
FabK蛋白質の3次元結晶構造、及びこれを利用したFabK蛋白質阻害剤開発方法
【課題】サーモトガ・マリティマ(Thermotogamaritima)菌株からFabK蛋白質(エノイル−ACP還元酵素)を分離精製して結晶化し、3次元結晶構造を究明して、活性部位の個別アミノ酸の変異実験を通じて活性にメカニズム的に寄与するアミノ酸を定義し、既存のFabIとは異なるFabKの構造を究明して、新たな抗生剤の開発に使用される技術を提供する。
【解決手段】FabK蛋白質溶液を緩衝液、塩および沈澱剤を含む貯水槽溶液と接触させて濃度平衡法を行う段階を含むFabK蛋白質の結晶化方法。FabK−FMN(フラビンモノヌクレオチド)複合体結晶。FabK−FMN複合体の3次元結晶構造情報保存媒体。FabK−FMN複合体の3次元結晶構造情報を使用する、FabK蛋白質活性阻害剤開発方法。
【解決手段】FabK蛋白質溶液を緩衝液、塩および沈澱剤を含む貯水槽溶液と接触させて濃度平衡法を行う段階を含むFabK蛋白質の結晶化方法。FabK−FMN(フラビンモノヌクレオチド)複合体結晶。FabK−FMN複合体の3次元結晶構造情報保存媒体。FabK−FMN複合体の3次元結晶構造情報を使用する、FabK蛋白質活性阻害剤開発方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、好熱性真正細菌サーモトガ・マリティマ(Thermotoga maritima)菌株に由来するFabK(エノイル−ACP還元酵素)蛋白質に関し、前記蛋白質の活性部位及び3次元結晶構造を究明し、これを利用して新たなFabK蛋白質阻害物質をスクリーニングして、既存の抗生剤に耐性を示す菌株に対して優れた抗菌活性を示す新たな活性調節物質を開発する技術に関する。
【背景技術】
【0002】
トリクロサンは、包括的な抗菌作用を有する抗微生物剤であって、約30年間多様な製品に抗生及び抗菌活性を付与するために使用されてきた。例えば、トリクロサンは、石鹸、シャンプー、洗濯剤はもちろん、歯磨き粉や化粧品などに含まれているだけでなく、子供のおもちゃ、じゅうたんなどにも含まれている。そのような意味で、トリクロサンの使用範疇は、紫外線または漂白剤に相当するといっても過言でない。
【0003】
トリクロサンは、エノイル−ACP還元酵素(エノイル−[アシルキャリアー蛋白質]レダクターゼ(E.C.1.3.1.9)NADH−依存性 トランス−2−エノイル−ACP還元酵素、ENR)またはFabIといわれる酵素の活性を抑制する作用がある。前記FabIは、ほぼ全てのバクテリア及び真菌類に存在して、バクテリアの細胞膜形成に必須の脂肪酸の生合成に関与する酵素のうちの1つであって、微生物の生存に必須の酵素である。したがって、トリクロサンは、FabIの活性を阻害する活性を有するので、広範囲の殺菌剤として使用することができる。
【0004】
好気性細菌であるマイコバクテリア類(mycobacteria)の場合には、InhAという酵素が脂肪酸の生合成中にFabIのようにエノイル−ACP還元酵素として作用して反応段階を活性化し、この場合、結核の治療剤であるイソニアジドがトリクロサンと同一の方法でInhAの活性を抑制する役割を担うということが知られている。これら2つの酵素であるInhA及びFabIを比較する時、これらのアミノ酸配列の相同性は、異なる種類のFabI間のアミノ酸配列の相同性より相対的に低いが、活性に必要な主要アミノ酸は同一であることが明らかになった。
【0005】
一方、脂質は、細胞膜を構成する主成分であって、脂質の生合成に関与する酵素は、バクテリアはもちろん、すべての生物体になくてはならない必須の酵素である。哺乳類での脂質の生合成は、FAS(脂肪酸合成酵素)といわれる巨大分子ホモダイマーが複数の段階の反応を各々異なる部位で順次に循環して行うI型脂肪酸合成経路を通じて行われる反面、バクテリアなどの微生物での脂質の生合成は、哺乳類とは異なったII型脂肪酸合成経路を通じて行われる。前記II型脂肪酸合成経路は、各段階の反応が各々異なる独立した酵素によって行われるという特徴がある。FabIは、前記段階のうちの最後の段階に相当するエノイル−ACPを還元させてアシル−ACPに転換させる段階を担当する。
【0006】
前記のように、FabIは、脂質の生合成の最後の段階で、脂質の伸長を触媒する作用をすることによって、脂肪酸の生合成における必須の役割を担っている。FabIは、SDR(短鎖アルコール脱水素酵素/還元酵素)スーパーファミリーに属して、他のファミリーメンバーと共に4量体を形成しており、大腸菌から分離されたFabIはNADH(ニコチンアミドアデニンジヌクレオチド)という補酵素を使用する。(Egan,A.F.and Russel,R.R.B.,Genet.Res.,21,3603−3611(1973))。最近明らかになったFabI−トリクロサン複合体構造によれば、トリクロサンは、NADH及びNAD上に位置し、酵素の活性部位を占めて非共有性であるが、非常に強い相互作用で基質が入ってこられなくする方法で酵素の活性を阻害する(Heath,R.J.,etal.,J.Biol.Chem.,274,11110−11114(1999))。黄色ブドウ球菌(Staphyloccus aureus)の場合、FabIがNADHでなくNADPHという補酵素を使用することが明らかになり、この場合にも、トリクロサンが同様に相互作用して、酵素の活性を阻害することが明らかになった。したがって、現在、製薬会社はもちろん、大学及び各種研究団体で開発しているFabI阻害剤は、トリクロサンと類似したメカニズムを有する化合物である。
【0007】
しかし、最近、肺炎球菌(Streptococcus pneumoniae)などの一部の主要病原菌がトリクロサンに耐性を示すことが報告された(Heath,R.J.and Rock,C.O.,Nature,406,145−146(2000))。これら耐性を示す菌株にはFabI酵素が存在しないが、遺伝子群にFabKという遺伝子が存在して、この遺伝子が発現して生成されるFabK蛋白質がFabI酵素の役割を代わりに担うことが明らかになった。フェカリス菌(Enterococcus faecalis)及びサーモトガ・マリティマ(Thermotoga maritima)も、脂肪酸の生合成経路にFabI蛋白質の代わりにFabK蛋白質が関与すると報告された。これらFabK蛋白質は、現在まで知られているFabI蛋白質とはコード遺伝子配列が全く異なり、SDRスーパーファミリーの他の蛋白質ともコード遺伝子配列に類似性がない。フェカリス菌及びサーモトガ・マリティマなどの菌株に存在するFabKは、そのアミノ酸配列において、肺炎球菌のFabKと各々68%及び48%の同一性を示している(Marrakchi,H.,etal.,Biochem.J.,370,1055−1062(2003))。FabKはフラビン蛋白質であり、活性のために補酵素であるNADHを必要として、トリクロサンによって不活性化されない特性を有する蛋白質である。
【0008】
現在の結核治療剤は、大部分がFabI/InhAと関連したものであるため、前記FabI蛋白質を使用せずに前記結核治療剤に対して耐性を示す菌株が次から次へ発見されている。このような耐性を示す菌株は、FabIの代わりにFabKを使用するものである。例えば、結核治療剤に対して耐性を示す呼吸器疾患の主要菌株である肺炎球菌またはバンコマイシンに耐性を示すフェカリス菌の場合も、FabIの代わりにFabKを使用することが明らかになった。つまり、これら耐性を示す菌株に対して抗菌活性を有するためには、FabIでなくFabKの活性を阻害する新たな化合物が必要である。現在までに報告されたところによれば、FabKは、その作動メカニズムが比較的よく知られているFabIとは異なっていることが明らかになり、その構造及び機能に対する研究が切実に要求されている。
【発明の開示】
【発明が解決しようとする課題】
【0009】
このような要求に応じるために、本発明の発明者は、サーモトガ・マリティマからFabK蛋白質を分離精製して結晶化して、3次元結晶構造を究明して、活性部位の個別アミノ酸の変異実験を通じて活性にメカニズム的に寄与するアミノ酸を定義し、既存のFabIとは異なるFabKの構造を究明して、新たな抗生剤の開発に有用に使用される技術を提供することによって、本発明を完成した。
【0010】
そのために、本発明の目的は、前記FabK蛋白質の活性部位を究明することにある。
【0011】
本発明の他の目的は、前記FabK蛋白質の3次元結晶構造を究明することにある。
【0012】
本発明の他の目的は、前記で究明されたFabK蛋白質の活性部位及び/または3次元結晶構造情報を利用して、FabK蛋白質と相互作用して阻害活性を有する化合物をスクリーニングする方法を提供することにある。
【課題を解決するための手段】
【0013】
前記課題を解決するために、本発明は、サーモトガ・マリティマ菌株に由来するFabK蛋白質の活性部位及び3次元結晶構造の究明に関する。
【0014】
一実施形態として、本発明はFabK蛋白質溶液を緩衝液、塩および沈澱剤を含む 貯水槽溶液と接触させて濃度平衡法を行う段階を含み、前記FabK蛋白質溶液の濃度は3〜30mg/mlであり、前記緩衝液はpH4〜9.5で、貯水槽溶液内濃度は0.005〜1.5Mであり、前記塩は全ての金属塩およびアンモニウム塩からなる群から選択された1種以上のもので、貯水槽溶液内濃度は0.05〜2Mであり、前記沈澱剤は炭素数1〜4のアルコールおよびポリエチレングリコールからなる群から選択された1種以上のものであり、貯水槽溶液内濃度は1%(v/v)〜40%(v/v)である、FabK蛋白質の結晶化方法に関する。
【0015】
別の実施形態では、本発明は後述の表1のX線回折パターンデータ、または後述の表2に記載の2389個の原子座標で示された3次元結晶構造を有する、FabK−FMN(フラビンモノヌクレオチド)複合体結晶に関する。
【0016】
他の実施形態では、本発明は後述の表1のX線回折パターンデータ、または後述の表2に記載の2389個の原子座標情報から選択された1つ以上のFabK−FMN複合体の3次元結晶構造情報またはこれらの全てを保存する、保存媒体に関する。
【0017】
さらに別の実施形態では、本発明は後述の表2に記載の2389個の原子座標情報から選択された1つ以上のFabK−FMN複合体の3次元結晶構造情報を使用する、FabK蛋白質活性阻害剤開発方法に関する。
【発明の効果】
【0018】
本発明は、抗生剤の標的蛋白質として知られているFabK蛋白質の活性部位及び3次元結晶構造に関するもので、本発明によるサーモトガ・マリティマ菌株に由来したFabK蛋白質結晶は、結晶化程度が優れているだけでなく、X線回析が容易であり、類似した蛋白質であるFabI蛋白質とは異なる構造的特徴を有しているので、このような結晶の3次元構造は、現在まで開発されたFabI阻害剤とは異なる新たな活性調節物質の開発に有用に使用される。
【発明を実施するための最良の形態】
【0019】
以下、本発明を詳細に説明する。
【0020】
まず、本発明は、サーモトガ・マリティマから分離及び精製されたFabK蛋白質を提供する。前記蛋白質は、配列番号1のアミノ酸配列を有する蛋白質であって、本発明では、前記アミノ酸配列がFabK蛋白質の酵素の機能を有するものであるということを最初に究明した。前記のように、FabK蛋白質は、多くのバクテリア及び真菌類でFabIに代わって使用される酵素であって、FabI阻害剤に対して耐性を示す菌株に対する新たな抗生剤の開発において、標的蛋白質として非常に有用である。本発明によるFabK蛋白質は水溶性であって、適切な方法で結晶性を有することができるため、その3次元構造の分析が容易で、新たな抗生剤の開発のための新たな標的蛋白質として有用である。
【0021】
また、本発明は、前記配列番号1のアミノ酸配列を有するサーモトガ・マリティマから分離及び精製されたFabK蛋白質のコード塩基配列;及び前記コード塩基配列と作動可能に連結されたプロモーター及びターミネーター;を含むFabK蛋白質の発現ベクターを提供する。前記発現ベクターは、カナマイシン抵抗遺伝子、アンピシリン抵抗遺伝子、テトラサイクリン抵抗遺伝子、クロラムフェニコール抵抗遺伝子などの通常の選別マーカーを追加的に含むことができる。また、本発明は、前記FabK蛋白質の発現ベクターによって形質転換された形質転換体を提供する。前記形質転換体に使用される宿主としては、原生細菌であり、複製起源がpBR322を有し、T7 ポリメラーゼを生成することができるものであればいずれも使用することができ、例えば大腸菌などを使用することができる。また、本発明は、前記FabK蛋白質の発現ベクターを使用して宿主を形質転換させて、前記発現ベクターを含む形質転換体を製作して、前記製作された形質転換体を培養してFabK蛋白質を生成する段階を含むFabK蛋白質の製造方法を提供する。
【0022】
本発明の発現ベクターの製造のために、アミノ酸の欠失が全くなく、配列番号1に記載されたように1番のメチオニン末端(N−末端)から314番のアミノ酸であるグルタミン酸までのアミノ酸配列を有するサーモトガ・マリティマ由来のFabK蛋白質のコード塩基配列を利用して、生成された蛋白質の精製を容易にするために、N−末端に6個のヒスチジンタグを結合させて使用することができる。
【0023】
本発明による発現ベクター、形質転換体、及びこれを利用したFabK蛋白質の製造方法をより具体的に説明すると下記の通りであるが、下記の内容は、本発明の内容を例示して説明するための具体例にすぎず、本発明の範囲はこれに限定されない。
【0024】
まず、サーモトガ・マリティマのゲノムDNAからFabK蛋白質をコードする遺伝子を増幅するために、FabK蛋白質をコードする遺伝子の5´−末端及び3´−末端に対応するPCR反応用プライマーを考案して合成する。この時、前記5´−末端及び3´−末端に対応するプライマーは、各々に対応するクローニングされるベクターに存在する制限酵素認知部位と同一な制限酵素認知部位が存在するように考案して、前記ベクターのクローニングを容易にする。
【0025】
サーモトガ・マリティマのゲノムDNAを鋳型として、前記のように製造されたプライマーを使用してPCR反応を行って、FabK遺伝子を増幅する。増幅されたFabK遺伝子を適切な制限酵素で切断して得られた遺伝子断片を適切な発現ベクター、例えば大腸菌ベクターに挿入して、再組み合わせFabK遺伝子を含む発現ベクターを製造する。使用可能な制限酵素部位はNcoI NdeI、NheI、BamHI、EcoRI、SalI、HindIII、NotI、及びXhoIである。本発明の具体的な例としては、NdeI及びXhoI制限酵素部位を使用した。前記のように、前記発現ベクターは、宿主ゲノムへの挿入の有無の確認を容易にするために、カナマイシン抵抗遺伝子、アンピシリン抵抗遺伝子、テトラサイクリン抵抗遺伝子、クロラムフェニコール抵抗遺伝子などの通常の選別マーカーを含むことができる。このように製造された組換え発現ベクターを使用して、適切な宿主、例えば大腸菌を形質転換させた後、前記形質転換体を遺伝子の発現に適切な条件で培養する。この時、培養条件は、使用された宿主によって異なり、約15から40℃で、3から15時間培養することができ、前記条件は、比較的高い温度では培養時間を短くし、比較的低い温度では培養時間を長くして調節することができる。
【0026】
