
Eph受容体活性を調節する新規薬剤
Eph受容体に結合する新規薬剤を記載する。これらの薬剤を使用してEph受容体の活性を調節し、アポトーシスを活性化し、および治療剤を送達する方法も記載する。また、Eph受容体に選択的に結合できる薬剤のスクリーニング方法も記載する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、Eph受容体機能を調節する新規リガンドに関する。
【0002】
[米国政府の所有権]
本発明は、米国国立衛生研究所により与えられた助成金番号CA82713およびNS43029ならびに米国防総省により与えられた助成金番号DAMD17−01−1−0168のもと、米国政府の援助によりなされた。米国政府は、本発明において一定の権利を有する。
【0003】
[関連技術の記載]
Eph受容体は、そのエフリンリガンドに結合すると活発にシグナル伝達を行なう、密接に関連する膜貫通型チロシンキナーゼの大きな一ファミリーを構成する(フラナガン(Flanagan) & バンデルヘーゲン(Vanderhaeghen) 1998 Ann Rev Neurosci 21:309-345); マニング(Manning)他2002 Trends Biochem Sci 27:514-520; ムライ(Murai), K. & パスクワレ(Pasquale), E. 2003 J. Cell Sci 116:2823-2832)。16個のEph受容体は、配列相同性に基づいて2つのサブグループ(EphAおよびEphB)に分けられる。EphA受容体は、GPI結合エフリン−Aリガンドと結合するが、EphB受容体は、膜貫通型エフリン−Bリガンドと結合する。
【0004】
もともとは神経発達の調節因子として同定された、Ephファミリーの受容体チロシンキナーゼおよびそのエフリンリガンドはまた、血管発達および病的な血管新生にも重要である(フラナガン, J. G. & バンデルヘーゲン P. 1998 Ann Rev Neurosci 21:309-345); ドデレット(Dodelet), V. C. & パスクワレ, E. B. 2000 Oncogene 19:5614-5619; ヤンコポウラス(Yancopoulos), G. D.他2000 Nature 407:242-248)。例えば、EphA2受容体およびエフリン−A1(EphA2のリガンド)は、ヒト腫瘍およびヒト癌細胞から成長させたマウス異種移植腫瘍の血管系において、協調的に発現する(オガワ(Ogawa),
K.他2000 Oncogene 19:6043-6052)。EphA2受容体は、血管内皮成長因子(VEGF)および腫瘍壊死因子α(TNFα)誘導性血管新生において重要な役割を果たす。これは、VEGFおよびTNFαがエフリン−A1をアップレギュレートし、このことが、血管における受容体活性化を引き起こすからである(パンデイ(Pandey), A.他1995 Science 268:567-569; チェン(Cheng), N.他2002 Mol Cancer Res 1:2-11)。同様に、ホメオボックス転写因子Hox B3は、エフリン−A1をアップレギュレートすることにより、血管新生を促進する(マイアーズ(Myers), C.他2000 J Cell Biol 148:343-351)。さらにまた、EphA2シグナル伝達は、インビトロ法での内皮毛細血管の形成に必要とされ(オガワ, K.他2000 Oncogene 19:6043-6052; ダニエル(Daniel), T. O.他1996 Kidney Int Suppl 57:S73-81)、黒色腫細胞による血管様構造の形成を促進する(ヘス(Hess), A. R.他2001 Cancer Res 61:3250-3255)。EphA2の発現は、受容体が胚血管または成体の沈静状態血管のいずれにおいても検出されていないため、「活性化された」成体血管に限定されているようである(オガワ, K.他2000 Oncogene 19:6043-6052; ルイス(Ruiz), J.
C. & ロバートソン(Robertson), E. J. 1994 Mech Dev 46:87-100; ガンジュ(Ganju), P.他1994 Oncogene 9:1613-1624)。エフリン−A1もまた成体血管においては検出されていないが、胎児の血管構造には存在する(マクブライド(McBride), J. L. & ルイス, J. C. 1998 Mech Dev 77:201-204)。
【0005】
腫瘍内皮細胞に存在する以外に、EphA2およびエフリン−A1は、乳癌、前立腺癌、結腸癌、皮膚癌および食道癌を含む多種多様な腫瘍の形質転換細胞内でアップレギュレートされる(オガワ, K.他2000 Oncogene 19:6043-6052; ゼリンスキー(Zelinski), D. P
.他2001 Cancer Res 61:2301-2306; ウォーカー−ダニエル(Walker-Daniels), J.他1999 Prostate 41:275-28; イースティー(Easty), D. J.他1995 Int J Cancer 60:129-136; ネモト(Nemoto), T.他1997 Pathobiology 65:195-203)。癌遺伝子H−Ras、E−カドヘリン、転写調節因子のp53ファミリーメンバー、DNA損傷、ならびにエストロゲン受容体およびc−Mycの減少を含む多くの因子が、癌細胞におけるEphA2発現を増加させる(アンドレス(Andres), A. C.他1994 Oncogene 9:1461-1467; ドーン(Dohn), M他2001 Oncogene 20:6503-6515; ゼリンスキー, D. P.他2002 J Cell Biochem 85:714-720)。
【0006】
腫瘍血管系は、本質的に不連続で漏出性であるため、血管と腫瘍細胞の両方に癌根絶剤を送達するのにEphA2およびエフリン−A1のアップレギュレートを利用することが可能である(ドボラク(Dvorak), H. F.他1988 Am J Pathol 133:95-109)。実際、全身投与された生物学製剤は、血液循環から腫瘍に容易に浸透する(エスラー(Essler), M. & ルオスラーティ(Ruoslahti), E. 2002 PNAS USA 99:2252-2257)。しかしながら、EphA2およびエフリン−A1を選択的に標的化することは、これらのタンパク質が、密接に関連するタンパク質から成る大きなファミリーに属するものであるため、困難な課題である(Eph-Nomenclature-Committee Unified nomenclature for Eph family receptors and
their ligands, the ephrins 1997 Cell 90:403-404)。
【0007】
EphA4は、Ephファミリーの受容体チロシンキナーゼの別のメンバーである。これは、発達中および成体の神経系において重要な機能を有する。神経発達時のその既知の発現パターンの他に(モリ(Mori), T.他1995 Brain Res Mol Brain Res 29:325-335; ニエト(Nieto), M. A.他1992 Development 116:1137-1150; オオタ(Ohta), K.他1996 Mechanisms of Development 54:59-69; ソーンズ(Soans), C.他1994 Oncogene 9:3353-3361)、EphA4は、広範なシナプス再構築を示す脳領域で発現している(ムライ, K.他2003 Nature Neurosci 6:153-169)。成体では、EphA4は、学習と記憶に重要な脳組織である海馬および皮質において富んでいる。
【0008】
EphA5およびEphA7は、Eph4と密接に関連する受容体であるが、発達中の神経系および成体神経系において異なった形で発現する2つのEphA受容体である(エリス(Ellis), J.他1995 Mechanisms of Development 52:319-341; モリ, T.他1995 Brain
Res Mol Brain Res 34:154-160; オリビエリ(Olivieri), G. & ミーシャー(Miescher), G. C.他1999 J Histochem Cytochem 47:855-861; チャン(Zhang), J. H.他1997 Brain Res Mol Brain Res 47:202-214)。例えば、EphA5受容体は、多くのヒト神経膠腫および神経膠芽腫細胞株において過剰発現する(ブルース(Bruce), V.他1999 Brain Res 821:169-176; ミーシャー, G. C.他1997 Mol Brain Res 46:17-24)。
【0009】
EphB2およびEphB4受容体は、ともに、或る種の腫瘍組織において過剰発現する。EphB4過剰発現は、主に、悪性度の高い浸潤性乳管癌腫において見られるが(ベルクラス(Berclaz), G.他1996 Biochem Biophys Res Commun 226:869-875)、EphB2は、たいていの胃腫瘍において過剰発現する(キヨカワ(Kiyuokawa), E.他1994 Cancer Res 54:3645-3650 )。両受容体は、多くの腫瘍細胞株でも同様に過剰発現している(ベルクラス, G.他1996 Biochem Biophys Res Commun 226:869-875; キヨカワ, E.他1994 Cancer Res 54:3645-3650; ベネット(Bennett), B. D.他1995 PNAS USA 92:1866-1870)。また、両EphB2およびEphB4は、結腸癌腫組織においてアップレギュレートされる(リウ(Liu), W.他2002 Cancer 94:934-939; ステフェンソン(Stephenson), S.他2001 BMC Mol Biol 2:15-23)。さらに、EphB2およびEphB4はまた、胚およびおそらく腫瘍における血管系発達に重要である(ワン(Wang), H. U.他1998 Cell 93:741-753; ゲレティ(Gerety), S. S.他1999 Mol Cell 4:403-414)。
【0010】
異なるアゴニスト活性および薬物標的化活性を有するEph受容体結合ペプチドが同定されれば、重要な治療法になろう。例えば、生体活性型エフリン擬似ペプチドは、Eph受容体発現組織に薬剤を選択的に送達し、また、癌、病的血管新生、神経再生および認知障害の治療におけるEphシグナル伝達を変更するために使用し得る。同定されたEph結合ペプチドの他の考えられ得る適用としては、負傷による慢性的な痛みの治療(バッタリア(Battaglia), A. A.他2003 Nature Neurosci 6:339-340)、成体脳における神経芽細胞の増殖の刺激(コノバー(Conover), J. C.他2000 Nature Neurosci 3:1091-1097)、および学習および記憶において重要な役割を有すると考えられるシナプス可塑性の調節(ムライ, K. K. & パスクワレ, E. B. 2002 Neuron 33:159-162)が挙げられる。
【発明の開示】
【0011】
本発明の一実施形態は、Eph受容体ファミリーのメンバーすなわちこれら受容体のサブセットに選択的に結合する化合物である。
【0012】
本発明の別の実施形態は、単離されたペプチド、擬似ペプチド、またはEph受容体ファミリーのメンバーすなわちこれら受容体のサブセットに選択的に結合する低分子である。
【0013】
本発明の別の実施形態は、ペプチド、擬似ペプチド、またはEph受容体ファミリーメンバーに選択的に結合する低分子を含むEph受容体結合化合物の有効量と細胞を接触させることによる、細胞中のEph受容体活性の調節方法である。Eph受容体結合化合物は、アゴニストまたはアンタゴニストのいずれかであり得る。或る特定の実施形態では、EphA2、EphA4、EphA5、EphA7、EphB2およびEphB4を含むEph受容体が、インビトロまたはインビボのいずれかで調節され得る。
【0014】
本発明の別の実施形態は、哺乳動物に、プログラム細胞死(アポトーシス)を活性化するのに充分な量のEph受容体アゴニストまたはアンタゴニストを投与することによる、プログラム細胞死を促進する方法である。Eph受容体アゴニストまたはアンタゴニストとしては、ペプチド、擬似ペプチド、またはEph受容体ファミリーメンバーに選択的に結合する低分子が含まれる。Eph受容体アゴニストまたはアンタゴニストは、正常細胞および腫瘍性細胞の両方に送達され得る。或る実施形態では、アゴニストがEph受容体のリン酸化を活性化する一方、アンタゴニストがEph受容体のリン酸化を阻害する。別の実施形態では、アゴニストまたはアンタゴニストが、エフリンリガンドのEph受容体への結合を阻害する。
【0015】
本発明の別の実施形態は、神経再生の活性化および促進、ならびに外傷性損傷、精神遅滞および神経変性疾患によるニューロン変性の改善を行なう方法である。この方法は、哺乳動物に、該哺乳動物において神経再生を促進するのに有効な量のEph受容体アゴニストを投与することを含み、該アゴニストとしては、ペプチド、擬似ペプチド、またはEph受容体ファミリーメンバーに選択的に結合する低分子が含まれる。
【0016】
本発明の別の実施形態は、哺乳動物に、認知機能を調節するのに有効な量のEph受容体アゴニストまたはアンタゴニストを投与することによる、認知機能の調節方法であり、ここで、アゴニストまたはアンタゴニストには、ペプチド、擬似ペプチド、またはEph受容体ファミリーメンバーに選択的に結合する低分子が含まれる。
【0017】
本発明の別の実施形態は、哺乳動物に、血液凝固プロセスを変更するのに有効な量のEph受容体アゴニストを投与することによる、血液凝固プロセスを変更する方法であり、ここで、上記アゴニストとしては、ペプチド、擬似ペプチド、またはEph受容体ファミリーメンバーに選択的に結合する低分子が含まれる。
【0018】
本発明のさらなる実施形態は、細胞への治療剤の送達方法である。該治療剤は、ペプチド、擬似ペプチド、またはEph受容体ファミリーメンバーに選択的に結合する低分子を含むEph受容体結合化合物と連結されている。
【0019】
本発明のさらに別の実施形態は、Eph受容体ファミリーメンバーに選択的に結合するEph受容体結合化合物と連結された治療剤を含有するコンジュゲートである。或る実施形態では、治療剤はイメージング剤であり得る。
【0020】
本発明の別の実施形態は、固定化したEph受容体に対して、ランダム配列を有するペプチドをコードするファージ封入(phage-encapsulated)核酸を含むファージディスプレイライブラリーをパニング(panning)することによる、Eph受容体結合化合物の同定方法である。次いで、固定化した受容体に結合するファージクローンを同定する。
【0021】
本発明の別の実施形態は、Eph受容体に結合しているEph受容体結合因子を提供し、試験化合物または試験化合物のライブラリーを提供し、Eph受容体からEph受容体結合因子を解離させることができる化合物を同定することによる、Eph受容体結合リガンドの同定方法である。この方法により同定された化合物を、さらに他のEph受容体およびEph受容体結合因子に対してスクリーニングし、選択的Eph受容体結合リガンドを同定してもよい。
【0022】
本発明の他の実施形態は、Eph受容体への化合物の結合を測定するための分光技法を用いて化合物をスクリーニングする方法を提供する。化合物は、アルカリホスファターゼアッセイ、核磁気共鳴(NMR)結合アッセイ、蛍光偏光アッセイ(FPA)およびコンピュータによる結合の予測(Computational-Docking)試験の組合せを用いることにより、スクリーニングしてもよい。
【0023】
[発明の詳細な説明]
Eph受容体チロシンキナーゼのファミリーは、病変組織と正常組織において異なった形で発現するため、有望な疾患標的である。例えば、EphA2受容体は、形質転換細胞および腫瘍の血管系においてアップレギュレートされ、ここで、癌の病原性に寄与すると思われる。EphB4は、種々の黒色腫および癌腫において過剰発現する。EphA4は、血管疾患および場合によっては慢性関節リウマチの発症に、或る役割を果たし得る(プレボー(Prevost)他, 2002. PNAS 99:9219-9224参照)。
【0024】
Eph受容体を治療標的として利用するため、ファージディスプレイを用いて、Eph受容体ファミリーの特定のメンバーに選択的に結合するペプチドを同定した。かかるペプチドのいくつかを本明細書に記載する。例えば、EphA2に高親和性(マイクロモル未満のKD)で選択的に結合するペプチドを単離した。EphB2、EphB4、EphA4、EphA5およびEphA7に選択的に結合する別のペプチドを見出した。或る場合において、これらのペプチドは、単一の受容体でなくEph受容体のサブセットに結合する。EphA2の場合、2種類のペプチドがEphA2のリガンド結合ドメインを標的化し、結合に関してエフリンリガンドと競合することがわかった。これらのペプチドは、EphA2チロシンリン酸化および受容体による下流シグナル伝達を刺激するという点で、エフリン様活性を有する。さらにまた、これらのペプチドの1つは、EphA2を発現する内皮および腫瘍細胞にファージ粒子を送達することができる。これに対し、エフリンリガンドの受容体相互作用部分に相当するペプチドは、多くのEph受容体に対し、弱く、無差別に結合する。3種類のEphA4結合ペプチドおよびEphB2結合ペプチドもまた、これらの受容体への結合に関してエフリンリガンドと競合した。しかしながら、これらのペプチドは、受容体を活性化せず、したがって、アンタゴニストとして作用するよう
である。生体活性型エフリン擬似ペプチドを用いて、Eph受容体発現組織に薬剤を選択的に送達すること、および、Ephシグナル伝達を変更して、癌、病的血管新成および神経変性の治療に利用できる。
【0025】
定義
以下の定義は、本明細書に記載した本発明を説明するために使用した種々の用語の意味および範囲を例示および明確にするために記載する。
【0026】
本明細書で使用する場合、「アゴニスト」は、その相補的な生体活性受容体に結合して受容体を活性化し、該受容体において生物学的応答を引き起こすか、または該受容体の既存の生体活性を増強するかのいずれかである生物活性リガンドをいう。
【0027】
本明細書で使用する場合、「アンタゴニスト」は、その相補的な生体活性受容体に結合し、該受容体の生理学的応答を阻害する生物活性リガンドをいう。
【0028】
本明細書で使用する場合、「製薬的に許容され得る塩」は、製薬業界で一般的に使用されている無毒性のアルカリ金属、アルカリ土類金属およびアンモニウム塩をいい、ナトリウム、カリウム、リチウム、カルシウム、マグネシウム、バリウム、アンモニウムおよびプロタミン亜鉛の塩が挙げられ、これらは、当該技術分野で既知の方法により調製される。また、この用語は、本発明の化合物を適当な有機酸または無機酸と反応させることにより一般的に調製される無毒性の酸付加塩も包含する。代表的な塩としては、塩酸塩、臭化水素酸塩、硫酸塩、重硫酸塩、酢酸塩、シュウ酸塩、吉草酸塩、オレイン酸塩、ラウリン酸塩、ホウ酸塩、安息香酸塩、乳酸塩、リン酸塩、トシル酸塩、クエン酸塩、マレイン酸塩、フマル酸塩、コハク酸塩、酒石酸塩、ナプシル酸塩などが挙げられる。
【0029】
本明細書で使用する場合、「製薬的に許容され得る酸付加塩」は、遊離塩基の生体有効性および特性を維持し、かつ生物学的またはその他の点で望ましくない点のないものであり、塩酸、臭化水素酸、硫酸、硝酸、リン酸などの無機酸、および酢酸、プロピオン酸、グリコール酸、ピルビン酸、シュウ酸、リンゴ酸、マロン酸、コハク酸、マレイン酸、フマル酸、酒石酸、クエン酸、安息香酸、桂皮酸、マンデル酸、メタンスルホン酸(menthanesulfonic acid)、エタンスルホン酸、p−トルエンスルホン酸、サリチル酸などの有機酸と形成された塩をいう。プロドラッグとしての製薬的に許容され得る酸付加塩の説明については、バンガード(Bundgaard), H.編, 1985 Design of Prodrugs, Elsevier Science
Publishers,(アムステルダム)を参照のこと。
【0030】
本明細書で使用する場合、「製薬的に許容され得るエステル」は、エステル結合の加水分解時に、カルボン酸またはアルコールの生体有効性および特性を維持し、生物学的またはその他の点で望ましくないものでないエステルをいう。プロドラッグとしての製薬的に許容され得るエステルの説明については、バンガード, H.編, 1985 Design of Prodrugs,
Elsevier Science Publishers (アムステルダム)を参照のこと。これらのエステルは、通常、対応するカルボン酸と、アルコールとから形成される。一般的に、エステルの形成は、従来の合成技法によりなされ得る。例えば、1992年3月, Advanced Organic Chemistry, 第4版, John Wiley & Sons (ニューヨーク), 第393−396頁およびそこに挙げられる参考文献、ならびにマーク(Mark)他1980 Encyclopedia of Chemical Technology, John Wiley & Sons (ニューヨーク)を参照のこと。エステルのアルコール成分としては、一般的に、(i)1つ以上の二重結合を含んでいても含んでいなくてもよく、分枝炭素を含んでいても含んでいなくてもよいC2−C12脂肪族アルコール、または(ii)C7−C12芳香族アルコールもしくは複素環芳香族アルコールが含まれる。また、本発明は、本明細書に記載したエステルであると同時に製薬的に許容され得るその酸付加塩でもあるこれらの組成物の使用も意図している。
【0031】
本明細書で使用する場合、「製薬的に許容され得るアミド」は、アミド結合の加水分解時に、カルボン酸またはアミンの生体有効性および特性を維持し、生物学的またはその他の点で望ましくない点のないアミドをいう。プロドラッグとしての製薬的に許容され得るアミドの説明については、バンガード, H.編, 1985 Design of Prodrugs, Elsevier Science Publishers (アムステルダム)を参照のこと。これらのアミドは、通常、対応するカルボン酸と、アミンとから形成される。一般的に、アミドの形成は、従来の合成技法によりなされ得る。例えば、1992年3月, Advanced Organic Chemistry, 第4版, John Wiley & Sons (ニューヨーク), 第393頁およびマーク他1980 Encyclopedia of Chemical Technology, John Wiley & Sons (ニューヨーク)を参照のこと。また、本発明では、本明細書に記載したアミドであると同時に製薬的に許容され得るその酸付加塩でもあるこれらの組成物の使用も意図される。
【0032】
本明細書で使用する場合、「製薬的または治療的に許容され得る担体」は、活性成分の生体活性の有効性を妨げず、かつ宿主または患者に毒性を持たない担体をいう。
【0033】
本明細書で使用する場合、「立体異性体」は、別の分子と同じ分子量、化学組成および結合順を有するが、1つ以上のキラル中心に関して空間的配置の異なる原子を有する化合物をいう。すなわち、同じ化学式を有する立体異性体は、少なくとも1つのキラル中心に関しては、空間的に異なる位置にある同一の化学物質部分を含む。純粋な場合、立体異性体は、平面偏光を回転させる能力を有する。しかしながら、一部の純粋な立体異性体は、非常にわずかな旋光性しか持たないため既存の器機では検出できない場合がある。本発明の化合物は、1つ以上の不斉炭素原子を有していてもよく、したがって、種々の立体異性体を包含する。すべての立体異性体は、本発明の範囲内に含まれる。
【0034】
本明細書で使用する場合、本発明の組成物に適用される「治療的有効量または製薬的有効量」は、所望の生物学的結果を誘導するのに充分な組成物の量をいう。ここでいう結果とは、徴候、症状もしくは疾患の原因の緩和、または生体系のあらゆる所望の改変であり得る。本発明では、この成果は、例えば、癌性細胞成長の阻害および反転(reversal)を含む。
【0035】
本明細書で使用する場合、用語「ペプチド化合物」および「ペプチド構造」は、天然型L−アミノ酸からなるペプチド、ならびに天然型L−アミノ酸構造のペプチド誘導体、ペプチドアナログおよび擬似ペプチドを含むものとする。用語「ペプチドアナログ」、「ペプチド誘導体」および「擬似ペプチド」は、本明細書で使用する場合、ペプチドの化学構造を擬態し、そのペプチドの機能的特性を保持した分子を含むものとする。ペプチドのアナログ、誘導体および擬似物を設計するためのアプローチは、当該技術分野で既知である。例えば、Drug Design E. J. アリエンス(Ariens)編 Academic Press(ニューヨーク),
1980, 第10巻, 第119−143頁のファーマー(Farmer), P. S.; ボール(Ball) J. B. & アレウッド(Alewood), P. F. 1990 J Mol Recognition 3:55; モーガン(Morgan), B. A. & ゲイナー(Gainor), J. A. 1989 Ann Rep Med Chem 24:243; およびフライディンガー(Freidinger), R. M. 1989 Trends Pharmacol Sci 10:270; ルスマン(Luthman)他1996 A TextBook of Drug Design and Development, 14:386-406, 第2版, Harwood Academic Publihers; ヨアキム グランテ(Joachim Grante), アンギュー(Angew). 1994 Chem Int Ed Engl 33:1699-1720; フォーシェア(Fauchere). J. 1986 Adv Drug Res 15:29; ベベール(Veber)およびフライディンガー 1985 TINS第392頁; エバンス(Evans)他1987 J Med Chem 30:229を参照のこと。治療的に有用なペプチドに構造的に類似する擬似ペプチドは、同等または向上した治療効果または予防効果を得るために使用され得る。一般的に、擬似ペプチドは、天然型受容体結合ポリペプチドなどの模範(paradigm)ポリペプチド(すなわち、生物学的または薬理学的活性を有するポリペプチド)に構造的に類似するが、当
該技術分野で既知の方法により、−CH2NH−、−CH2S−、−CH2−CH2−、−CH=CH−(シスおよびトランス)、−COCH2−、−CH(OH)CH2−および−CH2SO−からなる群より選択される結合で、任意に置換した1つ以上のペプチド結合を有する。以下にさらに参考文献を挙げる:Chemistry and Biochemistry of Amino Acids, Peptides, and Proteins, B. ウェインスタイン(Weinstein)編, Marcel Dekker, Inc.(ニューヨーク)第267頁のスパトラ(Spatola), A. F. 1983; スパトラ, A. F. 1983 Vega Data, 第1巻, 第3号, Peptide Backbone Modification (一般概説); モーリー(Morley), 1980 Trends Pharm Sci 第463−468頁(一般概説); ハドソン(Hudson)他1979 Int J Pept Prot Res 14:177-185(−CH2NH−、−CH2CH2−、); スパトラ他1986 Life Sci 38:1243-1249(−CH2−S); ハン(Hann), 1982 J Chem Soc Perkin Trans I 307-314(−CH−CH−、シスおよびトランス); アルムキスト(Almquist)他1980 J Med Chem 23:1392-1398(−COCH2−); ジェニングス-ホワイト(Jennings-White)他1982 Tetrahedron Lett 23:2533(−COCH2−); セルケ(Szelke)他1982 欧州特許出願EP45665(−CH(OH)CH2−); ホラデイ(Holladay)他1983 Tetrahedron Lett 24:4401-4404(−C(OH)CH2−);およびルビー(Hruby), 1982 Life Sci 31:189-199(−CH2−S)。かかる擬似ペプチドは、ポリペプチドでの実施形態よりも大きな利点を有し、例えば、より経済的な生産、より高い化学的安定性、薬理学的特性(半減期、吸収、効能、効力など)の向上、特異性の改変(例えば、広い生物活性)、抗原性の低下などが挙げられる。擬似ペプチドの標識化は、通常、1つ以上の標識を、直接またはスペーサー(例えば、アミド基)を介して、擬似ペプチドの定量的構造活性データおよび分子モデリングにより予測した非妨害性位置(複数可)に共有結合させることを伴う。かかる非妨害性位置は、一般的に、擬似ペプチドが結合して治療効果を生じる巨大分子(複数可)(受容体分子等)と直接接触しない位置である。擬似ペプチドの誘導体化(例えば、標識化)は、その擬似ペプチドの所望の生物学的または薬理学的活性を実質的に妨げないのがよい。一般的に、受容体結合ペプチドの擬似ペプチドは、高親和性で受容体に結合し、検出可能な生体活性を有する(すなわち、1つ以上の受容体媒介性表現型変化に対して作用性または拮抗性である)。
【0036】
より安定なペプチドを作製するため、コンセンサス配列の1個以上のアミノ酸を、同じ型のD−アミノ酸で系統的(systematic)に置換してもよい(例えば、L−リシンの代わりにD−リシン)を用いてもよい。また、コンセンサス配列または実質的に同一のコンセンサス配列変異を含有する束縛(constrained)ペプチドを、当該技術分野で既知の方法(リゾ(Rizo)他1992 Ann Rev Biochem 61:387)、例えば、ペプチドを環化する分子内ジスルフィド結合を形成することができる分子内システイン残基を付加することにより作製してもよい。
【0037】
合成または非天然型アミノ酸は、インビボでは天然に存在しないが、それでも、本明細書に記載したペプチド構造に組み込まれ得るアミノ酸をいう。好ましい合成アミノ酸は、天然型L−α−アミノ酸のD−α−アミノ酸、ならびに式H2NCHR5COOH(式中、R5は、1)低級アルキル基、2)炭素数が3−7のシクロアルキル基、3)炭素数が3−7で1−2個のヘテロ原子が酸素、イオウおよび窒素からなる群より選択される複素環、4)芳香核上に、ヒドロキシル、低級アルコキシ、アミノおよびカルボキシルからなる群より任意に選択される1−3個の置換基を有する炭素数が6−10の芳香族残基、5)−アルキレン−Y(式中、アルキレンは、炭素数が1−7のアルキレン基であり、Yは、(a)ヒドロキシ、(b)アミノ、(c)炭素数が3−7のシクロアルキルおよびシクロアルケニル、(d)芳香核上に、ヒドロキシル、低級アルコキシ、アミノおよびカルボキシルからなる群より任意に選択される1−3個の置換基を有する炭素数が6−10のアリール、(e)炭素数が3−7で1−2個のヘテロ原子が酸素、イオウおよび窒素からなる群より選択される複素環、(f)−C(O)R2(式中、R2は、水素、ヒドロキシ、低級アルキル、低級アルコキシおよび−NR3R4(式中、R3およびR4は、独立に、
水素および低級アルキルからなる群より選択される)からなる群より選択される)、(g)−S(O)nR6(式中、nは1−2の整数であり、R6は、低級アルキルである)、からなる群より選択される)である。ただし、R5は、天然型アミノ酸の側鎖を規定するものではない)で表される非天然型D−およびL−α−アミノ酸である。
【0038】
他の好ましい合成アミノ酸としては、β−アラニン、γ−アミノ酪酸など、アミノ基がカルボキシル基と2つ以上の炭素原子で分断されているアミノ酸が挙げられる。
【0039】
特に好ましい合成アミノ酸としては、一例として、天然型L−アミノ酸のD−アミノ酸、L−(1−ナフチル)−アラニン、L−(2−ナフチル)−アラニン、L−シクロヘキシルアラニン、L−2−アミノイソ酪酸、メチオニンのスルホキシドおよびスルホン誘導体(すなわち、HOOC−(H2NCH))−CH2CH2−S(O)nR6(式中、nおよびR6は、上記のとおりである))ならびにメチオニンの低級アルコキシ誘導体(すなわち、HOOC−(H2NCH))−CH2CH2−OR6(式中、R6は、上記のとおりである))が挙げられる。
【0040】
本明細書で使用する場合、化合物X(例えば、ペプチドまたはアミノ酸)の「誘導体」は、この化合物内の1つ以上の反応性基が置換基によって誘導体化されているXの形態をいう。ペプチド誘導体の例としては、アミノ酸側鎖、ペプチド主鎖またはアミノ末端もしくはカルボキシ末端が誘導体化されているペプチドが挙げられる(例えば、メチル化アミド結合を有するペプチド性化合物)。
【0041】
本明細書で使用する場合、化合物Xの「アナログ」は、Xの機能的活性に必要なXの化学構造を保持しているが、Xとは異なる或る特定の化学構造も含む化合物をいう。天然型ペプチドのアナログの一例は、1つ以上の非天然型アミノ酸を含むペプチドである。本明細書で使用する場合、化合物Xの「擬似物」は、Xの機能的活性に必要なXの化学構造が、Xの立体配座を擬態する別の化学構造と置換されている化合物をいう。擬似ペプチドの例としては、以下にさらに記載するような、ペプチド主鎖が1つ以上のベンゾジアゼピン分子で置換されているペプチド様化合物(例えば、ジェームズ(James), G. L.他1993 Science 260:1937-1942参照)、全てのL−アミノ酸が対応するD−アミノ酸で置換されたペプチドおよび「retro-inverso」型ペプチド(シスト(Sisto)による米国特許第4,522,752号参照)が挙げられる。
【0042】
擬似物という用語、特に擬似ペプチドは、同配体(isostere)を含むものとする。用語「同配体」は、本明細書で使用する場合、第2の化学構造で置換され得る化学構造を含むものとする。これは、第1の構造の立体配座が第2の構造に特異的な結合部位に適合しているからである。この用語には、具体的には、当業者に既知のペプチド主鎖の修飾(すなわち、アミド結合擬似物)が含まれる。かかる修飾としては、アミド窒素、α−炭素、アミドカルボニルの修飾、アミド結合の完全な置換、伸長、欠失または架橋が挙げられる。いくつかのペプチド主鎖の修飾は既知であり、Ψ[CH2S]、Ψ[CH2NH]、Ψ[CSNH2]、Ψ[NHCO]、Ψ[COCH2]、Ψ[(E)または(Z)CH=CH]が挙げられる。上記に使用した述語において、Ψは、アミド結合の不在を示す。アミド基と置き換わる構造を、括弧内に特定する。同配体の他の例としては、1つ以上のベンゾジアゼピン分子で置換されたペプチドが挙げられる(例えば、ジェームズ, G. L.他1993 Science 260:1937-1942参照)。
【0043】
他の考えられ得る修飾としては、N−アルキル(またはアリール)置換(Ψ[CONR])、主鎖の架橋によるラクタムおよび他の環状構造の構築、化合物内のすべてのL−アミノ酸のD−アミノ酸への置換(「inverso」型化合物)、または「retro-inverso」型アミノ酸の組み込み(Ψ[NHCO])が挙げられる。「inverso」とは、配列のL−アミ
ノ酸をD−アミノ酸で置換することを意味し、「retro-inverso」または「enantio-retro」とはアミノ酸の配列の逆転(「retro」)およびL−アミノ酸のD−アミノ酸での置換を意味する。例えば、親ペプチドがThr−Ala−Tryである場合、retro修飾型は、Try−Ala−Thrであり、inverso型は、thr−ala−try(小文字は、D−アミノ酸を示す)であり、retro-inverso型は、try−ala−thrである。親ペプチドと比較すると、retro-inversoペプチドは、逆転した主鎖を有するが、側鎖の元の空間的配座は実質的に保持しており、親ペプチドに非常に似たトポロジーを有するretro-inverso異性体がもたらされる。グッドマン(Goodman)他1981 Perspectives in Peptide
Chemistry第283−294頁を参照のこと。また、「retro-inverso」ペプチドのさらなる説明については、シストによる米国特許第4,522,752号も参照のこと。他の誘導体としては、C末端ヒドロキシルメチル誘導体、O−修飾誘導体(例えば、C末端ヒドロキシルメチルベンジルエーテル)ならびにアルキルアミドおよびヒドラジドなどの置換アミドを含むN末端修飾誘導体が挙げられる。
【0044】
本明細書で使用する場合、用語「アミノ酸構造」(「ロイシン構造」、「フェニルアラニン構造」または「グルタミン構造」など)は、アミノ酸、ならびに化合物の機能的活性を維持しているそのアミノ酸のアナログ、誘導体および擬似物を含むものとする。例えば、用語「フェニルアラニン構造」は、フェニルアラニンならびにピリジルアラニンおよびホモフェニルアラニンを含むものとする。用語「ロイシン構造」は、ロイシン、ならびにノルロイシンなどの、バリンまたは脂肪族側鎖を有する他の天然型もしくは非天然型アミノ酸での置換を含むものとする。
【0045】
本明細書に開示するペプチド化合物のアミノ末端およびカルボキシ末端は、修飾されていない(すなわち、Y1およびY2は独立に)水素であってもよい。あるいはまた、ペプチド化合物のアミノ末端およびカルボキシ末端は、誘導体基で置換されていてもよい。ペプチド化合物のN末端に存在し得る(すなわちY1であり得る)アミノ誘導体基としては、アセチル、アリール、アラルキル、アシル、エポキシサクシニルおよびコレステリル基が挙げられる。ペプチド化合物のC末端に存在し得る(すなわちY2であり得る)カルボキシ誘導体基としては、アルコール、アルデヒド、エポキシコハク酸、酸ハロゲン化物、カルボニル、ハロメタンおよびジアゾメタン基が挙げられる。
【0046】
本明細書で使用する場合、「検出可能な標識」または「イメージング剤」は、化合物に共有結合すると、Eph受容体結合因子が投与された患者におけるインビボでの検出を含む(これに限定されない)この化合物の検出を可能にする物質をいう。好適な検出可能な標識は、当該技術分野で既知であり、一例として、放射性同位体、蛍光標識(例えば、フルオレセイン)などが挙げられる。使用する具体的な検出可能な標識は重要ではなく、使用する標識の量および使用する標識の量における標識の毒性に関連して選択される。かかる因子に関連した標識の選択は、十分に、当該技術分野の技術範囲内である。
【0047】
ペプチドまたは擬似ペプチドへの検出可能な標識の共有結合は、当該技術分野で既知の従来の方法によりなされる。例えば、125I放射性同位体を検出可能な標識として用いる場合、ペプチドまたは擬似ペプチドへの125Iの共有結合は、ペプチドまたは擬似ペプチドにアミノ酸チロシンを組み込み、次いで、このペプチドをヨウ化物にすることにより、なされ得る(例えば、ウィーナー(Weaner)他1994 Synthesis and Applications of Isotopically Labelled Compounds,第137−140頁を参照)。チロシンがペプチドまたは擬似ペプチド内に存在しない場合、ペプチドまたは擬似ペプチドのNまたはC末端へのチロシンの組み込みは、既知の化学反応によりなされ得る。同様に、32Pを、従来の化学反応を用いて、例えば、ペプチドまたは擬似ペプチド上のヒドロキシル基を介して、リン酸部分としてペプチドまたは擬似ペプチド上に組み込むことができる。
【0048】
「選択的に」とは、1つまたは数個のEph受容体ファミリーメンバーに対して、他の既知のEph受容体ファミリーメンバーに対する結合親和性よりもかなり大きな結合親和性を有することを意味する。選択的結合親和性に関連して使用する場合、「実質的に大きい」は、受容体に結合したリガンドの量の少なくとも2倍、少なくとも3倍、少なくとも4倍、少なくとも5倍、少なくとも6倍、少なくとも7倍、少なくとも8倍、少なくとも9倍、少なくとも10倍、少なくとも15倍、少なくとも20倍、少なくとも30倍、少なくとも40倍、少なくとも50倍または少なくとも100倍の増加を意味する。
【0049】
本明細書で使用する場合、「Eph受容体結合因子」または「Eph受容体結合リガンド」は、Eph受容体に結合する化合物である。この化合物は、1つ以上のEph受容体に結合することができる任意の分子を包含する。或る場合において、1つ以上のEph受容体に結合することができる分子は、ペプチドまたは擬似ペプチドである。かかるペプチドまたは擬似ペプチドは、10個未満、11個未満、12個未満、13個未満、14個未満、15個未満、20個未満、25個未満、30個未満、35個未満、40個未満、45個未満、50個未満、75個未満、100個未満、200個未満、300個未満、400個未満または500個未満の長さの残基を有し得る。用語「Eph受容体結合因子」および「Eph受容体結合リガンド」は、互換的に使用され得る。
【0050】
本明細書で使用する場合、「エフリン−A」としては、エフリン−Aリガンドサブクラスのメンバーである任意のエフリンが挙げられる。
【0051】
本明細書で使用する場合、「エフリン−B」としては、エフリン−Bリガンドサブクラスのメンバーである任意のエフリンが挙げられる。
【0052】
本明細書で使用する場合、用語「治療剤」は、抗癌剤、神経保護剤またはある特定の疾患の兆候に所望の治療効果を生じ得るその他の薬剤を意味する。
【0053】
本明細書に記載される抗癌剤は、細胞傷害剤または癌化学療法剤であってもよい。非限定的な例として、DNA関連過程を標的とする細胞傷害剤には、シクロホスファミド、メルファラン、マイトマイシンC、ビゼレシン、シスプラチン、ドキソルビシン、エトポシド、ミトキサントロン、SN38、Et743、アクチノマイシンD、ブレオマイシンおよびTLK286が包含される。癌化学療法剤は、限定されないが、ドセタキセルなどのタキサン、ドキソルビシンなどのアントラサイクリン、アルキル化剤、ビンカアルカロイド、代謝拮抗物質、シスプラチンもしくはカルボプラチンなどの白金剤、メトトレキセートなどのステロイド、アドリアマイシンなどの抗生物質、イソファミド、または選択的エストロゲン受容体調節物質、トラスツズマブなどの抗体であり得る。
【0054】
タキサンは、本発明の併用治療に有用な化学療法剤である。有用なタキサンとしては、限定されないが、ドセタキセル(Taxotere; Aventis Pharmaceuticals, Inc.; パーシッパニー, ニュージャージー州)およびパクリタキセル(Taxol; Bristol Myers Squibb; プリンストン, ニュージャージー州)が挙げられる。例えば、チャン(Chan)他1999 J Clin Oncol 17:2341 2354およびパリデンス(Paridaens)他2000 J Clin Oncol 18:724を参照のこと。
【0055】
本発明の併用治療に有用な別の癌化学療法剤は、ドキソルビシン、イダルビシンまたはダウノルビシンなどのアントラサイクリンである。ドキソルビシンは、一般的に使用されている癌化学療法剤であり、例えば、乳癌の標的化に有用であり得る(スチュワート(Stewart)およびラタイン(Ratain)著「Cancer: Principles and Practice of Oncology」第5版、第19章、デビタ(DeVita), Jr.他編,; J. P. リッピンコット(Lippincott) 1997; ハリス(Harris)他著「Cancer: Principles and practice of oncology」前掲, 1997)。
また、ドキソルビシンは、抗血管新生活性を有し(フォルクマン(Folkman), 1997 Nature
Biotechnology 15:510;スタイナー(Steiner)他著「Angiogenesis: Key principles Science, technology and medicine」第449 454頁, スタイナー他編 Birkhauser Verlag, 1992)、これは癌の治療におけるその有効性に寄与し得る。
