
GANP遺伝子導入トランスジェニック哺乳動物及びその利用
【課題】各種疾患の診断薬及び治療薬として有効な高親和性抗体、当該抗体を産生するためのトランスジェニック哺乳動物、該高親和性抗体又は高親和性抗体産生細胞を用いた薬剤の提供。
【解決手段】GANP遺伝子を導入したトランスジェニック哺乳動物、その子孫又はそれらの一部、並びにそれを用いた高親和性抗体の産生方法。
【解決手段】GANP遺伝子を導入したトランスジェニック哺乳動物、その子孫又はそれらの一部、並びにそれを用いた高親和性抗体の産生方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、GANP遺伝子を導入したトランスジェニック哺乳動物及びその利用に関する。より詳細には、本発明は、GANPを高発現し、高親和性抗体を産生することができるトランスジェニック哺乳動物、該トランスジェニック哺乳動物を用いて高親和性抗体を産生する方法、及び得られた高親和性抗体の利用に関する。
【背景技術】
【0002】
免疫系の機能は、T細胞の効果を主とする細胞性免疫反応に基づく機能と抗体の効果を主とする液性免疫に基づく機能とに分類される。実際は、この両者の機能は協調して免疫応答が行われる。抗体は骨髄で生まれるB細胞の細胞表面レセプターとして表出している。生体で形成される最初の抗体が認識する抗原の多様性は109〜1011個のオーダーに上ると言われ、そのような抗体(抗原レセプター)は、環境に存在しうるあらゆる抗原決定基を認識する。しかしながら、この多様な抗原レセプターは抗原に対して結合する能力は概して低く、低親和性の抗体が産生されることが多いが、これでは十分な免疫応答とはならない。
【0003】
リンパ球、特にB細胞/免疫グロブリン(抗体)は、その免疫反応に基づく各種用途、例えば病原体等の抗原検出のためのキット、診断薬、治療薬として利用されている。このような抗原検出薬、あるいは各種疾患治療薬における抗体として、抗原に対する反応性が高い抗体を使用すると、抗原に対する感度が優れ、かつ同一投与量での治療薬としての性能が優れる。しかしながら、これまでの所、抗体の親和性を高める手段は知られていない。
【0004】
ところで、生体は病原体や異物が体内に侵入すると、それらを抗原として認識して、末梢のリンパ組織において、抗原と直接結合する抗体のV領域の遺伝子に高頻度の体細胞突然変異を誘導する。その変化にはT細胞の刺激を必要としており、胚中心(Germinal center)領域で活性化T細胞から刺激を受けると考えられる。近年、本発明者らはこの領域の活性化B細胞で選択的に発現が上昇する分子GANPを見出している(特許文献1)。この分子はDNAへリカーゼ活性を有するMCM(minichromosome maintenance)と呼ばれる分子と直接結合し、さらにRNAプライマーゼ活性を有することから、DNA複製に関連することが示唆されている。しかしながら、免疫系におけるGANPの機能については解明されていなかった。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】国際公開第00/50611号
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は、各種疾患の診断薬及び治療薬として有効な高親和性抗体、当該抗体を産生するためのトランスジェニック哺乳動物、該高親和性抗体又は高親和性抗体産生細胞を用いた薬剤を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明者は、上記課題を解決するため鋭意研究を行った結果、GANP遺伝子を導入したトランスジェニック動物を作製し、抗原で免疫すると、このトランスジェニック動物は高親和性抗体を産生し得ることを見出し、本発明を完成するに至った。
【0008】
すなわち、本発明は以下の通りである。
(1) GANP遺伝子を導入したトランスジェニック哺乳動物又はその子孫。
導入したGANP遺伝子はB細胞で発現することができる。また、本発明のトランスジェニック哺乳動物又はその子孫は、GANP遺伝子をトランスフェクトしたES細胞から発生させることが可能である。哺乳動物としては、例えばマウスが挙げられる。
(2) 前記トランスジェニック哺乳動物又はその子孫の一部。
(3) 前記トランスジェニック哺乳動物又はその子孫に抗原を投与し、得られる動物又は子孫から抗体を採取することを特徴とする高親和性抗体の製造方法。
【0009】
(4) 前記(3)記載の方法により得られる高親和性抗体又はその断片。
本発明の抗体は、親和性が1×10-7 (M) 以下で示されるものである。本発明の抗体は、ポリクローナル抗体であってもモノクローナル抗体であってもよい。
(5) 前記抗体又はその断片のV領域を含む、ヒト型化抗体若しくはヒト抗体又はそれらの断片。
(6) 前記抗体又はその断片、及び前記ヒト型化抗体若しくはヒト抗体又はそれらの断片からなる群から選択される少なくとも1つを含有する医薬組成物。
(7) 抗原を投与した請求項1〜4のいずれかに記載のトランスジェニック哺乳動物又はその子孫から採取される、高親和性抗体産生細胞。
【発明の効果】
【0010】
本発明によるGANPを過剰発現したマウスを用いることによって、従来では得られることのできなかったウイルス抗原に特異的で高親和性の抗体を速やかに作製することができる。そのため、エイズやC型肝炎のように遷延する感染による病状悪化を当該のウイルス抗原の変異に遅れないで特異的で強力な抗体を速やかに得ることが可能となると期待される。また、これによって、感染患者からのウイルスの抗原の変異に対応するオーダーメードの特異的抗体を作製することができる。抗体の作製に必要な免疫期間は10日程度で十分であり、そのうち高親和性の変異を持つ抗体の産生効率は60%近くに上る。ベッドサイドの患者サンプルを用いる高親和性抗体の産生プロトコールはワクチン療法に変わる新しい免疫療法となると期待される。
【図面の簡単な説明】
【0011】
【図1】抗GANPモノクローナル抗体およびALP結合抗ラットIg抗体を用いた免疫組織化学分析の結果を示す。スケールバーは100μm。
【図2】雌NZBマウスの膝窩のリンパ節中のGANPhi細胞の出現速度を示す。スケールバーは100μm。
【図3】雌NZBマウスの脾臓中のGANPhi細胞の出現速度を示す。スケールバーは100μm。
【図4】複数系統のマウス由来の脾臓切片を抗GANPモノクローナル抗体で染色した結果を示す。RP; 赤脾髄, F; 濾胞。スケールバーは100μm。
【図5】脾臓の赤脾髄におけるGANPhi 細胞の同定を示す。
【0012】
【図6】GANPhi 細胞における形質細胞マーカーの同定を示す。スケールバーは100μm。
【図7】TD-Ag免疫によるC57BL/6マウスの脾臓の赤脾髄領域におけるGANPhi細胞の発現を示す。スケールバーは100μm。
【図8A】Daudi細胞にマウスGANPを安定的に発現させたトランスフェクタントの体細胞突然変異を示す。
【図8B】Daudi細胞にマウスGANPを安定的に発現させたトランスフェクタントの体細胞突然変異を示す。
【図8C】Daudi細胞にマウスGANPを安定的に発現させたトランスフェクタントの体細胞突然変異を示す。
【0013】
【図9A】B細胞でGANPを過剰発現させたトランスジェニックマウスの作製の概要を示す。
【図9B】B細胞でGANPを過剰発現させたトランスジェニックマウスの作製の概要を示す。
【図9C】B細胞でGANPを過剰発現させたトランスジェニックマウスの作製の概要を示す。
【図10】GANP過剰発現トランスジェニック(Tg)マウス又は野生型マウスにおける体細胞突然変異を解析した結果を示す。
【図10−1】GANP過剰発現トランスジェニック(Tg)マウス又は野生型マウスにおける体細胞突然変異を解析した結果を示す。
【図10−2】GANP過剰発現トランスジェニック(Tg)マウス又は野生型マウスにおける体細胞突然変異を解析した結果を示す。
【図10−3】GANP過剰発現トランスジェニック(Tg)マウス又は野生型マウスにおける体細胞突然変異を解析した結果を示す。
【図10−4】GANP過剰発現トランスジェニック(Tg)マウス又は野生型マウスにおける体細胞突然変異を解析した結果を示す。
【図10−5】GANP過剰発現トランスジェニック(Tg)マウス又は野生型マウスにおける体細胞突然変異を解析した結果を示す。
【0014】
【図11A】B細胞特異的GANP欠損マウス(B-GANP-/-)の作製の概要を示す。
【図11B】B細胞特異的GANP欠損マウス(B-GANP-/-)の作製の概要を示す。
【図11C】B細胞特異的GANP欠損マウス(B-GANP-/-)の作製の概要を示す。
【図11D】B細胞特異的GANP欠損マウス(B-GANP-/-)の作製の概要を示す。
【図11E】B細胞特異的GANP欠損マウス(B-GANP-/-)の作製の概要を示す。
【図12】B細胞特異的GANP欠損マウス(B-GANP-/-)を用いた細胞表面染色の結果(フローサイトメトリー)を示す。
【図13】B細胞の増殖アッセイの結果を示す。ほとんど差はなかったが、抗CD40抗体刺激による増殖のみが約1/2に減少していた。
【0015】
【図14】免疫をしていないCre-flox/+マウス及びB-GANP-/-マウス血清中の抗体価を示す。各種アイソタイプの抗体価に差はなかった。
【図15】B-GANP-/-マウスにおける抗体産生を測定した結果を示す。
【図16】GCをピーナッツアグルチニンで染色した結果を示す。
【図17】B-GANP-/-マウスにおける抗原特異的抗体産生を測定した結果を示す。
【図18】100μgのNP-CGを免疫し、免疫後14日及び35日目における親和性の成熟の度合いをディファレンシャルELISAにより測定した結果を示す。
【図19】GC-B細胞のフローサイトメトリーの結果を示す。
【0016】
【図20A】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Aから図20Fに順に続く)。
【図20B】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Aから図20Fに順に続く)。
【図20C】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Aから図20Fに順に続く)。
【図20D】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Aから図20Fに順に続く)。
【図20E】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Aから図20Fに順に続く)。
【図20F】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Aから図20Fに順に続く)。
【図20G】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Gから図20Lに順に続く)。
【図20H】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Gから図20Lに順に続く)。
【図20I】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Gから図20Lに順に続く)。
【図20J】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Gから図20Lに順に続く)。
【図20K】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Gから図20Lに順に続く)。
【図20L】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Gから図20Lに順に続く)。
【0017】
【図21】Cre-flox/+及びB-GANP-/-マウスにおけるIgG1の変異の頻度を示す。
【図22】VH186.2の33番目のWをLに変異させたときの変異の頻度を示す。
【図23】活性化誘導細胞死(AICD)の測定結果及びアポトーシスの抑制結果を示す。
【図24】抗CD40及び抗CD95刺激に対する細胞のアポトーシス感受性を測定した結果を示す。
【図25】TUNELアッセイによりアポトーシス細胞を検出した結果を示す。
【図26】TUNELアッセイによりアポトーシス細胞を検出した結果を示す。
【図27】アポトーシス抑制に関与するBcl-2ファミリーのRNA発現レベルを示す。
【図28】GANPトランスジェニックマウスを用いて高親和性抗体を産生した結果を示す。
【図29】GANPトランスジェニックマウス由来のハイブリドーマクローンを用いて高親和性抗体を産生した結果を示す。
【図30】GANPトランスジェニックマウス由来のハイブリドーマクローン培養上清を用いたBiacoreによる結合解離曲線を示す。
【0018】
【図31】GANPトランスジェニックマウス由来のハイブリドーマクローン培養上清を用いたBiacoreによる結合解離曲線を示す。
【図32】GANP-GST融合タンパク質の構造の概略を示す。
【図33】MCMと直接結合するGANPの領域を決定するためのプルダウンアッセイの結果を示す。左に大きさのスタンダードの位置を示す。
【図34】in vitro翻訳されたMCMを用いたプルダウンアッセイの結果を示す。
【図35】GANP各構築物とMCMの結合を免疫沈降によって示す。
【図36A】GANP各構築物とMCMの結合を免疫沈降によって示す。
【図36B】GANP各構築物とMCMの結合を免疫沈降によって示す。
【図37】GANP構築物の構造の概要及び、構築物の細胞内での分布を示す。
【0019】
【図38】GANP構築物の細胞内での分布を示す。
【図39】MCM3の核における局在化を示す。
【図40】GANPの発現によって誘導されるMCM3の細胞質局在化を示す。
【図41】核に局在する対照タンパク質を示す。
【図42】MCM3変異体の局在化におけるGANP構築物の効果を示す。
【図43】ヘテロカリオンアッセイによって検出されるMCM3の核-細胞質間シャトリングを示す。
【図44】細胞周期の間のGANPの局在化を示す。
【発明を実施するための形態】
【0020】
以下、本発明を詳細に説明する。
本発明は、GANP遺伝子を非ヒト哺乳動物に導入してトランスジェニック動物を作製し、そのようなトランスジェニック動物を抗原で免疫すると、高親和性の抗体が得られる知見に基づいて完成されたものである。
【0021】
1.GANP
GANPは、胚中心結合核タンパク質(Germinal center-associated nuclear protein)と呼ばれており、酵母Sac3タンパク質とホモロジーを有する210kDaの核タンパク質である(WO00/50611号公報)。そして、SAC3はアクチン形成の抑制物質として特徴づけられている。また、GANPは、濾胞樹状細胞(follicular dendritic cells: FDC)により囲まれる胚中心(germinal center, GC)B細胞において選択的にアップレギュレートされ、リン酸化依存性RNA-プライマーゼ活性を有し、B細胞の細胞周期調節に関与しているタンパク質である (Kuwahara, K. et al., (2000) Blood 95, 2321-2328)。
本発明においては、GANPタンパク質のアミノ酸配列を、マウスについて配列番号2に、ヒトについて配列番号4に示す。また、GANPタンパク質をコードする遺伝子(GANP遺伝子という)の塩基配列を、マウスについて配列番号1に、ヒトについて配列番号3に示す。なお、上記アミノ酸配列及び塩基配列は、国際公開WO00/50611号公報にも記載されている。
【0022】
またGANPタンパク質は変異体でもよく、配列番号2又は4に記載のアミノ酸配列において1又は複数のアミノ酸が欠失、置換又は付加されたアミノ酸配列であってRNAプライマーゼ活性を有するタンパク質であってもよい。例えば、配列番号2又は4に示すアミノ酸配列のうち1若しくは複数個(好ましくは1個又は数個(例えば1個〜10個、さらに好ましくは1個〜5個))のアミノ酸が欠失しており、1若しくは複数個(好ましくは1個又は数個(例えば1個〜10個、さらに好ましくは1個〜5個))のアミノ酸が他のアミノ酸で置換されており、及び/又は1若しくは複数個(好ましくは1個又は数個(例えば1個〜10個、さらに好ましくは1個〜5個))の他のアミノ酸が付加されたアミノ酸配列からなり、かつ上記GANPタンパク質と同様のRNAプライマーゼ活性を有するGANP変異型タンパク質を使用することもできる。
【0023】
「RNAプライマーゼ活性」とは、RNA複製において、5'→3'方向に進む鎖の伸長とは逆向きの鎖(ラギング鎖)を合成する際に、伸長の開始点となる短いプライマーのRNAを合成する酵素活性を意味する。通常はαプライマーゼと呼ばれるDNAポリメラーゼαと結合する分子が用いられるが、胚中心B細胞では第二のプライマーゼであるGANPプライマーゼも誘導されている。
GANPタンパク質は、上記配列番号2若しくは4に示すアミノ酸配列又はこれらの変異型アミノ酸配列のほか、N末端側の一部の配列(例えば配列番号2に示すアミノ酸配列の1〜600番、好ましくは139〜566番)又はこれらの変異型アミノ酸配列を有するものも含まれる。
【0024】
本発明において、動物に導入するためのGANP遺伝子は、上記GANPタンパク質、N末側の一部の配列、又は変異型タンパク質をコードする遺伝子が挙げられる。そのような遺伝子として、例えば配列番号1又は3に示す塩基配列を有するものを使用することができる。配列番号1又は3に示す塩基配列のうち、コード領域のみの塩基配列であってもよい。また、上記配列番号1又は3に示す塩基配列に相補的な配列と、ストリンジェントな条件下でハイブリダイズし、かつ、RNAプライマーゼ活性を有するタンパク質をコードする遺伝子を使用することも可能である。
「ストリンジェントな条件」とは、ハイブリダイズさせた後の洗浄時の条件であって塩(ナトリウム)濃度が150〜900mMであり、温度が55〜75℃、好ましくは塩(ナトリウム)濃度が250〜450 mMであり、温度が68℃での条件をいう。
【0025】
遺伝子に変異を導入するには、Kunkel法や Gapped duplex法等の公知手法により、例えば部位特異的突然変異誘発法を利用した変異導入用キット、例えばGeneTailorTM Site-Directed Mutagenesis System(インビトロジェン社製)、TaKaRa Site-Directed Mutagenesis System(Mutan-K、Mutan-Super Express Km等:タカラバイオ社製)を用いて行うことができる。
変異遺伝子の詳細並びに取得方法は国際公開WO00/50611号公報にも記載されている。
【0026】
なお、抗μ抗体及び抗CD-40モノクローナル抗体でB細胞をin vitro刺激すると、GANP発現のアップレギュレーションのみならず、GANPタンパク質のアミノ酸配列のうち特定のセリン残基(例えば502番目のセリン: S502)のリン酸化を引き起こす。この反応は、GANPのRNA-プライマーゼ活性についてキーとなる反応である(Kuwahara, K. et al. (2001) Proc. Natl. Acad. Sci. USA, 98, 10279-10283)。GANPタンパク質のN末端側のRNA-プライマーゼドメインはセリン残基を含んでおり、そのリン酸化はin vitroにおいてCdk2によって触媒される。C末端側ドメインにより、GANPはMCM3複製ライセンシング因子に結合する(Kuwahara, K. et al., (2000) Blood 95, 2321-2328; Abe, E. et al. (2000) Gene 255, 219-227)。
【0027】
2.GANP遺伝子を導入したトランスジェニック哺乳動物
本発明は、GANP遺伝子を導入したトランスジェニック哺乳動物に関するものであり、当該トランスジェニック哺乳動物は、好ましくは、導入したGANP遺伝子をB細胞で発現することができる。
【0028】
(1) GANP遺伝子とその関連分子
GANP遺伝子とその関連分子で形成される複合体は、遺伝子に変異を誘導するプロセスで直接および間接的に必要な分子である。GANPタンパク質は、遺伝子変異を修復する際に、高親和性の抗体が得られるようにV領域の変異の誘導を促す能力を保有していることから、本発明のトランスジェニック哺乳動物は、このGANP遺伝子又はその変異遺伝子の導入によって、獲得性免疫の高親和性抗体産生を促進することができる。また、この遺伝子を過剰に発現するトランスジェニック非ヒト哺乳動物は、速やかに抗原に対する結合力の高い抗体を産生することができる。従って、上記トランスジェニック非ヒト哺乳動物を所定の抗原で免疫することで、従来では得られないような高親和性の抗体を簡便に得ることができる。その結果、難治性の病原微生物や異物を排除できるポリクローナル抗体又はモノクローナル抗体を得ることができる。また、本発明のトランスジェニック哺乳動物を用いてヒト型化抗体を作製することによって、あるいは、本発明のトランスジェニック哺乳動物が産生する抗体のV領域を含む一本鎖抗体を作製することによって、抗体療法の効力を飛躍的に高めることが可能となる。
【0029】
本発明のトランスジェニック哺乳動物は、GANP又はその変異遺伝子の導入によって、B細胞で高親和性抗体の産生を促進することができ、前記高親和性抗体産生細胞はアポトーシスを誘導するシグナルに対して抵抗性を有する。
本発明者らは、GANPが獲得免疫応答の抗体産生に機能している分子であることを確認するため、先ずGANP遺伝子の欠損マウスをB細胞選択的に欠損するように企図して作製した。その結果、GANP遺伝子が欠損しても、免疫系の細胞の発達、分化、増殖には影響が見られず、また抗体の産生総和には大きな変化は見られないことが判明した。
【0030】
ここで、抗原と反応したB細胞がそのまま増殖し、抗体産生細胞に分化するものは、ある種の抗原の場合に限られており、通常の抗原に対する抗体産生についてはT細胞の共存を必要とする。T細胞が存在しなくても抗体が産生されるような抗原をT細胞非依存性抗原という。これに対し、T細胞非依存性抗原以外の一般の抗原をT細胞依存性抗原という。T細胞依存性抗原の場合は、B細胞の抗体産生細胞への分化がヘルパーT細胞により補助される。
病原ウイルスの抗原決定基(抗原エピトープとも言う)の多くはそれ自体免疫原性が弱く、T細胞によって認識されるキャリアタンパク質のペプチド抗原によって活性化を受ける。
【0031】
本発明においては、通常の動物では、強力な抗体を産生できないような可溶性の抗原に対する抗体産生応答に対して、GANP遺伝子導入動物では高頻度に高親和性の抗体を産生することができることを調べるため、ハプテンとして解析が進んでいるニトロフェニル基 (NP基)をニワトリ-ガンマグロブリンと結合させて抗原(NP-CGという)を作製し、T細胞性依存性抗原の反応を調べた。
ここで、C57BL/6マウスのNPに対する反応は単一のV領域で行われることが知られている。この反応は、抗体のIgG重鎖のV領域(VH186.2という)とL鎖ラムダ1遺伝子によってのみ形成されるというものである。このシステムを使えば、アイソタイプがIgG1の抗体について、VH186.2のアミノ酸配列を調べることによって高親和性抗体の遺伝子変異を調べることが可能である。しかも、最も強い親和性は、重鎖V領域(VH186.2)のアミノ酸配列のうち、第33番目のアミノ酸であるトリプトファン(W)がロイシン(L)に変異(W33-L)したときに生じることが報告されている。
【0032】
そこで、本発明者は、GANP遺伝子欠損マウスでW33-L変異を起こさせたときに、高親和性抗体が産生されるか否かを調べた。その結果、対照であるCre-flox/+マウスに比べて高親和性抗体産生はほとんど見られなかった。従って、GANP遺伝子は、高親和性抗体を産生するための重要な機能を有する遺伝子であることが判明した。このことをさらに検証するために、GANP遺伝子を過剰に発現するマウスを作製した。GANP遺伝子の過剰発現は、マウス免疫グロブリンプロモーター部分とヒト免疫グロブリン遺伝子イントロンエンハンサー部分をGANP遺伝子の5'側に連結して、B細胞で選択的に発現させることによって行った。
