
CDDOメチルエステルの新規形態
【課題】CDDOメチルエステルの新規形態を提供する。
【解決手段】トリテルペノイド化合物であるメチル2−シアノ−3,12−ジオキソレアナ−1,9(11)−ジエン−28−オエート(CDDOメチルエステル)は、例えば飽和メタノール溶液から製造することができる、非結晶質のガラス質固体形態及び非水和結晶質形態を有する。そのガラス質形態は、非水和結晶質形態と比べ、高い生物学的利用能を示す。CDDOメチルエステルの各形態は、一般に固体剤形で、一般に炎症に関連した種々の疾患状態の治療に使用される優れた候補物質である。
【解決手段】トリテルペノイド化合物であるメチル2−シアノ−3,12−ジオキソレアナ−1,9(11)−ジエン−28−オエート(CDDOメチルエステル)は、例えば飽和メタノール溶液から製造することができる、非結晶質のガラス質固体形態及び非水和結晶質形態を有する。そのガラス質形態は、非水和結晶質形態と比べ、高い生物学的利用能を示す。CDDOメチルエステルの各形態は、一般に固体剤形で、一般に炎症に関連した種々の疾患状態の治療に使用される優れた候補物質である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、CDDOメチルエステルの新規形態に関する。
【背景技術】
【0002】
トリテルペノイドは、スクアレンの環化によって植物で生合成されている。これらの天然分子は、医薬用途のための候補物質であるが、比較的弱い生物活性を示す。従って、化学者は、高い効力を有する類似体を合成しようとしている(Hondaら,1997&1998)。
【0003】
いくつかの合成類似体は、IFN−γ又はLPSによって刺激されたマクロファージにおけるiNOS及びCOX−2のde novo生成を抑制することが報告されている(Suhら,1998;Hondaら,2002)。他の合成トリテルペノイド、2−シアノ−3,12−ジオキソレアナ−1,9(11)−ジエン−28−オエート(CDDO)は、抗炎症活性及び抗増殖活性を示す(Hondaら,1998&2000)。
【0004】
CDDOのメチルエステル(これは、メチル2−シアノ−3,12−ジオキソレアナ−1,9(11)−ジエン−28−オエート(CDDOメチルエステル)である)の研究により、Boreら(2002)は結晶構造を決定した。水和した形態において、水は、特定の結晶充填及び構造を発生させる相互作用を調整する。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Honda, et al. , Bioorganic & Medicinal Chemistry Letters 1997, 7, 1623
【非特許文献2】Honda, et al , loc. cit. 1998, 8, 2711-14
【非特許文献3】Honda, et al., Bioorganic & Medicinal Chemistry Letters 2002, 12, 1027
【非特許文献4】Honda, et al., J. Med. Chem. 2000, 43, 1866
【非特許文献5】Bore, et al., Acta Cryst. 2002, C58, o199-o200
【発明の概要】
【0006】
本発明の1つの実施形態において、CDDOメチルエステルの非水和結晶形が提供される。非水和結晶形は、好ましくは、単位格子寸法a=14.2Å、b=14.2Å、及びc=81.6Åを有するP43212の空間群を有する。本発明は、(i)治療有効量のCDDOメチルエステルの非水和結晶形及び(ii)食用担体を含んで成る固体投与形態の医薬組成物も意図する。
【0007】
さらに、本発明は、図2Cに示されるように、約13.5°2θにおいてハローピークを有する粉末X線回折パターン、及びガラス転移温度(Tg)を有するCDDOメチルエステルのガラス質固体形態においても具体化される。特定の実施形態において、Tgは約120℃〜135℃である。他の実施形態において、Tgは約125℃〜約130℃である。CDDOメチルエステルのガラス質固体形態は、図28と同様の約5Å〜約20Åにおけるピークを有するPDFスペクトルを有しうる。
【0008】
さらに本発明は、(i)治療有効量のCDDOメチルエステルのガラス質固体形態、及び(ii)食用担体を含んで成る固体剤形の医薬組成物も提供する。これに関連して、本発明は、そのような医薬組成物を癌患者に投与することを含んで成る癌患者の治療法も意図する。本発明は、他の抗癌剤と組み合わせてCDDOメチルエステルのガラス質形態を投与することも意図する。例えば、抗癌剤はゲムシタビンであってよく、癌は膵臓癌であってよい。本発明は同様に、急性又は慢性の酸化性ストレス及び炎症に関係した疾患又は障害、特に、誘導型一酸化窒素シンターゼ(iNOS)又は誘導シクロオキシゲナーゼ(COX 2)の過剰発現を部分的に特徴とする疾患又は障害の治療法も含む。
【0009】
さらに、本発明は、表18に示されている特徴的なピークを有する粉末X線回折パターン及び図24に示されるDSCパターンを有するCDDOメチルエステルのジメタノール溶媒和形態にも関する。本発明により、ジメタノール溶媒和形態は、CDDOメチルエステルのガラス質固体形態を製造するための中間体として使用しうる。ジメタノール溶媒和形態を介したCDDOメチルエステルのガラス質固体形態の製造法は、CDDOメチルエステルのジメタノール溶媒和形態の調製及び該ジメタノール溶媒和形態の乾燥を含んで成る。
【0010】
他の実施形態によれば、本発明は、CDDOメチルエステルジメタノレートの結晶を成長させる方法に関し、該方法は、無水温メタノール中の精製CDDOメチルエステルの溶液を調製し、その温溶液を冷メタノールの容器に添加し、得られた結晶を濾過することを含んで成る。
【0011】
他の実施形態によれば、本発明は、(i)治療有効量のCDDOメチルエステル、及び(ii)ガラス形成剤である賦形剤を含んで成り、Tgを有する医薬組成物に関する。
【0012】
賦形剤は、例えば、(A)炭水化物、炭水化物誘導体又は炭水化物ポリマー、(B)合成有機ポリマー、(C)有機酸塩、(D)タンパク質、ポリペプチド又はペプチド及び(E)高分子量多糖から成る群より選択しうる。合成有機ポリマー賦形剤の種類の例は、ヒドロキシプロピルメチルセルロース、例えば、ヒドロキシプロピルメチルセルロースフタレートエステル、ポリ[1−(2−オキソ−1−ピロリジニル)エチレン又はそのコポリマー、例えばPVP/VA、及びメタクリル酸コポリマー、例えばメタクリル酸−エチルアクリレートコポリマー(1:1)である。
【0013】
これに関する他の賦形剤は、1−ビニル−2−ピロリドン−ビニルアセテートコポリマー(3:2)であるコポビドンである。
【図面の簡単な説明】
【0014】
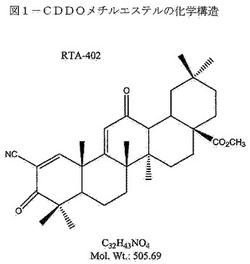
【図1】図1は、CDDOメチルエステルの化学構造を示す。
【図2】図2は、形態A(上)及び形態B(下)のXRPDパターンを示す。上から下へ:非微粉化形態A、微粉化形態A、及び形態B。
【図3】図3は、CDDOメチルエステル(形態A)のDSC及びTG曲線を示す。
【図4−1】図4は、形態A(非微粉化)のホットステージ分析(hot stage analysis)を示す。
【図4−2】図4は、形態A(非微粉化)のホットステージ分析(hot stage analysis)を示す。
【図5】図5は、形態A(非微粉化)の動的蒸気収着等温線を示す。
【図6−1】図6は、形態A(非微粉化)のSEM画像を示す。
【図6−2】図6は、形態A(非微粉化)のSEM画像を示す。
【図7】図7は、195℃でのストレスの前(上)及び後(下)の形態Aを示す。
【図8】図8は、形態A(非微粉化)のNMRスペクトルを示す。
【図9】図9は、形態BのCDDOメチルエステルのMDSC曲線を示す。
【図10】図10は、60分間の200℃/周囲相対湿度での熱ストレスの前(上)及び後(下)の形態BのCDDOメチルエステルを示す。
【図11】図11は、形態BのCDDOメチルエステルのNMRスペクトルを示す。
【図12】図12は、標識を有する単一の形態A分子のORTEP図を示す。原子は、50%確率の異方性熱振動楕円体で示されている。
【図13】図13は、形態A結晶の非対称単位の含有物のORTEP図を示す。原子は、50%確率の異方性熱振動楕円体で示されている。
【図14】図14は、結晶a軸に沿って見た形態A結晶の充填図を示す。
【図15】図15は、結晶b軸に沿って見た形態A結晶の充填図を示す。
【図16】図16は、結晶c軸に沿って見た形態A結晶の充填図を示す。
【図17】図17は、形態Aの計算による粉末X線パターンを示す。
【図18】図18は、形態Aの実験から得られたXRPDを示す。
【図19】図19は、形態AのCDDOメチルエステルの計算及び実験によるXRPDパターンの比較を示す。
【図20】図20は、カニクイザルへの4.1mg/kg経口投与後の、形態A及び形態Bの曲線下面積の代表的プロットを示す。各データ点は、8匹の動物におけるCDDOメチルエステルの平均血漿濃度を示す。エラーバーは、試料採取母集団内の標準偏差を示す。
【図21】図21は、動物#505M(上のグラフ)及び動物#507F(下のグラフ)における、 CDDOメチルエステルの形態B対形態Aの血漿濃度の比較を示す。
【図22】図22は、動物#508F(上のグラフ)及び動物#502M(下のグラフ)における、CDDOメチルエステルの形態B対形態Aの血漿濃度の比較を示す。
【図23】図23は、CDDOメチルエステルヘミベンゼン溶媒和物のサーモグラムを示す。
【図24】図24は、CDDOメチルエステルジメタノール溶媒和物のサーモグラムを示す。
【図25】図25は、CDDOメチルエステルジメタノール溶媒和物に関するTGIRデータを示す。
【図26】図26は、TGIR分析(140℃まで)の前(上)及び後(下)の、CDDOメチルエステルジメタノール溶媒和物のXRPDパターンを示す。
【図27】図27は、形態A対形態BのPDFデータのオーバーレイ表示である。約5Å〜約20Åにおいて、部分的にオーダーが類似している。
【図28】図28は、形態Bの種々の調製物についてのX線非結晶パターンのオーバーレイ表示であり、調製物間の実質的均一性を示す。
【図29】図29は、空間群P43212(#96)の概略図である。
【図30】図30は、雄カニクイザルへのCDDOメチルエステルカプセルの単一経口投与後の、CDDOメチルエステルの平均血中濃度を示す(段階2及び3)。
【発明を実施するための形態】
【0015】
前記のように、iNOS活性の抑制剤としての、特にNO生成の阻害における、トリテルペノイドの研究は、CDDO及びCDDOメチルエステルの高い有効性を示している(IC50<1nMレベル)。Hondaら(2000)参照のこと。これらの研究は、可溶化CDDOメチルエステルに焦点を当てており、CDDOメチルエステルの固体についてはほとんど特性評価していない。Boreら(2002)の研究は、CDDOメチルエステルの単一溶媒和結晶形の構造(トリテルペノイドについて最初に公表された構造)を解明した。
【0016】
図1(化学構造)及び図12(ORTEP図)に示されているCDDOメチルエステルの治療における潜在能力を理解するために、本発明者らは、好適な薬物動態を有する医薬製剤の開発に有益な、より高い水溶性及び化学安定性等の特性を有する該化合物の他の形態について研究した。その結果、本発明者らは、かかる特性を有し、従ってそれ自体で薬剤開発の候補物質となる、Boreら(2002)によって解明された結晶形とは異なる、CDDOメチルエステルの2つの形態を発見した。
【0017】
本発明のCDDOメチルエステルの「形態A」は、溶媒和されず(非水和)、図29に示される空間群P43212(no.96)、単位格子寸法a=14.2Å、b=14.2Å及びc=81.6Åを有する特有の結晶構造を特徴とし、かつ、図14〜16に示される充填構造を特徴とし、3個の分子が結晶b軸に沿って螺旋状に充填されている。下記の表10は、結晶データ収集パラメータと共に、形態Aの追加の結晶データを列記している。
【0018】
本発明の他の「形態B」は、単一相であるが、形態Aのような明確な結晶構造を欠いている。むしろ、形態Bは、形態Aとは異なる粉末X線回折(XRPD)スペクトルによって象徴される(特に図2参照)。さらに、形態Bは、形態Aより驚くほど高い生物学的利用能を示す(実施例7参照)。
【0019】
CDDOメチルエステルの合成方法は既に公表されている。米国特許第6,326,507号明細書、Hondaら(1998)、及びHondaら(2000)参照のこと。本発明者らは、CDDOメチルエステルの形態A及び形態Bが共に、下記の表3〜5に列記されているものによって例示される種々の化合物溶液から容易に調製されることを発見した。特に形態Bは、MTBE、THF、トルエン又は酢酸エチル中での急速蒸発又は低速蒸発によって調製できる。同様に、形態Aもエタノール又はメタノール中のCDDOメチルエステル溶液の急速蒸発、低速蒸発又は低速冷却によって調製できる。アセトン中のCDDOメチルエステルの調製物は、急速蒸発を使用して形態Aを生じうるか、又は低速蒸発を使用して形態Bを生じうる。別の製造法は下記に示され、その表も含まれる。
【0020】
形態Bは、明確な結晶構造を有さないので、形態Aに特徴的な明確なXRPDピークを欠き、それに代わって、全般的な「ハロー」XRPDパターンを特徴とする。特に、非結晶質形態Bは、そのXRPDパターンが3つ又はそれ以下の主要な回折ハローを示すので、「X線非晶質」固体に分類される(例えば、図10参照)。この分類において、形態Bは「ガラス質」材料である。PDFによって示されるように、最近接原子−原子相互作用は、結晶質形態Aにおいて観察されるものと一致するが、明白な長距離秩序が存在しないので、平均単位格子の概念が適用されない。
【0021】
従って、形態Aと異なり、形態Bの試料は、長距離の分子相関を示さない(即ち、約20Å以上において)(図27参照)。さらに、形態Bの試料の熱分析は、ガラス転移温度(Tg)を示す。これに対して、規則性のないナノ結晶材料はTgを示さないが、その代わりに融解温度(Tm)(それより高い温度で結晶構造が液体になる)を示すのみである。
【0022】
本明細書は、形態Bを製造するために使用できるCDDOメチルエステルジメタノール溶媒和物形態の特性を明らかにする(実施例9参照)。CDDOメチルエステルヘミベンゼネート(hemibenzenate)形態の特性も本明細書で明らかにされる(実施例8参照)。
【0023】
他の結晶材料の微粉化はXRPDスペクトルに影響を及ぼすことが見出されているが、微粉化形態AのXRPD分析は、非微粉化形態Aに類似したスペクトルを生じる。非微粉化形態A、微粉化形態A、及び形態BのCDDOメチルエステルを並列して比較した図2を参照のこと。
【0024】
種々の特性評価の方法を併用して、CDDOメチルエステルの形態A及び形態Bを、互いに、またCDDOメチルエステルの他の形態と区別することができる。この目的に好適な方法の例は、固体状態核磁気共鳴(NMR)、粉末X線回折、X線結晶構造解析、示差走査熱量測定(DSC)、動的蒸気収着/脱離(DVS)、カールフィッシャー分析(KF)、ホットステージ顕微鏡法、変調示差走査熱量測定、FT−IR、及びラマン分光法である。
【0025】
特に、XRPD及びDSCデータの分析によって、CDDOメチルエステルの形態A、形態B及びヘミベンゼネート形態を区別することができる。
【0026】
本発明のCDDOメチルエステル形態の特性は、前記のように特有であり、かつ、薬剤としての使用に有用である。例えば、形態B及び形態AのCDDOメチルエステルの生物学的利用能は、等用量のその2つの形態をゼラチンカプセルに入れて経口的に投与したサルで変化していた。実施例7参照のこと。さらに、新たに同定されたCDDOメチルエステル形態の安定性は、医薬組成物の製造に有用である。CDDOメチルエステルの形態A及び形態Bを、互いに、またCDDOメチルエステルの他の形態から区別するのと同様に、以下に詳細に記載するように「X線非晶質」特性を保持しているCDDOメチルエステル分散物を、XRPD及びDSC分析等の種々の方法によって、結晶質形態AのCDDOメチルエステルを含む分散物から区別することができる。即ち、結晶質CDDOメチルエステルの形態Aを含む分散物は、一般に、純粋形態AのCDDOメチルエステルに特徴的な明確なピークを示す(特に、約13.35及び8.78(°2θ)において生じるピーク)(例えば、下記の表17参照)。
【0027】
本発明のCDDOメチルエステルポリマー賦形剤分散物の特性は、特有であり、かつ、薬剤としての使用に有用である。例えば、付加的不活性添加剤と共に処方した、選択したCDDOメチルエステル分散物の生物学的利用能は、サルに等用量の該分散物をゼラチンカプセルに入れて与えた場合、変化した。下記の実施例7、研究段階2及び3を参照のこと。いくつかの例において、CDDOメチルポリマー賦形剤分散物を含む処方物は、CDDOメチルエステルの純粋形態Bから製造された処方物と比較した場合でも、生物学的利用能において、驚くべき増加を生じた。
【0028】
医薬固形物における、多型を包含する多様な形態の存在は、例えば、Cuiら(Int’l J. Pharmaceutics,2007,339,3−18)によって以前に記載されている。化合物の結晶形及び非晶形は、異なる物理的及び化学的特性を示しうる。例えば、非晶形は結晶形と比較して、より高い溶解性を有しうる。しかし、あらゆる化合物はこの点で特有であり、非晶質材料が結晶状態と異なる度合は個別に調査すべきであり、演繹的に予測することはできない。さらに、いくつかの非晶質材料は再結晶しやすい。
【0029】
本発明に関して、データ収集における変動が、多くの要因によって生じうる。従って、本明細書は、CDDOメチルエステル形態を説明するために使用されるデータにおける変動を示すために、「約」又は「ほぼ」という用語を使用する。例えば、融解温度は、計器装備又は条件によって変化しうる。測定の精度に関して、USP<891>は、「融解の場合、「開始」及び「ピーク」温度の両方は、客観的及び再現的に、多くの場合十分の数度以内で測定されうる」と記載している。実際の実験は、このことが材料のTgの測定については当てはまらないことを示す。Tgは、多くの要因に依存する:試料の調製方法;試料の熱履歴(緩和);Tg前に蒸発するか又はしない残留溶媒;装置;試料調製物(試料質量、粒度、充填、希釈剤);Tgの測定に使用されるパラメータ(特に走査速度);Tgの位置を決定するために使用されるパラメータ(開始温度、中間点温度、変曲点温度、又はオフセット温度);緩和による吸熱がTgにおいて存在するかどうか;及び他の要因。いくつかの要因はTgを低下させる(残留水/溶媒による可塑化)が、他の要素はTgを上昇させ(より速い走査速度、緩和)、10〜15℃も上昇させる場合がある。Tgにおける熱容量(ΔCp)の変化は、Zhouら、J.Pharmaceutical Sciences 91:1863−72(2002)によって報告されているように重要である。
【0030】
本明細書は、それらの「特徴的」ピークに関する種々のパターンについて記載する。そのようなピークの集合又は群は、個々の装置及び実験条件にそれぞれ起因しうる不確実性の範囲内で、所定の多型形態に特有である。
【0031】
各結晶形について、5つの特徴的ピークの群が下記の表17〜19に示されている。一般的な変動は±0.1°2θであるが、いくつかの実験において、ピーク位置は±0.2°2θ又はそれ以上まで変化しうる。
【表1】

【表2】
【表3】
【0032】
ガラス質材料(形態B)のXRPDパターンは、約13.5°2θにおいて幅広いハローピークを示し、これは形態Bに特徴的であると考えられる。他のハローは明確ではなく、このパターンの形/位置は、装置及び実験条件の関数として変化しうる。この幅広いピーク位置の変化は、それぞれの結晶形の特徴的ピークの変化より大きい。特に、形態Bの幅広いピークについて±1°2θまでの変動性が、特定の装置において予測できる。
【0033】
CDDOメチルエステル賦形剤分散物として製造されたガラス質材料のXRPDパターンも、一般に約13.5°2θを中心とする幅広いハローピークを示す。これらの材料は、変調示差走査熱量測定(mDSC)によってTgも示す。純粋形態BのCDDOメチルエステルの試料と同様に、賦形剤分散物のXRPDパターンの形及び位置は、使用された装置、実験条件、及び分散物を製造するのに使用された特定賦形剤の関数として変化しうる。
【0034】
本発明は、さらに、癌性疾患及び中枢神経系を冒す種々の病状等の炎症関連疾患を治療するための、CDDOメチルエステルの形態A、形態B及びガラス質XRPD−非晶質賦形剤分散物のそれぞれの使用にも関する。本発明によれば、これらの疾患の治療は、それを必要とする患者に、本明細書に列挙した新規CDDOメチルエステル形態の有効量を投与することを含んで成る。これらの化合物は、癌、アルツハイマー病(AD)、パーキンソン病(PD)、多発性硬化症(MS)、筋委縮性側索硬化症(ALS)、慢性関節リウマチ(RA)及び他の自己免疫疾患、炎症性腸疾患、並びに酸化窒素又はプロスタグランジンの過剰生成に関係した他の病理学的症状の病因に関連した炎症を改善又は予防するのに有用である。
【0035】
前記のように、シクロオキシゲナーゼ−2(COX−2)又は誘導型一酸化窒素シンターゼ(iNOS)の異常又は過剰発現が、結腸における発癌等の多くの疾患過程の病因に関与している。CDDOメチルエステル等のトリテルペノイドのいくつかの合成類似体が、iNOSの発現を抑制することが報告されている。関連した研究は、IFN−γ又はLPSによって刺激されたマクロファージにおいてiNOS及びCOX−2の発現がトリテルペノイドにより抑制されたことを示している(Suhら、1998;Hondaら、2002)。従って、CDDOメチルエステル形態を投与する治療は、iNOS及びCOX−2の抑制に作用すると考えられる。
【0036】
