
CRTH2受容体アンタゴニストを用いたニューロパシー性疼痛処置方法
本発明は、ニューロパシー性疼痛の処置用の薬剤の製造におけるCRTH2受容体アンタゴニストの使用、およびCRTH2受容体アンタゴニストを用いてニューロパシー性疼痛を処置する方法に関する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ニューロパシー性疼痛の処置用の薬剤の製造におけるCRTH2受容体アンタゴニストの使用、およびCRTH2受容体アンタゴニストを用いてニューロパシー性疼痛を処置する方法に関する。
【背景技術】
【0002】
1999年、Nagata et al は、白血球化学誘引物質受容体ファミリーに属する新規なGタンパク質共役型受容体(GPCR)である、GPR44としても従来知られているCRTH2(Th2細胞で発現された化学誘引物質受容体相同分子)を識別した(Nagata et al., FEBS Letters (1999) 459(2):195-9)。CRTH2受容体は、マウスの場合、脳、肺およびリンパ系器官を含めた広範囲の組織から選択的に発現される(Abe et al., Gene (1999) 227(1):71-7)。CRTH2受容体は、ヒトの場合、Th2細胞、好酸球および好塩基球で選択的に発現されるが、Th1細胞、B細胞およびNK細胞では発現されない(Nagata et al., FEBS Letters (1999) 459(2):195-9)。
【0003】
Bauer et al,(EP1170594A2号を参照されたい)は、プロスタグランジンD2(PGD2)を、CRTH2受容体のアゴニストである内因性リガンドとして識別した。PGD2は、免疫学的に刺激されたマスト細胞およびTh2細胞から放出される。CRTH2とPGD2との相互作用は、アレルギー性炎症の標的組織におけるTh2細胞のアレルゲンで誘発される動員に臨界的役割を果たすことが知られている。更に、CRTH2は、血中好酸球および好塩基球のPGD2依存性細胞移動を媒介する。したがって、CRTH2受容体は、炎症およびアレルギー性反応を与える分子イベントにおいて活性な役割を果たすことが分かった。CRTH2のPGD2依存性活性と相互作用する化合物は、免疫系の異常な活性化に関連したアレルギー性疾患状態および炎症の処置に有用であると考えられる。
【0004】
Torisu et al.,(WO03022814号を参照されたい)は、PGD2受容体、特に、DP受容体に特異的に結合するインドール誘導体を識別した。その化合物は、CRTH2受容体に結合し且つ生物学的活性に拮抗すると考えられるので、疼痛の防止および/または処置に有用であると考えられる。
【0005】
Pairaudeau et al.,(WO2004089884号を参照されたい)は、呼吸器障害を処置するのに有用な医薬化合物としての置換されたフェノキシ酢酸;それらを含有する医薬組成物;およびそれらの製造方法を開示した。化合物は、薬剤としての、具体的には、CRTH2受容体活性のモジュレーターとしての活性を有すると考えられ、したがって、PGD2およびその代謝産物の過度のまたは未調節の生産によって引き起こされるまたは悪化するヒトおよび非ヒト動物の状態/疾患の(治療的または予防的)処置に用いうると考えられ;この文書によれば、このような状態の例には、ニューロパシー性疼痛症候群が含まれる。
【0006】
驚くべきことに、本発明者は、CRTH2受容体のアンタゴニストである化合物が、ニューロパシー性疼痛の処置に有効であるということを発見した。
多数の異なった疼痛状態、例えば、慢性疼痛、ニューロパシー性疼痛、炎症性疼痛、侵害受容性疼痛、内臓痛、背痛、および疾患および変性に関連した疼痛がある。当業者は、更に、これら疼痛タイプが、臨床的におよび機械論的に異なるということを承知している。このような状態は、多数の疼痛症状のために、しばしば、臨床的に処置するのが困難である。例えば、(糖尿病性ニューロパシー、または末梢神経またはCNSへの外傷などの疾患に起因することがありうる状態である)ニューロパシー性疼痛の患者は、しばしば、痛覚過敏(hyperlagesia)(有害刺激への過大疼痛)、過感作、異痛症(以前の無害刺激による疼痛)、更には、継続している疼痛を含めた多数の疼痛症状を示す。更に、ニューロパシー性疼痛は、防御的役割がないので、病的である。それは、しばしば、最初の原因が消散した後によく現れる。
【0007】
侵害受容性疼痛は、組織損傷によって、または損傷を引き起こす可能性のある強い刺激によって誘発される。疼痛求心性神経(afferents)は、損傷部位にある侵害受容器による刺激の伝達によって活性化され、それらの終末レベルで脊髄を感作する。次に、これは、脊髄路を脳へと中継され、そこで疼痛が知覚される(Meyer et al., 1994 Textbook of Pain 13-44)。侵害受容器の活性化は、二つのタイプの求心性神経線維を活性化する。有髄A−δ線維は、急速伝達し、鋭く且つ刺すような痛覚に関与するが、無髄C線維は、より遅い速度で伝達し、鈍いまたはうずく痛みを伝える。損傷が修復された時、疼痛は止む。中程度〜重症の急性侵害受容性疼痛は、挫傷/捻挫による疼痛、術後疼痛(いずれかのタイプの外科的処置後の疼痛)、外傷後疼痛、熱傷、心筋梗塞、急性膵炎および腎疝痛の顕著な特徴であるが、これに制限されるわけではない。更に、癌関連急性疼痛症候群は、一般的には、化学療法毒性、免疫療法、ホルモン療法および放射線療法などの治療的相互作用のためである。中程度〜重症の急性侵害受容性疼痛は、腫瘍関連疼痛でありうる癌疼痛(例えば、骨痛、頭痛および顔面痛、内臓痛)または癌療法に関連した疼痛(例えば、化学療法後症候群、慢性術後疼痛症候群、照射後症候群)、ヘルニア様のまたは断裂した椎間円板によるまたは腰椎間関節、仙腸関節、傍脊柱筋または後縦靱帯の異常によることがありうる背痛の顕著な特徴であるが、これに制限されるわけではない。
【0008】
炎症過程は、組織損傷または異物の存在に応答して活性化される一連の複雑な生化学的および細胞性のイベントであり、それが、腫脹および疼痛を引き起こす(Levine and Taiwo 1994: Textbook of Pain 45-56)。関節炎痛は、炎症性疼痛集団の大部分を構成している。リウマチ様疾患は、先進諸国において最も一般的な慢性炎症性状態の一つであり、関節リウマチは、障害の共通原因である。RAの正確な病因は未知であるが、現在の仮説は、遺伝的および微生物学的双方の因子が重要でありうるということを示唆している(Grennan & Jayson 1994 Textbook of Pain 397-407)。ほぼ1600万人の米国人が、症候性変形性関節症(OA)または変性関節疾患を有すると推定されたが、その大多数は60歳を超えているので、その集団の年齢が増加するにつれて、これは4000万人へと増加して、これをきわめて大きな公衆衛生問題にすると考えられる(Houge & Mersfelder 2002 Ann Pharmacother. 36: 679-686; McCarthy et al., 1994 Textbook of Pain 387-395)。大部分のOA患者は、疼痛のために、医療行為を探し求めている。関節炎は、心理・社会的および身体的機能に有意の影響力があり、晩年における障害の主原因であることが知られている。他のタイプの炎症性疼痛には、炎症性腸疾患(IBD)が含まれるが、これに制限されるわけではない。
【0009】
対照的に、ニューロパシー性疼痛の臨床的特性は、神経病理学的過程自体の機構、場所および重症度によって優先的に決定される。ニューロパシー性疼痛は、神経系の一次病変または機能不全によって開始するまたは引き起こされる疼痛として定義される(IASP定義)。神経損傷は、外傷および疾患によって引き起こされることがありうるので、「ニューロパシー性疼痛」という用語は、多様な病因を有する多数の障害を包含する。これらには、糖尿病性ニューロパシー、帯状疱疹後神経痛、背痛、癌ニューロパシー、HIVニューロパシー、幻想肢痛、手根管症候群、慢性アルコール症、甲状腺機能低下症、三叉神経痛、尿毒症またはビタミン欠乏症が含まれるが、これに制限されるわけではない。ニューロパシー性疼痛は、防御的役割がないので、病的である。それは、しばしば、最初の原因が消散した後によく現れ、一般的に何年間も続き、患者の生活の質を有意に低下させる(Woolf and Mannion 1999 Lancet 353:1959-1964)。ニューロパシー性疼痛の症状は、しばしば、同じ疾患の患者間でさえも異種であるので、処置するのが困難である(Woolf & Decosterd 1999 Pain Supp. 6: S141-S147; Woolf and Mannion 1999 Lancet 353:1959-1964)。それらには、継続することがありうる自発痛、または痛覚過敏(有害刺激への増加した感受性)および異痛症(通常は無害な刺激への感受性)などの発作性で且つ異常な誘発痛が含まれる。
【0010】
更に、抗炎症薬およびアヘン薬のような、侵害受容性疼痛を処置するのに慣用的に用いられる薬物は、慢性ニューロパシー性疼痛患者への効力が限られている。したがって、抗痙攣薬および三環系抗うつ薬は、しばしば、不十分に耐性であるにもかかわらず、ニューロパシーへの主な鎮痛薬である。侵害受容性疼痛および炎症性疼痛とは対照的に、ニューロパシー性疼痛は、公然として処置するのが困難であり、慢性経過をたどるが;それは、非ステロイド性抗炎症薬およびアセトアミノフェンのような、侵害受容性疼痛の処置に有効である標準的な鎮痛療法には全くまたはきわめて不十分にしか応答しないし;そして侵害受容性疼痛状態の場合よりも、予想通りに且つ確実にオピオイドに応答することに乏しい。侵害受容性疼痛に有効な処置は、ニューロパシー性疼痛へと広げられるとは考えられない。例えば、Gabapentin(Neurontin(登録商標))および Pregabalin(Lyrica(登録商標))は、慢性狭窄性坐骨神経損傷(CCI)およびストレプトゾシンで誘発される糖尿病(STZ)のラットモデルにおいて静的および動的双方の異痛症を逆転させるが、モルヒネは、CCIラットモデルにおいて、動的ではなく静的異痛症を逆転させる(Field MJ, et al, 1999, Pain, 83: 303-311)。更に、慢性疼痛の場合の非ステロイド性抗炎症薬(NSAID)およびコルチコステロイド類(デキサメタゾンおよびプレドニゾン)の効能は疑わしく、齧歯類動物かまたは患者での一貫した薬理学的証拠によって支持されない。同様に、臨床データは、ニューロパシー性疼痛疾患におけるこれら薬剤の限られた使用を示し、潜在的に、これら化合物を用いた比較的少数の齧歯類動物研究を説明する。Schafers(2004, Experimental Neurology, 185: 160-168)は、ラットのCCIで誘発される疼痛を逆転させる場合の非選択的(イブプロフェン)および選択的(Celebrex(登録商標)TM)COX2阻害剤の有意の作用を示さなかった。
【0011】
これら理由、臨床的特性の相違、機構の相違、および処置への従順性の相違のために、ニューロパシー性疼痛は、当業者の判断で、侵害受容性疼痛および炎症性疼痛とは明らかに異なると考えられるであろう。
【0012】
したがって、これら疼痛状態の原因となるニューロパシー性疼痛過程のキー段階を妨げる薬学的に活性な化合物を識別することが、救急医学的に要求されている。
したがって、中枢神経系(CNS)において中心的に発現される疼痛経路に関与する標的受容体を識別すること、およびCNSおよび関連組織において中枢作用することによって鎮痛作用を及ぼす薬学的に活性な化合物を識別することは、好都合である。CRTH2受容体は、皮質、視床、扁桃および脊髄が含まれるが、必ずしもこれに制限されるわけではないCNS組織において中心的に発現されること、更には、多数の末梢組織でも発現されることが分かった(Nagata and Hirai (2003) prostaglandins, leukotrienes and Essential Fatty acids 69: 169-177)。CRTH2は、ヒトの脳および脊髄でも発現される。
【0013】
支持組織分布データは、Abe et al., (1999) Gene 227: 71-77 に記載のようにマウスにおいて、そして更には、Shichijo et al., (2003) JPET 307: 518-525 に記載のようにラットにおいて、CRTH2受容体について提供された。
【発明の開示】
【課題を解決するための手段】
【0014】
発明の簡単な説明
本発明は、ニューロパシー性疼痛の処置用の薬剤の製造におけるCRTH2受容体アンタゴニストの使用に関する。
【0015】
本発明は、更に、哺乳動物対象のニューロパシー性疼痛を処置する方法であって、その対象に、治療的有効量のCRTH2受容体アンタゴニストを投与することを含む方法を提供する。
【0016】
本発明は、更に、ニューロパシー性疼痛の処置用のCRTH2受容体アンタゴニストを提供する。
発明の詳細な説明
「CRTH2リガンド」または「CRTH2受容体リガンド」という用語は、CRTH2受容体に結合する化合物を意味する。このような化合物は、有機または無機の化合物類似体またはそれらの立体異性体;または天然または合成の他の化学的または生物学的な化合物、例えば、天然プロスタグランジン;ペプチド;ポリペプチド;抗体および抗体リガンド結合性ドメインを含めたタンパク質;ホルモン;ヌクレオチド;DNAまたはRNAなどの核酸であってよく、そして更には、化合物または立体異性体の薬学的に許容しうる塩、化合物または立体異性体のプロドラッグ、またはプロドラッグの薬学的に許容しうる塩を包含する。CRTH2受容体リガンドは、CRTH2受容体アンタゴニストでもありうる。
【0017】
本明細書中で用いられる「CRTH2受容体アンタゴニスト」という用語は、CRTH2受容体の活性化をブロックするように作用する化合物を意味する。適するアンタゴニストの例には、天然プロスタグランジンなどの有機化合物またはそれらの類似体、または他の化合物、有機または無機の分子、ペプチド、抗体および抗体のリガンド結合性ドメインを含めたタンパク質、DNAまたはRNAなどの核酸が含まれる。CRTH2受容体アンタゴニストの適する例は、例えば、有機化合物、またはペプチドまたはタンパク質、抗体およびそれらのフラグメント、ペプチド模擬有機化合物であって、例えば、CRTH2受容体の細胞外ドメイン(ECD)に結合し且つ天然リガンドによって引き起こされる活性を阻害するものでありうる。更に、有機化合物、ペプチド、抗体またはそれらのフラグメントであって、CRTH2受容体のECD(またはその一部分)が共有結合するものは、更に、PGD2に結合し、したがって、それを「中和する」ことがありうる。アンタゴニストという用語は、ランダムペプチドライブラリー(例えば、Lam et al., 1991, Nature 354:82-84; Houghten et al., 1991, Nature 354:84-86 を参照されたい)およびDおよび/またはL立体配置アミノ酸より構成される組合せ化学由来分子ライブラリーのメンバーが含まれるがこれに制限されるわけではないペプチドおよび可溶性ペプチド;ホスホペプチド(ランダムまたは部分変性に支配されたホスホペプチドライブラリーのメンバーが含まれるがこれに制限されるわけではない;例えば、Songyang et al., 1993, Cell 72:767-778 を参照されたい);抗体(多クローン性、単クローン性、ヒト化、抗イディオタイプ、キメラまたは短鎖の抗体、およびFab、F(ab’)2およびFAb発現ライブラリーフラグメントおよびそれらのエピトープ結合性フラグメントが含まれるがこれに制限されるわけではない);および有機または無機の小分子を包含する。適するアンタゴニストは、更に、ランダムまたは組合せのペプチドまたは非ペプチドなどの多様性ライブラリーに由来してよく、用いることができるライブラリー、例えば、化学的に合成されたライブラリー、リコンビナント(例えば、ファージ表示ライブラリー)および in vitro 翻訳に基づくライブラリーはいずれも、当該技術分野において知られている。化学的に合成されたライブラリーの例は、Fodor et al., 1991, Science 251:767-773; Houghten et al., 1991, Nature 354:84-88; Lam et al., 1991, Nature 354:82-84; Medynski, 1994, Bio/Technology 12:709-710; Gallop et al., 1994, J. Medicinal Chemistry 37(9):1233-1251; Ohlmeyer et al., 1993, Proc. Natl. Acad. Sci. USA 90:10922-10926; Erb et al., 1994, Proc. Natl. Acad. Sci. USA 91:11422-11426; Houghten et al., 1992, Biotechniques 13:412; Jayawickreme et al., 1994, Proc. Natl. Acad. Sci. USA 91:1614-1618; Salmon et al., 1993, Proc. Natl. Acad. Sci. USA 90:11708-11712; PCT公開WO93/20242号;および Brenner and Lerner, 1992, Proc. Natl. Acad. Sci. USA 89:5381-5383 に記載されている。
【0018】
ファージ表示ライブラリーの例は、Scott & Smith, 1990, Science 249:386-390; Devlin et al., Science 249:404-406; Christian, et al., 1992, J. Mol. Biol. 227:711-718; Lenstra, 1992, J. Immunol. Meth. 152:149-157; Kay et al., 1993, Gene 128:59-65; および1994年8月18日付のPCT公開WO94/18318号に記載されている。
【0019】
非ペプチドライブラリーの例として、ベンゾジアゼピンライブラリー(例えば、Bunin et al., 1994, Proc. Natl. Acad. Sci. USA 91:4708-4712 を参照されたい)は、使用に適応することができる。ペプトイドライブラリー(Simon et al., 1992, Proc. Natl. Acad. Sci. USA 89:9367-9371)を用いることもできる。用いることができるライブラリーの別の例であって、ペプチド中のアミド官能基を過メチル化して、化学変換された組合せライブラリーを生じたものは、Ostresh et al.(1994, Proc. Natl. Acad. Sci. USA 91:11138-11142)によって記載されている。
【0020】
それらライブラリーをスクリーニングすることは、種々の一般的に知られている方法のいずれによっても行うことができる。例えば、ペプチドライブラリーのスクリーニングを開示している次の参考文献:Parmley & Smith, 1989, Adv. Exp. Med. Biol. 251:215-218; Scott & Smith, 1990, Science 249:386-390; Fowlkes et al., 1992; BioTechniques 13:422-427; Oldenburg et al., 1992, Proc. Natl. Acad. Sci. USA 89:5393-5397; Yu et al., 1994, Cell 76:933-945; Staudt et al., 1988, Science 241:577-580; Bock et al., 1992, Nature 355:564-566; Tuerk et al., 1992, Proc. Natl. Acad. Sci. USA 89:6988-6992; Ellington et al., 1992, Nature 355:850-852; 全て Ladner et al. による米国特許第5,096,815号、米国特許第5,223,409号および米国特許第5,198,346号;Rebar & Pabo, 1993, Science 263:671-673; およびPCT公開WO94/18318号を参照されたい。
【0021】
CRTH2受容体アンタゴニストである化合物は、CRTH2受容体上の、PGD2が通常結合する同じ部位で結合し且つ作用することができるが、それは、PGD2結合部位に遠いCRTH2上の部位で作用することができる。CRTH2受容体のアンタゴニストは、CRTH2受容体活性化を、いずれか適する手段によって、例えば、CRTH2受容体にまたはPGD2またはいずれか他の活性化リガンドに結合することなどによってブロックするように作用し、それによって、PGD2または活性化リガンドとCRTH2受容体との結合を阻害することができる。このようなアンタゴニストは、CRTH2受容体においてPGD2の代わりに作用することができるし、またはPGD2と相互作用する、PGD2と一緒になる、またはそれ以外には、PGD2を修飾することによって、CRTH2受容体においてそれが作用する方法に影響を与えることができる。或いは、アンタゴニストは、CRTH2受容体下流活性を、例えば、CRTH2受容体シグナル伝達のモジュレーションによってブロックするように作用して、下流シグナリングイベントに影響を与えることができるが、この活性は、例えば、PGD2によってまたはCRTH2受容体のいずれか他の活性化リガンドによって活性化されるようなシグナルの伝達を妨げることができるGタンパク質の阻害剤に共通している。或いは、アンタゴニストは、CRTH2受容体活性を、CRTH2受容体遺伝子発現に影響を与えることによってブロックするように作用することができるが、このようなアンタゴニストには、例えば、分子、タンパク質または小有機分子、またはDNAまたはRNAが含まれるが、それは、転写に影響を与えるまたはスプライシングイベントを妨げるので、完全長または切断された形のCRTH2受容体の発現に影響することがありうる。したがって、このようなCRTH2受容体アンタゴニストには、アンチセンスRNAおよびsRNA産物(サイレンス妨害RNA)も含まれうる。
【0022】
本発明での使用に適するCRTH2受容体アンタゴニストの例には、Annex 1に添付の特許出願PCT/IB03/04505号に概括的ににまたは具体的に開示された化合物、特に、cis−N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドという化合物およびその薬学的に許容しうる塩および溶媒和化合物が含まれる。
【0023】
本発明での使用に適するCRTH2受容体アンタゴニストの更に別の例には、特許出願WO−2004007451号に概括的ににまたは具体的に開示された化合物、3−スルホニルインドール誘導体およびそれらの塩、特に、2−[5−クロロ−3−(4−クロロフェニルスルホニル)−2−メチル−1H−インドール−1−イル]酢酸という化合物が含まれる。
【0024】
更に、特許出願WO03066047号は、インドール−3−酢酸誘導体およびそれらの塩である、本発明での使用に適するCRTH2受容体アンタゴニストの更に別の例、特に、2−[1−(2,6−ジフェノキシピリミジン−4−イル)−2,5−ジメチル−1H−インドール−3−イル]酢酸という化合物を開示している。特許出願WO03101981号は、更に適するCRTH2受容体アンタゴニスト、置換されたインドール−1−イル酢酸誘導体、特に、3−(1,2−ベンゾイソチアゾール−3−イル)−5−フルオロ−2−メチル−1H−インドール−1−酢酸という化合物を開示している。WO03101961号は、適するCRTH2受容体アンタゴニストの追加の例、置換されたインドール化合物、特に、3−[(3−メトキシフェニル)チオ]−2,5−ジメチル−1H−インドール−1−酢酸という化合物を開示し、そしてWO03066046号は、適するCRTH2受容体アンタゴニストの追加の例、インドール−3−酢酸誘導体、特に、1−(7−クロロキナゾリン−4−イル)−2−メチル−5−(1−メチルエチル)−1H−インドール−3−酢酸という化合物を開示している。適するCRTH2受容体アンタゴニストの更に別の例には、特許出願WO03097042号に概括的ににまたは具体的に開示された化合物、特に、Ramatroban という化合物、そしてWO03097598号の、特に、(3−[1−(4−フルオロベンゼンスルホニル)ピロリジン−3−イル]インドール−1−イル)酢酸という化合物が含まれる。適するCRTH2受容体アンタゴニストの更に別の例には、CRTH2受容体への抗体または抗体サブドメイン、具体的には、抗CRTH2受容体単クローン性抗体または抗体サブドメイン、例えば、CRTH2受容体に特異的な抗体またはサブドメイン、またはPGD2によって一部分与えられたエピトープに特異的な抗体またはサブドメインが含まれる。
【0025】
好ましくは、本発明によるCRTH2受容体アンタゴニストは、中枢作用性である。中枢作用性であるためには、このような化合物は、血液脳関門を貫通することができるはずである。
【0026】
本発明での使用に好適なCRTH2受容体アンタゴニストは、一般式(I):
【0027】
【化1】
【0028】
(式中、R1は、H、(C1〜C4)アルキル、(C2〜C4)アルケニル、(C2〜C4)アルキニルまたは(CH2)mRxであり;
Rxは、het1,フェニルまたは(C3〜C6)シクロアルキルであり、このhet1,フェニルおよび(C3〜C6)シクロアルキルは、1個またはそれを超えるQ1基または(C1〜C4)アルキル基で置換されていてよく、この(C1〜C4)アルキルは、1個またはそれを超えるQ1基で置換されていてよく;
Q1は、ハロゲン、NO2、CN、SO2CH3、SO2NR9R10、OR9、COOR9、C(=O)NR9R10、NR9R10、NR9SO2R10、NR9C(=O)R10またはC(=O)R9であり、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
mは、0、1および2より選択される整数であり;
R2は、(C1〜C4)アルキルであり、ここにおいて、アルキル基は、ハロゲン、OR9、NR9R10、COOR9、C(=O)NR9R10、NHSO2R9およびC(=O)(C1〜C4)アルキルより選択される1個またはそれを超える置換基で置換されていてよく、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
R3は、(C3〜C6)シクロアルキルまたは−A−Ryであり;
Aは、結合、直鎖または分岐状の(C1〜C3)アルキレンまたは(C2〜C3)アルケニレンであり;
Ryは、(C6〜C12)アリールまたはhet2であり、ここにおいて、アリール基およびhet2基は、(C6〜C12)アリール、het1、Q2および(C1〜C4)アルキルより選択される1個またはそれを超える置換基で置換されていてよく、この(C1〜C4)アルキルは、1個またはそれを超えるQ2基であって、同じであるかまたは異なる基で置換されていてよく;
Q2は、ハロゲン、NO2、CN、SO2CH3、SO2NR9R10、OR9、SR9、OCH2CF3、COOR9、C(=O)NR9R10、NR9R10、NR9SO2R10、NR9C(=O)R10またはC(=O)R9であり、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
R4は、Hまたは(C1〜C4)アルキルであり;
R5、R6、R7およびR8は、同じであるかまたは異なり、H、Q3および(C1〜C4)アルキルより選択され、この(C1〜C4)アルキルは、1個またはそれを超えるQ3基であって、同じであるかまたは異なる基で置換されていてよく;
Q3は、ハロゲン、NO2、CN、SO2CH3、SO2NR9R10、OR9、SR9、COOR9、C(=O)NR9R10、NR9R10、NR9SO2R10、NR9C(=O)R10またはC(=O)R9であり、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
het1は、酸素、硫黄および窒素より選択される1〜4個のヘテロ原子を有する5〜10員芳香族複素環であり;そして
het2は、酸素、硫黄および窒素より選択される1〜4個のヘテロ原子を有する5〜10員の飽和、不飽和または部分飽和複素環式基である)
を有する化合物またはその薬学的に許容しうる塩または溶媒和化合物である。
【0029】
het1およびhet2は各々、好ましくは、酸素、硫黄および窒素より選択される1〜3個のヘテロ原子を含有する5員または6員芳香族複素環である。
具体的には、het1およびhet2について好適な定義は、イソオキサゾリル、オキサゾリル、チエニル、ピラゾリル、ピロリル、トリアゾリル、テトラゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、ピリジニル、ピラジニル、ベンゾオキサジアゾリルまたはピラゾロピリジニル、キノリニルおよびキノキサリニルである。
【0030】
(C6〜C12)アリールは、6〜12個の炭素原子を含有する芳香族炭素環を意味すると理解される。好適なアリール基はフェニルである。
CRTH2受容体をコードしているヌクレオチド配列およびアミノ酸は、当業者に知られており、受託番号AB008535として GenBank で見出されうる。
【0031】
好ましくは、本発明で用いるためのCRTH2受容体アンタゴニストは、選択的CRTH2受容体アンタゴニストである。
「選択的」という用語は、リガンドまたはアンタゴニストが、ある特定の受容体に、別の受容体へのリガンドまたはアンタゴニストの結合親和性と比較した場合、より大きい親和性で結合するということを意味する。好ましくは、第一受容体へのアンタゴニストの結合親和性は、第二受容体への結合親和性より約50%またはそれを超えて大きい。より好ましくは、第一受容体へのアンタゴニストの結合親和性は、第二受容体への結合親和性より約75%またはそれを超えて大きい。最も好ましくは、第一受容体へのアンタゴニストの結合親和性は、第二受容体への結合親和性より約90%またはそれを超えて大きい。本発明の好ましい態様において、アンタゴニストは、CRTH2受容体への一層大きい結合親和性を示す。具体的には、好適なアンタゴニストは、ケモカイン受容体ファミリーのメンバー、例えば、C3a、C5a、FMLP、LTB4、GPCR0269、GPCR0232またはGPCR0288受容体のような、またはDタイププロスタノイド受容体(DP)のような、またはプロスタノイド受容体ファミリー、例えば、プロスタグランジンE2受容体サブタイプEP1〜EP4、プロスタグランジンF受容体、トロンボキサンA2受容体、最も好ましくは、DPのような別の受容体への結合と比較した場合、より大きい親和性でCRTH受容体に結合するものである。好適なアンタゴニストは、CRTH2受容体をマイクロモルまたはそれより大きい親和性で結合すると考えられる。より好適なアンタゴニストは、CRTH2受容体をナノモルまたはそれより大きい親和性で結合する。本発明の好適なCRTH2受容体アンタゴニストには、CRTH2受容体の選択的アンタゴニストであるリガンドまたは化合物が含まれる。選択性は、カルシウム動員などの機能性終点に基づいて決定することもできる。
【0032】
CRTH2受容体リガンドは、例えば、化合物ライブラリーをスクリーニングすることによって識別することができる。受容体のアンタゴニストを識別する方法は、当業者に周知である。CRTH2受容体リガンドを識別するのに用いることができる具体的な手順を、下に示す。
【0033】
生理学的疼痛は、外部環境からの潜在的に有害な刺激による危険を警告するように設計された重要な防御機構である。そのシステムは、ある一定の組の一次感覚ニューロンによって作動し、末梢伝達機構を経た有害刺激によって独占的に活性化される(統合的概説については、Millan 1999 Prog. Neurobio. 57:1-164)。これら感覚線維は、侵害受容器として知られ、遅い伝導速度を有する小径軸策を特徴とする。侵害受容器は、有害刺激の強さ、持続時間および質を、そして脊髄へのそれらの局所解剖学的に組織化された投射路によって、その刺激の場所をコードする。それら侵害受容器は、A−δ線維(有髄)およびC線維(無髄)という二つの主要タイプがある侵害受容神経線維上に見出される。侵害受容器入力によって生じる活性は、後角における複雑なプロセシング後、直接的にかまたは脳幹中継核(brain stem relay nyclei)を経て、腹底側視床へ、そして次に皮質へと移送され、そこで、痛みの感覚を生じる。
【0034】
強い急性痛および慢性痛は、病態生理学的過程によって駆動される同じ経路を含み、それ自体、防御機構を与えることを止め、代わりに、広範囲の疾患状態に関連した症状を弱くする原因となることがありうる。疼痛は、多数の外傷および疾患状態の特徴である。疾患または外傷によって体組織への実質的な損傷が起こった場合、侵害受容器活性化の特性は変化する。末梢では、損傷の周りの局所におよび侵害受容器が終端となるところでは中枢に鋭敏化が存在する。これは、損傷部位および付近の正常組織に過敏性をもたらす。急性痛の場合、これら機構は、修復過程が起こることを可能にしうるし且つそれに有用でありうるが、過敏性は、損傷がいったん治癒すると、正常に戻る。しかしながら、多数の慢性痛状態では、過敏性は、治癒過程をはるかに長く続け、通常は、神経系損傷のためである。この損傷は、しばしば、求心性線維の不適応をもたらす(Woolf & Salter 2000 Science 288: 1765-1768)。患者の症状の中で、不快および異常な感受性が特徴となっている場合、臨床的疼痛が存在する。患者は、全く異種である傾向があり、いろいろな疼痛症状を示すことがありうる。多数の典型的な疼痛サブタイプが存在する。(1)鈍い、焼けるようまたは刺すようでありうる自発痛;(2)有害刺激への過大疼痛応答(痛覚過敏);(3)通常は無害な刺激によって疼痛が生じる(異痛症)(Meyer et al., 1994 Textbook of Pain 13-44)。背痛、関節炎痛、CNS外傷またはニューロパシー性痛を有する患者は、類似した症状を有することがありうるが、根本的な機構は異なるので、異なった処置戦略を必要とすることがありうる。したがって、疼痛は、異なった病態生理学ゆえに、多数の異なった領域に分けることができ、これらには、侵害受容性疼痛、炎症性疼痛、ニューロパシー性疼痛等が含まれる。何等かのタイプの疼痛は、多数の病因を有するので、二つ以上の領域に分類することができるということは注目されるべきであり、例えば、背痛、癌性疼痛は、侵害受容性およびニューロパシー性双方の成分を有する。
【0035】
ニューロパシー性疼痛は、神経系の一次病変または機能不全によって開始するまたは引き起こされる疼痛として定義される(IASP定義)。神経損傷は、外傷および疾患によって引き起こされることがありうるので、「ニューロパシー性疼痛」という用語は、多様な病因を有する多数の障害を包含する。これらには、糖尿病性ニューロパシー、帯状疱疹後神経痛、背痛、癌ニューロパシー、HIVニューロパシー、幻想肢痛、手根管症候群、慢性アルコール症、甲状腺機能低下症、三叉神経痛、尿毒症またはビタミン欠乏症が含まれるが、これに制限されるわけではない。ニューロパシー性疼痛は、防御的役割がないので、病的である。それは、しばしば、最初の原因が消散した後によく現れ、一般的に何年間も続き、患者の生活の質を有意に低下させる(Woolf and Mannion 1999 Lancet 353: 1959-1964)。ニューロパシー性疼痛の症状は、しばしば、同じ疾患の患者間でさえも異種であるので、処置するのが困難である(Woolf & Decosterd 1999 Pain Supp. 6: S141-S147; Woolf and Mannion 1999 Lancet 353: 1959-1964)。それらには、継続することがありうる自発痛、または痛覚過敏(有害刺激への増加した感受性)および異痛症(通常は無害な刺激への感受性)などの発作性で且つ異常な誘発痛が含まれる。