【0033】
上記GANP過剰発現マウスも正常に生まれ、リンパ組織の発達、分化、増殖には変化が見られないが、NP-CGに対する反応では、著しく高親和性型のV領域遺伝子(W33-L)が増加していた。RNAプライマーゼ活性がこの際にどのような機能的な役割を担っているかについてはまだ確定されていないが、(i) GANP分子のプライマーゼ活性をみる指標となる502番目のセリン残基のリン酸化が胚中心の高親和性B細胞の生み出される領域の細胞(セントロサイト)に高いこと、(ii) Daudi細胞へのganp遺伝子導入実験によって誘導されるV領域の突然変異の頻度が高いことから、GANP分子のRNA プライマーゼ活性あるいはそれに関わる502番目のセリン残基のリン酸化が高親和性抗体産生に関連していると考えている。この結果は、GANP分子を高発現させること、及びRNAプライマーゼ活性を賦活化することが、免疫応答による高親和性抗体の産生に必要であることを示すものである。
【0034】
(2) GANP遺伝子導入用哺乳動物
本発明における「哺乳動物」とは、ウシ、ウマ、ブダ、ヤギ、ウサギ、イヌ、ネコ、マウス、ラット、ハムスター及びモルモット等の任意の非ヒト哺乳動物を意味し、好ましくはマウス、ウサギ、ラットまたはハムスターであり、特に好ましくはマウスである。
本発明のトランスジェニック哺乳動物は、未受精卵、受精卵、精子およびその始原細胞を含む胚芽細胞などに対して、好ましくは、非ヒト哺乳動物の発生における胚発生の段階(さらに好ましくは、単細胞または受精卵細胞の段階でかつ一般に8細胞期以前)の細胞に対して、リン酸カルシウム法、電気パルス法、リポフェクション法、凝集法、マイクロインジェクション法、パーティクルガン法、DEAE-デキストラン法などにより、GANP遺伝子を導入することにより作製することができる。また、上記遺伝子導入方法により、体細胞、生体の臓器、組織細胞などに目的とするGANP遺伝子を転移させ、細胞培養、組織培養などに利用することもできる。さらに、これら細胞を上述の胚芽細胞と公知の細胞融合法により融合させることにより、トランスジェニック哺乳動物を作製することもできる。
【0035】
GANP遺伝子を対象動物に導入させる際、当該遺伝子を対象となる動物の細胞で発現させうるプロモーターの下流に連結した遺伝子構築物として導入することが好ましい。具体的には、目的とするGANP遺伝子を有する各種哺乳動物由来のGANP遺伝子を発現させうる各種プロモーターの下流に、GANP遺伝子を連結したベクターを、対象となる哺乳動物の受精卵(例えば、マウス受精卵)にマイクロインジェクションすることによって、目的とするGANP遺伝子を高発現するトランスジェニック哺乳動物を作製することができる。
【0036】
(3) 発現ベクター
GANP遺伝子の発現ベクターとしては、大腸菌由来のプラスミド、枯草菌由来のプラスミド、酵母由来のプラスミド、λファージなどのバクテリオファージ、モロニー白血病ウイルスなどのレトロウイルス、ワクシニアウイルス又はバキュロウイルスなどの動物又は昆虫ウイルスなどが用いられる。
遺伝子発現の調節を行うプロモーターとしては、たとえばウイルス由来遺伝子のプロモーター、各種哺乳動物(ヒト、ウサギ、イヌ、ネコ、モルモット、ハムスター、ラット、マウスなど)および鳥類(ニワトリなど)由来遺伝子のプロモーターなどを使用することが可能である。
【0037】
ウイルス由来遺伝子のプロモーターとしては、例えばサイトメガロウイルス、モロニー白血病ウイルス、JCウイルス、乳癌ウイルス等由来遺伝子のプロモーターが挙げられる。
各種哺乳動物及び鳥類由来遺伝子のプロモーターとしては、例えば、アルブミン、インスリンII、エリスロポエチン、エンドセリン、オステオカルシン、筋クレアチンキナーゼ、血小板由来成長因子β、ケラチンK1,K10およびK14、コラーゲンI型およびII型、心房ナトリウム利尿性因子、ドーパミンβ-水酸化酵素、内皮レセプターチロシンキナーゼ、ナトリウムカリウムアデノシン3リン酸化酵素、ニューロフィラメント軽鎖、メタロチオネインI及びIIA、メタロプロティナーゼ1組織インヒビター、MHCクラスI抗原、平滑筋αアクチン、ポリペプチド鎖延長因子1α(EF-1α)、βアクチン、α及びβミオシン重鎖、ミオシン軽鎖1及び2、ミエリン基礎タンパク、血清アミロイドPコンポーネント、ミオグロビン、レニンなどの遺伝子のプロモーターが挙げられる。
【0038】
上記ベクターは、トランスジェニック哺乳動物において目的とするメッセンジャーRNAの転写を終結するターミネターを有していてもよい。その他、GANP遺伝子をさらに高発現させる目的で、各遺伝子のスプライシングシグナル、エンハンサー領域、真核生物遺伝子のイントロンの一部をプロモーター領域の5'上流、プロモーター領域と翻訳領域間、あるいは翻訳領域の3'下流 に連結することも所望により可能である。
本発明の好ましい態様では、免疫グロブリンプロモーターの下流にGANP遺伝子を連結することにより、あるいはヒト免疫グロブリン遺伝子イントロンエンハンサー部分をGANP遺伝子の5'側に連結することにより、GANP遺伝子をB細胞で選択的に発現させることができる。
【0039】
(4) GANP遺伝子の導入
受精卵細胞段階におけるGANP遺伝子の導入は、対象の哺乳動物の胚芽細胞および体細胞の全てに過剰に存在するように確保することが好ましい。遺伝子導入後の作出動物の胚芽細胞においてGANP遺伝子が過剰に存在することは、作出動物の子孫が全てその胚芽細胞および体細胞の全てにGANP遺伝子を過剰に有することを意味する。そして、遺伝子を受け継いだこの種の動物の子孫は、その胚芽細胞および体細胞の全てにGANP蛋白質を過剰に有する。
本発明においては、導入遺伝子を相同染色体の一方に持つヘテロ接合体を取得し、ヘテロ接合体同士を交配することで導入遺伝子を相同染色体の両方に持つホモ接合体を取得し、この雌雄の動物を交配することによりすべての子孫が導入されたGANP遺伝子を安定に保持する。そして、GANP遺伝子を過剰に有することを確認して、通常の飼育環境で繁殖継代することができる。
【0040】
トランスジェニック対象動物が有する内在性の遺伝子とは異なる遺伝子である外来性GANP遺伝子を対象非ヒト哺乳動物(好ましくはマウスなど)、又はその先祖の受精卵(バッククロス)に転移する際に用いられる受精卵は、同種の雄哺乳動物と雌哺乳動物を交配させることによって得られる。
受精卵は自然交配によっても得られるが、雌哺乳動物の性周期を人工的に調節した後、雄哺乳動物と交配させる方法が好ましい。雌哺乳動物の性周期を人工的に調節する方法としては、例えば、初めに卵胞刺激ホルモン(妊馬血清性性腺刺激ホルモン(PMSG))、次いで黄体形成ホルモン(ヒト絨毛性性腺刺激ホルモン(hCG))を、例えば腹腔注射などにより投与する方法が好ましい。
【0041】
得られた受精卵に、前述の方法により外来性GANP遺伝子を導入した後、雌哺乳動物に人工的に移植・着床することにより、外来性遺伝子を組み込んだDNAを有する非ヒト哺乳動物が得られる。雌哺乳動物に黄体形成ホルモン放出ホルモン(LHRH)を投与後、雄哺乳動物と交配させることにより受精能を誘起された偽妊娠雌哺乳動物に、受精卵を人工的に移植・着床させる方法が好ましい。遺伝子を導入する全能性細胞としては、マウスの場合、受精卵や初期胚を用いることができる。また培養細胞への遺伝子導入法としては、トランスジェニック哺乳動物個体の産出効率や次代への導入遺伝子の伝達効率を考慮した場合、DNAのマイクロインジェクションが好ましい。
遺伝子を注入した受精卵は、次に仮親の卵管に移植され、個体まで発生し出生した動物を里親につけて飼育させたのち、体の一部(マウスの場合には、例えば、尾部先端)からDNAを抽出し、サザン解析やPCR法により導入遺伝子の存在を確認することができる。導入遺伝子の存在が確認された個体を初代(Founder)とすれば、導入遺伝子はその子(F1)の50%に伝達される。さらに、このF1個体を野生型動物または他のF1動物と交配させることにより、2倍体染色体の片方(ヘテロ接合)または両方(ホモ接合)に導入遺伝子を有する個体(F2)を作製することができる。
【0042】
あるいは、GANP蛋白質高発現トランスジェニック哺乳動物は、上記したGANP遺伝子をES細胞(embryonic stem cell)に導入することによって作製することもできる。例えば、正常マウス胚盤胞(blastocyst)に由来するHPRT陰性(ヒポキサンチングアニン・フォスフォリボシルトランスフェラーゼ遺伝子を欠いている)ES細胞に、GANP遺伝子を導入する。当該GANP遺伝子がマウス内在性遺伝子上に相同組み換えを起こさせ、インテグレートされたES細胞をHATセレクション法により選別する。次いで、選別したES細胞を、別の正常マウスから取得した受精卵(胚盤胞)にマイクロインジェクションする。得られた胚盤胞を、仮親としての別の正常マウスの子宮に移植する。その後、仮親マウスからキメラトランスジェニックマウスが生まれる。生まれたキメラトランスジェニックマウスを正常マウスと交配させることにより、ヘテロトランスジェニックマウスを得ることができる。そして、ヘテロトランスジェニックマウス同士を交配することにより、ホモトランスジェニックマウスが得られる。
【0043】
本発明においては、上記したトランスジェニック哺乳動物に限らず、その子孫、並びにトランスジェニック哺乳動物又はその子孫の一部も本発明の範囲内である。トランスジェニック哺乳動物の一部としては、当該トランスジェニック哺乳動物又はその子孫の組織、器官及び細胞などが挙げられ、器官または組織としては、脾臓、胸腺、リンパ節、骨髄あるいは扁桃腺などが挙げられ、細胞としてはB細胞などが挙げられる。
本発明のトランスジェニック哺乳動物は、B細胞をさらに活性化する哺乳動物と交配することも可能であり、これによりさらに高親和性抗体を産生することが可能である。
【0044】
最近、MRL/lprマウスでB細胞が末梢のリンパ節での活性化の際に胚中心を経過した後、T細胞領域でさらにV領域の突然変異誘導が亢進していることが報告されている。また、本発明者らもMRL/lprマウスにおいてGANP遺伝子がIgプロモーター、エンハンサーの下流に結合して作成したganpトランスジェニックマウスに見られるのと同等の高い発現が、非免疫の状態で見られることを見出している。このことは、正常では自己の抗原に対しては高親和性の抗体はできないのに対して、この自己免疫疾患マウスでは、GANP分子の異常な活性化が起こるために、自己の抗原に対しての高親和性抗体が産生されることとなる可能性が示唆される。
【0045】
そこで、上記B細胞をさらに活性化する動物として、自己免疫疾患マウスであるとされるMRL/lpr, NZB, (NZB x NZW)F1などを用いれば、さらに高い変異誘導を期待できる。
以上のことを利用したMLR/lprマウスのGANPトランスジェニックマウスを作製することによって、スーパー高親和性抗体産生マウスを作出できる可能性がある。すなわち、本発明のGANP遺伝子過剰発現トランスジェニック哺乳動物とさまざまな自己免疫疾患モデル動物との交配により、高親和性抗体を産生できる哺乳動物を作製することができる。
【0046】
3.高親和性抗体の作製
本発明でいう抗体とは、抗原と特異的に結合する活性を有する蛋白質を意味し、好ましくはB細胞が産生するものである。本発明では、抗原に対する反応性が高い抗体のことを高親和性抗体と言う。「高親和性」とは、抗体が抗原と結合する結合能が高いことを意味し、本発明においては、抗体の結合能が一般のマウスなどの動物を用いて作製した抗体と比較して高く、また逆に当該の抗原から解離することが遅い抗体のことをいう。これは、抗原決定基(エピトープ)に対して、立体的に密接して結合する能力が高く特異的であることを意味するとともに、抗体が結合することによって抗原決定基のみならずその抗原の構造の変換をきたすことによって結果的に強力な活性(毒性の中和、ウイルスなどの感染性阻止、病原体の不活性化、生体内からの排除の促進、抗原分子の変性を引き起こす等、生物活性を示すもの)を示すことも包含している。
【0047】
また、抗体の結合能(親和性)は、スキャッチャード解析やBiacore と呼ばれる表面プラズモン共鳴センサーにより、解離定数(KD)、解離速度定数(Kdiss)、結合速度定数(Kass)として測定することができる。Biacore装置は、センサーチップ、マイクロ流路系、SPR検出系の3つの技術を統合して分子結合の強さ、速さ、選択性を測定するというものであり、標識を使わずにリアルタイムで生体分子の検出と複数個の分子間での相互作用のモニタリングを行うことができる。Biacore装置としては、例えばBiacore 3000、Biacore 2000、Biacore X、Biacore J、Biacore Q(いずれもBiacore社)などが挙げられる。
【0048】
上記Biacore によって、抗体の親和性を示すパラメーター、すなわち解離定数(KD)、解離速度定数(Kdiss) [1/Sec] 及び結合速度定数(Kass) [1/M.Sec]を測定する。
抗体は、解離定数(KD値)が小さい値であるほど親和性が高いという点で好ましい。抗体の結合能(親和性)は、Kdiss及びKassの2つのパラメーターにより決定され、
KD[M]= Kdiss/Kass
により表わされる。
抗原の種類等複数の要因によって、得られる抗体の親和性は異なるが、一般には、KD値は1×10-7(M) 以下であることが好ましく、例えば1×10-8(M) 以下、1×10-10(M)以下、あるいは1×10-11(M)以下のものが挙げられる。
【0049】
本発明においては、作製された抗体が上記いずれかの作用又は性質を発揮する抗体であるときに、「高親和性」であると判断される。
抗体分子の親和性亢進は抗体遺伝子の可変領域(V領域)遺伝子に体細胞突然変異(SHM)が誘導することによって生まれる。抗体の抗原に対する特異性は、抗原を生体に免疫した当初から認められるが、初期の抗体の多くはIgMクラスであり、また抗原に対する結合親和性は高くはなく、病原体や異物を除去したり、不活化する能力は低い。しかし、抗原を生体に投与して数回の追加免疫を行うと抗体の抗原に対する結合親和性が高まる。この際、B細胞はT細胞からの刺激が必要であり、末梢のリンパ組織の胚中心領域でその活性化が行われるとされている。V領域遺伝子の突然変異誘導に必要な分子としては、最近、胚中心で発現するRNAエディティング分子AIDであると報告されている。さらに、ウラシルDNAグリコシダーゼ、またDNA複製に必要なDNAポリメラーゼとしてミスを生じやすいDNAポリメラーゼゼータ(ζ)とアイオタ(ι)が関与していることが報告されているが、これらの機能を制御する分子はまだ明らかにされていない。GANP分子は新たなSHM誘導分子としてその機能が明らかにされたものであり、その分子の発現上昇がSHM誘導に重要な鍵を握る。とりわけ、高親和性抗体を産生するために重要であることが明らかになった。
【0050】
C57BL/6マウスにハプテン・キャリアの抗原としてニトロフェニル-ニワトリγグロブリンを免疫することにより誘導される抗体は、H鎖はVH186.2ローカスを用い、L鎖はλ1である。この際、追加免疫をして得られる抗体はIgG1抗体であり、そのうち特に結合親和性の高い抗体のV領域配列に誘導される突然変異は、33番目のトリプトファンがロイシンに変異したものであることが知られている。本明細書の実施例では、このモデルシステムで高い高親和性型のV領域突然変異が誘導されており、これは、高親和性抗体が誘導されたことを示す分子レベルでの明らかな証拠であると言える。
従って、上記トランスジェニック哺乳動物又はその子孫に抗原を投与して抗体を産生することによって、高親和性抗体を得ることができる。即ち、GANP蛋白質を高発現させた動物に目的とする抗原を常法によって投与し、感作された動物の血液又は脾臓等の組織(これらの組織に限定されない)のリンパ球より高親和性抗体を調製することができる。高親和性抗体はポリクローナル抗体でもモノクローナル抗体でもよい。
【0051】
ポリクローナル抗体を産生する方法としては、例えば、抗原を本発明のトランスジェニック哺乳動物に投与して免疫し、免疫された哺乳動物から血液を採取し、採取した血液から抗体を分離・精製することにより得ることができる。
免疫感作の方法は当業者に公知であり、例えば抗原を1回以上投与することにより行うことができる。
抗原の種類は特に限定されるものではなく、抗原決定基としての立体構造を持ちうる物質すべてが該当し、タンパク質、酵素、ペプチド、糖、脂質、DNA, RNA,プリオンなどのあらゆる生体成分のほか、癌抗原、ウイルス抗原、有機、無機合成抗原など任意のものを使用することができる。
【0052】
抗原投与は、例えば7〜30日間隔で2〜3回投与すればよい。投与量は1回につき、例えば抗原約0.05〜2mg程度とすることができる。投与経路も特に限定されず、皮下投与、皮内投与、腹膜腔内投与、静脈内投与、筋肉内投与等を適宜選択することができるが、静脈内、腹膜腔内もしくは皮下に注射することにより投与することが好ましい。また、抗原は適当な緩衝液、例えば完全フロイントアジュバント又は水酸化アルミニウム等の通常用いられるアジュバントを含有する適当な緩衝液に溶解して用いることができるが、投与経路や条件等に応じてアジュバントを使用しない場合もある。
免疫感作した哺乳動物を一定期間飼育した後、哺乳動物の血清をサンプリングし、抗体価を測定する。抗体価が上昇してきたときに、例えば100μg〜1000μgの抗原を用いて追加免疫を行なうことができる。最後の投与から1〜2ケ月後に免疫感作した哺乳動物から血液を採取して、その血液をタンパク質の分離に採用される各種常法、例えば遠心分離、硫酸アンモニウム又はポリエチレングリコールを用いた沈澱、ゲルろ過クロマトグラフィー、イオン交換クロマトグラフィー、アフィニティークロマトグラフィー等のクロマトグラフィー等によって、ポリクローナル抗血清として、ポリクローナル抗体を得ることができる。
【0053】
モノクローナル抗体を産生する方法としては、ハイブリドーマ法を挙げることができる。先ず、アジュバント中に目的とする抗原を構成するペプチドを懸濁し、得られる懸濁液を、免疫動物(即ち、本発明のトランスジェニック哺乳動物)の皮下または真皮内に投与する。抗原の種類は前記と同様である。用いられるアジュバントとしては、フロイントの完全アジュバント、フロイントの不完全アジュバント、BCG、トレハロースダイマイコレート(TDM)、リポ多糖(LPS)、ミョウバンアジュバント、シリカアジュバント等が挙げられるが、抗体の誘導能等の関係から、フロイントの完全アジュバント(CFA)とフロイントの不完全アジュバント(IFA)とを組み合わせて使用することが好ましい。
モノクローナル抗体の産生において、免疫動物は抗原の初回免疫後、更に、追加免疫を数回行い、適当な日数を経過した後に部分採血を行い、抗体価を測定することが好ましい。本発明の方法で産生される抗体は高親和性抗体であるため、上記免疫は初回のみで十分である可能性がある。なお、抗体価は、例えば酵素イムノアッセイ(以下「ELISA」という)法等、公知の方法により測定することができる。
【0054】
次いで、感作の終了した免疫動物から脾臓を摘出し、B細胞を得る。この際、抗原に結合するB細胞を得ることが、その後のスクリーニングを軽減できる点で好ましい。ここで得られるB細胞は高親和性抗体産生細胞であり、これをそのまま免疫賦活剤として使用することもできる。また、B細胞から直接V領域遺伝子を得て、そのV領域の体細胞突然変異を測定することもできる。
次いで、B細胞を常法に従いミエローマ細胞と融合させて抗体産生ハイブリドーマを作製する。例えば、マウスの場合であれば、脾臓を摘出し、摘出した脾臓を、例えばハンクスの平衡塩溶液(HBSS)中に置き、ピンセットで細胞を押し出して脾臓リンパ球(B細胞)を得る。得られた脾臓リンパ球は、トリパンブルー等の染色液で染めて生細胞数をカウントし、ミエローマ細胞と融合させてハイブリドーマとする。
細胞融合に用いられるミエローマ細胞は特に限定されず、公知のものを使用できる。例えば、P3-X63.Ag8 (X63)、P3-X63.Ag8.U1 (P3U1)、P3/NS I/1-Ag4-1(NSI)、Sp2/0-Ag14(Sp2/0)等を挙げることができる。ミエローマ細胞の選択にあたっては、抗体産生細胞との適合性を適宜考慮する。
【0055】
細胞融合は、血清を含まないDMEM、RPMI-1640培地などの動物細胞培養用培地中で、1×106〜1×107個/mlの抗体産生細胞と2×105〜2×106個/mlのミエローマ細胞とを混合し(抗体産生細胞とミエローマ細胞との細胞比5:1が好ましい)、細胞融合促進剤存在のもとで融合反応を行う。
細胞の融合方法は、センダイウイルス法、ポリエチレングリコール法、プロトプラスト法等、当該分野で公知の方法を任意に選択して用いることができるが、特にポリエチレングリコール法が、細胞毒性が比較的少なく、融合操作も簡単であるという理由から好ましい。細胞融合促進剤としてのポリエチレングリコールは、平均分子量1000〜6000ダルトンのものを使用することができる。なお、抗体を大量に作り出したい場合は、ビニルピリジン誘導体で刺激した抗体産生細胞と骨髄腫細胞を細胞融合させたハイブリドーマを用いることが好ましい。
【0056】
得られたハイブリドーマは、常法に従い、HAT培地(ヒポキサンチン、アミノプテリン、およびチミジン含有培地)中で適当な期間培養し、ハイブリドーマの選択を行う。次いで、目的とする抗体産生ハイブリドーマのスクリーニングを行った後、ハイブリドーマのクローニングを行う。
スクリーニング法としては、公知の抗体検出方法を用いることができ、例えば、ELISA法、ラジオイムノアッセイ(以下「RIA」という)法、プラーク法、凝集反応法等を用いることができる。また、クローニング法としては、当該分野で公知の方法を用いることができ、例えば、限界希釈法、軟寒天法およびFACS法等を用いることができる。得られたハイブリドーマは、適当な培養液中で培養するか、あるいはハイブリドーマと適合性のある、例えばマウス腹腔内に投与する。こうして得られる培養液中または腹水中から、塩析、イオン交換クロマトグラフィー、ゲル濾過、アフィニティークロマトグラフィー等により、所望のモノクローナル抗体を単離精製することができる。
【0057】
また、上記した抗体の断片及びV領域の一本鎖抗体も本発明の範囲内である。抗体の断片としては、前述したポリクローナル抗体又はモノクローナル抗体の一部分の領域を意味し、具体的にはF(ab')2、Fab'、Fab、Fv(variable fragment of antibody)、sFv、dsFv(disulphide stabilized Fv)あるいはdAb(single domain antibody)等が挙げられる。ここで、「F(ab')2」及び「Fab'」とは、イムノグロブリン(モノクローナル抗体)を、それぞれ蛋白分解酵素であるペプシン、パパイン等で処理することにより製造され、ヒンジ領域中の2本のH鎖間に存在するジスルフィド結合の前後で消化されて生成される抗体フラグメントを意味する。例えば、IgGをパパインで処理すると、ヒンジ領域中の2本のH鎖間に存在するジスルフィド結合の上流で切断されてVL(L鎖可変領域)とCL(L鎖定常領域)からなるL鎖、及びVH(H鎖可変領域)とCHγ1(H鎖定常領域中のγ1領域)とからなるH鎖フラグメントがC末端領域でジスルフィド結合により結合した相同な2つの抗体フラグメントを製造することができる。これら2つの相同な抗体フラグメントを各々Fab'という。またIgGをペプシンで処理すると、ヒンジ領域中の2本のH鎖間に存在するジスルフィド結合の下流で切断されて前記2つのFab'がヒンジ領域でつながったものよりやや大きい抗体フラグメントを製造することができる。この抗体フラグメントをF(ab')2という。一本鎖抗体は、VL とVHをリンカーでつないだ構造を持つ。
【0058】