COX−2の遺伝子の過剰発現は、結腸発癌における早期かつ中心的事象である(Prescott及びWhite,1996;Duboisら、1996)。APC(大腸腺腫性ポリポーシス)遺伝子における欠損を有するマウスは、早期に多数の腸ポリープを発生させ、COX−2酵素レベルの顕著な増加がこれらのポリープにおいて見出されている。これらの動物を用いた知見は、多くのヒト原発性結腸癌及び結腸癌細胞系においてCOX−2 mRNA及びタンパク質が高レベルとなる知見と相互に関係しており(Prescott及びWhite,1996)、このCOX−2の増加が、通常は前新生物細胞の死に導くアポトーシスの抑制を生じさせると考えられる(Tsujii及びDuBois,1996)。腸腫瘍発生に対するCOX−2の機能的関連性は、COX−2遺伝子のノックアウトによって示されている(Oshimaら、1996)。このノックアウトを有するマウスを、APC遺伝子において損傷を有するポリープ形成マウスと交配させたところ、COX−2ノックアウトは、子孫におけるポリープの数を劇的に減少させた。さらに、選択的COX−2阻害剤又は非選択的COX−1/COX−2阻害剤を実験動物に処置することは、腸癌の化学的予防に有効な方法であることが報告されている(Marnett,1992;Oshimaら、1996;Boolbolら、1996;Reddyら、1996;Shengら、1997)。発癌におけるiNOSの役割に関して、NOは強力な突然変異原であり(Tamir及びTannebaum,1996)、酸化窒素はCOX−2を活性化することもできる(Salveminiら、1993,1994)ことが明らかである。発癌物質アゾキシメタンによって誘発されたラット結腸腫瘍においても、iNOSの顕著な増加が見られる(Takahashiら、1997)。同様に、ヒト腫瘍におけるiNOSの過剰発現は、悪い予後因子として報告されている(例えば、Ekemekciogluら、2006)。
【0037】
アンギオテンシンIIによって誘発されるような、炎症シグナル経路及び他の疾患関連シグナル経路は、スーパーオキシド、過酸化水素、酸化窒素及びパーオキシナイトライト等の活性酸素又は窒素種(RONS)の過剰生成を刺激する場合が多い。CDDOメチルエステルは、抗酸化活性の有効な誘発物質、及び種々の細胞型における炎症過程の有効な阻害物質であることが示されている(Dinkova−Kostovaら、2005;Libyら、2006;Ahmadら、2006;Shishodiaら、2006)。感染、外傷、火傷及び化学暴露等の種々の原因による重篤な急性炎症は、生命にかかわり、肝不全、腎不全、呼吸不全又は心不全を生じる場合がある。慢性炎症及び関連酸化性ストレスは、下記に挙げられる多くの重大疾患の病状の一因となる:自己免疫疾患(例えば、慢性関節リウマチ、狼瘡、乾癬及び多発性硬化症)、心血管疾患(例えば、アテローム性動脈硬化症及び心不全)、糖尿病(I型及びII型)、呼吸器疾患(例えば、慢性閉塞性肺疾患及び喘息)、慢性腎臓病、腎不全、肝不全、及び疼痛症候群(例えば、神経障害性疼痛、線維筋痛症及び片頭痛)。さらに、トリテルペノイドは、マクロファージにおけるHIV−1の複製を阻害することが示されており(Vazquezら、2005)、従って、ウイルス性疾患、特に、器官又は組織の炎症によって重大な病的状態を生じるウイルス性疾患(例えば、ウイルス性肝炎、インフルエンザ、単純ヘルペス)の治療に有用になりうる。
【0038】
MSは、中枢神経系の炎症状態であることが既知である(Williams,Ulvestad及びHickey,1994;Merrill及びBeneviste,1996;Genain及びNauser,1997)。炎症、酸化又は免疫機構が、MS、AD、PD及びALSの病因に関与していると考えられる(Bagasraら、1995;Griffinら、1995;McGeer及びMcGeer,1995;Goodら、1996;Simonian及びCoyle,1996;Kaltschmidtら、1997)。反応性星状細胞及び活性化小グリア細胞は共に、NDD/NIDの原因に関連付けられている。NO及びプロスタグランジンの両方を、それぞれの酵素、iNOS及びCOX−2の生成物として合成する細胞として、小グリア細胞が特に重要視されている。これらの酵素のde novo生成は、炎症サイトカイン、例えば、インターフェロンガンマ又はインターロイキン−1によって引き起こされうる。次に、NOの過剰生成は、多くの器官の細胞及び組織(神経系のニューロン及び乏突起神経膠芽細胞等)における炎症カスケード及び/又は酸化損傷を生じ、その結果、AD及びMS、並びにおそらくはPD及びALSの症状発現を生じうる(Coyle及びPuttfarcken,1993;Goodwinら、1995;Beal,1996;Goodら、1996;Merrill及びBenvenist,1996;Simonian及びCoyle,1996;Vodovotzら、1996)。疫学的データは、アラキドネートからのプロスタグランジンの合成を阻害するNSAIDの慢性使用が、AD発症のリスクを顕著に減少させることを示している(McGeerら、1996;Stewartら、1997)。従って、CDDOメチルエステルの形態A及び形態Bは、NO及びプロスタグランジンの生成を阻害する薬剤として、NDDを治療又は予防する治療的方法に有用であると考えうる。
【0039】
前記のように、種々の前臨床研究において、CDDO−Meは、COX−2及びiNOS(炎症及び発癌の両方に関係した酵素)の発現を阻害する能力を示している。CDDO−Meは、核性因子−カッパB(NF−κB)、並びに転写3のシグナルトランスデューサー及びアクチベーター(STAT3)(炎症、腫瘍進行及び腫瘍の治療耐性に関連した転写因子)の活性を阻害することも示されている。初期の研究は、CDDO−Meが多くの癌細胞系の成長を阻害することを明示しており、NCI−60腫瘍細胞株パネルにおけるCDDO−Meの平均IC50値は約35nMであった。生体内試験において、CDDO−Meが、げっ歯類にインプラントしたヒト腫瘍細胞系又はげっ歯類にインプラントした同系癌細胞系によって形成された腫瘍の成長を効果的に阻害することが確認された(表16)。これらの試験に使用された投与量は、種、株及び投与方法によって変わり、一般に10〜100mg/kg/日であった。
【0040】
以下に詳述する研究は、癌疾患の患者におけるCDDOメチルエステルの有利な作用を示すヒトデータを与える。実施例10参照のこと。
【0041】
前記に照らして、本発明は、CDDOメチルエステル形態を含有する安定な徐放性放出剤形を包含する。本発明では、1日1回の投与で、遅延放出又はパルス放出の形態による剤形が可能となることから、薬物動態性能を薬力学的要求に適合させることによって治療法を最適化することができる。
【0042】
形態B、形態A、及びCDDOメチルエステルの賦形剤分散物を含有する処方物はいずれも経口投与しうる。活性化合物は、該化合物を不活性化しうる酸の作用及び他の自然条件から該化合物を保護する材料でコーティングしてもよい。他の投与法、例えば、局所、皮下、静脈内及び腹腔内投与も本発明の一部である。
【0043】
治療用化合物を投与するために、その不活性化を防止する材料を用いて該化合物をコーティングするか、又は該材料と共に同時投与することが必要な場合がある。即ち、形態B又は形態AのCDDOメチルエステルは、適切な担体、例えばリポソームで、又は希釈剤で患者に投与してもよい。医薬的に許容される希釈剤は、生理食塩水及び水性緩衝液を包含する。リポソームは、水中油中水型CGFエマルジョンだけでなく通常のリポソームも包含する。例えばStrejanら、J.Neuroimmunol.7:27(1984)参照のこと。
【0044】
治療用化合物は、医薬組成物の調製用の不活性希釈剤、添加剤又は食用担体と共に、経口投与することができる。この目的のために、本発明の治療用化合物を、他の成分と共に、硬質又は軟質ゼラチンカプセルに封入するか、錠剤に圧縮するか、又は患者の食事に直接入れてもよい。経口治療投与のために、形態A又は形態Bを、賦形剤と混合し、飲み込み可能な錠剤、口腔錠剤、トローチ剤、カプセル剤、エリキシル剤、懸濁剤、シロップ剤、カシェ剤等の形態で使用できる。同様に、本発明の賦形剤分散物を、種々の剤形(形態A及び形態Bに関して本明細書に記載した剤形を包含する)で与えることができる。組成物及び調製物における治療用化合物の割合は、活性剤の好適投与を達成するために、慣例に従って変化させることができる。
【0045】
さらに、本発明は、有効量の形態BのCDDOメチルエステル又は形態Aを、1以上の非毒性の医薬的に許容される担体及び/又は希釈剤並びに所望であれば他の活性成分と共に含んで成る医薬組成物にも関する。前記のように、活性化合物は、形態B又は形態Aから出発して、均質な賦形剤分散物として製造することができる。そのようなCDDOメチルエステル賦形剤分散物は、固溶体であり、分子レベルにおいて均質分散物とみなすことができる。有利なことに、そのような分散物を、他の医薬的に許容される添加剤と共に処方して、活性化合物を安定化させることができ、生物学的利用能をさらに向上させることも場合により可能である。
【0046】
賦形剤分散物としてCDDOメチルエステルを処方する場合において、分散物用の賦形剤の選択は、賦形剤が優れた「ガラス形成剤」であり、医薬的に許容されるという両基準に依存する。より一般的には、賦形剤は、一般的な周囲温度貯蔵条件より高いTgを与えることによって分散物を安定化させる安定で均質なガラス質マトリックスを形成すべきである。これに関する別の基準は、分散物に使用される賦形剤が、所望の機能特性を与えるように最終処方物に使用できる他の添加剤、例えば、結合剤、充填剤、潤滑剤、グリダント(glidant滑剤)等と化学的に適合性を有するべきであるという基準である。
【0047】
これらの基準を満たすために、賦形剤は、本発明によって、適切には高いTg値を特徴とする下記のような多くの化合物から選択できる:(A)炭水化物、炭水化物誘導体及び炭水化物ポリマー、(B)合成有機ポリマー、(C)有機酸塩、(D)タンパク質、ポリペプチド及びペプチド、並びに(E)高分子量多糖、例えば、ヘパリン(これは硫酸化多糖である)及びヒアルロン酸、ムコ多糖。
【0048】
種類(A)の例は下記の物質である:セルロース誘導体、例えば、ヒドロキシプロピルセルロース(HPC)、ヒドロキシプロピルメチルセルロース(HPMC)及びエチルセルロース;多糖、例えば、ラフィノース、マルトトリオース(maltotriose)、スタキオース、デキストリン(特にマルトデキストリン及びシクロデキストリン等)、デキストラン及び可溶性デンプン;アルジトール、例えば、マンニトール、キシリトール及びソルビトール;並びに、二糖、例えば、ラクトース、トレハロース、マルトース及びスクロース。この種類の好ましい賦形剤は、ヒドロキシプロピルメチルセルロースフタル酸エステル(HPMC−P)である。
【0049】
種類(B)の例は、可変分子量の、ポリ[1−(2−オキソ−1−ピロリジニル)エチレン、a/k/aポビドン又はポリビニルピロリドン(PVP)及び関連コポリマー、例えばPVP/VAである。コポリマーのメタクリル酸ファミリー、例えばメタクリル酸コポリマーC型(USP/NF)もこの種類に包含される。
【0050】
種類(C)の例は、塩、例えば、乳酸、アスコルビン酸、マレイン酸、シュウ酸、マロン酸、リンゴ酸、コハク酸、クエン酸、グルコン酸及びグルタミン酸の、それぞれのナトリウム、カリウム、カルシウム及びマグネシウム塩である。即ち、これに関する代表的な塩は、クエン酸ナトリウム、乳酸ナトリウム、マレイン酸ナトリウム、グルコン酸マグネシウム及びアスコルビン酸ナトリウムである。
【0051】
種類(D)賦形剤の例は下記の物質である:ヒト血清アルブミン;ポリアミノ酸、例えば、ポリアラニン、ポリアルギニン、ポリグリシン及びポリグルタミン酸;カゼイン;コラーゲン;ゼラチン及び精製ゼラチンタンパク質;並びにある種の薬理学的活性化合物、例えばインスリン。
【0052】
前記のように、賦形剤は、医薬処方物のいくつかの物理的特性を変化させることができる。例えば、種々のポリマー賦形剤中の分散物は、処方物の測定されるTgを減少させうる。一般に、Tgは、含まれている材料の比率に基づいて加成性である。従って、非晶形Bより低いTg値を有するポリマーを使用した場合、分散物(混合物)の測定されるTgが低下すると予測される。さらに、湿分又は微量な残留有機溶媒が存在する場合が多く、これらも同様にTgを低下させる。固体CDDO−Me分散物を生成するために、賦形剤の最適選択は、一般に、実験により決定すべきである。例えば、賦形剤のポリエチレングリコール(PEG)ファミリー、例えばPEG6000を使用して、ガラス質XRPD−非晶質分散物を製造した場合、形態AのCDDOメチルエステルに関連した特徴的ピークを有する混合物が生じた。同様の結果が、ビタミンE−TPGS、d−アルファ−トコフェリル酸スクシネートをポリエチレングリコール1000でエステル化することによって製造された賦形剤を使用した場合、並びにエチレンオキシド−プロピレンオキシドコポリマー、例えばPluronic(登録商標)を使用した場合に得られた。下記の例が示すように、本発明によってCDDO−Meとの分散物を生成するために使用された特定のポリマー賦形剤は、純粋形態B薬剤物質と比較して、驚くべき経口生物学的利用能の向上を示す。
【0053】
医薬的に許容される賦形剤を使用してCDDO−Meの均質なガラス質X線非晶質分散物を製造ために方法を変えることができ、本明細書に示す例は、そのような分散物を製造するために噴霧乾燥を使用している。他の製造法を使用して、同等の特性及び有効性を有する本発明の分散物を製造できる。Repkaら,Hot−melt extrusion technology In:ENCLOPEDIA OF PHARMACEUTICAL TECHNOLOGY,2nded(Marcel Dekker,2002),pages203−06及びそれに引用されている文献を参照のこと。そのような他の方法は、溶媒蒸発及び押出、例えば熱溶融押出を包含するが、それらに限定されない。
【0054】
賦形剤の他に、他の添加剤を含有させることにより、活性成分の安定性を補助し、pHを調節し(即ち、緩衝剤)、分散性を向上させ、送達均一性を与えるのを補助し、医薬処方物に所望される他の特性を得ることができる。
【0055】
本発明の化合物又は組成物の投与量は、患者及び投与方法によって異なり、任意の有効量にすることができる。
【0056】
本発明の組成物を投与する所定の治療計画は、標準的及び日常的な前臨床及び臨床試験によって作成することができ、その詳細は、他の因子のうち治療適応症に応じて変わる。投与される活性剤の量は幅広い範囲で変化させてもよく、それによって、単位投与量において、患者体重に基づく1日当たりの薬理学的有効量を与えて、所望の効果を得ることができる。所望の投与量は、治療される症状によっても変化しうる。例えば、急性の癌の治療は、関節炎のような炎症性疾患の治療より有意に多い投与量を必要としうる。
【0057】
特に本発明の組成物は、単位投与量として投与され、好ましくは、1日に1〜3回、最も好ましくは1日に1回投与されて、所望の効果を与える。
【0058】
さらに、本発明の組成物は、2日おき、3日おき、4日おき、5日おき、6日おき、又は1週間に1回投与しうる。
【0059】
本発明の組成物は、患者の具体的なニーズに基づいて、単独で、又は他の薬剤と組み合わせて投与してもよい。特に、本発明の組成物は、治療計画の一部として、抗癌剤と共に投与しうる。例えば、膵臓癌等の癌の治療中に、CDDOメチルエステルを、ゲムシタビン、又は他の薬剤と共に投与できる。
【0060】
一般に、本発明の医薬組成物は、通常の材料及び方法、例えば、混合、調合等を使用して製造される。さらに、形態A又は形態Bを含有する薬剤は、好適な佐剤、担体、賦形剤及び安定剤等を包含するがそれらに限定されない他の成分も含有しうる。本発明の治療処方物は、好ましくは固体であるが、原則として液体、例えば懸濁液又は乳濁液でありうる。
【0061】
本発明により、経口維持投与量は、一般に、約0.1mg〜約1000mgであり、好ましくは1日に1回投与される。その投与量は、変えることができ、患者の体重に適合させることができる。一般的な投与量は、錠剤及びカプセル剤等の好ましい単位剤形を用いて約0.01mg/kg〜100mg/kgでありうる。
【実施例】
【0062】
下記の実施例は、例示するものにすぎず、本発明を限定するものではない。実施例に使用した材料及び方法を以下に概説する。
【0063】
a.材料
溶媒及び他の試薬は、販売業者から購入し、HPLCグレード又はACSグレードであった。
【0064】
b.実験方法
i.近似溶解度−溶媒添加法(Approximate Solubility--Solvent Addition Method)
計量した試料を、室温にて試験溶媒のアリコートで処理した。試験材料が完全に溶解したことを、目視検査によって判定した。溶解度は、完全に溶解させるために使用された合計溶媒に基づいて推定した。実溶解度は、溶媒のアリコートの使用が多すぎた場合か、又は遅い溶解速度によって、計算値より高くなりうる。溶解度は、実験中に溶解が生じなかった場合に「より低い」として表わされる。ただ1つのアリコートを添加して完全に溶解した場合は、溶解度は「より高い」として表わされる。
【0065】
ii.多型の選別
熱力学的及び動力学的結晶化法を共に使用した。これらの方法を以下により詳しく記載する。結晶化により固体試料を得た時点で、それらの形態について顕微鏡下で検査するか、又は肉眼で観察した。あらゆる結晶形について留意したが、粒度が小さいため、形態を判別できない固形物もあった。次に、固体試料をXRPDによって分析し、パターンを相互に比較して、新しい結晶形又は非晶形を同定した。
【0066】
(i)低温沈殿(CP)
種々の溶媒で温度を上昇させて溶液を調製した。次に、室温以下で0.2−μmのナイロン又はPTFEフィルターによりその溶液を濾過して、貧溶媒に添加した。その際、固形物が存在するか否かを確認した。固形物を確認できない場合、又は固形物の量がXRPD分析のために少なすぎると判断した場合は、そのバイアルを冷凍庫に入れた。得られた固形物を濾過によって分離し、分析前に乾燥させた。
【0067】
(ii)高速蒸発(FE)
種々の溶媒で溶液を調製し、溶解しやすくするため、アリコートの添加と添加の間に超音波処理した。目視検査で混合物が完全に溶解したと判断したら、0.2−μmナイロンフィルターでその溶液を濾過した。蓋をしていないバイアル中において、室温でその濾過した溶液を蒸発させた。生成した固形物を分離し、分析した。
【0068】
(iii)凍結乾燥(FD)
1,4−ジオキサン溶液を調製し、0.2−μmナイロンフィルターで濾過し、ドライアイスで凍結させた。続いて、その凍結試料をFTSシステムFlexi−Dryを使用して凍結乾燥した。凍結乾燥温度は制御しなかった。
【0069】
(iv)微粉化
材料の微粉化は流体エネルギーミル(fluid energy mill)で行うことができ、粒度を1〜20ミクロンに減少させることができる。これらの過程の詳しい記載は、PERRY‘S CHEMICAL ENGINEERS’ HANDBOOK、第7版(McGraw Hill,1998)に見出すことができる。
【0070】
(v)粉砕
ステンレス製ミルローターに固体試料を小金属球と共に投入した。試料に少量の水を添加した場合もあった(湿式粉砕)。次に、その試料をRetesh MM220型ミキサーミルで30Hzにおいて約20分間粉砕した。得られた固形物を分離し、分析した。
【0071】
(vi)低温粉砕
粉砕棒を有するステンレス製粉砕ジャーに固体試料を投入した。次に、その試料をSPEX Certiprep 6750型クライオミルで、15Hzにおいて、一定時間粉砕した。実験中、粉砕ジャーを液体窒素浴に浸した。その固形物を分離し、分析した。
【0072】
(vii)溶融/急冷
顕微鏡スライドガラスに固体試料を載せ、平らにした。次に、固形物が溶融するまで、スライドガラスを設定温度でホットプレートに載せた。溶融後、ホットプレートからスライドガラスを取り除き、冷たいカウンタートップに載せて、急速に冷却した。得られた固形物を窒素下にて乾燥し、分析した。
【0073】
(viii)回転蒸発
種々の溶媒で溶液を調製し、0.2−μmナイロンフィルターで濾過した。試料をロータリーエバポレーターに載せ、乾燥したら取り除いた。得られた固形物を分離し、分析した。
【0074】
(ix)低速蒸発(SE)
種々の溶媒で溶液を調製し、溶解しやすくするため、アリコートの添加と添加の間に超音波処理した。目視検査で混合物が完全に溶解したことを確認し、溶液を0.2−μmナイロンフィルターで濾過した。濾過した溶液を、小さい穴をあけたアルミ箔で覆ったバイアル中で、室温又は窒素下で蒸発させた。このようにして生成した固形物を分離し、分析した。
【0075】
(x)低速冷却(SC)
約60℃にて種々の溶媒で飽和溶液を調製し、温かいうちに、0.2−μmナイロンフィルターで濾過して、開放バイアルに入れた。そのバイアルを覆い、室温へゆっくり冷却した。固形物が存在するか否かを確認し、確認できない場合、又は固形物の量がXRPD分析のために少なすぎると判断された場合、バイアルを冷蔵庫に入れた。再び、固形物の有無を確認し、確認できない場合は、バイアルを冷凍庫に入れた。生成した固形物を濾過によって分離し、分析前に乾燥させた。
【0076】
(xi)スラリー実験
過剰の固形物が存在するように充分な固形物を所定の溶媒に添加して溶液を調製した。次に、混合物を密閉バイアル中にて室温で撹拌した。7日又は10日後に、固形物を真空濾過によって分離し、分析した。
【0077】
(xii)ストレス実験
種々の温度及び/又は相対湿度(RH)環境下で、計測の間、固形物にストレスを加えた。飽和塩溶液を含有する密閉室又はESPEC温度及び湿度室に試料を入れることによって、特定のRH値を得た。塩溶液は、ASTM規格の手順に従い選択し調製した。試料をストレス環境から取り出して、直ちにXRPDによって分析した。
【0078】
iii単結晶構造決定
(i)試料調製
約60℃でメタノール中のCDDOメチルエステルの飽和溶液を調製し、温かいうちに、0.2−μmフィルターで濾過して開放バイアルに入れた。バイアルを覆い、室温へゆっくり冷却した。錐体状の小塊が1日後に観察された。
【0079】
(ii)データ収集
