
FVIIIの特定部位の変更
【課題】向上した薬物動態学的特性及び治療的特性を有する生物適合性ポリマー−複合機能性FVIIIポリペプチドを提供する。
【解決手段】Bドメインにおいて1個もしくはそれ以上の生物適合性ポリマーに共有結合した全長因子VIIIを含んでなる単離された複合体。機能性因子VIIIポリペプチドをコードするヌクレオチド配列を突然変異させてあらかじめ規定される部位においてシステイン残基に関するコード配列に置換し;突然変異したヌクレオチド配列を発現させてシステイン増強突然変異タンパク質を生産し;突然変異タンパク質を精製し;実質的に導入されたシステイン残基のみにおいてポリペプチドと反応するように活性化された生物適合性ポリマーと突然変異タンパク質を反応させて複合体を形成する。
【解決手段】Bドメインにおいて1個もしくはそれ以上の生物適合性ポリマーに共有結合した全長因子VIIIを含んでなる単離された複合体。機能性因子VIIIポリペプチドをコードするヌクレオチド配列を突然変異させてあらかじめ規定される部位においてシステイン残基に関するコード配列に置換し;突然変異したヌクレオチド配列を発現させてシステイン増強突然変異タンパク質を生産し;突然変異タンパク質を精製し;実質的に導入されたシステイン残基のみにおいてポリペプチドと反応するように活性化された生物適合性ポリマーと突然変異タンパク質を反応させて複合体を形成する。
【発明の詳細な説明】
【関連出願へのクロスリファレンス】
【0001】
本出願は、2004年11月12日に申請された米国特許出願第60/627,277号明細書への優先権の利益を請求し、その出願は引用することによりその記載事項全体が本明細書の内容となる。
【技術分野】
【0002】
発明の分野
本発明は、規定される部位において1個もしくはそれより多い生物適合性ポリマー、例えばポリエチレングリコールへのカップリングを許す因子VIII(FVIII)突然変異タンパク質に関する。さらに、治療目的のための関連する調製物、投薬物(dosages)及びその投与の方法を提供する。これらの変更されたFVIII突然変異体ならびに関連する組成物及び方法は、血友病Aに苦しむ患者のための注入頻度が低下し、且つ免疫原性反応が低下した処置の選択肢を与えるのに有用である。
【背景技術】
【0003】
発明の背景
血友病Aは最も普通の遺伝性凝固障害であり、5000人の男性当たり1人の発生率と見積もられる。それは、血液凝固の固有の経路の決定的な構成成分であるFVIIIにおける欠失又は構造的欠陥により引き起こされる。血友病Aのための現在の処置は、ヒトFVIIIの静脈内注射を含む。ヒトFVIIIは約300kDの一本鎖分子として組換え的に生産されてきた。それは構造ドメインA1−A2−B−A3−C1−C2から成る(非特許文献1)。前駆体生成物はゴルジ装置において200kD(重)及び80kD(軽)の2つのポリペプチド鎖にプロセシングされ、2つの鎖は金属イオンにより一緒に保持される(非特許文献2;非特許文献3)。
【0004】
FVIIIのB−ドメインはB−ドメイン欠失FVIII(BDD,90kD A1−A2重鎖プラス80kD軽鎖)として省略可能であると思われ、血友病Aのための置換療法として有効であることも示された。B−ドメイン欠失FVIII配列は、B−ドメインの14個のアミノ酸以外のすべての欠失を含有する。
【0005】
血友病A患者は現在、必要時の、又は1週に数回投与される予防的治療としてのFVIIIの静脈内投与により処置される。予防的処置のために、体重のkg当たり15〜25IUの因子VIIIが1週に3回投与される。患者においてそれは常に必要である。人間におけるFVIIIの短い半減期の故に、それは頻繁に投与されねばならない。全長タンパク質の場合の300kDを超えるその大きなサイズにかかわらず、FVIIIは人間中でわずか約11時間の半減期を有する。(非特許文献4)。頻繁な静脈内注射の必要性は、患者のコンプライアンスにとってものすごい障壁を作る。より長い半減期を有し、従って必要な投与がより頻繁でないFVIII製品を開発できたら、それは患者にとってもっと簡便であろう。さらに、半減期が向上したら、その場合はより少ない用量が必要となり得るので、処置の経費を減少させることができる。
【0006】
現在の治療へのさらなる欠点は、患者の約25〜30%がFVIII活性を阻害する抗体を発現することである(非特許文献5)。阻害性抗体の主なエピトープはA2ドメイン内で残基484−508に、A3ドメイン内で残基1811−1818に、及びC2ドメイン内に位置する。抗体発現は置換療法としてのFVIIIの使用を妨げ、この群の患者に高い用量の組換え因子VIIaを用いるさらにもっと高価な処置及び免疫寛容治療(immune tolerance therapy)を追求することを強いる。
【0007】
以下の研究は、阻害性抗体のFVIIIエピトープを同定した。25個の阻害性血漿試料の研究において、11個はトロンビン生成73kD軽鎖フラグメントA3C1C2に、4個はA2ドメインに、そして10個は両方に結合することが見出された(非特許文献6)。別の研究において、患者からの8個のA2ドメイン阻害物質(inhibitors)中の6個は組換えA2ポリペプチドにより中和された(非特許文献7)。患者からの9個の阻害物質中の6個に関するエピトープは、A2残基379−538にマッピングされた(非特許文献8)。18個の重−鎖阻害物質に関するエピトープは、A2ドメインの同じN−末端18.3kD領域中に位置決定された(非特許文献9)。
【0008】
ヒトA2ドメイン残基387−604を相同ブタ配列で置換することにより形成される活性な組換えハイブリッドヒト/ブタFVIII分子は、患者のA2阻害物質に抵抗性であり(非特許文献10)、A2への結合に関して患者のA2阻害物質と競合するネズミモノクローナル抗体mAB 413 IgGに抵抗性(非特許文献11)であった。このA2ドメインエピトープはさらに、mAB 413 IgG及び4個の患者の阻害物質が、A2ドメイン残基484−508がブタのそれで置換されたハイブリッドヒト/ブタFVIIIを阻害しないことを実験が示した時に、A2ドメイン残基484−508に位置決定された(非特許文献12)。このハイブリッドFVIIIは、スクリーニングされた23個の患者の血漿の少なくとも半分により抵抗性でもあった(非特許文献13)。アラニン走査突然変異誘発(alanine scanning mutagenesis)は、試験された5個のすべての患者の阻害物質への結合に関して残基487が決定的であるが、残基484、487、489及び492はすべてmAB 413 IgGとの相互作用に重要であることを同定した(非特許文献14)。R484A/R489A/P492A突然変異体を与えられるがR484A/R489突然変異体を与えられないマウスにおける阻害性抗体力価は、標準ヒトBDD FVIIIを与えられるマウスにおけるより有意に低かった(非特許文献15)。要するに、A2ドメインの484−508領域はFVIII活性の阻害物質のための結合部位であると思われる。
【0009】
FVIIIに対する免疫反応の出現に加え、通常の治療を用いる別の問題は、生体内におけるFVIIIの短い半減期の故にそれが頻繁な投薬を必要とすることである。循環からのFVIIIのクリアランスに関する機構が研究された。
【0010】
循環からのFVIIIクリアランスは、部分的に低−密度リポタンパク質受容体−関連タンパク質(LRP)、広いリガンド特性を有する肝臓クリアランス受容体への特異的な結合に帰せられた(非特許文献16)。最近、低−密度リポタンパク質(LDL)受容体も、例えばFVIIIの血漿レベルの調節においてLRPと協働することにより、FVIIIクリアランスにおいて役割を果たすことが示された(非特許文献17)。両相互作用は、細胞−表面ヘパリン硫酸プロテオグリカン(HSPGs)に結合することにより助長される。マウスにおける血漿半減期は、LRPが遮断されると3.3倍に延長され得るか、あるいはLRP及び細胞−表面HSPGsの両方が遮断されると5.5倍に延長され得る(非特許文献18)。HSPGsはFVIIIを細胞表面上に濃縮し、それをLRPに与えると仮定される。FVIII上のLRP結合部位はA2残基484−509(非特許文献19)、A3残基1811−1818(非特許文献20)及びC2ドメイン中のエピトープ(非特許文献21)に位置決定された。
【0011】
FVIIIはプロテアーゼの作用によっても循環からクリアランスされる。この効果を理解するためには、FVIIIが血液凝固に含まれる機構を理解しなければならない。FVIIIはvWFに結合した重鎖及び軽鎖のヘテロ二量体として循環する。VWF結合はFVIII残基1649−1689(非特許文献22)ならびにC1(非特許文献23)及びC2ドメイン(非特許文献24)の一部を含む。FVIIIはトロンビンにより活性
化され、それは残基372、740及び1689の後でペプチド結合を切断し、A1、A2及びA3−C1−C2ドメインのヘテロ三量体を形成する(非特許文献25)。FVIIIは、活性化されるとvWFから解離し、リン脂質に結合することにより血小板の細胞表面に濃縮される。リン脂質結合はFVIII残基2199、2200、2251及び2252を含む(非特許文献26)。そこでそれはFVIII残基558−565(非特許文献27)及び1811−1818(非特許文献28)との相互作用を介してFIXに、ならびにFVIII残基349−372との相互作用を介してFXに(非特許文献29)結合し、固有の凝固経路の必須の成分であるFXのFIX活性化のための補因子として働く。活性化されたFVIII(FVIIIa)は、プロテアーゼ活性化タンパク質C(APC)により、FVIII残基336及び562の後の切断を介して部分的に不活性化される(非特許文献30)。しかしながら、不活性化の主な決定因子は、A1及びA3−C1−C2からのA2ドメインの解離である(非特許文献31)。
【0012】
タンパク質の生体内半減期を向上させることが示された1つの方法はPEG化である。PEG化は、タンパク質又は他の分子への長−鎖ポリエチレングリコール(PEG)分子の共有結合である。PEGは直鎖状形態又は分枝鎖状形態にあり、種々の特徴を有する種々の分子を形成することができる。ペプチド又はタンパク質の半減期を向上させることの他に、PEG化は抗体発現を減少させ、プロテアーゼ消化からタンパク質を保護し、且つ腎臓濾液(kidney filtrate)の外に材料を保持するために用いられてきた(非特許文献32)。さらに、PEG化はタンパク質の全体的な安定性及び溶解度も向上させることができる。最後に、PEG化されたタンパク質の持続的な血漿濃度は、薬剤の谷からピークまでのレベル(the trough to peak level)を低下させることにより不利な副作用の程度を低下させ、かくして初期の時点にタンパク質の超−生理学的レベルを導入する必要を除去することができる。
【0013】
PEG及びデキストランのような大きなポリマーを用いて第1級アミン(N−末端及びリシン)を標的とすることによるFVIIIの無作為な変更は、種々の成功の程度を以って試みられてきた(特許文献1、特許文献2、特許文献3)。1994年の特許出願(特許文献1)において公開された最も劇的な進歩は、全−長FVIIIを50−倍モル過剰のPEGと反応させた後、4−倍の半減期の向上を示すが、2分の1への活性の損失(2−fold activity loss)を犠牲にする。特許文献4は、無作為な変更を介して作られるFVIIIとポリエチレングリコールの複合体を開示している。無作為にPEG化されたタンパク質、例えばインターフェロン−アルファ(非特許文献33)は、過去に療薬として承認された。
【0014】
しかしながらこの無作為法は、ヘテロ二量体性FVIIIにとってもっとずっと問題が多い。FVIIIは、158個のリシン、2個のN−末端及び多数のヒスチジン、セリン、トレオニン及びチロシンを含む何百のPEG化可能な部位を有し、それらのすべてはおそらく主に第1級アミンを標的とする試薬を用いてPEG化され得る。例えばPEG化されたインターフェロンアルファ−2bに関する主な位置異性体はヒスチジンであることが示された(非特許文献34)。さらに、全長FVIIIの不均一なプロセシングは出発材料の混合物を生じ得、それはPEG化された生成物におけるさらなる複雑性に導く。PEGが決定的な活性部位もしくはその近辺で結合するべき場合、特に1個より多いPEG又は1個の大きなPEGをFVIIIに複合させる場合、FVIII上のPEG化の部位を制御しないさらなる欠点は活性の低下の可能性である。無作為なPEG化は必ず多量の多重にPEG化された生成物を生産するので、モノ−PEG化生成物のみを得るための精製は全体的収率を非常に低下させるであろう。最後に、生成物分布におけるものすごい不均一性は、各ロットの一貫した合成及び特性化をほとんど不可能にするであろう。優れた製造は一貫した十分に特性化される生成物を必要とするので、生成物の不均一性は商品化への障壁である。これらの理由のすべてのために、FVIIIをPEG化するためのもっと
特異的な方法が望ましい。
【0015】
種々の特定部位のタンパク質PEG化戦略が最近の総説にまとめられている(非特許文献35)。1つの方法は、化学的合成又は組換え発現によるタンパク質中への不自然なアミノ酸の導入及びそれに続く不自然なアミノ酸と特異的に反応するであろうPEG誘導体の添加を含む。例えば不自然なアミノ酸は本来のタンパク質中に見出されないケト基を含有するものであることができる。しかしながらタンパク質の化学的合成は、FVIIIのような大きなタンパク質には実行できない。ペプチド合成の現在の限界は約50個の残基である。数個のペプチドを連結させてもっと大きな1片のポリペプチドを作ることができるが、B−ドメイン欠失FVIIIを生産することさえ20回より多い連結を必要とし、それは理想的な反応条件下でさえ1%より低い回収率を生ずる。不自然なアミノ酸を有するタンパク質の組換え発現はこれまで、主に非−哺乳類発現系に限られてきた。この方法は、哺乳類系で発現される必要があるFVIIIのような大きく且つ複雑なタンパク質にとって問題が多いと思われる。
【0016】
タンパク質の部位−特異的PEG化への他の方法は、PEG−アルデヒドを用いてN−末端主鎖アミンを標的とすることによる。しかしながら、他のアミン基を超える特異性を達成するためにこの方法の下で必要な低いpHは、FVIIIの安定性のために必要な狭い近−中性pH範囲と適合しない(非特許文献36)。さらに、FVIIIのN−末端PEG化は、この領域が血漿クリアランスに含まれない場合、血漿半減期の向上を生ずることができない。事実、FVIII軽鎖のN−末端領域は、循環中のFVIII生存のために決定的に重要である担体タンパク質であるフォンビルブラント因子(vWF)への結合に関係があるとされてきた。因子VIIIのN−末端変更により、決定的に重要なvWFとの会合が崩壊するか、又は弱くなり得る。かくしてFVIIIのN−末端PEG化はFVIIIの血漿半減期を低下させる反対の効果を有し得る。
【0017】
特許文献5は、システインを挿入するか、又は他のアミノ酸の代わりに置換し、次いでスルフヒドリル反応性基を有するリガンドを加えることによるヒトIL−3、顆粒球コロニー刺激因子及びエリトロポエチンポリペプチドの部位−特異的変更を開示している。リガンドはシステイン残基に選択的にカップリングする。FVIII又はその変異体の変更は開示されていない。
【0018】
上記の理由のために、機能的活性を保持しながら生体内におけるより長い作用の持続時間及び低下した免疫原性を有する改良されたFVIII変異体に対する要求がある。さらに、そのようなタンパク質が一貫したやり方で均一な生成物として生産されるのが望ましい。
【先行技術文献】
【特許文献】
【0019】
【特許文献1】国際公開第94/15625号パンフレット
【特許文献2】米国特許第4970300号明細書
【特許文献3】米国特許第6048720号明細書
【特許文献4】国際公開第2004/075923号パンフレット
【特許文献5】国際公開第90/12874号パンフレット
【非特許文献】
【0020】
【非特許文献1】Thompson著,Semin.Hematol.29,2003年,11−22
【非特許文献2】Kaufman et al.著,J.Biol.Chem.263,1988年,p.6352
【非特許文献3】Andersson et al.著,Proc.Natl.Acad.Sci.83,1986年,p.2979
【非特許文献4】Ewenstein et al.著,Semin,Hematol.41,2004年,pp.1−16
【非特許文献5】Saenko et al.著,Haemophilia 8,2002年,pp.1−11
【非特許文献6】Fulcher,C.et al.著,Proc.Natl.Acad.Sci.2(22),1985年,pp.7728−32
【非特許文献7】Scandella,D.et al.著,Blood 82(6),1993年,pp.1767−75
【非特許文献8】Scandella,D.et al.著,Proc.Natl.Acad.Sci.85(16),1988年,pp.6152−6
【非特許文献9】Scandella,D.et al.著,Blood 74(5),1989年,pp.1618−26
【非特許文献10】Lubin,I.et al.著,J.Biol.Chem.269(12),1994年,pp.8639−41
【非特許文献11】Scandella,D.et al.著,Thromb Haemost.67(6),1992年,pp.665−71
【非特許文献12】Healey,J.et al.著,J.Biol.Chem.270(24),1995年,pp.14505−9
【非特許文献13】Barrow,R.et al.著,Blood 95(2),2000年,pp.564−8
【非特許文献14】Lubin,I.著,J.Biol.Chem.272(48),pp.30191−5
【非特許文献15】Parker,E.et al.著,Blood 104(3),2004年,pp.704−10
【非特許文献16】Oldenburg et al.著,Haemophilia 10 Suppl 4,2004年,pp.133−139
【非特許文献17】Bovenschen et al.著,Blood 106,2005年,pp.906−910
【非特許文献18】Sarafanov et al.著,J.Biol.Chem.276,2001年,pp.11970−11979
【非特許文献19】Saenko et al.著,J.Biol.Chem.274,1999年,pp.37685−37692
【非特許文献20】Bovenschen et al.著,J.Biol.Chem.278,2003年,pp.9370−9377
【非特許文献21】Lenting et al.著,J.Biol.Chem.274,1999年,pp.23734−23739
【非特許文献22】Foster et al.著,J.Biol.Chem.263,1988年,pp.5230−5234
【非特許文献23】Jacquemin et al.著,Blood 96,2000年,pp.958−965
【非特許文献24】Spiegel,P.et al.著,J.Biol.Chem.279(51),2004年,pp.53691−8
【非特許文献25】Pittman et al.著,Proc.Natl.Acad.Sci.276,2001年,pp.12434−12439
【非特許文献26】Gilbert et al.著,J.Biol.Chem.277,2002年,pp.6374−6381
【非特許文献27】Fay et al.著,J.Biol.Chem.269,1994年,pp.20522−20527
【非特許文献28】Lenting et al.著,J.Biol.Chem.271,1996年,pp.1935−1940
【非特許文献29】Nogami et al.著,J.Biol.Chem.279,2004年,pp.15763−15771
【非特許文献30】Regan et al.著,J.Biol.Chem.271,1996年,pp.3982−3987
【非特許文献31】Fay et al.著,J.Biol.Chem.266,1991年,pp.8957−8962
【非特許文献32】Harris et al.著,Clinical Pharmacokinetics 40,2001年,pp.539−51
【非特許文献33】Kozlowski et al.著,Biodrugs 15,2001年,pp.419−429
【非特許文献34】Wang et al.著,Biochemistry 39,2000年,pp.10634−10640
【非特許文献35】Kochendoerfer,G.著,Curr.Opin.Chem.Biol.2005,of Oct.15,2005,direct object identifier doi:10.1016/j.cbpa.2005.10.007としてオンラインで入手可能)
【非特許文献36】Wang et al.著,International J.Pharmaceutics 259,2003年,pp.1−15
【発明の概要】
【発明が解決しようとする課題】
【0021】
本発明の目的は、向上した薬物動態学的特性及び治療的特性を有する生物適合性ポリマー−複合機能性FVIIIポリペプチドを提供することである。
【0022】
本発明の他の目的は、向上した薬物動態学的性質を有する生物適合性ポリマー−複合Bドメイン欠失FVIIIタンパク質を提供することである。
【0023】
本発明のさらに別の目的は、低−密度リポタンパク質受容体−関連タンパク質(LRP)、低−密度リポタンパク質(LDL)受容体、ヘパラン硫酸プロテオグリカン(HSPGs)及び/又はFVIIIに対する阻害性抗体への結合が減少した生物適合性ポリマー−複合機能性FVIIIポリペプチドを提供することである。
【0024】
本発明のさらに別の目的は、生体内におけるより長い作用の持続時間及び低下した免疫原性を有し、一貫したやり方で均一な生成物として生産され得る改良されたFVIII変異体を提供することである。
【課題を解決するための手段】
【0025】
本発明の1つの側面において、ポリペプチド上のあらかじめ規定される1個もしくはそれより多い部位において1個もしくはそれより多い生物適合性ポリマーに共有結合した機能性因子VIIIポリペプチドを含んでなる因子VIIIプロコアグラント活性を有する複合体であって、あらかじめ規定される部位がN−末端アミンではない複合体を提供する。本発明は、機能性因子VIIIポリペプチドをコードするヌクレオチド配列を突然変異させてあらかじめ規定される部位においてシステイン残基に関するコード配列に置換し;突然変異したヌクレオチド配列を発現させてシステイン増強突然変異タンパク質を生産し;突然変異タンパク質を精製し;実質的に導入されたシステイン残基のみにおいてポリペプチドと反応するように活性化された生物適合性ポリマーと突然変異タンパク質を反応させて複合体を形成し;そして複合体を精製することを含んでなる、この複合体の調製方法も含む。本発明は、複合体及び製薬学的に許容され得る添加剤を含んでなる製薬学的組成
物ならびに必要のある患者にこれらの製薬学的組成物の治療的に有効な量を投与することによる、血友病の処置方法も目的とする。
【0026】
本発明は、(a)特定部位の因子VIII突然変異タンパク質を発現させ、ここで突然変異タンパク質は因子VIII突然変異タンパク質の露出された表面上のアミノ酸残基に代わるシステイン置換を有し、且つそのシステインはキャッピングされており;(b)システイン突然変異タンパク質を穏やかに還元し、且つキャップを放出する条件下でシステイン突然変異タンパク質を還元剤と接触させ;(c)システイン突然変異タンパク質からキャップ及び還元剤を除去し;そして(d)還元剤の除去から少なくとも約5分後に、PEG化された因子VIII突然変異タンパク質が生産されるような条件下で、スルフヒドリルカップリング部分を含んでなるPEGでシステイン突然変異タンパク質を処理することを含んでなる、因子VIII突然変異タンパク質の特定部位のPEG化のための方法にも関する。
【0027】
図面の簡単な記述
図1.PEG突然変異タンパク質のためのベクターマップ及び突然変異誘発戦略。
【0028】
図2.モノクローナルFVIII抗体クロマトグラフィーカラム上で精製されたPEG2タンパク質の場合の、時間に関する280nmにおけるUV吸光度分布。クロマトグラフィーは、Amersham BioscienceからのAKTAR Explorer 100クロマトグラフィーシステムを用いて行なわれた。
【0029】
図3.3−段階特定部位PEG化法。PEGはPEG−マレイミドのようなシステイン−反応性PEGを示す。閉じられた棒はジスルフィド形成を示し、開放された棒は還元されたシステインを示す。
【0030】
図4.PEG2の特定部位のPEG化。
【0031】
図5.PEG6の特定部位のPEG化。
【0032】
図6a.BDD、PEG2、4、5及び6の特定部位のPEG化。上のパネルは重(H)鎖抗体で染色され、下のパネルは軽(L)鎖抗体で染色された。“U”はH及びLの両方を含有するプロセシングされない材料である。
【0033】
図6b.PEG2及びPEG6を標準として用いるPEG15及びPEG7のPEG化。TCEPを用いて開始精製PEG突然変異タンパク質(“S”)を還元し、還元剤(“R”)の除去の後に12kD(“12”)又は22kD(“22”)PEGを用いてPEG化する。試料を6% Tris−グリシン SDS PAGE上で移動させ、左のパネル上で重鎖(“HC”)抗体又は右のパネル上で軽鎖(“LC”)抗体を用いて染色した。“U”はHC及びLCの両方を含有するプロセシングされない材料である。PEG化されたバンドを点により強調する。
【0034】
図6c.PEG2及びPEG6を標準として用いるPEG2+6のPEG化。TCEPを用いてPEG2、PEG6又はPEG2+6を還元し、還元剤(“R”)の除去の後に5kD(“5”)又は43kD(“43”)PEGを用いてPEG化する。12、22及び33kD PEGsを用いてもPEG2+6をPEG化した。試料を6% Tris−グリシン SDS PAGE上で移動させ、左でタンパク質に関してクーマシーを用いて、あるいは重鎖(H)又は軽鎖(L)抗体を用いて染色した。“U”はH及びLの両方を含有するプロセシングされない材料である。PEG化されたバンドを点により強調する。
【0035】
図6d.PEG2を標準として用いる野生型全長FVIII(KG−2)のPEG化。左のゲルをタンパク質に関してクーマシー染色を用いて染色し、右のゲルをPEGに関してヨウ素を用いて染色した。“BDD U”はH及びLの両方を含有するプロセシングされないBDD材料である。PEG化されたバンドを点により強調する。
【0036】
図7.PEG化されたPEG2のトロンビン切断。A2ドメインのN−末端半分を青く着色し、C−末端半分を緑に着色し、R8B12抗体エピトープを暗緑色に強調した(右FVIIIモデル)。PEG2(列1)及び22kD PEG化PEG2(列2)をトロンビンで処理し(それぞれ列3及び4)、次いで7% Tris−アセテートゲル(Invitrogen)上で移動させ、R8B12抗体で染色した。各列は約50ngのFVIIIを含有する。
【0037】
図8.PEG化された野生型全長FVIII(KG−2)のトロンビン切断。“S”=出発KG−2材料。“R”=還元されたKG−2及び除去された還元剤。“P”=43kD PEGを用いてPEG化された“R”。“Pure”=過剰のPEGから精製された“P”。“L”=軽鎖。PEG化されたバンドを点により強調する。
【0038】
図9.PEG化されたPEG2のヨウ素染色。22又は43kD PEG化PEG2を6%Trisグリシンゲル上で移動させ、R8B12 FVIII抗体(列1及び2)又はヨウ素(列3及び4)を用いて染色した。2つの染色をそれらの分子量マーカーの列に従って整列させた。列1及び2はそれぞれ約30ngのFVIIIを含有するが、列3及び4は約2μgを含有する。
【0039】
図10.PEG化及び非PEG化PEG2のMALDI質量分析。MALDI質量分析はPEG2(図10a)又は22kD PEG化PEG2(図10b)について行なわれた。PEG化されると、PEG2の重(H)鎖ピークは非常に減少し、111kD(22kD PEG+89kD 重鎖)を中心とする新しいピーク(H+PEG)が現れる。100kD(22kD PEG+83kD 軽鎖)を中心とすることが予測されるPEG化された軽(L)鎖のピークは検出されない。
【0040】
図11.トロンビン切断の後のPEG化及び非PEG化PEG2のMALDI質量分析。
【0041】
図12.トロンビン切断の前後のPEG化PEG6のMALDI質量分析。
【0042】
図13.サイズ−排除カラム上で精製されたPEG化PEG2の280nmにおけるUV吸収分布。
【0043】
図14.カチオン交換カラム上で精製されたPEG化及び非PEG化PEG6の280nmにおけるUV吸収分布。
【0044】
図15.サイズ−排除カラム上で精製されたPEG化及び非PEG化PEG6の280nmにおけるUV吸収分布。
【0045】
図16.発色アッセイ及び凝固アッセイにより測定される場合のPEG化タンパク質の活性を非PEG化タンパク質の活性と比較する。精製された全長FVIIIをKG−2と示す。報告されるパーセント活性は、還元及び還元剤の除去後にPEGで処理された試料の値を、PEG化の収率を考慮しながら緩衝液標準で処理された試料の値で割ることにより決定された。
【0046】
図17.PEG2と比較されるPEG化PEG2のウサギPK研究。
【0047】
図18.BDD及びPEG2と比較されるPEG化PEG2のウサギPK研究。P−値はPEG化PEGとBDDの間の比較である。
【0048】
図19.BDD及びPEG6と比較されるPEG化PEG6のウサギPK研究。
【0049】
図20.非変更全長FVIIIと比較されるPEG化野生型全長(“fl”)FVIIIのウサギPK研究。
【0050】
図21.PEG6及びBDDと比較されるPEG化PEG6の血友病マウスPK研究。
【0051】
図22.BDDと比較される22及び43kD PEG化PEG2の正常マウスPK研究。
【0052】
図23.時間経過中ずっと(full time course)、BDDと比較される22kD PEG化PEG2の正常マウスPK研究。
【0053】
図24.血友病マウスアッセイにおけるBDD因子VIIIの2つの種の半減期の薬物動態学的(PK)評価を描く、血友病マウス(BDD)因子VIII回収率ヒストグラム。
【0054】
図25.BDDと比較される22kD PEG化PEG2の血友病マウス腎臓裂傷研究。ビヒクル処置されたマウスは体重のグラム当たり25uLの血液損失を有する。
