
DNAディスプレイを利用した核酸送達ビヒクルの同定方法
本発明は、例えば核酸分子の細胞内送達を促進する分子を同定するための方法および組成物をその特徴とする。本発明の方法および組成物は、ライブラリー(例えば小分子またはタンパク質のライブラリー)を、そのライブラリーの各メンバーをコードする核酸(例えば、RNAまたはDNA)と結合させる任意のディスプレイ法を利用するものである。
【発明の詳細な説明】
【背景技術】
【0001】
発明の背景
効率的な細胞質送達は核酸療法の開発において依然として重要な課題である。細胞内への核酸の標的化送達は、細胞表面の分子と結合してそれにより内部移行されるリガンド(例えば、抗体、抗体フラグメント、抗体模倣体、ペプチド、又は小分子)と核酸とを結合することによって実現可能である。標準的なディスプレイ技術は、定向進化プロセスを推進するための選択圧力として標的結合を利用しており、そのため標的と最も高い親和性で結合するリガンドライブラリーのメンバーは、さらに高ストリンジェントな選択条件下でも生き残り、濃縮されていくという、選択上、有利な点を有する。しかしながら、標的と最も高い親和性で結合するメンバーが機能的に最も関連性の高いライブラリーのメンバーであるとは限らない場合もある。例えば、細胞内に容易に侵入し、細胞質に到達することが可能なライブラリーのメンバーは、核酸送達用の標的ビヒクルとして最も効果的である可能性が高い。
【0002】
したがって、内部移行の機序とは無関係に、細胞内に侵入して細胞質に到達可能な核酸と結合したリガンドを直接的に選択するために細胞質への侵入を利用するディスプレイ技術が求められている。
【発明の概要】
【0003】
本発明は、例えば核酸分子の細胞内送達を促進する分子を同定するための方法及び組成物をその特徴とする。本発明の方法及び組成物は、ライブラリー(例えば、小分子またはタンパク質のライブラリー)と、ライブラリーの各メンバーをコードまたはタグする核酸(例えば、RNAまたはDNA)とが結合する、任意のディスプレイ法を利用する。
【0004】
したがって第1の態様において、本発明は、核酸ディスプレイライブラリーを含む組成物であって、該核酸ディスプレイライブラリーのメンバーと、細胞内読み出しシグナルを発生する分子とが連結する組成物をその特徴とする。別の態様において、本発明は、核酸ディスプレイライブラリーを含む組成物であって、該核酸ディスプレイライブラリーのメンバーとストレプトアビジン分子とが連結し、該ストレプトアビジン分子と、細胞内読み出しシグナルを発生する分子とが更に連結する組成物をその特徴とする。いずれの態様においても、細胞内読み出しシグナルを発生する分子は、例えば核酸(例えば、レポーター遺伝子、転写因子遺伝子、RNA、またはアンチセンス遺伝子)、タンパク質(例えば、緑色蛍光タンパク質(GFP))、ペプチド、または小分子(例えば、フルオロフォア)であってよい。本明細書に記載の組成物の核酸ディスプレイライブラリーの核酸分子は、外因性ポリメラーゼ(例えば、T7 RNAポリメラーゼ)プロモーターの制御下で細胞内で発現させることができる。
【0005】
別の態様において、本発明は、分枝鎖リンカーを介してライブラリーのメンバーに多量体小分子種が結合した、DNAによりコードされた小分子ライブラリーを含む組成物をその特徴とする。
【0006】
本発明によれば、更に、リンカー種によって修飾されたDNA塩基を介してライブラリーのDNAに2個以上の小分子(例えば、2個、3個、4個、5個、6個、7個、8個、9個、10個、またはそれよりも多い小分子)が結合した、DNAによりコードされた小分子ライブラリーを含む組成物が提供される。
【0007】
更なる一態様において、本発明は、核酸の細胞内送達を促進する分子を同定するための方法であって、前記分子が核酸ディスプレイライブラリーのメンバーに連結されており、かつ前記核酸ライブラリーの前記メンバーが更にある遺伝子に連結されている方法を、その特徴とする。この方法では、細胞を核酸ディスプレイライブラリーと接触させ、前記細胞内への前記核酸の送達を促進する分子に連結された前記核酸ディスプレイライブラリーのメンバーを、前記核酸ライブラリーのメンバーに連結された前記遺伝子の発現を監視することによって同定する。一局面において、前記核酸ライブラリーのメンバーに連結された前記遺伝子の発現は外因性RNAポリメラーゼプロモーターの制御下にあり、前記細胞は細胞の細胞質中でRNAポリメラーゼ(例えば、T7 RNAポリメラーゼ)を発現する。別の局面において、細胞は、細胞内に送達される前記核酸ライブラリーのメンバーを修飾することが可能な1種類以上の酵素(例えば、DNAメチルトランスフェラーゼ)を発現する。
【0008】
本発明は更に、核酸の細胞内送達を促進する分子を同定するための方法であって、前記分子が核酸ディスプレイライブラリーのメンバーに連結されており、前記核酸ライブラリーの前記メンバーが更にRNAポリメラーゼ結合部位に連結されている方法を、その特徴とする。この方法では、細胞を前記核酸ディスプレイライブラリーと接触させ、前記細胞内への前記核酸の送達を促進する分子に連結された前記核酸ディスプレイライブラリーのメンバーを、前記核酸ライブラリーのメンバーの核酸部分の細胞内における転写を監視および解読することによって同定する。コードするライブラリーがdsDNA由来のものである場合には、前記細胞内に存在するRNAポリメラーゼ(例えば、T7 RNAポリメラーゼ)が転写を触媒する。ライブラリーがssRNAである別の例では、前記細胞内に存在するRNA依存性RNAポリメラーゼが転写を触媒する(例えば、ポリオウイルス3Dpol、水疱性口内炎ウイルスL、およびC型肝炎NS5bタンパク質)。あるいは、前記細胞内に存在する逆転写酵素がDNAの重合を触媒する。ライブラリーがssDNAである別の態様では、前記細胞内に存在するssDNA依存性RNAポリメラーゼが転写を触媒する(例えば、N4バクテリオファージssDNA依存性RNAポリメラーゼ)。本発明においてssDNAからなるライブラリーに対して使用することが可能なssDNA依存性DNAポリメラーゼも存在する。
【0009】
上記に述べた方法においては、核酸の細胞内送達を促進する分子は核酸分子であってよい(例えば、RNAi、miRNA、アンチセンス核酸分子、または遺伝子)。あるいは、この分子はタンパク質、ペプチド、または小分子であってもよい。
【0010】
更なる一態様において、本発明は、第2の分子の細胞内送達を促進する第1の分子を同定するための方法であって、前記第1および第2の分子が核酸ライブラリーのメンバーに連結されている方法をその特徴とする。この方法は、細胞を前記核酸ディスプレイライブラリーと接触させる工程と、前記細胞内に存在する1種類以上の酵素による前記核酸ライブラリーのメンバーの修飾を監視することによって、前記細胞内への前記第2の分子の送達を促進する前記第1の分子に連結された前記核酸ディスプレイライブラリーのメンバーを同定する工程とを含む。この態様では、前記第1または第2の分子は、核酸分子(例えば、RNAi、miRNA、アンチセンス核酸分子、または遺伝子)、タンパク質、ペプチド、または小分子である。
【0011】
本明細書に記載の態様のいずれにおいても、核酸ディスプレイライブラリーはdsDNAディスプレイライブラリーであってよい(例えば、CISディスプレイライブラリー、ピューロマイシン媒介dsDNAディスプレイライブラリー、CDTディスプレイライブラリー、小分子に結合させたdsDNAライブラリー、およびストレプトアビジンディスプレイライブラリー)。
【0012】
「フルオロフォア」とは、分子を蛍光性とする分子の構成要素または官能基を意味する。例示的なフルオロフォアとしては、フルオロセイン、緑色蛍光タンパク質(GFP)、黄色蛍光タンパク質(YFP)、Alexa Fluor色素、Cy色素(GE Healthcare)、核酸プローブ(例えば、DAPI、臭化エチジウム、アクリジンオレンジ、またはヨウ化プロピジウム)、ヒドロキシクマリン、アミノクマリン、エトキシクマリン、ローダミン、BODIPY-FL、テキサスレッド、またはTRITCが挙げられる。
【0013】
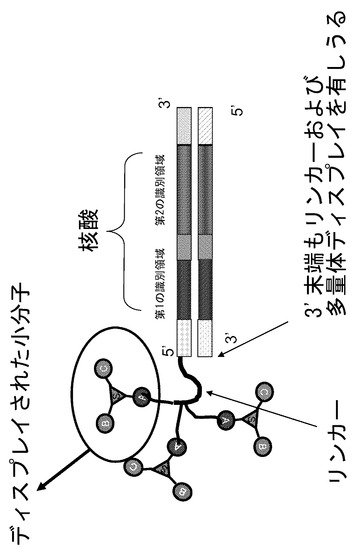
「リンカー」とは、ライブラリーの核酸部分を機能性のディスプレイされた化学種と連結する分子を意味する。このようなリンカーは当技術分野においては周知であり、ライブラリー合成において使用することが可能なものとしては、これらに限定されるものではないが、5'-O-ジメトキシトリチル-l',2'-ジデオキシリボース-3'-[(2-シアノエチル)-(N,N-ジイソプロピル)]-ホスホロアミダイト、9-O-ジメトキシトリチル-トリエチレングリコール,1-[(2-シアノエチル)-(N,N-ジイソプロピル)]-ホスホロアミダイト、3-(4,4'-ジメトキシトリチルオキシ)プロピル-1-[(2-シアノエチル)-(N,N-ジイソプロピル)]-ホスホロアミダイト、および18-O-ジメトキシトリチルヘキサエチレングリコール,1-[(2-シアノエチル)-(N,N-ジイソプロピル)]-ホスホロアミダイトが挙げられる。このようなリンカーは異なる組み合わせで互いに縦に付加して、異なる所望の長さのリンカーを生成することができる。「分枝鎖リンカー」とは、ライブラリーの核酸部分をライブラリーの2以上の同一の官能性の化学種と連結する分子を意味する。分枝鎖リンカーは当技術分野では周知のものであり、その例は、対称的もしくは非対称的ダブラー(doubler)(1)および(2)、または対称的トレブラー(trebler)(3)から構成されうる。例として、Newcome et al., Dendritic Molecules: Concepts, Synthesis, Perspectives, VCR Publishers (1996); Boussif et al., Proc. Natl. Acad. Sci. USA 92: 7297-7301 (1995); および Jansen et al., Science 266: 1226 (1994)を参照されたい。
【0014】
「核酸ディスプレイライブラリー」とは、所望の標的に結合可能な分子を生成するためのインビトロでのタンパク質および/もしくはペプチド進化、ならびに/または小分子および/もしくは核酸進化(例えば、ssRNAまたはssDNA)の発見に使用されるディスプレイ技術を意味する。修飾ペプチドを含むタンパク質およびペプチドの場合では、このプロセスによりピューロマイシン連結部を介してmRNA前駆体またはdsDNAと結合した翻訳されたペプチドまたはタンパク質が得られる。一部の核酸ディスプレイの場合では、タンパク質またはペプチドは、核酸と共有結合または非共有結合により会合するタンパク質を介してmRNA、ssDNA、またはdsDNAと結合される。小分子ディスプレイの場合では、核酸は小分子と共有結合する。核酸ディスプレイの場合では、ssRNAまたはssDNAのランダム化された領域を直接使用する。次いで、核酸ライブラリー複合体が選択工程(例えば、アフィニティークロマトグラフィー)において、固定された標的と結合する。次いで、十分に結合した核酸コンジュゲートを回収し、ポリメラーゼ連鎖反応により増幅する。その結果、対象とする分子に対する所望の性質(例えば、親和性または特異性)を有する結合分子をコードしたヌクレオチド配列が得られる。核酸ディスプレイライブラリーにはdsDNAディスプレイライブラリーが含まれうる。例示的なdsDNAディスプレイライブラリーとしては、CIS dsDNAディスプレイライブラリー、ピューロマイシン媒介dsDNAディスプレイライブラリー、CDT dsDNAディスプレイライブラリー、小分子に結合されたdsDNAライブラリー、およびストレプトアビジンdsDNAディスプレイライブラリーが挙げられる。例として、Odegrip et al., Proc. Natl. Acad. Sci. USA 101: 2806-2810 (2004); Kurz et al., Chembiochem. 2: 666-672 (2001); Fitzgerald, Drug Discov. Today 5: 253-258 (2000); およびClark et al., Nat. Chem. Biol. 5: 647-654 (2009) を参照されたい。
【0015】
「核酸」とは、モノマーヌクレオチド(例えば、5個以上のヌクレオチド)からなる巨大分子を意味する。核酸にはデオキシリボ核酸(DNA)(例えば、cDNA、mtDNA、および2本鎖DNA(dsDNA))およびリボ核酸(RNA)(例えばmiRNA、siRNA、snRNA、snoRNA、shRNA、RNAiおよびmRNA)が含まれる。核酸は2本鎖、1本鎖、または単離されたもの(例えば部分精製されたもの、本質的に純粋なもの、合成されたもの、組み換えにより生成したもの)であってよい。核酸は1個以上のヌクレオチドの付加、欠失、置換、および/または改変によって改変させることができる。こうした改変には、核酸の末端または内部への(1個以上のヌクレオチドにおける)非ヌクレオチド物質の付加が含まれうる。本発明の核酸分子中のヌクレオチドには、天然に存在しないヌクレオチドなどの非標準的ヌクレオチドも含まれうる。
【0016】
「タンパク質」、「ポリペプチド」、「ポリペプチドフラグメント」、または「ペプチド」とは、天然に存在するポリペプチドまたはペプチドの全体または一部を構成する、あるいは、天然に存在しないポリペプチドまたはペプチドを構成する、翻訳後修飾(例えばグリコシル化またはリン酸化)によらない、2個以上のアミノ酸の任意の鎖を意味する。
【0017】
「小分子」とは、約1000ダルトンよりも低い分子量を有する分子を意味する。小分子は有機または無機であってよく、例えば化合物ライブラリーまたは天然の供給源から単離するか、あるいは既知の化合物の誘導体化によって得ることができる。
【0018】
本発明の他の特徴および利点は、以下の詳細な説明、図面、実施例および特許請求の範囲より明らかとなる。
【図面の簡単な説明】
【0019】
【図1】2個以上の化学分子が、多官能性リンカー部分を用いて(例えば、デンドリマーディスプレイとして)ライブラリーの核酸部分に結合されるように例示的なライブラリーが生成されることを示す模式図である。
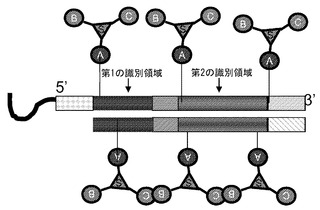
【図2】2個以上の化学分子が、多官能性リンカー部分を用いてライブラリーの核酸部分の両方の鎖に結合されるように例示的なライブラリーが生成されることを示す模式図である。
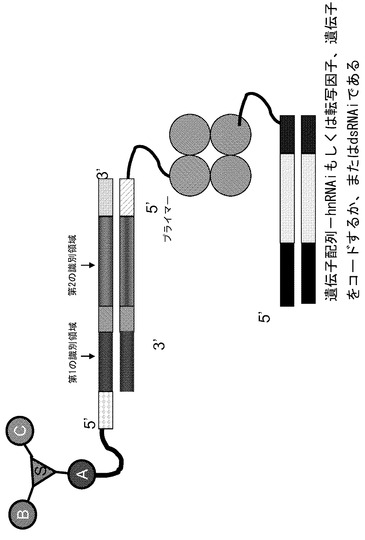
【図3】核酸ライブラリーに結合し、更に発現遺伝子またはdsRNAiに結合したストレプトアビジン(4量体)を示す模式図である。
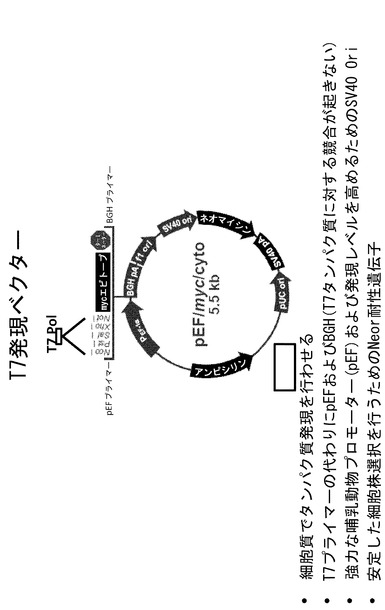
【図4】T7発現ベクターの代表例を示す模式図である。
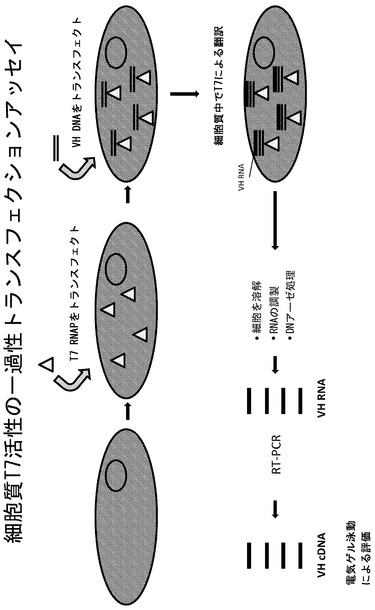
【図5】細胞質T7活性を検出するための一過性トランスフェクションアッセイを説明した模式図である。
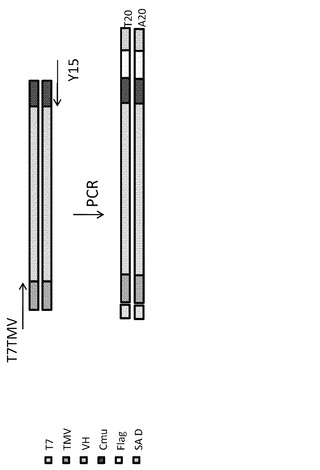
【図6】VH抗体ドメインのコード領域の上流にT7プロモーターを含むPCRフラグメントの構成要素を示す模式図である。
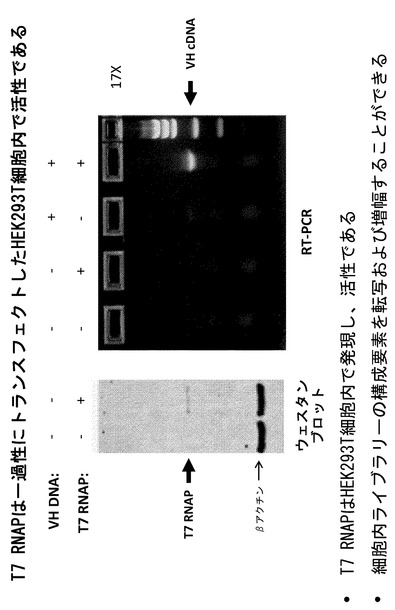
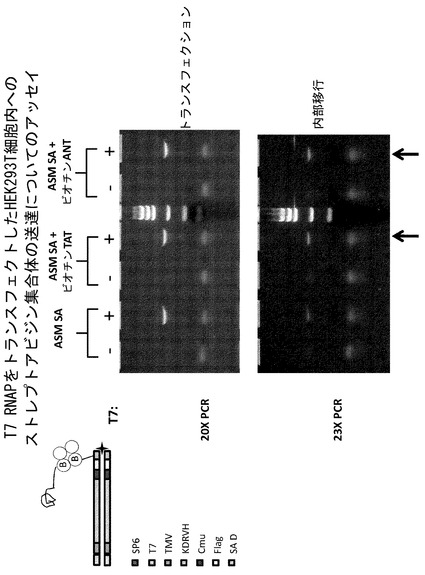
【図7】T7 RNAポリメラーゼ(RNAP)が、一過性にトランスフェクトされたHEK293T細胞内で活性であることを示すウェスタンブロットおよびRT-PCRアッセイである。細胞溶解物をSDS-PAGEにより分離し、T7ポリメラーゼに対するモノクローナル抗体を用いてウェスタンブロット分析に供した。抗T7ポリメラーゼ抗体は、このタンパク質の予想される分子量(約99kDa)と一致するバンドを認識する。RT-PCRアッセイは、VH PCR鋳型がHEK293T細胞内でT7ポリメラーゼによって転写されることを示している。コントロールレーンは、RT-PCR活性がT7ポリメラーゼの発現に依存しており、T7ポリメラーゼプロモーターを含む鋳型の存在下においてのみ生ずることを示している。
