
FGFR融合タンパク質によって疾患を治療するための組成物および方法
【課題】増殖性疾患治療のために有用なFGFR融合タンパク質を提供すること。
【解決手段】本発明は、FGFR融合タンパク質、その作製方法、ならびに癌および血管形成障害を含めた増殖性障害を治療するためのその使用方法を提供する。FGFR融合分子はCHO細胞中で作製することができ、FGFRの細胞外ドメイン中に、その安定性を向上させる欠失突然変異を含み得る。これらの融合タンパク質は、in vitroおよびin vivoで癌細胞の成長および生存度を阻害する。これらの受容体がそのリガンドFGFに対して比較的高い親和性を有することと、これらの囮受容体が腫瘍成長を阻害する能力が実証されていることとの組合せは、本明細書中に提供する組成物および方法の臨床的価値の指標である。
【解決手段】本発明は、FGFR融合タンパク質、その作製方法、ならびに癌および血管形成障害を含めた増殖性障害を治療するためのその使用方法を提供する。FGFR融合分子はCHO細胞中で作製することができ、FGFRの細胞外ドメイン中に、その安定性を向上させる欠失突然変異を含み得る。これらの融合タンパク質は、in vitroおよびin vivoで癌細胞の成長および生存度を阻害する。これらの受容体がそのリガンドFGFに対して比較的高い親和性を有することと、これらの囮受容体が腫瘍成長を阻害する能力が実証されていることとの組合せは、本明細書中に提供する組成物および方法の臨床的価値の指標である。
Notice: Undefined index: DEJ in /mnt/www/gzt_disp.php on line 298
【特許請求の範囲】
【請求項1】
明細書中に記載の発明。
【請求項1】
明細書中に記載の発明。
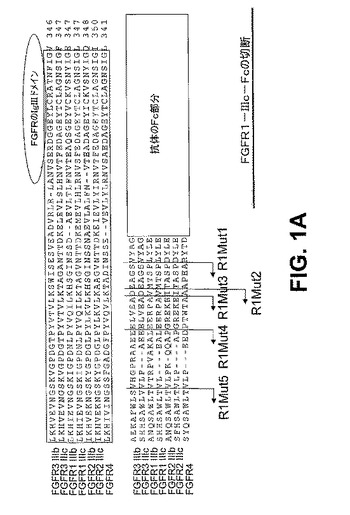
【図1A】


