
Gタンパク質共役型受容体アゴニストおよびアンタゴニストならびに使用方法

【課題】Gタンパク質共役型受容体アゴニストおよびアンタゴニストならびに使用方法の提供。
【解決手段】本発明は、概ね、Gタンパク質共役型受容体(GPCR)、および詳しくはGPCRアゴニストおよびアンタゴニスト、これらの化合物およびそれらの医薬組成物の使用、例えば、GPCRに関連する生理的状態の処置、変調、および予防もしくはその一方、例えばケモカイン受容体が役割を果たす状態(例えば、敗血症、関節炎、炎症、および自己免疫疾患)の処置での使用に関する。本発明は、GPCRの第1の細胞内ループ構造から誘導されるペプチドに付着する細胞貫通または細胞係留部分を含むペプデュシン(pepducin)類と呼ばれる修飾ペプチドの発見にもとづいている。
【解決手段】本発明は、概ね、Gタンパク質共役型受容体(GPCR)、および詳しくはGPCRアゴニストおよびアンタゴニスト、これらの化合物およびそれらの医薬組成物の使用、例えば、GPCRに関連する生理的状態の処置、変調、および予防もしくはその一方、例えばケモカイン受容体が役割を果たす状態(例えば、敗血症、関節炎、炎症、および自己免疫疾患)の処置での使用に関する。本発明は、GPCRの第1の細胞内ループ構造から誘導されるペプチドに付着する細胞貫通または細胞係留部分を含むペプデュシン(pepducin)類と呼ばれる修飾ペプチドの発見にもとづいている。
【発明の詳細な説明】
【技術分野】
【0001】
発明の分野
本発明は、概ねGタンパク質共役型受容体(GPCR)に関し、詳しくは、GPCRアゴニストおよびアンタゴニスト、これらの化合物およびそれらの医薬組成物の使用、例えば、ケモカイン受容体が一因となる状態(例えば、敗血症、関節炎、炎症、および自己免疫性疾患)を処置する際等のGPCRが関与する生理的状態の処置、調節、および予防もしくはその一方における使用に関する。
【背景技術】
【0002】
発明の背景
種々のホルモン類、神経伝達物質、および生物活性物質は、細胞膜に位置する特異的な受容体を介して、生体の機能を制御、調節、または調整する。これらの受容体の多くは、受容体が連結する活性化グアニンヌクレオチド結合タンパク質(Gタンパク質)によって、細胞内シグナルの伝達をもたらす。この受容体は、Gタンパク質共役型受容体(「GPCR」)と一般的に呼ばれる。GPCRに対する特異的なシグナル伝達分子の結合は、受容体に構造変化を引き起し、その結果としてGタンパク質に対する結合およびGタンパク質の活性化が可能である形状となり、最終的には生物反応に導く細胞内現象のカスケードを誘発する。概して、GPCRはGタンパク質と相互作用して、細胞内第二メッセンジャー(例えば、サイクリックAMP、イノシトールリン酸、ジアシルグリセロール、およびカルシウムイオン)の合成を調節する。
【0003】
ケモカインは白血球誘引物質であって、白血球遊走を含む免疫プロセスに貢献する。白血球細胞内輸送は高度に調整されている、根底にある制御機構の破壊は強調された先天性免疫活性化(例えば、全身性炎症反応症候群または自己免疫性疾患)に関与する可能性がある。ケモカイン誘発シグナル伝達は、GPCRによってもたらされ、定義上、ケモカインの特徴は白血球の化学誘引性である。また、ケモカインは、細胞生存、ウイルス宿主相互作用、腫瘍成長および転移、器官形成、および脈管形成と同様に、白血球遊走性とは無関係である細胞性応答である。
【0004】
GPCRは、細胞の代謝、細胞増殖および運動性、癒着、炎症、神経シグナル伝達、ならびに血液凝固を制御するシグナル伝達プロセスにおいて、不可欠な役割を果たす。GPCRタンパク質にも、生身の機能を制御、調節、または調整する種々のシグナル伝達分子の標的として非常に重要な役割を有する。当該技術分野で周知であるように、GPCRは多種多様な障害に関係している。新規のGPCRモジュレータ(例えば、アゴニスト、部分的アゴニスト、インバースアゴニスト、およびアンタゴニスト)の開発には、敗血症、関節炎、炎症、および自己免疫性疾患等のGPCR関障害を処置するために、治療に応用可能である。
【発明の概要】
【課題を解決するための手段】
【0005】
発明の要旨
本発明は、GPCRの第1の細胞内ループ構造から誘導されるペプチドに付着する細胞貫通または細胞係留部分を含むペプデュシン(pepducin)類と呼ばれる修飾ペプチドの発見にもとづいている。ペプデュシン類はキメラのペプチド/ポリペチドと考え、受容体−Gタンパク質シグナル伝達のアゴニストおよびアンタゴニストもしくはその一方である。これらの組成物は、それらの同族の受容体に対して選択性を示す。
【0006】
したがって、本発明はペプデュシン組成物、すなわちGPCRの第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と第1の領域に結合した第2の領域を含むキメラポリペプチドを提供する。第2の領域は、天然に存在するまたは天然に存在しない細胞膜透過性および膜係留性疎水性部分もしくはその一方である。第1の領域は、好ましくはGPCR固有の細胞外部分を含まない。本発明のペプデュシンは、望ましくは第1の領域が誘導される同族のGPCRと結合する。
【0007】
第1の領域(Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメント)は、黄体形成ホルモン受容体、卵胞刺激ホルモン受容体、甲状腺刺激ホルモン受容体、カルシトニン受容体、グルカゴン受容体、グルカゴン様ペプチド1受容体(GLP−1)、向代謝性グルタミン酸受容体、副甲状腺ホルモン受容体、脈管活性腸幹ペプチド受容体、セクレチン受容体、成長ホルモン放出因子(GRF)受容体、プロテアーゼ活性受容体(PAR)、コレシストキニン受容器、ソマトスタチン受容体、メラノコルチン受容体、ADP受容体、アデノシンレセプター、トロンボキサン受容体、血小板活性化因子受容体、アドレナリン作動性受容体、5−HT受容体、ケモカイン受容体、神経ペプチド受容体、オピオイド受容体、副甲状腺ホルモン(PTH)受容体、または脈管活性腸幹ペプチド(VIP)受容体のアミノ酸配列を含む。
【0008】
例えば、第1の領域(Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメント)は、プロテアーゼ活性受容体(PAR)またはケモカイン受容体のアミノ酸配列を含む。プロテアーゼ活性受容体は、例えば、PARl、PAR2、PAR3、またはPAR4であってもよい。ケモカイン受容体は、CCまたはCXC受容体であってもよく、例えば、それぞれCCRl、CCR2、CCR3、CCR4、CCR5、CCR6、CCR7、CCR8、またはCCR9、あるいはCXCRl、CXCR2、CXCR3、CXCR4、CXCR5、CXCR6、またはCX3CR1が挙げられる。別の実施形態において、第1の領域(Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメント)が、例えば、コレシストキニンAおよびB(CCKA、CCKB)、ソマトスタチン−2(SSTR2)、メラノコルチン−4(MC4R)、グルカゴン様ペプチド−1受容体(GLP−1R)、P2Y12ADP受容体、あるいは「非定型」ケモカイン受容体(例えば、NKl、NK2、GRP/ボンベシン受容体、FPR1、FPRL−1、C3aRまたはC5aR)に由来するものであってよい。特定の実施形態において、本発明のペプデュシン類は、PAR2、CXCRl、CXCR2、CXCR4、およびCCR5ケモカイン受容体に対するものを含む。
【0009】
第2の領域(細胞膜透過性および膜係留性疎水性部分もしくはその一方)は、第1の領域のN末端、C末端、C末端アミノ酸とN末端アミノ酸との間のアミノ酸、またはN末端およびC末端の両方で結合する。望ましくは、細胞膜透過性および膜係留性疎水性部分もしくはその一方は、直鎖脂肪酸、例えばノナノイル(C9)、カプリル(C10)、ウンデカノイル(C11)、ラウロイル(C12)、トリデカノイル(C13)、ミリストイル(C14)、ペンタデカノイル(C15)、パルミトイル(C16)、フィタノイル(メチル置換C16)、ヘプタデカノイル(C17)、ステアロイル(C18)、ノナデカノイル(C19)、アラキドイル(C20)、ヘニエコサノイル(C21)、ベヘノイル(C22)、トルシサノイル(trucisanoyl)(C23)、およびリグノセロイル(C24)部分である。細胞膜透過性および膜係留性部分もしくはその一方は、アミド結合、スルフヒドリル、アミン、アルコール、フェノール基、または炭素−炭素結合によって、キメラ化合物に結合したものであってもよい。特定の実施形態は、疎水性部分としてパルミトイルまたはリトコール酸(またはその塩類)を含む。他の細胞膜透過性および膜係留性疎水性部分もしくはその一方が、コレステロール、リン脂質、ステロイド、スフィンゴシン、セラミド、オクチル−グリシン、2−シクロヘキシルアラニン、ベンゾリルフェニルアラニン、C1またはC2アシル基、あるいはC3〜C8の脂肪酸が挙げられる。
【0010】
本発明は、さらに、本発明のペプデュシン組成物と薬学的に許容される担体とを含む医薬組成物に関し、また一つ以上の容器にこれらの医薬組成物が含まれるキットに関する。
【0011】
本発明は、Gタンパク質共役型受容体(GPCR)の第1の細胞内細胞内ループ(i1ループ)の第1の領域またはそのフラグメントとこの第1の領域に結合する第2の領域とを含むペプデュシンを投与することによって、哺乳類被検体等で敗血症の処置、敗血症重篤度の低減、または敗血症の予防をおこなうための方法を含む。第2の領域は、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である。被検体は、敗血症を発症またはそのリスクがあると診断されている。
【0012】
組成物はまた、炎症および脈管形成もしくはその一方の処置、その重篤度の減少、あるいは予防するために、用いられる。炎症および脈管形成もしくはその一方を処置または予防する方法は、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)の第1の領域またはそのフラグメントと、第1の領域に結合し、かつ天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である第2の領域とから構成されるキメラポリペプチドを含むケモカイン阻害性ペプデュシンを投与することによって実施される。
【0013】
組成物はまた、癌の重篤度を処置する、または減少させるために、用いられる。癌の重篤度を処置する、または減少させる方法は、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)の第1の領域またはそのフラグメントと、第1の領域に結合し、かつ天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である第2の領域とから構成されるキメラポリペプチドを含むペプデュシンを投与することによって実施される。
【0014】
組成物は、血栓症、例えば冠状動脈、動脈性および静脈(例えば、深静脈または腸間膜)血栓症の重篤度を処置または低減させるためにも用いられる。血栓症の処置または予防の方法は、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)の第1の領域またはそのフラグメントと、第1の領域に結合し、かつ天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である第2の領域とから構成されるキメラポリペプチドを含むペプデュシンを投与することによって実施される。
【0015】
本発明はまた、炎症または障害を処置する、または予防する方法に関し、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)の第1の領域またはそのフラグメントと、第1の領域に結合し、かつ天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である第2の領域とを含むペプデュシンを、必要に応じて被検体に投与することによって実施される。処置に適した炎症性疾患として、慢性閉塞性肺疾患(COPD)、強直性脊椎炎(alkylosing spondylitis)、頸部関節炎(cervical arthritis)、線維筋痛症、虚血再灌流障害、腸管虚血、若年性関節リウマチ、腰仙骨関節炎変形性関節症(lumbosacral
arthritis osteoarthritis)、骨粗鬆症、乾癬性関節炎、リウマチ病、関節リウマチ、湿疹、乾癬、皮膚炎、ブドウ膜炎および結膜炎、喘息および気管支炎、潰瘍、歯肉炎、クローン病、萎縮性胃炎、胃炎バリアロフォルム(gastritis varialoforme)、潰瘍性大腸炎、セリアック病、限局性回腸炎、消化性潰瘍、発熱(pyresis)、膀胱刺激症状および膀胱炎、中枢神経系または末梢神経系の炎症性神経障害、多発性硬化症、エイズの炎症性ニューロパシーおよび神経性合併症、自己免疫性炎症または外科手術による外傷が挙げられる。本明細書中に述べられる組成物は、敗血症および関連病理学(例えば、播種性血管内凝固(DIC)、フィブリン溶解反応、および全身性炎症反応(SIRS)もしくはその一方)の重篤度の予防、逆転、または減少をおこなう。
【0016】
本発明のペプデュシン組成物は、GPCRの広範囲に及ぶ活性を活性化または阻害させるのに有用である。本発明にもとづくペプデュシンとして、CXCRl、CXCR2、CXCR3、CXCR4、CXCR5、CXCR6、およびCX3CR1を含むケモカインCXC受容体、CCRl、CCR2、CCR3、CCR4、CCR5、CCR6、CCR7、CCR8、およびCCR9を含むケモカインCC受容体、プロテアーゼ活性受容体(PAR)(例えば、PARl、PAR2、PAR4)、コレシストキニンAおよびB受容体(CCKA、1CCKB)、ソマトスタチン−2(SSTR2)受容体、メラノコルチン−4(MC4R)受容体、グルカゴン様ペプチド−1受容体(GLP−IR)、スフィンゴシン1−リン酸(SlP)受容体(例えば、亜型SlPlおよびS1P3)、EDG受容体、エンドセリン(ET)受容体(例えば、亜型ET−I、ET−2、ET−3、ETA、ETB)、EDG受容体(例えば、亜型EDG−I、EDG−2、EDG−3、EDG−4、EDG−5、EDG−6)、ならびにP2Y12 ADP受容体に作用するものが挙げられる。また、NKl、NK2、GRP/ボンベシン受容体、FPR−l、FPRLI、C3aR、およびC5aR等の「非定型」ケモカイン受容体のためのペプデュシンは、本発明の範囲内である。特定の実施形態において、本発明のペプデュシンは、PAR2、CXCRl、CXCR2、CXCR4とCCR5ケモカイン受容体に対するものを含む。
【0017】
投与は、例えば全身性炎症、COPDおよび敗血症もしくはその一方の場合、全身的に、例えば静注によって実施される。あるいは、組成物の送達は、経皮、経口、経鼻腔(例えば、喘息を処置するため)により、または局所的に、例えば、粘着パッチの形態、またはクリーム、泡、軟膏(例えば、皮膚炎、乾癬または他の皮膚炎症性条件の症状の緩和のために)の形態で行う。
例えば、本願発明は以下の項目を提供する。
(項目1)
キメラポリペプチドであって、
a)Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、
b)前記第1の領域に結合した第2の領域であって、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、第2の領域とを含み、
前記第1の領域が前記GPCRの天然の細胞外部分を含まない、キメラポリペプチド。
(項目2)
前記キメラポリペプチドがその同族のGPCRに結合する、項目1のキメラポリペプチド。
(項目3)
前記疎水性部分が前記第1の領域のN末端、C末端、C末端アミノ酸とN末端アミノ酸との間のアミノ酸、またはN末端およびC末端の両方に結合している、項目1に記載のキメラポリペプチド。
(項目4)
前記疎水性部分が脂質である、項目1に記載のキメラポリペプチド。
(項目5)
前記疎水性部分が、ノナノイル(C9)、カプリル(C10)、ウンデカノイル(C11)、ラウロイル(C12)、トリデカノイル(C13)、ミリストイル(C14)、ペンタデカノイル(C15)、パルミトイル(C16)、フィタノイル(メチル置換C16)、ヘプタデカノイル(C17)、ステアロイル(C18)、ノナデカノイル(C19)、アラキドイル(C20)、ヘニエコサノイル(C21)、ベヘノイル(C22)、トルシサノイル(C23)、およびリグノセロイル(C24)部分からなる群から選択され、前記疎水性部分がアミド結合、スルフヒドリル、アミン、アルコール、フェノール基、または炭素−炭素結合によって、キメラポリペプチドに結合している、項目4に記載のキメラポリペプチド。
(項目6)
前記疎水性部分がパラミトイル部分である、項目4に記載のキメラポリペプチド。
(項目7)
前記疎水性部分がリトコール酸またはその塩である、項目4に記載のキメラポリペプチド。
(項目8)
前記疎水性部分がコレステロール、リン脂質、ステロイド、スフィンゴシン、セラミド、オクチル−グリシン、2−シクロヘキシルアラニン、ベンゾリルフェニルアラニン、C1またはC2アシル基、およびC3〜C8の脂肪酸からなる群から選択される、項目4に記載のキメラポリペプチド。
(項目9)
前記第1の領域がプロテアーゼ活性化受容体(PAR)またはケモカイン受容体を含み、前記第2の領域が脂質部分を含む、項目1に記載のキメラポリペプチド。
(項目10)
前記プロテアーゼ活性化受容体がPAR1、PAR2、PAR3、およびPAR4からなる群から選択される、項目9に記載のキメラポリペプチド。
(項目11)
前記ケモカイン受容体が、CCRまたはCXCR受容体からなる群から選択される、項目9に記載のキメラポリペプチド。
(項目12)
前記CCR受容体がCCR1、CCR2、CCR3、CCR4、CCR5、CCR6、CCR7、CCR8、およびCCR9からなる群から選択される、項目11のキメラポリペプチド。
(項目13)
前記CCR受容体が、CXCR1、CXCR2、CXCR3、CXCR4、CXCR5、CXCR6、およびCX3CR1からなる群から選択される、項目11に記載のキメラポリペプチド。
(項目14)
前記ケモカイン受容体がNK1、NK2、MLT2、GRP/ボンベシン、FPR1、FPRL−1、C3Ar、OR受容体からなる群から選択される、項目9に記載のキメラポリペプチド。
(項目15)
前記Gタンパク質共役型受容体またはそのフラグメントが、黄体形成ホルモン受容体、卵胞刺激ホルモン受容体、甲状腺刺激ホルモン受容体、カルシトニン受容体、EDG受容体、エンドセリン(ET)受容体、グルカゴン受容体、グルカゴン様ペプチド1受容体(GLP−I)、代謝調節型グルタミン酸受容体、副甲状腺ホルモン受容体、血管作動性腸管ペプチド受容体、セクレチン受容体、成長ホルモン放出因子(GRF)受容体、プロテアーゼ活性化受容体(PAR)、コレシストキニン受容体、ソマトスタチン受容体、メラノコルチン受容体、ADP受容体、アデノシン受容体、トロンボキサン受容体、血小板活性化因子受容体、アドレナリン作動性受容体、5−HT受容体、CXCR4、CCR5、ケモカイン受容体、ニューロペプチド受容体、オピオイド受容体、副甲状腺ホルモン(PTH)受容体、スフィンゴシン1−リン酸(SlP)受容体、および血管作動性腸管ペプチド(VIP)受容体からなる群から選択される、項目1に記載のキメラポリペプチド。
(項目16)
前記フラグメントが単離された細胞内フラグメントである、項目1に記載のキメラポリペプチド。
(項目17)
前記フラグメントが単離された細胞外フラグメントである、項目1に記載のキメラポリペプチド。
(項目18)
医薬組成物であって、項目1に記載のキメラポリペプチドと薬学的に許容される担体とを含む、医薬組成物。
(項目19)
キットであって、1つ以上の容器に項目18に記載の医薬組成物を含む、キット。
(項目20)
前記Gタンパク質共役型受容体が哺乳類のGタンパク質共役型受容体である、項目1に記載のキメラポリペプチド。
(項目21)
前記Gタンパク質共役型受容体がCCKA、CCKB、SSTR2およびSubP受容体からなる群から選択される、項目1に記載のキメラポリペプチド。
(項目22)
前記疎水性部分がステロイドである、項目1に記載のキメラポリペプチド。
(項目23)
前記疎水性部分が、リン脂質、ステロイド、スフィンゴシン、セラミド、オクチル−グリシン、2−シクロヘキシルアラニン、およびベンゾリルフェニルアラニンからなる群から選択される、項目1に記載のキメラポリペプチド。
(項目24)
哺乳類被検体の敗血症を処置または予防する方法であって、
処置を必要としている患者を診断すること、および
Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチドをそれを必要とする患者に投与すること
を含む、方法。
(項目25)
炎症を処置または予防する方法であって、
処置を必要とする患者を診断すること、および
Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチドをそれを必要とする患者に投与すること
を含む、方法。
(項目26)
血管新生を処置または予防する方法であって、
処置を必要とする患者を診断すること、および
Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分であるケモカイン阻害性ペプデュシンをそれを必要とする患者に投与すること
を含む、方法。
(項目27)
炎症性疾患を処置または予防する方法であって、
処置を必要とする患者を診断すること、および
Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチドをそれを必要とする患者に投与すること
を含む、方法。
(項目28)
前記炎症性疾患が、強直性脊椎炎、頸部関節炎、線維筋痛症、腸管虚血、若年性関節リウマチ、腰仙骨関節炎変形性関節症、骨粗鬆症、乾癬性関節炎、リウマチ病、関節リウマチ、湿疹、乾癬、皮膚炎、ブドウ膜炎および結膜炎、喘息および気管支炎、潰瘍、歯肉炎、クローン病、萎縮性胃炎、胃炎バリアロフォルム、潰瘍性大腸炎、セリアック病、限局性回腸炎、消化性潰瘍、発熱、膀胱刺激症状および膀胱炎、中枢神経系または末梢神経系の炎症性神経障害、多発性硬化症、エイズの炎症性ニューロパシーおよび神経性合併症、自己免疫性炎症、ならびに外科手術による外傷からなる群から選択される、項目27に記載の方法。
(項目29)
虚血再灌流障害を処置または予防する方法であって、
処置を必要とする患者を診断すること、および
Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチドをそれを必要とする患者に投与すること
を含む、方法。
(項目30)
癌の重篤度を処置または減少させる方法であって、
処置を必要とする患者を診断すること、およびGタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチドをそれを必要とする患者に投与することを含む、方法。
(項目31)
血栓症の重篤度を処置または減少させる方法であって、
処置を必要とする患者を診断すること、およびGタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチドをそれを必要とする患者に投与することを含む、方法。
(項目32)
キメラポリペプチドであって、
D1(ILKMKVKKPAV)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目33)
キメラポリペプチドであって、
D1(ATQAPRLPST)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目34)
キメラポリペプチドであって、
D1(VLATGAPRLPST)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目35)
キメラポリペプチドであって、
D1(ATGAPRLPST)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目36)
キメラポリペプチドであって、
D1(FLFRTKKKHPAV)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目37)
キメラポリペプチドであって、
D1(ILYSRVGRSVTD)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目38)
キメラポリペプチドであって、
D1(YSRVGRSVTD)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目39)
キメラポリペプチドであって、
D1(YQKKLRSMTD)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目40)
キメラポリペプチドであって、
D1(MGYQKKLRSMTD)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目41)
キメラポリペプチドであって、
D1(KRLKSMTD)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目42)
キメラポリペプチドであって、
D1(LINCKRLKSMTD)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目43)
キメラポリペプチドであって、
D1(VLATQAPRLPST)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目44)
敗血症を処置または予防するための薬剤の製造でのキメラポリペプチドの使用であって、前記キメラポリペプチドが、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、使用。
(項目45)
炎症を処置または予防するための薬剤の製造でのキメラポリペプチドの使用であって、前記キメラポリペプチドが、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、使用。
(項目46)
血管新生を処置または予防するための薬剤の製造でのキメラポリペプチドを含むケモカイン阻害性ペプデュシンの使用であって、前記キメラポリペプチドが、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、使用。
(項目47)
炎症性疾患を処置または予防するための薬剤の製造でのキメラポリペプチドの使用であって、前記キメラポリペプチドが、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、使用。
(項目48)
前記炎症性疾患が、強直性脊椎炎、頸部関節炎、線維症、虚血再灌流障害、腸管虚血、若年性関節炎、腰仙骨関節炎変形性関節症、骨粗鬆症、乾癬性関節炎、リウマチ性疾患、関節リウマチ、湿疹、乾癬、皮膚炎、ブドウ膜炎および結膜炎、喘息および気管支炎、潰瘍、歯肉炎、クローン病、萎縮性胃炎、胃炎バリアロフォルム、潰瘍性大腸炎、セリアック病、限局性回腸炎、消化性潰瘍、発熱、膀胱刺激症状および膀胱炎、中枢または末梢神経系の炎症性神経障害、多発性硬化症、エイズの炎症性ニューロパシーおよび神経性合併症、自己免疫性炎症、ならびに外科手術による外傷からなる群から選択される、項目47に記載の使用。
(項目49)
癌の重篤度を処置、予防、または減少するための薬剤の製造でのキメラポリペプチドの使用であって、前記キメラポリペプチドが、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、使用。
(項目50)
血栓症の重篤度を処置、予防、または減少するための薬剤の製造でのキメラポリペプチドの使用であって、前記キメラポリペプチドが、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、使用。
【図面の簡単な説明】
【0018】
【図1】図1は、GPCRトポロジーを示す図である。i1ループをこの図に示す。
【図2】図2は、構造の類似性を説明するために整列されている膜貫通型フランキング部を含むCXCR1およびCXCR2i1ループ配列を示す図である。
【図3】図3は、本発明にもとづくペプデュシンが作用するケモカイン受容体、各々のリガンド、および受容体が見出される各々の細胞型を示す図である。
【図4】図4は、本発明にもとづくペプデュシンが作用するさらに別のケモカイン受容体、各々のリガンド、および受容体が見出される各々の細胞型を示す図である。
【図5】図5は、実施例1に詳述した実験におけるCLP時のペプデュシン療法に対する動物の反応を示す線グラフである。
【図6】図6は、CLP後8時間で開始されたペプデュシン療法の結果が示されている、実施例2で詳述した実験の結果を示す線グラフである。
【図7】図7Aないし図7Cは、実施例3にさらに詳述したように、ペプデュシン処置後の肺への白血球浸潤の抑制と肺でのTNF−α産生および繊維素沈着とを示す。図7Aおよび図7Bは棒グラフであり、図7Cは顕微鏡写真である。
【図8】図8Aないし図8Dは、ペプデュシン処置後、敗血マウスでの出血時間短縮を示す棒グラフである。
【図9】図9は、実施例5により詳しく示されるように、ヒト好中球走化性がどのようにしてペプデュシン処置によって阻害されるかについて示すグラフである。
【図10】図10は、実施例6により詳しく示されるように、生体内でのペプデュシン処置によって、どのようにして白血球化学走性が阻害されるかについて示しているグラフである。
【図11】図11は、実施例7でより詳しく示されるように、実験の結果を示す線グラフである。
【図12】図12は、実施例8により詳しく示されているように、ヒト好中球走化性の抑制を示ことによって、どのようにして本発明のペプデュシンが炎症を選択的に減少させるかを示すグラフである。
【図13】図13は、実施例9により詳しく示されているように、ヒト単核細胞化学走性の抑制を示すことによって、どのようにして本発明のペプデュシンが炎症を選択的に減少させるかを示すグラフである。
【図14】図14は、本発明にもとづいてペプデュシンで使用することが可能な膜貫通フランキング部を含むCXCR4i1ループ配列を示す図である。
【図15】図15は、本発明にもとづくペプデュシンで使用することが可能な膜貫通フランキング部を含むCCR1i1ループ配列を示す図である。
【図16】図16は、本発明にもとづくペプデュシンで使用することが可能な膜貫通フランキング部を含むCCR2i1ループ配列を示す図である。
【図17】図17は、本発明にもとづくペプデュシンで使用することが可能な膜貫通フランキング部を含むCCR4i1ループ配列を示す図である。
【図18】図18は、本発明にもとづくペプデュシンに使用することが可能な膜貫通型フランキング部を含むCCR5i1ループ配列を示す図である。
【図19】図19は、本発明にもとづくペプデュシンに使用することが可能な膜貫通型フランキング部を含むPAR1i1ループ配列を示す図である。
【図20】図20は、本発明にもとづくペプデュシンに使用することが可能な膜貫通型フランキング部を含むEDGi1ループ配列を示す図である。
【図21】図21は、本発明にもとづくペプデュシンに使用することが可能な膜貫通型フランキング部を含むNK1−Ri1ループ配列を示す図である。
【図22】図22は、実施例10により詳しく述べられる実験の結果を示す棒グラフである。
【図23】図23は、実施例11により詳しく述べられる実験の結果を示すグラフである。
【図24A】図24Aは、HEK細胞を示す棒グラフであり、PAR1およびPAR4もしくはその一方を過渡的に形質移入され、24時間にわたり300μMのP4pa1−i1の存在または非存在下で0.5μMのTFLLRN(配列番号33)または500μMのAYPGKF(配列番号34)に向けて移行することができるHEK細胞を示す棒グラフである。
【図24B】図24Bは、160μMのAYPGKFを添加する前に3μMのP4pal−i1または緩衝液(未処理)で2分間、あらかじめインキュベートしたヒト血小板を示すグラフである。
【図24C】図24Cは、13μMのSFLLRNを添加に先立って3μMのP4pal−i1または緩衝液(未処理)で2分間、あらかじめインキュベートしたヒト血小板を示すグラフである。
【図24D】図24Dは3μMトロンビン(T)の添加する前に1μMのRWJ−56110、5μMのP4pal−i1、または1μMのRWJ−56110プラス5μMのP4pal−i1で2分間、あらかじめインキュベートした血小板を示す図である。これらの図に示すデータは、実施例12に詳述する実験の結果である。
【図25A】図25Aは、ビバリルジンとP4a1−i1阻害トロンビン依存型凝集との組み合わせを示すグラフである。ヒト血小板を、20pMないし20nMトロンビンの添加に先立って、緩衝液(未処理)、ビバリルジン(200nM)、RWJ−56110(1μM)、およびPAR1−Ab(74μg/ml)もしくはその一方とともに、2分間にわたり、事前のインキュベーションをおこなった。
【図25B】図25Bは、示された濃度のトロンビンの添加に先立って、血小板を2分間にわたり、200nMのビバリルジン、プラスまたはマイナス5μMP4pal−i1と事前のインキュベーションをおこなったことを示すグラフである。これらの図に示すデータは、実施例13に詳述する実験の結果である。
【図26】図26は、モルモットでの頸動脈閉鎖症を妨げているビバリルジンとP4pal−i1との組み合わせを示す棒グラフである。モルモットに対して、実施例14に述べられるように、FeCl3による頸動脈の損傷に対して5分間先だって、ビバリルジン、P4pal−i1、およびP1pal−7(各々の処置群についてn=3ないし5)もしくはその一方による処理をおこなった。ビヒクル処置に関連するP値を一番下に示す。
【図27】図27は、乳癌細胞および卵巣癌細胞の化学走性をi1ループ・ペプデュシンがどのようにして阻害しているかを示すグラフである。NIH3T3から条件培地を調製した。化学組成アッセイ(20時間)を、8mm孔ニトロセルロース・フィルターを備えた48ブラインドウェル・マイクロケモタキシス・チャンバー(Neuroprobe)を用いて、OVACAR−4ヒト卵巣癌細胞およびMDA−MB−231ヒト乳癌細胞に対しておこなった。データを化学走性指数として表し、この指数はRPMI培地単独への移動とNIH3T条件培地への移動との距離の比である。
【発明を実施するための形態】
【0019】
発明の詳細な説明
定義
便宜上、明細書、実施例、および添付の特許請求の範囲に使用した特定の用語をここに集める。
【0020】
「処置する」とは、状態、疾患、障害等、またはその症状の改善をもたらす任意の作用、例えば、軽減、減少、変調、または排除を含む。
【0021】
「GPCRフラグメント」は、GPCRの全ての天然のアミノ酸配列よりも少ないGPCRタンパク質配列の一部を有するペプチドを含む。「単離GPCRフラグメント」は、全ての配列よりも少ないGPCRタンパク質配列の一部を有するが、天然のフランキング領域は含まないペプチドを含む。単離されたGPCRフラグメントは、天然の分子にある参照フラグメントを直ちにフランキングする1つ以上のアミノ酸が欠けている。
【0022】
「単離細胞内GPCRフラグメント」は、GPCRタンパク質の細胞内i1ループのアミノ酸配列を有するが、細胞外ループまたは細胞内i1ループをフランキングする膜貫通型ヘリクス配列に由来する配列を持たないペプチドを含む。「単離細胞外GPCRフラグメント」は、GPCRタンパク質の細胞外ループのアミノ酸配列を有するが、細胞外ループまたは細胞外ループの領域をフランキングする膜貫通型ヘリクス配列に由来するアミノ酸を持たないペプチドを含む。
【0023】
「連結」とは、結合を意味する。例えば、ペプチドと細胞貫通または係留部分は、連結(すなわち共有結合)を経て、ペプデュシンと各々結合する。好ましくは、連結は不安定な結合(例えば、チオールまたはエステル結合)である。不安定な連結を有するペプデュシン化合物は、不安定ではない連結の化合物と比べて体内組織での蓄積が少ないことから、有利である。被験者に対する投与後の身体組織での蓄積が減少することは、被験者での副作用の減少をともなう。
【0024】
「GPCRアゴニスト」は、その受容体に特異的な内因性シグナル伝達分子の作用を模倣するためにGPCRを活性化する組成物を含む。「GPCRアンタゴニスト」は、GPCR活性を阻害する組成物を含む。GPCR活性の測定を、Gタンパク質等のエフェクター・シグナル伝達分子と結合する能力でおこなう。「活性GPCR」は、Gタンパク質と相互作用して活性化することができる。自己抑制的な受容体は、細胞外リガンドと結合し、およびGタンパク質と生産的に相互作用して、もしくはその一方を行い、このGタンパク質を活性化させる能力を有する。
【0025】
「細胞膜透過性部分」は、細胞外間隙から細胞の細胞内区画まで物質の転送を媒介する化合物または官能基を含む。細胞膜透過性成分は、細胞質内または細胞膜の細胞質空間へ連結物質(例えば、本発明のGPCRペプチドまたはフラグメント)を往復させる。例えば、細胞膜透過性部分は疎水性部分である。疎水性部分は、少なくとも約11アミノ酸長の、例えば、混合配列ペプチドまたはホモポリマー・ペプチド(ポリロイシンまたはポリアルギニン)である。この物質は、本発明のGPCRフラグメントまたはペプチド模倣薬であってもよい。細胞膜透過性部分として、少なくとも10個の隣接アミノ酸、例えばGPCR膜貫通型ヘリクス領域の1ないし15アミノ酸を挙げることができる。
【0026】
「膜係留性部分」として、細胞膜に関係または結合する化合物または官能基が挙げられる。したがって、膜係留性部分は、標的細胞の膜近傍で膜係留性部分が付着する物質(すなわち、本発明のGPCRフラグメントまたはペプチド模倣薬)をもたらす。細胞膜は真核生物または原核生物である。膜係留性部分は、望ましくは疎水性部分である。疎水性部分は、アミノ酸長が10アミノ酸未満であるポリロイシンまたはポリアルギニン等のホモポリマー・ペプチドまたは混合配列ペプチドを含むことができる。膜係留性部分は、GPCR膜貫通型ヘリクス領域の少なくとも1〜7つの隣接アミノ酸を含むことができる。好ましくは、膜係留性部分は、GPCR膜貫通型領域の少なくとも10個の隣接アミノ酸(しかし、16未満のアミノ酸)である。より好ましくは、膜係留性部分は、GPCR膜貫通型領域の少なくとも15個の隣接アミノ酸である。膜係留性部分は、コレステロール、リン脂質、ステロイド、スフィンゴシン、セラミド、オクチル−グリシン、2−シクロヘキシルアラニン、またはベンゾリルフェニルアラニンも含む。他の膜係留性部分は、C1またはC2アシル基またはC3〜C8脂肪酸成分、例えばプロピオノイル(C3)、ブタノイル(C4)、ペンタノイル(C5)、カプロイル(C6)、ヘプタノイル(C7)、およびカプリロイル(C8)を含む。膜係留性部分は、ペプデュシンのGPCRフラグメントのC末端アミノ酸、N末端アミノ酸、またはN末端アミノ酸とC末端アミノ酸との間のアミノ酸に結合することが可能である。
【0027】
「製薬的または薬理的に許容される」は、動物、またはヒトに対して投与した場合に、それぞれに見合った副作用、アレルギー反応、または他の有害反応を生ずることのない分子実体または組成物を包含する。「製薬的に許容される担体」は、任意または全ての溶媒、分散媒、コーティング、抗菌、および抗真菌剤、等張および吸収遅延剤、その他を包含する。製薬活性物質のためのそのような培地および薬剤の使用は、周知の技術である。任意の従来の培地または薬剤が主成分と適合しない場合を除いて、治療用組成物でのその使用が検討される。補助的活性成分も組成物に取り込むことができる。
【0028】
「低分子」は、約5kD未満、最も好ましくは約4kD未満の分子量を持つ組成物を含む。低分子を、核酸、ペプチド、ポリペチド、ペプチド模倣薬、炭水化物、脂質、または他の有機分子あるいは無機分子とすることができる。化学的および生物学的混合物もしくはその一方(例えば、菌類、細菌、または藻類抽出物)のライブラリは、当該技術分野で知られており、本発明のアッセイのいずれかによってスクリーニングすることができる。
【0029】
「標的分子」として、GPCRタンパク質が本来結合または相互作用する分子、例えばGPCR相互作用タンパク質を発現する細胞表面上の分子、第二の細胞表面上の分子、細胞外環境にある分子、細胞膜の内面に結合した分子、または細胞質分子が挙げられる。GPCR標的分子を非GPCR分子または本発明のGPCRペプチドとすることができる。一実施形態では、GPCR標的分子は、例えば膜結合GPCRに対する化合物の結合によって生ずるシグナル等の、細胞膜を介して細胞内への細胞外シグナル伝達を促すシグナル伝達経路の構成要素である。標的は、下流のシグナル伝達分子とGPCRとの結合を促進する触媒活性またはタンパク質を有している第二の細胞間タンパク質である。
【0030】
「併用療法」(または「共同療法」)は、本発明のペプデュシンの投与と特異的な治療計画の一部としての少なくとも第2の薬剤とを含み、これらの治療薬の相互作用による有益な効果を提供することを意図している。併用の有益な効果は、治療薬の併用から生ずる薬動力学的または薬力学的相互作用を含むが、これに限定されるものではない。これらの治療薬の併用投与は、所定の時間(通常、選択される組合せによって、分、時間、日または週)にわたって、概して実施される。「併用療法」は、一般的ではないが、本発明の併用を偶然または任意にもたらす別々の単独療法計画の一部として、これらの治療薬を二種類以上投与することを包含することを意図していることもある。「併用療法」は、これらの治療薬の投与を順番におこなうことを意図している。すなわち、これらの治療薬の投与と同様に、各々の治療薬、または治療薬のうちの少なくとも2つを、異なる時間に投与するとともに、これらの治療薬の投与と同様に、または少なくとも2つの治療薬含むことを目的とする。実質的な同時投与は、例えば、被検体に対して、一定の比率の各々の治療薬を含む単一のカプセルまたは治療薬の各々に対応した単一のカプセルを複数、投与することによって達成し得る。各治療薬の経時的投与または実質的な同時投与は、限定されるものではないが、経口経路、静脈経路、筋内経路、および粘膜組織を通しての直接吸収を含む適当な経路によっておこなうことができる。治療薬を同一の経路または異なる経路で投与することができる。選択された組み合わせの第1の治療薬を静脈内注射により投与し、この組み合わせの他の治療薬を経口投与してもよい。あるいは、例えば、すべての治療薬を経口投与することも可能であり、またはすべての治療薬は静脈内注射によって投与することも可能である。治療薬が投与される順番は、かなりクリティカルであるというものではない。「併用療法」も、他の生物学的に活性成分と非薬物療法(例えば、手術または放射線処置)とのさらなる併用で、先に述べたように治療薬の投与を含むことができる。併用療法が非薬物処置をさらに含む場合、治療薬と非薬物処置とを組合せることによる相互作用から有益な効果が達成される限り、非薬物処置を任意の適当な時刻におこなうことができる。例えば、適当な症例では、非薬物処置が治療薬の投与から一時的に除外される場合、おそらく数日、さもなければ数週間まで、有益な効果がさらに達成される。
【0031】
「相同アミノ酸配列」が意味することは、アミノ酸配列、すなわち、本発明のペプデュシンでは、1つ以上(例えば、1、2、3、4、または5)の保存的アミノ酸置換によって、またはポリペプチドの活性に副作用を及ぼさない位置での1つ以上(例えば、1、2、3、4、または5)非保存的アミノ酸置換、欠失、または付加によって、参照アミノ酸配列とは異なる。好ましくは、そのような配列は、参照アミノ酸配列に対して少なくとも75%、80%、85%、90%、または95%同一である。
【0032】
相同アミノ酸配列として、参照アミノ酸配列と同一または実質的に同一であるペプチド配列が挙げられる。「実質的に同一であるアミノ酸配列」は、参照のアミノ酸配列に対して、少なくとも90%、好ましくは95%、より好ましくは97%、さらに最も好ましくは99%同一であり、好ましくは参照の配列とは、たとえ保存的アミノ酸置換の大部分によるものでも、異なる。
【0033】
保存的アミノ酸置換として、主として、同一クラスのアミノ酸間での置換が挙げられる。これらのクラスとして、例えば、(a)無電荷極性側鎖(例えば、アスパラギン、グルタミン、セリン、スレオニン、およびチロシン)、(b)塩基性側鎖を持つアミノ酸(例えば、リシン、アルギニン、およびヒスチジン)、(c)酸性側鎖を持つアミノ酸(例えば、アスパラギン酸およびグルタミン酸)、ならびに(d)非極性側鎖を有するアミノ酸、例えばグリシン、アラニン、バリン、ロイシン、イソロイシン、プロリン、フェニルアラニン、メチオニン、トリプトファン、ならびにシステインが挙げられる。
【0034】
相同は、主として、配列分析用ソフトウェアを用いて測定される(例えば、ウィシコンシン大学ジェネティック・コンピュータ・グループ(Genetics Computer Group,University of Wisconsin Biotechnology Center,1710 University Avenue,Madison,WI 53705)の配列分析用ソフトウェア・パッケージ(Sequence
Analysis Software Package))。最大限の相同(すなわち同一性)を得るために、類似のアミノ酸配列を整列させる。この目的のために、人工的に隙間を配列に導入することが必要と考えられる。一旦最適なアラインメントが設定されれば、相同(すなわち同一性)の度合いは位置の全てを記録することによって確立され、全位置数に対して、両方の配列のアミノ酸は同一である。
【0035】
類似性因子として、同程度の大きさ、形状、および電荷が挙げられる。アミノ酸類似度を決定する特に好ましい方法の一つは、ダイホフ他(Dayhoff et al.,5
ATLAS OF PROTEIN SEQUENCE AND STRUCTURE
345−352(1978 & Suppl.))(本明細書中に援用)で述べられるPAM250マトリックスである。類似度のスコアは、整列配置された対合アミノ酸類似度のスコアの合計として、最初に算出される。挿入および欠失は、相同率および同一率の目的にとって無視される。したがって、ギャップ・ペナルティはこの算出で使われない。生スコアを、候補化合物と参照配列のスコアの幾何平均によって分ることで正規化する。幾何平均は、これらのスコアの製品の二乗根である。正規化された生スコアは、相同率である。
【0036】
好ましくは、相同配列は参照アミノ酸配列に対して少なくとも45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、または95%同一である。この明細書(例えば、表3)に示す配列の一つに対して配列相同性を持つポリペプチドとして、天然の対立遺伝子変異体が挙げられ、同様に、機能に関して類似している突然変異体および変異体または任意の他の非天然の変異体(例えば、ペプチド模倣薬)が挙げられる。
【0037】
この出願は、「Gタンパク質結合受容体アゴニストおよびアンタゴニストならびにそれを用いたGタンパク質結合受容体の活性化および阻害」と題された同時係属米国特許出願第10/251,703号(この特許出願の内容全体を本明細書中に援用する)に関連している。
【0038】
本発明は、GPCRi1ループから誘導されるペプチドに結合した細胞貫通または係留部分を含むペプデュシンと呼ばれる改質ペプチドの発見に基づく。ペプデュシンはキメラのペプチド/ポリペチドと考えられ、受容体−Gタンパク質シグナル伝達のアゴニストおよびアンタゴニストもしくはその一方であって、それらの同族の受容体に対して選択性を示す。
【0039】
本発明のペプデュシンは、GPCRの第1の細胞内ループ(i1)またはそのフラグメントに由来するGPCR部分と、標的細胞の脂質二重層内またはそれを横断して複合体を区分けする細胞貫通または膜係留部分とを含み、また本発明のペプデュシンをGPCR媒介症状の処置に用いる方法を含む。細胞膜透過性部分は、例えば、GPCRフラグメント自体の疎水性部分である。細胞貫通または膜係留部分は脂質二重層(または細胞表面)で複合体を係留することで、細胞内受容体の近傍(例えば、受容体−Gタンパク質界面)で複合体の有効なモル濃度を上昇させる。ペプデュシンの外因性GPCR部分は、受容体−Gタンパク質相互作用を崩壊させて、シグナル伝達活性化および阻害(すなわちアゴニストまたはアンタゴニスト活性)もしくはその一方を生じる。
【0040】
ペプデュシンは、受容体の細胞内表面を目標とすることによって、受容体修飾剤として働く。例えば、本発明のペプデュシンは、生体内条件下での抗うっ血作用および抗血栓作用に対するPARlおよびPAR4ベースのアンタゴニストを含む。トロンビンが血小板で最も強力な賦活体であるので、標的としてPARl(Vu et al.Cell 64,1057(1991))およびPAR4(Xu et al.Proc.Natl.Acad.Sci.(USA)95,6642(1998);Covic et al.Biochemistry 39,5458(2000)および Covic et al.Thromb.Haemost.87,722(2002))が選択された。これらの2つの受容体のアンタゴニストは、敗血症を含む急性冠動脈症候群の血栓および増殖合併症を妨げるために役立つ。
【0041】
長期間に及ぶ虚血後に血流が回復する際に、虚血再灌流障害が起こる。それは、心筋梗塞、脳卒中、腸管虚血、心肺バイパス等の状態での羅病率および死亡率の一般的な原因であることから、しばしばそれに対する特異的療法がみあたらない。本発明のペプデュシンを静注投与することで、心筋の実験的再灌流障害に対する有意な保護が得られる。
【0042】
心筋に対する動脈血の供給が限られていることによって促される心筋梗塞は、心不全および死亡の主因である。したがって、正常な血行の早急な回復は、酸素圧低下から心臓組織の不可逆的損傷の予防のために重要である。正常な血流の再建は、特定薬剤を有する心筋梗塞(MI)患者の処置後、自然または人工的に起こり得る。しかし、多くの場合、梗塞部の再灌流は、心臓細胞の損害を促進し、それによって回復する血行の陽性作用を減らす炎症反応を引き起こす。有意に再潅流された組織の低酸素領域に補充される白血球がこの病理学的炎症反応に関与することが証明された。
【0043】
臓器移植は、2001年には米国で22,000件以上行われており、現在一般的であり、最も一般的に移植された臓器は腎臓、肝臓、および心臓である。現在実施されている多数の移植体は、ある程度は、急性拒絶を制御する免疫調節性剤の能力に依拠している。遅延移植機能(虚血−再灌流障害に起因する)と宿主対移植反応とは、移植の後の急性拒絶の主要な機序である。免疫抑制剤の使用は、急性拒絶に対して成功裏に対処しており、免疫抑制剤投薬量が非常に低レベルになる場合であっても、一般に異型移植が長期わたって生存する。
【0044】
しかし、免疫抑制剤はすべての免疫反応を抑制することから、激しい感染症が移植レシピエントの主要な死因となる。免疫抑制剤は、重症毒性とも関係している。移植患者のために使用される薬物で最も有意な合併症として、腎毒性、神経毒性、新規発症移植後真正糖尿病、高脂血症、および高血圧が挙げられる。これらの副作用は、現在使用される免疫抑制剤の分子標的が普遍的に発現することから、部分的に生ずる。本発明のペプデュシンを含む治療的な戦略は、特に虚血−再灌流障害と単核細胞のその後の浸潤(急性拒絶の主因)を対象とすることが可能である。
【0045】
重篤な敗血症は急性の入院の主要な原因であって、しばしば、他の疾患の処置がおこなわれている患者の臨床経過を難しくする。敗血症発症の際、細菌の存在とエンドトキシン等の細菌産生物とが免疫防衛機制を刺激する。侵入病原体の除去に失敗すると、全身性炎症反応症候群(SIRS)と称され、かつサイトカインおよびケモカインによってもたらされる機能亢進性炎症反応が開始される。ケモカインを、炎症の治療標的として調べる。CXCRlおよびCXCR2は、好中球、内皮、上皮、マクロファージ、および他の細胞の活性化にかかわる重要なケモカイン受容体である。唯一のIL−8受容体であるCXCR2の遺伝子削除によってIL−8シグナル伝達が欠けているマウスは、敗血症の発現から保護される。
【0046】
ヒトで、全身性IL−8のレベルが高いと、内皮細胞機能不全と内皮の正常な抗凝固状態とが失われる。凝固障害は敗血患者の30ないし50%に起こり、明白な播種性血管内凝固(DIC)の発生は不吉な予後徴候である。CXCRl/2の作用に対抗して、CXCR4は老好中球の除去をもたらし、骨髄で未成熟な白血球を保持する。CXCR4もリンパ球および癌細胞のホーミング機構において顕著な役割を果たすが、急性炎症におけるCXCR4およびそのリガンドSDF−lαの機能は謎のままである。
【0047】
抗生物質の投与は別として、敗血症および敗血症性ショックの処置は、主に、支えとなる戦略に限られている。CXCRl/2に特異的である低分子阻害剤が再灌流障害および肺損傷に対する保護をおこなうことができるけれども、敗血症の処置に利用可能なケモカイン受容体に対して特異的な治療的介入は、いまのところ存在しない。本発明のペプデュシンを、確立された全身性炎症および血管障害の抑制に用いることができ、同様に宿主防御に対する干渉なしで、凝固カスケードの活性化を妨げる。
【0048】
ヒトPARとして、PAR1(Genbankアクセッション番号AF019616)、PAR2(Genbankアクセッション番号XM_003671)、PAR3(Genbankアクセッション番号 NM_004101)、およびPAR4(Genbankアクセッション番号NM_003950.1)が挙げられ、これらの配列を本明細書中で援用する。
【0049】
ペプデュシンが受容体−Gタンパク質間シグナル伝達の活性化および阻害をおこなう二部位機構は、本明細書中に述べられているが、本発明者は、本発明をこの機構またはその根拠となる理論によって制限されるとは意図していない。この機構は、アゴニストの二相活性化および抑制とアンタゴニストの抑制とに適応する。ペプデュシンは、その疎水性係留によって、急速に形質膜を変換して、周囲膜質界面で有効性の高いモル濃度を成し遂げる。ペプデュシン・アゴニストは、GPCRの細胞内表面で、最初に高親和性部位を占める。結合したアゴニストは結合させられたGタンパク質をオンにするために受容体の活性化状態を安定化または誘導する。この第1の部位が飽和したあと、より高濃度のペプデュシンが親和性のより低い第2の阻害部位を占め始める。この第2の阻害部位は、優位なかたちでGタンパク質に対するシグナル転移を、おそらくGタンパク質でGPCR(例えば、受容体i1ループ)基底状態相互作用を模倣することによって、遮断する。ペプデュシン・アンタゴニストによる阻害は、アゴニストの阻害相と一致することから、アンタゴニストはこの低親和性部位で結合もすると思われる。ペプデュシンによる受容体の外因性活性化または阻害は、潜在的二量体化様式を反映することができ、それによって1つの受容体がその細胞内ループを近隣の受容体に提供する。異なったシグナル伝達性質(Milligan,Science 288,65−67(2000))を生ずる受容体ダイマーに関するいくつかの実施例があり、EPO受容体等のサイトカイン/GPCRが含まれるが(Guillard et al.,J.Biol.Chem.(2001)276,2007−2013)、受容体間変調機構は知られていない。
【0050】
天然のGPCRは、少なくとも一回細胞膜を越える細胞表面モジュールである。例えば、多くの天然のGPCRは7回細胞膜を越えて、いくつかの細胞内領域を含む。本発明の単離GPCRフラグメントは、GPCRの第1の細胞内ループ領域を含む。細胞内部分は、サイトカインGPCRの第1の細胞内ループ領域の1−膜貫通型領域Gタンパク質結合受容体またはそのフラグメント、あるいは、マルチ・ポリペチド−GPCRの第1の細胞内ループ領域(例えば、GPIb/v/IX受容体またはコラーゲン受容体)から選択される。
【0051】
本発明は、可溶性ペプデュシンも包含するもので、第2の領域(細胞貫通または膜係留部分)は、任意に、細胞外または細胞内フラグメントに隣接した膜貫通型領域から自然に生じる隣接アミノ酸を含む。例えば、コンストラクトは、GPCR膜貫通型ヘリクス領域の隣接アミノ酸を少なくとも3個(しかし16個未満)含むものであってもよい。膜貫通型領域は、CXCR4の膜貫通型領域1〜7、CCKA受容体の膜貫通型領域1〜7、またはCCR5受容体の膜貫通型2でない。実施形態において、第2の領域は細胞外または細胞内フラグメントに直隣接して天然の膜貫通型ヘリクス領域の1〜15の隣接アミノ酸を含む。
【0052】
ペプチド・ベースのペプデュシンに加えて、発明はGPCRフラグメントがペプチド模倣薬を含む組成物を含む。例えば、本発明は一つ以上のペプチド結合が他のタイプの共有結合に置き換えられたペプデュシン化合物を含み、その結合はペプチダーゼ(「ペプチド・ミメティック」または「ペプチド模倣薬」)による裂開の影響を受けにくい。被験者への注射後のペプチドのタンパク質分解が問題である一方で、非切断型ペプチド・ミメティックによる特に感受性が高いペプチド結合の置換は結果として生じるペプチドをより安定にすることから、より役立つ治療法である。このミメティックとそれらをペプチドに組み込む方法は、周知の技術である。同様に、Lアミノ酸残基の置換(例えば、D−アミノ酸による)は、プロテオリシスによるペプチドへの影響をより少なくする。
【0053】
本発明のペプデュシン化合物を逆配列およびキラリティーのペプチドを含むレトロ・インベルソ異性体として合成することができる。例えば、Jameson et al.Nature 368:744−746(1994)and Brady et al.Nature 368:692−693(1994)を参照せよ。D−エナンチオマーと逆合成との組み合わせによる正味の結果は、各アミド結合でのカルボニル基の位置とアミノ基との位置が交換され、その一方で各α炭素の側鎖基の位置が保存されるということである。例えば、ペプチド・モデルが配列ABCのLアミノ酸から形成されるペプチドである場合、D−アミノ酸から形成されるレトロ・インベルソ・ペプチドの類似体は配列CBAを持つと思われる。レトロ・インベルソ・ペプチドを形成するためにD−アミノ酸の連鎖を合成するための手法は、当該技術分野で知られている。
【0054】
また、有用なアミノ末端ブロック基として、t−ブチロキシカルボニル、アセチル、セイル、サクシニル、メトキシ・サクシニル、スべリル、アジピル、アゼライル、ダンシル、ベンジルオキシカルボニル、フルオレニルメトキシカルボニル、メトキシアゼライル、メトキシアジピル基、メトキシスベリル、および2,4−ジニトロフェニルが挙げられる。ペプチドの荷電アミノ末端およびカルボキシ末端を阻害することは、疎水性細胞膜を介し、かつ細胞内に向けたペプチドの通過を高めるという付加的な利益がある。
【0055】
単離されたGPCRフラグメントは、クラスAのGPCRまたはクラスBのGPCRの配列から誘導してもよい。単離されたGPCRフラグメントは、任意の既知または未知のGPCR由来のフラグメントであり、限定されるものではないが、プロテアーゼ活性化受容体(PAR、例えばトロンビン受容器)、黄体形成ホルモン受容体、卵胞刺激ホルモン受容体、甲状腺刺激ホルモン受容体、カルシトニン受容体、グルカゴン受容器、グルカゴン様ペプチド1受容体(GLP−1)、向代謝性グルタミン酸受容体、副甲状腺ホルモン受容体、脈管活性腸幹ペプチド(VIP)受容体、セクレチン受容体、成長ホルモン放出因子(GRF)受容体、コレシストキニン受容器、ソマトスタチン受容体、メラノコルチン受容体、ヌクレオチド(例えば、ADP受容体)、アデノシンレセプター、トロンボキサン受容体、血小板活性化因子受容体、アドレナリン作動受容器、5−ヒドロキシトリプタミン(5−HT)受容体、ケモカイン受容体(例えば、CXCR4、CCR5)、神経ペプチド受容体、オピオイド受容体、エリスロポエチン受容体、および副甲状腺ホルモン(PTH)受容体が挙げられる。
【0056】
好ましい実施形態において、GPCRはプロテアーゼ活性化受容体、ペプチド受容体、またはヌクレオチド受容体である。特定の実施形態において、GPCRはPAR1、PAR2、PAR3、またはPAR4受容体である。他の実施形態において、GPCRはグルカゴン様受容体、ヌクレオチド受容体、例えばP2Y−12ADP受容体、MC4肥満受容体、CXCR受容体(例えば、CXCR4)もしくはCCR5ケモカイン受容体、CCKA、またはCCKBである。
【0057】
単離されたGPCRフラグメントは、GPCR由来の少なくとも50未満の隣接アミノ酸であり、GPCRの固有の細胞外リガンドを含まない。例えば、このフラグメントはGPCRの3ないし30の隣接アミノ酸を含むものであってもよい。好ましい実施形態において、GPCRフラグメントは7と24との間(7および24を含む)の隣接アミノ酸であるGPCRフラグメントを含む。例えば、フラグメントは、GPCRの7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23または24の隣接アミノ酸を含む。
【0058】
任意には、GPCRのアミノ酸配列は、天然のアミノ酸配列と異なる。例えば、所定の領域(例えば、膜貫通型ヘリクス・ループ、細胞外ループ、または細胞内ループ)由来の個々の残基を突然変異または修飾アミノ酸に置換によってペプデュシンの活性を改善する。好ましくは、そのようなGPCR類似体のアミノ酸配列は、保存的アミノ酸置換(すなわち同一クラスの他方のものに対する一方のアミノ酸の置換によって、またはタンパク質の機能を破壊しない場所に位置した非保存的置換、欠失、または挿入)によって単独で異なる。
【0059】
本発明のペプデュシンのGPCR部分は、ヒトまたは他の生物(例えば、モルモット、ラット、マウス、チキン、ウサギ、ブタ、ヒツジ、牛、サル、ウイルス、真菌、昆虫、植物、バクテリア、その他)の任意の細胞に由来するもので、このような細胞の例として、脾細胞、神経細胞、グリア細胞、膵ベータ細胞、骨髄細胞、糸球体間質細胞、ランゲルハンス細胞、表皮細胞、上皮細胞、血管内皮細胞、フィブロプラスト、フィブロサイト、筋細胞、脂肪細胞、免疫細胞(例えば、マクロファージ、T細胞、B細胞、ナチュラルキラー細胞、マストセル、好中球、好塩基、好酸球、単核細胞、その他)、巨核球、滑膜細胞、軟骨細胞、骨細胞、骨芽細胞、破骨細胞、乳房腺細胞、肝細胞または間質細胞、あるいはそれらの前駆体細胞、幹細胞等、または癌細胞等、さらにそのような細胞を含んでいる任意の組織、例えば脳、脳の種々の部分(例えば、嗅球、扁桃体、脳幹神経節、海馬、視床、視床下部、視索上核、大脳皮質、延髄、小脳、後頭極、前頭葉、果核、尾状核、脳梁、黒質)、脊髄、下垂体、胃、膵臓、腎臓、肝臓、生殖器、甲状腺、胆嚢、骨髄、副腎、皮膚、筋肉、肺、消化管、血管、心臓、胸腺、脾臓、顎下腺、末梢血液、末梢血白血球、腸管、前立腺、睾丸、精巣、卵巣、胎盤、子宮、骨、関節、小腸、大腸、骨格筋等、特に脳および脳の種々の部分が挙げられる。
【0060】
細胞膜透過性部分は、脂質、コレステロール、リン脂質、ステロイド、スフィンゴシン、セラミドまたは脂肪酸部分を含む。脂肪酸部分は、例えば、少なくとも8つの炭素を含む任意の脂肪酸である。例えば、脂肪酸を、例えば、ノナノイル(C9)、カプリル(C10)、ウンデカノイル(C11)、ラウロイル(C12)、トリデカノイル(C13)、ミリストイル(C14)、ペンタデカノイル(C15)、パルミトイル(C16)、フィタノイル(メチル置換C16)、ヘプタデカノイル(C17)、ステアロイル(C18)、ノナデカノイル(C19)、アラキドイル(C20)、ヘニエコサノイル(C21)、ベヘノイル(C22)、トルシサノイル(C23)、またはリグノセロイル(C24)部分とすることができる。細胞膜透過性部分は、オクチル−グリシン、2−シクロヘキシルアラニンまたはベンゾリルフェニルアラニンのマルティマー(例えば、一つ以上の単位を含んでいる組成物)を含むこともできる。細胞膜透過性部分は、非置換またはハロゲン置換(例えば、クロロ)ビフェニル部分を含む。非置換されたビフェニルによる化合物と比較すると、置換されたビフェニルは、生体組織で蓄積の減少を伴う。被験者に対する投与後の身体組織での蓄積減少は、被験者における副作用の減少を伴う。
【0061】
好ましくは、細胞膜透過性部分は天然または非天然のパルミトイル部分である。C15よりも長い脂肪酸(例えば、パルミトイル)部分を持つペプデュシンは、生体内で驚くほど長命であるように思われ、また皮下適用に適していると思われる。別の実施形態において、C15よりも短い部分(例えば、ミリストイル)を有するペプデュシンは、短期間の適用(例えば、外科適用)に適しているように思われ、ここでは静脈内投与が有用である。
【0062】
細胞貫通または膜係留部分は、GPCRフラグメントのC末端アミノ酸、N末端アミノ酸、またはN末端とC末端アミノ酸との間のアミノ酸に結合するものであってもよい。
【0063】
また、本発明の範囲内にあるものは、細胞貫通または膜係留部分に連結する配列番号1〜14のアミノ酸配列を持つポリペチドまたはその一部分を含む組成物である。本発明の特に適したペプデュシンを、以下の表1に示す。
【0064】
【表1】