【0056】
メルファランまたはクロラムブチルなどのアルキル化剤は、本発明の併用治療に有用な癌化学療法剤である。同様に、ビンデシン、ビンブラスチンもしくはビノレルビンなどのビンカアルカロイド、または5フルオロウラシル、5フルオロウリジンもしくはその誘導体などの代謝拮抗物質は、本発明の併用治療に有用な癌化学療法剤である。
【0057】
白金剤は、本発明の併用治療に有用な癌化学療法剤である。かかる白金剤は、例えば、クラウン(Crown), 2001 Seminars in Oncol 28:28-37に記載されているような、シスプラチンもしくはカルボプラチンなどであり得る。本発明の併用治療に有用な他の癌化学療法剤としては、限定されないが、メトトレキセート、マイトマイシンC、アドリアマイシン、イホスファミドおよびアンサマイシンが挙げられる。
【0058】
選択的エストロゲン受容体調節物質または抗エストロゲンなどの、乳癌およびその他のホルモン依存性癌の治療に使用される癌化学療法剤もまた、エストロゲンの作用を拮抗阻害する薬剤として使用することができる。選択的エストロゲン受容体調節物質であるタモキシフェンは、乳癌の治療のために本発明の併用治療に使用され得る癌化学療法剤である(フィッシャー(Fisher)他1998 J Natl Cancer Instit 90:1371 1388)。
【0059】
本発明の併用治療に有用な治療剤は、ヒト化モノクローナル抗体などの抗体であってもよい。一例として、抗表皮成長因子受容体2(HER2)抗体であるトラスツズマブ(Herceptin; Genentech; サウスサンフランシスコ, カリフォルニア州)は、HER2/neu過剰発現乳癌(ホワイト(White)他2001 Ann Rev Med 52:125-141)の治療用の本発明のコンジュゲートにおいて有用な治療剤である。
【0060】
また、本発明に有用な別の治療剤は、本発明で使用されるような、直接または間接的に細胞死を促進する任意の分子である細胞傷害剤であってもよい。本発明に有用な細胞傷害剤としては、限定されないが、低分子、ポリペプチド、ペプチド、擬似ペプチド、核酸分子、細胞およびウイルスが挙げられる。非限定的な例として、本発明に有用な細胞傷害剤としては、ドキソルビシン、ドセタキセルまたはトラスツズマブなどの細胞傷害性低分子;以下にさらに記載するようなものなどの抗菌性ペプチド;カスパーゼ、および毒素、例えば、カスパーゼ8などのプロアポトーシスポリペプチド;ジフテリア毒素A鎖、シュードモナス外毒素A、コレラ毒素、DAB389EGFなどのリガンド融合毒素、ヒマ(ricinus communis)毒素(リシン);ならびに細胞傷害性T細胞などの細胞傷害性細胞が挙げられる。例えば、マーチン(Martin)他2000 Cancer Res 60:3218-3224; クライトマン(Kreitman)およびパスタン(Pastan) 1997 Blood 90:252-259; アラム(Allam)他1997 Cancer Res 57:2615-2618; オズボーン(Osborne)およびコロナド・ハインソン(Coronado Heinsohn) 1996 Cancer J Sci Am 2:175を参照のこと。当業者であれば、本明細書に記載した、または当該技術分野で既知のこれらの及びさらなる細胞傷害剤が、本発明の治療剤として有用であり得ることがわかる。
【0061】
神経保護剤は、当該技術分野で既知であり、神経細胞の死を抑制または遅滞させる化合物であり得る。非限定的な例として、神経保護剤は、低分子薬物、ペプチド、タンパク質、抗体またはこれらの組合せなどの抗アポトーシス化合物であり得る。神経保護剤は、1つ以上のアポトーシス経路または壊死経路の妨害、神経成長ホルモン受容体の活性化またはイオンチャネルの調節により作用し得る。当業者であれば、本明細書に記載した、または当該技術分野で既知のこれらおよびさらなる神経保護剤が、本発明の治療剤として有用
であり得ることがわかる。
【0062】
Eph受容体結合因子
本発明の実施形態は、Eph受容体に結合する薬剤を提供する。本明細書に記載した化合物の多くは、Eph受容体ファミリーの16個の既知受容体のうち、1つだけ、または限られた数の受容体に選択的に結合する。Eph受容体結合因子は、低分子薬剤、ペプチドまたは擬似ペプチドであり得る。Eph受容体結合因子は、天然化合物であってもよく、合成化合物であってもよい。また、本明細書に記載した化合物の多くは、Eph受容体に高親和性で結合し、Eph受容体アゴニストまたはアンタゴニストとしての機能を果たし得る。本明細書に記載した化合物は、「リード」化合物、およびリード化合物と同じかまたは類似の分子構造または形状を有するが、加水分解もしくはタンパク質分解に対する感受性のいずれかに関して、および受容体に対する親和性の増加もしくは標的Eph受容体とは無関係の生物学的特性をさらに有することなどの他の生物学的特性に関してリード化合物とは異なるように構成された「誘導体」化合物を含む。
【0063】
ペプチドおよび擬似ペプチドの調製
1.固相合成
本明細書に記載するペプチドは、当該技術分野で既知の古典的な方法により、例えば、標準的な固相技法により調製することができる。標準的な方法としては、排他的固相合成、部分固相合成法、フラグメント濃縮(fragment condensation)、古典的溶液合成が挙げられ、組換えDNA技術によるものでさえ含まれる。例えば、メリフィールド(Merrifield), 1963 J Am Chem Soc 85:2149を参照のこと。固相では、合成は、通常α−アミノ保護樹脂を用いてペプチドのC末端から開始される。適当な出発物質は、例えば、必要なα−アミノ酸をクロロメチル化樹脂、ヒドロキシメチル樹脂またはベンズヒドリルアミン樹脂に結合することにより調製することができる。かかるクロロメチル化樹脂の一例は、BioRad Laboratories(リッチモンド、カリフォルニア州)により商品名BIO−BEADS SX−1で販売されているものであり、ヒドロキシメチル樹脂の調製は、ボドンスキー(Bodonszky)他1966 Chem Ind (ロンドン) 38:1597に記載されている。ベンズヒドリルアミン(BHA)樹脂は、ピエッタ(Pietta)およびマーシャル(Marshall), 1970 Chem Commn 650に記載されており、塩酸塩の形態でBeckman Instruments, Inc.(パロ・アルト, カリフォルニア州)から市販されている。
【0064】
このように、化合物は、ギシン(Gisin), 1973 Helv Chim Acta 56:1467に記載の方法にしたがって、例えば重炭酸セシウム触媒の補助により、α−アミノ保護アミノ酸をクロロメチル化樹脂に結合させることにより調製することができる。最初の結合後、α−アミノ保護基を、トリフルオロ酢酸(TFA)または塩酸(HCl)溶液などの選択した試薬により、有機溶媒中、室温で除去する。
【0065】
α−アミノ保護基は、ペプチドの段階的合成の技術分野において有用であることが知られているものであり、アシル型保護基(例えば、ホルミル、トリフルオロアセチル、アセチル)、芳香族ウレタン型保護基(例えば、ベンジルオキシカルボニル(carboyl)(Cbz)および置換Cbz)、脂肪族ウレタン保護基(例えば、t−ブチルオキシカルボニル(Boc)、イソプロピルオキシカルボニル、シクロヘキシルオキシカルボニル)およびアルキル型保護基(例えば、ベンジル、トリフェニルメチル)が挙げられる。BocおよびFmocは、好ましい保護基である。側鎖保護基は、結合反応中は損傷を受けず、アミノ末端保護基の脱保護中または結合反応中に分離しないものである。側鎖保護基は、最終ペプチドの合成完了時、かつ標的ペプチドを改変しない反応条件下で除去可能でなければならない。
【0066】
Tyrの側鎖保護基としては、テトラヒドロピラニル、tert−ブチル、トリチル、
ベンジル、Cbz、Z−Br−Cbzおよび2,5−ジクロロベンジルが挙げられる。Aspの側鎖保護基としては、ベンジル、2,6−ジクロロベンジル、メチル、エチルおよびシクロヘキシルが挙げられる。ThrおよびSerの側鎖保護基としては、アセチル、ベンゾイル、トリチル、テトラヒドロピラニル、ベンジル、2,6−ジクロロベンジルおよびCbzが挙げられる。ThrおよびSerの側鎖保護基はベンジルである。Argの側鎖保護基としては、ニトロ、トシル(Tos)、Cbz、アダマンチルオキシカルボニルメシトイルスルホニル(Mts)またはBocが挙げられる。Lysの側鎖保護基としては、Cbz、2−クロロベンジルオキシカルボニル(2Cl−Cbz)、2−ブロモベンジルオキシカルボニル(2BrCbz)、TosまたはBocが挙げられる。
【0067】
α−アミノ保護基の除去後、残留する保護アミノ酸を、所望の順序で段階的に結合させる。一般的には、過剰の各保護アミノ酸を、ジシクロヘキシルカルボジイミド(DCC)などの適切なカルボキシル基活性化剤とともに溶液中、例えば塩化メチレン(CH2Cl2)、ジメチルホルムアミド(DMF)混合物中で使用する。
【0068】
所望のアミノ酸配列が完成した後、この所望のペプチドを樹脂支持体から、樹脂からペプチドを切断するだけでなく、すべての残留側鎖保護基をも切断するトリフルオロ酢酸またはフッ化水素(HF)などの試薬での処理により取り出す。クロロメチル化樹脂を使用した場合、フッ化水素処理により、遊離ペプチド酸の形成がもたらされる。ベンズヒドリルアミン樹脂を使用した場合は、フッ化水素処理により、遊離ペプチドアミドが直接もたらされる。あるいはまた、クロロメチル化樹脂を用いた場合、側鎖保護ペプチドは、ペプチド樹脂を、アンモニアで処理して所望の側鎖保護アミドを得るか、またはアルキルアミンで処理して側鎖保護されたアルキルアミドまたはジアルキルアミドにすることにより、取り出すことができる。次いで、側鎖保護を、フッ化水素で処理して遊離アミド、アルキルアミドまたはジアルキルアミドにすることによる常套的な方法で解除する。
【0069】
これらの固相ペプチド合成手順は、当該技術分野で既知であり、J. M. スチュワートおよびJ. D. ヤング(Young), 1984 Solid Phase Peptide Syntheses第2版, Pierce Chemical Companyにさらに記載されている。
【0070】
1990年3月7日に出願された米国特許出願第07/492,462号、1990年12月6日に出願された同第07/624,120号および1991年12月6日に出願された同第07/805,727号に記載されている「コード(encoded)合成ライブラリー」または「巨大(very large)規模固定化ポリマー合成」系を用いると、かかる活性を有する最小サイズのペプチドを決定できるだけでなく、好ましいモチーフ(またはそのモチーフの最小サイズ)と、1つ、2つまたはそれ以上の残基において異なるペプチド群を構成するあらゆるペプチドを作製することができる。次いで、この収集ペプチドを、EphA2、EphA4、EphA5、EphA7、EphB2およびEphB4(これらに限定されない)を含む、Eph受容体ファミリーメンバーに結合する能力に関してスクリーニングし得る。この固定化ポリマー合成系または他のペプチド合成法はまた、本発明のペプチド化合物すべての切断型アナログおよび欠失型アナログならびに切断型と欠失型のアナログの組合せを合成するためにも使用できることを理解されたい。
【0071】
2.合成アミノ酸
これらの手順はまた、本発明のいずれかの化合物の1つ、2つまたはそれ以上の位置において、遺伝子にコードされた20個の天然型アミノ酸以外のアミノ酸に置換されているペプチドを合成するためにも使用することができる。例えば、トリプトファンをナフチルアラニンに置換すると、合成が容易になる。置換して本発明のペプチドに取り込み得る他の合成アミノ酸としては、L−ヒドロキシプロピル、L−3,4−ジヒドロキシフェニルアラニル、L−d−ヒドロキシリシルおよびL−d−メチルアラニルなどのdアミノ酸、
L−α−メチルアラニル、βアミノ酸、ならびにイソキノリルが挙げられる。Dアミノ酸および非天然型合成アミノ酸もまた、本発明のペプチドに組み込むことができる(例えば、ロバーツ(Roberts)他1983 Unusual Amino/Acids in Peptide Synthesis 5:341-449を参照のこと)。
【0072】
20個の遺伝子にコードされたアミノ酸(またはDアミノ酸)の天然型側鎖を、他の側鎖、例えば、アルキル、低級アルキル、環状の四、五、六−七員環アルキル、アミド、アミド低級アルキル、アミドジ(低級アルキル)、低級アルコキシ、ヒドロキシ、カルボキシおよびその低級エステル誘導体などの基で、ならびに四、五、六−七員環の複素環で置き換えてもよい。特に、プロリン残基の環の大きさを五員環から四、六または七員環に変えたプロリンのアナログを使用するのがよい。環式基は、飽和であってもよく、不飽和であってもよく、不飽和の場合、芳香族系であってもよく、非芳香族系であってもよい。複素環式基は、好ましくは、1個以上の窒素、酸素およびイオウのヘテロ原子を含む。かかる基の例としては、フラザニル、フリル、イミダゾリジニル、イミダゾリル、イミダゾリニル、イソチアゾリル、イソキサゾリル、モルホリニル(例えば、モルホリノ)、オキサゾリル、ピペラジニル(例えば、1−ピペラジニル)、ピペリジル(例えば、1−ピペリジル、ピペリジノ)、ピラニル、ピラジニル、ピラゾリジニル、ピラゾリニル、ピラゾリル、ピリダジニル、ピリジル、ピリミジニル、ピロリジニル(例えば、1−ピロリジニル)、ピロリニル、ピロリル、チアジアゾリル、チアゾリル、チエニル、チオモルホリニル(例えば、チオモルホリノ)およびトリアゾリルが挙げられる。これらの複素環式基は、置換されていても、置換されていなくてもよい。基が置換されている場合、置換基は、アルキル、アルコキシ、ハロゲン、酸素、または置換もしくは非置換のフェニルであり得る。
【0073】
また、リン酸化により、本発明のペプチドを容易に修飾することができ(例えば、W. バンワース(Bannwarth)他1996 Biorganic and Medicinal Chemistry Letters 6:2141-2146を参照のこと)、本発明の化合物であるペプチド誘導体を作製するための他の方法は、ルビー他1990 Biochem J 268:249-262に記載されている。このように、本発明のペプチド化合物はまた、類似の生体活性を有する擬似ペプチドを調製するための基剤として使用される。
【0074】
3.末端修飾
当業者であれば、対応するペプチド化合物と同じかまたは類似する所望の生体活性を有するが、溶解性、安定性ならびに加水分解およびタンパク質分解への感受性に関して、このペプチドよりもより好都合な活性を有する擬似ペプチドを構築するために、種々の技法が利用可能であることが認識されよう。例えば、モーガン他1989 Ann Rep Med Chem 24:243-252を参照のこと。以下は、N末端アミノ基、C末端カルボキシル基が修飾された擬似ペプチドを調製するための方法およびペプチドのアミド結合の1つ以上を非アミド結合に変化させるための方法を記載したものである。2つ以上のかかる修飾を、1つの擬似ペプチド構造に結合させ得ること(例えば、C末端カルボキシル基の修飾およびペプチドの2つのアミノ酸間に−CH2−カルバメート結合を含めること)は、理解されよう。
【0075】
1)N末端修飾
ペプチドは、通常、遊離酸として合成されるが、上記のように、アミドまたはエステルとして容易に調製され得る。また、ペプチド化合物のアミノ末端およびカルボキシ末端を修飾して、他の有用な化合物を作製することができる。アミノ末端修飾としては、メチル化(すなわち、−NHCH3または−NH(CH3)2)、アセチル化、ベンジルオキシカルボニル基の付加、またはRCOO−(式中、Rは、ナフチル、アクリジニル、ステロイジルおよび同様の基からなる群より選択される)で規定されるカルボン酸官能基を含有する任意のブロック基によるアミノ末端のブロックが挙げられる。カルボキシ末端修飾と
しては、遊離酸のカルボキシアミド基での置換、構造的制約を導入するためのカルボキシ末端での環状ラクタムの形成が挙げられる。
【0076】
アミノ末端修飾は、上記のとおりであり、アルキル化、アセチル化、カルボベンゾイル基の付加、スクシニミド基の形成などが挙げられる(例えば、マーレイ(Murray)他1995 Burger’s Medicinal Chemistry and Drug Discovery第5版, 第1巻, マンフレッド(Manfred) E. ウルフ(Wolf)編, John Wiley & Sons Inc.を参照のこと)。このとき、具体的には、N末端アミノ基を、以下のようにして反応させることができる。
【0077】
(a)酸ハロゲン化物[例えば、RC(O)Cl]または対称な無水物との反応により、式RC(O)NH−(式中、Rは上記の通りである)のアミド基を形成する。通常、この反応は、好ましくは、反応中に生成する酸を除去するためのジイソプロピルエチルアミンなどの過剰(例えば、約10当量)の第三級アミンを含有する不活性希釈剤(例えば、ジクロロメタン)中で、ほぼ等モル量か過剰量(例えば、約5当量)の酸ハロゲン化物をペプチドと接触させることにより行なうことができる。反応条件は、これ以外の点では従来通りである(例えば、室温で30分間)。末端アミノをアルキル化して低級アルキルN−置換し、次いで上記のように酸ハロゲン化物との反応を行うことにより、式RC(O)NR−のN−アルキルアミド基が得られる;
【0078】
(b)無水コハク酸との反応によりスクシンイミド基を形成する。上記のように、ほぼ等モル量か過剰量の無水コハク酸(例えば、約5当量)を用いるのがよく、適当な不活性溶媒(例えば、ジクロロメタン)中での過剰(例えば、10当量)のジイソプロピルエチルアミンなどの第三級アミンの使用を含む、当該技術分野で既知の方法により、アミノ基をスクシンイミドに変換する。例えば、ウォレンバーグ(Wollenberg)他, 米国特許第4,612,132号を参照のこと。ペプチドのN末端が置換されたスクシンイミドを得るために、コハク酸基を、例えば、C2−C6アルキル、または慣用法で調製される−SR置換基で置き換えてもよいことを理解されたい。かかるアルキル置換基は、ウォレンバーグ他(前掲)により記載された方法で低級オレフィン(C2−C6)と無水マレイン酸との反応により調製され、−SR置換基は、RSH(式中、Rは上記のとおりである)と無水マレイン酸との反応により調製される;
【0079】
(c)好ましくは反応中に生成する酸を除去するための第三級アミンを含有する適当な不活性希釈剤(例えば、ジクロロメタン)中で、ほぼ等モル量か過剰のCBZ−Cl(すなわち、塩化ベンジルオキシカルボニル)または置換CBZ−Clとの反応により、ベンジルオキシカルボニル−NH−または置換ベンジルオキシカルボニル−NH−基を形成する;
【0080】
(d)適当な不活性希釈剤(例えば、ジクロロメタン)中で、等モル量か過剰(例えば、約5当量)のR−S(O)2Cl(式中、Rは上記の通りである)と反応させて末端アミンをスルホンアミドに変換することにより、スルホンアミド基を形成する。好ましくは、不活性希釈剤は、反応中に生成する酸を除去するための、ジイソプロピルエチルアミンなどの過剰(例えば、約10当量)の第三級アミンを含有する。反応条件は、これ以外の点では従来通りである(例えば、室温で30分間);
【0081】
(e)適当な不活性希釈剤(例えば、ジクロロメタン)中で、等モル量か過剰(例えば、約5当量)のR−OC(O)ClまたはR−OC(O)OC6H4−p−NO2(式中、Rは上記の通りである)と反応させて末端アミンをカルバメートに変換することにより、カルバメート基を形成する。好ましくは、不活性希釈剤は、反応中に生成する酸を除去するための、ジイソプロピルエチルアミンなどの過剰(例えば、約10当量)の第三級アミンを含有する。反応条件は、これ以外の点では従来通りである(例えば、室温で30分
間);ならびに
【0082】
(f)適当な不活性希釈剤(例えば、ジクロロメタン)中で、等モル量か過剰(例えば、約5当量)のR−N=C=O(式中、Rは上記の通りである)と反応させて末端アミンをウレア(すなわち、RNHC(O)NH−)基に変換することにより、ウレア基を形成する。好ましくは、不活性希釈剤は、ジイソプロピルエチルアミンなどの過剰(例えば、約10当量)の第三級アミンを含有する。反応条件は、これ以外の点では従来通りである(例えば、室温で30分間)。
【0083】
2)C末端修飾
C末端カルボキシル基がエステルに置き換えられた(すなわち、−C(O)OR(式中、Rは上記の通りである))擬似ペプチドの調製において、ペプチド酸を調製するために樹脂が用いられ、側鎖保護ペプチドを基剤および適切なアルコール(例えば、メタノール)で切断する。次いで、側鎖保護基を、フッ化水素で処理して所望のエステルを得ることによる常套法で除去する。
【0084】
C末端カルボキシル基がアミド−C(O)NR3R4で置き換えられた擬似ペプチドの調製において、ベンズヒドリルアミン樹脂を、ペプチド合成用固体支持体として使用する。合成の完了時、支持体からペプチドを切り離すためのフッ化水素処理により、直接、遊離ペプチドアミド(すなわち、C末端が−C(O)NH2)がもたらされる。あるいはまた、ペプチド合成でのクロロメチル化樹脂の使用と併せて、支持体から側鎖保護ペプチドを切断するためのアンモニアとの反応により遊離ペプチドアミドが得られ、アルキルアミンまたはジアルキルアミンとの反応により、側鎖保護アルキルアミドまたはジアルキルアミド(すなわち、C末端が−C(O)NRR1(式中、RおよびR1は上記の通りである))が得られる。次いで、側鎖保護を、フッ化水素で処理して遊離アミド、アルキルアミドまたはジアルキルアミドにすることによる常法により解除する。
【0085】
また別の実施形態では、C末端カルボキシル基またはC末端エステルは、カルボキシル基またはエステルのそれぞれの−OH基またはエステル(―OR)をN末端アミノ基と分子内で置き換えることにより環化して、環状ペプチドを形成するように誘導してもよい。例えば、合成および切断によりペプチド酸を得た後、溶液中、例えば塩化メチレン(CH2Cl2)、ジメチルホルムアミド(DMF)混合液中で、ジシクロカルボジイミド(DCC)などの適切なカルボキシル基活性化剤により、遊離酸を活性化エステルに変換する。次いで、活性化エステルをN末端アミンで分子内置換することにより、環状ペプチドを形成する。重合とは対照的に、内部環化は、高希釈溶液の使用により、増強され得る。かかる方法は、当該技術分野において既知である。
【0086】
また、本発明のペプチドを環化し、または該ペプチドの末端にデスアミノまたはデスカルボキシ(descarboxyl)残基を組み込むことにより、末端のアミノまたはカルボキシル基をなくし、プロテアーゼに対する感受性を低減する、またはペプチドの立体構造を制限することも可能である。本発明の化合物のC末端の官能基としては、アミド、アミド低級アルキル、アミドジ(低級アルキル)、低級アルコキシ、ヒドロキシル、カルボキシおよびその低級エステル誘導体ならびにその製薬的に許容され得る塩が挙げられる。
【0087】
前記のN末端およびC末端の修飾に加え、擬似ペプチドを含む本明細書に記載するペプチド化合物は、種々の親水性ポリマーのうちの1種類以上により、またはこれらとの共有結合により有利に修飾され得る。ペプチド化合物は、親水性ポリマーで誘導体化すると、その溶解度および循環半減期が増加し、その免疫原性が遮蔽されることがわかった。かなり驚くべきことには、前述のことは、その結合活性の低減(あるとしても)がほとんどない状態で達成できることがわかった。使用に好適な非タンパク質系ポリマーとしては、ポ
リエチレングリコールおよびポリプロピレングリコールが例となるポリアルキルエーテル、ポリ乳酸、ポリグリコール酸、ポリオキシアルケン、ポリビニルアルコール、ポリビニルピロリドン、セルロースおよびセルロース誘導体、デキストランおよびデキストラン誘導体などが挙げられるが、これらに限定されない。一般的に、かかる親水性ポリマーは、約500−約100,000ダルトン、より好ましくは約2,000−約40,000ダルトン、さらにより好ましくは約5,000−約20,000ダルトンの範囲の平均分子量を有する。好ましい実施形態では、かかる親水性ポリマーは、約5,000ダルトン、約10,000ダルトン、および約20,000ダルトンの平均分子量を有する。
【0088】
ペプチド化合物は、ゼリプスキー(Zallipsky), S. 1995 Bioconjugate Chem 6:150-165; モンファルジーニ(Monfardini), C他1995 Bioconjugate Chem 6:62-69; 米国特許第4,640,835号、同第4,496,689号、同第4,301,144号、同第4,670,417号、同第4,791,192号、同第4,179,337号または国際公開第WO 95/34326号に記載された方法のいずれかを用いて、かかるポリマーにより、またはこれとの結合により誘導体化することができる。
【0089】
一実施形態において、ペプチド化合物はポリエチレングリコール(PEG)により誘導体化される。PEGは、2つの末端ヒドロキシル基を有する、エチレンオキシド繰り返し単位の線状の水溶性ポリマーである。PEGは、その分子量により分類され、通常500ダルトン−約40,000ダルトンの範囲である。現時点で好ましい実施形態では、使用されるPEGは、5,000ダルトン−約20,000ダルトンの範囲の分子量を有する。本発明のペプチド化合物に連結させるPEGは、分枝であっても非分枝であってもよい。(例えば、モンファルジーニ, C他1995 Bioconjugate Chem 6:62-69参照)。PEGは、Shearwater Polymers, Inc.(ハンツビル, アラバマ州)、Sigma Chemical Co.および他社から市販されている。かかるPEGとしては、モノメトキシポリエチレングリコール(MePEG−OH)、モノメトキシポリエチレングリコール−スクシネート(MePEG−S)、モノメトキシポリエチレングリコール−スクシンイミジルスクシネート(MePEG−S−NHS)、モノメトキシポリエチレングリコール−アミン(MePEG−NH2)、モノメトキシポリエチレングリコール−トレシレート(MePEG−TRES)およびモノメトキシポリエチレングリコール−イミダゾリル−カルボニル(MePEG−IM)が挙げられるが、これらに限定されない。
【0090】
簡単には、一例の実施形態において、使用される親水性ポリマー、例えばPEGは、その一端が、好ましくは、メトキシまたはエトキシ基などの非反応性基でキャップされている。その後、ポリマーの他端を、ハロゲン化シアヌル(例えば、塩化、臭化またはフッ化シアヌル、)、ジイミダゾール、無水物試薬(例えば、ジブロモコハク酸無水物などのジハロコハク酸無水物)、アシルアジド、p−ジアゾイウムベンジル(diazoiumbenzyl)エーテル、3−(p−ジアゾニウムフェノキシ)−2−ヒドロキシプロピルエーテル)などの適当な活性化剤との反応で活性化する。次いで、活性化したポリマーを、本明細書に記載するペプチド化合物と反応させ、ポリマーで誘導体化されたペプチド化合物を作製する。あるいはまた、本発明のペプチド化合物内の官能基を、ポリマーとの反応のために活性化してもよく、または既知のカップリング方法を用いた協奏カップリング反応にて2つの官能基を結合させてもよい。本発明のペプチド化合物は、当業者に既知であり、当業者により使用されている他の無数の反応スキームを用いてPEGで誘導体化させ得ることは、容易に理解されよう。
【0091】
或る実施形態では、誘導体化されたペプチドは、非修飾ペプチドの約0.1−約0.01倍の活性を有する。また他の実施形態では、誘導体化されたペプチドは、非修飾ペプチドの約0.1−約1倍の活性を有する。さらにまた他の実施形態では、誘導体化されたペプチドは、非修飾ペプチドよりも大きな活性を有する。
【0092】
本実施形態における使用に好適なペプチドとしては、一般的に、EphA2、EphA4、EphA5、EphA7、EphB2およびEphB4(これらに限定されない)を含む、Eph受容体ファミリーメンバーに結合するペプチド、すなわちリガンドが挙げられる。かかるペプチドは、通常約50個以下のアミノ酸残基、より好ましくは約20個以下のアミノ酸残基を有する。本発明における使用に好適な親水性ポリマーとしては、ポリエチレングリコールおよびポリプロピレングリコールが例示されるポリアルキルエーテル、ポリ乳酸、ポリグリコール酸、ポリオキシアルケン、ポリビニルアルコール、ポリビニルピロリドン、セルロースおよびセルロース誘導体、デキストランおよびデキストラン誘導体などが挙げられるが、これらに限定されない。一般的に、かかる親水性ポリマーは、約500−約100,000ダルトン、より好ましくは約2,000−約40,000ダルトン、さらにより好ましくは約5,000−約20,000ダルトンの範囲の平均分子量を有する。或る実施形態では、かかる親水性ポリマーは、約5,000ダルトン、約10,000ダルトン、および約20,000ダルトンの平均分子量を有する。ペプチド化合物は、上記およびそこに挙げられた参考文献に記載された方法を用いて誘導体化することができる。
【0093】
4.主鎖修飾
化合物のペプチド誘導体を作製するための他の方法は、ルビー他1990 Biochem J 268(2):249-262に記載されている。このように、ペプチド化合物はまた、類似の生体活性を有する非ペプチド化合物の構造モデルとして使用される。当業者であれば、リードペプチド化合物と同じかまたは類似する所望の生体活性を有するが、溶解性、安定性ならびに加水分解およびタンパク質分解への感受性に関して、リードよりもより好都合な活性を有する化合物を構築するために、種々の技法が利用可能であることが認識されよう。モーガン他1989 Ann Rep Med Chem 24:243-252を参照のこと。これらの技法としては、ペプチド主鎖を、ホスホネート、アミデート、カルバメート、スルホンアミド、第二級アミンおよびN−メチルアミノ酸から構成される主鎖と置き換えることが挙げられる。
【0094】
ペプチジル結合[−C(O)NH−]の1つ以上が、−CH2−カルバメート結合、ホスホネート結合、−CH2−スルホンアミド結合、ウレア結合、第二級アミン(−CH2NH−)結合およびアルキル化ペプチジル結合[−C(O)NR6(式中、R6は、低級アルキルである)]などの結合で置き換えられている擬似ペプチドは、従来のペプチド合成の際に、合成の適切な時点で、アミノ酸試薬のかわりに適当に保護されたアミノ酸アナログを 用いるだけで調製される。
【0095】
好適な試薬としては、例えば、アミノ酸のカルボキシル基が、上記の結合のうちの1つを形成するのに適当な部分で置き換えられているアミノ酸アナログが挙げられる。例えば、ペプチド内の−C(O)NR−結合を−CH2−カルバメート結合(−CH2OC(O)NR−)で置き換えることが所望される場合、適切に保護されたアミノ酸のカルボキシル(−COOH)基をまず還元して−CH2OH基とし、次いで、これを、慣用法により−OC(O)Cl官能基またはp−ニトロカルボネート−OC(O)O−C6H4−p−NO2官能基に変換する。かかる官能基のいずれかと、固体支持体上に見られる部分的に組み立てられた(fabricated)ペプチドのN末端の遊離アミンまたはアルキル化アミンとの反応により、−CH2OC(O)NR−結合の形成がもたらされる。かかる−CH2−カルバメート結合の形成に関するより詳細な説明については、チョー(Cho)他1993 Science 261:1303-1305を参照のこと。
【0096】
同様に、ペプチド内のアミド結合のホスホネート結合での置き換えは、米国特許出願第07/943,805号、同第08/081,577号および第08/119,700号に記載のようにしてなされ得る。
【0097】
ペプチド内のアミド結合の−CH2−スルホンアミド結合での置き換えは、慣用法により、適当に保護されたアミノ酸のカルボキシル(−COOH)基を−CH2OH基に還元し、次いで、ヒドロキシル基をトシル基などの適当な脱離基に変換することによりなされ得る。トシル化誘導体と、例えばチオ酢酸との反応後、加水分解および酸化的塩素化することにより、―CH2−S(O)2Cl官能基が得られ、これは、カルボキシル基以外が適当に保護されたアミノ酸のカルボキシル基と置き変わる。ペプチド合成において、この適当に保護されたアミノ酸アナログを使用することにより、−CH2−S(O)2NR−結合の内包がもたらされてペプチド内のアミド結合と置き換わり、それにより擬似ペプチドが得られる。−CH2−S(O)2Cl基を得るためのアミノ酸のカルボキシル基の変換に関するより完全な説明については、例えば、ウェインスタイン, B., 1983 Chemistry
& Biochemistry of Amino Acids, Peptides and Proteins, 第7巻, 第267−357頁, Marcel Dekker, Inc.(ニューヨーク)を参照のこと。
【0098】
ペプチド内のアミド結合のウレア結合での置き換えは、米国特許出願第08/147,805号に記載のようにしてなされ得る。
【0099】
−CH2NH−結合がペプチド内のアミド結合と置き換わる第二級アミン結合は、例えば、アミド結合のカルボニル結合が、慣用法によりCH2基に還元されている、適当に保護されたジペプチドアナログを用いることにより調製することができる。例えば、ジグリシンの場合、アミドをアミンへ還元して脱保護すると、H2NCH2CH2NHCH2COOHになり、これは、後で、次のカップリング反応でのN−保護形態で使用される。ジペプチド内のアミド結合のカルボニル基の還元によるかかるアナログの調製は、当該技術分野で既知である(例えば、M. W. レミントン(Remington) 1994 Meth Mol Bio 35:241-247を参照のこと)。
【0100】
適当に保護されたアミノ酸アナログは、対応するアミノ酸の場合と同様にして、慣用のペプチド合成に使用される。例えば、通常約3当量の保護されたアミノ酸アナログが、この反応に使用される。塩化メチレンまたはDMFなどの不活性な有機系希釈剤を使用するが、反応副生成物として酸が生じる場合、通常、反応溶媒は、反応中に生成する酸を除去するための過剰量の第三級アミンを含有する。特に好ましい第三級アミンの1つは、ジイソプロピルエチルアミンであり、これは、通常約10倍過剰で用いる。この反応により、擬似ペプチドへの非ペプチジル結合を有するアミノ酸アナログの組み込みがもたらされる。かかる置換は、所望により、ペプチド内のアミド結合の0−すべてが非アミド結合で置き換えられるように、反復してもよい。
【0101】
また、ペプチドを環化しても、該ペプチドの末端にデスアミノまたはデスカルボキシ残基を組み込んでもよく、それにより末端のアミノまたはカルボキシル基をなくし、プロテアーゼに対する感受性を低減したり、またはペプチドの立体構造を制限したりしてもよい。、化合物のC末端の官能基としては、アミド、アミド低級アルキル、アミドジ(低級アルキル)、低級アルコキシ、ヒドロキシ、カルボキシおよびその低級エステル誘導体ならびにその製薬的に許容され得る塩が挙げられる。
【0102】
5.ジスルフィド結合形成
化合物は、システインのチオール基間の分子内ジスルフィド結合により環化された形態で存在してもよい。あるいはまた、システインのチオール基間の分子間ジスルフィド結合により、二量体(またはより多いオリゴマー)化合物を得るように作製することができる。また、システイン残基の1つ以上が、ホモシステインで置換してもよい。
【0103】
本発明の他の実施形態としては、イオウの1個が、CH2基またはイオウの他の同配体
と置換されている、これらのジスルフィド誘導体のアナログが挙げられる。これらのアナログは、当該技術分野で既知の方法を用いる分子内置換または分子間置換により作製され得る。
【0104】
あるいはまた、ペプチドのアミノ末端を、α−置換酢酸でキャップしてもよく、ここで、α置換基は、α−ハロ酢酸、例えば、α−クロロ酢酸、α−ブロモ酢酸またはα−ヨード酢酸などの脱離基である。脱離基をシステイン残基またはホモシステイン残基のイオウに置き換えることにより、本発明の化合物を環化または二量体化してもよい。例えば、アンドリュー(Andreu)他1994 Meth Mol Bio 35(7):91-169; バーカー(Barker)他1992 J Med
Chem 35:2040-2048;およびオア(Or)他1991 J Org Chem 56:3146-3149を参照のこと。
【0105】
また、ペプチドは、当該技術分野で既知の組換えDNA技法によっても調製され得る。
【0106】
Eph受容体のEph受容体結合化合物による調節
本明細書に記載するEph受容体結合化合物は、細胞におけるEph活性を調節することができるものである。Eph活性を調節するEph受容体結合化合物には、Eph受容体ファミリーの1つ以上のメンバーに結合する、本明細書に記載したペプチドまたは擬似ペプチドが含まれる。本発明の或る実施形態では、Eph受容体結合化合物は、本明細書に記載した単一のペプチドまたは擬似ペプチドのみを含む。本発明の或る実施形態では、細胞を、Eph受容体のリン酸化を引き起こし、それにより下流シグナル伝達事象を活性化するのに有効な量のEph受容体結合化合物と接触させる。或る実施形態では、細胞を、エフリンによるEph受容体のリン酸化を抑制し、それにより受容体活性を阻害するのに有効な量のEph受容体結合化合物と接触させる。他の実施形態では、細胞を、下流シグナル伝達事象を活性化または不活化するのに有効な量のEph受容体結合化合物と接触させ得る。下流シグナル伝達事象を活性化または不活化するのに有効なEph受容体結合化合物の量としては、少なくとも0.05μM、少なくとも0.1μM、少なくとも0.2μM、少なくとも0.3μM、少なくとも0.4μM、少なくとも0.5μM、少なくとも0.6μM、少なくとも0.7μM、少なくとも0.8μM、少なくとも0.9μM、少なくとも1μM、少なくとも5μM、少なくとも10μM、少なくとも20μM、少なくとも30μM、少なくとも40μM、少なくとも50μM、少なくとも60μM、少なくとも70μM、少なくとも80μM、少なくとも90μM、少なくとも100μMまたは少なくとも200μMの濃度が挙げられる。本明細書に記載していない他の有効濃度の測定は、当業者であれば容易に行える。
【0107】
本発明の或る実施形態では、目的のEph受容体は、細胞において、インビトロおよびインビボのいずれでも調節される。いずれの適用に関しても、細胞は、Ephファミリーの受容体の少なくとも1つのメンバーを発現する任意の細胞であればよく、ヒト細胞が挙げられるが、これに限定されない。
【0108】
本発明の或る実施形態では、EphAサブファミリーの受容体が調節される。かかる受容体としては、EphA1、EphA2、EphA3、EphA4、EphA5、EphA6、EphA7、EphA8、EphA9またはEphA10が挙げられる。或る特定の実施形態では、EphA2受容体を発現する細胞を、該受容体の活性および後続の下流シグナル伝達事象が調節されるように、本明細書に記載するペプチド、擬似ペプチドまたは低分子の有効量と接触させる。他の実施形態では、EphA4受容体を発現する細胞を、該受容体の活性および後続の下流シグナル伝達事象が調節されるように、本明細書に記載するペプチド、擬似ペプチドまたは低分子の有効量と接触させる。さらに他の実施形態では、EphA5またはEphA7受容体を発現する細胞を、該受容体の活性および後続の下流シグナル伝達事象が調節されるように、本明細書に記載するペプチド、擬似ペプチドまたは低分子の有効量と接触させる。
【0109】
他の実施形態では、EphBサブファミリーの受容体が調節される。かかる受容体としては、EphB1、EphB2、EphB3、EphB4、EphB5およびEphB6が挙げられる。或る特定の実施形態では、EphB4受容体を発現する細胞を、該受容体の活性および後続の下流シグナル伝達事象が調節されるように、本明細書に記載するペプチド、擬似ペプチドまたは低分子の有効量と接触させる。
【0110】
Ephファミリー受容体の特定のメンバーを刺激することは、アポトーシス(プログラム細胞死)の活性化に関係しているとみなされている。したがって、Eph受容体を過剰発現する或る特定の細胞(例えば、或る特定の型の腫瘍性細胞)においてプログラム細胞死を活性化することは、望ましくない細胞集団の選択的死滅に好都合であり得る。さらにまた、プログラム細胞死に関して標的化した細胞型において過剰発現する特定のEph受容体の選択的アゴニストとして機能するEph受容体結合化合物は、非標的化細胞を死滅させることなく、標的細胞を排除するための方法を提供し得る。
【0111】
本発明の或る実施形態では、Eph受容体ファミリーの特定のメンバーの選択的アゴニストまたはアンタゴニストとしての機能を果たすEph受容体結合化合物を投与する方法が意図される。或る実施形態では、本明細書に記載するペプチド、擬似ペプチドまたは低分子などの選択的アゴニストを用いて、ペプチド、擬似ペプチドまたは低分子の有効量を、哺乳動物(ヒトを含む)に投与することにより、プログラム細胞死を活性化することができる。或る特定の実施形態では、アゴニストはEphA2に結合し、それにより、この受容体へのエフリン−A1の結合を競合的に阻害する。他の実施形態では、アゴニストの結合により、受容体のリン酸化が活性化される。
【0112】
哺乳動物に投与されるアゴニストの有効量は、約0.001mg−約50mg/kg体重/日の範囲であり得る。有効量は、アゴニストが投与される経路、アゴニストの結合親和性、標的細胞内でのEph受容体発現レベルおよび非標的細胞内でのEph受容体発現レベル(これらに限定されない)などの因子によって決まる。しかしながら、アゴニストの有効量の決定は、当業者により容易に決定され得ることを理解されたい。
【0113】
治療剤および治療剤送達剤としてのEph受容体結合化合物
本明細書に記載するEph受容体結合化合物はまた、インビボでEph受容体を調節するために、ヒトを含む温血動物にも投与することができる。例えば、本明細書に開示する或る特定のペプチドが、EphA2を選択的に活性化するため、またはEphA4を選択的に阻害するために使用され得る。したがって、本発明は、インビボでEph受容体を活性化または阻害するのに十分な量のかかる化合物を投与することを含む、Eph関連疾患の治療上の処置のための方法を包含する。
【0114】
また、Eph受容体を標的化することにより、癌および他の疾患の治療行為が可能になる。本明細書に記載するEph受容体結合化合物は、細胞傷害剤を疾患組織の血管に送達するために使用され得る。実際、化学療法剤、毒素またはプロアポトーシスペプチドに結合させた血管標的ペプチドは、腫瘍成長を低減し、臨床的関節炎を抑制し、または前立腺組織を破壊し得る(アラップ(Arap), W他1998 Science 279:377-380; オルソン(Olson), T. A.他1997 Int J Cancer 73:865-870; エルビー(Ellerby) H.M.他1999 Nat Med 5:1032-1038; アラップ, W他2002 PNAS USA 99:1527-1531; ゲルラク(Gerlag), D. M.他2001 Arthritis Research 3:357-361)。例えば、アゴニストにより引き起こされるEphA2のチロシンリン酸化は、受容体およびアゴニストのインターナリゼーションを媒介し(ザンテック(Zantek), N. D.他1999 Cell Growth Differ 10:629-638; カルレス−キンチ(Carles-Kinch), K他2002 Cancer Res 62:2840-2847; ファン・デル・ギア(Van der Geer), P他1994 Annu Rev Cell Biol 10:251-337)、したがって、毒性物質またはプロアポトーシ
