
DNAなどの分離精製機構
【課題】核酸、就中フラグメントDNAの分離精製を極めて効率化し、再現性を高く行うものであって、高濃度の塩による溶出を行わず、溶出精製の必要がない簡単な機構、方法により高純度のフラグメントを得る。
【解決手段】この機構はモノリス構造体を使用し、核酸、就中フラグメントDNAを精製するための機構であって、ガラスやシリカにより形成されるモノリス構造体、即ち、細孔が上端から下端まで連通した開放構造を有する一体が多孔質体であって、核酸大きさ35bp(mer)から100Kbp(mer)に対応する通孔が設けられている。
【解決手段】この機構はモノリス構造体を使用し、核酸、就中フラグメントDNAを精製するための機構であって、ガラスやシリカにより形成されるモノリス構造体、即ち、細孔が上端から下端まで連通した開放構造を有する一体が多孔質体であって、核酸大きさ35bp(mer)から100Kbp(mer)に対応する通孔が設けられている。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、DNAなどの分離精製機構に関するものである。
【背景技術】
【0002】
今まで、核酸含有物からの核酸の精製分離法は、核酸混合物をカオトロピックの塩の存在下でガラスやシリカゲルの粒子、ガラス、石英ウール、シリカ、ガラス膜、ポリマーなどに吸着させることはよく知られている。核酸含有物としては、培養細胞・組織、血液・血清・尿・糞便などの体液、バクテリア・ヒト結核菌などの細菌、HIV・B型肝炎・C型肝炎などのウイルスなどの生物学的原料物質が主なもので、プラスミドDNA、ゲノムDNA、染色体DNA、RNA、ミトコンドリアDNA、フラグメントDNAなどが分離精製可能である。
【0003】
フラグメントDNAの精製は、分子生物学的研究において、非常に頻繁に用いられる技術で、PCR、クローニング、シークエンシング、制限酵素消化、その他酵素的作用などのアプリケーションに先立って行われている。
例えば、組換M13ファージからDNAを単離する方法があり、ガラス繊維フィルター上でカオトロピック物質の添加により、M13ファージDNAを結合させ、次いで分離洗浄乾燥を経て溶離する単離方法がNucleic Acids Research Vol.15 5507-5516(1987)(非特許文献1)に示されている。
【0004】
又、ガラスパウダーを添加してDNAをガラスパウダーに結合させ、遠心分離して、ガラスパウダーを集め、洗浄、溶離し単離する方法がPvoc. Natl. Acad.Sci.USA Vol.76,615-619.(1979)(非特許文献2)に示されている。又、同様の方法は、特開昭59−227744号公報(特許文献1)、Analytical Biochemistry Vol.121.382-387.(1982)(非特許文献3)、Molecular cloning: A Laboratory Manual 188-190.(1982)(非特許文献4)等に記載されている。
【0005】
又、複合性生物出発材料、カオトロピック物質及びシリカ又はその誘導体を含む核酸結合性固相を混合し、 核酸を結合した固相を液体から分離し、洗浄し、核酸を溶離する方法が特許第2680462号公報に提案されている。
カオトロピック試薬の存在下でDNAやRNAを吸着させる物理的メカニズムについては詳しくは明らかになっていないが、負に帯電した担体と、核酸との間にカチオン交換反応が起こると考えられている。従って精製の効率は、担体表面と生体試料の接触の効率とイコールと考えることができる。
【0006】
前記のどの担体を使用するにしろ、吸着させる担体を容器(カートリッジ、チップなど)に保持し、その容器に生体試料を通液し、吸着バッファー液で担体に核酸を吸着させ、その後洗浄液で核酸成分以外の夾雑物をカートリッジ外に追い出し、更にその後、溶出液を通液して核酸成分をその液と共に取り出す手順が一般的である。
【0007】
その他に、電気泳動やさまざまな抽出によってフラグメントDNAをアガロースゲルから精製することもよく行われるが、この方法は時間がかかり、得られたDNAも極めて希薄となり、塩や有機溶媒が含まれているため、更にエタノール沈殿で脱塩や濃縮する必要が生じるものである。又、ゲル濾過精製テクノロジーのような従来法では、分子量の類似した分子同士を分離することは非常に困難である。
【0008】
担体を使用するこれらの分離方法は、高濃度の塩類を使う必要があるため、DNAのガラスまたはシリカゲルの粒子表面での分解若しくは変性をもたらす現象が確認されている。高濃度のカオトロピック塩の存在下では、この吸着は略定量的に生じるが、吸着したDNAの溶出は、塩類の緩衝液の存在下で行われる。DNAの断片の処理は100塩基対(bp)〜10,000塩基対(bp)の範囲が限度で、100塩基対(bp)以下のDNA断片や10,000塩基対(bp)以上の100,000塩基対(bp)までのDNAの定量的な分離や精製をすることは不可能であった。
【0009】
担体として、ガラス粉末をベースにした調整方法では、その表面と核酸との接触効率を上げるために、ビーズを小さくする、量を増やすと云うことが考えられる。然し、通液時の圧力が上がってしまい、操作性が著しく落ちると共に、ビーズ間空隙が小さくなり、核酸分子は分子量が大きいため、その空隙に入り込めず、かえって効率が落ちると云う問題が生じる。又、分離の向上を目的に容器の長さを長くすると、圧力が上がることに加えて溶出溶媒の量も増え、濃縮効率が落ちることになり、簡便な処理からは遠ざかってしまう。更に、ビーズやウールなどを使用すると微量ながらそのかけらや粒が溶出液に入り込んでしまうので、後のアプリケーションに問題となる。又、容器に充填する際の充填法が一定しないと分離時間やパターンが変わってしまうので、分離の安定性が悪いと云う問題も生じる。
【0010】
担体として膜やフィルターを使用する方法は、使い勝手よく加工できるメリットがあるが、分離に適当な孔を制御して作成することが難しく、実用性に乏しい。
又、ポリマーを使用する方法は、そのポリマーの性質によって核酸と特異的に反応する働きが充分でなかったり、又その部分以外に影響を与える部分が存在したりと分離系が複雑になり、単純なプロトコルでの精製は不可能である。
何れも高純度なフラグメントDNAを精製することは不可能である。又、粉末シリカ樹脂や懸濁液が持つ取扱い難さや、その後のアプリケーション反応を妨げる虞がある等の欠点がある。
【0011】
特許文献2の発明に於ける如く、複雑な出発材料から、前処理なしで核酸を直接単離する考え方は、所謂「消化」と「精製」を同時に行うものである。そのために過激な反応条件が必要となり、且つ適用できる核酸の分子量範囲が狭くなると云う弊害が生じる。
核酸混合物を、高濃度の塩(イオン強度)を含み、且つ脂肪族アルコールやポリエチレングリコール等の有機酸を含む吸着水溶液からシリカゲル、ガラス等の多孔質、非多孔質の無機基体上に吸着させ、洗浄させ、次いでより低濃度の塩(イオン強度)を含む溶液で溶出して核酸を得る方法が特表平8−5011321号公報(特許文献2)に提案されている。
【0012】
然し、この方法に於いては、高濃度の塩を含む吸着水溶液から、核酸混合物を無機体上に吸着させること及び低濃度ではあるが、塩を含む溶液で核酸を溶出するため、例えばDNAサンプルが大きなアガロース片に含まれる場合には、複数のカラムを使っての処理が必要であり、溶出したフラグメントDNAをプールし、更に得られたフラグメントDNAに塩や有機溶媒が含まれるので、濃縮や脱塩等の操作工程が必要であり、その上エタノール沈殿中に精製DNAを損失する虞もある。
【先行技術文献】
【特許文献】
【0013】
【特許文献1】特開昭59−227744号公報
【特許文献2】特表平8−5011321号公報
【非特許文献】
【0014】
【非特許文献1】Nucleic Acids Research Vol.15 5507-5516(1987)
【非特許文献2】Pvoc. Natl. Acad.Sci.USA Vol.76,615-619.(1979)
【非特許文献3】Analytical Biochemistry Vol.121.382-387.(1982)
【非特許文献4】Molecular cloning: A Laboratory Manual 188-190.(1982)
【発明の概要】
【発明が解決しようとする課題】
【0015】
そこで本発明に於いては、先行技術の上述した点を改良し、吸着や溶出分離が極めて簡単且つ容易に行えると共に、高濃度の塩による溶出を行わず、溶出精製の必要がなく、核酸就中フラグメントDNAなどの分離精製が著しく効率化された再現性の高い方法を提案せんとするもので、
【課題を解決するための手段】
【0016】
一体型モリノス構造体であって、一端から他端まで連続した通孔を形成させ、かつ核酸大きさが35bp(mer)から100Kbp(mer)に対応する通孔が設けられ、分離すべき核酸を含有する溶液を通過させることにより、該通孔に対応する核酸が夫々保持できるように構成したことを特徴とし、モノリス構造体は、ガラス、シリカ等の無機物又は無機物に有機物を含有するハイブリッド体であって、上面から下面まで貫通しているマクロ細孔を持つ多孔質体を使用することを特徴とし、モノリス構造体の多孔質体はマクロ細孔の内部にミクロ細孔を有することを特徴とし、モノリス構造体の多孔質体はマクロ細孔1〜100μm、ミクロ細孔0〜100nmであることを特徴とし、カラムチューブにモノリス構造体により形成されるディスクを配置することにより、モノリス固相カラムを構成することを特徴とし、上下を開放した筒状体に、モノリス構造体により構成した基体を着脱自在に装着して形成したモノリス固相カラムを使用することを特徴とする。
【発明の効果】
【0017】
本発明によれば、100bp(mer)以下のDNA断片や10000bp(mer)以上の100Kbp(mer)までの広範囲のDNA断片の定量的な分離や効率的な精製が可能である。又、過敏な反応条件を使用しないで核酸の吸着分離が可能となり、高濃度の塩による溶解、吸着が必要なく、濃縮や脱塩の操作が必要なく、核酸精製就中DNAフラグメントの精製が容易である。又、塩を含まない溶液又は無菌水で溶出でき、高純度フラグメントDNAなどが容易に得られる。
【図面の簡単な説明】
【0018】
【図1】本発明による実施例1の従来法との対比図
【図2】本発明による実施例1の従来法との対比図
【図3】本発明実施例1のHPLCによる評価図
【図4】本発明による実施例2のDNAフラグメント精製分離図
【図5】本発明による実施例4のDNAフラグメント精製分離図
【図6】本発明による実施例5のDNAフラグメント精製分離図
【図7】本発明による実施例6の1本鎖PCR増幅産物分離図
【図8】本発明による実施例7のナトリウムとカリウムによる精製比較図
【図9】本発明一実施例カラムチューブ説明斜面図
【図10】同上ディスク説明斜面図
【図11】同上コレクションチューブ説明斜面図
【図12】同上モノリス固相カラム説明斜面図
【発明を実施するための形態】
【0019】
本発明者らは、効率のよいモノリス構造体を核酸精製に使用し、バッファー条件を整えると、これまで常識とされていた塩濃度の高いバッファーを使用しなくてもよいことを見出した。核酸保存のために加えるトリス塩酸EDTAのほかには水だけで、吸着していた担体から結合が外れて溶出する。核酸を吸着させる際にも、分離溶媒として働くイソプロパノールとシラノール基と反応すると思われるイオンを供給する塩化合物、そしてアガロースゲルを溶解させるためのカオトロピック塩であるグアニジン塩酸塩などを吸着溶媒とする。このとき陽イオンになりやすい、アルカリ金属塩が存在すると、容易に脱水素反応を起こし、それによりその陽イオンがカチオン架橋反応を核酸のリン酸部分と起こし、核酸を吸着すると考えられる。アルカリ金属は、例えばリチウム、ナトリウム、カリウム、ルビジウム、セシウム、フランシウムなどが該当する。中でも電気陰性度が小さく、容易に陽イオンとなりカチオン架橋反応をするカリウム塩は反応が起こりやすく、最後に、核酸成分と一緒にイオン状態で溶出されるが、後のアプリケーションの妨害をしないので有用である。尚、ナトリウム塩はその後のアプリケーションの妨害となるので、脱塩操作を行わないと使用できずあまり有用ではない。換言すれば、この装置及び方法であれば、脱塩操作やアルコール沈殿などが必要なく、得られた精製液をそのまま後の操作(PCR、クローニング、シークエンシング、酵素的操作)に持っていけることになる。操作手順が簡便化することは、核酸の損傷を防ぐために非常に重要な事項である。
【0020】
本発明の目的は、精製効率のよいモノリス構造体のシリカやガラスを使用し、より簡単な緩衝液を使用することで、その後のクローニングなどの各種アプリケーション操作に影響を与えず、核酸を精製することができる方法を提供することにある。
【0021】
モノリス構造体とは、上端から下端まで連通した開放構造のマクロ細孔を持つ、一体型多孔質体のことを指し、そのマクロ細孔に更にミクロ細孔を持つものが多く合成されている。
モノリス構造体は、主に、ゾル−ゲル法で作成することができる。即ち、金属アルコキシドを部分的に加水分解して反応性モノマーを作り、このモノマーを重縮合してコロイド状オリゴマーを作り(ゾルの生成)、更に加水分解して重合と架橋を促進させ、三次元構造を作る(ゲルの生成)ことで合成される。
【0022】
