
DNAへのランダム変異導入方法
【課題】 複数塩基単位の置換、挿入、欠失といった変異をDNAに簡便に導入する新たな手法を提供すること。
【解決手段】 以下の工程を含むDNAへのランダム変異導入方法。
(a) 変異導入の対象とするDNA鎖をランダムな部位で切断し、DNA断片を得る工程
(b) 得られたDNA断片の3'末端に複数個の塩基を付加する工程
(c) 塩基を付加した3'末端から、外来の鋳型DNA鎖の存在下または非存在下において、DNAポリメラーゼによりDNA鎖を伸長させる工程
【解決手段】 以下の工程を含むDNAへのランダム変異導入方法。
(a) 変異導入の対象とするDNA鎖をランダムな部位で切断し、DNA断片を得る工程
(b) 得られたDNA断片の3'末端に複数個の塩基を付加する工程
(c) 塩基を付加した3'末端から、外来の鋳型DNA鎖の存在下または非存在下において、DNAポリメラーゼによりDNA鎖を伸長させる工程
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、DNAへのランダム変異導入方法、詳しくは、置換、挿入、欠失といった変異を複数塩基単位で簡便に導入することのできるDNAへのランダム変異導入方法に関する。
【背景技術】
【0002】
酵素をコードする遺伝子へのランダム変異導入技術は、その酵素の機能を改変する上で重要な技術のひとつである。酵素をコードする遺伝子にランダム変異を導入すると、その変異に応じて機能がさまざまに変化した変異酵素が得られるので、その中から適当な選抜系にて目的とする機能をもった酵素を選抜することが可能となる。たとえば、ランダム変異を導入した酵素のライブラリーから、熱処理しても活性の残っている酵素を同定することで、耐熱性の向上した変異酵素を得ることができる。さらに、ランダム変異と選抜の操作を繰り返せば、有効な変異が蓄積でき、酵素の機能をさらに向上させることができる。変異と選抜の操作を繰り返して酵素の機能を向上させる技術は、生物が進化するさまに似ていることから、「進化分子工学」と呼ばれている。
【0003】
自然界における変異の種類は、大別して、別の種類の塩基への置き換え(置換)、新たな塩基の挿入(挿入)、塩基の脱落(欠失)の三つに分けられる。このうち、進化分子工学の分野では、主に置換変異が利用される。まず、エラープローンPCR 法(非特許文献1)などで遺伝子にランダムな1塩基置換変異を導入し、機能の変化した変異体を選抜する。取得した変異体には、さらに置換変異を導入するか、変異部位のコドンを部位特異的にランダムに改変するか(サチュレーション変異導入;非特許文献2)、いくつかの変異体の遺伝子をかけあわせて(DNA シャッフリング;非特許文献3)、変異の組み合わせを検討する。そして、これらの作業をルーチン化して、機能をできるだけ向上させるというのが、従来の進化分子工学研究の進め方であった。ただ、機能が無限に向上し続けることはなく、数世代の実験で進化が頭打ちになることも多い。置換変異のバリエーションは、300アミノ酸残基からなるタンパク質において1アミノ酸残基を置換する場合、19×300=5700通りである。このバリエーションの中に、機能を向上させる変異がなければ、置換変異による進化は起こらない。たとえ置換変異の頻度を上げたとしても、それ以上の進化が望めるとは限らない。なぜなら、置換変異はおおむね相加性を持っているが、相乗効果を生む例はまれで、予想外の進化が得られる可能性は低いからである。従って、この5700種の置換変異を探索しつくした時点で、進化は頭打ちになると予想される。
【0004】
一方、もし挿入や欠失をランダムに導入できるなら、置換とはまったく異なる変異体のバリエーションを作製できるため、たとえ進化が頭打ちになっても、さらなる進化を起こさせる可能性がある。例えば、300アミノ酸残基からなるタンパク質の場合、nアミノ酸残基欠失のライブラリーの数は(301-n)通り、nアミノ酸残基挿入のライブラリーの数は(20のn乗×301)通り存在することから、非常に大きなライブラリーを得ることができる。従って、置換変異による進化が限界でも、挿入や欠失を利用することで進化の頭打ちに対してブレークスルーをもたらすこともできると考えられる。
【0005】
しかしながら、挿入や欠失をタンパク質の機能改変に用いた例はきわめて少なく、現代の構造生物学および分子進化学の分野において、挿入や欠失に関しては、いまだ未知の部分が多い。これは、構造変化の予想が難しいからである。もし、挿入や欠失を含む変異体を作成し、構造や機能に関する情報を蓄積できれば、進化分子工学のみならず、構造生物学や分子進化学にも大きな影響を与える可能性を秘めている。
【0006】
挿入や欠失は、置換に比べて構造の変化が大きいため、酵素の構造を著しく壊すかもしれず、タンパク質工学には応用しにくいと考えられがちである。ところが、酵素のアミノ酸配列のデータベースを調査したところ、10アミノ酸残基以下の挿入や欠失は、進化の過程でかなりの確率でおきていることがわかった(非特許文献4)。とりわけ、5アミノ酸残基以下の挿入欠失は頻繁に起こるとされている。これは、挿入や欠失が、タンパク質の機能改変に応用できる可能性を示唆している。
【0007】
これまでDNAに挿入や欠失をランダムに導入する方法は、いくつか開発されている。たとえば、認識配列の外側を切断する特殊な制限酵素を用いて挿入や欠失を導入した例(非特許文献5)、コドン単位で DNA を合成して欠失を導入した例(非特許文献6)、トランスポゾンを利用して特定の15塩基をランダムに挿入した例(非特許文献7)、エンドヌクレアーゼを用いて欠失やリピート配列を導入した例(非特許文献8)、などが挙げられる。これらの方法のうちいくつかは、実際に酵素や機能性タンパク質の向上を実現しており、挿入欠失変異の有効性を示している。しかしながら、いずれの方法も、操作が複雑であったり、導入できる変異の種類や長さが限られていたり、特殊な技術や設備が必要であったりするなどの問題がある。従って、より簡便にDNAに挿入欠失変異を導入する方法が求められていた。
【非特許文献1】Leung, D. W., Chen, E. and Goeddel, D. W., A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction. Techniques, 1, 11-15 (1989).
【非特許文献2】Miyazaki, K. and Arnold, F. H., Exploring nonnatural evolutionary pathways by saturation mutagenesis: Rapid improvement of protein function. J. Mol. Evol., 49, 716-720 (1999).
【非特許文献3】Stemmer, W. P. C., Rapid evolution of a protein in-vitro by DNA shuffling. Nature, 370, 389-391 (1994).
【非特許文献4】Pascarella, S. and Argos, P., Analysis of Insertions Deletions in Protein Structures. J. Mol. Biol., 224, 461-471 (1992).
【非特許文献5】Murakami, H., Hohsaka, T. and Sisido, M., Random insertion and deletion of arbitrary number of bases for codon-based random mutation of DNAs. Nat. Biotechnol., 20, 76-81 (2002).
【非特許文献6】Osuna, J., Yanez, J., Soberon, X. and Gaytan, P., Protein evolution by codon-based random deletions. Nucleic Acids Res., 32, e136 (2004).