【0055】
図26.増加する量のFVIII抗体の存在下におけるPEG化PEG2及びBDDの発色活性。抗体エピトープを括弧内に示す。
【0056】
図27.増加する量のFVIII mAB 413抗体の存在下におけるPEG化PEGの発色活性。
【0057】
図28.FVIIIへの阻害物質を発現した患者に由来するヒト血漿の存在下におけるBDD、43kD PEG化PEG2、33kD PEG化PEG6及び33kD ジPEG化PEG2+6の発色活性。阻害物質の力価及び血液収集の日付を上に記す。上の2つのパネルは、5−〜405−倍の患者血漿希釈において集められたデータを含む。下の左のパネルは、患者HRF−828血漿に関する1:15−倍希釈に焦点を当てている。下の右のパネルは、上の2つのパネルにおいて各FVIII試料のために用いられた0.064IU/mLが飽和用量でなかったことを確証する。
【0058】
図29.PEG化スクリーニング法及び正当化。上のパネルは、一過的に発現されるPEG突然変異タンパク質のPEG化スクリーニングの略図を示す。下のパネルは、重鎖(“H”)−特異的抗体(左)又は軽−鎖(“L”)特異的抗体(右)を用いるPEG化生成物のウェスタン分析を示す。PEG化されたバンドを点により強調する。“U”はH及びLの両方を含有するプロセシングされない材料である。
【0059】
図30.PEG15〜17のPEG化スクリーニング。重鎖(“H”)−特異的抗体(R8B12及び58.12)又は軽−鎖(“L”)特異的抗体(C7F7及びGM)を用いるPEG化生成物のウェスタン分析。3つのすべての突然変異タンパク質は重鎖に関して選択的であり、PEG15−PEG16>PEG17の相対的なPEG化効率を有する。PEG化されたバンドを点により強調する。“U”はH及びLの両方を含有するプロセ
シングされない材料である。
【0060】
図31.還元剤濃度の関数としてPEG2+14のPEG化を示すゲル。PEG2+14を4℃において30分間、67〜670uMのTCEPを用いて処理した。スピン−カラム(spin−column)により還元剤を除去し、続いて12kD PEGを用いてPEG化した。FVIIIの重及び軽鎖をそれぞれ“H”及び“L”により強調する。2つの点はPEG化された重及び軽鎖を指し示す。
【0061】
図32.67〜670uMのTCEPで処理され、続いて還元剤が除去されたPEG2+14のデコンボリューションされた質量スペクトル。
【発明を実施するための形態】
【0062】
発明の詳細な記述
本発明は、FVIII活性を有するポリペプチドをあらかじめ規定される部位において生物適合性ポリマーに共有結合させることができ、それはN−末端アミンにおいてではなく、且つそのようなポリペプチドは実質的にそれらの凝固剤活性を保持しているという発見に基づく。さらに、これらのポリペプチド複合体は増加した循環時間及び低下した抗原性を有する。本発明の複合体は、FVIIIへの無作為ポリマー結合又はN−末端における結合を有した先行技術の複合体を超えて有利である。特定部位の結合は、生物学的活性のために必要な領域を避ける変更を設計し、それにより実質的なFVIII活性を保持することを可能にする。それは、ポリマーを結合させてFVIIIクリアランスに含まれる部位において結合を遮断するように設計することも可能にする。特定部位の結合は、無作為なポリマーカップリングによる技術において生産される不均一な複合体ではなくて均一な生成物も可能にする。軽鎖のN−末端アミンにおける結合を避けることにより、本発明の複合体は、FVIIIポリペプチドの活性部位におけるリガンドの結合から可能な活性の損失を避ける。軽鎖のN−末端領域は、vWF因子のFVIIIへの会合に含まれると思われ、それは循環における安定化会合である。
【0063】
定義
生物適合性ポリマー。生物適合性ポリマーにはポリアルキレンオキシド、例えばこれらに限られないがポリエチレングリコール(PEG)、デキストラン、コロミン酸又は他の炭水化物に基づくポリマー、アミノ酸のポリマー、ビオチン誘導体、ポリビニルアルコール(PVA)、ポリカルボキシレート、ポリビニルピロリドン、ポリエチレン−コ−無水マレイン酸、ポリスチレン−コ−無水リンゴ酸、ポリオキサゾリン、ポリアクリロイルモルホリン、ヘパリン、アルブミン、セルロース、キトサンの加水分解産物、デンプン、例えばヒドロキシエチル−デンプン及びヒドロキシプロピル−デンプン、グリコゲン、アガロース及びその誘導体、グアゴム、プルラン、イヌリン、キサンタンゴム、カラゲナン、ペクチン、アルギン酸加水分解産物、他の生体高分子ならびにそれらの同等物が含まれる。好ましいのはポリエチレングリコールであり、さらにもっと好ましくはメトキシポリエチレングリコール(mPEG)である。他の有用なポリアルキレングリコール化合物はポリプロピレングリコール(PPG)、ポリブチレングリコール(PBG)、PEG−グリシジルエーテル(Epox−PEG)、PEG−オキシカルボニルイミダゾール(CDI−PEG)、分枝鎖状ポリエチレングリコール、直鎖状ポリエチレングリコール、フォーク状ポリエチレングリコール(forked polyethylene glycols)及び多−分枝もしくは「超分枝鎖状」ポリエチレングリコール(star−PEG)である。
【0064】
ポリエチレングリコール(PEG)。本明細書で用いられる「PEG」及び「ポリエチレングリコール」は互換性であり、いずれの水溶性ポリ(エチレンオキシド)も含む。典型的には、本発明に従う使用のためのPEGsは以下の構造、「−(OCH2CH2)n
−」を含み、ここで(n)は2〜4000である。本明細書で用いられる場合、PEGは、末端酸素が置き換えられているかもしくはいないかに依存して、「−CH2CH2−O(CH2CH2O)n−CH2CH2−」及び「−(OCH2CH2)nO−」も含む。明細書及び請求項全体を通じて、「PEG」という用語は種々の末端もしくは「末端キャッピング」基、例えば制限ではないがヒドロキシル又はC1−20アルコキシ基を有する構造を含むことを思い出さねばならない。「PEG」という用語は、大部分、すなわち50%より多くの−OCH2CH2−繰り返しサブユニットを含有するポリマーも意味する。特定の形態に関し、PEGは、多様な分子量ならびに構造又は形状、例えば分枝鎖状、直鎖状、フォーク状及び多官能基性のいくつを帯びることもできる。
【0065】
PEG化。PEG化は、ポリエチレングリコール(PEG)がタンパク質のような分子に共有結合する過程である。
【0066】
活性化もしくは活性官能基。生物適合性ポリマーのような官能基が活性化されたと記述される場合、官能基は他の分子上の求電子基(electrophile)又は求核基(nucloephile)と容易に反応する。
【0067】
Bドメイン欠失FVIII(BDD)。本明細書で用いられる場合、BDDは、FVIIIのB−ドメインの14個のアミノ酸以外のすべての欠失を含有するアミノ酸配列を有することを特徴とする。B−ドメインの最初の4個のアミノ酸(SFSQ,配列番号:1)はB−ドメインの最後の10個の残基(NPPVLKRHQR,配列番号:2)に連結している。(Lind,P.et al.著,Eur.J.Biochem.232,1995年,pp.19−27)。本明細書で用いられるBDDは配列番号:3のアミノ酸配列を有する。
【0068】
FVIII。血液凝固因子VIII(FVIII)は、肝臓により合成されて血流中に放出される糖タンパク質である。循環血中で、それはフォンビルブラント因子(vWF、因子VIII−関連抗原としても既知)に結合して、安定な複合体を形成する。トロンビンにより活性化されると、それは複合体から解離し、凝固カスケードにおいて他の凝固因子と相互作用し、それは結局血栓の形成を生ずる。ヒト全長FVIIIは配列番号:4のアミノ酸配列を有するが、対立遺伝子変異体が可能である。
【0069】
機能性因子VIIIポリペプチド。本明細書で用いられる場合、機能性因子VIIIポリペプチドは、生体内もしくは試験管内で、例えば血友病Aにより特徴付けられるヒト因子VIII欠失を修正することができる機能性ポリペプチド又はポリペプチドの組み合わせを示す。因子VIIIは自然の状態で複数の分解もしくはプロセシング形態を有する。これらは本明細書で示される通り、1個の鎖状タンパク質(one chain protein)である前駆体からタンパク質分解的に誘導される。機能性因子VIIIポリペプチドはそのような1個の鎖状タンパク質を含み、且つヒト因子VIII欠失を修正する生物学的活性を有するこれらの種々の分解産物も示す。対立遺伝子変異がおそらく存在する。機能性因子VIIIポリペプチドは、因子VIIIの誘導体を生ずるそのような対立遺伝子変異、グリコシル化形、変更及びフラグメントのすべてを、それらがヒト因子VIIIの機能性セグメントを含有し、必須の特徴的なヒト因子VIII機能的活性が本質的に留まっている限り含む。必要な機能的活性を保有する因子VIIIの誘導体は、本明細書に記載される直接的な試験管内試験により容易に同定され得る。さらに、機能性因子VIIIポリペプチドは、因子IXa、カルシウム及びリン脂質の存在下で因子XのXaへの転換を触媒することができ、ならびに血友病Aに冒された患者に由来する血漿における凝固欠陥を修正することができる。本明細書におけるヒト因子VIIIアミノ酸配列及び機能性領域の配列の開示から、DNAの制限酵素切断を介して、あるいはヒト因子VIIIタンパク質のタンパク質分解的もしくは他の分解を介して誘導され得るフラグメントは
、当該技術分野における熟練者に明らかになるであろう。
【0070】
FIX。本明細書で用いられる場合、FIXは凝固因子IXを意味し、それはヒト凝固因子IX又は血漿トロンボプラスチン成分としても既知である。
【0071】
FX。本明細書で用いられる場合、FXは凝固因子Xを意味し、それはヒト凝固因子Xの名前及び名祖(eponym)Stuart−Prower因子によっても既知である。
【0072】
薬物動態学。「薬物動態学」(「PK」)は、体内における薬剤の吸収、分布、代謝及び除去の性質を記述するために用いられる用語である。薬剤の薬物動態学の向上は、薬剤を生体内で治療薬としてより有効にする特性、特に体内におけるその有用な持続時間における向上を意味する。
【0073】
突然変異タンパク質。突然変異タンパク質は、タンパク質又はポリペプチドへの実験室誘導突然変異の結果として生ずる遺伝子操作されたタンパク質である。
【0074】
タンパク質。本明細書で用いられる場合、タンパク質とポリペプチドは同義語である。
【0075】
FVIIIクリアランス受容体。本明細書で用いられるFVIIIクリアランス受容体は、1種もしくはそれより多い他の分子と結合又は会合して循環からのFVIIIクリアランスを生ずる機能性FVIIIポリペプチド上の受容体領域を意味する。因子VIIIクリアランス受容体には、制限ではないがLRP、LDL受容体及び/又はHSPGに結合するFVIII分子の領域が含まれる。
【0076】
議論
本発明の方法に従い、いずれの機能性因子VIIIポリペプチドもあらかじめ決められた部位で突然変異させ、次いでその部位において生物適合性ポリマーに共有結合させることができると考えられる。有用なポリペプチドには、配列番号:4に示されるアミノ酸配列を有する全長因子VIII及び配列番号:3に示されるアミノ酸配列を有するBDD FVIIIが含まれるが、これらに限られない。好ましいのはBDD FVIIIである。
【0077】
本発明の複合体において用いられる生物適合性ポリマーは、上記のポリマーのいずれであることもできる。生物適合性ポリマーは、薬物動態学における所望の向上を与えるように選ばれる。例えばポリマーの正体、サイズ及び構造は、活性における許容され得ない低下なしでFVIII活性を有するポリペプチドの循環半減期を向上させるように、あるいはポリペプチドの抗原性を低下させるように選ばれる。好ましくは、ポリマーはPEGを含み、さらにもっと好ましくはその分子量の少なくとも50%をPEGとして有する。1つの態様において、ポリマーは末端−キャッピング部分、例えばヒドロキシル、アルコキシ、置換アルコキシ、アルケノキシ、置換アルケノキシ、アルキノキシ、置換アルキノキシ、アリールオキシ及び置換アリールオキシで末端がキャッピングされたポリエチレングリコールである。さらにもっと好ましいのは、メトキシポリエチレングリコールを含んでなるポリマーである。さらにもっと好ましいのは、3kD〜100kD、そしてより好ましくは5kD〜64kD又は5kD〜43kDのサイズ範囲を有するメトキシポリエチレングリコールを含んでなるポリマーである。
【0078】
好ましくは、ポリマーは反応性部分を有する。例えば1つの態様において、ポリマーはスルフヒドリル反応性部分を有し、それは機能性因子VIIIポリペプチド上の遊離のシステインと反応して共有結合を形成することができる。そのようなスルフヒドリル反応性
部分にはチオール、トリフレート、トレシレート、アジリジン、オキシラン、S−ピリジル又はマレイミド部分が含まれる。好ましいのはマレイミド部分である。1つの態様において、ポリマーは直鎖状であり、一方の末端にスルフヒドリルに対して強く反応性でない「キャップ」(例えばメトキシ)を、且つ他方の末端にスルフヒドリル反応性部分を有する。好ましい態様において、複合体はPEG−マレイミドを含み、5kD〜64kDのサイズ範囲を有する。
【0079】
有用な生物適合性ポリマーの選択のためのさらなる指針は、下記の実施例に示される。
【0080】
FVIII活性を有するポリペプチドをコードするヌクレオチド配列の特定部位の突然変異は、当該技術分野において既知のいずれの方法によっても起こり得る。好ましい方法は、ポリマーの共有結合のために選ばれる部位にシステインコドンを導入するための突然変異誘発を含む。これは、商業的に入手可能な特定部位の突然変異誘発キット、例えばStratagene cQuickChangeTM II特定部位の突然変異誘発キット、Clontech Transformer特定部位の突然変異誘発キット no.K1600−1、Invitrogen GenTaylor特定部位の突然変異誘発システム no.12397014、Promega Altered Sites II
試験管内突然変異誘発システムキット no.Q6210又はTakara Mirus Bio LA PCR突然変異誘発キット no.TAK RR016を用いて行なうことができる。
【0081】
最初に機能性FVIIIポリペプチドの表面上の1個もしくはそれより多いアミノ酸に関するコドンをシステインに関するコドンで置換して組換え発現系においてシステイン突然変異タンパク質を生産し、突然変異タンパク質をシステイン−特異的ポリマー試薬と反応させ、突然変異タンパク質を精製することにより、本発明の複合体を調製することができる。
【0082】
この系において、システイン部位におけるポリマーの付加は、ポリマー上のマレイミド活性官能基を介して行なうことができる。この方法の例を下記に示す。用いられるスルフヒドリル反応性ポリマーの量は、誘導体化されるべきシステインのモル量に少なくとも等モル量でなければならず、好ましくは過剰に存在する。好ましくは、少なくとも5−倍モル過剰のスルフヒドリル反応性ポリマーが用いられ、さらにもっと好ましくは、少なくとも10−倍過剰のそのようなポリマーが用いられる。共有結合に有用な他の条件は、当該技術分野における者の熟練の範囲内である。
【0083】
下記の実施例において、突然変異タンパク質は当該技術分野において通常の方法で命名される。突然変異体の命名のための約定(convention)は、配列番号:4に示される成熟全長因子VIIIに関するアミノ酸配列に基づく。分泌タンパク質として、FVIIIは翻訳過程の間にタンパク質分解により切断されるシグナル配列を含有する。19個のアミノ酸のシグナル配列の除去に続き、分泌されるFVIII生成物の最初のアミノ酸はアラニンである。
【0084】
通常であり且つ本明細書で用いられる通り、BDD FVIII中の突然変異アミノ酸に言及する場合、突然変異アミノ酸は全長FVIIIの配列中のその位置により称される。例えば下記で議論されるPEG6突然変異タンパク質はK1808Cと称され、それは、それが全長配列中の1808に類似の位置においてリシン(K)がシステイン(C)に変更されているからである。
【0085】
ポリマーの共有結合のためにあらかじめ規定される部位は、FVIII活性に含まれないか、又は生体内でFVIIIを安定化する他の機構、例えばvWFへの結合に含まれな
い、ポリペプチドの表面上に露出されている部位から最適に選ばれる。そのような部位は、FVIIIが不活性化されるか、又は循環からクリアランスされる機構に含まれることが知られている部位からも最適に選ばれる。これらの部位の選択は、下記で詳細に議論される。好ましい部位には、(a)低密度リポタンパク質受容体関連タンパク質、(b)ヘパリン硫酸プロテオグリカン、(c)低密度リポタンパク質受容体及び/又は(d)因子VIII阻害性抗体に関する結合部位内もしくはそれらの近辺のアミノ酸残基が含まれる。「結合部位内もしくはそれらの近辺」により、結合部位に十分に近く、部位への生物適合性ポリマーの共有結合が結合部位の立体障害を生ずるであろう残基を意味する。そのような部位は、例えば結合部位の20Å以内であると思われる。
【0086】
本発明の1つの態様において、生物適合性ポリマーは(a)上記で定義された因子VIIIクリアランス受容体、(b)因子VIIIを分解することができるプロテアーゼのための結合部位及び/又は(c)因子VIII阻害性抗体のための結合部位内又はそれらの近辺のアミノ酸残基において機能性因子VIIIポリペプチドに共有結合する。プロテアーゼは活性化タンパク質C(APC)であることができる。他の態様において、生物適合性ポリマーは機能性因子VIIIポリペプチド上のあらかじめ規定される部位において共有結合し、ポリペプチドへの低−密度リポタンパク質受容体関連タンパク質の結合は、ポリペプチドが複合されない場合のそれへの結合より少なく、好ましくは2分の1より少ない。1つの態様において、生物適合性ポリマーは機能性因子VIIIポリペプチド上のあらかじめ規定される部位において共有結合し、ヘパリン硫酸プロテオグリカンのポリペプチドへの結合は、ポリペプチドが複合されない場合のそれへの結合より少なく、好ましくは2分の1より少ない。さらに別の態様において、生物適合性ポリマーは機能性因子VIIIポリペプチド上のあらかじめ規定される部位において共有結合し、因子VIII阻害性抗体のポリペプチドへの結合は、ポリペプチドが複合されない場合のそれへの結合より少なく、好ましくはポリペプチドが複合されない場合のそれへの結合の2分の1より少ない。別の態様において、生物適合性ポリマーは機能性因子VIIIポリペプチド上のあらかじめ規定される部位において共有結合し、低密度リポタンパク質受容体のポリペプチドへの結合は、ポリペプチドが複合されない場合のそれへの結合より少なく、好ましくは2分の1より少ない。別の態様において、生物適合性ポリマーは機能性因子VIIIポリペプチド上のあらかじめ規定される部位において共有結合し、血漿プロテアーゼがポリペプチドを分解する程度は、ポリペプチドが複合されない場合より低い。さらに別の態様において、血漿プロテアーゼによるポリペプチドの分解は、同じ条件下で同じ時間に及んで測定される場合に、ポリペプチドが複合されない場合のその分解の2分の1より少ない。
【0087】
FVIIIに関するLRP、LDL受容体又はHSPG結合親和性は、表面プラスモン共鳴法(Biacore)を用いて決定することができる。例えばFVIIIを直接又はFVIII抗体を介して間接的にBlacoreTMチップにコーティングし、種々の濃度のLRPをチップ上に通過させて相互作用の開始速度(on−rate)及び終了速度(off−rate)の両方を測定することができる(Bovenschen N.et
al.著,J.Biol.Chem.278(11),2003年,pp.9370−7)。2つの速度の比率は親和性の尺度を与える。PEG化した場合の2分の1、好ましくは5分の1、より好ましくは10分の1、そしてさらにもっと好ましくは30分の1への親和性における低下が望ましい。
【0088】
プロテアーゼAPCによるFVIIIの分解は、当該技術分野における熟練者に既知のいずれの方法によっても測定することができる。
【0089】
1つの態様において、生物適合性ポリマーは因子VIIIアミノ酸位置81、129、377、378、468、487、491、504、556、570、711、1648、1795、1796、1803、1804、1808、1810、1864、1903
、1911、2091、2118及び2284の1個もしくはそれより多くにおいてポリペプチドに共有結合する。別の態様において、生物適合性ポリマーは因子VIIIアミノ酸位置377、378、468、491、504、556、1795、1796、1803、1804、1808、1810、1864、1903、1911及び2284の1個もしくはそれより多くにおいてポリペプチドに共有結合し、且つ(1)低−密度リポタンパク質受容体関連タンパク質への複合体の結合は低−密度リポタンパク質受容体関連タンパク質への非複合ポリペプチドの結合より少ないか;(2)低−密度リポタンパク質受容体への複合体の結合は低−密度リポタンパク質受容体への非複合ポリペプチドの結合より少ないか;あるいは(3)低−密度リポタンパク質受容体関連タンパク質及び低−密度リポタンパク質受容体の両方への複合体の結合は低−密度リポタンパク質受容体関連タンパク質及び低−密度リポタンパク質受容体への非複合ポリペプチドの結合より少ない。
【0090】
さらに別の態様において、生物適合性ポリマーは因子VIIIアミノ酸位置377、378、468、491、504、556及び711の1個もしくはそれより多くにおいてポリペプチドに共有結合し、且つヘパリン硫酸プロテオグリカンへの複合体の結合はヘパリン硫酸プロテオグリカンへの非複合ポリペプチドの結合より少ない。さらに別の態様において、生物適合性ポリマーは因子VIIIアミノ酸位置81、129、377、378、468、487、491、504、556、570、711、1648、1795、1796、1803、1804、1808、1810、1864、1903、1911、2091、2118及び2284の1個もしくはそれより多くにおいてポリペプチドに共有結合し、且つ複合体は非複合ポリペプチドより少ない因子VIII阻害性抗体への結合を有する。さらに別の態様において、生物適合性ポリマーは因子VIIIアミノ酸位置81、129、377、378、468、487、491、504、556、570、711、1648、1795、1796、1803、1804、1808、1810、1864、1903、1911、2091、2118及び2284の1個もしくはそれより多くにおいて、そして好ましくは位置377、378、468、491、504、556及び711の1個もしくはそれより多くにおいてポリペプチドに共有結合し、且つ複合体は、非複合ポリペプチドが有するより少ない因子VIIIを分解できる血漿プロテアーゼからの分解を有する。より好ましくは、血漿プロテアーゼは活性化タンパク質Cである。
【0091】
さらに別の態様において、生物適合性ポリマーはアミノ酸位置129、491、1804及び/又は1808において、より好ましくは491又は1808においてB−ドメイン欠失因子VIIIに共有結合する。さらに別の態様において、生物適合性ポリマーは因子VIIIアミノ酸位置1804においてポリペプチドに結合し、且つポリエチレングリコールを含む。好ましくは、生物適合性ポリマー結合のための1個もしくはそれより多いあらかじめ規定される部位は、部位特異的システイン突然変異により制御される。
【0092】
機能性因子VIIIポリペプチド上の1個もしくはそれより多い部位、好ましくは1もしくは2個の部位がポリマー結合のためにあらかじめ規定される部位であることができる。特定の態様において、ポリペプチドはモノ−PEG化又はジPEG化される。
【0093】
本発明は、機能性因子VIIIポリペプチドをコードするヌクレオチド配列を突然変異させてあらかじめ規定される部位においてシステイン残基に関するコード配列に置換し;突然変異したヌクレオチド配列を発現させてシステイン増強突然変異タンパク質を生産し;突然変異タンパク質を精製し;実質的に還元システイン残基のみにおいてポリペプチドと反応するように活性化された生物適合性ポリマーと突然変異タンパク質を反応させて複合体を形成し;そして複合体を精製することを含んでなる、複合体の調製方法にも関する。他の態様において、本発明は:(a)特定部位の因子VIII突然変異タンパク質を発現させ、ここで突然変異タンパク質は因子VIII突然変異タンパク質の露出された表面上のアミノ酸残基に代わるシステイン置換を有し、且つそのシステインはキャッピングさ
れており;(b)システイン突然変異タンパク質を穏やかに還元し、且つキャップを放出する条件下でシステイン突然変異タンパク質を還元剤と接触させ;(c)システイン突然変異タンパク質からキャップ及び還元剤を除去し;そして(d)還元剤の除去から少なくとも約5分、そして好ましくは少なくとも15分、さらにもっと好ましくは少なくとも30分後に、PEG化された因子VIII突然変異タンパク質が生産されるような条件下で、スルフヒドリルカップリング部分を含んでなるPEGでシステイン突然変異タンパク質を処理することを含んでなる、因子VIII突然変異タンパク質の特定部位のPEG化のための方法を提供する。PEGのスルフヒドリルカップリング部分は、チオール、トリフレート、トレシレート、アジリジン、オキシラン、S−ピリジル及びマレイミド部分より成る群から選ばれ、好ましくはマレイミドである。
【0094】
本発明は、本発明の複合体の治療的に有効な量及び製薬学的に許容され得る添加剤を含んで成る非経口的投与のための製薬学的組成物にも関する。製薬学的に許容され得る添加剤は、調製物の調製を助けるか、又は安定化するために活性成分に加えることができ、且つ患者に有意な毒物学的悪影響を起こさない物質である。そのような添加剤の例は当該技術分野における熟練者に周知であり、水、糖類、例えばマルトースもしくはスクロース、アルブミン、塩類などが含まれる。他の添加剤は、例えばE.W.MartinによるRemington’s Pharmaceutical Sciencesに記載されている。そのような組成物は本明細書の複合体の有効量を、宿主への有効な投与に適した製薬学的に許容され得る組成物を調製するために適した量のビヒクルと一緒に含有するであろう。例えば血友病Aに苦しむ患者に、出血事例の重度とともに変わり得る投薬量で非経口的に複合体を投与することができる。静脈内に投与される平均投薬量は、術前適用のためにキログラム当たり40単位、少ない出血のためにキログラム当たり15〜20単位及び保持投薬のためにキログラム当たり8−時間に及んで投与される20〜40単位の範囲内である。
【0095】
1つの態様において、本発明の方法は1個もしくはそれより多い表面BDDアミノ酸をシステインで置換し、哺乳類発現系においてシステイン突然変異タンパク質を生産し、生育培地からのシステインにより発現の間にキャッピングされたシステインを還元し、還元剤を除去してBDDジスルフィドを再生させ、システイン−特異的な生物適合性ポリマー試薬、例えばPEG−マレイミドと反応させることを含む。そのような試薬の例は、Nektar Therapeutics of San Carios,CAからそれぞれNektarカタログ番号2D2M0H01 mPEG−MAL分子量5,000Da、2D2M0P01 mPEG−MAL分子量20kD、2D3X0P01 mPEG2−MAL分子量40kDの下に入手可能な5、22又は43kDあるいはNOF Corporation,Tokyo,JapanからそれぞれNOFカタログ番号Sunbright ME−120MA及びSunbright ME−300MAの下に入手可能な12又は33kDのようなPEGサイズを有するPEG−マレイミドである。PEG化生成物は、未反応PEGの除去のためにイオン−交換クロマトグラフィーを用いて、及び未反応BDDの除去のためにサイズ−排除クロマトグラフィーを用いて精製される。この方法を用いてFVIIIとの望ましくない相互作用、例えば受容体−媒介クリアランス、阻害性抗体結合及びタンパク質分解酵素による分解を同定し、且つ選択的に遮蔽することができる。我々は、Nektar又はNOFにより5kDとして供給されるPEG試薬が我々の実験室で6kDとして判断され(tested)、類似して我々の実験室では直鎖状20kDとして供給されるPEG試薬が22kDとして判断され、40kDとして供給されるものが43kDとして判断され、60kDとして供給されるものが64kDとして判断されたことに注意した。混乱を避けるために、我々は本明細書の議論において、5kD
PEGを除いて我々の実験室で判断された通りの分子量を用い、5kD PEGは製造者がそれを同定した通りに5kDとして報告する。
【0096】
BDDの位置491及び1808におけるシステイン突然変異(上記で開示された)の他に、位置487、496、504、468、1810、1812、1813、1815、1795、1796、1803及び1804をシステインに突然変異させ、PEG化された時におそらくLRP結合の遮断を可能にした。位置377、378及び556もシステインに突然変異させ、PEG化された時にLRP及びHSPG結合の両方の遮断を可能にした。位置81、129、422、523、570、1864、1911、2091及び2284はBDD上で等しく空間的に隔たるように選ばれ、これらの位置における大きなPEGs(>40kD)を用いる特定部位のPEG化は本来のグリコシル化部位(41、239及び2118)及びLRP結合部位におけるPEG化と一緒になってBDDの表面を完全に覆い、BDDに関する新規なクリアランス機構を同定するはずである。
【0097】
1つの態様において、細胞培養培地はシステインを含有し、それはジスルフィド結合の形成により突然変異タンパク質のシステイン残基を「キャッピング」する。複合体の調製において、組換え系で生産されるシステイン突然変異タンパク質は培地からのシステインでキャッピングされ、このキャップはシステイン−特異的ポリマー試薬の添加の前に、キャップを放出する穏やかな還元により除去される。当該技術分野における熟練者に明らかな通り、FVIIIの部位−特異的突然変異のための当該技術において既知の他の方法も用いることができる。
【実施例】
【0098】
FVIIIの構造活性関連性分析。FVIII及びBDD FVIIIは、生物学的反応に含まれる多種の部位を有する非常に大きな複合(complex)分子である。薬物動態学的性質を向上させるためにそれらを共有結合により変更する以前の試みは、混同した結果を有した。分子を特異的に突然変異させ、次いで部位−特異的なやり方でポリマーを加えることができることは驚くべきことである。さらに、非特異的な付加及び活性の低下を引き起こす過去のポリマー複合体の場合の問題を考えると、向上した薬物動態学的性質及び保持される活性の結果も驚くべきことである。
