウイルス性ベクター、アデノ-随伴ウイルス性ベクター、ヘルペスウイルス性ベクターおよびレトロウイルス性ベクターからなる群から選択されるウイルス性ベクターである。組み換えウイルス性ベクター、および特に、アデノウイルス性ベクターを用いる幾つかの実施態様では、キャプシド、例えば、アデノウイルス性キャプシドのヘキソンタンパク質の免疫原性は、当分野に既知の方法により低下し得るが、本明細書で詳述の改善の観点からその修飾はもはや必要ではない。
【0046】
“遺伝子治療送達ビヒクル”は、以下に限らないが、ウイルス性ベクターおよび非-ウイルス性ベクターを含む、上記のような発現ベクターを含む組成物を意味する。
【0047】
“阻害する”は、ベクター-関連抗原に対するおよび/または標的細胞-特異的抗原に対する、直接的または間接的、部分的または完全な、先天性または後天性免疫反応(細胞性(例えば、白血球供給)または体液性の何れか)の阻害および/または低下を意味する。ベクター-関連抗原は、例えば、核酸担体またはエンベロープ(例えば、ウイルス性被覆タンパク質など)から得られる抗原ならびにベクター遺伝子(例えば、細菌性またはウイルス性核酸およびタンパク質)から得られる抗原および/またはベクターに含まれる任意の治療導入遺伝子(例えば、哺乳類核酸および/またはタンパク質)を含む。
【0048】
“特異的免疫性阻害”または “抗原-特異的免疫性阻害”は、抗原-特異的でない一般的な免疫性阻害とは対照的に、ベクター-関連抗原のような抗原に対する免疫反応の阻害を意味する。このため、一例として、他の外来の抗原に対するインビボ免疫適格性の証拠と組み合わせた、ベクター-関連抗原に対する宿主細胞性および/または体液性免疫反応の非存在が、ベクター-関連抗原の特異的免疫性阻害を証明する。
【0049】
“免疫反応”は、好ましくは、細胞性または体液性免疫反応のような後天性免疫反応を意味する。
【0050】
“接触させること”は、ベクターと細胞との間で物理的に接触するような方法および量で細胞に遺伝子治療発現ベクターを投与することを意味する。ベクターが組み換え体ウイルス性粒子ならば、望ましくは、ウイルス性ベクターへの結合およびそれによる細胞の感染はそのような物理的接触により生ずる。ウイルス性ベクターが、非カプセル化ウイルス性核酸または他の核酸のような組み換え体ウイルス性粒子以外ならば、望ましくは、核酸による細胞の感染が生ずる。
【0051】
その “接触させること”は、当業者に既知であり本明細書に記載の任意の手段によって行い得、それによって、ベクターと標的細胞とが明かに接触することまたは相互の接触が生じ得る。所望により、アデノウイルス性ベクターのようなベクターは、更に、二重特異性または多特異性分子(例えば、抗体またはそのフラグメント)と複合体となり得、その場合、 “接触させること”は、ベクターおよび二重特異性または多特異性分子と標的細胞との複合で明かに接触することまたは相互の接触を含む。例えば、ベクターおよび二重特異性(多特異性)分子は、例えば、当業者に既知の化学的手段、または他の手段により、共有結合し得る。好ましくは、ベクターおよび二重特異性(多特異性)分子は、非共有結合的相互作用(例えば、イオン結合、水素結合、ファン・デル・ワールス力、および/または非極性相互作用)により、連結し得る。ベクターおよび二重特異性(多特異性)分子は、小体積の同一溶液を混合することにより接触し得るが、例えば、該複合体が宿主(例えば、ヒト)に投与され、該複合体が血流により標的細胞に移動し、選択的に標的細胞に結合し、標的細胞中に入るような場合、標的細胞および複合体は小体積で接触させる必要はない。ベクターと二重特異性(多特異性)分子とを接触させることは、好ましくは、標的細胞がベクターおよび二重特異性(多特異性)分子の複合体と接触する前に行う。
【0052】
“導入遺伝子”は遺伝子を意味し、それは、導入遺伝子を含む発現ベクターと接触した細胞中で発現し得、その発現は、望ましくは、細胞または組織、器官、器官系、生体または細胞が一部を形成する細胞培養物にとって予防的または治療的利点がある。このため、導入遺伝子は治療遺伝子であり得、例えば、目的の治療遺伝子であり得る。治療遺伝子は、RNAまたはタンパク質のレベルでその効果を発揮するものであり得る。例えば、治療遺伝子によりコードされるタンパク質が遺伝性の疾患の治療、例えば、嚢胞性繊維症の治療における嚢胞性繊維症膜貫通コンダクタンスレギュレーター(cystic fibrosis transmembrane conductance regulator)をコードするcDNAの使用において用いられ得る。
【0053】
更に、治療遺伝子は、例えば、アンチセンスメッセージまたはリボザイム、当分野に既知のようなsiRNA、選択的RNAスプライシングアクセプターまたはドナー、スプライシングまたは3'プロセッシング(例えば、ポリアデニル化) に影響を及ぼすタンパク質、または細胞内での他の遺伝子の発現のレベルに影響を及ぼすタンパク質(すなわち、遺伝子発現が、プロセッシングされたタンパク質の産生により転写の当初からのすべての工程を含むと概ね考えられる場合)をコードすることにより、恐らく、他の場合には、mRNA累積の変化率、mRNA輸送の変化、および/または転写後調節の変化を仲介することにより、RNAのレベルでその効果を発揮し得る。
【0054】
好ましい本発明の態様により、発現ベクターは、所望により、本明細書に記載のCD8ポリペプチドと共に目的の治療分子をコードする1以上の導入遺伝子を含む。本発明により治療され得る疾患には、以下に限らないが、フェニルケトン尿症(フェニルアラニン-L-モノオキシゲナーゼ)、嚢胞性繊維症(嚢胞性繊維症コンダクタンスレギュレーター)、オルニチンカラミルトランスフェラーゼ欠損(OTC)、血友病(XI因子-欠損、VIII因子-欠損)、テイ・サックス病(N-アセチル-ヘキソサミニダーゼ A)および他の脂質蓄積症などのような、よくある遺伝的疾患がある。更に、エリトロポイエチン(EPO)をコードする遺伝子が使用され得る。EPOは、胎児の肝臓および成体の腎臓で産生される糖タンパク質ホルモンであり、それは、骨髄および他の造血組織において前駆細胞で作用し、赤血球細胞の形成を刺激する。ヒトおよび他の哺乳類 EPOをコードする遺伝子は、クローニングされ、配列決定され、発現させ、そしてそれは、種間でのコーディング領域において高い配列相同性がある。Wen et al. (1993) Blood 82:1507-1516. 天然のヒト EPOをコードする遺伝子の配列、およびそれを得るための方法は、例えば、米国特許番号4,954,437および4,703,008に記載されており、それらは、引用により本明細書にすべて含める。EPOを用いる遺伝子治療の方法は、米国特許番号6,610,290に開示されており、それらは、引用により本明細書に特に含める。
【0055】
更にまた、リソソーム酵素酸α-グルコシダーゼ (GAA)をコードするヌクレオチド配列を使用し得る。GAAが機能し、リソソームグリコーゲンのα-1,4およびα-1,6結合を切断し、モノサッカライドを放出する。ヒト GAAをコードする遺伝子の配列、およびそれを得るための方法は以前に記載されており(GenBank 受け入れ番号: M34424 および Y00839; Martiniuk et al. (1990) DNA Cell Biol. 9:85-94; Martiniuk et al. (1986) Proc. Natl. Acad. Sci. USA 83:9641-9644; Hoefsloot et al. (1988) Eur. Mol. Biol. Organ. 7:1697-1704)、これらは、引用により本明細書に特に含める。
【0056】
本明細書で開示の方法および組成物により治療し得る好ましい疾患を以下の表1に開示する。受け入れ番号で提供する配列は引用により本明細書に特に含める。
表 1
【表1】
【0057】
免疫修飾 CD8 分子が、治療導入遺伝子を含みそれを発現するベクターとは別のベクターに含まれる遺伝子によりコードされるならば、CD8 分子を含む該ベクターは、該遺伝子を含みそれを発現するベクターと細胞との接触前、該接触と同時または該接触後に、該細胞と接触し得る。これは、類似または同一の型のベクターを用い、そして、接触(contact effects)のタイミングが該細胞と接触するベクターに対する免疫反応を十分に阻害する場合に限られる。
【0058】
“標的細胞”は、単一の存在として存在し得るか、または細胞のより巨大な集合物の一部であり得る。その “細胞のより巨大な集合物”は、例えば、細胞培養物(混合したものかまたは純粋なものか何れか)、組織(例えば、上皮または他の組織)、器官(例えば、心臓、肺、肝臓、胆嚢、膀胱、目または他の器官)、器官系(例えば、循環系、呼吸系、胃腸系、泌尿系、神経系、外皮系または他の器官系)、または生体(例えば、鳥類、哺乳類、特にヒトなど)を含み得る。好ましくは、標的とする器官/組織/細胞は、循環系(例えば、以下に限らないが、心臓、血管、および血液を含む)、呼吸系(例えば、鼻、咽頭、喉頭、気管、気管支、細気管支、肺など)、胃腸系(例えば、口、咽頭、食道、胃、腸、唾液腺、膵臓、肝臓、胆嚢、および他を含む)、泌尿系(例えば、腎臓、尿管、膀胱、尿道など)、神経系(例えば、以下に限らないが、脳および脊髄、および目のような特殊感覚器を含む)および外皮系(例えば、皮膚)を含み得る。更により好ましくは、該細胞は、心臓、血管、肺、肝臓、胆嚢、膀胱、目の細胞および幹細胞からなる群から選択される。幹細胞を培養し用いる方法は、米国特許番号5,672,346、6,143,292および6,534,052に詳細に開示されており、それは引用により本明細書に含める。
【0059】
幾つかの実施態様では、ウイルス性ベクターまたはプラスミドのような発現ベクターが接触する標的細胞は、接触した標的細胞は発現ベクターにより標的とされ得る特定の細胞-表面結合部位を含むという点で、他の細胞とは異なる。"特定の細胞-表面結合部位"は、細胞の表面上に存在する任意の部位(すなわち、分子または分子の組合せ)を意味し、該ベクター、例えば、アデノウイルス性ベクターは、該細胞と結合し、それにより、該細胞中に入るように相互作用し得る。それ故、特定の細胞-表面結合部位は、細胞-表面レセプターを包含し、好ましくは、それは、タンパク質(修飾タンパク質を含む)、炭水化物、糖タンパク質、プロテオグリカン、脂質、ムチン分子またはムコタンパク質などである。可能性ある細胞-表面結合部位の例には、以下に限らないが、グリコサミノグリカンで見られるヘパリンおよびコンドロイチン硫酸部分; ムチン、糖タンパク質、およびガングリオシドで見られるシアル酸部分; 主要組織適合遺伝子複合体 I(MHC I)糖タンパク質; マンノース、N-アセチル-ガラクトサミン、N-アセチル-グルコサミン、フコース、およびガラクトースを含む膜糖タンパク質中に見られる共通炭水化物分子; ICAM-1、VCAM、E-セレクチン、P-セレクチン、L-セレクチン、およびインテグリン分子のような糖タンパク質; および例えばMUC-1 腫瘍-特異的エピトープのような、癌性細胞に存在する腫瘍-特異的抗原がある。しかし、細胞に対しアデノウイルスのような発現ベクターを標的化することは、細胞性相互作用(すなわち、所定の細胞-表面結合部位との相互作用)の何れかの特定の機構に制限されない。
【0060】
本明細書で使用するように、および更に以下で定義するように、 “ポリヌクレオチド”または “核酸”とは、DNAまたはRNAの何れか、またはデオキシ-およびリボヌクレオチドの両方を含む分子を言い得る。核酸には、ゲノムDNA、cDNA および オリゴヌクレオチドがあり、センスおよびアンチ-センス核酸が含まれる。その核酸はまた、リボース-ホスフェートバックボーンに修飾を含み得、それにより、生理学的環境下におけるその分子の安定性および半減期が増大する。
【0061】
該核酸は、二本鎖、一本鎖であり得るか、または二本鎖または一本鎖配列の両方の部分を含み得る。当業者が認識するように、一本鎖( “Watson”)を表すと、他の鎖( “Crick”)の配列もまた決定される; このため、図2、4および6に示す配列は、配列の相補体をも含む。本明細書の用語 “組み換え核酸”は、エンドヌクレアーゼによる核酸の操作により、一般的に、本来的にインビトロで形成され、通常天然では見られない形の核酸を意味する。このため、直線状の単離核酸、または通常は結合していないDNA 分子をライゲーションすることによりインビトロで形成された発現ベクターは、両方とも、本発明の目的の組み換え体と見なされる。一旦、組み換え核酸を作成し、宿主細胞または生体中に再導入すると、それは、非組み換え的に、すなわち、インビトロではない宿主細胞のインビボ細胞性機構または染色体外操作を用い複製し得ると理解される; しかし、一旦組み換え的に作成されたその核酸もまた、その後に非組み換え的に複製されるけれども、発明の目的の組み換え体と見なされる。
【0062】
用語 “ポリペプチド”および “タンパク質”は、本出願において同義的に使用され、少なくとも2つの共有結合したアミノ酸を意味し得、タンパク質、ポリペプチド、オリゴペプチドおよびペプチドを含む。該タンパク質は、天然に生ずるアミノ酸およびペプチド結合、または合成ペプチド模倣構造で形成され得る。このため、本明細書で使用するように、 “アミノ酸”、または “ペプチド残基”は、天然に生ずるアミノ酸および合成アミノ酸の両方を意味する。例えば、ホモ-フェニルアラニン、シトルリンおよびノルロイシンは、本発明の目的のアミノ酸と見なされる。 “アミノ酸”はまた、プロリンおよびヒドロキシプロリンのようなイミノ酸残基を含む。側鎖は、(R)または(S)配置の何れかであり得る。好ましい実施態様では、アミノ酸は、(S)またはL-配置である。天然に生じない側鎖が使用されるならば、非-アミノ酸置換基が使用され得、例えば、インビボ分解を妨げるか、または遅延させる。導入遺伝子のターゲッティングまたは発現のために変異体タンパク質およびペプチドを作成する天然アミノ酸配列の変化は、例えば、当業者に既知の種々の手段によって行い得る。変異体ペプチドは、実質的に所定のペプチドに相同性であるが、そのペプチドとは異なるアミノ酸配列を有するペプチドである。相同性の程度(すなわち、同一性のパーセント)は、例えば、比較用に最適化されたコンピュータープログラム(例えば、GAPコンピュータープログラム、バージョン6.0またはより高いバージョン、Devereux et al. (Nucleic Acids Res., 12, 387 (1984))により記載、を用い、それは、University of Wisconsin Genetics Computer Group (UWGCG)から自由に入手可能である) を用い配列情報を比較することにより、決定され得る。変異体タンパク質および/またはペプチドの活性は、当業者に既知の他の方法を用い評価し得る。
【0063】
変異体タンパク質(ペプチド)と参照タンパク質(ペプチド)との間で同一でないアミノ酸残基に関して、変異体タンパク質(ペプチド)は、好ましくは、保存アミノ酸置換を含み、すなわち、所定のアミノ酸は、類似の大きさ、荷電密度、疎水性/親水性、および/または配置の他のアミノ酸により置換される(例えば、ValとPhe)。変異体部位-特異的変異は、発現ベクター中に、修飾部位を含む合成オリゴヌクレオチドをライゲーションすることにより、導入され得る。更にまた、オリゴヌクレオチド-指定部位-特異的突然変異誘発手順が使用され得、それは、例えば、Walder et al., Gene, 42:133 (1986); Bauer et al., Gene, 37:73 (1985); Craik, Biotechniques, January 1995, pp. 12-19; および米国特許番号 4,518,584 および 4,737,462に開示のものである。
【0064】
免疫修飾分子
本明細書に照らして、 “免疫修飾分子”は、抗原-特異的形式において標的細胞に対する細胞性および/または体液性宿主免疫反応を修飾するすなわち、増加または減少させるポリペプチド分子であり、好ましくは、宿主免疫反応を減少させるものである。一般的に、本発明の教示により、免疫修飾分子は、標的細胞表面膜と結合し、例えば、本明細書に記載のベクターからの発現後、細胞表面膜に挿入されるかまたはそれに共有結合的または非共有結合的に結合する。
【0065】
好ましい実施態様では、免疫修飾分子は、CD8 タンパク質のすべてまたは機能部分、またより好ましくは、CD8 α鎖のすべてまたは機能部分を含む。ヒト CD8 コーディング配列では、Leahy, Faseb J. 9:17-25 (1995); Leahy et al., Cell 68:1145-62 (1992); Nakayama et al., Immunogenetics 30:393-7 (1989)参照。CD8 タンパク質およびポリペプチドに関する"機能部分"は、本明細書に記載のようにveto活性を保持するCD8 α-鎖の部分、より特にCD8 α-鎖のHLA-結合活性を保持する部分、および特にCD8 α-鎖の細胞外領域中のIg-様ドメインを意味する。典型的な変異体 CD8 ポリペプチドが、Gao and Jakobsen, Immunology Today 21:630-636 (2000)に記載されており、それは引用により本明細書に含める。ある実施態様では、完全長CD8 α-鎖を用いる。しかし、幾つかの実施態様では、細胞質ドメインは欠失している。好ましくは、膜貫通ドメインおよび細胞外ドメインが保持されている。
【0066】
当業者が認識するように、CD8 α-鎖の膜貫通ドメインは、必要ならば、他の分子の膜貫通ドメインと交換され得、細胞外ドメインと標的細胞表面との関係を修飾し得る。この実施態様では、CD8 α-鎖の細胞外ドメインをコードする核酸は、膜貫通ドメインをコードする核酸に作動可能なように連結されている。任意の膜貫通タンパク質の膜貫通ドメインが、本発明に使用し得る。更にまた、膜貫通は、膜貫通タンパク質に見られることが知られていないものでもよい。この実施態様では、 “合成膜貫通ドメイン”は、約20から25までの疎水性アミノ酸を、その後に、少なくとも1つおよび好ましくは2つの荷電アミノ酸を含む。幾つかの実施態様では、CD8 細胞外ドメインは、当分野の通常の技術により、標的細胞膜に連結する。好ましいCD8 α-鎖配列を図 1に開示する。それは、ヒト、マウス、ラット、オランウータン、クモザル、モルモット、ウシ、Hispidコットンラット、飼育ブタおよびネコを含む種由来のアミノ酸配列または完全長CD8 α-鎖をコードする核酸配列の何れかの完全長配列を含む。
【0067】
好ましい実施態様では、CD8 α-鎖は、融合タンパク質ではなく、むしろトランケーションタンパク質であり、ここで、細胞内ドメインは欠失されている。図2に表したように、ヒトCD8 α-鎖遺伝子は、235のアミノ酸のタンパク質を発現する。該タンパク質は、以下のドメインに分けられると考えられ得る(該ポリペプチドのアミノ末端で始まり、カルボキシ末端で終わる): シグナルペプチド(アミノ酸1から21); 免疫グロブリン(Ig)-様ドメイン(およそアミノ酸22-136); 膜隣接ストーク領域(membrane proximal stalk region)(アミノ酸137-181); 膜貫通ドメイン(アミノ酸183-210)および細胞質ドメイン(アミノ酸211-235)。これらの種々のドメインをコードするコーディング配列の該ヌクレオチドは、シグナルペプチドをコードする1-63、細胞外ドメインをコードする64-546、細胞内ドメインをコードする約547-621および細胞内ドメインをコードする約622-708を含む。同様に、マウス配列は、以下のようにドメインに分け得る。該ポリペプチドは、アミノ酸1-27を含むシグナル配列、アミノ酸約28から194を含む細胞外ドメイン、アミノ酸約195-222を含む膜貫通ドメインおよびアミノ酸約223-310を含む細胞内ドメインに分け得る。同様に、これらのドメインをコードするコーディング配列の該ヌクレオチドは、シグナルペプチドをコードする核酸 1-81、細胞外ドメインをコードする約 82-582、膜貫通ドメインをコードする約 583-666および細胞外ドメインをコードする約 667-923を含む。
【0068】
幾つかの実施態様では、該全長タンパク質をコードする核酸は、遺伝子導入ビヒクルに含まれる。他の実施態様では、細胞内ドメインをコードする核酸は、遺伝子導入ビヒクル中のポリヌクレオチドには含まれず、これにより、膜アンカー型タンパク質は、細胞内ドメインを欠くこととなる。対応するドメインがまた、好ましい実施態様においてはマウスを含む他の種において同定され得る。
【0069】
当業者はまた、上記ポリペプチドにかなりの相同性を有する免疫修飾分子は、本発明における有利な使用を見出し得ると認識するだろう。従って、例えば、 “CD8 ポリペプチド”にはまた、図2に示されるヌクレオチドによりコードされるポリペプチドと少なくとも約80%の配列同一性、通常、少なくとも約85%の配列同一性、好ましくは、少なくとも約90%の配列同一性、より好ましくは、少なくとも約95%の配列同一性および最も好ましくは、少なくとも約98%の配列同一性を有する相同性ポリペプチドが含まれる。
【0070】
“CD8をコードする核酸分子”、およびそれと文法的に同等の用語は、図2に示すようなヒト CD8のヌクレオチド配列、ならびに図2に示す配列、および図1に開示するような配列を有するポリペプチドをコードする図2に示すヌクレオチドと少なくとも約80%の配列同一性、通常、少なくとも約85%の配列同一性、好ましくは、少なくとも約90%の配列同一性、より好ましくは、少なくとも約95%の配列同一性および最も好ましくは、少なくとも約98%の配列同一性を有するヌクレオチド配列を意味する。
【0071】
前記のように、多数の種々のプログラムが用いられ、タンパク質または核酸が既知配列と配列同一性または類似性を有するかどうか同定され得る。配列同一性および/または類似性は、以下に限らないが、Smith & Waterman, Adv. Appl. Math. 2:482 (1981)の局所的配列同一性アルゴリズムを含む当分野に既知の標準技術を用い、Needleman & Wunsch, J. Mol. Biol. 48:443 (1970)の配列同一性アライメントアルゴリズムにより、Pearson & Lipman, PNAS USA 85:2444 (1988)の類似性方法用のサーチにより、これらのアルゴリズム(GAP、BESTFIT、FASTA、およびTFASTA、Wisconsin Genetics Software Package, Genetics Computer Group, 575 Science Drive, Madison, WI)のコンピューター化インプリメンテーションにより、Devereux et al., Nucl. Acid Res. 12:387-395 (1984)に記載のBest Fit sequence programにより、好ましくは、セッティングされているディフォルトかまたはインスペクションにより、決定される。好ましくは、同一性の割合は、以下のパラメーターに基づきFastDBにより算出する: ミスマッチペナルティー 1; ギャップペナルティー 1; ギャップサイズペナルティー 0.33; およびジョイニングペナルティー 30、 “Current Methods in Sequence Comparison and Analysis,” Macromolecule Sequencing and Synthesis, Selected Methods and Applications, pp 127-149 (1988), Alan R. Liss, Inc.
【0072】
有用なアルゴリズムの例はPILEUPである。PILEUPは、連続的、対となっているアライメントを用い、関連の配列のグループから複数の配列アライメントを作成する。それはまた、アライメントの作成に用いるクラスタリングの関係を示すツリー(tree)をプロットし得る。PILEUPは、Feng & Doolittle, J. Mol. Evol. 35:351-360 (1987)の単純化した連続アライメント方法を使用する; 該方法は、Higgins & Sharp CABIOS 5:151-153 (1989) に記載のものに類似する。有用なPILEUPパラメーターは、ディフォルトギャップウエイト 3.00、ディフォルトギャップレングスウエイト 0.10、およびウエイテッドエンドギャップを含む。
【0073】
有用なアルゴリズムの他の例は、BLAST アルゴリズムであり、それは、Altschul et al., J. Mol. Biol. 215, 403-410, (1990)およびKarlin et al., PNAS USA 90:5873-5787 (1993)に記載されている。特に有用なBLASTプログラムは、Altschul et al., Methods in Enzymology, 266: 460-480 (1996); http://blast.wustl/edu/blast/ README.htmlから得たWU-BLAST-2 プログラムである。WU-BLAST-2は、幾つかのサーチパラメーター、それらの殆どは、ディフォルト値にセットされている、を用いる。調整可能パラメーターは、以下の値にセットする: オーバーラップスパン =1、オーバーラップフラクション = 0.125、ワードスレショウルド(T) = 11。HSP S および HSP S2 パラメーターは、動力学的な値であり、目的の配列がサーチされる特定の配列の組成および特定のデータベースの組成に依存してプラグラム自体によって決定される; しかし、それらの値は、感度が増大するように調整され得る。
【0074】
更なる有用なアルゴリズムは、Altschul et al. Nucleic Acids Res. 25:3389-3402により報告されているようなギャップ化BLASTである。ギャップ化BLASTは、BLOSUM-62置換スコアを用いる; スレショウルド T パラメーターは9にセットする; 非ギャップ化伸張を生ずるツー-ヒット法; チャージギャップレングス k、コスト 10+k; Xuは16にセットし、そしてXgはデータベースサーチステージ用に40にセットし、アルゴリズムのアウトプットステージ用に67にセットする。ギャップ化アライメントは、〜22 ビットに相当するスコアにより得られる。
【0075】
% アミノ酸または核酸配列同一性値は、マッチングする同一残基の数を、アライメント領域中の"より長い"配列の残基の合計数で割ることにより決定される。 “より長い”配列は、アライメントした領域中に最大数の実際の残基を有するものである(WU-Blast-2により導入され、アライメントスコアが最大化するギャップは無視される)。
【0076】
該アライメントには、アライメントされる配列におけるギャップの導入を含み得る。更に、図11で示されるヌクレオチドによりコードされるポリペプチドのアミノ酸配列よりも多いかまたは少ないか何れかのアミノ酸を含む配列では、ある実施態様では、配列同一性の割合は、アミノ酸の合計数に対する同一アミノ酸の数に基づき決定されることが理解される。このため、例えば、図11のヌクレオチドによりコードされるポリペプチドの配列よりも短い配列の配列同一性は、以下に考察するように、ある実施態様では、より短い配列中のアミノ酸数を用い決定される。同一性の割合の算出において、相対的重さは、挿入、欠失、置換などのような配列変化の種々の表示(manifestation)にわりあてられない。
【0077】
ある実施態様では、同一のみがプラス(+1)としてスコアされ、ギャップを含むすべての型の配列変化は値 “0”となり、ウエイテッドスケールまたは下記のような配列類似性算出用のパラメーターの必要を排除する。配列同一性の割合は、例えば、マッチングする同一残基数を、アライメント領域中の “より短い”配列の合計残基数により割り、100を掛けることにより、算出され得る。 “より長い”配列は、アライメントした領域中に最大数の実際の残基を有するものである。
【0078】
図2のヌクレオチドによりコードされるポリペプチドと100%未満の配列同一性を有するCD8は、一般的に、ヒト以外の種由来の天然CD8 ヌクレオチド配列、およびヒトまたは非-ヒト源由来の天然CD8 ヌクレオチド配列の変異体から作成される。これに関して、多くの技術が当分野に既知であり、それらは日常的に用いられ、天然CD8 配列のヌクレオチド配列変異体が作成され、天然CD8 ポリペプチドと通常関連する少なくとも1つの活性の存在に関してそれらの変異体のポリペプチド産物をアッセイし得る。好ましい実施態様では、CD8 α-鎖はヒト由来であるが、図1に示すように、ラット、マウスおよび霊長類由来のCD8 α-鎖が知られており、本発明における使用が見出される。
【0079】
CD8 活性を有するポリペプチドは、図2で示したヌクレオチドによりコードされるポリペプチドよりも短いかまたは長くてもよい。このため、好ましい実施態様では、CD8 ポリペプチドの定義に含まれるものは、図2のヌクレオチドによりコードされるポリペプチドの部分またはフラグメントである。本明細書の1つの実施態様でおいて、図2のヌクレオチドによりコードされるポリペプチドのフラグメントは、a)それらが、少なくとも、示した配列同一性を有し、そしてb)好ましくは、上記のように天然に生ずるCD8の生物学的活性を有するならば、CD8 ポリペプチドであると見なされる。
【0080】
更に、以下により十分に概略するように、CD8 α-鎖は、図2のヌクレオチドによりコードされるポリペプチドよりも長くつくられ得る; 例えば、他の融合配列の付加、または更なるコーディングおよび非-コーディング配列の解明によってつくられ得る。
【0081】
CD8 ポリペプチドは、好ましくは、組み換え体である。 “組み換えポリペプチド”は、組み換え技術を用い、すなわち、下記のような組み換え核酸の発現により、作成されたポリペプチドである。好ましい実施態様では、本発明のCD8は、図2に示す核酸配列、またはそのフラグメントの発現により作成される。組み換えポリペプチドは、少なくとも1以上の特性により天然に生ずるタンパク質とは区別される。例えば、ポリペプチドは、通常、その野生型宿主と関連する幾つかまたはすべてのタンパク質および化合物から単離されるかまたは精製され得、このため、実質的に精製され得る。例えば、単離ポリペプチドは、通常、その天然状態と関連する少なくとも幾つかの物質を伴っておらず、好ましくは、所定のサンプル中の合計タンパク質の少なくとも約 0.5重量%、より好ましくは、少なくとも約 5重量%を構成する。実質的に精製したポリペプチドは、合計ポリペプチドの少なくとも約 75重量%を構成し、少なくとも約 80重量%が好ましく、そして少なくとも約 90重量%が特に好ましい。その定義は、種々の生体または宿主細胞中、1つの生体からのCD8 ポリペプチドの産生を含む。
【0082】
更にまた、ポリペプチドは、誘導性プロモーターまたは高発現プロモーターの使用により、通常見られる濃度よりも顕著により高い濃度で作成され得、それによって、該ポリペプチドは高濃度のレベルで作成される。更にまた、ポリペプチドは、以下に考察するような、アミノ酸置換、挿入および欠失に加えて、天然では通常見られない形態であり得る。
【0083】
1つの実施態様では、本発明は、核酸 CD8 変異体を提供する。これらの変異体は、3つのクラスのうち1以上の中に入る: 置換、挿入または欠失変異体。これらの変異体は、通常、カセットまたはPCR 変異誘発または当分野に既知の他の技術を用い、図2のヌクレオチド中のヌクレオチドの部位特異的変異誘発により作成し、該変異体をコードするDNAを作成し、遺伝子治療ベクター中に該変異体を含ませ、その後、そのDNAを発現させる。アミノ酸配列変異体は、事前に決定した変異の性質、CD8 アミノ酸配列の天然に生ずる対立遺伝子または異種間変異とは異なるものとする特徴により特性解析される。変異体は、典型的に、天然に生ずる類似体と同質的な生物学的活性を示すが、変異体はまた、以下により十分に概略するように、修飾された特性を有するように選択され得る。
【0084】
配列変異を導入するための部位または領域は事前に決定するが、変異自体は事前に決定する必要はない。例えば、所定の部位での変異のパフォーマンスを最適化するために、ランダムの変異誘発は、標的コドンまたは領域で行われ得、発現した変異体は、最適化された所望の活性についてスクリーニングされる。既知配列を有するDNA中の事前に決定した部位での置換変異を作るための技術は既知であり、例えば、M13 プライマー変異誘発およびPCR 変異誘発である。変異体を作るための技術の他の例は、遺伝子シャッフリングの方法であり、これにより、ヌクレオチド配列の類似変異体のフラグメントは組み換えられ、新規変異体の組合せを作成することができる。その技術の例は、米国特許番号 5,605,703; 5,811,238; 5,873,458; 5,830,696; 5,939,250; 5,763,239; 5,965,408; および 5,945,325に見られ、それらは各々、引用により本明細書にすべて含める。
【0085】
アミノ酸置換は、典型的に単一残基である; 挿入は、通常、約 1から20 アミノ酸のオーダーであるが、相当に大きな挿入物が許容され得る。欠失は、約 1から約 20残基の範囲であるが、幾つかの場合、欠失は、かなり大きくなり得、細胞質ドメインまたはそのフラグメントを含み得る。
【0086】
置換、欠失、挿入またはそれらの任意の組合せを用い、最終の誘導体に到達し得る。一般的に、これらの変化は、数アミノ酸で行われ、分子の変化を最小化する。しかし、より大きな変化が、特定の状況下で許容され得る。CD8の特性において変化が小さいほうが望ましいとき、置換は、一般的に、以下のチャートにより作成される:
チャート1
もとの残基 例示的な置換
【表2】
【0087】
機能または免疫学的同一性における実質的な変化は、チャート1に示す置換ほどにはあまり保存されていない置換を選択することにより生ずる。例えば、置換は、より顕著な影響を与えるように作成され得る: 変化領域におけるポリペプチドバックボーンの構造、例えば、α-ヘリックスまたはβ-シート構造; 標的部位での分子の荷電または疎水性; または側鎖の体積。一般的に、ポリペプチドの特性に最も大きな変化を生ずると予測される該置換は、(a)親水性残基、例えば、セリル(seryl)またはスレオニルが、疎水性残基、例えば、ロイシル、イソロイシル、フェニルアラニル、バリルまたはアラニルに置換される; (b) システインまたはプロリンが、任意の他の残基に置換される; (c) 正に荷電した側鎖を有する残基、例えば、リシル、アルギニル、またはヒスチジル、負に荷電した残基、例えば、グルタミルまたはアスパルチルに置換される; または(d) 体積の大きい(bulky)側鎖を有する残基、例えば、フェニルアラニンは、側鎖を有しないもの、例えば、グリシンに置換される場合である。
【0088】
該変異体は、典型的に、同質の生物学的活性を示し、天然に生ずる類似体と同じ免疫反応を顕在化させるが、変異体はまた、所望によりCD8の特性を修飾するように選択される。更にまた、該変異体は、タンパク質の生物学的活性が変わるように設計され得る。
【0089】
本発明内に含まれるポリペプチドの1つのタイプの共有結合的修飾は、ポリペプチドの天然のグリコシル化パターンを変えることを含む。 “天然のグリコシル化パターンを変えること”は、本明細書の目的を意図し、天然配列のCD8 ポリペプチドに見られる1以上の炭水化物部分を欠失させること、および/または天然配列のポリペプチドには存在しない1以上のグリコシル化部位を加えることを意味する。
【0090】
ポリペプチドへのグリコシル化部位の付加は、そのアミノ酸配列を変えることにより達し得る。その変化は、例えば、1以上のセリンまたはトレオニン残基の付加、またはそれらによる天然配列のポリペプチドへの置換により為し得る(O-結合型グリコシル化部位の場合)。該アミノ酸配列は、所望により、DNAレベルでの変化により変え得る。特に、事前に選択した塩基においてポリペプチドをコードするDNAを変異させることにより、そのコドンは、望ましいアミノ酸に翻訳されるように生ずる。
【0091】
ポリペプチドに存在する炭水化物部分の除去は、グリコシル化のための標的としての役割をするアミノ酸残基をコードするコドンの変異置換により達し得る。
【0092】
一旦、天然の源から単離され、例えば、プラスミドまたは他のベクター内に含まれ、またはそれらから直鎖状核酸セグメントとして切除されると、組み換え核酸は、更に、プローブとして使用され得、他の核酸を同定および単離し得る。それはまた、 “前駆体”核酸として使用し得、修飾または変異核酸およびタンパク質を作り得る。それはまた、本明細書に記載のように、標的細胞を処置するためにベクターまたは他の送達ビヒクル中に組み込まれ得る。
【0093】
遺伝子治療発現ベクター
本発明に照らして、任意の適当な遺伝子治療発現ベクターが使用され得る。 “ベクター”は、その用語が、当業者に理解されている通り、遺伝子転移のためのビヒクルである。本発明による該ベクターは、以下に限らないが、プラスミド、ファージ、ウイルス、リポソームなどを含む。本発明による発現ベクターは、好ましくは、更なる配列および変異を含む。特に、本発明による発現ベクターは、本明細書に定義のように、免疫修飾分子、特にCD8 α-鎖をコードする導入遺伝子を含む核酸を含み、所望により、目的の治療分子をコードする少なくとも1つの更なる導入遺伝子を更に含む。該核酸は、全体的または部分的に合成的に作成されたコーディングまたは他の遺伝的配列またはゲノムまたは相補DNA(cDNA)配列を含み得、そして、DNAまたはRNAの何れかの形態で提供され得る。
【0094】
導入遺伝子および/または免疫修飾および/または治療分子をコードする遺伝子は、mRNAの発現およびタンパク質の産生のため、および他の生化学的特性の評価のため、ウイルス性ベクターからバキュロウイルスまたは適当な原核性または真核性発現ベクター中へ移動し得る。
【0095】
本発明によるベクター(組み換えアデノウイルス性ベクターおよび転移ベクターを含む)の作成に関して、そのベクターは、当業者に既知のように、標準的な分子的および遺伝的技術を用い構成され得る。ビリオンまたはウイルス性粒子を含むベクター(例えば、組み換えアデノウイルス性ベクター)は、適当な細胞系においてウイルス性ベクターを用い作成され得る。同様に、1以上のキメラコートタンパク質を含む粒子が、標準的な細胞系、例えば、目下、アデノウイルス性ベクターに使用されるもので作成され得る。次いで、これらの得られた粒子は、望ましいならば、特異的細胞に標的化され得る。
【0096】