【非特許文献7】Hallet, B., Sherratt, D. J. and Hayes, F., Pentapeptide scanning mutagenesis: Random insertion of a variable five amino acid cassette in a target protein. Nucleic Acids Res., 25, 1866-1867 (1997).
【非特許文献8】Pikkemaat, M. G. and Janssen, D. B., Generating segmental mutations in haloalkane dehalogenase: a novel part in the directed evolution toolbox. Nucleic Acids Res., 30, e35 (2002).
【発明の開示】
【発明が解決しようとする課題】
【0008】
本発明は、置換変異のみならず、挿入欠失変異をDNAに簡便に導入する新たな手法を提供することにある。
【課題を解決するための手段】
【0009】
本発明者らは、上記課題を解決すべく鋭意研究を重ねた結果、対象とするDNA鎖をランダムに切断し、その切断断片の3'末端に複数個のランダムな塩基を付加した後、DNAポリメラーゼを用いて該3'末端から伸長反応を行うことで、前記DNA鎖中に複数塩基単位の置換、挿入、および欠失をランダムに導入できることを見出した。本発明はかかる知見により完成されたものである。
【0010】
即ち、本発明は以下の発明を包含する。
(1) 以下の工程を含むDNAへのランダム変異導入方法。
(a) 対象とするDNA鎖をランダムな部位で切断し、DNA断片を得る工程
(b) 得られたDNA断片の3'末端に複数個の塩基を付加する工程
(c) 塩基を付加した3'末端から、外来の鋳型DNA鎖の存在下または非存在下において、DNAポリメラーゼによりDNA鎖を伸長させる工程
(2) 外来の鋳型DNA鎖を除去する工程をさらに含む、(1)に記載の方法。
(3) 外来の鋳型DNA鎖が、含ウラシルDNA、メチル化DNA、末端ビオチン化 DNAのいずれかを含むDNA鎖である、(2)に記載の方法。
(4) 対象となるDNA鎖が、二本鎖または一本鎖である、(1)〜(3)のいずれに記載の方法。
(5) DNA断片の3'末端に付加する塩基が、デオキシリボ核酸またはデオキシリボ核酸類縁体である、(1)〜(4)のいずれかに記載の方法。
(6) DNA断片の3'末端に付加する塩基数が5〜50塩基である、(1)〜(5)のいずれかに記載の方法。
(7) DNA断片の3'末端への塩基の付加が、ターミナルデオキシヌクレオチジルトランスフェラーゼ、DNAポリメラーゼ、DNAリガーゼ、またはRNAリガーゼのいずれかを用いて行われる、(1)〜(6)のいずれかに記載の方法。
【発明の効果】
【0011】
本発明によれば、置換、挿入、欠失といった変異を、わずかに数ステップの簡便な操作で、特別な試薬や機器を使うことなくDNAに導入できる方法が提供される。本発明の方法によれば、挿入や欠失といった、従来の方法では導入することが難しい変異を自在に導入することができ、また、その挿入欠失部位に連続して複数塩基の置換が導入されることが多いため、単なる挿入欠失以上にランダム性が高い変異を導入することができる。また、本発明の方法によれば、進化分子工学に適当な頻度とされる1〜3塩基/kb程度の1塩基置換変異が導入でき、また、複数塩基単位の置換も導入できる。その上、本発明の方法は、DNAを断片化して再構築する操作を含むことから、DNAシャッフリング(もしくはファミリーシャッフリング)の能力も備えている。従って、本発明の方法は、酵素の改変など、タンパク質の機能改変に非常に有用である。
【発明を実施するための最良の形態】
【0012】
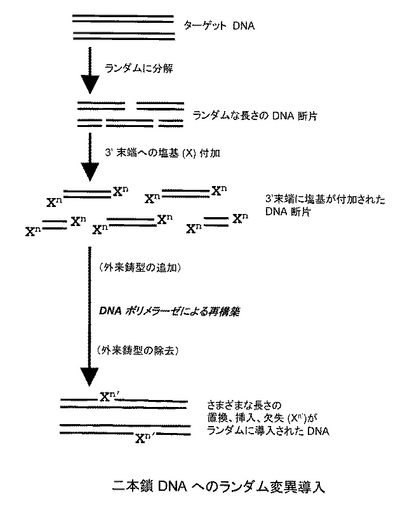
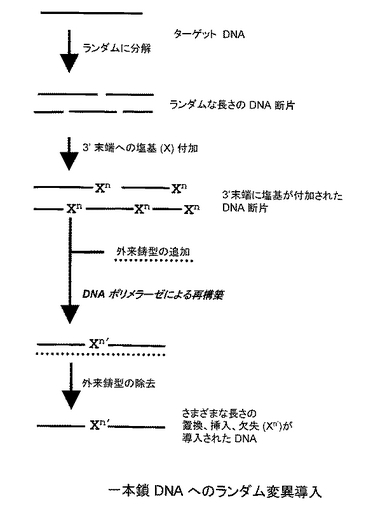
本発明のDNAへのランダム変異導入方法は、(a)対象とするDNA鎖をランダムな部位で切断し、DNA断片を得る工程、(b)得られたDNA断片の3'末端に複数個の塩基を付加する工程、(c)塩基を付加した3'末端から、外来の鋳型DNA鎖の存在下または非存在下において、DNAポリメラーゼによりDNA鎖を伸長させる工程を含む(図1A:二本鎖DNAへのランダム変異導入、図1B:一本鎖DNAへのランダム変異導入参照)。以下、各工程について説明する。
【0013】
工程(a)では、変異導入の対象とするDNA鎖をランダムな部位で切断し、DNA断片を得る。切断する方法としては、ヌクレアーゼ処理または超音波破砕処理などが挙げられる。ヌクレアーゼ処理としては、エンドヌクレアーゼを使う方法(DNaseIやエンドヌクレアーゼVなど)、エキソヌクレアーゼを使う方法のいずれであってもよい。
【0014】
対象とするDNA鎖は一本鎖、二本鎖どちらでもよい。二本鎖DNAの場合は平滑末端が望ましいので、T4 DNAポリメラーゼなどで平滑化するか、Mnイオンを加えた条件でDNaseI処理を行うことが好ましい。断片の大きさは特に限定はされないが、10bp以上、好ましくは50bp以上で、対象とするDNA全長の半分以下であることが好ましい。
【0015】
本工程の具体例として、例えばDNaseIを用いたDNA鎖の切断は、以下のようにして行うことができる。以後、濃度はすべて最終濃度を示す。まず、対象とするDNA鎖を、Tris-HCl(pH 6〜8)、1〜100 mM MnCl2中で、1×10-5〜1×10-3 U/μL DNaseIと1〜100 分間反応させる。反応終了後、MnCl2に対して過剰量の EDTA 溶液を加え、反応を停止させた後、キアゲンの MinElute Reaction Cleanup Kit などを用いて精製する。
【0016】
続いて、工程(b)では、工程(a)で得られたDNA断片の3'末端に複数個の塩基を付加する。付加する塩基数は 5〜50塩基が好ましい。DNA断片の3'末端に塩基を付加するための方法としては、ターミナルデオキシヌクレオチジルトランスフェラーゼ(TdT)、DNAポリメラーゼ、DNAリガーゼ(T4 DNAリガーゼなど)、RNAリガーゼ(T4 RNAリガーゼなど)を用いる方法などが挙げられる。ターミナルデオキシヌクレオチジルトランスフェラーゼ(TdT)を用いて3'末端に塩基を付加する場合は、まず、アガロースゲル電気泳動などでDNA断片の平均鎖長を計算し、DNA断片のおよそのモル濃度を計算してから、その10倍〜100倍のdNTPをTdTの反応溶液に加えることで、理論上、平均5〜50bpの塩基を3'末端に付加することができる。また、T4 RNAリガーゼを利用する場合は、対象とするDNA断片を脱リン酸化してから、3'末端と 5'末端をリン酸化した 5〜50 塩基からなるランダムオリゴヌクレオチドを結合させる。
【0017】
