
CEP−18770とボルテゾミブまたはメルファランを併用する多発性骨髄腫を治療するための併用療法
本発明は対象に化合物1とボルテゾミブを併用投与する工程を含む、対象の多発性骨髄腫を治療する方法を提供する。本発明はさらに、対象に化合物1とメルファランを併用投与する工程を含む、対象の多発性骨髄腫を治療する方法を提供する。

【発明の詳細な説明】
【技術分野】
【0001】
多発性骨髄腫の多剤併用療法。
【背景技術】
【0002】
多発性骨髄腫(MM)、即ち形質細胞新生物は、血液悪性腫瘍全体の約10%を構成する(1)。プロテアソーム阻害剤(PI)であるボルテゾミブのMMにおける臨床成果により、ユビキチン−プロテアソーム系(UPS)が薬物開発の有力な標的となることが証明された(2)。プロテアソームは、ミスフォールドし損傷したタンパク質ならびに細胞内シグナル伝達中間体の分解を担う、複数のサブユニットからなるタンパク質複合体である(3)。シグナル伝達経路の調節異常のため、新生物細胞はUPSに大きく依存し、従ってプロテアソーム阻害に対する感受性が特に高い(4)。プロテアソーム阻害後のMM細胞のアポトーシスは、生存促進性NF−κBシグナル伝達のダウンレギュレーション、血管新生の阻害、ミスフォールドしたタンパク質によるストレス応答の活性化、内因性および外因性細胞死経路の誘導、ならびに骨髄間質細胞へのMM細胞の接着の阻害を含む複数の機構によって起こる(5−8)。
【0003】
臨床開発における最初のPIであるボルテゾミブ(PS−341または[(1R)−3−メチル−1−({(2S)−3−フェニル−2−[(ピラジン−2−イルカルボニル)アミノ]プロパノイル}アミノ)ブチル]ボロン酸としても知られる)は、再発MMにおける2つの単剤第II相臨床試験の成功後、2003年にFDAによって認可された(9、10)。ボルテゾミブは他剤と併用した場合でも顕著な活性を示す。前臨床試験において、中毒量未満の(subtoxic)濃度のボルテゾミブは、メルファラン、ドキソルビシン、またはミトキサントロンを含む化学療法薬に対するMM細胞の耐性を克服した(11−13)。さらに、ボルテゾミブは、レナリドミド、三酸化二ヒ素、およびヒストン脱アセチル化酵素またはPKCの阻害剤、ならびに第2世代のPIを含む新規なMM治療薬の活性を相乗的に増強する(14−8)。ボルテゾミブをベースにする併用療法はin vitroで相乗作用が認められたが、それを試験する臨床試験においてin vivoでも高い有効性が認められた。ボルテゾミブ(V)を加えた場合または加えない場合のメルファランとプレドニゾンの併用(MP)を評価する第III相VISTA臨床試験において、MP療法では3年全生存率が59%であったのに対し、VMPでは3年全生存率が72%という結果が得られた(P=0.003)(19)。特に、投与計画にボルテゾミブを加えると、場合によっては、奏功しなかった治療薬に対して患者が再び感受性を示すことがある。例えば、第II相臨床試験において、メルファランで治療した後に再発したMM患者の60%は、その後、ボルテゾミブ/メルファラン併用療法に反応した(20)。同様に、ボルテゾミブをサリドマイドおよびデキサメタゾンと併用すると、再発MM患者集団(その73%はサリドマイドによる治療歴があった)において63%の全奏功率が得られた(21)。
【0004】
ボルテゾミブの認可によってMMの治療が変化したが、かなりの割合の患者がボルテゾミブ療法に反応しない。最近の研究結果から、プロテアソームの発現や活性レベルの差により、PIでの治療に対するMM腫瘍の感受性が異なり得ることが示唆された(22)。さらに、最初はボルテゾミブに反応した患者でも、ほぼ確実に再発が起こる。薬物耐性癌幹細胞の小集団が寛解後のMMの再発の原因となっている可能性があることを示唆する証拠が増加している(23−26)。これらの細胞は、正常な記憶B細胞の表面抗原特性を発現し、形質細胞マーカーCD138が欠損し、抗体を分泌しない(24)。さらに、一般的に使用されている抗骨髄腫薬(例えば、デキサメタゾン、レナリドミド、シクロホスファミド)で攻撃した場合、CD138陰性幹細胞集団は、残りの悪性細胞集団より高い薬物耐性を示す(24)。単剤のボルテゾミブは、例えば、多量の免疫グロブリンを産生するMM細胞に対して活性を有するが、CD138陰性MM細胞の増殖に対してはほとんど効果がない(24)。これらのデータから、癌幹細胞、ならびに腫瘍集団内の残りの悪性形質細胞サブタイプを標的とする新規なMM療法が必要とされていることが明らかである。
【0005】
新規で、より強力な、またはより忍容性の高いPIの探求の結果、化合物1([(1R)−1−[[(2S,3R)−3−ヒドロキシ−2−[6−フェニル−ピリジン−2−カルボニル)アミノ]−1−オキソブチル]アミノ]−3−メチルブチルボロン酸としても知られる;Bernardiniら、米国特許出願公開第2005/0107307号明細書)が合成された。ボルテゾミブと同様に、化合物1はペプチドボロン酸の部類の可逆性PIである(28)。静脈内(IV)ボーラス投与されるボルテゾミブとは対照的に、化合物1は、前臨床試験で経口製剤として活性を有している(28、29)。さらに、化合物1は、ボルテゾミブと比較して、in vitroの初代培養MM形質細胞とin vivoのRPMI8226MM異種移植片の両方で、同様のまたはより優れた単剤抗腫瘍活性を示す(29)。化合物1は、次の化学構造を有する:
【化1】
【0006】
多発性骨髄腫患者に最良の長期結果をもたらし得る治療選択肢が依然として必要とされている。とりわけ、再発または難治性疾患を有する患者に対する新規な療法が緊急に必要とされている。本明細書で研究が開示されるまで、化合物1とボルテゾミブまたはメルファランとの併用療法は研究されなかった。これらの併用療法は、再発または難治性疾患を有する患者を含む、MM患者に魅力的な治療選択肢を提供する。
【0007】
引用される参照文献は全て、参照により本明細書に援用される。
【発明の概要】
【課題を解決するための手段】
【0008】
化合物1で対象(subject)の多発性骨髄腫を治療する方法を提供する。一実施形態では、対象に化合物1とボルテゾミブを併用投与する。好ましくは、ボルテゾミブをプロドラッグとして投与する。好ましくは、ボルテゾミブを静脈内投与または経口投与する。
【0009】
好ましくは、ボルテゾミブを約0.5mg/m2〜約2mg/m2の範囲の用量で投与する。好ましくは、ボルテゾミブを約0.7mg/m2〜約1.3mg/m2の範囲の用量で投与する。
【0010】
好ましくは、ボルテゾミブを3〜7日毎に2〜4週間投与した後、ボルテゾミブを投与しない約7〜21日間の休薬期間を設ける、計画された投与サイクルに従ってボルテゾミブを投与する。好ましくは、ボルテゾミブを21日サイクルの1日目、4日目、8日目および11日目に投与する、計画された投与サイクルに従ってボルテゾミブを投与する。好ましくは、ボルテゾミブを28日サイクルの1日目、4日目、8日目および11日目に投与する、計画された投与サイクルに従ってボルテゾミブを投与する。好ましくは、計画されたサイクルを少なくとも1回繰り返す。
【0011】
別の実施形態では、対象に化合物1とメルファランを併用投与する。好ましくは、メルファランをプロドラッグとして投与する。好ましくは、メルファランを経口投与または静脈内投与する。
【0012】
好ましくは、メルファランを約0.025mg/kg〜約0.5mg/kgの範囲の用量で投与する。好ましくは、メルファランを約0.025mg/kg〜約0.3mg/kgの範囲の用量で投与する。
【0013】
好ましくは、メルファランを3〜7日毎に1〜2週間投与した後、メルファランを投与しない約4〜6週間の休薬期間を設ける、計画された投与サイクルに従ってメルファランを投与する。好ましくは、メルファランを1日1回、約4〜約7日間投与した後、約4〜6週間の休薬期間を設ける、計画された投与サイクルに従ってメルファランを投与する。好ましくは、メルファランを1日1回、約4〜約5日間投与した後、約4〜6週間の休薬期間を設ける、計画された投与サイクルに従ってメルファランを投与する。好ましくは、計画されたサイクルを少なくとも1回繰り返す。
【0014】
好ましくは、化合物1をプロドラッグとして投与する。好ましくは、化合物1のプロドラッグは、化合物1の薬学的に許容されるエステルの形態である。好ましくは、化合物1を静脈内投与または経口投与する。
【0015】
好ましくは、化合物1を約0.5mg/m2〜約5mg/m2の範囲の用量で投与する。好ましくは、化合物1を約1mg/m2〜約3mg/m2の範囲の用量で投与する。好ましくは、化合物1を3〜14日毎に2〜4週間投与した後、化合物1を投与しない約7〜21日間の休薬期間を設ける、計画された投与サイクルに従って化合物1を投与する。
【0016】
好ましくは、化合物1を21日サイクルの1日目、4日目、8日目および11日目に投与する、計画された投与サイクルに従って化合物1を投与する。好ましくは、化合物1を、28日サイクルの1日目、4日目、8日目および11日目に投与する、計画された投与サイクルに従って化合物1を投与する。好ましくは、化合物1を21日サイクルの1日目と15日目に投与する、計画された投与サイクルに従って化合物1を投与する。好ましくは、化合物1を28日サイクルの1日目と15日目に投与する、計画された投与サイクルに従って化合物1を投与する。好ましくは、計画されたサイクルを少なくとも1回繰り返す。
【0017】
好ましくは、化合物1を21日サイクルまたは28日サイクルの1日目、5日目および9日目に投与し、ボルテゾミブを21日サイクルまたは28日サイクルの3日目、8日目、および12日目に投与する。好ましくは、ボルテゾミブを21日サイクルまたは28日サイクルの1日目、5日目および9日目に投与し、化合物1を21日サイクルまたは28日サイクルの3日目、8日目、および12日目に投与する。
【図面の簡単な説明】
【0018】
【図1A】
【図1B】
【図1C】化合物1は、単剤の場合または他の抗MM治療薬と併用した場合、MM細胞株の生存率を低下させることを示す図である。A,MM1S(三角形)およびRPMI8226(正方形)を、表示した濃度の化合物1と共に48時間インキュベートした後、その生存率をMTSアッセイで評価した。B,MM1S細胞を溶媒対照(黒色の棒グラフ)、表示した濃度の化合物1(白色の棒グラフ)、ボルテゾミブ(縞模様の棒グラフ)、または両方の薬剤(線影の付いた棒グラフ)で48時間処置し、生存率をMTSアッセイで定量化した。C,RPMI8226細胞を溶媒対照(黒色の棒グラフ)、メルファラン(40μM)(白色の棒グラフ)、化合物1(表示した濃度)(縞模様の棒グラフ)、または両方の薬剤(線影の付いた棒グラフ)で48時間処置し、生存率をMTSアッセイで定量化した。グラフ化したデータは、6つの反復試験を使用した平均値±平均値の標準誤差(SEM)である。BおよびCでは、線影の付いた棒グラフの上に併用指数(CI)を示す。0.9未満のCI値は相乗作用を示し;0.9〜1.1のCI値は相加作用を示し、1.1超のCI値は拮抗作用を示す。
【図2A】
【図2B】
【図2C】
【図2D】化合物1は、単独の場合またはボルテゾミブと併用した場合、正常な末梢血単核細胞(PBMC)の生存率を低下させないことを示す図である。A,健常被験者から採取したPBMCを溶媒対照(黒色の棒グラフ)、化合物1単独(3.0nM)(白色の棒グラフ)、ボルテゾミブ単独(表示した濃度)(縞模様の棒グラフ)、または化合物1(3.0nM)+ボルテゾミブ(表示した濃度)(線影の付いた棒グラフ)と共に48時間インキュベートした後、細胞生存率をMTSアッセイで測定した。B,Aと同じ被験者から採取したPBMCを溶媒対照(黒色の棒グラフ)、ボルテゾミブ単独(3.0nM)(白色の棒グラフ)、化合物1単独(表示した濃度)(縞模様の棒グラフ)、または化合物1(表示した濃度)+ボルテゾミブ(3.0nM)(線影の付いた棒グラフ)と共にインキュベートした後、細胞生存率をMTSアッセイを使用して測定した。C,別の被験者から採取したPBMCを溶媒対照(黒色の棒グラフ)、化合物1単独(3.0nM)(白色の棒グラフ)、ボルテゾミブ単独(表示した濃度)(縞模様の棒グラフ)、または化合物1(3.0nM)+ボルテゾミブ(表示した濃度)(線影の付いた棒グラフ)と共に48時間インキュベートした後、細胞生存率をMTSアッセイで測定した。D,3人の健常被験者から採取したPBMCを様々な濃度の化合物1と共に48時間インキュベートした後、細胞生存率をMTSアッセイで測定した。各グラフ(A−D)は3つの独立した実験を表す。
【図3A】
【図3B】
【図3C】
【図3D】化合物1をボルテゾミブと併用すると、MM細胞のアポトーシスが誘導されることを示す図である。RPMI8226細胞を(A)溶媒対照、(B)化合物1(2.5nM)、(C)ボルテゾミブ(2.5nM)、または(D)化合物1(2.5nM)+ボルテゾミブ(2.5nM)と共に30時間インキュベートし、ヨウ化プロピジウム(PrI)およびアネキシンVに関して陽性の染色率を、フローサイトメトリー分析を使用して定量化した。初期アポトーシスの細胞は、PrI陰性、アネキシンV陽性である。
【図4A】
【図4B】
【図4C】化合物1がヒトMM腫瘍の増殖を阻害することを示す図である。A,B,14日目から、ヒトLAGκ−1A腫瘍を有するマウスに溶媒対照または表示された用量の化合物1をIV投与によりまたは強制経口投与により試験期間中週2回投与した。その後毎週、血清ヒト免疫グロブリン(IgG)濃度をELISAで評価し(A)、腫瘍体積をカリパスで測定した(B)。C,21日目から、ヒトLAGκ−1B腫瘍を有するマウスを試験期間中週2回、溶媒対照または表示した濃度の化合物1で処置し、腫瘍体積を毎週評価した。データは、1群当たりマウス7〜8匹の平均値±平均値の標準誤差として表す。
【図5A】
【図5B】
【図5C】
【図5D】
【図5E】
【図5F】化合物1をボルテゾミブまたはメルファランと併用すると、骨髄腫腫瘍の増殖が著しく阻害されることを示す図である。A,B,化合物1をボルテゾミブと併用すると、LAGκ−1A腫瘍の増殖が防止される。7日目から、LAGκ−1A腫瘍を有するマウスを週2回、溶媒対照、化合物1単独(1mg/kg)、ボルテゾミブ単独(0.5mg/kg)、または化合物1(1mg/kg IV)+ボルテゾミブ(0.5mg/kg IV)で処置した。その後毎週、血清ヒトIgG濃度(A)および腫瘍体積(B)を測定した。C,化合物1をボルテゾミブと併用すると、LAGκ−1B腫瘍体積の進行が100%遅延される。7日目から、LAGκ−1B腫瘍を有するマウスを週2回、溶媒対照、化合物1単独(1mg/kg)、ボルテゾミブ単独(0.5mg/kg)、または化合物1(1mg/kg IV)+ボルテゾミブ(0.5mg/kg IV)で処置した。その後毎週、腫瘍体積を測定した。D,E,化合物1をメルファランと併用すると、LAGκ−1A腫瘍の増殖が阻害される。7日目から、LAGκ−1A腫瘍を有するマウスを溶媒対照で週2回、化合物1単独で週2回(1mg/kg)、メルファラン単独で毎週(1mg/kg)、または週2回化合物1(1mg/kg IV)+毎週メルファラン(1mg/kg IP)で処置した。その後毎週、血清ヒトIgG濃度(D)および腫瘍体積(E)を測定した。F,化合物1をメルファランと併用すると、いずれかの薬剤を単独で使用した場合より、LAGκ−1B腫瘍体積の減少が大きかった。7日目から、LAGκ−1B腫瘍を有するマウスを溶媒対照で週2回、化合物1単独で週2回(1mg/kg)、メルファラン単独で毎週(3mg/kg)、または週2回化合物1(1mg/kg IV)+毎週メルファラン(3mg/kg IP)で処置した。その後毎週、腫瘍体積を測定した。A〜Fのデータは、1群当たりマウス7〜8匹の平均値±平均値の標準誤差として表す。
【図6A】
【図6B】
【図6C】
【図6D】
【図6E】
【図6F】
【図6G】
【図6H】化合物1およびボルテゾミブで処置したLAGκ−1B腫瘍が、アポトーシスマーカーであるアポトーシス誘導因子(AIF)の発現の増加を示すことを示す図である。LAGκ−1Bを有するマウスを溶媒対照(A)、化合物1(1mg/kg)単独(B)、ボルテゾミブ(0.5mg/kg)単独(C)、または両方の薬剤(D)で処置した後、そのマウスから切除した腫瘍を薄切し、AIFを測定するために染色した。E〜H,アイソタイプ対照でのA〜Dと同じ腫瘍から得られた染色された切片。スライドガラスは同時に染色した。
【図7】経口化合物1がヒトMM腫瘍の増殖を阻害することを示す図である。腫瘍体積の大きさに対する経口投与化合物1の効果。
【図8】経口化合物1がヒトMM腫瘍の増殖を阻害することを示す図である。IgG濃度に対する経口投与化合物1の効果。
【発明を実施するための形態】
【0019】
量および持続時間等の測定可能な値に言及する場合、本明細書で使用する「約」という用語は、特定の値から±10%の偏差を包含することを意味する。例えば、「約50%」という句は、50の±10%、即ち、45%〜55%を含む。
【0020】
本明細書で使用する場合、「対象」という用語は、温血動物、好ましくはヒトを含む哺乳動物を含む。好ましい実施形態では、対象は霊長類である。さらにより好ましい実施形態では、対象はヒトである。
【0021】
対象の多発性骨髄腫を治療する方法を提供する。一実施形態では、対象に化合物1とボルテゾミブを併用投与する。化合物1とボルテゾミブを併用する本発明の多発性骨髄腫の治療方法は、多発性骨髄腫の治療効果を相乗的に高めることが分かった。化合物1とボルテゾミブは両方とも、外因性および内因性アポトーシスシグナル伝達経路の活性化により細胞死を誘導する可逆的ボロン酸プロテアソーム阻害剤である(7、29)ため、これは意外である。さらに、薬剤は両方とも主に、プロテアソームのキモトリプシン様触媒活性を標的とし、カスパーゼ様活性をあまり阻害せず、トリプシン様活性をほとんど阻害しない(29、34)。従って、化合物1とボルテゾミブは、類似の作用機構を有するものと思われる。さらに、これらの化合物の化学構造は非常に類似している。
【0022】
ボルテゾミブ([(1R)−3−メチル−1−({(2S)−3−フェニル−2−[(ピラジン−2−イルカルボニル)アミノ]プロパノイル}アミノ)ブチル]ボロン酸;Millennium PharmaceuticalsによりVelcade(登録商標)の商標名で販売)は、次の化学構造を有する:
【化2】
【0023】
従って、化合物1とボルテゾミブが併用時に、in vitroでのMM細胞と、in vivoでの腫瘍、特に非分泌性腫瘍に対して高い活性を誘導する手段は不明である。
【0024】
別の実施形態では、対象に化合物1とメルファランを併用投与する。化合物1とメルファランを併用する本発明の多発性骨髄腫の治療方法は、多発性骨髄腫の治療効果を相乗的に高めることが分かった。
【0025】
メルファラン(4−[ビス(2−クロロエチル)アミノ]−L−フェニルアラニン;GlaxoSmithKlineによりAlkeran(登録商標)の商標名で販売)は、次の化学構造を有する:
【化3】
【0026】
本発明に使用される化合物1、ボルテゾミブおよび/またはメルファランは、親化合物の薬学的に許容される塩の形態および/または薬学的に許容されるエステルの形態などのプロドラッグを含む任意の適した化学的形態で投与されてもよい。好ましくは、親化合物の薬学的に許容される塩またはエステル誘導体は、投与後直ぐに、親化合物に変換される。本明細書で使用する場合、「薬学的に許容される塩」は、親化合物がその酸塩または塩基塩を生成することにより修飾されている親化合物の誘導体を指す。薬学的に許容される塩の例としては、アミンなどの塩基性基の鉱酸塩または有機酸塩;カルボン酸またはボロン酸などの酸性基のアルカリ塩または有機塩等が挙げられるが、これらに限定されるものではない。本明細書で使用する場合、「薬学的に許容されるエステル」は、酸基がそのエステルを生成することにより修飾されている親化合物の誘導体を指す。薬学的に許容されるエステルの例としては、例えば、ボロン酸エステル、即ち、ボロン酸化合物のエステル誘導体、および環状ボロン酸エステルが挙げられる。環状ボロン酸エステルの例としては、ピナンジオールボロン酸エステル、ピナコールボロン酸エステル、1,2−エタンジオールボロン酸エステル、1,3−プロパンジオールボロン酸エステル、1,2−プロパンジオールボロン酸エステル、2,3−ブタンジオールボロン酸エステル、1,1,2,2−テトラメチルエタンジオールボロン酸エステル、1,2−ジイソプロピルエタンジオールボロン酸エステル、5,6−デカンジオールボロン酸エステル、1,2−ジシクロヘキシルエタンジオールボロン酸エステル、ビシクロへキシル−1,1’−ジオール、および1,2−ジフェニル−1,2−エタンジオールボロン酸エステルが挙げられるが、これらに限定されるものではない。
【0027】
従って、特定の実施形態では、化合物1および/またはボルテゾミブは親化合物のボロン酸エステル誘導体として投与される。一実施形態では、化合物1は化合物1のボロン酸エステル誘導体として投与される。一実施形態では、ボルテゾミブは、ボルテゾミブのボロン酸エステル誘導体として投与される。
【0028】
任意の適した投与方法を使用してもよい。例としては、注射(皮下、静脈内、非経口、腹腔内、髄空内等)、経口投与、吸入、および経皮投与が挙げられる。注射により投与する場合、注射はボーラスであっても、または持続注入であってもよい。化合物1とボルテゾミブは対象に別々に(例えば、順次注射、注射と経口投与、または別々の経口投与として)投与されても、または混合物として一緒に(例えば、単回注射、または、化合物1とボルテゾミブを両方含有する単一錠剤の投与などによる単回経口投与で)投与されてもよい。同様に化合物1とメルファランも対象に別々に投与されても、または混合物として一緒に投与されてもよい。医薬組成物中の本発明の化合物の割合または濃度は、用量、化学的特性(例えば、疎水性)、および投与経路を含む多くの要因によって変わり得る。
【0029】
例えば、ボルテゾミブは経口投与または静脈内注射に適している。例えば、ボルテゾミブは、Millennium PharmaceuticalsからVelcade(登録商標)の商標名で、ボルテゾミブ3.5mgおよび増量剤であるマンニトール35mgを含有する使い捨てバイアルに入った滅菌凍結乾燥粉末として入手可能である。粉末は、臨床医により0.9%NaCl3.5mLで注射用に再構成される。ボルテゾミブは、Velcade凍結乾燥製剤中にマンニトールボロン酸エステルとして存在し、再構成後は、親ボロン酸と平衡状態にあるマンニトールボロン酸エステルとして存在する(42)。従って、一実施形態では、ボルテゾミブは静脈内(IV)注射により投与される。別の実施形態では、ボルテゾミブは経口投与、好ましくは錠剤またはカプセルで投与される。一実施形態では、ボルテゾミブは、ボロン酸エステルなどのプロドラッグの形態で注射により投与される。一実施形態では、ボルテゾミブは、ボロン酸エステルなどのプロドラッグの形態で経口投与される。
【0030】
例えば、メルファランは、経口投与または静脈内注射に適している。例えば、メルファランは、GlaxoSmithKlineからAlkeran(登録商標)の商標名で、経口投与用のフィルムコート錠または使い捨てバイアルに入った滅菌凍結乾燥粉末として入手可能である。フィルムコート錠は、メルファラン2mg、ならびに賦形剤であるコロイド状二酸化ケイ素、クロスポピドン、ヒプロメロース、マクロゴール/PEG400、ステアリン酸マグネシウム、微結晶セルロース、および二酸化チタンを含有する。凍結乾燥粉末は、メルファラン50mgと同等のメルファラン塩酸塩、およびポビドン20mgを含有する。粉末は、クエン酸ナトリウム0.2g、プロピレングリコール6.0mL、エタノール(96%)0.52mL、および注射用蒸留水を合計10mLとなるように含有する、提供される滅菌希釈剤のバイアルを使用して注射用に再構成される(43)。従って、一実施形態では、メルファランは、塩酸塩として静脈内(IV)注射により投与される。別の実施形態では、メルファランは経口投与、好ましくは錠剤またはカプセルで投与される。
【0031】
例えば、化合物1は、IV注射により、または、錠剤またはカプセルなどの経口剤形で投与するのに適している(28、29)。例えば、化合物1は、現在、固形腫瘍または非ホジキンリンパ腫を有する患者で最初のヒト第1相臨床試験で評価中である。第1相臨床試験では、化合物1は、化合物1、4mg、増量剤であるヒドロキシプロピル−β−シクロデキストリン196mg、および増量剤であるマンニトール156.8mgを含有する、使い捨てバイアルに入った滅菌凍結乾燥粉末として提供される。粉末は、注射前に、注射用滅菌水、0.9%NaClまたは5%マンニトールのいずれか5mLまたは10mL(目的の用量による)で再構成される。従って、一実施形態では、化合物1は静脈内(IV)注射により投与される。別の実施形態では、化合物1は経口投与、好ましくは錠剤またはカプセルで投与される。一実施形態では、化合物1は、ボロン酸エステルなどのプロドラッグの形態で注射により投与される。一実施形態では、化合物1は、ボロン酸エステルなどのプロドラッグの形態で経口投与される。
【0032】
