
Colerbrookeaoppositifoliaからの肝保護剤アクテオシドの単離
植物Colerbrookea oppositifoliaから高肝保護のアクテオシドを単離するプロセスであって、このプロセスは、この植物の空中部分を乾燥させ、その部分を粉砕し、この粉末に水またはエタノールを浸透させて抽出し、この抽出物をろ過して上清を得て、この上清から残渣を得て、この残渣をクロロホルム、酢酸エチルおよびブタノールを連続的に用いて分画し、ブタノール画分にメタノールを添加した後、この画分を吸着クロマトグラフィーに供し、この吸着した画分をガラスカラムに充填し、このカラムを、メタノール:クロロホルムの溶媒で溶出して、さらなる画分を得て、この画分をカラムクロマトグラフィーに供して画分を得て、この画分を濃縮してアクテオシドを得る工程を包含する、プロセス;ならびに上記植物由来の純粋なアクテオシドの低用量を被験体に投与して被験体を有効に肝保護する方法。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の分野)
植物Colerbrookea oppositifoliaからの高肝保護の純粋なアクテオシドの単離プロセスであって、このプロセスは、この植物の空中部分を乾燥させる工程、この乾燥させた部分を粉末に粉砕する工程、この粉末に水またはエタノールを3〜4回浸透させて抽出物を得る工程、この抽出物を、懸濁した粒子を取り除くためにろ過して上清を得る工程、この上清を約45℃〜55℃で乾燥させて残渣を得る工程、この残渣をクロロホルム、酢酸エチルおよびブタノールを連続的に用いて分画する工程、ブタノール画分にメタノールを添加した後、この画分をSiO2の吸着クロマトグラフィーに供する工程、この吸着した画分をガラスカラムに充填する工程、このカラムを、次第に増加する極性のメタノール:クロロホルムの溶媒で溶出して、さらなる画分を得る工程、およびこのプロセスをもう1回繰り返す工程、この画分をカラムクロマトグラフィーに供して画分を得る工程、この画分を減圧下で濃縮してアクテオシドを残渣として得る工程を包含する、プロセス;ならびに植物Colerbrookea oppositifolia由来の純粋なアクテオシドを用いて被験体を有効に肝保護する方法であって、適切な低用量のアクテオシドを被験体に投与する工程を包含する上記方法。
【背景技術】
【0002】
(発明の背景)
フェニルエタノイドは、Cistanche deserticolaおよびフジウツギ種のような植物に存在すると報告されているクラスの化合物である(Quanbo Xing、Koji Haseら、Planta Medica、64、120〜125(1998);Peter J.HoughtonおよびHiroshi Hikino.Planta Medica、55巻、123〜126(1989))。
【0003】
フェニルエタノイド−ベルバスコシド(アクテオシドまたはクサギニン(kusaginin)とも呼ばれる)が、著者らによりColebrookea oppositifoliaから単離され、ラットおよびマウスにおいて非常に低い用量(1.25mg/kgと2.5mg/kgとの間)で非常に強い抗肝毒性/肝保護性活性を保有することが見出されたのは、初めてである。
【0004】
Colerbrookea oppositifolia由来の多数のフラボノイドおよび糖フラボノイドの存在が、これまでに文献に報告されている(S.Aziz Ahmed、S.A.SiddiquiおよびAsif Zaman、Indian J.Chemistry 12 1327〜28(1974);S.A.PatwardhanおよびA.S.Gupta、Indian J.Chemistry、20B、627、(1981);Fan Yank、Xing−Li、HAN−Qing WangおよびChong−Ren Yang、Phytochemistry 42、867〜69(1996))。しかし、この植物由来のアクテオシドの存在は、初めて報告される。
【0005】
特に、精巣細胞集団動力学に関して、この植物の雄性ラットにおける避妊活性が、以前に報告されている(R.S.Gupta、R.J.Yadav、V.P.DixitおよびM.P.Dobhal、Fitoterapia、72、236〜45(2001))。
【0006】
他の二種(すなわち、ニクジュヨウ種およびフジウツギ種)から単離されたアクテオシド(ベルバスコシド)の肝保護/抗肝毒性活性は報告されている(Quanbo Xing、Koji Haseら、Planta Medica、64、120〜125(1998);Peter J.HoughtonおよびHiroshi Hikino、Planta Medica、55巻、123〜126(1989))が、本発明は、別の供給源、すなわち、Colerbrookea oppositifoliaを報告するだけでなく、ラットにおいて、1.5mg/kg〜2.5mg/kgの間の非常に低い用量で所望の活性が達成されることも報告する。
【0007】
既に上記に記載したように、アクテオシドの肝臓保護活性/抗肝毒性活性が、インビトロおよびインビボの両方で報告されている(Quanbo Xing,Koji Haseら、Planta Medica,64,120〜125(1998))。しかし、本発明は、上記化合物が、先行技術において既に報告されたもの(Quanbo Xiang,Koji Haseら、Planta Medica,64.120〜125(1998);Peter J.HoughtonおよびHiroshi Hikino.Planta Medica,Vol.55,123〜126(1989))のほぼ12分の1〜25分の1の用量で抗肝毒性活性/肝臓保護活性を示す様式での、これまでに報告されていなかった供給源からのアクテオシドの単離を記載する。さらに、先行技術において評価されたパラメーターは、ALT(インビボ)ならびにASTおよびMDA(インビトロ)に限定されるが、一方で、本発明は、任意の肝臓保護生成物または抗肝毒性生成物の効力に関する明確な決定をもたらす全ての重要なパラメーターにおいて、実用的にアクテオシドの影響を記載する。
【0008】
先行技術において報告された用量と比較したそのような低い用量でのアクテオシドの活性の理由は、その具体的単離方法が、より高純度の生成物を生じることが明確であることに起因し得る。植物化学物質中の混入物は、純粋な活性化合物の活性を深刻に脅かすことが充分に実証されているが、その相乗作用もまた存在し得る。しかし、その相乗作用について、先行技術においては何の言及も指示も存在しない。
【発明の開示】
【発明が解決しようとする課題】
【0009】
(本発明の目的)
本発明の主要な目的は、植物Colerbrookea oppositifoliaから肝臓保護物質を単離することである。
【0010】
本発明の別の目的は、植物Colerbrookea oppositifoliaからのアクテオシドの単離プロセスを開発することである。
【0011】
本発明のなお別の目的は、Colerbrookea oppositifoliaを使用して、被験体の肝臓を保護する方法を開発することである。
【課題を解決するための手段】
【0012】
(発明の要旨)
植物Colerbrookea oppositifoliaからの高肝保護の純粋なアクテオシドの単離プロセスであって、そのプロセスは、
その植物の空中部分を乾燥させる工程、
その乾燥させた部分を粉末へと粉砕する工程、
その粉末に水またはエタノールを3〜4回浸透させて、抽出物を得る工程、
その抽出物を、懸濁した粒子を取り除くためにろ過して、上清を得る工程、
その上清を約45℃〜55℃で乾燥させて、残渣を得る工程、
その残渣を、クロロホルム、酢酸エチルおよびブタノールを連続的に用いて分画する工程、
ブタノール画分にメタノールを添加した後、ブタノール画分をSiO2の吸着クロマトグラフィーに供する工程、
その吸着した画分をガラスカラムに充填する工程、
そのカラムを、極性が漸増するメタノール:クロロホルムの溶媒で溶出して、さらなる画分を得る工程、およびこのプロセスを1回以上繰り返す工程、
それらの画分をカラムクロマトグラフィーに供して画分を得る工程、
それらの画分を減圧下で濃縮してアクテオシドを残渣として得る工程、
を包含する、プロセス。植物Colerbrookea oppositifolia由来の純粋なアクテオシドを使用して、有効に被験体の肝臓を保護する方法であって、この方法は、その被験体に、適切な低用量の上記アクテオシドを投与する工程を包含する、方法。
【0013】
(本発明の詳細な説明)
植物Colerbrookea oppositifoliaから高肝保護の純粋なアクテオシドの単離プロセスであって、そのプロセスは、
