
D−セリン合成活性を有する酵素を利用したD−セリンの製造方法
【課題】グリシンとホルムアルデヒドからD−セリンを製造する際に、L−セリンの副生を抑制する方法を提供する。
【解決手段】ホルムアルデヒドとグリシンからD−セリンを合成する活性を有する新規な酵素をコードするDNA、該DNAをベクターに組み込んでなる組換え体DNA、該組換え体DNAで形質転換した形質転換体、ホルムアルデヒドとグリシンからD−セリンを合成する活性を有する新規な酵素及び該酵素を利用したホルムアルデヒドとグリシンからD−セリンを製造する。
【解決手段】ホルムアルデヒドとグリシンからD−セリンを合成する活性を有する新規な酵素をコードするDNA、該DNAをベクターに組み込んでなる組換え体DNA、該組換え体DNAで形質転換した形質転換体、ホルムアルデヒドとグリシンからD−セリンを合成する活性を有する新規な酵素及び該酵素を利用したホルムアルデヒドとグリシンからD−セリンを製造する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ホルムアルデヒドとグリシンからD−セリンを合成する活性を有する新規な酵素をコードするDNA、該DNAをベクターに組み込んでなる組換え体DNA、該組換え体DNAで形質転換した形質転換体、ホルムアルデヒドとグリシンからD−セリンを合成する活性を有する新規な酵素及び該酵素を利用したホルムアルデヒドとグリシンからD−セリンを製造する方法に関する。
【背景技術】
【0002】
D−セリンはD−サイクロセリン等の医薬品の合成中間体として有用な化合物であることが知られている。
【0003】
従来、ホルムアルデヒドとグリシンとからD−セリンを合成する活性を有する酵素としては、アリスロバクター(Arthrobacter)sp.DK−19に由来するD−スレオニンアルドラーゼ(以下、「DTA」と略記する。)が知られているに過ぎない(特許文献1:特開昭58−116690号公報を参照)。
【0004】
このDTAは、ホルムアルデヒド及びグリシンをそれぞれ50mmol使用して30℃で40時間反応させた後のD−セリンの生成量は2.5mmol(ホルムアルデヒドに対する収率は僅かに5%)であったことが報告されている。
【0005】
これに対し、同じアリスロバクター属の微生物である、アリスロバクターsp.DK−38に由来するDTAは、D−スレオニン、D−β−ヒドロキシフェニルセリン、D−β−ヒドロキシ−α−アミノ吉草酸等に反応する広い基質特異性を有しているにもかかわらず、D−セリンには反応しないことが報告されている(非特許文献1:Eur.J.Biochem.,1997,248,p385−393を参照)。
【0006】
また、キサントモナス属(Xanthomonas)由来のDTAを用いることにより、グリシンとアルデヒド化合物からD−β−ヒドロキシアミノ酸類を製造できることが報告されているが、キサントモナス属由来のDTAを用いてホルムアルデヒドとグリシンとからD−セリンが合成できるとの報告はなされていない(特許文献2:特開平5−168484号公報を参照)。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開昭58−116690号公報
【特許文献2】特開平5−168484号公報
【非特許文献】
【0008】
【非特許文献1】Eur.J.Biochem.,1997,248,p385−393
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明は、前記の背景技術に鑑み、ホルムアルデヒドとグリシンからD−セリンを合成する活性を有する新規な酵素をコードするDNA、該DNAをベクターに組み込んでなる組換え体DNA、該組換え体DNAで形質転換した形質転換体、ホルムアルデヒドとグリシンからD−セリンを合成する活性を有する新規な酵素、及び該酵素を利用したホルムアルデヒドとグリシンからD−セリンを製造する方法を提供することを目的とする。
【課題を解決するための手段】
【0010】
本発明者らは、公知のDTAと類似の構造を有する未知の酵素のなかに、ホルムアルデヒドとグリシンとからD−セリンを合成する能力を有する酵素が存在する可能性があると考え、公知のDTAのアミノ酸配列に関する情報に関する調査を行った。その結果、キサントモナス属(GenBank accession No.E05055)、アクロモバクター(Achromobacter)属(GenBank accession No.AB026892)、及びアリスロバクター属(GenBank accession No.AB010956)に由来するDTAのアミノ酸配列に関する情報を見出した。これらのアミノ酸配列を比較検討した結果、DTAのN末端領域及びC末端領域に相当するアミノ酸配列に高い相同性を持つことを見出した。
【0011】
本発明者らは、これらのアミノ酸配列からN末端領域とC末端領域を基にプライマーを設計し、このプライマーを用いて各種の微生物の染色体DNAからPCRによる増幅を試みたところ、アクロモバクター属の微生物からホルムアルデヒドとグリシンとからD−セリンを合成する活性を有する酵素をコードするDNAの増幅に成功した。
【0012】
このDNAに相当するアミノ酸配列と公知のアクロモバクター属に由来するDTAのアミノ酸配列との相同性は約50%程度であり、公知のアクロモバクター属に由来するDTAのアミノ酸配列とはかなり異なるものであった。一方、公知のキサントモナス属に由来するDTAのアミノ酸配列との相同性は約90%であった。
【0013】
本発明者らは次に、前記の新規なDNAをベクターに組み込んでなる組換え体DNAで形質転換された組換えEscherichia coliを用いてホルムアルデヒドとグリシンとを反応させてD−セリンの合成を試みたところ、驚くべきことに従来知られているDTAとは異なり、100mMのグリシンとホルムアルデヒドから反応収率70%以上という高収率でD−セリンを合成できることを見出した。
【0014】
更に、この組換えEscherichia coliを用いてD−セリンの蓄積反応を行うと、僅かながらL−セリンが生成することが認められた。
【0015】
D−セリンを医薬中間体等の高純度が要求される分野に用いる場合、D−セリンへのL−セリンの混入は好ましくない。D−セリンの製造時にL−セリンが副生することは、D−セリンの溶解度よりDL−セリンの溶解度が低いことからD−セリンの精製収率を大幅に低下させる。
【0016】
本発明者らは、この問題を解決するために、更に検討した結果、2価金属イオンの存在下、有機溶媒処理及び/又は加熱処理を施すことでL−セリンの副生を抑制できることを見出した。また、このような処理を施さなくとも反応中のホルムアルデヒド濃度を150mM以上に維持することでL−セリンの副生を抑制できることを見出した。更に、D−セリン合成に利用するDSAを生産する宿主としてL−セリン合成酵素遺伝子が欠失している微生物を利用することでL−セリンの副生を抑制できることを見出した。
【0017】
以上の知見を基に、本発明を完成した。すなわち、本発明は以下のとおりである。
(1)以下の(a)又は(b)のタンパク質をコードするDNA。
(a)配列番号4、6、8の何れかに記載のアミノ酸配列を含むタンパク質
(b)(a)のアミノ酸配列において1もしくは数個のアミノ酸が欠失、置換、挿入もしくは付加されたアミノ酸配列を含み、かつグリシンとホルムアルデヒドからD−セリンを合成する酵素活性を有するタンパク質
(2)以下の(a)又は(b)のDNA。
(a)配列表の配列番号3、5、7の何れかに記載の塩基配列又はそれらの相補配列からなるDNA
(b)配列表の配列番号3、5、7の何れかに記載の塩基配列又はそれらの相補配列中の少なくとも20塩基の連続したDNA配列を含むDNA断片とストリンジェントな条件下でハイブリダイズし、かつグリシンとホルムアルデヒドからD−セリンを合成する活性を有する酵素をコードするDNA
(3)前記(1)又は(2)に記載のDNAをベクターに組み込んでなる組換え体DNA。
【0018】
(4)前記(3)に記載の組換え体DNAを用いて宿主細胞を形質転換して得られた形質転換体。
(5)形質転換される宿主細胞が微生物である前記(4)に記載の形質転換体。
(6)形質転換される微生物がD−セリンデアミナーゼの欠損した微生物である前記(5)に記載の形質転換体。
【0019】
(7)以下の(a)又は(b)のタンパク質。
(a)配列番号4、6、8の何れかに記載のアミノ酸配列を含むタンパク質
(b)(a)のアミノ酸配列において1もしくは数個のアミノ酸が欠失、置換、挿入もしくは付加されたアミノ酸配列を含み、かつグリシンとホルムアルデヒドからD−セリンを合成する酵素活性を有するタンパク質
【0020】
(8)前記(4)から(6)のいずれかに記載の形質転換体を培養し、得られた培養物からグリシンとホルムアルデヒドからD−セリンを合成する酵素活性を有するタンパク質を採取することを特徴とする、グリシンとホルムアルデヒドからD−セリンを合成する活性を有する酵素の製造方法。
(9)前記(4)から(6)のいずれかに記載の形質転換体又はその処理物の存在下でグリシンとホルムアルデヒドを反応させることを含むD−セリンの製造方法。
(10)前記(7)に記載のタンパク質の存在下でグリシンとホルムアルデヒドを反応させることを含むD−セリンの製造方法。
【0021】
(11)グリシンとホルムアルデヒドからD−セリンを合成する活性を有する微生物又はその処理物の存在下で、グリシンとホルムアルデヒドとを反応させてD−セリンを合成するD−セリンの製造方法であって、該反応の反応液中のL−セリンの副生をD−セリンに対して1.5モル%以下に抑制させる以下の(i)〜(iv)の一つ以上の手段を有するD−セリンの製造方法。
(i)グリシンとホルムアルデヒドからD−セリンを合成する活性を有する微生物を有機溶媒処理及び/又は加熱処理する方法、
(ii)反応液中のホルムアルデヒド濃度を、a)前記(i)の方法により有機溶媒処理及び/又は加熱処理を施す場合には2M以下に制御し、b)有機溶媒処理及び/又は加熱処理を施さない場合には、150mM以上2M以下に制御する方法、
(iii)グリシンとホルムアルデヒドからD−セリンを合成する活性を有する酵素を含有し、かつ、L−セリン合成酵素遺伝子が欠失している微生物を触媒として利用する方法、
(iv)L−セリンデアミナーゼ活性を有する酵素を含有する微生物を反応液中に添加する方法。
【0022】
(12)有機溶媒がホルムアルデヒド、ベンズアルデヒド、ジクロロエタン及びイソプロピルアルコールから選ばれる一種以上のものである前記(11)に記載の製造方法。
(13)有機溶媒処理及び/又は加熱処理を2価の金属イオンの存在下で行う前記(11)又は(12)に記載の製造方法。
(14)2価の金属がマグネシウム、マンガン、亜鉛、ニッケル、コバルト及び鉄から選ばれる一種以上の金属である前記(13)に記載の製造方法。
(15)グリシンとホルムアルデヒドからD−セリンを合成する活性を有する微生物が前記(5)又は(6)に記載の形質転換体である前記(11)に記載の製造方法。
【発明の効果】
【0023】
本発明によれば、公知のDTAを用いたD−セリンの製造方法に比べ、グリシンとホルムアルデヒドからD−セリンを収率よく製造できる。
【図面の簡単な説明】
【0024】
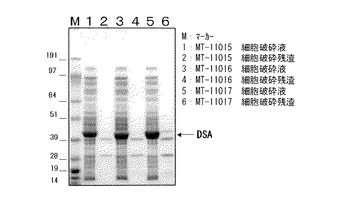
【図1】図1は、形質転換体から得た破砕液をSDS−ポリアクリルアミド電気泳動により分析した結果を示す図である。
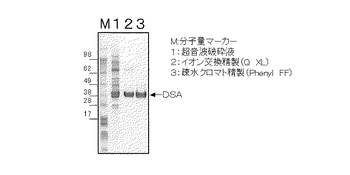
【図2】図2は、精製されたDSAのSDS−ポリアクリルアミド電気泳動写真である。
【発明を実施するための形態】
【0025】
1.ホルムアルデヒドとグリシンからD−セリンを合成する活性を有する新規な酵素をコードするDNAの遺伝子取得
配列表の配列番号4、6、8の何れかに記載のアミノ酸配列を含み、かつ、グリシンとホルムアルデヒドからD−セリンを合成する活性を有する酵素(以下、グリシンとホルムアルデヒドからD−セリンを合成する活性を有する酵素を「DSA」と略記する。)をコードするDNAは、例えば、12301 Parklawn Drive, Rockville, Maryland 20852, U.S.A.のAmerican Type Culture Collectionより入手可能なアクロモバクター・キシロソキシダンス(Achromobacter xylosoxidans)(ATCC9220)、独立行政法人 製品評価技術基盤機構(National Institute of Technology and Evaluation)の生物遺伝資源部門(NITE Biological Resource Center)(日本国千葉県木更津市かずさ鎌足2−5−8)より入手可能なアクロモバクター・キシロソキシダンス(NBRC13495)及びアクロモバクター・デニトリフィカンス(Achromobacter denitrificans)(Synonym: Achromobacter xylosoxidans subsp. denitrificans)(NBRC15125)から染色体DNAを抽出し、配列表の配列番号1及び2に示した塩基配列を有するプライマーを用いてPCRを実施することにより得ることができる。
【0026】
PCRを実施するに際して、若干のミスマッチを持つ遺伝子も増幅することができるように増幅時の条件を改良することが有効である。例えば、アニール温度を低めに抑える方法、PCR反応液中にジメチルスルホキシドを5%程度添加する方法、あるいはGCリッチな遺伝子を増幅しやすいように工夫されたPCRキット(例えばGC−RICH PCRSystem(Roche社製))を使用する方法、更には前記の方法を組み合わせる方法などにより増幅が困難な遺伝子を増幅できることがある。
【0027】
DSAをコードするDNAの具体的な例としてアクロモバクター・キシロソキシダンス(ATCC9220)由来DTA遺伝子のDNA塩基配列を配列番号3に、該塩基配列より翻訳されるアミノ酸配列を配列番号4に例示できる。アクロモバクター・キシロソキシダンス(NBRC13495)由来DTA遺伝子のDNA塩基配列を配列番号5に、該塩基配列より翻訳されるアミノ酸配列を配列番号6に例示できる。アクロモバクター・デニトリフィカンス(NBRC15125)由来DTA遺伝子のDNA塩基配列を配列番号7に、該塩基配列より翻訳されるアミノ酸配列を配列番号8に例示できる。
【0028】
DSAをコードするDNAとしては前記したものの他に、配列表の配列番号3、5、7の何れかに記載の塩基配列又はそれらの相補配列中の少なくとも20塩基の連続したDNA配列を含むDNA断片とストリンジェントな条件下でハイブリダイズし、かつDSA活性を有するタンパク質をコードするDNAであればどのような生物由来であっても構わない。例えば、その生物中で機能を有していないサイレントなDNAであっても単離したDNAを適当な発現ベクターに連結することでDSAを産生することができればそれでも構わない。
【0029】
更に生物を特定しなくても、土壌などを直接、鋳型DNAとして配列表の配列番号1及び2の塩基配列をもつプライマーを用いたPCRに付すことでDSAをコードするDNAを得ることも可能である。
【0030】
DSAをコードするDNAの具体的な取得方法として、例えばD−セリンを含有する培地で生育する微生物の染色体DNAを鋳型とし、配列表の配列番号1及び2に記載の塩基配列を有するプライマーを用いてPCRを行い増幅したDNAを例示することができる。
【0031】
また、配列表の配列番号3、5、7の何れかに記載の塩基配列もしくは相補配列がストリンジェントな条件下でハイブリダイズし得る範囲内でかつ、コードされる酵素の活性に影響を及ぼさない範囲内で部位特異的変異導入法(Nucleic Acid Res., 10, pp. 6487 (1982); Nucleic Acid Res., 13, pp. 4431 (1985); Methods in Enzymol., 100, pp. 448(1983); Molecular Cloning 2ndEdt., Cold Spring Harbor Laboratory Press(1989); PCR A Practical Approach IRL Press pp. 200 (1991); Current Protocols in Molecular Biology, John Wiley &;Sons (1987-1997); Proc. Natl. Acad. Sci., USA, 79, pp. 6409 (1982); Gene, 34, 315 (1985); Proc. Natl. Acad. Sci., USA, 82, pp.488 (1985))などを用いて、適宜欠失、置換、挿入、及び/又は付加変異を導入することにより、配列番号4、6、8の何れかに記載のアミノ酸配列において1もしくは数個のアミノ酸が欠失、置換、挿入もしくは付加されたアミノ酸配列を含み、かつDSA活性を有するタンパク質をコードするDNAを得ることも可能である。
【0032】
本明細書中の、アミノ酸の欠失、置換、挿入もしくは付加は、前記の出願前周知技術である部位特異的変異導入法により実施することができ、また、1もしくは数個のアミノ酸とは、部位特異的変異導入法により欠失、置換、挿入もしくは付加できる程度の数、例えば1〜5個のアミノ酸、好ましくは1〜3個のアミノ酸を意味する。
【0033】
ハイブリダイゼーションのための条件とは、特定のハイブリダイゼーションシグナルを検出するために当業者が一般的に用いている条件を例示できる。好ましくは、ストリンジェントなハイブリダイゼーション条件とストリンジェントな洗浄条件を意味する。具体的には、例えば、6×SSC(1×SSCの組成:0.15M NaCl、0.015Mクエン酸ナトリウム、pH7.0)、0.5%SDS、5×デンハート及び100mg/mlニシン精子DNAを含む溶液中プローブとともに55℃で一晩保温するという条件等が挙げられる。ついでフィルターを0.2×SSC中42℃で洗浄するなどを例示することができる。ストリンジェントな条件としては、フィルターの洗浄工程における0.1×SSC、50℃の条件であり、更にストリンジェントな条件としては、同工程における0.1×SSC、65℃の条件を挙げることができる。