この反応時に種々の有機モノマーを添加すると、有機・無機ハイブリッドモノリスも簡単に作成できる。従って、添加する有機モノマーの種類によって、化学的特性を加えることも可能となる。例えば、親水基を持つ有機モノマーを添加することによって、水系試料成分の吸水性を向上することができる。又、選択的な化学作用を示す官能基を持つ有機モノマーを添加することによって、精製における不純物となる特性成分の吸着に利用し、その不純物を固相に残すことで、核酸の精製効率をアップすることができるようになる。又、弾性率の高いポリマーをゾル−ゲル工程中に入れることにより、モノリス構造体に弾力性を持たせることも出来る。更に基本的には、有機・無機を混在させることによって、モノリス構造体の化学的安定を上げることも可能となる。
【0023】
これらのことは、ゾル−ゲル法にて作成される有機・無機ハイブリッドモノリス構造体は、添加有機モノマーの種類で目的に応じた化学表面を構成したり、化学的安定性を向上したりと云う性質を付加できることを意味し、DNA前処理に於いて有効なモノリス特性を目的に応じて自由に改善できることを示している。更に、ゾル−ゲル方法から作成したモノリスは、本発明のDNA用固相として金属含有が少ないと云う点で適している。一般的なシリカゲルなどは、ケイ酸ナトリウムなどから作成され、多量の金属が残る。確かに、一部では精製したケイ酸ナトリウムからの作成やゾル−ゲル法からの高純度のシリカゲルもあるが、それらは高価であり、通常使用態様である使い捨てには適さない。又、安価に出来たとしても、粒子作成時点ではバッチ式合成であり、合成雰囲気から金属濃縮が生じる可能性が大きい。
【0024】
ゾル−ゲル法におけるモノリス構造の合成に於いては、連続行程により作成され、金属のコンタミネーションは全くない。従来、シリカゲルでは、塩酸や硝酸等で洗浄するなどの工夫や、金属影響を減らすようなEDTAなどの添加があったが、本発明固相では全く必要ない。
【0025】
もう一法として、ガラス分相によってもモノリス構造固相を作成できる。基本的には、ゾルーゲル法からのモノリス構造の合成と同様の有効性があるが、ゾルーゲルモノリスよりもマクロ細孔を大きく作る事ができるので、2次ミクロ細孔を内部表面に作る場合に有効である。更に、ガラス分相は、その組成より耐アルカリ性が高く、アルカリ洗浄による再生が出来ると言うメリットがある。
【0026】
以下、図に示す実施例により、本発明を詳細に説明する。
本発明に於いて最も基本的な構成は、フラグメントDNAをアガロースゲル、PCR反応物、制限酵素処理DNA、1本鎖DNA及びRNA等から精製するために、カオトロピック塩の存在下、ガラスやシリカに吸着させること、殊に優れた分離能力を有するガラスやシリカにより形成されるモノリス構造体、即ち、細孔が上端から下端まで連通した開放構造を有する一体型多孔質体を使用し、これに吸着させることにある。
フラグメントDNAをカオトロピック塩の存在下ガラスやシリカに吸着させることは、前記の如く既に提案され実施されている。
【0027】
然し、従来の方法は全て充填材を使用していた。このため、充填材の充填にばらつきがあり、均一にならないこと、また充填材通過の後に粒がDNA液に残留してしまうこと、液との接触面積が小さく反応効率が悪いこと、通液圧力が大きく扱いにくいこと等の欠点が残るものであった。
【0028】
本発明は、カオトロピック塩の存在下に於いて、アガロースゲル、PCR反応液からガラスやシリカゲル粒子に吸着したものを抽出する点に於いて、従来の技術とある部分共通するところがあるものである。
この従来の技術は、高濃度のカオトロピック塩の存在下で吸着を定量的に生じさせ、吸着した核酸の溶出はより低い塩濃度で行なうものであった。
【0029】
然も、特表平8−5011321号公報の方法は、分離すべき核酸を予備的精製行程なしに1つの操作行程で、核酸分画を行う方法である。このために高濃度の塩のバッファーによる過激な反応条件が必要となり、適用できる核酸の分子量範囲が狭くなる。更に精製DNAは極めて希薄で、塩や有機溶媒があるため、更にエタノール沈殿や濃縮する必要がある。
【0030】
本発明はこれらの欠点を改良するもので、高濃度の塩での吸着や溶出は行わず、水で溶出するものである。この結果、非常に高濃度で更なる精製の必要のないサンプルが得られる。
本発明はアガロースゲルからのDNA断片とPCR増幅反応DNA液、酵素反応液からのフラグメントDNAなどの精製を1つの目的とするものである。
【0031】
本発明は、Tris酢酸(TAE)バッファー又はTrisホウ酸(TBE)バッファーで作成した標準アガロースゲルや低沸点アガロースゲルからの35bp〜100KbpのDNA断片の抽出、精製が可能で60〜80%の回収率を得ることが出来る。又ゲルからの抽出のほかにもPCR増幅反応液からも35bp〜100kbpのPCR産物を直接精製することができ、80〜95%の回収率を得ることが出来る。得られたフラグメントDNAに塩や有機溶媒が含まないため、エタノール沈殿、脱塩や濃縮する必要がない。本製品はモノリスベースのシステムであり、DNA結合能は最大5〜8μgで、単離したフラグメントDNAを僅か5分で回収することができる。
【0032】
ゲルから精製する場合は、電気泳動後にゲルから目的のDNAバンドを切出し、グアニジンチオシアン酸(溶解、吸着バッフアー)の存在下で溶解する。PCR増幅後の精製の場合は、吸着バッファーを増幅反応液に直接添加する。溶解したゲル片は、マイクロ遠心機又は吸引装置を用いてモノリス固相カラムを通過させる。その際、目的のフラグメントDNAはシリカモノリスやガラスモノリスの表面に結合し、結合したフラグメントDNA断片を洗浄バッファーで洗浄後、DNAを水で溶出する(溶出バッファー)。
【0033】
このバッファーとして下記が使用される。
A バッフアー(溶解、吸着バッファー)
B バッフアー(洗浄バッファー)
C バッフアー(溶出バッファー又無菌水)
これを更に詳述すると
A1 バッフアー PCR反応液用(グアニジン塩酸塩1〜8M、酢酸カリウム0.1〜1M未満、2−プロパノール1〜70%)
A2 バッフアー アガロースゲル用(グアニジンチオシアン酸1〜8M、酢酸カリウム0.1〜1M未満、2−プロパノール1〜70%)
B バッフアー(酢酸カリウム0.1〜1M未満、エタノール1〜80%)
C バッフアー(pH=8〜8.5 Tris−HCI10mM、EDTA1mM、又は無菌DNA、RNA free水)
【0034】
A1バッファーに於いて、グアニジン塩酸塩はグアニジンチオシアン酸が好適で、1〜8Mがより好ましい。2−プロパノールは40%以上が効率的である。
A2バッファーに於いても以上の条件が当て嵌まる。
溶出バッファー液Cに於いては、水による溶出が可能であるが、雑菌の混入を防ぐために、限外濾過膜処理やディエチルピロカーネートで処理などを行ったRNaseフリーな水を使用する事が推奨される。又、精製したDNAを保管する場合、菌の混入を防ぐため、溶出バッファー液Cとして、先にEDTAバッファーを添加した水を使用してもよい。分離機構から見てもEDTAバッファーの有無で精製効率が変わることは無い。
【0035】
本願発明に於いて、基体にモノリス構造体を使用することにより、核酸のリン酸基とシラノール基との反応を効率的に進行させることが出来る。又、粒状の充填材を使用する場合に避けられないサンプルにシリカ粒子が付着したまま精製され、所謂アプリケーション反応を妨げる虞のあるシリカのキャリーオーバーの問題を防ぐことができる。更に、分離時における圧力も低く抑えられ、又充填材を使用する場合と異なり、封止剤も不要である等の利点を有する。
【0036】
このモノリス構造体は、例えば、シリカゲル等の無機質多孔質体を、重合可能な低分子化合物ゾルを精製し、最終的に凝集体や重合体のゲルを得る、所謂ゾルゲル法によって作製することが出来る。この方法は、一般的に中心細孔径1〜100μmであるが、その後の技術進歩により数nmも可能である。
【0037】
又、ゾル−ゲル過程におけるスピノールダル分解を利用し、2種類の細孔を有するシリカ骨格中に細孔の存在するモノリス構造体も作成できる。
又、上面から下面まで貫通しているマクロ細孔の内部に開放構造を有するミクロ細孔を持つ多孔質体が充填された構造の多孔質体も作成できる。因みに、この多孔質体はマクロ細孔の直径1〜100μmであり、ミクロ細孔が0〜100nmである。
【0038】
この他、ガラス、シリカが含有されて形成されるものであれば、適宜のモノリス構造体が使用しうること勿論である。
このモノリス構造体は、その製法により、多孔質体の貫通孔は所望の通り形成できるので、適宜選択して形成するが、1〜100μm程度が使用しやすい。然し、この選択は、使用する原料、例えばアガロースゲルやPCR反応液、バッファーにより、又その目的により決められる。
【0039】
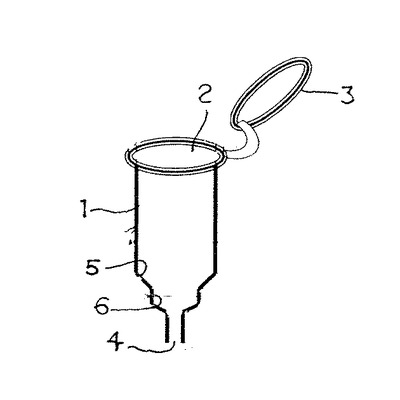
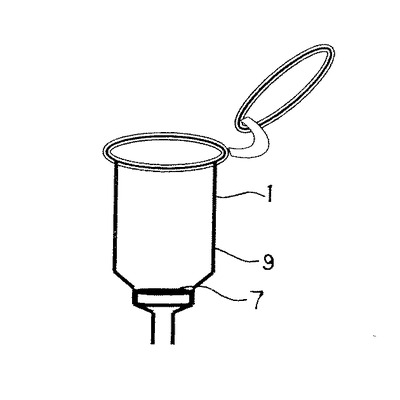
このモノリス構造体により構成される基体の一使用形態について述べると、筒型のカラムチューブ1を形成し、上端開放部2には密閉可能な蓋3を着脱自在に設ける。下端には細口径の出口4を構成し、出口4上部には段差5を設けて中口径部6を形成してある。該段差5にはモノリス構造体により形成された基体としてのディスク7が載置乃至嵌合できるようにしてある。該ディスク7は中口径部6と略同形で所望厚さ例えば0.1〜10mm程度の円盤状或は所望に応じ、円錐状を形成してもよい。このカラムチューブ1とディスク7により、モノリス固相カラム9を構成する。8はコレクションチューブで、カラムチューブ1の挿通が可能の径に構成され、上部にカラムチューブ1の上端縁が係止可能に構成してある。
【0040】
前記カラムチューブ1やコレクションチューブ8については、ポリプロピレン製が使用されるが、核酸に影響を与えない有機ポリマー、例えばポリエチレン、ポリエチレンテレフタレート、ポリスチレンや無機化合物、例えばガラス、シリカ等視認性がよくある程度強度があれば使用できる。
又、モノリス構造体の基体としてディスク7が挙げられているが、これに限らず皿状や筒状等溶液の通過の自在である型で使用できる。
【0041】
DNAの分子サイズは、10塩基対あたり、約3.4nm程度と云われている。例えば、35bp〜300bpのDNAに於いて、10nm〜100n程度のマクロ細孔、300bp〜3KbpのDNAに於いて、100nm〜1μm程度のマクロ細孔、30Kbp300KbpのDNAに於いては、1μm〜10μm程度のマクロ細孔、30Kbp300〜KbpのDNAに於いては、10μm〜100μm程度のマクロ細孔を持てば、DNA分子を傷つけたり、壊したりすることなく、DNA分子を通過させることが可能と考えられる。
【0042】
又、不純物などとの相互作用を高め、より効率的に精製するためには、ミクロ細孔の付加が行われる。各種試験、実験などの経験から、ミクロ細孔が数10nm前後であれば分子量数10万の化合物、10nm前後であれば分子量数百〜数万の化合物と相互作用があることが確認されており、更に大きな分子量の化合物ならば、その分子の破壊を助長すると云われるミクロ細穴が限りなくない状態、つまり0nmがよいことが判っている。
【0043】
マクロ細孔にミクロ細孔を付加した種々の一体型モノリス構造体を数種用意し、DNAの種類及び不純物除去などの目的ごとに使い分けることも可能である。ちなみに、数種類の一体型モノリス構造体を作成するには、マクロ細孔を予め作成し、その後にミクロ細孔を形成させる方法の方が合成上便利である。
【0044】
現状必要とされている精製に於いては、プライマーやアガロースゲル分解物などの低分子からの分離精製が中心であり、1種類のモノリス構造体でも充分分離可能である。
1〜100μm、好ましくは20μm程度のマクロ細孔と、0〜100nm、好ましくは10nm程度のミクロ細孔を形成したモノリス構造体を用いれば、実施例にあるような、35bp(mer)〜100Kbp(mer)の広範囲のDNAの精製が充分可能である。
【0045】
A.モノリス固相カラムを用いてPCR産物を生成するのには次の方法がある。
(1)マイクロ遠心法
このプロトコールは、PCR反応液から2本鎖DNAフラグメントを精製する目的でデザインされ、1つのモノリス固相カラムと精製用バッファーを用いれば、遠心操作により、35bp〜100Kbpのフラグメントがプライマー、ヌクレオチド、ポリメラーゼ、塩等から分離可能である。
【0046】
PCR反応液10μlに対して、100μlのバッファーA1(吸着バッファー)を添加する。バッファーBは約300μl(洗浄バッファー)で洗浄する。
全ての遠心操作は一般的な卓上マイクロ遠心機で〜10,000rpmで行う。
【0047】
1.PCR反応液に10倍容量のバッファーA1を加えて混合する。ミネラルオイルを除去する必要はない。例えば、50μlのPCR反応溶液(オイルを含まない量)には500μlバッファーA1を加える。
【0048】
2.コレクションチューブ8にモノリス固相カラム9を挿入し、調整したサンプルをモノリス固相カラムにアプライする。高回収率を得るため、サンプル液を残さないでモノリス固相カラム9に添加する。
【0049】
3.モノリス固相カラム9を10,000rpm、30秒間遠心した後、モノリス固相カラム9を取外し、コレクションチューブ8内の液体を除去。モノリス固相カラム9を、コレクションチューブに再度挿入する。