その植物の空中部分を乾燥させる工程、
その乾燥させた部分を粉末へと粉砕する工程、
その粉末に水またはエタノールを3〜4回浸透させて、抽出物を得る工程、
その抽出物を、懸濁した粒子を取り除くためにろ過して、上清を得る工程、
その上清を約45℃〜55℃で乾燥させて、残渣を得る工程、
その残渣を、クロロホルム、酢酸エチルおよびブタノールを連続的に用いて分画する工程、
ブタノール画分にメタノールを添加した後、ブタノール画分をSiO2の吸着クロマトグラフィーに供する工程、
その吸着した画分をガラスカラムに充填する工程、
そのカラムを、極性が漸増するメタノール:クロロホルムの溶媒で溶出して、さらなる画分を得る工程、およびこのプロセスを1回以上繰り返す工程、
それらの画分をカラムクロマトグラフィーに供して画分を得る工程、
それらの画分を減圧下で濃縮してアクテオシドを残渣として得る工程、
を包含する、プロセス。植物Colerbrookea oppositifolia由来の純粋なアクテオシドを使用して、有効に被験体の肝臓を保護する方法であって、この方法は、その被験体に、適切な低用量の上記アクテオシドを投与する工程を包含する。
【0014】
本発明の一実施形態において、植物Colerbrookea oppositifolia由来の純粋なアクテオシドの単離プロセスであって、そのプロセスは、
その植物の空中部分を乾燥させる工程、
その乾燥させた部分を粉末へと粉砕する工程、
その粉末に水またはエタノールを3〜4回浸透させて、抽出物を得る工程、
その抽出物を、懸濁した粒子を取り除くためにろ過して、上清を得る工程、
その上清を約45℃〜55℃で乾燥させて、残渣を得る工程、
その残渣を、クロロホルム、酢酸エチルおよびブタノールを連続的に用いて分画する工程、
ブタノール画分にメタノールを添加した後、ブタノール画分をSiO2の吸着クロマトグラフィーに供する工程、
その吸着した画分をガラスカラムに充填する工程、
そのカラムを、極性が漸増するメタノール:クロロホルムの溶媒で溶出して、さらなる画分を得る工程、およびこのプロセスを1回以上繰り返す工程、
それらの画分をカラムクロマトグラフィーに供して画分を得る工程、
それらの画分を減圧下で濃縮してアクテオシドを残渣として得る工程、
を包含する。
【0015】
本発明の別の実施形態において、上記プロセスによって得たアクテオシドは、他の供給源から得たアクテオシドと比較して12分の1〜25分の1の低投薬量で肝保護性活性を示す。
【0016】
本発明のなお別の実施形態において、上記抽出物は、水性抽出物およびアルコール抽出物である。
【0017】
本発明のなお別の実施形態において、上記アクテオシドは、全抽出物の約1.0(重量)%である。
【0018】
本発明のなお別の実施形態において、植物Colerbrookea oppositifolia由来の純粋なアクテオシドを用いて、被験体を有効に肝保護する方法であって、この方法は、その被験体に、適切な低用量の上記アクテオシドを投与する工程を包含する、方法。
【0019】
本発明のなお別の実施形態において、上記投薬量は、0.5〜10.0mg/kg体重の範囲である。
【0020】
本発明のなお別の実施形態において、上記アクテオシドは、経口経路を介して投与される。
【0021】
本発明のなお別の実施形態において、上記アクテオシドは、異常に増加した血清グルタミントランスフェラーゼ(GPT)レベルを低減させる。
【0022】
本発明のなお別の実施形態において、上記アクテオシドは、異常に増加した血清グルタミントランスフェラーゼ(GOT)レベルを低減させる。
【0023】
本発明のなお別の実施形態において、上記アクテオシドは、異常に増加した血清アルカリホスファターゼ(ALP)レベルを低減させる。
【0024】
本発明のなお別の実施形態において、上記アクテオシドは、異常に増加した血清ビリルビンレベルを低減させる。
【0025】
本発明のなお別の実施形態において、上記アクテオシドは、異常に増加した血清トリグリセリド(TG)レベルを低減させる。
【0026】
本発明のなお別の実施形態において、上記アクテオシドは、異常に増加した脂質ペルオキシダーゼ(LP)レベルを低減させる。
【0027】
本発明のなお別の実施形態において、上記アクテオシドは、異常に減少したアルブミンレベルを増加させる。
【0028】
本発明のなお別の実施形態において、上記アクテオシドは、異常に減少した還元型グルタチオンレベルを増加させる。
【0029】
本発明のなお別の実施形態において、上記アクテオシドは、市販の肝保護剤と比較して約10〜20倍有効である。
【0030】
本発明のなお別の実施形態において、上記アクテオシドは、肝毒性に対して約40〜85%の保護を提供する。
【0031】
本発明は、Colerbrookea oppositifoliaと命名された植物から最初に単離され、そして報告された、ベルバスコシドと命名された(アクテオシドまたはクサギニンともまた称される)フェニルエタノイドの、普通でなく非常に低い投薬量の抗肝毒性/肝保護性活性に関する。本発明は、特定の様式でのベルバスコシドの単離を記載し、そして異なる毒素に対する非常に低い用量での抗肝毒性/肝保護性活性を実証する。
【0032】
活性化合物である、植物Colerbrookea oppositifolia由来のアクテオシド(ベルバスコシド)は、本発明の著者らによって、最初に報告される。本著者らによって単離されたアクテオシドの確実性は、文献(Phytochemistry 21,1123−1127(1982))において報告されたものと類似の1H NMRデータおよび13C NMRデータによって確認された。
【発明を実施するための最良の形態】
【0033】
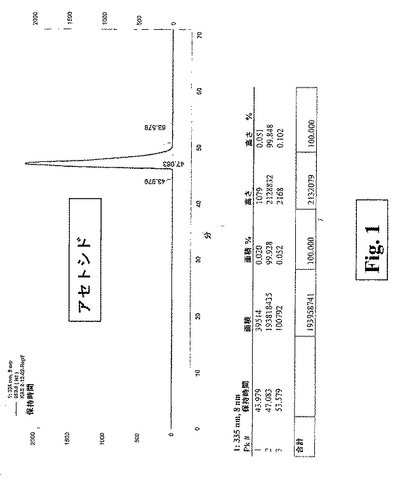
植物の異なる抽出物(95% alc.、50% alc.および水性)からのアクテオシドの単離のプロセスが、別紙1として本明細書に添付された実施例において記載される。純度を確認するHPLCグラフが、図1として含まれる。
【0034】
この化合物は、別紙2として本明細書に添付された総合的な研究設計を使用することによって、肝保護性/抗肝毒性活性について生物的に評価された。生物効力および最適な用量を確立するために、この生物評価の間に研究されたパラメータは、キット(Clonital,ItalyおよびAccurex Biomedical Pvt.Ltd Thane)を使用して、好ましかった。これらのパラメータの列挙が、本明細書に添付される別紙2に与えられる。シリマリンおよびグリシルリジンを含む標準的な薬物と比較した、異なるパラメータに関する用量応答グラフが、本明細書に添付される別紙3に与えられる。
【0035】
参照材料(すなわち、シリマリン、グリシルリジン)および必要とされるコントロールと比較した場合の、アクテオシドの各用量についての生物評価データが、表1に与えられ、この表は、本発明に添付されている。図2および表1から明らかであるように、肝毒素CCl4の投与後に比較された全てのパラメータは、1.25mg/kgから5mg/kgまでのアクテオシドの用量の正常レベルまで戻る、非常によい傾向を有する。本発明において研究されたすべてのパラメータを参照して、アクテオシドによって提供された全体の保護の百分率は、一般に、40〜80%であり、そして10〜20倍の投薬量のアクテオシドにおいてさえも、シリマリンによって得られるより良好である。
【0036】
本発明のアクテオシドを用いてこの保護を達成するために最適な用量は、1.25mg/kgと2.5mg/kgとの間である。1.25mg/kgの用量のデータが5mg/kgの用量と比較される場合を除いて、統計的には、3つの用量(すなわち、1.25mg/kg、2.5mg/kgおよび5mg/kg)の間には、有意な差は観察されない(表2)。しかし、より高い用量の間(すなわち、2.5mg/kgと5mg/kgとの間)では、統計学的有意性は観察されない。同時に、類似の観察が、1.25mg/kgと2.5mg/kgとの間での用量の影響について、十分によく維持される。
【0037】
上記発明は、実施例の形態でさらに詳細に調べられ、そして本発明の範囲を限定するとは解釈されるべきではない。
【実施例】
【0038】
(実施例1)
