
DACHYP組成物および方法
【課題】改良特性のある新しいダクリズマブ分子を提供すること。
【解決手段】本開示は、皮下投与に適したダクリズマブの組成物およびその製造方法に関する。
【解決手段】本開示は、皮下投与に適したダクリズマブの組成物およびその製造方法に関する。
【発明の詳細な説明】
【背景技術】
【0001】
ダクリズマブ(DAC)は、活性化されているが休止していないTリンパ球およびBリンパ球の表面で発現されるヒト高親和性インターロイキン−2(IL−2)受容体のアルファサブユニット(CD25またはTac)に結合するヒト化IgG1モノクローナル抗体である。活性化された細胞上のCD25が結合すると、DACは高親和性IL−2受容体複合体の形成を阻止し、それによって活性化された細胞のIL−2誘導増殖を阻止する。
【0002】
PHA芽細胞上での直接結合アッセイにおいて測定された場合、DACは0.3nMの近似の結合親和性(KD)でCD25に結合し、用量依存の形でPHA芽細胞の増殖を抑制する(Hakimi et al.、1993年、J.Immunol.151(2):1075−85頁)。最適以下の用量のIL−2(2.5ng/mL)では、15nMのDACはIL−2依存性細胞系統Kit225/K6の増殖を50%抑制する(Pilson et al.、1997年、J.Immunol.159(3):1543−56頁)。IL−2依存性抗原誘導T細胞増殖アッセイでは、0.5−1μg/mL(3−7nM)の範囲のDACを用いると増殖の50%抑制が観察された(Junghans et al.、1990年、Cancer Res.50(5):1495−502頁)。
【0003】
DACの1バージョンは、腎臓移植患者における急性同種移植片拒絶反応の治療のために、シクロスポリンおよびコルチコステロイドを含む免疫抑制療法の補助剤としてZENAPAX(商標)の商品名でHoffman−La Roche社から以前市販されていた。ZENAPAXは、さらなる希釈および静脈内投与のために濃縮製剤として供給されていた。濃縮製剤のバイアルごとに、5mg/mLのDAC、3.6mg/mLのリン酸ナトリウム一塩基一水和物、11mg/mLのリン酸ナトリウム二塩基七水和物、4.6mg/mLの塩化ナトリウム、0.2mg/mLのポリソルベート80ならびにpHをpH6.9に調整するのに十分なHClおよび/またはNaOHを含有する溶液を5mL含有していた。成人患者にも小児患者にも推奨される用量は1.0mg/kgであり、25mg/5mLのZENAPAX濃縮製剤の計算された容積を50mLの無菌0.9%塩化ナトリウム溶液で希釈することにより調製され、15分間かけて末梢静脈または中心静脈を介して静脈内投与された。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】米国特許第7,258,859号明細書
【非特許文献】
【0005】
【非特許文献1】Hakimi et al.、1993年、J.Immunol.151(2):1075−85頁
【非特許文献2】Pilson et al.、1997年、J.Immunol.159(3):1543−56頁
【非特許文献3】Junghans et al.、1990年、Cancer Res.50(5):1495−502頁
【非特許文献4】Nussenblatt et al.、2004年、FOCIS 2004 meeting;7月18日−23日、Montreal、QC.Abstract 4688
【非特許文献5】Nussenblatt et al.、2003年、J.Autoimmun.21:283−93頁
【非特許文献6】Bielekova et al.、2004年、Proc.Nat’l.Acad.Sci.USA101(23):8705−8708頁
【非特許文献7】Rose et al.、2007年、Neurology 69:785−789頁
【発明の概要】
【発明が解決しようとする課題】
【0006】
DACは、ブドウ膜(Nussenblatt et al.、2004年、FOCIS 2004 meeting;7月18日−23日、Montreal、QC.Abstract 4688;Nussenblatt et al.、2003年、J.Autoimmun.21:283−93頁)および多発性硬化症(たとえば、Bielekova et al.、2004年、Proc.Nat’l.Acad.Sci.USA 101(23):8705−8708頁;Rose et al.、2007年、Neurology 69:785−789頁;米国特許第7,258,859号参照)の治療にも有効性を示しており、現在、多発性硬化症の治療のために進行中の臨床試験の主題となっている。DACは安全で効果的であることが判明しているが、長い有効期限を有し、さらに処方するまたは操作することなく都合よく投与することが可能な高濃度液状製剤の他にも、ZENAPAX DACと比べて増強された安全性などの改良特性のある新しいダクリズマブ分子があれば望ましい。
【課題を解決するための手段】
【0007】
背景技術のセクションで述べられたように、ダクリズマブは、リンパ球活性化の重要な媒介物であるヒトインターロイキン−2受容体(IL−2R)のアルファサブユニット(CD25またはTacとも呼ばれる)に特異的に結合するヒト化IgG1抗体である。ZENAPAX(商標)の商品名でHoffman−La Roche社から以前市販されていたダクリズマブの一バージョンは、シクロスポリンおよびコルチコステロイドを含む免疫抑制療法の補助剤として使用される場合、腎同種移植片拒絶反応の治療において安全性および有効性を実証しており(たとえば、the European Medicines Agency(“EMEA”)market authorization for ZENAPAX参照)、多発性硬化症においても有効性を実証している(たとえば、Bielekova et al.、2004年、Proc.Nat’l.Acad.Sci.USA 101(23):8705−8708頁;Rose et al.、2007年、Neurology 69:785−789頁;米国特許第7,258,859号参照)。EMEAによれば、ZENAPAX DACはGS−NS0(マウス骨髄腫)細胞において発現され、Q−セファロースクロマトグラフィー、S−セファロースクロマトグラフィー、ダイアフィルトレーション、Q−セファロースIIクロマトグラフィー、限外濾過、S−300ゲル濾過クロマトグラフィーおよび限外濾過を含む方法を使用して精製される。無血清、無コレステロールおよび他の無動物産物培養物で増殖するように適応されており、異なる方法により単離されるNS0細胞系統において発現されるダクリズマブは、ZENAPAXダクリズマブ(「ZENAPAX DAC」)とは異なり、いくつかの例ではZENAPAXダクリズマブより優れている特徴および特性を有することが現在では見出されている。この新しいダクリズマブは、本明細書ではDAC HYPと呼ばれるが、ZENAPAX DACとは異なるアイソフォームプロファイル(陽イオン交換クロマトグラフィーにより決定される場合);両方の形態のダクリズマブはNS0細胞において発現されるが、ZENAPAX DACとは異なるN連結グリコシル化プロファイル;および生物学的アッセイにおいてZENAPAX DACよりも少ないADCC細胞傷害性を有する。
【0008】
たとえば、重鎖N末端およびC末端での不均一性のために、ダクリズマブのアイソフォームが可能になる。ダクリズマブの成熟VH鎖のアミノ酸配列は、図2に示されるアミノ酸配列の20位で始まる(配列番号4)。成熟VH鎖のN末端グルタミン(Q)(図2では太字で下線付きのテキストで)は環化され、ピログルタミン酸(pE)を形成することができる。いくつかの例では、シグナルペプチド配列はトランケートし、バリン−ヒスチジン−セリン(VHS)配列を成熟VH鎖のN末端グルタミン残基に結合したままにしておくことがある。各ダクリズマブ分子は2つのVH鎖を含有するために、ダクリズマブの様々なN末端アイソフォームは、(1)2つのグルタミン残基(Q/Q)、(2)1つのグルタミン残基と1つのVHS配列(Q/VHSまたはVHS/Q)、(3)2つのVHS配列(VHS/VHS)、(4)1つのグルタミン残基と1つのピログルタミン酸残基(Q/pEまたはpE/Q)、(5)1つのピログルタミン酸残基と1つのVHS配列(pE/VHSまたはVHS/pE)および(6)2つのピログルタミン酸残基(pE/pE)を含有する形態を含むことができる。異なるC末端アイソフォームも可能であり、アイソフォームは0、1つまたは2つのC末端リジン(K)残基(0K、1Kまたは2K)のいずれかを含有し、複雑なアイソフォームプロファイルを生じる。
【0009】
非常に驚くべきことに、ZENAPAX DACのVH鎖のN末端グルタミンは完全に環化されてピログルタミン酸になるが、完全な環化はDAC HYPでは実現されない。その結果、DAC HYPの陽イオン交換クロマトグラムは、pE/QアイソフォームピークおよびQ/VHSアイソフォームピークにより特徴付けられる。いかなる理論にも縛られるつもりはないが、これら独特のpE/QおよびQ/VHSアイソフォームは、DAC HYPを発現するのに使用されるリーダー配列に影響される可能性があると考えられている。したがって、一態様では、本開示は、陽イオン交換クロマトグラフィーにより決定される場合、pE/QアイソフォームがN末端アイソフォームのうちの3%−17%、3%−15%、5%−15%、さらに好ましくは、5%−12%もしくは7%−12%に及ぶ、および/またはQ/VHSアイソフォームがN末端アイソフォームのうちの1%−15%、さらに好ましくは3%−12%に及ぶダクリズマブ組成物を提供する。
【0010】
いくつかの実施形態では、ダクリズマブ組成物は、図18または図23のDAC HYPプロファイルに実質的に類似する陽イオン交換クロマトグラフィープロファイルにより特徴付けられる。
【0011】
ダクリズマブはN連結オリゴ糖が重鎖残基Asn296に結合している。これらN連結オリゴ糖が、アミダーゼPNGアーゼFを使用して放出され、HPLCによって分析されると、DAC HYPは、両方ともNS0細胞系統において組換え的に産生されるという事実にもかかわらず、ZENAPAX DACとは異なるグリコシル化プロファイルを示す。実際、DAC HYPのグリコシル化プロファイルは著しく均質である。図21の上パネルを参照すると、ZENAPAX DACのグリコシル化プロファイルは、オリゴ糖G0−GlcNAc、G0、G1、Man5、G2、Man6、Man7およびシアル酸付加オリゴ糖を表すピークにより特徴付けられる。図21の下パネルは、DAC HYPのグリコシル化プロファイルが、G0−GlcNAcグリコフォームおよびG0グリコフォームに一致する2つの主要ピークならびにG1グリコフォームに一致する小さいほうのピークにより特徴付けられることを示している。G0−GlcNAcグリコフォームは、AUCの約5%から約20%、さらに典型的にはAUCの約7.2%から14.6%に及ぶことが可能である。G0グリコフォームは、AUCの70%から99.2%、さらに典型的にはAUCの80.9%から99.2%に及ぶことが可能である。G1グリコフォームは、AUCの1%から9%、さらに典型的にはAUCの1.4%から3.8%に及ぶことが可能である。シアル酸付加オリゴ糖は総AUCの1.0%以下である。
【0012】
免疫原性および高レベルのエフェクター機能は、長期的に投与される薬物には問題となることがある。さらに、急速なクリアランス速度は薬物有効性を低下させ得る。当業者には周知であるように、治療抗体のグリコシル化パターンの違いは免疫原性の違いを生じ得る。DAC HYPのように高度に均質なグリコシル化パターンを有する抗体は、有益な免疫原性プロファイル、ADCCレベルおよびクリアランス速度を提供し得る。さらに、より均質なグリコシル化パターンを有する生物製剤であれば、バッチによる変動を減少させ、一貫性および安定性を改善することが可能である。
【0013】
したがって、別の態様では、本開示は、均質なN連結グリコシル化プロファイルにより特徴付けられるダクリズマブ組成物を提供する。一実施形態では、ダクリズマブ組成物は、HPLCにより測定された場合、G0−GlcNAcグリコフォームの総AUCのおよそ5−20%、いくつかの実施形態では、G0−GlcNAcグリコフォームの総AUCのおよそ5%−18%またはおよそ7−15%(たとえば、7.2%−14.6%もしくは6.9%から14.7%)(いくつかの特定の実施形態では、G0−GlcNAcグリコフォームの総AUCの7.3%)、ならびにG0グリコフォームの総AUCのおよそ70%−99.2%、およびいくつかの実施形態では、G0グリコフォームの総AUCのおよそ75%−90%、およそ75−92%またはおよそ81−88%(いくつかの特定の実施形態では、G0グリコフォームの総AUCの86%)を含むN連結グリコシル化プロファイルにより特徴付けられる。場合によって、G1ピークは、総AUCの約10%未満、総AUCの約5%未満、約4%未満または約3%未満であり、ある種の実施形態では、約1%から約4%(たとえば、1.4%から3.8%)または約1%から約3%に及ぶ。Man5グリコフォームは好ましくは、総AUCの約3%以下である。他の実施形態では、ダクリズマブ組成物は、図19に示されるプロファイルに実質的に類似するHPLC N連結グリコフォームプロファイルにより特徴付けられる。
【0014】
ある種の態様では、本開示のダクリズマブ組成物は、2つまたはこれよりも多いグリコフォームピークの総数により特徴付けられる。ある種の実施形態では、本開示のダクリズマブ組成物は、(a)合わせると総AUCの約75%から約100%、約80%から約100%、もしくは約85%から約100%に及ぶG0−GlcNAcグリコフォームおよびG0グリコフォームに一致する2つの主要なピーク、ならびに/または(b)合わせると総AUCの約6%以下であるMan5、Man6およびMan7グリコフォームに一致するピーク、ならびに/または(c)合わせると総AUCの約2%以下であるMan6およびMan7グリコフォームに一致するピークにより特徴付けられる。そのような実施形態では、G0−GlcNAc G0、G1および/またはMan5の百分率は、前の段落に記載されている量で存在し得る。
【0015】
DAC HYPの結合および抑制特性の他にT細胞のIL−2誘導増殖の抑制を測定するアッセイにおいて評価されるDAC HYPの機能的効力は、ZENAPAX DACの特性および効力に類似している。しかし、非常に驚くべきことに、DAC HYPが示すADCC細胞傷害性はZENAPAX DACよりも著しく小さいが、これはおそらく、少なくとも一部は、その非フコシル化マンノースグリコシル化レベルの違いによる(図21参照)。図22Aおよび図22Bにおいて示されるように、細胞アッセイにおいて測定される場合、DAC HYPが示すADCC細胞傷害性は、ZENAPAX DACよりも少なくとも25%小さい。当業者であれば認識されることになるように、DAC HYPの減少したADCC細胞傷害性は、たとえば、多発性硬化症またはブドウ膜炎の治療のためには細胞死が望ましくない長期投与に関連する兆候には有益である可能性がある。たとえば、多発性硬化症および他の非腫瘍学兆候の治療においてなどの治療が慢性的に適用されるこれらの状況では、DAC HYP治療はZENAPAX(商標)を用いる治療よりも安全でありうる。
【0016】
したがって、別の態様では、本開示は、たとえば、標的細胞としてKit225/K6を使用する場合および/または3体以上、6体以上、10体以上または50体以上の健康なドナー由来のPBMCエフェクター細胞を使用する場合、エフェクター対標的細胞比25対1、40対1、50対1または60対1を使用するインビトロアッセイにおいて測定される場合、濃度1μg/mLで約30%、25%、20%、15%、10%、5%未満またはこれよりもずっと低いADCC細胞傷害性を示すことにより特徴付けられるダクリズマブ組成物を提供する。特定の実施形態では、本開示は、たとえば、標的細胞としてKit225/K6を使用する場合および/または3体以上、6体以上、10体以上または50体以上の健康なドナー由来のPBMCエフェクター細胞を使用する場合、エフェクター対標的細胞比25対1、40対1、50対1または60対1を使用するインビトロアッセイにおいて測定される場合、濃度1μg/mLで、5−30%、10−30%、15−30%、15−30%、5−25%、10−25%、20−30%、15−25%、15−35%または20−35%に及ぶADCC細胞傷害性を示すことにより特徴付けられるダクリズマブ組成物を提供する。DAC HYPはIgG1免疫グロブリンであり、ADCC細胞傷害性を低下させることが知られているフレームワーク変異を含有しないことを考えると、ZENAPAX DACと比べた場合DAC HYPを用いて観察されるさらに低いレベルのADCC細胞傷害性は驚きである。
【0017】
ZENAPAX DACと比べた場合のDAC HYPの安全性プロファイルは、ウシ血清アルブミン(BSA)のない高度に純粋な生産物の生産を可能にする高収率無血清方法の使用によりさらに改善され得る。したがって、本開示は、BSAがないおよび/またはBSAが存在しない細胞培養方法の生産物であるダクリズマブ組成物を提供する。
【0018】
上で考察された特性のうちの1つまたは複数により特徴付けられるダクリズマブ組成物(DAC HYP組成物)は、哺乳動物細胞における組換え発現により都合よく入手することが可能である。いかなる特定の動作原理にも縛られるつもりはないが、上で考察された独特の特徴および/または特性のうちの1つまたは複数は、少なくとも一部は、高生産性組換え発現系の使用による可能性があると考えられている。これは、DHFRを使用する、または好ましくは、対象のタンパク質(好ましくは、分泌されたタンパク質)の発現を推進する強いプロモーターと組み合わせて、弱いプロモーターの制御下にある選択可能なマーカー遺伝子を使用する遺伝子増幅によるなどのいかなる方法によっても達成することが可能である。理論に縛られずに、弱いプロモーターの制御下にあるマーカーの選択は、発現ベクターが転写的に活性である染色体領域に組み込まれ、高発現レベルの対象のタンパク質を産生する、安定なトランスフェクタントの同定を促進すると考えられている。一実施形態では、選択可能なマーカーの発現を促進する弱いプロモーターは、1つまたは両方のエンハンサー領域の活性が、部分的なまたは完全な欠失によるなどで減少しているまたは除去されているSV40プロモーター(Reddy et al.、1978年、Science 200:494−502頁)であり、場合によって、CMV IEプロモーター(Boshart et al.、1985年、Cell 41(2):521−30頁)などの強いプロモーターと組み合わせて、対象のタンパク質の発現を推進する。
【0019】
したがって、別の態様では、本開示は、DAC HYPなどの高レベルのダクリズマブを安定的に発現し、選択マーカーの発現が1つまたは両方のエンハンサー配列の部分的欠失によるなどでエンハンサー機能が減少されているSV40プロモーター(dE−SV40と命名されている)の制御下にある組換え細胞系統を作製するのに有用なベクターを提供する。安定した発現細胞系統を生産するのに使用することが可能な特定のdE−SV40プロモーター配列は、図3A−図3Dに、および図3Eに示されているベクターpHAT.IgG1.rg.dE(配列番号5)の6536位−6735位にある(配列番号12)。安定した発現細胞系統を生産するのに使用することが可能な特定のベクターの様々な実施形態は、2011年11月30日提出の米国特許出願第61/565,419号および2011年11月30日提出の国際特許出願第PCT/US11/62720号に記載されており、これら特許文献は参照により本明細書に組み込む。
【0020】
一般に、DAC HYPなどのダクリズマブを発現するのに有用なベクターは、pHAT.IgG1.rg.dEにより例証される、プロモーターなどの特徴(下のセクション5.1に記載されている。)のうちの1つまたは複数を含むことになる。ダクリズマブの2つの鎖は別々の転写制御下に置くことが可能であるが、好ましくは同一ベクター上にあり、そのコード領域はcDNAまたはイントロンとエキソンを含有するゲノムDNAであることが可能である。別々の転写制御の代案として、2つの鎖は、単一転写物または単一オープンリーディングフレームとして発現されることが可能であり、そのコード領域は配列内リボソーム進入部位または自己切断インテイン配列により分離されており、その場合には重鎖および軽鎖コード配列は単一プロモーターの制御下にある。例となるプロモーターはCMV IEプロモーターおよびエンハンサーである(pHAT.IgG1.rg.dE(配列番号5)の0001位−0623位および3982位−4604位)。追加の特徴には、転写開始部位(選ばれるプロモーターに不在であれば)、転写終結部位および複製の起点が含まれる。そのような特徴の例は表1に例示されており、この表はpHAT.IgG1.rg.dEの成分を概説している。
【0021】
NS0細胞において、単一の外来性核酸からDAC HYPなどのダクリズマブの重鎖と軽鎖の両方を発現するのに有用な特定の実施形態は、ネオマイシンホスホトランスフェラーゼ(neor)、ハイグロマイシンBホスホトランスフェラーゼ(hygr)、ハイグロマイシンBホスホトランスフェラーゼ(Hph)、ピューロマイシン−N−アセチルトランスフェラーゼ(puror)、ブラストサイジンSデアミナーゼ(bsrr)、キサンチン/グアニンホスホリボシルトランスフェラーゼ(gpt)、グルタミンシンテターゼ(GS)または単純ヘルペスウイルスチミジンキナーゼ(HSV−tk)などの、哺乳動物細胞において作動可能な選択マーカーを利用する。好ましい実施形態では、本開示のベクターにおける選択可能マーカーは、そのコード配列が図3A−図3Dに示されるpHAT.IgG1.rg.dE(配列番号5)の6935位−7793位に見出すことが可能なエンハンサーレスSV40プロモーターの制御下の大腸菌(E.coli)グアニンホスホリボシルトランスフェラーゼ選択可能マーカーである。
【0022】
別の態様では、本開示は、たとえば、DAC HYPなどのダクリズマブを組換え的に生産するのに有用なベクターをトランスフェクトされている宿主細胞を提供する。宿主細胞は、たとえば、チャイニーズハムスター卵巣(CHO)細胞、NS0マウス骨髄腫細胞、Sp2/0細胞、PER.C6細胞、ベロ細胞、BHK細胞、HT1080細胞、COS7細胞、WI38細胞、CV−1/EBNA細胞、L細胞、3T3細胞、HEPG2細胞、MDCK細胞および293細胞を含むいかなる哺乳動物細胞でもよい。ベクターは、トランスフェクトされると、ゲノムに組み込まれて安定した産生細胞系統を生じ得る。当業者であれば、ヒトへの投与のために指定された組成物中に動物産物を含むのは望ましくないことを認識する。したがって、増殖のために血清または他の動物産物(たとえば、コレステロールなど)を必要としない宿主細胞が好まれる。そのような動物産物を必要とする宿主細胞は、無血清および他の無動物産物培地を利用するように適応させることが可能である。無血清および無コレステロール培地で増殖するようにマウス骨髄腫NS0細胞を適応させるための方法は、Hartman et al.、2007年、Biotech.&Bioeng.96(2):294−306頁およびBurky et al.、2007年、Biotech.&Bioeng.96(2):281−293頁に記載されている。無血清および無コレステロール培地で増殖するように適応されたNS0細胞の特定の系統は、DAC HYPを生産するのに使用することが可能な上記のベクターを安定的にトランスフェクトされてきた(クローン7A11−5H7−14−43)。
【0023】
当業者であれば認識することになるように、組換えタンパク質産生のために細胞を培養するのに使用される基本培地および流加培地の他に、フィーディング計画、増殖速度、温度および酸素レベルなどの他の変数は発現されるタンパク質の収率および質に影響を及ぼすことが可能である。これらの条件を最適化する方法は当業者の範囲内であり、例となる条件は本開示の例となる実施形態に記載されている。好ましくは、細胞は、コレステロール由来、血清由来および他の動物由来の成分のない培地で増殖するように適応され、そのような場合には、基本培地および流加培地は、好ましくはそのような成分の代わりとなる定義された化学物質を含む。高レベルのグルコース、たとえば、10−35g/Lのグルコースを含有する培地は、細胞培養生産性を有利に増加することも見出されている。特定の実施形態では、基本培地は約10−20g/L、さらに好ましくは、約15g/Lのグルコースを有し、および/または流加培地は22−35g/L、さらに好ましくは、約28g/Lのグルコースを有している。当技術分野では公知のように、流加培地を、8−15日間、9−13日間または、もっとも好ましくは10−13日間の期間にわたり、加速するフィーディング計画に従って細胞に追加することが可能である。
【0024】
NS0産生系統7A11−5H7−14−43において発現されるDAC HYPでは、増殖および流加培地の成分ならびに発現と産生に影響を及ぼす他の変数は最適化されている。したがって、本開示は、最適化された基本培地、流加培地、フィーディング計画および他の培養法ならびにダクリズマブを高収率および純度で生産するのに有用な条件も提供する。これらの培地ならびに培養パラメータおよび方法は、セクション5.3にさらに詳細に記載されている。
【0025】
ある種のクロマトグラフィーステップの組合せを利用して細胞培養物からダクリズマブを精製すると、精製されたダクリズマブおよびDAC HYP原薬組成物ならびに高濃度で、典型的には少なくとも約100mg/mL±10−15%の、およびいくつかの実施形態では150mg/mL±10−15%の名目上のダクリズマブまたはDAC HYP濃度(紫外分光または屈折率により測定した場合)で液状の貯蔵安定である液体ダクリズマブおよびDAC HYP薬物製剤を生じることも見出されている。
【0026】
安定した高濃度ダクリズマブ薬物製剤は、一般に、濃縮されたダクリズマブ製剤を約267−327mOsm/kgの範囲の(たとえば、270−310mOsm/kg)の浸透圧および25℃で約pH5.8−6.2の範囲の(たとえば、25℃で5.9−6.1)のpHを有する交換緩衝液を用いて交換して、中間製剤を生じ、次に中間製剤をポリソルベート希釈緩衝液で希釈して、紫外分光または屈折率により測定した場合、約100mg/mL±10%のダクリズマブ(たとえば、DAC HYP)、いくつかの実施形態では少なくとも約150mg/mLのダクリズマブ(たとえば、DAC HYP)を含む安定な高濃度液体製剤を生じることにより調製される。希釈緩衝液は交換緩衝液と同じであるが、約0−10%(w/v)のポリソルベート80を含み、最終の安定な高濃度ダクリズマブ製剤が0.02−0.04%の範囲で、いくつかの実施形態では約0.03%(w/v)で計算されたポリソルベート80濃度(名目濃度)を有するような量で使用される。様々な異なる緩衝剤および賦形剤は、定義された範囲内の浸透圧およびpHを達成するよう交換および希釈緩衝液中に含まれることが可能である。安定で高濃度液体ダクリズマブおよびDAC HYP薬物製剤を処方するのに適した交換緩衝液の特定の非限定的例は、約40mMのコハク酸および約100mMのNaClを含有し25℃で約6.0のpHを有する。この交換緩衝液を用いて使用するのに適した希釈緩衝液の特定の非限定的例は、約40mMのコハク酸、約100mMのNaClおよび約1%(w/v)のポリソルベート80を含有し25℃で約6.0のpHを有する。最終製剤のpHは酸または塩基を用いて調整して25℃で約6.0の実際のpHを生じることが可能である。
【0027】
安定で高濃度液体ダクリズマブ製剤は、低レベルのアグリゲーション、典型的には、サイズ排除クロマトグラフィーにより測定される場合、少なくとも95%の単量体および3%未満のアグリゲート、ときには1.5%未満のアグリゲート、より一般的には、99%を超える単量体および0.8%未満のアグリゲートを含有することにより特徴付けられる。高濃度液体ダクリズマブ薬物製剤の他の純度の特徴はセクション5.6にさらに詳細に記載されている。
【0028】
高濃度ダクリズマブ薬物製剤は、長い貯蔵期間、すなわち、2−8℃で貯蔵される場合、最長54ヶ月以上、たとえば、少なくとも5年間の期間、促進的条件(23−27℃/60±5%の相対湿度)下で貯蔵される場合、最長9ヶ月の期間およびストレス条件(38−42℃/75±5%の相対湿度)下で貯蔵される場合、最長3ヶ月の期間、5%を超える分解および3%を超えるアグリゲートの形成(それぞれSDS−PAGEおよびサイズ排除クロマトグラフィーにより測定される場合)に対して安定であることにも特徴付けられる。
【0029】
上記のように、安定な高濃度液体ダクリズマブ製剤は、中間製剤をポリソルベート希釈緩衝液で希釈し、完成したダクリズマブ薬物製剤を生じることにより調製することが可能である。したがって、別の態様では、本開示は、ポリソルベート希釈緩衝液で希釈して本明細書に記載される安定な高濃度ダクリズマブ液体薬物製剤を生じることが可能な、少なくとも約150mg/mLのダクリズマブ、いくつかの実施形態では約170−190mg/mLのダクリズマブを含有する無ポリソルベート精製されたダクリズマブ(好ましくはDAC HYP)中間製剤を提供する。特定の実施形態では、濃縮された無ポリソルベート中間製剤は、約155mg/mLまたは約180mg/mLのダクリズマブ(好ましくはDAC HYP)、約40mMのクエン酸ナトリウムおよび約100mMのNaCl、25℃でpH6.0を名目上含有する。特定の実施形態では、濃縮された無ポリソルベート中間製剤は、約155mg/mLまたは約180mg/mLのダクリズマブ(好ましくはDAC HYP)、約40mMのコハク酸ナトリウムおよび約100mMのNaCl、25℃でpH6.0を名目上含有する。ダクリズマブ組成物は、下でさらに記載される低レベルのアグリゲートにより特徴付けられる。
【0030】
ダクリズマブを限外濾過により濃縮すると、アグリゲートが誘導されて形成され、容認できない(たとえば、>3%)レベルのアグリゲートを含有する高濃度ダクリズマブ薬物製剤が生じ得ることが見出されている。したがって、アグリゲートを取り除くためにダクリズマブ原薬を濃縮することに先立って「研磨」ステップを利用するのが好ましい。濃縮に先立って容認できるアグリゲートのレベルは、濃縮されるダクリズマブ原薬の濃度、最終ダクリズマブ薬物製剤中の望ましい濃度および最終ダクリズマブ薬物製剤中の容認できるレベルのアグリゲートに依存することになる。たとえば、3%未満のアグリゲートを含有する150mg/mLのダクリズマブ製剤が望ましく、この完成したダクリズマブ製剤を達成するためにはダクリズマブ原薬を10倍から30倍(たとえば、20倍)濃縮しなければならない場合は、濃縮されるダクリズマブ組成物は、<0.3%のアグリゲート、好ましくは<0.2%のアグリゲート、好ましくはさらに低いレベル、たとえば、約0.1%のアグリゲートを含有するほうがよい。
【0031】
たとえば、強陽イオン交換クロマトグラフィーおよび疎水性相互作用クロマトグラフィーを含む、様々な公知の技法を使用して、本明細書に記載される濃縮されたダクリズマブ中間物および最終製剤に濃縮するための容認できるレベルのアグリゲートを含有する出発ダクリズマブ原薬組成物を得ることが可能である。しかし、驚くべきことに、弱陽イオン交換クロマトグラフィーは、4−12mg/mLのダクリズマブおよび最大2.5%のアグリゲートから極度に低いレベル、典型的には、約0.1%のアグリゲートの範囲で含有するダクリズマブ組成物のアグリゲートのレベルを低下させることが見出されている。アグリゲートを取り除くために弱陽イオン交換を使用するのは、窒素含有溶液(たとえば、硫酸アンモニウム溶液)を利用する疎水性相互作用クロマトグラフィーよりも環境にやさしい。
【0032】
したがって、別の態様では、本開示は、得られる研磨された組成物が、一般に、約4から15mg/mLのダクリズマブを含有し、サイズ排除クロマトグラフィーにより測定される場合、0.3%以下(たとえば、0.2%以下または0.1%以下)がアグリゲート形態であるように、ダクリズマブ組成物を研磨しアグリゲートを取り除く方法を提供する。方法は、一般に、約4−10mg/mL、典型的には約8−9mg/mL、好ましくは約8.5mg/mLのダクリズマブおよび>0.5%のアグリゲートを含有するダクリズマブ組成物を、適切な緩衝液中の弱陽イオン交換樹脂上に通過させてダクリズマブを吸着させ、吸着されたダクリズマブを溶出緩衝液で溶出することを含む。有用な弱陽イオン交換樹脂には、CM−650M(Tosoh Biosciences)、CM−セファロース、CM−HyperDが含まれるが、これらに限定されない。平衡化、洗浄および溶出緩衝液の成分は、使用される弱陽イオン交換樹脂に依存することになり、当業者には明らかとなる。CM−650M樹脂(Tosoh Biosciences、品番101392)では、約20mMのクエン酸ナトリウム、pH4.5を含有する平衡化と洗浄緩衝液および20mMのクエン酸ナトリウムと75mMの硫酸ナトリウム、pH4.5を含有する溶出緩衝液がうまく機能する。使用される流速は樹脂とカラムのサイズの選択に依存することになる。約10−30cmの範囲(たとえば、17−19cm)の床高を有するCM−650M樹脂の円筒形カラムおよび約50−200cm/時の範囲(たとえば、90−110cm/時、好ましくは約100cm/時)の流速では、クロマトグラフィーを十分用いた作業は、室温で、またはもっと低い温度、たとえば、4℃から、10℃、15℃、20℃もしくは25℃の範囲の温度で、実施することが可能である。典型的で有用な温度範囲は18−25℃(たとえば、18−22℃)である。
【0033】
ZENAPAX EMEAに従えば、ZENAPAX DACのための精製方法は、以下の12のステップ:
(i)培養液濃度;
(ii)Q−セファロースクロマトグラフィー;
(iii)S−セファロースクロマトグラフィー;
(iv)ウイルス不活化のための低pH処置;
(v)濃縮/限外濾過;
(vi)ウイルス除去のためのDV50濾過;
(vii)Q−セファロースIIクロマトグラフィー;
(viii)ウイルス除去のためのバイアソルブ(viresolve)クロマトグラフィー;
(ix)限外濾過による濃縮;
(x)S−300ゲル濾過クロマトグラフィー;
(xi)限外濾過による濃縮;
(Xii)バイアルの無菌充填
を含む。
【0034】
この方法は非効率的であり、提供する精製収率は低い。ステップはもっと少ないが、同時にもっと高い純度を生じる方法を用いてもっと高い収率を達成することが可能であることが見出されており、これを使えば、得られるダクリズマブ原薬を上記の高濃度薬物製剤に処方することが可能になる。したがって、本開示は、ダクリズマブ原薬と高濃度薬物製剤の両方を単離するおよび/または精製するための改良された方法も提供する。該方法は、強い陰イオン交換(Q−セファロース)クロマトグラフィーおよび弱陽イオン交換(CM−650M)クロマトグラフィーと併せてプロテインA親和性クロマトグラフィーを利用し、それにより方法中間物を希釈しない連続フロー処理が可能になる。精製されたダクリズマブ原薬を得るための改良された方法は、以下のステップ:
(i)ダクリズマブを他の細胞培養成分から単離するためのプロテインA親和性クロマトグラフィー;
(ii)低pHウイルス不活化;
(iii)DNAを取り除くための強い陰イオン交換(Q−セファロース)クロマトグラフィー;
(iv)アグリゲートを減らすための弱陽イオン交換(CM−650M)クロマトグラフィー;および
(v)ウイルスを取り除くための濾過
を含む。
【0035】
当技術分野で周知のように、正確な容積、カラムサイズおよび操作用パラメータは、一部、精製の規模に依存することになる。大規模精製に有用な比容積、カラムサイズおよび操作用パラメータはセクション5.4に記載されている。
【0036】
精製され、場合によって上記の方法により処方される未精製ダクリズマブは、様々な従来の方法、たとえば、マイクロフィルター法、遠心分離および直接バイオリアクターからのデプス濾過を使用して細胞培養物から収穫することが可能である。しかし、未精製ダクリズマブは、15℃未満の温度で細胞培養物のpHをおよそpH5まで下げて、細胞を凝結させ、この細胞を遠心分離により取り除くことができることにより都合よく収穫することが可能であることはすでに見出されている。特定の実施形態では、未精製ダクリズマブは、細胞培養物のpHをおよそpH5まで下げ、培養物を30−90分間15℃未満、たとえば、4℃まで冷却し、得られた懸濁物を遠心分離にかけて細胞を取り除くことにより収穫される。この方法は、一般に、組換えタンパク質を培養培地中に分泌するいかなる細胞培養物にも適用可能であり、ダクリズマブまたは治療抗体を産生する培養物に限定されてはいない。培養物のpHは、弱い有機酸もしくは強い有機酸、または弱い無機酸もしくは強い無機酸を含む様々な異なる酸を使用して調整することが可能である。ダクリズマブ培養物では、クエン酸がうまく機能することが見出されている。濃縮されたクエン酸溶液、たとえば、0.5M−2M溶液を、収穫に先立って培養物のpHを調整するために使用することが可能である。
【0037】
DAC HYPの精製は、3つのクロマトグラフィーステップ、すなわち、ウイルス不活化、ウイルス濾過および最終の限外濾過の使用により実現される。プロテインA親和性クロマトグラフィーは、精製方法の最初のステップであり、大部分のプロセス関連不純物を取り除く。プロテインA親和性カラムの再利用を可能にするためには、カラムを再生し衛生化しなければならない。NaOH水溶液はカラム再生と衛生化の両方を実現するのに効果的であることが見出されている。しかし、NaOH溶液を使用するとプロテインA樹脂を分解することがあり、全体的な生産コストを増やしてしまう。プロテインA親和性クロマトグラフィー樹脂をNaOHおよびベンジルアルコールを含有する溶液で衛生化すると良好な結果を生じ、精製サイクルの回数を著しく増やすことも見出されている。したがって、本開示は、プロテインA親和性カラムおよび樹脂を再生し衛生化するための衛生化溶液および方法も提供する。該緩衝液は、一般に約100から500mMのクエン酸ナトリウム、約10から30mMのNaOHおよび約0.5から3%(v/v)のベンジルアルコールを含み、約pH10から13の範囲のpHを有する。該緩衝液は、場合によって、たとえば、塩および/または洗剤などの他の成分も含み得る。クエン酸ナトリウムとベンジルアルコールの両方はプロテインA樹脂をNaOHにより破壊されることから保護し、殺菌作用を増強するために重要である。特定の実施形態では、プロテインA衛生化緩衝液は、約200mMのクエン酸ナトリウム、約20mMのNaOHおよび約1%(v/v)のベンジルアルコールを含有する。セクション5.4.2において記載されるように、ベンジルアルコールおよび水酸化ナトリウムを含有する衛生化溶液は有益な抗菌効果を有し、これを使用して、いかなる抗体についても精製方法におけるプロテインAカラムを衛生化することができる。
【0038】
衛生化緩衝液を使用して、バッチ式方法においてプロテインAクロマトグラフィー樹脂を衛生化することができ、樹脂は過剰な(たとえば、1.5−2×容積)衛生化緩衝液で洗浄され、続いて過剰な(たとえば、1.5−2×容積)衛生化緩衝液中で約30−45分間インキュベートし、続いて平衡化緩衝液または貯蔵緩衝液で平衡化される。衛生化緩衝液を使用して、カラムを適切な流速(たとえば、約110−190cm/時または135−165cm/時の幅がある)で過剰な(たとえば、1.5−2×カラム容積)衛生化緩衝液で洗浄し、カラムを約30−40分間、流速0の条件下に保持し、次にカラムを平衡化緩衝液または貯蔵緩衝液で洗浄することにより、調製されたプロテインAクロマトグラフィーカラムを衛生化することが可能である。適切な平衡化緩衝液および貯蔵緩衝液はセクション5.5に記載されている。
【0039】
本明細書に記載される衛生化緩衝液を用いてプロテインAカラムを衛生化すると、単一バッチの樹脂を使用することができる精製の回数を著しく増加させる。たとえば、単一バッチのプロテインA樹脂は、従来のNaOH緩衝液(たとえば、50mMのNaOH、0.5MのNaCl)で衛生化される場合、典型的には約30精製サイクルしかもちこたえないが、本明細書に記載される衛生化緩衝液で衛生化されたプロテインAカラムは、100回を超える精製サイクルに使用することが可能である。いかなる動作原理にも縛られるつもりはないが、本明細書に記載される衛生化緩衝液は、固定化されたプロテインAをNaOH誘導分解から一部保護し、それによって樹脂の耐用期間を増加すると考えられている。したがって、NaOH分解に耐性であるよう設計されたプロテインAの変異系統を利用する樹脂(たとえば、MabSuRe樹脂)を含むあらゆるプロテインA樹脂には改良が予想されるが、非改変の固定化されたプロテインAまたはNaOH安定であるように操作されていないプロテインAを利用してプロテインA樹脂およびカラムを衛生化するのに使用される場合、本明細書に記載される衛生化緩衝液は特に有益である。本開示は、精製実行を行い、樹脂を本明細書に開示される衛生化溶液で洗浄することを含む、30回を超える、35回を超えるまたは40回を超える抗体精製実行、いくつかの例では最大50または最大100のタンパク質精製サイクルにプロテインA親和性樹脂を使用することを含む方法をさらに提供する。
【0040】
上記のように、ダクリズマブは、活性化されており休止していないTリンパ球およびBリンパ球上で発現されるCD25に特異的に結合し、CD25へのIL−2の結合をブロックして、それによって高親和性IL−2受容体複合体の形成を阻害し、活性化されたT細胞およびB細胞の増殖を抑制する。本明細書に記載されるDAC組成物および製剤ならびに特にDAC HYP組成物および製剤は、同様にCD25に特異的に結合し、類似の生物学的特性を示す。したがって、本明細書に記載されるDAC組成物および製剤ならびに特にDAC HYPは、ダクリズマブ全般、具体的にはZENAPAXについて記載されているアッセイおよび治療法のいずれにおいても有用である。したがって、本開示は、同種移植片拒絶の治療および予防、ブドウ膜炎の治療ならびに多発性硬化症の治療などの、活性化されたT細胞および/またはB細胞増殖が関与している疾患の治療に向けた治療手段として、インビトロ適用でもインビボ適用でも活性化されたT細胞およびB細胞の増殖を抑制するために、本明細書に記載されるDAC組成物および製剤ならびに特にDAC HYP組成物および高濃度安定液体製剤を使用する方法もさらに提供する。
【0041】
該方法は、一般的には、活性化されたT細胞および/またはB細胞を、その増殖を抑制するのに十分な量の本明細書に記載されるダクリズマブ組成物または製剤に接触させることを含む。
【0042】
治療法では、該方法は、一般的には、治療効果を与える量のダクリズマブ組成物、たとえば、本明細書に記載されるDAC HYP組成物または高濃度DAC製剤を対象に投与することを含む。特定の実施形態では、ダクリズマブ組成物および製剤を使用して、単独でまたはインターフェロンベータなどの他の薬物と組み合わせて、多発性硬化症を治療することができる。本明細書に記載されるDAC組成物は、75mgから300mg(たとえば、75mg、100mg、125mg、150mg、175mg、200mg、225mg、250mg、275mgもしくは300mg)の範囲のまたは1mg/kgから4mg/kgの範囲の用量で、患者に毎週から毎月(たとえば、毎週、2週間ごとに、月2回、4週ごとにもしくは毎月)皮下投与することが可能である。該組成物は、好ましくは、100mg/mL±10−15%または150mg/mL±10−15%の名目上のダクリズマブ濃度で、皮下使用に便利な充填済みのシリンジで提供することが可能である。濃縮されたDAC組成物は、静脈内投与のために希釈することも可能である。
【図面の簡単な説明】
【0043】
【図1】DAC−HYP軽鎖cDNA(配列番号1)および翻訳アミノ酸(配列番号2)配列を提供する図である。太字の下線を引いたアスパラギン酸(D)残基は、適切にプロセッシングされた成熟タンパク質における第1のアミノ酸であり、この残基の上流のアミノ酸配列はシグナル配列に対応する。
【図2】DAC−HYP重鎖cDNA(配列番号3)および翻訳アミノ酸配列(配列番号4)を提供する図である。太字の下線を引いたグルタミン(Q)残基は、適切にプロセッシングされた成熟タンパク質における第1のアミノ酸であり、この残基の上流のアミノ酸配列はシグナル配列に対応する。
【図3A】ベクターpHAT.IgG1.rg.dE(配列番号5)に関する完全ヌクレオチド配列を提供する図である。
【図3B】ベクターpHAT.IgG1.rg.dE(配列番号5)に関する完全ヌクレオチド配列を提供する図である。
【図3C】ベクターpHAT.IgG1.rg.dE(配列番号5)に関する完全ヌクレオチド配列を提供する図である。
【図3D】ベクターpHAT.IgG1.rg.dE(配列番号5)に関する完全ヌクレオチド配列を提供する図である。
【図3E】高収量の生産株の選択に使用できる、dESV40プロモーター(配列番号12)の具体的実施形態を提供する図である。
【図4A】ベクターpHAT.IgG1.rg.dEの略図であり、これはpABX.gptに由来する。
【図4B】任意の重鎖遺伝子および軽鎖遺伝子またはさらに非抗体ポリペプチドを発現するように適合させ得るベクターである、pABX.gptの略図である。
【図5】DAC HYPの例示的生産方法を提供する図である。
【図6】プロテインAの親和性クロマトグラフィーの間の生産物分画をモニターする、UV(280nm)、pHおよび導電率を明示する図である。
【図7】Q−セファロースクロマトグラフィーの間の生産物分画をモニターする、UV(280nm)、pHおよび導電率を明示する図である。
【図8】CM陽イオン交換クロマトグラフィーの間の生産物分画をモニターする、UV(280nm)、pHおよび導電率を明示する図である。
【図9】DAC HYP限外濾過系の略図である。
【図10】0−60分のDAC HYPのペプチドマップクロマトグラムを提供する図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。
【図11】55−115分のDAC HYPのペプチドマップクロマトグラムを提供する図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。
【図12】110−170分のDAC HYPのペプチドマップクロマトグラムを提供する図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。
【図13】DAC HYPの150mg/mlのロットのバッチ1およびバッチ2の円偏光二色性スペクトルを重ね合わせた図である。参照は、100mg/mlのDAC HYP調製物である。
【図14A】重ね合わせたゼロ次紫外スペクトルを提供する図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製品物に対応する。3種すべてのスペクトルが存在するが、互いに重ね合わせたところ単一のスペクトルのように見える。
【図14B】重ね合わせた二次微分紫外スペクトルを提供する図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。3種すべてのスペクトルが存在するが、互いに重ね合わせたところ単一のスペクトルのように見える。
【図15A】フルスケールのサイズ排除クロマトグラムを提供する図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。
【図15B】拡大スケールのサイズ排除クロマトグラムを提供する図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。
【図16】時間の関数としてのDAC HYPの凝集のプロットである。
【図17】還元型および非還元型SDS−PAGE(それぞれ、左および右のパネル)を示す図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。
【図18】DAC HYPの陽イオン交換クロマトグラムを示す図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。ピークラベルは、さまざまなN末端およびC末端のアイソフォームに対応する。
【図19】DAC HYPから酵素的に切断された、N連結オリゴ糖のHPLCクロマトグラムを示す図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。