【0050】
4.バッファーB(洗浄バッファー)500μlを添加し、モノリス固相カラム9を10,000rpm、30秒間遠心。更に10,000rpm、1分間遠心する。
コレクションチューブ8内の液体を捨てた後に、バッファー由来の残留エタノールを完全に除去するためには再遠心操作を行うことが必要である。
【0051】
5.モノリス固相カラム9を新しい1.5ml遠心サンプリングチューブに移し、バッフアーC(溶出バッファー)10〜50μlをモノリス表面の中央に添加し、モノリス固相カラム9を1分間室温でインキュベートした後、10,000rpm、1分間遠心する。遠心チューブ内に溶出されたDNAは精製されたDNAで、−20℃で保存する。又は後の操作にそのまま使用する。
モノリスに結合したDNAが完全に溶出されるように、モノリス表面中央部分に溶出バッファーCを添加する。10μlの溶出バッファーCを用いた場合は溶出液量は9μlである。
溶出効率はpHが8〜8.5の間で最大となる。溶出に滅菌水系を用いる場合には、pHがこの範囲であることを確認するのがよい。
【0052】
(2)吸引マニホールド法
モノリス固相カラムは、一般的なルアーアダプターを含む吸引マニホールドにより操作を行うことが出来る。このプロトコールは、PCR反応液から2本鎖DNAフラグメントを精製する目的でデザインされている。1つのモノリス固相カラムと精製用バッファーを用いれば、吸引装置によるサンプル処理操作により、35bp〜100Kbpのフラグメントがプライマー、ヌクレオチド、ポリメラーゼ、塩等から分離可能である。
【0053】
PCR反応液10μlに対して、100μlのバッファーA1(吸着バッファー)を添加する。バッファーBは約300μl(洗浄バッファー)で洗浄する。
一定で安定した吸引が行われるよう、各操作ステップ毎に一端吸引スイッチを切る。
【0054】
1.PCR反応液に10倍量のバッフアーA1を加えて混合する。ミネラルオイルを除去する必要はない。例えば、50μlのPCR反応溶液(オイルを含まない量)には500μlバッファーA1を加える。
【0055】
2.吸引マニホールドとモノリス固相カラムを準備する。
ルアーアダプター吸引マニホールドを吸引装置に接続する。
【0056】
3.吸引マニホールド上のポートに取付けたバキュームアダプターを装着。モノリス固相カラム9をバキュームアダプターに挿入する。
【0057】
4.調整したPCRサンプルをピペットによりモノリス固相カラム9にアプライし、DNAを結合させるために吸引する。溶液がモノリス固相カラム9を完全に通過するまで吸引する。サンプルがカラムを通過した後、吸引を止める。
高回収率を得るため、サンプル液を残さないでモノリス固相カラム9に添加する。添加最大容量は800μlで、800μlよりサンプル量が多い場合には、数回に分けて添加する。
【0058】
5.バッファーB(洗浄バッファー)500μlをモノリス固相カラム9に添加し、液体がモノリス固相カラムを通過するまで吸引する。
【0059】
6.モノリス固相カラム9をマニホールドから外し、コレクションチューブ8に移す。10,000rpmで1分間遠心する。
バッファー由来の残留エタノールを完全に除去するために遠心操作が必要である。(液体がモノリス固相カラムを通過するまで吸引し、乾燥させる。モノリス固相カラム9に残っている洗浄バッファーを完全に除去するための必要手段である。)
【0060】
7.モノリス固相カラム9を新しい1.5ml遠心サンプリングチューブに移し、バッファーC(溶出バッファー)10〜50μlをモノリス表面の中央に添加し、カラムを1分間室温でインキュベータした後、10,000rpm、1分間遠心する。遠心チューブ内の溶出されたDNAは精製されたDNAで、−20℃で保存する。又は後の操作にそのまま使用する。
モノリスに結合したDNAが完全に溶出されるように、モノリス表面中央部分に溶出バッファーCを添加する。10μlの溶出バッファーCを用いた場合は溶出液量は9μlである。
溶出効率はpHが8〜8.5の間で最大となる。溶出に滅菌水系を用いる場合には、pHがこの範囲であることを確認するのがよい。
【0061】
B.モノリス固相カラム9を用いてアガロースゲルを精製するのには次の方法がある。
(1)マイクロ遠心法
このプロトコールは、標準的又は低温融解アガロースゲル(TE又はTEBバッファー使用)から、DNAフラグメントを精製する目的でデザインされ、1つのモノリスカラムと精製用バッファーを用いれば、遠心操作により、35bp〜100Kbpのフラグメントがプライマー、ヌクレオチド、ポリメラーゼ、塩等から分離可能である。1個のモノリスカラムにつき、最大1000mgのアガロースの処理が可能である。
アガロースゲル10mgに対し10μlのバッファーA(溶解、吸着バッファー)を添加する。バッファーBは約500μl(洗浄バッファー)で洗浄する。
全ての遠心操作は一般的な卓上マイクロ遠心機で〜10,000rpmで行う。
【0062】
1.清潔なカミソリやメスで目的のバンドを切取り、1.5ml遠心チューブに入れる。余分なゲルを取除いて、ゲルスライスのサイズを最小になるようにする。
【0063】
2.バッファーA2(溶解、吸着バッファー)をゲルスライス100mgに対し100μl添加する。
100mgのゲルには、バッファーA2を100μl添加するが、濃度が2%以上のアガロースゲルを用いる場合は、バッファーBを600μl添加する。1個のモノリスカラムで処理できるゲル量は1,000mgであるので、ゲル量が1,000mgを超える場合は2個以上のモノリスカラムを使用する。
【0064】
3.60℃で5分間又はゲルスライスが完全に溶解するまでインキュベーションする。インキュベーション中、2回チューブをボルテックスにかけて溶液を混合する。アガロースを完全に溶解させる。2%以上のゲルを用いる場合は、インキュベーション時間を長くすると回収率がアップする。
【0065】
以下の操作は、前記A.モノリス固相カラムを用いたPCR反応液の精製(1)マイクロ遠心法と同じであるから省略する。
(2)吸引マニホールド法
モノリスカラムは、ルアーアダプターを含む吸引マニホールドにより操作を行うことが出来る。このプロトコールは、標準的又は低温融解アガロースゲル(TE又はTBEバッファー使用)から、DNAフラグメントを精製する目的でデザインされた。1つのモノリスカラムと精製用バッファーを用いれば、吸引装置によるサンプル処理操作により、35bp〜100Kbpのフラグメントがプライマー、ヌクレオチド、ポリメラーゼ、塩等から分離可能である。
【0066】
アガロースゲル10mg対し10μlのバッファーA2(溶解、吸着バッファー)を添加する。バッファーBは約500μl(洗浄バッファー)で洗浄する。
溶出は遠心操作により卓上マイクロ遠心機で〜10,000rpmで行う。
一定で安定した吸引が行われるよう、各操作ステップ毎に一端吸引スイッチを切る。
【0067】
1.清潔なカミソリやメスで目的のバンドを切取り、1.5ml遠心チューブに入れる。余分なゲルを取除いて、ゲルスライスのサイズを最小になるようにする。
【0068】
2.バッファーA2(溶解、吸着バッファー)をゲルスライス100mgに対し100μl添加する。
100mgのゲルには、バッファーA2を100μl添加するが、濃度が2%以上のアガロースゲルを用いる場合は、バッファーA1を600μl添加する。1個のモノリスカラムで処理できるゲル量は600mgであるので、ゲル量が600mgを超える場合は2個以上のモノリスカラムを使用する。
【0069】
3.60℃で5分間又はゲルスライスが完全に溶解するまでインキュベーションする。インキュベーション中、2回チューブをボルテックスにかけて溶液を混合する。アガロースを完全に溶解させる。2%以上のゲルを用いる場合は、インキュベーション時間を長くすると回収率がアップする。
【0070】
以下の操作は、前記A.モノリス固相カラムを用いたPCR反応液の精製(2)吸引マニホールド法と同じであるから省略する。
C.モノリス固相カラムを用いて酵素反応液を精製するのには次の方法がある。
(1)マイクロ遠心法
このプロトコールは、制限酵素分解や標識反応などの酵素反応液から2本鎖DNAフラグメントを精製する目的でデザインされた。1つのモノリスカラムと精製用バッファーを用いれば、遠心操作により、35bp〜100Kbpのフラグメントが酵素、プライマー、ヌクレオチド、塩等から分離可能である。
【0071】
酵素反応液10μlに対し、30μlのバッファーA1(吸着バッファー)を添加する。バッファーBは約300μl(洗浄バッファー)で洗浄する。
全ての遠心操作は一般的な卓上マイクロ遠心機で〜10,000rpmで行う。
【0072】
1.酵素反応液に3倍容量のバッファーA1を加えて混合する。モノリスカラムで処理できる酵素反応液の最大容量は100μlである。
例えば、100μlの酵素反応液には300μlバッファーA1を加える。
【0073】
以下の操作は、前記A.モノリス固相カラムを用いたPCR反応液の精製(1)マイクロ遠心法と同じであるから省略する。
(2)吸引マニホールド法
モノリスカラムは、ルアーアダプターを含む吸引マニホールドにより操作を行うことが出来る。このプロトコールは、制限酵素分解や標識反応などの酵素反応液から2本鎖DNAフラグメントを精製する目的でデザインされた。1つのモノリスカラムと精製用バッファーを用いれば、吸引装置によるサンプル処理装置により35bp〜35Kbpのフラグメントが酵素、プライマー、ヌクレオチド、塩等から分離可能である。
【0074】
例えば、酵素反応液10μlに対し30μlのバッファーA1(吸着バッファー)を添加する。バッファーBは約300μl(洗浄バッファー)で洗浄する。
全ての遠心操作は一般的な卓上マイクロ遠心機で〜10,000rpmで行う。
一定で安定した吸引が行われるよう、各操作ステップ毎に一端吸引スイッチを切る。
【0075】
1.酵素反応液に3倍容量のバッファーA1を加えて混合する。モノリスカラムで処理できる酵素反応液は最大容量は100μlである。
例えば、100μlの酵素反応液には300μlバッファーA1を加える。
【0076】
以下の操作は、前記A.モノリス固相カラムを用いたPCR反応液の精製(2)吸引マニホールド法と同じであるから省略する。
この本発明の吸着や溶出分離が極めて容易に行え、高濃度の塩による溶出は必要なく、核酸の精製が極めて効率的に行える利点は、核酸成分をモノリス構造体に吸着させる点に基因する。
【0077】
従来タイプの方法では、シリカゲル粒子、ガラス粒子、それらをフィルター状にしたものが使用されている。それら全てに於いて、液が通る空間は、粒子表面を通ることになり、粒子にぶつかり、乱流が生じ、不均一な流れとなってしまう。そのため、全て表面に均一に触れることはできない。モノリス構造は、一体構造で内部に連続孔があるので、粒子内部を通るイメージとなる。即ち、全ての液が均一に接触する。又、粒子に比べると、骨格が小さく、液がぶつかった後の乱流の生ずることなく均一な流れとなる。
【0078】
即ち、従来の固相タイプでは、粒子における乱流が生じ、表面との接触が不均一となり、低分子側DNAの吸着が生ぜずに抜けてしまうことになる。
その抜けを防止させるために、従来のタイプでは、カオトロピック塩濃度を増やすことにより、反応を起こし易くしている。然し、この場合塩沈殿が生じ、限界が生じるため、低分子DNA捕集には限界が生じる。
【0079】
本発明モノリスタイプでは、均一な液の流れが保障されており、より低分子DNAの吸着が可能となる。
当然、洗浄工程でも同じことが起きる。従来の方法では洗浄液の乱流が生じてゲル表面を洗浄することが困難となる。実施例1に記載されているように、1回目洗浄後でも従来の方法では目的としないプライマーなどがかなり残っているが、本発明の方法では殆ど残らない。
繊維に粒子を埋め込んだフィルターや繊維そのものを用いたものでも乱流が生ずるのはやはり同じことになる。
【0080】
従来例として、入口と出口を備えた円筒状の中空体の出口付近に無機基体材料が配置されており、その無機基体材料は、きつく押込められたポリエチレンフリットの間にはさまれて保持されている例がある。(特許文献3)
この場合、分離に寄与する部分は、その無機基体材料部分であり、上下フリットは無機体を中空体の保持されるために用いられている。
いくらきつく押込められても、フリットと無機体の間には空間が生じることになり、その空間部分に液は残存してしまう。その空間部分の液の追出しや置換は困難となる。特に、上記の如き減圧方法では、一部に気相部分ができてしまうと、その部分が優先的に流れることになり、均一に液を抜くことができなきなる。試料付加、洗浄工程に於いては、液体の置換が成され難くなってしまう。
【0081】
又、最後の溶出に於いては、液が残ってしまい、回収が悪くなってしまう。
本発明のモノリス構造では、液体の流れるマクロ細孔は連続体となり、流れの方向に対して均一に液体が変わってゆくことになる。即ち、液体の置換効率が大幅に上昇する。実施例1のように、この従来タイプでは、2回目の溶出でも試料成分が多く残る原因の1つになっていると考えられる。本発明の連続体のモノリス構造に於いては、液体の置換効率が高いので、1回の溶出で充分であり、2回目溶出の残存が殆どないことがわかる。
【0082】
又、従来タイプでは、押込み具合で、その空間は変化し、中空体への押込み時のロット間のバラツキも出易くなる。本発明に於いて、モノリス構造は一体構造であり、中空体への押込み時のバラツキは全くない。