DNA断片の3'末端に付加する塩基は、デオキシリボ核酸またはデオキシリボ核酸類縁体のいずれであってもよい。デオキシリボ核酸類縁体としては、例えば、dITPや7-deaza-dGTPなどが挙げられる。
【0018】
本工程の具体例として、例えばTdTを用いた塩基の付加は、以下のようにして行うことができる。1〜100 mM カコジル酸ナトリウム(pH 6〜8)、0.1〜10 mM CoCl2(またはMnCl2)、0.02〜2 mM ジチオスレイトール、0.1〜10 U/μL TdT、0.01〜10 pmol/μL の工程(a)のDNA断片、該DNA断片の5〜50倍濃度のdNTP、を加え、20〜40℃で10分〜10時間放置する。その後、DNAを精製して、工程(b)の最終産物とする。
【0019】
工程(c)では、工程(b)で得られた産物(複数個の塩基を3’末端に付加したDNA断片)の3'末端から、外来の鋳型DNA鎖の存在下または非存在下において、DNAポリメラーゼによりDNA鎖を伸長させる。
【0020】
ここで用いるDNA ポリメラーゼとしては、特に限定はされないが、Taq DNA ポリメラーゼや Deep Vent exo-DNAポリメラーゼなど、3'-5' エキソヌクレアーゼ活性を持たない DNA ポリメラーゼが望ましい。
【0021】
上記の伸長反応は、通常のPCRの手法に従えばよく、PCRの条件(温度設定、サイクル数)も通常採用される条件でよい。
【0022】
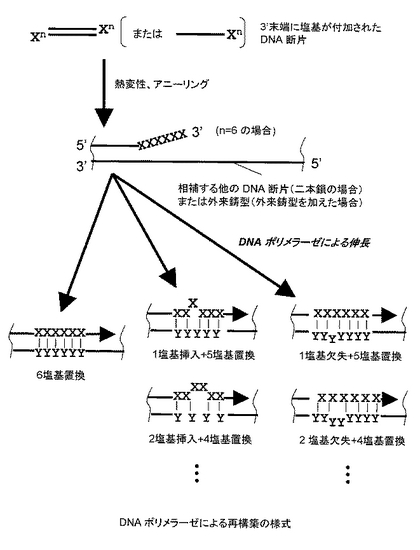
対象となるDNA鎖が二本鎖の場合は、以下の操作を行う。まず、工程(b)で得られた3’末端に複数個の塩基(Xn)が付加されたDNA断片を、熱変性により一本鎖DNA断片とする。次に、温度を下げ、産生された任意の一本鎖DNA断片(「X断片」とする)を、それに相補的な配列を有する他の任意の一本鎖DNA断片(「Y断片」とする)に結合(アニーリング)させる。その後、DNA ポリメラーゼを作用させると、Y断片を鋳型としてX断片の3'末端からDNA鎖を伸長させることができる。
【0023】
あるいは、別の態様として、X断片が鋳型となるDNA断片と結合(アニーリング)できる割合を上げるため、外来の鋳型DNA鎖をあらかじめ加えておいてもよい。外来の鋳型DNA鎖としては、対象となる二本鎖DNAの全配列または部分配列からなるDNA鎖を用いる。この場合は、X断片を外来の鋳型DNA鎖にアニーリングさせ、その後、DNAポリメラーゼを作用させると、該鋳型DNA鎖を鋳型としてX断片の3'末端からDNA鎖を伸長させることができる。
【0024】
対象となるDNA鎖が一本鎖の場合は、外来の鋳型DNA鎖の添加が必須となる以外は、上記と同様の操作で伸長反応を行う。まず、工程(b)で得られた3’末端に複数個の塩基が付加された任意の一本鎖DNA断片(「Z断片」とする)に対して相補的な配列を有する外来の鋳型DNA鎖を加える。その後、Z断片をその外来の鋳型DNA鎖にアニーリングさせ、DNA ポリメラーゼを作用させると、該鋳型DNA鎖を鋳型として、Z断片の3'末端からDNA鎖を伸長させることができる。
【0025】
上記の外来の鋳型DNA鎖は、伸長反応終了後に除去できる形状にしておく。このような鋳型DNA鎖として、例えば、含ウラシルDNA、メチル化DNA、ビオチン修飾DNAを含むDNA鎖などを用いることができ、その除去は、それぞれ、ウラシルDNAグリコシダーゼ処理、メチル化DNAを分解する菌体の形質転換、アビジンカラムによる精製より行うことができる。
【0026】
本工程の具体例として、例えば耐熱性DNAポリメラーゼ(New England Biolabs 社のDeep vent exo-DNAポリメラーゼ)を用いた伸長反応は、以下のようにして行うことができる。まず、1〜100 ng/μLの工程(b)の産物を、反応溶液(1〜100 mM KCl, 1〜100 mM (NH4)2SO4, 2〜200 mM Tris-HCl pH 7〜10, 0.2〜20 mM MgSO4, 0.01〜1% Triton X-100, 0.2〜20 mM dNTP, 0.02〜20 U/μL Deep vent exo-DNA ポリメラーゼ)中で、サーマルサイクリング反応を行う。温度条件は、90〜99℃, 10秒〜2分→30〜70℃, 10秒〜2分→60〜80℃, 10秒〜10分で、このサイクルを1〜60 回繰り返す。
【0027】
工程(c)では、3’末端に複数個の塩基が付加されたDNA断片が互いにプライマーとなるようにPCR反応させるので、その3'末端の付加塩基(Xn)と鋳型とのミスマッチに起因する変異(置換、挿入、欠失)導入と、異なったDNA断片どうしの再結合(シャッフリング)が同時に達成できる。
【0028】
工程(c)で得られた産物は通常の手法でクローニングすることができる。得られた産物は、さまざまな長さの置換、挿入、欠失(Xn')が導入されたDNAが含まれる。例えば、DNAポリメラーゼによるDNA鎖の伸長反応の際、3'末端の付加塩基(Xn)に存在するミスマッチ塩基の数に応じて、置換変異が導入される。さらに、このミスマッチ塩基は、相補鎖のさまざまな部分と結合するので、相補する部位にしたがって、欠失や挿入となる。すなわち、相補鎖側が長い場合は欠失、ミスマッチ側が長い場合は挿入になる(図2参照)。
【実施例】
【0029】
以下、実施例によって本発明を更に具体的に説明するが、これらの実施例は本発明を限定するものでない。
(実施例1)DNA 配列へのランダム変異導入
(1) ターゲット遺伝子の調製
pUC19 の2661-2686-1-480 位のDNA配列をターゲット遺伝子として、ランダム変異導入を行った。まず、プラスミドpUC19 上の2661-2686-1-480位のDNA配列をPCR法で増幅した。反応溶液の組成は、20 pg/μL pUC19, 0.02 U/μL TOYOBO社製 KOD plus DNA ポリメラーゼ, 1×添付バッファー, 0.2 mM dNTP, 1 mM MgSO4, 0.3 pmol/μLプライマー1:5'-TAGGCGTATCACGAGGCCCTTTCGTC-3'(配列番号1)およびプライマー2:5'-ATTACGCCAAGCTAGCATGCCTGCAGGTCG-3'(配列番号2)とした。なお、プライマー1は、pUC19の2661-2686位のDNA配列の相補鎖に相当し、プライマー2は、pUC19の431-460位のDNA配列の相補鎖に相当する(ただし447位にA-T変異を持つ)。
【0030】
反応溶液を100 μLずつ10 本の PCR チューブに分注し、PE アプライドバイオシステムス社の Gene Amp 9700 を用いて、94℃, 2分 のプレヒート後、 94℃, 15 秒 → 60℃, 30秒→ 68℃, 30秒の温度条件で 25 回のサーマルサイクリング反応を行った。生成物の100 μL ずつを、キアゲン社の MinElute Reaction Cleanup kit を用いて精製した。DNA 濃度を260 nm の吸光度から算出したところ、670ng/μLであった。