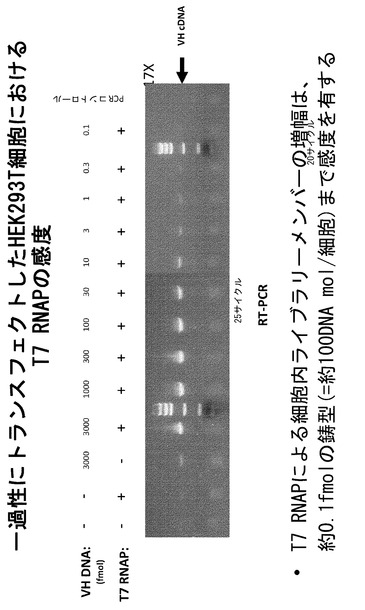
【図8】VH PCR鋳型の量を一過性トランスフェクション中に滴定することによってT7ポリメラーゼRT-PCRアッセイの感度を試験するRT-PCRアッセイである。
【図9】T7 RNAPが、一過性にトランスフェクトされたVCaP前立腺癌細胞内で活性であることを示すRT-PCRアッセイである。
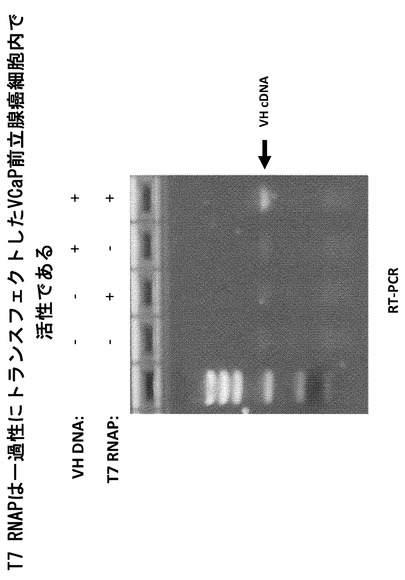
【図10】安定的な前立腺癌細胞株からのT7 RNAP転写産物の検出を示すRT-PCRアッセイである。
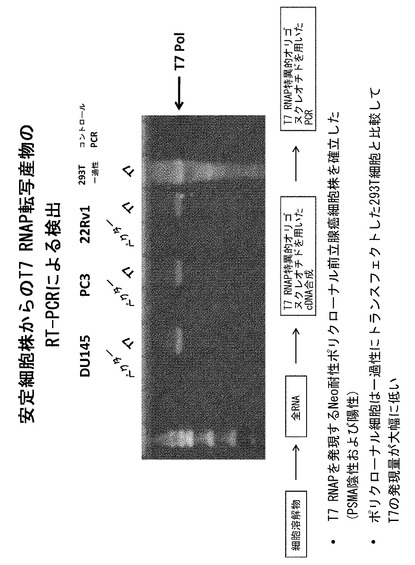
【図11】22rV1_T7細胞株が活性T7ポリメラーゼを発現することを示すRT-PCRアッセイである。
【図12】ビオチン化ペプチドまたはVH結合物質とストレプトアビジンとの複合体の集合を示す模式図である。
【図13】集合したVHまたはペプチド複合体、またはビオチン化ペプチドもしくはタンパク質を有さない集合体のHEK293T細胞内への一過性トランスフェクションを示す模式図である。
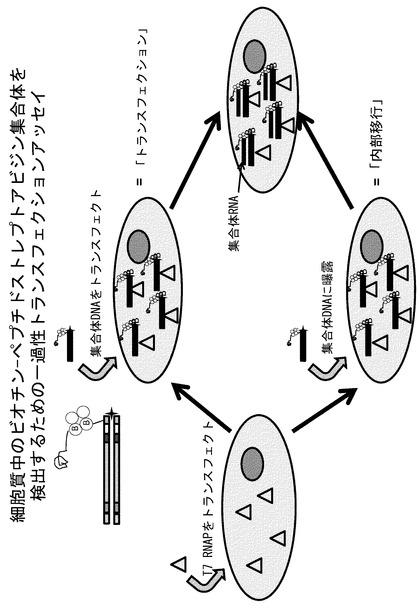
【図14】T7 RNAPをトランスフェクトしたHEK293T細胞内へのストレプトアビジン集合体の送達を示すアッセイである。
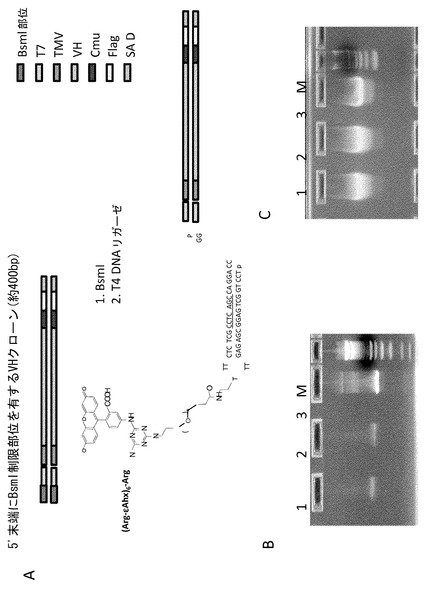
【図15】図15Aは、ペプチド-dsDNAコンストラクトの合成の模式図である。VHクローンをPCR増幅してBsmI部位をT7プロモーターの5'側の上流に付与した。制限酵素による消化および精製の後、コンストラクトをHP-1-DATF-R7(DTAFおよび(-Arg-εAhx)6-Argペプチドにより修飾したヘッドピース)にライゲートした。図15Bは、ライゲーション反応の電気泳動ゲルである(レーン1および2: VHにライゲートされた異なるHP-1試料; レーン3: ライゲートしていないVHPCR産物; M: マーカー)。図15Cは、T7プロモーター活性の確認を示すゲルである。このゲルは、図15Bのレーン1〜3からの試料を用いたT7 Megascript (Ambion)反応を示している。
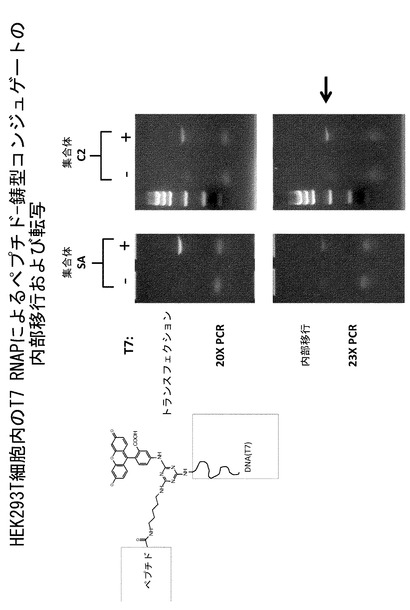
【図16】HEK293T細胞内におけるT7 RNAPによるペプチド-鋳型コンジュゲートの内部移行および転写を示すアッセイである。
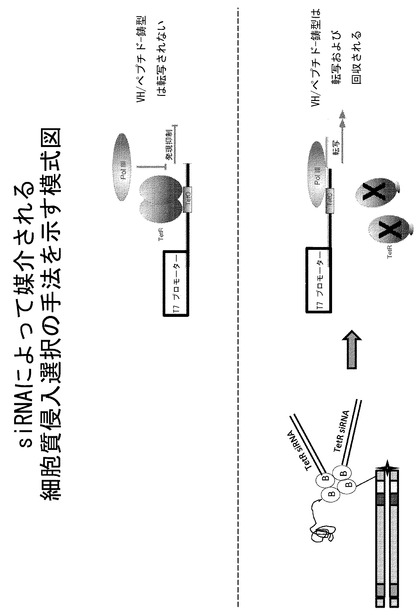
【図17】siRNAによって媒介される細胞質侵入選択の手法を示す模式図である。
【発明を実施するための形態】
【0020】
発明の詳細な説明
本発明は細胞内に核酸を送達する分子を同定するための方法をその特徴とする。
【0021】
本発明の組成物および方法は、核酸アプタマーライブラリーまたは任意のディスプレイ法を利用するものであり、ライブラリー(例えば小分子またはタンパク質のライブラリー)を、例えばライブラリーの各メンバーをコードする核酸(例えばRNAまたはDNA)に(例えば遺伝子型-表現型結合を介して、または共有結合若しくは非共有結合による相互作用を介して)結合させるものである。例えば、Lipovsek et aI., J. Immunol. Methods 290: 51-67 (2004); Bertschinger et al., Protein Eng. Des. Sel. 17: 699-707 (2004); Yonezawa et al., Nucleic Acids Res. 31: e118 (2003); Tabuchi et al., FEBS Lett. 508: 309-312 (2001); Odegrip et al., Proc. Natl. Acad. Sci. USA 101: 2806-2810 (2004);およびFujita et al., J. Med. Chem. 45: 1598-1606 (2002)を参照されたい。一態様においては、例えばフルオロフォア、重要な遺伝子を標的としたRNAi分子、RNAi分子もしくはアンチセンス配列をコードしかつ発現可能なdsDNA配列、レポーター遺伝子(例えばGFP)を発現可能なdsDNA配列、タンパク質(例えばポリメラーゼ、転写因子、もしくはリプレッサー)を結合するdsDNA配列、小分子、または細胞内読み出しシグナルを発生するタンパク質またはペプチドを発現可能なdsDNAなどの細胞内読み出しシグナルを発生する分子とRNAまたはDNAとを更に連結する。
【0022】
細胞を本発明のライブラリーと接触させることもできる。細胞内への所望の分子(例えば核酸分子)の送達を促進する分子(例えば小分子またはペプチド)に連結されたライブラリーのメンバーは、例えば、細胞内にエンドサイトーシスによって取り込まれる。細胞表面に結合したままの核酸ライブラリーのメンバーは、例えばイオン強度、pH、界面活性剤、またはプロテアーゼ処理によって除去する。この後、細胞を溶解し、内部移行した物質を増幅して(例えばPCR)、送達を促進させる分子を同定することができる。
【0023】
リンカー部分(例えば多官能性リンカー部分)を用いて2個、3個、4個またはそれ以上の化学分子が(例えばデンドリマーディスプレイとして)核酸ディスプレイライブラリーのメンバーに結合されたライブラリーを生成することができる(図1)。この手法を用いて例えば細胞上の複数の受容体を刺激して内部移行を生じさせることができる。モノマー化学種を用いる他の方法では、受容体が効果的な内部移行を生じず、シグナルが非常に低いかあるいは存在しない場合がある。このため、本発明のライブラリーの設計の1つ(例えばDNAに結合された小分子)は、リンカーの一方の端部にDNA識別領域が、他方の端部に複数のアミン(または他の反応性化学種)が結合されたリンカーを有するものとして生成される。複数のアミンまたは他の反応性化学種を用いて、合成された小分子の複数のコピーをDNA上に生成及び結合させる。一態様においては、両方の鎖が小分子をディスプレイしてもよい(図2)。また、核酸ディスプレイライブラリーのメンバーのDNA分子の長さに沿って複数の小分子をディスプレイするように、ライブラリー合成によって容易に官能化されるアミンまたは他の分子を、例えばウリジンまたはシトシンのC5位を介して識別領域の複数の位置に沿って単独でまたはマルチマーとして組み込むことも可能である。DNAの両方の鎖を修飾することができる。更に、疎水性残基(例えば5-メチルC、C5アルキル置換、C5アルキニル置換など)の付加などのように、細胞内への侵入が促進されるようにDNAの塩基を修飾することができる。
【0024】
核酸ディスプレイ法(例えば、mRNAディスプレイ、ストレプトアビジンディスプレイ、共有結合DNAディスプレイ、非共有結合DNAディスプレイなど)によってドメイン同士が連結されるように、タンパク質またはペプチドのドメイン(例えば抗体のVHドメイン)からなるライブラリーが生成される。核酸はタンパク質またはペプチド結合ドメインをコードしており、更に、例えばレポーター遺伝子(例えばGFP)、RNAi遺伝子(例えばhnRNAi)、転写因子、または転写因子結合部位をコードしている。1つの例では、GFPの遺伝子に結合したVHドメインのライブラリーを細胞と接触させ、GFPを発現する細胞を単離し、VHドメインのアイデンティティーをPCRおよび配列決定によって決定する。この方法を用いることにより、細胞にdsDNAを送達する特異的なVH結合物質を新規な送達ビヒクルとして同定することができる。
【0025】
別の態様では、ストレプトアビジンを核酸ライブラリーに結合させ(例えば核酸がビオチン分子を含み、ビオチン結合部位を介してストレプトアビジンと結合する)、発現遺伝子(例えば発現遺伝子がビオチンを含み、ストレプトアビジンは4量体であるために、第2のビオチン結合部位を介してストレプトアビジンと結合する)またはdsRNAi(例えばdsRNAiがビオチンを含み、第2のビオチン結合部位を介してストレプトアビジンと結合する)に更に結合させる(図3)。
【0026】
本発明は更に、核酸、または他のペイロード(例えば小分子またはペプチド)を細胞内に送達する新規な分子を同定するための一般的な方法をその特徴とする。この方法では、小分子ライブラリーまたはタンパク質ライブラリーのいずれかを、ライブラリーの各メンバーをコードし、更にRNAポリメラーゼのプロモーター領域(例えばT7 RNAポリメラーゼ)を含むdsDNAと(例えば、遺伝子型-表現型結合を介して、または共有結合若しくは非共有結合による相互作用を介して)結合させるディスプレイ法を利用する。この後、このライブラリーを、細胞質中に適切なRNAポリメラーゼを発現している細胞とインキュベートする。この後、細胞の細胞質中に局在化したライブラリーのメンバーはRNAポリメラーゼによって転写されうる。次いで細胞を溶解し、RNAを単離してRT-PCRに供することにより細胞質中に存在するdsDNAを同定する。この後、このdsDNAを同定することにより、細胞内への送達を媒介した、dsDNAと結合した分子が同定される。
【0027】
細胞内への核酸の送達は、アンチセンス、miRNA、RNAiまたは遺伝子治療的アプローチのいずれであるかによらず、このクラスの分子の治療的開発において依然として重要な課題の1つである。送達物質の発見における大きな障害の1つとして、細胞内への核酸の放出につながる、発生頻度が希な事象を検出する能力がある。細胞内への1個の分子の放出を検出できることが理想的であるが、これには超高感度の読み出しシステムを必要とする。1個の分子の送達が実現されれば、送達法を更に最適化することが可能となる。
【0028】
dsDNAは、ポリメラーゼによる増幅により細胞内の1個の分子を検出することが可能である。例えば、dsDNAの個別の分子の細胞核内へのマイクロインジェクションは、対象とする遺伝子を有する、制限酵素で消化された直鎖状のフラグメントであっても、発現タンパク質の免疫蛍光染色によって検出されるような当該遺伝子の転写および翻訳を生じる。これに対して、dsDNAの細胞質中へのマイクロインジェクションは、高濃度で導入した場合でも、遺伝子の発現が生じることはほとんどない。しかしながら、細胞質中に局在するT7 RNAポリメラーゼを発現する細胞を使用することにより、dsDNAを細胞質中で発現させることができる。
【0029】
ディスプレイライブラリーとして、小分子、ペプチド、またはタンパク質をdsDNAと結合させるための方法が幾つか存在している。一般に、遺伝子型(dsDNA)とディスプレイされる分子との間にはコード関係が存在するため、遺伝子型の配列を決定することによってディスプレイされる分子が同定される。本明細書では、新規な選択技術をディスプレイライブラリーとともに用いて細胞への分子の送達の媒介物質を同定するための方法について述べる。
【0030】
核酸の細胞内送達を媒介するタンパク質または小分子を同定するためのある一態様では、RNAポリメラーゼ結合部位を含むdsDNAディスプレイライブラリーを、細胞質中でRNAポリメラーゼを発現している細胞とインキュベートする。インキュベート後、細胞を溶解し、RT-PCRに供して細胞質中に送達されたdsDNAライブラリーのメンバーから生じたあらゆるRNA転写産物を増幅する。
【0031】
別の態様では、小分子またはペプチドライブラリーのスクリーニングによるアプローチを利用し、タグが付与されていない分子をdsDNAライブラリーによってスクリーニングすることによって送達を促進する物質を探索する。この態様では、RNAポリメラーゼ結合部位を含むdsDNAディスプレイライブラリーを、細胞質中でRNAポリメラーゼを発現している細胞とインキュベートする。この後、小分子またはペプチドを細胞に加えてdsDNA分子の放出を促進する。
【0032】
更に別の態様では、いかなる小分子、ペプチドまたはタンパク質もディスプレイされていない、RNAポリメラーゼ結合部位を含むdsDNAを、細胞質中でRNAポリメラーゼを発現している細胞とインキュベートする。この後、細胞に小分子またはペプチドを加えてdsDNA分子の放出を促進する。上記の方法をハイスループットのスクリーニングモードにおいて使用することによって細胞へのdsDNAの送達の促進物質を同定することができる。
【0033】
更に別の態様では、ライブラリーのdsDNAを、dsDNAメチルトランスフェラーゼを発現する細胞に加える。ライブラリーのメンバーが細胞に入ると、細胞内のdsDNAはすべてメチル化され、回収され、かつメチル化特異的PCRに供される。
【0034】
哺乳動物細胞中へのバクテリオファージT7 RNAポリメラーゼ(T7 RNAP)の細胞質局在化については、例えば参照により本明細書に組み入れられるElroy-Stein and Moss; Proc. Natl. Acad. Sci. USA 87: 6743-6747 (1990) および Wang et al., Analytical Biochem. 375: 97-104 (2008)に述べられている。
【0035】
本明細書に記載の方法を使用することにより、例えば任意の形態の核酸、タンパク質、ペプチド、小分子、リポソーム、またはナノ粒子などの(ただしこれらに限定されない)、幅広い送達性を潜在的に有する、組織または細胞への送達を媒介する一般的な物質を同定することが可能である。
【実施例】
【0036】
以下の実施例は本明細書を説明することを目的としたものである。これらの実施例はいかなる意味においても本発明を限定しようとするものではない。
【0037】
実施例1 T7 RNAポリメラーゼ細胞株の構築
下記のPCRプライマーを用いてBL21細胞からバクテリオファージT7 RNAポリメラーゼ(RNAP)を増幅した(NcoI部位を太字、開始ATGを斜体、Kozak配列を下線、NotI部位を小文字、タンデム停止コドン(TTATTA)を斜体で示す)。

【0038】
増幅した遺伝子産物をNcoI/NotIフラグメントとしてpEF/myc/cyto発現ベクター(Invitrogen # V890-20)に方向を決めてクローニングした(図4)。このベクターは、強力なEF-1aプロモーターおよび安定した細胞株選択のためのネオマイシン耐性遺伝子を有する、細胞質での発現用に設計されたものである。このベクターは、マルチクローニング部位であるポリリンカーの上流に通常見られるT7プロモーターを欠いているため、T7 RNAPが発現する際にプロモーター活性が競合する可能性がない。
【0039】
実施例2 一過性にトランスフェクトしたHEK293T細胞におけるT7 RNAPの活性
細胞質T7が哺乳動物細胞においてポリメラーゼとして活性を有しうることを調べるため、本発明者らはHEK293T細胞において一過性トランスフェクションアッセイを開発した(図5)。細胞を24ウェル皿に350,000細胞/ウェルで播種し、10%ウシ胎児血清(FBS)を添加したイーグル最小必須培地中で一晩インキュベートした。VH抗体ドメインのコード領域の上流にT7プロモーターを含むDNA鋳型をPCRにより以下のようにして調製した。インビトロの転写および翻訳シグナルであるT7TMVをコードする5'末端側のオリゴヌクレオチド
と、C末端のVHDNAのCmuおよびSAリンカー領域Y15とアニールする3'末端側のオリゴヌクレオチド
を用いて、ナイーブなヒトVHライブラリーからのVHDNAをPCR増幅した(Strategene 600312)。PCR産物をゲルで精製した。PCRフラグメントの構成要素を図6にまとめる。このVHDNAフラグメントを、T7 RNAポリメラーゼを発現している細胞にトランスフェクトすると、T7 RNAPはこのDNAに作用してDNAをRNAに転写する。この特定のRNAをRT-PCRによって検出および増幅する。
【0040】