市販品としては高価であり、実在しないが、フリットと無機体を更に押し潰し一体にすることも可能となるが、この方法でも押し潰した界面部分に異なる層ができてしまう。やはり均一な連続孔を持つモノリス構造に比べると、液体の流れは阻害される。
【0083】
更に、本発明では、シリカゲルである無機物とフリット材料であるポリエチレンなどの有機物を、ゾル−ゲル行程で混合すれば、シリカとポリエチレンの性質を持つハイブリッドの均一相での作成も可能である。
無機体が粒子状の物では、それを止めるために、上下フリットは不可欠となり、上記説明のような置換効率の問題が生じる。
【0084】
シリカ繊維やケル粒子を埋込んだシリカゲル薄膜(例えば3M社のエムポアディスク(登録商標))を無機体とした場合でも、やはり物理的な硬さが無く、急激な減圧や高回転の遠心分離により、変形して繊維の一部や粒子が溶出してしまうことがある。僅かな変形に於てでも、空間容積は変化することになり、バラツキ要因になってしまう。モノリス構造では、固い骨格の内部にある連続孔による分離のため、圧力変動などでも変形しないので、再現性も得られる。
【0085】
例え、フリットで止める必要のない固い繊維膜やシリカゲル薄膜を形成できたとしても、液の流れは、繊維やゲル表面を流れることにより、乱流が生じ、均一な分離が得られず、本発明のモノリス構造体を用いた場合ほどの分離は期待できない。
乱流の生じないモノリス構造体であることにより、初めて低分子DNAの吸着及び高い洗浄効果を達成できる。
【0086】
更に、従来タイプの粒子における流路と本発明のモノリス構造体におけるミクロ細孔とは液流れに対して接触と云う点で異なる。粒子タイプなどでは、液が入ってくる側と液が抜ける側とは、液抵抗における圧力の均一性がなく、細孔内部への液の接触が異なってしまう。HPLCのような加圧系では均一な圧力にできるので、その影響は少なくなるが、本発明分野で用いられる減圧系では、入口側は常圧であり、出口側では負圧になり、粒子1個における細孔内部への出入りが不均一となる。即ち、同じ成分でも成分分子によって、細孔内部まで入るものと、入らないものが生じ、トータル的には溶出時における幅が大きくなることになる。そのため洗浄時に除去したい成分だけを排除するのが難しく、最終的に溶出された成分は、実施例1に示す従来例のように低分子側のプライマーが残ってしまう。本発明のモノリス構造体に於いては、液の流れるマクロ細孔表面にミクロ細孔があるので、全て均一な入り込みが生じる。そのため、低分子不純物であるプライマーの除去が簡単に行える。
【0087】
従来法に於いて、大きなDNAに於いては、細穴へ入込み難くなり、更に、成分が含まれる液の粘性も上がるため、接触が不均一になり、2種現象が同時に起こることになり、吸着されない部分が多いと考えられる。又、乱流により、高分子DNAへの物理的なダメージが生じ、破壊してしまう可能性も高まる。
【0088】
基本的には、カオトロピック塩濃度を増やすことにより、それらの現象は軽減できるが、洗浄時でも取除かれず、溶出時に溶出してきてしまう。即ち、精製後の試料成分に多量の塩が入ることになる。これは、後の使用に於いては大きな問題となる。
【0089】
モノリス構造体を用いた本発明方法では、塩濃度を下げることができ、上記の諸問題は解決でき非常に有効である。更に、よりカチオン交換作用を持つカリウム塩を合せて使用するとより効果的になる。カリウム塩はカチオン交換作用が強いので、核酸の表面への吸着に寄与するが、それが故に、溶出時に基体表面に残ってしまうと目的とする精製DNAが溶出しないと云う問題が生じるため、確実な洗浄が不可決である。粒子タイプでは、洗浄時にも乱流が生じ、更に細孔への入込みが不均一のため、どうしてもカリウム塩が高濃度で基体に残存する部分が生じる。対策として、溶出液にカリウムを除けるバッファー、即ち他の塩を加えたりすればよいことになるが、やはり後のアプリケーション目的に適さないことになる。
モノリス構造体では、均一な液の流れと均一な細孔への入込みが可能となるため、カリウム塩が有効に作用する。
【実施例1】
【0090】
PCR反応液(フラグメントDNA)の精製では、PCR増幅反応物50μlをバッファーA1(1Mグアニジン塩酸塩、0.2M酢酸カリウム、50%2−プロパノール)300μlに混合する。シリカモノリス固相カラム9をコレクションチューブ8に挿入し、混合物をシリカモノリス固相カラム9に注入、1.5mlの遠心管内で遠心分離する。シリカモノリス固相カラム9をBバッファー(0.2M酢酸カリウム、50%エタノール)での洗浄処理により塩を含まないようにする。
【0091】
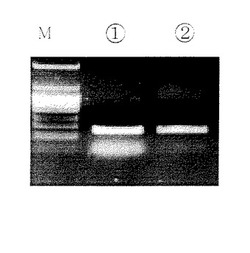
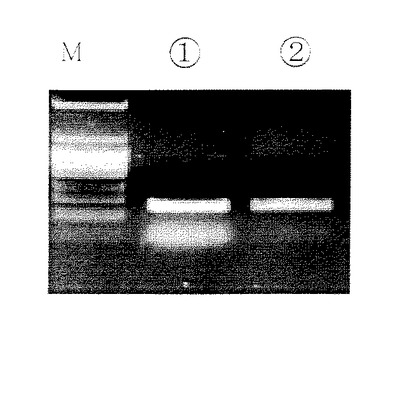
溶出には溶出用バッファーC(EDTA4mM、Tris−HCI10mM、pH8又は無菌DNA、RNA free水)20μlを、別の1.5mlの遠心管で遠心用カラムに通して遠心分離する。こうして精製されたPCR産物(フラグメントDNA)にはプライマー、dNTPs、ポリメラーゼ及び塩が含まれず、後の操作に直接用いることができる。(図1,図2参照)図1中M:分子量マーカー、(1)従来法で精製したサンプル、(2)本発明で精製したサンプル。
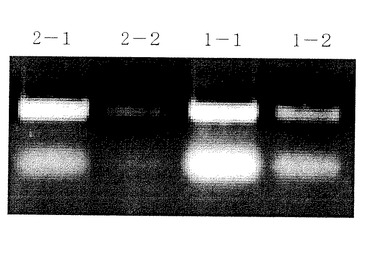
【0092】
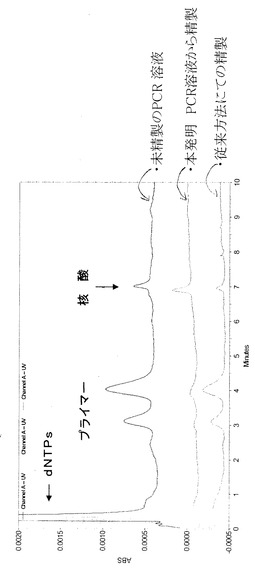
(1)が従来の特許文献3の方法で精製した試料(400bp)での電気泳動による評価である。(2)が本発明方法である。(1)では、低分子側(下)部分が多く残っているが、(2)では殆ど残っておらず、高い精製効率が得られている。又、図2に於いて1−1及び1−2は従来方法で2回行い、残存を見たもので、2−1,2−2は本発明のものである。本発明の場合1回目で殆ど残っておらず、高い精製効率が得られることが分かる。図3はHPLCを使用しての評価である。HPLC条件は以下の通り
HPLC条件
カラム:CIM DEAE
溶離液:
A:20mM Tris-HCl Ph7.4
B:A+1M NaCl
A/B=50/50-(10MIN)-0/100 Flow rate:3ml/min
検出:UV260nm
【0093】
図3に於いて未精製のPCR溶液のHPLC評価データは、1番上のクロマトグラムであり、従来の特許文献3法による精製のクロマトグラム(一番下)と比べてパターンに変化がなく、dNTPs及びプライマーは、殆ど除かれていないことがわかる。本発明方法では、2番目のクロマトのようにdNTPs及びプライマーの2つのピークが大幅に取除かれ、目的とする核酸が高く精製されていることがわかる。(図3参照)
【実施例2】
【0094】
アガロースゲルからのDNA断片の精製ではPCR増幅産物を標準的又は低融点アガロースゲル(TE又はTBEバッファー使用)を用いて電気泳動し、DNAをアガロースゲル(TE又はTBE0.5%)中で分離する。清潔なカミソリやメスで単離すべきDNA断片をゲルから切出して、1.5ml遠心チューブに入れる。バッファーA2(2Mグアニジンチオシアン酸、0.4M酢酸カリウム、30%の2−プロパノール)300μlに混合し、60℃で5分間又はゲルスライスが完全に溶解するまでインキュベートする。
この溶解液を実施例1に従って、モノリス固相カラム9を、コレクションチューブ8に挿入し、混合物をモノリス固相カラム9に注入、1.5mlの遠心管内で遠心分離する。モノリス固相カラム9をバッファーB(0.2M酢酸カリウム、50%エタノール)での処理により洗って塩を含まないようにする。
【0095】
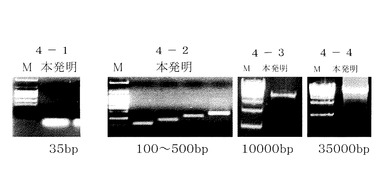
溶出にはバッファーC(EDTA1mM、Tris−HCI10mM、pH8又は無菌DNA、RNA free水)20μlを、別の1.5mlの遠心管で遠心用カラムに通して遠心分離する。図4のうち図4−1は35bpの、図4−2は100〜500bpの、図4−3は10,000bpの、図4−4は35,000bpの電気泳動による評価である。
【0096】
本発明方法では、低分子の35bpから約100Kbpまで回収されており、アガロースゲルからでも広い範囲のDNAが精製できることがわかる。溶出バッファーとして、EDTAバッファーを含まない水でも同様の結果が得られた。
【実施例3】
【0097】
制限酵素反応後のDNA100μgを制限酵素で処理する。このDNA制限反応溶液を実施例1に従ってバッファーA1,300μlに混合し、そしてそれ以後の処理は実施例1と同様に行った。溶出後、得られた精製DNAには制限酵素及び塩が含まれず、吸光度測定比率(mm)260/280の比率が1.8と良好なものであった。
【実施例4】
【0098】
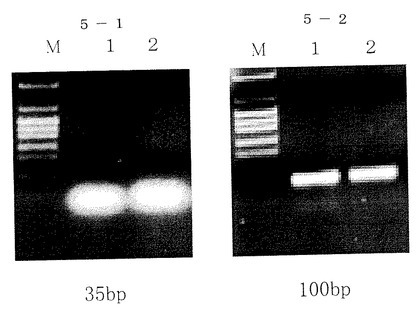
分子サイズの小さい、35bpのPCR増幅産物の精製ではPCR増幅反応物10μlを実施例1に従ってバッファーA1,100μlに混合し、それ以後の処理は実施例1と同様に行った。(図5参照)図中M:分子量マーカー、1:精製前のサンプル、2:本発明で精製したサンプル。
100bpや100bp以下の小さなDNAでもここでは35bpのDNAのPCR反応液からの精製が出来た。
【実施例5】
【0099】
分子サイズの大きな(100bp以上100,000bpまで)PCR増幅産物の精製ではPCR増幅反応溶液20μlを実施例1に従ってバッファーA1,200μlに混合し、それ以後の処理は実施例1と同様に行った。(図6参照)図中M:分子量マーカー、1:精製前のサンプル、2:本発明で精製したサンプル3:従来法で精製したサンプル。
1000bp〜100KbpまでのDNAでもPCR反応液からの精製が出来た。
【実施例6】
【0100】
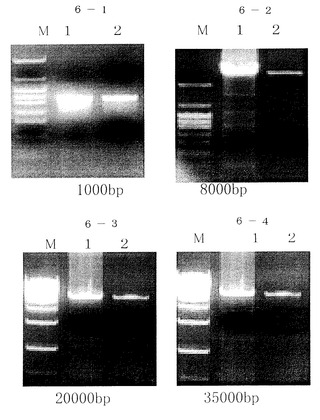
1本鎖DNA溶液の精製では溶液20μlを実施例1に従ってバッファーA1,200μlに混合し、それ以後の処理は実施例1と同様に行った。(図7参照)図中M:分子量マーカー、1:精製前のサンプル、2:本発明で精製したサンプル。
1本鎖DNA35merは、従来方法では回収できていない(3)が、本発明では(2)再現性よく2回とも回収されている。
【実施例7】
【0101】
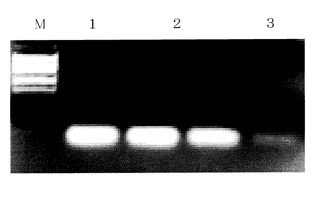
ナトリウムとカリウムによる精製比較(保持メカニズム)(図8参照)図中M:分子量マーカー、1:ガラスモノリス、カリウムによる精製後サンプル、2:シリカモノリス、カリウムによる精製後サンプル、3:シリカモノリス、ナトリウムによる精製後サンプル、
従来からよく用いられるナトリウムと、本発明の効果があるカリウムとの精製比較を行なった。ナトリウムでは、保持が殆ど得られず、カリウムでは高い精製効率が得られた。
又、シリカモノリスと同様にガラスモノリスでも同じくカリウムの方が高い生成効率が得られた。
【0102】
本発明を実効あらしめるために下記の方法を実施することは意義がある。
核酸を含有する溶液をアルカリ金属塩を介在させることにより、一体型モノリス構造体の通孔に夫々対応する核酸を吸着させ、洗浄液で洗浄後、溶出させることを特徴とするDNA分離精製方法。
アルカリ金属塩は酢酸カリウムであることを特徴とするDNAの分離精製方法。
酢酸カリウムは0.1〜1M未満含有する溶解吸着バッファーを使用することを特徴とする分離精製方法。
グアニジン塩又は、酢酸カリウムなどのカリウム塩を含有する溶解、吸着バッファーにより、溶解吸着を行うことを特徴とするDNAの分離精製方法。
Tris−HCl、EDTAを含有する水により溶出を行うことを特徴とするDNAの分離精製方法。
溶解、吸着、分離、洗浄操作を1つのモノリス固相カラムを用いて行なうことを特徴とするDNAの分離精製方法。
溶解吸着バッファー、水及び分離精製機構よりなるキット。
【産業上の利用可能性】
【0103】
以上のように本発明に係るDNAなどの分離生成機構は、分子生物学的研究に於いて、非常に頻繁に用いられ、P.C.R、クローニング、シークエンシンク、制限酵素消化、その他酵素作用などのアプリケーションに先立って行われるフラグメントDNAなどの精製に特に有用で35bp(mer)から100Kbp(mer)以下の広範なDNAの定量的な分離や効率的な精製ができ、幅広い核酸の精製に対応できる。
【技術分野】
【0001】
本発明は、DNAなどの分離精製機構に関するものである。
【背景技術】
【0002】
今まで、核酸含有物からの核酸の精製分離法は、核酸混合物をカオトロピックの塩の存在下でガラスやシリカゲルの粒子、ガラス、石英ウール、シリカ、ガラス膜、ポリマーなどに吸着させることはよく知られている。核酸含有物としては、培養細胞・組織、血液・血清・尿・糞便などの体液、バクテリア・ヒト結核菌などの細菌、HIV・B型肝炎・C型肝炎などのウイルスなどの生物学的原料物質が主なもので、プラスミドDNA、ゲノムDNA、染色体DNA、RNA、ミトコンドリアDNA、フラグメントDNAなどが分離精製可能である。
【0003】
フラグメントDNAの精製は、分子生物学的研究において、非常に頻繁に用いられる技術で、PCR、クローニング、シークエンシング、制限酵素消化、その他酵素的作用などのアプリケーションに先立って行われている。