2辺が約0.01x0.01mmの寸法を有するCDDO−Me(C32H43NO4)の無色の小板を、ランダムの配向のグラスファイバー上にマウントした。グラファイト結晶の入射ビームモノクロメータを備えたNonius KappaCCD回折計によってMo Kα線(λ=0.71073Å)により予備実験及びデータ収集を行った。精密化は、SHELX97(Sheldrick,1997)を使用してLINUX PCで行った。
【0080】
データ収集のための格子定数及び方位マトリックスは、2°<θ<22°の範囲で46742の反射を有するような設定角を使用して、最小二乗精密化(least−squares refinement)から得た。DENZO/SCALEPACK(Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307)による精密化したモザイシティ(mosaicity)は0.32°であり、優れた結晶品質を示した。空間群は、XPREPプログラム(Bruker, XPREP in SHELXTL v. 6.12., Bruker AXS Inc., Madison, WI(2002))によって決定した。条件h00 h=2n,00l l=4nの系統的存在、及びそれに続く最小二乗精密化から、空間群はP43212(no.96)と決定された。
【0081】
データは、150±1Kの温度で、44.43°の最大2θ値まで収集した。
【0082】
(iii)データ整理
フレームはDENZO−SMN(Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307)で積分した。合計46742の反射を収集し、そのうち9168が固有であった。ローレンツ偏光補正をデータに適用した。線吸収係数はMo Kα線について0.074mm−1である。SCALEPACK(Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307)により実験的吸収補正を適用した。透過率は0.9995〜0.9999であった。等価な反射の強度を平均化した。平均化のためのアグリーメントファクター(agreement factor)は、強度に基づいて9.3%であった。
【0083】
(iv)構造解析及び精密化
SHELXS97(Sheldrick, G. M. SHELX97, A PROGRAM FOR CRYSTAL STRUCTURE REFINEMENT, University of Gottingen, Germany(1997))による直接法によって構造を解析した。残っている原子を、次の差フーリエ合成で配置した。水素原子は精密化に含まれたが、水素原子が結合している原子上に拘束された。以下の関数を最小にすることによって、フルマトリックス最小二乗において構造を精密化した:
【数1】
【0084】
重みwは1/[σ2(Fo2)+(0.0176P)2+(0.0000P)]として定義され、ここでP=(Fo2+2Fc2)/3である。
【0085】
散乱因子は、「International Tables for Crystallography」(INTERNATIONAL TABLES FOR CRYSTALLOGRAPHY, Vol. C, Tables 4.2.6.8 and 6.1.1.4, Kluwer Academic Publishers:Dordrecht, The Netherlands(1992))から得た。精密化に使用した9168の反射のうち、Fo2>2σ(Fo2)を有する反射だけをRの計算に使用した。合計5421の反射を計算に使用した。精密化の最終サイクルは1024個の変数パラメータを含み、下記の非加重及び加重したアグリーメントファクターで収束した(最大パラメータシフトは、その推定標準偏差の<0.01倍であった):
【数2】
【0086】
単位重量の観測の標準偏差は1.05であった。最終の差フーリエにおける最高ピークは0.22e/Å3の高さであった。最小負ピークは−0.25e/Å3の高さであった。
【0087】
(v)計算した粉末X線回折(XRPD)パターン
計算XRPDパターンは、PowderCell 2.3(Kraus, W., and G. Nolze, POWDERCELL FOR WINDOWS VERSION 2.3, Federal Institute for Materials Research and Testing, Berlin(1999))、並びに単結晶データからの原子座標、空間群及び単位格子パラメータを使用して、Cu線について生成した。
【0088】
(vi)ORTEP図及び充填図
ORTEP図は、ORTEP III(Johnson, C. K. ORTEPIII, Report ORNL-6895, Oak Ridge National Laboratory, TN, U.S.A. 1996)(OPTEP-3 for Windows V1.05 ,. Farrugia, L.J., J. Appl. Cryst. 1997, 30, 565)を使用して作成した。原子は50%確率の異方性熱振動楕円体で表わされている。充填図は、CAMERON(Watkin, D. J.; Prout, C .K.; Pearce, L. J. Cameron, CHEMICAL CRYSTALLOGRAPHY LABORATORY, University of Oxford, Oxford, 1996)モデリングソフトウエアを使用して作成した。別の図及びBFDH形態予測は、Mercury1.4.1(Bruno, et al., Acta Crystallogr. 2002, B58, 389)を使用して生成した。
【0089】
c.計測方法
i.示差走査熱量測定(DSC)
TA示差走査熱量計2920又はQ1000で分析を行った。該熱量計は、インジウムを標準物質として使用して較正した。試料を、圧着蓋(crimped lid)形状を有する標準アルミニウムDSC皿に入れ、重量を正確に記録した。試料セルを25℃で平衡にし、窒素パージ下にて10℃/分の速度で最終温度250℃まで加熱した。
【0090】
ii.動的蒸気収着/脱離(DVS)
湿分収着/脱離データを、VTI SGA−100蒸気収着分析器で収集した。収着及び脱離データを、5%〜95%相対湿度(RH)の範囲で、10%RH間隔で窒素パージ下で収集した。試料は分析前に乾燥しなかった。分析に使用した平衡基準は、5分間で0.010%未満の重量変化であり、重量基準が満たされない場合は3時間の最大平衡時間であった。試料の初期湿分についてデータを補正しなかった。塩化ナトリウム及びポリビニルピロリジンを較正標準として使用した。
【0091】
iii.カールフィッシャー(KF)
Mettler Toledo DL39カールフィッシャー滴定器を使用して、水分測定のための電量的カールフィッシャー(KF)分析を行った。Hydranal−Coulomat ADを含有するKF滴定容器に、約24〜32mgの試料を入れた。次に、電気化学的酸化によってヨウ素を生じる発生電極によって、試料を滴定した:
2I−⇒I2+2e。再現性を確実にするために3回反復実験を行った。
【0092】
iv.ホットステージ顕微鏡法
Leica DM LP顕微鏡に載せたLinkamホットステージ(FTIR 600型)を使用して、ホットステージ顕微鏡法を行った。交差偏光子(CP)及びラムダ(λ)補償子を有する20×対物レンズを使用して、試料を観測した。試料をカバーガラスに載せた。次に第二のカバーガラスを試料上に置いた。ステージが加熱されると共に、各試料を目視観測した。SPOT Software v.4.5.9.を有するSPOT Insight(商標)カラーデジタルカメラを使用して、画像を取り込んだ。USP融点標準品を使用して、ホットステージを較正した。
【0093】
v.変調示差走査熱量測定(MDSC)
冷蔵冷却システム(RCS)を備えたTA Instruments示差走査熱量計によって、変調示差走査熱量測定データを得た。試料をアルミニウムDSC皿に入れ、重量を正確に記録した。皿を蓋で覆い、圧着した。変調振幅+/−0.8℃及び60秒周期を使用し、−25℃から250℃に2℃/分の平均昇温速度で、MDSCデータを得た。較正標準としてインジウム金属及びサファイアを使用して、それぞれ温度及び熱容量を較正した。記録されるガラス転移温度は、可逆熱流対温度曲線における階段状変化の変曲から得る。
【0094】
vi.核磁気共鳴(NMR)
溶液相1H NMRスペクトルはSpectra Data Services,Inc.によって収集された。取得パラメータは各スペクトルに記載されている。スペクトルはテトラメチルシランを0.0ppmにおける内部基準とした。
【0095】
vii.光学顕微鏡法
光学顕微鏡法による観測は、Wolfe偏光光学顕微鏡によって倍率4xで収集した。交差偏光子(CP)を使用して、試料における複屈折を観測した。
【0096】
viii.走査型電子顕微鏡法(SEM)
走査型電子顕微鏡法(SEM)は、FEI Quanta200走査型電子顕微鏡を使用して行った。高真空モード下に、固体状態後方散乱(Etd)検出器を使用した。ビーム電圧は5.0kVであった。Cressington 108auto Sputter Coaterを使用して、約20mA及び約0.13mbar(Ar)においてAu/Pdを用いて75秒間で、試料をスパッタコーティングした。アルミニウム試料台に固定した両面粘着テープ上に少量を載せることによって、分析のために試料を調製した。NIST標準品を使用して、倍率について器具を較正した。xTm(v.2.01)、ビルド番号1564を使用してデータを収集し、XT Docu(v.3.2)を使用して分析した。SEM画像について示されている倍率は、初期データ収集時に計算した。各画像の下部に示されているスケールバーは、画像のサイズ変更する上で正確であり、サイズ測定を行う際に使用すべきである。
【0097】
ix.熱重量分析(TG)
TA Instruments2950熱重量分析器を使用して、分析を行った。較正標準品はニッケル及びAlumel(商標)であった。各試料をアルミニウム試料皿に入れ、TG炉に挿入した。25℃で試料を先ず平衡させ、次に、窒素気流下に10℃/分の加熱速度で、特に指定されないかぎり最終温度350℃に加熱した。
【0098】
x.粉末X線回折(XRPD)
(i)Inel XRG−3000
120°の2θレンジを有する湾曲した位置感受性検出器を備えたInel XRG−3000回折計で、粉末X線回折分析も行った。0.03°2θの分解能でCu Kα線を使用して、実時間データを収集した。管電圧及びアンペア数を、それぞれ40kV及び30mAに設定した。パターンを直接比較しやすくするために2.5から40°2θのパターンを表示する。薄壁ガラスキャピラリーに試料を詰めることによって分析用の試料を調製した。データ収集中にキャピラリーを回転させるように動力化されたゴニオメータヘッドに、各キャピラリーを取り付けた。器具の較正は、ケイ素参照標準品を使用して毎日行った。
【0099】
d.別の計算法
i.PDF
X線非晶質データのコンピュータ分析に使用される方法の1つは、対相関関数(PDF)である。名前が示すように、PDFは、材料内の全てのコヒーレントな原子−原子相互作用の線形和からなる。不完全な(無秩序な)材料は、結晶相と同じ原子−原子相互作用を示すが、それは短い長さスケールにおいてである。従って、そのような材料は、最初の数ナノメートルにおけるPDFのピークを調べることによって、親結晶材料と比較することができる。0Å〜約5Åの範囲のスペクトルの比較は、この領域のアーチファクトのため困難である。
【0100】
ガラス質材料の形成時では、分子が結晶ピークの位置を緩和させるため、PDFピークは結晶ピークの位置からいくらか移動する。このことは、熱膨張/収縮事象に似ており、その場合、PDFピークは少し移動するが、それでも相対的なピーク強度は元の結晶質材料に関係していることが確認できる。材料が熱力学的非晶質状態になると、いくらかの点群対称関係が失われ、複雑度の減少したPDFを与える。ピーク移動するものも存在する。ガラス質/非晶質材料では、PDFが2〜3最近接(NN)距離において急速にゼロに低下する。
【0101】
X線パターンにおけるバックグラウンドを最小限にする測定条件を使用し、測定したX線データからPDFを計算するアルゴリズムを使用する。全試料について測定データの全範囲を使用し、PatternMatch v2.2.1を使用して、PDFを計算した。
【実施例1】
【0102】
溶解度推定値
種々の溶媒中において近似溶解度を室温で測定し、その結果を表2に示す。CDDOメチルエステルは、使用した有機溶媒の大半で高い溶解度を示している。水への溶解度は、0.1mg/mL未満であると考えられる。
【実施例2】
【0103】
多型の選別結果
約50個の多型の選別実験を行った。形態Aは約50%の試料から観測された。形態Aの形成は、特定の結晶化条件に限定されず、種々の実験及び溶媒において調製した。形態Bは、凍結乾燥、溶融/急冷、及びいくつかの蒸発実験から調製した。
【0104】
使用した溶媒のアルファベット順で表3〜5に多型の選別試料を示す。形態A及び形態Bの代表的なXRPDパターンを図2において比較する。形態の特性評価のデータを、以下の実施例において概説する。
【実施例3】
【0105】
CDDOメチルエステル−形態A(非微粉化)の特性評価
形態Aは非溶媒和である(表6)。形態Aの単結晶構造を、前記の方法に基づいて決定した。CDDOメチルエステルの結晶を成長させ、単結晶構造を分析した。結晶構造を単結晶X線回折によって決定した。CDDOメチルエステルの提示された構造を図1に示す。
【0106】
正方晶のセルパラメータ及び計算された体積は次の通りである:a=14.21620(10)Å,b=14.21620(10)Å,c=81.5875(12)Å,α=90.00°,β=90.00°,γ=90.00°,V=16488.9(3)Å3。CDDOメチルエステルの分子量は、505.70g/mol(Z=24)であり、計算された密度は1.222gcm−3であった。空間群はP43212(no.96)であった。結晶データ及び結晶学的データの収集パラメータの概要を、表10に示す。
【0107】
R値0.051(5.1%)によって示されるように、得られた構造の質は高い。一般に、0.02〜0.06の範囲のR値は最も信頼できる決定構造であるとみなされる。
【0108】
単一CDDOメチルエステル分子のORTEP図を図12に示す。図13に示されている非対称単位は3個のCDDOメチルエステル分子を含有している。分子は、図1の提示された構造と同じである。
【0109】
結晶軸a、b、及びcに沿って見た充填図をそれぞれ図14〜16に示す。水素結合を有さず、結晶構造は多くのファンデルワールス相互作用を有する。結晶b軸に沿って見た図(図15)は、正方晶螺旋軸の充填される配置の螺旋性、及び予測されるBFDH形態を強調表示している。予測される形態は、データ収集に使用した単結晶で観測された性質とよく一致している。
【0110】
単結晶データから生じたCDDOメチルエステルの計算によるXRPDパターンを図17に示す。CDDOメチルエステルの実験により得られたXRPDパターンを図18に示す。形態AのXRPDパターンにおける特徴的ピークを表17に示す。計算及び実験により得られたXRPDパターンの比較(図19)によって、実験によるパターンの全てのピークが、計算によるXRPDパターンに示されていることが明らかである。このことは、バルク体がおそらく単一相であることを示している。ピーク位置において見られる共通のわずかなシフトは、実験の粉末パターンを室温で収集し、単結晶データを150°Kで収集したことによるものと考えられる。構造の質を向上させるために、単結晶分析に低温を使用する。
【0111】
要約すれば、CDDOメチルエステルの形態Aの単結晶構造は、提示された分子構造と一致することが確認された。空間群はP43212(no.96)と確認された。CDDOメチルエステルの構造は、結晶b軸に沿って螺旋状に充填された3個の分子から成る。実験によるパターンの全ピークが計算によるXRPDパターンに示されており、バルク体がおそらく単一相であることが示唆されている。
【0112】
形態Aの熱データを図3に示す。そのDSC曲線は、約157℃におけるベースラインシフト、及び約222℃の開始温度での吸熱(約224℃においてシグナルが最大)を示している。約224℃における事象は、ホットステージ顕微鏡法による溶融であることが確認された(図4)。熱重量分析(TG)曲線は、150℃まで0.34%の無視しうる重量減少が生じ、続いて150〜210℃において1.2%の重量減少が生じたことを示す。カールフィッシャーデータは、材料が約0.38%の残留水を含有することを示しており、このことは、TGによって観測された初期重量の減少と一致する。
【0113】
DVSデータは、形態Aが吸湿性でないことを示す(図5)。実験を通して、材料の重量変化はわずかであることが示された。得られた材料はXRPDで分析され、形態Aである。
【0114】
SEM画像を図6に示す。錐体、タブレット及び板状等のいくつかの結晶の性質が観察される。
【0115】
種々の条件において、形態Aの物理的安定性を調べた(表7)。25℃/60%RH又は40℃/75%RHで7日間ストレスを加えた試料は共に、わずかな重量変化(それぞれ、0.6%の減少及び0.2%の増加)を示し、このことは、形態Aが吸湿性でないことを示している。2つの試料を、1つは乾燥状態で、もう1つは少量の水と共に、ボールミルで約20分間粉砕した。全試料をXRPDにより再分析したところ、形態Aを維持していた。195℃で15分間試料にストレスを加え、2%の重量減少を示した。得られた材料のXRPDは、形態Aのものと同様であるが、ベースラインのノイズの増加は顕著である(図7)。
【0116】
溶液NMRスペクトルを図8に示す。スペクトルは、CDDOメチルエステルの構造と一致している。約1.6及び7.3ppmにおけるピークは、それぞれ水及びクロロホルム(交換による)を表している。
【0117】
形態Aは非溶媒和、かつ、非吸湿性であることから、ホットステージ顕微鏡法の際に分析者の観測に基づくと約228℃で溶融する。
【実施例4】
【0118】
CDDOメチルエステル−形態A(微粉化)の特性評価
XRPDによって、微粉化した形態AのCDDOメチルエステルが、形態Aであることを確認した(図2、表1)。微粉化した材料は、当技術分野で周知の従来法、例えばエアジェットミリングによって製造できる。これらの知見は、微粉化がそのXRPDパターンを変えるような形態Aへの影響を及ぼさないことを示していると考えられる。
【実施例5】
【0119】
CDDOメチルエステル−形態Bの特性評価
形態B材料は、表3に示すように、凍結乾燥、溶融/急冷、及び他のいくつかの蒸発実験から調製できる。
【0120】
変調DSC(MDSC)データを図9に示す。可逆曲線は、約125℃におけるガラス転移温度(Tg)を示している。不可逆曲線は、195℃で最大となるシグナルを有する発熱、及び223℃で最大となるシグナルを有する吸熱を示している。不可逆事象は、形態B材料の結晶化(発熱)が生じ、続いて結晶化した材料の溶融(吸熱)が生じたことによって起こった可能性が非常に高い。
【0121】
種々の条件における形態B材料の物理的安定性を調べた(表9)。22℃/97%RH、40℃/75%RH、80℃/0%RH、及び195℃/周囲RHにおいてストレスを加えた試料は、形態Bのままであった。200℃/周囲RHで60分間にわたって材料にストレスを加えることによって、形態A及び少量の形態B材料を生じた(図10)。
【0122】
溶液NMRスペクトルを図11に示す。スペクトルは、CDDOメチルエステルの構造と一致している。約1.6、5.3及び7.3ppmにおけるピークは、それぞれ、水、ジクロロメタン及びクロロホルムを表す。
【0123】
形態Bは、吸湿性でなく、約200℃で形態Aに結晶化し、約125℃〜130℃のガラス転移温度(Tg)を有する。
【0124】
要約すれば、形態Bは吸湿性でない。MDSCデータは、形態Bのガラス転移温度(Tg)が、約125℃〜130℃であることを示している。形態B材料は、約200℃でストレスを加えた場合に形態Aに結晶化する。
【実施例6】
【0125】
形態BのCDDOメチルエステル及びCDDOメチルエステルポリマー賦形剤分散物の安定性試験
(i)精製した形態Bの試験
形態BのCDDOメチルエステルを、種々のストレス条件に付した。表15は、これらの試験のいくつかの結果を示している。溶媒として酢酸エチルを使用した、有用ではあるが好ましくない本発明の実施形態によって調製した形態BのCDDOメチルエステルは、かなりの安定性を示す。それにもかかわらず、酢酸エチルの存在下で調製した形態B試料の試験では、60℃及びそれ以上の温度で28日間保存した後に、形態Aが形成することが明らかとなった。これに対して、本発明の好ましい実施形態(下記の実施例11はその例である)によって調製した全ての試料は、特に厳しい条件下でのストレス試験後でも、非晶質特性を保持していた(表15参照)。これらの試験は、特に前記の好ましい実施形態によって製造された場合に、形態B材料の驚くべき安定性を示している。
【0126】
さらに、種々の条件下で調製した形態B試料が同様の化学特性を有するかについて分析した。前記の通り、低温粉砕、溶融急冷、及び噴霧乾燥法によって形態B試料を調製した。さらに、微粉化していない形態Bを微粉化して、微粉化形態Bを調製した。試料のPDF分析を行い(図28)、その試料が性質上、ガラス質であることを確認した。
【0127】
(ii)CDDO−Me分散調製物を用いた試験
噴霧乾燥によって調製した、形態Bの種々のポリマー固体分散物の性能を比較するために、試験を行った。試験した生成物の特性は、安定性及び薬剤溶解プロファイルを含んでいた。
【0128】
活性医薬成分(API)対ポリマーの3種の比率が、20:80、40:60及び60:40%w/wとなる種々のポリマーを使用した。
【0129】
下記の3つのポリマーを選択した:
メタクリル酸−エチルアクリレートコポリマー(1:1);
コポビドン(1−ビニル−2−ピロリドン−ビニルアセテートコポリマー(3:2));
ヒプロメロースフタレート。
【0130】