【0034】
本明細書中で用いる「配列番号3、5、7の何れかに記載の塩基配列又はそれらの相補配列中の少なくとも20塩基の連続したDNA配列を含むDNA断片」とは、配列表の配列番号3、5、7の何れかに記載の又はそれらの相補配列中の任意の少なくとも20塩基、好ましくは少なくとも30塩基、例えば40、60又は100塩基の連続した配列を一つ又は複数選択したDNAの断片のことである。
【0035】
2.該タンパク質の遺伝子を持つ組換えDNAの作製
前記のDSAをコードするDNAをベクターに組込むことにより組換え体DNAを得ることができる。
クローニングする際のベクターとしては、宿主微生物内で自律的に増殖し得るファージ又はプラスミドから遺伝子組換え用として構築されたものが適している。ファージとしては、例えばEscherichia coliを宿主微生物とする場合にはLambda gt10、Lambda gt11などが例示することができる。また、プラスミドとしては、例えばEschenchia coliを宿主微生物とする場合には、pBTrp2、pBTac1、pBTac2(いずれもBoehringer Mannheim社製)、pKK233−2(Pharmacia社製)、pSE280(Invitrogen社製)、pGEMEX−1(Promega社製)、pQE−8(QIAGEN社製)、pQE−30(QIAGEN社製)、pBluescriptII SK+、pBluescriptII SK(−)(Stratagene社製)、pET−3(Novagen社製)、pUC18(宝酒造社製)、pSTV28(宝酒造社製)、pSTV29(宝酒造社製)、pUC118(宝酒造社製)等を例示することができる。
【0036】
プロモーターとしては、宿主細胞中で発現できるものであればいかなるものでもよい。例えば、trpプロモーター(Ptrp)、lacプロモーター(Plac)、PLプロモーター、PHプロモーター、PSEプロモーター等の、Escherichia coliやファージ等に由来するプロモーターを挙げることができる。またtacプロモーター、lacT7プロモーターのように人為的に設計改変されたプロモーター等も用いることができる。更にバチルス属細菌中で発現させるためにNpプロモーター(特公平8−24586号公報)なども用いることができる。
【0037】
リボソーム結合配列としては、宿主細胞中で発現できるものであればいかなるものでもよいが、シャイン−ダルガノ(Shine−Dalgarno)配列と開始コドンとの間を適当な距離(例えば6〜18塩基)に調節したプラスミドを用いることが好ましい。
【0038】
転写・翻訳を効率的に行うため、該タンパク質活性を有するタンパク質のN末端又はその一部を欠失したタンパク質と発現ベクターのコードするタンパク質のN末端部分を融合させたタンパク質を発現させてもよい。
【0039】
目的とするタンパク質の発現には転写終結配列は必ずしも必要ではないが、構造遺伝子直下に転写終結配列を配置することが望ましい。
【0040】
クローニングの際、前記のようなベクターを、上述したDSAをコードするDNAの切断に使用した制限酵素で切断してベクター断片を得ることができるが、必ずしも該DNAの切断に使用した制限酵素と同一の制限酵素を用いる必要はない。該DNA断片とベクターDNA断片とを結合させる方法は、公知のDNAリガーゼを用いる方法であればよく、例えば該DNA断片の付着末端とベクター断片の付着末端とのアニーリングの後、適当なDNAリガーゼの使用により該DNA断片とベクターDNA断片との組換えベクターを作製する。必要に応じて、アニーリングの後、微生物等の宿主に移入して生体内のDNAリガーゼを利用し組換えベクターを作製することもできる。
【0041】
3.DSAをコードするDNAをベクターに組み込んでなる組換え体DNAによる形質転換体の作製
前記の組換え体DNAを宿主細胞に導入することにより、前記の組換え体DNAで形質転換された形質転換体を得ることができる。
宿主細胞としては、組換えベクターが安定であり、かつ自律増殖可能で外来性遺伝子の形質発現できるものであれば特に制限されない。宿主細胞としては、微生物、例えば細菌を挙げることができ、そのようなものとしてEscherichia coli DH5α,XL−1BlueなどのEscherichia coliが例示される。また、宿主細胞としてその他酵母等の微生物や昆虫細胞を利用することも可能である。
【0042】
微生物に組換え体DNAを移入する方法としては、例えば微生物がEscherichia coliの場合には、カルシウム処理によるコンピテントセル法やエレクトロポーレーション法などを用いることができる。
【0043】
宿主細胞として、生成物であるD−セリンの分解を抑制する目的でD−セリンデアミナーゼ活性の低い微生物又はD−セリンデアミナーゼ活性の欠損した微生物を用いることができる。D−セリンデアミナーゼ活性の欠損した微生物の具体的な例として、例えば実施例6に記載のD−セリンデアミナーゼ遺伝子を組換えたEscherichia coli等が挙げられる。
【0044】
また、D−セリン合成反応におけるL−セリンの生成を抑制する目的で、L−セリンデアミナーゼ活性の高い微生物を用いることにより生成したL−セリンを分解することも可能である。L−セリンデアミナーゼ活性の高い微生物として実施例15に記載のL−セリンデアミナーゼ遺伝子を組換えたEscherichia coli等を用いることもできる。
【0045】
微生物として、アラニンラセマーゼ、セリンヒドロキシメチルトランスフェラーゼ、L−スレオニンアルドラーゼ等のL−セリンの合成に関与する酵素活性が低い微生物又はこれらの酵素活性が欠損した微生物を用いることは、L−セリンが生成しないため好適である。
【0046】
更に好ましくは、これらの酵素が全て欠損した微生物を宿主細胞として用いることが望ましい。
【0047】
4.DSAの生産
前記の形質転換体を培養し、次に得られたDSAを採取することにより、DSAを製造することができる。
形質転換体の培養は、宿主細胞の培養に用いられる通常の方法に従って行うことができる。形質転換体がEscherichia coli等の原核微生物、酵母菌等の真核微生物である場合、これら微生物を培養する培地は、該微生物が資化し得る炭素源、窒素源、無機塩類等を含有し、形質転換体の培養を効率的に行える培地であれば天然培地、合成培地のいずれでもよい。
【0048】
炭素源としては、形質転換体が資化し得るものであればよく、グルコース、フラクトース、スクロース、これらを含有する糖蜜、デンプンあるいはデンプン加水分解物等の炭水化物、酢酸、プロピオン酸等の有機酸、エタノール、プロパノールなどのアルコール類が用いられる。
【0049】
窒素源としては、アンモニア、塩化アンモニウム、硫酸アンモニウム、酢酸アンモニウム、リン酸アンモニウム等の各種無機酸や有機酸のアンモニウム塩、その他含窒素化合物、並びに、ペプトン、肉エキス、酵母エキス、コーンスチープリカー、カゼイン加水分解物、大豆粕、大豆粕加水分解物、各種発酵菌体及びその消化物等が用いられる。
【0050】
無機塩としては、リン酸第一カリウム、リン酸第二カリウム、リン酸マグネシウム、硫酸マグネシウム、塩化ナトリウム、硫酸第一鉄、硫酸マンガン、硫酸銅、炭酸カルシウム等が用いられる。
【0051】
培養は、振盪培養又は深部通気攪拌培養などの好気的条件下で行う。培養温度は15〜50℃がよく、培養時間は、通常16時間〜5日間である。培養中pHは、3.0〜9.0に保持する。pHの調整は、無機あるいは有機の酸、アルカリ溶液、尿素、炭酸カルシウム、アンモニアなどを用いて行う。また培養中必要に応じて、アンピシリンやテトラサイクリン等の抗生物質を培地に添加してもよい。
【0052】
プロモーターとして誘導性のプロモーターを用いた発現ベクターで形質転換した微生物を培養するときには、必要に応じてインデューサーを培地に添加してもよい。例えば、lacプロモーターを用いた発現ベクターで形質転換した微生物を培養するときにはイソプロピル−β−D−チオガラクトピラノシド(IPTG)等を、trpプロモーターを用いた発現ベクターで形質転換した微生物を培養するときにはインドール酢酸(IAA)等をそれぞれ培地に添加してもよい。
【0053】
形質転換体は、これを含む培養液を遠心分離、ろ過等の手段を用いて分離し回収することができる。
【0054】
形質転換体処理物は、細胞の破壊を目的として、形質転換体を機械的破壊、超音波処理、凍結融解処理、乾燥処理、加圧又は減圧処理、浸透圧処理、自己消化、界面活性剤処理、酵素処理したもの、及びこれらの処理により得られるDSAを含む画分の固定化物、形質転換体の固定化物として得ることができる。
【0055】
なお、本発明において微生物の処理物も前記形質転換体処理物と同様の処理をしたものを指す。
【0056】
このような形質転換体又は形質転換体処理物からDSAを精製するためには、形質転換細胞を破砕し細胞破砕液をイオン交換樹脂やゲルろ過等のクロマトグラフィーによる分画や硫酸アンモニウムなどによる塩析などを組み合わせることで精製酵素を得ることができる。
【0057】
5.D−セリンの製造方法
前記の形質転換体又はその形質転換体処理物の存在下でグリシンとホルムアルデヒドを反応させることによりD−セリンを製造することができる。
D−セリンの製造は、pH6.0〜9.0、温度20〜60℃で振盪、もしくは攪拌条件下で行うのが好ましい。
【0058】
形質転換体又はその形質転換体処理物の使用量は、ホルムアルデヒドとグリシンとの反応が十分に進行すれば特に制限されないが、通常グリシン1gに対して少なくても10unit、好ましくは50unit以上のDSA活性となる量を添加することが望ましい。形質転換体又はその形質転換体処理物の添加方法は反応開始時に一括で添加しても構わないし、反応中に分割して又は連続して添加しても構わない。
【0059】
DSA活性は、1μモルのD−セリンを1分間に合成できる能力を1unitと定義する。
DSA活性は、100mMグリシン、0.1mMピリドキサールリン酸、10mM塩化マグネシウム、5mMホルムアルデヒドを含むpH8.0の200mMのトリス塩酸緩衝液中に酵素液を加えて30℃で保温し、D−セリンの生成量を測定して算出する。
【0060】
反応液中のグリシンの濃度は、100mM以上であり、好ましくは1M以上5M以下である。グリシンの添加方法については反応開始時に一括添加しても、反応の進行に伴い分割して又は連続して添加してもよい。
【0061】
ホルムアルデヒドは、その気体を反応液中に供給することができるが、水溶液又はアルコール溶液として供給することもできる。また、ホルムアルデヒドの供給源としてパラホルムアルデヒドを使用することもできるが、37%程度のホルムアルデヒドの水溶液の使用が好適である。
【0062】
ホルムアルデヒドの添加方法としては、一括添加又は反応の進行に伴い分割又は連続的に添加する方法で行えるが、反応液中のホルムアルデヒド濃度はDSA活性を阻害しない程度の濃度に制御することが好ましい。DSA活性を阻害しない程度の濃度とは通常5M以下、好ましくは2M以下、更に好ましくは500mM以下、特に好ましくは300mM以下である。
【0063】
反応液中のホルムアルデヒド濃度を制御する方法として(1)反応液に一定速度で添加する方法、(2)ホルムアルデヒド濃度を定量し、酵素活性が失活しない程度の濃度になってから分割添加する方法、(3)パラホルムアルデヒドを添加し、パラホルムアルデヒドがホルムアルデヒドに遊離する速度以上の酵素量を反応系に添加しておくことで実質的にホルムアルデヒドによる阻害を回避する方法、(4)反応の進行に伴い反応液中に生成物のD−セリンが蓄積し、原料のグリシンが減少する。D−セリンとグリシンの等電点がそれぞれ、5.68と5.97であるため、反応の進行に伴い反応液のpHの低下が起こる。反応液のpHの低下を補正する量以上のアルカリを反応液に添加し、そのpH上昇分を補正できる量のホルムアルデヒドを添加するという方法でホルムアルデヒドを添加することも可能である。
【0064】
反応液中のDSA量を予め設定したアルカリの添加速度、即ちD−セリンの合成速度以上にしておけば、ホルムアルデヒドの濃度を測定するなどの煩雑な操作を用いずとも反応液中のホルムアルデヒド濃度を制御することが可能となる。
【0065】
反応液に添加するアルカリとしては、水酸化リチウム、水酸化ナトリウム、水酸化カリウムなどのアルカリ金属水酸化物の他、水酸化アンモニウム、水酸化カルシウム、リン酸二カリウム、リン酸二ナトリウム、ピロリン酸カリウム、アンモニアなど水に溶解して、液性を塩基性とするものであればよい。
【0066】
反応液の媒体としては、水もしくは水性媒体、有機溶媒又は水もしくは水性媒体と有機溶媒の混合液が用いられる。水性媒体としては、例えばリン酸緩衝液、HEPES(N−2−ヒドロキシエチルピペラジン−N−エタンスルホン酸)緩衝液、トリス[トリス(ヒドロキシメチル)アミノメタン]塩酸緩衝液等の緩衝液が用いられる。有機溶媒としては反応を阻害しないものであればいずれでもよく、例えばアセトン、酢酸エチル、ジメチルスルホキシド、キシレン、メタノール、エタノール、ブタノール等が用いられる。
【0067】
上述の反応において、2価の金属イオンを有する化合物、2−メルカプトエタノール、ジチオスレイトール、亜硫酸水素ナトリウムなどの還元剤や補酵素であるピリドキサールリン酸、アンモニウム塩等を添加することにより反応収率が向上することがある。
【0068】
2価の金属イオンを有する化合物としては、例えば塩化マグネシウム、塩化マンガン、酢酸コバルト、硫酸第1鉄、塩化カルシウムなどが挙げられる。
これら化合物の反応液中の濃度としては通常0.1mMから100mM、好ましくは0.1mMから10mMである。
【0069】
6.反応液中のD−セリンの光学純度を高める方法
前記の形質転換体又はその形質転換体処理物を加熱処理して得られる処理物、又は前記の形質転換体又はその形質転換体処理物を有機溶媒で処理して得られる処理物の存在下で、グリシンとホルムアルデヒドを反応させることにより、L−セリンの副生を抑制することができる。
【0070】
加熱処理の条件としては加熱により大幅にDSA活性を低減させず、L−セリンを生成する活性を低減又は消失させることが可能であればどのような条件でも構わない。具体的な例として、例えばpH6.0から9.0、温度40℃から70℃で10分間から6時間程度攪拌する方法を挙げることができる。
また、有機溶媒処理と加熱処理を組み合わせて行うことも可能である。
【0071】
有機溶媒処理の条件としては大幅にDSA活性を低減させず、L−セリンを生成する活性を低減又は消失させることが可能であればどのような条件でも構わない。有機溶媒処理の条件としては、有機溶媒の濃度は通常20mM以上2M以下、好ましくは20mM以上1M以下、更に好ましくは50mMから1000mM、特に好ましくは50mMから300mM程度である。有機溶媒はDSA活性を大幅に低減させないものであれば制限はないが、ホルムアルデヒド、アセトアルデヒド、ベンズアルデヒド等のアルデヒド類、メタノール、エタノール、イソプロピルアルコールなどのアルコール類、アセトンなどのケトン類、ジクロロエタンなどのハロゲン化炭化水素類などの有機溶媒を用いることが好ましい。中でもホルムアルデヒドは酵素反応の基質であるため、最も好適である。D−セリンを添加することで該酵素反応によりホルムアルデヒドが生成するため、ホルムアルデヒドの変わりにD−セリンを添加する方法でも構わない。D−セリンの添加濃度としては100mMから5M程度が望ましい。
【0072】
有機溶媒処理の温度は10℃から50℃の範囲であり、pHは6.0から9.0の範囲が望ましい。有機溶媒処理中は、処理液のpHや有機溶媒濃度が均一となるように攪拌することが望ましい。
【0073】
また、D−セリン製造の反応開始時にホルムアルデヒド濃度を高めることでL−セリンの生成活性を低減又は失活させることも可能である。この場合、反応中のホルムアルデヒド濃度を0.1%から5%程度で30分間から3時間程度保った後、通常の反応を行えばよい。
【0074】
上述の処理を行う際に、2価の金属イオンを有する化合物、2−メルカプトエタノール、ジチオスレイトール、亜硫酸水素ナトリウムなどの還元剤や補酵素であるピリドキサールリン酸、アンモニウム塩等を添加することにより酵素活性が更に安定化することがある。
【0075】
2価の金属イオンを有する化合物としては、例えば、塩化マグネシウム、塩化マンガン、酢酸コバルト、硫酸第1鉄、塩化カルシウムなどが挙げられる。
【0076】
これら化合物の処理液中の濃度としては通常0.1mMから100mM、好ましくは0.1mMから10mMである。
【0077】
D−セリン合成反応終了後の反応液にL−セリンセアミナーゼ生産菌を加え、生成したL−セリンを分解させることにより最終製品であるD−セリンの光学純度を高めることもできる。L−セリンデアミナーゼ生産菌としてはD−セリンデアミナーゼを欠損した微生物を用いることが望ましい。
【0078】
7.D−セリンの採取方法
反応液からのD−セリンの採取は、通常の有機合成化学で用いられる方法、例えば、有機溶媒による抽出、結晶化、薄層クロマトグラフィー、高速液体クロマトグラフィー等により行うことができる。
本明細書は、本願の優先権の基礎である特願2004−298344の明細書及び/又は図面に記載された内容を包含する。
【実施例】
【0079】
以下に本発明の実施例を示すが、本発明はこれらの実施例に限定されるものではない。なお、D−セリン、L−セリン及びグリシンは高速液体クロマトグラフィーにより定量した。これらの分析条件及び、酵素(DSA及びDTA)活性の測定方法は次のとおりである。
【0080】
(1)D−セリン及びL−セリンの分析条件
カラム;TSK−GEL ENANTIO L1 4.6x250(東ソー株式会社)
カラム温度;45℃
ポンプ流速;0.8ml/min.