(2) プラスミドベクターの調製
上記のターゲット遺伝子をプラスミドに組み込むためのベクターとして、pUC19 の 431-2680 位のDNA配列をPCR増幅した。PCRの条件としては、プライマー1および2の代わりにプライマー3:5'-ATTACGCCAAGCTAGCATGCCTGCAGGTCG-3'(配列番号3)とプライマー4:5'-AGGGCCTCGTGATACGCCTATTTTTATAGG-3'(配列番号4)を用いる点、温度条件中の68℃で保持する時間を2分にする以外は、前記のターゲット遺伝子の増幅と同じであった。なお、プライマー3は、pUC19の431-460位のDNA配列の相補鎖に相当し(ただし447位にA-T変異を持つ)、プライマー4は、pUC19の2651-2680位のDNA配列の相補鎖に相当する。生成物をキアゲン社の QIAprep PCR Purification kit を用いて精製した。DNA 濃度は64ng/μLであった。
(3) ターゲット遺伝子への変異導入
前記のターゲット遺伝子(pUC19の2661-2686-1-480 位のDNA配列)の増幅産物22μgを、1mLのDNaseI溶液(50 mM Tris-HCl (pH 7.5), 10 mM MnCl2, 0.5 U/mL TaKaRa 社製 DNaseI)に加え、16℃で10分インキュベートすることにより断片化した。反応後、0.5 M EDTAを40μL加えて反応を停止して、MinElute Reaction Cleanup kit を用いて精製し、200μLの精製産物を得た。DNA濃度は12ng/μLであった。2% アガロースゲル上で電気泳動することにより断片の長さを調べたところ、平均 100bp 程度であった。従って、生成物のモル濃度は、約0.18 pmol/μLと推定された。
【0031】
次に、上記で得られた断片(DNaseI 断片)の3'末端に、TdTを用いてランダムな塩基を付加した。反応は、20μLの反応溶液(0.1 pmol/μL DNaseI 断片、1 pmol/μL dNTP、0.25 U/μL TOYOBO社製 TdT、1×添付バッファー)を37℃で1時間インキュベートすることにより行った。反応後、生成物をMinElute Reaction Cleanup kit を用いて精製した。DNA濃度は7ng/μLであった。
【0032】
さらに、この塩基が付加されたDNA断片を用いてDNAポリメラーゼにより伸長反応を行った(1st PCR)。反応は、10μLの反応溶液(5ng/μL 塩基付加断片、0.2U/μL New England Biolabs 社製 Deep Vent exo-DNA ポリメラーゼ、0.2 mM dNTP, 1×添付バッファー)に対し、Gene Amp 9700 を用いて96℃, 2分のプレヒート後、96℃, 30秒→ 60℃, 30秒→75℃, 30秒を1サイクルとするサーマルサイクリングを40回繰り返すことによって行った。
【0033】
次に、1st PCR 産物を1μL分取し、PCR増幅を行った(2nd PCR)。反応は、50μLの反応溶液(2% 1st PCR産物, 0.3 pmol/μL プライマー1および2、0.02 U/μL KOD plus DNA ポリメラーゼ, 1×添付バッファー, 0.2 mM dNTP, 1 mM MgSO4)に対し、Gene Amp 9700 を用いて96℃, 2分のプレヒート後、96℃, 15秒→ 60℃, 30秒→ 68℃, 30秒を1サイクルとするサーマルサイクリングを25回繰り返すことによって行った。その後、2% アガロースゲル電気泳動により、目的産物のバンド (500 bp 付近)を分画し、キアゲン社の MinElute Gel Extraction kit を用いて精製した。この結果、350ng/μLのDNA産物10μLを得た。
【0034】
この 2nd PCR 産物を、オーバーラップ伸長PCR法によりベクターに組み込み、大腸菌を形質転換して、変異体をクローニングした。100μLの反応溶液(10% の2nd PCR産物, 0.2 ng/μL 431-2680 位のDNA配列のPCR 産物、0.02 U/μL KOD plus DNA ポリメラーゼ, 1×添付バッファー, 0.2 mM dNTP, 1 mM MgSO4)を、Gene Amp 9700 を用いて96℃, 2分のプレヒート後、96℃, 15秒→ 60℃, 30秒→ 68℃, 30秒を1サイクルとするサーマルサイクリングを25回繰り返した。その後、この産物1μLを用いて、大腸菌TOP10をエレクロトポレーション法で形質転換し、LB プレート上で、37℃で18時間培養した。
【0035】
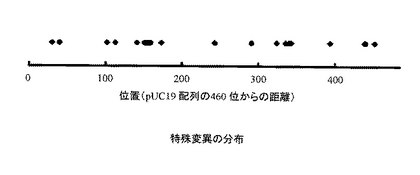
形成されたコロニーをピックアップして、アマシャムバイオサイエンス社の TempliPhi Amplification Kit によりプラスミドを増幅し、2661-2686-1-480 位 のDNA 配列を解析した。その結果、全88サンプル中、計22個の特殊な変異(二塩基以上の欠失、挿入、置換)が導入されていることが確認された(表1、2)。また、これらの特殊な変異は、ターゲット遺伝子の配列中で広く分布することがわかった(図3)。従って、本方法によれば、さまざまな長さの挿入、欠失、置換をDNAにランダムに導入できることが確認できた。
【0036】
【表1】

【0037】
【表2】
【図面の簡単な説明】
【0038】
【図1A】本発明のDNAへのランダム変異導入方法の概略(二本鎖DNAの場合)を示す。
【図1B】本発明のDNAへのランダム変異導入方法の概略(一本鎖DNAの場合)を示す。
【図2】本発明のDNAへのランダム変異導入方法におけるDNAポリメラーゼによる再構築の様式を示す。
【図3】本発明方法によりターゲット遺伝子に導入した特殊変異の分布を示す。
【技術分野】
【0001】
本発明は、DNAへのランダム変異導入方法、詳しくは、置換、挿入、欠失といった変異を複数塩基単位で簡便に導入することのできるDNAへのランダム変異導入方法に関する。
【背景技術】
【0002】
酵素をコードする遺伝子へのランダム変異導入技術は、その酵素の機能を改変する上で重要な技術のひとつである。酵素をコードする遺伝子にランダム変異を導入すると、その変異に応じて機能がさまざまに変化した変異酵素が得られるので、その中から適当な選抜系にて目的とする機能をもった酵素を選抜することが可能となる。たとえば、ランダム変異を導入した酵素のライブラリーから、熱処理しても活性の残っている酵素を同定することで、耐熱性の向上した変異酵素を得ることができる。さらに、ランダム変異と選抜の操作を繰り返せば、有効な変異が蓄積でき、酵素の機能をさらに向上させることができる。変異と選抜の操作を繰り返して酵素の機能を向上させる技術は、生物が進化するさまに似ていることから、「進化分子工学」と呼ばれている。