形態A結晶質材料又は形態B材料のCDDO−Me及び試験用に選択したポリマーの溶液を、一般に溶液中10〜20wt%の固形物を与えるのに適切な重量比で、好適な溶媒に溶解させた。一般に使用した溶媒はアセトンであった。乾燥及びキャリヤーガスとして窒素を使用して、二流体ノズルを備えた実験室スケールの噴霧乾燥器(BUCHI、B−290型)によって、その得られた溶液を噴霧乾燥した。キャリヤーガス入口温度は65〜85℃を、出口温度は50〜60℃を一般に使用した。固形物を収集し、噴霧乾燥粉末を真空下で再び乾燥させ、有機溶媒の度合いをさらに減少させた。
【0131】
残留有機溶媒、ガラス転移温度(Tg)及び嵩密度について、噴霧乾燥粉末を分析した。再度乾燥させた後に、その粉末について、純度、水含有量、平均粒度、粉末X線回折(XRPD)による結晶質材料の不存在、及び溶解プロファイルも分析した。
【0132】
分散物の物理化学的特性を、短期のストレス後(40℃/75%RHで5日後)に、XRPD及び変調示差走査熱量測定(mDSC)によって評価した。
【0133】
これらの試験によって、ガラス転移温度Tgが、おそらくは短期のストレスを加えている間の湿分の吸収によって、ストレス負荷中に低下することが分かった。その低下は、低いCCDOメチルエステル対ポリマーの比率を有する処方物を使用した場合に、より顕著であった。短期のストレス前の試料について、1つ又は2つの吸熱転移がより高い温度で観測されたが、これらの温度は初期に観測された温度より幾分低い。これらの転移に関係したエンタルピーは、ポリマー含有量の減少に伴って減少した。このことは、その転移がポリマーと関係しており、結晶形の溶融とは関係していない可能性が高いことを示している。実際に、PVP/VAを使用して調製した分散物について、この吸熱転移の温度は、製造業者から受け取った純粋な賦形剤について観測されたものと同様であった。各場合に、ストレス後のXRPDプロファイルでは、約13.5°2θを中心とする特徴的なハローパターンが続き、結晶形に関連したピークは検出されなかった。
【0134】
より大規模な噴霧乾燥装置を使用して、2つの処方物についてさらに噴霧乾燥試験を行った。その場合、Niroパイロットスケール乾燥器PSD−1型(mobile minor2000)を使用した。前記と同等のノズル及び噴霧乾燥条件も使用した。表20及び21は、噴霧乾燥用に調製した溶液、及び噴霧乾燥後の溶液の特性を要約している。処方物は、形態Bより低いTgを有するポリマーを含有するよう処方したため、純粋形態Bと比較してより低いTgを示した。
【実施例7】
【0135】
CDDOメチルエステルの投与:カニクイザルにおける形態B対形態A
この試験の段階1において、純粋で微粉化した形態AのCDDO−Me又は純粋で微粉化したCDDO−Meの形態Bを含有する多数の硬質ゼラチンカプセルを、下記のように調製した:(i)適切に計量した薬剤物質の純粋形態を、サイズ1の硬質ゼラチンカプセルに添加し、(ii)カプセルを密閉した。別の賦形剤は使用しなかった。CDDOメチルエステルの形態B又は形態Aを、ゼラチンカプセルでカニクイザルに経口投与した(全ての場合で、投与量は4.1mg/kg)。
【0136】
CDDOメチルエステルの形態Bの投与は、サルにおいて、CDDOメチルエステルの形態Aを同じ投与量で投与した場合に比べ約520%高い暴露量の中央値を与えた。表11は、各動物の薬剤暴露を比較している。この試験において、「n」値を増加させ、データ信頼性を高めるために、交差法を行った。交差の間に、1週間の洗い流し期間を組み込んだ。図20は、試料採取した母集団における、時間の経過に対する、CDDOメチルエステルの両形態の得られた血漿濃度を示す。図21は動物♯505M及び♯507Fの比較したCDDOメチルエステル血漿濃度を示す。図22は、動物♯508F及び♯502Mの比較したCDDOメチルエステル血漿濃度を示す。
【0137】
さらに、CDDOメチルエステルの経口剤形(CDDOメチルエステル賦形剤分散物を含有する経口剤形を包含する)の比較生物学的利用能を評価するために、下記のように2つの別の試験段階2及び3を行った。これらの試験は、いくつかのCDDO−Meの形態B、即ち実施例6で記載したポリマー分散物を含んだ。全ての処方物は、一般に使用される処方添加剤を含有していた。
【0138】
段階2:
微粉化した形態A材料の試料から開始して、ナノ結晶質CDDO−Meの水性懸濁液を調製した。平均寸法2mmのジルコニアボールを含有するRetsch(登録商標)Planatory Ball Mill PM 400型に、25gmの微粉化したCDDO−Me(平均粒度分布6.1uM)、5gmのドキュセートナトリウム、1gmのTween80、及び68.3gmの水を装填した。約400RPMで粉砕を開始し、2時間継続した。レーザー光粒度計を使用した粒度分布(PSD)の測定によって、0.37μMの平均PSDが得られたことが示された。この濃厚懸濁液に、1gmの微結晶性セルロース及び0.2gmのキサンタンガムを添加し、短時間混合し、懸濁液を冷却保存した。
【0139】
スプレーノズルサイズが0.4mmのトップスプレーアセンブリを使用して、実験室規模のAeromatic Strea 1流動層において、乾燥賦形剤ブレンドにボールミル粉砕したナノ懸濁液を吹付け塗布した。入口温度を55℃に設定した。吹付けの間の出口温度は32〜35℃であった。得られた顆粒を、約5分間、出口温度が38℃に達するまで乾燥させた(添付書類(Attachment)3−5)。被覆物の組成を下記に示す。
【表4】
【0140】
得られた乾燥顆粒をHPLC分析にかけて、活性成分を定量したところ、14.4%(wt/wt)となり、理論値(5.7%)よりかなり高かった。HPLC分析に基づいて、カプセルは、正味のCDDO−Me含有量が30mgになるように充填した。
【0141】
微結晶性セルロース、アルファ化デンプン、クロスポビドン(崩壊剤として機能する)、コロイド状二酸化ケイ素及び植物等級ステアリン酸マグネシウムを添加剤として使用して、結晶質の微粉化した形態A及び非晶質の微粉化した形態Bの処方物を、通常の乾燥粉末ブレンド法によって調製した。平均PSDが6.1μMの微粉化したCDDO−Meの形態Aを、形態Aの処方物に使用し、平均PSDが10.8μMの微粉化したCDDO−Meの形態Bを、対応するCDDO−Meの形態Bの処方物に使用した。下記の表は、両方の処方物の定量的組成を示す。
【表5】
【0142】
15匹の雄のカニクイザル(Macaca fascicularis)のそれぞれに、目標投与量が10mg/kgとなるように、CDDOメチルエステル(3種類の処方物、1処方物につき5匹のサル)を単一経口投与した。サルは年齢1〜3才であり、体重2.5〜3.5kgであった。血液試料を、投与後72時間まで収集した。
【0143】
段階3:
微結晶性セルロース、ラクトース一水和物、クロスポビドン(崩壊剤として機能する)及びラウリル硫酸ナトリウムを添加剤として使用して、実施例6に記載したCDDO−Me賦形剤分散物をさらに、通常の乾燥粉末ブレンド法によって処方した。各処方物の定量的組成を下記に示す。
【表6】
【0144】
好適な洗い流し期間(7〜10日間)の後、段階2に使用した同一の15匹の雄カニクイザルのそれぞれに、目標投与量が10mg/kgとなるように、CDDOメチルエステル(3種類の処方物、1処方物につき5匹のサル)を単一経口投与した。血液試料を、投与後72時間まで収集した。
【0145】
下記の表は、試験の各段階を要約している
【表7】
【0146】
雄のカニクイザルに投与したCDDOメチルエステルの平均静脈内投与量及び平均経口投与量並びに平均濃度を下記に要約する:
【表8】
【0147】
段階2においては、サイズ2のゼラチンカプセルを使用して処方物を送達し、段階3においては、サイズ1のゼラチンカプセルを送達に使用した。各カプセルにおける正味薬剤含有量は30mgであり、それは、各サルの体重が3kgであるという仮定に基づいた10mg/kgの薬剤投与量に対応する。カプセルを経管栄養器具に加え、動物に経管栄養器具を取り付け、空の注射器による空気圧によって経管栄養器具の末端からカプセルを放出させた。最後のカプセルの投与後に、少量の水(約10mL)を経口的に与えた。
【0148】
下記の各時点(実際の時間を記録した)で、各動物の大腿静脈又は動脈から、血液試料(約1mL)を逐次採取し、K2−EDTAを含有する試験管に移した:
段階2 投与前、投与後1、2、4、8、16、24、48及び72時間
段階3 投与前、投与後1、2、4、8、16、24、48及び72時間
収集後、どの試料も充分に混合し、約4℃で冷蔵する前にぬれた氷上に置いた。血液中のCDDOメチルエステル濃度は、HPLC−MS/MSによって分析した。
【0149】
結果を表22及び図30に示す。段階2について、形態Bは、試験した2つの形態Aの処方物より有意に高い生物学的利用能を示した。段階3の結果は、各CDDO−Me、即ちポリマー分散物に基づく処方物が、微粉化した形態A又はナノ結晶質形態Aの処方物のいずれよりも極めて高い生物学的利用能を有することを示した。メタクリル酸コポリマーC型及びHPMC−P処方物は、被験サルにおいて最も高い生物学的利用能を示した。
【実施例8】
【0150】
CDDOメチルエステルのヘミベンゼネート形態の特性評価
CDDOメチルエステル合成の最後の回収段階を繰り返す種々の実験を行った。Hondaら、2000を参照のこと。その目的は、ベンゼン/アセトン(10:1)の溶液混合物から結晶質材料を分離することであった。
【0151】
約100mgのCDDOメチルエステルを、300μLのベンゼン/アセトン(10:1)に溶解し、0.2−μmナイロンフィルターで濾過した。次に、超音波処理装置により溶液を10分間音波処理し、蓋を取ったバイアル中において室温で一晩蒸発させた。透明なゲルが形成され、100μLのベンゼン/アセトン(10:1)を添加した。その溶液を、超音波処理装置で約30分間、超音波処理したところ、白色沈殿物が形成した。固形物を風乾した。
【0152】
他の実験において、約200mgのCDDOメチルエステルを、0.8mLのベンゼン/アセトン(10:1)に溶解させ、0.2−μmナイロンフィルターで濾過した。次に、その溶液を2つの1−ドラムバイアルに均等に分けた。次に、試料A及びBを室温で数時間、急速蒸発させた。試料Aに蓋をし、冷凍庫に入れた。試料が凍結した後、室温で解凍した。ヘラを用いて小さい引っ掻きを導入し、試料を室温で蒸発させた。白色固形物が形成され、風乾した。試料Bに蓋をし、室温で置き、室温で一晩おいた後に透明溶液になった。ヘラを用いて小さい引っ掻きを導入し、試料を室温で蒸発させた。白色固形物が形成され、風乾した。
【0153】
いくつかのこれらの実験から、ヘミベンゼネートであることが確認された結晶質材料を得た。前記のように、軽微な妨害作用、例えば、回収容器内での音波処理又は単なる小さい引っ掻きの導入が、ベンゼン溶媒和物の結晶化を促進する(表12)。
【0154】
ヘミベンゼネートの特性評価データを表13に要約する。ヘミベンゼネートXRPDパターンの特徴的なピークが、表19に示されている。DSC曲線は、図23のTGサーモグラフにおける約7.0%の重量減少に関連して、133℃付近の幅広い吸熱を示している。重量減少はおそらく、ベンゼンが揮発したからであり(下記のNMR考察を参照)、CDDOメチルエステルの各モルにつき0.5molのベンゼンに相当する。223℃付近で観測されたDSC吸熱は、脱溶媒和した材料の溶融によって生じる可能性が極めて高い。
【0155】
これらのデータは、CDDOメチルエステルの以前に分離された形態と本発明のその形態との区別を明らかにしている。
【実施例9】
【0156】
CDDOメチルエステルの新規ジメタノレート形態の特性評価
CDDOメチルエステルジメタノール溶媒和物を、下記の手順に従い調製した。約500mgのCDDOメチルエステルを、20mLのメタノールに60℃で溶解させた。次に、その溶液を、20mLの冷メタノールに−10℃で撹拌しながらゆっくり添加した。白色固形物を真空濾過によって収集した後、冷凍庫に保存した。
【0157】
特性評価データを表14に要約する。ジメタノレートXRPDパターンの特徴的なピークが、表18に示されている。
【0158】
DSC曲線は、TGサーモグラフにおける約11%の重量減少に関連して、102℃付近の幅広い吸熱を示している(図24)。TGIRデータは、重量減少が約2.0molのメタノールの揮発によることを裏付けている(図25)。TGIR実験から得られた物質を回収したところ、XRPDにより非晶質であることが分かった(図26)。約130℃におけるベースラインシフト、203℃付近の幅広い発熱、それに続く鋭い吸熱(開始:223℃)も、DSC曲線において見られる。これらの事象は、ジメタノール溶媒和物の脱溶媒が生じ、非晶質材料が形態Aに結晶化し、その結晶質材料が溶融したことによって得られた非晶質材料(形態B)のTgを示している可能性が極めて高い。
【0159】
溶液プロトンNMRスペクトルを得た。ケミカルシフトの帰属は行わなかったが、CDDOメチルエステルの化学構造と一致していると考えられる。約3.51ppmにおけるピークは、メタノールを表し、約1.7molに相当する。この結果は、前記の熱データと一致している。
【実施例10】
【0160】
CDDOメチルエステルを用いた臨床試験
臨床開発のために、微粉化形態Aを使用して処方したCDDO−Meを選択し、以前の治療に対して充分に反応しなかった進行癌を有する患者において、第I相安全性指向試験を先ず行った。この第I相用量増加試験において、種々の形態の進行(転移)癌を有する21人の成人患者に、CDDO−Meを投与した。患者にCDDO−Meカプセルの1日量を、5〜900mg/日(特に、5、10、20、40、80、150、300、600又は900mg/日)の用量で投与した。患者が容認できない毒性を経験するか、又は疾患進行の徴候を示すまで繰り返される「サイクル」で、CDDO−Meを投与した。この試験において、CDDO−Meの1サイクルは21日間の連続投与に続く7日間の休止期間から成り、その後に、患者は次のサイクルを開始することができた。
【0161】
CDDO−Meの安全性及び抗腫瘍活性を調査した。さらに、CDDO−Meの生物学的作用も特性評価した。CDDO−Meは、これらの患者において非常によく許容され、有意な薬剤関連有害事象も報告されなかった。第二治療サイクルの終了後の第一評価時点において、幾人かの患者(評価可能患者の約75%)が、安定疾患を有していると考えられた(標準的な放射線及び臨床基準に基づく)。第二サイクルの終了前に進行疾患の徴候を有することが見い出された患者は、形式的に評価されず、評価可能患者の群に含まれなかった。メラノーマ及び腎細胞癌の患者を含む5人の患者は、安定疾患を示し続け、治療の4サイクル後に、個々の腫瘍病変が退縮する徴候を示した。4人の患者は、治療の少なくとも6サイクル後に、安定疾患を有すると考えられた。処方計画により1日当たり少なくとも40mgのCDDO−Meの用量を投与されたいずれの患者においても、新たな転移は生じなかった。
【0162】
CDDO−Meの周知の抗炎症特性に基づいて、第I相試験で患者の循環炎症性サイトカインを評価した。5mg/日と同じくらい低い投与量で、MMP−9、TNFα、IL−8及びVEGF等のいくつかの循環炎症誘発性サイトカイン及びケモカインの減少が見られた。特に、TNFα(慢性関節リウマチ等の疾患の炎症過程において重要な役割を果たしていることが知られている)は、上昇したTNFベースライン値を有する3人の患者において、実質的に、又は検出限界未満に減少した(各患者につき、10、20及び40mg/日の治療投与量)。標的に結合して不活性にする抗TNFモノクローナル抗体と異なり、CDDO−Meは、TNFαの産生を減少させ、その結果としてTNFαの循環レベルを減少させる。
【0163】
さらに、抗酸化酵素及び解毒酵素を包含する第2相遺伝子産物を、第I相試験における患者の末梢血単核細胞において測定した。第2相転写活性のマーカーであるNQO1(NAD(P)H:キノンオキシドレダクターゼ)の顕著な誘導が、10mg/日及びそれ以上の投与量において観測された。
【0164】
CDDO−Meによる2サイクルの治療後に数人の患者から採取した腫瘍生検データは、シクロオキシゲナーゼ−2(COX−2)、誘導型一酸化窒素シンターゼ(iNOS)及びホスホリル化STAT3(pSTAT3)の腫瘍組織レベルにおける顕著な減少を示した。これらの各タンパク質の高レベルの発現は、腫瘍進行及び低い臨床結果に相関していることが知られている。数人の患者における腫瘍生検データは、CDDO−Meによる2サイクルの治療後に腫瘍細胞死が顕著に生じることも示した。血清クレアチニンのレベルは、この試験における80%より多い患者において、治療前のベースライン値と比較した場合、21日目において有意に低かった。複数サイクルによる治療を継続した多くの患者は、血清クレアチニンの継続的な減少を示した。血清クレアチニンは広く使用されている腎機能の指標であるので、これらの観測は、CDDO−Meによる治療が腎機能を向上させることを示している。
【0165】
これらの試験は、癌患者におけるCDDOメチルエステルの有利な作用を示すヒト癌患者データを提供する。データはさらに、腎機能不全等の他の炎症関連疾患の患者においてCDDO−Meが臨床的に有利な作用を有する可能性が高いことも示している。
【実施例11】
【0166】
ジメタノール溶媒和物中間体を使用した形態Bの大規模生成
1kgの形態AのCDDO−Meを、60±5℃メタノールに溶解させて、完全な溶液を得た。−5℃〜−15℃の冷メタノールの入った容器に、得られたCDDO−Meの温溶液を添加し、添加の間中、撹拌及び−5℃〜−15℃の温度を維持した。得られたCDDO−Meの結晶質ジメタノール溶媒和物の懸濁液を濾過した。図26に示されているXRPDパターンと一致したパターン(TGIR分析前)を示す得られた固形物を、オーブンで70±5℃で乾燥させた。XRPDプロファイルが結晶質物質に特徴的な反射を示さなくなるまで、乾燥を続けた。得られたXRPD非晶質CDDO−Me固形物を篩にかけ、包装した。生成物回収率は65〜95%である。
【実施例12】
【0167】
低温粉砕した形態A及び形態B
形態Aを低温粉砕し、分析した。低温粉砕(2時間)によって得られた試料の測定X線データは、約13.5°2θにおけるピークの幾分かの広がりを示した。低温粉砕した形態AのPDF分析は、形態B分析と同様の結果を生じた。これらの結果は、低温粉砕した形態Aがガラス質材料であり、低温粉砕が形態Bを生成する代替法を与え得ることを示す。
【0168】
形態Bを低温粉砕し分析したところ、低温粉砕(1時間)によって得られた試料の測定X線データは、形態B材料から開始したものと同様であった。これらの結果は、形態Bが安定であり、低温粉砕によって形態が変化しないことを示す。
【表9】
【表10】
【表11】
【表12】
【表13】
【表14】
【表15】
【表16】
【表17】
【表18】
【表19】
【表20】
【表21】
【表22】
【表23】
【表24】
【表25】
【技術分野】
【0001】
本発明は、CDDOメチルエステルの新規形態に関する。
【背景技術】
【0002】
トリテルペノイドは、スクアレンの環化によって植物で生合成されている。これらの天然分子は、医薬用途のための候補物質であるが、比較的弱い生物活性を示す。従って、化学者は、高い効力を有する類似体を合成しようとしている(Hondaら,1997&1998)。
【0003】
いくつかの合成類似体は、IFN−γ又はLPSによって刺激されたマクロファージにおけるiNOS及びCOX−2のde novo生成を抑制することが報告されている(Suhら,1998;Hondaら,2002)。他の合成トリテルペノイド、2−シアノ−3,12−ジオキソレアナ−1,9(11)−ジエン−28−オエート(CDDO)は、抗炎症活性及び抗増殖活性を示す(Hondaら,1998&2000)。
【0004】
CDDOのメチルエステル(これは、メチル2−シアノ−3,12−ジオキソレアナ−1,9(11)−ジエン−28−オエート(CDDOメチルエステル)である)の研究により、Boreら(2002)は結晶構造を決定した。水和した形態において、水は、特定の結晶充填及び構造を発生させる相互作用を調整する。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Honda, et al. , Bioorganic & Medicinal Chemistry Letters 1997, 7, 1623
【非特許文献2】Honda, et al , loc. cit. 1998, 8, 2711-14
【非特許文献3】Honda, et al., Bioorganic & Medicinal Chemistry Letters 2002, 12, 1027
【非特許文献4】Honda, et al., J. Med. Chem. 2000, 43, 1866
【非特許文献5】Bore, et al., Acta Cryst. 2002, C58, o199-o200
【発明の概要】
【0006】
本発明の1つの実施形態において、CDDOメチルエステルの非水和結晶形が提供される。非水和結晶形は、好ましくは、単位格子寸法a=14.2Å、b=14.2Å、及びc=81.6Åを有するP43212の空間群を有する。本発明は、(i)治療有効量のCDDOメチルエステルの非水和結晶形及び(ii)食用担体を含んで成る固体投与形態の医薬組成物も意図する。
【0007】
さらに、本発明は、図2Cに示されるように、約13.5°2θにおいてハローピークを有する粉末X線回折パターン、及びガラス転移温度(Tg)を有するCDDOメチルエステルのガラス質固体形態においても具体化される。特定の実施形態において、Tgは約120℃〜135℃である。他の実施形態において、Tgは約125℃〜約130℃である。CDDOメチルエステルのガラス質固体形態は、図28と同様の約5Å〜約20Åにおけるピークを有するPDFスペクトルを有しうる。
【0008】
さらに本発明は、(i)治療有効量のCDDOメチルエステルのガラス質固体形態、及び(ii)食用担体を含んで成る固体剤形の医薬組成物も提供する。これに関連して、本発明は、そのような医薬組成物を癌患者に投与することを含んで成る癌患者の治療法も意図する。本発明は、他の抗癌剤と組み合わせてCDDOメチルエステルのガラス質形態を投与することも意図する。例えば、抗癌剤はゲムシタビンであってよく、癌は膵臓癌であってよい。本発明は同様に、急性又は慢性の酸化性ストレス及び炎症に関係した疾患又は障害、特に、誘導型一酸化窒素シンターゼ(iNOS)又は誘導シクロオキシゲナーゼ(COX 2)の過剰発現を部分的に特徴とする疾患又は障害の治療法も含む。
