
CIS系薄膜太陽電池
【課題】直列抵抗を増加させることなく、曲線因子FFを向上させ、高効率なCIS系薄膜太陽電池を得る。
【解決手段】CIS系薄膜太陽電池は、CIS系光吸収層13、バッファ層14、透明導電膜15の順に積層された構造を備えたCIS系薄膜太陽電池であって、前記バッファ層14が、第1のバッファ層141、第2のバッファ層142、第3のバッファ層143の順に積層された3層の積層構造からなり、前記第1のバッファ層141が、厚さ1乃至3nmのZnSからなり、前記第2のバッファ層142が、厚さ20nm以下であって、前記CIS系光吸収層13から前記透明導電膜15へ向かってZnOからZnSに連続的に組成が変化する薄膜からなり、前記第3のバッファ層143が、厚さ100nm以上のZnOからなる。
【解決手段】CIS系薄膜太陽電池は、CIS系光吸収層13、バッファ層14、透明導電膜15の順に積層された構造を備えたCIS系薄膜太陽電池であって、前記バッファ層14が、第1のバッファ層141、第2のバッファ層142、第3のバッファ層143の順に積層された3層の積層構造からなり、前記第1のバッファ層141が、厚さ1乃至3nmのZnSからなり、前記第2のバッファ層142が、厚さ20nm以下であって、前記CIS系光吸収層13から前記透明導電膜15へ向かってZnOからZnSに連続的に組成が変化する薄膜からなり、前記第3のバッファ層143が、厚さ100nm以上のZnOからなる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、CIS系薄膜太陽電池に関する。
【背景技術】
【0002】
従来、CIS系薄膜太陽電池を製造する際、CuInSe2系薄膜からなる光吸収層上に高抵抗バッファ層として、硫化カドミウム(CdS)層を成長させることにより変換効率の向上が見込まれることが分かっていた。高抵抗バッファ層としては、CdS層の他に、ZnSと酸素との混晶なども有効であると考えられていた。
【0003】
更に、特許文献1には、バッファ層を2層構造で構成し、1層目のバッファ層としてCBD法(Chemical Bath Deposition:溶液成長法)により10nm以下の硫黄含有亜鉛混晶化合物(以下、「CBDバッファ層」ともいう)を製膜し、2層目のバッファ層としてMOCVD法(Metal Organic Chemical Vapor Deposition:有機金属化学的気相成長法)により100nm以上の酸化亜鉛系薄膜(以下、「MOCVDバッファ層」ともいう)を製膜することが開示されている。この特許文献1に開示された技術は、バッファ層をCBDバッファ層とMOCVDバッファ層との2層構造にすることにより、直列抵抗を増加させることなくリーク抑制が可能としている。
【0004】
一方、特許文献2には、透明導電膜を酸化亜鉛系薄膜とした場合に、CIS系光吸収層と透明導電膜との間に形成されるバッファ層の組成が、光吸収層から透明導電膜に向かって、ZnS(硫化亜鉛)からZnO(酸化亜鉛)に連続的に変化する構成が開示されている。この特許文献2に開示された技術は、バッファ層の透明導電膜側の組成がZnOとなるように変化させることで、バッファ層と透明導電膜との間の障壁をなくし接合界面特性を向上させている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】国際公開第2009/110092号
【特許文献2】特開2004−47916
【発明の概要】
【発明が解決しようとする課題】
【0006】
ここで、本件出願にかかる発明者は、更なる太陽電池特性の向上、特に、曲線因子(FF:Fill Factor)の向上を目指し、上記特許文献1に開示された構成に対して、特許文献2に記載された接合界面特性を向上させる技術を組み合わせたバッファ層を実験により作製したが、そのバッファ層を備えるCIS系薄膜太陽電池では、十分に高い曲線因子(FF)が得られなかった。
【0007】
本発明は、上記課題を解決するためになされたものであって、直列抵抗を増加させることなく、曲線因子FFを向上させ、高効率なCIS系薄膜太陽電池を得ることを目的とする。
【課題を解決するための手段】
【0008】
上記目的を達成するため、本願発明のある態様にかかるCIS系薄膜太陽電池は、CIS系光吸収層、バッファ層、透明導電膜の順に積層された構造を備えたCIS系薄膜太陽電池であって、前記バッファ層が、第1のバッファ層、第2のバッファ層、第3のバッファ層の順に積層された3層の積層構造からなり、前記第1のバッファ層が、厚さ1乃至3nmのZnSからなり、前記第2のバッファ層が、厚さ20nm以下であって、前記CIS系光吸収層から前記透明導電膜へ向かってZnSからZnOに連続的に組成が変化する薄膜からなり、前記第3のバッファ層が、厚さ100nm以上のZnOからなる
ことを特徴とする。
【0009】
また、本願発明のある態様にかかるCIS系薄膜太陽電池の積層構造は、上記CIS系光吸収層が、銅、インジウム、ガリウム、セレン、硫黄から構成されていることを特徴とする。
【図面の簡単な説明】
【0010】
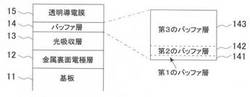
【図1】本願発明の好ましい実施形態によるCIS系薄膜太陽電池の積層構造の概略図を示す。
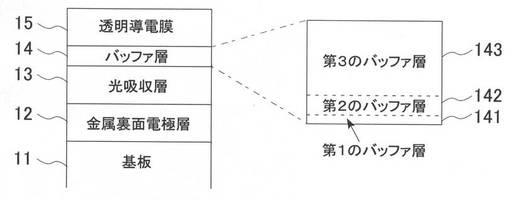
【図2】本願発明の好ましい実施形態によるCIS系薄膜太陽電池のバッファ層を作製するフローチャートの概略を示す。
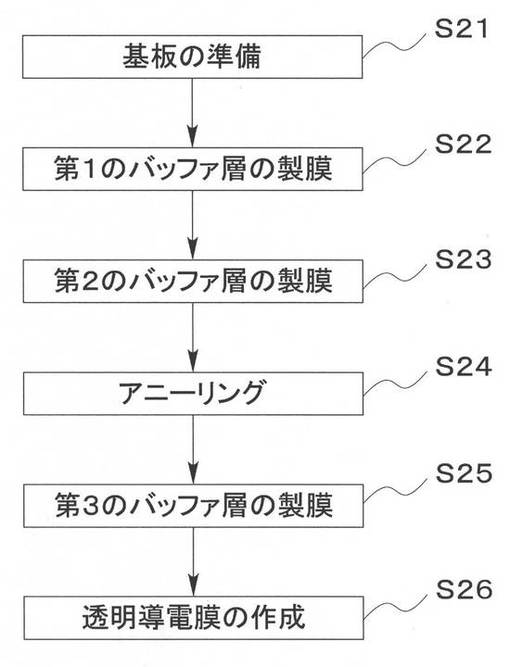