目的とする蛋白質の生成有無は、SDS−ポリアクリルアミドゲル電気泳動またはウエスタンブロッティング法などの通常の蛋白質同定方法によって確認することができる。組換えFabK遺伝子を含む発現ベクターに形質転換された形質転換体をアンピシリン、テトラサイクリン、カナマイシン、クロラムフェニコールなどの選別マーカーに対応する抗生剤が含まれている培地(例えばLB培地)で振盪培養し、前記培養した培地の光学密度(O.D.)が一定の細胞濃度(例えば600nmでのO.D.が0.5から0.7)になった時、IPTG(イソプロピル−β−D−ガラクトピラノシド)を添加して、組換えFabK遺伝子の発現を誘導する。前記培養液を遠心分離して形質転換体を沈殿させ、これを適切な緩衝液に懸濁させた後で破砕する。前記破砕物を遠心分離して蛋白質が溶解している上層液を分離した後、イオン交換クロマトグラフィー、アフィニティークロマトグラフィー、ゲルろ過クロマトグラフィーなどの通常の蛋白質分離及び精製手段によって製造された組換えFabK蛋白質を分離及び精製する。この時、前記組換えFabK蛋白質は、精製を容易にするために、ヒスチジンタグ融合蛋白質形態で生成される。
【0027】
また、他の側面から、本発明は、前記FabK蛋白質の3次元構造及び活性部位を究明して、FabK蛋白質を標的とする抑制化合物のスクリーニングにより有用な情報を提供することを特徴とする。
【0028】
また、他の側面から、本発明は、後述する表1または表2の原子座標情報から選択された1以上の原子座標情報で表現される3次元構造を有するFabK−FMN複合体結晶を提供する。FMNはFabK蛋白質の活性に必須の補助因子であり、インビトロ(in vitro)実験でFabKと複合体を形成することが観察され、生体内でもFabK蛋白質とFMN分子が複合体形態で作用することと考えられる。したがって、本発明で提供されるFabK−FMN複合体の結晶構造はFabK抑制剤探索および開発に非常に効果的に適用される。
【0029】
また、本発明は、下記のように究明されたFabK蛋白質の3次元結晶構造及び/またはFabK及びFMNが結合する特異的位置的特性及び前記活性部位で活性に影響を与える個別アミノ酸の役割に基づいて、FabK蛋白質の活性を阻害するFabK蛋白質阻害剤をスクリーニングすることおよび新たな抑制化合物を設計することを特徴とする、FabK蛋白質の阻害活性を有するFabK阻害剤開発方法を提供する。
【0030】
本発明の具体例において、前記FabK阻害剤開発方法は、配列番号1のアミノ酸配列を有するFabK蛋白質、またはFabK−FMN複合体、及び候補化合物を反応させる段階、及び前記候補化合物から前記FabK蛋白質またはFabK−FMN複合体と相互作用する化合物を選択して、FabK蛋白質の活性を阻害する化合物をスクリーニングする段階を含む。
【0031】
また、他の具体例において、本発明のFabK阻害剤開発方法は、使用されたFabK−FMN複合体の後述の表1に記載のX線回折パターン、または後述の表2に記載の2389個の原子座標から選択された1つ以上の3次元結晶構造情報を利用して、FabK蛋白質またはFabK−FMN複合体と結合可能な新たな化合物を新規設計することを特徴とする。
【0032】
また、他の具体例において、本発明のFabK阻害剤開発方法は、使用されたFabK−FMN複合体の後述の表1に記載のX線回折パターン、または後述の表2に記載の2389個の原子座標から選択された1つ以上の3次元結晶構造情報及び候補化合物の3次元結晶構造情報に基づく仮想スクリーニング法を使用して、FabK蛋白質またはFabK−FMN複合体と結合可能な化合物をスクリーニングすることを特徴とする。
【0033】
前記FabK蛋白質と相互作用(結合)する化合物の選択時に、下記のような3次元結晶構造及び/または活性部位に対する情報を使用して、より容易で正確にFabK蛋白質の活性を阻害する化合物を選択することができる。
【0034】
本発明のスクリーニング方法によって選択されたFabK蛋白質阻害剤は、抗生剤として作用し、前記抗生剤が抗生活性を有する菌株は、FabK蛋白質及びこれと類似した活性部位を有する蛋白質を必須で必要とする全ての微生物であり、例えば肺炎球菌、フェカリス菌、サーモトガ・マリティマ、化膿性連鎖球菌(Streptococcus pyogenes)、ネオリケッチャ・センネツ(Neorickettsia sennetsu)、エーリキア・シャフェンシス(Ehrlichia chaffeensis)、アナプラズマ・ファゴサイトフィルム(Anaplasma phagocytophilum)、好熱性一酸化炭素酸化細菌(Carboxydothermus hydrogenoformans)、ストレプトコッカス・アガラクチア(Streptococcus agalactiae)、及びクロストリジウム・ディフィシル(Clostridium difficile)からなる群から選択されたものである。
これをより詳細に説明すると、下記の通りである。
【0035】
まず、本発明は、FabK蛋白質の結晶化方法を提供する。新たな抗生剤の開発のための標的蛋白質の3次元結晶構造の究明においては、前記標的蛋白質が水溶性であって結晶性であることが重要である。本発明によるFabK蛋白質は水溶性であるので、その3次元結晶構造の究明のためには、結晶性を有するようにするための結晶化段階が必要である。このような結晶化段階には、一般に、X線結晶化法が使用され、このようなX線結晶化法を使用するための前処理段階として多様な結晶化方法が行われなければならない。本発明の具体例において、このような結晶化方法として通常の濃度平衡を利用した方法を使用でき、例えば、蒸気平衡法(例えば、シッティングまたはハンギングドロップ蒸気平衡法(Sitting−or Hanging−drop vapour diffusion method:Jancarik J.etal.,Appl.Cryst.,24,409−411(1991)参照)または透析法(連続式または回分式:Bunick C. and Stubbs G., Acta Cryst. D56, 1430−1431(2000))などが使用される。
【0036】
シッティングまたはハンギングドロップ蒸気平衡法の結晶化の原理及び過程は、下記の通りである。密閉された空間に母液の小さい滴及びこれよりはるかに大きい規模の貯水槽溶液が分離されたまま共存する時、これらの間で水または他の揮発性物質の移動が起こるようになる。一方、蛋白質の溶液条件のうち、熱力学的に準安定状態である過飽和状態で沈澱剤の種類によって蛋白質の沈澱が起こり、沈澱速度が遅くなって安定した結晶化状態となる。これに使用可能な沈澱剤はよく知られており、それらは濃縮状態の蛋白質溶液の溶解度を減少させる役割を担う。この時、蛋白質分子の周囲の相対的な吸着層を減少させるために、蛋白質分子が互いに凝集して結晶を生成するようになる。したがって、貯水槽溶液は、このような沈澱剤をはじめとして、緩衝液、塩、界面活性剤などが多様な濃度で混合され、蛋白質溶液及びこのような多様な条件の貯水槽溶液が通常の場合は約1:1の比率で混合されて滴を形成するようになる。このように得られた滴をシリコンでコーティングされたガラススライドに載せてひっくり返した状態で、準備されたプレートに載せて密封する。初期状態では、滴内の蛋白質の濃度が貯水槽溶液の濃度と異なるために蛋白質が結晶状態で存在しないが、密封された状態で置いておくと徐々に平衡が保たれるようになり、この時に前記で説明した原理によって特定の状態の条件で結晶が生成されるのである。このようなシッティングまたはハンギングドロップ蒸気平衡法において、貯水槽溶液内の沈澱剤だけでなく、塩、緩衝液、及び界面活性剤の種類、適正濃度、溶液のpH、及び実験温度は蛋白質の種類によって異なって選択され、場合によっては蛋白質結晶の生成においてとても重要な要素となる。
【0037】
本発明で、水溶性FabK蛋白質を結晶化させるためにFabK蛋白質溶液および貯水槽溶液を濃度平衡法、例えばシッティングまたはハンギングドロップ蒸気平衡法、透析法などを使用して反応させて結晶化を行う。
【0038】
前記貯水槽溶液は緩衝液、塩および沈澱剤を含むものを使用する。前記緩衝液としては、pHを調節して蛋白質を安定化させるためのものであって、pH4〜9.5、好ましくはpH4.6〜9、さらに好ましくは6.2〜8.5である全ての緩衝液が使用可能であり、例えば、PIPES(ピペラジン−1,4−ビス(2−エタンスルホン酸))、ビシン、トリス、酢酸ナトリウム、コハク酸ナトリウム、ビス−トリス緩衝液からなる群から選択された1種以上のものを使用することができる。
【0039】
前記塩は構造分析が容易な結晶形態を有するようにする役割を果たすものであって、全ての金属塩およびアンモニウム塩からなる群から選択されたものを使用することができる。前記アンモニウム塩は硫酸アンモニウム、塩化アンモニウム、リン酸アンモニウムなどからなる群から選択されたものを使用することができる。前記金属塩は全てのアルカリ金属(例えば、リチウム、ナトリウムなど)塩、アルカリ土類金属(例えば、マグネシウムなど)塩、および遷移金属(例えば、マンガン、亜鉛など)塩からなる群から選択された1種以上のものを使用することができ、好ましくは塩化物、シアン化物、チオシアン酸化物、酸化物、硝酸化物、水酸化物、硫酸化物などから選択された1種以上を使用することができ、例えば、塩化リチウム、塩化マグネシウム、チオシアン酸ナトリウムまたは硫酸アンモニウムなどを使用することができる。
【0040】
前記沈澱剤は結晶化のために沈澱を形成する物質であって、炭素数1〜4の直鎖または分枝鎖アルコールおよびポリエチレングリコール(PEG)(重量平均分子量200以上、好ましくは400〜20,000、さらに好ましくは550〜10,000)からなる群から選択された1種以上のものを使用することができる。
【0041】
貯水槽溶液全体での緩衝液、塩および沈澱剤の濃度は、使用される蛋白質溶液の濃度によって適切に調節することができる。具体的に、貯水槽溶液全体でのPIPES、ビシン、トリスなどの緩衝液の濃度は0.005〜1.5M、好ましくは0.01〜0.2Mであることができ、塩化リチウム、塩化マグネシウム、チオシアン酸ナトリウムまたは硫酸アンモニウムなどの塩濃度は0.05〜2M、より好ましくは0.05〜0.5Mであることができ、ポリエチレングリコールなどの沈澱剤の濃度は1〜40%(v/v)、好ましくは5〜30%(v/v)であるのが好ましい。貯水槽溶液内での沈殿剤及び塩の濃度が前記範囲より高い場合には、蛋白質が沈殿する現象が起こることがあり、前記範囲より低い場合には、結晶が生成されないので好ましくない。また、前記緩衝液は主に貯水槽溶液内での蛋白質の安定性に関連するものであり、その濃度が前記範囲を逸脱する場合には、貯水槽溶液内で適切な蛋白質の安定性を得ることができず、所望の結晶を得るのも困難である。前記貯水槽溶液は、このような成分以外にも、通常使用可能な塩、緩衝液、及び界面活性剤などを追加的に含むことができる。
【0042】
本発明に使用されるFabK蛋白質溶液はFabK蛋白質を適切な緩衝液に溶解したものであって、FabK蛋白質可溶性の全ての緩衝液を使用することができ、例えば、PIPES、ビシンおよびトリス、酢酸ナトリウム、コハク酸ナトリウム、ビス−トリス、HEPES(N−(2−ヒドロキシエチル)ピペラジン−N’−(2−エタンスルホン酸))、イミダゾール、リン酸ナトリウム、リン酸カリウム、MES(2−モルホリノ エタンスルホン酸)、ADA(N−(2−アセトアミド)イミノ酢酸)、カコジル酸ナトリウム、クエン酸三ナトリウムなどからなる群から選択された1種以上のものを使用することができる。蛋白質結晶を容易に得て構造分析が容易な結晶を得るために、FabK蛋白質溶液濃度は3〜30mg/ml、好ましくは5〜20mg/ml、さらに好ましくは 8〜15mg/mlであるのが良い。
【0043】
本発明に使用されるFabK蛋白質溶液と貯水槽溶液内の沈澱剤濃度は大体反比例するように使用するのが良い。つまり、FabK蛋白質溶液の濃度が高い場合には比較的に結晶化が容易であるので、貯水槽溶液内の沈澱剤の濃度は相対的に低くして使用し、FabK蛋白質溶液の濃度が高い場合には結晶化のために相対的に沈澱剤(例えば、ポリエチレングリコール)濃度が高い貯水槽溶液を使用するのが良い。例えば、蛋白質溶液の濃度が約10mg/mlである時には沈澱剤濃度を約15〜20%(v/v)として使用することができる。
【0044】
また、前記濃度平衡法は、pH8.0〜9.0で行われるのが好ましい。前記範囲は実験的に決定されたものであり、前記範囲のpH条件下で反応する時に、最も優れた結晶を得ることができる。シッティングまたはハンギングドロップ蒸気平衡法による結晶生成反応の反応温度は、4〜26℃であるのが好ましく、約20〜24℃であるのがより好ましく、反応時間は、1日〜20日であるのが好ましく、1週間〜2週間であるのがより好ましい。前記反応温度及び反応時間範囲は、X線分析が容易な最適のデータを有する結晶を得ることができる条件として、実験的に決定された範囲である。
【0045】
製造された結晶性FabK蛋白質の構造を分析するためには、X線分析を利用することができる。しかし、蛋白質結晶がX線の高いエネルギーに露出されると、その寿命が短くなり、データ強度も弱くなって、構造分析に良くない結果が発生する。このような欠点を防止するために、X線分析前にFabK蛋白質結晶を高速で窒素冷却する高速窒素冷却法を行うのが好ましい。高速窒素冷却時に、効果的なFabK蛋白質結晶の保護のために、結晶性条件に加えてエチレングリコール及びパラトン−Nを使用するのが好ましい。エチレングリコールを使用する場合、20〜35%(v/v)、好ましくは25〜30%(v/v)の濃度で使用するのが好ましい。パラトン−Nは、結晶に上載せして使用するのが好ましい。前記高速窒素冷却法を行う温度は、50〜200K、好ましくは80〜120Kである。前記濃度及び温度範囲は、FabK蛋白質がその結晶性に影響なく、窒素ストリームに耐えられて、高速窒素内で保管が容易な範囲として、実験的に決定されるものである。
【0046】
また、本発明は、X線結晶化方法によって得られた前記のような方法で結晶化されたFabK蛋白質の3次元結晶構造を提供する。本発明の具体例において、前記結晶の3次元構造を知るために、X線映像プレートを使用して回折パターンを得て、多波長異常分散法(MAD:multiwavelength anomalous dispersion)で位相情報を得ることができる。前記のように得られたFabK蛋白質結晶のX線回折パターン及び位相情報から電子密度地図を作成し、これから再び原子座標を導き出すことによって、3次元構造情報を得ることができる。
【0047】
X線を使用した回折パターンのデータ収集方法は、X線の供給源によって一般実験室で得る方法及びシンクロトロンを利用する方法に分けられる。このうちのシンクロトロンを利用する方法は、結晶サイズが50μm程度の小さい結晶まで可能であり、1つの波長だけでなく多様な波長でデータを収集することができるため、迅速な構造を得ることができる。したがって、本発明の好ましい具体例では、シンクロトロンを利用して回折パターンのデータを収集することができ、このように得られた本発明によるFabK蛋白質結晶の回折パターンは、下記の表1に示した通りである。
【0048】
【表1】

【0049】
位相情報は、多重重原子同型置換法(Multiple isomorphous replacement)、多波長異常分散法、構造置換法(Molecular replacement)などによって得ることができ、本発明の好ましい具体例では、多波長異常分散法を利用して、FabK蛋白質結晶の位相情報を得ることができる(Modern X−ray Analysis on Single Crystals,Peter Luger参照)。具体的には、前記多波長異常分散法は、重金属を有する蛋白質結晶で互いに異なる三個の波長を通じて異常回折パターン(anomalous diffraction pattern)を利用して位相を計算するのである。このような多波長異常分散法を利用するために、位相を計算してこれによる初期モデルの計算を行わなければならず、このような計算に使用することができる全てのプログラムを使用することができる。本発明の好ましい具体例では、SOLVE及びRESOLVE(ロスアラモス研究所、米国)を使用することができ、その後の精密化段階及び標準化段階のために、CNS(エール大学)、CCP4(ケンブリッジ大学)などのプログラムを使用することができる。