化合物1とボルテゾミブ、または化合物1とメルファランは、好ましくは、多発性骨髄腫を治療するのに有効な量で、例えば、疾患の症状を予防、緩和、または寛解する、治療を受ける対象の生存期間を延長する、望ましくない細胞増殖を防止する、または対象の既存の良性細胞塊もしくは悪性腫瘍のサイズを減少させるのに有効な量で、併用投与される。本明細書に記載の詳細な開示内容および実施例に鑑みて、当業者は、併用する各薬剤の有効量をうまく決定することができる。有効量は、処置または阻害される細胞増殖のタイプ、対象のサイズ、癌細胞増殖または腫瘍の重症度、投与頻度(例えば、毎日か数日に1回か)、化合物の投与方式(manner of administration)、患者の健康状態および共存症の状態、処方する医師の判断および経験(例えば、同じまたは類似の薬物に関する)、投与方法(mode of administration)、投与される剤形のバイオアベイラビリティ特性、選択される投与計画、および併用療法の種類(例えば、他の化学療法剤)などの要因に応じて変わり得る。例えば、米国特許第5,427,916号明細書は、個々の患者における抗腫瘍療法の有効性を予測する方法を記載し、本発明の治療プロトコルと併用できる特定の方法を説明している。例えば、有効量は、in vitroまたは動物モデル試験系から得られる用量−反応曲線から外挿することができ、患者の表面積に基づいてもよい。
【0033】
治療は、化合物の最適用量より少ない比較的低用量で開始してもよい。その後、その状況における最適効果が達せられるまで、少しずつ漸増することにより用量を増加してもよい。必要に応じて全1日量を分割し、1日の間に少量ずつ投与してもよい。投与計画を最適化するため、抗癌治療を受けている患者から得られる2つ以上の時点における腫瘍測定値を比較することによって、多発性骨髄腫を治療するための化合物1とボルテゾミブの併用、または化合物1とメルファランの併用の有効性をモニターすることができる。一般に、療法を開始する前の患者の全身腫瘍組織量の初期評価と、治療中の異なる時点での1つ以上の追加の評価を得ることが好ましい。このような使用では、療法を開始する前に全身腫瘍組織量のベースラインを測定した後、療法を行っている間に癌の量の変化を測定する。或いは、治療前の全身腫瘍組織量のベースラインを測定する必要なく、2回以上連続測定を行ってもよい。このような使用では、全身腫瘍組織量の増減を測定するためのベースラインレベルとして、対象の全身腫瘍組織量の最初の評価を行わなければならない。
【0034】
化合物1とボルテゾミブ、または化合物1とメルファランの投与計画、例えば、投与のタイミングおよび/または順番は、各剤形の薬物動態、処置または阻害される細胞増殖のタイプ、対象のサイズ、癌細胞増殖または腫瘍の重症度、および有効量などの要因によって変わり得る。治療医師は、上記考慮事項および本発明の詳細な開示内容に鑑みて、有効性を最適化し副作用を最小限にするように、化合物1とボルテゾミブ、または化合物1とメルファランの投与のタイミングおよび/または順番を容易に変えることができる
【0035】
本発明の化合物1、ボルテゾミブ、およびメルファランの投与計画は柔軟性が高い。特定の実施形態では、これらの薬物に適していることが知られている投与計画を改良して投与計画を立ててもよい。例えば、表1に示すように、ボルテゾミブ(1.3mg/m2)を3〜5秒のボーラスIV注射として、経口メルファラン(9mg/m2)および経口プレドニゾン(60mg/m2)と併用投与する6週間の治療サイクルを9サイクル行うことにより、前治療歴のない多発性骨髄腫を治療することが認可されている。サイクル1〜4では、ボルテゾミブを週2回、1日目、4日目、8日目、11日目、22日目、25日目、29日目および32日目に投与する。サイクル5〜9では、ボルテゾミブを週1回、1日目、8日目、22日目および29日目に投与する(42)。
【0036】
【表1】
【0037】
治療計画中に薬物に関連する著しい毒性(例えば、血液毒性)が認められた場合、その後のボルテゾミブの用量をスキップするおよび/または減少することができる(例えば、1.3mg/m2から1mg/m2に減少する、および0.7mg/m2まで減少することが可能)。さらに、または代わりに、次のサイクルではメルファランの用量を25%減少させることができる(42)。
【0038】
別の例として、ボルテゾミブを3〜5秒のボーラスIV注射として3週間サイクルの1日目、4日目、8日目、および11日目に投与した後、10日間の休薬期間(12日目〜21日目)を設けることにより、再発または難治性多発性骨髄腫を治療することが認可されている。8サイクルを超える長期療法では、ボルテゾミブは、標準的な計画で投与されても、または週1回4週間(1日目、8日目、15日目および22日目)投与した後、13日間の休薬期間(23日目〜35日目)を設ける維持計画で投与されてもよい(42)。
【0039】
治療計画中に薬物に関連する著しい毒性(例えば、血液毒性、神経因性疼痛および/または末梢神経障害)が認められた場合、その後のボルテゾミブの用量をスキップするおよび/または減少することができる(例えば、1.3mg/m2から1mg/m2に減少する、および0.7mg/m2まで減少することが可能)(42)。
【0040】
本発明の併用療法では、ボルテゾミブの投与計画は、前述のものを含む認可された多発性骨髄腫の投与計画と類似していても、または異なっていてもよい。例えば、ボルテゾミブを認可された投与計画より高頻度または低頻度で投与してもよく、任意選択により、比較的高用量または低用量で投与してもよい。
【0041】
ボルテゾミブは、化合物1と任意の適した用量で併用投与されてもよい。適したボルテゾミブの用量は、約0.5mg/m2〜約7mg/m2の範囲、例えば、約0.5mg/m2〜約5mg/m2、例えば、約0.5mg/m2〜約3mg/m2の範囲であってもよい。適したボルテゾミブの用量は、典型的には約0.5mg/m2〜約2mg/m2の範囲となるであろう。好ましくは、ボルテゾミブの用量は約0.6mg/m2〜約1.5mg/m2の範囲である。より好ましくは、ボルテゾミブの用量は約0.7mg/m2〜約1.3mg/m2の範囲である。好ましいボルテゾミブの用量としては、0.7mg/m2、1mg/m2、または1.3mg/m2が挙げられるが、これらに限定されるものではない。前述の用量はどのボルテゾミブ投与方法にも適しており、とりわけ皮下投与または静脈内投与に適しており、静脈内投与が好ましい。ボルテゾミブの経口用量は、典型的には前述の範囲の上限(high end)にあり、例えば、約1mg/m2〜約5mg/m2、約1.5mg/m2〜約4mg/m2、または約2mg/m2〜約3mg/m2となるであろう。
【0042】
ボルテゾミブは、前述の用量で化合物1と任意の適した計画に従って併用投与されてもよい。ボルテゾミブの投与量は一定であっても、または投与計画内で変えてもよい。薬物に関連する著しい毒性が認められない限り、好ましくはボルテゾミブの用量は投与計画中、一定のレベルに維持されるが、薬物に関連する著しい毒性が認められた場合、その後の用量を例えば約20〜30%減少させることができる。ボルテゾミブは、化合物1と同じ日に投与されても、または異なる日に投与されてもよい。一実施形態では、ボルテゾミブと化合物1は、投与計画中、同じ日に投与される。適したボルテゾミブ投与計画は、典型的には1日1回投与〜週1回投与またはさらには月1回投与の範囲となるであろう。好ましくは、ボルテゾミブは1日1回より低頻度で、例えば、2〜14日に1回投与される。好ましくは、ボルテゾミブは3〜7日毎に、例えば、3〜4日毎に投与される。好ましくは、投与計画は、1週間以上、例えば、2、3または4週間ボルテゾミブで治療した後、ボルテゾミブを投与しない期間を少なくとも5日間、例えば、約7〜21日間含む。好ましくは、休薬期間は約10〜17日間、例えば、約10日間または約17日間である。例えば、ボルテゾミブを21日サイクルの1日目、4日目、8日目および11日目に投与してもよく、12〜21日目は休薬期間である。別の例として、ボルテゾミブを28日サイクルの1日目、4日目、8日目および11日目に投与してもよく、12〜28日目は休薬期間である。別の例として、ボルテゾミブを週1回4週間(例えば、35日サイクルの1日目、8日目、15日目および22日目に)投与した後、13日間の休薬期間(例えば、35日サイクルの23日目〜35日目)を設けてもよい。計画された投与サイクルを1回以上繰り返してもよい。例えば、最大反応が認められるまで、1または2サイクル追加して、計画されたサイクルを繰り返してもよい。別の例として、計画されたサイクルを6〜12サイクル繰り返してもよい。任意選択により、最初のサイクルを完了した後、ボルテゾミブを最初の計画より低頻度で、例えば、週1回または2週間に1回投与する「維持計画」を使用してもよい。維持計画を一定期間、一般的に1〜2年継続しても、または患者が進行の徴候を示さない状態が続き、著しい毒性がなく治療に耐える限り無期限に継続してもよい。
【0043】
化合物1は、任意の適した用量でボルテゾミブと併用投与されてもよい。適した化合物1の用量は、約0.5mg/m2〜約10mg/m2の範囲、例えば、約0.5mg/m2〜約5mg/m2、または約0.5mg/m2〜約3mg/m2の範囲であってもよい。適した化合物1の用量は、典型的には約0.5mg/m2〜約3mg/m2の範囲となるであろう。好ましくは、化合物1の用量は、約1mg/m2〜約3mg/m2の範囲である。より好ましくは、化合物1の用量は、約1.5mg/m2〜約2.5mg/m2の範囲である。好ましい化合物1の用量としては、1.5mg/m2、1.8mg/m2、2.1mg/m2、または2.4mg/m2が挙げられるが、これらに限定されるものではない。前述の用量はどの化合物1投与方法にも適しており、とりわけ皮下投与または静脈内投与に適しており、静脈内投与が好ましい。化合物1の経口用量は、典型的には前述の範囲の上限にあり、例えば、約1mg/m2〜約7mg/m2となるであろう。一実施形態では、化合物1の経口用量は、約2mg/m2〜約6mg/m2、例えば、約3mg/m2〜約5mg/m2である。例示的な化合物1の経口用量としては、2mg/m2、3mg/m2、4mg/m2、5mg/m2または6mg/m2が挙げられるが、これらに限定されるものではない。
【0044】
化合物1は、前述の用量でボルテゾミブと任意の適した計画に従って併用投与されてもよい。化合物1の投与量は一定であっても、または投与計画内で変えてもよい。薬物に関連する著しい毒性が認められない限り、好ましくは、化合物1の用量は投与計画中、一定のレベルに維持されるが、薬物に関連する著しい毒性が認められた場合、その後の用量を例えば約20〜30%減少させることができる。化合物1は、ボルテゾミブと同じ日に投与されても、または異なる日に投与されてもよい。一実施形態では、化合物1とボルテゾミブは、投与計画中、同じ日に投与される。適した化合物1の投与計画は、典型的には1日1回投与〜週1回投与またはさらには月1回投与の範囲となるであろう。好ましくは、化合物1は1日1回より低頻度で、例えば、2〜14日に1回投与される。好ましくは、化合物1は、3〜28日毎に、例えば、7〜21日毎に投与される。例えば、化合物1を週2回投与してもよい。別の例では、化合物1を週1回投与してもよい。別の例では、化合物1を2週間に1回投与してもよい。好ましくは、投与計画は、1週間以上、例えば、2、3または4週間、化合物1で治療した後、化合物1を投与しない期間を少なくとも5日間、例えば、約7〜21日間含む。好ましくは、休薬期間は約10〜17日間、例えば、約10日間または約17日間である。例えば、化合物1を21日サイクルの1日目、4日目、8日目および11日目に投与してもよく、12〜21日目は休薬期間である。別の実施形態では、化合物1を28日サイクルの1日目、4日目、8日目、および11日目に投与してもよく、12〜28日目は休薬期間である。別の実施形態では、化合物1を21日サイクルの1日目と8日目に投与してもよく、12〜21日目は休薬期間である。別の実施形態では、化合物1を28日サイクルの1日目と8日目に投与してもよく、12〜28日目は休薬期間である。別の実施形態では、化合物1を21日サイクルの1日目と15日目に投与してもよい。別の実施形態では、化合物1を28日サイクルの1日目と15日目に投与してもよい。前述のように、ボルテゾミブは投与計画の同じ日に投与されても、または異なる日に投与されてもよい。例えば、化合物1とボルテゾミブを両方とも21日サイクルの1日目、4日目、8日目および11日目に投与してもよい。別の実施形態では、化合物1とボルテゾミブを両方とも、28日サイクルの1日目、4日目、8日目、および11日目に投与してもよい。別の実施形態では、ボルテゾミブを21日サイクルの1日目、4日目、8日目および11日目に投与し、化合物1を21日サイクルの1日目と8日目に投与してもよい。別の実施形態では、ボルテゾミブを28日サイクルの1日目、4日目、8日目および11日目に投与し、化合物1を28日サイクルの1日目と8日目に投与してもよい。別の実施形態では、ボルテゾミブを21日サイクルの1日目、4日目、8日目、および11日目に投与し、化合物1を21日サイクルの1日目と15日目に投与してもよい。別の実施形態では、ボルテゾミブを28日サイクルの1日目、4日目、8日目、および11日目に投与し、化合物1を28日サイクルの1日目と15日目に投与してもよい。別の実施形態では、化合物1を21日サイクルまたは28日サイクルの1日目、4日目、8日目および11日目に投与し、ボルテゾミブを21日サイクルまたは28日サイクルの2日目、5日目、9日目および12日目に投与してもよい。別の実施形態では、ボルテゾミブを21日サイクルまたは28日サイクルの1日目、4日目、8日目および11日目に投与し、化合物1を21日サイクルまたは28日サイクルの2日目、5日目、9日目および12日目に投与してもよい。別の実施形態では、化合物1を21日サイクルまたは28日サイクルの1日目と8日目投与し、ボルテゾミブを21日サイクルまたは28日サイクルの2日目、5日目、9日目および12日目に投与してもよい。別の実施形態では、ボルテゾミブを21日サイクルまたは28日サイクルの1日目、4日目、8日目および11日目に投与し、化合物1を21日サイクルまたは28日サイクルの2日目と9日目に投与してもよい。別の実施形態では、化合物1を21日サイクルまたは28日サイクルの1日目と15日目投与し、ボルテゾミブを21日サイクルまたは28日サイクルの2日目、5日目、9日目、および12日目に投与してもよい。別の実施形態では、ボルテゾミブを21日サイクルまたは28日サイクルの1日目、4日目、8日目、および11日目に投与し、化合物1を21日サイクルまたは28日サイクルの2日目と16日目に投与してもよい。別の実施形態では、化合物1を21日サイクルまたは28日サイクルの1日目と8日目投与し、ボルテゾミブを21日サイクルまたは28日サイクルの4日目と11日目に投与してもよい。別の実施形態では、ボルテゾミブを21日サイクルまたは28日サイクルの1日目と8日目に投与し、化合物1を21日サイクルまたは28日サイクルの4日目と11日目に投与してもよい。別の実施形態では、化合物1を21日サイクルまたは28日サイクルの1日目、5日目および9日目に投与し、ボルテゾミブを21日サイクルまたは28日サイクルの3日目、8日目、および12日目に投与してもよい。別の実施形態では、ボルテゾミブを21日サイクルまたは28日サイクルの1日目、5日目および9日目に投与し、化合物1を21日サイクルまたは28日サイクルの3日目、8日目、および12日目に投与してもよい。別の実施形態では、化合物1を21日サイクルまたは28日サイクルの1日目と15日目に投与し、ボルテゾミブを21日サイクルまたは28日サイクルの1日目、6日目および11日目に投与してもよい。別の実施形態では、ボルテゾミブを21日サイクルまたは28日サイクルの1日目と11日目に投与し、化合物1を21日サイクルまたは28日サイクルの5日目と15日目に投与してもよい。計画された投与サイクルを1回以上繰り返してもよい。例えば、最大反応が認められるまで、1または2サイクル追加して、計画されたサイクルを繰り返してもよい。別の例として、計画されたサイクルを6〜12サイクル繰り返してもよい。任意選択により、最初のサイクルを完了した後、ボルテゾミブと化合物1を最初の計画より低頻度で、例えば、週1回、2週間に1回、3週間に1回、または4週間に1回投与する「維持計画」を使用してもよい。維持計画を一定期間、一般的に1〜2年継続しても、または、患者が進行の徴候を示さない状態が続き、著しい毒性がなく治療に耐える限り無期限に継続してもよい。
【0045】
前述のように、本発明の化合物1とメルファランの投与計画は柔軟性が高い。特定の実施形態では、これらの薬物に適していることが知られている投与計画を改良して投与計画を立てることができる。例えば、上記表1に示すように、経口メルファラン(9mg/m2)を、ボルテゾミブ(1.3mg/m2)および経口プレドニゾン(60mg/m2)と併用投与する6週間の治療サイクルを9サイクル行うことにより、前治療歴のない多発性骨髄腫を治療することが認可されている。メルファランは各6週間のサイクルの1日目、2日目、3日目、および4日目に投与される(42)。
【0046】
経口メルファランは、通常、単剤として毎日6mgの用量で投与される。約1週間の間隔で行われる血球算定に基づき、必要に応じて用量を調節する。2〜3週間治療した後、最大4週間投薬を中止し、その間、血球算定を慎重に追跡しなければならない。白血球数と血小板数が上昇している場合、毎日2mgの維持量を定めてもよい(43)。
【0047】
本発明の併用療法では、メルファランの投与計画は、前述のものを含む認可された多発性骨髄腫の投与計画と類似していても、または異なってもよい。例えば、メルファランを認可された投与計画より高頻度または低頻度で投与してもよく、任意選択により、比較的高用量または低用量で投与してもよい。
【0048】
メルファランは、化合物1と任意の適した用量で併用投与されてもよい。適したメルファランの用量は、約0.025mg/kg〜約0.5mg/kgの範囲、例えば、約0.05mg/kg〜約0.3mg/kgの範囲であってもよい。適したメルファランの用量は、典型的には約0.025mg/kg〜約0.3mg/kgの範囲となるであろう。好ましくは、メルファランの用量は、約0.05mg/kg〜約0.25mg/kgの範囲である。より好ましくは、メルファランの用量は、約0.1mg/kg〜約0.2mg/kgの範囲である。好ましいメルファランの用量としては、0.1mg/kg、0.15mg/kg、0.2mg/kg、または0.25mg/kgが挙げられるが、これらに限定されるものではない。前述の用量は、どのメルファラン投与方法にも適しており、とりわけ皮下投与、静脈内投与または経口投与に適しており、経口投与が好ましい。
【0049】
メルファランは、前述の用量で化合物1と任意の適した計画に従って併用投与されてもよい。メルファランの投与量は一定であっても、または投与計画内で変えてもよい。薬物に関連する著しい毒性が認められない限り、好ましくは、メルファランの用量は投与計画中、一定のレベルに維持されるが、薬物に関連する著しい毒性が見られた場合、その後の用量を例えば約20〜30%減少させることができる。メルファランは、化合物1と同じ日に投与されても、または異なる日に投与されてもよい。適したメルファランの投与計画では、期間中、連日投与した後、休薬期間が設けられるであろう。好ましくは、メルファランを約3〜約7日間、1日1回投与した後、約1〜6週間の休薬期間を設ける。好ましくは、メルファランを約4〜約7日間、1日1回投与した後、約4〜6週間の休薬期間を設ける。好ましくは、メルファランを約4〜約5日間、1日1回投与した後、約4〜6週間の休薬期間を設ける。計画を1回以上繰り返してもよい。
【0050】
化合物1は、メルファランと任意の適した用量で併用投与されてもよい。適した化合物1の用量は、約0.5mg/m2〜約10mg/m2の範囲、例えば、約0.5mg/m2〜約5mg/m2、または約0.5mg/m2〜約3mg/m2の範囲であってもよい。適した化合物1の用量は、典型的には約0.5mg/m2〜約3mg/m2の範囲となるであろう。好ましくは、化合物1の用量は、約1mg/m2〜約3mg/m2の範囲である。より好ましくは、化合物1の用量は、約1.5mg/m2〜約2.5mg/m2の範囲である。好ましい化合物1の用量としては、1.5mg/m2、1.8mg/m2、2.1mg/m2、または2.4mg/m2が挙げられるが、これらに限定されるものではない。前述の用量は、どの化合物1投与方法にも適しており、とりわけ皮下投与または静脈内投与に適しており、静脈内投与が好ましい。化合物1の経口用量は、典型的には前述の範囲の上限にあり、例えば、約1mg/m2〜約7mg/m2となるであろう。一実施形態では、化合物1の経口用量は、約2mg/m2〜約6mg/m2、例えば、約3mg/m2〜約5mg/m2である。例示的な化合物1の経口用量としては、2mg/m2、3mg/m2、4mg/m2、5mg/m2または約6mg/m2が挙げられるが、これらに限定されるものではない。
【0051】
化合物1は、前述の用量でメルファランと任意の適した計画に従って併用投与されてもよい。化合物1の投与量は一定であっても、または投与計画内で変えてもよい。薬物に関連する著しい毒性が認められない限り、好ましくは、化合物1の用量は投与計画中、一定のレベルに維持されるが、薬物に関連する著しい毒性が見られた場合、その後の用量を例えば約20〜30%減少させることができる。化合物1は、メルファランと同じ日に投与されても、または異なる日に投与されてもよい。適した化合物1の投与計画は、典型的には1日1回投与〜週1回投与またはさらには月1回投与の範囲となるであろう。好ましくは、化合物1を1日1回より低頻度で、例えば、2〜14日に1回投与する。好ましくは、化合物1を3〜28日毎に、例えば、7〜21日毎に投与する。例えば、化合物1を週2回投与してもよい。別の例では、化合物1を週1回投与してもよい。別の例では、化合物1を2週間に1回投与してもよい。好ましくは、投与計画は、化合物1を投与しない期間を少なくとも5日間、例えば、約7〜21日間含む。好ましくは、休薬期間は約10〜17日間、例えば、約10日間または約17日間である。例えば、化合物1を21日サイクルの1日目、4日目、8日目および11日目に投与してもよく、12〜21日目は休薬期間である。別の実施形態では、化合物1を28日サイクルの1日目、4日目、8日目、および11日目に投与してもよく、12〜28日目は休薬期間である。別の実施形態では、化合物1を21日サイクルの1日目と8日目に投与してもよく、12〜21日目は休薬期間である。別の実施形態では、化合物1を28日サイクルの1日目と8日目に投与してもよく、12〜28日目は休薬期間である。別の実施形態では、化合物1を21日サイクルの1日目と15日目に投与してもよい。別の実施形態では、化合物1を28日サイクルの1日目と15日目に投与してもよい。一実施形態では、化合物1を42日サイクルの1日目、4日目、8日目、11日目、22日目、25日目、29日目および32日目に投与し、メルファランを42日サイクルの1日目、2日目、3日目および4日目に投与してもよい。別の実施形態では、化合物1を84日サイクルの1日目、4日目、8日目、11日目、22日目、25日目、29日目、32日目、43日目、50日目、64日目、および71日目に投与し、メルファランを84日サイクルの1日目、2日目、3日目、4日目、43日目、44日目、45日目、および46日目に投与してもよい。別の実施形態では、メルファランを28日サイクルの1日目、2日目、3日目、および4日目に投与し、化合物1を28日サイクルの1日目と15日目に投与してもよい。別の実施形態では、メルファランを42日サイクルの1日目、2日目、3日目、4日目、および5日目に投与し、化合物1を42日サイクルの1日目、8日目、22日目および29日目に投与してもよい。計画を1回以上繰り返してもよい。
【0052】
他の1種類以上の癌治療薬を、化合物1とボルテゾミブ、または化合物1とメルファランの投与と併用してもよい。このような治療薬としては、ボルテゾミブ、メルファラン、デキサメタゾンおよび他のステロイド、ドキソルビシン、シクロホスファミド、サリドマイド、レナリドミド、三酸化ヒ素、およびヒストン脱アセチル化酵素阻害剤を含む抗癌剤が挙げられるが、これらに限定されるものではない。これらの薬剤の適切な用量は当該技術分野で周知である。本発明の別の態様では、追加的な薬剤はフィルグラスチムなどの顆粒球コロニー形成刺激因子(G−CSF)であってもよい。好ましい実施形態では、フィルグラスチムを6日目から、好中球数がANC>1000に回復するまで約5μg/kg/日の用量でSC投与する。ANCは、「好中球絶対数」の略語である。
【0053】
本発明の併用療法は、癌性増殖の一部または全部を外科的に除去する試みをさらに含む治療過程の一部として使用されてもよい。例えば、対象の外科的治療後に、残存する新生物細胞または転移細胞を治療するため、併用療法を施してもよい。また、腫瘍のサイズを小さくし、切除する組織の量を減少させるために手術の前に治療を行い、それによって手術の侵襲性と外傷性を低くしてもよい。
【0054】
本明細書に開示される対象である併用療法による多発性骨髄腫の治療は、DNA損傷を誘導する放射線治療手段を用いた1つ以上の治療過程を含んでもよい。放射線治療手段としては、例えば、γ線照射、X線、紫外線照射、マイクロ波、電子放出、および放射性同位体等が挙げられる。局在する腫瘍部位に前述の形態の放射線を照射することにより療法を達成することができる。
【0055】
本発明の別の態様は、骨髄細胞を本発明の併用療法で処置することにより、骨髄をパージする、即ち、骨髄から癌細胞を除去する方法に関する。その後、パージした骨髄を、骨髄を取り出した対象に再移植しても、または異なる対象に移植してもよい。