300fmolのT7 DNA鋳型および2.5μgのT7発現コンストラクトとの組み合わせを細胞にトランスフェクトし、同時にコントロール試料には以下をトランスフェクトした。すなわち、鋳型なし + 2.5μgのT7_pEF/myc/cytoベクター; 300fmolのT7 DNA鋳型 + 2.5μgのpEF/myc/cytoベクター; または鋳型なし + 2.5μgのpEF/myc/cytoベクター。細胞は、Lipofectamine2000(Invitrogen)を使用し、製造者のプロトコールにしたがって(2μl Lipofectamine/トランスフェクション)トランスフェクトした。細胞を一晩インキュベートした後、溶解した細胞からRT-PCRによる分析用にRNAを調製した。T7 RNAPの活性を試験するため、特異的かつ高感度のRT-PCRアッセイを開発した。RNeasy Mini Kit(Qiagen 74104)を使用し、上記実験から回収した細胞溶解物から細胞質RNAを調製した。簡単に述べると、細胞溶解物を遠心分離により沈降させて不溶性タンパク質を除去した。清澄化した溶解物をグアニジン塩およびエタノールを含むRLTバッファーと混合し、結合カラムに加えた。カラムをRWIバッファーで洗った。RNase-free DNase kit(Qiagen 79254)によってカラムにDNアーゼ処理を行ってキャリーオーバーDNAを除去した。カラムを更にRPEバッファー、70%エタノールで洗浄し、乾燥した。RNAを30μlのヌクレアーゼを含まない水で溶出した。下流のPCRのためにRNA試料からDNAを完全に除去するため、RNA溶出液を37℃で1時間、10単位のDNアーゼIで消化した(Ambion AM2222)。次いで製造者の推奨するプロトコールにしたがってRNAをRNeasy MiniElute Cleanup Kit(Qiagen 74204)で精製し、20μlのH2O中で溶出した。SuperscriptII逆転写酵素(Invitrogen 18064-014)を用いて逆転写を行った。簡単に述べると、10μlのRNA、10nmolの各dNTP、および5pmolのVH特異的3'Cmuオリゴヌクレオチド
の混合物を65℃で5分間インキュベートし、4℃に冷却してRNAの2次構造およびプライマーのアニーリングを低減させた。5X ファーストストランドcDNA合成バッファー、0.1M DTT (10X)、および200単位の逆転写酵素を最終容量20μlになるまで加えた。反応液を42℃で50分間インキュベートし、70℃で15分間、熱失活させた。1μlのファーストストランドcDNAを25μlの容量中、Herculaseバッファー、200μMのdNTP、0.2μMのT7TMV、S6-1
オリゴヌクレオチド、および1.25単位のHerculase(Strategene 600312)の存在下で15〜25サイクル増幅した。PCR産物を2%アガロースゲル(Invitrogen G5018-02)上で可視化した。
【0041】
T7ポリメラーゼの発現を確認するため、平行ウェルを上記のようにトランスフェクトし、NP-40界面活性細胞溶解バッファー中で溶解した。細胞溶解物をSDS-PAGEにより分離し、T7ポリメラーゼに対するモノクローナル抗体(EMD Bioscience #70566-3)を用いてウェスタンブロット分析に供した。抗T7ポリメラーゼ抗体は、このタンパク質の予想される分子量(約99kDa)と一致するバンドを認識した(図7)。RT-PCRアッセイにより、VHPCR鋳型がHEK293T細胞内においてT7ポリメラーゼによって転写されることが示された。コントロールレーンは、RT-PCR活性がT7ポリメラーゼの発現に依存し、T7ポリメラーゼプロモーターを含む鋳型の存在下においてのみ生ずることを示した(図7)。
【0042】
実施例3 一過性にトランスフェクトしたHEK293T細胞におけるT7 RNAPの感度
選択用の開始ライブラリーは極めて多様である(最大で約1014種類の異なる配列が存在する)ことから、細胞質に到達するライブラリーのメンバーの捕捉および増幅は極めて高感度でなければならない。本発明者らは、ある量のVHPCR鋳型を一過性トランスフェクション中に滴定することによってT7ポリメラーゼRT-PCRアッセイの感度を試験した。上記に概略を述べたトランスフェクションプロトコールにしたがって、3pmol/試料〜0.1fmol/試料の範囲のVHPCR鋳型をT7_pEF/myc/cyto発現コンストラクトとコトランスフェクトした。回収された試料のRT-PCRは、細胞当たり0.1fmolの鋳型であっても細胞質のT7ポリメラーゼによって転写され、検出可能なレベルにまで増幅可能であることを示している(図8)。
【0043】
実施例4 一過性にトランスフェクトしたVCaP前立腺癌細胞におけるT7 RNAPの活性
本発明の目的は、様々な種類の細胞において細胞質への取り込みの選択を可能にするプラットフォームを確立することにある。最近、幾つかのグループが、前立腺癌細胞の前立腺特異的膜抗原を標的とした試薬とsiRNAのコンジュゲートを作製し、標的化送達によってmRNAがノックダウンされることを実証することに成功した。例えば、Chu et al., Nucleic Acids Res. 34: e73 (2006); McNamara et al., Nat. Biotechnol. 24: 1005-1015 (2006), Dassie et al., Nat. Biotechnol. 27: 839-846 (2009)を参照されたい。そこで本発明者らは、概念実証実験を拡張して様々な前立腺癌細胞株における細胞質T7ポリメラーゼの活性を調べた。VCaP細胞は、細胞表面に前立腺特異的膜抗原(PSMA)を発現する前立腺癌細胞である。T7_pEF/myc/cyto発現コンストラクトおよびVHPCR鋳型によるVCaP細胞の一過性のトランスフェクションにより、T7ポリメラーゼがこれらの細胞の細胞質中において転写活性を有し、一過性にトランスフェクトしたHEK293T細胞において観察されたのと同様のT7および鋳型依存性が見られることが示された(図9)。
【0044】
実施例5 安定的な前立腺癌細胞株からのT7 RNAP転写産物の検出
T7ポリメラーゼRT-PCRアッセイを更に試験するため、本発明者らは、T7_pEF/myc/cytoコンストラクトを使用してネオマイシンに対する薬物耐性によりT7ポリメラーゼの発現について安定的に選択された他の前立腺癌細胞株を確立した。PC3およびDU-145細胞はPSMAを発現しない前立腺癌細胞株であり、LnCaPおよび22rV1はPSMAを発現する前立腺癌細胞株である。4種類の細胞のすべてにT7_pEF/myc/cytoまたはpEF/myc/cyto空ベクターのいずれかをトランスフェクトし、750μg/mlのネオマイシン(Geneticin, Gibco)の存在下での増殖について選択した。ネオマイシン耐性細胞をプールし、RT-PCRによりT7の発現についてアッセイした。約10 X 106個の細胞を回収、溶解し、RNAをRNeasy Mini Kit(Quiagen 74104)により上記に述べたようにして単離した。T7 RNAP転写産物を、3'T7 RNAP特異的オリゴヌクレオチド
を逆転写反応に、5'T7 RNAP特異的オリゴヌクレオチド
および同じ3'オリゴヌクレオチドをPCR増幅に使用することにより検出した。T7_pEF/myc/cytoコンストラクトをトランスフェクトすることにより安定的に薬物選択された細胞のみで、T7ポリメラーゼの増幅された領域とサイズが一致するcDNAバンドが見られた(図10)。比較のため、T7_pEF/myc/cytoコンストラクトを一過性にトランスフェクトしたHEK293T細胞由来の同量の全RNAを同じT7特異的オリゴヌクレオチドを用いたRT-PCRに供した。データは、安定的なT7前立腺癌細胞株では、一過性にトランスフェクトしたHEK293T細胞で見られるよりもT7ポリメラーゼの発現量が低いことを示している(図10)。
【0045】
実施例6 PSMA発現癌細胞株におけるT7 RNAPの活性
安定的に薬物選択された細胞において発現されるT7ポリメラーゼが活性を有するかを調べるため、本発明者らは改変されたRT-PCRアッセイを確立した。このアッセイは、すでに存在する安定的なT7の発現を考慮し、T7を一過性に発現させるための最初のトランスフェクションを省略した点以外は、上記に述べたアッセイと同様のものである。代わりに、VHPCR産物をT7の安定的な発現の存在下または非存在下で前立腺癌細胞株にトランスフェクトし、RT-PCRアッセイを上記に述べたように行った。図11は、VH cDNAの存在によって示されているように、22rV1_T7細胞株が活性なT7ポリメラーゼを発現することを示している。対照的に、陰性コントロール細胞株(22rV1_ベクター)はT7依存性のRT-PCR活性を示さない。
【0046】
実施例7 ビオチン-ストレプトアビジン集合体に対する細胞質T7ポリメラーゼ活性
細胞質への侵入の選択が支障なく行われるためには、オリゴヌクレオチドおよびコードされたペプチドまたはタンパク質のビオチン-ストレプトアビジン集合体はT7ポリメラーゼ鋳型と同様に効果的でなければならない。ストレプトアビジンと集合したビオチン化ペプチドまたはVH結合物質の複合体を細胞にトランスフェクトし、T7 RNAPによって転写させ、RT-PCRによって検出することが可能であるかを試験するため、上記のVHDNAを、5'末端にSP6およびT7プロモーターの両方の配列
を有し、3'末端にCmu-flag-SA-polyA配列を有するように改変した(図12)。次いでこのDNAをSP6 transcription kit (Ambion AM1330)によってインビトロでRNAに転写した。RNAeasy MiniElute Cleanup Kit (Qiagen 74204)によりRNAを精製した。SAディスプレイ用のビオチン化RNA/DNAリンカーをRNAに1:1の比でアニールし、RNA鋳型とUV架橋させた。リンカー上のビオチンと1:1のモル比で相互作用させることにより、ライゲートしたRNA上にストレプトアビジンを負荷した。次いでこの集合複合体をオリゴdT精製に供した。集合複合体はRNAのpolyAテールを介してオリゴdTと結合し、遊離SAを洗い流した。ライゲートしたDNAリンカーをプライマーとして使用してオリゴdTセルロース上で逆転写を行い、superscriptII(Invitrogen 18064-014)によって37℃で1時間、伸長した。次いでRNAとファーストストランドcDNAとのハイブリッドをRNアーゼH(Invitrogen 18021-014)によって1時間消化してRNA鎖を開裂させた。溶液中のファーストストランドDNAをオリゴdTセルロースを遠心分離して回収した。SP6T7オリゴヌクレオチドをプライマーとして用いてセカンドストランドDNAを合成し、superscriptII(Invitrogen 18021-014)によって伸長した。次いでビオチン化されたVHまたはペプチドの分子をdsDNA-SA複合体と2:1のモル比でインキュベートしてdsDNA-SA-VHまたはdsDNA-SA-ペプチドを生成した(図12)。
【0047】
概念実証実験として、細胞貫通ペプチド(CPP)としてこれまでに述べられている2種類の異なるペプチドを試験した。すなわち、TATペプチド
およびアンテナペディア(Antennapedia)ペプチド
である。図13に概略を示すように、集合したVHまたはペプチド複合体、またはビオチン化ペプチドもしくはタンパク質を有さない集合体をHEK293T細胞に一過性にトランスフェクトし、T7ポリメラーゼをコトランスフェクトした細胞中でRT-PCRについてアッセイした。その結果、本発明者らのライブラリーにおいて利用されるビオチン:ストレプトアビジン結合手法を介してペプチドまたはVHタンパク質と集合したオリゴヌクレオチドは、細胞質のT7ポリメラーゼによって転写されうることが確認された(図14、上側のパネル)。したがって、細胞質T7ポリメラーゼシステムを、細胞に基づいた選択を行う際に細胞質中への侵入の選択圧力として利用することが可能である。
【0048】
この選択手法を更に検証するため、本発明者らは、トランスフェクションとは独立した細胞質侵入によるT7ポリメラーゼ活性を実証することを試みた。TATおよびANTペプチドはこれまでにCPPとして同定されていることから、これらのペプチドとビオチン:ストレプトアビジンの集合体も細胞質に侵入して、トランスフェクション試薬の非存在下で細胞質T7ポリメラーゼの鋳型として機能しうる可能性がある。40pmolのペプチドが結合した集合体、またはペプチドを有さない集合体を、T7_pEF/myc/cyto発現コンストラクトを一過性にトランスフェクトしたHEK293T細胞に加えた。18時間後に細胞を溶解し、上記に述べたようにRT-PCR活性についてアッセイした。その結果は、ペプチド集合体の細胞への取り込みが起こり、これらの複合体に対する細胞質T7ポリメラーゼ活性の結果としてのRT-PCRを容易に測定できることを示している(図14、下側のパネル)。コントロール集合体も同様に細胞質への侵入能を獲得したようであるが、これらのコントロール鋳型からのRT-PCR活性は、CPP集合体のRT-PCR活性と比較して大幅に低くなっている。
【0049】
実施例8 T7 RNAPを発現している細胞内に送達するためのペプチド-dsDNAコンストラクトの合成
リン酸化オリゴヌクレオチドHP:
をIDT DNAにより合成した。このDNAはオーバーハングを有するヘアピンへと折り畳まれ、制限酵素BbvCIおよびこの酵素のニック型である、トップストランドまたはボトムストランドのいずれかを切断しうるNb.BbvCIまたはNt.BbvCI (New England Biolabs, Inc.)の開裂部位CCTC AGCを含んでいる。ヘアピンループの中央には、アミノ-PEGリンカー(PEG2000、約45個のエチレングリコール単位)の結合に用いられる側鎖としてC5アミノ修飾dT(dT-C6-NH、C6は6位の炭素リンカーを指す)が挿入されている。
【0050】
10nmolのオリゴ「HP」を50μlの水に溶解した。20倍のモル過剰量のFmoc-アミノ-PEG2000-カルボキシル-NHSエステル(Jen-Kem)を50μlのDMFに溶解し、室温で2時間の間に2回に分けてオリゴ溶液に加えた(最終的な溶媒組成は、50% DMF-50% 水)。この後、60μlの1M Tris HCL、pH7.0(最終濃度200mM)を加えて過剰量のNHSエステルをクエンチした。この溶液を室温で更に30分間インキュベートした。得られた反応混合物を水で500μlに希釈し、NAP-5カラム(Sephadex-25, GE)に通して脱塩した。
【0051】
得られた物質を凍結乾燥し、100μlの水に溶解した。20μlのピペリジン(最終濃度20%)を加え、室温で2時間インキュベートした。アミンの脱保護および非水溶性のFmoc基の放出により濁った沈殿が生じた。次いで反応液を0.2μmスピンフィルター(Millipore)に通して濾過し、エタノールを3回加えることによって300mM酢酸ナトリウムから沈殿させた。高い結合効率のため、得られたヘッドピースHP-1を更に精製することなく使用した。
【0052】
モデル化合物として5-(4,6-ジクロロトリアジニルアミノフルオレセイン)(DTAF)(Anaspec)をHP-1のアミノ基と結合させた。DTAFは、1個のアミノ化合物が結合したトリクロロトリアジンのスキャフォールドを構造的に表している。ライブラリーを形成するには、トリクロロトリアジンのスキャフォールドを3個の塩素の位置のそれぞれにおいて、多様な構成単位によって誘導体化することができる。これにより、モデルライブラリーに蛍光標識も提供される。反応液(10μl)は以下のように構成した。5μlの400μM HP-1水溶液に、2μlの750mMホウ酸塩バッファー(pH9.5)、および1μlのDMFを加えた。DTAFを50mMにまでDMFに溶解し、2μlを反応液に加えた。HP-1およびDTAFの最終濃度はそれぞれ200μMおよび10mMであった(DTAFの50倍過剰)。DMFの最終濃度は30%であった。HP-1は最大で90%のDMFにまで溶解することが分かり、DMFなどの有機溶媒に溶解しうることが示唆された。反応を4℃で16〜20時間進行させた。次いで反応混合物を水で30〜50μlに希釈し、Zebaスピンカラム(Pierce)上で脱塩した。更なる精製は行わなかった。
【0053】