検出;UV254nm、溶離液;0.25mM硫酸銅:メタノール=9:1(V/V)
(2)セリン及びグリシンの分析条件
カラム;Shodex RSpak NN−814 8x250(昭和電工株式会社)
カラム温度;40℃
溶離液;10mM リン酸カリウム(pH3.0)
ポンプ流速;0.8ml/min
検出はオルトフタルアルデヒド(OPA)を用いたポストカラム誘導体化法〔J.Chromatogr.,83,353−355(1973)〕を用いた。
(3)酵素活性の測定方法
100mMグリシン、0.1mMピリドキサールリン酸、10mM塩化マグネシウム、5mMホルムアルデヒドを含むpH8.0の200mMのトリス塩酸緩衝液0.9mLに菌体懸濁液を超音波破砕した酵素液を適当に希釈して0.1mLを加え、30℃で15分間反応した。
生成したD−セリンをHPLCにて分析し活性を測定した。活性の単位は1分間に1μmolのD−セリンを生成する活性を1unitとした。
【0081】
[実施例1]
(DSAをコードする遺伝子の取得)
アクロモバクター・キシロソキシダンス(ATCC9220)、アクロモバクター・デニトリフィカンス(NBRC15125)、アクロモバクター・キシロソキシダンス(NBRC13495)を50mlのLB培地に接種した。30℃で一夜培養した後、集菌し、リゾチーム1mg/mlを含む溶菌液で溶菌した。溶菌液をフェノール処理した後、通常の方法によりエタノール沈殿によりDNAを沈殿させた。生じたDNAの沈殿は、ガラス棒に巻き付けて回収した後、洗浄し、PCRに用いた。
【0082】
PCR用のプライマーには、公知のDTA遺伝子に基づいて設計した配列番号1及び2に示す塩基配列を有するオリゴヌクレオチド(北海道システム・サイエンス株式会社に委託して合成した)を用いた。これらのプライマーは、5’末端付近及び3’末端付近に、それぞれKpnI及びHindIIIの制限酵素認識配列を有する。
【0083】
前記微生物の染色体DNA6ng/μl及びプライマー各3μMを含む0.025mlのPCR反応液を用いて、変性:96℃、1分間、アニーリング:55℃、30秒間、伸長反応:68℃、1分15秒間からなる反応サイクルを、35サイクルの条件でPCRを行った。
【0084】
PCR反応産物及びプラスミドpUC18(宝酒造(株))を、KpnI及びHindIIIで消化し、ライゲーション・ハイ(東洋紡(株))を用いて連結した後、得られた組換えプラスミドを用いて、Escherichia coli DH5αを形質転換した。形質転換株を、アンピシリン(Am)50μg/ml及びX−Gal(5−ブロモ−4−クロロ−3−インドリル−β−D−ガラクトシド)を含むLB寒天培地で培養し、Am耐性で且つ白色コロニーとなった形質転換株を得た。このようにして得られた形質転換株よりプラスミドを抽出した。プラスミドに導入されたDNA断片の塩基配列を通常の塩基配列の決定法に従い塩基配列を確認した。
【0085】
得られたDSAをコードするDNAから予想されるアミノ酸配列の分子量は何れもほぼ40kDaであった。
【0086】
得られたアクロモバクター・キシロソキシダンス(ATCC9220)由来のDSAをコードするDNAを持つプラスミドをpAcDTA1と命名した。
【0087】
アクロモバクター・キシロソキシダンス(NBRC13495)由来のDSAをコードするDNAを持つプラスミドをpAcDTA2と命名した。アクロモバクター・デニトリフィカンス(NBRC15125)由来のDSAをコードするDNAを持つプラスミドをpAcDTA3と命名した。
【0088】
(形質転換体の作製)
pAcDTA1、pAcDTA2、pAcDTA3を用いてEscherichia coli DH5αを通常の方法で形質転換し、得られた形質転換体をMT−11015、MT−11016、MT−11017と命名した。
【0089】
各々の組換え微生物を500mLのバッフル付き三角フラスコにAm50μg/mlを含むLB培地100mLに接種し、30℃にてOD660が0.6になるまで培養し、IPTG(イソプロピル−β−チオガラクトピラノシド)が1mMとなるように添加し、更に16時間振盪培養した。培養液を13000rpmで10分間遠心分離し、得られた菌体を5mLの1mM塩化マグネシウムを含む100mMトリス塩酸緩衝液(pH8.0)に懸濁し、−20℃にて凍結保存した。
【0090】
[実施例2]
(DSAの製造法)
実施例1で作製した形質転換体の懸濁液0.5mLをバイオラピュター(オリンパス社製)を用いて、氷水中で5分間破砕した。形質転換体の破砕液を遠心分離し、破砕液を調製した。破砕液をSDS−ポリアクリルアミド電気泳動により分析した結果を図1に示した。
沈殿物に100mMトリス塩酸緩衝液(pH8.0)を0.5mL加えて細胞残渣として同様に分析した。
【0091】
何れの形質転換体も可溶性画分におよそ40kDaの位置に発現するタンパク質が見出され、不溶性画分には見出されなかった。この分子量はそれぞれの遺伝子から推測されるアミノ酸配列の分子量とほぼ一致した。
【0092】
[実施例3]
(DSAの精製と精製DSAによるD−セリンの合成)
実施例2と同様の方法で作製したMT−11015の破砕液10mLを10000rpmで20分間遠心分離し、細胞残渣を除いた酵素液を調製した。酵素液を陰イオン交換樹脂(HiTrap Q−XL、アマシャム社製)に吸着させ、10mM塩化マグネシウム及び50mM塩化ナトリウムを含む100mMトリス塩酸緩衝液(pH8.0)から10mM塩化マグネシウム及び500mM塩化ナトリウムを含む100mMトリス塩酸緩衝液(pH8.0)へのリニアーグラジエントで溶出させた。活性画分を疎水クロマト樹脂(HiTrap Phenyl FF、アマシャム社製)に吸着させ、硫酸アンモニウムで飽和させた10mM塩化マグネシウムを含む100mMトリス塩酸緩衝液(pH8.0)から10mMの塩化マグネシウムを含む100mMトリス塩酸緩衝液(pH8.0)へのリニア−グラジエントで溶出させた。なお、以上の操作は、約10℃で操作した。
【0093】
超音波破砕液、イオン交換クロマトグラフィー処理した後の活性画分及び疎水クロマトグラフィー処理した後の活性画分をSDS−ポリアクリルアミド電気泳動により分析した結果を図2に示した。精製DSAのモノマーの分子量は40000±5000であった。
【0094】
100mMホルムアルデヒド、100mMグリシン、0.1mM PLP、10mM塩化マグネシウム、200mMトリス塩酸緩衝液(pH8.0)100mLよりなる基質溶液に精製DSA酵素液150unitを加え30℃で20時間反応させた。
D−セリンの反応収率は95%であった。
【0095】
[実施例4]
(D−セリン合成能力とD−スレオニン合成能力の比較)
アルデヒド源として、100mMのホルムアルデヒド又はアセトアルデヒドを含み、100mMグリシン、0.1mM PLP、10mM塩化マグネシウム、200mMトリス塩酸緩衝液(pH8.0)100mLよりなる基質溶液に実施例1で作製したMT−11015株の超音波破砕酵素液150unitを加え30℃で20時間反応させた。
アルデヒド源としてホルムアルデヒドは用いたときは、収率90%であったがアセトアルデヒドを用いたときは10%であった。
【0096】
[実施例5]
(ホルムアルデヒド100mM濃度でのD−セリン合成反応)
ホルムアルデヒド100mM、グリシン100mM、0.1mM PLP、10mM塩化マグネシウムを含むpH8.0の200mMトリス塩酸緩衝液100mLよりなる基質溶液に実施例1で作製した組換え微生物の超音波破砕酵素液150unitを加え30℃で20時間反応させた。結果を表1に示した。
【0097】
【表1】

【0098】
[実施例6]
(D−セリンデアミナーゼ欠損Escherichia coliの作製)
Escherichia coliのゲノムDNAの全塩基配列は公知であり(GenBanak accession number U00096)、Escherichia coliのD−セリンデアミナーゼのアミノ酸配列と遺伝子(以下、dsdAと略することがある)の塩基配列もすでに報告されている(GenBank accession number J01603)。Escherichia coli W3110株のゲノムDNAのdsdA近傍領域の遺伝子情報に基づいて作製された、配列番号9及び10、配列番号11及び12に示す塩基配列を有するオリゴヌクレオチドを用いてEscherichia coli W3110株(ATCC27325)のゲノムDNAをテンプレートとしてPCRを行った。得られたDNAフラグメントをそれぞれ、制限酵素PstIとXbaI、XbaIとKpnIで消化することにより、それぞれ約900bp、800bpのフラグメントを得た。このDNAフラグメントを温度感受性クローニングベクターpTH18cs1(GenBank accession number AB019610)〔Hashimoto−Gotoh, T., Gene, 241, 185−191 (2000)〕をPstI、KpnIで消化して得られるフラグメントと混合し、リガーゼを用いて結合した後、DH5α株に30℃で形質転換し、クロラムフェニコール10μg/mlを含むLB寒天プレートに生育する形質転換体を得た。得られたコロニーをクロラムフェニコール10μg/mlを含むLB液体培地で30℃で一晩培養し、得られた菌体からプラスミドを回収した。回収したプラスミドをXbaIで消化し、T4DNAポリメラーゼで平滑末端処理を行った後、pUC4Kプラスミド(Pharmacia)由来のカナマイシン耐性遺伝子と連結した。
【0099】
こうして得られたプラスミドをEscherichia coli W3110株(ATCC27325)に30℃で形質転換し、クロラムフェニコール10μg/mlとカナマイシン50μg/mlを含むLB寒天プレートに30℃で一晩培養し、形質転換体を得た。得られた形質転換体をカナマイシン50μg/mlを含むLB液体培地に接種し、30℃で一晩培養した。次にこれらの培養菌体が得られるようにカナマイシン50μg/mlを含むLB寒天プレートに塗布し、42℃で生育するコロニーを得た。得られたコロニーをカナマイシン50μg/mlを含むLB液体培地で30℃で一晩培養し、更にカナマイシン50μg/mlを含むLB寒天プレートに塗布して42℃で生育するコロニーを得た。
【0100】
出現したコロニーの中から無作為に100コロニーをピックアップしてそれぞれをカナマイシン50μg/mlを含むLB寒天プレートとクロラムフェニコール10μg/mlを含むLB寒天プレートに生育させ、カナマイシンを含むLB寒天プレートにのみ生育するクロラムフェニコール感受性のクローンを選んだ。更にこれらの目的クローンの染色体DNAからPCRによりdsdA近傍領域の約3.0kb断片を増幅させ、dsdAがカナマイシン耐性遺伝子に置換されている株を選抜し、得られた株をW3110dsdA欠失株(以下ΔdsdAと略することがある)と命名した。実施例1で作製したプラスミドを用いて形質転換し、前記と同様に凍結保存した菌体を作製し、同様の反応を行ったところD−セリンの分解は殆ど確認できなかった。
【0101】
[実施例7]
(形質転換体MT−11016を使用したD−セリンの製造:Mg無添加による製造)
7.5gのグリシンと9.4gの0.026重量%のピリドキサールリン酸に蒸留水53.1gを加え、水酸化ナトリウムにてpHを8.0に調整した。実施例1で得たMT−11016の菌体懸濁液(活性として1500unit相当)を加えた。反応温度30℃にて20重量%のホルムアルデヒド20.8gを反応液中のホルムアルデヒド濃度をAHMT法(衛生化学(1976)、Vol22、p39)にて定量し、50mMから300mMとなるように添加した。反応液のpHは水酸化ナトリウムにてpH8.0に調整した。72時間後の反応収率は85%に達していた。
【0102】
[実施例8]
(形質転換体MT−11017を使用したD−セリンの製造:Mg無添加による製造)
7.5gのグリシンと9.4gの0.026重量%のピリドキサールリン酸に蒸留水53.1gを加え、水酸化ナトリウムにてpHを8.0に調整した。実施例1で得たMT−11017菌体懸濁液(活性として1500unit相当)を加えた。反応温度30℃にて20重量%のホルムアルデヒド20.8gを0.8g/15分の速度で15分間添加し、ホルムアルデヒドの添加を45分間停止するというサイクルを繰り返して反応液に添加した。反応液のpHは反応中水酸化ナトリウムにてpH8.0に調整した。24時間後のD−セリンの反応収率は95%に達していた。
一方、ホルムアルデヒドを一括で添加して反応させたところ反応収率は20%であった。
【0103】
[実施例9]
(ホルムアルデヒド処理した形質転換体によるD−セリンの製造:Mg無添加による製造)
実施例1で作製したMT−11015の凍結菌体の溶解液にホルムアルデヒドを100mMとなるように添加し、30℃で1時間緩やかに攪拌した。攪拌中はpHを8.0に水酸化ナトリウムにて調整した。該菌体験濁液を用いて実施例8と同じ反応を行った。ホルムアルデヒド処理を行わなかった場合はL−セリンが2モル%生成したが、ホルムアルデヒド処理を行った場合はL−セリンを検出できなかった。
【0104】
前記の反応液に6N−塩酸を加えpHを4.1に調整した。活性炭(50%含水率)0.97gを加え、60℃で1時間攪拌し、ろ過により活性炭と菌体成分を除去した。次いで、ろ液を30gまで濃縮し、イソプロピルアルコール13gを徐々に加え氷中で1時間緩やかに攪拌しながらD−セリンを析出させた。晶析液をろ過し、結晶を冷却した40%イソプロピルアルコール13mLで洗浄して乾燥させた。回収率は60%で白色のD−セリン結晶が得られ、原料であるグリシンは検出されなかった。結晶の光学純度は99.8%eeであった。
【0105】
[実施例10]
[10−1](キサントモナスのDTAクローニングとベクター構築)
東京大学 分子細胞生物学研究所より入手可能なキサントモナス・オリゼー(IAM1657)を50mlのLB培地に接種した。30℃で一夜培養した後、集菌し、リゾチーム1mg/mlを含む溶菌液で溶菌した。溶菌液をフェノール処理した後、通常の方法によりエタノール沈殿によりDNAを沈殿させた。生じたDNAの沈殿は、ガラス棒に巻き付けて回収した後、洗浄し、PCRに用いた。
【0106】
PCR用のプライマーには、公知のキサントモナス・オリゼーのDTA遺伝子(GenBanak accession number E05055)に基づいて設計した配列番号13及び14に示す塩基配列を有するオリゴヌクレオチド(北海道システム・サイエンス株式会社に委託して合成した)を用いた。これらのプライマーは、5’末端付近及び3’末端付近に、それぞれKpnI及びHindIIIの制限酵素認識配列を有する。
【0107】
前記微生物の染色体DNA6ng/μl及びプライマー各3μMを含む0.025mlのPCR反応液を用いて、変性:96℃、1分間、アニーリング:55℃、30秒間、伸長反応:68℃、1分15秒間からなる反応サイクルを、35サイクルの条件でPCRを行った。
【0108】
PCR反応産物及びプラスミドpUC18(宝酒造(株))を、KpnI及びHindIIIで消化し、ライゲーション・ハイ(東洋紡(株))を用いて連結した後、得られた組換えプラスミドを用いて、Escherichia coli DH5αを形質転換した。形質転換株を、アンピシリン(Am)50μg/ml及びX−Gal(5−ブロモ−4−クロロ−3−インドリル−β−D−ガラクトシド)を含むLB寒天培地で培養し、Am耐性で且つ白色コロニーとなった形質転換株を得た。このようにして得られた形質転換株よりプラスミドを抽出した。プラスミドに導入されたDNA断片の塩基配列を通常の塩基配列の決定法に従い、公知のキサントモナス・オリゼーのDTAと同じ配列であることを確認した。得られた発現プラスミドをpXDTA1と命名した。