【0036】
「治療的有効量」という用語は、疾患を処置する;特定の疾患の一つまたはそれを超える症状を改善する、減衰させるまたは除去する;または疾患の一つまたはそれを超える症状の開始を防止するまたは遅らせる化合物または化合物組合せの量を意味する。
【0037】
「患者」という用語は、イヌ、ネコ、ウシ、ウマ、ヒツジ、ガチョウおよびヒトなどの動物を意味する。具体的には、好適な患者は、双方の性のヒトを含めた哺乳動物である。
「薬学的に許容しうる」という用語は、物質または組成物が、製剤の他の成分と相溶性であるべきであり且つ患者に有害であってはならないということを意味する。
【0038】
「処置すること」、「処置する」または「処置」という用語は、防止的または予防的および待期的処置を包含する。
一次結合検定
CRTH2受容体リガンドおよびアンタゴニストは、例えば、化合物ライブラリーをスクリーニングすることによっておよびいろいろなスクリーニング技術を用いることによって識別することができる。受容体のリガンドおよびアンタゴニストを識別する方法は知られている。CRTH2受容体リガンドおよびアンタゴニストを識別するのに用いることができる具体的な手順は、下に示されるが、本明細書中に援用される欧州特許出願第01305857.3号(公開番号EP1170594号)に公表されている。
【0039】
CRTH2受容体のリガンドを識別する結合検定は、直接結合検定の形でかまたは競合結合検定として行うことができる。直接結合検定の場合、試験化合物を、CRTH2受容体への結合について調べる。もう一方において、競合結合検定は、CRTH2受容体への結合について、プロスタグランジンD2(PGD2)またはそのファミリーの他の適するリガンドと競合する試験化合物の能力を評価する。
【0040】
直接結合検定の場合、CRTH2受容体を、CRTH2受容体に試験化合物を結合させる条件下において試験化合物と接触させる。その結合は、溶液中または固体表面上で起こりうる。好ましくは、試験化合物を、検出用に予め標識する。発光体、蛍光体、または放射性同位体またはそれを含有する基、または酵素または染料などの非同位体標識のような検出可能な基はいずれも、標識するのに用いることができるが、これに制限されるわけではない。結合が起こるのに充分なインキュベーション時間後、その反応を、過度にまたは非特異的に結合した試験化合物を除去する操作および条件に暴露する。典型的には、これは、適当な緩衝液で洗浄することを伴う。最後に、CRTH2受容体−試験化合物複合体の存在を検出する。
【0041】
競合結合検定の場合、試験化合物を、CRTH2受容体へのPGD2の結合を破壊するまたは促進するそれらの能力について検定する。標識されたPGD2を、CRTH2受容体またはそのフラグメントまたは誘導体と混合し、そしてそれらの間の相互作用が、通常は生じると考えられる条件下に、試験化合物の添加を伴ってまたは伴うことなく置くことができる。CRTH2受容体を結合する標識PGD2の量は、試験化合物の存在下または不存在下において結合した量と比較することができる。
【0042】
好ましい態様において、複合体の形成および検出を容易にするために、結合検定は、固体表面上に固定された一つまたはそれを超える成分で行われる。いろいろな態様において、その固体支持体は、ポリカーボネート、ポリスチレン、ポリプロピレン、ポリエチレン、ガラス、ニトロセルロース、デキストラン、ナイロン、ポリアクリルアミドおよびアガロースでありうるが、これに制限されるわけではない。その支持体形状には、ビーズ;メンブラン;微粒子;微量滴定プレート、試験管または他の反応容器などの反応容器の内部表面が含まれうる。CRTH2受容体または他の成分の固定は、共有結合または非共有結合の付着によって達成されうる。一つの態様において、その付着は、間接的であってよい、すなわち、付着抗体を介していてよい。別の態様において、CRTH2受容体および負の対照は、グルタチオンS−トランスフェラーゼ(GST)などのエピトープで標識されるので、固体表面への付着は、抗GST(Santa Cruz Biotechnology)などの商業的に入手可能な抗体によって媒介されうる。例えば、このような親和性結合検定は、固体支持体に固定されるCRTH2受容体を用いて行うことができる。典型的に、結合反応の非固定成分、この場合はPGD2かまたは試験化合物を、検出可能であるように標識する。発光体、発色団、蛍光体、または放射性同位体または基の検出、または酵素または染料などの非同位体標識の検出のような種々の標識方法が、利用可能であり且つ用いることができる。一つの好ましい態様において、試験化合物を、フルオレセインイソチオシアネート(FITC,Sigma Chemicals, St. Louis より入手可能)などの発蛍光団で標識する。次に、標識された試験化合物、または試験化合物を加えたPGD2を、特異的結合を生じさせる条件下において固体支持体と接触させる。結合反応が起こった後、未結合のおよび非特異的に結合した試験化合物を、表面を洗浄することによって分離する。固相への結合パートナーの付着は、当業者に知られているいろいろな方法で行うことができ、化学架橋;プラスチック表面への非特異的接着;固相に付着した抗体との相互作用;結合パートナーに付着したリガンド(ビオチンなど)と、固相に付着したリガンド結合性タンパク質(アビジンまたはストレプトアビジンなど)との間の相互作用等が含まれるが、これに制限されるわけではない。最後に、固体表面上に残留する標識は、当該技術分野において知られているいずれかの検出方法によって検出することができる。例えば、試験化合物が発蛍光団で標識されている場合、蛍光計を用いて複合体を検出することができる。
【0043】
好ましい態様において、結合検定は、次のように行うことができる:
(a)CRTH2受容体を発現する細胞を、ペレットにし、そして室温において検定緩衝液(Ca2+およびMg2+を包含し且つHEPESおよび重炭酸ナトリウムを補足した Hank's 平衡生理食塩水)で2回洗浄する。それら細胞を、2x107個/mlの細胞濃度で再懸濁させる。96ウェルU底微量滴定皿を用いて、検定を次のように設定する(150μl容量中);
(b)50μlのビヒクル(検定緩衝液中0.3%DMSOとして、完全ウェル);または50μlの30μM冷PGD2であって、10μMの最終検定濃度を生じるもの[冷PGD2の原液を、DMSO中に10mMの原液濃度で溶解させ、−20℃で貯蔵した後、それを、使用するために3:1000に希釈して、30μMの最終原液濃度とした];50μlの細胞(106個/ウェルのための2x107個/ml);50μlの6nM[3H]−PGD2を、2nMの最終濃度のために加える(Amersham;162Ci/mmol,メタノール:水:アセトニトリル(3:2:1)中の0.1Ci/ml,617nMを6nMの濃度のために10μl/mlの検定緩衝液に希釈)。
【0044】
(c)プレートを、室温で20分間インキュベートさせた後、遠心分離を行う(2800rpm,Sorval RT6000,5分間,4℃)。上澄みを捨てて、非特異的結合を減少させる。プレート(Packard Unifilter プレートGF/C,予め3%PEI中に少なくとも1時間浸漬)を、冷検定緩衝液で、150μl/ウェルの緩衝液洗浄で6回洗浄することによって採取する。プレートを一晩乾燥させる。50μlのシンチレーション液の添加後、プレートをシンチレーション計数器で計数する(1分/ウェル)。(好ましくは、CRTH2受容体は、CRTH2受容体を発現する無傷の細胞の形で、またはCRTH2受容体を含有する単離された細胞膜として、結合検定に加える。したがって、CRTH2受容体へのリガンドの直接結合、またはPGD2−CRTH2受容体複合体をモジュレーションする試験化合物の能力は、培養物中の無傷の細胞において、試験化合物の存在下および/または不存在下で検定することができる)。CRTH2受容体を発現する細胞には、M.G. Reth et al., Nature, 317(6035), pp. 353-365, 1985 に開示されたような、CRTH2受容体を発現する300−19細胞(形質転換されたプレBリンパ球)が含まれる。CRTH2受容体は、アンピシリンおよびネオマイシンの耐性マーカーを含有するプラスミドから発現させることができ、CMVプロモーターによって駆動する。プロラク(prolac)シグナリングペプチドは、遺伝子インサートの膜発現を可能にし、N末端にある Flag ペプチド標識が、発現された分子の好都合な検出を可能にする。CRTH2受容体の好ましい発現レベルは、約40,000分子/細胞表面である。被標識PGD2は、CRTH2受容体を発現する細胞と、またはこのような細胞から得られる粗製抽出物と混合することができ、そして試験化合物を加えることができる。単離された膜は、CRTH2受容体と相互作用する化合物を識別するのに用いることができる。例えば、単離された膜を用いた典型的な実験では、細胞を遺伝子操作して、CRTH2受容体を発現させることができる。膜は、標準的な技法によって採取し、そして in vitro 結合検定で用いることができる。被標識リガンド(例えば、125I標識PGD2)を、膜に結合させ、比活性について検定し;そして特異的結合を、過剰の未標識(冷)リガンドの存在下で行われた結合検定との比較によって決定する。或いは、可溶性CRTH2受容体を、組換え発現させ、非細胞結合検定に利用して、CRTH2受容体に結合する化合物を識別することができる。一つまたは複数の組換え発現されたCRTH2受容体ポリペプチド、またはCRTH2受容体の一つまたはそれを超える細胞外ドメインを含有する融合タンパク質は、非細胞基材スクリーニング検定に用いることができる。或いは、CRTH2受容体の一つまたはそれを超える細胞外ドメインに該当するペプチド、またはCRTH2受容体の一つまたはそれを超える細胞外ドメインを含有する融合タンパク質は、非細胞基材検定システムで用いて、CRTH2受容体の細胞質部分に結合する化合物を識別することができるが;このような化合物は、CRTH2受容体のシグナル伝達経路をモジュレーションするのに有用でありうる。非細胞基材検定の場合、組換え発現されたCRTH2受容体を、当業者に知られている手段によって、試験管、微量滴定ウェルまたはカラムなどの固体支持体に付着させる。次に、試験化合物を、CRTH2受容体に結合するそれらの能力について検定する。
【0045】
或いは、結合反応は、溶液中で行うことができる。この検定では、被標識成分を、溶液中において一つまたは複数の結合パートナーと相互作用させる。被標識成分とその一つまたは複数の結合パートナーとの間のサイズ差が、このような分離を可能にする場合、その分離は、限外濾過器の細孔が、未結合の被標識成分を通過させるが、その一つまたは複数の結合パートナーまたはその一つまたは複数のパートナーに結合した被標識成分を通過させない限外濾過器を介して結合反応生成物を通過させることによって行うことができる。分離は、結合パートナーに対する抗体、結合パートナーに予め付着したリガンドと相互作用しうるリガンド結合性タンパク質等のような、被標識成分の結合パートナーを溶液から捕捉することが可能ないずれかの試薬を用いて行うこともできる。
【0046】
本発明の化合物は、CRTH2受容体アンタゴニスト、好ましくは、選択的CRTH2受容体アンタゴニストである。これら化合物は、典型的には、少なくとも100nM、好ましくは、10nM未満、より好ましくは、1nM未満の低いIC50値を有する。
【0047】
CRTH2受容体アンタゴニストの力価(下に記載の機能性検定において受容体の機能活性の値の半分を生じるアンタゴニストの濃度として定義することができるIC50力価に基づく)は、好ましくは、ヒト受容体(リコンビナントおよび/または天然)において、少なくとも100nMのIC50、より好ましくは、10nM未満、そして更に好ましくは、1nM未満である。例えば、機能性細胞基材検定の場合、IC50は、例えば、プロスタグランジンD2(または他の小分子アゴニスト)に応答して、ヒトCRTH2受容体の最大活性化を50%阻害するアンタゴニストのモル濃度である。結合検定の場合、IC50は、3H標識プロスタグランジンD2(または他の適当なリガンド)の特異的結合の50%を置き換えるアンタゴニストのモル濃度である。
【0048】
CRTH2受容体アンタゴニストの選択性は、好ましくは、CRTH2受容体について、他のGPCR、特に、Dタイプのプロスタノイド受容体(DP受容体)にまさって、そして或いは、化学誘引物質受容体サブファミリーの関連メンバー、例えば、補体受容体C3a、C5a、FMLP(FMet−Leu−Phe受容体)FMLP受容体IおよびII、ロイコトリエンB4(LTB4)、GPCR0269、GPCR0232、GPCR0288受容体に対して、少なくとも10倍選択的であり、好ましくは、それは、少なくとも100倍選択的、そして更に好ましくは、少なくとも1000倍選択的であるはずである。選択性は、概して、目的の受容体に適当なリガンドについての、二つの受容体サブタイプ間の化合物の相対力価である。
【0049】
CRTH2受容体リガンドまたはアンタゴニストは、CRTH2受容体への選択性について、DPと比較して調べることができる。この検定では、3H−PGD2の結合について競合する試験化合物各々の能力を、CRTH2およびDP双方の受容体において測定し、そしてIC50値を(μMで)決定する。対照は、3H−PGD2と競合する冷PGD2を用いて設定することができる。上述の結合検定手順はいずれも、用いることができる。CRTH2受容体アンタゴニストの選択性は、他のGPCR、特に、Dタイプのプロスタノイド受容体またはDP受容体と比較して少なくとも10倍であるはずであり(N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドは、>50倍選択的である)、好ましくは、それは、少なくとも100倍選択的、そしてなお一層好ましくは、少なくとも1000倍選択的であるはずであり、或いは、アンタゴニストは、CRTH2受容体について、C3a、C5a、FMLP、LTB4、GPCR0269、GPCR0232またはGPCR0288のいずれの受容体にもまさって選択的であるはずである。
【0050】
機能性検定
機能性検定方法は、CRTH2受容体に媒介される過程をモジュレーションする化合物およびCRTH2受容体のアンタゴニストである化合物を識別することについて知られている。それら方法は、概して、(a)CRTH2受容体発現性細胞と試験化合物とを、場合により、PGD2または別のCRTH2受容体活性化リガンドの存在下において接触させること;および(b)得られたCRTH2受容体活性レベル、または細胞中のCRTH2受容体の発現レベルを、この測定された活性レベルまたは発現レベルが、試験化合物の不存在下で測定されたレベルと異なる場合に、CRTH2受容体−PGD2に媒介される過程をモジュレーションする化合物が識別されるように測定すること含む工程を包含する。測定されるCRTH2受容体活性は、PGD2と相互作用する能力、またはPGD2への細胞の走化性応答、または細胞内Ca2+濃度/動員、または反応性酸素種の放出、アデニル酸シクラーゼ/サイクリックAMP生産の阻害、またはアクチン重合でありうる。機能性検定のための実施例プロトコールを下に示す。
【0051】
カルシウム動員は、フローサイトメトリーによって、および細胞内に捕捉される蛍光染料で標識することによって検出し且つ測定することができる。例えば、インドール−1染料は、カルシウム結合時の発光スペクトルの変化を示す。カルシウム結合染料によって生じる蛍光対未結合染料によって生じる蛍光の比率を用いて、細胞内カルシウム濃度を算定する。実施例方法では、CRTH2受容体を発現する細胞を集め、そしてカルシウムフラックス検定を行う前日に、新鮮培地中に約2x105/mlで再懸濁させる。細胞を37℃で20〜30分以下の間インキュベート後、回転沈降させ、そしてインドール−1AMを含有する、37℃に予熱された50mlの新鮮PT1緩衝液(Hank's Buffer,pH7.2〜7.4;10mM Hepes;1.6mM CaCl2)中に、1000万個/mlの濃度で再懸濁させる。細胞を刺激し、そして蛍光計(Photon Technology Corporation, International)を用いて蛍光を測定する。読みが安定した後、時間軸をリセットし、そしてPGD2を、特定の時点(例えば、20秒)に加える。応答後、次の試薬を次の順序でキュベットに加えて、全カルシウムを放出させ且つキレート化する。20μlの18% Triton X−100、20μlの3M Tris,pH8.5および20μlの0.5M EDTA,pH8.5。その実験を、試験化合物の存在下および不存在下で繰り返す。試験化合物の不存在下では、PGD2は、CRTH2受容体を発現する細胞において15nMのEC50で、増加した([Ca2+])を生じる。したがって、アンタゴニスト試験化合物の存在下において、EC50は減少するであろうと考えられる。
【0052】
アクチン重合検定は、化合物の活性を特性決定する二次スクリーニングとして用いることができる。アクチン重合は、重合したアクチン線維を結合するアクチン特異的蛍光標識であるニトロベンゾオキサジアゾール(NBD)−ファラシジン(phallacidin)を用いて検定することができる。その検定は、次のように行うことができる。細胞標品を、10mM HEPES、100/10の Pen/Strep および0.5%FCSを加えたRPMI1640中に、5〜10x106個/mlで再懸濁させる。その細胞懸濁液を、96ウェルU底ポリプロピレン微量滴定プレート中に小分けする(100μL/ウェル)。50ulの適当な刺激物質(PGD2または試験化合物、またはPGD2および試験化合物双方)を、8チャンネルピペットを用いて加えた後、正確に25秒後、リゾホスホチジルコリン(0.5mg/ml)、Hank's 平衡塩類溶液(100μl,10x)、16%ホルムアルデヒド(800ul)および6.6uM NBD−ファラシジンをMEOH(100ul)中に含有する50ulの停止用溶液を加える。そのプレートを、室温で15分間放置する。次に、プレートを1000rpmで5分間遠心分離し、上澄みを振り落とし(flicked off)、そして細胞ペレットを、2%FCSおよび0.2%アジ化ナトリウムを加えた250ulのPBS中に再懸濁させる。次に、各々の試料をFACS Calliber 装置で読み取る。細胞は、リンパ球領域中の正分散/副分散データを用いてゲート処理する。応答は、ビヒクルで処理された細胞と刺激物質で処理された細胞との間のFL−1蛍光中央値の変化によって測定する。試験化合物は、PGD2の存在下および不存在下で検定し、PGD2単独を含有する試料と比較することができる。CRTH2受容体細胞のPGD2で誘発されるアクチン重合を減少させる化合物は、CRTH2受容体アンタゴニスト候補として識別される。
【0053】
in vivo 手順
CRTH2受容体アンタゴニストの鎮痛作用は、選択された疼痛状態の動物モデルを用いて in vivo で決定することができる。いくつかの疼痛状態モデルが知られているが、CRTH2受容体アンタゴニストの鎮痛作用を決定するのに用いることができる具体的な手順を下に示す。
【0054】
ある一つの疼痛モデルは、ラットのニューロパシー性疼痛を有するストレプトゾシンで誘発される糖尿病モデルである。この手順は、糖尿病を誘発するために、Charles River Sprague dawley ラット(225〜250g)などの被験動物への単回用量でのストレプトゾシン(50mg/kg,i.p.)の投与を行う。被験動物を、投与から2週間後に、静的および動的異痛症試験を用いて評価し、そしてニューロパシー性疼痛が確認された場合、それらを用いて、ニューロパシー性疼痛への化合物の作用について更に評価する。
【0055】
ラットの疼痛の慢性狭窄性損傷(CCI)モデルは、坐骨神経周囲を弛緩性結紮糸で縛ることを行い、Charles River Sprague dawley 雄ラット(175〜200g)を、麻酔室に入れ、2%イソフルオラン(isofluorane)O2混合物で麻酔する。右後大腿を剃毛し、1%ヨウ素を綿棒で塗る。次に、被験動物を、鼻用コーンによって手術中維持される麻酔および手順の間、恒温ブランケットに移す。皮膚を、大腿骨の線に沿って切開する。総坐骨神経を、大腿二頭筋を介する鈍的剥離によって大腿の中央に露出させる。坐骨三分岐に近位の約7mmの神経を、神経下に鉗子を挿入することによって自由にし、その神経を、静かに大腿から持ち上げる。神経からの筋膜の除去を助けるように、鉗子を静かに数回開閉する。縫合糸を、鉗子を用いて神経下に引き寄せ、僅かな抵抗が感じられるまで簡単な結び目で縛った後、二重に結ぶ。その手順を、4本の結紮糸(4−0絹)で、約1mm間隔で神経周囲を緩く縛るまで繰り返す。切開を層状に閉じる。手術から14日後、被験動物の静的異痛症、動的異痛症または体重支持不足について評価する。
【0056】
* 疼痛状態の別の動物モデルには、坐骨神経の部分緊密結紮である Seltzer モデル(Seltzer, Z. (1995). Sem. Neurosci, 8: pp.34-39)または坐骨神経の二つの脊髄神経の一方の緊密結紮である Chung's モデル(Kim SH, Chung JM. Pain (1992); 50: pp.355-63)または慢性狭窄性損傷モデル(CCI)(Bennett GJ, Xie Y-K. Pain (1988); 33: pp.87-107)が含まれる。更に別の動物疼痛モデルには、疼痛誘発剤、例えば、カプサイシン(Dirks J, Petersen KL, Rowbotham MC, Dahl JB, Anesthesiology. 2002 Jul;97(1):pp.102-107);またはホルマリン(Tjolsen, A. et al (1992), Pain 51, pp.5-17);または完全フロイントアジュバント(Abdi S, Vilassova N, Decosterd I, et al, Anesth Analog 2000; 91: pp.955-99);またはカラゲナン(Itoh, M., Takasaki, I., Andoh, T., Nojima, H., Tominaga, M. & Kuraishi, Y. (2001) Neurosci. Res., 40, pp.227-233);またはタキソール(Polomano RC. Mannes AJ. Clark US. Bennett GJ, (2001) Pain. 94(3): pp.293-304);またはビンカアルカロイド、ビンクリスチン(Aley KO, Reichling DB, Levine JD, Neuroscience (1996); 73: pp.259-65);または内臓痛のためのテルペンチン(Koster, R., Anderson, M. and De Beer, E.J., Acetic acid for analgesic screening, Fed. Proc., 18 (1959) 412./ Mogil, J.S., Kest, B., Sadowski, B. and Belknap, J.K., Differential genetic mediation of sensitivity to morphine in genetic models of opiate anti-nociception: influence of nociceptive assay, J. Pharmacol. Exp. Ther., 276 (1996a) 532-544./ Ness TJ, Gebhart GF, Pain (1990);41: pp.167-234 および McMahon SB, Agents Actions (1988); 25: pp.231-233)の投与が含まれる。更に別の動物モデルは、その被験動物に、有害な身体的刺激を、例えば、有害な熱刺激の投与(Malmberg, A.B., and Bannon, A.W. Models of neciception: hot-plate, tall-flick, and formalin tests in rodents. Current Protocols in Neuroscience 1999; pp 8.9.1-8.9.15);または有害な寒冷刺激または有害な圧力刺激の投与またはUV照射(S.J. Boxall, A. Berthele, D.J. Laurie, B. Sommer, W. Zieglgansberger, L. Urban and T.R. Tolle, Enhanced expression of metabotropic glutamate receptor 3 messenger RNA in the rat spinal cord during ultraviolet irradiation induced peripheral inflammation Neuroscience (1988) 82(2): pp.591-602)によって与えることを行うことができる。
【0057】
疼痛状態の別の動物モデルは、関節炎またはHIVまたはヘルペスまたは癌または糖尿病のような有痛性疾患状態を生まれつき有する動物の選択を行うことができる。或いは、その動物を、関節炎またはHIVまたはヘルペスまたは癌または糖尿病のような疼痛誘発性疾患状態を有するための動物の修飾により、疼痛状態を経験するように手配することができる。被験動物は、疾患による疼痛状態を有するように、いろいろな方法で、例えば、糖尿病性ニューロパシーを誘発するストレプトゾシンの投与(Courteix,C., Eschalier,A., Lavarenne,J., Pain, 53 (1993) pp.81-88.);またはHIV関連ニューロパシー性疼痛を引き起こすウイルスタンパク質の投与(Herzberg U. Sagen J., Journal of Neuroimmunology. (2001 May 1), 116(1): pp.29-39);または関節炎および炎症性疼痛を誘発する完全フロイントアジュバントまたはモノヨードアセテートの投与(Rikard Holmdahl, Johnny C. Lorentzen, Shemin Lu, Peter Olofsson, Lena Wester, Jens Holmberg, Ulf Pettersson Immunological Reviews (2001) Volume 184, Issue 1, pp.184);またはヘルペスおよびヘルペス後神経痛(post hepatic neuralgia)を引き起こす水痘−帯状疱疹ウイルスの投与(Fleetwood-Walker SM. Quinn JP. Wallace C. Blackburn-Munro G. Kelly BG. Fiskerstrand CE. Nash AA. Dalziel RG., Journal of General Virology. 80 (Pt 9):2433-6, 1999 Sep.);または癌を引き起こすための動物への発癌物質または癌細胞の投与(Shimoyama M. Tanaka K. Hasue F. Shimoyama N, Pain. 99(1-2): pp.167-74, 2002 Sep)によって修飾することができる。
【0058】
動的異痛症は、被験動物の後足底表面を綿棒で軽くたたくことによって評価することができる。全身運動活性を記録することがないように、活発でない充分に慣らされたラットでこの手順を行うように注意する。各々の時点で、少なくとも2回の測定値を得、その平均が、足引っ込め潜伏時間(PWL)である。15秒以内に反応が示されない場合、その手順を終え、被験動物をこの引っ込め時間に割り当てる。したがって、15秒は、事実上、引っ込めなしである。引っ込め反応は、しばしば、繰り返し足を縮めることまたは舐めることを伴う。動的異痛症は、被験動物が、たたくことを開始して8秒以内に綿棒刺激に応答する場合、存在すると考えられる。
【0059】
ベースライン評価後、被験動物に、鎮痛性評価用の化合物を、次の経路、すなわち、経口投与、皮下、腹腔内、静脈内または鞘内の一つによって投与することができる。PWLは、次の時点、すなわち、30分、1時間、2時間、3時間、4時間、5時間、6時間、7時間、24時間の全部またはいくつかにおいて再評価する。被験動物は、それらのベースライン値によって、各々の化合物群に無作為に割り当てられる。平均および標準誤差平均を、各々の化合物について各々の時点で計算する。動的異痛症の測度を、一元ANOVAを用いてそれぞれの対照と比較後、Dunnett's t検定を用いて、各々の時点において化合物とビヒクルを比較する。一群当たりの最小被験動物数は、6匹である。
【0060】
静的異痛症は、後足底表面への力の昇順(0.6g、1g、1.4g、2g、4g、6g、8g、10g、15gおよび26g)での von Frey 毛(Stoelting, Wood Dale, Illinois, USA)の適用によって評価することができる。被験動物は、異痛症評価の前に、ワイヤ底試験ケージに慣らす。各々の von Frey 毛を、最大6秒間または引っ込め応答が起こるまで、足に適用する。von Frey 毛への引っ込め応答が確かめられたら、その足を再試験し、引っ込めを生じるものより下のフィラメントで開始し、そしてその後、引っ込めが起こらなくなるまで、残りのフィラメントを力の降順で用いる。最高の26gの力は、足を持ち上げ、更には、応答を引き出し、したがって、中止点である。各々の被験動物の両後足をこの方式で調べられる。応答を引き出すのに必要な力の最低量を、足引っ込め閾値(PWT)としてグラムで記録する。静的異痛症は、被験動物が、普通のラットには無害である4gまたはそれ未満の刺激に応答した場合、存在すると定義される。
【0061】
ベースライン評価後、被験動物に、鎮痛性評価用の化合物を、次の経路、すなわち、経口、皮下、腹腔内、静脈内または鞘内の一つによって投与し、そしてPWTを、次の時点、すなわち、30分、1時間、2時間、3時間、4時間、5時間、6時間、7時間、24時間の全部またはいくつかにおいて再評価する。静的異痛症測定値を、ノンパラメトリック結果についての Kruskall-Wallis 検定後、Mann Whitney's U検定を用いて、ビヒクル群に対して分析する。一群当たりの最小被験動物数は、6匹である。
【0062】
温熱性痛覚過敏は、ラット足底試験(Ugo Basile, Italy)を用いて、Hargreaves et al., (1988) Pain 32:77-88 の変法にしたがって評価する。ラットは、高さのあるガラステーブル上の3個のパースペックスボックスから成る装置に慣らす。移動輻射熱源を、テーブルの下に置き、後足に集中させ、そして足引っ込め潜伏時間(PWL)を記録する。組織損傷を防止するために、22.5秒に自動中止点がある。PWLを、各々の被験動物の両後足について2〜3回得るが、その平均が、左右後足についてのベースラインである。装置は、約10秒のPWLを与えるように検量する。PWLを、カラゲナン投与から2時間後に再評価する。鎮痛性評価用の化合物の投与後、PWLを、1時間毎に6時間まで再評価する。化合物群のPWLを、一元ANOVA後、Dunnett's t検定を用いて、それぞれの対照と比較する。一群当たりの最小被験動物数は、6匹である。
【0063】
体重支持不足は、Bove SE, et al. Weight bearing as a measure of disease progression and efficacy of anti-inflammatory compounds in a model of monosodium iodoacetate-induced osteoarthritis. Osteoarthritis Cartilage. 2003 Nov;11(11):821-30 の方法にしたがって測定することができる。開放現場試験は、Prut L and Belzung, C. The open field as a paradigm to measure the effects of compounds on anxiety-like behaviors: a review. Eur J Pharmacol. 2003;463::3-33 の方法にしたがって行うことができる。運動試験は、Salmi P and Ahlenius S- Sedative effects of the dopamine D1 receptor agonist A 68930 on rat open-field behavior. Neuroreport. 2000 Apr 27;11(6):1269-72 の方法にしたがって行うことができる。
【0064】
組合せ
CRTH2受容体アンタゴニストは、有用に、具体的には、疼痛の処置において、別の薬理学的に活性な化合物と、または二つまたはそれを超える他の薬理学的に活性な化合物と組み合わせることができる。例えば、CRTH2受容体アンタゴニスト、具体的には、上に定義の一般式(I)の化合物またはその薬学的に許容しうる塩または溶媒和化合物は、次より選択される一つまたはそれを超える薬剤と組み合わせて、同時に、逐次的にまたは別々に投与することができる。
【0065】
(i)オピオイド鎮痛薬、例えば、モルヒネ、ヘロイン、ヒドロモルホン、オキシモルホン、レボルファノール、レバロルファン、メタドン、メペリジン、フェンタニール、コカイン、コデイン、ジヒドロコデイン、オキシコドン、ヒドロコドン、プロポキシフェン、ナルメフェン(nalmefene)、ナロルフィン、ナロキソン、ナルトレキソン、ブプレノルフィン、ブトルファノール、ナルブフェンまたはペンタゾシン;
(ii)非ステロイド性抗炎症薬(NSAID)、例えば、アスピリン、ジクロフェナク、ジフルシナル(diflusinal)、エトドラク(etodolac)、フェンブフェン、フェノプロフェン、フルフェニサル(flufenisal)、フルルビプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、ケトロラク、メクロフェナム酸、メフェナム酸、ナブメトン(nabumetone)、ナプロキセン、オキサプロジン、フェニルブタゾン、ピロキシカム、スリンダク、トルメチンまたはゾメピラック、またはその薬学的に許容しうる塩;
(iii)バルビツレート鎮静薬、例えば、アモバルビタール、アプロバルビタール、ブタバルビタール、ブタビタール(butabital)、メフォバルビタール、メタルビタール、メトヘキシタール、ペントバルビタール、フェノバルビタール、セコバルビタール、タルブタール、テアミラル(theamylal)またはチオペンタール、またはその薬学的に許容しうる塩;
(iv)鎮静作用を有するベンゾジアゼピン、例えば、クロルジアゼポキシド、クロラゼペート、ジアゼパム、フルラゼパム、ロラゼパム、オキサゼパム、テマゼパムまたはトリアゾラム、またはその薬学的に許容しうる塩;
(v)鎮静作用を有するH1アンタゴニスト、例えば、ジフェンヒドラミン、ピリラミン、プロメタジン、クロルフェニラミンまたはクロルシクリジン、またはその薬学的に許容しうる塩;
(vi)鎮静薬であって、グルテチミド、メプロバメート、メタクアロンまたはジクロラルフェナゾン、またはその薬学的に許容しうる塩のようなもの;
(vii)骨格筋弛緩薬、例えば、バクロフェン、カリソプロドール、クロルゾキサゾン、シクロベンザプリン、メトカルバモルまたはオルフレナジン(orphrenadine)、またはその薬学的に許容しうる塩;
(viii)NMDA受容体アンタゴニスト、例えば、デキストロメトルファン((+)−3−ヒドロキシ−N−メチルモルフィナン)またはその代謝産物デキストロルファン((+)−3−ヒドロキシ−N−メチルモルフィナン)、ケタミン、メマンチン、ピロロキノリンキノンまたは cis−4−(ホスホノメチル)−2−ピペリジンカルボン酸、またはその薬学的に許容しうる塩;