【図20】DAC HYPのADCC反応曲線を示す図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。
【図21】それらのさまざまなグリコシル化プロファイルを例示する、DAC HYPから放出されたN連結オリゴ糖のHPLCクロマトグラム(下のパネル)およびZENAPAX DACから放出されたN連結オリゴ糖のHPLCクロマトグラム(上のパネル)を示す図である。
【図22A】エフェクター対標的細胞比を可変としたADCCアッセイフォーマットを使用した、2種のDAC HYP調製物(DAC HYP バッチ3およびDAC HYP バッチ4と称される)、DAC PenzburgおよびZENAPAX DACのADCC活性間の比較を提供する図である。
【図22B】抗体濃度を可変としたADCCアッセイフォーマットを使用した、2種のDAC HYP調製物(DAC HYP バッチ3およびDAC HYP バッチ4と称される)、DAC PenzburgおよびZENAPAX DACのADCC活性間の比較を提供する図である。
【図23】DAC HYP、DAC PenzbergおよびZENAPAX DACの電荷アイソフォームの比較を提供する図である。
【発明を実施するための形態】
【0044】
4.詳細な説明
本開示は、たとえば、活性化されたT細胞および/またはB細胞の増殖を抑制し、たとえば、多発性硬化症などの活性化されたT細胞および/またはB細胞媒介疾患を治療するおよび/または予防するために、とりわけ、特定の特性を有するDAC組成物、ある種の投与様式に特に有用であり異なる温度で貯蔵安定である高濃度DAC製剤、DAC組成物を生産するのに有用なベクターおよび宿主細胞、DAC組成物を生産するのに有用な最適化された培養液および培養条件、DAC組成物および高濃度製剤を精製するための方法ならびにDAC組成物および高濃度製剤を使用する方法を提供する。
【0045】
本明細書において使用されるダクリズマブ(DAC)とは、図1に例示される軽(VL)鎖配列(配列番号2の21位−233位)および図2に例示される重(VH)鎖配列(配列番号4の20位から465位)を有するヒト化IgG1モノクローナル抗体のことである。DACのCDR配列は以下の通りである。
【0046】
【化1】

【0047】
ある種のダクリズマブ分子は文献中に報告されており、DACの特定のバージョンは、シクロスポリンおよびコルチコステロイドを含む免疫治療の補助剤として腎臓移植患者における同種移植片拒絶の予防のためにHoffman−La RocheによりZENAPAXの商品名ですでに市販されている。ZENAPAXの商品名で販売されているバージョンのDACは本明細書においては「ZENAPAX DAC」と呼ばれる。
【0048】
DACの別のバージョンは、ドイツのペンツベルグの施設で製造されており、一度も市販されたことはないが、ある種の臨床試験で使用されたことがある。DACのこのバージョンは本明細書では「DACペンツベルグ」と呼ばれる。
【0049】
本明細書に記載されるように、本開示は、一部には、ZENAPAX DACおよびDACペンツベルグの特徴および特性とは異なる、いくつかの例では、これらより優れている特徴および特性を有する新しいバージョンのDACに関する。したがって、本開示は、一部には、新しいDAC組成物に関する。新しいDAC組成物は、発明の概要においてより完全に記載されている以下の特色:
(1)特徴的なpE/Qおよび/またはQ/VHS N末端アイソフォーム;
(2)2つの主要なピークおよび小ピークにより特徴付けられる均質なN連結オリゴ糖プロファイル;
(3)ZENAPAX DACおよびDACペンツベルグと比べて減少したADCC細胞傷害性;ならびに
(4)150±10−15%もの名目上の濃度で処方される場合の低レベルのアグリゲート形態(<3%)
のうちの1つまたは複数により特徴付けられる。
【0050】
これらの特徴および/または特性のうちの1つまたは複数を有するDAC組成物は、本明細書において「DAC HYP」組成物と呼ばれる。本明細書に記載される発明の様々な態様および特色を例証する目的で、上記4つの特性すべてを有する特定のDAC HYPが、この生産および精製のための特定の組成物および方法と同じように、記載される。しかし、DAC HYP組成物は、本開示の範囲に収まる上記4つの特徴をすべて有する必要はないことは理解されるべきである。特定の実施形態では、DAC HYPは、上記(1)から(4)までの特徴うち少なくとも2つ(たとえば、少なくとも(1)および(2);(1)および(3);(1)および(4);(2)および(3);(2)および(4)、もしくは(3)および(4)の組合せ)または上記(1)から(4)までの特徴うち少なくとも3つ(たとえば、少なくとも(1)、(2)および(3);(1)、(2)および(4);(1)、(3)および(4)、ならびに(2)、(3)、(4)の組合せ)を有する。そのようなDAC HYP組成物は、100mg±10−15%またはさらに150±10−15%の濃度で処方される場合、<3%のアグリゲート、<2%のアグリゲートおよびこれよりはるかに低いレベル、たとえば、<1%のアグリゲートも有し得る。
【0051】
さらに、本明細書に記載される発明のある種の態様および実施形態はDAC HYPを用いて例示され例証されるが、本発明はDAC HYPに限定されておらず、ダクリズマブ組成物全般に有用であり、DACに類似する特定のCD25結合特性を有するIgG2、IgG3およびIgG4抗CD25抗体、ならびにヒトに投与するのに適しておりまだヒト化されていない抗CD25抗体にも限定されていないことは当業者であれば認識することになる。これら様々な異なる抗CD25抗体は本明細書において「DAC類似体」と呼ばれる。そのようなDAC類似体は、通常上記の6つのDAC CDRを含み得るが、他のCDR配列を含み得る。
【0052】
DAC HYP組成物の特徴および特性は、標準アッセイ及び方法を使用して確認することが可能である。たとえば、N末端およびC末端アイソフォームプロファイルは、220nmで検出する陽イオン交換クロマトグラフィーを使用して評価することができる。特定の方法では、100μLの試験試料(緩衝液Aに溶解した1mg/mLの抗体)は、以下の分離勾配(カラムは緩衝液Aを用いて平衡化されている。):
【0053】
【表1】
を使用してProPac WCX−10Gガードカラム(Dionex Corporation)を備えたProPac WCX−10カラム(Dionex Coporation)上、室温で分離される。
【0054】
N連結グルコシル化プロファイルは、アミダーゼPNGアーゼFを用いてN連結オリゴ糖を切断し、オリゴ糖を蛍光標識を用いて誘導体化し、得られた混合物を蛍光検出付きの順相HPLCにより分析することにより評価することが可能である。特定の方法では、アントラニル酸誘導体化切断N連結グリカンは、以下の溶出勾配(100μLの試料注入量を使用して;カラムは、85%緩衝液A/15%緩衝液Bで平衡化されている。):
【0055】
【表2】
を使用して、250×4.6mm重合体アミン結合Asahipak Amino NH2P−504Eカラム(5μm粒子サイズ、Phenomenex、カタログ番号CHO−2628)上、50℃で分離される。
【0056】
純度は、還元SDS−PAGE(プレキャスト14%Trisグリシン勾配ミニゲル、Invitrogen 品番601632)およびコロイドブルー染色、ならびに/または280nmでの検出付きサイズ排除クロマトグラフィーを使用して確認することが可能である。特に、15μLの試験試料(溶出緩衝液中20mg/mL抗体)は、流速1mL/分で、均一濃度勾配の溶出緩衝液(200mMのKPO4、150mMのKCl、pH6.9)を使用する0.5μmのプレカラムフィルター(Upchurch、品番A−102X)を備えた7.8mm×30cmTSK G3000SWXLカラム(Tosoh Biosciences、品番601342)上、室温で分離することが可能である。
【0057】
本明細書に記載される安定な高濃度液体DAC製剤などの、本明細書に記載されるDAC HYP組成物および他のDAC製剤は、少なくとも一部は、たとえば、同種移植の拒絶および多発性硬化症を含む、活性化されたT細胞および/またはB細胞により媒介されると考えられる様々な障害および状態を治療するのに有用である。特定の患者集団、製剤、投与様式ならびに同種移植片拒絶を治療するまたは予防するのに有用な投与量および投与計画は、米国特許第6,013,256号に記載されており、参照により本明細書に組み込む。特定の患者集団、製剤、投与様式ならびに多発性硬化症に罹った患者を治療するのに有用な投与量および投与計画は、米国特許第7,258,859号に記載されており、参照により本明細書に組み込む。これらの製剤、投与様式、投与量および投与計画すべて、の他に開示された特定の患者集団および組合せ療法は、本明細書に記載されるDAC HYP組成物および、適用可能な場合には、高濃度DAC製剤に等しく適している。
【0058】
本明細書に記載されるDAC HYP組成物および製剤は、治療効果を与える量で投与される。治療効果には、根底にある障害の治療が含まれるが、これに限定されない。治療効果には、標準診断検査および他の検査を使用して評価される特定の疾患の症状または副作用を改善するまたは寛解させることも含まれ得る。多発性硬化症では、たとえば、脳病変を評価する磁気共鳴画像法の使用を含む治療効果を評価するおよび/または能力障害への進行を評価する様々な手段は米国特許第7,258,859号に記載されており、参照により本明細書に組み込む。これらの様々な検査法すべてを使用して、多発性硬化症に罹っている患者という状況で治療効果を評価することが可能である。
【0059】
安定な高濃度DAC製剤は、DAC HYPで作製されるものでも、DAC全般で作製されるものでも、DAC類似体で作製されるものでも、多発性硬化症などの慢性疾患の治療における皮下投与に特に有用である。該製剤は、単回ボーラス皮下注射として都合よく投与されるまたは静脈投与のために希釈されることが可能である。該製剤は、75mgから300mg(たとえば、75mg、100mg、125mg、150mg、175mg、200mg、225mg、250mg、275mgもしくは300mg)の範囲のまたは1mg/kgから4mg/kgの範囲の用量で、患者に毎週から毎月(たとえば、毎週、2週間ごとに、月2回、4週ごとにもしくは毎月)皮下投与することが可能である。該組成物は、皮下使用に便利な充填済みのシリンジで提供することが可能である。希釈された製剤は、皮下投与と同じ回数、適切な投与量で静脈内投与することが可能である。
【0060】
5.例示的実施形態
本明細書に記載の発明のさまざまな態様および特徴は、以下の例示的実施形態を用いてさらに記載される。例示的実施形態が、特定の細胞培養培地、細胞培養条件、カラムクロマトグラフィー樹脂および平衡化緩衝液、洗浄緩衝液および溶出緩衝液を利用する限り、日常の変更は実施可能であると認められる。さらに、さまざまな細胞培養方法が特定の生産株(クローン7A11−5H7−14−43)により例示される限り、日常の最適化を用いて、または用いずに、他のDACまたはDAC類似体生産株が良好に使用され得ることが期待される。さらに、(上記の要約または以下の例示的実施形態のいずれにおいても)特定の実施形態を伴って記載される特徴は、本開示の方法および組成物の望ましい特性に実質的に影響を与えずに逸脱可能であり、さらにその異なる実施形態は、これらが明確に互いに排他的でない限り、さまざまな方法で一緒に組み合わせ、使用することができる。したがって、以下に提供される例示的実施形態は例示を意図し、限定することを意図するものではないと理解されるべきであり、これらの実施形態に従った特許請求の範囲を限定すると解釈するべきではない。
【0061】
以下に例示された製造方法は、DAC HYP製剤原料を150mg/mLで生産するために使用された。DAC HYP製剤原料を100mg/mLで作るために、小さなプロセス変更が導入される:
DAC HYP(セクション5.3を参照されたい)を100mg/mLで生産するために使用される細胞培養は、消泡剤エマルジョンを含まず、一方、DAC HYPが150mg/mLの細胞培養は、発泡を最小に抑えるために、10,000Lバイオリアクターにおいて低濃度のDow Corning Antifoam Cを使用する。最終抗体濃度100mg/mLを有するDAC HYP製剤を生産する場合、CM−650Mカラム(セクション5.4.5を参照されたい)は、0.5MのNaOH、0.5Mの硫酸ナトリウムの緩衝液を用いて衛生化され、最終抗体濃度150mg/mLを有するDAC HYP製剤を生産する場合、硫酸ナトリウムは衛生化緩衝液から省かれる。DAC HYPを100mg/mLで作るために、ワンステップの限外濾過/ダイアフィルトレーション(UF/DF)が、ポリソルベート80の添加および製剤原料の最終容積への希釈の直前の、下流プロセスの終わりに使用され(セクション5.4.7を参照されたい)、一方、製剤原料を濃度150mg/mLに作るためには、ツーステップのUF/DFが使用される。以下の実施例は、さまざまなロットの、100mg/mLおよび150mg/mLのDAC HYPの比較分析を示す。いくつかの研究において、150mg/mLのDAC HYPのバッチが、以下に参照標準ロットRS0801と称される、10,000L規模で製造された、1種のロットのDAC HYP100mg/mLと比較された。
【0062】
5.1.DAC HYP発現構築体
抗Tac、マウスIgG2aモノクローナル抗体を産生するハイブリドーマを、マウス骨髄腫細胞系NS−1と、T細胞白血病患者から生じたヒトT細胞系を用いて免疫化されたマウス由来の脾臓細胞とを融合することによって発生させた(Uchiyamaら、1981年、J.Immunol.126(4):1393−7頁)。抗Tacは、活性化T細胞とのその反応性に関して選択されたが、静止T細胞または静止B細胞とは反応性がない。抗Tacは、後に、ヒトIL−2受容体のアルファサブユニットと反応することが示された(Leonardら、1982年、Nature 300(5889):267−9頁)。
【0063】
マウス抗Tacの軽鎖および重鎖の可変領域に関するアミノ酸配列は、それぞれのcDNAから決定された(Queenら、1989年、Proc.Nat’l Acad.Sci.USA86(24):10029−33頁)。マウス抗Tacの結合親和性は、Queenらが記載したヒト化形態中に保有された。マウス抗Tacの相補性決定領域(CDR)は、まず、ヒト抗体Euのアクセプターフレームワークにグラフトされた。三次元モデルの助けを借りて、CDRの立体構造に重要な、カギとなるマウスフレームワーク残基、したがって結合親和性が特定され、アクセプターフレームワーク中のヒトカウンターパートに置換された。さらに、アクセプターフレームワーク中の非定型アミノ酸が、対応する位置のヒトコンセンサス残基と置き換えられ、潜在的免疫原性が排除された。
【0064】
DAC HYPのVLおよびVH遺伝子は、Queen(1989)らにより記載されたように、重複オリゴヌクレオチドのアニーリングおよび伸長によってミニエクソンとして構築された。IgG1形態におけるDAC HYPの発現のために、結果として得られたVLおよびVH遺伝子が、Coleら、(1997年、J.Immunol.159(7):3613−21頁)およびKostelnyら(2001年、Int.J.Cancer 93(4):556−65頁)に概説されたように、単一発現ベクター中にクローニングされ、pHAT.IgG1.rg.dEが構築された(図3および図4Aを参照されたい)。プラスミドpHAT.IgG1.rg.dEは、ダクリズマブのIgG1重鎖およびカッパ軽鎖の両方に関する遺伝子を含有し、各々がヒトサイトメガロウイルス(CMV)プロモーターの調節下にある。このプラスミドは、選択可能なマーカーとして、E.コリ(E.coli)のグアニン・ホスホリボシルトランスフェラーゼ(gpt)遺伝子を含有する。pHAT.IgG1.rg.dE中のこの遺伝成分を、以下の表1に記載する。
【0065】
【表3】
【0066】
dESV40プロモーターは、pHAT.IgG1.rg.dEの6536−6735位に及ぶ(6536−6562は72bpのエンハンサーAの27残基であり、6566−6629は3つの21−bpの反復である。6536−6735はGenBank:J02400.1(シミアンウイルス(Simian Virus)40完全ゲノム))中の5172−1および1−133の逆相補配列(reverse complement)である。発現ベクター中のDAC HYP軽鎖遺伝子および重鎖遺伝子のヌクレオチド配列は、DNA配列決定により確認された。
【0067】
5.2.DAC HYP安定細胞系
マウス骨髄腫細胞系NS0は、European Collection of Cell Cultures(ECACCカタログ番号85110503、Salisbury、Wiltshire、UK)から得られた。これらのNS0細胞のバイアルを、10%FBSを添加されたDMEM中で解凍した。細胞は、加湿インキュベーターにおいて37℃および7.5%CO2において維持された。細胞は、次いで、1mg/mLのBSAを添加された基本培地SFM−3において培養された。SFM−3は、10mg/mLインスリンおよび10μg/mLトランスフェリンを添加された、DMEMおよびHam’sF−12の1:1混合物である。およそ3ヶ月にわたって、NS0細胞を、それが排除されるまで培養培地中のFBSの量を徐々に減少させることによって、添加なしのSFM−3に適応させ、その後、単一ステップにおいてBSAを最終的に除去する。得られた宿主細胞系は、SFM−3において15−20回継代され、凍結バンクが調製された。
【0068】
SFM−3適応細胞に、(FspI酵素(New England Biolabs、カタログ番号R0135L、ロット43)により線形化された)pHAT.IgG1.rg.dEを、エレクトロポレーションにより形質移入した。簡潔に言うと、30−40μgのpHAT.IgG1.rg.dEを、1×107の対数的に増殖する適応NS0細胞に加え、1.5kV、25μFで、Gene Pulser装置(BioRad、Richmond、CA)を使用して2回パルスをかけた。エレクトロポレーション後、細胞を5つの96−ウェルプレート中のDMEM±10%FBSに、ミコフェノール酸(「MPA」)選択後に単一コロニー/ウェルを支持する密度である20,000細胞/ウェルでプレーティングした。Hartmanら、2007年、Biotech.&Bioeng96(2):294−306頁に記載のように、ベクターに安定に統合された形質移入体を、ミコフェノール酸の存在下で選択した。高レベルのDAC HYPを産生するNS0安定形質移入体から出発し、連続3ラウンドのサブクローニングを、限界希釈クローニングまたは蛍光標識細胞分取(FACS)によって、2.5%または5%いずれかのウシ胎児血清(FBS;HyClone、Logan、UT)を含有するPFBM−1中に実施した。サブクローニングの各ラウンドにおいて、最も優れた生産株の1種を次のラウンドのサブクローニングに使用した。第3ラウンドのサブクローニング後、最終生産細胞系(7A11−5H7−14−43)を選択した。その後、最終生産細胞系のシードバンクを、1mLの90%FBS/10%DMSO(Sigma、St.Louis、MO)中1×107細胞/バイアルを凍結することによって調製した。
【0069】
5.3.DAC HYP組換体の生産
5.3.1.細胞の培養および回収
単一細胞バンクバイアルから細胞を解凍し、T−フラスコ、ローラーボトル、スピナーフラスコおよびバイオリアクター内に、生産規模が達成されるまで、徐々により大きな容積に拡大する。生産培養が完了次第、細胞培養液を遠心分離および深層濾過により浄化し、収穫保持タンクに移送した。生産培養期間はおよそ10日である。
【0070】
細胞の培養および回収を、さまざまな異なる細胞培養設備において標準的な機器を使用して、当技術分野において公知のように実施することができる。別の実施例において、細胞を、単一細胞バンクバイアルから解凍し、振とうフラスコおよびバイオリアクター内に、生産規模が達成されるまで、徐々により大きな容積に拡大する。生産培養が完了次第、細胞培養液を遠心分離および深層濾過により浄化し、収穫保持タンクに移送する。生産培養期間はおよそ10日である。
【0071】
5.3.1.1.接種材料の調製
単一細胞バンクバイアルを解凍することによって生産バッチを開始する。細胞を、既知組成培地である無タンパク質基本培地−2(PFBM−2)を含有するTフラスコに移す。NaCl、フェノールレッド、トランスフェリンおよびインスリンを含まず、再構成した場合に、5mg/Lの濃度を得る量のEDTA鉄(III)ナトリウム塩を含み、再構成した場合に、それらの濃度が再構成されたHybridoma−SFMと同じであるように調整された残りの成分量を有するHybridoma−SFM培地粉末を要求することにより、PFBM−2を作るためのカスタム粉末をInvitrogenから注文することができる。調製されたPFBM−2培地は、以下の成分を含有する:8g/Lのカスタム粉末、2.45g/Lの重炭酸ナトリウム、3.15g/LのNaClおよび16.5g/LのD−グルコース一水和物(15g/Lグルコース)。
【0072】
細胞を、その後2日ごとに、ローラーボトルまたはスピナーフラスコ内で連続継代することによって拡大する。T−フラスコ、ローラーボトルおよびスピナーフラスコは設定温度37℃において、T−フラスコおよびローラーボトルに関しては7.5%CO2およびスピナーフラスコに関しては5%CO2の雰囲気下で動作するインキュベーターに配置する。
【0073】
スピナーフラスコに、細胞培養液の容積に依存して、ヘッドスペースに重ねること、または培養液内への散布のいずれかによって、5%CO2を添加し、インペラ速度は、一定の回転数/分(RPM)に調節される。全ての接種材料の拡大継代における標的シーディング密度は、およそ2.5×l05生存細胞/mLである。
【0074】
さらに、接種材料の調製を、当技術分野において公知の方法に従って、さまざまな標準的培養槽、容積および条件を使用して、実施することができる。例えば、生産バッチは、単一細胞バンクバイアルを解凍することによって開始できる。細胞は、既知組成培地である無タンパク質基本培地2(Protein Free Basal Medium−2)(PFBM−2)を含有する振とうフラスコに移すことができる。NaCl、フェノールレッド、トランスフェリンおよびインスリンを含まず、再構成した場合に、5mg/Lの濃度を得る量のEDTA鉄(III)ナトリウム塩を含み、再構成した場合に、それらの濃度が再構成されたHybridoma−SFMと同じであるように調整された残りの成分量を有するHybridoma−SFM培地粉末を要求することにより、PFBM−2を作るためのカスタム粉末をInvitrogenから注文することができる。調製されたPFBM−2培地は以下の成分を含有する:8g/Lのカスタム粉末、2.45g/Lの重炭酸ナトリウム、3.15g/LのNaClおよび16.5g/LのD−グルコース一水和物(15g/Lグルコース)。場合により、バイオリアクター段階において、例えば、0.04mg/Lの濃度で硫酸銅七水和物を加えてもよい。
【0075】
細胞を、その後2日ごとに、振とうフラスコ内で連続継代することによって拡大する。振とうフラスコは、設定温度37℃において、7.5%CO2の雰囲気下で動作するインキュベーターに配置する。
【0076】
振とうフラスコは、インキュベーター内の振とうプラットホームにおいて、一定の回転数/分(RPM)でかき混ぜられる。全ての接種材料の拡大継代における標的シーディング密度は、およそ2.2−2.5×l05生存細胞/mLである。
【0077】
細胞バンクの解凍後およそ14日、十分な数の生存細胞が生産された時、最初のいくつか、通常3または4のステンレス製撹拌タンクのシードバイオリアクターに接種する。使用前に、これらのシードバイオリアクターは、定置洗浄され、定置蒸気滅菌され、適切な容積のPFBM−2培養培地をロードされる。pHプローブおよび溶存酸素プローブは、バイオリアクターが定置蒸気滅菌される前に較正する。最初のシードバイオリアクターに、2.0−2.5×105の生存細胞/mLの初期細胞密度を標的とするための十分な数の細胞を接種する。より大きな容積(通常、100Lから300Lおよびその後1,000Lのシードバイオリアクターまたは60Lから235L、950Lおよび3750Lのシードバイオリアクター)への連続移送を、各リアクターにおける増殖のおよそ2日後および標的初期細胞密度2.0−2.5×105生存細胞/mLに実施する。培養のpHは、自動制御を介してCO2ガスまたは1Mの炭酸ナトリウム(Na2CO3)の添加により維持される。シードバイオリアクターおよび生産バイオリアクターの標的動作条件は、設定温度37℃、pH7.0および(空気飽和の百分率として)30%の溶存酸素を含む。100L、300Lおよび1,000Lのバイオリアクターは、それぞれ100rpm、80rpmおよび70rpmでかき混ぜられる。いくつかの場合、シードバイオリアクターおよび生産バイオリアクターの標的動作条件は、設定温度37℃、CO2散布および塩基の添加による調節を用いた7.0のpHおよび(空気飽和の百分率として)30%の溶存酸素を含む。より大きな容積のバイオリアクターは、100rpm、80rpm、70rpmまたは40rpmの速度でかき混ぜることができる。
【0078】
5.3.2.細胞培養液生産バイオリアクター
1,000Lのシードバイオリアクターにおいておよそ2日後、接種材料をステンレス製撹拌タンクの生産バイオリアクターに移送する。生産バイオリアクターは、およそ10,000Lの作業容積を有する。使用前に、バイオリアクターは、定置洗浄され、定置蒸気滅菌され、およそ4,000LのPFBM−2培地をロードされる。pHプローブおよび溶存酸素プローブは、バイオリアクターが定置蒸気滅菌される前に較正する。
【0079】
別の実施例において、接種材料は、3750Lのシードバイオリアクターにおいて増殖され、その後、およそ15,000Lの作業容積を有するステンレス製撹拌タンクの生産バイオリアクターへ移送され、このステンレス製撹拌タンク生産バイオリアクターは、使用前に、定置洗浄され、定置蒸気滅菌され、およそ4,000−7,000LのPFBM−2培地をロードされる。
【0080】
生産バイオリアクターの標的シーディング密度は、2.0−2.5×105生存細胞/mLの範囲である。既知組成の無タンパク質流加培地濃度(PFFM−3)(PFFM3小成分1および2、L−グルタミン、D−グルコース、リン酸ナトリウム二塩基性七水和物、L−チロシン、葉酸、塩酸および水酸化ナトリウムを再構成することによって作られた、既知組成濃縮流加培地)を、培養の間に加える。PFFM3は、表4に示す成分を含有する。
【0081】
【表4】
【0082】
PFFM3 小成分1は、以下の表5に示す成分を含有する。
【0083】
【表5】
【0084】
PFFM3小成分2は以下の表6に示す成分を含有する。
【0085】
【表6】
【0086】
培養液へのPFFM−3の添加のタイミングおよび量は、以下の表7に示すように行う。
【0087】
【表7】
【0088】
培養のpHは、CO2ガスおよび1Mの炭酸ナトリウム(Na2CO3)の添加の自動制御により、およそpH7.0、好ましくはpH7.0およびpH7.1の間に維持される。溶存酸素含有量は、空気飽和のおよそ30%まで下がってもよい。酸素/空気混合物を、培養液中に散布し、一定の合計ガスの流速を達成し、溶存酸素は、必要に応じて空気対酸素ガスの比率を調整することによって、および最大の酸素対空気の比率に達した後のかき混ぜ速度を増加することによって調節される。別の実施例において、かき混ぜは、力/容積の一定比率を維持するように調整される。シメチコン系消泡剤エマルジョンを、発泡レベルに基づき、必要に応じてバイオリアクターに加える。試料を定期的に採取し、細胞密度、細胞の生存能力、生産物濃度、グルコースおよび乳酸塩濃度、溶存O2、溶存CO2、pHおよび浸透圧に関して試験する。バイオリアクター培養液は、接種後およそ10日で収穫される。収穫前に、バイオリアクターの内容物を未処理バルクとしてサンプリングする。
【0089】
5.3.3. 収穫および細胞除去
収穫の直前に、生産バイオリアクターをまず<15℃に冷やし、pHを、0.5Mまたは1Mまたは2Mのクエン酸を使用して5.0±0.1に調整し、およそ30−90または45−60分の期間そのまま保持し、収穫槽に移送する前に細胞および細胞残渣を凝集させる。pHを調整した収穫は、その後、バッチ記録書類に定義のボウル速度および流速に関する所定のパラメーターの下で動作された連続遠心分離によって浄化される。
【0090】
遠心分離液を、デプスフィルターを介してろ過し、その後0.22μmのメンブレンフィルターを介してろ過し、あらかじめ滅菌されたタンクに回収する。無細胞収穫を、1−2MのTris溶液を使用しておよそ6.4のpHに調整し、さらなる処理のために2−8℃で貯蔵する。いくつかの場合、このpH調整は、pH5.0への元のバイオリアクターのpH調整の12時間以内に行う。
【0091】
5.4.DAC HYPの精製
5.4.1.概説
DAC HYPの精製および製剤化方法は、ZENAPAX生産方法と比較して有効性を改良し、生産物関連不純物およびプロセス関連不純物の一貫したクリアランスを確実にするように設計された。以下のサブセクションは精製方法を記載する。精製は、低pHウイルス不活性化、ウイルスのろ過、限外濾過/ダイアフィルトレーションおよび製剤化ステップと組み合わせた、3種のクロマトグラフィー技術に基づく(プロテインA親和性クロマトグラフィー、Qセファロース陰イオン交換クロマトグラフィーおよびCM−650(M)陽イオン交換クロマトグラフィー)。すべてのステップは、閉鎖型装置において行われる。DAC HYPのための精製方法の概要を、図5に提示し、以下に記載する。
【0092】
5.4.2.プロテインAクロマトグラフィー
プロテインA親和性クロマトグラフィーステップは、一連の下流動作において第1の精製ステップである。このステップは、カラムのサイズに依存して1回または複数回のサイクル、通常、表8Aに記載のカラムに関して2回または3回のサイクルで行われる(すなわち、無細胞の収穫を2つのアリコートに分割し、その後各アリコートをプロテインAカラムに別々にロードし、溶出する)。組換えタンパク質A親和性クロマトグラフィー樹脂はIgGに特異的に結合し、細胞培養液の収穫の他の成分から抗体を分離する。
【0093】
平衡化緩衝液を用いたプロテインAカラムの平衡化の後、カラム樹脂に抗体を結合させるために、中和された、無細胞の収穫をカラムに通過させる。平衡化緩衝液は20mMのクエン酸ナトリウム、150mMの塩化ナトリウム、pH7.0である。カラムに、35グラムの抗体(タンパク質)/充填された樹脂のリットルを超えない容量をロードする。ロード後、平衡化緩衝液を用いてカラムを洗浄し、未結合および緩く結合した不純物を樹脂から除去し、クエン酸塩緩衝液を用いて溶出前の洗浄を行い、カラムのクエン酸塩濃度および塩化ナトリウム濃度を調整する。クエン酸塩緩衝液は、10mMのクエン酸ナトリウムpH7.0である。次いで、結合した抗体を、pH3.5の10mMのクエン酸ナトリウムの溶出緩衝液を使用するpHの変更ステップにより、カラムから溶出する。プロテインAクロマトグラフィーの条件の要約を、表8Aに述べる。
【0094】
【表8】
【0095】
生産物溶出液がカラムから離れたら、流出物の吸光度を波長280nmにおいてモニターし、生産物分画の回収のガイドに使用した(図6を参照されたい)。
【0096】
水酸化ナトリウムおよびベンジルアルコールを含有する衛生化緩衝液の使用は、広範囲の微生物を有利に殺すが、プロテインA樹脂の品質に与える影響が最小である。このことを例示するために、さまざまな衛生化溶液をさまざまな微生物にスパイクし、一定期間にわたってインキュベートした。インキュベーション時間の異なる間隔において、スパイクされた衛生化溶液の一部は中和され、微生物の力価を測定し、対照と比較した。殺菌作用は、一定期間にわたる微生物のlog減少として表す。表8Bは、衛生化緩衝液20mMの水酸化ナトリウム、200mMのクエン酸ナトリウムおよび1%のベンジルアルコールとの接触時間の関数としての、微生物力価の減少を示す。
【0097】
【表9】
【0098】
表8Cは、異なる衛生化溶液による微生物の減少を示す。
【0099】
【表10】
【0100】
前述のデータは、ベンジルアルコールおよび水酸化ナトリウムを含有する衛生化溶液が、グラム陰性細菌およびグラム陽性細菌、芽胞形成細菌、酵母ならびに真菌を含む、広範囲の微生物を殺すために非常に有効であることを示す。通常の衛生化の30分後、E.コリ、スタフィロコッカス・アウレウス、シュードモナス・アルギノーザおよびカンジダ・アルビカンスに関して5log10を超える減少が観察された。真菌(A.ニガー)を殺すためにはさらに時間がかかるが、細胞培養液において真菌の感染は稀である。バイオテクノロジー設備において単離される最も一般的な微生物は、バチルス、シュードモナスおよびスタフィロコッカスである。これらは、衛生化溶液によって、接触時間の30分後に有効に殺される。比較すると、水酸化ナトリウムまたはベンジルアルコール単独では、すべての微生物を殺すためには有効ではない。さらに、水酸化ナトリウム衛生化溶液は、芽胞形成バチルスは殺さない。
【0101】
5.4.3.ウイルス不活性化のための低pHの保持
このステップは、低pH感受性の内因性ウイルス様粒子およびウイルスを不活性化するように設計されている。各プロテインAサイクルからのプロテインA溶出液は、回収タンク内に溶出され、ここに、0.5MのHClをpH3.5±0.1に達するまで加える。生産物は保持タンクに移送され、ここで別のpHメーターによってpHが確認される。低pH保持ステップは、pH3.5±0.1または±0.2(例えば、pH3.35−3.64)に、30−120分または30−240分の間厳密に制御される。30−120分の保持後、ウイルス不活性化溶出液を、1MのTris塩基を使用して、pH7.8±0.1または±0.3(例えば、pH8.05−8.34)に中和し、その後、0.22μmのフィルターを介して生産物プールタンクに移送する。低pHウイルス不活性化条件の要約を、表9に述べる。
【0102】
【表11】
【0103】
5.4.4.Qセファロース陰イオン交換フロースルークロマトグラフィー
Qセファロース陰イオン交換クロマトグラフィーステップを使用し、生産物関連不純物およびプロセス関連不純物(例えば、核酸、宿主細胞タンパク質、生産物アグリゲート、浸出されたプロテインAリガンドなど)を減少させ、さらなるウイルスクリアランス能力を精製方法に提供する。ロードの電導率およびpHを、抗体がカラムをフロースルーし宿主細胞タンパク質および細胞DNAなどの負に帯電した不純物が正に帯電した樹脂と結合するような様式で選択する。
【0104】
陰イオン交換カラムを、20mMのTris、20mMの塩化ナトリウム、pH7.8の平衡化緩衝液を用いて平衡化する。低pH保持ステップによるpHが調製された生産物をカラムに、抗体(タンパク質)の60グラムを超えない容量、または抗体(タンパク質)の30−60グラム/充填された樹脂のリットルを超えない容量までロードする。ローディングの完了後、未結合の抗体および不純物を、平衡化緩衝液を用いてカラムから除去する。
【0105】
生産物の回収を、波長280nmにおいて流出液の吸光度をモニターすることによってガイドする(図7を参照されたい)。
【0106】
衛生化流速は、100cm/時であり保持時間は60分である。
【0107】
Q−セファロースクロマトグラフィーの条件の要約を表10に述べる。
【0108】
【表12】
【0109】
いくつかの使用に関して、貯蔵緩衝液容積は3に設定し、カラム貯蔵温度は5℃−25℃に設定する。
【0110】
5.4.5.CM−650(M)陽イオン交換クロマトグラフィー
このクロマトグラフィーステップは、微量レベルのプロセス関連不純物および生産物関連不純物を減少させるために、方法において使用する最終ステップである。抗体のアグリゲートおよび切断断片に加えて、このステップは、宿主細胞の核酸およびタンパク質などのプロセス関連不純物ならびに浸出したプロテインAもまた減少させる。
【0111】
カラムは、20mMのクエン酸ナトリウム、pH4.5の平衡化緩衝液を用いて平衡化される。陰イオン交換溶出液のプールを、0.5Mのクエン酸を使用して4.5±0.1または±0.2(例えば、4.35−4.64)のpHに調整し、25または30グラムの抗体(タンパク質)/充填樹脂のリットルを超えない標的ローディング容量までカラムにロードする。結合ステップ後、カラムを平衡化緩衝液で洗浄し、任意の未結合または緩く結合した不純物を樹脂から除去する。その後、結合した抗体を、20mMクエン酸ナトリウム、75mM硫酸ナトリウム、pH4.5の溶出緩衝液を用いて溶出様式のステップにおいてカラムから溶出する。ピークの回収を、波長280nmにおいて流出液の吸光度をモニターすることによってガイドする(図8を参照されたい)。
【0112】
CM−650(M)クロマトグラフィーの条件の要約を表11に述べる。
【0113】
【表13】
【0114】
5.4.6.ナノ濾過
ナノ濾過ステップの目的は、精製方法に追加のウイルスクリアランス能力を提供することである。このステップにおけるウイルスおよびウイルス様粒子の除去は、サイズ排除機序を介して行われる。フィルターの細孔は、抗体が細孔を通過するが、ウイルス様粒子およびウイルスはフィルターの上流側に保有されるように設計される。
【0115】
0.22μmまたは0.1μmのフィルターを介して濾過された陽イオン交換溶出液は、小型のウイルス保有ナノフィルターを通過し、その後ポリソルベート80を含まないDAC HYP製剤化緩衝液(40mMのコハク酸塩、100mMの塩化ナトリウム、pH6.0)を用いたフィルターフラッシュが続く。緩衝液フラッシュステップを適用し、ラインおよびフィルターのハウジングに残存する抗体を回収する。
【0116】
ナノ濾過のパラメーターの要約を、表12に述べる。
【0117】
【表14】
【0118】
5.4.7.限外濾過/ダイアフィルトレーション(UF/DF)
この方法ステップは、生産物を濃縮し、生産物中の緩衝液を、ポリソルベート80を含まないDAC HYP製剤化緩衝液と交換するように設計されている。この方法ステップは、30kDaの名目分画分子量のメンブレンを使用するタンジェンシャルフロー様式で動作する。最終濃度の期待生産物容量および各UF系の相対的保持容積のため、2つの限外濾過/ダイアフィルトレーション段階を使用し、150mg/mLの製剤を生産する。
【0119】
第1の段階は、大型のUF系を使用して処理される(図9を参照されたい)。ナノ濾過のろ液を、まずおよそ30mg/mLまで濃縮し、その後、交換緩衝液(ポリソルベート80を含まない製剤化緩衝液)にダイアフィルトレーションする。ダイアフィルトレーションされた抗体溶液を、およそ100mg/mLまでさらに濃縮し、その後、UF/DF系からおよそ55mg/mLの濃度で回収し、0.22μmのフィルターを介して移送する。ダイアフィルトレーションされた抗体溶液は、およそ20−60mg/mLの濃度で回収することもできる。第1段階のパラメーターの要約を、表13に述べる。
【0120】
【表15】
【0121】
第2段階は、より小さいUF系であるが、同じ30kDaのカットオフのメンブレンを使用して処理される。DAC HYP(通常55mg/mL)溶液を、およそ180mg/mLに濃縮し、UF系から回収し、その後0.22μmのフィルターを介して移送する。UF系を、ポリソルベート80を含まない製剤化緩衝液を用いてすすぎ、0.22μmのフィルターを介して移送し、およそ170mg/mLまたはおよそ150−170mg/mLの精製された製剤原料を得る。第2段階のパラメーターの要約を、表14に述べる。
【0122】
【表16】
【0123】
5.5.DAC HYPの製剤化
最終方法ステップは、精製された製剤原料の150または100mg/L±10%の最終標的濃度、すなわち、適切な濃度のポリソルベート80を含有する緩衝液中、150±15mg/mL(150mg/mLの製剤化の場合)または100±10mg/mL(100mg/mLの製剤化の場合)の最終標的濃度への希釈である。製剤化は、段階的に実施される。
【0124】
例えば、まず、ポリソルベート80を含まない製剤化緩衝液(40mMのコハク酸ナトリウム、100mMの塩化ナトリウム、pH6.0)を、製剤化された製剤原料の90%標的容積に達するように、精製された製剤原料に加える。その後、計算された量のポリソルベート80希釈緩衝液(40mMのコハク酸塩、100mMの塩化ナトリウム、1%ポリソルベート80、pH6.0)を、最終製剤中0.03%(w/v)のポリソルベート80の標的濃度に達するように加える。最終的に、ポリソルベート80を含まない製剤化緩衝液(150mg/mLの製剤のためにコハク酸塩およびコハク酸ならびに100mg/mLの製剤のためにコハク酸塩およびHClで作られる)を使用して生産物容積を調整し、150±15mg/mL(好ましくは150±8mg/mL)の最終抗体濃度を達成する。100mg/mLの薬剤生産物は、同様の様式で、最終濃度100±10mg/mL(好ましくは100±5mg/mL)に製剤化される。
【0125】
製剤化された製剤原料を、0.22μmのフィルターを介して、半剛性の円筒状の支持体内部に配置されたBioProcess Container TM(BPC(登録商標))のバッグ(または同等物)内に濾過する。支持体は、BPCと蓋とを包囲し、柔軟性のバッグおよび外部環境の間の防護壁を提供する。製剤化された製剤原料は、薬剤生産物の充填/仕上げ操作のために、アクセス制御クーラー中で2−8℃において貯蔵される。
【0126】
製剤化条件の要約を表15に述べる。
【0127】
【表17】
【0128】
1mLの薬剤生産物を、バイアルまたは注射器に充填する。仕上がった150mg/mLおよび100mg/mL生産物の成分の要約を、表16に示す(すべての量は名目値である。)。
【0129】
【表18】
【0130】
5.6.DAC HYP製剤原料の特徴付け
DAC HYPを、両方の重鎖サブユニットのアミノ酸296においてグリコシル化し、主要なオリゴ糖形態は、末端のガラクトースを欠いた、コアがフコシル化された二分岐構造として存在する。
【0131】
DAC HYP重鎖のN末端は、3つの主要な形態:1)(DNA配列から予測される)N−末端グルタミン、2)N−末端ピログルタミン酸(N−末端グルタミンの環化による)および3)(シグナルペプチドの不完全な切断によりもたらされる)予測されるN末端グルタミン残基に加えてN−末端バリン、ヒスチジンおよびセリン残基として存在する。
【0132】
DAC HYP重鎖のC末端は、C末端リジン残基を含んで、または含まずに存在する。主要形態はC末端リジン残基を欠き、C末端グリシンをもたらす。
【0133】
DAC HYPは、ヌクレオチド配列により定義された一次アミノ酸組成に基づき、分子量144kDaと計算された。縮小された重鎖の対応する分子量は48.9kDaであり、縮小された軽鎖は23.2kDaであり、これらの重量は、炭水化物含有量または翻訳後修飾を占めない。
【0134】
DAC HYPの結合はCD25に高度に特異的であり、CD25は活性化されたTリンパ球およびBリンパ球に発現されるが、静止Tリンパ球および静止Bリンパ球には発現されない。これらの活性化細胞におけるDAC HYPとCD25との結合は、IL−2とCD25との結合およびその後の高親和性IL−2受容体複合体の形成を遮断する。その結果として、活性化細胞のIL−2誘導性増殖が遮断される。DAC HYPの観察された治療有効性および潜在的治療有効性は、活性化された自己反応性T細胞の増殖についてのその阻害効果の大部分を静止すると考えられる。しかし、DAC HYPはまた、好酸球などの他のCD25産生細胞型に関してその遮断効果を介して治療効果を発揮すると思われる。
【0135】
高濃度の150mg/ml DAC HYP製剤が臨床試験に適していることを確認するために、包括的な物理化学的および生物学的評価を特徴付けのために実施し、本明細書においてバッチ1およびバッチ2(またはそれぞれ、バッチ150−1もしくはバッチ150−2)と称される、DAC HYP 150mg/mL製剤原料の2種のバッチを、10,000L規模で製造されたDAC HYP 100mg/mLの1種のロットである参照標準ロットRS0801と比較した。
【0136】
結果は、DAC HYP薬剤生産物の150mg/mLロットは純度が高く、100mg/mLロットに匹敵し、臨床研究における使用に適していることを実証している。これらの特徴の要約を表17に示す。
【0137】
【表19】
【0138】
5.6.1.色、外観および透明度
DAC HYP製剤原料の外観は、色の視覚的検査ならびに拡大なしの黒色背景および白色背景に対する直射光における溶液の透明度によって査定する。溶液は、目に見える粒子の存在に関してもまた評価する。さまざまなロットのDAC HYP薬剤生産物の通常の外観を表17に記載する。
【0139】
5.6.2.pH決定
DAC HYPのpHは、U.S.Pharmacopeia Protocol No.(791)に従って決定する。DAC HYP薬剤生産物のさまざまなロットのpH範囲を、表17に要約する。
【0140】
5.6.3.UV分光器による生産物濃度
DAC HYPの濃度は、UV分光器により決定する。DAC HYP試料を、緩衝液を用いて重量測定法で希釈する。各希釈試料溶液のUV吸光度を、緩衝液ブランクに対して278nmにおいて測定する。試料のタンパク質濃度を、DAC HYPに関する吸光係数を使用して計算する。DAC HYP薬剤生産物のさまざまなロットのタンパク質濃度を、表17に要約する。
【0141】
5.6.4.N末端配列決定
DAC HYP150mg/mLロットを、N末端配列決定によって評価した。試料を、自動Edman分解配列決定器を使用して分析した。
【0142】
最初の15残基DIQMTQSPSTLSASV(配列番号13)を介して、軽鎖の予想アミノ酸配列を、すべての試料に関して確認した。
【0143】
DAC HYP中の重鎖の大部分は、N末端重鎖配列を生じないピログルタミン酸(pE)残基によって遮断される。DAC HYP中で次に最も高頻度で見られるN末端重鎖配列は、バリン、ヒスチジン、セリン(VHS)配列で始まり、重鎖シグナルペプチドの3個の末端残基のプロセッシングを欠いたことによりもたらされる。最初の15個のN末端残基のうちの14個は、すべての試料において、VHS重鎖配列(VHSQVQLVQSGAEVK(配列番号14))が確認された。先行する配列決定サイクルにおいてLCから検出された大量のグルタミンのため、4番目の残基であるグルタミンは確認できなかった。N末端グルタミンを有する重鎖の証拠もまた、すべての試料中に存在した。この配列は、天然のN末端重鎖グルタミン残基が、ピログルタミン酸形態への環化を受けなかった結果である。150mg/mLロットに関するN末端配列決定の結果は、重鎖および軽鎖のコーディング配列から予測された配列と一致する。同様の結果を、100mg/mLロットに関して得た。
【0144】
5.6.5.重鎖および軽鎖の質量分析
DAC HYP150mg/mLロットの重鎖および軽鎖ならびに参照標準RS0801の分子量を液体クロマトグラフィー質量分析(LC−MS)によって評価した。すべてのロットを、アミダーゼPNGaseFを用いて脱グリコシル化させ、ジチオスレイトールを用いて還元し、ヨード酢酸を用いてアルキル化し、逆相クロマトグラフィーにより分離した。理論的な重鎖および軽鎖の質量を、タンパク質配列から計算した。試料の観察された質量は、以下の表18に示すように計算質量の1Da以内であった。
【0145】
【表20】
【0146】
先行するサブセクションに記載のように、DAC HYP重鎖の2種の最も高頻度で見られる形態は、N末端ピログルタミン酸(pE)残基またはバリン、ヒスチジン、セリン(VHS)配列を含有し、C末端リジンを欠くことが公知である。150mg/mLロット中の2種の優勢な重鎖変異体および軽鎖に関して得られた分子量は、参照標準RS0801の分子量と同様であり、タンパク質配列から予測された質量と一致した。
【0147】
以下のサブセクションに提示したペプチドマッピングの結果と一緒に、重鎖および軽鎖の質量結果により、DAC HYP150mg/mLロット中の予想される軽鎖および重鎖の一次構造の存在を確認する。