【0109】
[10−2](キサントモナス属のDTA発現Escherichia coliの取得)
pXDTA1を用いてEscherichia coli W3110ΔdsdAを通常の方法で形質転換し、得られた形質転換体をMT−11028と命名した。
また、実施例1で作製したpAcDTA1、pAcDTA2、pAcDTA3を用いてEscherichia coli W3110ΔdsdAを通常の方法で形質転換し、得られた形質転換体をMT−11015W、MT−11016W、MT−11017Wと命名した。
【0110】
[10−3](Jar培養による菌体の取得)
MT−11015W、MT−11016W、MT−11017W、MT−11028の組換えEscherichia coliを500mLのバッフル付き三角フラスコにAm50μg/mlを含むLB培地100mLに接種し、30℃にてOD660が1.0になるまで培養した。
次いで10L容量のBMS10(エイブル社製)で培養を行った。培養の条件は、撹拌:700rpm、温度:30℃、pH(NH3で維持):7.2、通気:1vvm、容量:5L、培養時間:48時間の条件で操作した。培地は、特記しない限り、水1Lに対してポリペプトン(大日本製薬)7g、硫酸第一鉄・7水和物0.09g、硫酸アンモニウム1.5g、硫酸マグネシウム・6水和物2g、リン酸水素1カリウム2g、リン酸水素2カリウム2g、アデカノールLG126(旭電化工業)0.6gの培地組成を用いた。
【0111】
接種前に、20g/Lの濃度となるようにグルコースを添加し、前記のバッフル付き三角フラスコの培養液50mLを接種した。前記の条件で最初のグルコースが完全に枯渇した後、残りの時間を0.1g/L未満となるような可変速度でグルコースを全量で200gを供給した。培養液から遠心分離により菌体を集めて−20℃にて凍結した。
【0112】
[10−4](有機溶媒処理した微生物のL−セリン副生抑制)
[L−セリン副生酵素活性の測定方法]
MT−11028の凍結菌体60g(固形分率およそ10%)に塩化マグネシウム・6水和物を1.0g加え各種有機溶媒を所定濃度となるように加え、35℃で1時間攪拌した。
前記の処理菌体液から乾燥菌体として0.22g分を測りとり、酵素活性測定液9gを加えて35℃で20時間攪拌し、生成したL−セリンと残存するD−セリンの比率を測定した。結果を表2にまとめて示した。
【0113】
(L−セリン生成活性測定液)
D−セリン10.84g、PLP6mgをpH7.0の0.5Mリン酸カリウム緩衝液で100gに溶解した。
【0114】
【表2】
【0115】
[実施例11]
(有機溶媒処理した微生物によるD−セリン合成)
実施例10で得た湿菌体83.4g(固定分率約10%)に塩化マグネシウム・6水和物1.85gと37重量%のホルムアルデヒド1.2gを加え、水を加えてホルムアルデヒド濃度0.5%に調整し、35℃で1時間撹拌した。
【0116】
水280gにグリシン80gと35重量%の塩化マグネシウム6水和物4g、37重量%のホルムアルデヒド3.1gを加え、水酸化ナトリウムでpHを7.5に調整した。
0.38重量%のPLP溶液を3.2g添加し、前記の有機溶媒処理菌体を湿菌体として30gを加えて反応を開始した。反応中にpHが7.3を上回ったらホルムアルデヒドを添加し、pHを7.3に制御した。反応中のホルムアルデヒド濃度は、添加したホルムアルデヒドからHPLCで定量したD−セリンの生成量を差し引くことで求めた。反応液中のホルムアルデヒド濃度は、およそ80mMから100mMに制御されていた。反応終了後のセリンをHPLCにて分析した結果、対Gly収率95モル%で、光学純度は99.9%eeであった。
【0117】
[実施例12]
(高ホルムアルデヒド濃度の反応実施例)
水280gにグリシン80g、35重量%の塩化マグネシウム6水和物を4g加え溶解させた。ホルムアルデヒドを各濃度となるように添加し、水酸化ナトリウムでpHを7.5に調節した。0.38重量%のPLPを3.2g加え、実施例10で得た凍結菌体を30g加えて反応を開始した。反応中のpHが7.3以上となったらホルムアルデヒドを添加し、pHを7.3に制御した。結果を表3にまとめて示した。
【0118】
【表3】
【0119】
[実施例13]
[13−1](glyA遺伝子破壊Escherichia coliの作出とDSA生産菌の作製)
Escherichia coliのゲノムDNAの全塩基配列は公知であり(GenBanak accession number U00096)、Escherichia coliのセリンヒドロキシメチルトランスフェラーゼのアミノ酸配列と遺伝子(以下、glyAと略することがある)の塩基配列もすでに報告されている(GenBank accession number J01620)。Escherichia coli W3110株のゲノムDNAのglyA近傍領域の遺伝子情報に基づいて作製された、配列番号15及び16、配列番号17及び18に示す塩基配列を有するオリゴヌクレオチドを用いてEscherichia coli W3110株(ATCC27325)のゲノムDNAをテンプレートとしてPCRを行った。得られたDNAフラグメントをそれぞれ、制限酵素BamHIとPstI、PstIとHindIIIで消化することにより、それぞれ約850bp、750bpのフラグメントを得た。このDNAフラグメントを温度感受性クローニングベクターpTH18cs1(GenBank accession number AB019610)〔Hashimoto−Gotoh,T.,Gene,241,185−191(2000)〕をBamHI、HindIIIで消化して得られるフラグメントと混合し、リガーゼを用いて結合した後、DH5α株に30℃で形質転換し、クロラムフェニコール10μg/mlを含むLB寒天プレートに生育する形質転換体を得た。得られたコロニーをクロラムフェニコール10μg/mlを含むLB液体培地で30℃で一晩培養し、得られた菌体からプラスミドを回収した。回収したプラスミドをPstIで消化し、T4DNAポリメラーゼで平滑末端処理を行った後、トランスポゾンTn10由来のテトラサイクリン耐性遺伝子と連結した。
【0120】
こうして得られたプラスミドをEscherichia coli W3110dsdA欠失株に30℃で形質転換し、クロラムフェニコール10μg/mlとテトラサイクリン50μg/mlを含むLB寒天プレートに30℃で一晩培養し、形質転換体を得た。得られた形質転換体をテトラサイクリン50μg/mlを含むLB液体培地に接種し、30℃で一晩培養した。次にこれらの培養菌体が得られるようにテトラサイクリン50μg/mlを含むLB寒天プレートに塗布し、42℃で生育するコロニーを得た。得られたコロニーをテトラサイクリン50μg/mlを含むLB液体培地で30℃で一晩培養し、更にテトラサイクリン50μg/mlを含むLB寒天プレートに塗布して42℃で生育するコロニーを得た。
【0121】
出現したコロニーの中から無作為に100コロニーをピックアップしてそれぞれをテトラサイクリン50μg/mlを含むLB寒天プレートとクロラムフェニコール10μg/mlを含むLB寒天プレートに生育させ、テトラサイクリンを含むLB寒天プレートにのみ生育するクロラムフェニコール感受性のクローンを選んだ。更にこれらの目的クローンの染色体DNAからPCRによりglyA近傍領域の約3.6kbpの断片を増幅させ、glyAがテトラサイクリン耐性遺伝子に置換されている株を選抜し、得られた株をW3110dsdA・glyA欠失株と命名した。
【0122】
[13−2](glyA遺伝子破壊Escherichia coliの効果)
このEscherichia coliをプラスミドpXDTA1で形質転換し、実施例10と同じ方法で培養した。但し、フラスコ培養ではグリシンを20mg/L添加し、培養槽での培養では、グリシンを2g/L添加し、培養の途中にグリシンを分割添加したものを用いた。
実施例9と同じ方法でL−セリンの生成活性を調べたところD−セリンの光学純度は97%であった。
【0123】
[実施例14]
(金属塩類の効果)
実施例10で得たMT−11028の凍結菌体100gに300mMのEDTA(pH7.5)3.5gを添加し、4℃にて1時間攪拌した。懸濁液10gを塩化マンガン、硫酸亜鉛、塩化コバルト、塩化ニッケル、塩化カルシウム、塩化第一鉄を各々20mM含むpH7.0の0.5Mリン酸カリウム緩衝液10gに懸濁し、4℃にて1時間攪拌した。
【0124】
次いで前記懸濁液に、ホルムアルデヒドを0.5%となるように加え、35℃で1時間攪拌した。前記の処理菌体液から乾燥菌体として0.22g分を測りとり、実施例10に記載の酵素活性測定液9gを加えて35℃で20時間攪拌し、生成したL−セリンと残存するD−セリンの比率を測定した。
何れの金属塩を用いた場合もD−セリンの光学純度は96%以上であった。また、ホルムアルデヒドとグリシンからD−セリンを合成する酵素活性は何れの金属塩でも有機溶媒処理前の活性に対して50%以上保持していた。
【0125】
[実施例15]
(L−セリンデアミナーゼ発現Escherichia coliによる光学純度を高める方法)
Escherichia coli K−12株を50mlのLB培地に接種した。30℃で一夜培養した後、集菌し、リゾチーム1mg/mlを含む溶菌液で溶菌した。溶菌液をフェノール処理した後、通常の方法によりエタノール沈殿によりDNAを沈殿させた。生じたDNAの沈殿は、ガラス棒に巻き付けて回収した後、洗浄し、PCRに用いた。
【0126】
PCR用のプライマーには、公知のEscherichia coliのL−セリンデアミナーゼ遺伝子(GenBanak accession number M28695)に基づいて設計した配列番号19及び20に示す塩基配列を有するオリゴヌクレオチド(北海道システム・サイエンス株式会社に委託して合成した)を用いた。これらのプライマーは、5’末端付近及び3’末端付近に、それぞれEcoRI及びHindIIIの制限酵素認識配列を有する。
【0127】
前記微生物の染色体DNA6ng/μl及びプライマー各3μMを含む0.025mlのPCR反応液を用いて、変性:96℃、1分間、アニーリング:55℃、30秒間、伸長反応:68℃、1分30秒間からなる反応サイクルを、35サイクルの条件でPCRを行った。
【0128】
PCR反応産物及びプラスミドpUC18(宝酒造(株))を、EcoRI及びHindIIIで消化し、ライゲーション・ハイ(東洋紡(株))を用いて連結した後、得られた組換えプラスミドを用いて、Escherichia coli DH5αを形質転換した。形質転換株を、アンピシリン(Am)50μg/ml及びX−Gal(5−ブロモ−4−クロロ−3−インドリル−β−D−ガラクトシド)を含むLB寒天培地で培養し、Am耐性で且つ白色コロニーとなった形質転換株を得た。このようにして得られた形質転換株よりプラスミドを抽出した。プラスミドに導入されたDNA断片の塩基配列を通常の塩基配列の決定法に従い、公知のEscherichia coliのL−セリンデアミナーゼと同じ配列であることを確認した。得られた発現プラスミドをpSDA1と命名した。
【0129】
pSDA1を用いてEscherichia coli W3110dsdA・glyA欠失株を通常の方法で形質転換し、得られた形質転換体を実施例13と同じ方法で培養槽による培養を行った。
【0130】
実施例12の比較例で行った反応と同じ方法で、反応液に前記の菌体を10g加えて反応を行った。反応液をHPLCで分析したところ、反応液中のD−セリンの光学純度は99.9%であった。
【0131】
本明細書中で引用した全ての刊行物、特許及び特許出願をそのまま参考として本明細書中にとり入れるものとする。
【産業上の利用可能性】
【0132】
本発明は、グリシンとホルムアルデヒドとからD−セリンを製造する方法として有用である。また、本発明の製造方法により得られるD−セリンは結核薬として有用なD−サイクロセリンの原料等の医薬中間体として有用である。
【技術分野】
【0001】
本発明は、ホルムアルデヒドとグリシンからD−セリンを合成する活性を有する新規な酵素をコードするDNA、該DNAをベクターに組み込んでなる組換え体DNA、該組換え体DNAで形質転換した形質転換体、ホルムアルデヒドとグリシンからD−セリンを合成する活性を有する新規な酵素及び該酵素を利用したホルムアルデヒドとグリシンからD−セリンを製造する方法に関する。
【背景技術】
【0002】
D−セリンはD−サイクロセリン等の医薬品の合成中間体として有用な化合物であることが知られている。
【0003】
従来、ホルムアルデヒドとグリシンとからD−セリンを合成する活性を有する酵素としては、アリスロバクター(Arthrobacter)sp.DK−19に由来するD−スレオニンアルドラーゼ(以下、「DTA」と略記する。)が知られているに過ぎない(特許文献1:特開昭58−116690号公報を参照)。
【0004】
このDTAは、ホルムアルデヒド及びグリシンをそれぞれ50mmol使用して30℃で40時間反応させた後のD−セリンの生成量は2.5mmol(ホルムアルデヒドに対する収率は僅かに5%)であったことが報告されている。
【0005】
これに対し、同じアリスロバクター属の微生物である、アリスロバクターsp.DK−38に由来するDTAは、D−スレオニン、D−β−ヒドロキシフェニルセリン、D−β−ヒドロキシ−α−アミノ吉草酸等に反応する広い基質特異性を有しているにもかかわらず、D−セリンには反応しないことが報告されている(非特許文献1:Eur.J.Biochem.,1997,248,p385−393を参照)。
【0006】
また、キサントモナス属(Xanthomonas)由来のDTAを用いることにより、グリシンとアルデヒド化合物からD−β−ヒドロキシアミノ酸類を製造できることが報告されているが、キサントモナス属由来のDTAを用いてホルムアルデヒドとグリシンとからD−セリンが合成できるとの報告はなされていない(特許文献2:特開平5−168484号公報を参照)。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開昭58−116690号公報
【特許文献2】特開平5−168484号公報
【非特許文献】
【0008】
【非特許文献1】Eur.J.Biochem.,1997,248,p385−393
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明は、前記の背景技術に鑑み、ホルムアルデヒドとグリシンからD−セリンを合成する活性を有する新規な酵素をコードするDNA、該DNAをベクターに組み込んでなる組換え体DNA、該組換え体DNAで形質転換した形質転換体、ホルムアルデヒドとグリシンからD−セリンを合成する活性を有する新規な酵素、及び該酵素を利用したホルムアルデヒドとグリシンからD−セリンを製造する方法を提供することを目的とする。
【課題を解決するための手段】
【0010】
本発明者らは、公知のDTAと類似の構造を有する未知の酵素のなかに、ホルムアルデヒドとグリシンとからD−セリンを合成する能力を有する酵素が存在する可能性があると考え、公知のDTAのアミノ酸配列に関する情報に関する調査を行った。その結果、キサントモナス属(GenBank accession No.E05055)、アクロモバクター(Achromobacter)属(GenBank accession No.AB026892)、及びアリスロバクター属(GenBank accession No.AB010956)に由来するDTAのアミノ酸配列に関する情報を見出した。これらのアミノ酸配列を比較検討した結果、DTAのN末端領域及びC末端領域に相当するアミノ酸配列に高い相同性を持つことを見出した。