(ix)αアドレナリン作動性薬、例えば、ドキサゾシン、タムスロシン(tamsulosin)、クロニジンまたは4−アミノ−6,7−ジメトキシ−2−(5−メタンスルホンアミド−1,2,3,4−テトラヒドロイソキノール−2−イル)−5−(2−ピリジル)キナゾリン;
(x)三環系抗うつ薬、例えば、デシプラミン、イミプラミン、アミトリプチリン(amytriptiline)またはノルトリプチリン;
(xi)抗痙攣薬、例えば、カルバマゼピンまたはバルプロエート;
(xii)タキキニン(NK)アンタゴニスト、具体的には、NK−3、NK−2またはNK−1アンタゴニスト、例えば、(αR,9R)−7−[3,5−ビス(トリフルオロメチル)ベンジル]−8,9,10,11−テトラヒドロ−9−メチル−5−(4−メチルフェニル)−7H−[1,4]ジアゾシノ[2,1−g][1,7]ナフトリジン−6−13−ジオン(TAK−637)、5−[[(2R,3S)−2−[(1R)−1−[3,5−ビス(トリフルオロメチル)フェニル]エトキシ−3−(4−フルオロフェニル)−4−モルホリニル]メチル]−1,2−ジヒドロ−3H−1,2,4−トリアゾール−3−オン(MK−869)、ラネピタント(lanepitant)、ダピタント(dapitant)または3−[[2−メトキシ−5−(トリフルオロメトキシ)フェニル]メチルアミノ]−2−フェニルピペリジン(2S,3S);
(xiii)ムスカリン様アンタゴニスト、例えば、オキシブチン(oxybutin)、トルテロジン(tolterodine)、プロピベリン(propiverine)、塩化トロプシウム(tropsium chloride)またはダリフェナシン(darifenacin);
(xiv)COX−2阻害剤、例えば、セレコキシブ(celecoxib)、ロフェコキシブ(rofecoxib)またはバルデコキシブ(valdecoxib);
(xv)非選択的COX阻害剤(好ましくは、GI防御を伴う)、例えば、ニトロフルルビプロフェン(HCT−1026);
(xvi)コールタール鎮痛薬、特に、パラセタモール;
(xvii)神経遮断薬であって、ドロペリドールのようなもの;
(xviii)バニロイド受容体アゴニスト(例えば、レシンフェラトキシン(resinferatoxin))またはアンタゴニスト(例えば、カプサゼピン(capsazepine));
(xix)βアドレナリン作動性薬であって、プロプラノロールのようなもの;
(xx)局所麻酔薬であって、メキシレチンのようなもの;
(xxi)コルチコステロイドであって、デキサメタゾンのようなもの;
(xxii)セロトニン受容体アゴニストまたはアンタゴニスト;
(xxiii)コリン作動性(ニコチン性)鎮痛薬;
(xxiv)Tramadol(登録商標);
(xxv)PDEV阻害剤であって、シルデナフィル(sildenafil)、バルデナフィル(vardenafil)またはタラダフィル(taladafil)のようなもの;
(xxvi)α−2−δリガンドであって、ガバペンチン(gabapentin)またはプレガバリン(pregabalin)のようなもの;および
(xxvii)カナビノイド(canabinoid)。
【0066】
CRTH2受容体アンタゴニストは、治療的有効量で患者に投与される。CRTH2受容体アンタゴニストは、単独でまたは薬学的に許容しうる組成物の一部分として投与することができる。
【0067】
薬物物質
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、薬学的に許容しうる塩、例えば、酸付加塩または塩基塩の形で投与することができる。
【0068】
適する酸付加塩は、無毒性塩を形成する酸から形成される。例には、酢酸塩、アスパラギン酸塩、安息香酸塩、ベシル酸塩、重炭酸塩/炭酸塩、重硫酸塩/硫酸塩、ホウ酸塩、カムシレート塩、クエン酸塩、エジシレート塩、エシレート塩、ギ酸塩、フマル酸塩、グルセプテート塩、グルコン酸塩、グルクロン酸塩、ヘキサフルオロリン酸塩、ヒベンゼート(hibenzate)塩、塩酸塩/塩素塩、臭化水素酸塩/臭素塩、ヨウ化水素酸塩/ヨウ素塩、イセチオン酸塩、乳酸塩、リンゴ酸塩、マレイン酸塩、マロン酸塩、メシレート塩、メチル硫酸塩、ナフチル酸塩、2−ナプシレート塩、ニコチン酸塩、硝酸塩、オロチン酸塩、シュウ酸塩、パルミチン酸塩、パモ酸塩、リン酸塩/リン酸水素塩、リン酸塩/リン酸二水素塩、リン酸塩、糖酸塩、ステアリン酸塩、コハク酸塩、酒石酸塩、トシレート塩およびトリフルオロ酢酸塩が含まれる。
【0069】
適する塩基塩は、無毒性塩を形成する塩基から形成される。例には、アルミニウム塩、アルギニン塩、ベンザチン塩、カルシウム塩、コリン塩、ジエチルアミン塩、ジオールアミン塩、グリシン塩、リシン塩、マグネシウム塩、メグルミン塩、オラミン(olamine)塩、カリウム塩、ナトリウム塩、トロメタミン塩および亜鉛塩が含まれる。
【0070】
酸および塩基のヘミ塩、例えば、ヘミ硫酸塩およびヘミカルシウム塩も形成することができる。
適する塩の概説については、Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth(Wiley-VCH, Weinheim, Germany, 2002)を参照されたい。
【0071】
薬学的に許容しうる塩は、次の3種類の内の一つまたはそれを超える方法によって製造することができる。
(i)化合物を、所望の酸または塩基と反応させることによる;
(ii)化合物の適する前駆体から、酸または塩基に反応活性な保護基を除去することによる、または適する環状前駆体、例えば、ラクトンまたはラクタムを、所望の酸または塩基を用いて開環することによる;または
(iii)化合物の一つの塩を、適当な酸または塩基との反応によってまたは適するイオン交換カラムによって、別の塩へと変換することによる。
【0072】
3種類の反応は全て、典型的に、溶液中で行われる。得られた塩は、沈殿するので、濾過によって集めることができるし、または溶媒の蒸発によって回収することができる。得られた塩のイオン化の程度は、完全にイオン化された状態〜ほとんどイオン化されていない状態でありうる。
【0073】
本発明の化合物は、非溶媒和および溶媒和双方の形で存在しうる。「溶媒和化合物」という用語は、本明細書中において、本発明の化合物と、化学量論的量の一つまたはそれを超える薬学的に許容しうる溶媒分子、例えば、エタノールを含む分子複合体を記載するのに用いられる。「水和物」という用語は、この溶媒が水である場合に用いられる。
【0074】
本発明の範囲内に包含されるのは、クラスレート、薬物−宿主包接複合体などの複合体であるが、この場合、前述の溶媒和化合物とは対照的に、薬物および宿主は、化学量論的量または非化学量論的量で存在する。更に包含されるのは、化学量論的量であってよいしまたは非化学量論的量であってよい二つまたはそれを超える有機および/または無機成分を含有する薬物の複合体である。得られた複合体は、イオン化、部分イオン化または非イオン化の状態であってよい。このような複合体についての概説については、Halebllan (august 1975) J Pharm Sci, 64(8), 1269-1288 を参照されたい。
【0075】
以下、本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物という意味は全て、その塩、溶媒和化合物および複合体、およびそれら塩の溶媒和化合物および複合体の意味を包含する。
【0076】
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、プロドラッグの形で投与することができる。プロドラッグは、それ自体はほとんどまたは全く薬理活性が無くてよいが、体内または上に投与された場合、例えば、加水分解的開裂により、所望の活性を有する化合物へと変換されうる化合物である。プロドラッグの使用に関する追加の情報は、Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series(T. Higuchi and W. Stella)および Bioreversible Carries in Drug Design, Pergamon Press, 1987(ed. E. B. Roche, American Pharmaceutical Association)に見出されうる。
【0077】
プロドラッグは、例えば、化合物中に存在する適当な官能基を、例えば、Design of Prodrugs by H. Bundgaard(Elsevier, 1985)に記載の「プロ残基(pro-moieties)」のような、当業者に知られているある種の残基で置き換えることによって製造することができる。
【0078】
プロドラッグの若干の例には、次が含まれる。
(i)化合物が、カルボン酸官能基(−COOH)を含有する場合、そのエステル、例えば、式(I)の化合物のカルボン酸官能基の水素が、(C1〜C8)アルキルで置き換えられている化合物;
(ii)化合物が、アルコール官能基(−OH)を含有する場合、そのエーテル、例えば、その化合物のアルコール官能基の水素が、(C1〜C6)アルカノイルオキシメチルで置き換えられている化合物;および
(iii)化合物が、第一級または第二級アミノ官能基(−NH2または−NHR、ここにおいて、R≠H)を含有する場合、そのアミド、例えば、その化合物のアミノ官能基の、場合により一つまたは双方の水素が、(C1〜C10)アルカノイルで置き換えられている化合物。
【0079】
前述の例および他のプロドラッグタイプの例による置換基の更に別の例は、前述の参考文献に見出されうる。
更に、ある種の化合物は、それ自体、他の化合物のプロドラッグとして作用することができる。
【0080】
本発明の範囲内に更に包含されるのは、本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物の代謝産物、すなわち、薬物の投与時に in vivo で形成される化合物である。本発明による代謝産物の若干の例には、次が含まれる。
【0081】
(i)化合物が、メチル基を含有する場合、そのヒドロキシメチル誘導体(−CH3→−CH2OH);
(ii)化合物が、アルコキシ基を含有する場合、そのヒドロキシ誘導体(−OR→−OH);
(iii)化合物が、第三級アミノ基を含有する場合、その第二級アミノ誘導体(−NR1R2→−NHR1または−NHR2);
(iv)化合物が、第二級アミノ基を含有する場合、その第一級誘導体(−NHR1→−NH2);
(v)化合物が、フェニル残基を含有する場合、そのフェノール誘導体(−Ph→−PhOH);および
(vi)化合物が、アミド基を含有する場合、そのカルボン酸誘導体(−CONH2→−COOH)。
【0082】
一つまたはそれを超える不斉炭素原子を含有する本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、二つまたはそれを超える立体異性体として存在しうる。化合物が、アルケニル基またはアルケニレン基を含有する場合、幾何的 cis/trans(またはZ/E)異性体が可能である。構造異性体が、低エネルギー障壁によって相互変換できる場合、互変異性体の異性(「互変異性」)が存在しうる。これは、例えば、イミノ基、ケト基またはオキシム基を含有する式Iの化合物におけるプロトン互変異性の形、または芳香族残基を含有する化合物におけるいわゆる原子価互変異性の形をとりうる。単一の化合物は、1より多くのタイプの異性を示すことがありうるということである。
【0083】
cis/trans 異性体は、当業者に周知の慣用的な技法、例えば、クロマトグラフィーおよび分別結晶によって分離することができる。
個々の鏡像異性体の製造/分離のための慣用的な技法には、適する光学的に純粋な前駆体からのキラル合成、または例えば、キラル高速液体クロマトグラフィー(HPLC)を用いたラセミ体(または塩または誘導体のラセミ体)の分割が含まれる。
【0084】
或いは、ラセミ体(またはラセミ前駆体)は、適する光学活性化合物、例えば、アルコールと反応することができるし、または式Iの化合物が、酸性または塩基性の残基を含有する場合、1−フェニルエチルアミンまたは酒石酸のような塩基または酸と反応することができる。得られたジアステレオマー混合物は、クロマトグラフィーおよび/または分別結晶によって分離することができ、それらジアステレオ異性体の一方または双方は、当業者に周知の手段によって、その一つまたは複数の該当する純粋な鏡像異性体へと変換することができる。
【0085】
キラル化合物(およびそれらのキラル前駆体)は、クロマトグラフィー、典型的には、不斉樹脂上の、0〜50容量%、典型的には、2%〜20%のイソプロパノールおよび0〜5容量%のアルキルアミン、典型的には、0.1%のジエチルアミンを含有する炭化水素、典型的には、ヘプタンまたはヘキサンから成る移動相でのHPLCを用いて、鏡像異性体が豊富な形で得ることができる。溶出液の濃縮は、豊富な混合物を与える。
【0086】
立体異性体集合体は、当業者に知られている慣用的な技法によって分離することができ、例えば、Stereochemistry of Organic Compounds by E. L. Eliel and S. H. Wilen (Wiley, New York, 1994) を参照されたい。
【0087】
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、1個またはそれを超える炭素原子が、同じ原子番号を有するが、天然において優先的に存在する原子質量または質量数とは異なった原子質量または質量数を有する原子で置き換えられている一つまたはそれを超える同位体で存在してよい。
【0088】
同位体の例には、2Hおよび3Hなどの水素、11C、13Cおよび14Cなどの炭素、36Clなどの塩素、18Fなどのフッ素、123Iおよび125Iなどのヨウ素、13Nおよび15Nなどの窒素、15O、17Oおよび18Oなどの酸素、32Pなどのリン、および35Sなどの硫黄の同位体が含まれる。
【0089】
ある種の同位体標識された化合物、例えば、放射性同位体を包含するものは、薬物および/または基質の組織分布研究において有用である。放射性同位体トリチウム、すなわち、3Hおよび炭素14、すなわち、14Cは、それらの包含の容易さおよび容易な検出手段のために、この目的に特に有用である。
【0090】
ジュウテリウム、すなわち、2Hのような、より重い同位体での置換は、より大きい代謝安定性、例えば、増加した in vivo 半減期または減少した投薬要件によって生じるある種の治療的利点を与えることがあり、したがって、何等かの状況において好適でありうる。
【0091】
11C、18F、15Oおよび13Nのような、陽電子放出性同位体での置換は、基質受容体占有を調べるための陽電子放出トポグラフィー(PET)研究に有用でありうる。
同位体標識された化合物は、概して、慣用的な技法によって製造することができる。
【0092】
本発明による薬学的に許容しうる溶媒和化合物には、結晶化の溶媒が、同位体置換されていてよいもの、例えば、D2O、d6−アセトン、d6−DMSOが含まれる。
薬物製品
薬学的使用を予定した本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、結晶性または非晶質製品として投与することができる。それは、例えば、固形プラグ、粉末または薄膜として、沈殿、結晶化、凍結乾燥、噴霧乾燥または蒸発乾燥などの方法によって得ることができる。マイクロ波または高周波乾燥は、この目的に用いることができる。
【0093】
それは、単独で、または一つまたはそれを超える本発明の他の化合物との組合せで、または一つまたはそれを超える他の薬物との組合せで(またはいずれかそれらの組合せとして)投与することができる。概して、それは、一つまたはそれを超える薬学的に許容しうる賦形剤と一緒の製剤として投与されるであろう。「賦形剤」という用語は、本明細書中において、本発明の一つまたは複数の化合物以外のいずれかの成分を記載するのに用いられる。賦形剤の選択は、具体的な投与様式、溶解度および安定性への賦形剤の作用、および剤形の性状などの因子に大きく依存するであろう。
【0094】
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物の送達に適する医薬組成物、およびその製造方法は、当業者に容易に明らかであろう。このような組成物およびその製造方法は、例えば、Remington's Pharmaceutical Sciences, 19th Edition (Macl Publishing Company, 1995) に見出されうる。
【0095】
経口投与
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、経口投与することができる。経口投与は、化合物が胃腸管に入るように飲み込むことを必要とすることがありうるし、または化合物が口から直接的に血流に入る口腔内投与または舌下投与を用いることができる。
【0096】
経口投与に適する製剤には、錠剤、粒状剤を含有するカプセル剤、液剤または散剤、ロゼンジ(液体入りを含めた)、咀嚼剤、多粒状剤およびナノ粒状剤、ゲル剤、固溶体、リポソーム、薄膜剤、小卵剤、噴霧剤および液状製剤などの固形製剤が含まれる。
【0097】
液状製剤には、懸濁剤、液剤、シロップ剤およびエリキシル剤が含まれる。このような製剤は、軟または硬カプセル剤中の充填剤として用いることができ、典型的には、担体、例えば、水、エタノール、ポリエチレングリコール、プロピレングリコール、メチルセルロースまたは適する油、および一つまたはそれを超える乳化剤および/または懸濁化剤を含むことができる。液状製剤は、固体の再構成によって、例えば、サシェ剤から製造することもできる。
【0098】
本発明のCRTH2受容体アンタゴニスト、例えば、本発明の一般式Iの化合物は、Liang and Chen (2001) によって Expert Opinion in Therapeutic Patents, 11(6), 981-986 に記載のものなどの、急速溶解性、急速崩壊性剤形で用いることもできる。
【0099】
錠剤剤形については、用量に依存して、薬物は、剤形の1重量%〜80重量%、より典型的には、剤形の5重量%〜60重量%を構成してよい。薬物に加えて、錠剤は、概して、崩壊剤を含有する。崩壊剤の例には、ナトリウムデンプングリコラート、ナトリウムカルボキシメチルセルロース、カルシウムカルボキシメチルセルロース、クロスカルメロースナトリウム、クロスポビドン、ポリビニルピロリドン、メチルセルロース、微結晶性セルロース、低級アルキル置換ヒドロキシプロピルセルロース、デンプン、プレゲル化デンプンおよびアルギン酸ナトリウムが含まれる。概して、崩壊剤は、剤形の1重量%〜25重量%、好ましくは、5重量%〜20重量%を構成するであろう。
【0100】
結合剤は、概して、錠剤製剤に凝集質を与えるのに用いられる。適する結合剤には、微結晶性セルロース、ゼラチン、糖類、ポリエチレングリコール、天然および合成のガム、ポリビニルピロリドン、プレゲル化デンプン、ヒドロキシプロピルセルロースおよびヒドロキシプロピルメチルセルロースが含まれる。錠剤は、更に、ラクトース(一水和物、噴霧乾燥一水和物、無水物等)、マンニトール、キシリトール、デキストロース、スクロース、ソルビトール、微結晶性セルロース、デンプンおよび第二リン酸カルシウム二水和物などの希釈剤を含有してよい。
【0101】
錠剤は、更に、ラウリル硫酸ナトリウムおよびポリソルベート80などの界面活性剤、および二酸化ケイ素およびタルクなどの滑剤を含んでもよい。存在する場合、界面活性剤は、錠剤の0.2重量%〜5重量%を構成してよく、そして滑剤は、錠剤の0.2重量%〜1重量%を構成してよい。
【0102】
錠剤は、概して、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸亜鉛、ステアリルフマル酸ナトリウム、およびステアリン酸マグネシウムとラウリルリル硫酸ナトリウムとの混合物などの滑沢剤も含有する。滑沢剤は、概して、錠剤の0.25重量%〜10重量%、好ましくは、0.5重量%〜3重量%を構成する。
【0103】
他の可能性のある成分には、酸化防止剤、着色剤、着香剤、保存剤および風味マスキング剤が含まれる。
代表的な錠剤は、約80%までの薬物;約10重量%〜約90重量%の結合剤;約0重量%〜約85重量%の希釈剤;約2重量%〜約10重量%の崩壊剤;および約0.25重量%〜約10重量%の滑沢剤を含有する。
【0104】
錠剤ブレンドは、直接的にまたはローラーによって圧縮して、錠剤を成形することができる。或いは、錠剤ブレンドまたはブレンドの一部分は、錠剤成形する前に、湿式、乾式または溶融造粒する、溶融凝固させる、または押出することができる。最終製剤は、一つまたはそれを超える層を構成していてよく、コーティングされていてよいしまたは未コーティングであってよく;それは、カプセル封入されていてもよい。
【0105】
錠剤の製剤は、H. Lieberman and L. Lachman (Marcel Decker, New York, 1990) による Pharmaceutical Dosage Forms: Tablets, Vol. 1 に論じられている。
ヒトまたは獣医学的使用への消耗経口薄膜剤は、典型的に、急速溶解性または粘膜接着性であってよい柔軟な水溶性または水膨潤性の薄膜剤形であり、典型的には、式Iの化合物、薄膜形成性ポリマー、結合剤、溶媒、保湿剤、可塑剤、安定化剤または乳化剤、粘度調整剤および溶媒を含む。製剤の若干の成分は、二つ以上の機能を果たすことができる。
【0106】
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、水溶性であってよいしまたは不溶性であってよい。水溶性化合物は、典型的に、溶質の1重量%〜80重量%、より典型的には、20重量%〜50重量%を構成する。あまり可溶性でない化合物は、より大きい組成比、典型的に、溶質の88重量%までを構成してよい。或いは、本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、多粒状ビーズの形であってよい。
【0107】
薄膜形成性ポリマーは、天然多糖類、タンパク質または合成ヒドロコロイドより選択されてよく、典型的には、0.01〜99重量%の範囲内、より典型的には、30〜80重量%の範囲内で存在する。
【0108】
他の可能性のある成分には、酸化防止剤、着色剤、着香剤およびフレーバーエンハンサー、保存剤、唾液刺激剤、冷却剤、補助溶媒(油を含めた)、皮膚軟化薬、増量剤、消泡剤、界面活性剤および風味マスキング剤が含まれる。
【0109】
本発明による薄膜は、典型的に、可剥性支持剤または裏紙上にコーティングされた水性薄膜の蒸発乾燥によって製造される。これは、乾燥用オーブンまたはトンネル中で、典型的には、併用コーター・ドライヤー中で、または凍結乾燥または真空にすることによって行うことができる。
【0110】
経口投与用の固形製剤は、即時放出および/または修飾放出であるように製剤化することができる。修飾放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的放出およびプログラム化放出が含まれる。
【0111】
本発明の目的に適する修飾放出製剤は、米国特許第6,106,864号に記載されている。高エネルギー分散系および浸透圧性およびコーティング粒子などの他の適する放出技術の詳細は、Verma et al (2001) による Pharmaceutical Technology On-line, 25(2), 1-14 に見出されるはずである。制御放出を達成するチューインガムの使用は、WO00/35298号に記載されている。
【0112】
非経口投与
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、血流中、筋肉中または内臓中に直接投与することもできる。非経口投与に適する手段には、静脈内、動脈内、腹腔内、鞘内、脳室内、尿道内、胸骨内、頭蓋内、筋肉内および皮下が含まれる。非経口投与に適するデバイスには、針(顕微針を含めた)注射器、無針注射器および注入法が含まれる。
【0113】
非経口製剤は、典型的に、塩類、炭水化物および緩衝剤(好ましくは、3〜9のpHへ)などの賦形剤を含有してよい水性液剤であるが、若干の用途については、それらは、無菌の非水溶液として、または滅菌発熱物質不含水などの適するビヒクルと一緒に用いられる乾燥した形として、一層好適に製剤化することができる。
【0114】
無菌条件下における、例えば、凍結乾燥による非経口製剤の製造は、当業者に周知の標準的な製剤技術を用いて容易に達成することができる。
非経口液剤の製造に用いられる本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物の溶解度は、溶解促進剤の包含のような適当な製剤化技術の使用によって増加させることができる。
【0115】
非経口投与用製剤は、即時放出および/または修飾放出であるように製剤化することができる。修飾放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的放出およびプログラム化放出が含まれる。したがって、本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、活性化合物を修飾放出する植込みデポ剤としての投与用に、固体、半固体またはチキソトロピー性液体として製剤化することができる。このような製剤の例には、薬物コーティングステントおよびポリ(dl−乳酸コグリコール酸)(PGLA)ミクロスフェアが含まれる。
【0116】
局所投与
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、皮膚または粘膜への局所に、すなわち、外皮にまたは経皮で投与することもできる。この目的に典型的な製剤には、ゲル剤、ヒドロゲル剤、ローション剤、液剤、クリーム剤、軟膏剤、散布剤、包帯剤、フォーム剤、薄膜剤、皮膚パッチ、ウェファー、植込剤、スポンジ、ファイバー、バンデージおよびミクロエマルジョンが含まれる。リポソームを用いてもよい。典型的な担体には、アルコール、水、鉱油、流動パラフィン、白色ワセリン、グリセリン、ポリエチレングリコールおよびプロピレングリコールが含まれる。浸透促進剤を包含してよい。例えば、Finn and Morgan (October 1999) による J Pharm Sci, 88(10), 955-958 を参照されたい。
【0117】
他の局所投与手段には、エレクトロポレーション、イオン導入法、フォノフォレシス(phonophoresis)、ソノフォレシス(sonophoresis)および顕微針または無針(例えば、PowderjectTM、BiojectTM等)注射による送達が含まれる。
【0118】
局所投与用製剤は、即時放出および/または修飾放出であるように製剤化することができる。修飾放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的放出およびプログラム化放出が含まれる。
【0119】
吸入/鼻腔内投与
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、鼻腔内にまたは吸入によって、典型的には、乾燥粉末の形で(単独でか、混合物として、例えば、ラクトースとのドライブレンド中でかまたは、例えば、ホスファチジルコリンなどのリン脂質と混合された混合成分粒子として)乾燥粉末吸入器から、または加圧容器、ポンプ、スプレー、アトマイザー(好ましくは、電気流体力学を用いて微細な霧を生じるアトマイザー)またはネブライザーからのエアゾルスプレーとして、1,1,1,2−テトラフルオロエタンまたは1,1,1,2,3,3,3−ヘプタフルオロプロパンなどの適する噴射剤を伴ってまたは伴うことなく投与することもできる。鼻腔内使用のための粉末は、生体接着剤、例えば、キトサンまたはシクロデキストリンを含んでよい。
【0120】
加圧容器、ポンプ、スプレー、アトマイザーまたはネブライザーは、例えば、溶媒としてのエタノール、水性エタノール、または活性な一つまたは複数の噴射剤を分散させる、可溶化するまたは延長放出するのに適する別の薬剤と、ソルビタントリオレアート、オレイン酸またはオリゴ乳酸などの任意の界面活性剤を含む、本発明の一つまたは複数の化合物の溶液または懸濁液を含有する。
【0121】
乾燥粉末または懸濁液製剤での使用前に、薬物製品は、吸入による送達に適するサイズ(典型的に、5ミクロン未満)へと超微粉砕する。これは、スパイラルジェットミリング、流動床ジェットミリング、ナノ粒子を形成する超臨界流動処理、高圧ホモジナイゼーションまたは噴霧乾燥のような、いずれか適当な微粉砕法によって行うことができる。
【0122】
吸入器または吹き入れ器で用いるためのカプセル剤(例えば、ゼラチンまたはヒドロキシプロピルメチルセルロースから製造される)、ブリスターおよびカートリッジは、本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物;ラクトースまたはデンプンなどの適する粉末基剤;およびl−ロイシン、マンニトールまたはステアリン酸マグネシウムなどの性能調整剤の粉末配合物を含有するように製剤化することができる。ラクトースは、無水物であってよいしまたは一水和物の形であってよいが、好ましくは、後者である。他の適する賦形剤には、デキストラン、グルコース、マルトース、ソルビトール、キシリトール、フルクトース、スクロースおよびトレハロースが含まれる。
【0123】
電気流体力学を用いて微細な霧を生じるアトマイザーでの使用に適する溶液製剤は、1作動につき1μg〜20mgの本発明の化合物を含有してよく、その作動容量は、1μl〜100μlであってよい。典型的な製剤は、本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物、プロピレングリコール、滅菌水、エタノールおよび塩化ナトリウムを含むことができる。プロピレングリコールの代わりに用いることができる別の溶媒には、グリセロールおよびポリエチレングリコールが含まれる。
【0124】
メントールおよびレボメントールなどの適するフレーバー、またはサッカリンまたはサッカリンナトリウムなどの甘味剤は、吸入/鼻腔内投与を予定した本発明の製剤に加えることができる。
【0125】
吸入/鼻腔内投与用製剤は、即時放出および/または修飾放出であるように、例えば、PGLAを用いて製剤化することができる。修飾放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的放出およびプログラム化放出が含まれる。
【0126】
乾燥粉末吸入剤およびエアゾル剤の場合、投薬量単位は、一定の計測量を送達するバルブによって決定される。全1日用量は、単回用量で、またはより通常は、当日中に分割用量として投与することができる。
【0127】
直腸/膣内投与
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、直腸または膣に、例えば、坐剤、膣坐剤または浣腸剤の形で投与することができる。カカオ脂は、伝統的な坐剤基剤であるが、いろいろな代用物を、適宜用いることができる。
【0128】
直腸/膣内投与用製剤は、即時放出および/または修飾放出であるように製剤化することができる。修飾放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的放出およびプログラム化放出が含まれる。
【0129】
眼/耳投与
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、眼または耳に直接的に、典型的には、等張のpH調整済み滅菌生理食塩水中の超微粉懸濁液または溶液の小滴の形で投与することもできる。眼および耳投与に適する他の製剤には、軟膏剤、生物分解性(例えば、吸収性ゲルスポンジ、コラーゲン)および非生物分解性(例えば、シリコーン)植込剤、ウェファー、レンズおよび粒状物または、ニオソーム(niosomes)またはリポソームなどの小胞システムが含まれる。架橋ポリアクリル酸、ポリビニルアルコール、ヒアルロン酸、セルロース系ポリマー、例えば、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルセルロースまたはメチルセルロース、またはヘテロ多糖ポリマー、例えば、ゲランガム(gelan gum)は、塩化ベンザルコニウムなどの保存剤と一緒に包含されてよい。このような製剤は、イオン導入法によって送達することもできる。
【0130】
眼/耳投与用製剤は、即時放出および/または修飾放出であるように製剤化することができる。修飾放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的放出またはプログラム化放出が含まれる。
【0131】
他の技法
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、前述のいずれかの投与様式で用いるためのそれらの溶解度、溶解速度、風味マスキング、バイオアベイラビリティーおよび/または安定性を改善するために、シクロデキストリンおよびその適する誘導体またはポリエチレングリコール含有ポリマーなどの可溶性高分子物質と組み合わせることができる。
【0132】
例えば、薬物−シクロデキストリン複合体は、概して、大部分の剤形および投与経路に有用であることが判っている。包接および非包接双方の複合体を用いることができる。薬物との直接複合体形成に代わるものとして、シクロデキストリンは、補助添加剤として、すなわち、担体、希釈剤または可溶化剤として用いることができる。これら目的に最も一般的に用いられるのは、α−、β−およびγ−シクロデキストリンであり、その例は、国際特許出願WO91/11172号、WO94/02518号およびWO98/55148号に見出されうる。
【0133】
キット部材
活性化合物の組合せを、例えば、特定の疾患または状態を処置する目的で投与することが望ましいことがありうる限りにおいて、二つまたはそれを超える医薬組成物の少なくとも一つが、本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物を含有する医薬組成物を、それら組成物の共投与に適するキットの形で好都合に組み合わせることができるということは、本発明の範囲内である。
【0134】
したがって、本発明のキットは、二つまたはそれを超える別々の医薬組成物の少なくとも一つが、本発明のCRTH2受容体アンタゴニスト、例えば、本発明による一般式Iの化合物を含有する医薬組成物と、それら組成物を別々に保持するための、容器、分割ボトルまたは分割ホイルパケットなどの手段を含む。このようなキットの例は、錠剤、カプセル剤等の包装に用いられる公知のブリスターパックである。
【0135】
本発明のキットは、異なった剤形を、例えば、経口および非経口投与するのに;異なった投薬間隔で別々の組成物を投与するのに;または別々の組成物を互いに対して滴定するのに、特に適している。コンプライアンスを助けるために、キットは、典型的に、投与の手引きを含むが、いわゆる記憶を助けるものと一緒に与えられてよい。
【0136】
投薬量
ヒト患者への投与について、本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物の全1日用量は、当然ながら、投与様式に依存して、典型的に、0.1mg〜1gの範囲内である。医薬製剤の基本は、好ましくは、単位剤形である。このような形での製剤は、適当な量の活性成分を含有する単位用量へと小分けされる。その単位剤形は、包装された製剤でありうるが、その包装は、バイアルまたはアンプル中の包装された錠剤、カプセル剤および散剤のような、不連続的量の製剤を含有する。更に、単位剤形は、それ自体がカプセル剤、錠剤、カシェ剤またはロゼンジでありうるし、またはそれは、包装された形でのいずれかこれらの適当な数でありうる。単位用量製剤中の活性成分の量は、具体的な用途および活性成分の力価にしたがって、0.1mg〜1gに変更または調整することができる。医学的使用において、薬物は、例えば、100mgまたは300mgのカプセル剤として1日1〜3回投与することができる。治療的使用において、本発明の薬学的方法で利用される化合物は、約0.01mg〜約100mg/kgの初期投薬量で毎日投与される。約0.01mg〜約100mg/kgの1日用量範囲が好適である。全1日用量は、単回用量または分割用量で投与することができ、しかも医師の裁量において、本明細書中に与えられた典型的な範囲外であってよい。
