
DNAグリコシラーゼ/リアーゼおよびAPエンドヌクレアーゼ基質
APエンドヌクレアーゼ、および酵素のグリコシラーゼ/リアーゼファミリーの一員の核酸基質の新たなクラスについて記述する。各々のファミリーの代表、それぞれ酵素Nfoおよびfpgは、様々な標識部分が取り付けられているリンカーによって塩基が置換される位置で核酸主鎖を開裂する。これらの合成基質をオリゴヌクレオチド内に埋め込んで使用すると、多くの用途に有用である。一実施形態において、標的核酸の存在または不在を検出するプロセスが提供され、このプロセスは、(a)オリゴヌクレオチドプローブを標的核酸に接触させ、それによりプローブと標的核酸との間に複合体を形成するステップと、(b)APエンドヌクレアーゼ、DNAグリコシラーゼ/リアーゼおよびDNAグリコシラーゼからなる群より選択したヌクレアーゼにプローブ/核酸複合体を接触させるステップと、(c)切除または開裂が行われたかどうかを検出するステップとを含む。

【発明の詳細な説明】
【技術分野】
【0001】
関連出願
この出願は、2009年5月20日に出願された米国仮特許出願第61/179,793号(これは、参考として本明細書に援用される)に対する優先権の利益を主張する。
【0002】
発明の分野
本発明は、オリゴヌクレオチド基質およびその使用、たとえば核酸増幅反応のためのプローブに関する。
【背景技術】
【0003】
一次配列、主鎖構造、または基本特性(多くの場合、傷害塩基または修飾塩基)により指定される方法で核酸を代謝する酵素は、生物工学的用途に有用である。このような酵素のいくつかのファミリーは、核酸ベースの技術に普通に使用されており、制限エンドヌクレアーゼ、ポリメラーゼ、リガーゼおよびエクソヌクレアーゼが含まれる。さらに、特定の配列ストリングに依存するのではなく、普通でない塩基、傷害塩基または欠落塩基の認識に依存する種々のシングルサブユニットの(非制限)エンドヌクレアーゼについては、これまで長年にわたって記述されてきた。これらの酵素は、おおまかに2つのグループ、つまり一例として大腸菌エンドヌクレアーゼIV(Nfo)および大腸菌エクソヌクレアーゼIIIがあるAPエンドヌクレアーゼと、一例として大腸菌fpg、MUGおよびNthがあるDNAグリコシラーゼ/リアーゼファミリーとに分割することができる。
【0004】
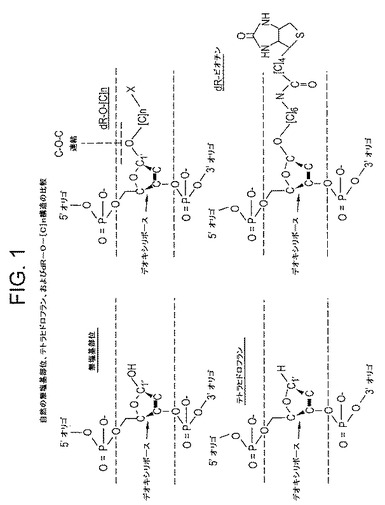
APエンドヌクレアーゼは、二重鎖DNAに関連して発見された場合、無塩基部位(その他の酵素活性も存在し得る)で糖−リン酸主鎖を認識し、開裂する能力に特徴がある。無塩基部位における認識および切開は、グリコシラーゼ/リアーゼファミリーに独特な生化学的な方法で行われ、ベータ脱離またはベータ/デルタ脱離によって行われるのではない。したがって、これらは、真の無塩基部位だけでなく、糖環の’1炭素に酸素原子が欠けているテトラヒドロフラン部分を含むその他の基質も攻撃する。(非特許文献1)(図1の化学構造を参照)。
【0005】
対照的に、fpgタンパク質(8−オキソグアニンDNAグリコシラーゼ、大腸菌fpg、および哺乳動物のOGG1)、またはNthタンパク質(大腸菌のエンドヌクレアーゼIII、ヒトのNth1など)を含むグリコシラーゼ/リアーゼ酵素は、第1に、傷害塩基が、タンパク質とDNAとの間のシッフ塩基の形成を介して認識されて切除され、第2に、このように生成された無塩基部位が、APエンドヌクレアーゼに独特な方法でベータ脱離またはベータ/デルタ脱離によって処理される2段階の触媒作用方法で機能する。この場合、C1’酸素原子はこの無塩基部位の模擬は存在せず、このような位置に酸素が欠如している糖は攻撃に耐性があるため、テトラヒドロフラン(THF)残基はリアーゼ活性の基質ではない(非特許文献1)(図1)。
【0006】
分子生物学的技術におけるAPエンドヌクレアーゼおよびグリコシラーゼ/リアーゼの使用について説明した。1つの用途は、インビトロDNA増幅反応の際に生成される基質を処理するためにこれらの酵素を使用するか、または類似の用途であり、特に、合成オリゴヌクレオチドが、サンプル中の分子に特異的にハイブリダイズする場合に、酵素の基質になる可能性がある修飾糖または塩基を含む合成「プローブ」オリゴヌクレオチドが提供された場合の用途である。このような用途の一例は、特許文献1および非特許文献2に記載されており、テトラヒドロフラン修飾オリゴヌクレオチドは、DNAの増幅を測定する方法で、大腸菌Nfo(エンドヌクレアーゼIV)タンパク質の基質として使用される(Nfoは、大腸菌の2つのAPエンドヌクレアーゼの1つである)。
【0007】
グリコシラーゼ/リアーゼを類似の方法に用途することも想定可される。fpgタンパク質が、反応監視のためのDNA増幅反応の範囲内で8−オキソグアニンなどの修飾塩基を同様に処理する能力について記載されている(特許文献1)。さらに、fpgおよびNthなどのグリコシラーゼ/リアーゼ酵素は3’伸長可能末端を離れないが、むしろブロック3’末端を離れる(触媒モードの違いにより)という事実は、処理プローブがポリメラーゼの対応基質にも、3’ヒドロキシル部分に依存するその他の活性にもなり得ないことを確実にすることが望まれる状況で特に有用であると思われる。
【0008】
これらの酵素は、その電位にも関わらず、ある用途に使用するには適さなくなる特定の特徴を有する。特に、THF残基と違って、主鎖に必要な真の無塩基部位 − DNAリアーゼの切開活性は、生理学的条件下で安定せず、水溶液中で急速に加水分解するため、最も分子的な手順に使用するのには実際的ではない。その代わり、特定の傷害塩基は、リアーゼ活性による主鎖加水分解の前に、無塩基部位を一時的に生成するために、グリコシラーゼ活性のための一次基質として包含して使用することができる。しかし、残念ながら、8−オキソグアニン(fpg)またはチミジングリコール(Nth)などの代表的な傷害塩基類似体は、理想的には、特異塩基の反対側で逆鎖上で対にする必要があるため、むしろ合成が高く付き、プローブに配列要件を課すことになる。原則として、APエンドヌクレアーゼに使用できる一般的なTHF残基に類似する安定した基質を有するものの、グリコシラーゼ/リアーゼ酵素のリアーゼ活性との反応性を維持するとはるかに好都合であると思われる。
【0009】
本明細書では、塩基が欠如しているが、炭素ベースのリンカー[C]nに共有結合する1’酸素原子を含む様々な基質を含むDNA主鎖をfpgタンパク質、および大腸菌(Nfo)のAPエンドヌクレアーゼIVが効果的に開裂することを証明する。このリンカーは、その他の部分、たとえばビオチン、蛍光体およびその他の結合基を結合するために単独で使用することができ、アミン末端リンカーを使用して様々な物質を結合できる場合に特に有用である。意外なことに、この配列を有し、一般にdR−O−[C]nと呼ばれるヌクレオチドは、多くの状況でfpgタンパク質の良好な基質であり、エンドヌクレアーゼIVタンパク質の基質でもあると思われるが、大腸菌エクソヌクレアーゼIIIには比較的不良な基質であると思われる。多くの状況で、特に、核酸検出方法のための検出策の部分など、特にインビトロ反応の範囲内で、dR−O−[C]n基を基質として含むオリゴヌクレオチドを使用することが想定される。この研究に使用するリンカーの長さは、市販の一定のヌクレオチドで使用可能であるように6炭素原子であるが、多様な炭素鎖の長さを使用することができ、後続の立体容積が小さく、酵素に対する十分な可塑性をこれらの構造に与える炭素−酸素−炭素構造であると想定される。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】米国特許第7,435,561号明細書
【非特許文献】
【0011】
【非特許文献1】Takeshita等、J Biol Chem.(1987年)262(21):1017l
【非特許文献2】Piepenburgら、PlosBiology、(2006年)4(7):e204
【発明の概要】
【課題を解決するための手段】
【0012】
本発明は一部には、APエンドヌクレアーゼ、DNAグリコシラーゼ、DNAグリコシラーゼ/リアーゼ、たとえばfpgおよびNfoタンパク質が、塩基が糖のC1’位に存在しないが、当該位置に酸素原子を保持しているdR−O−[C]n残基を含む部位でDNA主鎖の切断を触媒することが可能であるという発見に関する。酸素原子は、n個(たとえば1〜8)の炭素原子(つまり、[C]n)の炭素リンカーの炭素原子に糖を架橋する。したがって、市販のホスホルアミダイトを使用することにより、dR−O−[C]n残基を含む核酸プローブを構成することができ、相補的核酸と二本鎖を形成する場合、APエンドヌクレアーゼおよびDNAグリコシラーゼ/リアーゼ酵素のための基質でよい。様々な部分は、蛍光体、および特定の標的核酸が存在する証拠であるプローブの上手くいった処理を検出するための多くの方法を提示するその他の標識を含むdR−O−[C]nのリンカー部分に結合することができる。出願人は、蛍光分子およびクエンチャー(quencher)を使用し、fpgもしくはNfo、またはその他の可能なAPエンドヌクレアーゼもしくはリアーゼの標的部位としてdR−O−[C]nを使用してどのようにプローブを構成することができるかを示す。出願人は、dR−O−[C]n基質のその他の用途を他の検出計画で想定している。たとえば、dR−O−[C]n残基は、ヌクレアーゼの活性が標識を遊離させた直後、または後続のプロセスを介するか、もしくは結合体化状態と遊離状態との測定可能な相違を介して検出することが可能な検出可能な標識に結合体化し得る。
【0013】
一態様では、二本鎖をAPエンドヌクレアーゼ、DNAグリコシラーゼ、またはDNAグリコシラーゼ/リアーゼから選択されるヌクレアーゼと接触させることにより、核酸とともに二本鎖を形成するdR−O−[C]n残基を含むオリゴヌクレオチドを開裂するプロセスを本明細書で提供する。実施態様によっては、ヌクレアーゼは、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である。実施態様によっては、リンカーは3〜6炭素原子リンカー(たとえば、6炭素原子リンカー)である。実施態様によっては、オリゴヌクレオチドはその3’末端でブロックされて、ポリメラーゼ伸長を防止する。実施態様によっては、リンカーは、検出可能な標識(たとえば、ビオチン、ジオキシゲニン、ペプチド、蛍光体、クエンチャー、抗体または量子ドット)に結合体化する。
【0014】
実施態様によっては、このプロセスは、オリゴヌクレオチドと核酸とを接触させて、オリゴヌクレオチド/核酸二本鎖を形成するステップをさらに含む。実施態様によっては、これは、オリゴヌクレオチドを核酸にハイブリダイズさせることを含む。実施態様によっては、これは、(i)オリゴヌクレオチドをリコンビナーゼと接触させて、リコンビナーゼ/オリゴヌクレオチド複合体を形成するステップと、(ii)リコンビナーゼ/オリゴヌクレオチド複合体を核酸に接触させて、オリゴヌクレオチド/核酸二本鎖を形成するステップとを含む。実施態様によっては、核酸は、核酸増幅反応(たとえば、リコンビナーゼポリメラーゼ増幅(RPA)プロセスまたはポリメラーゼ鎖反応(PCR))の生成物である。
【0015】
実施態様によっては、このプロセスは、オリゴヌクレオチドの開裂を検出するステップをさらに含む。実施態様によっては、この検出はリアルタイムで監視される。実施態様によっては、実施態様によっては、検出は、反応の終点で監視される。
【0016】
実施態様によっては、オリゴヌクレオチドは蛍光体およびクエンチャーを含み、蛍光体またはクエンチャーの一方は炭素リンカーに結合体化する。ヌクレアーゼ活性は、結合体化した蛍光体またはクエンチャーをオリゴヌクレオチドから切除し、検出ステップは、結合体化状態と遊離状態との間の蛍光性に差がある場合、その差の測定を含む。
【0017】
別の態様では、標的核酸の存在または不在を検出するプロセスを本明細書で提供する。このプロセスは、以下のステップを含む。(a)dR−O−[C]n残基またはヌクレオチドを含むオリゴヌクレオチドプローブを標的核酸と接触させて、プローブ/核酸二本鎖を形成する、(b)二本鎖をAPエンドヌクレアーゼ、DNAグリコシラーゼ、またはDNAグリコシラーゼ/リアーゼから選択したヌクレアーゼと接触させて、リンカーを複合体から切除するか、および/またはプローブをdR−O−[C]nヌクレオチドにおいて特異的に開裂する、並びに(c)こうした切除または開裂が行われたかどうかを検出する。実施態様によっては、核酸は、核酸増幅反応(たとえば、リコンビナーゼポリメラーゼ増幅(RPA)プロセスまたはポリメラーゼ鎖反応(PCR)の生成物である。実施態様によっては、増幅反応はリアルタイムで監視される。実施態様によっては、増幅反応は反応の終点で監視される。
【0018】
実施態様によっては、二本鎖は、プローブを核酸にハイブリダイズさせることによって形成される。実施態様によっては、二本鎖は(i)プローブをリコンビナーゼに接触させて、リコンビナーゼ/プローブ複合体を形成する、(ii)リコンビナーゼ/プローブ複合体を核酸に接触させて、プローブ/核酸二本鎖を形成する、ことによって形成される。
【0019】
実施態様によっては、ヌクレアーゼは、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である。実施態様によっては、リンカーは、3〜6炭素原子リンカー(たとえば、6炭素原子リンカー)である。実施態様によっては、オリゴヌクレオチドは、3’末端でブロックされて、ポリメラーゼ伸長を防止する。実施態様によっては、リンカーは、検出可能な標識(たとえばビオチン、ジオキシゲニン、ペプチド、蛍光体、クエンチャー、抗体または量子ドット)に結合体化される。
【0020】
実施態様によっては、プローブは蛍光体およびクエンチャーを含み、蛍光体またはクエンチャーの一方が炭素リンカーに結合体化される。たとえば、蛍光体およびクエンチャーは、プローブ中の4〜6個の塩基によって分離される。実施態様によっては、炭素リンカーに結合体化されない蛍光体またはクエンチャーは、プローブの末端(たとえば、5’末端)に結合体化される。ヌクレアーゼ活性は、dR−O−[C]n残基に関連して結合体化された蛍光体またはクエンチャーをプローブから切除して遊離させ、検出ステップは、結合体化状態と遊離状態との間の蛍光性に差がある場合、その差を測定することを含む。
【0021】
