
DNA構築物、ならびにそれを用いた組換えdhfr遺伝子欠損CHO細胞およびその製造方法
【課題】
dhfr遺伝子欠損CHO細胞における、hprt遺伝子組換え細胞の効率的な製造に有用なDNA構築物の提供。
【解決手段】
5’末端から3’末端に向かって、第一の相同DNA断片、目的タンパク質遺伝子、および第二の相同DNA断片を含んでなるDNA構築物であって、第一の相同DNA断片および第二の相同DNA断片が、dhfr遺伝子欠損CHO細胞ゲノムのヒポキサンチン−ホスホリボシルトランスフェラーゼ酵素(hprt)遺伝子座の一部と相同組換え可能な相同性を有し、かつ1kbp以上の鎖長を有するものであり、選択マーカー遺伝子がdhfr遺伝子であることを特徴とするDNA構築物。
dhfr遺伝子欠損CHO細胞における、hprt遺伝子組換え細胞の効率的な製造に有用なDNA構築物の提供。
【解決手段】
5’末端から3’末端に向かって、第一の相同DNA断片、目的タンパク質遺伝子、および第二の相同DNA断片を含んでなるDNA構築物であって、第一の相同DNA断片および第二の相同DNA断片が、dhfr遺伝子欠損CHO細胞ゲノムのヒポキサンチン−ホスホリボシルトランスフェラーゼ酵素(hprt)遺伝子座の一部と相同組換え可能な相同性を有し、かつ1kbp以上の鎖長を有するものであり、選択マーカー遺伝子がdhfr遺伝子であることを特徴とするDNA構築物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、dhfr遺伝子欠損CHO細胞における、組換え細胞の効率的な製造に有用なDNA構築物、およびそれを用いた組換え細胞の製造方法に関する。
【背景技術】
【0002】
組換えタンパク質生産システムにおいて、原核生物や真核生物を宿主とした種々の方法が知られている。哺乳類動物細胞を宿主とした組換えタンパク質生産システムは、ヒトを始めとする高等動物由来のタンパク質に対し、糖鎖付加、フォールディング、リン酸化などの翻訳後修飾をより生体で作られるものと同じように施すことが可能である。
【0003】
この翻訳後修飾は、タンパク質の本来有する生理活性を組換えタンパク質で再現するために必要なものであり、そのような生理活性が特に必要とされる医薬品などに用いられる組換えタンパク質の生産系には、哺乳類動物細胞を宿主としたタンパク質生産システムがよく用いられている。
【0004】
現在、工業的生産に用いられている哺乳類動物細胞による主なタンパク質生産システムとしては、CHO−DHFRシステムとGS−NS0システムの二つが挙げられる。これらの生産システムでは、プラスミドベクターに含まれる選択マーカーと適切な薬剤選択プロセスを組み合わせることで、染色体に組み込まれたプラスミドベクターのコピー数が増幅したクローンを選択する。特にCHO−DHFRシステムは、選択薬剤メトトレキセートによる2段階の選抜工程により、発現レベルが数十倍に増大した細胞クローンを選択しうる。CHO−DHFRシステムでは、dhfr遺伝子を欠損したCHO細胞の使用が必須である。
【0005】
しかしながら、上記タンパク質生産システムには、単純に増幅されたプラスミドベクターのコピー数に比例して目的タンパク質の発現レベルが必ずしも増大しないこと、発現レベルが増大した細胞クローンを選択するまでにかかる時間が長いことなどの問題があることが知られている。さらに、発現レベルが増大した細胞クローンの選択後に、選択薬剤を含まない培地で選択細胞クローンの培養を継続することで、ほとんどのクローンにおいて発現レベルの減少または消失が確認されることが報告されている(特許文献1:特表2002−541854公報、非特許文献1:Kim, N. S. (1998) Biotechnol. Bioeng.,60, 679−688.)。
【0006】
また、上記のような哺乳類動物細胞による目的タンパク質生産システムは、一般的に、目的タンパク質遺伝子を含むベクターを宿主細胞に導入し、該ベクターが染色体へ組み込まれた細胞クローンを選択し、さらに該細胞クローンを適切な培養条件で培養することにより製造されている。
【0007】
この染色体への組み込みはランダムな位置で起こりうる事象であり、得られる細胞クローンによって目的タンパク質の発現レベルが異なる。また細胞クローンによっては目的タンパク質を発現しないなどの問題がある。これについては、多数のクローンを組換えタンパク質の発現レベルにより選抜して、好ましいクローンを選択するという方法が採られている。しかしながら、このスクリーニングプロセスは非常に手間と労力を要する。このような手間を回避して迅速に好ましいクローンを選択するための種々のプロセスが報告されている。
【0008】
例えば、マウス細胞の特定染色体位置にベクターを組み込む技術が開示されている(特許文献2:特表平9−510865公報)。免疫グロブリンγ2A座と相同な塩基配列を有する配列を搭載したベクターにより、組換え細胞クローンのプール中に相同的組換え細胞クローンを生じさせる。標的となる染色体位置は、外来遺伝子が組み込まれた際にランダム組み込みと比較して高い発現レベルを与えうる位置としてあらかじめ同定されている。したがって、スクリーニング対象となる組換え細胞クローンのプール中に、高い発現レベルを有する相同的組換え細胞クローンが一定の頻度で存在することにより、発現レベルによるスクリーニングの際の労力を低減することが可能となる。
【0009】
また、マーキングプラスミドの利用技術についても開示されている(特許文献3:特表2001−516221公報)。あらかじめマーキングプラスミドをランダム組換えした細胞クローン集団の中からマーキングプラスミド内に存在するマーカー遺伝子の発現レベルが高いクローンを選択しておく。次に目的タンパク質遺伝子を有するプラスミドベクターとランダムに組み込まれたマーキングプラスミド配列との間で部位特異的組換えが引き起こされた細胞クローンを選択することにより、マーカー遺伝子の発現レベルを継承した目的タンパク質産生クローンが得られる。
【0010】
上記技術は、発現レベルの高いクローンの選択に要する労力削減において有利である。しかしながら、例えば、組換え細胞を選択薬剤非添加で継続的に培養した際、得られたクローンが長期的に発現レベルを安定に維持しうるかについては依然として予測できない。
【0011】
医薬タンパク質の工業的生産においては、タンパク質の発現が高いレベルでかつ安定に維持されることが重要である。特に発現レベルが安定に維持されることに関しては、コスト面のみならず、医薬タンパク質としての同一性および安全性を証明するためにも重要である。組換えタンパク質生産細胞を工業的スケールでの生産に用いるためには生産細胞クローンの培養スケールの拡大を図る必要がある。これには通常少なくとも樹立直後のクローンから約60回の細胞分裂を経なければならないと見積られており(非特許文献2:Brown, M. E. et al. (1992) Cytotechnology, 9, 231−236.)、この発現レベルが一定に保たれていなければならない。
【0012】
また、選択薬剤は培養コストを上昇させることのみならず、医薬品への異物混入リスク回避のために行われる精製プロセスのコストも上昇させる。したがって、選択薬剤を添加することなく発現レベルを安定に維持できる細胞クローンの作製技術の開発が強く望まれている。
【0013】
上記のような事情があるにも関わらず、目的タンパク質発現レベルの安定性に関して充分な技術的検討が行なわれてきたとはいえない。これまでのところ、多くのタンパク質産生系の製造方法においては、長期培養時における成長速度ならびに生産性に関して蓄積されたデータを元に、経験的にクローン選択が行なわれている。しかしながら、この経験的な方法では発現レベルが安定な細胞クローンを実際に取得できるケースは稀である(非特許文献3:Barnes, L. M. et al. (2003) Biotechnology and Bioengineering, 81, 631−639.)。
【0014】
近年、細胞ゲノム中の標的遺伝子座に目的タンパク質遺伝子を特異的な組み込み、長期間、安定にタンパク質を発現しうる組換え細胞を効率的に取得する手法が検討されている。
【0015】
この標的遺伝子の一例としては、hprt遺伝子が挙げられる。hprt遺伝子は、ヒト等のX染色体長腕に存在するハウスキーピング遺伝子の一つとして知られており、hprt遺伝子のノックアウト後の細胞は、薬剤6−Thioguanime (6TG)に対して耐性を示すため、陰性選択が容易に行われる。
【0016】
そして、本発明者らの一部は、ヒトの雄由来のHT1080細胞株のhprt遺伝子座へ目的タンパク質遺伝子を組換えベクターにより導入し、選択薬剤非存在下で長期間、安定にタンパク質を発現しうる組換え細胞を取得したことを報告している(特許文献5:特開2007−325571公報、非特許文献4:Koyama Y Et Al.,(2006) Biotechnology And Bioengineering,95,1052−1060)。この実施例においては、各々約1kbpの第一および第二の相同DNA断片をホモロジーアームとして用いたところ、107細胞あたり10クローン程度の組換え細胞を取得したことが報告されている。
【0017】
また、hprt遺伝子座へのターゲッティングの別の例としては、マウスES細胞に対してターゲッティングが行われたことが報告されている(特許文献6:特表平5−507853公報)。
【0018】
しかしながら、同じ遺伝子座を標的としても、培養細胞の種類によっては、ジーンターゲッティング頻度が大きく異なる場合がある。例えば、Porter C. G. Itzhaki J. E, Eur. J. Biochem 218, 273−281(非特許文献5)、および広島大学原爆放射線医科学研究所 年報第44号 (2003)(非特許文献6)では、ES細胞と、HT1080細胞とを比較すると、ジーンターゲッティング頻度が異なり、体細胞由来の培養細胞では、ジーンターゲッティング頻度が極めて低いことが報告されている。
【0019】
一方、dhfr遺伝子欠損CHO細胞は、DHFR−MTXシステムによる抗体医薬タンパク質産生系の宿主細胞として利用されており、dhfr遺伝子欠損CHO細胞を用いて高レベルで安定なタンパク質生産システムを製造することが求められている。
【0020】
しかしながら、dhfr遺伝子欠損CHO細胞のhprt遺伝子座に対してジーンターゲッティングが行われたことは報告されていない。dhfr遺伝子欠損CHO細胞の親株である、CHO細胞の一方の染色体上のaprt遺伝子を欠損した特殊な細胞株を用いた研究例が存在するのみである(非特許文献8:PNAS,88, 9488−9502(1991)、非特許文献9:Somatic Cell. Mol. Genet., 19, 363−375、非特許文献10:PNAS,86,4574−4578(1989))。これらの実施例では、一方の染色体上のみにaprt遺伝子が存在する細胞株を宿主とし、2.6kbp〜4kbpの相同DNA断片をホモロジーアームとして用いた結果、107細胞あたり数クローン〜15クローン程度の相同組換え体を取得している。
【0021】
また、dhfr遺伝子欠損CHO細胞のhprt遺伝子座のゲノムの配列はエキソン以外は何ら報告されていない。したがって、dhfr遺伝子欠損CHO細胞のhprt遺伝子座に対する特異的なジーンターゲッティングを行うためには、エキソン以外の塩基配列を解析し、取得しなければならないという問題がある。
【0022】
さらに、CHO細胞は、雌由来の細胞であることから、X染色体を2本有しており、そのhprt遺伝子座も2つ有している。そのため、両方の染色体のhprt遺伝子座に外来遺伝子が組み込まれることで、CHO細胞は6TG等の選択薬剤耐性となる。しかしながら、このように、2本の染色体で組換えが生じる確率は、雄由来細胞の場合と比べて低くなるのが通常である。例えば、Koyama Y Et Al., (2006) Biotechnology And Bioengineering,95,1052−1060(非特許文献4)における雄由来HT1080細胞株のhprt遺伝子座への組換え効率を参考として染色体1本で組換えが生じる確率を10−6と仮定すると、染色体2本で同時に生じる理論上の確率は10−12となる。
【0023】
したがって、目的タンパク質遺伝子を発現する組換えCHO細胞を効率的に製造する技術の創出が依然として必要とされている。
【0024】
また、dhfr欠損CHO細胞は、DNAのde novo合成能を欠いた細胞株であり、培地中のヒポキサンチン、チミヂンをサルベージすることで自らのDNAをまかなって生存している。サルベージ経路にはhprt遺伝子機能が必須であるため、hprt遺伝子座へのベクター組み込みは致死となる(非特許文献7:Carcinogenesis, 20, 215−220(1999))。したがって、hprt遺伝子座を外来遺伝子組み込みの標的部位として使用できないという問題がある。
【0025】
ところで、陽性選択遺伝子と陰性選択遺伝子を融合させた二機能選択可能融合遺伝子を発現させる方法が公開されており、陽性選択遺伝子の例としてdhfr遺伝子が、陰性選択遺伝子としてhprt遺伝子が例示されている(特許文献7:特表平6−504432)。
【0026】
この方法を適応した場合、dhfr−hprt融合遺伝子をベクターに搭載して細胞に導入することになる。ところが、dhfr遺伝子欠損CHO細胞では染色体上のhprtが元来活性な状態にあるため、組換え細胞の選択はヒポキサンチン非要求性でしか行えない。CHO細胞では、ランダムな挿入が相同組換えのおよそ4千倍の頻度で生じる(非特許文献10:PNAS,86,4574−4578(1989))。したがって、染色体hprt遺伝子座と相同性をもつホモロジーアームとともにdhfr−hprt融合遺伝子をdhfr遺伝子欠損CHO細胞に導入しても、hprt遺伝子座への相同組換え細胞が取得できる見込みはほとんどない。
【0027】
したがって、dhfr遺伝子欠損CHO細胞のhprt遺伝子座への目的タンパク質遺伝子組み込み細胞を選択可能にする技術の創出が依然として必要とされている。
【先行技術文献】
【特許文献】
【0028】
【特許文献1】特表2002−541854公報
【特許文献2】特表平9−510865公報
【特許文献3】特表2001−516221公報
【特許文献4】WO2004/022741公報
【特許文献5】特開2007−325571公報
【特許文献6】特表平5−507853公報
【特許文献7】特表平6−504432公報
【非特許文献】
【0029】
【非特許文献1】Kim, N. S. (1998) Biotechnol. Bioeng., 60, 679−688.
【非特許文献2】Brown, M. E. et al. (1992) Cytotechnology, 9, 231−236.
【非特許文献3】Barnes, L. M. et al. (2003) Biotechnology and Bioengineering , 81, 631−639.