本発明の高親和性抗体はヒト型化抗体やヒト抗体でもよい。これらの抗体は、免疫系をヒトのものと入れ換えた哺乳動物を用いて、該哺乳動物を免疫して、通常のモノクローナル抗体と同様に直接ヒト抗体を作製することができる。
ヒト型化抗体を作製する場合は、マウス抗体の可変領域から相補性決定領域(complementarity determining region;CDR)をヒト可変領域に移植して、フレームワーク領域(FR)はヒト由来のものを、CDRはマウス由来のものからなる再構成した可変領域を作製する。
次に、これらのヒト型化された再構成ヒト可変領域をヒト定常領域に連結する。最終的に再構成されたヒト型化抗体のヒト以外のアミノ酸配列に由来する部分は、CDR及び極く一部のFRのみである。CDRは超可変アミノ酸配列により構成されており、これらは種特異的配列を示さないため、マウスCDRを有するヒト型化抗体を使用することが可能である。ヒト型化抗体の作製法は、当分野において周知である。
【0059】
ヒト抗体は、一般にV領域の抗原結合部位、すなわち超過変領域(Hyper Variable region)についてはその特異性と結合親和性が問題となるが、構造的にどの動物で作製してもかまわない(マウス、ラット等)。一方、V領域のそのほかの部分や定常領域の構造はヒトの抗体と同じ構造をしていることが望ましい。ヒトに共通の遺伝子配列については遺伝子工学的手法によって作成する方法が確立されている。
本発明の抗体のアイソタイプは特に限定されず、例えば、IgG(IgG1、IgG2、IgG3、IgG4)、IgM、IgA(IgA1、IgA2)、IgDまたはIgEの任意のアイソタイプを有することができる。
【0060】
4.高親和性抗体の利用
本発明の高親和性抗体は、疾患の診断、治療又は予防のための薬剤として有用である。
【0061】
(1) 疾患の診断
本発明の抗体を用いた各種疾患の診断方法は、各種疾患の疑いのある被験者から採取した検体、例えば血清等と本発明の抗体とを抗原抗体反応によって結合させ、結合した抗体量により検体中の目的とする抗原の量を検出することにより行う。抗体量の検出は、公知の免疫学的測定法に従って行えばよく、例えば、免疫沈降法、免疫凝集法、標識免疫測定法、免疫比ろう法、免疫比濁法等を用いることができる。特に標識免疫測定法が簡便かつ高感度という点で好ましい。標識免疫測定法では、検体中の抗体価は標識抗体を用いて直接検出した標識量で表すほか、既知濃度あるいは既知抗体価の抗体を標準液として用いて相対的に表してもよい。すなわち、標準液と検体を同測定系にて同時に測定し、標準液の値を基準にして検体中の抗体価を相対的に表すことができる。
【0062】
標識免疫測定法としては、公知の測定法、例えば、ELISA法、RIA法、蛍光免疫測定法、化学発光免疫測定法等を任意に利用することができる。用いる標識物質は、上記測定法に応じて、酵素、放射性同位体、蛍光化合物、および化学発光化合物等を適宜選択すればよい。前記酵素としては、例えば、ペルオキシダーゼ、アルカリホスファターゼ、酸性ホスファターゼ、グルコースオキシダーゼ等を挙げることができる。上記標識物質はアビジン−ビオチン複合体を用いることにより、標識物質の検出感度を向上させることも可能である。また、放射性同位体としては、主に125Iが、蛍光化合物としては、フルオレセインイソチオシアネート(FITC)やテトラメチルローダミンイソチオシアネート(TRITC)等が挙げられる。化学発光化合物としては、ロフィン、ルミノール、ルシゲニン等が挙げられる。上記標識物質による抗体の標識は、常法に従って行うことができる。以下、標識抗体を用いた標識免疫測定法について説明する。
【0063】
標識免疫測定法による、各種疾患を検出する方法としては、公知の非競合反応系あるいは競合反応系を用いて行うことができる。非競合反応系においては、固相が必要である(固相法)。競合反応系においては、必ずしも固相を必要としない(液相法)が、固相を用いた方が、測定操作が簡便になるため好ましい。固相の材質としては、例えば、ポリスチレン、ナイロン、ガラス、シリコンラバー、セルロース等が挙げられ、固相の形状としては、球状、ウェル状、チューブ状、シート状等が挙げられるが、これらに限定されず、標識免疫測定法に用いられる公知のものを任意に用いることができる。
【0064】
非競合反応系の場合、測定操作は、検体または本発明の抗体を固相化した後、本発明の抗体または検体と反応させ、次いであらかじめ標識しておいた抗免疫グロブリン抗体(二次抗体)を加えて固相化した検体と反応している抗体と反応させる。この二次抗体の標識により、検体に結合した抗体量を検出することができる。検出された標識化二次抗体の量は、検体中の目的とする抗原の量と正相関するので、これにより検体中の目的とする抗原の量を求めることができる。
競合反応系では、一定量の抗体に対して、検体と一定量の目的とする抗原を競合的に結合させる。例えば、検体を固相化した後に、あらかじめ目的とする抗原を添加し反応させた本発明の抗体と反応させる。次に、固相化された検体と反応した抗体を、あらかじめ標識しておいた抗免疫グロブリン抗体(二次抗体)と反応させ、標識物質によって抗体の量を検出することができる。検出される標識量は、添加された目的とする抗原の量と逆相関する。そのほかの競合反応系としては、本発明の抗体を固相化して、これに検体と反応させた後、あらかじめ標識しておいた目的とする抗原を反応させる。検出される標識量は、抗体と結合した検体中のGANP蛋白質量と逆相関する。
【0065】
前記した抗原または抗体の固相化法としては、物理的吸着法、共有結合法、イオン結合法、架橋法等、公知の方法を使用できる。特に、物理的吸着法が簡便という点で好ましい。また、抗免疫グロブリン抗体(二次抗体)としては、例えば、抗IgG抗体、抗IgM抗体等を用いることができる。これらの抗体は、抗体分子をそのまま使用してもよいし、あるいは抗体を酵素処理して得られる抗原結合部位を含む抗体フラグメントであるFab、Fab'、F(ab')2を使用してもよい。さらに、標識した抗免疫グロブリン抗体の代わりに、抗体分子に特異的な親和性をもつ物質、例えばIgGに特異的な親和性をもつプロテインA等を標識して使用してもよい。
【0066】
前記標識免疫測定法の好適な例として、酵素を標識とした免疫測定法、ELISA法を挙げることができる。ELISA法は、例えば、96穴プレートに検体またはその希釈液を入れて、4℃〜室温で一晩、または37℃で1〜3時間程度静置して検出すべきGANP蛋白質を吸着させて固相化する。次に、本発明の抗体を反応させ、次いであらかじめ酵素を結合させた抗免疫グロブリン抗体(二次抗体)を反応させる。最後に酵素と反応する適当な発色性の基質(例えば、酵素がホスファターゼの場合はp-ニトロフェニルリン酸等)を加え、この発色によって抗体を検出する。
また、本発明の高親和性抗体を利用することにより、各種疾患の治療薬の薬効評価を行うことができる。本発明の高親和性抗体を利用した薬効評価方法は、各種疾患患者あるいは各種疾患モデル動物に対して薬剤を投与後、これら生体中のウイルス等の抗原の量を本発明の抗体を用いて検出し、その量を比較することにより、生体中の抗原の量を通して各種疾患の治療薬としての薬効を評価することができる。
【0067】
本発明の高親和性抗体は、各種疾患診断用キットの形態で提供することができる。該キットは、本発明の診断方法や本発明の薬効評価方法に使用することができる。本発明のキットは以下の(a)及び(b)から選ばれる少なくとも一つ以上を含む。
(a)本発明の抗体またはその標識物
(b)前項(a)記載の抗体またはその標識物を固定した固相化試薬
ここで、抗体の標識物とは、酵素、放射性同位体、蛍光化合物、または化学発光化合物によって標識されたものを意味する。
また本発明のキットにおける抗体、もしくはこれらの標識物を固定する固相の材質としては、ポリスチレン、ナイロン、ガラス、シリコンラバー、セルロース等が挙げられ、固相の形状としては、球状、ウェル状、チューブ状、シート状等が挙げられるが、これらに限定されない。該固相化試薬の代わりに、固相と固相化に必要な固相化試薬を添付したものでもよい。固相化試薬として、例えば物理的吸着による固相化の場合は、50mM炭酸塩緩衝液(pH 9.6)、10 mMトリス-塩酸緩衝液(pH8.5、100mM塩化ナトリウム含有)、PBS等のコーティング液と、さらに必要に応じてコーティング液に0.5%のゼラチン等を含有させたブロッキング液が挙げられる。
【0068】
また、本発明のキットにおける抗体は、PBS等に溶解させた状態、あるいはゲルに結合させた状態(以下、「吸収用ゲル」と略す)であってもよい。前記吸収用ゲルはさらに適量を、バッチ法による吸収処理用に0.5〜2ml程度のマイクロ遠沈チューブに予めパッケージングされた状態であってもよく、あるいはカラム法による吸収処理用にカラム容量が0.1 〜5 mlのミニカラムに予め充填された状態であってもよい。
本発明のキットは、上記した構成要素のほか、本発明の検出を実施するための他の試薬、例えば標識物が酵素標識物の場合は、酵素基質(発色性基質等)、酵素基質溶解液、酵素反応停止液、あるいは検体用希釈液等を含んでいてもよい。前記検体用希釈液としては、例えばPBS(生理的リン酸緩衝液、pH7.4)、137mM塩化ナトリウムおよび3mM塩化カリウムを含むpH7.4かつ20mMのトリス-塩酸緩衝液(以下、「TBS」と略す)、0.05%Tween20、0.1〜1%のBSAを含有させたPBS、あるいはTBS等を挙げることができる。該検体用希釈液は、検体希釈以外、例えば抗体の希釈等に用いてもよい。
【0069】
(2) 疾患の治療又は予防用医薬組成物
本発明の高親和性抗体は、疾患の病原となる抗原の活性を中和させる作用を有するものであれば、疾患の治療又は予防のための医薬組成物として有用である。本発明の医薬組成物は、本発明の高親和性抗体またはその断片を有効成分として含み、さらに薬学的に許容される担体を含む医薬組成物の形態で提供することが好ましい。
ここで「薬学的に許容され得る担体」とは、賦形剤、希釈剤、増量剤、崩壊剤、安定剤、保存剤、緩衝剤、乳化剤、芳香剤、着色剤、甘味剤、粘稠剤、矯味剤、溶解補助剤あるいはその他の添加剤等が挙げられる。そのような担体の一つ以上を用いることにより、錠剤、丸剤、散剤、顆粒剤、注射剤、液剤、カプセル剤、トローチ剤、エリキシル剤、懸濁剤、乳剤あるいはシロップ剤等の形態の医薬組成物を調製することができる。これらの医薬組成物は、経口あるいは非経口的に投与することができる。非経口投与のためのその他の形態としては、一つまたはそれ以上の活性物質を含み、常法により処方される外用液剤、腸溶内投与のための坐剤およびペッサリーなどが含まれる。
【0070】
本発明の薬剤の投与量は、患者の年齢、性別、体重及び症状、治療効果、投与方法、処理時間、あるいは該薬剤に含有される活性成分である高親和性抗体の種類などにより異なるが、通常成人一人当たり、一回につき10μgから1000mg、好ましくは10μgから100mgの範囲で投与することができるが、この範囲に限定されるものではない。
【0071】
例えば、注射剤の場合には、例えば生理食塩水あるいは市販の注射用蒸留水等の薬学的に許容される担体中に0.1μg抗体/ml担体〜10mg抗体/ml担体の濃度となるように溶解または懸濁することにより製造することができる。このようにして製造された注射剤は、処置を必要とするヒト患者に対し、1回の投与において1kg体重あたり、1μg〜100mgの割合で、好ましくは50μg〜50mgの割合で、1日あたり1回〜数回投与することができる。投与の形態としては、静脈内注射、皮下注射、皮内注射、筋肉内注射あるいは腹腔内注射などが挙げられるが、好ましくは静脈内注射である。また、注射剤は、場合により、非水性の希釈剤(例えばプロピレングリコール、ポリエチレングリコール、オリーブ油のような植物油、エタノールのようなアルコール類など)、懸濁剤あるいは乳濁剤として調製することもできる。そのような注射剤の無菌化は、バクテリア保留フィルターを通す濾過滅菌、殺菌剤の配合または照射により行うことができる。注射剤は、用時調製の形態として製造することができる。即ち、凍結乾燥法などによって無菌の固体組成物とし、使用前に無菌の注射用蒸留水または他の溶媒に溶解して使用することができる。
【0072】
5.本発明の応用
本発明者らは、B細胞腫瘍株にGANPの過剰発現を誘導した上で解析を行った結果、B細胞腫瘍株はGANP遺伝子導入によって、飛躍的なV領域遺伝子の体細胞突然変異誘導効果を有することを示した。この効果は、GANPのRNAプライマーゼ活性に必要な502番目のセリンのリン酸化が起こらないような変異遺伝子を用いた場合には見られないことから、V領域遺伝子の体細胞突然変異の飛躍的な誘導には、RNAプライマーゼ活性が必要であることを示している。この結果は、臨床的な補助免疫賦活剤として、GANPが特異的抗体産生の増強効果を有することを示すものである。
【0073】
ベクターとしてレトロウイルスベクターを使用し、CD40、BAFFなどのTNFファミリー分子を介する刺激をGANPと併用することも、臨床的な補助免疫賦活作用にとって効果的である。また、この遺伝子導入を骨髄細胞レベルで行うことによって、T細胞における高親和性結合の誘導も期待できる。エイズ、C型肝炎ウイルス、成人T細胞白血病、狂牛病などの高親和性抗体が得られない場合や、あるいは得られたとしてもすぐに抗原の変異が起こるために十分に高親和性抗体の産生が持続できない場合には、この遺伝子導入は優れた効果を発揮すると期待できる。
【0074】
本発明のGANP遺伝子過剰発現哺乳動物は、生物学研究試薬、臨床検査試薬作製に有効なモノクローナル抗体の開発に有効である。例えば、特定のシグナル伝達分子に対するモノクローナル抗体を機能ドメインや機能モチーフに特異的にそして結合力の高い高親和性抗体を簡便に作製することは非常に活用される範囲が広い。多くの抗体はそれほど多くのスクリーニングをかけないため、ウエスタン解析と免疫沈降に用いることができない場合がある。この場合、本発明のトランスジェニック哺乳動物を用いれば、比較的少ないクローンの抗体から高親和性抗体産生細胞を短時間で選別することができ、経費、時間、労力の削減する効果は大きい。特にリン酸化抗体、遺伝子変異部分に対する特異抗体の作製は診断薬、あるいは抗体を用いた薬物の選択的注入法に応用できる。また遺伝子の配列やヌクレオチド部分に選択的に結合する高親和性抗体の産生も可能となる。
【0075】
無機物、炭水化物、化学合成物など、任意の物質の立体構造の一部は、抗原モチーフとして認識される。従来には高親和性抗体は得られていないが、自己免疫疾患マウスとの交配で作製されるマウスは、あらゆる抗原に対して高親和性抗体を得るために有効である。この方法で結合力が10-11Mオーダーの高親和性抗体ができる可能性があり、ELISA法の技術開発を導入することにより、微量物質の検出を簡便に行う技術を開発することが可能である。
また、本発明によれば、RNAプライマーゼ不活性型GANP遺伝子を含む、アレルギー疾患又は自己免疫疾患のための遺伝子治療剤を提供することも可能である。「RNAプライマーゼ不活性型GANP遺伝子」とは、RNAプライマーゼドメイン欠損、又はRNAプライマーゼドメインが変異した遺伝子を意味し、502番目のセリン残基の変異を含む近傍の遺伝子の変異によってGANP分子の構造および機能的な変化を生じた遺伝子のことを意味する。
【0076】
本発明の遺伝子治療剤は、RNAプライマーゼ不活性型GANP遺伝子を含む組み換えベクターを、遺伝子治療剤に用いる基剤と一緒に配合することにより製造することができる。組み換えベクターの構築の際に用いるベクターとしては、ウイルスベクターとしてレトロウイルスベクター、アデノウイルスベクター、アデノ随伴ウイルスベクター、バキュロウイルスベクター、ワクシニアウイルスベクターなどが挙げられ、あるいは動物発現用プラスミドを使用することもできる。ベクターは好ましくはウイルスベクターである。RNAプライマーゼ不活性型GANP遺伝子をウイルスベクターに組み込んだ場合は、組換え体DNAを含有するウイルス粒子を調製し、遺伝子治療剤に用いる基剤と一緒に配合することにより遺伝子治療剤を製造することができる。
【0077】
遺伝子治療剤に用いる基剤としては、通常注射剤に用いる基剤を使用することができ、例えば、蒸留水、塩化ナトリウム又は塩化ナトリウムと無機塩との混合物等の塩溶液、マンニトール、ラクトース、デキストラン、グルコース等の溶液、グリシン、アルギニン等のアミノ酸溶液、有機酸溶液又は塩溶液とグルコース溶液との混合溶液等が挙げられる。あるいはまた、当業者に既知の常法に従って、これらの基剤に浸透圧調整剤、pH調整剤、植物油、もしくは界面活性剤等の助剤を用いて、溶液、懸濁液、分散液として注射剤を調製することもできる。これらの注射剤は、粉末化、凍結乾燥等の操作により用時溶解用製剤として調製することもできる。
本発明の遺伝子治療剤の投与形態としては、通常の静脈内、動脈内等の全身投与でもよいし、局所注射又は経口投与等の局所投与を行ってもよい。本発明の遺伝子治療剤の投与量は、年齢、性別、症状、投与経路、投与回数、剤型によって異なるが、一般に、成人では一日当たり組み換え遺伝子の重量として1μg/kgから1000mg/kg程度の範囲であり、好ましくは10μg/kgから100mg/kg程度の範囲である。投与回数は特に限定されない。
【0078】
実施例
以下の実施例により本発明をさらに具体的に説明するが、本発明は実施例によって限定されるものではない。
【実施例1】
【0079】
自己免疫疾患モデル動物におけるGANPの発現とその機能
(材料及び方法)
【0080】
1.動物
NZB、NZW、B/WF1、MRL/lpr、及びBXSB マウスはJapan SLC Co.から購入した。
C57BL/6 及びBALB/cマウスはCharles River Japanから購入した。NODマウスは大阪大学大学院の宮崎博士から供与された。
【0081】
2.抗体及び試薬
マウスB220(RA3-6B2)、マウスIgM (AM/3) 及びマウスIgD (CS/15)に対するラットモノクローナル抗体はハイブリドーマの培養上清から精製し、D-ビオチン-N-ヒドロキシスクシンイミドエステル(Roche diagnostics, Branchburg, NJ)で標識した。ビオチン標識ラット抗マウスSyndecan-1及び抗マウスCD5モノクローナル抗体は購入した(BD PharMingen, San Diego, CA)。ビオチン標識ピーナッツアグルチニン(PNA)はVector Laboratories (Burlingame, CA)から購入した。
【0082】
3.免疫
トリニトロフェニル-キーホールリンペットヘモシアニン(Trinitrophenyl keyhole limpet hemocyanin (TNP-KLH)及びTNP-FicollはBiosearch Technologies (Novato, CA)から購入した。完全フロイントアジュバンドに乳化した100μgのTNP-KLHまたはPBS中の25μgのTNP-Ficollをマウスの腹腔内に注入した。14日後、リンパ器官を取得し、免疫組織分析用にOCT 化合物とともに凍結した。
【0083】
4.免疫組織分析
6μmの凍結切片をアセトンで5分間固定し、PBS中の3% BSAで15分間ブロッキングし、ラット抗マウスGANPモノクローナル抗体(42-23) [Kuwahara K., 他、2000, Blood 95: 2321-2328]またはラット抗-pSer502 GANPモノクローナル抗体 (PG/103) [Kuwahara K.,他、2001, Proc. Natl. Acad. Sci. USA 98: 10279-10283]と一緒に1時間インキュベートした。切片をのせたスライドグラスをPBSで数回洗浄し、アルカリホスファターゼ (ALP)-結合ヤギ抗ラットIgG 抗体(ICN Pharmaceuticals, Costa Mesa, CA)と一緒にインキュベートした。発色はVector Blue kit (Vector)を用いて行った。二重染色のために、反応はビオチン標識抗体と西洋ワサビペルオキシダーゼ(HRP)-結合ストレプトアビジン(Kirkegaard & Perry Laboratories, Gaithersburg, MD)を組み合わせて行った。3-3'-ジアミノベンジジンテトラヒドロクロライド(DAB; 同仁化学)による発色後、切片をPBS中1%グルタルアルデヒドで1分間固定した。Aquatex (Merck, Darmstadt, Germany)をマウンティングのために用いた。インビボで増殖活性のある細胞を検出するために、ブロモデオキシウリジン (BrdU) (Sigma Chemicals Co., St. Louis, MO; 1 mg/マウス)を屠殺する2時間前に静脈内に注入した。DNA合成を行う細胞を抗BrdUモノクローナル抗体 (BD PharMingen)とALP-結合ヤギ抗マウスIg 抗体(Sigma)とを組み合わせて染色しVector Red (Vector)により発色させて検出した。PAS染色は既報の通り行った [Jiang Y.他、1997, J. Immunol. 158: 992-997]。
【0084】
5.結果
(1) MRL/lpr マウスのリンパ節におけるGANPhi細胞の出現
GANP発現は自己免疫傾向の高活性B細胞で高発現している。高レベルのGANPを発現するリンパ細胞(GANPhi 細胞)は、MRL/lprマウスの末梢リンパ節において非免疫状態において自発的に出現する。
抗GANPモノクローナル抗体およびALP結合抗ラットIg抗体を用いて、自己免疫疾患モデルMRL/lpr、NZBおよび正常C57BL/6雌マウス由来の膝窩リンパ節について免疫組織化学分析を行った。
結果を図1に示す。Vector Blue(ALP基質)で染色されたGANPhi細胞は7週目にMRL/lprマウスのリンパ節で観察されたのに対し、同年齢のNZBマウスでは観察されず、40週目に出現した(図1)。正常C57BL/6マウスでは、極少数のGANPhi細胞が全期間を通じて観察された。
【0085】
自己免疫疾患モデルマウスは、C57BL/6 マウスと比較してリンパ細胞の増加は顕著であり、非免疫条件下ではGANPhi細胞は示されない(図1)。そのようなGANPhi細胞の出現を、加齢の間少しずつ自己免疫状態を引き起こすNZBマウスのリンパ節で調べた。若いNZBマウス(7週齢)は、膝窩リンパ節にGANPhi 細胞を有さないが、加齢したNZB (40 週齢)は多数のGANPhi 細胞を有した。
GANP RNAプライマーゼ活性はB細胞の活性化と分化において重要な役割を担っている可能性がある。そこで、NZBマウスで抗pSer502 モノクローナル抗体を用いて、RNAプライマーゼ活性の重要なリン酸化部位であるSer502のリン酸化状態を比較した。
NZBマウスのリンパ節中のGANPおよびpSer502 GANPの発現を比較した。pSer502 GANPは抗pSer502 GANP(PG/103)モノクローナル抗体(青)で検出し、全切片をビオチン標識抗B220モノクローナル抗体で染色した後、HRP結合ストレプトアビジンとDAB(茶色)を組み合わせて検出した。2回の独立した実験から代表的データを示した(図2)。
【0086】
図2において、下段の図(グラフ)は、加齢の間に濾胞外領域のGANPhi(黒の棒)およびpSer502 GANPhi(斜線の棒)の細胞数を示す。
GANPの発現は8週目で顕著であり、GANPhi 細胞が32週までの全期間を通じて検出された (図2;上図)。対照的に、pSer502-陽性細胞は8週目に最大であったが、その後、陽性細胞は顕著に減少した(図2;真中の図)。 ピーク年齢に基づく顕微鏡観察での反応性細胞数を図に示す(図2;下図)。これらの結果から、GANP発現は最初にRNAプライマーゼ活性を伴うが、この活性は長期間に渡っては調節されていないことが分かる。
【0087】
(2) 自己免疫傾向マウスの脾臓の赤脾髄におけるGANPhi細胞の自発的出現
自己免疫傾向NZBマウスの膝窩リンパ節で検出されたGANPhi細胞が非免疫状況下の脾臓に出現するかどうかを調べた。
免疫染色は前記(1)の操作(図2)と同様に行った。3回の独立した実験からの代表的データを示した(図3)。