【0003】
自然界における変異の種類は、大別して、別の種類の塩基への置き換え(置換)、新たな塩基の挿入(挿入)、塩基の脱落(欠失)の三つに分けられる。このうち、進化分子工学の分野では、主に置換変異が利用される。まず、エラープローンPCR 法(非特許文献1)などで遺伝子にランダムな1塩基置換変異を導入し、機能の変化した変異体を選抜する。取得した変異体には、さらに置換変異を導入するか、変異部位のコドンを部位特異的にランダムに改変するか(サチュレーション変異導入;非特許文献2)、いくつかの変異体の遺伝子をかけあわせて(DNA シャッフリング;非特許文献3)、変異の組み合わせを検討する。そして、これらの作業をルーチン化して、機能をできるだけ向上させるというのが、従来の進化分子工学研究の進め方であった。ただ、機能が無限に向上し続けることはなく、数世代の実験で進化が頭打ちになることも多い。置換変異のバリエーションは、300アミノ酸残基からなるタンパク質において1アミノ酸残基を置換する場合、19×300=5700通りである。このバリエーションの中に、機能を向上させる変異がなければ、置換変異による進化は起こらない。たとえ置換変異の頻度を上げたとしても、それ以上の進化が望めるとは限らない。なぜなら、置換変異はおおむね相加性を持っているが、相乗効果を生む例はまれで、予想外の進化が得られる可能性は低いからである。従って、この5700種の置換変異を探索しつくした時点で、進化は頭打ちになると予想される。
【0004】
一方、もし挿入や欠失をランダムに導入できるなら、置換とはまったく異なる変異体のバリエーションを作製できるため、たとえ進化が頭打ちになっても、さらなる進化を起こさせる可能性がある。例えば、300アミノ酸残基からなるタンパク質の場合、nアミノ酸残基欠失のライブラリーの数は(301-n)通り、nアミノ酸残基挿入のライブラリーの数は(20のn乗×301)通り存在することから、非常に大きなライブラリーを得ることができる。従って、置換変異による進化が限界でも、挿入や欠失を利用することで進化の頭打ちに対してブレークスルーをもたらすこともできると考えられる。
【0005】
しかしながら、挿入や欠失をタンパク質の機能改変に用いた例はきわめて少なく、現代の構造生物学および分子進化学の分野において、挿入や欠失に関しては、いまだ未知の部分が多い。これは、構造変化の予想が難しいからである。もし、挿入や欠失を含む変異体を作成し、構造や機能に関する情報を蓄積できれば、進化分子工学のみならず、構造生物学や分子進化学にも大きな影響を与える可能性を秘めている。
【0006】
挿入や欠失は、置換に比べて構造の変化が大きいため、酵素の構造を著しく壊すかもしれず、タンパク質工学には応用しにくいと考えられがちである。ところが、酵素のアミノ酸配列のデータベースを調査したところ、10アミノ酸残基以下の挿入や欠失は、進化の過程でかなりの確率でおきていることがわかった(非特許文献4)。とりわけ、5アミノ酸残基以下の挿入欠失は頻繁に起こるとされている。これは、挿入や欠失が、タンパク質の機能改変に応用できる可能性を示唆している。
【0007】
これまでDNAに挿入や欠失をランダムに導入する方法は、いくつか開発されている。たとえば、認識配列の外側を切断する特殊な制限酵素を用いて挿入や欠失を導入した例(非特許文献5)、コドン単位で DNA を合成して欠失を導入した例(非特許文献6)、トランスポゾンを利用して特定の15塩基をランダムに挿入した例(非特許文献7)、エンドヌクレアーゼを用いて欠失やリピート配列を導入した例(非特許文献8)、などが挙げられる。これらの方法のうちいくつかは、実際に酵素や機能性タンパク質の向上を実現しており、挿入欠失変異の有効性を示している。しかしながら、いずれの方法も、操作が複雑であったり、導入できる変異の種類や長さが限られていたり、特殊な技術や設備が必要であったりするなどの問題がある。従って、より簡便にDNAに挿入欠失変異を導入する方法が求められていた。
【非特許文献1】Leung, D. W., Chen, E. and Goeddel, D. W., A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction. Techniques, 1, 11-15 (1989).
【非特許文献2】Miyazaki, K. and Arnold, F. H., Exploring nonnatural evolutionary pathways by saturation mutagenesis: Rapid improvement of protein function. J. Mol. Evol., 49, 716-720 (1999).
【非特許文献3】Stemmer, W. P. C., Rapid evolution of a protein in-vitro by DNA shuffling. Nature, 370, 389-391 (1994).
【非特許文献4】Pascarella, S. and Argos, P., Analysis of Insertions Deletions in Protein Structures. J. Mol. Biol., 224, 461-471 (1992).
【非特許文献5】Murakami, H., Hohsaka, T. and Sisido, M., Random insertion and deletion of arbitrary number of bases for codon-based random mutation of DNAs. Nat. Biotechnol., 20, 76-81 (2002).
【非特許文献6】Osuna, J., Yanez, J., Soberon, X. and Gaytan, P., Protein evolution by codon-based random deletions. Nucleic Acids Res., 32, e136 (2004).
【非特許文献7】Hallet, B., Sherratt, D. J. and Hayes, F., Pentapeptide scanning mutagenesis: Random insertion of a variable five amino acid cassette in a target protein. Nucleic Acids Res., 25, 1866-1867 (1997).
【非特許文献8】Pikkemaat, M. G. and Janssen, D. B., Generating segmental mutations in haloalkane dehalogenase: a novel part in the directed evolution toolbox. Nucleic Acids Res., 30, e35 (2002).