植物材料Colerbrookea oppositifoliaの乾燥した空中部分(500g)を、粗い粉末に粉砕した。この粉末を、95%エタノールで14時間浸出した。抽出プロセスを、合計9リットルの95%エタノールを使用して、4回繰り返した(3.0l+2.0l+2.0l+2.0lで4回の抽出)。4つの抽出物の全てをプールし、そして濾過して、懸濁した粒子を清澄化した。その上清を、ワイプトフィルムエバポレーター(wiped film evaporator)で、50±5℃で、乾固するまでエバポレートした。得られた残渣は60.0gであり(RJM/0862/P08/A001と称した)、抽出値は12%であった。
【0039】
この抽出物を、CHCl3、EtOAcおよびn−BuOH(各々2×1リットル)で連続的に分画した。15.0gのn−BuOH抽出物を、最少量のMeOHに溶解し、SiO2ゲル(100〜200メッシュ(100g))に吸着させた後に吸着クロマトグラフィーに供した。溶媒を完全に除去して、自由に流動する材料を得た。この吸着された抽出物を、内径37.5mmのガラスカラムに充填した。このカラムを、CHCl3中のMeOHの百分率を次第に増加させることによって、溶媒で溶出した。各100mlの105の画分を収集し、そして移動相としてEtOAc:HCOOH:H2O::8:1:1を使用することによって確認したTLCパターンに基づいて、プールした。スポットを、新たに調製したホウ酸−PEG 4000溶液[MeOH中2−アミノエチルジフェニルボリネートの1%溶液およびEtOH中ポリエチレングリコール4000の5%溶液(スプレーする前に1:1 v/vで混合した)]をスプレーすることによって、可視化した。
【0040】
同じTLCパターンを示す画分のうちの7つをプールし、乾燥させ、そして100〜200メッシュのSiO2ゲルカラム(1:20比)を使用する、次第に増加する極性のCHCl3:MeOH混合物で溶出する再クロマトグラフィーに供した。各200mlの60の画分すべてにおいて、収集した。これらの画分のうちの8つを、TLCに基づいてプールし、乾燥し、そして再度、カラムクロマトグラフィーに供した。各100mlの30の画分を収集した。6つの画分を、減圧下で濃縮した。残渣をMeOH/CHCl3から無色の非晶質粉末として結晶化させ、MeOHに溶解した。Rf 0.42(溶媒系EtOAc:HCOOH:H2O::8:1:1)において見出された化合物を、862−IIと称した。862−IIの純度を、以下の操作条件を使用するHPLCに基づいて確証した。
カラム:RP−18e(E−Merck,5μm、4.6×250mm)、
移動相:アセトニトリル(B):水中1.5%酢酸(A)
流速:1ml/分
λmax:335nm。
【0041】
この化合物を、NMR(1Hおよび13C)ならびにFABMSのデータに基づいて、ベルバスコシドとして確証した。
【0042】
(実施例2)
Colerbrookea oppositifoliaの乾燥した空中部分(500g)を粉砕し、そして50%水性エタノールで4回(50%EtOH、4×2.5l)、それぞれ14時間浸出した。4つの抽出物の全てをプールした。プールした水性抽出物を遠心分離し、透明な上清を、ワイプトフィルムエバポレーターで、50±5℃で、乾固するまでエバポレートした。その残渣(90g)を、RJM/0862/P08/A002と称し(抽出値18%)、CHCl3、EtOAcおよびn−BuOHで連続的に分画した。
【0043】
n−BuOH抽出物を、CHCl3中MeOHの勾配で溶出するシリカゲル(60〜120メッシュ)のカラムで、クロマトグラフィーで分離した。CHCl3:MeOH(5:1)での溶出物を、溶媒としてCHCl3−MeOH:H2O(6:1:0.1を使用するシリカゲル(100〜200メッシュ)カラムで再クロマトグラフィーで分離した。TLC上で類似の画分をプールし、乾燥させ、そしてセファデックスLH−20カラムに充填し、MeOHで溶出して、各500mlの2つの画分を生じた。主として目標化合物(862−II)を含有する第二の画分を、セファデックスLH−20でのさらなる精製に供した。カラムをMeOH:H2O(3:2)で溶出して、画分を得、この画分は、MeOH/CHCl3からの結晶化の際に、無色の非晶質粉末(MeOHに可溶、Rf 0.42(溶媒系EtOAc:HCOOH:H2O::8:1:1))を生じた。
【0044】
抽出されたRJM/0862/P08/A002の標定を、862−IIに基づいて、以下の操作条件を使用するHPLCによって実施した:
カラム:RP−18e(E−Merck,5μm、4.6×250mm)、
移動相:アセトニトリル(B):水中1.5%酢酸(A)
流速:1ml/分
λmax:335nm。
【0045】
この化合物を、NMR(1Hおよび13C)ならびにFABMSのデータに基づいて、ベルバスコシドとして確証した。
【0046】
抽出物RJM/0862/P08/A002中の862−IIの百分率は、0.86であることがわかった。
【0047】
(実施例3)
Colebrookea oppositifolia気生部分(500g)を、粗い粉末に粉砕し、次いで、2時間、98±1℃で、脱イオン水で抽出した。抽出プロセスを、合計の水(1+3×0.7リットル、4回の抽出)を使用して、4回繰り返した。4回の抽出物全てをプールした。プールした抽出物を遠心分離し、透明な濾液を凍結乾燥して、淡黄色無定形粉末を得た(収量95gm)。水性抽出残渣をRJM/0862/P08/A003としてコード化し(抽出値19%)、CHCl3、EtOAcおよびn−BuOHで連続的に分画した。
【0048】
n−BuOH抽出物をSiO2ゲル、60−120メッシュ(150gm)の吸着クロマトグラフィーに供した。溶媒を完全に除去して、自由に流動する物質を得た。1.5インチ直径のガラスカラムに、100gm SiO2ゲル、60−120メッシュ(EtOH中)を詰めた。吸着された物質をカラムに充填した。カラムをEtOAcで、MeOHの割合を次第に増加させて溶出した。各々100mlの全ての120の画分を集め、TLCパターンに基づいてプールした(展開溶媒として、EtOAc:HCOOH:H2O::8:1:1)。新たに調製したBorinate−PEG4000溶液[MeOH中の2−アミノエチルジフェニルボリネートの1%溶液およびEtOH中のポリエチレングリコール4000の5%溶液(噴霧する前に1:1v/vに混合した)]を噴霧することによってスポットを可視化した。
【0049】
EtOAcおよび10%MeOH中に溶出した画分は、同じTLCパターンを示した。これらの画分をプールし、乾燥し、次いで、最小量のMeOH中に溶解した。結晶化をメタノール溶液に少量ずつCHCl3を添加することによって行い、862−IIとして特徴付けられる無色の無定形粉末を得た。
【0050】
抽出物RJM/0862/P08/A003の標準化を、以下の操作条件を使用するHPLCによって、862−IIに基づいて行った:
カラム :RP−18e(E−Merck,5μm、4.6×250mm)において
移動相 :アセトニトリル(B):1.5%酢酸(水中)(A)
流速 :1ml/分
λmax:335nm
この化合物をNMR(1Hおよび13C)ならびにFABMSデータに基づいて、ベルバスコシドとして確立した。
【0051】
抽出物RJM/0862/P08/A003中の862−IIのパーセンテージが0.22であることを見出した。
【0052】
862−II、無定形粉末、融点145〜146℃、[α]21−85.6[C0.5%MeOH]、MS:FABMS、[M+Na]+m/z647が、公知のフェニルプロパノイド、すなわち、ベルバスコシド(アクテオシド);[β−(3’,4’−ジヒドロキシフェニル)エチル−O−α−L−ラムノピラノシル(1→3)−β−D−(4−O−カフェオイル)−グルコピラノシド]であることを見出した。構造を、文献(Andary C.,Wylde,R.Laffite C.,Privat G.およびWinternitz F.;Laboratie de Botanique et Cryptogamie,Faculte de Pharmacie.34000 Montpellier.France;Phytochemistry,21(5),1123−1127(1982))に報告されるデータと類似の1H NMRデータおよび13C NMRデータによって最終的に確認した。
【0053】
研究設計
1.動物:シロネズミ(Wistar,150〜180g)いずれかの性別
シロマウス(Swiss,25〜30g)いずれかの性別
2.肝細胞毒素:四塩化炭素(CCl4)
3.研究:予防
4.処置スケジュール:毒素の48時間前、24時間前、02時間前、および毒素の06時間後;試験物質/参照標準の最後の処置の18時間後に血液および肝臓サンプルを収集
5.用量:
アクテオシド:0.625mg/kg、1.25mg/kg、2.5mg/kg、および5.0mg/kg、p.o.