【0011】
本発明者らは、これらのアミノ酸配列からN末端領域とC末端領域を基にプライマーを設計し、このプライマーを用いて各種の微生物の染色体DNAからPCRによる増幅を試みたところ、アクロモバクター属の微生物からホルムアルデヒドとグリシンとからD−セリンを合成する活性を有する酵素をコードするDNAの増幅に成功した。
【0012】
このDNAに相当するアミノ酸配列と公知のアクロモバクター属に由来するDTAのアミノ酸配列との相同性は約50%程度であり、公知のアクロモバクター属に由来するDTAのアミノ酸配列とはかなり異なるものであった。一方、公知のキサントモナス属に由来するDTAのアミノ酸配列との相同性は約90%であった。
【0013】
本発明者らは次に、前記の新規なDNAをベクターに組み込んでなる組換え体DNAで形質転換された組換えEscherichia coliを用いてホルムアルデヒドとグリシンとを反応させてD−セリンの合成を試みたところ、驚くべきことに従来知られているDTAとは異なり、100mMのグリシンとホルムアルデヒドから反応収率70%以上という高収率でD−セリンを合成できることを見出した。
【0014】
更に、この組換えEscherichia coliを用いてD−セリンの蓄積反応を行うと、僅かながらL−セリンが生成することが認められた。
【0015】
D−セリンを医薬中間体等の高純度が要求される分野に用いる場合、D−セリンへのL−セリンの混入は好ましくない。D−セリンの製造時にL−セリンが副生することは、D−セリンの溶解度よりDL−セリンの溶解度が低いことからD−セリンの精製収率を大幅に低下させる。
【0016】
本発明者らは、この問題を解決するために、更に検討した結果、2価金属イオンの存在下、有機溶媒処理及び/又は加熱処理を施すことでL−セリンの副生を抑制できることを見出した。また、このような処理を施さなくとも反応中のホルムアルデヒド濃度を150mM以上に維持することでL−セリンの副生を抑制できることを見出した。更に、D−セリン合成に利用するDSAを生産する宿主としてL−セリン合成酵素遺伝子が欠失している微生物を利用することでL−セリンの副生を抑制できることを見出した。
【0017】
以上の知見を基に、本発明を完成した。すなわち、本発明は以下のとおりである。
(1)以下の(a)又は(b)のタンパク質をコードするDNA。
(a)配列番号4、6、8の何れかに記載のアミノ酸配列を含むタンパク質
(b)(a)のアミノ酸配列において1もしくは数個のアミノ酸が欠失、置換、挿入もしくは付加されたアミノ酸配列を含み、かつグリシンとホルムアルデヒドからD−セリンを合成する酵素活性を有するタンパク質
(2)以下の(a)又は(b)のDNA。
(a)配列表の配列番号3、5、7の何れかに記載の塩基配列又はそれらの相補配列からなるDNA
(b)配列表の配列番号3、5、7の何れかに記載の塩基配列又はそれらの相補配列中の少なくとも20塩基の連続したDNA配列を含むDNA断片とストリンジェントな条件下でハイブリダイズし、かつグリシンとホルムアルデヒドからD−セリンを合成する活性を有する酵素をコードするDNA
(3)前記(1)又は(2)に記載のDNAをベクターに組み込んでなる組換え体DNA。
【0018】
(4)前記(3)に記載の組換え体DNAを用いて宿主細胞を形質転換して得られた形質転換体。
(5)形質転換される宿主細胞が微生物である前記(4)に記載の形質転換体。
(6)形質転換される微生物がD−セリンデアミナーゼの欠損した微生物である前記(5)に記載の形質転換体。
【0019】
(7)以下の(a)又は(b)のタンパク質。
(a)配列番号4、6、8の何れかに記載のアミノ酸配列を含むタンパク質
(b)(a)のアミノ酸配列において1もしくは数個のアミノ酸が欠失、置換、挿入もしくは付加されたアミノ酸配列を含み、かつグリシンとホルムアルデヒドからD−セリンを合成する酵素活性を有するタンパク質
【0020】
(8)前記(4)から(6)のいずれかに記載の形質転換体を培養し、得られた培養物からグリシンとホルムアルデヒドからD−セリンを合成する酵素活性を有するタンパク質を採取することを特徴とする、グリシンとホルムアルデヒドからD−セリンを合成する活性を有する酵素の製造方法。
(9)前記(4)から(6)のいずれかに記載の形質転換体又はその処理物の存在下でグリシンとホルムアルデヒドを反応させることを含むD−セリンの製造方法。
(10)前記(7)に記載のタンパク質の存在下でグリシンとホルムアルデヒドを反応させることを含むD−セリンの製造方法。
【0021】
(11)グリシンとホルムアルデヒドからD−セリンを合成する活性を有する微生物又はその処理物の存在下で、グリシンとホルムアルデヒドとを反応させてD−セリンを合成するD−セリンの製造方法であって、該反応の反応液中のL−セリンの副生をD−セリンに対して1.5モル%以下に抑制させる以下の(i)〜(iv)の一つ以上の手段を有するD−セリンの製造方法。
(i)グリシンとホルムアルデヒドからD−セリンを合成する活性を有する微生物を有機溶媒処理及び/又は加熱処理する方法、
(ii)反応液中のホルムアルデヒド濃度を、a)前記(i)の方法により有機溶媒処理及び/又は加熱処理を施す場合には2M以下に制御し、b)有機溶媒処理及び/又は加熱処理を施さない場合には、150mM以上2M以下に制御する方法、
(iii)グリシンとホルムアルデヒドからD−セリンを合成する活性を有する酵素を含有し、かつ、L−セリン合成酵素遺伝子が欠失している微生物を触媒として利用する方法、
(iv)L−セリンデアミナーゼ活性を有する酵素を含有する微生物を反応液中に添加する方法。
【0022】
(12)有機溶媒がホルムアルデヒド、ベンズアルデヒド、ジクロロエタン及びイソプロピルアルコールから選ばれる一種以上のものである前記(11)に記載の製造方法。
(13)有機溶媒処理及び/又は加熱処理を2価の金属イオンの存在下で行う前記(11)又は(12)に記載の製造方法。
(14)2価の金属がマグネシウム、マンガン、亜鉛、ニッケル、コバルト及び鉄から選ばれる一種以上の金属である前記(13)に記載の製造方法。
(15)グリシンとホルムアルデヒドからD−セリンを合成する活性を有する微生物が前記(5)又は(6)に記載の形質転換体である前記(11)に記載の製造方法。
【発明の効果】
【0023】
本発明によれば、公知のDTAを用いたD−セリンの製造方法に比べ、グリシンとホルムアルデヒドからD−セリンを収率よく製造できる。
【図面の簡単な説明】
【0024】
【図1】図1は、形質転換体から得た破砕液をSDS−ポリアクリルアミド電気泳動により分析した結果を示す図である。
【図2】図2は、精製されたDSAのSDS−ポリアクリルアミド電気泳動写真である。
【発明を実施するための形態】
【0025】
1.ホルムアルデヒドとグリシンからD−セリンを合成する活性を有する新規な酵素をコードするDNAの遺伝子取得
配列表の配列番号4、6、8の何れかに記載のアミノ酸配列を含み、かつ、グリシンとホルムアルデヒドからD−セリンを合成する活性を有する酵素(以下、グリシンとホルムアルデヒドからD−セリンを合成する活性を有する酵素を「DSA」と略記する。)をコードするDNAは、例えば、12301 Parklawn Drive, Rockville, Maryland 20852, U.S.A.のAmerican Type Culture Collectionより入手可能なアクロモバクター・キシロソキシダンス(Achromobacter xylosoxidans)(ATCC9220)、独立行政法人 製品評価技術基盤機構(National Institute of Technology and Evaluation)の生物遺伝資源部門(NITE Biological Resource Center)(日本国千葉県木更津市かずさ鎌足2−5−8)より入手可能なアクロモバクター・キシロソキシダンス(NBRC13495)及びアクロモバクター・デニトリフィカンス(Achromobacter denitrificans)(Synonym: Achromobacter xylosoxidans subsp. denitrificans)(NBRC15125)から染色体DNAを抽出し、配列表の配列番号1及び2に示した塩基配列を有するプライマーを用いてPCRを実施することにより得ることができる。
【0026】
PCRを実施するに際して、若干のミスマッチを持つ遺伝子も増幅することができるように増幅時の条件を改良することが有効である。例えば、アニール温度を低めに抑える方法、PCR反応液中にジメチルスルホキシドを5%程度添加する方法、あるいはGCリッチな遺伝子を増幅しやすいように工夫されたPCRキット(例えばGC−RICH PCRSystem(Roche社製))を使用する方法、更には前記の方法を組み合わせる方法などにより増幅が困難な遺伝子を増幅できることがある。
【0027】
DSAをコードするDNAの具体的な例としてアクロモバクター・キシロソキシダンス(ATCC9220)由来DTA遺伝子のDNA塩基配列を配列番号3に、該塩基配列より翻訳されるアミノ酸配列を配列番号4に例示できる。アクロモバクター・キシロソキシダンス(NBRC13495)由来DTA遺伝子のDNA塩基配列を配列番号5に、該塩基配列より翻訳されるアミノ酸配列を配列番号6に例示できる。アクロモバクター・デニトリフィカンス(NBRC15125)由来DTA遺伝子のDNA塩基配列を配列番号7に、該塩基配列より翻訳されるアミノ酸配列を配列番号8に例示できる。
【0028】
DSAをコードするDNAとしては前記したものの他に、配列表の配列番号3、5、7の何れかに記載の塩基配列又はそれらの相補配列中の少なくとも20塩基の連続したDNA配列を含むDNA断片とストリンジェントな条件下でハイブリダイズし、かつDSA活性を有するタンパク質をコードするDNAであればどのような生物由来であっても構わない。例えば、その生物中で機能を有していないサイレントなDNAであっても単離したDNAを適当な発現ベクターに連結することでDSAを産生することができればそれでも構わない。
【0029】
更に生物を特定しなくても、土壌などを直接、鋳型DNAとして配列表の配列番号1及び2の塩基配列をもつプライマーを用いたPCRに付すことでDSAをコードするDNAを得ることも可能である。
【0030】
DSAをコードするDNAの具体的な取得方法として、例えばD−セリンを含有する培地で生育する微生物の染色体DNAを鋳型とし、配列表の配列番号1及び2に記載の塩基配列を有するプライマーを用いてPCRを行い増幅したDNAを例示することができる。
【0031】
また、配列表の配列番号3、5、7の何れかに記載の塩基配列もしくは相補配列がストリンジェントな条件下でハイブリダイズし得る範囲内でかつ、コードされる酵素の活性に影響を及ぼさない範囲内で部位特異的変異導入法(Nucleic Acid Res., 10, pp. 6487 (1982); Nucleic Acid Res., 13, pp. 4431 (1985); Methods in Enzymol., 100, pp. 448(1983); Molecular Cloning 2ndEdt., Cold Spring Harbor Laboratory Press(1989); PCR A Practical Approach IRL Press pp. 200 (1991); Current Protocols in Molecular Biology, John Wiley &;Sons (1987-1997); Proc. Natl. Acad. Sci., USA, 79, pp. 6409 (1982); Gene, 34, 315 (1985); Proc. Natl. Acad. Sci., USA, 82, pp.488 (1985))などを用いて、適宜欠失、置換、挿入、及び/又は付加変異を導入することにより、配列番号4、6、8の何れかに記載のアミノ酸配列において1もしくは数個のアミノ酸が欠失、置換、挿入もしくは付加されたアミノ酸配列を含み、かつDSA活性を有するタンパク質をコードするDNAを得ることも可能である。
【0032】
本明細書中の、アミノ酸の欠失、置換、挿入もしくは付加は、前記の出願前周知技術である部位特異的変異導入法により実施することができ、また、1もしくは数個のアミノ酸とは、部位特異的変異導入法により欠失、置換、挿入もしくは付加できる程度の数、例えば1〜5個のアミノ酸、好ましくは1〜3個のアミノ酸を意味する。
【0033】
ハイブリダイゼーションのための条件とは、特定のハイブリダイゼーションシグナルを検出するために当業者が一般的に用いている条件を例示できる。好ましくは、ストリンジェントなハイブリダイゼーション条件とストリンジェントな洗浄条件を意味する。具体的には、例えば、6×SSC(1×SSCの組成:0.15M NaCl、0.015Mクエン酸ナトリウム、pH7.0)、0.5%SDS、5×デンハート及び100mg/mlニシン精子DNAを含む溶液中プローブとともに55℃で一晩保温するという条件等が挙げられる。ついでフィルターを0.2×SSC中42℃で洗浄するなどを例示することができる。ストリンジェントな条件としては、フィルターの洗浄工程における0.1×SSC、50℃の条件であり、更にストリンジェントな条件としては、同工程における0.1×SSC、65℃の条件を挙げることができる。
【0034】
本明細書中で用いる「配列番号3、5、7の何れかに記載の塩基配列又はそれらの相補配列中の少なくとも20塩基の連続したDNA配列を含むDNA断片」とは、配列表の配列番号3、5、7の何れかに記載の又はそれらの相補配列中の任意の少なくとも20塩基、好ましくは少なくとも30塩基、例えば40、60又は100塩基の連続した配列を一つ又は複数選択したDNAの断片のことである。
【0035】
2.該タンパク質の遺伝子を持つ組換えDNAの作製
前記のDSAをコードするDNAをベクターに組込むことにより組換え体DNAを得ることができる。
クローニングする際のベクターとしては、宿主微生物内で自律的に増殖し得るファージ又はプラスミドから遺伝子組換え用として構築されたものが適している。ファージとしては、例えばEscherichia coliを宿主微生物とする場合にはLambda gt10、Lambda gt11などが例示することができる。また、プラスミドとしては、例えばEschenchia coliを宿主微生物とする場合には、pBTrp2、pBTac1、pBTac2(いずれもBoehringer Mannheim社製)、pKK233−2(Pharmacia社製)、pSE280(Invitrogen社製)、pGEMEX−1(Promega社製)、pQE−8(QIAGEN社製)、pQE−30(QIAGEN社製)、pBluescriptII SK+、pBluescriptII SK(−)(Stratagene社製)、pET−3(Novagen社製)、pUC18(宝酒造社製)、pSTV28(宝酒造社製)、pSTV29(宝酒造社製)、pUC118(宝酒造社製)等を例示することができる。
【0036】
プロモーターとしては、宿主細胞中で発現できるものであればいかなるものでもよい。例えば、trpプロモーター(Ptrp)、lacプロモーター(Plac)、PLプロモーター、PHプロモーター、PSEプロモーター等の、Escherichia coliやファージ等に由来するプロモーターを挙げることができる。またtacプロモーター、lacT7プロモーターのように人為的に設計改変されたプロモーター等も用いることができる。更にバチルス属細菌中で発現させるためにNpプロモーター(特公平8−24586号公報)なども用いることができる。