【0056】
材料および方法
試薬
化合物1(4mg;Cephalon,Frazer,PA)をプロピレングリコール(800μL)に溶解し、5%マンニトールに添加して、最終保存液濃度1mg/mLにし;処置直前に化合物1の保存溶液を表示した濃度に希釈した。ボルテゾミブ(Millennium Pharmaceuticals,Cambridge,MA)を1mg/mLで得、0.9%塩化ナトリウムを使用して規定どおり希釈した。メルファラン(Sigma,St.Louis,MO)を酸−EtOH(酸−EtOH:濃HCl47μL+100%EtOH1mL)100μLに溶解し、リン酸緩衝生理食塩水で1mLに希釈した。製剤は毎週調製した。
【0057】
細胞株および初代培養細胞
アメリカ培養細胞系統保存機関(American Type Culture Collection)(Rockville,MD)からヒト骨髄腫細胞株RPMI8226を得た。MM1S骨髄腫細胞株は、Steven Rosen博士(Northwestern University,Chicago,IL)によって提供された。正常な末梢血単核細胞(PBMC)をHistopaque(登録商標)密度勾配遠心分離法で、製造業者のプロトコル(Sigma−Aldrich,St.Louis,MO)に従って分離した。骨髄腫細胞株およびPBMCを、5%二酸化炭素(CO2)雰囲気中37℃で、10%牛胎児血清、2mM L−グルタミン、100IU/mLペニシリン、100μg/mLストレプトマイシン、および必須アミノ酸を添加したRPMI 1640(Omega Scientific,Tarzana,CA)中で培養した。
【0058】
細胞生存率アッセイ(MTSアッセイ)
細胞を105細胞/100μL/ウェルで96穴プレートに播種し、24時間インキュベートした。RPMI8226細胞およびMM1S細胞を溶媒、化合物1、ボルテゾミブ、メルファラン、化合物1+ボルテゾミブ、または化合物1+メルファランの存在下で48時間培養した。インキュベート時間後に、CellTiter 96(登録商標)AQueous Non−Radioactive Cell Proliferation Assay(Promega,Madison,WI)を使用して細胞生存率を定量化した。各ウェルをMTS(3−(4, 5−ジメチルチアゾール−2−イル)−5−(3−カルボキシメトキシフェニル)−2−(4−スルホフェニル)−2H−テトラゾリウム、分子内塩)で1〜4時間処理した後、490nmで吸光度を記録した。490nmにおける吸光度で測定されるホルマザン生成物の量は、培地内の生細胞数に正比例する。
【0059】
ChouおよびTalalayの半有効(median effect)法を使用して、化合物1とボルテゾミブまたはメルファランとのin vitro相乗作用を評価した(30)。各組み合わせについて別々に併用指数(CI)を算出した。薬物相互作用は、CIが0.9未満の場合は相乗的であると判定され、またはCIが1.1より大きい場合は拮抗的であると判定された。CIが0.9〜1.1の場合は薬物相加効果を示すものと見なされた(31)。
【0060】
アネキシンVおよびヨウ化プロピジウム染色によるアポトーシスアッセイ
薬物療法に反応したアポトーシスを定量化するため、RPMI8226細胞(1ウェル当たり5×105細胞)を溶媒またはPIと共に37℃、5%CO2で30時間インキュベートした。陽性対照として、細胞をアクチノマイシンD250ng/mLと共に24時間または48時間インキュベートした。次いで、細胞を2回リン酸緩衝生理食塩水で洗浄し、結合緩衝液(1.4M NaClおよび25mM CaCl2を含有する、100mM HEPES/NaOH、pH7.5)に再懸濁させ、製造業者のプロトコル(BioVision,Mountain View,CA)に従って、フルオレッセインイソチオシアネート(FITC)結合アネキシンV、および蛍光色素ヨウ化プロピジウム(PrI)で染色した。各薬物療法について、ゲートをかけた1×105の事象を記録した。PI染色とアネキシンV染色の両方について陰性の細胞を生存と見なし;アネキシンV−陽性、PrI−陰性の細胞を初期アポトーシスと見なし;アネキシンV−陽性、PrI−陽性の細胞を後期アポトーシスと見なした。Cytomics CXPソフトウェアを用いたBeckman Coulter FC500サイトメーター(Beckman Coulter,Fullerton,CA)を使用してフローサイトメトリー分析を行った。
【0061】
SCIDマウス
Jackson Laboratory(Bar Harbor,ME)から6〜8週齢の雄性SCIDマウスを得、動物資源施設の特定病原菌を排除した領域で飼育した。動物試験は全て、動物実験委員会(Institutional Animal Care and Use Committee)により認可されたプロトコルに従って行った。手術前にケタミン、キシラジンおよびイソフルランで動物に麻酔をかけ、腫瘍が直径2cmに達したとき安楽死させた。
【0062】
筋肉内腫瘍異種移植モデル
LAGκ−1A腫瘍(ボルテゾミブおよびメルファランに対して感受性)を形成するため、レナリドミドで治療中に進行した女性MM患者から骨髄生検試料を得た。この生検の直後に、患者はメルファランとボルテゾミブを併用して治療され、反応を示した。麻酔したSCIDマウスの後肢に生検組織を外科的に移植し、継代した(32)。LAGκ−1Aと同じ患者に由来するLAGκ−1B腫瘍(ボルテゾミブおよびメルファランに対して耐性)は、患者がボルテゾミブおよびメルファランで治療中に進行しているときに採取した生検試料から得られた(32)。
【0063】
麻酔したドナーマウスから骨髄腫腫瘍(LAGκ−1AまたはLAGκ−1B)を切除し、20〜40mm3片に薄切し、麻酔した未処置SCIDマウスの左浅臀筋に外科的に移植した。免疫活性をさらに抑制するため、レシピエントマウスに毎週、抗アシアロGM1ウサギ血清(Wako Bioproducts,Richmond,VA)を注射した。マウスを実験群の1つに無作為に割り付け、腫瘍移植7〜21日後に処置を開始した。化合物1は、規定通り、週2回(水、金)、静脈内注射(0.5〜3.0mg/kg)または強制経口投与(10mg/kg)により投与した。メルファラン(1mg/kg)は毎週(水)、腹腔内(IP)注射により投与した。ボルテゾミブ(0.5mg/kg)は週2回(火、木)IV注射により投薬した。対照処置は、化合物1の希釈剤(5%マンニトール3.2mLおよびプロピレングリコール800μL)のみからなった。
【0064】
ヒト免疫グロブリンG(hIgG)酵素結合免疫吸着検定法(ELISA)
LAGκ−1A腫瘍により分泌されたhIgGの血清中濃度(LAGκ−1B腫瘍は異常タンパク質を分泌しない)をELISAにより腫瘍増殖のタンパク質マーカーとして定量化した。毎週、MM腫瘍を有するマウスの後眼窩静脈叢から採血した。得られた試料を13,000rpmで30分間遠心分離して、血清を得た。製造業者の仕様書に従って、hIgG ELISAキット(Bethyl Laboratories,Montgomery,TX)を使用した。KC Juniorソフトウェアを用いたμQuantマイクロプレート分光光度計(Bio−Tek Instruments,Winooski,VT)で、450nmにおける吸光度(参照波長550mm)を測定した。グラフ化したデータは、n=マウス7〜8匹/群での平均値±SEMである。
【0065】
腫瘍体積の測定
腫瘍増殖の直接測定として、毎週、カリパスを使用して腫瘍体積を計測し、楕円体の体積に関する式を適用した(4/3π×[幅/2]2×[長さ/2])。
【0066】
腫瘍細胞におけるアポトーシス誘導因子(AIF)発現の免疫組織化学分析
LAGκ−1B腫瘍を4%パラホルムアルデヒド中に固定し、5μmの切片に切断した。 簡潔に言えば、0.05%Tween−20(TBST)および3%BSAを有するトリス緩衝生理食塩水で、室温で1時間切片をブロッキングした後、AIFに対するウサギ抗体(Sigma,St.Louis,MO)と共に終夜インキュベートした。切片をTBSTで3回洗浄し、西洋ワサビペルオキシダーゼ(ARH)結合抗ウサギ抗体(KPL,Gaithersburg,MD)をTBSTで1:500に希釈したもので、室温で2時間処理した。スライドガラスはTBST中で3回洗浄し、3−アミノ−9−エチルカルバゾール(AEC)緩衝液に5分間入れ、AECキット(Dako,Glostrup,Denmark)を使用して色を検出した。染色はOlympus BX51顕微鏡(Olympus Imaging America Inc.,Center Valley,PA)を使用して記録し、Microsuite Biological Suiteプログラム (Olympus BX51)で分析した。
【0067】
統計解析
腫瘍増殖およびhIgG濃度を処置群の平均値および標準誤差に関して解析した。Studentのt検定を使用して、処置群間の統計学的有意差を検定した。最小有意水準は、P<0.05であった。
【実施例】
【0068】
次の実施例に示すデータから、本発明の併用療法は、化合物1とボルテゾミブまたは化合物1とメルファランを低用量で併用した場合、標準用量の単剤療法と比較して、MMにおける有効性が同様であるかまたは高い可能性があることが示唆された。このようにして、ボルテゾミブに関する末梢神経障害およびメルファランに関する骨髄抑制などの薬物に関連する毒性が低減し得るか、または回避され得る(40、41)。本明細書に記載の実験では、併用療法で処置したマウスは処置に対する忍容性が高く、腫瘍の進行がほとんどまたは全くなかった。
【0069】
実施例1.化合物1はMM細胞に対する細胞毒性があり、in vitroで抗MM剤と併用した場合、相乗作用を示す
RPMI8226細胞およびMM1S細胞を増加する濃度(0.1〜10nM)の化合物1の存在下で培養した。48時間後、MTSアッセイで細胞生存率を評価した。化合物1は、両方の細胞株で濃度依存的な生存率の低下を誘導した(図1A)。細胞を化合物1で24時間または72時間処置した場合、結果は同様であった(データ不示)。
【0070】
次に、化合物1+ボルテゾミブ(PI)またはメルファラン(化学療法剤)の存在下における細胞生存率を調べた。まず、MM1S細胞を一定濃度の化合物1(1.75nM)および増加する濃度(0.5〜2.5nM)のボルテゾミブと共に48時間インキュベートした。1.5nM以下のボルテゾミブ濃度では、化合物1の細胞毒性効果が増強した。例えば、単剤の場合、化合物1(1.75nM)と最低濃度(0.5nM)のボルテゾミブはそれぞれ細胞生存率を約16%低下させた。しかし、化合物1(1.75nM)をボルテゾミブ(0.5nM)と併用した場合、細胞生存率は約43%低下した(図1B)。Chou−Talalayの式を使用して、この組み合わせの相乗作用を確認した(CI、0.74〜0.85)(30、31)。RPMI8226細胞で実験を繰り返した場合、同様の結果が得られた(データ不示)。
【0071】
RPMI8226細胞をメルファラン(40μM)および6.0nM以上の濃度の化合物1(IC50=8.5nM)と共にインキュベートした場合、相乗的な生存率の低下が認められた(CI、0.78〜0.87)。例えば、細胞生存率は単剤のメルファラン(40μM)の存在下では約30%低下し、単剤の化合物1(9.0nM)の存在下では64%低下した。両方の薬物を同時に使用した場合、細胞生存率は90%低下した(図1C)。総合的に、これらの結果から、化合物1をボルテゾミブまたはメルファランと併用すると、MM細胞の生存率を相乗的に抑制できることが分かる。
【0072】
実施例2.化合物1とボルテゾミブは新生物細胞に対して選択的な細胞毒性を有する
2種類以上のPIを用いた療法がin vivoで実施可能であるためには、この組み合わせが非新生物細胞に損傷を与えてはならない。従って、正常なPBMCの生存率に対する化合物1+ボルテゾミブの効果を試験した。健常なドナーのPBMCを化合物1単独、ボルテゾミブ単独、または両方の薬剤の存在下で48時間培養し、細胞生存率をMTSアッセイで定量化した。どちらかのPIをそのIC50付近で用いたMM細胞における単独療法では、PBMCの生存率はあまり低下しなかった(PBMCを9nMの化合物1と9nMのボルテゾミブでそれぞれ処置した場合、生細胞は約75%および85%であった)(図2A、B)。両方のPIと共に共インキュベートした場合、どちらかの薬剤を単独で投与した場合と比較して、細胞生存率はそれ以上低下しなかったか、または僅かに上昇した(PIを両方とも投与した場合、試験した全ての濃度で細胞生存率が3%〜23%低下した)(図2A、B)。別の健常なドナーに由来するPBMCで類似の結果が得られた(図2C)。PBMCが高濃度のPIによって損傷を受けるかどうかを調べるため、PBMCを120nMまでの化合物1と共に培養した。対照と比較した場合、試験したどの濃度でも細胞生存率の顕著な差は認められなかった(図2D)。PBMCを120nMまでの濃度のボルテゾミブと共にインキュベートした場合、同様の結果が得られた(データ不示)。従って、本発明の併用療法はMMに対する有効性が高く、正常な細胞に対する毒性が増加しない。
【0073】
実施例3.化合物1とボルテゾミブの併用によりMM細胞のアポトーシスが誘導される
化合物1とボルテゾミブでMM細胞を処置した後に認められる細胞生存率の低下がアポトーシスによるものかどうかを調べるため、RPMI8226細胞を両方の薬剤(どちらの薬物も2.5nM)と共に30時間インキュベートし、生存率測定用色素であるPrIおよびアポトーシスマーカーであるアネキシンVで染色された細胞の割合を測定した。初期アポトーシス(PrI−/アネキシンV+)の細胞の割合は、どちらかの薬剤単独で処置した後(それぞれ2.5nMの化合物1または2.5nMのボルテゾミブで処置した細胞の10.4%および17.5%)より、両方のPIで処置した後の方が大きかった(細胞の38.9%)(図3)。後期アポトーシス(PrI+/アネキシンV+)または壊死(PrI+/アネキシンV−)の細胞の割合は、この時点では処置群間で差がなかった。
【0074】
実施例4.単剤の化合物1はin vivoでヒトMM腫瘍の増殖を阻害した
化合物1は、単剤の場合も併用した場合もin vitroで強力な抗MM効果を示すため、次に一連のin vivo試験を行った。これらの実験では、LAGκ−1A(ボルテゾミブ感受性およびメルファラン感受性)腫瘍およびLAGκ−1B(ボルテゾミブ耐性およびメルファラン耐性)腫瘍を有するマウスを使用したが、これらの腫瘍は両方とも元々MM患者の骨髄生検試料に由来した。これらの腫瘍はヒトのMMによく似ており、マウスで継代され、一貫した増殖パターンおよび化学療法抵抗性パターンを有した。腫瘍組織を筋肉内移植した後、化合物1を0.1〜3mg/kgの範囲の様々な用量でIV投与することにより、または10mg/kg経口投与することによりマウスを週2回処置した。対照群のマウスには化合物1の希釈剤を投与した。
【0075】
単剤の化合物1をIV投与すると、LAGκ−1A腫瘍からの異常タンパク質の分泌が用量依存的に減少した。比較的低用量の化合物1では腫瘍hIgG分泌が減少し、比較的高用量では血清hIgG濃度が本質的に検出不可能となった(薬物療法の28日目に対照と比較した場合、IV化合物1、1mg/kgではP=0.0001、3mg/kgではP=0.0002)(図4A)。ボルテゾミブと異なり、化合物1は経口製剤としても活性を示す(28、29)。経口化合物1で処置して2週間内に、血清hIgG濃度は、対照で処置した動物より著しく低下した(P=0.0007)。経口化合物1で処置して28日までに、血清hIgG濃度はごく僅かになった(対照で処置した動物と比較した場合、P=0.0001)(図4A)。
【0076】
異常タンパク質濃度に対する効果に加え、単剤の化合物1は、溶媒で処置したマウスと比較して、LAGκ−1A腫瘍体積の増加を遅延させた。4週間薬物療法を行った後、化合物1を1mg/kgまたは3mg/kg、IV投与したものは、対照で処置した異種移植片と比較して同時点で腫瘍体積が約15倍減少した(対照と比較した場合、各用量でP=0.0001)(図4B)。化合物1を経口送達すると、腫瘍増殖も阻害された。経口化合物1で処置してわずか14日後に、対照で処置した腫瘍と比較して、腫瘍体積の著しい減少が認められ(P=0.0002)、試験期間中ずっとその差があった(図4B)。
【0077】
また、腫瘍体積に対する化合物1の効果を、ボルテゾミブ耐性非分泌性LAGκ−1B腫瘍を有するマウスでも試験した(図4C)。LAGκ−1A腫瘍の場合と同様に、化合物1は、IV注射と経口製剤の両方で腫瘍増殖を阻害した。対照で処置したマウスと比較して、3mg/kg IV化合物1または10mg/kg経口化合物1で処置したマウスの腫瘍は、14日間処置した後、約8〜12倍小さかった(それぞれ、P=0.0008およびP=0.0028)(図4C)。この実験に使用されるLAGκ−1B腫瘍は非分泌性であるため、これらの腫瘍を有するマウスの血清hIgG濃度は試験しなかった。
【0078】
実施例5.化合物1をボルテゾミブと併用するとボルテゾミブ感受性LAGκ−1A MM腫瘍の増殖が阻害される
化合物1をボルテゾミブと併用するとin vitroでMM細胞の相乗的アポトーシスが誘導されるため、in vivoにおいてヒトMM腫瘍でこの組み合わせを試験した。最適以下の単剤抗腫瘍活性を有する薬物濃度を選択した。単独療法の場合、化合物1(1mg/kg IV)とボルテゾミブ(0.5mg/kg IV)は両方とも、溶媒対照と比較して血清hIgG濃度およびLAGκ−1A腫瘍体積を部分的にしか低減しなかった(図5A、B)。しかし、異常タンパク質分泌と腫瘍体積の両方で測定した場合、単剤の化合物1またはボルテゾミブで処置したにもかかわらず、腫瘍増殖は徐々に進行し続けた。対照的に、化合物1とボルテゾミブを同じ用量で併用投与すると、検出可能な異常タンパク質分泌および腫瘍体積の増加がなくなった。対照で処置したLAGκ−1A腫瘍と併用療法で処置したLAGκ−1A腫瘍との増殖の差は、まず、療法開始28日後に顕著になった(血清hIgG濃度ではP=0.0028、腫瘍体積ではP=0.0265)(図5A、B)。腫瘍の進行は実験期間中ずっと(110日間)完全に阻害され続けた。さらに、化合物1+ボルテゾミブで処置したマウスを安楽死させたとき、切除した後肢を検査すると、筋肉塊だけが認められ、腫瘍組織は認められなかった。試験開始時に移植された腫瘍は完全に退縮していた。特に、併用療法は忍容性が高く、明らかな毒性の徴候は認められなかった。
【0079】
以前の研究では、hIgG分泌骨髄腫腫瘍体積の変化は、血清ヒト異常タンパク質濃度の変化と密接に相関した(15、32)。しかし、これらの実験では、単剤の化合物1またはボルテゾミブで処置した腫瘍からの異常タンパク質分泌はプラトーに達した後、処置63日目(試験70日目)から低下し;対照的に、腫瘍体積は試験期間中ずっと増加し続けた(図5A、B)。従って、単独療法の場合、各薬剤は、腫瘍体積より血清hIgG濃度の上昇をより効果的に抑制した。
【0080】
これらの結果を検証するため、70日目以降の試料をELISAで再試験し、hIgG濃度の低下を確認した。hIgG濃度と腫瘍体積の間の反比例関係は、おそらく癌幹細胞に由来する非分泌性、薬物耐性MM細胞集団が存在することを示唆する。従って、ボルテゾミブまたは化合物1は単独では、主に、MMの抗体を分泌する成熟形質細胞成分に対して作用し(22)、腫瘍増殖の遅延の原因となる小さい幹細胞集団に影響を及ぼさない可能性がある(24)。
【0081】
対照的に、化合物1とボルテゾミブを併用投与したLAGκ−1Aを有するマウスは、hIgGと腫瘍体積の測定の両方で評価した場合、110日間の試験中ずっと、腫瘍増殖の顕著で持続的な抑制が認められた。これらのデータから、単剤のPIの存在下で異常タンパク質を産生することなく増殖するMM細胞は、化合物1とボルテゾミブの組み合わせに対して感受性があることが分かる。
【0082】
実施例6.化合物1とボルテゾミブの併用療法はボルテゾミブ耐性LAGκ−1B腫瘍の薬物耐性を克服する
LAGκ−1B腫瘍は、ボルテゾミブに対して耐性があり;実際、どちらかのPI単独 (0.5mg/kg IVボルテゾミブまたは1mg/kg IV化合物1)ではこれらの腫瘍の増殖はあまり抑制されない。対照的に、化合物1+ボルテゾミブで処置したLAGκ−1Bを有するマウスは、21日間処置した後、溶媒で処置したマウスよりも形成した腫瘍が著しく小さかった(P=0.0014)。さらに、28日間処置した後、併用療法を受けたマウスの腫瘍はまた、どちらかのPI単独で処置したマウスの腫瘍より小さかった(それぞれ化合物1単独およびボルテゾミブ単独と比較した場合、P=0.0039およびP< 0.0001)(図5C)。時間対腫瘍の進行について調べるため、併用療法群のマウスの投与を継続した。単剤のPIで処置した腫瘍と比較して、併用療法群の腫瘍体積の進行は100%遅延された(単剤のPIで処置したマウスでは腫瘍の進行まで35日であったのに対して、両方のPIで処置したマウスでは腫瘍の進行まで70日であった)。最後に、各処置群の全生存期間を記録した。溶媒で処置したマウスと比較して、併用療法を受けたマウスは、150%長く生存した(データ不示)。どちらかのPI単独で処置したマウスは、溶媒で処置したマウスより20%長く生存した(データ不示)。他のマウス生存期間データから、両方のPIを用いた併用療法の忍容性は単独療法と同様であることが分かった(データ不示)。総合的に、これらのデータから、化合物1をボルテゾミブと併用するとin vivoにおけるヒトMMのボルテゾミブ耐性を克服できることが分かる。
【0083】
実施例7.化合物1をメルファランと併用するとLAGκ−1A腫瘍およびLAGκ−1B腫瘍の増殖が阻害される
化合物1はメルファランと共同して、培養されたMM細胞の生存率を低下させ、ボルテゾミブは実験室(11)と臨床試験(33)の両方でメルファランの抗MM効果を増強するため、in vivoにおけるこのアルキル化剤と化合物1の併用の有効性を評価した。低用量(1mg/kg IP)での単剤のメルファランによる処置は、LAGκ−1Aを有するマウスの血清hIgG濃度または腫瘍体積に対して効果がなかった。同様に、単剤の化合物1(1mg/kg IV)を投与した結果、異常タンパク質分泌と腫瘍体積は両方ともあまり減少しなかった。しかし、3週間処置した後、化合物1とメルファランの両方で処置した腫瘍は、溶媒で処置した腫瘍と比較して、hIgG分泌(P=0.0012)と腫瘍体積(P=0.032)が両方とも著しく減少した(図5D、E)。ボルテゾミブ耐性およびメルファラン耐性のLAGκ−1B腫瘍を有するマウスで、同様の結果が得られた。3mg/kg IPの単剤メルファラン(LAGκ−1Aを有するマウスに投与した用量より3倍高い)または1mg/kg IVの化合物1(LAGκ−1Aを有するマウスに投与したのと同じ用量)では、腫瘍体積の増加が部分的に抑制されたが、化合物1をメルファランと併用した場合、腫瘍体積は事実上検出不可能なレベルまで減少した(図5F)。
【0084】
単剤療法と対照的に、どちらかのタイプの腫瘍を有するマウスで併用療法を継続する限り(LAGκ−1Aマウスでは63日間の処置、およびLAGκ−1Bマウスでは49日間の処置)、腫瘍増殖は防止された。さらに、併用療法の忍容性は、各薬剤単独の忍容性と同様であった(データ不示)。
【0085】
実施例8.化合物1とボルテゾミブで処置したLAGκ−1Bマウスの腫瘍ではアポトーシス誘導因子の高い発現が認められる
LAGκ−1Bを有するマウスの腫瘍を処置後に切除し、アポトーシスマーカーであるAIFに関して染色した。単剤の化合物1またはボルテゾミブで処置した腫瘍では、溶媒で処置した腫瘍と比較して、高いAIF発現が認められた。しかし、AIF発現は、両方のPIで処置した動物から採取した腫瘍ではさらに増加する(図6)。化合物1+ボルテゾミブが投与されたLAGκ−1Aを有するマウスの腫瘍は、この処置群では腫瘍が完全に退縮した後、使用可能な異種移植片試料がなかったため、組織分析に使用できなかった。
【0086】
実施例9.単剤の経口化合物1はin vivoでヒトMM腫瘍の増殖を阻害した
これらの実験は、実施例4に記載のように、元々MM患者の骨髄生検材料に由来するLAGκ−1A腫瘍を有するマウスを使用して行った。腫瘍組織を筋肉内に移植した後、マウスを毎日または週2回化合物1で、経口投与により0.5〜5mg/kgの範囲の用量で毎日、または経口投与により5〜10mg/kgの範囲の用量で週2回、処置した。対照群のマウスには化合物1の希釈剤を投与した。
【0087】
化合物1は、毎日3mg/kg経口投与すると、ヒトIgG濃度と腫瘍体積の両方に関して中程度の抗骨髄腫活性を有する。化合物1は、5mg/kgで毎日経口投与した場合、統計学的に有意な抗骨髄腫活性(IgG濃度および腫瘍体積)を有する(図7および8)。この計画および用量(毎日5mg/kg)では、マウスの87%が42日目まで生存した。また、化合物1を10mg/kgで週2回経口投与すると、統計学的に有意な抗骨髄腫活性(IgG濃度および腫瘍体積)が得られ、マウスの100%が42日目まで生存する(図7および8)。
【0088】
参考文献
1.Jemal A,Siegel R,Ward E,et al.Cancer statistics,2008.CA Cancer J Clin 2008;58:71−96.