別の態様では、dR−O−[C]n残基を含むオリゴヌクレオチドプローブを本明細書で提供する。実施態様によっては、プローブは、30〜60ヌクレオチド長であり、10ヌクレオチド以下(たとえば、4〜6ヌクレオチド)で分離された蛍光体クエンチャー対を含み、蛍光体またはクエンチャーの何れかがdR−O−[C]n残基に結合体化する。実施態様によっては、リンカーは、3〜6炭素原子リンカー(たとえば、6炭素原子リンカー)である。実施態様によっては、オリゴヌクレオチドは、3’末端でブロックされて、ポリメラーゼ伸長を防止する。実施態様によっては、dR−O−[C]n残基に結合体化しない蛍光体またはクエンチャーは、プローブの末端(たとえば、5’末端)に結合体化する。実施態様によっては、プローブは、30〜40ヌクレオチド(たとえば、35ヌクレオチド)長である。
【0022】
さらに別の態様では、(i)dR−O−[C]n残基を含むオリゴヌクレオチドと、(ii)APエンドヌクレアーゼ、DNAグリコシラーゼ、またはDNAグリコシラーゼ/リアーゼから選択したヌクレアーゼとを含むキットを本明細書で提供する。実施態様によっては、ヌクレアーゼは、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である。
【0023】
さらに他の態様では、dR−O−[C]n残基およびAPエンドヌクレアーゼ、DNAグリコシラーゼ、またはDNAグリコシラーゼ/リアーゼから選択したヌクレアーゼ(たとえば、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg))を含有するオリゴヌクレオチドを含む反応混合物を本明細書で提供する。実施態様によっては、リンカーは、3〜6炭素原子リンカー(たとえば、6炭素原子リンカー)である。実施態様によっては、オリゴヌクレオチドは、3’末端でブロックされてポリメラーゼ伸長を防止する。実施態様によっては、リンカーは、検出可能な標識(たとえば、ビオチン、ジオキシゲニン、ペプチド、蛍光体、クエンチャー、抗体または量子ドット)に結合体化する。実施態様によっては、反応混合物は冷凍乾燥、つまり凍結乾燥される。
【0024】
実施態様によっては、反応混合物は容器をさらに含む。たとえば、反応混合物は、チューブまたは多ウェル容器のウェル内に含むことができる。反応混合物は乾燥させて、ビードもしくはストリップなどの可動性固体支持体、またはウェル上に取り付けることができる。
【0025】
実施態様によっては、反応混合物は、オリゴヌクレオチドに対して相補的である配列を含む標的またはテンプレート核酸をさらに含む。
【0026】
本発明のその他の実施態様、目的、態様、特徴および利点は、添付の明細書および請求の範囲から明らかになる。実施態様が、本発明の異なる態様に関して説明されている場合でも、適切であれば、本発明の任意の実施態様を本発明の1つまたは複数の他の実施態様と結合できることを意図する。
【図面の簡単な説明】
【0027】
【図1】図1は、以下:1’炭素位置にヒドロキシルを含む通常の無塩基部位の化学構造、1’位に水素を含むテトラヒドロフラン(THF)残基の化学構造、DNAリボース基のC1’と、結合したマーカー部分に対するリンカーとの間の炭素−酸素−炭素架橋の位置を示す、一般的なdR−O−[C]n基の化学構造、および最後に、実施例1に使用され、かつdR−O−[C]n構造に適合するdR−ビオチンヌクレオチドの化学構造を記載する。
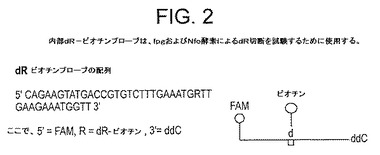
【図2】図2は、dR−ビオチンプローブの設計である:プローブ配列に一致する標的配列が増幅されるRPA反応の際に、Nfoおよびfpgタンパク質の開裂活性を評価するために使用されるオリゴヌクレオチドプローブの配列および配置図。オリゴヌクレオチドの配列が指示されている。プライマーは、5’末端にFAM蛍光体で標識付けされ、配列本体内にdR−ビオチンを含み、2’、3’ジオキシチジン残基によりブロックされる。
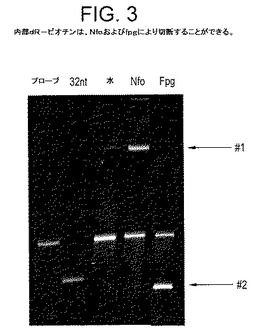
【図3】図3は、ヌクレアーゼがないか、またはNfoもしくはfpg酵素を含む増幅反応の比較である:両方の酵素が、開裂生成物、Nfoの場合、開裂生成物の伸長によって生じる生成物を迅速に移動させるdR−ビオチン部分を処理することができることを明らかにする。
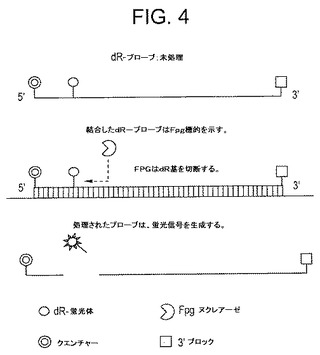
【図4】図4は、オリゴヌクレオチドプローブの設計である:オリゴヌクレオチド本体(この場合、35ヌクレオチド長)、5’クエンチャー修飾(この場合、5’−BHQ1)、クエンチャーに近接する内部dR−蛍光体ヌクレオチド(この場合、dR−FAMオリゴヌクレオチド位置6)、および3’ポリメラーゼ伸長ブロックを含むプローブ設計の例。
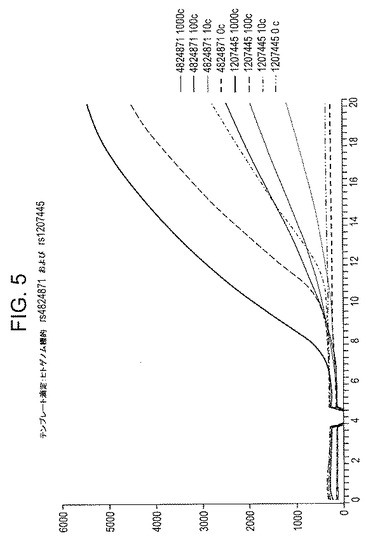
【図5】図5は、感受性および特性を示す:DNA dR−プローブを使用して、指示されたヒトゲノム標的について2回のテンプレート滴定実験をリアルタイムで蛍光監視した結果。どちらの場合も、蛍光信号の増加(0〜3分の基準線に対して)は、テンプレートを含む反応でのみ観察され、非テンプレート対照では観察されない。信号増加の開始時間は、開始テンプレートの量(1000、100または10コピー)と相関する。反応時間は分(X軸)、蛍光は任意の蛍光単位(Y軸)である。
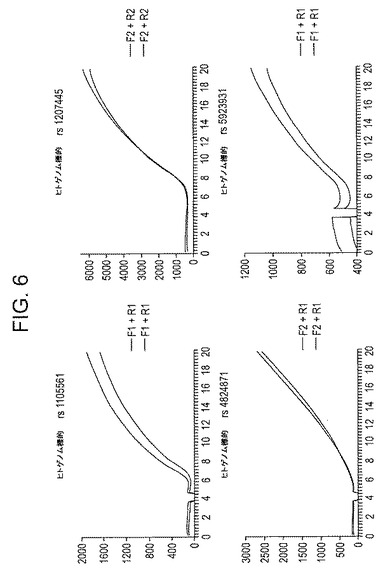
【図6】図6は、様々なプローブの性能である:図2に概略を示す設計のDNAオリゴヌクレオチドプローブを用いて、指示されたヒトゲノム標的に関するRPA反応の4セット(重複)をリアルタイムで監視した結果。6〜8分の蛍光の増加は、プローブのdR基(この場合、dR−FAM)のfpg依存処理から生じ、DNA増幅が進行しており、その結果標的DNAテンプレートの存在を示す。反応時間は分(X軸)、蛍光は任意の蛍光単位(Y軸)である。
【発明を実施するための形態】
【0028】
本発明の1つまたは複数の実施態様の詳細は、付随する以下の説明に記載する。本明細書に記載されている方法および材料に類似するか、または等価な任意の方法および材料を実際に、または本発明の試験に使用することができるが、この方法および材料について説明する。本発明のその他の特徴、目的および利点は、本明細書から明らかであろう。本明細書では、単数形は、文脈上そうではないことが明らかではない限り、複数形も含む。特記しない限り、本明細書で使用する技術用語および科学用語はすべて、本明細書が属する技術分野の当業者が一般に理解する意味と同じ意味を有する。矛盾する場合、本明細書が優先される。
【0029】
実験室で分析および操作に使用する酵素と合成基質の組合せは先行技術で公知である。グリコシラーゼfpgおよびNthなどのDNA修復エンドヌクレアーゼ、並びに大腸菌エクソヌクレアーゼIIIおよびエンドヌクレアーゼIVなどのAPエンドヌクレアーゼは、この組合せの良い例である。現在のオリゴヌクレオチド合成法、およびDNAプライマーに組み込まれる既存の多様な合成ヌクレオチドを使用することにより、これらのDNA修復酵素のための合成基質を生成することは容易である。これらのDNA修復酵素は様々な目的で容易に使用することができ、最近開拓された1つの目的は、等温リコンビナーゼポリメラーゼ増幅(RPA)反応の監視を支援する剤として使用することである。
【0030】
RPAは、DNAに対するオリゴヌクレオチドのリコンビナーゼ媒介標的が、ポリメラーゼによりDNA合成に結合するプロセスである(米国特許第7,270,981 B2号;米国特許第7,399,590号;米国特許第7,435,561 B2号;米国特許第7,485,428 B2号;米国特許第 7,666,598 B2号および海外の対応特許)。RPAは、細胞DNA複製および修復機構の構成要素によって決まり、特定のクラウディング剤の存在で達成される高レベルの再結合活性を維持する適切な速度のリコンビナーゼのロードおよびアンロードの両方を有する「動的」再結合環境の確立に依存する。RPAは、PCRの感受性、特異性およびその他ほとんどの特徴を結合し、熱サイクリングの必要がなく、異例の速度、および異常温度設定に対する頑健性という利点を有する。RPAは、熱安定性の等価物が必要であるか、または単鎖DNA結合タンパク質などの付属タンパク質、RPA反応の天然成分がない場合は調節が不良であることが実証されているため、他のプロセスには利用されていなかった多様な核酸処理酵素、たとえば公知の修復エンドヌクレアーゼの潜在的な使用から既に利益が得られている。
【0031】
簡潔に述べると、RPAは以下のステップを含む:第1に、リコンビナーゼ因子が第1および第2核酸プライマーと接触して、第1および第2核タンパク質プライマーを形成する。第2に、第1および第2核タンパク質プライマーは、二本鎖標的配列と接触して、前記第1鎖の第1部分に第1二本鎖構造を形成し、前記第2鎖の第2部分に二本鎖構造を形成し、よって前記第1核酸プライマーおよび前記第2核酸プライマーの3’末端は、所定のテンプレートDNA分子上で互いに対して方向付けられる。第3に、前記第1および第2核タンパク質プライマーの3’末端はDNAポリメラーゼによって伸長され、第1および第2二本鎖核酸、および核酸の第1および第2変位鎖を生成する。最後に、第2および第3ステップは、所望の程度の増幅に達するまで繰り返される。
【0032】
以前の研究は、RPA法のためのプローブシステムの開発で、合成ヌクレオチドテトラヒドロフラン(THF)がきわめて有用であることを実証している(Piepenburg等、2006年;米国特許第7,435,561 B2号)。この塩基類似体は、しばしば無塩基部位を模倣するために使用され、安定しているという自然な利点を有し、天然の無塩基部位の1’ヒドロキシルを水素原子に置き換えることによりヌクレオチドは安定し、瞬間的な開環およびオリゴヌクレオチドの分裂が生じなくなる。この類似体は容易に入手可能であり、オリゴヌクレオチドに組み込むのも安価である。しかし、生化学的機序の違いにより、大腸菌APエンドヌクレアーゼNfoおよびExoIIIは合成プライマーのこうしたTHF残基で開裂することが可能である一方、他のDNAグリコシラーゼ/リアーゼは開裂することができない。これらの後者の酵素は通常、傷害塩基(グリコシラーゼ活性)および/またはヒドロキシル基が糖の1’位(リアーゼ活性)に存在する必要があり、THFはその酵素活性に対して完全に不活性である。これらのグリコシラーゼ/リアーゼ酵素は、プローブがRPA、またはその他の状況および方法で標的DNA蓄積に応じて処理される反応などのインビトロ反応にも有用なツールになり得るため、多少厄介になる。より自然な基質、たとえばfpg用の8−オキソグアニンは、オリゴヌクレオチド内に挿入して、これらのグリコシラーゼ/リアーゼの開裂部位を生成することができるが、これらの修飾は一般に高価であり、さらに、修飾ヌクレオチドに対向することが可能な塩基を制限することが多い。比較的安価で、さらに一般的であり、これらの酵素の基質であるヌクレオチド修飾は、非常に有用であると思われる。
【0033】
多くの異常な塩基類似体のDNA修復酵素の基質としての効果を調査するために、完全に塩基が欠如しているが、糖の1’位に炭素−酸素−炭素連結を保持するヌクレオチドを含有するオリゴヌクレオチドを合成した。このようなヌクレオチド試薬は容易に入手可能で安価であり、一般に、蛍光体またはビオチンなどの標識基をオリゴヌクレオチドの本体内の複数のオリゴヌクレオチド内に組み込むために使用される。一般に、酸素を介して糖の1’炭素に結合する炭素原子は、多くの場合、最終的に標識基、アミン、または試薬が容易に結合し得るその他の化学的部分(たとえば、チオ)で終端するリンカーの第1炭素原子である。こうした試薬は、文献にはdR−Xと記述されることが多く、この場合、dRはデオキシリボースを意味し、Xはリンカーアミン、リンカー蛍光体、リンカービオチン、またはいくつかのその他の基または標識である。修復エンドヌクレアーゼが、塩基は欠如しているが、炭素−酸素−炭素共有結合を1’糖位置に保持する構造を認識するかどうか、以前に調査されたことはなかった。ヒドロキシルの欠如は、リアーゼの開環プロセスが、リンカー基を事前に処理し、それに関連して切除することなく実施すべきではないことを意味する。公知のグリコシラーゼは、通常、異常な炭素リンカーではなく傷害塩基に作用するので、これらのdR−O−[C]n基はfpgなどのDNAグリコシラーゼ/リアーゼの基質であろうという先例はなかった。
【0034】
図1は、dR−O−[C]n基の一般的な構造、特に、本明細書で使用され、ベルギーのEurogentecが市販するオリゴヌクレオチド中に組み込まれるdR−ビオチン試薬の構造を示す。本明細書で使用するこれらの試薬は、1’糖と、オリゴヌクレオチド合成の前または後にその他の試薬を結合するために使用されることが多い窒素原子との間に一般的な6炭素原子リンカーを有する。この研究では、dR−ビオチンオリゴヌクレオチドのビオチン部分は、この窒素原子を介してアミド結合として結合し、さらに4炭素原子リンカーを通して結合する。この研究に使用されるその他の標識試薬 − dR−FAMおよびdR−Texas Red − は同様に配置され、この場合、蛍光体は6炭素原子リンカーの末端でアミド結合を通して結合する。
【0035】
検出可能な標識は、現在の方法で検出できる任意の部分と定義される。これらの標識は、少なくとも蛍光体(蛍光分子、蛍光色素とも呼ばれる)、酵素、クエンチャー、酵素抑制物質、放射性標識、結合対の一員、ジオキシゲニン残基、ペプチド、およびこれらの組合せを含む。
【0036】
「結合対の一員」とは、第1部分および第2部分の一方を意味し、前記第1部分および第2部分は互いに特異的な結合親和性を有する。本発明に使用するのに適する結合対としては、抗原/抗体(たとえば、ジゴキシゲニン/抗−ジゴキシゲニン、ジニトロフェニル (DNP)/抗−DNP、ダンシル−X−抗−ダンシル、フルオロセイン/抗−フルオロセイン、ルシファーイエロー/抗−ルシファーイエロー、ペプチド/抗−ペプチド、リガンド/受容体およびローダミン/抗−ローダミン)、ビオチン/アビジン(またはビオチン/ストレプトアビジン)およびカルモジュリン結合タンパク質(CBP)/カルモジュリンが挙げられるが、これらだけに限らない。その他の適切な結合対としては、ポリペプチド、たとえばFLAG−ペプチド(DYKDDDDK)[Hopp 等、BioTechnology,6:1204 1210(1988年);KT3エピトープペプチド(Martin 等、Science 255:192 194(1992年));チューブリンエピトープペプチド(Skinner等、J.Biol.Chem 266:15163 15166(1991年));およびT7遺伝子10タンパク質ペプチドタグ(Lutz−Freyermuth 等、Proc.Natl.Acad.Sci.USA,87:6393 6397(1990年))並びにこれらに対する抗体が挙げられる。一般に、好ましい実施態様では、結合対パートナーのより小さい方は、立体配置に関する考察が重要な場合があるため、検出可能な標識として役立つ。
【0037】