任意の適当な適当な発現ベクター(例えば、Pouwels et al., Cloning Vectors: A Laboratory Manual (Elsevior, N.Y.: 1985)に記載のような)および対応する適当な宿主細胞は、宿主細胞中における組み換えペプチドまたはタンパク質の作成に用い得る。発現宿主は、以下に限らないが、エシェリキア(Escherichia)、バチラス(Bacillus)、シュードモナス(Pseudomonas)、サルモネラ(Salmonella)属内の細菌性種、哺乳類またはバキュロウイルス性システム(例えば、Luckow et al., Bio/Technology, 6, 47 (1988)に記載のような)を含む昆虫宿主細胞システム、およびCOS-7、C127、3T3、CHO、HeLa、BHKなどのような樹立細胞系を含む。本発明によるキメラタンパク質(ペプチド)を調製するための特に好ましい発現システムは、バキュロウイルス性発現システムであり、それは、Trichoplusia ni、Tn 5B1-4 昆虫細胞、または他の適当な昆虫細胞を用い、高レベルの組み換えタンパク質を産生する。当業者はもちろん、発現宿主の選択は、産生するペプチドのタイプに影響することを知っている。例えば、酵母または哺乳類細胞(例えば、COS-7 細胞)内で産生されたペプチドのグリコシル化は、大腸菌のような細菌性細胞内で産生されたペプチドのそれとは異なる。
【0097】
好ましい実施態様では、タンパク質は、哺乳類細胞内で発現する。哺乳類発現システムはまた、当分野で既知であり、レトロウイルス性システムを含む。哺乳類プロモーターは、哺乳類 RNA ポリメラーゼと結合し、あるタンパク質のコーディング配列の、mRNAへの下流域(3')転写を開始することができる任意のDNA 配列である。プロモーターは、転写開始領域を有し、それは、通常、コーディング配列の5’末端、およびTATAボックス(転写開始部位の上流に位置づけられた25-30塩基対を用いる)に隣接して位置する。該TATAボックスは、正しい部位でRNA 合成を開始することをRNA ポリメラーゼ IIに指示すると考えられている。哺乳類プロモーターはまた、上流プロモーターエレメント(エンハンサーエレメント)を含み、典型的に、TATAボックスの上流の100から200塩基対内に位置する。転写が開始され、何れかの方向に機能し得る、上流プロモーターエレメントが速度を決定する。哺乳類プロモーターとして使用するのに特に好ましいのは、哺乳類ウイルス性遺伝子由来のプロモーターである。そのウイルス性遺伝子はしばしば非常に高発現され、宿主範囲が広範だからである。例には、SV40 初期プロモーター、マウス乳腺腫瘍ウイルス LTR プロモーター、アデノウイルス主要後期プロモーター、単純ヘルペスウイルスプロモーター、およびCMV プロモーターがある。
【0098】
典型的に、哺乳類細胞により認識される転写終結およびポリアデニル化配列は、翻訳ストップコドンに対し3'側に位置する調節領域であり、このため、プロモーターエレメントと共に、コーディング配列に隣接する。成熟mRNAの3'末端は、部位-特異的な翻訳後開裂およびポリアデニル化により形成される。転写ターミネーターおよびポリアデニル化シグナルの例には、SV40由来のものがある。
【0099】
哺乳類宿主に、および他の宿主に外因性核酸を導入する方法は、当業者に既知であり、用いる宿主細胞により変化する。技術には、デキストラン-仲介トランスフェクション、リン酸カルシウム沈殿、ポリブレン仲介トランスフェクション、プロトプラスト融合、エレクトロポレーション、ウイルス性感染、リポソーム中へのポリヌクレオチドのカプセル化、および核中へのDNAの直接のマイクロインジェクションがある。
【0100】
タンパク質はまた、当分野に既知の技術を用い、融合タンパク質として作成され得る。このため、例えば、該タンパク質は、発現を増加するか、他の理由のために融合タンパク質として作成され得る。例えば、タンパク質がペプチドであるとき、該ペプチドをコードする核酸は、発現目的のために、他の核酸と連結させ得る。
【0101】
CD8の試験のために、タンパク質は、発現後に精製するかまたは単離する。タンパク質は、サンプル中に存在する他の成分に応じて、当業者に既知の種々の方法で単離し得るかまたは精製し得る。標準的な精製方法には、電気泳動的、分子的、免疫学的およびクロマトグラフィー的な技術があり、イオン交換、疎水性、親和性、および逆相HPLCクロマトグラフィー、およびクロマト分画が含まれる。例えば、CD8 タンパク質は、標準的な抗-CD8 抗体カラムを用い精製し得る。タンパク質濃度と関連する限外濾過およびダイアフィルトレーション技術もまた有用である。適当な精製技術の一般的な手引きとして、Scopes, R., Protein Purification, Springer-Verlag, NY (1982) 参照。必要な純度は、CD8 タンパク質の用途に応じて変化する。幾つかの場合には、精製しないことが必要である。幾つかの場合には、CD8 発現は、抗体結合および蛍光による検出かまたは蛍光活性化細胞選別装置(FACS)により、例えば、細胞表面で検出される。
【0102】
CD8をコードする核酸分子およびCD8 核酸分子のコーディングまたは非-コーディング鎖の何れかから得られる任意の核酸分子は、当分野に既知であり通常よく用いられる種々の方法で標的の細胞と接触され得、ここで、接触させることは、エキソビボまたはインビボであり得る。
【0103】
ウイルス性結合、侵入および遺伝子発現は、所望のタンパク質を発現する組み換えウイルスまたはRNAおよびβ-ガラクトシダーゼのようなマーカー遺伝子を生ずる目的の挿入物を含むアデノウイルス性ベクターを用いることにより最初に評価され得る。β-ガラクトシダーゼ遺伝子(Ad-LacZ)を含むアデノウイルスで感染された細胞におけるβ-ガラクトシダーゼ発現は、細胞にAd-Glucを加えた後、二時間ぐらいの短時間で検出され得る。この手順は、組み換えウイルスの細胞侵入および遺伝子発現の迅速で効果的な分析を提供し、通常の技術を用いる当業者により容易に実行される。
【0104】
タンパク質をコードする本発明の核酸を用いると、種々の発現ベクターが作成され得る。該発現ベクターは、自己複製する染色体外ベクターであるかまたは宿主ゲノム中に組み込まれるベクターの何れかであり得る。一般的に、これらの発現ベクターは、該タンパク質をコードする核酸に作動可能なように連結した転写性および翻訳性調節核酸を含む。用語 “コントロール配列”は、特定の宿主生体中において作動可能なように連結したコーディング配列の発現に必要なDNA 配列をいう。原核生物に適当なコントロール配列には、例えば、プロモーター、所望により、オペレーター配列、およびリボゾーム結合部位がある。真核生物細胞では、プロモーター、ポリアデニル化シグナル、およびエンハンサーを利用することが知られる。
【0105】
核酸は、他の核酸配列との機能的関係に置かれるときに、"作動可能なように連結"される。例えば、プレ配列(presequence)または分泌リーダー(secretory leader)のDNAは、ポリペプチドがポリペプチドの分泌に関係するプレタンパク質(preprotein)として発現するならば、ポリペプチドのDNAに作動可能なように連結されている; プロモーターまたはエンハンサーは、配列の転写に影響するならば、コーディング配列に作動可能なように連結されている; またはリボソーム結合部位は、翻訳を促進するように位置するならば、コーディング配列に作動可能なように連結されている。他の例として、作動可能なように連結とは、連続するように、および分泌リーダーの場合には、連続しかつ読み取り枠(reading phase)内にあるように連結したDNA 配列をいう。しかし、エンハンサーは、連続している必要はない。連結は、通常の制限部位での連結により達成される。そのような部位が存在しないならば、合成オリゴヌクレオチドアダプターまたはリンカーは、通常の実施により使用する。転写性および翻訳性調節核酸は、一般的に、CD8の発現に使用する宿主細胞に適当である;例えば、ヒト転写性および翻訳性調節核酸配列は、好ましくは、ヒト細胞におけるCD8の発現に使用する。多くのタイプの適当な発現ベクター、および適当な調節配列が、種々の宿主細胞用として当分野に既知である。
【0106】
一般的に、転写性および翻訳性調節配列には、以下に限らないが、プロモーター配列、リボゾーム結合部位、転写開始および停止配列、翻訳開始および停止配列、およびエンハンサーまたはアクチベーター配列があり得る。好ましい実施態様では、調節配列には、プロモーターおよび転写開始および停止配列がある。
【0107】
プロモーター配列は、構成性または誘導性プロモーターの何れかをコードする。プロモーターは、天然に生ずるプロモーターまたはハイブリッドプロモーターの何れかであり得る。1を超えるプロモーターのエレメントを組み合わせるハイブリッドプロモーターがまた、当分野に既知であり、本発明に有用である。
【0108】
加えて、発現ベクターは、更なるエレメントを含み得る。例えば、発現ベクターは、2つの複製システムを有し得、このため、2つの生体で、例えば、発現用に哺乳類または昆虫細胞で、およびクローニングおよび増幅用に原核生物宿主で保持され得る。更に、発現ベクターを組み込むため、該発現ベクターは、宿主細胞ゲノムに相同な少なくとも1つの配列、および好ましくは、発現コンストラクトに隣接する2つの相同配列を含む。該組み込みベクターは、ベクターに包含させるための適当な相同性配列を選択することにより宿主細胞中の特定の座に向けられ得る。ベクターを組み込むためのコンストラクトは当分野に既知である。
【0109】
更なる実施態様では、発現ベクターは、形質転換した宿主細胞を選択し得る選択マーカー遺伝子含み得る。選択遺伝子は、当分野に既知であり、使用する宿主細胞により変化する。
【0110】
好ましくは、該ベクターは、とりわけ、アデノウイルス性ベクター、アデノ-随伴ウイルス性ベクター、ヘルペスベクターまたはレトロウイルス性ベクターのようなウイルス性ベクターである。最も好ましくは、ウイルス性ベクターは、アデノウイルス性ベクターである。アデノウイルス性ベクターは、任意のアデノウイルスから得られ得る。"アデノウイルス"は、アデノウイルス科ファミリーの任意のウイルスであり、望ましくは、マストアデノウイルス(例えば、哺乳類アデノウイルス)またはアビアデノウイルス(例えば、エビアンアデノウイルス)属である。アデノウイルスは、任意の血清型である。アデノウイルスの源として用い得るアデノウイルス性ストックは、アデノウイルス性血清型 1 ないし 47から増幅し得、それは、現在、American Type Culture Collection (ATCC, Rockville, Md.)から入手可能であり、または任意の他の血清型のアデノウイルスから増幅し得、それは、任意の他の源から入手可能である。例えば、アデノウイルスは、サブグループ A (例えば、血清型 12、18、および31)、サブグループ B (例えば、血清型 3、7、11、14、16、21、34、および35)、サブグループ C (例えば、血清型 1、2、5、および6)、サブグループ D (例えば、血清型 8、9、10、13、15、17、19、20、22-30、32、33、36-39、および42-47)、サブグループ E (血清型 4)、サブグループ F (血清型 40 および 41)、または任意の他のアデノウイルス性血清型であり得る。しかしながら、好ましくは、アデノウイルスは、血清型 2、5 または 9である。望ましくは、アデノウイルスは、同一血清型の被覆タンパク質(例えば、ペントンベース、ヘキソン、および/または ファイバー)を含む。しかしながら、また好ましくは、例えば、すべてまたは一部の所定の被覆タンパク質が他の血清型由来であり得るという意味で1以上の被覆タンパク質は、キメラであり得る。
【0111】
好ましくは、アデノウイルス性ベクターであるウイルス性ベクターは、複製-能があり得るが、好ましくは、ウイルス性ベクターは、複製-欠損であるか、または条件付きで複製-欠損である。例えば、好ましくは、アデノウイルス性ベクターであるウイルス性ベクターは、ウイルス複製-欠損にする、少なくとも1つの修飾を有するゲノムを含む。ウイルス性ゲノムに対する修飾には、以下に限らないが、DNA セグメントの欠失、DNA セグメントの付加、DNA セグメントの再配列、DNA セグメントの置換、またはDNA 損傷の導入がある。DNA セグメントは、1ヌクレオチドぐらいに小さくてもよいし、または36 キロベース対、すなわち、およそアデノウイルス性ゲノムの大きさ、または38 キロベース対、それは、アデノウイルス性ビリオン中にパッケージされ得る最大の大きさ、ぐらいに大きくてもよい。
【0112】
ウイルス性、特に、アデノウイルス性、ゲノムに対する好ましい修飾には、ウイルス複製-欠損にする修飾に加えて、本明細書に定義の免疫修飾分子をコードする導入遺伝子、および更にはおよび好ましくは、目的の治療分子をコードする少なくとも1つの導入遺伝子の挿入がある。アデノウイルスのようなウイルスはまた、好ましくは、共組み込み(cointegrate)され得、すなわち、アデノウイルス性ゲノム配列のようなウイルスと、プラスミド、ファージまたは他のウイルスのもののような他の配列とのライゲーションである。
【0113】
アデノウイルス性ベクター(特に複製-欠損アデノウイルス性ベクター)に関して、そのようなベクターは、中実キャプシド(complete capsid)(すなわち、アデノウイルス性ゲノムのようなウイルス性ゲノムを含む)または中空キャプシド(empty capsid)(すなわち、そのウイルス性ゲノムは、欠いているか、例えば、物理的または化学的手段により破壊されている)の何れかのものを含み得る。好ましくは、ウイルス性ベクターは、中実キャプシド、すなわち、免疫修飾分子をコードする導入遺伝子および所望により好ましくは、阻害する手段をコードする少なくとも1つの導入遺伝子を運ぶ手段として含む。更にまた、好ましくは、導入遺伝子は、アデノウイルス性キャプシドの外側のセルに運ばれ得る。
【0114】
アデノウイルスのようなウイルスを特定の細胞に標的とさせるのに好ましいかまたは望ましい程度に、該ウイルスは、プラスミド DNAの、細胞中への転移において本質的にエンドソモリティック剤(endosomolytic agent)として用いられ得、それは、マーカー遺伝子を含み、トランスフェリンのような細胞-結合リガンドと共有結合したポリリジンと複合となりそれにより凝縮する(Cotten et al., PNAS (USA), 89, 6094-6098 (1992); および Curiel et al., PNAS (USA), 88, 8850-8854 (1991))。トランスフェリン-ポリリジン/DNA複合体およびアデノウイルス(例えば、トランスグルタミナーゼを伴う、アデノウイルスに対する抗体の手段によるか、またはビオチン/ストレプトアビジン結合(bridge)による)のカップリングは、実質的に、遺伝子転移を促進することが示されている(Wagner et al., PNAS (USA), 89, 6099-6103 (1992))。
【0115】
更にまた、アデノウイルス性ファイバーのような1以上のウイルス性被覆タンパク質は、例えば、細胞-表面レセプターに対するリガンド用の配列または二重特異性抗体(すなわち、ファイバーに対し特異性を有する一方の端、細胞-表面レセプターに対し特異性を有する他方の端を有する分子)に結合し得る配列の何れかの組み込みにより修飾され得る(PCT国際特許出願番号WO 95/26412('412 出願)およびWatkins et al., "Targeting Adenovirus-Mediated Gene Delivery with Recombinant Antibodies," Abst. No. 336)。両方の場合において、典型的なファイバー/細胞-表面レセプター相互作用が抑制され、アデノウイルスのようなウイルスは、そのファイバーを用いて新規細胞-表面レセプターに再度向けられる(redirect)。
【0116】
更にまた、選択した細胞タイプに特異的に結合できる標的化エレメントは、高親和性結合対の第一の分子に結合し得、宿主細胞に投与され得る(PCT国際特許出願番号WO 95/31566)。次いで、高親和性結合対の第二の分子に結合した遺伝子導入ビヒクルが、宿主細胞に投与され得、ここで、その第二の分子は、第一の分子に特異的に結合でき、そのため、遺伝子導入ビヒクルは、選択した細胞タイプを標的とする。
【0117】
同じ方針で、いくつかの方法(例えば、エレクトロポレーション、形質転換、三親性接合(triparental mating)の接合、(共-)トランスフェクション、(共-)感染、膜融合、微粒子銃(microprojectiles)の使用、リン酸カルシウム-DNA沈殿を伴うインキュベーション、ダイレクトのマイクロインジェクションなど)がウイルス、プラスミド、および核酸配列形態(すなわち、RNA または DNA)のファージの転移に利用可能であるため、ベクターは、同様に、キャプシドタンパク質のような任意の関連タンパク質の非存在下で、および任意のエンベロープ脂質の非存在下でRNA または DNAを含み得る。
【0118】
同様に、リポソームが、細胞膜との融合により細胞侵入を行うため、ベクターは、被覆タンパク質をコードする構成核酸を有するリポソームを含み得る。そのリポソームは、市場で入手可能であり、例えば、Life Technologies, Bethesda, Md.から入手でき、製造者の推奨に従い使用できる。更に、リポソームを使用し、遺伝子導入でき、転移能が増大しおよび/またはインビボで毒性が減少したリポソームが、使用し得る。可溶性キメラ被覆タンパク質(本明細書に記載の方法を用い作成したとき)は、製造者の指示に従い該リポソームを調製した後か、または該リポソームの調製中か何れかにおいて、リポソームに添加され得る。
【0119】
本発明によるベクターは、本発明の方法に用い得るものに制限されないが、遺伝子転移ベクターの構成において用い得る中間体-型ベクター(例えば、"転移ベクター")をも含む。
【0120】
1以上の核酸配列のインビボ送達のための好ましい方法の1つは、アデノウイルス発現ベクターの使用を含む。"アデノウイルス発現ベクター"は、(a)コンストラクトのパッケージングをサポートするのに、および(b)センスまたはアンチセンス方向で本発明でクローニングされたポリヌクレオチドを発現するのに十分なアデノウイルス配列を含むコンストラクトを含むことを意味する。もちろん、アンチセンスコンストラクトとの関連で、発現は、遺伝子産物が合成されることを必要としない。
【0121】
該発現ベクターは、遺伝子工学型のアデノウイルスを含む。アデノウイルスの遺伝的構成、36 kb、直鎖状、二本鎖 DNA ウイルスという知識により、アデノウイルス性 DNAの大きな断片が7 kbまでの外来配列と置換できる(Grunhaus and Horwitz, 1992)。レトロウイルスとは対照的に、宿主細胞のアデノウイルス性感染は、染色体への組み込みを生じない。アデノウイルス性 DNAが、エピソームの手法(episomal manner)で、遺伝毒性の可能性もなしに複製し得るためである。また、アデノウイルスは構造的に安定であり、ゲノム再配列は、広範囲な増幅後、検出されていない。アデノウイルスは、細胞周期の段階にかかわらず、すべての上皮細胞に実質的に感染し得る。今のところ、アデノウイルス性感染は、ヒトの急性呼吸器疾患のような軽い疾患にのみ関連していると思われる。
【0122】
アデノウイルスは、その中程度の大きさのゲノム、操作の容易さ、高力価、広範の標的-細胞範囲および高い感染性のため、遺伝子転移ベクターとしての使用に特に適当である。ウイルス性ゲノムの両端は、100-200 塩基対の逆方向反復(ITR)を含み、それらは、ウイルス性 DNA 複製およびパッケージングに必要なシスエレメントである。ゲノムの初期(E)および後期(L)領域は、ウイルス性 DNA 複製の開始により分けられる種々の転写単位を含む。E1 領域 (E1A および E1B)は、ウイルス性ゲノムおよび幾つかの細胞性遺伝子の転写の調節に関与するタンパク質コードする。E2 領域 (E2A および E2B)の発現は、ウイルス性 DNA 複製用のタンパク質の合成を生ずる。これらのタンパク質は、DNA 複製、後期遺伝子発現および宿主細胞停止(shut-off)に関与する(Renan, 1990)。多数のウイルス性キャプシドタンパク質を含む後期遺伝子の産物は、主要後期プロモーター(major late promoter)(MLP)により生ずる単一の一次転写産物の重要なプロセッシング後のみ発現する。該MLP(16.8 m.u.に位置する)は、特に、感染の後期フェーズの間に重要であり、このプロモーターから生ずるすべてのmRNAは、5'-三分裂リーダー(tripartite leader)(TPL)配列を有し、その配列により、翻訳に好ましいmRNAとなる。
【0123】
現在の系では、組み換えアデノウイルスは、シャトルベクターとプロウイルスベクターとの間の相同組み換えから生ずる。2つのプロウイルスベクター間での可能な組み換えのため、野生型アデノウイルスは、このプロセスから生じ得る。そのため、各プラークからウイルスの単一クローンを単離し、そのゲノム構造を調査することが重要である。
【0124】
複製欠損のアデノウイルスベクターの作成および増幅は、特有のヘルパー細胞系に依存する。天然では、アデノウイルスは、野生型ゲノムのおよそ105%をパッケージし得(Ghosh-Choudhury et al., 1987)、約 2 kB余のDNAを収容する。E1 および E3 領域で置換可能であるおよそ 5.5 kBのDNAと組み合わせると、現在のアデノウイルスベクターの最大収容は、7.5 kB未満、または該ベクターの全長の約 15%未満である。80%を超えるアデノウイルスウイルス性ゲノムが、ベクターバックボーン内にあり、ベクターから生ずる(vector-borne)細胞毒性の源である。また、E1-欠失ウイルスの複製欠損は不完全である。例えば、ウイルス性遺伝子発現の漏れ(leakage)が、高い感染効率(MOI)の現在利用可能なベクターで観察されている(Mulligan, 1993)。
【0125】
ヘルパー細胞系は、ヒト胎児腎臓細胞、筋細胞、造血細胞または他のヒト胎児間葉もしくは上皮細胞のようなヒト細胞から得られ得る。更にまた、ヘルパー細胞は、ヒトアデノウイルスにとって許容状態にある他の哺乳類種の細胞から得られ得る。その細胞には、例えば、ベロ細胞または他のサル胎児間葉もしくは上皮細胞がある。上記のように、現在、好ましいヘルパー細胞系は293である。
【0126】
最近、Racher et al. (1995)は、293 細胞を培養し、アデノウイルスを増殖させるための改善した方法を開示した。1つの形態では、天然の細胞凝集物は、100-200 mlの培地を含む1 リットルのシリコナイズしたスピナーフラスコ(Techne, Cambridge, UK)に各細胞を接種することにより培養する。40 rpmで撹拌後、細胞バイアビリティーを、トリパンブルーで評価する。他の形態では、Fibra-Cel マイクロキャリア (Bibby Sterlin, Stone, UK) (5 g/l)は以下のように用いる。5 ml の培地に再懸濁した細胞接種材料を、250 ml Erlenmeyer flask中の該キャリア(50 ml)に加え、時折撹拌(agitation)しつつ1から4時間静置させる。次いで、該培地は、50 ml の新しい培地と取り換え、撹拌を開始する。ウイルス産物の場合、細胞は、約 80% コンフルエンスにまで増殖させ得、その時間後、その培地は取り換え(最終体積の25%に)、アデノウイルスをMOI 0.05で加える。培養物は一夜静置し、その後、その体積は、100%に増加し、撹拌を更に72時間行う。
【0127】
好ましい実施態様では、アデノウイルスは、当分野に既知のように “弱い”アデノウイルスである。その"弱い"アデノウイルスベクターは、アデノウイルス性遺伝子導入のために最近開発されたシステムである。アデノウイルスの複製には、ヘルパーウイルス、およびE1a および Creの両方を発現する特別のヒト 293 細胞系が必要であり、天然環境下にはない条件である。今日最も効果的なシステムでは、E1-欠損ヘルパーウイルスを、バクテリオファージ P1 loxP 部位 ("loxPが導入された")が隣接するパッケージングシグナルと共に用いる。loxPが導入されたパッケージングシグナルを有するヘルパーウイルスと共に該弱いウイルスでCre リコンビナーゼを発現するヘルパー細胞の感染は、パッケージングシグナルをヘルパーウイルスのDNAから欠損させるため、弱い rAVを生ずるのみである。しかし、293-に基づくヘルパー細胞を用いるならば、そのヘルパーウイルス DNAは、ヘルパー細胞 DNA中に組み込まれるAd5 DNAと組み換えられ得る。結果として、野生型パッケージングシグナル、およびE1 領域は回復する。このため、また、293-(または911-)に基づくヘルパー細胞における弱いrAVの産物は、E1-欠損ヘルパーウイルスを用いるならば、RCAを生じ得る。
【0128】
ベクターは、すべてのウイルス性遺伝子が取り除かれる。このため、ベクターは、非-免疫原性であり、必要であれは、繰り返し使用し得る。その"弱い"アデノウイルスベクターはまた、導入遺伝子を収容するための36 kb のスペースを含み、このため、細胞中への大多数の遺伝子の共-送達をし得る。RGD モチーフのような特定配列モチーフは、アデノウイルスベクターのH-I ループ中に挿入し得、その感染性を促進し得る。アデノウイルス組み換え体は、本明細書の記載のようにおよび当分野に既知のように、任意のアデノウイルスベクター中へ、特定の導入遺伝子または導入遺伝子のフラグメントをクローニングすることにより構成される。アデノウイルス組み換え体は、免疫剤としての使用のため、非-侵入形態において脊椎動物の上皮細胞を形質導入するのに使用し得る。
【0129】
"弱い"アデノウイルスの使用は、特に、異種性 DNAの巨大挿入物の挿入に利点がある(レビューとしてYeh. and Perricaudet, FASEB J. 11:615 (1997)参照)。この文献は引用により本明細書に含める。加えて、弱いアデノウイルス性ベクターおよびそれを作成および使用する方法は、米国特許番号6,156,497 および 6,228,646により詳細に記載されており、それらの両方は、引用により本明細書に明示的に含める。
【0130】
アデノウイルスベクターは複製欠損であるか、または少なくとも条件付きで欠損であるという必要以外に、アデノウイルスベクターの性質が、本発明の実施の成功に重要であるとは考えられない。アデノウイルスは、42 種の既知の血清型またはサブグループ A-Fの何れかであり得る。サブグループ Cのアデノウイルス5型は、本発明での使用用の条件付き複製-欠損アデノウイルスベクターを得るための好ましい出発物質である。アデノウイルス5型は、多くの生化学的および遺伝的情報が知られているヒトアデノウイルスであるためである。そして、それは、ベクターとしてアデノウイルスを用いる殆どのコンストラクションとして歴史的に使用されている。
【0131】
上記のように、本発明による典型的なベクターは、複製欠損であり、アデノウイルス E1 領域を有しない。このため、E1-コーディング配列が除かれた位置に免疫修飾分子および/または目的の更なる治療タンパク質をコードする導入遺伝子を導入することが最も都合がよい。しかしながら、アデノウイルス配列内の発現コンストラクトの挿入の位置は、本発明に重要ではない。目的の導入遺伝子はまた、Karlsson et al. (1986)に記載のようにE3 置換ベクター中の欠失E3 領域のかわりに、またはE4 領域(ヘルパー細胞系またはヘルパーウイルスはE4 欠損を相補する)中に挿入し得る。
【0132】
アデノウイルスは、増殖および操作が容易であり、インビトロおよびインビボで広範の宿主範囲を示す。このグループのウイルスは、高力価、例えば、mlあたり109-1011のプラーク-形成単位(109 -1011 plaque-forming units per ml)で得られ得、それらは、高い感染力がある。アデノウイルスのライフサイクルは、宿主細胞ゲノム中への組み込みを必要としない。アデノウイルスベクターにより運ばれる外来遺伝子はエピソームであり、そのため、宿主細胞に対する遺伝毒性は低い。野生型アデノウイルスによるワクチン接種の研究では、副作用は報告されておらず(Couch et al., 1963; Top et al., 1971)、この文献は、インビボの遺伝子転移ベクターとしてその安全性および治療可能性を示している。
【0133】
アデノウイルスベクターは、真核性遺伝子発現(Levrero et al., 1991; Gomez-Foix et al., 1992)およびワクチン開発に使用されている(Grunhaus and Horwitz, 1992; Graham and Prevec, 1992)。最近、動物の研究は、組み換えアデノウイルスは遺伝子治療に使用され得ることを示唆する(Stratford-Perricaudet and Perricaudet, 1991; Stratford-Perricaudet et al., 1990; Rich et al., 1993)。組み換えアデノウイルスを種々の組織に投与する研究には、気管注入 (Rosenfeld et al., 1991; Rosenfeld et al., 1992)、筋肉注射 (Ragot et al., 1993)、末梢静脈注射 (Herz and Gerard, 1993) および脳中への定位固定接種(stereotactic inoculation)(Le Gal La Salle et al., 1993)がある。
【0134】
従って、好ましい実施態様では、本明細書で使用する発現ベクターは、アデノウイルス性ベクターである。適当なアデノウイルス性ベクターはAd2 または Ad5のようなヒトアデノウイルスの修飾を含み、ここで、インビボで複製するウイルスに必要な遺伝的エレメントは、取り除かれている; 例えば、E1 領域、およびアデノウイルス性ゲノムに挿入された目的の外因性遺伝子をコードする発現カセット。
【0135】
加えて、上記のように、好ましい発現ベクターシステムは、一般的にPCT/US97/01019 および PCT/US97/01048(それらは両方とも引用により本明細書に明示的に含まれる)に記載されているようなレトロウイルス性ベクターシステムである。
【0136】
レトロウイルスは、逆-転写のプロセスにより、感染細胞においてそのRNAを二本鎖DNAに変換する能力が特徴的である一本鎖 RNA ウイルスのグループである(Coffin, 1990)。次いで、生ずるDNAは、安定して、細胞性染色体中にプロウイルスとして組み込まれ、ウイルス性タンパク質の合成を指示する。その組み込みは、レシピエント細胞およびその子孫においてウイルス性遺伝子配列の保持を生ずる。レトロウイルス性ゲノムは、キャプシドタンパク質、ポリメラーゼ酵素、およびエンベロープコンポーネントをそれぞれコードする3つの遺伝子、gag、pol、およびenvを含む。gag 遺伝子の上流にある配列は、ゲノムをビリオン中にパッケージングするためのシグナルを含む。2つの長い末端反復(LTR)配列は、ウイルス性ゲノムの5' および 3' 末端に位置する。これらは、強力なプロモーターおよびエンハンサー配列を含み、また、宿主細胞ゲノムにおける組み込みに必要である(Coffin, 1990)。
【0137】
レトロウイルス性ベクターを構成するため、目的の1以上のオリゴヌクレオチドまたはポリヌクレオチド配列をコードする核酸は、複製-欠損であるウイルスを生ずる特定のウイルス性配列の代わりにウイルス性ゲノム中に挿入される。ビリオンを作成するため、gag、pol、およびenv 遺伝子を含むが、LTR およびパッケージングコンポーネントはないパッケージング細胞系を構成する(Mann et al., 1983)。レトロウイルス性 LTR およびパッケージング配列と共にcDNAを含む組み換えプラスミドが、この細胞系中に導入されるとき(例えば、リン酸カルシウム沈殿により)、該パッケージング配列は、組み換えプラスミドのRNA 転写物をウイルス性粒子中にパッケージし得、次いで、それは、培養培地中に分泌される(Nicolas and Rubenstein, 1988; Temin, 1986; Mann et al., 1983)。次いで、組み換えレトロウイルスを含む該培地を回収し、所望により濃縮し、そして遺伝子転移に使用する。レトロウイルス性ベクターは、広範の種々の細胞タイプに感染可能である。しかしながら、組み込みおよび安定な発現は、宿主細胞の分裂を必要とする(Paskind et al., 1975)。
【0138】
レトロウイルスベクターの特異的ターゲッティングができるように設計した新規アプローチは、最近、ウイルス性エンベロープに対するラクトース残基の化学的付加による、レトロウイルスの化学修飾に基づき開発された。修飾は、シアロ糖タンパク質レセプターを介する肝細胞の特異的感染を許容し得る。
【0139】
組み換えレトロウイルスのターゲッティングに対する種々のアプローチは、レトロウイルス性エンベロープタンパク質に対するおよび特異的細胞レセプターに対するビオチン化抗体が使用されるように設計された。該抗体は、ストレプトアビジンを用いることによりビオチンコンポーネントを介して結合した(Roux et al., 1989)。主要組織適合遺伝子複合体クラスIおよびクラスII抗原に対する抗体を用い、それらは、インビトロでエコトロピックウイルスによる表面抗原を生ずる種々のヒト細胞の感染を示した(Roux et al., 1989)。適当なレトロウイルス性ベクターは、LNL6、LXSN、およびLNCXを含む(レビューとしてByun et al., Gene Ther. 3(9):780-8 (1996)参照)。
【0140】
AAV (Ridgeway, 1988; Hermonat and Muzycska, 1984)は、パルボウイルスであり、アデノウイルス性ストックのコンタミネーションとして発見された。それは、任意の疾患とは関連していない広範に分布するウイルス(抗体は、USの人口の85%にある)である。それはまた、デペンドウイルス(dependovirus)として分類される。その複製が、アデノウイルスのようなヘルパーウイルスの存在に依存するためである。5つの血清型が単離されており、そのうち、AAV-2が最もよく特性解析されている。AAV は、キャプシドタンパク質 VP1、VP2 および VP3にカプセル化され、直径20から24 nmの二十面体ビリオンを形成する一本鎖直鎖状DNAを有する(Muzyczka and McLaughlin, 1988)。
【0141】
AAV DNAは、およそ 4.7 キロベース長である。それは、2つのオープンリーディングフレームを含み、2つのITRに隣接している。AAV ゲノム中に2つの主要な遺伝子: rep および capがある。該rep 遺伝子はウイルス性複製に関与するタンパク質をコードし、その一方、capはキャプシドタンパク質 VP1-3をコードする。各々のITRは、T-形ヘアピン構造を形成する。これらの末端反復は、AAVの染色体組み込みのために唯一の必須のcis コンポーネントである。そのため、AAVは、送達用の遺伝子のカセットにより除かれ置換されるすべてのウイルス性コーディング配列を有するベクターとして使用し得る。3つのウイルス性プロモーターは、同定されており、地図の位置によりp5、p19、およびp40と名付けられている。p5 および p19からの転写は、rep タンパク質の産物を生じ、p40からの転写は、キャプシドタンパク質を生ずる(Hermonat and Muzyczka, 1984)。
【0142】
AAVはまた、その安全性のため、送達ビヒクルとしてよい選択である。比較的複雑なレスキュー機構がある: 野生型アデノウイルスだけでなく、AAV 遺伝子もまた、rAAVを起動させることを必要とする。同様に、AAVは、病原性ではなく、任意の疾患とは関連しない。ウイルス性コーディング配列の除去は、ウイルス性遺伝子発現に対する免疫性反応を最小化し、それ故、rAAVは、炎症性反応を引き起こさない。AAVに関連する他の開示は、米国特許番号6,531,456に開示されている。その文献は、引用により本明細書に明示的に含める。
【0143】
他のウイルス性ベクターは、宿主細胞への免疫修飾分子の送達ために本発明で発現ベクターとして用いられ得る。ワクシニアウイルス (Ridgeway, 1988; Coupar et al., 1988)、レンチウイルス、ポリオウイルスおよびヘルペスウイルスのようなウイルスから得られるベクターが用いられ得る。それらは、種々の哺乳類細胞に幾つかの魅力的な特徴を供する(Friedmann, 1989; Ridgeway, 1988; Coupar et al., 1988; Horwich et al., 1990)。
【0144】
発現ベクターの送達
免疫修飾分子(例えば、CD8 α-鎖)および/または更なる治療タンパク質の発現を行うために、発現ベクターは、細胞中に送達されなければならない。この送達は、細胞系を形質転換する実験手順などでインビトロで、または特定の病状の治療などでインビボまたはエキソビボで行い得る。上記のように、送達のための1つの好ましい機構は、感染を介し、この場合、核酸は、組み換えウイルス性粒子中にカプセル化される。
【0145】
一旦、発現ベクターが細胞中に送達されると、望ましいオリゴヌクレオチドまたはポリヌクレオチド配列をコードする核酸は、種々の部位に位置し、発現し得る。特定の実施態様では、コンストラクトをコードする核酸は、安定して、細胞のゲノム中に組み込まれ得る。この組み込みは、相同組み換えによる特定の位置および方向であり得るか(遺伝子置換)またはそれは、ランダムに、非-特異的な位置に組み込まれ得る(遺伝子増大)。更に、および好ましい実施態様では、核酸は、DNAのエピソームセグメントとして別々に細胞中に安定して保持され得る。その核酸セグメントまたは"エピソーム"は、宿主細胞サイクルとは独立したまたはそれと同期した保持および複製を許容するのに十分な配列コードする。どのように発現コンストラクトが細胞に送達されるのかおよび細胞中のどこに核酸が保持されるのかは、用いる発現ベクターのタイプに依存する。
【0146】