【発明の開示】
【発明が解決しようとする課題】
【0008】
本発明は、置換変異のみならず、挿入欠失変異をDNAに簡便に導入する新たな手法を提供することにある。
【課題を解決するための手段】
【0009】
本発明者らは、上記課題を解決すべく鋭意研究を重ねた結果、対象とするDNA鎖をランダムに切断し、その切断断片の3'末端に複数個のランダムな塩基を付加した後、DNAポリメラーゼを用いて該3'末端から伸長反応を行うことで、前記DNA鎖中に複数塩基単位の置換、挿入、および欠失をランダムに導入できることを見出した。本発明はかかる知見により完成されたものである。
【0010】
即ち、本発明は以下の発明を包含する。
(1) 以下の工程を含むDNAへのランダム変異導入方法。
(a) 対象とするDNA鎖をランダムな部位で切断し、DNA断片を得る工程
(b) 得られたDNA断片の3'末端に複数個の塩基を付加する工程
(c) 塩基を付加した3'末端から、外来の鋳型DNA鎖の存在下または非存在下において、DNAポリメラーゼによりDNA鎖を伸長させる工程
(2) 外来の鋳型DNA鎖を除去する工程をさらに含む、(1)に記載の方法。
(3) 外来の鋳型DNA鎖が、含ウラシルDNA、メチル化DNA、末端ビオチン化 DNAのいずれかを含むDNA鎖である、(2)に記載の方法。
(4) 対象となるDNA鎖が、二本鎖または一本鎖である、(1)〜(3)のいずれに記載の方法。
(5) DNA断片の3'末端に付加する塩基が、デオキシリボ核酸またはデオキシリボ核酸類縁体である、(1)〜(4)のいずれかに記載の方法。
(6) DNA断片の3'末端に付加する塩基数が5〜50塩基である、(1)〜(5)のいずれかに記載の方法。
(7) DNA断片の3'末端への塩基の付加が、ターミナルデオキシヌクレオチジルトランスフェラーゼ、DNAポリメラーゼ、DNAリガーゼ、またはRNAリガーゼのいずれかを用いて行われる、(1)〜(6)のいずれかに記載の方法。
【発明の効果】
【0011】
本発明によれば、置換、挿入、欠失といった変異を、わずかに数ステップの簡便な操作で、特別な試薬や機器を使うことなくDNAに導入できる方法が提供される。本発明の方法によれば、挿入や欠失といった、従来の方法では導入することが難しい変異を自在に導入することができ、また、その挿入欠失部位に連続して複数塩基の置換が導入されることが多いため、単なる挿入欠失以上にランダム性が高い変異を導入することができる。また、本発明の方法によれば、進化分子工学に適当な頻度とされる1〜3塩基/kb程度の1塩基置換変異が導入でき、また、複数塩基単位の置換も導入できる。その上、本発明の方法は、DNAを断片化して再構築する操作を含むことから、DNAシャッフリング(もしくはファミリーシャッフリング)の能力も備えている。従って、本発明の方法は、酵素の改変など、タンパク質の機能改変に非常に有用である。
【発明を実施するための最良の形態】
【0012】
本発明のDNAへのランダム変異導入方法は、(a)対象とするDNA鎖をランダムな部位で切断し、DNA断片を得る工程、(b)得られたDNA断片の3'末端に複数個の塩基を付加する工程、(c)塩基を付加した3'末端から、外来の鋳型DNA鎖の存在下または非存在下において、DNAポリメラーゼによりDNA鎖を伸長させる工程を含む(図1A:二本鎖DNAへのランダム変異導入、図1B:一本鎖DNAへのランダム変異導入参照)。以下、各工程について説明する。
【0013】
工程(a)では、変異導入の対象とするDNA鎖をランダムな部位で切断し、DNA断片を得る。切断する方法としては、ヌクレアーゼ処理または超音波破砕処理などが挙げられる。ヌクレアーゼ処理としては、エンドヌクレアーゼを使う方法(DNaseIやエンドヌクレアーゼVなど)、エキソヌクレアーゼを使う方法のいずれであってもよい。
【0014】
対象とするDNA鎖は一本鎖、二本鎖どちらでもよい。二本鎖DNAの場合は平滑末端が望ましいので、T4 DNAポリメラーゼなどで平滑化するか、Mnイオンを加えた条件でDNaseI処理を行うことが好ましい。断片の大きさは特に限定はされないが、10bp以上、好ましくは50bp以上で、対象とするDNA全長の半分以下であることが好ましい。
【0015】
本工程の具体例として、例えばDNaseIを用いたDNA鎖の切断は、以下のようにして行うことができる。以後、濃度はすべて最終濃度を示す。まず、対象とするDNA鎖を、Tris-HCl(pH 6〜8)、1〜100 mM MnCl2中で、1×10-5〜1×10-3 U/μL DNaseIと1〜100 分間反応させる。反応終了後、MnCl2に対して過剰量の EDTA 溶液を加え、反応を停止させた後、キアゲンの MinElute Reaction Cleanup Kit などを用いて精製する。
【0016】
続いて、工程(b)では、工程(a)で得られたDNA断片の3'末端に複数個の塩基を付加する。付加する塩基数は 5〜50塩基が好ましい。DNA断片の3'末端に塩基を付加するための方法としては、ターミナルデオキシヌクレオチジルトランスフェラーゼ(TdT)、DNAポリメラーゼ、DNAリガーゼ(T4 DNAリガーゼなど)、RNAリガーゼ(T4 RNAリガーゼなど)を用いる方法などが挙げられる。ターミナルデオキシヌクレオチジルトランスフェラーゼ(TdT)を用いて3'末端に塩基を付加する場合は、まず、アガロースゲル電気泳動などでDNA断片の平均鎖長を計算し、DNA断片のおよそのモル濃度を計算してから、その10倍〜100倍のdNTPをTdTの反応溶液に加えることで、理論上、平均5〜50bpの塩基を3'末端に付加することができる。また、T4 RNAリガーゼを利用する場合は、対象とするDNA断片を脱リン酸化してから、3'末端と 5'末端をリン酸化した 5〜50 塩基からなるランダムオリゴヌクレオチドを結合させる。
【0017】
DNA断片の3'末端に付加する塩基は、デオキシリボ核酸またはデオキシリボ核酸類縁体のいずれであってもよい。デオキシリボ核酸類縁体としては、例えば、dITPや7-deaza-dGTPなどが挙げられる。
【0018】
本工程の具体例として、例えばTdTを用いた塩基の付加は、以下のようにして行うことができる。1〜100 mM カコジル酸ナトリウム(pH 6〜8)、0.1〜10 mM CoCl2(またはMnCl2)、0.02〜2 mM ジチオスレイトール、0.1〜10 U/μL TdT、0.01〜10 pmol/μL の工程(a)のDNA断片、該DNA断片の5〜50倍濃度のdNTP、を加え、20〜40℃で10分〜10時間放置する。その後、DNAを精製して、工程(b)の最終産物とする。
【0019】
工程(c)では、工程(b)で得られた産物(複数個の塩基を3’末端に付加したDNA断片)の3'末端から、外来の鋳型DNA鎖の存在下または非存在下において、DNAポリメラーゼによりDNA鎖を伸長させる。
【0020】
ここで用いるDNA ポリメラーゼとしては、特に限定はされないが、Taq DNA ポリメラーゼや Deep Vent exo-DNAポリメラーゼなど、3'-5' エキソヌクレアーゼ活性を持たない DNA ポリメラーゼが望ましい。
【0021】
上記の伸長反応は、通常のPCRの手法に従えばよく、PCRの条件(温度設定、サイクル数)も通常採用される条件でよい。
【0022】