一態様では、二本鎖をAPエンドヌクレアーゼ、DNAグリコシラーゼ、またはDNAグリコシラーゼ/リアーゼから選択したヌクレアーゼに接触させることによって、核酸とともに二本鎖を形成するdR−O−[C]n残基を含むオリゴヌクレオチドを開裂するプロセスを本明細書で提供する。実施態様によっては、ヌクレアーゼは、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である。
実施態様によっては、リンカーは、3〜6炭素原子リンカー(たとえば、6炭素原子リンカー)である。実施態様によっては、オリゴヌクレオチドは、3’末端でブロックされて、ポリメラーゼ伸長を防止する。実施態様によっては、リンカーは、検出可能な標識(たとえばビオチン、ジオキシゲニン、ペプチド、蛍光体、クエンチャー、抗体または量子ドット)に結合体化される。
【0038】
実施態様によっては、このプロセスは、オリゴヌクレオチドを核酸と接触させてオリゴヌクレオチド/核酸二本鎖を形成するステップをさらに含む。実施態様によっては、これは、オリゴヌクレオチドを核酸にハイブリダイズさせることを含む。実施態様によっては、これは、(i)オリゴヌクレオチドをリコンビナーゼと接触させてリコンビナーゼ/オリゴヌクレオチド複合体を形成するステップと、(ii)リコンビナーゼ/オリゴヌクレオチド複合体を核酸と接触させて、オリゴヌクレオチド/核酸二本鎖を形成するステップとを含む。実施態様によっては、核酸は、核酸増幅反応(たとえば、リコンビナーゼポリメラーゼ増幅(RPA)プロセスまたはポリメラーゼ鎖反応(PCR))の生成物である。
【0039】
実施態様によっては、このプロセスは、オリゴヌクレオチドの開裂を検出するステップをさらに含む。実施態様によっては、検出はリアルタイムで監視される。実施態様によっては、検出は、反応の終点で監視される。
【0040】
実施態様によっては、オリゴヌクレオチドは蛍光体およびクエンチャーを含み、蛍光体またはクエンチャーの一方は炭素リンカーに結合体化する。ヌクレアーゼ活性は、結合体化した蛍光体またはクエンチャーをオリゴヌクレオチドから切除し、検出ステップは、結合体化状態と遊離状態との間の蛍光性に差がある場合、その差を測定することを含む。
【0041】
別の態様では、標的核酸の存在または不在を検出するプロセスを本明細書で提供する。このプロセスは、以下のステップを含む:(a)dR−O−[C]n残基またはヌクレオチドを含むオリゴヌクレオチドプローブを標的核酸に接触させて、プローブ/核酸二本鎖を形成するステップと、(b)二本鎖をAPエンドヌクレアーゼ、DNAグリコシラーゼ、またはDNAグリコシラーゼ/リアーゼから選択したヌクレアーゼと接触させて、リンカーを複合体から切除するか、および/またはプローブをdR−O−[C]nヌクレオチドで特異的に開裂するステップと、(c)こうした切除または開裂が生じたかどうかを検出するステップ。実施態様によっては、核酸は、核酸増幅反応(たとえば、リコンビナーゼポリメラーゼ増幅(RPA)プロセスまたはポリメラーゼ鎖反応(PCR))の生成物である。実施態様によっては、増幅反応はリアルタイムで監視される。実施態様によっては、増幅反応は反応の終点で監視される。
【0042】
実施態様によっては、二本鎖は、プローブを核酸にハイブリダイズさせることによって形成される。実施態様によっては、二本鎖は、(i)プローブをリコンビナーゼに接触させて、リコンビナーゼ/プローブ複合体を形成する、(ii)リコンビナーゼ/プローブ複合体を核酸に接触させて、プローブ/核酸二本鎖を形成する、ことによって形成される。
【0043】
実施態様によっては、ヌクレアーゼは、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である。実施態様によっては、リンカーは、3〜6炭素原子リンカー(たとえば、6炭素原子リンカー)である。実施態様によっては、オリゴヌクレオチドは、3’末端でブロックされて、ポリメラーゼ伸長を防止する。実施態様によっては、リンカーは、検出可能な標識(たとえばビオチン、ジオキシゲニン、ペプチド、蛍光体、クエンチャー、抗体または量子ドット)に結合体化される。
【0044】
実施態様によっては、プローブは蛍光体およびクエンチャーを含み、蛍光体またはクエンチャーの一方が炭素リンカーに結合体化される。たとえば、蛍光体およびクエンチャーは、プローブ中の4〜6個の塩基によって分離される。実施態様によっては、炭素リンカーに結合体化されない蛍光体またはクエンチャーは、プローブの末端(たとえば、5’末端)に結合体化される。ヌクレアーゼ活性は、dR−O−[C]n残基に関連して結合体化された蛍光体またはクエンチャーをプローブから切除して遊離させ、検出ステップは、結合体化状態と遊離状態との間の蛍光性に差がある場合、その差を測定することを含む。
【0045】
別の態様では、dR−O−[C]n残基を含むオリゴヌクレオチドプローブを本明細書で提供する。実施態様によっては、プローブは、30〜60ヌクレオチド長であり、10ヌクレオチド以下(たとえば、4〜6ヌクレオチド)で分離された蛍光体クエンチャー対を含み、蛍光体またはクエンチャーの何れかがdR−O−[C]n残基に結合体化する。実施態様によっては、リンカーは、3〜6炭素原子リンカー(たとえば、6炭素原子リンカー)である。実施態様によっては、オリゴヌクレオチドは、3’末端でブロックされて、ポリメラーゼ伸長を防止する。実施態様によっては、dR−O−[C]n残基に結合体化されない蛍光体またはクエンチャーは、プローブの末端(たとえば、5’末端)に結合体化される。実施態様によっては、プローブは、30〜40ヌクレオチド(たとえば、35ヌクレオチド)長である。
【0046】
さらに別の態様では、(i)dR−O−[C]n残基を含むオリゴヌクレオチドと、(ii)APエンドヌクレアーゼ、DNAグリコシラーゼ、またはDNAグリコシラーゼ/リアーゼから選択したヌクレアーゼとを含むキットを本明細書で提供する。実施態様によっては、ヌクレアーゼは、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である。
【0047】
さらに他の態様では、dR−O−[C]n残基、およびAPエンドヌクレアーゼ、DNAグリコシラーゼ、またはDNAグリコシラーゼ/リアーゼから選択したヌクレアーゼ(たとえば、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg))を含有するオリゴヌクレオチドを含む反応混合物を本明細書で提供する。実施態様によっては、リンカーは、3〜6炭素原子リンカー(たとえば、6炭素原子リンカー)である。実施態様によっては、オリゴヌクレオチドは、3’末端でブロックされて、ポリメラーゼ伸長を防止する。実施態様によっては、リンカーは、検出可能な標識(たとえばビオチン、ジオキシゲニン、ペプチド、蛍光体、クエンチャー、抗体または量子ドット)に結合体化される。実施態様によっては、反応混合物は冷凍乾燥、つまり凍結乾燥される。
【0048】
実施態様によっては、反応混合物は容器をさらに含む。たとえば、反応混合物は、チューブまたは多ウェル容器のウェル内に含むことができる。反応混合物は乾燥させるか、またはビードもしくはストリップなどの可動性固体支持体、またはウェル上に取り付けることができる。
【0049】
実施態様によっては、反応混合物は、オリゴヌクレオチドに対して相補的である配列を含む標的またはテンプレート核酸をさらに含む。
【0050】
図2は、プローブに指定される配列を含むDNA分子を標的として使用して、RPAのDNA増幅反応に使用するために生成されるdR−ビオチンプローブの一次配列、およびおおまかなその性質の両方を示す。プローブはブロックされ(増幅段階でポリメラーゼ伸長を防止するため)、内部dR−ビオチンを酵素の試験基質として含む。プローブは、5’−FAMも含む。したがって、原則として、DNAが、このプローブを含む反応で増幅される場合、プローブが、増幅時に形成された相補的単鎖に対する「従来の」ハイブリダイゼーションにより、またはリコンビナーゼ媒介プロセスにより、増幅されたDNAと特異的に結合して相互作用する可能性がある。こうした実験の結果を図3に示し、以下の実施例1で説明する。
【0051】
第2セットの実験は、この開裂活性の普遍性を調査するために実施され、この場合、蛍光試薬を使用し、dR−O−[C]nヌクレオチドは、図4に示すように蛍光体に結合する。この場合、dR−蛍光体は、オリゴヌクレオチドプローブの5’末端付近に位置し、その5’末端に結合するクエンチャーにきわめて接近して位置する。従来どおり、プローブの3’末端は異常な伸長またはプライマーのアーティファクトを防止するために適切にブロックされる。図4に示すように、プローブは、相補的増幅材料とハイブリダイズする場合、fpg(またはNfo)の基質になる可能性があり、この場合、この位置で主鎖を開裂することができる(そして、グリコシラーゼ活性がfpgまたはその他の非APエンドヌクレアーゼ酵素中に存在する場合、オリゴヌクレオチドの断片から分離した水媒体中に潜在的に蛍光体を直接遊離させる)。開裂が生じる場合、蛍光体およびクエンチャーが物理的に分離し、その結果、大腸菌NfoまたはexoIIIタンパク質を使用してTHFベースの蛍光プローブに関して前に説明した方法に類似する方法で、検出可能な蛍光が増加する。図5および図6はこうした実験の結果を示し、公知の単一ヌクレオチド多型(SNP)領域を特異的に対象としたプライマーおよびプローブを使用して、RPA反応をヒトゲノム標的に関して実施した実施例2で説明されている。
【0052】
これらの実験は、dR−O−[C]n基はNfoおよびfpgヌクレアーゼの基質であることを全体的に明確に証明する。さらに、相補的核酸鎖が蓄積して二本鎖の形成が可能になり、それによって、増幅が蛍光またはその他の機序によって生じたかどうかの判断を可能にする状況でのみ、プローブのヌクレアーゼ活性が生じるような基を含むプローブを構成することが可能である。したがって、これらのdR−O−[C]nヌクレオチド試薬は、fpg、Nfoまたはグリコシラーゼ/リアーゼ、および多様な用途の等価な酵素と組み合わせて広く応用することができる。
【0053】
配列に関するすべての引用、言及、特許、特許出願、その他の引用文献は、引用することにより本明細書に包含する。
【実施例】
【0054】
本発明について、以下の実施例でさらに定義する。これらの実施例は、本発明の好ましい実施態様を示しているが、具体的に説明するために記載されているにすぎないと考えるべきである。上記の説明およびこれらの実施例から、当業者は、本発明の本質的な特徴を確認し、本発明の精神および範囲から逸脱することなく、本発明に様々な変更および修正を加えて様々な用途および条件に適合させることができる。
【0055】
実施例1
内部dR−ビオチンを含むオリゴヌクレオチドプローブは、Nfoおよびfpgにより切断される。
【0056】
この実施例では、図2に大まかに示され、内部dR−ビオチンを含むオリゴヌクレオチドプローブは、Nfoおよびfpgにより切断することができる。この反応(全量150μL)は新鮮な試薬から混合し、75分間にわたって37℃で培養する。使用した条件は、50mMのトリスアセテート(pH 7.9)、14mMのMg−アセテート、100 mMのカリウムアセテート、2mMのDTT、それぞれ200nMのdNTP、6%PEG 35,000、3mMのATP、50mMのクレアチンリン酸、900ng/μLのT4gp32、120ng/μLのT4uvsX、30ng/μLのT4uvsY、360ng/μLの枯草菌DNAポリメラーゼIであった。DNAテンプレート3000コピーまたは水(負の対照として)は、指示どおりに加えた。ヌクレアーゼ、200ng/μLのNfoまたは50ng/μLのfpgは、指示どおりに加えた。プライマー、K2およびJ1は、それぞれ480nMで含め、プローブ、FpgProb1は120nMで加え、これらの配列は以下のとおりである。サンプルは、1容積の2%SDS/1容積のフェノール中で急冷し、20分間にわたって65℃で培養した。その後、サンプルは、標準の分子生物学的技術によりフェノール/クロロフォルム抽出し、2回エタノール沈殿させた。次に、各々のサンプルの半分は、16.5%変性ポリアクリルアミドゲル(尿素)上でホルムアミドのローディングバッファーに再懸濁させ、標準のプロトコルに従って視覚化した(FAM蛍光を使用して)。マーカーは、2pmolのプローブおよび2pmolの32ntマーカーオリゴヌクレオチドであった。
【0057】
【化1】
プローブおよび何らかの生物は、本明細書では、紫外線照射して活性化した場合、可視光を放出するFAM部分により視覚化した。標的DNA、2つの適切な増幅プライマー、dR−ビオチンプローブ、およびヌクレアーゼなし、Nfoタンパク質またはfpgタンパク質のいずれかを含む増幅反応(RPA)は、培養後に洗浄し、変性アクリルアミドゲル上でサイズ別に分離した後、紫外線に曝露した。次に、5’−FAMを保持するプローブまたは何らかの派生物が視覚化される(図3)。添加ヌクレアーゼが存在しない場合、42ヌクレオチド長のプローブはほとんど、32ヌクレオチド(指示)の対照標識付きプライマーより緩慢にその予想位置に移動する。わずかに緩慢に移動する(長めの)断片も、RPA中で培養されない適切なプローブ(♯1)と比べて観察される。これはおそらく、酵素調合物の一部に存在すると考えられるヌクレアーゼ(3’末端で徐々に減少する)によって、プローブのブロックが徐々に解除され、一度ブロックが解除されると、増幅標的に対するハイブリダイゼーション後に拡張可能であり、この種のネスト化した複製配列を形成するために生じる。しかし、Nfoが存在すると、この現象は予想よりはるかに顕著になり、プローブの大部分が伸長する。さらに、少量の比較的迅速に移動するプローブDNA(♯2)も見え、その後の伸長を伴わないdR−O−[C]n位置における開裂を示すため、Nfoタンパク質は、3’末端を単に「研磨する」のではなく、実際にdR−O−[C]n残基を攻撃していた。最後に、fpgタンパク質が反応環境に含まれる場合、fpgは、開裂後にブロック3’−末端を離れ、その結果、混合物中に存在するポリメラーゼ酵素によって伸長しないため、比較的迅速に移動する開裂プローブの大部分が見え、伸長した材料は検出されない。
【0058】
実施例2
内部dR蛍光体を含むオリゴヌクレオチドプローブによるDNA増幅
この実施例では、RPAは、dR−O−[C]nヌクレオチドが図4に示すように蛍光体に結合する蛍光試薬を使用して実験する。この場合、dR蛍光体は、オリゴヌクレオチドプローブの5’末端付近に位置し、その5’末端に結合するクエンチャーにきわめて接近して位置する。従来の実施例のとおり、プローブの3’末端は異常な伸長またはプライマーのアーティファクトを防止するために適切にブロックされる。