組成物は、異常なGPCR活性によって特徴づけられる疾患および状態を伴う一つ以上症状(重篤度を減らす)を処置、予防、または改善するのに用いられる。そのような疾患および状態として、血栓症、心臓発作、脳卒中、過剰な出血、喘息、炎症、疼痛、炎症性疼痛、内臓痛、神経性疼痛、関節炎、糖尿病、HIV感染症、不安神経症、鬱病、肺動脈弁閉鎖不全症、および種々の癌が挙げられる。この方法は、ペプデュシンGPCRアンタゴニストに細胞(病理学的にGPCRを過剰発現させる)を接触させることによって実行される。例えば、方法は被検体(例えば、ヒト患者)に投与することが必要であり、被験者で病理学の重篤度を減らすのに十分な量のペプデュシンを処置または予防に必要とされる。本発明は、ペプデュシン組成物および製薬的に許容される担体のいずれかを有する医薬組成物も含む。本発明はまた、医薬組成物を含有するキットも含む。本発明は、本発明のいずれかのポリペチドの投与を通して哺乳動物における病理学的状態を処置する方法をさらに含む。
【0065】
コンストラクトは、腫瘍増殖および移動を阻害するのにも用いられる。乳癌細胞浸潤は、複雑なプロセスであり、細胞移動、組織基底膜のタンパク分解性修飾、およびマトリックス・メタロプロテアーゼ(MMP)による細胞外マトリックスの低下が起こる。MMPlによるPARlの活性化は、乳癌細胞の浸潤と腫瘍形成において重要な役割を果たす(Boire et al.Cell,120(3),303(2005))。PAR1i1ループ・ペプデュシン・アンタゴニスト(例えば、Plilpal11)が、単独で、またはアジュバント癌治療(例えば、タキソテール(登録商標)等のドセタキセル化合物とともに投与される場合)として使われる。好ましくは、相乗的抗腫瘍効果が達成される。PAR1の細胞内阻害の作用は、転移性ヒト乳癌細胞株MDA−MB−231を用いることで示される。単独または併用療法(例えば、タキソテールと共に)におけるペプデュシン有効性を、MTTアッセイおよび異種移植片マウス・モデルを用いて分析する。組成物が別々または同時に投与されたあと、ペプデュシンPlilpal11およびタキソテールのIC50を測定する。アイソボログラム(isoborogram)技術と組み合わせインデックス(CI)法を用いて、相乗効果の程度を定量化する。異種移植片データは、腫瘍成長速度がタキソテールだけで処置されたマウスと比較した際に、同時にPlilpal11およびタキソテールで処置されるマウスで抑制されたかどうか決定するのに用いられる。この化合物は、全ての移動が腫瘍成長(例えば、腫瘍成長の後期段階と同様に腫瘍成長の初期)の病期分類で腫瘍増殖を低下させる。
【0066】
例えば、図27に示すように、CXCRl/2(xl/2pal−i1)に基づくI1−ループ・ペプデュシンおよびCXCR4(x4pal−i1)ケモカイン受容体は、胸部と卵巣癌の化学走性の移動を妨げる。
【0067】
発明の特定のペプデュシンは、血小板活性化阻害剤である。阻害剤は、GPCRポリペチドに連結する細胞膜透過性部分連結とプロテアーゼ活性化受容体の単離フラグメントとを含む。一部の実施形態において、プロテアーゼ活性化受容体はトロンビン受容器、トリプシン受容体、凝固因子Xa受容体、活性プロテインC受容体、トリプターゼ受容体、またはプラスミン受容体である。トロンビン受容器は、好ましくはPAR−4またはPAR−1である。
【0068】
本発明は、先に述べたように細胞膜透過性部分に連結したプロテアーゼ活性化受容体の単離フラグメントの組成物で血小板を接触させることによって血小板凝集を阻害する方法も含む。例えば、プロテアーゼ活性化受容体はトロンビン受容体(例えば、PAR−1受容体またはPAR−4受容体)である。また、本発明の範囲内にあるものは、細胞膜透過性部分に連結したトロンビンの単離されたフラグメントを含む本発明の組成物を哺乳動物投与することによってこの哺乳動物で血栓形成を阻害する方法である。
【0069】
本発明の方法は、血管内腔(例えば、頸静脈、末梢静脈または血管周囲隙)に発明の抑制性組成物を注入することによって実行される。末梢静脈は、例えば、端(例えば、手、手首または足)にある静脈である。いくつかの実施形態では、組成物を、例えば噴霧質として哺乳動物の肺に注入する。他の実施形態では、本発明の組成物を注射によって投与する。種々の実施形態において、哺乳類動物の腹腔内、真皮下または皮下に注射することができる。本発明の組成物を経皮的に投与することもできる。他の実施形態では、本発明の組成物を経膣投与または直腸投与する。本発明の組成物が被覆または注入された坐剤または充填材料を損傷部分に移植することによって、投与することができる。
【0070】
凝血塊構造または血小板凝集の阻害剤を、医療器具に用いることができる(例えば、コーティングとして)。例えば、脈管内人工装置(例えば、スクリーン、ステントまたはカテーテル)は細胞膜透過性部分に連結したトロンビンの単離されたフラグメントである血栓形成の阻害剤を含む。組成物を装置内に含浸させ、組織との接触または装置の移植に応じて身体の組織に広まる。あるいは、装置をペプデュシンで覆う。
【0071】
ペプデュシンは、細胞膜透過性部分に連結したプロテアーゼ活性化受容体の単離フラグメントと腫瘍細胞とを接触させることによって腫瘍細胞の移動および浸潤を阻害するのにも用いられる。プロテアーゼ活性化受容体は、PAR−4、PAR−2、またはPAR−1受容体である。腫瘍細胞の転移を阻害する方法は、細胞膜透過性部分に連結したプロテアーゼ活性化受容体の単離フラグメントと腫瘍細胞とを接触させることによって実行される。腫瘍細胞は、メラノーマ細胞、乳癌細胞、腎臓癌細胞、前立腺癌細胞、肺癌細胞、大腸癌細胞、中枢神経系(CNS)癌細胞、肝癌細胞、胃癌細胞、肉腫細胞、白血病細胞、またはリンパ腫細胞である。
【0072】
喘息症状の軽減は、トロンビンまたはトリプシン/トリプターゼGPCR系ペプデュシンの投与によってなされる、したがって、喘息抑制方法は細胞膜透過性部分にトロンビンまたはトリプシン/トリプターゼGPCR連結の単離されたフラグメントを含んでいる組成物を投与することによって実施される。好ましくは、トリプシン/トリプターゼ受容体は、PAR−1、PAR−2またはPAR−4受容体である。種々の実施形態において、組成物を血管内腔(例えば、末梢静脈)に注入、哺乳動物の肺に注入(例えば、吸入(例えば、煙霧質として)、または経皮経路によって投与する。
【0073】
血小板活性化の阻害剤は、細胞膜透過性部分に連結したP2Y12受容体等のヌクレオチド活性GPCRの単離フラグメントを含む。この組成物は、血小板凝集を阻害する方法に役立つ。
【0074】
さらにもう一つの態様において、本発明は哺乳動物に対して細胞膜透過性部分に連結したヌクレオチド活性受容体の単離フラグメントを含む組成物を投与することによって、哺乳動物で血栓形成を阻害する方法を含む。いくつかの実施形態では、トロンビン受容体はP2Y12受容体である。血管内腔(例えば、頸静脈、末梢静脈)に本発明の阻害組成物を注入することによって、方法を実施することができる。末梢静脈は、例えば、四肢(例えば、手、手首または足)にある静脈であってもよい。いくつかの実施形態では、組成物を例えば煙霧質として哺乳動物の肺に注入する。他の実施形態では、発明の組成物を、注射によって投与する。種々の実施形態において、注射が哺乳動物(真皮下または皮下)の腹腔にあることができる。他の実施形態において、発明の組成物は、経皮的に投与される。種々の実施形態では、哺乳類動物の腹腔内に真皮下または皮下注射することができる。本発明の組成物を経皮投与することもできる。組成物の投与は、本発明の組成物をコーティングまたは含浸させた創傷充填材または坐剤を埋め込むことによっておこなうことができる。
【0075】
別の態様では、本発明は血栓形成の阻害剤(細胞膜透過性部分と連結したヌクレオチド受容体の単離フラグメント)を含む脈管内人工装置を包含する。種々の実施形態では、この装置は、例えばステントまたはカテーテルである。いくつかの実施形態では、この装置に阻害剤を被覆または含浸させる。
【0076】
本発明の一つ以上の実施形態の詳細を、下記の付随的説明で述べる。本明細書中に述べられる方法および材料と類似または等価な任意の方法および材料を本発明の実施または試験で使用することができるが、好ましい方法および材料をここで説明する。本発明の他の特徴、目的、および長所は、説明から明らかである。本明細書と添付の特許請求の範囲において、文脈をはっきりと述べないかぎり、単数形も複数を含む。定義されない限り、本明細書中に使用されるすべての専門用語および科学用語は、本発明が属する当該技術分野の当業者が理解する意味と同じ意味を持つ。不一致の場合、定義を含む本明細書を規制する。また、加えて、材料、方法、および実施例は、限定することを意図したものではなく、説明を目的とするものである。この明細書で引用されるすべての特許および刊行物は、参照によって本明細書中に組み込まれる。
【0077】
Gタンパク質結合受容体
Gタンパク質結合受容体は、複数の受容体からなる大きなスーパーファミリーを含む内因性膜タンパク質である。Gタンパク質結合受容体(GPCR)のファミリーは、少なくとも250のメンバーを有する(Strader et al.FASEB J.,9:745−754,1995;Strader et al.Annu.Rev.Biochem.,63:101−32,1994)。1パーセントのヒト遺伝子がGPCRをコードすると推定されている。多くのGPCRは、一般の分子構築と一般のシグナル伝達機構とを共有する。歴史的に、GPCRは、本来、無関係であると思われた6つのファミリーに分類され、それのうちの3つは脊椎動物で見つかっている。最近の研究では、いくつかの新規のGCPRファミリーが確認され、それらの全てについて進化上の起源が共通する可能性があることを示唆している。
【0078】
多くのGPCRは、7つの膜貫通型ヘリカル領域の一般の構造モチーフを共有する。しかし、いくつかのGPCRは、7つの膜貫通型ヘリカル領域を持たず、その代わりに単スパン膜貫通型受容体である。
【0079】
単スパンGPCRは、サイトカイン(例えば、エリスロポエチン、EGF、インシュリン、インスリン様増殖因子IおよびII、ならびにTGF)のために、受容体を含む。
【0080】
GPCRファミリーとして、クラスAロドプシン様、クラスBセクレチン様、クラスC代謝調節型グルタメート/フェロモン、クラスD真菌フェロモン、クラスEcAMP受容体(タマホコリカビ(Dictyostelium))、および縮れた/平滑化されたファミリーが挙げられる。推定上のファミリーは、眼白子症タンパク質、ショウジョウバエ・ニオイ物質受容体、植物Mlo受容体、ネマトーダ化学受容体、鋤鼻受容体(V1R及びV3R)を含む。
【0081】
クラスAロドプシン様受容体は以下を含む:アミン受容器:アセチルコリン、αアドレナリン受容体、βアドレナリン受容体、ドーパミン、ヒスタミン、セロトニン、オクトパミン、および微量アミン;ペプチド受容体:アンギオテンシン、ボンベシン、ブラジキニン、C5aアナフィラトキシン、Fmet−leu−phe、APJ様、インターロイキン‐8、ケモカイン受容体(C−Cケモカイン、C−X−Cケモカイン、ボンゾー受容体(CXC6R)、C−X3−Cケモカイン、およびXCケモカイン)、CCK受容体、エンドセリン受容体、メラノコルチン受容体、ニューロペプチドY受容体、ニューロテンシン受容器、オピオイド受容体、ソマトスタチン受容体、タキキニン受容体、(P物質(NKl)、サブスタンスK(NK2)、ニューロメジンK(NK3)、タヒキニン様1およびタヒキニン様2)、バソプレシン様受容体(バソプレシン、オキシトシン、およびコノプレシン)、ガラニン様受容体(ガラニン、アラトスタチン、およびGPCR54)、プロテイナーゼ活性化様受容体(例えば、トロンビン)、オレキシン及び神経ペプチドFF、ウロテンシンII受容体、アドレノメデュリン(Gl0D)受容体、GPR37/エンドセリンB様受容体、ケモカイン受容体様受容体、およびニューロメジンU受容体、ホルモンタンパク質受容体:卵胞刺激ホルモン、ルトロピン−絨毛膜性腺刺激性ホルモン、チロトロピンとゴナドトロピン;(Rhod)オプシン受容体;きゅう覚受容体;プロスタノイド受容体:プロスタグランジン、プロスタサイクリン、およびスロンボキサン;ヌクレオチド様受容体:アデノシンとプリノセプター;大麻受容体;血小板活性化因子受容体;ゴナドトロピン放出ホルモン受容体;甲状腺刺激ホルモン放出ホルモンおよび分泌促進受容体:甲状腺刺激ホルモン放出ホルモン、成長ホルモン分泌促進物質および成長ホルモン分泌促進物質同類;メラトニン受容体;ウイルス受容器;リゾスフィンゴ脂質およびLPA(EDG)受容体;ロイコトリエンB4受容体:ロイコトリエンB4受容体BLTlおよびロイコトリエンB4受容体BLT2;ならびにクラスA オーファン/他受容体:血小板ADPおよびKI01受容体、SREB、マス・プロトオンコジーン、RDCl、ORPH、LGR様(ホルモン受容体)、GPR、GPR45様、システイニルロイコトリエン、Mas関連受容体(MRG)、およびGP40受容体。
【0082】
GPCRのクラスB(セクレチン受容体ファミリーまたは「ファミリー2」)は、より小さいが、タンパク質の構造的または機能的に異なった基であり、ポリペプチドホルモン用受容体(カルシトニン、コルチコトロピン放出因子、胃抑制ペプチド、グルカゴン、グルカゴン様ペプチド−1、−2、成長ホルモン放出ホルモン、副甲状腺ホルモン、PACAP、セクレチン、血管作動性腸管ポリペプチド、利尿ホルモン、EMRl、ラトロフィリン)、ストレス応答および長命を調節する形質膜(脳特異的脈管形成阻害剤(BAI))と一群のショウジョウバエ・タンパク質(メトセラ様タンパク質)で細胞間相互作用を媒介すると思わる分子が挙げられる。
【0083】
クラスC代謝調節型グルタメート/フェロモン受容体は、代謝調節型グルタミン酸塩、代謝調節型グルタミン酸塩群I、代謝調節型グルタミン酸塩群II、代謝調節型グルタミン酸塩群III、代謝調節型グルタミン酸塩他、細胞外カルシウムを感知する推定上のフェロモン受容体、GABA−B、GABA−B亜型1、GABA−B亜型2、およびオーファンGPRC5受容体を含む。
【0084】
GPCRは、潜在的にGタンパク質を経てアウトサイド・イン・シグナル伝達示すGPIb−V−IXまたはコラーゲン受容体等のマルチ・ポリペチド受容体である。
【0085】
何百ものGタンパク質結合受容体遺伝子またはcDNAがクローン化されたにもかかわらず、GPCRとまだ認められなかった多くの特徴のないGタンパク質結合受容体がまだあると考えられる。
【0086】
GPCRは、細胞の代謝、細胞増殖および運動性(癒着、炎症、神経シグナル伝達、および血液凝固)を制御するシグナル伝達プロセスにおいて、不可欠な役割を果たす。Gタンパク質結合受容体タンパク質にも、生体の機能を制御、調節、または調整する種々のシグナル伝達分子の標的としての非常に重要な役割がある。シグナル伝達種は、内因性分子(例えば、神経伝達物質またはホルモン類)、外因性分子(例えば、ニオイ物質)または、視覚変換の場合には光である。
【0087】
例えば、GPCRは、生体アミン受容体、例えばドーパミン、エピネフリン、ヒスタミン、グルタミン酸塩(代謝調節型作用)、アセチルコリン(ムスカリン様作用)、およびセロトニンのための受容体と、炎症(例えば、プロスタグランジン、血小板活性化因子、およびロイコトリエン)の脂質性化学伝達物質のための受容体と、ペプチドホルモン(例えば、カルシトニン、C5aアナフィラトキシン、卵胞刺激ホルモン、性腺刺激ホルモン放出ホルモン、ニューロキニン、オキシトシン)のための受容体と、プロテアーゼ(例えば、トロンビン、トリプシン、トリプターゼ、活性プロテインCと因子Vlla/Xa)のための受容体と、感覚信号メディエイタ(例えば、レチナール光色素と嗅覚刺激性の分子)のための受容体とを含む。各分子は受容体タンパク質に特異的であり、個々の生理活性物質(特異的な的状赤血球と臓器を含む)、特異的な薬理作用、特異的な作用長、作用時間、その他に対して特異的である。このように、GPCRは薬物作用および開発の主な目標である。
【0088】
リガンド結合の際に、GPCRはグアニンヌクレオチド結合タンパク質(Gタンパク質)を活性化することによって、細胞内シグナル伝達経路を調節する。GPCRの領域構造は、GPCRファミリーのメンバー間で保存される。TMヘリクス領域、細胞内ループ領域、およびGPCRSの細胞外領域の領域境界は、当該技術分野で知られている。例えば、本明細書中に援用するPalczewski et al.,Science 289:739(2000)に述べられているように、マッピングされていないGPCRの構造は、既知の方法を使用して、原型GPCR(ロドプシン)との比較によって決定される。
【0089】
大部分のGPCRの特徴の1つは、疎水性アミノ酸残基からなる7つのクラスター、すなわち膜貫通型領域(TM、7つの膜貫通型領域を、TMl、TM2、TM3、TM4、TM5、TM6、およびTM7とする)は一次構造内に位置しており、その各領域で細胞膜を貫通する(スパン)(図1)。領域は、3つの細胞内ループ(i1、i2、およびi3)、3つの細胞外ループ(e1、e2、およびe3)、ならびにアミノ(N)−カルボキシル(C)−末端領域によって接続される膜貫通型α−ヘリクスを表すと考えられている(Palczewski et al.,Science 289,739−45(2000))。大部分のGPCRは、機能性タンパク質構造を安定させると考えられているS‐S結合を形成する最初の2つの細胞外ループの各々で、一つの保存されたシステイン残基を持つ。上に詳述されるこれらの構造がGタンパク質結合受容体タンパク質間に共通すること、また領域に対応しているアミノ酸配列(タンパク質は、膜スパニング領域に近い膜(膜スパニング領域または膜貫通型領域)およびアミノ酸配列を通過する)は、受容体の間でしばしば高度に保存されることは、周知である。このように、GPCRの相同が高度であるため、新しいGPCRの同定は、そのような新しいメンバーの細胞内および細胞外部分の同定と同様に、当業者によって容易に達成される。
【0090】
Gタンパク質結合受容体の小さなリガンドに対する結合部は、いくつかのGPCR膜貫通型領域によって形成される細胞外表面の近傍に位置する親水性ソケットを含むと考えられている。親水性ソケットは、Gタンパク質結合受容体の疎水性残基によって囲まれる。各Gタンパク質結合受容体膜貫通型耳輪の親水性側は、内部に向かって、極性結合部を形成するために要求される。TM3は、TM3アスパルテート残基を含むリガンド結合部位があることから、いくつかのGPCRに関係した。複数のTM5セリン、TM6アスパラギン、およびTM6またはTM7フェニルアラニンあるいはチロシンもまた、リガンド結合に関係している。ペプチドホルモン受容体のためのリガンド結合部位と、糖タンパク質(例えば、黄体形成ホルモン、卵胞刺激ホルモン、ヒト絨毛性ゴナドトロピン、甲状腺刺激ホルモン(チロトロピン))等の他のより大きなリガンドによる受容体と、受容体のCa2+/グルタミン酸塩/GABA(γ‐アミノ酪酸)クラスとは、おそらく細胞外領域およびループにある。
【0091】
不活性から活性受容体への転換のための鍵となるイベントは、GPCRの膜貫通型ヘリクス3(TM3)および6(TM6)のリガンドによって誘発された構造変化であり、7つの膜貫通型スパン・ヘリクスを有する(Gether and Kolbilka,J.Biol.Chem.273,17979− 17982(1998))。これらのらせん状の動きは、付随するヘテロ三量体のGタンパク質の活性化を促進するために、次々に受容体の細胞内ループのコンフォメーションを変える。突然変異生成に関する研究(Cotecchia et al.,J.Biol.Chem.267:1633−1639(1992);Kostenis et al.,Biochemistry 36:1487−1495(1997);Kjelsberg et al.,J.Biol.Chem.267:1430−1433(1992))、第3の細胞内ループ(i3)が受容体とGタンパク質との間に継手の大部分を媒介することを証明した。ミニ遺伝子が直接アドレナリン作動性受容体とGq結合とが争うことも示されたように、13ループが発現された(Luttrel et al.,Science 259:1453− 1457(1993))、あるいは、無細胞条件(Okamoto et al.,Cell
67,723−730(1991))の可溶性ペプチドとして、Gタンパク質を活性化することができる。
【0092】
GPCRの1つの特定のクラスは、プロテアーゼ活性化受容体(PAR)である。プロテアーゼ活性化受容体(PAR)は、細胞外セリンプロテアーゼのタンパク分解性活性によって細胞シグナル伝達を開始するGタンパク質結合受容体スーパーファミリーのメンバーである。PARは、トロンビン(PAR−1、−3、および−4)とトリプシン/トリプターゼ(PAR−2および−4)を含む内因性プロテアーゼによって、受容体のアミノ末端のタンパク分解性裂開の後、活性化される。これらのうち、PAR2(Nystedt et al.,Proc.Natl.Acad.Sci.(USA)91:9208−9212(1994))は、炎症と疼痛で重要であるトリプシン/トリプターゼ活性化受容体であり、PAR4(Xu et al.,Proc.Natl.Acad.Sci.(USA)95:6642−6646(1998);Kahn et al.,Nature(London)394:690−694(1998))は、第2のトロンビン受容体であり、血小板凝集で固有の役割を演ずる(Covic et al.,Biochemistry 39,5458−5467(2000))。
【0093】
トロンビン、トリプシン、およびトリプターゼが炎症を起こした気道に存在することから、PARはおそらく気道炎症で主な役割を果たすことになっている。Knight et al.,J.Allergy Clin.Immunol.108:797−803(2001)。
【0094】
止血でのその重要な役割に加えて、トロンビンは特異的な細胞表面レセプター(PAR)のタンパク分解性開裂を経て血小板および血管平滑筋細胞等の種々の細胞型を活性化する。なお、原型はPAR−1である。トロンビン受容体活性化は、おそらく、心臓血管疾患(例えば、血栓症、アテローム性動脈硬化と再狭窄)で重要な役割を演じることから、トロンビン受容体アンタゴニストには、これらの障害の処置で、潜在的有用性がなければならない。Chackalamannil,Curr.Opin.Drug Discov.Devel.4:417−27(2001)。
【0095】
トロンビンは、哺乳類の中枢神経系(CNS)に対する障害の後、機能的な損失に関係していると考えられる。PAR−1のダウンレギュレーションはCNA神経の外傷後生存を増加させることが示され、またトロンビンの外傷後毒性はPAR−1受容体の適当なモジュレーションによって調節されるダウンレギュレーションであると考えられる。Friedmann et al.,Neuroimmunol.,121:12−21(2001)。
【0096】
PARSは、種々の癌、細胞増殖、および疼痛を含む種々の他の疾患または表示に関与してもいる。
【0097】
GPCR領域
大部分のGPCRは、疎水性アミノ酸残基または膜貫通型領域(TM、7つの膜貫通型領域は、TMl、TM2、TM3、TM4、TM5、TM6、およびTM7とする)の7つのクラスターによって特徴づけられ、これらは一次構造に位置して、細胞膜を貫通する(スパン)(図1A)。TM領域は、3つの細胞内ループ(i1、i2、およびi3)、3つの細胞外ループ(e1、e2、およびe3)によって接続される膜貫通型α−ヘリクスを表すと考えられている。GPCRはまた、アミノ(N)−カルボキシル(C)−末端領域(Palczewski et al.,Science 289,739−45(2000))を含む。膜貫通型領域の間の配列はGPCRループに対応し、細胞の範囲内のループの位置はそれが菌体内または細胞外のループであるかどうか決定する。大部分のGPCRは、機能性タンパク質構造を安定させると考えられているS‐S結合を形成する最初の2つの細胞外ループの各々で、一つの保存されたシステイン残基を持つ。PAR1
GPCRの膜貫通型とループ領域の略図を、図IAに示す。
【0098】
GPCRの1つの実施例は、CXCR4受容体(配列番号15として表2で示される)である。7本の下線が引かれた配列は、GPCRの7つの膜貫通型領域に対応する。したがって、配列IFLPTIYSIIFLTGIVGNGLVILV(配列番号16)は、第1の膜貫通型領域(TMl)に対応する。
【0099】
【表2】
単離GPCRフラグメントは、完全長タンパク質より少ないGPCRの任意の部分である。単離GPCRフラグメントを含んでいるペプチドは、自然に生じるアミノ酸配列以外のGPCR配列に、アミノ酸配列N末端およびC末端もしくはその一方を含むものであってもよい。GPCRの単離膜貫通型配列を含んでいるペプチドは、GPCRのその膜貫通型領域に対応している配列だけを含むと考えられる。あるいは、自然に生じるフランキング配列でない膜貫通型配列にアミノ酸配列のN末端およびC末端もしくはその一方を含むものであってもよい(すなわち、天然のGPCR配列でその領域に隣接してあるループ配列でない)。
【0100】
このように、CXCR4受容体の単離された膜貫通型領域を含むペプチドは任意のペプチドであり、それは表2で示される配列の下線を引かれた領域の隣接アミノ酸のいずれかまたは全てを含む。このペプチドは、天然のGPCR配列のTM領域に隣接してあるループ配列に対応する自然に生じる(下線なし)フランキング配列のいずれも含まない。
【0101】
同様に、CXCR4受容体の単離された(細胞内であるか細胞外の)ループ領域を含むペプチドは、表2に示す配列の非下線領域の任意または全ての隣接アミノ酸を含む任意のペプチドである。このペプチドは任意の自然に生じる膜貫通型配列を含まず、表2で下線が引かれたフランキング配列として示されており、天然のGPCR配列にあるループ領域に隣接してある。
【0102】
単離細胞外領域または単離細胞内領域を含むペプチドは、任意の(細胞外または細胞内)ループおよびN末端領域もしくはその一方、またはC末端領域に由来するアミノ酸配列を含むことができる。このペプチドは、天然のGPCR配列でその細胞外領域または細胞内領域に隣接してある膜貫通型領域に由来する配列をいっさい含まない。
【0103】
医薬組成物
本発明のペプデュシン(ここでは「活性化合物」とも称する)と、その誘導体、フラグメント、類似体、および相同体とを、投与にふさわしい医薬組成物に組み込むことができる。この組成物は、ペプデュシンと製薬的に許容される担体を概して含む。
【0104】
発明の医薬組成物は、その意図された投与経路と互換性を持つために処方される。投与経路の例として、非経口投与、例えば静脈(例として、末梢静脈(四肢で見出される末梢静脈)、腹腔内、内皮、皮下、真皮下、経口、鼻腔、煙霧質(例えば、吸入)、経皮(例えば、局所)、経粘膜、膣、子宮内、および直腸(例えば、坐剤)投与が挙げられる。本発明の活性化合物を含んでいる注射可能溶液は、血管内腔(例えば、大動脈または頸静脈)に投与してもよい。あるいは、本発明の活性化合物を、装置(例えば、ステントまたはカテーテル)を介して、活性化合物を含浸または被覆によって投与してもよい。
【0105】
投与のために使用される溶液または懸濁液(例えば、非経口投与薬)は以下の構成要素を含むものであってもよい。すなわち、注射用蒸留水等の無菌の希釈剤、食塩水、不揮発性油、ポリエチレングリコール、グリセリン、プロピレングリコールまたは他の合成溶媒;抗菌因子(例えば、ベンジルアルコールまたはメチル・パラベン);抗酸化剤(例えば、アスコルビン酸または亜硫酸水素ナトリウム);エチレンジアミン四酢酸(EDTA)等のキレート剤;緩衝液(例えば、酢酸塩、クエン酸塩、またはリン酸塩);および塩化ナトリウムまたはデキストラン等の等張性を調製するための薬剤である。pHを、酸または塩基(例えば、塩酸または水酸化ナトリウム)で調整することができる。本発明の医薬組成物の製剤を、ガラスまたはプラスチックでできているアンプル、使い捨て注射器または多人数用バイアルに入れられることができる。
【0106】
注射用途にふさわしい医薬組成物が、無菌の水溶液(水溶性の場合)または注射可能な滅菌溶液もしくは分散の一時しのぎの製剤のための分散および無菌の粉を含む。静脈内投与のための適切な担体として、生理食塩液、静菌性の水、クレモホルEL(BASF社、パーシパニー、ニュージャージー州)またはリン酸緩衝食塩水(PBS)を含む。全ての場合において、簡単なシリンジアビリティが存在する程度まで、組成物は無菌でなければならず、また流動的でなければならない。それは、製造及び貯蔵の条件下に安定でなければならず、また細菌および菌類等の微生物の汚染作用に対して保存されなければならない。担体を、溶媒または分散媒とすることができ、それらは、例えば水、エチルアルコール、ポリオール(例えば、グリセリン、プロピレングリコール、および液体ポリエチレングリコール等)、ならびにそれらの適当な混合物を含む。適する流動性は、例えばレシチンのようなコーティングの使用、分散液の場合には必要とする粒子寸法の維持及び界面活性剤の使用により保持しうる。微生物の作用を防止することは種々の抗菌および抗かび剤、例えば、パラベン、クロロブタノール、フェノール、アスコルビン酸、チメロサール等によって達成可能である。多くの場合、等張剤、たとえば糖、ポリアルコール、たとえばマンニトール、ソルビトール、塩化ナトリウムなどが組成物中に含まれているのが好ましいであろう。注射用組成物の持続吸収は、吸収を遅らせる薬剤、たとえばモノステアリン酸アルミニウムおよびゼラチン等を組成物中に配合することにより行うことができる。
【0107】
滅菌注射用溶液を活性化合物を所望の量で、必要に応じて上記したようにして、1つの成分または複数の成分の組み合わせによって、適当な溶媒に取り込み、続いて濾過滅菌する。通常、分散は活性化合物を基本的な分散媒を含む無菌の媒体に取り込むことによって調製されている、それらからの必要な他の成分を上記に列挙した。注射可能な滅菌溶液の製剤のための無菌粉末の場合、調整法は事前に滅菌ろ過された溶液から主成分+任意の添加された所望の成分の粉を産生する真空乾燥と凍結乾燥である。
【0108】
経口組成物は、一般に不活性の希釈剤または食用の担体を含む。それらをゼラチンカプセルに入れ、あるいは圧縮して錠剤にすることができる。経口治療的投与を目的として、活性化合物に賦形剤を取り込ませることができ、錠剤、トローチ剤、またはカプセルの形で使うことができる。経口組成物を、うがい薬として使用するために、流体担体を用いて調製することもでき、ここでは、流体担体の化合物は経口適用され、口内洗浄し、吐き出し、または飲み込むことができる。製薬的に互換の結着剤およびアジュバント材料もしくはその一方を、組成物の一部として含ませることができる。錠剤、ピル、カプセル、トローチ剤は、以下の成分のいずれかまたは類似の性質の化合物を含むことができる。すなわち、結着剤(例えば、微結晶性セルロース、トラガカントガム、またはゼラチン);賦形剤(例えば、澱粉または乳糖)、崩壊剤(例えば、アルギン酸)、プリモゲルまたはトウモロコシ澱粉);潤滑油(例えば、ステアリン酸マグネシウムまたはステロート);コロイド状二酸化ケイ素等の滑走剤;甘味料(例えば、ショ糖またはサッカリン);または香料(例えば、セイヨウハッカ、メチルサリチレートまたはオレンジ調味料)。
【0109】
吸入または鼻内投与による投与のために、化合物は適切な推進体(例えば、ガス(例として、二酸化炭素)または噴霧器(例えば、肺に対する送達))を含む加圧された容器またはディスペンサからエアゾール・スプレーの形で送達される。
【0110】
全身投与を経粘膜または経皮手段でおこなうこともできる。経粘膜または経皮投与のために、透過すべきバリヤーに対する適当な浸透剤をこの配合物に用いる。この浸透剤は通常、当該技術分野で知られており、例えば、経粘膜投与に対しては、洗剤、胆汁酸塩、およびフシジン酸誘導体が挙げられる。経粘膜投与を、鼻内噴霧または坐剤を用いることにより達成することができる。経皮投与のために、一般に当該技術分野で知られているように、活性化合物を軟膏、軟膏、ゲル類またはクリームに処方する。
【0111】
活性化合物を、坐剤(例えば、従来の坐剤基剤(例えば、カカオ脂と他のグリセリド))、坐剤コーティング、または直腸送達のための保持浣腸の形で調製することもできる。活性化合物を、腟内または子宮内投与のために、同じように調製することができる。活性化合物を傷パッキングに含浸または被覆し投与してもよい(例えば、出血を減少させるために)。
【0112】
一実施形態において、活性化合物は担体(例えば、移植片とマイクロカプセルに入れられた投射手段を含む徐放製剤)から化合物を迅速な排泄から保護する担体で調製されている。エチレンビニルアセテート、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、及びポリ乳酸等の、生分解性の生体適合性ポリマーを使用することができる。そのような製剤の調製は、当業者にとってあきらかである。材料は、Alza CorporationおよびNova Pharmaceuticals,Inc.から商業的に入手することができ、リポソーム懸濁液(ウイルス抗原に対するモノクローナル抗体を持つ感染細胞を標的としたリポソームを含む))が製薬的に許容される担体として使われることもできる。米国特許第4,522,811号で述べられるように、これらは、例えば、当業者に知られている方法によって調製することができる。
【0113】
投与の容易さと投薬量の均一性のために用量単位の形態で経口または非経口組成物を処方することは、特に有利である。ここで用いられる用量単位の形態は、処置される被検体に対して一体的投薬量として適している物理的離散性単位を参照する。活性化合物の所定量を含む各単位を、必要な製薬担体に関連して所望の治療効果を発生するために算出した。本発明の用量単位形態についての明細書を、活性成分の独特の特性および達成すべき特定の治療効果、ならびに個体処置のためにこの活性化合物を配合する当該技術分野に本来ある限界によって、また直接依存して、記述した。
【0114】
ペプデュシンおよびGPCRペプチドは、医薬組成物の形で種々の障害を処置するために投与される。そのような組成物の調製に関係している原理および考慮点は、構成要素の選択の際のガイダンスと同様に提供される。例えば、Remington:The Science And Practice Of Pharmacy 19th ed.(Alfonso R.Gennaro,et al.,editors)Mack Pub.Co.,Easton,Pa.:1995;Drug Absorption Enhancement:Concepts,Possibilities,Limitations,And Trends,Harwood Academic Publishers,Langhorne,Pa.,1994;およびPeptide And Protein Drug Delivery(Advances In Parenteral Sciences,Vol.4),1991,M.Dekker,New York。製剤は、本明細書中に処置されている(好ましくは各々に悪影響を与えない補完的な活性を持つもの)特定の徴候のために、必要に応じて一つ以上の活性化合物を含有することもできる。あるいは、または加えて、組成物はその機能を高める薬剤(例えば、細胞毒性剤、サイトカイン、化学療法剤または増殖阻害性薬剤)を含むことができる。この分子は、意図される目的のために効果的である量での組合せで十分に存在する。主成分は、例えば、コアセルベーション技術によって、または、界面重合法によって調製されるマイクロカプセルにトラップすることもできる。例えば、ヒドロキシメチルセルロースまたはゼラチン−マイクロカプセル、ポリ(メタアクリル酸メチル)マイクロカプセルを、それぞれ含むコロイド状薬物送達系(例えば、リポソーム、アルブミン・ミクロスフィア、マイクロ・エマルジョン類、ナノ粒子、およびナノ・カプセル)、あるいはマクロエマルジョンである。
【0115】
生体内での投与のために使用される製剤は、無菌でなければならない。これは、濾過滅菌法膜を通して濾過によって容易に達成される。
【0116】
持続放出性製剤を調製することができる。持続放出性製剤の適切な実施例は抗体を含んでいる固形疎水性ポリマーの半透性マトリックスを含む。そして、マトリックスは造形品(例えば、フィルムまたはマイクロカプセル)の形である。徐放性マトリックスの実施例は、ポリエステル類、ヒドロゲル(例えば、ポリ(2−ヒドロキシエチルメタクリレート)またはポリ(ビニルアルコール))、ポリ乳酸(米国特許第3,773,919号)、L−グルタミン酸とエチル−L−グルタミン酸塩のコポリマー、非分解性エチレン−酢酸ビニル、LUPPRON DEPOT(登録商標)(乳酸グリコール酸コポリマーと酢酸ロイプロリドから成る注射可能ミクロスフィア)等の分解可能な乳酸グリコール酸コポリマーとポリD−(−)−3−ヒドロキシ酪酸を含む。ポリマー(例えば、エチレン−酢酸ビニルおよび乳酸グリコール酸)が100日以上間の分子の放出を可能にする一方で、特定のヒドロゲル放出タンパク質がより短い時間にわたって放出する。
【0117】
活性化合物の徐放は、種々の技術を利用することができる。装置(例えば、ステントまたはカテーテル)は、活性剤の不均一な溶液および活性剤もしくはその一方の分散を高分子物質に取り入れているモノリシックな層またはコーティングを有していることが知られており、薬剤がポリマー流体界面にポリマーを通して拡散して、その周囲の液体に放出されるにつれて、治療薬の拡散速度が制限される。活性化合物は、適当な重合物質で溶解または分散させることで、材料が溶けた後に付加的な孔またはチェンネルが残る。マトリックス装置は、通常、同様に拡散性が制限されてはいるが、流体への薬剤の放出でもチャンネルまたは装置の内部ジオメトリーが役割を果たす。チャネルは、放出された薬剤または他の可溶性物質によって取り残される既存のチャネルまたはチャネルである。
【0118】
浸食または分解可能な装置は、概して物理的に活性化合物がポリマーに固定してもよい。活性化合物を、重合物質の全体を通じて溶解および分散もしくはその一方を行うことができる。重合物質は不安定な結合の加水分解を通して時間とともに加水分解的に分解され、ポリマーが液体に侵食することがあり、液体に活性剤を放出する。親水性ポリマーは、疎水性ポリマーと関連して一般により速い侵食率を持つ。疎水性ポリマーはほとんど単に活性剤の表面拡散を持つと考えられて、表面内部から浸食を持つ。親水性ポリマーは水をポリマーの表面に侵入させると考えられ、表面の下で不安定な結合の加水分解を可能にすることで、ポリマーの浸食が均一または大きくなり得る。
【0119】
医薬組成物は、投与のための指示書と共に、容器、パックまたはディスペンサに入れられる。
【0120】
本発明にもとづくペプデュシンによるアプローチによって、細胞内受容体構造の豊かな多様性を新規の治療薬の生成、さらには生体内条件下での受容体−Gタンパク質結合の機構を描写するために利用することができる。この戦略によって発見されるペプデュシンは、ペプデュシンが主にGタンパク質よりむしろ受容体を対象とするという範囲で、より選択的であることが明らかである。加えて、多くの受容体はゲノムおよび遺伝学的なアプローチによって種々の疾患プロセスに重要であると確認されたが、既知のリガンドを持たない、いわゆるオーファンレセプタである。ペプデュシン・アゴニストおよびアンタゴニストは、発生し、これらの受容体にテーラードで、どのシグナル伝達経路がその固有の環境の状況でオーファンレセプタによって起動するかを決定することに役立つかもしれない。このように、ポストゲノムの時代には、ペプデュシン・アプローチは膜タンパク質のターゲッティングに広く適用可能であり、従来の分子技術に従わないシステムで、新規の実験的な道が開けられる。
【0121】
本発明の化合物および他の薬理活性剤を、同時、経時的、または併用して患者に投与してもよい。発明の組合せを使用する場合、本発明の化合物と他の薬理活性剤が同じ製薬的に許容される担体であると考えて同時に投与する。それらを同時に取られる従来の経口剤形のような別々の製薬担体内にあってもよい。それらは、別々の製薬担体(例えば、同時にされる従来の経口剤形)であってもよい。用語「組合せ(併用)」は、化合物の剤形が別々で、経時的に投与されるという場合にも言及する。
【0122】
発明の化合物は、最適製薬有効性を提供する投薬量で、この処置を必要とする患者(動物とヒト)に投与される。いうまでもないことだが、任意の特定用途用に必要な用量は、特定の化合物または組成物の選択のみならず、投与経路、処置されている状態の性質、患者の年齢と状態、並列の薬物または患者が従う特別食、さらに当業者が認識する他の要素、最後に付き添いの医師の裁量である適切な投薬に従って、患者間で変える。
【0123】
状態の処置において、適当な投薬量レベルは通常、患者体重約0.001〜50mg/kg/日である。また、投薬は1回または複数回であってもよい。好ましくは、投薬量レベルは、1日約0.01〜約25mg/kg、より好ましくは1日0.05〜約10mg/kgである。例えば、適切な投薬量レベルは、1日約0.001〜10mg/kg、好ましくは1日約0.005〜5mg/kg、特に1日約0.01〜1mg/kgである。
【0124】
例えば、急性心筋梗塞患者において、適切なペプデュシン(例えば、x1/2pal−i1)を大量瞬時投与量として直ちに静脈投与することができ、それに続いて心筋梗塞後2〜3日にわたって、毎日1,2度追加して静脈注射をおこなう。投薬量は、約0.1〜0.5mg/kgである。
【0125】
任意の処置用に必要な発明の化合物の量が選択される特定の化合物または組成物でだけでなく投与経路、処置されている状態の性質、および年齢と患者の状態でも変化して、最後に付き添いの医師の裁量で変わることはいうまでもない。
【0126】
本発明の組成物および併用療法は種々の医薬賦形剤と組み合わせて投与することができ、本明細書中に述べられるように、安定化剤、担体およびカプセル化法製剤もしくはその一方を含む。
【0127】
本発明の水性組成物は、発明のペプチドの有効な量を含有、溶解し、製薬的に許容される担体または水溶媒質で分散した。
【0128】
「製薬的または薬理的に許容される」は、動物またはヒトに投与されるとき、副作用、アレルギー反応、または他の有害反応を発生しない分子実体と組成物を必要に応じて包含する。「製薬的に許容される担体」は、任意または全ての溶媒、分散媒、コーティング、抗菌、および抗真菌剤、等張および吸収遅延剤、その他を含む。製薬活性物質のためのこのような培地および薬剤の使用は、周知の技術である。任意の従来の培地または剤が主成分と適合しないこと以外は、治療用組成物のその使用は考察される。補助主成分を、組成物に組み込むこともできる。
【0129】
ヒト投与のために、製剤は、FDA局の生物学標準の要求に従って、生殖不能、発熱原性、一般安全、および純度の基準を満たすものとする。
【0130】
次に、発明の組成物および併用療法を、通常、非経口投与、例えば、静脈、筋内、皮下、病巣内、腹腔内経路を経て、注射のために処方される。発明の組成物または活性構成要素または成分を含む水組成物の製剤は、本開示を考慮して当該技術分野の当業者に知られている。典型例として、液溶体または懸濁液として、そのような組成物は、注射剤として調製されている。注射前に液体の追加に応じて溶液または懸濁液を調製するための使用に適した固形も、調製することができ、また製剤を乳化させることもできる。
【0131】
注射用途にふさわしい剤型として、無菌の水溶液または分散;ゴマ油、落花生油または水プロピレングリコールを含む製剤;ならびに注射可能な滅菌溶液または分散の即席の製剤のための滅菌粉末が挙げられる。全ての剤形は、無菌であり、かつ容易に注射器に流入できる程度に流体でなければならない。製造及び貯蔵の条件下に安定でなければならず、バクテリアおよび菌類のような微生物の汚染作用に対して保存されなければならない。