ス物質が、細胞内に送達され、選択的に細胞を死滅させ得る(エルビー, H. M.他1999 Nat Med 5:1032-1038)。さらにまた、本明細書に記載するEph受容体結合化合物により誘導されるEphA2の活性化により、EphA2発現癌細胞の増殖、浸潤性および転移性が低減され得る(ザンテック, N. D.他1999 Cell Growth Cell Differ 10:629-638; カルレス−キンチ, K他2002 Cancer Res 62:2840-2847; ミャオ(Miao), H.他Nature 2000 Cell Biol 2:62-69)。EphA2活性化は、乳癌細胞および前立腺癌細胞の悪性度の低減と相関し、EphA2過剰発現の形質転換効果を逆転させることは、当該技術分野では既知である(ゼリンスキー, D. P.他2002 J Cell Biochem 85:714-720; ザンテック, N. D.他1999 Cell Growth Differ 10:629-638; カルレス−キンチ, K他2002 Cancer Res 62:2840-2847)。細胞傷害剤を送達するために本明細書に記載するEph受容体結合化合物を使用した場合、本明細書に開示する組成物によるEphA2の活性化により、アポトーシス刺激に対して細胞を感受性とし得る(ドーン(Dohn), M.他, 2001 Oncogene 20:6503-6515)。
【0115】
本発明の或る実施形態は、本明細書に記載するペプチド、擬似ペプチドまたは低分子などのEph受容体結合化合物に連結させた治療剤を含有するコンジュゲートを意図する。かかるコンジュゲートは、治療を必要とする動物に適切なコンジュゲートを投与することにより、適切なEph受容体を発現する標的細胞に送達され得る。或る実施形態では、治療剤は治療を担う。他の実施形態では、治療剤およびEph受容体結合化合物の両方が治療に寄与する。或る実施形態では、治療剤は、イメージング剤である。
【0116】
目的のEph受容体に結合するEph受容体結合化合物は、リンカーにより治療剤と連結させる。リンカーは、Eph受容体結合化合物と治療剤の両者が同じ領域、組織または細胞に標的化されることを許容するものであれば、任意の結合、低分子、またはその他のビヒクルのいずれであってもよい。好ましくは、リンカーが切断可能なものである。
【0117】
一実施形態において、リンカーは、1つ以上のEph受容体結合化合物と1つ以上の治療剤との間の化学結合である。したがって、該結合は、共有結合またはイオン結合であり得る。リンカーが化学結合であるコンジュゲートの一例は、融合タンパク質ということになる。一実施形態において、化学結合は、pH感受性結合である。あるいはまた、該結合は、pH感受性でなくてもよいが、標的部位の微環境において、後で付加されるか、または元々存在する特定の酵素または化学物質により切断可能であるのがよい。あるいはまた、該結合は、還元条件下で切断される結合(例えば、ジスルフィド結合)であってもよい。あるいはまた、該結合は、切断可能でなくてもよい。
【0118】
任意の種類のpH切断性またはpH感受性リンカーが使用され得る。酸切断性結合の例としては、シス−ポリカルボン酸アルケンとして知られる部類の有機酸が挙げられるが、これらに限定されない。この部類の分子は、少なくとも1つの二重結合を含む炭素鎖に結合した少なくとも3個のカルボン酸基(COOH)を含む。これらの分子、ならびにこれらをどのように作製および使用するかは、シェン(Shen)他(米国特許第4,631,190号)に開示されている。あるいはまた、アミノ−スルフヒドリル架橋剤などの穏和な酸性条件下で切断可能な分子を使用してもよい。これらの分子は、ブラットラー(Blattler)他の米国特許第4,569,789号に開示されている。
【0119】
あるいはまた、切断可能なリンカーは、生分解性、加水分解性結合などの徐放性結合であってもよい。通常の生分解性担体結合としては、エステル、アミドまたはウレタン結合が挙げられるため、通常の担体は、ポリエステル、ポリアミド、ポリウレタン、および約5,000−1,000,000の分子量を有する他の縮合重合体である。これらの担体/結合の例は、ピーターソン(Peterson)他の米国特許第4,356,166号に示されている。他の酸切断性リンカーは、米国特許第4,569,789号および同第4,631
,190号またはブラットラー他1985 Biochemistry 24:1517-1525で確認できる。リンカーは、自然な酸性条件により切断され、あるいはまた、酸性条件は、エイブラムズ(Abrams)他の米国特許第4,171,563号に説明してあるようにして標的部位にて誘導してもよい。
【0120】
切断可能なジスルフィド結合(還元性結合)を含む連結試薬の例としては、「DPDPB」(1,4−ジ−[3’−(2’−ピリジルジチオ)プロピオンアミド]ブタン)、「SADP」(N−スクシンイミジル(4−アジドフェニル)1,3−ジチオプロピオネート)、「スルホ−SADP」(スルホスクシンイミジル(4−アジドフェニルジチオ)プロピオネート)、「DSP」(ジチオビス(スクシンイミジルプロピオネート))、「DTSSP」(3,3’−ジチオビス(スルホスクシンイミジルプロピオネート))、「DTBP」(ジメチル3,3’−ジチオビスプロピオンイミデート−2HCl))(これらは、すべて、Pierce Chemicals(ロックフォード、イリノイ州)から入手可能である)が挙げられるが、これらに限定されない。
【0121】
酸化により切断され得る連結試薬の例としては「DST」(ジスクシンイミジルタルタレート)および「スルホ−DST」(ジスクシンイミジルタルタレート)である。これらのリンカーもまた、Pierce Chemicalsから入手可能である。
【0122】
切断可能でないリンカーの例は、「スルホ−LC−SMPT」(スルホスクシンイミジル6−[α−メチル−α−(2−ピリジルチオ)トルアミド]ヘキサノエート)、「SMPT」、「ABH」(アジドベンゾイルヒドラジド)、「NHS−ASA」(N−ヒドロキシスクシンイミジル−4−アジドサリチル酸)、「SASD」(スルホスクシンイミジル2−(p−アジドサリチルアミド)エチル−1,3−ジチオプロピオネート)、「APDP」(N−[4−(p−アジドサリチルアミド)ブチル]−3’(2’−ピリジルジチオ)プロピオンアミド)、「BASED」(ビス−[β−(4−アジドサリチルアミド)エチル]ジスルフィド)、「HSAB」(N−ヒドロキシスクシンイミジル−4アジドベンゾエート)、「APG」(p−アジドフェニルグリオキサル一水和物)、「SANPAH」(N−スクシンイミジル−6(4’−アジド−2’−ミトロ(mitro)フェニル−アミド)ヘキサノエート)、「スルホ−SANPAH」(スルホスクシンイミジル6−(4’−アジド−2’−ニトロフェニルアミド)ヘキサノエート)、「ANB−NOS」(N−5−アジド−2−ニトロベンジオイル(bezyoyl)オキシスクシンイミド)、「SAND」(スルホスクシンイミジル−2−(m−アジド−o−ミトロ(mitro)ベンズアミド)−エチル−1,3’−ジチオプロピオネート)、「PNP−DTP」(p−ニトロフェニル−2−ジアゾ−3,3,3−トリフルオロプロピオネート)、「SMCC」(スクシンイミジル4−(N−マレイミドメチル)シクロヘキサン−1−カルボキシレート)、「スルホ−SMCC」(スルホスクシンイミジル4−(N−マレイミドメチル)シクロヘキサン−1−カルボキシレート)、「MBS」(m−マレイミドベンゾイル−N−ヒドロキシスクシンイミドエステル)、「スルホ−MBS」(m−マレイミドベンゾイル−N−ヒドロキシスルホスクシンイミドエステル)、「SIAB」(N−スクシンイミジル(4−ヨードアセチル)アミノベンゾエート)、「スルホ−SIAB」(N−スルホスクシンイミジル(4−ヨードアセチル)アミノベンゾエート)、「SMPB」(スクシンイミジル4−(p−マレン(malen)イミドフェニル)ブチレート)、「スルホ−SMPB」(スルホスクシンイミジル4−(p−マレンイミドフェニル)ブチレート)、「DSS」(ジスクシンイミジルスベレート)、「BSSS」(ビス(スルホスクシンイミジルスベレート))、「BMH」(ビスマレイミドヘキサン)、「DFDNB」(1,5−ジフルオロ−2,4−ジニトロベンゼン)、「DMA」(ジメチルアジピミデート(adipimidate)2HCl)、「DMP」(ジメチルピメルイミデート(pimelimidate)−2HCl)、「DMS」(ジメチルスベルイミデート(suberimidate)−2−HCl)、「SPDP」(N−スクシンイミジル−3−(2−ピリジルチオ)プロピオネート)、「スルホ−HSAB」(スルホス
クシンイミジル4−(p−アジドフェニル)ブチレート)、「スルホ−SAPB」(スルホスクシンイミジル4−(p−アジドフェニルブチレート)、「ASIB」(1−9p−アジドサリチルアミド)−4−(ヨードアセトアミド)ブタン)、「ASBA」(4−(p−アジドサリチルアミド)ブチルアミン)である。これらのリンカーは、すべて、Pierce Chemicalsから入手可能である。
【0123】
別の実施形態では、リンカーは、ペプチドリンカーなどの低分子である。一実施形態では、ペプチドリンカーは、切断可能ではない。さらなる実施形態では、ペプチドリンカーは、還元条件下での塩基により、または特定の酵素により切断可能である。一実施形態では、酵素は内生のもの(indigenous)である。あるいはまた、低分子ペプチドは、治療用コンジュゲートの後またはこれに加えて投与される内生でない酵素により、切断可能であってもよい。あるいはまた、低分子ペプチドは、例えばジスルフィド結合を含む場合には還元条件下で切断できる。。あるいはまた、低分子ペプチドはpH感受性であってもよい。ペプチドリンカーの例としては、ポリ(L−Gly)(ポリL−グリシンリンカー)、ポリ(L−Glu)(ポリL−グルタミンリンカー)、ポリ(L−Lys)(ポリL−リシンリンカー)が挙げられる。一実施形態では、ペプチドリンカーは、式(アミノ酸)nを有し、式中、nは、2−100の整数であり、好ましくは、この場合のペプチドは、1個以上のアミノ酸のポリマーを含有する。
【0124】
さらなる実施形態では、ペプチドリンカーは、プロテイナーゼにより切断可能である(スズキ(Suzuki)他1998 J Biomed Mater Res 42:112-6)。或る実施形態では、リンカーは、ポリ(エチレングリコール)(PEG)、およびジペプチドであるL−アラニル−L−バリン(Ala−Val)(Goyal他2000 Biochem J 345:247-254)を含む、酵素サーモリシンにより切断可能なリンカーである。
【0125】
化学物質リンカーおよびペプチドリンカーは、コンジュゲート合成のための当該技術分野で既知の技法により、すなわち、遺伝子操作を用いることにより、または化学的に、Eph受容体結合化合物と治療剤とを結合するものであり得る。コンジュゲート合成は、タンパク質の適切な官能基での他の構成部分との古典的なカップリング反応により、適切な抗体を介して化学的になされ得る。タンパク質に存在し、化学的カップリング反応に通常用いられる官能基の例を以下に概説する。糖質(carbohydrate)構造を、アルデヒド基に酸化し、これを、H2NNH−R基を含有する化合物(式中、Rは当該化合物)と反応させ、C=NH−NH−R基を形成してもよい。チオール基(タンパク質中のシステイン)を、チオール反応性基を含有する化合物と反応させて、チオエーテル基またはジスルフィド基を形成してもよい。アミノ酸残基中(タンパク質のアミノ末端またはリシン上)の遊離アミノ基を、活性化カルボキシ基などの求電子基を含有する化合物と反応させて、アミド基を形成してもよい。アミノ酸残基中の遊離カルボキシル基を反応性カルボキシル基に変換し、次いで、アミノ基を含有する化合物と反応させてアミド基を形成してもよい。
【0126】
Eph受容体結合化合物と連結させる治療剤は、所望の効果をもたらす任意の化学物質、分子またはコンジュゲートであり得る。例としては、抗生物質、抗腫瘍剤、免疫抑制剤、ホルモンなどの従来の医薬剤、1種類以上の遺伝子、アンチセンスオリゴヌクレオチド、低分子干渉性RNA、造影剤、タンパク質、毒素、放射性分子もしくは原子、界面活性タンパク質、ナノ粒子または凝固タンパク質が挙げられるが、これらに限定されない。治療剤は、親油性(標的細胞内に進入するのを補助する性質)であってもよい。
【0127】
造影剤は、当業者に既知の任意の型の造影剤であり得る。最も一般的な造影剤は、基本的に、4つの群のうちの1つに分類される;X線試薬、X線撮影試薬、磁気共鳴画像形成剤、量子ドット、ナノ粒子および超音波剤。X線試薬としては、イオン性ヨウ素含有試薬ならびにOmnipaque(Nycomed)およびUltravist(Schering)などの非イ
オン性試薬が挙げられる。X線撮影試薬としては、以下に開示するものなどの放射性同位体が挙げられる。磁気共鳴画像形成剤としては、ガドリニウムおよび鉄酸化物キレートなどの磁気剤(magnetic agent)が挙げられる。超音波剤としては、気体の超微粒気泡およびいくつかの気泡放出製剤が挙げられる。
【0128】
放射性核種は、診断用であってもよく、治療用であってもよい。一般的に医学的に有用な放射性核種の例としては、90Y、111Ln、67Cu、77Lu、99Tcなど、好ましくは90Yおよび111Lnなどの三価のカチオンなどの、Y、Ln、Cu、Lu、Tc、Re、Co、Feなどが挙げられる。
【0129】
診断用γシンチレーション測光法による器官および組織のインビボでの画像形成に好適な放射性核種としては、以下のもの:γ放出放射性核種:111Ln、113mLn、67Ga、68Ga、99mTc、51Cr、197Hg、203Hg、169Yb、85Srおよび87Srが挙げられる。Fab’断片による結合に好適なキレート化放射性核種の調製は、米国特許第4,658,839号(ニコレッティ(Nicoletti)他)に教示されている。
【0130】
MRIにおける画像形成剤としての使用に好適な常磁性体金属イオンとしては、原子番号57−70のランタニド系元素または原子番号21−29、42または44の遷移金属が挙げられる。米国特許第4,647,447号(グリース(Gries)他)には、キレート化常磁性体金属イオンによるMRI画像形成が教示されている。
【0131】
治療用放射性核種の例は、β放出体である。好適なβ放出体としては、67Cu、186Rh、188Rh、189Rh、153Sm、90Yおよび111Lnが挙げられる。
【0132】
アンチセンスオリゴヌクレオチドは、正常遺伝子の過剰発現、または異常遺伝子の発現により引き起こされる任意の疾患の治療における使用の可能性を有する。アンチセンスオリゴヌクレオチドは、かかる遺伝子の発現を低減または停止するために使用され得る。アンチセンス技術により治療され得る癌遺伝子の例および使用され得る具体的なアンチセンス分子を教示する参考文献としては、c−JunおよびcFos(米国特許第5,985,558号)、HER−2(米国特許第5,968,748号)、E2F−1(ポポフ(Popoff)ら 米国特許第6,187,587号)、SMAD1−7(米国特許第6,159,697号;同第6,013,788号;同第6,013,787号;同第6,013,522号;および同第6,037,142号)、ならびにFas(ディーン(Dean)ら 米国特許第6,204,055号)が挙げられる。
【0133】
また、正常遺伝子の過剰発現、または異常遺伝子の発現により引き起こされる任意の疾患の治療におけるRNA干渉方法における使用のための二本鎖RNA分子を提供する。RNA干渉(RNAi)は、転写後RNA分解による配列特異的遺伝子サイレンシングプロセスであり、これは、発現抑制される遺伝子と配列が相同的な二本鎖RNA(dsRNA)により開始される。RNAiに好適な二本鎖RNA(dsRNA)は、標的対象遺伝子に対応する19個のRNA塩基対で構成され、各3’末端の2つのヌクレオチドを突出端(overhang)とした約21個の連続ヌクレオチドのセンスおよびアンチセンス鎖を含む(エルバシル(Elbashir)ら 2001 Nature 411: 494-498;バス(Bass)、2001 Nature 411: 428-429;ザモア(Zamore)、2001 Nat Struct Biol 8: 746-750)。約25−30個のヌクレオチドのdsRNAもまた、RNAiに成功裡に使用されている(カラビノス(Karabinos)ら2001 PNAS 98: 7863-7868)。dsRNAは、当該技術分野で既知の方法により、インビトロで合成し、細胞内に導入することができる。かかる方法により、標的ポリペプチドの翻訳を低減させ得る。
【0134】
治療剤として使用され得るタンパク質としては、細胞内に存在させるとアポトーシスを誘導するpRBおよびp53(スー(Xu)他米国特許第5,912,236号)などのアポトーシス誘導剤、およびエリスロポエチンなどの、疾患時において消失または発現が低下するタンパク質(スイトコウスキー(Sytkowski)他米国特許第6,048,971号)が挙げられる。
【0135】
治療剤は、アルキル化剤(ナイトロジェンマスタード、エチレンイミン、アルキルスルホネート、ニトロソウレアおよびトリアゼン)、代謝拮抗物質(メトトレキセートなどの葉酸アナログ、ピリミジンアナログおよびプリンアナログ)、天然産物およびその誘導体(抗生物質、アルカロイド、酵素)、ホルモンおよびアンタゴニスト(副腎皮質ステロイド、プロゲスチン、エストロゲン)などの腫瘍性疾患のための任意の化学療法剤であり得ることを理解されたい。あるいはまた、治療剤は、抗腫瘍剤としての機能を果たすアンチセンスオリゴヌクレオチド、または腫瘍性細胞でのアポトーシスを活性化するタンパク質であり得る。
【0136】
治療剤は、任意の型の神経奏効物質であってもよく、例えば、神経伝達物質または神経伝達物質アンタゴニストを、その使用に伴って一般的に経験する広範な副作用なしで、必要とされる領域に標的化し得る。
【0137】
治療剤は、痛みの領域に特異的に標的化され得る、オピオイドなどの麻酔薬であってもよい。吐気などの副作用は、オピオイド鎮痛剤を使用している患者が一般的に経験する。本発明の方法により、外科的創傷または関節炎の場合における関節などの必要な領域への薬物の非常に特異的な局在が可能になり、これは、副作用を低減し得る。
【0138】
治療剤は、ヒスタミン、H1−受容体アンタゴニストおよびブラジキニンなどの抗炎症剤であってもよい。あるいはまた、抗炎症剤は、サリチル酸誘導体、インドールおよびインデン酢酸、ならびにアルカノンなどの非ステロイド系抗炎症剤であってもよい。あるいはまた、抗炎症剤は、コルチコステロイド、クロモリンナトリウムおよびネドクロミルなどの喘息の治療用のものであってもよい。抗炎症剤は、B2選択的アドレナリン作用薬およびテオフィリンなどの気管支拡張薬とともに、または単独で投与され得る。
【0139】
治療剤は、利尿剤、バソプレシンアゴニストもしくはアンタゴニスト、アンギオテンシンまたは患者の血圧に特異的に作用するレニンであってもよい。
【0140】
治療剤は、心臓疾患の治療に使用される任意の医薬であり得る。かかる医薬としては、有機系亜硝酸塩(アミル亜硝酸塩、ニトログリセリン、イソソルビド二硝酸塩)、カルシウムチャネル遮断薬、抗血小板剤および抗血栓剤、血管拡張薬、血管抑制薬、抗ジギタリス抗体ならびに結節遮断薬が挙げられるが、これらに限定されない。
【0141】
治療剤は、テトラサイクリン、クリンダマイシン、キニン、クロロキン、メフロキン、トリメトプリムスルファメトキサゾール、メトロニダゾールおよびオラミンなどの、原生動物感染の治療に使用される任意の医薬であり得る。標的医薬または他の治療薬を原生動物感染の領域に標的化できることは、これらの抗生物質医薬によって、一般に非常に重篤な副作用を経験することから、特に重要である。
【0142】
治療剤は、スルホンアミド、キノロン、ペニシリン、セファロスポリン、アミノグリコシド、テトラサイクリン、クロラムフェニコール、エリスロマイシン、イソニアジドおよびリファンピンなどの任意の抗菌薬であり得る。
【0143】
治療剤は、アンフォテリシン、フルシトシン、ミコナゾールおよびフルコナゾールなど
の、真菌感染の治療に使用される任意の医薬であり得る。
【0144】
治療剤は、アシクロビル、ビダラビン、インターフェロン、リバビリン、ジドブジン、ザルシタビン、逆転写酵素インヒビター、およびプロテアーゼインヒビターなどの、ウイルス感染の治療に使用される任意の医薬であり得る。また、毒素、放射性原子およびアポトーシス誘導剤などの他の治療剤を用いてウイルス感染細胞を標的化し、死滅させることも想定され得る。
【0145】
治療剤は、種々の抗凝血剤、抗血栓薬および抗血小板薬から選択され得る。
【0146】
かかる治療剤をホルモン(成長ホルモン、アンドロゲン、エストロゲン、性腺刺激ホルモン放出ホルモン、甲状腺ホルモン、副腎皮質ステロイド、インスリンおよびグルカゴン)として使用し、ホルモンの過剰産生または産生不良に起因する疾患を治療し得ることを理解されたい。あるいはまた、ホルモンが過剰産生される場合、そのホルモンに対するアンタゴニストまたは抗体を治療剤として使用し得る。
【0147】
他の考えられ得る種々の治療剤としては、ビタミン、酵素、ならびに、産生の不十分な他の細胞成分、およびジフテリア毒素またはボツリヌス毒素などの毒素が挙げられる。
【0148】
あるいはまた、治療剤は、インビトロ診断で通常使用されるものであってもよい。したがって、リガンドおよびリンカーを慣用法により標識し、シグナル生成系のすべてまたは一部を形成する。リガンドおよびリンカーは、当該技術分野で既知の方法により、トリチウム、炭素14、リン32、ヨウ素125およびヨウ素131などの放射性同位体に共有結合させることができる。例えば、125Iは、クロラミン−T法などの手順、ラクトペルオキシダーゼ法により酵素的に、または予備標識Bolton-Hunter法により導入され得る。これらおよびその他の技法は、H. バン・ブナキス(Van Vunakis)およびJ. J. ランゴン(Langone)編、Methods in Enzymology、第70巻、パートA、1980において考察されている。また、放射性標識のさらなる例については、米国特許第3,646,346号および同第4,062,733号を参照のこと。
【0149】
あるいはまた、治療剤は、化学的環境の変化により、または酵素などの別の分子薬剤の作用により、対応する医薬剤に変換されるプロドラッグまたはプロ分子であってもよい。好ましくは、治療剤は、プロ分子の変換に必要な特定の分子とともに投与される。あるいはまた、プロ分子は、標的組織の微環境において見られる天然分子により切断され得る。あるいはまた、プロドラッグは、pH感受性であり、血管から細胞または細胞内小胞に環境が変化すると、変換される(グレコ(Greco)他2001 J Cell Physiol 187: 22-36)。
【0150】
哺乳動物に投与されるコンジュゲートの有効量は、約0.001mg−約50mg/kg体重/日の範囲であり得る。有効量は、コンジュゲートが投与される経路、コンジュゲートの結合親和性、標的細胞内でのEph受容体発現レベルおよび非標的細胞内でのEph受容体発現レベル(これらに限定されない)などの因子によって決まる。しかしながら、アゴニストの有効量の決定は、当業者により容易に決定され得ることを理解されたい。
【0151】
本発明の別の態様としては、活性成分として本明細書に開示するペプチド、擬似ペプチド、低分子の少なくとも1種類を、医薬用担体または希釈剤との組合せで含有する医薬組成物が挙げられる。これらの化合物は、経口、肺系、非経口(筋肉内、腹腔内、静脈内もしくは皮下への注射)、吸入(微粉末製剤もしくはエーロゾル)、経皮的、鼻腔内、膣内、直腸内、または舌下経路の投与により投与でき、各投与経路に適切な投薬形態に製剤化され得る。例えば、バーンスタイン(Bernstein)他のPCT特許公開公報第WO 93/25221;ピット(Pitt)他のPCT特許公開公報第WO 94/17784;およびピ
ット他の欧州特許出願第613,683号を参照のこと。
【0152】
経口投与のための固形投薬形態としては、カプセル、錠剤、丸剤、散剤および顆粒剤が挙げられる。かかる固形投薬形態では、活性化合物は、スクロース、ラクトースまたはデンプンなどの少なくとも1種類の不活性な製薬的に許容され得る担体と混合する。また、かかる投薬形態は、通常の粒子のように、不活性希釈剤以外の添加剤物質、例えば、ステアリン酸マグネシウムなどの潤沢剤を含有し得る。カプセル、錠剤および丸剤の場合は、投薬形態は、緩衝剤も含有し得る。錠剤および丸剤は、さらに、腸溶性コートを用いて調製してもよい。
【0153】
経口投与のための液状投薬形態としては、製薬的に許容され得るエマルジョン、液剤、懸濁液、シロップが挙げられ、水などの当該技術分野で一般的に使用される不活性希釈剤を含むエリキシル剤を含む。かかる不活性希釈剤の他に、組成物はまた、湿潤剤、乳化剤および懸濁剤ならびに甘味料、香味料および香料などの佐剤を含み得る。
【0154】
非経口投与用の本発明の調製物としては、滅菌された水性または非水性溶液、懸濁液またはエマルジョンが挙げられる。非水性溶剤またはビヒクルの例は、プロピレングリコール、ポリエチレングリコール、オリーブ油およびコーン油などの植物油、ゼラチン、およびオレイン酸エチルなどの注射用有機エステルである。かかる投薬形態はまた、保存剤、湿潤剤、乳化剤、および分散剤などの佐剤を含み得る。これらは、例えば、細菌保持フィルターによるろ過により、組成物に滅菌剤を組み込むことにより、組成物に放射線照射することにより、または組成物を加熱することにより、滅菌し得る。これらはまた、使用直前に、滅菌水、またはいくつかの他の滅菌注射用媒体を用いて製造され得る。
【0155】
膣内または直腸内投与のための組成物は、好ましくは、活性物質に加えてココアバターまたは坐剤用ワックスなどの賦形剤を含んでいてもよい坐剤である。また、鼻腔内または舌下投与のための組成物は、当該技術分野で既知の標準的な賦形剤を用いて調製される。
【0156】
化合物を含有する組成物は、予防的および治療上の処置のために投与され得る。治療的な適用では、組成物は、上記のような疾患に既に苦しんでいる患者に、該疾患の症状およびその合併症を治癒するか、または少なくとも一部抑止するのに充分な量で投与される。これを成し遂げるのに充分な量を「治療的有効投与量」と定義する。この使用に有効な量は、疾患の重篤度および患者の体重および全身状態により決まる。
【0157】
本明細書に記載する組成物はまた、例えば、タイス(Tice)およびビビ(Bibi)の方法(Treatise on Controlled Drug Delivery, A. キドニエウス(Kydonieus)編、Marcel Dekker(ニューヨーク)1992、315−339頁)によりマイクロカプセル封入してもよい。
【0158】
予防的な適用では、本明細書に開示する化合物を含む組成物は、特定の疾患に感受性か、または罹患の危険性がある患者に投与される。かかる量を「予防的有効投与量」と定義する。この用法においてもまた、正確な量は、患者の健康状態および体重により決まり、当業者は容易に決定し得る。
【0159】
有効な治療に必要なEph受容体アゴニストの量は、投与手段、標的部位、患者の生理学的状態および他に受けている薬物治療を含む多くの種々の因子により決まる。したがって、治療投薬は、安全性および有効性を最適化するために滴定されなければならない。通常、インビトロで使用される投薬量により、これらの試薬のインサイチュ投薬に有用な量についての有用な指針をもたらされる。具体的な障害の治療のための有効投与量の動物試験により、ヒトへの投薬量のさらなる予測の目安が提供される。種々の考慮事項が、例えば、ギルマン(Gilman)他編、1990 Goodman and Gilman’s: The Pharmacological Basis
of Therapeutics第8版、Pergamon Press;およびRemington’s Pharmaceutical Sciences、第7版、Mack Publishing Co.(イーストン、ペンシルベニア州)(1985)に記載されている。
【0160】
本明細書に記載するペプチドおよび擬似ペプチドは、約0.001mg−約50mg/kg体重/日の範囲の投薬量で投与すると、Eph受容体媒介性症状の治療に有効である。使用される具体的な投与量は、治療対象の具体的な状態、投与経路により、および状態の重篤度、患者の年齢および全身状態などの因子に応じて担当医の判断により調節される。かかる投与量は、当業者により容易に決定され得る。
【0161】
非経口投与には、ペプチドを、製薬的に許容され得る非経口用ビヒクルとの組合せで、例えば、液剤、懸濁液、エマルジョンまたは凍結乾燥粉末剤に製剤化すればよい。かかるビヒクルの例は、水、生理食塩水、リンゲル液、デキストロース溶液、および5%ヒト血清アルブミンである。また、リポソームおよび固定油などの非水性ビヒクルを使用してもよい。ビヒクルまたは凍結乾燥粉末は、等張性を維持する添加剤(例えば、塩化ナトリウム、マンニトール)、化学的安定性を維持する添加剤(例えば、緩衝剤および保存剤)を含み得る。製剤は、一般的に使用されている技法により滅菌する。例えば、注射による投与に適した非経口用組成物は、1.5重量%の活性成分を0.9%塩化ナトリウム溶液に溶解することにより調製される。
【0162】
本明細書に記載する医薬組成物は、単回投与で投与してもよく、反復投与で投与してもよく、単独の治療剤として、または他の治療剤と併用のいずれかで投与しても、従来からの治療法と組み合わせてもよく、逐次投与しても、同時投与してもよい。
【0163】
化合物は、徐放製剤の状態、例えば、低速放出ポリマーを含む組成物の状態で投与してもよい。活性化合物は、埋込み、およびマイクロカプセル封入型送達システムを含む、制御放出製剤などの、該化合物を急速放出から保護する担体を用いて調製することができる。エチレン酢酸ビニル、ポリ酸無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、ポリ乳酸およびポリ乳酸系、ポリグリコールコポリマー(PLG)などの生分解性生体適合性ポリマーが使用され得る。かかる製剤の調製のための多くの方法が、一般的に、当業者に既知である。
【0164】
本明細書に記載するEph受容体結合化合物は、該化合物を唯一の活性剤として含む医薬組成物に製剤化してもよい。あるいはまた、医薬組成物は、さらなる活性剤を含んでいてもよい。例えば、本明細書に記載するEph受容体結合化合物の2種類以上を組み合わせて使用してもよい。さらにまた、本ペプチド化合物を、Eph受容体活性に対して調節効果を有する1種類以上の他の薬剤と組み合わせてもよい。
【0165】
Ephファミリーメンバーに特異的に結合するペプチドを同定するためのファージディスプレイの使用
16種類の既知のEph受容体の各々に特異的に結合するペプチドを単離するためにファージディスプレイを使用し得る。本明細書に記載するように、EphA2、EphA4、EphA5、EphA7、EphB2またはEphB4に特異的に結合する、数個のファージディスプレイペプチドを単離し、それらの多くは、特異的に結合する。したがって、目的のEph受容体に選択的に結合するペプチドを得るため、Eph受容体ファミリーの16種類の既知のメンバーのいずれかに対するパニング用ランダムペプチドライブラリーを使用し得る。クローンは、当該技術分野で既知の配列決定技法により同定することができる。高結合選択性および高結合親和性の両方を有するペプチドを得るために、ペプチドライブラリーに含まれるペプチドの長さを調節してもよい。
【0166】
他の有用性
本明細書に記載する化合物は、エフリンリガンドの産生およびその受容体結合プロセスに影響する、およびこれらに影響されると考えられる多くの要因の評価を含む、Eph受容体の生物学的役割を理解するための独自のツールとして、インビトロで有用である。本発明の化合物はまた、Eph受容体に結合し、活性化する他の化合物の開発にも有用である。それは、本発明の化合物が、かかる開発を容易にするための構造と活性との関係に関する重要な情報を提供するものだからである。
【0167】
本化合物はまた、新しいEph受容体アゴニストをスクリーニングするためのアッセイにおける競合的結合体としても有用である。かかるアッセイの実施形態において、本明細書に記載する化合物は、修飾なしで使用してもよく、種々の方法、例えば、検出可能なシグナルを直接的または間接的に提供する部分と共有結合または非共有結合で連結するなどして標識することにより修飾してもよい。任意のこのようなアッセイにおいて、これに対する物質は、直接または間接的のいずれかで標識され得る。直接標識として考えられ得るものは、125Iなどの放射性標識、ペルオキシダーゼおよびアルカリホスファターゼなどの酵素(米国特許第3,645,090号)、ならびに蛍光強度の変化、波長シフトまたは蛍光分極をモニターできる蛍光標識(米国特許第3,940,475号)などの標識基が挙げられる。間接標識として考えられ得るものは、一成分をビオチン化した後、上記の標識基の1つを連結させたアビジンに結合させることが挙げられる。また、本化合物を固相支持体に結合させる場合、本化合物はスペーサーまたはリンカーを含み得る。
【0168】
核磁気共鳴(NMR)分光法は、巨大分子の構造を解明できることが知られており、標的分子へのリガンドの結合の静止時の特性および一時的な特性の両方を調べるための技法である(ペレッキア(Pellecchia)他2002 Nature Rev Drug Disc 1: 211)。NRM分光法は、標的分子へのリガンドの結合を測定するための有用なツールであり、タンパク質機能の予備知識を必要とせずに、高感度で相互作用を検知し、数量化することができるという利点を有する。さらにまた、NMR分光法は、標的およびリガンドの両方の構造の情報を提供し、後で、低結合性の候補物(hit)を高親和性リード化合物(leads)に最適化するのを補助し得る。
【0169】
均一に標識されている標的生体分子からの第1および第2核磁気共鳴相関スペクトルを得ることにより、標的生体分子へのリガンド化合物の結合を検出する方法は、米国特許第5,698,401号および同第5,804,390号に報告されている。第1スペクトルは、リガンドの不在下で標的物質に関して収集したデータから得、第2スペクトルは、1種類以上のリガンドの存在下で得る。2つのスペクトルの比較により、推定リガンドの混合物中のどの化合物が標的生体分子に結合するのかを調べることが可能になる。
【0170】
Eph受容体は、1個以上のアミノ酸残基の側鎖への1H、13C、15Nおよび19Fの組込みにより、選択的に標識してもよい。Eph受容体結合リガンドに結合したEph受容体の選択的に標識されたコンジュゲートを、第2分子に曝露してもよく、任意の分子相互作用をNMR分光法により調べることができる。例えば、分子相互作用を検知するため、および任意のコンジュゲートの解離定数を測定するために、2D、13C、1H−HMQC(heteronuclear multiple quantum coherence)および13C−エディット(edited)1H、1H−NOESY NMR実験を使用することができる。また、標的の三次元構造に基づいて、および標識された側鎖に対するリガンドの相対位置から、予想モデルを創製することができる。単一の選択的に標識された標的分子において数個の異なる標識側鎖を使用することにより、分解能およびモデルの予想性状が改善される。
【0171】
非ペプチド低分子は、臨床開発用ペプチドよりも好適であり得るため、Eph−エフリンコンジュゲートを分解する低分子の化学物質ライブラリーをスクリーニングするために
ハイスループットスクリーニングを使用し得る。このアッセイは、エフリン−アルカリホスファターゼ融合タンパク質とのコンジュゲートの状態にある固定化したEph受容体エクトドメインを使用する。結合したアルカリホスファターゼ活性を低下させる能力により、Eph−エフリン相互作用の低分子インヒビターを同定する。
【0172】
さらにまた、本明細書に記載するペプチドは、Eph受容体に選択的に結合する能力に基づいて、生細胞上、固定細胞上、生体液中、組織ホモジネート中、精製天然生体物質中などでEph受容体を選択的に検出するための試薬として使用することができる。例えば、本明細書に記載するペプチドを標識することにより、EphA2、EphA4、EphA5、EphA7、EphB2またはEphB4などの受容体を表面上に有する細胞を選択的に同定することができる。また、本ペプチドは、Eph受容体に結合する能力に基づいて、インサイチュ染色、FACS(蛍光標示式細胞分取)、ウエスタンブロッティング、ELISAなどに使用することができる。また、本ペプチドは、Eph受容体に選択的に結合できる能力に基づいて、受容体精製において、または細胞表面上(または浸透性にした細胞内部)に特定のEph受容体のみを発現する細胞を精製する際に使用することができる。
【0173】
本明細書に記載する化合物はまた、種々の医学的研究および診断的用途のための市販の試薬としても利用され得る。かかる用途としては、(1)種々の機能アッセイにおける候補Ephアゴニストの活性を定量するための較正標品としての用途、(2)Eph依存性細胞株の増殖および成長を維持するための使用、(3)共結晶化によるEph受容体リガンド結合の界面構造解析における使用、(4)Ephシグナル伝達/受容体活性化の機構を調べるための使用、(5)Eph受容体が好ましくは活性化されるか、またはかかる活性化が既知量のEphアゴニストに対して簡便に較正される研究および診断的適用など、ならびに(6)Eph受容体が好ましくは阻害されるか、またはかかる阻害が既知量のEphアンタゴニストに対して簡便に較正される研究および診断的適用など、が挙げられるが、これらに限定されない。
【実施例】
【0174】
以下の実施例は、EphA2、EphA4、EphA5、EphA7、EphB2およびEphB4受容体を標的化するペプチドを同定するに使用した方法を記載するものである。
【0175】
合成ペプチド
以下の実施例において、カルボキシ末端GSGSK(配列番号:30)リンカーを含むビオチン化ペプチドは、Fmoc化学反応を用いて合成し、HPLCにより精製し、MALDI−TOF質量分析法により確認した。本明細書に記載するペプチドおよび擬似ペプチドの調製に関するさらなる情報を、以下に提供する。
【0176】
プラスミド
エフリン−A5−APおよびエフリン−A6−APプラスミドは、記載されている(メンゼル(Menzel), P.他2001 Dev Biol 230: 74-88)。EphA2 AP(EphA2−AP)プラスミドを構築するため、ヒトEphA2の球状アミノ末端領域(aa1−219、GenBank受託番号M36395)を、PCRにより増幅し、APtag−2ベクター(チェン, H. J. & フラナガン(Flanagan), J. G. 1994 Cell 79: 157-168)にクローニングした。発現プラスミドを、Superfectトランスフェクション試薬(Qiagen)を用いて293T細胞に一過的にトランスフェクトした。AP融合タンパク質を含有する細胞培養上澄みを遠心分離し、細胞残屑を除き、20mM Hepesを添加して−20℃で冷凍保存した。
【0177】
上記のものと同様の方法を用いて、Eph−AP融合タンパク質をコードする核酸を含む類似の発現プラスミドを構築してもよく、コードされた融合タンパク質の発現がなされ得ることを理解されたい。
<実施例1>
【0178】
EphA2を標的化するペプチドの同定
本実施例では、EphA2に結合するペプチドを得るために使用される方法を説明する。ランダムな12量体ペプチドをディスプレイするM13ファージライブラリー(New England Biolabs、ベバリー、マサチューセッツ州)を、EphA2のパニングに使用した。ヒスチジンタグを有するマウスEphA2 Fc融合タンパク質(R & D Systems、ミネアポリス、ミネソタ州)を、ニッケル−ニトリロ三酢酸(Ni−NTA)コートELISAプレート内で、Tris緩衝生理食塩水(TBS)(150mM NaCl、50mM Tris−HCl、pH7.5)中1−10μg/mlの濃度で、一晩4℃でインキュベートした。ウェルを、0.5%ウシ血清アルブミン(BSA)含有TBSでブロックし、結合バッファー(TBS、1mM CaCl2、0.1% Tween 20)でリンスした。対照ウェルを、上記の方法を用いてウシ血清アルブミン(BSA)でコートした。
【0179】
EphA2パニングの第1回目の操作(round)では、100μlの結合バッファー中の1.7×1011プラーク形成単位(PFU)のファージライブラリーを、EphA2コートウェル内で室温で1時間インキュベートした。洗浄後も結合したままのファージを100μlの0.2Mグリセリン−HCl(pH2.2)または100μgのエフリン−A1 Fcで溶出した。溶出液全部を用いて、初期対数期のER2738宿主菌に感染させ、増幅させた。ファージを濃縮し、製造者の推奨にしたがって保存した。第2および第3回目の操作では、前回分からの2×1011PFUの増幅ファージプールを、EphA2
FcコートウェルおよびBSAコート対照ウェルに添加した。ファージは、洗浄バッファー中のTween濃度を0.5%とした以外は、第1回目について記載したと同様にパニングし、富化を評価するために溶出ファージを滴定した。
【0180】
図1Aおよび図1Bは、EphA2に選択的かつ高親和性で結合する2つのペプチドが同定された試験の結果を示す。図1Aは、低pH溶液で結合ファージを溶出すると、ファージ回収が最大になることを示し、一方、図1Bは、エフリン−A1で結合ファージを溶出すると、EphA2のリガンド結合部位と相互作用するペプチドの回収が向上することを示す。EphA2の選択に関する数回の操作の後、このスクリーニングにより、BSA結合ファージに対し、約17倍(低pH溶出)および115倍(エフリン−A1溶出)のEphA2結合ファージの富化が得られた。これに対し、エフリン−A1に関するパニングでは、ファージ富化はもたらされず、さらに続行はしなかった(結果は示さず)。
【0181】
低pHで溶出したプール由来の20個の別個のファージクローンのうち19個は、ネガティブコントロールとして使用したエフリン−A1 Fcと比べると、EphA2 Fcに特異的に結合することがわかった。19個のすべてのクローンは、同じペプチド:SWLAYPGAVSYR(SWLペプチド)(配列番号:1)をディスプレイすることがわかった。さらにまた、エフリン-A1で溶出したプール由来10個のファージクローンのうち9個が、EphA2に特異的に結合した。これらのクローンのうち7個がSWLペプチドをディスプレイし、2個がペプチドYSAYPDSVPMMS(YSAペプチド)(配列番号:2)をディスプレイした。
【0182】
また、上記の方法を用いて、適切な固定化受容体Fc融合体に対してファージライブラリーをパニングすることにより、EphA4、EphA5、EphA7、EphB2およびEphB4に結合するペプチドを見出した。かかるパニング実験の結果を以下の実施例
に記載する。
<実施例2>
【0183】
YSAおよびSWLペプチドはEphA2に特異的に結合する
この実施例では、種々のEphA受容体のFc融合タンパク質を捕捉するために、固定化したSWLおよびYSAペプチドを使用した。ビオチン化ペプチドを、ストレプトアビジンコートマイクロタイタープレート(Pierce、ロックフォード、イリノイ州)上に捕捉し、Eph受容体Fc融合タンパク質とともにインキュベートした。結合Eph受容体を、アルカリホスファターゼ(AP)に連結した抗ヒトFc抗体(Promega、マジソン、ウィスコンシン州)で検出した。あるいはまた、固定化したビオチン化ペプチドを、EphA2−APを含有する希釈細胞培養上澄みとともにインキュベートした。基質は、ホースラディッシュ・ペルオキシダーゼには2,2’−アジノ−ビス(3−エチルベンズチアゾリン−6−スルホン酸)、APにはp−ニトロフェニルホスフェートとした。405または450nmにおける吸光度を、ELISAプレートリーダーを用いて測定した。
【0184】
対応する合成SWLおよびYSAペプチドは、EphA2に特異的に結合することがわかった。特に、SWLおよびYSAは、ともにEphA2に結合したが、他のEphA受容体には結合せず(図2)、さらにこれらのペプチドは、EphBファミリーの受容体に結合する能力を欠くことが示された。
<実施例3>