【0050】
精密化段階は、X線回折パターンから得られた電子密度をO(Alwyn Jones,http://web.mac.com/alwyn_47/OInTheWorks/OInTheWorks.htmlを参照)及びCOOT(Paul Emsley, http://www.ysbl.york.ac.uk/~emsley/coot/を参照)プログラムでコンピュータモニターに表示して、そこに最も適した構造に修正することによって行われる。精密化段階を行った結果、本発明によるFabK蛋白質結晶は、FMN及び24個の水分子を含み、下記の表2に記載の3次元結晶構造を有する原子モデルで表現されることが明らかになった。FabK蛋白質を標的とするFabK阻害剤開発において、FabK蛋白質を単独で使用したり、このようなFabK−FMN複合体の形態で使用し、特に、このような複合体の形態で3次元構造の結晶性が優れていて、FabK阻害剤開発に非常に有用である。
【0051】
FabK−FMN複合体の3次元上の原子座標
【表2】
【0052】
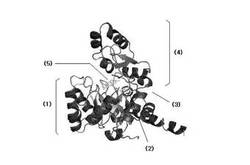
前記表2のうち、A:原子、B:原子数、C:原子名、D:残基名、E:残基数、F:x軸情報、G:y軸情報、H:z軸情報、I:占有因子、J:温度因子である。また、WATは水分子を表す。
【0053】
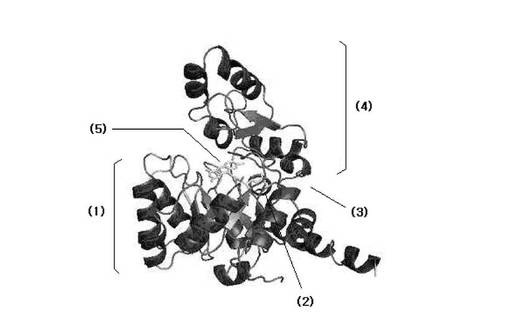
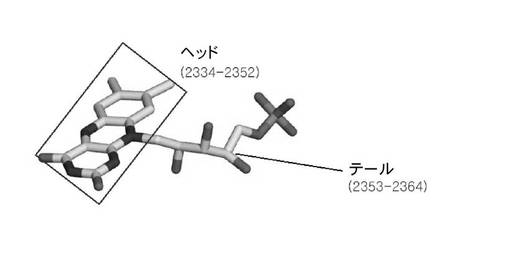
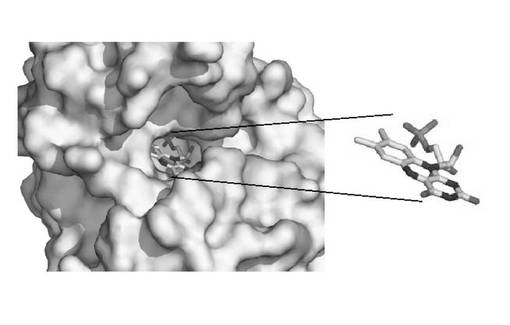
精密化段階後、前記得られた原子モデルから多様な情報を得るために分析段階を追加的に行うことができる。例えば、グラフィック上に3次元空間のFabK蛋白質の原子モデルを表示しておいて活性部位の形態を観察することによって各原子間の距離及び空間を測定したり、重要残基の空間的位置と異なる残基との相互作用を観察して、適切な阻害剤を見つけるモデリング段階を経ることができる。前記分析段階を行った結果得られたFabK−FMN複合体の構造を図9、図11aおよび11bに示した。これら図面から確認できるように、FabK蛋白質でFMNが結合している部位は、TIMバレル(TIM barrel;G.K.Farber and G.A.Petsko:The evolution of a/b barrel enzymes,TIBS1990;228−234参照)の上側に位置するループで構成されたところであり、これよりこの部位がFabK蛋白質の活性部位であるといえる。より具体的に、FMN分子(5)は前記表2の原子番号2334から2364までの原子からなり、そのうちの原子番号2334から2352までがヘッド(HEAD)部分を構成し、原子番号2353から2364までがテール(TAIL)部分を構成する。このような構造を有するFMN分子はFabK蛋白質のTIMバレル(1)の上側のループ部位(2)(ループ1:表2の原子番号1015から1103まで、ループ2:原子番号141から206まで)に結合しており、その結合部位上側にFabK活性に必要な補酵素であるNADPおよび基質が結合する。FabK蛋白質のTIMバレルと残りの部分(以下、蓋部位(4))を連結するヒンジ部位(3)(原子番号1428から1485;すなわち198から205までのアミノ酸配列)が存在し、流動性を付与する。つまり、前記ヒンジ部位が動いてNADPおよび基質が結合する空間を形成してFabKが作用するようにする。したがって、前記ヒンジ部位はFabK活性部位のうちの一つであり得る。また、NADPおよび基質が結合する空間を提供するTIMバレルの上のFMN分子が位置している部位の上側部分、つまり、FabK蛋白質に結合されたFMNのヘッド部分水平面を半球の平らな底面とし、ヒンジ(部位および蓋部分を含む半球部分もFabK活性部位である。より具体的に、前記TIMバレル上側活性部位はTIMバレルの上に結合されたFMN分子のヘッド水平面を半球底面とする半径15Å以内、好ましくは半径6〜12Åの半球内に位置する部位であり得る。前記半球部分はループ1部分、ヒンジ部分を含む原子番号1380から2137までを含む。
【0054】
また、突然変異実験を通じてFabK蛋白質の活性に影響を与えるアミノ酸配列を確認し、このために、配列番号1のアミノ酸配列のうちの1つを標的となるアミノ酸として選定した後、前記アミノ酸のコード塩基配列を他のアミノ酸、例えばアラニンをコードするように変異させて、プライマーを製造する。この時、5´末端プライマー及び3´末端プライマーを全て使用し、これらプライマーの中間に変異させるアミノ酸をコードする塩基配列を有するように構成して、前記PCR方法で所望の組換えクローンを得る。このように得られた組換えクローンは、DpnIなどの制限酵素の処理を通じて大腸菌から得られた元のベクターを完全に分解するようにする。この時、制限酵素(DpnI)の反応時間は30分〜2時間であるのが好ましい。その後、組換えクローンだけを大腸菌に導入して、所望の形質を有する突然変異FabK蛋白質を形成する。
【0055】
このような方法で行われた突然変異実験を通じて、本発明によるFabK蛋白質(配列番号1)の208番のチロシン(Y208)および209番のリシン(K209)をアラニンに置換させた場合、その活性は各々増加し、一方で前記FabK蛋白質(配列番号1)の211番のリシン(K211)、214番のリシン(K214)、229番のヒスチジン(H229)および261番のロイシン(L261)をアラニンに置換させた場合、その活性は各々減少し、特に前記FabK蛋白質(配列番号1)の229番のヒスチジン(H229)及び276番のメチオニン(M276)をアラニンに置換させた場合、活性が大幅に減少することが明らかになり、前記アミノ酸がFabK蛋白質の活性に影響を与える重要なアミノ酸であると考えられる。また、本発明によるFabK蛋白質(配列番号1)の208番から215番までの8個のアミノ酸から構成されたへリックス及び278番から283番までの6個のアミノ酸から構成されたへリックスの欠失がFabK蛋白質の活性の大幅な減少を招くことが観察され、前記2個のへリックス部位も、本発明によるFabK蛋白質の活性に影響を与える重要な部位であることが明らかになった。
【0056】
したがって、本発明によるFabK蛋白質(配列番号1)のTIMバレルの上側に位置するループ部位;FabK蛋白質のTIMバレルの上に結合されたFMN分子のヘッド)水平面を平らな半球底面とする半径15Å以内、好ましくは半径6から12Åの半球内に位置する1つ以上のアミノ酸;198から205までのアミノ酸配列を含むヒンジ部位;208番のチロシン(Y208);209番のリシン(K209);211番のリシン(K211);214番のリシン(K214);229番のヒスチジン(H229);261番のロイシン(L261);276番のメチオニン(M276);208番から215番までの8個のアミノ酸から構成されたへリックス、;及び278番から283番までの6個のアミノ酸から構成されたへリックス;からなる群から選択された1つまたはそれ以上の部位及び候補物質の相互作用の有無を検出して、より効果的なFabK蛋白質阻害剤を選択することができる。
【0057】
したがって、本発明によるFabK蛋白質の阻害活性を有するFabK阻害剤開発において、前記表2に記載の2389個の原子座標のうちの前記活性部位に相当する原子座標を選択的に使用してFabK蛋白質阻害剤をスクリーニングしたり、FabK蛋白質の阻害活性を有する化合物を設計したり仮想スクリーニングして、より容易で効果的にFabK阻害剤開発を行うことができる。
【0058】
また、本発明は、前記で究明されたFabK蛋白質の3次元結晶構造に対する情報がコンピュータ読取可能に保存された保存媒体、例えばフロッピー(登録商標)ディスク、ハードディスクなどを提供する。前記媒体に保存された3次元結晶構造情報は、前記表2に記載の原子座標の全部または一部(特に、前記活性部位に相当する原子座標)を含み、前記活性部位のアミノ酸及び前記突然変異アミノ酸の残基を含む部位に対する情報を含む。
【0059】
前記で究明されたFabK蛋白質の3次元結晶構造、及びFabK及びFMNが結合した特異な位置的特性及び前記活性部位で活性に影響を与える個別アミノ酸の役割に基づいて、本発明はFabK蛋白質の活性を阻害するFabK蛋白質阻害剤をスクリーニングする方法を提供する。本発明によるFabK蛋白質阻害剤のスクリーニング方法は、配列番号1のアミノ酸配列を有するFabK蛋白質及び候補化合物を反応させる段階、及び前記候補化合物から前記FabK蛋白質と相互作用する化合物を選択する段階を含む。
【0060】
前記FabK蛋白質は、前記のような結晶化方法によって結晶化されたものである。前記FabK蛋白質と相互作用する化合物の選択時に、前記記載されたような3次元結晶構造及び/または活性部位に対する情報を使用して、より容易で正確にFabK蛋白質の活性を阻害する化合物を選択することができる。また、本発明のスクリーニング方法は、このような保存媒体を使用するものである。
【0061】
以下、本発明を下記の実施例によって詳細に説明する。但し、下記の実施例は本発明を例示するためのもので、本発明が下記の実施例によって限定されるのではない。
【実施例】
【0062】
実施例1:サーモトガ・マリティマ FabK蛋白質の発現及び精製
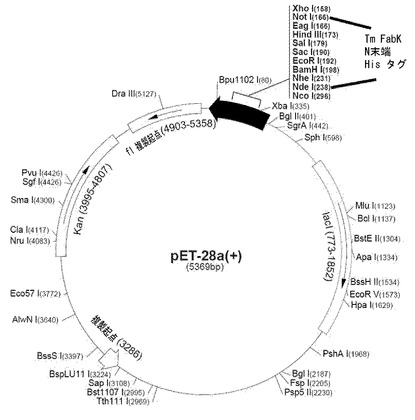
サーモトガ・マリティマ FabK(配列番号1)の1番アミノ酸のメチオニン(N−末端)から312番アミノ酸のグルタミン酸(C−末端)までのFabKをコードする遺伝子(Genbank Accession No.AE000512)を、PCR反応を通じて増幅した。PCR反応に使用されるプライマーとして、配列番号2及び3のオリゴヌクレオチドを使用した。配列番号2及び3のプライマーは、各々NdeI及びXhoI制限酵素認識部位を有する。
【0063】
鋳型ゲノムDNAを1μl、2.5mMdNTP溶液を5μl、各100pmolの配列番号2及び配列番号3のプライマーを各々0.3μl、PfuTaq DNA重合酵素(5U/μl、Stratagene社、米国)を1μl、及び10倍濃度PCR反応溶液(Stratagene社、米国)を5μl混合し、さらに蒸溜水を最終的に50μlになるように加えて反応溶液を作成した後、この反応溶液を95℃で5分、95℃で30秒、55℃で30秒、及び72℃で1分の反応を30回繰り返した。その後、PCR反応溶液を0.8%アガロースゲルで電気泳動して、約970塩基長のFabK遺伝子を分離した。分離されたFabK遺伝子をNdeI及びXhoIで処理した後、電気泳動して、FabK遺伝子の切片を抽出した後、50μlの蒸溜水溶液に溶存させ、これをFabKN/Xと命名した。
【0064】
プラスミドpET−28a(Novagene社、米国)は、N−末端に6個のヒスチジン残基を発現するが、これをNdeI及びXhoI制限酵素で処理した後、電気泳動して、約5400bpの大きさのDNA切片を分離して、これをpET−28aN/Xと命名した。0.5μgのFabKN/Xを0.1μgのpET−28aN/Xと共に反応チューブに入れた後、2μlの10倍連結反応溶液(50mMのトリス塩酸(pH7.8)、100mMの塩化マグネシウム、100mMのDTT(ジチオトレイオール)、及び10mMのATP(アデノシン三リン酸))、10UのT4 DNAリガーゼを入れて総体積が20μlになるように蒸溜水を加えた後、16℃で12時間反応させた。この反応溶液を大腸菌BL21(DE3)コンピテント細胞(Novagene 社,米国)に添加して形質転換させた後、50μg/mlのカナマイシンを含むLB寒天培地(1% バクトトリプトン、0.5%酵母抽出物、1%塩化ナトリウム)にプレーティングして、大腸菌形質転換体を選別した。この形質転換体からプラスミドを抽出し、制限酵素及び塩基配列分析によって組換えプラスミドpET−28a−FabKを得た。
【0065】
前記で得られた再組み合わせプラスミドにクローニングされたFabK遺伝子の塩基配列は、Big−Dye Cycle Sequeincing System(Applied Biosystem社、米国)を利用して、ABI 377 DNA塩基配列分析器によって確認した。
【0066】
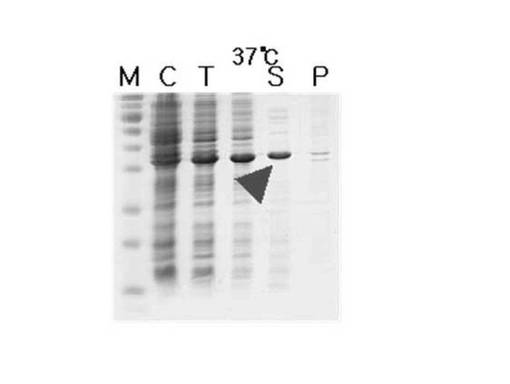
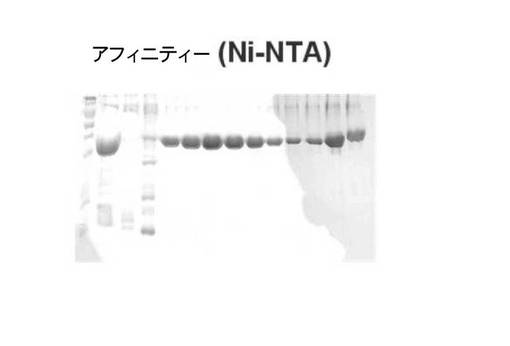
前記形質転換された大腸菌菌株を50μg/mlのカナマイシンが含まれているLB培地で12時間振盪培養した後、1mlの培養液を50μg/mlのカナマイシンが含まれている100mlのLB培地に移して、37℃で培養液のO.D.が600nmで約0.6程度になった時点で、IPTGを最終濃度が0.5mMになるように添加した。IPTG添加4時間後に、10,000gで30分間遠心分離して、各々細胞沈殿物を回収した。細胞沈殿物を20mMのトリス塩酸(pH8.0)、0.1Mの塩化ナトリウム、1mM のTCEP(トリス(2−カルボキシエチル)ホスフィン塩酸)溶液に懸濁させて、氷上で超音波で細胞を破砕した。細胞破砕液を遠心分離して、上層液をニッケル親和性カラムで、(ファーマシア社、スウェーデン)に通過させ、その後、BLUEカラムファーマシア社、スウェーデン)に通過させて、通過溶液をスーパーデックス−75(Superdex−75)ゲルろ過クロマトグラフィーカラム(ファーマシア社、スウェーデン)を使用して精製して、FabK蛋白質を得た。
【0067】
実施例2:シッティングまたはハンギングドロップ蒸気平衡法によるFabK蛋白質の結晶化
前記実施例1で得られたFabK蛋白質を下記のようなシッティングまたはハンギングドロップ蒸気平衡法によって結晶化した。
【0068】
20mMのトリス塩酸(pH8.0)、300mMの塩化ナトリウム、1mMのジチオトレイオール、50mMの塩化アンモニウムを含む溶液内のFabKの濃度が11mg/mlである蛋白質溶液を製造した。最終蛋白質濃度は、ブラッドフォード法(Current Protocols in Protein Science,3.4.10)によって決定した。11mg/mlに濃縮された蛋白質溶液に補助因子であるNADH及びFMNを添加して、最終蛋白質溶液を製造し、この時、FabK蛋白質に対する、前記補助因子のモル当量比は1:5になるようにした。
【0069】
前記FabK蛋白質溶液の結晶条件を検索するために、初期スクリーン溶液(Hampton research Inc.)を使用し、自動化されたスクリーニングのために、Hydra II−plus one system(Matrix technologies社、米国)を利用した。0.2μlの蛋白質溶液及び0.2μlの貯水槽溶液を混合した後、22℃で2日から20日が経過した後で結晶の生成有無を確認した。その結果、0.1Mのビシン緩衝液(pH9.0)、及び1M塩化リチウム及び20%(v/v)ポリエチレングリコール6,000沈澱剤を含む貯水槽溶液、0.1Mのトリス塩酸緩衝液(pH8.0)、及び0.2Mの塩化マグネシウム、及び20%(v/v)ポリエチレングリコール20,000沈澱剤を含む貯水槽溶液を使用した場合に、X線回折能力が最も優れた結晶が得られた。
【0070】
1μlの最終蛋白質溶液及び1μlの前記貯水槽溶液を混合した滴をシリコンでコーティングされたガラススライドの表面に滴下し、このスライドを貯水槽溶液0.5mlが入っているプレート上に覆って、22℃の恒温状態に置いた。一日で種結晶が生じ、一週間後にその結晶の大きさが0.1×0.1×0.2mmに成長した。
【0071】
実施例3:高速窒素冷却法を追加的に行ったFabK蛋白質の結晶化
前記実施例2で得られたFabK蛋白質結晶を直接高エネルギーのX線に露出させる場合に発生する問題点を解決するために、得られたFabK蛋白質結晶のX線分析前に、下記のような高速窒素冷却法による冷却を行った。