【非特許文献4】Koyama Y Et Al., (2006) Biotechnology And Bioengineering, 95, 1052−1060
【非特許文献5】Porter C. G. Itzhaki J. E, Eur. J. Biochem 218, 273−281
【非特許文献6】広島大学原爆放射線医科学研究所 年報第44号 (2003)
【非特許文献7】Carcinogenesis, 20, 215−220(1999)
【非特許文献8】PNAS,88, 9488−9502(1991)
【非特許文献9】Somatic Cell. Mol. Genet., 19, 363−375
【非特許文献10】PNAS,86,4574−4578(1989)
【発明の概要】
【0030】
本発明者らは、今般、dhfr遺伝子欠損CHO細胞におけるhprt遺伝子の相同DNA断片と選択マーカー遺伝子としてジヒドロ葉酸還元酵素遺伝子(dhrf)を有する特定のDNA構築物により目的タンパク質遺伝子をdhfr遺伝子欠損CHO細胞に導入したところ、組換え細胞を顕著な高頻度で製造しうることを見出した。本発明は、かかる知見に基づくものである。
【0031】
したがって、本発明は、dhfr遺伝子欠損CHOにおける組換え細胞の効率的な製造に有用なDNA構築物およびそれを用いた組換え細胞製造方法の提供をその目的とする。
【0032】
そして、本発明によるDNA構築物は、5’末端から3’末端に向かって、第一の相同DNA断片、目的タンパク質遺伝子、選択マーカー遺伝子および第二の相同DNA断片を含んでなるDNA構築物であって、前記第一の相同DNA断片および第二の相同DNA断片が、dhfr遺伝子欠損CHO細胞ゲノムのヒポキサンチン−ホスホリボシルトランスフェラーゼ酵素(hprt)遺伝子座の一部と相同組換え可能な相同性を有し、かつ1kbp以上の鎖長を有するものであり、前記選択マーカー遺伝子がジヒドロ葉酸還元酵素遺伝子(dhfr)であることを特徴とするものである。
【0033】
また、本発明によるdhfr遺伝子欠損CHOにおける組換え細胞の製造方法は、上記DNA構築物を含んでなるベクターをdhfr遺伝子欠損CHO細胞に導入することを含んでなる。
【0034】
本発明によれば、目的タンパク質遺伝子を発現する組換えCHO細胞を顕著な高頻度で取得することができる。
【図面の簡単な説明】
【0035】
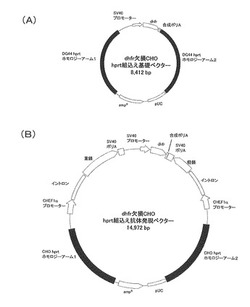
【図1】実施例1で用いられたdhfr欠損CHO細胞hprt相同組換え用ベクターの模式図である。(A)はdhfr欠損CHO細胞のhprt遺伝子に由来する相同DNA断片を有し、選択マーカー遺伝子としてdhfr遺伝子を有するベクターである。(B)はdhfr遺伝子欠損CHO細胞のhprt遺伝子に由来する相同DNA断片を有し、選択マーカー遺伝子としてdhfr遺伝子を有し、抗体発現カセットを含むベクターである。
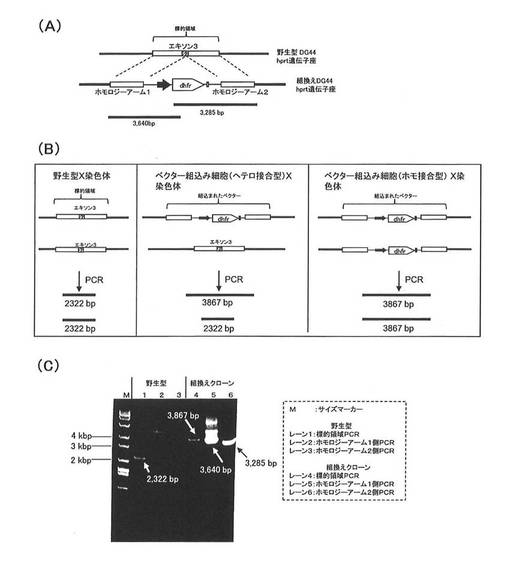
【図2】(A)は、dhfr遺伝子欠損CHO細胞のhprt遺伝子領域、相同組換え用ベクター、および実施例3において組換え細胞のゲノムPCRの指標となったDNAの関係を示す模式図である。(B)は、組換えdhfr遺伝子欠損CHO細胞中の野生型hprt遺伝子の残存を確認するためのゲノムPCRの詳細を説明する模式図である。左図は、野生型のdhfr遺伝子欠損CHO細胞におけるPCRを示し、中央図は、ヘテロ接合型の組換え細胞におけるPCRを示し、右図は、ホモ接合型の組換え細胞におけるPCRを示す。(C)は、実施例3のPCRの結果を示す。
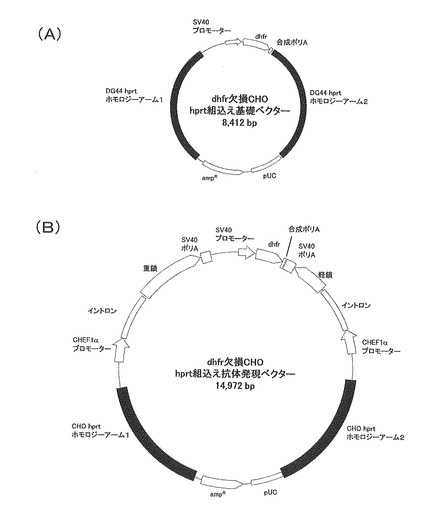
【図3】dhfr欠損CHO細胞のhprt遺伝子座の模式図である。番号1から9まではhprt遺伝子のエクソン1〜9を示し、各エクソン間の配列は、イントロン1〜8を示す。
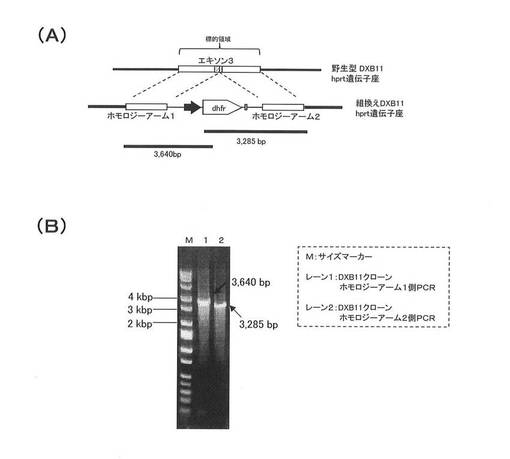
【図4】(A)は、DXB11のhprt遺伝子領域、相同組換え用ベクター、および実施例4において組換え細胞のゲノムPCRの指標となったDNAの関係を示す模式図である。(C)は、実施例4のPCRの結果を示す。
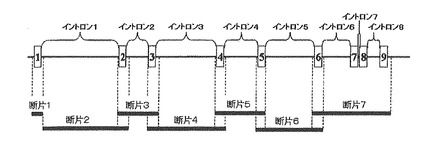
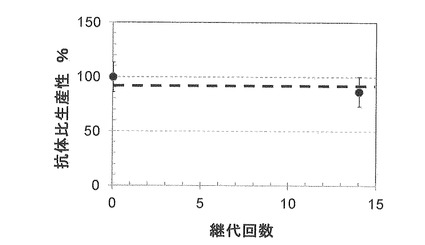
【図5】は、実施例5で作製したdhfr欠損CHO hprt組換え抗体発現クローンによって抗体を生産した結果を示す。
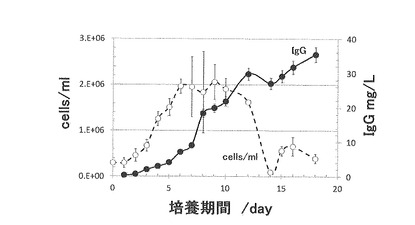
【図6】は、実施例5で作製したdhfr欠損CHO hprt組換え抗体発現クローンの抗体比生産性の長期安定性試験結果を示す。
【発明を実施するための形態】
【0036】
DNA構築物
本発明によるDNA構築物は、CHO細胞ゲノム中のhprt遺伝子座の一部と相同組換え可能な相同性を有し、各々が1kbp以上の鎖長を有する、二つの相同DNA断片を含んでなることを一つの特徴とする。上記DNA構築物によれば、CHO細胞が2本のX染色体を有するにもかかわらず、顕著な高頻度で組換えCHO細胞を取得しうるのは意外な事実である。本発明によるDNA構築物によれば、後述の実施例3に示される通り、2x10−6あたり4個の組換え細胞が得られている。この結果は、本願出願日当時の技術状況において、染色体2本のhprt遺伝子座において同時に組換えが生じる理論上の確率が10−12と予測されていたことを勘案すると、驚くべき事実といえる。
【0037】
また、本発明によるDNA構築物は、陽性選択マーカー遺伝子としてジヒドロ葉酸還元酵素遺伝子(dhfr)を含んでなることを一つの特徴とする。dhfr遺伝子は、相同組換えを妨げない限り、DNA構築物中に適宜配置することができる。dhfr遺伝子を用いることにより、de novo合成能とhprt遺伝子の不活化が合わさることによる致死性を回避することが可能となる。さらに、組換えCHO細胞の選択において、ヒポキサンチン、チミジン非要求性を指標とした陽性選択と、hprt遺伝子の不活化による陰性選択の両者を適用でき、偽陽性クローンを大幅に低減する上で有利である。
【0038】
本発明によるDNA構築物は、hprt遺伝子座の一部を相同組換えの標的領域とするものである。hprt遺伝子座の一部を標的領域とすることは、目的タンパク質遺伝子を高レベルで安定に発現する上で有利である。また、かかる標的領域に、DNA構築物を組み込むことは、hprt遺伝子の転写・発現を阻害してhprt遺伝子の機能を不活化し、6−TG等を用いる陰性選択によって効率的に組換え細胞を取得する上でも好ましい。
【0039】
また、本発明の標的領域は、目的タンパク質遺伝子の発現を妨げない限り、hprt遺伝子座中において適宜決定してよい。しかしながら、相同組換えに必要な相同DNA断片の鎖長等を勘案すれば、上記標的領域は、hprt遺伝子のイントロンの少なくとも一部を含む領域が好ましい。上記イントロンは、本発明者らにより今般、塩基配列が決定されたものであり、具体的には、配列番号35〜42のいずれかで表される塩基配列を有する、後述する図3のイントロン1〜8が挙げられる。
【0040】
また、本発明の標的領域は、上記イントロンと隣接するエクソンの全部または一部を含んでいてもよい。かかるエクソンとしては、具体的には、図3のエクソン1〜9が挙げられ、これらの塩基配列は、例えば、アメリカ合衆国国立バイオテクノロジー情報センター(National Center for Biotechnology Information)等の公知のデータベースにアクセスすることにより取得することができる。
【0041】
この組み込みの態様としては、(1)DNA構築物の組み込みによってエクソンが分断されるタイプ、(2)DNA構築物の組み込みによってエクソンが1つ以上欠失するタイプ、(3)DNA構築物の組み込みによってエクソンが2個に増幅し、その間にDNA構築物が挿入されるタイプ、(4)DNA構築物の組み込みによってイントロンが分断されるタイプ、(5)DNA構築物の組み込みによってイントロンが1つ以上欠失するタイプ、(6)DNA構築物の組み込みによってイントロンが2個に増幅し、その間にDNA構築物が挿入されるタイプが挙げられる。
【0042】
また、本発明のDNA断片は、相同組換えに必要な鎖長を勘案すると、上述の通り、hprt遺伝子のイントロンの少なくとも一部を含む領域と相同性を有するものが好ましい。したがって、本発明の一つの態様によれば、本発明の第一の相同DNA断片または第二の相同DNA断片は、配列番号35〜42のいずれかに記載の塩基配列またはその部分配列を含んでなるものとされる。
【0043】
また、上記部分配列の鎖長の下限は1bpであり、その上限は配列番号35〜42のいずれかに記載の塩基配列の鎖長の範囲内において、適宜調節することができる。
【0044】
また、上記相同DNA断片と、hprt遺伝子座との相同性は、相同組換えの効率を勘案して適宜決定されるが、好ましくは99.0%以上、より好ましくは99.9%以上、さらに好ましくは100%である。かかる相同性は、例えば、DNAシーケンサー等を用いて解析することにより適宜決定することができる。
【0045】
また、上記相同DNA断片は1kbp以上の鎖長を有するものとされており、かかる鎖長は、DNA構築物の顕著な高頻度での相同組換えを達成する上で好ましい。さらに、上記相同DNA断片の鎖長は、組換え効率の向上等を勘案して適宜調整してよく、より好ましくは2.5kbp以上である。また、上記相同DNA断片の鎖長の上限は、目的タンパク質遺伝子の効率的な導入を妨げないという観点から、好ましくは7.5kbp以下であり、最も好ましくは5kbp以下である。相同DNA断片の鎖長の範囲は上記の上限値、下限値を適宜組み合わせることができる。具体的には好ましくは1kbp以上7.5kbp以下、より好ましくは1kbp以上5kbp以下、最も好ましくは2.5kbp以上5kbp以下である。鎖長がこの範囲であれば、DNA構築物の顕著な高頻度での相同組換えを達成しながら、組換え細胞の取得頻度を良好に保つことが可能となる。
【0046】
また、本発明によるDNA構築物において、目的タンパク質遺伝子は、医薬として有用なタンパク質をコードしていることが好ましい。目的タンパク質遺伝子は、cDNAに由来する配列であっても、ゲノムDNAに由来する天然イントロンを含む構造遺伝子であっても、好適に利用可能である。具体的には、目的タンパク質としては、抗体、酵素、サイトカイン、ホルモン、凝固因子、調節タンパク質、レセプター等が挙げられるが、好ましくは、モノクローナル抗体、ポリクローナル抗体、エリスロポエチン、組織特異的プラスミノーゲン活性化因子または顆粒球コロニー活性化因子等である。
【0047】
また、上記目的タンパク質遺伝子は、プロモーター配列や転写終結シグナル配列等の発現に必要な要素を含む発現ユニットとしてhprt遺伝子座に組み込まれることが好ましい。したがって、本発明の一つの態様によれば、目的タンパク質遺伝子は、少なくともプロモーター配列および転写終結シグナル配列を含んだ発現ユニットとしてDNA構築物中に配置される。もっとも、発現に必要な要素としてCHO細胞の内在性のものを用いるように構成してもよく、本発明にはかかる態様も包含される。
【0048】
また、上記プロモーターや転写終結シグナルは、目的タンパク質遺伝子の種類、性質等に応じて適宜決定してよく、かかるプロモーター配列の好適な例としては、CMVプロモーター、SV40プロモーター等が挙げられる。また、転写終結シグナル配列の好適な例としては、BGHポリAシグナル配列、SV40ポリAシグナル配列等が挙げられる。
【0049】
また、プロモーター配列、転写終結シグナル配列の他の発現に必要な要素としては、例えば、目的遺伝子を効率的に発現させるための調節エレメント(例えば、エンハンサー、IRES(internal ribosome entry site)配列、LoxP配列およびFRT配列等の組換え酵素認識配列)等を適宜選択して用いてよい。調節エレメントはその性質に応じて、発現ユニットにおける適切な位置に配置することが可能である。これら発現に必要な要素は、目的タンパク質の生産性等を勘案して適宜選択される。
【0050】
ベクター
本発明によるDNA構築物は、ベクターに組み込んでdhfr遺伝子欠損CHO細胞ゲノムに導入することができる。かかるベクターシステムとしては、相同組換え反応によりDNA構築物を細胞ゲノムに組み込みうる限り特に限定されないが、好ましくは、プラスミドベクター、コスミドベクター、ファージベクターまたは人工染色体ベクター等が挙げられる。
【0051】
本発明によるDNA構築物およびこれを含むベクターは、制限酵素切断反応およびライゲーション反応を組み合わせて好適に構築される。例えば、各構成ユニットの両末端に制限酵素の認識配列を含有させ、該認識配列用の制限酵素で切断反応を行い、不要なDNA配列(大腸菌内での操作用配列等)をゲル切り出し等の処理によって除去し、得られたユニットのライゲーション反応を行うことにより、DNA構築物およびこれを含むベクターを構築しうる。
【0052】
ライゲーション反応により構築したベクターDNAは、フェノール・クロロホルム抽出等により精製し、ベクターの種類等を勘案して選択した大腸菌、酵母等の宿主細胞において増殖させることができる。
【0053】
本発明によるベクターは、CHEF1αプロモーターを、目的タンパク質遺伝子に作動可能に連結して搭載していることが好ましい。CHO hprt遺伝子座に組込んだ外来遺伝子の発現はプロモーターの不活化によって生じる。目的タンパク質遺伝子をCHO内在性のCHEF1αプロモーターで発現させるベクター構造にすることで、CHO
hprt遺伝子座での安定発現が可能となる。
【0054】
dhfr欠損CHO細胞株
本発明の対象となる細胞株はdhfr遺伝子を欠損したチャイニーズハムスターの細胞株であり、DG44、DXB11などが挙げられる。
【0055】
組換えdhfr欠損CHO細胞/製造方法