【0099】
1つの態様において本発明は、PEG−マレイミドのようなシステイン−特異的リガンドを用いる特定部位の突然変異誘発に関する。非−突然変異BDDはPEG−マレイミドと反応するために利用できるシステインを有しておらず、従って突然変異システイン位置のみがPEG化の部位となるであろう。さらに特定的に、BDD FVIIIは19個のシステインを有し、その中の16個はジスルフィドを形成し、その中の他の3個は遊離のシステインである(McMullen et al.著,Protein Sci.4,1995年,pp.740−746)。BDDの構造モデルは、3個の遊離のシステインのすべてが埋もれていることを示唆している(Stoliova−McPhie et al.著,Blood 99,2002年,pp.1215−1223)。酸化されたシステインはPEG−マレイミドによりPEG化され得ないので、BDD中でジスルフィドを形成する16個のシステインは、最初に還元されずにはPEG化され得ない。BDDの構造モデルに基づき、BDD中の3個の遊離のシステインは、最初にタンパク質を変性させ、これらのシステインをPEG試薬に露出せずにはPEG化され得ない。かくしてBDD構造を劇的に変えることなく、本来のシステイン残基におけるPEG化によりBDDの特異的なPEG化を達成することは実行可能であると思われず、BDDの構造を劇的に変えることは、おそらくその機能を破壊するであろう。
【0100】
全長FVIIIのBドメイン中の4個のシステインのレドックス状態は未知である。Bドメイン中の4個のシステインのPEG化は、それらがジスルフィドを形成しておらず、表面に露出されていれば可能であり得る。しかしながら、全長FVIII及びBDDは類似の薬物動態学的(PK)側面及び生体内における類似の半減期を有するので(Gruppo et al.著,Haemophilia 9,2003年,pp.251−26
0)、BドメインPEG化は、PEGが偶然非−Bドメイン領域をも保護しなければ、向上した血漿半減期を生ずるとは思われない。
【0101】
FVIII活性を有するポリペプチド上のポリマー結合のためのあらかじめ規定される部位であって、因子VIII活性を保持し、且つ薬物動態学を向上させる部位を決定するために、BDD FVIIIに基づいて以下の指針を示す。変更はクリアランス、不活性化及び免疫原性機構、例えばLRP、HSPG、APCならびに阻害性抗体結合部位を標的としなければならない。Stoilova−McPhie,S.et al.著,Blood 99(4),2002年,pp.1215−23はBDDの構造を示している。例えば半減期を延長するために、1個のPEGをA2残基484−509及びA3残基1811−1818中のLRP結合部位もしくはその近辺における特異的な部位に導入することができる。これらの部位における嵩高いPEGの導入は、FVIIIがLRPに結合する能力を崩壊させ、循環からのFVIIIのクリアランスを減少させるはずである。活性に有意に影響せずに半減期を延長するために、PEGを残基1648において導入することができるとも思われ、それは全長分子中のBドメインとA3ドメインの連結部分にあり、BDDではA2及びA3ドメインの間の14−アミノ酸中にある(in the 14−amino acid liker I the BDD between the
A2 and A3 domains)。
【0102】
PEG化の特異性は、組換えDNA突然変異誘発法を用いて1個のシステイン残基をA2又はA3ドメイン中に工作し(engineering)、続いて導入されたシステインをPEG−マレイミドのようなシステイン−特異的PEG試薬を用いて部位−特異的にPEG化することにより達成され得る。484−509及び1811−1818におけるPEG化の他の利点は、これらの2つのエピトープが患者における阻害性抗原部位の3つの主な種類の2つに相当することである。循環半減期の向上及び免疫原性反応の低下の最大の効果を達成するために、A2及びA3 LRP結合部位の両方をPEG化し、ジPEG化生成物を与えることができる。1811−1818領域内におけるPEG化は、この領域がFIX結合にも含まれるために、活性の有意な損失を生じ得ることに注意しなければならない。558−565内における特定部位のPEG化はHSPG結合を廃止するが、この領域はFIXにも結合するので、活性も低下させ得る。
【0103】
FVIIIの新規なクリアランス機構を同定するために、さらに別の表面部位をPEG化することができる。A2ドメインのPEG化は、活性化されるとA2ドメインがFVIIIから解離し、おそらくそのより小さいサイズの故にFVIII分子の残りの部分より速く循環から除去される点で追加の利点を与えることができる。他方、PEG化されたA2は、腎臓クリアランスを逃れるのに十分に大きく、FVIIIの残りの部分と同等の血漿半減期を有することができ、かくして生体内で活性FVIIIを再構築することができる。
【0104】
A2及びA3領域内のPEG化部位の同定。推定A2 LRP結合領域もしくはその近辺における5個の位置(PEG1−5位置に対応するY487、L491、K496、L504及びQ468)を、高い表面露出及びそれらのCαからCβへの軌道(trajectory)の外側への向きに基づき、特定部位のPEG化のための例として選んだ。さらに、これらの残基は分子の三−次元構造において互いから大体同じ距離にあり、それらは一緒になってこの領域全体を表す(represent)ことができる。推定A3 LRP結合領域もしくはその近辺における8個の位置(PEG6−14に対応する1808、1810、1812、1813、1815、1795、1796、1803、1804)を特定部位のPEG化のための例として選んだ。PEG6(K1808)は1811−1818及び1810における自然のN−結合グリコシル化部位に隣接している。位置1810(PEG7)におけるPEG化は糖をPEGで置換するであろう。PEG8位置T
1812における突然変異もグリコシル化部位を廃止するであろう。PEG9位置(K1813)は内側に向いていると予測されたが、構造モデルが正しくない場合にそれは選ばれた。PEG10(Y1815)はLRP結合ループ内の嵩高い疎水性アミノ酸であり、疎水性アミノ酸は典型的にはタンパク質−タンパク質相互作用の中心に存在するので、決定的な相互作用残基であり得る。1811−1818領域はLRP及びFIX結合の両方に含まれると報告されているので、このループ内におけるPEG化はおそらく活性の低下を生ずると考えられた。かくしてPEG11−PEG14(1795、1796、1803、1804)は1811−1818ループ近辺にあるがループ内にないように設計され、種々のPEGサイズを用いてLRP及びFIX結合を解離させる(dissociate)ことができるようにした。
【0105】
両LRP結合部位を同時に遮断するために、例えばPEG2及びPEG6位置における二重PEG化を発生させることができる。
【0106】
558−565領域はHSPG及びFIXの両方に結合することが示されているので、この領域内で部位は設計されなかった。代わりに、結合PEGが両方の相互作用を妨げ、且つそれらの間のあり得る相互作用を崩壊させることができるように、PEG15−PEG17(377、378及び556)がA2 LRP及びHSPG結合領域の間に設計された。表面露出され且つ外側に向いているさらに別の部位をLRP及びHPSG結合領域内もしくはその近辺で選ぶこともできた。新規なクリアランス機構を同定するために、FVIIIを系統的にPEG化することができる。PEG1−17の他に、3個の他の自然のグリコシル化部位、すなわちPEG18−20に対応するN41、N239及びN2118をPEG化のための結合点(tethering points)として用いることができ、それはそれらが表面露出されているはずだからである。PEG2、PEG6及び4個のグリコシル化部位のCβ原子から20オングストローム半径内の表面領域は、vWF、FIX、FX、リン脂質及びトロンビンに関する機能的相互作用部位の他にBDDモデル上にマッピングされた。
【0107】
Y81、F129、K422、K523、K570、N1864、T1911、Q2091及びQ2284に対応するPEG21−29は次いで、それらのCβ原子のそれぞれからの20オングストローム半径で残るBDD表面のほとんど全体を覆うそれらの能力に基づいて選択された。これらの位置は、それらが完全に露出されており、外側に向いており、且つ自然のシステインからずっと離れていて正しくないジスルフィド形成の可能性を最小にする故にも選択された。20オングストローム半径は、64kD分枝鎖状PEGのような大きなPEGが約20オングストローム半径を有する球を覆う可能性を有すると思われる故に選ばれる。PEG2及びPEG6ならびにグリコシル化部位PEG18、19及び20と一緒のPEG21−29のPEG化は、FVIIIの非−機能性(non−functional)表面のほとんど全体を保護するようである。
【0108】
PK側面の向上、より高い安定性又は免疫原性の低下のような強化された性質に導くPEG化位置を組み合わせて、最大に強化された性質を有する多重−PEG化生成物を形成することができる。PEG30及びPEG31は、それぞれA2及びA3ドメイン中の露出されたジスルフィドを除去することにより設計された。PEG30又はC630Aは、そのジスルフィドパートナーC711をPEG化のために遊離させるはずである。同様に、PEG31、C1899AはC1903がPEG化されるのを可能にするはずである。
【0109】
突然変異誘発。PEG化のために選ばれる部位においてシステインコドンを導入することにより、FVIIIの特定部位のPEG化のための基質を形成することができる。PEG突然変異体のすべての生産のためにStratagene cQuickChangeTM II特定部位の突然変異誘発キットを用いた(Stratagene Corpo
ration,La Jolla,CAからのStratageneキット200523)。cQuikChangeTM特定部位の突然変異誘発法は、Pfu TurboR DNAポリメラーゼ及び温度サイクラーを用いて行なわれる。所望の突然変異を含有する2個の相補的オリゴヌクレオチドプライマーを、Pfu Turboを用いて伸張させ、それはプライマーを置き換えないであろう。野生型FVIII遺伝子を含有するdsDNAを鋳型として用いる。複数の伸張サイクルに続き、メチル化DNAに関して特異的なDpnlエンドヌクレアーゼを用いて生成物を消化する。突然変異を含有する新しく合成されたDNAはメチル化されないが、親の野生型DNAはメチル化される。次いで消化されたDNAを用いてXL−1 Blue超−感応細胞を形質転換する。
【0110】
突然変異誘発の効率はほとんど80%である。突然変異誘発反応はpSK207+BDD C2.6又はpSK207+BDDにおいて行なわれた(図1)。DNA配列決定により成功した突然変異誘発を確かめ、突然変異を含有する適したフラグメントを哺乳類発現ベクターpSS207+BDD中のFVIII主鎖中に転移させた。転移の後、突然変異のすべてを再び配列−確認した。A3突然変異タンパク質PEG6、7、8、9及び10の場合、ベクターpSK207+BDD C2.6において突然変異誘発を行なった。配列決定により確かめた後、突然変異体フラグメント、Kpnl/PmeをpSK207+BDD中にサブクローニングした。次いでBDD突然変異タンパク質をpSS207+BDD発現ベクター中にサブクローニングした。A3突然変異タンパク質PEG11、12、13、14の場合、突然変異誘発をベクターpSK207+BDD中で直接行い、配列−確認突然変異体BDDを次いでpSS207+BDD中にサブクローニングした。A2突然変異タンパク質PEG1、2、3、4、5の場合、突然変異誘発をpSK207+BDD C2.6ベクター中で行なった。配列が確認された突然変異体をpSK207+BDD中に、及び次いでpSS207+BDD中にサブクローニングした。
【0111】
突然変異誘発に用いられるプライマー(センス鎖(sense stand)のみ)を各反応に関して挙げる:
PEG1,Y487C:GATGTCCGTCCTTTGTGCTCAAGGAGATTACCA(配列番号:5)
PEG2,L491C:TTGTATTCAAGGAGATGCCCAAAAGGTGTAAAAC(配列番号:6)
PEG3,K496C:TTACCAAAAGGTGTATGCCATTTGAAGGATTTTC(配列番号:7)
PEG4,L504C:AAGGATTTTCCAATTTGCCCAGGAGAAATATTC(配列番号:8)
PEG5,Q468C:GATTATATTTAAGAATTGCGCAAGCAGACCATAT(配列番号:9)
PEG6,K1808C:TAGAAAAAACTTTGTCTGCCCTAATGAAACCAAAAC(配列番号:10)
PEG7,N1810C:AACTTTGTCAAGCCTTGCGAAACCAAAACTTAC(配列番号:11)
PEG8,T1812C:GTCAAGCCTAATGAATGCAAAACTTACTTTTGGA(配列番号:12)
PEG9,K1813C:CAAGCCTAATGAAACCTGCACTTACTTTTGGAAAG(配列番号:13)
PEG10,Y1815C:CTAATGAAACCAAAACTTGCTTTTGGAAAGTGCAAC(配列番号:14)
PEG11,D1795C:ATTTCTTATGAGGAATGCCAGAGGCAAGGAGCA(配列番号:15)
PEG12,Q1796C:TCTTATGAGGAAGATTGCAGGCAAGGA
GCAGGA(配列番号:16)
PEG13,R1803C:CAAGGAGCAGAACCTTGCAAAAACTTTGTCAAGCCT(配列番号:17)
PEG14,K1804C:GGAGGAGAACCTAGATGCAACTTTGTCAAGCCT(配列番号:18)
PEG15,K377C:CGCTCAGTTGCCAAGTGTCATCCTAAAACTTGG(配列番号:19)
PEG16,H378C:TCAGTTGCCAAGAAGTGTCCTAAAACTTGGGTA(配列番号:20)
PEG17,K556C:CTCCTCATCTGCTACTGCGAATCTGTAGATCAA(配列番号:21)
PEG18,N41C:CAAAATCTTTTCCATTCTGCACCTCAGTCGTGTAC(配列番号:22)
PEH19,N239C:GTCAATGGTTATGTATGCAGGTCTCTGCCAGGT(配列番号:23)
PEG20,N2118C:CAGACTTATGCAGGATGTTCCACTGGAACCTTA(配列番号:24)
PEG21,Y81C:ATCCAGGCTGAGGTTTGTGATACAGTGGTCATT(配列番号:25)
PEG22,F129C:GAAGATGATAAAGTCTGTCCTGGTGGAAGCCAT(配列番号:26)
PEG23,K422C:CAGCGGATTGGTAGGTGTTACAAAAAAGTCCGA(配列番号:27)
PEG24,K523C:GAAGATGGGCCAACTTGCTCAGATCCTCGGTGC(配列番号:28)
PEG25,K570C:CAGATAATGTCAGACTGCAGGAATGTCATCCTG(配列番号:29)
PEG26,N1864C:CACACTAACACACTGTGTCCTGCTCATGGGAGA(配列番号:30)
PEG27,T1911C:CAGATGGAAGATCCCTGCTTTAAAGAGAATTAT(配列番号:31)
PEG28,Q2091C:ACCCAGGGTGCCCGTTGCAAGTTCTCCAGCCTC(配列番号:32)
PEG29,Q2284C:AAAGTAAAGGTTTTTTGCGGAAATCAAGACTCC(配列番号:33)
PEG30,C630A:TTGCAGTTGTCAGTTGCTTTGCATGAGGTGGCA(配列番号:34)
PEG31,C1899A:AATATGGAAAGAAACGCTAGGGCTCCCTGCAAT(配列番号:35)
【0112】
突然変異タンパク質発現。ハイグロマイシンBに対する耐性を与えるベクターにおける挿入の後、PEG突然変異タンパク質を、製造者の指示に従って293 フェクチントランスフェクション試薬(Fectin Transfection Reagent)(Invitrogen Corp.Cat#12347−019)と複合させたHKB11細胞中にトランスフェクションした(米国特許第6,136,599号明細書)。トランスフェクションから3日後におけるFVIII発現をCoatest発色アッセイにより評価した(Chromogenix Corp.Cat#821033,実施例12発色アッセイを参照されたい)(表1)。トランスフェクションされた細胞を次いで、5%FBSが補足された生育培地中で、50□g/mlのHyg Bを用いる選択圧下に置いた。Hyg B−耐性コロニーが現れたら、それらを手で採集し、Coatest発色ア
ッセイによりFVIII発現に関してスクリーニングした。FVIII発現安定細胞を次いでHPPS補足物質を含有する培地に適応させた。細胞を拡大し、新しい培地を有する振盪フラスコ中にml当たり1X106個の細胞において播種した。3日後に収穫される組織培養液(TCF)をFVIII BDD突然変異タンパク質の精製のために用いた。TCFのFVIII活性をCoatestにより検定した(表1)。
【0113】
【表1】

【0114】
突然変異タンパク質精製。分泌される突然変異タンパク質FVIIIタンパク質を含有する細胞培養上澄み液を集めたら、0.2ミクロンの膜フィルターを介して上澄み液を濾過し、残る細胞を除去する。次いで限外濾過又はアニオン交換により上澄み液を濃縮する。次いでそれを免疫親和性カラム(immunoaffinity column)に適用し、そこで細胞培養培地成分及び宿主細胞タンパク質不純物の大部分を除去する。免疫親和性カラム溶出液を次いでスクロースを含有する調製緩衝液中へのダイアフィルトレーションにより緩衝液交換し、凍結する。発色アッセイにより、モノクローナルFVIII抗体カラムを通過する(across)タンパク質の収率及び回収率を評価した。クロマトグラフィーランの負荷物、通過流(flow through)、種々の溶出液画分、ストリップ及びダイアフィルトレーションされた溶出液の試料をFVIII活性に関して検定した(表2)。表2はモノクローナル抗体カラムからのPEG2突然変異タンパク質の回収率を示す。抗体はC7F7抗体である。表2におけるパーセント回収率は発色アッ
セイにより決定される。最終的な収率は73%であった。図2に示されるのは、モノクローナルFVIII抗体クロマトグラフィーカラム上で精製されたPEG2タンパク質の場合の、時間に関する280nmにおけるUV吸光度のプロットである。クロマトグラフィーは、Amersham BioscienceからのAKTAR Explorer 100クロマトグラフィーシステムを用いて行なわれた。この機器は多重−波長UV−可視モニター及び2mmのフローセルを用いる。PEG2突然変異タンパク質は高濃度の塩(high salt)の存在下でカラムから溶出し、溶出ピークは280nmにおける吸光度及びFVIII活性アッセイの両方により示される。
【0115】
【表2】
【0116】
PEG化。100倍を超える過剰のPEG:タンパク質比において、本来の全長FVIII又はBDDを還元及び変性なしでシステイン−特異的PEGによりPEG化することはできず(データは示されていない)、すべての本来のシステインがジスルフィドを形成しているか、又はFVIII内に埋もれているというBDD構造モデルに基づく仮説を確証している。上記で挙げた標準的な案を用いて発現し且つ精製されたFVIIIシステイン突然変異タンパク質をシステイン−特異的PEGマレイミド試薬を用いてPEG化することはできず、おそらくそれは導入されたFVIIIシステインが細胞生育培地中に存在するシステイン及びβ−メルカプトエタノールのようなスルフヒドリル基との反応により「キャッピングされている」からである。この問題はおそらく培地からシステイン及びβ−メルカプトエタノールを除去することにより解決され得るが、これはより低いFVIII生産を生じ得、細胞により放出されるスルフヒドリルが導入されたFVIIIシステインを遮断するのを妨げないであろう。
【0117】
本発明の他の側面において、FVIIIの部位−特異的PEG化を可能にする3−段階法を開発した(図3)。段階1において、約1μMにおける精製されたFVIIIシステイン突然変異タンパク質を約0.7mM トリス(2−カルボキシエチル)ホスフィン(TCEP)又は0.07mM ジチオトレイトール(DTT)のような還元剤を用い、4℃において30分間穏やかに還元し、「キャップ」を放出させる。段階2に、回転カラム(BioRadR)を介して試料を移動させるようなサイズ−排除クロマトグラフィー(SEC)法により「キャップ」と一緒に還元剤を除去し、導入されたシステインを遊離且つ還元された状態にしながらFVIIIジスルフィドの再形成を可能にする。段階3において、還元剤の除去から少なくとも30分後に、遊離されたFVIIIシステイン突然変異タンパク質を5〜64kDの範囲のサイズを有する少なくとも10−倍モル過剰のPEG−マレイミド(Nektar Therapeutics及びN.O.F.Corporation)を用い、4℃で少なくとも1時間処理する。この方法は、異なる個人により繰り返される数十回の反応に関し、再現性の良いデータを有する高度に一貫した生成物分布を与える。
【0118】
TCEPの除去のための回転カラム法は計ることができないので、ゲル濾過脱塩クロマトグラフィーが選ばれた。しかしながら、TCEPのスパイクサンプル(spike s
ample)を用いてこの方法を調べると、TCEPがカラム空隙において測定可能なレベルで溶出し、その低い分子量を有する分子から予測される塩画分中のみで溶出するのではないことが示された。ウェスタンブロットアッセイは、おそらくTCEPの不完全な除去の故の有意なバックグラウンドPEG化を示した。そうしている間に別の実験は、塩勾配と組み合わされたアニオン交換クロマトグラフィー媒体を用いて、C7F7精製材料を他のタンパク質不純物からさらに有意に精製できることを示した。そこで上記のTCEPを用いてC7F7材料を還元し、次いでアニオン交換カラム上で材料を処理する(process)ことが決定された。電荷の相違のために、FVIIIタンパク質は保持されるが、TCEPはカラムを通過して流れ、保持されない。同時に勾配塩溶出の間に、FVIIIタンパク質は残るタンパク質不純物の大部分から精製された。これは、後に起こるPEG化が、より純粋な出発材料を用いて理論的により均一であろうことを意味した。しかしながら、TCEPのスパイクサンプルを用いて調べると、勾配中でFVIIIと一緒に溶出する測定可能なレベルのTCEPが見出されることが示された。従って、アニオン交換クロマトグラフィーの後にゲル濾過脱塩クロマトグラフィーを行なうことが決定され、これらの2つの段階は順番に用いられると、TCEPの完全な除去及び非−特異的PEG化の除去を生じた。
【0119】
SDS PAGE及びウェスタンブロットによるPEG化分析。還元性6%TrisGlycine SDSポリアクリルアミドゲル(Invitrogen)上の電気泳動により、PEG化された生成物を分析することができる。電気泳動に続き、クーマシーブルーを用いてゲルを染色してすべてのタンパク質を同定するか、あるいは標準的なウェスタンブロット案に供してFVIIIの種々の領域上のPEG化パターンを同定することができる。それぞれFVIII重鎖のC−末端領域又はVIII軽鎖のN−末端領域に対して構成される(raised)マウスモノクローナルR8B12又はC7F7抗体を用いるブロットの染色は、それぞれの鎖のPEG化を同定するはずである。FVIIIの484−509領域に対する413抗体を用いる染色は、PEG1−4のような突然変異タンパク質に関してPEG化が実際に部位−特異的であるか否かを決定するであろう。同様に、FVIIIの1801−1823領域を認識するCLB−CAg A抗体を用いる染色は、PEG6−10のような突然変異タンパク質に関してPEG化が部位−特異的であるか否かを決定するであろう。
【0120】
PEG2(L491C)PEG化は、軽鎖より重鎖に関して選択的であり、特に484−509領域に関して選択的であることが示されたが(図4)、PEG6(K1808C)は重鎖より軽鎖に関して選択的であることが示された(図5)。
【0121】
図4に描かれる研究のために、TCEPを用いてPEG2突然変異タンパク質(列1及び8)を還元し、続いてTCEPを除去し(列2及び9)、5、12、22、33又は43kD PEG−マレイミドで処理する(列3−7及び10−14)。非PEG化FVIIIは非プロセシング(H+L)ならびにプロセシング重(H)及び軽(L)鎖バンドとして移動する。3つのすべてのバンドはクーマシーブルー染色されたゲル上で検出可能であるが(下の右)、鎖特異的抗体を用いるウェスタン染色はプロセシングされない対応する鎖のみを明らかにする。R8B12染色(上の左)を用いると、PEG2をPEG−マレイミドで処理する場合の重鎖(H)バンドは強度が劇的に低下し、新しいバンドが形成され、それはPEGのサイズに比例して親Hバンドより高く移動する。C7F7染色(下の左)を用いると、軽鎖(L)バンド(不均一なグリコシル化の故に多重バンド)は強度を変えない。両染色に関する非プロセシングH+Lバンドは、H鎖が非プロセシングFVIIIの一部であるために移動する。クーマシー染色も軽鎖よりずっと多い重鎖のPEG化、すなわちHバンド強度の低下を確証する。最後にPEG化バンドは、おそらく484−509への413抗体の結合を遮断する491の部位−特異的PEG化の故に、PEGサイズ−依存的やり方で、R8B12染色より413抗体染色上(上の右)で相対的によ
り強度を失う。列当たりに負荷されるFVIIIの量は2つの左のゲルの場合に約30ngであり、上の右のゲルの場合に約1000ngであり、下の右のゲルの場合に約2000ngである。
【0122】
還元及びそれに続く還元剤の除去は、FVIIIの移動を変えない(列1対2及び8対9)。PEG2への22kD PEGの付加は413抗体の結合を遮断し、491位置における特異的なPEG化と一致する(図4の上の右のゲル)。これはPEG化されたPEG2が人間においてより低い免疫原性を有するであろうことも示唆しており、それは、413抗体がヒトA2阻害性抗体と同じエピトープを共有することが示されているからである(Scandella et al.著,Thromb.Haemost.67,1992年,pp.665−71)。
【0123】
図5に描かれる研究のために、TCEPを用いてPEG6突然変異タンパク質を還元し、続いてTCEPを除去し(列1及び6)、5、12、22又は33kD PEG−マレイミドで処理する(列2−5及び7−10)。非PEG化FVIIIは非プロセシング(H+L)ならびにプロセシング重(H)及び軽(L)鎖バンドとして移動する。PEG6(K1808)突然変異は軽鎖上にあるので、PEG化は軽鎖上のみで検出され、重鎖上では検出されなかった。列当たりに負荷されるFVIIIの量は左のゲルの場合に約100ngであり、右のゲルの場合に約30ngである。
【0124】
標準として移動させたBDDは、上記の還元及び還元剤除去法の後でさえ、100−倍を超えるモル過剰のPEG−マレイミドを用いて処理した時に有意なPEG化を示さなかった(図6a)。同じ方法をPEG4及びPEG5にも適用した(図6a)。PEG2と比較して、これらの突然変異タンパク質は有効にPEG化されなかったが、それらはPEG2(L491C)に類似して重鎖に関して選択的であった。PEG6(K1080C)のPEG化効率は比較的低く、それはおそらくそれがN1810におけるN−結合グリコシル化部位に非常に近接しており、それが位置1808におけるPEG化を遮断し得るからである。かくして我々は、1810における本来のグリコシル化部位を除去するためにPEG7(N1810C)を設計した。頭−頭比較(head−to−head comparison)においてPEG7はPEG6と比較して向上したPEG化効率を示す(図6b)。類似して、PEG15はPEG2よりわずかに良いPEG化効率を示す。BDDの二重突然変異体であるPEG2+6は、PEG2が重鎖システイン突然変異であり、PEG6が軽鎖突然変異であるために、重鎖及び軽鎖の両方の上でPEG化され得る(図6c)。この方法を野生型全長FVIIIにも適用した(図6d)。PEG化は、A1、A2及びBドメインのほとんどを含む重鎖の最大のフラグメントに関して検出された。PEG化パターンはモノPEG化を示唆し、1個のPEG化されたシステインのみがあることを示唆した。
【0125】
トロンビン切断及びウェスタンブロットによるPEG化分析。PEG化された生成物を37℃において30分間トロンビン(FVIIIのug当たり40IU)で処理することができる。用いられるトロンビンは汚染物としてAPCも含有する。トロンビン切断は重鎖から50kD A1及び43kD A2ドメインを形成するが、APC切断はA2ドメインをさらに21及び22kDのフラグメントに割るであろう(図7)。重鎖のC−末端を認識するR8B12抗体を用いる染色は、無損傷のA2ドメイン及び21kD C−末端フラグメントのみを同定するであろう(FVIII562−740)。かくしてPEG2のPEG化が位置491に関して特異的であったら、43kD A2ドメインはPEG化されるが21kD C−末端フラグメントはPEG化されないはずである。これは実際に、図7に示される22kD PEG化PEG2に関するウェスタンブロットにより確証された。かくして除去により、PEG2のPEG化は、A2ドメインのN−末端22kDフラグメント(FVIII373−561)に位置決定された。PEG−マレイミドはp
H6.8においてシステインに関して完全に選択的であり、373−561内の唯一の本来のFVIIIシステインは528−554の間の埋もれたジスルフィドに由来するので、PEG2は位置491における導入されたシステイン上でPEG化されるのが非常にありそうなことである。FVIII重鎖N−末端抗体を用いるトロンビン−処理されたPEG化PEG2のウェスタン染色は、A1ドメインのPEG化を示さなかった(データは示されない)。トロンビン切断法を用いるPEG2の選択的PEG化は、5、12、33及び43kDのPEGsに関しても確証された(データは示されない)。PEG化された野生型全長FVIIIのトロンビン切断は、BドメインのみがPEG化されることを示す(図8)。