【0185】
EphA2と結合するペプチド間の結合相互作用の特徴付け
この実施例では、BIAcoreシステムを用いて、EphA2と、実施例1で見出されたペプチドとの間の結合相互作用を特徴付けした。EphA2 Fcを、活性化バイオセンサーチップに共有結合により結合し、種々の濃度でのペプチドの結合の平衡を、BIAcore3000を用いて表面プラスモン共鳴における変化を測定することにより調べた。チップは、1M Na2CO3(pH10.5)で洗浄することにより再生した。表面プラスモン共鳴により得られた結合平衡データにより、YSAペプチドは、SWLペプチド(KD=678nM±23)よりも高親和性(KD=186nM±7)でEphA2に結合することが示された(図3Aおよび図3Bを比較のこと)。
<実施例4>
【0186】
YSAおよびSWLペプチドは、細胞表面上のEphA2を標的化する
この実施例は、YSAペプチドおよびSWLペプチドの両方が、培養中の細胞の表面上のEphA2に結合することを実証する。試験した細胞型の各々において、EphA2に対するYSAペプチドの親和性は、SWLペプチドよりもずっと大きい。
【0187】
等量の野生型対象ファージ(WT)ならびにYSAおよびSWLペプチドをディスプレイするファージを、強化緑色蛍光タンパク質(EGFP)(MDA EphA2−EGFP)(オガワ, K.他2000 Oncogene 19: 6043-6052)、非トランスフェクトMDA−MB−435細胞(MDA WT)またはヒト臍帯静脈内皮(HUVE)接着細胞と融合させたEphA2の細胞外ドメインおよび膜貫通型ドメインを過剰発現するMDA−MB−435ヒト乳癌細胞とともにインキュベートした。結合は、懸濁状態のMDA細胞、および接着HUVE細胞を用いて行なった。特に、細胞に結合したファージは、1×109PFUを60−90分間37℃で、1×106個のMDA−MB−435細胞の0.5ml懸濁液とともにインキュベートするか、または1×1010PFUを直接、24ウェル組織培養プレート中のコンフルエントな単層のHUVE細胞に添加するかのいずれかにより定量した。ファージを、1%BSAを含むダルベッコ改変イーグル培地(MDA−MB−435細胞)または1%BSA、10mM Hepesを含む内皮細胞基本培地−2(EBM−2)(Clonetics Products, BioWhittaker, Inc.、ウォーカースビル、メリーランド
州)(HUVE細胞)のいずれかで希釈した。
【0188】
YSAファージは、表面上にEphA2細胞外ドメインを過剰発現するMDA−MB−435ヒト乳癌細胞に対して、野生型ファージよりも、50倍高い結合性を示した。一方、SWLファージは、7倍高い結合性を示した(図4A)。内因性EphA2を低レベルでのみ発現する非トランスフェクトMDA−MB−435細胞の場合、YSAファージは野生型ファージよりも2.5倍高い結合性を示すが、SWLファージは、特異的結合性を示さない(図4B)。また、YSAファージは、中程度レベルのEphA2を発現するヒト臍帯静脈内皮(HUVE)細胞に対して、野生型ファージよりも12倍高い結合性を示す。これに対し、SWLファージは、これらの細胞に対して特異的に結合しない(図4C)。
<実施例5>
【0189】
YSAペプチドは、EphA2のチロシンリン酸化を促進し、下流シグナル伝達を活性化する
この実施例は、YSAペプチドが、EphA2のチロシンリン酸化を促進し、この受容体の活性化が寄与する下流シグナル伝達事象を活性化することを示す。
【0190】
YSAペプチドの存在下または不在下で、2μg/mlのエフリン−A1 FcまたはFcタンパク質で刺激するに先がけて、ヒト臍帯静脈内皮(HUVE)細胞を、10%FCSを含む微小血管内皮細胞培地−2(EGM−2 MV)(Clonetics)および無血清 (serum-starved)EGM−2で2時間成長させた。エフリン−A1 Fcを、0.2μg/mlの抗ヒトFc抗体と、氷上で30分間プレインキュベーションすることにより架橋した。刺激後、細胞を、改変RIPAバッファー(オガワ, K.他2000 Oncogene 19: 6043-6052)中で溶解した。細胞溶解物を5μgの抗EphA2抗体(Upstate、レークプラシッド、ニューヨーク州)で免疫沈降させ、SDSポリアクリルアミドゲル電気泳動により分離し、ペルオキシダーゼ結合抗ホスホチロシン抗体(Transduction Laboratories、サンディエゴ、カリフォルニア州)、または抗EphA2抗体と抗マウスIgGペルオキシダーゼ結合二次抗体(Amersham)を用いる免疫ブロッティングによりプローブした。
【0191】
別の方法では、非血清枯渇HUVE細胞を、YSAペプチドの存在下または不在下で、2μg/mlのエフリン−A1 FcまたはFcタンパク質で刺激した。細胞溶解物を、上記のようにしてEphA2を免疫沈降させるために使用し、または抗リン酸p44/p42MAPK抗体(Cell Signaling Technology、ベバリー、マサチューセッツ州)と抗マウスIgGペルオキシダーゼ結合抗体(Amersham)とを用いる免疫ブロッティングによりプローブし、かつ、抗ERK2抗体(Santa Cruz Biotechnology、サンタクルス、カリフォルニア州)と抗ウサギIgGペルオキシダーゼ結合二次抗体(Amersham)とを用いて再プローブした。Erk2(p42 MAPキナーゼ)が、存在する主なリン酸化形態である。EphA2を、免疫沈降(IP)させ、抗ホスホチロシン(PTyr)抗体または抗EphA2抗体を用いる免疫ブロッティングによりプローブ検索した。
【0192】
HUVE細胞におけるEphA2への結合に加え、YSAペプチドは、エフリンの不在下で受容体のチロシンリン酸化および下流シグナル伝達を刺激するが、エフリンの存在下において、EphA2リン酸化を低減させない(図5)。さらにまた、YSAペプチドは、以前に報告されたMAPキナーゼ活性化を抑制するEphA2シグナル伝達経路を活性化することがわかった(EphA2シグナル伝達経路によるMAPキナーゼ活性化の抑制の考察については、ミャオ他、2001 Nature Cell Biol 3: 527-530を参照のこと)。YSAペプチドによる刺激に応答したMAPキナーゼの抑制を図6Aに示し、これに対応するYSAペプチドに応答したEphA2のリン酸化を図Bに示す。したがって、YSAペプチドは、EphA2のアゴニストである。同様の結果がSWLペプチドについて得られ、
したがって、これもまたEphA2のアゴニストである。
<実施例6>
【0193】
YSAおよびSWLペプチドは、EphA2へのエフリン−Aの結合を阻害する
この実施例は、YSAおよびSWLペプチドがエフリン−A5およびエフリン−A6の結合を濃度依存的に阻害することを示す。Eph受容体コートプレートへのエフリンの結合を、エフリン−A5およびエフリン−A6のアルカリホスファターゼ(AP)融合タンパク質を用いて定量した(これらのAP融合体をコードするプラスミドは上記のものである)。エフリン−A5−APおよびエフリン−A6−APを含有する希釈細胞培養上澄みを、EphA2 FcまたはEphA4 Fcでコートしたマイクロタイターウェル内でペプチドとともに同時インキュベートした。洗浄後も結合したままのエフリンを、AP活性を測定することにより検出した。
【0194】
YSAおよびSWLペプチドは、濃度依存的に、固定化したEphA2へのA−エフリンの結合を阻害するが、EphA4への結合は阻害しない(図7Aおよび図7B)。これは、該ペプチドは、エフリンと相互作用するEphA2の表面に結合することを示す。ELISAアッセイにより、YSAおよびSWLペプチドが、EphA2の球状リガンド結合ドメインに結合することを確認した。球状リガンド結合ドメインは、その受容体のアミノ末端に2つの異なるエフリン結合領域を含むことが示されている(ラブラドル(Labrador), J. P.他1997 EMBO J 16: 3889-3897;ヒマネン(Himanen), J. P.他1998 Nature 396:
486-491)。
<実施例7>
【0195】
SWLおよびYSAは、EphA2上の同じかまたは重複する部位に結合する
この実施例では、YSAおよびSWLペプチドの結合部位を、ファージディスプレイペプチドとの競合実験を用いて特徴付けした。Eph受容体コートプレートに対するファージの結合を、ホースラディッシュペルオキシダーゼに連結した抗ファージ抗体(M13ファージ検出キット、Amersham Pharmacia Biotech.、ピスカタウェイ、ニュージャージー州)を用いて定量した。ファージクローンを用いるペプチド競合アッセイのため、EphA2 FcでコートしたNi−NTAマイクロタイターウェルを、結合バッファー中で1:600−1:9000に希釈したファージクローン(100μl/ウェル)とともに、室温で1時間インキュベートした。非結合ファージを洗い流し、競合ペプチドを1時間添加した。あるいはまた、ペプチドおよびファージを一緒に同時インキュベートした。
【0196】
SWLおよびYSAペプチドは、同じかまたは重複する部位に対する親和性を有することが示された。合成SWLペプチドは、固定されたEphA2に結合したYSAファージと競合し、逆にYSAペプチドは、SWLファージと競合する(図8)。したがって、これらの結果は、YSAおよびSWLペプチドが、EphA2上の同じかまたは重複する部位に結合することを示す。
<実施例8>
【0197】
エフリン配列に相当するペプチドは、Eph受容体に無差別に弱く結合する
この実施例は、SWLおよびYSAペプチドの両方との類似性を有する他の配列が、種々の異なるEph受容体に低親和性で結合することを示す。図9Aは、関連する配列を有するYSAおよびSWLペプチドが、高親和性Eph受容体結合インターフェース(A−エフリンのG−Hループ)(ヒマネン, J. P.他2001 Nature 414: 933-938を参照のこと)に関して或る程度の類似性を有することを示す。SWLおよびYSAペプチドの配列が逆の順序であるとみなせば、低親和性受容体結合インターフェース(A−A’β鎖)との類似性が示される(図9B)。エフリン−A3のG−Hループに相当する12量体合成ペプチド(A3ペプチド、図9A)およびエフリン−A5のA−A’β鎖に相当する12量
体合成ペプチド(A5ペプチド、図9B)は、YSAペプチドよりも弱いが、実際にEphA2に結合する(図10Aおよび図10B)。さらなる受容体結合残基(VADRYAVYWNSSNPR)(配列番号:19)を含むより長いA5ペプチドでさえ、同様の弱い結合性を示す。興味深いことに、A3およびA5ペプチドは、A−エフリンと同様に、すべてのEphA受容体に無差別に結合し、EphB4にも結合する(図11Aおよび図11B)(Eph-Nomenclature-Committee Unified nomenclature for Eph family receptors and their ligands, the ephrins 1997 Cell 90: 403-404)。
【0198】
実施例7で考察した競合方法を用いて、或る特定のエフリン関連配列の結合親和性を調べた。特に、A3ペプチドはファージ結合を阻害しないことがわかり(図14)、これは、おそらく高親和性エフリン結合部位に結合するA3ペプチドは、結合が弱すぎて競合できない可能性を示す。実際、A3ペプチドはまた、EphA2への結合に関してエフリン−A5とも競合しなかった。これに対し、A5ペプチドは、EphA2に対するファージとエフリンA5の両方の結合を増強した。総合すると、これらの結果は、EphA2の低親和性エフリン結合部位へのA5ペプチドの結合により、エフリンならびにYSAおよびSWLペプチドの高親和性部位への結合が増強されることを示す。
<実施例9>
【0199】
Eph受容体に結合するペプチドの同定
この実施例は、ファージディスプレイを用いて、種々の分類型のEph受容体に結合するさらなるペプチドを成功裡に見出すことができることを示す。実施例1に記載の方法を用いて、EphA4、EphA5、EphA7、EphB4およびEphB2に結合し得る他のペプチドを同定した。以下の表は、ペプチド名、受容体親和性、ペプチド配列、およびEphAまたはEphBファミリー受容体に結合することがこれまでわかっている各々の12量体ペプチドの配列番号を示す。
【0200】
【表1】

<実施例10>
【0201】
EphB4に対するDAL、IPWおよびSGHペプチドの結合
この実施例では、先の結合特性実験で使用したものと同様の方法を用いて、DAL、IPWまたはSGHペプチドをディスプレイするファージクローンがEphB4 Fcに結合したことを示した。図17は、これらのファージディスプレイペプチドの各々が、EphA2より6−10倍高い親和性でEphB4に結合することを示す。さらにまた、ファージディスプレイペプチドの各々は、試験したいずれのEphAファミリー受容体(Eph1−Eph7)よりも高い親和性でEphB4に結合した。
<実施例11>
【0202】
EphB4に対するDALペプチドの特異性
この実施例では、実施例2で記載した方法を用いて、DAL合成ペプチドがEphB4受容体に特異的に結合することを示した。EphBファミリー受容体を固定化したDALペプチドとインキュベートすると、DALペプチドは、他のEphBファミリー受容体と比べて、4倍多くEphB4に結合することが観察された。同様の特異性が、EphAファミリー受容体およびEphB4を、固定化したDALペプチドに対して試験した場合にも観察された。
<実施例12>
【0203】
EphA4に結合するペプチドの同定
この実施例では、ファージディスプレイを用いて、EphA4受容体に結合するペプチドを同定した。ランダムな12量体ペプチドをディスプレイするM13ファージライブラリーを、ヒトFc領域に融合させた固定化EphA4受容体細胞外ドメインに関してパニングした(図12A)。EphA4に結合したままのファージクローンを、低pH溶液で溶出し、増幅した。EphA4の選択に関して4回操作した後、BSAネガティブコントロールと比べて、約80倍のEphA4結合ファージの富化が検出された。
【0204】
個別に試験した38個のクローンのうち20個がEphA4 Fcに結合したが、エフリンA1 Fcネガティブコントロールには結合しなかった。EphA4結合クローンは、4つの異なるペプチド配列を示した(図12B−図12E)。これらのペプチドを、APY(APYCVYRGSWSC)(配列番号:20)、KYL(KYLPYWPVLSSL)(配列番号:21)、VTM(VTMEAINLAFPG)(配列番号:22)およびNHW(NHWLDTLFPMHM)(配列番号:46)とよぶ。これらのペプチドをディスプレイするファージクローンは、すべて、他のEphAまたはEphB4受容体と比べて優先的にEphA4に結合した(図12B−図12E)。しかしながら、NHWファージは、試験した他のEph受容体に対してもまた実質的な結合を示した。したがって、EphA4に対してより選択性であると思われる3つのペプチド(APY、KYLおよびVTM)を、その特異性および機能的特性を調べるために合成した。
<実施例13>
【0205】
3つの合成ペプチドは、EphA4に高親和性で優先的に結合する
この実施例では、ペプチド断片をディスプレイするファージクローンと同様、対応する合成ペプチドが、他のEphA受容体と比べて、EphA4に優先的に結合することがわかった。VTMペプチドは、EphA4に対して高度に選択的であるが、APYおよびKYLペプチドは、他のEphAおよびEphB受容体に対して或る程度の結合性を示した(図13)。固定化したペプチドに対する可溶性EphA4 Fc融合タンパク質の結合の平衡は、EphA4が、VTMペプチドよりもKYLおよびAPYペプチドに対してより良好に結合することを示す。ペプチドに対する二量体EphA4 Fcタンパク質の見かけ上の結合親和性は、低ナノモル濃度範囲においてであり、これは、EphA4−ペプ
チドの相互作用が高親和性であることを示す。
<実施例14>
【0206】
APY、KYLおよびVTMペプチドは、EphA4への結合に対して互いに競合する
この実施例では、アミノ酸配列において密接に関連しないAPYペプチドおよびKYLペプチドが、モチーフΦXXΦ(式中、Φは、芳香族アミノ酸を示し、Xは任意のアミノ酸である)を共有することが示された(図19)。VTMペプチドの配列は、APYペプチドおよびKYLペプチドの両方と異なっており、ΦXXΦモチーフを含まない。EphA4の多ドメイン細胞外領域全体をパニングに用いたと考えると、ペプチドは、受容体の異なる領域に結合し得ることが考えられた。しかしながら、3種類のペプチドを用いて、固定化したEphA4へのVTMペプチドディスプレイファージの結合に関して競合させた実験では、3種類すべてのペプチドが、VTMファージクローンおよびAPYファージクローンの結合を拮抗阻害し得ることが示された(図14)。したがって、3種類すべてのペプチドは、EphA4上の同じか、または一部重複する部位に結合するようである。<実施例15>
【0207】
APY、KYLおよびVTMペプチドは、EphA4へのエフリンの結合を拮抗阻害し、受容体のエフリン誘導性活性化を阻害する
この実施例では、APYおよびKYLペプチドのΦXXΦモチーフは、エフリン−Aリガンドの高親和性受容体結合部位、および別のEphA受容体へのエフリンの結合を阻害する先に単離した2つのペプチドにも存在することがわかった(上記および図19参照)。これは、これらのEphA4結合ペプチドもエフリンの結合を阻害し得ることを示した。配列が異なるにもかかわらず、3種類すべてのEphA4結合ペプチドは、アルカリホスファターゼ標識を有するエフリン−A5(エフリン−A5 AP)のEphA4への結合を、投与量依存的に阻害した(図15)。KYLペプチドが最も有効であり、1μMの濃度でほぼ完全にエフリンの結合を阻害した。
【0208】
同様のレベルの阻害を達成するには、APYペプチドは約10μM、VTMペプチドは約100μM必要であった。EphA4結合曲線と同様、これらの阻害実験は、KYLペプチドが最も高い結合親和性を有し、VTMペプチドが最も低いことを示す。さらにまた、異なるエフリン濃度でのさらなるペプチド阻害曲線により、これらのペプチドが受容体のエフリンの結合を競合的に阻害することを確認した。したがって、これらのペプチドは、EphA4のエフリン結合部位に結合し、マイクロモル濃度で使用しても、この受容体へのエフリンの結合を阻害し得る。これらのペプチドはまた、他のEphA受容体と比べて、EphA4へのエフリン−A5 APの結合を、優先的に(APYおよびKYL)または選択的に(VTM)にブロックすることがわかった(図16)。しかしながら、図16で使用したペプチド濃度(50μM)では、EphA4に対する選択性は、3種類すべてのペプチドで十分であった。
【0209】
EphA4結合ペプチドが、エフリンによるEphA4活性化を阻害するか、あるいはまたEphA4を活性化するか否かを調べるため、これらのペプチドを、エフリン−A3
Fcとともに海馬薄片に投与した(ムライ, K. K.他2003 Nat Neurosci 6: 153-160)。3種類すべてのペプチドは、マイクロモル濃度で、内因性EphA4のエフリン−A3誘導性チロシンリン酸化を拮抗阻害することがわかった(図17)。また、これらのペプチドは、自身の受容体を活性化する本来の能力は示さなかった。KYLペプチドは、APYおよびVTMペプチドよりも低濃度でEphA4チロシンリン酸化をブロックし、その相対的に高い結合親和性と一致する。
<実施例16>
【0210】
KYLペプチドは、神経冠細胞の分節間遊走(segmental migration)に必要なエフリン/
EphA4相互作用を撹乱する
この実施例では、EphA4結合ペプチドが内生エフリン−EphA4生理学的機能を阻害し得るか否かを検討するため、神経冠細胞遊走アッセイ(マックレナン(McLennan), R. & クルル(Krull), C. E. 2002 Gene Expr 10: 295-305)を利用した。遊走前(premigratory)神経冠細胞を含むひな鳥神経幹外植片を、10μg/mlの対照Fc、10μg/mlのEphA4−Fc(陽性対照)、13μM KYLペプチドまたは13μMの対照ペプチドを加えた培地内で24時間成長させた。神経冠細胞の遊走は、神経冠細胞についてHNK−1を染色し、エフリン−B1を染色して尾側椎板(caudal sclerotome)を標識することにより調べた。KYLペプチドは、その見かけ上の高親和性により選択した。神経冠細胞は、対照Fcで処理した外植片内の吻側椎板内に適正に遊走した。しかしながら、EphA4−Fc処理では、既報(マックレナン, R. & クルル, C. E. 2002 Gene Expr 10: 295-305)のように、神経冠細胞の尾側半椎板への異常な遊走が引き起こされた。KYLペプチドでの処理でも同様に、神経冠細胞の尾側半椎板への異所性遊走がもたらされ、一方、EphA4に結合しない対照ペプチドは、検出可能な作用はなかった。したがって、KYLペプチドは、適正な神経冠細胞遊走を可能にする内生EphA4/エフリンコンジュゲートの生物学的機能に影響を及ぼす可能性がある。
<実施例17>
【0211】
成体脳で発現する他の2つのEphA受容体:EphA5およびEphA7に結合するペプチドの同定
この実施例では、さらなるファージパニング実験を行ない、EphA5およびEphA7に結合するペプチドを同定した(図18A)。EphA5およびEphA7は、EphA4と密接に関連するが、発達中の神経系および成体神経系において発現する点で異なる2つのEphA受容体である(エリス, J.他1995 Mechanisms of Development 52: 319-341;モリ, T.他1995 Brain Res Mol Brain Res 34: 154-160;オリビエリ, G. & ミーシャー, G. C.他1999 J Histochem Cytochem 47: 855-861;チャン, J. H.他1997 Brain Res Mol Brain Res 47: 202-214)。各受容体に結合する3つの異なるペプチド配列を同定した。EphA5に関して単離したペプチドのうち2つ((SLRDTYMRAKVL、配列番号:27)および(WDCNGPYCHWLG、配列番号:28))は、この受容体に対して選択的結合性を示したが、もう1つのペプチド(WTFPVLWDDKHP、配列番号:29)はEphA6にも結合する。EphA7結合ペプチドのうち、1つ目(WASHAPYWPHPP、配列番号:61)は、EphA3にも結合し、2つ目(SVSVGMKPSPRP、配列番号:25)はEphA3およびEphA6にも結合し、3つ目(KHLPFYPHPTSP、配列番号:62)はEphA3、EphA5およびEphA6にも結合する。
【0212】
【表2】
【0213】
【表3】
【0214】
【表4】
【0215】
受容体チロシンキナーゼのEphAサブクラスの異なるメンバーを標的化する一群の新規ペプチドを同定した。これは、エフリンリガンドとは対照的であり、同じサブクラス内のほとんどのEph受容体に無差別に結合する(フラナガン & バンデルヘーゲン 1998 A
nn Rev Neurosci 21: 309-345)。3種類のEphA4結合ペプチドをさらに特徴付けすると、EphA4へのエフリンの結合を拮抗阻害し、エフリン誘導性EphA4の活性化および生物学的機能をブロックすることがわかった。これらのペプチドは、エフリン−Eph受容体相互作用に干渉するため、それぞれ、EphA4およびそのリガンドの下流に伝達される順方向および逆方向の両方のシグナルを阻害すると考えられる。
【0216】
最近解明されたエフリン/Eph受容体コンジュゲートの結晶構造は、エフリンの受容体結合領域が、αヘリックスG−H間に延長された15アミノ酸ループを含むことを示す(ヒマネン, J. P.他2001 Nature 414: 933-938)。このG−Hループの配列アラインメントは、いくつかのアミノ酸がすべてのエフリン間で保存されているが、A−エフリンとB−エフリンとで異なるものもあり、おそらく、EphAまたはEphB受容体に対する特異性に寄与していることを示す(図19)。興味深いことに、EphA4結合ペプチドのうちの2つ(APYおよびKYLペプチド)およびEphA5結合ペプチドのうちの1つ(WDCペプチド)が、2つの非保存アミノ酸により分断された2つの芳香族残基からなるA−エフリンのG−Hループ内に見られるモチーフ(ΦXXΦ)を含んでいた(図19)。
【0217】
このモチーフは、EphA2受容体に結合したペプチドにも存在することがわかった。相当数の同定されたEphB受容体結合ペプチドは、ΦXXXΦまたはΦXXXXΦモチーフを有した。異なるEph受容体に結合したペプチド間でのΦXnΦ(n=2−4)モチーフの保存は、2つの芳香族残基が、これらの受容体との相互作用に重要であり、Eph受容体のエフリン結合ポケットの保存領域内に入り込み得る(EphA受容体ではΦXXΦが有利であり、EphB受容体ではΦXXXΦまたはΦXXXXΦが有利である)ことを示す。芳香族残基に隣接する残基が、特定のEphAまたはEphB受容体への結合に対する特異性をもたらすと考えられる。
【0218】
他のアミノ酸残基もまた、受容体への結合および特異性に重要であると考えられる。APY EphA4結合ペプチドおよびWDC EphA5結合ペプチドは、これらのペプチドが分子内ジスルフィド結合を形成するのを可能にする2つのシステイン残基を含む。立体構造上束縛のある環状構造は、ペプチドの高結合親和性および受容体結合選択性の決定において重要であり得る。また、ペプチドおよびエフリンG−Hループ内に存在するプロリンは、Eph受容体結合において或る役割を果たし得る。
【0219】
EphA4結合ペプチドのうち1つ(VTMペプチド)は、他の2つのEphA4結合ペプチドとの明白な配列類似性を示さなかった。しかしながら、EphA4は、A−エフリンに加えてエフリン−B2およびエフリン−B3に結合するため、例外的なリガンド特異性を有する。したがって、一部のEphA4結合ペプチドは、B−エフリンのG−Hループに特徴的なアミノ酸を含み得る。実際、VTMペプチドは、エフリン−B2およびエフリン−B3のG−Hループに見られる(が、EphA4に結合しないエフリン−B1には見られず)配列、アスパラギン−ロイシン(NL)を含む(図19)。したがって、NLモチーフは、B−エフリンとEphA4との接触の重要な部位であると考えられる。あるいはまた、VTMペプチド内のフェニルアラニン(F)は、A−エフリンおよびB−エフリンの両方で保存されているΦXnΦモチーフの第2芳香族残基に相当し得る。それにもかかわらず、VTMペプチドは、エフリンの結合を競合的に阻害し、これは、該ペプチドがEphA4のリガンド結合ポケットに結合することを示す。
<実施例18>
【0220】
Eph受容体結合因子を同定するためのハイスループットスクリーニング方法
ハイスループットスクリーニングアッセイは、Eph受容体−エフリンコンジュゲートを分解する低分子の化学物質ライブラリーをスクリーニングするために使用されている。
このアッセイでは、エフリン−アルカリホスファターゼ融合タンパク質とのコンジュゲートの状態の固定化したEph受容体エクトドメインを使用する。結合したアルカリホスファターゼ活性を低下させる能力を利用して、Eph−エフリン相互作用の低分子インヒビターを同定する。
【0221】
個々のEph受容体を標的化し、生物活性特性を有する低分子は、受容体の機能を解明するため、およびその特定の受容体が富化した組織の生理機能を選択的に撹乱するための両方に有用である。例えば、ペプチドの1つは、内因的に発現しているEphA4とそのエフリンリガンドとの相互作用を阻害し、生物学的影響をもたらした。EphA4シグナル伝達を選択的に阻害すると、この特定の受容体が高度に発現する海馬および大脳皮質において、ニューロンの形状発達、再構築または再生を方向付ける反発的(repulsive)リガンド−受容体相互作用を低減し得る。実際、EphA4シグナル伝達は、海馬における樹状突起棘の形態および組織化を維持することがわかっている(ムライ他2003 Nat Neurosci 6: 153-160)。これは、おそらく、エフリン−A3陽性星状膠細胞との一過的な反発的接触により起こるだろう。興味深いことに、ペプチドは、海馬薄片においてエフリン−A3 Fcによる内生EphA4の活性化をブロックする。したがって、これらは、突起棘の拡張を制限するインビボリガンド−受容体相互作用に干渉し得る。例えば、脳損傷は神経膠症を誘導し、これは、神経膠エフリン−A3によるEphA4の活性化により起こり得る樹状突起棘の大きさの縮小と相関している(トンプソン(Thompson)、2003 Nat Neurosci 6:103-104)。EphA4結合ペプチドは、阻害性シグナルの一部を打ち消し、機能回路の再生を促進すると考えられる。他方において、EphA4を活性化できる人工的ペプチドは、精神遅滞を伴う患者における伸長樹状突起棘の正常形状を回復させると予想される。
【0222】
反発因子および成長阻害因子の存在により、中枢神経系の再生能力が制限されると考えられている。これは、外傷性脳損傷またはニューロン変性後、非許容シグナルをブロックし、構造的可塑性を促進することが所望される場合、特に重要であろう。興味深いことに、EphA4結合ペプチドは、神経幹細胞が、失われたニューロンの置き換えを生涯にわたって行なう歯状回において新たに生成したニューロンのより高速な成熟および統合(integration)を促進し得る。
【0223】
また、同定したペプチドのうちいくつかは、脊椎内の損傷軸索の再生を補助すると考えられる。数個のエフリンならびにEphA4およびEphA7を含むEph受容体は、脊椎損傷または求心路遮断後にアップレギュレートされる(ミランダ(Miranda)他1999 Exp Neurol 156: 218-222;ウィルソン(Willson)他2002 Cell Transplantation 11: 229-239)。また、EphA4は、発達中の運動軸索の誘導のため(エバーハート(Eberhart)他2002 Dev Biol 247:89-101)および皮質脊髄線維の正確な軌道(trajectory)を確立するため(ドットーリ(Dottori)他1998 PNAS USA 95: 13248-13253;クランダー(Kullander)他2001 Nat Rev Mol Cell Biol 3: 475-486)に重要である。これらの軸索において、EphA4は、反発的誘導事象を媒介すると考えられている。EphA4媒介性反発は、中枢神経系における軸索の再成長を制限することが可能で、かかる相互作用の阻害は、再生を促進し得る。
【0224】
また、EphA受容体結合ペプチドは、限られた特有の再生能力を示す中枢神経系の障害領域を含む、特定の組織に試薬を送り届ける(escort)のを補助すると考えられる。これらのペプチドに医薬剤を連結させることにより、変性しつつあるニューロンに栄養を補助すると同時に成長に対する阻害的拘束を減弱する分子の標的化送達が可能になる。EphA4結合ペプチドは、神経疾患を有する患者の解剖学的および認知機能を改善するための海馬および皮質などの脳組織への選択的送達に有用である。また、EphA5結合ペプチドは、黒質および小脳に試薬を送達するため(ユー(Yue)他1999 J Neurosci 19: 2090-21
01)およびこれらの脳領域に影響する神経疾患を治療するために有用である。
【0225】
神経系以外では、血小板に対するEphA4のペプチド阻害は、血液凝固プロセスを変更するために適用できると考えられる(プレボー(Prevost)他2002 PNAS USA 99: 9219-9224)。さらにまた、これらのペプチドは、発達研究のため、インビボまたはインビトロでEphA4機能を選択的に撹乱させるのに有用である。これには、本明細書に記載した神経冠細胞の移動が含まれる。
【0226】
EphA受容体結合ペプチドは、機能的に万能な試薬であり、種々の疾患および病状を治療するための重要なツールである。これらは、細胞遊走、軸索誘導およびシナプス可塑性におけるEphA受容体の役割の解析を目的とした発達研究の面において、ならびに中枢神経系への障害を伴い得る異常EphA受容体シグナル伝達の調節のためにも有用である。
【0227】
本発明を、明瞭にしかつ理解する目的で、或る程度詳細に記載したが、当業者であれば、本発明の真の意図から逸脱することなく、形態および詳細において種々の変更がなされ得ることが理解されよう。
【図面の簡単な説明】
【0228】
【図1−1】図1Aは、固定化したEphA2 Fcまたは固定化したウシ血清アルブミン(BSA)のいずれかに対する連続パニング操作(R1−R3)で得られた、ランダム12量体ファージディスプレイライブラリー由来のファージの力価(pfu/μl)を示す棒グラフである。結合したファージを低pHで溶出した。得られたEphA2結合クローンのペプチド配列を示す。
【図1−2】図1Bは、固定化したEphA2 Fcまたは固定化したウシ血清アルブミン(BSA)のいずれかに対する連続パニング操作(R1−R2)で得られた、ランダム12量体ファージディスプレイライブラリー由来のファージの力価(pfu/μl)を示す棒グラフである。結合したファージをエフリン−A1で溶出した。得られたEphA2結合クローンのペプチド配列を示す。
【図2】固定化したSWLおよびYSAペプチド間での、EphA2 Fc対他のEphA受容体Fc融合タンパク質の結合特異性を示す棒グラフである。
【図3】種々の濃度のYSAペプチド(図3A)またはSWLペプチド(図3B)について測定したときの、EphA2 FcでコートしたBIACOREバイオセンサーチップの表面上の表面プラスモン共鳴単位における変化を示す線グラフである。解離定数は、非線形回帰により求めた。
【図4】強化緑色蛍光タンパク質(MDA EphA2−EGFP)(図4A)、非トランスフェクトMDA−MB−435細胞(MDA WT)(図4B)、またはヒト臍帯静脈内皮(HUVE)接着細胞(図4C)と融合させたEphA2の細胞外ドメインおよび膜貫通型ドメインを過剰発現するMDA−MB−435ヒト乳癌細胞に結合した野生型M13ファージ(WT)、YSAペプチドをディスプレイするファージ(YSA)、およびSWLペプチドをディスプレイするファージ(SWL)の数を比較する棒グラフである。
【図5】Fc(レーン1−3)、エフリン−A1−Fc抗Fc架橋コンジュゲート(レーン4−6)およびエフリン−A1−Fc(レーン7−9)での処理前に、0、10または50μMいずれかのYSAペプチドで処理したHUVE細胞におけるEphA2のリン酸化を示すイムノブロットである。EphA2は、免疫沈降(IP)させ、抗ホスホチロシン(PTyr)または抗EphA2抗体を用いるイムノブロッティング(IB)によりプローブした。
【図6】Fc処理前にYSAペプチドで処理したHUVE細胞におけるMAPキナーゼ(MAPK)のリン酸化(図6A)(レーン3および4)および対応するEphA2のリン酸化(図6B)(レーン3および4)の阻害を示すイムノブロットである。IBは免疫沈降を示し、IPはイムノブロット前の免疫沈降を示す。
【図7】SWLおよびYSAペプチドが、固定化したEphA2へのエフリン−A5(図7A)およびエフリン−A6(図7B)の結合を濃度依存的に拮抗阻害するが、EphA4ではそうでないことを示すグラフである。
【図8】SWLおよびYSAペプチドが、EphA2上の同じかまたは重複する部位に結合することを示すグラフである。
【図9】図9Aでは、SWLおよびYSAペプチド配列のアラインメント、A−エフリンのG−Hループ、およびこのアラインメントに基づいて合成した「A3ペプチド」を示す。同一アミノ酸を濃い灰色で示し、類似した特徴を有するアミノ酸を薄い灰色で示す。図9Bでは、SWLおよびYSAペプチドの逆配列のアラインメント、エフリンのA−A’β鎖、およびこのアラインメントに基づいて合成した「A5ペプチド」を示す。同一アミノ酸を濃い灰色で示し、類似した特徴を有するアミノ酸を薄い灰色で示す。
【図10】ビオチン化したA3、A5と、EphA2 Fcを捕捉するために使用したストレプトアビジンコートウェル上に固定化したYSAペプチドとの間の結合の結果を示す棒グラフである。5分(図10A)および20分(図10B)におけるODの目盛りの値を示す。
【図11】ストレプトアビジンコートウェル上に固定化した、ビオチン化したA3(図11A)、およびA5(図11B)ペプチドと、EphA受容体のFc融合タンパク質との間の結合の結果を示す棒グラフである。すべてのパネルにおけるエラーバーは、2回の測定値の標準偏差を示す。
【図12−1】EphA4受容体を標的化するファージクローンの同定および結合選択性を示す棒グラフである。ランダムな12量体ペプチドをディスプレイするM13ファージがEphA4 FcまたはBSA(対照)に結合したことを示す(図12A)。
【図12−2】EphA4受容体を標的化するファージクローンの同定および結合選択性を示す棒グラフである。APY(図12B)、KYL(図12C)、VTM(図12D)、およびNHW(図12E)で始まるペプチドをディスプレイするファージクローンがEphA4に優先的に結合したことを示す。
【図13−1】合成EphA4結合ペプチドの結合選択性を示す棒グラフである。ストレプトアビジンコートプレート上に固定化した、ビオチン化したAPY(図13A)、KYL(図13B)、およびVTM(図13C)ペプチドが、EphA受容体Fcタンパク質を捕捉したことを示す棒グラフである。
【図13−2】合成EphA4結合ペプチドの結合選択性を示す棒グラフである。ストレプトアビジンコートプレート上に固定化した、ビオチン化したAPY(図13D)、KYL(図13E)、およびVTM(図13F)ペプチドが、EphB受容体に対して低結合能力を示したことを示す棒グラフである。
【図13−3】合成EphA4結合ペプチドの結合選択性を示す棒グラフである。固定化したペプチドに対する二量体EphA4 Fcの相対的結合親和力を示す棒グラフである(図13G、13H、13I)。
【図14】APY、KYLおよびVTMペプチドが、EphA4上の同じか、または一部重複する部位に結合することを示すグラフである。
【図15−1】図15Aは、APYペプチドが、EphA4へのエフリン−A5結合を濃度依存的に(対照ペプチド、白四角形)拮抗阻害することを示すグラフである。
【図15−2】図15Bは、KYLペプチドが、EphA4へのエフリン−A5結合を濃度依存的に(対照ペプチド、白四角形)拮抗阻害することを示すグラフである。
【図15−3】図15Cは、VTMペプチドが、EphA4へのエフリン−A5結合を濃度依存的に(対照ペプチド、白四角形)拮抗阻害することを示すグラフである。
【図16】対照(図16A)、APY(図16B)、KYL(図16C)、およびVTM(図16D)ペプチドが、その他のEphA受容体と比べ、EphA4へのエフリン−A5結合を優先的に阻害することを示す棒グラフである。
【図17】APY(図17A)、KYL(図17B)、およびVTM(図17C)ペプチドが、マウス海馬薄片において、内因性EphA4のエフリン−A3誘導型活性化を拮抗阻害することを示すグラフである。
【図18−1】EphA5結合ペプチドおよびEphA7結合ペプチドの同定および結合選択性を示す棒グラフである。ランダムな12量体ペプチドをディスプレイするM13ファージがEphA5 Fc、EphA7 Fc、またはBSA(対照)に結合したことを示す(図18A、18B)。
【図18−2】EphA5結合ペプチドおよびEphA7結合ペプチドの同定および結合選択性を示す棒グラフである。EphA5に関するパニングにより単離されたファージクローンは、EphA5に選択的に結合するペプチドSLR(図18C)およびWDC(図18D)をディスプレイするが、ファージクローンの1つWTF(図18E)は、EphA5およびEphA6の両方に結合するペプチドをディスプレイすることを示す。
【図18−3】EphA5結合ペプチドおよびEphA7結合ペプチドの同定および結合選択性を示す棒グラフである。EphA7に関するパニングにより単離された3つのファージクローンは、EphA7を含むEphA受容体の種々のサブセットに結合するペプチドをディスプレイする(図18F、18G)。
【図19−1】Eph受容体結合ペプチドの、A−エフリンおよびB−エフリンのG−Hループに対するアラインメントである(図19A)。
【図19−2】Eph受容体結合ペプチドの、A−エフリンおよびB−エフリンのG−Hループに対するアラインメントである(図19B)。
【配列表】
【技術分野】
【0001】
本発明は、Eph受容体機能を調節する新規リガンドに関する。
【0002】
[米国政府の所有権]
本発明は、米国国立衛生研究所により与えられた助成金番号CA82713およびNS43029ならびに米国防総省により与えられた助成金番号DAMD17−01−1−0168のもと、米国政府の援助によりなされた。米国政府は、本発明において一定の権利を有する。
【0003】
[関連技術の記載]
Eph受容体は、そのエフリンリガンドに結合すると活発にシグナル伝達を行なう、密接に関連する膜貫通型チロシンキナーゼの大きな一ファミリーを構成する(フラナガン(Flanagan) & バンデルヘーゲン(Vanderhaeghen) 1998 Ann Rev Neurosci 21:309-345); マニング(Manning)他2002 Trends Biochem Sci 27:514-520; ムライ(Murai), K. & パスクワレ(Pasquale), E. 2003 J. Cell Sci 116:2823-2832)。16個のEph受容体は、配列相同性に基づいて2つのサブグループ(EphAおよびEphB)に分けられる。EphA受容体は、GPI結合エフリン−Aリガンドと結合するが、EphB受容体は、膜貫通型エフリン−Bリガンドと結合する。
【0004】
もともとは神経発達の調節因子として同定された、Ephファミリーの受容体チロシンキナーゼおよびそのエフリンリガンドはまた、血管発達および病的な血管新生にも重要である(フラナガン, J. G. & バンデルヘーゲン P. 1998 Ann Rev Neurosci 21:309-345); ドデレット(Dodelet), V. C. & パスクワレ, E. B. 2000 Oncogene 19:5614-5619; ヤンコポウラス(Yancopoulos), G. D.他2000 Nature 407:242-248)。例えば、EphA2受容体およびエフリン−A1(EphA2のリガンド)は、ヒト腫瘍およびヒト癌細胞から成長させたマウス異種移植腫瘍の血管系において、協調的に発現する(オガワ(Ogawa),
K.他2000 Oncogene 19:6043-6052)。EphA2受容体は、血管内皮成長因子(VEGF)および腫瘍壊死因子α(TNFα)誘導性血管新生において重要な役割を果たす。これは、VEGFおよびTNFαがエフリン−A1をアップレギュレートし、このことが、血管における受容体活性化を引き起こすからである(パンデイ(Pandey), A.他1995 Science 268:567-569; チェン(Cheng), N.他2002 Mol Cancer Res 1:2-11)。同様に、ホメオボックス転写因子Hox B3は、エフリン−A1をアップレギュレートすることにより、血管新生を促進する(マイアーズ(Myers), C.他2000 J Cell Biol 148:343-351)。さらにまた、EphA2シグナル伝達は、インビトロ法での内皮毛細血管の形成に必要とされ(オガワ, K.他2000 Oncogene 19:6043-6052; ダニエル(Daniel), T. O.他1996 Kidney Int Suppl 57:S73-81)、黒色腫細胞による血管様構造の形成を促進する(ヘス(Hess), A. R.他2001 Cancer Res 61:3250-3255)。EphA2の発現は、受容体が胚血管または成体の沈静状態血管のいずれにおいても検出されていないため、「活性化された」成体血管に限定されているようである(オガワ, K.他2000 Oncogene 19:6043-6052; ルイス(Ruiz), J.