【0059】
この反応(全量50μL)は、冷凍−乾燥反応に関する標準RPAプロトコルに従って実施された。簡潔に述べると、凍結乾燥試薬はPEG、マグネシウム−アセテートおよびテンプレートと混合し、蛍光光度計内で38℃で20分間にわたって培養した(Twistaプロトタイプ;ESE GmbH、ドイツ)。使用した条件は、50mMのトリス/アセテート(pH 8.3)、14mMのマグネシウム−アセテート、100mMのカリウム−アセテート、5mMのDTT、それぞれ240nMのdNTP、5%のPEG 35,000、4%トレハロース2.5mM ATP、50mMのホスホ−クレアチン、300ng/μLのrb69gp32、273ng/μLのuvsX、120ng/μL uvsY、50ng/μLの黄色ブドウ状球菌DNAポリメラーゼIであった。図5の実験では、1000、100、10 または0コピーのDNAテンプレートを図に示されているように包含し、図6の実験では1000コピーのDNAテンプレートを包含した。25ng/μLのfpgヌクレアーゼを包含した。プライマーは、それぞれ360nMで包含し、プローブは120nMで包含し、それぞれの配列は以下のとおりであった。蛍光は20秒ごとに測定した(励起470nM、発光520nM)。サンプルは、4分間の培養時間で短時間の混合/回転を行って培養器から取り出し、培養器/蛍光光度計に戻した。分の時間に対して任意の蛍光単位をプロットした。
【0060】
ヒトゲノムの遺伝子座rs4824871の場合、使用したプライマーの配列F2およびR1並びにプローブは、以下のとおりであった。
【0061】
【化2】
ヒトゲノムの遺伝子座rsl207445の場合、使用したプライマーの配列F2およびR2並びにプローブは、以下の通りであった。
【0062】
【化3】
ヒトゲノムの遺伝子座rsl105561の場合、使用したプライマーの配列F1およびR1並びにプローブは、以下の通りであった。
【0063】
【化4】
ヒトゲノムの遺伝子座rs5923931の場合、使用したプライマーの配列F1およびR1並びにプローブは、以下の通りであった。
【0064】
【化5】
図5および6は、上記の実験の結果を示し、RPA反応は、公知のSNP領域を特異的に対象としたプライマーおよびプローブを使用して、ヒトゲノム標的に関して実施した。図5では、このような2つのゲノム領域は、fpgタンパク質の封入と共に、RPAおよび図4に示されている一般的な構造を持つプローブを使用して増幅した。標的ゲノムDNAは、合計標的コピー数を1000、100、10またはゼロ標的分子にするために追加し、こうして、標的に一致する単位複製配列の特異的な蓄積要件が確保される。6〜12分後(標的およびコピー数に応じて)、標的を含むこれらのサンプルの蛍光は明らかに上昇するが、標的が欠如しているサンプルは事実上蛍光は安定していることに注意する。図6では、4つのヒトゲノムDNA標的/プローブのセット(そのうち2つは図5にも使用されている)に関して類似のデータが示されており、何れの場合も、蛍光はDNA増幅のほぼ予想時間に上昇している。上記およびその他の好結果のプローブのほかに、RPA増幅/検出システムで十分に作用しなかったと思われるプローブが時折みられたが(おそらく、分析したプローブの10〜20%)、こうした欠陥の原因は未だ明らかではなく、場合によってはプローブの欠陥ではなく、可能性として他の場合のプローブの欠陥の結果としてRPA増幅の欠陥を表していることが考えられる。場合によっては、近接塩基の位置および性質、またはdR−O−[C]n基に対向する塩基の性質が、プローブの効果に影響した可能性があると考えられる。
【0065】
本明細書で特定の実施態様について詳細に開示しているが、これは、具体的に示すために一例として説明したのであって、添付の請求の範囲の範囲に関して制限することを意図しているのではない。特に、発明人は、請求の範囲により定義される本発明の精神および範囲を逸脱することなく、本発明に様々な置き換え、変更および修飾が施されることを意図している。その他の態様、利点および修正は、添付の請求項の範囲内と考えられる。提示される請求の範囲は、本明細書で開示されている本発明を表す。請求されていないその他の発明も想定されている。出願人は、かかる発明を後の請求の範囲で追跡する権利を留保する。
【技術分野】
【0001】
関連出願
この出願は、2009年5月20日に出願された米国仮特許出願第61/179,793号(これは、参考として本明細書に援用される)に対する優先権の利益を主張する。
【0002】
発明の分野
本発明は、オリゴヌクレオチド基質およびその使用、たとえば核酸増幅反応のためのプローブに関する。
【背景技術】
【0003】
一次配列、主鎖構造、または基本特性(多くの場合、傷害塩基または修飾塩基)により指定される方法で核酸を代謝する酵素は、生物工学的用途に有用である。このような酵素のいくつかのファミリーは、核酸ベースの技術に普通に使用されており、制限エンドヌクレアーゼ、ポリメラーゼ、リガーゼおよびエクソヌクレアーゼが含まれる。さらに、特定の配列ストリングに依存するのではなく、普通でない塩基、傷害塩基または欠落塩基の認識に依存する種々のシングルサブユニットの(非制限)エンドヌクレアーゼについては、これまで長年にわたって記述されてきた。これらの酵素は、おおまかに2つのグループ、つまり一例として大腸菌エンドヌクレアーゼIV(Nfo)および大腸菌エクソヌクレアーゼIIIがあるAPエンドヌクレアーゼと、一例として大腸菌fpg、MUGおよびNthがあるDNAグリコシラーゼ/リアーゼファミリーとに分割することができる。
【0004】
APエンドヌクレアーゼは、二重鎖DNAに関連して発見された場合、無塩基部位(その他の酵素活性も存在し得る)で糖−リン酸主鎖を認識し、開裂する能力に特徴がある。無塩基部位における認識および切開は、グリコシラーゼ/リアーゼファミリーに独特な生化学的な方法で行われ、ベータ脱離またはベータ/デルタ脱離によって行われるのではない。したがって、これらは、真の無塩基部位だけでなく、糖環の’1炭素に酸素原子が欠けているテトラヒドロフラン部分を含むその他の基質も攻撃する。(非特許文献1)(図1の化学構造を参照)。
【0005】
対照的に、fpgタンパク質(8−オキソグアニンDNAグリコシラーゼ、大腸菌fpg、および哺乳動物のOGG1)、またはNthタンパク質(大腸菌のエンドヌクレアーゼIII、ヒトのNth1など)を含むグリコシラーゼ/リアーゼ酵素は、第1に、傷害塩基が、タンパク質とDNAとの間のシッフ塩基の形成を介して認識されて切除され、第2に、このように生成された無塩基部位が、APエンドヌクレアーゼに独特な方法でベータ脱離またはベータ/デルタ脱離によって処理される2段階の触媒作用方法で機能する。この場合、C1’酸素原子はこの無塩基部位の模擬は存在せず、このような位置に酸素が欠如している糖は攻撃に耐性があるため、テトラヒドロフラン(THF)残基はリアーゼ活性の基質ではない(非特許文献1)(図1)。
【0006】
分子生物学的技術におけるAPエンドヌクレアーゼおよびグリコシラーゼ/リアーゼの使用について説明した。1つの用途は、インビトロDNA増幅反応の際に生成される基質を処理するためにこれらの酵素を使用するか、または類似の用途であり、特に、合成オリゴヌクレオチドが、サンプル中の分子に特異的にハイブリダイズする場合に、酵素の基質になる可能性がある修飾糖または塩基を含む合成「プローブ」オリゴヌクレオチドが提供された場合の用途である。このような用途の一例は、特許文献1および非特許文献2に記載されており、テトラヒドロフラン修飾オリゴヌクレオチドは、DNAの増幅を測定する方法で、大腸菌Nfo(エンドヌクレアーゼIV)タンパク質の基質として使用される(Nfoは、大腸菌の2つのAPエンドヌクレアーゼの1つである)。
【0007】
グリコシラーゼ/リアーゼを類似の方法に用途することも想定可される。fpgタンパク質が、反応監視のためのDNA増幅反応の範囲内で8−オキソグアニンなどの修飾塩基を同様に処理する能力について記載されている(特許文献1)。さらに、fpgおよびNthなどのグリコシラーゼ/リアーゼ酵素は3’伸長可能末端を離れないが、むしろブロック3’末端を離れる(触媒モードの違いにより)という事実は、処理プローブがポリメラーゼの対応基質にも、3’ヒドロキシル部分に依存するその他の活性にもなり得ないことを確実にすることが望まれる状況で特に有用であると思われる。
【0008】
これらの酵素は、その電位にも関わらず、ある用途に使用するには適さなくなる特定の特徴を有する。特に、THF残基と違って、主鎖に必要な真の無塩基部位 − DNAリアーゼの切開活性は、生理学的条件下で安定せず、水溶液中で急速に加水分解するため、最も分子的な手順に使用するのには実際的ではない。その代わり、特定の傷害塩基は、リアーゼ活性による主鎖加水分解の前に、無塩基部位を一時的に生成するために、グリコシラーゼ活性のための一次基質として包含して使用することができる。しかし、残念ながら、8−オキソグアニン(fpg)またはチミジングリコール(Nth)などの代表的な傷害塩基類似体は、理想的には、特異塩基の反対側で逆鎖上で対にする必要があるため、むしろ合成が高く付き、プローブに配列要件を課すことになる。原則として、APエンドヌクレアーゼに使用できる一般的なTHF残基に類似する安定した基質を有するものの、グリコシラーゼ/リアーゼ酵素のリアーゼ活性との反応性を維持するとはるかに好都合であると思われる。
【0009】
本明細書では、塩基が欠如しているが、炭素ベースのリンカー[C]nに共有結合する1’酸素原子を含む様々な基質を含むDNA主鎖をfpgタンパク質、および大腸菌(Nfo)のAPエンドヌクレアーゼIVが効果的に開裂することを証明する。このリンカーは、その他の部分、たとえばビオチン、蛍光体およびその他の結合基を結合するために単独で使用することができ、アミン末端リンカーを使用して様々な物質を結合できる場合に特に有用である。意外なことに、この配列を有し、一般にdR−O−[C]nと呼ばれるヌクレオチドは、多くの状況でfpgタンパク質の良好な基質であり、エンドヌクレアーゼIVタンパク質の基質でもあると思われるが、大腸菌エクソヌクレアーゼIIIには比較的不良な基質であると思われる。多くの状況で、特に、核酸検出方法のための検出策の部分など、特にインビトロ反応の範囲内で、dR−O−[C]n基を基質として含むオリゴヌクレオチドを使用することが想定される。この研究に使用するリンカーの長さは、市販の一定のヌクレオチドで使用可能であるように6炭素原子であるが、多様な炭素鎖の長さを使用することができ、後続の立体容積が小さく、酵素に対する十分な可塑性をこれらの構造に与える炭素−酸素−炭素構造であると想定される。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】米国特許第7,435,561号明細書
【非特許文献】
【0011】
【非特許文献1】Takeshita等、J Biol Chem.(1987年)262(21):1017l
【非特許文献2】Piepenburgら、PlosBiology、(2006年)4(7):e204
【発明の概要】
【課題を解決するための手段】
【0012】
本発明は一部には、APエンドヌクレアーゼ、DNAグリコシラーゼ、DNAグリコシラーゼ/リアーゼ、たとえばfpgおよびNfoタンパク質が、塩基が糖のC1’位に存在しないが、当該位置に酸素原子を保持しているdR−O−[C]n残基を含む部位でDNA主鎖の切断を触媒することが可能であるという発見に関する。酸素原子は、n個(たとえば1〜8)の炭素原子(つまり、[C]n)の炭素リンカーの炭素原子に糖を架橋する。したがって、市販のホスホルアミダイトを使用することにより、dR−O−[C]n残基を含む核酸プローブを構成することができ、相補的核酸と二本鎖を形成する場合、APエンドヌクレアーゼおよびDNAグリコシラーゼ/リアーゼ酵素のための基質でよい。様々な部分は、蛍光体、および特定の標的核酸が存在する証拠であるプローブの上手くいった処理を検出するための多くの方法を提示するその他の標識を含むdR−O−[C]nのリンカー部分に結合することができる。出願人は、蛍光分子およびクエンチャー(quencher)を使用し、fpgもしくはNfo、またはその他の可能なAPエンドヌクレアーゼもしくはリアーゼの標的部位としてdR−O−[C]nを使用してどのようにプローブを構成することができるかを示す。出願人は、dR−O−[C]n基質のその他の用途を他の検出計画で想定している。たとえば、dR−O−[C]n残基は、ヌクレアーゼの活性が標識を遊離させた直後、または後続のプロセスを介するか、もしくは結合体化状態と遊離状態との測定可能な相違を介して検出することが可能な検出可能な標識に結合体化し得る。
【0013】
一態様では、二本鎖をAPエンドヌクレアーゼ、DNAグリコシラーゼ、またはDNAグリコシラーゼ/リアーゼから選択されるヌクレアーゼと接触させることにより、核酸とともに二本鎖を形成するdR−O−[C]n残基を含むオリゴヌクレオチドを開裂するプロセスを本明細書で提供する。実施態様によっては、ヌクレアーゼは、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である。実施態様によっては、リンカーは3〜6炭素原子リンカー(たとえば、6炭素原子リンカー)である。実施態様によっては、オリゴヌクレオチドはその3’末端でブロックされて、ポリメラーゼ伸長を防止する。実施態様によっては、リンカーは、検出可能な標識(たとえば、ビオチン、ジオキシゲニン、ペプチド、蛍光体、クエンチャー、抗体または量子ドット)に結合体化する。
【0014】
実施態様によっては、このプロセスは、オリゴヌクレオチドと核酸とを接触させて、オリゴヌクレオチド/核酸二本鎖を形成するステップをさらに含む。実施態様によっては、これは、オリゴヌクレオチドを核酸にハイブリダイズさせることを含む。実施態様によっては、これは、(i)オリゴヌクレオチドをリコンビナーゼと接触させて、リコンビナーゼ/オリゴヌクレオチド複合体を形成するステップと、(ii)リコンビナーゼ/オリゴヌクレオチド複合体を核酸に接触させて、オリゴヌクレオチド/核酸二本鎖を形成するステップとを含む。実施態様によっては、核酸は、核酸増幅反応(たとえば、リコンビナーゼポリメラーゼ増幅(RPA)プロセスまたはポリメラーゼ鎖反応(PCR))の生成物である。
【0015】
実施態様によっては、このプロセスは、オリゴヌクレオチドの開裂を検出するステップをさらに含む。実施態様によっては、この検出はリアルタイムで監視される。実施態様によっては、実施態様によっては、検出は、反応の終点で監視される。
【0016】