2.Richardson PG,Sonneveld P,Schuster A,et al.Bortezomib demonstrates superior survival compared with high−dose dexamethasone and higher response rates after extended follow−up in the APEX trial in relapsed multiple myeloma.Presented at:11th Congress of the European Hematology Association;June 15−18,2006;Amsterdam,the Netherlands.Abstract 224.
3.Chauhan D,Hideshima T,Anderson KC.Targeting proteasomes as therapy in multiple myeloma.Adv Exp Med Biol 2008;615:251−60.
4.McConkey DJ,Zhu K.Mechanisms of proteasome inhibitor action and resistance in cancer.Drug Resist Updat 2008;11:164−79.
5.Hideshima T,Richardson P,Chauhan D,et al.The proteasome inhibitor PS−341 inhibits growth,induces apoptosis,and overcomes drug resistance in human multiple myeloma cells.Cancer Res 2001;61:3071−6.
6.Obeng EA,Carlson LM,Gutman DM,Harrington Jr WJ,Lee KP,Boise LH.Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells.Blood 2006;107:4907−16.
7.Chauhan D,Anderson KC.Mechanisms of cell death and survival in multiple myeloma(MM):Therapeutic implications.Apoptosis 2003;8:337−43.
8.Roccaro AM,Hideshima T,Raje N,et al.Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells.Cancer Res 2006;66:184−91.
9.Richardson PG,Barlogie B,Berenson J,et al.A phase 2 study of bortezomib in relapsed,refractory myeloma.N Engl J Med 2003;348:2609−17.
10.Jagannath S,Barlogie B,Berenson J,et al.A phase 2 study of two doses of bortezomib in relapsed or refractory myeloma.Br J Haematol 2004;127:165−72.
11.Ma MH,Yang HH,Parker K,et al.The proteasome inhibitor PS−341 markedly enhances sensitivity of multiple myeloma tumor cells to chemotherapeutic agents.Clin Cancer Res 2003;9:1136−44.
12.Mitsiades N,Mitsiades CS,Richardson PG,et al.The proteasome inhibitor PS−341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents:therapeutic applications.Blood 2003;101:2377−80.
13.Baumann P,Mandl−Weber S,Oduncu F,Schmidmaier R.Alkylating agents induce activation of NFkappaB in multiple myeloma cells.Leuk Res 2008;32:1144−7.
14.Mitsiades N,Mitsiades CS,Poulaki V,et al.Apoptotic signaling induced by immunomodulatory thalidomide analogs in human multiple myeloma cells:therapeutic implications.Blood 2002;99:4525−30.
15.Campbell RA,Sanchez E,Steinberg JA,et al.Antimyeloma effects of arsenic trioxide are enhanced by melphalan,bortezomib and ascorbic acid.Br J Haematol 2007;138:467−78.
16.Podar K,Raab MS,Zhang J,et al.Targeting PKC in multiple myeloma:in vitro and in vivo effects of the novel,orally available small−molecule inhibitor enzastaurin(LY317615.HCl).Blood 2007;109:1669−77.
17.Chauhan D,Singh A,Brahmandam M,et al.Combination of proteasome inhibitors bortezomib and NPI−0052 trigger in vivo synergistic cytotoxicity in multiple myeloma.Blood 2008;111:1654−64.
18.Pei XY,Dai Y,Grant S.Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors.Clin Cancer Res 2004;10:3839−52.
19.San Miguel JF,Schlag R,Khuageva NK,et al.Updated follow−up and results of subsequent therapy in the phase III VISTA trial:bortezomib plus melphalan−prednisone versus melphalan−prednisone in newly diagnosed multiple myeloma.Blood(ASH Annual Meeting Abstracts)2008;112:650.
20.Berenson JR,Yang HH,Vescio RA,et al.Safety and efficacy of bortezomib and melphalan combination in patients with relapsed or refractory multiple myeloma:updated results of a phase 1/2 study after longer follow−up.Ann Hematol 2008;87:623−31.
21.Pineda−Roman M,Zangari M,van Rhee F,et al.VTD combination therapy with bortezomib−thalidomide−dexamethasone is highly effective in advanced and refractory multiple myeloma.Leukemia 2008;22:1419−27.
22.Bianchi G,Oliva L,Cascio P,et al.The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition.Blood 2009;113:3040−9.
23.Matsui W,Huff CA,Wang Q,et al.Characterization of clonogenic multiple myeloma cells.Blood 2004;103:2332−6.
24.Matsui W,Wang Q,Barber JP,et al.Clonogenic multiple myeloma progenitors,stem cell properties,and drug resistance.Cancer Res 2008;68:190−7.
25.Hamburger A,Salmon SE.Primary bioassay of human myeloma stem cells.J Clin Invest 1977;60:846−54.
26.Pilarski LM,Seeberger K,Coupland RW,et al.Leukemic B cells clonally identical to myeloma plasma cells are myelomagenic in NOD/SCID mice.Exp Hematol 2002;30:221−8.
27.Meister S,Schubert U,Neubert K,et al.Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition.Cancer Res 2007;67:1783−92.
28.Dorsey BD, Iqbal M, Chatterjee S, et al.Discovery of a potent,selective,and orally active proteasome inhibitor for the treatment of cancer.J Med Chem 2008;51:1068−72.
29.Piva R,Ruggeri B,Williams M,et al.CEP−18770:a novel orally−active proteasome inhibitor with a tumor−selective pharmacological profile competitive with bortezomib.Blood 2008;111:2765−75.
30.Chou TC,Talalay P.Quantitative analysis of dose−effect relationships:the combined effects of multiple drugs or enzyme inhibitors.Adv Enzyme Regul 1984;22:27−55.
31.Chou TC.Theoretical basis,experimental design,and computerized simulation of synergism and antagonism in drug combination studies.Pharmacol Rev 2006;58:621−81.
32.Campbell RA,Berenson JR.Animal models of multiple myeloma and their utility in drug discovery.In:Current Protocols in Pharmacology,vol.40,unit 49.Hoboken,NJ:John Wiley & Sons,Inc.;2008:14.9.1−22.
33.Berenson JR,Yang HH,Sadler K,et al.Phase I/II trial assessing bortezomib and melphalan combination therapy for the treatment of patients with relapsed or refractory multiple myeloma.J Clin Oncol 2006;24:937−44.
34.Demo SD,Kirk CJ,Aujay MA,et al.Antitumor activity of PR−171,a novel irreversible inhibitor of the proteasome.Cancer Res 2007;67:6383−91.
35.Crawford LJ,Walker B,Ovaa H,et al.Comparative selectivity and specificity of the proteasome inhibitors BzLLLCOCHO,PS−341,and MG−132.Cancer Res 2006;66:6379−86.
36.Sunters A,Springer CJ,Bagshawe KD,Souhami RL,Hartley JA.The cytotoxicity,DNA crosslinking ability and DNA sequence selectivity of the aniline mustards melphalan,chlorambucil and 4−[bis(2−chloroethyl)amino]benzoic acid.Biochem Pharmacol 1992;44:59−64.
37.Ahn KS,Sethi G,Chao TH,et al.Salinosporamide A(NPI−0052)potentiates apoptosis,suppresses osteoclastogenesis,and inhibits invasion through down−modulation of NF−kappaB regulated gene products.Blood 2007;110:2286−95.
38.Chen Q,Van der Sluis PC,Boulware D,Hazlehurst LA,Dalton WS.The FA/BRCA pathway is involved in melphalan−induced DNA interstrand cross−link repair and accounts for melphalan resistance in multiple myeloma cells.Blood 2005;106:698−705.
39.Jacquemont C,Taniguchi T.Proteasome function is required for DNA damage response and fanconi anemia pathway activation.Cancer Res 2007;67:7395−405.
40.Zweegman S,Huijgens PC.Treatment of myeloma:recent developments.Anticancer Drugs 2002;13:339−51.
41.Argyriou AA,Iconomou G,Kalofonos HP.Bortezomib−induced peripheral neuropathy in multiple myeloma:a comprehensive review of the literature.Blood 2008;112:1593−9.
42.VELCADER(登録商標)Full Prescribing Information
43.ALKERANR(登録商標)Tablets Prescribing Information;ALKERANR(登録商標)for Injection Prescribing Information
【技術分野】
【0001】
多発性骨髄腫の多剤併用療法。
【背景技術】
【0002】
多発性骨髄腫(MM)、即ち形質細胞新生物は、血液悪性腫瘍全体の約10%を構成する(1)。プロテアソーム阻害剤(PI)であるボルテゾミブのMMにおける臨床成果により、ユビキチン−プロテアソーム系(UPS)が薬物開発の有力な標的となることが証明された(2)。プロテアソームは、ミスフォールドし損傷したタンパク質ならびに細胞内シグナル伝達中間体の分解を担う、複数のサブユニットからなるタンパク質複合体である(3)。シグナル伝達経路の調節異常のため、新生物細胞はUPSに大きく依存し、従ってプロテアソーム阻害に対する感受性が特に高い(4)。プロテアソーム阻害後のMM細胞のアポトーシスは、生存促進性NF−κBシグナル伝達のダウンレギュレーション、血管新生の阻害、ミスフォールドしたタンパク質によるストレス応答の活性化、内因性および外因性細胞死経路の誘導、ならびに骨髄間質細胞へのMM細胞の接着の阻害を含む複数の機構によって起こる(5−8)。
【0003】
臨床開発における最初のPIであるボルテゾミブ(PS−341または[(1R)−3−メチル−1−({(2S)−3−フェニル−2−[(ピラジン−2−イルカルボニル)アミノ]プロパノイル}アミノ)ブチル]ボロン酸としても知られる)は、再発MMにおける2つの単剤第II相臨床試験の成功後、2003年にFDAによって認可された(9、10)。ボルテゾミブは他剤と併用した場合でも顕著な活性を示す。前臨床試験において、中毒量未満の(subtoxic)濃度のボルテゾミブは、メルファラン、ドキソルビシン、またはミトキサントロンを含む化学療法薬に対するMM細胞の耐性を克服した(11−13)。さらに、ボルテゾミブは、レナリドミド、三酸化二ヒ素、およびヒストン脱アセチル化酵素またはPKCの阻害剤、ならびに第2世代のPIを含む新規なMM治療薬の活性を相乗的に増強する(14−8)。ボルテゾミブをベースにする併用療法はin vitroで相乗作用が認められたが、それを試験する臨床試験においてin vivoでも高い有効性が認められた。ボルテゾミブ(V)を加えた場合または加えない場合のメルファランとプレドニゾンの併用(MP)を評価する第III相VISTA臨床試験において、MP療法では3年全生存率が59%であったのに対し、VMPでは3年全生存率が72%という結果が得られた(P=0.003)(19)。特に、投与計画にボルテゾミブを加えると、場合によっては、奏功しなかった治療薬に対して患者が再び感受性を示すことがある。例えば、第II相臨床試験において、メルファランで治療した後に再発したMM患者の60%は、その後、ボルテゾミブ/メルファラン併用療法に反応した(20)。同様に、ボルテゾミブをサリドマイドおよびデキサメタゾンと併用すると、再発MM患者集団(その73%はサリドマイドによる治療歴があった)において63%の全奏功率が得られた(21)。
【0004】
ボルテゾミブの認可によってMMの治療が変化したが、かなりの割合の患者がボルテゾミブ療法に反応しない。最近の研究結果から、プロテアソームの発現や活性レベルの差により、PIでの治療に対するMM腫瘍の感受性が異なり得ることが示唆された(22)。さらに、最初はボルテゾミブに反応した患者でも、ほぼ確実に再発が起こる。薬物耐性癌幹細胞の小集団が寛解後のMMの再発の原因となっている可能性があることを示唆する証拠が増加している(23−26)。これらの細胞は、正常な記憶B細胞の表面抗原特性を発現し、形質細胞マーカーCD138が欠損し、抗体を分泌しない(24)。さらに、一般的に使用されている抗骨髄腫薬(例えば、デキサメタゾン、レナリドミド、シクロホスファミド)で攻撃した場合、CD138陰性幹細胞集団は、残りの悪性細胞集団より高い薬物耐性を示す(24)。単剤のボルテゾミブは、例えば、多量の免疫グロブリンを産生するMM細胞に対して活性を有するが、CD138陰性MM細胞の増殖に対してはほとんど効果がない(24)。これらのデータから、癌幹細胞、ならびに腫瘍集団内の残りの悪性形質細胞サブタイプを標的とする新規なMM療法が必要とされていることが明らかである。
【0005】
新規で、より強力な、またはより忍容性の高いPIの探求の結果、化合物1([(1R)−1−[[(2S,3R)−3−ヒドロキシ−2−[6−フェニル−ピリジン−2−カルボニル)アミノ]−1−オキソブチル]アミノ]−3−メチルブチルボロン酸としても知られる;Bernardiniら、米国特許出願公開第2005/0107307号明細書)が合成された。ボルテゾミブと同様に、化合物1はペプチドボロン酸の部類の可逆性PIである(28)。静脈内(IV)ボーラス投与されるボルテゾミブとは対照的に、化合物1は、前臨床試験で経口製剤として活性を有している(28、29)。さらに、化合物1は、ボルテゾミブと比較して、in vitroの初代培養MM形質細胞とin vivoのRPMI8226MM異種移植片の両方で、同様のまたはより優れた単剤抗腫瘍活性を示す(29)。化合物1は、次の化学構造を有する:
【化1】
【0006】
多発性骨髄腫患者に最良の長期結果をもたらし得る治療選択肢が依然として必要とされている。とりわけ、再発または難治性疾患を有する患者に対する新規な療法が緊急に必要とされている。本明細書で研究が開示されるまで、化合物1とボルテゾミブまたはメルファランとの併用療法は研究されなかった。これらの併用療法は、再発または難治性疾患を有する患者を含む、MM患者に魅力的な治療選択肢を提供する。
【0007】
引用される参照文献は全て、参照により本明細書に援用される。
【発明の概要】
【課題を解決するための手段】
【0008】
化合物1で対象(subject)の多発性骨髄腫を治療する方法を提供する。一実施形態では、対象に化合物1とボルテゾミブを併用投与する。好ましくは、ボルテゾミブをプロドラッグとして投与する。好ましくは、ボルテゾミブを静脈内投与または経口投与する。
【0009】
好ましくは、ボルテゾミブを約0.5mg/m2〜約2mg/m2の範囲の用量で投与する。好ましくは、ボルテゾミブを約0.7mg/m2〜約1.3mg/m2の範囲の用量で投与する。
【0010】
好ましくは、ボルテゾミブを3〜7日毎に2〜4週間投与した後、ボルテゾミブを投与しない約7〜21日間の休薬期間を設ける、計画された投与サイクルに従ってボルテゾミブを投与する。好ましくは、ボルテゾミブを21日サイクルの1日目、4日目、8日目および11日目に投与する、計画された投与サイクルに従ってボルテゾミブを投与する。好ましくは、ボルテゾミブを28日サイクルの1日目、4日目、8日目および11日目に投与する、計画された投与サイクルに従ってボルテゾミブを投与する。好ましくは、計画されたサイクルを少なくとも1回繰り返す。
【0011】
別の実施形態では、対象に化合物1とメルファランを併用投与する。好ましくは、メルファランをプロドラッグとして投与する。好ましくは、メルファランを経口投与または静脈内投与する。
【0012】
好ましくは、メルファランを約0.025mg/kg〜約0.5mg/kgの範囲の用量で投与する。好ましくは、メルファランを約0.025mg/kg〜約0.3mg/kgの範囲の用量で投与する。
【0013】
好ましくは、メルファランを3〜7日毎に1〜2週間投与した後、メルファランを投与しない約4〜6週間の休薬期間を設ける、計画された投与サイクルに従ってメルファランを投与する。好ましくは、メルファランを1日1回、約4〜約7日間投与した後、約4〜6週間の休薬期間を設ける、計画された投与サイクルに従ってメルファランを投与する。好ましくは、メルファランを1日1回、約4〜約5日間投与した後、約4〜6週間の休薬期間を設ける、計画された投与サイクルに従ってメルファランを投与する。好ましくは、計画されたサイクルを少なくとも1回繰り返す。
【0014】
好ましくは、化合物1をプロドラッグとして投与する。好ましくは、化合物1のプロドラッグは、化合物1の薬学的に許容されるエステルの形態である。好ましくは、化合物1を静脈内投与または経口投与する。
【0015】
好ましくは、化合物1を約0.5mg/m2〜約5mg/m2の範囲の用量で投与する。好ましくは、化合物1を約1mg/m2〜約3mg/m2の範囲の用量で投与する。好ましくは、化合物1を3〜14日毎に2〜4週間投与した後、化合物1を投与しない約7〜21日間の休薬期間を設ける、計画された投与サイクルに従って化合物1を投与する。
【0016】
好ましくは、化合物1を21日サイクルの1日目、4日目、8日目および11日目に投与する、計画された投与サイクルに従って化合物1を投与する。好ましくは、化合物1を、28日サイクルの1日目、4日目、8日目および11日目に投与する、計画された投与サイクルに従って化合物1を投与する。好ましくは、化合物1を21日サイクルの1日目と15日目に投与する、計画された投与サイクルに従って化合物1を投与する。好ましくは、化合物1を28日サイクルの1日目と15日目に投与する、計画された投与サイクルに従って化合物1を投与する。好ましくは、計画されたサイクルを少なくとも1回繰り返す。
【0017】
好ましくは、化合物1を21日サイクルまたは28日サイクルの1日目、5日目および9日目に投与し、ボルテゾミブを21日サイクルまたは28日サイクルの3日目、8日目、および12日目に投与する。好ましくは、ボルテゾミブを21日サイクルまたは28日サイクルの1日目、5日目および9日目に投与し、化合物1を21日サイクルまたは28日サイクルの3日目、8日目、および12日目に投与する。
【図面の簡単な説明】
【0018】
【図1A】
【図1B】
【図1C】化合物1は、単剤の場合または他の抗MM治療薬と併用した場合、MM細胞株の生存率を低下させることを示す図である。A,MM1S(三角形)およびRPMI8226(正方形)を、表示した濃度の化合物1と共に48時間インキュベートした後、その生存率をMTSアッセイで評価した。B,MM1S細胞を溶媒対照(黒色の棒グラフ)、表示した濃度の化合物1(白色の棒グラフ)、ボルテゾミブ(縞模様の棒グラフ)、または両方の薬剤(線影の付いた棒グラフ)で48時間処置し、生存率をMTSアッセイで定量化した。C,RPMI8226細胞を溶媒対照(黒色の棒グラフ)、メルファラン(40μM)(白色の棒グラフ)、化合物1(表示した濃度)(縞模様の棒グラフ)、または両方の薬剤(線影の付いた棒グラフ)で48時間処置し、生存率をMTSアッセイで定量化した。グラフ化したデータは、6つの反復試験を使用した平均値±平均値の標準誤差(SEM)である。BおよびCでは、線影の付いた棒グラフの上に併用指数(CI)を示す。0.9未満のCI値は相乗作用を示し;0.9〜1.1のCI値は相加作用を示し、1.1超のCI値は拮抗作用を示す。
【図2A】
【図2B】
【図2C】