C. & ロバートソン(Robertson), E. J. 1994 Mech Dev 46:87-100; ガンジュ(Ganju), P.他1994 Oncogene 9:1613-1624)。エフリン−A1もまた成体血管においては検出されていないが、胎児の血管構造には存在する(マクブライド(McBride), J. L. & ルイス, J. C. 1998 Mech Dev 77:201-204)。
【0005】
腫瘍内皮細胞に存在する以外に、EphA2およびエフリン−A1は、乳癌、前立腺癌、結腸癌、皮膚癌および食道癌を含む多種多様な腫瘍の形質転換細胞内でアップレギュレートされる(オガワ, K.他2000 Oncogene 19:6043-6052; ゼリンスキー(Zelinski), D. P
.他2001 Cancer Res 61:2301-2306; ウォーカー−ダニエル(Walker-Daniels), J.他1999 Prostate 41:275-28; イースティー(Easty), D. J.他1995 Int J Cancer 60:129-136; ネモト(Nemoto), T.他1997 Pathobiology 65:195-203)。癌遺伝子H−Ras、E−カドヘリン、転写調節因子のp53ファミリーメンバー、DNA損傷、ならびにエストロゲン受容体およびc−Mycの減少を含む多くの因子が、癌細胞におけるEphA2発現を増加させる(アンドレス(Andres), A. C.他1994 Oncogene 9:1461-1467; ドーン(Dohn), M他2001 Oncogene 20:6503-6515; ゼリンスキー, D. P.他2002 J Cell Biochem 85:714-720)。
【0006】
腫瘍血管系は、本質的に不連続で漏出性であるため、血管と腫瘍細胞の両方に癌根絶剤を送達するのにEphA2およびエフリン−A1のアップレギュレートを利用することが可能である(ドボラク(Dvorak), H. F.他1988 Am J Pathol 133:95-109)。実際、全身投与された生物学製剤は、血液循環から腫瘍に容易に浸透する(エスラー(Essler), M. & ルオスラーティ(Ruoslahti), E. 2002 PNAS USA 99:2252-2257)。しかしながら、EphA2およびエフリン−A1を選択的に標的化することは、これらのタンパク質が、密接に関連するタンパク質から成る大きなファミリーに属するものであるため、困難な課題である(Eph-Nomenclature-Committee Unified nomenclature for Eph family receptors and
their ligands, the ephrins 1997 Cell 90:403-404)。
【0007】
EphA4は、Ephファミリーの受容体チロシンキナーゼの別のメンバーである。これは、発達中および成体の神経系において重要な機能を有する。神経発達時のその既知の発現パターンの他に(モリ(Mori), T.他1995 Brain Res Mol Brain Res 29:325-335; ニエト(Nieto), M. A.他1992 Development 116:1137-1150; オオタ(Ohta), K.他1996 Mechanisms of Development 54:59-69; ソーンズ(Soans), C.他1994 Oncogene 9:3353-3361)、EphA4は、広範なシナプス再構築を示す脳領域で発現している(ムライ, K.他2003 Nature Neurosci 6:153-169)。成体では、EphA4は、学習と記憶に重要な脳組織である海馬および皮質において富んでいる。
【0008】
EphA5およびEphA7は、Eph4と密接に関連する受容体であるが、発達中の神経系および成体神経系において異なった形で発現する2つのEphA受容体である(エリス(Ellis), J.他1995 Mechanisms of Development 52:319-341; モリ, T.他1995 Brain
Res Mol Brain Res 34:154-160; オリビエリ(Olivieri), G. & ミーシャー(Miescher), G. C.他1999 J Histochem Cytochem 47:855-861; チャン(Zhang), J. H.他1997 Brain Res Mol Brain Res 47:202-214)。例えば、EphA5受容体は、多くのヒト神経膠腫および神経膠芽腫細胞株において過剰発現する(ブルース(Bruce), V.他1999 Brain Res 821:169-176; ミーシャー, G. C.他1997 Mol Brain Res 46:17-24)。
【0009】
EphB2およびEphB4受容体は、ともに、或る種の腫瘍組織において過剰発現する。EphB4過剰発現は、主に、悪性度の高い浸潤性乳管癌腫において見られるが(ベルクラス(Berclaz), G.他1996 Biochem Biophys Res Commun 226:869-875)、EphB2は、たいていの胃腫瘍において過剰発現する(キヨカワ(Kiyuokawa), E.他1994 Cancer Res 54:3645-3650 )。両受容体は、多くの腫瘍細胞株でも同様に過剰発現している(ベルクラス, G.他1996 Biochem Biophys Res Commun 226:869-875; キヨカワ, E.他1994 Cancer Res 54:3645-3650; ベネット(Bennett), B. D.他1995 PNAS USA 92:1866-1870)。また、両EphB2およびEphB4は、結腸癌腫組織においてアップレギュレートされる(リウ(Liu), W.他2002 Cancer 94:934-939; ステフェンソン(Stephenson), S.他2001 BMC Mol Biol 2:15-23)。さらに、EphB2およびEphB4はまた、胚およびおそらく腫瘍における血管系発達に重要である(ワン(Wang), H. U.他1998 Cell 93:741-753; ゲレティ(Gerety), S. S.他1999 Mol Cell 4:403-414)。
【0010】
異なるアゴニスト活性および薬物標的化活性を有するEph受容体結合ペプチドが同定されれば、重要な治療法になろう。例えば、生体活性型エフリン擬似ペプチドは、Eph受容体発現組織に薬剤を選択的に送達し、また、癌、病的血管新生、神経再生および認知障害の治療におけるEphシグナル伝達を変更するために使用し得る。同定されたEph結合ペプチドの他の考えられ得る適用としては、負傷による慢性的な痛みの治療(バッタリア(Battaglia), A. A.他2003 Nature Neurosci 6:339-340)、成体脳における神経芽細胞の増殖の刺激(コノバー(Conover), J. C.他2000 Nature Neurosci 3:1091-1097)、および学習および記憶において重要な役割を有すると考えられるシナプス可塑性の調節(ムライ, K. K. & パスクワレ, E. B. 2002 Neuron 33:159-162)が挙げられる。
【発明の開示】
【0011】
本発明の一実施形態は、Eph受容体ファミリーのメンバーすなわちこれら受容体のサブセットに選択的に結合する化合物である。
【0012】
本発明の別の実施形態は、単離されたペプチド、擬似ペプチド、またはEph受容体ファミリーのメンバーすなわちこれら受容体のサブセットに選択的に結合する低分子である。
【0013】
本発明の別の実施形態は、ペプチド、擬似ペプチド、またはEph受容体ファミリーメンバーに選択的に結合する低分子を含むEph受容体結合化合物の有効量と細胞を接触させることによる、細胞中のEph受容体活性の調節方法である。Eph受容体結合化合物は、アゴニストまたはアンタゴニストのいずれかであり得る。或る特定の実施形態では、EphA2、EphA4、EphA5、EphA7、EphB2およびEphB4を含むEph受容体が、インビトロまたはインビボのいずれかで調節され得る。
【0014】
本発明の別の実施形態は、哺乳動物に、プログラム細胞死(アポトーシス)を活性化するのに充分な量のEph受容体アゴニストまたはアンタゴニストを投与することによる、プログラム細胞死を促進する方法である。Eph受容体アゴニストまたはアンタゴニストとしては、ペプチド、擬似ペプチド、またはEph受容体ファミリーメンバーに選択的に結合する低分子が含まれる。Eph受容体アゴニストまたはアンタゴニストは、正常細胞および腫瘍性細胞の両方に送達され得る。或る実施形態では、アゴニストがEph受容体のリン酸化を活性化する一方、アンタゴニストがEph受容体のリン酸化を阻害する。別の実施形態では、アゴニストまたはアンタゴニストが、エフリンリガンドのEph受容体への結合を阻害する。
【0015】
本発明の別の実施形態は、神経再生の活性化および促進、ならびに外傷性損傷、精神遅滞および神経変性疾患によるニューロン変性の改善を行なう方法である。この方法は、哺乳動物に、該哺乳動物において神経再生を促進するのに有効な量のEph受容体アゴニストを投与することを含み、該アゴニストとしては、ペプチド、擬似ペプチド、またはEph受容体ファミリーメンバーに選択的に結合する低分子が含まれる。
【0016】
本発明の別の実施形態は、哺乳動物に、認知機能を調節するのに有効な量のEph受容体アゴニストまたはアンタゴニストを投与することによる、認知機能の調節方法であり、ここで、アゴニストまたはアンタゴニストには、ペプチド、擬似ペプチド、またはEph受容体ファミリーメンバーに選択的に結合する低分子が含まれる。
【0017】
本発明の別の実施形態は、哺乳動物に、血液凝固プロセスを変更するのに有効な量のEph受容体アゴニストを投与することによる、血液凝固プロセスを変更する方法であり、ここで、上記アゴニストとしては、ペプチド、擬似ペプチド、またはEph受容体ファミリーメンバーに選択的に結合する低分子が含まれる。
【0018】
本発明のさらなる実施形態は、細胞への治療剤の送達方法である。該治療剤は、ペプチド、擬似ペプチド、またはEph受容体ファミリーメンバーに選択的に結合する低分子を含むEph受容体結合化合物と連結されている。
【0019】
本発明のさらに別の実施形態は、Eph受容体ファミリーメンバーに選択的に結合するEph受容体結合化合物と連結された治療剤を含有するコンジュゲートである。或る実施形態では、治療剤はイメージング剤であり得る。
【0020】
本発明の別の実施形態は、固定化したEph受容体に対して、ランダム配列を有するペプチドをコードするファージ封入(phage-encapsulated)核酸を含むファージディスプレイライブラリーをパニング(panning)することによる、Eph受容体結合化合物の同定方法である。次いで、固定化した受容体に結合するファージクローンを同定する。
【0021】
本発明の別の実施形態は、Eph受容体に結合しているEph受容体結合因子を提供し、試験化合物または試験化合物のライブラリーを提供し、Eph受容体からEph受容体結合因子を解離させることができる化合物を同定することによる、Eph受容体結合リガンドの同定方法である。この方法により同定された化合物を、さらに他のEph受容体およびEph受容体結合因子に対してスクリーニングし、選択的Eph受容体結合リガンドを同定してもよい。
【0022】
本発明の他の実施形態は、Eph受容体への化合物の結合を測定するための分光技法を用いて化合物をスクリーニングする方法を提供する。化合物は、アルカリホスファターゼアッセイ、核磁気共鳴(NMR)結合アッセイ、蛍光偏光アッセイ(FPA)およびコンピュータによる結合の予測(Computational-Docking)試験の組合せを用いることにより、スクリーニングしてもよい。
【0023】
[発明の詳細な説明]
Eph受容体チロシンキナーゼのファミリーは、病変組織と正常組織において異なった形で発現するため、有望な疾患標的である。例えば、EphA2受容体は、形質転換細胞および腫瘍の血管系においてアップレギュレートされ、ここで、癌の病原性に寄与すると思われる。EphB4は、種々の黒色腫および癌腫において過剰発現する。EphA4は、血管疾患および場合によっては慢性関節リウマチの発症に、或る役割を果たし得る(プレボー(Prevost)他, 2002. PNAS 99:9219-9224参照)。
【0024】
Eph受容体を治療標的として利用するため、ファージディスプレイを用いて、Eph受容体ファミリーの特定のメンバーに選択的に結合するペプチドを同定した。かかるペプチドのいくつかを本明細書に記載する。例えば、EphA2に高親和性(マイクロモル未満のKD)で選択的に結合するペプチドを単離した。EphB2、EphB4、EphA4、EphA5およびEphA7に選択的に結合する別のペプチドを見出した。或る場合において、これらのペプチドは、単一の受容体でなくEph受容体のサブセットに結合する。EphA2の場合、2種類のペプチドがEphA2のリガンド結合ドメインを標的化し、結合に関してエフリンリガンドと競合することがわかった。これらのペプチドは、EphA2チロシンリン酸化および受容体による下流シグナル伝達を刺激するという点で、エフリン様活性を有する。さらにまた、これらのペプチドの1つは、EphA2を発現する内皮および腫瘍細胞にファージ粒子を送達することができる。これに対し、エフリンリガンドの受容体相互作用部分に相当するペプチドは、多くのEph受容体に対し、弱く、無差別に結合する。3種類のEphA4結合ペプチドおよびEphB2結合ペプチドもまた、これらの受容体への結合に関してエフリンリガンドと競合した。しかしながら、これらのペプチドは、受容体を活性化せず、したがって、アンタゴニストとして作用するよう
である。生体活性型エフリン擬似ペプチドを用いて、Eph受容体発現組織に薬剤を選択的に送達すること、および、Ephシグナル伝達を変更して、癌、病的血管新成および神経変性の治療に利用できる。
【0025】
定義
以下の定義は、本明細書に記載した本発明を説明するために使用した種々の用語の意味および範囲を例示および明確にするために記載する。
【0026】
本明細書で使用する場合、「アゴニスト」は、その相補的な生体活性受容体に結合して受容体を活性化し、該受容体において生物学的応答を引き起こすか、または該受容体の既存の生体活性を増強するかのいずれかである生物活性リガンドをいう。
【0027】
本明細書で使用する場合、「アンタゴニスト」は、その相補的な生体活性受容体に結合し、該受容体の生理学的応答を阻害する生物活性リガンドをいう。
【0028】
本明細書で使用する場合、「製薬的に許容され得る塩」は、製薬業界で一般的に使用されている無毒性のアルカリ金属、アルカリ土類金属およびアンモニウム塩をいい、ナトリウム、カリウム、リチウム、カルシウム、マグネシウム、バリウム、アンモニウムおよびプロタミン亜鉛の塩が挙げられ、これらは、当該技術分野で既知の方法により調製される。また、この用語は、本発明の化合物を適当な有機酸または無機酸と反応させることにより一般的に調製される無毒性の酸付加塩も包含する。代表的な塩としては、塩酸塩、臭化水素酸塩、硫酸塩、重硫酸塩、酢酸塩、シュウ酸塩、吉草酸塩、オレイン酸塩、ラウリン酸塩、ホウ酸塩、安息香酸塩、乳酸塩、リン酸塩、トシル酸塩、クエン酸塩、マレイン酸塩、フマル酸塩、コハク酸塩、酒石酸塩、ナプシル酸塩などが挙げられる。
【0029】
本明細書で使用する場合、「製薬的に許容され得る酸付加塩」は、遊離塩基の生体有効性および特性を維持し、かつ生物学的またはその他の点で望ましくない点のないものであり、塩酸、臭化水素酸、硫酸、硝酸、リン酸などの無機酸、および酢酸、プロピオン酸、グリコール酸、ピルビン酸、シュウ酸、リンゴ酸、マロン酸、コハク酸、マレイン酸、フマル酸、酒石酸、クエン酸、安息香酸、桂皮酸、マンデル酸、メタンスルホン酸(menthanesulfonic acid)、エタンスルホン酸、p−トルエンスルホン酸、サリチル酸などの有機酸と形成された塩をいう。プロドラッグとしての製薬的に許容され得る酸付加塩の説明については、バンガード(Bundgaard), H.編, 1985 Design of Prodrugs, Elsevier Science
Publishers,(アムステルダム)を参照のこと。
【0030】
本明細書で使用する場合、「製薬的に許容され得るエステル」は、エステル結合の加水分解時に、カルボン酸またはアルコールの生体有効性および特性を維持し、生物学的またはその他の点で望ましくないものでないエステルをいう。プロドラッグとしての製薬的に許容され得るエステルの説明については、バンガード, H.編, 1985 Design of Prodrugs,
Elsevier Science Publishers (アムステルダム)を参照のこと。これらのエステルは、通常、対応するカルボン酸と、アルコールとから形成される。一般的に、エステルの形成は、従来の合成技法によりなされ得る。例えば、1992年3月, Advanced Organic Chemistry, 第4版, John Wiley & Sons (ニューヨーク), 第393−396頁およびそこに挙げられる参考文献、ならびにマーク(Mark)他1980 Encyclopedia of Chemical Technology, John Wiley & Sons (ニューヨーク)を参照のこと。エステルのアルコール成分としては、一般的に、(i)1つ以上の二重結合を含んでいても含んでいなくてもよく、分枝炭素を含んでいても含んでいなくてもよいC2−C12脂肪族アルコール、または(ii)C7−C12芳香族アルコールもしくは複素環芳香族アルコールが含まれる。また、本発明は、本明細書に記載したエステルであると同時に製薬的に許容され得るその酸付加塩でもあるこれらの組成物の使用も意図している。
【0031】
本明細書で使用する場合、「製薬的に許容され得るアミド」は、アミド結合の加水分解時に、カルボン酸またはアミンの生体有効性および特性を維持し、生物学的またはその他の点で望ましくない点のないアミドをいう。プロドラッグとしての製薬的に許容され得るアミドの説明については、バンガード, H.編, 1985 Design of Prodrugs, Elsevier Science Publishers (アムステルダム)を参照のこと。これらのアミドは、通常、対応するカルボン酸と、アミンとから形成される。一般的に、アミドの形成は、従来の合成技法によりなされ得る。例えば、1992年3月, Advanced Organic Chemistry, 第4版, John Wiley & Sons (ニューヨーク), 第393頁およびマーク他1980 Encyclopedia of Chemical Technology, John Wiley & Sons (ニューヨーク)を参照のこと。また、本発明では、本明細書に記載したアミドであると同時に製薬的に許容され得るその酸付加塩でもあるこれらの組成物の使用も意図される。
【0032】
本明細書で使用する場合、「製薬的または治療的に許容され得る担体」は、活性成分の生体活性の有効性を妨げず、かつ宿主または患者に毒性を持たない担体をいう。
【0033】
本明細書で使用する場合、「立体異性体」は、別の分子と同じ分子量、化学組成および結合順を有するが、1つ以上のキラル中心に関して空間的配置の異なる原子を有する化合物をいう。すなわち、同じ化学式を有する立体異性体は、少なくとも1つのキラル中心に関しては、空間的に異なる位置にある同一の化学物質部分を含む。純粋な場合、立体異性体は、平面偏光を回転させる能力を有する。しかしながら、一部の純粋な立体異性体は、非常にわずかな旋光性しか持たないため既存の器機では検出できない場合がある。本発明の化合物は、1つ以上の不斉炭素原子を有していてもよく、したがって、種々の立体異性体を包含する。すべての立体異性体は、本発明の範囲内に含まれる。
【0034】
本明細書で使用する場合、本発明の組成物に適用される「治療的有効量または製薬的有効量」は、所望の生物学的結果を誘導するのに充分な組成物の量をいう。ここでいう結果とは、徴候、症状もしくは疾患の原因の緩和、または生体系のあらゆる所望の改変であり得る。本発明では、この成果は、例えば、癌性細胞成長の阻害および反転(reversal)を含む。
【0035】
本明細書で使用する場合、用語「ペプチド化合物」および「ペプチド構造」は、天然型L−アミノ酸からなるペプチド、ならびに天然型L−アミノ酸構造のペプチド誘導体、ペプチドアナログおよび擬似ペプチドを含むものとする。用語「ペプチドアナログ」、「ペプチド誘導体」および「擬似ペプチド」は、本明細書で使用する場合、ペプチドの化学構造を擬態し、そのペプチドの機能的特性を保持した分子を含むものとする。ペプチドのアナログ、誘導体および擬似物を設計するためのアプローチは、当該技術分野で既知である。例えば、Drug Design E. J. アリエンス(Ariens)編 Academic Press(ニューヨーク),
1980, 第10巻, 第119−143頁のファーマー(Farmer), P. S.; ボール(Ball) J. B. & アレウッド(Alewood), P. F. 1990 J Mol Recognition 3:55; モーガン(Morgan), B. A. & ゲイナー(Gainor), J. A. 1989 Ann Rep Med Chem 24:243; およびフライディンガー(Freidinger), R. M. 1989 Trends Pharmacol Sci 10:270; ルスマン(Luthman)他1996 A TextBook of Drug Design and Development, 14:386-406, 第2版, Harwood Academic Publihers; ヨアキム グランテ(Joachim Grante), アンギュー(Angew). 1994 Chem Int Ed Engl 33:1699-1720; フォーシェア(Fauchere). J. 1986 Adv Drug Res 15:29; ベベール(Veber)およびフライディンガー 1985 TINS第392頁; エバンス(Evans)他1987 J Med Chem 30:229を参照のこと。治療的に有用なペプチドに構造的に類似する擬似ペプチドは、同等または向上した治療効果または予防効果を得るために使用され得る。一般的に、擬似ペプチドは、天然型受容体結合ポリペプチドなどの模範(paradigm)ポリペプチド(すなわち、生物学的または薬理学的活性を有するポリペプチド)に構造的に類似するが、当
該技術分野で既知の方法により、−CH2NH−、−CH2S−、−CH2−CH2−、−CH=CH−(シスおよびトランス)、−COCH2−、−CH(OH)CH2−および−CH2SO−からなる群より選択される結合で、任意に置換した1つ以上のペプチド結合を有する。以下にさらに参考文献を挙げる:Chemistry and Biochemistry of Amino Acids, Peptides, and Proteins, B. ウェインスタイン(Weinstein)編, Marcel Dekker, Inc.(ニューヨーク)第267頁のスパトラ(Spatola), A. F. 1983; スパトラ, A. F. 1983 Vega Data, 第1巻, 第3号, Peptide Backbone Modification (一般概説); モーリー(Morley), 1980 Trends Pharm Sci 第463−468頁(一般概説); ハドソン(Hudson)他1979 Int J Pept Prot Res 14:177-185(−CH2NH−、−CH2CH2−、); スパトラ他1986 Life Sci 38:1243-1249(−CH2−S); ハン(Hann), 1982 J Chem Soc Perkin Trans I 307-314(−CH−CH−、シスおよびトランス); アルムキスト(Almquist)他1980 J Med Chem 23:1392-1398(−COCH2−); ジェニングス-ホワイト(Jennings-White)他1982 Tetrahedron Lett 23:2533(−COCH2−); セルケ(Szelke)他1982 欧州特許出願EP45665(−CH(OH)CH2−); ホラデイ(Holladay)他1983 Tetrahedron Lett 24:4401-4404(−C(OH)CH2−);およびルビー(Hruby), 1982 Life Sci 31:189-199(−CH2−S)。かかる擬似ペプチドは、ポリペプチドでの実施形態よりも大きな利点を有し、例えば、より経済的な生産、より高い化学的安定性、薬理学的特性(半減期、吸収、効能、効力など)の向上、特異性の改変(例えば、広い生物活性)、抗原性の低下などが挙げられる。擬似ペプチドの標識化は、通常、1つ以上の標識を、直接またはスペーサー(例えば、アミド基)を介して、擬似ペプチドの定量的構造活性データおよび分子モデリングにより予測した非妨害性位置(複数可)に共有結合させることを伴う。かかる非妨害性位置は、一般的に、擬似ペプチドが結合して治療効果を生じる巨大分子(複数可)(受容体分子等)と直接接触しない位置である。擬似ペプチドの誘導体化(例えば、標識化)は、その擬似ペプチドの所望の生物学的または薬理学的活性を実質的に妨げないのがよい。一般的に、受容体結合ペプチドの擬似ペプチドは、高親和性で受容体に結合し、検出可能な生体活性を有する(すなわち、1つ以上の受容体媒介性表現型変化に対して作用性または拮抗性である)。
【0036】
より安定なペプチドを作製するため、コンセンサス配列の1個以上のアミノ酸を、同じ型のD−アミノ酸で系統的(systematic)に置換してもよい(例えば、L−リシンの代わりにD−リシン)を用いてもよい。また、コンセンサス配列または実質的に同一のコンセンサス配列変異を含有する束縛(constrained)ペプチドを、当該技術分野で既知の方法(リゾ(Rizo)他1992 Ann Rev Biochem 61:387)、例えば、ペプチドを環化する分子内ジスルフィド結合を形成することができる分子内システイン残基を付加することにより作製してもよい。
【0037】
合成または非天然型アミノ酸は、インビボでは天然に存在しないが、それでも、本明細書に記載したペプチド構造に組み込まれ得るアミノ酸をいう。好ましい合成アミノ酸は、天然型L−α−アミノ酸のD−α−アミノ酸、ならびに式H2NCHR5COOH(式中、R5は、1)低級アルキル基、2)炭素数が3−7のシクロアルキル基、3)炭素数が3−7で1−2個のヘテロ原子が酸素、イオウおよび窒素からなる群より選択される複素環、4)芳香核上に、ヒドロキシル、低級アルコキシ、アミノおよびカルボキシルからなる群より任意に選択される1−3個の置換基を有する炭素数が6−10の芳香族残基、5)−アルキレン−Y(式中、アルキレンは、炭素数が1−7のアルキレン基であり、Yは、(a)ヒドロキシ、(b)アミノ、(c)炭素数が3−7のシクロアルキルおよびシクロアルケニル、(d)芳香核上に、ヒドロキシル、低級アルコキシ、アミノおよびカルボキシルからなる群より任意に選択される1−3個の置換基を有する炭素数が6−10のアリール、(e)炭素数が3−7で1−2個のヘテロ原子が酸素、イオウおよび窒素からなる群より選択される複素環、(f)−C(O)R2(式中、R2は、水素、ヒドロキシ、低級アルキル、低級アルコキシおよび−NR3R4(式中、R3およびR4は、独立に、
水素および低級アルキルからなる群より選択される)からなる群より選択される)、(g)−S(O)nR6(式中、nは1−2の整数であり、R6は、低級アルキルである)、からなる群より選択される)である。ただし、R5は、天然型アミノ酸の側鎖を規定するものではない)で表される非天然型D−およびL−α−アミノ酸である。
【0038】
他の好ましい合成アミノ酸としては、β−アラニン、γ−アミノ酪酸など、アミノ基がカルボキシル基と2つ以上の炭素原子で分断されているアミノ酸が挙げられる。
【0039】
特に好ましい合成アミノ酸としては、一例として、天然型L−アミノ酸のD−アミノ酸、L−(1−ナフチル)−アラニン、L−(2−ナフチル)−アラニン、L−シクロヘキシルアラニン、L−2−アミノイソ酪酸、メチオニンのスルホキシドおよびスルホン誘導体(すなわち、HOOC−(H2NCH))−CH2CH2−S(O)nR6(式中、nおよびR6は、上記のとおりである))ならびにメチオニンの低級アルコキシ誘導体(すなわち、HOOC−(H2NCH))−CH2CH2−OR6(式中、R6は、上記のとおりである))が挙げられる。
【0040】
本明細書で使用する場合、化合物X(例えば、ペプチドまたはアミノ酸)の「誘導体」は、この化合物内の1つ以上の反応性基が置換基によって誘導体化されているXの形態をいう。ペプチド誘導体の例としては、アミノ酸側鎖、ペプチド主鎖またはアミノ末端もしくはカルボキシ末端が誘導体化されているペプチドが挙げられる(例えば、メチル化アミド結合を有するペプチド性化合物)。
【0041】
本明細書で使用する場合、化合物Xの「アナログ」は、Xの機能的活性に必要なXの化学構造を保持しているが、Xとは異なる或る特定の化学構造も含む化合物をいう。天然型ペプチドのアナログの一例は、1つ以上の非天然型アミノ酸を含むペプチドである。本明細書で使用する場合、化合物Xの「擬似物」は、Xの機能的活性に必要なXの化学構造が、Xの立体配座を擬態する別の化学構造と置換されている化合物をいう。擬似ペプチドの例としては、以下にさらに記載するような、ペプチド主鎖が1つ以上のベンゾジアゼピン分子で置換されているペプチド様化合物(例えば、ジェームズ(James), G. L.他1993 Science 260:1937-1942参照)、全てのL−アミノ酸が対応するD−アミノ酸で置換されたペプチドおよび「retro-inverso」型ペプチド(シスト(Sisto)による米国特許第4,522,752号参照)が挙げられる。
【0042】
擬似物という用語、特に擬似ペプチドは、同配体(isostere)を含むものとする。用語「同配体」は、本明細書で使用する場合、第2の化学構造で置換され得る化学構造を含むものとする。これは、第1の構造の立体配座が第2の構造に特異的な結合部位に適合しているからである。この用語には、具体的には、当業者に既知のペプチド主鎖の修飾(すなわち、アミド結合擬似物)が含まれる。かかる修飾としては、アミド窒素、α−炭素、アミドカルボニルの修飾、アミド結合の完全な置換、伸長、欠失または架橋が挙げられる。いくつかのペプチド主鎖の修飾は既知であり、Ψ[CH2S]、Ψ[CH2NH]、Ψ[CSNH2]、Ψ[NHCO]、Ψ[COCH2]、Ψ[(E)または(Z)CH=CH]が挙げられる。上記に使用した述語において、Ψは、アミド結合の不在を示す。アミド基と置き換わる構造を、括弧内に特定する。同配体の他の例としては、1つ以上のベンゾジアゼピン分子で置換されたペプチドが挙げられる(例えば、ジェームズ, G. L.他1993 Science 260:1937-1942参照)。
【0043】
他の考えられ得る修飾としては、N−アルキル(またはアリール)置換(Ψ[CONR])、主鎖の架橋によるラクタムおよび他の環状構造の構築、化合物内のすべてのL−アミノ酸のD−アミノ酸への置換(「inverso」型化合物)、または「retro-inverso」型アミノ酸の組み込み(Ψ[NHCO])が挙げられる。「inverso」とは、配列のL−アミ
ノ酸をD−アミノ酸で置換することを意味し、「retro-inverso」または「enantio-retro」とはアミノ酸の配列の逆転(「retro」)およびL−アミノ酸のD−アミノ酸での置換を意味する。例えば、親ペプチドがThr−Ala−Tryである場合、retro修飾型は、Try−Ala−Thrであり、inverso型は、thr−ala−try(小文字は、D−アミノ酸を示す)であり、retro-inverso型は、try−ala−thrである。親ペプチドと比較すると、retro-inversoペプチドは、逆転した主鎖を有するが、側鎖の元の空間的配座は実質的に保持しており、親ペプチドに非常に似たトポロジーを有するretro-inverso異性体がもたらされる。グッドマン(Goodman)他1981 Perspectives in Peptide
Chemistry第283−294頁を参照のこと。また、「retro-inverso」ペプチドのさらなる説明については、シストによる米国特許第4,522,752号も参照のこと。他の誘導体としては、C末端ヒドロキシルメチル誘導体、O−修飾誘導体(例えば、C末端ヒドロキシルメチルベンジルエーテル)ならびにアルキルアミドおよびヒドラジドなどの置換アミドを含むN末端修飾誘導体が挙げられる。
【0044】
本明細書で使用する場合、用語「アミノ酸構造」(「ロイシン構造」、「フェニルアラニン構造」または「グルタミン構造」など)は、アミノ酸、ならびに化合物の機能的活性を維持しているそのアミノ酸のアナログ、誘導体および擬似物を含むものとする。例えば、用語「フェニルアラニン構造」は、フェニルアラニンならびにピリジルアラニンおよびホモフェニルアラニンを含むものとする。用語「ロイシン構造」は、ロイシン、ならびにノルロイシンなどの、バリンまたは脂肪族側鎖を有する他の天然型もしくは非天然型アミノ酸での置換を含むものとする。
【0045】
本明細書に開示するペプチド化合物のアミノ末端およびカルボキシ末端は、修飾されていない(すなわち、Y1およびY2は独立に)水素であってもよい。あるいはまた、ペプチド化合物のアミノ末端およびカルボキシ末端は、誘導体基で置換されていてもよい。ペプチド化合物のN末端に存在し得る(すなわちY1であり得る)アミノ誘導体基としては、アセチル、アリール、アラルキル、アシル、エポキシサクシニルおよびコレステリル基が挙げられる。ペプチド化合物のC末端に存在し得る(すなわちY2であり得る)カルボキシ誘導体基としては、アルコール、アルデヒド、エポキシコハク酸、酸ハロゲン化物、カルボニル、ハロメタンおよびジアゾメタン基が挙げられる。
【0046】
本明細書で使用する場合、「検出可能な標識」または「イメージング剤」は、化合物に共有結合すると、Eph受容体結合因子が投与された患者におけるインビボでの検出を含む(これに限定されない)この化合物の検出を可能にする物質をいう。好適な検出可能な標識は、当該技術分野で既知であり、一例として、放射性同位体、蛍光標識(例えば、フルオレセイン)などが挙げられる。使用する具体的な検出可能な標識は重要ではなく、使用する標識の量および使用する標識の量における標識の毒性に関連して選択される。かかる因子に関連した標識の選択は、十分に、当該技術分野の技術範囲内である。
【0047】
ペプチドまたは擬似ペプチドへの検出可能な標識の共有結合は、当該技術分野で既知の従来の方法によりなされる。例えば、125I放射性同位体を検出可能な標識として用いる場合、ペプチドまたは擬似ペプチドへの125Iの共有結合は、ペプチドまたは擬似ペプチドにアミノ酸チロシンを組み込み、次いで、このペプチドをヨウ化物にすることにより、なされ得る(例えば、ウィーナー(Weaner)他1994 Synthesis and Applications of Isotopically Labelled Compounds,第137−140頁を参照)。チロシンがペプチドまたは擬似ペプチド内に存在しない場合、ペプチドまたは擬似ペプチドのNまたはC末端へのチロシンの組み込みは、既知の化学反応によりなされ得る。同様に、32Pを、従来の化学反応を用いて、例えば、ペプチドまたは擬似ペプチド上のヒドロキシル基を介して、リン酸部分としてペプチドまたは擬似ペプチド上に組み込むことができる。
【0048】
「選択的に」とは、1つまたは数個のEph受容体ファミリーメンバーに対して、他の既知のEph受容体ファミリーメンバーに対する結合親和性よりもかなり大きな結合親和性を有することを意味する。選択的結合親和性に関連して使用する場合、「実質的に大きい」は、受容体に結合したリガンドの量の少なくとも2倍、少なくとも3倍、少なくとも4倍、少なくとも5倍、少なくとも6倍、少なくとも7倍、少なくとも8倍、少なくとも9倍、少なくとも10倍、少なくとも15倍、少なくとも20倍、少なくとも30倍、少なくとも40倍、少なくとも50倍または少なくとも100倍の増加を意味する。
【0049】
本明細書で使用する場合、「Eph受容体結合因子」または「Eph受容体結合リガンド」は、Eph受容体に結合する化合物である。この化合物は、1つ以上のEph受容体に結合することができる任意の分子を包含する。或る場合において、1つ以上のEph受容体に結合することができる分子は、ペプチドまたは擬似ペプチドである。かかるペプチドまたは擬似ペプチドは、10個未満、11個未満、12個未満、13個未満、14個未満、15個未満、20個未満、25個未満、30個未満、35個未満、40個未満、45個未満、50個未満、75個未満、100個未満、200個未満、300個未満、400個未満または500個未満の長さの残基を有し得る。用語「Eph受容体結合因子」および「Eph受容体結合リガンド」は、互換的に使用され得る。
【0050】
本明細書で使用する場合、「エフリン−A」としては、エフリン−Aリガンドサブクラスのメンバーである任意のエフリンが挙げられる。
【0051】
本明細書で使用する場合、「エフリン−B」としては、エフリン−Bリガンドサブクラスのメンバーである任意のエフリンが挙げられる。
【0052】
本明細書で使用する場合、用語「治療剤」は、抗癌剤、神経保護剤またはある特定の疾患の兆候に所望の治療効果を生じ得るその他の薬剤を意味する。
【0053】
本明細書に記載される抗癌剤は、細胞傷害剤または癌化学療法剤であってもよい。非限定的な例として、DNA関連過程を標的とする細胞傷害剤には、シクロホスファミド、メルファラン、マイトマイシンC、ビゼレシン、シスプラチン、ドキソルビシン、エトポシド、ミトキサントロン、SN38、Et743、アクチノマイシンD、ブレオマイシンおよびTLK286が包含される。癌化学療法剤は、限定されないが、ドセタキセルなどのタキサン、ドキソルビシンなどのアントラサイクリン、アルキル化剤、ビンカアルカロイド、代謝拮抗物質、シスプラチンもしくはカルボプラチンなどの白金剤、メトトレキセートなどのステロイド、アドリアマイシンなどの抗生物質、イソファミド、または選択的エストロゲン受容体調節物質、トラスツズマブなどの抗体であり得る。
【0054】
タキサンは、本発明の併用治療に有用な化学療法剤である。有用なタキサンとしては、限定されないが、ドセタキセル(Taxotere; Aventis Pharmaceuticals, Inc.; パーシッパニー, ニュージャージー州)およびパクリタキセル(Taxol; Bristol Myers Squibb; プリンストン, ニュージャージー州)が挙げられる。例えば、チャン(Chan)他1999 J Clin Oncol 17:2341 2354およびパリデンス(Paridaens)他2000 J Clin Oncol 18:724を参照のこと。
【0055】
本発明の併用治療に有用な別の癌化学療法剤は、ドキソルビシン、イダルビシンまたはダウノルビシンなどのアントラサイクリンである。ドキソルビシンは、一般的に使用されている癌化学療法剤であり、例えば、乳癌の標的化に有用であり得る(スチュワート(Stewart)およびラタイン(Ratain)著「Cancer: Principles and Practice of Oncology」第5版、第19章、デビタ(DeVita), Jr.他編,; J. P. リッピンコット(Lippincott) 1997; ハリス(Harris)他著「Cancer: Principles and practice of oncology」前掲, 1997)。
また、ドキソルビシンは、抗血管新生活性を有し(フォルクマン(Folkman), 1997 Nature
Biotechnology 15:510;スタイナー(Steiner)他著「Angiogenesis: Key principles Science, technology and medicine」第449 454頁, スタイナー他編 Birkhauser Verlag, 1992)、これは癌の治療におけるその有効性に寄与し得る。
【0056】
メルファランまたはクロラムブチルなどのアルキル化剤は、本発明の併用治療に有用な癌化学療法剤である。同様に、ビンデシン、ビンブラスチンもしくはビノレルビンなどのビンカアルカロイド、または5フルオロウラシル、5フルオロウリジンもしくはその誘導体などの代謝拮抗物質は、本発明の併用治療に有用な癌化学療法剤である。
【0057】
白金剤は、本発明の併用治療に有用な癌化学療法剤である。かかる白金剤は、例えば、クラウン(Crown), 2001 Seminars in Oncol 28:28-37に記載されているような、シスプラチンもしくはカルボプラチンなどであり得る。本発明の併用治療に有用な他の癌化学療法剤としては、限定されないが、メトトレキセート、マイトマイシンC、アドリアマイシン、イホスファミドおよびアンサマイシンが挙げられる。
【0058】
選択的エストロゲン受容体調節物質または抗エストロゲンなどの、乳癌およびその他のホルモン依存性癌の治療に使用される癌化学療法剤もまた、エストロゲンの作用を拮抗阻害する薬剤として使用することができる。選択的エストロゲン受容体調節物質であるタモキシフェンは、乳癌の治療のために本発明の併用治療に使用され得る癌化学療法剤である(フィッシャー(Fisher)他1998 J Natl Cancer Instit 90:1371 1388)。
【0059】
本発明の併用治療に有用な治療剤は、ヒト化モノクローナル抗体などの抗体であってもよい。一例として、抗表皮成長因子受容体2(HER2)抗体であるトラスツズマブ(Herceptin; Genentech; サウスサンフランシスコ, カリフォルニア州)は、HER2/neu過剰発現乳癌(ホワイト(White)他2001 Ann Rev Med 52:125-141)の治療用の本発明のコンジュゲートにおいて有用な治療剤である。
【0060】
また、本発明に有用な別の治療剤は、本発明で使用されるような、直接または間接的に細胞死を促進する任意の分子である細胞傷害剤であってもよい。本発明に有用な細胞傷害剤としては、限定されないが、低分子、ポリペプチド、ペプチド、擬似ペプチド、核酸分子、細胞およびウイルスが挙げられる。非限定的な例として、本発明に有用な細胞傷害剤としては、ドキソルビシン、ドセタキセルまたはトラスツズマブなどの細胞傷害性低分子;以下にさらに記載するようなものなどの抗菌性ペプチド;カスパーゼ、および毒素、例えば、カスパーゼ8などのプロアポトーシスポリペプチド;ジフテリア毒素A鎖、シュードモナス外毒素A、コレラ毒素、DAB389EGFなどのリガンド融合毒素、ヒマ(ricinus communis)毒素(リシン);ならびに細胞傷害性T細胞などの細胞傷害性細胞が挙げられる。例えば、マーチン(Martin)他2000 Cancer Res 60:3218-3224; クライトマン(Kreitman)およびパスタン(Pastan) 1997 Blood 90:252-259; アラム(Allam)他1997 Cancer Res 57:2615-2618; オズボーン(Osborne)およびコロナド・ハインソン(Coronado Heinsohn) 1996 Cancer J Sci Am 2:175を参照のこと。当業者であれば、本明細書に記載した、または当該技術分野で既知のこれらの及びさらなる細胞傷害剤が、本発明の治療剤として有用であり得ることがわかる。
【0061】
神経保護剤は、当該技術分野で既知であり、神経細胞の死を抑制または遅滞させる化合物であり得る。非限定的な例として、神経保護剤は、低分子薬物、ペプチド、タンパク質、抗体またはこれらの組合せなどの抗アポトーシス化合物であり得る。神経保護剤は、1つ以上のアポトーシス経路または壊死経路の妨害、神経成長ホルモン受容体の活性化またはイオンチャネルの調節により作用し得る。当業者であれば、本明細書に記載した、または当該技術分野で既知のこれらおよびさらなる神経保護剤が、本発明の治療剤として有用
であり得ることがわかる。
【0062】
Eph受容体結合因子
本発明の実施形態は、Eph受容体に結合する薬剤を提供する。本明細書に記載した化合物の多くは、Eph受容体ファミリーの16個の既知受容体のうち、1つだけ、または限られた数の受容体に選択的に結合する。Eph受容体結合因子は、低分子薬剤、ペプチドまたは擬似ペプチドであり得る。Eph受容体結合因子は、天然化合物であってもよく、合成化合物であってもよい。また、本明細書に記載した化合物の多くは、Eph受容体に高親和性で結合し、Eph受容体アゴニストまたはアンタゴニストとしての機能を果たし得る。本明細書に記載した化合物は、「リード」化合物、およびリード化合物と同じかまたは類似の分子構造または形状を有するが、加水分解もしくはタンパク質分解に対する感受性のいずれかに関して、および受容体に対する親和性の増加もしくは標的Eph受容体とは無関係の生物学的特性をさらに有することなどの他の生物学的特性に関してリード化合物とは異なるように構成された「誘導体」化合物を含む。
【0063】
ペプチドおよび擬似ペプチドの調製
1.固相合成
本明細書に記載するペプチドは、当該技術分野で既知の古典的な方法により、例えば、標準的な固相技法により調製することができる。標準的な方法としては、排他的固相合成、部分固相合成法、フラグメント濃縮(fragment condensation)、古典的溶液合成が挙げられ、組換えDNA技術によるものでさえ含まれる。例えば、メリフィールド(Merrifield), 1963 J Am Chem Soc 85:2149を参照のこと。固相では、合成は、通常α−アミノ保護樹脂を用いてペプチドのC末端から開始される。適当な出発物質は、例えば、必要なα−アミノ酸をクロロメチル化樹脂、ヒドロキシメチル樹脂またはベンズヒドリルアミン樹脂に結合することにより調製することができる。かかるクロロメチル化樹脂の一例は、BioRad Laboratories(リッチモンド、カリフォルニア州)により商品名BIO−BEADS SX−1で販売されているものであり、ヒドロキシメチル樹脂の調製は、ボドンスキー(Bodonszky)他1966 Chem Ind (ロンドン) 38:1597に記載されている。ベンズヒドリルアミン(BHA)樹脂は、ピエッタ(Pietta)およびマーシャル(Marshall), 1970 Chem Commn 650に記載されており、塩酸塩の形態でBeckman Instruments, Inc.(パロ・アルト, カリフォルニア州)から市販されている。
【0064】
このように、化合物は、ギシン(Gisin), 1973 Helv Chim Acta 56:1467に記載の方法にしたがって、例えば重炭酸セシウム触媒の補助により、α−アミノ保護アミノ酸をクロロメチル化樹脂に結合させることにより調製することができる。最初の結合後、α−アミノ保護基を、トリフルオロ酢酸(TFA)または塩酸(HCl)溶液などの選択した試薬により、有機溶媒中、室温で除去する。
【0065】
α−アミノ保護基は、ペプチドの段階的合成の技術分野において有用であることが知られているものであり、アシル型保護基(例えば、ホルミル、トリフルオロアセチル、アセチル)、芳香族ウレタン型保護基(例えば、ベンジルオキシカルボニル(carboyl)(Cbz)および置換Cbz)、脂肪族ウレタン保護基(例えば、t−ブチルオキシカルボニル(Boc)、イソプロピルオキシカルボニル、シクロヘキシルオキシカルボニル)およびアルキル型保護基(例えば、ベンジル、トリフェニルメチル)が挙げられる。BocおよびFmocは、好ましい保護基である。側鎖保護基は、結合反応中は損傷を受けず、アミノ末端保護基の脱保護中または結合反応中に分離しないものである。側鎖保護基は、最終ペプチドの合成完了時、かつ標的ペプチドを改変しない反応条件下で除去可能でなければならない。