【0072】
グリセロール、ギ酸ナトリウム、エチレングリコール、スクロース、パラトン−Nなどの多様な冷却溶液を多様な濃度で検索した後、高速窒素冷却法の最適条件を得た。その結果、25%(v/v)エチレングリコール、0.1Mのビシン緩衝液(pH9.0)、1Mの塩化リチウム、20%(v/v)ポリエチレングリコール6,000を含む高速冷却用溶液に結晶を数秒以内で浸漬して取り出すと、FabK蛋白質結晶に全く害を及ぼさず、100Kの液体窒素ストリームに最もよく耐えることが明らかになった。
【0073】
冷却溶液に浸漬した結晶を約0.3mmのナイロン結晶回収ツール(Hampton research社、米国)を利用して取り出して直ちに100Kの窒素ストリームに入れた。
【0074】
本実施例では、実験中によく発生する水枠現象(高速窒素冷却が完全でない場合に水が凍って現れる現象)も見られなかった。
【0075】
実施例4:放射光加速器を通じたFabK蛋白質結晶のX線回折パターンのデータ収集及び分析
前記実施例3で得られたFabK蛋白質結晶を利用して、フォトンファクトリー(茨城県つくば市大穂1-1)の放射光加速器のAR−NW12ラインで回折パターンのデータを収集する実験を行った。結晶データの限界は2.3Åであり、データはDENZO及びSCALEPACK(Otwinowski,Z.and Minor,W.Methods Enzymol.,276,461−472(1997))で処理した。結晶データの収集及び精密化の結果を表3に示した。
【0076】
【表3】
【0077】
実施例5:FabK蛋白質結晶のX線回折パターンのデータ解釈及び構造計算
FabK蛋白質の構造は、多波長異常分散法を通じて究明した。多重波長分析法は、初期位相情報を求めるために、SOLVE(Terwilliger T.C.and BerendzenJ.,Acta Crystallogr.D.Biol.Crystallogr..55,849−861(1999))プログラムを利用し、電子密度の修正のために、RESOLVE(Terwilliger T.C.,ActaCrystallogr.D.Biol.Crystallogr..56,965−972(2000))プログラムを利用した。使用した回折パターンのデータは、2.3Åの解像度を有して行った。
【0078】
精密化段階では、CNSプログラム(Brunger,AT.et al.,ActaCryst.D.,54,905−921(1998))を使用した。精密化段階は、CNSプログラムのシミュレーションアニーリング方法を利用して行った。シミュレーション開始温度は1500℃であり、各段階で100℃ずつ冷却して、25℃まで冷却させて行い、最終R因子及びRfree因子は各々24.3%及び24.2%であった。その後、Oプログラムを使用して最適構造を導き出した。最適構造を導き出すために、精密化段階を行ったが、これはX線回折パターンのデータから導き出された電子密度をOプログラムでコンピュータモニターに表示して、そこに最も適した構造に修正する作業を繰り返すことによって行った。
【0079】
精密化段階を行った結果、本発明によるFabK蛋白質の3次元結晶構造は、以前までのFabI蛋白質の構造とは明確に異なるTIMバレル形態の構造及びTIMバレルの上側のループが構成する活性部位を示し、ここにFMNが結合していることが明らかになった。
【0080】
実施例6:FabK蛋白質の活性部位を中心とする個別アミノ酸残基の突然変異体の作成
前記実施例5でFabK蛋白質の3次元結晶構造を得た後、FabK蛋白質の活性部位の個別アミノ酸の役割を調べるために、突然変異体を作成して活性に対する影響を観察した。
【0081】
個別残基の突然変異体は、チロシン208番残基をアラニン(Y208A:配列番号:4及び5)に、リシン209番残基をアラニン(K209A:配列番号:6及び7)に、リシン211番残基をアラニン(K211A:配列番号:8及び9)に、リシン214番残基をアラニン(K214A:配列番号:10及び11)に、ヒスチジン229番残基をアラニン(H229A:配列番号:12及び13)に、ロイシン261番残基をアラニン(L261A:配列番号:14及び15)に、メチオニン276番残基をアラニン(M276A:配列番号16:及び17)にした。
【0082】
FabK蛋白質の活性部位に影響を与えるへリックスの欠失に対する効果を観察するために、208番から215番残基まで(8個のアミノ酸:配列番号:18及び19、以下ではD208とする)及び278番から283番まで(6個のアミノ酸:配列番号:20及び21、以下ではD278とする)のアミノ酸が欠損した突然変異蛋白質を前記PCR方法を利用して形成した。
【0083】
個別アミノ酸突然変異体及びD208及びD278突然変異蛋白質を形成するために、配列番号4から配列番号21までのヌクレオチドオリゴマーを合成した後、前記PCR方法で増幅した。pET−28a−FabKクローン化されたプラスミドを10ng、2.5mMのdNTPを5μl、前記配列プライマーを0.2μl、10倍濃度のPCR反応溶液(100mM 塩化カリウム、100mM 硫酸アンモニウム、200mM トリス塩酸(pH8.8)、20mM 硫酸マグネシウム)を5μlを入れ、反応液が全体で50μlになるように蒸溜水を入れた。95℃で1分、95℃で50秒、60℃で50秒、68℃で6分の反応を18回繰り返した。その後、DpnI(Stratagene社、米国)を0.5μl(10U)を入れて37℃の恒温器で1時間反応させた。DpnI制限酵素によって大腸菌から由来したpET−28a−FabKプラスミドは分解され、前記PCR反応を通じて生成された突然変異pET−28a−FabKプラスミドだけが残るようになる。このような反応生成物を大腸菌DH5αコンピテント細胞に添加して形質転換させた後、実施例1で既述の50μg/mlのカナマイシンを含むLB寒天培地にプレーティングして、大腸菌の形質転換体を選別した。これらからプラスミドを抽出して、塩基配列分析を通じて突然変異pET−28a−FabKプラスミドを得た。
【0084】
得られた突然変異pET−28a−FabKプラスミドは、発現宿主である大腸菌BL21(DE3)に形質転換させた後、実施例1の精製方法を通じて精製した。
【0085】
生成された突然変異体は、図7で、SDS−アクリルアミドゲル電気泳動で分離程度を示す。
【0086】
実施例7:FabK蛋白質の活性部位を中心とする個別アミノ酸残基の突然変異研究を通じた重要残基及び活性の影響
実施例6で精製された蛋白質は、エノイル−ACP還元酵素活性測定方式を通じて各突然変異体の活性を測定した。
【0087】
具体的には、100mMリン酸緩衝液(pH 7.5)中に、NADHまたはNADPHを終濃度175μM、FabK蛋白質を終量5μg、酵素基質(トランス−2−オクテノイル−N−アセチルシステラミン(trans−2−octenoly−N−acetylcysteramine))のDMSO溶液を10μl加え、蒸留水を加えて最終体積を200μlにした。室温で0−60分反応後、溶液の340nmの吸光度を測定して、酵素活性を測定した。酵素活性は、野生型FabKの活性を100とした時の各変異体の比活性(%)で測定した。
【0088】
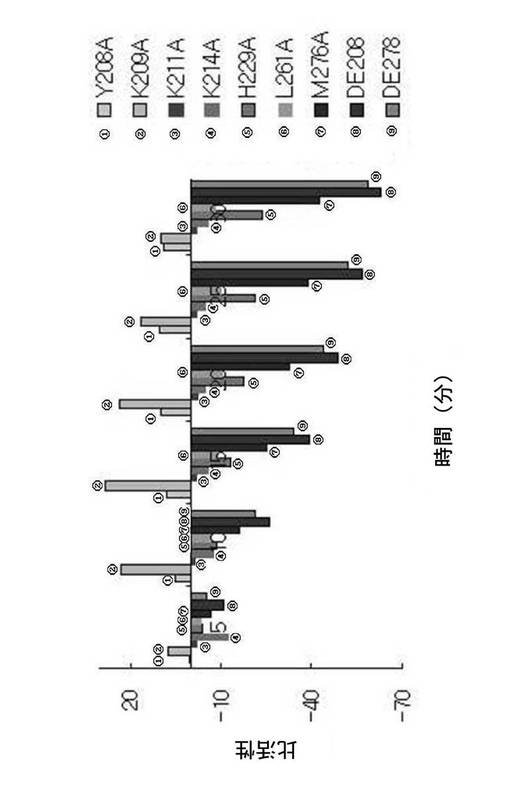
活性の加減を図10に示す。Y208A及びK209Aの場合、活性の増加を示しており、これら残基はFabK蛋白質の活性を阻害する残基として現れ、その他のFabK変異体K211A、K214A、H229A、L261A、M276A、D208、D278の場合、活性の減少を示している。特に、H229A及びM276Aの場合、大幅な活性の減少を示し、D208及びD278のへリックスを除去した突然変異体は、活性の減少がより著しいことが分かる。これは、ヒスチジン229番及びメチオニン276番、そして構造を支持するへリックスである208番から215番、278番から283番がFabKの蛋白質の活性を維持するのに重要な作用をすることを示している。
【図面の簡単な説明】
【0089】
【図1】FabK発現ベクター及びクローニング情報を示した図面である。
【図2】FabK蛋白質の発現程度のテスト結果を示した図面である。図中、Mは分子量マーカー、Cは粗抽出物、Tは全細胞溶解液、Sは可溶性画分、Pは不溶性画分(封入体)をそれぞれ表す。
【図3】蛋白質発現細胞を破砕した上清をニッケル親和性カラムで精製したサンプルのゲル電気泳動図である。
【図4】図3の精製サンプルを、BLUEカラムに通過させたサンプルのゲル電気泳動図である。
【図5】図4の精製サンプルを、スーパーデックス−75ゲルろ過クロマトグラフィーカラムで精製したサンプルのゲル電気泳動図である。
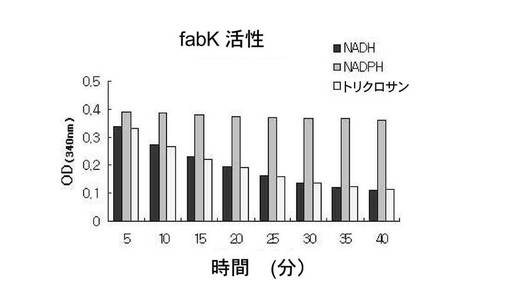
【図6】NADH、NADPH依存によるFabK蛋白質の活性度を示した図面である。
【図7】FabK蛋白質の活性部位のアミノ酸突然変異体の精製結果を示した図面である。
【図8】FabK蛋白質結晶構造を示した図面である。
【図9】FabK蛋白質の骨格構造をCアルファ(Calpha)リボンで示した図面である。
【図10】FabK変異体の活性度を示した図面である。
【図11a】FMNの構造を模式的に示したものである。
【図11b】FabK−FMN複合体でのFMN位置を示したものである。
【符号の説明】
【0090】
(1) TIMバレル部位
(2) ループ部位
(3) ヒンジ部位
(4) 蓋部位
(5) FMN分子
【技術分野】
【0001】
本発明は、好熱性真正細菌サーモトガ・マリティマ(Thermotoga maritima)菌株に由来するFabK(エノイル−ACP還元酵素)蛋白質に関し、前記蛋白質の活性部位及び3次元結晶構造を究明し、これを利用して新たなFabK蛋白質阻害物質をスクリーニングして、既存の抗生剤に耐性を示す菌株に対して優れた抗菌活性を示す新たな活性調節物質を開発する技術に関する。
【背景技術】
【0002】
トリクロサンは、包括的な抗菌作用を有する抗微生物剤であって、約30年間多様な製品に抗生及び抗菌活性を付与するために使用されてきた。例えば、トリクロサンは、石鹸、シャンプー、洗濯剤はもちろん、歯磨き粉や化粧品などに含まれているだけでなく、子供のおもちゃ、じゅうたんなどにも含まれている。そのような意味で、トリクロサンの使用範疇は、紫外線または漂白剤に相当するといっても過言でない。
【0003】
トリクロサンは、エノイル−ACP還元酵素(エノイル−[アシルキャリアー蛋白質]レダクターゼ(E.C.1.3.1.9)NADH−依存性 トランス−2−エノイル−ACP還元酵素、ENR)またはFabIといわれる酵素の活性を抑制する作用がある。前記FabIは、ほぼ全てのバクテリア及び真菌類に存在して、バクテリアの細胞膜形成に必須の脂肪酸の生合成に関与する酵素のうちの1つであって、微生物の生存に必須の酵素である。したがって、トリクロサンは、FabIの活性を阻害する活性を有するので、広範囲の殺菌剤として使用することができる。
【0004】
好気性細菌であるマイコバクテリア類(mycobacteria)の場合には、InhAという酵素が脂肪酸の生合成中にFabIのようにエノイル−ACP還元酵素として作用して反応段階を活性化し、この場合、結核の治療剤であるイソニアジドがトリクロサンと同一の方法でInhAの活性を抑制する役割を担うということが知られている。これら2つの酵素であるInhA及びFabIを比較する時、これらのアミノ酸配列の相同性は、異なる種類のFabI間のアミノ酸配列の相同性より相対的に低いが、活性に必要な主要アミノ酸は同一であることが明らかになった。
【0005】
一方、脂質は、細胞膜を構成する主成分であって、脂質の生合成に関与する酵素は、バクテリアはもちろん、すべての生物体になくてはならない必須の酵素である。哺乳類での脂質の生合成は、FAS(脂肪酸合成酵素)といわれる巨大分子ホモダイマーが複数の段階の反応を各々異なる部位で順次に循環して行うI型脂肪酸合成経路を通じて行われる反面、バクテリアなどの微生物での脂質の生合成は、哺乳類とは異なったII型脂肪酸合成経路を通じて行われる。前記II型脂肪酸合成経路は、各段階の反応が各々異なる独立した酵素によって行われるという特徴がある。FabIは、前記段階のうちの最後の段階に相当するエノイル−ACPを還元させてアシル−ACPに転換させる段階を担当する。
【0006】
前記のように、FabIは、脂質の生合成の最後の段階で、脂質の伸長を触媒する作用をすることによって、脂肪酸の生合成における必須の役割を担っている。FabIは、SDR(短鎖アルコール脱水素酵素/還元酵素)スーパーファミリーに属して、他のファミリーメンバーと共に4量体を形成しており、大腸菌から分離されたFabIはNADH(ニコチンアミドアデニンジヌクレオチド)という補酵素を使用する。(Egan,A.F.and Russel,R.R.B.,Genet.Res.,21,3603−3611(1973))。最近明らかになったFabI−トリクロサン複合体構造によれば、トリクロサンは、NADH及びNAD上に位置し、酵素の活性部位を占めて非共有性であるが、非常に強い相互作用で基質が入ってこられなくする方法で酵素の活性を阻害する(Heath,R.J.,etal.,J.Biol.Chem.,274,11110−11114(1999))。黄色ブドウ球菌(Staphyloccus aureus)の場合、FabIがNADHでなくNADPHという補酵素を使用することが明らかになり、この場合にも、トリクロサンが同様に相互作用して、酵素の活性を阻害することが明らかになった。したがって、現在、製薬会社はもちろん、大学及び各種研究団体で開発しているFabI阻害剤は、トリクロサンと類似したメカニズムを有する化合物である。
【0007】
しかし、最近、肺炎球菌(Streptococcus pneumoniae)などの一部の主要病原菌がトリクロサンに耐性を示すことが報告された(Heath,R.J.and Rock,C.O.,Nature,406,145−146(2000))。これら耐性を示す菌株にはFabI酵素が存在しないが、遺伝子群にFabKという遺伝子が存在して、この遺伝子が発現して生成されるFabK蛋白質がFabI酵素の役割を代わりに担うことが明らかになった。フェカリス菌(Enterococcus faecalis)及びサーモトガ・マリティマ(Thermotoga maritima)も、脂肪酸の生合成経路にFabI蛋白質の代わりにFabK蛋白質が関与すると報告された。これらFabK蛋白質は、現在まで知られているFabI蛋白質とはコード遺伝子配列が全く異なり、SDRスーパーファミリーの他の蛋白質ともコード遺伝子配列に類似性がない。フェカリス菌及びサーモトガ・マリティマなどの菌株に存在するFabKは、そのアミノ酸配列において、肺炎球菌のFabKと各々68%及び48%の同一性を示している(Marrakchi,H.,etal.,Biochem.J.,370,1055−1062(2003))。FabKはフラビン蛋白質であり、活性のために補酵素であるNADHを必要として、トリクロサンによって不活性化されない特性を有する蛋白質である。
【0008】
現在の結核治療剤は、大部分がFabI/InhAと関連したものであるため、前記FabI蛋白質を使用せずに前記結核治療剤に対して耐性を示す菌株が次から次へ発見されている。このような耐性を示す菌株は、FabIの代わりにFabKを使用するものである。例えば、結核治療剤に対して耐性を示す呼吸器疾患の主要菌株である肺炎球菌またはバンコマイシンに耐性を示すフェカリス菌の場合も、FabIの代わりにFabKを使用することが明らかになった。つまり、これら耐性を示す菌株に対して抗菌活性を有するためには、FabIでなくFabKの活性を阻害する新たな化合物が必要である。現在までに報告されたところによれば、FabKは、その作動メカニズムが比較的よく知られているFabIとは異なっていることが明らかになり、その構造及び機能に対する研究が切実に要求されている。