【0132】
遊離塩基または薬理的に受け入れられる塩類としての活性化合物の溶液は、界面活性剤(例えば、ヒドロキシプロピルセルロース)を十分に混ぜ合わせて水中で調製される。グリセロール、液体ポリエチレングリコール、およびそれら混合物中、および油中で分散液を調製することもできる。通常の貯蔵または使用の条件下で、これらの調製物は微生物の生長を防止するための保存剤を含む。
【0133】
本発明の治療的または薬理学的組成物は、一般に、製薬的に許容される媒体に溶解または分散した併用療法の構成要素の有効量を含む。製薬的に許容される媒体または担体として、任意または全ての溶媒、分散媒、コーティング、抗菌、および抗真菌剤、等張および吸収遅延剤、その他が挙げられる。製薬活性物質のためのこのような培地および薬剤の使用は、周知の技術である。補助主成分は、本発明の治療用組成物に組み込むこともできる。
【0134】
製薬または薬理学組成物の製剤は、本開示を考慮して当業者に知られている。典型的には、この組成物を、注射可能なものとして調製、液溶体または懸濁液のいずれかとして、注射に先立って溶液中または懸濁液中に含ませることに適した固形として、経口投与のための錠剤または他の固体として、限時解放カプセルとして、あるいは現在利用されている他のいずれかの形態(クリーム、ローション剤、うがい薬、吸入剤等)として、調製することが可能である。
【0135】
滅菌注射用溶液の調製は、必要量の活性成分を適当な溶媒に、必要に応じて上記に列挙した種々の他の成分とともに、取り込んだ後、濾過滅菌することでおこなった。一般に、分散は、種々の滅菌化された活性成分をビヒクルに取り込むことによって調製され、このビヒクルには基本的な分散媒体および上記に列挙された他の必要とされる成分を含む。注射可能な滅菌溶液の調製に用いられる無菌粉末の場合、好ましい調製法は、真空乾燥および凍結乾燥技術であり、これらの技術は活性成分の粉末に加えて、既に濾過滅菌したそれの溶液から所望の任意の成分を追加して得られる。
【0136】
筋肉内注射用のより多くまたは高濃度の溶液を調製することも検討される。この点に関しては、溶媒としてのDMSOの使用が好ましく、なぜならこのことが極度に迅速な浸透をもたらすことからであり、活性化合物または活性薬剤が小さな領域に送達される。
【0137】
オペレーティング・フィールドの特定域を浄化するために外科医、内科医、または医療従事者による滅菌製剤(例えば、食塩水ベースの洗浄液)の使用もまた、特に有用であると考えられる。本発明にもとづく治療用製剤は、うがい薬の剤形、または抗真菌性試薬とともに再構成してもよい。吸入剤の形態も、想定される。本発明の治療用製剤を、局所投与に適した剤形、例えばクリームおよびローション剤に調製することも可能である。
【0138】
このような溶液に適した防腐剤として、ベンザルコニウムクロリド、塩化ベンゼトニウム、クロロブタノール、チメロサール等が挙げられる。適当な緩衝液として、pH6ないしpH8、好ましくはpH7ないしpH7.5に保たれる量のホウ酸、ナトリウムおよび重炭酸カリウム、ナトリウムおよびカリウム・ホウ酸塩、ナトリウムおよびカリウム炭酸塩、酢酸ソーダ、燐酸ナトリウム等が挙げられる。適当な緊張剤は、点眼剤の食塩相当量が0.9プラスまたはマイナス0.2%の範囲内であるように、デキストラン40、デキストラン70、デキストロース、グリセリン、塩化カリウム、プロピレングリコール、塩化ナトリウム等である。適当な抗酸化剤と安定剤として、亜硫酸水素ナトリウム、メタ重亜硫酸ナトリウム、チオ硫酸ナトリウム、チオ尿素等が挙げられる。適当な湿潤剤および清澄剤として、ポリソルベート80、ポリソルベート20、ポリオキサマー282、およびチロキサポールが挙げられる。適当な粘度増加性剤として、デキストラン40、デキストラン70、ゼラチン、グリセリン、ヒドロキシエチルセルロース、ヒドロキシメチルプロピルセルロース、ラノリン、メチルセルロース、ワセリン、ポリエチレングリコール、ポリビニルアルコール、ポリビニルピロリドン、カルボキシメチルセルロース等が挙げられる。
【0139】
製剤に応じて、薬理的に効果的であるように、投薬量処方と互換性を持つ方法で、療法が施される。製剤は、種々の投薬形態、例えば上記したような注射溶液のタイプで容易に投与される。製剤を種々の投薬形態(例えば、上記した注射可能な溶液)で容易に投薬することができる。
【0140】
この文脈において、投与される組成物の主成分および容積は、処置される宿主動物に依存している。投与に必要な活性化合物の正確な量は、開業医の判断に依存し、各個人に特有である。
【0141】
活性化合物を分散させることが要求される組成物の最小量を、概して利用する。投与のための適当な投与計画も、可変的ではあるが、初めに化合物を投与して結果をモニターした後、さらに間隔を置いてさらに制御された用量を与えることによって代表される。例えば、非経口投与を目的として、適当に緩衝され、また必要に応じて等張である水溶液を調製し、静脈、筋肉内、皮下、または腹腔内投与に使用される。1つの投薬量は1mlのNaCl等張溶液に溶解し、1000mlの皮下注入液に添加または輸液の予定部位に注入される(例えば、Remington’s Pharmaceutical Sciences 15th Edition,pages 1035−1038 and 1570−1580)。
【0142】
特定の実施形態では、活性化合物を経口投与してもよい。このことは、消化酵素によるタンパク質分解に対して耐性、または耐性となった薬剤に対して検討される。そのような化合物は、化学的に設計された薬剤または改質された薬剤、右旋性ペプチド、ならびにペプチドおよびリポソーム型製剤(ペプチダーゼおよびリパーゼによる分解を回避するために徐放性カプセルに入れられている)であることが考えられる。
【0143】
製薬的に許容される塩類として、酸付加塩が挙げられ、該酸付加塩は、無機酸(例えば、塩酸、臭化水素酸、ホウ酸、リン酸、硫酸またはリン酸類)、あるいは有機酸(例えば、酢酸、シュウ酸、酒石酸、マレイン酸、フマル酸、クエン酸、コハク酸、メシル酸、マンデリン酸、コハク酸、安息香酸、アスコルビン酸、メタンスルホン酸、α−ケトグルタル酸、α−グリセロリン酸、グルコース−1−リン酸等)により形成される塩類である。遊離カルボキシル基で形成される塩類を、無機塩基(例えば、ナトリウム、カリウム、アンモニウム、カルシウム、マグネシウムまたは水酸化第二鉄)と有機塩基(例えば、イソプロピルアミン、トリメチルアミン、ヒスチジン、プロカインなど)とから誘導することもできる。製薬的に許容される塩類の他の例として、式(I)の化合物の四級誘導体が挙げられ、例えば、化合物Rx−Tによって四級化した化合物であり、式中RxはC1−6アルキル、フェニル−C1−6アルキル、またはC5−7シクロアルキル、Tは酸のアニオンに対応するラジカルである。Rxの適当な例として、メチル、エチル、ならびにn−およびイソ−プロピル、ならびにベンジルおよびフェネチルが挙げられる。Tの適当な例として、ハロゲン化合物(例えば、塩化物、臭化物、またはヨウ化物)が挙げられる。製薬的に許容される塩類のさらなる別の例もまた、N−酸化物等の内錯塩も含まれる。
【0144】
担体は、例えば、水、エチルアルコール、ポリオール(例えば、グリセリン、プロピレングリコール、および液体ポリエチレングリコール等)、それらの適当な混合物、ならびに植物油を含む溶媒または分散媒でもあり得る。適当な流動性を、例えばレシチン等のコーティング、分散の場合ならば所望の粒径に保つこと、ならびに界面活性剤の使用によって、保つことができる。微生物の影響からの保護は、種々の抗菌剤及び抗真菌剤、例えばパラベン、クロロブタノール、フェノール、ソルビン酸、チメロサール等により達成される。多くの場合、等張剤、例えば糖又は塩化ナトリウムを含むことが好ましい。注射用組成物の長期にわたる吸収は、吸収を遅延させる薬剤、例えばモノステアリン酸アルミニウムおよびゼラチンを組成物中で用いることにより達成される。
【0145】
滅菌注射用溶液を、上記列挙された種々の他の成分を必要に応じて含む溶媒に必要量の活性化合物を取り込み、続いて濾過滅菌することで調製する。通常、分散を、滅菌された種々の活性成分を滅菌されたビヒクル(基本分散媒および上記列挙したもののなかから選ばれる所望の他の成分を含む)に取り込むことによって調製する。注射可能な滅菌溶液の調製に用いられる滅菌粉末の場合、調製の好ましい方法は、真空乾燥および凍結乾燥技術であり、これによって、既に濾過滅菌した溶液から活性成分プラス任意の付加的な所望の成分の粉末が得られる。
【0146】
直接注射のためのより多くの、または高濃度の溶液の調製も検討する。ここで、溶媒としてDMSOを使用することで極度に迅速な貫通がなされ、小さな領域に活性剤が高濃度に送達される。
【0147】
処方に際して、投薬処方と互換性を持つ方法で、かつ治療的に有効であるような量で、溶液の投与をおこなう。製剤の投与を、種々の剤形(例えば、上記タイプの注射可能溶液)で容易に行えるが、薬放出カプセル等を使用することもできる。
【0148】
水溶液状態での非経口投与をおこなうために、例えば、その溶液を必要ならば適当に緩衝させるべきであり、また液体希釈剤が先ず十分な生理食塩液またはグルコースにより等張にされるものとする。これらの特定の水溶液は、特に静脈、筋内、皮下、および腹腔内投与に適している。この点について、使用可能な滅菌水性培地は、本開示を鑑みることで当業者に知られている。
【0149】
非経口投与(例えば、静脈内または筋内注射)用に処方される化合物に加えて、他の製薬的に受け入れられる剤形として、例えば、内服のための錠剤または他の固体;リポソーム型製剤;徐放性カプセル;および現在使用されている任意の他の剤形(クリームを含む)が挙げられる。
【0150】
投与の他の形態に適した別の製剤として、坐剤が挙げられる。坐剤のために、従来の結着剤および担体として、ポリアルキレン・グリコールまたはトリグリセライドを挙げることが可能であり、そのような坐剤は、0.5%ないし10%、好ましくは1%ないし2%の範囲内で活性成分を含む混合物から形成することが可能である。
【0151】
例えば、経口製剤として、製薬等級のマンニトール、乳糖、澱粉、ステアリン酸マグネシウム、ナトリウム・サッカリン、セルロース、炭酸マグネシウム等の通常使用される賦形剤が挙げられる。これらの組成物は、溶液、懸濁液、錠剤、ピル、カプセル、徐放性の製剤または粉末の剤形をとる。
【0152】
明確な特定の実施形態では、経口医薬組成物は、不活性の希釈剤または吸収消化可能な食用の担体を含み、またはそれらが固いまたは柔らかいシェル・ゼラチンカプセルに入れられたものであってもよく、または圧縮されて錠剤になったものであってもよく、または規定食に直接取り込まれたものであってもよい。経口治療投与用に、有効化合物に添加剤を組み込んでもよく、摂取可能な錠剤、口腔錠、トローチ、カプセル、エリキシル剤、懸濁剤、シロップ、ウエハース等の剤形で使用することが可能である。このような組成物および調剤は少なくとも0.1%の活性化合物を含有するものとする。組成物および製剤の割合が、もちろん変動する場合もあり、便宜上、単位重量の約2〜約75%、または好ましくは25〜60%の間であってもよい。このような治療上役立つ組成物の活性化合物の量は、適当な投薬量が得られるような量である。
【0153】
錠剤、トローチ剤、ピル、カプセル等は以下のようなものを含むものであってもよい。すなわち、結着剤(例えば、トラガカント、アカシア、コーンスターチまたはゼラチン)、賦形剤(例えば、リン酸二カルシウム)、崩壊剤(例えば、トウモロコシ澱粉、ジャガイモ澱粉、アルギン酸等)、潤滑油(例えば、ステアリン酸マグネシウム)、および甘味料(例えば、ショ糖、乳糖またはサッカリン)を添加してもよく、あるいは香料(例えば、セイヨウハッカ、ウィンターグリーン油またはチェリー香料)である。単位投与剤形がカプセル剤である場合は、前述の物質に加えて、液体担体を含むことができる。様々な他の物質がコーティングとして存在し、さもなければ投薬単位の物理的形態を修飾するために存在してもよい。例えば、錠剤、ピルまたはカプセルを、シェラック、糖、またはこの両方で被覆することができる。エリキシルのシロップは、防腐剤、染料、および香料(例えば、サクランボまたはオレンジ味)として、甘味料(メチルおよびプロピルパラベン)として活性化合物(ショ糖)を含有してもよい。
【0154】
本発明の医薬組成物を、例えば固体、半流動または液体状態の製剤の形態で使用することが可能であり、本発明の化合物の一つ以上を主成分として含み、外用、腸内適用、または非経口適用に適した有機もしくは無機の担体または賦形剤と混合された状態で、本発明の化合物の一つ以上を主成分として含む。主成分は、例えば、錠剤、ペレット、カプセル、坐剤、溶液、エマルジョン類、懸濁液、および使用に適した任意の他の剤形用の通常非中毒性で製薬的に許容される担体であってもよい。使用し得る担体として、補助剤、安定化剤、増粘剤、および着色剤に加えて、固体、半流動、または液体状態にある水、ブドウ糖、乳糖、アカシアゴム、ゼラチン、マンニトール、デンプンのり、三ケイ酸マグネシウム、滑石、トウモロコシ澱粉、ケラチン、コロイダルシリカ、ジャガイモ澱粉、尿素、および製剤製造での使用に適している他の担体が挙げられ、また香水を使用してもよい。アクティブ・オブジェクト化合物は、プロセスまたは病状に所望の効果を発生するのに十分な量で、医薬組成物に含まれる。
【0155】
錠剤等の固形組成物を調製するために、主要な活性成分を、製薬担体、例えば従来の錠剤化成分(トウモロコシ澱粉、乳糖、ショ糖、ソルビトール、滑石、ステアリン酸、ステアリン酸マグネシウム、リン酸二カルシウムまたはガム)および他の製薬希釈剤(例えば、水)とを混合して、本発明の化合物の均質混合物またはその無毒性で製薬的に許容される塩を含む固形予備処方組成物を形成する。これらの予備処方組成物をホモジェナスと称する場合、組成物が等しく有効な単位剤形(例えば、錠剤、ピルとカプセル)に容易に再分割されないように、主成分が組成物の全体を通じて均一に分散することを意味する。次に、この固形予備処方組成物は、本発明の主成分が0.1ないし約500mg含まれる上述のタイプの単位剤形に再分割される。新規組成物の錠剤またはピルを被覆、さもなければ化合して、持続性作用の長所を産みだす剤形を提供することができる。例えば、錠剤またはピルは内側投薬量構成要素と外側投薬量構成要素とを含むことができ、後者が前者を包む膜状となる。2つの構成要素を腸層によって分離することができ、この腸層によって胃の崩壊に耐え、内側構成要素が十二指腸を無傷に通過することが可能であり、または放出が遅れる。種々の材料を、このような腸層またはコーティングに使うことができ、この材料として、いくつかの高分子酸とシェラック、セチルアルコール、およびセルロースアセテート等の材料と高分子との混合物とが挙げられる。
【0156】
経口的にまたは注射によって投与するために本発明の組成物が取り込まれる液体の剤形として、水溶液、十分に風味をつけたシロップ剤、水性懸濁液または油性懸濁液、および許容される油(例えば、綿実油、ゴマ油、ヤシ油、または落花生油)または静注に適した可溶化または乳化剤によるエマルジョン類、同様にエリキシルおよび類似の賦形薬が挙げられる。水性懸濁液に適した分散剤または沈澱防止剤として、合成および天然のガム(例えば、トラガカンタ、アカシア、アルギン酸塩、デキストラン、ナトリウムカルボキシメチルセルロース、メチルセルロース、ポリビニルピロリドンまたはゼラチン)が挙げられる。
【0157】
吸入または吹送のための組成物として、溶液および懸濁液を含む製薬的に許容される水性または有機性の溶媒またはその混合物、および粉末が挙げられる。上述したように、液体または固体組成物は適当な製薬的に許容される賦形薬を含むものであってもよい。好ましくは、組成物は局所的または全身的な効果を得るために経口または鼻呼吸経路によって投与される。好ましくは製薬的に許容される滅菌溶媒の組成物は、不活性ガスの使用によって霧状にしたものであってもよい。霧状にされた溶液を噴霧器から直接呼吸してもよく、または、噴霧器をフェースマスク、テント、または間欠的陽圧呼吸機械に接続してもよい。溶液、懸濁液、または粉組成物は、好ましくは経口的または経鼻的に、適切な方法で製剤を送達する装置から投与することが可能である。
【0158】
上記の臨床症状および疾患を処置するために、本発明の化合物を従来の無毒性製薬的に許容される担体、アジュバント、およびビヒクルを含む用量単位製剤で、経口的、局所的、非経口的、吸入噴霧、または直腸的に投与してもよい。本文中で使用される非経口なる用語は、皮下注射、静注、筋肉内注射、胸骨内注射、または輸液法を包含する。
【0159】
スクリーニング法および検出法
本発明の組成物を、GPCR活性または発現を調整する薬物または化合物のスクリーニングに用いられるとともに、GPCRタンパク質の不十分または過剰な生産あるいはGPCR野生型タンパク質と比較して減少または異常活性を呈するGPCRタンパク質形態の生産によって特徴付けられる疾患を処置するために用いられる。
【0160】
本発明は、方法(また、ここでは「スクリーニング・アッセイ」と称する)を、モジュレータ、例えばGPCRに結合またはGPCRタンパク質発現もしくはGPCR活性に対して刺激または阻害効果を呈する候補または試験化合物または薬剤(例えば、ペプチド、ペプチド模倣薬、低分子、または他の薬物)を確認するために提供する。本発明はまた、本明細書中に述べられるスクリーニング・アッセイで同定される化合物を含む。
【0161】
本発明は、ペプデュシン−GPCR複合体の膜結合形態またはその生物学的活性部分の活性を調整またはそれに結合する候補または試験化合物をスクリーニングするためのアッセイを提供する。本発明の試験化合物を、数多くのアプローチのいずれかを当該技術分野で知られている組み合わせライブラリ、例えば生物学的ライブラリ、空間的にアドレス可能な平行固相または溶液相ライブラリ、デコンボリューションを必要とする合成ライブラリ方法、「1−ビード1−化合物」ライブラリ方法、およびアフィニティークロマトグラフィー選択を使用する合成ライブラリ方法で用いることによって、得ることができる。生物学的ライブラリー・アプローチは、ペプチド・ライブラリーに限られており、一方その他の4つのアプローチがペプチド、非ペプチド・オリゴマー、または化合物の低分子ライブラリに適用できる。例えば、Lam,1997.Anticancer Drug Design 12:145を参照せよ。
【0162】
分子ライブラリの合成に関する方法の例は、当該技術分野、例えば、DeWitt,et al.,1993.Proc.Natl.Acad.Sci.U.S.A.90:6909;Erb,et al.,1994.Proc.Natl.Acad.Sci.U.S.A.91:11422;Zuckermann,et al.,1994.J.Med.Chem.37:2678;Cho,et al.,1993.Science 261 :1303;Carrell,et al.,1994.Angew.Chem.Int.Ed.Engl.33:2059;Carell,et al.,1994.Angew.Chem.Int.Ed.Engl.33:2061;およびGallop,et al.,1994.J.Med.Chem.37:1233で見出すことができる。
【0163】
化合物ライブラリは溶液状態で存在(例えば、Houghten,1992.Biotechniques 13:412−421)、またはビーズ(Lam,1991.Nature 354:82−84),on chips(Fodor,1993.Nature 364:555−556)、細菌(Ladner,U.S.Patent No.5,223,409),胞子(Ladner,U.S.Patent 5,233,409)、プラスミド(Cull,et al.,1992.Proc.Natl.Acad.Sci.USA 89:1865− 1869)あるいは、ファージ(Scott and Smith,1990.Science 249:386−390;Devlin,1990.Science 249:404−406;Cwirla,et al.,1990.Proc.Natl.Acad.Sci.U.S.A.87:6378−6382;Felici,1991.J.MoL Biol.222:301−310;Ladner,U.S.Patent No.5,233,409)に存在するものであってもよい。
【0164】
アッセイは、細胞ベースのアッセイであり、GPCRの膜結合形態または細胞表面上でその生物学的活性部分、プラスペプデュシンを発現する細胞を試験化合物と接触させ、GPCRと結合してペプデュシンを移動させる試験化合物の能力を決定する。試験化合物は、GPCR標的の細胞外表面、膜貫通型領域または細胞内表面で結合することができ、GPCRのペプデュシン活性化を阻害することができ、あるいは高めることが可能である。細胞は、例えば、哺乳類起源または酵母細胞である。ペプデュシンをGPCRタンパク質から移す試験化合物の能力の決定は、試験化合物の結合がGPCRからペプデュシンまたはその生物学的活性部分を移動させるように、例えば、ペプデュシンを放射性同位元素または酵素ラベルに連結させることによって達成される。あるいは、直接または間接的に、試験化合物を125I、135S、4Cまたは3Hで標識することができ、ペプデュシンは放射性同位元素標識試験化合物をGPCRから移すことが可能であり、さらに遊離放射性同位元素標識試験化合物が、放射能放出の直接的計数によって、またはシンチレーション・カウントによって検出される。あるいは、例えば、試験化合物をワサビペルオキシダーゼ、アルカリホスファターゼまたはルシフェラーゼによって酵素標識することができ、酵素ラベルはペプデュシンの追加に応じて製品に対する適当な基質の転換での減少または増加によって検出される。
【0165】
別の実施形態では、アッセイは、細胞膜上のGPCRタンパク質の膜結合形態またはその生物学的活性部分を試験化合物と接触させること、およびGPCRに関してのペプデュシンの結合、活性を変調(例えば、刺激または阻害)する試験化合物の能力を決定することを含む細胞ベースのアッセイである。
【0166】
GPCR標的分子と相互作用する試験分子の能力を決定することは、直接結合を決定するための上記方法のうちの1つによって達成される。一実施形態では、GPCR標的分子とのGPCRペプチド相互作用を阻害する試験分子の能力を決定することは、標的GCPR−ペプデュシン複合体の活性を決定することによって達成される。例えば、標的分子の活性は、GPCR標的の細胞の二次メッセンジャーのGPCRペプチド誘導を阻害すること(細胞内Ca2+、ジアシルグリセロール、IP3、その他)、GPCR活性化または阻害に依存している触媒/酵素活性を検出すること、リポーター遺伝子(検出可能な標識(例えば、ルシフェラーゼ)をコードしている核酸に対して有効に連結するGPCR反応性調節因子を含む)の誘導または阻害を検出すること、または、細胞性応答(例えば、細胞生存、細胞分化、または細胞増殖)を検出することを含む。
【0167】
あるいは、本発明のアッセイは、試験化合物をGPCRペプチドまたは生物学的活性部分に接触させること、GPCRタンパク質またはその生物学的活性部分の活性を調節(例えば、刺激または阻害)またはそれに結合する試験化合物の能力を決定することを含む無細胞アッセイである。
【0168】
GPCRに対する試験化合物の結合を、先に述べたように、直接または間接的に決定することができる。例えば、アッセイは、アッセイ混合物を形成するために、ペプデュシンとGPCRまたはその生物学的活性部分を、GPCRと結合する既知の化合物と接触させること、アッセイ混合物を試験化合物と接触させること、およびGPCRタンパク質と相互作用する試験化合物の能力を決定することを含み、GPCRタンパク質と相互作用する試験化合物の能力を決定することは、既知の化合物と比較すると優先してGPCRと結合する試験化合物の能力またはその生物学的活性部分を決定することを含む。
【0169】
GPCRの活性を変調する試験化合物の能力を決定することは、例えば、直接結合を決定する上記の方法の1つによって、GPCR標的分子に対して結合するGPCRペプチドの能力を決定することによって、達成し得る。あるいは、GPCRの活性を変調する試験化合物の能力ペプチドを決定することは、CPCR標的分子をさらに変調するGPCRペプチドの能力を決定することによって達成し得る。例えば、適当な基質上での標的分子の触媒的/酵素的活性を上記のように決定することができる。
【0170】
無細胞アッセイは、GPCRペプチドまたはその生物学的活性部分をGPCRに結合してアッセイ混合物を形成する既知の化合物と接触させること、アッセイ混合物を試験化合物と接触させること、およびCPCRと相互作用する試験化合物の能力を測定することを含み、GPCRと相互作用する試験化合物の能力を決定することは、GPCR標的分子の活性を優先的に変調またはそれに結合するGPCRペプチドの能力を決定することを含む。
【0171】
本発明の無細胞アッセイは、GPCRタンパク質の可溶性の形状または膜結合形態の両方の使用にしたがう。GPCRタンパク質の膜結合形態を含む無細胞アッセイの場合、GPCRタンパク質の膜結合形態が溶液で維持されるように、溶解剤を利用することは、望ましいと考えられる。この溶解剤の実施例は、非イオン性界面活性剤、例えばn−オクチルグルコシド、n−ドデシルグルコシド、n−ドデシルマルトシド、オクタノイル−N−メチルグルカミド、デカノイル−N−メチルグルカミド、トリトン(登録商標)X−100、トリトンX−(登録商標)114、セシト((登録商標)、イソトリデシポリ(エチレングリコールエーテル)n、N−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネート、3−(3−コラミドプロピル)ジメチルアミニオル−1−プロパンスルホネート(CHAPS)、または3−(3−コラミドプロピル)ジメチルアミニオル−2−ヒドロキシ−1−プロパンスルホネート(CHAPSO)が挙げられる。
【0172】
GPCRペプチドまたはその標的分子のいずれかを固定して、該タンパク質の一方または両方の未複合体化形態から複合体を形成したものを分離することを促進させ、同様にアッセイのオートメーションを適応させることが望ましいかもしれない。候補化合物の存在または非存在下でのGPCRタンパク質に対する試験化合物の結合またはGPCRタンパク質とペプデュシンとの相互作用は、反応物の含有に適した任意の容器で達成される。そのような容器の例として、マイクロタイター・プレート、試験管、およびマイクロ遠心管が挙げられる。一実施形態では、タンパク質の一方または両方がマトリックスに結合するのを可能にする領域を加える融合タンパクが、提供される。例えば、GST−GPCR融合ペプチドまたはGST−標的融合タンパクをグルタチオン・セファロース・ビード(シグマ・ケミカル社、セントルイス、ミズーリ州)またはグルタチン誘導体化マイクロタイター・プレートに吸着させることができ、試験化合物、または該試験化合物と非吸着標的タンパク質またはGPCRペプチドのいずれかと組み合わせることで、混合物を、複合体形成(例えば、塩およびpHの生理学的条件で)に貢献する条件下でインキュベートする。インキュベーション後、ビードまたはマイクロタイター・プレート・ウェルを、任意の未結合構成要素を除去するために洗い、ビードの場合、マトリックスが固定される、複合体が直接または間接的に、例えば、上述のようにして、決定される。あるいは、複合体をマトリックスから分離し、GPCRペプチド結合または活性のレベルを標準技術を用いて決定した。
【0173】
マトリックス上での非流動化タンパク質の他の技術が、本発明のスクリーニング・アッセイでも使われる。例えば、GPCRペプチドまたはその標的分子は、ビオチンとストレプトアビジンとの複合体形成を利用して固定することができる。ビオチニル化GPCRペプチドまたは標的分子は、当該技術分野(例えば、ビオチン化キット、ピアスケミカル社、ロックフォード、イリノイ州)の中で周知の技術を使用しているビオチン−NHS(N−ヒドロキシ−スクシニミド)から調製することができ、ストレプトアビジン被覆の96穴プレート(ピアスケミカル社)のウエルで固定することができる。GPCRペプチドまたは標的分子と反応性はあるが、GPCRペプチドと同族GPCRとの結合を妨害しない抗体を、プレートのウエルに誘導体化させることができ、未結合標的またはGPCRペプチドは抗体結合によってウエルに捕捉することができる。GPCRペプチドまたは標的分子と反応性はあるが、GPCRペプチドと同族GPCRとの結合を妨害しない抗体を、プレートのウエルに誘導体化させることができ、未結合標的またはGPCRペプチドは抗体結合によってウエルに捕捉することができる。このような複合体を検出する方法として、GST固定複合体に関する上記のものに加えて、GPCRペプチドまたは標的分子に対するGPCRペプチドと反応する抗体を用いた複合体の免疫学的検出法、同様にGPCRペプチドまたは標的分子に関連する酵素活性の検出に依存する酵素結合アッセイが挙げられる。
【0174】
GPCRタンパク質発現のモジュレータは、細胞が候補化合物で接触する方法で特定され、細胞内でのGPCRmRNAまたはタンパク質の発現が決定される。候補化合物の存在下でのGPCRmRNAまたはタンパク質の発現レベルを候補化合物が存在しない場合のGPCRmRNAまたはタンパク質の発現と比較する。この比較にもとづいて、候補化合物がGPCRmRNAまたはタンパク質の発現のモジュレータとして同定される。例えば、GPCRmRNAまたはタンパク質の発現が候補化合物の存在下でその不在の場合よりも大きい(すなわち、統計学的に有意により大きい)場合、候補化合物は、GPCRmRNAまたはタンパク質発現の刺激因子であると同定される。あるいは、GPCRmRNAまたはタンパク質の発現が候補化合物の存在下で、不在の場合よりもより少ない(統計学的に有意により少ない)場合、候補化合物は、GPCRmRNAまたはタンパク質発現の阻害剤と確認される。細胞のGPCRmRNAまたはタンパク質発現のレベルは、GPCRmRNAまたはタンパク質を検出するために本明細書中に述べられる方法によって測定することができる。
【0175】
本明細書で議論されるペプチド配列を以下の表3に示す。
【0176】
【表3−1】
【0177】
【表3−2】
【0178】
【表3−3】
【0179】
【表3−4】
【0180】
【表3−5】
ペプデュシン(例えば、i1ループから)のペプチドの部分は、結果として意図された有益な効果をもたらす任意の適当な長さであってもよく、一般に5〜15、または、例えば、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29または30である。
【0181】
本発明のペプデュシン化合物を調製する方法を、以下の合成スキームおよび実施例で例示する。以下のスキーム、実施例、および生物学データは、発明の範囲または精神を制限するためのものではなく、発明を例示するために与えられる。
【0182】
ペプデュシン化合物の製造および特徴づけ
一般にCovic et al.PNAS 99:643−648(2002)に述べられるように、パルミトイル化されたペプデュシン・ペプチドの標準Fmoc固相合成物方法および調製による合成をおこなった。ペプデュシンをC18またはC4逆相クロマトグラフィーによって>95%純度に精製し、DMSOに溶した。
【0183】
出血時間は、キシラジン(10mg/kg)+塩酸ケタミン(50mg/kg)の腹腔内投与で6〜8週間目の成体雄CF−1マウスを麻酔しておこなった。内頸静脈にカニューレを挿入し、0.28×1.52mmゲージ・カテーテルおよびPlpal−12(3μmoles/L)、P4pal−10(3μmoles/L)またはビヒクル(DMSO)単独で、100μLの総容積で、1分にわたって注入した。実験は、注射された物質に対して盲検下でおこなった。5分後に、尾端から2mmのところで尾部の切断をおこなった。尾部を、37℃に維持されるリン酸塩緩衝食塩水のビーカーに浸漬させ、出血を視覚的に追跡し、時間を計った。出血が5分以内に再開する場合、それを再ブリードとして記録されて、前述のとおり、不安定な止血現象を採用した(Law et al.,Nature 401,808(1999)。許される最大出血時間は、10分間であり、その後、尾部を焼灼した。
【実施例】
【0184】
実施例1
敗血症がどのように本発明のペプデュシンによって効果的に処置されるかについて示すために、6〜8週間目の雌のCF−1マウスを、20%のDMSOまたはビヒクル(20%DMSO)中2.5mg/kgのペプデュシン製剤で処置した。処置のために選択される時点は、盲端連結反応および穿刺(CLP)による非致死的敗血症の誘導である。2.5mg/kgの初期量の後、1mg/kgのペプデュシン(またはビヒクル)をマウスに対して6日間にわたり毎日皮下注射した。ビヒクル処置群からの4匹のマウスを、6日後に犠牲にした。xl/2i1LCA10ペプデュシンで処置される群からの1匹のマウスを6日目に犠牲にし、全ての残りの生存マウスを17日目に犠牲にした。図5は、結果を表す。CLPの時点で、左側パネルでは、マウスをxl/2pal16、xl/2i1LCA10またはビヒクル(20%のDMSO)で処置した。右側のパネルは、マウスの体重を毎日、モニターした。ビヒクルを処置されたマウスは重篤な体重損失を示したが、ペプデュシン処置されたマウスは最初の体重損失の後、体重が増えた。
【0185】
実施例2
全身性炎症反応症候群の発症の後でさえ、敗血症の処置が充分である報告は、ほんのわずかしかない。敗血症が起こったかなり後に、本発明のペプデュシンによって、敗血症が効果的に処置されることを示すために、マウスを、40%DMSOまたはビヒクル(40%のDMSO)中5.0mg/kgのペプデュシン製剤で処置した。処置のために選択される時点は、盲腸の結紮および穿刺(CLP)による非致死的敗血症の誘導時の8時間である。5.0mg/kgの初回量の後、2.5mg/kgのペプデュシン(またはビヒクル)を2日目から3日目までは毎日皮下注射し、3日目から6日まで毎日、1.0mg/kgをマウスに注射した。CLPの8時間後に、マウスはすでに敗血症に特有の全身性疾患の徴候(例えば、乱れた毛皮、減少した運動能、減少した食物および水摂取、ならびに閉じた炎症を起こした眼)を示した。図6は、結果を表す。左側パネル(*)で:ビヒクル処置群からの2匹のマウスを、6日目に犠牲にした。実験を生き残ったマウスを、17日目に犠牲にした。右側パネルにおいて、マウスの体重を毎日、モニターした。
【0186】
実施例3
炎症性疾患のペプデュシン処置の有用性を示すために、マウスを本発明のペプデュシンで処置した。図7は、結果を表す。成人性呼吸反応症候群(ARDS)は、肺への過剰な好中性浸入とそれらの活性化(線維素沈着に至る)から生じる。敗血症によってARDSが起きる場合があり、それは高い致死性を持つ。パネルAとB:マウスをCLPの24時間後に犠牲にし、BALを実行した。好中球をギムザ染色した後に計数した、TNFをELISAで測定した。*p<0.02対ビヒクル処置群。C.肺組織学。マウスを、CLPの48時間後に犠牲にした。
【0187】
実施例4
この実施例は、ペプデュシン処置がどのように敗血症のもう一つの態様を軽減するのに用いられるかについて示す。全体的な炎症反応、DICの凝固と線維素溶解の錯乱は、いくつかの炎症誘発性サイトカインによって媒介される。出血時間の変化とD−二量体の増加(それは、過剰な対抗制御的な線維素溶解の産物)は、播種性血管内血液凝固の特徴であり、血行によるフィブリンの広範囲にわたる堆積に至り、敗血症の患者における多臓器不全と死亡に関与する。図8は、結果を表す。A.出血時間をCLPの24時間後と処置開始直後に測定した。出血時間は、ビヒクルを処置されたマウスで短くなり、その一方で、i1−ループ・ペプデュシンx1/2i1LCA10−またはi3ループ・ペプデュシンx1/2pal16−処置マウスは、正常な出血時間を示した。B.D−二量体濃度は、CLPの48時間後に測定した。線維素沈着量の代用の標識として、CXCR2ペプデュシン処置マウスでよりビヒクルを処置されたマウスにおけるより高い範囲に、D−二量体がCLPの後に増加した。C.CLPの8時間後に処置されたマウスの出血時間を表す。D.CLPの8時間後に処置されるマウスのD−二量体濃度を表す。
【0188】
実施例5
炎症性疾患のペプデュシン処置の有用性を示すために、走化性アッセイを、修飾48ウエル・マイクロ走化性チャンバーで行った。1ミオ(mio)好中球/mLは、より低いウエル内の化学遊走物質に向けての移動が可能であった。好中球を、0.2%DMSOまたはペプデュシンのいずれでも前処理したN=3。図9は、結果を表す。
【0189】
実施例6
生体内で炎症性疾患のペプデュシン処置の有用性を示すために、走化性アッセイを、マウスで行った。マウスに対して、腹腔内に3%チオグリコール酸塩1mLを注射した。チオグリコール酸塩は分泌サイトカインに腹腔マクロファージを誘導し、白血球を腹腔に補充させる。ペプデュシンを、示された用量で静脈注射した。4時間後に、細胞を腹腔洗浄によって採取し、ギムザ染色を施し、40倍で計数した。N=60。図10は、結果を表す。
【0190】
実施例7
SDF−lαとCXCR4が未熟好中球を骨髄から補充することに関係していることが示された。CXCR4発現と活性化の高水準は骨髄に老化の好中球のホーミングに至るが、ケモカインが好中球のCXCR2受容体に作用する。すなわち、KC、Gro−αまたはIL−8がさらなる好中球の移動を導く。血液をCLP後1、3、5、7、および17日に採血し、白血球の計数をおこなった。ビヒクルを処置されたマウスにおける白血球の最初の減少が、観察された。しかし、これらのマウスは末梢白血球の高レベルを示し、実験の終わりまで高レベルで残った。xl/2pal16およびxl/2i1LCA10−処置されたマウスにおいて、白血球レベルは、正常範囲のままだった。x4i1pal12を処置されたマウスにおいて、白血球レベルは直ちに増加したが、非常に低いレベルまでCLPの5日後に減少した。この知見は、CXCR4のブロッキングまたはCXCR2の刺激がマウスの末梢血で好中球増多に至るという以前の報告に一致する。x4i1pal12処置後の末梢血の白血球の増加は、CXCR2遮断が末梢血好中球増多を予防することを示す。図11は、結果を表す。
【0191】
実施例8
走化性アッセイを、修飾された48穴マイクロ化学走性チャンバーで行った。1ミオ(mio)好中球/mLは、より低いウエル内の化学遊走物質に向けての移動が可能であった。好中球を、指示濃度で0.2%DMSOまたはペプデュシンのいずれかで前処理した。N=3。図12は、本発明のi1−ループ・ペプデュシンには強力および選択的な消炎作用があることを示す。
【0192】
実施例9
走化性アッセイを、修飾48穴マイクロ化学走性チャンバーで行った。1ミオ(mio)好中球/mLは、より低いウエル内のランテス(Rantes)−CCRl、CCR3、およびCCR5(20ng/mL)のためのリガンドに向けての移動が可能であった。好中球を、指示濃度で0.2%DMSOまたはペプデュシンのいずれかによって前処理した。N=3。図13は、本発明のペプデュシンi1ループが強力かつ選択的な消炎作用を持つことを示している。
【0193】
実施例10
臓器不全がSIRS患者の結果を決定するので、肝機能を調べた。マウスをCLPの24時間後に犠牲にし、ALT(肝機能の損失で増加する酵素)の血漿レベルを測定した。疑似処置マウス(すなわち盲腸の結紮と穿刺のない開腹)において、ALTの上昇は観察されなかった。ビヒクルとx4pal12処置されたマウスはALTの増加を明らかにした、xl/2pal16とxl/2LCA10処置されたマウスで、増加が観察された。しかし、それはビヒクルを処置されたマウスと比較して著しく少なかった。図22は、CXCR2ペプデュシンが肝機能を改善するのに用いることができるという結果を表す。
【0194】
実施例11
GLPを、6〜8週間目の雌のCF−Iマウスに対して行われた。右側パネルにおいて、CLPの時点に、x4i1pal12またはビヒクル(20%のDMSO)でマウスを処置された。2.5mg/kgの初回量の後、動物に1mg/kgのペプデュシンまたはビヒクルを6日にわたり毎日皮下注射した。左側パネルにおいて、マウスの体重を毎日、モニターした。図23は、結果を表す。
【0195】
実施例12
受容体の第1の細胞内ループ(i1)に基づくより選択的なPAR4ペプデュシンを開発した。i1ループは、i3ループと関連した受容体の反対側の上にあることから、PAR4に基づくI1ループ・ペプデュシンは、PARlの減少または非相互抑制を示す。PAR4は、PARlで単独または組み合わせで、HEK細胞上で発現した。PAR4i1ペプデュシン(P4pal−i1)は、完全にHEK細胞の上でPAR4の走化性反応を妨げて、図24Aおよび図24Bで示すようにそのペプチド・リガンド(AYPGKF(配列番号34))に対する血小板凝集を予防した。P4pal−i1は、PAR4に対して選択的であり、P4pal−i1はPARlの走化性反応を阻害しなかったし、図24Aないし図24Cで見られるように、それはPARlペプチド・リガンド(SFLLRN(配列番号35))に対する血小板凝集を明らかに阻害しなかった。P4pal−i1は、PARlとPAR4とが複合体を形成する場合、PAR4の結合したi1ペプデュシンがPARlからシグナル伝達にこれといって影響を及ぼさないことを示しているPAR4で、同時発現さえもPARlを阻害しなかった。同様に、単独で使用される場合、トロンビンもPARl(図24D)を活性化することから、P4pal−i1には3nMトロンビンに対する血小板凝集上で軽度の作用だけがあった。しかし、PARlアンタゴニスト(RWJ−56110)と結合して使われる場合に、P4pal−i1は3nMトロンビンに対する凝集を阻害した。このように、PARlまたはPAR4単独を標的とすることは、トロンビンに対する凝集に限られた作用を持つ。PARlおよびPAR4の同時阻害はトロンビンに対する反応を妨げることに効果的であり、それによって血栓症を減らすか、阻害する。
【0196】
実施例13
血小板PARlおよびPAR4反応に対するビバリルジンの作用は調べず、また血小板凝集に対するPAR4遮断と組み合わせたトロンビン−PARl相互作用を阻害する有効性について評価した。PARlのHir部位ビバリルジン(別名Hirulog(登録商標)、Angiomax(登録商標))に対するトロンビンの結合を抑制するために、ナノモルの親和性によるトロンビンとの結合を用いた。患者を急性冠動脈症候群で処置することでのその広範囲にわたる使用にもかかわらず、PARlおよびPAR4に依存する血小板活性化に対するビバリルジンの作用は、本発明に先だって決定されることはなかった。ビバリルジン単独またはビバリルジン・プラスRWJ−56110は、PAR4に依存する凝集のトロンビン活性化と同程度の50%有効濃度(10〜11nM)がPARl−抗体(図25A)で示された。11nMを超える濃度で、RWJ−56110がPARl−阻止抗体+ビバリルジンで補充された場合でも、トロンビンはPAR4に依存する凝集の完全な活性化を回復した。これらのデータは、Hir−遮断薬のどちらも血小板PAR4で完全にトロンビンの活性部位の相互作用を予防しなかったことを示す。しかし、PAR4ペプデュシン、P4pal−i1、のビバリルジンを処置された血小板への添加は、非常に高いトロンビン(12−16nM)濃度(図25B)であっても、凝集の広がりを著しく遅らせ、かつ阻害した。これらのデータは、PARlのHir−モチーフにドッキングしたトロンビンがPAR4の活性化を高めるが、高度に十分なトロンビン濃度で、PAR4ペプデュシンによって下流に封鎖されない限り、PAR4はトロンビンによって活性化される。