トリアジンスキャフォールド上の最後の塩素反応基の修飾に用いるため、高アルギニンペプチドR7、すなわちH(-Arg-εAhx)6-Arg-OH (Bachem)を選択した。これは、細胞内への化合物の送達に使用されるアルギニン-アミノヘキサン酸細胞膜透過性ペプチドである。反応液は上記と同様に構成した。すなわち、20μlの反応液は、150mMホウ酸塩バッファー(pH9.5)に溶解された約200pmolのHP-1-DTAF(工程1)、および10nmolのR7ペプチドを含んでいた。これらの条件下ではアルギニンの側鎖は反応せず、ペプチド内の唯一の反応性アミンはN末端である。反応を75℃で12時間進行させ、Zebaスピンカラム上で脱塩することによって精製した。
【0054】
細胞内送達実験で使用したVHDNAコンストラクトは、分子の5'末端にT7プロモーター領域を、3'末端の近傍にCmu領域を有する約400bpのVHDNAの単一のクローンのPCR産物から調製した。VHDNAコンストラクトをモデル化学ライブラリーの修飾されたヘッドピースに連結するため、クローンのPCR増幅によってBsmI制限部位をT7プロモーター領域の上流に組み込んだ。BsmI制限酵素による消化物は、ヘッドピース(3'CCオーバーハング)とのライゲーションを可能にする3'GGオーバーハングを生じた。BsmI部位(下線で示される)を有する5'プライマーをIDT DNAにより合成した。すなわち、
である。PCR増幅後、PCR purificatin kit (Invitrogen)を使用してVHDNAコンストラクトを精製し、得られたDNAを、65℃でNEBバッファー4中、2時間、250UのBsmI(NEB)により消化した。DNAを2%アガロースゲル上で精製した。ライゲーション反応液(30μl)は、BsmIで消化された2pmolの各VHDNAコンストラクト、ならびに1 X T4 DNAリガーゼバッファーに加えたHP-1-DTAF-R7(アルギニン-アミノヘキサン酸ペプチド)および60Weiss単位のT4 DNAリガーゼ(NEB)を含んでいた。反応液を16℃で20時間インキュベートした。高いライゲーションの効率のため、この物質を更に精製を行うことなく細胞内送達/T7 RNAP実験に更に使用した。結果を図15にまとめて示す。
【0055】
上記に述べた実験と対応して、DNAリンカーを介してVHDNAとライゲートされた細胞貫通小分子C2は、トランスフェクションおよびトランスフェクションとは独立した細胞質侵入により同様の結果を示した(図16)。
【0056】
実施例9 高活性T7ポリメラーゼ細胞株のクローン誘導
ポリクローナル集団よりも高いT7ポリメラーゼ活性を有するクローン細胞株を単離するため、T7-蛍光レポーターコンストラクトを作製して、トランスフェクションまたはウイルス感染により上記に述べたように前立腺癌ポリクローナル安定細胞株に導入した。最も高いT7ポリメラーゼ活性のレベルを有する細胞は最大の蛍光を発生し、Mo-Flo単一細胞分別FACS装置により単一細胞の集団として捕捉される。次いで個々のクローン細胞株を蛍光についてスクリーニングし、続いて、FACSにより報告される最も高いT7活性を有する細胞をRT-PCRアッセイにより二次スクリーニングする。
【0057】
実施例10 核酸送達用の高親和性VH/ペプチド試薬の機能的選択
本明細書に記載のシステムの用途の1つに、機能性細胞に基づいた選択による核酸送達用の高親和性VH/ペプチド/小分子試薬の同定がある。SP6およびT7プロモーターを有するVH/ペプチドをコードするDNAライブラリーを、SP6転写酵素によってRNAライブラリーに転写し、ビオチン化ストレプトアビジン(SA)ディスプレイリンカーにライゲートする。次いでライゲートしたRNA上にSAを負荷し、3'末端にピューロマイシン様分子を有する別のビオチン化リンカーと集合させる。集合したVH/ペプチドライブラリーをインビトロで翻訳するとRNAライブラリーとそれらがコードするVH/ペプチドライブラリーとの融合体を形成する。本明細書に記載のように、オリゴdT精製、逆転写、RNアーゼH消化、およびセカンドストランドcDNA合成を行う。特定の標的によって媒介される細胞侵入を行うために、精製されたdsDNA-VH/ペプチド融合ライブラリーを、標的を発現していない一致した陰性細胞株とライブラリーを何度も接触させてバックグラウンドの結合物質を除去することによって対抗選択する。予めきれいにしたDNA融合ライブラリーと標的発現細胞とを接触させ、内部移行の機序とは無関係に、細胞に結合、侵入させ、細胞質に到達させる。細胞を洗って非特異的結合物質を除去し、細胞表面の内部移行されていない結合物質を取り除く。細胞質に侵入したライブラリーのメンバーのみがT7 RNAPによって認識され、転写される。細胞質に到達してT7ポリメラーゼによって転写されたライブラリーのメンバーは、上記に概略を述べた概念実証実験によって規定される条件を適用して、RT-PCRによって回収される。濃縮された結合物質は次のラウンドの選択に供する。複数ラウンドの選択および濃縮によって細胞質侵入プールが生成され、当技術分野では周知の標準的方法を用いた濃縮化集団のサブクローニングおよび配列決定によって媒介物質が同定される。
【0058】
一態様においては、細胞内に送達される分子を有する細胞を同定するための更なる基準として、ビオチン化siRNAをSAと集合させてDNA、SA、VH/ペプチド、およびsiRNAの複合体を生成する。SAは4つの結合部位を有しているため、各成分をSAと1:1のモル比で加えることによって結合部位のそれぞれに対象とする試薬を負荷することができる。この複合体を、機能的読み出しシグナルによって、標的遺伝子のmRNAノックダウン、および標的遺伝子により媒介される細胞機能の阻害についてスクリーニングする。
【0059】
標的特異的な選択以外に、上記に概略を述べた手法は、異なる細胞起源または細胞内状態(例えば、対抗選択用細胞型としての肝細胞および陽性細胞型としての心筋細胞; 対抗選択用の形質転換されていない細胞型および陽性選択用の形質転換された細胞型; または、対抗選択用の未分化細胞および陽性選択用の分化細胞)を構成する陽性および陰性細胞株を使用することによって細胞特異的な細胞質侵入ビヒクルを同定するためにも適用される。また、細胞質侵入選択は、複数の細胞型または細胞表面の標的を標的としうる送達ビヒクルを単離するために、対抗選択の手法を用いずに特定の細胞型に適用される。
【0060】
細胞質侵入選択には、選択圧力としてオリゴヌクレオチド(例えば、siRNA、miRNA、転写因子/リプレッサー滴定、またはアンチセンスオリゴヌクレオチド)送達によって媒介されるmRNAノックダウンを利用して更なる改良が加えられる。テトラサイクリン調節性T7ポリメラーゼ遺伝子を、選択した細胞株にTetリプレッサータンパク質のcDNA(TetR)とともに導入する。これにより、ビオチン:ストレプトアビジンライブラリー複合体は、ビオチン化DNAをコードしたペプチド/VH+ストレプトアビジン+ビオチン化ペプチド/VH+TetR mRNAに対するビオチン化siRNAを含むことになる(図17)。いずれかのライブラリーメンバーが細胞質に到達し、siRNAがmRNA分解に関与するRSC複合体に到達すると、TetR mRNAが除去され、その結果TetRタンパク質が失われ、T7ポリメラーゼ活性が誘導される。これにより細胞質取り込みを媒介するVH/ペプチドがT7によって転写され、その後、RT-PCRによって増幅および回収される。
【0061】
実施例11 核酸送達ビヒクルのインビボ選択
核酸のインビボ送達を媒介する物質の同定は、細胞質ポリメラーゼを有する形質転換マウスを作製することによって実現される。例えば、T7 RNAPを発現する形質転換マウスを作製する。次いで、標準的投与法(例えば、尾静脈注射)を用いてdsDNAライブラリーをマウスに投与する。注射後、組織または細胞を単離して溶解する。RNAを単離してRT-PCR、サブクローニング、および配列決定を行って所望の組織および/または細胞への侵入を媒介するコードされた分子を同定する。場合により、このプロセスを繰り返して、侵入可能な化学種を濃縮する。化学種が同定された後、対象とする分子を核酸、タンパク質、または小分子の任意の形態に結合させて組織および/または細胞特異的送達について試験する。
【0062】
実施例12 DNAメチルトランスフェラーゼを用いた核酸ディスプレイライブラリーのメンバーの同定
DNAのメチル化用のPCRプライマーの最適な結合部位を含むdsDNAライブラリーを調製する。このdsDNAライブラリーをDNAメチルトランスフェラーゼ(DNMT1)を過剰発現している細胞とインキュベートし、特異的なライブラリーメンバーを特定の細胞内に内部移行させる。細胞質内に侵入するとライブラリーからのdsDNAタグはDNMT1の基質となり、選択的にメチル化される。この後、細胞を溶解し、dsDNAタグを単離してメチル化特異的PCRのための標準的プロトコール(Herman et al, Proc. Natl. Acad Sci. USA 93: 9821-9826 (1996))を用いて亜硫酸水素ナトリウムで処理する。次いでメチル化されたdsDNAタグを、メチル化特異的プライマーを用いて選択的に増幅する。PCRの後、DNA産物を配列決定することで、細胞の細胞質内への選択的取り込みを媒介する分子をライブラリーから同定することが可能である。
【0063】
実施例13 ssRNAアプタマーライブラリー
ssRNA依存性RNAポリメラーゼ(例えば、ポリオウイルス3Dpol、水疱性口内炎ウイルスL、およびC型肝炎NS5bタンパク質)の重合部位を含むssRNAアプタマーライブラリー(当技術分野で周知のように非修飾または修飾塩基)を調製する。ssRNAアプタマーライブラリーを、ssRNA依存性RNAポリメラーゼを過剰発現している細胞とインキュベートし、特異的なライブラリーメンバーを特定の細胞内に内部移行させる。細胞質内に侵入するとそのライブラリーメンバーはポリメラーゼの基質となる。RNAを細胞から回収し、重合反応で得られたRNA産物に特異的なプライマーを使用して細胞内に侵入可能な分子を逆転写し、PCRし、かつ配列決定する。
【0064】
他の態様
上記の明細書で述べたすべての刊行物、特許、および特許出願は、参照によって本明細書に組み入れられる。記載される本発明の方法およびシステムの様々な改変および変形が、本発明の範囲および趣旨から逸脱することなく当業者には明らかとなろう。以上、本発明を特定の態様に関連して述べたが、特許請求されるところの本発明はこのような特定の態様に不要に限定されるべきではない点は理解されるはずである。実際、当業者にとって明らかな、本発明を実施するための記載された態様の様々な改変は、本発明の範囲内に含まれるものとする。
【0065】
その他の態様は、添付の特許請求の範囲内にある。
【背景技術】
【0001】
発明の背景
効率的な細胞質送達は核酸療法の開発において依然として重要な課題である。細胞内への核酸の標的化送達は、細胞表面の分子と結合してそれにより内部移行されるリガンド(例えば、抗体、抗体フラグメント、抗体模倣体、ペプチド、又は小分子)と核酸とを結合することによって実現可能である。標準的なディスプレイ技術は、定向進化プロセスを推進するための選択圧力として標的結合を利用しており、そのため標的と最も高い親和性で結合するリガンドライブラリーのメンバーは、さらに高ストリンジェントな選択条件下でも生き残り、濃縮されていくという、選択上、有利な点を有する。しかしながら、標的と最も高い親和性で結合するメンバーが機能的に最も関連性の高いライブラリーのメンバーであるとは限らない場合もある。例えば、細胞内に容易に侵入し、細胞質に到達することが可能なライブラリーのメンバーは、核酸送達用の標的ビヒクルとして最も効果的である可能性が高い。
【0002】
したがって、内部移行の機序とは無関係に、細胞内に侵入して細胞質に到達可能な核酸と結合したリガンドを直接的に選択するために細胞質への侵入を利用するディスプレイ技術が求められている。
【発明の概要】
【0003】
本発明は、例えば核酸分子の細胞内送達を促進する分子を同定するための方法及び組成物をその特徴とする。本発明の方法及び組成物は、ライブラリー(例えば、小分子またはタンパク質のライブラリー)と、ライブラリーの各メンバーをコードまたはタグする核酸(例えば、RNAまたはDNA)とが結合する、任意のディスプレイ法を利用する。
【0004】
したがって第1の態様において、本発明は、核酸ディスプレイライブラリーを含む組成物であって、該核酸ディスプレイライブラリーのメンバーと、細胞内読み出しシグナルを発生する分子とが連結する組成物をその特徴とする。別の態様において、本発明は、核酸ディスプレイライブラリーを含む組成物であって、該核酸ディスプレイライブラリーのメンバーとストレプトアビジン分子とが連結し、該ストレプトアビジン分子と、細胞内読み出しシグナルを発生する分子とが更に連結する組成物をその特徴とする。いずれの態様においても、細胞内読み出しシグナルを発生する分子は、例えば核酸(例えば、レポーター遺伝子、転写因子遺伝子、RNA、またはアンチセンス遺伝子)、タンパク質(例えば、緑色蛍光タンパク質(GFP))、ペプチド、または小分子(例えば、フルオロフォア)であってよい。本明細書に記載の組成物の核酸ディスプレイライブラリーの核酸分子は、外因性ポリメラーゼ(例えば、T7 RNAポリメラーゼ)プロモーターの制御下で細胞内で発現させることができる。
【0005】
別の態様において、本発明は、分枝鎖リンカーを介してライブラリーのメンバーに多量体小分子種が結合した、DNAによりコードされた小分子ライブラリーを含む組成物をその特徴とする。
【0006】
本発明によれば、更に、リンカー種によって修飾されたDNA塩基を介してライブラリーのDNAに2個以上の小分子(例えば、2個、3個、4個、5個、6個、7個、8個、9個、10個、またはそれよりも多い小分子)が結合した、DNAによりコードされた小分子ライブラリーを含む組成物が提供される。
【0007】
更なる一態様において、本発明は、核酸の細胞内送達を促進する分子を同定するための方法であって、前記分子が核酸ディスプレイライブラリーのメンバーに連結されており、かつ前記核酸ライブラリーの前記メンバーが更にある遺伝子に連結されている方法を、その特徴とする。この方法では、細胞を核酸ディスプレイライブラリーと接触させ、前記細胞内への前記核酸の送達を促進する分子に連結された前記核酸ディスプレイライブラリーのメンバーを、前記核酸ライブラリーのメンバーに連結された前記遺伝子の発現を監視することによって同定する。一局面において、前記核酸ライブラリーのメンバーに連結された前記遺伝子の発現は外因性RNAポリメラーゼプロモーターの制御下にあり、前記細胞は細胞の細胞質中でRNAポリメラーゼ(例えば、T7 RNAポリメラーゼ)を発現する。別の局面において、細胞は、細胞内に送達される前記核酸ライブラリーのメンバーを修飾することが可能な1種類以上の酵素(例えば、DNAメチルトランスフェラーゼ)を発現する。
【0008】
本発明は更に、核酸の細胞内送達を促進する分子を同定するための方法であって、前記分子が核酸ディスプレイライブラリーのメンバーに連結されており、前記核酸ライブラリーの前記メンバーが更にRNAポリメラーゼ結合部位に連結されている方法を、その特徴とする。この方法では、細胞を前記核酸ディスプレイライブラリーと接触させ、前記細胞内への前記核酸の送達を促進する分子に連結された前記核酸ディスプレイライブラリーのメンバーを、前記核酸ライブラリーのメンバーの核酸部分の細胞内における転写を監視および解読することによって同定する。コードするライブラリーがdsDNA由来のものである場合には、前記細胞内に存在するRNAポリメラーゼ(例えば、T7 RNAポリメラーゼ)が転写を触媒する。ライブラリーがssRNAである別の例では、前記細胞内に存在するRNA依存性RNAポリメラーゼが転写を触媒する(例えば、ポリオウイルス3Dpol、水疱性口内炎ウイルスL、およびC型肝炎NS5bタンパク質)。あるいは、前記細胞内に存在する逆転写酵素がDNAの重合を触媒する。ライブラリーがssDNAである別の態様では、前記細胞内に存在するssDNA依存性RNAポリメラーゼが転写を触媒する(例えば、N4バクテリオファージssDNA依存性RNAポリメラーゼ)。本発明においてssDNAからなるライブラリーに対して使用することが可能なssDNA依存性DNAポリメラーゼも存在する。
【0009】
上記に述べた方法においては、核酸の細胞内送達を促進する分子は核酸分子であってよい(例えば、RNAi、miRNA、アンチセンス核酸分子、または遺伝子)。あるいは、この分子はタンパク質、ペプチド、または小分子であってもよい。