【図2D】化合物1は、単独の場合またはボルテゾミブと併用した場合、正常な末梢血単核細胞(PBMC)の生存率を低下させないことを示す図である。A,健常被験者から採取したPBMCを溶媒対照(黒色の棒グラフ)、化合物1単独(3.0nM)(白色の棒グラフ)、ボルテゾミブ単独(表示した濃度)(縞模様の棒グラフ)、または化合物1(3.0nM)+ボルテゾミブ(表示した濃度)(線影の付いた棒グラフ)と共に48時間インキュベートした後、細胞生存率をMTSアッセイで測定した。B,Aと同じ被験者から採取したPBMCを溶媒対照(黒色の棒グラフ)、ボルテゾミブ単独(3.0nM)(白色の棒グラフ)、化合物1単独(表示した濃度)(縞模様の棒グラフ)、または化合物1(表示した濃度)+ボルテゾミブ(3.0nM)(線影の付いた棒グラフ)と共にインキュベートした後、細胞生存率をMTSアッセイを使用して測定した。C,別の被験者から採取したPBMCを溶媒対照(黒色の棒グラフ)、化合物1単独(3.0nM)(白色の棒グラフ)、ボルテゾミブ単独(表示した濃度)(縞模様の棒グラフ)、または化合物1(3.0nM)+ボルテゾミブ(表示した濃度)(線影の付いた棒グラフ)と共に48時間インキュベートした後、細胞生存率をMTSアッセイで測定した。D,3人の健常被験者から採取したPBMCを様々な濃度の化合物1と共に48時間インキュベートした後、細胞生存率をMTSアッセイで測定した。各グラフ(A−D)は3つの独立した実験を表す。
【図3A】
【図3B】
【図3C】
【図3D】化合物1をボルテゾミブと併用すると、MM細胞のアポトーシスが誘導されることを示す図である。RPMI8226細胞を(A)溶媒対照、(B)化合物1(2.5nM)、(C)ボルテゾミブ(2.5nM)、または(D)化合物1(2.5nM)+ボルテゾミブ(2.5nM)と共に30時間インキュベートし、ヨウ化プロピジウム(PrI)およびアネキシンVに関して陽性の染色率を、フローサイトメトリー分析を使用して定量化した。初期アポトーシスの細胞は、PrI陰性、アネキシンV陽性である。
【図4A】
【図4B】
【図4C】化合物1がヒトMM腫瘍の増殖を阻害することを示す図である。A,B,14日目から、ヒトLAGκ−1A腫瘍を有するマウスに溶媒対照または表示された用量の化合物1をIV投与によりまたは強制経口投与により試験期間中週2回投与した。その後毎週、血清ヒト免疫グロブリン(IgG)濃度をELISAで評価し(A)、腫瘍体積をカリパスで測定した(B)。C,21日目から、ヒトLAGκ−1B腫瘍を有するマウスを試験期間中週2回、溶媒対照または表示した濃度の化合物1で処置し、腫瘍体積を毎週評価した。データは、1群当たりマウス7〜8匹の平均値±平均値の標準誤差として表す。
【図5A】
【図5B】
【図5C】
【図5D】
【図5E】
【図5F】化合物1をボルテゾミブまたはメルファランと併用すると、骨髄腫腫瘍の増殖が著しく阻害されることを示す図である。A,B,化合物1をボルテゾミブと併用すると、LAGκ−1A腫瘍の増殖が防止される。7日目から、LAGκ−1A腫瘍を有するマウスを週2回、溶媒対照、化合物1単独(1mg/kg)、ボルテゾミブ単独(0.5mg/kg)、または化合物1(1mg/kg IV)+ボルテゾミブ(0.5mg/kg IV)で処置した。その後毎週、血清ヒトIgG濃度(A)および腫瘍体積(B)を測定した。C,化合物1をボルテゾミブと併用すると、LAGκ−1B腫瘍体積の進行が100%遅延される。7日目から、LAGκ−1B腫瘍を有するマウスを週2回、溶媒対照、化合物1単独(1mg/kg)、ボルテゾミブ単独(0.5mg/kg)、または化合物1(1mg/kg IV)+ボルテゾミブ(0.5mg/kg IV)で処置した。その後毎週、腫瘍体積を測定した。D,E,化合物1をメルファランと併用すると、LAGκ−1A腫瘍の増殖が阻害される。7日目から、LAGκ−1A腫瘍を有するマウスを溶媒対照で週2回、化合物1単独で週2回(1mg/kg)、メルファラン単独で毎週(1mg/kg)、または週2回化合物1(1mg/kg IV)+毎週メルファラン(1mg/kg IP)で処置した。その後毎週、血清ヒトIgG濃度(D)および腫瘍体積(E)を測定した。F,化合物1をメルファランと併用すると、いずれかの薬剤を単独で使用した場合より、LAGκ−1B腫瘍体積の減少が大きかった。7日目から、LAGκ−1B腫瘍を有するマウスを溶媒対照で週2回、化合物1単独で週2回(1mg/kg)、メルファラン単独で毎週(3mg/kg)、または週2回化合物1(1mg/kg IV)+毎週メルファラン(3mg/kg IP)で処置した。その後毎週、腫瘍体積を測定した。A〜Fのデータは、1群当たりマウス7〜8匹の平均値±平均値の標準誤差として表す。
【図6A】
【図6B】
【図6C】
【図6D】
【図6E】
【図6F】
【図6G】
【図6H】化合物1およびボルテゾミブで処置したLAGκ−1B腫瘍が、アポトーシスマーカーであるアポトーシス誘導因子(AIF)の発現の増加を示すことを示す図である。LAGκ−1Bを有するマウスを溶媒対照(A)、化合物1(1mg/kg)単独(B)、ボルテゾミブ(0.5mg/kg)単独(C)、または両方の薬剤(D)で処置した後、そのマウスから切除した腫瘍を薄切し、AIFを測定するために染色した。E〜H,アイソタイプ対照でのA〜Dと同じ腫瘍から得られた染色された切片。スライドガラスは同時に染色した。
【図7】経口化合物1がヒトMM腫瘍の増殖を阻害することを示す図である。腫瘍体積の大きさに対する経口投与化合物1の効果。
【図8】経口化合物1がヒトMM腫瘍の増殖を阻害することを示す図である。IgG濃度に対する経口投与化合物1の効果。
【発明を実施するための形態】
【0019】
量および持続時間等の測定可能な値に言及する場合、本明細書で使用する「約」という用語は、特定の値から±10%の偏差を包含することを意味する。例えば、「約50%」という句は、50の±10%、即ち、45%〜55%を含む。
【0020】
本明細書で使用する場合、「対象」という用語は、温血動物、好ましくはヒトを含む哺乳動物を含む。好ましい実施形態では、対象は霊長類である。さらにより好ましい実施形態では、対象はヒトである。
【0021】
対象の多発性骨髄腫を治療する方法を提供する。一実施形態では、対象に化合物1とボルテゾミブを併用投与する。化合物1とボルテゾミブを併用する本発明の多発性骨髄腫の治療方法は、多発性骨髄腫の治療効果を相乗的に高めることが分かった。化合物1とボルテゾミブは両方とも、外因性および内因性アポトーシスシグナル伝達経路の活性化により細胞死を誘導する可逆的ボロン酸プロテアソーム阻害剤である(7、29)ため、これは意外である。さらに、薬剤は両方とも主に、プロテアソームのキモトリプシン様触媒活性を標的とし、カスパーゼ様活性をあまり阻害せず、トリプシン様活性をほとんど阻害しない(29、34)。従って、化合物1とボルテゾミブは、類似の作用機構を有するものと思われる。さらに、これらの化合物の化学構造は非常に類似している。
【0022】
ボルテゾミブ([(1R)−3−メチル−1−({(2S)−3−フェニル−2−[(ピラジン−2−イルカルボニル)アミノ]プロパノイル}アミノ)ブチル]ボロン酸;Millennium PharmaceuticalsによりVelcade(登録商標)の商標名で販売)は、次の化学構造を有する:
【化2】
【0023】
従って、化合物1とボルテゾミブが併用時に、in vitroでのMM細胞と、in vivoでの腫瘍、特に非分泌性腫瘍に対して高い活性を誘導する手段は不明である。
【0024】
別の実施形態では、対象に化合物1とメルファランを併用投与する。化合物1とメルファランを併用する本発明の多発性骨髄腫の治療方法は、多発性骨髄腫の治療効果を相乗的に高めることが分かった。
【0025】
メルファラン(4−[ビス(2−クロロエチル)アミノ]−L−フェニルアラニン;GlaxoSmithKlineによりAlkeran(登録商標)の商標名で販売)は、次の化学構造を有する:
【化3】
【0026】
本発明に使用される化合物1、ボルテゾミブおよび/またはメルファランは、親化合物の薬学的に許容される塩の形態および/または薬学的に許容されるエステルの形態などのプロドラッグを含む任意の適した化学的形態で投与されてもよい。好ましくは、親化合物の薬学的に許容される塩またはエステル誘導体は、投与後直ぐに、親化合物に変換される。本明細書で使用する場合、「薬学的に許容される塩」は、親化合物がその酸塩または塩基塩を生成することにより修飾されている親化合物の誘導体を指す。薬学的に許容される塩の例としては、アミンなどの塩基性基の鉱酸塩または有機酸塩;カルボン酸またはボロン酸などの酸性基のアルカリ塩または有機塩等が挙げられるが、これらに限定されるものではない。本明細書で使用する場合、「薬学的に許容されるエステル」は、酸基がそのエステルを生成することにより修飾されている親化合物の誘導体を指す。薬学的に許容されるエステルの例としては、例えば、ボロン酸エステル、即ち、ボロン酸化合物のエステル誘導体、および環状ボロン酸エステルが挙げられる。環状ボロン酸エステルの例としては、ピナンジオールボロン酸エステル、ピナコールボロン酸エステル、1,2−エタンジオールボロン酸エステル、1,3−プロパンジオールボロン酸エステル、1,2−プロパンジオールボロン酸エステル、2,3−ブタンジオールボロン酸エステル、1,1,2,2−テトラメチルエタンジオールボロン酸エステル、1,2−ジイソプロピルエタンジオールボロン酸エステル、5,6−デカンジオールボロン酸エステル、1,2−ジシクロヘキシルエタンジオールボロン酸エステル、ビシクロへキシル−1,1’−ジオール、および1,2−ジフェニル−1,2−エタンジオールボロン酸エステルが挙げられるが、これらに限定されるものではない。
【0027】
従って、特定の実施形態では、化合物1および/またはボルテゾミブは親化合物のボロン酸エステル誘導体として投与される。一実施形態では、化合物1は化合物1のボロン酸エステル誘導体として投与される。一実施形態では、ボルテゾミブは、ボルテゾミブのボロン酸エステル誘導体として投与される。
【0028】
任意の適した投与方法を使用してもよい。例としては、注射(皮下、静脈内、非経口、腹腔内、髄空内等)、経口投与、吸入、および経皮投与が挙げられる。注射により投与する場合、注射はボーラスであっても、または持続注入であってもよい。化合物1とボルテゾミブは対象に別々に(例えば、順次注射、注射と経口投与、または別々の経口投与として)投与されても、または混合物として一緒に(例えば、単回注射、または、化合物1とボルテゾミブを両方含有する単一錠剤の投与などによる単回経口投与で)投与されてもよい。同様に化合物1とメルファランも対象に別々に投与されても、または混合物として一緒に投与されてもよい。医薬組成物中の本発明の化合物の割合または濃度は、用量、化学的特性(例えば、疎水性)、および投与経路を含む多くの要因によって変わり得る。
【0029】
例えば、ボルテゾミブは経口投与または静脈内注射に適している。例えば、ボルテゾミブは、Millennium PharmaceuticalsからVelcade(登録商標)の商標名で、ボルテゾミブ3.5mgおよび増量剤であるマンニトール35mgを含有する使い捨てバイアルに入った滅菌凍結乾燥粉末として入手可能である。粉末は、臨床医により0.9%NaCl3.5mLで注射用に再構成される。ボルテゾミブは、Velcade凍結乾燥製剤中にマンニトールボロン酸エステルとして存在し、再構成後は、親ボロン酸と平衡状態にあるマンニトールボロン酸エステルとして存在する(42)。従って、一実施形態では、ボルテゾミブは静脈内(IV)注射により投与される。別の実施形態では、ボルテゾミブは経口投与、好ましくは錠剤またはカプセルで投与される。一実施形態では、ボルテゾミブは、ボロン酸エステルなどのプロドラッグの形態で注射により投与される。一実施形態では、ボルテゾミブは、ボロン酸エステルなどのプロドラッグの形態で経口投与される。
【0030】
例えば、メルファランは、経口投与または静脈内注射に適している。例えば、メルファランは、GlaxoSmithKlineからAlkeran(登録商標)の商標名で、経口投与用のフィルムコート錠または使い捨てバイアルに入った滅菌凍結乾燥粉末として入手可能である。フィルムコート錠は、メルファラン2mg、ならびに賦形剤であるコロイド状二酸化ケイ素、クロスポピドン、ヒプロメロース、マクロゴール/PEG400、ステアリン酸マグネシウム、微結晶セルロース、および二酸化チタンを含有する。凍結乾燥粉末は、メルファラン50mgと同等のメルファラン塩酸塩、およびポビドン20mgを含有する。粉末は、クエン酸ナトリウム0.2g、プロピレングリコール6.0mL、エタノール(96%)0.52mL、および注射用蒸留水を合計10mLとなるように含有する、提供される滅菌希釈剤のバイアルを使用して注射用に再構成される(43)。従って、一実施形態では、メルファランは、塩酸塩として静脈内(IV)注射により投与される。別の実施形態では、メルファランは経口投与、好ましくは錠剤またはカプセルで投与される。
【0031】
例えば、化合物1は、IV注射により、または、錠剤またはカプセルなどの経口剤形で投与するのに適している(28、29)。例えば、化合物1は、現在、固形腫瘍または非ホジキンリンパ腫を有する患者で最初のヒト第1相臨床試験で評価中である。第1相臨床試験では、化合物1は、化合物1、4mg、増量剤であるヒドロキシプロピル−β−シクロデキストリン196mg、および増量剤であるマンニトール156.8mgを含有する、使い捨てバイアルに入った滅菌凍結乾燥粉末として提供される。粉末は、注射前に、注射用滅菌水、0.9%NaClまたは5%マンニトールのいずれか5mLまたは10mL(目的の用量による)で再構成される。従って、一実施形態では、化合物1は静脈内(IV)注射により投与される。別の実施形態では、化合物1は経口投与、好ましくは錠剤またはカプセルで投与される。一実施形態では、化合物1は、ボロン酸エステルなどのプロドラッグの形態で注射により投与される。一実施形態では、化合物1は、ボロン酸エステルなどのプロドラッグの形態で経口投与される。
【0032】
化合物1とボルテゾミブ、または化合物1とメルファランは、好ましくは、多発性骨髄腫を治療するのに有効な量で、例えば、疾患の症状を予防、緩和、または寛解する、治療を受ける対象の生存期間を延長する、望ましくない細胞増殖を防止する、または対象の既存の良性細胞塊もしくは悪性腫瘍のサイズを減少させるのに有効な量で、併用投与される。本明細書に記載の詳細な開示内容および実施例に鑑みて、当業者は、併用する各薬剤の有効量をうまく決定することができる。有効量は、処置または阻害される細胞増殖のタイプ、対象のサイズ、癌細胞増殖または腫瘍の重症度、投与頻度(例えば、毎日か数日に1回か)、化合物の投与方式(manner of administration)、患者の健康状態および共存症の状態、処方する医師の判断および経験(例えば、同じまたは類似の薬物に関する)、投与方法(mode of administration)、投与される剤形のバイオアベイラビリティ特性、選択される投与計画、および併用療法の種類(例えば、他の化学療法剤)などの要因に応じて変わり得る。例えば、米国特許第5,427,916号明細書は、個々の患者における抗腫瘍療法の有効性を予測する方法を記載し、本発明の治療プロトコルと併用できる特定の方法を説明している。例えば、有効量は、in vitroまたは動物モデル試験系から得られる用量−反応曲線から外挿することができ、患者の表面積に基づいてもよい。
【0033】
治療は、化合物の最適用量より少ない比較的低用量で開始してもよい。その後、その状況における最適効果が達せられるまで、少しずつ漸増することにより用量を増加してもよい。必要に応じて全1日量を分割し、1日の間に少量ずつ投与してもよい。投与計画を最適化するため、抗癌治療を受けている患者から得られる2つ以上の時点における腫瘍測定値を比較することによって、多発性骨髄腫を治療するための化合物1とボルテゾミブの併用、または化合物1とメルファランの併用の有効性をモニターすることができる。一般に、療法を開始する前の患者の全身腫瘍組織量の初期評価と、治療中の異なる時点での1つ以上の追加の評価を得ることが好ましい。このような使用では、療法を開始する前に全身腫瘍組織量のベースラインを測定した後、療法を行っている間に癌の量の変化を測定する。或いは、治療前の全身腫瘍組織量のベースラインを測定する必要なく、2回以上連続測定を行ってもよい。このような使用では、全身腫瘍組織量の増減を測定するためのベースラインレベルとして、対象の全身腫瘍組織量の最初の評価を行わなければならない。
【0034】
化合物1とボルテゾミブ、または化合物1とメルファランの投与計画、例えば、投与のタイミングおよび/または順番は、各剤形の薬物動態、処置または阻害される細胞増殖のタイプ、対象のサイズ、癌細胞増殖または腫瘍の重症度、および有効量などの要因によって変わり得る。治療医師は、上記考慮事項および本発明の詳細な開示内容に鑑みて、有効性を最適化し副作用を最小限にするように、化合物1とボルテゾミブ、または化合物1とメルファランの投与のタイミングおよび/または順番を容易に変えることができる
【0035】
本発明の化合物1、ボルテゾミブ、およびメルファランの投与計画は柔軟性が高い。特定の実施形態では、これらの薬物に適していることが知られている投与計画を改良して投与計画を立ててもよい。例えば、表1に示すように、ボルテゾミブ(1.3mg/m2)を3〜5秒のボーラスIV注射として、経口メルファラン(9mg/m2)および経口プレドニゾン(60mg/m2)と併用投与する6週間の治療サイクルを9サイクル行うことにより、前治療歴のない多発性骨髄腫を治療することが認可されている。サイクル1〜4では、ボルテゾミブを週2回、1日目、4日目、8日目、11日目、22日目、25日目、29日目および32日目に投与する。サイクル5〜9では、ボルテゾミブを週1回、1日目、8日目、22日目および29日目に投与する(42)。
【0036】
【表1】
【0037】
治療計画中に薬物に関連する著しい毒性(例えば、血液毒性)が認められた場合、その後のボルテゾミブの用量をスキップするおよび/または減少することができる(例えば、1.3mg/m2から1mg/m2に減少する、および0.7mg/m2まで減少することが可能)。さらに、または代わりに、次のサイクルではメルファランの用量を25%減少させることができる(42)。
【0038】
別の例として、ボルテゾミブを3〜5秒のボーラスIV注射として3週間サイクルの1日目、4日目、8日目、および11日目に投与した後、10日間の休薬期間(12日目〜21日目)を設けることにより、再発または難治性多発性骨髄腫を治療することが認可されている。8サイクルを超える長期療法では、ボルテゾミブは、標準的な計画で投与されても、または週1回4週間(1日目、8日目、15日目および22日目)投与した後、13日間の休薬期間(23日目〜35日目)を設ける維持計画で投与されてもよい(42)。
【0039】
治療計画中に薬物に関連する著しい毒性(例えば、血液毒性、神経因性疼痛および/または末梢神経障害)が認められた場合、その後のボルテゾミブの用量をスキップするおよび/または減少することができる(例えば、1.3mg/m2から1mg/m2に減少する、および0.7mg/m2まで減少することが可能)(42)。
【0040】
本発明の併用療法では、ボルテゾミブの投与計画は、前述のものを含む認可された多発性骨髄腫の投与計画と類似していても、または異なっていてもよい。例えば、ボルテゾミブを認可された投与計画より高頻度または低頻度で投与してもよく、任意選択により、比較的高用量または低用量で投与してもよい。
【0041】
ボルテゾミブは、化合物1と任意の適した用量で併用投与されてもよい。適したボルテゾミブの用量は、約0.5mg/m2〜約7mg/m2の範囲、例えば、約0.5mg/m2〜約5mg/m2、例えば、約0.5mg/m2〜約3mg/m2の範囲であってもよい。適したボルテゾミブの用量は、典型的には約0.5mg/m2〜約2mg/m2の範囲となるであろう。好ましくは、ボルテゾミブの用量は約0.6mg/m2〜約1.5mg/m2の範囲である。より好ましくは、ボルテゾミブの用量は約0.7mg/m2〜約1.3mg/m2の範囲である。好ましいボルテゾミブの用量としては、0.7mg/m2、1mg/m2、または1.3mg/m2が挙げられるが、これらに限定されるものではない。前述の用量はどのボルテゾミブ投与方法にも適しており、とりわけ皮下投与または静脈内投与に適しており、静脈内投与が好ましい。ボルテゾミブの経口用量は、典型的には前述の範囲の上限(high end)にあり、例えば、約1mg/m2〜約5mg/m2、約1.5mg/m2〜約4mg/m2、または約2mg/m2〜約3mg/m2となるであろう。
【0042】
ボルテゾミブは、前述の用量で化合物1と任意の適した計画に従って併用投与されてもよい。ボルテゾミブの投与量は一定であっても、または投与計画内で変えてもよい。薬物に関連する著しい毒性が認められない限り、好ましくはボルテゾミブの用量は投与計画中、一定のレベルに維持されるが、薬物に関連する著しい毒性が認められた場合、その後の用量を例えば約20〜30%減少させることができる。ボルテゾミブは、化合物1と同じ日に投与されても、または異なる日に投与されてもよい。一実施形態では、ボルテゾミブと化合物1は、投与計画中、同じ日に投与される。適したボルテゾミブ投与計画は、典型的には1日1回投与〜週1回投与またはさらには月1回投与の範囲となるであろう。好ましくは、ボルテゾミブは1日1回より低頻度で、例えば、2〜14日に1回投与される。好ましくは、ボルテゾミブは3〜7日毎に、例えば、3〜4日毎に投与される。好ましくは、投与計画は、1週間以上、例えば、2、3または4週間ボルテゾミブで治療した後、ボルテゾミブを投与しない期間を少なくとも5日間、例えば、約7〜21日間含む。好ましくは、休薬期間は約10〜17日間、例えば、約10日間または約17日間である。例えば、ボルテゾミブを21日サイクルの1日目、4日目、8日目および11日目に投与してもよく、12〜21日目は休薬期間である。別の例として、ボルテゾミブを28日サイクルの1日目、4日目、8日目および11日目に投与してもよく、12〜28日目は休薬期間である。別の例として、ボルテゾミブを週1回4週間(例えば、35日サイクルの1日目、8日目、15日目および22日目に)投与した後、13日間の休薬期間(例えば、35日サイクルの23日目〜35日目)を設けてもよい。計画された投与サイクルを1回以上繰り返してもよい。例えば、最大反応が認められるまで、1または2サイクル追加して、計画されたサイクルを繰り返してもよい。別の例として、計画されたサイクルを6〜12サイクル繰り返してもよい。任意選択により、最初のサイクルを完了した後、ボルテゾミブを最初の計画より低頻度で、例えば、週1回または2週間に1回投与する「維持計画」を使用してもよい。維持計画を一定期間、一般的に1〜2年継続しても、または患者が進行の徴候を示さない状態が続き、著しい毒性がなく治療に耐える限り無期限に継続してもよい。
【0043】
化合物1は、任意の適した用量でボルテゾミブと併用投与されてもよい。適した化合物1の用量は、約0.5mg/m2〜約10mg/m2の範囲、例えば、約0.5mg/m2〜約5mg/m2、または約0.5mg/m2〜約3mg/m2の範囲であってもよい。適した化合物1の用量は、典型的には約0.5mg/m2〜約3mg/m2の範囲となるであろう。好ましくは、化合物1の用量は、約1mg/m2〜約3mg/m2の範囲である。より好ましくは、化合物1の用量は、約1.5mg/m2〜約2.5mg/m2の範囲である。好ましい化合物1の用量としては、1.5mg/m2、1.8mg/m2、2.1mg/m2、または2.4mg/m2が挙げられるが、これらに限定されるものではない。前述の用量はどの化合物1投与方法にも適しており、とりわけ皮下投与または静脈内投与に適しており、静脈内投与が好ましい。化合物1の経口用量は、典型的には前述の範囲の上限にあり、例えば、約1mg/m2〜約7mg/m2となるであろう。一実施形態では、化合物1の経口用量は、約2mg/m2〜約6mg/m2、例えば、約3mg/m2〜約5mg/m2である。例示的な化合物1の経口用量としては、2mg/m2、3mg/m2、4mg/m2、5mg/m2または6mg/m2が挙げられるが、これらに限定されるものではない。
【0044】