【0197】
実施例14
モルモットにおける動脈血栓症は、ビバリルジンとPAR4ペプデュシンの組合せを使用して予防される。モルモットで動脈血栓に関してビバリルジンとPAR4ペプデュシンの同時投与の有効性を評価する標準頸動脈障害モデルが、使われた。それらの血小板上でPARlがないマウスとは異なり、モルモットは機能性類似度をヒト血小板と共有し、PARlおよびPAR4を発現する。ヒルジンを使用している初期の結果と整合して、これが有意でなかったにもかかわらず、ビバリルジン(0.18mg/kg)単独は13分から20分まで平均動脈の閉塞時間を延長させた(図26)。P4pal−i1(0.13mg/kg)は、同程度の閉塞時間を延長した。ビバリルジンさらにP4pal−i1の共投与は、急性の動脈の閉塞に対して有意な(p=0.0001)保護を生じた。RWJ−58259で示されたように、PARlペプデュシン(Plpal−7)で1つのPARlの遮断は動脈血栓の部分的な保護だけを生じた。P4pal−i1に対する比較において、ビバリルジンによるPlpal−7のサプルメントは、動脈の閉塞時間の付加的な延長をしなかった。同時に、これらの生体内データは、PAR4も血栓症に対して有意な保護を達成するために遮断されなければならないことを示す。
【0198】
これらの実験は、PARlとPAR4ペプデュシン・アンタゴニストと阻止抗体とによる以前の知見をサポートする。PARlおよびPAR4だけを標的とすることは、治療的有効性を制限してしまうと思われる。代わりの治療的戦略として、トロンビンでPARlHirのモチーフの相互作用を予防することは、直接PARlを阻害して、間接的にPAR4を阻害するという二重利点を持つ。これらのデータは、広く使われている抗血栓症薬(ビバリルジン)がPARlおよびPAR4に依存する血小板凝集のトロンビン活性化を遮断する際に有効だったことを初めて証明する。PARlペプデュシンでなくPAR4ペプデュシンと結合して、ビバリルジンは急性の動脈血栓症を予防することが可能だった。
【0199】
実施例15
宿主防御に対する干渉なしで凝固カスケードの活性化をかなり予防するにつれて、本発明のペプデュシンは確立した全身性炎症および脈管損害を妨げるのに用いられる。例えば、本発明のi1ペプデュシン(CXCRl/2に基づく)は、CXCR4を相互抑制しない。i1ペプデュシンは、ヒトおよびマウス好中球上で完全にそれらの同族の受容体の走化性反応を妨げる。xl/2LCA−i1はCXCRl/2IL−8受容体に対して選択的で、SDF−1αの方へヒト、マウス白血球の移動を阻害しない。同様に、CXCRlまたはCXCR2で共同発現される場合でも、xl/2LCA−i1はCXCR4を阻害しない。これらのデータは、CXCRlとCXCR4受容体が複合体を形成する場合、CXCRlの結合したi1ペプデュシンがCXCR4からのシグナル伝達に影響を及ぼさないことを示す。
【0200】
正常マウスにおける対炎症誘発性条件下の好中性ホメオスタシスに対するCXCRl/2とCXCR4ペプデュシンの効果を評価した。白血球補充を、チオグリコール酸塩腹膜炎モデルで最初に評価した。それぞれ、xl/2pal−i3およびxl/2LCA−i1−ペプデュシンのIC50値は0.03mg/kgおよび0.15mg/kgであり、完全に好中球の腹腔への移住を阻害した。逆に言えば、CXCR4アンタゴニスト、x4pal−i1(EC50−0.1mg/kg)で処置されるマウスは、腹膜好中球の実質的な増加を示した。
【0201】
末梢的な白血球数のケモカイン・ペプデュシンの長期の作用を、健康な無処置のマウスで評価した。ペプデュシンを1日1回、サブQ(2.5mg/kg/1日目、1.0mg/kg/2〜6日目)注射し、循環する白血球レベルを1週間にわたって測定した。健康なマウスにおいて、xl/2pal−i3もxl/2LCA−i1も、ビヒクルを処置されたマウスと比較すると白血球数を変えなかった。対照的に、x4pal−i1は末梢血で白血球増加症を引き起こし、先行の研究と整合していた。末梢的な白血球数のケモカイン・ペプデュシンの作用は、盲腸の結紮と穿刺(CLP)を受ける敗血症のマウスに関して、全く同様だった。ビヒクルを処置されたマウスは、CLP後の24時間、また3日目の白血球増加症で最初の白血球減少症を示した。一日1回のxl/2pal−i3またはxl/2LCA−i1投与は、一度は正常範囲の中でCLP−マウスの好中球数を保ったが、x4pal−i1は1日目の早期白血球増加症と7日目までの白血球減少症に至った。同時に、これらのデータは、CXCR2およびCXCR4受容体がマウスで正常および炎症誘発性状態で好中性ホメオスタシスにおいて対向した役割を果たすことを示す。
【0202】
ケモカイン受容体ペプデュシンを、マウスにおいて致死CLP腹膜炎の進行を保護または潜在的に逆転させる能力について試験した。実験の第1セットには、ペプデュシンを2.5mg/kgの用量で、CLP手法直後に与えた。その後の用量は、処置が止められた6日目まで1mg/kgとした。マウスのいずれも、抗生物質療法を受けなかった。高度に有意な減少は、xl/2LCA−i1およびxl/2pal−i3ペプデュシンについて、敗血症誘導死亡率で観察された。著しく、17日間の観察期間にわたって、1/34ペプデュシン処置マウスだけは死亡したが、無処置のマウスの17/17は9日目で死亡した。6日目のCXCRl/2ペデュシン療法の中断にもかかわらず、生存はその後ほぼ100%残った。
【0203】
敗血症の診断はしばしば遅れ、また薬剤を予防様式で容易に投与することができないことから、ペプデュシン処置をCLP手法の8時間後まで保留とした。遅延処置であっても、死亡率の高度に有意な縮小は、無処置のマウスと比較すると見られた。総生存率は、無処置のマウスで0/20であったのに対して、遅延CXCRl/2ペプデュシン処置マウスでは26/30であった。CXCR4ペプデュシン(x4pal−i1)の投与には、生存に関しての効果はなかった。CXCRl/2−ペプデュシン処置マウスは、体重が増え、活性化し、ペデュシン療法が8時間遅れたときでも、1日目後に正常な身繕い行動を維持した。
【0204】
ペプデュシンによるCXCRl/2シグナル伝達の遮断は、敗血症のマウスで確立したSIRSのいくつかの基準を逆転させた。第一に、全身KCの上のCXCRl/2ペプデュシンの作用はフラットになり、マウスIL−8相同分子種を調べた。無処置のマウスにおいて、KCレベルはCLPの後、16時間にわたって上昇し、少なくとも48時間上昇するままだった。8時間の時点でのCXCRl/2ペプデュシンの投与の後で、全身KCレベルは、急速に低下して、その後低いままだった。無処置のCLPマウスの多数は24時間という早い時期に頻脈および低酸素血症ようになり、このことは十分に死亡率と相関した。敗血症の間、菌体内毒素は肺上皮を刺激して白血球を補充し、かつ活性化させるIL−8を隠すようにする。結果として生じる好中性縁のあることは、最終的には肺の損傷に至る。無処置のマウスの気管支肺胞洗浄(BAL)液体の好中球の数は、CLPの後、4時間という早い時期に100倍まで増加し、8時間後に200〜300倍まで上昇した。l/2pal−i3による処置または8時間のxl/2LCA−i1は、低レベルにとどまったBAL好中球の急速な低下を引き起こした。無処置のCLPマウスで48時間に収集される肺の組織学的分析は、崩壊した肺胞、白血球浸入と広範囲な線維素沈着を明らかにした。xl/2pal−i3およびxl/2LCA−i1は線維素沈着に対して有意な保護を与え、肺胞が組織学的に正常に見えた。CXCRl/2ペプデュシンも、LPSをシミュレートした上皮および内皮単分子層全体で完全に白血球の移動を妨害した。同様に、CXCRl/2ペプデュシンは、予想通りの単球でなくヒト大食細胞のIL−8に依存する走化性を抑えた。CXCRl/2ペプデュシンは、腹腔に有意に生体内マウス大食細胞移動を阻害した。同時に、これらのデータは、ペプデュシンは、多数の細胞型上でCXCRl/2受容体を封鎖することによってそれらの治療的作用を奏することを示している。
【0205】
肝不全は、重篤な敗血症の一般の後遺症である。肝酵素濃度(AST、ALT)で52−87%の減少によって明示されるように、CLPの直後のCXCRl/2ペプデュシンによる処置は肝障害を減らし、一方CXCR4ペプデュシン(x4pal−i1)には、作用がなかった。CXCRl/2ペプデュシン処置がCLPの8時間後に開始された場合、血漿ASTおよびALTレベルの上昇は16時間の時点までに停止し、そこから正常レベルに落ちた。同時に、これらのデータは、ペプデュシンによるCXCRl/2シグナル伝達の遮断が敗血症のマウスにおける確立したSIRSのいくつかの基準を逆転させることを示し、このアプローチが重症をセットする際に有益であることが示される。
【0206】
特発出血と血栓の発症は無処置のCLPマウスの多数で観察され、播種性血管内血液凝固(DIC)の発症と整合していた。したがって、全身血小板活性化は、血小板数およびD−二量体濃度、増加したフィブリン産生の標識、DIC間中に起こるその後の線維素溶解を測定することによって観察された。ペプデュシン処置がCLPの後、8時間まで遅れた場合でも、CXCRl/2ペプデュシンは24時間に有意に血小板減少症から保護した。D−二量体濃度が、無処置のマウスでCLP手法の48時間後に高度に上昇するとわかった。しかし、xl/2pal−i3およびxl/2LCA−i1−処置されたマウスは、D−二量体の濃度の有意な減少を示した。CXCRl/2ペプデュシン処置がCLP手法の後、8時間まで遅れたとき、D−二量体濃度は依然として60〜70%減少した。これらのデータは、明白な敗血症で発現する凝固障害がCXCR2の遮断によって改善される第1の実証である。
【0207】
我々の研究で観察されるより3〜4倍高い死亡率ではあるが、機能的なCXCR2受容体がないマウスを使用している最近の研究はCLP敗血症モデルで生存の増加を示した。CXCR2の欠失の作用と関連してペプデュシン処置に続いている死亡率の改善された結果は、いくつかの因子に起因することができた。CXCR2(−/−)マウスは、それらの全ての寿命の間にCXCR2がなくて、異常な骨髄造血に起因している急性のCXCR2特異的な作用と補償機構を区別することを難しくしているそれらの適応可能な免疫系に、更なる障害がある。使用されるCF−1マウスは、CLPから重度の免疫原性的取り組みの状況で生存利益を与えるかもしれない非近交系の野生型動物である。抗体CXCRl/2ペプデュシンが細菌fMLP等の他のケモカインの方へ白血球遊走を抑制しないことは注目に値する、したがって、ペプデュシン作用は免疫抑制性であるというよりはむしろ免疫調節性であると考慮されるかもしれない。さらにまた、ペプデュシン技術の用途は、遺伝学的なノックアウトから結果を確認するのを助けて、敗血症のような複合体疾患の病因と処置に対するさらなる洞察を提供する。
【0208】
等価物
当業者は、日常的実験、本明細書中に述べられる特異的な手法に対する多数の等価物しか使用しないことを理解し、または確かめることができる。そのような等価物は発明の範囲内であると考えられ、以下の請求の範囲によってカバーされる。請求の範囲によって定義されるように、種々の置換、変更、および修正は発明の精神および範囲から逸脱することなく発明が実施される。他の態様、利点、および変形は、発明の範囲内である。この明細書の全体を通じて引用されるすべての参考文献、出された特許と公開された特許出願の内容は、参照によってここに組み込まれる。適当な構成要素、プロセスとそれらの特許、明細書と他の文書の方法は、その発明と実施形態のために選択することが可能である。
【技術分野】
【0001】
発明の分野
本発明は、概ねGタンパク質共役型受容体(GPCR)に関し、詳しくは、GPCRアゴニストおよびアンタゴニスト、これらの化合物およびそれらの医薬組成物の使用、例えば、ケモカイン受容体が一因となる状態(例えば、敗血症、関節炎、炎症、および自己免疫性疾患)を処置する際等のGPCRが関与する生理的状態の処置、調節、および予防もしくはその一方における使用に関する。
【背景技術】
【0002】
発明の背景
種々のホルモン類、神経伝達物質、および生物活性物質は、細胞膜に位置する特異的な受容体を介して、生体の機能を制御、調節、または調整する。これらの受容体の多くは、受容体が連結する活性化グアニンヌクレオチド結合タンパク質(Gタンパク質)によって、細胞内シグナルの伝達をもたらす。この受容体は、Gタンパク質共役型受容体(「GPCR」)と一般的に呼ばれる。GPCRに対する特異的なシグナル伝達分子の結合は、受容体に構造変化を引き起し、その結果としてGタンパク質に対する結合およびGタンパク質の活性化が可能である形状となり、最終的には生物反応に導く細胞内現象のカスケードを誘発する。概して、GPCRはGタンパク質と相互作用して、細胞内第二メッセンジャー(例えば、サイクリックAMP、イノシトールリン酸、ジアシルグリセロール、およびカルシウムイオン)の合成を調節する。
【0003】
ケモカインは白血球誘引物質であって、白血球遊走を含む免疫プロセスに貢献する。白血球細胞内輸送は高度に調整されている、根底にある制御機構の破壊は強調された先天性免疫活性化(例えば、全身性炎症反応症候群または自己免疫性疾患)に関与する可能性がある。ケモカイン誘発シグナル伝達は、GPCRによってもたらされ、定義上、ケモカインの特徴は白血球の化学誘引性である。また、ケモカインは、細胞生存、ウイルス宿主相互作用、腫瘍成長および転移、器官形成、および脈管形成と同様に、白血球遊走性とは無関係である細胞性応答である。
【0004】
GPCRは、細胞の代謝、細胞増殖および運動性、癒着、炎症、神経シグナル伝達、ならびに血液凝固を制御するシグナル伝達プロセスにおいて、不可欠な役割を果たす。GPCRタンパク質にも、生身の機能を制御、調節、または調整する種々のシグナル伝達分子の標的として非常に重要な役割を有する。当該技術分野で周知であるように、GPCRは多種多様な障害に関係している。新規のGPCRモジュレータ(例えば、アゴニスト、部分的アゴニスト、インバースアゴニスト、およびアンタゴニスト)の開発には、敗血症、関節炎、炎症、および自己免疫性疾患等のGPCR関障害を処置するために、治療に応用可能である。
【発明の概要】
【課題を解決するための手段】
【0005】
発明の要旨
本発明は、GPCRの第1の細胞内ループ構造から誘導されるペプチドに付着する細胞貫通または細胞係留部分を含むペプデュシン(pepducin)類と呼ばれる修飾ペプチドの発見にもとづいている。ペプデュシン類はキメラのペプチド/ポリペチドと考え、受容体−Gタンパク質シグナル伝達のアゴニストおよびアンタゴニストもしくはその一方である。これらの組成物は、それらの同族の受容体に対して選択性を示す。
【0006】
したがって、本発明はペプデュシン組成物、すなわちGPCRの第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と第1の領域に結合した第2の領域を含むキメラポリペプチドを提供する。第2の領域は、天然に存在するまたは天然に存在しない細胞膜透過性および膜係留性疎水性部分もしくはその一方である。第1の領域は、好ましくはGPCR固有の細胞外部分を含まない。本発明のペプデュシンは、望ましくは第1の領域が誘導される同族のGPCRと結合する。
【0007】
第1の領域(Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメント)は、黄体形成ホルモン受容体、卵胞刺激ホルモン受容体、甲状腺刺激ホルモン受容体、カルシトニン受容体、グルカゴン受容体、グルカゴン様ペプチド1受容体(GLP−1)、向代謝性グルタミン酸受容体、副甲状腺ホルモン受容体、脈管活性腸幹ペプチド受容体、セクレチン受容体、成長ホルモン放出因子(GRF)受容体、プロテアーゼ活性受容体(PAR)、コレシストキニン受容器、ソマトスタチン受容体、メラノコルチン受容体、ADP受容体、アデノシンレセプター、トロンボキサン受容体、血小板活性化因子受容体、アドレナリン作動性受容体、5−HT受容体、ケモカイン受容体、神経ペプチド受容体、オピオイド受容体、副甲状腺ホルモン(PTH)受容体、または脈管活性腸幹ペプチド(VIP)受容体のアミノ酸配列を含む。
【0008】
例えば、第1の領域(Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメント)は、プロテアーゼ活性受容体(PAR)またはケモカイン受容体のアミノ酸配列を含む。プロテアーゼ活性受容体は、例えば、PARl、PAR2、PAR3、またはPAR4であってもよい。ケモカイン受容体は、CCまたはCXC受容体であってもよく、例えば、それぞれCCRl、CCR2、CCR3、CCR4、CCR5、CCR6、CCR7、CCR8、またはCCR9、あるいはCXCRl、CXCR2、CXCR3、CXCR4、CXCR5、CXCR6、またはCX3CR1が挙げられる。別の実施形態において、第1の領域(Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメント)が、例えば、コレシストキニンAおよびB(CCKA、CCKB)、ソマトスタチン−2(SSTR2)、メラノコルチン−4(MC4R)、グルカゴン様ペプチド−1受容体(GLP−1R)、P2Y12ADP受容体、あるいは「非定型」ケモカイン受容体(例えば、NKl、NK2、GRP/ボンベシン受容体、FPR1、FPRL−1、C3aRまたはC5aR)に由来するものであってよい。特定の実施形態において、本発明のペプデュシン類は、PAR2、CXCRl、CXCR2、CXCR4、およびCCR5ケモカイン受容体に対するものを含む。
【0009】
第2の領域(細胞膜透過性および膜係留性疎水性部分もしくはその一方)は、第1の領域のN末端、C末端、C末端アミノ酸とN末端アミノ酸との間のアミノ酸、またはN末端およびC末端の両方で結合する。望ましくは、細胞膜透過性および膜係留性疎水性部分もしくはその一方は、直鎖脂肪酸、例えばノナノイル(C9)、カプリル(C10)、ウンデカノイル(C11)、ラウロイル(C12)、トリデカノイル(C13)、ミリストイル(C14)、ペンタデカノイル(C15)、パルミトイル(C16)、フィタノイル(メチル置換C16)、ヘプタデカノイル(C17)、ステアロイル(C18)、ノナデカノイル(C19)、アラキドイル(C20)、ヘニエコサノイル(C21)、ベヘノイル(C22)、トルシサノイル(trucisanoyl)(C23)、およびリグノセロイル(C24)部分である。細胞膜透過性および膜係留性部分もしくはその一方は、アミド結合、スルフヒドリル、アミン、アルコール、フェノール基、または炭素−炭素結合によって、キメラ化合物に結合したものであってもよい。特定の実施形態は、疎水性部分としてパルミトイルまたはリトコール酸(またはその塩類)を含む。他の細胞膜透過性および膜係留性疎水性部分もしくはその一方が、コレステロール、リン脂質、ステロイド、スフィンゴシン、セラミド、オクチル−グリシン、2−シクロヘキシルアラニン、ベンゾリルフェニルアラニン、C1またはC2アシル基、あるいはC3〜C8の脂肪酸が挙げられる。
【0010】
本発明は、さらに、本発明のペプデュシン組成物と薬学的に許容される担体とを含む医薬組成物に関し、また一つ以上の容器にこれらの医薬組成物が含まれるキットに関する。
【0011】
本発明は、Gタンパク質共役型受容体(GPCR)の第1の細胞内細胞内ループ(i1ループ)の第1の領域またはそのフラグメントとこの第1の領域に結合する第2の領域とを含むペプデュシンを投与することによって、哺乳類被検体等で敗血症の処置、敗血症重篤度の低減、または敗血症の予防をおこなうための方法を含む。第2の領域は、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である。被検体は、敗血症を発症またはそのリスクがあると診断されている。
【0012】
組成物はまた、炎症および脈管形成もしくはその一方の処置、その重篤度の減少、あるいは予防するために、用いられる。炎症および脈管形成もしくはその一方を処置または予防する方法は、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)の第1の領域またはそのフラグメントと、第1の領域に結合し、かつ天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である第2の領域とから構成されるキメラポリペプチドを含むケモカイン阻害性ペプデュシンを投与することによって実施される。
【0013】
組成物はまた、癌の重篤度を処置する、または減少させるために、用いられる。癌の重篤度を処置する、または減少させる方法は、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)の第1の領域またはそのフラグメントと、第1の領域に結合し、かつ天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である第2の領域とから構成されるキメラポリペプチドを含むペプデュシンを投与することによって実施される。
【0014】
組成物は、血栓症、例えば冠状動脈、動脈性および静脈(例えば、深静脈または腸間膜)血栓症の重篤度を処置または低減させるためにも用いられる。血栓症の処置または予防の方法は、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)の第1の領域またはそのフラグメントと、第1の領域に結合し、かつ天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である第2の領域とから構成されるキメラポリペプチドを含むペプデュシンを投与することによって実施される。
【0015】
本発明はまた、炎症または障害を処置する、または予防する方法に関し、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)の第1の領域またはそのフラグメントと、第1の領域に結合し、かつ天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である第2の領域とを含むペプデュシンを、必要に応じて被検体に投与することによって実施される。処置に適した炎症性疾患として、慢性閉塞性肺疾患(COPD)、強直性脊椎炎(alkylosing spondylitis)、頸部関節炎(cervical arthritis)、線維筋痛症、虚血再灌流障害、腸管虚血、若年性関節リウマチ、腰仙骨関節炎変形性関節症(lumbosacral
arthritis osteoarthritis)、骨粗鬆症、乾癬性関節炎、リウマチ病、関節リウマチ、湿疹、乾癬、皮膚炎、ブドウ膜炎および結膜炎、喘息および気管支炎、潰瘍、歯肉炎、クローン病、萎縮性胃炎、胃炎バリアロフォルム(gastritis varialoforme)、潰瘍性大腸炎、セリアック病、限局性回腸炎、消化性潰瘍、発熱(pyresis)、膀胱刺激症状および膀胱炎、中枢神経系または末梢神経系の炎症性神経障害、多発性硬化症、エイズの炎症性ニューロパシーおよび神経性合併症、自己免疫性炎症または外科手術による外傷が挙げられる。本明細書中に述べられる組成物は、敗血症および関連病理学(例えば、播種性血管内凝固(DIC)、フィブリン溶解反応、および全身性炎症反応(SIRS)もしくはその一方)の重篤度の予防、逆転、または減少をおこなう。
【0016】
本発明のペプデュシン組成物は、GPCRの広範囲に及ぶ活性を活性化または阻害させるのに有用である。本発明にもとづくペプデュシンとして、CXCRl、CXCR2、CXCR3、CXCR4、CXCR5、CXCR6、およびCX3CR1を含むケモカインCXC受容体、CCRl、CCR2、CCR3、CCR4、CCR5、CCR6、CCR7、CCR8、およびCCR9を含むケモカインCC受容体、プロテアーゼ活性受容体(PAR)(例えば、PARl、PAR2、PAR4)、コレシストキニンAおよびB受容体(CCKA、1CCKB)、ソマトスタチン−2(SSTR2)受容体、メラノコルチン−4(MC4R)受容体、グルカゴン様ペプチド−1受容体(GLP−IR)、スフィンゴシン1−リン酸(SlP)受容体(例えば、亜型SlPlおよびS1P3)、EDG受容体、エンドセリン(ET)受容体(例えば、亜型ET−I、ET−2、ET−3、ETA、ETB)、EDG受容体(例えば、亜型EDG−I、EDG−2、EDG−3、EDG−4、EDG−5、EDG−6)、ならびにP2Y12 ADP受容体に作用するものが挙げられる。また、NKl、NK2、GRP/ボンベシン受容体、FPR−l、FPRLI、C3aR、およびC5aR等の「非定型」ケモカイン受容体のためのペプデュシンは、本発明の範囲内である。特定の実施形態において、本発明のペプデュシンは、PAR2、CXCRl、CXCR2、CXCR4とCCR5ケモカイン受容体に対するものを含む。
【0017】
投与は、例えば全身性炎症、COPDおよび敗血症もしくはその一方の場合、全身的に、例えば静注によって実施される。あるいは、組成物の送達は、経皮、経口、経鼻腔(例えば、喘息を処置するため)により、または局所的に、例えば、粘着パッチの形態、またはクリーム、泡、軟膏(例えば、皮膚炎、乾癬または他の皮膚炎症性条件の症状の緩和のために)の形態で行う。
例えば、本願発明は以下の項目を提供する。
(項目1)
キメラポリペプチドであって、
a)Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、
b)前記第1の領域に結合した第2の領域であって、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、第2の領域とを含み、
前記第1の領域が前記GPCRの天然の細胞外部分を含まない、キメラポリペプチド。
(項目2)
前記キメラポリペプチドがその同族のGPCRに結合する、項目1のキメラポリペプチド。
(項目3)
前記疎水性部分が前記第1の領域のN末端、C末端、C末端アミノ酸とN末端アミノ酸との間のアミノ酸、またはN末端およびC末端の両方に結合している、項目1に記載のキメラポリペプチド。
(項目4)
前記疎水性部分が脂質である、項目1に記載のキメラポリペプチド。
(項目5)
前記疎水性部分が、ノナノイル(C9)、カプリル(C10)、ウンデカノイル(C11)、ラウロイル(C12)、トリデカノイル(C13)、ミリストイル(C14)、ペンタデカノイル(C15)、パルミトイル(C16)、フィタノイル(メチル置換C16)、ヘプタデカノイル(C17)、ステアロイル(C18)、ノナデカノイル(C19)、アラキドイル(C20)、ヘニエコサノイル(C21)、ベヘノイル(C22)、トルシサノイル(C23)、およびリグノセロイル(C24)部分からなる群から選択され、前記疎水性部分がアミド結合、スルフヒドリル、アミン、アルコール、フェノール基、または炭素−炭素結合によって、キメラポリペプチドに結合している、項目4に記載のキメラポリペプチド。
(項目6)
前記疎水性部分がパラミトイル部分である、項目4に記載のキメラポリペプチド。
(項目7)
前記疎水性部分がリトコール酸またはその塩である、項目4に記載のキメラポリペプチド。
(項目8)
前記疎水性部分がコレステロール、リン脂質、ステロイド、スフィンゴシン、セラミド、オクチル−グリシン、2−シクロヘキシルアラニン、ベンゾリルフェニルアラニン、C1またはC2アシル基、およびC3〜C8の脂肪酸からなる群から選択される、項目4に記載のキメラポリペプチド。
(項目9)
前記第1の領域がプロテアーゼ活性化受容体(PAR)またはケモカイン受容体を含み、前記第2の領域が脂質部分を含む、項目1に記載のキメラポリペプチド。
(項目10)
前記プロテアーゼ活性化受容体がPAR1、PAR2、PAR3、およびPAR4からなる群から選択される、項目9に記載のキメラポリペプチド。
(項目11)
前記ケモカイン受容体が、CCRまたはCXCR受容体からなる群から選択される、項目9に記載のキメラポリペプチド。
(項目12)
前記CCR受容体がCCR1、CCR2、CCR3、CCR4、CCR5、CCR6、CCR7、CCR8、およびCCR9からなる群から選択される、項目11のキメラポリペプチド。
(項目13)
前記CCR受容体が、CXCR1、CXCR2、CXCR3、CXCR4、CXCR5、CXCR6、およびCX3CR1からなる群から選択される、項目11に記載のキメラポリペプチド。
(項目14)
前記ケモカイン受容体がNK1、NK2、MLT2、GRP/ボンベシン、FPR1、FPRL−1、C3Ar、OR受容体からなる群から選択される、項目9に記載のキメラポリペプチド。
(項目15)
前記Gタンパク質共役型受容体またはそのフラグメントが、黄体形成ホルモン受容体、卵胞刺激ホルモン受容体、甲状腺刺激ホルモン受容体、カルシトニン受容体、EDG受容体、エンドセリン(ET)受容体、グルカゴン受容体、グルカゴン様ペプチド1受容体(GLP−I)、代謝調節型グルタミン酸受容体、副甲状腺ホルモン受容体、血管作動性腸管ペプチド受容体、セクレチン受容体、成長ホルモン放出因子(GRF)受容体、プロテアーゼ活性化受容体(PAR)、コレシストキニン受容体、ソマトスタチン受容体、メラノコルチン受容体、ADP受容体、アデノシン受容体、トロンボキサン受容体、血小板活性化因子受容体、アドレナリン作動性受容体、5−HT受容体、CXCR4、CCR5、ケモカイン受容体、ニューロペプチド受容体、オピオイド受容体、副甲状腺ホルモン(PTH)受容体、スフィンゴシン1−リン酸(SlP)受容体、および血管作動性腸管ペプチド(VIP)受容体からなる群から選択される、項目1に記載のキメラポリペプチド。
(項目16)
前記フラグメントが単離された細胞内フラグメントである、項目1に記載のキメラポリペプチド。
(項目17)
前記フラグメントが単離された細胞外フラグメントである、項目1に記載のキメラポリペプチド。
(項目18)
医薬組成物であって、項目1に記載のキメラポリペプチドと薬学的に許容される担体とを含む、医薬組成物。
(項目19)
キットであって、1つ以上の容器に項目18に記載の医薬組成物を含む、キット。
(項目20)
前記Gタンパク質共役型受容体が哺乳類のGタンパク質共役型受容体である、項目1に記載のキメラポリペプチド。
(項目21)
前記Gタンパク質共役型受容体がCCKA、CCKB、SSTR2およびSubP受容体からなる群から選択される、項目1に記載のキメラポリペプチド。
(項目22)
前記疎水性部分がステロイドである、項目1に記載のキメラポリペプチド。
(項目23)
前記疎水性部分が、リン脂質、ステロイド、スフィンゴシン、セラミド、オクチル−グリシン、2−シクロヘキシルアラニン、およびベンゾリルフェニルアラニンからなる群から選択される、項目1に記載のキメラポリペプチド。
(項目24)
哺乳類被検体の敗血症を処置または予防する方法であって、
処置を必要としている患者を診断すること、および
Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチドをそれを必要とする患者に投与すること
を含む、方法。
(項目25)
炎症を処置または予防する方法であって、
処置を必要とする患者を診断すること、および
Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチドをそれを必要とする患者に投与すること
を含む、方法。
(項目26)
血管新生を処置または予防する方法であって、
処置を必要とする患者を診断すること、および
Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分であるケモカイン阻害性ペプデュシンをそれを必要とする患者に投与すること
を含む、方法。
(項目27)
炎症性疾患を処置または予防する方法であって、
処置を必要とする患者を診断すること、および
Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチドをそれを必要とする患者に投与すること
を含む、方法。
(項目28)
前記炎症性疾患が、強直性脊椎炎、頸部関節炎、線維筋痛症、腸管虚血、若年性関節リウマチ、腰仙骨関節炎変形性関節症、骨粗鬆症、乾癬性関節炎、リウマチ病、関節リウマチ、湿疹、乾癬、皮膚炎、ブドウ膜炎および結膜炎、喘息および気管支炎、潰瘍、歯肉炎、クローン病、萎縮性胃炎、胃炎バリアロフォルム、潰瘍性大腸炎、セリアック病、限局性回腸炎、消化性潰瘍、発熱、膀胱刺激症状および膀胱炎、中枢神経系または末梢神経系の炎症性神経障害、多発性硬化症、エイズの炎症性ニューロパシーおよび神経性合併症、自己免疫性炎症、ならびに外科手術による外傷からなる群から選択される、項目27に記載の方法。
(項目29)
虚血再灌流障害を処置または予防する方法であって、
処置を必要とする患者を診断すること、および
Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチドをそれを必要とする患者に投与すること
を含む、方法。
(項目30)
癌の重篤度を処置または減少させる方法であって、
処置を必要とする患者を診断すること、およびGタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチドをそれを必要とする患者に投与することを含む、方法。
(項目31)
血栓症の重篤度を処置または減少させる方法であって、
処置を必要とする患者を診断すること、およびGタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチドをそれを必要とする患者に投与することを含む、方法。
(項目32)
キメラポリペプチドであって、
D1(ILKMKVKKPAV)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目33)
キメラポリペプチドであって、
D1(ATQAPRLPST)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目34)
キメラポリペプチドであって、
D1(VLATGAPRLPST)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目35)
キメラポリペプチドであって、
D1(ATGAPRLPST)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目36)
キメラポリペプチドであって、
D1(FLFRTKKKHPAV)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目37)
キメラポリペプチドであって、
D1(ILYSRVGRSVTD)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目38)
キメラポリペプチドであって、
D1(YSRVGRSVTD)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目39)
キメラポリペプチドであって、
D1(YQKKLRSMTD)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目40)
キメラポリペプチドであって、
D1(MGYQKKLRSMTD)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目41)
キメラポリペプチドであって、
D1(KRLKSMTD)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目42)
キメラポリペプチドであって、
D1(LINCKRLKSMTD)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目43)
キメラポリペプチドであって、
D1(VLATQAPRLPST)を含み、D1がポリペプチド配列のいずれかの端部に結合した天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、キメラポリペプチド。
(項目44)
敗血症を処置または予防するための薬剤の製造でのキメラポリペプチドの使用であって、前記キメラポリペプチドが、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、使用。
(項目45)
炎症を処置または予防するための薬剤の製造でのキメラポリペプチドの使用であって、前記キメラポリペプチドが、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、使用。
(項目46)
血管新生を処置または予防するための薬剤の製造でのキメラポリペプチドを含むケモカイン阻害性ペプデュシンの使用であって、前記キメラポリペプチドが、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、使用。
(項目47)
炎症性疾患を処置または予防するための薬剤の製造でのキメラポリペプチドの使用であって、前記キメラポリペプチドが、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、使用。
(項目48)
前記炎症性疾患が、強直性脊椎炎、頸部関節炎、線維症、虚血再灌流障害、腸管虚血、若年性関節炎、腰仙骨関節炎変形性関節症、骨粗鬆症、乾癬性関節炎、リウマチ性疾患、関節リウマチ、湿疹、乾癬、皮膚炎、ブドウ膜炎および結膜炎、喘息および気管支炎、潰瘍、歯肉炎、クローン病、萎縮性胃炎、胃炎バリアロフォルム、潰瘍性大腸炎、セリアック病、限局性回腸炎、消化性潰瘍、発熱、膀胱刺激症状および膀胱炎、中枢または末梢神経系の炎症性神経障害、多発性硬化症、エイズの炎症性ニューロパシーおよび神経性合併症、自己免疫性炎症、ならびに外科手術による外傷からなる群から選択される、項目47に記載の使用。
(項目49)
癌の重篤度を処置、予防、または減少するための薬剤の製造でのキメラポリペプチドの使用であって、前記キメラポリペプチドが、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、使用。
(項目50)
血栓症の重篤度を処置、予防、または減少するための薬剤の製造でのキメラポリペプチドの使用であって、前記キメラポリペプチドが、Gタンパク質共役型受容体(GPCR)の第1の細胞内ループ(i1ループ)またはそのフラグメントの第1の領域と、前記第1の領域に結合した第2の領域とを含み、前記第2の領域が、天然に存在するまたは天然に存在しない細胞膜透過性の膜係留性疎水性部分である、使用。
【図面の簡単な説明】
【0018】
【図1】図1は、GPCRトポロジーを示す図である。i1ループをこの図に示す。
【図2】図2は、構造の類似性を説明するために整列されている膜貫通型フランキング部を含むCXCR1およびCXCR2i1ループ配列を示す図である。
【図3】図3は、本発明にもとづくペプデュシンが作用するケモカイン受容体、各々のリガンド、および受容体が見出される各々の細胞型を示す図である。
【図4】図4は、本発明にもとづくペプデュシンが作用するさらに別のケモカイン受容体、各々のリガンド、および受容体が見出される各々の細胞型を示す図である。
【図5】図5は、実施例1に詳述した実験におけるCLP時のペプデュシン療法に対する動物の反応を示す線グラフである。
【図6】図6は、CLP後8時間で開始されたペプデュシン療法の結果が示されている、実施例2で詳述した実験の結果を示す線グラフである。
【図7】図7Aないし図7Cは、実施例3にさらに詳述したように、ペプデュシン処置後の肺への白血球浸潤の抑制と肺でのTNF−α産生および繊維素沈着とを示す。図7Aおよび図7Bは棒グラフであり、図7Cは顕微鏡写真である。
【図8】図8Aないし図8Dは、ペプデュシン処置後、敗血マウスでの出血時間短縮を示す棒グラフである。
【図9】図9は、実施例5により詳しく示されるように、ヒト好中球走化性がどのようにしてペプデュシン処置によって阻害されるかについて示すグラフである。
【図10】図10は、実施例6により詳しく示されるように、生体内でのペプデュシン処置によって、どのようにして白血球化学走性が阻害されるかについて示しているグラフである。
【図11】図11は、実施例7でより詳しく示されるように、実験の結果を示す線グラフである。
【図12】図12は、実施例8により詳しく示されているように、ヒト好中球走化性の抑制を示ことによって、どのようにして本発明のペプデュシンが炎症を選択的に減少させるかを示すグラフである。
【図13】図13は、実施例9により詳しく示されているように、ヒト単核細胞化学走性の抑制を示すことによって、どのようにして本発明のペプデュシンが炎症を選択的に減少させるかを示すグラフである。
【図14】図14は、本発明にもとづいてペプデュシンで使用することが可能な膜貫通フランキング部を含むCXCR4i1ループ配列を示す図である。
【図15】図15は、本発明にもとづくペプデュシンで使用することが可能な膜貫通フランキング部を含むCCR1i1ループ配列を示す図である。
【図16】図16は、本発明にもとづくペプデュシンで使用することが可能な膜貫通フランキング部を含むCCR2i1ループ配列を示す図である。
【図17】図17は、本発明にもとづくペプデュシンで使用することが可能な膜貫通フランキング部を含むCCR4i1ループ配列を示す図である。
【図18】図18は、本発明にもとづくペプデュシンに使用することが可能な膜貫通型フランキング部を含むCCR5i1ループ配列を示す図である。
【図19】図19は、本発明にもとづくペプデュシンに使用することが可能な膜貫通型フランキング部を含むPAR1i1ループ配列を示す図である。
【図20】図20は、本発明にもとづくペプデュシンに使用することが可能な膜貫通型フランキング部を含むEDGi1ループ配列を示す図である。
【図21】図21は、本発明にもとづくペプデュシンに使用することが可能な膜貫通型フランキング部を含むNK1−Ri1ループ配列を示す図である。
【図22】図22は、実施例10により詳しく述べられる実験の結果を示す棒グラフである。
【図23】図23は、実施例11により詳しく述べられる実験の結果を示すグラフである。
【図24A】図24Aは、HEK細胞を示す棒グラフであり、PAR1およびPAR4もしくはその一方を過渡的に形質移入され、24時間にわたり300μMのP4pa1−i1の存在または非存在下で0.5μMのTFLLRN(配列番号33)または500μMのAYPGKF(配列番号34)に向けて移行することができるHEK細胞を示す棒グラフである。
【図24B】図24Bは、160μMのAYPGKFを添加する前に3μMのP4pal−i1または緩衝液(未処理)で2分間、あらかじめインキュベートしたヒト血小板を示すグラフである。
【図24C】図24Cは、13μMのSFLLRNを添加に先立って3μMのP4pal−i1または緩衝液(未処理)で2分間、あらかじめインキュベートしたヒト血小板を示すグラフである。
【図24D】図24Dは3μMトロンビン(T)の添加する前に1μMのRWJ−56110、5μMのP4pal−i1、または1μMのRWJ−56110プラス5μMのP4pal−i1で2分間、あらかじめインキュベートした血小板を示す図である。これらの図に示すデータは、実施例12に詳述する実験の結果である。
【図25A】図25Aは、ビバリルジンとP4a1−i1阻害トロンビン依存型凝集との組み合わせを示すグラフである。ヒト血小板を、20pMないし20nMトロンビンの添加に先立って、緩衝液(未処理)、ビバリルジン(200nM)、RWJ−56110(1μM)、およびPAR1−Ab(74μg/ml)もしくはその一方とともに、2分間にわたり、事前のインキュベーションをおこなった。