例えば、組換M13ファージからDNAを単離する方法があり、ガラス繊維フィルター上でカオトロピック物質の添加により、M13ファージDNAを結合させ、次いで分離洗浄乾燥を経て溶離する単離方法がNucleic Acids Research Vol.15 5507-5516(1987)(非特許文献1)に示されている。
【0004】
又、ガラスパウダーを添加してDNAをガラスパウダーに結合させ、遠心分離して、ガラスパウダーを集め、洗浄、溶離し単離する方法がPvoc. Natl. Acad.Sci.USA Vol.76,615-619.(1979)(非特許文献2)に示されている。又、同様の方法は、特開昭59−227744号公報(特許文献1)、Analytical Biochemistry Vol.121.382-387.(1982)(非特許文献3)、Molecular cloning: A Laboratory Manual 188-190.(1982)(非特許文献4)等に記載されている。
【0005】
又、複合性生物出発材料、カオトロピック物質及びシリカ又はその誘導体を含む核酸結合性固相を混合し、 核酸を結合した固相を液体から分離し、洗浄し、核酸を溶離する方法が特許第2680462号公報に提案されている。
カオトロピック試薬の存在下でDNAやRNAを吸着させる物理的メカニズムについては詳しくは明らかになっていないが、負に帯電した担体と、核酸との間にカチオン交換反応が起こると考えられている。従って精製の効率は、担体表面と生体試料の接触の効率とイコールと考えることができる。
【0006】
前記のどの担体を使用するにしろ、吸着させる担体を容器(カートリッジ、チップなど)に保持し、その容器に生体試料を通液し、吸着バッファー液で担体に核酸を吸着させ、その後洗浄液で核酸成分以外の夾雑物をカートリッジ外に追い出し、更にその後、溶出液を通液して核酸成分をその液と共に取り出す手順が一般的である。
【0007】
その他に、電気泳動やさまざまな抽出によってフラグメントDNAをアガロースゲルから精製することもよく行われるが、この方法は時間がかかり、得られたDNAも極めて希薄となり、塩や有機溶媒が含まれているため、更にエタノール沈殿で脱塩や濃縮する必要が生じるものである。又、ゲル濾過精製テクノロジーのような従来法では、分子量の類似した分子同士を分離することは非常に困難である。
【0008】
担体を使用するこれらの分離方法は、高濃度の塩類を使う必要があるため、DNAのガラスまたはシリカゲルの粒子表面での分解若しくは変性をもたらす現象が確認されている。高濃度のカオトロピック塩の存在下では、この吸着は略定量的に生じるが、吸着したDNAの溶出は、塩類の緩衝液の存在下で行われる。DNAの断片の処理は100塩基対(bp)〜10,000塩基対(bp)の範囲が限度で、100塩基対(bp)以下のDNA断片や10,000塩基対(bp)以上の100,000塩基対(bp)までのDNAの定量的な分離や精製をすることは不可能であった。
【0009】
担体として、ガラス粉末をベースにした調整方法では、その表面と核酸との接触効率を上げるために、ビーズを小さくする、量を増やすと云うことが考えられる。然し、通液時の圧力が上がってしまい、操作性が著しく落ちると共に、ビーズ間空隙が小さくなり、核酸分子は分子量が大きいため、その空隙に入り込めず、かえって効率が落ちると云う問題が生じる。又、分離の向上を目的に容器の長さを長くすると、圧力が上がることに加えて溶出溶媒の量も増え、濃縮効率が落ちることになり、簡便な処理からは遠ざかってしまう。更に、ビーズやウールなどを使用すると微量ながらそのかけらや粒が溶出液に入り込んでしまうので、後のアプリケーションに問題となる。又、容器に充填する際の充填法が一定しないと分離時間やパターンが変わってしまうので、分離の安定性が悪いと云う問題も生じる。
【0010】
担体として膜やフィルターを使用する方法は、使い勝手よく加工できるメリットがあるが、分離に適当な孔を制御して作成することが難しく、実用性に乏しい。
又、ポリマーを使用する方法は、そのポリマーの性質によって核酸と特異的に反応する働きが充分でなかったり、又その部分以外に影響を与える部分が存在したりと分離系が複雑になり、単純なプロトコルでの精製は不可能である。
何れも高純度なフラグメントDNAを精製することは不可能である。又、粉末シリカ樹脂や懸濁液が持つ取扱い難さや、その後のアプリケーション反応を妨げる虞がある等の欠点がある。
【0011】
特許文献2の発明に於ける如く、複雑な出発材料から、前処理なしで核酸を直接単離する考え方は、所謂「消化」と「精製」を同時に行うものである。そのために過激な反応条件が必要となり、且つ適用できる核酸の分子量範囲が狭くなると云う弊害が生じる。
核酸混合物を、高濃度の塩(イオン強度)を含み、且つ脂肪族アルコールやポリエチレングリコール等の有機酸を含む吸着水溶液からシリカゲル、ガラス等の多孔質、非多孔質の無機基体上に吸着させ、洗浄させ、次いでより低濃度の塩(イオン強度)を含む溶液で溶出して核酸を得る方法が特表平8−5011321号公報(特許文献2)に提案されている。
【0012】
然し、この方法に於いては、高濃度の塩を含む吸着水溶液から、核酸混合物を無機体上に吸着させること及び低濃度ではあるが、塩を含む溶液で核酸を溶出するため、例えばDNAサンプルが大きなアガロース片に含まれる場合には、複数のカラムを使っての処理が必要であり、溶出したフラグメントDNAをプールし、更に得られたフラグメントDNAに塩や有機溶媒が含まれるので、濃縮や脱塩等の操作工程が必要であり、その上エタノール沈殿中に精製DNAを損失する虞もある。
【先行技術文献】
【特許文献】
【0013】
【特許文献1】特開昭59−227744号公報
【特許文献2】特表平8−5011321号公報
【非特許文献】
【0014】
【非特許文献1】Nucleic Acids Research Vol.15 5507-5516(1987)
【非特許文献2】Pvoc. Natl. Acad.Sci.USA Vol.76,615-619.(1979)
【非特許文献3】Analytical Biochemistry Vol.121.382-387.(1982)
【非特許文献4】Molecular cloning: A Laboratory Manual 188-190.(1982)
【発明の概要】
【発明が解決しようとする課題】
【0015】
そこで本発明に於いては、先行技術の上述した点を改良し、吸着や溶出分離が極めて簡単且つ容易に行えると共に、高濃度の塩による溶出を行わず、溶出精製の必要がなく、核酸就中フラグメントDNAなどの分離精製が著しく効率化された再現性の高い方法を提案せんとするもので、
【課題を解決するための手段】
【0016】
一体型モリノス構造体であって、一端から他端まで連続した通孔を形成させ、かつ核酸大きさが35bp(mer)から100Kbp(mer)に対応する通孔が設けられ、分離すべき核酸を含有する溶液を通過させることにより、該通孔に対応する核酸が夫々保持できるように構成したことを特徴とし、モノリス構造体は、ガラス、シリカ等の無機物又は無機物に有機物を含有するハイブリッド体であって、上面から下面まで貫通しているマクロ細孔を持つ多孔質体を使用することを特徴とし、モノリス構造体の多孔質体はマクロ細孔の内部にミクロ細孔を有することを特徴とし、モノリス構造体の多孔質体はマクロ細孔1〜100μm、ミクロ細孔0〜100nmであることを特徴とし、カラムチューブにモノリス構造体により形成されるディスクを配置することにより、モノリス固相カラムを構成することを特徴とし、上下を開放した筒状体に、モノリス構造体により構成した基体を着脱自在に装着して形成したモノリス固相カラムを使用することを特徴とする。
【発明の効果】
【0017】
本発明によれば、100bp(mer)以下のDNA断片や10000bp(mer)以上の100Kbp(mer)までの広範囲のDNA断片の定量的な分離や効率的な精製が可能である。又、過敏な反応条件を使用しないで核酸の吸着分離が可能となり、高濃度の塩による溶解、吸着が必要なく、濃縮や脱塩の操作が必要なく、核酸精製就中DNAフラグメントの精製が容易である。又、塩を含まない溶液又は無菌水で溶出でき、高純度フラグメントDNAなどが容易に得られる。
【図面の簡単な説明】
【0018】
【図1】本発明による実施例1の従来法との対比図
【図2】本発明による実施例1の従来法との対比図
【図3】本発明実施例1のHPLCによる評価図
【図4】本発明による実施例2のDNAフラグメント精製分離図
【図5】本発明による実施例4のDNAフラグメント精製分離図
【図6】本発明による実施例5のDNAフラグメント精製分離図
【図7】本発明による実施例6の1本鎖PCR増幅産物分離図
【図8】本発明による実施例7のナトリウムとカリウムによる精製比較図
【図9】本発明一実施例カラムチューブ説明斜面図
【図10】同上ディスク説明斜面図
【図11】同上コレクションチューブ説明斜面図
【図12】同上モノリス固相カラム説明斜面図
【発明を実施するための形態】
【0019】
本発明者らは、効率のよいモノリス構造体を核酸精製に使用し、バッファー条件を整えると、これまで常識とされていた塩濃度の高いバッファーを使用しなくてもよいことを見出した。核酸保存のために加えるトリス塩酸EDTAのほかには水だけで、吸着していた担体から結合が外れて溶出する。核酸を吸着させる際にも、分離溶媒として働くイソプロパノールとシラノール基と反応すると思われるイオンを供給する塩化合物、そしてアガロースゲルを溶解させるためのカオトロピック塩であるグアニジン塩酸塩などを吸着溶媒とする。このとき陽イオンになりやすい、アルカリ金属塩が存在すると、容易に脱水素反応を起こし、それによりその陽イオンがカチオン架橋反応を核酸のリン酸部分と起こし、核酸を吸着すると考えられる。アルカリ金属は、例えばリチウム、ナトリウム、カリウム、ルビジウム、セシウム、フランシウムなどが該当する。中でも電気陰性度が小さく、容易に陽イオンとなりカチオン架橋反応をするカリウム塩は反応が起こりやすく、最後に、核酸成分と一緒にイオン状態で溶出されるが、後のアプリケーションの妨害をしないので有用である。尚、ナトリウム塩はその後のアプリケーションの妨害となるので、脱塩操作を行わないと使用できずあまり有用ではない。換言すれば、この装置及び方法であれば、脱塩操作やアルコール沈殿などが必要なく、得られた精製液をそのまま後の操作(PCR、クローニング、シークエンシング、酵素的操作)に持っていけることになる。操作手順が簡便化することは、核酸の損傷を防ぐために非常に重要な事項である。
【0020】
本発明の目的は、精製効率のよいモノリス構造体のシリカやガラスを使用し、より簡単な緩衝液を使用することで、その後のクローニングなどの各種アプリケーション操作に影響を与えず、核酸を精製することができる方法を提供することにある。
【0021】
モノリス構造体とは、上端から下端まで連通した開放構造のマクロ細孔を持つ、一体型多孔質体のことを指し、そのマクロ細孔に更にミクロ細孔を持つものが多く合成されている。
モノリス構造体は、主に、ゾル−ゲル法で作成することができる。即ち、金属アルコキシドを部分的に加水分解して反応性モノマーを作り、このモノマーを重縮合してコロイド状オリゴマーを作り(ゾルの生成)、更に加水分解して重合と架橋を促進させ、三次元構造を作る(ゲルの生成)ことで合成される。
【0022】
この反応時に種々の有機モノマーを添加すると、有機・無機ハイブリッドモノリスも簡単に作成できる。従って、添加する有機モノマーの種類によって、化学的特性を加えることも可能となる。例えば、親水基を持つ有機モノマーを添加することによって、水系試料成分の吸水性を向上することができる。又、選択的な化学作用を示す官能基を持つ有機モノマーを添加することによって、精製における不純物となる特性成分の吸着に利用し、その不純物を固相に残すことで、核酸の精製効率をアップすることができるようになる。又、弾性率の高いポリマーをゾル−ゲル工程中に入れることにより、モノリス構造体に弾力性を持たせることも出来る。更に基本的には、有機・無機を混在させることによって、モノリス構造体の化学的安定を上げることも可能となる。
【0023】
これらのことは、ゾル−ゲル法にて作成される有機・無機ハイブリッドモノリス構造体は、添加有機モノマーの種類で目的に応じた化学表面を構成したり、化学的安定性を向上したりと云う性質を付加できることを意味し、DNA前処理に於いて有効なモノリス特性を目的に応じて自由に改善できることを示している。更に、ゾル−ゲル方法から作成したモノリスは、本発明のDNA用固相として金属含有が少ないと云う点で適している。一般的なシリカゲルなどは、ケイ酸ナトリウムなどから作成され、多量の金属が残る。確かに、一部では精製したケイ酸ナトリウムからの作成やゾル−ゲル法からの高純度のシリカゲルもあるが、それらは高価であり、通常使用態様である使い捨てには適さない。又、安価に出来たとしても、粒子作成時点ではバッチ式合成であり、合成雰囲気から金属濃縮が生じる可能性が大きい。
【0024】
ゾル−ゲル法におけるモノリス構造の合成に於いては、連続行程により作成され、金属のコンタミネーションは全くない。従来、シリカゲルでは、塩酸や硝酸等で洗浄するなどの工夫や、金属影響を減らすようなEDTAなどの添加があったが、本発明固相では全く必要ない。
【0025】