【0066】
Tyrの側鎖保護基としては、テトラヒドロピラニル、tert−ブチル、トリチル、
ベンジル、Cbz、Z−Br−Cbzおよび2,5−ジクロロベンジルが挙げられる。Aspの側鎖保護基としては、ベンジル、2,6−ジクロロベンジル、メチル、エチルおよびシクロヘキシルが挙げられる。ThrおよびSerの側鎖保護基としては、アセチル、ベンゾイル、トリチル、テトラヒドロピラニル、ベンジル、2,6−ジクロロベンジルおよびCbzが挙げられる。ThrおよびSerの側鎖保護基はベンジルである。Argの側鎖保護基としては、ニトロ、トシル(Tos)、Cbz、アダマンチルオキシカルボニルメシトイルスルホニル(Mts)またはBocが挙げられる。Lysの側鎖保護基としては、Cbz、2−クロロベンジルオキシカルボニル(2Cl−Cbz)、2−ブロモベンジルオキシカルボニル(2BrCbz)、TosまたはBocが挙げられる。
【0067】
α−アミノ保護基の除去後、残留する保護アミノ酸を、所望の順序で段階的に結合させる。一般的には、過剰の各保護アミノ酸を、ジシクロヘキシルカルボジイミド(DCC)などの適切なカルボキシル基活性化剤とともに溶液中、例えば塩化メチレン(CH2Cl2)、ジメチルホルムアミド(DMF)混合物中で使用する。
【0068】
所望のアミノ酸配列が完成した後、この所望のペプチドを樹脂支持体から、樹脂からペプチドを切断するだけでなく、すべての残留側鎖保護基をも切断するトリフルオロ酢酸またはフッ化水素(HF)などの試薬での処理により取り出す。クロロメチル化樹脂を使用した場合、フッ化水素処理により、遊離ペプチド酸の形成がもたらされる。ベンズヒドリルアミン樹脂を使用した場合は、フッ化水素処理により、遊離ペプチドアミドが直接もたらされる。あるいはまた、クロロメチル化樹脂を用いた場合、側鎖保護ペプチドは、ペプチド樹脂を、アンモニアで処理して所望の側鎖保護アミドを得るか、またはアルキルアミンで処理して側鎖保護されたアルキルアミドまたはジアルキルアミドにすることにより、取り出すことができる。次いで、側鎖保護を、フッ化水素で処理して遊離アミド、アルキルアミドまたはジアルキルアミドにすることによる常套的な方法で解除する。
【0069】
これらの固相ペプチド合成手順は、当該技術分野で既知であり、J. M. スチュワートおよびJ. D. ヤング(Young), 1984 Solid Phase Peptide Syntheses第2版, Pierce Chemical Companyにさらに記載されている。
【0070】
1990年3月7日に出願された米国特許出願第07/492,462号、1990年12月6日に出願された同第07/624,120号および1991年12月6日に出願された同第07/805,727号に記載されている「コード(encoded)合成ライブラリー」または「巨大(very large)規模固定化ポリマー合成」系を用いると、かかる活性を有する最小サイズのペプチドを決定できるだけでなく、好ましいモチーフ(またはそのモチーフの最小サイズ)と、1つ、2つまたはそれ以上の残基において異なるペプチド群を構成するあらゆるペプチドを作製することができる。次いで、この収集ペプチドを、EphA2、EphA4、EphA5、EphA7、EphB2およびEphB4(これらに限定されない)を含む、Eph受容体ファミリーメンバーに結合する能力に関してスクリーニングし得る。この固定化ポリマー合成系または他のペプチド合成法はまた、本発明のペプチド化合物すべての切断型アナログおよび欠失型アナログならびに切断型と欠失型のアナログの組合せを合成するためにも使用できることを理解されたい。
【0071】
2.合成アミノ酸
これらの手順はまた、本発明のいずれかの化合物の1つ、2つまたはそれ以上の位置において、遺伝子にコードされた20個の天然型アミノ酸以外のアミノ酸に置換されているペプチドを合成するためにも使用することができる。例えば、トリプトファンをナフチルアラニンに置換すると、合成が容易になる。置換して本発明のペプチドに取り込み得る他の合成アミノ酸としては、L−ヒドロキシプロピル、L−3,4−ジヒドロキシフェニルアラニル、L−d−ヒドロキシリシルおよびL−d−メチルアラニルなどのdアミノ酸、
L−α−メチルアラニル、βアミノ酸、ならびにイソキノリルが挙げられる。Dアミノ酸および非天然型合成アミノ酸もまた、本発明のペプチドに組み込むことができる(例えば、ロバーツ(Roberts)他1983 Unusual Amino/Acids in Peptide Synthesis 5:341-449を参照のこと)。
【0072】
20個の遺伝子にコードされたアミノ酸(またはDアミノ酸)の天然型側鎖を、他の側鎖、例えば、アルキル、低級アルキル、環状の四、五、六−七員環アルキル、アミド、アミド低級アルキル、アミドジ(低級アルキル)、低級アルコキシ、ヒドロキシ、カルボキシおよびその低級エステル誘導体などの基で、ならびに四、五、六−七員環の複素環で置き換えてもよい。特に、プロリン残基の環の大きさを五員環から四、六または七員環に変えたプロリンのアナログを使用するのがよい。環式基は、飽和であってもよく、不飽和であってもよく、不飽和の場合、芳香族系であってもよく、非芳香族系であってもよい。複素環式基は、好ましくは、1個以上の窒素、酸素およびイオウのヘテロ原子を含む。かかる基の例としては、フラザニル、フリル、イミダゾリジニル、イミダゾリル、イミダゾリニル、イソチアゾリル、イソキサゾリル、モルホリニル(例えば、モルホリノ)、オキサゾリル、ピペラジニル(例えば、1−ピペラジニル)、ピペリジル(例えば、1−ピペリジル、ピペリジノ)、ピラニル、ピラジニル、ピラゾリジニル、ピラゾリニル、ピラゾリル、ピリダジニル、ピリジル、ピリミジニル、ピロリジニル(例えば、1−ピロリジニル)、ピロリニル、ピロリル、チアジアゾリル、チアゾリル、チエニル、チオモルホリニル(例えば、チオモルホリノ)およびトリアゾリルが挙げられる。これらの複素環式基は、置換されていても、置換されていなくてもよい。基が置換されている場合、置換基は、アルキル、アルコキシ、ハロゲン、酸素、または置換もしくは非置換のフェニルであり得る。
【0073】
また、リン酸化により、本発明のペプチドを容易に修飾することができ(例えば、W. バンワース(Bannwarth)他1996 Biorganic and Medicinal Chemistry Letters 6:2141-2146を参照のこと)、本発明の化合物であるペプチド誘導体を作製するための他の方法は、ルビー他1990 Biochem J 268:249-262に記載されている。このように、本発明のペプチド化合物はまた、類似の生体活性を有する擬似ペプチドを調製するための基剤として使用される。
【0074】
3.末端修飾
当業者であれば、対応するペプチド化合物と同じかまたは類似する所望の生体活性を有するが、溶解性、安定性ならびに加水分解およびタンパク質分解への感受性に関して、このペプチドよりもより好都合な活性を有する擬似ペプチドを構築するために、種々の技法が利用可能であることが認識されよう。例えば、モーガン他1989 Ann Rep Med Chem 24:243-252を参照のこと。以下は、N末端アミノ基、C末端カルボキシル基が修飾された擬似ペプチドを調製するための方法およびペプチドのアミド結合の1つ以上を非アミド結合に変化させるための方法を記載したものである。2つ以上のかかる修飾を、1つの擬似ペプチド構造に結合させ得ること(例えば、C末端カルボキシル基の修飾およびペプチドの2つのアミノ酸間に−CH2−カルバメート結合を含めること)は、理解されよう。
【0075】
1)N末端修飾
ペプチドは、通常、遊離酸として合成されるが、上記のように、アミドまたはエステルとして容易に調製され得る。また、ペプチド化合物のアミノ末端およびカルボキシ末端を修飾して、他の有用な化合物を作製することができる。アミノ末端修飾としては、メチル化(すなわち、−NHCH3または−NH(CH3)2)、アセチル化、ベンジルオキシカルボニル基の付加、またはRCOO−(式中、Rは、ナフチル、アクリジニル、ステロイジルおよび同様の基からなる群より選択される)で規定されるカルボン酸官能基を含有する任意のブロック基によるアミノ末端のブロックが挙げられる。カルボキシ末端修飾と
しては、遊離酸のカルボキシアミド基での置換、構造的制約を導入するためのカルボキシ末端での環状ラクタムの形成が挙げられる。
【0076】
アミノ末端修飾は、上記のとおりであり、アルキル化、アセチル化、カルボベンゾイル基の付加、スクシニミド基の形成などが挙げられる(例えば、マーレイ(Murray)他1995 Burger’s Medicinal Chemistry and Drug Discovery第5版, 第1巻, マンフレッド(Manfred) E. ウルフ(Wolf)編, John Wiley & Sons Inc.を参照のこと)。このとき、具体的には、N末端アミノ基を、以下のようにして反応させることができる。
【0077】
(a)酸ハロゲン化物[例えば、RC(O)Cl]または対称な無水物との反応により、式RC(O)NH−(式中、Rは上記の通りである)のアミド基を形成する。通常、この反応は、好ましくは、反応中に生成する酸を除去するためのジイソプロピルエチルアミンなどの過剰(例えば、約10当量)の第三級アミンを含有する不活性希釈剤(例えば、ジクロロメタン)中で、ほぼ等モル量か過剰量(例えば、約5当量)の酸ハロゲン化物をペプチドと接触させることにより行なうことができる。反応条件は、これ以外の点では従来通りである(例えば、室温で30分間)。末端アミノをアルキル化して低級アルキルN−置換し、次いで上記のように酸ハロゲン化物との反応を行うことにより、式RC(O)NR−のN−アルキルアミド基が得られる;
【0078】
(b)無水コハク酸との反応によりスクシンイミド基を形成する。上記のように、ほぼ等モル量か過剰量の無水コハク酸(例えば、約5当量)を用いるのがよく、適当な不活性溶媒(例えば、ジクロロメタン)中での過剰(例えば、10当量)のジイソプロピルエチルアミンなどの第三級アミンの使用を含む、当該技術分野で既知の方法により、アミノ基をスクシンイミドに変換する。例えば、ウォレンバーグ(Wollenberg)他, 米国特許第4,612,132号を参照のこと。ペプチドのN末端が置換されたスクシンイミドを得るために、コハク酸基を、例えば、C2−C6アルキル、または慣用法で調製される−SR置換基で置き換えてもよいことを理解されたい。かかるアルキル置換基は、ウォレンバーグ他(前掲)により記載された方法で低級オレフィン(C2−C6)と無水マレイン酸との反応により調製され、−SR置換基は、RSH(式中、Rは上記のとおりである)と無水マレイン酸との反応により調製される;
【0079】
(c)好ましくは反応中に生成する酸を除去するための第三級アミンを含有する適当な不活性希釈剤(例えば、ジクロロメタン)中で、ほぼ等モル量か過剰のCBZ−Cl(すなわち、塩化ベンジルオキシカルボニル)または置換CBZ−Clとの反応により、ベンジルオキシカルボニル−NH−または置換ベンジルオキシカルボニル−NH−基を形成する;
【0080】
(d)適当な不活性希釈剤(例えば、ジクロロメタン)中で、等モル量か過剰(例えば、約5当量)のR−S(O)2Cl(式中、Rは上記の通りである)と反応させて末端アミンをスルホンアミドに変換することにより、スルホンアミド基を形成する。好ましくは、不活性希釈剤は、反応中に生成する酸を除去するための、ジイソプロピルエチルアミンなどの過剰(例えば、約10当量)の第三級アミンを含有する。反応条件は、これ以外の点では従来通りである(例えば、室温で30分間);
【0081】
(e)適当な不活性希釈剤(例えば、ジクロロメタン)中で、等モル量か過剰(例えば、約5当量)のR−OC(O)ClまたはR−OC(O)OC6H4−p−NO2(式中、Rは上記の通りである)と反応させて末端アミンをカルバメートに変換することにより、カルバメート基を形成する。好ましくは、不活性希釈剤は、反応中に生成する酸を除去するための、ジイソプロピルエチルアミンなどの過剰(例えば、約10当量)の第三級アミンを含有する。反応条件は、これ以外の点では従来通りである(例えば、室温で30分
間);ならびに
【0082】
(f)適当な不活性希釈剤(例えば、ジクロロメタン)中で、等モル量か過剰(例えば、約5当量)のR−N=C=O(式中、Rは上記の通りである)と反応させて末端アミンをウレア(すなわち、RNHC(O)NH−)基に変換することにより、ウレア基を形成する。好ましくは、不活性希釈剤は、ジイソプロピルエチルアミンなどの過剰(例えば、約10当量)の第三級アミンを含有する。反応条件は、これ以外の点では従来通りである(例えば、室温で30分間)。
【0083】
2)C末端修飾
C末端カルボキシル基がエステルに置き換えられた(すなわち、−C(O)OR(式中、Rは上記の通りである))擬似ペプチドの調製において、ペプチド酸を調製するために樹脂が用いられ、側鎖保護ペプチドを基剤および適切なアルコール(例えば、メタノール)で切断する。次いで、側鎖保護基を、フッ化水素で処理して所望のエステルを得ることによる常套法で除去する。
【0084】
C末端カルボキシル基がアミド−C(O)NR3R4で置き換えられた擬似ペプチドの調製において、ベンズヒドリルアミン樹脂を、ペプチド合成用固体支持体として使用する。合成の完了時、支持体からペプチドを切り離すためのフッ化水素処理により、直接、遊離ペプチドアミド(すなわち、C末端が−C(O)NH2)がもたらされる。あるいはまた、ペプチド合成でのクロロメチル化樹脂の使用と併せて、支持体から側鎖保護ペプチドを切断するためのアンモニアとの反応により遊離ペプチドアミドが得られ、アルキルアミンまたはジアルキルアミンとの反応により、側鎖保護アルキルアミドまたはジアルキルアミド(すなわち、C末端が−C(O)NRR1(式中、RおよびR1は上記の通りである))が得られる。次いで、側鎖保護を、フッ化水素で処理して遊離アミド、アルキルアミドまたはジアルキルアミドにすることによる常法により解除する。
【0085】
また別の実施形態では、C末端カルボキシル基またはC末端エステルは、カルボキシル基またはエステルのそれぞれの−OH基またはエステル(―OR)をN末端アミノ基と分子内で置き換えることにより環化して、環状ペプチドを形成するように誘導してもよい。例えば、合成および切断によりペプチド酸を得た後、溶液中、例えば塩化メチレン(CH2Cl2)、ジメチルホルムアミド(DMF)混合液中で、ジシクロカルボジイミド(DCC)などの適切なカルボキシル基活性化剤により、遊離酸を活性化エステルに変換する。次いで、活性化エステルをN末端アミンで分子内置換することにより、環状ペプチドを形成する。重合とは対照的に、内部環化は、高希釈溶液の使用により、増強され得る。かかる方法は、当該技術分野において既知である。
【0086】
また、本発明のペプチドを環化し、または該ペプチドの末端にデスアミノまたはデスカルボキシ(descarboxyl)残基を組み込むことにより、末端のアミノまたはカルボキシル基をなくし、プロテアーゼに対する感受性を低減する、またはペプチドの立体構造を制限することも可能である。本発明の化合物のC末端の官能基としては、アミド、アミド低級アルキル、アミドジ(低級アルキル)、低級アルコキシ、ヒドロキシル、カルボキシおよびその低級エステル誘導体ならびにその製薬的に許容され得る塩が挙げられる。
【0087】
前記のN末端およびC末端の修飾に加え、擬似ペプチドを含む本明細書に記載するペプチド化合物は、種々の親水性ポリマーのうちの1種類以上により、またはこれらとの共有結合により有利に修飾され得る。ペプチド化合物は、親水性ポリマーで誘導体化すると、その溶解度および循環半減期が増加し、その免疫原性が遮蔽されることがわかった。かなり驚くべきことには、前述のことは、その結合活性の低減(あるとしても)がほとんどない状態で達成できることがわかった。使用に好適な非タンパク質系ポリマーとしては、ポ
リエチレングリコールおよびポリプロピレングリコールが例となるポリアルキルエーテル、ポリ乳酸、ポリグリコール酸、ポリオキシアルケン、ポリビニルアルコール、ポリビニルピロリドン、セルロースおよびセルロース誘導体、デキストランおよびデキストラン誘導体などが挙げられるが、これらに限定されない。一般的に、かかる親水性ポリマーは、約500−約100,000ダルトン、より好ましくは約2,000−約40,000ダルトン、さらにより好ましくは約5,000−約20,000ダルトンの範囲の平均分子量を有する。好ましい実施形態では、かかる親水性ポリマーは、約5,000ダルトン、約10,000ダルトン、および約20,000ダルトンの平均分子量を有する。
【0088】
ペプチド化合物は、ゼリプスキー(Zallipsky), S. 1995 Bioconjugate Chem 6:150-165; モンファルジーニ(Monfardini), C他1995 Bioconjugate Chem 6:62-69; 米国特許第4,640,835号、同第4,496,689号、同第4,301,144号、同第4,670,417号、同第4,791,192号、同第4,179,337号または国際公開第WO 95/34326号に記載された方法のいずれかを用いて、かかるポリマーにより、またはこれとの結合により誘導体化することができる。
【0089】
一実施形態において、ペプチド化合物はポリエチレングリコール(PEG)により誘導体化される。PEGは、2つの末端ヒドロキシル基を有する、エチレンオキシド繰り返し単位の線状の水溶性ポリマーである。PEGは、その分子量により分類され、通常500ダルトン−約40,000ダルトンの範囲である。現時点で好ましい実施形態では、使用されるPEGは、5,000ダルトン−約20,000ダルトンの範囲の分子量を有する。本発明のペプチド化合物に連結させるPEGは、分枝であっても非分枝であってもよい。(例えば、モンファルジーニ, C他1995 Bioconjugate Chem 6:62-69参照)。PEGは、Shearwater Polymers, Inc.(ハンツビル, アラバマ州)、Sigma Chemical Co.および他社から市販されている。かかるPEGとしては、モノメトキシポリエチレングリコール(MePEG−OH)、モノメトキシポリエチレングリコール−スクシネート(MePEG−S)、モノメトキシポリエチレングリコール−スクシンイミジルスクシネート(MePEG−S−NHS)、モノメトキシポリエチレングリコール−アミン(MePEG−NH2)、モノメトキシポリエチレングリコール−トレシレート(MePEG−TRES)およびモノメトキシポリエチレングリコール−イミダゾリル−カルボニル(MePEG−IM)が挙げられるが、これらに限定されない。
【0090】
簡単には、一例の実施形態において、使用される親水性ポリマー、例えばPEGは、その一端が、好ましくは、メトキシまたはエトキシ基などの非反応性基でキャップされている。その後、ポリマーの他端を、ハロゲン化シアヌル(例えば、塩化、臭化またはフッ化シアヌル、)、ジイミダゾール、無水物試薬(例えば、ジブロモコハク酸無水物などのジハロコハク酸無水物)、アシルアジド、p−ジアゾイウムベンジル(diazoiumbenzyl)エーテル、3−(p−ジアゾニウムフェノキシ)−2−ヒドロキシプロピルエーテル)などの適当な活性化剤との反応で活性化する。次いで、活性化したポリマーを、本明細書に記載するペプチド化合物と反応させ、ポリマーで誘導体化されたペプチド化合物を作製する。あるいはまた、本発明のペプチド化合物内の官能基を、ポリマーとの反応のために活性化してもよく、または既知のカップリング方法を用いた協奏カップリング反応にて2つの官能基を結合させてもよい。本発明のペプチド化合物は、当業者に既知であり、当業者により使用されている他の無数の反応スキームを用いてPEGで誘導体化させ得ることは、容易に理解されよう。
【0091】
或る実施形態では、誘導体化されたペプチドは、非修飾ペプチドの約0.1−約0.01倍の活性を有する。また他の実施形態では、誘導体化されたペプチドは、非修飾ペプチドの約0.1−約1倍の活性を有する。さらにまた他の実施形態では、誘導体化されたペプチドは、非修飾ペプチドよりも大きな活性を有する。
【0092】
本実施形態における使用に好適なペプチドとしては、一般的に、EphA2、EphA4、EphA5、EphA7、EphB2およびEphB4(これらに限定されない)を含む、Eph受容体ファミリーメンバーに結合するペプチド、すなわちリガンドが挙げられる。かかるペプチドは、通常約50個以下のアミノ酸残基、より好ましくは約20個以下のアミノ酸残基を有する。本発明における使用に好適な親水性ポリマーとしては、ポリエチレングリコールおよびポリプロピレングリコールが例示されるポリアルキルエーテル、ポリ乳酸、ポリグリコール酸、ポリオキシアルケン、ポリビニルアルコール、ポリビニルピロリドン、セルロースおよびセルロース誘導体、デキストランおよびデキストラン誘導体などが挙げられるが、これらに限定されない。一般的に、かかる親水性ポリマーは、約500−約100,000ダルトン、より好ましくは約2,000−約40,000ダルトン、さらにより好ましくは約5,000−約20,000ダルトンの範囲の平均分子量を有する。或る実施形態では、かかる親水性ポリマーは、約5,000ダルトン、約10,000ダルトン、および約20,000ダルトンの平均分子量を有する。ペプチド化合物は、上記およびそこに挙げられた参考文献に記載された方法を用いて誘導体化することができる。
【0093】
4.主鎖修飾
化合物のペプチド誘導体を作製するための他の方法は、ルビー他1990 Biochem J 268(2):249-262に記載されている。このように、ペプチド化合物はまた、類似の生体活性を有する非ペプチド化合物の構造モデルとして使用される。当業者であれば、リードペプチド化合物と同じかまたは類似する所望の生体活性を有するが、溶解性、安定性ならびに加水分解およびタンパク質分解への感受性に関して、リードよりもより好都合な活性を有する化合物を構築するために、種々の技法が利用可能であることが認識されよう。モーガン他1989 Ann Rep Med Chem 24:243-252を参照のこと。これらの技法としては、ペプチド主鎖を、ホスホネート、アミデート、カルバメート、スルホンアミド、第二級アミンおよびN−メチルアミノ酸から構成される主鎖と置き換えることが挙げられる。
【0094】
ペプチジル結合[−C(O)NH−]の1つ以上が、−CH2−カルバメート結合、ホスホネート結合、−CH2−スルホンアミド結合、ウレア結合、第二級アミン(−CH2NH−)結合およびアルキル化ペプチジル結合[−C(O)NR6(式中、R6は、低級アルキルである)]などの結合で置き換えられている擬似ペプチドは、従来のペプチド合成の際に、合成の適切な時点で、アミノ酸試薬のかわりに適当に保護されたアミノ酸アナログを 用いるだけで調製される。
【0095】
好適な試薬としては、例えば、アミノ酸のカルボキシル基が、上記の結合のうちの1つを形成するのに適当な部分で置き換えられているアミノ酸アナログが挙げられる。例えば、ペプチド内の−C(O)NR−結合を−CH2−カルバメート結合(−CH2OC(O)NR−)で置き換えることが所望される場合、適切に保護されたアミノ酸のカルボキシル(−COOH)基をまず還元して−CH2OH基とし、次いで、これを、慣用法により−OC(O)Cl官能基またはp−ニトロカルボネート−OC(O)O−C6H4−p−NO2官能基に変換する。かかる官能基のいずれかと、固体支持体上に見られる部分的に組み立てられた(fabricated)ペプチドのN末端の遊離アミンまたはアルキル化アミンとの反応により、−CH2OC(O)NR−結合の形成がもたらされる。かかる−CH2−カルバメート結合の形成に関するより詳細な説明については、チョー(Cho)他1993 Science 261:1303-1305を参照のこと。
【0096】
同様に、ペプチド内のアミド結合のホスホネート結合での置き換えは、米国特許出願第07/943,805号、同第08/081,577号および第08/119,700号に記載のようにしてなされ得る。
【0097】
ペプチド内のアミド結合の−CH2−スルホンアミド結合での置き換えは、慣用法により、適当に保護されたアミノ酸のカルボキシル(−COOH)基を−CH2OH基に還元し、次いで、ヒドロキシル基をトシル基などの適当な脱離基に変換することによりなされ得る。トシル化誘導体と、例えばチオ酢酸との反応後、加水分解および酸化的塩素化することにより、―CH2−S(O)2Cl官能基が得られ、これは、カルボキシル基以外が適当に保護されたアミノ酸のカルボキシル基と置き変わる。ペプチド合成において、この適当に保護されたアミノ酸アナログを使用することにより、−CH2−S(O)2NR−結合の内包がもたらされてペプチド内のアミド結合と置き換わり、それにより擬似ペプチドが得られる。−CH2−S(O)2Cl基を得るためのアミノ酸のカルボキシル基の変換に関するより完全な説明については、例えば、ウェインスタイン, B., 1983 Chemistry
& Biochemistry of Amino Acids, Peptides and Proteins, 第7巻, 第267−357頁, Marcel Dekker, Inc.(ニューヨーク)を参照のこと。
【0098】
ペプチド内のアミド結合のウレア結合での置き換えは、米国特許出願第08/147,805号に記載のようにしてなされ得る。
【0099】
−CH2NH−結合がペプチド内のアミド結合と置き換わる第二級アミン結合は、例えば、アミド結合のカルボニル結合が、慣用法によりCH2基に還元されている、適当に保護されたジペプチドアナログを用いることにより調製することができる。例えば、ジグリシンの場合、アミドをアミンへ還元して脱保護すると、H2NCH2CH2NHCH2COOHになり、これは、後で、次のカップリング反応でのN−保護形態で使用される。ジペプチド内のアミド結合のカルボニル基の還元によるかかるアナログの調製は、当該技術分野で既知である(例えば、M. W. レミントン(Remington) 1994 Meth Mol Bio 35:241-247を参照のこと)。
【0100】
適当に保護されたアミノ酸アナログは、対応するアミノ酸の場合と同様にして、慣用のペプチド合成に使用される。例えば、通常約3当量の保護されたアミノ酸アナログが、この反応に使用される。塩化メチレンまたはDMFなどの不活性な有機系希釈剤を使用するが、反応副生成物として酸が生じる場合、通常、反応溶媒は、反応中に生成する酸を除去するための過剰量の第三級アミンを含有する。特に好ましい第三級アミンの1つは、ジイソプロピルエチルアミンであり、これは、通常約10倍過剰で用いる。この反応により、擬似ペプチドへの非ペプチジル結合を有するアミノ酸アナログの組み込みがもたらされる。かかる置換は、所望により、ペプチド内のアミド結合の0−すべてが非アミド結合で置き換えられるように、反復してもよい。
【0101】
また、ペプチドを環化しても、該ペプチドの末端にデスアミノまたはデスカルボキシ残基を組み込んでもよく、それにより末端のアミノまたはカルボキシル基をなくし、プロテアーゼに対する感受性を低減したり、またはペプチドの立体構造を制限したりしてもよい。、化合物のC末端の官能基としては、アミド、アミド低級アルキル、アミドジ(低級アルキル)、低級アルコキシ、ヒドロキシ、カルボキシおよびその低級エステル誘導体ならびにその製薬的に許容され得る塩が挙げられる。
【0102】
5.ジスルフィド結合形成
化合物は、システインのチオール基間の分子内ジスルフィド結合により環化された形態で存在してもよい。あるいはまた、システインのチオール基間の分子間ジスルフィド結合により、二量体(またはより多いオリゴマー)化合物を得るように作製することができる。また、システイン残基の1つ以上が、ホモシステインで置換してもよい。
【0103】
本発明の他の実施形態としては、イオウの1個が、CH2基またはイオウの他の同配体
と置換されている、これらのジスルフィド誘導体のアナログが挙げられる。これらのアナログは、当該技術分野で既知の方法を用いる分子内置換または分子間置換により作製され得る。
【0104】
あるいはまた、ペプチドのアミノ末端を、α−置換酢酸でキャップしてもよく、ここで、α置換基は、α−ハロ酢酸、例えば、α−クロロ酢酸、α−ブロモ酢酸またはα−ヨード酢酸などの脱離基である。脱離基をシステイン残基またはホモシステイン残基のイオウに置き換えることにより、本発明の化合物を環化または二量体化してもよい。例えば、アンドリュー(Andreu)他1994 Meth Mol Bio 35(7):91-169; バーカー(Barker)他1992 J Med
Chem 35:2040-2048;およびオア(Or)他1991 J Org Chem 56:3146-3149を参照のこと。
【0105】
また、ペプチドは、当該技術分野で既知の組換えDNA技法によっても調製され得る。
【0106】
Eph受容体のEph受容体結合化合物による調節
本明細書に記載するEph受容体結合化合物は、細胞におけるEph活性を調節することができるものである。Eph活性を調節するEph受容体結合化合物には、Eph受容体ファミリーの1つ以上のメンバーに結合する、本明細書に記載したペプチドまたは擬似ペプチドが含まれる。本発明の或る実施形態では、Eph受容体結合化合物は、本明細書に記載した単一のペプチドまたは擬似ペプチドのみを含む。本発明の或る実施形態では、細胞を、Eph受容体のリン酸化を引き起こし、それにより下流シグナル伝達事象を活性化するのに有効な量のEph受容体結合化合物と接触させる。或る実施形態では、細胞を、エフリンによるEph受容体のリン酸化を抑制し、それにより受容体活性を阻害するのに有効な量のEph受容体結合化合物と接触させる。他の実施形態では、細胞を、下流シグナル伝達事象を活性化または不活化するのに有効な量のEph受容体結合化合物と接触させ得る。下流シグナル伝達事象を活性化または不活化するのに有効なEph受容体結合化合物の量としては、少なくとも0.05μM、少なくとも0.1μM、少なくとも0.2μM、少なくとも0.3μM、少なくとも0.4μM、少なくとも0.5μM、少なくとも0.6μM、少なくとも0.7μM、少なくとも0.8μM、少なくとも0.9μM、少なくとも1μM、少なくとも5μM、少なくとも10μM、少なくとも20μM、少なくとも30μM、少なくとも40μM、少なくとも50μM、少なくとも60μM、少なくとも70μM、少なくとも80μM、少なくとも90μM、少なくとも100μMまたは少なくとも200μMの濃度が挙げられる。本明細書に記載していない他の有効濃度の測定は、当業者であれば容易に行える。
【0107】
本発明の或る実施形態では、目的のEph受容体は、細胞において、インビトロおよびインビボのいずれでも調節される。いずれの適用に関しても、細胞は、Ephファミリーの受容体の少なくとも1つのメンバーを発現する任意の細胞であればよく、ヒト細胞が挙げられるが、これに限定されない。
【0108】
本発明の或る実施形態では、EphAサブファミリーの受容体が調節される。かかる受容体としては、EphA1、EphA2、EphA3、EphA4、EphA5、EphA6、EphA7、EphA8、EphA9またはEphA10が挙げられる。或る特定の実施形態では、EphA2受容体を発現する細胞を、該受容体の活性および後続の下流シグナル伝達事象が調節されるように、本明細書に記載するペプチド、擬似ペプチドまたは低分子の有効量と接触させる。他の実施形態では、EphA4受容体を発現する細胞を、該受容体の活性および後続の下流シグナル伝達事象が調節されるように、本明細書に記載するペプチド、擬似ペプチドまたは低分子の有効量と接触させる。さらに他の実施形態では、EphA5またはEphA7受容体を発現する細胞を、該受容体の活性および後続の下流シグナル伝達事象が調節されるように、本明細書に記載するペプチド、擬似ペプチドまたは低分子の有効量と接触させる。
【0109】
他の実施形態では、EphBサブファミリーの受容体が調節される。かかる受容体としては、EphB1、EphB2、EphB3、EphB4、EphB5およびEphB6が挙げられる。或る特定の実施形態では、EphB4受容体を発現する細胞を、該受容体の活性および後続の下流シグナル伝達事象が調節されるように、本明細書に記載するペプチド、擬似ペプチドまたは低分子の有効量と接触させる。
【0110】
Ephファミリー受容体の特定のメンバーを刺激することは、アポトーシス(プログラム細胞死)の活性化に関係しているとみなされている。したがって、Eph受容体を過剰発現する或る特定の細胞(例えば、或る特定の型の腫瘍性細胞)においてプログラム細胞死を活性化することは、望ましくない細胞集団の選択的死滅に好都合であり得る。さらにまた、プログラム細胞死に関して標的化した細胞型において過剰発現する特定のEph受容体の選択的アゴニストとして機能するEph受容体結合化合物は、非標的化細胞を死滅させることなく、標的細胞を排除するための方法を提供し得る。
【0111】
本発明の或る実施形態では、Eph受容体ファミリーの特定のメンバーの選択的アゴニストまたはアンタゴニストとしての機能を果たすEph受容体結合化合物を投与する方法が意図される。或る実施形態では、本明細書に記載するペプチド、擬似ペプチドまたは低分子などの選択的アゴニストを用いて、ペプチド、擬似ペプチドまたは低分子の有効量を、哺乳動物(ヒトを含む)に投与することにより、プログラム細胞死を活性化することができる。或る特定の実施形態では、アゴニストはEphA2に結合し、それにより、この受容体へのエフリン−A1の結合を競合的に阻害する。他の実施形態では、アゴニストの結合により、受容体のリン酸化が活性化される。
【0112】
哺乳動物に投与されるアゴニストの有効量は、約0.001mg−約50mg/kg体重/日の範囲であり得る。有効量は、アゴニストが投与される経路、アゴニストの結合親和性、標的細胞内でのEph受容体発現レベルおよび非標的細胞内でのEph受容体発現レベル(これらに限定されない)などの因子によって決まる。しかしながら、アゴニストの有効量の決定は、当業者により容易に決定され得ることを理解されたい。
【0113】
治療剤および治療剤送達剤としてのEph受容体結合化合物
本明細書に記載するEph受容体結合化合物はまた、インビボでEph受容体を調節するために、ヒトを含む温血動物にも投与することができる。例えば、本明細書に開示する或る特定のペプチドが、EphA2を選択的に活性化するため、またはEphA4を選択的に阻害するために使用され得る。したがって、本発明は、インビボでEph受容体を活性化または阻害するのに十分な量のかかる化合物を投与することを含む、Eph関連疾患の治療上の処置のための方法を包含する。
【0114】
また、Eph受容体を標的化することにより、癌および他の疾患の治療行為が可能になる。本明細書に記載するEph受容体結合化合物は、細胞傷害剤を疾患組織の血管に送達するために使用され得る。実際、化学療法剤、毒素またはプロアポトーシスペプチドに結合させた血管標的ペプチドは、腫瘍成長を低減し、臨床的関節炎を抑制し、または前立腺組織を破壊し得る(アラップ(Arap), W他1998 Science 279:377-380; オルソン(Olson), T. A.他1997 Int J Cancer 73:865-870; エルビー(Ellerby) H.M.他1999 Nat Med 5:1032-1038; アラップ, W他2002 PNAS USA 99:1527-1531; ゲルラク(Gerlag), D. M.他2001 Arthritis Research 3:357-361)。例えば、アゴニストにより引き起こされるEphA2のチロシンリン酸化は、受容体およびアゴニストのインターナリゼーションを媒介し(ザンテック(Zantek), N. D.他1999 Cell Growth Differ 10:629-638; カルレス−キンチ(Carles-Kinch), K他2002 Cancer Res 62:2840-2847; ファン・デル・ギア(Van der Geer), P他1994 Annu Rev Cell Biol 10:251-337)、したがって、毒性物質またはプロアポトーシ
ス物質が、細胞内に送達され、選択的に細胞を死滅させ得る(エルビー, H. M.他1999 Nat Med 5:1032-1038)。さらにまた、本明細書に記載するEph受容体結合化合物により誘導されるEphA2の活性化により、EphA2発現癌細胞の増殖、浸潤性および転移性が低減され得る(ザンテック, N. D.他1999 Cell Growth Cell Differ 10:629-638; カルレス−キンチ, K他2002 Cancer Res 62:2840-2847; ミャオ(Miao), H.他Nature 2000 Cell Biol 2:62-69)。EphA2活性化は、乳癌細胞および前立腺癌細胞の悪性度の低減と相関し、EphA2過剰発現の形質転換効果を逆転させることは、当該技術分野では既知である(ゼリンスキー, D. P.他2002 J Cell Biochem 85:714-720; ザンテック, N. D.他1999 Cell Growth Differ 10:629-638; カルレス−キンチ, K他2002 Cancer Res 62:2840-2847)。細胞傷害剤を送達するために本明細書に記載するEph受容体結合化合物を使用した場合、本明細書に開示する組成物によるEphA2の活性化により、アポトーシス刺激に対して細胞を感受性とし得る(ドーン(Dohn), M.他, 2001 Oncogene 20:6503-6515)。
【0115】
本発明の或る実施形態は、本明細書に記載するペプチド、擬似ペプチドまたは低分子などのEph受容体結合化合物に連結させた治療剤を含有するコンジュゲートを意図する。かかるコンジュゲートは、治療を必要とする動物に適切なコンジュゲートを投与することにより、適切なEph受容体を発現する標的細胞に送達され得る。或る実施形態では、治療剤は治療を担う。他の実施形態では、治療剤およびEph受容体結合化合物の両方が治療に寄与する。或る実施形態では、治療剤は、イメージング剤である。
【0116】
目的のEph受容体に結合するEph受容体結合化合物は、リンカーにより治療剤と連結させる。リンカーは、Eph受容体結合化合物と治療剤の両者が同じ領域、組織または細胞に標的化されることを許容するものであれば、任意の結合、低分子、またはその他のビヒクルのいずれであってもよい。好ましくは、リンカーが切断可能なものである。
【0117】
一実施形態において、リンカーは、1つ以上のEph受容体結合化合物と1つ以上の治療剤との間の化学結合である。したがって、該結合は、共有結合またはイオン結合であり得る。リンカーが化学結合であるコンジュゲートの一例は、融合タンパク質ということになる。一実施形態において、化学結合は、pH感受性結合である。あるいはまた、該結合は、pH感受性でなくてもよいが、標的部位の微環境において、後で付加されるか、または元々存在する特定の酵素または化学物質により切断可能であるのがよい。あるいはまた、該結合は、還元条件下で切断される結合(例えば、ジスルフィド結合)であってもよい。あるいはまた、該結合は、切断可能でなくてもよい。
【0118】
任意の種類のpH切断性またはpH感受性リンカーが使用され得る。酸切断性結合の例としては、シス−ポリカルボン酸アルケンとして知られる部類の有機酸が挙げられるが、これらに限定されない。この部類の分子は、少なくとも1つの二重結合を含む炭素鎖に結合した少なくとも3個のカルボン酸基(COOH)を含む。これらの分子、ならびにこれらをどのように作製および使用するかは、シェン(Shen)他(米国特許第4,631,190号)に開示されている。あるいはまた、アミノ−スルフヒドリル架橋剤などの穏和な酸性条件下で切断可能な分子を使用してもよい。これらの分子は、ブラットラー(Blattler)他の米国特許第4,569,789号に開示されている。
【0119】
あるいはまた、切断可能なリンカーは、生分解性、加水分解性結合などの徐放性結合であってもよい。通常の生分解性担体結合としては、エステル、アミドまたはウレタン結合が挙げられるため、通常の担体は、ポリエステル、ポリアミド、ポリウレタン、および約5,000−1,000,000の分子量を有する他の縮合重合体である。これらの担体/結合の例は、ピーターソン(Peterson)他の米国特許第4,356,166号に示されている。他の酸切断性リンカーは、米国特許第4,569,789号および同第4,631
,190号またはブラットラー他1985 Biochemistry 24:1517-1525で確認できる。リンカーは、自然な酸性条件により切断され、あるいはまた、酸性条件は、エイブラムズ(Abrams)他の米国特許第4,171,563号に説明してあるようにして標的部位にて誘導してもよい。
【0120】
切断可能なジスルフィド結合(還元性結合)を含む連結試薬の例としては、「DPDPB」(1,4−ジ−[3’−(2’−ピリジルジチオ)プロピオンアミド]ブタン)、「SADP」(N−スクシンイミジル(4−アジドフェニル)1,3−ジチオプロピオネート)、「スルホ−SADP」(スルホスクシンイミジル(4−アジドフェニルジチオ)プロピオネート)、「DSP」(ジチオビス(スクシンイミジルプロピオネート))、「DTSSP」(3,3’−ジチオビス(スルホスクシンイミジルプロピオネート))、「DTBP」(ジメチル3,3’−ジチオビスプロピオンイミデート−2HCl))(これらは、すべて、Pierce Chemicals(ロックフォード、イリノイ州)から入手可能である)が挙げられるが、これらに限定されない。
【0121】
酸化により切断され得る連結試薬の例としては「DST」(ジスクシンイミジルタルタレート)および「スルホ−DST」(ジスクシンイミジルタルタレート)である。これらのリンカーもまた、Pierce Chemicalsから入手可能である。
【0122】
切断可能でないリンカーの例は、「スルホ−LC−SMPT」(スルホスクシンイミジル6−[α−メチル−α−(2−ピリジルチオ)トルアミド]ヘキサノエート)、「SMPT」、「ABH」(アジドベンゾイルヒドラジド)、「NHS−ASA」(N−ヒドロキシスクシンイミジル−4−アジドサリチル酸)、「SASD」(スルホスクシンイミジル2−(p−アジドサリチルアミド)エチル−1,3−ジチオプロピオネート)、「APDP」(N−[4−(p−アジドサリチルアミド)ブチル]−3’(2’−ピリジルジチオ)プロピオンアミド)、「BASED」(ビス−[β−(4−アジドサリチルアミド)エチル]ジスルフィド)、「HSAB」(N−ヒドロキシスクシンイミジル−4アジドベンゾエート)、「APG」(p−アジドフェニルグリオキサル一水和物)、「SANPAH」(N−スクシンイミジル−6(4’−アジド−2’−ミトロ(mitro)フェニル−アミド)ヘキサノエート)、「スルホ−SANPAH」(スルホスクシンイミジル6−(4’−アジド−2’−ニトロフェニルアミド)ヘキサノエート)、「ANB−NOS」(N−5−アジド−2−ニトロベンジオイル(bezyoyl)オキシスクシンイミド)、「SAND」(スルホスクシンイミジル−2−(m−アジド−o−ミトロ(mitro)ベンズアミド)−エチル−1,3’−ジチオプロピオネート)、「PNP−DTP」(p−ニトロフェニル−2−ジアゾ−3,3,3−トリフルオロプロピオネート)、「SMCC」(スクシンイミジル4−(N−マレイミドメチル)シクロヘキサン−1−カルボキシレート)、「スルホ−SMCC」(スルホスクシンイミジル4−(N−マレイミドメチル)シクロヘキサン−1−カルボキシレート)、「MBS」(m−マレイミドベンゾイル−N−ヒドロキシスクシンイミドエステル)、「スルホ−MBS」(m−マレイミドベンゾイル−N−ヒドロキシスルホスクシンイミドエステル)、「SIAB」(N−スクシンイミジル(4−ヨードアセチル)アミノベンゾエート)、「スルホ−SIAB」(N−スルホスクシンイミジル(4−ヨードアセチル)アミノベンゾエート)、「SMPB」(スクシンイミジル4−(p−マレン(malen)イミドフェニル)ブチレート)、「スルホ−SMPB」(スルホスクシンイミジル4−(p−マレンイミドフェニル)ブチレート)、「DSS」(ジスクシンイミジルスベレート)、「BSSS」(ビス(スルホスクシンイミジルスベレート))、「BMH」(ビスマレイミドヘキサン)、「DFDNB」(1,5−ジフルオロ−2,4−ジニトロベンゼン)、「DMA」(ジメチルアジピミデート(adipimidate)2HCl)、「DMP」(ジメチルピメルイミデート(pimelimidate)−2HCl)、「DMS」(ジメチルスベルイミデート(suberimidate)−2−HCl)、「SPDP」(N−スクシンイミジル−3−(2−ピリジルチオ)プロピオネート)、「スルホ−HSAB」(スルホス
クシンイミジル4−(p−アジドフェニル)ブチレート)、「スルホ−SAPB」(スルホスクシンイミジル4−(p−アジドフェニルブチレート)、「ASIB」(1−9p−アジドサリチルアミド)−4−(ヨードアセトアミド)ブタン)、「ASBA」(4−(p−アジドサリチルアミド)ブチルアミン)である。これらのリンカーは、すべて、Pierce Chemicalsから入手可能である。
【0123】
別の実施形態では、リンカーは、ペプチドリンカーなどの低分子である。一実施形態では、ペプチドリンカーは、切断可能ではない。さらなる実施形態では、ペプチドリンカーは、還元条件下での塩基により、または特定の酵素により切断可能である。一実施形態では、酵素は内生のもの(indigenous)である。あるいはまた、低分子ペプチドは、治療用コンジュゲートの後またはこれに加えて投与される内生でない酵素により、切断可能であってもよい。あるいはまた、低分子ペプチドは、例えばジスルフィド結合を含む場合には還元条件下で切断できる。。あるいはまた、低分子ペプチドはpH感受性であってもよい。ペプチドリンカーの例としては、ポリ(L−Gly)(ポリL−グリシンリンカー)、ポリ(L−Glu)(ポリL−グルタミンリンカー)、ポリ(L−Lys)(ポリL−リシンリンカー)が挙げられる。一実施形態では、ペプチドリンカーは、式(アミノ酸)nを有し、式中、nは、2−100の整数であり、好ましくは、この場合のペプチドは、1個以上のアミノ酸のポリマーを含有する。
【0124】
さらなる実施形態では、ペプチドリンカーは、プロテイナーゼにより切断可能である(スズキ(Suzuki)他1998 J Biomed Mater Res 42:112-6)。或る実施形態では、リンカーは、ポリ(エチレングリコール)(PEG)、およびジペプチドであるL−アラニル−L−バリン(Ala−Val)(Goyal他2000 Biochem J 345:247-254)を含む、酵素サーモリシンにより切断可能なリンカーである。
【0125】