本発明によるdhfr欠損CHOの組換え細胞は、上記ベクターをdhfr欠損CHO細胞に導入することにより好適に製造することができる。したがって、本発明によるdhfr欠損CHOの組換え細胞は、hprt座に外来性の目的タンパク質遺伝子が組み込まれてなる。また、本発明の好ましい態様によれば、組換えdhfr欠損CHO細胞はhprt遺伝子の機能が不活化している。かかる組換えdhfr欠損CHO細胞は、抗体等の目的タンパク質を高レベルで安定に産生する上で有利である。
【0056】
ベクターの導入
上記ベクターの導入方法としては、一般的に用いられる方法が好適に利用できる。例えば、リン酸カルシウム法、エレクトロポレーション法、マイクロインジェクション法、DEAE−デキストラン法、リポソーム試薬を用いる方法、カチオン性脂質を用いたリポフェクション法などが挙げられる。ここで、ベクターが環状である場合、公知の方法により線状化して細胞に導入されてもよい。
【0057】
また、ベクターや目的タンパク質の性質によっては、組換え細胞の取得効率等を勘案して、Cre/LoxPシステムやFlp/FRTシステムなどのような、組換え酵素を利用した部位特異的導入システムも適宜適用してよく、本発明にはかかる態様も包含される。
【0058】
細胞株の選択
また、本発明による製造方法にあっては、上記導入後、組換え細胞の選択を行うことが好ましい。選択工程は、hprt遺伝子の不活化に基づく陰性選択により行うことができる。また、dhfr遺伝子を陽性マーカー遺伝子として、組換え細胞に導入している場合には、陰性選択と、ヒポキサンチン、チミジン非要求性による陽性選択とを組み合わせて高い精度で細胞選択を行うことが可能となる。
【0059】
また、上記選択方法の他、プロモータートラップ法やポリAトラップ法なども適宜組み合わせて利用してよい。
【0060】
また、本発明による製造方法にあっては、組換え細胞を取得後、Chemical Defined 培地などに馴化させることにより無血清下での培養も可能である。かかる馴化条件は、組換え細胞の状態に応じて適宜決定することができる。
【0061】
目的タンパク質の製造方法
また、本発明の別の態様によれば、上記組換え細胞を用意し、該細胞を培養して目的タンパク質を産生することを含んでなる、目的タンパク質の製造方法が提供される。上記方法によれば、目的タンパク質を効率的かつ安定に取得することができる。
【0062】
上記培養工程における培地としては、組換え細胞の状態に応じて公知の培地を適宜選択してよいが、無血清培地が好ましい。また、上記培地は、培養コストおよび精製コストを勘案すれば、選択薬剤を添加しないものが好ましい。上記培地の好適な例としては、Chemical Defined培地等が挙げられる。
【0063】
上記培養方法としては、バッチカルチャー法、フェッド−バッチカルチャー法、還流培養法など公知の方法が適用可能である。
【0064】
なお、組換え細胞の製造に使用される種々の手法のさらなる詳細は、例えば、F.M.Ausubel,Current Protocols in Molecular Biology,John Wiley & Sons,New York,(1989)に記載されており、上記文献の全開示内容は引用することにより本明細書の一部とされる。
【実施例】
【0065】
以下、本発明を実施例によって具体的に説明するが、本発明はこれら実施例に限定されるものではない。
【0066】
なお、以下の実験において、制限酵素による反応、PCR反応、ライゲーション反応等の各反応条件は、メーカーの推奨する反応条件、あるいは、Molecular Cloning 2nd Edition; Sambrook et al., Cold Spring Harbor Laboratory Press に記載の方法に従って設定した。また、得られた種々のプラスミドベクターDNAについては、DNAシーケンサー(310 Genetic Analyser Applied Bio Systems, Inc.)を用いてDNA配列を決定した。また、以下に示すホモロジーアーム1および2はそれぞれ、本発明の第一および第二の相同DNA断片に対応している。
【0067】
実施例1: dhfr欠損CHO hprt組換えベクターの構築
dhfr欠損CHO hprt組換え基礎ベクターの構築
以下に記載の手法により、図1(A)で表されるベクター(dhfr欠損CHO hprt組換え基礎ベクター)を構築した。
【0068】
インビトロジェン社から入手したdhfr欠損チャイニーズハムスター卵母細胞株DG44細胞株をAMEM培地(組成;Advanced MEM(GIBCO)、5%[v/v]FBS、1× GlutaMAX(インビトロジェン)、1× HTサプリメント(インビトロジェン))を用いて、CO2インキュベーター(37℃、5% CO2)で培養した。得られた培養液を遠心分離し、DG44細胞ペレットを得た。このペレットをDNA Isolation Kit for Cells and Tissues (Roche Diagnostics K.K.)により処理し、ゲノムDNAを得た。次に、このゲノムDNAを鋳型とし、PCR(KOD−Plus ver.2、TOYOBO)によって、DG44細胞のhprt遺伝子座のDNA断片を2つ得た。プライマー配列は以下に示す通りである。
【0069】
ホモロジーアーム1センスプライマー:(配列番号1)
5’−TCGCGAgtctgtgtgtatgtttgtgataggc−3’
ホモロジーアーム1アンチセンスプライマー:(配列番号2)
5’−ACGCGTtgataaaatctacagtcatggg−3’
ホモロジーアーム2センスプライマー:(配列番号3)
5’−GGATCCgactgaagagctactgtgta−3’
ホモロジーアーム2アンチセンスプライマー:(配列番号4)
5’−TCGCGAtgaaggttatagagcataggggacc−3’
【0070】
上記PCR反応においては、ホモロジーアーム1センスプライマーの5’末端には、制限酵素Nru Iの認識サイトを付加した。同様に、ホモロジーアーム1アンチセンスプライマーの5’末端にはMlu Iの認識サイト、ホモロジーアーム2センスプライマーの5’末端にはBam HIの認識サイト、ホモロジーアーム2アンチセンスプライマーの5’末端にはNru Iの認識サイトをそれぞれ付加した。
【0071】
pQBI25プラスミドベクター(和光純薬株式会社)のDNAから、大腸菌内での複製起点であるori配列およびアンピシリン耐性遺伝子を含むDNA配列を、PCR反応によりクローニングした。PCR反応に用いたプライマー配列は以下に示す通りである。このPCR反応において、センスプライマー、アンチセンスプライマーの5’末端に、制限酵素Nru Iの認識サイトをそれぞれ付加した。
【0072】
Ecoliセンスプライマー:(配列番号5)
5’−TCGCGACCTGCAGGTCCGGAGCGGCCGCcatgtgagcaaaaggcca−3’
Ecoliアンチセンスプライマープライマー:(配列番号6)
5’−TCGCGACCTGCAGGTCCGGAGCGGCCGCaggtggcacttttcgggg−3’
【0073】
次に、リンカーsオリゴとリンカーaオリゴをアニールさせたDNAリンカーを準備した。
【0074】
リンカーsオリゴ:(配列番号7)
5’−CGCGTatcTCTAGAataATCGATagaAAGCTTacaG−3’
リンカーaオリゴ:(配列番号8)
5’− GATCCtgtAAGCTTtctATCGATtatTCTAGAgatA−3’
【0075】
ホモロジーアーム1、ホモロジーアーム2、およびori配列およびアンピシリン耐性遺伝子を含むDNA配列をそれぞれ、PCR反応によりクローニングした。これら3つのDNA配列を制限酵素Nru I、Mlu IおよびBam HIにより切断し、前記DNAリンカーとともにライゲーション反応を行うことによりpHA12プラスミドベクターを得た。
【0076】
dhfr発現カセットの作製
また、pQBI15プラスミドベクター(和光純薬株式会社)のDNAから、SV40プロモーター断片を、PCR反応により増幅した。PCR反応に用いたプライマー配列は以下の通りである。このPCR反応において、センスプライマーの5’末端に制限酵素Xba Iの認識サイトを、アンチセンスプライマーの5’末端にPci Iの認識サイトを付加した。
【0077】
SV40センスプライマー:(配列番号9)
5’−TCTAGActtctgaggcggaaagaacc−3’
SV40アンチセンスプライマー:(配列番号10)
5’−ACATGTcgaaacgatcctcatcctgt−3’
【0078】
次に、pSV2−dhfrプラスミド(ATCC、37146)のDNAから、dhfr断片をPCR反応により増幅した。PCR反応に用いたプライマー配列は以下の通りである。このPCR反応において、センスプライマーの5’末端に制限酵素Nco Iの認識サイトを、アンチセンスプライマーの5’末端にポリAシグナル配列およびCla Iの認識サイトを付加した。
【0079】
dhfrセンスプライマー:(配列番号11)
5’−CCatggttcgaccattgaactg−3’
dhfrアンチセンスプライマー:(配列番号12)
5’−ATCGATtCACACAAAAAACCAACACACAGATGTAATGAAAATAAAGATATTTTATTttagtctttcttctcgtagact−3’
【0080】
PCR増幅したSV40プロモーター断片をPci Iで、dhfr断片をNco Iでそれぞれ切断し、ライゲーション反応を行うことにより連結し、dhfr遺伝子カセットを得た。
【0081】
次に、pHA12プラスミドを制限酵素Xba I、Cla Iで切断し、前記dhfr遺伝子カセットを挿入し、dhfr欠損CHO hprt組換え基礎ベクターを得た。
【0082】
dhfr欠損CHO hprt組換え抗体発現ベクターの構築
以下に記載の手法により、抗体遺伝子をhprt遺伝子座に対して組み込むため、抗体遺伝子を含む、図1(B)で表されるベクター(dhfr欠損CHO hprt組換え抗体ベクター)を構築した。
【0083】
抗体重鎖発現カセットの構築
重鎖可変領域を保持したプラスミド(「Cloning of cDNA and Characterization of Anti−RNase A Monoclonal Antibody 3A21」:Journal of Fermentation and Bioengineering. Vol.82, No.3, pp. 312−314,1999)を鋳型とし、可変領域をIGH−3A21s、IGH−3A21a1プライマーセットでPCR増幅し、可変領域断片を得た。
【0084】
IGH−3A21sプライマー:(配列番号13)
5’− TAAAAGGTGTCCAGGATGTGCAGTTTCAGG −3’
IGH−3A21a1プライマー:(配列番号14)
5’− GAGGCCGATGAAACAGTGACCAGAGTCCCT −3’
【0085】
RPMI8226培養細胞からISOGEN(日本ジーン)で抽出したRNAを鋳型とし、One−step RT−PCR kit(QIAGEN)を用いてRT−PCRを行い、得られたmRNAを鋳型として定常領域をIGH−Cs、IGH−CaプライマーセットでPCR増幅し、定常領域断片を得た。
【0086】
IGH−Csプライマー:(配列番号15)
5’− CATCGGCCTCCACCAAGGGCCCATCGGTCT −3’
IGH−Caプライマー:(配列番号16)
5’− TTAAGCGGCCGCTCATTTACCCGGAGACAG −3’
【0087】
次に、IGH−LS、IGH−Vaプライマーセットで、可変領域断片を鋳型としてPCR増幅を行い、分泌シグナル付加可変領域断片を得た。
【0088】
IGH−LSプライマー:(配列番号17)
5’−GGTCGCCACCATGGAGTTTGGACTGAGCTGGGTTTTCCTTGTTGCTATTTTAAAAGGTGTCCAGGATGTG −3’
IGH−Vaプライマー:(配列番号18)
5’− GCCCTTGGTGGAGGCCGATGAAACAGTGAC −3’
【0089】
分泌シグナル付加可変領域断片および定常領域断片を鋳型とし、IGH−Vs、IGH−CaプライマーセットでアセンブルPCRを行い、両配列を連結させた抗体重鎖配列を得た。
【0090】
IGH−Vsプライマー:(配列番号19)
5’− TTCCGGTACCGGTCGCCACCATGGAGTTTG −3’
【0091】
次に、DG44ゲノムDNAを鋳型とし、EF1aP−MluI−F/EF1aP−XbaI−RプライマーセットでCHEF1αプロモーターを増幅した。
【0092】
EF1aP−MluI−Fプライマー:(配列番号20)
5’−AGAACGCGTCCACACAATCAGAACCACA−3’
EF1aP−XbaI−Rプライマー:(配列番号21)
5’−GACGATCTAGAGGTGGTTTTCACAACA−3’
【0093】
次に、得られたCHEF1αプロモーター、抗体重鎖配列およびSV40ポリAを連結し、両末端にMlu I認識配列を付加することにより、抗体重鎖発現カセットを得た。
【0094】
抗体軽鎖発現カセットの構築
同様に、軽鎖可変領域を保持したプラスミドを鋳型とし、可変領域をIGL−3A21s1、IGL−3A21a1プライマーセットでPCR増幅し、可変領域断片を得た。
【0095】
IGL−3A21s1プライマー:(配列番号22)
5’−CCCAGGTGCCAGATGTGACATCAAGATGAC−3’
IGL−3A21a1プライマー:(配列番号23)
5’−GCCACAGTTCGTTTTATTTCCAACTTTGTC−3’
【0096】
RPMI8226培養細胞からISOGENで抽出したRNAを鋳型とし、One−step RT−PCR kitを用いてRT−PCRを行い、得られたmRNAを鋳型として定常領域をIGL−Cs、IGL−CaプライマーセットでPCR増幅し、定常領域断片を得た。
【0097】
IGL−Csプライマー:(配列番号24)
5’−TGGAAATAAAACGAACTGTGGCTGCACCAT−3’
IGL−Caプライマー:(配列番号25)