【図1B】

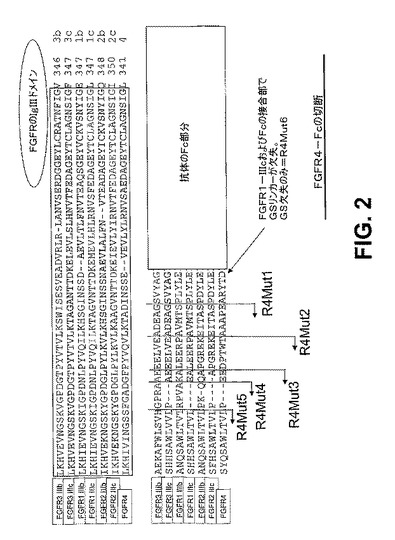
【図2】
【図3A】

【図3B】

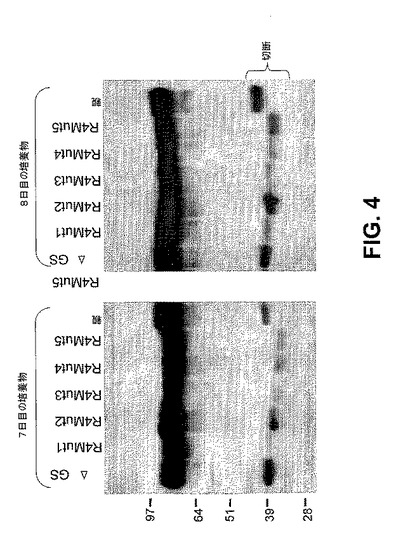
【図4】
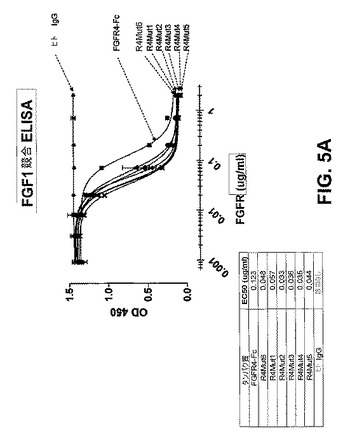
【図5A】
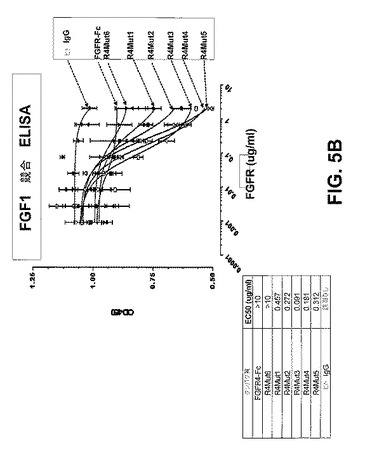
【図5B】
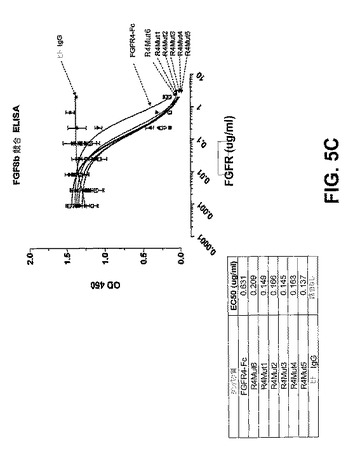
【図5C】
【図6】

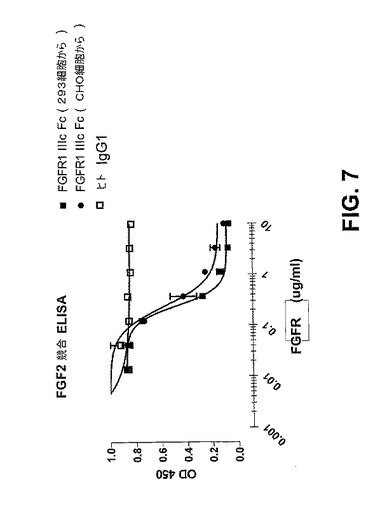
【図7】
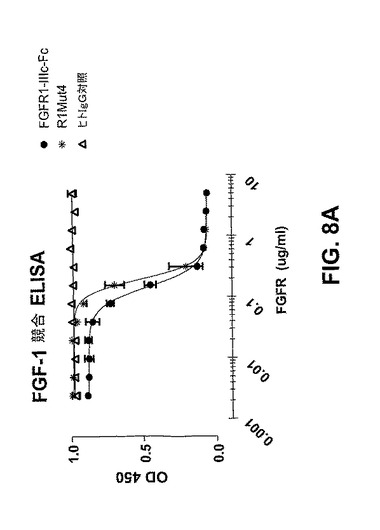
【図8A】
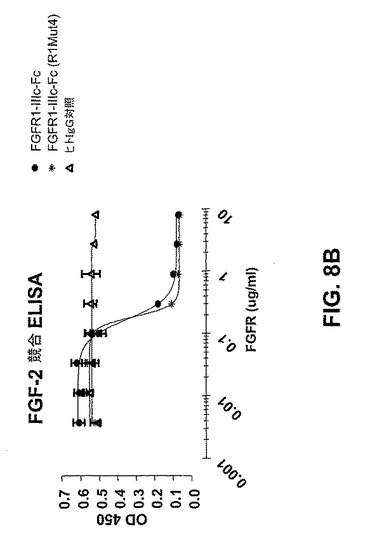
【図8B】
【図9】