対象となるDNA鎖が二本鎖の場合は、以下の操作を行う。まず、工程(b)で得られた3’末端に複数個の塩基(Xn)が付加されたDNA断片を、熱変性により一本鎖DNA断片とする。次に、温度を下げ、産生された任意の一本鎖DNA断片(「X断片」とする)を、それに相補的な配列を有する他の任意の一本鎖DNA断片(「Y断片」とする)に結合(アニーリング)させる。その後、DNA ポリメラーゼを作用させると、Y断片を鋳型としてX断片の3'末端からDNA鎖を伸長させることができる。
【0023】
あるいは、別の態様として、X断片が鋳型となるDNA断片と結合(アニーリング)できる割合を上げるため、外来の鋳型DNA鎖をあらかじめ加えておいてもよい。外来の鋳型DNA鎖としては、対象となる二本鎖DNAの全配列または部分配列からなるDNA鎖を用いる。この場合は、X断片を外来の鋳型DNA鎖にアニーリングさせ、その後、DNAポリメラーゼを作用させると、該鋳型DNA鎖を鋳型としてX断片の3'末端からDNA鎖を伸長させることができる。
【0024】
対象となるDNA鎖が一本鎖の場合は、外来の鋳型DNA鎖の添加が必須となる以外は、上記と同様の操作で伸長反応を行う。まず、工程(b)で得られた3’末端に複数個の塩基が付加された任意の一本鎖DNA断片(「Z断片」とする)に対して相補的な配列を有する外来の鋳型DNA鎖を加える。その後、Z断片をその外来の鋳型DNA鎖にアニーリングさせ、DNA ポリメラーゼを作用させると、該鋳型DNA鎖を鋳型として、Z断片の3'末端からDNA鎖を伸長させることができる。
【0025】
上記の外来の鋳型DNA鎖は、伸長反応終了後に除去できる形状にしておく。このような鋳型DNA鎖として、例えば、含ウラシルDNA、メチル化DNA、ビオチン修飾DNAを含むDNA鎖などを用いることができ、その除去は、それぞれ、ウラシルDNAグリコシダーゼ処理、メチル化DNAを分解する菌体の形質転換、アビジンカラムによる精製より行うことができる。
【0026】
本工程の具体例として、例えば耐熱性DNAポリメラーゼ(New England Biolabs 社のDeep vent exo-DNAポリメラーゼ)を用いた伸長反応は、以下のようにして行うことができる。まず、1〜100 ng/μLの工程(b)の産物を、反応溶液(1〜100 mM KCl, 1〜100 mM (NH4)2SO4, 2〜200 mM Tris-HCl pH 7〜10, 0.2〜20 mM MgSO4, 0.01〜1% Triton X-100, 0.2〜20 mM dNTP, 0.02〜20 U/μL Deep vent exo-DNA ポリメラーゼ)中で、サーマルサイクリング反応を行う。温度条件は、90〜99℃, 10秒〜2分→30〜70℃, 10秒〜2分→60〜80℃, 10秒〜10分で、このサイクルを1〜60 回繰り返す。
【0027】
工程(c)では、3’末端に複数個の塩基が付加されたDNA断片が互いにプライマーとなるようにPCR反応させるので、その3'末端の付加塩基(Xn)と鋳型とのミスマッチに起因する変異(置換、挿入、欠失)導入と、異なったDNA断片どうしの再結合(シャッフリング)が同時に達成できる。
【0028】
工程(c)で得られた産物は通常の手法でクローニングすることができる。得られた産物は、さまざまな長さの置換、挿入、欠失(Xn')が導入されたDNAが含まれる。例えば、DNAポリメラーゼによるDNA鎖の伸長反応の際、3'末端の付加塩基(Xn)に存在するミスマッチ塩基の数に応じて、置換変異が導入される。さらに、このミスマッチ塩基は、相補鎖のさまざまな部分と結合するので、相補する部位にしたがって、欠失や挿入となる。すなわち、相補鎖側が長い場合は欠失、ミスマッチ側が長い場合は挿入になる(図2参照)。
【実施例】
【0029】
以下、実施例によって本発明を更に具体的に説明するが、これらの実施例は本発明を限定するものでない。
(実施例1)DNA 配列へのランダム変異導入
(1) ターゲット遺伝子の調製
pUC19 の2661-2686-1-480 位のDNA配列をターゲット遺伝子として、ランダム変異導入を行った。まず、プラスミドpUC19 上の2661-2686-1-480位のDNA配列をPCR法で増幅した。反応溶液の組成は、20 pg/μL pUC19, 0.02 U/μL TOYOBO社製 KOD plus DNA ポリメラーゼ, 1×添付バッファー, 0.2 mM dNTP, 1 mM MgSO4, 0.3 pmol/μLプライマー1:5'-TAGGCGTATCACGAGGCCCTTTCGTC-3'(配列番号1)およびプライマー2:5'-ATTACGCCAAGCTAGCATGCCTGCAGGTCG-3'(配列番号2)とした。なお、プライマー1は、pUC19の2661-2686位のDNA配列の相補鎖に相当し、プライマー2は、pUC19の431-460位のDNA配列の相補鎖に相当する(ただし447位にA-T変異を持つ)。
【0030】
反応溶液を100 μLずつ10 本の PCR チューブに分注し、PE アプライドバイオシステムス社の Gene Amp 9700 を用いて、94℃, 2分 のプレヒート後、 94℃, 15 秒 → 60℃, 30秒→ 68℃, 30秒の温度条件で 25 回のサーマルサイクリング反応を行った。生成物の100 μL ずつを、キアゲン社の MinElute Reaction Cleanup kit を用いて精製した。DNA 濃度を260 nm の吸光度から算出したところ、670ng/μLであった。
(2) プラスミドベクターの調製
上記のターゲット遺伝子をプラスミドに組み込むためのベクターとして、pUC19 の 431-2680 位のDNA配列をPCR増幅した。PCRの条件としては、プライマー1および2の代わりにプライマー3:5'-ATTACGCCAAGCTAGCATGCCTGCAGGTCG-3'(配列番号3)とプライマー4:5'-AGGGCCTCGTGATACGCCTATTTTTATAGG-3'(配列番号4)を用いる点、温度条件中の68℃で保持する時間を2分にする以外は、前記のターゲット遺伝子の増幅と同じであった。なお、プライマー3は、pUC19の431-460位のDNA配列の相補鎖に相当し(ただし447位にA-T変異を持つ)、プライマー4は、pUC19の2651-2680位のDNA配列の相補鎖に相当する。生成物をキアゲン社の QIAprep PCR Purification kit を用いて精製した。DNA 濃度は64ng/μLであった。
(3) ターゲット遺伝子への変異導入
前記のターゲット遺伝子(pUC19の2661-2686-1-480 位のDNA配列)の増幅産物22μgを、1mLのDNaseI溶液(50 mM Tris-HCl (pH 7.5), 10 mM MnCl2, 0.5 U/mL TaKaRa 社製 DNaseI)に加え、16℃で10分インキュベートすることにより断片化した。反応後、0.5 M EDTAを40μL加えて反応を停止して、MinElute Reaction Cleanup kit を用いて精製し、200μLの精製産物を得た。DNA濃度は12ng/μLであった。2% アガロースゲル上で電気泳動することにより断片の長さを調べたところ、平均 100bp 程度であった。従って、生成物のモル濃度は、約0.18 pmol/μLと推定された。
【0031】