5’−TTAAGCGGCCGCCTAACACTCTCCCCTGT−3’
【0098】
IGL−LS、IGL−3A21aプライマーセットで、DNA配列9を鋳型としてPCR増幅を行い、分泌シグナル付加可変領域断片を得た。
【0099】
IGL−LSプライマー:(配列番号26)
5’−GGTCGCCACCATGGACATGAGGGTCCCCGCTCAGCTCCTGGGGCTCCTGCTGCTCTGGCTCCCAGGTGCCAGATGTGACA −3’
IGL−3A21aプライマー:(配列番号27)
5’−GCCACAGTTCGTTTTATTTCCAACTTTGTC−3’
【0100】
分泌シグナル付加可変領域断片および定常領域断片を鋳型とし、IGL−Vs、IGL−CaプライマーセットでアセンブルPCRを行って両産物を連結させた抗体軽鎖配列を得た。
【0101】
IGL−Vsプライマー:(配列番号28)
5’− CCTTGGTACCGGTCGCCACCATGGACATGA −3’
【0102】
次に、CHEF1αプロモーター、抗体軽鎖配列およびSV40ポリAを連結し、両末端にHind III認識配列を付加することにより、抗体軽鎖発現カセットを得た。
【0103】
dhfr欠損CHO hprt組換え基礎ベクターを制限酵素Mlu Iで切断して抗体重鎖発現カセットを挿入し、次いで制限酵素Hind IIIで切断して抗体軽鎖カセットを挿入し、dhfr欠損CHO hprt組換え抗体発現ベクターを得た。
【0104】
実施例2:細胞へのベクターの導入
ベクター線状化
dhfr欠損CHO hprt組換え基礎ベクター、dhfr欠損CHO hprt組換え抗体発現ベクターは、Endofree Plasmid Maxi kit(QIAGEN社製)を用いて精製し、Nru Iにて切断した。2g/Lの濃度になるように滅菌水に溶解し、以下のトランスフェクション実験に用いた。
【0105】
トランスフェクションおよびスクリーニング
DG44細胞をNucleofection T solution(amaxa)で1×106細胞に調製し、線状化したプラスミドベクター2μgと混合した。次に、得られた混合液を用いて、NuceofectorII(amaxa)を用い、プログラムU−023で電圧印加した。トランスフェクションは各ベクターにつき、2回施行した。トランスフェクトした細胞は、100 細胞/ウェルにて96ウェルプレートに播種し、インキュベーター中37 ℃、5 % CO2にて培養した(培地: Advanced MEM(GIBCO社)に5% FBS、1x Glutamax(インビトロジェン社)、1x HTサプリメント(インビトロジェン社)を添加したもの)
【0106】
トランスフェクション後、HTサプリメントを除去し、6−TG(最終濃度; 50μM)(和光純薬工業)を添加した培地に交換し、培養を継続した。培養後、全ウェルを確認し、HTサプリメント非要求性/6TG耐性コロニーを単離した。
【0107】
結果、dhfr欠損CHO hprt組換え基礎ベクターによって得られたHTサプリメント非要求性/6TG耐性クローンは、2×106細胞あたり4クローンであった。
【0108】
実施例3:ゲノムPCRによる解析
DNA Isolation kit(ロシュ)を用い、dhfr欠損CHO hprt組換え基礎ベクターのトランスフェクションにより得たHTサプリメント非要求性および6TGに耐性のクローンからゲノムDNAを抽出し、このゲノムDNAを鋳型とした以下に示すPCR反応により、標的hprt遺伝子座への位置特異的な組換えの確認を行った。
【0109】
図2(A)は、ゲノムPCRによる相同組換え反応の解析の詳細を説明する模式図である。hprt遺伝子の相同組換えの標的領域は、エクソン3を含んで設定されており、この領域にベクターDNA中のホモロジーアーム1、ホモロジーアーム2およびこれに挟まれたdhfr遺伝子発現カセットは相同組換えにより組込まれる。
【0110】
ホモロジーアーム1と、dhfr遺伝子発現カセットの一部とを含むDNA(3640bpのDNA)および、ホモロジーアーム2と、dhfr遺伝子発現カセットの一部とを含むDNA(3285bpのDNA)は、相同組換えによってのみゲノムから取得することができ、相同組換えの指標となる。
【0111】
よって、ホモロジーアーム1と標的領域との相同組換えの指標として、図2(A)に示す3640bpのDNAを設定し、このDNAは、以下に示すプライマーCHA1seq−s11/dhfr−seq−a1によるPCR反応により検出した。一方、ホモロジーアームと標的hprt遺伝子座との相同組換えの指標として、図2(A)に示す3285bpのDNAを設定し、このDNAは、以下に示すプライマーNcoI−dhfr−s/ CHA2−seq−a4によるPCR反応により検出した。
【0112】
CHA1seq−s11プライマー:(配列番号29)
5’−GACACATGCAGACAGAACAG−3’
CHA2−seq−a4プライマー:(配列番号30)
5’−GTTTGCTAACACCCCTTCTC−3’
dhfr−seq−a1プライマー:(配列番号31)
5’−ctcaggaatggagaaccagg−3’
NcoI−dhfr−sプライマー:(配列番号32)
5’−ttaaCCatggttcgaccattgaactg−3’
【0113】
次に、得られたPCR増幅産物を1.0%アガロースゲル電気泳動により解析した。結果は、図2(C)に示される通りであった。図2(C)において、取得したクローンにおいてのみ、3640bpおよび3285bpの両方のPCR増幅産物が確認され、相同組換え反応が確認された。
【0114】
次に、dhfr欠損CHO hprt組換え基礎ベクターのトランスフェクションにより得たHTサプリメント非要求性および6TGに耐性のクローンのゲノムDNAについて、以下に示すPCR反応により、野生型hprt遺伝子が残存しているのかを確認した。
【0115】
図2(B)は、野生型hprt遺伝子の残存を確認するためのゲノムPCRの詳細を説明する模式である。まず、図2(B)左図は2本のX染色体を有する野生型DG44細胞におけるPCRを示す。hprt遺伝子の相同組換えの標的領域は、エクソン3を含んで設定されている。ベクター非導入の野生型細胞の場合、標的領域のDNA(2322bp)は、PCR反応によって増幅することができ、これを指標として野生型hprt遺伝子が残存していることを確認することができる。
【0116】
図2(B)の中央図は、1本のX染色体のhprt遺伝子座にベクターが組込まれたヘテロ接合型の相同組換え細胞におけるPCRを示す。一方の染色体では、標的領域中にベクターのDNAは相同組換えによりに組込まれているため、3867bpのPCR増幅産物が生じる。一方、もう1本の染色体は、標的領域を保持しており、標的領域のDNA(2322bp)もまた、PCR反応によって増幅することができ、野生型hprt遺伝子が残存していることを確認できる。
【0117】
一方、図2(B)の右図に示されるとおり、両方のX染色体にベクターが組込まれたホモ接合型の相同組換え細胞においては、組換えDNAに由来する3867bpのPCR増幅産物のみが発現し、野生型hprt遺伝子に由来するDNA(2322bp)の増幅は生じない。
【0118】
以上の理解から、図2(B)に示す2322bpのDNAを上記指標として設定し、このDNAを検出するため、プライマーCHPRTs/CHA2−seq−a1を用いてPCR反応を行った。
【0119】
次に、得られたPCR増幅産物を1.0%アガロースゲル電気泳動により解析した。結果は、図2(C)に示される通りであった。
【0120】
CHPRTsプライマー:(配列番号33)
5’−TGTTCCTGTGCATACTAGGC−3’
CHA2−seq−a1プライマー:(配列番号34)
5’−CCAGAGAAATTATTTGCCACCAGC−3’
【0121】
図2(C)において、野生型細胞からは2322bpのPCR増幅産物が確認され、野生型hprt遺伝子の存在が確認された。一方、組換えクローンにおいては2322bpのPCR増幅産物が確認されず、3867bpの産物のみが確認されたことから、組換え細胞は両方のX染色体にベクターが組込まれたホモ接合型であった。
【0122】
この結果から、本発明によるベクターを用いると、2本のX染色体の両hprt遺伝子座に目的タンパク質遺伝子が組込まれた組換え細胞クローンが高頻度で取得されていることが確認された。
【0123】
実施例4:DXB11(dhfr欠損CHO)hprt組換えクローン作製
DXB11へのベクター導入およびスクリーニング
実施例1で作製した、dhfr欠損CHO hprt組換え基礎ベクターを、実施例2と同様に精製および線状化し、3×106個のDXB11細胞にトランスフェクションし、スクリーニングを実施した。
【0124】
結果、HTサプリメント非要求性/6TG耐性のDXB11クローンを1クローン取得した。
【0125】
ゲノムPCRによる、DXB11のhprt遺伝子座へのベクター組込みの確認
実施例3と同様に、dhfr欠損CHO hprt組換え基礎ベクターのトランスフェクションにより得たHTサプリメント非要求性および6TGに耐性のDXB11クローンからゲノムDNAを抽出し、このゲノムDNAを鋳型とした以下に示すPCR反応により、標的hprt遺伝子座への位置特異的な組換えの確認を行った。
【0126】
図4(A)は、ゲノムPCRによる相同組換え反応の解析の詳細を説明する模式図である。
【0127】
実施例3でのDG44の場合と全く同様に、DXB11のhprt遺伝子の相同組換えの標的領域は、エクソン3を含んで設定されており、この領域にベクターDNA中のホモロジーアーム1、ホモロジーアーム2およびこれに挟まれたdhfr遺伝子発現カセットは相同組換えにより組込まれる。
【0128】
ホモロジーアーム1と、dhfr遺伝子発現カセットの一部とを含むDNA(3640bpのDNA)および、ホモロジーアーム2と、dhfr遺伝子発現カセットの一部とを含むDNA(3285bpのDNA)は、相同組換えによってのみゲノムから取得することができ、相同組換えの指標となる。これらのDNAは、取得したDXB11クローンのゲノムDNAを鋳型とした、実施例3と同一のプライマーセットによるPCR反応によって検出した。
【0129】
PCR増幅産物を1.0%アガロースゲル電気泳動により解析した結果は、図4(B)に示される通りであり、取得したDXB11クローンにおいて3640bpおよび3285bpの両方のPCR増幅産物が確認され、相同組換え反応が確認された。
【0130】
以上の結果から、DXB11のhprt遺伝子座にベクターが組み込まれたクローンが取得できたことが確認できた。
【0131】
実施例5:dhfr欠損CHO hprt組換え抗体発現ベクタークローン作製および抗体生産
dhfr欠損CHO hprt組換え抗体発現クローン作製
実施例1で作製した、dhfr欠損CHO hprt組換え抗体発現ベクターを、実施例2と同様に精製および線状化し、3×106個のDG44細胞にトランスフェクションし、スクリーニングを実施した。
【0132】
結果、dhfr欠損CHO hprt組換え抗体発現ベクターによって、HTサプリメント非要求性/6TG耐性クローンを1クローン取得した。
【0133】
取得クローンからゲノムDNAを抽出し、このゲノムDNAを鋳型としたPCRを実施例2と同様に実施し、hprt遺伝子座にdhfr欠損CHO hprt組換え抗体発現ベクターが組み込まれていることを確認した。
【0134】
dhfr欠損CHO hprt組換え抗体発現クローンの無血清浮遊馴化
100mmディッシュに捲き込んだDG44 dhfr欠損CHO hprt組換え抗体発現ベクタークローンを、70%コンフルエントまで培養した後、トリプシン処理によって剥離した。
【0135】
1000rpm、5分間の遠心によって細胞を沈殿させ、上澄みを破棄した後、完全CD OptiCHO培地(CD OptiCHO(インビトロジェン社)に1x Glutamax(インビトロジェン社)を添加したもの)で3×105cells/mlに調整してフラスコに移し、インキュベーター中37℃、8%CO2にて、121rpmで振とう培養した。
【0136】
細胞密度が1×106cells/mlを超えた時点で、完全CD OptiCHOで3×105cells/mlに希釈継代し、培養を継続した。
【0137】
上記継代を3回繰り返した時点で、無血清浮遊培養への馴化を完了した。
【0138】
dhfr欠損CHO hprt組換え抗体発現クローンによる抗体生産
無血清浮遊培養に馴化したDG44 dhfr欠損CHO hprt組換え抗体発現ベクタークローンを、1000rpm、5分間の遠心によってペレット化し、新鮮な完全CD OptiCHO培地に懸濁した。この細胞懸濁液を完全CD OptiCHO培地で希釈し、60mlの3×105cells/ml細胞液を調整し、フラスコに移した。
【0139】
インキュベーター中37℃、8%CO2にて、121rpmで振とう培養し、24時間ごとに細胞密度を測定して記録した。また、1.5mlの培地を回収し、遠心した上澄みを−80℃で保存した。
【0140】
18日間のサンプリングを実施後、回収しておいた培地サンプル回収した培地中のIgG量を、Human IgG EIA Kit (precoated)(宝酒造製)により、450nmの吸光度を測定することにより定量した。
【0141】
その結果、図5に示すように、培地上澄1Lあたり35mgのIgG生産が認められた。
【0142】
dhfr欠損CHO hprt組換え抗体発現クローンの発現の長期安定性確認
無血清浮遊培養に馴化したDG44 dhfr欠損CHO hprt組換え抗体発現ベクタークローンを、14回継代培養して維持した。継代操作は1週間あたり2回実施した。その後、細胞集団の一部を、新鮮な完全CD OptiCHO培地60ml中に3×105cells/mlになるように調整し、フラスコに移し、培養を開始した。
【0143】
培養開始後、7日目に培地中の細胞密度を測定した後、培地上澄1.5mlを採取し、遠心分離した上澄みをー80℃保存した。24時間後、細胞密度を測定し、培地上澄1.5mlを同様に保存した。