GANPhi細胞は4週目に脾臓に出現し、細胞数は12週目に最大値に達したが、24週後に消失した(図3;上図)。pSer502 GANPの発現も8週目と12週目に検出された (図3;真中の図)。赤脾髄の相対細胞数と比較した結果、脾臓に出現したGANPhi細胞は 12週後には末梢リンパ節に移動していることが分かる。GANPhi の増加は、自己免疫疾患の発症に先行する自己抗体の産生量に比例している (図2及び図3; Theofilopoulos A.N.,他、1985, Adv. Immunol. 37: 269-390)。
【0088】
GANPhi細胞の出現は自己免疫傾向マウスにおけるB細胞の異常と関連している可能性があるので、非免疫条件下で各種の自己免疫傾向マウス(8週齢)におけるGANPhi細胞の出現を調べた。
結果を図4に示す。GANPhi 細胞はMRL/lpr、NZB及びB/WF1の赤脾髄で顕著に出現した。
GANPhi細胞の数はSLE-モデルマウスのBXSB 及びNODの脾臓にはそれほど増加しなかったが、対照のBALB/cマウス(図4) 及びC57BL/6マウス (図1)と比較すれば増加していた。脾臓の切片は、GC様構造としてPNA+ B細胞の連想又は未熟の会合を示した。GC様領域でのGANPの発現は、正常C57BL/6マウス及びBALB/cマウスに、T細胞依存性抗原(T cell-dependent Ags: TD-Ags)を免疫することによって作製したGCでのGANPの発現と比較して高くない。しかし、GANPhi 細胞は、自己免疫傾向マウスの赤脾髄領域で顕著に出現していた(図4)。
【0089】
さらに、GANPhi 細胞集団を、リンパ系細胞のマーカー分析によって解析した。
ビオチン標識抗B220モノクローナル抗体、ビオチン標識抗Syndecan-1モノクローナル抗体、ビオチン標識抗IgMモノクローナル抗体、及び抗IgG抗体を用いて、NZBマウスの脾臓切片について二重染色を行い、GANPhi 細胞を同定した。
結果を図5に示す。図5に示すパネルの左の列は、上から下に順にビオチン標識抗IgMモノクローナル抗体、抗IgG抗体、ビオチン標識抗B220モノクローナル抗体及びビオチン標識抗Syndecan-1モノクローナル抗体を用いたときの図である。中央の列は、上記それぞれの抗体を用いたのと同じ切片でのGANPの発現を示す。右の列は左の列と中央の列を重ね合わせた図である。右列の二重に染色された細胞は、GANPhi 細胞がB220-Syndecan1+IgM+であることを示す。GANP発現は、IgM、IgG及びB200の場合は赤色で示し、Syndecan-1の場合は緑色で示す。マーカーは、IgM、IgG及びB200は緑色で示し、Syndecan-1の場合は赤色で示す。
【0090】
GANPhi 細胞はB220-Syndecan-1+ の表現型を発現し、多量のIgM を細胞中に発現する(図5)。CR1、Thy-1、GL-7、CD23及びPNAについては陰性であり、これらの結果から、GANPhi 細胞がB系細胞の後期成熟段階、おそらく形質細胞であることが示される。このGANPhi 細胞が増殖性形質芽細胞であるかどうかを調べるために、NZBマウスにBrdU (1 mg/マウス)を投与(静脈内注入)し、インビボでBrdUを取り込ませるために2時間インキュベートした。その後、マウスから脾臓切片を調製した。
切片を抗GANPモノクローナル抗体(青)及び抗BrdUモノクローナル抗体(赤)で二重染色した。PAS染色を常法に従って行った。
結果を図6に示す。GCは胚中心を示す(左)。GANP単一陽性GANPhi 細胞は矢印で示し、PAS単一陽性細胞を矢頭で示す(中央)。
【0091】
また、切片をビオチン標識抗CD-5モノクローナル抗体で染色した。PALS領域は、リンパ節動脈周囲鞘を示す(右)。図6は、3回の独立した実験から代表的データを示した。
GANPhi 細胞はBrdU 取り込みについて陽性ではないことから(図6)、これらの細胞は増殖性ではなく、形質芽細胞段階より成熟していることが示唆された。
B-1細胞の異常な分化として、Mott細胞形成が自己免疫傾向マウスで観察される。Mott細胞は形質細胞の異常な形態であり、多量のIgM 分子が、PAS染色により細胞質内Russell小体として検出される粗面小胞体結合小胞に蓄積している [Jiang Y.,他、1997, J. Immunol. 158: 992-997]。GANPhi 細胞はPAS-染色で染色されず(図6)、これによりGANPhi 細胞をB-1 細胞由来形質細胞のMott細胞と区別することができる。脾臓GANPhi 集団はCD5発現が陰性であり(図6)、NZBマウス(12週)から得た腹膜細胞は GANPhi 細胞について陰性であったことから、B-1細胞は多量のGANPを発現していないことが示される。これらの結果から、GANPhi 細胞は自己免疫状態で高活性のB細胞に分類され、この集団がB-1細胞の起源とは異なる起源の系統であることが示唆される。
【0092】
(3) TD-Agでの免疫による正常マウスにおけるGANPhi細胞の誘導
二次リンパ器官におけるGANPhi形質細胞の出現が、自己免疫傾向マウスに限られているかどうかを調べた。
雌C57BL/6 マウス(7週齢)を、TNP-Ficoll (TI-2-Ag)又はTNP-KLH (TD-Ag)で腹腔内免疫し、14日後に脾臓を得た。TNP-Ficollで免疫したマウスは、ビオチン標識抗IgDモノクローナル抗体で対比染色した場合、GANPhi 細胞を赤脾髄領域で示さなかった(図7左)。TNP-KLHで免疫したマウスは 、GANPhi細胞の誘導を赤脾髄領域で示した(図7右)。図7において、GANPhi細胞は矢印で示す。WPは白脾髄領域を示す。
GANPhi 形質細胞集団は、数は非常に少ないが、TD-Agsによる免疫によって正常C57BL/6及びBALB/c マウスの脾臓においても誘導される(図7)。T細胞非依存性Ag (T cell-independent Ag: TI-Ag) による免疫はそのような細胞の誘導において効果は小さい。GANPhi 細胞集団はB220loIgMhiIgDloGL-7loPNAlo CD5loCD40lo と同様の表現型を示したが、Syndecan-1+を示した。
【0093】
これらの結果から、自己免疫傾向マウスにおけるGANPhi 形質細胞の生成は、TD-Agに対する免疫応答のために提供されるのと同様の刺激によって誘導されることが示される。GCで増殖と分化を経たAg駆動B細胞は、GANPを発現しながらより長い間、形質細胞段階として赤脾髄領域に局在化している可能性がある。
【実施例2】
【0094】
GANPの過剰発現
(方法)
1.Daudi細胞への安定なトランスフェクション
10μgの線状化したpCXN-2マウスGANP又はGANPS/A502 cDNAをDaudi細胞に、Gene Pulser II (Bio-Rad)を用いてエレクトロポレーションを行った。48時間後、G418 (Promega; 1 mg/ml)により選択を開始して、マウスGANPを安定に発現するDaudi細胞を得た。
【0095】
2.DaudiトランスフェクタントのIg VH 転写物の分析
全RNAを全細胞からTrizol (Invitrogen)を用いて抽出した。cDNAを既報の通り取得した(Kuwahara, K. et al., Blood 95, 2321-2328 (2000))。LVH3- CH1Cμ転写物を以下のプライマー及び反応液を用いて増幅した。増幅は、Pfu Turbo (Stratagene)を用いた。
【0096】
5'-LVH3 プライマー:
5'-CTATAACCATGGACCATGGACATACTTTGTTCC-3' (配列番号5)
3'-XbaI-CH1-Cμプライマー :
5'-TGCATGCATTCTAGAGTTGCCGTTGGGGTGCTGGAC-3' (配列番号6)
【0097】
反応液組成:
【表1】

【0098】
反応条件:
94℃ 1 min
[94℃ 1 min; 62℃ 1 min ; 72℃ 1 min] ×35 サイクル
72℃ 10 min
4℃
【0099】
PCR産物をNcoI及びXbaIで消化し、ゲルで精製し、NcoI-XbaIで消化したプラスミドとライゲーションした。コンピテント細菌に形質転換後、QIAprepキット(QIAGEN)を用いて調製した少量のプラスミドDNAの塩基配列を自動シークエンサー(Applied Biosystems)により決定した。
【0100】
3.GANP-トランスジェニック(Tg)マウスの作製
導入遺伝子は、pLGベクターのXhoIサイトに5.6 kbのマウスGANP遺伝子を導入して作製した。このベクターはヒト免疫グロブリンイントロンエンハンサー領域(2kb EcoRIフラグメント)を持ち、B細胞での強力な発現を行う、特異的ベクターである。この遺伝子を直線化してマウスに遺伝子導入を行った。マウスGANP全長cDNAを含む線状化したpLG vector(Koike, M. et al. Int. Immunol. 7, 21-30 (1995))をC57BL/6 マウスの受精卵にマイクロインジェクションした。マウスの尾のゲノムDNAおよび以下のプライマー及び反応液を用いて導入遺伝子の存在についてスクリーニングした。
【0101】
1-5' プライマー:
5'-TCCCGCCTTCCAGCT GTGAC-3'(配列番号7)
1-3'プライマー:
5'-GTGCTGCTGTGTTATGTCCT-3' (配列番号8)
【0102】
反応液組成:
【表2】
【0103】
反応条件:
[98℃ 5 sec; 59℃ 5 sec ; 72℃ 10 sec] ×35 サイクル
4℃
【0104】
4.RT-PCR
全RNAは、脾臓又は脾臓B細胞からTrizol(Invitrogen)を用いて抽出し、RT-PCRは、2種のプライマー1-5'及び 1-3'を用いて行い、cDNAを合成した(Kuwahara, K. et al., Blood 95, 2321-2328 (2000))。GANP 転写物はアガロースゲル電気泳動により検出した。β-アクチン転写物は対照として用いた。
【0105】
5.結果
(1) Daudi細胞にマウスGANPを安定的に発現させたトランスフェクタントのV領域遺伝子の体細胞突然変異(SHM)
GANP遺伝子を、インビトロでSHMの分析に使用される各種ヒトBリンパ球細胞に導入した(Rogozin, I. B., et al., Nat. Immunol. 2, 530-536 (2001); Kuwahara, K. et al. Blood 95, 2321-2328 (2000); 及びDenepoux, S. et al., Immunity 6, 35-46 (1997))。多くのB細胞株にはトランスフェクションできなかったが、維持中はSHMを通常は生成しないAIDを発現するDaudi B細胞にはGANP遺伝子を導入できた。
このクローンは、野生型及び偽トランスフェクションした細胞と比較して高頻度のSHM (5 x 10-4/bp)をV領域で示した。
VH3-CH1Cμの断片をPCRで増幅してプラスミドにサブクローニングし、シークエンスした。
【0106】
体細胞変異の模式図を図8A〜Cに示す。縦線(「|」)はサイレントミューテーション(アミノ酸が変わらない)、もう一つの記号(縦線に●印を付したもの)にはアミノ酸が置換している変異を示す。Daudi/mockではほとんど変異が入っていないが、Daudi/GANP-14, 15, 17, 21の4クローンは、程度の差はあるが変異を多く認める。DNAプライマーゼ活性の制御に関係する502番目のセリンをアラニンに置換した変異体(GANP S/A)を導入したトランスフェクタントでは変異の入る効率が減少する。
SHMは、定常領域遺伝子には誘導されなかった(図8A〜C)。GANPのRNAプライマーゼ活性はS502でのリン酸化により調節され、これは特異的モノクローナル抗体により検出できる(Kuwahara, K. et al., Proc. Natl. Acad. Sci. USA 98, 10279-10283 (2001))。インビトロ及びインビボでのB細胞の刺激は共にS502のリン酸化を誘導するので(Kuwahara, K. et al., Proc. Natl. Acad. Sci. USA 98, 10279-10283 (2001))、このリン酸化がDaudi B細胞でのSHMの生成に関与するかどうかを調べた。
非リン酸化GANP変異体(GANP-S502A)を導入した場合、SHMは誘発されなかったことから(図8A)、S502のリン酸化が、GC-B細胞でのSHMの生成に重要であることが示唆された。
【0107】
(2) B細胞でGANPを過剰発現させたトランスジェニックマウス
免疫応答におけるGANPの関与を調べるために、ヒトIgエンハンサー及びプロモーターの制御下にマウスGANPを過剰発現させたGANP-トランスジェニック (Tg)マウスを作製した(図9A及びB)。GANP mRNAの発現の亢進は、RT-PCRで確認した。
このマウスは、B細胞でGANPの発現の増加を示し(図9C)、骨髄、脾臓及びリンパ節の細胞の表層マーカー分析において、B系細胞の通常の分化を示した。
SHMにおけるGANPのインビボでの役割を調べるために、TD-AgであるNP-CGの免疫後におけるVH186.2 領域について調べた。すなわち、GANP過剰発現トランスジェニック(Tg)マウスに50μgのNP-CGを2週間おきに3回免疫して、VH186.2をPCRで増幅し、体細胞突然変異を解析した。
【0108】
結果を図10に示す。変異の数はTgでやや増加しているが、高親和性を示す33番目のWがLに変わる変異(SHM)は、Tgで約3倍に増加していた。なお、CDRは相補鎖決定領域を示す。
VH186.2ローカスは、高親和性IgG(γ1λ1)NP-応答について特有のパターンのSHMを示す。NP-CGでの免疫後における全脾臓B細胞の配列分析により、野生型マウスと比較してGANP-Tg マウスではSHMの頻度が僅かに増加していることが示された(図10)。
この変異は、ハプテン特異的B細胞の親和性成熟に重要であることが以前に示されている(Allen, D. et al., EMBO J. 1995-2001 (1988))。
【実施例3】
【0109】
B細胞特異的GANP欠損マウス(B-GANP-/-マウス)の作製
(方法)
1.CD19-Cre/+GANP flox マウスの樹立
GANPゲノムDNAを用いて、エクソンIIの下流にネオマイシン耐性遺伝子(neo)を挿入することによってターゲッティングベクターを調製した。loxP部位は、neoの3’側の隣接領域とエクソンI及びIIの間のイントロンに導入した。
つまり、エクソンIIをloxP配列で挿んだfloxマウスを作製し、これとCD19-Creマウスとを交配させて、B細胞でGANPが欠損するマウスを樹立した(図11A及びB)。
直線化したターゲッティングベクターをTT2 ES 細胞(Yagi, T. et al. Anal. Biochem. 214, 70-76 (1993))にエレクトロポレーションによりトランスフェクションした。G418で選択後、ES コロニーを拾い上げ、プロテナーゼKと一緒にインキュベートした。相同組換え体を下記neo2プライマー及びCGK3'-2プライマーによりスクリーニングした。
【0110】
neo2プライマー:
5'-GCCTGCTTGCCGAATATCATGGTGGAAAAT-3' (配列番号9)
CGK3'-2プライマー
5'-GGCACCAAGCATGCACGGAGTACACAGA-3' (配列番号10)
【0111】
相同組み換えは、BamHIで消化したESクローンのDNAをプローブAを用いてサザンブロット分析することにより確認した。4kbのバンドを示す3個の陽性クローンを用いて、ICR胚盤胞へのマイクロインジェクションによりキメラ初代マウスを作製した。なお、B細胞でGANPの発現が見られないことは、サザンブロッティング、RT-PCRと細胞染色で確認した(図11C、D及びE)。
GANP flox/+マウスは少なくとも10回、C57BL/6マウスと戻し交配した。B細胞におけるGANP遺伝子を欠失させるために、GANP-floxed マウスとCD19-Cre ノックインマウス(Rickert, R. C., et al., Nucleic Acids Res. 25, 1317-1318 (1997))とを交配した。
【0112】
2.FACS分析
リンパ器官由来の単一細胞懸濁物を、各ビオチン標識モノクローナル抗体により氷上で1時間染色した。染色バッファーで洗浄後、細胞をFITC結合ストレプトアビジン (Amersham Bioscience)及びPE結合モノクローナル抗体で1時間インキュベートした。リンパ細胞を、CellQuestソフトウエアを用いて FACScan (Becton Dickinson)により分析した。
【0113】
3.B細胞の精製
脾臓細胞をCre-flox/+マウス及び B-GANP-/-マウス(7から8週齢) から単離し、0.15 M 塩化アンモニウム緩衝液で処理して赤血球を除いた。プラスチック皿上で37℃で30分間インキュベーションした後、未接着細胞をリンパ球として回収し、T細胞をDynabeads-anti-mouse Thy1.2 モノクローナル抗体(Dynal)を添付のプロトコールに従って使用して除去した。B細胞の純度(90%以上)をFITC結合抗B220モノクローナル抗体(BD Pharmingen)を用いた細胞表層染色により確認した。
【0114】
4.インビトロ増殖アッセイ
精製したB細胞を、10%熱不働化FCS(JRH Biosciences)、2 mMのL-グルタミン及び5×10-5 Mの2-メルカプトエタノールを含むRPMI-1640 培地(分裂促進剤を含むものと含まないもの)中において、96穴マイクロタイタープレートで2×105 細胞/ウエルで48時間インキュベートした。細胞を、0.2μCi/ウエルの[3H]-チミジン(ICN)で16時間パルスしてから回収し、取り込まれた放射活性をシンチレーションカウンターで測定した。
分裂促進剤は、アフィニティー精製したヤギ抗マウスμ鎖特異的抗体(10μg/ml)[F(ab')2] (ICN)、ラット抗マウスCD40モノクローナル抗体(LB429; 10μg/ml)及びLPS(Sigma; 10μg/ml)を用いた。
【0115】
5.抗原及び免疫
TNP-KLH、TNP-Ficoll及びニトロフェニル-ニワトリγグロブリン(NP-CG) (23:1)は、Biosearch Technologiesから購入した。50μgのTNP-KLH及びNP-CG (アルミニウムで沈殿)、又は25μgのTNP-Ficoll(PBSに溶解)をCre-flox/+マウス及びB-GANP-/-マウスの腹腔内に注入した。
【0116】
6.抗原特異的抗体産生の測定
抗原投与の10日後と14日後に、免疫したマウスから血清を回収した。5μg/ウエルのTNP-BSA (Biosearch Technology) をELISAプレートに被覆した。各ウエルをPBS中の3% BSAでブッロキングし、段階希釈した血清とインキュベートした。PBS-0.1% Tween 20で洗浄後、ウエルをビオチン結合isotype特異的モノクローナル抗体及びアルカリホスファターゼ(ALP)結合ストレプトアビジン(Southern Biotechnology)とともにインキュベートした。発色は基質の存在下で行った。
血清中のNP結合抗体の親和性を求めるために、NP2結合抗体のNP25結合抗体に対する割合を、被覆抗原にNP2-BSA(1分子あたり2個のNPがBSAに結合したもの)及びNP25-BSA(1分子あたり25個のNPがBSAに結合したもの)(Biosearch Technology)を用いたディファレンシャルELISAにより計算した。
【0117】
7.免疫組織化学
免疫したマウスからの8μmの脾臓切片をアセトンで軽く固定した。サンプルをPBS-Tween 20中の3% BSAでブロックし、抗IgDモノクローナル抗体及びALP-結合抗ラットIgG(ICN)抗体とともにインキュベートした。第一の発色はVector Blueキット(Vector)を用いて行った。第二の発色は、サンプルをビオチン結合ピーナッツアグルチニン(PNA)(Vecor)及び西洋わさびペルオキシダーゼ結合ストレプトアビジン(Kirkegaard & Perry)とともにインキュベートし、次に3,3'-ジアミノベンジジンテトラ塩酸塩(Dojindo)とともにインキュベートした。PBS中の1%グルタルアルデヒドを用いてサンプルを固定した後、Aquatex(Merck)によりマウンティングを行った。
【0118】
8.VH186.2 遺伝子の配列分析
NP-CGで免疫したCre-flox/+及びB-GANP-/-マウスからのNP-結合IgG1dullCD38low B細胞を、(4-ヒドロキシ-5-ヨード-3-ニトロフェニル)アセチル (NIP)を用いてFACS Vantage (Becton Dickinson Biosciences)で分画し、プロテナーゼKと一緒に37℃で一晩インキュベートした。ライセートを用いて、PCRを既報の通り(Takahashi, Y., et al. Immunity 14, 181-192 (2001))2回実施した。VH186.2 遺伝子DNAをpBluescriptにライゲーションし、自動シークエンサーにより配列を決定した。
【0119】
9.アポトーシス細胞の検出
Cre-flox/+及びB-GANP-/-マウスから精製したB細胞を40時間、種々の試薬で刺激した(Watanabe, N. et al. (1998) Scand. J. Immunol. 47, 541-547)。AICDには、24ウエルプレートに抗μ抗体(50μg/ml)を固定した。そのほかの場合には、精製したB細胞を種々の刺激物質で48時間刺激し、続いて抗Fasモノクローナル抗体(Jo2;BD Pharmingen)で4時間インキュベートした(Wang, J. et al. (1996) J. Exp. Med. 184, 831-838)。細胞を、プロピジウムアイオダイド(PI)溶液(50μg/ml PI, 0.1% Triton X-100, 0.1% クエン酸ナトリウム)を用い、室温で1時間インキュベートし、アポトーシス細胞(%)をFACScanでG1以下領域として計算した。なお、アポトーシス細胞は、トリパンブルーによる染色後、顕微鏡下での確認も行った。
【0120】
10.TUNELアッセイ
Cre-flox/+及びB-GANP-/-マウスをSRBC(ヒツジ赤血球細胞)で免役した後、それぞれの脾臓切片を調製し、PBS中の4%パラホルムアルデヒドで凍結切片を固定した。切片サンプルを、MEBSTAIN Apoptosis Kit II(MBL)で処理し、PIで対比染色した。TdT媒介dUTP-ビオチンニック-エンド標識(TUNEL)アッセイと共に行う実験用に、切片は抗IgG1 モノクローナル抗体(BD Pharmingen)及びAlexa546-結合ヤギ抗ラットIgG抗体(Molecular Probes)を用いた染色も行った。陽性シグナルを検出し、結果を蛍光顕微鏡(BX51; Olympus)により確認した。
【0121】
11.結果
(1) RNAプライマーゼGANPの役割
RNAプライマーゼGANPの役割を調べるために、Cre-loxP システムを用いてCD19+ B細胞にしてGANP遺伝子を欠失させた B-GANP-/-マウスを作製した(図11A及びB)。B-GANP-/-マウス遺伝子は、エクソンIIをほとんど欠損していた(図11C)。B-GANP-/-細胞はGANP mRNAを発現せず(図11D)、また免疫染色によればタンパク質をほとんど発現しなかった(図11E)。B-GANP-/-マウスは正常に成育し、骨髄、脾臓、胸腺、リンパ節で正常な数のリンパ細胞を示した。フローサイトメトリー分析では、B-GANP-/-は、骨髄、脾臓及びリンパ節の細胞上に、対照であるCre-flox/+マウスと同様の表面マーカープロフィールを示し(図12)、Cre-flox/+(対照)と差はなかった。