化合物1は、前述の用量でボルテゾミブと任意の適した計画に従って併用投与されてもよい。化合物1の投与量は一定であっても、または投与計画内で変えてもよい。薬物に関連する著しい毒性が認められない限り、好ましくは、化合物1の用量は投与計画中、一定のレベルに維持されるが、薬物に関連する著しい毒性が認められた場合、その後の用量を例えば約20〜30%減少させることができる。化合物1は、ボルテゾミブと同じ日に投与されても、または異なる日に投与されてもよい。一実施形態では、化合物1とボルテゾミブは、投与計画中、同じ日に投与される。適した化合物1の投与計画は、典型的には1日1回投与〜週1回投与またはさらには月1回投与の範囲となるであろう。好ましくは、化合物1は1日1回より低頻度で、例えば、2〜14日に1回投与される。好ましくは、化合物1は、3〜28日毎に、例えば、7〜21日毎に投与される。例えば、化合物1を週2回投与してもよい。別の例では、化合物1を週1回投与してもよい。別の例では、化合物1を2週間に1回投与してもよい。好ましくは、投与計画は、1週間以上、例えば、2、3または4週間、化合物1で治療した後、化合物1を投与しない期間を少なくとも5日間、例えば、約7〜21日間含む。好ましくは、休薬期間は約10〜17日間、例えば、約10日間または約17日間である。例えば、化合物1を21日サイクルの1日目、4日目、8日目および11日目に投与してもよく、12〜21日目は休薬期間である。別の実施形態では、化合物1を28日サイクルの1日目、4日目、8日目、および11日目に投与してもよく、12〜28日目は休薬期間である。別の実施形態では、化合物1を21日サイクルの1日目と8日目に投与してもよく、12〜21日目は休薬期間である。別の実施形態では、化合物1を28日サイクルの1日目と8日目に投与してもよく、12〜28日目は休薬期間である。別の実施形態では、化合物1を21日サイクルの1日目と15日目に投与してもよい。別の実施形態では、化合物1を28日サイクルの1日目と15日目に投与してもよい。前述のように、ボルテゾミブは投与計画の同じ日に投与されても、または異なる日に投与されてもよい。例えば、化合物1とボルテゾミブを両方とも21日サイクルの1日目、4日目、8日目および11日目に投与してもよい。別の実施形態では、化合物1とボルテゾミブを両方とも、28日サイクルの1日目、4日目、8日目、および11日目に投与してもよい。別の実施形態では、ボルテゾミブを21日サイクルの1日目、4日目、8日目および11日目に投与し、化合物1を21日サイクルの1日目と8日目に投与してもよい。別の実施形態では、ボルテゾミブを28日サイクルの1日目、4日目、8日目および11日目に投与し、化合物1を28日サイクルの1日目と8日目に投与してもよい。別の実施形態では、ボルテゾミブを21日サイクルの1日目、4日目、8日目、および11日目に投与し、化合物1を21日サイクルの1日目と15日目に投与してもよい。別の実施形態では、ボルテゾミブを28日サイクルの1日目、4日目、8日目、および11日目に投与し、化合物1を28日サイクルの1日目と15日目に投与してもよい。別の実施形態では、化合物1を21日サイクルまたは28日サイクルの1日目、4日目、8日目および11日目に投与し、ボルテゾミブを21日サイクルまたは28日サイクルの2日目、5日目、9日目および12日目に投与してもよい。別の実施形態では、ボルテゾミブを21日サイクルまたは28日サイクルの1日目、4日目、8日目および11日目に投与し、化合物1を21日サイクルまたは28日サイクルの2日目、5日目、9日目および12日目に投与してもよい。別の実施形態では、化合物1を21日サイクルまたは28日サイクルの1日目と8日目投与し、ボルテゾミブを21日サイクルまたは28日サイクルの2日目、5日目、9日目および12日目に投与してもよい。別の実施形態では、ボルテゾミブを21日サイクルまたは28日サイクルの1日目、4日目、8日目および11日目に投与し、化合物1を21日サイクルまたは28日サイクルの2日目と9日目に投与してもよい。別の実施形態では、化合物1を21日サイクルまたは28日サイクルの1日目と15日目投与し、ボルテゾミブを21日サイクルまたは28日サイクルの2日目、5日目、9日目、および12日目に投与してもよい。別の実施形態では、ボルテゾミブを21日サイクルまたは28日サイクルの1日目、4日目、8日目、および11日目に投与し、化合物1を21日サイクルまたは28日サイクルの2日目と16日目に投与してもよい。別の実施形態では、化合物1を21日サイクルまたは28日サイクルの1日目と8日目投与し、ボルテゾミブを21日サイクルまたは28日サイクルの4日目と11日目に投与してもよい。別の実施形態では、ボルテゾミブを21日サイクルまたは28日サイクルの1日目と8日目に投与し、化合物1を21日サイクルまたは28日サイクルの4日目と11日目に投与してもよい。別の実施形態では、化合物1を21日サイクルまたは28日サイクルの1日目、5日目および9日目に投与し、ボルテゾミブを21日サイクルまたは28日サイクルの3日目、8日目、および12日目に投与してもよい。別の実施形態では、ボルテゾミブを21日サイクルまたは28日サイクルの1日目、5日目および9日目に投与し、化合物1を21日サイクルまたは28日サイクルの3日目、8日目、および12日目に投与してもよい。別の実施形態では、化合物1を21日サイクルまたは28日サイクルの1日目と15日目に投与し、ボルテゾミブを21日サイクルまたは28日サイクルの1日目、6日目および11日目に投与してもよい。別の実施形態では、ボルテゾミブを21日サイクルまたは28日サイクルの1日目と11日目に投与し、化合物1を21日サイクルまたは28日サイクルの5日目と15日目に投与してもよい。計画された投与サイクルを1回以上繰り返してもよい。例えば、最大反応が認められるまで、1または2サイクル追加して、計画されたサイクルを繰り返してもよい。別の例として、計画されたサイクルを6〜12サイクル繰り返してもよい。任意選択により、最初のサイクルを完了した後、ボルテゾミブと化合物1を最初の計画より低頻度で、例えば、週1回、2週間に1回、3週間に1回、または4週間に1回投与する「維持計画」を使用してもよい。維持計画を一定期間、一般的に1〜2年継続しても、または、患者が進行の徴候を示さない状態が続き、著しい毒性がなく治療に耐える限り無期限に継続してもよい。
【0045】
前述のように、本発明の化合物1とメルファランの投与計画は柔軟性が高い。特定の実施形態では、これらの薬物に適していることが知られている投与計画を改良して投与計画を立てることができる。例えば、上記表1に示すように、経口メルファラン(9mg/m2)を、ボルテゾミブ(1.3mg/m2)および経口プレドニゾン(60mg/m2)と併用投与する6週間の治療サイクルを9サイクル行うことにより、前治療歴のない多発性骨髄腫を治療することが認可されている。メルファランは各6週間のサイクルの1日目、2日目、3日目、および4日目に投与される(42)。
【0046】
経口メルファランは、通常、単剤として毎日6mgの用量で投与される。約1週間の間隔で行われる血球算定に基づき、必要に応じて用量を調節する。2〜3週間治療した後、最大4週間投薬を中止し、その間、血球算定を慎重に追跡しなければならない。白血球数と血小板数が上昇している場合、毎日2mgの維持量を定めてもよい(43)。
【0047】
本発明の併用療法では、メルファランの投与計画は、前述のものを含む認可された多発性骨髄腫の投与計画と類似していても、または異なってもよい。例えば、メルファランを認可された投与計画より高頻度または低頻度で投与してもよく、任意選択により、比較的高用量または低用量で投与してもよい。
【0048】
メルファランは、化合物1と任意の適した用量で併用投与されてもよい。適したメルファランの用量は、約0.025mg/kg〜約0.5mg/kgの範囲、例えば、約0.05mg/kg〜約0.3mg/kgの範囲であってもよい。適したメルファランの用量は、典型的には約0.025mg/kg〜約0.3mg/kgの範囲となるであろう。好ましくは、メルファランの用量は、約0.05mg/kg〜約0.25mg/kgの範囲である。より好ましくは、メルファランの用量は、約0.1mg/kg〜約0.2mg/kgの範囲である。好ましいメルファランの用量としては、0.1mg/kg、0.15mg/kg、0.2mg/kg、または0.25mg/kgが挙げられるが、これらに限定されるものではない。前述の用量は、どのメルファラン投与方法にも適しており、とりわけ皮下投与、静脈内投与または経口投与に適しており、経口投与が好ましい。
【0049】
メルファランは、前述の用量で化合物1と任意の適した計画に従って併用投与されてもよい。メルファランの投与量は一定であっても、または投与計画内で変えてもよい。薬物に関連する著しい毒性が認められない限り、好ましくは、メルファランの用量は投与計画中、一定のレベルに維持されるが、薬物に関連する著しい毒性が見られた場合、その後の用量を例えば約20〜30%減少させることができる。メルファランは、化合物1と同じ日に投与されても、または異なる日に投与されてもよい。適したメルファランの投与計画では、期間中、連日投与した後、休薬期間が設けられるであろう。好ましくは、メルファランを約3〜約7日間、1日1回投与した後、約1〜6週間の休薬期間を設ける。好ましくは、メルファランを約4〜約7日間、1日1回投与した後、約4〜6週間の休薬期間を設ける。好ましくは、メルファランを約4〜約5日間、1日1回投与した後、約4〜6週間の休薬期間を設ける。計画を1回以上繰り返してもよい。
【0050】
化合物1は、メルファランと任意の適した用量で併用投与されてもよい。適した化合物1の用量は、約0.5mg/m2〜約10mg/m2の範囲、例えば、約0.5mg/m2〜約5mg/m2、または約0.5mg/m2〜約3mg/m2の範囲であってもよい。適した化合物1の用量は、典型的には約0.5mg/m2〜約3mg/m2の範囲となるであろう。好ましくは、化合物1の用量は、約1mg/m2〜約3mg/m2の範囲である。より好ましくは、化合物1の用量は、約1.5mg/m2〜約2.5mg/m2の範囲である。好ましい化合物1の用量としては、1.5mg/m2、1.8mg/m2、2.1mg/m2、または2.4mg/m2が挙げられるが、これらに限定されるものではない。前述の用量は、どの化合物1投与方法にも適しており、とりわけ皮下投与または静脈内投与に適しており、静脈内投与が好ましい。化合物1の経口用量は、典型的には前述の範囲の上限にあり、例えば、約1mg/m2〜約7mg/m2となるであろう。一実施形態では、化合物1の経口用量は、約2mg/m2〜約6mg/m2、例えば、約3mg/m2〜約5mg/m2である。例示的な化合物1の経口用量としては、2mg/m2、3mg/m2、4mg/m2、5mg/m2または約6mg/m2が挙げられるが、これらに限定されるものではない。
【0051】
化合物1は、前述の用量でメルファランと任意の適した計画に従って併用投与されてもよい。化合物1の投与量は一定であっても、または投与計画内で変えてもよい。薬物に関連する著しい毒性が認められない限り、好ましくは、化合物1の用量は投与計画中、一定のレベルに維持されるが、薬物に関連する著しい毒性が見られた場合、その後の用量を例えば約20〜30%減少させることができる。化合物1は、メルファランと同じ日に投与されても、または異なる日に投与されてもよい。適した化合物1の投与計画は、典型的には1日1回投与〜週1回投与またはさらには月1回投与の範囲となるであろう。好ましくは、化合物1を1日1回より低頻度で、例えば、2〜14日に1回投与する。好ましくは、化合物1を3〜28日毎に、例えば、7〜21日毎に投与する。例えば、化合物1を週2回投与してもよい。別の例では、化合物1を週1回投与してもよい。別の例では、化合物1を2週間に1回投与してもよい。好ましくは、投与計画は、化合物1を投与しない期間を少なくとも5日間、例えば、約7〜21日間含む。好ましくは、休薬期間は約10〜17日間、例えば、約10日間または約17日間である。例えば、化合物1を21日サイクルの1日目、4日目、8日目および11日目に投与してもよく、12〜21日目は休薬期間である。別の実施形態では、化合物1を28日サイクルの1日目、4日目、8日目、および11日目に投与してもよく、12〜28日目は休薬期間である。別の実施形態では、化合物1を21日サイクルの1日目と8日目に投与してもよく、12〜21日目は休薬期間である。別の実施形態では、化合物1を28日サイクルの1日目と8日目に投与してもよく、12〜28日目は休薬期間である。別の実施形態では、化合物1を21日サイクルの1日目と15日目に投与してもよい。別の実施形態では、化合物1を28日サイクルの1日目と15日目に投与してもよい。一実施形態では、化合物1を42日サイクルの1日目、4日目、8日目、11日目、22日目、25日目、29日目および32日目に投与し、メルファランを42日サイクルの1日目、2日目、3日目および4日目に投与してもよい。別の実施形態では、化合物1を84日サイクルの1日目、4日目、8日目、11日目、22日目、25日目、29日目、32日目、43日目、50日目、64日目、および71日目に投与し、メルファランを84日サイクルの1日目、2日目、3日目、4日目、43日目、44日目、45日目、および46日目に投与してもよい。別の実施形態では、メルファランを28日サイクルの1日目、2日目、3日目、および4日目に投与し、化合物1を28日サイクルの1日目と15日目に投与してもよい。別の実施形態では、メルファランを42日サイクルの1日目、2日目、3日目、4日目、および5日目に投与し、化合物1を42日サイクルの1日目、8日目、22日目および29日目に投与してもよい。計画を1回以上繰り返してもよい。
【0052】
他の1種類以上の癌治療薬を、化合物1とボルテゾミブ、または化合物1とメルファランの投与と併用してもよい。このような治療薬としては、ボルテゾミブ、メルファラン、デキサメタゾンおよび他のステロイド、ドキソルビシン、シクロホスファミド、サリドマイド、レナリドミド、三酸化ヒ素、およびヒストン脱アセチル化酵素阻害剤を含む抗癌剤が挙げられるが、これらに限定されるものではない。これらの薬剤の適切な用量は当該技術分野で周知である。本発明の別の態様では、追加的な薬剤はフィルグラスチムなどの顆粒球コロニー形成刺激因子(G−CSF)であってもよい。好ましい実施形態では、フィルグラスチムを6日目から、好中球数がANC>1000に回復するまで約5μg/kg/日の用量でSC投与する。ANCは、「好中球絶対数」の略語である。
【0053】
本発明の併用療法は、癌性増殖の一部または全部を外科的に除去する試みをさらに含む治療過程の一部として使用されてもよい。例えば、対象の外科的治療後に、残存する新生物細胞または転移細胞を治療するため、併用療法を施してもよい。また、腫瘍のサイズを小さくし、切除する組織の量を減少させるために手術の前に治療を行い、それによって手術の侵襲性と外傷性を低くしてもよい。
【0054】
本明細書に開示される対象である併用療法による多発性骨髄腫の治療は、DNA損傷を誘導する放射線治療手段を用いた1つ以上の治療過程を含んでもよい。放射線治療手段としては、例えば、γ線照射、X線、紫外線照射、マイクロ波、電子放出、および放射性同位体等が挙げられる。局在する腫瘍部位に前述の形態の放射線を照射することにより療法を達成することができる。
【0055】
本発明の別の態様は、骨髄細胞を本発明の併用療法で処置することにより、骨髄をパージする、即ち、骨髄から癌細胞を除去する方法に関する。その後、パージした骨髄を、骨髄を取り出した対象に再移植しても、または異なる対象に移植してもよい。
【0056】
材料および方法
試薬
化合物1(4mg;Cephalon,Frazer,PA)をプロピレングリコール(800μL)に溶解し、5%マンニトールに添加して、最終保存液濃度1mg/mLにし;処置直前に化合物1の保存溶液を表示した濃度に希釈した。ボルテゾミブ(Millennium Pharmaceuticals,Cambridge,MA)を1mg/mLで得、0.9%塩化ナトリウムを使用して規定どおり希釈した。メルファラン(Sigma,St.Louis,MO)を酸−EtOH(酸−EtOH:濃HCl47μL+100%EtOH1mL)100μLに溶解し、リン酸緩衝生理食塩水で1mLに希釈した。製剤は毎週調製した。
【0057】
細胞株および初代培養細胞
アメリカ培養細胞系統保存機関(American Type Culture Collection)(Rockville,MD)からヒト骨髄腫細胞株RPMI8226を得た。MM1S骨髄腫細胞株は、Steven Rosen博士(Northwestern University,Chicago,IL)によって提供された。正常な末梢血単核細胞(PBMC)をHistopaque(登録商標)密度勾配遠心分離法で、製造業者のプロトコル(Sigma−Aldrich,St.Louis,MO)に従って分離した。骨髄腫細胞株およびPBMCを、5%二酸化炭素(CO2)雰囲気中37℃で、10%牛胎児血清、2mM L−グルタミン、100IU/mLペニシリン、100μg/mLストレプトマイシン、および必須アミノ酸を添加したRPMI 1640(Omega Scientific,Tarzana,CA)中で培養した。
【0058】
細胞生存率アッセイ(MTSアッセイ)
細胞を105細胞/100μL/ウェルで96穴プレートに播種し、24時間インキュベートした。RPMI8226細胞およびMM1S細胞を溶媒、化合物1、ボルテゾミブ、メルファラン、化合物1+ボルテゾミブ、または化合物1+メルファランの存在下で48時間培養した。インキュベート時間後に、CellTiter 96(登録商標)AQueous Non−Radioactive Cell Proliferation Assay(Promega,Madison,WI)を使用して細胞生存率を定量化した。各ウェルをMTS(3−(4, 5−ジメチルチアゾール−2−イル)−5−(3−カルボキシメトキシフェニル)−2−(4−スルホフェニル)−2H−テトラゾリウム、分子内塩)で1〜4時間処理した後、490nmで吸光度を記録した。490nmにおける吸光度で測定されるホルマザン生成物の量は、培地内の生細胞数に正比例する。
【0059】
ChouおよびTalalayの半有効(median effect)法を使用して、化合物1とボルテゾミブまたはメルファランとのin vitro相乗作用を評価した(30)。各組み合わせについて別々に併用指数(CI)を算出した。薬物相互作用は、CIが0.9未満の場合は相乗的であると判定され、またはCIが1.1より大きい場合は拮抗的であると判定された。CIが0.9〜1.1の場合は薬物相加効果を示すものと見なされた(31)。
【0060】
アネキシンVおよびヨウ化プロピジウム染色によるアポトーシスアッセイ
薬物療法に反応したアポトーシスを定量化するため、RPMI8226細胞(1ウェル当たり5×105細胞)を溶媒またはPIと共に37℃、5%CO2で30時間インキュベートした。陽性対照として、細胞をアクチノマイシンD250ng/mLと共に24時間または48時間インキュベートした。次いで、細胞を2回リン酸緩衝生理食塩水で洗浄し、結合緩衝液(1.4M NaClおよび25mM CaCl2を含有する、100mM HEPES/NaOH、pH7.5)に再懸濁させ、製造業者のプロトコル(BioVision,Mountain View,CA)に従って、フルオレッセインイソチオシアネート(FITC)結合アネキシンV、および蛍光色素ヨウ化プロピジウム(PrI)で染色した。各薬物療法について、ゲートをかけた1×105の事象を記録した。PI染色とアネキシンV染色の両方について陰性の細胞を生存と見なし;アネキシンV−陽性、PrI−陰性の細胞を初期アポトーシスと見なし;アネキシンV−陽性、PrI−陽性の細胞を後期アポトーシスと見なした。Cytomics CXPソフトウェアを用いたBeckman Coulter FC500サイトメーター(Beckman Coulter,Fullerton,CA)を使用してフローサイトメトリー分析を行った。
【0061】
SCIDマウス
Jackson Laboratory(Bar Harbor,ME)から6〜8週齢の雄性SCIDマウスを得、動物資源施設の特定病原菌を排除した領域で飼育した。動物試験は全て、動物実験委員会(Institutional Animal Care and Use Committee)により認可されたプロトコルに従って行った。手術前にケタミン、キシラジンおよびイソフルランで動物に麻酔をかけ、腫瘍が直径2cmに達したとき安楽死させた。
【0062】
筋肉内腫瘍異種移植モデル
LAGκ−1A腫瘍(ボルテゾミブおよびメルファランに対して感受性)を形成するため、レナリドミドで治療中に進行した女性MM患者から骨髄生検試料を得た。この生検の直後に、患者はメルファランとボルテゾミブを併用して治療され、反応を示した。麻酔したSCIDマウスの後肢に生検組織を外科的に移植し、継代した(32)。LAGκ−1Aと同じ患者に由来するLAGκ−1B腫瘍(ボルテゾミブおよびメルファランに対して耐性)は、患者がボルテゾミブおよびメルファランで治療中に進行しているときに採取した生検試料から得られた(32)。
【0063】
麻酔したドナーマウスから骨髄腫腫瘍(LAGκ−1AまたはLAGκ−1B)を切除し、20〜40mm3片に薄切し、麻酔した未処置SCIDマウスの左浅臀筋に外科的に移植した。免疫活性をさらに抑制するため、レシピエントマウスに毎週、抗アシアロGM1ウサギ血清(Wako Bioproducts,Richmond,VA)を注射した。マウスを実験群の1つに無作為に割り付け、腫瘍移植7〜21日後に処置を開始した。化合物1は、規定通り、週2回(水、金)、静脈内注射(0.5〜3.0mg/kg)または強制経口投与(10mg/kg)により投与した。メルファラン(1mg/kg)は毎週(水)、腹腔内(IP)注射により投与した。ボルテゾミブ(0.5mg/kg)は週2回(火、木)IV注射により投薬した。対照処置は、化合物1の希釈剤(5%マンニトール3.2mLおよびプロピレングリコール800μL)のみからなった。
【0064】
ヒト免疫グロブリンG(hIgG)酵素結合免疫吸着検定法(ELISA)
LAGκ−1A腫瘍により分泌されたhIgGの血清中濃度(LAGκ−1B腫瘍は異常タンパク質を分泌しない)をELISAにより腫瘍増殖のタンパク質マーカーとして定量化した。毎週、MM腫瘍を有するマウスの後眼窩静脈叢から採血した。得られた試料を13,000rpmで30分間遠心分離して、血清を得た。製造業者の仕様書に従って、hIgG ELISAキット(Bethyl Laboratories,Montgomery,TX)を使用した。KC Juniorソフトウェアを用いたμQuantマイクロプレート分光光度計(Bio−Tek Instruments,Winooski,VT)で、450nmにおける吸光度(参照波長550mm)を測定した。グラフ化したデータは、n=マウス7〜8匹/群での平均値±SEMである。
【0065】
腫瘍体積の測定
腫瘍増殖の直接測定として、毎週、カリパスを使用して腫瘍体積を計測し、楕円体の体積に関する式を適用した(4/3π×[幅/2]2×[長さ/2])。
【0066】
腫瘍細胞におけるアポトーシス誘導因子(AIF)発現の免疫組織化学分析
LAGκ−1B腫瘍を4%パラホルムアルデヒド中に固定し、5μmの切片に切断した。 簡潔に言えば、0.05%Tween−20(TBST)および3%BSAを有するトリス緩衝生理食塩水で、室温で1時間切片をブロッキングした後、AIFに対するウサギ抗体(Sigma,St.Louis,MO)と共に終夜インキュベートした。切片をTBSTで3回洗浄し、西洋ワサビペルオキシダーゼ(ARH)結合抗ウサギ抗体(KPL,Gaithersburg,MD)をTBSTで1:500に希釈したもので、室温で2時間処理した。スライドガラスはTBST中で3回洗浄し、3−アミノ−9−エチルカルバゾール(AEC)緩衝液に5分間入れ、AECキット(Dako,Glostrup,Denmark)を使用して色を検出した。染色はOlympus BX51顕微鏡(Olympus Imaging America Inc.,Center Valley,PA)を使用して記録し、Microsuite Biological Suiteプログラム (Olympus BX51)で分析した。
【0067】
統計解析
腫瘍増殖およびhIgG濃度を処置群の平均値および標準誤差に関して解析した。Studentのt検定を使用して、処置群間の統計学的有意差を検定した。最小有意水準は、P<0.05であった。
【実施例】
【0068】
次の実施例に示すデータから、本発明の併用療法は、化合物1とボルテゾミブまたは化合物1とメルファランを低用量で併用した場合、標準用量の単剤療法と比較して、MMにおける有効性が同様であるかまたは高い可能性があることが示唆された。このようにして、ボルテゾミブに関する末梢神経障害およびメルファランに関する骨髄抑制などの薬物に関連する毒性が低減し得るか、または回避され得る(40、41)。本明細書に記載の実験では、併用療法で処置したマウスは処置に対する忍容性が高く、腫瘍の進行がほとんどまたは全くなかった。
【0069】
実施例1.化合物1はMM細胞に対する細胞毒性があり、in vitroで抗MM剤と併用した場合、相乗作用を示す
RPMI8226細胞およびMM1S細胞を増加する濃度(0.1〜10nM)の化合物1の存在下で培養した。48時間後、MTSアッセイで細胞生存率を評価した。化合物1は、両方の細胞株で濃度依存的な生存率の低下を誘導した(図1A)。細胞を化合物1で24時間または72時間処置した場合、結果は同様であった(データ不示)。
【0070】
次に、化合物1+ボルテゾミブ(PI)またはメルファラン(化学療法剤)の存在下における細胞生存率を調べた。まず、MM1S細胞を一定濃度の化合物1(1.75nM)および増加する濃度(0.5〜2.5nM)のボルテゾミブと共に48時間インキュベートした。1.5nM以下のボルテゾミブ濃度では、化合物1の細胞毒性効果が増強した。例えば、単剤の場合、化合物1(1.75nM)と最低濃度(0.5nM)のボルテゾミブはそれぞれ細胞生存率を約16%低下させた。しかし、化合物1(1.75nM)をボルテゾミブ(0.5nM)と併用した場合、細胞生存率は約43%低下した(図1B)。Chou−Talalayの式を使用して、この組み合わせの相乗作用を確認した(CI、0.74〜0.85)(30、31)。RPMI8226細胞で実験を繰り返した場合、同様の結果が得られた(データ不示)。
【0071】
RPMI8226細胞をメルファラン(40μM)および6.0nM以上の濃度の化合物1(IC50=8.5nM)と共にインキュベートした場合、相乗的な生存率の低下が認められた(CI、0.78〜0.87)。例えば、細胞生存率は単剤のメルファラン(40μM)の存在下では約30%低下し、単剤の化合物1(9.0nM)の存在下では64%低下した。両方の薬物を同時に使用した場合、細胞生存率は90%低下した(図1C)。総合的に、これらの結果から、化合物1をボルテゾミブまたはメルファランと併用すると、MM細胞の生存率を相乗的に抑制できることが分かる。
【0072】
実施例2.化合物1とボルテゾミブは新生物細胞に対して選択的な細胞毒性を有する
2種類以上のPIを用いた療法がin vivoで実施可能であるためには、この組み合わせが非新生物細胞に損傷を与えてはならない。従って、正常なPBMCの生存率に対する化合物1+ボルテゾミブの効果を試験した。健常なドナーのPBMCを化合物1単独、ボルテゾミブ単独、または両方の薬剤の存在下で48時間培養し、細胞生存率をMTSアッセイで定量化した。どちらかのPIをそのIC50付近で用いたMM細胞における単独療法では、PBMCの生存率はあまり低下しなかった(PBMCを9nMの化合物1と9nMのボルテゾミブでそれぞれ処置した場合、生細胞は約75%および85%であった)(図2A、B)。両方のPIと共に共インキュベートした場合、どちらかの薬剤を単独で投与した場合と比較して、細胞生存率はそれ以上低下しなかったか、または僅かに上昇した(PIを両方とも投与した場合、試験した全ての濃度で細胞生存率が3%〜23%低下した)(図2A、B)。別の健常なドナーに由来するPBMCで類似の結果が得られた(図2C)。PBMCが高濃度のPIによって損傷を受けるかどうかを調べるため、PBMCを120nMまでの化合物1と共に培養した。対照と比較した場合、試験したどの濃度でも細胞生存率の顕著な差は認められなかった(図2D)。PBMCを120nMまでの濃度のボルテゾミブと共にインキュベートした場合、同様の結果が得られた(データ不示)。従って、本発明の併用療法はMMに対する有効性が高く、正常な細胞に対する毒性が増加しない。
【0073】
実施例3.化合物1とボルテゾミブの併用によりMM細胞のアポトーシスが誘導される
化合物1とボルテゾミブでMM細胞を処置した後に認められる細胞生存率の低下がアポトーシスによるものかどうかを調べるため、RPMI8226細胞を両方の薬剤(どちらの薬物も2.5nM)と共に30時間インキュベートし、生存率測定用色素であるPrIおよびアポトーシスマーカーであるアネキシンVで染色された細胞の割合を測定した。初期アポトーシス(PrI−/アネキシンV+)の細胞の割合は、どちらかの薬剤単独で処置した後(それぞれ2.5nMの化合物1または2.5nMのボルテゾミブで処置した細胞の10.4%および17.5%)より、両方のPIで処置した後の方が大きかった(細胞の38.9%)(図3)。後期アポトーシス(PrI+/アネキシンV+)または壊死(PrI+/アネキシンV−)の細胞の割合は、この時点では処置群間で差がなかった。