【図3】本願発明の好ましい実施形態によるCIS系薄膜太陽電池と比較例のCIS系薄膜太陽電池とのRs値の比較結果を示すグラフである。
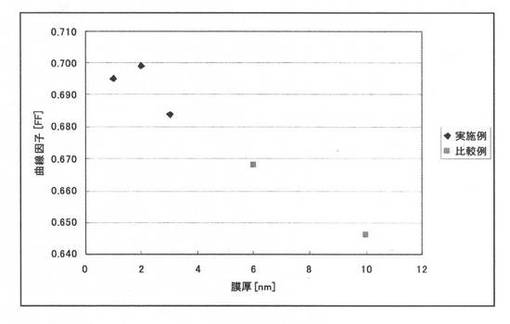
【図4】本願発明の好ましい実施形態によるCIS系薄膜太陽電池と比較例のCIS系薄膜太陽電池との曲線因子FFの比較結果を示すグラフである。
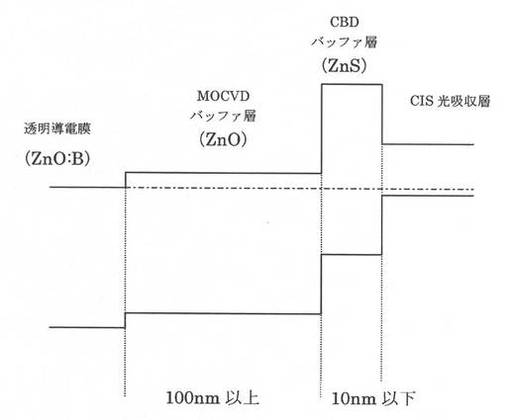
【図5】従来技術による積層構造を備えたCIS系薄膜太陽電池のエネルギーバンド図である。
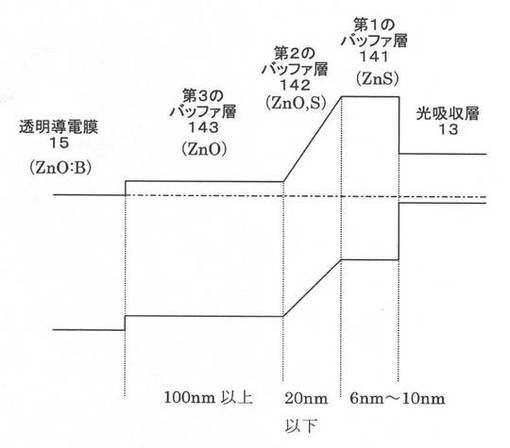
【図6】比較例による積層構造を備えたCIS系薄膜太陽電池のエネルギーバンド図である。
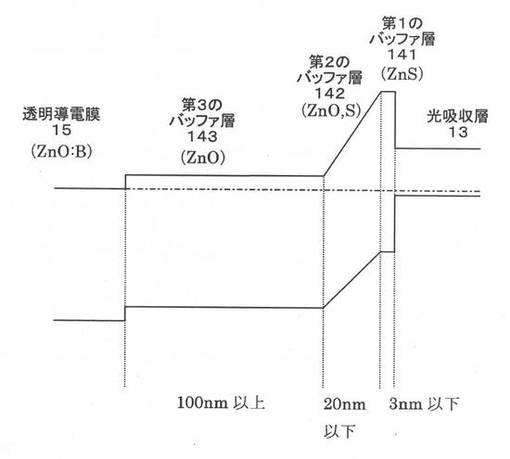
【図7】本願発明の好ましい実施形態による積層構造を備えたCIS系薄膜太陽電池のエネルギーバンド図である。
【図8】本願発明の好ましい実施形態によるCIS系薄膜太陽電池の積層構造の断面画像の一例を示す。
【符号の説明】
【0011】
11 基板
12 金属裏面電極層
13 光吸収層
14 バッファ層
141 第1のバッファ層
142 第2のバッファ層
143 第3のバッファ層
15 透明導電膜
【発明を実施するための形態】
【0012】
本願発明にかかるCIS系薄膜太陽電池の積層構造について、以下に説明する。
<基本構成>
図1に示すように、本実施形態にかかるCIS系薄膜太陽電池は、基板11、金属裏面電極層12、p型CIS系光吸収層(以下、単に「光吸収層」ともいう。)13、高抵抗バッファ層(以下、バッファ層と称す)14、n型の透明導電膜15の順に積層されたpnヘテロ接合デバイスを構成している。
【0013】
<実施例>
以下に、本実施形態の一例となる実施例(以下、本実施例とする。)の詳細な構成および作製方法を説明する。
【0014】
<金属裏面電極層12>
まず、ガラス基板からなる基板11上に、Mo(モリブデン)を材料としてDCスパッタ法等によって、膜厚200乃至500nmの金属裏面電極層12を製膜する。なお、本実施例においては、基板11にガラス基板を用いたが、本発明はこれに限らず、ステンレス板等の金属基板、ボリイミド膜等の樹脂基板を用いても良い。また、図示していないが、基板11と金属裏面電極層12との間に、SiOx等からなるアルカリ制御層を製膜してもよい。このアルカリ制御層を設けることにより、ガラス基板中に含まれるアルカリ金属(Na等)が、光吸収層13へ熱拡散することを制御できる。
【0015】
さらに、金属裏面電極層12の材料として、Mo以外にTi(チタン)やCr(クロム)等の、耐セレン腐食性に優れた高融点の金属を適用してもよい。
<光吸収層13>
次に、金属裏面電極層12上に、Cu−Ga合金を材料としてDCスパッタ法によってCuGa膜を製膜した後、その上に、Inを材料としてDCスパッタ法によってIn膜を積層することで、金属プリカーサー膜を形成する。この金属プリカーサー膜をセレン化および硫化することにより、光吸収層13が製膜される。本実施例では、InおよびGaのIII族元素の原子数に対するCuの原子数の比率(Cu/III族比)を0.85〜0.95とし、III族元素の原子数に占めるGaの原子数の比率(Ga/III族比)を0.15〜0.4とし、セレン化を350℃〜600℃、硫化を550℃〜650℃の条件で実行することにより、1.0〜2.0μmの光吸収層13を製膜した。
【0016】
本実施例の光吸収層13は、セレン化だけでなく硫化も行って製膜されているため、この光吸収層13の表面(概ね表面より200nmまで)における硫黄濃度が0.5atoms%以上となる。これにより、受光面側(高抵抗バッファ層側)での禁制帯幅を増大させることができ、結果、より効果的に光を吸収させることができる。
【0017】
なお、本実施例では、光吸収層13として、セレン化および硫化によって、Cu(InGa)(SeS)2を製膜したが、本発明はこれに限らず、セレン化又は硫化のいずれか一方によって、例えばCuInSe2、Cu(InGa)Se2、CuGaSe2、CuInS2、Cu(InGa)S2、CuGaS2、等の光吸収層13で構成されてもよい。
【0018】
さらに、本実施例においては、金属プリカーサー膜として、CuGa膜上にIn膜を積層したが、Cu−Ga−In合金膜、Cu−In合金膜や、Cu/Inの積層膜等であってもよい。
【0019】
また、光吸収層13の他の製造方法として、セレン化/硫化以外に多元同時蒸着法がある。多元同時蒸着法では、500℃程度以上に加熱した金属裏面電極層12が形成されたガラス基板11上に、銅(Cu)、インジウム(In)、ガリウム(Ga)、セレン(Se)、を含む原料を適当な組み合わせで同時に蒸着することによって光吸収層13を製膜することができる。本発明に係るCIS系薄膜太陽電池は、多元同時蒸着法によって作製された光吸収層を備える構成であってもよい。
【0020】
<透明導電膜15>
本実施例の透明導電膜15として、光吸収層13上に高抵抗バッファ層14(詳細は後述)を製膜した後、この高抵抗バッファ層14上にMOCVD法によって、厚さ0.5〜2.5μmのZnO:Bを製膜する。
【0021】
なお、透明導電膜15は、ZnO:B以外に、ZnO:Al、ZnO:Ga等の酸化亜鉛系薄膜や、ITO(Indium Tin Oxide)等であってもよい。さらに、MOCVD法の代わりにスパッタ法によっても製膜可能である。
【0022】
<高抵抗バッファ層14>