【0010】
更なる一態様において、本発明は、第2の分子の細胞内送達を促進する第1の分子を同定するための方法であって、前記第1および第2の分子が核酸ライブラリーのメンバーに連結されている方法をその特徴とする。この方法は、細胞を前記核酸ディスプレイライブラリーと接触させる工程と、前記細胞内に存在する1種類以上の酵素による前記核酸ライブラリーのメンバーの修飾を監視することによって、前記細胞内への前記第2の分子の送達を促進する前記第1の分子に連結された前記核酸ディスプレイライブラリーのメンバーを同定する工程とを含む。この態様では、前記第1または第2の分子は、核酸分子(例えば、RNAi、miRNA、アンチセンス核酸分子、または遺伝子)、タンパク質、ペプチド、または小分子である。
【0011】
本明細書に記載の態様のいずれにおいても、核酸ディスプレイライブラリーはdsDNAディスプレイライブラリーであってよい(例えば、CISディスプレイライブラリー、ピューロマイシン媒介dsDNAディスプレイライブラリー、CDTディスプレイライブラリー、小分子に結合させたdsDNAライブラリー、およびストレプトアビジンディスプレイライブラリー)。
【0012】
「フルオロフォア」とは、分子を蛍光性とする分子の構成要素または官能基を意味する。例示的なフルオロフォアとしては、フルオロセイン、緑色蛍光タンパク質(GFP)、黄色蛍光タンパク質(YFP)、Alexa Fluor色素、Cy色素(GE Healthcare)、核酸プローブ(例えば、DAPI、臭化エチジウム、アクリジンオレンジ、またはヨウ化プロピジウム)、ヒドロキシクマリン、アミノクマリン、エトキシクマリン、ローダミン、BODIPY-FL、テキサスレッド、またはTRITCが挙げられる。
【0013】
「リンカー」とは、ライブラリーの核酸部分を機能性のディスプレイされた化学種と連結する分子を意味する。このようなリンカーは当技術分野においては周知であり、ライブラリー合成において使用することが可能なものとしては、これらに限定されるものではないが、5'-O-ジメトキシトリチル-l',2'-ジデオキシリボース-3'-[(2-シアノエチル)-(N,N-ジイソプロピル)]-ホスホロアミダイト、9-O-ジメトキシトリチル-トリエチレングリコール,1-[(2-シアノエチル)-(N,N-ジイソプロピル)]-ホスホロアミダイト、3-(4,4'-ジメトキシトリチルオキシ)プロピル-1-[(2-シアノエチル)-(N,N-ジイソプロピル)]-ホスホロアミダイト、および18-O-ジメトキシトリチルヘキサエチレングリコール,1-[(2-シアノエチル)-(N,N-ジイソプロピル)]-ホスホロアミダイトが挙げられる。このようなリンカーは異なる組み合わせで互いに縦に付加して、異なる所望の長さのリンカーを生成することができる。「分枝鎖リンカー」とは、ライブラリーの核酸部分をライブラリーの2以上の同一の官能性の化学種と連結する分子を意味する。分枝鎖リンカーは当技術分野では周知のものであり、その例は、対称的もしくは非対称的ダブラー(doubler)(1)および(2)、または対称的トレブラー(trebler)(3)から構成されうる。例として、Newcome et al., Dendritic Molecules: Concepts, Synthesis, Perspectives, VCR Publishers (1996); Boussif et al., Proc. Natl. Acad. Sci. USA 92: 7297-7301 (1995); および Jansen et al., Science 266: 1226 (1994)を参照されたい。
【0014】
「核酸ディスプレイライブラリー」とは、所望の標的に結合可能な分子を生成するためのインビトロでのタンパク質および/もしくはペプチド進化、ならびに/または小分子および/もしくは核酸進化(例えば、ssRNAまたはssDNA)の発見に使用されるディスプレイ技術を意味する。修飾ペプチドを含むタンパク質およびペプチドの場合では、このプロセスによりピューロマイシン連結部を介してmRNA前駆体またはdsDNAと結合した翻訳されたペプチドまたはタンパク質が得られる。一部の核酸ディスプレイの場合では、タンパク質またはペプチドは、核酸と共有結合または非共有結合により会合するタンパク質を介してmRNA、ssDNA、またはdsDNAと結合される。小分子ディスプレイの場合では、核酸は小分子と共有結合する。核酸ディスプレイの場合では、ssRNAまたはssDNAのランダム化された領域を直接使用する。次いで、核酸ライブラリー複合体が選択工程(例えば、アフィニティークロマトグラフィー)において、固定された標的と結合する。次いで、十分に結合した核酸コンジュゲートを回収し、ポリメラーゼ連鎖反応により増幅する。その結果、対象とする分子に対する所望の性質(例えば、親和性または特異性)を有する結合分子をコードしたヌクレオチド配列が得られる。核酸ディスプレイライブラリーにはdsDNAディスプレイライブラリーが含まれうる。例示的なdsDNAディスプレイライブラリーとしては、CIS dsDNAディスプレイライブラリー、ピューロマイシン媒介dsDNAディスプレイライブラリー、CDT dsDNAディスプレイライブラリー、小分子に結合されたdsDNAライブラリー、およびストレプトアビジンdsDNAディスプレイライブラリーが挙げられる。例として、Odegrip et al., Proc. Natl. Acad. Sci. USA 101: 2806-2810 (2004); Kurz et al., Chembiochem. 2: 666-672 (2001); Fitzgerald, Drug Discov. Today 5: 253-258 (2000); およびClark et al., Nat. Chem. Biol. 5: 647-654 (2009) を参照されたい。
【0015】
「核酸」とは、モノマーヌクレオチド(例えば、5個以上のヌクレオチド)からなる巨大分子を意味する。核酸にはデオキシリボ核酸(DNA)(例えば、cDNA、mtDNA、および2本鎖DNA(dsDNA))およびリボ核酸(RNA)(例えばmiRNA、siRNA、snRNA、snoRNA、shRNA、RNAiおよびmRNA)が含まれる。核酸は2本鎖、1本鎖、または単離されたもの(例えば部分精製されたもの、本質的に純粋なもの、合成されたもの、組み換えにより生成したもの)であってよい。核酸は1個以上のヌクレオチドの付加、欠失、置換、および/または改変によって改変させることができる。こうした改変には、核酸の末端または内部への(1個以上のヌクレオチドにおける)非ヌクレオチド物質の付加が含まれうる。本発明の核酸分子中のヌクレオチドには、天然に存在しないヌクレオチドなどの非標準的ヌクレオチドも含まれうる。
【0016】
「タンパク質」、「ポリペプチド」、「ポリペプチドフラグメント」、または「ペプチド」とは、天然に存在するポリペプチドまたはペプチドの全体または一部を構成する、あるいは、天然に存在しないポリペプチドまたはペプチドを構成する、翻訳後修飾(例えばグリコシル化またはリン酸化)によらない、2個以上のアミノ酸の任意の鎖を意味する。
【0017】
「小分子」とは、約1000ダルトンよりも低い分子量を有する分子を意味する。小分子は有機または無機であってよく、例えば化合物ライブラリーまたは天然の供給源から単離するか、あるいは既知の化合物の誘導体化によって得ることができる。
【0018】
本発明の他の特徴および利点は、以下の詳細な説明、図面、実施例および特許請求の範囲より明らかとなる。
【図面の簡単な説明】
【0019】
【図1】2個以上の化学分子が、多官能性リンカー部分を用いて(例えば、デンドリマーディスプレイとして)ライブラリーの核酸部分に結合されるように例示的なライブラリーが生成されることを示す模式図である。
【図2】2個以上の化学分子が、多官能性リンカー部分を用いてライブラリーの核酸部分の両方の鎖に結合されるように例示的なライブラリーが生成されることを示す模式図である。
【図3】核酸ライブラリーに結合し、更に発現遺伝子またはdsRNAiに結合したストレプトアビジン(4量体)を示す模式図である。
【図4】T7発現ベクターの代表例を示す模式図である。
【図5】細胞質T7活性を検出するための一過性トランスフェクションアッセイを説明した模式図である。
【図6】VH抗体ドメインのコード領域の上流にT7プロモーターを含むPCRフラグメントの構成要素を示す模式図である。
【図7】T7 RNAポリメラーゼ(RNAP)が、一過性にトランスフェクトされたHEK293T細胞内で活性であることを示すウェスタンブロットおよびRT-PCRアッセイである。細胞溶解物をSDS-PAGEにより分離し、T7ポリメラーゼに対するモノクローナル抗体を用いてウェスタンブロット分析に供した。抗T7ポリメラーゼ抗体は、このタンパク質の予想される分子量(約99kDa)と一致するバンドを認識する。RT-PCRアッセイは、VH PCR鋳型がHEK293T細胞内でT7ポリメラーゼによって転写されることを示している。コントロールレーンは、RT-PCR活性がT7ポリメラーゼの発現に依存しており、T7ポリメラーゼプロモーターを含む鋳型の存在下においてのみ生ずることを示している。
【図8】VH PCR鋳型の量を一過性トランスフェクション中に滴定することによってT7ポリメラーゼRT-PCRアッセイの感度を試験するRT-PCRアッセイである。
【図9】T7 RNAPが、一過性にトランスフェクトされたVCaP前立腺癌細胞内で活性であることを示すRT-PCRアッセイである。
【図10】安定的な前立腺癌細胞株からのT7 RNAP転写産物の検出を示すRT-PCRアッセイである。
【図11】22rV1_T7細胞株が活性T7ポリメラーゼを発現することを示すRT-PCRアッセイである。
【図12】ビオチン化ペプチドまたはVH結合物質とストレプトアビジンとの複合体の集合を示す模式図である。
【図13】集合したVHまたはペプチド複合体、またはビオチン化ペプチドもしくはタンパク質を有さない集合体のHEK293T細胞内への一過性トランスフェクションを示す模式図である。
【図14】T7 RNAPをトランスフェクトしたHEK293T細胞内へのストレプトアビジン集合体の送達を示すアッセイである。
【図15】図15Aは、ペプチド-dsDNAコンストラクトの合成の模式図である。VHクローンをPCR増幅してBsmI部位をT7プロモーターの5'側の上流に付与した。制限酵素による消化および精製の後、コンストラクトをHP-1-DATF-R7(DTAFおよび(-Arg-εAhx)6-Argペプチドにより修飾したヘッドピース)にライゲートした。図15Bは、ライゲーション反応の電気泳動ゲルである(レーン1および2: VHにライゲートされた異なるHP-1試料; レーン3: ライゲートしていないVHPCR産物; M: マーカー)。図15Cは、T7プロモーター活性の確認を示すゲルである。このゲルは、図15Bのレーン1〜3からの試料を用いたT7 Megascript (Ambion)反応を示している。
【図16】HEK293T細胞内におけるT7 RNAPによるペプチド-鋳型コンジュゲートの内部移行および転写を示すアッセイである。
【図17】siRNAによって媒介される細胞質侵入選択の手法を示す模式図である。
【発明を実施するための形態】
【0020】
発明の詳細な説明
本発明は細胞内に核酸を送達する分子を同定するための方法をその特徴とする。
【0021】
本発明の組成物および方法は、核酸アプタマーライブラリーまたは任意のディスプレイ法を利用するものであり、ライブラリー(例えば小分子またはタンパク質のライブラリー)を、例えばライブラリーの各メンバーをコードする核酸(例えばRNAまたはDNA)に(例えば遺伝子型-表現型結合を介して、または共有結合若しくは非共有結合による相互作用を介して)結合させるものである。例えば、Lipovsek et aI., J. Immunol. Methods 290: 51-67 (2004); Bertschinger et al., Protein Eng. Des. Sel. 17: 699-707 (2004); Yonezawa et al., Nucleic Acids Res. 31: e118 (2003); Tabuchi et al., FEBS Lett. 508: 309-312 (2001); Odegrip et al., Proc. Natl. Acad. Sci. USA 101: 2806-2810 (2004);およびFujita et al., J. Med. Chem. 45: 1598-1606 (2002)を参照されたい。一態様においては、例えばフルオロフォア、重要な遺伝子を標的としたRNAi分子、RNAi分子もしくはアンチセンス配列をコードしかつ発現可能なdsDNA配列、レポーター遺伝子(例えばGFP)を発現可能なdsDNA配列、タンパク質(例えばポリメラーゼ、転写因子、もしくはリプレッサー)を結合するdsDNA配列、小分子、または細胞内読み出しシグナルを発生するタンパク質またはペプチドを発現可能なdsDNAなどの細胞内読み出しシグナルを発生する分子とRNAまたはDNAとを更に連結する。
【0022】
細胞を本発明のライブラリーと接触させることもできる。細胞内への所望の分子(例えば核酸分子)の送達を促進する分子(例えば小分子またはペプチド)に連結されたライブラリーのメンバーは、例えば、細胞内にエンドサイトーシスによって取り込まれる。細胞表面に結合したままの核酸ライブラリーのメンバーは、例えばイオン強度、pH、界面活性剤、またはプロテアーゼ処理によって除去する。この後、細胞を溶解し、内部移行した物質を増幅して(例えばPCR)、送達を促進させる分子を同定することができる。
【0023】
リンカー部分(例えば多官能性リンカー部分)を用いて2個、3個、4個またはそれ以上の化学分子が(例えばデンドリマーディスプレイとして)核酸ディスプレイライブラリーのメンバーに結合されたライブラリーを生成することができる(図1)。この手法を用いて例えば細胞上の複数の受容体を刺激して内部移行を生じさせることができる。モノマー化学種を用いる他の方法では、受容体が効果的な内部移行を生じず、シグナルが非常に低いかあるいは存在しない場合がある。このため、本発明のライブラリーの設計の1つ(例えばDNAに結合された小分子)は、リンカーの一方の端部にDNA識別領域が、他方の端部に複数のアミン(または他の反応性化学種)が結合されたリンカーを有するものとして生成される。複数のアミンまたは他の反応性化学種を用いて、合成された小分子の複数のコピーをDNA上に生成及び結合させる。一態様においては、両方の鎖が小分子をディスプレイしてもよい(図2)。また、核酸ディスプレイライブラリーのメンバーのDNA分子の長さに沿って複数の小分子をディスプレイするように、ライブラリー合成によって容易に官能化されるアミンまたは他の分子を、例えばウリジンまたはシトシンのC5位を介して識別領域の複数の位置に沿って単独でまたはマルチマーとして組み込むことも可能である。DNAの両方の鎖を修飾することができる。更に、疎水性残基(例えば5-メチルC、C5アルキル置換、C5アルキニル置換など)の付加などのように、細胞内への侵入が促進されるようにDNAの塩基を修飾することができる。
【0024】
核酸ディスプレイ法(例えば、mRNAディスプレイ、ストレプトアビジンディスプレイ、共有結合DNAディスプレイ、非共有結合DNAディスプレイなど)によってドメイン同士が連結されるように、タンパク質またはペプチドのドメイン(例えば抗体のVHドメイン)からなるライブラリーが生成される。核酸はタンパク質またはペプチド結合ドメインをコードしており、更に、例えばレポーター遺伝子(例えばGFP)、RNAi遺伝子(例えばhnRNAi)、転写因子、または転写因子結合部位をコードしている。1つの例では、GFPの遺伝子に結合したVHドメインのライブラリーを細胞と接触させ、GFPを発現する細胞を単離し、VHドメインのアイデンティティーをPCRおよび配列決定によって決定する。この方法を用いることにより、細胞にdsDNAを送達する特異的なVH結合物質を新規な送達ビヒクルとして同定することができる。
【0025】