もう一法として、ガラス分相によってもモノリス構造固相を作成できる。基本的には、ゾルーゲル法からのモノリス構造の合成と同様の有効性があるが、ゾルーゲルモノリスよりもマクロ細孔を大きく作る事ができるので、2次ミクロ細孔を内部表面に作る場合に有効である。更に、ガラス分相は、その組成より耐アルカリ性が高く、アルカリ洗浄による再生が出来ると言うメリットがある。
【0026】
以下、図に示す実施例により、本発明を詳細に説明する。
本発明に於いて最も基本的な構成は、フラグメントDNAをアガロースゲル、PCR反応物、制限酵素処理DNA、1本鎖DNA及びRNA等から精製するために、カオトロピック塩の存在下、ガラスやシリカに吸着させること、殊に優れた分離能力を有するガラスやシリカにより形成されるモノリス構造体、即ち、細孔が上端から下端まで連通した開放構造を有する一体型多孔質体を使用し、これに吸着させることにある。
フラグメントDNAをカオトロピック塩の存在下ガラスやシリカに吸着させることは、前記の如く既に提案され実施されている。
【0027】
然し、従来の方法は全て充填材を使用していた。このため、充填材の充填にばらつきがあり、均一にならないこと、また充填材通過の後に粒がDNA液に残留してしまうこと、液との接触面積が小さく反応効率が悪いこと、通液圧力が大きく扱いにくいこと等の欠点が残るものであった。
【0028】
本発明は、カオトロピック塩の存在下に於いて、アガロースゲル、PCR反応液からガラスやシリカゲル粒子に吸着したものを抽出する点に於いて、従来の技術とある部分共通するところがあるものである。
この従来の技術は、高濃度のカオトロピック塩の存在下で吸着を定量的に生じさせ、吸着した核酸の溶出はより低い塩濃度で行なうものであった。
【0029】
然も、特表平8−5011321号公報の方法は、分離すべき核酸を予備的精製行程なしに1つの操作行程で、核酸分画を行う方法である。このために高濃度の塩のバッファーによる過激な反応条件が必要となり、適用できる核酸の分子量範囲が狭くなる。更に精製DNAは極めて希薄で、塩や有機溶媒があるため、更にエタノール沈殿や濃縮する必要がある。
【0030】
本発明はこれらの欠点を改良するもので、高濃度の塩での吸着や溶出は行わず、水で溶出するものである。この結果、非常に高濃度で更なる精製の必要のないサンプルが得られる。
本発明はアガロースゲルからのDNA断片とPCR増幅反応DNA液、酵素反応液からのフラグメントDNAなどの精製を1つの目的とするものである。
【0031】
本発明は、Tris酢酸(TAE)バッファー又はTrisホウ酸(TBE)バッファーで作成した標準アガロースゲルや低沸点アガロースゲルからの35bp〜100KbpのDNA断片の抽出、精製が可能で60〜80%の回収率を得ることが出来る。又ゲルからの抽出のほかにもPCR増幅反応液からも35bp〜100kbpのPCR産物を直接精製することができ、80〜95%の回収率を得ることが出来る。得られたフラグメントDNAに塩や有機溶媒が含まないため、エタノール沈殿、脱塩や濃縮する必要がない。本製品はモノリスベースのシステムであり、DNA結合能は最大5〜8μgで、単離したフラグメントDNAを僅か5分で回収することができる。
【0032】
ゲルから精製する場合は、電気泳動後にゲルから目的のDNAバンドを切出し、グアニジンチオシアン酸(溶解、吸着バッフアー)の存在下で溶解する。PCR増幅後の精製の場合は、吸着バッファーを増幅反応液に直接添加する。溶解したゲル片は、マイクロ遠心機又は吸引装置を用いてモノリス固相カラムを通過させる。その際、目的のフラグメントDNAはシリカモノリスやガラスモノリスの表面に結合し、結合したフラグメントDNA断片を洗浄バッファーで洗浄後、DNAを水で溶出する(溶出バッファー)。
【0033】
このバッファーとして下記が使用される。
A バッフアー(溶解、吸着バッファー)
B バッフアー(洗浄バッファー)
C バッフアー(溶出バッファー又無菌水)
これを更に詳述すると
A1 バッフアー PCR反応液用(グアニジン塩酸塩1〜8M、酢酸カリウム0.1〜1M未満、2−プロパノール1〜70%)
A2 バッフアー アガロースゲル用(グアニジンチオシアン酸1〜8M、酢酸カリウム0.1〜1M未満、2−プロパノール1〜70%)
B バッフアー(酢酸カリウム0.1〜1M未満、エタノール1〜80%)
C バッフアー(pH=8〜8.5 Tris−HCI10mM、EDTA1mM、又は無菌DNA、RNA free水)
【0034】
A1バッファーに於いて、グアニジン塩酸塩はグアニジンチオシアン酸が好適で、1〜8Mがより好ましい。2−プロパノールは40%以上が効率的である。
A2バッファーに於いても以上の条件が当て嵌まる。
溶出バッファー液Cに於いては、水による溶出が可能であるが、雑菌の混入を防ぐために、限外濾過膜処理やディエチルピロカーネートで処理などを行ったRNaseフリーな水を使用する事が推奨される。又、精製したDNAを保管する場合、菌の混入を防ぐため、溶出バッファー液Cとして、先にEDTAバッファーを添加した水を使用してもよい。分離機構から見てもEDTAバッファーの有無で精製効率が変わることは無い。
【0035】
本願発明に於いて、基体にモノリス構造体を使用することにより、核酸のリン酸基とシラノール基との反応を効率的に進行させることが出来る。又、粒状の充填材を使用する場合に避けられないサンプルにシリカ粒子が付着したまま精製され、所謂アプリケーション反応を妨げる虞のあるシリカのキャリーオーバーの問題を防ぐことができる。更に、分離時における圧力も低く抑えられ、又充填材を使用する場合と異なり、封止剤も不要である等の利点を有する。
【0036】
このモノリス構造体は、例えば、シリカゲル等の無機質多孔質体を、重合可能な低分子化合物ゾルを精製し、最終的に凝集体や重合体のゲルを得る、所謂ゾルゲル法によって作製することが出来る。この方法は、一般的に中心細孔径1〜100μmであるが、その後の技術進歩により数nmも可能である。
【0037】
又、ゾル−ゲル過程におけるスピノールダル分解を利用し、2種類の細孔を有するシリカ骨格中に細孔の存在するモノリス構造体も作成できる。
又、上面から下面まで貫通しているマクロ細孔の内部に開放構造を有するミクロ細孔を持つ多孔質体が充填された構造の多孔質体も作成できる。因みに、この多孔質体はマクロ細孔の直径1〜100μmであり、ミクロ細孔が0〜100nmである。
【0038】
この他、ガラス、シリカが含有されて形成されるものであれば、適宜のモノリス構造体が使用しうること勿論である。
このモノリス構造体は、その製法により、多孔質体の貫通孔は所望の通り形成できるので、適宜選択して形成するが、1〜100μm程度が使用しやすい。然し、この選択は、使用する原料、例えばアガロースゲルやPCR反応液、バッファーにより、又その目的により決められる。
【0039】
このモノリス構造体により構成される基体の一使用形態について述べると、筒型のカラムチューブ1を形成し、上端開放部2には密閉可能な蓋3を着脱自在に設ける。下端には細口径の出口4を構成し、出口4上部には段差5を設けて中口径部6を形成してある。該段差5にはモノリス構造体により形成された基体としてのディスク7が載置乃至嵌合できるようにしてある。該ディスク7は中口径部6と略同形で所望厚さ例えば0.1〜10mm程度の円盤状或は所望に応じ、円錐状を形成してもよい。このカラムチューブ1とディスク7により、モノリス固相カラム9を構成する。8はコレクションチューブで、カラムチューブ1の挿通が可能の径に構成され、上部にカラムチューブ1の上端縁が係止可能に構成してある。
【0040】
前記カラムチューブ1やコレクションチューブ8については、ポリプロピレン製が使用されるが、核酸に影響を与えない有機ポリマー、例えばポリエチレン、ポリエチレンテレフタレート、ポリスチレンや無機化合物、例えばガラス、シリカ等視認性がよくある程度強度があれば使用できる。
又、モノリス構造体の基体としてディスク7が挙げられているが、これに限らず皿状や筒状等溶液の通過の自在である型で使用できる。
【0041】
DNAの分子サイズは、10塩基対あたり、約3.4nm程度と云われている。例えば、35bp〜300bpのDNAに於いて、10nm〜100n程度のマクロ細孔、300bp〜3KbpのDNAに於いて、100nm〜1μm程度のマクロ細孔、30Kbp300KbpのDNAに於いては、1μm〜10μm程度のマクロ細孔、30Kbp300〜KbpのDNAに於いては、10μm〜100μm程度のマクロ細孔を持てば、DNA分子を傷つけたり、壊したりすることなく、DNA分子を通過させることが可能と考えられる。
【0042】
又、不純物などとの相互作用を高め、より効率的に精製するためには、ミクロ細孔の付加が行われる。各種試験、実験などの経験から、ミクロ細孔が数10nm前後であれば分子量数10万の化合物、10nm前後であれば分子量数百〜数万の化合物と相互作用があることが確認されており、更に大きな分子量の化合物ならば、その分子の破壊を助長すると云われるミクロ細穴が限りなくない状態、つまり0nmがよいことが判っている。
【0043】
マクロ細孔にミクロ細孔を付加した種々の一体型モノリス構造体を数種用意し、DNAの種類及び不純物除去などの目的ごとに使い分けることも可能である。ちなみに、数種類の一体型モノリス構造体を作成するには、マクロ細孔を予め作成し、その後にミクロ細孔を形成させる方法の方が合成上便利である。
【0044】
現状必要とされている精製に於いては、プライマーやアガロースゲル分解物などの低分子からの分離精製が中心であり、1種類のモノリス構造体でも充分分離可能である。
1〜100μm、好ましくは20μm程度のマクロ細孔と、0〜100nm、好ましくは10nm程度のミクロ細孔を形成したモノリス構造体を用いれば、実施例にあるような、35bp(mer)〜100Kbp(mer)の広範囲のDNAの精製が充分可能である。
【0045】
A.モノリス固相カラムを用いてPCR産物を生成するのには次の方法がある。
(1)マイクロ遠心法
このプロトコールは、PCR反応液から2本鎖DNAフラグメントを精製する目的でデザインされ、1つのモノリス固相カラムと精製用バッファーを用いれば、遠心操作により、35bp〜100Kbpのフラグメントがプライマー、ヌクレオチド、ポリメラーゼ、塩等から分離可能である。
【0046】
PCR反応液10μlに対して、100μlのバッファーA1(吸着バッファー)を添加する。バッファーBは約300μl(洗浄バッファー)で洗浄する。
全ての遠心操作は一般的な卓上マイクロ遠心機で〜10,000rpmで行う。
【0047】
1.PCR反応液に10倍容量のバッファーA1を加えて混合する。ミネラルオイルを除去する必要はない。例えば、50μlのPCR反応溶液(オイルを含まない量)には500μlバッファーA1を加える。
【0048】
2.コレクションチューブ8にモノリス固相カラム9を挿入し、調整したサンプルをモノリス固相カラムにアプライする。高回収率を得るため、サンプル液を残さないでモノリス固相カラム9に添加する。
【0049】
3.モノリス固相カラム9を10,000rpm、30秒間遠心した後、モノリス固相カラム9を取外し、コレクションチューブ8内の液体を除去。モノリス固相カラム9を、コレクションチューブに再度挿入する。
【0050】
4.バッファーB(洗浄バッファー)500μlを添加し、モノリス固相カラム9を10,000rpm、30秒間遠心。更に10,000rpm、1分間遠心する。
コレクションチューブ8内の液体を捨てた後に、バッファー由来の残留エタノールを完全に除去するためには再遠心操作を行うことが必要である。
【0051】
5.モノリス固相カラム9を新しい1.5ml遠心サンプリングチューブに移し、バッフアーC(溶出バッファー)10〜50μlをモノリス表面の中央に添加し、モノリス固相カラム9を1分間室温でインキュベートした後、10,000rpm、1分間遠心する。遠心チューブ内に溶出されたDNAは精製されたDNAで、−20℃で保存する。又は後の操作にそのまま使用する。
モノリスに結合したDNAが完全に溶出されるように、モノリス表面中央部分に溶出バッファーCを添加する。10μlの溶出バッファーCを用いた場合は溶出液量は9μlである。
溶出効率はpHが8〜8.5の間で最大となる。溶出に滅菌水系を用いる場合には、pHがこの範囲であることを確認するのがよい。
【0052】
(2)吸引マニホールド法
モノリス固相カラムは、一般的なルアーアダプターを含む吸引マニホールドにより操作を行うことが出来る。このプロトコールは、PCR反応液から2本鎖DNAフラグメントを精製する目的でデザインされている。1つのモノリス固相カラムと精製用バッファーを用いれば、吸引装置によるサンプル処理操作により、35bp〜100Kbpのフラグメントがプライマー、ヌクレオチド、ポリメラーゼ、塩等から分離可能である。
【0053】
PCR反応液10μlに対して、100μlのバッファーA1(吸着バッファー)を添加する。バッファーBは約300μl(洗浄バッファー)で洗浄する。
一定で安定した吸引が行われるよう、各操作ステップ毎に一端吸引スイッチを切る。
【0054】
1.PCR反応液に10倍量のバッフアーA1を加えて混合する。ミネラルオイルを除去する必要はない。例えば、50μlのPCR反応溶液(オイルを含まない量)には500μlバッファーA1を加える。
【0055】
2.吸引マニホールドとモノリス固相カラムを準備する。
ルアーアダプター吸引マニホールドを吸引装置に接続する。