本発明の特定の実施態様では、発現ベクターは、単に、裸の組み換えDNAまたはプラスミドからなり得る。ベクターの転移は、上記の任意の方法により行い得、それは、物理的または化学的に細胞膜を透過処理する。これは、特に、インビトロの転移に適用可能であるが、それは、インビボの使用に同様に適用され得る。Dubensky et al. (1984)は、成体および新生マウスの肝臓および脾臓中への、リン酸カルシウム沈殿の形態でのポリオーマウイルスDNAの注入に成功し、活動性のウイルス性複製および急性の感染を示した。Benvenisty and Reshef (1986)はまた、リン酸カルシウム-沈殿化プラスミドの直接の腹膜内注射が、トランスフェクトした遺伝子の発現を生ずることを示した。目的の遺伝子をコードするDNAがまた、類似の方法でインビボで転移され得、遺伝子産物を発現し得ることが想定される。
【0147】
裸のDNA発現コンストラクトを細胞中に転移するための本発明の他の実施態様は、微粒子銃を含み得る。この方法は、DNA-被覆マイクロプロジェクチルを高速にまで加速し、細胞膜を貫通し、細胞を殺すことなく細胞に入ることができる能力に依存する(Klein et al., 1987)。小粒子を加速させるための幾つかのデバイスが開発されている。そのようなデバイスは、電流を生じる高圧放電に依存し、次に、原動力を供する(Yang et al., 1990)。使用する該マイクロプロジェクチルは、一般的に、タングステンまたは金ビーズのような生物学的に不活性な物質からなる。
【0148】
ラットおよびマウスの肝臓、皮膚、および筋組織を含む選択した器官は、インビボで発射される(Yang et al., 1990; Zelenin et al., 1991)。これは、銃と標的器官との間に介在する任意の組織を排除する、組織または細胞の外科的露出、すなわち、エキソビボ治療を必要とし得る。更に、特定の遺伝子をコードするDNAは、この方法を介して送達され得、また、本発明により組み込まれる。
【0149】
本発明の1つの実施態様では、核酸分子は、リポソーム-仲介核酸転移により標的細胞中に導入される。これに関して、多くのリポソーム-ベースの試薬が当分野に既知であり、市場で入試可能であり、標的の細胞中に核酸分子を導入するために日常的に用い得る。本発明の特定の実施態様は、Lipofectamine または Lipofectin (Life Technologies)、陽イオンコレステロール誘導体(DC コレステロール)を伴うジオレオイルホスファチジルエタノールアミン(DOPE)、N[1-(2,3-ジオレイルオキシ)プロピル]-N,N,N-トリメチル塩化アンモニウム (DOTMA) (Sioud et al., J. Mol. Biol. 242:831-835 (1991))、DOSPA:DOPE、DOTAP、DMRIE:コレステロール、DDAB:DOPEなどのような陽イオン脂質転移ビヒクルを用いる。リポソーム-カプセル化核酸の生成物は、当分野に既知であり、典型的に、脂質および核酸の組合せを約 1:1の割合で含む。
【0150】
本発明の使用
上記のように、本明細書に記載され本明細書により可能となる方法および組成物には、例えば、遺伝子治療プロトコールでの使用のための、発現ベクターに対する宿主免疫反応を阻止する一般的な利用がある。すなわち、殆どの遺伝子治療プロトコールでよくある通常の問題は、ベクター-関連抗原に対する宿主免疫反応である。しかし、本発明により、この困難な問題は、遺伝子治療ベクターにおいて対象 CD8 ポリペプチドをコードする核酸の包含により克服される。すなわち、キメラベクターは、目的の治療分子をコードする核酸配列を、CD8 ポリペプチドとともに含むように使用される。ベクター-関連抗原と関連して細胞表面上で生じるCD8 ポリペプチドの発現は、アデノウイルス性ベクター中に存在するウイルス性被覆タンパク質のようなベクター-関連抗原に対する宿主免疫反応の効果的で特異的な阻害を生ずる。すなわち、ウイルス性タンパク質およびCD8が同一細胞内で発現するとき、CD8は、感染した細胞において宿主免疫反応を阻害し得、それにより、遺伝子治療ベクターによる治療処置を持続し得る。
【0151】
理論に制約されることなく、標的細胞におけるCD8の発現は、宿主免疫系において"veto効果"を誘発する能力を標的細胞に供与すると考えられる。すなわち、上記のように、CD8を発現する細胞が、宿主 T 細胞と接触するとき、該 T 細胞は、ダウンレギュレートされるかまたは死滅される。従って、 “veto効果” または “古典的veto”は、標的細胞に対する免疫反応をダウンレギュレートする標的細胞の能力を意味する。CD8 分子は、veto効果の誘発または転移に必要であると考えられる。 “veto効果の転移”は、veto効果は、通常、veto効果を誘発しない細胞に転移されることを意味する。すなわち、標的細胞に対するT 細胞反応を減少するまたはダウンレギュレートする能力が、CD8の発現を誘発するかまたは増加することにより、標的細胞に供与される。
【0152】
従って、本発明は、veto効果を誘発することにより、遺伝子治療送達ビヒクルおよび/または標的細胞に対する免疫反応を低減する使用を見出す。これは、他に標的細胞を認識するT 細胞のダウンレギュレートおよび欠失を生ずる。同様に、これは、体液性免疫反応の低下を生ずる。
【0153】
本発明の発現ベクターは、更に、インビトロでの利用を有する。そのベクターは、ウイルスクリアランスおよび持続の研究においておよび免疫反応を回避する手段の効果を評価する方法において研究ツールとして使用し得る。同様に、発現ベクター、好ましくは、組み換え発現ベクター、特にウイルス性またはアデノウイルス性ベクター(導入遺伝子および免疫修飾分子をコードする少なくとも1つの遺伝子を含む)は、インビボで用い得る。
【0154】
インビボ送達には、以下に限らないが、カテーテルを介するか、または他の手段である潅流による器官中への直接の注入がある。核酸は、標的器官に隣接する位置で血管内に投与され得るかまたは全身的に投与され得る。当業者は、各形態の送達の利点および不利点を認識する。例えば、直接の注入は、核酸の力価を最も高くし得るが、核酸の分布は、標的全体で不均一となるようである。標的に隣接する核酸の導入は、一般的に、器官の細胞とのよりよい接触を生ずるが、全身的投与は、一般的に、より簡単である。
【0155】
特に、本発明の組み換えアデノウイルス性ベクターのような発現ベクターを使用し、矯正 DNA、例えば、非存在であるかまたは欠損であるか何れかである機能をコードするDNAを細胞に送達することにより、多数の疾患のうちの何れか1つを治療し得る。その治療の候補となる疾患には、例えば、癌、例えば、黒色腫または神経膠腫、嚢胞性繊維症、遺伝性疾患、およびHIV 感染を含む病原性の感染がある。
【0156】
同種免疫および自己免疫反応を特異的に阻害する対象の組成物および方法の使用は、同時係属米国特許出願番号XXに記載されており、その開示は、引用により本明細書にすべて含める。本発明の方法および組成物の他の用途は、当業者に明かであろう。
【0157】
発現ベクターを投与するための組成物および方法
当業者は、発現ベクター(特にアデノウイルス性ベクター)を投与する多くの適当な方法および動物に対する本発明の免疫反応を阻害する手段(例えば、Rosenfeld et al., Science, 252, 431-434 (1991); Jaffe et al., Clin. Res., 39(2), 302A (1991); Rosenfeld et al., Clin. Res., 39(2), 311A (1991); Berkner, BioTechniques, 6, 616-629 (1988)参照)が利用可能であり、そして1を超える経路が、投与に使用され得るが、特定の経路が、他の経路よりもより早くより効果的な反応を提供し得ることを認識する。発現ベクターの投与に使用のための医薬に許容される賦形剤および/または免疫反応を阻害する手段も当業者に既知であり、容易に利用できる。賦形剤の選択は、発現ベクターの投与に使用される特定の方法によりおよび免疫反応を阻害する手段として幾分決定される。従って、本発明は、免疫修飾タンパク質(例えば、CD8 α-鎖) をコードする発現ベクターを、単独で、または導入遺伝子との更なる組合せで、適当な担体中に含む組成物を提供し、そして、広範の種々の、本発明と関連する使用に適当な製剤が存在する。特に、本発明は、CD8のα 鎖(またはその機能性フラグメント)をコードする遺伝子を含む発現ベクターおよびその担体を含む組成物を提供する。好ましい実施態様では、発現ベクターは、目的の治療分子またはタンパク質をコードする導入遺伝子を更に含む。その組成物は、治療薬または予防薬のような他の活性剤(active agents)および/または 当分野に知られるような免疫抑制剤を更に含み得る。以下の方法および賦形剤は、単なる例示であり、それらに制限されるものでは全くない。
【0158】
経口投与に適当な製剤は、以下からなり得る:(a)水、生理食塩水、またはオレンジジュースのような、希釈剤に溶解した有効量の化合物のような、溶液; (b) カプセル、サチェット(sachet)または錠剤、それぞれ、事前に決定した量の有効成分を、固体または顆粒として含んでいる; (c) 適当な液体中の懸濁液; および (d) 適当なエマルジョン。錠剤形は、1以上のラクトース、マンニトール、コーンスターチ、ジャガイモデンプン、微晶質セルロース、アカシア、ゼラチン、コロイド状二酸化ケイ素、クロスカルメロース(croscarmellose)ナトリウム、タルク、ステアリン酸マグネシウム、ステアリン酸、および他の賦形剤、色素、希釈剤、緩衝剤、湿潤剤、保存料、香料添加剤、および薬理学的に融和する賦形剤を含み得る。トローチ形は、香味料、通常、スクロースおよびアカシアまたはトラガカントゴム中に有効成分、ならびに該有効成分に加えて当分野に既知のようなその賦形剤を含むゼラチンおよびグリセリン、エマルジョン、ゲルなどのような不活性基剤中、有効成分を含む錠剤(pastilles)を含み得る。
【0159】
エアロゾール製剤は、吸入による投与のために作成され得る。これらのエアロゾール製剤は、ジクロロジフルオロメタン、プロパン、窒素などのような加圧型可能高圧ガス(pressurized acceptable propellants)中に置き得る。それらはまた、例えば、ネブライザーまたはアトマイザー中の非加圧型製剤用の医薬として製剤し得る。
【0160】
非経口的投与に適当な製剤には以下のものがある:水性および非-水性、等張性滅菌注入液溶液、それは、抗-酸化剤、緩衝剤、静菌剤および目的レシピエントの血液と製剤とを等張とする溶質を含み得る、および水性および非-水性滅菌懸濁液、それは、懸濁化剤、可溶化剤、増粘剤、安定剤、および保存料を含み得る。該製剤は、単位用量または複用量(multi-dose)でシールしたコンテナー、例えば、アンプルおよびバイアル中に存在し得、フリーズドライした(凍結乾燥した)状態、使用直前の注入用の滅菌液体賦形剤、例えば、水の添加のみを必要とする状態で保存し得る。即時注入の溶液および懸濁液は、上記種類の滅菌パウダー、顆粒、および錠剤から調製し得る。更に、座剤は、乳状化基材または水可溶性基材のような種々の基材の使用により作成され得る。膣内投与に適当な製剤は、ペッサリー、タンポン、クリーム、ゲル、ペースト、泡、またはスプレー製剤、これらの有効成分とともに、適当であると当分野で既知であるような担体を含む該製剤として存在し得る。
【0161】
本発明との関連で動物、特にヒトに投与する用量は、目的の治療導入遺伝子、ベクターの源および/または免疫修飾分子の性質、用いた組成物、投与の方法、および治療する特定の部位および生体により変わる。しかし、好ましくは、有効量のベクター(例えば、本発明によるアデノウイルス性ベクター)に相当する用量を用いる。"有効量"は、宿主において望ましい効果を生ずるのに十分な量であり、それは、当業者に既知の幾つかのエンドポイントを用いモニターし得る。例えば、1つの望ましい効果は、宿主細胞への核酸転移である。その転移は、以下に限らないが、治療効果(例えば、治療する疾患、病状、異常または症候群と関連する幾つかの徴候の緩和)を含む種々の手段により、または転移遺伝子またはコーディング配列または宿主内でのその発現(例えば、宿主細胞中の核酸を検出するポリメラーゼ連鎖反応、ノーザンもしくはサザンハイブリダイゼーション、または転写アッセイを用いるか、または転移した核酸によりコードされるタンパク質もしくはポリペプチド、またはその転移に起因するレベルまたは機能に影響するタンパク質もしくはポリペプチドを検出するイムノブロット分析、抗体-仲介検出、または詳細に述べられているアッセイを用いる)の証明によりモニターされ得る。記載されているこれらの方法は、決して包括的というわけではなく、そして特定の適用に適当な更なる方法は、当業者に明らかである。これに関して、ウイルス性ベクター、特に、アデノウイルス性ベクターのようなベクター、ならびに免疫反応を阻害する手段をコードするベクターの導入に対する宿主の反応は、投与されたウイルスの用量、送達の部位、およびベクターの遺伝的性質(makeup)ならびに導入遺伝子および免疫反応を阻害する手段により変化し得ることに注意すべきである。
【0162】
一般的に、本発明のベクターの有効な転移を確実とするために、所定の経路の投与を考慮し、接触するおおよその細胞数に基づき、細胞あたり約 1から約 5,000コピーの本発明によるベクターを用い接触させることことが好ましく、各細胞への約 3から約 300 pfuの導入が更により好ましい。しかし、これは、単なる一般的なガイドラインであり、それは、インビトロまたはインビボの何れかの特定の適用を保証し得るような、より高い量またはより少ない量の使用を決して排除するものではない。同様に、免疫反応を阻害する手段の量は、タンパク質を含む組成物の形態ならば、導入遺伝子を含む組み換えベクターに対する免疫反応の阻害に十分であるべきである。例えば、実際の用量およびスケジュールは、いずれの組成物を他の医薬組成物と組み合わせて投与するかにより、または薬物動力学、薬物動態、および代謝における個人間の相違により、変わり得る。同様に、量は、標的とする特定の細胞タイプまたはベクターが転移される手段により、インビトロの適応で変わり得る。当業者は、特定の状況の必要性に応じて任意に必要な調節をすることが容易であり得る。
【0163】
本発明は、好ましい実施態様に準拠して記載されているが、当業者は、本発明の精神および範囲を逸脱することなく形式および詳細を変え得ると認識するだろう。本明細書で特定している、特許、刊行物および他の引用文献、それぞれは、引用により本明細書にすべて明示的に含める。
【実施例】
【0164】
実施例1 veto作用−ベクターを用いた研究
a. 繊維芽細胞をveto細胞として操作するプラスミド発現ベクターの使用
繊維芽細胞を、ヒトまたはマウスの何れかのCD8-鎖をその表面において発現するように操作した。繊維芽細胞を、pCMVhCD8 プラスミドまたはpCMVmCD8 プラスミドでトランスフェクトし、それによって、CD8 α-鎖の発現は、CMV 前初期プロモーター/エンハンサー(Invitrogen)により生じた。CD8-鎖 トランスフェクト化繊維芽細胞 (H-2b)を、混合リンパ球培養物(BALB/c; H-2d 抗-C57BL/6; H-2b)に加えると、CD8-鎖を発現する系のみがCTL反応を抑制した。図 3A および Bに示すように、マウスまたはヒト CD8-鎖の何れかを発現するMC57T 繊維芽細胞の添加は、CTLの誘導を完全に抑制した。対照的に、非-トランスフェクト化繊維芽細胞の添加は、T-リンパ球の活性化に影響しなかった。CD8 α-鎖の阻害機能を確認することに加えて、これらの実験は、マウス T-リンパ球は、ヒト CD8 α-鎖によりvetoされ得ることをも示していた。それ故、マウスモデルは、臨床的使用用に設計したvetoを試験することに有用である。
【0165】
操作したveto細胞のインビボ機能
操作したvetoが動物で機能するかどうかを決定した。トランスフェクトし、CD8 α-鎖を発現するC57BL/6 (H-2b)-由来繊維芽細胞を、Balb/c (H-2d) マウスに注射した。対照動物に、非-トランスフェクト化繊維芽細胞を注射した。脾臓細胞は、8から40日後、回収し、スティミュレーター細胞としてのC57BL/6 (H-2b)脾臓細胞と共にMLC培養物中に導入した。5日後、培養物を回収し、EL4 (C57BL/6, H-2b)標的細胞を溶解する能力について試験した。抗-H-2b CTL反応の誘発は、CD8 -鎖を発現する繊維芽細胞を注射された動物では完全に抑制された(図 4)。抗-H-2b T 細胞の阻害は、非常に特異的であった。これらマウス由来のT 細胞はまた、第三のグループH-2k アロ-MHC 分子群に対する反応をマウントした。これらの実験で、操作したveto細胞は、古典的veto細胞と同様にインビボで免疫反応を特異的に抑制したこと、および非古典的veto細胞を操作するとveto細胞となったことを確認した。言い換えれば、操作した細胞は、これらの細胞において保有される抗原に対し動物を負に免疫した。
【0166】
CD8-鎖の発現が、完全に活性化されたT 細胞の機能を妨げるかどうか試験した。この目的のため、CD8 α-鎖を発現する標的細胞を、完全に活性化されたCTLによる溶菌に対する感受性について試験した。2種のT 細胞群をこれらの研究のために選択した。それは、MLCにおいて刺激されたアロ-反応性CTLおよび活性化ペプチド-特異的CTLであった。図 5に示したように、CD8 α-鎖を発現する標的は、アロ反応性 T 細胞の群により効果的に溶解されたが、抗原-特異的 T 細胞では溶解されなかった。これらの結果は、操作したvetoは、自己免疫反応で見られるような、継続的な抗原特異的免疫反応を妨げることができたことを示唆していた。
【0167】
b. veto細胞として繊維芽細胞を操作するためのウイルス転移ベクター
アデノウイルス転移ベクターm-CD8のveto機能: 複製-欠損ベクターアデノウイルス転移ベクター(mAdCD8α)は、マウス CD8 α-鎖を保有するように開発した。mAdCDB veto転移ベクターで感染されたマウス繊維芽細胞(MC57)は、日2に高レベルのマウス CD8 α-鎖を発現した。これらの早い増殖細胞において、マウス CD8 α-鎖の発現は、日5までに顕著に減少した。mAdCD8はまた、低効率であるにも関わらず、EL4のような他のマウス細胞系に感染した(データ示さず)。
【0168】
その後の実験において、mAdCD8 α-感染性MC57 繊維芽細胞 (H-2b)を、Balb/C (H-2d) 抗-C57B1/6 (H-2b) MLCに添加した。5 日後、培養物を回収し、抗-H-2b CTLの存在について試験した。繊維芽細胞に感染したMLCを加え、もはや抗-H-2b CTLを含まなかった(図 12)。これらの実験は、免疫抑制を仲介するveto転移ベクターの能力を確認した。
【0169】
加えて、アデノウイルス性ベクターのヒト CD8-版を作成した。また、マウス CD8 α-鎖を発現するアデノウイルス関連ウイルスを作成した。これらのウイルスは、各CD8 鎖の発現を誘導することが示された。マウスまたはヒトの何れかのCD8 α-鎖を発現するアデノウイルス性vetoベクターは、キラー T 細胞の誘導の完全阻害を仲介した(図 7参照)。
【0170】
mAdCD8 veto転移ベクターによる負の免疫化: 2種の実験を設定し、mAdCD8は、免疫反応をインビボで抑制するかどうかを決定した。第一の実験では、C57Bl/6 マウスを、等用量の、mAdCD8 veto転移ベクターまたは同様のアデノウイルス対照ベクター(マウス CD8 α-鎖の代わりにβ-ガラクトシダーゼをコードする(Adβgal))の何れかで感染させた。免疫化7日後、これらの動物を屠殺した。その脾臓細胞の単一細胞の懸濁液を、Adβgalウイルスの存在下、5日間、培養した。次いで、その培養物を回収し、増殖能を評価した。図 7に示したように、Adβgalに力強く増殖したT 細胞は、Adβgalで免疫したマウスから回収し、これは増殖性の高いCD4+ T 細胞の存在を示す。対照的に、mAdCD8-注射した動物から回収したT 細胞は、増加しなかった。
【0171】
第二の工程では、我々は、これらの培養物が、機能性 CD8+ CTLを含むかどうかを試験し、Adβgal-感染性標的細胞(EL4, H-2b) を溶解する能力を試験した。CTLは、Adβgalを注射したマウスから作成した培養物中に見られ得るのみであった(図 8)。この第一の実験は、AdCD8αが、恐らく、CD8 α-鎖の発現によってアデノウイルス性抗原に対する反応を誘発しなかったことを示唆する。しかしながら、別の理由のため、AdCD8は、免疫反応を誘発しなかったという可能性があった。AdCD8は、幾つかの不確定な方法において非-機能性であるか、またはマウスは、mAdCD8には見られなかったβ-ガラクトシダーゼタンパク質と反応し得るのみであった。
【0172】
種々の結論の妥当性を試験するために、C57Bl/6 マウスは、mAdCD8またはAdβgalの何れかで一旦注射され、その後、7日後、Adβgalで2回目の注射を行った。7日後、マウスを屠殺し、そして5-日の脾臓細胞培養物を、Adβgalの存在下、作成した。その対応するT 細胞は、Adβgal-感染性標的細胞に対する溶菌能について試験した(図 8)。確かに、Adβgalに対する2つの暴露は、免疫化を改善した。これらの研究はまた、AdCD8注射後、マウスはもはやAdβgalに反応せず、Adβgalは、主として、排他的でないならば、ベクターの両方に共通のアデノウイルス性タンパク質に対するCTL反応を誘発することを示していた。この一連の実験は、これらのベクターに保持される遺伝子に対する反応に対し負の免疫化ができる遺伝子治療ウイルス性ベクターを作成することが可能であることを強力に示唆する。
【0173】
vetoによるCD4+ T リンパ球の阻害: veto転移ベクターを用い、CD4+ T リンパ球の誘導を阻害し得るかどうか試験するために、以下の実験系を確立した。C57Bl/6-由来繊維芽細胞スティミュレーターを形質転換し、同種性MHCクラスII 分子 (H-2Ek)および免疫刺激性CD80を発現させた。これら遅く増殖する繊維芽細胞であって、放射線を照射せず刺激能を完全に保持している該細胞は、mAdCD8またはAdβgal転移ベクターの何れかで形質導入され、任意抽出のC57B1/6 脾臓細胞に添加された。4日後、これらの培養物を回収し、活性化、すなわち、ブラスティング(blasting)、CD4+ T リンパ球の存在について表面の免疫蛍光により分析した(図 9)。通常のまたはAdβgal-形質導入化スティミュレーター細胞とともに培養した任意抽出のC57Bl/6 脾臓細胞は、非常に多くのCD4+ T リンパ芽球を有していることが判った。対照的に、mAdCD8-感染性スティミュレーターが添加された培養物で、ほんの僅かなCD4+T リンパ芽球が検出された。この研究では、vetoがCD4+ T リンパ球を阻害し、そして加えて、ウイルス性veto転移ベクターがこの目的に使用され得ることを確認した。
【0174】
種々のウイルスコンストラクトによる感染後のマウスおよびヒト CD8 α-鎖の表面発現
染色プロトコール:
mAdCD8:
MC57Tは、偽-感染するか、またはmAdCD8によりおよそ104の感染効率で、修飾したIMDMにおいて3日間で感染される。感染した細胞を回収し、FITC(Pharmingen)で直接標識した抗-マウス CD8 α-鎖 抗体を伴うCD8 α-鎖の表面発現について染色した。表面蛍光の程度を、蛍光活性化細胞アナライザー(FACScan, Beckton-Dickinson)で測定した(図 10)。
【0175】
骨髄細胞は、Balb/c マウスの大腿骨の腔からから回収した。該細胞は、β-ガラクトシダーゼを発現するアデノウイルス性対照ベクター (AdLacZ)またはmAdCD8により104の感染効率で、修飾したIMDMにおいて3日間の培養で感染された。感染した細胞を回収し、FITCで直接標識した抗-マウス CD8 α-鎖 抗体によってCD8 α-鎖の表面発現について染色した。表面蛍光の程度を測定した(図 10C)。加えて、CD34+ 骨髄細胞、すなわち、幹細胞プール内の細胞を含む幾つかの細胞タイプを効率的に形質導入することを決定した(表 2)。
【表3】
表2
hAdCD8:
MC57Tを偽-感染した。hAdCD8のウイルス力価は知られていない。100μlのそのストック溶液を用い、3 x 105 細胞を3日間、感染した。その感染した細胞を回収し、そしてFITC (Pharmingen)で直接標識した抗-ヒト CD8 α-鎖 抗体によってCD8 α-鎖の表面発現について染色した。表面蛍光の程度を、蛍光活性化細胞アナライザーで測定した(図 10)。
【0176】
AAVに基づくvetoベクター
AAVに基づくvetoベクターを、Strategene /Avigen システムを用い平行して作成した。これらのコンストラクトにおいて、ヒトおよびマウス CD8 α-鎖は、同じCMV 中間体初期プロモーター/エンハンサーから得られた。2つのウイルス、mAAVCD8 および hAAVCD8を、HEK 293 パッケージング細胞系にパッケージした。その用いた系は、ヘルパーウイルスを用いない。mAAVCD8 および hAAVCD8は、効率的に、マウス繊維芽細胞(MC57T)に感染し、マウスまたはヒト CD8 α-鎖、それぞれについて高レベルの発現を生じた。蛍光の程度を、蛍光活性化細胞アナライザーで測定した(図 10D)。高レベルのCD8 α-鎖 発現が、形質導入後、36 時間以内に見られることが興味深い。この発見は、他者による観察とは対照的であった。それらでは、AAV-誘導化遺伝子発現は、顕著なレベルに到達するまで数日かかった(PH Schmelck, PrimeBiotech)。AAV vetoベクターによる更なる研究で、それらを用い免疫反応を抑制し得るという我々の先の発見が繰り返し得られた。ここで、標準的なMLC プロトコールを用いた(図 6)。
【0177】
実施例 2: インビトロ阻害研究 - 混合リンパ球培養物
脾臓細胞を、Balb/c (H-2d) および C57BL/6 (H-2b) マウスから回収した。単一の細胞懸濁液を調製した。C57BL/6 脾臓細胞を、3,000 rad (Mark 1 Cesium Irradiator)で照射した。4 x 106 Balb/c 脾臓細胞(レスポンダー/エフェクター細胞)は、10% 胎児ウシ血清(FCS)(Sigma)、HEPES、ペニシリンG、硫酸ストレプトマイシン、硫酸ゲンタマイシン、L-グルタミン、2-メルカプトエタノール、非必須アミノ酸(Sigma)、ピルビン酸ナトリウムおよび炭酸水素ナトリウムを含むIMDM(Sigma)(修飾IMDM)において、24-ウェルプレート(TPP, Midwest Scientific, Inc.)中のウェルあたり4 x 106 照射 C57BL/6 脾臓細胞(スティミュレーター細胞)と共に培養した。CO2 インキュベーター(Forma Scientific)中での培養で5日後、培養物をすべて回収し、C57BL/6-由来標的細胞(H-2b)を溶解する能力について試験した。
【0178】
幾つかのこれらの培養物に、4 x 105 MC57T 繊維芽細胞(H-2d)を加え、12,000 radを照射した。阻害培養において、4 x 105 MC57T 細胞を含み、mAdCD8によりおよそ104の感染効率で、1〜2日間で感染された。
【0179】
細胞毒性 T リンパ球キラーアッセイ
混合リンパ球培養物から回収した細胞を、活性化T リンパ球のインディケーターとして芽細胞の数についてカウントした。これらのエフェクター細胞を、U-底の96-ウェルプレート中の単一のウェルに加えた。ウェルあたりのエフェクターの数を、ウェルあたり3 x 106 または 1 x 105 エフェクターから出発する3-倍力価(titration)工程で測定した(titrated)。これらのエフェクター細胞に、ウェルあたり1 x 104 標的細胞 EL4 (H-2b)、MC57T (H-2b) または P815 (H-2d)を加えた。標的細胞は、先に、51Cr (Na-Chromate, Perkin-Elmer)で標識していた。1 x 106 標的細胞を、修飾IMDM中、およそ 500μlの体積で、90分間、100 μCiと共にインキュベートした。その後、組み込まれていない51Crを、修飾IMDMで数回洗浄することにより取り除いた。
【0180】
エフェクターおよび標的細胞を、合計体積 200 μlで、4時間、CO2 インキュベーター中でインキュベートした。その後、プレートを、1,500 rpmで3 分間、スピンさせ遠心分離した(Centra CJ35R, International Equipment Company)。100 mlの培地を、各ウェルから取り除き、標的細胞から放出される51Crの量をModel 4000 Gamma counter (Beckman Instruments)でカウントした。対照培養物をセットし、そのエフェクター細胞について、バックグラウンド放射を測定するのは省略した。標的細胞中への合計の51Cr取り込みをウェル中で測定し、Triton X100 (Sigma)の1% 溶液 (w/v)をエフェクター細胞と置換した。
【0181】
特異的な溶菌の量を以下のように%で決定した:
% = (特異的放射 - バックグラウンド放射) / (合計放射 - バックグラウンド放射) x 100
【0182】
mAdCD8のインビトロでの活性
混合化リンパ球培養物をセットした(Balb/c 抗-C57BL/6)。これらの培養物に、MC57T 繊維芽細胞を加え(示したように)、12,000 radで照射し、mAdCD8を感染させた。培養の5日後、その培養物を回収し、種々の標的に対するエフェクターの比(E/T)においてEL4 (H-2b) 標的細胞を溶解する能力について試験した(図 4参照)。
【0183】
図に見られるように、混合化リンパ球培養物においてさえ、CD8を発現する細胞は、溶菌 T リンパ球の誘導を阻害した。
【0184】
mAdCD8 および hAdCD8の作成
両アデノウイルス性ベクターを、BiogeneのAdEasy(商標)システムの助けにより作成した。ここで、マウスおよびヒト CD8 α-鎖 cDNAは、転移ベクターに組み込まれる(工程 1)。Ad5ΔE1/ΔE3 ベクターの組み換えは、BJ5183 EC 細菌において達成する(工程 2)。次いで、その組み換えベクターは、E1A および E1B アデノウイルス 5 ウイルス遺伝子(組み換えアデノウイルス中のこの本質的な領域の欠失を相補する)を含むQBI-HEK 293A 細胞に転移する。このため、これらの細胞中で作成されるhAdCD8 および mAdCD8は、複製欠損である。
【0185】
細菌性 LacZ 遺伝子 (β-ガラクトシダーゼ)を発現するコントロールベクターとして、Qbiogeneは、QBI-Infect+ Viral Particle(Ad5.CMVLacZΔE1/ΔE3)を提供していた。マウスCD8 α-鎖配列を用いた。この配列は、公開されているマウス配列に類似する:
タンパク質-配列:
実際の配列: MASPLTRFLS LNLLLMGESI ILGSGEAKPQAPELRIFPKK MDAELGQKVD LVCEVLGSVS QGCSWLFQNS SSKLPQPTFVVYMASSHNKI TWDEKLNSSK LFSAVRDTNN KYVLTLNKFS KENEGYYFCSVISNSVMYFS SVVPVLQKVN STTTKPVLRT PSPVHPTGTS QPQRPEDCRPRGSVKGTGLD FACDIYIWAP LAGICVAPLL SLIITLICYH RSRKRVCKCPRPLVRQEGKP RPSEKIV
【0186】
ヒト CD8 α-鎖配列を用いた。この配列は、示したように、公開されているヒト配列と比較するとサイレント変異を有する。
実際の配列: MALPVTALLL PLALLLHAAR PSQFRVSPLDRTWNLGWTVE LKCQVLLSNP TSGCSWLFQP RGAAASPTFL LYLSQNKPKAAEGLDTQRFS GKRLGDTFVL TLSDFRRENE GYYFCSALSN SIMYFSHFVPVFLPAKPTTT PAPRPPTPAP TIASQPLSLR PEACRPAAGG AGNRRRVCKCPRPVVKSGDK PSLARYV
【0187】
pAAV-mCD8 および pAAV-hCD8の作成
これらのベクターは、StratageneのAAV Helper-Free システムの助けにより作成した。そのシステムは、マウスおよびヒト配列をpAAV-MCS クローニングベクター中に挿入することにより機能する。次いで、このプラスミドは、ヘルパープラスミド(必要なアデノウイルス性タンパク質を含む)およびpAAV-RCベクター(キャプシド遺伝子を含む)と共に、HEK 293 細胞中に共-トランスフェクトし、組み換えAAV 粒子を産生する。
【0188】
実施例 3: 動物モデルにおける、操作したveto
我々は、どのように動物がmAdCD8の大用量の注射に対し反応するかを調査した。第一のセットの実験では、Balb/c マウス (各群で2匹のマウス)に、等用量のmAdCD8 または β-ガラクトシダーゼ(AdLacZ)をコードするアデノウイルス性コントロールベクターを静脈注射した。7日後、その動物は屠殺した。それらの脾臓細胞を、AdLacZの存在下、5日間、培養した。次いで、それらを、AdLacZ-感染性標的細胞(P815, Balb/c-由来)を溶解する能力について試験した。図 13に示すように、特異的溶菌能を有するCTLは、AdLacZで免疫されたBalb/c マウスから発展し得るが、mAdCD8を受けたマウスからは発展し得なかった。この結果は、AdCD8は、CD8 α-鎖の発現によってアデノウイルス性抗原に対する免疫反応を誘導しなかったことを示唆した。
【0189】
第二の設定では、C57Bl/6 マウスを、等用量のmAdCD8 (2 マウス) または AdLacZ (2 マウス) で免疫した。免疫化して7日後、各群の動物を1匹屠殺した。その脾臓細胞を、AdLacZの存在下、5日間、細胞懸濁液中で培養した。次いで、それらを、特異的にAdLacZ-感染性標的細胞(EL-4, C57Bl/6-由来)を溶解する能力について試験した。再び、AdLacZの注射は、低頻度での特定のキラー細胞の発展を誘導したにもかかわらず、その一方、mAdCD8は、そうならなかった(図 14)。
【0190】
この実験の第二のフェーズでは、mAdCD8 または AdLacZの何れかを受けた残りのC57BL/6 マウスは、第一のウイルス性注射7日後に第二の用量のAdLacZを受けた。7日後、マウスを屠殺し、5日後、脾臓細胞培養をAdLacZの存在下行った。反応するT 細胞を、再び、AdLacZ-感染性EL4-標的細胞に対する溶菌能について試験した(図 8)。実際、AdLacZに対する2つの暴露が、幾らかの免疫化の改善を生ずる。しかしながら、先にmAdCD8を受けた動物はなお、反応をマウントできない。これらの実験は、AdCD8は、免疫反応を誘導できないだけでなく、それ自身に対する免疫反応の誘導を予防したことを示唆する。このため、mAdCD8は、免疫系を回避した。
【図面の簡単な説明】
【0191】
【図1−1】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−2】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−3】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−4】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−5】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−6】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−7】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−8】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−9】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−10】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−11】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−12】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−13】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−14】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−15】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−16】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−17】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図2−1】図2は、野生型 CD8 α-鎖のアミノ酸および核酸配列を示す。
【図2−2】図2は、野生型 CD8 α-鎖のアミノ酸および核酸配列を示す。
【図3】図3は、C57BL/6 脾臓細胞で刺激されたBalb/c 脾臓細胞を示す。培養物に、マウス(A)またはヒト(B)由来の正常の繊維芽細胞 (黒丸)、培地(黒四角)、またはCD8を有する繊維芽細胞 (黒三角)が供された。培養物は回収され、C57BL/6-由来標的細胞に対するその溶菌能について試験した。
【図4】図4は、対照繊維芽細胞(黒四角および黒三角)またはmCD8-トランスフェクトC57BL/6-(H-2b)由来(白丸および黒丸)繊維芽細胞を注射したBalb/c(H-2d)マウスを示す。2週間後、動物を屠殺し、脾臓細胞を回収し、C57BL/6(H-2b)(黒四角および白丸)またはCBA/J(H-2k)(黒丸および黒三角)脾臓細胞で刺激し、EL4 (H-2b)(黒四角および白丸)またはS.AKR (H-2k)(黒丸および黒三角)標的細胞に対する溶菌能について試験した。
【図5】図5は、異型反応性(alloreactive) T 細胞(A) によるかまたは抗原-特異的 CTL(B)による溶菌に対する感受性について試験した標的細胞(黒三角)またはCD8-発現標的(黒四角)を示す。
【図6】図6は、正常繊維芽細胞 (黒丸)およびmAdCD8 (A、黒三角)またはHAdCD8 (B、黒三角)で遺伝子導入された繊維芽細胞の存在下で設定したMLC(Balb/c 抗-C57B/6)を示す。繊維芽細胞は対照培養物(黒四角)には加えなかった。C57BL/6-由来標的に対するこれらの培養物の溶菌活性を培養期間の最後に測定した。