【0126】
ヨウ素染色によるPEG化分析。クーマシーブルー及びウェスタン染色の際に新しく作られるバンドが実際にPEG化されたバンドであることを確証するために、PEGに関して特異的であるバリウム−ヨウ素染色を用いた(図9)。PEG化されたPEG2を6%TrisGlycineゲル(Invitrogen)上で移動させ、R8B12重鎖抗体又はバリウム−ヨウ素溶液で染色した(Lee et al.著,Pharm Dev
Technol.4:1999年,269−275)。PEG化されたバンドは、それらを整列させるための分子量マーカーを用いて2つの染色の間で一致し、かくしてFVIII重鎖PEG化を確証した。
【0127】
MALDI−質量分析によるPEG化分析。重鎖中のA2ドメインのPEG化を確証するために、PEG化の前後のrFVIII試料をマトリックス−補助レーザー脱着/イオン化(MALDI)質量分析により分析した。30%アセトニトリル,0.1%TFA中のシナピン酸マトリックスを有するMALDI標的プレート(target plate)上で試料を混合し、結晶化させた。次いでそれらをVoyager DE−PRO分光計において、正の線状モードで分析した。図10に示される結果は、83kDを中心とするPEG2の軽鎖及び89kDにおける重鎖(HC)を示した。PEG化試料に関して取得されたスペクトルは、HCピークの低下及び111kDを中心とする新しいピークの形成を示した。これは、重鎖のPEG化を確証する。PEG化された軽鎖(105kDにおける)は、検出限界より上で観察されなかった。
【0128】
次いで試料を両方とも、FVIIIのmg当たり20単位のトロンビンにおいて37℃で30分間、トロンビン消化に供し、続いてアミノ酸分析によりFVIII濃度を決定した(Commonwealth Biothechnologies,Inc)。重鎖は46kD(A1)N−末端画分及び43kD(A2)画分に切断された。PEG化試料に関して取得されたMALDIスペクトル(図11)は、43kDピークの喪失及びPEG化されたA2ドメインの故の新しい65kDピークの出現を示す。LCのPEG化はやはり検出限界より上で観察されない。これらの結果は再び、FVIIIのA2ドメインのPEG化を確証している。同じ分析をPEG化されたPEG6に適用し、軽鎖A3C1C2フラグメントのPEG化を確証した(図12)。
【0129】
活性測定
凝固アッセイ。凝固FVIII:C試験法は、活性化部分トロンボプラスチン時間(aPTT)に基づく1−段階アッセイである。FVIIIは因子IXa、カルシウム及びリン脂質の存在下で、因子XからXaへの酵素的転換において補因子として働く。このアッセイでは、希釈された試験試料をFVIII欠失血漿基質及びaPTT試薬の混合物と一緒に37℃でインキュベーションする。インキュベーションされた混合物に塩化カルシウムを加え、凝固を開始させる。血餅が形成されるのに要する時間(秒)とFVIII:Cの濃度の対数の間には逆比例関係(inverse relationship)が存在する。種々の希釈度の試験材料の凝固時間を、一系列の希釈度の既知の活性の標準材料から構築される曲線と比較することにより、未知の試料に関する活性レベルを内挿し、mL
当たりの国際単位(IU/mL)において報告する。
【0130】
発色アッセイ。発色アッセイ法は2つの連続的段階から成り、その方法では色の強度がFVIII活性に比例する。第1段階において、因子Xが最適量のカルシウムイオン及びリン脂質の存在下でFIXaにより、その補因子FVIIIaを用いてFXaに活性化される。因子Xの活性化の速度がFVIIIの量のみに依存するように、過剰量の因子Xが存在する。第2段階において、因子Xaは発色基質を加水分解し、発色団を与え、色の強度を405nmにおいて測光的に読み取る。未知試料の力価を計算し、傾斜比統計法(slope−ratio statistical method)を用いてアッセイの正当性を調べる。活性をmL当たりの国際単位(IU/mL)において報告する。
【0131】
1811−1818ループはFIXへの結合に含まれるが、このループ内の個々の位置の重要性は決定されていない。PEG7−10突然変異タンパク質は、本来のFVIIIに関してほとんど同じ比発色活性を示す(表3)。表3は、BDDに関するPEG突然変異タンパク質及びPEG化されたPEG2又はPEG6のパーセント比活性(S.A.)を示す。S.A.は、発色、凝固又はvWF結合活性を全抗原ELISA(TAE)値で割ることにより決定された。次いでPEG化突然変異タンパク質のS.A.をBDDのS.A.(8IU/ug発色、5IU/ug凝固及び1vWF/TAE)で割り、100を掛けて、表3中に発色、凝固及びvWF/TAEの見出しの下に挙げられるパーセントS.A.を得る。
【0132】
【表3】
【0133】
表3中で用いられる場合、「PEG2 red」は、還元剤で処理され、続いて還元剤が除去されたPEG2突然変異タンパク質である。この還元法はFVIIIの3つの機能的活性を有意に変えなかった。5kD(PEG2−5kD)〜43kD(PEG2−43kD)の範囲のPEGsに複合したPEG2突然変異タンパク質は、有意な量の発色活性を失わなかったが、PEGサイズが5kDを超えて大きくなるとともに凝固活性を非常に低下させた。比較的大きいサイズのPEG化PEG2に関してvWF結合における中程度の減少もあり得る。
【0134】
全抗原ELISA(TAE)。ポリクローナルFVIII抗体がコーティングされたミクロタイタープレート上にFVIIIを捕獲する。ビオチニル化ポリクローナルrFVIII抗体及びストレプタビジンホースラディッシュペルオキシダーゼ(HRP)複合体を用いてFVIII結合を検出する。ペルオキシダーゼ−ストレプタビジン複合体は、テトラメチルベンジジン(TMB)基質を加えると発色反応を生ずる。4パラメーターフィットモデルを用いて標準曲線から試料濃度を内挿する。FVIIIの結果をμg/mLにおいて報告する。
【0135】
vWF結合ELISA。FVIIIを溶液中で重症血友病血漿(Severe Hem
ophilic Plasma)中のvWfに結合させる。次いでvWf−特異的モノクローナル抗体がコーティングされたミクロタイタープレート上でFVIII−vWf複合体を捕獲する。FVIIIポリクローナル抗体及びホースラディッシュペルオキシダーゼ−抗−ウサギ複合体を用いてvWfに結合したFVIIIを検出する。ペルオキシダーゼ−複合抗体複合体は、基質を添加すると発色反応を生ずる。4パラメーターフィットモデルを用いて標準曲線から試料濃度を内挿する。FVIII結合の結果をμg/mLにおいて報告する。PEG化された場合に活性のいずれにも有意な影響はなく、それはBドメインにおけるPEG化と一致した。
【0136】
【表4】
【0137】
イオン−交換クロマトグラフィーによるPEG化FVIIIの精製。PEG化されたFVIIIをアニオン交換カラム又はカチオン交換カラムに適用し、ここでタンパク質はカラムに結合するが、過剰の遊離のPEG試薬は結合せず、通過流中で除去される。次いで塩化ナトリウム勾配を用いてPEG突然変異タンパク質をカラムから溶出させる。負荷物、通過流及び勾配画分のバリウム−ヨウ素染色された4〜12%Bis−Trisゲルを用い、カラム溶出画分がPEG化突然変異タンパク質を有することを確証した。
【0138】
サイズ−排除クロマトグラフィーによるPEG化FVIIIの精製。PEG2突然変異タンパク質の大部分を含有するアニオン交換画分をプールし、限外濾過により濃縮し、次いでサイズ排除カラムに適用する。次いで調製緩衝液を用いてカラムを溶出させる。PEGがタンパク質に結合しているかどうかに依存するタンパク質のサイズ及び形における相違の故に、このカラムはPEG化PEG2突然変異タンパク質をPEG化されていない残るPEG2のそれから分離する。PEG化された突然変異タンパク質FVIII画分を、ほとんどのFVIII活性を有することに基づいてプールし、次いでその後の動物研究及び分子特性化のために凍結する。図13は、43kD PEG化PEG2突然変異タンパク質の溶出に対して非−PEG化PEG2突然変異タンパク質の溶出を比較する。PEG化PEG2は有意により初期に溶出し、それは共有結合したPEGからのそのサイズにおける増加及び形を示す。
【0139】
より低い、すなわち50%未満のPEG化の効率を示すPEG6のような突然変異タンパク質の場合、高度に純粋なモノ−PEG化生成物を与えるための最も有効な精製案は、カチオン交換クロマトグラフィーとそれに続くサイズ排除クロマトグラフィーの組み合わせを使用することである。例えばPEG6の場合、カチオン交換クロマトグラフィーはPEG化されたPEG6(より初期の溶出画分、図14)を非−PEG化PEG6の大部分(より後期の溶出画分、図15)から精製する。次いでサイズ排除クロマトグラフィーはPEG化されたタンパク質(より初期の溶出画分、図15)を非−PEG化タンパク質(より後期の溶出画分、図15)の残りから分離して仕上げを施す(polishes)。
【0140】
活性へのPEGサイズの影響。PEGサイズがPEG化された時のFVIIIの凝固及び発色活性の両方に影響を有するかどうかを調べるために、精製された全長FVIII、PEG2、PEG6及びPEG14をTCEPにより還元し、続いて還元剤を除去し、緩衝液標準又は6kD〜64kDの範囲のPEGsと反応させた。得られるPEG化FVIIIを過剰のPEG又は非PEG化FVIIIの除去なしで直接検定した。標準実験は、過剰のPEGがFVIII活性に影響を有していないことを示した。
【0141】
図16はこの研究の結果を示す。精製された全長FVIIIは図16中でKG−2として示される。図16中で報告されるパーセント活性は、還元及び還元剤の除去の後にPEGで処理された試料の値を、PEG化の収率を考慮しながら、緩衝液標準で処理された試料の値で割ることにより決定された。PEG化の収率は、いずれの与えられるFVIII構築物の場合にも、すべてのPEGsにわたり(across)同等であった。それらはKG−2、PEG2及びPEG14の場合に約80%であり、PEG6の場合に約40%である。例えば12kD PEG化PEG14試料の場合の3.2IU/mLに対して、緩衝液標準処理されたPEG14は6.8IU/mLの凝固活性を有する。しかしながら、PEG化の効率は約80%であり、3.2IU/mLが約80%のPEG化及び約20%の非PEG化の凝集体活性を示すことを意味する。非PEG化試料が緩衝液標準で処理されたPEG14と同じ活性を有すると仮定すると、PEG化PEG14の場合の非PEG化PEG14に対するパーセント活性は34%=(3.2−6.8x20%)/(6.8x80%)であると算定される。
【0142】
BDDのPEG2、PEG6又はPEG14位置におけるA2又はA3ドメイン内のPEG化は、PEGサイズが6kDを超えて増加すると凝固活性の劇的な喪失を生ずる。しかしながら、全長FVIIIのBドメイン内の本来のB−ドメインシステインにおけるPEG化は凝固活性に影響を有していなかった。興味深いことに、発色活性はすべてのPEG化構築物の場合に影響されなかった。これはアッセイの相違の故であり得る。小さい発色性ペプチド基質は、凝固活性で用いられるもっと大きなタンパク質基質よりPEG化FVIII/FIX/FX複合体に容易に近づけることがあり得る。あるいはまた、PEGは突然変異タンパク質の活性化に影響し得る。これは2−段階発色アッセイより1−段階凝固アッセイによって容易に検出されるであろう。
【0143】
PEG2、6及び14の凝固活性へのPEGの影響の観察を確証するために、いくつかのPEG化構築物を過剰のPEG及び非PEG化タンパク質(unPEGylated)から精製した。PEGは発色活性に影響を有していないので、発色活性対凝固活性の比率は凝固活性へのPEGの相対的影響についての良い評価となる(表5)。PEG2のような与えられる位置におけるより大きなPEGs及びPEG2+6構築物の場合におけるようなより多数のPEGsは、より多くの凝固活性の喪失を引き起こす。
【0144】
【表5】
【0145】
うさぎPK研究。FVIIIの薬物動態学(PK)へのPEG化の影響を理解するために、複数の種においてPK研究を行なった。研究のためにNZW SPFウサギを用いた:10匹の雌,グループあたり5匹のウサギ、2つのグループ(PEG2 FVIII及び22kD PEG化PEG2)。試料を100IU/mL(発色単位)の最終的濃度を以って無菌のPBS中に希釈した。各ウサギに耳縁静脈(marginal ear vein)を介して1ml/kg(100IU/kg)の用量の希釈された試験物質又は標準物質を与えた。注入から種々の時間の後に、投薬から後の規定された時点に耳中心動脈(central ear artery)から血液試料(1mL)を1mLシリンジ中に採取した(100μLの3.8%Na−クエン酸塩を装入)。血漿試料を、96−ウェルプレート上にコーティングされたR8B12重鎖抗体と一緒にインキュベーションし、投薬されたヒトFVIIIを特異的に捕獲した。捕獲されたFVIIIの活性を発色アッセイにより決定した(図17)。PEG化PEG2及びPEG化PEG6をBDDと比較もし(図18及び19)、PEG化された突然変異タンパク質はBDDと比較して血漿回収率における向上を示した。PEG化された野生型全長FVIIIは多くの向上を示すようではなかった(図20)。
【0146】
マウスPK研究。第2の種として、ICR正常もしくは血友病、FVIII欠失マウス(Taconic,Hudson,NY)をPK研究に用いた。1時点につきグループ当たり5匹のマウスで、正常なマウスを研究に用いた。試験材料を調製緩衝液中に、25IU/mLの公称最終濃度まで希釈した。各マウスに尾静脈を介して4mL/kg(約0.1mLの合計体積)の希釈された(dilute)試験材料を投与することができる。指示される時点に下大静脈から、血液試料(正常もしくは血友病マウス研究のためにそれぞれ0.45もしくは0.3mL)を1mLのシリンジ(正常もしくは血友病マウス研究のためにそれぞれ50もしくは30μLの3.8%Na−クエン酸塩が装入された)中に採取する(試料当たり1匹の動物)。上記の発色アッセイ法を用いて血漿試料をFVIII濃度に関して検定する。PEG化PEG6は、BDD又はPEG6と比較してより高い血漿回収率を示す(図21)。PEG化PEG2は、BDDと比較してより高い血漿回収率を示す(図22及び23)。
【0147】
【表6】
【0148】
【表7】
【0149】
血友病マウス(BDD)因子VIII回収率。図24に示される血友病マウス(BDD)因子VIII回収率ヒストグラムは、血友病マウスアッセイにおけるBDD因子VIIIの2つの種の半減期の薬物動態学的(PK)評価を描く。このアッセイは、マウスモデルにおいて静脈内投与から後の3つの時点に、BDD因子VIII(図24中で「wt」又は野生型BDD因子VIIIと呼ばれる)及びBDD因子VIIIのPEG2+6二重PEG化変異体(そして本明細書の他の箇所でBDD因子VIIIのL491C,K1808C二重変異体と同定される)の両方の血漿濃度を測定するように設計された。0.8及び4時間の両時点におけるPK評価は同等であったが、16時間の評価は特に注目する価値がある。16時間において、非−PEG化分子と比較する場合に約4倍(400%)もの多くの二重にPEG化されたBDD因子VIII変異体(PEG2+6)が投与から16時間後のマウス血漿中に留まった。
【0150】
腎臓裂傷モデル。PEG化FVIII突然変異タンパク質が血友病マウスにおいて出血を止めるのに有効であるかどうかを決定するために、腎臓裂傷モデルを用いた。血友病マ
ウス(破壊されたFVIII遺伝子を有するC57/BL6)をイソフルオラン(isofluorane)下で麻酔し、秤量した。下大静脈を露出し、31ゲージの針を用いて100ulの食塩水又はFVIIIを注入した。針を注意深く取り出し、出血を妨げるために注入の部位で30〜45秒間圧力を加えた。2分後、右腎臓を露出し、垂直軸に沿って鉗子間に保った。#15メスを用い、腎臓を3mmの深さまで水平に切断した。傷の均一な深さを保証するために、腎臓を中心で軽く持って鉗子の両側上の等しい組織を露出した。露出された腎臓の表面は鉗子の深さまで切断された。上記の通りに血液損失を定量した。マウスについて種々の用量のFVIIIを調べ、腎臓出血へのFVIIIの用量反応関係を特性化した。PEG化PEG2は、マウス腎臓傷害後の血液損失の減少においてBDDと同等の力価を示した(図25)。かくしてPEG化PEG2の凝固活性はBDDのそれより低いが、この腎臓裂傷モデルは、PEG化PEG2の生体内有効性がBDDと比較して測定可能なほど低下しないことを示し、発色アッセイデータと一致した。
【0151】
抗体阻害アッセイ。特異的に位置491(すなわちPEG2)においてポリエチレングリコール(PEG)のような高分子量ポリマーを付加することは、mAB 413へ、及び拡大により患者の大きな割合の阻害性抗体への結合及び感度を低下させるはずであり、それは、多くの患者が同じmAB 413エピトープに対する阻害性抗体(inhibitor antibodies)を発現するからである。これを調べるために、増加する量のmAB 413を非−飽和量(0.003IU/mL)のBDD又は43kD PEG化PEG2と一緒にインキュベーションし、発色アッセイにおいて機能的活性を調べた(図26)。非−阻害性抗体であるR8B12及びC2ドメインを標的とする阻害性抗体であるESH4を標準として用いた。PEG化PEG2は実際にBDDよりmAB 413阻害に対して抵抗性であり、491位置近辺で結合しない標準抗体の存在下で類似の阻害パターンを示す。さらに、mAB 413阻害に対するPEGの保護効果はPEGサイズに依存し、より大きなPEGsがより強い効果を有する(図27)。PEG化FVIIIが患者からの阻害性抗体により抵抗性であるかどうかを調べるために、FVIIIへの阻害物質を発現した血友病A患者に由来する1パネルの血漿の存在下で、発色活性を測定した。調べられた8人の患者の血漿の中で4人の患者の血漿試料において、43kD PEG化PEG2はBDDより患者の血漿阻害に抵抗性であった。例えばPEG化されたPEG2、PEG6又はPEG2+6は、1人の患者の血漿においてBDDより高い残留活性を示したが、他の血漿においては示さなかった(図28)。ジPEG化PEG2+6は、モノPEG化PEG2又はPEG6より抵抗性であると思われる。これらの結果は、PEG化PEG突然変異タンパク質がFVIIIへの阻害物質を発現する患者の処置においてより有効である得ることを示唆している。
【0152】
高処理量PEG化スクリーニング。特定のPEG突然変異タンパク質のPEG化効率は予測不可能であり、それは特にBDDの直接の構造的情報がないからである。例えばBDDの構造モデルに基づき、PEG4及びPEG5のPEG化効率がPEG2及びPEG15のそれに類似して非常に高いはずと予測され、それは構造に従って3個のすべての位置が表面露出され且つ外側に向いているからである。かくして系統的なPEG化を介して新規なクリアランス機構を探索するためにPEGを用いることは、多数の突然変異タンパク質をスクリーニングすることを必要とする。
【0153】
多数のPEG突然変異タンパク質を迅速にスクリーニングするために、新規な高処理量法が開発され、それは一過的にトランスフェクションされた突然変異タンパク質からのPEG化生成物のPEG化効率及び機能的活性を調べることができる。0.1〜0.2IU/mLもの低いFVIII発色値を有する5〜10mLもの少量の一過的に発現されたPEG突然変異タンパク質を、Amicon−centra Ultra device MWCO 30Kを用いて約50−倍濃縮し、FVIIIの濃度はFVIIIへの抗体の相互作用の親和性範囲に近い1nMより高くに達する。濃縮されたPEG突然変異タンパ
ク質(〜300uL)を〜30uLのC7F7 FVIII抗体樹脂と一緒に4℃で終夜インキュベーションし、洗浄し、溶出させ、透析し、還元する。還元剤を除去し、還元されたPEG突然変異タンパク質をPEG化し、上記のようなウェスタン分析上で移動させる(図29及び30)。一過的に発現されたPEG突然変異タンパク質の相対的なPEG化効率は、精製されたPEG突然変異タンパク質のそれと正確に一致する。
【0154】
この方法により、1〜2ヶ月内に数十個のPEG突然変異タンパク質をスクリーニングすることができる。例えばPEG14(K1804C BDD)は、12kD PEGを用いて少なくとも約80%の軽鎖のPEG化を有し、重鎖のPEG化はなく(データは示されない)、軽鎖上に位置決定されるK1804C突然変異と一致した。K1804とK1808(PEG6位置)の間のC□からC□への距離は、BDD構造に従うとわずか8.4オングストロームであり、この位置における43kD PEGの導入が、よりずっと高いPEG化収率の利点を以って、33kD PEG化PEG6と類似のPKにおける向上を有するであろうことを示唆している。調べられたすべてのPEG突然変異タンパク質に関する相対的なPEG化収率を表8にまとめる。重鎖中にシステインを有するすべての突然変異タンパク質は重鎖上のみでPEG化されるが、軽鎖中にシステインを有するすべての突然変異タンパク質は軽鎖上でPEG化される点で、PEG化はシステイン突然変異が導入される特定のFVIII鎖に関して高度に選択的であった。突然変異タンパク質番号2〜31は、挙げられる位置における本来のアミノ酸がシステインで置換されるBDDのシステイン突然変異を示す。PEG2+6は、位置491及び1808がシステインで置換されたBDDの二重突然変異タンパク質である。A1及びA2(ならびに全長FVIIIであるKG−2の場合にはBドメイン)は重鎖に属するが、A3、C1及びC2は軽鎖に属する。PEG化効率は、PEG化生成物をSDS PAGE上で移動させ、PEG化バンドの強度を非PEG化バンドと比較することから見積もられた:+++ >約80%PEG化収率,++ 約30〜70%収率,+ 約10〜30%収率及び− <約10%収率。
【0155】
【表8】
【0156】
還元されたPEG突然変異タンパク質の質量分析。PEG突然変異タンパク質又は全長FVIIIの直接のPEG化を妨げる「キャップ」の正体を決定するために、PEG2+14を67uM〜670uMの範囲の濃度におけるTCEPで還元した。PEG化収率はTCEPの増加する量に比例して向上した(図31)。同じ試料をPEG化の前に質量分析によっても分析した(図32)。直接研究できるタンパク質ドメインを得るために、FVIIIのmg当たり20単位の比率におけるトロンビンを用い、試料を37℃で30分間消化した。トロンビン切断は残基372−740を含み、占有されたグリコシル化部位を含まないA2フラグメントを生ずる。消化された試料をC4逆相液体クロマトグラフィーシステム上に注入し、カラムからの溶出物を、電子スプレーインターフェース(electrospray interface)を介して4極飛行時間質量分析計(quadrupole time−of −flight mass spectrometer)中に直接導入した。A2ドメインに相当するクロマトグラフィーピーク下からの質量スペクトルをデコンボリューションし、タンパク質の無損傷の質量値を与えた。還元の前に、PEG2+14のA2ドメインは、理論的に予測されるより118ダルトン大きい質量を与える。TCEP濃度が向上すると、A2ドメインの正確な予測質量を有する新しいピークが現れる。この新しいピークの割合は、TCEP濃度が向上するとともに増加する。118ダルトンの差は、システイン(119Da)とのジスルフィド形成を介する残基Cys 491におけるシステイン化及び機器の精度により説明され得る。かくしてこれは、PEG突然変異タンパク質がシステインによりキャッピングされ、それが直接のPEG化を妨げることを示す。
【0157】
本明細書で開示されるすべての引用文献は、引用することによりそれらの記載事項全体が本明細書の内容となる。
【図面の簡単な説明】
【0158】
【図1a】PEG突然変異タンパク質のためのベクターマップ及び突然変異誘発戦略。
【図1b】PEG突然変異タンパク質のためのベクターマップ及び突然変異誘発戦略。
【図2】モノクローナルFVIII抗体クロマトグラフィーカラム上で精製されたPEG2タンパク質の場合の、時間に関する280nmにおけるUV吸光度分布。
【図3】3−段階特定部位PEG化法。
【図4】PEG2の特定部位のPEG化。
【図5】PEG6の特定部位のPEG化。
【図6a】BDD、PEG2、4、5及び6の特定部位のPEG化。
【図6b】PEG2及びPEG6を標準として用いるPEG15及びPEG7のPEG化。
【図6c】PEG2及びPEG6を標準として用いるPEG2+6のPEG化。
【図6d】PEG2を標準として用いる野生型全長FVIII(KG−2)のPEG化。
【図7】PEG化されたPEG2のトロンビン切断。
【図8】PEG化された野生型全長FVIII(KG−2)のトロンビン切断。
【図9】PEG化されたPEG2のヨウ素染色。
【図10a】非PEG化PEG2のMALDI質量分析。
【図10b】22kD PEG化PEG2のMALDI質量分析。
【図11a】トロンビン切断の後の非PEG化PEG2のMALDI質量分析。
【図11b】トロンビン切断の後のPEG化PEG2のMALDI質量分析。
【図12】トロンビン切断の前後のPEG化PEG6のMALDI質量分析。
【図13】サイズ−排除カラム上で精製されたPEG化PEG2の280nmにおけるUV吸収分布。
【図14】カチオン交換カラム上で精製されたPEG化及び非PEG化PEG6の280nmにおけるUV吸収分布。
【図15】サイズ−排除カラム上で精製されたPEG化及び非PEG化PEG6の280nmにおけるUV吸収分布。
【図16】発色アッセイ及び凝固アッセイにより測定される場合のPEG化タンパク質の活性を非PEG化タンパク質の活性と比較するグラフ。
【図17】PEG2と比較されるPEG化PEG2のウサギPK研究。
【図18】BDD及びPEG2と比較されるPEG化PEG2のウサギPK研究。
【図19】BDD及びPEG6と比較されるPEG化PEG6のウサギPK研究。
【図20】非変更全長FVIIIと比較されるPEG化野生型全長(“fl”)FVIIIのウサギPK研究。
【図21】PEG6及びBDDと比較されるPEG化PEG6の血友病マウスPK研究。
【図22】BDDと比較される22及び43kD PEG化PEG2の正常マウスPK研究。
【図23】時間経過中ずっとBDDと比較される22kD PEG化PEG2の正常マウスPK研究。
【図24】血友病マウスアッセイにおけるBDD因子VIIIの2つの種の半減期の薬物動態学的(PK)評価を描く、血友病マウス(BDD)因子VIII回収率ヒストグラム。
【図25】BDDと比較される22kD PEG化PEG2の血友病マウス腎臓裂傷研究。
【図26】増加する量のFVIII抗体の存在下におけるPEG化PEG2及びBDDの発色活性。
【図27】増加する量のFVIII mAB 413抗体の存在下におけるPEG化PEGの発色活性。
【図28】FVIIIへの阻害物質を発現した患者に由来するヒト血漿の存在下におけるBDD、43kD PEG化PEG2、33kD PEG化PEG6及び33kD ジPEG化PEG2+6の発色活性。
【図29】PEG化スクリーニング法及び正当化。
【図30】PEG15〜17のPEG化スクリーニング。
【図31】還元剤濃度の関数としてPEG2+14のPEG化を示すゲル。
【図32】67〜670uMのTCEPで処理され、続いて還元剤が除去されたPEG2+14のデコンボリューションされた質量スペクトル。
【関連出願へのクロスリファレンス】
【0001】
本出願は、2004年11月12日に申請された米国特許出願第60/627,277号明細書への優先権の利益を請求し、その出願は引用することによりその記載事項全体が本明細書の内容となる。
【技術分野】
【0002】
発明の分野
本発明は、規定される部位において1個もしくはそれより多い生物適合性ポリマー、例えばポリエチレングリコールへのカップリングを許す因子VIII(FVIII)突然変異タンパク質に関する。さらに、治療目的のための関連する調製物、投薬物(dosages)及びその投与の方法を提供する。これらの変更されたFVIII突然変異体ならびに関連する組成物及び方法は、血友病Aに苦しむ患者のための注入頻度が低下し、且つ免疫原性反応が低下した処置の選択肢を与えるのに有用である。
【背景技術】
【0003】
発明の背景
血友病Aは最も普通の遺伝性凝固障害であり、5000人の男性当たり1人の発生率と見積もられる。それは、血液凝固の固有の経路の決定的な構成成分であるFVIIIにおける欠失又は構造的欠陥により引き起こされる。血友病Aのための現在の処置は、ヒトFVIIIの静脈内注射を含む。ヒトFVIIIは約300kDの一本鎖分子として組換え的に生産されてきた。それは構造ドメインA1−A2−B−A3−C1−C2から成る(非特許文献1)。前駆体生成物はゴルジ装置において200kD(重)及び80kD(軽)の2つのポリペプチド鎖にプロセシングされ、2つの鎖は金属イオンにより一緒に保持される(非特許文献2;非特許文献3)。
【0004】
FVIIIのB−ドメインはB−ドメイン欠失FVIII(BDD,90kD A1−A2重鎖プラス80kD軽鎖)として省略可能であると思われ、血友病Aのための置換療法として有効であることも示された。B−ドメイン欠失FVIII配列は、B−ドメインの14個のアミノ酸以外のすべての欠失を含有する。
【0005】
血友病A患者は現在、必要時の、又は1週に数回投与される予防的治療としてのFVIIIの静脈内投与により処置される。予防的処置のために、体重のkg当たり15〜25IUの因子VIIIが1週に3回投与される。患者においてそれは常に必要である。人間におけるFVIIIの短い半減期の故に、それは頻繁に投与されねばならない。全長タンパク質の場合の300kDを超えるその大きなサイズにかかわらず、FVIIIは人間中でわずか約11時間の半減期を有する。(非特許文献4)。頻繁な静脈内注射の必要性は、患者のコンプライアンスにとってものすごい障壁を作る。より長い半減期を有し、従って必要な投与がより頻繁でないFVIII製品を開発できたら、それは患者にとってもっと簡便であろう。さらに、半減期が向上したら、その場合はより少ない用量が必要となり得るので、処置の経費を減少させることができる。
【0006】
現在の治療へのさらなる欠点は、患者の約25〜30%がFVIII活性を阻害する抗体を発現することである(非特許文献5)。阻害性抗体の主なエピトープはA2ドメイン内で残基484−508に、A3ドメイン内で残基1811−1818に、及びC2ドメイン内に位置する。抗体発現は置換療法としてのFVIIIの使用を妨げ、この群の患者に高い用量の組換え因子VIIaを用いるさらにもっと高価な処置及び免疫寛容治療(immune tolerance therapy)を追求することを強いる。