【0144】
保存しておいた培地上澄サンプルを溶解し、Human IgG EIA KitでIgG量を定量し、細胞密度を用いて比生産性(pg/cell/day)を算出した。また、無血清浮遊培養に馴化した直後のDG44 dhfr欠損CHO hprt組換え抗体発現ベクタークローンについても、同様に比生産性を測定した。
【0145】
14回の継代操作後のクローンの比生産性は、図6に示すように全く低下していなかった。これは、実生産で最低限要求される50日間(約7週間)の安定性を満足する、好ましい結果と言える。
【技術分野】
【0001】
本発明は、dhfr遺伝子欠損CHO細胞における、組換え細胞の効率的な製造に有用なDNA構築物、およびそれを用いた組換え細胞の製造方法に関する。
【背景技術】
【0002】
組換えタンパク質生産システムにおいて、原核生物や真核生物を宿主とした種々の方法が知られている。哺乳類動物細胞を宿主とした組換えタンパク質生産システムは、ヒトを始めとする高等動物由来のタンパク質に対し、糖鎖付加、フォールディング、リン酸化などの翻訳後修飾をより生体で作られるものと同じように施すことが可能である。
【0003】
この翻訳後修飾は、タンパク質の本来有する生理活性を組換えタンパク質で再現するために必要なものであり、そのような生理活性が特に必要とされる医薬品などに用いられる組換えタンパク質の生産系には、哺乳類動物細胞を宿主としたタンパク質生産システムがよく用いられている。
【0004】
現在、工業的生産に用いられている哺乳類動物細胞による主なタンパク質生産システムとしては、CHO−DHFRシステムとGS−NS0システムの二つが挙げられる。これらの生産システムでは、プラスミドベクターに含まれる選択マーカーと適切な薬剤選択プロセスを組み合わせることで、染色体に組み込まれたプラスミドベクターのコピー数が増幅したクローンを選択する。特にCHO−DHFRシステムは、選択薬剤メトトレキセートによる2段階の選抜工程により、発現レベルが数十倍に増大した細胞クローンを選択しうる。CHO−DHFRシステムでは、dhfr遺伝子を欠損したCHO細胞の使用が必須である。
【0005】
しかしながら、上記タンパク質生産システムには、単純に増幅されたプラスミドベクターのコピー数に比例して目的タンパク質の発現レベルが必ずしも増大しないこと、発現レベルが増大した細胞クローンを選択するまでにかかる時間が長いことなどの問題があることが知られている。さらに、発現レベルが増大した細胞クローンの選択後に、選択薬剤を含まない培地で選択細胞クローンの培養を継続することで、ほとんどのクローンにおいて発現レベルの減少または消失が確認されることが報告されている(特許文献1:特表2002−541854公報、非特許文献1:Kim, N. S. (1998) Biotechnol. Bioeng.,60, 679−688.)。
【0006】
また、上記のような哺乳類動物細胞による目的タンパク質生産システムは、一般的に、目的タンパク質遺伝子を含むベクターを宿主細胞に導入し、該ベクターが染色体へ組み込まれた細胞クローンを選択し、さらに該細胞クローンを適切な培養条件で培養することにより製造されている。
【0007】
この染色体への組み込みはランダムな位置で起こりうる事象であり、得られる細胞クローンによって目的タンパク質の発現レベルが異なる。また細胞クローンによっては目的タンパク質を発現しないなどの問題がある。これについては、多数のクローンを組換えタンパク質の発現レベルにより選抜して、好ましいクローンを選択するという方法が採られている。しかしながら、このスクリーニングプロセスは非常に手間と労力を要する。このような手間を回避して迅速に好ましいクローンを選択するための種々のプロセスが報告されている。
【0008】
例えば、マウス細胞の特定染色体位置にベクターを組み込む技術が開示されている(特許文献2:特表平9−510865公報)。免疫グロブリンγ2A座と相同な塩基配列を有する配列を搭載したベクターにより、組換え細胞クローンのプール中に相同的組換え細胞クローンを生じさせる。標的となる染色体位置は、外来遺伝子が組み込まれた際にランダム組み込みと比較して高い発現レベルを与えうる位置としてあらかじめ同定されている。したがって、スクリーニング対象となる組換え細胞クローンのプール中に、高い発現レベルを有する相同的組換え細胞クローンが一定の頻度で存在することにより、発現レベルによるスクリーニングの際の労力を低減することが可能となる。
【0009】
また、マーキングプラスミドの利用技術についても開示されている(特許文献3:特表2001−516221公報)。あらかじめマーキングプラスミドをランダム組換えした細胞クローン集団の中からマーキングプラスミド内に存在するマーカー遺伝子の発現レベルが高いクローンを選択しておく。次に目的タンパク質遺伝子を有するプラスミドベクターとランダムに組み込まれたマーキングプラスミド配列との間で部位特異的組換えが引き起こされた細胞クローンを選択することにより、マーカー遺伝子の発現レベルを継承した目的タンパク質産生クローンが得られる。
【0010】
上記技術は、発現レベルの高いクローンの選択に要する労力削減において有利である。しかしながら、例えば、組換え細胞を選択薬剤非添加で継続的に培養した際、得られたクローンが長期的に発現レベルを安定に維持しうるかについては依然として予測できない。
【0011】
医薬タンパク質の工業的生産においては、タンパク質の発現が高いレベルでかつ安定に維持されることが重要である。特に発現レベルが安定に維持されることに関しては、コスト面のみならず、医薬タンパク質としての同一性および安全性を証明するためにも重要である。組換えタンパク質生産細胞を工業的スケールでの生産に用いるためには生産細胞クローンの培養スケールの拡大を図る必要がある。これには通常少なくとも樹立直後のクローンから約60回の細胞分裂を経なければならないと見積られており(非特許文献2:Brown, M. E. et al. (1992) Cytotechnology, 9, 231−236.)、この発現レベルが一定に保たれていなければならない。
【0012】
また、選択薬剤は培養コストを上昇させることのみならず、医薬品への異物混入リスク回避のために行われる精製プロセスのコストも上昇させる。したがって、選択薬剤を添加することなく発現レベルを安定に維持できる細胞クローンの作製技術の開発が強く望まれている。
【0013】
上記のような事情があるにも関わらず、目的タンパク質発現レベルの安定性に関して充分な技術的検討が行なわれてきたとはいえない。これまでのところ、多くのタンパク質産生系の製造方法においては、長期培養時における成長速度ならびに生産性に関して蓄積されたデータを元に、経験的にクローン選択が行なわれている。しかしながら、この経験的な方法では発現レベルが安定な細胞クローンを実際に取得できるケースは稀である(非特許文献3:Barnes, L. M. et al. (2003) Biotechnology and Bioengineering, 81, 631−639.)。
【0014】
近年、細胞ゲノム中の標的遺伝子座に目的タンパク質遺伝子を特異的な組み込み、長期間、安定にタンパク質を発現しうる組換え細胞を効率的に取得する手法が検討されている。
【0015】
この標的遺伝子の一例としては、hprt遺伝子が挙げられる。hprt遺伝子は、ヒト等のX染色体長腕に存在するハウスキーピング遺伝子の一つとして知られており、hprt遺伝子のノックアウト後の細胞は、薬剤6−Thioguanime (6TG)に対して耐性を示すため、陰性選択が容易に行われる。
【0016】
そして、本発明者らの一部は、ヒトの雄由来のHT1080細胞株のhprt遺伝子座へ目的タンパク質遺伝子を組換えベクターにより導入し、選択薬剤非存在下で長期間、安定にタンパク質を発現しうる組換え細胞を取得したことを報告している(特許文献5:特開2007−325571公報、非特許文献4:Koyama Y Et Al.,(2006) Biotechnology And Bioengineering,95,1052−1060)。この実施例においては、各々約1kbpの第一および第二の相同DNA断片をホモロジーアームとして用いたところ、107細胞あたり10クローン程度の組換え細胞を取得したことが報告されている。
【0017】
また、hprt遺伝子座へのターゲッティングの別の例としては、マウスES細胞に対してターゲッティングが行われたことが報告されている(特許文献6:特表平5−507853公報)。
【0018】
しかしながら、同じ遺伝子座を標的としても、培養細胞の種類によっては、ジーンターゲッティング頻度が大きく異なる場合がある。例えば、Porter C. G. Itzhaki J. E, Eur. J. Biochem 218, 273−281(非特許文献5)、および広島大学原爆放射線医科学研究所 年報第44号 (2003)(非特許文献6)では、ES細胞と、HT1080細胞とを比較すると、ジーンターゲッティング頻度が異なり、体細胞由来の培養細胞では、ジーンターゲッティング頻度が極めて低いことが報告されている。
【0019】
一方、dhfr遺伝子欠損CHO細胞は、DHFR−MTXシステムによる抗体医薬タンパク質産生系の宿主細胞として利用されており、dhfr遺伝子欠損CHO細胞を用いて高レベルで安定なタンパク質生産システムを製造することが求められている。
【0020】
しかしながら、dhfr遺伝子欠損CHO細胞のhprt遺伝子座に対してジーンターゲッティングが行われたことは報告されていない。dhfr遺伝子欠損CHO細胞の親株である、CHO細胞の一方の染色体上のaprt遺伝子を欠損した特殊な細胞株を用いた研究例が存在するのみである(非特許文献8:PNAS,88, 9488−9502(1991)、非特許文献9:Somatic Cell. Mol. Genet., 19, 363−375、非特許文献10:PNAS,86,4574−4578(1989))。これらの実施例では、一方の染色体上のみにaprt遺伝子が存在する細胞株を宿主とし、2.6kbp〜4kbpの相同DNA断片をホモロジーアームとして用いた結果、107細胞あたり数クローン〜15クローン程度の相同組換え体を取得している。
【0021】
また、dhfr遺伝子欠損CHO細胞のhprt遺伝子座のゲノムの配列はエキソン以外は何ら報告されていない。したがって、dhfr遺伝子欠損CHO細胞のhprt遺伝子座に対する特異的なジーンターゲッティングを行うためには、エキソン以外の塩基配列を解析し、取得しなければならないという問題がある。
【0022】
さらに、CHO細胞は、雌由来の細胞であることから、X染色体を2本有しており、そのhprt遺伝子座も2つ有している。そのため、両方の染色体のhprt遺伝子座に外来遺伝子が組み込まれることで、CHO細胞は6TG等の選択薬剤耐性となる。しかしながら、このように、2本の染色体で組換えが生じる確率は、雄由来細胞の場合と比べて低くなるのが通常である。例えば、Koyama Y Et Al., (2006) Biotechnology And Bioengineering,95,1052−1060(非特許文献4)における雄由来HT1080細胞株のhprt遺伝子座への組換え効率を参考として染色体1本で組換えが生じる確率を10−6と仮定すると、染色体2本で同時に生じる理論上の確率は10−12となる。
【0023】
したがって、目的タンパク質遺伝子を発現する組換えCHO細胞を効率的に製造する技術の創出が依然として必要とされている。
【0024】
また、dhfr欠損CHO細胞は、DNAのde novo合成能を欠いた細胞株であり、培地中のヒポキサンチン、チミヂンをサルベージすることで自らのDNAをまかなって生存している。サルベージ経路にはhprt遺伝子機能が必須であるため、hprt遺伝子座へのベクター組み込みは致死となる(非特許文献7:Carcinogenesis, 20, 215−220(1999))。したがって、hprt遺伝子座を外来遺伝子組み込みの標的部位として使用できないという問題がある。
【0025】
ところで、陽性選択遺伝子と陰性選択遺伝子を融合させた二機能選択可能融合遺伝子を発現させる方法が公開されており、陽性選択遺伝子の例としてdhfr遺伝子が、陰性選択遺伝子としてhprt遺伝子が例示されている(特許文献7:特表平6−504432)。
【0026】
この方法を適応した場合、dhfr−hprt融合遺伝子をベクターに搭載して細胞に導入することになる。ところが、dhfr遺伝子欠損CHO細胞では染色体上のhprtが元来活性な状態にあるため、組換え細胞の選択はヒポキサンチン非要求性でしか行えない。CHO細胞では、ランダムな挿入が相同組換えのおよそ4千倍の頻度で生じる(非特許文献10:PNAS,86,4574−4578(1989))。したがって、染色体hprt遺伝子座と相同性をもつホモロジーアームとともにdhfr−hprt融合遺伝子をdhfr遺伝子欠損CHO細胞に導入しても、hprt遺伝子座への相同組換え細胞が取得できる見込みはほとんどない。
【0027】
したがって、dhfr遺伝子欠損CHO細胞のhprt遺伝子座への目的タンパク質遺伝子組み込み細胞を選択可能にする技術の創出が依然として必要とされている。
【先行技術文献】
【特許文献】
【0028】
【特許文献1】特表2002−541854公報
【特許文献2】特表平9−510865公報
【特許文献3】特表2001−516221公報
【特許文献4】WO2004/022741公報
【特許文献5】特開2007−325571公報
【特許文献6】特表平5−507853公報
【特許文献7】特表平6−504432公報
【非特許文献】
【0029】
【非特許文献1】Kim, N. S. (1998) Biotechnol. Bioeng., 60, 679−688.
【非特許文献2】Brown, M. E. et al. (1992) Cytotechnology, 9, 231−236.
【非特許文献3】Barnes, L. M. et al. (2003) Biotechnology and Bioengineering , 81, 631−639.