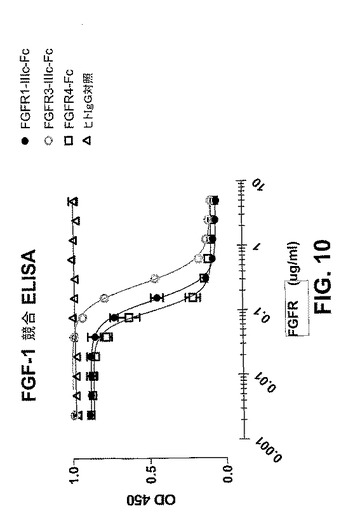
【図10】
【図11】

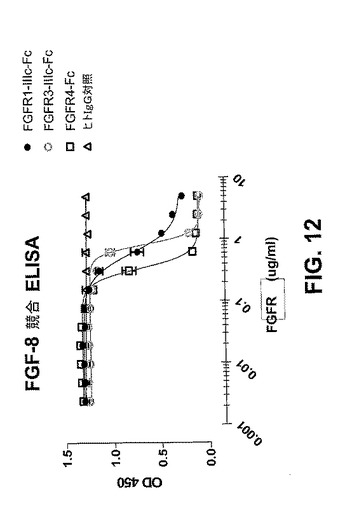
【図12】
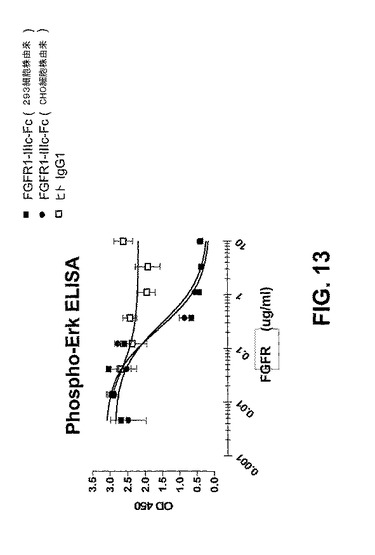
【図13】
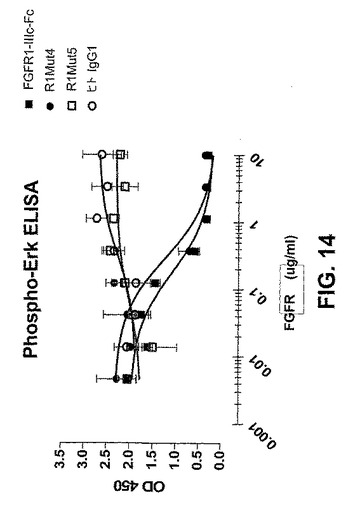
【図14】
【図15】

【図16】
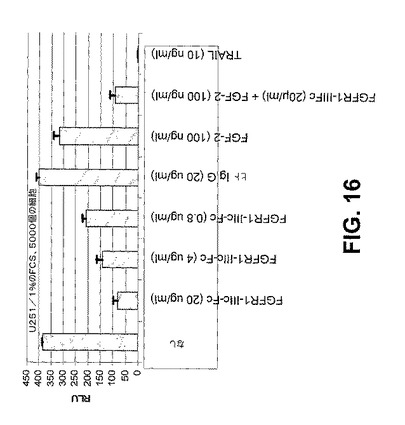
【図17】
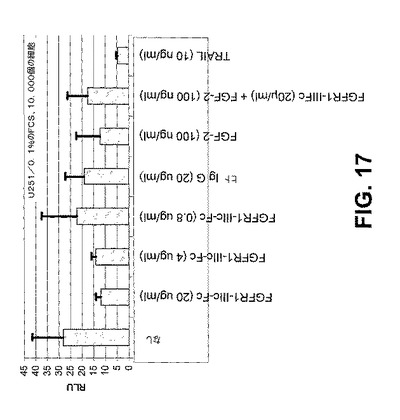
【図18】