【図7】図7は、アデノウイルス性veto転移ベクター、mAdCD8による免疫化を示す。C57BL/6 マウスは上記のベクターで感染させた。10日後、脾臓細胞を回収し、Adβgal ウイルスの存在下、培養した。芽細胞の数を示す。
【図8】図8 は、mAdCD8による陰性免疫化(negative immunization)を示す(A)。C57BL/6 マウスはAdβgalまたはmAdCD8により静脈注射で一度免疫化された。(A)のように処置した動物は、5日後、Adβgalで再免疫化した(B)。最後の注射の7日後、動物は屠殺し、その脾臓細胞はAdβgalの存在下、培養した。培養の5日後、細胞は、Adβgal-感染性同系標的細胞の溶菌能について試験した。
【図9】図9は、示したように形質導入した、1x106 (または全くなし)スティミュレーター細胞と共にインキュベートした3x106 C7Bl/6 脾臓細胞を示す。4日後、培養物を、免疫蛍光によりCD4+ T リンパ芽球の存在について分析した。
【図10−1】図10は、種々のウイルスコンストラクトによる感染後のマウスおよびヒト CD8 α-鎖の表面発現を示す。
【図10−2】図10は、種々のウイルスコンストラクトによる感染後のマウスおよびヒト CD8 α-鎖の表面発現を示す。
【図10−3】図10は、種々のウイルスコンストラクトによる感染後のマウスおよびヒト CD8 α-鎖の表面発現を示す。
【図10−4】図10は、種々のウイルスコンストラクトによる感染後のマウスおよびヒト CD8 α-鎖の表面発現を示す。
【図11】図11は、MLC (Balb/c 抗-C57BL/6)を、形質導入後、MLCに加えられる前、0 または 5 時間培養されたこれらの繊維芽細胞の存在下、設定したものを示す。培養の最後に、リンパ芽球の数を蛍光活性化細胞アナライザーで測定した。
【図12】図12は、veto転移ベクターによるインビトロ阻害を示す。
【図13】図13は、AdLacZ または mAdCD8で免疫化されたBalb/c マウスを示す。それらの脾臓細胞は、AdLacZの存在下で培養し、AdLacZ-感染性同系P815標的細胞に対する特異的溶菌活性について試験した。
【図14】図14は、AdLacZ (黒四角)またはmAdCD8 (黒三角)で免疫化した(A) C57BL/6 動物を示す。同系AdLacZ EL4 標的細胞に対する脾臓細胞の溶菌活性を試験した。(B)その動物は、AdLacz-感染化EL4 標的に対する溶菌活性を試験する前にAdLacZで再免疫化した。
【図15】図15は、ヘモグロビン βのmRNA 配列を示す。
【図16】図16は、GATA結合タンパク質のmRNA 配列を示す。
【図17−1】図17は、d-アミノエブリネート(aminoevulinate)シンターゼのmRNA 配列を示す。
【図17−2】図17は、d-アミノエブリネート(aminoevulinate)シンターゼのmRNA 配列を示す。
【図18−1】図18は、グルコース-6-ホスフェート-デヒドロゲナーゼのmRNA 配列を示す。
【図18−2】図18は、グルコース-6-ホスフェート-デヒドロゲナーゼのmRNA 配列を示す。
【図19−1】図19は、オルニチンカルバモイルトランスフェラーゼのmRNA 配列を示す。
【図19−2】図19は、オルニチンカルバモイルトランスフェラーゼのmRNA 配列を示す。
【図20−1】図20は、α-L-イズロニダーゼのmRNA 配列を示す。
【図20−2】図20は、α-L-イズロニダーゼのmRNA 配列を示す。
【図21−1】図21は、β-グルコシダーゼのmRNA 配列を示す。
【図21−2】図21は、β-グルコシダーゼのmRNA 配列を示す。
【図22】図22は、α-ガラクトシダーゼのmRNA 配列を示す。
【発明の詳細な説明】
【0001】
関連出願とのクロスレファレンス
この出願は、2003年3月19日出願の仮出願番号60/456,378の利益を主張する。
【0002】
本発明の分野
本発明は、一般的に遺伝子治療の分野に関係し、そしてより具体的には、遺伝子治療ベクターの免疫原性を低下させるための方法および組成物を提供する。
【0003】
発明の背景
遺伝子導入または遺伝子治療は後天性疾患および先天性疾患の治療のための見込みのある方法である。ますます多くの遺伝子(異常発現が生命を脅かすヒトの疾患と関連する遺伝子)が、クローニングされ、同定されている。ヒトにおいてそのようにクローニングされた遺伝子を発現する能力は、最終的に、多くの重要なヒトの疾患、現在の治療では不十分であるかまたは治療のない疾患の阻止および/または治癒を可能とする。例として、ヒト患者におけるコレステロール-調節遺伝子、HIV、または腫瘍抑制遺伝子の複製を選択的にブロックする遺伝子のインビボ発現は、心臓病、HIV、および癌、それぞれの治療を劇的に改善する。
【0004】
しかしながら、残念なことに、今日までに記載されている遺伝子治療プロトコールは、種々の問題により厄介であり、その中には、特に、ベクターからの遺伝子発現が短期間であることおよび同一ベクターを2回効果的に再投与できないことがあり、それらは、何れも、ベクターおよびその治療的ペイロードと関連する抗原に対する宿主免疫反応により生ずる。ウイルス性および/または治療遺伝子を組み込んだ組織は、CD8+ 細胞傷害性T細胞およびCD4+ ヘルパー T 細胞が仲介する宿主の細胞性免疫反応により最初に攻撃され、これは、ベクターからの遺伝子発現の持続を非常に制限する。更に、CD4+ T 細胞により仲介される宿主の体液性免疫反応は、更に、同一ベクターの再投与の成功を阻害することにより現在の遺伝子治療プロトコールの有効性を制限する。
【0005】
例えば、アデノウイルス性ベクターの最初の投与後、血清型-特異的抗体は、主要なウイルス性キャプシドタンパク質のエピトープ、すなわち、ペントン、ヘキソンおよびファイバーに対して生ずる。そのキャプシドタンパク質が、アデノウイルスそれ自身が細胞に結合しその後その細胞に感染する手段であるならば、次いでその抗体は、アデノウイルスの同一血清型により細胞の再感染を阻止するかまたは「中和」することができる。これは、遺伝子治療との関連で、その後に1以上の用量の外因性治療DNAを投与するために、アデノウイルスの異なる血清型を使用することを必要とする。加えて、治療およびウイルス性 遺伝子産物の両方が、標的細胞で発現し、それらは細胞性免疫反応に感受的となる。このため、それらは拒絶され、遺伝子治療の有益な効果が打ち消され、標的の器官または組織が破壊され得る。これら免疫が関連する障害の結果として、遺伝子治療プロトコールの進歩が妨げられている。
【0006】
従って、遺伝子治療発現ベクターおよびそのベクターによりトランスフェクトされた細胞に対する免疫反応を特異的に阻害する効果的な方法が当分野に非常に必要である。加えて、遺伝子治療ペイロードを投与しまたは導入するための改善された方法および組成物の必要がある。そのため、その遺伝子治療ベクターおよびその治療産物に対する細胞性および体液性免疫反応の両方を特異的に阻害し、それによって、そのベクターによりトランスフェクトされた細胞からの外因性遺伝子発現を増加させることが本発明の目的である。
【0007】
関連文献の要約
MHCクラスI-拘束(restricted)T 細胞(例えば、CD8+ CTL)の活性は、そのT 細胞レセプター複合体を通してシグナルを受け取るCTLがまたそのクラスI MHC 分子のα3 ドメインを通してシグナルを受け取るとき、抑制され得ることが知られている。これは、いわゆるvetoシグナルが、スティミュレーターまたは「veto」細胞により発現したCD8 分子により伝えられ得る。Sambhara and Miller, Science 252:1424-1427 (1991). 生じた免疫抑制は抗原-特異的でありかつMHC-拘束されており、それは、反応CTLによるveto細胞の一方向性認識から生ずるが、その逆は生じない。Rammensee et al., Eur. J. Immunol. 12:930-934 (1982); Fink et al., J. Exp. Med. 157:141-154 (1983); Rammensee et al., J. Immunol. 132:668-672 (1984). veto活性はCD8 α 鎖の存在と関連するため、それ故、CD8 α 鎖の発現のときにCD8の発現が消失し確認されるならばveto機能は喪失する。Hambor et al., J. Immunol. 145:1646-1652 (1990); Hambor et al., Intern. Immunol. 2:8856-8879 (1990); Kaplan et al., Proc. Natl. Acad. Sci. USA 86:8512-8515 (1989).
【0008】
多くのストラテジーが提唱され、望ましくない細胞傷害性T細胞反応を排除するこの抗原-特異的抑制経路が開発されている。1つのそのようなストラテジーは、CD8のveto活性を特異的な標的細胞に指示する第二リガンドに、CD8またはその機能ドメインを共有結合させるポリペプチド結合体(conjugate)の使用を含む。例えば、米国特許番号5,242,687、5,601,828および5,623,056参照。またあるいは、ハイブリッド抗体分子は、CD8 α 鎖の細胞外ドメインに結合するMHCクラスI 分子に対し特異性を有するモノクローナル抗体結合部位を有することが調べられている。Qi et al., J. Exp. Med. 183:1973-1980 (1996). しかし、その分子は、幾つかの欠点を有し、実際の臨床的有用性がまだ見い出されていない。
【0009】
更に最近では、WO 02/102852が、MHCクラスIに対する親和性が増加するように設計されたアミノ酸修飾を有する可溶性C8α 鎖変異体を用いるCTLの阻害を記載している。重要なことに、ここでは、提唱されたCD8α 組成物は、クラスI MHC 分子に特異的であり、それ故、CTLの反応のみを阻害すると予測されること、および更に他の免疫抑制剤との組合せが、細胞性および体液性免疫反応の他のエレメント、例えば、CD4+ T 細胞のようなMHCクラスII-拘束 T 細胞を含む状況で要求されることが教授されている。同文献 pp. 27-28.
【0010】
発明の概要
本発明は、CD8のような免疫修飾分子の標的化発現により仲介されるveto効果は、外因性遺伝的ペイロードを含む発現ベクターと関連する抗原に対する、およびトランスフェクトした標的細胞と関連する抗原に対する宿主免疫反応を効果的におよび特異的に阻害し得るという驚くべき発見に基づいている。本発明はまた、CD8αの標的化発現により仲介されるveto効果は、CD4+ T 細胞(MHCクラスII-拘束)およびCD8+ T 細胞(MHCクラスI-拘束)の反応を効果的におよび特異的に抑制し得るという更なる驚くべき発見、および該ベクター-関連抗原に対する宿主免疫反応の細胞性および体液性の両方のコンポーネントが阻害され得るという、得られた結論に基づいている。このため、本明細書で記載した方法および組成物を用いることにより、現在、ベクターからの遺伝子発現を制限し、遺伝子治療から最大の可能性を引き出すことを妨げるベクター-関連抗原に対する宿主免疫反応を阻害することにより遺伝子治療プロトコールは相乗的に促進され得る。
【0011】
従って、本発明は、発現ベクターおよびそのベクターでトランスフェクトされた標的細胞に対する宿主免疫反応を特異的に阻害するための組成物および方法を提供し、ここで、該ベクターは、veto効果を顕在化させることができる免疫修飾分子、好ましくは、CD8 ポリペプチド、より好ましくは、CD8 α-鎖、および最も好ましくは、CD8 α-鎖の細胞外および膜貫通ドメインの両方をコードする核酸配列を含む。対象の組成物および方法の性質、および上記CD8 α-鎖の従来技術の可溶性型の明かな不十分さを考慮すれば、CD8 α-鎖膜貫通ドメインまたは適当な代替の膜貫通領域の存在が重要であると思われる。
【0012】
1つの態様では、本発明は、発現ベクターに対する免疫反応を阻害するための方法であって、宿主標的細胞をインビボまたはエキソビボで、CD8 ポリペプチドのすべてまたは機能部分、好ましくは、CD8 α-鎖、および最も好ましくは、CD8 α-鎖の細胞外および膜貫通ドメインの両方をコードする発現ベクターと接触させること、ここで、該CD8 ポリペプチドは標的細胞の表面上で発現し、それにより、発現ベクターおよび標的細胞に対する免疫反応が特異的に阻害される、を含む該方法を提供する。組み換えベクターは、好ましくは、目的の治療タンパク質または分子をコードする1以上の更なる導入遺伝子を更にを含む。本明細書で記載および例示のように、免疫反応の体液性および細胞性コンポーネントの両方が、本発明の方法および組成物を使用することにより阻害される。
【0013】
更なる態様では、ベクター-関連抗原に対する宿主免疫反応の特異的阻害のための方法であって、CD8 ポリペプチドのすべてまたは機能部分、好ましくは、CD8 α-鎖、および最も好ましくは、CD8 α-鎖の細胞外および膜貫通ドメインの両方をコードする核酸を含む発現ベクターと宿主標的細胞をインビボまたはエキソビボで接触させること、ここで、CD8 ポリペプチドは標的細胞の表面上で発現し、それにより、ベクター-関連抗原に対する宿主免疫反応は特異的に阻害される、を含む該方法が提供される。
【0014】
更なる態様では、本発明は、宿主における治療導入遺伝子の発現を改善するのための方法であって、CD8 ポリペプチドのすべてまたは機能部分、好ましくは、CD8 α-鎖、および最も好ましくは、CD8 α-鎖の細胞外および膜貫通ドメインの両方をコードする核酸配列を含む発現ベクターを宿主に投与すること、ここで、CD8 ポリペプチドは宿主細胞の表面上で発現し、それにより、ベクター-関連抗原に対する宿主免疫反応が特異的に阻害される、を含む該方法を提供する。1つの実施態様では、治療導入遺伝子は、CD8 ポリペプチドと同一のベクターに含まれる。更に他の実施態様では、CD8 ポリペプチドおよび治療分子は別の発現ベクターによりコードされる。本明細書に記載のように、本発明の方法は、ベクター-関連抗原に対する宿主免疫反応の細胞性および体液性コンポーネントの両方を阻害し、それにより、宿主中の治療導入遺伝子を持続時間を延長させ、そしてその後の導入遺伝子発現のラウンドで発現ベクター再投与を可能とすることにより治療導入遺伝子の発現を改善する。
【0015】
更なる態様では、本発明は、免疫原性が減少した改善されたウイルス性発現ベクターを提供し、ここで、該発現ベクターは、本質的に、本明細書で開示したようなCD8 ポリペプチドをコードする核酸および目的の少なくとも1つの治療導入遺伝子をコードする核酸からなる非-ウイルス性核酸を含む。1つの実施態様では、治療導入遺伝子は免疫修飾分子以外のものである。好ましい実施態様では、CD8 ポリペプチドは、CD8 α-鎖のすべてまたは機能部分を含む。好ましくは、CD8 α-鎖の機能部分は、CD8 α-鎖の少なくとも細胞外ドメイン、およびより好ましくは、CD8 α-鎖の細胞外ドメインおよび膜貫通ドメインの両方を含む。一般的に、本明細書で提供する免疫修飾分子は、標的細胞のトランスフェクション後、標的細胞表面膜と結合し、例えば、該膜内に挿入されるか、またはそれと共有結合的または非共有結合的に結合される。
【0016】
本明細書での使用を意図する適当な発現ベクターは、組み換えおよび非-組み換えベクター、およびウイルス性(例えば、アデノウイルス性、レトロウイルス性、アデノ-随伴ウイルス性ベクターなど)および非-ウイルス性(例えば、細菌性プラスミド、ファージ、リポソームなど)ベクターを含む。ウイルスベクターが好ましく、アデノウイルス性ベクターが最も好ましい。
【0017】
多数の実施態様を開示しているが、本発明の他の実施態様が、本発明の例示となる実施態様を示し記載している、以下の詳細な説明から当業者には明らかとなるだろう。お解りのように、本発明は、種々の明らかな態様で修飾することができ、それらはすべて本発明の精神および範囲を逸脱することはない。従って、図面および詳細な説明は、事実上の例示であると見なされ、それらに限定すべきでない。
【0018】
図面の簡単な説明
図1は、様々な種由来のCD8 α-鎖 タンパク質および核酸配列を示す。示した配列の受け入れ番号もまた含まれている。
【0019】
図2A-Bは、野生型 CD8 α-鎖のアミノ酸および核酸配列を示し、ヒトおよびマウスのタンパク質の種々のドメインの区域(demarcation)が含まれている。
【0020】
図3は、C57BL/6 脾臓細胞で刺激されたBalb/c 脾臓細胞を示す。培養物に、マウス(A)またはヒト(B)由来の正常の繊維芽細胞 (黒丸)、培地(黒四角)、またはCD8を有する繊維芽細胞 (黒三角)が供された。培養物は回収され、C57BL/6-由来標的細胞に対するその溶菌能について試験した。
【0021】
図4は、対照繊維芽細胞(黒四角および黒三角)またはmCD8-トランスフェクトC57BL/6-(H-2b)由来(白丸および黒丸)繊維芽細胞を注射したBalb/c(H-2d)マウスを示す。2週間後、動物を屠殺し、脾臓細胞を回収し、C57BL/6(H-2b)(黒四角および白丸)またはCBA/J(H-2k)(黒丸および黒三角)脾臓細胞で刺激し、EL4 (H-2b)(黒四角および白丸)またはS.AKR (H-2k)(黒丸および黒三角)標的細胞に対する溶菌能について試験した。
【0022】
図5は、異型反応性(alloreactive) T 細胞(A) によるかまたは抗原-特異的 CTL(B)による溶菌に対する感受性について試験した標的細胞(黒三角)またはCD8-発現標的(黒四角)を示す。
【0023】
図6は、正常繊維芽細胞 (黒丸)およびmAdCD8 (A、黒三角)またはHAdCD8 (B、黒三角)で遺伝子導入された繊維芽細胞の存在下で設定したMLC(Balb/c 抗-C57B/6)を示す。繊維芽細胞は対照培養物(黒四角)には加えなかった。C57BL/6-由来標的に対するこれらの培養物の溶菌活性を培養期間の最後に測定した。
【0024】
図7は、アデノウイルス性veto転移ベクター、mAdCD8による免疫化を示す。C57BL/6 マウスは上記のベクターで感染させた。10日後、脾臓細胞を回収し、Adβgal ウイルスの存在下、培養した。芽細胞の数を示す。
【0025】
図8 は、mAdCD8による陰性免疫化(negative immunization)を示す(A)。C57BL/6 マウスはAdβgalまたはmAdCD8により静脈注射で一度免疫化された。(A)のように処置した動物は、5日後、Adβgalで再免疫化した(B)。最後の注射の7日後、動物は屠殺し、その脾臓細胞はAdβgalの存在下、培養した。培養の5日後、細胞は、Adβgal-感染性同系標的細胞の溶菌能について試験した。
【0026】
図9は、示したように形質導入した、1x106 (または全くなし)スティミュレーター細胞と共にインキュベートした3x106 C7Bl/6 脾臓細胞を示す。4日後、培養物を、免疫蛍光によりCD4+ T リンパ芽球の存在について分析した。
【0027】
図10A-Dは、種々のウイルスコンストラクトによる感染後のマウスおよびヒト CD8 α-鎖の表面発現を示す。A. 感染細胞: Mc57T 繊維芽細胞; パネル 1: 偽-感染; パネル 2: hAdCD8による感染. B. 感染細胞: MC57T 繊維芽細胞; パネル 1: 偽感染; パネル 2: mAdCD8による感染. C. 感染細胞: Balbc 非選択骨髄細胞; パネル 1: lacZ アデノウイルス性ベクター (AdLacZ)による感染; パネル l2: mAdCD8による感染. D. 感染細胞: MC57T 繊維芽細胞; パネル 1: 偽-感染; パネル 2: pAAV-mCD8による感染; パネル 3: pAAV-hCD8による感染.
【0028】
図11は、MLC (Balb/c 抗-C57BL/6)を、形質導入後、MLCに加えられる前、0 または 5 時間培養されたこれらの繊維芽細胞の存在下、設定したものを示す。培養の最後に、リンパ芽球の数を蛍光活性化細胞アナライザーで測定した。
【0029】
図12は、veto転移ベクターによるインビトロ阻害を示す。BALB/c 抗-C57BL/6混合リンパ球培養 (MLC)は、非感染またはmAdCD8-感染MC57繊維芽細胞 (H-2b)の非存在下または存在下で行った(X)。CTL反応は、EL4 (H-2b)標的細胞で測定した。
【0030】
図13は、AdLacZ または mAdCD8で免疫化されたBalb/c マウスを示す。それらの脾臓細胞は、AdLacZの存在下で培養し、AdLacZ-感染性同系P815標的細胞に対する特異的溶菌活性について試験した。
【0031】
図14は、AdLacZ (黒四角)またはmAdCD8 (黒三角)で免疫化した(A) C57BL/6 動物を示す。同系AdLacZ EL4 標的細胞に対する脾臓細胞の溶菌活性を試験した。(B)その動物は、AdLacz-感染化EL4 標的に対する溶菌活性を試験する前にAdLacZで再免疫化した。
【0032】
図15は、ヘモグロビン βのmRNA 配列を示す。
【0033】
図16は、GATA結合タンパク質のmRNA 配列を示す。
【0034】
図17は、d-アミノエブリネート(aminoevulinate)シンターゼのmRNA 配列を示す。
【0035】
図18は、グルコース-6-ホスフェート-デヒドロゲナーゼのmRNA 配列を示す。
【0036】
図19は、オルニチンカルバモイルトランスフェラーゼのmRNA 配列を示す。
【0037】
図20は、α-L-イズロニダーゼのmRNA 配列を示す。
【0038】
図21は、β-グルコシダーゼのmRNA 配列を示す。
【0039】
図22は、α-ガラクトシダーゼのmRNA 配列を示す。
【0040】
詳細な説明
発現ベクターと関連するタンパク質に対する宿主免疫反応は、遺伝子治療技術の開発に厄介であり、ここで、該反応の細胞性コンポーネントは、ベクター内に含まれる遺伝子の発現を厳格に制限し、該反応の体液性コンポーネントは、免疫能のある動物における同一ベクターの再投与を面倒にする。本発明の成功は、発現ベクターでトランスフェクトされた標的細胞におけるCD8のような免疫修飾分子の発現は、CD4+ T 細胞およびCD8+ T 細胞の両方の反応を抑制し、それにより、ベクター-関連抗原に対する宿主免疫反応の体液性および細胞性コンポーネントの両方を効果的におよび特異的に阻害するという驚くべき発見に起因する。
【0041】
このため、本明細書で記載する組成物および方法は、宿主細胞における発現ベクターの持続を向上させ、それにより、ベクター内に含まれる治療導入遺伝子の発現を改善させ、そして、同一ベクター(例えば、同一血清型の組み換えアデノウイルス性ベクター)による宿主細胞への再投与を成功させ得ることにより、インビボおよびエキソビボ遺伝子治療プロトコールを劇的に改善することができる。1つの実施態様では、免疫修飾分子、好ましくは、CD8 ポリペプチド、より好ましくは、CD8 α鎖、および最も好ましくは、CD8 α-鎖の細胞外ドメインおよび膜貫通ドメインの両方をコードする核酸、および目的の1以上の治療分子をコードする核酸配列を含む発現ベクターが提供される。更なる他の実施態様では、別々の発現ベクターを提供し、その1つはCD8 ポリペプチドをコードし、そしてその1つは、宿主に対する共-投与のための望ましい治療分子をコードする。
【0042】
本発明はまた、発現ベクター、特に、アデノウイルス性ベクター、アデノ-随伴ウイルス性ベクター、ヘルペスウイルス性ベクターまたはレトロウイルス性ベクターのような組み換えベクターに対する免疫反応を阻害するのための方法であって、標的細胞を、例えばインビボおよびエキソビボ遺伝子治療との関連で免疫修飾分子および目的の1以上の治療分子をコードする発現ベクターと接触させることを含む該方法を提供する。本明細書で記載および例示のように、本発明により達成される宿主免疫反応の抗原-特異的阻害は、細胞における発現ベクターのより持続的な存在、およびベクター内に含まれる治療導入遺伝子の付随的に改善した発現を可能とし、ならびに遺伝子治療を続けるための同一ベクターの再投与の成功が可能となる。
【0043】
従って、本発明は、遺伝子治療のための組成物および方法を提供し、ここで、遺伝子治療送達ビヒクルと関連する抗原に対する細胞性および体液性免疫反応は、完全になくなるか減少する。一般的に、本発明は、発現ベクター、遺伝子治療ベクター、遺伝子治療ベクターで感染させた標的細胞または標的細胞の子孫に対する細胞性および/または体液性免疫反応の両方を低下させるか減らすための方法および組成物を目的とする。
【0044】
“インビボ遺伝子治療”および “インビトロ遺伝子治療”は、エキソビボの応用を含む “遺伝子治療”の分野の当業者にとって通常既知のことおよび該当業者が引用することに関する、過去、現在および将来のすべての変更および修飾を含むことを意図する。
【0045】
“発現ベクター”は、標的細胞への核酸の送達のための任意のビヒクルを意味する。発現ベクターは、一般的に、ウイルス性ベクターおよび非-ウイルス性ベクターに分けることができる。ウイルス性ベクターは、以下に限定されないが、アデノウイルス性ベクター、アデノ-随伴ベクター、レトロウイルス性ベクター、レンチウイルス性ベクターなどを意味する。非-ウイルス性ベクターは、プラスミドベクター、裸のDNA、種々の担体と結合するかまたはリポソームもしくは他の脂質調製物と関連する裸のDNAを意味する。一般的に、発現ベクターは組み換え体であるが、幾つかの実施態様では、例えば、リポソームまたは細胞切除、例えば、遺伝子銃技術を用いるとき、それらは、組み換え体ではない。本明細書で使用のための好ましい組み換えベクターは、プラスミドベクター、およびアデノウイルス性ベクター、アデノ-随伴ウイルス性ベクター、ヘルペスウイルス性ベクターおよびレトロウイルス性ベクターからなる群から選択されるウイルス性ベクターである。組み換えウイルス性ベクター、および特に、アデノウイルス性ベクターを用いる幾つかの実施態様では、キャプシド、例えば、アデノウイルス性キャプシドのヘキソンタンパク質の免疫原性は、当分野に既知の方法により低下し得るが、本明細書で詳述の改善の観点からその修飾はもはや必要ではない。
【0046】
“遺伝子治療送達ビヒクル”は、以下に限らないが、ウイルス性ベクターおよび非-ウイルス性ベクターを含む、上記のような発現ベクターを含む組成物を意味する。
【0047】
“阻害する”は、ベクター-関連抗原に対するおよび/または標的細胞-特異的抗原に対する、直接的または間接的、部分的または完全な、先天性または後天性免疫反応(細胞性(例えば、白血球供給)または体液性の何れか)の阻害および/または低下を意味する。ベクター-関連抗原は、例えば、核酸担体またはエンベロープ(例えば、ウイルス性被覆タンパク質など)から得られる抗原ならびにベクター遺伝子(例えば、細菌性またはウイルス性核酸およびタンパク質)から得られる抗原および/またはベクターに含まれる任意の治療導入遺伝子(例えば、哺乳類核酸および/またはタンパク質)を含む。
【0048】
“特異的免疫性阻害”または “抗原-特異的免疫性阻害”は、抗原-特異的でない一般的な免疫性阻害とは対照的に、ベクター-関連抗原のような抗原に対する免疫反応の阻害を意味する。このため、一例として、他の外来の抗原に対するインビボ免疫適格性の証拠と組み合わせた、ベクター-関連抗原に対する宿主細胞性および/または体液性免疫反応の非存在が、ベクター-関連抗原の特異的免疫性阻害を証明する。
【0049】
“免疫反応”は、好ましくは、細胞性または体液性免疫反応のような後天性免疫反応を意味する。
【0050】
“接触させること”は、ベクターと細胞との間で物理的に接触するような方法および量で細胞に遺伝子治療発現ベクターを投与することを意味する。ベクターが組み換え体ウイルス性粒子ならば、望ましくは、ウイルス性ベクターへの結合およびそれによる細胞の感染はそのような物理的接触により生ずる。ウイルス性ベクターが、非カプセル化ウイルス性核酸または他の核酸のような組み換え体ウイルス性粒子以外ならば、望ましくは、核酸による細胞の感染が生ずる。
【0051】
その “接触させること”は、当業者に既知であり本明細書に記載の任意の手段によって行い得、それによって、ベクターと標的細胞とが明かに接触することまたは相互の接触が生じ得る。所望により、アデノウイルス性ベクターのようなベクターは、更に、二重特異性または多特異性分子(例えば、抗体またはそのフラグメント)と複合体となり得、その場合、 “接触させること”は、ベクターおよび二重特異性または多特異性分子と標的細胞との複合で明かに接触することまたは相互の接触を含む。例えば、ベクターおよび二重特異性(多特異性)分子は、例えば、当業者に既知の化学的手段、または他の手段により、共有結合し得る。好ましくは、ベクターおよび二重特異性(多特異性)分子は、非共有結合的相互作用(例えば、イオン結合、水素結合、ファン・デル・ワールス力、および/または非極性相互作用)により、連結し得る。ベクターおよび二重特異性(多特異性)分子は、小体積の同一溶液を混合することにより接触し得るが、例えば、該複合体が宿主(例えば、ヒト)に投与され、該複合体が血流により標的細胞に移動し、選択的に標的細胞に結合し、標的細胞中に入るような場合、標的細胞および複合体は小体積で接触させる必要はない。ベクターと二重特異性(多特異性)分子とを接触させることは、好ましくは、標的細胞がベクターおよび二重特異性(多特異性)分子の複合体と接触する前に行う。
【0052】
“導入遺伝子”は遺伝子を意味し、それは、導入遺伝子を含む発現ベクターと接触した細胞中で発現し得、その発現は、望ましくは、細胞または組織、器官、器官系、生体または細胞が一部を形成する細胞培養物にとって予防的または治療的利点がある。このため、導入遺伝子は治療遺伝子であり得、例えば、目的の治療遺伝子であり得る。治療遺伝子は、RNAまたはタンパク質のレベルでその効果を発揮するものであり得る。例えば、治療遺伝子によりコードされるタンパク質が遺伝性の疾患の治療、例えば、嚢胞性繊維症の治療における嚢胞性繊維症膜貫通コンダクタンスレギュレーター(cystic fibrosis transmembrane conductance regulator)をコードするcDNAの使用において用いられ得る。
【0053】
更に、治療遺伝子は、例えば、アンチセンスメッセージまたはリボザイム、当分野に既知のようなsiRNA、選択的RNAスプライシングアクセプターまたはドナー、スプライシングまたは3'プロセッシング(例えば、ポリアデニル化) に影響を及ぼすタンパク質、または細胞内での他の遺伝子の発現のレベルに影響を及ぼすタンパク質(すなわち、遺伝子発現が、プロセッシングされたタンパク質の産生により転写の当初からのすべての工程を含むと概ね考えられる場合)をコードすることにより、恐らく、他の場合には、mRNA累積の変化率、mRNA輸送の変化、および/または転写後調節の変化を仲介することにより、RNAのレベルでその効果を発揮し得る。
【0054】
好ましい本発明の態様により、発現ベクターは、所望により、本明細書に記載のCD8ポリペプチドと共に目的の治療分子をコードする1以上の導入遺伝子を含む。本発明により治療され得る疾患には、以下に限らないが、フェニルケトン尿症(フェニルアラニン-L-モノオキシゲナーゼ)、嚢胞性繊維症(嚢胞性繊維症コンダクタンスレギュレーター)、オルニチンカラミルトランスフェラーゼ欠損(OTC)、血友病(XI因子-欠損、VIII因子-欠損)、テイ・サックス病(N-アセチル-ヘキソサミニダーゼ A)および他の脂質蓄積症などのような、よくある遺伝的疾患がある。更に、エリトロポイエチン(EPO)をコードする遺伝子が使用され得る。EPOは、胎児の肝臓および成体の腎臓で産生される糖タンパク質ホルモンであり、それは、骨髄および他の造血組織において前駆細胞で作用し、赤血球細胞の形成を刺激する。ヒトおよび他の哺乳類 EPOをコードする遺伝子は、クローニングされ、配列決定され、発現させ、そしてそれは、種間でのコーディング領域において高い配列相同性がある。Wen et al. (1993) Blood 82:1507-1516. 天然のヒト EPOをコードする遺伝子の配列、およびそれを得るための方法は、例えば、米国特許番号4,954,437および4,703,008に記載されており、それらは、引用により本明細書にすべて含める。EPOを用いる遺伝子治療の方法は、米国特許番号6,610,290に開示されており、それらは、引用により本明細書に特に含める。
【0055】
更にまた、リソソーム酵素酸α-グルコシダーゼ (GAA)をコードするヌクレオチド配列を使用し得る。GAAが機能し、リソソームグリコーゲンのα-1,4およびα-1,6結合を切断し、モノサッカライドを放出する。ヒト GAAをコードする遺伝子の配列、およびそれを得るための方法は以前に記載されており(GenBank 受け入れ番号: M34424 および Y00839; Martiniuk et al. (1990) DNA Cell Biol. 9:85-94; Martiniuk et al. (1986) Proc. Natl. Acad. Sci. USA 83:9641-9644; Hoefsloot et al. (1988) Eur. Mol. Biol. Organ. 7:1697-1704)、これらは、引用により本明細書に特に含める。
【0056】
本明細書で開示の方法および組成物により治療し得る好ましい疾患を以下の表1に開示する。受け入れ番号で提供する配列は引用により本明細書に特に含める。
表 1
【表1】
【0057】
免疫修飾 CD8 分子が、治療導入遺伝子を含みそれを発現するベクターとは別のベクターに含まれる遺伝子によりコードされるならば、CD8 分子を含む該ベクターは、該遺伝子を含みそれを発現するベクターと細胞との接触前、該接触と同時または該接触後に、該細胞と接触し得る。これは、類似または同一の型のベクターを用い、そして、接触(contact effects)のタイミングが該細胞と接触するベクターに対する免疫反応を十分に阻害する場合に限られる。
【0058】
“標的細胞”は、単一の存在として存在し得るか、または細胞のより巨大な集合物の一部であり得る。その “細胞のより巨大な集合物”は、例えば、細胞培養物(混合したものかまたは純粋なものか何れか)、組織(例えば、上皮または他の組織)、器官(例えば、心臓、肺、肝臓、胆嚢、膀胱、目または他の器官)、器官系(例えば、循環系、呼吸系、胃腸系、泌尿系、神経系、外皮系または他の器官系)、または生体(例えば、鳥類、哺乳類、特にヒトなど)を含み得る。好ましくは、標的とする器官/組織/細胞は、循環系(例えば、以下に限らないが、心臓、血管、および血液を含む)、呼吸系(例えば、鼻、咽頭、喉頭、気管、気管支、細気管支、肺など)、胃腸系(例えば、口、咽頭、食道、胃、腸、唾液腺、膵臓、肝臓、胆嚢、および他を含む)、泌尿系(例えば、腎臓、尿管、膀胱、尿道など)、神経系(例えば、以下に限らないが、脳および脊髄、および目のような特殊感覚器を含む)および外皮系(例えば、皮膚)を含み得る。更により好ましくは、該細胞は、心臓、血管、肺、肝臓、胆嚢、膀胱、目の細胞および幹細胞からなる群から選択される。幹細胞を培養し用いる方法は、米国特許番号5,672,346、6,143,292および6,534,052に詳細に開示されており、それは引用により本明細書に含める。
【0059】
幾つかの実施態様では、ウイルス性ベクターまたはプラスミドのような発現ベクターが接触する標的細胞は、接触した標的細胞は発現ベクターにより標的とされ得る特定の細胞-表面結合部位を含むという点で、他の細胞とは異なる。"特定の細胞-表面結合部位"は、細胞の表面上に存在する任意の部位(すなわち、分子または分子の組合せ)を意味し、該ベクター、例えば、アデノウイルス性ベクターは、該細胞と結合し、それにより、該細胞中に入るように相互作用し得る。それ故、特定の細胞-表面結合部位は、細胞-表面レセプターを包含し、好ましくは、それは、タンパク質(修飾タンパク質を含む)、炭水化物、糖タンパク質、プロテオグリカン、脂質、ムチン分子またはムコタンパク質などである。可能性ある細胞-表面結合部位の例には、以下に限らないが、グリコサミノグリカンで見られるヘパリンおよびコンドロイチン硫酸部分; ムチン、糖タンパク質、およびガングリオシドで見られるシアル酸部分; 主要組織適合遺伝子複合体 I(MHC I)糖タンパク質; マンノース、N-アセチル-ガラクトサミン、N-アセチル-グルコサミン、フコース、およびガラクトースを含む膜糖タンパク質中に見られる共通炭水化物分子; ICAM-1、VCAM、E-セレクチン、P-セレクチン、L-セレクチン、およびインテグリン分子のような糖タンパク質; および例えばMUC-1 腫瘍-特異的エピトープのような、癌性細胞に存在する腫瘍-特異的抗原がある。しかし、細胞に対しアデノウイルスのような発現ベクターを標的化することは、細胞性相互作用(すなわち、所定の細胞-表面結合部位との相互作用)の何れかの特定の機構に制限されない。
【0060】
本明細書で使用するように、および更に以下で定義するように、 “ポリヌクレオチド”または “核酸”とは、DNAまたはRNAの何れか、またはデオキシ-およびリボヌクレオチドの両方を含む分子を言い得る。核酸には、ゲノムDNA、cDNA および オリゴヌクレオチドがあり、センスおよびアンチ-センス核酸が含まれる。その核酸はまた、リボース-ホスフェートバックボーンに修飾を含み得、それにより、生理学的環境下におけるその分子の安定性および半減期が増大する。
【0061】
該核酸は、二本鎖、一本鎖であり得るか、または二本鎖または一本鎖配列の両方の部分を含み得る。当業者が認識するように、一本鎖( “Watson”)を表すと、他の鎖( “Crick”)の配列もまた決定される; このため、図2、4および6に示す配列は、配列の相補体をも含む。本明細書の用語 “組み換え核酸”は、エンドヌクレアーゼによる核酸の操作により、一般的に、本来的にインビトロで形成され、通常天然では見られない形の核酸を意味する。このため、直線状の単離核酸、または通常は結合していないDNA 分子をライゲーションすることによりインビトロで形成された発現ベクターは、両方とも、本発明の目的の組み換え体と見なされる。一旦、組み換え核酸を作成し、宿主細胞または生体中に再導入すると、それは、非組み換え的に、すなわち、インビトロではない宿主細胞のインビボ細胞性機構または染色体外操作を用い複製し得ると理解される; しかし、一旦組み換え的に作成されたその核酸もまた、その後に非組み換え的に複製されるけれども、発明の目的の組み換え体と見なされる。
【0062】