【0056】
3.吸引マニホールド上のポートに取付けたバキュームアダプターを装着。モノリス固相カラム9をバキュームアダプターに挿入する。
【0057】
4.調整したPCRサンプルをピペットによりモノリス固相カラム9にアプライし、DNAを結合させるために吸引する。溶液がモノリス固相カラム9を完全に通過するまで吸引する。サンプルがカラムを通過した後、吸引を止める。
高回収率を得るため、サンプル液を残さないでモノリス固相カラム9に添加する。添加最大容量は800μlで、800μlよりサンプル量が多い場合には、数回に分けて添加する。
【0058】
5.バッファーB(洗浄バッファー)500μlをモノリス固相カラム9に添加し、液体がモノリス固相カラムを通過するまで吸引する。
【0059】
6.モノリス固相カラム9をマニホールドから外し、コレクションチューブ8に移す。10,000rpmで1分間遠心する。
バッファー由来の残留エタノールを完全に除去するために遠心操作が必要である。(液体がモノリス固相カラムを通過するまで吸引し、乾燥させる。モノリス固相カラム9に残っている洗浄バッファーを完全に除去するための必要手段である。)
【0060】
7.モノリス固相カラム9を新しい1.5ml遠心サンプリングチューブに移し、バッファーC(溶出バッファー)10〜50μlをモノリス表面の中央に添加し、カラムを1分間室温でインキュベータした後、10,000rpm、1分間遠心する。遠心チューブ内の溶出されたDNAは精製されたDNAで、−20℃で保存する。又は後の操作にそのまま使用する。
モノリスに結合したDNAが完全に溶出されるように、モノリス表面中央部分に溶出バッファーCを添加する。10μlの溶出バッファーCを用いた場合は溶出液量は9μlである。
溶出効率はpHが8〜8.5の間で最大となる。溶出に滅菌水系を用いる場合には、pHがこの範囲であることを確認するのがよい。
【0061】
B.モノリス固相カラム9を用いてアガロースゲルを精製するのには次の方法がある。
(1)マイクロ遠心法
このプロトコールは、標準的又は低温融解アガロースゲル(TE又はTEBバッファー使用)から、DNAフラグメントを精製する目的でデザインされ、1つのモノリスカラムと精製用バッファーを用いれば、遠心操作により、35bp〜100Kbpのフラグメントがプライマー、ヌクレオチド、ポリメラーゼ、塩等から分離可能である。1個のモノリスカラムにつき、最大1000mgのアガロースの処理が可能である。
アガロースゲル10mgに対し10μlのバッファーA(溶解、吸着バッファー)を添加する。バッファーBは約500μl(洗浄バッファー)で洗浄する。
全ての遠心操作は一般的な卓上マイクロ遠心機で〜10,000rpmで行う。
【0062】
1.清潔なカミソリやメスで目的のバンドを切取り、1.5ml遠心チューブに入れる。余分なゲルを取除いて、ゲルスライスのサイズを最小になるようにする。
【0063】
2.バッファーA2(溶解、吸着バッファー)をゲルスライス100mgに対し100μl添加する。
100mgのゲルには、バッファーA2を100μl添加するが、濃度が2%以上のアガロースゲルを用いる場合は、バッファーBを600μl添加する。1個のモノリスカラムで処理できるゲル量は1,000mgであるので、ゲル量が1,000mgを超える場合は2個以上のモノリスカラムを使用する。
【0064】
3.60℃で5分間又はゲルスライスが完全に溶解するまでインキュベーションする。インキュベーション中、2回チューブをボルテックスにかけて溶液を混合する。アガロースを完全に溶解させる。2%以上のゲルを用いる場合は、インキュベーション時間を長くすると回収率がアップする。
【0065】
以下の操作は、前記A.モノリス固相カラムを用いたPCR反応液の精製(1)マイクロ遠心法と同じであるから省略する。
(2)吸引マニホールド法
モノリスカラムは、ルアーアダプターを含む吸引マニホールドにより操作を行うことが出来る。このプロトコールは、標準的又は低温融解アガロースゲル(TE又はTBEバッファー使用)から、DNAフラグメントを精製する目的でデザインされた。1つのモノリスカラムと精製用バッファーを用いれば、吸引装置によるサンプル処理操作により、35bp〜100Kbpのフラグメントがプライマー、ヌクレオチド、ポリメラーゼ、塩等から分離可能である。
【0066】
アガロースゲル10mg対し10μlのバッファーA2(溶解、吸着バッファー)を添加する。バッファーBは約500μl(洗浄バッファー)で洗浄する。
溶出は遠心操作により卓上マイクロ遠心機で〜10,000rpmで行う。
一定で安定した吸引が行われるよう、各操作ステップ毎に一端吸引スイッチを切る。
【0067】
1.清潔なカミソリやメスで目的のバンドを切取り、1.5ml遠心チューブに入れる。余分なゲルを取除いて、ゲルスライスのサイズを最小になるようにする。
【0068】
2.バッファーA2(溶解、吸着バッファー)をゲルスライス100mgに対し100μl添加する。
100mgのゲルには、バッファーA2を100μl添加するが、濃度が2%以上のアガロースゲルを用いる場合は、バッファーA1を600μl添加する。1個のモノリスカラムで処理できるゲル量は600mgであるので、ゲル量が600mgを超える場合は2個以上のモノリスカラムを使用する。
【0069】
3.60℃で5分間又はゲルスライスが完全に溶解するまでインキュベーションする。インキュベーション中、2回チューブをボルテックスにかけて溶液を混合する。アガロースを完全に溶解させる。2%以上のゲルを用いる場合は、インキュベーション時間を長くすると回収率がアップする。
【0070】
以下の操作は、前記A.モノリス固相カラムを用いたPCR反応液の精製(2)吸引マニホールド法と同じであるから省略する。
C.モノリス固相カラムを用いて酵素反応液を精製するのには次の方法がある。
(1)マイクロ遠心法
このプロトコールは、制限酵素分解や標識反応などの酵素反応液から2本鎖DNAフラグメントを精製する目的でデザインされた。1つのモノリスカラムと精製用バッファーを用いれば、遠心操作により、35bp〜100Kbpのフラグメントが酵素、プライマー、ヌクレオチド、塩等から分離可能である。
【0071】
酵素反応液10μlに対し、30μlのバッファーA1(吸着バッファー)を添加する。バッファーBは約300μl(洗浄バッファー)で洗浄する。
全ての遠心操作は一般的な卓上マイクロ遠心機で〜10,000rpmで行う。
【0072】
1.酵素反応液に3倍容量のバッファーA1を加えて混合する。モノリスカラムで処理できる酵素反応液の最大容量は100μlである。
例えば、100μlの酵素反応液には300μlバッファーA1を加える。
【0073】
以下の操作は、前記A.モノリス固相カラムを用いたPCR反応液の精製(1)マイクロ遠心法と同じであるから省略する。
(2)吸引マニホールド法
モノリスカラムは、ルアーアダプターを含む吸引マニホールドにより操作を行うことが出来る。このプロトコールは、制限酵素分解や標識反応などの酵素反応液から2本鎖DNAフラグメントを精製する目的でデザインされた。1つのモノリスカラムと精製用バッファーを用いれば、吸引装置によるサンプル処理装置により35bp〜35Kbpのフラグメントが酵素、プライマー、ヌクレオチド、塩等から分離可能である。
【0074】
例えば、酵素反応液10μlに対し30μlのバッファーA1(吸着バッファー)を添加する。バッファーBは約300μl(洗浄バッファー)で洗浄する。
全ての遠心操作は一般的な卓上マイクロ遠心機で〜10,000rpmで行う。
一定で安定した吸引が行われるよう、各操作ステップ毎に一端吸引スイッチを切る。
【0075】
1.酵素反応液に3倍容量のバッファーA1を加えて混合する。モノリスカラムで処理できる酵素反応液は最大容量は100μlである。
例えば、100μlの酵素反応液には300μlバッファーA1を加える。
【0076】
以下の操作は、前記A.モノリス固相カラムを用いたPCR反応液の精製(2)吸引マニホールド法と同じであるから省略する。
この本発明の吸着や溶出分離が極めて容易に行え、高濃度の塩による溶出は必要なく、核酸の精製が極めて効率的に行える利点は、核酸成分をモノリス構造体に吸着させる点に基因する。
【0077】
従来タイプの方法では、シリカゲル粒子、ガラス粒子、それらをフィルター状にしたものが使用されている。それら全てに於いて、液が通る空間は、粒子表面を通ることになり、粒子にぶつかり、乱流が生じ、不均一な流れとなってしまう。そのため、全て表面に均一に触れることはできない。モノリス構造は、一体構造で内部に連続孔があるので、粒子内部を通るイメージとなる。即ち、全ての液が均一に接触する。又、粒子に比べると、骨格が小さく、液がぶつかった後の乱流の生ずることなく均一な流れとなる。
【0078】
即ち、従来の固相タイプでは、粒子における乱流が生じ、表面との接触が不均一となり、低分子側DNAの吸着が生ぜずに抜けてしまうことになる。
その抜けを防止させるために、従来のタイプでは、カオトロピック塩濃度を増やすことにより、反応を起こし易くしている。然し、この場合塩沈殿が生じ、限界が生じるため、低分子DNA捕集には限界が生じる。
【0079】
本発明モノリスタイプでは、均一な液の流れが保障されており、より低分子DNAの吸着が可能となる。
当然、洗浄工程でも同じことが起きる。従来の方法では洗浄液の乱流が生じてゲル表面を洗浄することが困難となる。実施例1に記載されているように、1回目洗浄後でも従来の方法では目的としないプライマーなどがかなり残っているが、本発明の方法では殆ど残らない。
繊維に粒子を埋め込んだフィルターや繊維そのものを用いたものでも乱流が生ずるのはやはり同じことになる。
【0080】
従来例として、入口と出口を備えた円筒状の中空体の出口付近に無機基体材料が配置されており、その無機基体材料は、きつく押込められたポリエチレンフリットの間にはさまれて保持されている例がある。(特許文献3)
この場合、分離に寄与する部分は、その無機基体材料部分であり、上下フリットは無機体を中空体の保持されるために用いられている。
いくらきつく押込められても、フリットと無機体の間には空間が生じることになり、その空間部分に液は残存してしまう。その空間部分の液の追出しや置換は困難となる。特に、上記の如き減圧方法では、一部に気相部分ができてしまうと、その部分が優先的に流れることになり、均一に液を抜くことができなきなる。試料付加、洗浄工程に於いては、液体の置換が成され難くなってしまう。
【0081】
又、最後の溶出に於いては、液が残ってしまい、回収が悪くなってしまう。
本発明のモノリス構造では、液体の流れるマクロ細孔は連続体となり、流れの方向に対して均一に液体が変わってゆくことになる。即ち、液体の置換効率が大幅に上昇する。実施例1のように、この従来タイプでは、2回目の溶出でも試料成分が多く残る原因の1つになっていると考えられる。本発明の連続体のモノリス構造に於いては、液体の置換効率が高いので、1回の溶出で充分であり、2回目溶出の残存が殆どないことがわかる。
【0082】
又、従来タイプでは、押込み具合で、その空間は変化し、中空体への押込み時のロット間のバラツキも出易くなる。本発明に於いて、モノリス構造は一体構造であり、中空体への押込み時のバラツキは全くない。
市販品としては高価であり、実在しないが、フリットと無機体を更に押し潰し一体にすることも可能となるが、この方法でも押し潰した界面部分に異なる層ができてしまう。やはり均一な連続孔を持つモノリス構造に比べると、液体の流れは阻害される。
【0083】
更に、本発明では、シリカゲルである無機物とフリット材料であるポリエチレンなどの有機物を、ゾル−ゲル行程で混合すれば、シリカとポリエチレンの性質を持つハイブリッドの均一相での作成も可能である。
無機体が粒子状の物では、それを止めるために、上下フリットは不可欠となり、上記説明のような置換効率の問題が生じる。
【0084】
シリカ繊維やケル粒子を埋込んだシリカゲル薄膜(例えば3M社のエムポアディスク(登録商標))を無機体とした場合でも、やはり物理的な硬さが無く、急激な減圧や高回転の遠心分離により、変形して繊維の一部や粒子が溶出してしまうことがある。僅かな変形に於てでも、空間容積は変化することになり、バラツキ要因になってしまう。モノリス構造では、固い骨格の内部にある連続孔による分離のため、圧力変動などでも変形しないので、再現性も得られる。
【0085】
例え、フリットで止める必要のない固い繊維膜やシリカゲル薄膜を形成できたとしても、液の流れは、繊維やゲル表面を流れることにより、乱流が生じ、均一な分離が得られず、本発明のモノリス構造体を用いた場合ほどの分離は期待できない。
乱流の生じないモノリス構造体であることにより、初めて低分子DNAの吸着及び高い洗浄効果を達成できる。
【0086】
更に、従来タイプの粒子における流路と本発明のモノリス構造体におけるミクロ細孔とは液流れに対して接触と云う点で異なる。粒子タイプなどでは、液が入ってくる側と液が抜ける側とは、液抵抗における圧力の均一性がなく、細孔内部への液の接触が異なってしまう。HPLCのような加圧系では均一な圧力にできるので、その影響は少なくなるが、本発明分野で用いられる減圧系では、入口側は常圧であり、出口側では負圧になり、粒子1個における細孔内部への出入りが不均一となる。即ち、同じ成分でも成分分子によって、細孔内部まで入るものと、入らないものが生じ、トータル的には溶出時における幅が大きくなることになる。そのため洗浄時に除去したい成分だけを排除するのが難しく、最終的に溶出された成分は、実施例1に示す従来例のように低分子側のプライマーが残ってしまう。本発明のモノリス構造体に於いては、液の流れるマクロ細孔表面にミクロ細孔があるので、全て均一な入り込みが生じる。そのため、低分子不純物であるプライマーの除去が簡単に行える。