【図19】

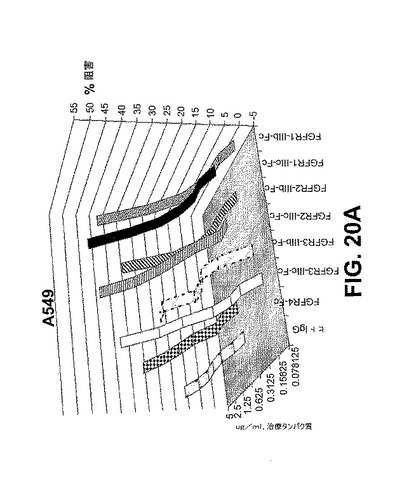
【図20A】
【図20B】
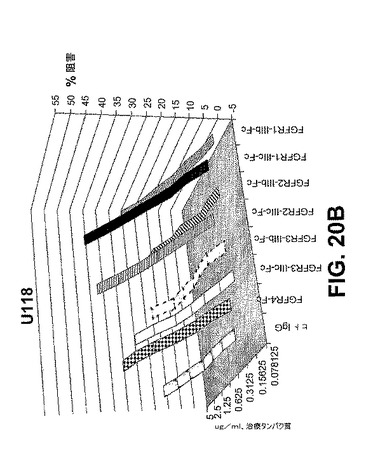
【図20C】
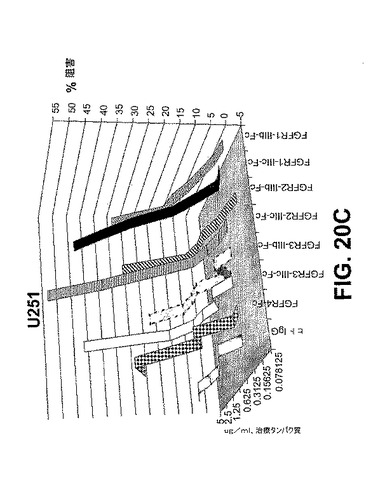
【図20D】
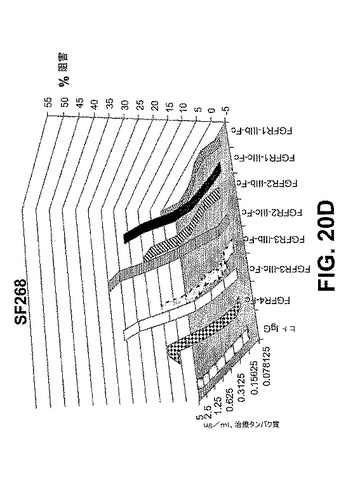
【図20E】

【図20F】

【図20G】

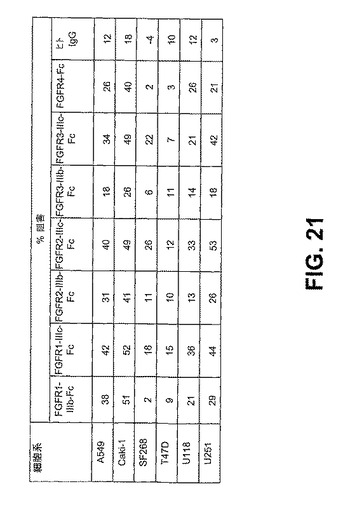
【図21】
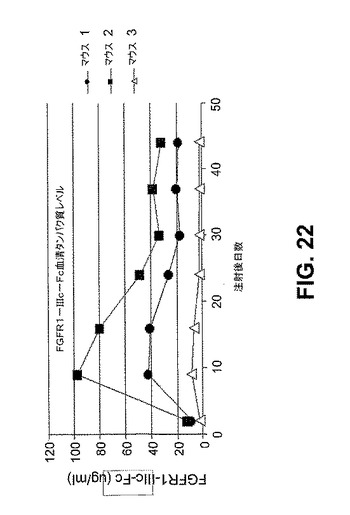
【図22】
【図23】
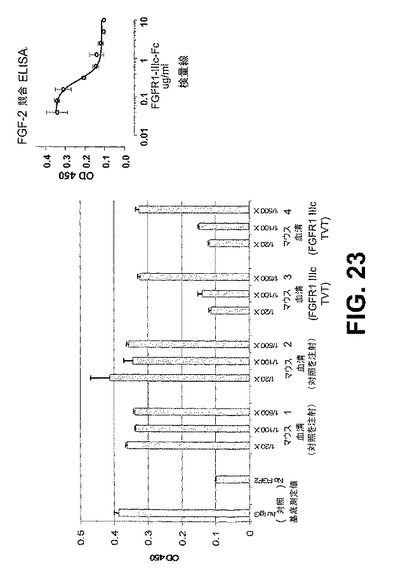
【図24】