【発明の開示】
【発明が解決しようとする課題】
【0009】
このような要求に応じるために、本発明の発明者は、サーモトガ・マリティマからFabK蛋白質を分離精製して結晶化して、3次元結晶構造を究明して、活性部位の個別アミノ酸の変異実験を通じて活性にメカニズム的に寄与するアミノ酸を定義し、既存のFabIとは異なるFabKの構造を究明して、新たな抗生剤の開発に有用に使用される技術を提供することによって、本発明を完成した。
【0010】
そのために、本発明の目的は、前記FabK蛋白質の活性部位を究明することにある。
【0011】
本発明の他の目的は、前記FabK蛋白質の3次元結晶構造を究明することにある。
【0012】
本発明の他の目的は、前記で究明されたFabK蛋白質の活性部位及び/または3次元結晶構造情報を利用して、FabK蛋白質と相互作用して阻害活性を有する化合物をスクリーニングする方法を提供することにある。
【課題を解決するための手段】
【0013】
前記課題を解決するために、本発明は、サーモトガ・マリティマ菌株に由来するFabK蛋白質の活性部位及び3次元結晶構造の究明に関する。
【0014】
一実施形態として、本発明はFabK蛋白質溶液を緩衝液、塩および沈澱剤を含む 貯水槽溶液と接触させて濃度平衡法を行う段階を含み、前記FabK蛋白質溶液の濃度は3〜30mg/mlであり、前記緩衝液はpH4〜9.5で、貯水槽溶液内濃度は0.005〜1.5Mであり、前記塩は全ての金属塩およびアンモニウム塩からなる群から選択された1種以上のもので、貯水槽溶液内濃度は0.05〜2Mであり、前記沈澱剤は炭素数1〜4のアルコールおよびポリエチレングリコールからなる群から選択された1種以上のものであり、貯水槽溶液内濃度は1%(v/v)〜40%(v/v)である、FabK蛋白質の結晶化方法に関する。
【0015】
別の実施形態では、本発明は後述の表1のX線回折パターンデータ、または後述の表2に記載の2389個の原子座標で示された3次元結晶構造を有する、FabK−FMN(フラビンモノヌクレオチド)複合体結晶に関する。
【0016】
他の実施形態では、本発明は後述の表1のX線回折パターンデータ、または後述の表2に記載の2389個の原子座標情報から選択された1つ以上のFabK−FMN複合体の3次元結晶構造情報またはこれらの全てを保存する、保存媒体に関する。
【0017】
さらに別の実施形態では、本発明は後述の表2に記載の2389個の原子座標情報から選択された1つ以上のFabK−FMN複合体の3次元結晶構造情報を使用する、FabK蛋白質活性阻害剤開発方法に関する。
【発明の効果】
【0018】
本発明は、抗生剤の標的蛋白質として知られているFabK蛋白質の活性部位及び3次元結晶構造に関するもので、本発明によるサーモトガ・マリティマ菌株に由来したFabK蛋白質結晶は、結晶化程度が優れているだけでなく、X線回析が容易であり、類似した蛋白質であるFabI蛋白質とは異なる構造的特徴を有しているので、このような結晶の3次元構造は、現在まで開発されたFabI阻害剤とは異なる新たな活性調節物質の開発に有用に使用される。
【発明を実施するための最良の形態】
【0019】
以下、本発明を詳細に説明する。
【0020】
まず、本発明は、サーモトガ・マリティマから分離及び精製されたFabK蛋白質を提供する。前記蛋白質は、配列番号1のアミノ酸配列を有する蛋白質であって、本発明では、前記アミノ酸配列がFabK蛋白質の酵素の機能を有するものであるということを最初に究明した。前記のように、FabK蛋白質は、多くのバクテリア及び真菌類でFabIに代わって使用される酵素であって、FabI阻害剤に対して耐性を示す菌株に対する新たな抗生剤の開発において、標的蛋白質として非常に有用である。本発明によるFabK蛋白質は水溶性であって、適切な方法で結晶性を有することができるため、その3次元構造の分析が容易で、新たな抗生剤の開発のための新たな標的蛋白質として有用である。
【0021】
また、本発明は、前記配列番号1のアミノ酸配列を有するサーモトガ・マリティマから分離及び精製されたFabK蛋白質のコード塩基配列;及び前記コード塩基配列と作動可能に連結されたプロモーター及びターミネーター;を含むFabK蛋白質の発現ベクターを提供する。前記発現ベクターは、カナマイシン抵抗遺伝子、アンピシリン抵抗遺伝子、テトラサイクリン抵抗遺伝子、クロラムフェニコール抵抗遺伝子などの通常の選別マーカーを追加的に含むことができる。また、本発明は、前記FabK蛋白質の発現ベクターによって形質転換された形質転換体を提供する。前記形質転換体に使用される宿主としては、原生細菌であり、複製起源がpBR322を有し、T7 ポリメラーゼを生成することができるものであればいずれも使用することができ、例えば大腸菌などを使用することができる。また、本発明は、前記FabK蛋白質の発現ベクターを使用して宿主を形質転換させて、前記発現ベクターを含む形質転換体を製作して、前記製作された形質転換体を培養してFabK蛋白質を生成する段階を含むFabK蛋白質の製造方法を提供する。
【0022】
本発明の発現ベクターの製造のために、アミノ酸の欠失が全くなく、配列番号1に記載されたように1番のメチオニン末端(N−末端)から314番のアミノ酸であるグルタミン酸までのアミノ酸配列を有するサーモトガ・マリティマ由来のFabK蛋白質のコード塩基配列を利用して、生成された蛋白質の精製を容易にするために、N−末端に6個のヒスチジンタグを結合させて使用することができる。
【0023】
本発明による発現ベクター、形質転換体、及びこれを利用したFabK蛋白質の製造方法をより具体的に説明すると下記の通りであるが、下記の内容は、本発明の内容を例示して説明するための具体例にすぎず、本発明の範囲はこれに限定されない。
【0024】
まず、サーモトガ・マリティマのゲノムDNAからFabK蛋白質をコードする遺伝子を増幅するために、FabK蛋白質をコードする遺伝子の5´−末端及び3´−末端に対応するPCR反応用プライマーを考案して合成する。この時、前記5´−末端及び3´−末端に対応するプライマーは、各々に対応するクローニングされるベクターに存在する制限酵素認知部位と同一な制限酵素認知部位が存在するように考案して、前記ベクターのクローニングを容易にする。
【0025】
サーモトガ・マリティマのゲノムDNAを鋳型として、前記のように製造されたプライマーを使用してPCR反応を行って、FabK遺伝子を増幅する。増幅されたFabK遺伝子を適切な制限酵素で切断して得られた遺伝子断片を適切な発現ベクター、例えば大腸菌ベクターに挿入して、再組み合わせFabK遺伝子を含む発現ベクターを製造する。使用可能な制限酵素部位はNcoI NdeI、NheI、BamHI、EcoRI、SalI、HindIII、NotI、及びXhoIである。本発明の具体的な例としては、NdeI及びXhoI制限酵素部位を使用した。前記のように、前記発現ベクターは、宿主ゲノムへの挿入の有無の確認を容易にするために、カナマイシン抵抗遺伝子、アンピシリン抵抗遺伝子、テトラサイクリン抵抗遺伝子、クロラムフェニコール抵抗遺伝子などの通常の選別マーカーを含むことができる。このように製造された組換え発現ベクターを使用して、適切な宿主、例えば大腸菌を形質転換させた後、前記形質転換体を遺伝子の発現に適切な条件で培養する。この時、培養条件は、使用された宿主によって異なり、約15から40℃で、3から15時間培養することができ、前記条件は、比較的高い温度では培養時間を短くし、比較的低い温度では培養時間を長くして調節することができる。
【0026】
目的とする蛋白質の生成有無は、SDS−ポリアクリルアミドゲル電気泳動またはウエスタンブロッティング法などの通常の蛋白質同定方法によって確認することができる。組換えFabK遺伝子を含む発現ベクターに形質転換された形質転換体をアンピシリン、テトラサイクリン、カナマイシン、クロラムフェニコールなどの選別マーカーに対応する抗生剤が含まれている培地(例えばLB培地)で振盪培養し、前記培養した培地の光学密度(O.D.)が一定の細胞濃度(例えば600nmでのO.D.が0.5から0.7)になった時、IPTG(イソプロピル−β−D−ガラクトピラノシド)を添加して、組換えFabK遺伝子の発現を誘導する。前記培養液を遠心分離して形質転換体を沈殿させ、これを適切な緩衝液に懸濁させた後で破砕する。前記破砕物を遠心分離して蛋白質が溶解している上層液を分離した後、イオン交換クロマトグラフィー、アフィニティークロマトグラフィー、ゲルろ過クロマトグラフィーなどの通常の蛋白質分離及び精製手段によって製造された組換えFabK蛋白質を分離及び精製する。この時、前記組換えFabK蛋白質は、精製を容易にするために、ヒスチジンタグ融合蛋白質形態で生成される。
【0027】
また、他の側面から、本発明は、前記FabK蛋白質の3次元構造及び活性部位を究明して、FabK蛋白質を標的とする抑制化合物のスクリーニングにより有用な情報を提供することを特徴とする。
【0028】
また、他の側面から、本発明は、後述する表1または表2の原子座標情報から選択された1以上の原子座標情報で表現される3次元構造を有するFabK−FMN複合体結晶を提供する。FMNはFabK蛋白質の活性に必須の補助因子であり、インビトロ(in vitro)実験でFabKと複合体を形成することが観察され、生体内でもFabK蛋白質とFMN分子が複合体形態で作用することと考えられる。したがって、本発明で提供されるFabK−FMN複合体の結晶構造はFabK抑制剤探索および開発に非常に効果的に適用される。
【0029】
また、本発明は、下記のように究明されたFabK蛋白質の3次元結晶構造及び/またはFabK及びFMNが結合する特異的位置的特性及び前記活性部位で活性に影響を与える個別アミノ酸の役割に基づいて、FabK蛋白質の活性を阻害するFabK蛋白質阻害剤をスクリーニングすることおよび新たな抑制化合物を設計することを特徴とする、FabK蛋白質の阻害活性を有するFabK阻害剤開発方法を提供する。
【0030】
本発明の具体例において、前記FabK阻害剤開発方法は、配列番号1のアミノ酸配列を有するFabK蛋白質、またはFabK−FMN複合体、及び候補化合物を反応させる段階、及び前記候補化合物から前記FabK蛋白質またはFabK−FMN複合体と相互作用する化合物を選択して、FabK蛋白質の活性を阻害する化合物をスクリーニングする段階を含む。
【0031】
また、他の具体例において、本発明のFabK阻害剤開発方法は、使用されたFabK−FMN複合体の後述の表1に記載のX線回折パターン、または後述の表2に記載の2389個の原子座標から選択された1つ以上の3次元結晶構造情報を利用して、FabK蛋白質またはFabK−FMN複合体と結合可能な新たな化合物を新規設計することを特徴とする。
【0032】
また、他の具体例において、本発明のFabK阻害剤開発方法は、使用されたFabK−FMN複合体の後述の表1に記載のX線回折パターン、または後述の表2に記載の2389個の原子座標から選択された1つ以上の3次元結晶構造情報及び候補化合物の3次元結晶構造情報に基づく仮想スクリーニング法を使用して、FabK蛋白質またはFabK−FMN複合体と結合可能な化合物をスクリーニングすることを特徴とする。
【0033】
前記FabK蛋白質と相互作用(結合)する化合物の選択時に、下記のような3次元結晶構造及び/または活性部位に対する情報を使用して、より容易で正確にFabK蛋白質の活性を阻害する化合物を選択することができる。
【0034】
本発明のスクリーニング方法によって選択されたFabK蛋白質阻害剤は、抗生剤として作用し、前記抗生剤が抗生活性を有する菌株は、FabK蛋白質及びこれと類似した活性部位を有する蛋白質を必須で必要とする全ての微生物であり、例えば肺炎球菌、フェカリス菌、サーモトガ・マリティマ、化膿性連鎖球菌(Streptococcus pyogenes)、ネオリケッチャ・センネツ(Neorickettsia sennetsu)、エーリキア・シャフェンシス(Ehrlichia chaffeensis)、アナプラズマ・ファゴサイトフィルム(Anaplasma phagocytophilum)、好熱性一酸化炭素酸化細菌(Carboxydothermus hydrogenoformans)、ストレプトコッカス・アガラクチア(Streptococcus agalactiae)、及びクロストリジウム・ディフィシル(Clostridium difficile)からなる群から選択されたものである。
これをより詳細に説明すると、下記の通りである。
【0035】
まず、本発明は、FabK蛋白質の結晶化方法を提供する。新たな抗生剤の開発のための標的蛋白質の3次元結晶構造の究明においては、前記標的蛋白質が水溶性であって結晶性であることが重要である。本発明によるFabK蛋白質は水溶性であるので、その3次元結晶構造の究明のためには、結晶性を有するようにするための結晶化段階が必要である。このような結晶化段階には、一般に、X線結晶化法が使用され、このようなX線結晶化法を使用するための前処理段階として多様な結晶化方法が行われなければならない。本発明の具体例において、このような結晶化方法として通常の濃度平衡を利用した方法を使用でき、例えば、蒸気平衡法(例えば、シッティングまたはハンギングドロップ蒸気平衡法(Sitting−or Hanging−drop vapour diffusion method:Jancarik J.etal.,Appl.Cryst.,24,409−411(1991)参照)または透析法(連続式または回分式:Bunick C. and Stubbs G., Acta Cryst. D56, 1430−1431(2000))などが使用される。
【0036】
シッティングまたはハンギングドロップ蒸気平衡法の結晶化の原理及び過程は、下記の通りである。密閉された空間に母液の小さい滴及びこれよりはるかに大きい規模の貯水槽溶液が分離されたまま共存する時、これらの間で水または他の揮発性物質の移動が起こるようになる。一方、蛋白質の溶液条件のうち、熱力学的に準安定状態である過飽和状態で沈澱剤の種類によって蛋白質の沈澱が起こり、沈澱速度が遅くなって安定した結晶化状態となる。これに使用可能な沈澱剤はよく知られており、それらは濃縮状態の蛋白質溶液の溶解度を減少させる役割を担う。この時、蛋白質分子の周囲の相対的な吸着層を減少させるために、蛋白質分子が互いに凝集して結晶を生成するようになる。したがって、貯水槽溶液は、このような沈澱剤をはじめとして、緩衝液、塩、界面活性剤などが多様な濃度で混合され、蛋白質溶液及びこのような多様な条件の貯水槽溶液が通常の場合は約1:1の比率で混合されて滴を形成するようになる。このように得られた滴をシリコンでコーティングされたガラススライドに載せてひっくり返した状態で、準備されたプレートに載せて密封する。初期状態では、滴内の蛋白質の濃度が貯水槽溶液の濃度と異なるために蛋白質が結晶状態で存在しないが、密封された状態で置いておくと徐々に平衡が保たれるようになり、この時に前記で説明した原理によって特定の状態の条件で結晶が生成されるのである。このようなシッティングまたはハンギングドロップ蒸気平衡法において、貯水槽溶液内の沈澱剤だけでなく、塩、緩衝液、及び界面活性剤の種類、適正濃度、溶液のpH、及び実験温度は蛋白質の種類によって異なって選択され、場合によっては蛋白質結晶の生成においてとても重要な要素となる。
【0037】
本発明で、水溶性FabK蛋白質を結晶化させるためにFabK蛋白質溶液および貯水槽溶液を濃度平衡法、例えばシッティングまたはハンギングドロップ蒸気平衡法、透析法などを使用して反応させて結晶化を行う。
【0038】