用語 “ポリペプチド”および “タンパク質”は、本出願において同義的に使用され、少なくとも2つの共有結合したアミノ酸を意味し得、タンパク質、ポリペプチド、オリゴペプチドおよびペプチドを含む。該タンパク質は、天然に生ずるアミノ酸およびペプチド結合、または合成ペプチド模倣構造で形成され得る。このため、本明細書で使用するように、 “アミノ酸”、または “ペプチド残基”は、天然に生ずるアミノ酸および合成アミノ酸の両方を意味する。例えば、ホモ-フェニルアラニン、シトルリンおよびノルロイシンは、本発明の目的のアミノ酸と見なされる。 “アミノ酸”はまた、プロリンおよびヒドロキシプロリンのようなイミノ酸残基を含む。側鎖は、(R)または(S)配置の何れかであり得る。好ましい実施態様では、アミノ酸は、(S)またはL-配置である。天然に生じない側鎖が使用されるならば、非-アミノ酸置換基が使用され得、例えば、インビボ分解を妨げるか、または遅延させる。導入遺伝子のターゲッティングまたは発現のために変異体タンパク質およびペプチドを作成する天然アミノ酸配列の変化は、例えば、当業者に既知の種々の手段によって行い得る。変異体ペプチドは、実質的に所定のペプチドに相同性であるが、そのペプチドとは異なるアミノ酸配列を有するペプチドである。相同性の程度(すなわち、同一性のパーセント)は、例えば、比較用に最適化されたコンピュータープログラム(例えば、GAPコンピュータープログラム、バージョン6.0またはより高いバージョン、Devereux et al. (Nucleic Acids Res., 12, 387 (1984))により記載、を用い、それは、University of Wisconsin Genetics Computer Group (UWGCG)から自由に入手可能である) を用い配列情報を比較することにより、決定され得る。変異体タンパク質および/またはペプチドの活性は、当業者に既知の他の方法を用い評価し得る。
【0063】
変異体タンパク質(ペプチド)と参照タンパク質(ペプチド)との間で同一でないアミノ酸残基に関して、変異体タンパク質(ペプチド)は、好ましくは、保存アミノ酸置換を含み、すなわち、所定のアミノ酸は、類似の大きさ、荷電密度、疎水性/親水性、および/または配置の他のアミノ酸により置換される(例えば、ValとPhe)。変異体部位-特異的変異は、発現ベクター中に、修飾部位を含む合成オリゴヌクレオチドをライゲーションすることにより、導入され得る。更にまた、オリゴヌクレオチド-指定部位-特異的突然変異誘発手順が使用され得、それは、例えば、Walder et al., Gene, 42:133 (1986); Bauer et al., Gene, 37:73 (1985); Craik, Biotechniques, January 1995, pp. 12-19; および米国特許番号 4,518,584 および 4,737,462に開示のものである。
【0064】
免疫修飾分子
本明細書に照らして、 “免疫修飾分子”は、抗原-特異的形式において標的細胞に対する細胞性および/または体液性宿主免疫反応を修飾するすなわち、増加または減少させるポリペプチド分子であり、好ましくは、宿主免疫反応を減少させるものである。一般的に、本発明の教示により、免疫修飾分子は、標的細胞表面膜と結合し、例えば、本明細書に記載のベクターからの発現後、細胞表面膜に挿入されるかまたはそれに共有結合的または非共有結合的に結合する。
【0065】
好ましい実施態様では、免疫修飾分子は、CD8 タンパク質のすべてまたは機能部分、またより好ましくは、CD8 α鎖のすべてまたは機能部分を含む。ヒト CD8 コーディング配列では、Leahy, Faseb J. 9:17-25 (1995); Leahy et al., Cell 68:1145-62 (1992); Nakayama et al., Immunogenetics 30:393-7 (1989)参照。CD8 タンパク質およびポリペプチドに関する"機能部分"は、本明細書に記載のようにveto活性を保持するCD8 α-鎖の部分、より特にCD8 α-鎖のHLA-結合活性を保持する部分、および特にCD8 α-鎖の細胞外領域中のIg-様ドメインを意味する。典型的な変異体 CD8 ポリペプチドが、Gao and Jakobsen, Immunology Today 21:630-636 (2000)に記載されており、それは引用により本明細書に含める。ある実施態様では、完全長CD8 α-鎖を用いる。しかし、幾つかの実施態様では、細胞質ドメインは欠失している。好ましくは、膜貫通ドメインおよび細胞外ドメインが保持されている。
【0066】
当業者が認識するように、CD8 α-鎖の膜貫通ドメインは、必要ならば、他の分子の膜貫通ドメインと交換され得、細胞外ドメインと標的細胞表面との関係を修飾し得る。この実施態様では、CD8 α-鎖の細胞外ドメインをコードする核酸は、膜貫通ドメインをコードする核酸に作動可能なように連結されている。任意の膜貫通タンパク質の膜貫通ドメインが、本発明に使用し得る。更にまた、膜貫通は、膜貫通タンパク質に見られることが知られていないものでもよい。この実施態様では、 “合成膜貫通ドメイン”は、約20から25までの疎水性アミノ酸を、その後に、少なくとも1つおよび好ましくは2つの荷電アミノ酸を含む。幾つかの実施態様では、CD8 細胞外ドメインは、当分野の通常の技術により、標的細胞膜に連結する。好ましいCD8 α-鎖配列を図 1に開示する。それは、ヒト、マウス、ラット、オランウータン、クモザル、モルモット、ウシ、Hispidコットンラット、飼育ブタおよびネコを含む種由来のアミノ酸配列または完全長CD8 α-鎖をコードする核酸配列の何れかの完全長配列を含む。
【0067】
好ましい実施態様では、CD8 α-鎖は、融合タンパク質ではなく、むしろトランケーションタンパク質であり、ここで、細胞内ドメインは欠失されている。図2に表したように、ヒトCD8 α-鎖遺伝子は、235のアミノ酸のタンパク質を発現する。該タンパク質は、以下のドメインに分けられると考えられ得る(該ポリペプチドのアミノ末端で始まり、カルボキシ末端で終わる): シグナルペプチド(アミノ酸1から21); 免疫グロブリン(Ig)-様ドメイン(およそアミノ酸22-136); 膜隣接ストーク領域(membrane proximal stalk region)(アミノ酸137-181); 膜貫通ドメイン(アミノ酸183-210)および細胞質ドメイン(アミノ酸211-235)。これらの種々のドメインをコードするコーディング配列の該ヌクレオチドは、シグナルペプチドをコードする1-63、細胞外ドメインをコードする64-546、細胞内ドメインをコードする約547-621および細胞内ドメインをコードする約622-708を含む。同様に、マウス配列は、以下のようにドメインに分け得る。該ポリペプチドは、アミノ酸1-27を含むシグナル配列、アミノ酸約28から194を含む細胞外ドメイン、アミノ酸約195-222を含む膜貫通ドメインおよびアミノ酸約223-310を含む細胞内ドメインに分け得る。同様に、これらのドメインをコードするコーディング配列の該ヌクレオチドは、シグナルペプチドをコードする核酸 1-81、細胞外ドメインをコードする約 82-582、膜貫通ドメインをコードする約 583-666および細胞外ドメインをコードする約 667-923を含む。
【0068】
幾つかの実施態様では、該全長タンパク質をコードする核酸は、遺伝子導入ビヒクルに含まれる。他の実施態様では、細胞内ドメインをコードする核酸は、遺伝子導入ビヒクル中のポリヌクレオチドには含まれず、これにより、膜アンカー型タンパク質は、細胞内ドメインを欠くこととなる。対応するドメインがまた、好ましい実施態様においてはマウスを含む他の種において同定され得る。
【0069】
当業者はまた、上記ポリペプチドにかなりの相同性を有する免疫修飾分子は、本発明における有利な使用を見出し得ると認識するだろう。従って、例えば、 “CD8 ポリペプチド”にはまた、図2に示されるヌクレオチドによりコードされるポリペプチドと少なくとも約80%の配列同一性、通常、少なくとも約85%の配列同一性、好ましくは、少なくとも約90%の配列同一性、より好ましくは、少なくとも約95%の配列同一性および最も好ましくは、少なくとも約98%の配列同一性を有する相同性ポリペプチドが含まれる。
【0070】
“CD8をコードする核酸分子”、およびそれと文法的に同等の用語は、図2に示すようなヒト CD8のヌクレオチド配列、ならびに図2に示す配列、および図1に開示するような配列を有するポリペプチドをコードする図2に示すヌクレオチドと少なくとも約80%の配列同一性、通常、少なくとも約85%の配列同一性、好ましくは、少なくとも約90%の配列同一性、より好ましくは、少なくとも約95%の配列同一性および最も好ましくは、少なくとも約98%の配列同一性を有するヌクレオチド配列を意味する。
【0071】
前記のように、多数の種々のプログラムが用いられ、タンパク質または核酸が既知配列と配列同一性または類似性を有するかどうか同定され得る。配列同一性および/または類似性は、以下に限らないが、Smith & Waterman, Adv. Appl. Math. 2:482 (1981)の局所的配列同一性アルゴリズムを含む当分野に既知の標準技術を用い、Needleman & Wunsch, J. Mol. Biol. 48:443 (1970)の配列同一性アライメントアルゴリズムにより、Pearson & Lipman, PNAS USA 85:2444 (1988)の類似性方法用のサーチにより、これらのアルゴリズム(GAP、BESTFIT、FASTA、およびTFASTA、Wisconsin Genetics Software Package, Genetics Computer Group, 575 Science Drive, Madison, WI)のコンピューター化インプリメンテーションにより、Devereux et al., Nucl. Acid Res. 12:387-395 (1984)に記載のBest Fit sequence programにより、好ましくは、セッティングされているディフォルトかまたはインスペクションにより、決定される。好ましくは、同一性の割合は、以下のパラメーターに基づきFastDBにより算出する: ミスマッチペナルティー 1; ギャップペナルティー 1; ギャップサイズペナルティー 0.33; およびジョイニングペナルティー 30、 “Current Methods in Sequence Comparison and Analysis,” Macromolecule Sequencing and Synthesis, Selected Methods and Applications, pp 127-149 (1988), Alan R. Liss, Inc.
【0072】
有用なアルゴリズムの例はPILEUPである。PILEUPは、連続的、対となっているアライメントを用い、関連の配列のグループから複数の配列アライメントを作成する。それはまた、アライメントの作成に用いるクラスタリングの関係を示すツリー(tree)をプロットし得る。PILEUPは、Feng & Doolittle, J. Mol. Evol. 35:351-360 (1987)の単純化した連続アライメント方法を使用する; 該方法は、Higgins & Sharp CABIOS 5:151-153 (1989) に記載のものに類似する。有用なPILEUPパラメーターは、ディフォルトギャップウエイト 3.00、ディフォルトギャップレングスウエイト 0.10、およびウエイテッドエンドギャップを含む。
【0073】
有用なアルゴリズムの他の例は、BLAST アルゴリズムであり、それは、Altschul et al., J. Mol. Biol. 215, 403-410, (1990)およびKarlin et al., PNAS USA 90:5873-5787 (1993)に記載されている。特に有用なBLASTプログラムは、Altschul et al., Methods in Enzymology, 266: 460-480 (1996); http://blast.wustl/edu/blast/ README.htmlから得たWU-BLAST-2 プログラムである。WU-BLAST-2は、幾つかのサーチパラメーター、それらの殆どは、ディフォルト値にセットされている、を用いる。調整可能パラメーターは、以下の値にセットする: オーバーラップスパン =1、オーバーラップフラクション = 0.125、ワードスレショウルド(T) = 11。HSP S および HSP S2 パラメーターは、動力学的な値であり、目的の配列がサーチされる特定の配列の組成および特定のデータベースの組成に依存してプラグラム自体によって決定される; しかし、それらの値は、感度が増大するように調整され得る。
【0074】
更なる有用なアルゴリズムは、Altschul et al. Nucleic Acids Res. 25:3389-3402により報告されているようなギャップ化BLASTである。ギャップ化BLASTは、BLOSUM-62置換スコアを用いる; スレショウルド T パラメーターは9にセットする; 非ギャップ化伸張を生ずるツー-ヒット法; チャージギャップレングス k、コスト 10+k; Xuは16にセットし、そしてXgはデータベースサーチステージ用に40にセットし、アルゴリズムのアウトプットステージ用に67にセットする。ギャップ化アライメントは、〜22 ビットに相当するスコアにより得られる。
【0075】
% アミノ酸または核酸配列同一性値は、マッチングする同一残基の数を、アライメント領域中の"より長い"配列の残基の合計数で割ることにより決定される。 “より長い”配列は、アライメントした領域中に最大数の実際の残基を有するものである(WU-Blast-2により導入され、アライメントスコアが最大化するギャップは無視される)。
【0076】
該アライメントには、アライメントされる配列におけるギャップの導入を含み得る。更に、図11で示されるヌクレオチドによりコードされるポリペプチドのアミノ酸配列よりも多いかまたは少ないか何れかのアミノ酸を含む配列では、ある実施態様では、配列同一性の割合は、アミノ酸の合計数に対する同一アミノ酸の数に基づき決定されることが理解される。このため、例えば、図11のヌクレオチドによりコードされるポリペプチドの配列よりも短い配列の配列同一性は、以下に考察するように、ある実施態様では、より短い配列中のアミノ酸数を用い決定される。同一性の割合の算出において、相対的重さは、挿入、欠失、置換などのような配列変化の種々の表示(manifestation)にわりあてられない。
【0077】
ある実施態様では、同一のみがプラス(+1)としてスコアされ、ギャップを含むすべての型の配列変化は値 “0”となり、ウエイテッドスケールまたは下記のような配列類似性算出用のパラメーターの必要を排除する。配列同一性の割合は、例えば、マッチングする同一残基数を、アライメント領域中の “より短い”配列の合計残基数により割り、100を掛けることにより、算出され得る。 “より長い”配列は、アライメントした領域中に最大数の実際の残基を有するものである。
【0078】
図2のヌクレオチドによりコードされるポリペプチドと100%未満の配列同一性を有するCD8は、一般的に、ヒト以外の種由来の天然CD8 ヌクレオチド配列、およびヒトまたは非-ヒト源由来の天然CD8 ヌクレオチド配列の変異体から作成される。これに関して、多くの技術が当分野に既知であり、それらは日常的に用いられ、天然CD8 配列のヌクレオチド配列変異体が作成され、天然CD8 ポリペプチドと通常関連する少なくとも1つの活性の存在に関してそれらの変異体のポリペプチド産物をアッセイし得る。好ましい実施態様では、CD8 α-鎖はヒト由来であるが、図1に示すように、ラット、マウスおよび霊長類由来のCD8 α-鎖が知られており、本発明における使用が見出される。
【0079】
CD8 活性を有するポリペプチドは、図2で示したヌクレオチドによりコードされるポリペプチドよりも短いかまたは長くてもよい。このため、好ましい実施態様では、CD8 ポリペプチドの定義に含まれるものは、図2のヌクレオチドによりコードされるポリペプチドの部分またはフラグメントである。本明細書の1つの実施態様でおいて、図2のヌクレオチドによりコードされるポリペプチドのフラグメントは、a)それらが、少なくとも、示した配列同一性を有し、そしてb)好ましくは、上記のように天然に生ずるCD8の生物学的活性を有するならば、CD8 ポリペプチドであると見なされる。
【0080】
更に、以下により十分に概略するように、CD8 α-鎖は、図2のヌクレオチドによりコードされるポリペプチドよりも長くつくられ得る; 例えば、他の融合配列の付加、または更なるコーディングおよび非-コーディング配列の解明によってつくられ得る。
【0081】
CD8 ポリペプチドは、好ましくは、組み換え体である。 “組み換えポリペプチド”は、組み換え技術を用い、すなわち、下記のような組み換え核酸の発現により、作成されたポリペプチドである。好ましい実施態様では、本発明のCD8は、図2に示す核酸配列、またはそのフラグメントの発現により作成される。組み換えポリペプチドは、少なくとも1以上の特性により天然に生ずるタンパク質とは区別される。例えば、ポリペプチドは、通常、その野生型宿主と関連する幾つかまたはすべてのタンパク質および化合物から単離されるかまたは精製され得、このため、実質的に精製され得る。例えば、単離ポリペプチドは、通常、その天然状態と関連する少なくとも幾つかの物質を伴っておらず、好ましくは、所定のサンプル中の合計タンパク質の少なくとも約 0.5重量%、より好ましくは、少なくとも約 5重量%を構成する。実質的に精製したポリペプチドは、合計ポリペプチドの少なくとも約 75重量%を構成し、少なくとも約 80重量%が好ましく、そして少なくとも約 90重量%が特に好ましい。その定義は、種々の生体または宿主細胞中、1つの生体からのCD8 ポリペプチドの産生を含む。
【0082】
更にまた、ポリペプチドは、誘導性プロモーターまたは高発現プロモーターの使用により、通常見られる濃度よりも顕著により高い濃度で作成され得、それによって、該ポリペプチドは高濃度のレベルで作成される。更にまた、ポリペプチドは、以下に考察するような、アミノ酸置換、挿入および欠失に加えて、天然では通常見られない形態であり得る。
【0083】
1つの実施態様では、本発明は、核酸 CD8 変異体を提供する。これらの変異体は、3つのクラスのうち1以上の中に入る: 置換、挿入または欠失変異体。これらの変異体は、通常、カセットまたはPCR 変異誘発または当分野に既知の他の技術を用い、図2のヌクレオチド中のヌクレオチドの部位特異的変異誘発により作成し、該変異体をコードするDNAを作成し、遺伝子治療ベクター中に該変異体を含ませ、その後、そのDNAを発現させる。アミノ酸配列変異体は、事前に決定した変異の性質、CD8 アミノ酸配列の天然に生ずる対立遺伝子または異種間変異とは異なるものとする特徴により特性解析される。変異体は、典型的に、天然に生ずる類似体と同質的な生物学的活性を示すが、変異体はまた、以下により十分に概略するように、修飾された特性を有するように選択され得る。
【0084】
配列変異を導入するための部位または領域は事前に決定するが、変異自体は事前に決定する必要はない。例えば、所定の部位での変異のパフォーマンスを最適化するために、ランダムの変異誘発は、標的コドンまたは領域で行われ得、発現した変異体は、最適化された所望の活性についてスクリーニングされる。既知配列を有するDNA中の事前に決定した部位での置換変異を作るための技術は既知であり、例えば、M13 プライマー変異誘発およびPCR 変異誘発である。変異体を作るための技術の他の例は、遺伝子シャッフリングの方法であり、これにより、ヌクレオチド配列の類似変異体のフラグメントは組み換えられ、新規変異体の組合せを作成することができる。その技術の例は、米国特許番号 5,605,703; 5,811,238; 5,873,458; 5,830,696; 5,939,250; 5,763,239; 5,965,408; および 5,945,325に見られ、それらは各々、引用により本明細書にすべて含める。
【0085】
アミノ酸置換は、典型的に単一残基である; 挿入は、通常、約 1から20 アミノ酸のオーダーであるが、相当に大きな挿入物が許容され得る。欠失は、約 1から約 20残基の範囲であるが、幾つかの場合、欠失は、かなり大きくなり得、細胞質ドメインまたはそのフラグメントを含み得る。
【0086】
置換、欠失、挿入またはそれらの任意の組合せを用い、最終の誘導体に到達し得る。一般的に、これらの変化は、数アミノ酸で行われ、分子の変化を最小化する。しかし、より大きな変化が、特定の状況下で許容され得る。CD8の特性において変化が小さいほうが望ましいとき、置換は、一般的に、以下のチャートにより作成される:
チャート1
もとの残基 例示的な置換
【表2】
【0087】
機能または免疫学的同一性における実質的な変化は、チャート1に示す置換ほどにはあまり保存されていない置換を選択することにより生ずる。例えば、置換は、より顕著な影響を与えるように作成され得る: 変化領域におけるポリペプチドバックボーンの構造、例えば、α-ヘリックスまたはβ-シート構造; 標的部位での分子の荷電または疎水性; または側鎖の体積。一般的に、ポリペプチドの特性に最も大きな変化を生ずると予測される該置換は、(a)親水性残基、例えば、セリル(seryl)またはスレオニルが、疎水性残基、例えば、ロイシル、イソロイシル、フェニルアラニル、バリルまたはアラニルに置換される; (b) システインまたはプロリンが、任意の他の残基に置換される; (c) 正に荷電した側鎖を有する残基、例えば、リシル、アルギニル、またはヒスチジル、負に荷電した残基、例えば、グルタミルまたはアスパルチルに置換される; または(d) 体積の大きい(bulky)側鎖を有する残基、例えば、フェニルアラニンは、側鎖を有しないもの、例えば、グリシンに置換される場合である。
【0088】
該変異体は、典型的に、同質の生物学的活性を示し、天然に生ずる類似体と同じ免疫反応を顕在化させるが、変異体はまた、所望によりCD8の特性を修飾するように選択される。更にまた、該変異体は、タンパク質の生物学的活性が変わるように設計され得る。
【0089】
本発明内に含まれるポリペプチドの1つのタイプの共有結合的修飾は、ポリペプチドの天然のグリコシル化パターンを変えることを含む。 “天然のグリコシル化パターンを変えること”は、本明細書の目的を意図し、天然配列のCD8 ポリペプチドに見られる1以上の炭水化物部分を欠失させること、および/または天然配列のポリペプチドには存在しない1以上のグリコシル化部位を加えることを意味する。
【0090】
ポリペプチドへのグリコシル化部位の付加は、そのアミノ酸配列を変えることにより達し得る。その変化は、例えば、1以上のセリンまたはトレオニン残基の付加、またはそれらによる天然配列のポリペプチドへの置換により為し得る(O-結合型グリコシル化部位の場合)。該アミノ酸配列は、所望により、DNAレベルでの変化により変え得る。特に、事前に選択した塩基においてポリペプチドをコードするDNAを変異させることにより、そのコドンは、望ましいアミノ酸に翻訳されるように生ずる。
【0091】
ポリペプチドに存在する炭水化物部分の除去は、グリコシル化のための標的としての役割をするアミノ酸残基をコードするコドンの変異置換により達し得る。
【0092】
一旦、天然の源から単離され、例えば、プラスミドまたは他のベクター内に含まれ、またはそれらから直鎖状核酸セグメントとして切除されると、組み換え核酸は、更に、プローブとして使用され得、他の核酸を同定および単離し得る。それはまた、 “前駆体”核酸として使用し得、修飾または変異核酸およびタンパク質を作り得る。それはまた、本明細書に記載のように、標的細胞を処置するためにベクターまたは他の送達ビヒクル中に組み込まれ得る。
【0093】
遺伝子治療発現ベクター
本発明に照らして、任意の適当な遺伝子治療発現ベクターが使用され得る。 “ベクター”は、その用語が、当業者に理解されている通り、遺伝子転移のためのビヒクルである。本発明による該ベクターは、以下に限らないが、プラスミド、ファージ、ウイルス、リポソームなどを含む。本発明による発現ベクターは、好ましくは、更なる配列および変異を含む。特に、本発明による発現ベクターは、本明細書に定義のように、免疫修飾分子、特にCD8 α-鎖をコードする導入遺伝子を含む核酸を含み、所望により、目的の治療分子をコードする少なくとも1つの更なる導入遺伝子を更に含む。該核酸は、全体的または部分的に合成的に作成されたコーディングまたは他の遺伝的配列またはゲノムまたは相補DNA(cDNA)配列を含み得、そして、DNAまたはRNAの何れかの形態で提供され得る。
【0094】
導入遺伝子および/または免疫修飾および/または治療分子をコードする遺伝子は、mRNAの発現およびタンパク質の産生のため、および他の生化学的特性の評価のため、ウイルス性ベクターからバキュロウイルスまたは適当な原核性または真核性発現ベクター中へ移動し得る。
【0095】
本発明によるベクター(組み換えアデノウイルス性ベクターおよび転移ベクターを含む)の作成に関して、そのベクターは、当業者に既知のように、標準的な分子的および遺伝的技術を用い構成され得る。ビリオンまたはウイルス性粒子を含むベクター(例えば、組み換えアデノウイルス性ベクター)は、適当な細胞系においてウイルス性ベクターを用い作成され得る。同様に、1以上のキメラコートタンパク質を含む粒子が、標準的な細胞系、例えば、目下、アデノウイルス性ベクターに使用されるもので作成され得る。次いで、これらの得られた粒子は、望ましいならば、特異的細胞に標的化され得る。
【0096】
任意の適当な適当な発現ベクター(例えば、Pouwels et al., Cloning Vectors: A Laboratory Manual (Elsevior, N.Y.: 1985)に記載のような)および対応する適当な宿主細胞は、宿主細胞中における組み換えペプチドまたはタンパク質の作成に用い得る。発現宿主は、以下に限らないが、エシェリキア(Escherichia)、バチラス(Bacillus)、シュードモナス(Pseudomonas)、サルモネラ(Salmonella)属内の細菌性種、哺乳類またはバキュロウイルス性システム(例えば、Luckow et al., Bio/Technology, 6, 47 (1988)に記載のような)を含む昆虫宿主細胞システム、およびCOS-7、C127、3T3、CHO、HeLa、BHKなどのような樹立細胞系を含む。本発明によるキメラタンパク質(ペプチド)を調製するための特に好ましい発現システムは、バキュロウイルス性発現システムであり、それは、Trichoplusia ni、Tn 5B1-4 昆虫細胞、または他の適当な昆虫細胞を用い、高レベルの組み換えタンパク質を産生する。当業者はもちろん、発現宿主の選択は、産生するペプチドのタイプに影響することを知っている。例えば、酵母または哺乳類細胞(例えば、COS-7 細胞)内で産生されたペプチドのグリコシル化は、大腸菌のような細菌性細胞内で産生されたペプチドのそれとは異なる。
【0097】
好ましい実施態様では、タンパク質は、哺乳類細胞内で発現する。哺乳類発現システムはまた、当分野で既知であり、レトロウイルス性システムを含む。哺乳類プロモーターは、哺乳類 RNA ポリメラーゼと結合し、あるタンパク質のコーディング配列の、mRNAへの下流域(3')転写を開始することができる任意のDNA 配列である。プロモーターは、転写開始領域を有し、それは、通常、コーディング配列の5’末端、およびTATAボックス(転写開始部位の上流に位置づけられた25-30塩基対を用いる)に隣接して位置する。該TATAボックスは、正しい部位でRNA 合成を開始することをRNA ポリメラーゼ IIに指示すると考えられている。哺乳類プロモーターはまた、上流プロモーターエレメント(エンハンサーエレメント)を含み、典型的に、TATAボックスの上流の100から200塩基対内に位置する。転写が開始され、何れかの方向に機能し得る、上流プロモーターエレメントが速度を決定する。哺乳類プロモーターとして使用するのに特に好ましいのは、哺乳類ウイルス性遺伝子由来のプロモーターである。そのウイルス性遺伝子はしばしば非常に高発現され、宿主範囲が広範だからである。例には、SV40 初期プロモーター、マウス乳腺腫瘍ウイルス LTR プロモーター、アデノウイルス主要後期プロモーター、単純ヘルペスウイルスプロモーター、およびCMV プロモーターがある。
【0098】
典型的に、哺乳類細胞により認識される転写終結およびポリアデニル化配列は、翻訳ストップコドンに対し3'側に位置する調節領域であり、このため、プロモーターエレメントと共に、コーディング配列に隣接する。成熟mRNAの3'末端は、部位-特異的な翻訳後開裂およびポリアデニル化により形成される。転写ターミネーターおよびポリアデニル化シグナルの例には、SV40由来のものがある。
【0099】
哺乳類宿主に、および他の宿主に外因性核酸を導入する方法は、当業者に既知であり、用いる宿主細胞により変化する。技術には、デキストラン-仲介トランスフェクション、リン酸カルシウム沈殿、ポリブレン仲介トランスフェクション、プロトプラスト融合、エレクトロポレーション、ウイルス性感染、リポソーム中へのポリヌクレオチドのカプセル化、および核中へのDNAの直接のマイクロインジェクションがある。
【0100】
タンパク質はまた、当分野に既知の技術を用い、融合タンパク質として作成され得る。このため、例えば、該タンパク質は、発現を増加するか、他の理由のために融合タンパク質として作成され得る。例えば、タンパク質がペプチドであるとき、該ペプチドをコードする核酸は、発現目的のために、他の核酸と連結させ得る。
【0101】
CD8の試験のために、タンパク質は、発現後に精製するかまたは単離する。タンパク質は、サンプル中に存在する他の成分に応じて、当業者に既知の種々の方法で単離し得るかまたは精製し得る。標準的な精製方法には、電気泳動的、分子的、免疫学的およびクロマトグラフィー的な技術があり、イオン交換、疎水性、親和性、および逆相HPLCクロマトグラフィー、およびクロマト分画が含まれる。例えば、CD8 タンパク質は、標準的な抗-CD8 抗体カラムを用い精製し得る。タンパク質濃度と関連する限外濾過およびダイアフィルトレーション技術もまた有用である。適当な精製技術の一般的な手引きとして、Scopes, R., Protein Purification, Springer-Verlag, NY (1982) 参照。必要な純度は、CD8 タンパク質の用途に応じて変化する。幾つかの場合には、精製しないことが必要である。幾つかの場合には、CD8 発現は、抗体結合および蛍光による検出かまたは蛍光活性化細胞選別装置(FACS)により、例えば、細胞表面で検出される。
【0102】
CD8をコードする核酸分子およびCD8 核酸分子のコーディングまたは非-コーディング鎖の何れかから得られる任意の核酸分子は、当分野に既知であり通常よく用いられる種々の方法で標的の細胞と接触され得、ここで、接触させることは、エキソビボまたはインビボであり得る。
【0103】
ウイルス性結合、侵入および遺伝子発現は、所望のタンパク質を発現する組み換えウイルスまたはRNAおよびβ-ガラクトシダーゼのようなマーカー遺伝子を生ずる目的の挿入物を含むアデノウイルス性ベクターを用いることにより最初に評価され得る。β-ガラクトシダーゼ遺伝子(Ad-LacZ)を含むアデノウイルスで感染された細胞におけるβ-ガラクトシダーゼ発現は、細胞にAd-Glucを加えた後、二時間ぐらいの短時間で検出され得る。この手順は、組み換えウイルスの細胞侵入および遺伝子発現の迅速で効果的な分析を提供し、通常の技術を用いる当業者により容易に実行される。
【0104】
タンパク質をコードする本発明の核酸を用いると、種々の発現ベクターが作成され得る。該発現ベクターは、自己複製する染色体外ベクターであるかまたは宿主ゲノム中に組み込まれるベクターの何れかであり得る。一般的に、これらの発現ベクターは、該タンパク質をコードする核酸に作動可能なように連結した転写性および翻訳性調節核酸を含む。用語 “コントロール配列”は、特定の宿主生体中において作動可能なように連結したコーディング配列の発現に必要なDNA 配列をいう。原核生物に適当なコントロール配列には、例えば、プロモーター、所望により、オペレーター配列、およびリボゾーム結合部位がある。真核生物細胞では、プロモーター、ポリアデニル化シグナル、およびエンハンサーを利用することが知られる。
【0105】
核酸は、他の核酸配列との機能的関係に置かれるときに、"作動可能なように連結"される。例えば、プレ配列(presequence)または分泌リーダー(secretory leader)のDNAは、ポリペプチドがポリペプチドの分泌に関係するプレタンパク質(preprotein)として発現するならば、ポリペプチドのDNAに作動可能なように連結されている; プロモーターまたはエンハンサーは、配列の転写に影響するならば、コーディング配列に作動可能なように連結されている; またはリボソーム結合部位は、翻訳を促進するように位置するならば、コーディング配列に作動可能なように連結されている。他の例として、作動可能なように連結とは、連続するように、および分泌リーダーの場合には、連続しかつ読み取り枠(reading phase)内にあるように連結したDNA 配列をいう。しかし、エンハンサーは、連続している必要はない。連結は、通常の制限部位での連結により達成される。そのような部位が存在しないならば、合成オリゴヌクレオチドアダプターまたはリンカーは、通常の実施により使用する。転写性および翻訳性調節核酸は、一般的に、CD8の発現に使用する宿主細胞に適当である;例えば、ヒト転写性および翻訳性調節核酸配列は、好ましくは、ヒト細胞におけるCD8の発現に使用する。多くのタイプの適当な発現ベクター、および適当な調節配列が、種々の宿主細胞用として当分野に既知である。
【0106】
一般的に、転写性および翻訳性調節配列には、以下に限らないが、プロモーター配列、リボゾーム結合部位、転写開始および停止配列、翻訳開始および停止配列、およびエンハンサーまたはアクチベーター配列があり得る。好ましい実施態様では、調節配列には、プロモーターおよび転写開始および停止配列がある。
【0107】
プロモーター配列は、構成性または誘導性プロモーターの何れかをコードする。プロモーターは、天然に生ずるプロモーターまたはハイブリッドプロモーターの何れかであり得る。1を超えるプロモーターのエレメントを組み合わせるハイブリッドプロモーターがまた、当分野に既知であり、本発明に有用である。
【0108】
加えて、発現ベクターは、更なるエレメントを含み得る。例えば、発現ベクターは、2つの複製システムを有し得、このため、2つの生体で、例えば、発現用に哺乳類または昆虫細胞で、およびクローニングおよび増幅用に原核生物宿主で保持され得る。更に、発現ベクターを組み込むため、該発現ベクターは、宿主細胞ゲノムに相同な少なくとも1つの配列、および好ましくは、発現コンストラクトに隣接する2つの相同配列を含む。該組み込みベクターは、ベクターに包含させるための適当な相同性配列を選択することにより宿主細胞中の特定の座に向けられ得る。ベクターを組み込むためのコンストラクトは当分野に既知である。
【0109】
更なる実施態様では、発現ベクターは、形質転換した宿主細胞を選択し得る選択マーカー遺伝子含み得る。選択遺伝子は、当分野に既知であり、使用する宿主細胞により変化する。
【0110】
好ましくは、該ベクターは、とりわけ、アデノウイルス性ベクター、アデノ-随伴ウイルス性ベクター、ヘルペスベクターまたはレトロウイルス性ベクターのようなウイルス性ベクターである。最も好ましくは、ウイルス性ベクターは、アデノウイルス性ベクターである。アデノウイルス性ベクターは、任意のアデノウイルスから得られ得る。"アデノウイルス"は、アデノウイルス科ファミリーの任意のウイルスであり、望ましくは、マストアデノウイルス(例えば、哺乳類アデノウイルス)またはアビアデノウイルス(例えば、エビアンアデノウイルス)属である。アデノウイルスは、任意の血清型である。アデノウイルスの源として用い得るアデノウイルス性ストックは、アデノウイルス性血清型 1 ないし 47から増幅し得、それは、現在、American Type Culture Collection (ATCC, Rockville, Md.)から入手可能であり、または任意の他の血清型のアデノウイルスから増幅し得、それは、任意の他の源から入手可能である。例えば、アデノウイルスは、サブグループ A (例えば、血清型 12、18、および31)、サブグループ B (例えば、血清型 3、7、11、14、16、21、34、および35)、サブグループ C (例えば、血清型 1、2、5、および6)、サブグループ D (例えば、血清型 8、9、10、13、15、17、19、20、22-30、32、33、36-39、および42-47)、サブグループ E (血清型 4)、サブグループ F (血清型 40 および 41)、または任意の他のアデノウイルス性血清型であり得る。しかしながら、好ましくは、アデノウイルスは、血清型 2、5 または 9である。望ましくは、アデノウイルスは、同一血清型の被覆タンパク質(例えば、ペントンベース、ヘキソン、および/または ファイバー)を含む。しかしながら、また好ましくは、例えば、すべてまたは一部の所定の被覆タンパク質が他の血清型由来であり得るという意味で1以上の被覆タンパク質は、キメラであり得る。
【0111】
好ましくは、アデノウイルス性ベクターであるウイルス性ベクターは、複製-能があり得るが、好ましくは、ウイルス性ベクターは、複製-欠損であるか、または条件付きで複製-欠損である。例えば、好ましくは、アデノウイルス性ベクターであるウイルス性ベクターは、ウイルス複製-欠損にする、少なくとも1つの修飾を有するゲノムを含む。ウイルス性ゲノムに対する修飾には、以下に限らないが、DNA セグメントの欠失、DNA セグメントの付加、DNA セグメントの再配列、DNA セグメントの置換、またはDNA 損傷の導入がある。DNA セグメントは、1ヌクレオチドぐらいに小さくてもよいし、または36 キロベース対、すなわち、およそアデノウイルス性ゲノムの大きさ、または38 キロベース対、それは、アデノウイルス性ビリオン中にパッケージされ得る最大の大きさ、ぐらいに大きくてもよい。
【0112】