【0007】
以下の研究は、阻害性抗体のFVIIIエピトープを同定した。25個の阻害性血漿試料の研究において、11個はトロンビン生成73kD軽鎖フラグメントA3C1C2に、4個はA2ドメインに、そして10個は両方に結合することが見出された(非特許文献6)。別の研究において、患者からの8個のA2ドメイン阻害物質(inhibitors)中の6個は組換えA2ポリペプチドにより中和された(非特許文献7)。患者からの9個の阻害物質中の6個に関するエピトープは、A2残基379−538にマッピングされた(非特許文献8)。18個の重−鎖阻害物質に関するエピトープは、A2ドメインの同じN−末端18.3kD領域中に位置決定された(非特許文献9)。
【0008】
ヒトA2ドメイン残基387−604を相同ブタ配列で置換することにより形成される活性な組換えハイブリッドヒト/ブタFVIII分子は、患者のA2阻害物質に抵抗性であり(非特許文献10)、A2への結合に関して患者のA2阻害物質と競合するネズミモノクローナル抗体mAB 413 IgGに抵抗性(非特許文献11)であった。このA2ドメインエピトープはさらに、mAB 413 IgG及び4個の患者の阻害物質が、A2ドメイン残基484−508がブタのそれで置換されたハイブリッドヒト/ブタFVIIIを阻害しないことを実験が示した時に、A2ドメイン残基484−508に位置決定された(非特許文献12)。このハイブリッドFVIIIは、スクリーニングされた23個の患者の血漿の少なくとも半分により抵抗性でもあった(非特許文献13)。アラニン走査突然変異誘発(alanine scanning mutagenesis)は、試験された5個のすべての患者の阻害物質への結合に関して残基487が決定的であるが、残基484、487、489及び492はすべてmAB 413 IgGとの相互作用に重要であることを同定した(非特許文献14)。R484A/R489A/P492A突然変異体を与えられるがR484A/R489突然変異体を与えられないマウスにおける阻害性抗体力価は、標準ヒトBDD FVIIIを与えられるマウスにおけるより有意に低かった(非特許文献15)。要するに、A2ドメインの484−508領域はFVIII活性の阻害物質のための結合部位であると思われる。
【0009】
FVIIIに対する免疫反応の出現に加え、通常の治療を用いる別の問題は、生体内におけるFVIIIの短い半減期の故にそれが頻繁な投薬を必要とすることである。循環からのFVIIIのクリアランスに関する機構が研究された。
【0010】
循環からのFVIIIクリアランスは、部分的に低−密度リポタンパク質受容体−関連タンパク質(LRP)、広いリガンド特性を有する肝臓クリアランス受容体への特異的な結合に帰せられた(非特許文献16)。最近、低−密度リポタンパク質(LDL)受容体も、例えばFVIIIの血漿レベルの調節においてLRPと協働することにより、FVIIIクリアランスにおいて役割を果たすことが示された(非特許文献17)。両相互作用は、細胞−表面ヘパリン硫酸プロテオグリカン(HSPGs)に結合することにより助長される。マウスにおける血漿半減期は、LRPが遮断されると3.3倍に延長され得るか、あるいはLRP及び細胞−表面HSPGsの両方が遮断されると5.5倍に延長され得る(非特許文献18)。HSPGsはFVIIIを細胞表面上に濃縮し、それをLRPに与えると仮定される。FVIII上のLRP結合部位はA2残基484−509(非特許文献19)、A3残基1811−1818(非特許文献20)及びC2ドメイン中のエピトープ(非特許文献21)に位置決定された。
【0011】
FVIIIはプロテアーゼの作用によっても循環からクリアランスされる。この効果を理解するためには、FVIIIが血液凝固に含まれる機構を理解しなければならない。FVIIIはvWFに結合した重鎖及び軽鎖のヘテロ二量体として循環する。VWF結合はFVIII残基1649−1689(非特許文献22)ならびにC1(非特許文献23)及びC2ドメイン(非特許文献24)の一部を含む。FVIIIはトロンビンにより活性
化され、それは残基372、740及び1689の後でペプチド結合を切断し、A1、A2及びA3−C1−C2ドメインのヘテロ三量体を形成する(非特許文献25)。FVIIIは、活性化されるとvWFから解離し、リン脂質に結合することにより血小板の細胞表面に濃縮される。リン脂質結合はFVIII残基2199、2200、2251及び2252を含む(非特許文献26)。そこでそれはFVIII残基558−565(非特許文献27)及び1811−1818(非特許文献28)との相互作用を介してFIXに、ならびにFVIII残基349−372との相互作用を介してFXに(非特許文献29)結合し、固有の凝固経路の必須の成分であるFXのFIX活性化のための補因子として働く。活性化されたFVIII(FVIIIa)は、プロテアーゼ活性化タンパク質C(APC)により、FVIII残基336及び562の後の切断を介して部分的に不活性化される(非特許文献30)。しかしながら、不活性化の主な決定因子は、A1及びA3−C1−C2からのA2ドメインの解離である(非特許文献31)。
【0012】
タンパク質の生体内半減期を向上させることが示された1つの方法はPEG化である。PEG化は、タンパク質又は他の分子への長−鎖ポリエチレングリコール(PEG)分子の共有結合である。PEGは直鎖状形態又は分枝鎖状形態にあり、種々の特徴を有する種々の分子を形成することができる。ペプチド又はタンパク質の半減期を向上させることの他に、PEG化は抗体発現を減少させ、プロテアーゼ消化からタンパク質を保護し、且つ腎臓濾液(kidney filtrate)の外に材料を保持するために用いられてきた(非特許文献32)。さらに、PEG化はタンパク質の全体的な安定性及び溶解度も向上させることができる。最後に、PEG化されたタンパク質の持続的な血漿濃度は、薬剤の谷からピークまでのレベル(the trough to peak level)を低下させることにより不利な副作用の程度を低下させ、かくして初期の時点にタンパク質の超−生理学的レベルを導入する必要を除去することができる。
【0013】
PEG及びデキストランのような大きなポリマーを用いて第1級アミン(N−末端及びリシン)を標的とすることによるFVIIIの無作為な変更は、種々の成功の程度を以って試みられてきた(特許文献1、特許文献2、特許文献3)。1994年の特許出願(特許文献1)において公開された最も劇的な進歩は、全−長FVIIIを50−倍モル過剰のPEGと反応させた後、4−倍の半減期の向上を示すが、2分の1への活性の損失(2−fold activity loss)を犠牲にする。特許文献4は、無作為な変更を介して作られるFVIIIとポリエチレングリコールの複合体を開示している。無作為にPEG化されたタンパク質、例えばインターフェロン−アルファ(非特許文献33)は、過去に療薬として承認された。
【0014】
しかしながらこの無作為法は、ヘテロ二量体性FVIIIにとってもっとずっと問題が多い。FVIIIは、158個のリシン、2個のN−末端及び多数のヒスチジン、セリン、トレオニン及びチロシンを含む何百のPEG化可能な部位を有し、それらのすべてはおそらく主に第1級アミンを標的とする試薬を用いてPEG化され得る。例えばPEG化されたインターフェロンアルファ−2bに関する主な位置異性体はヒスチジンであることが示された(非特許文献34)。さらに、全長FVIIIの不均一なプロセシングは出発材料の混合物を生じ得、それはPEG化された生成物におけるさらなる複雑性に導く。PEGが決定的な活性部位もしくはその近辺で結合するべき場合、特に1個より多いPEG又は1個の大きなPEGをFVIIIに複合させる場合、FVIII上のPEG化の部位を制御しないさらなる欠点は活性の低下の可能性である。無作為なPEG化は必ず多量の多重にPEG化された生成物を生産するので、モノ−PEG化生成物のみを得るための精製は全体的収率を非常に低下させるであろう。最後に、生成物分布におけるものすごい不均一性は、各ロットの一貫した合成及び特性化をほとんど不可能にするであろう。優れた製造は一貫した十分に特性化される生成物を必要とするので、生成物の不均一性は商品化への障壁である。これらの理由のすべてのために、FVIIIをPEG化するためのもっと
特異的な方法が望ましい。
【0015】
種々の特定部位のタンパク質PEG化戦略が最近の総説にまとめられている(非特許文献35)。1つの方法は、化学的合成又は組換え発現によるタンパク質中への不自然なアミノ酸の導入及びそれに続く不自然なアミノ酸と特異的に反応するであろうPEG誘導体の添加を含む。例えば不自然なアミノ酸は本来のタンパク質中に見出されないケト基を含有するものであることができる。しかしながらタンパク質の化学的合成は、FVIIIのような大きなタンパク質には実行できない。ペプチド合成の現在の限界は約50個の残基である。数個のペプチドを連結させてもっと大きな1片のポリペプチドを作ることができるが、B−ドメイン欠失FVIIIを生産することさえ20回より多い連結を必要とし、それは理想的な反応条件下でさえ1%より低い回収率を生ずる。不自然なアミノ酸を有するタンパク質の組換え発現はこれまで、主に非−哺乳類発現系に限られてきた。この方法は、哺乳類系で発現される必要があるFVIIIのような大きく且つ複雑なタンパク質にとって問題が多いと思われる。
【0016】
タンパク質の部位−特異的PEG化への他の方法は、PEG−アルデヒドを用いてN−末端主鎖アミンを標的とすることによる。しかしながら、他のアミン基を超える特異性を達成するためにこの方法の下で必要な低いpHは、FVIIIの安定性のために必要な狭い近−中性pH範囲と適合しない(非特許文献36)。さらに、FVIIIのN−末端PEG化は、この領域が血漿クリアランスに含まれない場合、血漿半減期の向上を生ずることができない。事実、FVIII軽鎖のN−末端領域は、循環中のFVIII生存のために決定的に重要である担体タンパク質であるフォンビルブラント因子(vWF)への結合に関係があるとされてきた。因子VIIIのN−末端変更により、決定的に重要なvWFとの会合が崩壊するか、又は弱くなり得る。かくしてFVIIIのN−末端PEG化はFVIIIの血漿半減期を低下させる反対の効果を有し得る。
【0017】
特許文献5は、システインを挿入するか、又は他のアミノ酸の代わりに置換し、次いでスルフヒドリル反応性基を有するリガンドを加えることによるヒトIL−3、顆粒球コロニー刺激因子及びエリトロポエチンポリペプチドの部位−特異的変更を開示している。リガンドはシステイン残基に選択的にカップリングする。FVIII又はその変異体の変更は開示されていない。
【0018】
上記の理由のために、機能的活性を保持しながら生体内におけるより長い作用の持続時間及び低下した免疫原性を有する改良されたFVIII変異体に対する要求がある。さらに、そのようなタンパク質が一貫したやり方で均一な生成物として生産されるのが望ましい。
【先行技術文献】
【特許文献】
【0019】
【特許文献1】国際公開第94/15625号パンフレット
【特許文献2】米国特許第4970300号明細書
【特許文献3】米国特許第6048720号明細書
【特許文献4】国際公開第2004/075923号パンフレット
【特許文献5】国際公開第90/12874号パンフレット
【非特許文献】
【0020】
【非特許文献1】Thompson著,Semin.Hematol.29,2003年,11−22
【非特許文献2】Kaufman et al.著,J.Biol.Chem.263,1988年,p.6352
【非特許文献3】Andersson et al.著,Proc.Natl.Acad.Sci.83,1986年,p.2979
【非特許文献4】Ewenstein et al.著,Semin,Hematol.41,2004年,pp.1−16
【非特許文献5】Saenko et al.著,Haemophilia 8,2002年,pp.1−11
【非特許文献6】Fulcher,C.et al.著,Proc.Natl.Acad.Sci.2(22),1985年,pp.7728−32
【非特許文献7】Scandella,D.et al.著,Blood 82(6),1993年,pp.1767−75
【非特許文献8】Scandella,D.et al.著,Proc.Natl.Acad.Sci.85(16),1988年,pp.6152−6
【非特許文献9】Scandella,D.et al.著,Blood 74(5),1989年,pp.1618−26
【非特許文献10】Lubin,I.et al.著,J.Biol.Chem.269(12),1994年,pp.8639−41
【非特許文献11】Scandella,D.et al.著,Thromb Haemost.67(6),1992年,pp.665−71
【非特許文献12】Healey,J.et al.著,J.Biol.Chem.270(24),1995年,pp.14505−9
【非特許文献13】Barrow,R.et al.著,Blood 95(2),2000年,pp.564−8
【非特許文献14】Lubin,I.著,J.Biol.Chem.272(48),pp.30191−5
【非特許文献15】Parker,E.et al.著,Blood 104(3),2004年,pp.704−10
【非特許文献16】Oldenburg et al.著,Haemophilia 10 Suppl 4,2004年,pp.133−139
【非特許文献17】Bovenschen et al.著,Blood 106,2005年,pp.906−910
【非特許文献18】Sarafanov et al.著,J.Biol.Chem.276,2001年,pp.11970−11979
【非特許文献19】Saenko et al.著,J.Biol.Chem.274,1999年,pp.37685−37692
【非特許文献20】Bovenschen et al.著,J.Biol.Chem.278,2003年,pp.9370−9377
【非特許文献21】Lenting et al.著,J.Biol.Chem.274,1999年,pp.23734−23739
【非特許文献22】Foster et al.著,J.Biol.Chem.263,1988年,pp.5230−5234
【非特許文献23】Jacquemin et al.著,Blood 96,2000年,pp.958−965
【非特許文献24】Spiegel,P.et al.著,J.Biol.Chem.279(51),2004年,pp.53691−8
【非特許文献25】Pittman et al.著,Proc.Natl.Acad.Sci.276,2001年,pp.12434−12439
【非特許文献26】Gilbert et al.著,J.Biol.Chem.277,2002年,pp.6374−6381
【非特許文献27】Fay et al.著,J.Biol.Chem.269,1994年,pp.20522−20527
【非特許文献28】Lenting et al.著,J.Biol.Chem.271,1996年,pp.1935−1940
【非特許文献29】Nogami et al.著,J.Biol.Chem.279,2004年,pp.15763−15771
【非特許文献30】Regan et al.著,J.Biol.Chem.271,1996年,pp.3982−3987
【非特許文献31】Fay et al.著,J.Biol.Chem.266,1991年,pp.8957−8962
【非特許文献32】Harris et al.著,Clinical Pharmacokinetics 40,2001年,pp.539−51
【非特許文献33】Kozlowski et al.著,Biodrugs 15,2001年,pp.419−429
【非特許文献34】Wang et al.著,Biochemistry 39,2000年,pp.10634−10640
【非特許文献35】Kochendoerfer,G.著,Curr.Opin.Chem.Biol.2005,of Oct.15,2005,direct object identifier doi:10.1016/j.cbpa.2005.10.007としてオンラインで入手可能)
【非特許文献36】Wang et al.著,International J.Pharmaceutics 259,2003年,pp.1−15
【発明の概要】
【発明が解決しようとする課題】
【0021】
本発明の目的は、向上した薬物動態学的特性及び治療的特性を有する生物適合性ポリマー−複合機能性FVIIIポリペプチドを提供することである。
【0022】
本発明の他の目的は、向上した薬物動態学的性質を有する生物適合性ポリマー−複合Bドメイン欠失FVIIIタンパク質を提供することである。
【0023】
本発明のさらに別の目的は、低−密度リポタンパク質受容体−関連タンパク質(LRP)、低−密度リポタンパク質(LDL)受容体、ヘパラン硫酸プロテオグリカン(HSPGs)及び/又はFVIIIに対する阻害性抗体への結合が減少した生物適合性ポリマー−複合機能性FVIIIポリペプチドを提供することである。
【0024】
本発明のさらに別の目的は、生体内におけるより長い作用の持続時間及び低下した免疫原性を有し、一貫したやり方で均一な生成物として生産され得る改良されたFVIII変異体を提供することである。
【課題を解決するための手段】
【0025】
本発明の1つの側面において、ポリペプチド上のあらかじめ規定される1個もしくはそれより多い部位において1個もしくはそれより多い生物適合性ポリマーに共有結合した機能性因子VIIIポリペプチドを含んでなる因子VIIIプロコアグラント活性を有する複合体であって、あらかじめ規定される部位がN−末端アミンではない複合体を提供する。本発明は、機能性因子VIIIポリペプチドをコードするヌクレオチド配列を突然変異させてあらかじめ規定される部位においてシステイン残基に関するコード配列に置換し;突然変異したヌクレオチド配列を発現させてシステイン増強突然変異タンパク質を生産し;突然変異タンパク質を精製し;実質的に導入されたシステイン残基のみにおいてポリペプチドと反応するように活性化された生物適合性ポリマーと突然変異タンパク質を反応させて複合体を形成し;そして複合体を精製することを含んでなる、この複合体の調製方法も含む。本発明は、複合体及び製薬学的に許容され得る添加剤を含んでなる製薬学的組成
物ならびに必要のある患者にこれらの製薬学的組成物の治療的に有効な量を投与することによる、血友病の処置方法も目的とする。
【0026】
本発明は、(a)特定部位の因子VIII突然変異タンパク質を発現させ、ここで突然変異タンパク質は因子VIII突然変異タンパク質の露出された表面上のアミノ酸残基に代わるシステイン置換を有し、且つそのシステインはキャッピングされており;(b)システイン突然変異タンパク質を穏やかに還元し、且つキャップを放出する条件下でシステイン突然変異タンパク質を還元剤と接触させ;(c)システイン突然変異タンパク質からキャップ及び還元剤を除去し;そして(d)還元剤の除去から少なくとも約5分後に、PEG化された因子VIII突然変異タンパク質が生産されるような条件下で、スルフヒドリルカップリング部分を含んでなるPEGでシステイン突然変異タンパク質を処理することを含んでなる、因子VIII突然変異タンパク質の特定部位のPEG化のための方法にも関する。
【0027】
図面の簡単な記述
図1.PEG突然変異タンパク質のためのベクターマップ及び突然変異誘発戦略。
【0028】
図2.モノクローナルFVIII抗体クロマトグラフィーカラム上で精製されたPEG2タンパク質の場合の、時間に関する280nmにおけるUV吸光度分布。クロマトグラフィーは、Amersham BioscienceからのAKTAR Explorer 100クロマトグラフィーシステムを用いて行なわれた。
【0029】
図3.3−段階特定部位PEG化法。PEGはPEG−マレイミドのようなシステイン−反応性PEGを示す。閉じられた棒はジスルフィド形成を示し、開放された棒は還元されたシステインを示す。
【0030】
図4.PEG2の特定部位のPEG化。
【0031】
図5.PEG6の特定部位のPEG化。
【0032】
図6a.BDD、PEG2、4、5及び6の特定部位のPEG化。上のパネルは重(H)鎖抗体で染色され、下のパネルは軽(L)鎖抗体で染色された。“U”はH及びLの両方を含有するプロセシングされない材料である。
【0033】
図6b.PEG2及びPEG6を標準として用いるPEG15及びPEG7のPEG化。TCEPを用いて開始精製PEG突然変異タンパク質(“S”)を還元し、還元剤(“R”)の除去の後に12kD(“12”)又は22kD(“22”)PEGを用いてPEG化する。試料を6% Tris−グリシン SDS PAGE上で移動させ、左のパネル上で重鎖(“HC”)抗体又は右のパネル上で軽鎖(“LC”)抗体を用いて染色した。“U”はHC及びLCの両方を含有するプロセシングされない材料である。PEG化されたバンドを点により強調する。
【0034】
図6c.PEG2及びPEG6を標準として用いるPEG2+6のPEG化。TCEPを用いてPEG2、PEG6又はPEG2+6を還元し、還元剤(“R”)の除去の後に5kD(“5”)又は43kD(“43”)PEGを用いてPEG化する。12、22及び33kD PEGsを用いてもPEG2+6をPEG化した。試料を6% Tris−グリシン SDS PAGE上で移動させ、左でタンパク質に関してクーマシーを用いて、あるいは重鎖(H)又は軽鎖(L)抗体を用いて染色した。“U”はH及びLの両方を含有するプロセシングされない材料である。PEG化されたバンドを点により強調する。
【0035】
図6d.PEG2を標準として用いる野生型全長FVIII(KG−2)のPEG化。左のゲルをタンパク質に関してクーマシー染色を用いて染色し、右のゲルをPEGに関してヨウ素を用いて染色した。“BDD U”はH及びLの両方を含有するプロセシングされないBDD材料である。PEG化されたバンドを点により強調する。
【0036】
図7.PEG化されたPEG2のトロンビン切断。A2ドメインのN−末端半分を青く着色し、C−末端半分を緑に着色し、R8B12抗体エピトープを暗緑色に強調した(右FVIIIモデル)。PEG2(列1)及び22kD PEG化PEG2(列2)をトロンビンで処理し(それぞれ列3及び4)、次いで7% Tris−アセテートゲル(Invitrogen)上で移動させ、R8B12抗体で染色した。各列は約50ngのFVIIIを含有する。
【0037】
図8.PEG化された野生型全長FVIII(KG−2)のトロンビン切断。“S”=出発KG−2材料。“R”=還元されたKG−2及び除去された還元剤。“P”=43kD PEGを用いてPEG化された“R”。“Pure”=過剰のPEGから精製された“P”。“L”=軽鎖。PEG化されたバンドを点により強調する。
【0038】
図9.PEG化されたPEG2のヨウ素染色。22又は43kD PEG化PEG2を6%Trisグリシンゲル上で移動させ、R8B12 FVIII抗体(列1及び2)又はヨウ素(列3及び4)を用いて染色した。2つの染色をそれらの分子量マーカーの列に従って整列させた。列1及び2はそれぞれ約30ngのFVIIIを含有するが、列3及び4は約2μgを含有する。
【0039】
図10.PEG化及び非PEG化PEG2のMALDI質量分析。MALDI質量分析はPEG2(図10a)又は22kD PEG化PEG2(図10b)について行なわれた。PEG化されると、PEG2の重(H)鎖ピークは非常に減少し、111kD(22kD PEG+89kD 重鎖)を中心とする新しいピーク(H+PEG)が現れる。100kD(22kD PEG+83kD 軽鎖)を中心とすることが予測されるPEG化された軽(L)鎖のピークは検出されない。
【0040】
図11.トロンビン切断の後のPEG化及び非PEG化PEG2のMALDI質量分析。
【0041】
図12.トロンビン切断の前後のPEG化PEG6のMALDI質量分析。
【0042】
図13.サイズ−排除カラム上で精製されたPEG化PEG2の280nmにおけるUV吸収分布。
【0043】
図14.カチオン交換カラム上で精製されたPEG化及び非PEG化PEG6の280nmにおけるUV吸収分布。
【0044】
図15.サイズ−排除カラム上で精製されたPEG化及び非PEG化PEG6の280nmにおけるUV吸収分布。
【0045】
図16.発色アッセイ及び凝固アッセイにより測定される場合のPEG化タンパク質の活性を非PEG化タンパク質の活性と比較する。精製された全長FVIIIをKG−2と示す。報告されるパーセント活性は、還元及び還元剤の除去後にPEGで処理された試料の値を、PEG化の収率を考慮しながら緩衝液標準で処理された試料の値で割ることにより決定された。
【0046】
図17.PEG2と比較されるPEG化PEG2のウサギPK研究。
【0047】
図18.BDD及びPEG2と比較されるPEG化PEG2のウサギPK研究。P−値はPEG化PEGとBDDの間の比較である。
【0048】
図19.BDD及びPEG6と比較されるPEG化PEG6のウサギPK研究。
【0049】
図20.非変更全長FVIIIと比較されるPEG化野生型全長(“fl”)FVIIIのウサギPK研究。
【0050】
図21.PEG6及びBDDと比較されるPEG化PEG6の血友病マウスPK研究。
【0051】
図22.BDDと比較される22及び43kD PEG化PEG2の正常マウスPK研究。
【0052】
図23.時間経過中ずっと(full time course)、BDDと比較される22kD PEG化PEG2の正常マウスPK研究。
【0053】
図24.血友病マウスアッセイにおけるBDD因子VIIIの2つの種の半減期の薬物動態学的(PK)評価を描く、血友病マウス(BDD)因子VIII回収率ヒストグラム。
【0054】
図25.BDDと比較される22kD PEG化PEG2の血友病マウス腎臓裂傷研究。ビヒクル処置されたマウスは体重のグラム当たり25uLの血液損失を有する。
【0055】
図26.増加する量のFVIII抗体の存在下におけるPEG化PEG2及びBDDの発色活性。抗体エピトープを括弧内に示す。
【0056】
図27.増加する量のFVIII mAB 413抗体の存在下におけるPEG化PEGの発色活性。
【0057】
図28.FVIIIへの阻害物質を発現した患者に由来するヒト血漿の存在下におけるBDD、43kD PEG化PEG2、33kD PEG化PEG6及び33kD ジPEG化PEG2+6の発色活性。阻害物質の力価及び血液収集の日付を上に記す。上の2つのパネルは、5−〜405−倍の患者血漿希釈において集められたデータを含む。下の左のパネルは、患者HRF−828血漿に関する1:15−倍希釈に焦点を当てている。下の右のパネルは、上の2つのパネルにおいて各FVIII試料のために用いられた0.064IU/mLが飽和用量でなかったことを確証する。
【0058】
図29.PEG化スクリーニング法及び正当化。上のパネルは、一過的に発現されるPEG突然変異タンパク質のPEG化スクリーニングの略図を示す。下のパネルは、重鎖(“H”)−特異的抗体(左)又は軽−鎖(“L”)特異的抗体(右)を用いるPEG化生成物のウェスタン分析を示す。PEG化されたバンドを点により強調する。“U”はH及びLの両方を含有するプロセシングされない材料である。
【0059】
図30.PEG15〜17のPEG化スクリーニング。重鎖(“H”)−特異的抗体(R8B12及び58.12)又は軽−鎖(“L”)特異的抗体(C7F7及びGM)を用いるPEG化生成物のウェスタン分析。3つのすべての突然変異タンパク質は重鎖に関して選択的であり、PEG15−PEG16>PEG17の相対的なPEG化効率を有する。PEG化されたバンドを点により強調する。“U”はH及びLの両方を含有するプロセ
シングされない材料である。
【0060】
図31.還元剤濃度の関数としてPEG2+14のPEG化を示すゲル。PEG2+14を4℃において30分間、67〜670uMのTCEPを用いて処理した。スピン−カラム(spin−column)により還元剤を除去し、続いて12kD PEGを用いてPEG化した。FVIIIの重及び軽鎖をそれぞれ“H”及び“L”により強調する。2つの点はPEG化された重及び軽鎖を指し示す。
【0061】
図32.67〜670uMのTCEPで処理され、続いて還元剤が除去されたPEG2+14のデコンボリューションされた質量スペクトル。
【発明を実施するための形態】
【0062】
発明の詳細な記述
本発明は、FVIII活性を有するポリペプチドをあらかじめ規定される部位において生物適合性ポリマーに共有結合させることができ、それはN−末端アミンにおいてではなく、且つそのようなポリペプチドは実質的にそれらの凝固剤活性を保持しているという発見に基づく。さらに、これらのポリペプチド複合体は増加した循環時間及び低下した抗原性を有する。本発明の複合体は、FVIIIへの無作為ポリマー結合又はN−末端における結合を有した先行技術の複合体を超えて有利である。特定部位の結合は、生物学的活性のために必要な領域を避ける変更を設計し、それにより実質的なFVIII活性を保持することを可能にする。それは、ポリマーを結合させてFVIIIクリアランスに含まれる部位において結合を遮断するように設計することも可能にする。特定部位の結合は、無作為なポリマーカップリングによる技術において生産される不均一な複合体ではなくて均一な生成物も可能にする。軽鎖のN−末端アミンにおける結合を避けることにより、本発明の複合体は、FVIIIポリペプチドの活性部位におけるリガンドの結合から可能な活性の損失を避ける。軽鎖のN−末端領域は、vWF因子のFVIIIへの会合に含まれると思われ、それは循環における安定化会合である。
【0063】
定義
生物適合性ポリマー。生物適合性ポリマーにはポリアルキレンオキシド、例えばこれらに限られないがポリエチレングリコール(PEG)、デキストラン、コロミン酸又は他の炭水化物に基づくポリマー、アミノ酸のポリマー、ビオチン誘導体、ポリビニルアルコール(PVA)、ポリカルボキシレート、ポリビニルピロリドン、ポリエチレン−コ−無水マレイン酸、ポリスチレン−コ−無水リンゴ酸、ポリオキサゾリン、ポリアクリロイルモルホリン、ヘパリン、アルブミン、セルロース、キトサンの加水分解産物、デンプン、例えばヒドロキシエチル−デンプン及びヒドロキシプロピル−デンプン、グリコゲン、アガロース及びその誘導体、グアゴム、プルラン、イヌリン、キサンタンゴム、カラゲナン、ペクチン、アルギン酸加水分解産物、他の生体高分子ならびにそれらの同等物が含まれる。好ましいのはポリエチレングリコールであり、さらにもっと好ましくはメトキシポリエチレングリコール(mPEG)である。他の有用なポリアルキレングリコール化合物はポリプロピレングリコール(PPG)、ポリブチレングリコール(PBG)、PEG−グリシジルエーテル(Epox−PEG)、PEG−オキシカルボニルイミダゾール(CDI−PEG)、分枝鎖状ポリエチレングリコール、直鎖状ポリエチレングリコール、フォーク状ポリエチレングリコール(forked polyethylene glycols)及び多−分枝もしくは「超分枝鎖状」ポリエチレングリコール(star−PEG)である。