【0148】
5.6.6.ペプチドマッピング
DAC HYP150mg/mLロットおよび参照標準RS0801(10,000L規模で製造された100mg/mLの製剤原料のロットから生産されたDAC HYP)を、逆相HPLCペプチドマッピングを使用して評価した。すべてのロットを、ジチオスレイトールを用いて還元し、ヨード酢酸を用いてアルキル化し、トリプシンを用いて酵素的に消化した。得られたペプチドを、逆相クロマトグラフィーにより分離し、215nmの紫外線吸光度によって検出し、ペプチドマップを作製した。
【0149】
一次アミノ酸配列を検証するために、150mg/mLロットのペプチドマップを、参照標準RS0801のペプチドマップと比較した。予想される重鎖および軽鎖の残基と98%対応するペプチドが、参照標準RS0801のペプチドマップにおける質量分析によってすでに特定されている。ペプチドマップに計上されない残基は、単一のアミノ酸である、または非常に極性のジペプチド中に存在し、逆相カラムに保有されることが予想されない。ピログルタミン酸、グルタミンおよび重鎖N末端ペプチドのN末端におけるVHS配列に一致する質量が、参照標準中に特定された。DAC HYPは、Asn296における重鎖のFc部分中のN連結グリコシル化に関するコンセンサス部位を含有し、連結複合体のコアが二分岐のオリゴ糖構造と一致する質量が、Asn296残基を含有するペプチドに関して特定された。
【0150】
DAC HYP150mg/mLロットと参照標準RS0801とを比較するペプチドマップを、図10(0から60分)、図11(55から115分)および図12(110から170分)に示す。DAC HYP150mg/mLロットのペプチドマップは、参照標準のペプチドマップと同様であり、150mg/mLロットにおける予想一次構造の存在が確認される。
【0151】
5.6.7.円偏光二色性分光法
DAC HYP150mg/mLロットおよび参照標準RS0801を、遠紫外円偏光二色性分光法(遠−UV CD)により分析し、二次構造を評価した。分析前に、試料を、最終タンパク質濃度0.2mg/mLに水を用いて希釈した。スペクトルを、0.1cmのセルを使用して195−260nmから入手し、得られたシグナルを、緩衝液を差し引いた後でモル楕円率に変換した。
【0152】
DAC HYP150mg/mLロットのバッチ1およびバッチ2ならびに参照標準RS0801の重ね合わせた遠−UV CDスペクトルを、図13に示す。すべてのロットのスペクトルは、およそ202nmにおいて正のバンドを示し、およそ217nmにおいて負のバンドを示す。217nmにおける負のバンドは、βシート立体構造の特徴であり、IgG分子の優勢な立体構造である。ロットの遠−UV CDスペクトルは同様であり、DAC HYP150mg/mLロットおよび参照標準RS0801の中の均一な二次構造を支持した。
【0153】
5.6.8.紫外分光法
DAC HYP150mg/mLロットおよび参照標準RS0801を、紫外(UV)分光法により分析し、三次構造を評価した。分析前に、試料を製剤化緩衝液(40mMのコハク酸塩、100mMの塩化ナトリウム、0.03%のポリソルベート80、pH6.0)で最終タンパク質濃度0.5mg/mlに希釈した。スペクトルを、1cmの路長の石英キュベットを使用して250から350nmから入手し、280nmにおける吸光度を1.0とし正規化した。
【0154】
重ね合わせたゼロ次および二次微分のUVスペクトル(平滑化したゼロ次のデータから計算)を、それぞれ図14Aおよび14Bに示す。評価したすべてのロットのゼロ次および二次微分スペクトルは、重ねることができ、DAC HYP150mg/mLロットのバッチ1およびバッチ2ならびに参照標準RS0801の中の均一な三次構造を支持した。
【0155】
5.6.9.サイズ排除クロマトグラフィー
サイズ排除クロマトグラフィー(SEC)は、水系移動相を有する多孔質シリカカラムおよび280nmの紫外吸光度を検出を用いて実施した。具体的には、15μLの試験試料(溶出緩衝液中20mg/mLの抗体)を、室温において、0.5μmプレカラムフィルター(Upchurch、part no.A−102X)を装備した7.8mm×30cmのTSK G3000SWXLカラム(Tosoh Biosciences、part no.601342)において、溶出緩衝液(200mMのKPO4、150mMのKCl、pH6.9)の均一濃度の勾配を使用して、1mL/分の流速で分析した。
【0156】
図15A(フルスケール)および図15B(拡大スケール)に示すように、DAC HYP150mg/mLロットのクロマトグラフィープロファイルは、参照標準RS0801のクロマトグラフィープロファイルと同様であった。すべてのロットは、ダクリズマブ単量体に一致する同じ主要なピークおよび小さなアグリゲートピークを示した。DAC HYP150mg/mLロットに関するアグリゲート結果は、以下の表19に示すように、DAC HYP100mg/mLロットのアグリゲート結果と同様であった。
【0157】
【表21】
【0158】
150mg/mLロットおよび参照標準RS0801を、多角度光散乱検出(SEC−MALS)を有するSECを使用して分析し、アグリゲートピークの分子量を決定した。すべてのロットに関して、アグリゲートピークに関して得られた分子量はおよそ300kDaであり、これは抗体二量体に一致する。
【0159】
DAC HYP中のアグリゲートの形成を、18か月の間にわたってモニターした。製剤中のアグリゲートのレベルは上昇したが、アグリゲートの百分率は横這いであり、5℃において18か月貯蔵した場合、およそ1.5%を超えなかった(図6を参照されたい)。新しいバッチの新しいデータは、5℃において貯蔵した場合、6週間でおよそ1.8−1.9%のアグリゲートが出現し得るが、試験したすべての試料に関しては、5℃において貯蔵した場合、5年間にわたって形成されたアグリゲートは3%未満であったことを示す。
【0160】
5.6.10.沈降速度超遠心分析法
DAC HYP150mg/mLおよび100mg/mLのロット中の単量体およびアグリゲートを、沈降速度超遠心分析法(SV−AUC)を使用して特徴付けた。単量体および個々のアグリゲートに関する沈降係数値および相対的存在度を、以下の表20および21に提示する。
【0161】
【表22】
【0162】
【表23】
【0163】
単量体は、個々のロットにおいて観察される主要な成分であった。単量体ピークの沈降係数は、単量体の立体構造が、150mg/mLおよび100mg/mLのロットの間で同様であることを示すロットと高度に一致した。150mg/mLロットの単量体の含有量は、100mg/mLのロットの単量体の含有量と同様であった。
【0164】
個々のロット中の優勢なアグリゲート種は、抗体の二量体と一致する沈降係数を有した。これはSEC−MALSの結果と一致し、SECアグリゲートピークが、主に抗体二量体から構成されることを示す(先行するサブセクションを参照されたい)。三量体および四量体と一致する沈降係数を有する、低レベルの2種のより大きなアグリゲートもまた、AUCにより個々のロットにおいて観察された。150mg/mLロットの二量体、三量体および四量体の含有量は、100mg/mLロットの二量体、三量体および四量体の含有量と同様であった。
【0165】
5.6.11.定量的還元型SDS−PAGE
純度を、4−20%(通常14%)のtris−グリシンゲルおよびコロイダルブルー染色を使用して、SDS−PAGEによって決定した。試料を、還元条件下で10μgの試料ロードを用いて分析した。純度を、密度測定によって測定された、重鎖および軽鎖のバンド面積の合計を全バンド面積で割ることによって計算した。
【0166】
以下の表22に示すように、150mg/mLロットは高純度であり、100mg/mLロットと同様である。
【0167】
【表24】
【0168】
5.6.12.定量的SDS−PAGE
DAC HYPの純度を、還元型および非還元型のゲル電気泳動の両方によって査定した。プレキャストの14%または8−16%のTris−グリシンゲルを分析に使用した。150mg/mL DAC HYP製剤の2種のバッチ由来のアリコートを、前述のように参照バッチと比較した。DAC HYPの純度を分析する還元型および非還元型ゲルを、図17に示す。150mg/mLロットのバンドパターンは、参照標準RS0801のバンドパターンと同様であり、150mg/mLロットに新しいバンドは検出されなかった。
【0169】
5.6.13.陽イオン交換クロマトグラフィー
DAC HYP150mg/mLロットおよび100mg/mLロットの荷電アイソフォームの分布を、陽イオン交換クロマトグラフィー(CEX)を使用して評価した。CEXを、非多孔質のカルボン酸塩官能化の、弱陽イオン交換カラムを使用して実施し、220nmにおいて検出した。100μLの試験試料(緩衝液Aに溶解した1mg/mLの抗体)を、室温で、ProPac WCX−10Gガードカラム(Dionex Corporation)を装備したProPac WCX−10カラム(Dionex Coporation)において以下の分離勾配を使用して(カラムは緩衝液Aを用いて平衡化される)分離した。
【0170】
【表25】
【0171】
図18に示すように、150mg/mLロットのCEXクロマトグラムは、参照標準RS0801のCEXクロマトグラムと一致し、150mg/mLロットにおいて新しい荷電アイソフォームは検出されなかった。CEXクロマトグラム中に存在する5種の主要なアイソフォームは、重鎖N末端における不均一のためであり、1)2つのピログルタミン酸残基(pE/pE);2)1つのピログルタミン酸残基および1つのグルタミン残基(pE/Q);3)1つのピログルタミン酸残基および1つのVHS配列(成熟重鎖のN末端グルタミン残基に先行するトランケート型VHSシグナルペプチド)(pE/VHS);4)1つのグルタミン残基および1つのVHS配列(Q/VHS);5)2つのVHS配列(VHS/VHS)を含む。C末端リジン(K)のアイソフォームも分離され、図18において特定される。上記の個々のN末端アイソフォームは、異なるC末端アイソフォーム(0、1または2つのK)として存在してもよい。混合物の複雑性のため、C末端リジンアイソフォームは、記載の方法を使用してのみ、明瞭に分離され、主要なpE/pEおよびpE/VHSのアイソフォームに関して測定可能である。
【0172】
N末端およびC末端アイソフォームの定量結果を、150mg/mLおよび100mg/mLのロットに関して、それぞれ図23および24に提供し、報告される百分率は、すべてのピークの全曲線下面積(AUC)と比較した特定のピークの曲線下面積の百分率に基づく。
【0173】
【表26】
【0174】
【表27】
【0175】
5.6.14.オリゴ糖マッピング
DAC HYP150mg/mLロットおよび100mg/mLロットのオリゴ糖の分布を、オリゴ糖マッピングにより評価した。N連結オリゴ糖は、重鎖Asn296からアミダーゼPNGaseFを使用して酵素的に放出された。オリゴ糖を、続いて蛍光標識(この場合、アンスラニル酸)を用いて誘導体化し、ナイロンメンブレンを介して抗体から分離した。誘導体化され、切断されたN連結グリカンを、50℃で、250×4.6mmの高分子アミン結合Asahipak Amino NH2P−504Eカラム(粒径5μm、Phenomenex、カタログ番号CHO−2628)において蛍光検出を用いて、以下の溶出勾配(試料注入容積100μL;カラムは85%の緩衝液A/15%の緩衝液Bを用いて平衡化する)を使用して分離した。
【0176】
【表28】
【0177】
150mg/mLロットと参照標準RS0801とを比較するクロマトグラムを、図19に示す。全ピーク面積の少なくとも1.0%を構成するオリゴ糖ピークを標識し、以下の表25に報告する。
【0178】
【表29】
【0179】
すべてのロットは、主にG0およびG0−GlcNAc(二分岐構造の1つのアームにGlcNAcを欠いたG0)からなり、これはDAC HYP方法を代表する。シアル化オリゴ糖は、およそ68分で溶出し、すべての被験ロットにおいて1.0%より低い。ピーク3と称される特徴付けされていないオリゴ糖は、すべての被験ロットにおいて同様の存在度で存在した。
【0180】
5.6.15.酸化
DAC HYP ロットを、ペプチドマップ中に存在する、酸化および非酸化トリプシンペプチドをモニターすることによって、潜在的なメチオニンの酸化を評価した。ペプチドを含有する個々のメチオニンの非酸化型および酸化型のピーク面積を、質量スペクトルにより抽出されたイオンクロマトグラムを使用して決定した。個々のメチオニン残基に関して、酸化メチオニンのパーセントを、酸化ペプチドの質量スペクトルピーク面積を、酸化および非酸化ペプチドのピーク面積の合計で割ることによって計算した。
【0181】
以下の表26に示すように、150mg/mLロットおよび5種の100mg/mLロットに関するメチオニン酸化の結果は同様であった。
【0182】
【表30】
【0183】
重鎖のMet251およびMet427は最も不安定であり、最も高い酸化程度を示す。比較可能性を評価するために同時に試験されたロットの中で、Met251およびMet427に関する酸化レベルは、それぞれ4.8%および1.8%を超えなかった。
【0184】
5.6.16.結合効力(CD25の結合)
DAC HYP150mg/mLおよび100mg/mLのロットを、放出試験の一部の効力の測定として、ELISAを介してIL−2受容体のアルファサブユニット(CD25)との結合に関して評価した。マイクロタイタープレートに可溶性CD25を固定し、さまざまな量のDAC HYPと一緒にインキュベートした。結合したDAC HYPを、ホースラッディシュペルオキシダーゼコンジュゲートヤギ抗ヒトIgG抗体と、3,3’,5,5’−テトラ−メチルベンジジン基質とを並行して使用して検出した。得られた吸光度値を、4−パラメーターフィットを使用してlog10のDAC HYP濃度に対してプロットし、相対効力値のパーセントを、平行線分析を使用して作製した。
【0185】
製剤原料の結果を以下の表に要約する。
【0186】
【表31】
【0187】
150mg/mLロットの結合効力の結果は、100mg/mLロットの結合効力の結果と同様であった。
【0188】
5.6.17.表面プラズモン共鳴(CD25の結合)
表面プラズモン共鳴分析を実施し、DAC HYPとIL−2受容体のアルファサブユニット(CD25)との結合相互作用に関する親和定数(KD)を決定した。
【0189】
ヤギ抗ヒトIgG Fc抗体をチップの表面に固定してDAC HYP試料を捕捉し、その後、可溶性CD25をさまざまな濃度で、捕捉されたDAC HYPの上に自動化法を使用して2連に注入した。結合データを回収し、参照フローセルおよび緩衝液ブランクを使用して修正し、平衡定数を得るために1:1 Langmuirモデルを使用してBIA Evaluationソフトウェアを用いてフィットさせた。
【0190】
DAC HYP150mg/mLロットおよび参照標準RS0801に関する結果を、表28に要約する。
【0191】
【表32】
【0192】
150mg/mLロットの会合定数(ka)、解離定数(kd)および親和定数(KD)の値は、参照標準RS0801のものと同様であった。
【0193】
5.6.18.機能的効力
DAC HYP150mg/mLおよび100mg/mLのロットを、放出試験の一部として機能的効力を評価した。機能的効力のアッセイは、DAC HYPがIL−2受容体のアルファサブユニット(CD25)と結合することによる、IL−2誘導性T細胞増殖の阻害を測定する。IL−2の存在下で、さまざまな量のDAC HYPをIL−2受容体を発現するKIT−225K6細胞(Horiら、1987年、Blood70:1069−1072頁)と一緒にインキュベートした。DAC HYPによるT細胞増殖の阻害を、アラマーブルーを使用して続けて検出した。得られた蛍光値を、4−パラメーターフィットを使用してlog10のDAC HYP濃度に対してプロットし、相対効力値のパーセントを、平行線分析を使用して作製した。
【0194】
機能的効力の結果を表29に要約する。
【0195】
【表33】
【0196】
150mg/mLロットの機能的効力の結果は100mg/mLロットの機能的効力と同様であった。
【0197】
5.6.19.抗体依存性細胞傷害性
DAC HYP150mg/mL製剤の2種のロットを、参照標準RS0801の100mg/mL DAC HYPのロットと比較して評価した。
【0198】
IL−2受容体発現KIT−225K6細胞を、51Crを用いて標識し、続けてDAC HYPと一緒にインキュベートした。ヒトエフェクター細胞(PBMC)をさまざまな量で加え、エフェクター対標的細胞(KIT−225K6)の異なる比率を達成した。Fc受容体産生単球は、DAC HYP Fcドメインと相互作用し、続いて標的細胞の溶解を起こす。細胞傷害性の程度は、標的細胞由来の51Crの放出を測定することにより決定し、最大細胞溶解の百分率として表した。
【0199】
多数のドナー由来のPBMCを、各試料に利用した。各ドナーに関して、試料のADCC活性のパーセントを、細胞傷害性のパーセントに基づき、参照標準RS0801のADCC活性に対して計算した。ADCCの結果を、以下の表30に要約する。
【0200】
【表34】
【0201】
150mg/mLロット、参照標準RS0801、陽性および陰性の対照抗体ならびに抗体を含まない対照(抗体独立性細胞傷害性またはAICCのため)に関する反応曲線を、図20に示す。
【0202】
150mg/mLロットのADCC活性は、参照標準RS0801のADCC活性と同様であった。
【0203】
5.6.20.残存プロテインA
残存プロテインAはELISA法によって決定でき、標準、試料対照、プレートブランクおよび試験試料を、変性緩衝液を用いて希釈し、プロテインAを解離させるために沸騰水のバスに配置し、変性させ、ダクリズマブを沈降させた。沸騰後、標準、対照および試料を冷却し、遠心分離にかけ、ポリクローナル抗プロテインA捕捉抗体をコーティングしたマイクロタイタープレートに加えた。試料中に存在する残存プロテインAを、その後、ビオチン化抗プロテインA抗体と並行してストレプトアビジンアルカリホスファターゼおよびP−ニトロフェニルリン酸塩(PNPP)基質を使用して検出する。プレートを、分光光度プレートリーダーにおいて分析し、log−log標準曲線を作製し、これに対してプロテインAの濃度を決定する。試験試料の結果を、百万分率(ppm)単位で報告する。百万分率の結果はng/mLのプロテインAの結果を、mg/mLの試験試料の抗体濃度で割ることによって計算する。
【0204】
5.6.21.DNA含有量
マウスDNAの検出を、契約研究所において定量的ポリメラーゼ連鎖反応(Q−PCR)試験法を使用して決定する。この方法において、試料をDNA抽出に供する。その後、試料抽出液を、マウスDNAの反復成分の特異的断片を増幅するためにマウス特異的プライマーおよびプローブを使用して、Q−PCRによって分析する。DNAの増幅は検出される蛍光シグナルをもたらす。試料中のDNAを、既知の量のマウスDNAを使用して作製した標準曲線と比較することによって測定する。結果は、DNAのピコグラム/抗体のミリグラムで表す。さまざまなロットのDAC HYP薬剤生産物中の平均DNA含有量を、表17に要約する。
【0205】
5.6.22.宿主細胞タンパク質(HCP)
生産物中の残存宿主細胞タンパク質を、市販のキットを使用して定量する。NS0細胞溶解物に対する、アフィニティ精製したヤギポリクローナル抗体を、NS0 HCPの捕捉および検出の両方に使用する。HCP標準を、疑似的生産工程から無細胞収穫材料を回収することによって生産する。HCP作業標準を使用して標準曲線を作製し、HCPを含有する試料を、標準曲線の範囲を標的として段階的に希釈する。標準、試料対照および試験試料を、抗NS0 HCPポリクローナル抗体コーティングプレートに加える。その後、宿主細胞タンパク質を、ホースラッディシュペルオキシダーゼ(HRP)にコンジュゲートした抗NS0 HCPポリクローナル抗体と並行して3,3’,5,5’−テトラ−メチルベンジジン(TMB)基質を用いて検出する。次いで、プレートを、分光光度プレートリーダーにおいて分析し4つのパラメーターカーブフィットを作製し、試料中のHCPの量を定量する。
【0206】
NS0 HCP ELISAアッセイに関する結果を、百万分率(ppm)単位で報告する。百万分率の結果はng/mLのHCPの結果を、mg/mLの抗体濃度で割ることによって計算する。DAC HYP薬剤生産物のさまざまなロットの平均HCPを表17に要約する。
【0207】
5.6.23.ポリソルベート80濃度
ポリソルベート80は、有色のチオシアン酸コバルトとポリソルベート80の複合体の形成に基づく分光光度方法を使用して定量する。標準曲線を、一連のポリソルベート80標準を使用して構築する。試料中のポリソルベート80濃度を、標準曲線から決定する。DAC HYP薬剤生産物のさまざまなロットのポリソルベート濃度範囲を、表17に要約する。
【0208】
5.6.24.浸透圧
浸透圧は、蒸気圧降下浸透圧計を使用して測定する。試料分析の前に、浸透圧計を、試料の予想浸透圧を一括する浸透圧標準を使用して較正する。DAC HYP薬剤生産物のさまざまなロットの浸透圧範囲を表17に要約する。
【0209】
5.6.25.結論
実施された物理化学的および生物学的分析は、DAC HYP150mg/mLおよびDAC HYP100mg/mL製剤の総合評価を提供する。今までのところ、すべての被験ロットの物理化学的および生物学的特徴は同様である。
【0210】
全てのDAC HYPロットに関して、N末端配列決定により決定された重鎖および軽鎖の最初の15アミノ酸、ペプチドマップおよび分子量分析は、ダクリズマブの遺伝子配列と一致した。
【0211】
SEC−MALSおよびSV−AUCにより決定した、すべての被験150mg/mLおよび100mg/mLのロット中のアグリゲートのレベルおよびアグリゲート種のサイズ分布は同様であり、ゲル電気泳動により試験したそれらの純度も同様であった。
【0212】
150mg/mLロットの電荷アイソフォーム分布は100mg/mLロットの電荷アイソフォーム分布と同様であり、pE/VHS(150mg/mLロット=31%pE/VHS;100mg/mLロット=34から42%pE/VHS)およびQ/VHS(150mg/mLロット=11から12%Q/VHS;100mg/mLロット=4から9%Q/VHS)N末端アイソフォームの相対量にわずかに差があっただけである。DAC HYPの特徴は以下のとおりである。
【0213】
CEXによるN末端アイソフォーム:
pE/pE:31−46%
pE/Q:7−12%
pE/VHS:31−42%
Q/VHS:3−12%
VHS/VHS:1−17%
CEXによるC末端アイソフォーム:
0K:53−80%
1K:14−28%
2K:5−19%
DAC HYPのN連結グリカンの分布は以下の通りである。
G0−G1cNAc:7.2−14.6%
G0:80.9−88.2%
ピーク3:1.3−1.7%
G1:1.4−3.8
DAC HYPに関して測定された酸化レベルは低かった。
【0214】
ELISAおよび表面プラズモン共鳴のCD25結合実験において確認されたように、DAC HYPは生物学的に活性であり、IL−2誘導性T細胞増殖の阻害に対して機能性である。DAC HYPはまた、貯蔵における低レベルのアグリゲーションを特徴とする。
【0215】
5.7.DAC HYPの安定性
高濃度のDAC HYP製剤は貯蔵において安定である。以下の表は、150mg/mL DAC HYP製剤原料ロットに関する安定性データを提供する。
【0216】
以下の表31は、推奨条件(2−8℃)における、50mLバッグ中での貯蔵後の安定性データを提供する。以下の表32は、23−27℃における50mLバッグ中での貯蔵の加速安定性データを提供する。以下の表33は、ストレス安定性データを提供する。
【0217】
【表35】
【0218】
【表36】
【0219】
【表37】
【0220】
5.8.ダクリズマブのさまざまな形態間の比較
Hoffman−La Roche,Inc.(「Roche」)は、ZENAPAX(商標)という商品名で販売される、製造中止となっている同種移植の拒絶反応治療用ダクリズマブの静注製剤を製造した。DAC Penzbergは、PDL BioPharmaによる臨床試験に使用されるダクリズマブの100mg/mlの皮下製剤である(以下のセクション5.9.1に記載のCHOICE研究を参照されたい)。
【0221】
DAC HYP、ZENAPAX DACおよびDAC Penzbergの製剤の間の比較を、表34に示す。表において、製剤化緩衝液はDACがダイアフィルトレーションされ、最終製剤を得た緩衝液である。したがって、記された濃度は名目濃度である。
【0222】
【表38】
【0223】
DAC HYPのさまざまな特徴をZENAPAX DACおよびDAC Penzbergのさまざまな特徴と比較した。
【0224】
DAC HYP対ZENAPAX DACのグリコシル化間の比較を図21に示す。ダクリズマブの3種の形態の例示的ロットの分析を表35に述べる。
【0225】
【表39】
【0226】
DAC HYPはまた、ZENAPAX DACより有意に低レベルのマンノースグリコシル(例えば、Man5、Man6、Man7)および低レベルのシアル化グリコシルも有する(例えば図21を参照されたい)。
【0227】
抗体依存性細胞介在性細胞傷害性(ADCC)は、Fc依存性活性および抗体−標的結合の潜在的細胞傷害作用を査定するために使用できる、インビトロアッセイである。エフェクター細胞として6例の健康なドナー由来の末梢血単核細胞(PBMC)および標的細胞としてCD25発現KIT225/K6細胞系を使用して、可変のエフェクター細胞対標的細胞比のフォーマットまたは可変抗体濃度フォーマットの両方において、さまざまなダクリズマブ調製物のADCC活性をアッセイした。
【0228】
可変のエフェクター細胞対標的細胞比のフォーマットに関して、51Cr標識KIT225/K6細胞(12,500細胞/ウェル)を、1μg/mLの抗体(最終濃度)と一緒に、30分間4℃において、V底96−ウェルプレート中で100μLの容積のADCCアッセイ培地(435mLのRPMI−1640、5.0mLのL−グルタミン、50mLの熱不活性化ウシ胎児血清、500μlの1000×2−メルカプトエタノール、5.0mLのペニシリン−ストレプトマイシン(100×)、および5.0mLのHEPES(1Mストック)/500mLを含有する)と一緒にプレインキュベートし、対照細胞は、その後の抗体独立性51Cr放出の決定のために、抗体の不在下でADCCアッセイ培地においてインキュベートした。
【0229】
PBMC(エフェクター)を、ADCCアッセイ培地において別々の96ウェルポリプロピレンプレート中に段階的に希釈し、6.25×105細胞、3.13×105細胞、1.56×105細胞、7.81×l04細胞または3.91×104細胞の細胞濃度/100μLを得た。100μL/ウェルの容積のPBMC懸濁液を、51Cr標識KIT225/K6および抗体を含有するプレートに加え、50:1、25:1、12.5:1、6.25:1および3.13:1のエフェクター対標的(E:T)比を得た。さらに、100μL/ウェルの容積のADCCアッセイ培地単独(エフェクターなし)を、51Cr標識KIT225/K6+mAbに加え、51Crの自然放出を決定した。アッセイプレートを、50RCFにおいて2分間回転させ、37℃で7.5%CO2インキュベーターにおいて4時間インキュベートした。
【0230】
4時間のインキュベーション終了の30分前、8%TritonX−100の25μLの容積を、適切な対照ウェルに加え、標的細胞由来の51Crの最大放出を決定した。4時間のインキュベーションの完了において、プレートを350RCFにおいて5分間回転させ、各ウェル由来の上清の100μLの容積を、ミニチューブに移した。各ミニチューブを、シンチレーションバイアルに挿入し、Beckman Gamma5500Bカウンターまたは同等物において1分間カウントした。
【0231】
可変抗体濃度のフォーマットに関して、51Cr標識KIT225/K6細胞(12,500細胞/ウェル;標的)を、mAb(最終濃度)のさまざまな用量の抗体(5、1、0.2、0.04、0.008および0.0016μg/mL)と一緒に、30分間4℃においてV底96−ウェルプレート中で100μLの容積のADCCアッセイ培地においてプレインキュベートした。対照細胞は、その後の抗体独立性51Cr放出の決定のために、ADCCアッセイ培地単独(mAbなし)と一緒にインキュベートした。
【0232】
PBMC(エフェクター)を、別々の96ウェルポリプロピレンプレートにおいて、3.13×105細胞/100μLの濃度にADCCアッセイ培地で段階的に希釈した。PBMC懸濁液の100μL/ウェルの容積を、51Cr標識KIT225/K6+mAbを含有するプレートに加え、25:1のエフェクター対標的(E:T)比を得た。さらに、100μL/ウェルの容積のADCCアッセイ培地単独(エフェクターなし)を、51Cr標識KIT225/K6+mAbに加え、51Crの自然放出を決定した。アッセイプレートを、50RCFにおいて2分間回転させ、37℃で7.5%CO2インキュベーターにおいて4時間インキュベートした。
【0233】
4時間のインキュベーション終了の30分前、8%TritonX−100の25μLの容積を、適切な対照ウェルに加え、標的細胞由来の51Crの最大放出を決定した。4時間のインキュベーションの完了において、プレートを350RCFにおいて5分間回転させ、各ウェル由来の上清の100μLの容積を、ミニチューブに移した。各ミニチューブを、シンチレーションバイアルに挿入し、Beckman Gamma5500Bカウンターまたは同等物において1分間カウントした。
【0234】
ADCCの結果を図22A(可変のエフェクター細胞対標的細胞比のフォーマット)および図22B(可変抗体濃度フォーマット)に示す。これらのデータは、段階的濃度において試験されたDAC HYPにより達成された最大ADCC活性が、同じ濃度のZENAPAX DACおよびDAC Penzbergにより発揮される活性よりおよそ30−40%低かったことを示す。
【0235】
DAC HYP対ZENAPAX DAC対DAC Penzbergの電荷アイソフォームプロファイル(N末端変異体と一致する)の比較を、図23に示す。
【0236】
5.9.DAC HYP臨床試験
5.9.1.CHOICE研究
CHOICE試験は、230例の再発性MS患者においてインターフェロンベータ療法に加えた、ダクリズマブの第2相、ランダム化、二重盲検、プラセボ対照試験であった。この試験は、皮下注射として投与した100mg/mlのDAC Penzberg(生産品の説明に関する上記のセクション5.8を参照されたい)の2種の投与計画:1mg/kgのダクリズマブを4週間ごとに投与、および2mg/kgダクリズマブを2週間ごとに投与、を試験した。この研究の結果は、インターフェロンベータ療法に2mg/kgで2週間ごとに投与するダクリズマブを加えることによって、インターフェロンベータ療法単独と比較して、新しい、または拡大したガドリニウム増強病巣が24週目に有意に縮小されることを示した。
【0237】
CHOICE研究の結果は、Wynnら、2010年、Lancet Neurol.9(4):381−90頁に記載されている。ダクリズマブ治療は、一般的に忍容性良好であった。一般的な有害事象は、すべての治療群において類似であった。グレード3の有害事象は、24パーセントのDAC/IFNβ治療患者および14%のプラセボ/IFNβ治療患者に観察された。最も頻度の高いグレード3有害事象は、感染症であり、7%のDAC/IFNβ治療患者および3パーセントのプラセボ/IFNβ治療患者に起こった。日和見感染または死亡は無く、すべての感染症は、標準的な療法で解消された。
【0238】
CHOICE試験は、IFNβ−al療法の経歴のあるMS患者において、ダクリズマブが忍容性良好であり、IFNβ−al単独と比較して、新しい/拡大したガドリニウム増強(Gd+)病巣において72%の用量依存性縮小を起こしたことを実証した。臨床的有効性は、免疫調節性CD56bright ナチュラルキラー(NK)細胞の著しい拡大を伴った。
【0239】
5.9.2.SELECT研究
ランダム化二重盲検プラセボ対照用量範囲探索試験(SELECT)を実施し、DAC HYPの2種の異なる投薬量レベルの安全性および有効性を決定した。
【0240】
概説。本研究は、チェコ共和国、ドイツ、ハンガリー、インド、ポーランド、ロシア、ウクライナおよび英国における76の施設で実施された。個々の患者のケアは、治療を行う神経科医、治療を行う看護師(または研究コーディネーター)、検査を行う神経科医、MRIの検査技師および薬剤師(または承認された被指名人)が関与する。集中型双方向音声応答システム(Interactive Voice Response System)を、すべての部位にわたるランダム化に使用した。プロトコルで規定された暫定無益分析を、150例の患者が24週の通院を完了した後に実施した。
【0241】
患者。臨床的に明確な再発寛解型多発性硬化症(2005年 McDonald基準1−4に従って;Polmanら、2005年Ann Neurol58:840−846頁を参照されたい)であり、ベースラインの総合障害度評価尺度(Expanded Disability Status Scale)(EDSS)が0−0.50(Kurtzke、1983年、Neurology33(11):1444−52頁)およびランダム化の前12か月に少なくとも1回MSが再発した、またはランダム化の前6週間以内に実施された脳MRIにおいて1つの新しいGd+病巣があった、適格基準に含まれる18−55歳の患者をランダム化し、DAC HYP(150mgまたは300mg)またはプラセボのいずれかを、4週ごとに1回の52週間の皮下注射として与えた。妊娠の可能性がある患者は、有効な避妊の実践を必要とした。
【0242】
患者が、原発性進行性、二次進行性もしくは進行性再発性のMS、悪性腫瘍の病歴、重症のアレルギー反応もしくはアナフィラキシー反応もしくは既知の薬剤過敏症または治験責任医師の立場から考えて、DAC HYPの投与を妨げる他の深刻な医学的条件を有する場合、その患者は排除した。さらなる排除基準は、DAC HYPまたはZENAPAX(商標)、全身リンパ節照射法、クラドリビン、ミトキサントロン、T−細胞もしくはT−細胞受容体のワクチン接種またはナタリズマブもしくはリツキシマブを除く任意の治療用mAbを用いた以前の治療を含む。ランダム化の時点において、患者は、前年にシクロホスファミドもしくはリツキシマブを用いた治療、6か月以内にナタリズマブ、シクロスポリン、アザチオプリン、メトトレキサート、静注用免疫グロブリン、血漿交換療法または細胞吸着除去療法を用いた治療、またはランダム化前の3か月以内に生ウイルスワクチン、酢酸グラチマー、IFNβ、インターフェロンアルファを用いた治療、または30日以内にコルチコステロイド、4−アミノピリジンまたは関連製品を用いた治療を受けることはできなかった。
【0243】
この群の特徴は以下の通りであった。
【0244】
【表40】
【0245】
エンドポイント。この研究の第1の目的は、DAC HYP単独療法が、52週において年換算再発率(ARR)で定義されたMSの再発を減少させるかどうかを決定することであった。再発は、検査を行う神経科医による査定において、新しい神経学的発見を伴う、24時間を超えて続く新しいまたは繰り返し起こる神経症状(熱または感染症を伴わない)として定義された。3人の盲検のMS神経科医からなる独立した神経学評価委員会(Independent Neurology Evaluation Committee)(INEC)は、すべての疑わしい再発を評価し、MS再発のプロトコル定義を満たすかどうかを判定した。INECが承認した再発だけがは、一次分析に含まれた。
【0246】
第2の目的は、患者のサブセットにおいて、8、12、16、20および24週に実施された脳MRIスキャンにおいて累積する新しいGd+病巣の数の減少、;52週における新しい、または新たに拡大したT2高強度病巣の数の減少;ベースラインと52週の間に再発する患者集団の減少;および29項目の多発性硬化症インパクトスケール(Multiple Sclerosis Impact Scale)(MSIS−29)(Hobartら、2001年、Brain 124(Pt5):962−73頁)の52週における身体的影響スコアのベースラインからの変化により測定される生活の質の改良(QoL)にDAC HYPが有効であるかどうかを決定することであった。確認された障害の進行を、ベースラインおよび52週の間のEDSSスコアの変化によって査定した(ベースラインEDSS≧1.0に関してEDSSは1.0ポイント上昇、または12週間持続したベースラインEDSS=0に関して1.5ポイント上昇)。EDSSの評価は、12週ごとならびに20、52、60および72週に実施した。
【0247】
追加のQoLエンドポイントは、EQ−視覚的アナログ尺度(Visual Analogue Scale)(EQ−VAS)(EuroQol−生活健康関連の質の測定のための新しい機関、2011年、17.11.11アクセス、http://www.euroqol.org/)により査定された対象の幸福の全体的査定であり、EQ−5D健康調査(EuroQol−生活健康関連の質の測定のための新しい機関、2011年、17.11.11アクセス、http://www.euroqol.org/);12項目の略式健康調査SF−12(Wareら、1996年、Medical Care34(3):220−33頁)および52週におけるMSIS−29の精神的スケール(Hobartら、2001年、Brain 124(Pt5):962−73頁)における変化であった。
【0248】
追加のMRIエンドポイントは、52週におけるGd+病巣の数、24および52週における全体および新しい、または新たに拡大したT2高強度病巣の容積、24および52週における全体および新しいT1低強度病巣「ブラックホール」(灰白質に対して等強度/低強度であり、ガドリニウム投与後に増強されなかった病巣として定義される)の容積ならびにSIENA法(Smithら、2001年、J Comput Assist Tomogr25(3):466−75頁)により査定された脳全体の容積における百分率の変化であった。
【0249】
リンパ球のサブセットは、検証されたFACSアッセイを使用して複数の時点で測定した。CD56brightNK細胞は、CD3−/CD16+/CD56brightリンパ球として定義された。DAC HYPに対する免疫原性を、抗薬剤抗体をスクリーニングするための標準的ELISAを使用して査定し、その後、細胞アッセイを使用して、すべての陽性試料における中和抗体に関して試験した。
【0250】
統計分析。サンプルサイズおよそ600例の患者を、DAC HYP治療群およびプラセボ群の間のARRにおいて50%の減少を検出するための90%の検出力を有するように選択し、10%の脱落率、5%の第1種過誤率および両側検定の負の2項分布と仮定するシミュレーションから推定した。プラセボ群におけるARRは、RRMS対象において最近完了した試験に基づき、0.476であると仮定された。報告されたすべてのp値は、両側である。
【0251】
一次分析により、各DAC HYP群対プラセボ間のARRの差が評価された。別のMS薬物を用いた救命治療後に起こった再発は打ち切りであった。差は、研究登録前の年の再発数に関して補正される負の二項回帰モデル、ベースラインEDSS(EDSS≦2.5対EDSS>2.5)およびベースライン年齢(≦35対>35歳)を使用して評価した。二次分析は、負の二項回帰(8および24週の間の新しいGd+病巣の数;新しい、または新たに拡大したT2高強度病巣の数)、Cox比例ハザードモデル(最初の再発までの時間、疾患の進行までの時間)および分散モデルの分析(EDSSにおける変化、新しい、または新たに拡大したT2病巣の容積、新しいT1低強度病巣の容積、QoL)ならびに比例オッズモデル(52週における新しいGd+病巣の数)を使用して治療差に関して試験した。再発なしの患者の割合は、Kaplan−Meierの生存率曲線分布から推定した。
【0252】
8および24週の間の新しいGd+病巣の累積数に関して、患者が1または2回の連続スキャンまたはすべてのスキャンを逃した場合、最後のベースラインではない観察をそれより先に実施するか、または各治療群内の病巣の平均数をそれぞれ使用した。他のMRIエンドポイントに関して、欠測データは、治療群内の平均を使用して転嫁した。MSIS−29に関して、患者が<10項目を逃した場合、非欠測項目の平均を使用してスコアに転嫁した。≧10項目および他のQoL測定を逃した患者に関して、ランダムスロープおよび切片モデルを使用して欠測データを推定した。
【0253】
有効性エンドポイントに関する統計的試験は、DAC HYP300mg群対プラセボおよびDAC HYP150mg群対プラセボの別々の比較を使用した。逐次的閉検定手順を使用して、多重比較による全体の第1種過誤率を調節した。
【0254】
有効性分析を、ランダム化を受けたすべての患者が含まれる治療意図(intent−to−treat)(ITT)集団において評価した。しかし、1つの研究施設からの21例の患者は、研究完了前に特定された、該施設における不正確な投薬の証拠ため(該施設の全ての患者は、積極的治療を受けていた)、研究完了前にITT集団からあらかじめ除外した。感度分析において、すべての一次および二次有効性分析を、すべてのランダム化した患者を使用して繰り返した。すべての安全性分析は、研究薬剤の少なくとも1回の投与を受け、少なくとも1回のランダム化後の査定を受けたすべての患者として定義される安全性集団に基づいた。
【0255】
あらかじめ計画された無益性分析を、150例の対象が24週の通院を完了した後に実施し、DAC HYPの仮定された効果が明白でなかった場合、停止する機会を提供した。有効性は、研究期間にわたって変化し得るので、無益性分析の時に、優位性の証拠のために研究を早期に停止する計画は無かった。無益性は、各投薬群に関する8および24週の間の新しいGd+病巣の累積数ならびにARRエンドポイントの両方に関する条件付き検出力を別々に推定することによって査定した。安全性モニタリング委員会(Safety Monitoring Committee)は、分析時のデータを検討し、データの全体的一貫性およびリスク便益の査定に基づき、研究を続けることを推奨した。
【0256】
結果の要約。
【0257】
適格な参加者を、2/15/2008から5/14/2010までランダム化した。ベースラインの特徴は、3種の治療群にわたって類似であるが、DAC HYP150mg群の患者がDAC HYP300mg群の患者より多くのT2およびGd+T1病巣を有する傾向があった。すべてのランダム化された患者にわたって、合計577例(93%)が、研究を完了したDAC HYPおよびプラセボ治療患者と類似の割合で治療期間を完了した。
【0258】
詳細な結果。臨床的有効性。52週におけるARR(一次エンドポイント)は、プラセボ(0.46;表36)と比較して、DAC HYP150mg(0.21)または300mg(0.23)にランダム化された患者に関してより低く、DAC HYP150mg(95%CI、31%から69%、p<0.0001)によりプラセボに対して54%の減少、およびDAC HYP300mg(95%CI、26%から66%、p=0.0002;表36)に関してプラセボに対して50%の減少を示した。52週にわたって、再発患者の割合は、プラセボ群において再発した36%に対してDAC HYP150mg(19%)および300mg(20%)の群において減少した(両方の比較に関してp≦0.001)(表36)。プラセボと比較して、52週における3か月持続する障害進行の危険性は、DAC HYP150mgにおいて57%(ハザード比=0.43;95%CI、0.21から0.88;p=0.021)およびDAC HYP300mg群において43%(ハザード比=0.57;95%CI、0.30から1.09;p=0.091)減少した。
【0259】
52週におけるMSIS−29身体的スコアで相対的に4.0の改良が、プラセボに対してDAC HYP150mgに観察され、DAC HYP300mg患者において著しい変化は低かった(p<0.0008およびp=0.1284対プラセボ、それぞれ;表36)。身体的および精神的機能両方ならびに全体の健康の測定を含む、生活の健康関連の質の他の測定についてもまた、類似の改良が観察された(表36)。
【0260】
【表41】
【0261】
MRI。DAC HYPは、MRIにより定義されたように、新しいMS病巣の活性を、
全研究集団および8から24週の間に実施される月1回のMRIによるサブセットの両方
において減少させた(表36)。臨床的エンドポイントと対照的に、有効性のこのポイントの推定値は、潜在的ベースラインの不均衡を調整後でも、150mg用量群と比較して300mg用量群においてわずかに強かった。縦モード解析は、Gd+病巣の活性が、治療の最初の2、3か月は300mg用量群と比較して150mg用量群において高かったが、52週には類似であったことを実証した(表36)。1つの排除研究現場由来の21例の患者を含んだ感度分析は、すべての有効性分析に関して類似の結果を得た。
【0262】
安全性。有害事象(AE)は、DAC HYP150mg(73%)、DAC HYP300mg(76%)およびプラセボ(79%)の群中の患者に類似の割合で起こった(表37)。重篤なAEは、プラセボ群の26%、DAC HYP150m群において15%およびDAC HYP300mg群において17%で起こった。MSの再発を除いて、SAEは各群の患者の6%、7%および9%で起こった(表37)。>5%のDAC HYP患者において起こったAEを、表37に示す。重篤な感染症の発生率は、DAC HYP治療患者中における2%対プラセボにおける0%であった。投薬が続いていた間に重篤な感染症になった7例の患者のうち、1例は重篤な感染症のため治療を中止し、6例は、感染症が解消後治療を再開した。皮膚事象の発生率は、DAC HYP150mg群において18%、DAC HYP300mg群において22%およびプラセボ群において13%であった(表37)。重篤な皮膚事象は、DAC HYP治療患者の1%において起こった。重篤な発疹から回復していた1例のDAC HYP治療患者は、腰筋膿瘍の合併症のため死亡した。剖検において、以前に診断未確定であった腰筋膿瘍は、腸間膜動脈に関与することが見いだされ、局所血栓症および急性虚血性大腸炎をもたらした。5例の悪性腫瘍が試験中に起こり、2例の子宮頸がん(プラセボ群およびDAC HYP150mg群において各1例);DAC HYP150mgにおける1例の甲状腺新生物は、重篤ではない甲状腺結節ではなく、DAC HYP300mg群における2例のメラノーマであった。メラノーマの症例は、局所切除により治療され、再発の報告はなかった。
【0263】
【表42】
【0264】
実験結果。DAC HYPを用いて治療された患者は、52週においてプラセボと比較して合計NK細胞カウント(細胞/mm3)が増加した(中央値:42.0(150mgDAC HYP);46.5(300mgDAC HYP);対−4.5プラセボ;p=<0.001)。合計NK細胞数の増加は、ベースラインにおける中央値7.77から治療終了時の44.84である、CD56brightNK細胞の選択的増加に関連していた。対照的に、CD56dimNK細胞はわずかに変化しただけであった(中央値の変化は122.68から123.70)。CD56brightNK細胞の拡大は、DAC HYP治療群対プラセボの両方において最初のベースライン後の時点(4週)で明らかであった(p<0.0001)。CD56brightNK細胞は、ベースラインのリンパ球の0.6%の中央値から52週の2.8%に拡大した。対照的に、DAC HYPを用いて治療された患者は、B細胞および合計リンパ球カウントがわずかに減少した(表38)。CD4+およびCD8+T細胞両方のカウントは、DAC HYP治療患者で52週においておよそ7−10%減少し、CD4+/CD8+比は、治療期間中一定を維持した。
【0265】
【表43】
【0266】