【0122】
B-GANP-/-のリンパ節では、sIgMlowsIgDhigh を発現する成熟B細胞(IgM+IgD+)の数が減少していた。B-GANP-/-マウス由来のB細胞は、インビトロで抗μ抗体、抗μ抗体+抗CD40モノクローナル抗体、またはリポ多糖による刺激後に通常の増殖応答を示した(図13:B-GANP-/-は白の棒、Cre-flox/+は黒の棒)。一方、B-GANP-/-マウス由来のB細胞は、抗CD40モノクローナル抗体(5, 10μg/mL)による刺激後に増殖活性が低下した(図13)。このことは、B-GANP-/-マウスのB細胞の増殖では、CD40/CD145相互作用への反応がわずかに損なわれていることを示す。血清Ig量はCre-flox/+マウスと同様であった(図14)。
【0123】
(2) B-GANP-/-マウスにおける抗原特異的抗体産生
TI-Ag及びTD-Agによる免疫後のB-GANP-/-マウスの免疫応答を調べた。TI抗原であるトリニトロフェニル(TNP)-Ficollを免疫して14日後に、抗TNP抗体価をELISAで測定した。その結果、(TNP)-FicollはB-GANP-/-マウス及びCre-flox/+マウスにおいて同様の応答を誘導し、特に差はなかった(図15)。
胚中心(GC)形成を調べてみると、TD-AgであるTNP-keyhole limpet hemocyanin (KLH)及びNP-CGなどのTD-Agsに応答して、変異体マウスはCre-flox/+マウスと比べて遅延したGC形成を示した。
【0124】
GC形成のピーク応答については、Cre-flox/+は、10日目にGC-B細胞のマーカーであるピーナッツアグルチニンで染色された大きな成熟したGCを示した(図16の矢印)。免疫後10日ではB-GANP-/-の方がGC形成がやや少なかった。しかし、14日後ではCre-flox/+に比べてB-GANP-/-の方が数が多くなっており、20日後でもGC形成が遷延していた(図16の矢印)。
B-GANP-/-マウスは14日目にGCの明白な形成を示したので、抗原特異的抗体応答を測定した(図17)。TD抗原であるTNP-KLHによる免疫では、B-GANP-/-マウスは10日目まで明確なGCを示さず、抗体価はほとんど差がなかったが、TNP-KLHによる免疫後の14日目には、B-GANP-/-でGCの段階的な増加と拡大を示した (図17)。変異体マウスはTNP-KLHに対してCre-flox/+マウスと同様の抗体応答を示した。
【0125】
(3) B-GANP-/-マウスにおける親和性成熟の障害
B-GANP-/-マウスにおけるGCの特徴(抗体応答が低親和性であること)をさらに調べるために、抗原特異的IgG1+ GC-B細胞をNP-CGによる免疫後に調べた。
異なる分子量を有するNPのハプテン/タンパク質コンジュゲートを用いたディファレンシャルELISAにより、マルチハプテンNP25-BSAコンジュゲートに対する応答と比較した。
B-GANP-/-マウスにおいて、NP2-BSAコンジュゲートに対する抗体応答は、NP-CG免疫後35日目において低親和性(13%)であった。この値は、Cre-flox/+マウスの値(42%)と比較して抗体反応は著しく減少していた(図18)。
【0126】
また、図19に示す通り、NP-特異的IgG1dullCD38low B細胞はB-GANP-/-マウスでは著しく減少した。すなわち、免疫後10日目において、Cre-flox/+マウスでは1,164細胞/106細胞であるのに対し、B-GANP-/-マウスでは88細胞/106細胞であった。また、14日目においては、Cre-flox/+が879細胞に対し、B-GANP-/-は83細胞であった。なお、この傾向は20日目も同様であった。
対照的に、IgG1highCD38high メモリーB細胞は減少しなかった。これらの結果は、GANP の無発現変異はIgG1highCD38low GC-B細胞段階でB細胞の分化に欠陥を生じさせていることを示す。
B-GANP-/-マウスにおける抗体の親和性成熟の減少を確認するため、NP-CGで免疫した後の脾臓B細胞のVH186.2領域の配列を調べた。
【0127】
SHMはB細胞分化のこの段階で生じるため、VH186.2 ローカスのSHMについて各種の精製したB細胞を調べた。なお、VH186.2 ローカスは、高親和性IgG (γ1λ1) NP-応答のために利用されている(Cumano, A. & Rajewsky, K. (1985) Eur. J. Immunol. 15, 512-520)。IgM ローカスはCre-flox/+と比較して差異がなかった。
次に、Cre-flox/+又はB-GANP-/-にNP-CGを免疫し、NP-結合IgG1弱陽性CD38弱陽性を示すGC-B細胞(Ag-結合IgG1 B細胞)についてソーティングした(図19)。また、ソーティング後の細胞からゲノムDNAを抽出し、VH186.2をPCRで増幅し、シークエンス解析を行い、VH186.2 の配列を比較した(図20A〜L)。図20A〜FはCre-flox/+のVH186.2 の配列を、図20G〜LはB-GANP-/-のVH186.2 の配列を比較したものである。
【0128】
B-GANP-/-マウスにおいて、IgV領域配列の全体の変異は14×10-3であり、Cre-flox/+マウス(21×10-3)と比較して変異の数が減少していた(図21)。さらに、W33からLへの高親和性型の変異(33番目のアミノ酸残基トリプトファンからリシンへの変異、C57BL/6 マウスで顕著に見られる)は13%(2/15 V領域)であり、変異型マウスでは、Cre-flox/+マウスの値(71%, 10/14 V領域)と比較してより顕著に減少し、親和性は1/3に低下した(図22)。
以上の結果より、GANPは抗体の親和性の成熟に必須であることが示された。
【0129】
(4) B-GANP-/-マウスB細胞におけるアポトーシスからの保護機能
B-GANP-/-マウスにおいて、高親和性抗体の産生が減少するのは、抗原刺激後のB細胞が不安定であるためであると考えられる。そこで、B細胞の感受性を調べるため、in vitroでのB細胞のアポトーシスを検討した。
正常なB細胞は、強力に架橋したB細胞抗原受容体により活性化誘導細胞死(AICD)が誘導され、AICDは、CD40の刺激によって阻止された。B-GANP-/- B細胞では、AICDの刺激に対する感受性は正常B細胞(対照)と同程度であったが、抗CD40を介するアポトーシスの抑制の程度については、Cre-flox/+対照B細胞よりも劣っていた(図23)。このことは、B-GANP-/-マウスは、GC形成期の間、抗原反応性B細胞の保護機能に欠けることを示す。
【0130】
GCでは、抗原刺激されたB細胞とCD40/CD154との相互作用によってFas/CD95の表面発現が誘導され、Fas誘導性アポトーシスに感受性を持つようになる。そこで、本発明者は、in vitroにおいてアポトーシスを誘導する抗CD95刺激に対するB-GANP-/-B細胞の感受性を測定した。
まず、脾臓B細胞を抗CD40モノクローナル抗体(LB429)、抗μ抗体+抗CD40モノクローナル抗体、IL-4+抗CD40モノクローナル抗体、及び抗μ抗体+IL-4+抗CD40モノクローナル抗体で48時間刺激し、次に抗CD95モノクローナル抗体を培地に添加した。
その結果、B-GANP-/-マウスのB細胞のアポトーシス応答は、Cre-flox/+マウスのB細胞の応答と同様であり、B-GANP-/-マウス及び Cre-flox/+マウスでは差がなかった(図24)。
【0131】
上記の通り、抗CD40(LB429)処理後に抗CD95で刺激しても、発現誘導は、B-GANP-/-マウス及び Cre-flox/+マウスでは差がなかった。このことは、B-GANP-/- マウスのB細胞においては、in vivoでGC-B細胞により通常受けるアポトーシス刺激に対して感受性を有することを示す。そこで、TUNELアッセイを、TD-AgとしてSRBCを用いて免疫した後の組織切片について行った。
その結果、TUNEL陽性細胞は、B-GANP-/-マウスのGC領域において増加しており、ほとんどの細胞は、IgG1を発現することも示された(図25、26)。これらの結果は、B-GANP-/-マウスのアポトーシス細胞は、ほとんどがGC-B細胞であることが示された(図25、26)
次に、種々の悪性リンパ腫細胞及び正常B細胞のCD40媒介性アポトーシス抑制に必要な分子であると認識されているBcl-2ファミリーメンバーのRNA発現を検討した。
【0132】
抗μ抗体+IL-4で刺激すると、Cre-flox/+ B細胞におけるbcl-2の転写の明らかな増加を誘導し、抗CD40 mAbはその発現をさらにアップレギュレートした(図27)。また、B-GANP-/- B細胞については、抗μ抗体によるbcl-2転写物と同様のアップレギュレーションを示したが、抗CD40 mAb(抗CD40 mAb単独又は抗μAb + IL-4 + 抗CD40 mAb)に対する応答は、Cre-flox/+のB細胞ほど高くはなかった(図27)。すなわち、アポトーシス抑制に関与するBcl-2ファミリーのRNA発現レベルは、抗CD40抗体で刺激したときのB-GANP-/-マウスB細胞において、対照よりも低下していた(図27)。
【0133】
変異B細胞中のbcl-XL、bax及びbadについては、Cre-flox/+ B細胞のレベルと同程度であった(図27)。
以上の結果は、GANPは、GC-B細胞においてCD-40を介したBcl-2の誘導に関する情報伝達を制御しており、in vivoでの高親和性BCR+ B細胞の生存に大きく寄与していることを示す。
【0134】
(5) まとめ
B-GANP-/-マウス及びGANP-Tg マウスの結果は、GANPがTD-Agによる免疫後における高親和性B細胞の生成に関与していることを実証している。GANPの役割としては、GC-B細胞でDNAポリメラーゼの効率的なリクルートと調節を仲介している可能性がある。V-領域SHMを有する GC-B細胞が高親和性BCRを取得すると、それらは選択されるべきであり、そしてさらにB細胞のV領域のSHM は抑制されて、インビボで高親和性抗体の産生が保証されるはずである。GC-B細胞でのAIDの発現はDNA変異を絶えず生成する可能性があるため、AID活性の調節がB細胞での高親和性BCRを維持するために必要であるかもしれない。B-GANP-/-マウスでの結果から、GANPがSHMプロセスに必要であることが示される。
【実施例4】
【0135】
GANPトランスジェニックマウスを用いた高親和性抗体の産生
1.ディファレンシャルELISAによる抗体価の比較
野生型(WT)マウスとGANPトランスジェニック(Tg)マウスの2匹ずつにNP-CG 100 μgを免疫し、28日後の血清を取り、ELISAを行った。20μg/mlのNP2-BSAとNP17-BSAをELISAプレートに4℃で1晩コーティングした。3% BSA/PBS-0.1% Tween20で1時間ブロックし、血清を1時間反応させた。PBS-0.1%Tween20で3回洗い、ビオチン標識抗マウスIgG1抗体(Southern Biotechnology)で1時間反応させた。PBS-0.1%Tween20で3回洗い、アルカリホスファターゼ標識ストレプトアビジン(Southern Biotechnology)で30分反応させた。PBS-0.1%Tween20で3回、TBSで1回洗い、p-Nitrophenyl phosphateを基質として発色させた。吸光度を405nmで測定した。
結果を図28に示す。図28の結果から、GANPトランスジェニックマウスを使用することにより高親和性抗体が産生されることが分かる。
【0136】
2.ELISA及びBiacoreを用いた抗原-抗体結合親和性解析
野生型(WT)マウスとGANPトランスジェニック(Tg)マウスに、免疫抗原としてNP-CGを免疫し、それぞれのマウスを用いて細胞融合を行い、得られた陽性ハイブリドーマの培養上清において、ELISA法及びBiacoreを用いて抗原と反応する抗体の結合曲線を得た。得られた結合曲線から、Tgマウスの有用性を導いた。
(1) 材料
(a) 動物
野生型(WT)マウスとGANPトランスジェニック(Tg)マウス。
(b) 抗体及び試薬
NP16-CG(一分子あたり16個のNPがCGニワトリイムノグロブリンに結合したもの)、NP2-BSA(一分子あたり2個のNPがBSA(牛血清アルブミン)に結合したもの)、NP17-BSA(一分子あたり17個のNPがBSAに結合したもの)、HRP標識抗マウスIgG、IgA及びIgMを用いた。
【0137】
(2) 方法及び結果
野生型(WT)マウスとGANPトランスジェニック(Tg)マウスの各5匹に、免疫抗原としてNP16-CGを、2週間をおきに3回免疫し、3回免疫後に採血し、抗血清を用いて抗体価の比較をしたところ、前記1項に記載の結果と同様にGANP-Tgマウスの有用性が確認された。
この中から、力価の高いマウスの脾細胞を用いて、P3U1ミエローマ細胞と細胞融合を実施し、GANP-Tgマウスの脾細胞数(6.0×107個)、WTの脾細胞数(4.8×107)に基づき、1×105/ウェルになるようにまきこみ、GANP-Tgマウス由来の細胞は600クローン、WTマウス由来の細胞は480クローンをHAT培地にて培養した。
HAT培養9日後の培養上清を用いて、NP2-BSA(1μg/mL)を固相化抗原としてELISA法を実施した。GANP-TgマウスおよびWTマウスの培養上清のそれぞれから、ELISA吸光度結果において高親和性抗体の産生を示す上位2.5%のクローンを選出し、HT培地にてクローニングを実施した。
【0138】
HT培養9日後の培養上清を用いて、NP2-BSA(1μg/mL)を固相化抗原としてELISA法を実施した結果、GANP-Tgは6クローン(クローン名:G2-6, G2-9, G2-12, G2-14, G2-15, G2-16)、WTは1クローン(クローン名:W2-7)のハイブリドーマを樹立した。
GANP-Tgマウス、WTマウスのそれぞれのクローンをRPMI培地にて培養し、以下の実験に用いるのに適した培養上清を1mLずつ確保した。この培養上清を用いて、以下の評価検討を実施した。
【0139】
(a) ELISA法
抗体価を評価するにあたり、抗原の性質の異なるもの、いわゆる、CG一分子あたりNP含有率の異なる物質を用いて、そのELISA反応性の比率をもって評価した。
本法は、NPの親和性を測るのに有用な測定法であり、簡便で多くの検査をすることができるので、一次スクリーニングとして適当で信頼できるものである。
【0140】
まず、NP2-BSA(1μg/mL)とNP17-BSA(1μg/mL)をそれぞれ固相化抗原として用い、4℃で一晩固相化した。固相化プレートをPBS-Tween20で洗浄した後、スキムミルクPBS-Tween20にてブロッキングを実施した。さらにプレートをPBS-Tween20で洗浄した後、GANP-Tgマウスの6クローン(クローン名:G2-6, G2-9, G2-12, G2-14, G2-15, G2-16)及びWTマウスの1クローン(クローン名:W2-7)のRPMI培養上清(原液〜256倍希釈液)を用いて、室温で1時間固相化抗原と反応させた。次に、プレートをPBS-Tween20で洗浄した後、HRP標識抗マウスIgG、IgA及びIgMを室温で1時間反応させた。さらに、プレートをPBS-Tween20で洗浄した後、オルトフェニレンジアミン(OPD)にて5分間発色し、2N硫酸を用いて反応を停止した。
吸光度は、ELISA READERを用いて、490nmにて測定した。
ELISAの結果を図29に示す。図29の結果よりGANP-Tgマウスを使用することにより、高親和性抗体が産生されることがわかる。
【0141】
(b) Biacoreを用いた高親和性解析
上記ELISAの結果で、一番親和性の高いと予想されるクローンについて、Biacoreを用いて物理化学的結合能を調べた。
Biacoreによる解析は、NP2-BSA(1μg/mL)をリガンドとしてBiacoreチップに結合させたものに、アナライト溶液として一番親和性の高いと思われるTg(G2-9)、一番低いと思われるTg(G2-15)、及びWT(W2-7)の3クローンのRPMI培養上清を用いて、それぞれの結合速度定数(k ass)、解離速度定数(k diss)、及び親和性の指標となる解離定数KD (KD = k diss/k ass)を算出した。KDが小さい程、親和性は高いと評価される。
【0142】
その結果として、G2-9(ELISA: 82.9% NP2/NP17比)のビアコアのパターンを図30に示す。図30に示す曲線(a)〜(e)は、抗体濃度がそれぞれ26.6、13.3、6.65、3.33及び1.66 nMのものである。上記結果から、結合速度定数(k ass)=1.48×105、解離速度定数(k diss) = 9.63×10-4、そして親和性の指標となる解離定数は、KD (KD = k diss/k ass)=6.50×10-9となった。
一方、ELISAの結果から比較的親和性の低いと予想されるG2-15(ELISA: 33.9% NP2/NP17比)のビアコアのパターンを図31に示す。図31に示す曲線(a)〜(e)は、抗体濃度がそれぞれ23.0、11.5、5.75、2.88及び1.44nMのものである。
【0143】
結合速度定数(k ass) = 5.33×104、解離速度定数(k diss) = 1.56×10-2、そして親和性の指標となる解離定数は、KD (KD = k diss/k ass)=2.92×10-7となった。この値は、ELISA法でも同等の親和性を示すW2-7(ELISA: 31.6% NP2/NP17比)の活性を持つKD値=1.67×10-7と近似していた。
以上をもって、GANPトランスジェニック(Tg)マウスを使用することにより、高親和性抗体が産生されることが明らかである。
【実施例5】
【0144】
GANPとMCM3の会合、細胞周期中の核/細胞質間の移動
1.概要
本実施例では、本発明者は部分切断型の変異体GANPを用いることによってGANPのMCM3結合領域を決定し、GANP特異的な領域における502番目のアミノ酸のリン酸化セリン(pSer502)に対する特異的なモノクローナル抗体を用いてNIH-3T3細胞におけるGANPの局在を特徴づけた。
プライマーゼとMCMとの結合は、一つの連動した機能であり、両者が結合した分子複合体はDNAを二重鎖から解く作用を有する。従って、GANP部分断片がMCMと結合すれば、当該GANP断片もプライマーゼ活性を発揮し、高親和性抗体の産生作用を有するものと考えられる。
そこで、GANP及びその部分断片、Map80及びMCM3の核/細胞質区画の局在を、cDNAトランスフェクションと細胞融合実験を用いて解析した。
得られたデータから、GANPはMCM3と結合することが示され、さらに、GANPの局在はMCM3の発現によって影響されることが示された。GANPはMap80の結合機序とは異なる結合機序によってMCM3と会合する。これらの結果は、MCM3に結合したGANPは、Map80/MCM3APの機能とは異なる独特の機能を介することを示す。
【0145】
2.材料および方法
2.1. 細胞および細胞培養
NIH-3T3、COS7、HelaおよびSwiss-3T3細胞を、10%熱不活化FCS(大日本製薬)、2 mM L-グルタミン(Biowhittaker)、100 μg/mlストレプトマイシン、100 U/mlペニシリン及び50 μM 2-メルカプトエタノールを補充したD-MEM培地(Invitrogen)中で、37℃で、5% CO2のもとで維持した(Takei, Y. et al., (1998) J. Biol. Chem. 273, 22177-22180、 Sakaguchi, N. et al., (1988) EMBO J. 7, 3457-3464、Kimura, H. et al., (1995) Nucl. Acids Res. 23, 2097-2104)。BAL17はRPMI-1640培地(Invitrogen)中で培養した。
【0146】
2.2. リン酸化GANPおよびMCM3の細胞内局在化
NIH-3T3をPBS (pH 7.4)中で3.7%パラホルムアルデヒドで5分間固定し、0.2% Triton X-100を用いて透過性とした(Kimura, H. et al., (1994) EMBO J. 13, 4311-4320)。ラット抗pSer502 GANPモノクローナル抗体(Kuwahara, K. et al., (2001) Proc. Natl. Acad. Sci. USA 98, 10279-10283)及びウサギ抗MCM3抗体Kimura, H. et al., (1994) EMBO J. 13, 4311-4320を一次抗体として使用した。次に二次抗体として、GANPに対してはAlexa488結合ヤギ抗ラットIgG抗体(Molecular Probes)を使用し、MCM3に対してはAlexa546結合ヤギ抗ウサギIgG抗体(Molecular Probes)を使用して、ヨウ化TOTO-3 (Molecular Probes)で対比染色し、そして共焦点レーザー顕微鏡(FV500; オリンパス)によって観察した。
【0147】
2.3. 発現のためのcDNA構築物
pSRα-MCM3-HAは文献に記載されている(Kimura, H. et al., (1995) Nucl. Acids Res. 23, 2097-2104)。SV40 T-Agの3つの核局在化シグナル(NLS)を担持するpECFP-NucベクターはClontechより購入した。下記の組み合わせの3'及び5'プライマーを用いて得たPCR断片をpGEX-4T-1 (Amersham)に導入し、それらを用いて異なる形のマウスganp cDNAをグルタチオンS-トランスフェラーゼ(GST)との融合タンパク質として調製した。
【0148】
GANP1-5':5'-GGGGATCCATACCCGG TGAACCCCTT-3' (配列番号11)
GANP1-3':5'-GGGTCGACGCGCACAGACTTTCCCCTGA-3' (配列番号12)
GANP2-5':5'-GGGAATTCTCCCGCCTTCCAGCTGTGAC-3' (配列番号13)
GANP2-3':5'-GGGTCGACGTGCTGCTGTGTTATGTCCT-3' (配列番号14)
GANP3-5':5'-GGGAATTCCATGAGCT GAGACCCTCAGC-3' (配列番号15)
GANP3-3':5'-GGGTCGACTGAGGATGCAGGAGGCGGCT -3' (配列番号16)
GANP4-5':5'-GGGAATTCTACGTTGGAGAGAGCCTGGC-3' (配列番号17)
GANP4-3':5'-GGGTCGACCATGCTGTCATCTCCTGTGA-3' (配列番号18)
GANP5-5':5'-GGGAATTCGAGAA CCTGGCCAAGGGTCT-3' (配列番号19)
GANP5-3':5'-GGGTCGACGAAAAACCGACGGCTGA ACT-3' (配列番号20)
GANP6-5':5'-GGGAATTCAAGCCCTTCCAGCCTGCCCT-3' (配列番号21)
GANP6-3':5'-GGGTCGACCGAGGGAACGTGGTATTTTC-3' (配列番号22)
GANP7-5':5'-GGCCCGGGCC CGTGGGATGACATCATCA-3' (配列番号23)
GANP7-3':5'-GGCTCGAGCATGTCCACCATCTC CAGCA-3' (配列番号24)
【0149】
cDNA構築物は、PCR断片をpSVEGFP pA に導入することにより緑色蛍光タンパク質(GFP)をタグしたGanp変異体として調製した(Kuwata, N. et al., (1999) J. Immunol. 163, 6355-6359)。次に、これらの構築物をpCXN2哺乳動物発現ベクター中に導入した(Niwa, H. et al., (1991) Gene 108, 193-200)。プライマー配列は、Ganpをコードするように下記の通り設計した。