次に、本発明のポイントとなる高抵抗バッファ層14について、特許文献1および特許文献2に開示された技術を組み合わせた比較例と、本実施例とを対比しながら説明する。
【0023】
特許文献1に開示された従来技術(以下、従来技術1とする)の高抵抗バッファ層は、CBDバッファ層とMOCVDバッファ層との2層から構成されているのに対して、本願発明に係る高抵抗バッファ層14では、第1のバッファ層141の上に第2のバッファ層142が積層され、該第2のバッファ層142の上に第3のバッファ層143が積層される3層から構成される。従来技術1のCBDバッファ層は、ZnSまたはZn(O,OH,S)からなる膜厚が10nm以下の薄膜であり、同MOCVDバッファ層は、ZnOからなる膜厚が100nm以上の薄膜である。また、特許文献2に開示された従来技術(以下、従来技術2とする)には、透明導電膜をZnO:Alとした場合に、CIS系光吸収層と透明導電膜との間に形成されるバッファ層の組成が、光吸収層から透明導電膜に向かって、ZnSからZnOに連続的に変化する構成が開示されている。
【0024】
そこで、本願発明者は、更なる太陽電池特性の向上、特に、曲線因子(FF:Fill Factor)の向上を目指し、従来技術1に開示された構成に対して、特許文献2に記載された接合界面特性を向上させる技術を組み合わせた高抵抗バッファ層を実験により作製した。かかる構成を、本明細書では比較例と称する。比較例および本実施例では、第1のバッファ層141、第2のバッファ層142、第3のバッファ層143が積層される3層から構成される。
【0025】
図2を参照して、本実施例および比較例による高抵抗バッファ層14の製造方法について説明する。
【0026】
図2のステップS21において、基板11上に金属裏面電極層12、光吸収層13を製膜した基板を準備する。
【0027】
ステップS22では、光吸収層13上に、本実施例の第1のバッファ層141として1〜3nmのZnS膜を、比較例の第1のバッファ層141として6および10nmのZnS膜を、CBD法によって製膜する。具体的には、酢酸亜鉛を所定の液温の水酸化アンモニウムに溶解して亜鉛アンモニウム錯塩を形成し、その溶液中に硫黄含有塩であるチオ尿素(チオウレア)を溶解し、この溶液に光吸収層13が製膜された基板11を所定の時間浸漬する。本実施例1乃至3、比較例1および2の各々の、溶液の液温および浸漬時間を表1に示す。なお、溶液中のアンモニアと酢酸亜鉛の濃度はそれぞれ7.5Mと0.16Mである。
【0028】
【表1】

【0029】
本実施例および比較例では、液温を70℃に固定して、浸漬時間を変化させることにより、第1のバッファ層の膜厚を調整した。なお、溶液の液温は60℃〜80℃の範囲であればよく、その場合、液温に応じて浸漬時間を調整することにより、第1のバッファ層の膜厚を制御可能となる。例として、液温が80℃の場合では、浸漬時間を5分にすることにより、2乃至3nmの第1のバッファ層141を製膜することができる。
【0030】
ステップS23では、基板を浸漬している溶液に対して、所定の時間間隔で酢酸を追加する。これにより、溶液中のpHが中性に近づき、製膜されるバッファ層(第2のバッファ層142)の組成が、ZnSからZnOに連続的に変化した硫黄含有亜鉛混晶化合物半導体薄膜が形成される。
【0031】
本実施例1〜3、比較例1および2におけるステップS23での浸漬時間は60分であり、その間に段階的に酢酸を追加した。なお、本実施例および比較例では、溶液のpHが11.0から9.0に変化するように酢酸を所定間隔で追加し、これにより、第2のバッファ層142のような、組成がZnSからZnOに連続的に変化した薄膜を形成可能となる。なお、ステップS23の浸漬時間について、ステップS22と同様に、溶液を変化させた場合は、浸漬時間を調整することにより、所望の膜厚の第2のバッファ層142を得ることが可能である。具体的には、本実施例では液温が70℃、浸漬時間が60分で第2のバッファ層142を形成したが、浸漬時間を短くすることにより、20nm未満の膜厚にすることが可能であり、さらに、液温を70℃よりも高くした場合は、浸漬時間が60分以下で膜厚20nm以下の第2のバッファ層142を形成可能性となる。
【0032】
他の実施形態では、第2のバッファ層142をCBDで製膜するにあたり、第1のバッファ層141を製膜した後、溶液中のアンモニアを蒸発させながら第2のバッファ層142を製膜することにより、層が成長する途中で、溶液のpHが中性に近づき、第2のバッファ層142の組成をZnSからZnOに変化させることができる。また、第2のバッファ層142の溶液に、段階的に酢酸亜鉛を添加することで、溶液のpHが中性に近づき、第2のバッファ層142の組成をZnSからZnOに変化させることができる。
【0033】
更に他の実施形態としては、ALD(Atomic Layer Deposition)法を用いてZnSからZnOに変化するように1原子層づつ堆積させる方法がある。具体的には、Zn源にジエチル亜鉛、S源に硫化水素、O源にH2Oを用いることで、組成がZnSからZnOに連続的に変化した第2のバッファ層142を製膜することが可能となる。
【0034】
次に、ステップS24では、第2のバッファ層142が製膜された基板を大気中で設定温度200℃で15分間アニールすることで乾燥し、かつ、膜中の水酸化亜鉛の一部を酸化亜鉛に転化すると同時に硫黄により改質する。これにより、第1のバッファ層141および第2のバッファ層142を高品質化できる。
【0035】