【0037】
リボソーム結合配列としては、宿主細胞中で発現できるものであればいかなるものでもよいが、シャイン−ダルガノ(Shine−Dalgarno)配列と開始コドンとの間を適当な距離(例えば6〜18塩基)に調節したプラスミドを用いることが好ましい。
【0038】
転写・翻訳を効率的に行うため、該タンパク質活性を有するタンパク質のN末端又はその一部を欠失したタンパク質と発現ベクターのコードするタンパク質のN末端部分を融合させたタンパク質を発現させてもよい。
【0039】
目的とするタンパク質の発現には転写終結配列は必ずしも必要ではないが、構造遺伝子直下に転写終結配列を配置することが望ましい。
【0040】
クローニングの際、前記のようなベクターを、上述したDSAをコードするDNAの切断に使用した制限酵素で切断してベクター断片を得ることができるが、必ずしも該DNAの切断に使用した制限酵素と同一の制限酵素を用いる必要はない。該DNA断片とベクターDNA断片とを結合させる方法は、公知のDNAリガーゼを用いる方法であればよく、例えば該DNA断片の付着末端とベクター断片の付着末端とのアニーリングの後、適当なDNAリガーゼの使用により該DNA断片とベクターDNA断片との組換えベクターを作製する。必要に応じて、アニーリングの後、微生物等の宿主に移入して生体内のDNAリガーゼを利用し組換えベクターを作製することもできる。
【0041】
3.DSAをコードするDNAをベクターに組み込んでなる組換え体DNAによる形質転換体の作製
前記の組換え体DNAを宿主細胞に導入することにより、前記の組換え体DNAで形質転換された形質転換体を得ることができる。
宿主細胞としては、組換えベクターが安定であり、かつ自律増殖可能で外来性遺伝子の形質発現できるものであれば特に制限されない。宿主細胞としては、微生物、例えば細菌を挙げることができ、そのようなものとしてEscherichia coli DH5α,XL−1BlueなどのEscherichia coliが例示される。また、宿主細胞としてその他酵母等の微生物や昆虫細胞を利用することも可能である。
【0042】
微生物に組換え体DNAを移入する方法としては、例えば微生物がEscherichia coliの場合には、カルシウム処理によるコンピテントセル法やエレクトロポーレーション法などを用いることができる。
【0043】
宿主細胞として、生成物であるD−セリンの分解を抑制する目的でD−セリンデアミナーゼ活性の低い微生物又はD−セリンデアミナーゼ活性の欠損した微生物を用いることができる。D−セリンデアミナーゼ活性の欠損した微生物の具体的な例として、例えば実施例6に記載のD−セリンデアミナーゼ遺伝子を組換えたEscherichia coli等が挙げられる。
【0044】
また、D−セリン合成反応におけるL−セリンの生成を抑制する目的で、L−セリンデアミナーゼ活性の高い微生物を用いることにより生成したL−セリンを分解することも可能である。L−セリンデアミナーゼ活性の高い微生物として実施例15に記載のL−セリンデアミナーゼ遺伝子を組換えたEscherichia coli等を用いることもできる。
【0045】
微生物として、アラニンラセマーゼ、セリンヒドロキシメチルトランスフェラーゼ、L−スレオニンアルドラーゼ等のL−セリンの合成に関与する酵素活性が低い微生物又はこれらの酵素活性が欠損した微生物を用いることは、L−セリンが生成しないため好適である。
【0046】
更に好ましくは、これらの酵素が全て欠損した微生物を宿主細胞として用いることが望ましい。
【0047】
4.DSAの生産
前記の形質転換体を培養し、次に得られたDSAを採取することにより、DSAを製造することができる。
形質転換体の培養は、宿主細胞の培養に用いられる通常の方法に従って行うことができる。形質転換体がEscherichia coli等の原核微生物、酵母菌等の真核微生物である場合、これら微生物を培養する培地は、該微生物が資化し得る炭素源、窒素源、無機塩類等を含有し、形質転換体の培養を効率的に行える培地であれば天然培地、合成培地のいずれでもよい。
【0048】
炭素源としては、形質転換体が資化し得るものであればよく、グルコース、フラクトース、スクロース、これらを含有する糖蜜、デンプンあるいはデンプン加水分解物等の炭水化物、酢酸、プロピオン酸等の有機酸、エタノール、プロパノールなどのアルコール類が用いられる。
【0049】
窒素源としては、アンモニア、塩化アンモニウム、硫酸アンモニウム、酢酸アンモニウム、リン酸アンモニウム等の各種無機酸や有機酸のアンモニウム塩、その他含窒素化合物、並びに、ペプトン、肉エキス、酵母エキス、コーンスチープリカー、カゼイン加水分解物、大豆粕、大豆粕加水分解物、各種発酵菌体及びその消化物等が用いられる。
【0050】
無機塩としては、リン酸第一カリウム、リン酸第二カリウム、リン酸マグネシウム、硫酸マグネシウム、塩化ナトリウム、硫酸第一鉄、硫酸マンガン、硫酸銅、炭酸カルシウム等が用いられる。
【0051】
培養は、振盪培養又は深部通気攪拌培養などの好気的条件下で行う。培養温度は15〜50℃がよく、培養時間は、通常16時間〜5日間である。培養中pHは、3.0〜9.0に保持する。pHの調整は、無機あるいは有機の酸、アルカリ溶液、尿素、炭酸カルシウム、アンモニアなどを用いて行う。また培養中必要に応じて、アンピシリンやテトラサイクリン等の抗生物質を培地に添加してもよい。
【0052】
プロモーターとして誘導性のプロモーターを用いた発現ベクターで形質転換した微生物を培養するときには、必要に応じてインデューサーを培地に添加してもよい。例えば、lacプロモーターを用いた発現ベクターで形質転換した微生物を培養するときにはイソプロピル−β−D−チオガラクトピラノシド(IPTG)等を、trpプロモーターを用いた発現ベクターで形質転換した微生物を培養するときにはインドール酢酸(IAA)等をそれぞれ培地に添加してもよい。
【0053】
形質転換体は、これを含む培養液を遠心分離、ろ過等の手段を用いて分離し回収することができる。
【0054】
形質転換体処理物は、細胞の破壊を目的として、形質転換体を機械的破壊、超音波処理、凍結融解処理、乾燥処理、加圧又は減圧処理、浸透圧処理、自己消化、界面活性剤処理、酵素処理したもの、及びこれらの処理により得られるDSAを含む画分の固定化物、形質転換体の固定化物として得ることができる。
【0055】
なお、本発明において微生物の処理物も前記形質転換体処理物と同様の処理をしたものを指す。
【0056】
このような形質転換体又は形質転換体処理物からDSAを精製するためには、形質転換細胞を破砕し細胞破砕液をイオン交換樹脂やゲルろ過等のクロマトグラフィーによる分画や硫酸アンモニウムなどによる塩析などを組み合わせることで精製酵素を得ることができる。
【0057】
5.D−セリンの製造方法
前記の形質転換体又はその形質転換体処理物の存在下でグリシンとホルムアルデヒドを反応させることによりD−セリンを製造することができる。
D−セリンの製造は、pH6.0〜9.0、温度20〜60℃で振盪、もしくは攪拌条件下で行うのが好ましい。
【0058】
形質転換体又はその形質転換体処理物の使用量は、ホルムアルデヒドとグリシンとの反応が十分に進行すれば特に制限されないが、通常グリシン1gに対して少なくても10unit、好ましくは50unit以上のDSA活性となる量を添加することが望ましい。形質転換体又はその形質転換体処理物の添加方法は反応開始時に一括で添加しても構わないし、反応中に分割して又は連続して添加しても構わない。
【0059】
DSA活性は、1μモルのD−セリンを1分間に合成できる能力を1unitと定義する。
DSA活性は、100mMグリシン、0.1mMピリドキサールリン酸、10mM塩化マグネシウム、5mMホルムアルデヒドを含むpH8.0の200mMのトリス塩酸緩衝液中に酵素液を加えて30℃で保温し、D−セリンの生成量を測定して算出する。
【0060】
反応液中のグリシンの濃度は、100mM以上であり、好ましくは1M以上5M以下である。グリシンの添加方法については反応開始時に一括添加しても、反応の進行に伴い分割して又は連続して添加してもよい。
【0061】
ホルムアルデヒドは、その気体を反応液中に供給することができるが、水溶液又はアルコール溶液として供給することもできる。また、ホルムアルデヒドの供給源としてパラホルムアルデヒドを使用することもできるが、37%程度のホルムアルデヒドの水溶液の使用が好適である。
【0062】
ホルムアルデヒドの添加方法としては、一括添加又は反応の進行に伴い分割又は連続的に添加する方法で行えるが、反応液中のホルムアルデヒド濃度はDSA活性を阻害しない程度の濃度に制御することが好ましい。DSA活性を阻害しない程度の濃度とは通常5M以下、好ましくは2M以下、更に好ましくは500mM以下、特に好ましくは300mM以下である。
【0063】
反応液中のホルムアルデヒド濃度を制御する方法として(1)反応液に一定速度で添加する方法、(2)ホルムアルデヒド濃度を定量し、酵素活性が失活しない程度の濃度になってから分割添加する方法、(3)パラホルムアルデヒドを添加し、パラホルムアルデヒドがホルムアルデヒドに遊離する速度以上の酵素量を反応系に添加しておくことで実質的にホルムアルデヒドによる阻害を回避する方法、(4)反応の進行に伴い反応液中に生成物のD−セリンが蓄積し、原料のグリシンが減少する。D−セリンとグリシンの等電点がそれぞれ、5.68と5.97であるため、反応の進行に伴い反応液のpHの低下が起こる。反応液のpHの低下を補正する量以上のアルカリを反応液に添加し、そのpH上昇分を補正できる量のホルムアルデヒドを添加するという方法でホルムアルデヒドを添加することも可能である。
【0064】
反応液中のDSA量を予め設定したアルカリの添加速度、即ちD−セリンの合成速度以上にしておけば、ホルムアルデヒドの濃度を測定するなどの煩雑な操作を用いずとも反応液中のホルムアルデヒド濃度を制御することが可能となる。
【0065】
反応液に添加するアルカリとしては、水酸化リチウム、水酸化ナトリウム、水酸化カリウムなどのアルカリ金属水酸化物の他、水酸化アンモニウム、水酸化カルシウム、リン酸二カリウム、リン酸二ナトリウム、ピロリン酸カリウム、アンモニアなど水に溶解して、液性を塩基性とするものであればよい。
【0066】
反応液の媒体としては、水もしくは水性媒体、有機溶媒又は水もしくは水性媒体と有機溶媒の混合液が用いられる。水性媒体としては、例えばリン酸緩衝液、HEPES(N−2−ヒドロキシエチルピペラジン−N−エタンスルホン酸)緩衝液、トリス[トリス(ヒドロキシメチル)アミノメタン]塩酸緩衝液等の緩衝液が用いられる。有機溶媒としては反応を阻害しないものであればいずれでもよく、例えばアセトン、酢酸エチル、ジメチルスルホキシド、キシレン、メタノール、エタノール、ブタノール等が用いられる。
【0067】
上述の反応において、2価の金属イオンを有する化合物、2−メルカプトエタノール、ジチオスレイトール、亜硫酸水素ナトリウムなどの還元剤や補酵素であるピリドキサールリン酸、アンモニウム塩等を添加することにより反応収率が向上することがある。
【0068】
2価の金属イオンを有する化合物としては、例えば塩化マグネシウム、塩化マンガン、酢酸コバルト、硫酸第1鉄、塩化カルシウムなどが挙げられる。
これら化合物の反応液中の濃度としては通常0.1mMから100mM、好ましくは0.1mMから10mMである。
【0069】
6.反応液中のD−セリンの光学純度を高める方法
前記の形質転換体又はその形質転換体処理物を加熱処理して得られる処理物、又は前記の形質転換体又はその形質転換体処理物を有機溶媒で処理して得られる処理物の存在下で、グリシンとホルムアルデヒドを反応させることにより、L−セリンの副生を抑制することができる。
【0070】
加熱処理の条件としては加熱により大幅にDSA活性を低減させず、L−セリンを生成する活性を低減又は消失させることが可能であればどのような条件でも構わない。具体的な例として、例えばpH6.0から9.0、温度40℃から70℃で10分間から6時間程度攪拌する方法を挙げることができる。
また、有機溶媒処理と加熱処理を組み合わせて行うことも可能である。
【0071】
有機溶媒処理の条件としては大幅にDSA活性を低減させず、L−セリンを生成する活性を低減又は消失させることが可能であればどのような条件でも構わない。有機溶媒処理の条件としては、有機溶媒の濃度は通常20mM以上2M以下、好ましくは20mM以上1M以下、更に好ましくは50mMから1000mM、特に好ましくは50mMから300mM程度である。有機溶媒はDSA活性を大幅に低減させないものであれば制限はないが、ホルムアルデヒド、アセトアルデヒド、ベンズアルデヒド等のアルデヒド類、メタノール、エタノール、イソプロピルアルコールなどのアルコール類、アセトンなどのケトン類、ジクロロエタンなどのハロゲン化炭化水素類などの有機溶媒を用いることが好ましい。中でもホルムアルデヒドは酵素反応の基質であるため、最も好適である。D−セリンを添加することで該酵素反応によりホルムアルデヒドが生成するため、ホルムアルデヒドの変わりにD−セリンを添加する方法でも構わない。D−セリンの添加濃度としては100mMから5M程度が望ましい。
【0072】
有機溶媒処理の温度は10℃から50℃の範囲であり、pHは6.0から9.0の範囲が望ましい。有機溶媒処理中は、処理液のpHや有機溶媒濃度が均一となるように攪拌することが望ましい。
【0073】
また、D−セリン製造の反応開始時にホルムアルデヒド濃度を高めることでL−セリンの生成活性を低減又は失活させることも可能である。この場合、反応中のホルムアルデヒド濃度を0.1%から5%程度で30分間から3時間程度保った後、通常の反応を行えばよい。
【0074】
上述の処理を行う際に、2価の金属イオンを有する化合物、2−メルカプトエタノール、ジチオスレイトール、亜硫酸水素ナトリウムなどの還元剤や補酵素であるピリドキサールリン酸、アンモニウム塩等を添加することにより酵素活性が更に安定化することがある。
【0075】
2価の金属イオンを有する化合物としては、例えば、塩化マグネシウム、塩化マンガン、酢酸コバルト、硫酸第1鉄、塩化カルシウムなどが挙げられる。
【0076】
これら化合物の処理液中の濃度としては通常0.1mMから100mM、好ましくは0.1mMから10mMである。
【0077】
D−セリン合成反応終了後の反応液にL−セリンセアミナーゼ生産菌を加え、生成したL−セリンを分解させることにより最終製品であるD−セリンの光学純度を高めることもできる。L−セリンデアミナーゼ生産菌としてはD−セリンデアミナーゼを欠損した微生物を用いることが望ましい。
【0078】
7.D−セリンの採取方法
反応液からのD−セリンの採取は、通常の有機合成化学で用いられる方法、例えば、有機溶媒による抽出、結晶化、薄層クロマトグラフィー、高速液体クロマトグラフィー等により行うことができる。
本明細書は、本願の優先権の基礎である特願2004−298344の明細書及び/又は図面に記載された内容を包含する。
【実施例】
【0079】
以下に本発明の実施例を示すが、本発明はこれらの実施例に限定されるものではない。なお、D−セリン、L−セリン及びグリシンは高速液体クロマトグラフィーにより定量した。これらの分析条件及び、酵素(DSA及びDTA)活性の測定方法は次のとおりである。
【0080】
(1)D−セリン及びL−セリンの分析条件
カラム;TSK−GEL ENANTIO L1 4.6x250(東ソー株式会社)
カラム温度;45℃
ポンプ流速;0.8ml/min.