【非特許文献4】Koyama Y Et Al., (2006) Biotechnology And Bioengineering, 95, 1052−1060
【非特許文献5】Porter C. G. Itzhaki J. E, Eur. J. Biochem 218, 273−281
【非特許文献6】広島大学原爆放射線医科学研究所 年報第44号 (2003)
【非特許文献7】Carcinogenesis, 20, 215−220(1999)
【非特許文献8】PNAS,88, 9488−9502(1991)
【非特許文献9】Somatic Cell. Mol. Genet., 19, 363−375
【非特許文献10】PNAS,86,4574−4578(1989)
【発明の概要】
【0030】
本発明者らは、今般、dhfr遺伝子欠損CHO細胞におけるhprt遺伝子の相同DNA断片と選択マーカー遺伝子としてジヒドロ葉酸還元酵素遺伝子(dhrf)を有する特定のDNA構築物により目的タンパク質遺伝子をdhfr遺伝子欠損CHO細胞に導入したところ、組換え細胞を顕著な高頻度で製造しうることを見出した。本発明は、かかる知見に基づくものである。
【0031】
したがって、本発明は、dhfr遺伝子欠損CHOにおける組換え細胞の効率的な製造に有用なDNA構築物およびそれを用いた組換え細胞製造方法の提供をその目的とする。
【0032】
そして、本発明によるDNA構築物は、5’末端から3’末端に向かって、第一の相同DNA断片、目的タンパク質遺伝子、選択マーカー遺伝子および第二の相同DNA断片を含んでなるDNA構築物であって、前記第一の相同DNA断片および第二の相同DNA断片が、dhfr遺伝子欠損CHO細胞ゲノムのヒポキサンチン−ホスホリボシルトランスフェラーゼ酵素(hprt)遺伝子座の一部と相同組換え可能な相同性を有し、かつ1kbp以上の鎖長を有するものであり、前記選択マーカー遺伝子がジヒドロ葉酸還元酵素遺伝子(dhfr)であることを特徴とするものである。
【0033】
また、本発明によるdhfr遺伝子欠損CHOにおける組換え細胞の製造方法は、上記DNA構築物を含んでなるベクターをdhfr遺伝子欠損CHO細胞に導入することを含んでなる。
【0034】
本発明によれば、目的タンパク質遺伝子を発現する組換えCHO細胞を顕著な高頻度で取得することができる。
【図面の簡単な説明】
【0035】
【図1】実施例1で用いられたdhfr欠損CHO細胞hprt相同組換え用ベクターの模式図である。(A)はdhfr欠損CHO細胞のhprt遺伝子に由来する相同DNA断片を有し、選択マーカー遺伝子としてdhfr遺伝子を有するベクターである。(B)はdhfr遺伝子欠損CHO細胞のhprt遺伝子に由来する相同DNA断片を有し、選択マーカー遺伝子としてdhfr遺伝子を有し、抗体発現カセットを含むベクターである。
【図2】(A)は、dhfr遺伝子欠損CHO細胞のhprt遺伝子領域、相同組換え用ベクター、および実施例3において組換え細胞のゲノムPCRの指標となったDNAの関係を示す模式図である。(B)は、組換えdhfr遺伝子欠損CHO細胞中の野生型hprt遺伝子の残存を確認するためのゲノムPCRの詳細を説明する模式図である。左図は、野生型のdhfr遺伝子欠損CHO細胞におけるPCRを示し、中央図は、ヘテロ接合型の組換え細胞におけるPCRを示し、右図は、ホモ接合型の組換え細胞におけるPCRを示す。(C)は、実施例3のPCRの結果を示す。
【図3】dhfr欠損CHO細胞のhprt遺伝子座の模式図である。番号1から9まではhprt遺伝子のエクソン1〜9を示し、各エクソン間の配列は、イントロン1〜8を示す。
【図4】(A)は、DXB11のhprt遺伝子領域、相同組換え用ベクター、および実施例4において組換え細胞のゲノムPCRの指標となったDNAの関係を示す模式図である。(C)は、実施例4のPCRの結果を示す。
【図5】は、実施例5で作製したdhfr欠損CHO hprt組換え抗体発現クローンによって抗体を生産した結果を示す。
【図6】は、実施例5で作製したdhfr欠損CHO hprt組換え抗体発現クローンの抗体比生産性の長期安定性試験結果を示す。
【発明を実施するための形態】
【0036】
DNA構築物
本発明によるDNA構築物は、CHO細胞ゲノム中のhprt遺伝子座の一部と相同組換え可能な相同性を有し、各々が1kbp以上の鎖長を有する、二つの相同DNA断片を含んでなることを一つの特徴とする。上記DNA構築物によれば、CHO細胞が2本のX染色体を有するにもかかわらず、顕著な高頻度で組換えCHO細胞を取得しうるのは意外な事実である。本発明によるDNA構築物によれば、後述の実施例3に示される通り、2x10−6あたり4個の組換え細胞が得られている。この結果は、本願出願日当時の技術状況において、染色体2本のhprt遺伝子座において同時に組換えが生じる理論上の確率が10−12と予測されていたことを勘案すると、驚くべき事実といえる。
【0037】
また、本発明によるDNA構築物は、陽性選択マーカー遺伝子としてジヒドロ葉酸還元酵素遺伝子(dhfr)を含んでなることを一つの特徴とする。dhfr遺伝子は、相同組換えを妨げない限り、DNA構築物中に適宜配置することができる。dhfr遺伝子を用いることにより、de novo合成能とhprt遺伝子の不活化が合わさることによる致死性を回避することが可能となる。さらに、組換えCHO細胞の選択において、ヒポキサンチン、チミジン非要求性を指標とした陽性選択と、hprt遺伝子の不活化による陰性選択の両者を適用でき、偽陽性クローンを大幅に低減する上で有利である。
【0038】
本発明によるDNA構築物は、hprt遺伝子座の一部を相同組換えの標的領域とするものである。hprt遺伝子座の一部を標的領域とすることは、目的タンパク質遺伝子を高レベルで安定に発現する上で有利である。また、かかる標的領域に、DNA構築物を組み込むことは、hprt遺伝子の転写・発現を阻害してhprt遺伝子の機能を不活化し、6−TG等を用いる陰性選択によって効率的に組換え細胞を取得する上でも好ましい。
【0039】
また、本発明の標的領域は、目的タンパク質遺伝子の発現を妨げない限り、hprt遺伝子座中において適宜決定してよい。しかしながら、相同組換えに必要な相同DNA断片の鎖長等を勘案すれば、上記標的領域は、hprt遺伝子のイントロンの少なくとも一部を含む領域が好ましい。上記イントロンは、本発明者らにより今般、塩基配列が決定されたものであり、具体的には、配列番号35〜42のいずれかで表される塩基配列を有する、後述する図3のイントロン1〜8が挙げられる。
【0040】
また、本発明の標的領域は、上記イントロンと隣接するエクソンの全部または一部を含んでいてもよい。かかるエクソンとしては、具体的には、図3のエクソン1〜9が挙げられ、これらの塩基配列は、例えば、アメリカ合衆国国立バイオテクノロジー情報センター(National Center for Biotechnology Information)等の公知のデータベースにアクセスすることにより取得することができる。
【0041】
この組み込みの態様としては、(1)DNA構築物の組み込みによってエクソンが分断されるタイプ、(2)DNA構築物の組み込みによってエクソンが1つ以上欠失するタイプ、(3)DNA構築物の組み込みによってエクソンが2個に増幅し、その間にDNA構築物が挿入されるタイプ、(4)DNA構築物の組み込みによってイントロンが分断されるタイプ、(5)DNA構築物の組み込みによってイントロンが1つ以上欠失するタイプ、(6)DNA構築物の組み込みによってイントロンが2個に増幅し、その間にDNA構築物が挿入されるタイプが挙げられる。
【0042】
また、本発明のDNA断片は、相同組換えに必要な鎖長を勘案すると、上述の通り、hprt遺伝子のイントロンの少なくとも一部を含む領域と相同性を有するものが好ましい。したがって、本発明の一つの態様によれば、本発明の第一の相同DNA断片または第二の相同DNA断片は、配列番号35〜42のいずれかに記載の塩基配列またはその部分配列を含んでなるものとされる。
【0043】
また、上記部分配列の鎖長の下限は1bpであり、その上限は配列番号35〜42のいずれかに記載の塩基配列の鎖長の範囲内において、適宜調節することができる。
【0044】
また、上記相同DNA断片と、hprt遺伝子座との相同性は、相同組換えの効率を勘案して適宜決定されるが、好ましくは99.0%以上、より好ましくは99.9%以上、さらに好ましくは100%である。かかる相同性は、例えば、DNAシーケンサー等を用いて解析することにより適宜決定することができる。
【0045】
また、上記相同DNA断片は1kbp以上の鎖長を有するものとされており、かかる鎖長は、DNA構築物の顕著な高頻度での相同組換えを達成する上で好ましい。さらに、上記相同DNA断片の鎖長は、組換え効率の向上等を勘案して適宜調整してよく、より好ましくは2.5kbp以上である。また、上記相同DNA断片の鎖長の上限は、目的タンパク質遺伝子の効率的な導入を妨げないという観点から、好ましくは7.5kbp以下であり、最も好ましくは5kbp以下である。相同DNA断片の鎖長の範囲は上記の上限値、下限値を適宜組み合わせることができる。具体的には好ましくは1kbp以上7.5kbp以下、より好ましくは1kbp以上5kbp以下、最も好ましくは2.5kbp以上5kbp以下である。鎖長がこの範囲であれば、DNA構築物の顕著な高頻度での相同組換えを達成しながら、組換え細胞の取得頻度を良好に保つことが可能となる。
【0046】
また、本発明によるDNA構築物において、目的タンパク質遺伝子は、医薬として有用なタンパク質をコードしていることが好ましい。目的タンパク質遺伝子は、cDNAに由来する配列であっても、ゲノムDNAに由来する天然イントロンを含む構造遺伝子であっても、好適に利用可能である。具体的には、目的タンパク質としては、抗体、酵素、サイトカイン、ホルモン、凝固因子、調節タンパク質、レセプター等が挙げられるが、好ましくは、モノクローナル抗体、ポリクローナル抗体、エリスロポエチン、組織特異的プラスミノーゲン活性化因子または顆粒球コロニー活性化因子等である。
【0047】
また、上記目的タンパク質遺伝子は、プロモーター配列や転写終結シグナル配列等の発現に必要な要素を含む発現ユニットとしてhprt遺伝子座に組み込まれることが好ましい。したがって、本発明の一つの態様によれば、目的タンパク質遺伝子は、少なくともプロモーター配列および転写終結シグナル配列を含んだ発現ユニットとしてDNA構築物中に配置される。もっとも、発現に必要な要素としてCHO細胞の内在性のものを用いるように構成してもよく、本発明にはかかる態様も包含される。
【0048】
また、上記プロモーターや転写終結シグナルは、目的タンパク質遺伝子の種類、性質等に応じて適宜決定してよく、かかるプロモーター配列の好適な例としては、CMVプロモーター、SV40プロモーター等が挙げられる。また、転写終結シグナル配列の好適な例としては、BGHポリAシグナル配列、SV40ポリAシグナル配列等が挙げられる。
【0049】
また、プロモーター配列、転写終結シグナル配列の他の発現に必要な要素としては、例えば、目的遺伝子を効率的に発現させるための調節エレメント(例えば、エンハンサー、IRES(internal ribosome entry site)配列、LoxP配列およびFRT配列等の組換え酵素認識配列)等を適宜選択して用いてよい。調節エレメントはその性質に応じて、発現ユニットにおける適切な位置に配置することが可能である。これら発現に必要な要素は、目的タンパク質の生産性等を勘案して適宜選択される。
【0050】
ベクター
本発明によるDNA構築物は、ベクターに組み込んでdhfr遺伝子欠損CHO細胞ゲノムに導入することができる。かかるベクターシステムとしては、相同組換え反応によりDNA構築物を細胞ゲノムに組み込みうる限り特に限定されないが、好ましくは、プラスミドベクター、コスミドベクター、ファージベクターまたは人工染色体ベクター等が挙げられる。
【0051】
本発明によるDNA構築物およびこれを含むベクターは、制限酵素切断反応およびライゲーション反応を組み合わせて好適に構築される。例えば、各構成ユニットの両末端に制限酵素の認識配列を含有させ、該認識配列用の制限酵素で切断反応を行い、不要なDNA配列(大腸菌内での操作用配列等)をゲル切り出し等の処理によって除去し、得られたユニットのライゲーション反応を行うことにより、DNA構築物およびこれを含むベクターを構築しうる。
【0052】
ライゲーション反応により構築したベクターDNAは、フェノール・クロロホルム抽出等により精製し、ベクターの種類等を勘案して選択した大腸菌、酵母等の宿主細胞において増殖させることができる。
【0053】
本発明によるベクターは、CHEF1αプロモーターを、目的タンパク質遺伝子に作動可能に連結して搭載していることが好ましい。CHO hprt遺伝子座に組込んだ外来遺伝子の発現はプロモーターの不活化によって生じる。目的タンパク質遺伝子をCHO内在性のCHEF1αプロモーターで発現させるベクター構造にすることで、CHO
hprt遺伝子座での安定発現が可能となる。
【0054】
dhfr欠損CHO細胞株
本発明の対象となる細胞株はdhfr遺伝子を欠損したチャイニーズハムスターの細胞株であり、DG44、DXB11などが挙げられる。
【0055】
組換えdhfr欠損CHO細胞/製造方法
本発明によるdhfr欠損CHOの組換え細胞は、上記ベクターをdhfr欠損CHO細胞に導入することにより好適に製造することができる。したがって、本発明によるdhfr欠損CHOの組換え細胞は、hprt座に外来性の目的タンパク質遺伝子が組み込まれてなる。また、本発明の好ましい態様によれば、組換えdhfr欠損CHO細胞はhprt遺伝子の機能が不活化している。かかる組換えdhfr欠損CHO細胞は、抗体等の目的タンパク質を高レベルで安定に産生する上で有利である。
【0056】
ベクターの導入
上記ベクターの導入方法としては、一般的に用いられる方法が好適に利用できる。例えば、リン酸カルシウム法、エレクトロポレーション法、マイクロインジェクション法、DEAE−デキストラン法、リポソーム試薬を用いる方法、カチオン性脂質を用いたリポフェクション法などが挙げられる。ここで、ベクターが環状である場合、公知の方法により線状化して細胞に導入されてもよい。
【0057】
また、ベクターや目的タンパク質の性質によっては、組換え細胞の取得効率等を勘案して、Cre/LoxPシステムやFlp/FRTシステムなどのような、組換え酵素を利用した部位特異的導入システムも適宜適用してよく、本発明にはかかる態様も包含される。
【0058】
細胞株の選択
また、本発明による製造方法にあっては、上記導入後、組換え細胞の選択を行うことが好ましい。選択工程は、hprt遺伝子の不活化に基づく陰性選択により行うことができる。また、dhfr遺伝子を陽性マーカー遺伝子として、組換え細胞に導入している場合には、陰性選択と、ヒポキサンチン、チミジン非要求性による陽性選択とを組み合わせて高い精度で細胞選択を行うことが可能となる。
【0059】
また、上記選択方法の他、プロモータートラップ法やポリAトラップ法なども適宜組み合わせて利用してよい。
【0060】
また、本発明による製造方法にあっては、組換え細胞を取得後、Chemical Defined 培地などに馴化させることにより無血清下での培養も可能である。かかる馴化条件は、組換え細胞の状態に応じて適宜決定することができる。
【0061】
目的タンパク質の製造方法
また、本発明の別の態様によれば、上記組換え細胞を用意し、該細胞を培養して目的タンパク質を産生することを含んでなる、目的タンパク質の製造方法が提供される。上記方法によれば、目的タンパク質を効率的かつ安定に取得することができる。
【0062】