ステップS25では、第2のバッファ層142上に、MOCVD法によって膜厚が100nm以上の酸化亜鉛系薄膜を、第3のバッファ層143として形成する。この第3のバッファ層143は、透明導電膜15に接して形成される。このため、第3のバッファ層143は、ドーピング不純物元素として、アルミニウム(Al)、ガリウム(Ga)、ホウ素(B)などを含むが、これらのドーピング不純物元素濃度を、1×1019atoms/cm3以下、より好ましくは1×1018atoms/cm3以下となるように調整することにより、バッファ層として好ましい高抵抗な膜となる。この第3のバッファ層143の抵抗率は、0.1Ωcm以上、より好ましくは1Ωcm以上となっている。
【0036】
なお、本実施例においては、MOCVD法によって第3のバッファ層143を製膜したが、本発明はこれに限らず、スパッタ法などにより形成することもできる。
【0037】
<実験結果>
上記各ステップにより形成された第1のバッファ層141の製膜結果は、比較例では第1のバッファ層141はZnSからなり、膜厚は6nm(比較例1)および10nm(比較例2)となった。これに対して、本願発明に係る実施例における第1のバッファ層141はZnSからなり、膜厚は1nm(実施例1)、2nm(実施例2)および3nm(実施例3)となった。第2のバッファ層142の組成はいずれも、金属裏面電極層12側から透明導電膜15側にかけてZnSからZnOと変化する組成となり、膜厚は20nm以下である。第3のバッファ層143はいずれも、ZnOからなり、膜厚は100nm以上である。
【0038】
上記比較例1,2と実施例1乃至3により作製した高抵抗バッファ層14を備えた太陽電池について特性を比較した結果を図3及び図4に示す。なお具体的な数値を表2に示す。
【0039】
【表2】
【0040】
図3、4及び表2に示したとおり、従来技術1および2を単に組み合わせただけの比較例1および2にかかる太陽電池については、十分に高い曲線因子(FF)が得られなかった。これに対して、第1のバッファ層141の膜厚を3nm以下とした本実施例1乃至3の太陽電池は、良好な曲線因子(FF)が確認できる。同様にRs[Ω cm2]についても、比較例1および2に比べ、第1のバッファ層141の膜厚を3nm以下とした本実施例1乃至3の太陽電池は良好な結果が得られた。
【0041】
ここで、従来技術1には、CBDバッファ層(本発明の第1のバッファ層に相当)の膜厚について10nm以下と記載されているにすぎず、10nm以下の範囲において、第1のバッファ層の膜厚によってFFが変化することに関する知見はなく、従来技術1の出願時において発明者もかかる知見を認識していなかった。また、従来技術1における実施形態では、80℃の溶液中に10分間接触させてCBDバッファ層141(本発明の第1のバッファ層に相当)を製膜することが開示されているが、この方法で製膜したCBDバッファ層141の膜厚は、6nm以上になる。これは、溶液の温度が80℃であり、本発明にかかる溶液の温度よりも高く、製膜速度が高いためである。
【0042】
また、従来技術2では、ZnSからなるバッファ層の膜厚については記載されておらず、第1段階の処理で製膜されたバッファ層が、本発明の第1のバッファ層141に相当すると見ても、この第1段階の処理では、基材(CIS系光吸収層が製膜された基板)を80℃の溶液に15分間投入しており、第1段階の処理で製膜されたバッファ層の膜厚は、10nm以上になる。
【0043】
このように、従来技術1および従来技術2を単に組み合わせた構成では、第1のバッファ層141の膜厚は6nm以上となり、言い換えれば、比較例1および2の太陽電池を得られるにすぎない。この比較例1および2に対して、本実施例1〜3の太陽電池に示したとおり、第1のバッファ層の膜厚を3nm以下とすることにより、太陽電池特性の一つである曲線因子を向上させることが可能となる。
【0044】
上記事実に鑑みて比較例と本願発明に係る実施例との比較をエネルギーバンド図にて検討する。対比として、参考までに従来技術1の条件で作成した層構造のエネルギーバンド図を図5に示し、比較例のエネルギーバンド図を図6に示す。さらに、図7に本願発明に係る実施例によるエネルギーバンド図を示す。
【0045】
図5に示した従来技術1におけるエネルギーバンド図からわかるように、CBDバッファ層とMOCVDバッファ層との界面でのバンド構造に障壁があるのがわかる。図6に示した比較例におけるエネルギーバンド図は、従来技術1の構造をベースに、従来技術2の技術的特徴(ZnO膜とZnS膜との間に、組成が連続して変化する層を設ける)を単に組み合わせただけである。上述の通り、かかる比較例では、FFの大きな向上には繋がらない。これに対して、第1のバッファ層141を3nm以下とした本願発明に係る実施例によるエネルギーバンド図は図7に示したようになり、その結果は上述したとおり、良好なFFが得られる。
【0046】
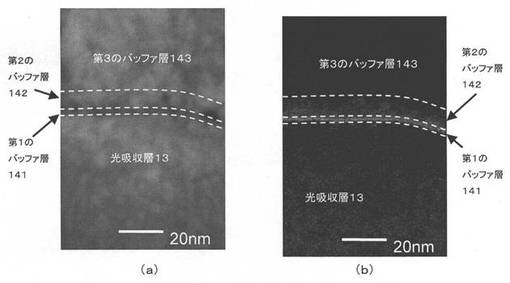
また最も好適なFFおよびRsの結果を示した、膜厚が2nmの第1のバッファ層141を備える実施例2を撮影した結果を図8に示す。図8(a)は走査透過電子顕微鏡(STEM:Scanning Transmission Electron Microscope)を使用した画像であり、図8(b)は電子エネルギー損失分光法(EELS:Electron Energy Loss Spectroscopy)を使用した硫黄マッピング像である。図8(b)において白く現れているのが硫黄であり、2nmの極薄膜の第1のバッファ層141が形成され、さらに、第2のバッファ層142において、CIS光吸収層13側から透明導電膜15側に向かって硫黄濃度が減少しているのが確認できる。
【技術分野】
【0001】