【0137】
これら投薬量は、約60kg〜70kgの体重を有する平均的なヒト対象に基づいている。医師は、体重がこの範囲外にある、乳児および高齢者などの対象のための用量を容易に決定することができる。
【0138】
疑わしさを免れるために、本明細書中の「処置」の意味は、治癒的処置、待期的処置および予防的処置の意味を包含する。
次の実施例は、本発明の態様および理論を詳しく説明し、そしてCRTH2受容体の強力且つ選択的なアンタゴニストであるN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドの使用を含む。アンタゴニスト N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドの構造を、図4に示す。
【実施例】
【0139】
in vivo モデル用の動物
Charles River(Manston, Kent, UK.)より入手した150〜400gの体重の雄 Sprague Dawley ラットを、三つの群に収容した。被験動物は全て、12時間明/暗サイクル下で(07時00分に照明)、飲食を自由にして維持した。実験は全て、処置のことを知らない観察者が、Home Office Animals (Scientific Procedures) Act 1986 にしたがって行った。
【0140】
ニューロパシー性疼痛の慢性狭窄損傷(CCI)ラットモデル
坐骨神経のCCIは、Bennett and Xie(Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain:33:87-107, 1988)によって以前に記載されたように行った。被験動物を、2%イソフルオラン/O2混合物で麻酔した。右後大腿を剃毛し、1%ヨウ素を綿棒で塗る。次に、被験動物を、手順および鼻用コーンによって手術中維持される麻酔の間、恒温ブランケットに移した。皮膚を、大腿骨の線に沿って切開した。総坐骨神経を、大腿二頭筋を介する鈍的剥離によって大腿の中央に露出させた。約7mmの神経を、坐骨三分岐に近位に、神経下に鉗子を挿入することによって自由にし、その神経を、静かに大腿から持ち上げた。縫合糸を、鉗子を用いて神経下に引き寄せ、僅かな抵抗が感じられるまで簡単な結び目で縛った後、二重に結んだ。その手順を、4本の結紮糸(4−0絹)で、約1mm間隔で神経周囲を緩く縛るまで繰り返した。切開を層状に閉じた。
【0141】
ラットのストレプトゾシン(STZ)で誘発される糖尿病ニューロパシー
糖尿病は、0.9%滅菌生理食塩水中に新たに溶解したストレプトゾシン(50mg/kg)の単回腹腔内注射によって誘発した。ストレプトゾシン注射は、再現性のある機械的異痛症を3週間以内に誘発し、少なくとも7週間持続する(Chen and Pan, (Chen SR and Pan HL. Hypersensitivity of Spinothalamic Tract Neurons Associated With Diabetic Neuropathic Pain in Rats. J Neurophysiol 87: 2726-2733, 2002)。
【0142】
ラットの静的および動的異痛症の評価
静的異痛症
被験動物は、異痛症評価の前に、ワイヤ底試験ケージに慣らした。静的異痛症は、後足底表面への力の昇順(0.6g、1g、1.4g、2g、4g、6g、8g、10g、15gおよび26g)での von Frey 毛(Stoelting, Wood Dale, Illinois, USA)の適用によって評価した。各々の von Frey 毛を、最大6秒間または引っ込め応答が起こるまで、足に適用した。von Frey 毛への引っ込め応答が確かめられたら、その足を再試験し、引っ込めを生じるものより下のフィラメントで開始し、そしてその後、引っ込めが起こらなくなるまで、残りのフィラメントを力の降順で用いた。最高の26gの力は、足を持ち上げ、更には、応答を引き出し、したがって、中止点であった。各々の被験動物の両後足をこの方式で調べた。応答を引き出すのに必要な力の最低量を、足引っ込め閾値(PWT)としてグラムで記録した。静的異痛症は、被験動物が、普通のラットには無害である4gまたはそれ未満の刺激に応答した場合、存在すると定義した(Field MJ, Bramwell S, Hughes J, Singh L. Detection of static and dynamic components of mechanical allodynia in rat models of neuropathic pain: are they signalled by distinct primary sensory neurones? Pain, 1999;83:303-11)。
【0143】
動的異痛症
動的異痛症は、後足底表面を綿棒で軽くたたくことによって評価した。全身運動活性を記録することがないように、活発でない充分に慣らされたラットでこの手順を行うように注意した。各々の時点で、少なくとも2回の測定値を得、その平均が、足引っ込め潜伏時間(PWL)であった。15秒以内に反応が示されなかった場合、その手順を終え、被験動物をこの引っ込め時間に割り当てた。疼痛引っ込め反応は、しばしば、繰り返し足を縮めることまたは舐めることを伴った。動的異痛症は、被験動物が、たたくことを開始して8秒以内に綿棒刺激に応答した場合、存在すると考えた(Field et al, 1999)。
【0144】
ラットのカラゲナンで誘発される温熱性痛覚過敏(CITH)
温熱性痛覚過敏は、ラット足底試験(Ugo Basile, Comerio, Italy)を用いて、Hargreaves et al., (1988) による変法にしたがって評価した。簡単にいうと、ラットは、ガラステーブル上の3個のパースペックスボックスから成る装置に慣らした。移動輻射熱源を、テーブルの下に置き、所望の足に集中させた。足引っ込め潜伏時間(PWL)を、各々の被験動物の両後足について3回記録し、その平均が、左右後足についてのベースラインであった。装置は、ナイーブラットにおいて約10秒のPWLを与えるように検量した。足底区域の組織損傷を防止するために、22.5秒中止を認めた。λカラゲナンを、右後足底内に注射し(100μl,20mg/ml)、PWLのベースライン記録を、投与から2時間後に行った。
【0145】
データ分析
実験は全て、盲検で行った。静的異痛症は、中央値として表し[UQ;LQ]、Mann Whitney U検定によって分析した。動的異痛症および温熱性痛覚過敏は、算術平均±SEMとして表し、ANOVAによって分析した。
【0146】
CCIで誘発される静的および動的異痛症へのN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドの作用
ナイーブラットは、von Frey 適用に約10gの足引っ込め閾値を示し、綿棒刺激の適用について完全に無害な状態を見いだす。神経損傷後、ラットは、これら刺激双方への増加した感受性を示し、静的および動的異痛症の発症を示す。外科手術後14日から、被験動物は、典型的な静的および動的異痛応答を示し、試験前に記録されたベースラインは、全ての被験動物において、それぞれ<4gおよび<4秒であった。これら異痛応答は、ビヒクル処置群において、実験の間中一貫したままであった。経口(PO)投与後、N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミド(12.5mg/kg、25mg/kgおよび50mg/kg)は、CCIで誘発される静的および動的異痛症の維持を、用量依存方式で逆転させた(図1Aおよび図1B)。MEDは、25mg/kgであり、静的および動的双方の異痛症において、投与後1時間にピーク作用を生じた。最高用量は、双方の行動試験において、投薬後30分から抗異痛作用を示した(ビヒクル処置群に対してp<0.01)。それは、静的異痛症を、ガバペンチン(100mg/kg,PO)に匹敵する曲線プロフィールで逆転させ、一方、動的異痛症でのその作用は、あまり強力ではないが、ビヒクル処置されたCCIラットとは有意に異なる(投与後2時間で11.8±1.0対3.5±0.7)。
【0147】
STZで誘発される静的および動的異痛症へのN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドの作用
ナイーブラットは、von Frey 適用に約10gの足引っ込め閾値を示し、綿棒刺激の適用について完全に無害な状態を見いだす。ストレプトゾシン注射後、ラットは、これら刺激双方への増加した感受性を示し、静的および動的異痛症の発症を示す。STZ注射後14日目から、ラットを、それらの疼痛様閾値(PWTおよびPWL)に基づいて選択し、薬理学的研究に用いた。全ての被験動物でのベースライン読みは、静的および動的異痛症について、それぞれ<4gおよび<5秒であった(図2Aおよび図2B)。これら異痛応答は、ビヒクル処置群において、実験の間中一貫したままであった。N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミド(25mg/kg,PO)投与後、STZで誘発される静的および動的双方の異痛症の維持は逆転した。ピーク作用は、化合物投与後1時間に認められ、2時間まで生物学的に関連性があった。正対照として実験に包含されたガバペンチン(100mg/kg,PO)は、静的および動的双方の異痛症終点の完全な逆転を生じた。
【0148】
ラットのCITHへのN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドの作用
ナイーブラットは、熱刺激に約10秒の足引っ込め潜伏時間(PWL)を示す。カラゲナンの片側足底内注射から2時間後に、ラットは、熱刺激への感受性を増加させて、同側足に温熱性痛覚過敏の発症を示した(反対側および同側の足それぞれについて11.0±0.5秒および4.1±0.3秒のベースラインの平均)。これらPWTは、ビヒクル処置群において、時間経過の間中一貫したままであった(図3B)。N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミド(25mg/kg,PO)は、温熱性痛覚過敏の維持を、投与後2時間にピーク作用を有して、完全に逆転させた(ビヒクル処置群の3.9±0.2に対して10.1±0.6)。この抗痛覚過敏作用は、化合物投与後5時間は一貫したままであり、反対側足には作用が認められなかった(図3A)。正対照として実験に包含されたモルヒネ(3mg/kg,SC)は、予想される鎮痛作用を生じた。それは、投薬から30分後に、ナイーブラットのベースライン値を上回って両後足でPWLを増加させた。
【0149】
考察
本研究は、選択的CRTH2受容体(CRTH2R)アンタゴニストが、ニューロパシーの慢性狭窄損傷およびSTZで誘発される糖尿病動物モデルにおいて静的および動的異痛症を逆転させることができるということを示している。更に、そのアンタゴニストは、CITHラットモデルにおいて長期持続性抗痛覚過敏作用を生じた。N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドを、経口投与後のいろいろな時点で、ラットの血液およびCSF中に投与した。注射から4時間後に、25mg/kgの化合物で経口処置されたナイーブ動物の脳脊髄液(CSF)中で、5Xを超えるIC50(rIC50=45nM)を測定した。したがって、CITHラットモデルで認められた抗痛覚過敏プロフィールが、中枢活性化合物の画像であり、その化合物は、血液脳関門を越えて、その受容体において中枢作用すると考えられる。
【0150】
CRTH2RアンタゴニストであるN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドは、ニューロパシー性疼痛の動物モデルにおいて効力を示す。
【0151】
結論として、CRTH2受容体アンタゴニストであるN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドは、ニューロパシーの2種類の齧歯類動物モデル、具体的には、坐骨神経の慢性狭窄損傷(CCI)ラットおよびストレプトゾシン(STZ)で誘発される糖尿病ラットにおける静的および動的異痛症を逆転させる(Field MJ, et al, 1999, Pain, 83: 303-311)。
【0152】
同じ動物モデルにおいて、N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドの作用は、ニューロパシー性疼痛の処置用に現在市場で主流の薬物である Gabapentin に匹敵する。
【0153】
この実験的証拠は、CRTH2受容体アンタゴニストが、ヒトニューロパシー性疼痛の処置に有効であるということを示唆している。
【図面の簡単な説明】
【0154】
【図1】図1は、CCIで誘発される(a)静的および(b)動的異痛症への、経口投与後のN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドおよびガバペンチン(gabapentin)の作用を示す。von Frey 毛への足引っ込め閾値(PWT)または綿棒刺激への足引っ込め潜伏時間(PWL)のベースライン(BL)を評価した。化合物投与後、PWLおよびPWT双方を、4時間まで再評価した。データは、6匹/群の被験動物より作成する。静的異痛症データは、中央値として表し(力,g)[UQ;LQ]、(Mann Whitney U検定)によって分析する。動的異痛症は、算術平均±SEMとして表し、(一元ANOVA後、Dunnett's t検定)によって分析する。各々の時点において、ビヒクル処置群に対して、*P<0.05、**P<0.01、***P<0.001。
【図2】図2は、STZで誘発される(a)静的および(b)動的異痛症への、経口投与後のN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドおよびガバペンチンの作用を示す。von Frey 毛への足引っ込め閾値(PWT)または綿棒刺激への足引っ込め潜伏時間(PWL)のベースライン(BL)を評価した。化合物投与後、PWLおよびPWT双方を、4時間まで再評価した。データは、6匹/群の被験動物より作成する。静的異痛症データは、中央値として表し(力,g)[UQ;LQ]、(Mann Whitney U検定)によって分析する。動的異痛症は、算術平均±SEMとして表し、(一元ANOVA後、Dunnett's t検定)によって分析する。各々の時点において、ビヒクル処置群に対して、*P<0.05、**P<0.01、***P<0.001。
【図3】図3は、カラゲナンで誘発される温熱性痛覚過敏へのN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドおよびモルヒネの作用を示す。3(a)反対側足、3(b)同側足。熱刺激への足引っ込め潜伏時間(PWL)ベースライン(BL)を、未処置ラットで評価した。PWLを、足底内カラゲナン投与後2時間に再評価した後、N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドまたはビヒクルを投与した。PWLを、カラゲナン投与後8時間まで再評価した。データは、6匹/群の被験動物の算術平均±SEMとして表す。各々の時点において、ビヒクル処置群に対して、*P<0.05、**P<0.01、***P<0.001(一元ANOVA後、Dunnett's t検定)。
【図4】図4は、MW=349.43のCRTH2受容体アンタゴニストであるN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミド(ラセミ体)である。
【技術分野】
【0001】
本発明は、ニューロパシー性疼痛の処置用の薬剤の製造におけるCRTH2受容体アンタゴニストの使用、およびCRTH2受容体アンタゴニストを用いてニューロパシー性疼痛を処置する方法に関する。
【背景技術】
【0002】
1999年、Nagata et al は、白血球化学誘引物質受容体ファミリーに属する新規なGタンパク質共役型受容体(GPCR)である、GPR44としても従来知られているCRTH2(Th2細胞で発現された化学誘引物質受容体相同分子)を識別した(Nagata et al., FEBS Letters (1999) 459(2):195-9)。CRTH2受容体は、マウスの場合、脳、肺およびリンパ系器官を含めた広範囲の組織から選択的に発現される(Abe et al., Gene (1999) 227(1):71-7)。CRTH2受容体は、ヒトの場合、Th2細胞、好酸球および好塩基球で選択的に発現されるが、Th1細胞、B細胞およびNK細胞では発現されない(Nagata et al., FEBS Letters (1999) 459(2):195-9)。
【0003】
Bauer et al,(EP1170594A2号を参照されたい)は、プロスタグランジンD2(PGD2)を、CRTH2受容体のアゴニストである内因性リガンドとして識別した。PGD2は、免疫学的に刺激されたマスト細胞およびTh2細胞から放出される。CRTH2とPGD2との相互作用は、アレルギー性炎症の標的組織におけるTh2細胞のアレルゲンで誘発される動員に臨界的役割を果たすことが知られている。更に、CRTH2は、血中好酸球および好塩基球のPGD2依存性細胞移動を媒介する。したがって、CRTH2受容体は、炎症およびアレルギー性反応を与える分子イベントにおいて活性な役割を果たすことが分かった。CRTH2のPGD2依存性活性と相互作用する化合物は、免疫系の異常な活性化に関連したアレルギー性疾患状態および炎症の処置に有用であると考えられる。
【0004】
Torisu et al.,(WO03022814号を参照されたい)は、PGD2受容体、特に、DP受容体に特異的に結合するインドール誘導体を識別した。その化合物は、CRTH2受容体に結合し且つ生物学的活性に拮抗すると考えられるので、疼痛の防止および/または処置に有用であると考えられる。
【0005】
Pairaudeau et al.,(WO2004089884号を参照されたい)は、呼吸器障害を処置するのに有用な医薬化合物としての置換されたフェノキシ酢酸;それらを含有する医薬組成物;およびそれらの製造方法を開示した。化合物は、薬剤としての、具体的には、CRTH2受容体活性のモジュレーターとしての活性を有すると考えられ、したがって、PGD2およびその代謝産物の過度のまたは未調節の生産によって引き起こされるまたは悪化するヒトおよび非ヒト動物の状態/疾患の(治療的または予防的)処置に用いうると考えられ;この文書によれば、このような状態の例には、ニューロパシー性疼痛症候群が含まれる。
【0006】
驚くべきことに、本発明者は、CRTH2受容体のアンタゴニストである化合物が、ニューロパシー性疼痛の処置に有効であるということを発見した。
多数の異なった疼痛状態、例えば、慢性疼痛、ニューロパシー性疼痛、炎症性疼痛、侵害受容性疼痛、内臓痛、背痛、および疾患および変性に関連した疼痛がある。当業者は、更に、これら疼痛タイプが、臨床的におよび機械論的に異なるということを承知している。このような状態は、多数の疼痛症状のために、しばしば、臨床的に処置するのが困難である。例えば、(糖尿病性ニューロパシー、または末梢神経またはCNSへの外傷などの疾患に起因することがありうる状態である)ニューロパシー性疼痛の患者は、しばしば、痛覚過敏(hyperlagesia)(有害刺激への過大疼痛)、過感作、異痛症(以前の無害刺激による疼痛)、更には、継続している疼痛を含めた多数の疼痛症状を示す。更に、ニューロパシー性疼痛は、防御的役割がないので、病的である。それは、しばしば、最初の原因が消散した後によく現れる。
【0007】
侵害受容性疼痛は、組織損傷によって、または損傷を引き起こす可能性のある強い刺激によって誘発される。疼痛求心性神経(afferents)は、損傷部位にある侵害受容器による刺激の伝達によって活性化され、それらの終末レベルで脊髄を感作する。次に、これは、脊髄路を脳へと中継され、そこで疼痛が知覚される(Meyer et al., 1994 Textbook of Pain 13-44)。侵害受容器の活性化は、二つのタイプの求心性神経線維を活性化する。有髄A−δ線維は、急速伝達し、鋭く且つ刺すような痛覚に関与するが、無髄C線維は、より遅い速度で伝達し、鈍いまたはうずく痛みを伝える。損傷が修復された時、疼痛は止む。中程度〜重症の急性侵害受容性疼痛は、挫傷/捻挫による疼痛、術後疼痛(いずれかのタイプの外科的処置後の疼痛)、外傷後疼痛、熱傷、心筋梗塞、急性膵炎および腎疝痛の顕著な特徴であるが、これに制限されるわけではない。更に、癌関連急性疼痛症候群は、一般的には、化学療法毒性、免疫療法、ホルモン療法および放射線療法などの治療的相互作用のためである。中程度〜重症の急性侵害受容性疼痛は、腫瘍関連疼痛でありうる癌疼痛(例えば、骨痛、頭痛および顔面痛、内臓痛)または癌療法に関連した疼痛(例えば、化学療法後症候群、慢性術後疼痛症候群、照射後症候群)、ヘルニア様のまたは断裂した椎間円板によるまたは腰椎間関節、仙腸関節、傍脊柱筋または後縦靱帯の異常によることがありうる背痛の顕著な特徴であるが、これに制限されるわけではない。
【0008】
炎症過程は、組織損傷または異物の存在に応答して活性化される一連の複雑な生化学的および細胞性のイベントであり、それが、腫脹および疼痛を引き起こす(Levine and Taiwo 1994: Textbook of Pain 45-56)。関節炎痛は、炎症性疼痛集団の大部分を構成している。リウマチ様疾患は、先進諸国において最も一般的な慢性炎症性状態の一つであり、関節リウマチは、障害の共通原因である。RAの正確な病因は未知であるが、現在の仮説は、遺伝的および微生物学的双方の因子が重要でありうるということを示唆している(Grennan & Jayson 1994 Textbook of Pain 397-407)。ほぼ1600万人の米国人が、症候性変形性関節症(OA)または変性関節疾患を有すると推定されたが、その大多数は60歳を超えているので、その集団の年齢が増加するにつれて、これは4000万人へと増加して、これをきわめて大きな公衆衛生問題にすると考えられる(Houge & Mersfelder 2002 Ann Pharmacother. 36: 679-686; McCarthy et al., 1994 Textbook of Pain 387-395)。大部分のOA患者は、疼痛のために、医療行為を探し求めている。関節炎は、心理・社会的および身体的機能に有意の影響力があり、晩年における障害の主原因であることが知られている。他のタイプの炎症性疼痛には、炎症性腸疾患(IBD)が含まれるが、これに制限されるわけではない。
【0009】
対照的に、ニューロパシー性疼痛の臨床的特性は、神経病理学的過程自体の機構、場所および重症度によって優先的に決定される。ニューロパシー性疼痛は、神経系の一次病変または機能不全によって開始するまたは引き起こされる疼痛として定義される(IASP定義)。神経損傷は、外傷および疾患によって引き起こされることがありうるので、「ニューロパシー性疼痛」という用語は、多様な病因を有する多数の障害を包含する。これらには、糖尿病性ニューロパシー、帯状疱疹後神経痛、背痛、癌ニューロパシー、HIVニューロパシー、幻想肢痛、手根管症候群、慢性アルコール症、甲状腺機能低下症、三叉神経痛、尿毒症またはビタミン欠乏症が含まれるが、これに制限されるわけではない。ニューロパシー性疼痛は、防御的役割がないので、病的である。それは、しばしば、最初の原因が消散した後によく現れ、一般的に何年間も続き、患者の生活の質を有意に低下させる(Woolf and Mannion 1999 Lancet 353:1959-1964)。ニューロパシー性疼痛の症状は、しばしば、同じ疾患の患者間でさえも異種であるので、処置するのが困難である(Woolf & Decosterd 1999 Pain Supp. 6: S141-S147; Woolf and Mannion 1999 Lancet 353:1959-1964)。それらには、継続することがありうる自発痛、または痛覚過敏(有害刺激への増加した感受性)および異痛症(通常は無害な刺激への感受性)などの発作性で且つ異常な誘発痛が含まれる。
【0010】
更に、抗炎症薬およびアヘン薬のような、侵害受容性疼痛を処置するのに慣用的に用いられる薬物は、慢性ニューロパシー性疼痛患者への効力が限られている。したがって、抗痙攣薬および三環系抗うつ薬は、しばしば、不十分に耐性であるにもかかわらず、ニューロパシーへの主な鎮痛薬である。侵害受容性疼痛および炎症性疼痛とは対照的に、ニューロパシー性疼痛は、公然として処置するのが困難であり、慢性経過をたどるが;それは、非ステロイド性抗炎症薬およびアセトアミノフェンのような、侵害受容性疼痛の処置に有効である標準的な鎮痛療法には全くまたはきわめて不十分にしか応答しないし;そして侵害受容性疼痛状態の場合よりも、予想通りに且つ確実にオピオイドに応答することに乏しい。侵害受容性疼痛に有効な処置は、ニューロパシー性疼痛へと広げられるとは考えられない。例えば、Gabapentin(Neurontin(登録商標))および Pregabalin(Lyrica(登録商標))は、慢性狭窄性坐骨神経損傷(CCI)およびストレプトゾシンで誘発される糖尿病(STZ)のラットモデルにおいて静的および動的双方の異痛症を逆転させるが、モルヒネは、CCIラットモデルにおいて、動的ではなく静的異痛症を逆転させる(Field MJ, et al, 1999, Pain, 83: 303-311)。更に、慢性疼痛の場合の非ステロイド性抗炎症薬(NSAID)およびコルチコステロイド類(デキサメタゾンおよびプレドニゾン)の効能は疑わしく、齧歯類動物かまたは患者での一貫した薬理学的証拠によって支持されない。同様に、臨床データは、ニューロパシー性疼痛疾患におけるこれら薬剤の限られた使用を示し、潜在的に、これら化合物を用いた比較的少数の齧歯類動物研究を説明する。Schafers(2004, Experimental Neurology, 185: 160-168)は、ラットのCCIで誘発される疼痛を逆転させる場合の非選択的(イブプロフェン)および選択的(Celebrex(登録商標)TM)COX2阻害剤の有意の作用を示さなかった。
【0011】
これら理由、臨床的特性の相違、機構の相違、および処置への従順性の相違のために、ニューロパシー性疼痛は、当業者の判断で、侵害受容性疼痛および炎症性疼痛とは明らかに異なると考えられるであろう。
【0012】
したがって、これら疼痛状態の原因となるニューロパシー性疼痛過程のキー段階を妨げる薬学的に活性な化合物を識別することが、救急医学的に要求されている。
したがって、中枢神経系(CNS)において中心的に発現される疼痛経路に関与する標的受容体を識別すること、およびCNSおよび関連組織において中枢作用することによって鎮痛作用を及ぼす薬学的に活性な化合物を識別することは、好都合である。CRTH2受容体は、皮質、視床、扁桃および脊髄が含まれるが、必ずしもこれに制限されるわけではないCNS組織において中心的に発現されること、更には、多数の末梢組織でも発現されることが分かった(Nagata and Hirai (2003) prostaglandins, leukotrienes and Essential Fatty acids 69: 169-177)。CRTH2は、ヒトの脳および脊髄でも発現される。
【0013】
支持組織分布データは、Abe et al., (1999) Gene 227: 71-77 に記載のようにマウスにおいて、そして更には、Shichijo et al., (2003) JPET 307: 518-525 に記載のようにラットにおいて、CRTH2受容体について提供された。
【発明の開示】
【課題を解決するための手段】
【0014】
発明の簡単な説明
本発明は、ニューロパシー性疼痛の処置用の薬剤の製造におけるCRTH2受容体アンタゴニストの使用に関する。
【0015】
本発明は、更に、哺乳動物対象のニューロパシー性疼痛を処置する方法であって、その対象に、治療的有効量のCRTH2受容体アンタゴニストを投与することを含む方法を提供する。
【0016】
本発明は、更に、ニューロパシー性疼痛の処置用のCRTH2受容体アンタゴニストを提供する。
発明の詳細な説明
「CRTH2リガンド」または「CRTH2受容体リガンド」という用語は、CRTH2受容体に結合する化合物を意味する。このような化合物は、有機または無機の化合物類似体またはそれらの立体異性体;または天然または合成の他の化学的または生物学的な化合物、例えば、天然プロスタグランジン;ペプチド;ポリペプチド;抗体および抗体リガンド結合性ドメインを含めたタンパク質;ホルモン;ヌクレオチド;DNAまたはRNAなどの核酸であってよく、そして更には、化合物または立体異性体の薬学的に許容しうる塩、化合物または立体異性体のプロドラッグ、またはプロドラッグの薬学的に許容しうる塩を包含する。CRTH2受容体リガンドは、CRTH2受容体アンタゴニストでもありうる。
【0017】
本明細書中で用いられる「CRTH2受容体アンタゴニスト」という用語は、CRTH2受容体の活性化をブロックするように作用する化合物を意味する。適するアンタゴニストの例には、天然プロスタグランジンなどの有機化合物またはそれらの類似体、または他の化合物、有機または無機の分子、ペプチド、抗体および抗体のリガンド結合性ドメインを含めたタンパク質、DNAまたはRNAなどの核酸が含まれる。CRTH2受容体アンタゴニストの適する例は、例えば、有機化合物、またはペプチドまたはタンパク質、抗体およびそれらのフラグメント、ペプチド模擬有機化合物であって、例えば、CRTH2受容体の細胞外ドメイン(ECD)に結合し且つ天然リガンドによって引き起こされる活性を阻害するものでありうる。更に、有機化合物、ペプチド、抗体またはそれらのフラグメントであって、CRTH2受容体のECD(またはその一部分)が共有結合するものは、更に、PGD2に結合し、したがって、それを「中和する」ことがありうる。アンタゴニストという用語は、ランダムペプチドライブラリー(例えば、Lam et al., 1991, Nature 354:82-84; Houghten et al., 1991, Nature 354:84-86 を参照されたい)およびDおよび/またはL立体配置アミノ酸より構成される組合せ化学由来分子ライブラリーのメンバーが含まれるがこれに制限されるわけではないペプチドおよび可溶性ペプチド;ホスホペプチド(ランダムまたは部分変性に支配されたホスホペプチドライブラリーのメンバーが含まれるがこれに制限されるわけではない;例えば、Songyang et al., 1993, Cell 72:767-778 を参照されたい);抗体(多クローン性、単クローン性、ヒト化、抗イディオタイプ、キメラまたは短鎖の抗体、およびFab、F(ab’)2およびFAb発現ライブラリーフラグメントおよびそれらのエピトープ結合性フラグメントが含まれるがこれに制限されるわけではない);および有機または無機の小分子を包含する。適するアンタゴニストは、更に、ランダムまたは組合せのペプチドまたは非ペプチドなどの多様性ライブラリーに由来してよく、用いることができるライブラリー、例えば、化学的に合成されたライブラリー、リコンビナント(例えば、ファージ表示ライブラリー)および in vitro 翻訳に基づくライブラリーはいずれも、当該技術分野において知られている。化学的に合成されたライブラリーの例は、Fodor et al., 1991, Science 251:767-773; Houghten et al., 1991, Nature 354:84-88; Lam et al., 1991, Nature 354:82-84; Medynski, 1994, Bio/Technology 12:709-710; Gallop et al., 1994, J. Medicinal Chemistry 37(9):1233-1251; Ohlmeyer et al., 1993, Proc. Natl. Acad. Sci. USA 90:10922-10926; Erb et al., 1994, Proc. Natl. Acad. Sci. USA 91:11422-11426; Houghten et al., 1992, Biotechniques 13:412; Jayawickreme et al., 1994, Proc. Natl. Acad. Sci. USA 91:1614-1618; Salmon et al., 1993, Proc. Natl. Acad. Sci. USA 90:11708-11712; PCT公開WO93/20242号;および Brenner and Lerner, 1992, Proc. Natl. Acad. Sci. USA 89:5381-5383 に記載されている。
【0018】
ファージ表示ライブラリーの例は、Scott & Smith, 1990, Science 249:386-390; Devlin et al., Science 249:404-406; Christian, et al., 1992, J. Mol. Biol. 227:711-718; Lenstra, 1992, J. Immunol. Meth. 152:149-157; Kay et al., 1993, Gene 128:59-65; および1994年8月18日付のPCT公開WO94/18318号に記載されている。
【0019】
非ペプチドライブラリーの例として、ベンゾジアゼピンライブラリー(例えば、Bunin et al., 1994, Proc. Natl. Acad. Sci. USA 91:4708-4712 を参照されたい)は、使用に適応することができる。ペプトイドライブラリー(Simon et al., 1992, Proc. Natl. Acad. Sci. USA 89:9367-9371)を用いることもできる。用いることができるライブラリーの別の例であって、ペプチド中のアミド官能基を過メチル化して、化学変換された組合せライブラリーを生じたものは、Ostresh et al.(1994, Proc. Natl. Acad. Sci. USA 91:11138-11142)によって記載されている。
【0020】
それらライブラリーをスクリーニングすることは、種々の一般的に知られている方法のいずれによっても行うことができる。例えば、ペプチドライブラリーのスクリーニングを開示している次の参考文献:Parmley & Smith, 1989, Adv. Exp. Med. Biol. 251:215-218; Scott & Smith, 1990, Science 249:386-390; Fowlkes et al., 1992; BioTechniques 13:422-427; Oldenburg et al., 1992, Proc. Natl. Acad. Sci. USA 89:5393-5397; Yu et al., 1994, Cell 76:933-945; Staudt et al., 1988, Science 241:577-580; Bock et al., 1992, Nature 355:564-566; Tuerk et al., 1992, Proc. Natl. Acad. Sci. USA 89:6988-6992; Ellington et al., 1992, Nature 355:850-852; 全て Ladner et al. による米国特許第5,096,815号、米国特許第5,223,409号および米国特許第5,198,346号;Rebar & Pabo, 1993, Science 263:671-673; およびPCT公開WO94/18318号を参照されたい。