【図25B】図25Bは、示された濃度のトロンビンの添加に先立って、血小板を2分間にわたり、200nMのビバリルジン、プラスまたはマイナス5μMP4pal−i1と事前のインキュベーションをおこなったことを示すグラフである。これらの図に示すデータは、実施例13に詳述する実験の結果である。
【図26】図26は、モルモットでの頸動脈閉鎖症を妨げているビバリルジンとP4pal−i1との組み合わせを示す棒グラフである。モルモットに対して、実施例14に述べられるように、FeCl3による頸動脈の損傷に対して5分間先だって、ビバリルジン、P4pal−i1、およびP1pal−7(各々の処置群についてn=3ないし5)もしくはその一方による処理をおこなった。ビヒクル処置に関連するP値を一番下に示す。
【図27】図27は、乳癌細胞および卵巣癌細胞の化学走性をi1ループ・ペプデュシンがどのようにして阻害しているかを示すグラフである。NIH3T3から条件培地を調製した。化学組成アッセイ(20時間)を、8mm孔ニトロセルロース・フィルターを備えた48ブラインドウェル・マイクロケモタキシス・チャンバー(Neuroprobe)を用いて、OVACAR−4ヒト卵巣癌細胞およびMDA−MB−231ヒト乳癌細胞に対しておこなった。データを化学走性指数として表し、この指数はRPMI培地単独への移動とNIH3T条件培地への移動との距離の比である。
【発明を実施するための形態】
【0019】
発明の詳細な説明
定義
便宜上、明細書、実施例、および添付の特許請求の範囲に使用した特定の用語をここに集める。
【0020】
「処置する」とは、状態、疾患、障害等、またはその症状の改善をもたらす任意の作用、例えば、軽減、減少、変調、または排除を含む。
【0021】
「GPCRフラグメント」は、GPCRの全ての天然のアミノ酸配列よりも少ないGPCRタンパク質配列の一部を有するペプチドを含む。「単離GPCRフラグメント」は、全ての配列よりも少ないGPCRタンパク質配列の一部を有するが、天然のフランキング領域は含まないペプチドを含む。単離されたGPCRフラグメントは、天然の分子にある参照フラグメントを直ちにフランキングする1つ以上のアミノ酸が欠けている。
【0022】
「単離細胞内GPCRフラグメント」は、GPCRタンパク質の細胞内i1ループのアミノ酸配列を有するが、細胞外ループまたは細胞内i1ループをフランキングする膜貫通型ヘリクス配列に由来する配列を持たないペプチドを含む。「単離細胞外GPCRフラグメント」は、GPCRタンパク質の細胞外ループのアミノ酸配列を有するが、細胞外ループまたは細胞外ループの領域をフランキングする膜貫通型ヘリクス配列に由来するアミノ酸を持たないペプチドを含む。
【0023】
「連結」とは、結合を意味する。例えば、ペプチドと細胞貫通または係留部分は、連結(すなわち共有結合)を経て、ペプデュシンと各々結合する。好ましくは、連結は不安定な結合(例えば、チオールまたはエステル結合)である。不安定な連結を有するペプデュシン化合物は、不安定ではない連結の化合物と比べて体内組織での蓄積が少ないことから、有利である。被験者に対する投与後の身体組織での蓄積が減少することは、被験者での副作用の減少をともなう。
【0024】
「GPCRアゴニスト」は、その受容体に特異的な内因性シグナル伝達分子の作用を模倣するためにGPCRを活性化する組成物を含む。「GPCRアンタゴニスト」は、GPCR活性を阻害する組成物を含む。GPCR活性の測定を、Gタンパク質等のエフェクター・シグナル伝達分子と結合する能力でおこなう。「活性GPCR」は、Gタンパク質と相互作用して活性化することができる。自己抑制的な受容体は、細胞外リガンドと結合し、およびGタンパク質と生産的に相互作用して、もしくはその一方を行い、このGタンパク質を活性化させる能力を有する。
【0025】
「細胞膜透過性部分」は、細胞外間隙から細胞の細胞内区画まで物質の転送を媒介する化合物または官能基を含む。細胞膜透過性成分は、細胞質内または細胞膜の細胞質空間へ連結物質(例えば、本発明のGPCRペプチドまたはフラグメント)を往復させる。例えば、細胞膜透過性部分は疎水性部分である。疎水性部分は、少なくとも約11アミノ酸長の、例えば、混合配列ペプチドまたはホモポリマー・ペプチド(ポリロイシンまたはポリアルギニン)である。この物質は、本発明のGPCRフラグメントまたはペプチド模倣薬であってもよい。細胞膜透過性部分として、少なくとも10個の隣接アミノ酸、例えばGPCR膜貫通型ヘリクス領域の1ないし15アミノ酸を挙げることができる。
【0026】
「膜係留性部分」として、細胞膜に関係または結合する化合物または官能基が挙げられる。したがって、膜係留性部分は、標的細胞の膜近傍で膜係留性部分が付着する物質(すなわち、本発明のGPCRフラグメントまたはペプチド模倣薬)をもたらす。細胞膜は真核生物または原核生物である。膜係留性部分は、望ましくは疎水性部分である。疎水性部分は、アミノ酸長が10アミノ酸未満であるポリロイシンまたはポリアルギニン等のホモポリマー・ペプチドまたは混合配列ペプチドを含むことができる。膜係留性部分は、GPCR膜貫通型ヘリクス領域の少なくとも1〜7つの隣接アミノ酸を含むことができる。好ましくは、膜係留性部分は、GPCR膜貫通型領域の少なくとも10個の隣接アミノ酸(しかし、16未満のアミノ酸)である。より好ましくは、膜係留性部分は、GPCR膜貫通型領域の少なくとも15個の隣接アミノ酸である。膜係留性部分は、コレステロール、リン脂質、ステロイド、スフィンゴシン、セラミド、オクチル−グリシン、2−シクロヘキシルアラニン、またはベンゾリルフェニルアラニンも含む。他の膜係留性部分は、C1またはC2アシル基またはC3〜C8脂肪酸成分、例えばプロピオノイル(C3)、ブタノイル(C4)、ペンタノイル(C5)、カプロイル(C6)、ヘプタノイル(C7)、およびカプリロイル(C8)を含む。膜係留性部分は、ペプデュシンのGPCRフラグメントのC末端アミノ酸、N末端アミノ酸、またはN末端アミノ酸とC末端アミノ酸との間のアミノ酸に結合することが可能である。
【0027】
「製薬的または薬理的に許容される」は、動物、またはヒトに対して投与した場合に、それぞれに見合った副作用、アレルギー反応、または他の有害反応を生ずることのない分子実体または組成物を包含する。「製薬的に許容される担体」は、任意または全ての溶媒、分散媒、コーティング、抗菌、および抗真菌剤、等張および吸収遅延剤、その他を包含する。製薬活性物質のためのそのような培地および薬剤の使用は、周知の技術である。任意の従来の培地または薬剤が主成分と適合しない場合を除いて、治療用組成物でのその使用が検討される。補助的活性成分も組成物に取り込むことができる。
【0028】
「低分子」は、約5kD未満、最も好ましくは約4kD未満の分子量を持つ組成物を含む。低分子を、核酸、ペプチド、ポリペチド、ペプチド模倣薬、炭水化物、脂質、または他の有機分子あるいは無機分子とすることができる。化学的および生物学的混合物もしくはその一方(例えば、菌類、細菌、または藻類抽出物)のライブラリは、当該技術分野で知られており、本発明のアッセイのいずれかによってスクリーニングすることができる。
【0029】
「標的分子」として、GPCRタンパク質が本来結合または相互作用する分子、例えばGPCR相互作用タンパク質を発現する細胞表面上の分子、第二の細胞表面上の分子、細胞外環境にある分子、細胞膜の内面に結合した分子、または細胞質分子が挙げられる。GPCR標的分子を非GPCR分子または本発明のGPCRペプチドとすることができる。一実施形態では、GPCR標的分子は、例えば膜結合GPCRに対する化合物の結合によって生ずるシグナル等の、細胞膜を介して細胞内への細胞外シグナル伝達を促すシグナル伝達経路の構成要素である。標的は、下流のシグナル伝達分子とGPCRとの結合を促進する触媒活性またはタンパク質を有している第二の細胞間タンパク質である。
【0030】
「併用療法」(または「共同療法」)は、本発明のペプデュシンの投与と特異的な治療計画の一部としての少なくとも第2の薬剤とを含み、これらの治療薬の相互作用による有益な効果を提供することを意図している。併用の有益な効果は、治療薬の併用から生ずる薬動力学的または薬力学的相互作用を含むが、これに限定されるものではない。これらの治療薬の併用投与は、所定の時間(通常、選択される組合せによって、分、時間、日または週)にわたって、概して実施される。「併用療法」は、一般的ではないが、本発明の併用を偶然または任意にもたらす別々の単独療法計画の一部として、これらの治療薬を二種類以上投与することを包含することを意図していることもある。「併用療法」は、これらの治療薬の投与を順番におこなうことを意図している。すなわち、これらの治療薬の投与と同様に、各々の治療薬、または治療薬のうちの少なくとも2つを、異なる時間に投与するとともに、これらの治療薬の投与と同様に、または少なくとも2つの治療薬含むことを目的とする。実質的な同時投与は、例えば、被検体に対して、一定の比率の各々の治療薬を含む単一のカプセルまたは治療薬の各々に対応した単一のカプセルを複数、投与することによって達成し得る。各治療薬の経時的投与または実質的な同時投与は、限定されるものではないが、経口経路、静脈経路、筋内経路、および粘膜組織を通しての直接吸収を含む適当な経路によっておこなうことができる。治療薬を同一の経路または異なる経路で投与することができる。選択された組み合わせの第1の治療薬を静脈内注射により投与し、この組み合わせの他の治療薬を経口投与してもよい。あるいは、例えば、すべての治療薬を経口投与することも可能であり、またはすべての治療薬は静脈内注射によって投与することも可能である。治療薬が投与される順番は、かなりクリティカルであるというものではない。「併用療法」も、他の生物学的に活性成分と非薬物療法(例えば、手術または放射線処置)とのさらなる併用で、先に述べたように治療薬の投与を含むことができる。併用療法が非薬物処置をさらに含む場合、治療薬と非薬物処置とを組合せることによる相互作用から有益な効果が達成される限り、非薬物処置を任意の適当な時刻におこなうことができる。例えば、適当な症例では、非薬物処置が治療薬の投与から一時的に除外される場合、おそらく数日、さもなければ数週間まで、有益な効果がさらに達成される。
【0031】
「相同アミノ酸配列」が意味することは、アミノ酸配列、すなわち、本発明のペプデュシンでは、1つ以上(例えば、1、2、3、4、または5)の保存的アミノ酸置換によって、またはポリペプチドの活性に副作用を及ぼさない位置での1つ以上(例えば、1、2、3、4、または5)非保存的アミノ酸置換、欠失、または付加によって、参照アミノ酸配列とは異なる。好ましくは、そのような配列は、参照アミノ酸配列に対して少なくとも75%、80%、85%、90%、または95%同一である。
【0032】
相同アミノ酸配列として、参照アミノ酸配列と同一または実質的に同一であるペプチド配列が挙げられる。「実質的に同一であるアミノ酸配列」は、参照のアミノ酸配列に対して、少なくとも90%、好ましくは95%、より好ましくは97%、さらに最も好ましくは99%同一であり、好ましくは参照の配列とは、たとえ保存的アミノ酸置換の大部分によるものでも、異なる。
【0033】
保存的アミノ酸置換として、主として、同一クラスのアミノ酸間での置換が挙げられる。これらのクラスとして、例えば、(a)無電荷極性側鎖(例えば、アスパラギン、グルタミン、セリン、スレオニン、およびチロシン)、(b)塩基性側鎖を持つアミノ酸(例えば、リシン、アルギニン、およびヒスチジン)、(c)酸性側鎖を持つアミノ酸(例えば、アスパラギン酸およびグルタミン酸)、ならびに(d)非極性側鎖を有するアミノ酸、例えばグリシン、アラニン、バリン、ロイシン、イソロイシン、プロリン、フェニルアラニン、メチオニン、トリプトファン、ならびにシステインが挙げられる。
【0034】
相同は、主として、配列分析用ソフトウェアを用いて測定される(例えば、ウィシコンシン大学ジェネティック・コンピュータ・グループ(Genetics Computer Group,University of Wisconsin Biotechnology Center,1710 University Avenue,Madison,WI 53705)の配列分析用ソフトウェア・パッケージ(Sequence
Analysis Software Package))。最大限の相同(すなわち同一性)を得るために、類似のアミノ酸配列を整列させる。この目的のために、人工的に隙間を配列に導入することが必要と考えられる。一旦最適なアラインメントが設定されれば、相同(すなわち同一性)の度合いは位置の全てを記録することによって確立され、全位置数に対して、両方の配列のアミノ酸は同一である。
【0035】
類似性因子として、同程度の大きさ、形状、および電荷が挙げられる。アミノ酸類似度を決定する特に好ましい方法の一つは、ダイホフ他(Dayhoff et al.,5
ATLAS OF PROTEIN SEQUENCE AND STRUCTURE
345−352(1978 & Suppl.))(本明細書中に援用)で述べられるPAM250マトリックスである。類似度のスコアは、整列配置された対合アミノ酸類似度のスコアの合計として、最初に算出される。挿入および欠失は、相同率および同一率の目的にとって無視される。したがって、ギャップ・ペナルティはこの算出で使われない。生スコアを、候補化合物と参照配列のスコアの幾何平均によって分ることで正規化する。幾何平均は、これらのスコアの製品の二乗根である。正規化された生スコアは、相同率である。
【0036】
好ましくは、相同配列は参照アミノ酸配列に対して少なくとも45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、または95%同一である。この明細書(例えば、表3)に示す配列の一つに対して配列相同性を持つポリペプチドとして、天然の対立遺伝子変異体が挙げられ、同様に、機能に関して類似している突然変異体および変異体または任意の他の非天然の変異体(例えば、ペプチド模倣薬)が挙げられる。
【0037】
この出願は、「Gタンパク質結合受容体アゴニストおよびアンタゴニストならびにそれを用いたGタンパク質結合受容体の活性化および阻害」と題された同時係属米国特許出願第10/251,703号(この特許出願の内容全体を本明細書中に援用する)に関連している。
【0038】
本発明は、GPCRi1ループから誘導されるペプチドに結合した細胞貫通または係留部分を含むペプデュシンと呼ばれる改質ペプチドの発見に基づく。ペプデュシンはキメラのペプチド/ポリペチドと考えられ、受容体−Gタンパク質シグナル伝達のアゴニストおよびアンタゴニストもしくはその一方であって、それらの同族の受容体に対して選択性を示す。
【0039】
本発明のペプデュシンは、GPCRの第1の細胞内ループ(i1)またはそのフラグメントに由来するGPCR部分と、標的細胞の脂質二重層内またはそれを横断して複合体を区分けする細胞貫通または膜係留部分とを含み、また本発明のペプデュシンをGPCR媒介症状の処置に用いる方法を含む。細胞膜透過性部分は、例えば、GPCRフラグメント自体の疎水性部分である。細胞貫通または膜係留部分は脂質二重層(または細胞表面)で複合体を係留することで、細胞内受容体の近傍(例えば、受容体−Gタンパク質界面)で複合体の有効なモル濃度を上昇させる。ペプデュシンの外因性GPCR部分は、受容体−Gタンパク質相互作用を崩壊させて、シグナル伝達活性化および阻害(すなわちアゴニストまたはアンタゴニスト活性)もしくはその一方を生じる。
【0040】
ペプデュシンは、受容体の細胞内表面を目標とすることによって、受容体修飾剤として働く。例えば、本発明のペプデュシンは、生体内条件下での抗うっ血作用および抗血栓作用に対するPARlおよびPAR4ベースのアンタゴニストを含む。トロンビンが血小板で最も強力な賦活体であるので、標的としてPARl(Vu et al.Cell 64,1057(1991))およびPAR4(Xu et al.Proc.Natl.Acad.Sci.(USA)95,6642(1998);Covic et al.Biochemistry 39,5458(2000)および Covic et al.Thromb.Haemost.87,722(2002))が選択された。これらの2つの受容体のアンタゴニストは、敗血症を含む急性冠動脈症候群の血栓および増殖合併症を妨げるために役立つ。
【0041】
長期間に及ぶ虚血後に血流が回復する際に、虚血再灌流障害が起こる。それは、心筋梗塞、脳卒中、腸管虚血、心肺バイパス等の状態での羅病率および死亡率の一般的な原因であることから、しばしばそれに対する特異的療法がみあたらない。本発明のペプデュシンを静注投与することで、心筋の実験的再灌流障害に対する有意な保護が得られる。
【0042】
心筋に対する動脈血の供給が限られていることによって促される心筋梗塞は、心不全および死亡の主因である。したがって、正常な血行の早急な回復は、酸素圧低下から心臓組織の不可逆的損傷の予防のために重要である。正常な血流の再建は、特定薬剤を有する心筋梗塞(MI)患者の処置後、自然または人工的に起こり得る。しかし、多くの場合、梗塞部の再灌流は、心臓細胞の損害を促進し、それによって回復する血行の陽性作用を減らす炎症反応を引き起こす。有意に再潅流された組織の低酸素領域に補充される白血球がこの病理学的炎症反応に関与することが証明された。
【0043】
臓器移植は、2001年には米国で22,000件以上行われており、現在一般的であり、最も一般的に移植された臓器は腎臓、肝臓、および心臓である。現在実施されている多数の移植体は、ある程度は、急性拒絶を制御する免疫調節性剤の能力に依拠している。遅延移植機能(虚血−再灌流障害に起因する)と宿主対移植反応とは、移植の後の急性拒絶の主要な機序である。免疫抑制剤の使用は、急性拒絶に対して成功裏に対処しており、免疫抑制剤投薬量が非常に低レベルになる場合であっても、一般に異型移植が長期わたって生存する。
【0044】
しかし、免疫抑制剤はすべての免疫反応を抑制することから、激しい感染症が移植レシピエントの主要な死因となる。免疫抑制剤は、重症毒性とも関係している。移植患者のために使用される薬物で最も有意な合併症として、腎毒性、神経毒性、新規発症移植後真正糖尿病、高脂血症、および高血圧が挙げられる。これらの副作用は、現在使用される免疫抑制剤の分子標的が普遍的に発現することから、部分的に生ずる。本発明のペプデュシンを含む治療的な戦略は、特に虚血−再灌流障害と単核細胞のその後の浸潤(急性拒絶の主因)を対象とすることが可能である。
【0045】
重篤な敗血症は急性の入院の主要な原因であって、しばしば、他の疾患の処置がおこなわれている患者の臨床経過を難しくする。敗血症発症の際、細菌の存在とエンドトキシン等の細菌産生物とが免疫防衛機制を刺激する。侵入病原体の除去に失敗すると、全身性炎症反応症候群(SIRS)と称され、かつサイトカインおよびケモカインによってもたらされる機能亢進性炎症反応が開始される。ケモカインを、炎症の治療標的として調べる。CXCRlおよびCXCR2は、好中球、内皮、上皮、マクロファージ、および他の細胞の活性化にかかわる重要なケモカイン受容体である。唯一のIL−8受容体であるCXCR2の遺伝子削除によってIL−8シグナル伝達が欠けているマウスは、敗血症の発現から保護される。
【0046】
ヒトで、全身性IL−8のレベルが高いと、内皮細胞機能不全と内皮の正常な抗凝固状態とが失われる。凝固障害は敗血患者の30ないし50%に起こり、明白な播種性血管内凝固(DIC)の発生は不吉な予後徴候である。CXCRl/2の作用に対抗して、CXCR4は老好中球の除去をもたらし、骨髄で未成熟な白血球を保持する。CXCR4もリンパ球および癌細胞のホーミング機構において顕著な役割を果たすが、急性炎症におけるCXCR4およびそのリガンドSDF−lαの機能は謎のままである。
【0047】
抗生物質の投与は別として、敗血症および敗血症性ショックの処置は、主に、支えとなる戦略に限られている。CXCRl/2に特異的である低分子阻害剤が再灌流障害および肺損傷に対する保護をおこなうことができるけれども、敗血症の処置に利用可能なケモカイン受容体に対して特異的な治療的介入は、いまのところ存在しない。本発明のペプデュシンを、確立された全身性炎症および血管障害の抑制に用いることができ、同様に宿主防御に対する干渉なしで、凝固カスケードの活性化を妨げる。
【0048】
ヒトPARとして、PAR1(Genbankアクセッション番号AF019616)、PAR2(Genbankアクセッション番号XM_003671)、PAR3(Genbankアクセッション番号 NM_004101)、およびPAR4(Genbankアクセッション番号NM_003950.1)が挙げられ、これらの配列を本明細書中で援用する。
【0049】
ペプデュシンが受容体−Gタンパク質間シグナル伝達の活性化および阻害をおこなう二部位機構は、本明細書中に述べられているが、本発明者は、本発明をこの機構またはその根拠となる理論によって制限されるとは意図していない。この機構は、アゴニストの二相活性化および抑制とアンタゴニストの抑制とに適応する。ペプデュシンは、その疎水性係留によって、急速に形質膜を変換して、周囲膜質界面で有効性の高いモル濃度を成し遂げる。ペプデュシン・アゴニストは、GPCRの細胞内表面で、最初に高親和性部位を占める。結合したアゴニストは結合させられたGタンパク質をオンにするために受容体の活性化状態を安定化または誘導する。この第1の部位が飽和したあと、より高濃度のペプデュシンが親和性のより低い第2の阻害部位を占め始める。この第2の阻害部位は、優位なかたちでGタンパク質に対するシグナル転移を、おそらくGタンパク質でGPCR(例えば、受容体i1ループ)基底状態相互作用を模倣することによって、遮断する。ペプデュシン・アンタゴニストによる阻害は、アゴニストの阻害相と一致することから、アンタゴニストはこの低親和性部位で結合もすると思われる。ペプデュシンによる受容体の外因性活性化または阻害は、潜在的二量体化様式を反映することができ、それによって1つの受容体がその細胞内ループを近隣の受容体に提供する。異なったシグナル伝達性質(Milligan,Science 288,65−67(2000))を生ずる受容体ダイマーに関するいくつかの実施例があり、EPO受容体等のサイトカイン/GPCRが含まれるが(Guillard et al.,J.Biol.Chem.(2001)276,2007−2013)、受容体間変調機構は知られていない。
【0050】
天然のGPCRは、少なくとも一回細胞膜を越える細胞表面モジュールである。例えば、多くの天然のGPCRは7回細胞膜を越えて、いくつかの細胞内領域を含む。本発明の単離GPCRフラグメントは、GPCRの第1の細胞内ループ領域を含む。細胞内部分は、サイトカインGPCRの第1の細胞内ループ領域の1−膜貫通型領域Gタンパク質結合受容体またはそのフラグメント、あるいは、マルチ・ポリペチド−GPCRの第1の細胞内ループ領域(例えば、GPIb/v/IX受容体またはコラーゲン受容体)から選択される。
【0051】
本発明は、可溶性ペプデュシンも包含するもので、第2の領域(細胞貫通または膜係留部分)は、任意に、細胞外または細胞内フラグメントに隣接した膜貫通型領域から自然に生じる隣接アミノ酸を含む。例えば、コンストラクトは、GPCR膜貫通型ヘリクス領域の隣接アミノ酸を少なくとも3個(しかし16個未満)含むものであってもよい。膜貫通型領域は、CXCR4の膜貫通型領域1〜7、CCKA受容体の膜貫通型領域1〜7、またはCCR5受容体の膜貫通型2でない。実施形態において、第2の領域は細胞外または細胞内フラグメントに直隣接して天然の膜貫通型ヘリクス領域の1〜15の隣接アミノ酸を含む。
【0052】
ペプチド・ベースのペプデュシンに加えて、発明はGPCRフラグメントがペプチド模倣薬を含む組成物を含む。例えば、本発明は一つ以上のペプチド結合が他のタイプの共有結合に置き換えられたペプデュシン化合物を含み、その結合はペプチダーゼ(「ペプチド・ミメティック」または「ペプチド模倣薬」)による裂開の影響を受けにくい。被験者への注射後のペプチドのタンパク質分解が問題である一方で、非切断型ペプチド・ミメティックによる特に感受性が高いペプチド結合の置換は結果として生じるペプチドをより安定にすることから、より役立つ治療法である。このミメティックとそれらをペプチドに組み込む方法は、周知の技術である。同様に、Lアミノ酸残基の置換(例えば、D−アミノ酸による)は、プロテオリシスによるペプチドへの影響をより少なくする。
【0053】
本発明のペプデュシン化合物を逆配列およびキラリティーのペプチドを含むレトロ・インベルソ異性体として合成することができる。例えば、Jameson et al.Nature 368:744−746(1994)and Brady et al.Nature 368:692−693(1994)を参照せよ。D−エナンチオマーと逆合成との組み合わせによる正味の結果は、各アミド結合でのカルボニル基の位置とアミノ基との位置が交換され、その一方で各α炭素の側鎖基の位置が保存されるということである。例えば、ペプチド・モデルが配列ABCのLアミノ酸から形成されるペプチドである場合、D−アミノ酸から形成されるレトロ・インベルソ・ペプチドの類似体は配列CBAを持つと思われる。レトロ・インベルソ・ペプチドを形成するためにD−アミノ酸の連鎖を合成するための手法は、当該技術分野で知られている。
【0054】
また、有用なアミノ末端ブロック基として、t−ブチロキシカルボニル、アセチル、セイル、サクシニル、メトキシ・サクシニル、スべリル、アジピル、アゼライル、ダンシル、ベンジルオキシカルボニル、フルオレニルメトキシカルボニル、メトキシアゼライル、メトキシアジピル基、メトキシスベリル、および2,4−ジニトロフェニルが挙げられる。ペプチドの荷電アミノ末端およびカルボキシ末端を阻害することは、疎水性細胞膜を介し、かつ細胞内に向けたペプチドの通過を高めるという付加的な利益がある。
【0055】
単離されたGPCRフラグメントは、クラスAのGPCRまたはクラスBのGPCRの配列から誘導してもよい。単離されたGPCRフラグメントは、任意の既知または未知のGPCR由来のフラグメントであり、限定されるものではないが、プロテアーゼ活性化受容体(PAR、例えばトロンビン受容器)、黄体形成ホルモン受容体、卵胞刺激ホルモン受容体、甲状腺刺激ホルモン受容体、カルシトニン受容体、グルカゴン受容器、グルカゴン様ペプチド1受容体(GLP−1)、向代謝性グルタミン酸受容体、副甲状腺ホルモン受容体、脈管活性腸幹ペプチド(VIP)受容体、セクレチン受容体、成長ホルモン放出因子(GRF)受容体、コレシストキニン受容器、ソマトスタチン受容体、メラノコルチン受容体、ヌクレオチド(例えば、ADP受容体)、アデノシンレセプター、トロンボキサン受容体、血小板活性化因子受容体、アドレナリン作動受容器、5−ヒドロキシトリプタミン(5−HT)受容体、ケモカイン受容体(例えば、CXCR4、CCR5)、神経ペプチド受容体、オピオイド受容体、エリスロポエチン受容体、および副甲状腺ホルモン(PTH)受容体が挙げられる。
【0056】
好ましい実施形態において、GPCRはプロテアーゼ活性化受容体、ペプチド受容体、またはヌクレオチド受容体である。特定の実施形態において、GPCRはPAR1、PAR2、PAR3、またはPAR4受容体である。他の実施形態において、GPCRはグルカゴン様受容体、ヌクレオチド受容体、例えばP2Y−12ADP受容体、MC4肥満受容体、CXCR受容体(例えば、CXCR4)もしくはCCR5ケモカイン受容体、CCKA、またはCCKBである。
【0057】
単離されたGPCRフラグメントは、GPCR由来の少なくとも50未満の隣接アミノ酸であり、GPCRの固有の細胞外リガンドを含まない。例えば、このフラグメントはGPCRの3ないし30の隣接アミノ酸を含むものであってもよい。好ましい実施形態において、GPCRフラグメントは7と24との間(7および24を含む)の隣接アミノ酸であるGPCRフラグメントを含む。例えば、フラグメントは、GPCRの7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23または24の隣接アミノ酸を含む。
【0058】
任意には、GPCRのアミノ酸配列は、天然のアミノ酸配列と異なる。例えば、所定の領域(例えば、膜貫通型ヘリクス・ループ、細胞外ループ、または細胞内ループ)由来の個々の残基を突然変異または修飾アミノ酸に置換によってペプデュシンの活性を改善する。好ましくは、そのようなGPCR類似体のアミノ酸配列は、保存的アミノ酸置換(すなわち同一クラスの他方のものに対する一方のアミノ酸の置換によって、またはタンパク質の機能を破壊しない場所に位置した非保存的置換、欠失、または挿入)によって単独で異なる。
【0059】
本発明のペプデュシンのGPCR部分は、ヒトまたは他の生物(例えば、モルモット、ラット、マウス、チキン、ウサギ、ブタ、ヒツジ、牛、サル、ウイルス、真菌、昆虫、植物、バクテリア、その他)の任意の細胞に由来するもので、このような細胞の例として、脾細胞、神経細胞、グリア細胞、膵ベータ細胞、骨髄細胞、糸球体間質細胞、ランゲルハンス細胞、表皮細胞、上皮細胞、血管内皮細胞、フィブロプラスト、フィブロサイト、筋細胞、脂肪細胞、免疫細胞(例えば、マクロファージ、T細胞、B細胞、ナチュラルキラー細胞、マストセル、好中球、好塩基、好酸球、単核細胞、その他)、巨核球、滑膜細胞、軟骨細胞、骨細胞、骨芽細胞、破骨細胞、乳房腺細胞、肝細胞または間質細胞、あるいはそれらの前駆体細胞、幹細胞等、または癌細胞等、さらにそのような細胞を含んでいる任意の組織、例えば脳、脳の種々の部分(例えば、嗅球、扁桃体、脳幹神経節、海馬、視床、視床下部、視索上核、大脳皮質、延髄、小脳、後頭極、前頭葉、果核、尾状核、脳梁、黒質)、脊髄、下垂体、胃、膵臓、腎臓、肝臓、生殖器、甲状腺、胆嚢、骨髄、副腎、皮膚、筋肉、肺、消化管、血管、心臓、胸腺、脾臓、顎下腺、末梢血液、末梢血白血球、腸管、前立腺、睾丸、精巣、卵巣、胎盤、子宮、骨、関節、小腸、大腸、骨格筋等、特に脳および脳の種々の部分が挙げられる。
【0060】
細胞膜透過性部分は、脂質、コレステロール、リン脂質、ステロイド、スフィンゴシン、セラミドまたは脂肪酸部分を含む。脂肪酸部分は、例えば、少なくとも8つの炭素を含む任意の脂肪酸である。例えば、脂肪酸を、例えば、ノナノイル(C9)、カプリル(C10)、ウンデカノイル(C11)、ラウロイル(C12)、トリデカノイル(C13)、ミリストイル(C14)、ペンタデカノイル(C15)、パルミトイル(C16)、フィタノイル(メチル置換C16)、ヘプタデカノイル(C17)、ステアロイル(C18)、ノナデカノイル(C19)、アラキドイル(C20)、ヘニエコサノイル(C21)、ベヘノイル(C22)、トルシサノイル(C23)、またはリグノセロイル(C24)部分とすることができる。細胞膜透過性部分は、オクチル−グリシン、2−シクロヘキシルアラニンまたはベンゾリルフェニルアラニンのマルティマー(例えば、一つ以上の単位を含んでいる組成物)を含むこともできる。細胞膜透過性部分は、非置換またはハロゲン置換(例えば、クロロ)ビフェニル部分を含む。非置換されたビフェニルによる化合物と比較すると、置換されたビフェニルは、生体組織で蓄積の減少を伴う。被験者に対する投与後の身体組織での蓄積減少は、被験者における副作用の減少を伴う。
【0061】
好ましくは、細胞膜透過性部分は天然または非天然のパルミトイル部分である。C15よりも長い脂肪酸(例えば、パルミトイル)部分を持つペプデュシンは、生体内で驚くほど長命であるように思われ、また皮下適用に適していると思われる。別の実施形態において、C15よりも短い部分(例えば、ミリストイル)を有するペプデュシンは、短期間の適用(例えば、外科適用)に適しているように思われ、ここでは静脈内投与が有用である。
【0062】
細胞貫通または膜係留部分は、GPCRフラグメントのC末端アミノ酸、N末端アミノ酸、またはN末端とC末端アミノ酸との間のアミノ酸に結合するものであってもよい。
【0063】
また、本発明の範囲内にあるものは、細胞貫通または膜係留部分に連結する配列番号1〜14のアミノ酸配列を持つポリペチドまたはその一部分を含む組成物である。本発明の特に適したペプデュシンを、以下の表1に示す。
【0064】
【表1】
組成物は、異常なGPCR活性によって特徴づけられる疾患および状態を伴う一つ以上症状(重篤度を減らす)を処置、予防、または改善するのに用いられる。そのような疾患および状態として、血栓症、心臓発作、脳卒中、過剰な出血、喘息、炎症、疼痛、炎症性疼痛、内臓痛、神経性疼痛、関節炎、糖尿病、HIV感染症、不安神経症、鬱病、肺動脈弁閉鎖不全症、および種々の癌が挙げられる。この方法は、ペプデュシンGPCRアンタゴニストに細胞(病理学的にGPCRを過剰発現させる)を接触させることによって実行される。例えば、方法は被検体(例えば、ヒト患者)に投与することが必要であり、被験者で病理学の重篤度を減らすのに十分な量のペプデュシンを処置または予防に必要とされる。本発明は、ペプデュシン組成物および製薬的に許容される担体のいずれかを有する医薬組成物も含む。本発明はまた、医薬組成物を含有するキットも含む。本発明は、本発明のいずれかのポリペチドの投与を通して哺乳動物における病理学的状態を処置する方法をさらに含む。
【0065】
コンストラクトは、腫瘍増殖および移動を阻害するのにも用いられる。乳癌細胞浸潤は、複雑なプロセスであり、細胞移動、組織基底膜のタンパク分解性修飾、およびマトリックス・メタロプロテアーゼ(MMP)による細胞外マトリックスの低下が起こる。MMPlによるPARlの活性化は、乳癌細胞の浸潤と腫瘍形成において重要な役割を果たす(Boire et al.Cell,120(3),303(2005))。PAR1i1ループ・ペプデュシン・アンタゴニスト(例えば、Plilpal11)が、単独で、またはアジュバント癌治療(例えば、タキソテール(登録商標)等のドセタキセル化合物とともに投与される場合)として使われる。好ましくは、相乗的抗腫瘍効果が達成される。PAR1の細胞内阻害の作用は、転移性ヒト乳癌細胞株MDA−MB−231を用いることで示される。単独または併用療法(例えば、タキソテールと共に)におけるペプデュシン有効性を、MTTアッセイおよび異種移植片マウス・モデルを用いて分析する。組成物が別々または同時に投与されたあと、ペプデュシンPlilpal11およびタキソテールのIC50を測定する。アイソボログラム(isoborogram)技術と組み合わせインデックス(CI)法を用いて、相乗効果の程度を定量化する。異種移植片データは、腫瘍成長速度がタキソテールだけで処置されたマウスと比較した際に、同時にPlilpal11およびタキソテールで処置されるマウスで抑制されたかどうか決定するのに用いられる。この化合物は、全ての移動が腫瘍成長(例えば、腫瘍成長の後期段階と同様に腫瘍成長の初期)の病期分類で腫瘍増殖を低下させる。
【0066】
例えば、図27に示すように、CXCRl/2(xl/2pal−i1)に基づくI1−ループ・ペプデュシンおよびCXCR4(x4pal−i1)ケモカイン受容体は、胸部と卵巣癌の化学走性の移動を妨げる。
【0067】
発明の特定のペプデュシンは、血小板活性化阻害剤である。阻害剤は、GPCRポリペチドに連結する細胞膜透過性部分連結とプロテアーゼ活性化受容体の単離フラグメントとを含む。一部の実施形態において、プロテアーゼ活性化受容体はトロンビン受容器、トリプシン受容体、凝固因子Xa受容体、活性プロテインC受容体、トリプターゼ受容体、またはプラスミン受容体である。トロンビン受容器は、好ましくはPAR−4またはPAR−1である。
【0068】
本発明は、先に述べたように細胞膜透過性部分に連結したプロテアーゼ活性化受容体の単離フラグメントの組成物で血小板を接触させることによって血小板凝集を阻害する方法も含む。例えば、プロテアーゼ活性化受容体はトロンビン受容体(例えば、PAR−1受容体またはPAR−4受容体)である。また、本発明の範囲内にあるものは、細胞膜透過性部分に連結したトロンビンの単離されたフラグメントを含む本発明の組成物を哺乳動物投与することによってこの哺乳動物で血栓形成を阻害する方法である。
【0069】
本発明の方法は、血管内腔(例えば、頸静脈、末梢静脈または血管周囲隙)に発明の抑制性組成物を注入することによって実行される。末梢静脈は、例えば、端(例えば、手、手首または足)にある静脈である。いくつかの実施形態では、組成物を、例えば噴霧質として哺乳動物の肺に注入する。他の実施形態では、本発明の組成物を注射によって投与する。種々の実施形態において、哺乳類動物の腹腔内、真皮下または皮下に注射することができる。本発明の組成物を経皮的に投与することもできる。他の実施形態では、本発明の組成物を経膣投与または直腸投与する。本発明の組成物が被覆または注入された坐剤または充填材料を損傷部分に移植することによって、投与することができる。
【0070】
凝血塊構造または血小板凝集の阻害剤を、医療器具に用いることができる(例えば、コーティングとして)。例えば、脈管内人工装置(例えば、スクリーン、ステントまたはカテーテル)は細胞膜透過性部分に連結したトロンビンの単離されたフラグメントである血栓形成の阻害剤を含む。組成物を装置内に含浸させ、組織との接触または装置の移植に応じて身体の組織に広まる。あるいは、装置をペプデュシンで覆う。
【0071】
ペプデュシンは、細胞膜透過性部分に連結したプロテアーゼ活性化受容体の単離フラグメントと腫瘍細胞とを接触させることによって腫瘍細胞の移動および浸潤を阻害するのにも用いられる。プロテアーゼ活性化受容体は、PAR−4、PAR−2、またはPAR−1受容体である。腫瘍細胞の転移を阻害する方法は、細胞膜透過性部分に連結したプロテアーゼ活性化受容体の単離フラグメントと腫瘍細胞とを接触させることによって実行される。腫瘍細胞は、メラノーマ細胞、乳癌細胞、腎臓癌細胞、前立腺癌細胞、肺癌細胞、大腸癌細胞、中枢神経系(CNS)癌細胞、肝癌細胞、胃癌細胞、肉腫細胞、白血病細胞、またはリンパ腫細胞である。
【0072】
喘息症状の軽減は、トロンビンまたはトリプシン/トリプターゼGPCR系ペプデュシンの投与によってなされる、したがって、喘息抑制方法は細胞膜透過性部分にトロンビンまたはトリプシン/トリプターゼGPCR連結の単離されたフラグメントを含んでいる組成物を投与することによって実施される。好ましくは、トリプシン/トリプターゼ受容体は、PAR−1、PAR−2またはPAR−4受容体である。種々の実施形態において、組成物を血管内腔(例えば、末梢静脈)に注入、哺乳動物の肺に注入(例えば、吸入(例えば、煙霧質として)、または経皮経路によって投与する。
【0073】
血小板活性化の阻害剤は、細胞膜透過性部分に連結したP2Y12受容体等のヌクレオチド活性GPCRの単離フラグメントを含む。この組成物は、血小板凝集を阻害する方法に役立つ。
【0074】
さらにもう一つの態様において、本発明は哺乳動物に対して細胞膜透過性部分に連結したヌクレオチド活性受容体の単離フラグメントを含む組成物を投与することによって、哺乳動物で血栓形成を阻害する方法を含む。いくつかの実施形態では、トロンビン受容体はP2Y12受容体である。血管内腔(例えば、頸静脈、末梢静脈)に本発明の阻害組成物を注入することによって、方法を実施することができる。末梢静脈は、例えば、四肢(例えば、手、手首または足)にある静脈であってもよい。いくつかの実施形態では、組成物を例えば煙霧質として哺乳動物の肺に注入する。他の実施形態では、発明の組成物を、注射によって投与する。種々の実施形態において、注射が哺乳動物(真皮下または皮下)の腹腔にあることができる。他の実施形態において、発明の組成物は、経皮的に投与される。種々の実施形態では、哺乳類動物の腹腔内に真皮下または皮下注射することができる。本発明の組成物を経皮投与することもできる。組成物の投与は、本発明の組成物をコーティングまたは含浸させた創傷充填材または坐剤を埋め込むことによっておこなうことができる。
【0075】
別の態様では、本発明は血栓形成の阻害剤(細胞膜透過性部分と連結したヌクレオチド受容体の単離フラグメント)を含む脈管内人工装置を包含する。種々の実施形態では、この装置は、例えばステントまたはカテーテルである。いくつかの実施形態では、この装置に阻害剤を被覆または含浸させる。
【0076】
本発明の一つ以上の実施形態の詳細を、下記の付随的説明で述べる。本明細書中に述べられる方法および材料と類似または等価な任意の方法および材料を本発明の実施または試験で使用することができるが、好ましい方法および材料をここで説明する。本発明の他の特徴、目的、および長所は、説明から明らかである。本明細書と添付の特許請求の範囲において、文脈をはっきりと述べないかぎり、単数形も複数を含む。定義されない限り、本明細書中に使用されるすべての専門用語および科学用語は、本発明が属する当該技術分野の当業者が理解する意味と同じ意味を持つ。不一致の場合、定義を含む本明細書を規制する。また、加えて、材料、方法、および実施例は、限定することを意図したものではなく、説明を目的とするものである。この明細書で引用されるすべての特許および刊行物は、参照によって本明細書中に組み込まれる。
【0077】
Gタンパク質結合受容体
Gタンパク質結合受容体は、複数の受容体からなる大きなスーパーファミリーを含む内因性膜タンパク質である。Gタンパク質結合受容体(GPCR)のファミリーは、少なくとも250のメンバーを有する(Strader et al.FASEB J.,9:745−754,1995;Strader et al.Annu.Rev.Biochem.,63:101−32,1994)。1パーセントのヒト遺伝子がGPCRをコードすると推定されている。多くのGPCRは、一般の分子構築と一般のシグナル伝達機構とを共有する。歴史的に、GPCRは、本来、無関係であると思われた6つのファミリーに分類され、それのうちの3つは脊椎動物で見つかっている。最近の研究では、いくつかの新規のGCPRファミリーが確認され、それらの全てについて進化上の起源が共通する可能性があることを示唆している。
【0078】
多くのGPCRは、7つの膜貫通型ヘリカル領域の一般の構造モチーフを共有する。しかし、いくつかのGPCRは、7つの膜貫通型ヘリカル領域を持たず、その代わりに単スパン膜貫通型受容体である。
【0079】
単スパンGPCRは、サイトカイン(例えば、エリスロポエチン、EGF、インシュリン、インスリン様増殖因子IおよびII、ならびにTGF)のために、受容体を含む。
【0080】
GPCRファミリーとして、クラスAロドプシン様、クラスBセクレチン様、クラスC代謝調節型グルタメート/フェロモン、クラスD真菌フェロモン、クラスEcAMP受容体(タマホコリカビ(Dictyostelium))、および縮れた/平滑化されたファミリーが挙げられる。推定上のファミリーは、眼白子症タンパク質、ショウジョウバエ・ニオイ物質受容体、植物Mlo受容体、ネマトーダ化学受容体、鋤鼻受容体(V1R及びV3R)を含む。
【0081】