【0009】
さらに、本発明は、表18に示されている特徴的なピークを有する粉末X線回折パターン及び図24に示されるDSCパターンを有するCDDOメチルエステルのジメタノール溶媒和形態にも関する。本発明により、ジメタノール溶媒和形態は、CDDOメチルエステルのガラス質固体形態を製造するための中間体として使用しうる。ジメタノール溶媒和形態を介したCDDOメチルエステルのガラス質固体形態の製造法は、CDDOメチルエステルのジメタノール溶媒和形態の調製及び該ジメタノール溶媒和形態の乾燥を含んで成る。
【0010】
他の実施形態によれば、本発明は、CDDOメチルエステルジメタノレートの結晶を成長させる方法に関し、該方法は、無水温メタノール中の精製CDDOメチルエステルの溶液を調製し、その温溶液を冷メタノールの容器に添加し、得られた結晶を濾過することを含んで成る。
【0011】
他の実施形態によれば、本発明は、(i)治療有効量のCDDOメチルエステル、及び(ii)ガラス形成剤である賦形剤を含んで成り、Tgを有する医薬組成物に関する。
【0012】
賦形剤は、例えば、(A)炭水化物、炭水化物誘導体又は炭水化物ポリマー、(B)合成有機ポリマー、(C)有機酸塩、(D)タンパク質、ポリペプチド又はペプチド及び(E)高分子量多糖から成る群より選択しうる。合成有機ポリマー賦形剤の種類の例は、ヒドロキシプロピルメチルセルロース、例えば、ヒドロキシプロピルメチルセルロースフタレートエステル、ポリ[1−(2−オキソ−1−ピロリジニル)エチレン又はそのコポリマー、例えばPVP/VA、及びメタクリル酸コポリマー、例えばメタクリル酸−エチルアクリレートコポリマー(1:1)である。
【0013】
これに関する他の賦形剤は、1−ビニル−2−ピロリドン−ビニルアセテートコポリマー(3:2)であるコポビドンである。
【図面の簡単な説明】
【0014】
【図1】図1は、CDDOメチルエステルの化学構造を示す。
【図2】図2は、形態A(上)及び形態B(下)のXRPDパターンを示す。上から下へ:非微粉化形態A、微粉化形態A、及び形態B。
【図3】図3は、CDDOメチルエステル(形態A)のDSC及びTG曲線を示す。
【図4−1】図4は、形態A(非微粉化)のホットステージ分析(hot stage analysis)を示す。
【図4−2】図4は、形態A(非微粉化)のホットステージ分析(hot stage analysis)を示す。
【図5】図5は、形態A(非微粉化)の動的蒸気収着等温線を示す。
【図6−1】図6は、形態A(非微粉化)のSEM画像を示す。
【図6−2】図6は、形態A(非微粉化)のSEM画像を示す。
【図7】図7は、195℃でのストレスの前(上)及び後(下)の形態Aを示す。
【図8】図8は、形態A(非微粉化)のNMRスペクトルを示す。
【図9】図9は、形態BのCDDOメチルエステルのMDSC曲線を示す。
【図10】図10は、60分間の200℃/周囲相対湿度での熱ストレスの前(上)及び後(下)の形態BのCDDOメチルエステルを示す。
【図11】図11は、形態BのCDDOメチルエステルのNMRスペクトルを示す。
【図12】図12は、標識を有する単一の形態A分子のORTEP図を示す。原子は、50%確率の異方性熱振動楕円体で示されている。
【図13】図13は、形態A結晶の非対称単位の含有物のORTEP図を示す。原子は、50%確率の異方性熱振動楕円体で示されている。
【図14】図14は、結晶a軸に沿って見た形態A結晶の充填図を示す。
【図15】図15は、結晶b軸に沿って見た形態A結晶の充填図を示す。
【図16】図16は、結晶c軸に沿って見た形態A結晶の充填図を示す。
【図17】図17は、形態Aの計算による粉末X線パターンを示す。
【図18】図18は、形態Aの実験から得られたXRPDを示す。
【図19】図19は、形態AのCDDOメチルエステルの計算及び実験によるXRPDパターンの比較を示す。
【図20】図20は、カニクイザルへの4.1mg/kg経口投与後の、形態A及び形態Bの曲線下面積の代表的プロットを示す。各データ点は、8匹の動物におけるCDDOメチルエステルの平均血漿濃度を示す。エラーバーは、試料採取母集団内の標準偏差を示す。
【図21】図21は、動物#505M(上のグラフ)及び動物#507F(下のグラフ)における、 CDDOメチルエステルの形態B対形態Aの血漿濃度の比較を示す。
【図22】図22は、動物#508F(上のグラフ)及び動物#502M(下のグラフ)における、CDDOメチルエステルの形態B対形態Aの血漿濃度の比較を示す。
【図23】図23は、CDDOメチルエステルヘミベンゼン溶媒和物のサーモグラムを示す。
【図24】図24は、CDDOメチルエステルジメタノール溶媒和物のサーモグラムを示す。
【図25】図25は、CDDOメチルエステルジメタノール溶媒和物に関するTGIRデータを示す。
【図26】図26は、TGIR分析(140℃まで)の前(上)及び後(下)の、CDDOメチルエステルジメタノール溶媒和物のXRPDパターンを示す。
【図27】図27は、形態A対形態BのPDFデータのオーバーレイ表示である。約5Å〜約20Åにおいて、部分的にオーダーが類似している。
【図28】図28は、形態Bの種々の調製物についてのX線非結晶パターンのオーバーレイ表示であり、調製物間の実質的均一性を示す。
【図29】図29は、空間群P43212(#96)の概略図である。
【図30】図30は、雄カニクイザルへのCDDOメチルエステルカプセルの単一経口投与後の、CDDOメチルエステルの平均血中濃度を示す(段階2及び3)。
【発明を実施するための形態】
【0015】
前記のように、iNOS活性の抑制剤としての、特にNO生成の阻害における、トリテルペノイドの研究は、CDDO及びCDDOメチルエステルの高い有効性を示している(IC50<1nMレベル)。Hondaら(2000)参照のこと。これらの研究は、可溶化CDDOメチルエステルに焦点を当てており、CDDOメチルエステルの固体についてはほとんど特性評価していない。Boreら(2002)の研究は、CDDOメチルエステルの単一溶媒和結晶形の構造(トリテルペノイドについて最初に公表された構造)を解明した。
【0016】
図1(化学構造)及び図12(ORTEP図)に示されているCDDOメチルエステルの治療における潜在能力を理解するために、本発明者らは、好適な薬物動態を有する医薬製剤の開発に有益な、より高い水溶性及び化学安定性等の特性を有する該化合物の他の形態について研究した。その結果、本発明者らは、かかる特性を有し、従ってそれ自体で薬剤開発の候補物質となる、Boreら(2002)によって解明された結晶形とは異なる、CDDOメチルエステルの2つの形態を発見した。
【0017】
本発明のCDDOメチルエステルの「形態A」は、溶媒和されず(非水和)、図29に示される空間群P43212(no.96)、単位格子寸法a=14.2Å、b=14.2Å及びc=81.6Åを有する特有の結晶構造を特徴とし、かつ、図14〜16に示される充填構造を特徴とし、3個の分子が結晶b軸に沿って螺旋状に充填されている。下記の表10は、結晶データ収集パラメータと共に、形態Aの追加の結晶データを列記している。
【0018】
本発明の他の「形態B」は、単一相であるが、形態Aのような明確な結晶構造を欠いている。むしろ、形態Bは、形態Aとは異なる粉末X線回折(XRPD)スペクトルによって象徴される(特に図2参照)。さらに、形態Bは、形態Aより驚くほど高い生物学的利用能を示す(実施例7参照)。
【0019】
CDDOメチルエステルの合成方法は既に公表されている。米国特許第6,326,507号明細書、Hondaら(1998)、及びHondaら(2000)参照のこと。本発明者らは、CDDOメチルエステルの形態A及び形態Bが共に、下記の表3〜5に列記されているものによって例示される種々の化合物溶液から容易に調製されることを発見した。特に形態Bは、MTBE、THF、トルエン又は酢酸エチル中での急速蒸発又は低速蒸発によって調製できる。同様に、形態Aもエタノール又はメタノール中のCDDOメチルエステル溶液の急速蒸発、低速蒸発又は低速冷却によって調製できる。アセトン中のCDDOメチルエステルの調製物は、急速蒸発を使用して形態Aを生じうるか、又は低速蒸発を使用して形態Bを生じうる。別の製造法は下記に示され、その表も含まれる。
【0020】
形態Bは、明確な結晶構造を有さないので、形態Aに特徴的な明確なXRPDピークを欠き、それに代わって、全般的な「ハロー」XRPDパターンを特徴とする。特に、非結晶質形態Bは、そのXRPDパターンが3つ又はそれ以下の主要な回折ハローを示すので、「X線非晶質」固体に分類される(例えば、図10参照)。この分類において、形態Bは「ガラス質」材料である。PDFによって示されるように、最近接原子−原子相互作用は、結晶質形態Aにおいて観察されるものと一致するが、明白な長距離秩序が存在しないので、平均単位格子の概念が適用されない。
【0021】
従って、形態Aと異なり、形態Bの試料は、長距離の分子相関を示さない(即ち、約20Å以上において)(図27参照)。さらに、形態Bの試料の熱分析は、ガラス転移温度(Tg)を示す。これに対して、規則性のないナノ結晶材料はTgを示さないが、その代わりに融解温度(Tm)(それより高い温度で結晶構造が液体になる)を示すのみである。
【0022】
本明細書は、形態Bを製造するために使用できるCDDOメチルエステルジメタノール溶媒和物形態の特性を明らかにする(実施例9参照)。CDDOメチルエステルヘミベンゼネート(hemibenzenate)形態の特性も本明細書で明らかにされる(実施例8参照)。
【0023】
他の結晶材料の微粉化はXRPDスペクトルに影響を及ぼすことが見出されているが、微粉化形態AのXRPD分析は、非微粉化形態Aに類似したスペクトルを生じる。非微粉化形態A、微粉化形態A、及び形態BのCDDOメチルエステルを並列して比較した図2を参照のこと。
【0024】
種々の特性評価の方法を併用して、CDDOメチルエステルの形態A及び形態Bを、互いに、またCDDOメチルエステルの他の形態と区別することができる。この目的に好適な方法の例は、固体状態核磁気共鳴(NMR)、粉末X線回折、X線結晶構造解析、示差走査熱量測定(DSC)、動的蒸気収着/脱離(DVS)、カールフィッシャー分析(KF)、ホットステージ顕微鏡法、変調示差走査熱量測定、FT−IR、及びラマン分光法である。
【0025】
特に、XRPD及びDSCデータの分析によって、CDDOメチルエステルの形態A、形態B及びヘミベンゼネート形態を区別することができる。
【0026】
本発明のCDDOメチルエステル形態の特性は、前記のように特有であり、かつ、薬剤としての使用に有用である。例えば、形態B及び形態AのCDDOメチルエステルの生物学的利用能は、等用量のその2つの形態をゼラチンカプセルに入れて経口的に投与したサルで変化していた。実施例7参照のこと。さらに、新たに同定されたCDDOメチルエステル形態の安定性は、医薬組成物の製造に有用である。CDDOメチルエステルの形態A及び形態Bを、互いに、またCDDOメチルエステルの他の形態から区別するのと同様に、以下に詳細に記載するように「X線非晶質」特性を保持しているCDDOメチルエステル分散物を、XRPD及びDSC分析等の種々の方法によって、結晶質形態AのCDDOメチルエステルを含む分散物から区別することができる。即ち、結晶質CDDOメチルエステルの形態Aを含む分散物は、一般に、純粋形態AのCDDOメチルエステルに特徴的な明確なピークを示す(特に、約13.35及び8.78(°2θ)において生じるピーク)(例えば、下記の表17参照)。
【0027】
本発明のCDDOメチルエステルポリマー賦形剤分散物の特性は、特有であり、かつ、薬剤としての使用に有用である。例えば、付加的不活性添加剤と共に処方した、選択したCDDOメチルエステル分散物の生物学的利用能は、サルに等用量の該分散物をゼラチンカプセルに入れて与えた場合、変化した。下記の実施例7、研究段階2及び3を参照のこと。いくつかの例において、CDDOメチルポリマー賦形剤分散物を含む処方物は、CDDOメチルエステルの純粋形態Bから製造された処方物と比較した場合でも、生物学的利用能において、驚くべき増加を生じた。
【0028】
医薬固形物における、多型を包含する多様な形態の存在は、例えば、Cuiら(Int’l J. Pharmaceutics,2007,339,3−18)によって以前に記載されている。化合物の結晶形及び非晶形は、異なる物理的及び化学的特性を示しうる。例えば、非晶形は結晶形と比較して、より高い溶解性を有しうる。しかし、あらゆる化合物はこの点で特有であり、非晶質材料が結晶状態と異なる度合は個別に調査すべきであり、演繹的に予測することはできない。さらに、いくつかの非晶質材料は再結晶しやすい。
【0029】
本発明に関して、データ収集における変動が、多くの要因によって生じうる。従って、本明細書は、CDDOメチルエステル形態を説明するために使用されるデータにおける変動を示すために、「約」又は「ほぼ」という用語を使用する。例えば、融解温度は、計器装備又は条件によって変化しうる。測定の精度に関して、USP<891>は、「融解の場合、「開始」及び「ピーク」温度の両方は、客観的及び再現的に、多くの場合十分の数度以内で測定されうる」と記載している。実際の実験は、このことが材料のTgの測定については当てはまらないことを示す。Tgは、多くの要因に依存する:試料の調製方法;試料の熱履歴(緩和);Tg前に蒸発するか又はしない残留溶媒;装置;試料調製物(試料質量、粒度、充填、希釈剤);Tgの測定に使用されるパラメータ(特に走査速度);Tgの位置を決定するために使用されるパラメータ(開始温度、中間点温度、変曲点温度、又はオフセット温度);緩和による吸熱がTgにおいて存在するかどうか;及び他の要因。いくつかの要因はTgを低下させる(残留水/溶媒による可塑化)が、他の要素はTgを上昇させ(より速い走査速度、緩和)、10〜15℃も上昇させる場合がある。Tgにおける熱容量(ΔCp)の変化は、Zhouら、J.Pharmaceutical Sciences 91:1863−72(2002)によって報告されているように重要である。
【0030】
本明細書は、それらの「特徴的」ピークに関する種々のパターンについて記載する。そのようなピークの集合又は群は、個々の装置及び実験条件にそれぞれ起因しうる不確実性の範囲内で、所定の多型形態に特有である。
【0031】
各結晶形について、5つの特徴的ピークの群が下記の表17〜19に示されている。一般的な変動は±0.1°2θであるが、いくつかの実験において、ピーク位置は±0.2°2θ又はそれ以上まで変化しうる。
【表1】
【表2】
【表3】
【0032】
ガラス質材料(形態B)のXRPDパターンは、約13.5°2θにおいて幅広いハローピークを示し、これは形態Bに特徴的であると考えられる。他のハローは明確ではなく、このパターンの形/位置は、装置及び実験条件の関数として変化しうる。この幅広いピーク位置の変化は、それぞれの結晶形の特徴的ピークの変化より大きい。特に、形態Bの幅広いピークについて±1°2θまでの変動性が、特定の装置において予測できる。
【0033】
CDDOメチルエステル賦形剤分散物として製造されたガラス質材料のXRPDパターンも、一般に約13.5°2θを中心とする幅広いハローピークを示す。これらの材料は、変調示差走査熱量測定(mDSC)によってTgも示す。純粋形態BのCDDOメチルエステルの試料と同様に、賦形剤分散物のXRPDパターンの形及び位置は、使用された装置、実験条件、及び分散物を製造するのに使用された特定賦形剤の関数として変化しうる。
【0034】
本発明は、さらに、癌性疾患及び中枢神経系を冒す種々の病状等の炎症関連疾患を治療するための、CDDOメチルエステルの形態A、形態B及びガラス質XRPD−非晶質賦形剤分散物のそれぞれの使用にも関する。本発明によれば、これらの疾患の治療は、それを必要とする患者に、本明細書に列挙した新規CDDOメチルエステル形態の有効量を投与することを含んで成る。これらの化合物は、癌、アルツハイマー病(AD)、パーキンソン病(PD)、多発性硬化症(MS)、筋委縮性側索硬化症(ALS)、慢性関節リウマチ(RA)及び他の自己免疫疾患、炎症性腸疾患、並びに酸化窒素又はプロスタグランジンの過剰生成に関係した他の病理学的症状の病因に関連した炎症を改善又は予防するのに有用である。
【0035】
前記のように、シクロオキシゲナーゼ−2(COX−2)又は誘導型一酸化窒素シンターゼ(iNOS)の異常又は過剰発現が、結腸における発癌等の多くの疾患過程の病因に関与している。CDDOメチルエステル等のトリテルペノイドのいくつかの合成類似体が、iNOSの発現を抑制することが報告されている。関連した研究は、IFN−γ又はLPSによって刺激されたマクロファージにおいてiNOS及びCOX−2の発現がトリテルペノイドにより抑制されたことを示している(Suhら、1998;Hondaら、2002)。従って、CDDOメチルエステル形態を投与する治療は、iNOS及びCOX−2の抑制に作用すると考えられる。
【0036】
COX−2の遺伝子の過剰発現は、結腸発癌における早期かつ中心的事象である(Prescott及びWhite,1996;Duboisら、1996)。APC(大腸腺腫性ポリポーシス)遺伝子における欠損を有するマウスは、早期に多数の腸ポリープを発生させ、COX−2酵素レベルの顕著な増加がこれらのポリープにおいて見出されている。これらの動物を用いた知見は、多くのヒト原発性結腸癌及び結腸癌細胞系においてCOX−2 mRNA及びタンパク質が高レベルとなる知見と相互に関係しており(Prescott及びWhite,1996)、このCOX−2の増加が、通常は前新生物細胞の死に導くアポトーシスの抑制を生じさせると考えられる(Tsujii及びDuBois,1996)。腸腫瘍発生に対するCOX−2の機能的関連性は、COX−2遺伝子のノックアウトによって示されている(Oshimaら、1996)。このノックアウトを有するマウスを、APC遺伝子において損傷を有するポリープ形成マウスと交配させたところ、COX−2ノックアウトは、子孫におけるポリープの数を劇的に減少させた。さらに、選択的COX−2阻害剤又は非選択的COX−1/COX−2阻害剤を実験動物に処置することは、腸癌の化学的予防に有効な方法であることが報告されている(Marnett,1992;Oshimaら、1996;Boolbolら、1996;Reddyら、1996;Shengら、1997)。発癌におけるiNOSの役割に関して、NOは強力な突然変異原であり(Tamir及びTannebaum,1996)、酸化窒素はCOX−2を活性化することもできる(Salveminiら、1993,1994)ことが明らかである。発癌物質アゾキシメタンによって誘発されたラット結腸腫瘍においても、iNOSの顕著な増加が見られる(Takahashiら、1997)。同様に、ヒト腫瘍におけるiNOSの過剰発現は、悪い予後因子として報告されている(例えば、Ekemekciogluら、2006)。
【0037】
アンギオテンシンIIによって誘発されるような、炎症シグナル経路及び他の疾患関連シグナル経路は、スーパーオキシド、過酸化水素、酸化窒素及びパーオキシナイトライト等の活性酸素又は窒素種(RONS)の過剰生成を刺激する場合が多い。CDDOメチルエステルは、抗酸化活性の有効な誘発物質、及び種々の細胞型における炎症過程の有効な阻害物質であることが示されている(Dinkova−Kostovaら、2005;Libyら、2006;Ahmadら、2006;Shishodiaら、2006)。感染、外傷、火傷及び化学暴露等の種々の原因による重篤な急性炎症は、生命にかかわり、肝不全、腎不全、呼吸不全又は心不全を生じる場合がある。慢性炎症及び関連酸化性ストレスは、下記に挙げられる多くの重大疾患の病状の一因となる:自己免疫疾患(例えば、慢性関節リウマチ、狼瘡、乾癬及び多発性硬化症)、心血管疾患(例えば、アテローム性動脈硬化症及び心不全)、糖尿病(I型及びII型)、呼吸器疾患(例えば、慢性閉塞性肺疾患及び喘息)、慢性腎臓病、腎不全、肝不全、及び疼痛症候群(例えば、神経障害性疼痛、線維筋痛症及び片頭痛)。さらに、トリテルペノイドは、マクロファージにおけるHIV−1の複製を阻害することが示されており(Vazquezら、2005)、従って、ウイルス性疾患、特に、器官又は組織の炎症によって重大な病的状態を生じるウイルス性疾患(例えば、ウイルス性肝炎、インフルエンザ、単純ヘルペス)の治療に有用になりうる。
【0038】
MSは、中枢神経系の炎症状態であることが既知である(Williams,Ulvestad及びHickey,1994;Merrill及びBeneviste,1996;Genain及びNauser,1997)。炎症、酸化又は免疫機構が、MS、AD、PD及びALSの病因に関与していると考えられる(Bagasraら、1995;Griffinら、1995;McGeer及びMcGeer,1995;Goodら、1996;Simonian及びCoyle,1996;Kaltschmidtら、1997)。反応性星状細胞及び活性化小グリア細胞は共に、NDD/NIDの原因に関連付けられている。NO及びプロスタグランジンの両方を、それぞれの酵素、iNOS及びCOX−2の生成物として合成する細胞として、小グリア細胞が特に重要視されている。これらの酵素のde novo生成は、炎症サイトカイン、例えば、インターフェロンガンマ又はインターロイキン−1によって引き起こされうる。次に、NOの過剰生成は、多くの器官の細胞及び組織(神経系のニューロン及び乏突起神経膠芽細胞等)における炎症カスケード及び/又は酸化損傷を生じ、その結果、AD及びMS、並びにおそらくはPD及びALSの症状発現を生じうる(Coyle及びPuttfarcken,1993;Goodwinら、1995;Beal,1996;Goodら、1996;Merrill及びBenvenist,1996;Simonian及びCoyle,1996;Vodovotzら、1996)。疫学的データは、アラキドネートからのプロスタグランジンの合成を阻害するNSAIDの慢性使用が、AD発症のリスクを顕著に減少させることを示している(McGeerら、1996;Stewartら、1997)。従って、CDDOメチルエステルの形態A及び形態Bは、NO及びプロスタグランジンの生成を阻害する薬剤として、NDDを治療又は予防する治療的方法に有用であると考えうる。