【0150】
Gp-gfp-5':
5'-GGGGATCCGAATTCCACCATGGCAGTCTTCAAACCGATA CC-3' (配列番号25)
Gp-gfp-3':
5'-GCAGGGGCTCCTCCTGATCT-3' (配列番号26)
Gsac-gfp-5':
5'-GGGGATC CGAATTCCACCATGTCCGAGGGCCTTGGTTCTTG-3' (配列番号27)
Gsac-gfp-3':
5'-CTGTCTT GTTTCTAAGCCGC-3' (配列番号28)
Gmap80-gfp-5':
5'-GGGGATCCGAATTCCACCATGGAGA ACCTGGCCAAGGGTCT-3' (配列番号29)
Gmap80-gfp-3':
5'-GAGGACTTGTAGATGTTTTCAC CATGG-3'(配列番号30)
【0151】
FLAGをタグしたGanp変異体については、以下のプライマーを用いてPCRにより得たcDNA断片をpCXN2に導入した。:
【0152】
FLAG-Gp-5':
5'-GGGAATTCCACCATGGATTACAAGGATGACGACGATAAGG CAGTCTTCAA CCGATACC-3' (配列番号31)
FLAG-Gp-3':
5'-GGGAATTCCTCCGGGTCTCCCTCAAGTA-3' (配列番号32)
FLAG-Gsac-5':
5'-GGGAATTCCACCATGGATTACAAGGATGACGACGATAAGTCCGAGGGCCTTGGTTCTTG-3' (配列番号33)
FLAGGsac-3':
5'-GGGAATTCGCTGTCTTGTTTCTAAGCCG-3' (配列番号34)
FLAG-Gmap-5':
5'-GGGAATTCCACCATGGATTACAAGGATGACGACGATAAGG AGAACCTGGCCAAGGGTCT-3' (配列番号35)
FLAG-Gmap-3':
5'-GGGAATTCTGAGGACTTG TAGATGTTTT-3' (配列番号36)
【0153】
内部欠失変異体GpΔNLS-GFP および I3変異体(MCMΔNLS-HA) は、文献に記述されているように調製した(Imai, Y. et al., (1991) Nucl. Acids Res. 19, 2785-2785)。全ての構築物は、ヌクレオチド配列の決定により配向が正しいこと、およびタグを付けた融合タンパク質としてコドンの読み枠が正しいことを確認し、品質を管理した。変異体RNA/DNAプライマーゼドメイン(PD) を有する発現ベクターは文献に記載されている(Ser502からAla [GpS502A]又はGlu [GpS502E]までのGp変異体)(Kuwahara, K. et al., (2001). Proc. Natl. Acad. Sci. USA 98, 10279-10283)。
【0154】
2.4. 導入した遺伝子産物の共焦点顕微鏡検査による検出
FuGENE 6 (Roche Diagnostics)を用いて、pCXN2-ganp-gfpおよび/またはpSRα-MCM3-HAによってNIH-3T3をトランスフェクトした。固定化の16時間前にレプトマイシンB (LMB;(Kudo, N. et al., (1999) Proc. Natl. Acad. Sci. USA 96, 9112-9117))を培養培地に添加した。共トランスフェクションの場合にはウサギ抗HA抗体 (Santa Cruz)およびAlexa546結合ヤギ抗ウサギIgG 抗体を用いて、そして単独トランスフェクションの場合には Alexa488結合ヤギ抗ウサギIgG 抗体 (Molecular Probes)を用いて、外因性MCM3タンパク質を染色した。核酸は、共トランスフェクション実験についてはヨウ化TOTO-3を用いて、単独トランスフェクション実験についてはヨウ化プロピジウム(PI; Sigma)を用いて対比染色した。
【0155】
2.5. GSTプルダウンアッセイ
GST融合タンパク質を文献に記述されているように精製した(Kuwahara, K. et al., (2000) Blood 95, 2321-2328)。グルタチオン-Sepharoseビーズ(Amersham)に固定した種々のGST融合タンパク質(5 μg)を、TNEバッファー(10 mM Tris-HCl [pH 7.8]、150 mM NaCl、1 mM EDTA、1% Nonidet P-40、10 μgアプロチニン、1 mMフェニルメチル-スルフォニルフルオリド[PMSF])を用いて調製したBAL17溶解物と共にインキュベートした。結合タンパク質を8% SDS-PAGEによって分離し、ニトロセルロースフィルターに転写し、ブロックした。次にウサギ抗マウスMCM3抗体(Kimura, H. et al., (1994) EMBO J. 13, 4311-4320)及びペルオキシダーゼ標識プロテインA (Amersham)と共に連続的にインキュベートし、最後にECL検出キット(Amersham)を用いてシグナルを可視化した。直接結合アッセイのためには、 [35S-メチオニン] (Amersham)を用いて、in vitro転写および翻訳結合システム(Novagen)を製造者の説明書に従って使用して放射性標識化MCM3を合成し、[35S]-標識MCM3をオートラジオグラフィーで検出した。
【0156】
2.6. 導入した遺伝子産物の免疫沈降およびウエスタンブロッティング
FuGENE 6を用いて、pCXN2-FLAG-ganpおよび/またはpSRα-MCM3-HAによってCOS7をトランスフェクトした。26時間後、TNEバッファーを用いて細胞を溶解し、得られた溶解物をプロテインA-Sepharose (Amersham)と組み合わせた抗HA抗体と共にインキュベートした。免疫沈降物を8% SDS-PAGEによって分離し、ニトロセルロースフィルターに転写し、ブロットをブロックし、マウス抗FLAG M2抗体(Stratagene)と共にインキュベートし、次にペルオキシダーゼ標識ヤギ抗マウスIgG (H+L)抗体(Zymed) と共にインキュベートした。Gp-GFPおよびその変異体の検出のためには、ブロッティングしたフィルターをウサギ抗GFP抗体(Santa Cruz)およびペルオキシダーゼ結合プロテインA(Zymed)により釣り上げた。
【0157】
2.7. ヘテロカリオンアッセイ
FuGENE 6を用いて、pSRα-MCM3-HAによってHela細胞をトランスフェクトした。20時間後、トランスフェクトしたHeLa細胞およびトランスフェクトしていないマウスSwiss-3T3細胞をトリプシン処理し、1:1の比で共に培養皿に播いた。24時間後、ポリエチレングリコール1500 (Roche diagnostics)を室温で2分間用いて細胞を融合させた(Schmidt-Zachmann, M.S. et al., (1993) Cell 74, 493-504)。培養皿を培地で4回洗浄した後、シクロヘキシミド含有培地を添加し(最終濃度20 μg/ml)、細胞をCO2インキュベーター中で 37℃で5時間インキュベートした。次に、250 mM HEPES-NaOH (pH 7.4)中で4%パラホルムアルデヒドを用いて細胞を20分間固定し、PBS中で0.5% Triton X-100を30分間用いて透過性とし、PBSで洗浄した。抗HA抗体(12CA5; Covence Research Products)およびCy3結合ロバ抗マウスIg抗体(Jackson)を用いて細胞を染色した。また、PBS中で100 ng/ml Hoechst 33342 (Sigma)を用いてDNAを20分間対比染色した。100x PlanNeofluar 位相差対物レンズ (NA 1.3) およびSpotII CCDを備えたZeiss Axioplanを用いて画像を集めた。
【0158】
3.結果および考察
3.1. GANPとMCM3の会合
これまでに、B細胞系統におけるGANPとMCM3の相互作用が、共免疫沈降によって実証されている(Kuwahara, K. et al., (2000) Blood 95, 2321-2328)。GANPのC末端領域は全Map80タンパク質と同一であるため、GANPは同一領域でMCM3と会合すると予想される。本発明者は、どの領域がMCM3と直接会合するのかを決定するため、図32に示す種々の末端切断型GANPを有するGST融合タンパク質を用いてプルダウンアッセイを実施した。GSTを図32に示される1から7及び5-7'と称した切断型GANPのN末端に融合した。図33の下のパネルにおいて、GANP5-7'と称するMap80領域は、以前に示されたように(Kimura, H. et al., (1994) EMBO J. 13, 4311-4320)、細胞抽出物からMCM3をプルダウンした。
驚くべきことに、GANPの部分断片であるGANP1およびGANP2もまたMCM3をプルダウンした(図33;上および下パネル)。
【0159】
次に、網状赤血球溶解物系により、in vitroで合成したMCM3を用いてこの結合を調べた(図34)。GST-GANP1 およびGST-GANP2 もまたin vitro翻訳カクテルから[35S]-MCM3 をプルダウンした。
GST単独を用いた陰性対照(最初のレーン)、又は、無関係なタンパク質と融合したGSTは、シグナルを示さなかった。そして、GST-GANP1との結合はMap80 領域 (GST-GANP5-7')との結合よりも強かった。この結合は、FLAGをタグした構築物を用いたDNAトランスフェクション実験により細胞においても確認された(図35)。
COS7細胞をpSRα-MCM3-HA又はpSRa-I3-HAと組み合わせて、pCXN2-FLAG-Ganp、pCXN2-FLAG-Gp及びpCXN2-FLAG-Gmap80によって共トランスフェクトした。抗HA抗体を用いた免疫沈降の後、抗FLAGモノクローナル抗体でウエスタンブロットを行った。FLAG標識タンパク質の予想サイズを、それぞれのレーンにおいて三角で示す。左及び右のパネルは、バンドの移動度(migration)が同様であるが、ECL検出の露光時間は左のパネルが1分、及び右のパネルは3分である(図35)。
【0160】
FLAG-Ganp、FLAG-Gp およびFLAG-Gmap80は野生型MCM3-HA (HAエピトープをタグしたMCM3)と結合した(図35; 左パネル)。FLAG-Gsac のみが結合しなかった。I3 変異体MCM3 (MCM3△NLS)との結合に関しては、FLAG-Gmap80 のみが陽性の結果を示した(図35;右パネル)。N末端NLSを担持するGp領域は、大量のMCM3を有する細胞において一貫してMCM3と会合している(図35;左パネル)。これらの結果は、GANPがGp領域を介してMCM3のNLS領域と会合していることを示している。
【0161】
本発明者はさらに、Gp領域のSer502のリン酸化の状態がMCM3との結合に影響するかどうかを検討した。プライマーゼ部位を欠損したGanp変異体(Ganp△PD-GFP)、及びGanpS502A又はGanpS502Eとして調製されるSer502におけるGanp変異体を、GFPと融合した(図36A)。細胞をpCXN2-Ganp-gfpとpSRα-MCM3-HAによって共トランスフェクトし、溶解液は抗HA抗体を用いた免疫沈降に用いた。
GFPシグナルは抗GFP抗体で検出され(図36B;上パネル)、このことは、GANPがMCMに結合したことを意味する。
Ganp-GFP変異体との共トランスフェクションも同様に行った。それぞれのタンパク質の予想位置を決定するために、溶解液をSDS-PAGE上で分離し、同じように抗GFP抗体でブロットした(図36B;下パネル)。
【0162】
非リン酸化性の変異体(GanpS502A-GFP)は、野生型Ganp-GFPまたはホスホセリンに良く似た変異体(GanpS502E-GFP)と同じくMCM3と結合した(図36B; 上パネル)。興味深いことに、Ganp△PD-GFP はMCM3-HAと共沈降しない (図36B; 上パネル)。
GANP分子全体としては、Map80領域の潜在的結合活性に関わりなく、MCM3との結合にはRNAプライマーゼ領域 (PD)が必要である。白抜き三角はGanp△PD-GFPの位置を示す。Ganp S502A-GFP およびGanp S502E-GFPのサイズと等しいGanp-GFPのサイズを黒三角で示す(図36B;下パネル)。これらの結果は、GANPのMCM3との結合はそのPD領域を介するが、Ser502 部位におけるリン酸化はこの結合に影響しないことを示している。
【0163】
末端切断型構築物を用いた実験によってGANPとMCM3との会合が広い領域にわたって示されたが、GANPのN末端の600個のアミノ酸領域が関与する全GANPとの結合にはMCM3のNLSが必要とされる。NLSを欠くMCM3変異体は、細胞中で全GANP分子と効果的に会合できなかった。Map80領域はNLS陰性MCM3と結合し、このことはGANPのMCM3との結合が主としてMap80との相互作用に必要とされる領域以外との結合であることを示している。Map80はMCM3インポート因子であると考えられているが、GANPはMCM3と共同して異なる役割を果たすのかもしれない。GANPは可能性のあるリン酸化部位を多数有し、そして細胞中に多くの会合成分をもつように思われる(Kuwahara, K. et al., (2000) Blood 95, 2321-2328)。したがって、その領域のリン酸化の状態がGANP/MCM3会合および細胞質と核コンパートメント間の輸送の調節に影響を及ぼす領域を特定することが必要であろう。
【0164】
3.2. トランスフェクションによって示されるMap80およびGanp変異体の細胞内局在化
GANPは2つの可能性のあるNLSを有する。1つはN末端プライマーゼ領域内にあり、もう1つはC末端Map80領域内にある。また、GANPは2つの核輸出シグナル(NES)様モチーフを有する。1つはSAC3相同領域とMap80領域との間にあり、もう1つはMap80領域内にある。
NIH-3T3細胞をpCXN2-Ganp-gfp又はpCXN2-Gmap80-gfpによってトランスフェクトし、48時間後に固定した。LMBを、固定の16時間前に添加した。核をPIで前染色し、共焦点顕微鏡を用いて画像を集積した。代表的な発現特性を図37に示す。異なる特性を持つ細胞数を計数し、パーセントとして表した(図37)。
【0165】
Ganp-GFP(GFPでタグしたほぼ全長GANP)は、核優性発現(NおよびN>C; 38%)又は細胞質優性発現(CおよびC>N; 31%)を示す細胞の割合に変動はあったが(合計500個の細胞から任意に計算された数の%)、細胞質および核コンパートメントの両方に存在することが見いだされた (図37; Ganp-GFP, LMB -)。対照的に、Gmap80-GFP は殆ど細胞質に見出され、発明者の分類による核優性発現は示さなかった (N>C, 0%; N=C, 35%; C and C>N, 65%)(図37; Gmap80-GFP, LMB -)。Ganp-GFPの局在化は、Gmap80-GFPの局在化とは異なる。
N末端NLSモチーフが機能性であるかどうかを確かめるため、RNA/DNAプライマーゼドメインおよびN末端NLSを担持する (ただしNES様モチーフは含まない) 5' 1-kb DNA 断片をGFPと融合させた (図38, Gp-GFP)。この Gp-GFP 産物は 核にのみ存在したが (NおよびN>C, 94%)(図38)、NLSを欠く変異体Gp-GFP (Gp△NLS-GFP; 図38に示すようにアミノ酸497から500が欠失している)は細胞質性であることを示し、N末端NLSが核局在化に関与していることが確認された。
【0166】
本発明者は、隣接するSer502 のアラニンへの突然変異(GpS502A-GFP; 非リン酸化型) 又はグルタミン酸への突然変異(GpS502E-GFP; ホスホセリンを模倣する型)がこの局在化に影響を及ぼすかどうかを検討した(図38)。そして、本発明者は、これらの突然変異がGpの局在化を変えないことを観察した。これは、N末端NLSがSer502 のリン酸化状態に関わりなく機能性であることを示唆している(図38)。対照的に、N末端NLSもC末端NLSも持たないGac-GFPは、殆どが細胞質中に存在するように思われる(N及びN>C, 0%; N=C, 3%; C及びC>N, 97%)(図38)。
これらの結果は、N末端NLSがGanpの核への移入に機能的な役割を有することを示唆している。しかし、NLSはGANP発現を核内に維持するほど強くはないのかもしれない。なぜなら、Ganp-GFPは細胞質中にも存在するからである(図37)。この問題をさらに検討するため、Crm1によって媒介される核への輸出を抑制するため、cDNAトランスフェクション後、細胞をレプトマイシンB (LMB)で処理した(Kudo, N. et al., (1999) Proc. Natl. Acad. Sci. USA 96, 9112-9117)。
【0167】
LMBで処理した細胞では、殆どのトランスフェクタントにおいてGanp-GFPは核に局在化した(図37)。優性な細胞質発現を示す細胞画分は31%から4% に減少し、対照的に、核において優性な発現を示す細胞が38%から81%に増大した。従って、Ganpの細胞質への移行はLMBによって抑制されるように思われる。
Gmap80-GFPの局在化もまたLMB処理後、劇的に変化した(図37)。すなわち、細胞質発現を示す細胞画分が65%から37%に減少し、優性な核発現を示す細胞画分が0%から41% に増大した。これらの所見は、COS7およびLtk- 細胞を含む他の細胞系においても再現され、核から細胞質へのGANP輸出がCrm1依存性経路によって調節されていることを示している。従って、GANP及びMap80は核と細胞質の間をシャトリング(往復)しているように思われ、そしてそれらの局在化は他の分子と共同している核移入および輸出機構の間のバランスに依存するようである。
【0168】
3.3. 共トランスフェクトした細胞におけるMCM3およびGANPの局在化
次に、本発明者はGANPの移行がMCM3発現と関連しているかどうかを検討した。哺乳動物のMCM3は、細胞周期中にクロマチンとの結合状態を変化させるが、間期の間を通して核にのみ存在する(Kimura, H. et al., (1994) EMBO J. 13, 4311-4320)。NIH-3T3細胞を、pSRα-MCM3-HA又はpSRα-I3-HAによってトランスフェクトし、固定し、抗HA抗体(Alexa488)で免疫標識し、PIで染色した。
トランスフェクトしたMCM3-HAは、典型的なNLSの存在と合致して(Kimura, H. et al., (1994) EMBO J. 13, 4311-4320、Takei, Y. et al., (1998) J. Biol. Chem. 273, 22177-22180)、核に局在化した(図39)。この核局在化はMCM3のNLSに依存した。なぜなら、このNLSを欠く変異体MCM3 (I3; MCM3△NLS-HA)は、細胞質にのみ発現されたからである (図39; 右パネル)。
【0169】
細胞を、pCXN2-Ganp-gfp又はpCXN2-Gmap80-gfp及びpSRα-MCM3-HAによって共トランスフェクトし、固定し、抗HA抗体(Alexa546)で免疫標識し、核をTOTO-3で前染色した(図40)。細胞数はパネルの下に示す(図40)。
興味深いことに、Ganp-GFP を用いて共トランスフェクションした場合、細胞の17%においてMCM3の細胞質局在化がもたらされた(図40, 白い矢印で示す)。Gmap80-GFPまたはGp-GFPを用いた共トランスフェクションの場合には、そのような結果はみられなかった。
MCM3へのGanpの効果が特異的であることを証明するために、核において出現するECFP-Nucの発現を、異なるganp-gfp構築物のトランスフェクション前後で調べた。Ganp-GFPでトランスフェクトした代表画像を示す(図41)。ECFP-Nucの核への局在化は、Ganp-GFP (図41)又はGmap80-GFP又はGp-GFPを用いた共トランスフェクションのいずれによっても影響されなかった。
【0170】
Ganp及びMCM3の共発現もまたGANP局在化の変更をもたらした。Ganp-GFP 単独(38%)及びGmap80-GFP単独(0%)のトランスフェクション(図37)と比較して、MCM3を用いた共トランスフェクションはGanp-GFP (74%)およびGmap80-GFP (64%) の核発現レベルを増大させた(図40)。MCM3は、GANPおよびMap80を核内に保持したが、Ganpの過剰発現のみがMCM3 の細胞質における出現を促進した(図40; Ganp-GFP発現によって17%)。他方、Gmap80 MCM3 の細胞質における出現を促進しなかった(Gmap80-GFP発現によって4%)。MCM3のNLSにおける突然変異(I3; MCM3△NLS-HA) (その結果、MCM3は細胞質に存在することになる)は、Ganp-GFPまたはGmap80-GFP の核への蓄積を引き起こさなかった(図42)。
野生型MCM3とは異なり、I3 変異体 (MCM3△NLS-HA) はGanp又はGpと会合しない(図35)という事実を考え合わせるならば、MCM3のNLSモチーフは、細胞においてGANPとの機能的会合および核と細胞質間のGANPの輸送に必要であることが示唆される。
【0171】
DNAトランスフェクション実験は、MCM3と共に導入された場合、Ganp-GFPが核コンパートメントに蓄積することを示し、核におけるGANP/MCM3複合体の形成を示唆した。MCM3 はCrm1によって認識されうる明らかな共通NES様モチーフを含まず、したがって核からのMCM3の輸出は、恐らく、NES様モチーフを有する他の結合分子または異なる輸出機構に依存するのであろう。GANP上の2つのNES様モチーフは、LMB感受性Crm1依存性輸出経路に関与しているように思われる(図37)。GANPによって担持される2つのNES様モチーフ(これらはCrm1によって認識されるのかもしれない)は、複合体の輸送に関与している可能性がある。
最近、GANP相同領域を担持する酵母SAC3が、あるタンパク質の核輸出に関与し、そして核膜孔複合体の成分と会合することが示された(Jones, A.L. et al., (2000) Proc. Natl. Acad. Sci. USA 97, 3224-3229)。GANPとの共発現は、MCM3の細胞質コンパートメントへの局在化を変化させた。
【0172】
細胞融合技法を用いて、MCM3の核-細胞質間シャトリング(往復)を調べた(Schmidt-Zachmann, M.S. et al., (1993) Cell 74, 493-504)。HeLa細胞を最初MCM3-HAでトランスフェクトし、次にトランスフェクトしていないマウスSwiss-3T3細胞と融合させた。タンパク質合成を抑制するためシクロヘキシミドの存在下で5時間インキュベートした後、細胞を固定し、MCM3-HA を免疫標識した。ヘキスト(Hoechst)染色によってヘテロカリオンを調べた。この染色は、「まだらの」ヘテロクロマチンを有するマウスの核(矢印)をヒトHeLa核と区別する。
図43に代表的なものを示すように、MCM3-HA はヘテロカリオン中にヒトおよびマウスの核の両方に見出された。融合されていないマウスの細胞は、そのような染色を示さない。このことは、MCM3-HAがHeLa核から細胞質へ輸出され、次にマウス核に移入されたことを示している。
【0173】