化学物質リンカーおよびペプチドリンカーは、コンジュゲート合成のための当該技術分野で既知の技法により、すなわち、遺伝子操作を用いることにより、または化学的に、Eph受容体結合化合物と治療剤とを結合するものであり得る。コンジュゲート合成は、タンパク質の適切な官能基での他の構成部分との古典的なカップリング反応により、適切な抗体を介して化学的になされ得る。タンパク質に存在し、化学的カップリング反応に通常用いられる官能基の例を以下に概説する。糖質(carbohydrate)構造を、アルデヒド基に酸化し、これを、H2NNH−R基を含有する化合物(式中、Rは当該化合物)と反応させ、C=NH−NH−R基を形成してもよい。チオール基(タンパク質中のシステイン)を、チオール反応性基を含有する化合物と反応させて、チオエーテル基またはジスルフィド基を形成してもよい。アミノ酸残基中(タンパク質のアミノ末端またはリシン上)の遊離アミノ基を、活性化カルボキシ基などの求電子基を含有する化合物と反応させて、アミド基を形成してもよい。アミノ酸残基中の遊離カルボキシル基を反応性カルボキシル基に変換し、次いで、アミノ基を含有する化合物と反応させてアミド基を形成してもよい。
【0126】
Eph受容体結合化合物と連結させる治療剤は、所望の効果をもたらす任意の化学物質、分子またはコンジュゲートであり得る。例としては、抗生物質、抗腫瘍剤、免疫抑制剤、ホルモンなどの従来の医薬剤、1種類以上の遺伝子、アンチセンスオリゴヌクレオチド、低分子干渉性RNA、造影剤、タンパク質、毒素、放射性分子もしくは原子、界面活性タンパク質、ナノ粒子または凝固タンパク質が挙げられるが、これらに限定されない。治療剤は、親油性(標的細胞内に進入するのを補助する性質)であってもよい。
【0127】
造影剤は、当業者に既知の任意の型の造影剤であり得る。最も一般的な造影剤は、基本的に、4つの群のうちの1つに分類される;X線試薬、X線撮影試薬、磁気共鳴画像形成剤、量子ドット、ナノ粒子および超音波剤。X線試薬としては、イオン性ヨウ素含有試薬ならびにOmnipaque(Nycomed)およびUltravist(Schering)などの非イ
オン性試薬が挙げられる。X線撮影試薬としては、以下に開示するものなどの放射性同位体が挙げられる。磁気共鳴画像形成剤としては、ガドリニウムおよび鉄酸化物キレートなどの磁気剤(magnetic agent)が挙げられる。超音波剤としては、気体の超微粒気泡およびいくつかの気泡放出製剤が挙げられる。
【0128】
放射性核種は、診断用であってもよく、治療用であってもよい。一般的に医学的に有用な放射性核種の例としては、90Y、111Ln、67Cu、77Lu、99Tcなど、好ましくは90Yおよび111Lnなどの三価のカチオンなどの、Y、Ln、Cu、Lu、Tc、Re、Co、Feなどが挙げられる。
【0129】
診断用γシンチレーション測光法による器官および組織のインビボでの画像形成に好適な放射性核種としては、以下のもの:γ放出放射性核種:111Ln、113mLn、67Ga、68Ga、99mTc、51Cr、197Hg、203Hg、169Yb、85Srおよび87Srが挙げられる。Fab’断片による結合に好適なキレート化放射性核種の調製は、米国特許第4,658,839号(ニコレッティ(Nicoletti)他)に教示されている。
【0130】
MRIにおける画像形成剤としての使用に好適な常磁性体金属イオンとしては、原子番号57−70のランタニド系元素または原子番号21−29、42または44の遷移金属が挙げられる。米国特許第4,647,447号(グリース(Gries)他)には、キレート化常磁性体金属イオンによるMRI画像形成が教示されている。
【0131】
治療用放射性核種の例は、β放出体である。好適なβ放出体としては、67Cu、186Rh、188Rh、189Rh、153Sm、90Yおよび111Lnが挙げられる。
【0132】
アンチセンスオリゴヌクレオチドは、正常遺伝子の過剰発現、または異常遺伝子の発現により引き起こされる任意の疾患の治療における使用の可能性を有する。アンチセンスオリゴヌクレオチドは、かかる遺伝子の発現を低減または停止するために使用され得る。アンチセンス技術により治療され得る癌遺伝子の例および使用され得る具体的なアンチセンス分子を教示する参考文献としては、c−JunおよびcFos(米国特許第5,985,558号)、HER−2(米国特許第5,968,748号)、E2F−1(ポポフ(Popoff)ら 米国特許第6,187,587号)、SMAD1−7(米国特許第6,159,697号;同第6,013,788号;同第6,013,787号;同第6,013,522号;および同第6,037,142号)、ならびにFas(ディーン(Dean)ら 米国特許第6,204,055号)が挙げられる。
【0133】
また、正常遺伝子の過剰発現、または異常遺伝子の発現により引き起こされる任意の疾患の治療におけるRNA干渉方法における使用のための二本鎖RNA分子を提供する。RNA干渉(RNAi)は、転写後RNA分解による配列特異的遺伝子サイレンシングプロセスであり、これは、発現抑制される遺伝子と配列が相同的な二本鎖RNA(dsRNA)により開始される。RNAiに好適な二本鎖RNA(dsRNA)は、標的対象遺伝子に対応する19個のRNA塩基対で構成され、各3’末端の2つのヌクレオチドを突出端(overhang)とした約21個の連続ヌクレオチドのセンスおよびアンチセンス鎖を含む(エルバシル(Elbashir)ら 2001 Nature 411: 494-498;バス(Bass)、2001 Nature 411: 428-429;ザモア(Zamore)、2001 Nat Struct Biol 8: 746-750)。約25−30個のヌクレオチドのdsRNAもまた、RNAiに成功裡に使用されている(カラビノス(Karabinos)ら2001 PNAS 98: 7863-7868)。dsRNAは、当該技術分野で既知の方法により、インビトロで合成し、細胞内に導入することができる。かかる方法により、標的ポリペプチドの翻訳を低減させ得る。
【0134】
治療剤として使用され得るタンパク質としては、細胞内に存在させるとアポトーシスを誘導するpRBおよびp53(スー(Xu)他米国特許第5,912,236号)などのアポトーシス誘導剤、およびエリスロポエチンなどの、疾患時において消失または発現が低下するタンパク質(スイトコウスキー(Sytkowski)他米国特許第6,048,971号)が挙げられる。
【0135】
治療剤は、アルキル化剤(ナイトロジェンマスタード、エチレンイミン、アルキルスルホネート、ニトロソウレアおよびトリアゼン)、代謝拮抗物質(メトトレキセートなどの葉酸アナログ、ピリミジンアナログおよびプリンアナログ)、天然産物およびその誘導体(抗生物質、アルカロイド、酵素)、ホルモンおよびアンタゴニスト(副腎皮質ステロイド、プロゲスチン、エストロゲン)などの腫瘍性疾患のための任意の化学療法剤であり得ることを理解されたい。あるいはまた、治療剤は、抗腫瘍剤としての機能を果たすアンチセンスオリゴヌクレオチド、または腫瘍性細胞でのアポトーシスを活性化するタンパク質であり得る。
【0136】
治療剤は、任意の型の神経奏効物質であってもよく、例えば、神経伝達物質または神経伝達物質アンタゴニストを、その使用に伴って一般的に経験する広範な副作用なしで、必要とされる領域に標的化し得る。
【0137】
治療剤は、痛みの領域に特異的に標的化され得る、オピオイドなどの麻酔薬であってもよい。吐気などの副作用は、オピオイド鎮痛剤を使用している患者が一般的に経験する。本発明の方法により、外科的創傷または関節炎の場合における関節などの必要な領域への薬物の非常に特異的な局在が可能になり、これは、副作用を低減し得る。
【0138】
治療剤は、ヒスタミン、H1−受容体アンタゴニストおよびブラジキニンなどの抗炎症剤であってもよい。あるいはまた、抗炎症剤は、サリチル酸誘導体、インドールおよびインデン酢酸、ならびにアルカノンなどの非ステロイド系抗炎症剤であってもよい。あるいはまた、抗炎症剤は、コルチコステロイド、クロモリンナトリウムおよびネドクロミルなどの喘息の治療用のものであってもよい。抗炎症剤は、B2選択的アドレナリン作用薬およびテオフィリンなどの気管支拡張薬とともに、または単独で投与され得る。
【0139】
治療剤は、利尿剤、バソプレシンアゴニストもしくはアンタゴニスト、アンギオテンシンまたは患者の血圧に特異的に作用するレニンであってもよい。
【0140】
治療剤は、心臓疾患の治療に使用される任意の医薬であり得る。かかる医薬としては、有機系亜硝酸塩(アミル亜硝酸塩、ニトログリセリン、イソソルビド二硝酸塩)、カルシウムチャネル遮断薬、抗血小板剤および抗血栓剤、血管拡張薬、血管抑制薬、抗ジギタリス抗体ならびに結節遮断薬が挙げられるが、これらに限定されない。
【0141】
治療剤は、テトラサイクリン、クリンダマイシン、キニン、クロロキン、メフロキン、トリメトプリムスルファメトキサゾール、メトロニダゾールおよびオラミンなどの、原生動物感染の治療に使用される任意の医薬であり得る。標的医薬または他の治療薬を原生動物感染の領域に標的化できることは、これらの抗生物質医薬によって、一般に非常に重篤な副作用を経験することから、特に重要である。
【0142】
治療剤は、スルホンアミド、キノロン、ペニシリン、セファロスポリン、アミノグリコシド、テトラサイクリン、クロラムフェニコール、エリスロマイシン、イソニアジドおよびリファンピンなどの任意の抗菌薬であり得る。
【0143】
治療剤は、アンフォテリシン、フルシトシン、ミコナゾールおよびフルコナゾールなど
の、真菌感染の治療に使用される任意の医薬であり得る。
【0144】
治療剤は、アシクロビル、ビダラビン、インターフェロン、リバビリン、ジドブジン、ザルシタビン、逆転写酵素インヒビター、およびプロテアーゼインヒビターなどの、ウイルス感染の治療に使用される任意の医薬であり得る。また、毒素、放射性原子およびアポトーシス誘導剤などの他の治療剤を用いてウイルス感染細胞を標的化し、死滅させることも想定され得る。
【0145】
治療剤は、種々の抗凝血剤、抗血栓薬および抗血小板薬から選択され得る。
【0146】
かかる治療剤をホルモン(成長ホルモン、アンドロゲン、エストロゲン、性腺刺激ホルモン放出ホルモン、甲状腺ホルモン、副腎皮質ステロイド、インスリンおよびグルカゴン)として使用し、ホルモンの過剰産生または産生不良に起因する疾患を治療し得ることを理解されたい。あるいはまた、ホルモンが過剰産生される場合、そのホルモンに対するアンタゴニストまたは抗体を治療剤として使用し得る。
【0147】
他の考えられ得る種々の治療剤としては、ビタミン、酵素、ならびに、産生の不十分な他の細胞成分、およびジフテリア毒素またはボツリヌス毒素などの毒素が挙げられる。
【0148】
あるいはまた、治療剤は、インビトロ診断で通常使用されるものであってもよい。したがって、リガンドおよびリンカーを慣用法により標識し、シグナル生成系のすべてまたは一部を形成する。リガンドおよびリンカーは、当該技術分野で既知の方法により、トリチウム、炭素14、リン32、ヨウ素125およびヨウ素131などの放射性同位体に共有結合させることができる。例えば、125Iは、クロラミン−T法などの手順、ラクトペルオキシダーゼ法により酵素的に、または予備標識Bolton-Hunter法により導入され得る。これらおよびその他の技法は、H. バン・ブナキス(Van Vunakis)およびJ. J. ランゴン(Langone)編、Methods in Enzymology、第70巻、パートA、1980において考察されている。また、放射性標識のさらなる例については、米国特許第3,646,346号および同第4,062,733号を参照のこと。
【0149】
あるいはまた、治療剤は、化学的環境の変化により、または酵素などの別の分子薬剤の作用により、対応する医薬剤に変換されるプロドラッグまたはプロ分子であってもよい。好ましくは、治療剤は、プロ分子の変換に必要な特定の分子とともに投与される。あるいはまた、プロ分子は、標的組織の微環境において見られる天然分子により切断され得る。あるいはまた、プロドラッグは、pH感受性であり、血管から細胞または細胞内小胞に環境が変化すると、変換される(グレコ(Greco)他2001 J Cell Physiol 187: 22-36)。
【0150】
哺乳動物に投与されるコンジュゲートの有効量は、約0.001mg−約50mg/kg体重/日の範囲であり得る。有効量は、コンジュゲートが投与される経路、コンジュゲートの結合親和性、標的細胞内でのEph受容体発現レベルおよび非標的細胞内でのEph受容体発現レベル(これらに限定されない)などの因子によって決まる。しかしながら、アゴニストの有効量の決定は、当業者により容易に決定され得ることを理解されたい。
【0151】
本発明の別の態様としては、活性成分として本明細書に開示するペプチド、擬似ペプチド、低分子の少なくとも1種類を、医薬用担体または希釈剤との組合せで含有する医薬組成物が挙げられる。これらの化合物は、経口、肺系、非経口(筋肉内、腹腔内、静脈内もしくは皮下への注射)、吸入(微粉末製剤もしくはエーロゾル)、経皮的、鼻腔内、膣内、直腸内、または舌下経路の投与により投与でき、各投与経路に適切な投薬形態に製剤化され得る。例えば、バーンスタイン(Bernstein)他のPCT特許公開公報第WO 93/25221;ピット(Pitt)他のPCT特許公開公報第WO 94/17784;およびピ
ット他の欧州特許出願第613,683号を参照のこと。
【0152】
経口投与のための固形投薬形態としては、カプセル、錠剤、丸剤、散剤および顆粒剤が挙げられる。かかる固形投薬形態では、活性化合物は、スクロース、ラクトースまたはデンプンなどの少なくとも1種類の不活性な製薬的に許容され得る担体と混合する。また、かかる投薬形態は、通常の粒子のように、不活性希釈剤以外の添加剤物質、例えば、ステアリン酸マグネシウムなどの潤沢剤を含有し得る。カプセル、錠剤および丸剤の場合は、投薬形態は、緩衝剤も含有し得る。錠剤および丸剤は、さらに、腸溶性コートを用いて調製してもよい。
【0153】
経口投与のための液状投薬形態としては、製薬的に許容され得るエマルジョン、液剤、懸濁液、シロップが挙げられ、水などの当該技術分野で一般的に使用される不活性希釈剤を含むエリキシル剤を含む。かかる不活性希釈剤の他に、組成物はまた、湿潤剤、乳化剤および懸濁剤ならびに甘味料、香味料および香料などの佐剤を含み得る。
【0154】
非経口投与用の本発明の調製物としては、滅菌された水性または非水性溶液、懸濁液またはエマルジョンが挙げられる。非水性溶剤またはビヒクルの例は、プロピレングリコール、ポリエチレングリコール、オリーブ油およびコーン油などの植物油、ゼラチン、およびオレイン酸エチルなどの注射用有機エステルである。かかる投薬形態はまた、保存剤、湿潤剤、乳化剤、および分散剤などの佐剤を含み得る。これらは、例えば、細菌保持フィルターによるろ過により、組成物に滅菌剤を組み込むことにより、組成物に放射線照射することにより、または組成物を加熱することにより、滅菌し得る。これらはまた、使用直前に、滅菌水、またはいくつかの他の滅菌注射用媒体を用いて製造され得る。
【0155】
膣内または直腸内投与のための組成物は、好ましくは、活性物質に加えてココアバターまたは坐剤用ワックスなどの賦形剤を含んでいてもよい坐剤である。また、鼻腔内または舌下投与のための組成物は、当該技術分野で既知の標準的な賦形剤を用いて調製される。
【0156】
化合物を含有する組成物は、予防的および治療上の処置のために投与され得る。治療的な適用では、組成物は、上記のような疾患に既に苦しんでいる患者に、該疾患の症状およびその合併症を治癒するか、または少なくとも一部抑止するのに充分な量で投与される。これを成し遂げるのに充分な量を「治療的有効投与量」と定義する。この使用に有効な量は、疾患の重篤度および患者の体重および全身状態により決まる。
【0157】
本明細書に記載する組成物はまた、例えば、タイス(Tice)およびビビ(Bibi)の方法(Treatise on Controlled Drug Delivery, A. キドニエウス(Kydonieus)編、Marcel Dekker(ニューヨーク)1992、315−339頁)によりマイクロカプセル封入してもよい。
【0158】
予防的な適用では、本明細書に開示する化合物を含む組成物は、特定の疾患に感受性か、または罹患の危険性がある患者に投与される。かかる量を「予防的有効投与量」と定義する。この用法においてもまた、正確な量は、患者の健康状態および体重により決まり、当業者は容易に決定し得る。
【0159】
有効な治療に必要なEph受容体アゴニストの量は、投与手段、標的部位、患者の生理学的状態および他に受けている薬物治療を含む多くの種々の因子により決まる。したがって、治療投薬は、安全性および有効性を最適化するために滴定されなければならない。通常、インビトロで使用される投薬量により、これらの試薬のインサイチュ投薬に有用な量についての有用な指針をもたらされる。具体的な障害の治療のための有効投与量の動物試験により、ヒトへの投薬量のさらなる予測の目安が提供される。種々の考慮事項が、例えば、ギルマン(Gilman)他編、1990 Goodman and Gilman’s: The Pharmacological Basis
of Therapeutics第8版、Pergamon Press;およびRemington’s Pharmaceutical Sciences、第7版、Mack Publishing Co.(イーストン、ペンシルベニア州)(1985)に記載されている。
【0160】
本明細書に記載するペプチドおよび擬似ペプチドは、約0.001mg−約50mg/kg体重/日の範囲の投薬量で投与すると、Eph受容体媒介性症状の治療に有効である。使用される具体的な投与量は、治療対象の具体的な状態、投与経路により、および状態の重篤度、患者の年齢および全身状態などの因子に応じて担当医の判断により調節される。かかる投与量は、当業者により容易に決定され得る。
【0161】
非経口投与には、ペプチドを、製薬的に許容され得る非経口用ビヒクルとの組合せで、例えば、液剤、懸濁液、エマルジョンまたは凍結乾燥粉末剤に製剤化すればよい。かかるビヒクルの例は、水、生理食塩水、リンゲル液、デキストロース溶液、および5%ヒト血清アルブミンである。また、リポソームおよび固定油などの非水性ビヒクルを使用してもよい。ビヒクルまたは凍結乾燥粉末は、等張性を維持する添加剤(例えば、塩化ナトリウム、マンニトール)、化学的安定性を維持する添加剤(例えば、緩衝剤および保存剤)を含み得る。製剤は、一般的に使用されている技法により滅菌する。例えば、注射による投与に適した非経口用組成物は、1.5重量%の活性成分を0.9%塩化ナトリウム溶液に溶解することにより調製される。
【0162】
本明細書に記載する医薬組成物は、単回投与で投与してもよく、反復投与で投与してもよく、単独の治療剤として、または他の治療剤と併用のいずれかで投与しても、従来からの治療法と組み合わせてもよく、逐次投与しても、同時投与してもよい。
【0163】
化合物は、徐放製剤の状態、例えば、低速放出ポリマーを含む組成物の状態で投与してもよい。活性化合物は、埋込み、およびマイクロカプセル封入型送達システムを含む、制御放出製剤などの、該化合物を急速放出から保護する担体を用いて調製することができる。エチレン酢酸ビニル、ポリ酸無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、ポリ乳酸およびポリ乳酸系、ポリグリコールコポリマー(PLG)などの生分解性生体適合性ポリマーが使用され得る。かかる製剤の調製のための多くの方法が、一般的に、当業者に既知である。
【0164】
本明細書に記載するEph受容体結合化合物は、該化合物を唯一の活性剤として含む医薬組成物に製剤化してもよい。あるいはまた、医薬組成物は、さらなる活性剤を含んでいてもよい。例えば、本明細書に記載するEph受容体結合化合物の2種類以上を組み合わせて使用してもよい。さらにまた、本ペプチド化合物を、Eph受容体活性に対して調節効果を有する1種類以上の他の薬剤と組み合わせてもよい。
【0165】
Ephファミリーメンバーに特異的に結合するペプチドを同定するためのファージディスプレイの使用
16種類の既知のEph受容体の各々に特異的に結合するペプチドを単離するためにファージディスプレイを使用し得る。本明細書に記載するように、EphA2、EphA4、EphA5、EphA7、EphB2またはEphB4に特異的に結合する、数個のファージディスプレイペプチドを単離し、それらの多くは、特異的に結合する。したがって、目的のEph受容体に選択的に結合するペプチドを得るため、Eph受容体ファミリーの16種類の既知のメンバーのいずれかに対するパニング用ランダムペプチドライブラリーを使用し得る。クローンは、当該技術分野で既知の配列決定技法により同定することができる。高結合選択性および高結合親和性の両方を有するペプチドを得るために、ペプチドライブラリーに含まれるペプチドの長さを調節してもよい。
【0166】
他の有用性
本明細書に記載する化合物は、エフリンリガンドの産生およびその受容体結合プロセスに影響する、およびこれらに影響されると考えられる多くの要因の評価を含む、Eph受容体の生物学的役割を理解するための独自のツールとして、インビトロで有用である。本発明の化合物はまた、Eph受容体に結合し、活性化する他の化合物の開発にも有用である。それは、本発明の化合物が、かかる開発を容易にするための構造と活性との関係に関する重要な情報を提供するものだからである。
【0167】
本化合物はまた、新しいEph受容体アゴニストをスクリーニングするためのアッセイにおける競合的結合体としても有用である。かかるアッセイの実施形態において、本明細書に記載する化合物は、修飾なしで使用してもよく、種々の方法、例えば、検出可能なシグナルを直接的または間接的に提供する部分と共有結合または非共有結合で連結するなどして標識することにより修飾してもよい。任意のこのようなアッセイにおいて、これに対する物質は、直接または間接的のいずれかで標識され得る。直接標識として考えられ得るものは、125Iなどの放射性標識、ペルオキシダーゼおよびアルカリホスファターゼなどの酵素(米国特許第3,645,090号)、ならびに蛍光強度の変化、波長シフトまたは蛍光分極をモニターできる蛍光標識(米国特許第3,940,475号)などの標識基が挙げられる。間接標識として考えられ得るものは、一成分をビオチン化した後、上記の標識基の1つを連結させたアビジンに結合させることが挙げられる。また、本化合物を固相支持体に結合させる場合、本化合物はスペーサーまたはリンカーを含み得る。
【0168】
核磁気共鳴(NMR)分光法は、巨大分子の構造を解明できることが知られており、標的分子へのリガンドの結合の静止時の特性および一時的な特性の両方を調べるための技法である(ペレッキア(Pellecchia)他2002 Nature Rev Drug Disc 1: 211)。NRM分光法は、標的分子へのリガンドの結合を測定するための有用なツールであり、タンパク質機能の予備知識を必要とせずに、高感度で相互作用を検知し、数量化することができるという利点を有する。さらにまた、NMR分光法は、標的およびリガンドの両方の構造の情報を提供し、後で、低結合性の候補物(hit)を高親和性リード化合物(leads)に最適化するのを補助し得る。
【0169】
均一に標識されている標的生体分子からの第1および第2核磁気共鳴相関スペクトルを得ることにより、標的生体分子へのリガンド化合物の結合を検出する方法は、米国特許第5,698,401号および同第5,804,390号に報告されている。第1スペクトルは、リガンドの不在下で標的物質に関して収集したデータから得、第2スペクトルは、1種類以上のリガンドの存在下で得る。2つのスペクトルの比較により、推定リガンドの混合物中のどの化合物が標的生体分子に結合するのかを調べることが可能になる。
【0170】
Eph受容体は、1個以上のアミノ酸残基の側鎖への1H、13C、15Nおよび19Fの組込みにより、選択的に標識してもよい。Eph受容体結合リガンドに結合したEph受容体の選択的に標識されたコンジュゲートを、第2分子に曝露してもよく、任意の分子相互作用をNMR分光法により調べることができる。例えば、分子相互作用を検知するため、および任意のコンジュゲートの解離定数を測定するために、2D、13C、1H−HMQC(heteronuclear multiple quantum coherence)および13C−エディット(edited)1H、1H−NOESY NMR実験を使用することができる。また、標的の三次元構造に基づいて、および標識された側鎖に対するリガンドの相対位置から、予想モデルを創製することができる。単一の選択的に標識された標的分子において数個の異なる標識側鎖を使用することにより、分解能およびモデルの予想性状が改善される。
【0171】
非ペプチド低分子は、臨床開発用ペプチドよりも好適であり得るため、Eph−エフリンコンジュゲートを分解する低分子の化学物質ライブラリーをスクリーニングするために
ハイスループットスクリーニングを使用し得る。このアッセイは、エフリン−アルカリホスファターゼ融合タンパク質とのコンジュゲートの状態にある固定化したEph受容体エクトドメインを使用する。結合したアルカリホスファターゼ活性を低下させる能力により、Eph−エフリン相互作用の低分子インヒビターを同定する。
【0172】
さらにまた、本明細書に記載するペプチドは、Eph受容体に選択的に結合する能力に基づいて、生細胞上、固定細胞上、生体液中、組織ホモジネート中、精製天然生体物質中などでEph受容体を選択的に検出するための試薬として使用することができる。例えば、本明細書に記載するペプチドを標識することにより、EphA2、EphA4、EphA5、EphA7、EphB2またはEphB4などの受容体を表面上に有する細胞を選択的に同定することができる。また、本ペプチドは、Eph受容体に結合する能力に基づいて、インサイチュ染色、FACS(蛍光標示式細胞分取)、ウエスタンブロッティング、ELISAなどに使用することができる。また、本ペプチドは、Eph受容体に選択的に結合できる能力に基づいて、受容体精製において、または細胞表面上(または浸透性にした細胞内部)に特定のEph受容体のみを発現する細胞を精製する際に使用することができる。
【0173】
本明細書に記載する化合物はまた、種々の医学的研究および診断的用途のための市販の試薬としても利用され得る。かかる用途としては、(1)種々の機能アッセイにおける候補Ephアゴニストの活性を定量するための較正標品としての用途、(2)Eph依存性細胞株の増殖および成長を維持するための使用、(3)共結晶化によるEph受容体リガンド結合の界面構造解析における使用、(4)Ephシグナル伝達/受容体活性化の機構を調べるための使用、(5)Eph受容体が好ましくは活性化されるか、またはかかる活性化が既知量のEphアゴニストに対して簡便に較正される研究および診断的適用など、ならびに(6)Eph受容体が好ましくは阻害されるか、またはかかる阻害が既知量のEphアンタゴニストに対して簡便に較正される研究および診断的適用など、が挙げられるが、これらに限定されない。
【実施例】
【0174】
以下の実施例は、EphA2、EphA4、EphA5、EphA7、EphB2およびEphB4受容体を標的化するペプチドを同定するに使用した方法を記載するものである。
【0175】
合成ペプチド
以下の実施例において、カルボキシ末端GSGSK(配列番号:30)リンカーを含むビオチン化ペプチドは、Fmoc化学反応を用いて合成し、HPLCにより精製し、MALDI−TOF質量分析法により確認した。本明細書に記載するペプチドおよび擬似ペプチドの調製に関するさらなる情報を、以下に提供する。
【0176】
プラスミド
エフリン−A5−APおよびエフリン−A6−APプラスミドは、記載されている(メンゼル(Menzel), P.他2001 Dev Biol 230: 74-88)。EphA2 AP(EphA2−AP)プラスミドを構築するため、ヒトEphA2の球状アミノ末端領域(aa1−219、GenBank受託番号M36395)を、PCRにより増幅し、APtag−2ベクター(チェン, H. J. & フラナガン(Flanagan), J. G. 1994 Cell 79: 157-168)にクローニングした。発現プラスミドを、Superfectトランスフェクション試薬(Qiagen)を用いて293T細胞に一過的にトランスフェクトした。AP融合タンパク質を含有する細胞培養上澄みを遠心分離し、細胞残屑を除き、20mM Hepesを添加して−20℃で冷凍保存した。
【0177】
上記のものと同様の方法を用いて、Eph−AP融合タンパク質をコードする核酸を含む類似の発現プラスミドを構築してもよく、コードされた融合タンパク質の発現がなされ得ることを理解されたい。
<実施例1>
【0178】
EphA2を標的化するペプチドの同定
本実施例では、EphA2に結合するペプチドを得るために使用される方法を説明する。ランダムな12量体ペプチドをディスプレイするM13ファージライブラリー(New England Biolabs、ベバリー、マサチューセッツ州)を、EphA2のパニングに使用した。ヒスチジンタグを有するマウスEphA2 Fc融合タンパク質(R & D Systems、ミネアポリス、ミネソタ州)を、ニッケル−ニトリロ三酢酸(Ni−NTA)コートELISAプレート内で、Tris緩衝生理食塩水(TBS)(150mM NaCl、50mM Tris−HCl、pH7.5)中1−10μg/mlの濃度で、一晩4℃でインキュベートした。ウェルを、0.5%ウシ血清アルブミン(BSA)含有TBSでブロックし、結合バッファー(TBS、1mM CaCl2、0.1% Tween 20)でリンスした。対照ウェルを、上記の方法を用いてウシ血清アルブミン(BSA)でコートした。
【0179】
EphA2パニングの第1回目の操作(round)では、100μlの結合バッファー中の1.7×1011プラーク形成単位(PFU)のファージライブラリーを、EphA2コートウェル内で室温で1時間インキュベートした。洗浄後も結合したままのファージを100μlの0.2Mグリセリン−HCl(pH2.2)または100μgのエフリン−A1 Fcで溶出した。溶出液全部を用いて、初期対数期のER2738宿主菌に感染させ、増幅させた。ファージを濃縮し、製造者の推奨にしたがって保存した。第2および第3回目の操作では、前回分からの2×1011PFUの増幅ファージプールを、EphA2
FcコートウェルおよびBSAコート対照ウェルに添加した。ファージは、洗浄バッファー中のTween濃度を0.5%とした以外は、第1回目について記載したと同様にパニングし、富化を評価するために溶出ファージを滴定した。
【0180】
図1Aおよび図1Bは、EphA2に選択的かつ高親和性で結合する2つのペプチドが同定された試験の結果を示す。図1Aは、低pH溶液で結合ファージを溶出すると、ファージ回収が最大になることを示し、一方、図1Bは、エフリン−A1で結合ファージを溶出すると、EphA2のリガンド結合部位と相互作用するペプチドの回収が向上することを示す。EphA2の選択に関する数回の操作の後、このスクリーニングにより、BSA結合ファージに対し、約17倍(低pH溶出)および115倍(エフリン−A1溶出)のEphA2結合ファージの富化が得られた。これに対し、エフリン−A1に関するパニングでは、ファージ富化はもたらされず、さらに続行はしなかった(結果は示さず)。
【0181】
低pHで溶出したプール由来の20個の別個のファージクローンのうち19個は、ネガティブコントロールとして使用したエフリン−A1 Fcと比べると、EphA2 Fcに特異的に結合することがわかった。19個のすべてのクローンは、同じペプチド:SWLAYPGAVSYR(SWLペプチド)(配列番号:1)をディスプレイすることがわかった。さらにまた、エフリン-A1で溶出したプール由来10個のファージクローンのうち9個が、EphA2に特異的に結合した。これらのクローンのうち7個がSWLペプチドをディスプレイし、2個がペプチドYSAYPDSVPMMS(YSAペプチド)(配列番号:2)をディスプレイした。
【0182】
また、上記の方法を用いて、適切な固定化受容体Fc融合体に対してファージライブラリーをパニングすることにより、EphA4、EphA5、EphA7、EphB2およびEphB4に結合するペプチドを見出した。かかるパニング実験の結果を以下の実施例
に記載する。
<実施例2>
【0183】
YSAおよびSWLペプチドはEphA2に特異的に結合する
この実施例では、種々のEphA受容体のFc融合タンパク質を捕捉するために、固定化したSWLおよびYSAペプチドを使用した。ビオチン化ペプチドを、ストレプトアビジンコートマイクロタイタープレート(Pierce、ロックフォード、イリノイ州)上に捕捉し、Eph受容体Fc融合タンパク質とともにインキュベートした。結合Eph受容体を、アルカリホスファターゼ(AP)に連結した抗ヒトFc抗体(Promega、マジソン、ウィスコンシン州)で検出した。あるいはまた、固定化したビオチン化ペプチドを、EphA2−APを含有する希釈細胞培養上澄みとともにインキュベートした。基質は、ホースラディッシュ・ペルオキシダーゼには2,2’−アジノ−ビス(3−エチルベンズチアゾリン−6−スルホン酸)、APにはp−ニトロフェニルホスフェートとした。405または450nmにおける吸光度を、ELISAプレートリーダーを用いて測定した。
【0184】
対応する合成SWLおよびYSAペプチドは、EphA2に特異的に結合することがわかった。特に、SWLおよびYSAは、ともにEphA2に結合したが、他のEphA受容体には結合せず(図2)、さらにこれらのペプチドは、EphBファミリーの受容体に結合する能力を欠くことが示された。
<実施例3>
【0185】
EphA2と結合するペプチド間の結合相互作用の特徴付け
この実施例では、BIAcoreシステムを用いて、EphA2と、実施例1で見出されたペプチドとの間の結合相互作用を特徴付けした。EphA2 Fcを、活性化バイオセンサーチップに共有結合により結合し、種々の濃度でのペプチドの結合の平衡を、BIAcore3000を用いて表面プラスモン共鳴における変化を測定することにより調べた。チップは、1M Na2CO3(pH10.5)で洗浄することにより再生した。表面プラスモン共鳴により得られた結合平衡データにより、YSAペプチドは、SWLペプチド(KD=678nM±23)よりも高親和性(KD=186nM±7)でEphA2に結合することが示された(図3Aおよび図3Bを比較のこと)。
<実施例4>
【0186】
YSAおよびSWLペプチドは、細胞表面上のEphA2を標的化する
この実施例は、YSAペプチドおよびSWLペプチドの両方が、培養中の細胞の表面上のEphA2に結合することを実証する。試験した細胞型の各々において、EphA2に対するYSAペプチドの親和性は、SWLペプチドよりもずっと大きい。
【0187】
等量の野生型対象ファージ(WT)ならびにYSAおよびSWLペプチドをディスプレイするファージを、強化緑色蛍光タンパク質(EGFP)(MDA EphA2−EGFP)(オガワ, K.他2000 Oncogene 19: 6043-6052)、非トランスフェクトMDA−MB−435細胞(MDA WT)またはヒト臍帯静脈内皮(HUVE)接着細胞と融合させたEphA2の細胞外ドメインおよび膜貫通型ドメインを過剰発現するMDA−MB−435ヒト乳癌細胞とともにインキュベートした。結合は、懸濁状態のMDA細胞、および接着HUVE細胞を用いて行なった。特に、細胞に結合したファージは、1×109PFUを60−90分間37℃で、1×106個のMDA−MB−435細胞の0.5ml懸濁液とともにインキュベートするか、または1×1010PFUを直接、24ウェル組織培養プレート中のコンフルエントな単層のHUVE細胞に添加するかのいずれかにより定量した。ファージを、1%BSAを含むダルベッコ改変イーグル培地(MDA−MB−435細胞)または1%BSA、10mM Hepesを含む内皮細胞基本培地−2(EBM−2)(Clonetics Products, BioWhittaker, Inc.、ウォーカースビル、メリーランド
州)(HUVE細胞)のいずれかで希釈した。
【0188】
YSAファージは、表面上にEphA2細胞外ドメインを過剰発現するMDA−MB−435ヒト乳癌細胞に対して、野生型ファージよりも、50倍高い結合性を示した。一方、SWLファージは、7倍高い結合性を示した(図4A)。内因性EphA2を低レベルでのみ発現する非トランスフェクトMDA−MB−435細胞の場合、YSAファージは野生型ファージよりも2.5倍高い結合性を示すが、SWLファージは、特異的結合性を示さない(図4B)。また、YSAファージは、中程度レベルのEphA2を発現するヒト臍帯静脈内皮(HUVE)細胞に対して、野生型ファージよりも12倍高い結合性を示す。これに対し、SWLファージは、これらの細胞に対して特異的に結合しない(図4C)。
<実施例5>
【0189】
YSAペプチドは、EphA2のチロシンリン酸化を促進し、下流シグナル伝達を活性化する
この実施例は、YSAペプチドが、EphA2のチロシンリン酸化を促進し、この受容体の活性化が寄与する下流シグナル伝達事象を活性化することを示す。
【0190】
YSAペプチドの存在下または不在下で、2μg/mlのエフリン−A1 FcまたはFcタンパク質で刺激するに先がけて、ヒト臍帯静脈内皮(HUVE)細胞を、10%FCSを含む微小血管内皮細胞培地−2(EGM−2 MV)(Clonetics)および無血清 (serum-starved)EGM−2で2時間成長させた。エフリン−A1 Fcを、0.2μg/mlの抗ヒトFc抗体と、氷上で30分間プレインキュベーションすることにより架橋した。刺激後、細胞を、改変RIPAバッファー(オガワ, K.他2000 Oncogene 19: 6043-6052)中で溶解した。細胞溶解物を5μgの抗EphA2抗体(Upstate、レークプラシッド、ニューヨーク州)で免疫沈降させ、SDSポリアクリルアミドゲル電気泳動により分離し、ペルオキシダーゼ結合抗ホスホチロシン抗体(Transduction Laboratories、サンディエゴ、カリフォルニア州)、または抗EphA2抗体と抗マウスIgGペルオキシダーゼ結合二次抗体(Amersham)を用いる免疫ブロッティングによりプローブした。
【0191】
別の方法では、非血清枯渇HUVE細胞を、YSAペプチドの存在下または不在下で、2μg/mlのエフリン−A1 FcまたはFcタンパク質で刺激した。細胞溶解物を、上記のようにしてEphA2を免疫沈降させるために使用し、または抗リン酸p44/p42MAPK抗体(Cell Signaling Technology、ベバリー、マサチューセッツ州)と抗マウスIgGペルオキシダーゼ結合抗体(Amersham)とを用いる免疫ブロッティングによりプローブし、かつ、抗ERK2抗体(Santa Cruz Biotechnology、サンタクルス、カリフォルニア州)と抗ウサギIgGペルオキシダーゼ結合二次抗体(Amersham)とを用いて再プローブした。Erk2(p42 MAPキナーゼ)が、存在する主なリン酸化形態である。EphA2を、免疫沈降(IP)させ、抗ホスホチロシン(PTyr)抗体または抗EphA2抗体を用いる免疫ブロッティングによりプローブ検索した。
【0192】
HUVE細胞におけるEphA2への結合に加え、YSAペプチドは、エフリンの不在下で受容体のチロシンリン酸化および下流シグナル伝達を刺激するが、エフリンの存在下において、EphA2リン酸化を低減させない(図5)。さらにまた、YSAペプチドは、以前に報告されたMAPキナーゼ活性化を抑制するEphA2シグナル伝達経路を活性化することがわかった(EphA2シグナル伝達経路によるMAPキナーゼ活性化の抑制の考察については、ミャオ他、2001 Nature Cell Biol 3: 527-530を参照のこと)。YSAペプチドによる刺激に応答したMAPキナーゼの抑制を図6Aに示し、これに対応するYSAペプチドに応答したEphA2のリン酸化を図Bに示す。したがって、YSAペプチドは、EphA2のアゴニストである。同様の結果がSWLペプチドについて得られ、
したがって、これもまたEphA2のアゴニストである。
<実施例6>
【0193】
YSAおよびSWLペプチドは、EphA2へのエフリン−Aの結合を阻害する
この実施例は、YSAおよびSWLペプチドがエフリン−A5およびエフリン−A6の結合を濃度依存的に阻害することを示す。Eph受容体コートプレートへのエフリンの結合を、エフリン−A5およびエフリン−A6のアルカリホスファターゼ(AP)融合タンパク質を用いて定量した(これらのAP融合体をコードするプラスミドは上記のものである)。エフリン−A5−APおよびエフリン−A6−APを含有する希釈細胞培養上澄みを、EphA2 FcまたはEphA4 Fcでコートしたマイクロタイターウェル内でペプチドとともに同時インキュベートした。洗浄後も結合したままのエフリンを、AP活性を測定することにより検出した。
【0194】
YSAおよびSWLペプチドは、濃度依存的に、固定化したEphA2へのA−エフリンの結合を阻害するが、EphA4への結合は阻害しない(図7Aおよび図7B)。これは、該ペプチドは、エフリンと相互作用するEphA2の表面に結合することを示す。ELISAアッセイにより、YSAおよびSWLペプチドが、EphA2の球状リガンド結合ドメインに結合することを確認した。球状リガンド結合ドメインは、その受容体のアミノ末端に2つの異なるエフリン結合領域を含むことが示されている(ラブラドル(Labrador), J. P.他1997 EMBO J 16: 3889-3897;ヒマネン(Himanen), J. P.他1998 Nature 396:
486-491)。
<実施例7>
【0195】
SWLおよびYSAは、EphA2上の同じかまたは重複する部位に結合する
この実施例では、YSAおよびSWLペプチドの結合部位を、ファージディスプレイペプチドとの競合実験を用いて特徴付けした。Eph受容体コートプレートに対するファージの結合を、ホースラディッシュペルオキシダーゼに連結した抗ファージ抗体(M13ファージ検出キット、Amersham Pharmacia Biotech.、ピスカタウェイ、ニュージャージー州)を用いて定量した。ファージクローンを用いるペプチド競合アッセイのため、EphA2 FcでコートしたNi−NTAマイクロタイターウェルを、結合バッファー中で1:600−1:9000に希釈したファージクローン(100μl/ウェル)とともに、室温で1時間インキュベートした。非結合ファージを洗い流し、競合ペプチドを1時間添加した。あるいはまた、ペプチドおよびファージを一緒に同時インキュベートした。
【0196】
SWLおよびYSAペプチドは、同じかまたは重複する部位に対する親和性を有することが示された。合成SWLペプチドは、固定されたEphA2に結合したYSAファージと競合し、逆にYSAペプチドは、SWLファージと競合する(図8)。したがって、これらの結果は、YSAおよびSWLペプチドが、EphA2上の同じかまたは重複する部位に結合することを示す。
<実施例8>
【0197】
エフリン配列に相当するペプチドは、Eph受容体に無差別に弱く結合する
この実施例は、SWLおよびYSAペプチドの両方との類似性を有する他の配列が、種々の異なるEph受容体に低親和性で結合することを示す。図9Aは、関連する配列を有するYSAおよびSWLペプチドが、高親和性Eph受容体結合インターフェース(A−エフリンのG−Hループ)(ヒマネン, J. P.他2001 Nature 414: 933-938を参照のこと)に関して或る程度の類似性を有することを示す。SWLおよびYSAペプチドの配列が逆の順序であるとみなせば、低親和性受容体結合インターフェース(A−A’β鎖)との類似性が示される(図9B)。エフリン−A3のG−Hループに相当する12量体合成ペプチド(A3ペプチド、図9A)およびエフリン−A5のA−A’β鎖に相当する12量
体合成ペプチド(A5ペプチド、図9B)は、YSAペプチドよりも弱いが、実際にEphA2に結合する(図10Aおよび図10B)。さらなる受容体結合残基(VADRYAVYWNSSNPR)(配列番号:19)を含むより長いA5ペプチドでさえ、同様の弱い結合性を示す。興味深いことに、A3およびA5ペプチドは、A−エフリンと同様に、すべてのEphA受容体に無差別に結合し、EphB4にも結合する(図11Aおよび図11B)(Eph-Nomenclature-Committee Unified nomenclature for Eph family receptors and their ligands, the ephrins 1997 Cell 90: 403-404)。
【0198】
実施例7で考察した競合方法を用いて、或る特定のエフリン関連配列の結合親和性を調べた。特に、A3ペプチドはファージ結合を阻害しないことがわかり(図14)、これは、おそらく高親和性エフリン結合部位に結合するA3ペプチドは、結合が弱すぎて競合できない可能性を示す。実際、A3ペプチドはまた、EphA2への結合に関してエフリン−A5とも競合しなかった。これに対し、A5ペプチドは、EphA2に対するファージとエフリンA5の両方の結合を増強した。総合すると、これらの結果は、EphA2の低親和性エフリン結合部位へのA5ペプチドの結合により、エフリンならびにYSAおよびSWLペプチドの高親和性部位への結合が増強されることを示す。
<実施例9>
【0199】
Eph受容体に結合するペプチドの同定
この実施例は、ファージディスプレイを用いて、種々の分類型のEph受容体に結合するさらなるペプチドを成功裡に見出すことができることを示す。実施例1に記載の方法を用いて、EphA4、EphA5、EphA7、EphB4およびEphB2に結合し得る他のペプチドを同定した。以下の表は、ペプチド名、受容体親和性、ペプチド配列、およびEphAまたはEphBファミリー受容体に結合することがこれまでわかっている各々の12量体ペプチドの配列番号を示す。
【0200】
【表1】
<実施例10>
【0201】
EphB4に対するDAL、IPWおよびSGHペプチドの結合
この実施例では、先の結合特性実験で使用したものと同様の方法を用いて、DAL、IPWまたはSGHペプチドをディスプレイするファージクローンがEphB4 Fcに結合したことを示した。図17は、これらのファージディスプレイペプチドの各々が、EphA2より6−10倍高い親和性でEphB4に結合することを示す。さらにまた、ファージディスプレイペプチドの各々は、試験したいずれのEphAファミリー受容体(Eph1−Eph7)よりも高い親和性でEphB4に結合した。
<実施例11>
【0202】
EphB4に対するDALペプチドの特異性
この実施例では、実施例2で記載した方法を用いて、DAL合成ペプチドがEphB4受容体に特異的に結合することを示した。EphBファミリー受容体を固定化したDALペプチドとインキュベートすると、DALペプチドは、他のEphBファミリー受容体と比べて、4倍多くEphB4に結合することが観察された。同様の特異性が、EphAファミリー受容体およびEphB4を、固定化したDALペプチドに対して試験した場合にも観察された。
<実施例12>
【0203】
EphA4に結合するペプチドの同定
この実施例では、ファージディスプレイを用いて、EphA4受容体に結合するペプチドを同定した。ランダムな12量体ペプチドをディスプレイするM13ファージライブラリーを、ヒトFc領域に融合させた固定化EphA4受容体細胞外ドメインに関してパニングした(図12A)。EphA4に結合したままのファージクローンを、低pH溶液で溶出し、増幅した。EphA4の選択に関して4回操作した後、BSAネガティブコントロールと比べて、約80倍のEphA4結合ファージの富化が検出された。
【0204】