別の態様では、ストレプトアビジンを核酸ライブラリーに結合させ(例えば核酸がビオチン分子を含み、ビオチン結合部位を介してストレプトアビジンと結合する)、発現遺伝子(例えば発現遺伝子がビオチンを含み、ストレプトアビジンは4量体であるために、第2のビオチン結合部位を介してストレプトアビジンと結合する)またはdsRNAi(例えばdsRNAiがビオチンを含み、第2のビオチン結合部位を介してストレプトアビジンと結合する)に更に結合させる(図3)。
【0026】
本発明は更に、核酸、または他のペイロード(例えば小分子またはペプチド)を細胞内に送達する新規な分子を同定するための一般的な方法をその特徴とする。この方法では、小分子ライブラリーまたはタンパク質ライブラリーのいずれかを、ライブラリーの各メンバーをコードし、更にRNAポリメラーゼのプロモーター領域(例えばT7 RNAポリメラーゼ)を含むdsDNAと(例えば、遺伝子型-表現型結合を介して、または共有結合若しくは非共有結合による相互作用を介して)結合させるディスプレイ法を利用する。この後、このライブラリーを、細胞質中に適切なRNAポリメラーゼを発現している細胞とインキュベートする。この後、細胞の細胞質中に局在化したライブラリーのメンバーはRNAポリメラーゼによって転写されうる。次いで細胞を溶解し、RNAを単離してRT-PCRに供することにより細胞質中に存在するdsDNAを同定する。この後、このdsDNAを同定することにより、細胞内への送達を媒介した、dsDNAと結合した分子が同定される。
【0027】
細胞内への核酸の送達は、アンチセンス、miRNA、RNAiまたは遺伝子治療的アプローチのいずれであるかによらず、このクラスの分子の治療的開発において依然として重要な課題の1つである。送達物質の発見における大きな障害の1つとして、細胞内への核酸の放出につながる、発生頻度が希な事象を検出する能力がある。細胞内への1個の分子の放出を検出できることが理想的であるが、これには超高感度の読み出しシステムを必要とする。1個の分子の送達が実現されれば、送達法を更に最適化することが可能となる。
【0028】
dsDNAは、ポリメラーゼによる増幅により細胞内の1個の分子を検出することが可能である。例えば、dsDNAの個別の分子の細胞核内へのマイクロインジェクションは、対象とする遺伝子を有する、制限酵素で消化された直鎖状のフラグメントであっても、発現タンパク質の免疫蛍光染色によって検出されるような当該遺伝子の転写および翻訳を生じる。これに対して、dsDNAの細胞質中へのマイクロインジェクションは、高濃度で導入した場合でも、遺伝子の発現が生じることはほとんどない。しかしながら、細胞質中に局在するT7 RNAポリメラーゼを発現する細胞を使用することにより、dsDNAを細胞質中で発現させることができる。
【0029】
ディスプレイライブラリーとして、小分子、ペプチド、またはタンパク質をdsDNAと結合させるための方法が幾つか存在している。一般に、遺伝子型(dsDNA)とディスプレイされる分子との間にはコード関係が存在するため、遺伝子型の配列を決定することによってディスプレイされる分子が同定される。本明細書では、新規な選択技術をディスプレイライブラリーとともに用いて細胞への分子の送達の媒介物質を同定するための方法について述べる。
【0030】
核酸の細胞内送達を媒介するタンパク質または小分子を同定するためのある一態様では、RNAポリメラーゼ結合部位を含むdsDNAディスプレイライブラリーを、細胞質中でRNAポリメラーゼを発現している細胞とインキュベートする。インキュベート後、細胞を溶解し、RT-PCRに供して細胞質中に送達されたdsDNAライブラリーのメンバーから生じたあらゆるRNA転写産物を増幅する。
【0031】
別の態様では、小分子またはペプチドライブラリーのスクリーニングによるアプローチを利用し、タグが付与されていない分子をdsDNAライブラリーによってスクリーニングすることによって送達を促進する物質を探索する。この態様では、RNAポリメラーゼ結合部位を含むdsDNAディスプレイライブラリーを、細胞質中でRNAポリメラーゼを発現している細胞とインキュベートする。この後、小分子またはペプチドを細胞に加えてdsDNA分子の放出を促進する。
【0032】
更に別の態様では、いかなる小分子、ペプチドまたはタンパク質もディスプレイされていない、RNAポリメラーゼ結合部位を含むdsDNAを、細胞質中でRNAポリメラーゼを発現している細胞とインキュベートする。この後、細胞に小分子またはペプチドを加えてdsDNA分子の放出を促進する。上記の方法をハイスループットのスクリーニングモードにおいて使用することによって細胞へのdsDNAの送達の促進物質を同定することができる。
【0033】
更に別の態様では、ライブラリーのdsDNAを、dsDNAメチルトランスフェラーゼを発現する細胞に加える。ライブラリーのメンバーが細胞に入ると、細胞内のdsDNAはすべてメチル化され、回収され、かつメチル化特異的PCRに供される。
【0034】
哺乳動物細胞中へのバクテリオファージT7 RNAポリメラーゼ(T7 RNAP)の細胞質局在化については、例えば参照により本明細書に組み入れられるElroy-Stein and Moss; Proc. Natl. Acad. Sci. USA 87: 6743-6747 (1990) および Wang et al., Analytical Biochem. 375: 97-104 (2008)に述べられている。
【0035】
本明細書に記載の方法を使用することにより、例えば任意の形態の核酸、タンパク質、ペプチド、小分子、リポソーム、またはナノ粒子などの(ただしこれらに限定されない)、幅広い送達性を潜在的に有する、組織または細胞への送達を媒介する一般的な物質を同定することが可能である。
【実施例】
【0036】
以下の実施例は本明細書を説明することを目的としたものである。これらの実施例はいかなる意味においても本発明を限定しようとするものではない。
【0037】
実施例1 T7 RNAポリメラーゼ細胞株の構築
下記のPCRプライマーを用いてBL21細胞からバクテリオファージT7 RNAポリメラーゼ(RNAP)を増幅した(NcoI部位を太字、開始ATGを斜体、Kozak配列を下線、NotI部位を小文字、タンデム停止コドン(TTATTA)を斜体で示す)。
【0038】
増幅した遺伝子産物をNcoI/NotIフラグメントとしてpEF/myc/cyto発現ベクター(Invitrogen # V890-20)に方向を決めてクローニングした(図4)。このベクターは、強力なEF-1aプロモーターおよび安定した細胞株選択のためのネオマイシン耐性遺伝子を有する、細胞質での発現用に設計されたものである。このベクターは、マルチクローニング部位であるポリリンカーの上流に通常見られるT7プロモーターを欠いているため、T7 RNAPが発現する際にプロモーター活性が競合する可能性がない。
【0039】
実施例2 一過性にトランスフェクトしたHEK293T細胞におけるT7 RNAPの活性
細胞質T7が哺乳動物細胞においてポリメラーゼとして活性を有しうることを調べるため、本発明者らはHEK293T細胞において一過性トランスフェクションアッセイを開発した(図5)。細胞を24ウェル皿に350,000細胞/ウェルで播種し、10%ウシ胎児血清(FBS)を添加したイーグル最小必須培地中で一晩インキュベートした。VH抗体ドメインのコード領域の上流にT7プロモーターを含むDNA鋳型をPCRにより以下のようにして調製した。インビトロの転写および翻訳シグナルであるT7TMVをコードする5'末端側のオリゴヌクレオチド
と、C末端のVHDNAのCmuおよびSAリンカー領域Y15とアニールする3'末端側のオリゴヌクレオチド
を用いて、ナイーブなヒトVHライブラリーからのVHDNAをPCR増幅した(Strategene 600312)。PCR産物をゲルで精製した。PCRフラグメントの構成要素を図6にまとめる。このVHDNAフラグメントを、T7 RNAポリメラーゼを発現している細胞にトランスフェクトすると、T7 RNAPはこのDNAに作用してDNAをRNAに転写する。この特定のRNAをRT-PCRによって検出および増幅する。
【0040】
300fmolのT7 DNA鋳型および2.5μgのT7発現コンストラクトとの組み合わせを細胞にトランスフェクトし、同時にコントロール試料には以下をトランスフェクトした。すなわち、鋳型なし + 2.5μgのT7_pEF/myc/cytoベクター; 300fmolのT7 DNA鋳型 + 2.5μgのpEF/myc/cytoベクター; または鋳型なし + 2.5μgのpEF/myc/cytoベクター。細胞は、Lipofectamine2000(Invitrogen)を使用し、製造者のプロトコールにしたがって(2μl Lipofectamine/トランスフェクション)トランスフェクトした。細胞を一晩インキュベートした後、溶解した細胞からRT-PCRによる分析用にRNAを調製した。T7 RNAPの活性を試験するため、特異的かつ高感度のRT-PCRアッセイを開発した。RNeasy Mini Kit(Qiagen 74104)を使用し、上記実験から回収した細胞溶解物から細胞質RNAを調製した。簡単に述べると、細胞溶解物を遠心分離により沈降させて不溶性タンパク質を除去した。清澄化した溶解物をグアニジン塩およびエタノールを含むRLTバッファーと混合し、結合カラムに加えた。カラムをRWIバッファーで洗った。RNase-free DNase kit(Qiagen 79254)によってカラムにDNアーゼ処理を行ってキャリーオーバーDNAを除去した。カラムを更にRPEバッファー、70%エタノールで洗浄し、乾燥した。RNAを30μlのヌクレアーゼを含まない水で溶出した。下流のPCRのためにRNA試料からDNAを完全に除去するため、RNA溶出液を37℃で1時間、10単位のDNアーゼIで消化した(Ambion AM2222)。次いで製造者の推奨するプロトコールにしたがってRNAをRNeasy MiniElute Cleanup Kit(Qiagen 74204)で精製し、20μlのH2O中で溶出した。SuperscriptII逆転写酵素(Invitrogen 18064-014)を用いて逆転写を行った。簡単に述べると、10μlのRNA、10nmolの各dNTP、および5pmolのVH特異的3'Cmuオリゴヌクレオチド
の混合物を65℃で5分間インキュベートし、4℃に冷却してRNAの2次構造およびプライマーのアニーリングを低減させた。5X ファーストストランドcDNA合成バッファー、0.1M DTT (10X)、および200単位の逆転写酵素を最終容量20μlになるまで加えた。反応液を42℃で50分間インキュベートし、70℃で15分間、熱失活させた。1μlのファーストストランドcDNAを25μlの容量中、Herculaseバッファー、200μMのdNTP、0.2μMのT7TMV、S6-1
オリゴヌクレオチド、および1.25単位のHerculase(Strategene 600312)の存在下で15〜25サイクル増幅した。PCR産物を2%アガロースゲル(Invitrogen G5018-02)上で可視化した。
【0041】
T7ポリメラーゼの発現を確認するため、平行ウェルを上記のようにトランスフェクトし、NP-40界面活性細胞溶解バッファー中で溶解した。細胞溶解物をSDS-PAGEにより分離し、T7ポリメラーゼに対するモノクローナル抗体(EMD Bioscience #70566-3)を用いてウェスタンブロット分析に供した。抗T7ポリメラーゼ抗体は、このタンパク質の予想される分子量(約99kDa)と一致するバンドを認識した(図7)。RT-PCRアッセイにより、VHPCR鋳型がHEK293T細胞内においてT7ポリメラーゼによって転写されることが示された。コントロールレーンは、RT-PCR活性がT7ポリメラーゼの発現に依存し、T7ポリメラーゼプロモーターを含む鋳型の存在下においてのみ生ずることを示した(図7)。
【0042】
実施例3 一過性にトランスフェクトしたHEK293T細胞におけるT7 RNAPの感度
選択用の開始ライブラリーは極めて多様である(最大で約1014種類の異なる配列が存在する)ことから、細胞質に到達するライブラリーのメンバーの捕捉および増幅は極めて高感度でなければならない。本発明者らは、ある量のVHPCR鋳型を一過性トランスフェクション中に滴定することによってT7ポリメラーゼRT-PCRアッセイの感度を試験した。上記に概略を述べたトランスフェクションプロトコールにしたがって、3pmol/試料〜0.1fmol/試料の範囲のVHPCR鋳型をT7_pEF/myc/cyto発現コンストラクトとコトランスフェクトした。回収された試料のRT-PCRは、細胞当たり0.1fmolの鋳型であっても細胞質のT7ポリメラーゼによって転写され、検出可能なレベルにまで増幅可能であることを示している(図8)。
【0043】
実施例4 一過性にトランスフェクトしたVCaP前立腺癌細胞におけるT7 RNAPの活性
本発明の目的は、様々な種類の細胞において細胞質への取り込みの選択を可能にするプラットフォームを確立することにある。最近、幾つかのグループが、前立腺癌細胞の前立腺特異的膜抗原を標的とした試薬とsiRNAのコンジュゲートを作製し、標的化送達によってmRNAがノックダウンされることを実証することに成功した。例えば、Chu et al., Nucleic Acids Res. 34: e73 (2006); McNamara et al., Nat. Biotechnol. 24: 1005-1015 (2006), Dassie et al., Nat. Biotechnol. 27: 839-846 (2009)を参照されたい。そこで本発明者らは、概念実証実験を拡張して様々な前立腺癌細胞株における細胞質T7ポリメラーゼの活性を調べた。VCaP細胞は、細胞表面に前立腺特異的膜抗原(PSMA)を発現する前立腺癌細胞である。T7_pEF/myc/cyto発現コンストラクトおよびVHPCR鋳型によるVCaP細胞の一過性のトランスフェクションにより、T7ポリメラーゼがこれらの細胞の細胞質中において転写活性を有し、一過性にトランスフェクトしたHEK293T細胞において観察されたのと同様のT7および鋳型依存性が見られることが示された(図9)。
【0044】
実施例5 安定的な前立腺癌細胞株からのT7 RNAP転写産物の検出
T7ポリメラーゼRT-PCRアッセイを更に試験するため、本発明者らは、T7_pEF/myc/cytoコンストラクトを使用してネオマイシンに対する薬物耐性によりT7ポリメラーゼの発現について安定的に選択された他の前立腺癌細胞株を確立した。PC3およびDU-145細胞はPSMAを発現しない前立腺癌細胞株であり、LnCaPおよび22rV1はPSMAを発現する前立腺癌細胞株である。4種類の細胞のすべてにT7_pEF/myc/cytoまたはpEF/myc/cyto空ベクターのいずれかをトランスフェクトし、750μg/mlのネオマイシン(Geneticin, Gibco)の存在下での増殖について選択した。ネオマイシン耐性細胞をプールし、RT-PCRによりT7の発現についてアッセイした。約10 X 106個の細胞を回収、溶解し、RNAをRNeasy Mini Kit(Quiagen 74104)により上記に述べたようにして単離した。T7 RNAP転写産物を、3'T7 RNAP特異的オリゴヌクレオチド
を逆転写反応に、5'T7 RNAP特異的オリゴヌクレオチド
および同じ3'オリゴヌクレオチドをPCR増幅に使用することにより検出した。T7_pEF/myc/cytoコンストラクトをトランスフェクトすることにより安定的に薬物選択された細胞のみで、T7ポリメラーゼの増幅された領域とサイズが一致するcDNAバンドが見られた(図10)。比較のため、T7_pEF/myc/cytoコンストラクトを一過性にトランスフェクトしたHEK293T細胞由来の同量の全RNAを同じT7特異的オリゴヌクレオチドを用いたRT-PCRに供した。データは、安定的なT7前立腺癌細胞株では、一過性にトランスフェクトしたHEK293T細胞で見られるよりもT7ポリメラーゼの発現量が低いことを示している(図10)。
【0045】
実施例6 PSMA発現癌細胞株におけるT7 RNAPの活性