クラスAロドプシン様受容体は以下を含む:アミン受容器:アセチルコリン、αアドレナリン受容体、βアドレナリン受容体、ドーパミン、ヒスタミン、セロトニン、オクトパミン、および微量アミン;ペプチド受容体:アンギオテンシン、ボンベシン、ブラジキニン、C5aアナフィラトキシン、Fmet−leu−phe、APJ様、インターロイキン‐8、ケモカイン受容体(C−Cケモカイン、C−X−Cケモカイン、ボンゾー受容体(CXC6R)、C−X3−Cケモカイン、およびXCケモカイン)、CCK受容体、エンドセリン受容体、メラノコルチン受容体、ニューロペプチドY受容体、ニューロテンシン受容器、オピオイド受容体、ソマトスタチン受容体、タキキニン受容体、(P物質(NKl)、サブスタンスK(NK2)、ニューロメジンK(NK3)、タヒキニン様1およびタヒキニン様2)、バソプレシン様受容体(バソプレシン、オキシトシン、およびコノプレシン)、ガラニン様受容体(ガラニン、アラトスタチン、およびGPCR54)、プロテイナーゼ活性化様受容体(例えば、トロンビン)、オレキシン及び神経ペプチドFF、ウロテンシンII受容体、アドレノメデュリン(Gl0D)受容体、GPR37/エンドセリンB様受容体、ケモカイン受容体様受容体、およびニューロメジンU受容体、ホルモンタンパク質受容体:卵胞刺激ホルモン、ルトロピン−絨毛膜性腺刺激性ホルモン、チロトロピンとゴナドトロピン;(Rhod)オプシン受容体;きゅう覚受容体;プロスタノイド受容体:プロスタグランジン、プロスタサイクリン、およびスロンボキサン;ヌクレオチド様受容体:アデノシンとプリノセプター;大麻受容体;血小板活性化因子受容体;ゴナドトロピン放出ホルモン受容体;甲状腺刺激ホルモン放出ホルモンおよび分泌促進受容体:甲状腺刺激ホルモン放出ホルモン、成長ホルモン分泌促進物質および成長ホルモン分泌促進物質同類;メラトニン受容体;ウイルス受容器;リゾスフィンゴ脂質およびLPA(EDG)受容体;ロイコトリエンB4受容体:ロイコトリエンB4受容体BLTlおよびロイコトリエンB4受容体BLT2;ならびにクラスA オーファン/他受容体:血小板ADPおよびKI01受容体、SREB、マス・プロトオンコジーン、RDCl、ORPH、LGR様(ホルモン受容体)、GPR、GPR45様、システイニルロイコトリエン、Mas関連受容体(MRG)、およびGP40受容体。
【0082】
GPCRのクラスB(セクレチン受容体ファミリーまたは「ファミリー2」)は、より小さいが、タンパク質の構造的または機能的に異なった基であり、ポリペプチドホルモン用受容体(カルシトニン、コルチコトロピン放出因子、胃抑制ペプチド、グルカゴン、グルカゴン様ペプチド−1、−2、成長ホルモン放出ホルモン、副甲状腺ホルモン、PACAP、セクレチン、血管作動性腸管ポリペプチド、利尿ホルモン、EMRl、ラトロフィリン)、ストレス応答および長命を調節する形質膜(脳特異的脈管形成阻害剤(BAI))と一群のショウジョウバエ・タンパク質(メトセラ様タンパク質)で細胞間相互作用を媒介すると思わる分子が挙げられる。
【0083】
クラスC代謝調節型グルタメート/フェロモン受容体は、代謝調節型グルタミン酸塩、代謝調節型グルタミン酸塩群I、代謝調節型グルタミン酸塩群II、代謝調節型グルタミン酸塩群III、代謝調節型グルタミン酸塩他、細胞外カルシウムを感知する推定上のフェロモン受容体、GABA−B、GABA−B亜型1、GABA−B亜型2、およびオーファンGPRC5受容体を含む。
【0084】
GPCRは、潜在的にGタンパク質を経てアウトサイド・イン・シグナル伝達示すGPIb−V−IXまたはコラーゲン受容体等のマルチ・ポリペチド受容体である。
【0085】
何百ものGタンパク質結合受容体遺伝子またはcDNAがクローン化されたにもかかわらず、GPCRとまだ認められなかった多くの特徴のないGタンパク質結合受容体がまだあると考えられる。
【0086】
GPCRは、細胞の代謝、細胞増殖および運動性(癒着、炎症、神経シグナル伝達、および血液凝固)を制御するシグナル伝達プロセスにおいて、不可欠な役割を果たす。Gタンパク質結合受容体タンパク質にも、生体の機能を制御、調節、または調整する種々のシグナル伝達分子の標的としての非常に重要な役割がある。シグナル伝達種は、内因性分子(例えば、神経伝達物質またはホルモン類)、外因性分子(例えば、ニオイ物質)または、視覚変換の場合には光である。
【0087】
例えば、GPCRは、生体アミン受容体、例えばドーパミン、エピネフリン、ヒスタミン、グルタミン酸塩(代謝調節型作用)、アセチルコリン(ムスカリン様作用)、およびセロトニンのための受容体と、炎症(例えば、プロスタグランジン、血小板活性化因子、およびロイコトリエン)の脂質性化学伝達物質のための受容体と、ペプチドホルモン(例えば、カルシトニン、C5aアナフィラトキシン、卵胞刺激ホルモン、性腺刺激ホルモン放出ホルモン、ニューロキニン、オキシトシン)のための受容体と、プロテアーゼ(例えば、トロンビン、トリプシン、トリプターゼ、活性プロテインCと因子Vlla/Xa)のための受容体と、感覚信号メディエイタ(例えば、レチナール光色素と嗅覚刺激性の分子)のための受容体とを含む。各分子は受容体タンパク質に特異的であり、個々の生理活性物質(特異的な的状赤血球と臓器を含む)、特異的な薬理作用、特異的な作用長、作用時間、その他に対して特異的である。このように、GPCRは薬物作用および開発の主な目標である。
【0088】
リガンド結合の際に、GPCRはグアニンヌクレオチド結合タンパク質(Gタンパク質)を活性化することによって、細胞内シグナル伝達経路を調節する。GPCRの領域構造は、GPCRファミリーのメンバー間で保存される。TMヘリクス領域、細胞内ループ領域、およびGPCRSの細胞外領域の領域境界は、当該技術分野で知られている。例えば、本明細書中に援用するPalczewski et al.,Science 289:739(2000)に述べられているように、マッピングされていないGPCRの構造は、既知の方法を使用して、原型GPCR(ロドプシン)との比較によって決定される。
【0089】
大部分のGPCRの特徴の1つは、疎水性アミノ酸残基からなる7つのクラスター、すなわち膜貫通型領域(TM、7つの膜貫通型領域を、TMl、TM2、TM3、TM4、TM5、TM6、およびTM7とする)は一次構造内に位置しており、その各領域で細胞膜を貫通する(スパン)(図1)。領域は、3つの細胞内ループ(i1、i2、およびi3)、3つの細胞外ループ(e1、e2、およびe3)、ならびにアミノ(N)−カルボキシル(C)−末端領域によって接続される膜貫通型α−ヘリクスを表すと考えられている(Palczewski et al.,Science 289,739−45(2000))。大部分のGPCRは、機能性タンパク質構造を安定させると考えられているS‐S結合を形成する最初の2つの細胞外ループの各々で、一つの保存されたシステイン残基を持つ。上に詳述されるこれらの構造がGタンパク質結合受容体タンパク質間に共通すること、また領域に対応しているアミノ酸配列(タンパク質は、膜スパニング領域に近い膜(膜スパニング領域または膜貫通型領域)およびアミノ酸配列を通過する)は、受容体の間でしばしば高度に保存されることは、周知である。このように、GPCRの相同が高度であるため、新しいGPCRの同定は、そのような新しいメンバーの細胞内および細胞外部分の同定と同様に、当業者によって容易に達成される。
【0090】
Gタンパク質結合受容体の小さなリガンドに対する結合部は、いくつかのGPCR膜貫通型領域によって形成される細胞外表面の近傍に位置する親水性ソケットを含むと考えられている。親水性ソケットは、Gタンパク質結合受容体の疎水性残基によって囲まれる。各Gタンパク質結合受容体膜貫通型耳輪の親水性側は、内部に向かって、極性結合部を形成するために要求される。TM3は、TM3アスパルテート残基を含むリガンド結合部位があることから、いくつかのGPCRに関係した。複数のTM5セリン、TM6アスパラギン、およびTM6またはTM7フェニルアラニンあるいはチロシンもまた、リガンド結合に関係している。ペプチドホルモン受容体のためのリガンド結合部位と、糖タンパク質(例えば、黄体形成ホルモン、卵胞刺激ホルモン、ヒト絨毛性ゴナドトロピン、甲状腺刺激ホルモン(チロトロピン))等の他のより大きなリガンドによる受容体と、受容体のCa2+/グルタミン酸塩/GABA(γ‐アミノ酪酸)クラスとは、おそらく細胞外領域およびループにある。
【0091】
不活性から活性受容体への転換のための鍵となるイベントは、GPCRの膜貫通型ヘリクス3(TM3)および6(TM6)のリガンドによって誘発された構造変化であり、7つの膜貫通型スパン・ヘリクスを有する(Gether and Kolbilka,J.Biol.Chem.273,17979− 17982(1998))。これらのらせん状の動きは、付随するヘテロ三量体のGタンパク質の活性化を促進するために、次々に受容体の細胞内ループのコンフォメーションを変える。突然変異生成に関する研究(Cotecchia et al.,J.Biol.Chem.267:1633−1639(1992);Kostenis et al.,Biochemistry 36:1487−1495(1997);Kjelsberg et al.,J.Biol.Chem.267:1430−1433(1992))、第3の細胞内ループ(i3)が受容体とGタンパク質との間に継手の大部分を媒介することを証明した。ミニ遺伝子が直接アドレナリン作動性受容体とGq結合とが争うことも示されたように、13ループが発現された(Luttrel et al.,Science 259:1453− 1457(1993))、あるいは、無細胞条件(Okamoto et al.,Cell
67,723−730(1991))の可溶性ペプチドとして、Gタンパク質を活性化することができる。
【0092】
GPCRの1つの特定のクラスは、プロテアーゼ活性化受容体(PAR)である。プロテアーゼ活性化受容体(PAR)は、細胞外セリンプロテアーゼのタンパク分解性活性によって細胞シグナル伝達を開始するGタンパク質結合受容体スーパーファミリーのメンバーである。PARは、トロンビン(PAR−1、−3、および−4)とトリプシン/トリプターゼ(PAR−2および−4)を含む内因性プロテアーゼによって、受容体のアミノ末端のタンパク分解性裂開の後、活性化される。これらのうち、PAR2(Nystedt et al.,Proc.Natl.Acad.Sci.(USA)91:9208−9212(1994))は、炎症と疼痛で重要であるトリプシン/トリプターゼ活性化受容体であり、PAR4(Xu et al.,Proc.Natl.Acad.Sci.(USA)95:6642−6646(1998);Kahn et al.,Nature(London)394:690−694(1998))は、第2のトロンビン受容体であり、血小板凝集で固有の役割を演ずる(Covic et al.,Biochemistry 39,5458−5467(2000))。
【0093】
トロンビン、トリプシン、およびトリプターゼが炎症を起こした気道に存在することから、PARはおそらく気道炎症で主な役割を果たすことになっている。Knight et al.,J.Allergy Clin.Immunol.108:797−803(2001)。
【0094】
止血でのその重要な役割に加えて、トロンビンは特異的な細胞表面レセプター(PAR)のタンパク分解性開裂を経て血小板および血管平滑筋細胞等の種々の細胞型を活性化する。なお、原型はPAR−1である。トロンビン受容体活性化は、おそらく、心臓血管疾患(例えば、血栓症、アテローム性動脈硬化と再狭窄)で重要な役割を演じることから、トロンビン受容体アンタゴニストには、これらの障害の処置で、潜在的有用性がなければならない。Chackalamannil,Curr.Opin.Drug Discov.Devel.4:417−27(2001)。
【0095】
トロンビンは、哺乳類の中枢神経系(CNS)に対する障害の後、機能的な損失に関係していると考えられる。PAR−1のダウンレギュレーションはCNA神経の外傷後生存を増加させることが示され、またトロンビンの外傷後毒性はPAR−1受容体の適当なモジュレーションによって調節されるダウンレギュレーションであると考えられる。Friedmann et al.,Neuroimmunol.,121:12−21(2001)。
【0096】
PARSは、種々の癌、細胞増殖、および疼痛を含む種々の他の疾患または表示に関与してもいる。
【0097】
GPCR領域
大部分のGPCRは、疎水性アミノ酸残基または膜貫通型領域(TM、7つの膜貫通型領域は、TMl、TM2、TM3、TM4、TM5、TM6、およびTM7とする)の7つのクラスターによって特徴づけられ、これらは一次構造に位置して、細胞膜を貫通する(スパン)(図1A)。TM領域は、3つの細胞内ループ(i1、i2、およびi3)、3つの細胞外ループ(e1、e2、およびe3)によって接続される膜貫通型α−ヘリクスを表すと考えられている。GPCRはまた、アミノ(N)−カルボキシル(C)−末端領域(Palczewski et al.,Science 289,739−45(2000))を含む。膜貫通型領域の間の配列はGPCRループに対応し、細胞の範囲内のループの位置はそれが菌体内または細胞外のループであるかどうか決定する。大部分のGPCRは、機能性タンパク質構造を安定させると考えられているS‐S結合を形成する最初の2つの細胞外ループの各々で、一つの保存されたシステイン残基を持つ。PAR1
GPCRの膜貫通型とループ領域の略図を、図IAに示す。
【0098】
GPCRの1つの実施例は、CXCR4受容体(配列番号15として表2で示される)である。7本の下線が引かれた配列は、GPCRの7つの膜貫通型領域に対応する。したがって、配列IFLPTIYSIIFLTGIVGNGLVILV(配列番号16)は、第1の膜貫通型領域(TMl)に対応する。
【0099】
【表2】
単離GPCRフラグメントは、完全長タンパク質より少ないGPCRの任意の部分である。単離GPCRフラグメントを含んでいるペプチドは、自然に生じるアミノ酸配列以外のGPCR配列に、アミノ酸配列N末端およびC末端もしくはその一方を含むものであってもよい。GPCRの単離膜貫通型配列を含んでいるペプチドは、GPCRのその膜貫通型領域に対応している配列だけを含むと考えられる。あるいは、自然に生じるフランキング配列でない膜貫通型配列にアミノ酸配列のN末端およびC末端もしくはその一方を含むものであってもよい(すなわち、天然のGPCR配列でその領域に隣接してあるループ配列でない)。
【0100】
このように、CXCR4受容体の単離された膜貫通型領域を含むペプチドは任意のペプチドであり、それは表2で示される配列の下線を引かれた領域の隣接アミノ酸のいずれかまたは全てを含む。このペプチドは、天然のGPCR配列のTM領域に隣接してあるループ配列に対応する自然に生じる(下線なし)フランキング配列のいずれも含まない。
【0101】
同様に、CXCR4受容体の単離された(細胞内であるか細胞外の)ループ領域を含むペプチドは、表2に示す配列の非下線領域の任意または全ての隣接アミノ酸を含む任意のペプチドである。このペプチドは任意の自然に生じる膜貫通型配列を含まず、表2で下線が引かれたフランキング配列として示されており、天然のGPCR配列にあるループ領域に隣接してある。
【0102】
単離細胞外領域または単離細胞内領域を含むペプチドは、任意の(細胞外または細胞内)ループおよびN末端領域もしくはその一方、またはC末端領域に由来するアミノ酸配列を含むことができる。このペプチドは、天然のGPCR配列でその細胞外領域または細胞内領域に隣接してある膜貫通型領域に由来する配列をいっさい含まない。
【0103】
医薬組成物
本発明のペプデュシン(ここでは「活性化合物」とも称する)と、その誘導体、フラグメント、類似体、および相同体とを、投与にふさわしい医薬組成物に組み込むことができる。この組成物は、ペプデュシンと製薬的に許容される担体を概して含む。
【0104】
発明の医薬組成物は、その意図された投与経路と互換性を持つために処方される。投与経路の例として、非経口投与、例えば静脈(例として、末梢静脈(四肢で見出される末梢静脈)、腹腔内、内皮、皮下、真皮下、経口、鼻腔、煙霧質(例えば、吸入)、経皮(例えば、局所)、経粘膜、膣、子宮内、および直腸(例えば、坐剤)投与が挙げられる。本発明の活性化合物を含んでいる注射可能溶液は、血管内腔(例えば、大動脈または頸静脈)に投与してもよい。あるいは、本発明の活性化合物を、装置(例えば、ステントまたはカテーテル)を介して、活性化合物を含浸または被覆によって投与してもよい。
【0105】
投与のために使用される溶液または懸濁液(例えば、非経口投与薬)は以下の構成要素を含むものであってもよい。すなわち、注射用蒸留水等の無菌の希釈剤、食塩水、不揮発性油、ポリエチレングリコール、グリセリン、プロピレングリコールまたは他の合成溶媒;抗菌因子(例えば、ベンジルアルコールまたはメチル・パラベン);抗酸化剤(例えば、アスコルビン酸または亜硫酸水素ナトリウム);エチレンジアミン四酢酸(EDTA)等のキレート剤;緩衝液(例えば、酢酸塩、クエン酸塩、またはリン酸塩);および塩化ナトリウムまたはデキストラン等の等張性を調製するための薬剤である。pHを、酸または塩基(例えば、塩酸または水酸化ナトリウム)で調整することができる。本発明の医薬組成物の製剤を、ガラスまたはプラスチックでできているアンプル、使い捨て注射器または多人数用バイアルに入れられることができる。
【0106】
注射用途にふさわしい医薬組成物が、無菌の水溶液(水溶性の場合)または注射可能な滅菌溶液もしくは分散の一時しのぎの製剤のための分散および無菌の粉を含む。静脈内投与のための適切な担体として、生理食塩液、静菌性の水、クレモホルEL(BASF社、パーシパニー、ニュージャージー州)またはリン酸緩衝食塩水(PBS)を含む。全ての場合において、簡単なシリンジアビリティが存在する程度まで、組成物は無菌でなければならず、また流動的でなければならない。それは、製造及び貯蔵の条件下に安定でなければならず、また細菌および菌類等の微生物の汚染作用に対して保存されなければならない。担体を、溶媒または分散媒とすることができ、それらは、例えば水、エチルアルコール、ポリオール(例えば、グリセリン、プロピレングリコール、および液体ポリエチレングリコール等)、ならびにそれらの適当な混合物を含む。適する流動性は、例えばレシチンのようなコーティングの使用、分散液の場合には必要とする粒子寸法の維持及び界面活性剤の使用により保持しうる。微生物の作用を防止することは種々の抗菌および抗かび剤、例えば、パラベン、クロロブタノール、フェノール、アスコルビン酸、チメロサール等によって達成可能である。多くの場合、等張剤、たとえば糖、ポリアルコール、たとえばマンニトール、ソルビトール、塩化ナトリウムなどが組成物中に含まれているのが好ましいであろう。注射用組成物の持続吸収は、吸収を遅らせる薬剤、たとえばモノステアリン酸アルミニウムおよびゼラチン等を組成物中に配合することにより行うことができる。
【0107】
滅菌注射用溶液を活性化合物を所望の量で、必要に応じて上記したようにして、1つの成分または複数の成分の組み合わせによって、適当な溶媒に取り込み、続いて濾過滅菌する。通常、分散は活性化合物を基本的な分散媒を含む無菌の媒体に取り込むことによって調製されている、それらからの必要な他の成分を上記に列挙した。注射可能な滅菌溶液の製剤のための無菌粉末の場合、調整法は事前に滅菌ろ過された溶液から主成分+任意の添加された所望の成分の粉を産生する真空乾燥と凍結乾燥である。
【0108】
経口組成物は、一般に不活性の希釈剤または食用の担体を含む。それらをゼラチンカプセルに入れ、あるいは圧縮して錠剤にすることができる。経口治療的投与を目的として、活性化合物に賦形剤を取り込ませることができ、錠剤、トローチ剤、またはカプセルの形で使うことができる。経口組成物を、うがい薬として使用するために、流体担体を用いて調製することもでき、ここでは、流体担体の化合物は経口適用され、口内洗浄し、吐き出し、または飲み込むことができる。製薬的に互換の結着剤およびアジュバント材料もしくはその一方を、組成物の一部として含ませることができる。錠剤、ピル、カプセル、トローチ剤は、以下の成分のいずれかまたは類似の性質の化合物を含むことができる。すなわち、結着剤(例えば、微結晶性セルロース、トラガカントガム、またはゼラチン);賦形剤(例えば、澱粉または乳糖)、崩壊剤(例えば、アルギン酸)、プリモゲルまたはトウモロコシ澱粉);潤滑油(例えば、ステアリン酸マグネシウムまたはステロート);コロイド状二酸化ケイ素等の滑走剤;甘味料(例えば、ショ糖またはサッカリン);または香料(例えば、セイヨウハッカ、メチルサリチレートまたはオレンジ調味料)。
【0109】
吸入または鼻内投与による投与のために、化合物は適切な推進体(例えば、ガス(例として、二酸化炭素)または噴霧器(例えば、肺に対する送達))を含む加圧された容器またはディスペンサからエアゾール・スプレーの形で送達される。
【0110】
全身投与を経粘膜または経皮手段でおこなうこともできる。経粘膜または経皮投与のために、透過すべきバリヤーに対する適当な浸透剤をこの配合物に用いる。この浸透剤は通常、当該技術分野で知られており、例えば、経粘膜投与に対しては、洗剤、胆汁酸塩、およびフシジン酸誘導体が挙げられる。経粘膜投与を、鼻内噴霧または坐剤を用いることにより達成することができる。経皮投与のために、一般に当該技術分野で知られているように、活性化合物を軟膏、軟膏、ゲル類またはクリームに処方する。
【0111】
活性化合物を、坐剤(例えば、従来の坐剤基剤(例えば、カカオ脂と他のグリセリド))、坐剤コーティング、または直腸送達のための保持浣腸の形で調製することもできる。活性化合物を、腟内または子宮内投与のために、同じように調製することができる。活性化合物を傷パッキングに含浸または被覆し投与してもよい(例えば、出血を減少させるために)。
【0112】
一実施形態において、活性化合物は担体(例えば、移植片とマイクロカプセルに入れられた投射手段を含む徐放製剤)から化合物を迅速な排泄から保護する担体で調製されている。エチレンビニルアセテート、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、及びポリ乳酸等の、生分解性の生体適合性ポリマーを使用することができる。そのような製剤の調製は、当業者にとってあきらかである。材料は、Alza CorporationおよびNova Pharmaceuticals,Inc.から商業的に入手することができ、リポソーム懸濁液(ウイルス抗原に対するモノクローナル抗体を持つ感染細胞を標的としたリポソームを含む))が製薬的に許容される担体として使われることもできる。米国特許第4,522,811号で述べられるように、これらは、例えば、当業者に知られている方法によって調製することができる。
【0113】
投与の容易さと投薬量の均一性のために用量単位の形態で経口または非経口組成物を処方することは、特に有利である。ここで用いられる用量単位の形態は、処置される被検体に対して一体的投薬量として適している物理的離散性単位を参照する。活性化合物の所定量を含む各単位を、必要な製薬担体に関連して所望の治療効果を発生するために算出した。本発明の用量単位形態についての明細書を、活性成分の独特の特性および達成すべき特定の治療効果、ならびに個体処置のためにこの活性化合物を配合する当該技術分野に本来ある限界によって、また直接依存して、記述した。
【0114】
ペプデュシンおよびGPCRペプチドは、医薬組成物の形で種々の障害を処置するために投与される。そのような組成物の調製に関係している原理および考慮点は、構成要素の選択の際のガイダンスと同様に提供される。例えば、Remington:The Science And Practice Of Pharmacy 19th ed.(Alfonso R.Gennaro,et al.,editors)Mack Pub.Co.,Easton,Pa.:1995;Drug Absorption Enhancement:Concepts,Possibilities,Limitations,And Trends,Harwood Academic Publishers,Langhorne,Pa.,1994;およびPeptide And Protein Drug Delivery(Advances In Parenteral Sciences,Vol.4),1991,M.Dekker,New York。製剤は、本明細書中に処置されている(好ましくは各々に悪影響を与えない補完的な活性を持つもの)特定の徴候のために、必要に応じて一つ以上の活性化合物を含有することもできる。あるいは、または加えて、組成物はその機能を高める薬剤(例えば、細胞毒性剤、サイトカイン、化学療法剤または増殖阻害性薬剤)を含むことができる。この分子は、意図される目的のために効果的である量での組合せで十分に存在する。主成分は、例えば、コアセルベーション技術によって、または、界面重合法によって調製されるマイクロカプセルにトラップすることもできる。例えば、ヒドロキシメチルセルロースまたはゼラチン−マイクロカプセル、ポリ(メタアクリル酸メチル)マイクロカプセルを、それぞれ含むコロイド状薬物送達系(例えば、リポソーム、アルブミン・ミクロスフィア、マイクロ・エマルジョン類、ナノ粒子、およびナノ・カプセル)、あるいはマクロエマルジョンである。
【0115】
生体内での投与のために使用される製剤は、無菌でなければならない。これは、濾過滅菌法膜を通して濾過によって容易に達成される。
【0116】
持続放出性製剤を調製することができる。持続放出性製剤の適切な実施例は抗体を含んでいる固形疎水性ポリマーの半透性マトリックスを含む。そして、マトリックスは造形品(例えば、フィルムまたはマイクロカプセル)の形である。徐放性マトリックスの実施例は、ポリエステル類、ヒドロゲル(例えば、ポリ(2−ヒドロキシエチルメタクリレート)またはポリ(ビニルアルコール))、ポリ乳酸(米国特許第3,773,919号)、L−グルタミン酸とエチル−L−グルタミン酸塩のコポリマー、非分解性エチレン−酢酸ビニル、LUPPRON DEPOT(登録商標)(乳酸グリコール酸コポリマーと酢酸ロイプロリドから成る注射可能ミクロスフィア)等の分解可能な乳酸グリコール酸コポリマーとポリD−(−)−3−ヒドロキシ酪酸を含む。ポリマー(例えば、エチレン−酢酸ビニルおよび乳酸グリコール酸)が100日以上間の分子の放出を可能にする一方で、特定のヒドロゲル放出タンパク質がより短い時間にわたって放出する。
【0117】
活性化合物の徐放は、種々の技術を利用することができる。装置(例えば、ステントまたはカテーテル)は、活性剤の不均一な溶液および活性剤もしくはその一方の分散を高分子物質に取り入れているモノリシックな層またはコーティングを有していることが知られており、薬剤がポリマー流体界面にポリマーを通して拡散して、その周囲の液体に放出されるにつれて、治療薬の拡散速度が制限される。活性化合物は、適当な重合物質で溶解または分散させることで、材料が溶けた後に付加的な孔またはチェンネルが残る。マトリックス装置は、通常、同様に拡散性が制限されてはいるが、流体への薬剤の放出でもチャンネルまたは装置の内部ジオメトリーが役割を果たす。チャネルは、放出された薬剤または他の可溶性物質によって取り残される既存のチャネルまたはチャネルである。
【0118】
浸食または分解可能な装置は、概して物理的に活性化合物がポリマーに固定してもよい。活性化合物を、重合物質の全体を通じて溶解および分散もしくはその一方を行うことができる。重合物質は不安定な結合の加水分解を通して時間とともに加水分解的に分解され、ポリマーが液体に侵食することがあり、液体に活性剤を放出する。親水性ポリマーは、疎水性ポリマーと関連して一般により速い侵食率を持つ。疎水性ポリマーはほとんど単に活性剤の表面拡散を持つと考えられて、表面内部から浸食を持つ。親水性ポリマーは水をポリマーの表面に侵入させると考えられ、表面の下で不安定な結合の加水分解を可能にすることで、ポリマーの浸食が均一または大きくなり得る。
【0119】
医薬組成物は、投与のための指示書と共に、容器、パックまたはディスペンサに入れられる。
【0120】
本発明にもとづくペプデュシンによるアプローチによって、細胞内受容体構造の豊かな多様性を新規の治療薬の生成、さらには生体内条件下での受容体−Gタンパク質結合の機構を描写するために利用することができる。この戦略によって発見されるペプデュシンは、ペプデュシンが主にGタンパク質よりむしろ受容体を対象とするという範囲で、より選択的であることが明らかである。加えて、多くの受容体はゲノムおよび遺伝学的なアプローチによって種々の疾患プロセスに重要であると確認されたが、既知のリガンドを持たない、いわゆるオーファンレセプタである。ペプデュシン・アゴニストおよびアンタゴニストは、発生し、これらの受容体にテーラードで、どのシグナル伝達経路がその固有の環境の状況でオーファンレセプタによって起動するかを決定することに役立つかもしれない。このように、ポストゲノムの時代には、ペプデュシン・アプローチは膜タンパク質のターゲッティングに広く適用可能であり、従来の分子技術に従わないシステムで、新規の実験的な道が開けられる。
【0121】
本発明の化合物および他の薬理活性剤を、同時、経時的、または併用して患者に投与してもよい。発明の組合せを使用する場合、本発明の化合物と他の薬理活性剤が同じ製薬的に許容される担体であると考えて同時に投与する。それらを同時に取られる従来の経口剤形のような別々の製薬担体内にあってもよい。それらは、別々の製薬担体(例えば、同時にされる従来の経口剤形)であってもよい。用語「組合せ(併用)」は、化合物の剤形が別々で、経時的に投与されるという場合にも言及する。
【0122】
発明の化合物は、最適製薬有効性を提供する投薬量で、この処置を必要とする患者(動物とヒト)に投与される。いうまでもないことだが、任意の特定用途用に必要な用量は、特定の化合物または組成物の選択のみならず、投与経路、処置されている状態の性質、患者の年齢と状態、並列の薬物または患者が従う特別食、さらに当業者が認識する他の要素、最後に付き添いの医師の裁量である適切な投薬に従って、患者間で変える。
【0123】
状態の処置において、適当な投薬量レベルは通常、患者体重約0.001〜50mg/kg/日である。また、投薬は1回または複数回であってもよい。好ましくは、投薬量レベルは、1日約0.01〜約25mg/kg、より好ましくは1日0.05〜約10mg/kgである。例えば、適切な投薬量レベルは、1日約0.001〜10mg/kg、好ましくは1日約0.005〜5mg/kg、特に1日約0.01〜1mg/kgである。
【0124】
例えば、急性心筋梗塞患者において、適切なペプデュシン(例えば、x1/2pal−i1)を大量瞬時投与量として直ちに静脈投与することができ、それに続いて心筋梗塞後2〜3日にわたって、毎日1,2度追加して静脈注射をおこなう。投薬量は、約0.1〜0.5mg/kgである。
【0125】
任意の処置用に必要な発明の化合物の量が選択される特定の化合物または組成物でだけでなく投与経路、処置されている状態の性質、および年齢と患者の状態でも変化して、最後に付き添いの医師の裁量で変わることはいうまでもない。
【0126】
本発明の組成物および併用療法は種々の医薬賦形剤と組み合わせて投与することができ、本明細書中に述べられるように、安定化剤、担体およびカプセル化法製剤もしくはその一方を含む。
【0127】
本発明の水性組成物は、発明のペプチドの有効な量を含有、溶解し、製薬的に許容される担体または水溶媒質で分散した。
【0128】
「製薬的または薬理的に許容される」は、動物またはヒトに投与されるとき、副作用、アレルギー反応、または他の有害反応を発生しない分子実体と組成物を必要に応じて包含する。「製薬的に許容される担体」は、任意または全ての溶媒、分散媒、コーティング、抗菌、および抗真菌剤、等張および吸収遅延剤、その他を含む。製薬活性物質のためのこのような培地および薬剤の使用は、周知の技術である。任意の従来の培地または剤が主成分と適合しないこと以外は、治療用組成物のその使用は考察される。補助主成分を、組成物に組み込むこともできる。
【0129】
ヒト投与のために、製剤は、FDA局の生物学標準の要求に従って、生殖不能、発熱原性、一般安全、および純度の基準を満たすものとする。
【0130】
次に、発明の組成物および併用療法を、通常、非経口投与、例えば、静脈、筋内、皮下、病巣内、腹腔内経路を経て、注射のために処方される。発明の組成物または活性構成要素または成分を含む水組成物の製剤は、本開示を考慮して当該技術分野の当業者に知られている。典型例として、液溶体または懸濁液として、そのような組成物は、注射剤として調製されている。注射前に液体の追加に応じて溶液または懸濁液を調製するための使用に適した固形も、調製することができ、また製剤を乳化させることもできる。
【0131】
注射用途にふさわしい剤型として、無菌の水溶液または分散;ゴマ油、落花生油または水プロピレングリコールを含む製剤;ならびに注射可能な滅菌溶液または分散の即席の製剤のための滅菌粉末が挙げられる。全ての剤形は、無菌であり、かつ容易に注射器に流入できる程度に流体でなければならない。製造及び貯蔵の条件下に安定でなければならず、バクテリアおよび菌類のような微生物の汚染作用に対して保存されなければならない。
【0132】
遊離塩基または薬理的に受け入れられる塩類としての活性化合物の溶液は、界面活性剤(例えば、ヒドロキシプロピルセルロース)を十分に混ぜ合わせて水中で調製される。グリセロール、液体ポリエチレングリコール、およびそれら混合物中、および油中で分散液を調製することもできる。通常の貯蔵または使用の条件下で、これらの調製物は微生物の生長を防止するための保存剤を含む。
【0133】
本発明の治療的または薬理学的組成物は、一般に、製薬的に許容される媒体に溶解または分散した併用療法の構成要素の有効量を含む。製薬的に許容される媒体または担体として、任意または全ての溶媒、分散媒、コーティング、抗菌、および抗真菌剤、等張および吸収遅延剤、その他が挙げられる。製薬活性物質のためのこのような培地および薬剤の使用は、周知の技術である。補助主成分は、本発明の治療用組成物に組み込むこともできる。
【0134】
製薬または薬理学組成物の製剤は、本開示を考慮して当業者に知られている。典型的には、この組成物を、注射可能なものとして調製、液溶体または懸濁液のいずれかとして、注射に先立って溶液中または懸濁液中に含ませることに適した固形として、経口投与のための錠剤または他の固体として、限時解放カプセルとして、あるいは現在利用されている他のいずれかの形態(クリーム、ローション剤、うがい薬、吸入剤等)として、調製することが可能である。
【0135】
滅菌注射用溶液の調製は、必要量の活性成分を適当な溶媒に、必要に応じて上記に列挙した種々の他の成分とともに、取り込んだ後、濾過滅菌することでおこなった。一般に、分散は、種々の滅菌化された活性成分をビヒクルに取り込むことによって調製され、このビヒクルには基本的な分散媒体および上記に列挙された他の必要とされる成分を含む。注射可能な滅菌溶液の調製に用いられる無菌粉末の場合、好ましい調製法は、真空乾燥および凍結乾燥技術であり、これらの技術は活性成分の粉末に加えて、既に濾過滅菌したそれの溶液から所望の任意の成分を追加して得られる。
【0136】
筋肉内注射用のより多くまたは高濃度の溶液を調製することも検討される。この点に関しては、溶媒としてのDMSOの使用が好ましく、なぜならこのことが極度に迅速な浸透をもたらすことからであり、活性化合物または活性薬剤が小さな領域に送達される。
【0137】
オペレーティング・フィールドの特定域を浄化するために外科医、内科医、または医療従事者による滅菌製剤(例えば、食塩水ベースの洗浄液)の使用もまた、特に有用であると考えられる。本発明にもとづく治療用製剤は、うがい薬の剤形、または抗真菌性試薬とともに再構成してもよい。吸入剤の形態も、想定される。本発明の治療用製剤を、局所投与に適した剤形、例えばクリームおよびローション剤に調製することも可能である。
【0138】
このような溶液に適した防腐剤として、ベンザルコニウムクロリド、塩化ベンゼトニウム、クロロブタノール、チメロサール等が挙げられる。適当な緩衝液として、pH6ないしpH8、好ましくはpH7ないしpH7.5に保たれる量のホウ酸、ナトリウムおよび重炭酸カリウム、ナトリウムおよびカリウム・ホウ酸塩、ナトリウムおよびカリウム炭酸塩、酢酸ソーダ、燐酸ナトリウム等が挙げられる。適当な緊張剤は、点眼剤の食塩相当量が0.9プラスまたはマイナス0.2%の範囲内であるように、デキストラン40、デキストラン70、デキストロース、グリセリン、塩化カリウム、プロピレングリコール、塩化ナトリウム等である。適当な抗酸化剤と安定剤として、亜硫酸水素ナトリウム、メタ重亜硫酸ナトリウム、チオ硫酸ナトリウム、チオ尿素等が挙げられる。適当な湿潤剤および清澄剤として、ポリソルベート80、ポリソルベート20、ポリオキサマー282、およびチロキサポールが挙げられる。適当な粘度増加性剤として、デキストラン40、デキストラン70、ゼラチン、グリセリン、ヒドロキシエチルセルロース、ヒドロキシメチルプロピルセルロース、ラノリン、メチルセルロース、ワセリン、ポリエチレングリコール、ポリビニルアルコール、ポリビニルピロリドン、カルボキシメチルセルロース等が挙げられる。
【0139】
製剤に応じて、薬理的に効果的であるように、投薬量処方と互換性を持つ方法で、療法が施される。製剤は、種々の投薬形態、例えば上記したような注射溶液のタイプで容易に投与される。製剤を種々の投薬形態(例えば、上記した注射可能な溶液)で容易に投薬することができる。
【0140】
この文脈において、投与される組成物の主成分および容積は、処置される宿主動物に依存している。投与に必要な活性化合物の正確な量は、開業医の判断に依存し、各個人に特有である。
【0141】
活性化合物を分散させることが要求される組成物の最小量を、概して利用する。投与のための適当な投与計画も、可変的ではあるが、初めに化合物を投与して結果をモニターした後、さらに間隔を置いてさらに制御された用量を与えることによって代表される。例えば、非経口投与を目的として、適当に緩衝され、また必要に応じて等張である水溶液を調製し、静脈、筋肉内、皮下、または腹腔内投与に使用される。1つの投薬量は1mlのNaCl等張溶液に溶解し、1000mlの皮下注入液に添加または輸液の予定部位に注入される(例えば、Remington’s Pharmaceutical Sciences 15th Edition,pages 1035−1038 and 1570−1580)。
【0142】
特定の実施形態では、活性化合物を経口投与してもよい。このことは、消化酵素によるタンパク質分解に対して耐性、または耐性となった薬剤に対して検討される。そのような化合物は、化学的に設計された薬剤または改質された薬剤、右旋性ペプチド、ならびにペプチドおよびリポソーム型製剤(ペプチダーゼおよびリパーゼによる分解を回避するために徐放性カプセルに入れられている)であることが考えられる。
【0143】
製薬的に許容される塩類として、酸付加塩が挙げられ、該酸付加塩は、無機酸(例えば、塩酸、臭化水素酸、ホウ酸、リン酸、硫酸またはリン酸類)、あるいは有機酸(例えば、酢酸、シュウ酸、酒石酸、マレイン酸、フマル酸、クエン酸、コハク酸、メシル酸、マンデリン酸、コハク酸、安息香酸、アスコルビン酸、メタンスルホン酸、α−ケトグルタル酸、α−グリセロリン酸、グルコース−1−リン酸等)により形成される塩類である。遊離カルボキシル基で形成される塩類を、無機塩基(例えば、ナトリウム、カリウム、アンモニウム、カルシウム、マグネシウムまたは水酸化第二鉄)と有機塩基(例えば、イソプロピルアミン、トリメチルアミン、ヒスチジン、プロカインなど)とから誘導することもできる。製薬的に許容される塩類の他の例として、式(I)の化合物の四級誘導体が挙げられ、例えば、化合物Rx−Tによって四級化した化合物であり、式中RxはC1−6アルキル、フェニル−C1−6アルキル、またはC5−7シクロアルキル、Tは酸のアニオンに対応するラジカルである。Rxの適当な例として、メチル、エチル、ならびにn−およびイソ−プロピル、ならびにベンジルおよびフェネチルが挙げられる。Tの適当な例として、ハロゲン化合物(例えば、塩化物、臭化物、またはヨウ化物)が挙げられる。製薬的に許容される塩類のさらなる別の例もまた、N−酸化物等の内錯塩も含まれる。
【0144】
担体は、例えば、水、エチルアルコール、ポリオール(例えば、グリセリン、プロピレングリコール、および液体ポリエチレングリコール等)、それらの適当な混合物、ならびに植物油を含む溶媒または分散媒でもあり得る。適当な流動性を、例えばレシチン等のコーティング、分散の場合ならば所望の粒径に保つこと、ならびに界面活性剤の使用によって、保つことができる。微生物の影響からの保護は、種々の抗菌剤及び抗真菌剤、例えばパラベン、クロロブタノール、フェノール、ソルビン酸、チメロサール等により達成される。多くの場合、等張剤、例えば糖又は塩化ナトリウムを含むことが好ましい。注射用組成物の長期にわたる吸収は、吸収を遅延させる薬剤、例えばモノステアリン酸アルミニウムおよびゼラチンを組成物中で用いることにより達成される。
【0145】
滅菌注射用溶液を、上記列挙された種々の他の成分を必要に応じて含む溶媒に必要量の活性化合物を取り込み、続いて濾過滅菌することで調製する。通常、分散を、滅菌された種々の活性成分を滅菌されたビヒクル(基本分散媒および上記列挙したもののなかから選ばれる所望の他の成分を含む)に取り込むことによって調製する。注射可能な滅菌溶液の調製に用いられる滅菌粉末の場合、調製の好ましい方法は、真空乾燥および凍結乾燥技術であり、これによって、既に濾過滅菌した溶液から活性成分プラス任意の付加的な所望の成分の粉末が得られる。
【0146】
直接注射のためのより多くの、または高濃度の溶液の調製も検討する。ここで、溶媒としてDMSOを使用することで極度に迅速な貫通がなされ、小さな領域に活性剤が高濃度に送達される。
【0147】
処方に際して、投薬処方と互換性を持つ方法で、かつ治療的に有効であるような量で、溶液の投与をおこなう。製剤の投与を、種々の剤形(例えば、上記タイプの注射可能溶液)で容易に行えるが、薬放出カプセル等を使用することもできる。
【0148】
水溶液状態での非経口投与をおこなうために、例えば、その溶液を必要ならば適当に緩衝させるべきであり、また液体希釈剤が先ず十分な生理食塩液またはグルコースにより等張にされるものとする。これらの特定の水溶液は、特に静脈、筋内、皮下、および腹腔内投与に適している。この点について、使用可能な滅菌水性培地は、本開示を鑑みることで当業者に知られている。
【0149】
非経口投与(例えば、静脈内または筋内注射)用に処方される化合物に加えて、他の製薬的に受け入れられる剤形として、例えば、内服のための錠剤または他の固体;リポソーム型製剤;徐放性カプセル;および現在使用されている任意の他の剤形(クリームを含む)が挙げられる。
【0150】
投与の他の形態に適した別の製剤として、坐剤が挙げられる。坐剤のために、従来の結着剤および担体として、ポリアルキレン・グリコールまたはトリグリセライドを挙げることが可能であり、そのような坐剤は、0.5%ないし10%、好ましくは1%ないし2%の範囲内で活性成分を含む混合物から形成することが可能である。
【0151】
例えば、経口製剤として、製薬等級のマンニトール、乳糖、澱粉、ステアリン酸マグネシウム、ナトリウム・サッカリン、セルロース、炭酸マグネシウム等の通常使用される賦形剤が挙げられる。これらの組成物は、溶液、懸濁液、錠剤、ピル、カプセル、徐放性の製剤または粉末の剤形をとる。
【0152】
明確な特定の実施形態では、経口医薬組成物は、不活性の希釈剤または吸収消化可能な食用の担体を含み、またはそれらが固いまたは柔らかいシェル・ゼラチンカプセルに入れられたものであってもよく、または圧縮されて錠剤になったものであってもよく、または規定食に直接取り込まれたものであってもよい。経口治療投与用に、有効化合物に添加剤を組み込んでもよく、摂取可能な錠剤、口腔錠、トローチ、カプセル、エリキシル剤、懸濁剤、シロップ、ウエハース等の剤形で使用することが可能である。このような組成物および調剤は少なくとも0.1%の活性化合物を含有するものとする。組成物および製剤の割合が、もちろん変動する場合もあり、便宜上、単位重量の約2〜約75%、または好ましくは25〜60%の間であってもよい。このような治療上役立つ組成物の活性化合物の量は、適当な投薬量が得られるような量である。
【0153】