【0064】
ポリエチレングリコール(PEG)。本明細書で用いられる「PEG」及び「ポリエチレングリコール」は互換性であり、いずれの水溶性ポリ(エチレンオキシド)も含む。典型的には、本発明に従う使用のためのPEGsは以下の構造、「−(OCH2CH2)n
−」を含み、ここで(n)は2〜4000である。本明細書で用いられる場合、PEGは、末端酸素が置き換えられているかもしくはいないかに依存して、「−CH2CH2−O(CH2CH2O)n−CH2CH2−」及び「−(OCH2CH2)nO−」も含む。明細書及び請求項全体を通じて、「PEG」という用語は種々の末端もしくは「末端キャッピング」基、例えば制限ではないがヒドロキシル又はC1−20アルコキシ基を有する構造を含むことを思い出さねばならない。「PEG」という用語は、大部分、すなわち50%より多くの−OCH2CH2−繰り返しサブユニットを含有するポリマーも意味する。特定の形態に関し、PEGは、多様な分子量ならびに構造又は形状、例えば分枝鎖状、直鎖状、フォーク状及び多官能基性のいくつを帯びることもできる。
【0065】
PEG化。PEG化は、ポリエチレングリコール(PEG)がタンパク質のような分子に共有結合する過程である。
【0066】
活性化もしくは活性官能基。生物適合性ポリマーのような官能基が活性化されたと記述される場合、官能基は他の分子上の求電子基(electrophile)又は求核基(nucloephile)と容易に反応する。
【0067】
Bドメイン欠失FVIII(BDD)。本明細書で用いられる場合、BDDは、FVIIIのB−ドメインの14個のアミノ酸以外のすべての欠失を含有するアミノ酸配列を有することを特徴とする。B−ドメインの最初の4個のアミノ酸(SFSQ,配列番号:1)はB−ドメインの最後の10個の残基(NPPVLKRHQR,配列番号:2)に連結している。(Lind,P.et al.著,Eur.J.Biochem.232,1995年,pp.19−27)。本明細書で用いられるBDDは配列番号:3のアミノ酸配列を有する。
【0068】
FVIII。血液凝固因子VIII(FVIII)は、肝臓により合成されて血流中に放出される糖タンパク質である。循環血中で、それはフォンビルブラント因子(vWF、因子VIII−関連抗原としても既知)に結合して、安定な複合体を形成する。トロンビンにより活性化されると、それは複合体から解離し、凝固カスケードにおいて他の凝固因子と相互作用し、それは結局血栓の形成を生ずる。ヒト全長FVIIIは配列番号:4のアミノ酸配列を有するが、対立遺伝子変異体が可能である。
【0069】
機能性因子VIIIポリペプチド。本明細書で用いられる場合、機能性因子VIIIポリペプチドは、生体内もしくは試験管内で、例えば血友病Aにより特徴付けられるヒト因子VIII欠失を修正することができる機能性ポリペプチド又はポリペプチドの組み合わせを示す。因子VIIIは自然の状態で複数の分解もしくはプロセシング形態を有する。これらは本明細書で示される通り、1個の鎖状タンパク質(one chain protein)である前駆体からタンパク質分解的に誘導される。機能性因子VIIIポリペプチドはそのような1個の鎖状タンパク質を含み、且つヒト因子VIII欠失を修正する生物学的活性を有するこれらの種々の分解産物も示す。対立遺伝子変異がおそらく存在する。機能性因子VIIIポリペプチドは、因子VIIIの誘導体を生ずるそのような対立遺伝子変異、グリコシル化形、変更及びフラグメントのすべてを、それらがヒト因子VIIIの機能性セグメントを含有し、必須の特徴的なヒト因子VIII機能的活性が本質的に留まっている限り含む。必要な機能的活性を保有する因子VIIIの誘導体は、本明細書に記載される直接的な試験管内試験により容易に同定され得る。さらに、機能性因子VIIIポリペプチドは、因子IXa、カルシウム及びリン脂質の存在下で因子XのXaへの転換を触媒することができ、ならびに血友病Aに冒された患者に由来する血漿における凝固欠陥を修正することができる。本明細書におけるヒト因子VIIIアミノ酸配列及び機能性領域の配列の開示から、DNAの制限酵素切断を介して、あるいはヒト因子VIIIタンパク質のタンパク質分解的もしくは他の分解を介して誘導され得るフラグメントは
、当該技術分野における熟練者に明らかになるであろう。
【0070】
FIX。本明細書で用いられる場合、FIXは凝固因子IXを意味し、それはヒト凝固因子IX又は血漿トロンボプラスチン成分としても既知である。
【0071】
FX。本明細書で用いられる場合、FXは凝固因子Xを意味し、それはヒト凝固因子Xの名前及び名祖(eponym)Stuart−Prower因子によっても既知である。
【0072】
薬物動態学。「薬物動態学」(「PK」)は、体内における薬剤の吸収、分布、代謝及び除去の性質を記述するために用いられる用語である。薬剤の薬物動態学の向上は、薬剤を生体内で治療薬としてより有効にする特性、特に体内におけるその有用な持続時間における向上を意味する。
【0073】
突然変異タンパク質。突然変異タンパク質は、タンパク質又はポリペプチドへの実験室誘導突然変異の結果として生ずる遺伝子操作されたタンパク質である。
【0074】
タンパク質。本明細書で用いられる場合、タンパク質とポリペプチドは同義語である。
【0075】
FVIIIクリアランス受容体。本明細書で用いられるFVIIIクリアランス受容体は、1種もしくはそれより多い他の分子と結合又は会合して循環からのFVIIIクリアランスを生ずる機能性FVIIIポリペプチド上の受容体領域を意味する。因子VIIIクリアランス受容体には、制限ではないがLRP、LDL受容体及び/又はHSPGに結合するFVIII分子の領域が含まれる。
【0076】
議論
本発明の方法に従い、いずれの機能性因子VIIIポリペプチドもあらかじめ決められた部位で突然変異させ、次いでその部位において生物適合性ポリマーに共有結合させることができると考えられる。有用なポリペプチドには、配列番号:4に示されるアミノ酸配列を有する全長因子VIII及び配列番号:3に示されるアミノ酸配列を有するBDD FVIIIが含まれるが、これらに限られない。好ましいのはBDD FVIIIである。
【0077】
本発明の複合体において用いられる生物適合性ポリマーは、上記のポリマーのいずれであることもできる。生物適合性ポリマーは、薬物動態学における所望の向上を与えるように選ばれる。例えばポリマーの正体、サイズ及び構造は、活性における許容され得ない低下なしでFVIII活性を有するポリペプチドの循環半減期を向上させるように、あるいはポリペプチドの抗原性を低下させるように選ばれる。好ましくは、ポリマーはPEGを含み、さらにもっと好ましくはその分子量の少なくとも50%をPEGとして有する。1つの態様において、ポリマーは末端−キャッピング部分、例えばヒドロキシル、アルコキシ、置換アルコキシ、アルケノキシ、置換アルケノキシ、アルキノキシ、置換アルキノキシ、アリールオキシ及び置換アリールオキシで末端がキャッピングされたポリエチレングリコールである。さらにもっと好ましいのは、メトキシポリエチレングリコールを含んでなるポリマーである。さらにもっと好ましいのは、3kD〜100kD、そしてより好ましくは5kD〜64kD又は5kD〜43kDのサイズ範囲を有するメトキシポリエチレングリコールを含んでなるポリマーである。
【0078】
好ましくは、ポリマーは反応性部分を有する。例えば1つの態様において、ポリマーはスルフヒドリル反応性部分を有し、それは機能性因子VIIIポリペプチド上の遊離のシステインと反応して共有結合を形成することができる。そのようなスルフヒドリル反応性
部分にはチオール、トリフレート、トレシレート、アジリジン、オキシラン、S−ピリジル又はマレイミド部分が含まれる。好ましいのはマレイミド部分である。1つの態様において、ポリマーは直鎖状であり、一方の末端にスルフヒドリルに対して強く反応性でない「キャップ」(例えばメトキシ)を、且つ他方の末端にスルフヒドリル反応性部分を有する。好ましい態様において、複合体はPEG−マレイミドを含み、5kD〜64kDのサイズ範囲を有する。
【0079】
有用な生物適合性ポリマーの選択のためのさらなる指針は、下記の実施例に示される。
【0080】
FVIII活性を有するポリペプチドをコードするヌクレオチド配列の特定部位の突然変異は、当該技術分野において既知のいずれの方法によっても起こり得る。好ましい方法は、ポリマーの共有結合のために選ばれる部位にシステインコドンを導入するための突然変異誘発を含む。これは、商業的に入手可能な特定部位の突然変異誘発キット、例えばStratagene cQuickChangeTM II特定部位の突然変異誘発キット、Clontech Transformer特定部位の突然変異誘発キット no.K1600−1、Invitrogen GenTaylor特定部位の突然変異誘発システム no.12397014、Promega Altered Sites II
試験管内突然変異誘発システムキット no.Q6210又はTakara Mirus Bio LA PCR突然変異誘発キット no.TAK RR016を用いて行なうことができる。
【0081】
最初に機能性FVIIIポリペプチドの表面上の1個もしくはそれより多いアミノ酸に関するコドンをシステインに関するコドンで置換して組換え発現系においてシステイン突然変異タンパク質を生産し、突然変異タンパク質をシステイン−特異的ポリマー試薬と反応させ、突然変異タンパク質を精製することにより、本発明の複合体を調製することができる。
【0082】
この系において、システイン部位におけるポリマーの付加は、ポリマー上のマレイミド活性官能基を介して行なうことができる。この方法の例を下記に示す。用いられるスルフヒドリル反応性ポリマーの量は、誘導体化されるべきシステインのモル量に少なくとも等モル量でなければならず、好ましくは過剰に存在する。好ましくは、少なくとも5−倍モル過剰のスルフヒドリル反応性ポリマーが用いられ、さらにもっと好ましくは、少なくとも10−倍過剰のそのようなポリマーが用いられる。共有結合に有用な他の条件は、当該技術分野における者の熟練の範囲内である。
【0083】
下記の実施例において、突然変異タンパク質は当該技術分野において通常の方法で命名される。突然変異体の命名のための約定(convention)は、配列番号:4に示される成熟全長因子VIIIに関するアミノ酸配列に基づく。分泌タンパク質として、FVIIIは翻訳過程の間にタンパク質分解により切断されるシグナル配列を含有する。19個のアミノ酸のシグナル配列の除去に続き、分泌されるFVIII生成物の最初のアミノ酸はアラニンである。
【0084】
通常であり且つ本明細書で用いられる通り、BDD FVIII中の突然変異アミノ酸に言及する場合、突然変異アミノ酸は全長FVIIIの配列中のその位置により称される。例えば下記で議論されるPEG6突然変異タンパク質はK1808Cと称され、それは、それが全長配列中の1808に類似の位置においてリシン(K)がシステイン(C)に変更されているからである。
【0085】
ポリマーの共有結合のためにあらかじめ規定される部位は、FVIII活性に含まれないか、又は生体内でFVIIIを安定化する他の機構、例えばvWFへの結合に含まれな
い、ポリペプチドの表面上に露出されている部位から最適に選ばれる。そのような部位は、FVIIIが不活性化されるか、又は循環からクリアランスされる機構に含まれることが知られている部位からも最適に選ばれる。これらの部位の選択は、下記で詳細に議論される。好ましい部位には、(a)低密度リポタンパク質受容体関連タンパク質、(b)ヘパリン硫酸プロテオグリカン、(c)低密度リポタンパク質受容体及び/又は(d)因子VIII阻害性抗体に関する結合部位内もしくはそれらの近辺のアミノ酸残基が含まれる。「結合部位内もしくはそれらの近辺」により、結合部位に十分に近く、部位への生物適合性ポリマーの共有結合が結合部位の立体障害を生ずるであろう残基を意味する。そのような部位は、例えば結合部位の20Å以内であると思われる。
【0086】
本発明の1つの態様において、生物適合性ポリマーは(a)上記で定義された因子VIIIクリアランス受容体、(b)因子VIIIを分解することができるプロテアーゼのための結合部位及び/又は(c)因子VIII阻害性抗体のための結合部位内又はそれらの近辺のアミノ酸残基において機能性因子VIIIポリペプチドに共有結合する。プロテアーゼは活性化タンパク質C(APC)であることができる。他の態様において、生物適合性ポリマーは機能性因子VIIIポリペプチド上のあらかじめ規定される部位において共有結合し、ポリペプチドへの低−密度リポタンパク質受容体関連タンパク質の結合は、ポリペプチドが複合されない場合のそれへの結合より少なく、好ましくは2分の1より少ない。1つの態様において、生物適合性ポリマーは機能性因子VIIIポリペプチド上のあらかじめ規定される部位において共有結合し、ヘパリン硫酸プロテオグリカンのポリペプチドへの結合は、ポリペプチドが複合されない場合のそれへの結合より少なく、好ましくは2分の1より少ない。さらに別の態様において、生物適合性ポリマーは機能性因子VIIIポリペプチド上のあらかじめ規定される部位において共有結合し、因子VIII阻害性抗体のポリペプチドへの結合は、ポリペプチドが複合されない場合のそれへの結合より少なく、好ましくはポリペプチドが複合されない場合のそれへの結合の2分の1より少ない。別の態様において、生物適合性ポリマーは機能性因子VIIIポリペプチド上のあらかじめ規定される部位において共有結合し、低密度リポタンパク質受容体のポリペプチドへの結合は、ポリペプチドが複合されない場合のそれへの結合より少なく、好ましくは2分の1より少ない。別の態様において、生物適合性ポリマーは機能性因子VIIIポリペプチド上のあらかじめ規定される部位において共有結合し、血漿プロテアーゼがポリペプチドを分解する程度は、ポリペプチドが複合されない場合より低い。さらに別の態様において、血漿プロテアーゼによるポリペプチドの分解は、同じ条件下で同じ時間に及んで測定される場合に、ポリペプチドが複合されない場合のその分解の2分の1より少ない。
【0087】
FVIIIに関するLRP、LDL受容体又はHSPG結合親和性は、表面プラスモン共鳴法(Biacore)を用いて決定することができる。例えばFVIIIを直接又はFVIII抗体を介して間接的にBlacoreTMチップにコーティングし、種々の濃度のLRPをチップ上に通過させて相互作用の開始速度(on−rate)及び終了速度(off−rate)の両方を測定することができる(Bovenschen N.et
al.著,J.Biol.Chem.278(11),2003年,pp.9370−7)。2つの速度の比率は親和性の尺度を与える。PEG化した場合の2分の1、好ましくは5分の1、より好ましくは10分の1、そしてさらにもっと好ましくは30分の1への親和性における低下が望ましい。
【0088】
プロテアーゼAPCによるFVIIIの分解は、当該技術分野における熟練者に既知のいずれの方法によっても測定することができる。
【0089】
1つの態様において、生物適合性ポリマーは因子VIIIアミノ酸位置81、129、377、378、468、487、491、504、556、570、711、1648、1795、1796、1803、1804、1808、1810、1864、1903
、1911、2091、2118及び2284の1個もしくはそれより多くにおいてポリペプチドに共有結合する。別の態様において、生物適合性ポリマーは因子VIIIアミノ酸位置377、378、468、491、504、556、1795、1796、1803、1804、1808、1810、1864、1903、1911及び2284の1個もしくはそれより多くにおいてポリペプチドに共有結合し、且つ(1)低−密度リポタンパク質受容体関連タンパク質への複合体の結合は低−密度リポタンパク質受容体関連タンパク質への非複合ポリペプチドの結合より少ないか;(2)低−密度リポタンパク質受容体への複合体の結合は低−密度リポタンパク質受容体への非複合ポリペプチドの結合より少ないか;あるいは(3)低−密度リポタンパク質受容体関連タンパク質及び低−密度リポタンパク質受容体の両方への複合体の結合は低−密度リポタンパク質受容体関連タンパク質及び低−密度リポタンパク質受容体への非複合ポリペプチドの結合より少ない。
【0090】
さらに別の態様において、生物適合性ポリマーは因子VIIIアミノ酸位置377、378、468、491、504、556及び711の1個もしくはそれより多くにおいてポリペプチドに共有結合し、且つヘパリン硫酸プロテオグリカンへの複合体の結合はヘパリン硫酸プロテオグリカンへの非複合ポリペプチドの結合より少ない。さらに別の態様において、生物適合性ポリマーは因子VIIIアミノ酸位置81、129、377、378、468、487、491、504、556、570、711、1648、1795、1796、1803、1804、1808、1810、1864、1903、1911、2091、2118及び2284の1個もしくはそれより多くにおいてポリペプチドに共有結合し、且つ複合体は非複合ポリペプチドより少ない因子VIII阻害性抗体への結合を有する。さらに別の態様において、生物適合性ポリマーは因子VIIIアミノ酸位置81、129、377、378、468、487、491、504、556、570、711、1648、1795、1796、1803、1804、1808、1810、1864、1903、1911、2091、2118及び2284の1個もしくはそれより多くにおいて、そして好ましくは位置377、378、468、491、504、556及び711の1個もしくはそれより多くにおいてポリペプチドに共有結合し、且つ複合体は、非複合ポリペプチドが有するより少ない因子VIIIを分解できる血漿プロテアーゼからの分解を有する。より好ましくは、血漿プロテアーゼは活性化タンパク質Cである。
【0091】
さらに別の態様において、生物適合性ポリマーはアミノ酸位置129、491、1804及び/又は1808において、より好ましくは491又は1808においてB−ドメイン欠失因子VIIIに共有結合する。さらに別の態様において、生物適合性ポリマーは因子VIIIアミノ酸位置1804においてポリペプチドに結合し、且つポリエチレングリコールを含む。好ましくは、生物適合性ポリマー結合のための1個もしくはそれより多いあらかじめ規定される部位は、部位特異的システイン突然変異により制御される。
【0092】
機能性因子VIIIポリペプチド上の1個もしくはそれより多い部位、好ましくは1もしくは2個の部位がポリマー結合のためにあらかじめ規定される部位であることができる。特定の態様において、ポリペプチドはモノ−PEG化又はジPEG化される。
【0093】
本発明は、機能性因子VIIIポリペプチドをコードするヌクレオチド配列を突然変異させてあらかじめ規定される部位においてシステイン残基に関するコード配列に置換し;突然変異したヌクレオチド配列を発現させてシステイン増強突然変異タンパク質を生産し;突然変異タンパク質を精製し;実質的に還元システイン残基のみにおいてポリペプチドと反応するように活性化された生物適合性ポリマーと突然変異タンパク質を反応させて複合体を形成し;そして複合体を精製することを含んでなる、複合体の調製方法にも関する。他の態様において、本発明は:(a)特定部位の因子VIII突然変異タンパク質を発現させ、ここで突然変異タンパク質は因子VIII突然変異タンパク質の露出された表面上のアミノ酸残基に代わるシステイン置換を有し、且つそのシステインはキャッピングさ
れており;(b)システイン突然変異タンパク質を穏やかに還元し、且つキャップを放出する条件下でシステイン突然変異タンパク質を還元剤と接触させ;(c)システイン突然変異タンパク質からキャップ及び還元剤を除去し;そして(d)還元剤の除去から少なくとも約5分、そして好ましくは少なくとも15分、さらにもっと好ましくは少なくとも30分後に、PEG化された因子VIII突然変異タンパク質が生産されるような条件下で、スルフヒドリルカップリング部分を含んでなるPEGでシステイン突然変異タンパク質を処理することを含んでなる、因子VIII突然変異タンパク質の特定部位のPEG化のための方法を提供する。PEGのスルフヒドリルカップリング部分は、チオール、トリフレート、トレシレート、アジリジン、オキシラン、S−ピリジル及びマレイミド部分より成る群から選ばれ、好ましくはマレイミドである。
【0094】
本発明は、本発明の複合体の治療的に有効な量及び製薬学的に許容され得る添加剤を含んで成る非経口的投与のための製薬学的組成物にも関する。製薬学的に許容され得る添加剤は、調製物の調製を助けるか、又は安定化するために活性成分に加えることができ、且つ患者に有意な毒物学的悪影響を起こさない物質である。そのような添加剤の例は当該技術分野における熟練者に周知であり、水、糖類、例えばマルトースもしくはスクロース、アルブミン、塩類などが含まれる。他の添加剤は、例えばE.W.MartinによるRemington’s Pharmaceutical Sciencesに記載されている。そのような組成物は本明細書の複合体の有効量を、宿主への有効な投与に適した製薬学的に許容され得る組成物を調製するために適した量のビヒクルと一緒に含有するであろう。例えば血友病Aに苦しむ患者に、出血事例の重度とともに変わり得る投薬量で非経口的に複合体を投与することができる。静脈内に投与される平均投薬量は、術前適用のためにキログラム当たり40単位、少ない出血のためにキログラム当たり15〜20単位及び保持投薬のためにキログラム当たり8−時間に及んで投与される20〜40単位の範囲内である。
【0095】
1つの態様において、本発明の方法は1個もしくはそれより多い表面BDDアミノ酸をシステインで置換し、哺乳類発現系においてシステイン突然変異タンパク質を生産し、生育培地からのシステインにより発現の間にキャッピングされたシステインを還元し、還元剤を除去してBDDジスルフィドを再生させ、システイン−特異的な生物適合性ポリマー試薬、例えばPEG−マレイミドと反応させることを含む。そのような試薬の例は、Nektar Therapeutics of San Carios,CAからそれぞれNektarカタログ番号2D2M0H01 mPEG−MAL分子量5,000Da、2D2M0P01 mPEG−MAL分子量20kD、2D3X0P01 mPEG2−MAL分子量40kDの下に入手可能な5、22又は43kDあるいはNOF Corporation,Tokyo,JapanからそれぞれNOFカタログ番号Sunbright ME−120MA及びSunbright ME−300MAの下に入手可能な12又は33kDのようなPEGサイズを有するPEG−マレイミドである。PEG化生成物は、未反応PEGの除去のためにイオン−交換クロマトグラフィーを用いて、及び未反応BDDの除去のためにサイズ−排除クロマトグラフィーを用いて精製される。この方法を用いてFVIIIとの望ましくない相互作用、例えば受容体−媒介クリアランス、阻害性抗体結合及びタンパク質分解酵素による分解を同定し、且つ選択的に遮蔽することができる。我々は、Nektar又はNOFにより5kDとして供給されるPEG試薬が我々の実験室で6kDとして判断され(tested)、類似して我々の実験室では直鎖状20kDとして供給されるPEG試薬が22kDとして判断され、40kDとして供給されるものが43kDとして判断され、60kDとして供給されるものが64kDとして判断されたことに注意した。混乱を避けるために、我々は本明細書の議論において、5kD
PEGを除いて我々の実験室で判断された通りの分子量を用い、5kD PEGは製造者がそれを同定した通りに5kDとして報告する。
【0096】
BDDの位置491及び1808におけるシステイン突然変異(上記で開示された)の他に、位置487、496、504、468、1810、1812、1813、1815、1795、1796、1803及び1804をシステインに突然変異させ、PEG化された時におそらくLRP結合の遮断を可能にした。位置377、378及び556もシステインに突然変異させ、PEG化された時にLRP及びHSPG結合の両方の遮断を可能にした。位置81、129、422、523、570、1864、1911、2091及び2284はBDD上で等しく空間的に隔たるように選ばれ、これらの位置における大きなPEGs(>40kD)を用いる特定部位のPEG化は本来のグリコシル化部位(41、239及び2118)及びLRP結合部位におけるPEG化と一緒になってBDDの表面を完全に覆い、BDDに関する新規なクリアランス機構を同定するはずである。
【0097】
1つの態様において、細胞培養培地はシステインを含有し、それはジスルフィド結合の形成により突然変異タンパク質のシステイン残基を「キャッピング」する。複合体の調製において、組換え系で生産されるシステイン突然変異タンパク質は培地からのシステインでキャッピングされ、このキャップはシステイン−特異的ポリマー試薬の添加の前に、キャップを放出する穏やかな還元により除去される。当該技術分野における熟練者に明らかな通り、FVIIIの部位−特異的突然変異のための当該技術において既知の他の方法も用いることができる。
【実施例】
【0098】
FVIIIの構造活性関連性分析。FVIII及びBDD FVIIIは、生物学的反応に含まれる多種の部位を有する非常に大きな複合(complex)分子である。薬物動態学的性質を向上させるためにそれらを共有結合により変更する以前の試みは、混同した結果を有した。分子を特異的に突然変異させ、次いで部位−特異的なやり方でポリマーを加えることができることは驚くべきことである。さらに、非特異的な付加及び活性の低下を引き起こす過去のポリマー複合体の場合の問題を考えると、向上した薬物動態学的性質及び保持される活性の結果も驚くべきことである。
【0099】
1つの態様において本発明は、PEG−マレイミドのようなシステイン−特異的リガンドを用いる特定部位の突然変異誘発に関する。非−突然変異BDDはPEG−マレイミドと反応するために利用できるシステインを有しておらず、従って突然変異システイン位置のみがPEG化の部位となるであろう。さらに特定的に、BDD FVIIIは19個のシステインを有し、その中の16個はジスルフィドを形成し、その中の他の3個は遊離のシステインである(McMullen et al.著,Protein Sci.4,1995年,pp.740−746)。BDDの構造モデルは、3個の遊離のシステインのすべてが埋もれていることを示唆している(Stoliova−McPhie et al.著,Blood 99,2002年,pp.1215−1223)。酸化されたシステインはPEG−マレイミドによりPEG化され得ないので、BDD中でジスルフィドを形成する16個のシステインは、最初に還元されずにはPEG化され得ない。BDDの構造モデルに基づき、BDD中の3個の遊離のシステインは、最初にタンパク質を変性させ、これらのシステインをPEG試薬に露出せずにはPEG化され得ない。かくしてBDD構造を劇的に変えることなく、本来のシステイン残基におけるPEG化によりBDDの特異的なPEG化を達成することは実行可能であると思われず、BDDの構造を劇的に変えることは、おそらくその機能を破壊するであろう。
【0100】
全長FVIIIのBドメイン中の4個のシステインのレドックス状態は未知である。Bドメイン中の4個のシステインのPEG化は、それらがジスルフィドを形成しておらず、表面に露出されていれば可能であり得る。しかしながら、全長FVIII及びBDDは類似の薬物動態学的(PK)側面及び生体内における類似の半減期を有するので(Gruppo et al.著,Haemophilia 9,2003年,pp.251−26
0)、BドメインPEG化は、PEGが偶然非−Bドメイン領域をも保護しなければ、向上した血漿半減期を生ずるとは思われない。
【0101】
FVIII活性を有するポリペプチド上のポリマー結合のためのあらかじめ規定される部位であって、因子VIII活性を保持し、且つ薬物動態学を向上させる部位を決定するために、BDD FVIIIに基づいて以下の指針を示す。変更はクリアランス、不活性化及び免疫原性機構、例えばLRP、HSPG、APCならびに阻害性抗体結合部位を標的としなければならない。Stoilova−McPhie,S.et al.著,Blood 99(4),2002年,pp.1215−23はBDDの構造を示している。例えば半減期を延長するために、1個のPEGをA2残基484−509及びA3残基1811−1818中のLRP結合部位もしくはその近辺における特異的な部位に導入することができる。これらの部位における嵩高いPEGの導入は、FVIIIがLRPに結合する能力を崩壊させ、循環からのFVIIIのクリアランスを減少させるはずである。活性に有意に影響せずに半減期を延長するために、PEGを残基1648において導入することができるとも思われ、それは全長分子中のBドメインとA3ドメインの連結部分にあり、BDDではA2及びA3ドメインの間の14−アミノ酸中にある(in the 14−amino acid liker I the BDD between the
A2 and A3 domains)。
【0102】
PEG化の特異性は、組換えDNA突然変異誘発法を用いて1個のシステイン残基をA2又はA3ドメイン中に工作し(engineering)、続いて導入されたシステインをPEG−マレイミドのようなシステイン−特異的PEG試薬を用いて部位−特異的にPEG化することにより達成され得る。484−509及び1811−1818におけるPEG化の他の利点は、これらの2つのエピトープが患者における阻害性抗原部位の3つの主な種類の2つに相当することである。循環半減期の向上及び免疫原性反応の低下の最大の効果を達成するために、A2及びA3 LRP結合部位の両方をPEG化し、ジPEG化生成物を与えることができる。1811−1818領域内におけるPEG化は、この領域がFIX結合にも含まれるために、活性の有意な損失を生じ得ることに注意しなければならない。558−565内における特定部位のPEG化はHSPG結合を廃止するが、この領域はFIXにも結合するので、活性も低下させ得る。
【0103】