次に、上記で得られた断片(DNaseI 断片)の3'末端に、TdTを用いてランダムな塩基を付加した。反応は、20μLの反応溶液(0.1 pmol/μL DNaseI 断片、1 pmol/μL dNTP、0.25 U/μL TOYOBO社製 TdT、1×添付バッファー)を37℃で1時間インキュベートすることにより行った。反応後、生成物をMinElute Reaction Cleanup kit を用いて精製した。DNA濃度は7ng/μLであった。
【0032】
さらに、この塩基が付加されたDNA断片を用いてDNAポリメラーゼにより伸長反応を行った(1st PCR)。反応は、10μLの反応溶液(5ng/μL 塩基付加断片、0.2U/μL New England Biolabs 社製 Deep Vent exo-DNA ポリメラーゼ、0.2 mM dNTP, 1×添付バッファー)に対し、Gene Amp 9700 を用いて96℃, 2分のプレヒート後、96℃, 30秒→ 60℃, 30秒→75℃, 30秒を1サイクルとするサーマルサイクリングを40回繰り返すことによって行った。
【0033】
次に、1st PCR 産物を1μL分取し、PCR増幅を行った(2nd PCR)。反応は、50μLの反応溶液(2% 1st PCR産物, 0.3 pmol/μL プライマー1および2、0.02 U/μL KOD plus DNA ポリメラーゼ, 1×添付バッファー, 0.2 mM dNTP, 1 mM MgSO4)に対し、Gene Amp 9700 を用いて96℃, 2分のプレヒート後、96℃, 15秒→ 60℃, 30秒→ 68℃, 30秒を1サイクルとするサーマルサイクリングを25回繰り返すことによって行った。その後、2% アガロースゲル電気泳動により、目的産物のバンド (500 bp 付近)を分画し、キアゲン社の MinElute Gel Extraction kit を用いて精製した。この結果、350ng/μLのDNA産物10μLを得た。
【0034】
この 2nd PCR 産物を、オーバーラップ伸長PCR法によりベクターに組み込み、大腸菌を形質転換して、変異体をクローニングした。100μLの反応溶液(10% の2nd PCR産物, 0.2 ng/μL 431-2680 位のDNA配列のPCR 産物、0.02 U/μL KOD plus DNA ポリメラーゼ, 1×添付バッファー, 0.2 mM dNTP, 1 mM MgSO4)を、Gene Amp 9700 を用いて96℃, 2分のプレヒート後、96℃, 15秒→ 60℃, 30秒→ 68℃, 30秒を1サイクルとするサーマルサイクリングを25回繰り返した。その後、この産物1μLを用いて、大腸菌TOP10をエレクロトポレーション法で形質転換し、LB プレート上で、37℃で18時間培養した。
【0035】
形成されたコロニーをピックアップして、アマシャムバイオサイエンス社の TempliPhi Amplification Kit によりプラスミドを増幅し、2661-2686-1-480 位 のDNA 配列を解析した。その結果、全88サンプル中、計22個の特殊な変異(二塩基以上の欠失、挿入、置換)が導入されていることが確認された(表1、2)。また、これらの特殊な変異は、ターゲット遺伝子の配列中で広く分布することがわかった(図3)。従って、本方法によれば、さまざまな長さの挿入、欠失、置換をDNAにランダムに導入できることが確認できた。
【0036】
【表1】
【0037】
【表2】
【図面の簡単な説明】
【0038】
【図1A】本発明のDNAへのランダム変異導入方法の概略(二本鎖DNAの場合)を示す。
【図1B】本発明のDNAへのランダム変異導入方法の概略(一本鎖DNAの場合)を示す。
【図2】本発明のDNAへのランダム変異導入方法におけるDNAポリメラーゼによる再構築の様式を示す。
【図3】本発明方法によりターゲット遺伝子に導入した特殊変異の分布を示す。
【特許請求の範囲】
【請求項1】
以下の工程を含むDNAへのランダム変異導入方法。
(a) 対象とするDNA鎖をランダムな部位で切断し、DNA断片を得る工程
(b) 得られたDNA断片の3'末端に複数個の塩基を付加する工程
(c) 塩基を付加した3'末端から、外来の鋳型DNA鎖の存在下または非存在下において、DNAポリメラーゼによりDNA鎖を伸長させる工程
【請求項2】
外来の鋳型DNA鎖を除去する工程をさらに含む、請求項1に記載の方法。
【請求項3】
外来の鋳型DNA鎖が、含ウラシルDNA、メチル化DNA、末端ビオチン化DNAのいずれかを含むDNA鎖である、請求項2に記載の方法。
【請求項4】
対象となるDNA鎖が、二本鎖または一本鎖である、請求項1〜3のいずれに記載の方法。
【請求項5】
DNA断片の3'末端に付加する塩基が、デオキシリボ核酸またはデオキシリボ核酸類縁体である、請求項1〜4のいずれかに記載の方法。
【請求項6】
DNA断片の3'末端に付加する塩基数が5〜50塩基である、請求項1〜5のいずれかに記載の方法。
【請求項7】
DNA断片の3'末端への塩基の付加が、ターミナルデオキシヌクレオチジルトランスフェラーゼ、DNAポリメラーゼ、DNAリガーゼ、またはRNAリガーゼのいずれかを用いて行われる、請求項1〜6のいずれかに記載の方法。
【請求項1】
以下の工程を含むDNAへのランダム変異導入方法。
(a) 対象とするDNA鎖をランダムな部位で切断し、DNA断片を得る工程
(b) 得られたDNA断片の3'末端に複数個の塩基を付加する工程
(c) 塩基を付加した3'末端から、外来の鋳型DNA鎖の存在下または非存在下において、DNAポリメラーゼによりDNA鎖を伸長させる工程
【請求項2】
外来の鋳型DNA鎖を除去する工程をさらに含む、請求項1に記載の方法。
【請求項3】
外来の鋳型DNA鎖が、含ウラシルDNA、メチル化DNA、末端ビオチン化DNAのいずれかを含むDNA鎖である、請求項2に記載の方法。
【請求項4】
対象となるDNA鎖が、二本鎖または一本鎖である、請求項1〜3のいずれに記載の方法。
【請求項5】
DNA断片の3'末端に付加する塩基が、デオキシリボ核酸またはデオキシリボ核酸類縁体である、請求項1〜4のいずれかに記載の方法。
【請求項6】
DNA断片の3'末端に付加する塩基数が5〜50塩基である、請求項1〜5のいずれかに記載の方法。
【請求項7】
DNA断片の3'末端への塩基の付加が、ターミナルデオキシヌクレオチジルトランスフェラーゼ、DNAポリメラーゼ、DNAリガーゼ、またはRNAリガーゼのいずれかを用いて行われる、請求項1〜6のいずれかに記載の方法。
【図1A】

【図1B】
【図2】
【図3】
【図1B】
【図2】
【図3】
【公開番号】特開2006−262838(P2006−262838A)
【公開日】平成18年10月5日(2006.10.5)
【国際特許分類】
【出願番号】特願2005−88811(P2005−88811)
【出願日】平成17年3月25日(2005.3.25)
【出願人】(501145295)独立行政法人食品総合研究所 (27)
【Fターム(参考)】
【公開日】平成18年10月5日(2006.10.5)
【国際特許分類】
【出願日】平成17年3月25日(2005.3.25)
【出願人】(501145295)独立行政法人食品総合研究所 (27)
【Fターム(参考)】
[ Back to top ]