錠剤、トローチ剤、ピル、カプセル等は以下のようなものを含むものであってもよい。すなわち、結着剤(例えば、トラガカント、アカシア、コーンスターチまたはゼラチン)、賦形剤(例えば、リン酸二カルシウム)、崩壊剤(例えば、トウモロコシ澱粉、ジャガイモ澱粉、アルギン酸等)、潤滑油(例えば、ステアリン酸マグネシウム)、および甘味料(例えば、ショ糖、乳糖またはサッカリン)を添加してもよく、あるいは香料(例えば、セイヨウハッカ、ウィンターグリーン油またはチェリー香料)である。単位投与剤形がカプセル剤である場合は、前述の物質に加えて、液体担体を含むことができる。様々な他の物質がコーティングとして存在し、さもなければ投薬単位の物理的形態を修飾するために存在してもよい。例えば、錠剤、ピルまたはカプセルを、シェラック、糖、またはこの両方で被覆することができる。エリキシルのシロップは、防腐剤、染料、および香料(例えば、サクランボまたはオレンジ味)として、甘味料(メチルおよびプロピルパラベン)として活性化合物(ショ糖)を含有してもよい。
【0154】
本発明の医薬組成物を、例えば固体、半流動または液体状態の製剤の形態で使用することが可能であり、本発明の化合物の一つ以上を主成分として含み、外用、腸内適用、または非経口適用に適した有機もしくは無機の担体または賦形剤と混合された状態で、本発明の化合物の一つ以上を主成分として含む。主成分は、例えば、錠剤、ペレット、カプセル、坐剤、溶液、エマルジョン類、懸濁液、および使用に適した任意の他の剤形用の通常非中毒性で製薬的に許容される担体であってもよい。使用し得る担体として、補助剤、安定化剤、増粘剤、および着色剤に加えて、固体、半流動、または液体状態にある水、ブドウ糖、乳糖、アカシアゴム、ゼラチン、マンニトール、デンプンのり、三ケイ酸マグネシウム、滑石、トウモロコシ澱粉、ケラチン、コロイダルシリカ、ジャガイモ澱粉、尿素、および製剤製造での使用に適している他の担体が挙げられ、また香水を使用してもよい。アクティブ・オブジェクト化合物は、プロセスまたは病状に所望の効果を発生するのに十分な量で、医薬組成物に含まれる。
【0155】
錠剤等の固形組成物を調製するために、主要な活性成分を、製薬担体、例えば従来の錠剤化成分(トウモロコシ澱粉、乳糖、ショ糖、ソルビトール、滑石、ステアリン酸、ステアリン酸マグネシウム、リン酸二カルシウムまたはガム)および他の製薬希釈剤(例えば、水)とを混合して、本発明の化合物の均質混合物またはその無毒性で製薬的に許容される塩を含む固形予備処方組成物を形成する。これらの予備処方組成物をホモジェナスと称する場合、組成物が等しく有効な単位剤形(例えば、錠剤、ピルとカプセル)に容易に再分割されないように、主成分が組成物の全体を通じて均一に分散することを意味する。次に、この固形予備処方組成物は、本発明の主成分が0.1ないし約500mg含まれる上述のタイプの単位剤形に再分割される。新規組成物の錠剤またはピルを被覆、さもなければ化合して、持続性作用の長所を産みだす剤形を提供することができる。例えば、錠剤またはピルは内側投薬量構成要素と外側投薬量構成要素とを含むことができ、後者が前者を包む膜状となる。2つの構成要素を腸層によって分離することができ、この腸層によって胃の崩壊に耐え、内側構成要素が十二指腸を無傷に通過することが可能であり、または放出が遅れる。種々の材料を、このような腸層またはコーティングに使うことができ、この材料として、いくつかの高分子酸とシェラック、セチルアルコール、およびセルロースアセテート等の材料と高分子との混合物とが挙げられる。
【0156】
経口的にまたは注射によって投与するために本発明の組成物が取り込まれる液体の剤形として、水溶液、十分に風味をつけたシロップ剤、水性懸濁液または油性懸濁液、および許容される油(例えば、綿実油、ゴマ油、ヤシ油、または落花生油)または静注に適した可溶化または乳化剤によるエマルジョン類、同様にエリキシルおよび類似の賦形薬が挙げられる。水性懸濁液に適した分散剤または沈澱防止剤として、合成および天然のガム(例えば、トラガカンタ、アカシア、アルギン酸塩、デキストラン、ナトリウムカルボキシメチルセルロース、メチルセルロース、ポリビニルピロリドンまたはゼラチン)が挙げられる。
【0157】
吸入または吹送のための組成物として、溶液および懸濁液を含む製薬的に許容される水性または有機性の溶媒またはその混合物、および粉末が挙げられる。上述したように、液体または固体組成物は適当な製薬的に許容される賦形薬を含むものであってもよい。好ましくは、組成物は局所的または全身的な効果を得るために経口または鼻呼吸経路によって投与される。好ましくは製薬的に許容される滅菌溶媒の組成物は、不活性ガスの使用によって霧状にしたものであってもよい。霧状にされた溶液を噴霧器から直接呼吸してもよく、または、噴霧器をフェースマスク、テント、または間欠的陽圧呼吸機械に接続してもよい。溶液、懸濁液、または粉組成物は、好ましくは経口的または経鼻的に、適切な方法で製剤を送達する装置から投与することが可能である。
【0158】
上記の臨床症状および疾患を処置するために、本発明の化合物を従来の無毒性製薬的に許容される担体、アジュバント、およびビヒクルを含む用量単位製剤で、経口的、局所的、非経口的、吸入噴霧、または直腸的に投与してもよい。本文中で使用される非経口なる用語は、皮下注射、静注、筋肉内注射、胸骨内注射、または輸液法を包含する。
【0159】
スクリーニング法および検出法
本発明の組成物を、GPCR活性または発現を調整する薬物または化合物のスクリーニングに用いられるとともに、GPCRタンパク質の不十分または過剰な生産あるいはGPCR野生型タンパク質と比較して減少または異常活性を呈するGPCRタンパク質形態の生産によって特徴付けられる疾患を処置するために用いられる。
【0160】
本発明は、方法(また、ここでは「スクリーニング・アッセイ」と称する)を、モジュレータ、例えばGPCRに結合またはGPCRタンパク質発現もしくはGPCR活性に対して刺激または阻害効果を呈する候補または試験化合物または薬剤(例えば、ペプチド、ペプチド模倣薬、低分子、または他の薬物)を確認するために提供する。本発明はまた、本明細書中に述べられるスクリーニング・アッセイで同定される化合物を含む。
【0161】
本発明は、ペプデュシン−GPCR複合体の膜結合形態またはその生物学的活性部分の活性を調整またはそれに結合する候補または試験化合物をスクリーニングするためのアッセイを提供する。本発明の試験化合物を、数多くのアプローチのいずれかを当該技術分野で知られている組み合わせライブラリ、例えば生物学的ライブラリ、空間的にアドレス可能な平行固相または溶液相ライブラリ、デコンボリューションを必要とする合成ライブラリ方法、「1−ビード1−化合物」ライブラリ方法、およびアフィニティークロマトグラフィー選択を使用する合成ライブラリ方法で用いることによって、得ることができる。生物学的ライブラリー・アプローチは、ペプチド・ライブラリーに限られており、一方その他の4つのアプローチがペプチド、非ペプチド・オリゴマー、または化合物の低分子ライブラリに適用できる。例えば、Lam,1997.Anticancer Drug Design 12:145を参照せよ。
【0162】
分子ライブラリの合成に関する方法の例は、当該技術分野、例えば、DeWitt,et al.,1993.Proc.Natl.Acad.Sci.U.S.A.90:6909;Erb,et al.,1994.Proc.Natl.Acad.Sci.U.S.A.91:11422;Zuckermann,et al.,1994.J.Med.Chem.37:2678;Cho,et al.,1993.Science 261 :1303;Carrell,et al.,1994.Angew.Chem.Int.Ed.Engl.33:2059;Carell,et al.,1994.Angew.Chem.Int.Ed.Engl.33:2061;およびGallop,et al.,1994.J.Med.Chem.37:1233で見出すことができる。
【0163】
化合物ライブラリは溶液状態で存在(例えば、Houghten,1992.Biotechniques 13:412−421)、またはビーズ(Lam,1991.Nature 354:82−84),on chips(Fodor,1993.Nature 364:555−556)、細菌(Ladner,U.S.Patent No.5,223,409),胞子(Ladner,U.S.Patent 5,233,409)、プラスミド(Cull,et al.,1992.Proc.Natl.Acad.Sci.USA 89:1865− 1869)あるいは、ファージ(Scott and Smith,1990.Science 249:386−390;Devlin,1990.Science 249:404−406;Cwirla,et al.,1990.Proc.Natl.Acad.Sci.U.S.A.87:6378−6382;Felici,1991.J.MoL Biol.222:301−310;Ladner,U.S.Patent No.5,233,409)に存在するものであってもよい。
【0164】
アッセイは、細胞ベースのアッセイであり、GPCRの膜結合形態または細胞表面上でその生物学的活性部分、プラスペプデュシンを発現する細胞を試験化合物と接触させ、GPCRと結合してペプデュシンを移動させる試験化合物の能力を決定する。試験化合物は、GPCR標的の細胞外表面、膜貫通型領域または細胞内表面で結合することができ、GPCRのペプデュシン活性化を阻害することができ、あるいは高めることが可能である。細胞は、例えば、哺乳類起源または酵母細胞である。ペプデュシンをGPCRタンパク質から移す試験化合物の能力の決定は、試験化合物の結合がGPCRからペプデュシンまたはその生物学的活性部分を移動させるように、例えば、ペプデュシンを放射性同位元素または酵素ラベルに連結させることによって達成される。あるいは、直接または間接的に、試験化合物を125I、135S、4Cまたは3Hで標識することができ、ペプデュシンは放射性同位元素標識試験化合物をGPCRから移すことが可能であり、さらに遊離放射性同位元素標識試験化合物が、放射能放出の直接的計数によって、またはシンチレーション・カウントによって検出される。あるいは、例えば、試験化合物をワサビペルオキシダーゼ、アルカリホスファターゼまたはルシフェラーゼによって酵素標識することができ、酵素ラベルはペプデュシンの追加に応じて製品に対する適当な基質の転換での減少または増加によって検出される。
【0165】
別の実施形態では、アッセイは、細胞膜上のGPCRタンパク質の膜結合形態またはその生物学的活性部分を試験化合物と接触させること、およびGPCRに関してのペプデュシンの結合、活性を変調(例えば、刺激または阻害)する試験化合物の能力を決定することを含む細胞ベースのアッセイである。
【0166】
GPCR標的分子と相互作用する試験分子の能力を決定することは、直接結合を決定するための上記方法のうちの1つによって達成される。一実施形態では、GPCR標的分子とのGPCRペプチド相互作用を阻害する試験分子の能力を決定することは、標的GCPR−ペプデュシン複合体の活性を決定することによって達成される。例えば、標的分子の活性は、GPCR標的の細胞の二次メッセンジャーのGPCRペプチド誘導を阻害すること(細胞内Ca2+、ジアシルグリセロール、IP3、その他)、GPCR活性化または阻害に依存している触媒/酵素活性を検出すること、リポーター遺伝子(検出可能な標識(例えば、ルシフェラーゼ)をコードしている核酸に対して有効に連結するGPCR反応性調節因子を含む)の誘導または阻害を検出すること、または、細胞性応答(例えば、細胞生存、細胞分化、または細胞増殖)を検出することを含む。
【0167】
あるいは、本発明のアッセイは、試験化合物をGPCRペプチドまたは生物学的活性部分に接触させること、GPCRタンパク質またはその生物学的活性部分の活性を調節(例えば、刺激または阻害)またはそれに結合する試験化合物の能力を決定することを含む無細胞アッセイである。
【0168】
GPCRに対する試験化合物の結合を、先に述べたように、直接または間接的に決定することができる。例えば、アッセイは、アッセイ混合物を形成するために、ペプデュシンとGPCRまたはその生物学的活性部分を、GPCRと結合する既知の化合物と接触させること、アッセイ混合物を試験化合物と接触させること、およびGPCRタンパク質と相互作用する試験化合物の能力を決定することを含み、GPCRタンパク質と相互作用する試験化合物の能力を決定することは、既知の化合物と比較すると優先してGPCRと結合する試験化合物の能力またはその生物学的活性部分を決定することを含む。
【0169】
GPCRの活性を変調する試験化合物の能力を決定することは、例えば、直接結合を決定する上記の方法の1つによって、GPCR標的分子に対して結合するGPCRペプチドの能力を決定することによって、達成し得る。あるいは、GPCRの活性を変調する試験化合物の能力ペプチドを決定することは、CPCR標的分子をさらに変調するGPCRペプチドの能力を決定することによって達成し得る。例えば、適当な基質上での標的分子の触媒的/酵素的活性を上記のように決定することができる。
【0170】
無細胞アッセイは、GPCRペプチドまたはその生物学的活性部分をGPCRに結合してアッセイ混合物を形成する既知の化合物と接触させること、アッセイ混合物を試験化合物と接触させること、およびCPCRと相互作用する試験化合物の能力を測定することを含み、GPCRと相互作用する試験化合物の能力を決定することは、GPCR標的分子の活性を優先的に変調またはそれに結合するGPCRペプチドの能力を決定することを含む。
【0171】
本発明の無細胞アッセイは、GPCRタンパク質の可溶性の形状または膜結合形態の両方の使用にしたがう。GPCRタンパク質の膜結合形態を含む無細胞アッセイの場合、GPCRタンパク質の膜結合形態が溶液で維持されるように、溶解剤を利用することは、望ましいと考えられる。この溶解剤の実施例は、非イオン性界面活性剤、例えばn−オクチルグルコシド、n−ドデシルグルコシド、n−ドデシルマルトシド、オクタノイル−N−メチルグルカミド、デカノイル−N−メチルグルカミド、トリトン(登録商標)X−100、トリトンX−(登録商標)114、セシト((登録商標)、イソトリデシポリ(エチレングリコールエーテル)n、N−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネート、3−(3−コラミドプロピル)ジメチルアミニオル−1−プロパンスルホネート(CHAPS)、または3−(3−コラミドプロピル)ジメチルアミニオル−2−ヒドロキシ−1−プロパンスルホネート(CHAPSO)が挙げられる。
【0172】
GPCRペプチドまたはその標的分子のいずれかを固定して、該タンパク質の一方または両方の未複合体化形態から複合体を形成したものを分離することを促進させ、同様にアッセイのオートメーションを適応させることが望ましいかもしれない。候補化合物の存在または非存在下でのGPCRタンパク質に対する試験化合物の結合またはGPCRタンパク質とペプデュシンとの相互作用は、反応物の含有に適した任意の容器で達成される。そのような容器の例として、マイクロタイター・プレート、試験管、およびマイクロ遠心管が挙げられる。一実施形態では、タンパク質の一方または両方がマトリックスに結合するのを可能にする領域を加える融合タンパクが、提供される。例えば、GST−GPCR融合ペプチドまたはGST−標的融合タンパクをグルタチオン・セファロース・ビード(シグマ・ケミカル社、セントルイス、ミズーリ州)またはグルタチン誘導体化マイクロタイター・プレートに吸着させることができ、試験化合物、または該試験化合物と非吸着標的タンパク質またはGPCRペプチドのいずれかと組み合わせることで、混合物を、複合体形成(例えば、塩およびpHの生理学的条件で)に貢献する条件下でインキュベートする。インキュベーション後、ビードまたはマイクロタイター・プレート・ウェルを、任意の未結合構成要素を除去するために洗い、ビードの場合、マトリックスが固定される、複合体が直接または間接的に、例えば、上述のようにして、決定される。あるいは、複合体をマトリックスから分離し、GPCRペプチド結合または活性のレベルを標準技術を用いて決定した。
【0173】
マトリックス上での非流動化タンパク質の他の技術が、本発明のスクリーニング・アッセイでも使われる。例えば、GPCRペプチドまたはその標的分子は、ビオチンとストレプトアビジンとの複合体形成を利用して固定することができる。ビオチニル化GPCRペプチドまたは標的分子は、当該技術分野(例えば、ビオチン化キット、ピアスケミカル社、ロックフォード、イリノイ州)の中で周知の技術を使用しているビオチン−NHS(N−ヒドロキシ−スクシニミド)から調製することができ、ストレプトアビジン被覆の96穴プレート(ピアスケミカル社)のウエルで固定することができる。GPCRペプチドまたは標的分子と反応性はあるが、GPCRペプチドと同族GPCRとの結合を妨害しない抗体を、プレートのウエルに誘導体化させることができ、未結合標的またはGPCRペプチドは抗体結合によってウエルに捕捉することができる。GPCRペプチドまたは標的分子と反応性はあるが、GPCRペプチドと同族GPCRとの結合を妨害しない抗体を、プレートのウエルに誘導体化させることができ、未結合標的またはGPCRペプチドは抗体結合によってウエルに捕捉することができる。このような複合体を検出する方法として、GST固定複合体に関する上記のものに加えて、GPCRペプチドまたは標的分子に対するGPCRペプチドと反応する抗体を用いた複合体の免疫学的検出法、同様にGPCRペプチドまたは標的分子に関連する酵素活性の検出に依存する酵素結合アッセイが挙げられる。
【0174】
GPCRタンパク質発現のモジュレータは、細胞が候補化合物で接触する方法で特定され、細胞内でのGPCRmRNAまたはタンパク質の発現が決定される。候補化合物の存在下でのGPCRmRNAまたはタンパク質の発現レベルを候補化合物が存在しない場合のGPCRmRNAまたはタンパク質の発現と比較する。この比較にもとづいて、候補化合物がGPCRmRNAまたはタンパク質の発現のモジュレータとして同定される。例えば、GPCRmRNAまたはタンパク質の発現が候補化合物の存在下でその不在の場合よりも大きい(すなわち、統計学的に有意により大きい)場合、候補化合物は、GPCRmRNAまたはタンパク質発現の刺激因子であると同定される。あるいは、GPCRmRNAまたはタンパク質の発現が候補化合物の存在下で、不在の場合よりもより少ない(統計学的に有意により少ない)場合、候補化合物は、GPCRmRNAまたはタンパク質発現の阻害剤と確認される。細胞のGPCRmRNAまたはタンパク質発現のレベルは、GPCRmRNAまたはタンパク質を検出するために本明細書中に述べられる方法によって測定することができる。
【0175】
本明細書で議論されるペプチド配列を以下の表3に示す。
【0176】
【表3−1】
【0177】
【表3−2】
【0178】
【表3−3】
【0179】
【表3−4】
【0180】
【表3−5】
ペプデュシン(例えば、i1ループから)のペプチドの部分は、結果として意図された有益な効果をもたらす任意の適当な長さであってもよく、一般に5〜15、または、例えば、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29または30である。
【0181】
本発明のペプデュシン化合物を調製する方法を、以下の合成スキームおよび実施例で例示する。以下のスキーム、実施例、および生物学データは、発明の範囲または精神を制限するためのものではなく、発明を例示するために与えられる。
【0182】
ペプデュシン化合物の製造および特徴づけ
一般にCovic et al.PNAS 99:643−648(2002)に述べられるように、パルミトイル化されたペプデュシン・ペプチドの標準Fmoc固相合成物方法および調製による合成をおこなった。ペプデュシンをC18またはC4逆相クロマトグラフィーによって>95%純度に精製し、DMSOに溶した。
【0183】
出血時間は、キシラジン(10mg/kg)+塩酸ケタミン(50mg/kg)の腹腔内投与で6〜8週間目の成体雄CF−1マウスを麻酔しておこなった。内頸静脈にカニューレを挿入し、0.28×1.52mmゲージ・カテーテルおよびPlpal−12(3μmoles/L)、P4pal−10(3μmoles/L)またはビヒクル(DMSO)単独で、100μLの総容積で、1分にわたって注入した。実験は、注射された物質に対して盲検下でおこなった。5分後に、尾端から2mmのところで尾部の切断をおこなった。尾部を、37℃に維持されるリン酸塩緩衝食塩水のビーカーに浸漬させ、出血を視覚的に追跡し、時間を計った。出血が5分以内に再開する場合、それを再ブリードとして記録されて、前述のとおり、不安定な止血現象を採用した(Law et al.,Nature 401,808(1999)。許される最大出血時間は、10分間であり、その後、尾部を焼灼した。
【実施例】
【0184】
実施例1
敗血症がどのように本発明のペプデュシンによって効果的に処置されるかについて示すために、6〜8週間目の雌のCF−1マウスを、20%のDMSOまたはビヒクル(20%DMSO)中2.5mg/kgのペプデュシン製剤で処置した。処置のために選択される時点は、盲端連結反応および穿刺(CLP)による非致死的敗血症の誘導である。2.5mg/kgの初期量の後、1mg/kgのペプデュシン(またはビヒクル)をマウスに対して6日間にわたり毎日皮下注射した。ビヒクル処置群からの4匹のマウスを、6日後に犠牲にした。xl/2i1LCA10ペプデュシンで処置される群からの1匹のマウスを6日目に犠牲にし、全ての残りの生存マウスを17日目に犠牲にした。図5は、結果を表す。CLPの時点で、左側パネルでは、マウスをxl/2pal16、xl/2i1LCA10またはビヒクル(20%のDMSO)で処置した。右側のパネルは、マウスの体重を毎日、モニターした。ビヒクルを処置されたマウスは重篤な体重損失を示したが、ペプデュシン処置されたマウスは最初の体重損失の後、体重が増えた。
【0185】
実施例2
全身性炎症反応症候群の発症の後でさえ、敗血症の処置が充分である報告は、ほんのわずかしかない。敗血症が起こったかなり後に、本発明のペプデュシンによって、敗血症が効果的に処置されることを示すために、マウスを、40%DMSOまたはビヒクル(40%のDMSO)中5.0mg/kgのペプデュシン製剤で処置した。処置のために選択される時点は、盲腸の結紮および穿刺(CLP)による非致死的敗血症の誘導時の8時間である。5.0mg/kgの初回量の後、2.5mg/kgのペプデュシン(またはビヒクル)を2日目から3日目までは毎日皮下注射し、3日目から6日まで毎日、1.0mg/kgをマウスに注射した。CLPの8時間後に、マウスはすでに敗血症に特有の全身性疾患の徴候(例えば、乱れた毛皮、減少した運動能、減少した食物および水摂取、ならびに閉じた炎症を起こした眼)を示した。図6は、結果を表す。左側パネル(*)で:ビヒクル処置群からの2匹のマウスを、6日目に犠牲にした。実験を生き残ったマウスを、17日目に犠牲にした。右側パネルにおいて、マウスの体重を毎日、モニターした。
【0186】
実施例3
炎症性疾患のペプデュシン処置の有用性を示すために、マウスを本発明のペプデュシンで処置した。図7は、結果を表す。成人性呼吸反応症候群(ARDS)は、肺への過剰な好中性浸入とそれらの活性化(線維素沈着に至る)から生じる。敗血症によってARDSが起きる場合があり、それは高い致死性を持つ。パネルAとB:マウスをCLPの24時間後に犠牲にし、BALを実行した。好中球をギムザ染色した後に計数した、TNFをELISAで測定した。*p<0.02対ビヒクル処置群。C.肺組織学。マウスを、CLPの48時間後に犠牲にした。
【0187】
実施例4
この実施例は、ペプデュシン処置がどのように敗血症のもう一つの態様を軽減するのに用いられるかについて示す。全体的な炎症反応、DICの凝固と線維素溶解の錯乱は、いくつかの炎症誘発性サイトカインによって媒介される。出血時間の変化とD−二量体の増加(それは、過剰な対抗制御的な線維素溶解の産物)は、播種性血管内血液凝固の特徴であり、血行によるフィブリンの広範囲にわたる堆積に至り、敗血症の患者における多臓器不全と死亡に関与する。図8は、結果を表す。A.出血時間をCLPの24時間後と処置開始直後に測定した。出血時間は、ビヒクルを処置されたマウスで短くなり、その一方で、i1−ループ・ペプデュシンx1/2i1LCA10−またはi3ループ・ペプデュシンx1/2pal16−処置マウスは、正常な出血時間を示した。B.D−二量体濃度は、CLPの48時間後に測定した。線維素沈着量の代用の標識として、CXCR2ペプデュシン処置マウスでよりビヒクルを処置されたマウスにおけるより高い範囲に、D−二量体がCLPの後に増加した。C.CLPの8時間後に処置されたマウスの出血時間を表す。D.CLPの8時間後に処置されるマウスのD−二量体濃度を表す。
【0188】
実施例5
炎症性疾患のペプデュシン処置の有用性を示すために、走化性アッセイを、修飾48ウエル・マイクロ走化性チャンバーで行った。1ミオ(mio)好中球/mLは、より低いウエル内の化学遊走物質に向けての移動が可能であった。好中球を、0.2%DMSOまたはペプデュシンのいずれでも前処理したN=3。図9は、結果を表す。
【0189】
実施例6
生体内で炎症性疾患のペプデュシン処置の有用性を示すために、走化性アッセイを、マウスで行った。マウスに対して、腹腔内に3%チオグリコール酸塩1mLを注射した。チオグリコール酸塩は分泌サイトカインに腹腔マクロファージを誘導し、白血球を腹腔に補充させる。ペプデュシンを、示された用量で静脈注射した。4時間後に、細胞を腹腔洗浄によって採取し、ギムザ染色を施し、40倍で計数した。N=60。図10は、結果を表す。
【0190】
実施例7
SDF−lαとCXCR4が未熟好中球を骨髄から補充することに関係していることが示された。CXCR4発現と活性化の高水準は骨髄に老化の好中球のホーミングに至るが、ケモカインが好中球のCXCR2受容体に作用する。すなわち、KC、Gro−αまたはIL−8がさらなる好中球の移動を導く。血液をCLP後1、3、5、7、および17日に採血し、白血球の計数をおこなった。ビヒクルを処置されたマウスにおける白血球の最初の減少が、観察された。しかし、これらのマウスは末梢白血球の高レベルを示し、実験の終わりまで高レベルで残った。xl/2pal16およびxl/2i1LCA10−処置されたマウスにおいて、白血球レベルは、正常範囲のままだった。x4i1pal12を処置されたマウスにおいて、白血球レベルは直ちに増加したが、非常に低いレベルまでCLPの5日後に減少した。この知見は、CXCR4のブロッキングまたはCXCR2の刺激がマウスの末梢血で好中球増多に至るという以前の報告に一致する。x4i1pal12処置後の末梢血の白血球の増加は、CXCR2遮断が末梢血好中球増多を予防することを示す。図11は、結果を表す。
【0191】
実施例8
走化性アッセイを、修飾された48穴マイクロ化学走性チャンバーで行った。1ミオ(mio)好中球/mLは、より低いウエル内の化学遊走物質に向けての移動が可能であった。好中球を、指示濃度で0.2%DMSOまたはペプデュシンのいずれかで前処理した。N=3。図12は、本発明のi1−ループ・ペプデュシンには強力および選択的な消炎作用があることを示す。
【0192】
実施例9
走化性アッセイを、修飾48穴マイクロ化学走性チャンバーで行った。1ミオ(mio)好中球/mLは、より低いウエル内のランテス(Rantes)−CCRl、CCR3、およびCCR5(20ng/mL)のためのリガンドに向けての移動が可能であった。好中球を、指示濃度で0.2%DMSOまたはペプデュシンのいずれかによって前処理した。N=3。図13は、本発明のペプデュシンi1ループが強力かつ選択的な消炎作用を持つことを示している。
【0193】
実施例10
臓器不全がSIRS患者の結果を決定するので、肝機能を調べた。マウスをCLPの24時間後に犠牲にし、ALT(肝機能の損失で増加する酵素)の血漿レベルを測定した。疑似処置マウス(すなわち盲腸の結紮と穿刺のない開腹)において、ALTの上昇は観察されなかった。ビヒクルとx4pal12処置されたマウスはALTの増加を明らかにした、xl/2pal16とxl/2LCA10処置されたマウスで、増加が観察された。しかし、それはビヒクルを処置されたマウスと比較して著しく少なかった。図22は、CXCR2ペプデュシンが肝機能を改善するのに用いることができるという結果を表す。
【0194】
実施例11
GLPを、6〜8週間目の雌のCF−Iマウスに対して行われた。右側パネルにおいて、CLPの時点に、x4i1pal12またはビヒクル(20%のDMSO)でマウスを処置された。2.5mg/kgの初回量の後、動物に1mg/kgのペプデュシンまたはビヒクルを6日にわたり毎日皮下注射した。左側パネルにおいて、マウスの体重を毎日、モニターした。図23は、結果を表す。
【0195】
実施例12
受容体の第1の細胞内ループ(i1)に基づくより選択的なPAR4ペプデュシンを開発した。i1ループは、i3ループと関連した受容体の反対側の上にあることから、PAR4に基づくI1ループ・ペプデュシンは、PARlの減少または非相互抑制を示す。PAR4は、PARlで単独または組み合わせで、HEK細胞上で発現した。PAR4i1ペプデュシン(P4pal−i1)は、完全にHEK細胞の上でPAR4の走化性反応を妨げて、図24Aおよび図24Bで示すようにそのペプチド・リガンド(AYPGKF(配列番号34))に対する血小板凝集を予防した。P4pal−i1は、PAR4に対して選択的であり、P4pal−i1はPARlの走化性反応を阻害しなかったし、図24Aないし図24Cで見られるように、それはPARlペプチド・リガンド(SFLLRN(配列番号35))に対する血小板凝集を明らかに阻害しなかった。P4pal−i1は、PARlとPAR4とが複合体を形成する場合、PAR4の結合したi1ペプデュシンがPARlからシグナル伝達にこれといって影響を及ぼさないことを示しているPAR4で、同時発現さえもPARlを阻害しなかった。同様に、単独で使用される場合、トロンビンもPARl(図24D)を活性化することから、P4pal−i1には3nMトロンビンに対する血小板凝集上で軽度の作用だけがあった。しかし、PARlアンタゴニスト(RWJ−56110)と結合して使われる場合に、P4pal−i1は3nMトロンビンに対する凝集を阻害した。このように、PARlまたはPAR4単独を標的とすることは、トロンビンに対する凝集に限られた作用を持つ。PARlおよびPAR4の同時阻害はトロンビンに対する反応を妨げることに効果的であり、それによって血栓症を減らすか、阻害する。
【0196】
実施例13
血小板PARlおよびPAR4反応に対するビバリルジンの作用は調べず、また血小板凝集に対するPAR4遮断と組み合わせたトロンビン−PARl相互作用を阻害する有効性について評価した。PARlのHir部位ビバリルジン(別名Hirulog(登録商標)、Angiomax(登録商標))に対するトロンビンの結合を抑制するために、ナノモルの親和性によるトロンビンとの結合を用いた。患者を急性冠動脈症候群で処置することでのその広範囲にわたる使用にもかかわらず、PARlおよびPAR4に依存する血小板活性化に対するビバリルジンの作用は、本発明に先だって決定されることはなかった。ビバリルジン単独またはビバリルジン・プラスRWJ−56110は、PAR4に依存する凝集のトロンビン活性化と同程度の50%有効濃度(10〜11nM)がPARl−抗体(図25A)で示された。11nMを超える濃度で、RWJ−56110がPARl−阻止抗体+ビバリルジンで補充された場合でも、トロンビンはPAR4に依存する凝集の完全な活性化を回復した。これらのデータは、Hir−遮断薬のどちらも血小板PAR4で完全にトロンビンの活性部位の相互作用を予防しなかったことを示す。しかし、PAR4ペプデュシン、P4pal−i1、のビバリルジンを処置された血小板への添加は、非常に高いトロンビン(12−16nM)濃度(図25B)であっても、凝集の広がりを著しく遅らせ、かつ阻害した。これらのデータは、PARlのHir−モチーフにドッキングしたトロンビンがPAR4の活性化を高めるが、高度に十分なトロンビン濃度で、PAR4ペプデュシンによって下流に封鎖されない限り、PAR4はトロンビンによって活性化される。
【0197】
実施例14
モルモットにおける動脈血栓症は、ビバリルジンとPAR4ペプデュシンの組合せを使用して予防される。モルモットで動脈血栓に関してビバリルジンとPAR4ペプデュシンの同時投与の有効性を評価する標準頸動脈障害モデルが、使われた。それらの血小板上でPARlがないマウスとは異なり、モルモットは機能性類似度をヒト血小板と共有し、PARlおよびPAR4を発現する。ヒルジンを使用している初期の結果と整合して、これが有意でなかったにもかかわらず、ビバリルジン(0.18mg/kg)単独は13分から20分まで平均動脈の閉塞時間を延長させた(図26)。P4pal−i1(0.13mg/kg)は、同程度の閉塞時間を延長した。ビバリルジンさらにP4pal−i1の共投与は、急性の動脈の閉塞に対して有意な(p=0.0001)保護を生じた。RWJ−58259で示されたように、PARlペプデュシン(Plpal−7)で1つのPARlの遮断は動脈血栓の部分的な保護だけを生じた。P4pal−i1に対する比較において、ビバリルジンによるPlpal−7のサプルメントは、動脈の閉塞時間の付加的な延長をしなかった。同時に、これらの生体内データは、PAR4も血栓症に対して有意な保護を達成するために遮断されなければならないことを示す。
【0198】
これらの実験は、PARlとPAR4ペプデュシン・アンタゴニストと阻止抗体とによる以前の知見をサポートする。PARlおよびPAR4だけを標的とすることは、治療的有効性を制限してしまうと思われる。代わりの治療的戦略として、トロンビンでPARlHirのモチーフの相互作用を予防することは、直接PARlを阻害して、間接的にPAR4を阻害するという二重利点を持つ。これらのデータは、広く使われている抗血栓症薬(ビバリルジン)がPARlおよびPAR4に依存する血小板凝集のトロンビン活性化を遮断する際に有効だったことを初めて証明する。PARlペプデュシンでなくPAR4ペプデュシンと結合して、ビバリルジンは急性の動脈血栓症を予防することが可能だった。
【0199】
実施例15
宿主防御に対する干渉なしで凝固カスケードの活性化をかなり予防するにつれて、本発明のペプデュシンは確立した全身性炎症および脈管損害を妨げるのに用いられる。例えば、本発明のi1ペプデュシン(CXCRl/2に基づく)は、CXCR4を相互抑制しない。i1ペプデュシンは、ヒトおよびマウス好中球上で完全にそれらの同族の受容体の走化性反応を妨げる。xl/2LCA−i1はCXCRl/2IL−8受容体に対して選択的で、SDF−1αの方へヒト、マウス白血球の移動を阻害しない。同様に、CXCRlまたはCXCR2で共同発現される場合でも、xl/2LCA−i1はCXCR4を阻害しない。これらのデータは、CXCRlとCXCR4受容体が複合体を形成する場合、CXCRlの結合したi1ペプデュシンがCXCR4からのシグナル伝達に影響を及ぼさないことを示す。
【0200】
正常マウスにおける対炎症誘発性条件下の好中性ホメオスタシスに対するCXCRl/2とCXCR4ペプデュシンの効果を評価した。白血球補充を、チオグリコール酸塩腹膜炎モデルで最初に評価した。それぞれ、xl/2pal−i3およびxl/2LCA−i1−ペプデュシンのIC50値は0.03mg/kgおよび0.15mg/kgであり、完全に好中球の腹腔への移住を阻害した。逆に言えば、CXCR4アンタゴニスト、x4pal−i1(EC50−0.1mg/kg)で処置されるマウスは、腹膜好中球の実質的な増加を示した。
【0201】
末梢的な白血球数のケモカイン・ペプデュシンの長期の作用を、健康な無処置のマウスで評価した。ペプデュシンを1日1回、サブQ(2.5mg/kg/1日目、1.0mg/kg/2〜6日目)注射し、循環する白血球レベルを1週間にわたって測定した。健康なマウスにおいて、xl/2pal−i3もxl/2LCA−i1も、ビヒクルを処置されたマウスと比較すると白血球数を変えなかった。対照的に、x4pal−i1は末梢血で白血球増加症を引き起こし、先行の研究と整合していた。末梢的な白血球数のケモカイン・ペプデュシンの作用は、盲腸の結紮と穿刺(CLP)を受ける敗血症のマウスに関して、全く同様だった。ビヒクルを処置されたマウスは、CLP後の24時間、また3日目の白血球増加症で最初の白血球減少症を示した。一日1回のxl/2pal−i3またはxl/2LCA−i1投与は、一度は正常範囲の中でCLP−マウスの好中球数を保ったが、x4pal−i1は1日目の早期白血球増加症と7日目までの白血球減少症に至った。同時に、これらのデータは、CXCR2およびCXCR4受容体がマウスで正常および炎症誘発性状態で好中性ホメオスタシスにおいて対向した役割を果たすことを示す。
【0202】
ケモカイン受容体ペプデュシンを、マウスにおいて致死CLP腹膜炎の進行を保護または潜在的に逆転させる能力について試験した。実験の第1セットには、ペプデュシンを2.5mg/kgの用量で、CLP手法直後に与えた。その後の用量は、処置が止められた6日目まで1mg/kgとした。マウスのいずれも、抗生物質療法を受けなかった。高度に有意な減少は、xl/2LCA−i1およびxl/2pal−i3ペプデュシンについて、敗血症誘導死亡率で観察された。著しく、17日間の観察期間にわたって、1/34ペプデュシン処置マウスだけは死亡したが、無処置のマウスの17/17は9日目で死亡した。6日目のCXCRl/2ペデュシン療法の中断にもかかわらず、生存はその後ほぼ100%残った。
【0203】
敗血症の診断はしばしば遅れ、また薬剤を予防様式で容易に投与することができないことから、ペプデュシン処置をCLP手法の8時間後まで保留とした。遅延処置であっても、死亡率の高度に有意な縮小は、無処置のマウスと比較すると見られた。総生存率は、無処置のマウスで0/20であったのに対して、遅延CXCRl/2ペプデュシン処置マウスでは26/30であった。CXCR4ペプデュシン(x4pal−i1)の投与には、生存に関しての効果はなかった。CXCRl/2−ペプデュシン処置マウスは、体重が増え、活性化し、ペデュシン療法が8時間遅れたときでも、1日目後に正常な身繕い行動を維持した。
【0204】
ペプデュシンによるCXCRl/2シグナル伝達の遮断は、敗血症のマウスで確立したSIRSのいくつかの基準を逆転させた。第一に、全身KCの上のCXCRl/2ペプデュシンの作用はフラットになり、マウスIL−8相同分子種を調べた。無処置のマウスにおいて、KCレベルはCLPの後、16時間にわたって上昇し、少なくとも48時間上昇するままだった。8時間の時点でのCXCRl/2ペプデュシンの投与の後で、全身KCレベルは、急速に低下して、その後低いままだった。無処置のCLPマウスの多数は24時間という早い時期に頻脈および低酸素血症ようになり、このことは十分に死亡率と相関した。敗血症の間、菌体内毒素は肺上皮を刺激して白血球を補充し、かつ活性化させるIL−8を隠すようにする。結果として生じる好中性縁のあることは、最終的には肺の損傷に至る。無処置のマウスの気管支肺胞洗浄(BAL)液体の好中球の数は、CLPの後、4時間という早い時期に100倍まで増加し、8時間後に200〜300倍まで上昇した。l/2pal−i3による処置または8時間のxl/2LCA−i1は、低レベルにとどまったBAL好中球の急速な低下を引き起こした。無処置のCLPマウスで48時間に収集される肺の組織学的分析は、崩壊した肺胞、白血球浸入と広範囲な線維素沈着を明らかにした。xl/2pal−i3およびxl/2LCA−i1は線維素沈着に対して有意な保護を与え、肺胞が組織学的に正常に見えた。CXCRl/2ペプデュシンも、LPSをシミュレートした上皮および内皮単分子層全体で完全に白血球の移動を妨害した。同様に、CXCRl/2ペプデュシンは、予想通りの単球でなくヒト大食細胞のIL−8に依存する走化性を抑えた。CXCRl/2ペプデュシンは、腹腔に有意に生体内マウス大食細胞移動を阻害した。同時に、これらのデータは、ペプデュシンは、多数の細胞型上でCXCRl/2受容体を封鎖することによってそれらの治療的作用を奏することを示している。
【0205】
肝不全は、重篤な敗血症の一般の後遺症である。肝酵素濃度(AST、ALT)で52−87%の減少によって明示されるように、CLPの直後のCXCRl/2ペプデュシンによる処置は肝障害を減らし、一方CXCR4ペプデュシン(x4pal−i1)には、作用がなかった。CXCRl/2ペプデュシン処置がCLPの8時間後に開始された場合、血漿ASTおよびALTレベルの上昇は16時間の時点までに停止し、そこから正常レベルに落ちた。同時に、これらのデータは、ペプデュシンによるCXCRl/2シグナル伝達の遮断が敗血症のマウスにおける確立したSIRSのいくつかの基準を逆転させることを示し、このアプローチが重症をセットする際に有益であることが示される。
【0206】
特発出血と血栓の発症は無処置のCLPマウスの多数で観察され、播種性血管内血液凝固(DIC)の発症と整合していた。したがって、全身血小板活性化は、血小板数およびD−二量体濃度、増加したフィブリン産生の標識、DIC間中に起こるその後の線維素溶解を測定することによって観察された。ペプデュシン処置がCLPの後、8時間まで遅れた場合でも、CXCRl/2ペプデュシンは24時間に有意に血小板減少症から保護した。D−二量体濃度が、無処置のマウスでCLP手法の48時間後に高度に上昇するとわかった。しかし、xl/2pal−i3およびxl/2LCA−i1−処置されたマウスは、D−二量体の濃度の有意な減少を示した。CXCRl/2ペプデュシン処置がCLP手法の後、8時間まで遅れたとき、D−二量体濃度は依然として60〜70%減少した。これらのデータは、明白な敗血症で発現する凝固障害がCXCR2の遮断によって改善される第1の実証である。
【0207】
我々の研究で観察されるより3〜4倍高い死亡率ではあるが、機能的なCXCR2受容体がないマウスを使用している最近の研究はCLP敗血症モデルで生存の増加を示した。CXCR2の欠失の作用と関連してペプデュシン処置に続いている死亡率の改善された結果は、いくつかの因子に起因することができた。CXCR2(−/−)マウスは、それらの全ての寿命の間にCXCR2がなくて、異常な骨髄造血に起因している急性のCXCR2特異的な作用と補償機構を区別することを難しくしているそれらの適応可能な免疫系に、更なる障害がある。使用されるCF−1マウスは、CLPから重度の免疫原性的取り組みの状況で生存利益を与えるかもしれない非近交系の野生型動物である。抗体CXCRl/2ペプデュシンが細菌fMLP等の他のケモカインの方へ白血球遊走を抑制しないことは注目に値する、したがって、ペプデュシン作用は免疫抑制性であるというよりはむしろ免疫調節性であると考慮されるかもしれない。さらにまた、ペプデュシン技術の用途は、遺伝学的なノックアウトから結果を確認するのを助けて、敗血症のような複合体疾患の病因と処置に対するさらなる洞察を提供する。
【0208】
等価物
当業者は、日常的実験、本明細書中に述べられる特異的な手法に対する多数の等価物しか使用しないことを理解し、または確かめることができる。そのような等価物は発明の範囲内であると考えられ、以下の請求の範囲によってカバーされる。請求の範囲によって定義されるように、種々の置換、変更、および修正は発明の精神および範囲から逸脱することなく発明が実施される。他の態様、利点、および変形は、発明の範囲内である。この明細書の全体を通じて引用されるすべての参考文献、出された特許と公開された特許出願の内容は、参照によってここに組み込まれる。適当な構成要素、プロセスとそれらの特許、明細書と他の文書の方法は、その発明と実施形態のために選択することが可能である。
【特許請求の範囲】
【請求項1】
本願明細書に記載された発明。
【請求項1】
本願明細書に記載された発明。
【図1】