FVIIIの新規なクリアランス機構を同定するために、さらに別の表面部位をPEG化することができる。A2ドメインのPEG化は、活性化されるとA2ドメインがFVIIIから解離し、おそらくそのより小さいサイズの故にFVIII分子の残りの部分より速く循環から除去される点で追加の利点を与えることができる。他方、PEG化されたA2は、腎臓クリアランスを逃れるのに十分に大きく、FVIIIの残りの部分と同等の血漿半減期を有することができ、かくして生体内で活性FVIIIを再構築することができる。
【0104】
A2及びA3領域内のPEG化部位の同定。推定A2 LRP結合領域もしくはその近辺における5個の位置(PEG1−5位置に対応するY487、L491、K496、L504及びQ468)を、高い表面露出及びそれらのCαからCβへの軌道(trajectory)の外側への向きに基づき、特定部位のPEG化のための例として選んだ。さらに、これらの残基は分子の三−次元構造において互いから大体同じ距離にあり、それらは一緒になってこの領域全体を表す(represent)ことができる。推定A3 LRP結合領域もしくはその近辺における8個の位置(PEG6−14に対応する1808、1810、1812、1813、1815、1795、1796、1803、1804)を特定部位のPEG化のための例として選んだ。PEG6(K1808)は1811−1818及び1810における自然のN−結合グリコシル化部位に隣接している。位置1810(PEG7)におけるPEG化は糖をPEGで置換するであろう。PEG8位置T
1812における突然変異もグリコシル化部位を廃止するであろう。PEG9位置(K1813)は内側に向いていると予測されたが、構造モデルが正しくない場合にそれは選ばれた。PEG10(Y1815)はLRP結合ループ内の嵩高い疎水性アミノ酸であり、疎水性アミノ酸は典型的にはタンパク質−タンパク質相互作用の中心に存在するので、決定的な相互作用残基であり得る。1811−1818領域はLRP及びFIX結合の両方に含まれると報告されているので、このループ内におけるPEG化はおそらく活性の低下を生ずると考えられた。かくしてPEG11−PEG14(1795、1796、1803、1804)は1811−1818ループ近辺にあるがループ内にないように設計され、種々のPEGサイズを用いてLRP及びFIX結合を解離させる(dissociate)ことができるようにした。
【0105】
両LRP結合部位を同時に遮断するために、例えばPEG2及びPEG6位置における二重PEG化を発生させることができる。
【0106】
558−565領域はHSPG及びFIXの両方に結合することが示されているので、この領域内で部位は設計されなかった。代わりに、結合PEGが両方の相互作用を妨げ、且つそれらの間のあり得る相互作用を崩壊させることができるように、PEG15−PEG17(377、378及び556)がA2 LRP及びHSPG結合領域の間に設計された。表面露出され且つ外側に向いているさらに別の部位をLRP及びHPSG結合領域内もしくはその近辺で選ぶこともできた。新規なクリアランス機構を同定するために、FVIIIを系統的にPEG化することができる。PEG1−17の他に、3個の他の自然のグリコシル化部位、すなわちPEG18−20に対応するN41、N239及びN2118をPEG化のための結合点(tethering points)として用いることができ、それはそれらが表面露出されているはずだからである。PEG2、PEG6及び4個のグリコシル化部位のCβ原子から20オングストローム半径内の表面領域は、vWF、FIX、FX、リン脂質及びトロンビンに関する機能的相互作用部位の他にBDDモデル上にマッピングされた。
【0107】
Y81、F129、K422、K523、K570、N1864、T1911、Q2091及びQ2284に対応するPEG21−29は次いで、それらのCβ原子のそれぞれからの20オングストローム半径で残るBDD表面のほとんど全体を覆うそれらの能力に基づいて選択された。これらの位置は、それらが完全に露出されており、外側に向いており、且つ自然のシステインからずっと離れていて正しくないジスルフィド形成の可能性を最小にする故にも選択された。20オングストローム半径は、64kD分枝鎖状PEGのような大きなPEGが約20オングストローム半径を有する球を覆う可能性を有すると思われる故に選ばれる。PEG2及びPEG6ならびにグリコシル化部位PEG18、19及び20と一緒のPEG21−29のPEG化は、FVIIIの非−機能性(non−functional)表面のほとんど全体を保護するようである。
【0108】
PK側面の向上、より高い安定性又は免疫原性の低下のような強化された性質に導くPEG化位置を組み合わせて、最大に強化された性質を有する多重−PEG化生成物を形成することができる。PEG30及びPEG31は、それぞれA2及びA3ドメイン中の露出されたジスルフィドを除去することにより設計された。PEG30又はC630Aは、そのジスルフィドパートナーC711をPEG化のために遊離させるはずである。同様に、PEG31、C1899AはC1903がPEG化されるのを可能にするはずである。
【0109】
突然変異誘発。PEG化のために選ばれる部位においてシステインコドンを導入することにより、FVIIIの特定部位のPEG化のための基質を形成することができる。PEG突然変異体のすべての生産のためにStratagene cQuickChangeTM II特定部位の突然変異誘発キットを用いた(Stratagene Corpo
ration,La Jolla,CAからのStratageneキット200523)。cQuikChangeTM特定部位の突然変異誘発法は、Pfu TurboR DNAポリメラーゼ及び温度サイクラーを用いて行なわれる。所望の突然変異を含有する2個の相補的オリゴヌクレオチドプライマーを、Pfu Turboを用いて伸張させ、それはプライマーを置き換えないであろう。野生型FVIII遺伝子を含有するdsDNAを鋳型として用いる。複数の伸張サイクルに続き、メチル化DNAに関して特異的なDpnlエンドヌクレアーゼを用いて生成物を消化する。突然変異を含有する新しく合成されたDNAはメチル化されないが、親の野生型DNAはメチル化される。次いで消化されたDNAを用いてXL−1 Blue超−感応細胞を形質転換する。
【0110】
突然変異誘発の効率はほとんど80%である。突然変異誘発反応はpSK207+BDD C2.6又はpSK207+BDDにおいて行なわれた(図1)。DNA配列決定により成功した突然変異誘発を確かめ、突然変異を含有する適したフラグメントを哺乳類発現ベクターpSS207+BDD中のFVIII主鎖中に転移させた。転移の後、突然変異のすべてを再び配列−確認した。A3突然変異タンパク質PEG6、7、8、9及び10の場合、ベクターpSK207+BDD C2.6において突然変異誘発を行なった。配列決定により確かめた後、突然変異体フラグメント、Kpnl/PmeをpSK207+BDD中にサブクローニングした。次いでBDD突然変異タンパク質をpSS207+BDD発現ベクター中にサブクローニングした。A3突然変異タンパク質PEG11、12、13、14の場合、突然変異誘発をベクターpSK207+BDD中で直接行い、配列−確認突然変異体BDDを次いでpSS207+BDD中にサブクローニングした。A2突然変異タンパク質PEG1、2、3、4、5の場合、突然変異誘発をpSK207+BDD C2.6ベクター中で行なった。配列が確認された突然変異体をpSK207+BDD中に、及び次いでpSS207+BDD中にサブクローニングした。
【0111】
突然変異誘発に用いられるプライマー(センス鎖(sense stand)のみ)を各反応に関して挙げる:
PEG1,Y487C:GATGTCCGTCCTTTGTGCTCAAGGAGATTACCA(配列番号:5)
PEG2,L491C:TTGTATTCAAGGAGATGCCCAAAAGGTGTAAAAC(配列番号:6)
PEG3,K496C:TTACCAAAAGGTGTATGCCATTTGAAGGATTTTC(配列番号:7)
PEG4,L504C:AAGGATTTTCCAATTTGCCCAGGAGAAATATTC(配列番号:8)
PEG5,Q468C:GATTATATTTAAGAATTGCGCAAGCAGACCATAT(配列番号:9)
PEG6,K1808C:TAGAAAAAACTTTGTCTGCCCTAATGAAACCAAAAC(配列番号:10)
PEG7,N1810C:AACTTTGTCAAGCCTTGCGAAACCAAAACTTAC(配列番号:11)
PEG8,T1812C:GTCAAGCCTAATGAATGCAAAACTTACTTTTGGA(配列番号:12)
PEG9,K1813C:CAAGCCTAATGAAACCTGCACTTACTTTTGGAAAG(配列番号:13)
PEG10,Y1815C:CTAATGAAACCAAAACTTGCTTTTGGAAAGTGCAAC(配列番号:14)
PEG11,D1795C:ATTTCTTATGAGGAATGCCAGAGGCAAGGAGCA(配列番号:15)
PEG12,Q1796C:TCTTATGAGGAAGATTGCAGGCAAGGA
GCAGGA(配列番号:16)
PEG13,R1803C:CAAGGAGCAGAACCTTGCAAAAACTTTGTCAAGCCT(配列番号:17)
PEG14,K1804C:GGAGGAGAACCTAGATGCAACTTTGTCAAGCCT(配列番号:18)
PEG15,K377C:CGCTCAGTTGCCAAGTGTCATCCTAAAACTTGG(配列番号:19)
PEG16,H378C:TCAGTTGCCAAGAAGTGTCCTAAAACTTGGGTA(配列番号:20)
PEG17,K556C:CTCCTCATCTGCTACTGCGAATCTGTAGATCAA(配列番号:21)
PEG18,N41C:CAAAATCTTTTCCATTCTGCACCTCAGTCGTGTAC(配列番号:22)
PEH19,N239C:GTCAATGGTTATGTATGCAGGTCTCTGCCAGGT(配列番号:23)
PEG20,N2118C:CAGACTTATGCAGGATGTTCCACTGGAACCTTA(配列番号:24)
PEG21,Y81C:ATCCAGGCTGAGGTTTGTGATACAGTGGTCATT(配列番号:25)
PEG22,F129C:GAAGATGATAAAGTCTGTCCTGGTGGAAGCCAT(配列番号:26)
PEG23,K422C:CAGCGGATTGGTAGGTGTTACAAAAAAGTCCGA(配列番号:27)
PEG24,K523C:GAAGATGGGCCAACTTGCTCAGATCCTCGGTGC(配列番号:28)
PEG25,K570C:CAGATAATGTCAGACTGCAGGAATGTCATCCTG(配列番号:29)
PEG26,N1864C:CACACTAACACACTGTGTCCTGCTCATGGGAGA(配列番号:30)
PEG27,T1911C:CAGATGGAAGATCCCTGCTTTAAAGAGAATTAT(配列番号:31)
PEG28,Q2091C:ACCCAGGGTGCCCGTTGCAAGTTCTCCAGCCTC(配列番号:32)
PEG29,Q2284C:AAAGTAAAGGTTTTTTGCGGAAATCAAGACTCC(配列番号:33)
PEG30,C630A:TTGCAGTTGTCAGTTGCTTTGCATGAGGTGGCA(配列番号:34)
PEG31,C1899A:AATATGGAAAGAAACGCTAGGGCTCCCTGCAAT(配列番号:35)
【0112】
突然変異タンパク質発現。ハイグロマイシンBに対する耐性を与えるベクターにおける挿入の後、PEG突然変異タンパク質を、製造者の指示に従って293 フェクチントランスフェクション試薬(Fectin Transfection Reagent)(Invitrogen Corp.Cat#12347−019)と複合させたHKB11細胞中にトランスフェクションした(米国特許第6,136,599号明細書)。トランスフェクションから3日後におけるFVIII発現をCoatest発色アッセイにより評価した(Chromogenix Corp.Cat#821033,実施例12発色アッセイを参照されたい)(表1)。トランスフェクションされた細胞を次いで、5%FBSが補足された生育培地中で、50□g/mlのHyg Bを用いる選択圧下に置いた。Hyg B−耐性コロニーが現れたら、それらを手で採集し、Coatest発色ア
ッセイによりFVIII発現に関してスクリーニングした。FVIII発現安定細胞を次いでHPPS補足物質を含有する培地に適応させた。細胞を拡大し、新しい培地を有する振盪フラスコ中にml当たり1X106個の細胞において播種した。3日後に収穫される組織培養液(TCF)をFVIII BDD突然変異タンパク質の精製のために用いた。TCFのFVIII活性をCoatestにより検定した(表1)。
【0113】
【表1】
【0114】
突然変異タンパク質精製。分泌される突然変異タンパク質FVIIIタンパク質を含有する細胞培養上澄み液を集めたら、0.2ミクロンの膜フィルターを介して上澄み液を濾過し、残る細胞を除去する。次いで限外濾過又はアニオン交換により上澄み液を濃縮する。次いでそれを免疫親和性カラム(immunoaffinity column)に適用し、そこで細胞培養培地成分及び宿主細胞タンパク質不純物の大部分を除去する。免疫親和性カラム溶出液を次いでスクロースを含有する調製緩衝液中へのダイアフィルトレーションにより緩衝液交換し、凍結する。発色アッセイにより、モノクローナルFVIII抗体カラムを通過する(across)タンパク質の収率及び回収率を評価した。クロマトグラフィーランの負荷物、通過流(flow through)、種々の溶出液画分、ストリップ及びダイアフィルトレーションされた溶出液の試料をFVIII活性に関して検定した(表2)。表2はモノクローナル抗体カラムからのPEG2突然変異タンパク質の回収率を示す。抗体はC7F7抗体である。表2におけるパーセント回収率は発色アッ
セイにより決定される。最終的な収率は73%であった。図2に示されるのは、モノクローナルFVIII抗体クロマトグラフィーカラム上で精製されたPEG2タンパク質の場合の、時間に関する280nmにおけるUV吸光度のプロットである。クロマトグラフィーは、Amersham BioscienceからのAKTAR Explorer 100クロマトグラフィーシステムを用いて行なわれた。この機器は多重−波長UV−可視モニター及び2mmのフローセルを用いる。PEG2突然変異タンパク質は高濃度の塩(high salt)の存在下でカラムから溶出し、溶出ピークは280nmにおける吸光度及びFVIII活性アッセイの両方により示される。
【0115】
【表2】
【0116】
PEG化。100倍を超える過剰のPEG:タンパク質比において、本来の全長FVIII又はBDDを還元及び変性なしでシステイン−特異的PEGによりPEG化することはできず(データは示されていない)、すべての本来のシステインがジスルフィドを形成しているか、又はFVIII内に埋もれているというBDD構造モデルに基づく仮説を確証している。上記で挙げた標準的な案を用いて発現し且つ精製されたFVIIIシステイン突然変異タンパク質をシステイン−特異的PEGマレイミド試薬を用いてPEG化することはできず、おそらくそれは導入されたFVIIIシステインが細胞生育培地中に存在するシステイン及びβ−メルカプトエタノールのようなスルフヒドリル基との反応により「キャッピングされている」からである。この問題はおそらく培地からシステイン及びβ−メルカプトエタノールを除去することにより解決され得るが、これはより低いFVIII生産を生じ得、細胞により放出されるスルフヒドリルが導入されたFVIIIシステインを遮断するのを妨げないであろう。
【0117】
本発明の他の側面において、FVIIIの部位−特異的PEG化を可能にする3−段階法を開発した(図3)。段階1において、約1μMにおける精製されたFVIIIシステイン突然変異タンパク質を約0.7mM トリス(2−カルボキシエチル)ホスフィン(TCEP)又は0.07mM ジチオトレイトール(DTT)のような還元剤を用い、4℃において30分間穏やかに還元し、「キャップ」を放出させる。段階2に、回転カラム(BioRadR)を介して試料を移動させるようなサイズ−排除クロマトグラフィー(SEC)法により「キャップ」と一緒に還元剤を除去し、導入されたシステインを遊離且つ還元された状態にしながらFVIIIジスルフィドの再形成を可能にする。段階3において、還元剤の除去から少なくとも30分後に、遊離されたFVIIIシステイン突然変異タンパク質を5〜64kDの範囲のサイズを有する少なくとも10−倍モル過剰のPEG−マレイミド(Nektar Therapeutics及びN.O.F.Corporation)を用い、4℃で少なくとも1時間処理する。この方法は、異なる個人により繰り返される数十回の反応に関し、再現性の良いデータを有する高度に一貫した生成物分布を与える。
【0118】
TCEPの除去のための回転カラム法は計ることができないので、ゲル濾過脱塩クロマトグラフィーが選ばれた。しかしながら、TCEPのスパイクサンプル(spike s
ample)を用いてこの方法を調べると、TCEPがカラム空隙において測定可能なレベルで溶出し、その低い分子量を有する分子から予測される塩画分中のみで溶出するのではないことが示された。ウェスタンブロットアッセイは、おそらくTCEPの不完全な除去の故の有意なバックグラウンドPEG化を示した。そうしている間に別の実験は、塩勾配と組み合わされたアニオン交換クロマトグラフィー媒体を用いて、C7F7精製材料を他のタンパク質不純物からさらに有意に精製できることを示した。そこで上記のTCEPを用いてC7F7材料を還元し、次いでアニオン交換カラム上で材料を処理する(process)ことが決定された。電荷の相違のために、FVIIIタンパク質は保持されるが、TCEPはカラムを通過して流れ、保持されない。同時に勾配塩溶出の間に、FVIIIタンパク質は残るタンパク質不純物の大部分から精製された。これは、後に起こるPEG化が、より純粋な出発材料を用いて理論的により均一であろうことを意味した。しかしながら、TCEPのスパイクサンプルを用いて調べると、勾配中でFVIIIと一緒に溶出する測定可能なレベルのTCEPが見出されることが示された。従って、アニオン交換クロマトグラフィーの後にゲル濾過脱塩クロマトグラフィーを行なうことが決定され、これらの2つの段階は順番に用いられると、TCEPの完全な除去及び非−特異的PEG化の除去を生じた。
【0119】
SDS PAGE及びウェスタンブロットによるPEG化分析。還元性6%TrisGlycine SDSポリアクリルアミドゲル(Invitrogen)上の電気泳動により、PEG化された生成物を分析することができる。電気泳動に続き、クーマシーブルーを用いてゲルを染色してすべてのタンパク質を同定するか、あるいは標準的なウェスタンブロット案に供してFVIIIの種々の領域上のPEG化パターンを同定することができる。それぞれFVIII重鎖のC−末端領域又はVIII軽鎖のN−末端領域に対して構成される(raised)マウスモノクローナルR8B12又はC7F7抗体を用いるブロットの染色は、それぞれの鎖のPEG化を同定するはずである。FVIIIの484−509領域に対する413抗体を用いる染色は、PEG1−4のような突然変異タンパク質に関してPEG化が実際に部位−特異的であるか否かを決定するであろう。同様に、FVIIIの1801−1823領域を認識するCLB−CAg A抗体を用いる染色は、PEG6−10のような突然変異タンパク質に関してPEG化が部位−特異的であるか否かを決定するであろう。
【0120】
PEG2(L491C)PEG化は、軽鎖より重鎖に関して選択的であり、特に484−509領域に関して選択的であることが示されたが(図4)、PEG6(K1808C)は重鎖より軽鎖に関して選択的であることが示された(図5)。
【0121】
図4に描かれる研究のために、TCEPを用いてPEG2突然変異タンパク質(列1及び8)を還元し、続いてTCEPを除去し(列2及び9)、5、12、22、33又は43kD PEG−マレイミドで処理する(列3−7及び10−14)。非PEG化FVIIIは非プロセシング(H+L)ならびにプロセシング重(H)及び軽(L)鎖バンドとして移動する。3つのすべてのバンドはクーマシーブルー染色されたゲル上で検出可能であるが(下の右)、鎖特異的抗体を用いるウェスタン染色はプロセシングされない対応する鎖のみを明らかにする。R8B12染色(上の左)を用いると、PEG2をPEG−マレイミドで処理する場合の重鎖(H)バンドは強度が劇的に低下し、新しいバンドが形成され、それはPEGのサイズに比例して親Hバンドより高く移動する。C7F7染色(下の左)を用いると、軽鎖(L)バンド(不均一なグリコシル化の故に多重バンド)は強度を変えない。両染色に関する非プロセシングH+Lバンドは、H鎖が非プロセシングFVIIIの一部であるために移動する。クーマシー染色も軽鎖よりずっと多い重鎖のPEG化、すなわちHバンド強度の低下を確証する。最後にPEG化バンドは、おそらく484−509への413抗体の結合を遮断する491の部位−特異的PEG化の故に、PEGサイズ−依存的やり方で、R8B12染色より413抗体染色上(上の右)で相対的によ
り強度を失う。列当たりに負荷されるFVIIIの量は2つの左のゲルの場合に約30ngであり、上の右のゲルの場合に約1000ngであり、下の右のゲルの場合に約2000ngである。
【0122】
還元及びそれに続く還元剤の除去は、FVIIIの移動を変えない(列1対2及び8対9)。PEG2への22kD PEGの付加は413抗体の結合を遮断し、491位置における特異的なPEG化と一致する(図4の上の右のゲル)。これはPEG化されたPEG2が人間においてより低い免疫原性を有するであろうことも示唆しており、それは、413抗体がヒトA2阻害性抗体と同じエピトープを共有することが示されているからである(Scandella et al.著,Thromb.Haemost.67,1992年,pp.665−71)。
【0123】
図5に描かれる研究のために、TCEPを用いてPEG6突然変異タンパク質を還元し、続いてTCEPを除去し(列1及び6)、5、12、22又は33kD PEG−マレイミドで処理する(列2−5及び7−10)。非PEG化FVIIIは非プロセシング(H+L)ならびにプロセシング重(H)及び軽(L)鎖バンドとして移動する。PEG6(K1808)突然変異は軽鎖上にあるので、PEG化は軽鎖上のみで検出され、重鎖上では検出されなかった。列当たりに負荷されるFVIIIの量は左のゲルの場合に約100ngであり、右のゲルの場合に約30ngである。
【0124】
標準として移動させたBDDは、上記の還元及び還元剤除去法の後でさえ、100−倍を超えるモル過剰のPEG−マレイミドを用いて処理した時に有意なPEG化を示さなかった(図6a)。同じ方法をPEG4及びPEG5にも適用した(図6a)。PEG2と比較して、これらの突然変異タンパク質は有効にPEG化されなかったが、それらはPEG2(L491C)に類似して重鎖に関して選択的であった。PEG6(K1080C)のPEG化効率は比較的低く、それはおそらくそれがN1810におけるN−結合グリコシル化部位に非常に近接しており、それが位置1808におけるPEG化を遮断し得るからである。かくして我々は、1810における本来のグリコシル化部位を除去するためにPEG7(N1810C)を設計した。頭−頭比較(head−to−head comparison)においてPEG7はPEG6と比較して向上したPEG化効率を示す(図6b)。類似して、PEG15はPEG2よりわずかに良いPEG化効率を示す。BDDの二重突然変異体であるPEG2+6は、PEG2が重鎖システイン突然変異であり、PEG6が軽鎖突然変異であるために、重鎖及び軽鎖の両方の上でPEG化され得る(図6c)。この方法を野生型全長FVIIIにも適用した(図6d)。PEG化は、A1、A2及びBドメインのほとんどを含む重鎖の最大のフラグメントに関して検出された。PEG化パターンはモノPEG化を示唆し、1個のPEG化されたシステインのみがあることを示唆した。
【0125】
トロンビン切断及びウェスタンブロットによるPEG化分析。PEG化された生成物を37℃において30分間トロンビン(FVIIIのug当たり40IU)で処理することができる。用いられるトロンビンは汚染物としてAPCも含有する。トロンビン切断は重鎖から50kD A1及び43kD A2ドメインを形成するが、APC切断はA2ドメインをさらに21及び22kDのフラグメントに割るであろう(図7)。重鎖のC−末端を認識するR8B12抗体を用いる染色は、無損傷のA2ドメイン及び21kD C−末端フラグメントのみを同定するであろう(FVIII562−740)。かくしてPEG2のPEG化が位置491に関して特異的であったら、43kD A2ドメインはPEG化されるが21kD C−末端フラグメントはPEG化されないはずである。これは実際に、図7に示される22kD PEG化PEG2に関するウェスタンブロットにより確証された。かくして除去により、PEG2のPEG化は、A2ドメインのN−末端22kDフラグメント(FVIII373−561)に位置決定された。PEG−マレイミドはp
H6.8においてシステインに関して完全に選択的であり、373−561内の唯一の本来のFVIIIシステインは528−554の間の埋もれたジスルフィドに由来するので、PEG2は位置491における導入されたシステイン上でPEG化されるのが非常にありそうなことである。FVIII重鎖N−末端抗体を用いるトロンビン−処理されたPEG化PEG2のウェスタン染色は、A1ドメインのPEG化を示さなかった(データは示されない)。トロンビン切断法を用いるPEG2の選択的PEG化は、5、12、33及び43kDのPEGsに関しても確証された(データは示されない)。PEG化された野生型全長FVIIIのトロンビン切断は、BドメインのみがPEG化されることを示す(図8)。
【0126】
ヨウ素染色によるPEG化分析。クーマシーブルー及びウェスタン染色の際に新しく作られるバンドが実際にPEG化されたバンドであることを確証するために、PEGに関して特異的であるバリウム−ヨウ素染色を用いた(図9)。PEG化されたPEG2を6%TrisGlycineゲル(Invitrogen)上で移動させ、R8B12重鎖抗体又はバリウム−ヨウ素溶液で染色した(Lee et al.著,Pharm Dev
Technol.4:1999年,269−275)。PEG化されたバンドは、それらを整列させるための分子量マーカーを用いて2つの染色の間で一致し、かくしてFVIII重鎖PEG化を確証した。
【0127】
MALDI−質量分析によるPEG化分析。重鎖中のA2ドメインのPEG化を確証するために、PEG化の前後のrFVIII試料をマトリックス−補助レーザー脱着/イオン化(MALDI)質量分析により分析した。30%アセトニトリル,0.1%TFA中のシナピン酸マトリックスを有するMALDI標的プレート(target plate)上で試料を混合し、結晶化させた。次いでそれらをVoyager DE−PRO分光計において、正の線状モードで分析した。図10に示される結果は、83kDを中心とするPEG2の軽鎖及び89kDにおける重鎖(HC)を示した。PEG化試料に関して取得されたスペクトルは、HCピークの低下及び111kDを中心とする新しいピークの形成を示した。これは、重鎖のPEG化を確証する。PEG化された軽鎖(105kDにおける)は、検出限界より上で観察されなかった。
【0128】
次いで試料を両方とも、FVIIIのmg当たり20単位のトロンビンにおいて37℃で30分間、トロンビン消化に供し、続いてアミノ酸分析によりFVIII濃度を決定した(Commonwealth Biothechnologies,Inc)。重鎖は46kD(A1)N−末端画分及び43kD(A2)画分に切断された。PEG化試料に関して取得されたMALDIスペクトル(図11)は、43kDピークの喪失及びPEG化されたA2ドメインの故の新しい65kDピークの出現を示す。LCのPEG化はやはり検出限界より上で観察されない。これらの結果は再び、FVIIIのA2ドメインのPEG化を確証している。同じ分析をPEG化されたPEG6に適用し、軽鎖A3C1C2フラグメントのPEG化を確証した(図12)。
【0129】
活性測定
凝固アッセイ。凝固FVIII:C試験法は、活性化部分トロンボプラスチン時間(aPTT)に基づく1−段階アッセイである。FVIIIは因子IXa、カルシウム及びリン脂質の存在下で、因子XからXaへの酵素的転換において補因子として働く。このアッセイでは、希釈された試験試料をFVIII欠失血漿基質及びaPTT試薬の混合物と一緒に37℃でインキュベーションする。インキュベーションされた混合物に塩化カルシウムを加え、凝固を開始させる。血餅が形成されるのに要する時間(秒)とFVIII:Cの濃度の対数の間には逆比例関係(inverse relationship)が存在する。種々の希釈度の試験材料の凝固時間を、一系列の希釈度の既知の活性の標準材料から構築される曲線と比較することにより、未知の試料に関する活性レベルを内挿し、mL
当たりの国際単位(IU/mL)において報告する。
【0130】
発色アッセイ。発色アッセイ法は2つの連続的段階から成り、その方法では色の強度がFVIII活性に比例する。第1段階において、因子Xが最適量のカルシウムイオン及びリン脂質の存在下でFIXaにより、その補因子FVIIIaを用いてFXaに活性化される。因子Xの活性化の速度がFVIIIの量のみに依存するように、過剰量の因子Xが存在する。第2段階において、因子Xaは発色基質を加水分解し、発色団を与え、色の強度を405nmにおいて測光的に読み取る。未知試料の力価を計算し、傾斜比統計法(slope−ratio statistical method)を用いてアッセイの正当性を調べる。活性をmL当たりの国際単位(IU/mL)において報告する。
【0131】
1811−1818ループはFIXへの結合に含まれるが、このループ内の個々の位置の重要性は決定されていない。PEG7−10突然変異タンパク質は、本来のFVIIIに関してほとんど同じ比発色活性を示す(表3)。表3は、BDDに関するPEG突然変異タンパク質及びPEG化されたPEG2又はPEG6のパーセント比活性(S.A.)を示す。S.A.は、発色、凝固又はvWF結合活性を全抗原ELISA(TAE)値で割ることにより決定された。次いでPEG化突然変異タンパク質のS.A.をBDDのS.A.(8IU/ug発色、5IU/ug凝固及び1vWF/TAE)で割り、100を掛けて、表3中に発色、凝固及びvWF/TAEの見出しの下に挙げられるパーセントS.A.を得る。