シリマリン(Silymarin):50mg/kg、p.o.
グリシルヒジン(Glycyrrhizin):100mg/kg、p.o.
CCl4:流動パラフィン中1ml/kg p.o.(1:1)
6.パラメータ
血清
ビリルビン:Clonital,24030−Carvico(BG)−Italyによって供給されるキットを使用することによるJendrassik法
トリグリセリド:Accurex BiomedicalPvt.Ltd.,Thaneによって供給されるキットによる酵素的GPO−POD法
アルブミン:Accurex BiomedicalPvt.Ltd.,Thaneによって供給されるキットによる比色B.C.G.法
トランスアミナーゼ(ALT,& AST):アミノ基転移反応によって形成されるピルベートを、2,4−ジニトロフェニルヒドラジンとの反応後に分光光度的に決定した(ReitmanおよびFrankel,1957)
ALP:アルカリ性溶媒中で形成されるp−ニトロフェノールを、基質としてリン酸p−ニトロフェニルを使用して分光光度的に測定した(WalerおよびSchutt,1974)。
肝臓ホモジネート:
GSH:これは、DTNBとの反応による除タンパク質後に決定した(David1987によって改変されたEllman1959)
脂質過酸化:チオバルビツール酸反応物質を、535nmで分光光度的に決定した。BuegeおよびAust(1978)の方法による。
肝臓保護活性:
肝臓保護活性(H)を以下の式によって計算した:
H=[1−(TC−V/VC−V)]×100
ここで、TC、VC、およびVは、それぞれ、薬物+毒素処置群の動物、ビヒクル+毒素の処置群の動物、およびビヒクル処置群の動物である。
【0054】
【表1−1】

【0055】
【表1−2】
【0056】
【表2】
【図面の簡単な説明】
【0057】
【図1】図1は、純度を確認するHPLCグラフを示す。
【図2】図2は、アクテオシド、シリマリン、およびグリシリジアの比較の肝保護性活性(%)を示す。
【技術分野】
【0001】
(発明の分野)
植物Colerbrookea oppositifoliaからの高肝保護の純粋なアクテオシドの単離プロセスであって、このプロセスは、この植物の空中部分を乾燥させる工程、この乾燥させた部分を粉末に粉砕する工程、この粉末に水またはエタノールを3〜4回浸透させて抽出物を得る工程、この抽出物を、懸濁した粒子を取り除くためにろ過して上清を得る工程、この上清を約45℃〜55℃で乾燥させて残渣を得る工程、この残渣をクロロホルム、酢酸エチルおよびブタノールを連続的に用いて分画する工程、ブタノール画分にメタノールを添加した後、この画分をSiO2の吸着クロマトグラフィーに供する工程、この吸着した画分をガラスカラムに充填する工程、このカラムを、次第に増加する極性のメタノール:クロロホルムの溶媒で溶出して、さらなる画分を得る工程、およびこのプロセスをもう1回繰り返す工程、この画分をカラムクロマトグラフィーに供して画分を得る工程、この画分を減圧下で濃縮してアクテオシドを残渣として得る工程を包含する、プロセス;ならびに植物Colerbrookea oppositifolia由来の純粋なアクテオシドを用いて被験体を有効に肝保護する方法であって、適切な低用量のアクテオシドを被験体に投与する工程を包含する上記方法。
【背景技術】
【0002】
(発明の背景)
フェニルエタノイドは、Cistanche deserticolaおよびフジウツギ種のような植物に存在すると報告されているクラスの化合物である(Quanbo Xing、Koji Haseら、Planta Medica、64、120〜125(1998);Peter J.HoughtonおよびHiroshi Hikino.Planta Medica、55巻、123〜126(1989))。
【0003】
フェニルエタノイド−ベルバスコシド(アクテオシドまたはクサギニン(kusaginin)とも呼ばれる)が、著者らによりColebrookea oppositifoliaから単離され、ラットおよびマウスにおいて非常に低い用量(1.25mg/kgと2.5mg/kgとの間)で非常に強い抗肝毒性/肝保護性活性を保有することが見出されたのは、初めてである。
【0004】
Colerbrookea oppositifolia由来の多数のフラボノイドおよび糖フラボノイドの存在が、これまでに文献に報告されている(S.Aziz Ahmed、S.A.SiddiquiおよびAsif Zaman、Indian J.Chemistry 12 1327〜28(1974);S.A.PatwardhanおよびA.S.Gupta、Indian J.Chemistry、20B、627、(1981);Fan Yank、Xing−Li、HAN−Qing WangおよびChong−Ren Yang、Phytochemistry 42、867〜69(1996))。しかし、この植物由来のアクテオシドの存在は、初めて報告される。
【0005】
特に、精巣細胞集団動力学に関して、この植物の雄性ラットにおける避妊活性が、以前に報告されている(R.S.Gupta、R.J.Yadav、V.P.DixitおよびM.P.Dobhal、Fitoterapia、72、236〜45(2001))。
【0006】
他の二種(すなわち、ニクジュヨウ種およびフジウツギ種)から単離されたアクテオシド(ベルバスコシド)の肝保護/抗肝毒性活性は報告されている(Quanbo Xing、Koji Haseら、Planta Medica、64、120〜125(1998);Peter J.HoughtonおよびHiroshi Hikino、Planta Medica、55巻、123〜126(1989))が、本発明は、別の供給源、すなわち、Colerbrookea oppositifoliaを報告するだけでなく、ラットにおいて、1.5mg/kg〜2.5mg/kgの間の非常に低い用量で所望の活性が達成されることも報告する。
【0007】
既に上記に記載したように、アクテオシドの肝臓保護活性/抗肝毒性活性が、インビトロおよびインビボの両方で報告されている(Quanbo Xing,Koji Haseら、Planta Medica,64,120〜125(1998))。しかし、本発明は、上記化合物が、先行技術において既に報告されたもの(Quanbo Xiang,Koji Haseら、Planta Medica,64.120〜125(1998);Peter J.HoughtonおよびHiroshi Hikino.Planta Medica,Vol.55,123〜126(1989))のほぼ12分の1〜25分の1の用量で抗肝毒性活性/肝臓保護活性を示す様式での、これまでに報告されていなかった供給源からのアクテオシドの単離を記載する。さらに、先行技術において評価されたパラメーターは、ALT(インビボ)ならびにASTおよびMDA(インビトロ)に限定されるが、一方で、本発明は、任意の肝臓保護生成物または抗肝毒性生成物の効力に関する明確な決定をもたらす全ての重要なパラメーターにおいて、実用的にアクテオシドの影響を記載する。
【0008】
先行技術において報告された用量と比較したそのような低い用量でのアクテオシドの活性の理由は、その具体的単離方法が、より高純度の生成物を生じることが明確であることに起因し得る。植物化学物質中の混入物は、純粋な活性化合物の活性を深刻に脅かすことが充分に実証されているが、その相乗作用もまた存在し得る。しかし、その相乗作用について、先行技術においては何の言及も指示も存在しない。
【発明の開示】
【発明が解決しようとする課題】
【0009】
(本発明の目的)
本発明の主要な目的は、植物Colerbrookea oppositifoliaから肝臓保護物質を単離することである。
【0010】
本発明の別の目的は、植物Colerbrookea oppositifoliaからのアクテオシドの単離プロセスを開発することである。
【0011】
本発明のなお別の目的は、Colerbrookea oppositifoliaを使用して、被験体の肝臓を保護する方法を開発することである。
【課題を解決するための手段】
【0012】
(発明の要旨)
植物Colerbrookea oppositifoliaからの高肝保護の純粋なアクテオシドの単離プロセスであって、そのプロセスは、
その植物の空中部分を乾燥させる工程、
その乾燥させた部分を粉末へと粉砕する工程、
その粉末に水またはエタノールを3〜4回浸透させて、抽出物を得る工程、
その抽出物を、懸濁した粒子を取り除くためにろ過して、上清を得る工程、
その上清を約45℃〜55℃で乾燥させて、残渣を得る工程、
その残渣を、クロロホルム、酢酸エチルおよびブタノールを連続的に用いて分画する工程、
ブタノール画分にメタノールを添加した後、ブタノール画分をSiO2の吸着クロマトグラフィーに供する工程、
その吸着した画分をガラスカラムに充填する工程、