ウイルス性、特に、アデノウイルス性、ゲノムに対する好ましい修飾には、ウイルス複製-欠損にする修飾に加えて、本明細書に定義の免疫修飾分子をコードする導入遺伝子、および更にはおよび好ましくは、目的の治療分子をコードする少なくとも1つの導入遺伝子の挿入がある。アデノウイルスのようなウイルスはまた、好ましくは、共組み込み(cointegrate)され得、すなわち、アデノウイルス性ゲノム配列のようなウイルスと、プラスミド、ファージまたは他のウイルスのもののような他の配列とのライゲーションである。
【0113】
アデノウイルス性ベクター(特に複製-欠損アデノウイルス性ベクター)に関して、そのようなベクターは、中実キャプシド(complete capsid)(すなわち、アデノウイルス性ゲノムのようなウイルス性ゲノムを含む)または中空キャプシド(empty capsid)(すなわち、そのウイルス性ゲノムは、欠いているか、例えば、物理的または化学的手段により破壊されている)の何れかのものを含み得る。好ましくは、ウイルス性ベクターは、中実キャプシド、すなわち、免疫修飾分子をコードする導入遺伝子および所望により好ましくは、阻害する手段をコードする少なくとも1つの導入遺伝子を運ぶ手段として含む。更にまた、好ましくは、導入遺伝子は、アデノウイルス性キャプシドの外側のセルに運ばれ得る。
【0114】
アデノウイルスのようなウイルスを特定の細胞に標的とさせるのに好ましいかまたは望ましい程度に、該ウイルスは、プラスミド DNAの、細胞中への転移において本質的にエンドソモリティック剤(endosomolytic agent)として用いられ得、それは、マーカー遺伝子を含み、トランスフェリンのような細胞-結合リガンドと共有結合したポリリジンと複合となりそれにより凝縮する(Cotten et al., PNAS (USA), 89, 6094-6098 (1992); および Curiel et al., PNAS (USA), 88, 8850-8854 (1991))。トランスフェリン-ポリリジン/DNA複合体およびアデノウイルス(例えば、トランスグルタミナーゼを伴う、アデノウイルスに対する抗体の手段によるか、またはビオチン/ストレプトアビジン結合(bridge)による)のカップリングは、実質的に、遺伝子転移を促進することが示されている(Wagner et al., PNAS (USA), 89, 6099-6103 (1992))。
【0115】
更にまた、アデノウイルス性ファイバーのような1以上のウイルス性被覆タンパク質は、例えば、細胞-表面レセプターに対するリガンド用の配列または二重特異性抗体(すなわち、ファイバーに対し特異性を有する一方の端、細胞-表面レセプターに対し特異性を有する他方の端を有する分子)に結合し得る配列の何れかの組み込みにより修飾され得る(PCT国際特許出願番号WO 95/26412('412 出願)およびWatkins et al., "Targeting Adenovirus-Mediated Gene Delivery with Recombinant Antibodies," Abst. No. 336)。両方の場合において、典型的なファイバー/細胞-表面レセプター相互作用が抑制され、アデノウイルスのようなウイルスは、そのファイバーを用いて新規細胞-表面レセプターに再度向けられる(redirect)。
【0116】
更にまた、選択した細胞タイプに特異的に結合できる標的化エレメントは、高親和性結合対の第一の分子に結合し得、宿主細胞に投与され得る(PCT国際特許出願番号WO 95/31566)。次いで、高親和性結合対の第二の分子に結合した遺伝子導入ビヒクルが、宿主細胞に投与され得、ここで、その第二の分子は、第一の分子に特異的に結合でき、そのため、遺伝子導入ビヒクルは、選択した細胞タイプを標的とする。
【0117】
同じ方針で、いくつかの方法(例えば、エレクトロポレーション、形質転換、三親性接合(triparental mating)の接合、(共-)トランスフェクション、(共-)感染、膜融合、微粒子銃(microprojectiles)の使用、リン酸カルシウム-DNA沈殿を伴うインキュベーション、ダイレクトのマイクロインジェクションなど)がウイルス、プラスミド、および核酸配列形態(すなわち、RNA または DNA)のファージの転移に利用可能であるため、ベクターは、同様に、キャプシドタンパク質のような任意の関連タンパク質の非存在下で、および任意のエンベロープ脂質の非存在下でRNA または DNAを含み得る。
【0118】
同様に、リポソームが、細胞膜との融合により細胞侵入を行うため、ベクターは、被覆タンパク質をコードする構成核酸を有するリポソームを含み得る。そのリポソームは、市場で入手可能であり、例えば、Life Technologies, Bethesda, Md.から入手でき、製造者の推奨に従い使用できる。更に、リポソームを使用し、遺伝子導入でき、転移能が増大しおよび/またはインビボで毒性が減少したリポソームが、使用し得る。可溶性キメラ被覆タンパク質(本明細書に記載の方法を用い作成したとき)は、製造者の指示に従い該リポソームを調製した後か、または該リポソームの調製中か何れかにおいて、リポソームに添加され得る。
【0119】
本発明によるベクターは、本発明の方法に用い得るものに制限されないが、遺伝子転移ベクターの構成において用い得る中間体-型ベクター(例えば、"転移ベクター")をも含む。
【0120】
1以上の核酸配列のインビボ送達のための好ましい方法の1つは、アデノウイルス発現ベクターの使用を含む。"アデノウイルス発現ベクター"は、(a)コンストラクトのパッケージングをサポートするのに、および(b)センスまたはアンチセンス方向で本発明でクローニングされたポリヌクレオチドを発現するのに十分なアデノウイルス配列を含むコンストラクトを含むことを意味する。もちろん、アンチセンスコンストラクトとの関連で、発現は、遺伝子産物が合成されることを必要としない。
【0121】
該発現ベクターは、遺伝子工学型のアデノウイルスを含む。アデノウイルスの遺伝的構成、36 kb、直鎖状、二本鎖 DNA ウイルスという知識により、アデノウイルス性 DNAの大きな断片が7 kbまでの外来配列と置換できる(Grunhaus and Horwitz, 1992)。レトロウイルスとは対照的に、宿主細胞のアデノウイルス性感染は、染色体への組み込みを生じない。アデノウイルス性 DNAが、エピソームの手法(episomal manner)で、遺伝毒性の可能性もなしに複製し得るためである。また、アデノウイルスは構造的に安定であり、ゲノム再配列は、広範囲な増幅後、検出されていない。アデノウイルスは、細胞周期の段階にかかわらず、すべての上皮細胞に実質的に感染し得る。今のところ、アデノウイルス性感染は、ヒトの急性呼吸器疾患のような軽い疾患にのみ関連していると思われる。
【0122】
アデノウイルスは、その中程度の大きさのゲノム、操作の容易さ、高力価、広範の標的-細胞範囲および高い感染性のため、遺伝子転移ベクターとしての使用に特に適当である。ウイルス性ゲノムの両端は、100-200 塩基対の逆方向反復(ITR)を含み、それらは、ウイルス性 DNA 複製およびパッケージングに必要なシスエレメントである。ゲノムの初期(E)および後期(L)領域は、ウイルス性 DNA 複製の開始により分けられる種々の転写単位を含む。E1 領域 (E1A および E1B)は、ウイルス性ゲノムおよび幾つかの細胞性遺伝子の転写の調節に関与するタンパク質コードする。E2 領域 (E2A および E2B)の発現は、ウイルス性 DNA 複製用のタンパク質の合成を生ずる。これらのタンパク質は、DNA 複製、後期遺伝子発現および宿主細胞停止(shut-off)に関与する(Renan, 1990)。多数のウイルス性キャプシドタンパク質を含む後期遺伝子の産物は、主要後期プロモーター(major late promoter)(MLP)により生ずる単一の一次転写産物の重要なプロセッシング後のみ発現する。該MLP(16.8 m.u.に位置する)は、特に、感染の後期フェーズの間に重要であり、このプロモーターから生ずるすべてのmRNAは、5'-三分裂リーダー(tripartite leader)(TPL)配列を有し、その配列により、翻訳に好ましいmRNAとなる。
【0123】
現在の系では、組み換えアデノウイルスは、シャトルベクターとプロウイルスベクターとの間の相同組み換えから生ずる。2つのプロウイルスベクター間での可能な組み換えのため、野生型アデノウイルスは、このプロセスから生じ得る。そのため、各プラークからウイルスの単一クローンを単離し、そのゲノム構造を調査することが重要である。
【0124】
複製欠損のアデノウイルスベクターの作成および増幅は、特有のヘルパー細胞系に依存する。天然では、アデノウイルスは、野生型ゲノムのおよそ105%をパッケージし得(Ghosh-Choudhury et al., 1987)、約 2 kB余のDNAを収容する。E1 および E3 領域で置換可能であるおよそ 5.5 kBのDNAと組み合わせると、現在のアデノウイルスベクターの最大収容は、7.5 kB未満、または該ベクターの全長の約 15%未満である。80%を超えるアデノウイルスウイルス性ゲノムが、ベクターバックボーン内にあり、ベクターから生ずる(vector-borne)細胞毒性の源である。また、E1-欠失ウイルスの複製欠損は不完全である。例えば、ウイルス性遺伝子発現の漏れ(leakage)が、高い感染効率(MOI)の現在利用可能なベクターで観察されている(Mulligan, 1993)。
【0125】
ヘルパー細胞系は、ヒト胎児腎臓細胞、筋細胞、造血細胞または他のヒト胎児間葉もしくは上皮細胞のようなヒト細胞から得られ得る。更にまた、ヘルパー細胞は、ヒトアデノウイルスにとって許容状態にある他の哺乳類種の細胞から得られ得る。その細胞には、例えば、ベロ細胞または他のサル胎児間葉もしくは上皮細胞がある。上記のように、現在、好ましいヘルパー細胞系は293である。
【0126】
最近、Racher et al. (1995)は、293 細胞を培養し、アデノウイルスを増殖させるための改善した方法を開示した。1つの形態では、天然の細胞凝集物は、100-200 mlの培地を含む1 リットルのシリコナイズしたスピナーフラスコ(Techne, Cambridge, UK)に各細胞を接種することにより培養する。40 rpmで撹拌後、細胞バイアビリティーを、トリパンブルーで評価する。他の形態では、Fibra-Cel マイクロキャリア (Bibby Sterlin, Stone, UK) (5 g/l)は以下のように用いる。5 ml の培地に再懸濁した細胞接種材料を、250 ml Erlenmeyer flask中の該キャリア(50 ml)に加え、時折撹拌(agitation)しつつ1から4時間静置させる。次いで、該培地は、50 ml の新しい培地と取り換え、撹拌を開始する。ウイルス産物の場合、細胞は、約 80% コンフルエンスにまで増殖させ得、その時間後、その培地は取り換え(最終体積の25%に)、アデノウイルスをMOI 0.05で加える。培養物は一夜静置し、その後、その体積は、100%に増加し、撹拌を更に72時間行う。
【0127】
好ましい実施態様では、アデノウイルスは、当分野に既知のように “弱い”アデノウイルスである。その"弱い"アデノウイルスベクターは、アデノウイルス性遺伝子導入のために最近開発されたシステムである。アデノウイルスの複製には、ヘルパーウイルス、およびE1a および Creの両方を発現する特別のヒト 293 細胞系が必要であり、天然環境下にはない条件である。今日最も効果的なシステムでは、E1-欠損ヘルパーウイルスを、バクテリオファージ P1 loxP 部位 ("loxPが導入された")が隣接するパッケージングシグナルと共に用いる。loxPが導入されたパッケージングシグナルを有するヘルパーウイルスと共に該弱いウイルスでCre リコンビナーゼを発現するヘルパー細胞の感染は、パッケージングシグナルをヘルパーウイルスのDNAから欠損させるため、弱い rAVを生ずるのみである。しかし、293-に基づくヘルパー細胞を用いるならば、そのヘルパーウイルス DNAは、ヘルパー細胞 DNA中に組み込まれるAd5 DNAと組み換えられ得る。結果として、野生型パッケージングシグナル、およびE1 領域は回復する。このため、また、293-(または911-)に基づくヘルパー細胞における弱いrAVの産物は、E1-欠損ヘルパーウイルスを用いるならば、RCAを生じ得る。
【0128】
ベクターは、すべてのウイルス性遺伝子が取り除かれる。このため、ベクターは、非-免疫原性であり、必要であれは、繰り返し使用し得る。その"弱い"アデノウイルスベクターはまた、導入遺伝子を収容するための36 kb のスペースを含み、このため、細胞中への大多数の遺伝子の共-送達をし得る。RGD モチーフのような特定配列モチーフは、アデノウイルスベクターのH-I ループ中に挿入し得、その感染性を促進し得る。アデノウイルス組み換え体は、本明細書の記載のようにおよび当分野に既知のように、任意のアデノウイルスベクター中へ、特定の導入遺伝子または導入遺伝子のフラグメントをクローニングすることにより構成される。アデノウイルス組み換え体は、免疫剤としての使用のため、非-侵入形態において脊椎動物の上皮細胞を形質導入するのに使用し得る。
【0129】
"弱い"アデノウイルスの使用は、特に、異種性 DNAの巨大挿入物の挿入に利点がある(レビューとしてYeh. and Perricaudet, FASEB J. 11:615 (1997)参照)。この文献は引用により本明細書に含める。加えて、弱いアデノウイルス性ベクターおよびそれを作成および使用する方法は、米国特許番号6,156,497 および 6,228,646により詳細に記載されており、それらの両方は、引用により本明細書に明示的に含める。
【0130】
アデノウイルスベクターは複製欠損であるか、または少なくとも条件付きで欠損であるという必要以外に、アデノウイルスベクターの性質が、本発明の実施の成功に重要であるとは考えられない。アデノウイルスは、42 種の既知の血清型またはサブグループ A-Fの何れかであり得る。サブグループ Cのアデノウイルス5型は、本発明での使用用の条件付き複製-欠損アデノウイルスベクターを得るための好ましい出発物質である。アデノウイルス5型は、多くの生化学的および遺伝的情報が知られているヒトアデノウイルスであるためである。そして、それは、ベクターとしてアデノウイルスを用いる殆どのコンストラクションとして歴史的に使用されている。
【0131】
上記のように、本発明による典型的なベクターは、複製欠損であり、アデノウイルス E1 領域を有しない。このため、E1-コーディング配列が除かれた位置に免疫修飾分子および/または目的の更なる治療タンパク質をコードする導入遺伝子を導入することが最も都合がよい。しかしながら、アデノウイルス配列内の発現コンストラクトの挿入の位置は、本発明に重要ではない。目的の導入遺伝子はまた、Karlsson et al. (1986)に記載のようにE3 置換ベクター中の欠失E3 領域のかわりに、またはE4 領域(ヘルパー細胞系またはヘルパーウイルスはE4 欠損を相補する)中に挿入し得る。
【0132】
アデノウイルスは、増殖および操作が容易であり、インビトロおよびインビボで広範の宿主範囲を示す。このグループのウイルスは、高力価、例えば、mlあたり109-1011のプラーク-形成単位(109 -1011 plaque-forming units per ml)で得られ得、それらは、高い感染力がある。アデノウイルスのライフサイクルは、宿主細胞ゲノム中への組み込みを必要としない。アデノウイルスベクターにより運ばれる外来遺伝子はエピソームであり、そのため、宿主細胞に対する遺伝毒性は低い。野生型アデノウイルスによるワクチン接種の研究では、副作用は報告されておらず(Couch et al., 1963; Top et al., 1971)、この文献は、インビボの遺伝子転移ベクターとしてその安全性および治療可能性を示している。
【0133】
アデノウイルスベクターは、真核性遺伝子発現(Levrero et al., 1991; Gomez-Foix et al., 1992)およびワクチン開発に使用されている(Grunhaus and Horwitz, 1992; Graham and Prevec, 1992)。最近、動物の研究は、組み換えアデノウイルスは遺伝子治療に使用され得ることを示唆する(Stratford-Perricaudet and Perricaudet, 1991; Stratford-Perricaudet et al., 1990; Rich et al., 1993)。組み換えアデノウイルスを種々の組織に投与する研究には、気管注入 (Rosenfeld et al., 1991; Rosenfeld et al., 1992)、筋肉注射 (Ragot et al., 1993)、末梢静脈注射 (Herz and Gerard, 1993) および脳中への定位固定接種(stereotactic inoculation)(Le Gal La Salle et al., 1993)がある。
【0134】
従って、好ましい実施態様では、本明細書で使用する発現ベクターは、アデノウイルス性ベクターである。適当なアデノウイルス性ベクターはAd2 または Ad5のようなヒトアデノウイルスの修飾を含み、ここで、インビボで複製するウイルスに必要な遺伝的エレメントは、取り除かれている; 例えば、E1 領域、およびアデノウイルス性ゲノムに挿入された目的の外因性遺伝子をコードする発現カセット。
【0135】
加えて、上記のように、好ましい発現ベクターシステムは、一般的にPCT/US97/01019 および PCT/US97/01048(それらは両方とも引用により本明細書に明示的に含まれる)に記載されているようなレトロウイルス性ベクターシステムである。
【0136】
レトロウイルスは、逆-転写のプロセスにより、感染細胞においてそのRNAを二本鎖DNAに変換する能力が特徴的である一本鎖 RNA ウイルスのグループである(Coffin, 1990)。次いで、生ずるDNAは、安定して、細胞性染色体中にプロウイルスとして組み込まれ、ウイルス性タンパク質の合成を指示する。その組み込みは、レシピエント細胞およびその子孫においてウイルス性遺伝子配列の保持を生ずる。レトロウイルス性ゲノムは、キャプシドタンパク質、ポリメラーゼ酵素、およびエンベロープコンポーネントをそれぞれコードする3つの遺伝子、gag、pol、およびenvを含む。gag 遺伝子の上流にある配列は、ゲノムをビリオン中にパッケージングするためのシグナルを含む。2つの長い末端反復(LTR)配列は、ウイルス性ゲノムの5' および 3' 末端に位置する。これらは、強力なプロモーターおよびエンハンサー配列を含み、また、宿主細胞ゲノムにおける組み込みに必要である(Coffin, 1990)。
【0137】
レトロウイルス性ベクターを構成するため、目的の1以上のオリゴヌクレオチドまたはポリヌクレオチド配列をコードする核酸は、複製-欠損であるウイルスを生ずる特定のウイルス性配列の代わりにウイルス性ゲノム中に挿入される。ビリオンを作成するため、gag、pol、およびenv 遺伝子を含むが、LTR およびパッケージングコンポーネントはないパッケージング細胞系を構成する(Mann et al., 1983)。レトロウイルス性 LTR およびパッケージング配列と共にcDNAを含む組み換えプラスミドが、この細胞系中に導入されるとき(例えば、リン酸カルシウム沈殿により)、該パッケージング配列は、組み換えプラスミドのRNA 転写物をウイルス性粒子中にパッケージし得、次いで、それは、培養培地中に分泌される(Nicolas and Rubenstein, 1988; Temin, 1986; Mann et al., 1983)。次いで、組み換えレトロウイルスを含む該培地を回収し、所望により濃縮し、そして遺伝子転移に使用する。レトロウイルス性ベクターは、広範の種々の細胞タイプに感染可能である。しかしながら、組み込みおよび安定な発現は、宿主細胞の分裂を必要とする(Paskind et al., 1975)。
【0138】
レトロウイルスベクターの特異的ターゲッティングができるように設計した新規アプローチは、最近、ウイルス性エンベロープに対するラクトース残基の化学的付加による、レトロウイルスの化学修飾に基づき開発された。修飾は、シアロ糖タンパク質レセプターを介する肝細胞の特異的感染を許容し得る。
【0139】
組み換えレトロウイルスのターゲッティングに対する種々のアプローチは、レトロウイルス性エンベロープタンパク質に対するおよび特異的細胞レセプターに対するビオチン化抗体が使用されるように設計された。該抗体は、ストレプトアビジンを用いることによりビオチンコンポーネントを介して結合した(Roux et al., 1989)。主要組織適合遺伝子複合体クラスIおよびクラスII抗原に対する抗体を用い、それらは、インビトロでエコトロピックウイルスによる表面抗原を生ずる種々のヒト細胞の感染を示した(Roux et al., 1989)。適当なレトロウイルス性ベクターは、LNL6、LXSN、およびLNCXを含む(レビューとしてByun et al., Gene Ther. 3(9):780-8 (1996)参照)。
【0140】
AAV (Ridgeway, 1988; Hermonat and Muzycska, 1984)は、パルボウイルスであり、アデノウイルス性ストックのコンタミネーションとして発見された。それは、任意の疾患とは関連していない広範に分布するウイルス(抗体は、USの人口の85%にある)である。それはまた、デペンドウイルス(dependovirus)として分類される。その複製が、アデノウイルスのようなヘルパーウイルスの存在に依存するためである。5つの血清型が単離されており、そのうち、AAV-2が最もよく特性解析されている。AAV は、キャプシドタンパク質 VP1、VP2 および VP3にカプセル化され、直径20から24 nmの二十面体ビリオンを形成する一本鎖直鎖状DNAを有する(Muzyczka and McLaughlin, 1988)。
【0141】
AAV DNAは、およそ 4.7 キロベース長である。それは、2つのオープンリーディングフレームを含み、2つのITRに隣接している。AAV ゲノム中に2つの主要な遺伝子: rep および capがある。該rep 遺伝子はウイルス性複製に関与するタンパク質をコードし、その一方、capはキャプシドタンパク質 VP1-3をコードする。各々のITRは、T-形ヘアピン構造を形成する。これらの末端反復は、AAVの染色体組み込みのために唯一の必須のcis コンポーネントである。そのため、AAVは、送達用の遺伝子のカセットにより除かれ置換されるすべてのウイルス性コーディング配列を有するベクターとして使用し得る。3つのウイルス性プロモーターは、同定されており、地図の位置によりp5、p19、およびp40と名付けられている。p5 および p19からの転写は、rep タンパク質の産物を生じ、p40からの転写は、キャプシドタンパク質を生ずる(Hermonat and Muzyczka, 1984)。
【0142】
AAVはまた、その安全性のため、送達ビヒクルとしてよい選択である。比較的複雑なレスキュー機構がある: 野生型アデノウイルスだけでなく、AAV 遺伝子もまた、rAAVを起動させることを必要とする。同様に、AAVは、病原性ではなく、任意の疾患とは関連しない。ウイルス性コーディング配列の除去は、ウイルス性遺伝子発現に対する免疫性反応を最小化し、それ故、rAAVは、炎症性反応を引き起こさない。AAVに関連する他の開示は、米国特許番号6,531,456に開示されている。その文献は、引用により本明細書に明示的に含める。
【0143】
他のウイルス性ベクターは、宿主細胞への免疫修飾分子の送達ために本発明で発現ベクターとして用いられ得る。ワクシニアウイルス (Ridgeway, 1988; Coupar et al., 1988)、レンチウイルス、ポリオウイルスおよびヘルペスウイルスのようなウイルスから得られるベクターが用いられ得る。それらは、種々の哺乳類細胞に幾つかの魅力的な特徴を供する(Friedmann, 1989; Ridgeway, 1988; Coupar et al., 1988; Horwich et al., 1990)。
【0144】
発現ベクターの送達
免疫修飾分子(例えば、CD8 α-鎖)および/または更なる治療タンパク質の発現を行うために、発現ベクターは、細胞中に送達されなければならない。この送達は、細胞系を形質転換する実験手順などでインビトロで、または特定の病状の治療などでインビボまたはエキソビボで行い得る。上記のように、送達のための1つの好ましい機構は、感染を介し、この場合、核酸は、組み換えウイルス性粒子中にカプセル化される。
【0145】
一旦、発現ベクターが細胞中に送達されると、望ましいオリゴヌクレオチドまたはポリヌクレオチド配列をコードする核酸は、種々の部位に位置し、発現し得る。特定の実施態様では、コンストラクトをコードする核酸は、安定して、細胞のゲノム中に組み込まれ得る。この組み込みは、相同組み換えによる特定の位置および方向であり得るか(遺伝子置換)またはそれは、ランダムに、非-特異的な位置に組み込まれ得る(遺伝子増大)。更に、および好ましい実施態様では、核酸は、DNAのエピソームセグメントとして別々に細胞中に安定して保持され得る。その核酸セグメントまたは"エピソーム"は、宿主細胞サイクルとは独立したまたはそれと同期した保持および複製を許容するのに十分な配列コードする。どのように発現コンストラクトが細胞に送達されるのかおよび細胞中のどこに核酸が保持されるのかは、用いる発現ベクターのタイプに依存する。
【0146】
本発明の特定の実施態様では、発現ベクターは、単に、裸の組み換えDNAまたはプラスミドからなり得る。ベクターの転移は、上記の任意の方法により行い得、それは、物理的または化学的に細胞膜を透過処理する。これは、特に、インビトロの転移に適用可能であるが、それは、インビボの使用に同様に適用され得る。Dubensky et al. (1984)は、成体および新生マウスの肝臓および脾臓中への、リン酸カルシウム沈殿の形態でのポリオーマウイルスDNAの注入に成功し、活動性のウイルス性複製および急性の感染を示した。Benvenisty and Reshef (1986)はまた、リン酸カルシウム-沈殿化プラスミドの直接の腹膜内注射が、トランスフェクトした遺伝子の発現を生ずることを示した。目的の遺伝子をコードするDNAがまた、類似の方法でインビボで転移され得、遺伝子産物を発現し得ることが想定される。
【0147】
裸のDNA発現コンストラクトを細胞中に転移するための本発明の他の実施態様は、微粒子銃を含み得る。この方法は、DNA-被覆マイクロプロジェクチルを高速にまで加速し、細胞膜を貫通し、細胞を殺すことなく細胞に入ることができる能力に依存する(Klein et al., 1987)。小粒子を加速させるための幾つかのデバイスが開発されている。そのようなデバイスは、電流を生じる高圧放電に依存し、次に、原動力を供する(Yang et al., 1990)。使用する該マイクロプロジェクチルは、一般的に、タングステンまたは金ビーズのような生物学的に不活性な物質からなる。
【0148】
ラットおよびマウスの肝臓、皮膚、および筋組織を含む選択した器官は、インビボで発射される(Yang et al., 1990; Zelenin et al., 1991)。これは、銃と標的器官との間に介在する任意の組織を排除する、組織または細胞の外科的露出、すなわち、エキソビボ治療を必要とし得る。更に、特定の遺伝子をコードするDNAは、この方法を介して送達され得、また、本発明により組み込まれる。
【0149】
本発明の1つの実施態様では、核酸分子は、リポソーム-仲介核酸転移により標的細胞中に導入される。これに関して、多くのリポソーム-ベースの試薬が当分野に既知であり、市場で入試可能であり、標的の細胞中に核酸分子を導入するために日常的に用い得る。本発明の特定の実施態様は、Lipofectamine または Lipofectin (Life Technologies)、陽イオンコレステロール誘導体(DC コレステロール)を伴うジオレオイルホスファチジルエタノールアミン(DOPE)、N[1-(2,3-ジオレイルオキシ)プロピル]-N,N,N-トリメチル塩化アンモニウム (DOTMA) (Sioud et al., J. Mol. Biol. 242:831-835 (1991))、DOSPA:DOPE、DOTAP、DMRIE:コレステロール、DDAB:DOPEなどのような陽イオン脂質転移ビヒクルを用いる。リポソーム-カプセル化核酸の生成物は、当分野に既知であり、典型的に、脂質および核酸の組合せを約 1:1の割合で含む。
【0150】
本発明の使用
上記のように、本明細書に記載され本明細書により可能となる方法および組成物には、例えば、遺伝子治療プロトコールでの使用のための、発現ベクターに対する宿主免疫反応を阻止する一般的な利用がある。すなわち、殆どの遺伝子治療プロトコールでよくある通常の問題は、ベクター-関連抗原に対する宿主免疫反応である。しかし、本発明により、この困難な問題は、遺伝子治療ベクターにおいて対象 CD8 ポリペプチドをコードする核酸の包含により克服される。すなわち、キメラベクターは、目的の治療分子をコードする核酸配列を、CD8 ポリペプチドとともに含むように使用される。ベクター-関連抗原と関連して細胞表面上で生じるCD8 ポリペプチドの発現は、アデノウイルス性ベクター中に存在するウイルス性被覆タンパク質のようなベクター-関連抗原に対する宿主免疫反応の効果的で特異的な阻害を生ずる。すなわち、ウイルス性タンパク質およびCD8が同一細胞内で発現するとき、CD8は、感染した細胞において宿主免疫反応を阻害し得、それにより、遺伝子治療ベクターによる治療処置を持続し得る。
【0151】
理論に制約されることなく、標的細胞におけるCD8の発現は、宿主免疫系において"veto効果"を誘発する能力を標的細胞に供与すると考えられる。すなわち、上記のように、CD8を発現する細胞が、宿主 T 細胞と接触するとき、該 T 細胞は、ダウンレギュレートされるかまたは死滅される。従って、 “veto効果” または “古典的veto”は、標的細胞に対する免疫反応をダウンレギュレートする標的細胞の能力を意味する。CD8 分子は、veto効果の誘発または転移に必要であると考えられる。 “veto効果の転移”は、veto効果は、通常、veto効果を誘発しない細胞に転移されることを意味する。すなわち、標的細胞に対するT 細胞反応を減少するまたはダウンレギュレートする能力が、CD8の発現を誘発するかまたは増加することにより、標的細胞に供与される。
【0152】
従って、本発明は、veto効果を誘発することにより、遺伝子治療送達ビヒクルおよび/または標的細胞に対する免疫反応を低減する使用を見出す。これは、他に標的細胞を認識するT 細胞のダウンレギュレートおよび欠失を生ずる。同様に、これは、体液性免疫反応の低下を生ずる。
【0153】
本発明の発現ベクターは、更に、インビトロでの利用を有する。そのベクターは、ウイルスクリアランスおよび持続の研究においておよび免疫反応を回避する手段の効果を評価する方法において研究ツールとして使用し得る。同様に、発現ベクター、好ましくは、組み換え発現ベクター、特にウイルス性またはアデノウイルス性ベクター(導入遺伝子および免疫修飾分子をコードする少なくとも1つの遺伝子を含む)は、インビボで用い得る。
【0154】
インビボ送達には、以下に限らないが、カテーテルを介するか、または他の手段である潅流による器官中への直接の注入がある。核酸は、標的器官に隣接する位置で血管内に投与され得るかまたは全身的に投与され得る。当業者は、各形態の送達の利点および不利点を認識する。例えば、直接の注入は、核酸の力価を最も高くし得るが、核酸の分布は、標的全体で不均一となるようである。標的に隣接する核酸の導入は、一般的に、器官の細胞とのよりよい接触を生ずるが、全身的投与は、一般的に、より簡単である。
【0155】
特に、本発明の組み換えアデノウイルス性ベクターのような発現ベクターを使用し、矯正 DNA、例えば、非存在であるかまたは欠損であるか何れかである機能をコードするDNAを細胞に送達することにより、多数の疾患のうちの何れか1つを治療し得る。その治療の候補となる疾患には、例えば、癌、例えば、黒色腫または神経膠腫、嚢胞性繊維症、遺伝性疾患、およびHIV 感染を含む病原性の感染がある。
【0156】
同種免疫および自己免疫反応を特異的に阻害する対象の組成物および方法の使用は、同時係属米国特許出願番号XXに記載されており、その開示は、引用により本明細書にすべて含める。本発明の方法および組成物の他の用途は、当業者に明かであろう。
【0157】
発現ベクターを投与するための組成物および方法
当業者は、発現ベクター(特にアデノウイルス性ベクター)を投与する多くの適当な方法および動物に対する本発明の免疫反応を阻害する手段(例えば、Rosenfeld et al., Science, 252, 431-434 (1991); Jaffe et al., Clin. Res., 39(2), 302A (1991); Rosenfeld et al., Clin. Res., 39(2), 311A (1991); Berkner, BioTechniques, 6, 616-629 (1988)参照)が利用可能であり、そして1を超える経路が、投与に使用され得るが、特定の経路が、他の経路よりもより早くより効果的な反応を提供し得ることを認識する。発現ベクターの投与に使用のための医薬に許容される賦形剤および/または免疫反応を阻害する手段も当業者に既知であり、容易に利用できる。賦形剤の選択は、発現ベクターの投与に使用される特定の方法によりおよび免疫反応を阻害する手段として幾分決定される。従って、本発明は、免疫修飾タンパク質(例えば、CD8 α-鎖) をコードする発現ベクターを、単独で、または導入遺伝子との更なる組合せで、適当な担体中に含む組成物を提供し、そして、広範の種々の、本発明と関連する使用に適当な製剤が存在する。特に、本発明は、CD8のα 鎖(またはその機能性フラグメント)をコードする遺伝子を含む発現ベクターおよびその担体を含む組成物を提供する。好ましい実施態様では、発現ベクターは、目的の治療分子またはタンパク質をコードする導入遺伝子を更に含む。その組成物は、治療薬または予防薬のような他の活性剤(active agents)および/または 当分野に知られるような免疫抑制剤を更に含み得る。以下の方法および賦形剤は、単なる例示であり、それらに制限されるものでは全くない。
【0158】
経口投与に適当な製剤は、以下からなり得る:(a)水、生理食塩水、またはオレンジジュースのような、希釈剤に溶解した有効量の化合物のような、溶液; (b) カプセル、サチェット(sachet)または錠剤、それぞれ、事前に決定した量の有効成分を、固体または顆粒として含んでいる; (c) 適当な液体中の懸濁液; および (d) 適当なエマルジョン。錠剤形は、1以上のラクトース、マンニトール、コーンスターチ、ジャガイモデンプン、微晶質セルロース、アカシア、ゼラチン、コロイド状二酸化ケイ素、クロスカルメロース(croscarmellose)ナトリウム、タルク、ステアリン酸マグネシウム、ステアリン酸、および他の賦形剤、色素、希釈剤、緩衝剤、湿潤剤、保存料、香料添加剤、および薬理学的に融和する賦形剤を含み得る。トローチ形は、香味料、通常、スクロースおよびアカシアまたはトラガカントゴム中に有効成分、ならびに該有効成分に加えて当分野に既知のようなその賦形剤を含むゼラチンおよびグリセリン、エマルジョン、ゲルなどのような不活性基剤中、有効成分を含む錠剤(pastilles)を含み得る。
【0159】
エアロゾール製剤は、吸入による投与のために作成され得る。これらのエアロゾール製剤は、ジクロロジフルオロメタン、プロパン、窒素などのような加圧型可能高圧ガス(pressurized acceptable propellants)中に置き得る。それらはまた、例えば、ネブライザーまたはアトマイザー中の非加圧型製剤用の医薬として製剤し得る。
【0160】
非経口的投与に適当な製剤には以下のものがある:水性および非-水性、等張性滅菌注入液溶液、それは、抗-酸化剤、緩衝剤、静菌剤および目的レシピエントの血液と製剤とを等張とする溶質を含み得る、および水性および非-水性滅菌懸濁液、それは、懸濁化剤、可溶化剤、増粘剤、安定剤、および保存料を含み得る。該製剤は、単位用量または複用量(multi-dose)でシールしたコンテナー、例えば、アンプルおよびバイアル中に存在し得、フリーズドライした(凍結乾燥した)状態、使用直前の注入用の滅菌液体賦形剤、例えば、水の添加のみを必要とする状態で保存し得る。即時注入の溶液および懸濁液は、上記種類の滅菌パウダー、顆粒、および錠剤から調製し得る。更に、座剤は、乳状化基材または水可溶性基材のような種々の基材の使用により作成され得る。膣内投与に適当な製剤は、ペッサリー、タンポン、クリーム、ゲル、ペースト、泡、またはスプレー製剤、これらの有効成分とともに、適当であると当分野で既知であるような担体を含む該製剤として存在し得る。
【0161】
本発明との関連で動物、特にヒトに投与する用量は、目的の治療導入遺伝子、ベクターの源および/または免疫修飾分子の性質、用いた組成物、投与の方法、および治療する特定の部位および生体により変わる。しかし、好ましくは、有効量のベクター(例えば、本発明によるアデノウイルス性ベクター)に相当する用量を用いる。"有効量"は、宿主において望ましい効果を生ずるのに十分な量であり、それは、当業者に既知の幾つかのエンドポイントを用いモニターし得る。例えば、1つの望ましい効果は、宿主細胞への核酸転移である。その転移は、以下に限らないが、治療効果(例えば、治療する疾患、病状、異常または症候群と関連する幾つかの徴候の緩和)を含む種々の手段により、または転移遺伝子またはコーディング配列または宿主内でのその発現(例えば、宿主細胞中の核酸を検出するポリメラーゼ連鎖反応、ノーザンもしくはサザンハイブリダイゼーション、または転写アッセイを用いるか、または転移した核酸によりコードされるタンパク質もしくはポリペプチド、またはその転移に起因するレベルまたは機能に影響するタンパク質もしくはポリペプチドを検出するイムノブロット分析、抗体-仲介検出、または詳細に述べられているアッセイを用いる)の証明によりモニターされ得る。記載されているこれらの方法は、決して包括的というわけではなく、そして特定の適用に適当な更なる方法は、当業者に明らかである。これに関して、ウイルス性ベクター、特に、アデノウイルス性ベクターのようなベクター、ならびに免疫反応を阻害する手段をコードするベクターの導入に対する宿主の反応は、投与されたウイルスの用量、送達の部位、およびベクターの遺伝的性質(makeup)ならびに導入遺伝子および免疫反応を阻害する手段により変化し得ることに注意すべきである。
【0162】