【0132】
【表3】
【0133】
表3中で用いられる場合、「PEG2 red」は、還元剤で処理され、続いて還元剤が除去されたPEG2突然変異タンパク質である。この還元法はFVIIIの3つの機能的活性を有意に変えなかった。5kD(PEG2−5kD)〜43kD(PEG2−43kD)の範囲のPEGsに複合したPEG2突然変異タンパク質は、有意な量の発色活性を失わなかったが、PEGサイズが5kDを超えて大きくなるとともに凝固活性を非常に低下させた。比較的大きいサイズのPEG化PEG2に関してvWF結合における中程度の減少もあり得る。
【0134】
全抗原ELISA(TAE)。ポリクローナルFVIII抗体がコーティングされたミクロタイタープレート上にFVIIIを捕獲する。ビオチニル化ポリクローナルrFVIII抗体及びストレプタビジンホースラディッシュペルオキシダーゼ(HRP)複合体を用いてFVIII結合を検出する。ペルオキシダーゼ−ストレプタビジン複合体は、テトラメチルベンジジン(TMB)基質を加えると発色反応を生ずる。4パラメーターフィットモデルを用いて標準曲線から試料濃度を内挿する。FVIIIの結果をμg/mLにおいて報告する。
【0135】
vWF結合ELISA。FVIIIを溶液中で重症血友病血漿(Severe Hem
ophilic Plasma)中のvWfに結合させる。次いでvWf−特異的モノクローナル抗体がコーティングされたミクロタイタープレート上でFVIII−vWf複合体を捕獲する。FVIIIポリクローナル抗体及びホースラディッシュペルオキシダーゼ−抗−ウサギ複合体を用いてvWfに結合したFVIIIを検出する。ペルオキシダーゼ−複合抗体複合体は、基質を添加すると発色反応を生ずる。4パラメーターフィットモデルを用いて標準曲線から試料濃度を内挿する。FVIII結合の結果をμg/mLにおいて報告する。PEG化された場合に活性のいずれにも有意な影響はなく、それはBドメインにおけるPEG化と一致した。
【0136】
【表4】
【0137】
イオン−交換クロマトグラフィーによるPEG化FVIIIの精製。PEG化されたFVIIIをアニオン交換カラム又はカチオン交換カラムに適用し、ここでタンパク質はカラムに結合するが、過剰の遊離のPEG試薬は結合せず、通過流中で除去される。次いで塩化ナトリウム勾配を用いてPEG突然変異タンパク質をカラムから溶出させる。負荷物、通過流及び勾配画分のバリウム−ヨウ素染色された4〜12%Bis−Trisゲルを用い、カラム溶出画分がPEG化突然変異タンパク質を有することを確証した。
【0138】
サイズ−排除クロマトグラフィーによるPEG化FVIIIの精製。PEG2突然変異タンパク質の大部分を含有するアニオン交換画分をプールし、限外濾過により濃縮し、次いでサイズ排除カラムに適用する。次いで調製緩衝液を用いてカラムを溶出させる。PEGがタンパク質に結合しているかどうかに依存するタンパク質のサイズ及び形における相違の故に、このカラムはPEG化PEG2突然変異タンパク質をPEG化されていない残るPEG2のそれから分離する。PEG化された突然変異タンパク質FVIII画分を、ほとんどのFVIII活性を有することに基づいてプールし、次いでその後の動物研究及び分子特性化のために凍結する。図13は、43kD PEG化PEG2突然変異タンパク質の溶出に対して非−PEG化PEG2突然変異タンパク質の溶出を比較する。PEG化PEG2は有意により初期に溶出し、それは共有結合したPEGからのそのサイズにおける増加及び形を示す。
【0139】
より低い、すなわち50%未満のPEG化の効率を示すPEG6のような突然変異タンパク質の場合、高度に純粋なモノ−PEG化生成物を与えるための最も有効な精製案は、カチオン交換クロマトグラフィーとそれに続くサイズ排除クロマトグラフィーの組み合わせを使用することである。例えばPEG6の場合、カチオン交換クロマトグラフィーはPEG化されたPEG6(より初期の溶出画分、図14)を非−PEG化PEG6の大部分(より後期の溶出画分、図15)から精製する。次いでサイズ排除クロマトグラフィーはPEG化されたタンパク質(より初期の溶出画分、図15)を非−PEG化タンパク質(より後期の溶出画分、図15)の残りから分離して仕上げを施す(polishes)。
【0140】
活性へのPEGサイズの影響。PEGサイズがPEG化された時のFVIIIの凝固及び発色活性の両方に影響を有するかどうかを調べるために、精製された全長FVIII、PEG2、PEG6及びPEG14をTCEPにより還元し、続いて還元剤を除去し、緩衝液標準又は6kD〜64kDの範囲のPEGsと反応させた。得られるPEG化FVIIIを過剰のPEG又は非PEG化FVIIIの除去なしで直接検定した。標準実験は、過剰のPEGがFVIII活性に影響を有していないことを示した。
【0141】
図16はこの研究の結果を示す。精製された全長FVIIIは図16中でKG−2として示される。図16中で報告されるパーセント活性は、還元及び還元剤の除去の後にPEGで処理された試料の値を、PEG化の収率を考慮しながら、緩衝液標準で処理された試料の値で割ることにより決定された。PEG化の収率は、いずれの与えられるFVIII構築物の場合にも、すべてのPEGsにわたり(across)同等であった。それらはKG−2、PEG2及びPEG14の場合に約80%であり、PEG6の場合に約40%である。例えば12kD PEG化PEG14試料の場合の3.2IU/mLに対して、緩衝液標準処理されたPEG14は6.8IU/mLの凝固活性を有する。しかしながら、PEG化の効率は約80%であり、3.2IU/mLが約80%のPEG化及び約20%の非PEG化の凝集体活性を示すことを意味する。非PEG化試料が緩衝液標準で処理されたPEG14と同じ活性を有すると仮定すると、PEG化PEG14の場合の非PEG化PEG14に対するパーセント活性は34%=(3.2−6.8x20%)/(6.8x80%)であると算定される。
【0142】
BDDのPEG2、PEG6又はPEG14位置におけるA2又はA3ドメイン内のPEG化は、PEGサイズが6kDを超えて増加すると凝固活性の劇的な喪失を生ずる。しかしながら、全長FVIIIのBドメイン内の本来のB−ドメインシステインにおけるPEG化は凝固活性に影響を有していなかった。興味深いことに、発色活性はすべてのPEG化構築物の場合に影響されなかった。これはアッセイの相違の故であり得る。小さい発色性ペプチド基質は、凝固活性で用いられるもっと大きなタンパク質基質よりPEG化FVIII/FIX/FX複合体に容易に近づけることがあり得る。あるいはまた、PEGは突然変異タンパク質の活性化に影響し得る。これは2−段階発色アッセイより1−段階凝固アッセイによって容易に検出されるであろう。
【0143】
PEG2、6及び14の凝固活性へのPEGの影響の観察を確証するために、いくつかのPEG化構築物を過剰のPEG及び非PEG化タンパク質(unPEGylated)から精製した。PEGは発色活性に影響を有していないので、発色活性対凝固活性の比率は凝固活性へのPEGの相対的影響についての良い評価となる(表5)。PEG2のような与えられる位置におけるより大きなPEGs及びPEG2+6構築物の場合におけるようなより多数のPEGsは、より多くの凝固活性の喪失を引き起こす。
【0144】
【表5】
【0145】
うさぎPK研究。FVIIIの薬物動態学(PK)へのPEG化の影響を理解するために、複数の種においてPK研究を行なった。研究のためにNZW SPFウサギを用いた:10匹の雌,グループあたり5匹のウサギ、2つのグループ(PEG2 FVIII及び22kD PEG化PEG2)。試料を100IU/mL(発色単位)の最終的濃度を以って無菌のPBS中に希釈した。各ウサギに耳縁静脈(marginal ear vein)を介して1ml/kg(100IU/kg)の用量の希釈された試験物質又は標準物質を与えた。注入から種々の時間の後に、投薬から後の規定された時点に耳中心動脈(central ear artery)から血液試料(1mL)を1mLシリンジ中に採取した(100μLの3.8%Na−クエン酸塩を装入)。血漿試料を、96−ウェルプレート上にコーティングされたR8B12重鎖抗体と一緒にインキュベーションし、投薬されたヒトFVIIIを特異的に捕獲した。捕獲されたFVIIIの活性を発色アッセイにより決定した(図17)。PEG化PEG2及びPEG化PEG6をBDDと比較もし(図18及び19)、PEG化された突然変異タンパク質はBDDと比較して血漿回収率における向上を示した。PEG化された野生型全長FVIIIは多くの向上を示すようではなかった(図20)。
【0146】
マウスPK研究。第2の種として、ICR正常もしくは血友病、FVIII欠失マウス(Taconic,Hudson,NY)をPK研究に用いた。1時点につきグループ当たり5匹のマウスで、正常なマウスを研究に用いた。試験材料を調製緩衝液中に、25IU/mLの公称最終濃度まで希釈した。各マウスに尾静脈を介して4mL/kg(約0.1mLの合計体積)の希釈された(dilute)試験材料を投与することができる。指示される時点に下大静脈から、血液試料(正常もしくは血友病マウス研究のためにそれぞれ0.45もしくは0.3mL)を1mLのシリンジ(正常もしくは血友病マウス研究のためにそれぞれ50もしくは30μLの3.8%Na−クエン酸塩が装入された)中に採取する(試料当たり1匹の動物)。上記の発色アッセイ法を用いて血漿試料をFVIII濃度に関して検定する。PEG化PEG6は、BDD又はPEG6と比較してより高い血漿回収率を示す(図21)。PEG化PEG2は、BDDと比較してより高い血漿回収率を示す(図22及び23)。
【0147】
【表6】
【0148】
【表7】
【0149】
血友病マウス(BDD)因子VIII回収率。図24に示される血友病マウス(BDD)因子VIII回収率ヒストグラムは、血友病マウスアッセイにおけるBDD因子VIIIの2つの種の半減期の薬物動態学的(PK)評価を描く。このアッセイは、マウスモデルにおいて静脈内投与から後の3つの時点に、BDD因子VIII(図24中で「wt」又は野生型BDD因子VIIIと呼ばれる)及びBDD因子VIIIのPEG2+6二重PEG化変異体(そして本明細書の他の箇所でBDD因子VIIIのL491C,K1808C二重変異体と同定される)の両方の血漿濃度を測定するように設計された。0.8及び4時間の両時点におけるPK評価は同等であったが、16時間の評価は特に注目する価値がある。16時間において、非−PEG化分子と比較する場合に約4倍(400%)もの多くの二重にPEG化されたBDD因子VIII変異体(PEG2+6)が投与から16時間後のマウス血漿中に留まった。
【0150】
腎臓裂傷モデル。PEG化FVIII突然変異タンパク質が血友病マウスにおいて出血を止めるのに有効であるかどうかを決定するために、腎臓裂傷モデルを用いた。血友病マ
ウス(破壊されたFVIII遺伝子を有するC57/BL6)をイソフルオラン(isofluorane)下で麻酔し、秤量した。下大静脈を露出し、31ゲージの針を用いて100ulの食塩水又はFVIIIを注入した。針を注意深く取り出し、出血を妨げるために注入の部位で30〜45秒間圧力を加えた。2分後、右腎臓を露出し、垂直軸に沿って鉗子間に保った。#15メスを用い、腎臓を3mmの深さまで水平に切断した。傷の均一な深さを保証するために、腎臓を中心で軽く持って鉗子の両側上の等しい組織を露出した。露出された腎臓の表面は鉗子の深さまで切断された。上記の通りに血液損失を定量した。マウスについて種々の用量のFVIIIを調べ、腎臓出血へのFVIIIの用量反応関係を特性化した。PEG化PEG2は、マウス腎臓傷害後の血液損失の減少においてBDDと同等の力価を示した(図25)。かくしてPEG化PEG2の凝固活性はBDDのそれより低いが、この腎臓裂傷モデルは、PEG化PEG2の生体内有効性がBDDと比較して測定可能なほど低下しないことを示し、発色アッセイデータと一致した。
【0151】
抗体阻害アッセイ。特異的に位置491(すなわちPEG2)においてポリエチレングリコール(PEG)のような高分子量ポリマーを付加することは、mAB 413へ、及び拡大により患者の大きな割合の阻害性抗体への結合及び感度を低下させるはずであり、それは、多くの患者が同じmAB 413エピトープに対する阻害性抗体(inhibitor antibodies)を発現するからである。これを調べるために、増加する量のmAB 413を非−飽和量(0.003IU/mL)のBDD又は43kD PEG化PEG2と一緒にインキュベーションし、発色アッセイにおいて機能的活性を調べた(図26)。非−阻害性抗体であるR8B12及びC2ドメインを標的とする阻害性抗体であるESH4を標準として用いた。PEG化PEG2は実際にBDDよりmAB 413阻害に対して抵抗性であり、491位置近辺で結合しない標準抗体の存在下で類似の阻害パターンを示す。さらに、mAB 413阻害に対するPEGの保護効果はPEGサイズに依存し、より大きなPEGsがより強い効果を有する(図27)。PEG化FVIIIが患者からの阻害性抗体により抵抗性であるかどうかを調べるために、FVIIIへの阻害物質を発現した血友病A患者に由来する1パネルの血漿の存在下で、発色活性を測定した。調べられた8人の患者の血漿の中で4人の患者の血漿試料において、43kD PEG化PEG2はBDDより患者の血漿阻害に抵抗性であった。例えばPEG化されたPEG2、PEG6又はPEG2+6は、1人の患者の血漿においてBDDより高い残留活性を示したが、他の血漿においては示さなかった(図28)。ジPEG化PEG2+6は、モノPEG化PEG2又はPEG6より抵抗性であると思われる。これらの結果は、PEG化PEG突然変異タンパク質がFVIIIへの阻害物質を発現する患者の処置においてより有効である得ることを示唆している。
【0152】
高処理量PEG化スクリーニング。特定のPEG突然変異タンパク質のPEG化効率は予測不可能であり、それは特にBDDの直接の構造的情報がないからである。例えばBDDの構造モデルに基づき、PEG4及びPEG5のPEG化効率がPEG2及びPEG15のそれに類似して非常に高いはずと予測され、それは構造に従って3個のすべての位置が表面露出され且つ外側に向いているからである。かくして系統的なPEG化を介して新規なクリアランス機構を探索するためにPEGを用いることは、多数の突然変異タンパク質をスクリーニングすることを必要とする。
【0153】
多数のPEG突然変異タンパク質を迅速にスクリーニングするために、新規な高処理量法が開発され、それは一過的にトランスフェクションされた突然変異タンパク質からのPEG化生成物のPEG化効率及び機能的活性を調べることができる。0.1〜0.2IU/mLもの低いFVIII発色値を有する5〜10mLもの少量の一過的に発現されたPEG突然変異タンパク質を、Amicon−centra Ultra device MWCO 30Kを用いて約50−倍濃縮し、FVIIIの濃度はFVIIIへの抗体の相互作用の親和性範囲に近い1nMより高くに達する。濃縮されたPEG突然変異タンパ
ク質(〜300uL)を〜30uLのC7F7 FVIII抗体樹脂と一緒に4℃で終夜インキュベーションし、洗浄し、溶出させ、透析し、還元する。還元剤を除去し、還元されたPEG突然変異タンパク質をPEG化し、上記のようなウェスタン分析上で移動させる(図29及び30)。一過的に発現されたPEG突然変異タンパク質の相対的なPEG化効率は、精製されたPEG突然変異タンパク質のそれと正確に一致する。
【0154】
この方法により、1〜2ヶ月内に数十個のPEG突然変異タンパク質をスクリーニングすることができる。例えばPEG14(K1804C BDD)は、12kD PEGを用いて少なくとも約80%の軽鎖のPEG化を有し、重鎖のPEG化はなく(データは示されない)、軽鎖上に位置決定されるK1804C突然変異と一致した。K1804とK1808(PEG6位置)の間のC□からC□への距離は、BDD構造に従うとわずか8.4オングストロームであり、この位置における43kD PEGの導入が、よりずっと高いPEG化収率の利点を以って、33kD PEG化PEG6と類似のPKにおける向上を有するであろうことを示唆している。調べられたすべてのPEG突然変異タンパク質に関する相対的なPEG化収率を表8にまとめる。重鎖中にシステインを有するすべての突然変異タンパク質は重鎖上のみでPEG化されるが、軽鎖中にシステインを有するすべての突然変異タンパク質は軽鎖上でPEG化される点で、PEG化はシステイン突然変異が導入される特定のFVIII鎖に関して高度に選択的であった。突然変異タンパク質番号2〜31は、挙げられる位置における本来のアミノ酸がシステインで置換されるBDDのシステイン突然変異を示す。PEG2+6は、位置491及び1808がシステインで置換されたBDDの二重突然変異タンパク質である。A1及びA2(ならびに全長FVIIIであるKG−2の場合にはBドメイン)は重鎖に属するが、A3、C1及びC2は軽鎖に属する。PEG化効率は、PEG化生成物をSDS PAGE上で移動させ、PEG化バンドの強度を非PEG化バンドと比較することから見積もられた:+++ >約80%PEG化収率,++ 約30〜70%収率,+ 約10〜30%収率及び− <約10%収率。
【0155】
【表8】
【0156】
還元されたPEG突然変異タンパク質の質量分析。PEG突然変異タンパク質又は全長FVIIIの直接のPEG化を妨げる「キャップ」の正体を決定するために、PEG2+14を67uM〜670uMの範囲の濃度におけるTCEPで還元した。PEG化収率はTCEPの増加する量に比例して向上した(図31)。同じ試料をPEG化の前に質量分析によっても分析した(図32)。直接研究できるタンパク質ドメインを得るために、FVIIIのmg当たり20単位の比率におけるトロンビンを用い、試料を37℃で30分間消化した。トロンビン切断は残基372−740を含み、占有されたグリコシル化部位を含まないA2フラグメントを生ずる。消化された試料をC4逆相液体クロマトグラフィーシステム上に注入し、カラムからの溶出物を、電子スプレーインターフェース(electrospray interface)を介して4極飛行時間質量分析計(quadrupole time−of −flight mass spectrometer)中に直接導入した。A2ドメインに相当するクロマトグラフィーピーク下からの質量スペクトルをデコンボリューションし、タンパク質の無損傷の質量値を与えた。還元の前に、PEG2+14のA2ドメインは、理論的に予測されるより118ダルトン大きい質量を与える。TCEP濃度が向上すると、A2ドメインの正確な予測質量を有する新しいピークが現れる。この新しいピークの割合は、TCEP濃度が向上するとともに増加する。118ダルトンの差は、システイン(119Da)とのジスルフィド形成を介する残基Cys 491におけるシステイン化及び機器の精度により説明され得る。かくしてこれは、PEG突然変異タンパク質がシステインによりキャッピングされ、それが直接のPEG化を妨げることを示す。
【0157】
本明細書で開示されるすべての引用文献は、引用することによりそれらの記載事項全体が本明細書の内容となる。
【図面の簡単な説明】
【0158】
【図1a】PEG突然変異タンパク質のためのベクターマップ及び突然変異誘発戦略。
【図1b】PEG突然変異タンパク質のためのベクターマップ及び突然変異誘発戦略。
【図2】モノクローナルFVIII抗体クロマトグラフィーカラム上で精製されたPEG2タンパク質の場合の、時間に関する280nmにおけるUV吸光度分布。
【図3】3−段階特定部位PEG化法。
【図4】PEG2の特定部位のPEG化。
【図5】PEG6の特定部位のPEG化。
【図6a】BDD、PEG2、4、5及び6の特定部位のPEG化。
【図6b】PEG2及びPEG6を標準として用いるPEG15及びPEG7のPEG化。
【図6c】PEG2及びPEG6を標準として用いるPEG2+6のPEG化。
【図6d】PEG2を標準として用いる野生型全長FVIII(KG−2)のPEG化。
【図7】PEG化されたPEG2のトロンビン切断。
【図8】PEG化された野生型全長FVIII(KG−2)のトロンビン切断。
【図9】PEG化されたPEG2のヨウ素染色。
【図10a】非PEG化PEG2のMALDI質量分析。
【図10b】22kD PEG化PEG2のMALDI質量分析。
【図11a】トロンビン切断の後の非PEG化PEG2のMALDI質量分析。
【図11b】トロンビン切断の後のPEG化PEG2のMALDI質量分析。
【図12】トロンビン切断の前後のPEG化PEG6のMALDI質量分析。
【図13】サイズ−排除カラム上で精製されたPEG化PEG2の280nmにおけるUV吸収分布。
【図14】カチオン交換カラム上で精製されたPEG化及び非PEG化PEG6の280nmにおけるUV吸収分布。
【図15】サイズ−排除カラム上で精製されたPEG化及び非PEG化PEG6の280nmにおけるUV吸収分布。
【図16】発色アッセイ及び凝固アッセイにより測定される場合のPEG化タンパク質の活性を非PEG化タンパク質の活性と比較するグラフ。
【図17】PEG2と比較されるPEG化PEG2のウサギPK研究。
【図18】BDD及びPEG2と比較されるPEG化PEG2のウサギPK研究。
【図19】BDD及びPEG6と比較されるPEG化PEG6のウサギPK研究。
【図20】非変更全長FVIIIと比較されるPEG化野生型全長(“fl”)FVIIIのウサギPK研究。
【図21】PEG6及びBDDと比較されるPEG化PEG6の血友病マウスPK研究。
【図22】BDDと比較される22及び43kD PEG化PEG2の正常マウスPK研究。
【図23】時間経過中ずっとBDDと比較される22kD PEG化PEG2の正常マウスPK研究。
【図24】血友病マウスアッセイにおけるBDD因子VIIIの2つの種の半減期の薬物動態学的(PK)評価を描く、血友病マウス(BDD)因子VIII回収率ヒストグラム。
【図25】BDDと比較される22kD PEG化PEG2の血友病マウス腎臓裂傷研究。
【図26】増加する量のFVIII抗体の存在下におけるPEG化PEG2及びBDDの発色活性。
【図27】増加する量のFVIII mAB 413抗体の存在下におけるPEG化PEGの発色活性。
【図28】FVIIIへの阻害物質を発現した患者に由来するヒト血漿の存在下におけるBDD、43kD PEG化PEG2、33kD PEG化PEG6及び33kD ジPEG化PEG2+6の発色活性。
【図29】PEG化スクリーニング法及び正当化。
【図30】PEG15〜17のPEG化スクリーニング。
【図31】還元剤濃度の関数としてPEG2+14のPEG化を示すゲル。
【図32】67〜670uMのTCEPで処理され、続いて還元剤が除去されたPEG2+14のデコンボリューションされた質量スペクトル。
【特許請求の範囲】
【請求項1】
Bドメインにおいて1個もしくはそれ以上の生物適合性ポリマーに共有結合した全長因子VIIIを含んでなる単離された複合体。
【請求項2】
全長因子VIIIがアミノ酸配列番号4又はその対立遺伝子変異体を含んでなる請求項1の複合体。
【請求項3】
生物適合性ポリマーがポリアルキレンオキシドを含んでなる請求項1の複合体。
【請求項4】
生物適合性ポリマーがポリエチレングリコールを含んでなる請求項1の複合体。
【請求項5】
ポリエチレングリコールがメトキシポリエチレングリコールを含んでなる請求項1の複合体。
【請求項6】
メトキシポリエチレングリコールが3kD〜100kDのサイズ範囲を有する請求項5の複合体。
【請求項7】
メトキシポリエチレングリコールが5kD〜64kDのサイズ範囲を有する請求項5の複合体。
【請求項8】
メトキシポリエチレングリコールが5kD〜43kDのサイズ範囲を有する請求項6の複合体。
【請求項9】
ポリエチレングリコールが5、12、22、30又は43kDのサイズを有する請求項4の複合体。
【請求項10】
請求項1〜9のいずれか1項の単離された複合体の治療的に有効な量及び製薬学的に許容され得る添加剤を含んでなる製薬学的組成物。
【請求項11】
請求項1〜9のいずれか1項の複合体を有効成分として含有することを特徴とする血友病の処置剤。
【請求項1】
Bドメインにおいて1個もしくはそれ以上の生物適合性ポリマーに共有結合した全長因子VIIIを含んでなる単離された複合体。
【請求項2】
全長因子VIIIがアミノ酸配列番号4又はその対立遺伝子変異体を含んでなる請求項1の複合体。
【請求項3】
生物適合性ポリマーがポリアルキレンオキシドを含んでなる請求項1の複合体。
【請求項4】
生物適合性ポリマーがポリエチレングリコールを含んでなる請求項1の複合体。
【請求項5】
ポリエチレングリコールがメトキシポリエチレングリコールを含んでなる請求項1の複合体。
【請求項6】
メトキシポリエチレングリコールが3kD〜100kDのサイズ範囲を有する請求項5の複合体。
【請求項7】
メトキシポリエチレングリコールが5kD〜64kDのサイズ範囲を有する請求項5の複合体。
【請求項8】
メトキシポリエチレングリコールが5kD〜43kDのサイズ範囲を有する請求項6の複合体。
【請求項9】
ポリエチレングリコールが5、12、22、30又は43kDのサイズを有する請求項4の複合体。
【請求項10】
請求項1〜9のいずれか1項の単離された複合体の治療的に有効な量及び製薬学的に許容され得る添加剤を含んでなる製薬学的組成物。
【請求項11】
請求項1〜9のいずれか1項の複合体を有効成分として含有することを特徴とする血友病の処置剤。
【図1a】

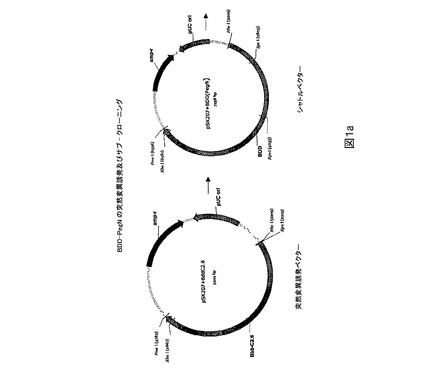
【図1b】
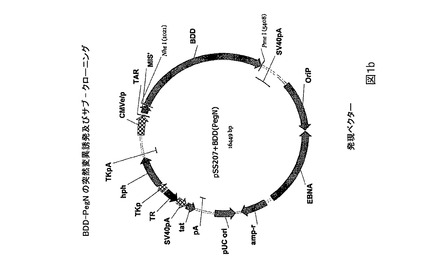
【図2】
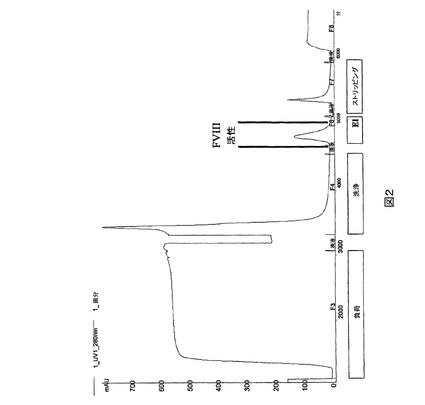
【図3】
【図4】

【図5】
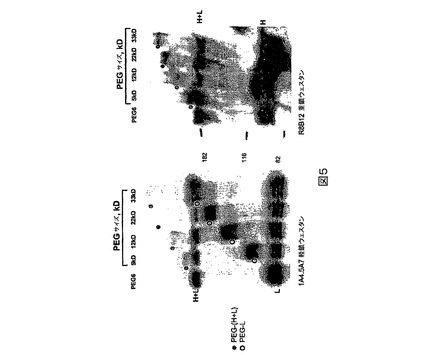
【図6a】
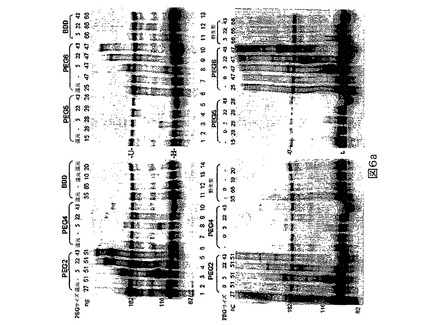
【図6b】
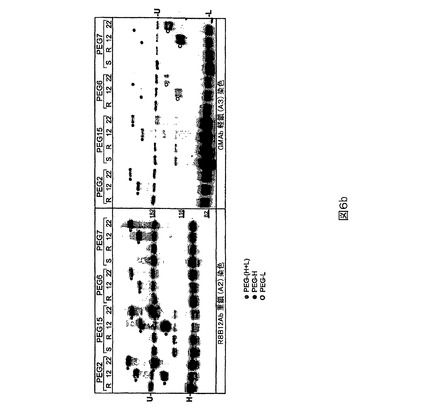
【図6c】
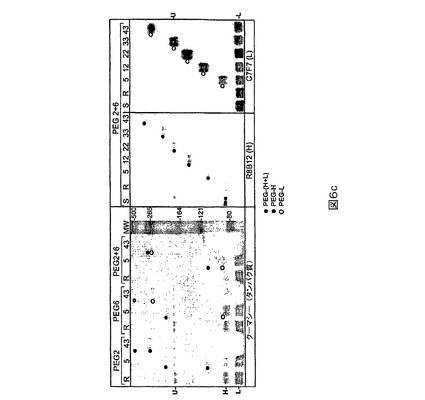
【図6d】
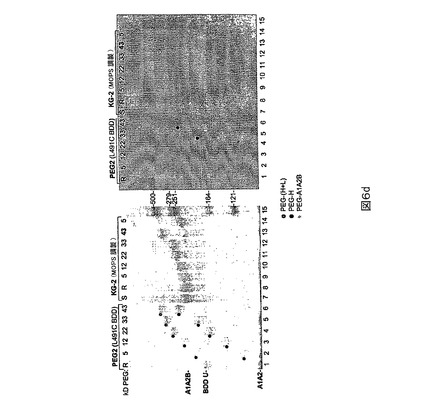
【図7】
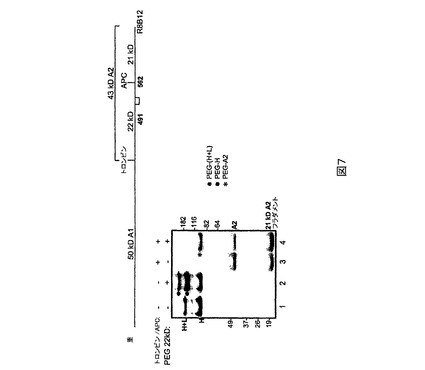
【図8】

【図9】
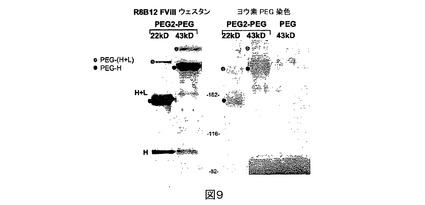
【図10a】
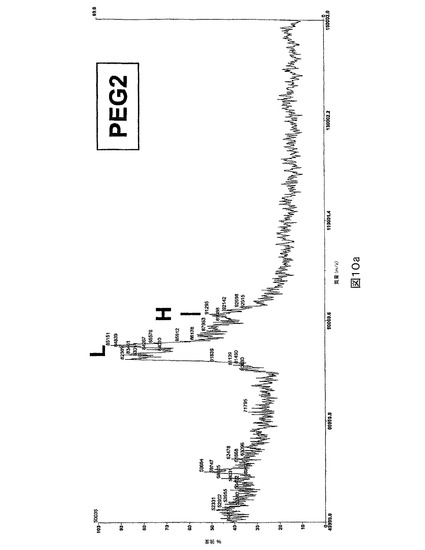
【図10b】
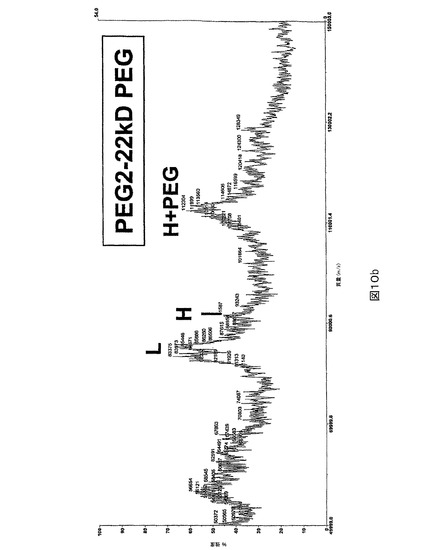
【図11a】
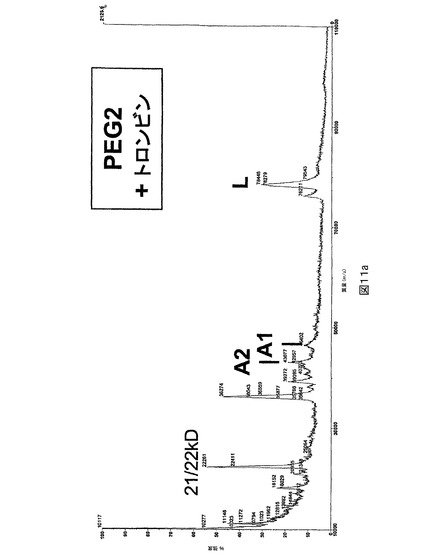
【図11b】
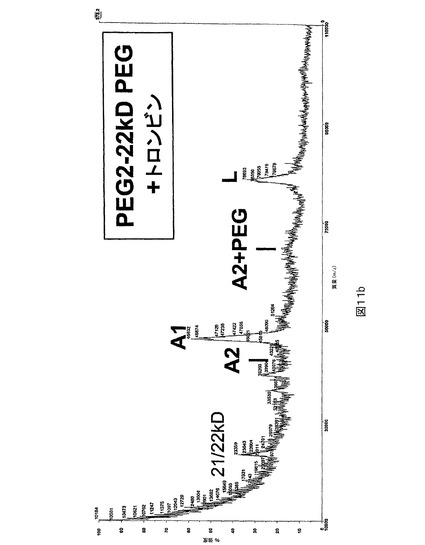
【図12】
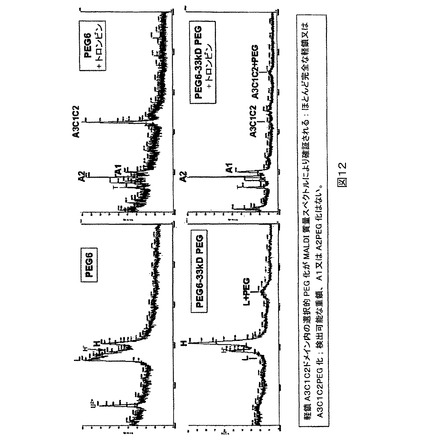
【図13】
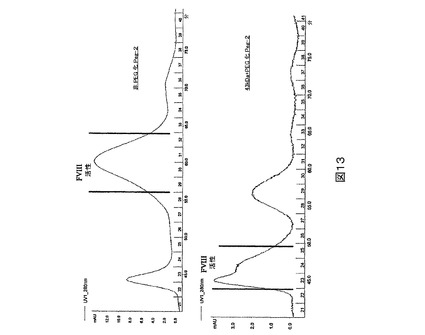
【図14】
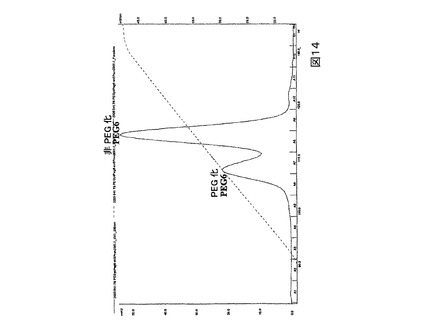
【図15】
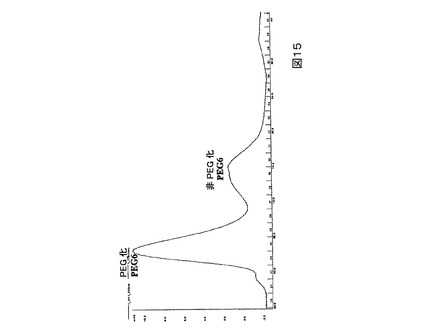
【図16】
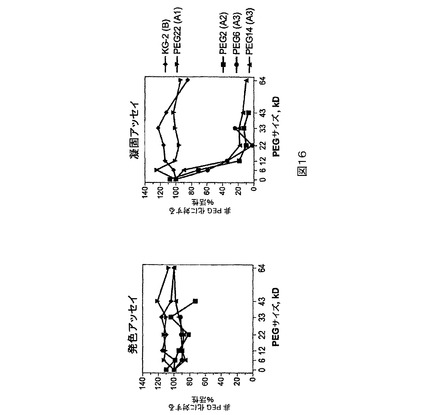
【図17】
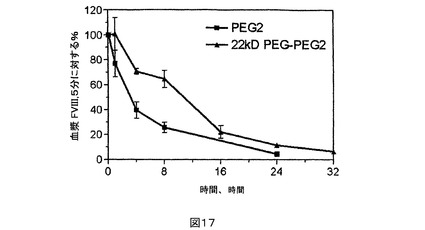
【図18】
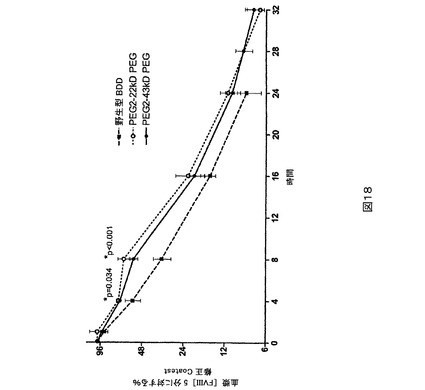
【図19】
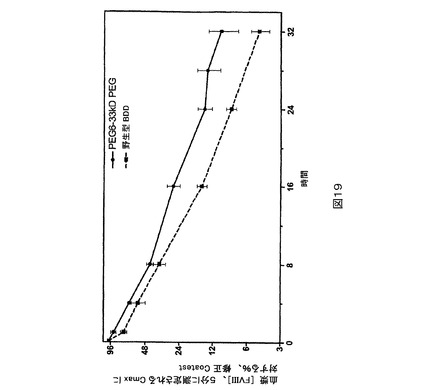
【図20】
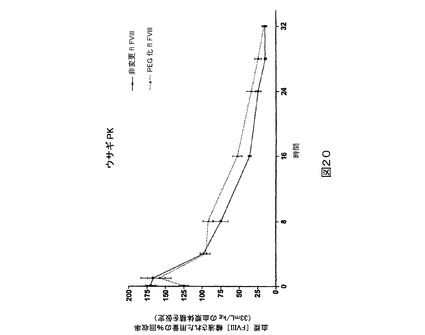
【図21】
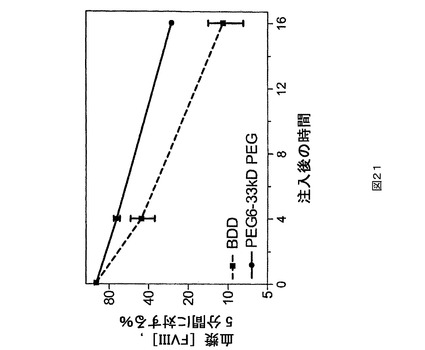
【図22】
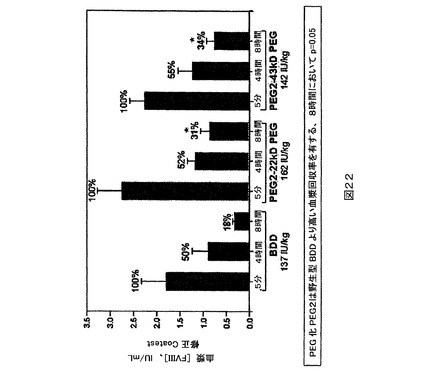
【図23】
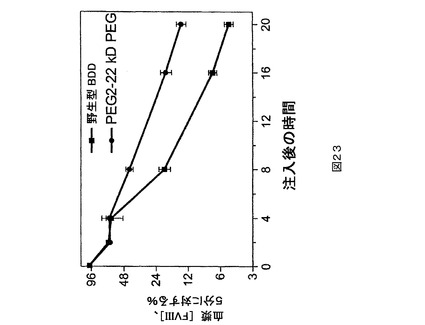
【図24】
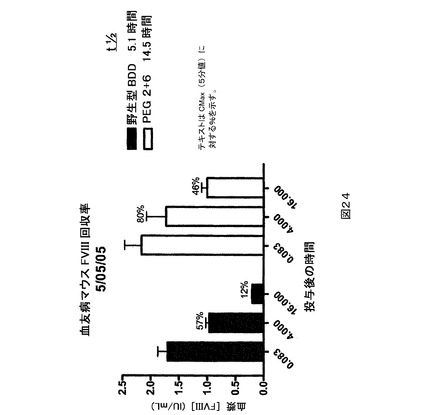
【図25】
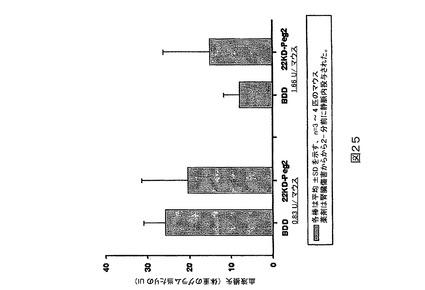
【図26】
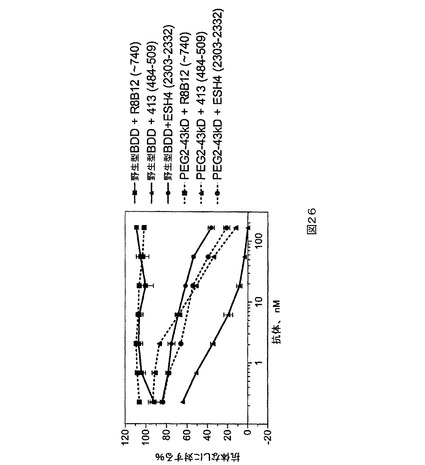
【図27】
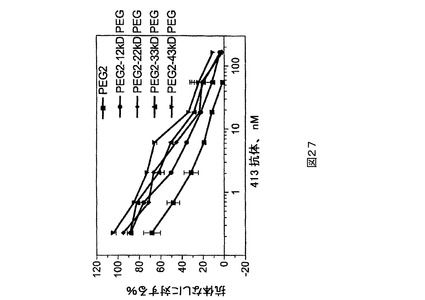
【図28】
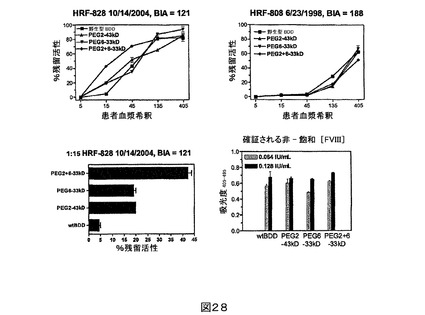
【図29】
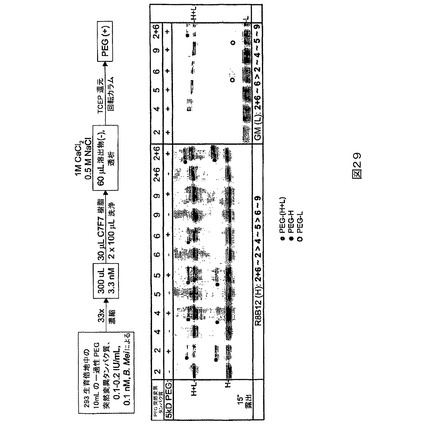
【図30】
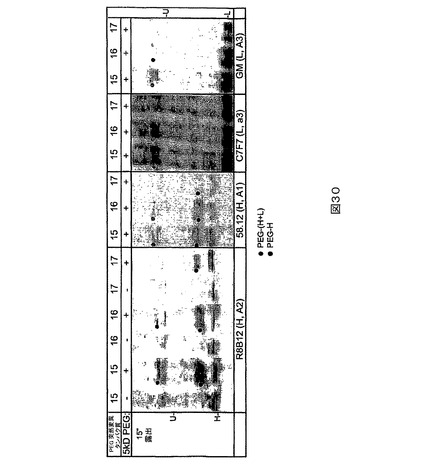
【図31】
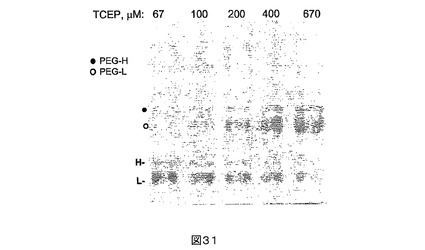
【図32】
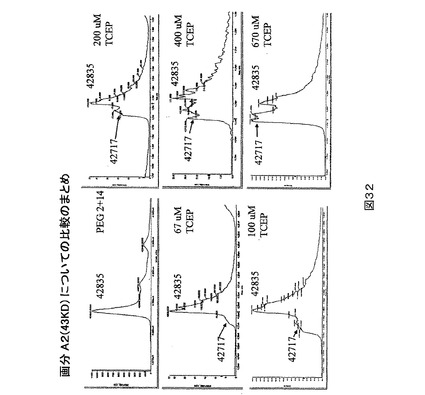
【図1b】
【図2】
【図3】
【図4】
【図5】
【図6a】
【図6b】
【図6c】
【図6d】
【図7】
【図8】
【図9】
【図10a】
【図10b】
【図11a】
【図11b】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【公開番号】特開2013−67621(P2013−67621A)
【公開日】平成25年4月18日(2013.4.18)
【国際特許分類】
【出願番号】特願2012−231926(P2012−231926)
【出願日】平成24年10月19日(2012.10.19)
【分割の表示】特願2007−541405(P2007−541405)の分割
【原出願日】平成17年11月14日(2005.11.14)
【出願人】(503106111)バイエル・ヘルスケア・エルエルシー (154)
【Fターム(参考)】
【公開日】平成25年4月18日(2013.4.18)
【国際特許分類】
【出願日】平成24年10月19日(2012.10.19)
【分割の表示】特願2007−541405(P2007−541405)の分割
【原出願日】平成17年11月14日(2005.11.14)
【出願人】(503106111)バイエル・ヘルスケア・エルエルシー (154)
【Fターム(参考)】
[ Back to top ]