【0039】
前記のように、種々の前臨床研究において、CDDO−Meは、COX−2及びiNOS(炎症及び発癌の両方に関係した酵素)の発現を阻害する能力を示している。CDDO−Meは、核性因子−カッパB(NF−κB)、並びに転写3のシグナルトランスデューサー及びアクチベーター(STAT3)(炎症、腫瘍進行及び腫瘍の治療耐性に関連した転写因子)の活性を阻害することも示されている。初期の研究は、CDDO−Meが多くの癌細胞系の成長を阻害することを明示しており、NCI−60腫瘍細胞株パネルにおけるCDDO−Meの平均IC50値は約35nMであった。生体内試験において、CDDO−Meが、げっ歯類にインプラントしたヒト腫瘍細胞系又はげっ歯類にインプラントした同系癌細胞系によって形成された腫瘍の成長を効果的に阻害することが確認された(表16)。これらの試験に使用された投与量は、種、株及び投与方法によって変わり、一般に10〜100mg/kg/日であった。
【0040】
以下に詳述する研究は、癌疾患の患者におけるCDDOメチルエステルの有利な作用を示すヒトデータを与える。実施例10参照のこと。
【0041】
前記に照らして、本発明は、CDDOメチルエステル形態を含有する安定な徐放性放出剤形を包含する。本発明では、1日1回の投与で、遅延放出又はパルス放出の形態による剤形が可能となることから、薬物動態性能を薬力学的要求に適合させることによって治療法を最適化することができる。
【0042】
形態B、形態A、及びCDDOメチルエステルの賦形剤分散物を含有する処方物はいずれも経口投与しうる。活性化合物は、該化合物を不活性化しうる酸の作用及び他の自然条件から該化合物を保護する材料でコーティングしてもよい。他の投与法、例えば、局所、皮下、静脈内及び腹腔内投与も本発明の一部である。
【0043】
治療用化合物を投与するために、その不活性化を防止する材料を用いて該化合物をコーティングするか、又は該材料と共に同時投与することが必要な場合がある。即ち、形態B又は形態AのCDDOメチルエステルは、適切な担体、例えばリポソームで、又は希釈剤で患者に投与してもよい。医薬的に許容される希釈剤は、生理食塩水及び水性緩衝液を包含する。リポソームは、水中油中水型CGFエマルジョンだけでなく通常のリポソームも包含する。例えばStrejanら、J.Neuroimmunol.7:27(1984)参照のこと。
【0044】
治療用化合物は、医薬組成物の調製用の不活性希釈剤、添加剤又は食用担体と共に、経口投与することができる。この目的のために、本発明の治療用化合物を、他の成分と共に、硬質又は軟質ゼラチンカプセルに封入するか、錠剤に圧縮するか、又は患者の食事に直接入れてもよい。経口治療投与のために、形態A又は形態Bを、賦形剤と混合し、飲み込み可能な錠剤、口腔錠剤、トローチ剤、カプセル剤、エリキシル剤、懸濁剤、シロップ剤、カシェ剤等の形態で使用できる。同様に、本発明の賦形剤分散物を、種々の剤形(形態A及び形態Bに関して本明細書に記載した剤形を包含する)で与えることができる。組成物及び調製物における治療用化合物の割合は、活性剤の好適投与を達成するために、慣例に従って変化させることができる。
【0045】
さらに、本発明は、有効量の形態BのCDDOメチルエステル又は形態Aを、1以上の非毒性の医薬的に許容される担体及び/又は希釈剤並びに所望であれば他の活性成分と共に含んで成る医薬組成物にも関する。前記のように、活性化合物は、形態B又は形態Aから出発して、均質な賦形剤分散物として製造することができる。そのようなCDDOメチルエステル賦形剤分散物は、固溶体であり、分子レベルにおいて均質分散物とみなすことができる。有利なことに、そのような分散物を、他の医薬的に許容される添加剤と共に処方して、活性化合物を安定化させることができ、生物学的利用能をさらに向上させることも場合により可能である。
【0046】
賦形剤分散物としてCDDOメチルエステルを処方する場合において、分散物用の賦形剤の選択は、賦形剤が優れた「ガラス形成剤」であり、医薬的に許容されるという両基準に依存する。より一般的には、賦形剤は、一般的な周囲温度貯蔵条件より高いTgを与えることによって分散物を安定化させる安定で均質なガラス質マトリックスを形成すべきである。これに関する別の基準は、分散物に使用される賦形剤が、所望の機能特性を与えるように最終処方物に使用できる他の添加剤、例えば、結合剤、充填剤、潤滑剤、グリダント(glidant滑剤)等と化学的に適合性を有するべきであるという基準である。
【0047】
これらの基準を満たすために、賦形剤は、本発明によって、適切には高いTg値を特徴とする下記のような多くの化合物から選択できる:(A)炭水化物、炭水化物誘導体及び炭水化物ポリマー、(B)合成有機ポリマー、(C)有機酸塩、(D)タンパク質、ポリペプチド及びペプチド、並びに(E)高分子量多糖、例えば、ヘパリン(これは硫酸化多糖である)及びヒアルロン酸、ムコ多糖。
【0048】
種類(A)の例は下記の物質である:セルロース誘導体、例えば、ヒドロキシプロピルセルロース(HPC)、ヒドロキシプロピルメチルセルロース(HPMC)及びエチルセルロース;多糖、例えば、ラフィノース、マルトトリオース(maltotriose)、スタキオース、デキストリン(特にマルトデキストリン及びシクロデキストリン等)、デキストラン及び可溶性デンプン;アルジトール、例えば、マンニトール、キシリトール及びソルビトール;並びに、二糖、例えば、ラクトース、トレハロース、マルトース及びスクロース。この種類の好ましい賦形剤は、ヒドロキシプロピルメチルセルロースフタル酸エステル(HPMC−P)である。
【0049】
種類(B)の例は、可変分子量の、ポリ[1−(2−オキソ−1−ピロリジニル)エチレン、a/k/aポビドン又はポリビニルピロリドン(PVP)及び関連コポリマー、例えばPVP/VAである。コポリマーのメタクリル酸ファミリー、例えばメタクリル酸コポリマーC型(USP/NF)もこの種類に包含される。
【0050】
種類(C)の例は、塩、例えば、乳酸、アスコルビン酸、マレイン酸、シュウ酸、マロン酸、リンゴ酸、コハク酸、クエン酸、グルコン酸及びグルタミン酸の、それぞれのナトリウム、カリウム、カルシウム及びマグネシウム塩である。即ち、これに関する代表的な塩は、クエン酸ナトリウム、乳酸ナトリウム、マレイン酸ナトリウム、グルコン酸マグネシウム及びアスコルビン酸ナトリウムである。
【0051】
種類(D)賦形剤の例は下記の物質である:ヒト血清アルブミン;ポリアミノ酸、例えば、ポリアラニン、ポリアルギニン、ポリグリシン及びポリグルタミン酸;カゼイン;コラーゲン;ゼラチン及び精製ゼラチンタンパク質;並びにある種の薬理学的活性化合物、例えばインスリン。
【0052】
前記のように、賦形剤は、医薬処方物のいくつかの物理的特性を変化させることができる。例えば、種々のポリマー賦形剤中の分散物は、処方物の測定されるTgを減少させうる。一般に、Tgは、含まれている材料の比率に基づいて加成性である。従って、非晶形Bより低いTg値を有するポリマーを使用した場合、分散物(混合物)の測定されるTgが低下すると予測される。さらに、湿分又は微量な残留有機溶媒が存在する場合が多く、これらも同様にTgを低下させる。固体CDDO−Me分散物を生成するために、賦形剤の最適選択は、一般に、実験により決定すべきである。例えば、賦形剤のポリエチレングリコール(PEG)ファミリー、例えばPEG6000を使用して、ガラス質XRPD−非晶質分散物を製造した場合、形態AのCDDOメチルエステルに関連した特徴的ピークを有する混合物が生じた。同様の結果が、ビタミンE−TPGS、d−アルファ−トコフェリル酸スクシネートをポリエチレングリコール1000でエステル化することによって製造された賦形剤を使用した場合、並びにエチレンオキシド−プロピレンオキシドコポリマー、例えばPluronic(登録商標)を使用した場合に得られた。下記の例が示すように、本発明によってCDDO−Meとの分散物を生成するために使用された特定のポリマー賦形剤は、純粋形態B薬剤物質と比較して、驚くべき経口生物学的利用能の向上を示す。
【0053】
医薬的に許容される賦形剤を使用してCDDO−Meの均質なガラス質X線非晶質分散物を製造ために方法を変えることができ、本明細書に示す例は、そのような分散物を製造するために噴霧乾燥を使用している。他の製造法を使用して、同等の特性及び有効性を有する本発明の分散物を製造できる。Repkaら,Hot−melt extrusion technology In:ENCLOPEDIA OF PHARMACEUTICAL TECHNOLOGY,2nded(Marcel Dekker,2002),pages203−06及びそれに引用されている文献を参照のこと。そのような他の方法は、溶媒蒸発及び押出、例えば熱溶融押出を包含するが、それらに限定されない。
【0054】
賦形剤の他に、他の添加剤を含有させることにより、活性成分の安定性を補助し、pHを調節し(即ち、緩衝剤)、分散性を向上させ、送達均一性を与えるのを補助し、医薬処方物に所望される他の特性を得ることができる。
【0055】
本発明の化合物又は組成物の投与量は、患者及び投与方法によって異なり、任意の有効量にすることができる。
【0056】
本発明の組成物を投与する所定の治療計画は、標準的及び日常的な前臨床及び臨床試験によって作成することができ、その詳細は、他の因子のうち治療適応症に応じて変わる。投与される活性剤の量は幅広い範囲で変化させてもよく、それによって、単位投与量において、患者体重に基づく1日当たりの薬理学的有効量を与えて、所望の効果を得ることができる。所望の投与量は、治療される症状によっても変化しうる。例えば、急性の癌の治療は、関節炎のような炎症性疾患の治療より有意に多い投与量を必要としうる。
【0057】
特に本発明の組成物は、単位投与量として投与され、好ましくは、1日に1〜3回、最も好ましくは1日に1回投与されて、所望の効果を与える。
【0058】
さらに、本発明の組成物は、2日おき、3日おき、4日おき、5日おき、6日おき、又は1週間に1回投与しうる。
【0059】
本発明の組成物は、患者の具体的なニーズに基づいて、単独で、又は他の薬剤と組み合わせて投与してもよい。特に、本発明の組成物は、治療計画の一部として、抗癌剤と共に投与しうる。例えば、膵臓癌等の癌の治療中に、CDDOメチルエステルを、ゲムシタビン、又は他の薬剤と共に投与できる。
【0060】
一般に、本発明の医薬組成物は、通常の材料及び方法、例えば、混合、調合等を使用して製造される。さらに、形態A又は形態Bを含有する薬剤は、好適な佐剤、担体、賦形剤及び安定剤等を包含するがそれらに限定されない他の成分も含有しうる。本発明の治療処方物は、好ましくは固体であるが、原則として液体、例えば懸濁液又は乳濁液でありうる。
【0061】
本発明により、経口維持投与量は、一般に、約0.1mg〜約1000mgであり、好ましくは1日に1回投与される。その投与量は、変えることができ、患者の体重に適合させることができる。一般的な投与量は、錠剤及びカプセル剤等の好ましい単位剤形を用いて約0.01mg/kg〜100mg/kgでありうる。
【実施例】
【0062】
下記の実施例は、例示するものにすぎず、本発明を限定するものではない。実施例に使用した材料及び方法を以下に概説する。
【0063】
a.材料
溶媒及び他の試薬は、販売業者から購入し、HPLCグレード又はACSグレードであった。
【0064】
b.実験方法
i.近似溶解度−溶媒添加法(Approximate Solubility--Solvent Addition Method)
計量した試料を、室温にて試験溶媒のアリコートで処理した。試験材料が完全に溶解したことを、目視検査によって判定した。溶解度は、完全に溶解させるために使用された合計溶媒に基づいて推定した。実溶解度は、溶媒のアリコートの使用が多すぎた場合か、又は遅い溶解速度によって、計算値より高くなりうる。溶解度は、実験中に溶解が生じなかった場合に「より低い」として表わされる。ただ1つのアリコートを添加して完全に溶解した場合は、溶解度は「より高い」として表わされる。
【0065】
ii.多型の選別
熱力学的及び動力学的結晶化法を共に使用した。これらの方法を以下により詳しく記載する。結晶化により固体試料を得た時点で、それらの形態について顕微鏡下で検査するか、又は肉眼で観察した。あらゆる結晶形について留意したが、粒度が小さいため、形態を判別できない固形物もあった。次に、固体試料をXRPDによって分析し、パターンを相互に比較して、新しい結晶形又は非晶形を同定した。
【0066】
(i)低温沈殿(CP)
種々の溶媒で温度を上昇させて溶液を調製した。次に、室温以下で0.2−μmのナイロン又はPTFEフィルターによりその溶液を濾過して、貧溶媒に添加した。その際、固形物が存在するか否かを確認した。固形物を確認できない場合、又は固形物の量がXRPD分析のために少なすぎると判断した場合は、そのバイアルを冷凍庫に入れた。得られた固形物を濾過によって分離し、分析前に乾燥させた。
【0067】
(ii)高速蒸発(FE)
種々の溶媒で溶液を調製し、溶解しやすくするため、アリコートの添加と添加の間に超音波処理した。目視検査で混合物が完全に溶解したと判断したら、0.2−μmナイロンフィルターでその溶液を濾過した。蓋をしていないバイアル中において、室温でその濾過した溶液を蒸発させた。生成した固形物を分離し、分析した。
【0068】
(iii)凍結乾燥(FD)
1,4−ジオキサン溶液を調製し、0.2−μmナイロンフィルターで濾過し、ドライアイスで凍結させた。続いて、その凍結試料をFTSシステムFlexi−Dryを使用して凍結乾燥した。凍結乾燥温度は制御しなかった。
【0069】
(iv)微粉化
材料の微粉化は流体エネルギーミル(fluid energy mill)で行うことができ、粒度を1〜20ミクロンに減少させることができる。これらの過程の詳しい記載は、PERRY‘S CHEMICAL ENGINEERS’ HANDBOOK、第7版(McGraw Hill,1998)に見出すことができる。
【0070】
(v)粉砕
ステンレス製ミルローターに固体試料を小金属球と共に投入した。試料に少量の水を添加した場合もあった(湿式粉砕)。次に、その試料をRetesh MM220型ミキサーミルで30Hzにおいて約20分間粉砕した。得られた固形物を分離し、分析した。
【0071】
(vi)低温粉砕
粉砕棒を有するステンレス製粉砕ジャーに固体試料を投入した。次に、その試料をSPEX Certiprep 6750型クライオミルで、15Hzにおいて、一定時間粉砕した。実験中、粉砕ジャーを液体窒素浴に浸した。その固形物を分離し、分析した。
【0072】
(vii)溶融/急冷
顕微鏡スライドガラスに固体試料を載せ、平らにした。次に、固形物が溶融するまで、スライドガラスを設定温度でホットプレートに載せた。溶融後、ホットプレートからスライドガラスを取り除き、冷たいカウンタートップに載せて、急速に冷却した。得られた固形物を窒素下にて乾燥し、分析した。
【0073】
(viii)回転蒸発
種々の溶媒で溶液を調製し、0.2−μmナイロンフィルターで濾過した。試料をロータリーエバポレーターに載せ、乾燥したら取り除いた。得られた固形物を分離し、分析した。
【0074】
(ix)低速蒸発(SE)
種々の溶媒で溶液を調製し、溶解しやすくするため、アリコートの添加と添加の間に超音波処理した。目視検査で混合物が完全に溶解したことを確認し、溶液を0.2−μmナイロンフィルターで濾過した。濾過した溶液を、小さい穴をあけたアルミ箔で覆ったバイアル中で、室温又は窒素下で蒸発させた。このようにして生成した固形物を分離し、分析した。
【0075】
(x)低速冷却(SC)
約60℃にて種々の溶媒で飽和溶液を調製し、温かいうちに、0.2−μmナイロンフィルターで濾過して、開放バイアルに入れた。そのバイアルを覆い、室温へゆっくり冷却した。固形物が存在するか否かを確認し、確認できない場合、又は固形物の量がXRPD分析のために少なすぎると判断された場合、バイアルを冷蔵庫に入れた。再び、固形物の有無を確認し、確認できない場合は、バイアルを冷凍庫に入れた。生成した固形物を濾過によって分離し、分析前に乾燥させた。
【0076】
(xi)スラリー実験
過剰の固形物が存在するように充分な固形物を所定の溶媒に添加して溶液を調製した。次に、混合物を密閉バイアル中にて室温で撹拌した。7日又は10日後に、固形物を真空濾過によって分離し、分析した。
【0077】
(xii)ストレス実験
種々の温度及び/又は相対湿度(RH)環境下で、計測の間、固形物にストレスを加えた。飽和塩溶液を含有する密閉室又はESPEC温度及び湿度室に試料を入れることによって、特定のRH値を得た。塩溶液は、ASTM規格の手順に従い選択し調製した。試料をストレス環境から取り出して、直ちにXRPDによって分析した。
【0078】
iii単結晶構造決定
(i)試料調製
約60℃でメタノール中のCDDOメチルエステルの飽和溶液を調製し、温かいうちに、0.2−μmフィルターで濾過して開放バイアルに入れた。バイアルを覆い、室温へゆっくり冷却した。錐体状の小塊が1日後に観察された。
【0079】
(ii)データ収集
2辺が約0.01x0.01mmの寸法を有するCDDO−Me(C32H43NO4)の無色の小板を、ランダムの配向のグラスファイバー上にマウントした。グラファイト結晶の入射ビームモノクロメータを備えたNonius KappaCCD回折計によってMo Kα線(λ=0.71073Å)により予備実験及びデータ収集を行った。精密化は、SHELX97(Sheldrick,1997)を使用してLINUX PCで行った。
【0080】
データ収集のための格子定数及び方位マトリックスは、2°<θ<22°の範囲で46742の反射を有するような設定角を使用して、最小二乗精密化(least−squares refinement)から得た。DENZO/SCALEPACK(Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307)による精密化したモザイシティ(mosaicity)は0.32°であり、優れた結晶品質を示した。空間群は、XPREPプログラム(Bruker, XPREP in SHELXTL v. 6.12., Bruker AXS Inc., Madison, WI(2002))によって決定した。条件h00 h=2n,00l l=4nの系統的存在、及びそれに続く最小二乗精密化から、空間群はP43212(no.96)と決定された。
【0081】
データは、150±1Kの温度で、44.43°の最大2θ値まで収集した。
【0082】
(iii)データ整理
フレームはDENZO−SMN(Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307)で積分した。合計46742の反射を収集し、そのうち9168が固有であった。ローレンツ偏光補正をデータに適用した。線吸収係数はMo Kα線について0.074mm−1である。SCALEPACK(Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307)により実験的吸収補正を適用した。透過率は0.9995〜0.9999であった。等価な反射の強度を平均化した。平均化のためのアグリーメントファクター(agreement factor)は、強度に基づいて9.3%であった。
【0083】
(iv)構造解析及び精密化
SHELXS97(Sheldrick, G. M. SHELX97, A PROGRAM FOR CRYSTAL STRUCTURE REFINEMENT, University of Gottingen, Germany(1997))による直接法によって構造を解析した。残っている原子を、次の差フーリエ合成で配置した。水素原子は精密化に含まれたが、水素原子が結合している原子上に拘束された。以下の関数を最小にすることによって、フルマトリックス最小二乗において構造を精密化した:
【数1】
【0084】
重みwは1/[σ2(Fo2)+(0.0176P)2+(0.0000P)]として定義され、ここでP=(Fo2+2Fc2)/3である。
【0085】
散乱因子は、「International Tables for Crystallography」(INTERNATIONAL TABLES FOR CRYSTALLOGRAPHY, Vol. C, Tables 4.2.6.8 and 6.1.1.4, Kluwer Academic Publishers:Dordrecht, The Netherlands(1992))から得た。精密化に使用した9168の反射のうち、Fo2>2σ(Fo2)を有する反射だけをRの計算に使用した。合計5421の反射を計算に使用した。精密化の最終サイクルは1024個の変数パラメータを含み、下記の非加重及び加重したアグリーメントファクターで収束した(最大パラメータシフトは、その推定標準偏差の<0.01倍であった):
【数2】
【0086】
単位重量の観測の標準偏差は1.05であった。最終の差フーリエにおける最高ピークは0.22e/Å3の高さであった。最小負ピークは−0.25e/Å3の高さであった。
【0087】
(v)計算した粉末X線回折(XRPD)パターン
計算XRPDパターンは、PowderCell 2.3(Kraus, W., and G. Nolze, POWDERCELL FOR WINDOWS VERSION 2.3, Federal Institute for Materials Research and Testing, Berlin(1999))、並びに単結晶データからの原子座標、空間群及び単位格子パラメータを使用して、Cu線について生成した。
【0088】
(vi)ORTEP図及び充填図