そのカラムを、極性が漸増するメタノール:クロロホルムの溶媒で溶出して、さらなる画分を得る工程、およびこのプロセスを1回以上繰り返す工程、
それらの画分をカラムクロマトグラフィーに供して画分を得る工程、
それらの画分を減圧下で濃縮してアクテオシドを残渣として得る工程、
を包含する、プロセス。植物Colerbrookea oppositifolia由来の純粋なアクテオシドを使用して、有効に被験体の肝臓を保護する方法であって、この方法は、その被験体に、適切な低用量の上記アクテオシドを投与する工程を包含する、方法。
【0013】
(本発明の詳細な説明)
植物Colerbrookea oppositifoliaから高肝保護の純粋なアクテオシドの単離プロセスであって、そのプロセスは、
その植物の空中部分を乾燥させる工程、
その乾燥させた部分を粉末へと粉砕する工程、
その粉末に水またはエタノールを3〜4回浸透させて、抽出物を得る工程、
その抽出物を、懸濁した粒子を取り除くためにろ過して、上清を得る工程、
その上清を約45℃〜55℃で乾燥させて、残渣を得る工程、
その残渣を、クロロホルム、酢酸エチルおよびブタノールを連続的に用いて分画する工程、
ブタノール画分にメタノールを添加した後、ブタノール画分をSiO2の吸着クロマトグラフィーに供する工程、
その吸着した画分をガラスカラムに充填する工程、
そのカラムを、極性が漸増するメタノール:クロロホルムの溶媒で溶出して、さらなる画分を得る工程、およびこのプロセスを1回以上繰り返す工程、
それらの画分をカラムクロマトグラフィーに供して画分を得る工程、
それらの画分を減圧下で濃縮してアクテオシドを残渣として得る工程、
を包含する、プロセス。植物Colerbrookea oppositifolia由来の純粋なアクテオシドを使用して、有効に被験体の肝臓を保護する方法であって、この方法は、その被験体に、適切な低用量の上記アクテオシドを投与する工程を包含する。
【0014】
本発明の一実施形態において、植物Colerbrookea oppositifolia由来の純粋なアクテオシドの単離プロセスであって、そのプロセスは、
その植物の空中部分を乾燥させる工程、
その乾燥させた部分を粉末へと粉砕する工程、
その粉末に水またはエタノールを3〜4回浸透させて、抽出物を得る工程、
その抽出物を、懸濁した粒子を取り除くためにろ過して、上清を得る工程、
その上清を約45℃〜55℃で乾燥させて、残渣を得る工程、
その残渣を、クロロホルム、酢酸エチルおよびブタノールを連続的に用いて分画する工程、
ブタノール画分にメタノールを添加した後、ブタノール画分をSiO2の吸着クロマトグラフィーに供する工程、
その吸着した画分をガラスカラムに充填する工程、
そのカラムを、極性が漸増するメタノール:クロロホルムの溶媒で溶出して、さらなる画分を得る工程、およびこのプロセスを1回以上繰り返す工程、
それらの画分をカラムクロマトグラフィーに供して画分を得る工程、
それらの画分を減圧下で濃縮してアクテオシドを残渣として得る工程、
を包含する。
【0015】
本発明の別の実施形態において、上記プロセスによって得たアクテオシドは、他の供給源から得たアクテオシドと比較して12分の1〜25分の1の低投薬量で肝保護性活性を示す。
【0016】
本発明のなお別の実施形態において、上記抽出物は、水性抽出物およびアルコール抽出物である。
【0017】
本発明のなお別の実施形態において、上記アクテオシドは、全抽出物の約1.0(重量)%である。
【0018】
本発明のなお別の実施形態において、植物Colerbrookea oppositifolia由来の純粋なアクテオシドを用いて、被験体を有効に肝保護する方法であって、この方法は、その被験体に、適切な低用量の上記アクテオシドを投与する工程を包含する、方法。
【0019】
本発明のなお別の実施形態において、上記投薬量は、0.5〜10.0mg/kg体重の範囲である。
【0020】
本発明のなお別の実施形態において、上記アクテオシドは、経口経路を介して投与される。
【0021】
本発明のなお別の実施形態において、上記アクテオシドは、異常に増加した血清グルタミントランスフェラーゼ(GPT)レベルを低減させる。
【0022】
本発明のなお別の実施形態において、上記アクテオシドは、異常に増加した血清グルタミントランスフェラーゼ(GOT)レベルを低減させる。
【0023】
本発明のなお別の実施形態において、上記アクテオシドは、異常に増加した血清アルカリホスファターゼ(ALP)レベルを低減させる。
【0024】
本発明のなお別の実施形態において、上記アクテオシドは、異常に増加した血清ビリルビンレベルを低減させる。
【0025】
本発明のなお別の実施形態において、上記アクテオシドは、異常に増加した血清トリグリセリド(TG)レベルを低減させる。
【0026】
本発明のなお別の実施形態において、上記アクテオシドは、異常に増加した脂質ペルオキシダーゼ(LP)レベルを低減させる。
【0027】
本発明のなお別の実施形態において、上記アクテオシドは、異常に減少したアルブミンレベルを増加させる。
【0028】
本発明のなお別の実施形態において、上記アクテオシドは、異常に減少した還元型グルタチオンレベルを増加させる。
【0029】
本発明のなお別の実施形態において、上記アクテオシドは、市販の肝保護剤と比較して約10〜20倍有効である。
【0030】
本発明のなお別の実施形態において、上記アクテオシドは、肝毒性に対して約40〜85%の保護を提供する。
【0031】
本発明は、Colerbrookea oppositifoliaと命名された植物から最初に単離され、そして報告された、ベルバスコシドと命名された(アクテオシドまたはクサギニンともまた称される)フェニルエタノイドの、普通でなく非常に低い投薬量の抗肝毒性/肝保護性活性に関する。本発明は、特定の様式でのベルバスコシドの単離を記載し、そして異なる毒素に対する非常に低い用量での抗肝毒性/肝保護性活性を実証する。
【0032】
活性化合物である、植物Colerbrookea oppositifolia由来のアクテオシド(ベルバスコシド)は、本発明の著者らによって、最初に報告される。本著者らによって単離されたアクテオシドの確実性は、文献(Phytochemistry 21,1123−1127(1982))において報告されたものと類似の1H NMRデータおよび13C NMRデータによって確認された。
【発明を実施するための最良の形態】
【0033】
植物の異なる抽出物(95% alc.、50% alc.および水性)からのアクテオシドの単離のプロセスが、別紙1として本明細書に添付された実施例において記載される。純度を確認するHPLCグラフが、図1として含まれる。
【0034】
この化合物は、別紙2として本明細書に添付された総合的な研究設計を使用することによって、肝保護性/抗肝毒性活性について生物的に評価された。生物効力および最適な用量を確立するために、この生物評価の間に研究されたパラメータは、キット(Clonital,ItalyおよびAccurex Biomedical Pvt.Ltd Thane)を使用して、好ましかった。これらのパラメータの列挙が、本明細書に添付される別紙2に与えられる。シリマリンおよびグリシルリジンを含む標準的な薬物と比較した、異なるパラメータに関する用量応答グラフが、本明細書に添付される別紙3に与えられる。
【0035】
参照材料(すなわち、シリマリン、グリシルリジン)および必要とされるコントロールと比較した場合の、アクテオシドの各用量についての生物評価データが、表1に与えられ、この表は、本発明に添付されている。図2および表1から明らかであるように、肝毒素CCl4の投与後に比較された全てのパラメータは、1.25mg/kgから5mg/kgまでのアクテオシドの用量の正常レベルまで戻る、非常によい傾向を有する。本発明において研究されたすべてのパラメータを参照して、アクテオシドによって提供された全体の保護の百分率は、一般に、40〜80%であり、そして10〜20倍の投薬量のアクテオシドにおいてさえも、シリマリンによって得られるより良好である。
【0036】
本発明のアクテオシドを用いてこの保護を達成するために最適な用量は、1.25mg/kgと2.5mg/kgとの間である。1.25mg/kgの用量のデータが5mg/kgの用量と比較される場合を除いて、統計的には、3つの用量(すなわち、1.25mg/kg、2.5mg/kgおよび5mg/kg)の間には、有意な差は観察されない(表2)。しかし、より高い用量の間(すなわち、2.5mg/kgと5mg/kgとの間)では、統計学的有意性は観察されない。同時に、類似の観察が、1.25mg/kgと2.5mg/kgとの間での用量の影響について、十分によく維持される。
【0037】
上記発明は、実施例の形態でさらに詳細に調べられ、そして本発明の範囲を限定するとは解釈されるべきではない。
【実施例】
【0038】
(実施例1)