肝機能検査(LFT)の異常は5X ULNを上回り、4%のDAC治療患者および<1%のプラセボ治療患者において起こった。これらの異常は、通常、治療期間の後期(中央値 発症+308日)に起こり、中央値にして62日で解消した。>5X ULNに上昇した17例のDAC HYP治療患者のうち、6例が、解消後少なくとも6か月間、DAC HYPを用いて治療を続け、または再開し、この期間中全員に再発はなかった。2例の患者において、LFTの上昇は感染症を伴った(1例がB型肝炎および1例がサイトメガロウイルス感染症)。
【0267】
免疫原性。24週において、DAC HYPに対する中和抗体を、6例(2%)のDAC HYP治療患者(150mg用量群中5例の患者および300mg用量群中1例の対象)において検出した。一部の患者において、これらの抗体は一時的であり、52週において、DAC HYPに対する中和抗体は、各DAC HYP用量群から1対象存在しただけであった。
【0268】
結論。毎月の、皮下DAC HYP単独療法によるCD25の拮抗作用は、例えば、再発率の減少、MRIにより定義された新しい病巣およびMS患者の優勢に治療された天然集団における障害の進行により測定して、MS疾患活性について1年にわたって強固で臨床的に意味のある効果を実証した。
【0269】
6.具体的な実施形態、参照による組み込み
本開示の様々な態様は、以下の番号付きパラグラフに述べられる実施形態に記載されている。
1.無血清および無コレステロール培地において増殖するよう適応されており、組換えタンパク質を発現するよう操作されている、無血清および無コレステロール培地において増殖される場合、10日間のフェドバッチ法において100Lの培養物中100mg/L/日の組換えタンパク質を超える容積生産性を達成することができる改変されたNS0細胞。
2.コレステロールおよび動物由来成分のない培地において増殖される場合、10日間のフェドバッチ法において1,000Lの培養物中100mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、実施形態1に記載の改変されたNS0細胞。
3.コレステロールおよび動物由来成分のない培地において増殖される場合、10日間のフェドバッチ法において16,000Lの培養物中100mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、実施形態1に記載の改変されたNS0細胞。
4.流加培地が、追加される容積が最初の細胞培養物容積の割合を表す、次のスケジュール:
【0270】
【表44】
に従って追加される、実施形態1−3のいずれか1つに記載の改変されたNS0細胞。
5.13日間のフェドバッチ法において少なくとも100Lの培養物中200mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、実施形態1に記載の改変されたNS0細胞。
6.無コレステロール培地において増殖される場合、13日間のフェドバッチ法において1,000Lの培養物中200mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、実施形態5に記載の改変されたNS0細胞。
7.無血清および無コレステロール培地において増殖される場合、10日間のフェドバッチ法において16,000Lの培養物中100mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、実施形態5に記載の改変されたNS0細胞。
8.抗CD25モノクローナル抗体を発現するのに有用な核酸で安定的にトランスフェクトされる、実施形態1に記載の改変されたNS0細胞。
9.抗CD25モノクローナル抗体が、配列番号2の21位から233位に配列が一致するVL鎖および配列番号4の20位から465位に配列が一致するVH鎖を含む、実施形態8に記載の改変されたNS0細胞。
10.ベクターpAbX.gptを用いて形質転換された、実施形態1に記載の改変されたNS0細胞。
11.ベクターpHAT.IgG1.rg.dEを用いて形質転換された、実施形態1に記載の改変されたNS0細胞。
12.クローン7A11−5H7−14−43で表される実施形態1に記載の改変されたNS0細胞。
13.実施形態1−12のいずれか1つに記載の改変されたNS0細胞を培養することを含む、組換えタンパク質を生産する方法。
14.10日間のフェドバッチ法において100L、1,000Lもしくは16,000Lの培養物中少なくとも100mg/L/日の組換えタンパク質を、または13日間のフェドバッチ法において100L、1,000Lもしくは16,000Lの培養物中少なくとも200mg/L/日の組換えタンパク質を生産する条件下で、改変されたNS0細胞が培養される、実施形態13に記載の方法。
15.改変されたNS0細胞が、血清およびコレステロールの非存在下で培養される、実施形態13または実施形態14に記載の方法。
16.改変されたNS0細胞が、トロポロンおよびヒドロコルチゾンの非存在下で培養される、実施形態15に記載の方法。
17.改変されたNS0細胞が、10−35g/Lグルコースを含有する基本および/または流加培地において培養される、実施形態13または実施形態14に記載の方法。
18.改変されたNS0細胞が、15g/Lグルコースを含有する基本培地および/または28g/Lグルコースを含有する流加培地において培養される、実施形態17に記載の方法。
19.基本培地が、PFBM2±10%成分で構成されている、実施形態18に記載の方法。
20.流加培地が、PFFM3±10%成分で構成されている、実施形態18に記載の方法。
21.細胞が基本培地において1−3日間、次に流加培地において10−13日間培養される実施形態19または実施形態20に記載の方法。
22.流加培地が、表7において概要が述べられているスケジュール±10%に従って追加される、実施形態18に記載の方法。
23.哺乳動物細胞において作動可能である選択可能なマーカーの発現を推進する弱いプロモーターおよび対象のタンパク質の発現を推進する強いプロモーターを含む、対象のタンパク質を組換え的に発現するのに有用なベクター。
24.対象のタンパク質が治療抗体である、実施形態23に記載のベクター。
25.治療抗体が抗CD25抗体である、実施形態24に記載のベクター。
26.CD25抗体がダクリズマブのCDRを含む、実施形態25に記載のベクター。
27.CD25抗体がダクリズマブである、実施形態26に記載のベクター。
28.細胞に実施形態23−27のいずれか1つに記載のベクターをトランスフェクトし、10日間のフェドバッチ法において100L、1,000Lもしくは16,000Lの培養物中少なくとも100mg/L/日対象のタンパク質を、または13日間のフェドバッチ法において100L、1,000Lもしくは16,000Lの培養物中少なくとも200mg/L/日の組換えタンパク質を生産することができる細胞を選択することを含む、対象のタンパク質の高い容積生産性を有する哺乳動物宿主細胞を得るための方法。
29.ダクリズマブがpE/Q重鎖N連結アイソフォームおよび/またはQ/VHS重鎖N末端アイソフォームの存在により特徴付けられる、ダクリズマブを含む組成物。
30.pE/Q重鎖N末端アイソフォームがダクリズマブのおよそ6−15%を構成する、実施形態29に記載の組成物。
31.pE/Q重鎖N末端アイソフォームがダクリズマブのおよそ7−12%を構成する、実施形態29に記載の組成物。
32.Q/VHS重鎖N末端アイソフォームがダクリズマブのおよそ1−15%を構成する、実施形態29−31のいずれか1つに記載の組成物。
33.Q/VHS重鎖N末端アイソフォームがダクリズマブのおよそ3−12%を構成する、実施形態32に記載の組成物。
34.ダクリズマブの重鎖が次のN末端アイソフォーム:
【0271】
【表45】
に存在する、実施形態29に記載の組成物。
35.ダクリズマブの重鎖が次のN末端アイソフォーム:
【0272】
【表46】
に存在する、実施形態29に記載の組成物。
36.ダクリズマブが、図18のアイソフォームプロファイルと実質的に類似する陽イオン交換クロマトグラフィーアイソフォームプロファイルにより特徴付けられる、実施形態29に記載の組成物。
37.ダクリズマブがDAC HYPである、実施形態29に記載の組成物。
38.1つがオリゴ糖G0−GlcNAcに一致し1つがオリゴ糖G0に一致する2つの主要ピークを含有するN連結グリコシル化HPLCプロファイルによりダクリズマブが特徴付けられ、これら2つのピークの合わせたAUCが全ピークの総AUCのうちの約88−99.5%を構成する、ダクリズマブを含む組成物。
39.G0−GlcNAcピークのAUCが全ピークの総AUCのうちの約5−18%を構成し、G0ピークのAUCが全ピークの総AUCのうちの約75−92%を構成する、実施形態38に記載の組成物。
40.G0−GlcNAcピークのAUCが全ピークの総AUCのうちの約6−16%を構成し、G0ピークのAUCが全ピークの総AUCのうちの約78−90%を構成する、実施形態39に記載の組成物。
41.N連結グリコシル化プロファイルが約3%未満のMan5を有する、実施形態38−40のいずれか1つに記載の組成物。
42.N連結グリコシル化プロファイルが約0.5%未満のG2、Man6および/またはMan7を有する、実施形態41に記載の組成物。
43.N連結グリコシル化プロファイルがシアル酸付加オリゴ糖に一致する第三のピークを含有し、シアル酸付加オリゴ糖ピークのAUCが全ピークの総AUCのうちの1%以下を構成する、実施形態38に記載の組成物。
44.N連結グリコシル化プロファイルがオリゴ糖G1に一致する第三のピークを含有し、G1ピークのAUCが全ピークの総AUCのうちの約1−5%を構成する、実施形態38に記載の組成物。
45.G1ピークのAUCが全ピークの総AUCのうちの約1−2%を構成する、実施形態44に記載の組成物。
46.ダクリズマブが、図19のプロファイルにまたは図21の下のパネルのプロファイルに実質的に類似しているN連結グリコシル化HPLCプロファイルを有する、実施形態38に記載の組成物。
47.ダクリズマブがDAC HYPである、実施形態38に記載の組成物。
48.1μg/mLのダクリズマブ濃度および約25対1のエフェクター細胞対標的細胞比で、少なくとも3体の健康なドナー由来のエフェクター細胞および標的細胞としてKit225 K6細胞を使用して、インビトロ細胞アッセイにおいて測定される場合、35%未満のADCC平均細胞傷害性を示すダクリズマブを含む組成物。
49.ダクリズマブが、前記アッセイにおいて10%から30%のADCC平均細胞傷害性を示す、実施形態48に記載の組成物。
50.前記アッセイが少なくとも6体の健康なドナー由来のエフェクター細胞を使用する、実施形態48または実施形態49に記載の組成物。
51.前記アッセイが少なくとも10体の健康なドナー由来のエフェクター細胞を使用する、実施形態48または実施形態49に記載の組成物。
52.ダクリズマブがDAC HYPである、実施形態48−51のいずれか1つに記載の組成物。
53.約150−190mg/mLのダクリズマブ、ならびに希釈緩衝液を用いた組成物の希釈により約85−165mg/mLのダクリズマブを含有し、約267−327mOsm/kgの範囲の浸透圧および25℃で約pH5.8−6.2の範囲のpHを有し、サイズ排除クロマトグラフィーにより測定される場合、ダクリズマブの少なくとも約95%が単量体形態である希釈組成物を生じるような量の賦形剤を含む、ダクリズマブ製剤を作製するのに有用な組成物。
54.希釈緩衝液で希釈される場合、希釈組成物が約85−115mg/mLのダクリズマブを含有するような量の賦形剤を含有する、実施形態53に記載の組成物。
55.希釈緩衝液で希釈される場合、希釈組成物が約150±15mg/mLのダクリズマブを含有するような量の賦形剤を含有する、実施形態53に記載の組成物。
56.ダクリズマブの0.1%以下がアグリゲート形態である、約4から15mg/mLのダクリズマブを含む組成物。
57.ダクリズマブの最大2.5%がアグリゲート形態である、約4から15mg/mLのダクリズマブを含むダクリズマブ組成物を、弱陽イオン交換樹脂上のカラムクロマトグラフィーにより精製することによって得られる、実施形態56に記載の組成物。
58.弱陽イオン交換樹脂がCM−650Mである、実施形態57に記載の組成物。
59.CM−650M樹脂が約20mMのクエン酸ナトリウムを含有する平衡化緩衝液、pH4.4−4.6と平衡化しており、ダクリズマブが約20mMのクエン酸ナトリウムおよび約75mMの硫酸ナトリウムを含有する溶出緩衝液、pH4.4−4.6を用いて溶出される、実施形態58に記載の組成物。
60.クロマトグラフィーが、約10−30cmまたは約17−19cmの高さを有する樹脂床を使用して円筒形カラムにおいて実施され、ダクリズマブが約4−22℃または約18−22℃の範囲の温度で、約50−200cm/時または90−110cm/時の範囲の流速で溶出される、実施形態59に記載の組成物。
61.約85−165mg/mLのダクリズマブおよび約0.02−0.04%(w/v)ポリソルベート80を含み、約267−327mOsm/kgの範囲の浸透圧および25℃で約pH5.8−6.2の範囲のpHを有し、サイズ排除クロマトグラフィーにより測定される場合、ダクリズマブの少なくとも約95%が単量体形態である、ヒトに投与するのに適している組成物。
62.サイズ排除クロマトグラフィーにより測定される場合、ダクリズマブの少なくとも約99%が単量体形態である、実施形態61に記載の組成物。
63.約85−115mg/mLのダクリズマブを含む、実施形態61に記載の組成物。
64.約100mg/mLのダクリズマブ、約40mMのコハク酸ナトリウム、約100mMの塩化ナトリウムおよび約0.03%(w/v)ポリソルベート80から本質的になり、25℃で約6.0のpHを有する、実施形態63に記載の組成物。
65.約135−165mg/mLのダクリズマブを含む、実施形態61に記載の組成物。
66.約150mg/mLのダクリズマブ、約40mMのコハク酸ナトリウム、約100mMの塩化ナトリウムおよび約0.03%(w/v)のポリソルベート80から本質的になり、25℃で約6.0のpHを有する、実施形態65に記載の組成物。
67.約4から15mg/mLのダクリズマブを含むダクリズマブ組成物を適切な緩衝液中での限外濾過により濃縮して、約85−180mg/mLの範囲のダクリズマブ濃度を達成するステップ、および、場合によって、濃縮された組成物を希釈緩衝液で希釈するステップを含む方法により得られる、実施形態65に記載の組成物。
68.約85−165mg/mLのダクリズマブを含み、約2−8℃の範囲の温度での約12ヶ月の期間の貯蔵後にアグリゲート形態中のダクリズマブの百分率が約3%を超えない、皮下投与に適している医薬組成物。
69.約85−115mg/mLのダクリズマブを含む、実施形態68に記載の医薬組成物。
70.約135−165mg/mLのダクリズマブを含む、実施形態68に記載の医薬組成物。
71.約2−8℃の範囲の温度での約12ヶ月の期間の貯蔵後にアグリゲート形態中のダクリズマブの百分率が約2%を超えない、実施形態69または実施形態70に記載の医薬組成物。
72.約2−8℃の範囲の温度での約18ヶ月の期間の貯蔵後にアグリゲート形態中のダクリズマブの百分率が約3%を超えない、実施形態69または実施形態70に記載の医薬組成物。
73.(i)組換えタンパク質を発現し分泌する細胞培養物のpHを、約pH4.5−5.5の範囲のpHに調整するステップ、
(ii)pH調整された細胞培養物を約4から15℃の範囲の温度で約30−90分間インキュベートするステップ、および
(iii)インキュベートされたpH調整細胞培養物を遠心分離して細胞残渣を除去するステップ
を含む、細胞培養物から組換えタンパク質を収穫するための方法。
74.(i)未精製ダクリズマブ調製物から親和性クロマトグラフィー樹脂上にダクリズマブを吸着させるステップ、
(ii)親和性クロマトグラフィー樹脂を洗浄緩衝液で洗浄して混入物を除去するステップ、
(iii)吸着したダクリズマブを溶出緩衝液で溶出させるステップ、
(iv)pHを約3−4の範囲のpHに調整することにより溶出液中のウイルスを不活化し、pH調整された溶出液を規定温度でウイルスを不活化するのに十分な期間インキュベートするステップ、
(v)ウイルス不活化溶出液を約pH7.7−7.9(25℃で測定される)の範囲のpHまで中和するステップ、
(vi)中和された溶出液を強陰イオン交換クロマトグラフィー樹脂に通して流すステップ、
(vii)ステップ(vi)の溶出液のダクリズマブを弱陽イオン交換クロマトグラフィー樹脂上に吸着させるステップ、および
(viii)吸着したダクリズマブを弱陽イオン交換クロマトグラフィー樹脂から溶出させるステップ
を含む、精製されたダクリズマブ組成物を生産するための方法。
75.未精製ダクリズマブ調製物が細胞培養物から収穫される、実施形態74に記載の方法。
76.未精製ダクリズマブ調製物は、ダクリズマブが培養培地中に分泌される条件下で宿主細胞7A11−5H7−14−43を培養することと、分泌されたダクリズマブを収穫することにより得られる、実施形態74に記載の方法。
77.ダクリズマブが実施形態73に記載の方法を使用して収穫される、実施形態76に記載の方法。
78.(ix)ステップ(viii)の溶出されたダクリズマブ組成物を濾過して、ウイルスを除去するステップ、および
(x)濾過された溶液を限外濾過により濃縮して、約85−180mg/mLのダクリズマブを含む精製されたダクリズマブ組成物を産出するステップ
をさらに含む、実施形態74に記載の方法。
79.約85−165mg/mLのダクリズマブおよび約0.02−0.04%(w/v)のポリソルベート80を含み、約267−327mOsm/kgの範囲の浸透圧および25℃で約pH5.8−6.2の範囲のpHを有し、サイズ排除クロマトグラフィーにより測定される場合、ダクリズマブの少なくとも約95%が単量体形態である組成物が得られるように、精製されたダクリズマブ組成物を希釈緩衝液で希釈するステップをさらに含む、実施形態78に記載の方法。
80.得られる組成物が、ダクリズマブの組換え供給源由来の50ppm未満の宿主細胞タンパク質、10ppm未満のプロテインAを有し、組成物中のダクリズマブの3%以下がアグリゲート形態である、実施形態79に記載の方法。
81.基本培地PFBM2。
82.流加培地PFFM3。
83.ダクリズマブが培養培地中に分泌される条件下で、実施形態1−11のいずれか1つに記載の宿主細胞を培養するステップを含む方法により得られる、ダクリズマブ組成物。
84.方法が、分泌されたダクリズマブを細胞培養培地から単離するステップをさらに含む、実施形態83に記載のダクリズマブ組成物。
85.約100−500mMのクエン酸ナトリウム、約10−30mMのNaOHおよび約0.5−3%(v/v)ベンジルアルコールを含む、プロテインA親和性クロマトグラフィー樹脂を衛生化するのに有用な緩衝液。
86.カラムを衛生化するのに十分な流速でおよび期間、実施形態85に記載の衛生化緩衝液を用いてカラムを洗浄することを含む、プロテインA親和性クロマトグラフィーカラムを衛生化する方法。
87.カラムを、約150cm/時の流速でおよそ1.8カラム容積の衛生化緩衝液を用いて洗浄し、洗浄されたカラムが約30−45分の期間流動なしでインキュベートされ、次に平衡化緩衝液を用いて平衡化される、実施形態86に記載の方法。
88.平衡化緩衝液が、約20mMのクエン酸ナトリウムおよび150mMのNaClを含み、約pH7(25℃で)のpHを有する、実施形態87に記載の方法。
89.治療効果を与えるのに十分な量のDAC HYP組成物を患者に投与することを含む、多発性硬化症に罹っている患者を治療する方法。
90.DAC HYP組成物が静脈内投与される、実施形態89に記載の方法。
91.DAC HYP組成物が、約0.8−0.9mg/kgのDAC HYPに一致する量で投与される、実施形態90に記載の方法。
92.DAC HYP組成物が、約1mg/kgのDAC HYPに一致する量で投与される、実施形態91に記載の方法。
93.DAC HYPが、少なくとも6週間、少なくとも12週間、少なくとも24週間の期間、週あたり1回投与される、実施形態89−92のいずれか1つに記載の方法。
94.DAC HYPが単独療法として投与される、実施形態89−93のいずれか1つに記載の方法。
95.患者がインターフェロンベータを用いた前治療に無効果であったかまたはインターフェロンベータを用いた前治療を中止している、実施形態94に記載の方法。
96.DAC HYPがインターフェロンベータに対して補助的に投与される、実施形態89から93のいずれか1つに記載の方法。
97.前記DAC HYP組成物が皮下投与される、実施形態89に記載の方法。
98.DAC HYP組成物が、約1mg/kgのDAC HYPに一致する量で投与される、実施形態97に記載の方法。
99.DAC HYP組成物が2週間に1回投与される、実施形態98に記載の方法。
100.DAC HYP組成物が計約24週間投与される、実施形態99に記載の方法。
101.DAC HYP組成物が、約2mg/kgのDAC HYPに一致する量で投与される、実施形態97に記載の方法。
102.DAC HYP組成物が、4週間に1回投与される、実施形態101に記載の方法。
103.DAC HYP組成物が、計約24週間投与される、実施形態102に記載の方法。
104.DAC HYP組成物が、75mgから300mgのDAC HYPに一致する量で投与される、実施形態103に記載の方法。
105.DAC HYP組成物が、150mgに一致する量で投与される、実施形態104に記載の方法。
106.DAC HYP組成物が、300mgに一致する量で投与される、実施形態104に記載の方法。
107.DAC HYP組成物が、4週間に1回投与される、実施形態103−106のいずれか1つに記載の方法。
108.DAC HYP組成物が、計少なくとも48週間投与される、実施形態107に記載の方法。
109.DAC HYPが単独療法として投与される、実施形態103−108のいずれか1つに記載の方法。
110.患者がインターフェロンベータを用いた前治療に無効果であったかまたはインターフェロンベータを用いた前治療を中止している、実施形態109に記載の方法。
111.DAC HYPがインターフェロンベータに対して補助的に投与される、実施形態103−108のいずれか1つに記載の方法。
112.(i)組換えタンパク質を発現し分泌する細胞培養物のpHを、約pH4.5−5.5の範囲のpHに調整するステップ、
(ii)pH調整された細胞培養物を約4から15℃の範囲の温度で約30−90分間インキュベートするステップ、および
(iii)インキュベートされたpH調整細胞培養物を遠心分離して細胞残渣を除去するステップ
を含む方法により細胞培養物から得られるまたは得られることが可能な、組換えタンパク質。
113.(i)未精製ダクリズマブ調製物から親和性クロマトグラフィー樹脂上にダクリズマブを吸着させるステップ、
(ii)親和性クロマトグラフィー樹脂を洗浄緩衝液で洗浄して混入物を除去するステップ、
(iii)吸着したダクリズマブを溶出緩衝液で溶出させるステップ、
(iv)pHを約3−4の範囲のpHに調整することにより溶出液中のウイルスを不活化し、pH調整された溶出液を規定温度でウイルスを不活化するのに十分な期間インキュベートするステップ、
(v)ウイルス不活化溶出液を約pH7.7−7.9(25℃で測定される)の範囲のpHまで中和するステップ、
(vi)中和された溶出液を強陰イオン交換クロマトグラフィー樹脂に通して流すステップ、
(vii)ステップ(vi)の溶出液のダクリズマブを弱陽イオン交換クロマトグラフィー樹脂上に吸着させるステップ、および
(viii)吸着したダクリズマブを弱陽イオン交換クロマトグラフィー樹脂から溶出させるステップ
を含む方法により得られるまたは得られることが可能な、精製されたダグリズマブ組成物。
114.実施形態74−80のいずれか1つに記載の方法により得られるまたは得られることが可能な、精製されたダグリズマブ組成物。
【0273】
本出願に引用されるすべての出版物、特許、特許出願および他の文書は、あたかも各個々の出版物、特許、特許出願または他の文書が、あらゆる目的のために参照により組み込むことを個別に指示されている場合と同じ程度に、これによってあらゆる目的のために参照によりその全体を組み込む。
【0274】
様々な具体的実施形態が例示され説明されたが、本発明(複数可)の精神と範囲から逸脱することなく様々な変化を加えることが可能であることは認識される。
【背景技術】
【0001】
ダクリズマブ(DAC)は、活性化されているが休止していないTリンパ球およびBリンパ球の表面で発現されるヒト高親和性インターロイキン−2(IL−2)受容体のアルファサブユニット(CD25またはTac)に結合するヒト化IgG1モノクローナル抗体である。活性化された細胞上のCD25が結合すると、DACは高親和性IL−2受容体複合体の形成を阻止し、それによって活性化された細胞のIL−2誘導増殖を阻止する。
【0002】
PHA芽細胞上での直接結合アッセイにおいて測定された場合、DACは0.3nMの近似の結合親和性(KD)でCD25に結合し、用量依存の形でPHA芽細胞の増殖を抑制する(Hakimi et al.、1993年、J.Immunol.151(2):1075−85頁)。最適以下の用量のIL−2(2.5ng/mL)では、15nMのDACはIL−2依存性細胞系統Kit225/K6の増殖を50%抑制する(Pilson et al.、1997年、J.Immunol.159(3):1543−56頁)。IL−2依存性抗原誘導T細胞増殖アッセイでは、0.5−1μg/mL(3−7nM)の範囲のDACを用いると増殖の50%抑制が観察された(Junghans et al.、1990年、Cancer Res.50(5):1495−502頁)。
【0003】
DACの1バージョンは、腎臓移植患者における急性同種移植片拒絶反応の治療のために、シクロスポリンおよびコルチコステロイドを含む免疫抑制療法の補助剤としてZENAPAX(商標)の商品名でHoffman−La Roche社から以前市販されていた。ZENAPAXは、さらなる希釈および静脈内投与のために濃縮製剤として供給されていた。濃縮製剤のバイアルごとに、5mg/mLのDAC、3.6mg/mLのリン酸ナトリウム一塩基一水和物、11mg/mLのリン酸ナトリウム二塩基七水和物、4.6mg/mLの塩化ナトリウム、0.2mg/mLのポリソルベート80ならびにpHをpH6.9に調整するのに十分なHClおよび/またはNaOHを含有する溶液を5mL含有していた。成人患者にも小児患者にも推奨される用量は1.0mg/kgであり、25mg/5mLのZENAPAX濃縮製剤の計算された容積を50mLの無菌0.9%塩化ナトリウム溶液で希釈することにより調製され、15分間かけて末梢静脈または中心静脈を介して静脈内投与された。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】米国特許第7,258,859号明細書
【非特許文献】
【0005】
【非特許文献1】Hakimi et al.、1993年、J.Immunol.151(2):1075−85頁
【非特許文献2】Pilson et al.、1997年、J.Immunol.159(3):1543−56頁
【非特許文献3】Junghans et al.、1990年、Cancer Res.50(5):1495−502頁
【非特許文献4】Nussenblatt et al.、2004年、FOCIS 2004 meeting;7月18日−23日、Montreal、QC.Abstract 4688
【非特許文献5】Nussenblatt et al.、2003年、J.Autoimmun.21:283−93頁
【非特許文献6】Bielekova et al.、2004年、Proc.Nat’l.Acad.Sci.USA101(23):8705−8708頁
【非特許文献7】Rose et al.、2007年、Neurology 69:785−789頁
【発明の概要】
【発明が解決しようとする課題】
【0006】
DACは、ブドウ膜(Nussenblatt et al.、2004年、FOCIS 2004 meeting;7月18日−23日、Montreal、QC.Abstract 4688;Nussenblatt et al.、2003年、J.Autoimmun.21:283−93頁)および多発性硬化症(たとえば、Bielekova et al.、2004年、Proc.Nat’l.Acad.Sci.USA 101(23):8705−8708頁;Rose et al.、2007年、Neurology 69:785−789頁;米国特許第7,258,859号参照)の治療にも有効性を示しており、現在、多発性硬化症の治療のために進行中の臨床試験の主題となっている。DACは安全で効果的であることが判明しているが、長い有効期限を有し、さらに処方するまたは操作することなく都合よく投与することが可能な高濃度液状製剤の他にも、ZENAPAX DACと比べて増強された安全性などの改良特性のある新しいダクリズマブ分子があれば望ましい。
【課題を解決するための手段】
【0007】
背景技術のセクションで述べられたように、ダクリズマブは、リンパ球活性化の重要な媒介物であるヒトインターロイキン−2受容体(IL−2R)のアルファサブユニット(CD25またはTacとも呼ばれる)に特異的に結合するヒト化IgG1抗体である。ZENAPAX(商標)の商品名でHoffman−La Roche社から以前市販されていたダクリズマブの一バージョンは、シクロスポリンおよびコルチコステロイドを含む免疫抑制療法の補助剤として使用される場合、腎同種移植片拒絶反応の治療において安全性および有効性を実証しており(たとえば、the European Medicines Agency(“EMEA”)market authorization for ZENAPAX参照)、多発性硬化症においても有効性を実証している(たとえば、Bielekova et al.、2004年、Proc.Nat’l.Acad.Sci.USA 101(23):8705−8708頁;Rose et al.、2007年、Neurology 69:785−789頁;米国特許第7,258,859号参照)。EMEAによれば、ZENAPAX DACはGS−NS0(マウス骨髄腫)細胞において発現され、Q−セファロースクロマトグラフィー、S−セファロースクロマトグラフィー、ダイアフィルトレーション、Q−セファロースIIクロマトグラフィー、限外濾過、S−300ゲル濾過クロマトグラフィーおよび限外濾過を含む方法を使用して精製される。無血清、無コレステロールおよび他の無動物産物培養物で増殖するように適応されており、異なる方法により単離されるNS0細胞系統において発現されるダクリズマブは、ZENAPAXダクリズマブ(「ZENAPAX DAC」)とは異なり、いくつかの例ではZENAPAXダクリズマブより優れている特徴および特性を有することが現在では見出されている。この新しいダクリズマブは、本明細書ではDAC HYPと呼ばれるが、ZENAPAX DACとは異なるアイソフォームプロファイル(陽イオン交換クロマトグラフィーにより決定される場合);両方の形態のダクリズマブはNS0細胞において発現されるが、ZENAPAX DACとは異なるN連結グリコシル化プロファイル;および生物学的アッセイにおいてZENAPAX DACよりも少ないADCC細胞傷害性を有する。
【0008】
たとえば、重鎖N末端およびC末端での不均一性のために、ダクリズマブのアイソフォームが可能になる。ダクリズマブの成熟VH鎖のアミノ酸配列は、図2に示されるアミノ酸配列の20位で始まる(配列番号4)。成熟VH鎖のN末端グルタミン(Q)(図2では太字で下線付きのテキストで)は環化され、ピログルタミン酸(pE)を形成することができる。いくつかの例では、シグナルペプチド配列はトランケートし、バリン−ヒスチジン−セリン(VHS)配列を成熟VH鎖のN末端グルタミン残基に結合したままにしておくことがある。各ダクリズマブ分子は2つのVH鎖を含有するために、ダクリズマブの様々なN末端アイソフォームは、(1)2つのグルタミン残基(Q/Q)、(2)1つのグルタミン残基と1つのVHS配列(Q/VHSまたはVHS/Q)、(3)2つのVHS配列(VHS/VHS)、(4)1つのグルタミン残基と1つのピログルタミン酸残基(Q/pEまたはpE/Q)、(5)1つのピログルタミン酸残基と1つのVHS配列(pE/VHSまたはVHS/pE)および(6)2つのピログルタミン酸残基(pE/pE)を含有する形態を含むことができる。異なるC末端アイソフォームも可能であり、アイソフォームは0、1つまたは2つのC末端リジン(K)残基(0K、1Kまたは2K)のいずれかを含有し、複雑なアイソフォームプロファイルを生じる。
【0009】
非常に驚くべきことに、ZENAPAX DACのVH鎖のN末端グルタミンは完全に環化されてピログルタミン酸になるが、完全な環化はDAC HYPでは実現されない。その結果、DAC HYPの陽イオン交換クロマトグラムは、pE/QアイソフォームピークおよびQ/VHSアイソフォームピークにより特徴付けられる。いかなる理論にも縛られるつもりはないが、これら独特のpE/QおよびQ/VHSアイソフォームは、DAC HYPを発現するのに使用されるリーダー配列に影響される可能性があると考えられている。したがって、一態様では、本開示は、陽イオン交換クロマトグラフィーにより決定される場合、pE/QアイソフォームがN末端アイソフォームのうちの3%−17%、3%−15%、5%−15%、さらに好ましくは、5%−12%もしくは7%−12%に及ぶ、および/またはQ/VHSアイソフォームがN末端アイソフォームのうちの1%−15%、さらに好ましくは3%−12%に及ぶダクリズマブ組成物を提供する。
【0010】
いくつかの実施形態では、ダクリズマブ組成物は、図18または図23のDAC HYPプロファイルに実質的に類似する陽イオン交換クロマトグラフィープロファイルにより特徴付けられる。
【0011】
ダクリズマブはN連結オリゴ糖が重鎖残基Asn296に結合している。これらN連結オリゴ糖が、アミダーゼPNGアーゼFを使用して放出され、HPLCによって分析されると、DAC HYPは、両方ともNS0細胞系統において組換え的に産生されるという事実にもかかわらず、ZENAPAX DACとは異なるグリコシル化プロファイルを示す。実際、DAC HYPのグリコシル化プロファイルは著しく均質である。図21の上パネルを参照すると、ZENAPAX DACのグリコシル化プロファイルは、オリゴ糖G0−GlcNAc、G0、G1、Man5、G2、Man6、Man7およびシアル酸付加オリゴ糖を表すピークにより特徴付けられる。図21の下パネルは、DAC HYPのグリコシル化プロファイルが、G0−GlcNAcグリコフォームおよびG0グリコフォームに一致する2つの主要ピークならびにG1グリコフォームに一致する小さいほうのピークにより特徴付けられることを示している。G0−GlcNAcグリコフォームは、AUCの約5%から約20%、さらに典型的にはAUCの約7.2%から14.6%に及ぶことが可能である。G0グリコフォームは、AUCの70%から99.2%、さらに典型的にはAUCの80.9%から99.2%に及ぶことが可能である。G1グリコフォームは、AUCの1%から9%、さらに典型的にはAUCの1.4%から3.8%に及ぶことが可能である。シアル酸付加オリゴ糖は総AUCの1.0%以下である。
【0012】
免疫原性および高レベルのエフェクター機能は、長期的に投与される薬物には問題となることがある。さらに、急速なクリアランス速度は薬物有効性を低下させ得る。当業者には周知であるように、治療抗体のグリコシル化パターンの違いは免疫原性の違いを生じ得る。DAC HYPのように高度に均質なグリコシル化パターンを有する抗体は、有益な免疫原性プロファイル、ADCCレベルおよびクリアランス速度を提供し得る。さらに、より均質なグリコシル化パターンを有する生物製剤であれば、バッチによる変動を減少させ、一貫性および安定性を改善することが可能である。
【0013】
したがって、別の態様では、本開示は、均質なN連結グリコシル化プロファイルにより特徴付けられるダクリズマブ組成物を提供する。一実施形態では、ダクリズマブ組成物は、HPLCにより測定された場合、G0−GlcNAcグリコフォームの総AUCのおよそ5−20%、いくつかの実施形態では、G0−GlcNAcグリコフォームの総AUCのおよそ5%−18%またはおよそ7−15%(たとえば、7.2%−14.6%もしくは6.9%から14.7%)(いくつかの特定の実施形態では、G0−GlcNAcグリコフォームの総AUCの7.3%)、ならびにG0グリコフォームの総AUCのおよそ70%−99.2%、およびいくつかの実施形態では、G0グリコフォームの総AUCのおよそ75%−90%、およそ75−92%またはおよそ81−88%(いくつかの特定の実施形態では、G0グリコフォームの総AUCの86%)を含むN連結グリコシル化プロファイルにより特徴付けられる。場合によって、G1ピークは、総AUCの約10%未満、総AUCの約5%未満、約4%未満または約3%未満であり、ある種の実施形態では、約1%から約4%(たとえば、1.4%から3.8%)または約1%から約3%に及ぶ。Man5グリコフォームは好ましくは、総AUCの約3%以下である。他の実施形態では、ダクリズマブ組成物は、図19に示されるプロファイルに実質的に類似するHPLC N連結グリコフォームプロファイルにより特徴付けられる。
【0014】
ある種の態様では、本開示のダクリズマブ組成物は、2つまたはこれよりも多いグリコフォームピークの総数により特徴付けられる。ある種の実施形態では、本開示のダクリズマブ組成物は、(a)合わせると総AUCの約75%から約100%、約80%から約100%、もしくは約85%から約100%に及ぶG0−GlcNAcグリコフォームおよびG0グリコフォームに一致する2つの主要なピーク、ならびに/または(b)合わせると総AUCの約6%以下であるMan5、Man6およびMan7グリコフォームに一致するピーク、ならびに/または(c)合わせると総AUCの約2%以下であるMan6およびMan7グリコフォームに一致するピークにより特徴付けられる。そのような実施形態では、G0−GlcNAc G0、G1および/またはMan5の百分率は、前の段落に記載されている量で存在し得る。
【0015】
DAC HYPの結合および抑制特性の他にT細胞のIL−2誘導増殖の抑制を測定するアッセイにおいて評価されるDAC HYPの機能的効力は、ZENAPAX DACの特性および効力に類似している。しかし、非常に驚くべきことに、DAC HYPが示すADCC細胞傷害性はZENAPAX DACよりも著しく小さいが、これはおそらく、少なくとも一部は、その非フコシル化マンノースグリコシル化レベルの違いによる(図21参照)。図22Aおよび図22Bにおいて示されるように、細胞アッセイにおいて測定される場合、DAC HYPが示すADCC細胞傷害性は、ZENAPAX DACよりも少なくとも25%小さい。当業者であれば認識されることになるように、DAC HYPの減少したADCC細胞傷害性は、たとえば、多発性硬化症またはブドウ膜炎の治療のためには細胞死が望ましくない長期投与に関連する兆候には有益である可能性がある。たとえば、多発性硬化症および他の非腫瘍学兆候の治療においてなどの治療が慢性的に適用されるこれらの状況では、DAC HYP治療はZENAPAX(商標)を用いる治療よりも安全でありうる。
【0016】
したがって、別の態様では、本開示は、たとえば、標的細胞としてKit225/K6を使用する場合および/または3体以上、6体以上、10体以上または50体以上の健康なドナー由来のPBMCエフェクター細胞を使用する場合、エフェクター対標的細胞比25対1、40対1、50対1または60対1を使用するインビトロアッセイにおいて測定される場合、濃度1μg/mLで約30%、25%、20%、15%、10%、5%未満またはこれよりもずっと低いADCC細胞傷害性を示すことにより特徴付けられるダクリズマブ組成物を提供する。特定の実施形態では、本開示は、たとえば、標的細胞としてKit225/K6を使用する場合および/または3体以上、6体以上、10体以上または50体以上の健康なドナー由来のPBMCエフェクター細胞を使用する場合、エフェクター対標的細胞比25対1、40対1、50対1または60対1を使用するインビトロアッセイにおいて測定される場合、濃度1μg/mLで、5−30%、10−30%、15−30%、15−30%、5−25%、10−25%、20−30%、15−25%、15−35%または20−35%に及ぶADCC細胞傷害性を示すことにより特徴付けられるダクリズマブ組成物を提供する。DAC HYPはIgG1免疫グロブリンであり、ADCC細胞傷害性を低下させることが知られているフレームワーク変異を含有しないことを考えると、ZENAPAX DACと比べた場合DAC HYPを用いて観察されるさらに低いレベルのADCC細胞傷害性は驚きである。
【0017】
ZENAPAX DACと比べた場合のDAC HYPの安全性プロファイルは、ウシ血清アルブミン(BSA)のない高度に純粋な生産物の生産を可能にする高収率無血清方法の使用によりさらに改善され得る。したがって、本開示は、BSAがないおよび/またはBSAが存在しない細胞培養方法の生産物であるダクリズマブ組成物を提供する。
【0018】
上で考察された特性のうちの1つまたは複数により特徴付けられるダクリズマブ組成物(DAC HYP組成物)は、哺乳動物細胞における組換え発現により都合よく入手することが可能である。いかなる特定の動作原理にも縛られるつもりはないが、上で考察された独特の特徴および/または特性のうちの1つまたは複数は、少なくとも一部は、高生産性組換え発現系の使用による可能性があると考えられている。これは、DHFRを使用する、または好ましくは、対象のタンパク質(好ましくは、分泌されたタンパク質)の発現を推進する強いプロモーターと組み合わせて、弱いプロモーターの制御下にある選択可能なマーカー遺伝子を使用する遺伝子増幅によるなどのいかなる方法によっても達成することが可能である。理論に縛られずに、弱いプロモーターの制御下にあるマーカーの選択は、発現ベクターが転写的に活性である染色体領域に組み込まれ、高発現レベルの対象のタンパク質を産生する、安定なトランスフェクタントの同定を促進すると考えられている。一実施形態では、選択可能なマーカーの発現を促進する弱いプロモーターは、1つまたは両方のエンハンサー領域の活性が、部分的なまたは完全な欠失によるなどで減少しているまたは除去されているSV40プロモーター(Reddy et al.、1978年、Science 200:494−502頁)であり、場合によって、CMV IEプロモーター(Boshart et al.、1985年、Cell 41(2):521−30頁)などの強いプロモーターと組み合わせて、対象のタンパク質の発現を推進する。
【0019】
したがって、別の態様では、本開示は、DAC HYPなどの高レベルのダクリズマブを安定的に発現し、選択マーカーの発現が1つまたは両方のエンハンサー配列の部分的欠失によるなどでエンハンサー機能が減少されているSV40プロモーター(dE−SV40と命名されている)の制御下にある組換え細胞系統を作製するのに有用なベクターを提供する。安定した発現細胞系統を生産するのに使用することが可能な特定のdE−SV40プロモーター配列は、図3A−図3Dに、および図3Eに示されているベクターpHAT.IgG1.rg.dE(配列番号5)の6536位−6735位にある(配列番号12)。安定した発現細胞系統を生産するのに使用することが可能な特定のベクターの様々な実施形態は、2011年11月30日提出の米国特許出願第61/565,419号および2011年11月30日提出の国際特許出願第PCT/US11/62720号に記載されており、これら特許文献は参照により本明細書に組み込む。
【0020】
一般に、DAC HYPなどのダクリズマブを発現するのに有用なベクターは、pHAT.IgG1.rg.dEにより例証される、プロモーターなどの特徴(下のセクション5.1に記載されている。)のうちの1つまたは複数を含むことになる。ダクリズマブの2つの鎖は別々の転写制御下に置くことが可能であるが、好ましくは同一ベクター上にあり、そのコード領域はcDNAまたはイントロンとエキソンを含有するゲノムDNAであることが可能である。別々の転写制御の代案として、2つの鎖は、単一転写物または単一オープンリーディングフレームとして発現されることが可能であり、そのコード領域は配列内リボソーム進入部位または自己切断インテイン配列により分離されており、その場合には重鎖および軽鎖コード配列は単一プロモーターの制御下にある。例となるプロモーターはCMV IEプロモーターおよびエンハンサーである(pHAT.IgG1.rg.dE(配列番号5)の0001位−0623位および3982位−4604位)。追加の特徴には、転写開始部位(選ばれるプロモーターに不在であれば)、転写終結部位および複製の起点が含まれる。そのような特徴の例は表1に例示されており、この表はpHAT.IgG1.rg.dEの成分を概説している。
【0021】
NS0細胞において、単一の外来性核酸からDAC HYPなどのダクリズマブの重鎖と軽鎖の両方を発現するのに有用な特定の実施形態は、ネオマイシンホスホトランスフェラーゼ(neor)、ハイグロマイシンBホスホトランスフェラーゼ(hygr)、ハイグロマイシンBホスホトランスフェラーゼ(Hph)、ピューロマイシン−N−アセチルトランスフェラーゼ(puror)、ブラストサイジンSデアミナーゼ(bsrr)、キサンチン/グアニンホスホリボシルトランスフェラーゼ(gpt)、グルタミンシンテターゼ(GS)または単純ヘルペスウイルスチミジンキナーゼ(HSV−tk)などの、哺乳動物細胞において作動可能な選択マーカーを利用する。好ましい実施形態では、本開示のベクターにおける選択可能マーカーは、そのコード配列が図3A−図3Dに示されるpHAT.IgG1.rg.dE(配列番号5)の6935位−7793位に見出すことが可能なエンハンサーレスSV40プロモーターの制御下の大腸菌(E.coli)グアニンホスホリボシルトランスフェラーゼ選択可能マーカーである。
【0022】
別の態様では、本開示は、たとえば、DAC HYPなどのダクリズマブを組換え的に生産するのに有用なベクターをトランスフェクトされている宿主細胞を提供する。宿主細胞は、たとえば、チャイニーズハムスター卵巣(CHO)細胞、NS0マウス骨髄腫細胞、Sp2/0細胞、PER.C6細胞、ベロ細胞、BHK細胞、HT1080細胞、COS7細胞、WI38細胞、CV−1/EBNA細胞、L細胞、3T3細胞、HEPG2細胞、MDCK細胞および293細胞を含むいかなる哺乳動物細胞でもよい。ベクターは、トランスフェクトされると、ゲノムに組み込まれて安定した産生細胞系統を生じ得る。当業者であれば、ヒトへの投与のために指定された組成物中に動物産物を含むのは望ましくないことを認識する。したがって、増殖のために血清または他の動物産物(たとえば、コレステロールなど)を必要としない宿主細胞が好まれる。そのような動物産物を必要とする宿主細胞は、無血清および他の無動物産物培地を利用するように適応させることが可能である。無血清および無コレステロール培地で増殖するようにマウス骨髄腫NS0細胞を適応させるための方法は、Hartman et al.、2007年、Biotech.&Bioeng.96(2):294−306頁およびBurky et al.、2007年、Biotech.&Bioeng.96(2):281−293頁に記載されている。無血清および無コレステロール培地で増殖するように適応されたNS0細胞の特定の系統は、DAC HYPを生産するのに使用することが可能な上記のベクターを安定的にトランスフェクトされてきた(クローン7A11−5H7−14−43)。
【0023】
当業者であれば認識することになるように、組換えタンパク質産生のために細胞を培養するのに使用される基本培地および流加培地の他に、フィーディング計画、増殖速度、温度および酸素レベルなどの他の変数は発現されるタンパク質の収率および質に影響を及ぼすことが可能である。これらの条件を最適化する方法は当業者の範囲内であり、例となる条件は本開示の例となる実施形態に記載されている。好ましくは、細胞は、コレステロール由来、血清由来および他の動物由来の成分のない培地で増殖するように適応され、そのような場合には、基本培地および流加培地は、好ましくはそのような成分の代わりとなる定義された化学物質を含む。高レベルのグルコース、たとえば、10−35g/Lのグルコースを含有する培地は、細胞培養生産性を有利に増加することも見出されている。特定の実施形態では、基本培地は約10−20g/L、さらに好ましくは、約15g/Lのグルコースを有し、および/または流加培地は22−35g/L、さらに好ましくは、約28g/Lのグルコースを有している。当技術分野では公知のように、流加培地を、8−15日間、9−13日間または、もっとも好ましくは10−13日間の期間にわたり、加速するフィーディング計画に従って細胞に追加することが可能である。