上記培養工程における培地としては、組換え細胞の状態に応じて公知の培地を適宜選択してよいが、無血清培地が好ましい。また、上記培地は、培養コストおよび精製コストを勘案すれば、選択薬剤を添加しないものが好ましい。上記培地の好適な例としては、Chemical Defined培地等が挙げられる。
【0063】
上記培養方法としては、バッチカルチャー法、フェッド−バッチカルチャー法、還流培養法など公知の方法が適用可能である。
【0064】
なお、組換え細胞の製造に使用される種々の手法のさらなる詳細は、例えば、F.M.Ausubel,Current Protocols in Molecular Biology,John Wiley & Sons,New York,(1989)に記載されており、上記文献の全開示内容は引用することにより本明細書の一部とされる。
【実施例】
【0065】
以下、本発明を実施例によって具体的に説明するが、本発明はこれら実施例に限定されるものではない。
【0066】
なお、以下の実験において、制限酵素による反応、PCR反応、ライゲーション反応等の各反応条件は、メーカーの推奨する反応条件、あるいは、Molecular Cloning 2nd Edition; Sambrook et al., Cold Spring Harbor Laboratory Press に記載の方法に従って設定した。また、得られた種々のプラスミドベクターDNAについては、DNAシーケンサー(310 Genetic Analyser Applied Bio Systems, Inc.)を用いてDNA配列を決定した。また、以下に示すホモロジーアーム1および2はそれぞれ、本発明の第一および第二の相同DNA断片に対応している。
【0067】
実施例1: dhfr欠損CHO hprt組換えベクターの構築
dhfr欠損CHO hprt組換え基礎ベクターの構築
以下に記載の手法により、図1(A)で表されるベクター(dhfr欠損CHO hprt組換え基礎ベクター)を構築した。
【0068】
インビトロジェン社から入手したdhfr欠損チャイニーズハムスター卵母細胞株DG44細胞株をAMEM培地(組成;Advanced MEM(GIBCO)、5%[v/v]FBS、1× GlutaMAX(インビトロジェン)、1× HTサプリメント(インビトロジェン))を用いて、CO2インキュベーター(37℃、5% CO2)で培養した。得られた培養液を遠心分離し、DG44細胞ペレットを得た。このペレットをDNA Isolation Kit for Cells and Tissues (Roche Diagnostics K.K.)により処理し、ゲノムDNAを得た。次に、このゲノムDNAを鋳型とし、PCR(KOD−Plus ver.2、TOYOBO)によって、DG44細胞のhprt遺伝子座のDNA断片を2つ得た。プライマー配列は以下に示す通りである。
【0069】
ホモロジーアーム1センスプライマー:(配列番号1)
5’−TCGCGAgtctgtgtgtatgtttgtgataggc−3’
ホモロジーアーム1アンチセンスプライマー:(配列番号2)
5’−ACGCGTtgataaaatctacagtcatggg−3’
ホモロジーアーム2センスプライマー:(配列番号3)
5’−GGATCCgactgaagagctactgtgta−3’
ホモロジーアーム2アンチセンスプライマー:(配列番号4)
5’−TCGCGAtgaaggttatagagcataggggacc−3’
【0070】
上記PCR反応においては、ホモロジーアーム1センスプライマーの5’末端には、制限酵素Nru Iの認識サイトを付加した。同様に、ホモロジーアーム1アンチセンスプライマーの5’末端にはMlu Iの認識サイト、ホモロジーアーム2センスプライマーの5’末端にはBam HIの認識サイト、ホモロジーアーム2アンチセンスプライマーの5’末端にはNru Iの認識サイトをそれぞれ付加した。
【0071】
pQBI25プラスミドベクター(和光純薬株式会社)のDNAから、大腸菌内での複製起点であるori配列およびアンピシリン耐性遺伝子を含むDNA配列を、PCR反応によりクローニングした。PCR反応に用いたプライマー配列は以下に示す通りである。このPCR反応において、センスプライマー、アンチセンスプライマーの5’末端に、制限酵素Nru Iの認識サイトをそれぞれ付加した。
【0072】
Ecoliセンスプライマー:(配列番号5)
5’−TCGCGACCTGCAGGTCCGGAGCGGCCGCcatgtgagcaaaaggcca−3’
Ecoliアンチセンスプライマープライマー:(配列番号6)
5’−TCGCGACCTGCAGGTCCGGAGCGGCCGCaggtggcacttttcgggg−3’
【0073】
次に、リンカーsオリゴとリンカーaオリゴをアニールさせたDNAリンカーを準備した。
【0074】
リンカーsオリゴ:(配列番号7)
5’−CGCGTatcTCTAGAataATCGATagaAAGCTTacaG−3’
リンカーaオリゴ:(配列番号8)
5’− GATCCtgtAAGCTTtctATCGATtatTCTAGAgatA−3’
【0075】
ホモロジーアーム1、ホモロジーアーム2、およびori配列およびアンピシリン耐性遺伝子を含むDNA配列をそれぞれ、PCR反応によりクローニングした。これら3つのDNA配列を制限酵素Nru I、Mlu IおよびBam HIにより切断し、前記DNAリンカーとともにライゲーション反応を行うことによりpHA12プラスミドベクターを得た。
【0076】
dhfr発現カセットの作製
また、pQBI15プラスミドベクター(和光純薬株式会社)のDNAから、SV40プロモーター断片を、PCR反応により増幅した。PCR反応に用いたプライマー配列は以下の通りである。このPCR反応において、センスプライマーの5’末端に制限酵素Xba Iの認識サイトを、アンチセンスプライマーの5’末端にPci Iの認識サイトを付加した。
【0077】
SV40センスプライマー:(配列番号9)
5’−TCTAGActtctgaggcggaaagaacc−3’
SV40アンチセンスプライマー:(配列番号10)
5’−ACATGTcgaaacgatcctcatcctgt−3’
【0078】
次に、pSV2−dhfrプラスミド(ATCC、37146)のDNAから、dhfr断片をPCR反応により増幅した。PCR反応に用いたプライマー配列は以下の通りである。このPCR反応において、センスプライマーの5’末端に制限酵素Nco Iの認識サイトを、アンチセンスプライマーの5’末端にポリAシグナル配列およびCla Iの認識サイトを付加した。
【0079】
dhfrセンスプライマー:(配列番号11)
5’−CCatggttcgaccattgaactg−3’
dhfrアンチセンスプライマー:(配列番号12)
5’−ATCGATtCACACAAAAAACCAACACACAGATGTAATGAAAATAAAGATATTTTATTttagtctttcttctcgtagact−3’
【0080】
PCR増幅したSV40プロモーター断片をPci Iで、dhfr断片をNco Iでそれぞれ切断し、ライゲーション反応を行うことにより連結し、dhfr遺伝子カセットを得た。
【0081】
次に、pHA12プラスミドを制限酵素Xba I、Cla Iで切断し、前記dhfr遺伝子カセットを挿入し、dhfr欠損CHO hprt組換え基礎ベクターを得た。
【0082】
dhfr欠損CHO hprt組換え抗体発現ベクターの構築
以下に記載の手法により、抗体遺伝子をhprt遺伝子座に対して組み込むため、抗体遺伝子を含む、図1(B)で表されるベクター(dhfr欠損CHO hprt組換え抗体ベクター)を構築した。
【0083】
抗体重鎖発現カセットの構築
重鎖可変領域を保持したプラスミド(「Cloning of cDNA and Characterization of Anti−RNase A Monoclonal Antibody 3A21」:Journal of Fermentation and Bioengineering. Vol.82, No.3, pp. 312−314,1999)を鋳型とし、可変領域をIGH−3A21s、IGH−3A21a1プライマーセットでPCR増幅し、可変領域断片を得た。
【0084】
IGH−3A21sプライマー:(配列番号13)
5’− TAAAAGGTGTCCAGGATGTGCAGTTTCAGG −3’
IGH−3A21a1プライマー:(配列番号14)
5’− GAGGCCGATGAAACAGTGACCAGAGTCCCT −3’
【0085】
RPMI8226培養細胞からISOGEN(日本ジーン)で抽出したRNAを鋳型とし、One−step RT−PCR kit(QIAGEN)を用いてRT−PCRを行い、得られたmRNAを鋳型として定常領域をIGH−Cs、IGH−CaプライマーセットでPCR増幅し、定常領域断片を得た。
【0086】
IGH−Csプライマー:(配列番号15)
5’− CATCGGCCTCCACCAAGGGCCCATCGGTCT −3’
IGH−Caプライマー:(配列番号16)
5’− TTAAGCGGCCGCTCATTTACCCGGAGACAG −3’
【0087】
次に、IGH−LS、IGH−Vaプライマーセットで、可変領域断片を鋳型としてPCR増幅を行い、分泌シグナル付加可変領域断片を得た。
【0088】
IGH−LSプライマー:(配列番号17)
5’−GGTCGCCACCATGGAGTTTGGACTGAGCTGGGTTTTCCTTGTTGCTATTTTAAAAGGTGTCCAGGATGTG −3’
IGH−Vaプライマー:(配列番号18)
5’− GCCCTTGGTGGAGGCCGATGAAACAGTGAC −3’
【0089】
分泌シグナル付加可変領域断片および定常領域断片を鋳型とし、IGH−Vs、IGH−CaプライマーセットでアセンブルPCRを行い、両配列を連結させた抗体重鎖配列を得た。
【0090】
IGH−Vsプライマー:(配列番号19)
5’− TTCCGGTACCGGTCGCCACCATGGAGTTTG −3’
【0091】
次に、DG44ゲノムDNAを鋳型とし、EF1aP−MluI−F/EF1aP−XbaI−RプライマーセットでCHEF1αプロモーターを増幅した。
【0092】
EF1aP−MluI−Fプライマー:(配列番号20)
5’−AGAACGCGTCCACACAATCAGAACCACA−3’
EF1aP−XbaI−Rプライマー:(配列番号21)
5’−GACGATCTAGAGGTGGTTTTCACAACA−3’
【0093】
次に、得られたCHEF1αプロモーター、抗体重鎖配列およびSV40ポリAを連結し、両末端にMlu I認識配列を付加することにより、抗体重鎖発現カセットを得た。
【0094】
抗体軽鎖発現カセットの構築
同様に、軽鎖可変領域を保持したプラスミドを鋳型とし、可変領域をIGL−3A21s1、IGL−3A21a1プライマーセットでPCR増幅し、可変領域断片を得た。
【0095】
IGL−3A21s1プライマー:(配列番号22)
5’−CCCAGGTGCCAGATGTGACATCAAGATGAC−3’
IGL−3A21a1プライマー:(配列番号23)
5’−GCCACAGTTCGTTTTATTTCCAACTTTGTC−3’
【0096】
RPMI8226培養細胞からISOGENで抽出したRNAを鋳型とし、One−step RT−PCR kitを用いてRT−PCRを行い、得られたmRNAを鋳型として定常領域をIGL−Cs、IGL−CaプライマーセットでPCR増幅し、定常領域断片を得た。
【0097】
IGL−Csプライマー:(配列番号24)
5’−TGGAAATAAAACGAACTGTGGCTGCACCAT−3’
IGL−Caプライマー:(配列番号25)
5’−TTAAGCGGCCGCCTAACACTCTCCCCTGT−3’
【0098】
IGL−LS、IGL−3A21aプライマーセットで、DNA配列9を鋳型としてPCR増幅を行い、分泌シグナル付加可変領域断片を得た。
【0099】
IGL−LSプライマー:(配列番号26)
5’−GGTCGCCACCATGGACATGAGGGTCCCCGCTCAGCTCCTGGGGCTCCTGCTGCTCTGGCTCCCAGGTGCCAGATGTGACA −3’
IGL−3A21aプライマー:(配列番号27)
5’−GCCACAGTTCGTTTTATTTCCAACTTTGTC−3’
【0100】
分泌シグナル付加可変領域断片および定常領域断片を鋳型とし、IGL−Vs、IGL−CaプライマーセットでアセンブルPCRを行って両産物を連結させた抗体軽鎖配列を得た。
【0101】
IGL−Vsプライマー:(配列番号28)
5’− CCTTGGTACCGGTCGCCACCATGGACATGA −3’
【0102】
次に、CHEF1αプロモーター、抗体軽鎖配列およびSV40ポリAを連結し、両末端にHind III認識配列を付加することにより、抗体軽鎖発現カセットを得た。
【0103】
dhfr欠損CHO hprt組換え基礎ベクターを制限酵素Mlu Iで切断して抗体重鎖発現カセットを挿入し、次いで制限酵素Hind IIIで切断して抗体軽鎖カセットを挿入し、dhfr欠損CHO hprt組換え抗体発現ベクターを得た。
【0104】
実施例2:細胞へのベクターの導入
ベクター線状化
dhfr欠損CHO hprt組換え基礎ベクター、dhfr欠損CHO hprt組換え抗体発現ベクターは、Endofree Plasmid Maxi kit(QIAGEN社製)を用いて精製し、Nru Iにて切断した。2g/Lの濃度になるように滅菌水に溶解し、以下のトランスフェクション実験に用いた。
【0105】
トランスフェクションおよびスクリーニング
DG44細胞をNucleofection T solution(amaxa)で1×106細胞に調製し、線状化したプラスミドベクター2μgと混合した。次に、得られた混合液を用いて、NuceofectorII(amaxa)を用い、プログラムU−023で電圧印加した。トランスフェクションは各ベクターにつき、2回施行した。トランスフェクトした細胞は、100 細胞/ウェルにて96ウェルプレートに播種し、インキュベーター中37 ℃、5 % CO2にて培養した(培地: Advanced MEM(GIBCO社)に5% FBS、1x Glutamax(インビトロジェン社)、1x HTサプリメント(インビトロジェン社)を添加したもの)
【0106】
トランスフェクション後、HTサプリメントを除去し、6−TG(最終濃度; 50μM)(和光純薬工業)を添加した培地に交換し、培養を継続した。培養後、全ウェルを確認し、HTサプリメント非要求性/6TG耐性コロニーを単離した。
【0107】
結果、dhfr欠損CHO hprt組換え基礎ベクターによって得られたHTサプリメント非要求性/6TG耐性クローンは、2×106細胞あたり4クローンであった。
【0108】
実施例3:ゲノムPCRによる解析
DNA Isolation kit(ロシュ)を用い、dhfr欠損CHO hprt組換え基礎ベクターのトランスフェクションにより得たHTサプリメント非要求性および6TGに耐性のクローンからゲノムDNAを抽出し、このゲノムDNAを鋳型とした以下に示すPCR反応により、標的hprt遺伝子座への位置特異的な組換えの確認を行った。
【0109】
図2(A)は、ゲノムPCRによる相同組換え反応の解析の詳細を説明する模式図である。hprt遺伝子の相同組換えの標的領域は、エクソン3を含んで設定されており、この領域にベクターDNA中のホモロジーアーム1、ホモロジーアーム2およびこれに挟まれたdhfr遺伝子発現カセットは相同組換えにより組込まれる。
【0110】
ホモロジーアーム1と、dhfr遺伝子発現カセットの一部とを含むDNA(3640bpのDNA)および、ホモロジーアーム2と、dhfr遺伝子発現カセットの一部とを含むDNA(3285bpのDNA)は、相同組換えによってのみゲノムから取得することができ、相同組換えの指標となる。
【0111】