【0021】
CRTH2受容体アンタゴニストである化合物は、CRTH2受容体上の、PGD2が通常結合する同じ部位で結合し且つ作用することができるが、それは、PGD2結合部位に遠いCRTH2上の部位で作用することができる。CRTH2受容体のアンタゴニストは、CRTH2受容体活性化を、いずれか適する手段によって、例えば、CRTH2受容体にまたはPGD2またはいずれか他の活性化リガンドに結合することなどによってブロックするように作用し、それによって、PGD2または活性化リガンドとCRTH2受容体との結合を阻害することができる。このようなアンタゴニストは、CRTH2受容体においてPGD2の代わりに作用することができるし、またはPGD2と相互作用する、PGD2と一緒になる、またはそれ以外には、PGD2を修飾することによって、CRTH2受容体においてそれが作用する方法に影響を与えることができる。或いは、アンタゴニストは、CRTH2受容体下流活性を、例えば、CRTH2受容体シグナル伝達のモジュレーションによってブロックするように作用して、下流シグナリングイベントに影響を与えることができるが、この活性は、例えば、PGD2によってまたはCRTH2受容体のいずれか他の活性化リガンドによって活性化されるようなシグナルの伝達を妨げることができるGタンパク質の阻害剤に共通している。或いは、アンタゴニストは、CRTH2受容体活性を、CRTH2受容体遺伝子発現に影響を与えることによってブロックするように作用することができるが、このようなアンタゴニストには、例えば、分子、タンパク質または小有機分子、またはDNAまたはRNAが含まれるが、それは、転写に影響を与えるまたはスプライシングイベントを妨げるので、完全長または切断された形のCRTH2受容体の発現に影響することがありうる。したがって、このようなCRTH2受容体アンタゴニストには、アンチセンスRNAおよびsRNA産物(サイレンス妨害RNA)も含まれうる。
【0022】
本発明での使用に適するCRTH2受容体アンタゴニストの例には、Annex 1に添付の特許出願PCT/IB03/04505号に概括的ににまたは具体的に開示された化合物、特に、cis−N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドという化合物およびその薬学的に許容しうる塩および溶媒和化合物が含まれる。
【0023】
本発明での使用に適するCRTH2受容体アンタゴニストの更に別の例には、特許出願WO−2004007451号に概括的ににまたは具体的に開示された化合物、3−スルホニルインドール誘導体およびそれらの塩、特に、2−[5−クロロ−3−(4−クロロフェニルスルホニル)−2−メチル−1H−インドール−1−イル]酢酸という化合物が含まれる。
【0024】
更に、特許出願WO03066047号は、インドール−3−酢酸誘導体およびそれらの塩である、本発明での使用に適するCRTH2受容体アンタゴニストの更に別の例、特に、2−[1−(2,6−ジフェノキシピリミジン−4−イル)−2,5−ジメチル−1H−インドール−3−イル]酢酸という化合物を開示している。特許出願WO03101981号は、更に適するCRTH2受容体アンタゴニスト、置換されたインドール−1−イル酢酸誘導体、特に、3−(1,2−ベンゾイソチアゾール−3−イル)−5−フルオロ−2−メチル−1H−インドール−1−酢酸という化合物を開示している。WO03101961号は、適するCRTH2受容体アンタゴニストの追加の例、置換されたインドール化合物、特に、3−[(3−メトキシフェニル)チオ]−2,5−ジメチル−1H−インドール−1−酢酸という化合物を開示し、そしてWO03066046号は、適するCRTH2受容体アンタゴニストの追加の例、インドール−3−酢酸誘導体、特に、1−(7−クロロキナゾリン−4−イル)−2−メチル−5−(1−メチルエチル)−1H−インドール−3−酢酸という化合物を開示している。適するCRTH2受容体アンタゴニストの更に別の例には、特許出願WO03097042号に概括的ににまたは具体的に開示された化合物、特に、Ramatroban という化合物、そしてWO03097598号の、特に、(3−[1−(4−フルオロベンゼンスルホニル)ピロリジン−3−イル]インドール−1−イル)酢酸という化合物が含まれる。適するCRTH2受容体アンタゴニストの更に別の例には、CRTH2受容体への抗体または抗体サブドメイン、具体的には、抗CRTH2受容体単クローン性抗体または抗体サブドメイン、例えば、CRTH2受容体に特異的な抗体またはサブドメイン、またはPGD2によって一部分与えられたエピトープに特異的な抗体またはサブドメインが含まれる。
【0025】
好ましくは、本発明によるCRTH2受容体アンタゴニストは、中枢作用性である。中枢作用性であるためには、このような化合物は、血液脳関門を貫通することができるはずである。
【0026】
本発明での使用に好適なCRTH2受容体アンタゴニストは、一般式(I):
【0027】
【化1】
【0028】
(式中、R1は、H、(C1〜C4)アルキル、(C2〜C4)アルケニル、(C2〜C4)アルキニルまたは(CH2)mRxであり;
Rxは、het1,フェニルまたは(C3〜C6)シクロアルキルであり、このhet1,フェニルおよび(C3〜C6)シクロアルキルは、1個またはそれを超えるQ1基または(C1〜C4)アルキル基で置換されていてよく、この(C1〜C4)アルキルは、1個またはそれを超えるQ1基で置換されていてよく;
Q1は、ハロゲン、NO2、CN、SO2CH3、SO2NR9R10、OR9、COOR9、C(=O)NR9R10、NR9R10、NR9SO2R10、NR9C(=O)R10またはC(=O)R9であり、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
mは、0、1および2より選択される整数であり;
R2は、(C1〜C4)アルキルであり、ここにおいて、アルキル基は、ハロゲン、OR9、NR9R10、COOR9、C(=O)NR9R10、NHSO2R9およびC(=O)(C1〜C4)アルキルより選択される1個またはそれを超える置換基で置換されていてよく、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
R3は、(C3〜C6)シクロアルキルまたは−A−Ryであり;
Aは、結合、直鎖または分岐状の(C1〜C3)アルキレンまたは(C2〜C3)アルケニレンであり;
Ryは、(C6〜C12)アリールまたはhet2であり、ここにおいて、アリール基およびhet2基は、(C6〜C12)アリール、het1、Q2および(C1〜C4)アルキルより選択される1個またはそれを超える置換基で置換されていてよく、この(C1〜C4)アルキルは、1個またはそれを超えるQ2基であって、同じであるかまたは異なる基で置換されていてよく;
Q2は、ハロゲン、NO2、CN、SO2CH3、SO2NR9R10、OR9、SR9、OCH2CF3、COOR9、C(=O)NR9R10、NR9R10、NR9SO2R10、NR9C(=O)R10またはC(=O)R9であり、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
R4は、Hまたは(C1〜C4)アルキルであり;
R5、R6、R7およびR8は、同じであるかまたは異なり、H、Q3および(C1〜C4)アルキルより選択され、この(C1〜C4)アルキルは、1個またはそれを超えるQ3基であって、同じであるかまたは異なる基で置換されていてよく;
Q3は、ハロゲン、NO2、CN、SO2CH3、SO2NR9R10、OR9、SR9、COOR9、C(=O)NR9R10、NR9R10、NR9SO2R10、NR9C(=O)R10またはC(=O)R9であり、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
het1は、酸素、硫黄および窒素より選択される1〜4個のヘテロ原子を有する5〜10員芳香族複素環であり;そして
het2は、酸素、硫黄および窒素より選択される1〜4個のヘテロ原子を有する5〜10員の飽和、不飽和または部分飽和複素環式基である)
を有する化合物またはその薬学的に許容しうる塩または溶媒和化合物である。
【0029】
het1およびhet2は各々、好ましくは、酸素、硫黄および窒素より選択される1〜3個のヘテロ原子を含有する5員または6員芳香族複素環である。
具体的には、het1およびhet2について好適な定義は、イソオキサゾリル、オキサゾリル、チエニル、ピラゾリル、ピロリル、トリアゾリル、テトラゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、ピリジニル、ピラジニル、ベンゾオキサジアゾリルまたはピラゾロピリジニル、キノリニルおよびキノキサリニルである。
【0030】
(C6〜C12)アリールは、6〜12個の炭素原子を含有する芳香族炭素環を意味すると理解される。好適なアリール基はフェニルである。
CRTH2受容体をコードしているヌクレオチド配列およびアミノ酸は、当業者に知られており、受託番号AB008535として GenBank で見出されうる。
【0031】
好ましくは、本発明で用いるためのCRTH2受容体アンタゴニストは、選択的CRTH2受容体アンタゴニストである。
「選択的」という用語は、リガンドまたはアンタゴニストが、ある特定の受容体に、別の受容体へのリガンドまたはアンタゴニストの結合親和性と比較した場合、より大きい親和性で結合するということを意味する。好ましくは、第一受容体へのアンタゴニストの結合親和性は、第二受容体への結合親和性より約50%またはそれを超えて大きい。より好ましくは、第一受容体へのアンタゴニストの結合親和性は、第二受容体への結合親和性より約75%またはそれを超えて大きい。最も好ましくは、第一受容体へのアンタゴニストの結合親和性は、第二受容体への結合親和性より約90%またはそれを超えて大きい。本発明の好ましい態様において、アンタゴニストは、CRTH2受容体への一層大きい結合親和性を示す。具体的には、好適なアンタゴニストは、ケモカイン受容体ファミリーのメンバー、例えば、C3a、C5a、FMLP、LTB4、GPCR0269、GPCR0232またはGPCR0288受容体のような、またはDタイププロスタノイド受容体(DP)のような、またはプロスタノイド受容体ファミリー、例えば、プロスタグランジンE2受容体サブタイプEP1〜EP4、プロスタグランジンF受容体、トロンボキサンA2受容体、最も好ましくは、DPのような別の受容体への結合と比較した場合、より大きい親和性でCRTH受容体に結合するものである。好適なアンタゴニストは、CRTH2受容体をマイクロモルまたはそれより大きい親和性で結合すると考えられる。より好適なアンタゴニストは、CRTH2受容体をナノモルまたはそれより大きい親和性で結合する。本発明の好適なCRTH2受容体アンタゴニストには、CRTH2受容体の選択的アンタゴニストであるリガンドまたは化合物が含まれる。選択性は、カルシウム動員などの機能性終点に基づいて決定することもできる。
【0032】
CRTH2受容体リガンドは、例えば、化合物ライブラリーをスクリーニングすることによって識別することができる。受容体のアンタゴニストを識別する方法は、当業者に周知である。CRTH2受容体リガンドを識別するのに用いることができる具体的な手順を、下に示す。
【0033】
生理学的疼痛は、外部環境からの潜在的に有害な刺激による危険を警告するように設計された重要な防御機構である。そのシステムは、ある一定の組の一次感覚ニューロンによって作動し、末梢伝達機構を経た有害刺激によって独占的に活性化される(統合的概説については、Millan 1999 Prog. Neurobio. 57:1-164)。これら感覚線維は、侵害受容器として知られ、遅い伝導速度を有する小径軸策を特徴とする。侵害受容器は、有害刺激の強さ、持続時間および質を、そして脊髄へのそれらの局所解剖学的に組織化された投射路によって、その刺激の場所をコードする。それら侵害受容器は、A−δ線維(有髄)およびC線維(無髄)という二つの主要タイプがある侵害受容神経線維上に見出される。侵害受容器入力によって生じる活性は、後角における複雑なプロセシング後、直接的にかまたは脳幹中継核(brain stem relay nyclei)を経て、腹底側視床へ、そして次に皮質へと移送され、そこで、痛みの感覚を生じる。
【0034】
強い急性痛および慢性痛は、病態生理学的過程によって駆動される同じ経路を含み、それ自体、防御機構を与えることを止め、代わりに、広範囲の疾患状態に関連した症状を弱くする原因となることがありうる。疼痛は、多数の外傷および疾患状態の特徴である。疾患または外傷によって体組織への実質的な損傷が起こった場合、侵害受容器活性化の特性は変化する。末梢では、損傷の周りの局所におよび侵害受容器が終端となるところでは中枢に鋭敏化が存在する。これは、損傷部位および付近の正常組織に過敏性をもたらす。急性痛の場合、これら機構は、修復過程が起こることを可能にしうるし且つそれに有用でありうるが、過敏性は、損傷がいったん治癒すると、正常に戻る。しかしながら、多数の慢性痛状態では、過敏性は、治癒過程をはるかに長く続け、通常は、神経系損傷のためである。この損傷は、しばしば、求心性線維の不適応をもたらす(Woolf & Salter 2000 Science 288: 1765-1768)。患者の症状の中で、不快および異常な感受性が特徴となっている場合、臨床的疼痛が存在する。患者は、全く異種である傾向があり、いろいろな疼痛症状を示すことがありうる。多数の典型的な疼痛サブタイプが存在する。(1)鈍い、焼けるようまたは刺すようでありうる自発痛;(2)有害刺激への過大疼痛応答(痛覚過敏);(3)通常は無害な刺激によって疼痛が生じる(異痛症)(Meyer et al., 1994 Textbook of Pain 13-44)。背痛、関節炎痛、CNS外傷またはニューロパシー性痛を有する患者は、類似した症状を有することがありうるが、根本的な機構は異なるので、異なった処置戦略を必要とすることがありうる。したがって、疼痛は、異なった病態生理学ゆえに、多数の異なった領域に分けることができ、これらには、侵害受容性疼痛、炎症性疼痛、ニューロパシー性疼痛等が含まれる。何等かのタイプの疼痛は、多数の病因を有するので、二つ以上の領域に分類することができるということは注目されるべきであり、例えば、背痛、癌性疼痛は、侵害受容性およびニューロパシー性双方の成分を有する。
【0035】
ニューロパシー性疼痛は、神経系の一次病変または機能不全によって開始するまたは引き起こされる疼痛として定義される(IASP定義)。神経損傷は、外傷および疾患によって引き起こされることがありうるので、「ニューロパシー性疼痛」という用語は、多様な病因を有する多数の障害を包含する。これらには、糖尿病性ニューロパシー、帯状疱疹後神経痛、背痛、癌ニューロパシー、HIVニューロパシー、幻想肢痛、手根管症候群、慢性アルコール症、甲状腺機能低下症、三叉神経痛、尿毒症またはビタミン欠乏症が含まれるが、これに制限されるわけではない。ニューロパシー性疼痛は、防御的役割がないので、病的である。それは、しばしば、最初の原因が消散した後によく現れ、一般的に何年間も続き、患者の生活の質を有意に低下させる(Woolf and Mannion 1999 Lancet 353: 1959-1964)。ニューロパシー性疼痛の症状は、しばしば、同じ疾患の患者間でさえも異種であるので、処置するのが困難である(Woolf & Decosterd 1999 Pain Supp. 6: S141-S147; Woolf and Mannion 1999 Lancet 353: 1959-1964)。それらには、継続することがありうる自発痛、または痛覚過敏(有害刺激への増加した感受性)および異痛症(通常は無害な刺激への感受性)などの発作性で且つ異常な誘発痛が含まれる。
【0036】
「治療的有効量」という用語は、疾患を処置する;特定の疾患の一つまたはそれを超える症状を改善する、減衰させるまたは除去する;または疾患の一つまたはそれを超える症状の開始を防止するまたは遅らせる化合物または化合物組合せの量を意味する。
【0037】
「患者」という用語は、イヌ、ネコ、ウシ、ウマ、ヒツジ、ガチョウおよびヒトなどの動物を意味する。具体的には、好適な患者は、双方の性のヒトを含めた哺乳動物である。
「薬学的に許容しうる」という用語は、物質または組成物が、製剤の他の成分と相溶性であるべきであり且つ患者に有害であってはならないということを意味する。
【0038】
「処置すること」、「処置する」または「処置」という用語は、防止的または予防的および待期的処置を包含する。
一次結合検定
CRTH2受容体リガンドおよびアンタゴニストは、例えば、化合物ライブラリーをスクリーニングすることによっておよびいろいろなスクリーニング技術を用いることによって識別することができる。受容体のリガンドおよびアンタゴニストを識別する方法は知られている。CRTH2受容体リガンドおよびアンタゴニストを識別するのに用いることができる具体的な手順は、下に示されるが、本明細書中に援用される欧州特許出願第01305857.3号(公開番号EP1170594号)に公表されている。
【0039】
CRTH2受容体のリガンドを識別する結合検定は、直接結合検定の形でかまたは競合結合検定として行うことができる。直接結合検定の場合、試験化合物を、CRTH2受容体への結合について調べる。もう一方において、競合結合検定は、CRTH2受容体への結合について、プロスタグランジンD2(PGD2)またはそのファミリーの他の適するリガンドと競合する試験化合物の能力を評価する。
【0040】
直接結合検定の場合、CRTH2受容体を、CRTH2受容体に試験化合物を結合させる条件下において試験化合物と接触させる。その結合は、溶液中または固体表面上で起こりうる。好ましくは、試験化合物を、検出用に予め標識する。発光体、蛍光体、または放射性同位体またはそれを含有する基、または酵素または染料などの非同位体標識のような検出可能な基はいずれも、標識するのに用いることができるが、これに制限されるわけではない。結合が起こるのに充分なインキュベーション時間後、その反応を、過度にまたは非特異的に結合した試験化合物を除去する操作および条件に暴露する。典型的には、これは、適当な緩衝液で洗浄することを伴う。最後に、CRTH2受容体−試験化合物複合体の存在を検出する。
【0041】
競合結合検定の場合、試験化合物を、CRTH2受容体へのPGD2の結合を破壊するまたは促進するそれらの能力について検定する。標識されたPGD2を、CRTH2受容体またはそのフラグメントまたは誘導体と混合し、そしてそれらの間の相互作用が、通常は生じると考えられる条件下に、試験化合物の添加を伴ってまたは伴うことなく置くことができる。CRTH2受容体を結合する標識PGD2の量は、試験化合物の存在下または不存在下において結合した量と比較することができる。
【0042】
好ましい態様において、複合体の形成および検出を容易にするために、結合検定は、固体表面上に固定された一つまたはそれを超える成分で行われる。いろいろな態様において、その固体支持体は、ポリカーボネート、ポリスチレン、ポリプロピレン、ポリエチレン、ガラス、ニトロセルロース、デキストラン、ナイロン、ポリアクリルアミドおよびアガロースでありうるが、これに制限されるわけではない。その支持体形状には、ビーズ;メンブラン;微粒子;微量滴定プレート、試験管または他の反応容器などの反応容器の内部表面が含まれうる。CRTH2受容体または他の成分の固定は、共有結合または非共有結合の付着によって達成されうる。一つの態様において、その付着は、間接的であってよい、すなわち、付着抗体を介していてよい。別の態様において、CRTH2受容体および負の対照は、グルタチオンS−トランスフェラーゼ(GST)などのエピトープで標識されるので、固体表面への付着は、抗GST(Santa Cruz Biotechnology)などの商業的に入手可能な抗体によって媒介されうる。例えば、このような親和性結合検定は、固体支持体に固定されるCRTH2受容体を用いて行うことができる。典型的に、結合反応の非固定成分、この場合はPGD2かまたは試験化合物を、検出可能であるように標識する。発光体、発色団、蛍光体、または放射性同位体または基の検出、または酵素または染料などの非同位体標識の検出のような種々の標識方法が、利用可能であり且つ用いることができる。一つの好ましい態様において、試験化合物を、フルオレセインイソチオシアネート(FITC,Sigma Chemicals, St. Louis より入手可能)などの発蛍光団で標識する。次に、標識された試験化合物、または試験化合物を加えたPGD2を、特異的結合を生じさせる条件下において固体支持体と接触させる。結合反応が起こった後、未結合のおよび非特異的に結合した試験化合物を、表面を洗浄することによって分離する。固相への結合パートナーの付着は、当業者に知られているいろいろな方法で行うことができ、化学架橋;プラスチック表面への非特異的接着;固相に付着した抗体との相互作用;結合パートナーに付着したリガンド(ビオチンなど)と、固相に付着したリガンド結合性タンパク質(アビジンまたはストレプトアビジンなど)との間の相互作用等が含まれるが、これに制限されるわけではない。最後に、固体表面上に残留する標識は、当該技術分野において知られているいずれかの検出方法によって検出することができる。例えば、試験化合物が発蛍光団で標識されている場合、蛍光計を用いて複合体を検出することができる。
【0043】
好ましい態様において、結合検定は、次のように行うことができる:
(a)CRTH2受容体を発現する細胞を、ペレットにし、そして室温において検定緩衝液(Ca2+およびMg2+を包含し且つHEPESおよび重炭酸ナトリウムを補足した Hank's 平衡生理食塩水)で2回洗浄する。それら細胞を、2x107個/mlの細胞濃度で再懸濁させる。96ウェルU底微量滴定皿を用いて、検定を次のように設定する(150μl容量中);
(b)50μlのビヒクル(検定緩衝液中0.3%DMSOとして、完全ウェル);または50μlの30μM冷PGD2であって、10μMの最終検定濃度を生じるもの[冷PGD2の原液を、DMSO中に10mMの原液濃度で溶解させ、−20℃で貯蔵した後、それを、使用するために3:1000に希釈して、30μMの最終原液濃度とした];50μlの細胞(106個/ウェルのための2x107個/ml);50μlの6nM[3H]−PGD2を、2nMの最終濃度のために加える(Amersham;162Ci/mmol,メタノール:水:アセトニトリル(3:2:1)中の0.1Ci/ml,617nMを6nMの濃度のために10μl/mlの検定緩衝液に希釈)。
【0044】
(c)プレートを、室温で20分間インキュベートさせた後、遠心分離を行う(2800rpm,Sorval RT6000,5分間,4℃)。上澄みを捨てて、非特異的結合を減少させる。プレート(Packard Unifilter プレートGF/C,予め3%PEI中に少なくとも1時間浸漬)を、冷検定緩衝液で、150μl/ウェルの緩衝液洗浄で6回洗浄することによって採取する。プレートを一晩乾燥させる。50μlのシンチレーション液の添加後、プレートをシンチレーション計数器で計数する(1分/ウェル)。(好ましくは、CRTH2受容体は、CRTH2受容体を発現する無傷の細胞の形で、またはCRTH2受容体を含有する単離された細胞膜として、結合検定に加える。したがって、CRTH2受容体へのリガンドの直接結合、またはPGD2−CRTH2受容体複合体をモジュレーションする試験化合物の能力は、培養物中の無傷の細胞において、試験化合物の存在下および/または不存在下で検定することができる)。CRTH2受容体を発現する細胞には、M.G. Reth et al., Nature, 317(6035), pp. 353-365, 1985 に開示されたような、CRTH2受容体を発現する300−19細胞(形質転換されたプレBリンパ球)が含まれる。CRTH2受容体は、アンピシリンおよびネオマイシンの耐性マーカーを含有するプラスミドから発現させることができ、CMVプロモーターによって駆動する。プロラク(prolac)シグナリングペプチドは、遺伝子インサートの膜発現を可能にし、N末端にある Flag ペプチド標識が、発現された分子の好都合な検出を可能にする。CRTH2受容体の好ましい発現レベルは、約40,000分子/細胞表面である。被標識PGD2は、CRTH2受容体を発現する細胞と、またはこのような細胞から得られる粗製抽出物と混合することができ、そして試験化合物を加えることができる。単離された膜は、CRTH2受容体と相互作用する化合物を識別するのに用いることができる。例えば、単離された膜を用いた典型的な実験では、細胞を遺伝子操作して、CRTH2受容体を発現させることができる。膜は、標準的な技法によって採取し、そして in vitro 結合検定で用いることができる。被標識リガンド(例えば、125I標識PGD2)を、膜に結合させ、比活性について検定し;そして特異的結合を、過剰の未標識(冷)リガンドの存在下で行われた結合検定との比較によって決定する。或いは、可溶性CRTH2受容体を、組換え発現させ、非細胞結合検定に利用して、CRTH2受容体に結合する化合物を識別することができる。一つまたは複数の組換え発現されたCRTH2受容体ポリペプチド、またはCRTH2受容体の一つまたはそれを超える細胞外ドメインを含有する融合タンパク質は、非細胞基材スクリーニング検定に用いることができる。或いは、CRTH2受容体の一つまたはそれを超える細胞外ドメインに該当するペプチド、またはCRTH2受容体の一つまたはそれを超える細胞外ドメインを含有する融合タンパク質は、非細胞基材検定システムで用いて、CRTH2受容体の細胞質部分に結合する化合物を識別することができるが;このような化合物は、CRTH2受容体のシグナル伝達経路をモジュレーションするのに有用でありうる。非細胞基材検定の場合、組換え発現されたCRTH2受容体を、当業者に知られている手段によって、試験管、微量滴定ウェルまたはカラムなどの固体支持体に付着させる。次に、試験化合物を、CRTH2受容体に結合するそれらの能力について検定する。
【0045】
或いは、結合反応は、溶液中で行うことができる。この検定では、被標識成分を、溶液中において一つまたは複数の結合パートナーと相互作用させる。被標識成分とその一つまたは複数の結合パートナーとの間のサイズ差が、このような分離を可能にする場合、その分離は、限外濾過器の細孔が、未結合の被標識成分を通過させるが、その一つまたは複数の結合パートナーまたはその一つまたは複数のパートナーに結合した被標識成分を通過させない限外濾過器を介して結合反応生成物を通過させることによって行うことができる。分離は、結合パートナーに対する抗体、結合パートナーに予め付着したリガンドと相互作用しうるリガンド結合性タンパク質等のような、被標識成分の結合パートナーを溶液から捕捉することが可能ないずれかの試薬を用いて行うこともできる。
【0046】
本発明の化合物は、CRTH2受容体アンタゴニスト、好ましくは、選択的CRTH2受容体アンタゴニストである。これら化合物は、典型的には、少なくとも100nM、好ましくは、10nM未満、より好ましくは、1nM未満の低いIC50値を有する。
【0047】
CRTH2受容体アンタゴニストの力価(下に記載の機能性検定において受容体の機能活性の値の半分を生じるアンタゴニストの濃度として定義することができるIC50力価に基づく)は、好ましくは、ヒト受容体(リコンビナントおよび/または天然)において、少なくとも100nMのIC50、より好ましくは、10nM未満、そして更に好ましくは、1nM未満である。例えば、機能性細胞基材検定の場合、IC50は、例えば、プロスタグランジンD2(または他の小分子アゴニスト)に応答して、ヒトCRTH2受容体の最大活性化を50%阻害するアンタゴニストのモル濃度である。結合検定の場合、IC50は、3H標識プロスタグランジンD2(または他の適当なリガンド)の特異的結合の50%を置き換えるアンタゴニストのモル濃度である。
【0048】
CRTH2受容体アンタゴニストの選択性は、好ましくは、CRTH2受容体について、他のGPCR、特に、Dタイプのプロスタノイド受容体(DP受容体)にまさって、そして或いは、化学誘引物質受容体サブファミリーの関連メンバー、例えば、補体受容体C3a、C5a、FMLP(FMet−Leu−Phe受容体)FMLP受容体IおよびII、ロイコトリエンB4(LTB4)、GPCR0269、GPCR0232、GPCR0288受容体に対して、少なくとも10倍選択的であり、好ましくは、それは、少なくとも100倍選択的、そして更に好ましくは、少なくとも1000倍選択的であるはずである。選択性は、概して、目的の受容体に適当なリガンドについての、二つの受容体サブタイプ間の化合物の相対力価である。
【0049】
CRTH2受容体リガンドまたはアンタゴニストは、CRTH2受容体への選択性について、DPと比較して調べることができる。この検定では、3H−PGD2の結合について競合する試験化合物各々の能力を、CRTH2およびDP双方の受容体において測定し、そしてIC50値を(μMで)決定する。対照は、3H−PGD2と競合する冷PGD2を用いて設定することができる。上述の結合検定手順はいずれも、用いることができる。CRTH2受容体アンタゴニストの選択性は、他のGPCR、特に、Dタイプのプロスタノイド受容体またはDP受容体と比較して少なくとも10倍であるはずであり(N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドは、>50倍選択的である)、好ましくは、それは、少なくとも100倍選択的、そしてなお一層好ましくは、少なくとも1000倍選択的であるはずであり、或いは、アンタゴニストは、CRTH2受容体について、C3a、C5a、FMLP、LTB4、GPCR0269、GPCR0232またはGPCR0288のいずれの受容体にもまさって選択的であるはずである。
【0050】
機能性検定
機能性検定方法は、CRTH2受容体に媒介される過程をモジュレーションする化合物およびCRTH2受容体のアンタゴニストである化合物を識別することについて知られている。それら方法は、概して、(a)CRTH2受容体発現性細胞と試験化合物とを、場合により、PGD2または別のCRTH2受容体活性化リガンドの存在下において接触させること;および(b)得られたCRTH2受容体活性レベル、または細胞中のCRTH2受容体の発現レベルを、この測定された活性レベルまたは発現レベルが、試験化合物の不存在下で測定されたレベルと異なる場合に、CRTH2受容体−PGD2に媒介される過程をモジュレーションする化合物が識別されるように測定すること含む工程を包含する。測定されるCRTH2受容体活性は、PGD2と相互作用する能力、またはPGD2への細胞の走化性応答、または細胞内Ca2+濃度/動員、または反応性酸素種の放出、アデニル酸シクラーゼ/サイクリックAMP生産の阻害、またはアクチン重合でありうる。機能性検定のための実施例プロトコールを下に示す。
【0051】
カルシウム動員は、フローサイトメトリーによって、および細胞内に捕捉される蛍光染料で標識することによって検出し且つ測定することができる。例えば、インドール−1染料は、カルシウム結合時の発光スペクトルの変化を示す。カルシウム結合染料によって生じる蛍光対未結合染料によって生じる蛍光の比率を用いて、細胞内カルシウム濃度を算定する。実施例方法では、CRTH2受容体を発現する細胞を集め、そしてカルシウムフラックス検定を行う前日に、新鮮培地中に約2x105/mlで再懸濁させる。細胞を37℃で20〜30分以下の間インキュベート後、回転沈降させ、そしてインドール−1AMを含有する、37℃に予熱された50mlの新鮮PT1緩衝液(Hank's Buffer,pH7.2〜7.4;10mM Hepes;1.6mM CaCl2)中に、1000万個/mlの濃度で再懸濁させる。細胞を刺激し、そして蛍光計(Photon Technology Corporation, International)を用いて蛍光を測定する。読みが安定した後、時間軸をリセットし、そしてPGD2を、特定の時点(例えば、20秒)に加える。応答後、次の試薬を次の順序でキュベットに加えて、全カルシウムを放出させ且つキレート化する。20μlの18% Triton X−100、20μlの3M Tris,pH8.5および20μlの0.5M EDTA,pH8.5。その実験を、試験化合物の存在下および不存在下で繰り返す。試験化合物の不存在下では、PGD2は、CRTH2受容体を発現する細胞において15nMのEC50で、増加した([Ca2+])を生じる。したがって、アンタゴニスト試験化合物の存在下において、EC50は減少するであろうと考えられる。
【0052】
アクチン重合検定は、化合物の活性を特性決定する二次スクリーニングとして用いることができる。アクチン重合は、重合したアクチン線維を結合するアクチン特異的蛍光標識であるニトロベンゾオキサジアゾール(NBD)−ファラシジン(phallacidin)を用いて検定することができる。その検定は、次のように行うことができる。細胞標品を、10mM HEPES、100/10の Pen/Strep および0.5%FCSを加えたRPMI1640中に、5〜10x106個/mlで再懸濁させる。その細胞懸濁液を、96ウェルU底ポリプロピレン微量滴定プレート中に小分けする(100μL/ウェル)。50ulの適当な刺激物質(PGD2または試験化合物、またはPGD2および試験化合物双方)を、8チャンネルピペットを用いて加えた後、正確に25秒後、リゾホスホチジルコリン(0.5mg/ml)、Hank's 平衡塩類溶液(100μl,10x)、16%ホルムアルデヒド(800ul)および6.6uM NBD−ファラシジンをMEOH(100ul)中に含有する50ulの停止用溶液を加える。そのプレートを、室温で15分間放置する。次に、プレートを1000rpmで5分間遠心分離し、上澄みを振り落とし(flicked off)、そして細胞ペレットを、2%FCSおよび0.2%アジ化ナトリウムを加えた250ulのPBS中に再懸濁させる。次に、各々の試料をFACS Calliber 装置で読み取る。細胞は、リンパ球領域中の正分散/副分散データを用いてゲート処理する。応答は、ビヒクルで処理された細胞と刺激物質で処理された細胞との間のFL−1蛍光中央値の変化によって測定する。試験化合物は、PGD2の存在下および不存在下で検定し、PGD2単独を含有する試料と比較することができる。CRTH2受容体細胞のPGD2で誘発されるアクチン重合を減少させる化合物は、CRTH2受容体アンタゴニスト候補として識別される。
【0053】
in vivo 手順
CRTH2受容体アンタゴニストの鎮痛作用は、選択された疼痛状態の動物モデルを用いて in vivo で決定することができる。いくつかの疼痛状態モデルが知られているが、CRTH2受容体アンタゴニストの鎮痛作用を決定するのに用いることができる具体的な手順を下に示す。
【0054】
ある一つの疼痛モデルは、ラットのニューロパシー性疼痛を有するストレプトゾシンで誘発される糖尿病モデルである。この手順は、糖尿病を誘発するために、Charles River Sprague dawley ラット(225〜250g)などの被験動物への単回用量でのストレプトゾシン(50mg/kg,i.p.)の投与を行う。被験動物を、投与から2週間後に、静的および動的異痛症試験を用いて評価し、そしてニューロパシー性疼痛が確認された場合、それらを用いて、ニューロパシー性疼痛への化合物の作用について更に評価する。
【0055】