【0024】
NS0産生系統7A11−5H7−14−43において発現されるDAC HYPでは、増殖および流加培地の成分ならびに発現と産生に影響を及ぼす他の変数は最適化されている。したがって、本開示は、最適化された基本培地、流加培地、フィーディング計画および他の培養法ならびにダクリズマブを高収率および純度で生産するのに有用な条件も提供する。これらの培地ならびに培養パラメータおよび方法は、セクション5.3にさらに詳細に記載されている。
【0025】
ある種のクロマトグラフィーステップの組合せを利用して細胞培養物からダクリズマブを精製すると、精製されたダクリズマブおよびDAC HYP原薬組成物ならびに高濃度で、典型的には少なくとも約100mg/mL±10−15%の、およびいくつかの実施形態では150mg/mL±10−15%の名目上のダクリズマブまたはDAC HYP濃度(紫外分光または屈折率により測定した場合)で液状の貯蔵安定である液体ダクリズマブおよびDAC HYP薬物製剤を生じることも見出されている。
【0026】
安定した高濃度ダクリズマブ薬物製剤は、一般に、濃縮されたダクリズマブ製剤を約267−327mOsm/kgの範囲の(たとえば、270−310mOsm/kg)の浸透圧および25℃で約pH5.8−6.2の範囲の(たとえば、25℃で5.9−6.1)のpHを有する交換緩衝液を用いて交換して、中間製剤を生じ、次に中間製剤をポリソルベート希釈緩衝液で希釈して、紫外分光または屈折率により測定した場合、約100mg/mL±10%のダクリズマブ(たとえば、DAC HYP)、いくつかの実施形態では少なくとも約150mg/mLのダクリズマブ(たとえば、DAC HYP)を含む安定な高濃度液体製剤を生じることにより調製される。希釈緩衝液は交換緩衝液と同じであるが、約0−10%(w/v)のポリソルベート80を含み、最終の安定な高濃度ダクリズマブ製剤が0.02−0.04%の範囲で、いくつかの実施形態では約0.03%(w/v)で計算されたポリソルベート80濃度(名目濃度)を有するような量で使用される。様々な異なる緩衝剤および賦形剤は、定義された範囲内の浸透圧およびpHを達成するよう交換および希釈緩衝液中に含まれることが可能である。安定で高濃度液体ダクリズマブおよびDAC HYP薬物製剤を処方するのに適した交換緩衝液の特定の非限定的例は、約40mMのコハク酸および約100mMのNaClを含有し25℃で約6.0のpHを有する。この交換緩衝液を用いて使用するのに適した希釈緩衝液の特定の非限定的例は、約40mMのコハク酸、約100mMのNaClおよび約1%(w/v)のポリソルベート80を含有し25℃で約6.0のpHを有する。最終製剤のpHは酸または塩基を用いて調整して25℃で約6.0の実際のpHを生じることが可能である。
【0027】
安定で高濃度液体ダクリズマブ製剤は、低レベルのアグリゲーション、典型的には、サイズ排除クロマトグラフィーにより測定される場合、少なくとも95%の単量体および3%未満のアグリゲート、ときには1.5%未満のアグリゲート、より一般的には、99%を超える単量体および0.8%未満のアグリゲートを含有することにより特徴付けられる。高濃度液体ダクリズマブ薬物製剤の他の純度の特徴はセクション5.6にさらに詳細に記載されている。
【0028】
高濃度ダクリズマブ薬物製剤は、長い貯蔵期間、すなわち、2−8℃で貯蔵される場合、最長54ヶ月以上、たとえば、少なくとも5年間の期間、促進的条件(23−27℃/60±5%の相対湿度)下で貯蔵される場合、最長9ヶ月の期間およびストレス条件(38−42℃/75±5%の相対湿度)下で貯蔵される場合、最長3ヶ月の期間、5%を超える分解および3%を超えるアグリゲートの形成(それぞれSDS−PAGEおよびサイズ排除クロマトグラフィーにより測定される場合)に対して安定であることにも特徴付けられる。
【0029】
上記のように、安定な高濃度液体ダクリズマブ製剤は、中間製剤をポリソルベート希釈緩衝液で希釈し、完成したダクリズマブ薬物製剤を生じることにより調製することが可能である。したがって、別の態様では、本開示は、ポリソルベート希釈緩衝液で希釈して本明細書に記載される安定な高濃度ダクリズマブ液体薬物製剤を生じることが可能な、少なくとも約150mg/mLのダクリズマブ、いくつかの実施形態では約170−190mg/mLのダクリズマブを含有する無ポリソルベート精製されたダクリズマブ(好ましくはDAC HYP)中間製剤を提供する。特定の実施形態では、濃縮された無ポリソルベート中間製剤は、約155mg/mLまたは約180mg/mLのダクリズマブ(好ましくはDAC HYP)、約40mMのクエン酸ナトリウムおよび約100mMのNaCl、25℃でpH6.0を名目上含有する。特定の実施形態では、濃縮された無ポリソルベート中間製剤は、約155mg/mLまたは約180mg/mLのダクリズマブ(好ましくはDAC HYP)、約40mMのコハク酸ナトリウムおよび約100mMのNaCl、25℃でpH6.0を名目上含有する。ダクリズマブ組成物は、下でさらに記載される低レベルのアグリゲートにより特徴付けられる。
【0030】
ダクリズマブを限外濾過により濃縮すると、アグリゲートが誘導されて形成され、容認できない(たとえば、>3%)レベルのアグリゲートを含有する高濃度ダクリズマブ薬物製剤が生じ得ることが見出されている。したがって、アグリゲートを取り除くためにダクリズマブ原薬を濃縮することに先立って「研磨」ステップを利用するのが好ましい。濃縮に先立って容認できるアグリゲートのレベルは、濃縮されるダクリズマブ原薬の濃度、最終ダクリズマブ薬物製剤中の望ましい濃度および最終ダクリズマブ薬物製剤中の容認できるレベルのアグリゲートに依存することになる。たとえば、3%未満のアグリゲートを含有する150mg/mLのダクリズマブ製剤が望ましく、この完成したダクリズマブ製剤を達成するためにはダクリズマブ原薬を10倍から30倍(たとえば、20倍)濃縮しなければならない場合は、濃縮されるダクリズマブ組成物は、<0.3%のアグリゲート、好ましくは<0.2%のアグリゲート、好ましくはさらに低いレベル、たとえば、約0.1%のアグリゲートを含有するほうがよい。
【0031】
たとえば、強陽イオン交換クロマトグラフィーおよび疎水性相互作用クロマトグラフィーを含む、様々な公知の技法を使用して、本明細書に記載される濃縮されたダクリズマブ中間物および最終製剤に濃縮するための容認できるレベルのアグリゲートを含有する出発ダクリズマブ原薬組成物を得ることが可能である。しかし、驚くべきことに、弱陽イオン交換クロマトグラフィーは、4−12mg/mLのダクリズマブおよび最大2.5%のアグリゲートから極度に低いレベル、典型的には、約0.1%のアグリゲートの範囲で含有するダクリズマブ組成物のアグリゲートのレベルを低下させることが見出されている。アグリゲートを取り除くために弱陽イオン交換を使用するのは、窒素含有溶液(たとえば、硫酸アンモニウム溶液)を利用する疎水性相互作用クロマトグラフィーよりも環境にやさしい。
【0032】
したがって、別の態様では、本開示は、得られる研磨された組成物が、一般に、約4から15mg/mLのダクリズマブを含有し、サイズ排除クロマトグラフィーにより測定される場合、0.3%以下(たとえば、0.2%以下または0.1%以下)がアグリゲート形態であるように、ダクリズマブ組成物を研磨しアグリゲートを取り除く方法を提供する。方法は、一般に、約4−10mg/mL、典型的には約8−9mg/mL、好ましくは約8.5mg/mLのダクリズマブおよび>0.5%のアグリゲートを含有するダクリズマブ組成物を、適切な緩衝液中の弱陽イオン交換樹脂上に通過させてダクリズマブを吸着させ、吸着されたダクリズマブを溶出緩衝液で溶出することを含む。有用な弱陽イオン交換樹脂には、CM−650M(Tosoh Biosciences)、CM−セファロース、CM−HyperDが含まれるが、これらに限定されない。平衡化、洗浄および溶出緩衝液の成分は、使用される弱陽イオン交換樹脂に依存することになり、当業者には明らかとなる。CM−650M樹脂(Tosoh Biosciences、品番101392)では、約20mMのクエン酸ナトリウム、pH4.5を含有する平衡化と洗浄緩衝液および20mMのクエン酸ナトリウムと75mMの硫酸ナトリウム、pH4.5を含有する溶出緩衝液がうまく機能する。使用される流速は樹脂とカラムのサイズの選択に依存することになる。約10−30cmの範囲(たとえば、17−19cm)の床高を有するCM−650M樹脂の円筒形カラムおよび約50−200cm/時の範囲(たとえば、90−110cm/時、好ましくは約100cm/時)の流速では、クロマトグラフィーを十分用いた作業は、室温で、またはもっと低い温度、たとえば、4℃から、10℃、15℃、20℃もしくは25℃の範囲の温度で、実施することが可能である。典型的で有用な温度範囲は18−25℃(たとえば、18−22℃)である。
【0033】
ZENAPAX EMEAに従えば、ZENAPAX DACのための精製方法は、以下の12のステップ:
(i)培養液濃度;
(ii)Q−セファロースクロマトグラフィー;
(iii)S−セファロースクロマトグラフィー;
(iv)ウイルス不活化のための低pH処置;
(v)濃縮/限外濾過;
(vi)ウイルス除去のためのDV50濾過;
(vii)Q−セファロースIIクロマトグラフィー;
(viii)ウイルス除去のためのバイアソルブ(viresolve)クロマトグラフィー;
(ix)限外濾過による濃縮;
(x)S−300ゲル濾過クロマトグラフィー;
(xi)限外濾過による濃縮;
(Xii)バイアルの無菌充填
を含む。
【0034】
この方法は非効率的であり、提供する精製収率は低い。ステップはもっと少ないが、同時にもっと高い純度を生じる方法を用いてもっと高い収率を達成することが可能であることが見出されており、これを使えば、得られるダクリズマブ原薬を上記の高濃度薬物製剤に処方することが可能になる。したがって、本開示は、ダクリズマブ原薬と高濃度薬物製剤の両方を単離するおよび/または精製するための改良された方法も提供する。該方法は、強い陰イオン交換(Q−セファロース)クロマトグラフィーおよび弱陽イオン交換(CM−650M)クロマトグラフィーと併せてプロテインA親和性クロマトグラフィーを利用し、それにより方法中間物を希釈しない連続フロー処理が可能になる。精製されたダクリズマブ原薬を得るための改良された方法は、以下のステップ:
(i)ダクリズマブを他の細胞培養成分から単離するためのプロテインA親和性クロマトグラフィー;
(ii)低pHウイルス不活化;
(iii)DNAを取り除くための強い陰イオン交換(Q−セファロース)クロマトグラフィー;
(iv)アグリゲートを減らすための弱陽イオン交換(CM−650M)クロマトグラフィー;および
(v)ウイルスを取り除くための濾過
を含む。
【0035】
当技術分野で周知のように、正確な容積、カラムサイズおよび操作用パラメータは、一部、精製の規模に依存することになる。大規模精製に有用な比容積、カラムサイズおよび操作用パラメータはセクション5.4に記載されている。
【0036】
精製され、場合によって上記の方法により処方される未精製ダクリズマブは、様々な従来の方法、たとえば、マイクロフィルター法、遠心分離および直接バイオリアクターからのデプス濾過を使用して細胞培養物から収穫することが可能である。しかし、未精製ダクリズマブは、15℃未満の温度で細胞培養物のpHをおよそpH5まで下げて、細胞を凝結させ、この細胞を遠心分離により取り除くことができることにより都合よく収穫することが可能であることはすでに見出されている。特定の実施形態では、未精製ダクリズマブは、細胞培養物のpHをおよそpH5まで下げ、培養物を30−90分間15℃未満、たとえば、4℃まで冷却し、得られた懸濁物を遠心分離にかけて細胞を取り除くことにより収穫される。この方法は、一般に、組換えタンパク質を培養培地中に分泌するいかなる細胞培養物にも適用可能であり、ダクリズマブまたは治療抗体を産生する培養物に限定されてはいない。培養物のpHは、弱い有機酸もしくは強い有機酸、または弱い無機酸もしくは強い無機酸を含む様々な異なる酸を使用して調整することが可能である。ダクリズマブ培養物では、クエン酸がうまく機能することが見出されている。濃縮されたクエン酸溶液、たとえば、0.5M−2M溶液を、収穫に先立って培養物のpHを調整するために使用することが可能である。
【0037】
DAC HYPの精製は、3つのクロマトグラフィーステップ、すなわち、ウイルス不活化、ウイルス濾過および最終の限外濾過の使用により実現される。プロテインA親和性クロマトグラフィーは、精製方法の最初のステップであり、大部分のプロセス関連不純物を取り除く。プロテインA親和性カラムの再利用を可能にするためには、カラムを再生し衛生化しなければならない。NaOH水溶液はカラム再生と衛生化の両方を実現するのに効果的であることが見出されている。しかし、NaOH溶液を使用するとプロテインA樹脂を分解することがあり、全体的な生産コストを増やしてしまう。プロテインA親和性クロマトグラフィー樹脂をNaOHおよびベンジルアルコールを含有する溶液で衛生化すると良好な結果を生じ、精製サイクルの回数を著しく増やすことも見出されている。したがって、本開示は、プロテインA親和性カラムおよび樹脂を再生し衛生化するための衛生化溶液および方法も提供する。該緩衝液は、一般に約100から500mMのクエン酸ナトリウム、約10から30mMのNaOHおよび約0.5から3%(v/v)のベンジルアルコールを含み、約pH10から13の範囲のpHを有する。該緩衝液は、場合によって、たとえば、塩および/または洗剤などの他の成分も含み得る。クエン酸ナトリウムとベンジルアルコールの両方はプロテインA樹脂をNaOHにより破壊されることから保護し、殺菌作用を増強するために重要である。特定の実施形態では、プロテインA衛生化緩衝液は、約200mMのクエン酸ナトリウム、約20mMのNaOHおよび約1%(v/v)のベンジルアルコールを含有する。セクション5.4.2において記載されるように、ベンジルアルコールおよび水酸化ナトリウムを含有する衛生化溶液は有益な抗菌効果を有し、これを使用して、いかなる抗体についても精製方法におけるプロテインAカラムを衛生化することができる。
【0038】
衛生化緩衝液を使用して、バッチ式方法においてプロテインAクロマトグラフィー樹脂を衛生化することができ、樹脂は過剰な(たとえば、1.5−2×容積)衛生化緩衝液で洗浄され、続いて過剰な(たとえば、1.5−2×容積)衛生化緩衝液中で約30−45分間インキュベートし、続いて平衡化緩衝液または貯蔵緩衝液で平衡化される。衛生化緩衝液を使用して、カラムを適切な流速(たとえば、約110−190cm/時または135−165cm/時の幅がある)で過剰な(たとえば、1.5−2×カラム容積)衛生化緩衝液で洗浄し、カラムを約30−40分間、流速0の条件下に保持し、次にカラムを平衡化緩衝液または貯蔵緩衝液で洗浄することにより、調製されたプロテインAクロマトグラフィーカラムを衛生化することが可能である。適切な平衡化緩衝液および貯蔵緩衝液はセクション5.5に記載されている。
【0039】
本明細書に記載される衛生化緩衝液を用いてプロテインAカラムを衛生化すると、単一バッチの樹脂を使用することができる精製の回数を著しく増加させる。たとえば、単一バッチのプロテインA樹脂は、従来のNaOH緩衝液(たとえば、50mMのNaOH、0.5MのNaCl)で衛生化される場合、典型的には約30精製サイクルしかもちこたえないが、本明細書に記載される衛生化緩衝液で衛生化されたプロテインAカラムは、100回を超える精製サイクルに使用することが可能である。いかなる動作原理にも縛られるつもりはないが、本明細書に記載される衛生化緩衝液は、固定化されたプロテインAをNaOH誘導分解から一部保護し、それによって樹脂の耐用期間を増加すると考えられている。したがって、NaOH分解に耐性であるよう設計されたプロテインAの変異系統を利用する樹脂(たとえば、MabSuRe樹脂)を含むあらゆるプロテインA樹脂には改良が予想されるが、非改変の固定化されたプロテインAまたはNaOH安定であるように操作されていないプロテインAを利用してプロテインA樹脂およびカラムを衛生化するのに使用される場合、本明細書に記載される衛生化緩衝液は特に有益である。本開示は、精製実行を行い、樹脂を本明細書に開示される衛生化溶液で洗浄することを含む、30回を超える、35回を超えるまたは40回を超える抗体精製実行、いくつかの例では最大50または最大100のタンパク質精製サイクルにプロテインA親和性樹脂を使用することを含む方法をさらに提供する。
【0040】
上記のように、ダクリズマブは、活性化されており休止していないTリンパ球およびBリンパ球上で発現されるCD25に特異的に結合し、CD25へのIL−2の結合をブロックして、それによって高親和性IL−2受容体複合体の形成を阻害し、活性化されたT細胞およびB細胞の増殖を抑制する。本明細書に記載されるDAC組成物および製剤ならびに特にDAC HYP組成物および製剤は、同様にCD25に特異的に結合し、類似の生物学的特性を示す。したがって、本明細書に記載されるDAC組成物および製剤ならびに特にDAC HYPは、ダクリズマブ全般、具体的にはZENAPAXについて記載されているアッセイおよび治療法のいずれにおいても有用である。したがって、本開示は、同種移植片拒絶の治療および予防、ブドウ膜炎の治療ならびに多発性硬化症の治療などの、活性化されたT細胞および/またはB細胞増殖が関与している疾患の治療に向けた治療手段として、インビトロ適用でもインビボ適用でも活性化されたT細胞およびB細胞の増殖を抑制するために、本明細書に記載されるDAC組成物および製剤ならびに特にDAC HYP組成物および高濃度安定液体製剤を使用する方法もさらに提供する。
【0041】
該方法は、一般的には、活性化されたT細胞および/またはB細胞を、その増殖を抑制するのに十分な量の本明細書に記載されるダクリズマブ組成物または製剤に接触させることを含む。
【0042】
治療法では、該方法は、一般的には、治療効果を与える量のダクリズマブ組成物、たとえば、本明細書に記載されるDAC HYP組成物または高濃度DAC製剤を対象に投与することを含む。特定の実施形態では、ダクリズマブ組成物および製剤を使用して、単独でまたはインターフェロンベータなどの他の薬物と組み合わせて、多発性硬化症を治療することができる。本明細書に記載されるDAC組成物は、75mgから300mg(たとえば、75mg、100mg、125mg、150mg、175mg、200mg、225mg、250mg、275mgもしくは300mg)の範囲のまたは1mg/kgから4mg/kgの範囲の用量で、患者に毎週から毎月(たとえば、毎週、2週間ごとに、月2回、4週ごとにもしくは毎月)皮下投与することが可能である。該組成物は、好ましくは、100mg/mL±10−15%または150mg/mL±10−15%の名目上のダクリズマブ濃度で、皮下使用に便利な充填済みのシリンジで提供することが可能である。濃縮されたDAC組成物は、静脈内投与のために希釈することも可能である。
【図面の簡単な説明】
【0043】
【図1】DAC−HYP軽鎖cDNA(配列番号1)および翻訳アミノ酸(配列番号2)配列を提供する図である。太字の下線を引いたアスパラギン酸(D)残基は、適切にプロセッシングされた成熟タンパク質における第1のアミノ酸であり、この残基の上流のアミノ酸配列はシグナル配列に対応する。
【図2】DAC−HYP重鎖cDNA(配列番号3)および翻訳アミノ酸配列(配列番号4)を提供する図である。太字の下線を引いたグルタミン(Q)残基は、適切にプロセッシングされた成熟タンパク質における第1のアミノ酸であり、この残基の上流のアミノ酸配列はシグナル配列に対応する。
【図3A】ベクターpHAT.IgG1.rg.dE(配列番号5)に関する完全ヌクレオチド配列を提供する図である。
【図3B】ベクターpHAT.IgG1.rg.dE(配列番号5)に関する完全ヌクレオチド配列を提供する図である。
【図3C】ベクターpHAT.IgG1.rg.dE(配列番号5)に関する完全ヌクレオチド配列を提供する図である。
【図3D】ベクターpHAT.IgG1.rg.dE(配列番号5)に関する完全ヌクレオチド配列を提供する図である。
【図3E】高収量の生産株の選択に使用できる、dESV40プロモーター(配列番号12)の具体的実施形態を提供する図である。
【図4A】ベクターpHAT.IgG1.rg.dEの略図であり、これはpABX.gptに由来する。
【図4B】任意の重鎖遺伝子および軽鎖遺伝子またはさらに非抗体ポリペプチドを発現するように適合させ得るベクターである、pABX.gptの略図である。
【図5】DAC HYPの例示的生産方法を提供する図である。
【図6】プロテインAの親和性クロマトグラフィーの間の生産物分画をモニターする、UV(280nm)、pHおよび導電率を明示する図である。
【図7】Q−セファロースクロマトグラフィーの間の生産物分画をモニターする、UV(280nm)、pHおよび導電率を明示する図である。
【図8】CM陽イオン交換クロマトグラフィーの間の生産物分画をモニターする、UV(280nm)、pHおよび導電率を明示する図である。
【図9】DAC HYP限外濾過系の略図である。
【図10】0−60分のDAC HYPのペプチドマップクロマトグラムを提供する図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。
【図11】55−115分のDAC HYPのペプチドマップクロマトグラムを提供する図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。
【図12】110−170分のDAC HYPのペプチドマップクロマトグラムを提供する図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。
【図13】DAC HYPの150mg/mlのロットのバッチ1およびバッチ2の円偏光二色性スペクトルを重ね合わせた図である。参照は、100mg/mlのDAC HYP調製物である。
【図14A】重ね合わせたゼロ次紫外スペクトルを提供する図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製品物に対応する。3種すべてのスペクトルが存在するが、互いに重ね合わせたところ単一のスペクトルのように見える。
【図14B】重ね合わせた二次微分紫外スペクトルを提供する図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。3種すべてのスペクトルが存在するが、互いに重ね合わせたところ単一のスペクトルのように見える。
【図15A】フルスケールのサイズ排除クロマトグラムを提供する図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。
【図15B】拡大スケールのサイズ排除クロマトグラムを提供する図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。
【図16】時間の関数としてのDAC HYPの凝集のプロットである。
【図17】還元型および非還元型SDS−PAGE(それぞれ、左および右のパネル)を示す図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。
【図18】DAC HYPの陽イオン交換クロマトグラムを示す図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。ピークラベルは、さまざまなN末端およびC末端のアイソフォームに対応する。
【図19】DAC HYPから酵素的に切断された、N連結オリゴ糖のHPLCクロマトグラムを示す図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。
【図20】DAC HYPのADCC反応曲線を示す図である。参照プロファイルは、100mg/mlのDAC HYP調製物であり、バッチ1およびバッチ2は150mg/mlのDAC HYP調製物に対応する。
【図21】それらのさまざまなグリコシル化プロファイルを例示する、DAC HYPから放出されたN連結オリゴ糖のHPLCクロマトグラム(下のパネル)およびZENAPAX DACから放出されたN連結オリゴ糖のHPLCクロマトグラム(上のパネル)を示す図である。
【図22A】エフェクター対標的細胞比を可変としたADCCアッセイフォーマットを使用した、2種のDAC HYP調製物(DAC HYP バッチ3およびDAC HYP バッチ4と称される)、DAC PenzburgおよびZENAPAX DACのADCC活性間の比較を提供する図である。
【図22B】抗体濃度を可変としたADCCアッセイフォーマットを使用した、2種のDAC HYP調製物(DAC HYP バッチ3およびDAC HYP バッチ4と称される)、DAC PenzburgおよびZENAPAX DACのADCC活性間の比較を提供する図である。
【図23】DAC HYP、DAC PenzbergおよびZENAPAX DACの電荷アイソフォームの比較を提供する図である。
【発明を実施するための形態】
【0044】
4.詳細な説明
本開示は、たとえば、活性化されたT細胞および/またはB細胞の増殖を抑制し、たとえば、多発性硬化症などの活性化されたT細胞および/またはB細胞媒介疾患を治療するおよび/または予防するために、とりわけ、特定の特性を有するDAC組成物、ある種の投与様式に特に有用であり異なる温度で貯蔵安定である高濃度DAC製剤、DAC組成物を生産するのに有用なベクターおよび宿主細胞、DAC組成物を生産するのに有用な最適化された培養液および培養条件、DAC組成物および高濃度製剤を精製するための方法ならびにDAC組成物および高濃度製剤を使用する方法を提供する。
【0045】
本明細書において使用されるダクリズマブ(DAC)とは、図1に例示される軽(VL)鎖配列(配列番号2の21位−233位)および図2に例示される重(VH)鎖配列(配列番号4の20位から465位)を有するヒト化IgG1モノクローナル抗体のことである。DACのCDR配列は以下の通りである。
【0046】
【化1】
【0047】
ある種のダクリズマブ分子は文献中に報告されており、DACの特定のバージョンは、シクロスポリンおよびコルチコステロイドを含む免疫治療の補助剤として腎臓移植患者における同種移植片拒絶の予防のためにHoffman−La RocheによりZENAPAXの商品名ですでに市販されている。ZENAPAXの商品名で販売されているバージョンのDACは本明細書においては「ZENAPAX DAC」と呼ばれる。
【0048】
DACの別のバージョンは、ドイツのペンツベルグの施設で製造されており、一度も市販されたことはないが、ある種の臨床試験で使用されたことがある。DACのこのバージョンは本明細書では「DACペンツベルグ」と呼ばれる。
【0049】
本明細書に記載されるように、本開示は、一部には、ZENAPAX DACおよびDACペンツベルグの特徴および特性とは異なる、いくつかの例では、これらより優れている特徴および特性を有する新しいバージョンのDACに関する。したがって、本開示は、一部には、新しいDAC組成物に関する。新しいDAC組成物は、発明の概要においてより完全に記載されている以下の特色:
(1)特徴的なpE/Qおよび/またはQ/VHS N末端アイソフォーム;
(2)2つの主要なピークおよび小ピークにより特徴付けられる均質なN連結オリゴ糖プロファイル;
(3)ZENAPAX DACおよびDACペンツベルグと比べて減少したADCC細胞傷害性;ならびに
(4)150±10−15%もの名目上の濃度で処方される場合の低レベルのアグリゲート形態(<3%)
のうちの1つまたは複数により特徴付けられる。
【0050】
これらの特徴および/または特性のうちの1つまたは複数を有するDAC組成物は、本明細書において「DAC HYP」組成物と呼ばれる。本明細書に記載される発明の様々な態様および特色を例証する目的で、上記4つの特性すべてを有する特定のDAC HYPが、この生産および精製のための特定の組成物および方法と同じように、記載される。しかし、DAC HYP組成物は、本開示の範囲に収まる上記4つの特徴をすべて有する必要はないことは理解されるべきである。特定の実施形態では、DAC HYPは、上記(1)から(4)までの特徴うち少なくとも2つ(たとえば、少なくとも(1)および(2);(1)および(3);(1)および(4);(2)および(3);(2)および(4)、もしくは(3)および(4)の組合せ)または上記(1)から(4)までの特徴うち少なくとも3つ(たとえば、少なくとも(1)、(2)および(3);(1)、(2)および(4);(1)、(3)および(4)、ならびに(2)、(3)、(4)の組合せ)を有する。そのようなDAC HYP組成物は、100mg±10−15%またはさらに150±10−15%の濃度で処方される場合、<3%のアグリゲート、<2%のアグリゲートおよびこれよりはるかに低いレベル、たとえば、<1%のアグリゲートも有し得る。
【0051】
さらに、本明細書に記載される発明のある種の態様および実施形態はDAC HYPを用いて例示され例証されるが、本発明はDAC HYPに限定されておらず、ダクリズマブ組成物全般に有用であり、DACに類似する特定のCD25結合特性を有するIgG2、IgG3およびIgG4抗CD25抗体、ならびにヒトに投与するのに適しておりまだヒト化されていない抗CD25抗体にも限定されていないことは当業者であれば認識することになる。これら様々な異なる抗CD25抗体は本明細書において「DAC類似体」と呼ばれる。そのようなDAC類似体は、通常上記の6つのDAC CDRを含み得るが、他のCDR配列を含み得る。
【0052】
DAC HYP組成物の特徴および特性は、標準アッセイ及び方法を使用して確認することが可能である。たとえば、N末端およびC末端アイソフォームプロファイルは、220nmで検出する陽イオン交換クロマトグラフィーを使用して評価することができる。特定の方法では、100μLの試験試料(緩衝液Aに溶解した1mg/mLの抗体)は、以下の分離勾配(カラムは緩衝液Aを用いて平衡化されている。):
【0053】
【表1】
を使用してProPac WCX−10Gガードカラム(Dionex Corporation)を備えたProPac WCX−10カラム(Dionex Coporation)上、室温で分離される。
【0054】
N連結グルコシル化プロファイルは、アミダーゼPNGアーゼFを用いてN連結オリゴ糖を切断し、オリゴ糖を蛍光標識を用いて誘導体化し、得られた混合物を蛍光検出付きの順相HPLCにより分析することにより評価することが可能である。特定の方法では、アントラニル酸誘導体化切断N連結グリカンは、以下の溶出勾配(100μLの試料注入量を使用して;カラムは、85%緩衝液A/15%緩衝液Bで平衡化されている。):
【0055】
【表2】
を使用して、250×4.6mm重合体アミン結合Asahipak Amino NH2P−504Eカラム(5μm粒子サイズ、Phenomenex、カタログ番号CHO−2628)上、50℃で分離される。
【0056】
純度は、還元SDS−PAGE(プレキャスト14%Trisグリシン勾配ミニゲル、Invitrogen 品番601632)およびコロイドブルー染色、ならびに/または280nmでの検出付きサイズ排除クロマトグラフィーを使用して確認することが可能である。特に、15μLの試験試料(溶出緩衝液中20mg/mL抗体)は、流速1mL/分で、均一濃度勾配の溶出緩衝液(200mMのKPO4、150mMのKCl、pH6.9)を使用する0.5μmのプレカラムフィルター(Upchurch、品番A−102X)を備えた7.8mm×30cmTSK G3000SWXLカラム(Tosoh Biosciences、品番601342)上、室温で分離することが可能である。
【0057】
本明細書に記載される安定な高濃度液体DAC製剤などの、本明細書に記載されるDAC HYP組成物および他のDAC製剤は、少なくとも一部は、たとえば、同種移植の拒絶および多発性硬化症を含む、活性化されたT細胞および/またはB細胞により媒介されると考えられる様々な障害および状態を治療するのに有用である。特定の患者集団、製剤、投与様式ならびに同種移植片拒絶を治療するまたは予防するのに有用な投与量および投与計画は、米国特許第6,013,256号に記載されており、参照により本明細書に組み込む。特定の患者集団、製剤、投与様式ならびに多発性硬化症に罹った患者を治療するのに有用な投与量および投与計画は、米国特許第7,258,859号に記載されており、参照により本明細書に組み込む。これらの製剤、投与様式、投与量および投与計画すべて、の他に開示された特定の患者集団および組合せ療法は、本明細書に記載されるDAC HYP組成物および、適用可能な場合には、高濃度DAC製剤に等しく適している。
【0058】
本明細書に記載されるDAC HYP組成物および製剤は、治療効果を与える量で投与される。治療効果には、根底にある障害の治療が含まれるが、これに限定されない。治療効果には、標準診断検査および他の検査を使用して評価される特定の疾患の症状または副作用を改善するまたは寛解させることも含まれ得る。多発性硬化症では、たとえば、脳病変を評価する磁気共鳴画像法の使用を含む治療効果を評価するおよび/または能力障害への進行を評価する様々な手段は米国特許第7,258,859号に記載されており、参照により本明細書に組み込む。これらの様々な検査法すべてを使用して、多発性硬化症に罹っている患者という状況で治療効果を評価することが可能である。
【0059】
安定な高濃度DAC製剤は、DAC HYPで作製されるものでも、DAC全般で作製されるものでも、DAC類似体で作製されるものでも、多発性硬化症などの慢性疾患の治療における皮下投与に特に有用である。該製剤は、単回ボーラス皮下注射として都合よく投与されるまたは静脈投与のために希釈されることが可能である。該製剤は、75mgから300mg(たとえば、75mg、100mg、125mg、150mg、175mg、200mg、225mg、250mg、275mgもしくは300mg)の範囲のまたは1mg/kgから4mg/kgの範囲の用量で、患者に毎週から毎月(たとえば、毎週、2週間ごとに、月2回、4週ごとにもしくは毎月)皮下投与することが可能である。該組成物は、皮下使用に便利な充填済みのシリンジで提供することが可能である。希釈された製剤は、皮下投与と同じ回数、適切な投与量で静脈内投与することが可能である。
【0060】
5.例示的実施形態
本明細書に記載の発明のさまざまな態様および特徴は、以下の例示的実施形態を用いてさらに記載される。例示的実施形態が、特定の細胞培養培地、細胞培養条件、カラムクロマトグラフィー樹脂および平衡化緩衝液、洗浄緩衝液および溶出緩衝液を利用する限り、日常の変更は実施可能であると認められる。さらに、さまざまな細胞培養方法が特定の生産株(クローン7A11−5H7−14−43)により例示される限り、日常の最適化を用いて、または用いずに、他のDACまたはDAC類似体生産株が良好に使用され得ることが期待される。さらに、(上記の要約または以下の例示的実施形態のいずれにおいても)特定の実施形態を伴って記載される特徴は、本開示の方法および組成物の望ましい特性に実質的に影響を与えずに逸脱可能であり、さらにその異なる実施形態は、これらが明確に互いに排他的でない限り、さまざまな方法で一緒に組み合わせ、使用することができる。したがって、以下に提供される例示的実施形態は例示を意図し、限定することを意図するものではないと理解されるべきであり、これらの実施形態に従った特許請求の範囲を限定すると解釈するべきではない。
【0061】
以下に例示された製造方法は、DAC HYP製剤原料を150mg/mLで生産するために使用された。DAC HYP製剤原料を100mg/mLで作るために、小さなプロセス変更が導入される:
DAC HYP(セクション5.3を参照されたい)を100mg/mLで生産するために使用される細胞培養は、消泡剤エマルジョンを含まず、一方、DAC HYPが150mg/mLの細胞培養は、発泡を最小に抑えるために、10,000Lバイオリアクターにおいて低濃度のDow Corning Antifoam Cを使用する。最終抗体濃度100mg/mLを有するDAC HYP製剤を生産する場合、CM−650Mカラム(セクション5.4.5を参照されたい)は、0.5MのNaOH、0.5Mの硫酸ナトリウムの緩衝液を用いて衛生化され、最終抗体濃度150mg/mLを有するDAC HYP製剤を生産する場合、硫酸ナトリウムは衛生化緩衝液から省かれる。DAC HYPを100mg/mLで作るために、ワンステップの限外濾過/ダイアフィルトレーション(UF/DF)が、ポリソルベート80の添加および製剤原料の最終容積への希釈の直前の、下流プロセスの終わりに使用され(セクション5.4.7を参照されたい)、一方、製剤原料を濃度150mg/mLに作るためには、ツーステップのUF/DFが使用される。以下の実施例は、さまざまなロットの、100mg/mLおよび150mg/mLのDAC HYPの比較分析を示す。いくつかの研究において、150mg/mLのDAC HYPのバッチが、以下に参照標準ロットRS0801と称される、10,000L規模で製造された、1種のロットのDAC HYP100mg/mLと比較された。
【0062】
5.1.DAC HYP発現構築体
抗Tac、マウスIgG2aモノクローナル抗体を産生するハイブリドーマを、マウス骨髄腫細胞系NS−1と、T細胞白血病患者から生じたヒトT細胞系を用いて免疫化されたマウス由来の脾臓細胞とを融合することによって発生させた(Uchiyamaら、1981年、J.Immunol.126(4):1393−7頁)。抗Tacは、活性化T細胞とのその反応性に関して選択されたが、静止T細胞または静止B細胞とは反応性がない。抗Tacは、後に、ヒトIL−2受容体のアルファサブユニットと反応することが示された(Leonardら、1982年、Nature 300(5889):267−9頁)。
【0063】
マウス抗Tacの軽鎖および重鎖の可変領域に関するアミノ酸配列は、それぞれのcDNAから決定された(Queenら、1989年、Proc.Nat’l Acad.Sci.USA86(24):10029−33頁)。マウス抗Tacの結合親和性は、Queenらが記載したヒト化形態中に保有された。マウス抗Tacの相補性決定領域(CDR)は、まず、ヒト抗体Euのアクセプターフレームワークにグラフトされた。三次元モデルの助けを借りて、CDRの立体構造に重要な、カギとなるマウスフレームワーク残基、したがって結合親和性が特定され、アクセプターフレームワーク中のヒトカウンターパートに置換された。さらに、アクセプターフレームワーク中の非定型アミノ酸が、対応する位置のヒトコンセンサス残基と置き換えられ、潜在的免疫原性が排除された。
【0064】
DAC HYPのVLおよびVH遺伝子は、Queen(1989)らにより記載されたように、重複オリゴヌクレオチドのアニーリングおよび伸長によってミニエクソンとして構築された。IgG1形態におけるDAC HYPの発現のために、結果として得られたVLおよびVH遺伝子が、Coleら、(1997年、J.Immunol.159(7):3613−21頁)およびKostelnyら(2001年、Int.J.Cancer 93(4):556−65頁)に概説されたように、単一発現ベクター中にクローニングされ、pHAT.IgG1.rg.dEが構築された(図3および図4Aを参照されたい)。プラスミドpHAT.IgG1.rg.dEは、ダクリズマブのIgG1重鎖およびカッパ軽鎖の両方に関する遺伝子を含有し、各々がヒトサイトメガロウイルス(CMV)プロモーターの調節下にある。このプラスミドは、選択可能なマーカーとして、E.コリ(E.coli)のグアニン・ホスホリボシルトランスフェラーゼ(gpt)遺伝子を含有する。pHAT.IgG1.rg.dE中のこの遺伝成分を、以下の表1に記載する。
【0065】
【表3】
【0066】
dESV40プロモーターは、pHAT.IgG1.rg.dEの6536−6735位に及ぶ(6536−6562は72bpのエンハンサーAの27残基であり、6566−6629は3つの21−bpの反復である。6536−6735はGenBank:J02400.1(シミアンウイルス(Simian Virus)40完全ゲノム))中の5172−1および1−133の逆相補配列(reverse complement)である。発現ベクター中のDAC HYP軽鎖遺伝子および重鎖遺伝子のヌクレオチド配列は、DNA配列決定により確認された。
【0067】
5.2.DAC HYP安定細胞系
マウス骨髄腫細胞系NS0は、European Collection of Cell Cultures(ECACCカタログ番号85110503、Salisbury、Wiltshire、UK)から得られた。これらのNS0細胞のバイアルを、10%FBSを添加されたDMEM中で解凍した。細胞は、加湿インキュベーターにおいて37℃および7.5%CO2において維持された。細胞は、次いで、1mg/mLのBSAを添加された基本培地SFM−3において培養された。SFM−3は、10mg/mLインスリンおよび10μg/mLトランスフェリンを添加された、DMEMおよびHam’sF−12の1:1混合物である。およそ3ヶ月にわたって、NS0細胞を、それが排除されるまで培養培地中のFBSの量を徐々に減少させることによって、添加なしのSFM−3に適応させ、その後、単一ステップにおいてBSAを最終的に除去する。得られた宿主細胞系は、SFM−3において15−20回継代され、凍結バンクが調製された。
【0068】
SFM−3適応細胞に、(FspI酵素(New England Biolabs、カタログ番号R0135L、ロット43)により線形化された)pHAT.IgG1.rg.dEを、エレクトロポレーションにより形質移入した。簡潔に言うと、30−40μgのpHAT.IgG1.rg.dEを、1×107の対数的に増殖する適応NS0細胞に加え、1.5kV、25μFで、Gene Pulser装置(BioRad、Richmond、CA)を使用して2回パルスをかけた。エレクトロポレーション後、細胞を5つの96−ウェルプレート中のDMEM±10%FBSに、ミコフェノール酸(「MPA」)選択後に単一コロニー/ウェルを支持する密度である20,000細胞/ウェルでプレーティングした。Hartmanら、2007年、Biotech.&Bioeng96(2):294−306頁に記載のように、ベクターに安定に統合された形質移入体を、ミコフェノール酸の存在下で選択した。高レベルのDAC HYPを産生するNS0安定形質移入体から出発し、連続3ラウンドのサブクローニングを、限界希釈クローニングまたは蛍光標識細胞分取(FACS)によって、2.5%または5%いずれかのウシ胎児血清(FBS;HyClone、Logan、UT)を含有するPFBM−1中に実施した。サブクローニングの各ラウンドにおいて、最も優れた生産株の1種を次のラウンドのサブクローニングに使用した。第3ラウンドのサブクローニング後、最終生産細胞系(7A11−5H7−14−43)を選択した。その後、最終生産細胞系のシードバンクを、1mLの90%FBS/10%DMSO(Sigma、St.Louis、MO)中1×107細胞/バイアルを凍結することによって調製した。
【0069】
5.3.DAC HYP組換体の生産
5.3.1.細胞の培養および回収
単一細胞バンクバイアルから細胞を解凍し、T−フラスコ、ローラーボトル、スピナーフラスコおよびバイオリアクター内に、生産規模が達成されるまで、徐々により大きな容積に拡大する。生産培養が完了次第、細胞培養液を遠心分離および深層濾過により浄化し、収穫保持タンクに移送した。生産培養期間はおよそ10日である。
【0070】
細胞の培養および回収を、さまざまな異なる細胞培養設備において標準的な機器を使用して、当技術分野において公知のように実施することができる。別の実施例において、細胞を、単一細胞バンクバイアルから解凍し、振とうフラスコおよびバイオリアクター内に、生産規模が達成されるまで、徐々により大きな容積に拡大する。生産培養が完了次第、細胞培養液を遠心分離および深層濾過により浄化し、収穫保持タンクに移送する。生産培養期間はおよそ10日である。
【0071】
5.3.1.1.接種材料の調製
単一細胞バンクバイアルを解凍することによって生産バッチを開始する。細胞を、既知組成培地である無タンパク質基本培地−2(PFBM−2)を含有するTフラスコに移す。NaCl、フェノールレッド、トランスフェリンおよびインスリンを含まず、再構成した場合に、5mg/Lの濃度を得る量のEDTA鉄(III)ナトリウム塩を含み、再構成した場合に、それらの濃度が再構成されたHybridoma−SFMと同じであるように調整された残りの成分量を有するHybridoma−SFM培地粉末を要求することにより、PFBM−2を作るためのカスタム粉末をInvitrogenから注文することができる。調製されたPFBM−2培地は、以下の成分を含有する:8g/Lのカスタム粉末、2.45g/Lの重炭酸ナトリウム、3.15g/LのNaClおよび16.5g/LのD−グルコース一水和物(15g/Lグルコース)。
【0072】
細胞を、その後2日ごとに、ローラーボトルまたはスピナーフラスコ内で連続継代することによって拡大する。T−フラスコ、ローラーボトルおよびスピナーフラスコは設定温度37℃において、T−フラスコおよびローラーボトルに関しては7.5%CO2およびスピナーフラスコに関しては5%CO2の雰囲気下で動作するインキュベーターに配置する。
【0073】
スピナーフラスコに、細胞培養液の容積に依存して、ヘッドスペースに重ねること、または培養液内への散布のいずれかによって、5%CO2を添加し、インペラ速度は、一定の回転数/分(RPM)に調節される。全ての接種材料の拡大継代における標的シーディング密度は、およそ2.5×l05生存細胞/mLである。
【0074】
さらに、接種材料の調製を、当技術分野において公知の方法に従って、さまざまな標準的培養槽、容積および条件を使用して、実施することができる。例えば、生産バッチは、単一細胞バンクバイアルを解凍することによって開始できる。細胞は、既知組成培地である無タンパク質基本培地2(Protein Free Basal Medium−2)(PFBM−2)を含有する振とうフラスコに移すことができる。NaCl、フェノールレッド、トランスフェリンおよびインスリンを含まず、再構成した場合に、5mg/Lの濃度を得る量のEDTA鉄(III)ナトリウム塩を含み、再構成した場合に、それらの濃度が再構成されたHybridoma−SFMと同じであるように調整された残りの成分量を有するHybridoma−SFM培地粉末を要求することにより、PFBM−2を作るためのカスタム粉末をInvitrogenから注文することができる。調製されたPFBM−2培地は以下の成分を含有する:8g/Lのカスタム粉末、2.45g/Lの重炭酸ナトリウム、3.15g/LのNaClおよび16.5g/LのD−グルコース一水和物(15g/Lグルコース)。場合により、バイオリアクター段階において、例えば、0.04mg/Lの濃度で硫酸銅七水和物を加えてもよい。
【0075】
細胞を、その後2日ごとに、振とうフラスコ内で連続継代することによって拡大する。振とうフラスコは、設定温度37℃において、7.5%CO2の雰囲気下で動作するインキュベーターに配置する。
【0076】
振とうフラスコは、インキュベーター内の振とうプラットホームにおいて、一定の回転数/分(RPM)でかき混ぜられる。全ての接種材料の拡大継代における標的シーディング密度は、およそ2.2−2.5×l05生存細胞/mLである。
【0077】