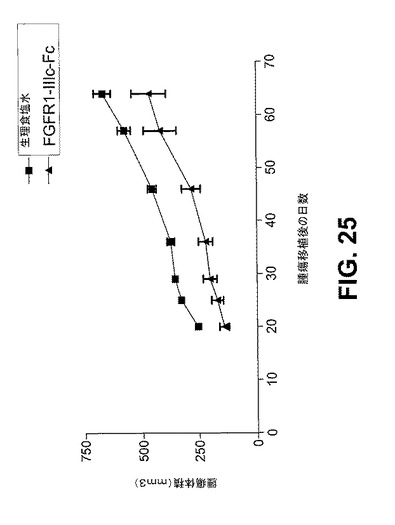
【図25】
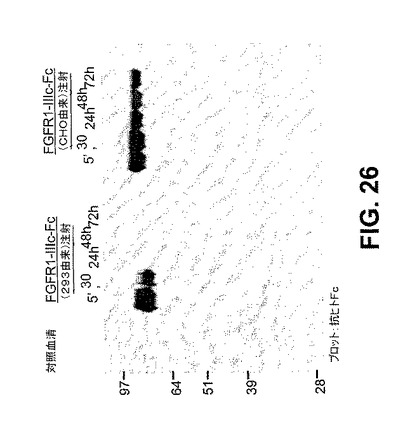
【図26】
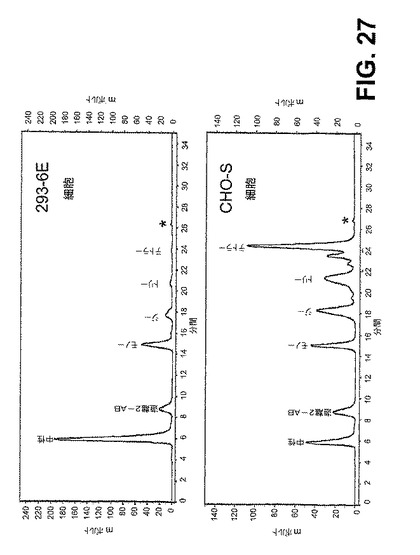
【図27】
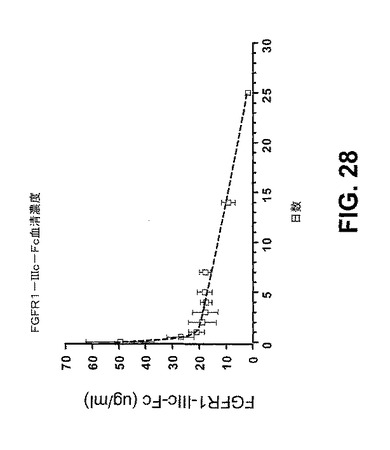
【図28】
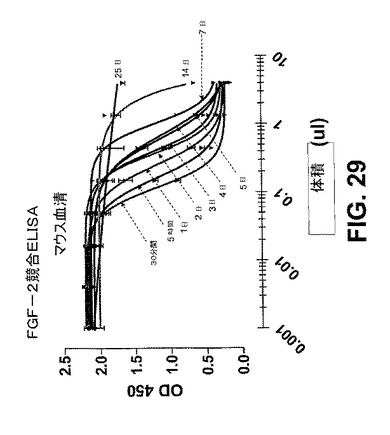
【図29】
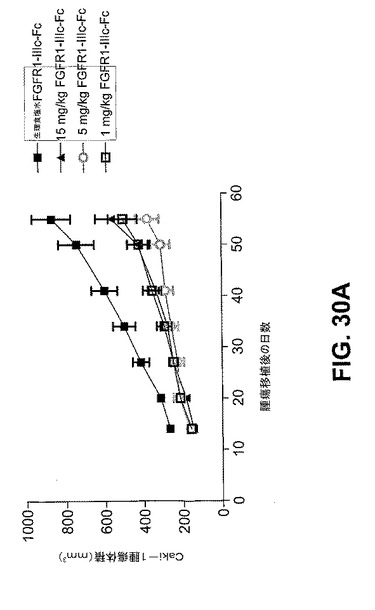
【図30A】
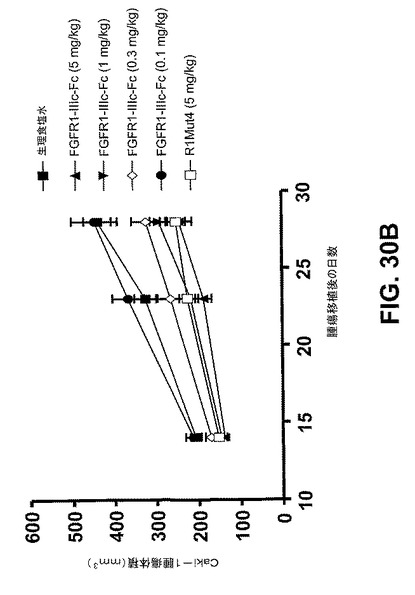
【図30B】
【図31】