ORTEP図は、ORTEP III(Johnson, C. K. ORTEPIII, Report ORNL-6895, Oak Ridge National Laboratory, TN, U.S.A. 1996)(OPTEP-3 for Windows V1.05 ,. Farrugia, L.J., J. Appl. Cryst. 1997, 30, 565)を使用して作成した。原子は50%確率の異方性熱振動楕円体で表わされている。充填図は、CAMERON(Watkin, D. J.; Prout, C .K.; Pearce, L. J. Cameron, CHEMICAL CRYSTALLOGRAPHY LABORATORY, University of Oxford, Oxford, 1996)モデリングソフトウエアを使用して作成した。別の図及びBFDH形態予測は、Mercury1.4.1(Bruno, et al., Acta Crystallogr. 2002, B58, 389)を使用して生成した。
【0089】
c.計測方法
i.示差走査熱量測定(DSC)
TA示差走査熱量計2920又はQ1000で分析を行った。該熱量計は、インジウムを標準物質として使用して較正した。試料を、圧着蓋(crimped lid)形状を有する標準アルミニウムDSC皿に入れ、重量を正確に記録した。試料セルを25℃で平衡にし、窒素パージ下にて10℃/分の速度で最終温度250℃まで加熱した。
【0090】
ii.動的蒸気収着/脱離(DVS)
湿分収着/脱離データを、VTI SGA−100蒸気収着分析器で収集した。収着及び脱離データを、5%〜95%相対湿度(RH)の範囲で、10%RH間隔で窒素パージ下で収集した。試料は分析前に乾燥しなかった。分析に使用した平衡基準は、5分間で0.010%未満の重量変化であり、重量基準が満たされない場合は3時間の最大平衡時間であった。試料の初期湿分についてデータを補正しなかった。塩化ナトリウム及びポリビニルピロリジンを較正標準として使用した。
【0091】
iii.カールフィッシャー(KF)
Mettler Toledo DL39カールフィッシャー滴定器を使用して、水分測定のための電量的カールフィッシャー(KF)分析を行った。Hydranal−Coulomat ADを含有するKF滴定容器に、約24〜32mgの試料を入れた。次に、電気化学的酸化によってヨウ素を生じる発生電極によって、試料を滴定した:
2I−⇒I2+2e。再現性を確実にするために3回反復実験を行った。
【0092】
iv.ホットステージ顕微鏡法
Leica DM LP顕微鏡に載せたLinkamホットステージ(FTIR 600型)を使用して、ホットステージ顕微鏡法を行った。交差偏光子(CP)及びラムダ(λ)補償子を有する20×対物レンズを使用して、試料を観測した。試料をカバーガラスに載せた。次に第二のカバーガラスを試料上に置いた。ステージが加熱されると共に、各試料を目視観測した。SPOT Software v.4.5.9.を有するSPOT Insight(商標)カラーデジタルカメラを使用して、画像を取り込んだ。USP融点標準品を使用して、ホットステージを較正した。
【0093】
v.変調示差走査熱量測定(MDSC)
冷蔵冷却システム(RCS)を備えたTA Instruments示差走査熱量計によって、変調示差走査熱量測定データを得た。試料をアルミニウムDSC皿に入れ、重量を正確に記録した。皿を蓋で覆い、圧着した。変調振幅+/−0.8℃及び60秒周期を使用し、−25℃から250℃に2℃/分の平均昇温速度で、MDSCデータを得た。較正標準としてインジウム金属及びサファイアを使用して、それぞれ温度及び熱容量を較正した。記録されるガラス転移温度は、可逆熱流対温度曲線における階段状変化の変曲から得る。
【0094】
vi.核磁気共鳴(NMR)
溶液相1H NMRスペクトルはSpectra Data Services,Inc.によって収集された。取得パラメータは各スペクトルに記載されている。スペクトルはテトラメチルシランを0.0ppmにおける内部基準とした。
【0095】
vii.光学顕微鏡法
光学顕微鏡法による観測は、Wolfe偏光光学顕微鏡によって倍率4xで収集した。交差偏光子(CP)を使用して、試料における複屈折を観測した。
【0096】
viii.走査型電子顕微鏡法(SEM)
走査型電子顕微鏡法(SEM)は、FEI Quanta200走査型電子顕微鏡を使用して行った。高真空モード下に、固体状態後方散乱(Etd)検出器を使用した。ビーム電圧は5.0kVであった。Cressington 108auto Sputter Coaterを使用して、約20mA及び約0.13mbar(Ar)においてAu/Pdを用いて75秒間で、試料をスパッタコーティングした。アルミニウム試料台に固定した両面粘着テープ上に少量を載せることによって、分析のために試料を調製した。NIST標準品を使用して、倍率について器具を較正した。xTm(v.2.01)、ビルド番号1564を使用してデータを収集し、XT Docu(v.3.2)を使用して分析した。SEM画像について示されている倍率は、初期データ収集時に計算した。各画像の下部に示されているスケールバーは、画像のサイズ変更する上で正確であり、サイズ測定を行う際に使用すべきである。
【0097】
ix.熱重量分析(TG)
TA Instruments2950熱重量分析器を使用して、分析を行った。較正標準品はニッケル及びAlumel(商標)であった。各試料をアルミニウム試料皿に入れ、TG炉に挿入した。25℃で試料を先ず平衡させ、次に、窒素気流下に10℃/分の加熱速度で、特に指定されないかぎり最終温度350℃に加熱した。
【0098】
x.粉末X線回折(XRPD)
(i)Inel XRG−3000
120°の2θレンジを有する湾曲した位置感受性検出器を備えたInel XRG−3000回折計で、粉末X線回折分析も行った。0.03°2θの分解能でCu Kα線を使用して、実時間データを収集した。管電圧及びアンペア数を、それぞれ40kV及び30mAに設定した。パターンを直接比較しやすくするために2.5から40°2θのパターンを表示する。薄壁ガラスキャピラリーに試料を詰めることによって分析用の試料を調製した。データ収集中にキャピラリーを回転させるように動力化されたゴニオメータヘッドに、各キャピラリーを取り付けた。器具の較正は、ケイ素参照標準品を使用して毎日行った。
【0099】
d.別の計算法
i.PDF
X線非晶質データのコンピュータ分析に使用される方法の1つは、対相関関数(PDF)である。名前が示すように、PDFは、材料内の全てのコヒーレントな原子−原子相互作用の線形和からなる。不完全な(無秩序な)材料は、結晶相と同じ原子−原子相互作用を示すが、それは短い長さスケールにおいてである。従って、そのような材料は、最初の数ナノメートルにおけるPDFのピークを調べることによって、親結晶材料と比較することができる。0Å〜約5Åの範囲のスペクトルの比較は、この領域のアーチファクトのため困難である。
【0100】
ガラス質材料の形成時では、分子が結晶ピークの位置を緩和させるため、PDFピークは結晶ピークの位置からいくらか移動する。このことは、熱膨張/収縮事象に似ており、その場合、PDFピークは少し移動するが、それでも相対的なピーク強度は元の結晶質材料に関係していることが確認できる。材料が熱力学的非晶質状態になると、いくらかの点群対称関係が失われ、複雑度の減少したPDFを与える。ピーク移動するものも存在する。ガラス質/非晶質材料では、PDFが2〜3最近接(NN)距離において急速にゼロに低下する。
【0101】
X線パターンにおけるバックグラウンドを最小限にする測定条件を使用し、測定したX線データからPDFを計算するアルゴリズムを使用する。全試料について測定データの全範囲を使用し、PatternMatch v2.2.1を使用して、PDFを計算した。
【実施例1】
【0102】
溶解度推定値
種々の溶媒中において近似溶解度を室温で測定し、その結果を表2に示す。CDDOメチルエステルは、使用した有機溶媒の大半で高い溶解度を示している。水への溶解度は、0.1mg/mL未満であると考えられる。
【実施例2】
【0103】
多型の選別結果
約50個の多型の選別実験を行った。形態Aは約50%の試料から観測された。形態Aの形成は、特定の結晶化条件に限定されず、種々の実験及び溶媒において調製した。形態Bは、凍結乾燥、溶融/急冷、及びいくつかの蒸発実験から調製した。
【0104】
使用した溶媒のアルファベット順で表3〜5に多型の選別試料を示す。形態A及び形態Bの代表的なXRPDパターンを図2において比較する。形態の特性評価のデータを、以下の実施例において概説する。
【実施例3】
【0105】
CDDOメチルエステル−形態A(非微粉化)の特性評価
形態Aは非溶媒和である(表6)。形態Aの単結晶構造を、前記の方法に基づいて決定した。CDDOメチルエステルの結晶を成長させ、単結晶構造を分析した。結晶構造を単結晶X線回折によって決定した。CDDOメチルエステルの提示された構造を図1に示す。
【0106】
正方晶のセルパラメータ及び計算された体積は次の通りである:a=14.21620(10)Å,b=14.21620(10)Å,c=81.5875(12)Å,α=90.00°,β=90.00°,γ=90.00°,V=16488.9(3)Å3。CDDOメチルエステルの分子量は、505.70g/mol(Z=24)であり、計算された密度は1.222gcm−3であった。空間群はP43212(no.96)であった。結晶データ及び結晶学的データの収集パラメータの概要を、表10に示す。
【0107】
R値0.051(5.1%)によって示されるように、得られた構造の質は高い。一般に、0.02〜0.06の範囲のR値は最も信頼できる決定構造であるとみなされる。
【0108】
単一CDDOメチルエステル分子のORTEP図を図12に示す。図13に示されている非対称単位は3個のCDDOメチルエステル分子を含有している。分子は、図1の提示された構造と同じである。
【0109】
結晶軸a、b、及びcに沿って見た充填図をそれぞれ図14〜16に示す。水素結合を有さず、結晶構造は多くのファンデルワールス相互作用を有する。結晶b軸に沿って見た図(図15)は、正方晶螺旋軸の充填される配置の螺旋性、及び予測されるBFDH形態を強調表示している。予測される形態は、データ収集に使用した単結晶で観測された性質とよく一致している。
【0110】
単結晶データから生じたCDDOメチルエステルの計算によるXRPDパターンを図17に示す。CDDOメチルエステルの実験により得られたXRPDパターンを図18に示す。形態AのXRPDパターンにおける特徴的ピークを表17に示す。計算及び実験により得られたXRPDパターンの比較(図19)によって、実験によるパターンの全てのピークが、計算によるXRPDパターンに示されていることが明らかである。このことは、バルク体がおそらく単一相であることを示している。ピーク位置において見られる共通のわずかなシフトは、実験の粉末パターンを室温で収集し、単結晶データを150°Kで収集したことによるものと考えられる。構造の質を向上させるために、単結晶分析に低温を使用する。
【0111】
要約すれば、CDDOメチルエステルの形態Aの単結晶構造は、提示された分子構造と一致することが確認された。空間群はP43212(no.96)と確認された。CDDOメチルエステルの構造は、結晶b軸に沿って螺旋状に充填された3個の分子から成る。実験によるパターンの全ピークが計算によるXRPDパターンに示されており、バルク体がおそらく単一相であることが示唆されている。
【0112】
形態Aの熱データを図3に示す。そのDSC曲線は、約157℃におけるベースラインシフト、及び約222℃の開始温度での吸熱(約224℃においてシグナルが最大)を示している。約224℃における事象は、ホットステージ顕微鏡法による溶融であることが確認された(図4)。熱重量分析(TG)曲線は、150℃まで0.34%の無視しうる重量減少が生じ、続いて150〜210℃において1.2%の重量減少が生じたことを示す。カールフィッシャーデータは、材料が約0.38%の残留水を含有することを示しており、このことは、TGによって観測された初期重量の減少と一致する。
【0113】
DVSデータは、形態Aが吸湿性でないことを示す(図5)。実験を通して、材料の重量変化はわずかであることが示された。得られた材料はXRPDで分析され、形態Aである。
【0114】
SEM画像を図6に示す。錐体、タブレット及び板状等のいくつかの結晶の性質が観察される。
【0115】
種々の条件において、形態Aの物理的安定性を調べた(表7)。25℃/60%RH又は40℃/75%RHで7日間ストレスを加えた試料は共に、わずかな重量変化(それぞれ、0.6%の減少及び0.2%の増加)を示し、このことは、形態Aが吸湿性でないことを示している。2つの試料を、1つは乾燥状態で、もう1つは少量の水と共に、ボールミルで約20分間粉砕した。全試料をXRPDにより再分析したところ、形態Aを維持していた。195℃で15分間試料にストレスを加え、2%の重量減少を示した。得られた材料のXRPDは、形態Aのものと同様であるが、ベースラインのノイズの増加は顕著である(図7)。
【0116】
溶液NMRスペクトルを図8に示す。スペクトルは、CDDOメチルエステルの構造と一致している。約1.6及び7.3ppmにおけるピークは、それぞれ水及びクロロホルム(交換による)を表している。
【0117】
形態Aは非溶媒和、かつ、非吸湿性であることから、ホットステージ顕微鏡法の際に分析者の観測に基づくと約228℃で溶融する。
【実施例4】
【0118】
CDDOメチルエステル−形態A(微粉化)の特性評価
XRPDによって、微粉化した形態AのCDDOメチルエステルが、形態Aであることを確認した(図2、表1)。微粉化した材料は、当技術分野で周知の従来法、例えばエアジェットミリングによって製造できる。これらの知見は、微粉化がそのXRPDパターンを変えるような形態Aへの影響を及ぼさないことを示していると考えられる。
【実施例5】
【0119】
CDDOメチルエステル−形態Bの特性評価
形態B材料は、表3に示すように、凍結乾燥、溶融/急冷、及び他のいくつかの蒸発実験から調製できる。
【0120】
変調DSC(MDSC)データを図9に示す。可逆曲線は、約125℃におけるガラス転移温度(Tg)を示している。不可逆曲線は、195℃で最大となるシグナルを有する発熱、及び223℃で最大となるシグナルを有する吸熱を示している。不可逆事象は、形態B材料の結晶化(発熱)が生じ、続いて結晶化した材料の溶融(吸熱)が生じたことによって起こった可能性が非常に高い。
【0121】
種々の条件における形態B材料の物理的安定性を調べた(表9)。22℃/97%RH、40℃/75%RH、80℃/0%RH、及び195℃/周囲RHにおいてストレスを加えた試料は、形態Bのままであった。200℃/周囲RHで60分間にわたって材料にストレスを加えることによって、形態A及び少量の形態B材料を生じた(図10)。
【0122】
溶液NMRスペクトルを図11に示す。スペクトルは、CDDOメチルエステルの構造と一致している。約1.6、5.3及び7.3ppmにおけるピークは、それぞれ、水、ジクロロメタン及びクロロホルムを表す。
【0123】
形態Bは、吸湿性でなく、約200℃で形態Aに結晶化し、約125℃〜130℃のガラス転移温度(Tg)を有する。
【0124】
要約すれば、形態Bは吸湿性でない。MDSCデータは、形態Bのガラス転移温度(Tg)が、約125℃〜130℃であることを示している。形態B材料は、約200℃でストレスを加えた場合に形態Aに結晶化する。
【実施例6】
【0125】
形態BのCDDOメチルエステル及びCDDOメチルエステルポリマー賦形剤分散物の安定性試験
(i)精製した形態Bの試験
形態BのCDDOメチルエステルを、種々のストレス条件に付した。表15は、これらの試験のいくつかの結果を示している。溶媒として酢酸エチルを使用した、有用ではあるが好ましくない本発明の実施形態によって調製した形態BのCDDOメチルエステルは、かなりの安定性を示す。それにもかかわらず、酢酸エチルの存在下で調製した形態B試料の試験では、60℃及びそれ以上の温度で28日間保存した後に、形態Aが形成することが明らかとなった。これに対して、本発明の好ましい実施形態(下記の実施例11はその例である)によって調製した全ての試料は、特に厳しい条件下でのストレス試験後でも、非晶質特性を保持していた(表15参照)。これらの試験は、特に前記の好ましい実施形態によって製造された場合に、形態B材料の驚くべき安定性を示している。
【0126】
さらに、種々の条件下で調製した形態B試料が同様の化学特性を有するかについて分析した。前記の通り、低温粉砕、溶融急冷、及び噴霧乾燥法によって形態B試料を調製した。さらに、微粉化していない形態Bを微粉化して、微粉化形態Bを調製した。試料のPDF分析を行い(図28)、その試料が性質上、ガラス質であることを確認した。
【0127】
(ii)CDDO−Me分散調製物を用いた試験
噴霧乾燥によって調製した、形態Bの種々のポリマー固体分散物の性能を比較するために、試験を行った。試験した生成物の特性は、安定性及び薬剤溶解プロファイルを含んでいた。
【0128】
活性医薬成分(API)対ポリマーの3種の比率が、20:80、40:60及び60:40%w/wとなる種々のポリマーを使用した。
【0129】
下記の3つのポリマーを選択した:
メタクリル酸−エチルアクリレートコポリマー(1:1);
コポビドン(1−ビニル−2−ピロリドン−ビニルアセテートコポリマー(3:2));
ヒプロメロースフタレート。
【0130】
形態A結晶質材料又は形態B材料のCDDO−Me及び試験用に選択したポリマーの溶液を、一般に溶液中10〜20wt%の固形物を与えるのに適切な重量比で、好適な溶媒に溶解させた。一般に使用した溶媒はアセトンであった。乾燥及びキャリヤーガスとして窒素を使用して、二流体ノズルを備えた実験室スケールの噴霧乾燥器(BUCHI、B−290型)によって、その得られた溶液を噴霧乾燥した。キャリヤーガス入口温度は65〜85℃を、出口温度は50〜60℃を一般に使用した。固形物を収集し、噴霧乾燥粉末を真空下で再び乾燥させ、有機溶媒の度合いをさらに減少させた。
【0131】
残留有機溶媒、ガラス転移温度(Tg)及び嵩密度について、噴霧乾燥粉末を分析した。再度乾燥させた後に、その粉末について、純度、水含有量、平均粒度、粉末X線回折(XRPD)による結晶質材料の不存在、及び溶解プロファイルも分析した。
【0132】
分散物の物理化学的特性を、短期のストレス後(40℃/75%RHで5日後)に、XRPD及び変調示差走査熱量測定(mDSC)によって評価した。
【0133】
これらの試験によって、ガラス転移温度Tgが、おそらくは短期のストレスを加えている間の湿分の吸収によって、ストレス負荷中に低下することが分かった。その低下は、低いCCDOメチルエステル対ポリマーの比率を有する処方物を使用した場合に、より顕著であった。短期のストレス前の試料について、1つ又は2つの吸熱転移がより高い温度で観測されたが、これらの温度は初期に観測された温度より幾分低い。これらの転移に関係したエンタルピーは、ポリマー含有量の減少に伴って減少した。このことは、その転移がポリマーと関係しており、結晶形の溶融とは関係していない可能性が高いことを示している。実際に、PVP/VAを使用して調製した分散物について、この吸熱転移の温度は、製造業者から受け取った純粋な賦形剤について観測されたものと同様であった。各場合に、ストレス後のXRPDプロファイルでは、約13.5°2θを中心とする特徴的なハローパターンが続き、結晶形に関連したピークは検出されなかった。
【0134】
より大規模な噴霧乾燥装置を使用して、2つの処方物についてさらに噴霧乾燥試験を行った。その場合、Niroパイロットスケール乾燥器PSD−1型(mobile minor2000)を使用した。前記と同等のノズル及び噴霧乾燥条件も使用した。表20及び21は、噴霧乾燥用に調製した溶液、及び噴霧乾燥後の溶液の特性を要約している。処方物は、形態Bより低いTgを有するポリマーを含有するよう処方したため、純粋形態Bと比較してより低いTgを示した。
【実施例7】
【0135】
CDDOメチルエステルの投与:カニクイザルにおける形態B対形態A
この試験の段階1において、純粋で微粉化した形態AのCDDO−Me又は純粋で微粉化したCDDO−Meの形態Bを含有する多数の硬質ゼラチンカプセルを、下記のように調製した:(i)適切に計量した薬剤物質の純粋形態を、サイズ1の硬質ゼラチンカプセルに添加し、(ii)カプセルを密閉した。別の賦形剤は使用しなかった。CDDOメチルエステルの形態B又は形態Aを、ゼラチンカプセルでカニクイザルに経口投与した(全ての場合で、投与量は4.1mg/kg)。
【0136】
CDDOメチルエステルの形態Bの投与は、サルにおいて、CDDOメチルエステルの形態Aを同じ投与量で投与した場合に比べ約520%高い暴露量の中央値を与えた。表11は、各動物の薬剤暴露を比較している。この試験において、「n」値を増加させ、データ信頼性を高めるために、交差法を行った。交差の間に、1週間の洗い流し期間を組み込んだ。図20は、試料採取した母集団における、時間の経過に対する、CDDOメチルエステルの両形態の得られた血漿濃度を示す。図21は動物♯505M及び♯507Fの比較したCDDOメチルエステル血漿濃度を示す。図22は、動物♯508F及び♯502Mの比較したCDDOメチルエステル血漿濃度を示す。
【0137】
さらに、CDDOメチルエステルの経口剤形(CDDOメチルエステル賦形剤分散物を含有する経口剤形を包含する)の比較生物学的利用能を評価するために、下記のように2つの別の試験段階2及び3を行った。これらの試験は、いくつかのCDDO−Meの形態B、即ち実施例6で記載したポリマー分散物を含んだ。全ての処方物は、一般に使用される処方添加剤を含有していた。
【0138】
段階2:
微粉化した形態A材料の試料から開始して、ナノ結晶質CDDO−Meの水性懸濁液を調製した。平均寸法2mmのジルコニアボールを含有するRetsch(登録商標)Planatory Ball Mill PM 400型に、25gmの微粉化したCDDO−Me(平均粒度分布6.1uM)、5gmのドキュセートナトリウム、1gmのTween80、及び68.3gmの水を装填した。約400RPMで粉砕を開始し、2時間継続した。レーザー光粒度計を使用した粒度分布(PSD)の測定によって、0.37μMの平均PSDが得られたことが示された。この濃厚懸濁液に、1gmの微結晶性セルロース及び0.2gmのキサンタンガムを添加し、短時間混合し、懸濁液を冷却保存した。