細胞バンクの解凍後およそ14日、十分な数の生存細胞が生産された時、最初のいくつか、通常3または4のステンレス製撹拌タンクのシードバイオリアクターに接種する。使用前に、これらのシードバイオリアクターは、定置洗浄され、定置蒸気滅菌され、適切な容積のPFBM−2培養培地をロードされる。pHプローブおよび溶存酸素プローブは、バイオリアクターが定置蒸気滅菌される前に較正する。最初のシードバイオリアクターに、2.0−2.5×105の生存細胞/mLの初期細胞密度を標的とするための十分な数の細胞を接種する。より大きな容積(通常、100Lから300Lおよびその後1,000Lのシードバイオリアクターまたは60Lから235L、950Lおよび3750Lのシードバイオリアクター)への連続移送を、各リアクターにおける増殖のおよそ2日後および標的初期細胞密度2.0−2.5×105生存細胞/mLに実施する。培養のpHは、自動制御を介してCO2ガスまたは1Mの炭酸ナトリウム(Na2CO3)の添加により維持される。シードバイオリアクターおよび生産バイオリアクターの標的動作条件は、設定温度37℃、pH7.0および(空気飽和の百分率として)30%の溶存酸素を含む。100L、300Lおよび1,000Lのバイオリアクターは、それぞれ100rpm、80rpmおよび70rpmでかき混ぜられる。いくつかの場合、シードバイオリアクターおよび生産バイオリアクターの標的動作条件は、設定温度37℃、CO2散布および塩基の添加による調節を用いた7.0のpHおよび(空気飽和の百分率として)30%の溶存酸素を含む。より大きな容積のバイオリアクターは、100rpm、80rpm、70rpmまたは40rpmの速度でかき混ぜることができる。
【0078】
5.3.2.細胞培養液生産バイオリアクター
1,000Lのシードバイオリアクターにおいておよそ2日後、接種材料をステンレス製撹拌タンクの生産バイオリアクターに移送する。生産バイオリアクターは、およそ10,000Lの作業容積を有する。使用前に、バイオリアクターは、定置洗浄され、定置蒸気滅菌され、およそ4,000LのPFBM−2培地をロードされる。pHプローブおよび溶存酸素プローブは、バイオリアクターが定置蒸気滅菌される前に較正する。
【0079】
別の実施例において、接種材料は、3750Lのシードバイオリアクターにおいて増殖され、その後、およそ15,000Lの作業容積を有するステンレス製撹拌タンクの生産バイオリアクターへ移送され、このステンレス製撹拌タンク生産バイオリアクターは、使用前に、定置洗浄され、定置蒸気滅菌され、およそ4,000−7,000LのPFBM−2培地をロードされる。
【0080】
生産バイオリアクターの標的シーディング密度は、2.0−2.5×105生存細胞/mLの範囲である。既知組成の無タンパク質流加培地濃度(PFFM−3)(PFFM3小成分1および2、L−グルタミン、D−グルコース、リン酸ナトリウム二塩基性七水和物、L−チロシン、葉酸、塩酸および水酸化ナトリウムを再構成することによって作られた、既知組成濃縮流加培地)を、培養の間に加える。PFFM3は、表4に示す成分を含有する。
【0081】
【表4】
【0082】
PFFM3 小成分1は、以下の表5に示す成分を含有する。
【0083】
【表5】
【0084】
PFFM3小成分2は以下の表6に示す成分を含有する。
【0085】
【表6】
【0086】
培養液へのPFFM−3の添加のタイミングおよび量は、以下の表7に示すように行う。
【0087】
【表7】
【0088】
培養のpHは、CO2ガスおよび1Mの炭酸ナトリウム(Na2CO3)の添加の自動制御により、およそpH7.0、好ましくはpH7.0およびpH7.1の間に維持される。溶存酸素含有量は、空気飽和のおよそ30%まで下がってもよい。酸素/空気混合物を、培養液中に散布し、一定の合計ガスの流速を達成し、溶存酸素は、必要に応じて空気対酸素ガスの比率を調整することによって、および最大の酸素対空気の比率に達した後のかき混ぜ速度を増加することによって調節される。別の実施例において、かき混ぜは、力/容積の一定比率を維持するように調整される。シメチコン系消泡剤エマルジョンを、発泡レベルに基づき、必要に応じてバイオリアクターに加える。試料を定期的に採取し、細胞密度、細胞の生存能力、生産物濃度、グルコースおよび乳酸塩濃度、溶存O2、溶存CO2、pHおよび浸透圧に関して試験する。バイオリアクター培養液は、接種後およそ10日で収穫される。収穫前に、バイオリアクターの内容物を未処理バルクとしてサンプリングする。
【0089】
5.3.3. 収穫および細胞除去
収穫の直前に、生産バイオリアクターをまず<15℃に冷やし、pHを、0.5Mまたは1Mまたは2Mのクエン酸を使用して5.0±0.1に調整し、およそ30−90または45−60分の期間そのまま保持し、収穫槽に移送する前に細胞および細胞残渣を凝集させる。pHを調整した収穫は、その後、バッチ記録書類に定義のボウル速度および流速に関する所定のパラメーターの下で動作された連続遠心分離によって浄化される。
【0090】
遠心分離液を、デプスフィルターを介してろ過し、その後0.22μmのメンブレンフィルターを介してろ過し、あらかじめ滅菌されたタンクに回収する。無細胞収穫を、1−2MのTris溶液を使用しておよそ6.4のpHに調整し、さらなる処理のために2−8℃で貯蔵する。いくつかの場合、このpH調整は、pH5.0への元のバイオリアクターのpH調整の12時間以内に行う。
【0091】
5.4.DAC HYPの精製
5.4.1.概説
DAC HYPの精製および製剤化方法は、ZENAPAX生産方法と比較して有効性を改良し、生産物関連不純物およびプロセス関連不純物の一貫したクリアランスを確実にするように設計された。以下のサブセクションは精製方法を記載する。精製は、低pHウイルス不活性化、ウイルスのろ過、限外濾過/ダイアフィルトレーションおよび製剤化ステップと組み合わせた、3種のクロマトグラフィー技術に基づく(プロテインA親和性クロマトグラフィー、Qセファロース陰イオン交換クロマトグラフィーおよびCM−650(M)陽イオン交換クロマトグラフィー)。すべてのステップは、閉鎖型装置において行われる。DAC HYPのための精製方法の概要を、図5に提示し、以下に記載する。
【0092】
5.4.2.プロテインAクロマトグラフィー
プロテインA親和性クロマトグラフィーステップは、一連の下流動作において第1の精製ステップである。このステップは、カラムのサイズに依存して1回または複数回のサイクル、通常、表8Aに記載のカラムに関して2回または3回のサイクルで行われる(すなわち、無細胞の収穫を2つのアリコートに分割し、その後各アリコートをプロテインAカラムに別々にロードし、溶出する)。組換えタンパク質A親和性クロマトグラフィー樹脂はIgGに特異的に結合し、細胞培養液の収穫の他の成分から抗体を分離する。
【0093】
平衡化緩衝液を用いたプロテインAカラムの平衡化の後、カラム樹脂に抗体を結合させるために、中和された、無細胞の収穫をカラムに通過させる。平衡化緩衝液は20mMのクエン酸ナトリウム、150mMの塩化ナトリウム、pH7.0である。カラムに、35グラムの抗体(タンパク質)/充填された樹脂のリットルを超えない容量をロードする。ロード後、平衡化緩衝液を用いてカラムを洗浄し、未結合および緩く結合した不純物を樹脂から除去し、クエン酸塩緩衝液を用いて溶出前の洗浄を行い、カラムのクエン酸塩濃度および塩化ナトリウム濃度を調整する。クエン酸塩緩衝液は、10mMのクエン酸ナトリウムpH7.0である。次いで、結合した抗体を、pH3.5の10mMのクエン酸ナトリウムの溶出緩衝液を使用するpHの変更ステップにより、カラムから溶出する。プロテインAクロマトグラフィーの条件の要約を、表8Aに述べる。
【0094】
【表8】
【0095】
生産物溶出液がカラムから離れたら、流出物の吸光度を波長280nmにおいてモニターし、生産物分画の回収のガイドに使用した(図6を参照されたい)。
【0096】
水酸化ナトリウムおよびベンジルアルコールを含有する衛生化緩衝液の使用は、広範囲の微生物を有利に殺すが、プロテインA樹脂の品質に与える影響が最小である。このことを例示するために、さまざまな衛生化溶液をさまざまな微生物にスパイクし、一定期間にわたってインキュベートした。インキュベーション時間の異なる間隔において、スパイクされた衛生化溶液の一部は中和され、微生物の力価を測定し、対照と比較した。殺菌作用は、一定期間にわたる微生物のlog減少として表す。表8Bは、衛生化緩衝液20mMの水酸化ナトリウム、200mMのクエン酸ナトリウムおよび1%のベンジルアルコールとの接触時間の関数としての、微生物力価の減少を示す。
【0097】
【表9】
【0098】
表8Cは、異なる衛生化溶液による微生物の減少を示す。
【0099】
【表10】
【0100】
前述のデータは、ベンジルアルコールおよび水酸化ナトリウムを含有する衛生化溶液が、グラム陰性細菌およびグラム陽性細菌、芽胞形成細菌、酵母ならびに真菌を含む、広範囲の微生物を殺すために非常に有効であることを示す。通常の衛生化の30分後、E.コリ、スタフィロコッカス・アウレウス、シュードモナス・アルギノーザおよびカンジダ・アルビカンスに関して5log10を超える減少が観察された。真菌(A.ニガー)を殺すためにはさらに時間がかかるが、細胞培養液において真菌の感染は稀である。バイオテクノロジー設備において単離される最も一般的な微生物は、バチルス、シュードモナスおよびスタフィロコッカスである。これらは、衛生化溶液によって、接触時間の30分後に有効に殺される。比較すると、水酸化ナトリウムまたはベンジルアルコール単独では、すべての微生物を殺すためには有効ではない。さらに、水酸化ナトリウム衛生化溶液は、芽胞形成バチルスは殺さない。
【0101】
5.4.3.ウイルス不活性化のための低pHの保持
このステップは、低pH感受性の内因性ウイルス様粒子およびウイルスを不活性化するように設計されている。各プロテインAサイクルからのプロテインA溶出液は、回収タンク内に溶出され、ここに、0.5MのHClをpH3.5±0.1に達するまで加える。生産物は保持タンクに移送され、ここで別のpHメーターによってpHが確認される。低pH保持ステップは、pH3.5±0.1または±0.2(例えば、pH3.35−3.64)に、30−120分または30−240分の間厳密に制御される。30−120分の保持後、ウイルス不活性化溶出液を、1MのTris塩基を使用して、pH7.8±0.1または±0.3(例えば、pH8.05−8.34)に中和し、その後、0.22μmのフィルターを介して生産物プールタンクに移送する。低pHウイルス不活性化条件の要約を、表9に述べる。
【0102】
【表11】
【0103】
5.4.4.Qセファロース陰イオン交換フロースルークロマトグラフィー
Qセファロース陰イオン交換クロマトグラフィーステップを使用し、生産物関連不純物およびプロセス関連不純物(例えば、核酸、宿主細胞タンパク質、生産物アグリゲート、浸出されたプロテインAリガンドなど)を減少させ、さらなるウイルスクリアランス能力を精製方法に提供する。ロードの電導率およびpHを、抗体がカラムをフロースルーし宿主細胞タンパク質および細胞DNAなどの負に帯電した不純物が正に帯電した樹脂と結合するような様式で選択する。
【0104】
陰イオン交換カラムを、20mMのTris、20mMの塩化ナトリウム、pH7.8の平衡化緩衝液を用いて平衡化する。低pH保持ステップによるpHが調製された生産物をカラムに、抗体(タンパク質)の60グラムを超えない容量、または抗体(タンパク質)の30−60グラム/充填された樹脂のリットルを超えない容量までロードする。ローディングの完了後、未結合の抗体および不純物を、平衡化緩衝液を用いてカラムから除去する。
【0105】
生産物の回収を、波長280nmにおいて流出液の吸光度をモニターすることによってガイドする(図7を参照されたい)。
【0106】
衛生化流速は、100cm/時であり保持時間は60分である。
【0107】
Q−セファロースクロマトグラフィーの条件の要約を表10に述べる。
【0108】
【表12】
【0109】
いくつかの使用に関して、貯蔵緩衝液容積は3に設定し、カラム貯蔵温度は5℃−25℃に設定する。
【0110】
5.4.5.CM−650(M)陽イオン交換クロマトグラフィー
このクロマトグラフィーステップは、微量レベルのプロセス関連不純物および生産物関連不純物を減少させるために、方法において使用する最終ステップである。抗体のアグリゲートおよび切断断片に加えて、このステップは、宿主細胞の核酸およびタンパク質などのプロセス関連不純物ならびに浸出したプロテインAもまた減少させる。
【0111】
カラムは、20mMのクエン酸ナトリウム、pH4.5の平衡化緩衝液を用いて平衡化される。陰イオン交換溶出液のプールを、0.5Mのクエン酸を使用して4.5±0.1または±0.2(例えば、4.35−4.64)のpHに調整し、25または30グラムの抗体(タンパク質)/充填樹脂のリットルを超えない標的ローディング容量までカラムにロードする。結合ステップ後、カラムを平衡化緩衝液で洗浄し、任意の未結合または緩く結合した不純物を樹脂から除去する。その後、結合した抗体を、20mMクエン酸ナトリウム、75mM硫酸ナトリウム、pH4.5の溶出緩衝液を用いて溶出様式のステップにおいてカラムから溶出する。ピークの回収を、波長280nmにおいて流出液の吸光度をモニターすることによってガイドする(図8を参照されたい)。
【0112】
CM−650(M)クロマトグラフィーの条件の要約を表11に述べる。
【0113】
【表13】
【0114】
5.4.6.ナノ濾過
ナノ濾過ステップの目的は、精製方法に追加のウイルスクリアランス能力を提供することである。このステップにおけるウイルスおよびウイルス様粒子の除去は、サイズ排除機序を介して行われる。フィルターの細孔は、抗体が細孔を通過するが、ウイルス様粒子およびウイルスはフィルターの上流側に保有されるように設計される。
【0115】
0.22μmまたは0.1μmのフィルターを介して濾過された陽イオン交換溶出液は、小型のウイルス保有ナノフィルターを通過し、その後ポリソルベート80を含まないDAC HYP製剤化緩衝液(40mMのコハク酸塩、100mMの塩化ナトリウム、pH6.0)を用いたフィルターフラッシュが続く。緩衝液フラッシュステップを適用し、ラインおよびフィルターのハウジングに残存する抗体を回収する。
【0116】
ナノ濾過のパラメーターの要約を、表12に述べる。
【0117】
【表14】
【0118】
5.4.7.限外濾過/ダイアフィルトレーション(UF/DF)
この方法ステップは、生産物を濃縮し、生産物中の緩衝液を、ポリソルベート80を含まないDAC HYP製剤化緩衝液と交換するように設計されている。この方法ステップは、30kDaの名目分画分子量のメンブレンを使用するタンジェンシャルフロー様式で動作する。最終濃度の期待生産物容量および各UF系の相対的保持容積のため、2つの限外濾過/ダイアフィルトレーション段階を使用し、150mg/mLの製剤を生産する。
【0119】
第1の段階は、大型のUF系を使用して処理される(図9を参照されたい)。ナノ濾過のろ液を、まずおよそ30mg/mLまで濃縮し、その後、交換緩衝液(ポリソルベート80を含まない製剤化緩衝液)にダイアフィルトレーションする。ダイアフィルトレーションされた抗体溶液を、およそ100mg/mLまでさらに濃縮し、その後、UF/DF系からおよそ55mg/mLの濃度で回収し、0.22μmのフィルターを介して移送する。ダイアフィルトレーションされた抗体溶液は、およそ20−60mg/mLの濃度で回収することもできる。第1段階のパラメーターの要約を、表13に述べる。
【0120】
【表15】
【0121】
第2段階は、より小さいUF系であるが、同じ30kDaのカットオフのメンブレンを使用して処理される。DAC HYP(通常55mg/mL)溶液を、およそ180mg/mLに濃縮し、UF系から回収し、その後0.22μmのフィルターを介して移送する。UF系を、ポリソルベート80を含まない製剤化緩衝液を用いてすすぎ、0.22μmのフィルターを介して移送し、およそ170mg/mLまたはおよそ150−170mg/mLの精製された製剤原料を得る。第2段階のパラメーターの要約を、表14に述べる。
【0122】
【表16】
【0123】
5.5.DAC HYPの製剤化
最終方法ステップは、精製された製剤原料の150または100mg/L±10%の最終標的濃度、すなわち、適切な濃度のポリソルベート80を含有する緩衝液中、150±15mg/mL(150mg/mLの製剤化の場合)または100±10mg/mL(100mg/mLの製剤化の場合)の最終標的濃度への希釈である。製剤化は、段階的に実施される。
【0124】
例えば、まず、ポリソルベート80を含まない製剤化緩衝液(40mMのコハク酸ナトリウム、100mMの塩化ナトリウム、pH6.0)を、製剤化された製剤原料の90%標的容積に達するように、精製された製剤原料に加える。その後、計算された量のポリソルベート80希釈緩衝液(40mMのコハク酸塩、100mMの塩化ナトリウム、1%ポリソルベート80、pH6.0)を、最終製剤中0.03%(w/v)のポリソルベート80の標的濃度に達するように加える。最終的に、ポリソルベート80を含まない製剤化緩衝液(150mg/mLの製剤のためにコハク酸塩およびコハク酸ならびに100mg/mLの製剤のためにコハク酸塩およびHClで作られる)を使用して生産物容積を調整し、150±15mg/mL(好ましくは150±8mg/mL)の最終抗体濃度を達成する。100mg/mLの薬剤生産物は、同様の様式で、最終濃度100±10mg/mL(好ましくは100±5mg/mL)に製剤化される。
【0125】
製剤化された製剤原料を、0.22μmのフィルターを介して、半剛性の円筒状の支持体内部に配置されたBioProcess Container TM(BPC(登録商標))のバッグ(または同等物)内に濾過する。支持体は、BPCと蓋とを包囲し、柔軟性のバッグおよび外部環境の間の防護壁を提供する。製剤化された製剤原料は、薬剤生産物の充填/仕上げ操作のために、アクセス制御クーラー中で2−8℃において貯蔵される。
【0126】
製剤化条件の要約を表15に述べる。
【0127】
【表17】
【0128】
1mLの薬剤生産物を、バイアルまたは注射器に充填する。仕上がった150mg/mLおよび100mg/mL生産物の成分の要約を、表16に示す(すべての量は名目値である。)。
【0129】
【表18】
【0130】
5.6.DAC HYP製剤原料の特徴付け
DAC HYPを、両方の重鎖サブユニットのアミノ酸296においてグリコシル化し、主要なオリゴ糖形態は、末端のガラクトースを欠いた、コアがフコシル化された二分岐構造として存在する。
【0131】
DAC HYP重鎖のN末端は、3つの主要な形態:1)(DNA配列から予測される)N−末端グルタミン、2)N−末端ピログルタミン酸(N−末端グルタミンの環化による)および3)(シグナルペプチドの不完全な切断によりもたらされる)予測されるN末端グルタミン残基に加えてN−末端バリン、ヒスチジンおよびセリン残基として存在する。
【0132】
DAC HYP重鎖のC末端は、C末端リジン残基を含んで、または含まずに存在する。主要形態はC末端リジン残基を欠き、C末端グリシンをもたらす。
【0133】
DAC HYPは、ヌクレオチド配列により定義された一次アミノ酸組成に基づき、分子量144kDaと計算された。縮小された重鎖の対応する分子量は48.9kDaであり、縮小された軽鎖は23.2kDaであり、これらの重量は、炭水化物含有量または翻訳後修飾を占めない。
【0134】
DAC HYPの結合はCD25に高度に特異的であり、CD25は活性化されたTリンパ球およびBリンパ球に発現されるが、静止Tリンパ球および静止Bリンパ球には発現されない。これらの活性化細胞におけるDAC HYPとCD25との結合は、IL−2とCD25との結合およびその後の高親和性IL−2受容体複合体の形成を遮断する。その結果として、活性化細胞のIL−2誘導性増殖が遮断される。DAC HYPの観察された治療有効性および潜在的治療有効性は、活性化された自己反応性T細胞の増殖についてのその阻害効果の大部分を静止すると考えられる。しかし、DAC HYPはまた、好酸球などの他のCD25産生細胞型に関してその遮断効果を介して治療効果を発揮すると思われる。
【0135】
高濃度の150mg/ml DAC HYP製剤が臨床試験に適していることを確認するために、包括的な物理化学的および生物学的評価を特徴付けのために実施し、本明細書においてバッチ1およびバッチ2(またはそれぞれ、バッチ150−1もしくはバッチ150−2)と称される、DAC HYP 150mg/mL製剤原料の2種のバッチを、10,000L規模で製造されたDAC HYP 100mg/mLの1種のロットである参照標準ロットRS0801と比較した。
【0136】
結果は、DAC HYP薬剤生産物の150mg/mLロットは純度が高く、100mg/mLロットに匹敵し、臨床研究における使用に適していることを実証している。これらの特徴の要約を表17に示す。
【0137】
【表19】
【0138】
5.6.1.色、外観および透明度
DAC HYP製剤原料の外観は、色の視覚的検査ならびに拡大なしの黒色背景および白色背景に対する直射光における溶液の透明度によって査定する。溶液は、目に見える粒子の存在に関してもまた評価する。さまざまなロットのDAC HYP薬剤生産物の通常の外観を表17に記載する。
【0139】
5.6.2.pH決定
DAC HYPのpHは、U.S.Pharmacopeia Protocol No.(791)に従って決定する。DAC HYP薬剤生産物のさまざまなロットのpH範囲を、表17に要約する。
【0140】
5.6.3.UV分光器による生産物濃度
DAC HYPの濃度は、UV分光器により決定する。DAC HYP試料を、緩衝液を用いて重量測定法で希釈する。各希釈試料溶液のUV吸光度を、緩衝液ブランクに対して278nmにおいて測定する。試料のタンパク質濃度を、DAC HYPに関する吸光係数を使用して計算する。DAC HYP薬剤生産物のさまざまなロットのタンパク質濃度を、表17に要約する。
【0141】
5.6.4.N末端配列決定
DAC HYP150mg/mLロットを、N末端配列決定によって評価した。試料を、自動Edman分解配列決定器を使用して分析した。
【0142】
最初の15残基DIQMTQSPSTLSASV(配列番号13)を介して、軽鎖の予想アミノ酸配列を、すべての試料に関して確認した。
【0143】
DAC HYP中の重鎖の大部分は、N末端重鎖配列を生じないピログルタミン酸(pE)残基によって遮断される。DAC HYP中で次に最も高頻度で見られるN末端重鎖配列は、バリン、ヒスチジン、セリン(VHS)配列で始まり、重鎖シグナルペプチドの3個の末端残基のプロセッシングを欠いたことによりもたらされる。最初の15個のN末端残基のうちの14個は、すべての試料において、VHS重鎖配列(VHSQVQLVQSGAEVK(配列番号14))が確認された。先行する配列決定サイクルにおいてLCから検出された大量のグルタミンのため、4番目の残基であるグルタミンは確認できなかった。N末端グルタミンを有する重鎖の証拠もまた、すべての試料中に存在した。この配列は、天然のN末端重鎖グルタミン残基が、ピログルタミン酸形態への環化を受けなかった結果である。150mg/mLロットに関するN末端配列決定の結果は、重鎖および軽鎖のコーディング配列から予測された配列と一致する。同様の結果を、100mg/mLロットに関して得た。
【0144】
5.6.5.重鎖および軽鎖の質量分析
DAC HYP150mg/mLロットの重鎖および軽鎖ならびに参照標準RS0801の分子量を液体クロマトグラフィー質量分析(LC−MS)によって評価した。すべてのロットを、アミダーゼPNGaseFを用いて脱グリコシル化させ、ジチオスレイトールを用いて還元し、ヨード酢酸を用いてアルキル化し、逆相クロマトグラフィーにより分離した。理論的な重鎖および軽鎖の質量を、タンパク質配列から計算した。試料の観察された質量は、以下の表18に示すように計算質量の1Da以内であった。
【0145】
【表20】
【0146】
先行するサブセクションに記載のように、DAC HYP重鎖の2種の最も高頻度で見られる形態は、N末端ピログルタミン酸(pE)残基またはバリン、ヒスチジン、セリン(VHS)配列を含有し、C末端リジンを欠くことが公知である。150mg/mLロット中の2種の優勢な重鎖変異体および軽鎖に関して得られた分子量は、参照標準RS0801の分子量と同様であり、タンパク質配列から予測された質量と一致した。
【0147】
以下のサブセクションに提示したペプチドマッピングの結果と一緒に、重鎖および軽鎖の質量結果により、DAC HYP150mg/mLロット中の予想される軽鎖および重鎖の一次構造の存在を確認する。
【0148】
5.6.6.ペプチドマッピング
DAC HYP150mg/mLロットおよび参照標準RS0801(10,000L規模で製造された100mg/mLの製剤原料のロットから生産されたDAC HYP)を、逆相HPLCペプチドマッピングを使用して評価した。すべてのロットを、ジチオスレイトールを用いて還元し、ヨード酢酸を用いてアルキル化し、トリプシンを用いて酵素的に消化した。得られたペプチドを、逆相クロマトグラフィーにより分離し、215nmの紫外線吸光度によって検出し、ペプチドマップを作製した。
【0149】
一次アミノ酸配列を検証するために、150mg/mLロットのペプチドマップを、参照標準RS0801のペプチドマップと比較した。予想される重鎖および軽鎖の残基と98%対応するペプチドが、参照標準RS0801のペプチドマップにおける質量分析によってすでに特定されている。ペプチドマップに計上されない残基は、単一のアミノ酸である、または非常に極性のジペプチド中に存在し、逆相カラムに保有されることが予想されない。ピログルタミン酸、グルタミンおよび重鎖N末端ペプチドのN末端におけるVHS配列に一致する質量が、参照標準中に特定された。DAC HYPは、Asn296における重鎖のFc部分中のN連結グリコシル化に関するコンセンサス部位を含有し、連結複合体のコアが二分岐のオリゴ糖構造と一致する質量が、Asn296残基を含有するペプチドに関して特定された。
【0150】
DAC HYP150mg/mLロットと参照標準RS0801とを比較するペプチドマップを、図10(0から60分)、図11(55から115分)および図12(110から170分)に示す。DAC HYP150mg/mLロットのペプチドマップは、参照標準のペプチドマップと同様であり、150mg/mLロットにおける予想一次構造の存在が確認される。
【0151】
5.6.7.円偏光二色性分光法
DAC HYP150mg/mLロットおよび参照標準RS0801を、遠紫外円偏光二色性分光法(遠−UV CD)により分析し、二次構造を評価した。分析前に、試料を、最終タンパク質濃度0.2mg/mLに水を用いて希釈した。スペクトルを、0.1cmのセルを使用して195−260nmから入手し、得られたシグナルを、緩衝液を差し引いた後でモル楕円率に変換した。
【0152】
DAC HYP150mg/mLロットのバッチ1およびバッチ2ならびに参照標準RS0801の重ね合わせた遠−UV CDスペクトルを、図13に示す。すべてのロットのスペクトルは、およそ202nmにおいて正のバンドを示し、およそ217nmにおいて負のバンドを示す。217nmにおける負のバンドは、βシート立体構造の特徴であり、IgG分子の優勢な立体構造である。ロットの遠−UV CDスペクトルは同様であり、DAC HYP150mg/mLロットおよび参照標準RS0801の中の均一な二次構造を支持した。
【0153】
5.6.8.紫外分光法
DAC HYP150mg/mLロットおよび参照標準RS0801を、紫外(UV)分光法により分析し、三次構造を評価した。分析前に、試料を製剤化緩衝液(40mMのコハク酸塩、100mMの塩化ナトリウム、0.03%のポリソルベート80、pH6.0)で最終タンパク質濃度0.5mg/mlに希釈した。スペクトルを、1cmの路長の石英キュベットを使用して250から350nmから入手し、280nmにおける吸光度を1.0とし正規化した。
【0154】
重ね合わせたゼロ次および二次微分のUVスペクトル(平滑化したゼロ次のデータから計算)を、それぞれ図14Aおよび14Bに示す。評価したすべてのロットのゼロ次および二次微分スペクトルは、重ねることができ、DAC HYP150mg/mLロットのバッチ1およびバッチ2ならびに参照標準RS0801の中の均一な三次構造を支持した。
【0155】
5.6.9.サイズ排除クロマトグラフィー
サイズ排除クロマトグラフィー(SEC)は、水系移動相を有する多孔質シリカカラムおよび280nmの紫外吸光度を検出を用いて実施した。具体的には、15μLの試験試料(溶出緩衝液中20mg/mLの抗体)を、室温において、0.5μmプレカラムフィルター(Upchurch、part no.A−102X)を装備した7.8mm×30cmのTSK G3000SWXLカラム(Tosoh Biosciences、part no.601342)において、溶出緩衝液(200mMのKPO4、150mMのKCl、pH6.9)の均一濃度の勾配を使用して、1mL/分の流速で分析した。
【0156】
図15A(フルスケール)および図15B(拡大スケール)に示すように、DAC HYP150mg/mLロットのクロマトグラフィープロファイルは、参照標準RS0801のクロマトグラフィープロファイルと同様であった。すべてのロットは、ダクリズマブ単量体に一致する同じ主要なピークおよび小さなアグリゲートピークを示した。DAC HYP150mg/mLロットに関するアグリゲート結果は、以下の表19に示すように、DAC HYP100mg/mLロットのアグリゲート結果と同様であった。
【0157】
【表21】
【0158】
150mg/mLロットおよび参照標準RS0801を、多角度光散乱検出(SEC−MALS)を有するSECを使用して分析し、アグリゲートピークの分子量を決定した。すべてのロットに関して、アグリゲートピークに関して得られた分子量はおよそ300kDaであり、これは抗体二量体に一致する。
【0159】
DAC HYP中のアグリゲートの形成を、18か月の間にわたってモニターした。製剤中のアグリゲートのレベルは上昇したが、アグリゲートの百分率は横這いであり、5℃において18か月貯蔵した場合、およそ1.5%を超えなかった(図6を参照されたい)。新しいバッチの新しいデータは、5℃において貯蔵した場合、6週間でおよそ1.8−1.9%のアグリゲートが出現し得るが、試験したすべての試料に関しては、5℃において貯蔵した場合、5年間にわたって形成されたアグリゲートは3%未満であったことを示す。
【0160】
5.6.10.沈降速度超遠心分析法
DAC HYP150mg/mLおよび100mg/mLのロット中の単量体およびアグリゲートを、沈降速度超遠心分析法(SV−AUC)を使用して特徴付けた。単量体および個々のアグリゲートに関する沈降係数値および相対的存在度を、以下の表20および21に提示する。
【0161】
【表22】
【0162】
【表23】
【0163】
単量体は、個々のロットにおいて観察される主要な成分であった。単量体ピークの沈降係数は、単量体の立体構造が、150mg/mLおよび100mg/mLのロットの間で同様であることを示すロットと高度に一致した。150mg/mLロットの単量体の含有量は、100mg/mLのロットの単量体の含有量と同様であった。
【0164】
個々のロット中の優勢なアグリゲート種は、抗体の二量体と一致する沈降係数を有した。これはSEC−MALSの結果と一致し、SECアグリゲートピークが、主に抗体二量体から構成されることを示す(先行するサブセクションを参照されたい)。三量体および四量体と一致する沈降係数を有する、低レベルの2種のより大きなアグリゲートもまた、AUCにより個々のロットにおいて観察された。150mg/mLロットの二量体、三量体および四量体の含有量は、100mg/mLロットの二量体、三量体および四量体の含有量と同様であった。
【0165】
5.6.11.定量的還元型SDS−PAGE
純度を、4−20%(通常14%)のtris−グリシンゲルおよびコロイダルブルー染色を使用して、SDS−PAGEによって決定した。試料を、還元条件下で10μgの試料ロードを用いて分析した。純度を、密度測定によって測定された、重鎖および軽鎖のバンド面積の合計を全バンド面積で割ることによって計算した。
【0166】
以下の表22に示すように、150mg/mLロットは高純度であり、100mg/mLロットと同様である。
【0167】
【表24】
【0168】
5.6.12.定量的SDS−PAGE
DAC HYPの純度を、還元型および非還元型のゲル電気泳動の両方によって査定した。プレキャストの14%または8−16%のTris−グリシンゲルを分析に使用した。150mg/mL DAC HYP製剤の2種のバッチ由来のアリコートを、前述のように参照バッチと比較した。DAC HYPの純度を分析する還元型および非還元型ゲルを、図17に示す。150mg/mLロットのバンドパターンは、参照標準RS0801のバンドパターンと同様であり、150mg/mLロットに新しいバンドは検出されなかった。
【0169】
5.6.13.陽イオン交換クロマトグラフィー
DAC HYP150mg/mLロットおよび100mg/mLロットの荷電アイソフォームの分布を、陽イオン交換クロマトグラフィー(CEX)を使用して評価した。CEXを、非多孔質のカルボン酸塩官能化の、弱陽イオン交換カラムを使用して実施し、220nmにおいて検出した。100μLの試験試料(緩衝液Aに溶解した1mg/mLの抗体)を、室温で、ProPac WCX−10Gガードカラム(Dionex Corporation)を装備したProPac WCX−10カラム(Dionex Coporation)において以下の分離勾配を使用して(カラムは緩衝液Aを用いて平衡化される)分離した。
【0170】
【表25】
【0171】
図18に示すように、150mg/mLロットのCEXクロマトグラムは、参照標準RS0801のCEXクロマトグラムと一致し、150mg/mLロットにおいて新しい荷電アイソフォームは検出されなかった。CEXクロマトグラム中に存在する5種の主要なアイソフォームは、重鎖N末端における不均一のためであり、1)2つのピログルタミン酸残基(pE/pE);2)1つのピログルタミン酸残基および1つのグルタミン残基(pE/Q);3)1つのピログルタミン酸残基および1つのVHS配列(成熟重鎖のN末端グルタミン残基に先行するトランケート型VHSシグナルペプチド)(pE/VHS);4)1つのグルタミン残基および1つのVHS配列(Q/VHS);5)2つのVHS配列(VHS/VHS)を含む。C末端リジン(K)のアイソフォームも分離され、図18において特定される。上記の個々のN末端アイソフォームは、異なるC末端アイソフォーム(0、1または2つのK)として存在してもよい。混合物の複雑性のため、C末端リジンアイソフォームは、記載の方法を使用してのみ、明瞭に分離され、主要なpE/pEおよびpE/VHSのアイソフォームに関して測定可能である。
【0172】
N末端およびC末端アイソフォームの定量結果を、150mg/mLおよび100mg/mLのロットに関して、それぞれ図23および24に提供し、報告される百分率は、すべてのピークの全曲線下面積(AUC)と比較した特定のピークの曲線下面積の百分率に基づく。
【0173】
【表26】
【0174】
【表27】
【0175】
5.6.14.オリゴ糖マッピング
DAC HYP150mg/mLロットおよび100mg/mLロットのオリゴ糖の分布を、オリゴ糖マッピングにより評価した。N連結オリゴ糖は、重鎖Asn296からアミダーゼPNGaseFを使用して酵素的に放出された。オリゴ糖を、続いて蛍光標識(この場合、アンスラニル酸)を用いて誘導体化し、ナイロンメンブレンを介して抗体から分離した。誘導体化され、切断されたN連結グリカンを、50℃で、250×4.6mmの高分子アミン結合Asahipak Amino NH2P−504Eカラム(粒径5μm、Phenomenex、カタログ番号CHO−2628)において蛍光検出を用いて、以下の溶出勾配(試料注入容積100μL;カラムは85%の緩衝液A/15%の緩衝液Bを用いて平衡化する)を使用して分離した。
【0176】
【表28】
【0177】
150mg/mLロットと参照標準RS0801とを比較するクロマトグラムを、図19に示す。全ピーク面積の少なくとも1.0%を構成するオリゴ糖ピークを標識し、以下の表25に報告する。
【0178】
【表29】
【0179】
すべてのロットは、主にG0およびG0−GlcNAc(二分岐構造の1つのアームにGlcNAcを欠いたG0)からなり、これはDAC HYP方法を代表する。シアル化オリゴ糖は、およそ68分で溶出し、すべての被験ロットにおいて1.0%より低い。ピーク3と称される特徴付けされていないオリゴ糖は、すべての被験ロットにおいて同様の存在度で存在した。
【0180】
5.6.15.酸化
DAC HYP ロットを、ペプチドマップ中に存在する、酸化および非酸化トリプシンペプチドをモニターすることによって、潜在的なメチオニンの酸化を評価した。ペプチドを含有する個々のメチオニンの非酸化型および酸化型のピーク面積を、質量スペクトルにより抽出されたイオンクロマトグラムを使用して決定した。個々のメチオニン残基に関して、酸化メチオニンのパーセントを、酸化ペプチドの質量スペクトルピーク面積を、酸化および非酸化ペプチドのピーク面積の合計で割ることによって計算した。
【0181】
以下の表26に示すように、150mg/mLロットおよび5種の100mg/mLロットに関するメチオニン酸化の結果は同様であった。
【0182】
【表30】
【0183】
重鎖のMet251およびMet427は最も不安定であり、最も高い酸化程度を示す。比較可能性を評価するために同時に試験されたロットの中で、Met251およびMet427に関する酸化レベルは、それぞれ4.8%および1.8%を超えなかった。
【0184】
5.6.16.結合効力(CD25の結合)
DAC HYP150mg/mLおよび100mg/mLのロットを、放出試験の一部の効力の測定として、ELISAを介してIL−2受容体のアルファサブユニット(CD25)との結合に関して評価した。マイクロタイタープレートに可溶性CD25を固定し、さまざまな量のDAC HYPと一緒にインキュベートした。結合したDAC HYPを、ホースラッディシュペルオキシダーゼコンジュゲートヤギ抗ヒトIgG抗体と、3,3’,5,5’−テトラ−メチルベンジジン基質とを並行して使用して検出した。得られた吸光度値を、4−パラメーターフィットを使用してlog10のDAC HYP濃度に対してプロットし、相対効力値のパーセントを、平行線分析を使用して作製した。
【0185】
製剤原料の結果を以下の表に要約する。
【0186】
【表31】
【0187】
150mg/mLロットの結合効力の結果は、100mg/mLロットの結合効力の結果と同様であった。
【0188】
5.6.17.表面プラズモン共鳴(CD25の結合)
表面プラズモン共鳴分析を実施し、DAC HYPとIL−2受容体のアルファサブユニット(CD25)との結合相互作用に関する親和定数(KD)を決定した。
【0189】
ヤギ抗ヒトIgG Fc抗体をチップの表面に固定してDAC HYP試料を捕捉し、その後、可溶性CD25をさまざまな濃度で、捕捉されたDAC HYPの上に自動化法を使用して2連に注入した。結合データを回収し、参照フローセルおよび緩衝液ブランクを使用して修正し、平衡定数を得るために1:1 Langmuirモデルを使用してBIA Evaluationソフトウェアを用いてフィットさせた。
【0190】
DAC HYP150mg/mLロットおよび参照標準RS0801に関する結果を、表28に要約する。
【0191】
【表32】
【0192】
150mg/mLロットの会合定数(ka)、解離定数(kd)および親和定数(KD)の値は、参照標準RS0801のものと同様であった。
【0193】
5.6.18.機能的効力
DAC HYP150mg/mLおよび100mg/mLのロットを、放出試験の一部として機能的効力を評価した。機能的効力のアッセイは、DAC HYPがIL−2受容体のアルファサブユニット(CD25)と結合することによる、IL−2誘導性T細胞増殖の阻害を測定する。IL−2の存在下で、さまざまな量のDAC HYPをIL−2受容体を発現するKIT−225K6細胞(Horiら、1987年、Blood70:1069−1072頁)と一緒にインキュベートした。DAC HYPによるT細胞増殖の阻害を、アラマーブルーを使用して続けて検出した。得られた蛍光値を、4−パラメーターフィットを使用してlog10のDAC HYP濃度に対してプロットし、相対効力値のパーセントを、平行線分析を使用して作製した。
【0194】
機能的効力の結果を表29に要約する。
【0195】
【表33】
【0196】
150mg/mLロットの機能的効力の結果は100mg/mLロットの機能的効力と同様であった。
【0197】
5.6.19.抗体依存性細胞傷害性
DAC HYP150mg/mL製剤の2種のロットを、参照標準RS0801の100mg/mL DAC HYPのロットと比較して評価した。
【0198】
IL−2受容体発現KIT−225K6細胞を、51Crを用いて標識し、続けてDAC HYPと一緒にインキュベートした。ヒトエフェクター細胞(PBMC)をさまざまな量で加え、エフェクター対標的細胞(KIT−225K6)の異なる比率を達成した。Fc受容体産生単球は、DAC HYP Fcドメインと相互作用し、続いて標的細胞の溶解を起こす。細胞傷害性の程度は、標的細胞由来の51Crの放出を測定することにより決定し、最大細胞溶解の百分率として表した。
【0199】
多数のドナー由来のPBMCを、各試料に利用した。各ドナーに関して、試料のADCC活性のパーセントを、細胞傷害性のパーセントに基づき、参照標準RS0801のADCC活性に対して計算した。ADCCの結果を、以下の表30に要約する。
【0200】
【表34】
【0201】
150mg/mLロット、参照標準RS0801、陽性および陰性の対照抗体ならびに抗体を含まない対照(抗体独立性細胞傷害性またはAICCのため)に関する反応曲線を、図20に示す。
【0202】
150mg/mLロットのADCC活性は、参照標準RS0801のADCC活性と同様であった。
【0203】
5.6.20.残存プロテインA
残存プロテインAはELISA法によって決定でき、標準、試料対照、プレートブランクおよび試験試料を、変性緩衝液を用いて希釈し、プロテインAを解離させるために沸騰水のバスに配置し、変性させ、ダクリズマブを沈降させた。沸騰後、標準、対照および試料を冷却し、遠心分離にかけ、ポリクローナル抗プロテインA捕捉抗体をコーティングしたマイクロタイタープレートに加えた。試料中に存在する残存プロテインAを、その後、ビオチン化抗プロテインA抗体と並行してストレプトアビジンアルカリホスファターゼおよびP−ニトロフェニルリン酸塩(PNPP)基質を使用して検出する。プレートを、分光光度プレートリーダーにおいて分析し、log−log標準曲線を作製し、これに対してプロテインAの濃度を決定する。試験試料の結果を、百万分率(ppm)単位で報告する。百万分率の結果はng/mLのプロテインAの結果を、mg/mLの試験試料の抗体濃度で割ることによって計算する。
【0204】
5.6.21.DNA含有量
マウスDNAの検出を、契約研究所において定量的ポリメラーゼ連鎖反応(Q−PCR)試験法を使用して決定する。この方法において、試料をDNA抽出に供する。その後、試料抽出液を、マウスDNAの反復成分の特異的断片を増幅するためにマウス特異的プライマーおよびプローブを使用して、Q−PCRによって分析する。DNAの増幅は検出される蛍光シグナルをもたらす。試料中のDNAを、既知の量のマウスDNAを使用して作製した標準曲線と比較することによって測定する。結果は、DNAのピコグラム/抗体のミリグラムで表す。さまざまなロットのDAC HYP薬剤生産物中の平均DNA含有量を、表17に要約する。
【0205】
5.6.22.宿主細胞タンパク質(HCP)
生産物中の残存宿主細胞タンパク質を、市販のキットを使用して定量する。NS0細胞溶解物に対する、アフィニティ精製したヤギポリクローナル抗体を、NS0 HCPの捕捉および検出の両方に使用する。HCP標準を、疑似的生産工程から無細胞収穫材料を回収することによって生産する。HCP作業標準を使用して標準曲線を作製し、HCPを含有する試料を、標準曲線の範囲を標的として段階的に希釈する。標準、試料対照および試験試料を、抗NS0 HCPポリクローナル抗体コーティングプレートに加える。その後、宿主細胞タンパク質を、ホースラッディシュペルオキシダーゼ(HRP)にコンジュゲートした抗NS0 HCPポリクローナル抗体と並行して3,3’,5,5’−テトラ−メチルベンジジン(TMB)基質を用いて検出する。次いで、プレートを、分光光度プレートリーダーにおいて分析し4つのパラメーターカーブフィットを作製し、試料中のHCPの量を定量する。
【0206】
NS0 HCP ELISAアッセイに関する結果を、百万分率(ppm)単位で報告する。百万分率の結果はng/mLのHCPの結果を、mg/mLの抗体濃度で割ることによって計算する。DAC HYP薬剤生産物のさまざまなロットの平均HCPを表17に要約する。
【0207】
5.6.23.ポリソルベート80濃度
ポリソルベート80は、有色のチオシアン酸コバルトとポリソルベート80の複合体の形成に基づく分光光度方法を使用して定量する。標準曲線を、一連のポリソルベート80標準を使用して構築する。試料中のポリソルベート80濃度を、標準曲線から決定する。DAC HYP薬剤生産物のさまざまなロットのポリソルベート濃度範囲を、表17に要約する。
【0208】
5.6.24.浸透圧
浸透圧は、蒸気圧降下浸透圧計を使用して測定する。試料分析の前に、浸透圧計を、試料の予想浸透圧を一括する浸透圧標準を使用して較正する。DAC HYP薬剤生産物のさまざまなロットの浸透圧範囲を表17に要約する。
【0209】
5.6.25.結論
実施された物理化学的および生物学的分析は、DAC HYP150mg/mLおよびDAC HYP100mg/mL製剤の総合評価を提供する。今までのところ、すべての被験ロットの物理化学的および生物学的特徴は同様である。
【0210】
全てのDAC HYPロットに関して、N末端配列決定により決定された重鎖および軽鎖の最初の15アミノ酸、ペプチドマップおよび分子量分析は、ダクリズマブの遺伝子配列と一致した。
【0211】
SEC−MALSおよびSV−AUCにより決定した、すべての被験150mg/mLおよび100mg/mLのロット中のアグリゲートのレベルおよびアグリゲート種のサイズ分布は同様であり、ゲル電気泳動により試験したそれらの純度も同様であった。
【0212】
150mg/mLロットの電荷アイソフォーム分布は100mg/mLロットの電荷アイソフォーム分布と同様であり、pE/VHS(150mg/mLロット=31%pE/VHS;100mg/mLロット=34から42%pE/VHS)およびQ/VHS(150mg/mLロット=11から12%Q/VHS;100mg/mLロット=4から9%Q/VHS)N末端アイソフォームの相対量にわずかに差があっただけである。DAC HYPの特徴は以下のとおりである。
【0213】
CEXによるN末端アイソフォーム:
pE/pE:31−46%
pE/Q:7−12%
pE/VHS:31−42%
Q/VHS:3−12%
VHS/VHS:1−17%
CEXによるC末端アイソフォーム:
0K:53−80%
1K:14−28%
2K:5−19%
DAC HYPのN連結グリカンの分布は以下の通りである。
G0−G1cNAc:7.2−14.6%
G0:80.9−88.2%
ピーク3:1.3−1.7%
G1:1.4−3.8
DAC HYPに関して測定された酸化レベルは低かった。
【0214】
ELISAおよび表面プラズモン共鳴のCD25結合実験において確認されたように、DAC HYPは生物学的に活性であり、IL−2誘導性T細胞増殖の阻害に対して機能性である。DAC HYPはまた、貯蔵における低レベルのアグリゲーションを特徴とする。
【0215】
5.7.DAC HYPの安定性
高濃度のDAC HYP製剤は貯蔵において安定である。以下の表は、150mg/mL DAC HYP製剤原料ロットに関する安定性データを提供する。
【0216】
以下の表31は、推奨条件(2−8℃)における、50mLバッグ中での貯蔵後の安定性データを提供する。以下の表32は、23−27℃における50mLバッグ中での貯蔵の加速安定性データを提供する。以下の表33は、ストレス安定性データを提供する。
【0217】
【表35】
【0218】
【表36】
【0219】
【表37】
【0220】
5.8.ダクリズマブのさまざまな形態間の比較
Hoffman−La Roche,Inc.(「Roche」)は、ZENAPAX(商標)という商品名で販売される、製造中止となっている同種移植の拒絶反応治療用ダクリズマブの静注製剤を製造した。DAC Penzbergは、PDL BioPharmaによる臨床試験に使用されるダクリズマブの100mg/mlの皮下製剤である(以下のセクション5.9.1に記載のCHOICE研究を参照されたい)。
【0221】