本発明は、CIS系薄膜太陽電池に関する。
【背景技術】
【0002】
従来、CIS系薄膜太陽電池を製造する際、CuInSe2系薄膜からなる光吸収層上に高抵抗バッファ層として、硫化カドミウム(CdS)層を成長させることにより変換効率の向上が見込まれることが分かっていた。高抵抗バッファ層としては、CdS層の他に、ZnSと酸素との混晶なども有効であると考えられていた。
【0003】
更に、特許文献1には、バッファ層を2層構造で構成し、1層目のバッファ層としてCBD法(Chemical Bath Deposition:溶液成長法)により10nm以下の硫黄含有亜鉛混晶化合物(以下、「CBDバッファ層」ともいう)を製膜し、2層目のバッファ層としてMOCVD法(Metal Organic Chemical Vapor Deposition:有機金属化学的気相成長法)により100nm以上の酸化亜鉛系薄膜(以下、「MOCVDバッファ層」ともいう)を製膜することが開示されている。この特許文献1に開示された技術は、バッファ層をCBDバッファ層とMOCVDバッファ層との2層構造にすることにより、直列抵抗を増加させることなくリーク抑制が可能としている。
【0004】
一方、特許文献2には、透明導電膜を酸化亜鉛系薄膜とした場合に、CIS系光吸収層と透明導電膜との間に形成されるバッファ層の組成が、光吸収層から透明導電膜に向かって、ZnS(硫化亜鉛)からZnO(酸化亜鉛)に連続的に変化する構成が開示されている。この特許文献2に開示された技術は、バッファ層の透明導電膜側の組成がZnOとなるように変化させることで、バッファ層と透明導電膜との間の障壁をなくし接合界面特性を向上させている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】国際公開第2009/110092号
【特許文献2】特開2004−47916
【発明の概要】
【発明が解決しようとする課題】
【0006】
ここで、本件出願にかかる発明者は、更なる太陽電池特性の向上、特に、曲線因子(FF:Fill Factor)の向上を目指し、上記特許文献1に開示された構成に対して、特許文献2に記載された接合界面特性を向上させる技術を組み合わせたバッファ層を実験により作製したが、そのバッファ層を備えるCIS系薄膜太陽電池では、十分に高い曲線因子(FF)が得られなかった。
【0007】
本発明は、上記課題を解決するためになされたものであって、直列抵抗を増加させることなく、曲線因子FFを向上させ、高効率なCIS系薄膜太陽電池を得ることを目的とする。
【課題を解決するための手段】
【0008】
上記目的を達成するため、本願発明のある態様にかかるCIS系薄膜太陽電池は、CIS系光吸収層、バッファ層、透明導電膜の順に積層された構造を備えたCIS系薄膜太陽電池であって、前記バッファ層が、第1のバッファ層、第2のバッファ層、第3のバッファ層の順に積層された3層の積層構造からなり、前記第1のバッファ層が、厚さ1乃至3nmのZnSからなり、前記第2のバッファ層が、厚さ20nm以下であって、前記CIS系光吸収層から前記透明導電膜へ向かってZnSからZnOに連続的に組成が変化する薄膜からなり、前記第3のバッファ層が、厚さ100nm以上のZnOからなる
ことを特徴とする。
【0009】
また、本願発明のある態様にかかるCIS系薄膜太陽電池の積層構造は、上記CIS系光吸収層が、銅、インジウム、ガリウム、セレン、硫黄から構成されていることを特徴とする。
【図面の簡単な説明】
【0010】
【図1】本願発明の好ましい実施形態によるCIS系薄膜太陽電池の積層構造の概略図を示す。
【図2】本願発明の好ましい実施形態によるCIS系薄膜太陽電池のバッファ層を作製するフローチャートの概略を示す。
【図3】本願発明の好ましい実施形態によるCIS系薄膜太陽電池と比較例のCIS系薄膜太陽電池とのRs値の比較結果を示すグラフである。
【図4】本願発明の好ましい実施形態によるCIS系薄膜太陽電池と比較例のCIS系薄膜太陽電池との曲線因子FFの比較結果を示すグラフである。
【図5】従来技術による積層構造を備えたCIS系薄膜太陽電池のエネルギーバンド図である。
【図6】比較例による積層構造を備えたCIS系薄膜太陽電池のエネルギーバンド図である。
【図7】本願発明の好ましい実施形態による積層構造を備えたCIS系薄膜太陽電池のエネルギーバンド図である。
【図8】本願発明の好ましい実施形態によるCIS系薄膜太陽電池の積層構造の断面画像の一例を示す。
【符号の説明】
【0011】
11 基板
12 金属裏面電極層
13 光吸収層
14 バッファ層
141 第1のバッファ層
142 第2のバッファ層
143 第3のバッファ層
15 透明導電膜
【発明を実施するための形態】
【0012】
本願発明にかかるCIS系薄膜太陽電池の積層構造について、以下に説明する。
<基本構成>
図1に示すように、本実施形態にかかるCIS系薄膜太陽電池は、基板11、金属裏面電極層12、p型CIS系光吸収層(以下、単に「光吸収層」ともいう。)13、高抵抗バッファ層(以下、バッファ層と称す)14、n型の透明導電膜15の順に積層されたpnヘテロ接合デバイスを構成している。
【0013】
<実施例>
以下に、本実施形態の一例となる実施例(以下、本実施例とする。)の詳細な構成および作製方法を説明する。
【0014】
<金属裏面電極層12>