実施態様によっては、オリゴヌクレオチドは蛍光体およびクエンチャーを含み、蛍光体またはクエンチャーの一方は炭素リンカーに結合体化する。ヌクレアーゼ活性は、結合体化した蛍光体またはクエンチャーをオリゴヌクレオチドから切除し、検出ステップは、結合体化状態と遊離状態との間の蛍光性に差がある場合、その差の測定を含む。
【0017】
別の態様では、標的核酸の存在または不在を検出するプロセスを本明細書で提供する。このプロセスは、以下のステップを含む。(a)dR−O−[C]n残基またはヌクレオチドを含むオリゴヌクレオチドプローブを標的核酸と接触させて、プローブ/核酸二本鎖を形成する、(b)二本鎖をAPエンドヌクレアーゼ、DNAグリコシラーゼ、またはDNAグリコシラーゼ/リアーゼから選択したヌクレアーゼと接触させて、リンカーを複合体から切除するか、および/またはプローブをdR−O−[C]nヌクレオチドにおいて特異的に開裂する、並びに(c)こうした切除または開裂が行われたかどうかを検出する。実施態様によっては、核酸は、核酸増幅反応(たとえば、リコンビナーゼポリメラーゼ増幅(RPA)プロセスまたはポリメラーゼ鎖反応(PCR)の生成物である。実施態様によっては、増幅反応はリアルタイムで監視される。実施態様によっては、増幅反応は反応の終点で監視される。
【0018】
実施態様によっては、二本鎖は、プローブを核酸にハイブリダイズさせることによって形成される。実施態様によっては、二本鎖は(i)プローブをリコンビナーゼに接触させて、リコンビナーゼ/プローブ複合体を形成する、(ii)リコンビナーゼ/プローブ複合体を核酸に接触させて、プローブ/核酸二本鎖を形成する、ことによって形成される。
【0019】
実施態様によっては、ヌクレアーゼは、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である。実施態様によっては、リンカーは、3〜6炭素原子リンカー(たとえば、6炭素原子リンカー)である。実施態様によっては、オリゴヌクレオチドは、3’末端でブロックされて、ポリメラーゼ伸長を防止する。実施態様によっては、リンカーは、検出可能な標識(たとえばビオチン、ジオキシゲニン、ペプチド、蛍光体、クエンチャー、抗体または量子ドット)に結合体化される。
【0020】
実施態様によっては、プローブは蛍光体およびクエンチャーを含み、蛍光体またはクエンチャーの一方が炭素リンカーに結合体化される。たとえば、蛍光体およびクエンチャーは、プローブ中の4〜6個の塩基によって分離される。実施態様によっては、炭素リンカーに結合体化されない蛍光体またはクエンチャーは、プローブの末端(たとえば、5’末端)に結合体化される。ヌクレアーゼ活性は、dR−O−[C]n残基に関連して結合体化された蛍光体またはクエンチャーをプローブから切除して遊離させ、検出ステップは、結合体化状態と遊離状態との間の蛍光性に差がある場合、その差を測定することを含む。
【0021】
別の態様では、dR−O−[C]n残基を含むオリゴヌクレオチドプローブを本明細書で提供する。実施態様によっては、プローブは、30〜60ヌクレオチド長であり、10ヌクレオチド以下(たとえば、4〜6ヌクレオチド)で分離された蛍光体クエンチャー対を含み、蛍光体またはクエンチャーの何れかがdR−O−[C]n残基に結合体化する。実施態様によっては、リンカーは、3〜6炭素原子リンカー(たとえば、6炭素原子リンカー)である。実施態様によっては、オリゴヌクレオチドは、3’末端でブロックされて、ポリメラーゼ伸長を防止する。実施態様によっては、dR−O−[C]n残基に結合体化しない蛍光体またはクエンチャーは、プローブの末端(たとえば、5’末端)に結合体化する。実施態様によっては、プローブは、30〜40ヌクレオチド(たとえば、35ヌクレオチド)長である。
【0022】
さらに別の態様では、(i)dR−O−[C]n残基を含むオリゴヌクレオチドと、(ii)APエンドヌクレアーゼ、DNAグリコシラーゼ、またはDNAグリコシラーゼ/リアーゼから選択したヌクレアーゼとを含むキットを本明細書で提供する。実施態様によっては、ヌクレアーゼは、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である。
【0023】
さらに他の態様では、dR−O−[C]n残基およびAPエンドヌクレアーゼ、DNAグリコシラーゼ、またはDNAグリコシラーゼ/リアーゼから選択したヌクレアーゼ(たとえば、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg))を含有するオリゴヌクレオチドを含む反応混合物を本明細書で提供する。実施態様によっては、リンカーは、3〜6炭素原子リンカー(たとえば、6炭素原子リンカー)である。実施態様によっては、オリゴヌクレオチドは、3’末端でブロックされてポリメラーゼ伸長を防止する。実施態様によっては、リンカーは、検出可能な標識(たとえば、ビオチン、ジオキシゲニン、ペプチド、蛍光体、クエンチャー、抗体または量子ドット)に結合体化する。実施態様によっては、反応混合物は冷凍乾燥、つまり凍結乾燥される。
【0024】
実施態様によっては、反応混合物は容器をさらに含む。たとえば、反応混合物は、チューブまたは多ウェル容器のウェル内に含むことができる。反応混合物は乾燥させて、ビードもしくはストリップなどの可動性固体支持体、またはウェル上に取り付けることができる。
【0025】
実施態様によっては、反応混合物は、オリゴヌクレオチドに対して相補的である配列を含む標的またはテンプレート核酸をさらに含む。
【0026】
本発明のその他の実施態様、目的、態様、特徴および利点は、添付の明細書および請求の範囲から明らかになる。実施態様が、本発明の異なる態様に関して説明されている場合でも、適切であれば、本発明の任意の実施態様を本発明の1つまたは複数の他の実施態様と結合できることを意図する。
【図面の簡単な説明】
【0027】
【図1】図1は、以下:1’炭素位置にヒドロキシルを含む通常の無塩基部位の化学構造、1’位に水素を含むテトラヒドロフラン(THF)残基の化学構造、DNAリボース基のC1’と、結合したマーカー部分に対するリンカーとの間の炭素−酸素−炭素架橋の位置を示す、一般的なdR−O−[C]n基の化学構造、および最後に、実施例1に使用され、かつdR−O−[C]n構造に適合するdR−ビオチンヌクレオチドの化学構造を記載する。
【図2】図2は、dR−ビオチンプローブの設計である:プローブ配列に一致する標的配列が増幅されるRPA反応の際に、Nfoおよびfpgタンパク質の開裂活性を評価するために使用されるオリゴヌクレオチドプローブの配列および配置図。オリゴヌクレオチドの配列が指示されている。プライマーは、5’末端にFAM蛍光体で標識付けされ、配列本体内にdR−ビオチンを含み、2’、3’ジオキシチジン残基によりブロックされる。
【図3】図3は、ヌクレアーゼがないか、またはNfoもしくはfpg酵素を含む増幅反応の比較である:両方の酵素が、開裂生成物、Nfoの場合、開裂生成物の伸長によって生じる生成物を迅速に移動させるdR−ビオチン部分を処理することができることを明らかにする。
【図4】図4は、オリゴヌクレオチドプローブの設計である:オリゴヌクレオチド本体(この場合、35ヌクレオチド長)、5’クエンチャー修飾(この場合、5’−BHQ1)、クエンチャーに近接する内部dR−蛍光体ヌクレオチド(この場合、dR−FAMオリゴヌクレオチド位置6)、および3’ポリメラーゼ伸長ブロックを含むプローブ設計の例。
【図5】図5は、感受性および特性を示す:DNA dR−プローブを使用して、指示されたヒトゲノム標的について2回のテンプレート滴定実験をリアルタイムで蛍光監視した結果。どちらの場合も、蛍光信号の増加(0〜3分の基準線に対して)は、テンプレートを含む反応でのみ観察され、非テンプレート対照では観察されない。信号増加の開始時間は、開始テンプレートの量(1000、100または10コピー)と相関する。反応時間は分(X軸)、蛍光は任意の蛍光単位(Y軸)である。
【図6】図6は、様々なプローブの性能である:図2に概略を示す設計のDNAオリゴヌクレオチドプローブを用いて、指示されたヒトゲノム標的に関するRPA反応の4セット(重複)をリアルタイムで監視した結果。6〜8分の蛍光の増加は、プローブのdR基(この場合、dR−FAM)のfpg依存処理から生じ、DNA増幅が進行しており、その結果標的DNAテンプレートの存在を示す。反応時間は分(X軸)、蛍光は任意の蛍光単位(Y軸)である。
【発明を実施するための形態】
【0028】
本発明の1つまたは複数の実施態様の詳細は、付随する以下の説明に記載する。本明細書に記載されている方法および材料に類似するか、または等価な任意の方法および材料を実際に、または本発明の試験に使用することができるが、この方法および材料について説明する。本発明のその他の特徴、目的および利点は、本明細書から明らかであろう。本明細書では、単数形は、文脈上そうではないことが明らかではない限り、複数形も含む。特記しない限り、本明細書で使用する技術用語および科学用語はすべて、本明細書が属する技術分野の当業者が一般に理解する意味と同じ意味を有する。矛盾する場合、本明細書が優先される。
【0029】
実験室で分析および操作に使用する酵素と合成基質の組合せは先行技術で公知である。グリコシラーゼfpgおよびNthなどのDNA修復エンドヌクレアーゼ、並びに大腸菌エクソヌクレアーゼIIIおよびエンドヌクレアーゼIVなどのAPエンドヌクレアーゼは、この組合せの良い例である。現在のオリゴヌクレオチド合成法、およびDNAプライマーに組み込まれる既存の多様な合成ヌクレオチドを使用することにより、これらのDNA修復酵素のための合成基質を生成することは容易である。これらのDNA修復酵素は様々な目的で容易に使用することができ、最近開拓された1つの目的は、等温リコンビナーゼポリメラーゼ増幅(RPA)反応の監視を支援する剤として使用することである。
【0030】
RPAは、DNAに対するオリゴヌクレオチドのリコンビナーゼ媒介標的が、ポリメラーゼによりDNA合成に結合するプロセスである(米国特許第7,270,981 B2号;米国特許第7,399,590号;米国特許第7,435,561 B2号;米国特許第7,485,428 B2号;米国特許第 7,666,598 B2号および海外の対応特許)。RPAは、細胞DNA複製および修復機構の構成要素によって決まり、特定のクラウディング剤の存在で達成される高レベルの再結合活性を維持する適切な速度のリコンビナーゼのロードおよびアンロードの両方を有する「動的」再結合環境の確立に依存する。RPAは、PCRの感受性、特異性およびその他ほとんどの特徴を結合し、熱サイクリングの必要がなく、異例の速度、および異常温度設定に対する頑健性という利点を有する。RPAは、熱安定性の等価物が必要であるか、または単鎖DNA結合タンパク質などの付属タンパク質、RPA反応の天然成分がない場合は調節が不良であることが実証されているため、他のプロセスには利用されていなかった多様な核酸処理酵素、たとえば公知の修復エンドヌクレアーゼの潜在的な使用から既に利益が得られている。
【0031】
簡潔に述べると、RPAは以下のステップを含む:第1に、リコンビナーゼ因子が第1および第2核酸プライマーと接触して、第1および第2核タンパク質プライマーを形成する。第2に、第1および第2核タンパク質プライマーは、二本鎖標的配列と接触して、前記第1鎖の第1部分に第1二本鎖構造を形成し、前記第2鎖の第2部分に二本鎖構造を形成し、よって前記第1核酸プライマーおよび前記第2核酸プライマーの3’末端は、所定のテンプレートDNA分子上で互いに対して方向付けられる。第3に、前記第1および第2核タンパク質プライマーの3’末端はDNAポリメラーゼによって伸長され、第1および第2二本鎖核酸、および核酸の第1および第2変位鎖を生成する。最後に、第2および第3ステップは、所望の程度の増幅に達するまで繰り返される。
【0032】
以前の研究は、RPA法のためのプローブシステムの開発で、合成ヌクレオチドテトラヒドロフラン(THF)がきわめて有用であることを実証している(Piepenburg等、2006年;米国特許第7,435,561 B2号)。この塩基類似体は、しばしば無塩基部位を模倣するために使用され、安定しているという自然な利点を有し、天然の無塩基部位の1’ヒドロキシルを水素原子に置き換えることによりヌクレオチドは安定し、瞬間的な開環およびオリゴヌクレオチドの分裂が生じなくなる。この類似体は容易に入手可能であり、オリゴヌクレオチドに組み込むのも安価である。しかし、生化学的機序の違いにより、大腸菌APエンドヌクレアーゼNfoおよびExoIIIは合成プライマーのこうしたTHF残基で開裂することが可能である一方、他のDNAグリコシラーゼ/リアーゼは開裂することができない。これらの後者の酵素は通常、傷害塩基(グリコシラーゼ活性)および/またはヒドロキシル基が糖の1’位(リアーゼ活性)に存在する必要があり、THFはその酵素活性に対して完全に不活性である。これらのグリコシラーゼ/リアーゼ酵素は、プローブがRPA、またはその他の状況および方法で標的DNA蓄積に応じて処理される反応などのインビトロ反応にも有用なツールになり得るため、多少厄介になる。より自然な基質、たとえばfpg用の8−オキソグアニンは、オリゴヌクレオチド内に挿入して、これらのグリコシラーゼ/リアーゼの開裂部位を生成することができるが、これらの修飾は一般に高価であり、さらに、修飾ヌクレオチドに対向することが可能な塩基を制限することが多い。比較的安価で、さらに一般的であり、これらの酵素の基質であるヌクレオチド修飾は、非常に有用であると思われる。
【0033】
多くの異常な塩基類似体のDNA修復酵素の基質としての効果を調査するために、完全に塩基が欠如しているが、糖の1’位に炭素−酸素−炭素連結を保持するヌクレオチドを含有するオリゴヌクレオチドを合成した。このようなヌクレオチド試薬は容易に入手可能で安価であり、一般に、蛍光体またはビオチンなどの標識基をオリゴヌクレオチドの本体内の複数のオリゴヌクレオチド内に組み込むために使用される。一般に、酸素を介して糖の1’炭素に結合する炭素原子は、多くの場合、最終的に標識基、アミン、または試薬が容易に結合し得るその他の化学的部分(たとえば、チオ)で終端するリンカーの第1炭素原子である。こうした試薬は、文献にはdR−Xと記述されることが多く、この場合、dRはデオキシリボースを意味し、Xはリンカーアミン、リンカー蛍光体、リンカービオチン、またはいくつかのその他の基または標識である。修復エンドヌクレアーゼが、塩基は欠如しているが、炭素−酸素−炭素共有結合を1’糖位置に保持する構造を認識するかどうか、以前に調査されたことはなかった。ヒドロキシルの欠如は、リアーゼの開環プロセスが、リンカー基を事前に処理し、それに関連して切除することなく実施すべきではないことを意味する。公知のグリコシラーゼは、通常、異常な炭素リンカーではなく傷害塩基に作用するので、これらのdR−O−[C]n基はfpgなどのDNAグリコシラーゼ/リアーゼの基質であろうという先例はなかった。