検出;UV254nm、溶離液;0.25mM硫酸銅:メタノール=9:1(V/V)
(2)セリン及びグリシンの分析条件
カラム;Shodex RSpak NN−814 8x250(昭和電工株式会社)
カラム温度;40℃
溶離液;10mM リン酸カリウム(pH3.0)
ポンプ流速;0.8ml/min
検出はオルトフタルアルデヒド(OPA)を用いたポストカラム誘導体化法〔J.Chromatogr.,83,353−355(1973)〕を用いた。
(3)酵素活性の測定方法
100mMグリシン、0.1mMピリドキサールリン酸、10mM塩化マグネシウム、5mMホルムアルデヒドを含むpH8.0の200mMのトリス塩酸緩衝液0.9mLに菌体懸濁液を超音波破砕した酵素液を適当に希釈して0.1mLを加え、30℃で15分間反応した。
生成したD−セリンをHPLCにて分析し活性を測定した。活性の単位は1分間に1μmolのD−セリンを生成する活性を1unitとした。
【0081】
[実施例1]
(DSAをコードする遺伝子の取得)
アクロモバクター・キシロソキシダンス(ATCC9220)、アクロモバクター・デニトリフィカンス(NBRC15125)、アクロモバクター・キシロソキシダンス(NBRC13495)を50mlのLB培地に接種した。30℃で一夜培養した後、集菌し、リゾチーム1mg/mlを含む溶菌液で溶菌した。溶菌液をフェノール処理した後、通常の方法によりエタノール沈殿によりDNAを沈殿させた。生じたDNAの沈殿は、ガラス棒に巻き付けて回収した後、洗浄し、PCRに用いた。
【0082】
PCR用のプライマーには、公知のDTA遺伝子に基づいて設計した配列番号1及び2に示す塩基配列を有するオリゴヌクレオチド(北海道システム・サイエンス株式会社に委託して合成した)を用いた。これらのプライマーは、5’末端付近及び3’末端付近に、それぞれKpnI及びHindIIIの制限酵素認識配列を有する。
【0083】
前記微生物の染色体DNA6ng/μl及びプライマー各3μMを含む0.025mlのPCR反応液を用いて、変性:96℃、1分間、アニーリング:55℃、30秒間、伸長反応:68℃、1分15秒間からなる反応サイクルを、35サイクルの条件でPCRを行った。
【0084】
PCR反応産物及びプラスミドpUC18(宝酒造(株))を、KpnI及びHindIIIで消化し、ライゲーション・ハイ(東洋紡(株))を用いて連結した後、得られた組換えプラスミドを用いて、Escherichia coli DH5αを形質転換した。形質転換株を、アンピシリン(Am)50μg/ml及びX−Gal(5−ブロモ−4−クロロ−3−インドリル−β−D−ガラクトシド)を含むLB寒天培地で培養し、Am耐性で且つ白色コロニーとなった形質転換株を得た。このようにして得られた形質転換株よりプラスミドを抽出した。プラスミドに導入されたDNA断片の塩基配列を通常の塩基配列の決定法に従い塩基配列を確認した。
【0085】
得られたDSAをコードするDNAから予想されるアミノ酸配列の分子量は何れもほぼ40kDaであった。
【0086】
得られたアクロモバクター・キシロソキシダンス(ATCC9220)由来のDSAをコードするDNAを持つプラスミドをpAcDTA1と命名した。
【0087】
アクロモバクター・キシロソキシダンス(NBRC13495)由来のDSAをコードするDNAを持つプラスミドをpAcDTA2と命名した。アクロモバクター・デニトリフィカンス(NBRC15125)由来のDSAをコードするDNAを持つプラスミドをpAcDTA3と命名した。
【0088】
(形質転換体の作製)
pAcDTA1、pAcDTA2、pAcDTA3を用いてEscherichia coli DH5αを通常の方法で形質転換し、得られた形質転換体をMT−11015、MT−11016、MT−11017と命名した。
【0089】
各々の組換え微生物を500mLのバッフル付き三角フラスコにAm50μg/mlを含むLB培地100mLに接種し、30℃にてOD660が0.6になるまで培養し、IPTG(イソプロピル−β−チオガラクトピラノシド)が1mMとなるように添加し、更に16時間振盪培養した。培養液を13000rpmで10分間遠心分離し、得られた菌体を5mLの1mM塩化マグネシウムを含む100mMトリス塩酸緩衝液(pH8.0)に懸濁し、−20℃にて凍結保存した。
【0090】
[実施例2]
(DSAの製造法)
実施例1で作製した形質転換体の懸濁液0.5mLをバイオラピュター(オリンパス社製)を用いて、氷水中で5分間破砕した。形質転換体の破砕液を遠心分離し、破砕液を調製した。破砕液をSDS−ポリアクリルアミド電気泳動により分析した結果を図1に示した。
沈殿物に100mMトリス塩酸緩衝液(pH8.0)を0.5mL加えて細胞残渣として同様に分析した。
【0091】
何れの形質転換体も可溶性画分におよそ40kDaの位置に発現するタンパク質が見出され、不溶性画分には見出されなかった。この分子量はそれぞれの遺伝子から推測されるアミノ酸配列の分子量とほぼ一致した。
【0092】
[実施例3]
(DSAの精製と精製DSAによるD−セリンの合成)
実施例2と同様の方法で作製したMT−11015の破砕液10mLを10000rpmで20分間遠心分離し、細胞残渣を除いた酵素液を調製した。酵素液を陰イオン交換樹脂(HiTrap Q−XL、アマシャム社製)に吸着させ、10mM塩化マグネシウム及び50mM塩化ナトリウムを含む100mMトリス塩酸緩衝液(pH8.0)から10mM塩化マグネシウム及び500mM塩化ナトリウムを含む100mMトリス塩酸緩衝液(pH8.0)へのリニアーグラジエントで溶出させた。活性画分を疎水クロマト樹脂(HiTrap Phenyl FF、アマシャム社製)に吸着させ、硫酸アンモニウムで飽和させた10mM塩化マグネシウムを含む100mMトリス塩酸緩衝液(pH8.0)から10mMの塩化マグネシウムを含む100mMトリス塩酸緩衝液(pH8.0)へのリニア−グラジエントで溶出させた。なお、以上の操作は、約10℃で操作した。
【0093】
超音波破砕液、イオン交換クロマトグラフィー処理した後の活性画分及び疎水クロマトグラフィー処理した後の活性画分をSDS−ポリアクリルアミド電気泳動により分析した結果を図2に示した。精製DSAのモノマーの分子量は40000±5000であった。
【0094】
100mMホルムアルデヒド、100mMグリシン、0.1mM PLP、10mM塩化マグネシウム、200mMトリス塩酸緩衝液(pH8.0)100mLよりなる基質溶液に精製DSA酵素液150unitを加え30℃で20時間反応させた。
D−セリンの反応収率は95%であった。
【0095】
[実施例4]
(D−セリン合成能力とD−スレオニン合成能力の比較)
アルデヒド源として、100mMのホルムアルデヒド又はアセトアルデヒドを含み、100mMグリシン、0.1mM PLP、10mM塩化マグネシウム、200mMトリス塩酸緩衝液(pH8.0)100mLよりなる基質溶液に実施例1で作製したMT−11015株の超音波破砕酵素液150unitを加え30℃で20時間反応させた。
アルデヒド源としてホルムアルデヒドは用いたときは、収率90%であったがアセトアルデヒドを用いたときは10%であった。
【0096】
[実施例5]
(ホルムアルデヒド100mM濃度でのD−セリン合成反応)
ホルムアルデヒド100mM、グリシン100mM、0.1mM PLP、10mM塩化マグネシウムを含むpH8.0の200mMトリス塩酸緩衝液100mLよりなる基質溶液に実施例1で作製した組換え微生物の超音波破砕酵素液150unitを加え30℃で20時間反応させた。結果を表1に示した。
【0097】
【表1】
【0098】
[実施例6]
(D−セリンデアミナーゼ欠損Escherichia coliの作製)
Escherichia coliのゲノムDNAの全塩基配列は公知であり(GenBanak accession number U00096)、Escherichia coliのD−セリンデアミナーゼのアミノ酸配列と遺伝子(以下、dsdAと略することがある)の塩基配列もすでに報告されている(GenBank accession number J01603)。Escherichia coli W3110株のゲノムDNAのdsdA近傍領域の遺伝子情報に基づいて作製された、配列番号9及び10、配列番号11及び12に示す塩基配列を有するオリゴヌクレオチドを用いてEscherichia coli W3110株(ATCC27325)のゲノムDNAをテンプレートとしてPCRを行った。得られたDNAフラグメントをそれぞれ、制限酵素PstIとXbaI、XbaIとKpnIで消化することにより、それぞれ約900bp、800bpのフラグメントを得た。このDNAフラグメントを温度感受性クローニングベクターpTH18cs1(GenBank accession number AB019610)〔Hashimoto−Gotoh, T., Gene, 241, 185−191 (2000)〕をPstI、KpnIで消化して得られるフラグメントと混合し、リガーゼを用いて結合した後、DH5α株に30℃で形質転換し、クロラムフェニコール10μg/mlを含むLB寒天プレートに生育する形質転換体を得た。得られたコロニーをクロラムフェニコール10μg/mlを含むLB液体培地で30℃で一晩培養し、得られた菌体からプラスミドを回収した。回収したプラスミドをXbaIで消化し、T4DNAポリメラーゼで平滑末端処理を行った後、pUC4Kプラスミド(Pharmacia)由来のカナマイシン耐性遺伝子と連結した。
【0099】
こうして得られたプラスミドをEscherichia coli W3110株(ATCC27325)に30℃で形質転換し、クロラムフェニコール10μg/mlとカナマイシン50μg/mlを含むLB寒天プレートに30℃で一晩培養し、形質転換体を得た。得られた形質転換体をカナマイシン50μg/mlを含むLB液体培地に接種し、30℃で一晩培養した。次にこれらの培養菌体が得られるようにカナマイシン50μg/mlを含むLB寒天プレートに塗布し、42℃で生育するコロニーを得た。得られたコロニーをカナマイシン50μg/mlを含むLB液体培地で30℃で一晩培養し、更にカナマイシン50μg/mlを含むLB寒天プレートに塗布して42℃で生育するコロニーを得た。
【0100】
出現したコロニーの中から無作為に100コロニーをピックアップしてそれぞれをカナマイシン50μg/mlを含むLB寒天プレートとクロラムフェニコール10μg/mlを含むLB寒天プレートに生育させ、カナマイシンを含むLB寒天プレートにのみ生育するクロラムフェニコール感受性のクローンを選んだ。更にこれらの目的クローンの染色体DNAからPCRによりdsdA近傍領域の約3.0kb断片を増幅させ、dsdAがカナマイシン耐性遺伝子に置換されている株を選抜し、得られた株をW3110dsdA欠失株(以下ΔdsdAと略することがある)と命名した。実施例1で作製したプラスミドを用いて形質転換し、前記と同様に凍結保存した菌体を作製し、同様の反応を行ったところD−セリンの分解は殆ど確認できなかった。
【0101】
[実施例7]
(形質転換体MT−11016を使用したD−セリンの製造:Mg無添加による製造)
7.5gのグリシンと9.4gの0.026重量%のピリドキサールリン酸に蒸留水53.1gを加え、水酸化ナトリウムにてpHを8.0に調整した。実施例1で得たMT−11016の菌体懸濁液(活性として1500unit相当)を加えた。反応温度30℃にて20重量%のホルムアルデヒド20.8gを反応液中のホルムアルデヒド濃度をAHMT法(衛生化学(1976)、Vol22、p39)にて定量し、50mMから300mMとなるように添加した。反応液のpHは水酸化ナトリウムにてpH8.0に調整した。72時間後の反応収率は85%に達していた。
【0102】
[実施例8]
(形質転換体MT−11017を使用したD−セリンの製造:Mg無添加による製造)
7.5gのグリシンと9.4gの0.026重量%のピリドキサールリン酸に蒸留水53.1gを加え、水酸化ナトリウムにてpHを8.0に調整した。実施例1で得たMT−11017菌体懸濁液(活性として1500unit相当)を加えた。反応温度30℃にて20重量%のホルムアルデヒド20.8gを0.8g/15分の速度で15分間添加し、ホルムアルデヒドの添加を45分間停止するというサイクルを繰り返して反応液に添加した。反応液のpHは反応中水酸化ナトリウムにてpH8.0に調整した。24時間後のD−セリンの反応収率は95%に達していた。
一方、ホルムアルデヒドを一括で添加して反応させたところ反応収率は20%であった。
【0103】
[実施例9]
(ホルムアルデヒド処理した形質転換体によるD−セリンの製造:Mg無添加による製造)
実施例1で作製したMT−11015の凍結菌体の溶解液にホルムアルデヒドを100mMとなるように添加し、30℃で1時間緩やかに攪拌した。攪拌中はpHを8.0に水酸化ナトリウムにて調整した。該菌体験濁液を用いて実施例8と同じ反応を行った。ホルムアルデヒド処理を行わなかった場合はL−セリンが2モル%生成したが、ホルムアルデヒド処理を行った場合はL−セリンを検出できなかった。
【0104】
前記の反応液に6N−塩酸を加えpHを4.1に調整した。活性炭(50%含水率)0.97gを加え、60℃で1時間攪拌し、ろ過により活性炭と菌体成分を除去した。次いで、ろ液を30gまで濃縮し、イソプロピルアルコール13gを徐々に加え氷中で1時間緩やかに攪拌しながらD−セリンを析出させた。晶析液をろ過し、結晶を冷却した40%イソプロピルアルコール13mLで洗浄して乾燥させた。回収率は60%で白色のD−セリン結晶が得られ、原料であるグリシンは検出されなかった。結晶の光学純度は99.8%eeであった。
【0105】
[実施例10]
[10−1](キサントモナスのDTAクローニングとベクター構築)
東京大学 分子細胞生物学研究所より入手可能なキサントモナス・オリゼー(IAM1657)を50mlのLB培地に接種した。30℃で一夜培養した後、集菌し、リゾチーム1mg/mlを含む溶菌液で溶菌した。溶菌液をフェノール処理した後、通常の方法によりエタノール沈殿によりDNAを沈殿させた。生じたDNAの沈殿は、ガラス棒に巻き付けて回収した後、洗浄し、PCRに用いた。
【0106】
PCR用のプライマーには、公知のキサントモナス・オリゼーのDTA遺伝子(GenBanak accession number E05055)に基づいて設計した配列番号13及び14に示す塩基配列を有するオリゴヌクレオチド(北海道システム・サイエンス株式会社に委託して合成した)を用いた。これらのプライマーは、5’末端付近及び3’末端付近に、それぞれKpnI及びHindIIIの制限酵素認識配列を有する。
【0107】
前記微生物の染色体DNA6ng/μl及びプライマー各3μMを含む0.025mlのPCR反応液を用いて、変性:96℃、1分間、アニーリング:55℃、30秒間、伸長反応:68℃、1分15秒間からなる反応サイクルを、35サイクルの条件でPCRを行った。
【0108】
PCR反応産物及びプラスミドpUC18(宝酒造(株))を、KpnI及びHindIIIで消化し、ライゲーション・ハイ(東洋紡(株))を用いて連結した後、得られた組換えプラスミドを用いて、Escherichia coli DH5αを形質転換した。形質転換株を、アンピシリン(Am)50μg/ml及びX−Gal(5−ブロモ−4−クロロ−3−インドリル−β−D−ガラクトシド)を含むLB寒天培地で培養し、Am耐性で且つ白色コロニーとなった形質転換株を得た。このようにして得られた形質転換株よりプラスミドを抽出した。プラスミドに導入されたDNA断片の塩基配列を通常の塩基配列の決定法に従い、公知のキサントモナス・オリゼーのDTAと同じ配列であることを確認した。得られた発現プラスミドをpXDTA1と命名した。
【0109】
[10−2](キサントモナス属のDTA発現Escherichia coliの取得)
pXDTA1を用いてEscherichia coli W3110ΔdsdAを通常の方法で形質転換し、得られた形質転換体をMT−11028と命名した。
また、実施例1で作製したpAcDTA1、pAcDTA2、pAcDTA3を用いてEscherichia coli W3110ΔdsdAを通常の方法で形質転換し、得られた形質転換体をMT−11015W、MT−11016W、MT−11017Wと命名した。
【0110】
[10−3](Jar培養による菌体の取得)
MT−11015W、MT−11016W、MT−11017W、MT−11028の組換えEscherichia coliを500mLのバッフル付き三角フラスコにAm50μg/mlを含むLB培地100mLに接種し、30℃にてOD660が1.0になるまで培養した。
次いで10L容量のBMS10(エイブル社製)で培養を行った。培養の条件は、撹拌:700rpm、温度:30℃、pH(NH3で維持):7.2、通気:1vvm、容量:5L、培養時間:48時間の条件で操作した。培地は、特記しない限り、水1Lに対してポリペプトン(大日本製薬)7g、硫酸第一鉄・7水和物0.09g、硫酸アンモニウム1.5g、硫酸マグネシウム・6水和物2g、リン酸水素1カリウム2g、リン酸水素2カリウム2g、アデカノールLG126(旭電化工業)0.6gの培地組成を用いた。
【0111】
接種前に、20g/Lの濃度となるようにグルコースを添加し、前記のバッフル付き三角フラスコの培養液50mLを接種した。前記の条件で最初のグルコースが完全に枯渇した後、残りの時間を0.1g/L未満となるような可変速度でグルコースを全量で200gを供給した。培養液から遠心分離により菌体を集めて−20℃にて凍結した。