よって、ホモロジーアーム1と標的領域との相同組換えの指標として、図2(A)に示す3640bpのDNAを設定し、このDNAは、以下に示すプライマーCHA1seq−s11/dhfr−seq−a1によるPCR反応により検出した。一方、ホモロジーアームと標的hprt遺伝子座との相同組換えの指標として、図2(A)に示す3285bpのDNAを設定し、このDNAは、以下に示すプライマーNcoI−dhfr−s/ CHA2−seq−a4によるPCR反応により検出した。
【0112】
CHA1seq−s11プライマー:(配列番号29)
5’−GACACATGCAGACAGAACAG−3’
CHA2−seq−a4プライマー:(配列番号30)
5’−GTTTGCTAACACCCCTTCTC−3’
dhfr−seq−a1プライマー:(配列番号31)
5’−ctcaggaatggagaaccagg−3’
NcoI−dhfr−sプライマー:(配列番号32)
5’−ttaaCCatggttcgaccattgaactg−3’
【0113】
次に、得られたPCR増幅産物を1.0%アガロースゲル電気泳動により解析した。結果は、図2(C)に示される通りであった。図2(C)において、取得したクローンにおいてのみ、3640bpおよび3285bpの両方のPCR増幅産物が確認され、相同組換え反応が確認された。
【0114】
次に、dhfr欠損CHO hprt組換え基礎ベクターのトランスフェクションにより得たHTサプリメント非要求性および6TGに耐性のクローンのゲノムDNAについて、以下に示すPCR反応により、野生型hprt遺伝子が残存しているのかを確認した。
【0115】
図2(B)は、野生型hprt遺伝子の残存を確認するためのゲノムPCRの詳細を説明する模式である。まず、図2(B)左図は2本のX染色体を有する野生型DG44細胞におけるPCRを示す。hprt遺伝子の相同組換えの標的領域は、エクソン3を含んで設定されている。ベクター非導入の野生型細胞の場合、標的領域のDNA(2322bp)は、PCR反応によって増幅することができ、これを指標として野生型hprt遺伝子が残存していることを確認することができる。
【0116】
図2(B)の中央図は、1本のX染色体のhprt遺伝子座にベクターが組込まれたヘテロ接合型の相同組換え細胞におけるPCRを示す。一方の染色体では、標的領域中にベクターのDNAは相同組換えによりに組込まれているため、3867bpのPCR増幅産物が生じる。一方、もう1本の染色体は、標的領域を保持しており、標的領域のDNA(2322bp)もまた、PCR反応によって増幅することができ、野生型hprt遺伝子が残存していることを確認できる。
【0117】
一方、図2(B)の右図に示されるとおり、両方のX染色体にベクターが組込まれたホモ接合型の相同組換え細胞においては、組換えDNAに由来する3867bpのPCR増幅産物のみが発現し、野生型hprt遺伝子に由来するDNA(2322bp)の増幅は生じない。
【0118】
以上の理解から、図2(B)に示す2322bpのDNAを上記指標として設定し、このDNAを検出するため、プライマーCHPRTs/CHA2−seq−a1を用いてPCR反応を行った。
【0119】
次に、得られたPCR増幅産物を1.0%アガロースゲル電気泳動により解析した。結果は、図2(C)に示される通りであった。
【0120】
CHPRTsプライマー:(配列番号33)
5’−TGTTCCTGTGCATACTAGGC−3’
CHA2−seq−a1プライマー:(配列番号34)
5’−CCAGAGAAATTATTTGCCACCAGC−3’
【0121】
図2(C)において、野生型細胞からは2322bpのPCR増幅産物が確認され、野生型hprt遺伝子の存在が確認された。一方、組換えクローンにおいては2322bpのPCR増幅産物が確認されず、3867bpの産物のみが確認されたことから、組換え細胞は両方のX染色体にベクターが組込まれたホモ接合型であった。
【0122】
この結果から、本発明によるベクターを用いると、2本のX染色体の両hprt遺伝子座に目的タンパク質遺伝子が組込まれた組換え細胞クローンが高頻度で取得されていることが確認された。
【0123】
実施例4:DXB11(dhfr欠損CHO)hprt組換えクローン作製
DXB11へのベクター導入およびスクリーニング
実施例1で作製した、dhfr欠損CHO hprt組換え基礎ベクターを、実施例2と同様に精製および線状化し、3×106個のDXB11細胞にトランスフェクションし、スクリーニングを実施した。
【0124】
結果、HTサプリメント非要求性/6TG耐性のDXB11クローンを1クローン取得した。
【0125】
ゲノムPCRによる、DXB11のhprt遺伝子座へのベクター組込みの確認
実施例3と同様に、dhfr欠損CHO hprt組換え基礎ベクターのトランスフェクションにより得たHTサプリメント非要求性および6TGに耐性のDXB11クローンからゲノムDNAを抽出し、このゲノムDNAを鋳型とした以下に示すPCR反応により、標的hprt遺伝子座への位置特異的な組換えの確認を行った。
【0126】
図4(A)は、ゲノムPCRによる相同組換え反応の解析の詳細を説明する模式図である。
【0127】
実施例3でのDG44の場合と全く同様に、DXB11のhprt遺伝子の相同組換えの標的領域は、エクソン3を含んで設定されており、この領域にベクターDNA中のホモロジーアーム1、ホモロジーアーム2およびこれに挟まれたdhfr遺伝子発現カセットは相同組換えにより組込まれる。
【0128】
ホモロジーアーム1と、dhfr遺伝子発現カセットの一部とを含むDNA(3640bpのDNA)および、ホモロジーアーム2と、dhfr遺伝子発現カセットの一部とを含むDNA(3285bpのDNA)は、相同組換えによってのみゲノムから取得することができ、相同組換えの指標となる。これらのDNAは、取得したDXB11クローンのゲノムDNAを鋳型とした、実施例3と同一のプライマーセットによるPCR反応によって検出した。
【0129】
PCR増幅産物を1.0%アガロースゲル電気泳動により解析した結果は、図4(B)に示される通りであり、取得したDXB11クローンにおいて3640bpおよび3285bpの両方のPCR増幅産物が確認され、相同組換え反応が確認された。
【0130】
以上の結果から、DXB11のhprt遺伝子座にベクターが組み込まれたクローンが取得できたことが確認できた。
【0131】
実施例5:dhfr欠損CHO hprt組換え抗体発現ベクタークローン作製および抗体生産
dhfr欠損CHO hprt組換え抗体発現クローン作製
実施例1で作製した、dhfr欠損CHO hprt組換え抗体発現ベクターを、実施例2と同様に精製および線状化し、3×106個のDG44細胞にトランスフェクションし、スクリーニングを実施した。
【0132】
結果、dhfr欠損CHO hprt組換え抗体発現ベクターによって、HTサプリメント非要求性/6TG耐性クローンを1クローン取得した。
【0133】
取得クローンからゲノムDNAを抽出し、このゲノムDNAを鋳型としたPCRを実施例2と同様に実施し、hprt遺伝子座にdhfr欠損CHO hprt組換え抗体発現ベクターが組み込まれていることを確認した。
【0134】
dhfr欠損CHO hprt組換え抗体発現クローンの無血清浮遊馴化
100mmディッシュに捲き込んだDG44 dhfr欠損CHO hprt組換え抗体発現ベクタークローンを、70%コンフルエントまで培養した後、トリプシン処理によって剥離した。
【0135】
1000rpm、5分間の遠心によって細胞を沈殿させ、上澄みを破棄した後、完全CD OptiCHO培地(CD OptiCHO(インビトロジェン社)に1x Glutamax(インビトロジェン社)を添加したもの)で3×105cells/mlに調整してフラスコに移し、インキュベーター中37℃、8%CO2にて、121rpmで振とう培養した。
【0136】
細胞密度が1×106cells/mlを超えた時点で、完全CD OptiCHOで3×105cells/mlに希釈継代し、培養を継続した。
【0137】
上記継代を3回繰り返した時点で、無血清浮遊培養への馴化を完了した。
【0138】
dhfr欠損CHO hprt組換え抗体発現クローンによる抗体生産
無血清浮遊培養に馴化したDG44 dhfr欠損CHO hprt組換え抗体発現ベクタークローンを、1000rpm、5分間の遠心によってペレット化し、新鮮な完全CD OptiCHO培地に懸濁した。この細胞懸濁液を完全CD OptiCHO培地で希釈し、60mlの3×105cells/ml細胞液を調整し、フラスコに移した。
【0139】
インキュベーター中37℃、8%CO2にて、121rpmで振とう培養し、24時間ごとに細胞密度を測定して記録した。また、1.5mlの培地を回収し、遠心した上澄みを−80℃で保存した。
【0140】
18日間のサンプリングを実施後、回収しておいた培地サンプル回収した培地中のIgG量を、Human IgG EIA Kit (precoated)(宝酒造製)により、450nmの吸光度を測定することにより定量した。
【0141】
その結果、図5に示すように、培地上澄1Lあたり35mgのIgG生産が認められた。
【0142】
dhfr欠損CHO hprt組換え抗体発現クローンの発現の長期安定性確認
無血清浮遊培養に馴化したDG44 dhfr欠損CHO hprt組換え抗体発現ベクタークローンを、14回継代培養して維持した。継代操作は1週間あたり2回実施した。その後、細胞集団の一部を、新鮮な完全CD OptiCHO培地60ml中に3×105cells/mlになるように調整し、フラスコに移し、培養を開始した。
【0143】
培養開始後、7日目に培地中の細胞密度を測定した後、培地上澄1.5mlを採取し、遠心分離した上澄みをー80℃保存した。24時間後、細胞密度を測定し、培地上澄1.5mlを同様に保存した。
【0144】
保存しておいた培地上澄サンプルを溶解し、Human IgG EIA KitでIgG量を定量し、細胞密度を用いて比生産性(pg/cell/day)を算出した。また、無血清浮遊培養に馴化した直後のDG44 dhfr欠損CHO hprt組換え抗体発現ベクタークローンについても、同様に比生産性を測定した。
【0145】
14回の継代操作後のクローンの比生産性は、図6に示すように全く低下していなかった。これは、実生産で最低限要求される50日間(約7週間)の安定性を満足する、好ましい結果と言える。
【特許請求の範囲】
【請求項1】
5’末端から3’末端に向かって、第一の相同DNA断片、目的タンパク質遺伝子、選択マーカー遺伝子、および第二の相同DNA断片を含んでなるDNA構築物であって、
前記第一の相同DNA断片および第二の相同DNA断片が、dhfr遺伝子欠損CHO細胞ゲノムのヒポキサンチン−ホスホリボシルトランスフェラーゼ酵素(hprt)遺伝子座の一部と相同組換え可能な相同性を有し、かつ1kbp以上の鎖長を有するものであり、前記選択マーカー遺伝子がジヒドロ葉酸還元酵素遺伝子(dhfr)であることを特徴とする、DNA構築物。
【請求項2】
前記第一の相同DNA断片および第二の相同DNA断片が1kbp以上7.5kbp以下の鎖長を有するものである、請求項1に記載のDNA構築物。
【請求項3】
前記第一の相同DNA断片および第二の相同DNA断片が1kbp以上5kbp以下の鎖長を有するものである、請求項1に記載のDNA構築物。
【請求項4】
前記hprt遺伝子座の一部が、hprt遺伝子のイントロンの少なくとも一部を含んでなる領域である、請求項1〜3の何れか一項に記載のDNA構築物。
【請求項5】
前記第一の相同DNA断片または第二の相同DNA断片が、配列番号35〜42のいずれかに記載の塩基配列またはその部分配列を含んでなるものである、請求項1〜4の何れか一項に記載のDNA構築物。
【請求項6】
前記目的タンパク質遺伝子に作動可能に連結されたCHEF−1α遺伝子プロモーターを含んでなる、請求項1〜6の何れか一項に記載のDNA構築物。
【請求項7】
請求項1〜6の何れか一項に記載のDNA構築物を含んでなる、ベクター。
【請求項8】
前記ベクターが、プラスミドベクター、コスミドベクター、ファージベクターまたは人工染色体ベクターである、請求項7に記載のベクター。
【請求項9】
請求項8に記載のベクターを、dhfr遺伝子を欠損したCHO細胞に導入し、ヒポキサンチン、チミジン非要求性および6−チオグアニン(6TG)耐性を指標として、hprt遺伝子座にベクターが組込まれた細胞を選択することを含んでなる、組換えCHO細胞の製造方法。
【請求項10】
請求項9に記載の製造方法により得られる、組換えCHO細胞。
【請求項11】
請求項10に記載の組換えCHO細胞を用意し、該細胞を培養して目的タンパク質を産生することを含んでなる、目的タンパク質の製造方法。
【請求項1】
5’末端から3’末端に向かって、第一の相同DNA断片、目的タンパク質遺伝子、選択マーカー遺伝子、および第二の相同DNA断片を含んでなるDNA構築物であって、
前記第一の相同DNA断片および第二の相同DNA断片が、dhfr遺伝子欠損CHO細胞ゲノムのヒポキサンチン−ホスホリボシルトランスフェラーゼ酵素(hprt)遺伝子座の一部と相同組換え可能な相同性を有し、かつ1kbp以上の鎖長を有するものであり、前記選択マーカー遺伝子がジヒドロ葉酸還元酵素遺伝子(dhfr)であることを特徴とする、DNA構築物。
【請求項2】
前記第一の相同DNA断片および第二の相同DNA断片が1kbp以上7.5kbp以下の鎖長を有するものである、請求項1に記載のDNA構築物。
【請求項3】
前記第一の相同DNA断片および第二の相同DNA断片が1kbp以上5kbp以下の鎖長を有するものである、請求項1に記載のDNA構築物。
【請求項4】
前記hprt遺伝子座の一部が、hprt遺伝子のイントロンの少なくとも一部を含んでなる領域である、請求項1〜3の何れか一項に記載のDNA構築物。
【請求項5】
前記第一の相同DNA断片または第二の相同DNA断片が、配列番号35〜42のいずれかに記載の塩基配列またはその部分配列を含んでなるものである、請求項1〜4の何れか一項に記載のDNA構築物。
【請求項6】
前記目的タンパク質遺伝子に作動可能に連結されたCHEF−1α遺伝子プロモーターを含んでなる、請求項1〜6の何れか一項に記載のDNA構築物。
【請求項7】
請求項1〜6の何れか一項に記載のDNA構築物を含んでなる、ベクター。
【請求項8】
前記ベクターが、プラスミドベクター、コスミドベクター、ファージベクターまたは人工染色体ベクターである、請求項7に記載のベクター。
【請求項9】
請求項8に記載のベクターを、dhfr遺伝子を欠損したCHO細胞に導入し、ヒポキサンチン、チミジン非要求性および6−チオグアニン(6TG)耐性を指標として、hprt遺伝子座にベクターが組込まれた細胞を選択することを含んでなる、組換えCHO細胞の製造方法。
【請求項10】
請求項9に記載の製造方法により得られる、組換えCHO細胞。
【請求項11】
請求項10に記載の組換えCHO細胞を用意し、該細胞を培養して目的タンパク質を産生することを含んでなる、目的タンパク質の製造方法。
【図1】


【図3】
【図5】
【図6】
【図2】
【図4】
【図3】
【図5】
【図6】
【図2】
【図4】
【公開番号】特開2012−196208(P2012−196208A)
【公開日】平成24年10月18日(2012.10.18)
【国際特許分類】
【出願番号】特願2012−51980(P2012−51980)
【出願日】平成24年3月8日(2012.3.8)
【出願人】(000010087)TOTO株式会社 (3,889)
【Fターム(参考)】
【公開日】平成24年10月18日(2012.10.18)
【国際特許分類】
【出願日】平成24年3月8日(2012.3.8)
【出願人】(000010087)TOTO株式会社 (3,889)
【Fターム(参考)】
[ Back to top ]