植物材料Colerbrookea oppositifoliaの乾燥した空中部分(500g)を、粗い粉末に粉砕した。この粉末を、95%エタノールで14時間浸出した。抽出プロセスを、合計9リットルの95%エタノールを使用して、4回繰り返した(3.0l+2.0l+2.0l+2.0lで4回の抽出)。4つの抽出物の全てをプールし、そして濾過して、懸濁した粒子を清澄化した。その上清を、ワイプトフィルムエバポレーター(wiped film evaporator)で、50±5℃で、乾固するまでエバポレートした。得られた残渣は60.0gであり(RJM/0862/P08/A001と称した)、抽出値は12%であった。
【0039】
この抽出物を、CHCl3、EtOAcおよびn−BuOH(各々2×1リットル)で連続的に分画した。15.0gのn−BuOH抽出物を、最少量のMeOHに溶解し、SiO2ゲル(100〜200メッシュ(100g))に吸着させた後に吸着クロマトグラフィーに供した。溶媒を完全に除去して、自由に流動する材料を得た。この吸着された抽出物を、内径37.5mmのガラスカラムに充填した。このカラムを、CHCl3中のMeOHの百分率を次第に増加させることによって、溶媒で溶出した。各100mlの105の画分を収集し、そして移動相としてEtOAc:HCOOH:H2O::8:1:1を使用することによって確認したTLCパターンに基づいて、プールした。スポットを、新たに調製したホウ酸−PEG 4000溶液[MeOH中2−アミノエチルジフェニルボリネートの1%溶液およびEtOH中ポリエチレングリコール4000の5%溶液(スプレーする前に1:1 v/vで混合した)]をスプレーすることによって、可視化した。
【0040】
同じTLCパターンを示す画分のうちの7つをプールし、乾燥させ、そして100〜200メッシュのSiO2ゲルカラム(1:20比)を使用する、次第に増加する極性のCHCl3:MeOH混合物で溶出する再クロマトグラフィーに供した。各200mlの60の画分すべてにおいて、収集した。これらの画分のうちの8つを、TLCに基づいてプールし、乾燥し、そして再度、カラムクロマトグラフィーに供した。各100mlの30の画分を収集した。6つの画分を、減圧下で濃縮した。残渣をMeOH/CHCl3から無色の非晶質粉末として結晶化させ、MeOHに溶解した。Rf 0.42(溶媒系EtOAc:HCOOH:H2O::8:1:1)において見出された化合物を、862−IIと称した。862−IIの純度を、以下の操作条件を使用するHPLCに基づいて確証した。
カラム:RP−18e(E−Merck,5μm、4.6×250mm)、
移動相:アセトニトリル(B):水中1.5%酢酸(A)
流速:1ml/分
λmax:335nm。
【0041】
この化合物を、NMR(1Hおよび13C)ならびにFABMSのデータに基づいて、ベルバスコシドとして確証した。
【0042】
(実施例2)
Colerbrookea oppositifoliaの乾燥した空中部分(500g)を粉砕し、そして50%水性エタノールで4回(50%EtOH、4×2.5l)、それぞれ14時間浸出した。4つの抽出物の全てをプールした。プールした水性抽出物を遠心分離し、透明な上清を、ワイプトフィルムエバポレーターで、50±5℃で、乾固するまでエバポレートした。その残渣(90g)を、RJM/0862/P08/A002と称し(抽出値18%)、CHCl3、EtOAcおよびn−BuOHで連続的に分画した。
【0043】
n−BuOH抽出物を、CHCl3中MeOHの勾配で溶出するシリカゲル(60〜120メッシュ)のカラムで、クロマトグラフィーで分離した。CHCl3:MeOH(5:1)での溶出物を、溶媒としてCHCl3−MeOH:H2O(6:1:0.1を使用するシリカゲル(100〜200メッシュ)カラムで再クロマトグラフィーで分離した。TLC上で類似の画分をプールし、乾燥させ、そしてセファデックスLH−20カラムに充填し、MeOHで溶出して、各500mlの2つの画分を生じた。主として目標化合物(862−II)を含有する第二の画分を、セファデックスLH−20でのさらなる精製に供した。カラムをMeOH:H2O(3:2)で溶出して、画分を得、この画分は、MeOH/CHCl3からの結晶化の際に、無色の非晶質粉末(MeOHに可溶、Rf 0.42(溶媒系EtOAc:HCOOH:H2O::8:1:1))を生じた。
【0044】
抽出されたRJM/0862/P08/A002の標定を、862−IIに基づいて、以下の操作条件を使用するHPLCによって実施した:
カラム:RP−18e(E−Merck,5μm、4.6×250mm)、
移動相:アセトニトリル(B):水中1.5%酢酸(A)
流速:1ml/分
λmax:335nm。
【0045】
この化合物を、NMR(1Hおよび13C)ならびにFABMSのデータに基づいて、ベルバスコシドとして確証した。
【0046】
抽出物RJM/0862/P08/A002中の862−IIの百分率は、0.86であることがわかった。
【0047】
(実施例3)
Colebrookea oppositifolia気生部分(500g)を、粗い粉末に粉砕し、次いで、2時間、98±1℃で、脱イオン水で抽出した。抽出プロセスを、合計の水(1+3×0.7リットル、4回の抽出)を使用して、4回繰り返した。4回の抽出物全てをプールした。プールした抽出物を遠心分離し、透明な濾液を凍結乾燥して、淡黄色無定形粉末を得た(収量95gm)。水性抽出残渣をRJM/0862/P08/A003としてコード化し(抽出値19%)、CHCl3、EtOAcおよびn−BuOHで連続的に分画した。
【0048】
n−BuOH抽出物をSiO2ゲル、60−120メッシュ(150gm)の吸着クロマトグラフィーに供した。溶媒を完全に除去して、自由に流動する物質を得た。1.5インチ直径のガラスカラムに、100gm SiO2ゲル、60−120メッシュ(EtOH中)を詰めた。吸着された物質をカラムに充填した。カラムをEtOAcで、MeOHの割合を次第に増加させて溶出した。各々100mlの全ての120の画分を集め、TLCパターンに基づいてプールした(展開溶媒として、EtOAc:HCOOH:H2O::8:1:1)。新たに調製したBorinate−PEG4000溶液[MeOH中の2−アミノエチルジフェニルボリネートの1%溶液およびEtOH中のポリエチレングリコール4000の5%溶液(噴霧する前に1:1v/vに混合した)]を噴霧することによってスポットを可視化した。
【0049】
EtOAcおよび10%MeOH中に溶出した画分は、同じTLCパターンを示した。これらの画分をプールし、乾燥し、次いで、最小量のMeOH中に溶解した。結晶化をメタノール溶液に少量ずつCHCl3を添加することによって行い、862−IIとして特徴付けられる無色の無定形粉末を得た。
【0050】
抽出物RJM/0862/P08/A003の標準化を、以下の操作条件を使用するHPLCによって、862−IIに基づいて行った:
カラム :RP−18e(E−Merck,5μm、4.6×250mm)において
移動相 :アセトニトリル(B):1.5%酢酸(水中)(A)
流速 :1ml/分
λmax:335nm
この化合物をNMR(1Hおよび13C)ならびにFABMSデータに基づいて、ベルバスコシドとして確立した。
【0051】
抽出物RJM/0862/P08/A003中の862−IIのパーセンテージが0.22であることを見出した。
【0052】
862−II、無定形粉末、融点145〜146℃、[α]21−85.6[C0.5%MeOH]、MS:FABMS、[M+Na]+m/z647が、公知のフェニルプロパノイド、すなわち、ベルバスコシド(アクテオシド);[β−(3’,4’−ジヒドロキシフェニル)エチル−O−α−L−ラムノピラノシル(1→3)−β−D−(4−O−カフェオイル)−グルコピラノシド]であることを見出した。構造を、文献(Andary C.,Wylde,R.Laffite C.,Privat G.およびWinternitz F.;Laboratie de Botanique et Cryptogamie,Faculte de Pharmacie.34000 Montpellier.France;Phytochemistry,21(5),1123−1127(1982))に報告されるデータと類似の1H NMRデータおよび13C NMRデータによって最終的に確認した。
【0053】
研究設計
1.動物:シロネズミ(Wistar,150〜180g)いずれかの性別
シロマウス(Swiss,25〜30g)いずれかの性別
2.肝細胞毒素:四塩化炭素(CCl4)
3.研究:予防
4.処置スケジュール:毒素の48時間前、24時間前、02時間前、および毒素の06時間後;試験物質/参照標準の最後の処置の18時間後に血液および肝臓サンプルを収集
5.用量:
アクテオシド:0.625mg/kg、1.25mg/kg、2.5mg/kg、および5.0mg/kg、p.o.