【0034】
図1は、dR−O−[C]n基の一般的な構造、特に、本明細書で使用され、ベルギーのEurogentecが市販するオリゴヌクレオチド中に組み込まれるdR−ビオチン試薬の構造を示す。本明細書で使用するこれらの試薬は、1’糖と、オリゴヌクレオチド合成の前または後にその他の試薬を結合するために使用されることが多い窒素原子との間に一般的な6炭素原子リンカーを有する。この研究では、dR−ビオチンオリゴヌクレオチドのビオチン部分は、この窒素原子を介してアミド結合として結合し、さらに4炭素原子リンカーを通して結合する。この研究に使用されるその他の標識試薬 − dR−FAMおよびdR−Texas Red − は同様に配置され、この場合、蛍光体は6炭素原子リンカーの末端でアミド結合を通して結合する。
【0035】
検出可能な標識は、現在の方法で検出できる任意の部分と定義される。これらの標識は、少なくとも蛍光体(蛍光分子、蛍光色素とも呼ばれる)、酵素、クエンチャー、酵素抑制物質、放射性標識、結合対の一員、ジオキシゲニン残基、ペプチド、およびこれらの組合せを含む。
【0036】
「結合対の一員」とは、第1部分および第2部分の一方を意味し、前記第1部分および第2部分は互いに特異的な結合親和性を有する。本発明に使用するのに適する結合対としては、抗原/抗体(たとえば、ジゴキシゲニン/抗−ジゴキシゲニン、ジニトロフェニル (DNP)/抗−DNP、ダンシル−X−抗−ダンシル、フルオロセイン/抗−フルオロセイン、ルシファーイエロー/抗−ルシファーイエロー、ペプチド/抗−ペプチド、リガンド/受容体およびローダミン/抗−ローダミン)、ビオチン/アビジン(またはビオチン/ストレプトアビジン)およびカルモジュリン結合タンパク質(CBP)/カルモジュリンが挙げられるが、これらだけに限らない。その他の適切な結合対としては、ポリペプチド、たとえばFLAG−ペプチド(DYKDDDDK)[Hopp 等、BioTechnology,6:1204 1210(1988年);KT3エピトープペプチド(Martin 等、Science 255:192 194(1992年));チューブリンエピトープペプチド(Skinner等、J.Biol.Chem 266:15163 15166(1991年));およびT7遺伝子10タンパク質ペプチドタグ(Lutz−Freyermuth 等、Proc.Natl.Acad.Sci.USA,87:6393 6397(1990年))並びにこれらに対する抗体が挙げられる。一般に、好ましい実施態様では、結合対パートナーのより小さい方は、立体配置に関する考察が重要な場合があるため、検出可能な標識として役立つ。
【0037】
一態様では、二本鎖をAPエンドヌクレアーゼ、DNAグリコシラーゼ、またはDNAグリコシラーゼ/リアーゼから選択したヌクレアーゼに接触させることによって、核酸とともに二本鎖を形成するdR−O−[C]n残基を含むオリゴヌクレオチドを開裂するプロセスを本明細書で提供する。実施態様によっては、ヌクレアーゼは、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である。
実施態様によっては、リンカーは、3〜6炭素原子リンカー(たとえば、6炭素原子リンカー)である。実施態様によっては、オリゴヌクレオチドは、3’末端でブロックされて、ポリメラーゼ伸長を防止する。実施態様によっては、リンカーは、検出可能な標識(たとえばビオチン、ジオキシゲニン、ペプチド、蛍光体、クエンチャー、抗体または量子ドット)に結合体化される。
【0038】
実施態様によっては、このプロセスは、オリゴヌクレオチドを核酸と接触させてオリゴヌクレオチド/核酸二本鎖を形成するステップをさらに含む。実施態様によっては、これは、オリゴヌクレオチドを核酸にハイブリダイズさせることを含む。実施態様によっては、これは、(i)オリゴヌクレオチドをリコンビナーゼと接触させてリコンビナーゼ/オリゴヌクレオチド複合体を形成するステップと、(ii)リコンビナーゼ/オリゴヌクレオチド複合体を核酸と接触させて、オリゴヌクレオチド/核酸二本鎖を形成するステップとを含む。実施態様によっては、核酸は、核酸増幅反応(たとえば、リコンビナーゼポリメラーゼ増幅(RPA)プロセスまたはポリメラーゼ鎖反応(PCR))の生成物である。
【0039】
実施態様によっては、このプロセスは、オリゴヌクレオチドの開裂を検出するステップをさらに含む。実施態様によっては、検出はリアルタイムで監視される。実施態様によっては、検出は、反応の終点で監視される。
【0040】
実施態様によっては、オリゴヌクレオチドは蛍光体およびクエンチャーを含み、蛍光体またはクエンチャーの一方は炭素リンカーに結合体化する。ヌクレアーゼ活性は、結合体化した蛍光体またはクエンチャーをオリゴヌクレオチドから切除し、検出ステップは、結合体化状態と遊離状態との間の蛍光性に差がある場合、その差を測定することを含む。
【0041】
別の態様では、標的核酸の存在または不在を検出するプロセスを本明細書で提供する。このプロセスは、以下のステップを含む:(a)dR−O−[C]n残基またはヌクレオチドを含むオリゴヌクレオチドプローブを標的核酸に接触させて、プローブ/核酸二本鎖を形成するステップと、(b)二本鎖をAPエンドヌクレアーゼ、DNAグリコシラーゼ、またはDNAグリコシラーゼ/リアーゼから選択したヌクレアーゼと接触させて、リンカーを複合体から切除するか、および/またはプローブをdR−O−[C]nヌクレオチドで特異的に開裂するステップと、(c)こうした切除または開裂が生じたかどうかを検出するステップ。実施態様によっては、核酸は、核酸増幅反応(たとえば、リコンビナーゼポリメラーゼ増幅(RPA)プロセスまたはポリメラーゼ鎖反応(PCR))の生成物である。実施態様によっては、増幅反応はリアルタイムで監視される。実施態様によっては、増幅反応は反応の終点で監視される。
【0042】
実施態様によっては、二本鎖は、プローブを核酸にハイブリダイズさせることによって形成される。実施態様によっては、二本鎖は、(i)プローブをリコンビナーゼに接触させて、リコンビナーゼ/プローブ複合体を形成する、(ii)リコンビナーゼ/プローブ複合体を核酸に接触させて、プローブ/核酸二本鎖を形成する、ことによって形成される。
【0043】
実施態様によっては、ヌクレアーゼは、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である。実施態様によっては、リンカーは、3〜6炭素原子リンカー(たとえば、6炭素原子リンカー)である。実施態様によっては、オリゴヌクレオチドは、3’末端でブロックされて、ポリメラーゼ伸長を防止する。実施態様によっては、リンカーは、検出可能な標識(たとえばビオチン、ジオキシゲニン、ペプチド、蛍光体、クエンチャー、抗体または量子ドット)に結合体化される。
【0044】
実施態様によっては、プローブは蛍光体およびクエンチャーを含み、蛍光体またはクエンチャーの一方が炭素リンカーに結合体化される。たとえば、蛍光体およびクエンチャーは、プローブ中の4〜6個の塩基によって分離される。実施態様によっては、炭素リンカーに結合体化されない蛍光体またはクエンチャーは、プローブの末端(たとえば、5’末端)に結合体化される。ヌクレアーゼ活性は、dR−O−[C]n残基に関連して結合体化された蛍光体またはクエンチャーをプローブから切除して遊離させ、検出ステップは、結合体化状態と遊離状態との間の蛍光性に差がある場合、その差を測定することを含む。
【0045】
別の態様では、dR−O−[C]n残基を含むオリゴヌクレオチドプローブを本明細書で提供する。実施態様によっては、プローブは、30〜60ヌクレオチド長であり、10ヌクレオチド以下(たとえば、4〜6ヌクレオチド)で分離された蛍光体クエンチャー対を含み、蛍光体またはクエンチャーの何れかがdR−O−[C]n残基に結合体化する。実施態様によっては、リンカーは、3〜6炭素原子リンカー(たとえば、6炭素原子リンカー)である。実施態様によっては、オリゴヌクレオチドは、3’末端でブロックされて、ポリメラーゼ伸長を防止する。実施態様によっては、dR−O−[C]n残基に結合体化されない蛍光体またはクエンチャーは、プローブの末端(たとえば、5’末端)に結合体化される。実施態様によっては、プローブは、30〜40ヌクレオチド(たとえば、35ヌクレオチド)長である。
【0046】
さらに別の態様では、(i)dR−O−[C]n残基を含むオリゴヌクレオチドと、(ii)APエンドヌクレアーゼ、DNAグリコシラーゼ、またはDNAグリコシラーゼ/リアーゼから選択したヌクレアーゼとを含むキットを本明細書で提供する。実施態様によっては、ヌクレアーゼは、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である。
【0047】
さらに他の態様では、dR−O−[C]n残基、およびAPエンドヌクレアーゼ、DNAグリコシラーゼ、またはDNAグリコシラーゼ/リアーゼから選択したヌクレアーゼ(たとえば、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg))を含有するオリゴヌクレオチドを含む反応混合物を本明細書で提供する。実施態様によっては、リンカーは、3〜6炭素原子リンカー(たとえば、6炭素原子リンカー)である。実施態様によっては、オリゴヌクレオチドは、3’末端でブロックされて、ポリメラーゼ伸長を防止する。実施態様によっては、リンカーは、検出可能な標識(たとえばビオチン、ジオキシゲニン、ペプチド、蛍光体、クエンチャー、抗体または量子ドット)に結合体化される。実施態様によっては、反応混合物は冷凍乾燥、つまり凍結乾燥される。
【0048】
実施態様によっては、反応混合物は容器をさらに含む。たとえば、反応混合物は、チューブまたは多ウェル容器のウェル内に含むことができる。反応混合物は乾燥させるか、またはビードもしくはストリップなどの可動性固体支持体、またはウェル上に取り付けることができる。
【0049】
実施態様によっては、反応混合物は、オリゴヌクレオチドに対して相補的である配列を含む標的またはテンプレート核酸をさらに含む。
【0050】
図2は、プローブに指定される配列を含むDNA分子を標的として使用して、RPAのDNA増幅反応に使用するために生成されるdR−ビオチンプローブの一次配列、およびおおまかなその性質の両方を示す。プローブはブロックされ(増幅段階でポリメラーゼ伸長を防止するため)、内部dR−ビオチンを酵素の試験基質として含む。プローブは、5’−FAMも含む。したがって、原則として、DNAが、このプローブを含む反応で増幅される場合、プローブが、増幅時に形成された相補的単鎖に対する「従来の」ハイブリダイゼーションにより、またはリコンビナーゼ媒介プロセスにより、増幅されたDNAと特異的に結合して相互作用する可能性がある。こうした実験の結果を図3に示し、以下の実施例1で説明する。
【0051】
第2セットの実験は、この開裂活性の普遍性を調査するために実施され、この場合、蛍光試薬を使用し、dR−O−[C]nヌクレオチドは、図4に示すように蛍光体に結合する。この場合、dR−蛍光体は、オリゴヌクレオチドプローブの5’末端付近に位置し、その5’末端に結合するクエンチャーにきわめて接近して位置する。従来どおり、プローブの3’末端は異常な伸長またはプライマーのアーティファクトを防止するために適切にブロックされる。図4に示すように、プローブは、相補的増幅材料とハイブリダイズする場合、fpg(またはNfo)の基質になる可能性があり、この場合、この位置で主鎖を開裂することができる(そして、グリコシラーゼ活性がfpgまたはその他の非APエンドヌクレアーゼ酵素中に存在する場合、オリゴヌクレオチドの断片から分離した水媒体中に潜在的に蛍光体を直接遊離させる)。開裂が生じる場合、蛍光体およびクエンチャーが物理的に分離し、その結果、大腸菌NfoまたはexoIIIタンパク質を使用してTHFベースの蛍光プローブに関して前に説明した方法に類似する方法で、検出可能な蛍光が増加する。図5および図6はこうした実験の結果を示し、公知の単一ヌクレオチド多型(SNP)領域を特異的に対象としたプライマーおよびプローブを使用して、RPA反応をヒトゲノム標的に関して実施した実施例2で説明されている。
【0052】
これらの実験は、dR−O−[C]n基はNfoおよびfpgヌクレアーゼの基質であることを全体的に明確に証明する。さらに、相補的核酸鎖が蓄積して二本鎖の形成が可能になり、それによって、増幅が蛍光またはその他の機序によって生じたかどうかの判断を可能にする状況でのみ、プローブのヌクレアーゼ活性が生じるような基を含むプローブを構成することが可能である。したがって、これらのdR−O−[C]nヌクレオチド試薬は、fpg、Nfoまたはグリコシラーゼ/リアーゼ、および多様な用途の等価な酵素と組み合わせて広く応用することができる。
【0053】
配列に関するすべての引用、言及、特許、特許出願、その他の引用文献は、引用することにより本明細書に包含する。
【実施例】
【0054】
本発明について、以下の実施例でさらに定義する。これらの実施例は、本発明の好ましい実施態様を示しているが、具体的に説明するために記載されているにすぎないと考えるべきである。上記の説明およびこれらの実施例から、当業者は、本発明の本質的な特徴を確認し、本発明の精神および範囲から逸脱することなく、本発明に様々な変更および修正を加えて様々な用途および条件に適合させることができる。
【0055】
実施例1
内部dR−ビオチンを含むオリゴヌクレオチドプローブは、Nfoおよびfpgにより切断される。
【0056】
この実施例では、図2に大まかに示され、内部dR−ビオチンを含むオリゴヌクレオチドプローブは、Nfoおよびfpgにより切断することができる。この反応(全量150μL)は新鮮な試薬から混合し、75分間にわたって37℃で培養する。使用した条件は、50mMのトリスアセテート(pH 7.9)、14mMのMg−アセテート、100 mMのカリウムアセテート、2mMのDTT、それぞれ200nMのdNTP、6%PEG 35,000、3mMのATP、50mMのクレアチンリン酸、900ng/μLのT4gp32、120ng/μLのT4uvsX、30ng/μLのT4uvsY、360ng/μLの枯草菌DNAポリメラーゼIであった。DNAテンプレート3000コピーまたは水(負の対照として)は、指示どおりに加えた。ヌクレアーゼ、200ng/μLのNfoまたは50ng/μLのfpgは、指示どおりに加えた。プライマー、K2およびJ1は、それぞれ480nMで含め、プローブ、FpgProb1は120nMで加え、これらの配列は以下のとおりである。サンプルは、1容積の2%SDS/1容積のフェノール中で急冷し、20分間にわたって65℃で培養した。その後、サンプルは、標準の分子生物学的技術によりフェノール/クロロフォルム抽出し、2回エタノール沈殿させた。次に、各々のサンプルの半分は、16.5%変性ポリアクリルアミドゲル(尿素)上でホルムアミドのローディングバッファーに再懸濁させ、標準のプロトコルに従って視覚化した(FAM蛍光を使用して)。マーカーは、2pmolのプローブおよび2pmolの32ntマーカーオリゴヌクレオチドであった。