一般的に、本発明のベクターの有効な転移を確実とするために、所定の経路の投与を考慮し、接触するおおよその細胞数に基づき、細胞あたり約 1から約 5,000コピーの本発明によるベクターを用い接触させることことが好ましく、各細胞への約 3から約 300 pfuの導入が更により好ましい。しかし、これは、単なる一般的なガイドラインであり、それは、インビトロまたはインビボの何れかの特定の適用を保証し得るような、より高い量またはより少ない量の使用を決して排除するものではない。同様に、免疫反応を阻害する手段の量は、タンパク質を含む組成物の形態ならば、導入遺伝子を含む組み換えベクターに対する免疫反応の阻害に十分であるべきである。例えば、実際の用量およびスケジュールは、いずれの組成物を他の医薬組成物と組み合わせて投与するかにより、または薬物動力学、薬物動態、および代謝における個人間の相違により、変わり得る。同様に、量は、標的とする特定の細胞タイプまたはベクターが転移される手段により、インビトロの適応で変わり得る。当業者は、特定の状況の必要性に応じて任意に必要な調節をすることが容易であり得る。
【0163】
本発明は、好ましい実施態様に準拠して記載されているが、当業者は、本発明の精神および範囲を逸脱することなく形式および詳細を変え得ると認識するだろう。本明細書で特定している、特許、刊行物および他の引用文献、それぞれは、引用により本明細書にすべて明示的に含める。
【実施例】
【0164】
実施例1 veto作用−ベクターを用いた研究
a. 繊維芽細胞をveto細胞として操作するプラスミド発現ベクターの使用
繊維芽細胞を、ヒトまたはマウスの何れかのCD8-鎖をその表面において発現するように操作した。繊維芽細胞を、pCMVhCD8 プラスミドまたはpCMVmCD8 プラスミドでトランスフェクトし、それによって、CD8 α-鎖の発現は、CMV 前初期プロモーター/エンハンサー(Invitrogen)により生じた。CD8-鎖 トランスフェクト化繊維芽細胞 (H-2b)を、混合リンパ球培養物(BALB/c; H-2d 抗-C57BL/6; H-2b)に加えると、CD8-鎖を発現する系のみがCTL反応を抑制した。図 3A および Bに示すように、マウスまたはヒト CD8-鎖の何れかを発現するMC57T 繊維芽細胞の添加は、CTLの誘導を完全に抑制した。対照的に、非-トランスフェクト化繊維芽細胞の添加は、T-リンパ球の活性化に影響しなかった。CD8 α-鎖の阻害機能を確認することに加えて、これらの実験は、マウス T-リンパ球は、ヒト CD8 α-鎖によりvetoされ得ることをも示していた。それ故、マウスモデルは、臨床的使用用に設計したvetoを試験することに有用である。
【0165】
操作したveto細胞のインビボ機能
操作したvetoが動物で機能するかどうかを決定した。トランスフェクトし、CD8 α-鎖を発現するC57BL/6 (H-2b)-由来繊維芽細胞を、Balb/c (H-2d) マウスに注射した。対照動物に、非-トランスフェクト化繊維芽細胞を注射した。脾臓細胞は、8から40日後、回収し、スティミュレーター細胞としてのC57BL/6 (H-2b)脾臓細胞と共にMLC培養物中に導入した。5日後、培養物を回収し、EL4 (C57BL/6, H-2b)標的細胞を溶解する能力について試験した。抗-H-2b CTL反応の誘発は、CD8 -鎖を発現する繊維芽細胞を注射された動物では完全に抑制された(図 4)。抗-H-2b T 細胞の阻害は、非常に特異的であった。これらマウス由来のT 細胞はまた、第三のグループH-2k アロ-MHC 分子群に対する反応をマウントした。これらの実験で、操作したveto細胞は、古典的veto細胞と同様にインビボで免疫反応を特異的に抑制したこと、および非古典的veto細胞を操作するとveto細胞となったことを確認した。言い換えれば、操作した細胞は、これらの細胞において保有される抗原に対し動物を負に免疫した。
【0166】
CD8-鎖の発現が、完全に活性化されたT 細胞の機能を妨げるかどうか試験した。この目的のため、CD8 α-鎖を発現する標的細胞を、完全に活性化されたCTLによる溶菌に対する感受性について試験した。2種のT 細胞群をこれらの研究のために選択した。それは、MLCにおいて刺激されたアロ-反応性CTLおよび活性化ペプチド-特異的CTLであった。図 5に示したように、CD8 α-鎖を発現する標的は、アロ反応性 T 細胞の群により効果的に溶解されたが、抗原-特異的 T 細胞では溶解されなかった。これらの結果は、操作したvetoは、自己免疫反応で見られるような、継続的な抗原特異的免疫反応を妨げることができたことを示唆していた。
【0167】
b. veto細胞として繊維芽細胞を操作するためのウイルス転移ベクター
アデノウイルス転移ベクターm-CD8のveto機能: 複製-欠損ベクターアデノウイルス転移ベクター(mAdCD8α)は、マウス CD8 α-鎖を保有するように開発した。mAdCDB veto転移ベクターで感染されたマウス繊維芽細胞(MC57)は、日2に高レベルのマウス CD8 α-鎖を発現した。これらの早い増殖細胞において、マウス CD8 α-鎖の発現は、日5までに顕著に減少した。mAdCD8はまた、低効率であるにも関わらず、EL4のような他のマウス細胞系に感染した(データ示さず)。
【0168】
その後の実験において、mAdCD8 α-感染性MC57 繊維芽細胞 (H-2b)を、Balb/C (H-2d) 抗-C57B1/6 (H-2b) MLCに添加した。5 日後、培養物を回収し、抗-H-2b CTLの存在について試験した。繊維芽細胞に感染したMLCを加え、もはや抗-H-2b CTLを含まなかった(図 12)。これらの実験は、免疫抑制を仲介するveto転移ベクターの能力を確認した。
【0169】
加えて、アデノウイルス性ベクターのヒト CD8-版を作成した。また、マウス CD8 α-鎖を発現するアデノウイルス関連ウイルスを作成した。これらのウイルスは、各CD8 鎖の発現を誘導することが示された。マウスまたはヒトの何れかのCD8 α-鎖を発現するアデノウイルス性vetoベクターは、キラー T 細胞の誘導の完全阻害を仲介した(図 7参照)。
【0170】
mAdCD8 veto転移ベクターによる負の免疫化: 2種の実験を設定し、mAdCD8は、免疫反応をインビボで抑制するかどうかを決定した。第一の実験では、C57Bl/6 マウスを、等用量の、mAdCD8 veto転移ベクターまたは同様のアデノウイルス対照ベクター(マウス CD8 α-鎖の代わりにβ-ガラクトシダーゼをコードする(Adβgal))の何れかで感染させた。免疫化7日後、これらの動物を屠殺した。その脾臓細胞の単一細胞の懸濁液を、Adβgalウイルスの存在下、5日間、培養した。次いで、その培養物を回収し、増殖能を評価した。図 7に示したように、Adβgalに力強く増殖したT 細胞は、Adβgalで免疫したマウスから回収し、これは増殖性の高いCD4+ T 細胞の存在を示す。対照的に、mAdCD8-注射した動物から回収したT 細胞は、増加しなかった。
【0171】
第二の工程では、我々は、これらの培養物が、機能性 CD8+ CTLを含むかどうかを試験し、Adβgal-感染性標的細胞(EL4, H-2b) を溶解する能力を試験した。CTLは、Adβgalを注射したマウスから作成した培養物中に見られ得るのみであった(図 8)。この第一の実験は、AdCD8αが、恐らく、CD8 α-鎖の発現によってアデノウイルス性抗原に対する反応を誘発しなかったことを示唆する。しかしながら、別の理由のため、AdCD8は、免疫反応を誘発しなかったという可能性があった。AdCD8は、幾つかの不確定な方法において非-機能性であるか、またはマウスは、mAdCD8には見られなかったβ-ガラクトシダーゼタンパク質と反応し得るのみであった。
【0172】
種々の結論の妥当性を試験するために、C57Bl/6 マウスは、mAdCD8またはAdβgalの何れかで一旦注射され、その後、7日後、Adβgalで2回目の注射を行った。7日後、マウスを屠殺し、そして5-日の脾臓細胞培養物を、Adβgalの存在下、作成した。その対応するT 細胞は、Adβgal-感染性標的細胞に対する溶菌能について試験した(図 8)。確かに、Adβgalに対する2つの暴露は、免疫化を改善した。これらの研究はまた、AdCD8注射後、マウスはもはやAdβgalに反応せず、Adβgalは、主として、排他的でないならば、ベクターの両方に共通のアデノウイルス性タンパク質に対するCTL反応を誘発することを示していた。この一連の実験は、これらのベクターに保持される遺伝子に対する反応に対し負の免疫化ができる遺伝子治療ウイルス性ベクターを作成することが可能であることを強力に示唆する。
【0173】
vetoによるCD4+ T リンパ球の阻害: veto転移ベクターを用い、CD4+ T リンパ球の誘導を阻害し得るかどうか試験するために、以下の実験系を確立した。C57Bl/6-由来繊維芽細胞スティミュレーターを形質転換し、同種性MHCクラスII 分子 (H-2Ek)および免疫刺激性CD80を発現させた。これら遅く増殖する繊維芽細胞であって、放射線を照射せず刺激能を完全に保持している該細胞は、mAdCD8またはAdβgal転移ベクターの何れかで形質導入され、任意抽出のC57B1/6 脾臓細胞に添加された。4日後、これらの培養物を回収し、活性化、すなわち、ブラスティング(blasting)、CD4+ T リンパ球の存在について表面の免疫蛍光により分析した(図 9)。通常のまたはAdβgal-形質導入化スティミュレーター細胞とともに培養した任意抽出のC57Bl/6 脾臓細胞は、非常に多くのCD4+ T リンパ芽球を有していることが判った。対照的に、mAdCD8-感染性スティミュレーターが添加された培養物で、ほんの僅かなCD4+T リンパ芽球が検出された。この研究では、vetoがCD4+ T リンパ球を阻害し、そして加えて、ウイルス性veto転移ベクターがこの目的に使用され得ることを確認した。
【0174】
種々のウイルスコンストラクトによる感染後のマウスおよびヒト CD8 α-鎖の表面発現
染色プロトコール:
mAdCD8:
MC57Tは、偽-感染するか、またはmAdCD8によりおよそ104の感染効率で、修飾したIMDMにおいて3日間で感染される。感染した細胞を回収し、FITC(Pharmingen)で直接標識した抗-マウス CD8 α-鎖 抗体を伴うCD8 α-鎖の表面発現について染色した。表面蛍光の程度を、蛍光活性化細胞アナライザー(FACScan, Beckton-Dickinson)で測定した(図 10)。
【0175】
骨髄細胞は、Balb/c マウスの大腿骨の腔からから回収した。該細胞は、β-ガラクトシダーゼを発現するアデノウイルス性対照ベクター (AdLacZ)またはmAdCD8により104の感染効率で、修飾したIMDMにおいて3日間の培養で感染された。感染した細胞を回収し、FITCで直接標識した抗-マウス CD8 α-鎖 抗体によってCD8 α-鎖の表面発現について染色した。表面蛍光の程度を測定した(図 10C)。加えて、CD34+ 骨髄細胞、すなわち、幹細胞プール内の細胞を含む幾つかの細胞タイプを効率的に形質導入することを決定した(表 2)。
【表3】
表2
hAdCD8:
MC57Tを偽-感染した。hAdCD8のウイルス力価は知られていない。100μlのそのストック溶液を用い、3 x 105 細胞を3日間、感染した。その感染した細胞を回収し、そしてFITC (Pharmingen)で直接標識した抗-ヒト CD8 α-鎖 抗体によってCD8 α-鎖の表面発現について染色した。表面蛍光の程度を、蛍光活性化細胞アナライザーで測定した(図 10)。
【0176】
AAVに基づくvetoベクター
AAVに基づくvetoベクターを、Strategene /Avigen システムを用い平行して作成した。これらのコンストラクトにおいて、ヒトおよびマウス CD8 α-鎖は、同じCMV 中間体初期プロモーター/エンハンサーから得られた。2つのウイルス、mAAVCD8 および hAAVCD8を、HEK 293 パッケージング細胞系にパッケージした。その用いた系は、ヘルパーウイルスを用いない。mAAVCD8 および hAAVCD8は、効率的に、マウス繊維芽細胞(MC57T)に感染し、マウスまたはヒト CD8 α-鎖、それぞれについて高レベルの発現を生じた。蛍光の程度を、蛍光活性化細胞アナライザーで測定した(図 10D)。高レベルのCD8 α-鎖 発現が、形質導入後、36 時間以内に見られることが興味深い。この発見は、他者による観察とは対照的であった。それらでは、AAV-誘導化遺伝子発現は、顕著なレベルに到達するまで数日かかった(PH Schmelck, PrimeBiotech)。AAV vetoベクターによる更なる研究で、それらを用い免疫反応を抑制し得るという我々の先の発見が繰り返し得られた。ここで、標準的なMLC プロトコールを用いた(図 6)。
【0177】
実施例 2: インビトロ阻害研究 - 混合リンパ球培養物
脾臓細胞を、Balb/c (H-2d) および C57BL/6 (H-2b) マウスから回収した。単一の細胞懸濁液を調製した。C57BL/6 脾臓細胞を、3,000 rad (Mark 1 Cesium Irradiator)で照射した。4 x 106 Balb/c 脾臓細胞(レスポンダー/エフェクター細胞)は、10% 胎児ウシ血清(FCS)(Sigma)、HEPES、ペニシリンG、硫酸ストレプトマイシン、硫酸ゲンタマイシン、L-グルタミン、2-メルカプトエタノール、非必須アミノ酸(Sigma)、ピルビン酸ナトリウムおよび炭酸水素ナトリウムを含むIMDM(Sigma)(修飾IMDM)において、24-ウェルプレート(TPP, Midwest Scientific, Inc.)中のウェルあたり4 x 106 照射 C57BL/6 脾臓細胞(スティミュレーター細胞)と共に培養した。CO2 インキュベーター(Forma Scientific)中での培養で5日後、培養物をすべて回収し、C57BL/6-由来標的細胞(H-2b)を溶解する能力について試験した。
【0178】
幾つかのこれらの培養物に、4 x 105 MC57T 繊維芽細胞(H-2d)を加え、12,000 radを照射した。阻害培養において、4 x 105 MC57T 細胞を含み、mAdCD8によりおよそ104の感染効率で、1〜2日間で感染された。
【0179】
細胞毒性 T リンパ球キラーアッセイ
混合リンパ球培養物から回収した細胞を、活性化T リンパ球のインディケーターとして芽細胞の数についてカウントした。これらのエフェクター細胞を、U-底の96-ウェルプレート中の単一のウェルに加えた。ウェルあたりのエフェクターの数を、ウェルあたり3 x 106 または 1 x 105 エフェクターから出発する3-倍力価(titration)工程で測定した(titrated)。これらのエフェクター細胞に、ウェルあたり1 x 104 標的細胞 EL4 (H-2b)、MC57T (H-2b) または P815 (H-2d)を加えた。標的細胞は、先に、51Cr (Na-Chromate, Perkin-Elmer)で標識していた。1 x 106 標的細胞を、修飾IMDM中、およそ 500μlの体積で、90分間、100 μCiと共にインキュベートした。その後、組み込まれていない51Crを、修飾IMDMで数回洗浄することにより取り除いた。
【0180】
エフェクターおよび標的細胞を、合計体積 200 μlで、4時間、CO2 インキュベーター中でインキュベートした。その後、プレートを、1,500 rpmで3 分間、スピンさせ遠心分離した(Centra CJ35R, International Equipment Company)。100 mlの培地を、各ウェルから取り除き、標的細胞から放出される51Crの量をModel 4000 Gamma counter (Beckman Instruments)でカウントした。対照培養物をセットし、そのエフェクター細胞について、バックグラウンド放射を測定するのは省略した。標的細胞中への合計の51Cr取り込みをウェル中で測定し、Triton X100 (Sigma)の1% 溶液 (w/v)をエフェクター細胞と置換した。
【0181】
特異的な溶菌の量を以下のように%で決定した:
% = (特異的放射 - バックグラウンド放射) / (合計放射 - バックグラウンド放射) x 100
【0182】
mAdCD8のインビトロでの活性
混合化リンパ球培養物をセットした(Balb/c 抗-C57BL/6)。これらの培養物に、MC57T 繊維芽細胞を加え(示したように)、12,000 radで照射し、mAdCD8を感染させた。培養の5日後、その培養物を回収し、種々の標的に対するエフェクターの比(E/T)においてEL4 (H-2b) 標的細胞を溶解する能力について試験した(図 4参照)。
【0183】
図に見られるように、混合化リンパ球培養物においてさえ、CD8を発現する細胞は、溶菌 T リンパ球の誘導を阻害した。
【0184】
mAdCD8 および hAdCD8の作成
両アデノウイルス性ベクターを、BiogeneのAdEasy(商標)システムの助けにより作成した。ここで、マウスおよびヒト CD8 α-鎖 cDNAは、転移ベクターに組み込まれる(工程 1)。Ad5ΔE1/ΔE3 ベクターの組み換えは、BJ5183 EC 細菌において達成する(工程 2)。次いで、その組み換えベクターは、E1A および E1B アデノウイルス 5 ウイルス遺伝子(組み換えアデノウイルス中のこの本質的な領域の欠失を相補する)を含むQBI-HEK 293A 細胞に転移する。このため、これらの細胞中で作成されるhAdCD8 および mAdCD8は、複製欠損である。
【0185】
細菌性 LacZ 遺伝子 (β-ガラクトシダーゼ)を発現するコントロールベクターとして、Qbiogeneは、QBI-Infect+ Viral Particle(Ad5.CMVLacZΔE1/ΔE3)を提供していた。マウスCD8 α-鎖配列を用いた。この配列は、公開されているマウス配列に類似する:
タンパク質-配列:
実際の配列: MASPLTRFLS LNLLLMGESI ILGSGEAKPQAPELRIFPKK MDAELGQKVD LVCEVLGSVS QGCSWLFQNS SSKLPQPTFVVYMASSHNKI TWDEKLNSSK LFSAVRDTNN KYVLTLNKFS KENEGYYFCSVISNSVMYFS SVVPVLQKVN STTTKPVLRT PSPVHPTGTS QPQRPEDCRPRGSVKGTGLD FACDIYIWAP LAGICVAPLL SLIITLICYH RSRKRVCKCPRPLVRQEGKP RPSEKIV
【0186】
ヒト CD8 α-鎖配列を用いた。この配列は、示したように、公開されているヒト配列と比較するとサイレント変異を有する。
実際の配列: MALPVTALLL PLALLLHAAR PSQFRVSPLDRTWNLGWTVE LKCQVLLSNP TSGCSWLFQP RGAAASPTFL LYLSQNKPKAAEGLDTQRFS GKRLGDTFVL TLSDFRRENE GYYFCSALSN SIMYFSHFVPVFLPAKPTTT PAPRPPTPAP TIASQPLSLR PEACRPAAGG AGNRRRVCKCPRPVVKSGDK PSLARYV
【0187】
pAAV-mCD8 および pAAV-hCD8の作成
これらのベクターは、StratageneのAAV Helper-Free システムの助けにより作成した。そのシステムは、マウスおよびヒト配列をpAAV-MCS クローニングベクター中に挿入することにより機能する。次いで、このプラスミドは、ヘルパープラスミド(必要なアデノウイルス性タンパク質を含む)およびpAAV-RCベクター(キャプシド遺伝子を含む)と共に、HEK 293 細胞中に共-トランスフェクトし、組み換えAAV 粒子を産生する。
【0188】
実施例 3: 動物モデルにおける、操作したveto
我々は、どのように動物がmAdCD8の大用量の注射に対し反応するかを調査した。第一のセットの実験では、Balb/c マウス (各群で2匹のマウス)に、等用量のmAdCD8 または β-ガラクトシダーゼ(AdLacZ)をコードするアデノウイルス性コントロールベクターを静脈注射した。7日後、その動物は屠殺した。それらの脾臓細胞を、AdLacZの存在下、5日間、培養した。次いで、それらを、AdLacZ-感染性標的細胞(P815, Balb/c-由来)を溶解する能力について試験した。図 13に示すように、特異的溶菌能を有するCTLは、AdLacZで免疫されたBalb/c マウスから発展し得るが、mAdCD8を受けたマウスからは発展し得なかった。この結果は、AdCD8は、CD8 α-鎖の発現によってアデノウイルス性抗原に対する免疫反応を誘導しなかったことを示唆した。
【0189】
第二の設定では、C57Bl/6 マウスを、等用量のmAdCD8 (2 マウス) または AdLacZ (2 マウス) で免疫した。免疫化して7日後、各群の動物を1匹屠殺した。その脾臓細胞を、AdLacZの存在下、5日間、細胞懸濁液中で培養した。次いで、それらを、特異的にAdLacZ-感染性標的細胞(EL-4, C57Bl/6-由来)を溶解する能力について試験した。再び、AdLacZの注射は、低頻度での特定のキラー細胞の発展を誘導したにもかかわらず、その一方、mAdCD8は、そうならなかった(図 14)。
【0190】
この実験の第二のフェーズでは、mAdCD8 または AdLacZの何れかを受けた残りのC57BL/6 マウスは、第一のウイルス性注射7日後に第二の用量のAdLacZを受けた。7日後、マウスを屠殺し、5日後、脾臓細胞培養をAdLacZの存在下行った。反応するT 細胞を、再び、AdLacZ-感染性EL4-標的細胞に対する溶菌能について試験した(図 8)。実際、AdLacZに対する2つの暴露が、幾らかの免疫化の改善を生ずる。しかしながら、先にmAdCD8を受けた動物はなお、反応をマウントできない。これらの実験は、AdCD8は、免疫反応を誘導できないだけでなく、それ自身に対する免疫反応の誘導を予防したことを示唆する。このため、mAdCD8は、免疫系を回避した。
【図面の簡単な説明】
【0191】
【図1−1】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−2】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−3】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−4】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−5】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−6】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−7】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−8】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−9】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−10】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−11】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−12】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−13】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−14】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−15】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−16】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図1−17】図1は、CD8 α-鎖 タンパク質および核酸配列を示す。
【図2−1】図2は、野生型 CD8 α-鎖のアミノ酸および核酸配列を示す。
【図2−2】図2は、野生型 CD8 α-鎖のアミノ酸および核酸配列を示す。
【図3】図3は、C57BL/6 脾臓細胞で刺激されたBalb/c 脾臓細胞を示す。培養物に、マウス(A)またはヒト(B)由来の正常の繊維芽細胞 (黒丸)、培地(黒四角)、またはCD8を有する繊維芽細胞 (黒三角)が供された。培養物は回収され、C57BL/6-由来標的細胞に対するその溶菌能について試験した。
【図4】図4は、対照繊維芽細胞(黒四角および黒三角)またはmCD8-トランスフェクトC57BL/6-(H-2b)由来(白丸および黒丸)繊維芽細胞を注射したBalb/c(H-2d)マウスを示す。2週間後、動物を屠殺し、脾臓細胞を回収し、C57BL/6(H-2b)(黒四角および白丸)またはCBA/J(H-2k)(黒丸および黒三角)脾臓細胞で刺激し、EL4 (H-2b)(黒四角および白丸)またはS.AKR (H-2k)(黒丸および黒三角)標的細胞に対する溶菌能について試験した。
【図5】図5は、異型反応性(alloreactive) T 細胞(A) によるかまたは抗原-特異的 CTL(B)による溶菌に対する感受性について試験した標的細胞(黒三角)またはCD8-発現標的(黒四角)を示す。
【図6】図6は、正常繊維芽細胞 (黒丸)およびmAdCD8 (A、黒三角)またはHAdCD8 (B、黒三角)で遺伝子導入された繊維芽細胞の存在下で設定したMLC(Balb/c 抗-C57B/6)を示す。繊維芽細胞は対照培養物(黒四角)には加えなかった。C57BL/6-由来標的に対するこれらの培養物の溶菌活性を培養期間の最後に測定した。
【図7】図7は、アデノウイルス性veto転移ベクター、mAdCD8による免疫化を示す。C57BL/6 マウスは上記のベクターで感染させた。10日後、脾臓細胞を回収し、Adβgal ウイルスの存在下、培養した。芽細胞の数を示す。
【図8】図8 は、mAdCD8による陰性免疫化(negative immunization)を示す(A)。C57BL/6 マウスはAdβgalまたはmAdCD8により静脈注射で一度免疫化された。(A)のように処置した動物は、5日後、Adβgalで再免疫化した(B)。最後の注射の7日後、動物は屠殺し、その脾臓細胞はAdβgalの存在下、培養した。培養の5日後、細胞は、Adβgal-感染性同系標的細胞の溶菌能について試験した。
【図9】図9は、示したように形質導入した、1x106 (または全くなし)スティミュレーター細胞と共にインキュベートした3x106 C7Bl/6 脾臓細胞を示す。4日後、培養物を、免疫蛍光によりCD4+ T リンパ芽球の存在について分析した。
【図10−1】図10は、種々のウイルスコンストラクトによる感染後のマウスおよびヒト CD8 α-鎖の表面発現を示す。
【図10−2】図10は、種々のウイルスコンストラクトによる感染後のマウスおよびヒト CD8 α-鎖の表面発現を示す。
【図10−3】図10は、種々のウイルスコンストラクトによる感染後のマウスおよびヒト CD8 α-鎖の表面発現を示す。
【図10−4】図10は、種々のウイルスコンストラクトによる感染後のマウスおよびヒト CD8 α-鎖の表面発現を示す。
【図11】図11は、MLC (Balb/c 抗-C57BL/6)を、形質導入後、MLCに加えられる前、0 または 5 時間培養されたこれらの繊維芽細胞の存在下、設定したものを示す。培養の最後に、リンパ芽球の数を蛍光活性化細胞アナライザーで測定した。
【図12】図12は、veto転移ベクターによるインビトロ阻害を示す。
【図13】図13は、AdLacZ または mAdCD8で免疫化されたBalb/c マウスを示す。それらの脾臓細胞は、AdLacZの存在下で培養し、AdLacZ-感染性同系P815標的細胞に対する特異的溶菌活性について試験した。
【図14】図14は、AdLacZ (黒四角)またはmAdCD8 (黒三角)で免疫化した(A) C57BL/6 動物を示す。同系AdLacZ EL4 標的細胞に対する脾臓細胞の溶菌活性を試験した。(B)その動物は、AdLacz-感染化EL4 標的に対する溶菌活性を試験する前にAdLacZで再免疫化した。
【図15】図15は、ヘモグロビン βのmRNA 配列を示す。
【図16】図16は、GATA結合タンパク質のmRNA 配列を示す。
【図17−1】図17は、d-アミノエブリネート(aminoevulinate)シンターゼのmRNA 配列を示す。
【図17−2】図17は、d-アミノエブリネート(aminoevulinate)シンターゼのmRNA 配列を示す。
【図18−1】図18は、グルコース-6-ホスフェート-デヒドロゲナーゼのmRNA 配列を示す。
【図18−2】図18は、グルコース-6-ホスフェート-デヒドロゲナーゼのmRNA 配列を示す。
【図19−1】図19は、オルニチンカルバモイルトランスフェラーゼのmRNA 配列を示す。
【図19−2】図19は、オルニチンカルバモイルトランスフェラーゼのmRNA 配列を示す。
【図20−1】図20は、α-L-イズロニダーゼのmRNA 配列を示す。
【図20−2】図20は、α-L-イズロニダーゼのmRNA 配列を示す。
【図21−1】図21は、β-グルコシダーゼのmRNA 配列を示す。
【図21−2】図21は、β-グルコシダーゼのmRNA 配列を示す。
【図22】図22は、α-ガラクトシダーゼのmRNA 配列を示す。
【特許請求の範囲】
【請求項1】
以下を含むポリヌクレオチド:
a)膜貫通ポリペプチドをコードする核酸に作動可能なように連結したCD8 α-鎖をコードする第一核酸; および
b)目的の治療遺伝子を含む第二の核酸; および
c)該第一および第二の核酸の発現を指示するための少なくとも第一の転写および翻訳制御エレメント。
【請求項2】
CD8 α-鎖をコードする核酸は、図1に記載のヒト CD8 α-鎖をコードする核酸に対して80%を超える配列同一性を有する、請求項1のポリヌクレオチド。
【請求項3】
CD8 α-鎖をコードする核酸は、図1に記載のマウス、ラット、またはブタ CD8 α-鎖をコードする核酸に対して80%を超える配列同一性を有する、請求項1のポリヌクレオチド。
【請求項4】
CD8 α-鎖をコードする核酸が、図1に記載のマウス、ラット、またはブタ CD8 α-鎖を含む、請求項3のポリヌクレオチド。
【請求項5】
CD8 α-鎖は、図1に記載の配列からなる群から選択される配列を含む、請求項1のポリヌクレオチド。
【請求項6】
CD8 α-鎖は、野生型 CD8 α-鎖の細胞内ドメインを欠失する、請求項1のポリヌクレオチド。
【請求項7】
目的の治療遺伝子は、ヘモグロビン-β、GATA-結合タンパク質、d-アミノエブリネートシンターゼ、グルコース-6-ホスフェート-デヒドロゲナーゼ、凝固VIII因子、凝固XI因子、嚢胞性繊維症膜貫通コンダクタンスレギュレーター、オルニチンカルバモイルトランスフェラーゼ、α-L-イズロニダーゼ、イズロネート-2-スルファターゼ、β-グルコシダーゼ、α-ガラクトシダーゼ、ガラクトシルセラミダーゼ、酸α-グルコシダーゼ、ヘキサミダーゼ A、フェニルアラニンヒドロキシラーゼ、コラーゲンIV型、α5、ブルーム病遺伝子産物、および低密度リポタンパク質レセプターからなる群から選択される、請求項1のポリヌクレオチド。
【請求項8】
ポリヌクレオチドがベクターを含む、請求項1から7の何れか1つのポリヌクレオチド。
【請求項9】
ベクターが、組み換えアデノウイルス、組み換えレトロウイルス、組み換えアデノ-随伴ウイルス、および組み換えヘルペスウイルスからなる群から選択される、請求項8のポリヌクレオチド。
【請求項10】
ベクターが複製欠損である、請求項9のポリヌクレオチド。
【請求項11】
リポソームを更に含む、請求項1、2、3、4、5、6または7の何れか1つのポリヌクレオチドを含む組成物。
【請求項12】
遺伝子治療送達システム由来の抗原に対する免疫反応を低下させるための方法であって、以下を含む該方法:
a)細胞を該遺伝子治療送達システムと接触させること、ここで、該遺伝子治療送達システムは以下を含む:
i)膜貫通ポリペプチドをコードする核酸に作動可能なように連結したCD8 α-鎖をコードする第一の核酸; および
ii)目的の治療遺伝子を含む第二の核酸; および
iii)該第一および第二の核酸の発現を指示するための少なくとも第一の転写および翻訳制御エレメント、これにより、該第一および第二の核酸が発現し、発現したCD8 α-鎖は該細胞の細胞膜と結合し、そして該細胞に対する宿主免疫反応は、CD8 α-鎖をコードする核酸を有さない細胞に対する免疫反応と比較すると低下する。
【請求項13】
遺伝子治療送達システムが、ウイルス性発現ベクター、プラスミドおよび裸の核酸発現ベクターからなる群から選択される、請求項12の方法。
【請求項14】
ウイルス性発現ベクターが、組み換えアデノウイルス、組み換えレトロウイルス、組み換えアデノ-随伴ウイルス、および組み換えヘルペスウイルスからなる群から選択される、請求項13の方法。
【請求項15】
目的の治療遺伝子は、ヘモグロビン-β、GATA-結合タンパク質、d-アミノエブリネートシンターゼ、グルコース-6-ホスフェート-デヒドロゲナーゼ、凝固VIII因子、凝固XI因子、嚢胞性繊維症膜貫通コンダクタンスレギュレーター、オルニチンカルバモイルトランスフェラーゼ、α-L-イズロニダーゼ、イズロネート-2-スルファターゼ、β-グルコシダーゼ、α-ガラクトシダーゼ、ガラクトシルセラミダーゼ、酸α-グルコシダーゼ、ヘキサミダーゼ A、フェニルアラニンヒドロキシラーゼ、コラーゲンIV型、α5、ブルーム病遺伝子産物、および低密度リポタンパク質レセプターからなる群から選択される、請求項12の方法。
【請求項16】
CD8 α-鎖をコードする核酸が、図11に記載の配列を含む、請求項12の方法。
【請求項17】
CD8 α-鎖をコードする核酸が、図10に記載の配列を有するタンパク質をコードする、請求項12の方法。
【請求項1】
以下を含むポリヌクレオチド:
a)膜貫通ポリペプチドをコードする核酸に作動可能なように連結したCD8 α-鎖をコードする第一核酸; および
b)目的の治療遺伝子を含む第二の核酸; および
c)該第一および第二の核酸の発現を指示するための少なくとも第一の転写および翻訳制御エレメント。
【請求項2】
CD8 α-鎖をコードする核酸は、図1に記載のヒト CD8 α-鎖をコードする核酸に対して80%を超える配列同一性を有する、請求項1のポリヌクレオチド。
【請求項3】
CD8 α-鎖をコードする核酸は、図1に記載のマウス、ラット、またはブタ CD8 α-鎖をコードする核酸に対して80%を超える配列同一性を有する、請求項1のポリヌクレオチド。
【請求項4】
CD8 α-鎖をコードする核酸が、図1に記載のマウス、ラット、またはブタ CD8 α-鎖を含む、請求項3のポリヌクレオチド。
【請求項5】
CD8 α-鎖は、図1に記載の配列からなる群から選択される配列を含む、請求項1のポリヌクレオチド。
【請求項6】
CD8 α-鎖は、野生型 CD8 α-鎖の細胞内ドメインを欠失する、請求項1のポリヌクレオチド。
【請求項7】
目的の治療遺伝子は、ヘモグロビン-β、GATA-結合タンパク質、d-アミノエブリネートシンターゼ、グルコース-6-ホスフェート-デヒドロゲナーゼ、凝固VIII因子、凝固XI因子、嚢胞性繊維症膜貫通コンダクタンスレギュレーター、オルニチンカルバモイルトランスフェラーゼ、α-L-イズロニダーゼ、イズロネート-2-スルファターゼ、β-グルコシダーゼ、α-ガラクトシダーゼ、ガラクトシルセラミダーゼ、酸α-グルコシダーゼ、ヘキサミダーゼ A、フェニルアラニンヒドロキシラーゼ、コラーゲンIV型、α5、ブルーム病遺伝子産物、および低密度リポタンパク質レセプターからなる群から選択される、請求項1のポリヌクレオチド。
【請求項8】
ポリヌクレオチドがベクターを含む、請求項1から7の何れか1つのポリヌクレオチド。
【請求項9】
ベクターが、組み換えアデノウイルス、組み換えレトロウイルス、組み換えアデノ-随伴ウイルス、および組み換えヘルペスウイルスからなる群から選択される、請求項8のポリヌクレオチド。
【請求項10】
ベクターが複製欠損である、請求項9のポリヌクレオチド。
【請求項11】
リポソームを更に含む、請求項1、2、3、4、5、6または7の何れか1つのポリヌクレオチドを含む組成物。
【請求項12】
遺伝子治療送達システム由来の抗原に対する免疫反応を低下させるための方法であって、以下を含む該方法:
a)細胞を該遺伝子治療送達システムと接触させること、ここで、該遺伝子治療送達システムは以下を含む:
i)膜貫通ポリペプチドをコードする核酸に作動可能なように連結したCD8 α-鎖をコードする第一の核酸; および
ii)目的の治療遺伝子を含む第二の核酸; および
iii)該第一および第二の核酸の発現を指示するための少なくとも第一の転写および翻訳制御エレメント、これにより、該第一および第二の核酸が発現し、発現したCD8 α-鎖は該細胞の細胞膜と結合し、そして該細胞に対する宿主免疫反応は、CD8 α-鎖をコードする核酸を有さない細胞に対する免疫反応と比較すると低下する。
【請求項13】
遺伝子治療送達システムが、ウイルス性発現ベクター、プラスミドおよび裸の核酸発現ベクターからなる群から選択される、請求項12の方法。
【請求項14】
ウイルス性発現ベクターが、組み換えアデノウイルス、組み換えレトロウイルス、組み換えアデノ-随伴ウイルス、および組み換えヘルペスウイルスからなる群から選択される、請求項13の方法。
【請求項15】
目的の治療遺伝子は、ヘモグロビン-β、GATA-結合タンパク質、d-アミノエブリネートシンターゼ、グルコース-6-ホスフェート-デヒドロゲナーゼ、凝固VIII因子、凝固XI因子、嚢胞性繊維症膜貫通コンダクタンスレギュレーター、オルニチンカルバモイルトランスフェラーゼ、α-L-イズロニダーゼ、イズロネート-2-スルファターゼ、β-グルコシダーゼ、α-ガラクトシダーゼ、ガラクトシルセラミダーゼ、酸α-グルコシダーゼ、ヘキサミダーゼ A、フェニルアラニンヒドロキシラーゼ、コラーゲンIV型、α5、ブルーム病遺伝子産物、および低密度リポタンパク質レセプターからなる群から選択される、請求項12の方法。
【請求項16】
CD8 α-鎖をコードする核酸が、図11に記載の配列を含む、請求項12の方法。
【請求項17】
CD8 α-鎖をコードする核酸が、図10に記載の配列を有するタンパク質をコードする、請求項12の方法。
【図1−1】