【0074】
実施例4.単剤の化合物1はin vivoでヒトMM腫瘍の増殖を阻害した
化合物1は、単剤の場合も併用した場合もin vitroで強力な抗MM効果を示すため、次に一連のin vivo試験を行った。これらの実験では、LAGκ−1A(ボルテゾミブ感受性およびメルファラン感受性)腫瘍およびLAGκ−1B(ボルテゾミブ耐性およびメルファラン耐性)腫瘍を有するマウスを使用したが、これらの腫瘍は両方とも元々MM患者の骨髄生検試料に由来した。これらの腫瘍はヒトのMMによく似ており、マウスで継代され、一貫した増殖パターンおよび化学療法抵抗性パターンを有した。腫瘍組織を筋肉内移植した後、化合物1を0.1〜3mg/kgの範囲の様々な用量でIV投与することにより、または10mg/kg経口投与することによりマウスを週2回処置した。対照群のマウスには化合物1の希釈剤を投与した。
【0075】
単剤の化合物1をIV投与すると、LAGκ−1A腫瘍からの異常タンパク質の分泌が用量依存的に減少した。比較的低用量の化合物1では腫瘍hIgG分泌が減少し、比較的高用量では血清hIgG濃度が本質的に検出不可能となった(薬物療法の28日目に対照と比較した場合、IV化合物1、1mg/kgではP=0.0001、3mg/kgではP=0.0002)(図4A)。ボルテゾミブと異なり、化合物1は経口製剤としても活性を示す(28、29)。経口化合物1で処置して2週間内に、血清hIgG濃度は、対照で処置した動物より著しく低下した(P=0.0007)。経口化合物1で処置して28日までに、血清hIgG濃度はごく僅かになった(対照で処置した動物と比較した場合、P=0.0001)(図4A)。
【0076】
異常タンパク質濃度に対する効果に加え、単剤の化合物1は、溶媒で処置したマウスと比較して、LAGκ−1A腫瘍体積の増加を遅延させた。4週間薬物療法を行った後、化合物1を1mg/kgまたは3mg/kg、IV投与したものは、対照で処置した異種移植片と比較して同時点で腫瘍体積が約15倍減少した(対照と比較した場合、各用量でP=0.0001)(図4B)。化合物1を経口送達すると、腫瘍増殖も阻害された。経口化合物1で処置してわずか14日後に、対照で処置した腫瘍と比較して、腫瘍体積の著しい減少が認められ(P=0.0002)、試験期間中ずっとその差があった(図4B)。
【0077】
また、腫瘍体積に対する化合物1の効果を、ボルテゾミブ耐性非分泌性LAGκ−1B腫瘍を有するマウスでも試験した(図4C)。LAGκ−1A腫瘍の場合と同様に、化合物1は、IV注射と経口製剤の両方で腫瘍増殖を阻害した。対照で処置したマウスと比較して、3mg/kg IV化合物1または10mg/kg経口化合物1で処置したマウスの腫瘍は、14日間処置した後、約8〜12倍小さかった(それぞれ、P=0.0008およびP=0.0028)(図4C)。この実験に使用されるLAGκ−1B腫瘍は非分泌性であるため、これらの腫瘍を有するマウスの血清hIgG濃度は試験しなかった。
【0078】
実施例5.化合物1をボルテゾミブと併用するとボルテゾミブ感受性LAGκ−1A MM腫瘍の増殖が阻害される
化合物1をボルテゾミブと併用するとin vitroでMM細胞の相乗的アポトーシスが誘導されるため、in vivoにおいてヒトMM腫瘍でこの組み合わせを試験した。最適以下の単剤抗腫瘍活性を有する薬物濃度を選択した。単独療法の場合、化合物1(1mg/kg IV)とボルテゾミブ(0.5mg/kg IV)は両方とも、溶媒対照と比較して血清hIgG濃度およびLAGκ−1A腫瘍体積を部分的にしか低減しなかった(図5A、B)。しかし、異常タンパク質分泌と腫瘍体積の両方で測定した場合、単剤の化合物1またはボルテゾミブで処置したにもかかわらず、腫瘍増殖は徐々に進行し続けた。対照的に、化合物1とボルテゾミブを同じ用量で併用投与すると、検出可能な異常タンパク質分泌および腫瘍体積の増加がなくなった。対照で処置したLAGκ−1A腫瘍と併用療法で処置したLAGκ−1A腫瘍との増殖の差は、まず、療法開始28日後に顕著になった(血清hIgG濃度ではP=0.0028、腫瘍体積ではP=0.0265)(図5A、B)。腫瘍の進行は実験期間中ずっと(110日間)完全に阻害され続けた。さらに、化合物1+ボルテゾミブで処置したマウスを安楽死させたとき、切除した後肢を検査すると、筋肉塊だけが認められ、腫瘍組織は認められなかった。試験開始時に移植された腫瘍は完全に退縮していた。特に、併用療法は忍容性が高く、明らかな毒性の徴候は認められなかった。
【0079】
以前の研究では、hIgG分泌骨髄腫腫瘍体積の変化は、血清ヒト異常タンパク質濃度の変化と密接に相関した(15、32)。しかし、これらの実験では、単剤の化合物1またはボルテゾミブで処置した腫瘍からの異常タンパク質分泌はプラトーに達した後、処置63日目(試験70日目)から低下し;対照的に、腫瘍体積は試験期間中ずっと増加し続けた(図5A、B)。従って、単独療法の場合、各薬剤は、腫瘍体積より血清hIgG濃度の上昇をより効果的に抑制した。
【0080】
これらの結果を検証するため、70日目以降の試料をELISAで再試験し、hIgG濃度の低下を確認した。hIgG濃度と腫瘍体積の間の反比例関係は、おそらく癌幹細胞に由来する非分泌性、薬物耐性MM細胞集団が存在することを示唆する。従って、ボルテゾミブまたは化合物1は単独では、主に、MMの抗体を分泌する成熟形質細胞成分に対して作用し(22)、腫瘍増殖の遅延の原因となる小さい幹細胞集団に影響を及ぼさない可能性がある(24)。
【0081】
対照的に、化合物1とボルテゾミブを併用投与したLAGκ−1Aを有するマウスは、hIgGと腫瘍体積の測定の両方で評価した場合、110日間の試験中ずっと、腫瘍増殖の顕著で持続的な抑制が認められた。これらのデータから、単剤のPIの存在下で異常タンパク質を産生することなく増殖するMM細胞は、化合物1とボルテゾミブの組み合わせに対して感受性があることが分かる。
【0082】
実施例6.化合物1とボルテゾミブの併用療法はボルテゾミブ耐性LAGκ−1B腫瘍の薬物耐性を克服する
LAGκ−1B腫瘍は、ボルテゾミブに対して耐性があり;実際、どちらかのPI単独 (0.5mg/kg IVボルテゾミブまたは1mg/kg IV化合物1)ではこれらの腫瘍の増殖はあまり抑制されない。対照的に、化合物1+ボルテゾミブで処置したLAGκ−1Bを有するマウスは、21日間処置した後、溶媒で処置したマウスよりも形成した腫瘍が著しく小さかった(P=0.0014)。さらに、28日間処置した後、併用療法を受けたマウスの腫瘍はまた、どちらかのPI単独で処置したマウスの腫瘍より小さかった(それぞれ化合物1単独およびボルテゾミブ単独と比較した場合、P=0.0039およびP< 0.0001)(図5C)。時間対腫瘍の進行について調べるため、併用療法群のマウスの投与を継続した。単剤のPIで処置した腫瘍と比較して、併用療法群の腫瘍体積の進行は100%遅延された(単剤のPIで処置したマウスでは腫瘍の進行まで35日であったのに対して、両方のPIで処置したマウスでは腫瘍の進行まで70日であった)。最後に、各処置群の全生存期間を記録した。溶媒で処置したマウスと比較して、併用療法を受けたマウスは、150%長く生存した(データ不示)。どちらかのPI単独で処置したマウスは、溶媒で処置したマウスより20%長く生存した(データ不示)。他のマウス生存期間データから、両方のPIを用いた併用療法の忍容性は単独療法と同様であることが分かった(データ不示)。総合的に、これらのデータから、化合物1をボルテゾミブと併用するとin vivoにおけるヒトMMのボルテゾミブ耐性を克服できることが分かる。
【0083】
実施例7.化合物1をメルファランと併用するとLAGκ−1A腫瘍およびLAGκ−1B腫瘍の増殖が阻害される
化合物1はメルファランと共同して、培養されたMM細胞の生存率を低下させ、ボルテゾミブは実験室(11)と臨床試験(33)の両方でメルファランの抗MM効果を増強するため、in vivoにおけるこのアルキル化剤と化合物1の併用の有効性を評価した。低用量(1mg/kg IP)での単剤のメルファランによる処置は、LAGκ−1Aを有するマウスの血清hIgG濃度または腫瘍体積に対して効果がなかった。同様に、単剤の化合物1(1mg/kg IV)を投与した結果、異常タンパク質分泌と腫瘍体積は両方ともあまり減少しなかった。しかし、3週間処置した後、化合物1とメルファランの両方で処置した腫瘍は、溶媒で処置した腫瘍と比較して、hIgG分泌(P=0.0012)と腫瘍体積(P=0.032)が両方とも著しく減少した(図5D、E)。ボルテゾミブ耐性およびメルファラン耐性のLAGκ−1B腫瘍を有するマウスで、同様の結果が得られた。3mg/kg IPの単剤メルファラン(LAGκ−1Aを有するマウスに投与した用量より3倍高い)または1mg/kg IVの化合物1(LAGκ−1Aを有するマウスに投与したのと同じ用量)では、腫瘍体積の増加が部分的に抑制されたが、化合物1をメルファランと併用した場合、腫瘍体積は事実上検出不可能なレベルまで減少した(図5F)。
【0084】
単剤療法と対照的に、どちらかのタイプの腫瘍を有するマウスで併用療法を継続する限り(LAGκ−1Aマウスでは63日間の処置、およびLAGκ−1Bマウスでは49日間の処置)、腫瘍増殖は防止された。さらに、併用療法の忍容性は、各薬剤単独の忍容性と同様であった(データ不示)。
【0085】
実施例8.化合物1とボルテゾミブで処置したLAGκ−1Bマウスの腫瘍ではアポトーシス誘導因子の高い発現が認められる
LAGκ−1Bを有するマウスの腫瘍を処置後に切除し、アポトーシスマーカーであるAIFに関して染色した。単剤の化合物1またはボルテゾミブで処置した腫瘍では、溶媒で処置した腫瘍と比較して、高いAIF発現が認められた。しかし、AIF発現は、両方のPIで処置した動物から採取した腫瘍ではさらに増加する(図6)。化合物1+ボルテゾミブが投与されたLAGκ−1Aを有するマウスの腫瘍は、この処置群では腫瘍が完全に退縮した後、使用可能な異種移植片試料がなかったため、組織分析に使用できなかった。
【0086】
実施例9.単剤の経口化合物1はin vivoでヒトMM腫瘍の増殖を阻害した
これらの実験は、実施例4に記載のように、元々MM患者の骨髄生検材料に由来するLAGκ−1A腫瘍を有するマウスを使用して行った。腫瘍組織を筋肉内に移植した後、マウスを毎日または週2回化合物1で、経口投与により0.5〜5mg/kgの範囲の用量で毎日、または経口投与により5〜10mg/kgの範囲の用量で週2回、処置した。対照群のマウスには化合物1の希釈剤を投与した。
【0087】
化合物1は、毎日3mg/kg経口投与すると、ヒトIgG濃度と腫瘍体積の両方に関して中程度の抗骨髄腫活性を有する。化合物1は、5mg/kgで毎日経口投与した場合、統計学的に有意な抗骨髄腫活性(IgG濃度および腫瘍体積)を有する(図7および8)。この計画および用量(毎日5mg/kg)では、マウスの87%が42日目まで生存した。また、化合物1を10mg/kgで週2回経口投与すると、統計学的に有意な抗骨髄腫活性(IgG濃度および腫瘍体積)が得られ、マウスの100%が42日目まで生存する(図7および8)。
【0088】
参考文献
1.Jemal A,Siegel R,Ward E,et al.Cancer statistics,2008.CA Cancer J Clin 2008;58:71−96.
2.Richardson PG,Sonneveld P,Schuster A,et al.Bortezomib demonstrates superior survival compared with high−dose dexamethasone and higher response rates after extended follow−up in the APEX trial in relapsed multiple myeloma.Presented at:11th Congress of the European Hematology Association;June 15−18,2006;Amsterdam,the Netherlands.Abstract 224.
3.Chauhan D,Hideshima T,Anderson KC.Targeting proteasomes as therapy in multiple myeloma.Adv Exp Med Biol 2008;615:251−60.
4.McConkey DJ,Zhu K.Mechanisms of proteasome inhibitor action and resistance in cancer.Drug Resist Updat 2008;11:164−79.
5.Hideshima T,Richardson P,Chauhan D,et al.The proteasome inhibitor PS−341 inhibits growth,induces apoptosis,and overcomes drug resistance in human multiple myeloma cells.Cancer Res 2001;61:3071−6.
6.Obeng EA,Carlson LM,Gutman DM,Harrington Jr WJ,Lee KP,Boise LH.Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells.Blood 2006;107:4907−16.
7.Chauhan D,Anderson KC.Mechanisms of cell death and survival in multiple myeloma(MM):Therapeutic implications.Apoptosis 2003;8:337−43.
8.Roccaro AM,Hideshima T,Raje N,et al.Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells.Cancer Res 2006;66:184−91.
9.Richardson PG,Barlogie B,Berenson J,et al.A phase 2 study of bortezomib in relapsed,refractory myeloma.N Engl J Med 2003;348:2609−17.
10.Jagannath S,Barlogie B,Berenson J,et al.A phase 2 study of two doses of bortezomib in relapsed or refractory myeloma.Br J Haematol 2004;127:165−72.
11.Ma MH,Yang HH,Parker K,et al.The proteasome inhibitor PS−341 markedly enhances sensitivity of multiple myeloma tumor cells to chemotherapeutic agents.Clin Cancer Res 2003;9:1136−44.
12.Mitsiades N,Mitsiades CS,Richardson PG,et al.The proteasome inhibitor PS−341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents:therapeutic applications.Blood 2003;101:2377−80.
13.Baumann P,Mandl−Weber S,Oduncu F,Schmidmaier R.Alkylating agents induce activation of NFkappaB in multiple myeloma cells.Leuk Res 2008;32:1144−7.
14.Mitsiades N,Mitsiades CS,Poulaki V,et al.Apoptotic signaling induced by immunomodulatory thalidomide analogs in human multiple myeloma cells:therapeutic implications.Blood 2002;99:4525−30.
15.Campbell RA,Sanchez E,Steinberg JA,et al.Antimyeloma effects of arsenic trioxide are enhanced by melphalan,bortezomib and ascorbic acid.Br J Haematol 2007;138:467−78.
16.Podar K,Raab MS,Zhang J,et al.Targeting PKC in multiple myeloma:in vitro and in vivo effects of the novel,orally available small−molecule inhibitor enzastaurin(LY317615.HCl).Blood 2007;109:1669−77.
17.Chauhan D,Singh A,Brahmandam M,et al.Combination of proteasome inhibitors bortezomib and NPI−0052 trigger in vivo synergistic cytotoxicity in multiple myeloma.Blood 2008;111:1654−64.
18.Pei XY,Dai Y,Grant S.Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors.Clin Cancer Res 2004;10:3839−52.
19.San Miguel JF,Schlag R,Khuageva NK,et al.Updated follow−up and results of subsequent therapy in the phase III VISTA trial:bortezomib plus melphalan−prednisone versus melphalan−prednisone in newly diagnosed multiple myeloma.Blood(ASH Annual Meeting Abstracts)2008;112:650.
20.Berenson JR,Yang HH,Vescio RA,et al.Safety and efficacy of bortezomib and melphalan combination in patients with relapsed or refractory multiple myeloma:updated results of a phase 1/2 study after longer follow−up.Ann Hematol 2008;87:623−31.
21.Pineda−Roman M,Zangari M,van Rhee F,et al.VTD combination therapy with bortezomib−thalidomide−dexamethasone is highly effective in advanced and refractory multiple myeloma.Leukemia 2008;22:1419−27.
22.Bianchi G,Oliva L,Cascio P,et al.The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition.Blood 2009;113:3040−9.
23.Matsui W,Huff CA,Wang Q,et al.Characterization of clonogenic multiple myeloma cells.Blood 2004;103:2332−6.
24.Matsui W,Wang Q,Barber JP,et al.Clonogenic multiple myeloma progenitors,stem cell properties,and drug resistance.Cancer Res 2008;68:190−7.
25.Hamburger A,Salmon SE.Primary bioassay of human myeloma stem cells.J Clin Invest 1977;60:846−54.
26.Pilarski LM,Seeberger K,Coupland RW,et al.Leukemic B cells clonally identical to myeloma plasma cells are myelomagenic in NOD/SCID mice.Exp Hematol 2002;30:221−8.
27.Meister S,Schubert U,Neubert K,et al.Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition.Cancer Res 2007;67:1783−92.
28.Dorsey BD, Iqbal M, Chatterjee S, et al.Discovery of a potent,selective,and orally active proteasome inhibitor for the treatment of cancer.J Med Chem 2008;51:1068−72.
29.Piva R,Ruggeri B,Williams M,et al.CEP−18770:a novel orally−active proteasome inhibitor with a tumor−selective pharmacological profile competitive with bortezomib.Blood 2008;111:2765−75.
30.Chou TC,Talalay P.Quantitative analysis of dose−effect relationships:the combined effects of multiple drugs or enzyme inhibitors.Adv Enzyme Regul 1984;22:27−55.
31.Chou TC.Theoretical basis,experimental design,and computerized simulation of synergism and antagonism in drug combination studies.Pharmacol Rev 2006;58:621−81.
32.Campbell RA,Berenson JR.Animal models of multiple myeloma and their utility in drug discovery.In:Current Protocols in Pharmacology,vol.40,unit 49.Hoboken,NJ:John Wiley & Sons,Inc.;2008:14.9.1−22.
33.Berenson JR,Yang HH,Sadler K,et al.Phase I/II trial assessing bortezomib and melphalan combination therapy for the treatment of patients with relapsed or refractory multiple myeloma.J Clin Oncol 2006;24:937−44.
34.Demo SD,Kirk CJ,Aujay MA,et al.Antitumor activity of PR−171,a novel irreversible inhibitor of the proteasome.Cancer Res 2007;67:6383−91.
35.Crawford LJ,Walker B,Ovaa H,et al.Comparative selectivity and specificity of the proteasome inhibitors BzLLLCOCHO,PS−341,and MG−132.Cancer Res 2006;66:6379−86.
36.Sunters A,Springer CJ,Bagshawe KD,Souhami RL,Hartley JA.The cytotoxicity,DNA crosslinking ability and DNA sequence selectivity of the aniline mustards melphalan,chlorambucil and 4−[bis(2−chloroethyl)amino]benzoic acid.Biochem Pharmacol 1992;44:59−64.
37.Ahn KS,Sethi G,Chao TH,et al.Salinosporamide A(NPI−0052)potentiates apoptosis,suppresses osteoclastogenesis,and inhibits invasion through down−modulation of NF−kappaB regulated gene products.Blood 2007;110:2286−95.
38.Chen Q,Van der Sluis PC,Boulware D,Hazlehurst LA,Dalton WS.The FA/BRCA pathway is involved in melphalan−induced DNA interstrand cross−link repair and accounts for melphalan resistance in multiple myeloma cells.Blood 2005;106:698−705.
39.Jacquemont C,Taniguchi T.Proteasome function is required for DNA damage response and fanconi anemia pathway activation.Cancer Res 2007;67:7395−405.
40.Zweegman S,Huijgens PC.Treatment of myeloma:recent developments.Anticancer Drugs 2002;13:339−51.