前記貯水槽溶液は緩衝液、塩および沈澱剤を含むものを使用する。前記緩衝液としては、pHを調節して蛋白質を安定化させるためのものであって、pH4〜9.5、好ましくはpH4.6〜9、さらに好ましくは6.2〜8.5である全ての緩衝液が使用可能であり、例えば、PIPES(ピペラジン−1,4−ビス(2−エタンスルホン酸))、ビシン、トリス、酢酸ナトリウム、コハク酸ナトリウム、ビス−トリス緩衝液からなる群から選択された1種以上のものを使用することができる。
【0039】
前記塩は構造分析が容易な結晶形態を有するようにする役割を果たすものであって、全ての金属塩およびアンモニウム塩からなる群から選択されたものを使用することができる。前記アンモニウム塩は硫酸アンモニウム、塩化アンモニウム、リン酸アンモニウムなどからなる群から選択されたものを使用することができる。前記金属塩は全てのアルカリ金属(例えば、リチウム、ナトリウムなど)塩、アルカリ土類金属(例えば、マグネシウムなど)塩、および遷移金属(例えば、マンガン、亜鉛など)塩からなる群から選択された1種以上のものを使用することができ、好ましくは塩化物、シアン化物、チオシアン酸化物、酸化物、硝酸化物、水酸化物、硫酸化物などから選択された1種以上を使用することができ、例えば、塩化リチウム、塩化マグネシウム、チオシアン酸ナトリウムまたは硫酸アンモニウムなどを使用することができる。
【0040】
前記沈澱剤は結晶化のために沈澱を形成する物質であって、炭素数1〜4の直鎖または分枝鎖アルコールおよびポリエチレングリコール(PEG)(重量平均分子量200以上、好ましくは400〜20,000、さらに好ましくは550〜10,000)からなる群から選択された1種以上のものを使用することができる。
【0041】
貯水槽溶液全体での緩衝液、塩および沈澱剤の濃度は、使用される蛋白質溶液の濃度によって適切に調節することができる。具体的に、貯水槽溶液全体でのPIPES、ビシン、トリスなどの緩衝液の濃度は0.005〜1.5M、好ましくは0.01〜0.2Mであることができ、塩化リチウム、塩化マグネシウム、チオシアン酸ナトリウムまたは硫酸アンモニウムなどの塩濃度は0.05〜2M、より好ましくは0.05〜0.5Mであることができ、ポリエチレングリコールなどの沈澱剤の濃度は1〜40%(v/v)、好ましくは5〜30%(v/v)であるのが好ましい。貯水槽溶液内での沈殿剤及び塩の濃度が前記範囲より高い場合には、蛋白質が沈殿する現象が起こることがあり、前記範囲より低い場合には、結晶が生成されないので好ましくない。また、前記緩衝液は主に貯水槽溶液内での蛋白質の安定性に関連するものであり、その濃度が前記範囲を逸脱する場合には、貯水槽溶液内で適切な蛋白質の安定性を得ることができず、所望の結晶を得るのも困難である。前記貯水槽溶液は、このような成分以外にも、通常使用可能な塩、緩衝液、及び界面活性剤などを追加的に含むことができる。
【0042】
本発明に使用されるFabK蛋白質溶液はFabK蛋白質を適切な緩衝液に溶解したものであって、FabK蛋白質可溶性の全ての緩衝液を使用することができ、例えば、PIPES、ビシンおよびトリス、酢酸ナトリウム、コハク酸ナトリウム、ビス−トリス、HEPES(N−(2−ヒドロキシエチル)ピペラジン−N’−(2−エタンスルホン酸))、イミダゾール、リン酸ナトリウム、リン酸カリウム、MES(2−モルホリノ エタンスルホン酸)、ADA(N−(2−アセトアミド)イミノ酢酸)、カコジル酸ナトリウム、クエン酸三ナトリウムなどからなる群から選択された1種以上のものを使用することができる。蛋白質結晶を容易に得て構造分析が容易な結晶を得るために、FabK蛋白質溶液濃度は3〜30mg/ml、好ましくは5〜20mg/ml、さらに好ましくは 8〜15mg/mlであるのが良い。
【0043】
本発明に使用されるFabK蛋白質溶液と貯水槽溶液内の沈澱剤濃度は大体反比例するように使用するのが良い。つまり、FabK蛋白質溶液の濃度が高い場合には比較的に結晶化が容易であるので、貯水槽溶液内の沈澱剤の濃度は相対的に低くして使用し、FabK蛋白質溶液の濃度が高い場合には結晶化のために相対的に沈澱剤(例えば、ポリエチレングリコール)濃度が高い貯水槽溶液を使用するのが良い。例えば、蛋白質溶液の濃度が約10mg/mlである時には沈澱剤濃度を約15〜20%(v/v)として使用することができる。
【0044】
また、前記濃度平衡法は、pH8.0〜9.0で行われるのが好ましい。前記範囲は実験的に決定されたものであり、前記範囲のpH条件下で反応する時に、最も優れた結晶を得ることができる。シッティングまたはハンギングドロップ蒸気平衡法による結晶生成反応の反応温度は、4〜26℃であるのが好ましく、約20〜24℃であるのがより好ましく、反応時間は、1日〜20日であるのが好ましく、1週間〜2週間であるのがより好ましい。前記反応温度及び反応時間範囲は、X線分析が容易な最適のデータを有する結晶を得ることができる条件として、実験的に決定された範囲である。
【0045】
製造された結晶性FabK蛋白質の構造を分析するためには、X線分析を利用することができる。しかし、蛋白質結晶がX線の高いエネルギーに露出されると、その寿命が短くなり、データ強度も弱くなって、構造分析に良くない結果が発生する。このような欠点を防止するために、X線分析前にFabK蛋白質結晶を高速で窒素冷却する高速窒素冷却法を行うのが好ましい。高速窒素冷却時に、効果的なFabK蛋白質結晶の保護のために、結晶性条件に加えてエチレングリコール及びパラトン−Nを使用するのが好ましい。エチレングリコールを使用する場合、20〜35%(v/v)、好ましくは25〜30%(v/v)の濃度で使用するのが好ましい。パラトン−Nは、結晶に上載せして使用するのが好ましい。前記高速窒素冷却法を行う温度は、50〜200K、好ましくは80〜120Kである。前記濃度及び温度範囲は、FabK蛋白質がその結晶性に影響なく、窒素ストリームに耐えられて、高速窒素内で保管が容易な範囲として、実験的に決定されるものである。
【0046】
また、本発明は、X線結晶化方法によって得られた前記のような方法で結晶化されたFabK蛋白質の3次元結晶構造を提供する。本発明の具体例において、前記結晶の3次元構造を知るために、X線映像プレートを使用して回折パターンを得て、多波長異常分散法(MAD:multiwavelength anomalous dispersion)で位相情報を得ることができる。前記のように得られたFabK蛋白質結晶のX線回折パターン及び位相情報から電子密度地図を作成し、これから再び原子座標を導き出すことによって、3次元構造情報を得ることができる。
【0047】
X線を使用した回折パターンのデータ収集方法は、X線の供給源によって一般実験室で得る方法及びシンクロトロンを利用する方法に分けられる。このうちのシンクロトロンを利用する方法は、結晶サイズが50μm程度の小さい結晶まで可能であり、1つの波長だけでなく多様な波長でデータを収集することができるため、迅速な構造を得ることができる。したがって、本発明の好ましい具体例では、シンクロトロンを利用して回折パターンのデータを収集することができ、このように得られた本発明によるFabK蛋白質結晶の回折パターンは、下記の表1に示した通りである。
【0048】
【表1】
【0049】
位相情報は、多重重原子同型置換法(Multiple isomorphous replacement)、多波長異常分散法、構造置換法(Molecular replacement)などによって得ることができ、本発明の好ましい具体例では、多波長異常分散法を利用して、FabK蛋白質結晶の位相情報を得ることができる(Modern X−ray Analysis on Single Crystals,Peter Luger参照)。具体的には、前記多波長異常分散法は、重金属を有する蛋白質結晶で互いに異なる三個の波長を通じて異常回折パターン(anomalous diffraction pattern)を利用して位相を計算するのである。このような多波長異常分散法を利用するために、位相を計算してこれによる初期モデルの計算を行わなければならず、このような計算に使用することができる全てのプログラムを使用することができる。本発明の好ましい具体例では、SOLVE及びRESOLVE(ロスアラモス研究所、米国)を使用することができ、その後の精密化段階及び標準化段階のために、CNS(エール大学)、CCP4(ケンブリッジ大学)などのプログラムを使用することができる。
【0050】
精密化段階は、X線回折パターンから得られた電子密度をO(Alwyn Jones,http://web.mac.com/alwyn_47/OInTheWorks/OInTheWorks.htmlを参照)及びCOOT(Paul Emsley, http://www.ysbl.york.ac.uk/~emsley/coot/を参照)プログラムでコンピュータモニターに表示して、そこに最も適した構造に修正することによって行われる。精密化段階を行った結果、本発明によるFabK蛋白質結晶は、FMN及び24個の水分子を含み、下記の表2に記載の3次元結晶構造を有する原子モデルで表現されることが明らかになった。FabK蛋白質を標的とするFabK阻害剤開発において、FabK蛋白質を単独で使用したり、このようなFabK−FMN複合体の形態で使用し、特に、このような複合体の形態で3次元構造の結晶性が優れていて、FabK阻害剤開発に非常に有用である。
【0051】
FabK−FMN複合体の3次元上の原子座標
【表2】
【0052】
前記表2のうち、A:原子、B:原子数、C:原子名、D:残基名、E:残基数、F:x軸情報、G:y軸情報、H:z軸情報、I:占有因子、J:温度因子である。また、WATは水分子を表す。
【0053】
精密化段階後、前記得られた原子モデルから多様な情報を得るために分析段階を追加的に行うことができる。例えば、グラフィック上に3次元空間のFabK蛋白質の原子モデルを表示しておいて活性部位の形態を観察することによって各原子間の距離及び空間を測定したり、重要残基の空間的位置と異なる残基との相互作用を観察して、適切な阻害剤を見つけるモデリング段階を経ることができる。前記分析段階を行った結果得られたFabK−FMN複合体の構造を図9、図11aおよび11bに示した。これら図面から確認できるように、FabK蛋白質でFMNが結合している部位は、TIMバレル(TIM barrel;G.K.Farber and G.A.Petsko:The evolution of a/b barrel enzymes,TIBS1990;228−234参照)の上側に位置するループで構成されたところであり、これよりこの部位がFabK蛋白質の活性部位であるといえる。より具体的に、FMN分子(5)は前記表2の原子番号2334から2364までの原子からなり、そのうちの原子番号2334から2352までがヘッド(HEAD)部分を構成し、原子番号2353から2364までがテール(TAIL)部分を構成する。このような構造を有するFMN分子はFabK蛋白質のTIMバレル(1)の上側のループ部位(2)(ループ1:表2の原子番号1015から1103まで、ループ2:原子番号141から206まで)に結合しており、その結合部位上側にFabK活性に必要な補酵素であるNADPおよび基質が結合する。FabK蛋白質のTIMバレルと残りの部分(以下、蓋部位(4))を連結するヒンジ部位(3)(原子番号1428から1485;すなわち198から205までのアミノ酸配列)が存在し、流動性を付与する。つまり、前記ヒンジ部位が動いてNADPおよび基質が結合する空間を形成してFabKが作用するようにする。したがって、前記ヒンジ部位はFabK活性部位のうちの一つであり得る。また、NADPおよび基質が結合する空間を提供するTIMバレルの上のFMN分子が位置している部位の上側部分、つまり、FabK蛋白質に結合されたFMNのヘッド部分水平面を半球の平らな底面とし、ヒンジ(部位および蓋部分を含む半球部分もFabK活性部位である。より具体的に、前記TIMバレル上側活性部位はTIMバレルの上に結合されたFMN分子のヘッド水平面を半球底面とする半径15Å以内、好ましくは半径6〜12Åの半球内に位置する部位であり得る。前記半球部分はループ1部分、ヒンジ部分を含む原子番号1380から2137までを含む。
【0054】
また、突然変異実験を通じてFabK蛋白質の活性に影響を与えるアミノ酸配列を確認し、このために、配列番号1のアミノ酸配列のうちの1つを標的となるアミノ酸として選定した後、前記アミノ酸のコード塩基配列を他のアミノ酸、例えばアラニンをコードするように変異させて、プライマーを製造する。この時、5´末端プライマー及び3´末端プライマーを全て使用し、これらプライマーの中間に変異させるアミノ酸をコードする塩基配列を有するように構成して、前記PCR方法で所望の組換えクローンを得る。このように得られた組換えクローンは、DpnIなどの制限酵素の処理を通じて大腸菌から得られた元のベクターを完全に分解するようにする。この時、制限酵素(DpnI)の反応時間は30分〜2時間であるのが好ましい。その後、組換えクローンだけを大腸菌に導入して、所望の形質を有する突然変異FabK蛋白質を形成する。
【0055】
このような方法で行われた突然変異実験を通じて、本発明によるFabK蛋白質(配列番号1)の208番のチロシン(Y208)および209番のリシン(K209)をアラニンに置換させた場合、その活性は各々増加し、一方で前記FabK蛋白質(配列番号1)の211番のリシン(K211)、214番のリシン(K214)、229番のヒスチジン(H229)および261番のロイシン(L261)をアラニンに置換させた場合、その活性は各々減少し、特に前記FabK蛋白質(配列番号1)の229番のヒスチジン(H229)及び276番のメチオニン(M276)をアラニンに置換させた場合、活性が大幅に減少することが明らかになり、前記アミノ酸がFabK蛋白質の活性に影響を与える重要なアミノ酸であると考えられる。また、本発明によるFabK蛋白質(配列番号1)の208番から215番までの8個のアミノ酸から構成されたへリックス及び278番から283番までの6個のアミノ酸から構成されたへリックスの欠失がFabK蛋白質の活性の大幅な減少を招くことが観察され、前記2個のへリックス部位も、本発明によるFabK蛋白質の活性に影響を与える重要な部位であることが明らかになった。
【0056】
したがって、本発明によるFabK蛋白質(配列番号1)のTIMバレルの上側に位置するループ部位;FabK蛋白質のTIMバレルの上に結合されたFMN分子のヘッド)水平面を平らな半球底面とする半径15Å以内、好ましくは半径6から12Åの半球内に位置する1つ以上のアミノ酸;198から205までのアミノ酸配列を含むヒンジ部位;208番のチロシン(Y208);209番のリシン(K209);211番のリシン(K211);214番のリシン(K214);229番のヒスチジン(H229);261番のロイシン(L261);276番のメチオニン(M276);208番から215番までの8個のアミノ酸から構成されたへリックス、;及び278番から283番までの6個のアミノ酸から構成されたへリックス;からなる群から選択された1つまたはそれ以上の部位及び候補物質の相互作用の有無を検出して、より効果的なFabK蛋白質阻害剤を選択することができる。
【0057】
したがって、本発明によるFabK蛋白質の阻害活性を有するFabK阻害剤開発において、前記表2に記載の2389個の原子座標のうちの前記活性部位に相当する原子座標を選択的に使用してFabK蛋白質阻害剤をスクリーニングしたり、FabK蛋白質の阻害活性を有する化合物を設計したり仮想スクリーニングして、より容易で効果的にFabK阻害剤開発を行うことができる。
【0058】
また、本発明は、前記で究明されたFabK蛋白質の3次元結晶構造に対する情報がコンピュータ読取可能に保存された保存媒体、例えばフロッピー(登録商標)ディスク、ハードディスクなどを提供する。前記媒体に保存された3次元結晶構造情報は、前記表2に記載の原子座標の全部または一部(特に、前記活性部位に相当する原子座標)を含み、前記活性部位のアミノ酸及び前記突然変異アミノ酸の残基を含む部位に対する情報を含む。