これらの結果から、MCM3-HAはシャトリングタンパク質であると結論される。なお、トランスジーン産物と同じような感度で内因性タンパク質の核から細胞質への移行を実証することはしばしば困難である(Kimura, H., Ohtomo, T.et al., (1996) Genes Cells 1, 977-993、Mizuno, T. et al., (1999) Mol. Cell. Biol. 19, 7886-7896)。本実施例に示す結果についても、そうであった。酵母のようなより原始的な細胞で達成されたとの同じような感度で、哺乳動物細胞における内因性MCMタンパク質の核から細胞質への移行を実証することもまた困難である。
しかしながら、本発明者の結果は、(実験はDNAトランスフェクション法によって行ったが)未操作細胞においてMCMタンパク質の核-細胞質間シャトリングが恐らく重要であることを示している。細胞周期の進行中におけるMCM複合体の核-細胞質間移動の明確な理解を容易にするには、さらなる成分の発見が必要であるかもしれない。
【0174】
3.4. 細胞周期の間のGANPの局在化
RNA/DNA プライマーゼドメインのGANPに特有のエピトープ(pSer502 GANP)に特異的なモノクローナル抗体を用いて、共焦点レーザー顕微鏡検査によってNIH-3T3細胞におけるGANPの局在化を調べた(Kuwahara, K. et al., (2001) Proc. Natl. Acad. Sci. USA 98, 10279-10283)。細胞周期の異なる時期にあるNIH-3T3細胞を、抗pSer502 GANP (Alexa488; 緑)及び抗MCM3 (Alexa546; 赤)抗体で免疫染色した。核をTOTO-3ヨウ化物(青)で前染色した。間期の間、上記モノクローナル抗体は核小体を除く核内のすべてで反応した(図44)。
抗MCM3抗体および核酸を染色するTOTO-3を用いた三重標識によって、有糸分裂の間のGANPの局在化を詳しく分析した。細胞が前中期から中期へ進むにつれ、GANPは濃縮クロマチンから解離されてくるように思われる(図44)。重ね合わせた画像の黄色いシグナルはGANPとMCM3との共局在化を示すが、若干の青い染色は前中期画像の中心部でGANPが単独でも見えることを表している。この段階では、GANP及びMCM3は濃縮された染色体と重なり合っていない。中期の細胞においては、GANPは紡錘体領域に検出される。そしてこのシグナルは後期に染色体が2つの娘細胞に分離する間に減少する。
【0175】
減数分裂後期においては、GANPの殆どは核が形成されるまで(終期)、細胞質コンパートメントに見出される。これらの結果は、GANPおよびMCM3の挙動が類似していて、これら2つは間期の間を通じて殆ど核に共局在化することを示している。このことは、これら2つの相互会合と一致している。しかし、有糸分裂の間に共焦点顕微鏡検査によって示されたように、GANPおよびMCM3は別々に存在することもできる(図44)。
第2型のRNA/DNAプライマーゼに関する核-細胞質間のGANPシャトリングの生物学的意味は、今後の解明を待たなければならない。それはDNA修復の最終段階におけるRNAプライマーの生成に関連するのかもしれない。正常細胞ではGANPの発現レベルは低いが、細胞が急速に増殖する胚中心においてはアップレギュレーションされる(Kuwahara, K.et al., (2000) Blood 95, 2321-2328 、Kuwahara, K. et al., (2001). Proc. Natl. Acad. Sci. USA 98, 10279-10283)。GANPはまた、速い細胞終期を有するある種の細胞においてより高レベルで発現される。このことは、MCM複合体との会合がDNA複製を刺激する可能性を示唆している(Kuwahara, K. et al., (2001). Proc. Natl. Acad. Sci. USA 98, 10279-10283)。RNA/DNAプライマーゼ活性、MCM3結合能、およびアセチルトランスフェラーゼドメイン(Takei, Y.et al., (2001) EMBO Rep. 2, 119-123を有するGANPの発現は、細胞周期進行の調節に関与しているのかもしれない。
【配列表フリーテキスト】
【0176】
配列番号5:プライマー
配列番号6:プライマー
配列番号7:プライマー
配列番号8:プライマー
配列番号9:プライマー
配列番号10:プライマー
配列番号11:プライマー
配列番号12:プライマー
配列番号13:プライマー
配列番号14:プライマー
配列番号15:プライマー
配列番号16:プライマー
配列番号17:プライマー
配列番号18:プライマー
配列番号19:プライマー
配列番号20:プライマー
配列番号21:プライマー
配列番号22:プライマー
配列番号23:プライマー
配列番号24:プライマー
配列番号25:プライマー
配列番号26:プライマー
配列番号27:プライマー
配列番号28:プライマー
配列番号29:プライマー
配列番号30:プライマー
配列番号31:プライマー
配列番号32:プライマー
配列番号33:プライマー
配列番号34:プライマー
配列番号35:プライマー
配列番号36:プライマー
【技術分野】
【0001】
本発明は、GANP遺伝子を導入したトランスジェニック哺乳動物及びその利用に関する。より詳細には、本発明は、GANPを高発現し、高親和性抗体を産生することができるトランスジェニック哺乳動物、該トランスジェニック哺乳動物を用いて高親和性抗体を産生する方法、及び得られた高親和性抗体の利用に関する。
【背景技術】
【0002】
免疫系の機能は、T細胞の効果を主とする細胞性免疫反応に基づく機能と抗体の効果を主とする液性免疫に基づく機能とに分類される。実際は、この両者の機能は協調して免疫応答が行われる。抗体は骨髄で生まれるB細胞の細胞表面レセプターとして表出している。生体で形成される最初の抗体が認識する抗原の多様性は109〜1011個のオーダーに上ると言われ、そのような抗体(抗原レセプター)は、環境に存在しうるあらゆる抗原決定基を認識する。しかしながら、この多様な抗原レセプターは抗原に対して結合する能力は概して低く、低親和性の抗体が産生されることが多いが、これでは十分な免疫応答とはならない。
【0003】
リンパ球、特にB細胞/免疫グロブリン(抗体)は、その免疫反応に基づく各種用途、例えば病原体等の抗原検出のためのキット、診断薬、治療薬として利用されている。このような抗原検出薬、あるいは各種疾患治療薬における抗体として、抗原に対する反応性が高い抗体を使用すると、抗原に対する感度が優れ、かつ同一投与量での治療薬としての性能が優れる。しかしながら、これまでの所、抗体の親和性を高める手段は知られていない。
【0004】
ところで、生体は病原体や異物が体内に侵入すると、それらを抗原として認識して、末梢のリンパ組織において、抗原と直接結合する抗体のV領域の遺伝子に高頻度の体細胞突然変異を誘導する。その変化にはT細胞の刺激を必要としており、胚中心(Germinal center)領域で活性化T細胞から刺激を受けると考えられる。近年、本発明者らはこの領域の活性化B細胞で選択的に発現が上昇する分子GANPを見出している(特許文献1)。この分子はDNAへリカーゼ活性を有するMCM(minichromosome maintenance)と呼ばれる分子と直接結合し、さらにRNAプライマーゼ活性を有することから、DNA複製に関連することが示唆されている。しかしながら、免疫系におけるGANPの機能については解明されていなかった。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】国際公開第00/50611号
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は、各種疾患の診断薬及び治療薬として有効な高親和性抗体、当該抗体を産生するためのトランスジェニック哺乳動物、該高親和性抗体又は高親和性抗体産生細胞を用いた薬剤を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明者は、上記課題を解決するため鋭意研究を行った結果、GANP遺伝子を導入したトランスジェニック動物を作製し、抗原で免疫すると、このトランスジェニック動物は高親和性抗体を産生し得ることを見出し、本発明を完成するに至った。
【0008】
すなわち、本発明は以下の通りである。
(1) GANP遺伝子を導入したトランスジェニック哺乳動物又はその子孫。
導入したGANP遺伝子はB細胞で発現することができる。また、本発明のトランスジェニック哺乳動物又はその子孫は、GANP遺伝子をトランスフェクトしたES細胞から発生させることが可能である。哺乳動物としては、例えばマウスが挙げられる。
(2) 前記トランスジェニック哺乳動物又はその子孫の一部。
(3) 前記トランスジェニック哺乳動物又はその子孫に抗原を投与し、得られる動物又は子孫から抗体を採取することを特徴とする高親和性抗体の製造方法。
【0009】
(4) 前記(3)記載の方法により得られる高親和性抗体又はその断片。
本発明の抗体は、親和性が1×10-7 (M) 以下で示されるものである。本発明の抗体は、ポリクローナル抗体であってもモノクローナル抗体であってもよい。
(5) 前記抗体又はその断片のV領域を含む、ヒト型化抗体若しくはヒト抗体又はそれらの断片。
(6) 前記抗体又はその断片、及び前記ヒト型化抗体若しくはヒト抗体又はそれらの断片からなる群から選択される少なくとも1つを含有する医薬組成物。
(7) 抗原を投与した請求項1〜4のいずれかに記載のトランスジェニック哺乳動物又はその子孫から採取される、高親和性抗体産生細胞。
【発明の効果】
【0010】
本発明によるGANPを過剰発現したマウスを用いることによって、従来では得られることのできなかったウイルス抗原に特異的で高親和性の抗体を速やかに作製することができる。そのため、エイズやC型肝炎のように遷延する感染による病状悪化を当該のウイルス抗原の変異に遅れないで特異的で強力な抗体を速やかに得ることが可能となると期待される。また、これによって、感染患者からのウイルスの抗原の変異に対応するオーダーメードの特異的抗体を作製することができる。抗体の作製に必要な免疫期間は10日程度で十分であり、そのうち高親和性の変異を持つ抗体の産生効率は60%近くに上る。ベッドサイドの患者サンプルを用いる高親和性抗体の産生プロトコールはワクチン療法に変わる新しい免疫療法となると期待される。
【図面の簡単な説明】
【0011】
【図1】抗GANPモノクローナル抗体およびALP結合抗ラットIg抗体を用いた免疫組織化学分析の結果を示す。スケールバーは100μm。
【図2】雌NZBマウスの膝窩のリンパ節中のGANPhi細胞の出現速度を示す。スケールバーは100μm。
【図3】雌NZBマウスの脾臓中のGANPhi細胞の出現速度を示す。スケールバーは100μm。
【図4】複数系統のマウス由来の脾臓切片を抗GANPモノクローナル抗体で染色した結果を示す。RP; 赤脾髄, F; 濾胞。スケールバーは100μm。
【図5】脾臓の赤脾髄におけるGANPhi 細胞の同定を示す。
【0012】
【図6】GANPhi 細胞における形質細胞マーカーの同定を示す。スケールバーは100μm。
【図7】TD-Ag免疫によるC57BL/6マウスの脾臓の赤脾髄領域におけるGANPhi細胞の発現を示す。スケールバーは100μm。
【図8A】Daudi細胞にマウスGANPを安定的に発現させたトランスフェクタントの体細胞突然変異を示す。
【図8B】Daudi細胞にマウスGANPを安定的に発現させたトランスフェクタントの体細胞突然変異を示す。
【図8C】Daudi細胞にマウスGANPを安定的に発現させたトランスフェクタントの体細胞突然変異を示す。
【0013】
【図9A】B細胞でGANPを過剰発現させたトランスジェニックマウスの作製の概要を示す。
【図9B】B細胞でGANPを過剰発現させたトランスジェニックマウスの作製の概要を示す。
【図9C】B細胞でGANPを過剰発現させたトランスジェニックマウスの作製の概要を示す。
【図10】GANP過剰発現トランスジェニック(Tg)マウス又は野生型マウスにおける体細胞突然変異を解析した結果を示す。
【図10−1】GANP過剰発現トランスジェニック(Tg)マウス又は野生型マウスにおける体細胞突然変異を解析した結果を示す。
【図10−2】GANP過剰発現トランスジェニック(Tg)マウス又は野生型マウスにおける体細胞突然変異を解析した結果を示す。
【図10−3】GANP過剰発現トランスジェニック(Tg)マウス又は野生型マウスにおける体細胞突然変異を解析した結果を示す。
【図10−4】GANP過剰発現トランスジェニック(Tg)マウス又は野生型マウスにおける体細胞突然変異を解析した結果を示す。
【図10−5】GANP過剰発現トランスジェニック(Tg)マウス又は野生型マウスにおける体細胞突然変異を解析した結果を示す。
【0014】
【図11A】B細胞特異的GANP欠損マウス(B-GANP-/-)の作製の概要を示す。
【図11B】B細胞特異的GANP欠損マウス(B-GANP-/-)の作製の概要を示す。
【図11C】B細胞特異的GANP欠損マウス(B-GANP-/-)の作製の概要を示す。
【図11D】B細胞特異的GANP欠損マウス(B-GANP-/-)の作製の概要を示す。
【図11E】B細胞特異的GANP欠損マウス(B-GANP-/-)の作製の概要を示す。
【図12】B細胞特異的GANP欠損マウス(B-GANP-/-)を用いた細胞表面染色の結果(フローサイトメトリー)を示す。
【図13】B細胞の増殖アッセイの結果を示す。ほとんど差はなかったが、抗CD40抗体刺激による増殖のみが約1/2に減少していた。
【0015】
【図14】免疫をしていないCre-flox/+マウス及びB-GANP-/-マウス血清中の抗体価を示す。各種アイソタイプの抗体価に差はなかった。
【図15】B-GANP-/-マウスにおける抗体産生を測定した結果を示す。
【図16】GCをピーナッツアグルチニンで染色した結果を示す。
【図17】B-GANP-/-マウスにおける抗原特異的抗体産生を測定した結果を示す。
【図18】100μgのNP-CGを免疫し、免疫後14日及び35日目における親和性の成熟の度合いをディファレンシャルELISAにより測定した結果を示す。
【図19】GC-B細胞のフローサイトメトリーの結果を示す。
【0016】
【図20A】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Aから図20Fに順に続く)。
【図20B】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Aから図20Fに順に続く)。
【図20C】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Aから図20Fに順に続く)。
【図20D】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Aから図20Fに順に続く)。
【図20E】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Aから図20Fに順に続く)。
【図20F】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Aから図20Fに順に続く)。
【図20G】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Gから図20Lに順に続く)。
【図20H】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Gから図20Lに順に続く)。
【図20I】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Gから図20Lに順に続く)。
【図20J】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Gから図20Lに順に続く)。
【図20K】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Gから図20Lに順に続く)。
【図20L】Cre-flox/+のVH186.2をPCRで増幅し、シークエンス解析を行った結果を示す(図20Gから図20Lに順に続く)。
【0017】
【図21】Cre-flox/+及びB-GANP-/-マウスにおけるIgG1の変異の頻度を示す。
【図22】VH186.2の33番目のWをLに変異させたときの変異の頻度を示す。
【図23】活性化誘導細胞死(AICD)の測定結果及びアポトーシスの抑制結果を示す。
【図24】抗CD40及び抗CD95刺激に対する細胞のアポトーシス感受性を測定した結果を示す。
【図25】TUNELアッセイによりアポトーシス細胞を検出した結果を示す。
【図26】TUNELアッセイによりアポトーシス細胞を検出した結果を示す。
【図27】アポトーシス抑制に関与するBcl-2ファミリーのRNA発現レベルを示す。
【図28】GANPトランスジェニックマウスを用いて高親和性抗体を産生した結果を示す。
【図29】GANPトランスジェニックマウス由来のハイブリドーマクローンを用いて高親和性抗体を産生した結果を示す。
【図30】GANPトランスジェニックマウス由来のハイブリドーマクローン培養上清を用いたBiacoreによる結合解離曲線を示す。
【0018】
【図31】GANPトランスジェニックマウス由来のハイブリドーマクローン培養上清を用いたBiacoreによる結合解離曲線を示す。
【図32】GANP-GST融合タンパク質の構造の概略を示す。
【図33】MCMと直接結合するGANPの領域を決定するためのプルダウンアッセイの結果を示す。左に大きさのスタンダードの位置を示す。
【図34】in vitro翻訳されたMCMを用いたプルダウンアッセイの結果を示す。
【図35】GANP各構築物とMCMの結合を免疫沈降によって示す。
【図36A】GANP各構築物とMCMの結合を免疫沈降によって示す。
【図36B】GANP各構築物とMCMの結合を免疫沈降によって示す。
【図37】GANP構築物の構造の概要及び、構築物の細胞内での分布を示す。
【0019】
【図38】GANP構築物の細胞内での分布を示す。
【図39】MCM3の核における局在化を示す。
【図40】GANPの発現によって誘導されるMCM3の細胞質局在化を示す。
【図41】核に局在する対照タンパク質を示す。
【図42】MCM3変異体の局在化におけるGANP構築物の効果を示す。
【図43】ヘテロカリオンアッセイによって検出されるMCM3の核-細胞質間シャトリングを示す。
【図44】細胞周期の間のGANPの局在化を示す。
【発明を実施するための形態】
【0020】
以下、本発明を詳細に説明する。
本発明は、GANP遺伝子を非ヒト哺乳動物に導入してトランスジェニック動物を作製し、そのようなトランスジェニック動物を抗原で免疫すると、高親和性の抗体が得られる知見に基づいて完成されたものである。
【0021】
1.GANP
GANPは、胚中心結合核タンパク質(Germinal center-associated nuclear protein)と呼ばれており、酵母Sac3タンパク質とホモロジーを有する210kDaの核タンパク質である(WO00/50611号公報)。そして、SAC3はアクチン形成の抑制物質として特徴づけられている。また、GANPは、濾胞樹状細胞(follicular dendritic cells: FDC)により囲まれる胚中心(germinal center, GC)B細胞において選択的にアップレギュレートされ、リン酸化依存性RNA-プライマーゼ活性を有し、B細胞の細胞周期調節に関与しているタンパク質である (Kuwahara, K. et al., (2000) Blood 95, 2321-2328)。
本発明においては、GANPタンパク質のアミノ酸配列を、マウスについて配列番号2に、ヒトについて配列番号4に示す。また、GANPタンパク質をコードする遺伝子(GANP遺伝子という)の塩基配列を、マウスについて配列番号1に、ヒトについて配列番号3に示す。なお、上記アミノ酸配列及び塩基配列は、国際公開WO00/50611号公報にも記載されている。
【0022】
またGANPタンパク質は変異体でもよく、配列番号2又は4に記載のアミノ酸配列において1又は複数のアミノ酸が欠失、置換又は付加されたアミノ酸配列であってRNAプライマーゼ活性を有するタンパク質であってもよい。例えば、配列番号2又は4に示すアミノ酸配列のうち1若しくは複数個(好ましくは1個又は数個(例えば1個〜10個、さらに好ましくは1個〜5個))のアミノ酸が欠失しており、1若しくは複数個(好ましくは1個又は数個(例えば1個〜10個、さらに好ましくは1個〜5個))のアミノ酸が他のアミノ酸で置換されており、及び/又は1若しくは複数個(好ましくは1個又は数個(例えば1個〜10個、さらに好ましくは1個〜5個))の他のアミノ酸が付加されたアミノ酸配列からなり、かつ上記GANPタンパク質と同様のRNAプライマーゼ活性を有するGANP変異型タンパク質を使用することもできる。
【0023】
「RNAプライマーゼ活性」とは、RNA複製において、5'→3'方向に進む鎖の伸長とは逆向きの鎖(ラギング鎖)を合成する際に、伸長の開始点となる短いプライマーのRNAを合成する酵素活性を意味する。通常はαプライマーゼと呼ばれるDNAポリメラーゼαと結合する分子が用いられるが、胚中心B細胞では第二のプライマーゼであるGANPプライマーゼも誘導されている。
GANPタンパク質は、上記配列番号2若しくは4に示すアミノ酸配列又はこれらの変異型アミノ酸配列のほか、N末端側の一部の配列(例えば配列番号2に示すアミノ酸配列の1〜600番、好ましくは139〜566番)又はこれらの変異型アミノ酸配列を有するものも含まれる。
【0024】
本発明において、動物に導入するためのGANP遺伝子は、上記GANPタンパク質、N末側の一部の配列、又は変異型タンパク質をコードする遺伝子が挙げられる。そのような遺伝子として、例えば配列番号1又は3に示す塩基配列を有するものを使用することができる。配列番号1又は3に示す塩基配列のうち、コード領域のみの塩基配列であってもよい。また、上記配列番号1又は3に示す塩基配列に相補的な配列と、ストリンジェントな条件下でハイブリダイズし、かつ、RNAプライマーゼ活性を有するタンパク質をコードする遺伝子を使用することも可能である。
「ストリンジェントな条件」とは、ハイブリダイズさせた後の洗浄時の条件であって塩(ナトリウム)濃度が150〜900mMであり、温度が55〜75℃、好ましくは塩(ナトリウム)濃度が250〜450 mMであり、温度が68℃での条件をいう。
【0025】
遺伝子に変異を導入するには、Kunkel法や Gapped duplex法等の公知手法により、例えば部位特異的突然変異誘発法を利用した変異導入用キット、例えばGeneTailorTM Site-Directed Mutagenesis System(インビトロジェン社製)、TaKaRa Site-Directed Mutagenesis System(Mutan-K、Mutan-Super Express Km等:タカラバイオ社製)を用いて行うことができる。
変異遺伝子の詳細並びに取得方法は国際公開WO00/50611号公報にも記載されている。
【0026】
なお、抗μ抗体及び抗CD-40モノクローナル抗体でB細胞をin vitro刺激すると、GANP発現のアップレギュレーションのみならず、GANPタンパク質のアミノ酸配列のうち特定のセリン残基(例えば502番目のセリン: S502)のリン酸化を引き起こす。この反応は、GANPのRNA-プライマーゼ活性についてキーとなる反応である(Kuwahara, K. et al. (2001) Proc. Natl. Acad. Sci. USA, 98, 10279-10283)。GANPタンパク質のN末端側のRNA-プライマーゼドメインはセリン残基を含んでおり、そのリン酸化はin vitroにおいてCdk2によって触媒される。C末端側ドメインにより、GANPはMCM3複製ライセンシング因子に結合する(Kuwahara, K. et al., (2000) Blood 95, 2321-2328; Abe, E. et al. (2000) Gene 255, 219-227)。
【0027】