【0057】
【化1】
プローブおよび何らかの生物は、本明細書では、紫外線照射して活性化した場合、可視光を放出するFAM部分により視覚化した。標的DNA、2つの適切な増幅プライマー、dR−ビオチンプローブ、およびヌクレアーゼなし、Nfoタンパク質またはfpgタンパク質のいずれかを含む増幅反応(RPA)は、培養後に洗浄し、変性アクリルアミドゲル上でサイズ別に分離した後、紫外線に曝露した。次に、5’−FAMを保持するプローブまたは何らかの派生物が視覚化される(図3)。添加ヌクレアーゼが存在しない場合、42ヌクレオチド長のプローブはほとんど、32ヌクレオチド(指示)の対照標識付きプライマーより緩慢にその予想位置に移動する。わずかに緩慢に移動する(長めの)断片も、RPA中で培養されない適切なプローブ(♯1)と比べて観察される。これはおそらく、酵素調合物の一部に存在すると考えられるヌクレアーゼ(3’末端で徐々に減少する)によって、プローブのブロックが徐々に解除され、一度ブロックが解除されると、増幅標的に対するハイブリダイゼーション後に拡張可能であり、この種のネスト化した複製配列を形成するために生じる。しかし、Nfoが存在すると、この現象は予想よりはるかに顕著になり、プローブの大部分が伸長する。さらに、少量の比較的迅速に移動するプローブDNA(♯2)も見え、その後の伸長を伴わないdR−O−[C]n位置における開裂を示すため、Nfoタンパク質は、3’末端を単に「研磨する」のではなく、実際にdR−O−[C]n残基を攻撃していた。最後に、fpgタンパク質が反応環境に含まれる場合、fpgは、開裂後にブロック3’−末端を離れ、その結果、混合物中に存在するポリメラーゼ酵素によって伸長しないため、比較的迅速に移動する開裂プローブの大部分が見え、伸長した材料は検出されない。
【0058】
実施例2
内部dR蛍光体を含むオリゴヌクレオチドプローブによるDNA増幅
この実施例では、RPAは、dR−O−[C]nヌクレオチドが図4に示すように蛍光体に結合する蛍光試薬を使用して実験する。この場合、dR蛍光体は、オリゴヌクレオチドプローブの5’末端付近に位置し、その5’末端に結合するクエンチャーにきわめて接近して位置する。従来の実施例のとおり、プローブの3’末端は異常な伸長またはプライマーのアーティファクトを防止するために適切にブロックされる。
【0059】
この反応(全量50μL)は、冷凍−乾燥反応に関する標準RPAプロトコルに従って実施された。簡潔に述べると、凍結乾燥試薬はPEG、マグネシウム−アセテートおよびテンプレートと混合し、蛍光光度計内で38℃で20分間にわたって培養した(Twistaプロトタイプ;ESE GmbH、ドイツ)。使用した条件は、50mMのトリス/アセテート(pH 8.3)、14mMのマグネシウム−アセテート、100mMのカリウム−アセテート、5mMのDTT、それぞれ240nMのdNTP、5%のPEG 35,000、4%トレハロース2.5mM ATP、50mMのホスホ−クレアチン、300ng/μLのrb69gp32、273ng/μLのuvsX、120ng/μL uvsY、50ng/μLの黄色ブドウ状球菌DNAポリメラーゼIであった。図5の実験では、1000、100、10 または0コピーのDNAテンプレートを図に示されているように包含し、図6の実験では1000コピーのDNAテンプレートを包含した。25ng/μLのfpgヌクレアーゼを包含した。プライマーは、それぞれ360nMで包含し、プローブは120nMで包含し、それぞれの配列は以下のとおりであった。蛍光は20秒ごとに測定した(励起470nM、発光520nM)。サンプルは、4分間の培養時間で短時間の混合/回転を行って培養器から取り出し、培養器/蛍光光度計に戻した。分の時間に対して任意の蛍光単位をプロットした。
【0060】
ヒトゲノムの遺伝子座rs4824871の場合、使用したプライマーの配列F2およびR1並びにプローブは、以下のとおりであった。
【0061】
【化2】
ヒトゲノムの遺伝子座rsl207445の場合、使用したプライマーの配列F2およびR2並びにプローブは、以下の通りであった。
【0062】
【化3】
ヒトゲノムの遺伝子座rsl105561の場合、使用したプライマーの配列F1およびR1並びにプローブは、以下の通りであった。
【0063】
【化4】
ヒトゲノムの遺伝子座rs5923931の場合、使用したプライマーの配列F1およびR1並びにプローブは、以下の通りであった。
【0064】
【化5】
図5および6は、上記の実験の結果を示し、RPA反応は、公知のSNP領域を特異的に対象としたプライマーおよびプローブを使用して、ヒトゲノム標的に関して実施した。図5では、このような2つのゲノム領域は、fpgタンパク質の封入と共に、RPAおよび図4に示されている一般的な構造を持つプローブを使用して増幅した。標的ゲノムDNAは、合計標的コピー数を1000、100、10またはゼロ標的分子にするために追加し、こうして、標的に一致する単位複製配列の特異的な蓄積要件が確保される。6〜12分後(標的およびコピー数に応じて)、標的を含むこれらのサンプルの蛍光は明らかに上昇するが、標的が欠如しているサンプルは事実上蛍光は安定していることに注意する。図6では、4つのヒトゲノムDNA標的/プローブのセット(そのうち2つは図5にも使用されている)に関して類似のデータが示されており、何れの場合も、蛍光はDNA増幅のほぼ予想時間に上昇している。上記およびその他の好結果のプローブのほかに、RPA増幅/検出システムで十分に作用しなかったと思われるプローブが時折みられたが(おそらく、分析したプローブの10〜20%)、こうした欠陥の原因は未だ明らかではなく、場合によってはプローブの欠陥ではなく、可能性として他の場合のプローブの欠陥の結果としてRPA増幅の欠陥を表していることが考えられる。場合によっては、近接塩基の位置および性質、またはdR−O−[C]n基に対向する塩基の性質が、プローブの効果に影響した可能性があると考えられる。
【0065】
本明細書で特定の実施態様について詳細に開示しているが、これは、具体的に示すために一例として説明したのであって、添付の請求の範囲の範囲に関して制限することを意図しているのではない。特に、発明人は、請求の範囲により定義される本発明の精神および範囲を逸脱することなく、本発明に様々な置き換え、変更および修飾が施されることを意図している。その他の態様、利点および修正は、添付の請求項の範囲内と考えられる。提示される請求の範囲は、本明細書で開示されている本発明を表す。請求されていないその他の発明も想定されている。出願人は、かかる発明を後の請求の範囲で追跡する権利を留保する。
【特許請求の範囲】
【請求項1】
標的核酸の存在または不在を検出するプロセスであって、
(a)オリゴヌクレオチドプローブを標的核酸に接触させ、それにより前記プローブと前記標的核酸との間に複合体を形成するステップであって、前記プローブは、塩基を欠き、かつ1’位に炭素がある糖を有するdR−O−[C]nヌクレオチドを含み、前記1’位の炭素が、n個の炭素原子を含むリンカーの炭素原子に酸素原子を介して共有結合する、ステップと、
(b)APエンドヌクレアーゼ、DNAグリコシラーゼ/リアーゼおよびDNAグリコシラーゼからなる群より選択したヌクレアーゼに前記プローブ/核酸複合体を接触させるステップであって、前記ヌクレアーゼが、前記リンカーを前記複合体から切除するか、および/または前記dR−O−[C]nヌクレオチドで前記プローブを特異的に開裂する、ステップと、
(c)前記切除または開裂が行われたかどうかを検出するステップと
を含むプロセス。
【請求項2】
前記標的核酸が核酸増幅反応の生成物である、請求項1に記載のプロセス。
【請求項3】
前記増幅反応がリアルタイムで監視される、請求項2に記載のプロセス。
【請求項4】
前記増幅反応が前記反応の終点で監視される、請求項2に記載のプロセス。
【請求項5】
前記増幅反応がリコンビナーゼポリメラーゼ増幅(RPA)プロセスである、請求項2〜5の何れか1項に記載のプロセス。
【請求項6】
ステップ(a)が、前記プローブを前記標的核酸にハイブリダイズさせるステップを含む、請求項1に記載のプロセス。
【請求項7】
ステップ(a)が、(i)前記プローブをリコンビナーゼに接触させて、リコンビナーゼ/プローブ複合体を形成するステップと、(ii)前記リコンビナーゼ/プローブ複合体を前記核酸に接触させて、前記プローブ/標的核酸複合体を形成するステップとを含む、請求項1に記載のプロセス。
【請求項8】
前記ヌクレアーゼが、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である、請求項1〜7の何れか1項に記載のプロセス。
【請求項9】
nが3〜6である、請求項1〜8の何れか1項に記載のプロセス。
【請求項10】
nが6である、請求項9に記載のプロセス。
【請求項11】
前記プローブが、その3’−末端でブロックされてポリメラーゼ伸長を防止する、請求項1に記載のプロセス。
【請求項12】
前記リンカーが検出可能な標識に結合体化する、請求項1〜11の何れか1項に記載のプロセス。
【請求項13】
前記検出可能な標識が、ビオチン、ジオキシゲニン、ペプチド、蛍光体、クエンチャー、抗体および量子ドットからなる群より選択される、請求項12に記載のプロセス。
【請求項14】
前記検出可能な標識が蛍光体であり、前記プローブがクエンチャーにさらに結合体化されるか、または前記検出可能な標識がクエンチャーであり、前記プローブが蛍光体にさらに結合体化される、請求項12に記載のプロセス。
【請求項15】
前記プローブにさらに結合体化する前記クエンチャーまたは蛍光体が、前記プローブの一方の端部に結合体化する、請求項14に記載のプロセス。
【請求項16】
前記プローブの前記結合体化した末端が5’末端である、請求項15に記載のプロセス。
【請求項17】
ヌクレアーゼ活性が、前記dR−O−[C]n残基に結合した前記蛍光体またはクエンチャーを切除して遊離させ、前記検出ステップが、結合体化状態と遊離状態との間の蛍光性に差がある場合、その差を測定することを含む、請求項14〜16の何れか1項に記載のプロセス。
【請求項18】
前記蛍光体およびクエンチャーが4〜6塩基離れている、請求項14〜17の何れか1項に記載のプロセス。
【請求項19】
核酸を含む複合体のオリゴヌクレオチドを開裂するプロセスであって、前記オリゴヌクレオチド/核酸複合体をAPエンドヌクレアーゼ、DNAグリコシラーゼ/リアーゼおよびDNAグリコシラーゼからなる群より選択されるヌクレアーゼに接触させるステップを含み、
前記オリゴヌクレオチドが、塩基を欠き、かつ1’位に炭素がある糖を有するdR−O−[C]nヌクレオチドを含み、前記1’位の炭素が、n個の炭素原子を含むリンカーの炭素原子に酸素原子を介して共有結合し、
前記ヌクレアーゼが、dR−O−[C]nヌクレオチドにおいて前記プローブを特異的に開裂するプロセス。
【請求項20】
前記ヌクレアーゼが、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である、請求項19に記載のプロセス。
【請求項21】
nが3〜6である、請求項19または請求項20に記載のプロセス。
【請求項22】
nが6である、請求項21に記載のプロセス。
【請求項23】
前記オリゴヌクレオチドが、3’末端でブロックされて、ポリメラーゼ伸長を防止する、請求項19〜22の何れか1項に記載のプロセス。
【請求項24】
前記リンカーが検出可能な標識に結合体化する、請求項19〜23の何れか1項に記載のプロセス。
【請求項25】
前記検出可能な標識が、ビオチン、ジオキシゲニン、ペプチド、蛍光体、クエンチャー、抗体および量子ドットからなる群より選択される、請求項24に記載のプロセス。
【請求項26】
前記プロセスが、前記オリゴヌクレオチドを前記核酸に接触させて、前記オリゴヌクレオチド/核酸複合体を形成するステップをさらに含む、請求項19〜25の何れかの1項
に記載のプロセス。
【請求項27】
前記標的核酸が核酸増幅反応の生成物である、請求項26に記載のプロセス。
【請求項28】
前記増幅反応がリコンビナーゼポリメラーゼ増幅(RPA)プロセスである、請求項27に記載のプロセス。
【請求項29】
前記オリゴヌクレオチドを前記核酸に接触させるステップが、前記オリゴヌクレオチドを前記核酸にハイブリダイズさせるステップを含む、請求項26に記載のプロセス。
【請求項30】
前記オリゴヌクレオチドを前記核酸に接触させるステップが、(i)前記オリゴヌクレオチドをリコンビナーゼに接触させてリコンビナーゼ/オリゴヌクレオチド複合体を形成するステップと、(ii)前記リコンビナーゼ/オリゴヌクレオチド複合体を前記核酸に接触させて、前記オリゴヌクレオチド/核酸複合体を形成するステップとを含む、請求項26に記載のプロセス。
【請求項31】
前記オリゴヌクレオチドの開裂を検出するステップをさらに含む、請求項19〜25の何れか1項に記載のプロセス。
【請求項32】
検出がリアルタイムで監視される、請求項31に記載のプロセス。
【請求項33】
検出が、前記反応の終点で監視される、請求項31に記載のプロセス。
【請求項34】
(i)前記リンカーが蛍光体に結合体化され、かつ前記オリゴヌクレオチドがクエンチャーにさらに結合体化されるか、または(ii)前記リンカーがクエンチャーに結合体化され、かつ前記オリゴヌクレオチドが蛍光体にさらに結合体化される、のいずれかである、請求項31〜33の何れか1項に記載のプロセス。
【請求項35】
ヌクレアーゼ活性が、前記dR−O−[C]n残基に結合した前記蛍光体またはクエンチャーを切除して遊離させ、前記検出ステップが結合体化状態と遊離状態との間の蛍光性に差がある場合、その差を測定することを含む、請求項34に記載のプロセス。
【請求項36】
30〜60ヌクレオチド長のオリゴヌクレオチドを含むオリゴヌクレオチドプローブであって、前記オリゴヌクレオチドが、塩基を欠き、かつ1’位に炭素がある糖を有するdR−O−[C]nヌクレオチドを含み、前記1’位の炭素が、n個の炭素原子を含むリンカーの炭素原子に酸素原子を介して共有結合し、前記オリゴヌクレオチドが、10ヌクレオチド以下離れた蛍光体−クエンチャーの対を含み、前記dR−O−[C]nヌクレオチドが、前記蛍光体または前記クエンチャーに結合体化される、オリゴヌクレオチドプローブ。
【請求項37】
nが3〜6である、請求項36に記載のプローブ。
【請求項38】