DAC HYP、ZENAPAX DACおよびDAC Penzbergの製剤の間の比較を、表34に示す。表において、製剤化緩衝液はDACがダイアフィルトレーションされ、最終製剤を得た緩衝液である。したがって、記された濃度は名目濃度である。
【0222】
【表38】
【0223】
DAC HYPのさまざまな特徴をZENAPAX DACおよびDAC Penzbergのさまざまな特徴と比較した。
【0224】
DAC HYP対ZENAPAX DACのグリコシル化間の比較を図21に示す。ダクリズマブの3種の形態の例示的ロットの分析を表35に述べる。
【0225】
【表39】
【0226】
DAC HYPはまた、ZENAPAX DACより有意に低レベルのマンノースグリコシル(例えば、Man5、Man6、Man7)および低レベルのシアル化グリコシルも有する(例えば図21を参照されたい)。
【0227】
抗体依存性細胞介在性細胞傷害性(ADCC)は、Fc依存性活性および抗体−標的結合の潜在的細胞傷害作用を査定するために使用できる、インビトロアッセイである。エフェクター細胞として6例の健康なドナー由来の末梢血単核細胞(PBMC)および標的細胞としてCD25発現KIT225/K6細胞系を使用して、可変のエフェクター細胞対標的細胞比のフォーマットまたは可変抗体濃度フォーマットの両方において、さまざまなダクリズマブ調製物のADCC活性をアッセイした。
【0228】
可変のエフェクター細胞対標的細胞比のフォーマットに関して、51Cr標識KIT225/K6細胞(12,500細胞/ウェル)を、1μg/mLの抗体(最終濃度)と一緒に、30分間4℃において、V底96−ウェルプレート中で100μLの容積のADCCアッセイ培地(435mLのRPMI−1640、5.0mLのL−グルタミン、50mLの熱不活性化ウシ胎児血清、500μlの1000×2−メルカプトエタノール、5.0mLのペニシリン−ストレプトマイシン(100×)、および5.0mLのHEPES(1Mストック)/500mLを含有する)と一緒にプレインキュベートし、対照細胞は、その後の抗体独立性51Cr放出の決定のために、抗体の不在下でADCCアッセイ培地においてインキュベートした。
【0229】
PBMC(エフェクター)を、ADCCアッセイ培地において別々の96ウェルポリプロピレンプレート中に段階的に希釈し、6.25×105細胞、3.13×105細胞、1.56×105細胞、7.81×l04細胞または3.91×104細胞の細胞濃度/100μLを得た。100μL/ウェルの容積のPBMC懸濁液を、51Cr標識KIT225/K6および抗体を含有するプレートに加え、50:1、25:1、12.5:1、6.25:1および3.13:1のエフェクター対標的(E:T)比を得た。さらに、100μL/ウェルの容積のADCCアッセイ培地単独(エフェクターなし)を、51Cr標識KIT225/K6+mAbに加え、51Crの自然放出を決定した。アッセイプレートを、50RCFにおいて2分間回転させ、37℃で7.5%CO2インキュベーターにおいて4時間インキュベートした。
【0230】
4時間のインキュベーション終了の30分前、8%TritonX−100の25μLの容積を、適切な対照ウェルに加え、標的細胞由来の51Crの最大放出を決定した。4時間のインキュベーションの完了において、プレートを350RCFにおいて5分間回転させ、各ウェル由来の上清の100μLの容積を、ミニチューブに移した。各ミニチューブを、シンチレーションバイアルに挿入し、Beckman Gamma5500Bカウンターまたは同等物において1分間カウントした。
【0231】
可変抗体濃度のフォーマットに関して、51Cr標識KIT225/K6細胞(12,500細胞/ウェル;標的)を、mAb(最終濃度)のさまざまな用量の抗体(5、1、0.2、0.04、0.008および0.0016μg/mL)と一緒に、30分間4℃においてV底96−ウェルプレート中で100μLの容積のADCCアッセイ培地においてプレインキュベートした。対照細胞は、その後の抗体独立性51Cr放出の決定のために、ADCCアッセイ培地単独(mAbなし)と一緒にインキュベートした。
【0232】
PBMC(エフェクター)を、別々の96ウェルポリプロピレンプレートにおいて、3.13×105細胞/100μLの濃度にADCCアッセイ培地で段階的に希釈した。PBMC懸濁液の100μL/ウェルの容積を、51Cr標識KIT225/K6+mAbを含有するプレートに加え、25:1のエフェクター対標的(E:T)比を得た。さらに、100μL/ウェルの容積のADCCアッセイ培地単独(エフェクターなし)を、51Cr標識KIT225/K6+mAbに加え、51Crの自然放出を決定した。アッセイプレートを、50RCFにおいて2分間回転させ、37℃で7.5%CO2インキュベーターにおいて4時間インキュベートした。
【0233】
4時間のインキュベーション終了の30分前、8%TritonX−100の25μLの容積を、適切な対照ウェルに加え、標的細胞由来の51Crの最大放出を決定した。4時間のインキュベーションの完了において、プレートを350RCFにおいて5分間回転させ、各ウェル由来の上清の100μLの容積を、ミニチューブに移した。各ミニチューブを、シンチレーションバイアルに挿入し、Beckman Gamma5500Bカウンターまたは同等物において1分間カウントした。
【0234】
ADCCの結果を図22A(可変のエフェクター細胞対標的細胞比のフォーマット)および図22B(可変抗体濃度フォーマット)に示す。これらのデータは、段階的濃度において試験されたDAC HYPにより達成された最大ADCC活性が、同じ濃度のZENAPAX DACおよびDAC Penzbergにより発揮される活性よりおよそ30−40%低かったことを示す。
【0235】
DAC HYP対ZENAPAX DAC対DAC Penzbergの電荷アイソフォームプロファイル(N末端変異体と一致する)の比較を、図23に示す。
【0236】
5.9.DAC HYP臨床試験
5.9.1.CHOICE研究
CHOICE試験は、230例の再発性MS患者においてインターフェロンベータ療法に加えた、ダクリズマブの第2相、ランダム化、二重盲検、プラセボ対照試験であった。この試験は、皮下注射として投与した100mg/mlのDAC Penzberg(生産品の説明に関する上記のセクション5.8を参照されたい)の2種の投与計画:1mg/kgのダクリズマブを4週間ごとに投与、および2mg/kgダクリズマブを2週間ごとに投与、を試験した。この研究の結果は、インターフェロンベータ療法に2mg/kgで2週間ごとに投与するダクリズマブを加えることによって、インターフェロンベータ療法単独と比較して、新しい、または拡大したガドリニウム増強病巣が24週目に有意に縮小されることを示した。
【0237】
CHOICE研究の結果は、Wynnら、2010年、Lancet Neurol.9(4):381−90頁に記載されている。ダクリズマブ治療は、一般的に忍容性良好であった。一般的な有害事象は、すべての治療群において類似であった。グレード3の有害事象は、24パーセントのDAC/IFNβ治療患者および14%のプラセボ/IFNβ治療患者に観察された。最も頻度の高いグレード3有害事象は、感染症であり、7%のDAC/IFNβ治療患者および3パーセントのプラセボ/IFNβ治療患者に起こった。日和見感染または死亡は無く、すべての感染症は、標準的な療法で解消された。
【0238】
CHOICE試験は、IFNβ−al療法の経歴のあるMS患者において、ダクリズマブが忍容性良好であり、IFNβ−al単独と比較して、新しい/拡大したガドリニウム増強(Gd+)病巣において72%の用量依存性縮小を起こしたことを実証した。臨床的有効性は、免疫調節性CD56bright ナチュラルキラー(NK)細胞の著しい拡大を伴った。
【0239】
5.9.2.SELECT研究
ランダム化二重盲検プラセボ対照用量範囲探索試験(SELECT)を実施し、DAC HYPの2種の異なる投薬量レベルの安全性および有効性を決定した。
【0240】
概説。本研究は、チェコ共和国、ドイツ、ハンガリー、インド、ポーランド、ロシア、ウクライナおよび英国における76の施設で実施された。個々の患者のケアは、治療を行う神経科医、治療を行う看護師(または研究コーディネーター)、検査を行う神経科医、MRIの検査技師および薬剤師(または承認された被指名人)が関与する。集中型双方向音声応答システム(Interactive Voice Response System)を、すべての部位にわたるランダム化に使用した。プロトコルで規定された暫定無益分析を、150例の患者が24週の通院を完了した後に実施した。
【0241】
患者。臨床的に明確な再発寛解型多発性硬化症(2005年 McDonald基準1−4に従って;Polmanら、2005年Ann Neurol58:840−846頁を参照されたい)であり、ベースラインの総合障害度評価尺度(Expanded Disability Status Scale)(EDSS)が0−0.50(Kurtzke、1983年、Neurology33(11):1444−52頁)およびランダム化の前12か月に少なくとも1回MSが再発した、またはランダム化の前6週間以内に実施された脳MRIにおいて1つの新しいGd+病巣があった、適格基準に含まれる18−55歳の患者をランダム化し、DAC HYP(150mgまたは300mg)またはプラセボのいずれかを、4週ごとに1回の52週間の皮下注射として与えた。妊娠の可能性がある患者は、有効な避妊の実践を必要とした。
【0242】
患者が、原発性進行性、二次進行性もしくは進行性再発性のMS、悪性腫瘍の病歴、重症のアレルギー反応もしくはアナフィラキシー反応もしくは既知の薬剤過敏症または治験責任医師の立場から考えて、DAC HYPの投与を妨げる他の深刻な医学的条件を有する場合、その患者は排除した。さらなる排除基準は、DAC HYPまたはZENAPAX(商標)、全身リンパ節照射法、クラドリビン、ミトキサントロン、T−細胞もしくはT−細胞受容体のワクチン接種またはナタリズマブもしくはリツキシマブを除く任意の治療用mAbを用いた以前の治療を含む。ランダム化の時点において、患者は、前年にシクロホスファミドもしくはリツキシマブを用いた治療、6か月以内にナタリズマブ、シクロスポリン、アザチオプリン、メトトレキサート、静注用免疫グロブリン、血漿交換療法または細胞吸着除去療法を用いた治療、またはランダム化前の3か月以内に生ウイルスワクチン、酢酸グラチマー、IFNβ、インターフェロンアルファを用いた治療、または30日以内にコルチコステロイド、4−アミノピリジンまたは関連製品を用いた治療を受けることはできなかった。
【0243】
この群の特徴は以下の通りであった。
【0244】
【表40】
【0245】
エンドポイント。この研究の第1の目的は、DAC HYP単独療法が、52週において年換算再発率(ARR)で定義されたMSの再発を減少させるかどうかを決定することであった。再発は、検査を行う神経科医による査定において、新しい神経学的発見を伴う、24時間を超えて続く新しいまたは繰り返し起こる神経症状(熱または感染症を伴わない)として定義された。3人の盲検のMS神経科医からなる独立した神経学評価委員会(Independent Neurology Evaluation Committee)(INEC)は、すべての疑わしい再発を評価し、MS再発のプロトコル定義を満たすかどうかを判定した。INECが承認した再発だけがは、一次分析に含まれた。
【0246】
第2の目的は、患者のサブセットにおいて、8、12、16、20および24週に実施された脳MRIスキャンにおいて累積する新しいGd+病巣の数の減少、;52週における新しい、または新たに拡大したT2高強度病巣の数の減少;ベースラインと52週の間に再発する患者集団の減少;および29項目の多発性硬化症インパクトスケール(Multiple Sclerosis Impact Scale)(MSIS−29)(Hobartら、2001年、Brain 124(Pt5):962−73頁)の52週における身体的影響スコアのベースラインからの変化により測定される生活の質の改良(QoL)にDAC HYPが有効であるかどうかを決定することであった。確認された障害の進行を、ベースラインおよび52週の間のEDSSスコアの変化によって査定した(ベースラインEDSS≧1.0に関してEDSSは1.0ポイント上昇、または12週間持続したベースラインEDSS=0に関して1.5ポイント上昇)。EDSSの評価は、12週ごとならびに20、52、60および72週に実施した。
【0247】
追加のQoLエンドポイントは、EQ−視覚的アナログ尺度(Visual Analogue Scale)(EQ−VAS)(EuroQol−生活健康関連の質の測定のための新しい機関、2011年、17.11.11アクセス、http://www.euroqol.org/)により査定された対象の幸福の全体的査定であり、EQ−5D健康調査(EuroQol−生活健康関連の質の測定のための新しい機関、2011年、17.11.11アクセス、http://www.euroqol.org/);12項目の略式健康調査SF−12(Wareら、1996年、Medical Care34(3):220−33頁)および52週におけるMSIS−29の精神的スケール(Hobartら、2001年、Brain 124(Pt5):962−73頁)における変化であった。
【0248】
追加のMRIエンドポイントは、52週におけるGd+病巣の数、24および52週における全体および新しい、または新たに拡大したT2高強度病巣の容積、24および52週における全体および新しいT1低強度病巣「ブラックホール」(灰白質に対して等強度/低強度であり、ガドリニウム投与後に増強されなかった病巣として定義される)の容積ならびにSIENA法(Smithら、2001年、J Comput Assist Tomogr25(3):466−75頁)により査定された脳全体の容積における百分率の変化であった。
【0249】
リンパ球のサブセットは、検証されたFACSアッセイを使用して複数の時点で測定した。CD56brightNK細胞は、CD3−/CD16+/CD56brightリンパ球として定義された。DAC HYPに対する免疫原性を、抗薬剤抗体をスクリーニングするための標準的ELISAを使用して査定し、その後、細胞アッセイを使用して、すべての陽性試料における中和抗体に関して試験した。
【0250】
統計分析。サンプルサイズおよそ600例の患者を、DAC HYP治療群およびプラセボ群の間のARRにおいて50%の減少を検出するための90%の検出力を有するように選択し、10%の脱落率、5%の第1種過誤率および両側検定の負の2項分布と仮定するシミュレーションから推定した。プラセボ群におけるARRは、RRMS対象において最近完了した試験に基づき、0.476であると仮定された。報告されたすべてのp値は、両側である。
【0251】
一次分析により、各DAC HYP群対プラセボ間のARRの差が評価された。別のMS薬物を用いた救命治療後に起こった再発は打ち切りであった。差は、研究登録前の年の再発数に関して補正される負の二項回帰モデル、ベースラインEDSS(EDSS≦2.5対EDSS>2.5)およびベースライン年齢(≦35対>35歳)を使用して評価した。二次分析は、負の二項回帰(8および24週の間の新しいGd+病巣の数;新しい、または新たに拡大したT2高強度病巣の数)、Cox比例ハザードモデル(最初の再発までの時間、疾患の進行までの時間)および分散モデルの分析(EDSSにおける変化、新しい、または新たに拡大したT2病巣の容積、新しいT1低強度病巣の容積、QoL)ならびに比例オッズモデル(52週における新しいGd+病巣の数)を使用して治療差に関して試験した。再発なしの患者の割合は、Kaplan−Meierの生存率曲線分布から推定した。
【0252】
8および24週の間の新しいGd+病巣の累積数に関して、患者が1または2回の連続スキャンまたはすべてのスキャンを逃した場合、最後のベースラインではない観察をそれより先に実施するか、または各治療群内の病巣の平均数をそれぞれ使用した。他のMRIエンドポイントに関して、欠測データは、治療群内の平均を使用して転嫁した。MSIS−29に関して、患者が<10項目を逃した場合、非欠測項目の平均を使用してスコアに転嫁した。≧10項目および他のQoL測定を逃した患者に関して、ランダムスロープおよび切片モデルを使用して欠測データを推定した。
【0253】
有効性エンドポイントに関する統計的試験は、DAC HYP300mg群対プラセボおよびDAC HYP150mg群対プラセボの別々の比較を使用した。逐次的閉検定手順を使用して、多重比較による全体の第1種過誤率を調節した。
【0254】
有効性分析を、ランダム化を受けたすべての患者が含まれる治療意図(intent−to−treat)(ITT)集団において評価した。しかし、1つの研究施設からの21例の患者は、研究完了前に特定された、該施設における不正確な投薬の証拠ため(該施設の全ての患者は、積極的治療を受けていた)、研究完了前にITT集団からあらかじめ除外した。感度分析において、すべての一次および二次有効性分析を、すべてのランダム化した患者を使用して繰り返した。すべての安全性分析は、研究薬剤の少なくとも1回の投与を受け、少なくとも1回のランダム化後の査定を受けたすべての患者として定義される安全性集団に基づいた。
【0255】
あらかじめ計画された無益性分析を、150例の対象が24週の通院を完了した後に実施し、DAC HYPの仮定された効果が明白でなかった場合、停止する機会を提供した。有効性は、研究期間にわたって変化し得るので、無益性分析の時に、優位性の証拠のために研究を早期に停止する計画は無かった。無益性は、各投薬群に関する8および24週の間の新しいGd+病巣の累積数ならびにARRエンドポイントの両方に関する条件付き検出力を別々に推定することによって査定した。安全性モニタリング委員会(Safety Monitoring Committee)は、分析時のデータを検討し、データの全体的一貫性およびリスク便益の査定に基づき、研究を続けることを推奨した。
【0256】
結果の要約。
【0257】
適格な参加者を、2/15/2008から5/14/2010までランダム化した。ベースラインの特徴は、3種の治療群にわたって類似であるが、DAC HYP150mg群の患者がDAC HYP300mg群の患者より多くのT2およびGd+T1病巣を有する傾向があった。すべてのランダム化された患者にわたって、合計577例(93%)が、研究を完了したDAC HYPおよびプラセボ治療患者と類似の割合で治療期間を完了した。
【0258】
詳細な結果。臨床的有効性。52週におけるARR(一次エンドポイント)は、プラセボ(0.46;表36)と比較して、DAC HYP150mg(0.21)または300mg(0.23)にランダム化された患者に関してより低く、DAC HYP150mg(95%CI、31%から69%、p<0.0001)によりプラセボに対して54%の減少、およびDAC HYP300mg(95%CI、26%から66%、p=0.0002;表36)に関してプラセボに対して50%の減少を示した。52週にわたって、再発患者の割合は、プラセボ群において再発した36%に対してDAC HYP150mg(19%)および300mg(20%)の群において減少した(両方の比較に関してp≦0.001)(表36)。プラセボと比較して、52週における3か月持続する障害進行の危険性は、DAC HYP150mgにおいて57%(ハザード比=0.43;95%CI、0.21から0.88;p=0.021)およびDAC HYP300mg群において43%(ハザード比=0.57;95%CI、0.30から1.09;p=0.091)減少した。
【0259】
52週におけるMSIS−29身体的スコアで相対的に4.0の改良が、プラセボに対してDAC HYP150mgに観察され、DAC HYP300mg患者において著しい変化は低かった(p<0.0008およびp=0.1284対プラセボ、それぞれ;表36)。身体的および精神的機能両方ならびに全体の健康の測定を含む、生活の健康関連の質の他の測定についてもまた、類似の改良が観察された(表36)。
【0260】
【表41】
【0261】
MRI。DAC HYPは、MRIにより定義されたように、新しいMS病巣の活性を、
全研究集団および8から24週の間に実施される月1回のMRIによるサブセットの両方
において減少させた(表36)。臨床的エンドポイントと対照的に、有効性のこのポイントの推定値は、潜在的ベースラインの不均衡を調整後でも、150mg用量群と比較して300mg用量群においてわずかに強かった。縦モード解析は、Gd+病巣の活性が、治療の最初の2、3か月は300mg用量群と比較して150mg用量群において高かったが、52週には類似であったことを実証した(表36)。1つの排除研究現場由来の21例の患者を含んだ感度分析は、すべての有効性分析に関して類似の結果を得た。
【0262】
安全性。有害事象(AE)は、DAC HYP150mg(73%)、DAC HYP300mg(76%)およびプラセボ(79%)の群中の患者に類似の割合で起こった(表37)。重篤なAEは、プラセボ群の26%、DAC HYP150m群において15%およびDAC HYP300mg群において17%で起こった。MSの再発を除いて、SAEは各群の患者の6%、7%および9%で起こった(表37)。>5%のDAC HYP患者において起こったAEを、表37に示す。重篤な感染症の発生率は、DAC HYP治療患者中における2%対プラセボにおける0%であった。投薬が続いていた間に重篤な感染症になった7例の患者のうち、1例は重篤な感染症のため治療を中止し、6例は、感染症が解消後治療を再開した。皮膚事象の発生率は、DAC HYP150mg群において18%、DAC HYP300mg群において22%およびプラセボ群において13%であった(表37)。重篤な皮膚事象は、DAC HYP治療患者の1%において起こった。重篤な発疹から回復していた1例のDAC HYP治療患者は、腰筋膿瘍の合併症のため死亡した。剖検において、以前に診断未確定であった腰筋膿瘍は、腸間膜動脈に関与することが見いだされ、局所血栓症および急性虚血性大腸炎をもたらした。5例の悪性腫瘍が試験中に起こり、2例の子宮頸がん(プラセボ群およびDAC HYP150mg群において各1例);DAC HYP150mgにおける1例の甲状腺新生物は、重篤ではない甲状腺結節ではなく、DAC HYP300mg群における2例のメラノーマであった。メラノーマの症例は、局所切除により治療され、再発の報告はなかった。
【0263】
【表42】
【0264】
実験結果。DAC HYPを用いて治療された患者は、52週においてプラセボと比較して合計NK細胞カウント(細胞/mm3)が増加した(中央値:42.0(150mgDAC HYP);46.5(300mgDAC HYP);対−4.5プラセボ;p=<0.001)。合計NK細胞数の増加は、ベースラインにおける中央値7.77から治療終了時の44.84である、CD56brightNK細胞の選択的増加に関連していた。対照的に、CD56dimNK細胞はわずかに変化しただけであった(中央値の変化は122.68から123.70)。CD56brightNK細胞の拡大は、DAC HYP治療群対プラセボの両方において最初のベースライン後の時点(4週)で明らかであった(p<0.0001)。CD56brightNK細胞は、ベースラインのリンパ球の0.6%の中央値から52週の2.8%に拡大した。対照的に、DAC HYPを用いて治療された患者は、B細胞および合計リンパ球カウントがわずかに減少した(表38)。CD4+およびCD8+T細胞両方のカウントは、DAC HYP治療患者で52週においておよそ7−10%減少し、CD4+/CD8+比は、治療期間中一定を維持した。
【0265】
【表43】
【0266】
肝機能検査(LFT)の異常は5X ULNを上回り、4%のDAC治療患者および<1%のプラセボ治療患者において起こった。これらの異常は、通常、治療期間の後期(中央値 発症+308日)に起こり、中央値にして62日で解消した。>5X ULNに上昇した17例のDAC HYP治療患者のうち、6例が、解消後少なくとも6か月間、DAC HYPを用いて治療を続け、または再開し、この期間中全員に再発はなかった。2例の患者において、LFTの上昇は感染症を伴った(1例がB型肝炎および1例がサイトメガロウイルス感染症)。
【0267】
免疫原性。24週において、DAC HYPに対する中和抗体を、6例(2%)のDAC HYP治療患者(150mg用量群中5例の患者および300mg用量群中1例の対象)において検出した。一部の患者において、これらの抗体は一時的であり、52週において、DAC HYPに対する中和抗体は、各DAC HYP用量群から1対象存在しただけであった。
【0268】
結論。毎月の、皮下DAC HYP単独療法によるCD25の拮抗作用は、例えば、再発率の減少、MRIにより定義された新しい病巣およびMS患者の優勢に治療された天然集団における障害の進行により測定して、MS疾患活性について1年にわたって強固で臨床的に意味のある効果を実証した。
【0269】
6.具体的な実施形態、参照による組み込み
本開示の様々な態様は、以下の番号付きパラグラフに述べられる実施形態に記載されている。
1.無血清および無コレステロール培地において増殖するよう適応されており、組換えタンパク質を発現するよう操作されている、無血清および無コレステロール培地において増殖される場合、10日間のフェドバッチ法において100Lの培養物中100mg/L/日の組換えタンパク質を超える容積生産性を達成することができる改変されたNS0細胞。
2.コレステロールおよび動物由来成分のない培地において増殖される場合、10日間のフェドバッチ法において1,000Lの培養物中100mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、実施形態1に記載の改変されたNS0細胞。
3.コレステロールおよび動物由来成分のない培地において増殖される場合、10日間のフェドバッチ法において16,000Lの培養物中100mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、実施形態1に記載の改変されたNS0細胞。
4.流加培地が、追加される容積が最初の細胞培養物容積の割合を表す、次のスケジュール:
【0270】
【表44】
に従って追加される、実施形態1−3のいずれか1つに記載の改変されたNS0細胞。
5.13日間のフェドバッチ法において少なくとも100Lの培養物中200mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、実施形態1に記載の改変されたNS0細胞。
6.無コレステロール培地において増殖される場合、13日間のフェドバッチ法において1,000Lの培養物中200mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、実施形態5に記載の改変されたNS0細胞。
7.無血清および無コレステロール培地において増殖される場合、10日間のフェドバッチ法において16,000Lの培養物中100mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、実施形態5に記載の改変されたNS0細胞。
8.抗CD25モノクローナル抗体を発現するのに有用な核酸で安定的にトランスフェクトされる、実施形態1に記載の改変されたNS0細胞。
9.抗CD25モノクローナル抗体が、配列番号2の21位から233位に配列が一致するVL鎖および配列番号4の20位から465位に配列が一致するVH鎖を含む、実施形態8に記載の改変されたNS0細胞。
10.ベクターpAbX.gptを用いて形質転換された、実施形態1に記載の改変されたNS0細胞。
11.ベクターpHAT.IgG1.rg.dEを用いて形質転換された、実施形態1に記載の改変されたNS0細胞。
12.クローン7A11−5H7−14−43で表される実施形態1に記載の改変されたNS0細胞。
13.実施形態1−12のいずれか1つに記載の改変されたNS0細胞を培養することを含む、組換えタンパク質を生産する方法。
14.10日間のフェドバッチ法において100L、1,000Lもしくは16,000Lの培養物中少なくとも100mg/L/日の組換えタンパク質を、または13日間のフェドバッチ法において100L、1,000Lもしくは16,000Lの培養物中少なくとも200mg/L/日の組換えタンパク質を生産する条件下で、改変されたNS0細胞が培養される、実施形態13に記載の方法。
15.改変されたNS0細胞が、血清およびコレステロールの非存在下で培養される、実施形態13または実施形態14に記載の方法。
16.改変されたNS0細胞が、トロポロンおよびヒドロコルチゾンの非存在下で培養される、実施形態15に記載の方法。
17.改変されたNS0細胞が、10−35g/Lグルコースを含有する基本および/または流加培地において培養される、実施形態13または実施形態14に記載の方法。
18.改変されたNS0細胞が、15g/Lグルコースを含有する基本培地および/または28g/Lグルコースを含有する流加培地において培養される、実施形態17に記載の方法。
19.基本培地が、PFBM2±10%成分で構成されている、実施形態18に記載の方法。
20.流加培地が、PFFM3±10%成分で構成されている、実施形態18に記載の方法。
21.細胞が基本培地において1−3日間、次に流加培地において10−13日間培養される実施形態19または実施形態20に記載の方法。
22.流加培地が、表7において概要が述べられているスケジュール±10%に従って追加される、実施形態18に記載の方法。
23.哺乳動物細胞において作動可能である選択可能なマーカーの発現を推進する弱いプロモーターおよび対象のタンパク質の発現を推進する強いプロモーターを含む、対象のタンパク質を組換え的に発現するのに有用なベクター。
24.対象のタンパク質が治療抗体である、実施形態23に記載のベクター。
25.治療抗体が抗CD25抗体である、実施形態24に記載のベクター。
26.CD25抗体がダクリズマブのCDRを含む、実施形態25に記載のベクター。
27.CD25抗体がダクリズマブである、実施形態26に記載のベクター。
28.細胞に実施形態23−27のいずれか1つに記載のベクターをトランスフェクトし、10日間のフェドバッチ法において100L、1,000Lもしくは16,000Lの培養物中少なくとも100mg/L/日対象のタンパク質を、または13日間のフェドバッチ法において100L、1,000Lもしくは16,000Lの培養物中少なくとも200mg/L/日の組換えタンパク質を生産することができる細胞を選択することを含む、対象のタンパク質の高い容積生産性を有する哺乳動物宿主細胞を得るための方法。
29.ダクリズマブがpE/Q重鎖N連結アイソフォームおよび/またはQ/VHS重鎖N末端アイソフォームの存在により特徴付けられる、ダクリズマブを含む組成物。
30.pE/Q重鎖N末端アイソフォームがダクリズマブのおよそ6−15%を構成する、実施形態29に記載の組成物。
31.pE/Q重鎖N末端アイソフォームがダクリズマブのおよそ7−12%を構成する、実施形態29に記載の組成物。
32.Q/VHS重鎖N末端アイソフォームがダクリズマブのおよそ1−15%を構成する、実施形態29−31のいずれか1つに記載の組成物。
33.Q/VHS重鎖N末端アイソフォームがダクリズマブのおよそ3−12%を構成する、実施形態32に記載の組成物。
34.ダクリズマブの重鎖が次のN末端アイソフォーム:
【0271】
【表45】
に存在する、実施形態29に記載の組成物。
35.ダクリズマブの重鎖が次のN末端アイソフォーム:
【0272】
【表46】
に存在する、実施形態29に記載の組成物。
36.ダクリズマブが、図18のアイソフォームプロファイルと実質的に類似する陽イオン交換クロマトグラフィーアイソフォームプロファイルにより特徴付けられる、実施形態29に記載の組成物。
37.ダクリズマブがDAC HYPである、実施形態29に記載の組成物。
38.1つがオリゴ糖G0−GlcNAcに一致し1つがオリゴ糖G0に一致する2つの主要ピークを含有するN連結グリコシル化HPLCプロファイルによりダクリズマブが特徴付けられ、これら2つのピークの合わせたAUCが全ピークの総AUCのうちの約88−99.5%を構成する、ダクリズマブを含む組成物。
39.G0−GlcNAcピークのAUCが全ピークの総AUCのうちの約5−18%を構成し、G0ピークのAUCが全ピークの総AUCのうちの約75−92%を構成する、実施形態38に記載の組成物。
40.G0−GlcNAcピークのAUCが全ピークの総AUCのうちの約6−16%を構成し、G0ピークのAUCが全ピークの総AUCのうちの約78−90%を構成する、実施形態39に記載の組成物。
41.N連結グリコシル化プロファイルが約3%未満のMan5を有する、実施形態38−40のいずれか1つに記載の組成物。
42.N連結グリコシル化プロファイルが約0.5%未満のG2、Man6および/またはMan7を有する、実施形態41に記載の組成物。
43.N連結グリコシル化プロファイルがシアル酸付加オリゴ糖に一致する第三のピークを含有し、シアル酸付加オリゴ糖ピークのAUCが全ピークの総AUCのうちの1%以下を構成する、実施形態38に記載の組成物。
44.N連結グリコシル化プロファイルがオリゴ糖G1に一致する第三のピークを含有し、G1ピークのAUCが全ピークの総AUCのうちの約1−5%を構成する、実施形態38に記載の組成物。
45.G1ピークのAUCが全ピークの総AUCのうちの約1−2%を構成する、実施形態44に記載の組成物。
46.ダクリズマブが、図19のプロファイルにまたは図21の下のパネルのプロファイルに実質的に類似しているN連結グリコシル化HPLCプロファイルを有する、実施形態38に記載の組成物。
47.ダクリズマブがDAC HYPである、実施形態38に記載の組成物。
48.1μg/mLのダクリズマブ濃度および約25対1のエフェクター細胞対標的細胞比で、少なくとも3体の健康なドナー由来のエフェクター細胞および標的細胞としてKit225 K6細胞を使用して、インビトロ細胞アッセイにおいて測定される場合、35%未満のADCC平均細胞傷害性を示すダクリズマブを含む組成物。
49.ダクリズマブが、前記アッセイにおいて10%から30%のADCC平均細胞傷害性を示す、実施形態48に記載の組成物。
50.前記アッセイが少なくとも6体の健康なドナー由来のエフェクター細胞を使用する、実施形態48または実施形態49に記載の組成物。
51.前記アッセイが少なくとも10体の健康なドナー由来のエフェクター細胞を使用する、実施形態48または実施形態49に記載の組成物。
52.ダクリズマブがDAC HYPである、実施形態48−51のいずれか1つに記載の組成物。
53.約150−190mg/mLのダクリズマブ、ならびに希釈緩衝液を用いた組成物の希釈により約85−165mg/mLのダクリズマブを含有し、約267−327mOsm/kgの範囲の浸透圧および25℃で約pH5.8−6.2の範囲のpHを有し、サイズ排除クロマトグラフィーにより測定される場合、ダクリズマブの少なくとも約95%が単量体形態である希釈組成物を生じるような量の賦形剤を含む、ダクリズマブ製剤を作製するのに有用な組成物。
54.希釈緩衝液で希釈される場合、希釈組成物が約85−115mg/mLのダクリズマブを含有するような量の賦形剤を含有する、実施形態53に記載の組成物。
55.希釈緩衝液で希釈される場合、希釈組成物が約150±15mg/mLのダクリズマブを含有するような量の賦形剤を含有する、実施形態53に記載の組成物。
56.ダクリズマブの0.1%以下がアグリゲート形態である、約4から15mg/mLのダクリズマブを含む組成物。
57.ダクリズマブの最大2.5%がアグリゲート形態である、約4から15mg/mLのダクリズマブを含むダクリズマブ組成物を、弱陽イオン交換樹脂上のカラムクロマトグラフィーにより精製することによって得られる、実施形態56に記載の組成物。
58.弱陽イオン交換樹脂がCM−650Mである、実施形態57に記載の組成物。
59.CM−650M樹脂が約20mMのクエン酸ナトリウムを含有する平衡化緩衝液、pH4.4−4.6と平衡化しており、ダクリズマブが約20mMのクエン酸ナトリウムおよび約75mMの硫酸ナトリウムを含有する溶出緩衝液、pH4.4−4.6を用いて溶出される、実施形態58に記載の組成物。
60.クロマトグラフィーが、約10−30cmまたは約17−19cmの高さを有する樹脂床を使用して円筒形カラムにおいて実施され、ダクリズマブが約4−22℃または約18−22℃の範囲の温度で、約50−200cm/時または90−110cm/時の範囲の流速で溶出される、実施形態59に記載の組成物。
61.約85−165mg/mLのダクリズマブおよび約0.02−0.04%(w/v)ポリソルベート80を含み、約267−327mOsm/kgの範囲の浸透圧および25℃で約pH5.8−6.2の範囲のpHを有し、サイズ排除クロマトグラフィーにより測定される場合、ダクリズマブの少なくとも約95%が単量体形態である、ヒトに投与するのに適している組成物。
62.サイズ排除クロマトグラフィーにより測定される場合、ダクリズマブの少なくとも約99%が単量体形態である、実施形態61に記載の組成物。
63.約85−115mg/mLのダクリズマブを含む、実施形態61に記載の組成物。
64.約100mg/mLのダクリズマブ、約40mMのコハク酸ナトリウム、約100mMの塩化ナトリウムおよび約0.03%(w/v)ポリソルベート80から本質的になり、25℃で約6.0のpHを有する、実施形態63に記載の組成物。
65.約135−165mg/mLのダクリズマブを含む、実施形態61に記載の組成物。
66.約150mg/mLのダクリズマブ、約40mMのコハク酸ナトリウム、約100mMの塩化ナトリウムおよび約0.03%(w/v)のポリソルベート80から本質的になり、25℃で約6.0のpHを有する、実施形態65に記載の組成物。
67.約4から15mg/mLのダクリズマブを含むダクリズマブ組成物を適切な緩衝液中での限外濾過により濃縮して、約85−180mg/mLの範囲のダクリズマブ濃度を達成するステップ、および、場合によって、濃縮された組成物を希釈緩衝液で希釈するステップを含む方法により得られる、実施形態65に記載の組成物。
68.約85−165mg/mLのダクリズマブを含み、約2−8℃の範囲の温度での約12ヶ月の期間の貯蔵後にアグリゲート形態中のダクリズマブの百分率が約3%を超えない、皮下投与に適している医薬組成物。
69.約85−115mg/mLのダクリズマブを含む、実施形態68に記載の医薬組成物。
70.約135−165mg/mLのダクリズマブを含む、実施形態68に記載の医薬組成物。
71.約2−8℃の範囲の温度での約12ヶ月の期間の貯蔵後にアグリゲート形態中のダクリズマブの百分率が約2%を超えない、実施形態69または実施形態70に記載の医薬組成物。
72.約2−8℃の範囲の温度での約18ヶ月の期間の貯蔵後にアグリゲート形態中のダクリズマブの百分率が約3%を超えない、実施形態69または実施形態70に記載の医薬組成物。
73.(i)組換えタンパク質を発現し分泌する細胞培養物のpHを、約pH4.5−5.5の範囲のpHに調整するステップ、
(ii)pH調整された細胞培養物を約4から15℃の範囲の温度で約30−90分間インキュベートするステップ、および
(iii)インキュベートされたpH調整細胞培養物を遠心分離して細胞残渣を除去するステップ
を含む、細胞培養物から組換えタンパク質を収穫するための方法。
74.(i)未精製ダクリズマブ調製物から親和性クロマトグラフィー樹脂上にダクリズマブを吸着させるステップ、
(ii)親和性クロマトグラフィー樹脂を洗浄緩衝液で洗浄して混入物を除去するステップ、
(iii)吸着したダクリズマブを溶出緩衝液で溶出させるステップ、
(iv)pHを約3−4の範囲のpHに調整することにより溶出液中のウイルスを不活化し、pH調整された溶出液を規定温度でウイルスを不活化するのに十分な期間インキュベートするステップ、
(v)ウイルス不活化溶出液を約pH7.7−7.9(25℃で測定される)の範囲のpHまで中和するステップ、
(vi)中和された溶出液を強陰イオン交換クロマトグラフィー樹脂に通して流すステップ、
(vii)ステップ(vi)の溶出液のダクリズマブを弱陽イオン交換クロマトグラフィー樹脂上に吸着させるステップ、および
(viii)吸着したダクリズマブを弱陽イオン交換クロマトグラフィー樹脂から溶出させるステップ
を含む、精製されたダクリズマブ組成物を生産するための方法。
75.未精製ダクリズマブ調製物が細胞培養物から収穫される、実施形態74に記載の方法。
76.未精製ダクリズマブ調製物は、ダクリズマブが培養培地中に分泌される条件下で宿主細胞7A11−5H7−14−43を培養することと、分泌されたダクリズマブを収穫することにより得られる、実施形態74に記載の方法。
77.ダクリズマブが実施形態73に記載の方法を使用して収穫される、実施形態76に記載の方法。
78.(ix)ステップ(viii)の溶出されたダクリズマブ組成物を濾過して、ウイルスを除去するステップ、および
(x)濾過された溶液を限外濾過により濃縮して、約85−180mg/mLのダクリズマブを含む精製されたダクリズマブ組成物を産出するステップ
をさらに含む、実施形態74に記載の方法。
79.約85−165mg/mLのダクリズマブおよび約0.02−0.04%(w/v)のポリソルベート80を含み、約267−327mOsm/kgの範囲の浸透圧および25℃で約pH5.8−6.2の範囲のpHを有し、サイズ排除クロマトグラフィーにより測定される場合、ダクリズマブの少なくとも約95%が単量体形態である組成物が得られるように、精製されたダクリズマブ組成物を希釈緩衝液で希釈するステップをさらに含む、実施形態78に記載の方法。
80.得られる組成物が、ダクリズマブの組換え供給源由来の50ppm未満の宿主細胞タンパク質、10ppm未満のプロテインAを有し、組成物中のダクリズマブの3%以下がアグリゲート形態である、実施形態79に記載の方法。
81.基本培地PFBM2。
82.流加培地PFFM3。
83.ダクリズマブが培養培地中に分泌される条件下で、実施形態1−11のいずれか1つに記載の宿主細胞を培養するステップを含む方法により得られる、ダクリズマブ組成物。
84.方法が、分泌されたダクリズマブを細胞培養培地から単離するステップをさらに含む、実施形態83に記載のダクリズマブ組成物。
85.約100−500mMのクエン酸ナトリウム、約10−30mMのNaOHおよび約0.5−3%(v/v)ベンジルアルコールを含む、プロテインA親和性クロマトグラフィー樹脂を衛生化するのに有用な緩衝液。
86.カラムを衛生化するのに十分な流速でおよび期間、実施形態85に記載の衛生化緩衝液を用いてカラムを洗浄することを含む、プロテインA親和性クロマトグラフィーカラムを衛生化する方法。
87.カラムを、約150cm/時の流速でおよそ1.8カラム容積の衛生化緩衝液を用いて洗浄し、洗浄されたカラムが約30−45分の期間流動なしでインキュベートされ、次に平衡化緩衝液を用いて平衡化される、実施形態86に記載の方法。
88.平衡化緩衝液が、約20mMのクエン酸ナトリウムおよび150mMのNaClを含み、約pH7(25℃で)のpHを有する、実施形態87に記載の方法。
89.治療効果を与えるのに十分な量のDAC HYP組成物を患者に投与することを含む、多発性硬化症に罹っている患者を治療する方法。
90.DAC HYP組成物が静脈内投与される、実施形態89に記載の方法。
91.DAC HYP組成物が、約0.8−0.9mg/kgのDAC HYPに一致する量で投与される、実施形態90に記載の方法。
92.DAC HYP組成物が、約1mg/kgのDAC HYPに一致する量で投与される、実施形態91に記載の方法。
93.DAC HYPが、少なくとも6週間、少なくとも12週間、少なくとも24週間の期間、週あたり1回投与される、実施形態89−92のいずれか1つに記載の方法。
94.DAC HYPが単独療法として投与される、実施形態89−93のいずれか1つに記載の方法。
95.患者がインターフェロンベータを用いた前治療に無効果であったかまたはインターフェロンベータを用いた前治療を中止している、実施形態94に記載の方法。
96.DAC HYPがインターフェロンベータに対して補助的に投与される、実施形態89から93のいずれか1つに記載の方法。
97.前記DAC HYP組成物が皮下投与される、実施形態89に記載の方法。
98.DAC HYP組成物が、約1mg/kgのDAC HYPに一致する量で投与される、実施形態97に記載の方法。
99.DAC HYP組成物が2週間に1回投与される、実施形態98に記載の方法。
100.DAC HYP組成物が計約24週間投与される、実施形態99に記載の方法。
101.DAC HYP組成物が、約2mg/kgのDAC HYPに一致する量で投与される、実施形態97に記載の方法。
102.DAC HYP組成物が、4週間に1回投与される、実施形態101に記載の方法。
103.DAC HYP組成物が、計約24週間投与される、実施形態102に記載の方法。
104.DAC HYP組成物が、75mgから300mgのDAC HYPに一致する量で投与される、実施形態103に記載の方法。
105.DAC HYP組成物が、150mgに一致する量で投与される、実施形態104に記載の方法。
106.DAC HYP組成物が、300mgに一致する量で投与される、実施形態104に記載の方法。
107.DAC HYP組成物が、4週間に1回投与される、実施形態103−106のいずれか1つに記載の方法。
108.DAC HYP組成物が、計少なくとも48週間投与される、実施形態107に記載の方法。
109.DAC HYPが単独療法として投与される、実施形態103−108のいずれか1つに記載の方法。
110.患者がインターフェロンベータを用いた前治療に無効果であったかまたはインターフェロンベータを用いた前治療を中止している、実施形態109に記載の方法。
111.DAC HYPがインターフェロンベータに対して補助的に投与される、実施形態103−108のいずれか1つに記載の方法。
112.(i)組換えタンパク質を発現し分泌する細胞培養物のpHを、約pH4.5−5.5の範囲のpHに調整するステップ、
(ii)pH調整された細胞培養物を約4から15℃の範囲の温度で約30−90分間インキュベートするステップ、および
(iii)インキュベートされたpH調整細胞培養物を遠心分離して細胞残渣を除去するステップ
を含む方法により細胞培養物から得られるまたは得られることが可能な、組換えタンパク質。
113.(i)未精製ダクリズマブ調製物から親和性クロマトグラフィー樹脂上にダクリズマブを吸着させるステップ、
(ii)親和性クロマトグラフィー樹脂を洗浄緩衝液で洗浄して混入物を除去するステップ、
(iii)吸着したダクリズマブを溶出緩衝液で溶出させるステップ、
(iv)pHを約3−4の範囲のpHに調整することにより溶出液中のウイルスを不活化し、pH調整された溶出液を規定温度でウイルスを不活化するのに十分な期間インキュベートするステップ、
(v)ウイルス不活化溶出液を約pH7.7−7.9(25℃で測定される)の範囲のpHまで中和するステップ、
(vi)中和された溶出液を強陰イオン交換クロマトグラフィー樹脂に通して流すステップ、
(vii)ステップ(vi)の溶出液のダクリズマブを弱陽イオン交換クロマトグラフィー樹脂上に吸着させるステップ、および
(viii)吸着したダクリズマブを弱陽イオン交換クロマトグラフィー樹脂から溶出させるステップ
を含む方法により得られるまたは得られることが可能な、精製されたダグリズマブ組成物。
114.実施形態74−80のいずれか1つに記載の方法により得られるまたは得られることが可能な、精製されたダグリズマブ組成物。
【0273】
本出願に引用されるすべての出版物、特許、特許出願および他の文書は、あたかも各個々の出版物、特許、特許出願または他の文書が、あらゆる目的のために参照により組み込むことを個別に指示されている場合と同じ程度に、これによってあらゆる目的のために参照によりその全体を組み込む。
【0274】
様々な具体的実施形態が例示され説明されたが、本発明(複数可)の精神と範囲から逸脱することなく様々な変化を加えることが可能であることは認識される。
【特許請求の範囲】
【請求項1】
無血清および無コレステロール培地において増殖するよう適応されており、組換えタンパク質を発現するよう操作されている改変されたNS0細胞であって、無血清および無コレステロール培地において増殖される場合、10日間のフェドバッチ法において100Lの培養物中100mg/L/日の組換えタンパク質を超える容積生産性を達成することができる改変されたNS0細胞。