シリマリン(Silymarin):50mg/kg、p.o.
グリシルヒジン(Glycyrrhizin):100mg/kg、p.o.
CCl4:流動パラフィン中1ml/kg p.o.(1:1)
6.パラメータ
血清
ビリルビン:Clonital,24030−Carvico(BG)−Italyによって供給されるキットを使用することによるJendrassik法
トリグリセリド:Accurex BiomedicalPvt.Ltd.,Thaneによって供給されるキットによる酵素的GPO−POD法
アルブミン:Accurex BiomedicalPvt.Ltd.,Thaneによって供給されるキットによる比色B.C.G.法
トランスアミナーゼ(ALT,& AST):アミノ基転移反応によって形成されるピルベートを、2,4−ジニトロフェニルヒドラジンとの反応後に分光光度的に決定した(ReitmanおよびFrankel,1957)
ALP:アルカリ性溶媒中で形成されるp−ニトロフェノールを、基質としてリン酸p−ニトロフェニルを使用して分光光度的に測定した(WalerおよびSchutt,1974)。
肝臓ホモジネート:
GSH:これは、DTNBとの反応による除タンパク質後に決定した(David1987によって改変されたEllman1959)
脂質過酸化:チオバルビツール酸反応物質を、535nmで分光光度的に決定した。BuegeおよびAust(1978)の方法による。
肝臓保護活性:
肝臓保護活性(H)を以下の式によって計算した:
H=[1−(TC−V/VC−V)]×100
ここで、TC、VC、およびVは、それぞれ、薬物+毒素処置群の動物、ビヒクル+毒素の処置群の動物、およびビヒクル処置群の動物である。
【0054】
【表1−1】
【0055】
【表1−2】
【0056】
【表2】
【図面の簡単な説明】
【0057】
【図1】図1は、純度を確認するHPLCグラフを示す。
【図2】図2は、アクテオシド、シリマリン、およびグリシリジアの比較の肝保護性活性(%)を示す。
【特許請求の範囲】
【請求項1】
植物Colerbrookea oppositifoliaからの高肝保護の純粋なアクテオシドの単離プロセスであって、該プロセスは、以下:
a.該植物の空中部分を乾燥させる工程、
b.該乾燥させた部分を粉末に粉砕する工程、
c.該粉末に水またはエタノールを3〜4回浸透させて、抽出物を得る工程、
d.該抽出物を、懸濁した粒子を取り除くためにろ過して、上清を得る工程、
e.該上清を約45℃〜55℃で乾燥させて、残渣を得る工程、
f.該残渣を、クロロホルム、酢酸エチルおよびブタノールを連続的に用いて分画する工程、
g.ブタノール画分にメタノールを添加した後、該画分をSiO2の吸着クロマトグラフィーに供する工程、
h.該吸着した画分をガラスカラムに充填する工程、
i.該カラムを、次第に増加する極性のメタノール:クロロホルムの溶媒で溶出して、さらなる画分を得る工程、およびこのプロセスをもう1回繰り返す工程、
j.該画分をカラムクロマトグラフィーに供して画分を得る工程、
k.該画分を減圧下で濃縮してアクテオシドを残渣として得る工程、
を包含する、プロセス。
【請求項2】
前記プロセスによって得た前記アクテオシドが、他の供給源から得たアクテオシドと比較して12分の1〜25分の1低い投薬量で肝保護性活性を示す、請求項1に記載のプロセス。
【請求項3】
前記抽出物が、水性抽出物およびアルコール抽出物である、請求項1に記載のプロセス。
【請求項4】
前記アクテオシドが、全抽出物の約1.0%(重量%)である、請求項1に記載のプロセス。
【請求項5】
植物Colerbrookea oppositifolia由来の純粋なアクテオシドを用いて、被験体を有効に肝保護する方法であって、該被験体に、適切な低用量のアクテオシドを投与する工程を包含する、方法。
【請求項6】
前記投薬量が約0.5〜10.0mg/kg体重の範囲である、請求項2に記載の方法。
【請求項7】
アクテオシドが、経口経路を介して投与される、請求項2に記載の方法。
【請求項8】
アクテオシドが、異常に増加したレベルの血清グルタミントランスフェラーゼ(GPT)を低減させる、請求項2に記載の方法。
【請求項9】
アクテオシドが、異常に増加したレベルの血清グルタミントランスフェラーゼ(GOT)を低減させる、請求項2に記載の方法。
【請求項10】
アクテオシドが、異常に増加したレベルの血清アルカリホスファターゼ(ALP)を低減させる、請求項2に記載の方法。
【請求項11】
アクテオシドが、異常に増加したレベルの血清ビリルビンを低減させる、請求項2に記載の方法。
【請求項12】
アクテオシドが、異常に増加したレベルの血清トリグリセリド(TG)を低減させる、請求項2に記載の方法。
【請求項13】
アクテオシドが、異常に増加したレベルの脂質ペルオキシダーゼ(LP)を低減させる、請求項2に記載の方法。
【請求項14】
アクテオシドが、異常に減少したレベルのアルブミンを増加させる、請求項2に記載の方法。
【請求項15】
アクテオシドが、異常に減少したレベルの還元型グルタチオンを増加させる、請求項2に記載の方法。
【請求項16】
アクテオシドが、市販の肝保護剤と比較して約10〜20倍有効である、請求項2に記載の方法。
【請求項17】
アクテオシドが、肝毒性に対して約40〜85%の保護を提供する、請求項2に記載の方法。
【特許請求の範囲】
【請求項1】
植物Colerbrookea oppositifoliaからの、高肝保護の純粋なアクテオシドの単離プロセスであって、該プロセスは、以下:
a.該植物の空中部分を乾燥させる工程、
b.該乾燥させた部分を粉末に粉砕する工程、
c.該粉末に水またはエタノールを3〜4回浸透させて、抽出物を得る工程、
d.該抽出物を、懸濁した粒子を取り除くためにろ過して、上清を得る工程、
e.該上清を約45℃〜55℃で乾燥させて、残渣を得る工程、
f.該残渣を、クロロホルム、酢酸エチルおよびブタノールを連続的に用いて分画する工程、
g.ブタノール画分にメタノールを添加した後、該画分をSiO2の吸着クロマトグラフィーに供する工程、
h.該吸着した画分をガラスカラムに充填する工程、
i.該カラムを、次第に増加する極性のメタノール:クロロホルムの溶媒で溶出して、さらなる画分を得る工程、およびこのプロセスをもう1回繰り返す工程、
j.該画分をカラムクロマトグラフィーに供して画分を得る工程、
k.該画分を減圧下で濃縮してアクテオシドを残渣として得る工程、
を包含する、プロセス。
【請求項2】