まず、ガラス基板からなる基板11上に、Mo(モリブデン)を材料としてDCスパッタ法等によって、膜厚200乃至500nmの金属裏面電極層12を製膜する。なお、本実施例においては、基板11にガラス基板を用いたが、本発明はこれに限らず、ステンレス板等の金属基板、ボリイミド膜等の樹脂基板を用いても良い。また、図示していないが、基板11と金属裏面電極層12との間に、SiOx等からなるアルカリ制御層を製膜してもよい。このアルカリ制御層を設けることにより、ガラス基板中に含まれるアルカリ金属(Na等)が、光吸収層13へ熱拡散することを制御できる。
【0015】
さらに、金属裏面電極層12の材料として、Mo以外にTi(チタン)やCr(クロム)等の、耐セレン腐食性に優れた高融点の金属を適用してもよい。
<光吸収層13>
次に、金属裏面電極層12上に、Cu−Ga合金を材料としてDCスパッタ法によってCuGa膜を製膜した後、その上に、Inを材料としてDCスパッタ法によってIn膜を積層することで、金属プリカーサー膜を形成する。この金属プリカーサー膜をセレン化および硫化することにより、光吸収層13が製膜される。本実施例では、InおよびGaのIII族元素の原子数に対するCuの原子数の比率(Cu/III族比)を0.85〜0.95とし、III族元素の原子数に占めるGaの原子数の比率(Ga/III族比)を0.15〜0.4とし、セレン化を350℃〜600℃、硫化を550℃〜650℃の条件で実行することにより、1.0〜2.0μmの光吸収層13を製膜した。
【0016】
本実施例の光吸収層13は、セレン化だけでなく硫化も行って製膜されているため、この光吸収層13の表面(概ね表面より200nmまで)における硫黄濃度が0.5atoms%以上となる。これにより、受光面側(高抵抗バッファ層側)での禁制帯幅を増大させることができ、結果、より効果的に光を吸収させることができる。
【0017】
なお、本実施例では、光吸収層13として、セレン化および硫化によって、Cu(InGa)(SeS)2を製膜したが、本発明はこれに限らず、セレン化又は硫化のいずれか一方によって、例えばCuInSe2、Cu(InGa)Se2、CuGaSe2、CuInS2、Cu(InGa)S2、CuGaS2、等の光吸収層13で構成されてもよい。
【0018】
さらに、本実施例においては、金属プリカーサー膜として、CuGa膜上にIn膜を積層したが、Cu−Ga−In合金膜、Cu−In合金膜や、Cu/Inの積層膜等であってもよい。
【0019】
また、光吸収層13の他の製造方法として、セレン化/硫化以外に多元同時蒸着法がある。多元同時蒸着法では、500℃程度以上に加熱した金属裏面電極層12が形成されたガラス基板11上に、銅(Cu)、インジウム(In)、ガリウム(Ga)、セレン(Se)、を含む原料を適当な組み合わせで同時に蒸着することによって光吸収層13を製膜することができる。本発明に係るCIS系薄膜太陽電池は、多元同時蒸着法によって作製された光吸収層を備える構成であってもよい。
【0020】
<透明導電膜15>
本実施例の透明導電膜15として、光吸収層13上に高抵抗バッファ層14(詳細は後述)を製膜した後、この高抵抗バッファ層14上にMOCVD法によって、厚さ0.5〜2.5μmのZnO:Bを製膜する。
【0021】
なお、透明導電膜15は、ZnO:B以外に、ZnO:Al、ZnO:Ga等の酸化亜鉛系薄膜や、ITO(Indium Tin Oxide)等であってもよい。さらに、MOCVD法の代わりにスパッタ法によっても製膜可能である。
【0022】
<高抵抗バッファ層14>
次に、本発明のポイントとなる高抵抗バッファ層14について、特許文献1および特許文献2に開示された技術を組み合わせた比較例と、本実施例とを対比しながら説明する。
【0023】
特許文献1に開示された従来技術(以下、従来技術1とする)の高抵抗バッファ層は、CBDバッファ層とMOCVDバッファ層との2層から構成されているのに対して、本願発明に係る高抵抗バッファ層14では、第1のバッファ層141の上に第2のバッファ層142が積層され、該第2のバッファ層142の上に第3のバッファ層143が積層される3層から構成される。従来技術1のCBDバッファ層は、ZnSまたはZn(O,OH,S)からなる膜厚が10nm以下の薄膜であり、同MOCVDバッファ層は、ZnOからなる膜厚が100nm以上の薄膜である。また、特許文献2に開示された従来技術(以下、従来技術2とする)には、透明導電膜をZnO:Alとした場合に、CIS系光吸収層と透明導電膜との間に形成されるバッファ層の組成が、光吸収層から透明導電膜に向かって、ZnSからZnOに連続的に変化する構成が開示されている。
【0024】
そこで、本願発明者は、更なる太陽電池特性の向上、特に、曲線因子(FF:Fill Factor)の向上を目指し、従来技術1に開示された構成に対して、特許文献2に記載された接合界面特性を向上させる技術を組み合わせた高抵抗バッファ層を実験により作製した。かかる構成を、本明細書では比較例と称する。比較例および本実施例では、第1のバッファ層141、第2のバッファ層142、第3のバッファ層143が積層される3層から構成される。
【0025】
図2を参照して、本実施例および比較例による高抵抗バッファ層14の製造方法について説明する。
【0026】
図2のステップS21において、基板11上に金属裏面電極層12、光吸収層13を製膜した基板を準備する。
【0027】
ステップS22では、光吸収層13上に、本実施例の第1のバッファ層141として1〜3nmのZnS膜を、比較例の第1のバッファ層141として6および10nmのZnS膜を、CBD法によって製膜する。具体的には、酢酸亜鉛を所定の液温の水酸化アンモニウムに溶解して亜鉛アンモニウム錯塩を形成し、その溶液中に硫黄含有塩であるチオ尿素(チオウレア)を溶解し、この溶液に光吸収層13が製膜された基板11を所定の時間浸漬する。本実施例1乃至3、比較例1および2の各々の、溶液の液温および浸漬時間を表1に示す。なお、溶液中のアンモニアと酢酸亜鉛の濃度はそれぞれ7.5Mと0.16Mである。