【図1−2】

【図1−3】

【図1−4】

【図1−5】

【図1−6】

【図1−7】

【図1−8】

【図1−9】

【図1−10】

【図1−11】

【図1−12】

【図1−13】

【図1−14】

【図1−15】

【図1−16】

【図1−17】

【図2−1】

【図2−2】

【図3】
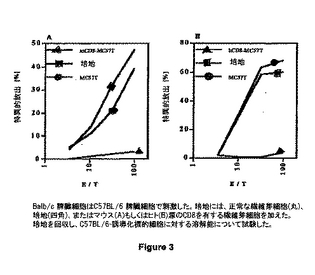
【図4】
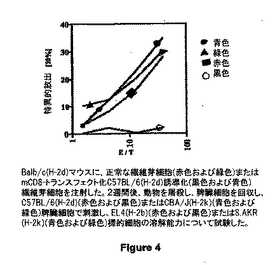
【図5】
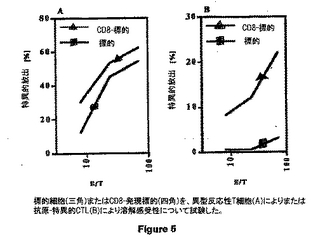
【図6】
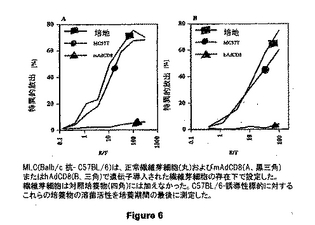
【図7】
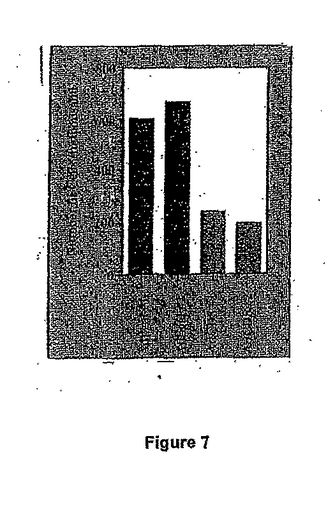
【図8】
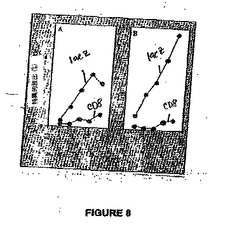
【図9】
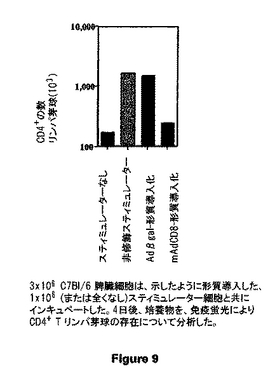
【図10−1】
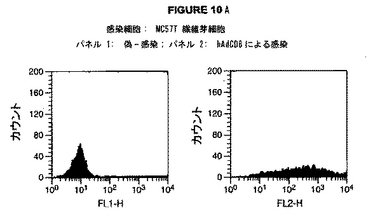
【図10−2】
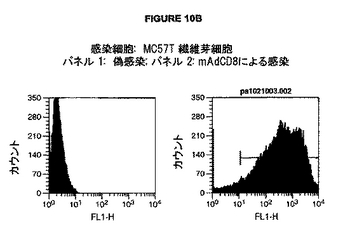
【図10−3】
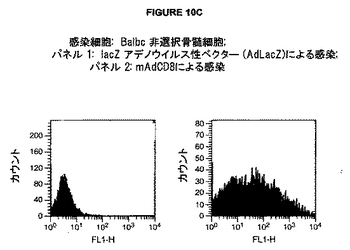
【図10−4】
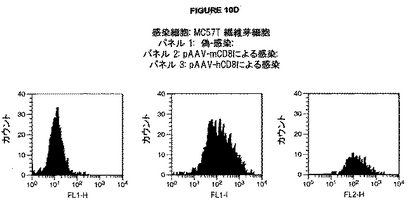
【図11】
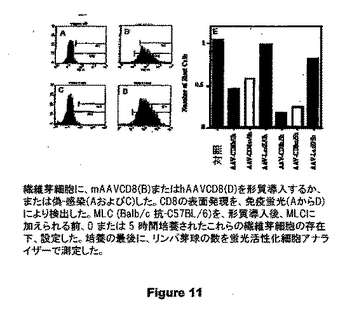
【図12】
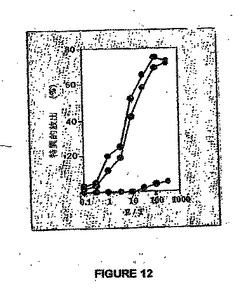
【図13】
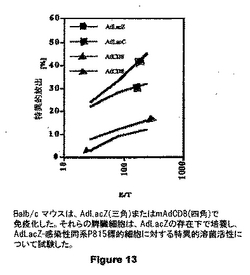
【図14】
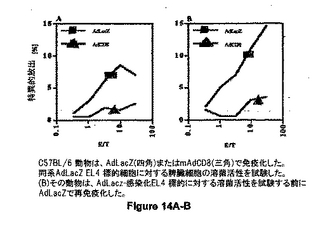
【図15】

【図16】

【図17−1】

【図17−2】

【図18−1】

【図18−2】

【図19−1】

【図19−2】

【図20−1】

【図20−2】

【図21−1】

【図21−2】

【図22】

【図1−2】
【図1−3】
【図1−4】
【図1−5】
【図1−6】
【図1−7】
【図1−8】
【図1−9】
【図1−10】
【図1−11】
【図1−12】
【図1−13】
【図1−14】
【図1−15】
【図1−16】
【図1−17】
【図2−1】
【図2−2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10−1】
【図10−2】
【図10−3】
【図10−4】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17−1】
【図17−2】
【図18−1】
【図18−2】
【図19−1】
【図19−2】
【図20−1】
【図20−2】
【図21−1】
【図21−2】
【図22】
【公表番号】特表2006−520604(P2006−520604A)
【公表日】平成18年9月14日(2006.9.14)
【国際特許分類】
【出願番号】特願2006−507410(P2006−507410)
【出願日】平成16年3月19日(2004.3.19)
【国際出願番号】PCT/US2004/008567
【国際公開番号】WO2004/083404
【国際公開日】平成16年9月30日(2004.9.30)
【出願人】(505353250)アイソジェニス・インコーポレイテッド (3)
【氏名又は名称原語表記】ISOGENIS, INC.
【Fターム(参考)】
【公表日】平成18年9月14日(2006.9.14)
【国際特許分類】
【出願日】平成16年3月19日(2004.3.19)
【国際出願番号】PCT/US2004/008567
【国際公開番号】WO2004/083404
【国際公開日】平成16年9月30日(2004.9.30)
【出願人】(505353250)アイソジェニス・インコーポレイテッド (3)
【氏名又は名称原語表記】ISOGENIS, INC.
【Fターム(参考)】
[ Back to top ]