41.Argyriou AA,Iconomou G,Kalofonos HP.Bortezomib−induced peripheral neuropathy in multiple myeloma:a comprehensive review of the literature.Blood 2008;112:1593−9.
42.VELCADER(登録商標)Full Prescribing Information
43.ALKERANR(登録商標)Tablets Prescribing Information;ALKERANR(登録商標)for Injection Prescribing Information
【特許請求の範囲】
【請求項1】
化合物1とボルテゾミブを対象に併用投与する工程を含む、対象の多発性骨髄腫を治療する方法。
【請求項2】
前記ボルテゾミブをプロドラッグとして投与する、請求項1に記載の方法。
【請求項3】
前記ボルテゾミブを静脈内投与する、請求項1または2に記載の方法。
【請求項4】
前記ボルテゾミブを経口投与する、請求項1または2に記載の方法。
【請求項5】
前記ボルテゾミブを約0.5mg/m2〜約2mg/m2の範囲の用量で投与する、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
前記ボルテゾミブを約0.7mg/m2〜約1.3mg/m2の範囲の用量で投与する、請求項5に記載の方法。
【請求項7】
ボルテゾミブを3〜7日毎に2〜4週間投与した後、前記ボルテゾミブを投与しない約7〜21日間の休薬期間を設ける、計画された投与サイクルに従って前記ボルテゾミブを投与する、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
ボルテゾミブを21日サイクルの1日目、4日目、8日目および11日目に投与する、計画された投与サイクルに従って前記ボルテゾミブを投与する、請求項7に記載の方法。
【請求項9】
ボルテゾミブを28日サイクルの1日目、4日目、8日目および11日目に投与する、計画された投与サイクルに従って前記ボルテゾミブを投与する、請求項7に記載の方法。
【請求項10】
前記計画されたサイクルを少なくとも1回繰り返す、請求項7〜9のいずれか1項に記載の方法。
【請求項11】
対象に化合物1とメルファランを併用投与する工程を含む、前記対象の多発性骨髄腫を治療する方法。
【請求項12】
前記メルファランをプロドラッグとして投与する、請求項11に記載の方法。
【請求項13】
前記メルファランを経口投与する、請求項11または12に記載の方法。
【請求項14】
前記メルファランを静脈内投与する、請求項11または12に記載の方法。
【請求項15】
前記メルファランを約0.025mg/kg〜約0.5mg/kgの範囲の用量で投与する、請求項11〜14のいずれか1項に記載の方法。
【請求項16】
前記メルファランを約0.025mg/kg〜約0.3mg/kgの範囲の用量で投与する、請求項15に記載の方法。
【請求項17】
メルファランを3〜7日毎に1〜2週間投与した後、前記メルファランを投与しない約4〜6週間の休薬期間を設ける、計画された投与サイクルに従って前記メルファランを投与する、請求項11〜16のいずれか1項に記載の方法。
【請求項18】
メルファランを1日1回約4〜約7日間投与した後、約4〜6週間の休薬期間を設ける、計画された投与サイクルに従って前記メルファランを投与する、請求項17に記載の方法。
【請求項19】
メルファランを1日1回約4〜約5日間投与した後、約4〜6週間の休薬期間を設ける、計画された投与サイクルに従って前記メルファランを投与する、請求項17に記載の方法。
【請求項20】
前記計画されたサイクルを少なくとも1回繰り返す、請求項17〜19のいずれか1項に記載の方法。
【請求項21】
前記化合物1をプロドラッグとして投与する、請求項1〜20のいずれか1項に記載の方法。
【請求項22】
前記化合物1のプロドラッグが、化合物1の薬学的に許容されるエステルの形態である、請求項21に記載の方法。
【請求項23】
前記化合物1を静脈内投与する、請求項1〜22のいずれか1項に記載の方法。
【請求項24】
前記化合物1を経口投与する、請求項1〜22のいずれか1項に記載の方法。
【請求項25】
前記化合物1を約0.5mg/m2〜約5mg/m2の範囲の用量で投与する、請求項1〜24のいずれか1項に記載の方法。
【請求項26】
前記化合物1を約1mg/m2〜約3mg/m2の範囲の用量で投与する、請求項25に記載の方法。
【請求項27】
化合物1を3〜14日毎に2〜4週間投与した後、前記化合物1を投与しない約7〜21日間の休薬期間を設ける、計画された投与サイクルに従って前記化合物1を投与する、請求項1〜26のいずれか1項に記載の方法。
【請求項28】
化合物1を21日サイクルの1日目、4日目、8日目および11日目に投与する、計画された投与サイクルに従って前記化合物1を投与する、請求項27に記載の方法。
【請求項29】
化合物1を28日サイクルの1日目、4日目、8日目および11日目に投与する、計画された投与サイクルに従って前記化合物1を投与する、請求項27に記載の方法。
【請求項30】
化合物1を21日サイクルの1日目と15日目に投与する、計画された投与サイクルに従って前記化合物1を投与する、請求項27に記載の方法。
【請求項31】
化合物1を28日サイクルの1日目と15日目に投与する、計画された投与サイクルに従って前記化合物1を投与する、請求項27に記載の方法。
【請求項32】
前記ボルテゾミブを21日サイクルの1日目、5日目および9日目に投与し、化合物1を前記21日サイクルの3日目、8日目および12日目に投与する、請求項1〜6のいずれか1項に記載の方法。
【請求項33】
前記ボルテゾミブを28日サイクルの1日目、5日目および9日目に投与し、化合物1を前記28日サイクルの3日目、8日目および12日目に投与する、請求項1〜6のいずれか1項に記載の方法。
【請求項34】
前記化合物1を21日サイクルの1日目、5日目および9日目に投与し、ボルテゾミブを前記21日サイクルの3日目、8日目、および12日目に投与する、請求項1〜6のいずれか1項に記載の方法。
【請求項35】
前記化合物1を28日サイクルの1日目、5日目および9日目に投与し、ボルテゾミブを前記28日サイクルの3日目、8日目、および12日目に投与する、請求項1〜6のいずれか1項に記載の方法。
【請求項36】
前記化合物1をプロドラッグとして投与する、請求項32〜35のいずれか1項に記載の方法。
【請求項37】
前記化合物1のプロドラッグが、化合物1の薬学的に許容されるエステルの形態である、請求項36に記載の方法。
【請求項38】
前記化合物1を静脈内投与する、請求項32〜37のいずれか1項に記載の方法。
【請求項39】
前記化合物1を経口投与する、請求項32〜37のいずれか1項に記載の方法。
【請求項40】
前記化合物1を約0.5mg/m2〜約5mg/m2の範囲の用量で投与する、請求項32〜39のいずれか1項に記載の方法。
【請求項41】
前記化合物1を約1mg/m2〜約3mg/m2の範囲の用量で投与する、請求項40に記載の方法。
【請求項42】
前記計画されたサイクルを少なくとも1回繰り返す、請求項27〜41のいずれか1項に記載の方法。
【請求項1】
化合物1とボルテゾミブを対象に併用投与する工程を含む、対象の多発性骨髄腫を治療する方法。
【請求項2】
前記ボルテゾミブをプロドラッグとして投与する、請求項1に記載の方法。
【請求項3】
前記ボルテゾミブを静脈内投与する、請求項1または2に記載の方法。
【請求項4】
前記ボルテゾミブを経口投与する、請求項1または2に記載の方法。
【請求項5】
前記ボルテゾミブを約0.5mg/m2〜約2mg/m2の範囲の用量で投与する、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
前記ボルテゾミブを約0.7mg/m2〜約1.3mg/m2の範囲の用量で投与する、請求項5に記載の方法。
【請求項7】
ボルテゾミブを3〜7日毎に2〜4週間投与した後、前記ボルテゾミブを投与しない約7〜21日間の休薬期間を設ける、計画された投与サイクルに従って前記ボルテゾミブを投与する、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
ボルテゾミブを21日サイクルの1日目、4日目、8日目および11日目に投与する、計画された投与サイクルに従って前記ボルテゾミブを投与する、請求項7に記載の方法。
【請求項9】
ボルテゾミブを28日サイクルの1日目、4日目、8日目および11日目に投与する、計画された投与サイクルに従って前記ボルテゾミブを投与する、請求項7に記載の方法。
【請求項10】
前記計画されたサイクルを少なくとも1回繰り返す、請求項7〜9のいずれか1項に記載の方法。
【請求項11】
対象に化合物1とメルファランを併用投与する工程を含む、前記対象の多発性骨髄腫を治療する方法。
【請求項12】
前記メルファランをプロドラッグとして投与する、請求項11に記載の方法。
【請求項13】
前記メルファランを経口投与する、請求項11または12に記載の方法。
【請求項14】
前記メルファランを静脈内投与する、請求項11または12に記載の方法。
【請求項15】
前記メルファランを約0.025mg/kg〜約0.5mg/kgの範囲の用量で投与する、請求項11〜14のいずれか1項に記載の方法。
【請求項16】
前記メルファランを約0.025mg/kg〜約0.3mg/kgの範囲の用量で投与する、請求項15に記載の方法。
【請求項17】
メルファランを3〜7日毎に1〜2週間投与した後、前記メルファランを投与しない約4〜6週間の休薬期間を設ける、計画された投与サイクルに従って前記メルファランを投与する、請求項11〜16のいずれか1項に記載の方法。
【請求項18】
メルファランを1日1回約4〜約7日間投与した後、約4〜6週間の休薬期間を設ける、計画された投与サイクルに従って前記メルファランを投与する、請求項17に記載の方法。
【請求項19】
メルファランを1日1回約4〜約5日間投与した後、約4〜6週間の休薬期間を設ける、計画された投与サイクルに従って前記メルファランを投与する、請求項17に記載の方法。
【請求項20】
前記計画されたサイクルを少なくとも1回繰り返す、請求項17〜19のいずれか1項に記載の方法。
【請求項21】
前記化合物1をプロドラッグとして投与する、請求項1〜20のいずれか1項に記載の方法。
【請求項22】
前記化合物1のプロドラッグが、化合物1の薬学的に許容されるエステルの形態である、請求項21に記載の方法。
【請求項23】
前記化合物1を静脈内投与する、請求項1〜22のいずれか1項に記載の方法。
【請求項24】
前記化合物1を経口投与する、請求項1〜22のいずれか1項に記載の方法。
【請求項25】
前記化合物1を約0.5mg/m2〜約5mg/m2の範囲の用量で投与する、請求項1〜24のいずれか1項に記載の方法。
【請求項26】
前記化合物1を約1mg/m2〜約3mg/m2の範囲の用量で投与する、請求項25に記載の方法。
【請求項27】
化合物1を3〜14日毎に2〜4週間投与した後、前記化合物1を投与しない約7〜21日間の休薬期間を設ける、計画された投与サイクルに従って前記化合物1を投与する、請求項1〜26のいずれか1項に記載の方法。
【請求項28】
化合物1を21日サイクルの1日目、4日目、8日目および11日目に投与する、計画された投与サイクルに従って前記化合物1を投与する、請求項27に記載の方法。
【請求項29】
化合物1を28日サイクルの1日目、4日目、8日目および11日目に投与する、計画された投与サイクルに従って前記化合物1を投与する、請求項27に記載の方法。
【請求項30】
化合物1を21日サイクルの1日目と15日目に投与する、計画された投与サイクルに従って前記化合物1を投与する、請求項27に記載の方法。
【請求項31】
化合物1を28日サイクルの1日目と15日目に投与する、計画された投与サイクルに従って前記化合物1を投与する、請求項27に記載の方法。
【請求項32】
前記ボルテゾミブを21日サイクルの1日目、5日目および9日目に投与し、化合物1を前記21日サイクルの3日目、8日目および12日目に投与する、請求項1〜6のいずれか1項に記載の方法。
【請求項33】
前記ボルテゾミブを28日サイクルの1日目、5日目および9日目に投与し、化合物1を前記28日サイクルの3日目、8日目および12日目に投与する、請求項1〜6のいずれか1項に記載の方法。
【請求項34】
前記化合物1を21日サイクルの1日目、5日目および9日目に投与し、ボルテゾミブを前記21日サイクルの3日目、8日目、および12日目に投与する、請求項1〜6のいずれか1項に記載の方法。
【請求項35】
前記化合物1を28日サイクルの1日目、5日目および9日目に投与し、ボルテゾミブを前記28日サイクルの3日目、8日目、および12日目に投与する、請求項1〜6のいずれか1項に記載の方法。
【請求項36】
前記化合物1をプロドラッグとして投与する、請求項32〜35のいずれか1項に記載の方法。
【請求項37】
前記化合物1のプロドラッグが、化合物1の薬学的に許容されるエステルの形態である、請求項36に記載の方法。
【請求項38】
前記化合物1を静脈内投与する、請求項32〜37のいずれか1項に記載の方法。
【請求項39】
前記化合物1を経口投与する、請求項32〜37のいずれか1項に記載の方法。
【請求項40】
前記化合物1を約0.5mg/m2〜約5mg/m2の範囲の用量で投与する、請求項32〜39のいずれか1項に記載の方法。
【請求項41】
前記化合物1を約1mg/m2〜約3mg/m2の範囲の用量で投与する、請求項40に記載の方法。
【請求項42】
前記計画されたサイクルを少なくとも1回繰り返す、請求項27〜41のいずれか1項に記載の方法。
【図1A】

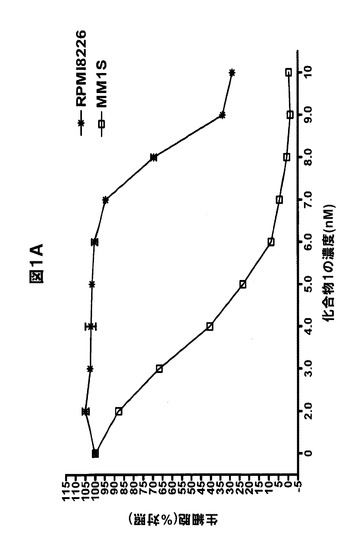
【図1B】
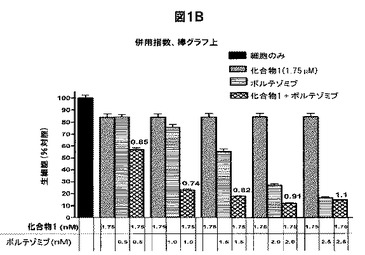
【図1C】

【図2A】
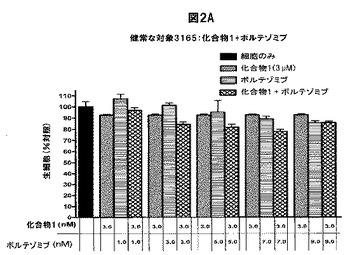
【図2B】
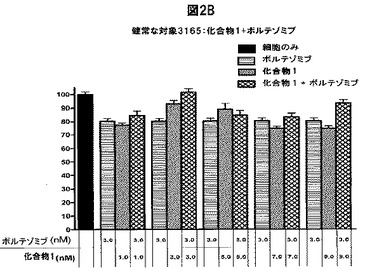
【図2C】
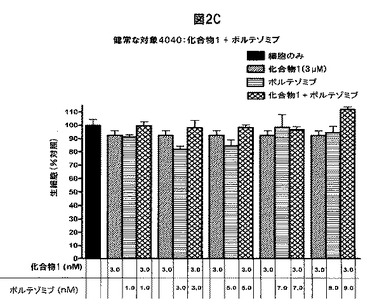
【図2D】

【図3A】
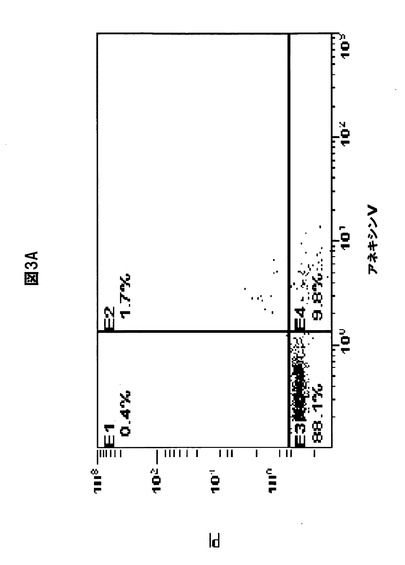
【図3B】

【図3C】
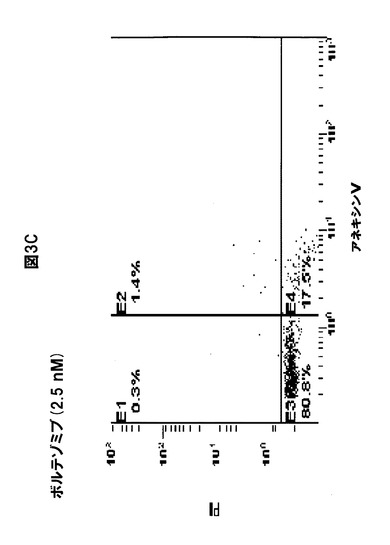
【図3D】

【図4A】
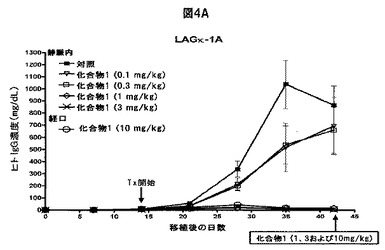
【図4B】
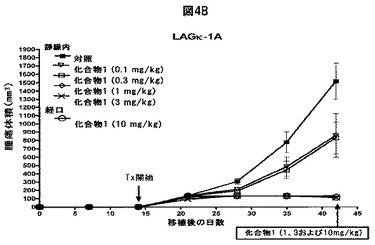
【図4C】
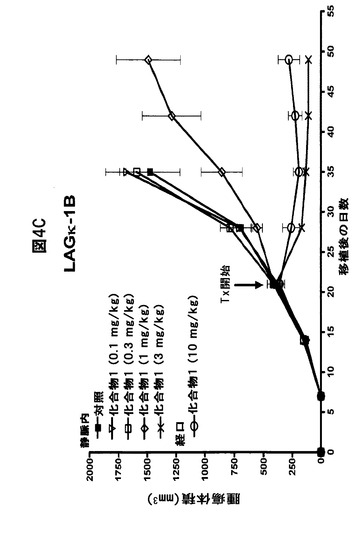
【図5A】
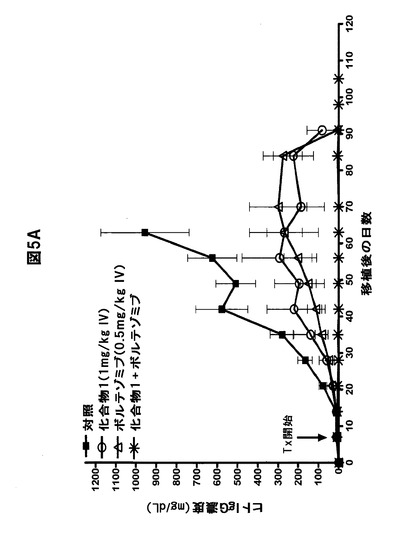
【図5B】
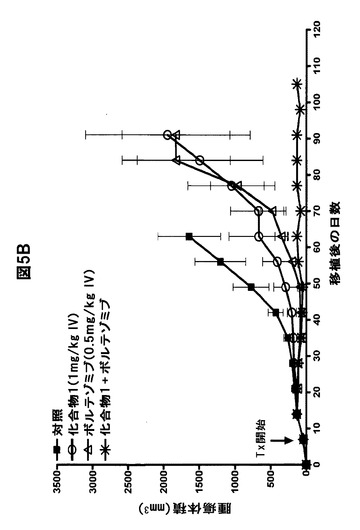
【図5C】
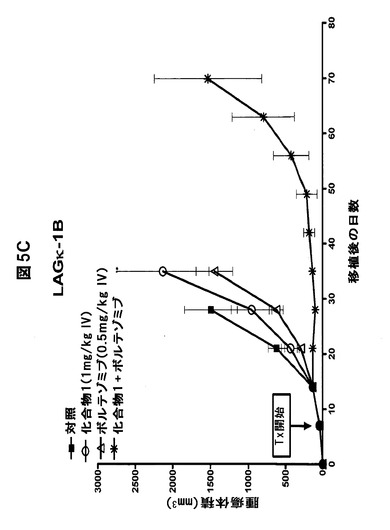
【図5D】
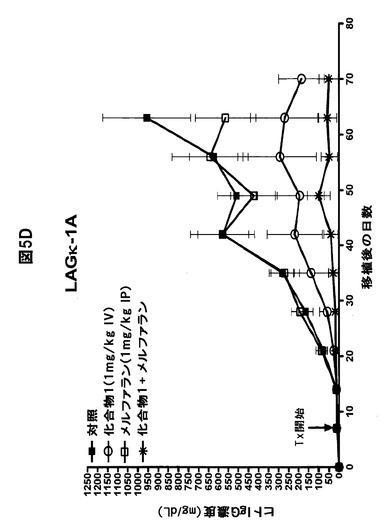
【図5E】

【図5F】
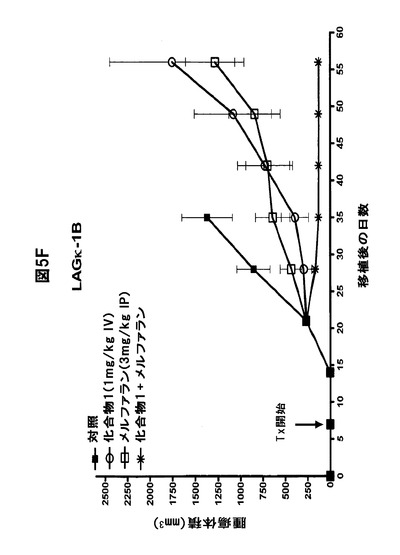
【図6A】

【図6B】

【図6C】

【図6D】

【図6E】

【図6F】
【図6G】

【図6H】

【図7】
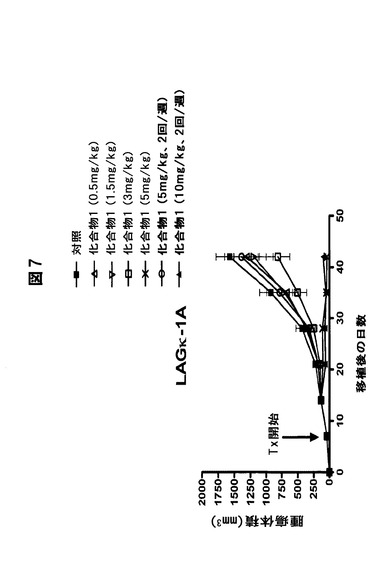
【図8】
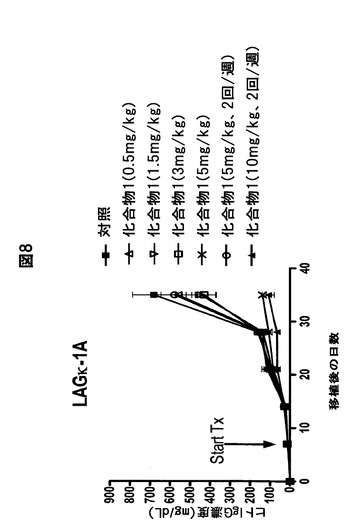
【図1B】
【図1C】
【図2A】
【図2B】
【図2C】
【図2D】
【図3A】
【図3B】
【図3C】
【図3D】
【図4A】
【図4B】
【図4C】
【図5A】
【図5B】
【図5C】
【図5D】
【図5E】
【図5F】
【図6A】
【図6B】
【図6C】
【図6D】
【図6E】
【図6F】
【図6G】
【図6H】
【図7】
【図8】
【公表番号】特表2012−528151(P2012−528151A)
【公表日】平成24年11月12日(2012.11.12)
【国際特許分類】
【出願番号】特願2012−513020(P2012−513020)
【出願日】平成21年6月9日(2009.6.9)
【国際出願番号】PCT/US2009/003467
【国際公開番号】WO2010/138101
【国際公開日】平成22年12月2日(2010.12.2)
【出願人】(509021085)セファロン、インク. (24)
【Fターム(参考)】
【公表日】平成24年11月12日(2012.11.12)
【国際特許分類】
【出願日】平成21年6月9日(2009.6.9)
【国際出願番号】PCT/US2009/003467
【国際公開番号】WO2010/138101
【国際公開日】平成22年12月2日(2010.12.2)
【出願人】(509021085)セファロン、インク. (24)
【Fターム(参考)】
[ Back to top ]