【0028】
【表1】
【0029】
本実施例および比較例では、液温を70℃に固定して、浸漬時間を変化させることにより、第1のバッファ層の膜厚を調整した。なお、溶液の液温は60℃〜80℃の範囲であればよく、その場合、液温に応じて浸漬時間を調整することにより、第1のバッファ層の膜厚を制御可能となる。例として、液温が80℃の場合では、浸漬時間を5分にすることにより、2乃至3nmの第1のバッファ層141を製膜することができる。
【0030】
ステップS23では、基板を浸漬している溶液に対して、所定の時間間隔で酢酸を追加する。これにより、溶液中のpHが中性に近づき、製膜されるバッファ層(第2のバッファ層142)の組成が、ZnSからZnOに連続的に変化した硫黄含有亜鉛混晶化合物半導体薄膜が形成される。
【0031】
本実施例1〜3、比較例1および2におけるステップS23での浸漬時間は60分であり、その間に段階的に酢酸を追加した。なお、本実施例および比較例では、溶液のpHが11.0から9.0に変化するように酢酸を所定間隔で追加し、これにより、第2のバッファ層142のような、組成がZnSからZnOに連続的に変化した薄膜を形成可能となる。なお、ステップS23の浸漬時間について、ステップS22と同様に、溶液を変化させた場合は、浸漬時間を調整することにより、所望の膜厚の第2のバッファ層142を得ることが可能である。具体的には、本実施例では液温が70℃、浸漬時間が60分で第2のバッファ層142を形成したが、浸漬時間を短くすることにより、20nm未満の膜厚にすることが可能であり、さらに、液温を70℃よりも高くした場合は、浸漬時間が60分以下で膜厚20nm以下の第2のバッファ層142を形成可能性となる。
【0032】
他の実施形態では、第2のバッファ層142をCBDで製膜するにあたり、第1のバッファ層141を製膜した後、溶液中のアンモニアを蒸発させながら第2のバッファ層142を製膜することにより、層が成長する途中で、溶液のpHが中性に近づき、第2のバッファ層142の組成をZnSからZnOに変化させることができる。また、第2のバッファ層142の溶液に、段階的に酢酸亜鉛を添加することで、溶液のpHが中性に近づき、第2のバッファ層142の組成をZnSからZnOに変化させることができる。
【0033】
更に他の実施形態としては、ALD(Atomic Layer Deposition)法を用いてZnSからZnOに変化するように1原子層づつ堆積させる方法がある。具体的には、Zn源にジエチル亜鉛、S源に硫化水素、O源にH2Oを用いることで、組成がZnSからZnOに連続的に変化した第2のバッファ層142を製膜することが可能となる。
【0034】
次に、ステップS24では、第2のバッファ層142が製膜された基板を大気中で設定温度200℃で15分間アニールすることで乾燥し、かつ、膜中の水酸化亜鉛の一部を酸化亜鉛に転化すると同時に硫黄により改質する。これにより、第1のバッファ層141および第2のバッファ層142を高品質化できる。
【0035】
ステップS25では、第2のバッファ層142上に、MOCVD法によって膜厚が100nm以上の酸化亜鉛系薄膜を、第3のバッファ層143として形成する。この第3のバッファ層143は、透明導電膜15に接して形成される。このため、第3のバッファ層143は、ドーピング不純物元素として、アルミニウム(Al)、ガリウム(Ga)、ホウ素(B)などを含むが、これらのドーピング不純物元素濃度を、1×1019atoms/cm3以下、より好ましくは1×1018atoms/cm3以下となるように調整することにより、バッファ層として好ましい高抵抗な膜となる。この第3のバッファ層143の抵抗率は、0.1Ωcm以上、より好ましくは1Ωcm以上となっている。
【0036】
なお、本実施例においては、MOCVD法によって第3のバッファ層143を製膜したが、本発明はこれに限らず、スパッタ法などにより形成することもできる。
【0037】
<実験結果>
上記各ステップにより形成された第1のバッファ層141の製膜結果は、比較例では第1のバッファ層141はZnSからなり、膜厚は6nm(比較例1)および10nm(比較例2)となった。これに対して、本願発明に係る実施例における第1のバッファ層141はZnSからなり、膜厚は1nm(実施例1)、2nm(実施例2)および3nm(実施例3)となった。第2のバッファ層142の組成はいずれも、金属裏面電極層12側から透明導電膜15側にかけてZnSからZnOと変化する組成となり、膜厚は20nm以下である。第3のバッファ層143はいずれも、ZnOからなり、膜厚は100nm以上である。
【0038】
上記比較例1,2と実施例1乃至3により作製した高抵抗バッファ層14を備えた太陽電池について特性を比較した結果を図3及び図4に示す。なお具体的な数値を表2に示す。
【0039】
【表2】
【0040】
図3、4及び表2に示したとおり、従来技術1および2を単に組み合わせただけの比較例1および2にかかる太陽電池については、十分に高い曲線因子(FF)が得られなかった。これに対して、第1のバッファ層141の膜厚を3nm以下とした本実施例1乃至3の太陽電池は、良好な曲線因子(FF)が確認できる。同様にRs[Ω cm2]についても、比較例1および2に比べ、第1のバッファ層141の膜厚を3nm以下とした本実施例1乃至3の太陽電池は良好な結果が得られた。
【0041】