ラットの疼痛の慢性狭窄性損傷(CCI)モデルは、坐骨神経周囲を弛緩性結紮糸で縛ることを行い、Charles River Sprague dawley 雄ラット(175〜200g)を、麻酔室に入れ、2%イソフルオラン(isofluorane)O2混合物で麻酔する。右後大腿を剃毛し、1%ヨウ素を綿棒で塗る。次に、被験動物を、鼻用コーンによって手術中維持される麻酔および手順の間、恒温ブランケットに移す。皮膚を、大腿骨の線に沿って切開する。総坐骨神経を、大腿二頭筋を介する鈍的剥離によって大腿の中央に露出させる。坐骨三分岐に近位の約7mmの神経を、神経下に鉗子を挿入することによって自由にし、その神経を、静かに大腿から持ち上げる。神経からの筋膜の除去を助けるように、鉗子を静かに数回開閉する。縫合糸を、鉗子を用いて神経下に引き寄せ、僅かな抵抗が感じられるまで簡単な結び目で縛った後、二重に結ぶ。その手順を、4本の結紮糸(4−0絹)で、約1mm間隔で神経周囲を緩く縛るまで繰り返す。切開を層状に閉じる。手術から14日後、被験動物の静的異痛症、動的異痛症または体重支持不足について評価する。
【0056】
* 疼痛状態の別の動物モデルには、坐骨神経の部分緊密結紮である Seltzer モデル(Seltzer, Z. (1995). Sem. Neurosci, 8: pp.34-39)または坐骨神経の二つの脊髄神経の一方の緊密結紮である Chung's モデル(Kim SH, Chung JM. Pain (1992); 50: pp.355-63)または慢性狭窄性損傷モデル(CCI)(Bennett GJ, Xie Y-K. Pain (1988); 33: pp.87-107)が含まれる。更に別の動物疼痛モデルには、疼痛誘発剤、例えば、カプサイシン(Dirks J, Petersen KL, Rowbotham MC, Dahl JB, Anesthesiology. 2002 Jul;97(1):pp.102-107);またはホルマリン(Tjolsen, A. et al (1992), Pain 51, pp.5-17);または完全フロイントアジュバント(Abdi S, Vilassova N, Decosterd I, et al, Anesth Analog 2000; 91: pp.955-99);またはカラゲナン(Itoh, M., Takasaki, I., Andoh, T., Nojima, H., Tominaga, M. & Kuraishi, Y. (2001) Neurosci. Res., 40, pp.227-233);またはタキソール(Polomano RC. Mannes AJ. Clark US. Bennett GJ, (2001) Pain. 94(3): pp.293-304);またはビンカアルカロイド、ビンクリスチン(Aley KO, Reichling DB, Levine JD, Neuroscience (1996); 73: pp.259-65);または内臓痛のためのテルペンチン(Koster, R., Anderson, M. and De Beer, E.J., Acetic acid for analgesic screening, Fed. Proc., 18 (1959) 412./ Mogil, J.S., Kest, B., Sadowski, B. and Belknap, J.K., Differential genetic mediation of sensitivity to morphine in genetic models of opiate anti-nociception: influence of nociceptive assay, J. Pharmacol. Exp. Ther., 276 (1996a) 532-544./ Ness TJ, Gebhart GF, Pain (1990);41: pp.167-234 および McMahon SB, Agents Actions (1988); 25: pp.231-233)の投与が含まれる。更に別の動物モデルは、その被験動物に、有害な身体的刺激を、例えば、有害な熱刺激の投与(Malmberg, A.B., and Bannon, A.W. Models of neciception: hot-plate, tall-flick, and formalin tests in rodents. Current Protocols in Neuroscience 1999; pp 8.9.1-8.9.15);または有害な寒冷刺激または有害な圧力刺激の投与またはUV照射(S.J. Boxall, A. Berthele, D.J. Laurie, B. Sommer, W. Zieglgansberger, L. Urban and T.R. Tolle, Enhanced expression of metabotropic glutamate receptor 3 messenger RNA in the rat spinal cord during ultraviolet irradiation induced peripheral inflammation Neuroscience (1988) 82(2): pp.591-602)によって与えることを行うことができる。
【0057】
疼痛状態の別の動物モデルは、関節炎またはHIVまたはヘルペスまたは癌または糖尿病のような有痛性疾患状態を生まれつき有する動物の選択を行うことができる。或いは、その動物を、関節炎またはHIVまたはヘルペスまたは癌または糖尿病のような疼痛誘発性疾患状態を有するための動物の修飾により、疼痛状態を経験するように手配することができる。被験動物は、疾患による疼痛状態を有するように、いろいろな方法で、例えば、糖尿病性ニューロパシーを誘発するストレプトゾシンの投与(Courteix,C., Eschalier,A., Lavarenne,J., Pain, 53 (1993) pp.81-88.);またはHIV関連ニューロパシー性疼痛を引き起こすウイルスタンパク質の投与(Herzberg U. Sagen J., Journal of Neuroimmunology. (2001 May 1), 116(1): pp.29-39);または関節炎および炎症性疼痛を誘発する完全フロイントアジュバントまたはモノヨードアセテートの投与(Rikard Holmdahl, Johnny C. Lorentzen, Shemin Lu, Peter Olofsson, Lena Wester, Jens Holmberg, Ulf Pettersson Immunological Reviews (2001) Volume 184, Issue 1, pp.184);またはヘルペスおよびヘルペス後神経痛(post hepatic neuralgia)を引き起こす水痘−帯状疱疹ウイルスの投与(Fleetwood-Walker SM. Quinn JP. Wallace C. Blackburn-Munro G. Kelly BG. Fiskerstrand CE. Nash AA. Dalziel RG., Journal of General Virology. 80 (Pt 9):2433-6, 1999 Sep.);または癌を引き起こすための動物への発癌物質または癌細胞の投与(Shimoyama M. Tanaka K. Hasue F. Shimoyama N, Pain. 99(1-2): pp.167-74, 2002 Sep)によって修飾することができる。
【0058】
動的異痛症は、被験動物の後足底表面を綿棒で軽くたたくことによって評価することができる。全身運動活性を記録することがないように、活発でない充分に慣らされたラットでこの手順を行うように注意する。各々の時点で、少なくとも2回の測定値を得、その平均が、足引っ込め潜伏時間(PWL)である。15秒以内に反応が示されない場合、その手順を終え、被験動物をこの引っ込め時間に割り当てる。したがって、15秒は、事実上、引っ込めなしである。引っ込め反応は、しばしば、繰り返し足を縮めることまたは舐めることを伴う。動的異痛症は、被験動物が、たたくことを開始して8秒以内に綿棒刺激に応答する場合、存在すると考えられる。
【0059】
ベースライン評価後、被験動物に、鎮痛性評価用の化合物を、次の経路、すなわち、経口投与、皮下、腹腔内、静脈内または鞘内の一つによって投与することができる。PWLは、次の時点、すなわち、30分、1時間、2時間、3時間、4時間、5時間、6時間、7時間、24時間の全部またはいくつかにおいて再評価する。被験動物は、それらのベースライン値によって、各々の化合物群に無作為に割り当てられる。平均および標準誤差平均を、各々の化合物について各々の時点で計算する。動的異痛症の測度を、一元ANOVAを用いてそれぞれの対照と比較後、Dunnett's t検定を用いて、各々の時点において化合物とビヒクルを比較する。一群当たりの最小被験動物数は、6匹である。
【0060】
静的異痛症は、後足底表面への力の昇順(0.6g、1g、1.4g、2g、4g、6g、8g、10g、15gおよび26g)での von Frey 毛(Stoelting, Wood Dale, Illinois, USA)の適用によって評価することができる。被験動物は、異痛症評価の前に、ワイヤ底試験ケージに慣らす。各々の von Frey 毛を、最大6秒間または引っ込め応答が起こるまで、足に適用する。von Frey 毛への引っ込め応答が確かめられたら、その足を再試験し、引っ込めを生じるものより下のフィラメントで開始し、そしてその後、引っ込めが起こらなくなるまで、残りのフィラメントを力の降順で用いる。最高の26gの力は、足を持ち上げ、更には、応答を引き出し、したがって、中止点である。各々の被験動物の両後足をこの方式で調べられる。応答を引き出すのに必要な力の最低量を、足引っ込め閾値(PWT)としてグラムで記録する。静的異痛症は、被験動物が、普通のラットには無害である4gまたはそれ未満の刺激に応答した場合、存在すると定義される。
【0061】
ベースライン評価後、被験動物に、鎮痛性評価用の化合物を、次の経路、すなわち、経口、皮下、腹腔内、静脈内または鞘内の一つによって投与し、そしてPWTを、次の時点、すなわち、30分、1時間、2時間、3時間、4時間、5時間、6時間、7時間、24時間の全部またはいくつかにおいて再評価する。静的異痛症測定値を、ノンパラメトリック結果についての Kruskall-Wallis 検定後、Mann Whitney's U検定を用いて、ビヒクル群に対して分析する。一群当たりの最小被験動物数は、6匹である。
【0062】
温熱性痛覚過敏は、ラット足底試験(Ugo Basile, Italy)を用いて、Hargreaves et al., (1988) Pain 32:77-88 の変法にしたがって評価する。ラットは、高さのあるガラステーブル上の3個のパースペックスボックスから成る装置に慣らす。移動輻射熱源を、テーブルの下に置き、後足に集中させ、そして足引っ込め潜伏時間(PWL)を記録する。組織損傷を防止するために、22.5秒に自動中止点がある。PWLを、各々の被験動物の両後足について2〜3回得るが、その平均が、左右後足についてのベースラインである。装置は、約10秒のPWLを与えるように検量する。PWLを、カラゲナン投与から2時間後に再評価する。鎮痛性評価用の化合物の投与後、PWLを、1時間毎に6時間まで再評価する。化合物群のPWLを、一元ANOVA後、Dunnett's t検定を用いて、それぞれの対照と比較する。一群当たりの最小被験動物数は、6匹である。
【0063】
体重支持不足は、Bove SE, et al. Weight bearing as a measure of disease progression and efficacy of anti-inflammatory compounds in a model of monosodium iodoacetate-induced osteoarthritis. Osteoarthritis Cartilage. 2003 Nov;11(11):821-30 の方法にしたがって測定することができる。開放現場試験は、Prut L and Belzung, C. The open field as a paradigm to measure the effects of compounds on anxiety-like behaviors: a review. Eur J Pharmacol. 2003;463::3-33 の方法にしたがって行うことができる。運動試験は、Salmi P and Ahlenius S- Sedative effects of the dopamine D1 receptor agonist A 68930 on rat open-field behavior. Neuroreport. 2000 Apr 27;11(6):1269-72 の方法にしたがって行うことができる。
【0064】
組合せ
CRTH2受容体アンタゴニストは、有用に、具体的には、疼痛の処置において、別の薬理学的に活性な化合物と、または二つまたはそれを超える他の薬理学的に活性な化合物と組み合わせることができる。例えば、CRTH2受容体アンタゴニスト、具体的には、上に定義の一般式(I)の化合物またはその薬学的に許容しうる塩または溶媒和化合物は、次より選択される一つまたはそれを超える薬剤と組み合わせて、同時に、逐次的にまたは別々に投与することができる。
【0065】
(i)オピオイド鎮痛薬、例えば、モルヒネ、ヘロイン、ヒドロモルホン、オキシモルホン、レボルファノール、レバロルファン、メタドン、メペリジン、フェンタニール、コカイン、コデイン、ジヒドロコデイン、オキシコドン、ヒドロコドン、プロポキシフェン、ナルメフェン(nalmefene)、ナロルフィン、ナロキソン、ナルトレキソン、ブプレノルフィン、ブトルファノール、ナルブフェンまたはペンタゾシン;
(ii)非ステロイド性抗炎症薬(NSAID)、例えば、アスピリン、ジクロフェナク、ジフルシナル(diflusinal)、エトドラク(etodolac)、フェンブフェン、フェノプロフェン、フルフェニサル(flufenisal)、フルルビプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、ケトロラク、メクロフェナム酸、メフェナム酸、ナブメトン(nabumetone)、ナプロキセン、オキサプロジン、フェニルブタゾン、ピロキシカム、スリンダク、トルメチンまたはゾメピラック、またはその薬学的に許容しうる塩;
(iii)バルビツレート鎮静薬、例えば、アモバルビタール、アプロバルビタール、ブタバルビタール、ブタビタール(butabital)、メフォバルビタール、メタルビタール、メトヘキシタール、ペントバルビタール、フェノバルビタール、セコバルビタール、タルブタール、テアミラル(theamylal)またはチオペンタール、またはその薬学的に許容しうる塩;
(iv)鎮静作用を有するベンゾジアゼピン、例えば、クロルジアゼポキシド、クロラゼペート、ジアゼパム、フルラゼパム、ロラゼパム、オキサゼパム、テマゼパムまたはトリアゾラム、またはその薬学的に許容しうる塩;
(v)鎮静作用を有するH1アンタゴニスト、例えば、ジフェンヒドラミン、ピリラミン、プロメタジン、クロルフェニラミンまたはクロルシクリジン、またはその薬学的に許容しうる塩;
(vi)鎮静薬であって、グルテチミド、メプロバメート、メタクアロンまたはジクロラルフェナゾン、またはその薬学的に許容しうる塩のようなもの;
(vii)骨格筋弛緩薬、例えば、バクロフェン、カリソプロドール、クロルゾキサゾン、シクロベンザプリン、メトカルバモルまたはオルフレナジン(orphrenadine)、またはその薬学的に許容しうる塩;
(viii)NMDA受容体アンタゴニスト、例えば、デキストロメトルファン((+)−3−ヒドロキシ−N−メチルモルフィナン)またはその代謝産物デキストロルファン((+)−3−ヒドロキシ−N−メチルモルフィナン)、ケタミン、メマンチン、ピロロキノリンキノンまたは cis−4−(ホスホノメチル)−2−ピペリジンカルボン酸、またはその薬学的に許容しうる塩;
(ix)αアドレナリン作動性薬、例えば、ドキサゾシン、タムスロシン(tamsulosin)、クロニジンまたは4−アミノ−6,7−ジメトキシ−2−(5−メタンスルホンアミド−1,2,3,4−テトラヒドロイソキノール−2−イル)−5−(2−ピリジル)キナゾリン;
(x)三環系抗うつ薬、例えば、デシプラミン、イミプラミン、アミトリプチリン(amytriptiline)またはノルトリプチリン;
(xi)抗痙攣薬、例えば、カルバマゼピンまたはバルプロエート;
(xii)タキキニン(NK)アンタゴニスト、具体的には、NK−3、NK−2またはNK−1アンタゴニスト、例えば、(αR,9R)−7−[3,5−ビス(トリフルオロメチル)ベンジル]−8,9,10,11−テトラヒドロ−9−メチル−5−(4−メチルフェニル)−7H−[1,4]ジアゾシノ[2,1−g][1,7]ナフトリジン−6−13−ジオン(TAK−637)、5−[[(2R,3S)−2−[(1R)−1−[3,5−ビス(トリフルオロメチル)フェニル]エトキシ−3−(4−フルオロフェニル)−4−モルホリニル]メチル]−1,2−ジヒドロ−3H−1,2,4−トリアゾール−3−オン(MK−869)、ラネピタント(lanepitant)、ダピタント(dapitant)または3−[[2−メトキシ−5−(トリフルオロメトキシ)フェニル]メチルアミノ]−2−フェニルピペリジン(2S,3S);
(xiii)ムスカリン様アンタゴニスト、例えば、オキシブチン(oxybutin)、トルテロジン(tolterodine)、プロピベリン(propiverine)、塩化トロプシウム(tropsium chloride)またはダリフェナシン(darifenacin);
(xiv)COX−2阻害剤、例えば、セレコキシブ(celecoxib)、ロフェコキシブ(rofecoxib)またはバルデコキシブ(valdecoxib);
(xv)非選択的COX阻害剤(好ましくは、GI防御を伴う)、例えば、ニトロフルルビプロフェン(HCT−1026);
(xvi)コールタール鎮痛薬、特に、パラセタモール;
(xvii)神経遮断薬であって、ドロペリドールのようなもの;
(xviii)バニロイド受容体アゴニスト(例えば、レシンフェラトキシン(resinferatoxin))またはアンタゴニスト(例えば、カプサゼピン(capsazepine));
(xix)βアドレナリン作動性薬であって、プロプラノロールのようなもの;
(xx)局所麻酔薬であって、メキシレチンのようなもの;
(xxi)コルチコステロイドであって、デキサメタゾンのようなもの;
(xxii)セロトニン受容体アゴニストまたはアンタゴニスト;
(xxiii)コリン作動性(ニコチン性)鎮痛薬;
(xxiv)Tramadol(登録商標);
(xxv)PDEV阻害剤であって、シルデナフィル(sildenafil)、バルデナフィル(vardenafil)またはタラダフィル(taladafil)のようなもの;
(xxvi)α−2−δリガンドであって、ガバペンチン(gabapentin)またはプレガバリン(pregabalin)のようなもの;および
(xxvii)カナビノイド(canabinoid)。
【0066】
CRTH2受容体アンタゴニストは、治療的有効量で患者に投与される。CRTH2受容体アンタゴニストは、単独でまたは薬学的に許容しうる組成物の一部分として投与することができる。
【0067】
薬物物質
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、薬学的に許容しうる塩、例えば、酸付加塩または塩基塩の形で投与することができる。
【0068】
適する酸付加塩は、無毒性塩を形成する酸から形成される。例には、酢酸塩、アスパラギン酸塩、安息香酸塩、ベシル酸塩、重炭酸塩/炭酸塩、重硫酸塩/硫酸塩、ホウ酸塩、カムシレート塩、クエン酸塩、エジシレート塩、エシレート塩、ギ酸塩、フマル酸塩、グルセプテート塩、グルコン酸塩、グルクロン酸塩、ヘキサフルオロリン酸塩、ヒベンゼート(hibenzate)塩、塩酸塩/塩素塩、臭化水素酸塩/臭素塩、ヨウ化水素酸塩/ヨウ素塩、イセチオン酸塩、乳酸塩、リンゴ酸塩、マレイン酸塩、マロン酸塩、メシレート塩、メチル硫酸塩、ナフチル酸塩、2−ナプシレート塩、ニコチン酸塩、硝酸塩、オロチン酸塩、シュウ酸塩、パルミチン酸塩、パモ酸塩、リン酸塩/リン酸水素塩、リン酸塩/リン酸二水素塩、リン酸塩、糖酸塩、ステアリン酸塩、コハク酸塩、酒石酸塩、トシレート塩およびトリフルオロ酢酸塩が含まれる。
【0069】
適する塩基塩は、無毒性塩を形成する塩基から形成される。例には、アルミニウム塩、アルギニン塩、ベンザチン塩、カルシウム塩、コリン塩、ジエチルアミン塩、ジオールアミン塩、グリシン塩、リシン塩、マグネシウム塩、メグルミン塩、オラミン(olamine)塩、カリウム塩、ナトリウム塩、トロメタミン塩および亜鉛塩が含まれる。
【0070】
酸および塩基のヘミ塩、例えば、ヘミ硫酸塩およびヘミカルシウム塩も形成することができる。
適する塩の概説については、Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth(Wiley-VCH, Weinheim, Germany, 2002)を参照されたい。
【0071】
薬学的に許容しうる塩は、次の3種類の内の一つまたはそれを超える方法によって製造することができる。
(i)化合物を、所望の酸または塩基と反応させることによる;
(ii)化合物の適する前駆体から、酸または塩基に反応活性な保護基を除去することによる、または適する環状前駆体、例えば、ラクトンまたはラクタムを、所望の酸または塩基を用いて開環することによる;または
(iii)化合物の一つの塩を、適当な酸または塩基との反応によってまたは適するイオン交換カラムによって、別の塩へと変換することによる。
【0072】
3種類の反応は全て、典型的に、溶液中で行われる。得られた塩は、沈殿するので、濾過によって集めることができるし、または溶媒の蒸発によって回収することができる。得られた塩のイオン化の程度は、完全にイオン化された状態〜ほとんどイオン化されていない状態でありうる。
【0073】
本発明の化合物は、非溶媒和および溶媒和双方の形で存在しうる。「溶媒和化合物」という用語は、本明細書中において、本発明の化合物と、化学量論的量の一つまたはそれを超える薬学的に許容しうる溶媒分子、例えば、エタノールを含む分子複合体を記載するのに用いられる。「水和物」という用語は、この溶媒が水である場合に用いられる。
【0074】
本発明の範囲内に包含されるのは、クラスレート、薬物−宿主包接複合体などの複合体であるが、この場合、前述の溶媒和化合物とは対照的に、薬物および宿主は、化学量論的量または非化学量論的量で存在する。更に包含されるのは、化学量論的量であってよいしまたは非化学量論的量であってよい二つまたはそれを超える有機および/または無機成分を含有する薬物の複合体である。得られた複合体は、イオン化、部分イオン化または非イオン化の状態であってよい。このような複合体についての概説については、Halebllan (august 1975) J Pharm Sci, 64(8), 1269-1288 を参照されたい。
【0075】
以下、本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物という意味は全て、その塩、溶媒和化合物および複合体、およびそれら塩の溶媒和化合物および複合体の意味を包含する。
【0076】
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、プロドラッグの形で投与することができる。プロドラッグは、それ自体はほとんどまたは全く薬理活性が無くてよいが、体内または上に投与された場合、例えば、加水分解的開裂により、所望の活性を有する化合物へと変換されうる化合物である。プロドラッグの使用に関する追加の情報は、Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series(T. Higuchi and W. Stella)および Bioreversible Carries in Drug Design, Pergamon Press, 1987(ed. E. B. Roche, American Pharmaceutical Association)に見出されうる。
【0077】
プロドラッグは、例えば、化合物中に存在する適当な官能基を、例えば、Design of Prodrugs by H. Bundgaard(Elsevier, 1985)に記載の「プロ残基(pro-moieties)」のような、当業者に知られているある種の残基で置き換えることによって製造することができる。
【0078】
プロドラッグの若干の例には、次が含まれる。
(i)化合物が、カルボン酸官能基(−COOH)を含有する場合、そのエステル、例えば、式(I)の化合物のカルボン酸官能基の水素が、(C1〜C8)アルキルで置き換えられている化合物;
(ii)化合物が、アルコール官能基(−OH)を含有する場合、そのエーテル、例えば、その化合物のアルコール官能基の水素が、(C1〜C6)アルカノイルオキシメチルで置き換えられている化合物;および
(iii)化合物が、第一級または第二級アミノ官能基(−NH2または−NHR、ここにおいて、R≠H)を含有する場合、そのアミド、例えば、その化合物のアミノ官能基の、場合により一つまたは双方の水素が、(C1〜C10)アルカノイルで置き換えられている化合物。
【0079】
前述の例および他のプロドラッグタイプの例による置換基の更に別の例は、前述の参考文献に見出されうる。
更に、ある種の化合物は、それ自体、他の化合物のプロドラッグとして作用することができる。
【0080】
本発明の範囲内に更に包含されるのは、本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物の代謝産物、すなわち、薬物の投与時に in vivo で形成される化合物である。本発明による代謝産物の若干の例には、次が含まれる。
【0081】
(i)化合物が、メチル基を含有する場合、そのヒドロキシメチル誘導体(−CH3→−CH2OH);
(ii)化合物が、アルコキシ基を含有する場合、そのヒドロキシ誘導体(−OR→−OH);
(iii)化合物が、第三級アミノ基を含有する場合、その第二級アミノ誘導体(−NR1R2→−NHR1または−NHR2);
(iv)化合物が、第二級アミノ基を含有する場合、その第一級誘導体(−NHR1→−NH2);
(v)化合物が、フェニル残基を含有する場合、そのフェノール誘導体(−Ph→−PhOH);および
(vi)化合物が、アミド基を含有する場合、そのカルボン酸誘導体(−CONH2→−COOH)。
【0082】
一つまたはそれを超える不斉炭素原子を含有する本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、二つまたはそれを超える立体異性体として存在しうる。化合物が、アルケニル基またはアルケニレン基を含有する場合、幾何的 cis/trans(またはZ/E)異性体が可能である。構造異性体が、低エネルギー障壁によって相互変換できる場合、互変異性体の異性(「互変異性」)が存在しうる。これは、例えば、イミノ基、ケト基またはオキシム基を含有する式Iの化合物におけるプロトン互変異性の形、または芳香族残基を含有する化合物におけるいわゆる原子価互変異性の形をとりうる。単一の化合物は、1より多くのタイプの異性を示すことがありうるということである。
【0083】
cis/trans 異性体は、当業者に周知の慣用的な技法、例えば、クロマトグラフィーおよび分別結晶によって分離することができる。
個々の鏡像異性体の製造/分離のための慣用的な技法には、適する光学的に純粋な前駆体からのキラル合成、または例えば、キラル高速液体クロマトグラフィー(HPLC)を用いたラセミ体(または塩または誘導体のラセミ体)の分割が含まれる。
【0084】
或いは、ラセミ体(またはラセミ前駆体)は、適する光学活性化合物、例えば、アルコールと反応することができるし、または式Iの化合物が、酸性または塩基性の残基を含有する場合、1−フェニルエチルアミンまたは酒石酸のような塩基または酸と反応することができる。得られたジアステレオマー混合物は、クロマトグラフィーおよび/または分別結晶によって分離することができ、それらジアステレオ異性体の一方または双方は、当業者に周知の手段によって、その一つまたは複数の該当する純粋な鏡像異性体へと変換することができる。
【0085】
キラル化合物(およびそれらのキラル前駆体)は、クロマトグラフィー、典型的には、不斉樹脂上の、0〜50容量%、典型的には、2%〜20%のイソプロパノールおよび0〜5容量%のアルキルアミン、典型的には、0.1%のジエチルアミンを含有する炭化水素、典型的には、ヘプタンまたはヘキサンから成る移動相でのHPLCを用いて、鏡像異性体が豊富な形で得ることができる。溶出液の濃縮は、豊富な混合物を与える。
【0086】
立体異性体集合体は、当業者に知られている慣用的な技法によって分離することができ、例えば、Stereochemistry of Organic Compounds by E. L. Eliel and S. H. Wilen (Wiley, New York, 1994) を参照されたい。
【0087】
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、1個またはそれを超える炭素原子が、同じ原子番号を有するが、天然において優先的に存在する原子質量または質量数とは異なった原子質量または質量数を有する原子で置き換えられている一つまたはそれを超える同位体で存在してよい。
【0088】
同位体の例には、2Hおよび3Hなどの水素、11C、13Cおよび14Cなどの炭素、36Clなどの塩素、18Fなどのフッ素、123Iおよび125Iなどのヨウ素、13Nおよび15Nなどの窒素、15O、17Oおよび18Oなどの酸素、32Pなどのリン、および35Sなどの硫黄の同位体が含まれる。
【0089】
ある種の同位体標識された化合物、例えば、放射性同位体を包含するものは、薬物および/または基質の組織分布研究において有用である。放射性同位体トリチウム、すなわち、3Hおよび炭素14、すなわち、14Cは、それらの包含の容易さおよび容易な検出手段のために、この目的に特に有用である。
【0090】
ジュウテリウム、すなわち、2Hのような、より重い同位体での置換は、より大きい代謝安定性、例えば、増加した in vivo 半減期または減少した投薬要件によって生じるある種の治療的利点を与えることがあり、したがって、何等かの状況において好適でありうる。
【0091】
11C、18F、15Oおよび13Nのような、陽電子放出性同位体での置換は、基質受容体占有を調べるための陽電子放出トポグラフィー(PET)研究に有用でありうる。
同位体標識された化合物は、概して、慣用的な技法によって製造することができる。
【0092】
本発明による薬学的に許容しうる溶媒和化合物には、結晶化の溶媒が、同位体置換されていてよいもの、例えば、D2O、d6−アセトン、d6−DMSOが含まれる。
薬物製品
薬学的使用を予定した本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、結晶性または非晶質製品として投与することができる。それは、例えば、固形プラグ、粉末または薄膜として、沈殿、結晶化、凍結乾燥、噴霧乾燥または蒸発乾燥などの方法によって得ることができる。マイクロ波または高周波乾燥は、この目的に用いることができる。
【0093】
それは、単独で、または一つまたはそれを超える本発明の他の化合物との組合せで、または一つまたはそれを超える他の薬物との組合せで(またはいずれかそれらの組合せとして)投与することができる。概して、それは、一つまたはそれを超える薬学的に許容しうる賦形剤と一緒の製剤として投与されるであろう。「賦形剤」という用語は、本明細書中において、本発明の一つまたは複数の化合物以外のいずれかの成分を記載するのに用いられる。賦形剤の選択は、具体的な投与様式、溶解度および安定性への賦形剤の作用、および剤形の性状などの因子に大きく依存するであろう。
【0094】
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物の送達に適する医薬組成物、およびその製造方法は、当業者に容易に明らかであろう。このような組成物およびその製造方法は、例えば、Remington's Pharmaceutical Sciences, 19th Edition (Macl Publishing Company, 1995) に見出されうる。
【0095】
経口投与
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、経口投与することができる。経口投与は、化合物が胃腸管に入るように飲み込むことを必要とすることがありうるし、または化合物が口から直接的に血流に入る口腔内投与または舌下投与を用いることができる。
【0096】
経口投与に適する製剤には、錠剤、粒状剤を含有するカプセル剤、液剤または散剤、ロゼンジ(液体入りを含めた)、咀嚼剤、多粒状剤およびナノ粒状剤、ゲル剤、固溶体、リポソーム、薄膜剤、小卵剤、噴霧剤および液状製剤などの固形製剤が含まれる。
【0097】
液状製剤には、懸濁剤、液剤、シロップ剤およびエリキシル剤が含まれる。このような製剤は、軟または硬カプセル剤中の充填剤として用いることができ、典型的には、担体、例えば、水、エタノール、ポリエチレングリコール、プロピレングリコール、メチルセルロースまたは適する油、および一つまたはそれを超える乳化剤および/または懸濁化剤を含むことができる。液状製剤は、固体の再構成によって、例えば、サシェ剤から製造することもできる。
【0098】
本発明のCRTH2受容体アンタゴニスト、例えば、本発明の一般式Iの化合物は、Liang and Chen (2001) によって Expert Opinion in Therapeutic Patents, 11(6), 981-986 に記載のものなどの、急速溶解性、急速崩壊性剤形で用いることもできる。
【0099】
錠剤剤形については、用量に依存して、薬物は、剤形の1重量%〜80重量%、より典型的には、剤形の5重量%〜60重量%を構成してよい。薬物に加えて、錠剤は、概して、崩壊剤を含有する。崩壊剤の例には、ナトリウムデンプングリコラート、ナトリウムカルボキシメチルセルロース、カルシウムカルボキシメチルセルロース、クロスカルメロースナトリウム、クロスポビドン、ポリビニルピロリドン、メチルセルロース、微結晶性セルロース、低級アルキル置換ヒドロキシプロピルセルロース、デンプン、プレゲル化デンプンおよびアルギン酸ナトリウムが含まれる。概して、崩壊剤は、剤形の1重量%〜25重量%、好ましくは、5重量%〜20重量%を構成するであろう。
【0100】
結合剤は、概して、錠剤製剤に凝集質を与えるのに用いられる。適する結合剤には、微結晶性セルロース、ゼラチン、糖類、ポリエチレングリコール、天然および合成のガム、ポリビニルピロリドン、プレゲル化デンプン、ヒドロキシプロピルセルロースおよびヒドロキシプロピルメチルセルロースが含まれる。錠剤は、更に、ラクトース(一水和物、噴霧乾燥一水和物、無水物等)、マンニトール、キシリトール、デキストロース、スクロース、ソルビトール、微結晶性セルロース、デンプンおよび第二リン酸カルシウム二水和物などの希釈剤を含有してよい。
【0101】
錠剤は、更に、ラウリル硫酸ナトリウムおよびポリソルベート80などの界面活性剤、および二酸化ケイ素およびタルクなどの滑剤を含んでもよい。存在する場合、界面活性剤は、錠剤の0.2重量%〜5重量%を構成してよく、そして滑剤は、錠剤の0.2重量%〜1重量%を構成してよい。
【0102】
錠剤は、概して、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸亜鉛、ステアリルフマル酸ナトリウム、およびステアリン酸マグネシウムとラウリルリル硫酸ナトリウムとの混合物などの滑沢剤も含有する。滑沢剤は、概して、錠剤の0.25重量%〜10重量%、好ましくは、0.5重量%〜3重量%を構成する。