【0112】
[10−4](有機溶媒処理した微生物のL−セリン副生抑制)
[L−セリン副生酵素活性の測定方法]
MT−11028の凍結菌体60g(固形分率およそ10%)に塩化マグネシウム・6水和物を1.0g加え各種有機溶媒を所定濃度となるように加え、35℃で1時間攪拌した。
前記の処理菌体液から乾燥菌体として0.22g分を測りとり、酵素活性測定液9gを加えて35℃で20時間攪拌し、生成したL−セリンと残存するD−セリンの比率を測定した。結果を表2にまとめて示した。
【0113】
(L−セリン生成活性測定液)
D−セリン10.84g、PLP6mgをpH7.0の0.5Mリン酸カリウム緩衝液で100gに溶解した。
【0114】
【表2】
【0115】
[実施例11]
(有機溶媒処理した微生物によるD−セリン合成)
実施例10で得た湿菌体83.4g(固定分率約10%)に塩化マグネシウム・6水和物1.85gと37重量%のホルムアルデヒド1.2gを加え、水を加えてホルムアルデヒド濃度0.5%に調整し、35℃で1時間撹拌した。
【0116】
水280gにグリシン80gと35重量%の塩化マグネシウム6水和物4g、37重量%のホルムアルデヒド3.1gを加え、水酸化ナトリウムでpHを7.5に調整した。
0.38重量%のPLP溶液を3.2g添加し、前記の有機溶媒処理菌体を湿菌体として30gを加えて反応を開始した。反応中にpHが7.3を上回ったらホルムアルデヒドを添加し、pHを7.3に制御した。反応中のホルムアルデヒド濃度は、添加したホルムアルデヒドからHPLCで定量したD−セリンの生成量を差し引くことで求めた。反応液中のホルムアルデヒド濃度は、およそ80mMから100mMに制御されていた。反応終了後のセリンをHPLCにて分析した結果、対Gly収率95モル%で、光学純度は99.9%eeであった。
【0117】
[実施例12]
(高ホルムアルデヒド濃度の反応実施例)
水280gにグリシン80g、35重量%の塩化マグネシウム6水和物を4g加え溶解させた。ホルムアルデヒドを各濃度となるように添加し、水酸化ナトリウムでpHを7.5に調節した。0.38重量%のPLPを3.2g加え、実施例10で得た凍結菌体を30g加えて反応を開始した。反応中のpHが7.3以上となったらホルムアルデヒドを添加し、pHを7.3に制御した。結果を表3にまとめて示した。
【0118】
【表3】
【0119】
[実施例13]
[13−1](glyA遺伝子破壊Escherichia coliの作出とDSA生産菌の作製)
Escherichia coliのゲノムDNAの全塩基配列は公知であり(GenBanak accession number U00096)、Escherichia coliのセリンヒドロキシメチルトランスフェラーゼのアミノ酸配列と遺伝子(以下、glyAと略することがある)の塩基配列もすでに報告されている(GenBank accession number J01620)。Escherichia coli W3110株のゲノムDNAのglyA近傍領域の遺伝子情報に基づいて作製された、配列番号15及び16、配列番号17及び18に示す塩基配列を有するオリゴヌクレオチドを用いてEscherichia coli W3110株(ATCC27325)のゲノムDNAをテンプレートとしてPCRを行った。得られたDNAフラグメントをそれぞれ、制限酵素BamHIとPstI、PstIとHindIIIで消化することにより、それぞれ約850bp、750bpのフラグメントを得た。このDNAフラグメントを温度感受性クローニングベクターpTH18cs1(GenBank accession number AB019610)〔Hashimoto−Gotoh,T.,Gene,241,185−191(2000)〕をBamHI、HindIIIで消化して得られるフラグメントと混合し、リガーゼを用いて結合した後、DH5α株に30℃で形質転換し、クロラムフェニコール10μg/mlを含むLB寒天プレートに生育する形質転換体を得た。得られたコロニーをクロラムフェニコール10μg/mlを含むLB液体培地で30℃で一晩培養し、得られた菌体からプラスミドを回収した。回収したプラスミドをPstIで消化し、T4DNAポリメラーゼで平滑末端処理を行った後、トランスポゾンTn10由来のテトラサイクリン耐性遺伝子と連結した。
【0120】
こうして得られたプラスミドをEscherichia coli W3110dsdA欠失株に30℃で形質転換し、クロラムフェニコール10μg/mlとテトラサイクリン50μg/mlを含むLB寒天プレートに30℃で一晩培養し、形質転換体を得た。得られた形質転換体をテトラサイクリン50μg/mlを含むLB液体培地に接種し、30℃で一晩培養した。次にこれらの培養菌体が得られるようにテトラサイクリン50μg/mlを含むLB寒天プレートに塗布し、42℃で生育するコロニーを得た。得られたコロニーをテトラサイクリン50μg/mlを含むLB液体培地で30℃で一晩培養し、更にテトラサイクリン50μg/mlを含むLB寒天プレートに塗布して42℃で生育するコロニーを得た。
【0121】
出現したコロニーの中から無作為に100コロニーをピックアップしてそれぞれをテトラサイクリン50μg/mlを含むLB寒天プレートとクロラムフェニコール10μg/mlを含むLB寒天プレートに生育させ、テトラサイクリンを含むLB寒天プレートにのみ生育するクロラムフェニコール感受性のクローンを選んだ。更にこれらの目的クローンの染色体DNAからPCRによりglyA近傍領域の約3.6kbpの断片を増幅させ、glyAがテトラサイクリン耐性遺伝子に置換されている株を選抜し、得られた株をW3110dsdA・glyA欠失株と命名した。
【0122】
[13−2](glyA遺伝子破壊Escherichia coliの効果)
このEscherichia coliをプラスミドpXDTA1で形質転換し、実施例10と同じ方法で培養した。但し、フラスコ培養ではグリシンを20mg/L添加し、培養槽での培養では、グリシンを2g/L添加し、培養の途中にグリシンを分割添加したものを用いた。
実施例9と同じ方法でL−セリンの生成活性を調べたところD−セリンの光学純度は97%であった。
【0123】
[実施例14]
(金属塩類の効果)
実施例10で得たMT−11028の凍結菌体100gに300mMのEDTA(pH7.5)3.5gを添加し、4℃にて1時間攪拌した。懸濁液10gを塩化マンガン、硫酸亜鉛、塩化コバルト、塩化ニッケル、塩化カルシウム、塩化第一鉄を各々20mM含むpH7.0の0.5Mリン酸カリウム緩衝液10gに懸濁し、4℃にて1時間攪拌した。
【0124】
次いで前記懸濁液に、ホルムアルデヒドを0.5%となるように加え、35℃で1時間攪拌した。前記の処理菌体液から乾燥菌体として0.22g分を測りとり、実施例10に記載の酵素活性測定液9gを加えて35℃で20時間攪拌し、生成したL−セリンと残存するD−セリンの比率を測定した。
何れの金属塩を用いた場合もD−セリンの光学純度は96%以上であった。また、ホルムアルデヒドとグリシンからD−セリンを合成する酵素活性は何れの金属塩でも有機溶媒処理前の活性に対して50%以上保持していた。
【0125】
[実施例15]
(L−セリンデアミナーゼ発現Escherichia coliによる光学純度を高める方法)
Escherichia coli K−12株を50mlのLB培地に接種した。30℃で一夜培養した後、集菌し、リゾチーム1mg/mlを含む溶菌液で溶菌した。溶菌液をフェノール処理した後、通常の方法によりエタノール沈殿によりDNAを沈殿させた。生じたDNAの沈殿は、ガラス棒に巻き付けて回収した後、洗浄し、PCRに用いた。
【0126】
PCR用のプライマーには、公知のEscherichia coliのL−セリンデアミナーゼ遺伝子(GenBanak accession number M28695)に基づいて設計した配列番号19及び20に示す塩基配列を有するオリゴヌクレオチド(北海道システム・サイエンス株式会社に委託して合成した)を用いた。これらのプライマーは、5’末端付近及び3’末端付近に、それぞれEcoRI及びHindIIIの制限酵素認識配列を有する。
【0127】
前記微生物の染色体DNA6ng/μl及びプライマー各3μMを含む0.025mlのPCR反応液を用いて、変性:96℃、1分間、アニーリング:55℃、30秒間、伸長反応:68℃、1分30秒間からなる反応サイクルを、35サイクルの条件でPCRを行った。
【0128】
PCR反応産物及びプラスミドpUC18(宝酒造(株))を、EcoRI及びHindIIIで消化し、ライゲーション・ハイ(東洋紡(株))を用いて連結した後、得られた組換えプラスミドを用いて、Escherichia coli DH5αを形質転換した。形質転換株を、アンピシリン(Am)50μg/ml及びX−Gal(5−ブロモ−4−クロロ−3−インドリル−β−D−ガラクトシド)を含むLB寒天培地で培養し、Am耐性で且つ白色コロニーとなった形質転換株を得た。このようにして得られた形質転換株よりプラスミドを抽出した。プラスミドに導入されたDNA断片の塩基配列を通常の塩基配列の決定法に従い、公知のEscherichia coliのL−セリンデアミナーゼと同じ配列であることを確認した。得られた発現プラスミドをpSDA1と命名した。
【0129】
pSDA1を用いてEscherichia coli W3110dsdA・glyA欠失株を通常の方法で形質転換し、得られた形質転換体を実施例13と同じ方法で培養槽による培養を行った。
【0130】
実施例12の比較例で行った反応と同じ方法で、反応液に前記の菌体を10g加えて反応を行った。反応液をHPLCで分析したところ、反応液中のD−セリンの光学純度は99.9%であった。
【0131】
本明細書中で引用した全ての刊行物、特許及び特許出願をそのまま参考として本明細書中にとり入れるものとする。
【産業上の利用可能性】
【0132】
本発明は、グリシンとホルムアルデヒドとからD−セリンを製造する方法として有用である。また、本発明の製造方法により得られるD−セリンは結核薬として有用なD−サイクロセリンの原料等の医薬中間体として有用である。
【特許請求の範囲】
【請求項1】
グリシンとホルムアルデヒドからD−セリンを合成する活性を有する微生物又はその処理物の存在下で、グリシンとホルムアルデヒドとを反応させてD−セリンを合成するD−セリンの製造方法であって、該反応の反応液中のL−セリンの副生をD−セリンに対して1.5モル%以下に抑制させる以下の(1)〜(4)の一つ以上の手段を有するD−セリンの製造方法。
(1)グリシンとホルムアルデヒドからD−セリンを合成する活性を有する微生物を有機溶媒処理及び/又は加熱処理する方法、
(2)反応液中のホルムアルデヒド濃度を、a)前記(1)の方法により有機溶媒処理及び/又は加熱処理を施す場合には2M以下に制御し、b)有機溶媒処理及び/又は加熱処理を施さない場合には、150mM以上2M以下に制御する方法、
(3)グリシンとホルムアルデヒドからD−セリンを合成する活性を有する酵素を含有し、かつ、L−セリン合成酵素遺伝子が欠失している微生物を触媒として利用する方法、
(4)L−セリンデアミナーゼ活性を有する酵素を含有する微生物を反応液中に添加する方法。
【請求項2】
グリシンとホルムアルデヒドからD−セリンを合成する活性を有する微生物が、反応液中410mMという高ホルムアルデヒド濃度下においてグリシンとホルムアルデヒドからD−セリンを合成する活性を有する請求項1記載の製造方法。
【請求項3】
グリシンとホルムアルデヒドからD−セリンを合成する活性を有する微生物が、配列番号4、6及び8に記載のアミノ酸配列並びにキサントモナス・オリゼー由来のD−スレオニンアルドラーゼのアミノ酸配列の共通配列を含むアミノ酸配列からなり、グリシンとホルムアルデヒドからD−セリンを合成する活性を有する酵素をコードするDNAをベクターに組み込んでなる組換え体DNAを用いて宿主細胞を形質転換して得られた形質転換体である請求項1又は2記載の製造方法。
【請求項4】
グリシンとホルムアルデヒドからD−セリンを合成する活性を有する微生物が、
以下の(a)もしくは(b)のタンパク質をコードするDNA、又は(c)のDNA:
(a)キサントモナス・オリゼー由来のD−スレオニンアルドラーゼ、
(b)(a)のキサントモナス・オリゼー由来のD−スレオニンアルドラーゼのアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換、挿入もしくは付加されたアミノ酸配列を含み、かつグリシンとホルムアルデヒドからD−セリンを合成する酵素活性を有するタンパク質、
(c)キサントモナス・オリゼー由来のD−スレオニンアルドラーゼをコードするDNA;
をベクターに組み込んでなる組換え体DNAを用いて宿主細胞を形質転換して得られた形質転換体である請求項1〜3のいずれか1項に記載の製造方法。
【請求項5】
有機溶媒がホルムアルデヒド、ベンズアルデヒド、ジクロロエタン及びイソプロピルアルコールから選ばれる一種以上のものである請求項1〜4のいずれか1項に記載の製造方法。
【請求項6】
有機溶媒処理及び/又は加熱処理を2価の金属イオンの存在下で行う請求項1〜5のいずれか1項に記載の製造方法。
【請求項7】
2価の金属がマグネシウム、マンガン、亜鉛、ニッケル、コバルト及び鉄から選ばれる一種以上の金属である請求項6記載の製造方法。
【請求項1】
グリシンとホルムアルデヒドからD−セリンを合成する活性を有する微生物又はその処理物の存在下で、グリシンとホルムアルデヒドとを反応させてD−セリンを合成するD−セリンの製造方法であって、該反応の反応液中のL−セリンの副生をD−セリンに対して1.5モル%以下に抑制させる以下の(1)〜(4)の一つ以上の手段を有するD−セリンの製造方法。
(1)グリシンとホルムアルデヒドからD−セリンを合成する活性を有する微生物を有機溶媒処理及び/又は加熱処理する方法、
(2)反応液中のホルムアルデヒド濃度を、a)前記(1)の方法により有機溶媒処理及び/又は加熱処理を施す場合には2M以下に制御し、b)有機溶媒処理及び/又は加熱処理を施さない場合には、150mM以上2M以下に制御する方法、
(3)グリシンとホルムアルデヒドからD−セリンを合成する活性を有する酵素を含有し、かつ、L−セリン合成酵素遺伝子が欠失している微生物を触媒として利用する方法、
(4)L−セリンデアミナーゼ活性を有する酵素を含有する微生物を反応液中に添加する方法。
【請求項2】
グリシンとホルムアルデヒドからD−セリンを合成する活性を有する微生物が、反応液中410mMという高ホルムアルデヒド濃度下においてグリシンとホルムアルデヒドからD−セリンを合成する活性を有する請求項1記載の製造方法。
【請求項3】
グリシンとホルムアルデヒドからD−セリンを合成する活性を有する微生物が、配列番号4、6及び8に記載のアミノ酸配列並びにキサントモナス・オリゼー由来のD−スレオニンアルドラーゼのアミノ酸配列の共通配列を含むアミノ酸配列からなり、グリシンとホルムアルデヒドからD−セリンを合成する活性を有する酵素をコードするDNAをベクターに組み込んでなる組換え体DNAを用いて宿主細胞を形質転換して得られた形質転換体である請求項1又は2記載の製造方法。
【請求項4】
グリシンとホルムアルデヒドからD−セリンを合成する活性を有する微生物が、
以下の(a)もしくは(b)のタンパク質をコードするDNA、又は(c)のDNA:
(a)キサントモナス・オリゼー由来のD−スレオニンアルドラーゼ、
(b)(a)のキサントモナス・オリゼー由来のD−スレオニンアルドラーゼのアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換、挿入もしくは付加されたアミノ酸配列を含み、かつグリシンとホルムアルデヒドからD−セリンを合成する酵素活性を有するタンパク質、
(c)キサントモナス・オリゼー由来のD−スレオニンアルドラーゼをコードするDNA;
をベクターに組み込んでなる組換え体DNAを用いて宿主細胞を形質転換して得られた形質転換体である請求項1〜3のいずれか1項に記載の製造方法。
【請求項5】
有機溶媒がホルムアルデヒド、ベンズアルデヒド、ジクロロエタン及びイソプロピルアルコールから選ばれる一種以上のものである請求項1〜4のいずれか1項に記載の製造方法。
【請求項6】
有機溶媒処理及び/又は加熱処理を2価の金属イオンの存在下で行う請求項1〜5のいずれか1項に記載の製造方法。
【請求項7】
2価の金属がマグネシウム、マンガン、亜鉛、ニッケル、コバルト及び鉄から選ばれる一種以上の金属である請求項6記載の製造方法。
【図1】

【図2】
【図2】
【公開番号】特開2011−172582(P2011−172582A)
【公開日】平成23年9月8日(2011.9.8)
【国際特許分類】
【出願番号】特願2011−87471(P2011−87471)
【出願日】平成23年4月11日(2011.4.11)
【分割の表示】特願2006−540976(P2006−540976)の分割
【原出願日】平成17年10月7日(2005.10.7)
【出願人】(000005887)三井化学株式会社 (2,318)
【Fターム(参考)】
【公開日】平成23年9月8日(2011.9.8)
【国際特許分類】
【出願日】平成23年4月11日(2011.4.11)
【分割の表示】特願2006−540976(P2006−540976)の分割
【原出願日】平成17年10月7日(2005.10.7)
【出願人】(000005887)三井化学株式会社 (2,318)
【Fターム(参考)】
[ Back to top ]