ここで、従来技術1には、CBDバッファ層(本発明の第1のバッファ層に相当)の膜厚について10nm以下と記載されているにすぎず、10nm以下の範囲において、第1のバッファ層の膜厚によってFFが変化することに関する知見はなく、従来技術1の出願時において発明者もかかる知見を認識していなかった。また、従来技術1における実施形態では、80℃の溶液中に10分間接触させてCBDバッファ層141(本発明の第1のバッファ層に相当)を製膜することが開示されているが、この方法で製膜したCBDバッファ層141の膜厚は、6nm以上になる。これは、溶液の温度が80℃であり、本発明にかかる溶液の温度よりも高く、製膜速度が高いためである。
【0042】
また、従来技術2では、ZnSからなるバッファ層の膜厚については記載されておらず、第1段階の処理で製膜されたバッファ層が、本発明の第1のバッファ層141に相当すると見ても、この第1段階の処理では、基材(CIS系光吸収層が製膜された基板)を80℃の溶液に15分間投入しており、第1段階の処理で製膜されたバッファ層の膜厚は、10nm以上になる。
【0043】
このように、従来技術1および従来技術2を単に組み合わせた構成では、第1のバッファ層141の膜厚は6nm以上となり、言い換えれば、比較例1および2の太陽電池を得られるにすぎない。この比較例1および2に対して、本実施例1〜3の太陽電池に示したとおり、第1のバッファ層の膜厚を3nm以下とすることにより、太陽電池特性の一つである曲線因子を向上させることが可能となる。
【0044】
上記事実に鑑みて比較例と本願発明に係る実施例との比較をエネルギーバンド図にて検討する。対比として、参考までに従来技術1の条件で作成した層構造のエネルギーバンド図を図5に示し、比較例のエネルギーバンド図を図6に示す。さらに、図7に本願発明に係る実施例によるエネルギーバンド図を示す。
【0045】
図5に示した従来技術1におけるエネルギーバンド図からわかるように、CBDバッファ層とMOCVDバッファ層との界面でのバンド構造に障壁があるのがわかる。図6に示した比較例におけるエネルギーバンド図は、従来技術1の構造をベースに、従来技術2の技術的特徴(ZnO膜とZnS膜との間に、組成が連続して変化する層を設ける)を単に組み合わせただけである。上述の通り、かかる比較例では、FFの大きな向上には繋がらない。これに対して、第1のバッファ層141を3nm以下とした本願発明に係る実施例によるエネルギーバンド図は図7に示したようになり、その結果は上述したとおり、良好なFFが得られる。
【0046】
また最も好適なFFおよびRsの結果を示した、膜厚が2nmの第1のバッファ層141を備える実施例2を撮影した結果を図8に示す。図8(a)は走査透過電子顕微鏡(STEM:Scanning Transmission Electron Microscope)を使用した画像であり、図8(b)は電子エネルギー損失分光法(EELS:Electron Energy Loss Spectroscopy)を使用した硫黄マッピング像である。図8(b)において白く現れているのが硫黄であり、2nmの極薄膜の第1のバッファ層141が形成され、さらに、第2のバッファ層142において、CIS光吸収層13側から透明導電膜15側に向かって硫黄濃度が減少しているのが確認できる。
【特許請求の範囲】
【請求項1】
CIS系光吸収層、バッファ層、透明導電膜の順に積層された構造を備えたCIS系薄膜太陽電池であって、
前記バッファ層が、第1のバッファ層、第2のバッファ層、第3のバッファ層の順に積層された3層の積層構造からなり、
前記第1のバッファ層が、厚さ1乃至3nmのZnSからなり、
前記第2のバッファ層が、厚さ20nm以下であって、前記CIS系光吸収層から前記透明導電膜へ向かってZnSからZnOに連続的に組成が変化する薄膜からなり、
前記第3のバッファ層が、厚さ100nm以上のZnOからなる
ことを特徴とするCIS系薄膜太陽電池。
【請求項2】
上記CIS系光吸収層が、銅、インジウム、ガリウム、セレン、硫黄から構成されていることを特徴とする、請求項1に記載のCIS系薄膜太陽電池。
【請求項1】
CIS系光吸収層、バッファ層、透明導電膜の順に積層された構造を備えたCIS系薄膜太陽電池であって、
前記バッファ層が、第1のバッファ層、第2のバッファ層、第3のバッファ層の順に積層された3層の積層構造からなり、
前記第1のバッファ層が、厚さ1乃至3nmのZnSからなり、
前記第2のバッファ層が、厚さ20nm以下であって、前記CIS系光吸収層から前記透明導電膜へ向かってZnSからZnOに連続的に組成が変化する薄膜からなり、
前記第3のバッファ層が、厚さ100nm以上のZnOからなる
ことを特徴とするCIS系薄膜太陽電池。
【請求項2】
上記CIS系光吸収層が、銅、インジウム、ガリウム、セレン、硫黄から構成されていることを特徴とする、請求項1に記載のCIS系薄膜太陽電池。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2012−4287(P2012−4287A)
【公開日】平成24年1月5日(2012.1.5)
【国際特許分類】
【出願番号】特願2010−137035(P2010−137035)
【出願日】平成22年6月16日(2010.6.16)
【出願人】(000186913)昭和シェル石油株式会社 (322)
【Fターム(参考)】
【公開日】平成24年1月5日(2012.1.5)
【国際特許分類】
【出願日】平成22年6月16日(2010.6.16)
【出願人】(000186913)昭和シェル石油株式会社 (322)
【Fターム(参考)】
[ Back to top ]