【0103】
他の可能性のある成分には、酸化防止剤、着色剤、着香剤、保存剤および風味マスキング剤が含まれる。
代表的な錠剤は、約80%までの薬物;約10重量%〜約90重量%の結合剤;約0重量%〜約85重量%の希釈剤;約2重量%〜約10重量%の崩壊剤;および約0.25重量%〜約10重量%の滑沢剤を含有する。
【0104】
錠剤ブレンドは、直接的にまたはローラーによって圧縮して、錠剤を成形することができる。或いは、錠剤ブレンドまたはブレンドの一部分は、錠剤成形する前に、湿式、乾式または溶融造粒する、溶融凝固させる、または押出することができる。最終製剤は、一つまたはそれを超える層を構成していてよく、コーティングされていてよいしまたは未コーティングであってよく;それは、カプセル封入されていてもよい。
【0105】
錠剤の製剤は、H. Lieberman and L. Lachman (Marcel Decker, New York, 1990) による Pharmaceutical Dosage Forms: Tablets, Vol. 1 に論じられている。
ヒトまたは獣医学的使用への消耗経口薄膜剤は、典型的に、急速溶解性または粘膜接着性であってよい柔軟な水溶性または水膨潤性の薄膜剤形であり、典型的には、式Iの化合物、薄膜形成性ポリマー、結合剤、溶媒、保湿剤、可塑剤、安定化剤または乳化剤、粘度調整剤および溶媒を含む。製剤の若干の成分は、二つ以上の機能を果たすことができる。
【0106】
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、水溶性であってよいしまたは不溶性であってよい。水溶性化合物は、典型的に、溶質の1重量%〜80重量%、より典型的には、20重量%〜50重量%を構成する。あまり可溶性でない化合物は、より大きい組成比、典型的に、溶質の88重量%までを構成してよい。或いは、本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、多粒状ビーズの形であってよい。
【0107】
薄膜形成性ポリマーは、天然多糖類、タンパク質または合成ヒドロコロイドより選択されてよく、典型的には、0.01〜99重量%の範囲内、より典型的には、30〜80重量%の範囲内で存在する。
【0108】
他の可能性のある成分には、酸化防止剤、着色剤、着香剤およびフレーバーエンハンサー、保存剤、唾液刺激剤、冷却剤、補助溶媒(油を含めた)、皮膚軟化薬、増量剤、消泡剤、界面活性剤および風味マスキング剤が含まれる。
【0109】
本発明による薄膜は、典型的に、可剥性支持剤または裏紙上にコーティングされた水性薄膜の蒸発乾燥によって製造される。これは、乾燥用オーブンまたはトンネル中で、典型的には、併用コーター・ドライヤー中で、または凍結乾燥または真空にすることによって行うことができる。
【0110】
経口投与用の固形製剤は、即時放出および/または修飾放出であるように製剤化することができる。修飾放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的放出およびプログラム化放出が含まれる。
【0111】
本発明の目的に適する修飾放出製剤は、米国特許第6,106,864号に記載されている。高エネルギー分散系および浸透圧性およびコーティング粒子などの他の適する放出技術の詳細は、Verma et al (2001) による Pharmaceutical Technology On-line, 25(2), 1-14 に見出されるはずである。制御放出を達成するチューインガムの使用は、WO00/35298号に記載されている。
【0112】
非経口投与
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、血流中、筋肉中または内臓中に直接投与することもできる。非経口投与に適する手段には、静脈内、動脈内、腹腔内、鞘内、脳室内、尿道内、胸骨内、頭蓋内、筋肉内および皮下が含まれる。非経口投与に適するデバイスには、針(顕微針を含めた)注射器、無針注射器および注入法が含まれる。
【0113】
非経口製剤は、典型的に、塩類、炭水化物および緩衝剤(好ましくは、3〜9のpHへ)などの賦形剤を含有してよい水性液剤であるが、若干の用途については、それらは、無菌の非水溶液として、または滅菌発熱物質不含水などの適するビヒクルと一緒に用いられる乾燥した形として、一層好適に製剤化することができる。
【0114】
無菌条件下における、例えば、凍結乾燥による非経口製剤の製造は、当業者に周知の標準的な製剤技術を用いて容易に達成することができる。
非経口液剤の製造に用いられる本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物の溶解度は、溶解促進剤の包含のような適当な製剤化技術の使用によって増加させることができる。
【0115】
非経口投与用製剤は、即時放出および/または修飾放出であるように製剤化することができる。修飾放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的放出およびプログラム化放出が含まれる。したがって、本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、活性化合物を修飾放出する植込みデポ剤としての投与用に、固体、半固体またはチキソトロピー性液体として製剤化することができる。このような製剤の例には、薬物コーティングステントおよびポリ(dl−乳酸コグリコール酸)(PGLA)ミクロスフェアが含まれる。
【0116】
局所投与
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、皮膚または粘膜への局所に、すなわち、外皮にまたは経皮で投与することもできる。この目的に典型的な製剤には、ゲル剤、ヒドロゲル剤、ローション剤、液剤、クリーム剤、軟膏剤、散布剤、包帯剤、フォーム剤、薄膜剤、皮膚パッチ、ウェファー、植込剤、スポンジ、ファイバー、バンデージおよびミクロエマルジョンが含まれる。リポソームを用いてもよい。典型的な担体には、アルコール、水、鉱油、流動パラフィン、白色ワセリン、グリセリン、ポリエチレングリコールおよびプロピレングリコールが含まれる。浸透促進剤を包含してよい。例えば、Finn and Morgan (October 1999) による J Pharm Sci, 88(10), 955-958 を参照されたい。
【0117】
他の局所投与手段には、エレクトロポレーション、イオン導入法、フォノフォレシス(phonophoresis)、ソノフォレシス(sonophoresis)および顕微針または無針(例えば、PowderjectTM、BiojectTM等)注射による送達が含まれる。
【0118】
局所投与用製剤は、即時放出および/または修飾放出であるように製剤化することができる。修飾放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的放出およびプログラム化放出が含まれる。
【0119】
吸入/鼻腔内投与
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、鼻腔内にまたは吸入によって、典型的には、乾燥粉末の形で(単独でか、混合物として、例えば、ラクトースとのドライブレンド中でかまたは、例えば、ホスファチジルコリンなどのリン脂質と混合された混合成分粒子として)乾燥粉末吸入器から、または加圧容器、ポンプ、スプレー、アトマイザー(好ましくは、電気流体力学を用いて微細な霧を生じるアトマイザー)またはネブライザーからのエアゾルスプレーとして、1,1,1,2−テトラフルオロエタンまたは1,1,1,2,3,3,3−ヘプタフルオロプロパンなどの適する噴射剤を伴ってまたは伴うことなく投与することもできる。鼻腔内使用のための粉末は、生体接着剤、例えば、キトサンまたはシクロデキストリンを含んでよい。
【0120】
加圧容器、ポンプ、スプレー、アトマイザーまたはネブライザーは、例えば、溶媒としてのエタノール、水性エタノール、または活性な一つまたは複数の噴射剤を分散させる、可溶化するまたは延長放出するのに適する別の薬剤と、ソルビタントリオレアート、オレイン酸またはオリゴ乳酸などの任意の界面活性剤を含む、本発明の一つまたは複数の化合物の溶液または懸濁液を含有する。
【0121】
乾燥粉末または懸濁液製剤での使用前に、薬物製品は、吸入による送達に適するサイズ(典型的に、5ミクロン未満)へと超微粉砕する。これは、スパイラルジェットミリング、流動床ジェットミリング、ナノ粒子を形成する超臨界流動処理、高圧ホモジナイゼーションまたは噴霧乾燥のような、いずれか適当な微粉砕法によって行うことができる。
【0122】
吸入器または吹き入れ器で用いるためのカプセル剤(例えば、ゼラチンまたはヒドロキシプロピルメチルセルロースから製造される)、ブリスターおよびカートリッジは、本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物;ラクトースまたはデンプンなどの適する粉末基剤;およびl−ロイシン、マンニトールまたはステアリン酸マグネシウムなどの性能調整剤の粉末配合物を含有するように製剤化することができる。ラクトースは、無水物であってよいしまたは一水和物の形であってよいが、好ましくは、後者である。他の適する賦形剤には、デキストラン、グルコース、マルトース、ソルビトール、キシリトール、フルクトース、スクロースおよびトレハロースが含まれる。
【0123】
電気流体力学を用いて微細な霧を生じるアトマイザーでの使用に適する溶液製剤は、1作動につき1μg〜20mgの本発明の化合物を含有してよく、その作動容量は、1μl〜100μlであってよい。典型的な製剤は、本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物、プロピレングリコール、滅菌水、エタノールおよび塩化ナトリウムを含むことができる。プロピレングリコールの代わりに用いることができる別の溶媒には、グリセロールおよびポリエチレングリコールが含まれる。
【0124】
メントールおよびレボメントールなどの適するフレーバー、またはサッカリンまたはサッカリンナトリウムなどの甘味剤は、吸入/鼻腔内投与を予定した本発明の製剤に加えることができる。
【0125】
吸入/鼻腔内投与用製剤は、即時放出および/または修飾放出であるように、例えば、PGLAを用いて製剤化することができる。修飾放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的放出およびプログラム化放出が含まれる。
【0126】
乾燥粉末吸入剤およびエアゾル剤の場合、投薬量単位は、一定の計測量を送達するバルブによって決定される。全1日用量は、単回用量で、またはより通常は、当日中に分割用量として投与することができる。
【0127】
直腸/膣内投与
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、直腸または膣に、例えば、坐剤、膣坐剤または浣腸剤の形で投与することができる。カカオ脂は、伝統的な坐剤基剤であるが、いろいろな代用物を、適宜用いることができる。
【0128】
直腸/膣内投与用製剤は、即時放出および/または修飾放出であるように製剤化することができる。修飾放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的放出およびプログラム化放出が含まれる。
【0129】
眼/耳投与
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、眼または耳に直接的に、典型的には、等張のpH調整済み滅菌生理食塩水中の超微粉懸濁液または溶液の小滴の形で投与することもできる。眼および耳投与に適する他の製剤には、軟膏剤、生物分解性(例えば、吸収性ゲルスポンジ、コラーゲン)および非生物分解性(例えば、シリコーン)植込剤、ウェファー、レンズおよび粒状物または、ニオソーム(niosomes)またはリポソームなどの小胞システムが含まれる。架橋ポリアクリル酸、ポリビニルアルコール、ヒアルロン酸、セルロース系ポリマー、例えば、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルセルロースまたはメチルセルロース、またはヘテロ多糖ポリマー、例えば、ゲランガム(gelan gum)は、塩化ベンザルコニウムなどの保存剤と一緒に包含されてよい。このような製剤は、イオン導入法によって送達することもできる。
【0130】
眼/耳投与用製剤は、即時放出および/または修飾放出であるように製剤化することができる。修飾放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的放出またはプログラム化放出が含まれる。
【0131】
他の技法
本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物は、前述のいずれかの投与様式で用いるためのそれらの溶解度、溶解速度、風味マスキング、バイオアベイラビリティーおよび/または安定性を改善するために、シクロデキストリンおよびその適する誘導体またはポリエチレングリコール含有ポリマーなどの可溶性高分子物質と組み合わせることができる。
【0132】
例えば、薬物−シクロデキストリン複合体は、概して、大部分の剤形および投与経路に有用であることが判っている。包接および非包接双方の複合体を用いることができる。薬物との直接複合体形成に代わるものとして、シクロデキストリンは、補助添加剤として、すなわち、担体、希釈剤または可溶化剤として用いることができる。これら目的に最も一般的に用いられるのは、α−、β−およびγ−シクロデキストリンであり、その例は、国際特許出願WO91/11172号、WO94/02518号およびWO98/55148号に見出されうる。
【0133】
キット部材
活性化合物の組合せを、例えば、特定の疾患または状態を処置する目的で投与することが望ましいことがありうる限りにおいて、二つまたはそれを超える医薬組成物の少なくとも一つが、本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物を含有する医薬組成物を、それら組成物の共投与に適するキットの形で好都合に組み合わせることができるということは、本発明の範囲内である。
【0134】
したがって、本発明のキットは、二つまたはそれを超える別々の医薬組成物の少なくとも一つが、本発明のCRTH2受容体アンタゴニスト、例えば、本発明による一般式Iの化合物を含有する医薬組成物と、それら組成物を別々に保持するための、容器、分割ボトルまたは分割ホイルパケットなどの手段を含む。このようなキットの例は、錠剤、カプセル剤等の包装に用いられる公知のブリスターパックである。
【0135】
本発明のキットは、異なった剤形を、例えば、経口および非経口投与するのに;異なった投薬間隔で別々の組成物を投与するのに;または別々の組成物を互いに対して滴定するのに、特に適している。コンプライアンスを助けるために、キットは、典型的に、投与の手引きを含むが、いわゆる記憶を助けるものと一緒に与えられてよい。
【0136】
投薬量
ヒト患者への投与について、本発明のCRTH2受容体アンタゴニスト、例えば、一般式Iの化合物の全1日用量は、当然ながら、投与様式に依存して、典型的に、0.1mg〜1gの範囲内である。医薬製剤の基本は、好ましくは、単位剤形である。このような形での製剤は、適当な量の活性成分を含有する単位用量へと小分けされる。その単位剤形は、包装された製剤でありうるが、その包装は、バイアルまたはアンプル中の包装された錠剤、カプセル剤および散剤のような、不連続的量の製剤を含有する。更に、単位剤形は、それ自体がカプセル剤、錠剤、カシェ剤またはロゼンジでありうるし、またはそれは、包装された形でのいずれかこれらの適当な数でありうる。単位用量製剤中の活性成分の量は、具体的な用途および活性成分の力価にしたがって、0.1mg〜1gに変更または調整することができる。医学的使用において、薬物は、例えば、100mgまたは300mgのカプセル剤として1日1〜3回投与することができる。治療的使用において、本発明の薬学的方法で利用される化合物は、約0.01mg〜約100mg/kgの初期投薬量で毎日投与される。約0.01mg〜約100mg/kgの1日用量範囲が好適である。全1日用量は、単回用量または分割用量で投与することができ、しかも医師の裁量において、本明細書中に与えられた典型的な範囲外であってよい。
【0137】
これら投薬量は、約60kg〜70kgの体重を有する平均的なヒト対象に基づいている。医師は、体重がこの範囲外にある、乳児および高齢者などの対象のための用量を容易に決定することができる。
【0138】
疑わしさを免れるために、本明細書中の「処置」の意味は、治癒的処置、待期的処置および予防的処置の意味を包含する。
次の実施例は、本発明の態様および理論を詳しく説明し、そしてCRTH2受容体の強力且つ選択的なアンタゴニストであるN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドの使用を含む。アンタゴニスト N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドの構造を、図4に示す。
【実施例】
【0139】
in vivo モデル用の動物
Charles River(Manston, Kent, UK.)より入手した150〜400gの体重の雄 Sprague Dawley ラットを、三つの群に収容した。被験動物は全て、12時間明/暗サイクル下で(07時00分に照明)、飲食を自由にして維持した。実験は全て、処置のことを知らない観察者が、Home Office Animals (Scientific Procedures) Act 1986 にしたがって行った。
【0140】
ニューロパシー性疼痛の慢性狭窄損傷(CCI)ラットモデル
坐骨神経のCCIは、Bennett and Xie(Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain:33:87-107, 1988)によって以前に記載されたように行った。被験動物を、2%イソフルオラン/O2混合物で麻酔した。右後大腿を剃毛し、1%ヨウ素を綿棒で塗る。次に、被験動物を、手順および鼻用コーンによって手術中維持される麻酔の間、恒温ブランケットに移した。皮膚を、大腿骨の線に沿って切開した。総坐骨神経を、大腿二頭筋を介する鈍的剥離によって大腿の中央に露出させた。約7mmの神経を、坐骨三分岐に近位に、神経下に鉗子を挿入することによって自由にし、その神経を、静かに大腿から持ち上げた。縫合糸を、鉗子を用いて神経下に引き寄せ、僅かな抵抗が感じられるまで簡単な結び目で縛った後、二重に結んだ。その手順を、4本の結紮糸(4−0絹)で、約1mm間隔で神経周囲を緩く縛るまで繰り返した。切開を層状に閉じた。
【0141】
ラットのストレプトゾシン(STZ)で誘発される糖尿病ニューロパシー
糖尿病は、0.9%滅菌生理食塩水中に新たに溶解したストレプトゾシン(50mg/kg)の単回腹腔内注射によって誘発した。ストレプトゾシン注射は、再現性のある機械的異痛症を3週間以内に誘発し、少なくとも7週間持続する(Chen and Pan, (Chen SR and Pan HL. Hypersensitivity of Spinothalamic Tract Neurons Associated With Diabetic Neuropathic Pain in Rats. J Neurophysiol 87: 2726-2733, 2002)。
【0142】
ラットの静的および動的異痛症の評価
静的異痛症
被験動物は、異痛症評価の前に、ワイヤ底試験ケージに慣らした。静的異痛症は、後足底表面への力の昇順(0.6g、1g、1.4g、2g、4g、6g、8g、10g、15gおよび26g)での von Frey 毛(Stoelting, Wood Dale, Illinois, USA)の適用によって評価した。各々の von Frey 毛を、最大6秒間または引っ込め応答が起こるまで、足に適用した。von Frey 毛への引っ込め応答が確かめられたら、その足を再試験し、引っ込めを生じるものより下のフィラメントで開始し、そしてその後、引っ込めが起こらなくなるまで、残りのフィラメントを力の降順で用いた。最高の26gの力は、足を持ち上げ、更には、応答を引き出し、したがって、中止点であった。各々の被験動物の両後足をこの方式で調べた。応答を引き出すのに必要な力の最低量を、足引っ込め閾値(PWT)としてグラムで記録した。静的異痛症は、被験動物が、普通のラットには無害である4gまたはそれ未満の刺激に応答した場合、存在すると定義した(Field MJ, Bramwell S, Hughes J, Singh L. Detection of static and dynamic components of mechanical allodynia in rat models of neuropathic pain: are they signalled by distinct primary sensory neurones? Pain, 1999;83:303-11)。
【0143】
動的異痛症
動的異痛症は、後足底表面を綿棒で軽くたたくことによって評価した。全身運動活性を記録することがないように、活発でない充分に慣らされたラットでこの手順を行うように注意した。各々の時点で、少なくとも2回の測定値を得、その平均が、足引っ込め潜伏時間(PWL)であった。15秒以内に反応が示されなかった場合、その手順を終え、被験動物をこの引っ込め時間に割り当てた。疼痛引っ込め反応は、しばしば、繰り返し足を縮めることまたは舐めることを伴った。動的異痛症は、被験動物が、たたくことを開始して8秒以内に綿棒刺激に応答した場合、存在すると考えた(Field et al, 1999)。
【0144】
ラットのカラゲナンで誘発される温熱性痛覚過敏(CITH)
温熱性痛覚過敏は、ラット足底試験(Ugo Basile, Comerio, Italy)を用いて、Hargreaves et al., (1988) による変法にしたがって評価した。簡単にいうと、ラットは、ガラステーブル上の3個のパースペックスボックスから成る装置に慣らした。移動輻射熱源を、テーブルの下に置き、所望の足に集中させた。足引っ込め潜伏時間(PWL)を、各々の被験動物の両後足について3回記録し、その平均が、左右後足についてのベースラインであった。装置は、ナイーブラットにおいて約10秒のPWLを与えるように検量した。足底区域の組織損傷を防止するために、22.5秒中止を認めた。λカラゲナンを、右後足底内に注射し(100μl,20mg/ml)、PWLのベースライン記録を、投与から2時間後に行った。
【0145】
データ分析
実験は全て、盲検で行った。静的異痛症は、中央値として表し[UQ;LQ]、Mann Whitney U検定によって分析した。動的異痛症および温熱性痛覚過敏は、算術平均±SEMとして表し、ANOVAによって分析した。
【0146】
CCIで誘発される静的および動的異痛症へのN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドの作用
ナイーブラットは、von Frey 適用に約10gの足引っ込め閾値を示し、綿棒刺激の適用について完全に無害な状態を見いだす。神経損傷後、ラットは、これら刺激双方への増加した感受性を示し、静的および動的異痛症の発症を示す。外科手術後14日から、被験動物は、典型的な静的および動的異痛応答を示し、試験前に記録されたベースラインは、全ての被験動物において、それぞれ<4gおよび<4秒であった。これら異痛応答は、ビヒクル処置群において、実験の間中一貫したままであった。経口(PO)投与後、N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミド(12.5mg/kg、25mg/kgおよび50mg/kg)は、CCIで誘発される静的および動的異痛症の維持を、用量依存方式で逆転させた(図1Aおよび図1B)。MEDは、25mg/kgであり、静的および動的双方の異痛症において、投与後1時間にピーク作用を生じた。最高用量は、双方の行動試験において、投薬後30分から抗異痛作用を示した(ビヒクル処置群に対してp<0.01)。それは、静的異痛症を、ガバペンチン(100mg/kg,PO)に匹敵する曲線プロフィールで逆転させ、一方、動的異痛症でのその作用は、あまり強力ではないが、ビヒクル処置されたCCIラットとは有意に異なる(投与後2時間で11.8±1.0対3.5±0.7)。
【0147】
STZで誘発される静的および動的異痛症へのN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドの作用
ナイーブラットは、von Frey 適用に約10gの足引っ込め閾値を示し、綿棒刺激の適用について完全に無害な状態を見いだす。ストレプトゾシン注射後、ラットは、これら刺激双方への増加した感受性を示し、静的および動的異痛症の発症を示す。STZ注射後14日目から、ラットを、それらの疼痛様閾値(PWTおよびPWL)に基づいて選択し、薬理学的研究に用いた。全ての被験動物でのベースライン読みは、静的および動的異痛症について、それぞれ<4gおよび<5秒であった(図2Aおよび図2B)。これら異痛応答は、ビヒクル処置群において、実験の間中一貫したままであった。N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミド(25mg/kg,PO)投与後、STZで誘発される静的および動的双方の異痛症の維持は逆転した。ピーク作用は、化合物投与後1時間に認められ、2時間まで生物学的に関連性があった。正対照として実験に包含されたガバペンチン(100mg/kg,PO)は、静的および動的双方の異痛症終点の完全な逆転を生じた。
【0148】
ラットのCITHへのN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドの作用
ナイーブラットは、熱刺激に約10秒の足引っ込め潜伏時間(PWL)を示す。カラゲナンの片側足底内注射から2時間後に、ラットは、熱刺激への感受性を増加させて、同側足に温熱性痛覚過敏の発症を示した(反対側および同側の足それぞれについて11.0±0.5秒および4.1±0.3秒のベースラインの平均)。これらPWTは、ビヒクル処置群において、時間経過の間中一貫したままであった(図3B)。N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミド(25mg/kg,PO)は、温熱性痛覚過敏の維持を、投与後2時間にピーク作用を有して、完全に逆転させた(ビヒクル処置群の3.9±0.2に対して10.1±0.6)。この抗痛覚過敏作用は、化合物投与後5時間は一貫したままであり、反対側足には作用が認められなかった(図3A)。正対照として実験に包含されたモルヒネ(3mg/kg,SC)は、予想される鎮痛作用を生じた。それは、投薬から30分後に、ナイーブラットのベースライン値を上回って両後足でPWLを増加させた。
【0149】
考察
本研究は、選択的CRTH2受容体(CRTH2R)アンタゴニストが、ニューロパシーの慢性狭窄損傷およびSTZで誘発される糖尿病動物モデルにおいて静的および動的異痛症を逆転させることができるということを示している。更に、そのアンタゴニストは、CITHラットモデルにおいて長期持続性抗痛覚過敏作用を生じた。N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドを、経口投与後のいろいろな時点で、ラットの血液およびCSF中に投与した。注射から4時間後に、25mg/kgの化合物で経口処置されたナイーブ動物の脳脊髄液(CSF)中で、5Xを超えるIC50(rIC50=45nM)を測定した。したがって、CITHラットモデルで認められた抗痛覚過敏プロフィールが、中枢活性化合物の画像であり、その化合物は、血液脳関門を越えて、その受容体において中枢作用すると考えられる。
【0150】
CRTH2RアンタゴニストであるN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドは、ニューロパシー性疼痛の動物モデルにおいて効力を示す。
【0151】
結論として、CRTH2受容体アンタゴニストであるN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドは、ニューロパシーの2種類の齧歯類動物モデル、具体的には、坐骨神経の慢性狭窄損傷(CCI)ラットおよびストレプトゾシン(STZ)で誘発される糖尿病ラットにおける静的および動的異痛症を逆転させる(Field MJ, et al, 1999, Pain, 83: 303-311)。
【0152】
同じ動物モデルにおいて、N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドの作用は、ニューロパシー性疼痛の処置用に現在市場で主流の薬物である Gabapentin に匹敵する。
【0153】
この実験的証拠は、CRTH2受容体アンタゴニストが、ヒトニューロパシー性疼痛の処置に有効であるということを示唆している。
【図面の簡単な説明】
【0154】
【図1】図1は、CCIで誘発される(a)静的および(b)動的異痛症への、経口投与後のN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドおよびガバペンチン(gabapentin)の作用を示す。von Frey 毛への足引っ込め閾値(PWT)または綿棒刺激への足引っ込め潜伏時間(PWL)のベースライン(BL)を評価した。化合物投与後、PWLおよびPWT双方を、4時間まで再評価した。データは、6匹/群の被験動物より作成する。静的異痛症データは、中央値として表し(力,g)[UQ;LQ]、(Mann Whitney U検定)によって分析する。動的異痛症は、算術平均±SEMとして表し、(一元ANOVA後、Dunnett's t検定)によって分析する。各々の時点において、ビヒクル処置群に対して、*P<0.05、**P<0.01、***P<0.001。
【図2】図2は、STZで誘発される(a)静的および(b)動的異痛症への、経口投与後のN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドおよびガバペンチンの作用を示す。von Frey 毛への足引っ込め閾値(PWT)または綿棒刺激への足引っ込め潜伏時間(PWL)のベースライン(BL)を評価した。化合物投与後、PWLおよびPWT双方を、4時間まで再評価した。データは、6匹/群の被験動物より作成する。静的異痛症データは、中央値として表し(力,g)[UQ;LQ]、(Mann Whitney U検定)によって分析する。動的異痛症は、算術平均±SEMとして表し、(一元ANOVA後、Dunnett's t検定)によって分析する。各々の時点において、ビヒクル処置群に対して、*P<0.05、**P<0.01、***P<0.001。
【図3】図3は、カラゲナンで誘発される温熱性痛覚過敏へのN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドおよびモルヒネの作用を示す。3(a)反対側足、3(b)同側足。熱刺激への足引っ込め潜伏時間(PWL)ベースライン(BL)を、未処置ラットで評価した。PWLを、足底内カラゲナン投与後2時間に再評価した後、N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミドまたはビヒクルを投与した。PWLを、カラゲナン投与後8時間まで再評価した。データは、6匹/群の被験動物の算術平均±SEMとして表す。各々の時点において、ビヒクル処置群に対して、*P<0.05、**P<0.01、***P<0.001(一元ANOVA後、Dunnett's t検定)。
【図4】図4は、MW=349.43のCRTH2受容体アンタゴニストであるN−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミド(ラセミ体)である。
【特許請求の範囲】
【請求項1】
ニューロパシー性疼痛の処置用の薬剤の製造におけるCRTH2受容体アンタゴニストの使用。
【請求項2】
CRTH2受容体アンタゴニストが、一般式(I):
【化1】
(式中、R1は、H、(C1〜C4)アルキル、(C2〜C4)アルケニル、(C2〜C4)アルキニルまたは(CH2)mRxであり;
Rxは、het1,フェニルまたは(C3〜C6)シクロアルキルであり、該het1,フェニルおよび(C3〜C6)シクロアルキルは、1個またはそれを超えるQ1基または(C1〜C4)アルキル基で置換されていてよく、該(C1〜C4)アルキルは、1個またはそれを超えるQ1基で置換されていてよく;
Q1は、ハロゲン、NO2、CN、SO2CH3、SO2NR9R10、OR9、COOR9、C(=O)NR9R10、NR9R10、NR9SO2R10、NR9C(=O)R10またはC(=O)R9であり、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
mは、0、1および2より選択される整数であり;
R2は、(C1〜C4)アルキルであり、ここにおいて、アルキル基は、ハロゲン、OR9、NR9R10、COOR9、C(=O)NR9R10、NHSO2R9およびC(=O)(C1〜C4)アルキルより選択される1個またはそれを超える置換基で置換されていてよく、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
R3は、(C3〜C6)シクロアルキルまたは−A−Ryであり;
Aは、結合、直鎖または分岐状の(C1〜C3)アルキレンまたは(C2〜C3)アルケニレンであり;
Ryは、(C6〜C12)アリールまたはhet2であり、ここにおいて、アリール基およびhet2基は、(C6〜C12)アリール、het1、Q2および(C1〜C4)アルキルより選択される1個またはそれを超える置換基で置換されていてよく、該(C1〜C4)アルキルは、1個またはそれを超えるQ2基であって、同じであるかまたは異なる基で置換されていてよく;
Q2は、ハロゲン、NO2、CN、SO2CH3、SO2NR9R10、OR9、SR9、OCH2CF3、COOR9、C(=O)NR9R10、NR9R10、NR9SO2R10、NR9C(=O)R10またはC(=O)R9であり、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
R4は、Hまたは(C1〜C4)アルキルであり;
R5、R6、R7およびR8は、同じであるかまたは異なり、H、Q3および(C1〜C4)アルキルより選択され、該(C1〜C4)アルキルは、1個またはそれを超えるQ3基であって、同じであるかまたは異なる基で置換されていてよく;
Q3は、ハロゲン、NO2、CN、SO2CH3、SO2NR9R10、OR9、SR9、COOR9、C(=O)NR9R10、NR9R10、NR9SO2R10、NR9C(=O)R10またはC(=O)R9であり、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
het1は、酸素、硫黄および窒素より選択される1〜4個のヘテロ原子を有する5〜10員芳香族複素環であり;そして
het2は、酸素、硫黄および窒素より選択される1〜4個のヘテロ原子を有する5〜10員の飽和、不飽和または部分飽和複素環式基である)
を有する化合物またはその薬学的に許容しうる塩または溶媒和化合物である、請求項1に記載の使用。
【請求項3】
CRTH2受容体アンタゴニストが、cis−N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミド、またはその薬学的に許容しうる塩または溶媒和化合物である、請求項2に記載の使用。
【請求項4】
CRTH2受容体アンタゴニストが、抗体、抗体リガンド結合性ドメインまたはポリヌクレオチドである、請求項1に記載の使用。
【請求項5】