【請求項2】
コレステロールおよび動物由来成分のない培地において増殖される場合、10日間のフェドバッチ法において1,000Lの培養物中100mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、請求項1に記載の改変されたNS0細胞。
【請求項3】
コレステロールおよび動物由来成分のない培地において増殖される場合、10日間のフェドバッチ法において16,000Lの培養物中100mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、請求項1に記載の改変されたNS0細胞。
【請求項4】
フェドバッチ法が、追加される容積が最初の細胞培養物容積の割合を表す、次のスケジュール:
【表1】
に従って追加される流加培地を添加することを含む、請求項1から3のいずれか一項に記載の改変されたNS0細胞。
【請求項5】
13日間のフェドバッチ法において少なくとも100Lの培養物中200mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、請求項1に記載の改変されたNS0細胞。
【請求項6】
無コレステロール培地において増殖される場合、13日間のフェドバッチ法において1,000Lの培養物中200mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、請求項5に記載の改変されたNS0細胞。
【請求項7】
無血清および無コレステロール培地において増殖される場合、10日間のフェドバッチ法において16,000Lの培養物中100mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、請求項5に記載の改変されたNS0細胞。
【請求項8】
抗CD25モノクローナル抗体を発現するのに有用な核酸で安定的にトランスフェクトされる、請求項1に記載の改変されたNS0細胞。
【請求項9】
抗CD25モノクローナル抗体が、配列番号2の21位から233位に配列が一致するVL鎖および配列番号4の20位から465位に配列が一致するVH鎖を含む、請求項8に記載の改変されたNS0細胞。
【請求項10】
ベクターpAbX.gptを用いて形質転換される、請求項1に記載の改変されたNS0細胞。
【請求項11】
ベクターpHAT.IgG1.rg.dEを用いて形質転換される、請求項1に記載の改変されたNS0細胞。
【請求項12】
請求項1から11のいずれか一項に記載の改変されたNS0細胞を培養することを含む、組換えタンパク質を生産する方法。
【請求項13】
請求項12に記載の方法であって、10日間のフェドバッチ法において100L、1,000Lもしくは16,000Lの培養物中少なくとも100mg/L/日の組換えタンパク質を、または13日間のフェドバッチ法において100L、1,000Lもしくは16,000Lの培養物中少なくとも200mg/L/日の組換えタンパク質を生産する条件下で、改変されたNS0細胞が培養される、請求項12に記載の方法。
【請求項14】
改変されたNS0細胞が、血清およびコレステロールの非存在下で培養される、請求項12または請求項13に記載の方法。
【請求項15】
改変されたNS0細胞が、トロポロンおよびヒドロコルチゾンの非存在下で培養される、請求項14に記載の方法。
【請求項16】
改変されたNS0細胞が、10−35g/Lグルコースを含有する基本および/または流加培地において培養される、請求項12または請求項13に記載の方法。
【請求項17】
改変されたNS0細胞が、15g/Lグルコースを含有する基本培地および/または28g/Lグルコースを含有する流加培地において培養される、請求項16に記載の方法。
【請求項18】
基本培地が、PFBM2±10%成分で構成されている、請求項17に記載の方法。
【請求項19】
流加培地が、PFFM3±10%成分で構成されている、請求項17に記載の方法。
【請求項20】
細胞が基本培地において1−3日間、次に流加培地において10−13日間培養される、請求項18または請求項19に記載の方法。
【請求項21】
流加培地が、表7において概要が述べられているスケジュール±10%に従って追加される、請求項18に記載の方法。
【請求項22】
哺乳動物細胞において作動可能である選択可能なマーカーの発現を推進する弱いプロモーターおよび対象のタンパク質の発現を推進する強いプロモーターを含む、対象のタンパク質を組換え的に発現するのに有用なベクター。
【請求項23】
対象のタンパク質が治療抗体である、請求項22に記載のベクター。
【請求項24】
治療抗体が抗CD25抗体である、請求項23に記載のベクター。
【請求項25】
CD25抗体がダクリズマブのCDRを含む、請求項24に記載のベクター。
【請求項26】
CD25抗体がダクリズマブである、請求項25に記載のベクター。
【請求項27】
対象のタンパク質の高い容積生産性を有する哺乳動物宿主細胞を得るための方法であって、細胞に請求項22から26のいずれか一項に記載のベクターをトランスフェクトし、10日間のフェドバッチ法において100L、1,000Lもしくは16,000Lの培養物中少なくとも100mg/L/日対象のタンパク質を、または13日間のフェドバッチ法において100L、1,000Lもしくは16,000Lの培養物中少なくとも200mg/L/日の組換えタンパク質を生産することができる細胞を選択することを含む、対象のタンパク質の高い容積生産性を有する哺乳動物宿主細胞を得るための方法。
【請求項28】
ダクリズマブがpE/Q重鎖N連結アイソフォームおよび/またはQ/VHS重鎖N末端アイソフォームの存在により特徴付けられる、ダクリズマブを含む組成物。
【請求項29】
pE/Q重鎖N末端アイソフォームがダクリズマブのおよそ3−17%または6−15%を構成する、請求項28に記載の組成物。
【請求項30】
pE/Q重鎖N末端アイソフォームがダクリズマブのおよそ5−12%または7−12%を構成する、請求項28に記載の組成物。
【請求項31】
Q/VHS重鎖N末端アイソフォームがダクリズマブのおよそ1−15%を構成する、請求項28から30のいずれか一項に記載の組成物。
【請求項32】
Q/VHS重鎖N末端アイソフォームがダクリズマブのおよそ3−12%を構成する、請求項31に記載の組成物。
【請求項33】
ダクリズマブの重鎖が次のN末端アイソフォーム:
【表2】
に存在する、請求項28に記載の組成物。
【請求項34】
ダクリズマブの重鎖が次のN末端アイソフォーム:
【表3】
に存在する、請求項28に記載の組成物。
【請求項35】
ダクリズマブが、図18のアイソフォームプロファイルと実質的に類似する陽イオン交換クロマトグラフィーアイソフォームプロファイルにより特徴付けられる、請求項28に記載の組成物。
【請求項36】
ダクリズマブがDAC HYPである、請求項28に記載の組成物。
【請求項37】
ダクリズマブを含む組成物であって、1つがオリゴ糖G0−GlcNAcに一致し1つがオリゴ糖G0に一致する2つの主要ピークを含有するN連結グリコシル化HPLCプロファイルによりダクリズマブが特徴付けられ、これら2つのピークの合わせたAUCが全ピークの総AUCのうちの約75−100%、約80−100%、約85−100%または約88−99.5%を構成する、ダクリズマブを含む組成物。
【請求項38】
G0−GlcNAcピークのAUCが全ピークの総AUCのうちの約5−20%、約5−18%を構成し、G0ピークのAUCが全ピークの総AUCのうちの約70−99%、約75−92%または約75−90%を構成する、請求項37に記載の組成物。
【請求項39】
G0−GlcNAcピークのAUCが全ピークの総AUCのうちの約6−16%または約7−15%を構成し、G0ピークのAUCが全ピークの総AUCのうちの約78−90%または約81−88%を構成する、請求項38に記載の組成物。
【請求項40】
N連結グリコシル化プロファイルが約3%未満のMan5を有する、請求項37から39のいずれか一項に記載の組成物。
【請求項41】
N連結グリコシル化プロファイルが約0.5%未満のG2、Man6および/またはMan7を有する、請求項40に記載の組成物。
【請求項42】
N連結グリコシル化プロファイルがシアル酸付加オリゴ糖に一致する第三のピークを含有し、シアル酸付加オリゴ糖ピークのAUCが全ピークの総AUCのうちの1%以下を構成する、請求項37に記載の組成物。
【請求項43】
N連結グリコシル化プロファイルがオリゴ糖G1に一致する第三のピークを含有し、G1ピークのAUCが全ピークの総AUCのうちの約10%未満、約5%未満、約4%未満、約3%未満または約1−5%、約1−4%もしくは約1−3%を構成する、請求項37に記載の組成物。
【請求項44】
G1ピークのAUCが全ピークの総AUCのうちの約1−2%を構成する、請求項43に記載の組成物。
【請求項45】
ダクリズマブが、図19のプロファイルにまたは図21の下のパネルのプロファイルに実質的に類似しているN連結グリコシル化HPLCプロファイルを有する、請求項37に記載の組成物。
【請求項46】
ダクリズマブがDAC HYPである、請求項37に記載の組成物。
【請求項47】
ダクリズマブを含む組成物であって、1μg/mLのダクリズマブ濃度および約25対1のエフェクター細胞対標的細胞比で、少なくとも3体の健康なドナー由来のエフェクター細胞および標的細胞としてKit225 K6細胞を使用して、インビトロ細胞アッセイにおいて測定される場合、30%未満、25%未満、20%未満、15%未満、10%未満または5%未満のADCC平均細胞傷害性を示すダクリズマブを含む組成物。
【請求項48】
ダクリズマブが、前記アッセイにおいて5%から30%、5%から25%、10%から30%、10%から25%または15%から25%のADCC平均細胞傷害性を示す、請求項47に記載の組成物。
【請求項49】
前記アッセイが少なくとも6体の健康なドナー由来のエフェクター細胞を使用する、請求項47または請求項48に記載の組成物。
【請求項50】
前記アッセイが少なくとも10体の健康なドナー由来のエフェクター細胞を使用する、請求項47または請求項48に記載の組成物。
【請求項51】
ダクリズマブがDAC HYPである、請求項47から50のいずれか一項に記載の組成物。
【請求項52】
ダクリズマブ製剤を作製するのに有用な組成物であって、約150−190mg/mLのダクリズマブ、ならびに希釈緩衝液を用いた組成物の希釈により約85−165mg/mLのダクリズマブを含有し、約267−327mOsm/kgの範囲の浸透圧および25℃で約pH5.8−6.2の範囲のpHを有し、サイズ排除クロマトグラフィーにより測定される場合、ダクリズマブの少なくとも約95%が単量体形態である希釈組成物を生じるような量の賦形剤を含む、ダクリズマブ製剤を作製するのに有用な組成物。
【請求項53】
希釈緩衝液で希釈される場合、希釈組成物が約85−115mg/mLのダクリズマブを含有するような量の賦形剤を含有する、請求項52に記載の組成物。
【請求項54】
希釈緩衝液で希釈される場合、希釈組成物が約150±15mg/mLのダクリズマブを含有するような量の賦形剤を含有する、請求項52に記載の組成物。
【請求項55】
ダクリズマブの0.1%以下がアグリゲート形態である、約4から15mg/mLのダクリズマブを含む組成物。
【請求項56】
ダクリズマブの最大2.5%がアグリゲート形態である、約4から15mg/mLのダクリズマブを含むダクリズマブ組成物を、弱陽イオン交換樹脂上のカラムクロマトグラフィーにより精製することによって得られる、請求項55に記載の組成物。
【請求項57】
弱陽イオン交換樹脂がCM−650Mである、請求項56に記載の組成物。
【請求項58】
CM−650M樹脂が約20mMのクエン酸ナトリウムを含有する平衡化緩衝液、pH4.4−4.6と平衡化しており、ダクリズマブが約20mMのクエン酸ナトリウムおよび約75mMの硫酸ナトリウムを含有する溶出緩衝液、pH4.4−4.6を用いて溶出される、請求項57に記載の組成物。
【請求項59】
クロマトグラフィーが、約10−30cmまたは約17−19cmの高さを有する樹脂床を使用して円筒形カラムにおいて実施され、ダクリズマブが約4−22℃または約18−22℃の範囲の温度で、約50−200cm/時または90−110cm/時の範囲の流速で溶出される、請求項58に記載の組成物。
【請求項60】
ヒト投与に適した組成物であって、約85−165mg/mLのダクリズマブおよび約0.02−0.04%(w/v)ポリソルベート80を含み、約267−327mOsm/kgの範囲の浸透圧および25℃で約pH5.8−6.2の範囲のpHを有し、サイズ排除クロマトグラフィーにより測定される場合、ダクリズマブの少なくとも約95%が単量体形態である、ヒト投与に適した組成物。
【請求項61】
サイズ排除クロマトグラフィーにより測定される場合、ダクリズマブの少なくとも約99%が単量体形態である、請求項60に記載の組成物。
【請求項62】
約85−115mg/mLのダクリズマブを含む、請求項60に記載の組成物。
【請求項63】
約100mg/mLのダクリズマブ、約40mMのコハク酸ナトリウム、約100mMの塩化ナトリウムおよび約0.03%(w/v)ポリソルベート80から本質的になり、25℃で約6.0のpHを有する、請求項62に記載の組成物。
【請求項64】
約135−165mg/mLのダクリズマブを含む、請求項60に記載の組成物。
【請求項65】
約150mg/mLのダクリズマブ、約40mMのコハク酸ナトリウム、約100mMの塩化ナトリウムおよび約0.03%(w/v)のポリソルベート80から本質的になり、25℃で約6.0のpHを有する、請求項64に記載の組成物。
【請求項66】
請求項64に記載の組成物であって、約4から15mg/mLのダクリズマブを含むダクリズマブ組成物を適切な緩衝液中での限外濾過により濃縮して、約85−180mg/mLの範囲のダクリズマブ濃度を達成するステップ、および、場合によって、濃縮された組成物を希釈緩衝液で希釈するステップを含む方法により得られる、請求項64に記載の組成物。
【請求項67】
約85−165mg/mLのダクリズマブを含み、約2−8℃の範囲の温度での約12ヶ月の期間の貯蔵後にアグリゲート形態中のダクリズマブの百分率が約3%を超えない、皮下投与に適した医薬組成物。
【請求項68】
約85−115mg/mLのダクリズマブを含む、請求項67に記載の医薬組成物。
【請求項69】
約135−165mg/mLのダクリズマブを含む、請求項68に記載の医薬組成物。
【請求項70】
約2−8℃の範囲の温度での約12ヶ月の期間の貯蔵後にアグリゲート形態中のダクリズマブの百分率が約2%を超えない、請求項68または請求項69に記載の医薬組成物。
【請求項71】
約2−8℃の範囲の温度での約18ヶ月の期間の貯蔵後にアグリゲート形態中のダクリズマブの百分率が約3%を超えない、請求項68または請求項69に記載の医薬組成物。
【請求項72】
細胞培養物から組換えタンパク質を収穫するための方法であって、
(i)組換えタンパク質を発現し分泌する細胞培養物のpHを、約pH4.5−5.5の範囲のpHに調整するステップ、
(ii)pH調整された細胞培養物を約4から15℃の範囲の温度で約30−90分間インキュベートするステップ、および
(iii)インキュベートされたpH調整細胞培養物を遠心分離して細胞残渣を除去するステップ
を含む、細胞培養物から組換えタンパク質を収穫するための方法。
【請求項73】
精製されたダクリズマブ組成物を生産するための方法であって、
(i)未精製ダクリズマブ調製物から親和性クロマトグラフィー樹脂上にダクリズマブを吸着させるステップ、
(ii)親和性クロマトグラフィー樹脂を洗浄緩衝液で洗浄して混入物を除去するステップ、
(iii)吸着したダクリズマブを溶出緩衝液で溶出させるステップ、
(iv)pHを約3−4の範囲のpHに調整することにより溶出液中のウイルスを不活化し、pH調整された溶出液を規定温度でウイルスを不活化するのに十分な期間インキュベートするステップ、
(v)ウイルス不活化溶出液を約pH7.7−7.9(25℃で測定される)の範囲のpHまで中和するステップ、
(vi)中和された溶出液を強陰イオン交換クロマトグラフィー樹脂に通して流すステップ、
(vii)ステップ(vi)の溶出液のダクリズマブを弱陽イオン交換クロマトグラフィー樹脂上に吸着させるステップ、および
(viii)吸着したダクリズマブを弱陽イオン交換クロマトグラフィー樹脂から溶出させるステップ
を含む、精製されたダクリズマブ組成物を生産するための方法。
【請求項74】
未精製ダクリズマブ調製物が細胞培養物から収穫される、請求項73に記載の方法。
【請求項75】
ダクリズマブが請求項72に記載の方法を使用して収穫される、請求項73に記載の方法。
【請求項76】
請求項73に記載の方法であって、
(ix)ステップ(viii)の溶出されたダクリズマブ組成物を濾過して、ウイルスを除去するステップ、および
(x)濾過された溶液を限外濾過により濃縮して、約85−180mg/mLのダクリズマブを含む精製されたダクリズマブ組成物を産出するステップ
をさらに含む、請求項73に記載の方法。
【請求項77】
請求項76に記載の方法であって、約85−165mg/mLのダクリズマブおよび約0.02−0.04%(w/v)のポリソルベート80を含み、約267−327mOsm/kgの範囲の浸透圧および25℃で約pH5.8−6.2の範囲のpHを有し、サイズ排除クロマトグラフィーにより測定される場合、ダクリズマブの少なくとも約95%が単量体形態である組成物が得られるように、精製されたダクリズマブ組成物を希釈緩衝液で希釈するステップをさらに含む、請求項76に記載の方法。
【請求項78】
得られる組成物が、ダクリズマブの組換え供給源由来の50ppm未満の宿主細胞タンパク質を有し、10ppm未満のプロテインAを有し、および組成物中のダクリズマブの3%以下がアグリゲート形態である、請求項77に記載の方法。
【請求項79】
基本培地PFBM2。
【請求項80】
流加培地PFFM3。
【請求項81】
ダクリズマブが培養培地中に分泌される条件下で、請求項1から11のいずれか一項に記載の宿主細胞を培養するステップを含む方法により得られる、ダクリズマブ組成物。
【請求項82】
請求項81に記載のダクリズマブ組成物であって、方法が、分泌されたダクリズマブを細胞培養培地から単離するステップをさらに含む、請求項81に記載のダクリズマブ組成物。
【請求項83】
約100−500mMのクエン酸ナトリウム、約10−30mMのNaOHおよび約0.5−3%(v/v)ベンジルアルコールを含む、プロテインA親和性クロマトグラフィー樹脂を衛生化するのに有用な緩衝液。
【請求項84】
カラムを衛生化するのに十分な流速でおよび期間、請求項83に記載の衛生化緩衝液を用いてカラムを洗浄することを含む、プロテインA親和性クロマトグラフィーカラムを衛生化する方法。
【請求項85】
カラムを、約150cm/時の流速でおよそ1.8カラム容積の衛生化緩衝液を用いて洗浄し、洗浄されたカラムが約30−45分の期間流動なしでインキュベートされ、次に平衡化緩衝液を用いて平衡化される、請求項84に記載の方法。
【請求項86】
平衡化緩衝液が、約20mMのクエン酸ナトリウムおよび150mMのNaClを含み、約pH7(25℃で)のpHを有する、請求項85に記載の方法。
【請求項87】
治療効果を与えるのに十分な量のDAC HYP組成物を患者に投与することを含む、多発性硬化症に罹っている患者を治療する方法。
【請求項88】
DAC HYP組成物が静脈内投与される、請求項87に記載の方法。
【請求項89】
DAC HYP組成物が、約0.8−0.9mg/kgのDAC HYPに一致する量で投与される、請求項88に記載の方法。
【請求項90】
DAC HYP組成物が、約1mg/kgのDAC HYPに一致する量で投与される、請求項89に記載の方法。
【請求項91】
DAC HYPが、少なくとも6週間、少なくとも12週間、少なくとも24週間の期間、週あたり1回投与される、請求項87から90のいずれか一項に記載の方法。
【請求項92】
DAC HYPが単独療法として投与される、請求項87から91のいずれか一項に記載の方法。
【請求項93】
請求項92に記載の方法であって、患者がインターフェロンベータを用いた前治療に無効果であったかまたはインターフェロンベータを用いた前治療を中止している、請求項92に記載の方法。
【請求項94】
DAC HYPがインターフェロンベータに対して補助的に投与される、請求項87から91のいずれか一項に記載の方法。
【請求項95】
前記DAC HYP組成物が皮下投与される、請求項87に記載の方法。
【請求項96】
DAC HYP組成物が、約1mg/kgのDAC HYPに一致する量で投与される、請求項95に記載の方法。
【請求項97】
DAC HYP組成物が2週間に1回投与される、請求項96に記載の方法。
【請求項98】
DAC HYP組成物が計約24週間投与される、請求項97に記載の方法。
【請求項99】
DAC HYP組成物が、約2mg/kgのDAC HYPに一致する量で投与される、請求項95に記載の方法。
【請求項100】
DAC HYP組成物が、4週間に1回投与される、請求項99に記載の方法。
【請求項101】
DAC HYP組成物が、計約24週間投与される、請求項100に記載の方法。
【請求項102】
DAC HYP組成物が、75mgから300mgのDAC HYPに一致する量で投与される、請求項101に記載の方法。
【請求項103】
DAC HYP組成物が、150mgに一致する量で投与される、請求項102に記載の方法。
【請求項104】
DAC HYP組成物が、300mgに一致する量で投与される、請求項102に記載の方法。
【請求項105】
DAC HYP組成物が、4週間に1回投与される、請求項101から104のいずれか一項に記載の方法。
【請求項106】
DAC HYP組成物が、計少なくとも48週間投与される、請求項105に記載の方法。
【請求項107】
DAC HYPが単独療法として投与される、請求項101から106のいずれか一項に記載の方法。
【請求項108】
請求項107に記載の方法であって、患者がインターフェロンベータを用いた前治療に無効果であったかまたはインターフェロンベータを用いた前治療を中止している、請求項107に記載の方法。
【請求項109】
DAC HYPがインターフェロンベータに対して補助的に投与される、請求項101から106のいずれか一項に記載の方法。
【請求項1】
無血清および無コレステロール培地において増殖するよう適応されており、組換えタンパク質を発現するよう操作されている改変されたNS0細胞であって、無血清および無コレステロール培地において増殖される場合、10日間のフェドバッチ法において100Lの培養物中100mg/L/日の組換えタンパク質を超える容積生産性を達成することができる改変されたNS0細胞。
【請求項2】
コレステロールおよび動物由来成分のない培地において増殖される場合、10日間のフェドバッチ法において1,000Lの培養物中100mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、請求項1に記載の改変されたNS0細胞。
【請求項3】
コレステロールおよび動物由来成分のない培地において増殖される場合、10日間のフェドバッチ法において16,000Lの培養物中100mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、請求項1に記載の改変されたNS0細胞。
【請求項4】
フェドバッチ法が、追加される容積が最初の細胞培養物容積の割合を表す、次のスケジュール:
【表1】
に従って追加される流加培地を添加することを含む、請求項1から3のいずれか一項に記載の改変されたNS0細胞。
【請求項5】
13日間のフェドバッチ法において少なくとも100Lの培養物中200mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、請求項1に記載の改変されたNS0細胞。
【請求項6】
無コレステロール培地において増殖される場合、13日間のフェドバッチ法において1,000Lの培養物中200mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、請求項5に記載の改変されたNS0細胞。
【請求項7】
無血清および無コレステロール培地において増殖される場合、10日間のフェドバッチ法において16,000Lの培養物中100mg/L/日の組換えタンパク質を超える容積生産性を達成することができる、請求項5に記載の改変されたNS0細胞。
【請求項8】
抗CD25モノクローナル抗体を発現するのに有用な核酸で安定的にトランスフェクトされる、請求項1に記載の改変されたNS0細胞。
【請求項9】
抗CD25モノクローナル抗体が、配列番号2の21位から233位に配列が一致するVL鎖および配列番号4の20位から465位に配列が一致するVH鎖を含む、請求項8に記載の改変されたNS0細胞。
【請求項10】
ベクターpAbX.gptを用いて形質転換される、請求項1に記載の改変されたNS0細胞。
【請求項11】
ベクターpHAT.IgG1.rg.dEを用いて形質転換される、請求項1に記載の改変されたNS0細胞。
【請求項12】
請求項1から11のいずれか一項に記載の改変されたNS0細胞を培養することを含む、組換えタンパク質を生産する方法。
【請求項13】
請求項12に記載の方法であって、10日間のフェドバッチ法において100L、1,000Lもしくは16,000Lの培養物中少なくとも100mg/L/日の組換えタンパク質を、または13日間のフェドバッチ法において100L、1,000Lもしくは16,000Lの培養物中少なくとも200mg/L/日の組換えタンパク質を生産する条件下で、改変されたNS0細胞が培養される、請求項12に記載の方法。
【請求項14】
改変されたNS0細胞が、血清およびコレステロールの非存在下で培養される、請求項12または請求項13に記載の方法。
【請求項15】
改変されたNS0細胞が、トロポロンおよびヒドロコルチゾンの非存在下で培養される、請求項14に記載の方法。
【請求項16】
改変されたNS0細胞が、10−35g/Lグルコースを含有する基本および/または流加培地において培養される、請求項12または請求項13に記載の方法。
【請求項17】
改変されたNS0細胞が、15g/Lグルコースを含有する基本培地および/または28g/Lグルコースを含有する流加培地において培養される、請求項16に記載の方法。
【請求項18】
基本培地が、PFBM2±10%成分で構成されている、請求項17に記載の方法。
【請求項19】
流加培地が、PFFM3±10%成分で構成されている、請求項17に記載の方法。
【請求項20】
細胞が基本培地において1−3日間、次に流加培地において10−13日間培養される、請求項18または請求項19に記載の方法。
【請求項21】
流加培地が、表7において概要が述べられているスケジュール±10%に従って追加される、請求項18に記載の方法。
【請求項22】
哺乳動物細胞において作動可能である選択可能なマーカーの発現を推進する弱いプロモーターおよび対象のタンパク質の発現を推進する強いプロモーターを含む、対象のタンパク質を組換え的に発現するのに有用なベクター。
【請求項23】
対象のタンパク質が治療抗体である、請求項22に記載のベクター。
【請求項24】
治療抗体が抗CD25抗体である、請求項23に記載のベクター。
【請求項25】
CD25抗体がダクリズマブのCDRを含む、請求項24に記載のベクター。
【請求項26】
CD25抗体がダクリズマブである、請求項25に記載のベクター。
【請求項27】
対象のタンパク質の高い容積生産性を有する哺乳動物宿主細胞を得るための方法であって、細胞に請求項22から26のいずれか一項に記載のベクターをトランスフェクトし、10日間のフェドバッチ法において100L、1,000Lもしくは16,000Lの培養物中少なくとも100mg/L/日対象のタンパク質を、または13日間のフェドバッチ法において100L、1,000Lもしくは16,000Lの培養物中少なくとも200mg/L/日の組換えタンパク質を生産することができる細胞を選択することを含む、対象のタンパク質の高い容積生産性を有する哺乳動物宿主細胞を得るための方法。
【請求項28】
ダクリズマブがpE/Q重鎖N連結アイソフォームおよび/またはQ/VHS重鎖N末端アイソフォームの存在により特徴付けられる、ダクリズマブを含む組成物。
【請求項29】
pE/Q重鎖N末端アイソフォームがダクリズマブのおよそ3−17%または6−15%を構成する、請求項28に記載の組成物。
【請求項30】
pE/Q重鎖N末端アイソフォームがダクリズマブのおよそ5−12%または7−12%を構成する、請求項28に記載の組成物。
【請求項31】
Q/VHS重鎖N末端アイソフォームがダクリズマブのおよそ1−15%を構成する、請求項28から30のいずれか一項に記載の組成物。
【請求項32】
Q/VHS重鎖N末端アイソフォームがダクリズマブのおよそ3−12%を構成する、請求項31に記載の組成物。
【請求項33】
ダクリズマブの重鎖が次のN末端アイソフォーム:
【表2】
に存在する、請求項28に記載の組成物。
【請求項34】
ダクリズマブの重鎖が次のN末端アイソフォーム:
【表3】
に存在する、請求項28に記載の組成物。
【請求項35】
ダクリズマブが、図18のアイソフォームプロファイルと実質的に類似する陽イオン交換クロマトグラフィーアイソフォームプロファイルにより特徴付けられる、請求項28に記載の組成物。
【請求項36】
ダクリズマブがDAC HYPである、請求項28に記載の組成物。
【請求項37】
ダクリズマブを含む組成物であって、1つがオリゴ糖G0−GlcNAcに一致し1つがオリゴ糖G0に一致する2つの主要ピークを含有するN連結グリコシル化HPLCプロファイルによりダクリズマブが特徴付けられ、これら2つのピークの合わせたAUCが全ピークの総AUCのうちの約75−100%、約80−100%、約85−100%または約88−99.5%を構成する、ダクリズマブを含む組成物。
【請求項38】
G0−GlcNAcピークのAUCが全ピークの総AUCのうちの約5−20%、約5−18%を構成し、G0ピークのAUCが全ピークの総AUCのうちの約70−99%、約75−92%または約75−90%を構成する、請求項37に記載の組成物。
【請求項39】
G0−GlcNAcピークのAUCが全ピークの総AUCのうちの約6−16%または約7−15%を構成し、G0ピークのAUCが全ピークの総AUCのうちの約78−90%または約81−88%を構成する、請求項38に記載の組成物。
【請求項40】
N連結グリコシル化プロファイルが約3%未満のMan5を有する、請求項37から39のいずれか一項に記載の組成物。
【請求項41】
N連結グリコシル化プロファイルが約0.5%未満のG2、Man6および/またはMan7を有する、請求項40に記載の組成物。
【請求項42】
N連結グリコシル化プロファイルがシアル酸付加オリゴ糖に一致する第三のピークを含有し、シアル酸付加オリゴ糖ピークのAUCが全ピークの総AUCのうちの1%以下を構成する、請求項37に記載の組成物。
【請求項43】
N連結グリコシル化プロファイルがオリゴ糖G1に一致する第三のピークを含有し、G1ピークのAUCが全ピークの総AUCのうちの約10%未満、約5%未満、約4%未満、約3%未満または約1−5%、約1−4%もしくは約1−3%を構成する、請求項37に記載の組成物。
【請求項44】
G1ピークのAUCが全ピークの総AUCのうちの約1−2%を構成する、請求項43に記載の組成物。
【請求項45】
ダクリズマブが、図19のプロファイルにまたは図21の下のパネルのプロファイルに実質的に類似しているN連結グリコシル化HPLCプロファイルを有する、請求項37に記載の組成物。
【請求項46】
ダクリズマブがDAC HYPである、請求項37に記載の組成物。
【請求項47】
ダクリズマブを含む組成物であって、1μg/mLのダクリズマブ濃度および約25対1のエフェクター細胞対標的細胞比で、少なくとも3体の健康なドナー由来のエフェクター細胞および標的細胞としてKit225 K6細胞を使用して、インビトロ細胞アッセイにおいて測定される場合、30%未満、25%未満、20%未満、15%未満、10%未満または5%未満のADCC平均細胞傷害性を示すダクリズマブを含む組成物。
【請求項48】
ダクリズマブが、前記アッセイにおいて5%から30%、5%から25%、10%から30%、10%から25%または15%から25%のADCC平均細胞傷害性を示す、請求項47に記載の組成物。
【請求項49】
前記アッセイが少なくとも6体の健康なドナー由来のエフェクター細胞を使用する、請求項47または請求項48に記載の組成物。
【請求項50】
前記アッセイが少なくとも10体の健康なドナー由来のエフェクター細胞を使用する、請求項47または請求項48に記載の組成物。
【請求項51】
ダクリズマブがDAC HYPである、請求項47から50のいずれか一項に記載の組成物。
【請求項52】
ダクリズマブ製剤を作製するのに有用な組成物であって、約150−190mg/mLのダクリズマブ、ならびに希釈緩衝液を用いた組成物の希釈により約85−165mg/mLのダクリズマブを含有し、約267−327mOsm/kgの範囲の浸透圧および25℃で約pH5.8−6.2の範囲のpHを有し、サイズ排除クロマトグラフィーにより測定される場合、ダクリズマブの少なくとも約95%が単量体形態である希釈組成物を生じるような量の賦形剤を含む、ダクリズマブ製剤を作製するのに有用な組成物。
【請求項53】
希釈緩衝液で希釈される場合、希釈組成物が約85−115mg/mLのダクリズマブを含有するような量の賦形剤を含有する、請求項52に記載の組成物。
【請求項54】
希釈緩衝液で希釈される場合、希釈組成物が約150±15mg/mLのダクリズマブを含有するような量の賦形剤を含有する、請求項52に記載の組成物。
【請求項55】
ダクリズマブの0.1%以下がアグリゲート形態である、約4から15mg/mLのダクリズマブを含む組成物。
【請求項56】
ダクリズマブの最大2.5%がアグリゲート形態である、約4から15mg/mLのダクリズマブを含むダクリズマブ組成物を、弱陽イオン交換樹脂上のカラムクロマトグラフィーにより精製することによって得られる、請求項55に記載の組成物。
【請求項57】
弱陽イオン交換樹脂がCM−650Mである、請求項56に記載の組成物。
【請求項58】
CM−650M樹脂が約20mMのクエン酸ナトリウムを含有する平衡化緩衝液、pH4.4−4.6と平衡化しており、ダクリズマブが約20mMのクエン酸ナトリウムおよび約75mMの硫酸ナトリウムを含有する溶出緩衝液、pH4.4−4.6を用いて溶出される、請求項57に記載の組成物。
【請求項59】
クロマトグラフィーが、約10−30cmまたは約17−19cmの高さを有する樹脂床を使用して円筒形カラムにおいて実施され、ダクリズマブが約4−22℃または約18−22℃の範囲の温度で、約50−200cm/時または90−110cm/時の範囲の流速で溶出される、請求項58に記載の組成物。
【請求項60】
ヒト投与に適した組成物であって、約85−165mg/mLのダクリズマブおよび約0.02−0.04%(w/v)ポリソルベート80を含み、約267−327mOsm/kgの範囲の浸透圧および25℃で約pH5.8−6.2の範囲のpHを有し、サイズ排除クロマトグラフィーにより測定される場合、ダクリズマブの少なくとも約95%が単量体形態である、ヒト投与に適した組成物。
【請求項61】
サイズ排除クロマトグラフィーにより測定される場合、ダクリズマブの少なくとも約99%が単量体形態である、請求項60に記載の組成物。
【請求項62】
約85−115mg/mLのダクリズマブを含む、請求項60に記載の組成物。
【請求項63】
約100mg/mLのダクリズマブ、約40mMのコハク酸ナトリウム、約100mMの塩化ナトリウムおよび約0.03%(w/v)ポリソルベート80から本質的になり、25℃で約6.0のpHを有する、請求項62に記載の組成物。
【請求項64】
約135−165mg/mLのダクリズマブを含む、請求項60に記載の組成物。
【請求項65】
約150mg/mLのダクリズマブ、約40mMのコハク酸ナトリウム、約100mMの塩化ナトリウムおよび約0.03%(w/v)のポリソルベート80から本質的になり、25℃で約6.0のpHを有する、請求項64に記載の組成物。
【請求項66】
請求項64に記載の組成物であって、約4から15mg/mLのダクリズマブを含むダクリズマブ組成物を適切な緩衝液中での限外濾過により濃縮して、約85−180mg/mLの範囲のダクリズマブ濃度を達成するステップ、および、場合によって、濃縮された組成物を希釈緩衝液で希釈するステップを含む方法により得られる、請求項64に記載の組成物。
【請求項67】
約85−165mg/mLのダクリズマブを含み、約2−8℃の範囲の温度での約12ヶ月の期間の貯蔵後にアグリゲート形態中のダクリズマブの百分率が約3%を超えない、皮下投与に適した医薬組成物。
【請求項68】
約85−115mg/mLのダクリズマブを含む、請求項67に記載の医薬組成物。
【請求項69】
約135−165mg/mLのダクリズマブを含む、請求項68に記載の医薬組成物。
【請求項70】
約2−8℃の範囲の温度での約12ヶ月の期間の貯蔵後にアグリゲート形態中のダクリズマブの百分率が約2%を超えない、請求項68または請求項69に記載の医薬組成物。
【請求項71】
約2−8℃の範囲の温度での約18ヶ月の期間の貯蔵後にアグリゲート形態中のダクリズマブの百分率が約3%を超えない、請求項68または請求項69に記載の医薬組成物。
【請求項72】
細胞培養物から組換えタンパク質を収穫するための方法であって、
(i)組換えタンパク質を発現し分泌する細胞培養物のpHを、約pH4.5−5.5の範囲のpHに調整するステップ、
(ii)pH調整された細胞培養物を約4から15℃の範囲の温度で約30−90分間インキュベートするステップ、および
(iii)インキュベートされたpH調整細胞培養物を遠心分離して細胞残渣を除去するステップ
を含む、細胞培養物から組換えタンパク質を収穫するための方法。
【請求項73】
精製されたダクリズマブ組成物を生産するための方法であって、
(i)未精製ダクリズマブ調製物から親和性クロマトグラフィー樹脂上にダクリズマブを吸着させるステップ、
(ii)親和性クロマトグラフィー樹脂を洗浄緩衝液で洗浄して混入物を除去するステップ、
(iii)吸着したダクリズマブを溶出緩衝液で溶出させるステップ、
(iv)pHを約3−4の範囲のpHに調整することにより溶出液中のウイルスを不活化し、pH調整された溶出液を規定温度でウイルスを不活化するのに十分な期間インキュベートするステップ、
(v)ウイルス不活化溶出液を約pH7.7−7.9(25℃で測定される)の範囲のpHまで中和するステップ、
(vi)中和された溶出液を強陰イオン交換クロマトグラフィー樹脂に通して流すステップ、
(vii)ステップ(vi)の溶出液のダクリズマブを弱陽イオン交換クロマトグラフィー樹脂上に吸着させるステップ、および
(viii)吸着したダクリズマブを弱陽イオン交換クロマトグラフィー樹脂から溶出させるステップ
を含む、精製されたダクリズマブ組成物を生産するための方法。
【請求項74】
未精製ダクリズマブ調製物が細胞培養物から収穫される、請求項73に記載の方法。
【請求項75】
ダクリズマブが請求項72に記載の方法を使用して収穫される、請求項73に記載の方法。
【請求項76】
請求項73に記載の方法であって、
(ix)ステップ(viii)の溶出されたダクリズマブ組成物を濾過して、ウイルスを除去するステップ、および
(x)濾過された溶液を限外濾過により濃縮して、約85−180mg/mLのダクリズマブを含む精製されたダクリズマブ組成物を産出するステップ
をさらに含む、請求項73に記載の方法。
【請求項77】
請求項76に記載の方法であって、約85−165mg/mLのダクリズマブおよび約0.02−0.04%(w/v)のポリソルベート80を含み、約267−327mOsm/kgの範囲の浸透圧および25℃で約pH5.8−6.2の範囲のpHを有し、サイズ排除クロマトグラフィーにより測定される場合、ダクリズマブの少なくとも約95%が単量体形態である組成物が得られるように、精製されたダクリズマブ組成物を希釈緩衝液で希釈するステップをさらに含む、請求項76に記載の方法。
【請求項78】
得られる組成物が、ダクリズマブの組換え供給源由来の50ppm未満の宿主細胞タンパク質を有し、10ppm未満のプロテインAを有し、および組成物中のダクリズマブの3%以下がアグリゲート形態である、請求項77に記載の方法。
【請求項79】
基本培地PFBM2。
【請求項80】
流加培地PFFM3。
【請求項81】
ダクリズマブが培養培地中に分泌される条件下で、請求項1から11のいずれか一項に記載の宿主細胞を培養するステップを含む方法により得られる、ダクリズマブ組成物。
【請求項82】
請求項81に記載のダクリズマブ組成物であって、方法が、分泌されたダクリズマブを細胞培養培地から単離するステップをさらに含む、請求項81に記載のダクリズマブ組成物。
【請求項83】
約100−500mMのクエン酸ナトリウム、約10−30mMのNaOHおよび約0.5−3%(v/v)ベンジルアルコールを含む、プロテインA親和性クロマトグラフィー樹脂を衛生化するのに有用な緩衝液。
【請求項84】
カラムを衛生化するのに十分な流速でおよび期間、請求項83に記載の衛生化緩衝液を用いてカラムを洗浄することを含む、プロテインA親和性クロマトグラフィーカラムを衛生化する方法。
【請求項85】
カラムを、約150cm/時の流速でおよそ1.8カラム容積の衛生化緩衝液を用いて洗浄し、洗浄されたカラムが約30−45分の期間流動なしでインキュベートされ、次に平衡化緩衝液を用いて平衡化される、請求項84に記載の方法。
【請求項86】
平衡化緩衝液が、約20mMのクエン酸ナトリウムおよび150mMのNaClを含み、約pH7(25℃で)のpHを有する、請求項85に記載の方法。
【請求項87】
治療効果を与えるのに十分な量のDAC HYP組成物を患者に投与することを含む、多発性硬化症に罹っている患者を治療する方法。
【請求項88】
DAC HYP組成物が静脈内投与される、請求項87に記載の方法。
【請求項89】
DAC HYP組成物が、約0.8−0.9mg/kgのDAC HYPに一致する量で投与される、請求項88に記載の方法。
【請求項90】
DAC HYP組成物が、約1mg/kgのDAC HYPに一致する量で投与される、請求項89に記載の方法。
【請求項91】
DAC HYPが、少なくとも6週間、少なくとも12週間、少なくとも24週間の期間、週あたり1回投与される、請求項87から90のいずれか一項に記載の方法。
【請求項92】
DAC HYPが単独療法として投与される、請求項87から91のいずれか一項に記載の方法。
【請求項93】
請求項92に記載の方法であって、患者がインターフェロンベータを用いた前治療に無効果であったかまたはインターフェロンベータを用いた前治療を中止している、請求項92に記載の方法。
【請求項94】
DAC HYPがインターフェロンベータに対して補助的に投与される、請求項87から91のいずれか一項に記載の方法。
【請求項95】
前記DAC HYP組成物が皮下投与される、請求項87に記載の方法。
【請求項96】
DAC HYP組成物が、約1mg/kgのDAC HYPに一致する量で投与される、請求項95に記載の方法。
【請求項97】
DAC HYP組成物が2週間に1回投与される、請求項96に記載の方法。
【請求項98】
DAC HYP組成物が計約24週間投与される、請求項97に記載の方法。
【請求項99】
DAC HYP組成物が、約2mg/kgのDAC HYPに一致する量で投与される、請求項95に記載の方法。
【請求項100】
DAC HYP組成物が、4週間に1回投与される、請求項99に記載の方法。
【請求項101】
DAC HYP組成物が、計約24週間投与される、請求項100に記載の方法。
【請求項102】
DAC HYP組成物が、75mgから300mgのDAC HYPに一致する量で投与される、請求項101に記載の方法。
【請求項103】
DAC HYP組成物が、150mgに一致する量で投与される、請求項102に記載の方法。
【請求項104】
DAC HYP組成物が、300mgに一致する量で投与される、請求項102に記載の方法。
【請求項105】
DAC HYP組成物が、4週間に1回投与される、請求項101から104のいずれか一項に記載の方法。
【請求項106】
DAC HYP組成物が、計少なくとも48週間投与される、請求項105に記載の方法。
【請求項107】
DAC HYPが単独療法として投与される、請求項101から106のいずれか一項に記載の方法。
【請求項108】
請求項107に記載の方法であって、患者がインターフェロンベータを用いた前治療に無効果であったかまたはインターフェロンベータを用いた前治療を中止している、請求項107に記載の方法。
【請求項109】
DAC HYPがインターフェロンベータに対して補助的に投与される、請求項101から106のいずれか一項に記載の方法。
【図1】


【図2】

【図3A】

【図3B】

【図3C】

【図3D】

【図3E】

【図4A】

【図4B】
【図5】
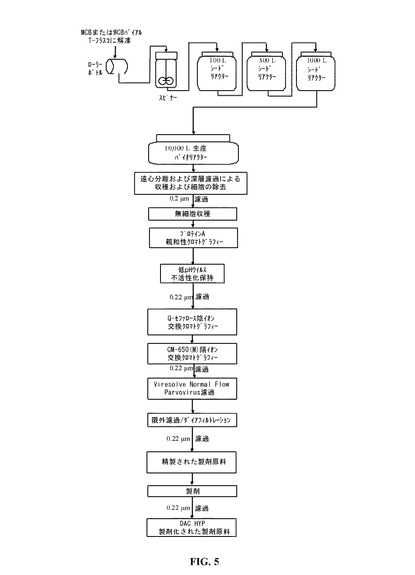
【図6】
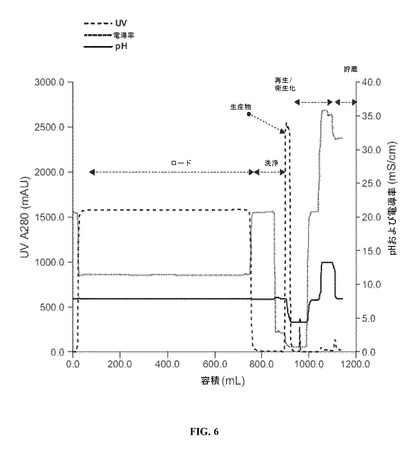
【図7】
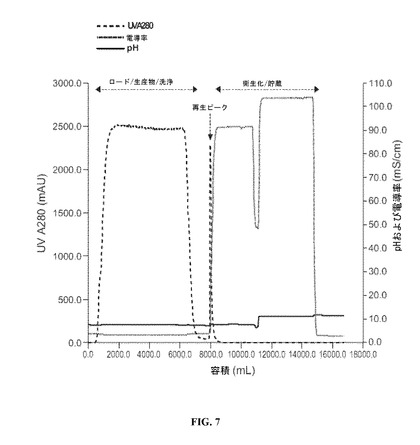
【図8】

【図9】
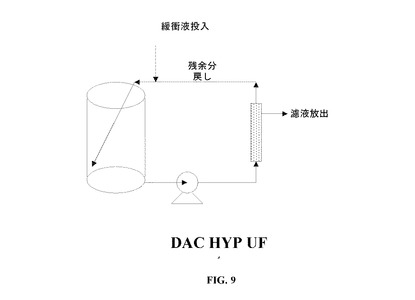
【図10】
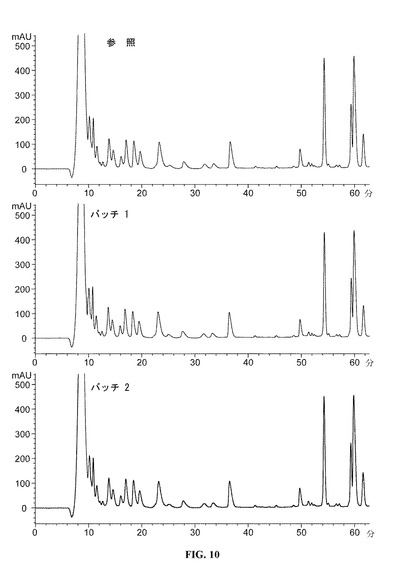
【図11】
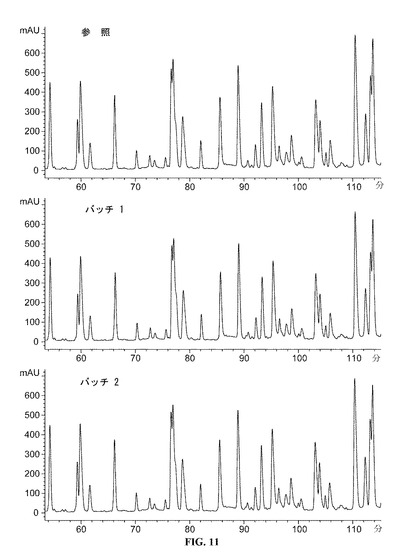
【図12】
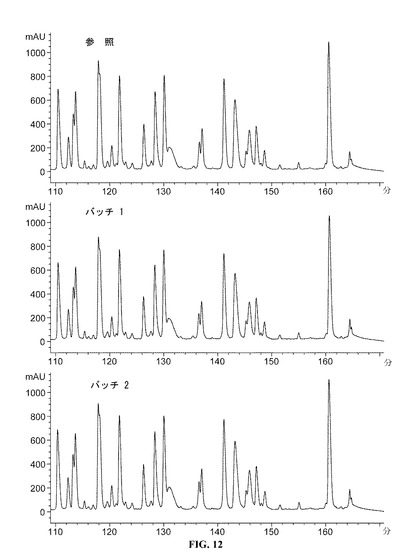
【図13】

【図14A】
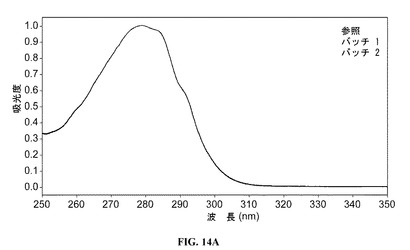
【図14B】

【図15A】
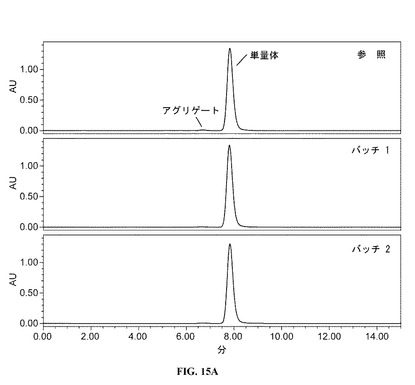
【図15B】
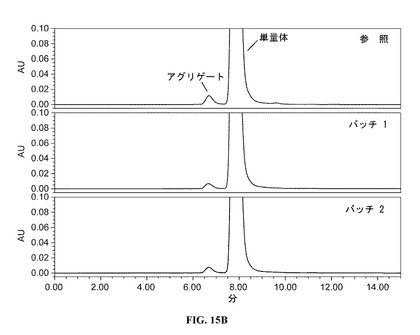
【図16】
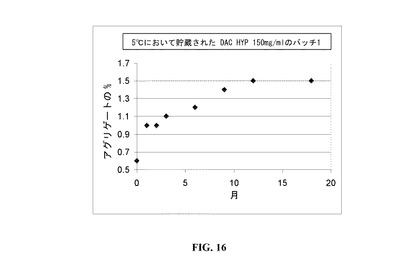
【図17】
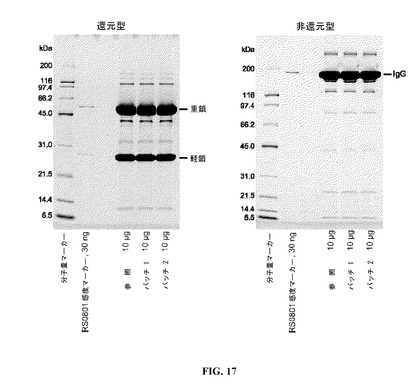
【図18】
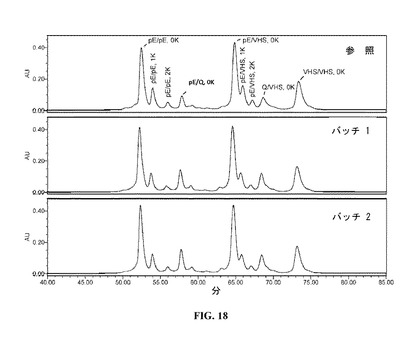
【図19】
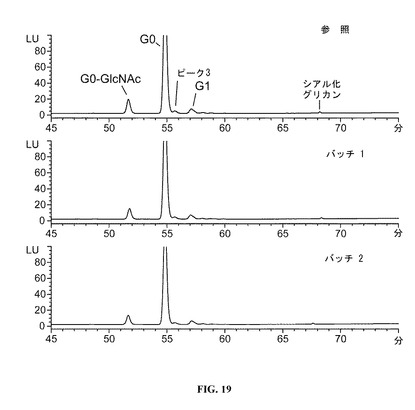
【図20】
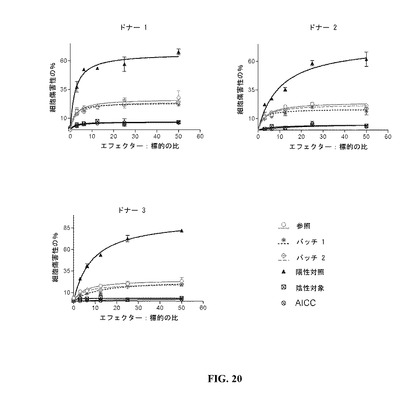
【図21】
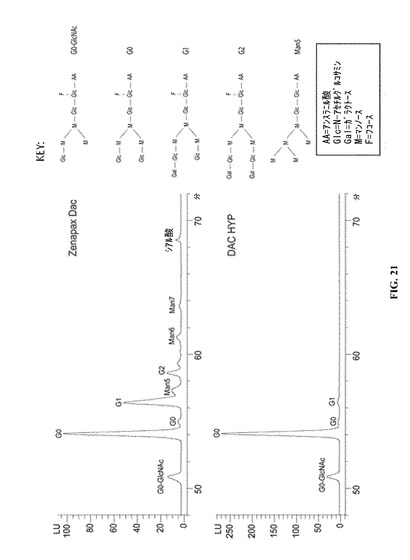
【図22A】

【図22B】
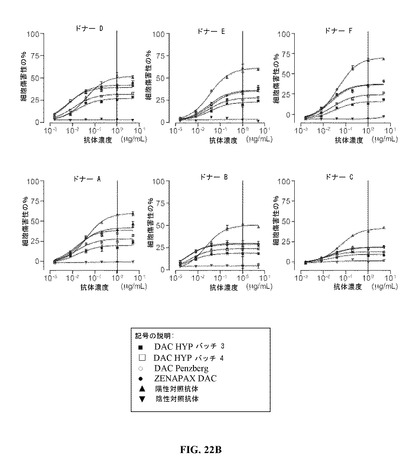
【図23】

【図2】
【図3A】
【図3B】
【図3C】
【図3D】
【図3E】
【図4A】
【図4B】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14A】
【図14B】
【図15A】
【図15B】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22A】
【図22B】
【図23】
【公開番号】特開2012−244993(P2012−244993A)
【公開日】平成24年12月13日(2012.12.13)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−119327(P2012−119327)
【出願日】平成24年5月25日(2012.5.25)
【出願人】(509189086)アボット バイオセラピューティクス コーポレイション (11)
【Fターム(参考)】
【公開日】平成24年12月13日(2012.12.13)
【国際特許分類】
【出願番号】特願2012−119327(P2012−119327)
【出願日】平成24年5月25日(2012.5.25)
【出願人】(509189086)アボット バイオセラピューティクス コーポレイション (11)
【Fターム(参考)】
[ Back to top ]