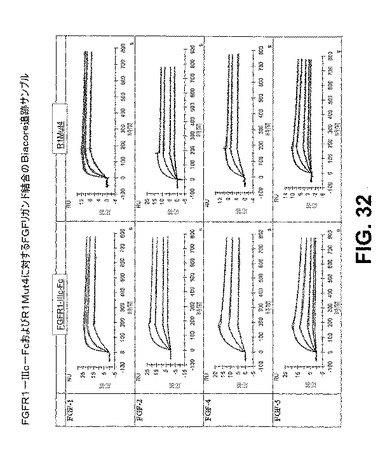
【図32】
【図1B】
【図2】
【図3A】
【図3B】
【図4】
【図5A】
【図5B】
【図5C】
【図6】
【図7】
【図8A】
【図8B】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20A】
【図20B】
【図20C】
【図20D】
【図20E】
【図20F】
【図20G】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30A】
【図30B】
【図31】
【図32】
【公開番号】特開2013−17484(P2013−17484A)
【公開日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願番号】特願2012−182376(P2012−182376)
【出願日】平成24年8月21日(2012.8.21)
【分割の表示】特願2008−33832(P2008−33832)の分割
【原出願日】平成18年7月24日(2006.7.24)
【出願人】(507150079)ファイブ プライム セラピューティクス, インコーポレイテッド (6)
【Fターム(参考)】
【公開日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願日】平成24年8月21日(2012.8.21)
【分割の表示】特願2008−33832(P2008−33832)の分割
【原出願日】平成18年7月24日(2006.7.24)
【出願人】(507150079)ファイブ プライム セラピューティクス, インコーポレイテッド (6)
【Fターム(参考)】
[ Back to top ]