安定的に薬物選択された細胞において発現されるT7ポリメラーゼが活性を有するかを調べるため、本発明者らは改変されたRT-PCRアッセイを確立した。このアッセイは、すでに存在する安定的なT7の発現を考慮し、T7を一過性に発現させるための最初のトランスフェクションを省略した点以外は、上記に述べたアッセイと同様のものである。代わりに、VHPCR産物をT7の安定的な発現の存在下または非存在下で前立腺癌細胞株にトランスフェクトし、RT-PCRアッセイを上記に述べたように行った。図11は、VH cDNAの存在によって示されているように、22rV1_T7細胞株が活性なT7ポリメラーゼを発現することを示している。対照的に、陰性コントロール細胞株(22rV1_ベクター)はT7依存性のRT-PCR活性を示さない。
【0046】
実施例7 ビオチン-ストレプトアビジン集合体に対する細胞質T7ポリメラーゼ活性
細胞質への侵入の選択が支障なく行われるためには、オリゴヌクレオチドおよびコードされたペプチドまたはタンパク質のビオチン-ストレプトアビジン集合体はT7ポリメラーゼ鋳型と同様に効果的でなければならない。ストレプトアビジンと集合したビオチン化ペプチドまたはVH結合物質の複合体を細胞にトランスフェクトし、T7 RNAPによって転写させ、RT-PCRによって検出することが可能であるかを試験するため、上記のVHDNAを、5'末端にSP6およびT7プロモーターの両方の配列
を有し、3'末端にCmu-flag-SA-polyA配列を有するように改変した(図12)。次いでこのDNAをSP6 transcription kit (Ambion AM1330)によってインビトロでRNAに転写した。RNAeasy MiniElute Cleanup Kit (Qiagen 74204)によりRNAを精製した。SAディスプレイ用のビオチン化RNA/DNAリンカーをRNAに1:1の比でアニールし、RNA鋳型とUV架橋させた。リンカー上のビオチンと1:1のモル比で相互作用させることにより、ライゲートしたRNA上にストレプトアビジンを負荷した。次いでこの集合複合体をオリゴdT精製に供した。集合複合体はRNAのpolyAテールを介してオリゴdTと結合し、遊離SAを洗い流した。ライゲートしたDNAリンカーをプライマーとして使用してオリゴdTセルロース上で逆転写を行い、superscriptII(Invitrogen 18064-014)によって37℃で1時間、伸長した。次いでRNAとファーストストランドcDNAとのハイブリッドをRNアーゼH(Invitrogen 18021-014)によって1時間消化してRNA鎖を開裂させた。溶液中のファーストストランドDNAをオリゴdTセルロースを遠心分離して回収した。SP6T7オリゴヌクレオチドをプライマーとして用いてセカンドストランドDNAを合成し、superscriptII(Invitrogen 18021-014)によって伸長した。次いでビオチン化されたVHまたはペプチドの分子をdsDNA-SA複合体と2:1のモル比でインキュベートしてdsDNA-SA-VHまたはdsDNA-SA-ペプチドを生成した(図12)。
【0047】
概念実証実験として、細胞貫通ペプチド(CPP)としてこれまでに述べられている2種類の異なるペプチドを試験した。すなわち、TATペプチド
およびアンテナペディア(Antennapedia)ペプチド
である。図13に概略を示すように、集合したVHまたはペプチド複合体、またはビオチン化ペプチドもしくはタンパク質を有さない集合体をHEK293T細胞に一過性にトランスフェクトし、T7ポリメラーゼをコトランスフェクトした細胞中でRT-PCRについてアッセイした。その結果、本発明者らのライブラリーにおいて利用されるビオチン:ストレプトアビジン結合手法を介してペプチドまたはVHタンパク質と集合したオリゴヌクレオチドは、細胞質のT7ポリメラーゼによって転写されうることが確認された(図14、上側のパネル)。したがって、細胞質T7ポリメラーゼシステムを、細胞に基づいた選択を行う際に細胞質中への侵入の選択圧力として利用することが可能である。
【0048】
この選択手法を更に検証するため、本発明者らは、トランスフェクションとは独立した細胞質侵入によるT7ポリメラーゼ活性を実証することを試みた。TATおよびANTペプチドはこれまでにCPPとして同定されていることから、これらのペプチドとビオチン:ストレプトアビジンの集合体も細胞質に侵入して、トランスフェクション試薬の非存在下で細胞質T7ポリメラーゼの鋳型として機能しうる可能性がある。40pmolのペプチドが結合した集合体、またはペプチドを有さない集合体を、T7_pEF/myc/cyto発現コンストラクトを一過性にトランスフェクトしたHEK293T細胞に加えた。18時間後に細胞を溶解し、上記に述べたようにRT-PCR活性についてアッセイした。その結果は、ペプチド集合体の細胞への取り込みが起こり、これらの複合体に対する細胞質T7ポリメラーゼ活性の結果としてのRT-PCRを容易に測定できることを示している(図14、下側のパネル)。コントロール集合体も同様に細胞質への侵入能を獲得したようであるが、これらのコントロール鋳型からのRT-PCR活性は、CPP集合体のRT-PCR活性と比較して大幅に低くなっている。
【0049】
実施例8 T7 RNAPを発現している細胞内に送達するためのペプチド-dsDNAコンストラクトの合成
リン酸化オリゴヌクレオチドHP:
をIDT DNAにより合成した。このDNAはオーバーハングを有するヘアピンへと折り畳まれ、制限酵素BbvCIおよびこの酵素のニック型である、トップストランドまたはボトムストランドのいずれかを切断しうるNb.BbvCIまたはNt.BbvCI (New England Biolabs, Inc.)の開裂部位CCTC AGCを含んでいる。ヘアピンループの中央には、アミノ-PEGリンカー(PEG2000、約45個のエチレングリコール単位)の結合に用いられる側鎖としてC5アミノ修飾dT(dT-C6-NH、C6は6位の炭素リンカーを指す)が挿入されている。
【0050】
10nmolのオリゴ「HP」を50μlの水に溶解した。20倍のモル過剰量のFmoc-アミノ-PEG2000-カルボキシル-NHSエステル(Jen-Kem)を50μlのDMFに溶解し、室温で2時間の間に2回に分けてオリゴ溶液に加えた(最終的な溶媒組成は、50% DMF-50% 水)。この後、60μlの1M Tris HCL、pH7.0(最終濃度200mM)を加えて過剰量のNHSエステルをクエンチした。この溶液を室温で更に30分間インキュベートした。得られた反応混合物を水で500μlに希釈し、NAP-5カラム(Sephadex-25, GE)に通して脱塩した。
【0051】
得られた物質を凍結乾燥し、100μlの水に溶解した。20μlのピペリジン(最終濃度20%)を加え、室温で2時間インキュベートした。アミンの脱保護および非水溶性のFmoc基の放出により濁った沈殿が生じた。次いで反応液を0.2μmスピンフィルター(Millipore)に通して濾過し、エタノールを3回加えることによって300mM酢酸ナトリウムから沈殿させた。高い結合効率のため、得られたヘッドピースHP-1を更に精製することなく使用した。
【0052】
モデル化合物として5-(4,6-ジクロロトリアジニルアミノフルオレセイン)(DTAF)(Anaspec)をHP-1のアミノ基と結合させた。DTAFは、1個のアミノ化合物が結合したトリクロロトリアジンのスキャフォールドを構造的に表している。ライブラリーを形成するには、トリクロロトリアジンのスキャフォールドを3個の塩素の位置のそれぞれにおいて、多様な構成単位によって誘導体化することができる。これにより、モデルライブラリーに蛍光標識も提供される。反応液(10μl)は以下のように構成した。5μlの400μM HP-1水溶液に、2μlの750mMホウ酸塩バッファー(pH9.5)、および1μlのDMFを加えた。DTAFを50mMにまでDMFに溶解し、2μlを反応液に加えた。HP-1およびDTAFの最終濃度はそれぞれ200μMおよび10mMであった(DTAFの50倍過剰)。DMFの最終濃度は30%であった。HP-1は最大で90%のDMFにまで溶解することが分かり、DMFなどの有機溶媒に溶解しうることが示唆された。反応を4℃で16〜20時間進行させた。次いで反応混合物を水で30〜50μlに希釈し、Zebaスピンカラム(Pierce)上で脱塩した。更なる精製は行わなかった。
【0053】
トリアジンスキャフォールド上の最後の塩素反応基の修飾に用いるため、高アルギニンペプチドR7、すなわちH(-Arg-εAhx)6-Arg-OH (Bachem)を選択した。これは、細胞内への化合物の送達に使用されるアルギニン-アミノヘキサン酸細胞膜透過性ペプチドである。反応液は上記と同様に構成した。すなわち、20μlの反応液は、150mMホウ酸塩バッファー(pH9.5)に溶解された約200pmolのHP-1-DTAF(工程1)、および10nmolのR7ペプチドを含んでいた。これらの条件下ではアルギニンの側鎖は反応せず、ペプチド内の唯一の反応性アミンはN末端である。反応を75℃で12時間進行させ、Zebaスピンカラム上で脱塩することによって精製した。
【0054】
細胞内送達実験で使用したVHDNAコンストラクトは、分子の5'末端にT7プロモーター領域を、3'末端の近傍にCmu領域を有する約400bpのVHDNAの単一のクローンのPCR産物から調製した。VHDNAコンストラクトをモデル化学ライブラリーの修飾されたヘッドピースに連結するため、クローンのPCR増幅によってBsmI制限部位をT7プロモーター領域の上流に組み込んだ。BsmI制限酵素による消化物は、ヘッドピース(3'CCオーバーハング)とのライゲーションを可能にする3'GGオーバーハングを生じた。BsmI部位(下線で示される)を有する5'プライマーをIDT DNAにより合成した。すなわち、
である。PCR増幅後、PCR purificatin kit (Invitrogen)を使用してVHDNAコンストラクトを精製し、得られたDNAを、65℃でNEBバッファー4中、2時間、250UのBsmI(NEB)により消化した。DNAを2%アガロースゲル上で精製した。ライゲーション反応液(30μl)は、BsmIで消化された2pmolの各VHDNAコンストラクト、ならびに1 X T4 DNAリガーゼバッファーに加えたHP-1-DTAF-R7(アルギニン-アミノヘキサン酸ペプチド)および60Weiss単位のT4 DNAリガーゼ(NEB)を含んでいた。反応液を16℃で20時間インキュベートした。高いライゲーションの効率のため、この物質を更に精製を行うことなく細胞内送達/T7 RNAP実験に更に使用した。結果を図15にまとめて示す。
【0055】
上記に述べた実験と対応して、DNAリンカーを介してVHDNAとライゲートされた細胞貫通小分子C2は、トランスフェクションおよびトランスフェクションとは独立した細胞質侵入により同様の結果を示した(図16)。
【0056】
実施例9 高活性T7ポリメラーゼ細胞株のクローン誘導
ポリクローナル集団よりも高いT7ポリメラーゼ活性を有するクローン細胞株を単離するため、T7-蛍光レポーターコンストラクトを作製して、トランスフェクションまたはウイルス感染により上記に述べたように前立腺癌ポリクローナル安定細胞株に導入した。最も高いT7ポリメラーゼ活性のレベルを有する細胞は最大の蛍光を発生し、Mo-Flo単一細胞分別FACS装置により単一細胞の集団として捕捉される。次いで個々のクローン細胞株を蛍光についてスクリーニングし、続いて、FACSにより報告される最も高いT7活性を有する細胞をRT-PCRアッセイにより二次スクリーニングする。
【0057】
実施例10 核酸送達用の高親和性VH/ペプチド試薬の機能的選択
本明細書に記載のシステムの用途の1つに、機能性細胞に基づいた選択による核酸送達用の高親和性VH/ペプチド/小分子試薬の同定がある。SP6およびT7プロモーターを有するVH/ペプチドをコードするDNAライブラリーを、SP6転写酵素によってRNAライブラリーに転写し、ビオチン化ストレプトアビジン(SA)ディスプレイリンカーにライゲートする。次いでライゲートしたRNA上にSAを負荷し、3'末端にピューロマイシン様分子を有する別のビオチン化リンカーと集合させる。集合したVH/ペプチドライブラリーをインビトロで翻訳するとRNAライブラリーとそれらがコードするVH/ペプチドライブラリーとの融合体を形成する。本明細書に記載のように、オリゴdT精製、逆転写、RNアーゼH消化、およびセカンドストランドcDNA合成を行う。特定の標的によって媒介される細胞侵入を行うために、精製されたdsDNA-VH/ペプチド融合ライブラリーを、標的を発現していない一致した陰性細胞株とライブラリーを何度も接触させてバックグラウンドの結合物質を除去することによって対抗選択する。予めきれいにしたDNA融合ライブラリーと標的発現細胞とを接触させ、内部移行の機序とは無関係に、細胞に結合、侵入させ、細胞質に到達させる。細胞を洗って非特異的結合物質を除去し、細胞表面の内部移行されていない結合物質を取り除く。細胞質に侵入したライブラリーのメンバーのみがT7 RNAPによって認識され、転写される。細胞質に到達してT7ポリメラーゼによって転写されたライブラリーのメンバーは、上記に概略を述べた概念実証実験によって規定される条件を適用して、RT-PCRによって回収される。濃縮された結合物質は次のラウンドの選択に供する。複数ラウンドの選択および濃縮によって細胞質侵入プールが生成され、当技術分野では周知の標準的方法を用いた濃縮化集団のサブクローニングおよび配列決定によって媒介物質が同定される。
【0058】
一態様においては、細胞内に送達される分子を有する細胞を同定するための更なる基準として、ビオチン化siRNAをSAと集合させてDNA、SA、VH/ペプチド、およびsiRNAの複合体を生成する。SAは4つの結合部位を有しているため、各成分をSAと1:1のモル比で加えることによって結合部位のそれぞれに対象とする試薬を負荷することができる。この複合体を、機能的読み出しシグナルによって、標的遺伝子のmRNAノックダウン、および標的遺伝子により媒介される細胞機能の阻害についてスクリーニングする。
【0059】
標的特異的な選択以外に、上記に概略を述べた手法は、異なる細胞起源または細胞内状態(例えば、対抗選択用細胞型としての肝細胞および陽性細胞型としての心筋細胞; 対抗選択用の形質転換されていない細胞型および陽性選択用の形質転換された細胞型; または、対抗選択用の未分化細胞および陽性選択用の分化細胞)を構成する陽性および陰性細胞株を使用することによって細胞特異的な細胞質侵入ビヒクルを同定するためにも適用される。また、細胞質侵入選択は、複数の細胞型または細胞表面の標的を標的としうる送達ビヒクルを単離するために、対抗選択の手法を用いずに特定の細胞型に適用される。
【0060】
細胞質侵入選択には、選択圧力としてオリゴヌクレオチド(例えば、siRNA、miRNA、転写因子/リプレッサー滴定、またはアンチセンスオリゴヌクレオチド)送達によって媒介されるmRNAノックダウンを利用して更なる改良が加えられる。テトラサイクリン調節性T7ポリメラーゼ遺伝子を、選択した細胞株にTetリプレッサータンパク質のcDNA(TetR)とともに導入する。これにより、ビオチン:ストレプトアビジンライブラリー複合体は、ビオチン化DNAをコードしたペプチド/VH+ストレプトアビジン+ビオチン化ペプチド/VH+TetR mRNAに対するビオチン化siRNAを含むことになる(図17)。いずれかのライブラリーメンバーが細胞質に到達し、siRNAがmRNA分解に関与するRSC複合体に到達すると、TetR mRNAが除去され、その結果TetRタンパク質が失われ、T7ポリメラーゼ活性が誘導される。これにより細胞質取り込みを媒介するVH/ペプチドがT7によって転写され、その後、RT-PCRによって増幅および回収される。
【0061】
実施例11 核酸送達ビヒクルのインビボ選択