【0059】
前記で究明されたFabK蛋白質の3次元結晶構造、及びFabK及びFMNが結合した特異な位置的特性及び前記活性部位で活性に影響を与える個別アミノ酸の役割に基づいて、本発明はFabK蛋白質の活性を阻害するFabK蛋白質阻害剤をスクリーニングする方法を提供する。本発明によるFabK蛋白質阻害剤のスクリーニング方法は、配列番号1のアミノ酸配列を有するFabK蛋白質及び候補化合物を反応させる段階、及び前記候補化合物から前記FabK蛋白質と相互作用する化合物を選択する段階を含む。
【0060】
前記FabK蛋白質は、前記のような結晶化方法によって結晶化されたものである。前記FabK蛋白質と相互作用する化合物の選択時に、前記記載されたような3次元結晶構造及び/または活性部位に対する情報を使用して、より容易で正確にFabK蛋白質の活性を阻害する化合物を選択することができる。また、本発明のスクリーニング方法は、このような保存媒体を使用するものである。
【0061】
以下、本発明を下記の実施例によって詳細に説明する。但し、下記の実施例は本発明を例示するためのもので、本発明が下記の実施例によって限定されるのではない。
【実施例】
【0062】
実施例1:サーモトガ・マリティマ FabK蛋白質の発現及び精製
サーモトガ・マリティマ FabK(配列番号1)の1番アミノ酸のメチオニン(N−末端)から312番アミノ酸のグルタミン酸(C−末端)までのFabKをコードする遺伝子(Genbank Accession No.AE000512)を、PCR反応を通じて増幅した。PCR反応に使用されるプライマーとして、配列番号2及び3のオリゴヌクレオチドを使用した。配列番号2及び3のプライマーは、各々NdeI及びXhoI制限酵素認識部位を有する。
【0063】
鋳型ゲノムDNAを1μl、2.5mMdNTP溶液を5μl、各100pmolの配列番号2及び配列番号3のプライマーを各々0.3μl、PfuTaq DNA重合酵素(5U/μl、Stratagene社、米国)を1μl、及び10倍濃度PCR反応溶液(Stratagene社、米国)を5μl混合し、さらに蒸溜水を最終的に50μlになるように加えて反応溶液を作成した後、この反応溶液を95℃で5分、95℃で30秒、55℃で30秒、及び72℃で1分の反応を30回繰り返した。その後、PCR反応溶液を0.8%アガロースゲルで電気泳動して、約970塩基長のFabK遺伝子を分離した。分離されたFabK遺伝子をNdeI及びXhoIで処理した後、電気泳動して、FabK遺伝子の切片を抽出した後、50μlの蒸溜水溶液に溶存させ、これをFabKN/Xと命名した。
【0064】
プラスミドpET−28a(Novagene社、米国)は、N−末端に6個のヒスチジン残基を発現するが、これをNdeI及びXhoI制限酵素で処理した後、電気泳動して、約5400bpの大きさのDNA切片を分離して、これをpET−28aN/Xと命名した。0.5μgのFabKN/Xを0.1μgのpET−28aN/Xと共に反応チューブに入れた後、2μlの10倍連結反応溶液(50mMのトリス塩酸(pH7.8)、100mMの塩化マグネシウム、100mMのDTT(ジチオトレイオール)、及び10mMのATP(アデノシン三リン酸))、10UのT4 DNAリガーゼを入れて総体積が20μlになるように蒸溜水を加えた後、16℃で12時間反応させた。この反応溶液を大腸菌BL21(DE3)コンピテント細胞(Novagene 社,米国)に添加して形質転換させた後、50μg/mlのカナマイシンを含むLB寒天培地(1% バクトトリプトン、0.5%酵母抽出物、1%塩化ナトリウム)にプレーティングして、大腸菌形質転換体を選別した。この形質転換体からプラスミドを抽出し、制限酵素及び塩基配列分析によって組換えプラスミドpET−28a−FabKを得た。
【0065】
前記で得られた再組み合わせプラスミドにクローニングされたFabK遺伝子の塩基配列は、Big−Dye Cycle Sequeincing System(Applied Biosystem社、米国)を利用して、ABI 377 DNA塩基配列分析器によって確認した。
【0066】
前記形質転換された大腸菌菌株を50μg/mlのカナマイシンが含まれているLB培地で12時間振盪培養した後、1mlの培養液を50μg/mlのカナマイシンが含まれている100mlのLB培地に移して、37℃で培養液のO.D.が600nmで約0.6程度になった時点で、IPTGを最終濃度が0.5mMになるように添加した。IPTG添加4時間後に、10,000gで30分間遠心分離して、各々細胞沈殿物を回収した。細胞沈殿物を20mMのトリス塩酸(pH8.0)、0.1Mの塩化ナトリウム、1mM のTCEP(トリス(2−カルボキシエチル)ホスフィン塩酸)溶液に懸濁させて、氷上で超音波で細胞を破砕した。細胞破砕液を遠心分離して、上層液をニッケル親和性カラムで、(ファーマシア社、スウェーデン)に通過させ、その後、BLUEカラムファーマシア社、スウェーデン)に通過させて、通過溶液をスーパーデックス−75(Superdex−75)ゲルろ過クロマトグラフィーカラム(ファーマシア社、スウェーデン)を使用して精製して、FabK蛋白質を得た。
【0067】
実施例2:シッティングまたはハンギングドロップ蒸気平衡法によるFabK蛋白質の結晶化
前記実施例1で得られたFabK蛋白質を下記のようなシッティングまたはハンギングドロップ蒸気平衡法によって結晶化した。
【0068】
20mMのトリス塩酸(pH8.0)、300mMの塩化ナトリウム、1mMのジチオトレイオール、50mMの塩化アンモニウムを含む溶液内のFabKの濃度が11mg/mlである蛋白質溶液を製造した。最終蛋白質濃度は、ブラッドフォード法(Current Protocols in Protein Science,3.4.10)によって決定した。11mg/mlに濃縮された蛋白質溶液に補助因子であるNADH及びFMNを添加して、最終蛋白質溶液を製造し、この時、FabK蛋白質に対する、前記補助因子のモル当量比は1:5になるようにした。
【0069】
前記FabK蛋白質溶液の結晶条件を検索するために、初期スクリーン溶液(Hampton research Inc.)を使用し、自動化されたスクリーニングのために、Hydra II−plus one system(Matrix technologies社、米国)を利用した。0.2μlの蛋白質溶液及び0.2μlの貯水槽溶液を混合した後、22℃で2日から20日が経過した後で結晶の生成有無を確認した。その結果、0.1Mのビシン緩衝液(pH9.0)、及び1M塩化リチウム及び20%(v/v)ポリエチレングリコール6,000沈澱剤を含む貯水槽溶液、0.1Mのトリス塩酸緩衝液(pH8.0)、及び0.2Mの塩化マグネシウム、及び20%(v/v)ポリエチレングリコール20,000沈澱剤を含む貯水槽溶液を使用した場合に、X線回折能力が最も優れた結晶が得られた。
【0070】
1μlの最終蛋白質溶液及び1μlの前記貯水槽溶液を混合した滴をシリコンでコーティングされたガラススライドの表面に滴下し、このスライドを貯水槽溶液0.5mlが入っているプレート上に覆って、22℃の恒温状態に置いた。一日で種結晶が生じ、一週間後にその結晶の大きさが0.1×0.1×0.2mmに成長した。
【0071】
実施例3:高速窒素冷却法を追加的に行ったFabK蛋白質の結晶化
前記実施例2で得られたFabK蛋白質結晶を直接高エネルギーのX線に露出させる場合に発生する問題点を解決するために、得られたFabK蛋白質結晶のX線分析前に、下記のような高速窒素冷却法による冷却を行った。
【0072】
グリセロール、ギ酸ナトリウム、エチレングリコール、スクロース、パラトン−Nなどの多様な冷却溶液を多様な濃度で検索した後、高速窒素冷却法の最適条件を得た。その結果、25%(v/v)エチレングリコール、0.1Mのビシン緩衝液(pH9.0)、1Mの塩化リチウム、20%(v/v)ポリエチレングリコール6,000を含む高速冷却用溶液に結晶を数秒以内で浸漬して取り出すと、FabK蛋白質結晶に全く害を及ぼさず、100Kの液体窒素ストリームに最もよく耐えることが明らかになった。
【0073】
冷却溶液に浸漬した結晶を約0.3mmのナイロン結晶回収ツール(Hampton research社、米国)を利用して取り出して直ちに100Kの窒素ストリームに入れた。
【0074】
本実施例では、実験中によく発生する水枠現象(高速窒素冷却が完全でない場合に水が凍って現れる現象)も見られなかった。
【0075】
実施例4:放射光加速器を通じたFabK蛋白質結晶のX線回折パターンのデータ収集及び分析
前記実施例3で得られたFabK蛋白質結晶を利用して、フォトンファクトリー(茨城県つくば市大穂1-1)の放射光加速器のAR−NW12ラインで回折パターンのデータを収集する実験を行った。結晶データの限界は2.3Åであり、データはDENZO及びSCALEPACK(Otwinowski,Z.and Minor,W.Methods Enzymol.,276,461−472(1997))で処理した。結晶データの収集及び精密化の結果を表3に示した。
【0076】
【表3】
【0077】
実施例5:FabK蛋白質結晶のX線回折パターンのデータ解釈及び構造計算
FabK蛋白質の構造は、多波長異常分散法を通じて究明した。多重波長分析法は、初期位相情報を求めるために、SOLVE(Terwilliger T.C.and BerendzenJ.,Acta Crystallogr.D.Biol.Crystallogr..55,849−861(1999))プログラムを利用し、電子密度の修正のために、RESOLVE(Terwilliger T.C.,ActaCrystallogr.D.Biol.Crystallogr..56,965−972(2000))プログラムを利用した。使用した回折パターンのデータは、2.3Åの解像度を有して行った。
【0078】
精密化段階では、CNSプログラム(Brunger,AT.et al.,ActaCryst.D.,54,905−921(1998))を使用した。精密化段階は、CNSプログラムのシミュレーションアニーリング方法を利用して行った。シミュレーション開始温度は1500℃であり、各段階で100℃ずつ冷却して、25℃まで冷却させて行い、最終R因子及びRfree因子は各々24.3%及び24.2%であった。その後、Oプログラムを使用して最適構造を導き出した。最適構造を導き出すために、精密化段階を行ったが、これはX線回折パターンのデータから導き出された電子密度をOプログラムでコンピュータモニターに表示して、そこに最も適した構造に修正する作業を繰り返すことによって行った。
【0079】
精密化段階を行った結果、本発明によるFabK蛋白質の3次元結晶構造は、以前までのFabI蛋白質の構造とは明確に異なるTIMバレル形態の構造及びTIMバレルの上側のループが構成する活性部位を示し、ここにFMNが結合していることが明らかになった。
【0080】
実施例6:FabK蛋白質の活性部位を中心とする個別アミノ酸残基の突然変異体の作成
前記実施例5でFabK蛋白質の3次元結晶構造を得た後、FabK蛋白質の活性部位の個別アミノ酸の役割を調べるために、突然変異体を作成して活性に対する影響を観察した。
【0081】
個別残基の突然変異体は、チロシン208番残基をアラニン(Y208A:配列番号:4及び5)に、リシン209番残基をアラニン(K209A:配列番号:6及び7)に、リシン211番残基をアラニン(K211A:配列番号:8及び9)に、リシン214番残基をアラニン(K214A:配列番号:10及び11)に、ヒスチジン229番残基をアラニン(H229A:配列番号:12及び13)に、ロイシン261番残基をアラニン(L261A:配列番号:14及び15)に、メチオニン276番残基をアラニン(M276A:配列番号16:及び17)にした。
【0082】
FabK蛋白質の活性部位に影響を与えるへリックスの欠失に対する効果を観察するために、208番から215番残基まで(8個のアミノ酸:配列番号:18及び19、以下ではD208とする)及び278番から283番まで(6個のアミノ酸:配列番号:20及び21、以下ではD278とする)のアミノ酸が欠損した突然変異蛋白質を前記PCR方法を利用して形成した。
【0083】
個別アミノ酸突然変異体及びD208及びD278突然変異蛋白質を形成するために、配列番号4から配列番号21までのヌクレオチドオリゴマーを合成した後、前記PCR方法で増幅した。pET−28a−FabKクローン化されたプラスミドを10ng、2.5mMのdNTPを5μl、前記配列プライマーを0.2μl、10倍濃度のPCR反応溶液(100mM 塩化カリウム、100mM 硫酸アンモニウム、200mM トリス塩酸(pH8.8)、20mM 硫酸マグネシウム)を5μlを入れ、反応液が全体で50μlになるように蒸溜水を入れた。95℃で1分、95℃で50秒、60℃で50秒、68℃で6分の反応を18回繰り返した。その後、DpnI(Stratagene社、米国)を0.5μl(10U)を入れて37℃の恒温器で1時間反応させた。DpnI制限酵素によって大腸菌から由来したpET−28a−FabKプラスミドは分解され、前記PCR反応を通じて生成された突然変異pET−28a−FabKプラスミドだけが残るようになる。このような反応生成物を大腸菌DH5αコンピテント細胞に添加して形質転換させた後、実施例1で既述の50μg/mlのカナマイシンを含むLB寒天培地にプレーティングして、大腸菌の形質転換体を選別した。これらからプラスミドを抽出して、塩基配列分析を通じて突然変異pET−28a−FabKプラスミドを得た。
【0084】
得られた突然変異pET−28a−FabKプラスミドは、発現宿主である大腸菌BL21(DE3)に形質転換させた後、実施例1の精製方法を通じて精製した。
【0085】
生成された突然変異体は、図7で、SDS−アクリルアミドゲル電気泳動で分離程度を示す。
【0086】
実施例7:FabK蛋白質の活性部位を中心とする個別アミノ酸残基の突然変異研究を通じた重要残基及び活性の影響
実施例6で精製された蛋白質は、エノイル−ACP還元酵素活性測定方式を通じて各突然変異体の活性を測定した。
【0087】
具体的には、100mMリン酸緩衝液(pH 7.5)中に、NADHまたはNADPHを終濃度175μM、FabK蛋白質を終量5μg、酵素基質(トランス−2−オクテノイル−N−アセチルシステラミン(trans−2−octenoly−N−acetylcysteramine))のDMSO溶液を10μl加え、蒸留水を加えて最終体積を200μlにした。室温で0−60分反応後、溶液の340nmの吸光度を測定して、酵素活性を測定した。酵素活性は、野生型FabKの活性を100とした時の各変異体の比活性(%)で測定した。
【0088】
活性の加減を図10に示す。Y208A及びK209Aの場合、活性の増加を示しており、これら残基はFabK蛋白質の活性を阻害する残基として現れ、その他のFabK変異体K211A、K214A、H229A、L261A、M276A、D208、D278の場合、活性の減少を示している。特に、H229A及びM276Aの場合、大幅な活性の減少を示し、D208及びD278のへリックスを除去した突然変異体は、活性の減少がより著しいことが分かる。これは、ヒスチジン229番及びメチオニン276番、そして構造を支持するへリックスである208番から215番、278番から283番がFabKの蛋白質の活性を維持するのに重要な作用をすることを示している。
【図面の簡単な説明】
【0089】
【図1】FabK発現ベクター及びクローニング情報を示した図面である。
【図2】FabK蛋白質の発現程度のテスト結果を示した図面である。図中、Mは分子量マーカー、Cは粗抽出物、Tは全細胞溶解液、Sは可溶性画分、Pは不溶性画分(封入体)をそれぞれ表す。
【図3】蛋白質発現細胞を破砕した上清をニッケル親和性カラムで精製したサンプルのゲル電気泳動図である。