個別に試験した38個のクローンのうち20個がEphA4 Fcに結合したが、エフリンA1 Fcネガティブコントロールには結合しなかった。EphA4結合クローンは、4つの異なるペプチド配列を示した(図12B−図12E)。これらのペプチドを、APY(APYCVYRGSWSC)(配列番号:20)、KYL(KYLPYWPVLSSL)(配列番号:21)、VTM(VTMEAINLAFPG)(配列番号:22)およびNHW(NHWLDTLFPMHM)(配列番号:46)とよぶ。これらのペプチドをディスプレイするファージクローンは、すべて、他のEphAまたはEphB4受容体と比べて優先的にEphA4に結合した(図12B−図12E)。しかしながら、NHWファージは、試験した他のEph受容体に対してもまた実質的な結合を示した。したがって、EphA4に対してより選択性であると思われる3つのペプチド(APY、KYLおよびVTM)を、その特異性および機能的特性を調べるために合成した。
<実施例13>
【0205】
3つの合成ペプチドは、EphA4に高親和性で優先的に結合する
この実施例では、ペプチド断片をディスプレイするファージクローンと同様、対応する合成ペプチドが、他のEphA受容体と比べて、EphA4に優先的に結合することがわかった。VTMペプチドは、EphA4に対して高度に選択的であるが、APYおよびKYLペプチドは、他のEphAおよびEphB受容体に対して或る程度の結合性を示した(図13)。固定化したペプチドに対する可溶性EphA4 Fc融合タンパク質の結合の平衡は、EphA4が、VTMペプチドよりもKYLおよびAPYペプチドに対してより良好に結合することを示す。ペプチドに対する二量体EphA4 Fcタンパク質の見かけ上の結合親和性は、低ナノモル濃度範囲においてであり、これは、EphA4−ペプ
チドの相互作用が高親和性であることを示す。
<実施例14>
【0206】
APY、KYLおよびVTMペプチドは、EphA4への結合に対して互いに競合する
この実施例では、アミノ酸配列において密接に関連しないAPYペプチドおよびKYLペプチドが、モチーフΦXXΦ(式中、Φは、芳香族アミノ酸を示し、Xは任意のアミノ酸である)を共有することが示された(図19)。VTMペプチドの配列は、APYペプチドおよびKYLペプチドの両方と異なっており、ΦXXΦモチーフを含まない。EphA4の多ドメイン細胞外領域全体をパニングに用いたと考えると、ペプチドは、受容体の異なる領域に結合し得ることが考えられた。しかしながら、3種類のペプチドを用いて、固定化したEphA4へのVTMペプチドディスプレイファージの結合に関して競合させた実験では、3種類すべてのペプチドが、VTMファージクローンおよびAPYファージクローンの結合を拮抗阻害し得ることが示された(図14)。したがって、3種類すべてのペプチドは、EphA4上の同じか、または一部重複する部位に結合するようである。<実施例15>
【0207】
APY、KYLおよびVTMペプチドは、EphA4へのエフリンの結合を拮抗阻害し、受容体のエフリン誘導性活性化を阻害する
この実施例では、APYおよびKYLペプチドのΦXXΦモチーフは、エフリン−Aリガンドの高親和性受容体結合部位、および別のEphA受容体へのエフリンの結合を阻害する先に単離した2つのペプチドにも存在することがわかった(上記および図19参照)。これは、これらのEphA4結合ペプチドもエフリンの結合を阻害し得ることを示した。配列が異なるにもかかわらず、3種類すべてのEphA4結合ペプチドは、アルカリホスファターゼ標識を有するエフリン−A5(エフリン−A5 AP)のEphA4への結合を、投与量依存的に阻害した(図15)。KYLペプチドが最も有効であり、1μMの濃度でほぼ完全にエフリンの結合を阻害した。
【0208】
同様のレベルの阻害を達成するには、APYペプチドは約10μM、VTMペプチドは約100μM必要であった。EphA4結合曲線と同様、これらの阻害実験は、KYLペプチドが最も高い結合親和性を有し、VTMペプチドが最も低いことを示す。さらにまた、異なるエフリン濃度でのさらなるペプチド阻害曲線により、これらのペプチドが受容体のエフリンの結合を競合的に阻害することを確認した。したがって、これらのペプチドは、EphA4のエフリン結合部位に結合し、マイクロモル濃度で使用しても、この受容体へのエフリンの結合を阻害し得る。これらのペプチドはまた、他のEphA受容体と比べて、EphA4へのエフリン−A5 APの結合を、優先的に(APYおよびKYL)または選択的に(VTM)にブロックすることがわかった(図16)。しかしながら、図16で使用したペプチド濃度(50μM)では、EphA4に対する選択性は、3種類すべてのペプチドで十分であった。
【0209】
EphA4結合ペプチドが、エフリンによるEphA4活性化を阻害するか、あるいはまたEphA4を活性化するか否かを調べるため、これらのペプチドを、エフリン−A3
Fcとともに海馬薄片に投与した(ムライ, K. K.他2003 Nat Neurosci 6: 153-160)。3種類すべてのペプチドは、マイクロモル濃度で、内因性EphA4のエフリン−A3誘導性チロシンリン酸化を拮抗阻害することがわかった(図17)。また、これらのペプチドは、自身の受容体を活性化する本来の能力は示さなかった。KYLペプチドは、APYおよびVTMペプチドよりも低濃度でEphA4チロシンリン酸化をブロックし、その相対的に高い結合親和性と一致する。
<実施例16>
【0210】
KYLペプチドは、神経冠細胞の分節間遊走(segmental migration)に必要なエフリン/
EphA4相互作用を撹乱する
この実施例では、EphA4結合ペプチドが内生エフリン−EphA4生理学的機能を阻害し得るか否かを検討するため、神経冠細胞遊走アッセイ(マックレナン(McLennan), R. & クルル(Krull), C. E. 2002 Gene Expr 10: 295-305)を利用した。遊走前(premigratory)神経冠細胞を含むひな鳥神経幹外植片を、10μg/mlの対照Fc、10μg/mlのEphA4−Fc(陽性対照)、13μM KYLペプチドまたは13μMの対照ペプチドを加えた培地内で24時間成長させた。神経冠細胞の遊走は、神経冠細胞についてHNK−1を染色し、エフリン−B1を染色して尾側椎板(caudal sclerotome)を標識することにより調べた。KYLペプチドは、その見かけ上の高親和性により選択した。神経冠細胞は、対照Fcで処理した外植片内の吻側椎板内に適正に遊走した。しかしながら、EphA4−Fc処理では、既報(マックレナン, R. & クルル, C. E. 2002 Gene Expr 10: 295-305)のように、神経冠細胞の尾側半椎板への異常な遊走が引き起こされた。KYLペプチドでの処理でも同様に、神経冠細胞の尾側半椎板への異所性遊走がもたらされ、一方、EphA4に結合しない対照ペプチドは、検出可能な作用はなかった。したがって、KYLペプチドは、適正な神経冠細胞遊走を可能にする内生EphA4/エフリンコンジュゲートの生物学的機能に影響を及ぼす可能性がある。
<実施例17>
【0211】
成体脳で発現する他の2つのEphA受容体:EphA5およびEphA7に結合するペプチドの同定
この実施例では、さらなるファージパニング実験を行ない、EphA5およびEphA7に結合するペプチドを同定した(図18A)。EphA5およびEphA7は、EphA4と密接に関連するが、発達中の神経系および成体神経系において発現する点で異なる2つのEphA受容体である(エリス, J.他1995 Mechanisms of Development 52: 319-341;モリ, T.他1995 Brain Res Mol Brain Res 34: 154-160;オリビエリ, G. & ミーシャー, G. C.他1999 J Histochem Cytochem 47: 855-861;チャン, J. H.他1997 Brain Res Mol Brain Res 47: 202-214)。各受容体に結合する3つの異なるペプチド配列を同定した。EphA5に関して単離したペプチドのうち2つ((SLRDTYMRAKVL、配列番号:27)および(WDCNGPYCHWLG、配列番号:28))は、この受容体に対して選択的結合性を示したが、もう1つのペプチド(WTFPVLWDDKHP、配列番号:29)はEphA6にも結合する。EphA7結合ペプチドのうち、1つ目(WASHAPYWPHPP、配列番号:61)は、EphA3にも結合し、2つ目(SVSVGMKPSPRP、配列番号:25)はEphA3およびEphA6にも結合し、3つ目(KHLPFYPHPTSP、配列番号:62)はEphA3、EphA5およびEphA6にも結合する。
【0212】
【表2】
【0213】
【表3】
【0214】
【表4】
【0215】
受容体チロシンキナーゼのEphAサブクラスの異なるメンバーを標的化する一群の新規ペプチドを同定した。これは、エフリンリガンドとは対照的であり、同じサブクラス内のほとんどのEph受容体に無差別に結合する(フラナガン & バンデルヘーゲン 1998 A
nn Rev Neurosci 21: 309-345)。3種類のEphA4結合ペプチドをさらに特徴付けすると、EphA4へのエフリンの結合を拮抗阻害し、エフリン誘導性EphA4の活性化および生物学的機能をブロックすることがわかった。これらのペプチドは、エフリン−Eph受容体相互作用に干渉するため、それぞれ、EphA4およびそのリガンドの下流に伝達される順方向および逆方向の両方のシグナルを阻害すると考えられる。
【0216】
最近解明されたエフリン/Eph受容体コンジュゲートの結晶構造は、エフリンの受容体結合領域が、αヘリックスG−H間に延長された15アミノ酸ループを含むことを示す(ヒマネン, J. P.他2001 Nature 414: 933-938)。このG−Hループの配列アラインメントは、いくつかのアミノ酸がすべてのエフリン間で保存されているが、A−エフリンとB−エフリンとで異なるものもあり、おそらく、EphAまたはEphB受容体に対する特異性に寄与していることを示す(図19)。興味深いことに、EphA4結合ペプチドのうちの2つ(APYおよびKYLペプチド)およびEphA5結合ペプチドのうちの1つ(WDCペプチド)が、2つの非保存アミノ酸により分断された2つの芳香族残基からなるA−エフリンのG−Hループ内に見られるモチーフ(ΦXXΦ)を含んでいた(図19)。
【0217】
このモチーフは、EphA2受容体に結合したペプチドにも存在することがわかった。相当数の同定されたEphB受容体結合ペプチドは、ΦXXXΦまたはΦXXXXΦモチーフを有した。異なるEph受容体に結合したペプチド間でのΦXnΦ(n=2−4)モチーフの保存は、2つの芳香族残基が、これらの受容体との相互作用に重要であり、Eph受容体のエフリン結合ポケットの保存領域内に入り込み得る(EphA受容体ではΦXXΦが有利であり、EphB受容体ではΦXXXΦまたはΦXXXXΦが有利である)ことを示す。芳香族残基に隣接する残基が、特定のEphAまたはEphB受容体への結合に対する特異性をもたらすと考えられる。
【0218】
他のアミノ酸残基もまた、受容体への結合および特異性に重要であると考えられる。APY EphA4結合ペプチドおよびWDC EphA5結合ペプチドは、これらのペプチドが分子内ジスルフィド結合を形成するのを可能にする2つのシステイン残基を含む。立体構造上束縛のある環状構造は、ペプチドの高結合親和性および受容体結合選択性の決定において重要であり得る。また、ペプチドおよびエフリンG−Hループ内に存在するプロリンは、Eph受容体結合において或る役割を果たし得る。
【0219】
EphA4結合ペプチドのうち1つ(VTMペプチド)は、他の2つのEphA4結合ペプチドとの明白な配列類似性を示さなかった。しかしながら、EphA4は、A−エフリンに加えてエフリン−B2およびエフリン−B3に結合するため、例外的なリガンド特異性を有する。したがって、一部のEphA4結合ペプチドは、B−エフリンのG−Hループに特徴的なアミノ酸を含み得る。実際、VTMペプチドは、エフリン−B2およびエフリン−B3のG−Hループに見られる(が、EphA4に結合しないエフリン−B1には見られず)配列、アスパラギン−ロイシン(NL)を含む(図19)。したがって、NLモチーフは、B−エフリンとEphA4との接触の重要な部位であると考えられる。あるいはまた、VTMペプチド内のフェニルアラニン(F)は、A−エフリンおよびB−エフリンの両方で保存されているΦXnΦモチーフの第2芳香族残基に相当し得る。それにもかかわらず、VTMペプチドは、エフリンの結合を競合的に阻害し、これは、該ペプチドがEphA4のリガンド結合ポケットに結合することを示す。
<実施例18>
【0220】
Eph受容体結合因子を同定するためのハイスループットスクリーニング方法
ハイスループットスクリーニングアッセイは、Eph受容体−エフリンコンジュゲートを分解する低分子の化学物質ライブラリーをスクリーニングするために使用されている。
このアッセイでは、エフリン−アルカリホスファターゼ融合タンパク質とのコンジュゲートの状態の固定化したEph受容体エクトドメインを使用する。結合したアルカリホスファターゼ活性を低下させる能力を利用して、Eph−エフリン相互作用の低分子インヒビターを同定する。
【0221】
個々のEph受容体を標的化し、生物活性特性を有する低分子は、受容体の機能を解明するため、およびその特定の受容体が富化した組織の生理機能を選択的に撹乱するための両方に有用である。例えば、ペプチドの1つは、内因的に発現しているEphA4とそのエフリンリガンドとの相互作用を阻害し、生物学的影響をもたらした。EphA4シグナル伝達を選択的に阻害すると、この特定の受容体が高度に発現する海馬および大脳皮質において、ニューロンの形状発達、再構築または再生を方向付ける反発的(repulsive)リガンド−受容体相互作用を低減し得る。実際、EphA4シグナル伝達は、海馬における樹状突起棘の形態および組織化を維持することがわかっている(ムライ他2003 Nat Neurosci 6: 153-160)。これは、おそらく、エフリン−A3陽性星状膠細胞との一過的な反発的接触により起こるだろう。興味深いことに、ペプチドは、海馬薄片においてエフリン−A3 Fcによる内生EphA4の活性化をブロックする。したがって、これらは、突起棘の拡張を制限するインビボリガンド−受容体相互作用に干渉し得る。例えば、脳損傷は神経膠症を誘導し、これは、神経膠エフリン−A3によるEphA4の活性化により起こり得る樹状突起棘の大きさの縮小と相関している(トンプソン(Thompson)、2003 Nat Neurosci 6:103-104)。EphA4結合ペプチドは、阻害性シグナルの一部を打ち消し、機能回路の再生を促進すると考えられる。他方において、EphA4を活性化できる人工的ペプチドは、精神遅滞を伴う患者における伸長樹状突起棘の正常形状を回復させると予想される。
【0222】
反発因子および成長阻害因子の存在により、中枢神経系の再生能力が制限されると考えられている。これは、外傷性脳損傷またはニューロン変性後、非許容シグナルをブロックし、構造的可塑性を促進することが所望される場合、特に重要であろう。興味深いことに、EphA4結合ペプチドは、神経幹細胞が、失われたニューロンの置き換えを生涯にわたって行なう歯状回において新たに生成したニューロンのより高速な成熟および統合(integration)を促進し得る。
【0223】
また、同定したペプチドのうちいくつかは、脊椎内の損傷軸索の再生を補助すると考えられる。数個のエフリンならびにEphA4およびEphA7を含むEph受容体は、脊椎損傷または求心路遮断後にアップレギュレートされる(ミランダ(Miranda)他1999 Exp Neurol 156: 218-222;ウィルソン(Willson)他2002 Cell Transplantation 11: 229-239)。また、EphA4は、発達中の運動軸索の誘導のため(エバーハート(Eberhart)他2002 Dev Biol 247:89-101)および皮質脊髄線維の正確な軌道(trajectory)を確立するため(ドットーリ(Dottori)他1998 PNAS USA 95: 13248-13253;クランダー(Kullander)他2001 Nat Rev Mol Cell Biol 3: 475-486)に重要である。これらの軸索において、EphA4は、反発的誘導事象を媒介すると考えられている。EphA4媒介性反発は、中枢神経系における軸索の再成長を制限することが可能で、かかる相互作用の阻害は、再生を促進し得る。
【0224】
また、EphA受容体結合ペプチドは、限られた特有の再生能力を示す中枢神経系の障害領域を含む、特定の組織に試薬を送り届ける(escort)のを補助すると考えられる。これらのペプチドに医薬剤を連結させることにより、変性しつつあるニューロンに栄養を補助すると同時に成長に対する阻害的拘束を減弱する分子の標的化送達が可能になる。EphA4結合ペプチドは、神経疾患を有する患者の解剖学的および認知機能を改善するための海馬および皮質などの脳組織への選択的送達に有用である。また、EphA5結合ペプチドは、黒質および小脳に試薬を送達するため(ユー(Yue)他1999 J Neurosci 19: 2090-21
01)およびこれらの脳領域に影響する神経疾患を治療するために有用である。
【0225】
神経系以外では、血小板に対するEphA4のペプチド阻害は、血液凝固プロセスを変更するために適用できると考えられる(プレボー(Prevost)他2002 PNAS USA 99: 9219-9224)。さらにまた、これらのペプチドは、発達研究のため、インビボまたはインビトロでEphA4機能を選択的に撹乱させるのに有用である。これには、本明細書に記載した神経冠細胞の移動が含まれる。
【0226】
EphA受容体結合ペプチドは、機能的に万能な試薬であり、種々の疾患および病状を治療するための重要なツールである。これらは、細胞遊走、軸索誘導およびシナプス可塑性におけるEphA受容体の役割の解析を目的とした発達研究の面において、ならびに中枢神経系への障害を伴い得る異常EphA受容体シグナル伝達の調節のためにも有用である。
【0227】
本発明を、明瞭にしかつ理解する目的で、或る程度詳細に記載したが、当業者であれば、本発明の真の意図から逸脱することなく、形態および詳細において種々の変更がなされ得ることが理解されよう。
【図面の簡単な説明】
【0228】
【図1−1】図1Aは、固定化したEphA2 Fcまたは固定化したウシ血清アルブミン(BSA)のいずれかに対する連続パニング操作(R1−R3)で得られた、ランダム12量体ファージディスプレイライブラリー由来のファージの力価(pfu/μl)を示す棒グラフである。結合したファージを低pHで溶出した。得られたEphA2結合クローンのペプチド配列を示す。
【図1−2】図1Bは、固定化したEphA2 Fcまたは固定化したウシ血清アルブミン(BSA)のいずれかに対する連続パニング操作(R1−R2)で得られた、ランダム12量体ファージディスプレイライブラリー由来のファージの力価(pfu/μl)を示す棒グラフである。結合したファージをエフリン−A1で溶出した。得られたEphA2結合クローンのペプチド配列を示す。
【図2】固定化したSWLおよびYSAペプチド間での、EphA2 Fc対他のEphA受容体Fc融合タンパク質の結合特異性を示す棒グラフである。
【図3】種々の濃度のYSAペプチド(図3A)またはSWLペプチド(図3B)について測定したときの、EphA2 FcでコートしたBIACOREバイオセンサーチップの表面上の表面プラスモン共鳴単位における変化を示す線グラフである。解離定数は、非線形回帰により求めた。
【図4】強化緑色蛍光タンパク質(MDA EphA2−EGFP)(図4A)、非トランスフェクトMDA−MB−435細胞(MDA WT)(図4B)、またはヒト臍帯静脈内皮(HUVE)接着細胞(図4C)と融合させたEphA2の細胞外ドメインおよび膜貫通型ドメインを過剰発現するMDA−MB−435ヒト乳癌細胞に結合した野生型M13ファージ(WT)、YSAペプチドをディスプレイするファージ(YSA)、およびSWLペプチドをディスプレイするファージ(SWL)の数を比較する棒グラフである。
【図5】Fc(レーン1−3)、エフリン−A1−Fc抗Fc架橋コンジュゲート(レーン4−6)およびエフリン−A1−Fc(レーン7−9)での処理前に、0、10または50μMいずれかのYSAペプチドで処理したHUVE細胞におけるEphA2のリン酸化を示すイムノブロットである。EphA2は、免疫沈降(IP)させ、抗ホスホチロシン(PTyr)または抗EphA2抗体を用いるイムノブロッティング(IB)によりプローブした。
【図6】Fc処理前にYSAペプチドで処理したHUVE細胞におけるMAPキナーゼ(MAPK)のリン酸化(図6A)(レーン3および4)および対応するEphA2のリン酸化(図6B)(レーン3および4)の阻害を示すイムノブロットである。IBは免疫沈降を示し、IPはイムノブロット前の免疫沈降を示す。
【図7】SWLおよびYSAペプチドが、固定化したEphA2へのエフリン−A5(図7A)およびエフリン−A6(図7B)の結合を濃度依存的に拮抗阻害するが、EphA4ではそうでないことを示すグラフである。
【図8】SWLおよびYSAペプチドが、EphA2上の同じかまたは重複する部位に結合することを示すグラフである。
【図9】図9Aでは、SWLおよびYSAペプチド配列のアラインメント、A−エフリンのG−Hループ、およびこのアラインメントに基づいて合成した「A3ペプチド」を示す。同一アミノ酸を濃い灰色で示し、類似した特徴を有するアミノ酸を薄い灰色で示す。図9Bでは、SWLおよびYSAペプチドの逆配列のアラインメント、エフリンのA−A’β鎖、およびこのアラインメントに基づいて合成した「A5ペプチド」を示す。同一アミノ酸を濃い灰色で示し、類似した特徴を有するアミノ酸を薄い灰色で示す。
【図10】ビオチン化したA3、A5と、EphA2 Fcを捕捉するために使用したストレプトアビジンコートウェル上に固定化したYSAペプチドとの間の結合の結果を示す棒グラフである。5分(図10A)および20分(図10B)におけるODの目盛りの値を示す。
【図11】ストレプトアビジンコートウェル上に固定化した、ビオチン化したA3(図11A)、およびA5(図11B)ペプチドと、EphA受容体のFc融合タンパク質との間の結合の結果を示す棒グラフである。すべてのパネルにおけるエラーバーは、2回の測定値の標準偏差を示す。
【図12−1】EphA4受容体を標的化するファージクローンの同定および結合選択性を示す棒グラフである。ランダムな12量体ペプチドをディスプレイするM13ファージがEphA4 FcまたはBSA(対照)に結合したことを示す(図12A)。
【図12−2】EphA4受容体を標的化するファージクローンの同定および結合選択性を示す棒グラフである。APY(図12B)、KYL(図12C)、VTM(図12D)、およびNHW(図12E)で始まるペプチドをディスプレイするファージクローンがEphA4に優先的に結合したことを示す。
【図13−1】合成EphA4結合ペプチドの結合選択性を示す棒グラフである。ストレプトアビジンコートプレート上に固定化した、ビオチン化したAPY(図13A)、KYL(図13B)、およびVTM(図13C)ペプチドが、EphA受容体Fcタンパク質を捕捉したことを示す棒グラフである。
【図13−2】合成EphA4結合ペプチドの結合選択性を示す棒グラフである。ストレプトアビジンコートプレート上に固定化した、ビオチン化したAPY(図13D)、KYL(図13E)、およびVTM(図13F)ペプチドが、EphB受容体に対して低結合能力を示したことを示す棒グラフである。
【図13−3】合成EphA4結合ペプチドの結合選択性を示す棒グラフである。固定化したペプチドに対する二量体EphA4 Fcの相対的結合親和力を示す棒グラフである(図13G、13H、13I)。
【図14】APY、KYLおよびVTMペプチドが、EphA4上の同じか、または一部重複する部位に結合することを示すグラフである。
【図15−1】図15Aは、APYペプチドが、EphA4へのエフリン−A5結合を濃度依存的に(対照ペプチド、白四角形)拮抗阻害することを示すグラフである。
【図15−2】図15Bは、KYLペプチドが、EphA4へのエフリン−A5結合を濃度依存的に(対照ペプチド、白四角形)拮抗阻害することを示すグラフである。
【図15−3】図15Cは、VTMペプチドが、EphA4へのエフリン−A5結合を濃度依存的に(対照ペプチド、白四角形)拮抗阻害することを示すグラフである。
【図16】対照(図16A)、APY(図16B)、KYL(図16C)、およびVTM(図16D)ペプチドが、その他のEphA受容体と比べ、EphA4へのエフリン−A5結合を優先的に阻害することを示す棒グラフである。
【図17】APY(図17A)、KYL(図17B)、およびVTM(図17C)ペプチドが、マウス海馬薄片において、内因性EphA4のエフリン−A3誘導型活性化を拮抗阻害することを示すグラフである。
【図18−1】EphA5結合ペプチドおよびEphA7結合ペプチドの同定および結合選択性を示す棒グラフである。ランダムな12量体ペプチドをディスプレイするM13ファージがEphA5 Fc、EphA7 Fc、またはBSA(対照)に結合したことを示す(図18A、18B)。
【図18−2】EphA5結合ペプチドおよびEphA7結合ペプチドの同定および結合選択性を示す棒グラフである。EphA5に関するパニングにより単離されたファージクローンは、EphA5に選択的に結合するペプチドSLR(図18C)およびWDC(図18D)をディスプレイするが、ファージクローンの1つWTF(図18E)は、EphA5およびEphA6の両方に結合するペプチドをディスプレイすることを示す。
【図18−3】EphA5結合ペプチドおよびEphA7結合ペプチドの同定および結合選択性を示す棒グラフである。EphA7に関するパニングにより単離された3つのファージクローンは、EphA7を含むEphA受容体の種々のサブセットに結合するペプチドをディスプレイする(図18F、18G)。
【図19−1】Eph受容体結合ペプチドの、A−エフリンおよびB−エフリンのG−Hループに対するアラインメントである(図19A)。
【図19−2】Eph受容体結合ペプチドの、A−エフリンおよびB−エフリンのG−Hループに対するアラインメントである(図19B)。
【配列表】
【特許請求の範囲】
【請求項1】
Eph受容体ファミリーメンバーに選択的に結合するEph受容体結合リガンド。
【請求項2】
単離されたペプチドまたはペプチド擬似化合物を含む、請求項1に記載のEph受容体結合リガンド。
【請求項3】
100残基未満の長さを有する、請求項2に記載の単離されたペプチド。
【請求項4】
50残基未満の長さを有する、請求項2に記載の単離されたペプチド。
【請求項5】
20残基未満の長さを有する、請求項2に記載の単離されたペプチド。
【請求項6】
芳香族アミノ酸Φ及び任意のアミノ酸Xから成るモチーフΦXXΦを含有する、請求項2に記載の単離されたペプチド。
【請求項7】
前記Eph受容体が、EphA2、EphA4、EphB4、EphA5、EphA7およびEphB2からなる群より選択される、請求項2に記載の単離されたペプチド。
【請求項8】
細胞を、Eph受容体ファミリーメンバーに選択的に結合する化合物を含むEph受容体結合リガンドの有効量と接触させることを含む、細胞におけるEph受容体活性の調節方法。
【請求項9】
前記Eph受容体結合リガンドが、単離されたペプチドまたはペプチド擬似化合物を含む、請求項8に記載の方法。
【請求項10】
前記Eph受容体ファミリーメンバーが、EphA2、EphA4、EphB4、EphA5、EphA7およびEphB2からなる群より選択される、請求項8に記載の方法。
【請求項11】
前記細胞がインビボである、請求項8に記載の方法。
【請求項12】
前記細胞がインビトロである、請求項8に記載の方法。
【請求項13】
哺乳動物に、該哺乳動物においてプログラム細胞死を活性化するのに有効な量のEph受容体アゴニストを投与することを含む方法であって、該アゴニストは、Eph受容体ファミリーメンバーに選択的に結合する化合物を含む、哺乳動物におけるプログラム細胞死の活性化方法。
【請求項14】
前記Eph受容体アゴニストが、単離されたペプチドまたはペプチド擬似化合物を含む、請求項13に記載の方法。
【請求項15】
前記哺乳動物がヒトである、請求項13に記載の方法。
【請求項16】
前記細胞が腫瘍性である、請求項13に記載の方法。
【請求項17】
前記アゴニストがEphA2受容体と結合する、請求項13に記載の方法。
【請求項18】
前記アゴニストが、EphA2受容体へのエフリン-Aの結合を阻害する、請求項13に記載の方法。
【請求項19】
前記アゴニストが、EphA2受容体のリン酸化を促進する、請求項13に記載の方法。
【請求項20】
哺乳動物に、該哺乳動物において神経再生を活性化するのに有効な量のEph受容体アゴニストを投与することを含み、該アゴニストは、Eph受容体ファミリーメンバーに選択的に結合する化合物を含む、神経再生の促進もしくはニューロン変性の改善、またはその両方を行なう方法。
【請求項21】
前記変性が、外傷性損傷、精神遅滞または神経変性疾患によるものである、請求項20に記載の方法。
【請求項22】
前記Eph受容体アゴニストが、単離されたペプチドまたはペプチド擬似化合物を含む、請求項20に記載の方法。
【請求項23】
細胞に治療剤を接触させることを含む、Eph受容体を発現する細胞への治療剤の送達方法であって、該治療剤は、Eph受容体ファミリーメンバーに選択的に結合する化合物を含むEph受容体結合リガンドと連結されていることを特徴とする方法。
【請求項24】
前記Eph受容体結合リガンドが、単離されたペプチドまたはペプチド擬似化合物を含む、請求項23に記載の方法。
【請求項25】
治療剤と連結されたEph受容体結合リガンドを含有するコンジュゲートであって、前記Eph受容体結合リガンドは、Eph受容体ファミリーメンバーに選択的に結合する、コンジュゲート。
【請求項26】
前記Eph受容体結合リガンドが、EphA2受容体に選択的に結合する、請求項25に記載のコンジュゲート。
【請求項27】
前記Eph受容体結合リガンドが、EphB4受容体に選択的に結合する、請求項25に記載のコンジュゲート。
【請求項28】
前記Eph受容体結合リガンドが、EphB2受容体に選択的に結合する、請求項25に記載のコンジュゲート。
【請求項29】
前記Eph受容体結合リガンドが、EphA4受容体に選択的に結合する、請求項25に記載のコンジュゲート。
【請求項30】
前記Eph受容体結合リガンドが、EphA5受容体に選択的に結合する、請求項25に記載のコンジュゲート。
【請求項31】
前記Eph受容体結合リガンドが、EphA7受容体に選択的に結合する、請求項25に記載のコンジュゲート。
【請求項32】
イメージング剤と連結されたEph受容体結合化合物を含有するコンジュゲートであって、該Eph受容体結合リガンドは、Eph受容体ファミリーメンバーに選択的に結合する、コンジュゲート。
【請求項33】
前記Eph受容体結合リガンドが、EphA2受容体に選択的に結合する、請求項32に記載のコンジュゲート。
【請求項34】
前記Eph受容体結合リガンドが、EphB4受容体に選択的に結合する、請求項32に記載のコンジュゲート。
【請求項35】
前記Eph受容体結合リガンドが、EphA4受容体に選択的に結合する、請求項32に記載のコンジュゲート。
【請求項36】
前記Eph受容体結合リガンドが、EphA5受容体に選択的に結合する、請求項32に記載のコンジュゲート。
【請求項37】
前記Eph受容体結合リガンドが、EphA7受容体に選択的に結合する、請求項32に記載のコンジュゲート。
【請求項38】
前記Eph受容体結合リガンドが、EphB2受容体に選択的に結合する、請求項32に記載のコンジュゲート。
【請求項39】
ランダムなペプチドをコードするファージディスプレイライブラリーを作製すること、
固定化したEph受容体に対して、前記ライブラリーをパニングすること、および
前記Eph受容体に結合するファージクローンを同定すること
を含む、Eph受容体結合リガンドの同定方法。
【請求項40】
いくつかの非保存アミノ酸により分断された2つの芳香族残基を含有するアミノ酸配列を有する、単離されたEph受容体結合リガンド。
【請求項41】
前記非保存アミノ酸の数が1−4個である、請求項40に記載の単離されたEph受容体結合化合物。
【請求項42】
Eph受容体に結合したEph受容体結合因子を検出すること、
前記Eph受容体を候補化合物と接触させること、および
前記Eph受容体からの前記Eph受容体結合因子の解離を検出すること
を含み、これにより、前記候補化合物がEph受容体結合リガンドであると同定する、Eph受容体結合リガンドの同定方法。
【請求項43】
前記Eph受容体が、EphA2、EphA4、EphA5、EphA7、EphB2およびEphB4からなる群より選択される、請求項42に記載の方法。
【請求項44】
前記Eph受容体結合リガンドが、EphA2受容体に選択的に結合する、請求項42に記載の方法。
【請求項45】
前記Eph受容体結合リガンドが、EphA4受容体に選択的に結合する、請求項42に記載の方法。
【請求項46】
前記Eph受容体結合リガンドが、EphA5受容体に選択的に結合する、請求項42に記載の同定方法。
【請求項47】
前記Eph受容体結合リガンドが、EphA7受容体に選択的に結合する、請求項42に記載の同定方法。
【請求項48】
前記Eph受容体結合リガンドが、EphB2受容体に選択的に結合する、請求項42に記載の同定方法。
【請求項49】
前記Eph受容体結合リガンドが、EphB4受容体に選択的に結合する、請求項42に記載の同定方法。
【請求項50】
前記Eph受容体結合リガンドが、検出可能な標識で標識されている、請求項42に記載の同定方法。
【請求項51】
前記Eph受容体は、そのトリプトファン残基が選択的に標識されている、請求項42に記載の同定方法。
【請求項52】
前記トリプトファン側鎖が、13Cおよび15Nおよび19Fおよび1Hで選択的に標識されている、請求項51に記載の方法。
【請求項53】
核磁気共鳴を用いて行なう、請求項52に記載の方法。
【請求項54】
前記Eph受容体結合リガンドがエフリンである、請求項42に記載の方法。
【請求項55】
前記エフリンが、エフリン−アルカリホスファターゼ融合タンパク質である、請求項54に記載の方法。
【請求項56】
外傷性損傷、精神遅滞および神経変性疾患の治療のための医薬の調製における、神経再生の促進およびニューロン変性の改善に有効であるEph受容体のアゴニストの使用。
【請求項57】
腫瘍性疾患の治療のための医薬の調製における、Eph受容体活性を調節し、プログラム細胞死を活性化するのに有効な化合物の使用。
【請求項58】
腫瘍性疾患、神経変性疾患、精神遅滞および外傷性損傷の治療のための医薬の調製における、Eph受容体ファミリーメンバーに選択的に結合する化合物の使用。
【請求項1】
Eph受容体ファミリーメンバーに選択的に結合するEph受容体結合リガンド。
【請求項2】
単離されたペプチドまたはペプチド擬似化合物を含む、請求項1に記載のEph受容体結合リガンド。
【請求項3】
100残基未満の長さを有する、請求項2に記載の単離されたペプチド。
【請求項4】
50残基未満の長さを有する、請求項2に記載の単離されたペプチド。
【請求項5】
20残基未満の長さを有する、請求項2に記載の単離されたペプチド。
【請求項6】
芳香族アミノ酸Φ及び任意のアミノ酸Xから成るモチーフΦXXΦを含有する、請求項2に記載の単離されたペプチド。
【請求項7】
前記Eph受容体が、EphA2、EphA4、EphB4、EphA5、EphA7およびEphB2からなる群より選択される、請求項2に記載の単離されたペプチド。
【請求項8】
細胞を、Eph受容体ファミリーメンバーに選択的に結合する化合物を含むEph受容体結合リガンドの有効量と接触させることを含む、細胞におけるEph受容体活性の調節方法。
【請求項9】
前記Eph受容体結合リガンドが、単離されたペプチドまたはペプチド擬似化合物を含む、請求項8に記載の方法。
【請求項10】
前記Eph受容体ファミリーメンバーが、EphA2、EphA4、EphB4、EphA5、EphA7およびEphB2からなる群より選択される、請求項8に記載の方法。
【請求項11】
前記細胞がインビボである、請求項8に記載の方法。
【請求項12】
前記細胞がインビトロである、請求項8に記載の方法。
【請求項13】
哺乳動物に、該哺乳動物においてプログラム細胞死を活性化するのに有効な量のEph受容体アゴニストを投与することを含む方法であって、該アゴニストは、Eph受容体ファミリーメンバーに選択的に結合する化合物を含む、哺乳動物におけるプログラム細胞死の活性化方法。
【請求項14】
前記Eph受容体アゴニストが、単離されたペプチドまたはペプチド擬似化合物を含む、請求項13に記載の方法。
【請求項15】
前記哺乳動物がヒトである、請求項13に記載の方法。
【請求項16】
前記細胞が腫瘍性である、請求項13に記載の方法。
【請求項17】
前記アゴニストがEphA2受容体と結合する、請求項13に記載の方法。
【請求項18】
前記アゴニストが、EphA2受容体へのエフリン-Aの結合を阻害する、請求項13に記載の方法。
【請求項19】
前記アゴニストが、EphA2受容体のリン酸化を促進する、請求項13に記載の方法。
【請求項20】
哺乳動物に、該哺乳動物において神経再生を活性化するのに有効な量のEph受容体アゴニストを投与することを含み、該アゴニストは、Eph受容体ファミリーメンバーに選択的に結合する化合物を含む、神経再生の促進もしくはニューロン変性の改善、またはその両方を行なう方法。
【請求項21】
前記変性が、外傷性損傷、精神遅滞または神経変性疾患によるものである、請求項20に記載の方法。
【請求項22】
前記Eph受容体アゴニストが、単離されたペプチドまたはペプチド擬似化合物を含む、請求項20に記載の方法。
【請求項23】
細胞に治療剤を接触させることを含む、Eph受容体を発現する細胞への治療剤の送達方法であって、該治療剤は、Eph受容体ファミリーメンバーに選択的に結合する化合物を含むEph受容体結合リガンドと連結されていることを特徴とする方法。
【請求項24】
前記Eph受容体結合リガンドが、単離されたペプチドまたはペプチド擬似化合物を含む、請求項23に記載の方法。
【請求項25】
治療剤と連結されたEph受容体結合リガンドを含有するコンジュゲートであって、前記Eph受容体結合リガンドは、Eph受容体ファミリーメンバーに選択的に結合する、コンジュゲート。
【請求項26】
前記Eph受容体結合リガンドが、EphA2受容体に選択的に結合する、請求項25に記載のコンジュゲート。
【請求項27】
前記Eph受容体結合リガンドが、EphB4受容体に選択的に結合する、請求項25に記載のコンジュゲート。
【請求項28】
前記Eph受容体結合リガンドが、EphB2受容体に選択的に結合する、請求項25に記載のコンジュゲート。
【請求項29】
前記Eph受容体結合リガンドが、EphA4受容体に選択的に結合する、請求項25に記載のコンジュゲート。
【請求項30】
前記Eph受容体結合リガンドが、EphA5受容体に選択的に結合する、請求項25に記載のコンジュゲート。
【請求項31】
前記Eph受容体結合リガンドが、EphA7受容体に選択的に結合する、請求項25に記載のコンジュゲート。
【請求項32】
イメージング剤と連結されたEph受容体結合化合物を含有するコンジュゲートであって、該Eph受容体結合リガンドは、Eph受容体ファミリーメンバーに選択的に結合する、コンジュゲート。
【請求項33】
前記Eph受容体結合リガンドが、EphA2受容体に選択的に結合する、請求項32に記載のコンジュゲート。
【請求項34】
前記Eph受容体結合リガンドが、EphB4受容体に選択的に結合する、請求項32に記載のコンジュゲート。
【請求項35】
前記Eph受容体結合リガンドが、EphA4受容体に選択的に結合する、請求項32に記載のコンジュゲート。
【請求項36】
前記Eph受容体結合リガンドが、EphA5受容体に選択的に結合する、請求項32に記載のコンジュゲート。
【請求項37】
前記Eph受容体結合リガンドが、EphA7受容体に選択的に結合する、請求項32に記載のコンジュゲート。
【請求項38】
前記Eph受容体結合リガンドが、EphB2受容体に選択的に結合する、請求項32に記載のコンジュゲート。
【請求項39】
ランダムなペプチドをコードするファージディスプレイライブラリーを作製すること、
固定化したEph受容体に対して、前記ライブラリーをパニングすること、および
前記Eph受容体に結合するファージクローンを同定すること
を含む、Eph受容体結合リガンドの同定方法。
【請求項40】
いくつかの非保存アミノ酸により分断された2つの芳香族残基を含有するアミノ酸配列を有する、単離されたEph受容体結合リガンド。
【請求項41】
前記非保存アミノ酸の数が1−4個である、請求項40に記載の単離されたEph受容体結合化合物。
【請求項42】
Eph受容体に結合したEph受容体結合因子を検出すること、
前記Eph受容体を候補化合物と接触させること、および
前記Eph受容体からの前記Eph受容体結合因子の解離を検出すること
を含み、これにより、前記候補化合物がEph受容体結合リガンドであると同定する、Eph受容体結合リガンドの同定方法。
【請求項43】
前記Eph受容体が、EphA2、EphA4、EphA5、EphA7、EphB2およびEphB4からなる群より選択される、請求項42に記載の方法。
【請求項44】
前記Eph受容体結合リガンドが、EphA2受容体に選択的に結合する、請求項42に記載の方法。
【請求項45】
前記Eph受容体結合リガンドが、EphA4受容体に選択的に結合する、請求項42に記載の方法。
【請求項46】
前記Eph受容体結合リガンドが、EphA5受容体に選択的に結合する、請求項42に記載の同定方法。
【請求項47】
前記Eph受容体結合リガンドが、EphA7受容体に選択的に結合する、請求項42に記載の同定方法。
【請求項48】
前記Eph受容体結合リガンドが、EphB2受容体に選択的に結合する、請求項42に記載の同定方法。
【請求項49】
前記Eph受容体結合リガンドが、EphB4受容体に選択的に結合する、請求項42に記載の同定方法。
【請求項50】
前記Eph受容体結合リガンドが、検出可能な標識で標識されている、請求項42に記載の同定方法。
【請求項51】
前記Eph受容体は、そのトリプトファン残基が選択的に標識されている、請求項42に記載の同定方法。
【請求項52】
前記トリプトファン側鎖が、13Cおよび15Nおよび19Fおよび1Hで選択的に標識されている、請求項51に記載の方法。
【請求項53】
核磁気共鳴を用いて行なう、請求項52に記載の方法。
【請求項54】
前記Eph受容体結合リガンドがエフリンである、請求項42に記載の方法。
【請求項55】
前記エフリンが、エフリン−アルカリホスファターゼ融合タンパク質である、請求項54に記載の方法。
【請求項56】
外傷性損傷、精神遅滞および神経変性疾患の治療のための医薬の調製における、神経再生の促進およびニューロン変性の改善に有効であるEph受容体のアゴニストの使用。
【請求項57】
腫瘍性疾患の治療のための医薬の調製における、Eph受容体活性を調節し、プログラム細胞死を活性化するのに有効な化合物の使用。
【請求項58】
腫瘍性疾患、神経変性疾患、精神遅滞および外傷性損傷の治療のための医薬の調製における、Eph受容体ファミリーメンバーに選択的に結合する化合物の使用。
【図1−1】

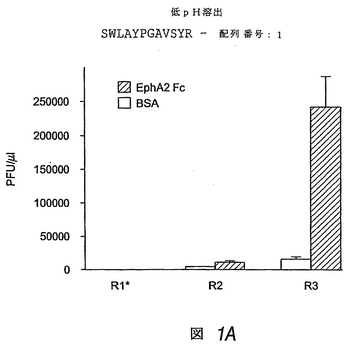
【図1−2】
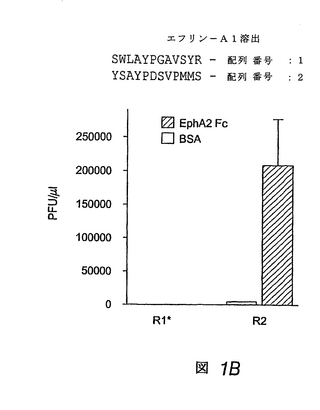
【図2】
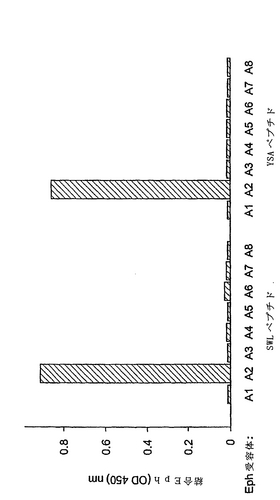
【図3】
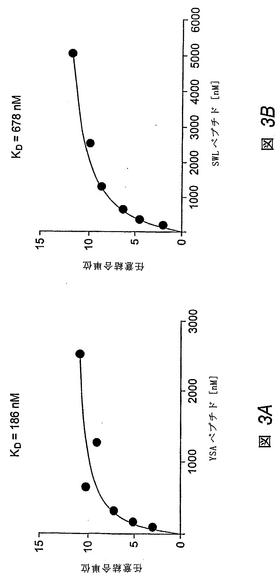
【図4】
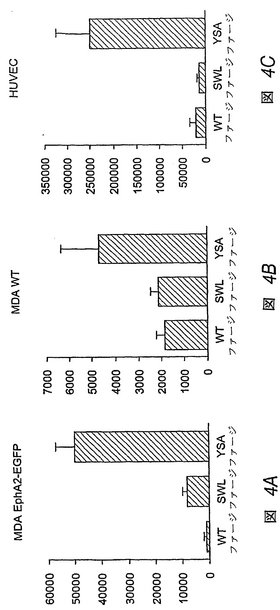
【図5】
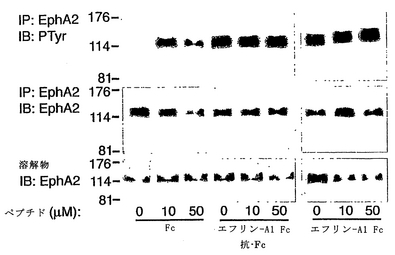
【図6】
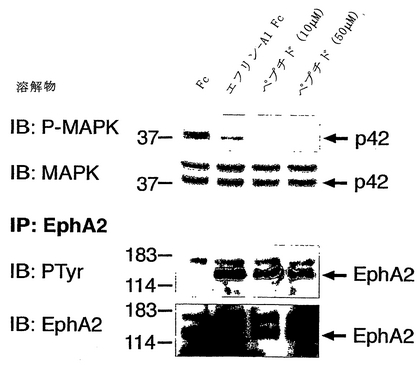
【図7】
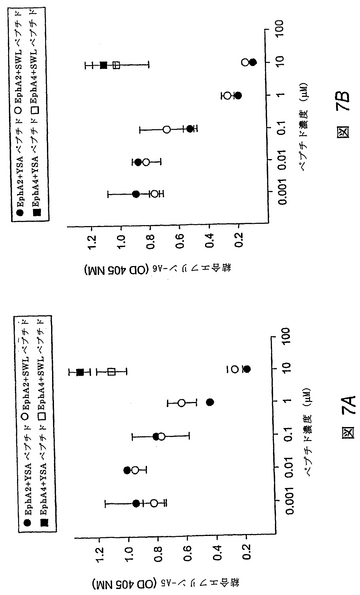
【図8】
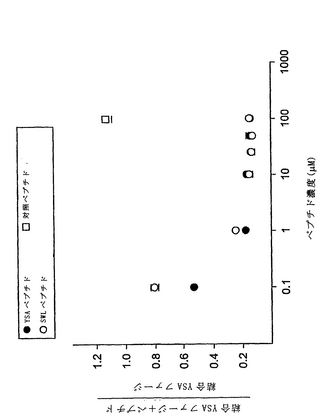
【図9】

【図10】
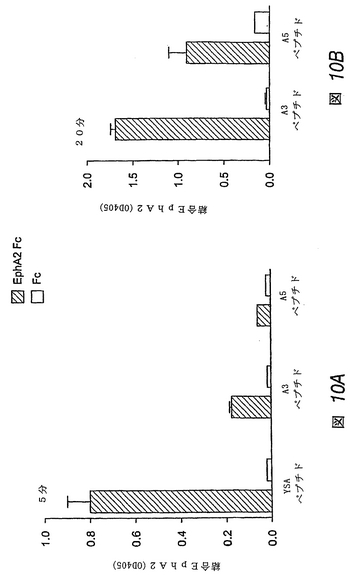
【図11】
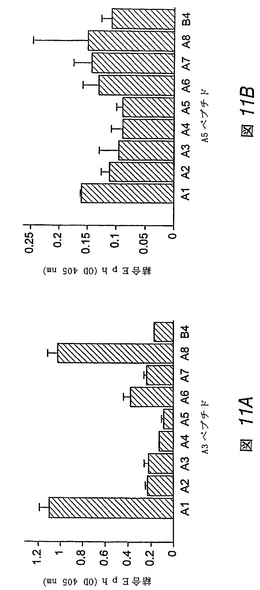
【図12−1】
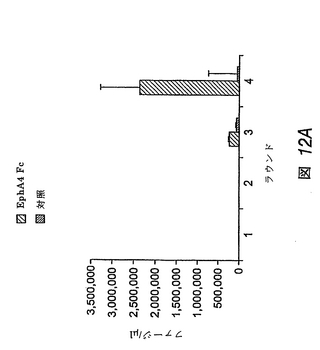
【図12−2】
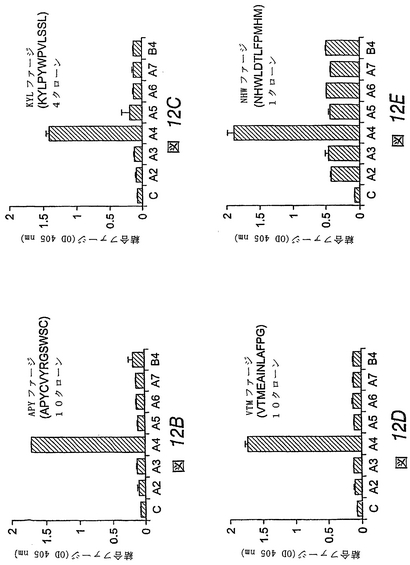
【図13−1】
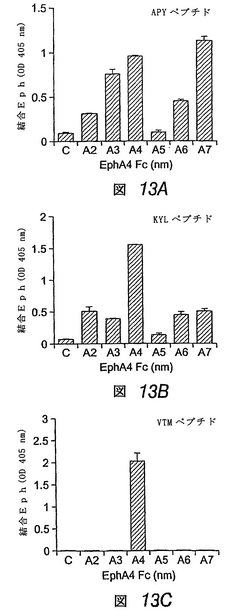
【図13−2】
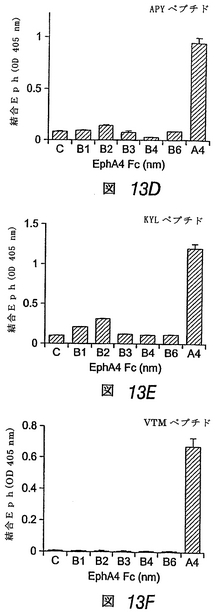
【図13−3】
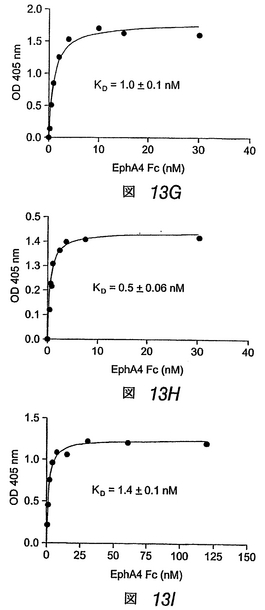
【図14】
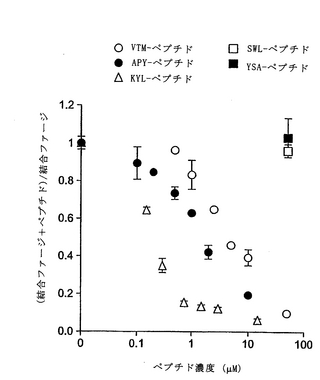
【図15−1】
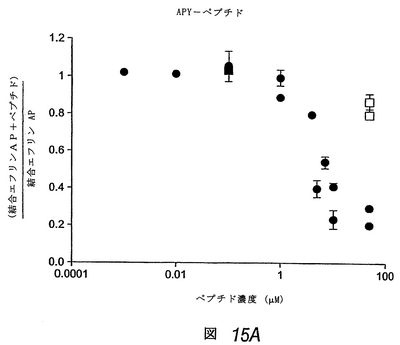
【図15−2】
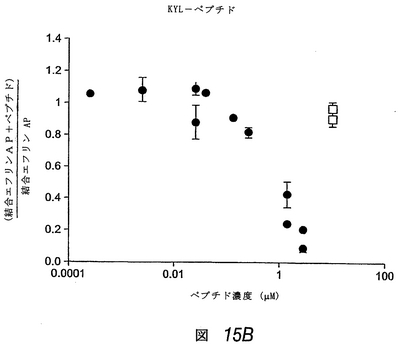
【図15−3】
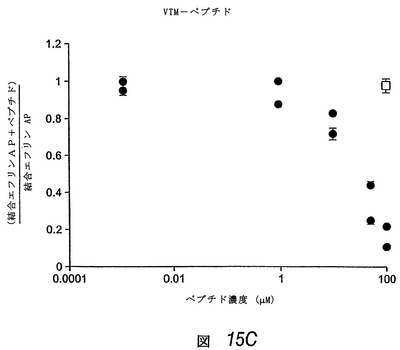
【図16】
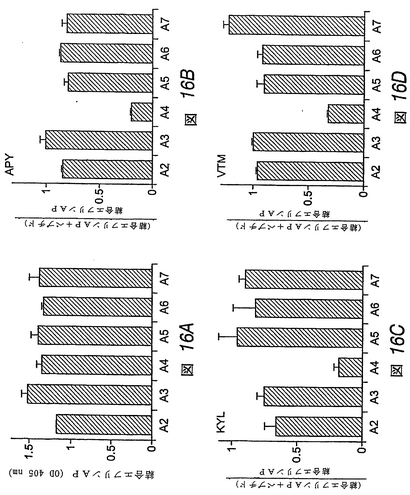
【図17】
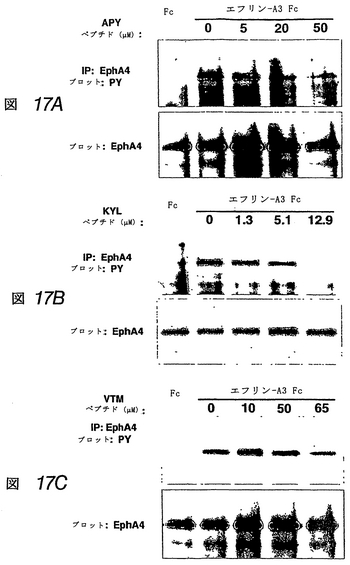
【図18−1】
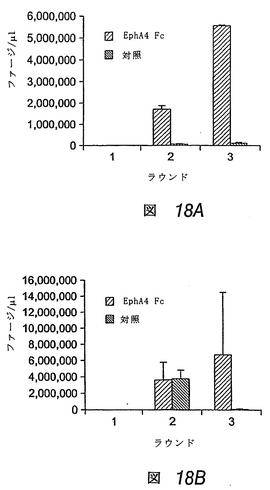
【図18−2】
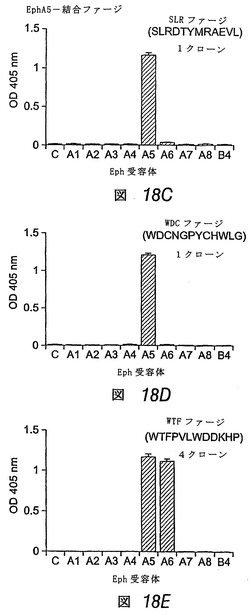
【図18−3】
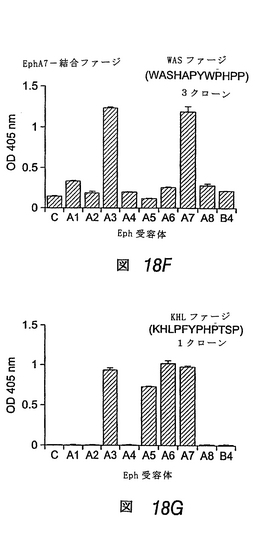
【図19】

【図19−1】
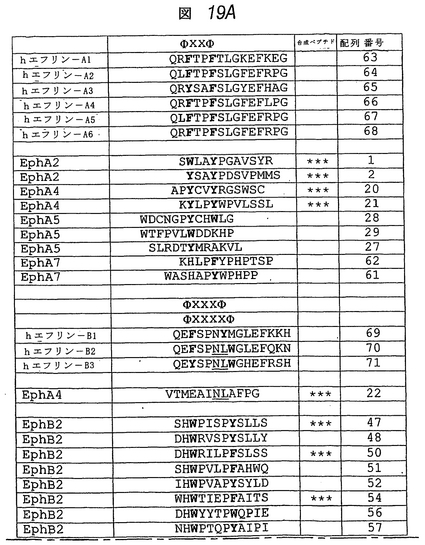
【図19−2】

【図1−2】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12−1】
【図12−2】
【図13−1】
【図13−2】
【図13−3】
【図14】
【図15−1】
【図15−2】
【図15−3】
【図16】
【図17】
【図18−1】
【図18−2】
【図18−3】
【図19】
【図19−1】
【図19−2】
【公表番号】特表2006−507256(P2006−507256A)
【公表日】平成18年3月2日(2006.3.2)
【国際特許分類】
【出願番号】特願2004−540038(P2004−540038)
【出願日】平成15年8月29日(2003.8.29)
【国際出願番号】PCT/US2003/027328
【国際公開番号】WO2004/028551
【国際公開日】平成16年4月8日(2004.4.8)
【出願人】(500114586)ザ バーナム インスティチュート (8)
【Fターム(参考)】
【公表日】平成18年3月2日(2006.3.2)
【国際特許分類】
【出願日】平成15年8月29日(2003.8.29)
【国際出願番号】PCT/US2003/027328
【国際公開番号】WO2004/028551
【国際公開日】平成16年4月8日(2004.4.8)
【出願人】(500114586)ザ バーナム インスティチュート (8)
【Fターム(参考)】
[ Back to top ]