【0087】
従来法に於いて、大きなDNAに於いては、細穴へ入込み難くなり、更に、成分が含まれる液の粘性も上がるため、接触が不均一になり、2種現象が同時に起こることになり、吸着されない部分が多いと考えられる。又、乱流により、高分子DNAへの物理的なダメージが生じ、破壊してしまう可能性も高まる。
【0088】
基本的には、カオトロピック塩濃度を増やすことにより、それらの現象は軽減できるが、洗浄時でも取除かれず、溶出時に溶出してきてしまう。即ち、精製後の試料成分に多量の塩が入ることになる。これは、後の使用に於いては大きな問題となる。
【0089】
モノリス構造体を用いた本発明方法では、塩濃度を下げることができ、上記の諸問題は解決でき非常に有効である。更に、よりカチオン交換作用を持つカリウム塩を合せて使用するとより効果的になる。カリウム塩はカチオン交換作用が強いので、核酸の表面への吸着に寄与するが、それが故に、溶出時に基体表面に残ってしまうと目的とする精製DNAが溶出しないと云う問題が生じるため、確実な洗浄が不可決である。粒子タイプでは、洗浄時にも乱流が生じ、更に細孔への入込みが不均一のため、どうしてもカリウム塩が高濃度で基体に残存する部分が生じる。対策として、溶出液にカリウムを除けるバッファー、即ち他の塩を加えたりすればよいことになるが、やはり後のアプリケーション目的に適さないことになる。
モノリス構造体では、均一な液の流れと均一な細孔への入込みが可能となるため、カリウム塩が有効に作用する。
【実施例1】
【0090】
PCR反応液(フラグメントDNA)の精製では、PCR増幅反応物50μlをバッファーA1(1Mグアニジン塩酸塩、0.2M酢酸カリウム、50%2−プロパノール)300μlに混合する。シリカモノリス固相カラム9をコレクションチューブ8に挿入し、混合物をシリカモノリス固相カラム9に注入、1.5mlの遠心管内で遠心分離する。シリカモノリス固相カラム9をBバッファー(0.2M酢酸カリウム、50%エタノール)での洗浄処理により塩を含まないようにする。
【0091】
溶出には溶出用バッファーC(EDTA4mM、Tris−HCI10mM、pH8又は無菌DNA、RNA free水)20μlを、別の1.5mlの遠心管で遠心用カラムに通して遠心分離する。こうして精製されたPCR産物(フラグメントDNA)にはプライマー、dNTPs、ポリメラーゼ及び塩が含まれず、後の操作に直接用いることができる。(図1,図2参照)図1中M:分子量マーカー、(1)従来法で精製したサンプル、(2)本発明で精製したサンプル。
【0092】
(1)が従来の特許文献3の方法で精製した試料(400bp)での電気泳動による評価である。(2)が本発明方法である。(1)では、低分子側(下)部分が多く残っているが、(2)では殆ど残っておらず、高い精製効率が得られている。又、図2に於いて1−1及び1−2は従来方法で2回行い、残存を見たもので、2−1,2−2は本発明のものである。本発明の場合1回目で殆ど残っておらず、高い精製効率が得られることが分かる。図3はHPLCを使用しての評価である。HPLC条件は以下の通り
HPLC条件
カラム:CIM DEAE
溶離液:
A:20mM Tris-HCl Ph7.4
B:A+1M NaCl
A/B=50/50-(10MIN)-0/100 Flow rate:3ml/min
検出:UV260nm
【0093】
図3に於いて未精製のPCR溶液のHPLC評価データは、1番上のクロマトグラムであり、従来の特許文献3法による精製のクロマトグラム(一番下)と比べてパターンに変化がなく、dNTPs及びプライマーは、殆ど除かれていないことがわかる。本発明方法では、2番目のクロマトのようにdNTPs及びプライマーの2つのピークが大幅に取除かれ、目的とする核酸が高く精製されていることがわかる。(図3参照)
【実施例2】
【0094】
アガロースゲルからのDNA断片の精製ではPCR増幅産物を標準的又は低融点アガロースゲル(TE又はTBEバッファー使用)を用いて電気泳動し、DNAをアガロースゲル(TE又はTBE0.5%)中で分離する。清潔なカミソリやメスで単離すべきDNA断片をゲルから切出して、1.5ml遠心チューブに入れる。バッファーA2(2Mグアニジンチオシアン酸、0.4M酢酸カリウム、30%の2−プロパノール)300μlに混合し、60℃で5分間又はゲルスライスが完全に溶解するまでインキュベートする。
この溶解液を実施例1に従って、モノリス固相カラム9を、コレクションチューブ8に挿入し、混合物をモノリス固相カラム9に注入、1.5mlの遠心管内で遠心分離する。モノリス固相カラム9をバッファーB(0.2M酢酸カリウム、50%エタノール)での処理により洗って塩を含まないようにする。
【0095】
溶出にはバッファーC(EDTA1mM、Tris−HCI10mM、pH8又は無菌DNA、RNA free水)20μlを、別の1.5mlの遠心管で遠心用カラムに通して遠心分離する。図4のうち図4−1は35bpの、図4−2は100〜500bpの、図4−3は10,000bpの、図4−4は35,000bpの電気泳動による評価である。
【0096】
本発明方法では、低分子の35bpから約100Kbpまで回収されており、アガロースゲルからでも広い範囲のDNAが精製できることがわかる。溶出バッファーとして、EDTAバッファーを含まない水でも同様の結果が得られた。
【実施例3】
【0097】
制限酵素反応後のDNA100μgを制限酵素で処理する。このDNA制限反応溶液を実施例1に従ってバッファーA1,300μlに混合し、そしてそれ以後の処理は実施例1と同様に行った。溶出後、得られた精製DNAには制限酵素及び塩が含まれず、吸光度測定比率(mm)260/280の比率が1.8と良好なものであった。
【実施例4】
【0098】
分子サイズの小さい、35bpのPCR増幅産物の精製ではPCR増幅反応物10μlを実施例1に従ってバッファーA1,100μlに混合し、それ以後の処理は実施例1と同様に行った。(図5参照)図中M:分子量マーカー、1:精製前のサンプル、2:本発明で精製したサンプル。
100bpや100bp以下の小さなDNAでもここでは35bpのDNAのPCR反応液からの精製が出来た。
【実施例5】
【0099】
分子サイズの大きな(100bp以上100,000bpまで)PCR増幅産物の精製ではPCR増幅反応溶液20μlを実施例1に従ってバッファーA1,200μlに混合し、それ以後の処理は実施例1と同様に行った。(図6参照)図中M:分子量マーカー、1:精製前のサンプル、2:本発明で精製したサンプル3:従来法で精製したサンプル。
1000bp〜100KbpまでのDNAでもPCR反応液からの精製が出来た。
【実施例6】
【0100】
1本鎖DNA溶液の精製では溶液20μlを実施例1に従ってバッファーA1,200μlに混合し、それ以後の処理は実施例1と同様に行った。(図7参照)図中M:分子量マーカー、1:精製前のサンプル、2:本発明で精製したサンプル。
1本鎖DNA35merは、従来方法では回収できていない(3)が、本発明では(2)再現性よく2回とも回収されている。
【実施例7】
【0101】
ナトリウムとカリウムによる精製比較(保持メカニズム)(図8参照)図中M:分子量マーカー、1:ガラスモノリス、カリウムによる精製後サンプル、2:シリカモノリス、カリウムによる精製後サンプル、3:シリカモノリス、ナトリウムによる精製後サンプル、
従来からよく用いられるナトリウムと、本発明の効果があるカリウムとの精製比較を行なった。ナトリウムでは、保持が殆ど得られず、カリウムでは高い精製効率が得られた。
又、シリカモノリスと同様にガラスモノリスでも同じくカリウムの方が高い生成効率が得られた。
【0102】
本発明を実効あらしめるために下記の方法を実施することは意義がある。
核酸を含有する溶液をアルカリ金属塩を介在させることにより、一体型モノリス構造体の通孔に夫々対応する核酸を吸着させ、洗浄液で洗浄後、溶出させることを特徴とするDNA分離精製方法。
アルカリ金属塩は酢酸カリウムであることを特徴とするDNAの分離精製方法。
酢酸カリウムは0.1〜1M未満含有する溶解吸着バッファーを使用することを特徴とする分離精製方法。
グアニジン塩又は、酢酸カリウムなどのカリウム塩を含有する溶解、吸着バッファーにより、溶解吸着を行うことを特徴とするDNAの分離精製方法。
Tris−HCl、EDTAを含有する水により溶出を行うことを特徴とするDNAの分離精製方法。
溶解、吸着、分離、洗浄操作を1つのモノリス固相カラムを用いて行なうことを特徴とするDNAの分離精製方法。
溶解吸着バッファー、水及び分離精製機構よりなるキット。
【産業上の利用可能性】
【0103】
以上のように本発明に係るDNAなどの分離生成機構は、分子生物学的研究に於いて、非常に頻繁に用いられ、P.C.R、クローニング、シークエンシンク、制限酵素消化、その他酵素作用などのアプリケーションに先立って行われるフラグメントDNAなどの精製に特に有用で35bp(mer)から100Kbp(mer)以下の広範なDNAの定量的な分離や効率的な精製ができ、幅広い核酸の精製に対応できる。
【特許請求の範囲】
【請求項1】
一体型モリノス構造体であって、一端から他端まで連続した通孔を形成させ、かつ核酸大きさに対応する大きさの通孔が設けられ、分離すべき核酸を含有する溶液を通過させることにより、該通孔に対応する核酸が夫々保持できるように構成したことを特徴とするDNAなどの分離精製機構。
【請求項2】
モノリス構造体は、ガラス、シリカ等の無機物又は無機物に有機物を含有するハイブリッド体であって、上面から下面まで貫通しているマクロ細孔を持つ多孔質体を使用することを特徴とする請求項1に記載のDNAなどの分離精製機構。
【請求項3】
モノリス構造体の多孔質体はマクロ細孔の内部にミクロ細孔を有することを特徴とする請求項1又は2の何れかの項に記載のDNAなどの分離精製機構。
【請求項4】
モノリス構造体の多孔質体はマクロ細孔1〜100μm、ミクロ細孔0〜100nmであることを特徴とする請求項1乃至3の何れかの項に記載のDNAなどの分離精製機構。
【請求項5】
カラムチューブにモノリス構造体により形成されるディスクを配置することにより、モノリス固相カラムを構成することを特徴とする請求項1乃至4の何れかの項に記載のDNAなどの分離精製機構。
【請求項6】
上下を開放した筒状体に、モノリス構造体により構成した基体を着脱自在に装着して形成したモノリス固相カラムを使用することを特徴とする請求項1乃至5の何れかの項に記載のDNAなどの分離精製機構。
【請求項1】
一体型モリノス構造体であって、一端から他端まで連続した通孔を形成させ、かつ核酸大きさに対応する大きさの通孔が設けられ、分離すべき核酸を含有する溶液を通過させることにより、該通孔に対応する核酸が夫々保持できるように構成したことを特徴とするDNAなどの分離精製機構。
【請求項2】
モノリス構造体は、ガラス、シリカ等の無機物又は無機物に有機物を含有するハイブリッド体であって、上面から下面まで貫通しているマクロ細孔を持つ多孔質体を使用することを特徴とする請求項1に記載のDNAなどの分離精製機構。
【請求項3】
モノリス構造体の多孔質体はマクロ細孔の内部にミクロ細孔を有することを特徴とする請求項1又は2の何れかの項に記載のDNAなどの分離精製機構。
【請求項4】
モノリス構造体の多孔質体はマクロ細孔1〜100μm、ミクロ細孔0〜100nmであることを特徴とする請求項1乃至3の何れかの項に記載のDNAなどの分離精製機構。
【請求項5】
カラムチューブにモノリス構造体により形成されるディスクを配置することにより、モノリス固相カラムを構成することを特徴とする請求項1乃至4の何れかの項に記載のDNAなどの分離精製機構。
【請求項6】
上下を開放した筒状体に、モノリス構造体により構成した基体を着脱自在に装着して形成したモノリス固相カラムを使用することを特徴とする請求項1乃至5の何れかの項に記載のDNAなどの分離精製機構。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公開番号】特開2010−233579(P2010−233579A)
【公開日】平成22年10月21日(2010.10.21)
【国際特許分類】
【出願番号】特願2010−138612(P2010−138612)
【出願日】平成22年6月17日(2010.6.17)
【分割の表示】特願2005−517842(P2005−517842)の分割
【原出願日】平成16年2月12日(2004.2.12)
【出願人】(390030188)ジーエルサイエンス株式会社 (37)
【Fターム(参考)】
【公開日】平成22年10月21日(2010.10.21)
【国際特許分類】
【出願日】平成22年6月17日(2010.6.17)
【分割の表示】特願2005−517842(P2005−517842)の分割
【原出願日】平成16年2月12日(2004.2.12)
【出願人】(390030188)ジーエルサイエンス株式会社 (37)
【Fターム(参考)】
[ Back to top ]