【図4】図3の精製サンプルを、BLUEカラムに通過させたサンプルのゲル電気泳動図である。
【図5】図4の精製サンプルを、スーパーデックス−75ゲルろ過クロマトグラフィーカラムで精製したサンプルのゲル電気泳動図である。
【図6】NADH、NADPH依存によるFabK蛋白質の活性度を示した図面である。
【図7】FabK蛋白質の活性部位のアミノ酸突然変異体の精製結果を示した図面である。
【図8】FabK蛋白質結晶構造を示した図面である。
【図9】FabK蛋白質の骨格構造をCアルファ(Calpha)リボンで示した図面である。
【図10】FabK変異体の活性度を示した図面である。
【図11a】FMNの構造を模式的に示したものである。
【図11b】FabK−FMN複合体でのFMN位置を示したものである。
【符号の説明】
【0090】
(1) TIMバレル部位
(2) ループ部位
(3) ヒンジ部位
(4) 蓋部位
(5) FMN分子
【特許請求の範囲】
【請求項1】
FabK蛋白質溶液を緩衝液、塩および沈澱剤を含む貯水槽溶液と接触させて濃度平衡法を行う段階を含み、
前記FabK蛋白質溶液の濃度は3〜30mg/mlであり、
前記緩衝液はpH4〜9.5で、貯水槽溶液内濃度は0.005〜1.5Mであり、
前記塩は金属塩およびアンモニウム塩からなる群から選択された1種以上のもので、貯水槽溶液内濃度は0.05〜2Mであり、
前記沈澱剤は炭素数1〜4のアルコールおよびポリエチレングリコールからなる群から選択された1種以上のものであり、貯水槽溶液内濃度は1%(v/v)〜40%(v/v)である、
FabK蛋白質の結晶化方法。
【請求項2】
前記濃度平衡法は、シッティングまたはハンギングドロップ蒸気平衡法、または透析法である、請求項1に記載のFabK蛋白質の結晶化方法。
【請求項3】
前記濃度平衡法は、8.0〜9.0の反応pH、4〜26℃の反応温度、及び1日〜20日の反応期間の反応条件下で行われる、請求項1に記載のFabK蛋白質の結晶化方法。
【請求項4】
前記濃度平衡法を行った後、高速窒素冷却法を追加的に行う、請求項1に記載のFabK蛋白質の結晶化方法。
【請求項5】
前記高速窒素冷却法は、50〜200Kの温度範囲で行われる、請求項4に記載のFabK蛋白質の結晶化方法。
【請求項6】
下記の表1のX線回折パターンデータ、または下記の表2に記載の2389個の原子座標で示された3次元結晶構造を有する、FabK−FMN(フラビンモノヌクレオチド)複合体結晶。
【表1】
【表2】
【請求項7】
下記の表1のX線回折パターンデータ、または表2に記載の2389個の原子座標情報から選択された1つ以上のFabK−FMN複合体の3次元結晶構造情報またはこれらの全てを保存する、保存媒体。
【表1】
【表2】
【請求項8】
前記表2に記載の2389個の原子座標情報から選択された1つ以上の3次元結晶構造情報は、FabK蛋白質(配列番号1)のTIMバレル上側に位置するループ部位;FabK蛋白質のTIMバレルの上に結合されたFMN分子のヘッド水平面を平らな半球底面とする半径15Å以内に位置する1つまたはそれ以上のアミノ酸;198から205までのアミノ酸配列を含むヒンジ部位;208番のチロシン(Y208)、209番のリシン(K209)、211番のリシン(K211)、214番のリシン(K214)、229番のヒスチジン(H229)、261番のロイシン(L261)、276番のメチオニン(M276)、208番から215番までの8個のアミノ酸から構成されたへリックス;および278番から283番までの6個のアミノ酸から構成されたへリックス;からなる群から選択される、1以上の該当する原子座標情報を含む、請求項7に記載の保存媒体。
【請求項9】
下記の表2に記載の2389個の原子座標情報から選択された1つ以上のFabK−FMN複合体の3次元結晶構造情報を使用する、FabK蛋白質活性阻害剤開発方法。
【表2】
【請求項10】
前記表2に記載の2389個の原子座標で示される3次元結晶構造を有するFabK−FMN複合体と候補化合物を反応させる段階;及び前記候補化合物から前記複合体と相互作用する化合物を選択して、FabK蛋白質の活性を阻害する化合物を決定する段階;を含む、請求項9に記載の方法。
【請求項11】
前記候補化合物及びFabK蛋白質のTIMバレルの上側に位置するループ部位;FabK蛋白質のTIMバレルの上に結合されたFMN分子のヘッド水平面を平らな半球底面とする半径15Å以内に位置する1つまたはそれ以上のアミノ酸;198から205までのアミノ酸配列を含むヒンジ部位;208番のチロシン(Y208);209番のリシン(K209);211番のリシン(K211);214番のリシン(K214);229番のヒスチジン(H229);261番のロイシン(L261);276番のメチオニン(M276);208番から215番までの8個のアミノ酸から構成されたへリックス;及び278番から283番までの6個のアミノ酸から構成されたへリックス;からなる群から選択された、1つ以上の部位の相互作用の有無を測定する、請求項10に記載の方法。
【請求項12】
請求項7に記載の保存媒体を使用して、FabK−FMN複合体と相互作用する化合物を選択する、請求項10に記載の方法。
【請求項13】
前記表2に記載のFabK−FMN複合体の2389個の原子座標情報から選択された1つ以上の3次元結晶構造情報を利用して、FabK−FMN複合体と結合可能な化合物を設計する、請求項9に記載の方法。
【請求項14】
前記2389個の原子座標から選択された1つ以上の3次元結晶構造情報が、FabK蛋白質のTIMバレルの上側に位置するループ部位;FabK蛋白質のTIMバレルの上に結合されたFMN分子のヘッド水平面を平らな半球底面とする半径15Å以内に位置する1つ以上のアミノ酸;198から205までのアミノ酸配列を含むヒンジ部位;208番のチロシン(Y208);209番のリシン(K209);211番のリシン(K211);214番のリシン(K214);229番のヒスチジン(H229);261番のロイシン(L261)、276番のメチオニン(M276);208番から215番までの8個のアミノ酸から構成されたへリックス;及び278番から283番までの6個のアミノ酸から構成されたへリックス;からなる群から選択された、1つまたはそれ以上の部位に相当する、請求項13に記載の方法。
【請求項15】
請求項7に記載の保存媒体を使用して、FabK−FMN複合体と相互作用する化合物を設計する、請求項13に記載の方法。
【請求項16】
前記2389個の座標から選択された1つ以上のFabK−FMN複合体の3次元結晶構造情報、及び候補化合物の3次元結晶構造情報をマッチングさせる仮想スクリーニング法を使用して、FabK−FMN複合体と結合可能な化合物をスクリーニングする、請求項9に記載の方法。
【請求項17】
前記FabK−FMN複合体の3次元結晶構造情報が、FabK蛋白質のTIMバレルの上側に位置するループ部位;FabK蛋白質のTIMバレルの上に結合されたFMN分子のヘッド水平面を平らな半球底面とする半径15Å以内に位置する1つ以上のアミノ酸;198から205までのアミノ酸配列を含むヒンジ部位;208番のチロシン(Y208);209番のリシン(K209);211番のリシン(K211);214番のリシン(K214);229番のヒスチジン(H229);261番のロイシン(L261);276番のメチオニン(M276);208番から215番までの8個のアミノ酸から構成されたヘリックス;及び278番から283番までの6個のアミノ酸から構成されたへリックス;からなる群から選択された、1つ以上の部位に相当する、請求項16に記載の方法。
【請求項18】
請求項7に記載の保存媒体を使用して仮想スクリーニングする、請求項16に記載の方法。
【請求項1】
FabK蛋白質溶液を緩衝液、塩および沈澱剤を含む貯水槽溶液と接触させて濃度平衡法を行う段階を含み、
前記FabK蛋白質溶液の濃度は3〜30mg/mlであり、
前記緩衝液はpH4〜9.5で、貯水槽溶液内濃度は0.005〜1.5Mであり、
前記塩は金属塩およびアンモニウム塩からなる群から選択された1種以上のもので、貯水槽溶液内濃度は0.05〜2Mであり、
前記沈澱剤は炭素数1〜4のアルコールおよびポリエチレングリコールからなる群から選択された1種以上のものであり、貯水槽溶液内濃度は1%(v/v)〜40%(v/v)である、
FabK蛋白質の結晶化方法。
【請求項2】
前記濃度平衡法は、シッティングまたはハンギングドロップ蒸気平衡法、または透析法である、請求項1に記載のFabK蛋白質の結晶化方法。
【請求項3】
前記濃度平衡法は、8.0〜9.0の反応pH、4〜26℃の反応温度、及び1日〜20日の反応期間の反応条件下で行われる、請求項1に記載のFabK蛋白質の結晶化方法。
【請求項4】
前記濃度平衡法を行った後、高速窒素冷却法を追加的に行う、請求項1に記載のFabK蛋白質の結晶化方法。
【請求項5】
前記高速窒素冷却法は、50〜200Kの温度範囲で行われる、請求項4に記載のFabK蛋白質の結晶化方法。
【請求項6】
下記の表1のX線回折パターンデータ、または下記の表2に記載の2389個の原子座標で示された3次元結晶構造を有する、FabK−FMN(フラビンモノヌクレオチド)複合体結晶。
【表1】
【表2】
【請求項7】
下記の表1のX線回折パターンデータ、または表2に記載の2389個の原子座標情報から選択された1つ以上のFabK−FMN複合体の3次元結晶構造情報またはこれらの全てを保存する、保存媒体。
【表1】
【表2】
【請求項8】
前記表2に記載の2389個の原子座標情報から選択された1つ以上の3次元結晶構造情報は、FabK蛋白質(配列番号1)のTIMバレル上側に位置するループ部位;FabK蛋白質のTIMバレルの上に結合されたFMN分子のヘッド水平面を平らな半球底面とする半径15Å以内に位置する1つまたはそれ以上のアミノ酸;198から205までのアミノ酸配列を含むヒンジ部位;208番のチロシン(Y208)、209番のリシン(K209)、211番のリシン(K211)、214番のリシン(K214)、229番のヒスチジン(H229)、261番のロイシン(L261)、276番のメチオニン(M276)、208番から215番までの8個のアミノ酸から構成されたへリックス;および278番から283番までの6個のアミノ酸から構成されたへリックス;からなる群から選択される、1以上の該当する原子座標情報を含む、請求項7に記載の保存媒体。
【請求項9】
下記の表2に記載の2389個の原子座標情報から選択された1つ以上のFabK−FMN複合体の3次元結晶構造情報を使用する、FabK蛋白質活性阻害剤開発方法。
【表2】
【請求項10】
前記表2に記載の2389個の原子座標で示される3次元結晶構造を有するFabK−FMN複合体と候補化合物を反応させる段階;及び前記候補化合物から前記複合体と相互作用する化合物を選択して、FabK蛋白質の活性を阻害する化合物を決定する段階;を含む、請求項9に記載の方法。
【請求項11】
前記候補化合物及びFabK蛋白質のTIMバレルの上側に位置するループ部位;FabK蛋白質のTIMバレルの上に結合されたFMN分子のヘッド水平面を平らな半球底面とする半径15Å以内に位置する1つまたはそれ以上のアミノ酸;198から205までのアミノ酸配列を含むヒンジ部位;208番のチロシン(Y208);209番のリシン(K209);211番のリシン(K211);214番のリシン(K214);229番のヒスチジン(H229);261番のロイシン(L261);276番のメチオニン(M276);208番から215番までの8個のアミノ酸から構成されたへリックス;及び278番から283番までの6個のアミノ酸から構成されたへリックス;からなる群から選択された、1つ以上の部位の相互作用の有無を測定する、請求項10に記載の方法。
【請求項12】
請求項7に記載の保存媒体を使用して、FabK−FMN複合体と相互作用する化合物を選択する、請求項10に記載の方法。
【請求項13】
前記表2に記載のFabK−FMN複合体の2389個の原子座標情報から選択された1つ以上の3次元結晶構造情報を利用して、FabK−FMN複合体と結合可能な化合物を設計する、請求項9に記載の方法。
【請求項14】
前記2389個の原子座標から選択された1つ以上の3次元結晶構造情報が、FabK蛋白質のTIMバレルの上側に位置するループ部位;FabK蛋白質のTIMバレルの上に結合されたFMN分子のヘッド水平面を平らな半球底面とする半径15Å以内に位置する1つ以上のアミノ酸;198から205までのアミノ酸配列を含むヒンジ部位;208番のチロシン(Y208);209番のリシン(K209);211番のリシン(K211);214番のリシン(K214);229番のヒスチジン(H229);261番のロイシン(L261)、276番のメチオニン(M276);208番から215番までの8個のアミノ酸から構成されたへリックス;及び278番から283番までの6個のアミノ酸から構成されたへリックス;からなる群から選択された、1つまたはそれ以上の部位に相当する、請求項13に記載の方法。
【請求項15】
請求項7に記載の保存媒体を使用して、FabK−FMN複合体と相互作用する化合物を設計する、請求項13に記載の方法。
【請求項16】
前記2389個の座標から選択された1つ以上のFabK−FMN複合体の3次元結晶構造情報、及び候補化合物の3次元結晶構造情報をマッチングさせる仮想スクリーニング法を使用して、FabK−FMN複合体と結合可能な化合物をスクリーニングする、請求項9に記載の方法。
【請求項17】
前記FabK−FMN複合体の3次元結晶構造情報が、FabK蛋白質のTIMバレルの上側に位置するループ部位;FabK蛋白質のTIMバレルの上に結合されたFMN分子のヘッド水平面を平らな半球底面とする半径15Å以内に位置する1つ以上のアミノ酸;198から205までのアミノ酸配列を含むヒンジ部位;208番のチロシン(Y208);209番のリシン(K209);211番のリシン(K211);214番のリシン(K214);229番のヒスチジン(H229);261番のロイシン(L261);276番のメチオニン(M276);208番から215番までの8個のアミノ酸から構成されたヘリックス;及び278番から283番までの6個のアミノ酸から構成されたへリックス;からなる群から選択された、1つ以上の部位に相当する、請求項16に記載の方法。
【請求項18】
請求項7に記載の保存媒体を使用して仮想スクリーニングする、請求項16に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】

【図9】
【図10】
【図11a】
【図11b】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11a】
【図11b】
【公開番号】特開2009−159939(P2009−159939A)
【公開日】平成21年7月23日(2009.7.23)
【国際特許分類】
【出願番号】特願2008−96480(P2008−96480)
【出願日】平成20年4月2日(2008.4.2)
【出願人】(591074116)韓国科学技術研究院 (17)
【氏名又は名称原語表記】KOREA INSTITUTE OF SCIENCE AND TECNOLOGY
【住所又は居所原語表記】39−1 Hawolgok−dong,Seongbuk−gu,Seoul 136−791KOREA
【Fターム(参考)】
【公開日】平成21年7月23日(2009.7.23)
【国際特許分類】
【出願日】平成20年4月2日(2008.4.2)
【出願人】(591074116)韓国科学技術研究院 (17)
【氏名又は名称原語表記】KOREA INSTITUTE OF SCIENCE AND TECNOLOGY
【住所又は居所原語表記】39−1 Hawolgok−dong,Seongbuk−gu,Seoul 136−791KOREA
【Fターム(参考)】
[ Back to top ]