CRTH2受容体アンタゴニストを、第二の薬理学的に活性な化合物と組み合わせて、別々に、逐次的にまたは同時に用いる、請求項1に記載の使用。
【請求項6】
第二の薬理学的に活性な化合物が、
(i)オピオイド鎮痛薬、例えば、モルヒネ、ヘロイン、ヒドロモルホン、オキシモルホン、レボルファノール、レバロルファン、メタドン、メペリジン、フェンタニール、コカイン、コデイン、ジヒドロコデイン、オキシコドン、ヒドロコドン、プロポキシフェン、ナルメフェン(nalmefene)、ナロルフィン、ナロキソン、ナルトレキソン、ブプレノルフィン、ブトルファノール、ナルブフェンまたはペンタゾシン;
(ii)非ステロイド性抗炎症薬(NSAID)、例えば、アスピリン、ジクロフェナク、ジフルシナル(diflusinal)、エトドラク(etodolac)、フェンブフェン、フェノプロフェン、フルフェニサル(flufenisal)、フルルビプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、ケトロラク、メクロフェナム酸、メフェナム酸、ナブメトン(nabumetone)、ナプロキセン、オキサプロジン、フェニルブタゾン、ピロキシカム、スリンダク、トルメチンまたはゾメピラック、またはその薬学的に許容しうる塩;
(iii)バルビツレート鎮静薬、例えば、アモバルビタール、アプロバルビタール、ブタバルビタール、ブタビタール(butabital)、メフォバルビタール、メタルビタール、メトヘキシタール、ペントバルビタール、フェノバルビタール、セコバルビタール、タルブタール、テアミラル(theamylal)またはチオペンタール、またはその薬学的に許容しうる塩;
(iv)鎮静作用を有するベンゾジアゼピン、例えば、クロルジアゼポキシド、クロラゼペート、ジアゼパム、フルラゼパム、ロラゼパム、オキサゼパム、テマゼパムまたはトリアゾラム、またはその薬学的に許容しうる塩;
(v)鎮静作用を有するH1アンタゴニスト、例えば、ジフェンヒドラミン、ピリラミン、プロメタジン、クロルフェニラミンまたはクロルシクリジン、またはその薬学的に許容しうる塩;
(vi)鎮静薬であって、グルテチミド、メプロバメート、メタクアロンまたはジクロラルフェナゾン、またはその薬学的に許容しうる塩のようなもの;
(vii)骨格筋弛緩薬、例えば、バクロフェン、カリソプロドール、クロルゾキサゾン、シクロベンザプリン、メトカルバモルまたはオルフレナジン(orphrenadine)、またはその薬学的に許容しうる塩;
(viii)NMDA受容体アンタゴニスト、例えば、デキストロメトルファン((+)−3−ヒドロキシ−N−メチルモルフィナン)またはその代謝産物デキストロルファン((+)−3−ヒドロキシ−N−メチルモルフィナン)、ケタミン、メマンチン、ピロロキノリンキノンまたは cis−4−(ホスホノメチル)−2−ピペリジンカルボン酸、またはその薬学的に許容しうる塩;
(ix)αアドレナリン作動性薬、例えば、ドキサゾシン、タムスロシン(tamsulosin)、クロニジンまたは4−アミノ−6,7−ジメトキシ−2−(5−メタンスルホンアミド−1,2,3,4−テトラヒドロイソキノール−2−イル)−5−(2−ピリジル)キナゾリン;
(x)三環系抗うつ薬、例えば、デシプラミン、イミプラミン、アミトリプチリン(amytriptiline)またはノルトリプチリン;
(xi)抗痙攣薬、例えば、カルバマゼピンまたはバルプロエート;
(xii)タキキニン(NK)アンタゴニスト、具体的には、NK−3、NK−2またはNK−1アンタゴニスト、例えば、(αR,9R)−7−[3,5−ビス(トリフルオロメチル)ベンジル]−8,9,10,11−テトラヒドロ−9−メチル−5−(4−メチルフェニル)−7H−[1,4]ジアゾシノ[2,1−g][1,7]ナフトリジン−6−13−ジオン(TAK−637)、5−[[(2R,3S)−2−[(1R)−1−[3,5−ビス(トリフルオロメチル)フェニル]エトキシ−3−(4−フルオロフェニル)−4−モルホリニル]メチル]−1,2−ジヒドロ−3H−1,2,4−トリアゾール−3−オン(MK−869)、ラネピタント(lanepitant)、ダピタント(dapitant)または3−[[2−メトキシ−5−(トリフルオロメトキシ)フェニル]メチルアミノ]−2−フェニルピペリジン(2S,3S);
(xiii)ムスカリン様アンタゴニスト、例えば、オキシブチン(oxybutin)、トルテロジン(tolterodine)、プロピベリン(propiverine)、塩化トロプシウム(tropsium chloride)またはダリフェナシン(darifenacin);
(xiv)COX−2阻害剤、例えば、セレコキシブ(celecoxib)、ロフェコキシブ(rofecoxib)またはバルデコキシブ(valdecoxib);
(xv)非選択的COX阻害剤(好ましくは、GI防御を伴う)、例えば、ニトロフルルビプロフェン(HCT−1026);
(xvi)コールタール鎮痛薬、特に、パラセタモール;
(xvii)神経遮断薬であって、ドロペリドールのようなもの;
(xviii)バニロイド受容体アゴニスト(例えば、レシンフェラトキシン(resinferatoxin))またはアンタゴニスト(例えば、カプサゼピン(capsazepine));
(xix)βアドレナリン作動性薬であって、プロプラノロールのようなもの;
(xx)局所麻酔薬であって、メキシレチンのようなもの;
(xxi)コルチコステロイドであって、デキサメタゾンのようなもの;
(xxii)セロトニン受容体アゴニストまたはアンタゴニスト;
(xxiii)コリン作動性(ニコチン性)鎮痛薬;
(xxiv)Tramadol(登録商標);
(xxv)PDEV阻害剤であって、シルデナフィル(sildenafil)、バルデナフィル(vardenafil)またはタラダフィル(taladafil)のようなもの;
(xxvi)α−2−δリガンドであって、ガバペンチン(gabapentin)またはプレガバリン(pregabalin)のようなもの;および
(xxvii)カナビノイド(canabinoid)
より選択される、請求項5に記載の使用。
【請求項7】
哺乳動物対象のニューロパシー性疼痛を処置する方法であって、該対象に、治療的有効量のCRTH2受容体アンタゴニストを投与することを含む方法。
【請求項8】
CRTH2受容体アンタゴニストが、一般式(I):
【化2】
(式中、R1は、H、(C1〜C4)アルキル、(C2〜C4)アルケニル、(C2〜C4)アルキニルまたは(CH2)mRxであり;
Rxは、het1,フェニルまたは(C3〜C6)シクロアルキルであり、該het1,フェニルおよび(C3〜C6)シクロアルキルは、1個またはそれを超えるQ1基または(C1〜C4)アルキル基で置換されていてよく、該(C1〜C4)アルキルは、1個またはそれを超えるQ1基で置換されていてよく;
Q1は、ハロゲン、NO2、CN、SO2CH3、SO2NR9R10、OR9、COOR9、C(=O)NR9R10、NR9R10、NR9SO2R10、NR9C(=O)R10またはC(=O)R9であり、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
mは、0、1および2より選択される整数であり;
R2は、(C1〜C4)アルキルであり、ここにおいて、アルキル基は、ハロゲン、OR9、NR9R10、COOR9、C(=O)NR9R10、NHSO2R9およびC(=O)(C1〜C4)アルキルより選択される1個またはそれを超える置換基で置換されていてよく、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
R3は、(C3〜C6)シクロアルキルまたは−A−Ryであり;
Aは、結合、直鎖または分岐状の(C1〜C3)アルキレンまたは(C2〜C3)アルケニレンであり;
Ryは、(C6〜C12)アリールまたはhet2であり、ここにおいて、アリール基およびhet2基は、(C6〜C12)アリール、het1、Q2および(C1〜C4)アルキルより選択される1個またはそれを超える置換基で置換されていてよく、該(C1〜C4)アルキルは、1個またはそれを超えるQ2基であって、同じであるかまたは異なる基で置換されていてよく;
Q2は、ハロゲン、NO2、CN、SO2CH3、SO2NR9R10、OR9、SR9、OCH2CF3、COOR9、C(=O)NR9R10、NR9R10、NR9SO2R10、NR9C(=O)R10またはC(=O)R9であり、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
R4は、Hまたは(C1〜C4)アルキルであり;
R5、R6、R7およびR8は、同じであるかまたは異なり、H、Q3および(C1〜C4)アルキルより選択され、該(C1〜C4)アルキルは、1個またはそれを超えるQ3基であって、同じであるかまたは異なる基で置換されていてよく;
Q3は、ハロゲン、NO2、CN、SO2CH3、SO2NR9R10、OR9、SR9、COOR9、C(=O)NR9R10、NR9R10、NR9SO2R10、NR9C(=O)R10またはC(=O)R9であり、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
het1は、酸素、硫黄および窒素より選択される1〜4個のヘテロ原子を有する5〜10員芳香族複素環であり;そして
het2は、酸素、硫黄および窒素より選択される1〜4個のヘテロ原子を有する5〜10員の飽和、不飽和または部分飽和複素環式基である)
を有する化合物またはその薬学的に許容しうる塩または溶媒和化合物である、請求項7に記載の処置方法。
【請求項9】
CRTH2受容体アンタゴニストが、cis−N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミド、またはその薬学的に許容しうる塩または溶媒和化合物である、請求項8に記載の処置方法。
【請求項10】
CRTH2受容体アンタゴニストが、抗体、抗体リガンド結合性ドメインまたはポリヌクレオチドである、請求項7に記載の処置方法。
【請求項11】
CRTH2受容体アンタゴニストを、第二の薬理学的に活性な化合物と組み合わせて、別々に、逐次的にまたは同時に用いる、請求項7に記載の処置方法。
【請求項12】
第二の薬理学的に活性な化合物が、
(xix)オピオイド鎮痛薬、例えば、モルヒネ、ヘロイン、ヒドロモルホン、オキシモルホン、レボルファノール、レバロルファン、メタドン、メペリジン、フェンタニール、コカイン、コデイン、ジヒドロコデイン、オキシコドン、ヒドロコドン、プロポキシフェン、ナルメフェン、ナロルフィン、ナロキソン、ナルトレキソン、ブプレノルフィン、ブトルファノール、ナルブフェンまたはペンタゾシン;
(xx)非ステロイド性抗炎症薬(NSAID)、例えば、アスピリン、ジクロフェナク、ジフルシナル、エトドラク、フェンブフェン、フェノプロフェン、フルフェニサル、フルルビプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、ケトロラク、メクロフェナム酸、メフェナム酸、ナブメトン、ナプロキセン、オキサプロジン、フェニルブタゾン、ピロキシカム、スリンダク、トルメチンまたはゾメピラック、またはその薬学的に許容しうる塩;
(xxi)バルビツレート鎮静薬、例えば、アモバルビタール、アプロバルビタール、ブタバルビタール、ブタビタール、メフォバルビタール、メタルビタール、メトヘキシタール、ペントバルビタール、フェノバルビタール、セコバルビタール、タルブタール、テアミラルまたはチオペンタール、またはその薬学的に許容しうる塩;
(xxii)鎮静作用を有するベンゾジアゼピン、例えば、クロルジアゼポキシド、クロラゼペート、ジアゼパム、フルラゼパム、ロラゼパム、オキサゼパム、テマゼパムまたはトリアゾラム、またはその薬学的に許容しうる塩;
(xxiii)鎮静作用を有するH1アンタゴニスト、例えば、ジフェンヒドラミン、ピリラミン、プロメタジン、クロルフェニラミンまたはクロルシクリジン、またはその薬学的に許容しうる塩;
(xxiv)鎮静薬であって、グルテチミド、メプロバメート、メタクアロンまたはジクロラルフェナゾン、またはその薬学的に許容しうる塩のようなもの;
(xxv)骨格筋弛緩薬、例えば、バクロフェン、カリソプロドール、クロルゾキサゾン、シクロベンザプリン、メトカルバモルまたはオルフレナジン、またはその薬学的に許容しうる塩;
(xxvi)NMDA受容体アンタゴニスト、例えば、デキストロメトルファン((+)−3−ヒドロキシ−N−メチルモルフィナン)またはその代謝産物デキストロルファン((+)−3−ヒドロキシ−N−メチルモルフィナン)、ケタミン、メマンチン、ピロロキノリンキノンまたは cis−4−(ホスホノメチル)−2−ピペリジンカルボン酸、またはその薬学的に許容しうる塩;
(xxvii)αアドレナリン作動性薬、例えば、ドキサゾシン、タムスロシン、クロニジンまたは4−アミノ−6,7−ジメトキシ−2−(5−メタンスルホンアミド−1,2,3,4−テトラヒドロイソキノール−2−イル)−5−(2−ピリジル)キナゾリン;
(xxviii)三環系抗うつ薬、例えば、デシプラミン、イミプラミン、アミトリプチリンまたはノルトリプチリン;
(xxix)抗痙攣薬、例えば、カルバマゼピンまたはバルプロエート;
(xxx)タキキニン(NK)アンタゴニスト、具体的には、NK−3、NK−2またはNK−1アンタゴニスト、例えば、(αR,9R)−7−[3,5−ビス(トリフルオロメチル)ベンジル]−8,9,10,11−テトラヒドロ−9−メチル−5−(4−メチルフェニル)−7H−[1,4]ジアゾシノ[2,1−g][1,7]ナフトリジン−6−13−ジオン(TAK−637)、5−[[(2R,3S)−2−[(1R)−1−[3,5−ビス(トリフルオロメチル)フェニル]エトキシ−3−(4−フルオロフェニル)−4−モルホリニル]メチル]−1,2−ジヒドロ−3H−1,2,4−トリアゾール−3−オン(MK−869)、ラネピタント、ダピタントまたは3−[[2−メトキシ−5−(トリフルオロメトキシ)フェニル]メチルアミノ]−2−フェニルピペリジン(2S,3S);
(xxxi)ムスカリン様アンタゴニスト、例えば、オキシブチン、トルテロジン、プロピベリン、塩化トロプシウムまたはダリフェナシン;
(xxxii)COX−2阻害剤、例えば、セレコキシブ、ロフェコキシブまたはバルデコキシブ;
(xxxiii)非選択的COX阻害剤(好ましくは、GI防御を伴う)、例えば、ニトロフルルビプロフェン(HCT−1026);
(xxxiv)コールタール鎮痛薬、特に、パラセタモール;
(xxxv)神経遮断薬であって、ドロペリドールのようなもの;
(xxxvi)バニロイド受容体アゴニスト(例えば、レシンフェラトキシン)またはアンタゴニスト(例えば、カプサゼピン);
(xix)βアドレナリン作動性薬であって、プロプラノロールのようなもの;
(xx)局所麻酔薬であって、メキシレチンのようなもの;
(xxi)コルチコステロイドであって、デキサメタゾンのようなもの;
(xxii)セロトニン受容体アゴニストまたはアンタゴニスト;
(xxiii)コリン作動性(ニコチン性)鎮痛薬;
(xxiv)Tramadol(登録商標);
(xxv)PDEV阻害剤であって、シルデナフィル、バルデナフィルまたはタラダフィルのようなもの;
(xxvi)α−2−δリガンドであって、ガバペンチンまたはプレガバリンのようなもの;および
(xxvii)カナビノイド
より選択される、請求項11に記載の処置方法。
【請求項1】
ニューロパシー性疼痛の処置用の薬剤の製造におけるCRTH2受容体アンタゴニストの使用。
【請求項2】
CRTH2受容体アンタゴニストが、一般式(I):
【化1】
(式中、R1は、H、(C1〜C4)アルキル、(C2〜C4)アルケニル、(C2〜C4)アルキニルまたは(CH2)mRxであり;
Rxは、het1,フェニルまたは(C3〜C6)シクロアルキルであり、該het1,フェニルおよび(C3〜C6)シクロアルキルは、1個またはそれを超えるQ1基または(C1〜C4)アルキル基で置換されていてよく、該(C1〜C4)アルキルは、1個またはそれを超えるQ1基で置換されていてよく;
Q1は、ハロゲン、NO2、CN、SO2CH3、SO2NR9R10、OR9、COOR9、C(=O)NR9R10、NR9R10、NR9SO2R10、NR9C(=O)R10またはC(=O)R9であり、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
mは、0、1および2より選択される整数であり;
R2は、(C1〜C4)アルキルであり、ここにおいて、アルキル基は、ハロゲン、OR9、NR9R10、COOR9、C(=O)NR9R10、NHSO2R9およびC(=O)(C1〜C4)アルキルより選択される1個またはそれを超える置換基で置換されていてよく、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
R3は、(C3〜C6)シクロアルキルまたは−A−Ryであり;
Aは、結合、直鎖または分岐状の(C1〜C3)アルキレンまたは(C2〜C3)アルケニレンであり;
Ryは、(C6〜C12)アリールまたはhet2であり、ここにおいて、アリール基およびhet2基は、(C6〜C12)アリール、het1、Q2および(C1〜C4)アルキルより選択される1個またはそれを超える置換基で置換されていてよく、該(C1〜C4)アルキルは、1個またはそれを超えるQ2基であって、同じであるかまたは異なる基で置換されていてよく;
Q2は、ハロゲン、NO2、CN、SO2CH3、SO2NR9R10、OR9、SR9、OCH2CF3、COOR9、C(=O)NR9R10、NR9R10、NR9SO2R10、NR9C(=O)R10またはC(=O)R9であり、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
R4は、Hまたは(C1〜C4)アルキルであり;
R5、R6、R7およびR8は、同じであるかまたは異なり、H、Q3および(C1〜C4)アルキルより選択され、該(C1〜C4)アルキルは、1個またはそれを超えるQ3基であって、同じであるかまたは異なる基で置換されていてよく;
Q3は、ハロゲン、NO2、CN、SO2CH3、SO2NR9R10、OR9、SR9、COOR9、C(=O)NR9R10、NR9R10、NR9SO2R10、NR9C(=O)R10またはC(=O)R9であり、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
het1は、酸素、硫黄および窒素より選択される1〜4個のヘテロ原子を有する5〜10員芳香族複素環であり;そして
het2は、酸素、硫黄および窒素より選択される1〜4個のヘテロ原子を有する5〜10員の飽和、不飽和または部分飽和複素環式基である)
を有する化合物またはその薬学的に許容しうる塩または溶媒和化合物である、請求項1に記載の使用。
【請求項3】
CRTH2受容体アンタゴニストが、cis−N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミド、またはその薬学的に許容しうる塩または溶媒和化合物である、請求項2に記載の使用。
【請求項4】
CRTH2受容体アンタゴニストが、抗体、抗体リガンド結合性ドメインまたはポリヌクレオチドである、請求項1に記載の使用。
【請求項5】
CRTH2受容体アンタゴニストを、第二の薬理学的に活性な化合物と組み合わせて、別々に、逐次的にまたは同時に用いる、請求項1に記載の使用。
【請求項6】
第二の薬理学的に活性な化合物が、
(i)オピオイド鎮痛薬、例えば、モルヒネ、ヘロイン、ヒドロモルホン、オキシモルホン、レボルファノール、レバロルファン、メタドン、メペリジン、フェンタニール、コカイン、コデイン、ジヒドロコデイン、オキシコドン、ヒドロコドン、プロポキシフェン、ナルメフェン(nalmefene)、ナロルフィン、ナロキソン、ナルトレキソン、ブプレノルフィン、ブトルファノール、ナルブフェンまたはペンタゾシン;
(ii)非ステロイド性抗炎症薬(NSAID)、例えば、アスピリン、ジクロフェナク、ジフルシナル(diflusinal)、エトドラク(etodolac)、フェンブフェン、フェノプロフェン、フルフェニサル(flufenisal)、フルルビプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、ケトロラク、メクロフェナム酸、メフェナム酸、ナブメトン(nabumetone)、ナプロキセン、オキサプロジン、フェニルブタゾン、ピロキシカム、スリンダク、トルメチンまたはゾメピラック、またはその薬学的に許容しうる塩;
(iii)バルビツレート鎮静薬、例えば、アモバルビタール、アプロバルビタール、ブタバルビタール、ブタビタール(butabital)、メフォバルビタール、メタルビタール、メトヘキシタール、ペントバルビタール、フェノバルビタール、セコバルビタール、タルブタール、テアミラル(theamylal)またはチオペンタール、またはその薬学的に許容しうる塩;
(iv)鎮静作用を有するベンゾジアゼピン、例えば、クロルジアゼポキシド、クロラゼペート、ジアゼパム、フルラゼパム、ロラゼパム、オキサゼパム、テマゼパムまたはトリアゾラム、またはその薬学的に許容しうる塩;
(v)鎮静作用を有するH1アンタゴニスト、例えば、ジフェンヒドラミン、ピリラミン、プロメタジン、クロルフェニラミンまたはクロルシクリジン、またはその薬学的に許容しうる塩;
(vi)鎮静薬であって、グルテチミド、メプロバメート、メタクアロンまたはジクロラルフェナゾン、またはその薬学的に許容しうる塩のようなもの;
(vii)骨格筋弛緩薬、例えば、バクロフェン、カリソプロドール、クロルゾキサゾン、シクロベンザプリン、メトカルバモルまたはオルフレナジン(orphrenadine)、またはその薬学的に許容しうる塩;
(viii)NMDA受容体アンタゴニスト、例えば、デキストロメトルファン((+)−3−ヒドロキシ−N−メチルモルフィナン)またはその代謝産物デキストロルファン((+)−3−ヒドロキシ−N−メチルモルフィナン)、ケタミン、メマンチン、ピロロキノリンキノンまたは cis−4−(ホスホノメチル)−2−ピペリジンカルボン酸、またはその薬学的に許容しうる塩;
(ix)αアドレナリン作動性薬、例えば、ドキサゾシン、タムスロシン(tamsulosin)、クロニジンまたは4−アミノ−6,7−ジメトキシ−2−(5−メタンスルホンアミド−1,2,3,4−テトラヒドロイソキノール−2−イル)−5−(2−ピリジル)キナゾリン;
(x)三環系抗うつ薬、例えば、デシプラミン、イミプラミン、アミトリプチリン(amytriptiline)またはノルトリプチリン;
(xi)抗痙攣薬、例えば、カルバマゼピンまたはバルプロエート;
(xii)タキキニン(NK)アンタゴニスト、具体的には、NK−3、NK−2またはNK−1アンタゴニスト、例えば、(αR,9R)−7−[3,5−ビス(トリフルオロメチル)ベンジル]−8,9,10,11−テトラヒドロ−9−メチル−5−(4−メチルフェニル)−7H−[1,4]ジアゾシノ[2,1−g][1,7]ナフトリジン−6−13−ジオン(TAK−637)、5−[[(2R,3S)−2−[(1R)−1−[3,5−ビス(トリフルオロメチル)フェニル]エトキシ−3−(4−フルオロフェニル)−4−モルホリニル]メチル]−1,2−ジヒドロ−3H−1,2,4−トリアゾール−3−オン(MK−869)、ラネピタント(lanepitant)、ダピタント(dapitant)または3−[[2−メトキシ−5−(トリフルオロメトキシ)フェニル]メチルアミノ]−2−フェニルピペリジン(2S,3S);
(xiii)ムスカリン様アンタゴニスト、例えば、オキシブチン(oxybutin)、トルテロジン(tolterodine)、プロピベリン(propiverine)、塩化トロプシウム(tropsium chloride)またはダリフェナシン(darifenacin);
(xiv)COX−2阻害剤、例えば、セレコキシブ(celecoxib)、ロフェコキシブ(rofecoxib)またはバルデコキシブ(valdecoxib);
(xv)非選択的COX阻害剤(好ましくは、GI防御を伴う)、例えば、ニトロフルルビプロフェン(HCT−1026);
(xvi)コールタール鎮痛薬、特に、パラセタモール;
(xvii)神経遮断薬であって、ドロペリドールのようなもの;
(xviii)バニロイド受容体アゴニスト(例えば、レシンフェラトキシン(resinferatoxin))またはアンタゴニスト(例えば、カプサゼピン(capsazepine));
(xix)βアドレナリン作動性薬であって、プロプラノロールのようなもの;
(xx)局所麻酔薬であって、メキシレチンのようなもの;
(xxi)コルチコステロイドであって、デキサメタゾンのようなもの;
(xxii)セロトニン受容体アゴニストまたはアンタゴニスト;
(xxiii)コリン作動性(ニコチン性)鎮痛薬;
(xxiv)Tramadol(登録商標);
(xxv)PDEV阻害剤であって、シルデナフィル(sildenafil)、バルデナフィル(vardenafil)またはタラダフィル(taladafil)のようなもの;
(xxvi)α−2−δリガンドであって、ガバペンチン(gabapentin)またはプレガバリン(pregabalin)のようなもの;および
(xxvii)カナビノイド(canabinoid)
より選択される、請求項5に記載の使用。
【請求項7】
哺乳動物対象のニューロパシー性疼痛を処置する方法であって、該対象に、治療的有効量のCRTH2受容体アンタゴニストを投与することを含む方法。
【請求項8】
CRTH2受容体アンタゴニストが、一般式(I):
【化2】
(式中、R1は、H、(C1〜C4)アルキル、(C2〜C4)アルケニル、(C2〜C4)アルキニルまたは(CH2)mRxであり;
Rxは、het1,フェニルまたは(C3〜C6)シクロアルキルであり、該het1,フェニルおよび(C3〜C6)シクロアルキルは、1個またはそれを超えるQ1基または(C1〜C4)アルキル基で置換されていてよく、該(C1〜C4)アルキルは、1個またはそれを超えるQ1基で置換されていてよく;
Q1は、ハロゲン、NO2、CN、SO2CH3、SO2NR9R10、OR9、COOR9、C(=O)NR9R10、NR9R10、NR9SO2R10、NR9C(=O)R10またはC(=O)R9であり、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
mは、0、1および2より選択される整数であり;
R2は、(C1〜C4)アルキルであり、ここにおいて、アルキル基は、ハロゲン、OR9、NR9R10、COOR9、C(=O)NR9R10、NHSO2R9およびC(=O)(C1〜C4)アルキルより選択される1個またはそれを超える置換基で置換されていてよく、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
R3は、(C3〜C6)シクロアルキルまたは−A−Ryであり;
Aは、結合、直鎖または分岐状の(C1〜C3)アルキレンまたは(C2〜C3)アルケニレンであり;
Ryは、(C6〜C12)アリールまたはhet2であり、ここにおいて、アリール基およびhet2基は、(C6〜C12)アリール、het1、Q2および(C1〜C4)アルキルより選択される1個またはそれを超える置換基で置換されていてよく、該(C1〜C4)アルキルは、1個またはそれを超えるQ2基であって、同じであるかまたは異なる基で置換されていてよく;
Q2は、ハロゲン、NO2、CN、SO2CH3、SO2NR9R10、OR9、SR9、OCH2CF3、COOR9、C(=O)NR9R10、NR9R10、NR9SO2R10、NR9C(=O)R10またはC(=O)R9であり、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
R4は、Hまたは(C1〜C4)アルキルであり;
R5、R6、R7およびR8は、同じであるかまたは異なり、H、Q3および(C1〜C4)アルキルより選択され、該(C1〜C4)アルキルは、1個またはそれを超えるQ3基であって、同じであるかまたは異なる基で置換されていてよく;
Q3は、ハロゲン、NO2、CN、SO2CH3、SO2NR9R10、OR9、SR9、COOR9、C(=O)NR9R10、NR9R10、NR9SO2R10、NR9C(=O)R10またはC(=O)R9であり、ここにおいて、R9およびR10は、同じであるかまたは異なり、Hおよび(C1〜C4)アルキルより選択され;
het1は、酸素、硫黄および窒素より選択される1〜4個のヘテロ原子を有する5〜10員芳香族複素環であり;そして
het2は、酸素、硫黄および窒素より選択される1〜4個のヘテロ原子を有する5〜10員の飽和、不飽和または部分飽和複素環式基である)
を有する化合物またはその薬学的に許容しうる塩または溶媒和化合物である、請求項7に記載の処置方法。
【請求項9】
CRTH2受容体アンタゴニストが、cis−N−シクロプロピル−N−[2−メチル−1−(ピリジン−3−カルボニル)−1,2,3,4−テトラヒドロキノリン−4−イル]−アセトアミド、またはその薬学的に許容しうる塩または溶媒和化合物である、請求項8に記載の処置方法。
【請求項10】
CRTH2受容体アンタゴニストが、抗体、抗体リガンド結合性ドメインまたはポリヌクレオチドである、請求項7に記載の処置方法。
【請求項11】
CRTH2受容体アンタゴニストを、第二の薬理学的に活性な化合物と組み合わせて、別々に、逐次的にまたは同時に用いる、請求項7に記載の処置方法。
【請求項12】
第二の薬理学的に活性な化合物が、
(xix)オピオイド鎮痛薬、例えば、モルヒネ、ヘロイン、ヒドロモルホン、オキシモルホン、レボルファノール、レバロルファン、メタドン、メペリジン、フェンタニール、コカイン、コデイン、ジヒドロコデイン、オキシコドン、ヒドロコドン、プロポキシフェン、ナルメフェン、ナロルフィン、ナロキソン、ナルトレキソン、ブプレノルフィン、ブトルファノール、ナルブフェンまたはペンタゾシン;
(xx)非ステロイド性抗炎症薬(NSAID)、例えば、アスピリン、ジクロフェナク、ジフルシナル、エトドラク、フェンブフェン、フェノプロフェン、フルフェニサル、フルルビプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、ケトロラク、メクロフェナム酸、メフェナム酸、ナブメトン、ナプロキセン、オキサプロジン、フェニルブタゾン、ピロキシカム、スリンダク、トルメチンまたはゾメピラック、またはその薬学的に許容しうる塩;
(xxi)バルビツレート鎮静薬、例えば、アモバルビタール、アプロバルビタール、ブタバルビタール、ブタビタール、メフォバルビタール、メタルビタール、メトヘキシタール、ペントバルビタール、フェノバルビタール、セコバルビタール、タルブタール、テアミラルまたはチオペンタール、またはその薬学的に許容しうる塩;
(xxii)鎮静作用を有するベンゾジアゼピン、例えば、クロルジアゼポキシド、クロラゼペート、ジアゼパム、フルラゼパム、ロラゼパム、オキサゼパム、テマゼパムまたはトリアゾラム、またはその薬学的に許容しうる塩;
(xxiii)鎮静作用を有するH1アンタゴニスト、例えば、ジフェンヒドラミン、ピリラミン、プロメタジン、クロルフェニラミンまたはクロルシクリジン、またはその薬学的に許容しうる塩;
(xxiv)鎮静薬であって、グルテチミド、メプロバメート、メタクアロンまたはジクロラルフェナゾン、またはその薬学的に許容しうる塩のようなもの;
(xxv)骨格筋弛緩薬、例えば、バクロフェン、カリソプロドール、クロルゾキサゾン、シクロベンザプリン、メトカルバモルまたはオルフレナジン、またはその薬学的に許容しうる塩;
(xxvi)NMDA受容体アンタゴニスト、例えば、デキストロメトルファン((+)−3−ヒドロキシ−N−メチルモルフィナン)またはその代謝産物デキストロルファン((+)−3−ヒドロキシ−N−メチルモルフィナン)、ケタミン、メマンチン、ピロロキノリンキノンまたは cis−4−(ホスホノメチル)−2−ピペリジンカルボン酸、またはその薬学的に許容しうる塩;
(xxvii)αアドレナリン作動性薬、例えば、ドキサゾシン、タムスロシン、クロニジンまたは4−アミノ−6,7−ジメトキシ−2−(5−メタンスルホンアミド−1,2,3,4−テトラヒドロイソキノール−2−イル)−5−(2−ピリジル)キナゾリン;
(xxviii)三環系抗うつ薬、例えば、デシプラミン、イミプラミン、アミトリプチリンまたはノルトリプチリン;
(xxix)抗痙攣薬、例えば、カルバマゼピンまたはバルプロエート;
(xxx)タキキニン(NK)アンタゴニスト、具体的には、NK−3、NK−2またはNK−1アンタゴニスト、例えば、(αR,9R)−7−[3,5−ビス(トリフルオロメチル)ベンジル]−8,9,10,11−テトラヒドロ−9−メチル−5−(4−メチルフェニル)−7H−[1,4]ジアゾシノ[2,1−g][1,7]ナフトリジン−6−13−ジオン(TAK−637)、5−[[(2R,3S)−2−[(1R)−1−[3,5−ビス(トリフルオロメチル)フェニル]エトキシ−3−(4−フルオロフェニル)−4−モルホリニル]メチル]−1,2−ジヒドロ−3H−1,2,4−トリアゾール−3−オン(MK−869)、ラネピタント、ダピタントまたは3−[[2−メトキシ−5−(トリフルオロメトキシ)フェニル]メチルアミノ]−2−フェニルピペリジン(2S,3S);
(xxxi)ムスカリン様アンタゴニスト、例えば、オキシブチン、トルテロジン、プロピベリン、塩化トロプシウムまたはダリフェナシン;
(xxxii)COX−2阻害剤、例えば、セレコキシブ、ロフェコキシブまたはバルデコキシブ;
(xxxiii)非選択的COX阻害剤(好ましくは、GI防御を伴う)、例えば、ニトロフルルビプロフェン(HCT−1026);
(xxxiv)コールタール鎮痛薬、特に、パラセタモール;
(xxxv)神経遮断薬であって、ドロペリドールのようなもの;
(xxxvi)バニロイド受容体アゴニスト(例えば、レシンフェラトキシン)またはアンタゴニスト(例えば、カプサゼピン);
(xix)βアドレナリン作動性薬であって、プロプラノロールのようなもの;
(xx)局所麻酔薬であって、メキシレチンのようなもの;
(xxi)コルチコステロイドであって、デキサメタゾンのようなもの;
(xxii)セロトニン受容体アゴニストまたはアンタゴニスト;
(xxiii)コリン作動性(ニコチン性)鎮痛薬;
(xxiv)Tramadol(登録商標);
(xxv)PDEV阻害剤であって、シルデナフィル、バルデナフィルまたはタラダフィルのようなもの;
(xxvi)α−2−δリガンドであって、ガバペンチンまたはプレガバリンのようなもの;および
(xxvii)カナビノイド
より選択される、請求項11に記載の処置方法。
【図1】

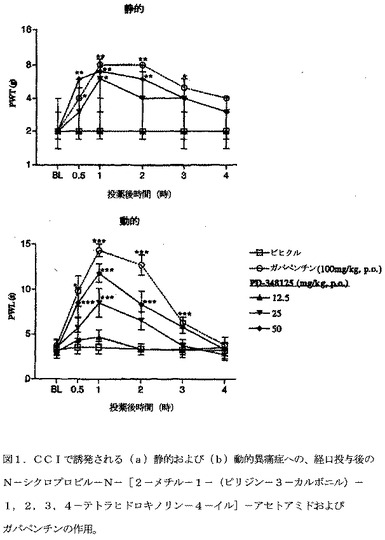
【図2】
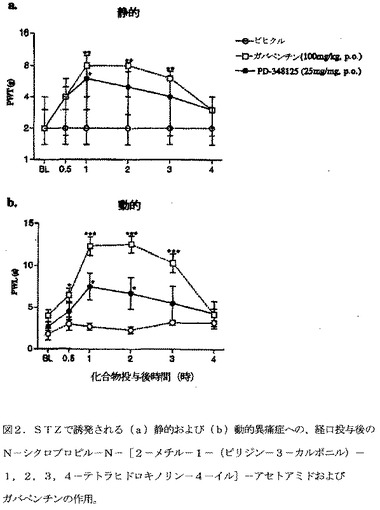
【図3】
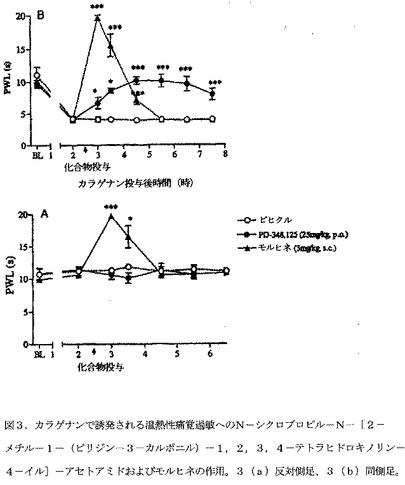
【図4】
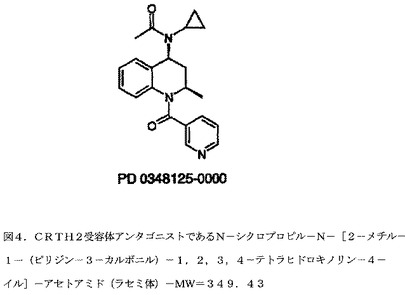
【図2】
【図3】
【図4】
【公表番号】特表2007−533725(P2007−533725A)
【公表日】平成19年11月22日(2007.11.22)
【国際特許分類】
【出願番号】特願2007−508995(P2007−508995)
【出願日】平成17年4月8日(2005.4.8)
【国際出願番号】PCT/IB2005/000992
【国際公開番号】WO2005/102338
【国際公開日】平成17年11月3日(2005.11.3)
【出願人】(593141953)ファイザー・インク (302)
【Fターム(参考)】
【公表日】平成19年11月22日(2007.11.22)
【国際特許分類】
【出願日】平成17年4月8日(2005.4.8)
【国際出願番号】PCT/IB2005/000992
【国際公開番号】WO2005/102338
【国際公開日】平成17年11月3日(2005.11.3)
【出願人】(593141953)ファイザー・インク (302)
【Fターム(参考)】
[ Back to top ]