nが6である、請求項37に記載のプローブ。
【請求項39】
前記プローブがその3’−末端でブロックされてポリメラーゼ伸長を防止する、請求項36に記載のプローブ。
【請求項40】
前記蛍光体およびクエンチャーが4〜6塩基離れている、請求項36に記載のプローブ。
【請求項41】
前記dR−O−[C]nヌクレオチドに結合体化されていない前記蛍光体またはクエンチャーが、前記プローブの一方の末端に結合体化している、請求項36に記載のプローブ。
【請求項42】
前記プローブの前記結合体化した末端が5’末端である、請求項41に記載のプローブ。
【請求項43】
キットであって、
(i)塩基を欠き、かつ1’位に炭素がある糖を有するdR−O−[C]nヌクレオチドを含むオリゴヌクレオチドであって、前記1’位の炭素が、n個の炭素原子を含むリンカーの炭素原子に酸素原子を介して共有結合する、オリゴヌクレオチド、ならびに
(ii)APエンドヌクレアーゼ、DNAグリコシラーゼ/リアーゼおよびDNAグリコシラーゼからなる群より選択されるヌクレアーゼを含む、キット。
【請求項44】
前記ヌクレアーゼが、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である、請求項43に記載のキット。
【請求項45】
塩基を欠き、かつ1’位に炭素がある糖を有するdR−O−[C]nヌクレオチドを包含するオリゴヌクレオチドであって、前記1’位の炭素が、酸素原子を介して、n個の炭素原子を含むリンカーの炭素原子に共有結合する、オリゴヌクレオチド、ならびに
APエンドヌクレアーゼ、DNAグリコシラーゼ/リアーゼおよびDNAグリコシラーゼからなる群より選択されるヌクレアーゼ
を含む、反応混合物。
【請求項46】
前記ヌクレアーゼが、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である、請求項45に記載の反応混合物。
【請求項47】
nが3〜6である、請求項45に記載の反応混合物。
【請求項48】
nが6である、請求項47に記載の反応混合物。
【請求項49】
前記プローブがその3’−末端でブロックされてポリメラーゼ伸長を防止する、請求項45に記載の反応混合物。
【請求項50】
前記リンカーが検出可能な標識に結合体化する、請求項45に記載の反応混合物。
【請求項51】
前記検出可能な標識が、ビオチン、ジオキシゲニン、ペプチド、蛍光体、クエンチャー、抗体および量子ドットからなる群より選択される、請求項50に記載の反応混合物。
【請求項52】
前記反応混合物が凍結乾燥される、請求項45〜51の何れか1項に記載の反応混合物。
【請求項53】
容器をさらに含む、請求項45〜52の何れか1項に記載の反応混合物。
【請求項54】
前記容器がチューブである、請求項53に記載の反応混合物。
【請求項55】
前記オリゴヌクレオチドに対して相補的な配列を含む標的核酸をさらに含む、請求項45〜54の何れか1項に記載の反応混合物。
【請求項1】
標的核酸の存在または不在を検出するプロセスであって、
(a)オリゴヌクレオチドプローブを標的核酸に接触させ、それにより前記プローブと前記標的核酸との間に複合体を形成するステップであって、前記プローブは、塩基を欠き、かつ1’位に炭素がある糖を有するdR−O−[C]nヌクレオチドを含み、前記1’位の炭素が、n個の炭素原子を含むリンカーの炭素原子に酸素原子を介して共有結合する、ステップと、
(b)APエンドヌクレアーゼ、DNAグリコシラーゼ/リアーゼおよびDNAグリコシラーゼからなる群より選択したヌクレアーゼに前記プローブ/核酸複合体を接触させるステップであって、前記ヌクレアーゼが、前記リンカーを前記複合体から切除するか、および/または前記dR−O−[C]nヌクレオチドで前記プローブを特異的に開裂する、ステップと、
(c)前記切除または開裂が行われたかどうかを検出するステップと
を含むプロセス。
【請求項2】
前記標的核酸が核酸増幅反応の生成物である、請求項1に記載のプロセス。
【請求項3】
前記増幅反応がリアルタイムで監視される、請求項2に記載のプロセス。
【請求項4】
前記増幅反応が前記反応の終点で監視される、請求項2に記載のプロセス。
【請求項5】
前記増幅反応がリコンビナーゼポリメラーゼ増幅(RPA)プロセスである、請求項2〜5の何れか1項に記載のプロセス。
【請求項6】
ステップ(a)が、前記プローブを前記標的核酸にハイブリダイズさせるステップを含む、請求項1に記載のプロセス。
【請求項7】
ステップ(a)が、(i)前記プローブをリコンビナーゼに接触させて、リコンビナーゼ/プローブ複合体を形成するステップと、(ii)前記リコンビナーゼ/プローブ複合体を前記核酸に接触させて、前記プローブ/標的核酸複合体を形成するステップとを含む、請求項1に記載のプロセス。
【請求項8】
前記ヌクレアーゼが、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である、請求項1〜7の何れか1項に記載のプロセス。
【請求項9】
nが3〜6である、請求項1〜8の何れか1項に記載のプロセス。
【請求項10】
nが6である、請求項9に記載のプロセス。
【請求項11】
前記プローブが、その3’−末端でブロックされてポリメラーゼ伸長を防止する、請求項1に記載のプロセス。
【請求項12】
前記リンカーが検出可能な標識に結合体化する、請求項1〜11の何れか1項に記載のプロセス。
【請求項13】
前記検出可能な標識が、ビオチン、ジオキシゲニン、ペプチド、蛍光体、クエンチャー、抗体および量子ドットからなる群より選択される、請求項12に記載のプロセス。
【請求項14】
前記検出可能な標識が蛍光体であり、前記プローブがクエンチャーにさらに結合体化されるか、または前記検出可能な標識がクエンチャーであり、前記プローブが蛍光体にさらに結合体化される、請求項12に記載のプロセス。
【請求項15】
前記プローブにさらに結合体化する前記クエンチャーまたは蛍光体が、前記プローブの一方の端部に結合体化する、請求項14に記載のプロセス。
【請求項16】
前記プローブの前記結合体化した末端が5’末端である、請求項15に記載のプロセス。
【請求項17】
ヌクレアーゼ活性が、前記dR−O−[C]n残基に結合した前記蛍光体またはクエンチャーを切除して遊離させ、前記検出ステップが、結合体化状態と遊離状態との間の蛍光性に差がある場合、その差を測定することを含む、請求項14〜16の何れか1項に記載のプロセス。
【請求項18】
前記蛍光体およびクエンチャーが4〜6塩基離れている、請求項14〜17の何れか1項に記載のプロセス。
【請求項19】
核酸を含む複合体のオリゴヌクレオチドを開裂するプロセスであって、前記オリゴヌクレオチド/核酸複合体をAPエンドヌクレアーゼ、DNAグリコシラーゼ/リアーゼおよびDNAグリコシラーゼからなる群より選択されるヌクレアーゼに接触させるステップを含み、
前記オリゴヌクレオチドが、塩基を欠き、かつ1’位に炭素がある糖を有するdR−O−[C]nヌクレオチドを含み、前記1’位の炭素が、n個の炭素原子を含むリンカーの炭素原子に酸素原子を介して共有結合し、
前記ヌクレアーゼが、dR−O−[C]nヌクレオチドにおいて前記プローブを特異的に開裂するプロセス。
【請求項20】
前記ヌクレアーゼが、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である、請求項19に記載のプロセス。
【請求項21】
nが3〜6である、請求項19または請求項20に記載のプロセス。
【請求項22】
nが6である、請求項21に記載のプロセス。
【請求項23】
前記オリゴヌクレオチドが、3’末端でブロックされて、ポリメラーゼ伸長を防止する、請求項19〜22の何れか1項に記載のプロセス。
【請求項24】
前記リンカーが検出可能な標識に結合体化する、請求項19〜23の何れか1項に記載のプロセス。
【請求項25】
前記検出可能な標識が、ビオチン、ジオキシゲニン、ペプチド、蛍光体、クエンチャー、抗体および量子ドットからなる群より選択される、請求項24に記載のプロセス。
【請求項26】
前記プロセスが、前記オリゴヌクレオチドを前記核酸に接触させて、前記オリゴヌクレオチド/核酸複合体を形成するステップをさらに含む、請求項19〜25の何れかの1項
に記載のプロセス。
【請求項27】
前記標的核酸が核酸増幅反応の生成物である、請求項26に記載のプロセス。
【請求項28】
前記増幅反応がリコンビナーゼポリメラーゼ増幅(RPA)プロセスである、請求項27に記載のプロセス。
【請求項29】
前記オリゴヌクレオチドを前記核酸に接触させるステップが、前記オリゴヌクレオチドを前記核酸にハイブリダイズさせるステップを含む、請求項26に記載のプロセス。
【請求項30】
前記オリゴヌクレオチドを前記核酸に接触させるステップが、(i)前記オリゴヌクレオチドをリコンビナーゼに接触させてリコンビナーゼ/オリゴヌクレオチド複合体を形成するステップと、(ii)前記リコンビナーゼ/オリゴヌクレオチド複合体を前記核酸に接触させて、前記オリゴヌクレオチド/核酸複合体を形成するステップとを含む、請求項26に記載のプロセス。
【請求項31】
前記オリゴヌクレオチドの開裂を検出するステップをさらに含む、請求項19〜25の何れか1項に記載のプロセス。
【請求項32】
検出がリアルタイムで監視される、請求項31に記載のプロセス。
【請求項33】
検出が、前記反応の終点で監視される、請求項31に記載のプロセス。
【請求項34】
(i)前記リンカーが蛍光体に結合体化され、かつ前記オリゴヌクレオチドがクエンチャーにさらに結合体化されるか、または(ii)前記リンカーがクエンチャーに結合体化され、かつ前記オリゴヌクレオチドが蛍光体にさらに結合体化される、のいずれかである、請求項31〜33の何れか1項に記載のプロセス。
【請求項35】
ヌクレアーゼ活性が、前記dR−O−[C]n残基に結合した前記蛍光体またはクエンチャーを切除して遊離させ、前記検出ステップが結合体化状態と遊離状態との間の蛍光性に差がある場合、その差を測定することを含む、請求項34に記載のプロセス。
【請求項36】
30〜60ヌクレオチド長のオリゴヌクレオチドを含むオリゴヌクレオチドプローブであって、前記オリゴヌクレオチドが、塩基を欠き、かつ1’位に炭素がある糖を有するdR−O−[C]nヌクレオチドを含み、前記1’位の炭素が、n個の炭素原子を含むリンカーの炭素原子に酸素原子を介して共有結合し、前記オリゴヌクレオチドが、10ヌクレオチド以下離れた蛍光体−クエンチャーの対を含み、前記dR−O−[C]nヌクレオチドが、前記蛍光体または前記クエンチャーに結合体化される、オリゴヌクレオチドプローブ。
【請求項37】
nが3〜6である、請求項36に記載のプローブ。
【請求項38】
nが6である、請求項37に記載のプローブ。
【請求項39】
前記プローブがその3’−末端でブロックされてポリメラーゼ伸長を防止する、請求項36に記載のプローブ。
【請求項40】
前記蛍光体およびクエンチャーが4〜6塩基離れている、請求項36に記載のプローブ。
【請求項41】
前記dR−O−[C]nヌクレオチドに結合体化されていない前記蛍光体またはクエンチャーが、前記プローブの一方の末端に結合体化している、請求項36に記載のプローブ。
【請求項42】
前記プローブの前記結合体化した末端が5’末端である、請求項41に記載のプローブ。
【請求項43】
キットであって、
(i)塩基を欠き、かつ1’位に炭素がある糖を有するdR−O−[C]nヌクレオチドを含むオリゴヌクレオチドであって、前記1’位の炭素が、n個の炭素原子を含むリンカーの炭素原子に酸素原子を介して共有結合する、オリゴヌクレオチド、ならびに
(ii)APエンドヌクレアーゼ、DNAグリコシラーゼ/リアーゼおよびDNAグリコシラーゼからなる群より選択されるヌクレアーゼを含む、キット。
【請求項44】
前記ヌクレアーゼが、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である、請求項43に記載のキット。
【請求項45】
塩基を欠き、かつ1’位に炭素がある糖を有するdR−O−[C]nヌクレオチドを包含するオリゴヌクレオチドであって、前記1’位の炭素が、酸素原子を介して、n個の炭素原子を含むリンカーの炭素原子に共有結合する、オリゴヌクレオチド、ならびに
APエンドヌクレアーゼ、DNAグリコシラーゼ/リアーゼおよびDNAグリコシラーゼからなる群より選択されるヌクレアーゼ
を含む、反応混合物。
【請求項46】
前記ヌクレアーゼが、エンドヌクレアーゼIV(Nfo)または8−オキソグアニンDNAグリコシラーゼ(fpg)である、請求項45に記載の反応混合物。
【請求項47】
nが3〜6である、請求項45に記載の反応混合物。
【請求項48】
nが6である、請求項47に記載の反応混合物。
【請求項49】
前記プローブがその3’−末端でブロックされてポリメラーゼ伸長を防止する、請求項45に記載の反応混合物。
【請求項50】
前記リンカーが検出可能な標識に結合体化する、請求項45に記載の反応混合物。
【請求項51】
前記検出可能な標識が、ビオチン、ジオキシゲニン、ペプチド、蛍光体、クエンチャー、抗体および量子ドットからなる群より選択される、請求項50に記載の反応混合物。
【請求項52】
前記反応混合物が凍結乾燥される、請求項45〜51の何れか1項に記載の反応混合物。
【請求項53】
容器をさらに含む、請求項45〜52の何れか1項に記載の反応混合物。
【請求項54】
前記容器がチューブである、請求項53に記載の反応混合物。
【請求項55】
前記オリゴヌクレオチドに対して相補的な配列を含む標的核酸をさらに含む、請求項45〜54の何れか1項に記載の反応混合物。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公表番号】特表2012−527239(P2012−527239A)
【公表日】平成24年11月8日(2012.11.8)
【国際特許分類】
【出願番号】特願2012−511958(P2012−511958)
【出願日】平成22年5月18日(2010.5.18)
【国際出願番号】PCT/US2010/035232
【国際公開番号】WO2010/135310
【国際公開日】平成22年11月25日(2010.11.25)
【出願人】(500204577)バイオサイト インコーポレイテッド (14)
【Fターム(参考)】
【公表日】平成24年11月8日(2012.11.8)
【国際特許分類】
【出願日】平成22年5月18日(2010.5.18)
【国際出願番号】PCT/US2010/035232
【国際公開番号】WO2010/135310
【国際公開日】平成22年11月25日(2010.11.25)
【出願人】(500204577)バイオサイト インコーポレイテッド (14)
【Fターム(参考)】
[ Back to top ]