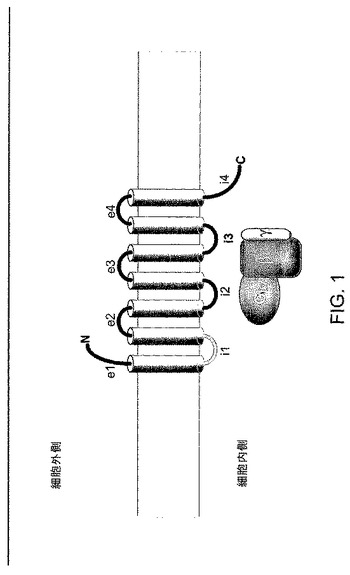
【図2】

【図3】
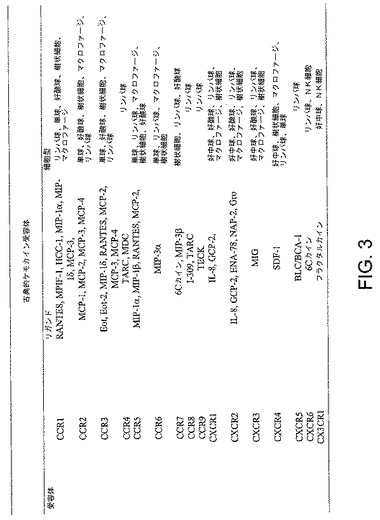
【図4】

【図5】

【図6】
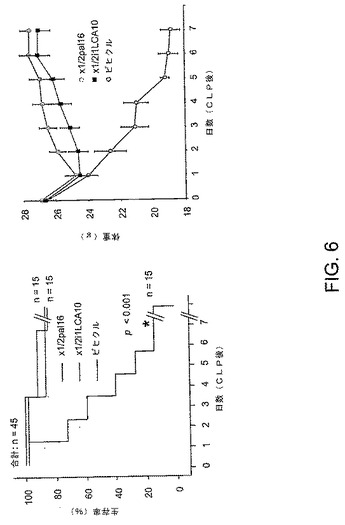
【図7】

【図8】
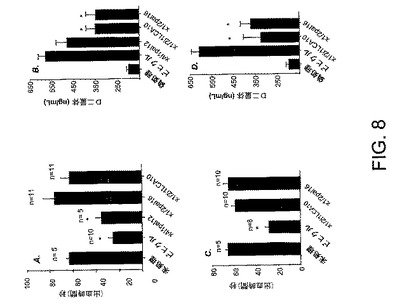
【図9】

【図10】
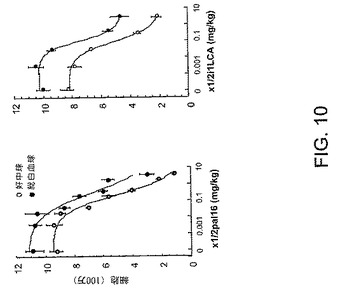
【図11】
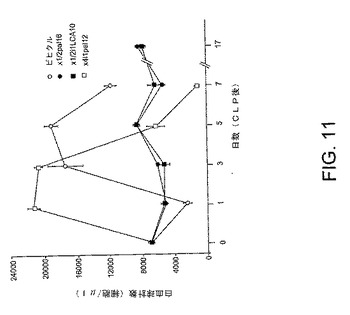
【図12】
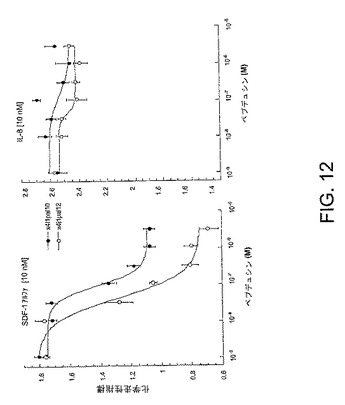
【図13】

【図14】

【図15】

【図16】

【図17】

【図18】

【図19】

【図20】

【図21】
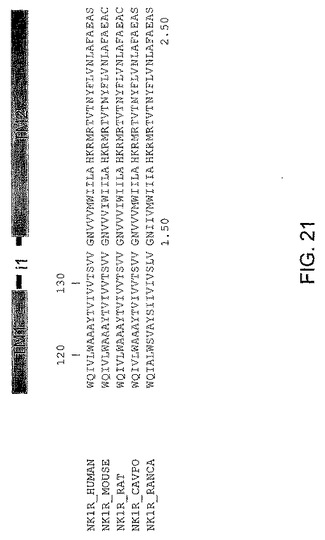
【図22】

【図23】
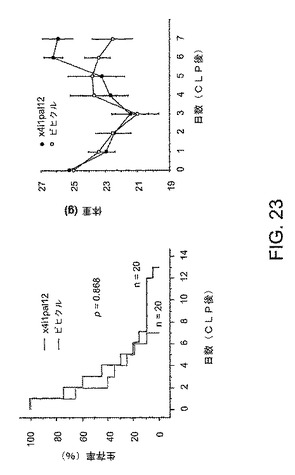
【図24A】
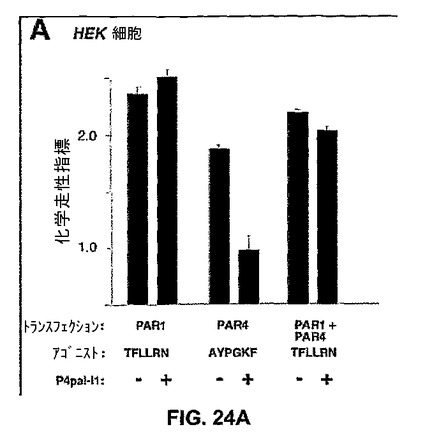
【図24B】

【図24C】

【図24D】

【図25A】

【図25B】

【図26】
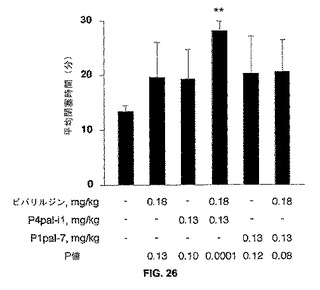
【図27】
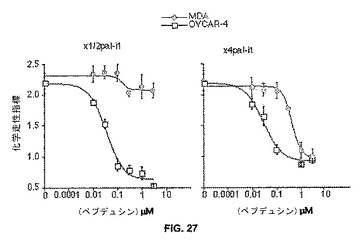
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24A】
【図24B】
【図24C】
【図24D】
【図25A】
【図25B】
【図26】
【図27】
【公開番号】特開2012−176985(P2012−176985A)
【公開日】平成24年9月13日(2012.9.13)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−135250(P2012−135250)
【出願日】平成24年6月14日(2012.6.14)
【分割の表示】特願2007−540067(P2007−540067)の分割
【原出願日】平成17年11月4日(2005.11.4)
【出願人】(504311419)タフツ メディカル センター インコーポレイテッド (10)
【Fターム(参考)】
【公開日】平成24年9月13日(2012.9.13)
【国際特許分類】
【出願番号】特願2012−135250(P2012−135250)
【出願日】平成24年6月14日(2012.6.14)
【分割の表示】特願2007−540067(P2007−540067)の分割
【原出願日】平成17年11月4日(2005.11.4)
【出願人】(504311419)タフツ メディカル センター インコーポレイテッド (10)
【Fターム(参考)】
[ Back to top ]