前記プロセスによって得た前記アクテオシドが、他の供給源から得たアクテオシドと比較して12分の1〜25分の1低い投薬量で肝保護性活性を示す、請求項1に記載のプロセス。
【請求項3】
前記抽出物が、水性抽出物およびアルコール抽出物である、請求項1に記載のプロセス。
【請求項4】
前記アクテオシドが、全抽出物の約1.0%(重量%)である、請求項1に記載のプロセス。
【請求項1】
植物Colerbrookea oppositifoliaからの高肝保護の純粋なアクテオシドの単離プロセスであって、該プロセスは、以下:
a.該植物の空中部分を乾燥させる工程、
b.該乾燥させた部分を粉末に粉砕する工程、
c.該粉末に水またはエタノールを3〜4回浸透させて、抽出物を得る工程、
d.該抽出物を、懸濁した粒子を取り除くためにろ過して、上清を得る工程、
e.該上清を約45℃〜55℃で乾燥させて、残渣を得る工程、
f.該残渣を、クロロホルム、酢酸エチルおよびブタノールを連続的に用いて分画する工程、
g.ブタノール画分にメタノールを添加した後、該画分をSiO2の吸着クロマトグラフィーに供する工程、
h.該吸着した画分をガラスカラムに充填する工程、
i.該カラムを、次第に増加する極性のメタノール:クロロホルムの溶媒で溶出して、さらなる画分を得る工程、およびこのプロセスをもう1回繰り返す工程、
j.該画分をカラムクロマトグラフィーに供して画分を得る工程、
k.該画分を減圧下で濃縮してアクテオシドを残渣として得る工程、
を包含する、プロセス。
【請求項2】
前記プロセスによって得た前記アクテオシドが、他の供給源から得たアクテオシドと比較して12分の1〜25分の1低い投薬量で肝保護性活性を示す、請求項1に記載のプロセス。
【請求項3】
前記抽出物が、水性抽出物およびアルコール抽出物である、請求項1に記載のプロセス。
【請求項4】
前記アクテオシドが、全抽出物の約1.0%(重量%)である、請求項1に記載のプロセス。
【請求項5】
植物Colerbrookea oppositifolia由来の純粋なアクテオシドを用いて、被験体を有効に肝保護する方法であって、該被験体に、適切な低用量のアクテオシドを投与する工程を包含する、方法。
【請求項6】
前記投薬量が約0.5〜10.0mg/kg体重の範囲である、請求項2に記載の方法。
【請求項7】
アクテオシドが、経口経路を介して投与される、請求項2に記載の方法。
【請求項8】
アクテオシドが、異常に増加したレベルの血清グルタミントランスフェラーゼ(GPT)を低減させる、請求項2に記載の方法。
【請求項9】
アクテオシドが、異常に増加したレベルの血清グルタミントランスフェラーゼ(GOT)を低減させる、請求項2に記載の方法。
【請求項10】
アクテオシドが、異常に増加したレベルの血清アルカリホスファターゼ(ALP)を低減させる、請求項2に記載の方法。
【請求項11】
アクテオシドが、異常に増加したレベルの血清ビリルビンを低減させる、請求項2に記載の方法。
【請求項12】
アクテオシドが、異常に増加したレベルの血清トリグリセリド(TG)を低減させる、請求項2に記載の方法。
【請求項13】
アクテオシドが、異常に増加したレベルの脂質ペルオキシダーゼ(LP)を低減させる、請求項2に記載の方法。
【請求項14】
アクテオシドが、異常に減少したレベルのアルブミンを増加させる、請求項2に記載の方法。
【請求項15】
アクテオシドが、異常に減少したレベルの還元型グルタチオンを増加させる、請求項2に記載の方法。
【請求項16】
アクテオシドが、市販の肝保護剤と比較して約10〜20倍有効である、請求項2に記載の方法。
【請求項17】
アクテオシドが、肝毒性に対して約40〜85%の保護を提供する、請求項2に記載の方法。
【特許請求の範囲】
【請求項1】
植物Colerbrookea oppositifoliaからの、高肝保護の純粋なアクテオシドの単離プロセスであって、該プロセスは、以下:
a.該植物の空中部分を乾燥させる工程、
b.該乾燥させた部分を粉末に粉砕する工程、
c.該粉末に水またはエタノールを3〜4回浸透させて、抽出物を得る工程、
d.該抽出物を、懸濁した粒子を取り除くためにろ過して、上清を得る工程、
e.該上清を約45℃〜55℃で乾燥させて、残渣を得る工程、
f.該残渣を、クロロホルム、酢酸エチルおよびブタノールを連続的に用いて分画する工程、
g.ブタノール画分にメタノールを添加した後、該画分をSiO2の吸着クロマトグラフィーに供する工程、
h.該吸着した画分をガラスカラムに充填する工程、
i.該カラムを、次第に増加する極性のメタノール:クロロホルムの溶媒で溶出して、さらなる画分を得る工程、およびこのプロセスをもう1回繰り返す工程、
j.該画分をカラムクロマトグラフィーに供して画分を得る工程、
k.該画分を減圧下で濃縮してアクテオシドを残渣として得る工程、
を包含する、プロセス。
【請求項2】
前記プロセスによって得た前記アクテオシドが、他の供給源から得たアクテオシドと比較して12分の1〜25分の1低い投薬量で肝保護性活性を示す、請求項1に記載のプロセス。
【請求項3】
前記抽出物が、水性抽出物およびアルコール抽出物である、請求項1に記載のプロセス。
【請求項4】
前記アクテオシドが、全抽出物の約1.0%(重量%)である、請求項1に記載のプロセス。
【図1】

【図2】
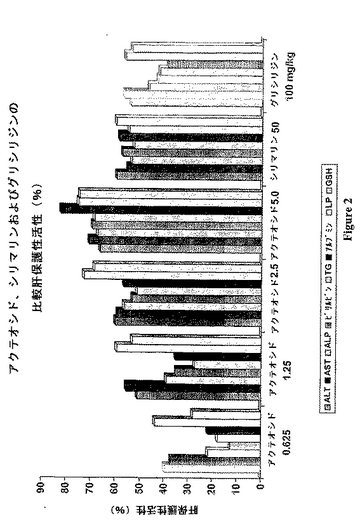
【図2】
【公表番号】特表2006−514675(P2006−514675A)
【公表日】平成18年5月11日(2006.5.11)
【国際特許分類】
【出願番号】特願2004−570051(P2004−570051)
【出願日】平成15年3月31日(2003.3.31)
【国際出願番号】PCT/IB2003/001180
【国際公開番号】WO2004/087184
【国際公開日】平成16年10月14日(2004.10.14)
【出願人】(599032822)カウンセル オブ サイエンティフィック アンド インダストリアル リサーチ (6)
【Fターム(参考)】
【公表日】平成18年5月11日(2006.5.11)
【国際特許分類】
【出願日】平成15年3月31日(2003.3.31)
【国際出願番号】PCT/IB2003/001180
【国際公開番号】WO2004/087184
【国際公開日】平成16年10月14日(2004.10.14)
【出願人】(599032822)カウンセル オブ サイエンティフィック アンド インダストリアル リサーチ (6)
【Fターム(参考)】
[ Back to top ]