核酸のインビボ送達を媒介する物質の同定は、細胞質ポリメラーゼを有する形質転換マウスを作製することによって実現される。例えば、T7 RNAPを発現する形質転換マウスを作製する。次いで、標準的投与法(例えば、尾静脈注射)を用いてdsDNAライブラリーをマウスに投与する。注射後、組織または細胞を単離して溶解する。RNAを単離してRT-PCR、サブクローニング、および配列決定を行って所望の組織および/または細胞への侵入を媒介するコードされた分子を同定する。場合により、このプロセスを繰り返して、侵入可能な化学種を濃縮する。化学種が同定された後、対象とする分子を核酸、タンパク質、または小分子の任意の形態に結合させて組織および/または細胞特異的送達について試験する。
【0062】
実施例12 DNAメチルトランスフェラーゼを用いた核酸ディスプレイライブラリーのメンバーの同定
DNAのメチル化用のPCRプライマーの最適な結合部位を含むdsDNAライブラリーを調製する。このdsDNAライブラリーをDNAメチルトランスフェラーゼ(DNMT1)を過剰発現している細胞とインキュベートし、特異的なライブラリーメンバーを特定の細胞内に内部移行させる。細胞質内に侵入するとライブラリーからのdsDNAタグはDNMT1の基質となり、選択的にメチル化される。この後、細胞を溶解し、dsDNAタグを単離してメチル化特異的PCRのための標準的プロトコール(Herman et al, Proc. Natl. Acad Sci. USA 93: 9821-9826 (1996))を用いて亜硫酸水素ナトリウムで処理する。次いでメチル化されたdsDNAタグを、メチル化特異的プライマーを用いて選択的に増幅する。PCRの後、DNA産物を配列決定することで、細胞の細胞質内への選択的取り込みを媒介する分子をライブラリーから同定することが可能である。
【0063】
実施例13 ssRNAアプタマーライブラリー
ssRNA依存性RNAポリメラーゼ(例えば、ポリオウイルス3Dpol、水疱性口内炎ウイルスL、およびC型肝炎NS5bタンパク質)の重合部位を含むssRNAアプタマーライブラリー(当技術分野で周知のように非修飾または修飾塩基)を調製する。ssRNAアプタマーライブラリーを、ssRNA依存性RNAポリメラーゼを過剰発現している細胞とインキュベートし、特異的なライブラリーメンバーを特定の細胞内に内部移行させる。細胞質内に侵入するとそのライブラリーメンバーはポリメラーゼの基質となる。RNAを細胞から回収し、重合反応で得られたRNA産物に特異的なプライマーを使用して細胞内に侵入可能な分子を逆転写し、PCRし、かつ配列決定する。
【0064】
他の態様
上記の明細書で述べたすべての刊行物、特許、および特許出願は、参照によって本明細書に組み入れられる。記載される本発明の方法およびシステムの様々な改変および変形が、本発明の範囲および趣旨から逸脱することなく当業者には明らかとなろう。以上、本発明を特定の態様に関連して述べたが、特許請求されるところの本発明はこのような特定の態様に不要に限定されるべきではない点は理解されるはずである。実際、当業者にとって明らかな、本発明を実施するための記載された態様の様々な改変は、本発明の範囲内に含まれるものとする。
【0065】
その他の態様は、添付の特許請求の範囲内にある。
【特許請求の範囲】
【請求項1】
核酸ディスプレイライブラリーを含む組成物であって、前記核酸ディスプレイライブラリーのメンバーが細胞内読み出しシグナルを発生する分子に連結されている、組成物。
【請求項2】
核酸ディスプレイライブラリーを含む組成物であって、前記核酸ディスプレイライブラリーのメンバーがストレプトアビジン分子に連結されており、かつ前記ストレプトアビジン分子が細胞内読み出しシグナルを発生する分子に更に連結されている、組成物。
【請求項3】
細胞内読み出しシグナルを発生する前記分子が、核酸、タンパク質、ペプチドまたは小分子である、請求項1または2に記載の組成物。
【請求項4】
細胞内読み出しシグナルを発生する前記核酸分子が、レポーター遺伝子、転写因子遺伝子、RNA、またはアンチセンス遺伝子をコードしている、請求項3に記載の組成物。
【請求項5】
前記タンパク質が緑色蛍光タンパク質(GFP)である、請求項3に記載の組成物。
【請求項6】
前記小分子がフルオロフォアである、請求項3に記載の組成物。
【請求項7】
前記核酸ディスプレイライブラリーの核酸分子が細胞内で発現される、請求項1〜6のいずれか一項に記載の組成物。
【請求項8】
前記核酸ディスプレイライブラリーの前記核酸分子の前記細胞内発現が外因性RNAポリメラーゼプロモーターの制御下にある、請求項7に記載の組成物。
【請求項9】
前記RNAポリメラーゼがT7 RNAポリメラーゼである、請求項8に記載の組成物。
【請求項10】
分枝鎖リンカーを介してライブラリーのメンバーに多量体小分子種が結合した、DNAによりコードされた小分子ライブラリーを含む組成物。
【請求項11】
リンカー種によって修飾されたDNA塩基を介してライブラリーのDNAに2個以上の小分子が結合した、DNAによりコードされた小分子ライブラリーを含む組成物。
【請求項12】
核酸の細胞内送達を促進する分子を同定するための方法であって、前記分子が核酸ディスプレイライブラリーのメンバーに連結されており、かつ前記核酸ライブラリーの前記メンバーが更にある遺伝子に連結されており、前記方法が、細胞を前記核酸ディスプレイライブラリーと接触させる工程と、前記核酸ライブラリーのメンバーに連結された前記遺伝子の発現を監視することによって、前記細胞内への前記核酸の送達を促進する前記分子に連結された前記核酸ディスプレイライブラリーのメンバーを同定する工程とを含む、方法。
【請求項13】
前記核酸ライブラリーのメンバーに連結された前記遺伝子の発現が、外因性RNAポリメラーゼプロモーターの制御下にある、請求項12に記載の方法。
【請求項14】
前記細胞が該細胞の細胞質中でRNAポリメラーゼを発現する、請求項12または13に記載の方法。
【請求項15】
前記RNAポリメラーゼがT7 RNAポリメラーゼである、請求項14に記載の方法。
【請求項16】
前記細胞が、細胞内に送達される前記核酸ライブラリーのメンバーを修飾することが可能な1種類以上の酵素を発現する、請求項12〜15のいずれか一項に記載の方法。
【請求項17】
前記酵素がDNAメチルトランスフェラーゼである、請求項16に記載の方法。
【請求項18】
前記DNAメチルトランスフェラーゼが、細胞内に送達される前記核酸ディスプレイライブラリーのメンバーを選択的にメチル化する、請求項17に記載の方法。
【請求項19】
核酸の細胞内送達を促進する分子を同定するための方法であって、前記分子が核酸ディスプレイライブラリーのメンバーに連結されており、かつ前記核酸ライブラリーの前記メンバーが更にRNAポリメラーゼ結合部位に連結されており、前記方法が、前記細胞を前記核酸ディスプレイライブラリーと接触させる工程と、前記核酸ライブラリーの前記メンバーの核酸部分の細胞内における転写を監視および解読することによって、前記細胞内への前記核酸の送達を促進する前記分子に連結された前記核酸ディスプレイライブラリーのメンバーを同定する工程とを含み、前記細胞内に存在するRNAポリメラーゼが前記転写を触媒する、方法。
【請求項20】
前記RNAポリメラーゼがT7 RNAポリメラーゼである、請求項19に記載の方法。
【請求項21】
前記分子が核酸分子である、請求項12〜20のいずれか一項に記載の方法。
【請求項22】
前記核酸分子が、RNAi、miRNA、アンチセンス核酸分子、または遺伝子である、請求項21に記載の方法。
【請求項23】
前記分子がタンパク質である、請求項12〜20のいずれか一項に記載の方法。
【請求項24】
前記分子がペプチドである、請求項12〜20のいずれか一項に記載の方法。
【請求項25】
前記分子が小分子である、請求項12〜20のいずれか一項に記載の方法。
【請求項26】
第2の分子の細胞内送達を促進する第1の分子を同定するための方法であって、前記第1および第2の分子が核酸ライブラリーのメンバーに連結されており、前記方法が、前記細胞を前記核酸ディスプレイライブラリーと接触させる工程と、前記細胞内に存在する1種類以上の酵素による前記核酸ライブラリーのメンバーの修飾を監視することによって、前記細胞内への前記第2の分子の送達を促進する前記第1の分子に連結された前記核酸ディスプレイライブラリーのメンバーを同定する工程とを含む、方法。
【請求項27】
前記第1および第2の分子が核酸分子である、請求項26に記載の方法。
【請求項28】
前記核酸分子が、RNAi、miRNA、アンチセンス核酸分子、または遺伝子である、請求項27に記載の方法。
【請求項29】
前記分子がタンパク質である、請求項26に記載の方法。
【請求項30】
前記分子がペプチドである、請求項26に記載の方法。
【請求項31】
前記分子が小分子である、請求項26に記載の方法。
【請求項32】
前記核酸ディスプレイライブラリーがdsDNAディスプレイライブラリーである、請求項1〜9および請求項12〜31のいずれか一項に記載の方法。
【請求項33】
前記dsDNAディスプレイライブラリーが、CISディスプレイライブラリー、ピューロマイシン媒介dsDNAディスプレイライブラリー、CDTディスプレイライブラリー、小分子に結合させたdsDNAライブラリー、およびストレプトアビジンディスプレイライブラリーである、請求項32に記載の方法。
【請求項1】
核酸ディスプレイライブラリーを含む組成物であって、前記核酸ディスプレイライブラリーのメンバーが細胞内読み出しシグナルを発生する分子に連結されている、組成物。
【請求項2】
核酸ディスプレイライブラリーを含む組成物であって、前記核酸ディスプレイライブラリーのメンバーがストレプトアビジン分子に連結されており、かつ前記ストレプトアビジン分子が細胞内読み出しシグナルを発生する分子に更に連結されている、組成物。
【請求項3】
細胞内読み出しシグナルを発生する前記分子が、核酸、タンパク質、ペプチドまたは小分子である、請求項1または2に記載の組成物。
【請求項4】
細胞内読み出しシグナルを発生する前記核酸分子が、レポーター遺伝子、転写因子遺伝子、RNA、またはアンチセンス遺伝子をコードしている、請求項3に記載の組成物。
【請求項5】
前記タンパク質が緑色蛍光タンパク質(GFP)である、請求項3に記載の組成物。
【請求項6】
前記小分子がフルオロフォアである、請求項3に記載の組成物。
【請求項7】
前記核酸ディスプレイライブラリーの核酸分子が細胞内で発現される、請求項1〜6のいずれか一項に記載の組成物。
【請求項8】
前記核酸ディスプレイライブラリーの前記核酸分子の前記細胞内発現が外因性RNAポリメラーゼプロモーターの制御下にある、請求項7に記載の組成物。
【請求項9】
前記RNAポリメラーゼがT7 RNAポリメラーゼである、請求項8に記載の組成物。
【請求項10】
分枝鎖リンカーを介してライブラリーのメンバーに多量体小分子種が結合した、DNAによりコードされた小分子ライブラリーを含む組成物。
【請求項11】
リンカー種によって修飾されたDNA塩基を介してライブラリーのDNAに2個以上の小分子が結合した、DNAによりコードされた小分子ライブラリーを含む組成物。
【請求項12】
核酸の細胞内送達を促進する分子を同定するための方法であって、前記分子が核酸ディスプレイライブラリーのメンバーに連結されており、かつ前記核酸ライブラリーの前記メンバーが更にある遺伝子に連結されており、前記方法が、細胞を前記核酸ディスプレイライブラリーと接触させる工程と、前記核酸ライブラリーのメンバーに連結された前記遺伝子の発現を監視することによって、前記細胞内への前記核酸の送達を促進する前記分子に連結された前記核酸ディスプレイライブラリーのメンバーを同定する工程とを含む、方法。
【請求項13】
前記核酸ライブラリーのメンバーに連結された前記遺伝子の発現が、外因性RNAポリメラーゼプロモーターの制御下にある、請求項12に記載の方法。
【請求項14】
前記細胞が該細胞の細胞質中でRNAポリメラーゼを発現する、請求項12または13に記載の方法。
【請求項15】
前記RNAポリメラーゼがT7 RNAポリメラーゼである、請求項14に記載の方法。
【請求項16】
前記細胞が、細胞内に送達される前記核酸ライブラリーのメンバーを修飾することが可能な1種類以上の酵素を発現する、請求項12〜15のいずれか一項に記載の方法。
【請求項17】
前記酵素がDNAメチルトランスフェラーゼである、請求項16に記載の方法。
【請求項18】
前記DNAメチルトランスフェラーゼが、細胞内に送達される前記核酸ディスプレイライブラリーのメンバーを選択的にメチル化する、請求項17に記載の方法。
【請求項19】
核酸の細胞内送達を促進する分子を同定するための方法であって、前記分子が核酸ディスプレイライブラリーのメンバーに連結されており、かつ前記核酸ライブラリーの前記メンバーが更にRNAポリメラーゼ結合部位に連結されており、前記方法が、前記細胞を前記核酸ディスプレイライブラリーと接触させる工程と、前記核酸ライブラリーの前記メンバーの核酸部分の細胞内における転写を監視および解読することによって、前記細胞内への前記核酸の送達を促進する前記分子に連結された前記核酸ディスプレイライブラリーのメンバーを同定する工程とを含み、前記細胞内に存在するRNAポリメラーゼが前記転写を触媒する、方法。
【請求項20】
前記RNAポリメラーゼがT7 RNAポリメラーゼである、請求項19に記載の方法。
【請求項21】
前記分子が核酸分子である、請求項12〜20のいずれか一項に記載の方法。
【請求項22】
前記核酸分子が、RNAi、miRNA、アンチセンス核酸分子、または遺伝子である、請求項21に記載の方法。
【請求項23】
前記分子がタンパク質である、請求項12〜20のいずれか一項に記載の方法。
【請求項24】
前記分子がペプチドである、請求項12〜20のいずれか一項に記載の方法。
【請求項25】
前記分子が小分子である、請求項12〜20のいずれか一項に記載の方法。
【請求項26】
第2の分子の細胞内送達を促進する第1の分子を同定するための方法であって、前記第1および第2の分子が核酸ライブラリーのメンバーに連結されており、前記方法が、前記細胞を前記核酸ディスプレイライブラリーと接触させる工程と、前記細胞内に存在する1種類以上の酵素による前記核酸ライブラリーのメンバーの修飾を監視することによって、前記細胞内への前記第2の分子の送達を促進する前記第1の分子に連結された前記核酸ディスプレイライブラリーのメンバーを同定する工程とを含む、方法。
【請求項27】
前記第1および第2の分子が核酸分子である、請求項26に記載の方法。
【請求項28】
前記核酸分子が、RNAi、miRNA、アンチセンス核酸分子、または遺伝子である、請求項27に記載の方法。
【請求項29】
前記分子がタンパク質である、請求項26に記載の方法。
【請求項30】
前記分子がペプチドである、請求項26に記載の方法。
【請求項31】
前記分子が小分子である、請求項26に記載の方法。
【請求項32】
前記核酸ディスプレイライブラリーがdsDNAディスプレイライブラリーである、請求項1〜9および請求項12〜31のいずれか一項に記載の方法。
【請求項33】
前記dsDNAディスプレイライブラリーが、CISディスプレイライブラリー、ピューロマイシン媒介dsDNAディスプレイライブラリー、CDTディスプレイライブラリー、小分子に結合させたdsDNAライブラリー、およびストレプトアビジンディスプレイライブラリーである、請求項32に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【公表番号】特表2012−517811(P2012−517811A)
【公表日】平成24年8月9日(2012.8.9)
【国際特許分類】
【出願番号】特願2011−550310(P2011−550310)
【出願日】平成22年2月16日(2010.2.16)
【国際出願番号】PCT/US2010/024296
【国際公開番号】WO2010/094027
【国際公開日】平成22年8月19日(2010.8.19)
【出願人】(511197822)エックス−ボディ インコーポレイテッド (2)
【Fターム(参考)】
【公表日】平成24年8月9日(2012.8.9)
【国際特許分類】
【出願日】平成22年2月16日(2010.2.16)
【国際出願番号】PCT/US2010/024296
【国際公開番号】WO2010/094027
【国際公開日】平成22年8月19日(2010.8.19)
【出願人】(511197822)エックス−ボディ インコーポレイテッド (2)
【Fターム(参考)】
[ Back to top ]