【0139】
スプレーノズルサイズが0.4mmのトップスプレーアセンブリを使用して、実験室規模のAeromatic Strea 1流動層において、乾燥賦形剤ブレンドにボールミル粉砕したナノ懸濁液を吹付け塗布した。入口温度を55℃に設定した。吹付けの間の出口温度は32〜35℃であった。得られた顆粒を、約5分間、出口温度が38℃に達するまで乾燥させた(添付書類(Attachment)3−5)。被覆物の組成を下記に示す。
【表4】
【0140】
得られた乾燥顆粒をHPLC分析にかけて、活性成分を定量したところ、14.4%(wt/wt)となり、理論値(5.7%)よりかなり高かった。HPLC分析に基づいて、カプセルは、正味のCDDO−Me含有量が30mgになるように充填した。
【0141】
微結晶性セルロース、アルファ化デンプン、クロスポビドン(崩壊剤として機能する)、コロイド状二酸化ケイ素及び植物等級ステアリン酸マグネシウムを添加剤として使用して、結晶質の微粉化した形態A及び非晶質の微粉化した形態Bの処方物を、通常の乾燥粉末ブレンド法によって調製した。平均PSDが6.1μMの微粉化したCDDO−Meの形態Aを、形態Aの処方物に使用し、平均PSDが10.8μMの微粉化したCDDO−Meの形態Bを、対応するCDDO−Meの形態Bの処方物に使用した。下記の表は、両方の処方物の定量的組成を示す。
【表5】
【0142】
15匹の雄のカニクイザル(Macaca fascicularis)のそれぞれに、目標投与量が10mg/kgとなるように、CDDOメチルエステル(3種類の処方物、1処方物につき5匹のサル)を単一経口投与した。サルは年齢1〜3才であり、体重2.5〜3.5kgであった。血液試料を、投与後72時間まで収集した。
【0143】
段階3:
微結晶性セルロース、ラクトース一水和物、クロスポビドン(崩壊剤として機能する)及びラウリル硫酸ナトリウムを添加剤として使用して、実施例6に記載したCDDO−Me賦形剤分散物をさらに、通常の乾燥粉末ブレンド法によって処方した。各処方物の定量的組成を下記に示す。
【表6】
【0144】
好適な洗い流し期間(7〜10日間)の後、段階2に使用した同一の15匹の雄カニクイザルのそれぞれに、目標投与量が10mg/kgとなるように、CDDOメチルエステル(3種類の処方物、1処方物につき5匹のサル)を単一経口投与した。血液試料を、投与後72時間まで収集した。
【0145】
下記の表は、試験の各段階を要約している
【表7】
【0146】
雄のカニクイザルに投与したCDDOメチルエステルの平均静脈内投与量及び平均経口投与量並びに平均濃度を下記に要約する:
【表8】
【0147】
段階2においては、サイズ2のゼラチンカプセルを使用して処方物を送達し、段階3においては、サイズ1のゼラチンカプセルを送達に使用した。各カプセルにおける正味薬剤含有量は30mgであり、それは、各サルの体重が3kgであるという仮定に基づいた10mg/kgの薬剤投与量に対応する。カプセルを経管栄養器具に加え、動物に経管栄養器具を取り付け、空の注射器による空気圧によって経管栄養器具の末端からカプセルを放出させた。最後のカプセルの投与後に、少量の水(約10mL)を経口的に与えた。
【0148】
下記の各時点(実際の時間を記録した)で、各動物の大腿静脈又は動脈から、血液試料(約1mL)を逐次採取し、K2−EDTAを含有する試験管に移した:
段階2 投与前、投与後1、2、4、8、16、24、48及び72時間
段階3 投与前、投与後1、2、4、8、16、24、48及び72時間
収集後、どの試料も充分に混合し、約4℃で冷蔵する前にぬれた氷上に置いた。血液中のCDDOメチルエステル濃度は、HPLC−MS/MSによって分析した。
【0149】
結果を表22及び図30に示す。段階2について、形態Bは、試験した2つの形態Aの処方物より有意に高い生物学的利用能を示した。段階3の結果は、各CDDO−Me、即ちポリマー分散物に基づく処方物が、微粉化した形態A又はナノ結晶質形態Aの処方物のいずれよりも極めて高い生物学的利用能を有することを示した。メタクリル酸コポリマーC型及びHPMC−P処方物は、被験サルにおいて最も高い生物学的利用能を示した。
【実施例8】
【0150】
CDDOメチルエステルのヘミベンゼネート形態の特性評価
CDDOメチルエステル合成の最後の回収段階を繰り返す種々の実験を行った。Hondaら、2000を参照のこと。その目的は、ベンゼン/アセトン(10:1)の溶液混合物から結晶質材料を分離することであった。
【0151】
約100mgのCDDOメチルエステルを、300μLのベンゼン/アセトン(10:1)に溶解し、0.2−μmナイロンフィルターで濾過した。次に、超音波処理装置により溶液を10分間音波処理し、蓋を取ったバイアル中において室温で一晩蒸発させた。透明なゲルが形成され、100μLのベンゼン/アセトン(10:1)を添加した。その溶液を、超音波処理装置で約30分間、超音波処理したところ、白色沈殿物が形成した。固形物を風乾した。
【0152】
他の実験において、約200mgのCDDOメチルエステルを、0.8mLのベンゼン/アセトン(10:1)に溶解させ、0.2−μmナイロンフィルターで濾過した。次に、その溶液を2つの1−ドラムバイアルに均等に分けた。次に、試料A及びBを室温で数時間、急速蒸発させた。試料Aに蓋をし、冷凍庫に入れた。試料が凍結した後、室温で解凍した。ヘラを用いて小さい引っ掻きを導入し、試料を室温で蒸発させた。白色固形物が形成され、風乾した。試料Bに蓋をし、室温で置き、室温で一晩おいた後に透明溶液になった。ヘラを用いて小さい引っ掻きを導入し、試料を室温で蒸発させた。白色固形物が形成され、風乾した。
【0153】
いくつかのこれらの実験から、ヘミベンゼネートであることが確認された結晶質材料を得た。前記のように、軽微な妨害作用、例えば、回収容器内での音波処理又は単なる小さい引っ掻きの導入が、ベンゼン溶媒和物の結晶化を促進する(表12)。
【0154】
ヘミベンゼネートの特性評価データを表13に要約する。ヘミベンゼネートXRPDパターンの特徴的なピークが、表19に示されている。DSC曲線は、図23のTGサーモグラフにおける約7.0%の重量減少に関連して、133℃付近の幅広い吸熱を示している。重量減少はおそらく、ベンゼンが揮発したからであり(下記のNMR考察を参照)、CDDOメチルエステルの各モルにつき0.5molのベンゼンに相当する。223℃付近で観測されたDSC吸熱は、脱溶媒和した材料の溶融によって生じる可能性が極めて高い。
【0155】
これらのデータは、CDDOメチルエステルの以前に分離された形態と本発明のその形態との区別を明らかにしている。
【実施例9】
【0156】
CDDOメチルエステルの新規ジメタノレート形態の特性評価
CDDOメチルエステルジメタノール溶媒和物を、下記の手順に従い調製した。約500mgのCDDOメチルエステルを、20mLのメタノールに60℃で溶解させた。次に、その溶液を、20mLの冷メタノールに−10℃で撹拌しながらゆっくり添加した。白色固形物を真空濾過によって収集した後、冷凍庫に保存した。
【0157】
特性評価データを表14に要約する。ジメタノレートXRPDパターンの特徴的なピークが、表18に示されている。
【0158】
DSC曲線は、TGサーモグラフにおける約11%の重量減少に関連して、102℃付近の幅広い吸熱を示している(図24)。TGIRデータは、重量減少が約2.0molのメタノールの揮発によることを裏付けている(図25)。TGIR実験から得られた物質を回収したところ、XRPDにより非晶質であることが分かった(図26)。約130℃におけるベースラインシフト、203℃付近の幅広い発熱、それに続く鋭い吸熱(開始:223℃)も、DSC曲線において見られる。これらの事象は、ジメタノール溶媒和物の脱溶媒が生じ、非晶質材料が形態Aに結晶化し、その結晶質材料が溶融したことによって得られた非晶質材料(形態B)のTgを示している可能性が極めて高い。
【0159】
溶液プロトンNMRスペクトルを得た。ケミカルシフトの帰属は行わなかったが、CDDOメチルエステルの化学構造と一致していると考えられる。約3.51ppmにおけるピークは、メタノールを表し、約1.7molに相当する。この結果は、前記の熱データと一致している。
【実施例10】
【0160】
CDDOメチルエステルを用いた臨床試験
臨床開発のために、微粉化形態Aを使用して処方したCDDO−Meを選択し、以前の治療に対して充分に反応しなかった進行癌を有する患者において、第I相安全性指向試験を先ず行った。この第I相用量増加試験において、種々の形態の進行(転移)癌を有する21人の成人患者に、CDDO−Meを投与した。患者にCDDO−Meカプセルの1日量を、5〜900mg/日(特に、5、10、20、40、80、150、300、600又は900mg/日)の用量で投与した。患者が容認できない毒性を経験するか、又は疾患進行の徴候を示すまで繰り返される「サイクル」で、CDDO−Meを投与した。この試験において、CDDO−Meの1サイクルは21日間の連続投与に続く7日間の休止期間から成り、その後に、患者は次のサイクルを開始することができた。
【0161】
CDDO−Meの安全性及び抗腫瘍活性を調査した。さらに、CDDO−Meの生物学的作用も特性評価した。CDDO−Meは、これらの患者において非常によく許容され、有意な薬剤関連有害事象も報告されなかった。第二治療サイクルの終了後の第一評価時点において、幾人かの患者(評価可能患者の約75%)が、安定疾患を有していると考えられた(標準的な放射線及び臨床基準に基づく)。第二サイクルの終了前に進行疾患の徴候を有することが見い出された患者は、形式的に評価されず、評価可能患者の群に含まれなかった。メラノーマ及び腎細胞癌の患者を含む5人の患者は、安定疾患を示し続け、治療の4サイクル後に、個々の腫瘍病変が退縮する徴候を示した。4人の患者は、治療の少なくとも6サイクル後に、安定疾患を有すると考えられた。処方計画により1日当たり少なくとも40mgのCDDO−Meの用量を投与されたいずれの患者においても、新たな転移は生じなかった。
【0162】
CDDO−Meの周知の抗炎症特性に基づいて、第I相試験で患者の循環炎症性サイトカインを評価した。5mg/日と同じくらい低い投与量で、MMP−9、TNFα、IL−8及びVEGF等のいくつかの循環炎症誘発性サイトカイン及びケモカインの減少が見られた。特に、TNFα(慢性関節リウマチ等の疾患の炎症過程において重要な役割を果たしていることが知られている)は、上昇したTNFベースライン値を有する3人の患者において、実質的に、又は検出限界未満に減少した(各患者につき、10、20及び40mg/日の治療投与量)。標的に結合して不活性にする抗TNFモノクローナル抗体と異なり、CDDO−Meは、TNFαの産生を減少させ、その結果としてTNFαの循環レベルを減少させる。
【0163】
さらに、抗酸化酵素及び解毒酵素を包含する第2相遺伝子産物を、第I相試験における患者の末梢血単核細胞において測定した。第2相転写活性のマーカーであるNQO1(NAD(P)H:キノンオキシドレダクターゼ)の顕著な誘導が、10mg/日及びそれ以上の投与量において観測された。
【0164】
CDDO−Meによる2サイクルの治療後に数人の患者から採取した腫瘍生検データは、シクロオキシゲナーゼ−2(COX−2)、誘導型一酸化窒素シンターゼ(iNOS)及びホスホリル化STAT3(pSTAT3)の腫瘍組織レベルにおける顕著な減少を示した。これらの各タンパク質の高レベルの発現は、腫瘍進行及び低い臨床結果に相関していることが知られている。数人の患者における腫瘍生検データは、CDDO−Meによる2サイクルの治療後に腫瘍細胞死が顕著に生じることも示した。血清クレアチニンのレベルは、この試験における80%より多い患者において、治療前のベースライン値と比較した場合、21日目において有意に低かった。複数サイクルによる治療を継続した多くの患者は、血清クレアチニンの継続的な減少を示した。血清クレアチニンは広く使用されている腎機能の指標であるので、これらの観測は、CDDO−Meによる治療が腎機能を向上させることを示している。
【0165】
これらの試験は、癌患者におけるCDDOメチルエステルの有利な作用を示すヒト癌患者データを提供する。データはさらに、腎機能不全等の他の炎症関連疾患の患者においてCDDO−Meが臨床的に有利な作用を有する可能性が高いことも示している。
【実施例11】
【0166】
ジメタノール溶媒和物中間体を使用した形態Bの大規模生成
1kgの形態AのCDDO−Meを、60±5℃メタノールに溶解させて、完全な溶液を得た。−5℃〜−15℃の冷メタノールの入った容器に、得られたCDDO−Meの温溶液を添加し、添加の間中、撹拌及び−5℃〜−15℃の温度を維持した。得られたCDDO−Meの結晶質ジメタノール溶媒和物の懸濁液を濾過した。図26に示されているXRPDパターンと一致したパターン(TGIR分析前)を示す得られた固形物を、オーブンで70±5℃で乾燥させた。XRPDプロファイルが結晶質物質に特徴的な反射を示さなくなるまで、乾燥を続けた。得られたXRPD非晶質CDDO−Me固形物を篩にかけ、包装した。生成物回収率は65〜95%である。
【実施例12】
【0167】
低温粉砕した形態A及び形態B
形態Aを低温粉砕し、分析した。低温粉砕(2時間)によって得られた試料の測定X線データは、約13.5°2θにおけるピークの幾分かの広がりを示した。低温粉砕した形態AのPDF分析は、形態B分析と同様の結果を生じた。これらの結果は、低温粉砕した形態Aがガラス質材料であり、低温粉砕が形態Bを生成する代替法を与え得ることを示す。
【0168】
形態Bを低温粉砕し分析したところ、低温粉砕(1時間)によって得られた試料の測定X線データは、形態B材料から開始したものと同様であった。これらの結果は、形態Bが安定であり、低温粉砕によって形態が変化しないことを示す。
【表9】
【表10】
【表11】
【表12】
【表13】
【表14】
【表15】
【表16】
【表17】
【表18】
【表19】
【表20】
【表21】
【表22】
【表23】
【表24】
【表25】
【特許請求の範囲】
【請求項1】
粉末X線回折パターンが、Cu Kα放射線を用いて回折計で得られるデータに基づいて8.78°2θ、12.94°2θ、13.35°2θ、14.18°2θ及び17.4°2θ±0.2°2θにおけるピークを含むことを特徴とする、メチル2−シアノ−3,12−ジオキソレアナ−1,9(11)−ジエン−28−オエート(CDDOメチルエステル)無水和物結晶。
【請求項2】
粉末X線回折パターンが図18で実質的に示されるパターンである、請求項1に記載のCDDOメチルエステル。
【請求項3】
示差走査熱量測定曲線が約157℃でのベースラインシフト及び約224℃での最大吸熱を含むことをさらに特徴とする、請求項1に記載のCDDOメチルエステル。
【請求項4】
示差走査熱量測定曲線が実質的に図3で示される曲線である、請求項3に記載のCDDOメチルエステル。
【請求項5】
治療有効量の請求項1に記載のCDDOメチルエステル及び1以上の医薬的に許容される固体担体を含む医薬組成物。
【請求項6】
CDDOメチルエステルが微粉化され、医薬的に許容される固体担体が食用担体である、請求項5に記載の医薬組成物。
【請求項7】
慢性炎症に罹る患者の慢性炎症状態を治療するための医薬の製造における、請求項1に記載のCDDOメチルエステルの使用。
【請求項8】
粉末X線回折パターンが、Cu Kα放射線を用いて回折計で得られるデータに基づいて9.25°2θ、14.17°2θ、14.62°2θ、16.32°2θ及び17.11°2θ±0.2°2θにおけるピークを含むことを特徴とする、CDDOメチルエステルヘミベンゼネート結晶。
【請求項9】
図23で実質的に示される示差走査熱量測定曲線を有することをさらに特徴とする、請求項9に記載のCDDOメチルエステルヘミベンゼネート結晶。
【請求項10】
請求項2に記載のCDDOメチルエステルを低温粉砕する工程を含む方法によって調製されるCDDOメチルエステル固体形態。
【請求項1】
粉末X線回折パターンが、Cu Kα放射線を用いて回折計で得られるデータに基づいて8.78°2θ、12.94°2θ、13.35°2θ、14.18°2θ及び17.4°2θ±0.2°2θにおけるピークを含むことを特徴とする、メチル2−シアノ−3,12−ジオキソレアナ−1,9(11)−ジエン−28−オエート(CDDOメチルエステル)無水和物結晶。
【請求項2】
粉末X線回折パターンが図18で実質的に示されるパターンである、請求項1に記載のCDDOメチルエステル。
【請求項3】
示差走査熱量測定曲線が約157℃でのベースラインシフト及び約224℃での最大吸熱を含むことをさらに特徴とする、請求項1に記載のCDDOメチルエステル。
【請求項4】
示差走査熱量測定曲線が実質的に図3で示される曲線である、請求項3に記載のCDDOメチルエステル。
【請求項5】
治療有効量の請求項1に記載のCDDOメチルエステル及び1以上の医薬的に許容される固体担体を含む医薬組成物。
【請求項6】
CDDOメチルエステルが微粉化され、医薬的に許容される固体担体が食用担体である、請求項5に記載の医薬組成物。
【請求項7】
慢性炎症に罹る患者の慢性炎症状態を治療するための医薬の製造における、請求項1に記載のCDDOメチルエステルの使用。
【請求項8】
粉末X線回折パターンが、Cu Kα放射線を用いて回折計で得られるデータに基づいて9.25°2θ、14.17°2θ、14.62°2θ、16.32°2θ及び17.11°2θ±0.2°2θにおけるピークを含むことを特徴とする、CDDOメチルエステルヘミベンゼネート結晶。
【請求項9】
図23で実質的に示される示差走査熱量測定曲線を有することをさらに特徴とする、請求項9に記載のCDDOメチルエステルヘミベンゼネート結晶。
【請求項10】
請求項2に記載のCDDOメチルエステルを低温粉砕する工程を含む方法によって調製されるCDDOメチルエステル固体形態。
【図1】

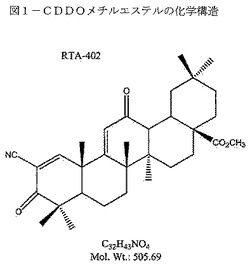
【図2】
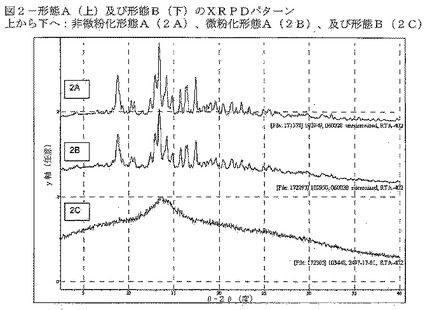
【図3】
【図4−1】

【図4−2】

【図5】
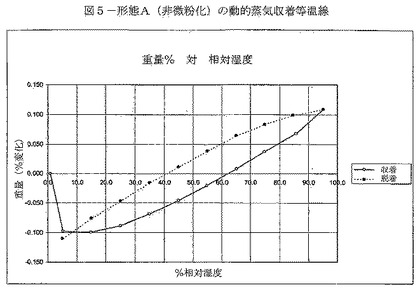
【図6−1】
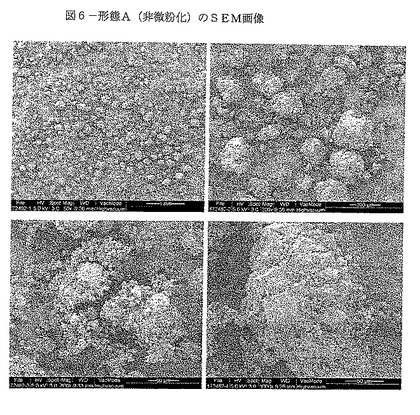
【図6−2】
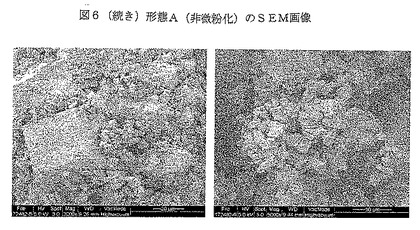
【図7】
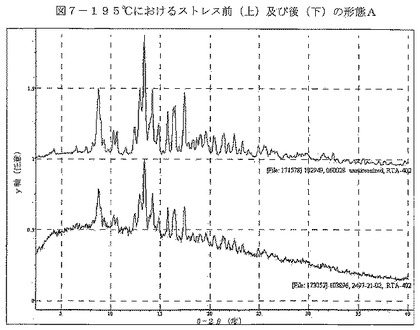
【図8】
【図9】
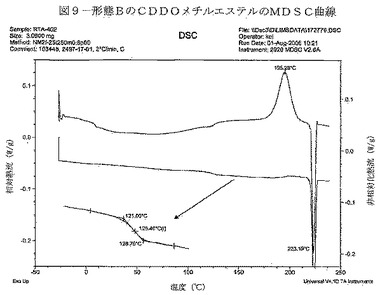
【図10】
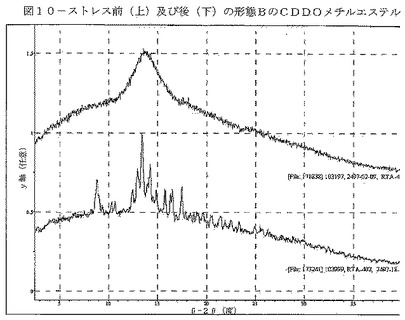
【図11】
【図12】
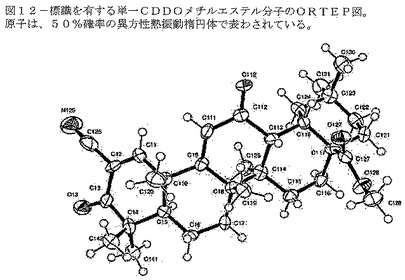
【図13】
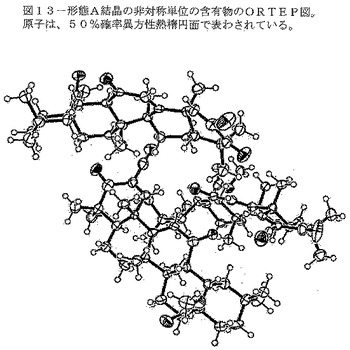
【図14】
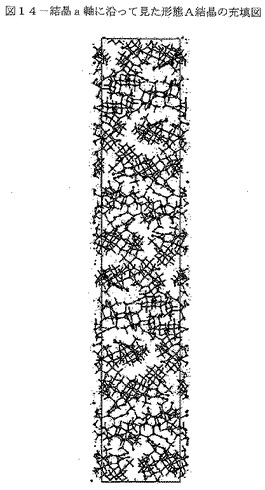
【図15】
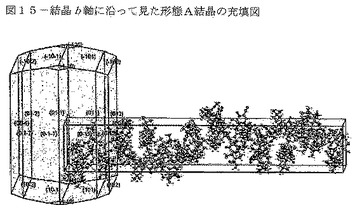
【図16】

【図17】
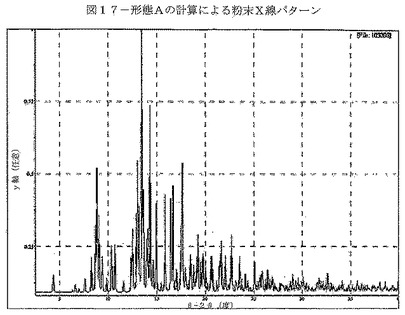
【図18】
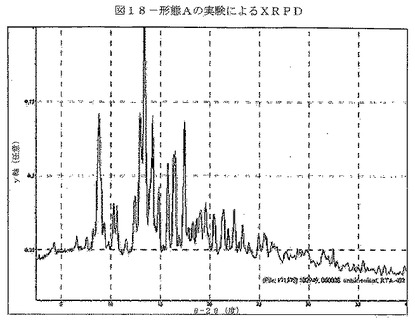
【図19】
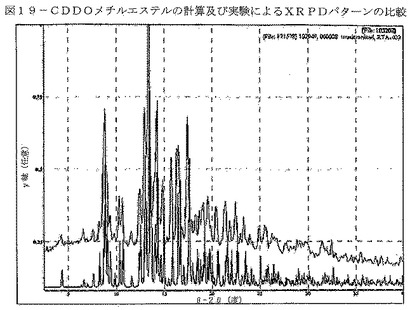
【図20】
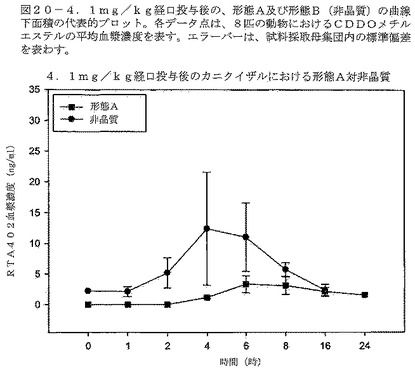
【図21】
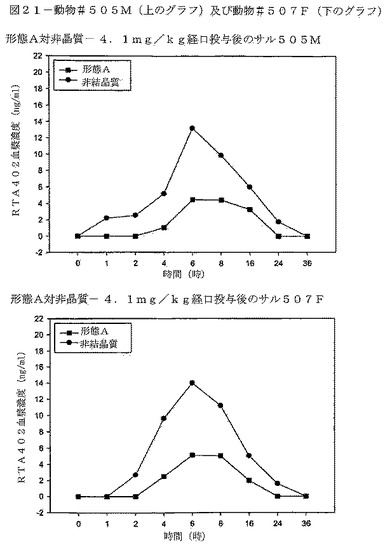
【図22】
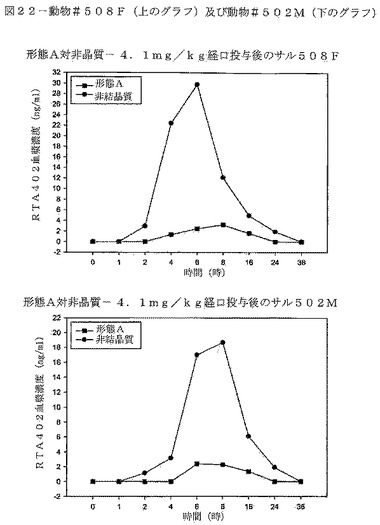
【図23】
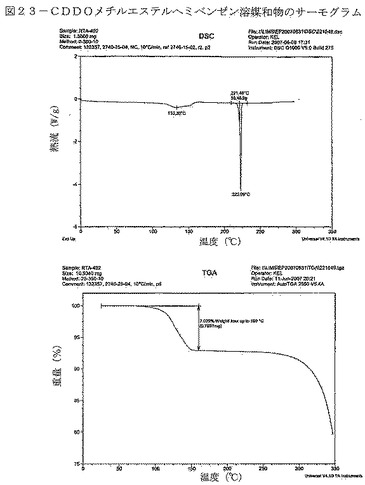
【図24】
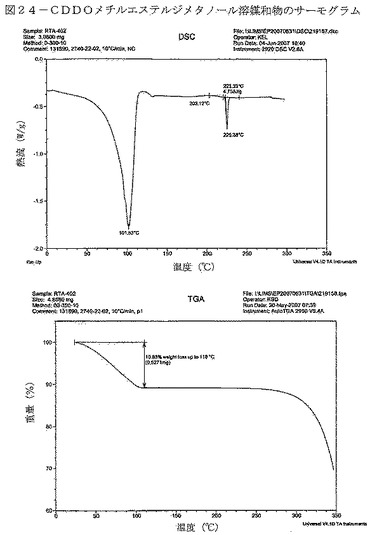
【図25】
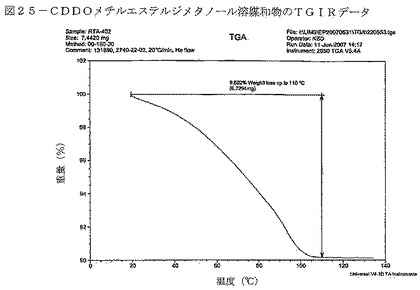
【図26】
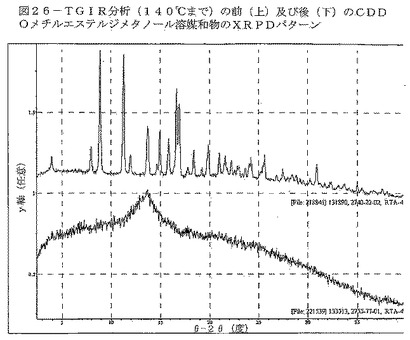
【図27】
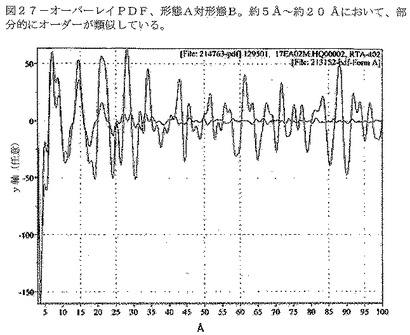
【図28】
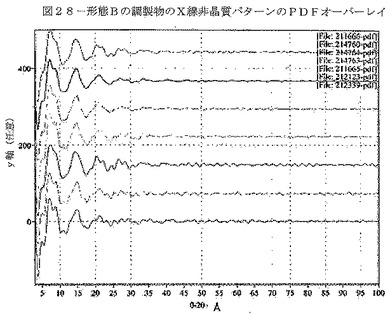
【図29】
【図30】
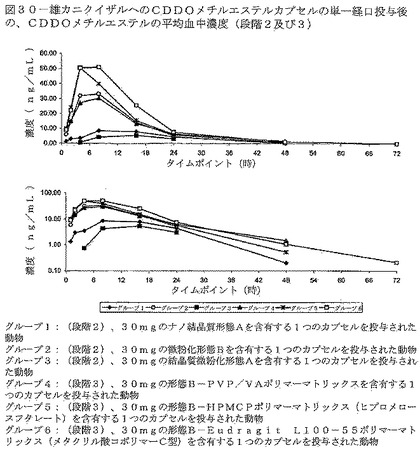
【図2】
【図3】
【図4−1】
【図4−2】
【図5】
【図6−1】
【図6−2】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【公開番号】特開2012−188438(P2012−188438A)
【公開日】平成24年10月4日(2012.10.4)
【国際特許分類】
【出願番号】特願2012−117773(P2012−117773)
【出願日】平成24年5月23日(2012.5.23)
【分割の表示】特願2010−521031(P2010−521031)の分割
【原出願日】平成20年8月14日(2008.8.14)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.Linux
2.WINDOWS
【出願人】(510037891)リアタ ファーマシューティカルズ,インク (3)
【Fターム(参考)】
【公開日】平成24年10月4日(2012.10.4)
【国際特許分類】
【出願日】平成24年5月23日(2012.5.23)
【分割の表示】特願2010−521031(P2010−521031)の分割
【原出願日】平成20年8月14日(2008.8.14)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.Linux
2.WINDOWS
【出願人】(510037891)リアタ ファーマシューティカルズ,インク (3)
【Fターム(参考)】
[ Back to top ]