2.GANP遺伝子を導入したトランスジェニック哺乳動物
本発明は、GANP遺伝子を導入したトランスジェニック哺乳動物に関するものであり、当該トランスジェニック哺乳動物は、好ましくは、導入したGANP遺伝子をB細胞で発現することができる。
【0028】
(1) GANP遺伝子とその関連分子
GANP遺伝子とその関連分子で形成される複合体は、遺伝子に変異を誘導するプロセスで直接および間接的に必要な分子である。GANPタンパク質は、遺伝子変異を修復する際に、高親和性の抗体が得られるようにV領域の変異の誘導を促す能力を保有していることから、本発明のトランスジェニック哺乳動物は、このGANP遺伝子又はその変異遺伝子の導入によって、獲得性免疫の高親和性抗体産生を促進することができる。また、この遺伝子を過剰に発現するトランスジェニック非ヒト哺乳動物は、速やかに抗原に対する結合力の高い抗体を産生することができる。従って、上記トランスジェニック非ヒト哺乳動物を所定の抗原で免疫することで、従来では得られないような高親和性の抗体を簡便に得ることができる。その結果、難治性の病原微生物や異物を排除できるポリクローナル抗体又はモノクローナル抗体を得ることができる。また、本発明のトランスジェニック哺乳動物を用いてヒト型化抗体を作製することによって、あるいは、本発明のトランスジェニック哺乳動物が産生する抗体のV領域を含む一本鎖抗体を作製することによって、抗体療法の効力を飛躍的に高めることが可能となる。
【0029】
本発明のトランスジェニック哺乳動物は、GANP又はその変異遺伝子の導入によって、B細胞で高親和性抗体の産生を促進することができ、前記高親和性抗体産生細胞はアポトーシスを誘導するシグナルに対して抵抗性を有する。
本発明者らは、GANPが獲得免疫応答の抗体産生に機能している分子であることを確認するため、先ずGANP遺伝子の欠損マウスをB細胞選択的に欠損するように企図して作製した。その結果、GANP遺伝子が欠損しても、免疫系の細胞の発達、分化、増殖には影響が見られず、また抗体の産生総和には大きな変化は見られないことが判明した。
【0030】
ここで、抗原と反応したB細胞がそのまま増殖し、抗体産生細胞に分化するものは、ある種の抗原の場合に限られており、通常の抗原に対する抗体産生についてはT細胞の共存を必要とする。T細胞が存在しなくても抗体が産生されるような抗原をT細胞非依存性抗原という。これに対し、T細胞非依存性抗原以外の一般の抗原をT細胞依存性抗原という。T細胞依存性抗原の場合は、B細胞の抗体産生細胞への分化がヘルパーT細胞により補助される。
病原ウイルスの抗原決定基(抗原エピトープとも言う)の多くはそれ自体免疫原性が弱く、T細胞によって認識されるキャリアタンパク質のペプチド抗原によって活性化を受ける。
【0031】
本発明においては、通常の動物では、強力な抗体を産生できないような可溶性の抗原に対する抗体産生応答に対して、GANP遺伝子導入動物では高頻度に高親和性の抗体を産生することができることを調べるため、ハプテンとして解析が進んでいるニトロフェニル基 (NP基)をニワトリ-ガンマグロブリンと結合させて抗原(NP-CGという)を作製し、T細胞性依存性抗原の反応を調べた。
ここで、C57BL/6マウスのNPに対する反応は単一のV領域で行われることが知られている。この反応は、抗体のIgG重鎖のV領域(VH186.2という)とL鎖ラムダ1遺伝子によってのみ形成されるというものである。このシステムを使えば、アイソタイプがIgG1の抗体について、VH186.2のアミノ酸配列を調べることによって高親和性抗体の遺伝子変異を調べることが可能である。しかも、最も強い親和性は、重鎖V領域(VH186.2)のアミノ酸配列のうち、第33番目のアミノ酸であるトリプトファン(W)がロイシン(L)に変異(W33-L)したときに生じることが報告されている。
【0032】
そこで、本発明者は、GANP遺伝子欠損マウスでW33-L変異を起こさせたときに、高親和性抗体が産生されるか否かを調べた。その結果、対照であるCre-flox/+マウスに比べて高親和性抗体産生はほとんど見られなかった。従って、GANP遺伝子は、高親和性抗体を産生するための重要な機能を有する遺伝子であることが判明した。このことをさらに検証するために、GANP遺伝子を過剰に発現するマウスを作製した。GANP遺伝子の過剰発現は、マウス免疫グロブリンプロモーター部分とヒト免疫グロブリン遺伝子イントロンエンハンサー部分をGANP遺伝子の5'側に連結して、B細胞で選択的に発現させることによって行った。
【0033】
上記GANP過剰発現マウスも正常に生まれ、リンパ組織の発達、分化、増殖には変化が見られないが、NP-CGに対する反応では、著しく高親和性型のV領域遺伝子(W33-L)が増加していた。RNAプライマーゼ活性がこの際にどのような機能的な役割を担っているかについてはまだ確定されていないが、(i) GANP分子のプライマーゼ活性をみる指標となる502番目のセリン残基のリン酸化が胚中心の高親和性B細胞の生み出される領域の細胞(セントロサイト)に高いこと、(ii) Daudi細胞へのganp遺伝子導入実験によって誘導されるV領域の突然変異の頻度が高いことから、GANP分子のRNA プライマーゼ活性あるいはそれに関わる502番目のセリン残基のリン酸化が高親和性抗体産生に関連していると考えている。この結果は、GANP分子を高発現させること、及びRNAプライマーゼ活性を賦活化することが、免疫応答による高親和性抗体の産生に必要であることを示すものである。
【0034】
(2) GANP遺伝子導入用哺乳動物
本発明における「哺乳動物」とは、ウシ、ウマ、ブダ、ヤギ、ウサギ、イヌ、ネコ、マウス、ラット、ハムスター及びモルモット等の任意の非ヒト哺乳動物を意味し、好ましくはマウス、ウサギ、ラットまたはハムスターであり、特に好ましくはマウスである。
本発明のトランスジェニック哺乳動物は、未受精卵、受精卵、精子およびその始原細胞を含む胚芽細胞などに対して、好ましくは、非ヒト哺乳動物の発生における胚発生の段階(さらに好ましくは、単細胞または受精卵細胞の段階でかつ一般に8細胞期以前)の細胞に対して、リン酸カルシウム法、電気パルス法、リポフェクション法、凝集法、マイクロインジェクション法、パーティクルガン法、DEAE-デキストラン法などにより、GANP遺伝子を導入することにより作製することができる。また、上記遺伝子導入方法により、体細胞、生体の臓器、組織細胞などに目的とするGANP遺伝子を転移させ、細胞培養、組織培養などに利用することもできる。さらに、これら細胞を上述の胚芽細胞と公知の細胞融合法により融合させることにより、トランスジェニック哺乳動物を作製することもできる。
【0035】
GANP遺伝子を対象動物に導入させる際、当該遺伝子を対象となる動物の細胞で発現させうるプロモーターの下流に連結した遺伝子構築物として導入することが好ましい。具体的には、目的とするGANP遺伝子を有する各種哺乳動物由来のGANP遺伝子を発現させうる各種プロモーターの下流に、GANP遺伝子を連結したベクターを、対象となる哺乳動物の受精卵(例えば、マウス受精卵)にマイクロインジェクションすることによって、目的とするGANP遺伝子を高発現するトランスジェニック哺乳動物を作製することができる。
【0036】
(3) 発現ベクター
GANP遺伝子の発現ベクターとしては、大腸菌由来のプラスミド、枯草菌由来のプラスミド、酵母由来のプラスミド、λファージなどのバクテリオファージ、モロニー白血病ウイルスなどのレトロウイルス、ワクシニアウイルス又はバキュロウイルスなどの動物又は昆虫ウイルスなどが用いられる。
遺伝子発現の調節を行うプロモーターとしては、たとえばウイルス由来遺伝子のプロモーター、各種哺乳動物(ヒト、ウサギ、イヌ、ネコ、モルモット、ハムスター、ラット、マウスなど)および鳥類(ニワトリなど)由来遺伝子のプロモーターなどを使用することが可能である。
【0037】
ウイルス由来遺伝子のプロモーターとしては、例えばサイトメガロウイルス、モロニー白血病ウイルス、JCウイルス、乳癌ウイルス等由来遺伝子のプロモーターが挙げられる。
各種哺乳動物及び鳥類由来遺伝子のプロモーターとしては、例えば、アルブミン、インスリンII、エリスロポエチン、エンドセリン、オステオカルシン、筋クレアチンキナーゼ、血小板由来成長因子β、ケラチンK1,K10およびK14、コラーゲンI型およびII型、心房ナトリウム利尿性因子、ドーパミンβ-水酸化酵素、内皮レセプターチロシンキナーゼ、ナトリウムカリウムアデノシン3リン酸化酵素、ニューロフィラメント軽鎖、メタロチオネインI及びIIA、メタロプロティナーゼ1組織インヒビター、MHCクラスI抗原、平滑筋αアクチン、ポリペプチド鎖延長因子1α(EF-1α)、βアクチン、α及びβミオシン重鎖、ミオシン軽鎖1及び2、ミエリン基礎タンパク、血清アミロイドPコンポーネント、ミオグロビン、レニンなどの遺伝子のプロモーターが挙げられる。
【0038】
上記ベクターは、トランスジェニック哺乳動物において目的とするメッセンジャーRNAの転写を終結するターミネターを有していてもよい。その他、GANP遺伝子をさらに高発現させる目的で、各遺伝子のスプライシングシグナル、エンハンサー領域、真核生物遺伝子のイントロンの一部をプロモーター領域の5'上流、プロモーター領域と翻訳領域間、あるいは翻訳領域の3'下流 に連結することも所望により可能である。
本発明の好ましい態様では、免疫グロブリンプロモーターの下流にGANP遺伝子を連結することにより、あるいはヒト免疫グロブリン遺伝子イントロンエンハンサー部分をGANP遺伝子の5'側に連結することにより、GANP遺伝子をB細胞で選択的に発現させることができる。
【0039】
(4) GANP遺伝子の導入
受精卵細胞段階におけるGANP遺伝子の導入は、対象の哺乳動物の胚芽細胞および体細胞の全てに過剰に存在するように確保することが好ましい。遺伝子導入後の作出動物の胚芽細胞においてGANP遺伝子が過剰に存在することは、作出動物の子孫が全てその胚芽細胞および体細胞の全てにGANP遺伝子を過剰に有することを意味する。そして、遺伝子を受け継いだこの種の動物の子孫は、その胚芽細胞および体細胞の全てにGANP蛋白質を過剰に有する。
本発明においては、導入遺伝子を相同染色体の一方に持つヘテロ接合体を取得し、ヘテロ接合体同士を交配することで導入遺伝子を相同染色体の両方に持つホモ接合体を取得し、この雌雄の動物を交配することによりすべての子孫が導入されたGANP遺伝子を安定に保持する。そして、GANP遺伝子を過剰に有することを確認して、通常の飼育環境で繁殖継代することができる。
【0040】
トランスジェニック対象動物が有する内在性の遺伝子とは異なる遺伝子である外来性GANP遺伝子を対象非ヒト哺乳動物(好ましくはマウスなど)、又はその先祖の受精卵(バッククロス)に転移する際に用いられる受精卵は、同種の雄哺乳動物と雌哺乳動物を交配させることによって得られる。
受精卵は自然交配によっても得られるが、雌哺乳動物の性周期を人工的に調節した後、雄哺乳動物と交配させる方法が好ましい。雌哺乳動物の性周期を人工的に調節する方法としては、例えば、初めに卵胞刺激ホルモン(妊馬血清性性腺刺激ホルモン(PMSG))、次いで黄体形成ホルモン(ヒト絨毛性性腺刺激ホルモン(hCG))を、例えば腹腔注射などにより投与する方法が好ましい。
【0041】
得られた受精卵に、前述の方法により外来性GANP遺伝子を導入した後、雌哺乳動物に人工的に移植・着床することにより、外来性遺伝子を組み込んだDNAを有する非ヒト哺乳動物が得られる。雌哺乳動物に黄体形成ホルモン放出ホルモン(LHRH)を投与後、雄哺乳動物と交配させることにより受精能を誘起された偽妊娠雌哺乳動物に、受精卵を人工的に移植・着床させる方法が好ましい。遺伝子を導入する全能性細胞としては、マウスの場合、受精卵や初期胚を用いることができる。また培養細胞への遺伝子導入法としては、トランスジェニック哺乳動物個体の産出効率や次代への導入遺伝子の伝達効率を考慮した場合、DNAのマイクロインジェクションが好ましい。
遺伝子を注入した受精卵は、次に仮親の卵管に移植され、個体まで発生し出生した動物を里親につけて飼育させたのち、体の一部(マウスの場合には、例えば、尾部先端)からDNAを抽出し、サザン解析やPCR法により導入遺伝子の存在を確認することができる。導入遺伝子の存在が確認された個体を初代(Founder)とすれば、導入遺伝子はその子(F1)の50%に伝達される。さらに、このF1個体を野生型動物または他のF1動物と交配させることにより、2倍体染色体の片方(ヘテロ接合)または両方(ホモ接合)に導入遺伝子を有する個体(F2)を作製することができる。
【0042】
あるいは、GANP蛋白質高発現トランスジェニック哺乳動物は、上記したGANP遺伝子をES細胞(embryonic stem cell)に導入することによって作製することもできる。例えば、正常マウス胚盤胞(blastocyst)に由来するHPRT陰性(ヒポキサンチングアニン・フォスフォリボシルトランスフェラーゼ遺伝子を欠いている)ES細胞に、GANP遺伝子を導入する。当該GANP遺伝子がマウス内在性遺伝子上に相同組み換えを起こさせ、インテグレートされたES細胞をHATセレクション法により選別する。次いで、選別したES細胞を、別の正常マウスから取得した受精卵(胚盤胞)にマイクロインジェクションする。得られた胚盤胞を、仮親としての別の正常マウスの子宮に移植する。その後、仮親マウスからキメラトランスジェニックマウスが生まれる。生まれたキメラトランスジェニックマウスを正常マウスと交配させることにより、ヘテロトランスジェニックマウスを得ることができる。そして、ヘテロトランスジェニックマウス同士を交配することにより、ホモトランスジェニックマウスが得られる。
【0043】
本発明においては、上記したトランスジェニック哺乳動物に限らず、その子孫、並びにトランスジェニック哺乳動物又はその子孫の一部も本発明の範囲内である。トランスジェニック哺乳動物の一部としては、当該トランスジェニック哺乳動物又はその子孫の組織、器官及び細胞などが挙げられ、器官または組織としては、脾臓、胸腺、リンパ節、骨髄あるいは扁桃腺などが挙げられ、細胞としてはB細胞などが挙げられる。
本発明のトランスジェニック哺乳動物は、B細胞をさらに活性化する哺乳動物と交配することも可能であり、これによりさらに高親和性抗体を産生することが可能である。
【0044】
最近、MRL/lprマウスでB細胞が末梢のリンパ節での活性化の際に胚中心を経過した後、T細胞領域でさらにV領域の突然変異誘導が亢進していることが報告されている。また、本発明者らもMRL/lprマウスにおいてGANP遺伝子がIgプロモーター、エンハンサーの下流に結合して作成したganpトランスジェニックマウスに見られるのと同等の高い発現が、非免疫の状態で見られることを見出している。このことは、正常では自己の抗原に対しては高親和性の抗体はできないのに対して、この自己免疫疾患マウスでは、GANP分子の異常な活性化が起こるために、自己の抗原に対しての高親和性抗体が産生されることとなる可能性が示唆される。
【0045】
そこで、上記B細胞をさらに活性化する動物として、自己免疫疾患マウスであるとされるMRL/lpr, NZB, (NZB x NZW)F1などを用いれば、さらに高い変異誘導を期待できる。
以上のことを利用したMLR/lprマウスのGANPトランスジェニックマウスを作製することによって、スーパー高親和性抗体産生マウスを作出できる可能性がある。すなわち、本発明のGANP遺伝子過剰発現トランスジェニック哺乳動物とさまざまな自己免疫疾患モデル動物との交配により、高親和性抗体を産生できる哺乳動物を作製することができる。
【0046】
3.高親和性抗体の作製
本発明でいう抗体とは、抗原と特異的に結合する活性を有する蛋白質を意味し、好ましくはB細胞が産生するものである。本発明では、抗原に対する反応性が高い抗体のことを高親和性抗体と言う。「高親和性」とは、抗体が抗原と結合する結合能が高いことを意味し、本発明においては、抗体の結合能が一般のマウスなどの動物を用いて作製した抗体と比較して高く、また逆に当該の抗原から解離することが遅い抗体のことをいう。これは、抗原決定基(エピトープ)に対して、立体的に密接して結合する能力が高く特異的であることを意味するとともに、抗体が結合することによって抗原決定基のみならずその抗原の構造の変換をきたすことによって結果的に強力な活性(毒性の中和、ウイルスなどの感染性阻止、病原体の不活性化、生体内からの排除の促進、抗原分子の変性を引き起こす等、生物活性を示すもの)を示すことも包含している。
【0047】
また、抗体の結合能(親和性)は、スキャッチャード解析やBiacore と呼ばれる表面プラズモン共鳴センサーにより、解離定数(KD)、解離速度定数(Kdiss)、結合速度定数(Kass)として測定することができる。Biacore装置は、センサーチップ、マイクロ流路系、SPR検出系の3つの技術を統合して分子結合の強さ、速さ、選択性を測定するというものであり、標識を使わずにリアルタイムで生体分子の検出と複数個の分子間での相互作用のモニタリングを行うことができる。Biacore装置としては、例えばBiacore 3000、Biacore 2000、Biacore X、Biacore J、Biacore Q(いずれもBiacore社)などが挙げられる。
【0048】
上記Biacore によって、抗体の親和性を示すパラメーター、すなわち解離定数(KD)、解離速度定数(Kdiss) [1/Sec] 及び結合速度定数(Kass) [1/M.Sec]を測定する。
抗体は、解離定数(KD値)が小さい値であるほど親和性が高いという点で好ましい。抗体の結合能(親和性)は、Kdiss及びKassの2つのパラメーターにより決定され、
KD[M]= Kdiss/Kass
により表わされる。
抗原の種類等複数の要因によって、得られる抗体の親和性は異なるが、一般には、KD値は1×10-7(M) 以下であることが好ましく、例えば1×10-8(M) 以下、1×10-10(M)以下、あるいは1×10-11(M)以下のものが挙げられる。
【0049】
本発明においては、作製された抗体が上記いずれかの作用又は性質を発揮する抗体であるときに、「高親和性」であると判断される。
抗体分子の親和性亢進は抗体遺伝子の可変領域(V領域)遺伝子に体細胞突然変異(SHM)が誘導することによって生まれる。抗体の抗原に対する特異性は、抗原を生体に免疫した当初から認められるが、初期の抗体の多くはIgMクラスであり、また抗原に対する結合親和性は高くはなく、病原体や異物を除去したり、不活化する能力は低い。しかし、抗原を生体に投与して数回の追加免疫を行うと抗体の抗原に対する結合親和性が高まる。この際、B細胞はT細胞からの刺激が必要であり、末梢のリンパ組織の胚中心領域でその活性化が行われるとされている。V領域遺伝子の突然変異誘導に必要な分子としては、最近、胚中心で発現するRNAエディティング分子AIDであると報告されている。さらに、ウラシルDNAグリコシダーゼ、またDNA複製に必要なDNAポリメラーゼとしてミスを生じやすいDNAポリメラーゼゼータ(ζ)とアイオタ(ι)が関与していることが報告されているが、これらの機能を制御する分子はまだ明らかにされていない。GANP分子は新たなSHM誘導分子としてその機能が明らかにされたものであり、その分子の発現上昇がSHM誘導に重要な鍵を握る。とりわけ、高親和性抗体を産生するために重要であることが明らかになった。
【0050】
C57BL/6マウスにハプテン・キャリアの抗原としてニトロフェニル-ニワトリγグロブリンを免疫することにより誘導される抗体は、H鎖はVH186.2ローカスを用い、L鎖はλ1である。この際、追加免疫をして得られる抗体はIgG1抗体であり、そのうち特に結合親和性の高い抗体のV領域配列に誘導される突然変異は、33番目のトリプトファンがロイシンに変異したものであることが知られている。本明細書の実施例では、このモデルシステムで高い高親和性型のV領域突然変異が誘導されており、これは、高親和性抗体が誘導されたことを示す分子レベルでの明らかな証拠であると言える。
従って、上記トランスジェニック哺乳動物又はその子孫に抗原を投与して抗体を産生することによって、高親和性抗体を得ることができる。即ち、GANP蛋白質を高発現させた動物に目的とする抗原を常法によって投与し、感作された動物の血液又は脾臓等の組織(これらの組織に限定されない)のリンパ球より高親和性抗体を調製することができる。高親和性抗体はポリクローナル抗体でもモノクローナル抗体でもよい。
【0051】
ポリクローナル抗体を産生する方法としては、例えば、抗原を本発明のトランスジェニック哺乳動物に投与して免疫し、免疫された哺乳動物から血液を採取し、採取した血液から抗体を分離・精製することにより得ることができる。
免疫感作の方法は当業者に公知であり、例えば抗原を1回以上投与することにより行うことができる。
抗原の種類は特に限定されるものではなく、抗原決定基としての立体構造を持ちうる物質すべてが該当し、タンパク質、酵素、ペプチド、糖、脂質、DNA, RNA,プリオンなどのあらゆる生体成分のほか、癌抗原、ウイルス抗原、有機、無機合成抗原など任意のものを使用することができる。
【0052】
抗原投与は、例えば7〜30日間隔で2〜3回投与すればよい。投与量は1回につき、例えば抗原約0.05〜2mg程度とすることができる。投与経路も特に限定されず、皮下投与、皮内投与、腹膜腔内投与、静脈内投与、筋肉内投与等を適宜選択することができるが、静脈内、腹膜腔内もしくは皮下に注射することにより投与することが好ましい。また、抗原は適当な緩衝液、例えば完全フロイントアジュバント又は水酸化アルミニウム等の通常用いられるアジュバントを含有する適当な緩衝液に溶解して用いることができるが、投与経路や条件等に応じてアジュバントを使用しない場合もある。
免疫感作した哺乳動物を一定期間飼育した後、哺乳動物の血清をサンプリングし、抗体価を測定する。抗体価が上昇してきたときに、例えば100μg〜1000μgの抗原を用いて追加免疫を行なうことができる。最後の投与から1〜2ケ月後に免疫感作した哺乳動物から血液を採取して、その血液をタンパク質の分離に採用される各種常法、例えば遠心分離、硫酸アンモニウム又はポリエチレングリコールを用いた沈澱、ゲルろ過クロマトグラフィー、イオン交換クロマトグラフィー、アフィニティークロマトグラフィー等のクロマトグラフィー等によって、ポリクローナル抗血清として、ポリクローナル抗体を得ることができる。
【0053】
モノクローナル抗体を産生する方法としては、ハイブリドーマ法を挙げることができる。先ず、アジュバント中に目的とする抗原を構成するペプチドを懸濁し、得られる懸濁液を、免疫動物(即ち、本発明のトランスジェニック哺乳動物)の皮下または真皮内に投与する。抗原の種類は前記と同様である。用いられるアジュバントとしては、フロイントの完全アジュバント、フロイントの不完全アジュバント、BCG、トレハロースダイマイコレート(TDM)、リポ多糖(LPS)、ミョウバンアジュバント、シリカアジュバント等が挙げられるが、抗体の誘導能等の関係から、フロイントの完全アジュバント(CFA)とフロイントの不完全アジュバント(IFA)とを組み合わせて使用することが好ましい。
モノクローナル抗体の産生において、免疫動物は抗原の初回免疫後、更に、追加免疫を数回行い、適当な日数を経過した後に部分採血を行い、抗体価を測定することが好ましい。本発明の方法で産生される抗体は高親和性抗体であるため、上記免疫は初回のみで十分である可能性がある。なお、抗体価は、例えば酵素イムノアッセイ(以下「ELISA」という)法等、公知の方法により測定することができる。
【0054】
次いで、感作の終了した免疫動物から脾臓を摘出し、B細胞を得る。この際、抗原に結合するB細胞を得ることが、その後のスクリーニングを軽減できる点で好ましい。ここで得られるB細胞は高親和性抗体産生細胞であり、これをそのまま免疫賦活剤として使用することもできる。また、B細胞から直接V領域遺伝子を得て、そのV領域の体細胞突然変異を測定することもできる。
次いで、B細胞を常法に従いミエローマ細胞と融合させて抗体産生ハイブリドーマを作製する。例えば、マウスの場合であれば、脾臓を摘出し、摘出した脾臓を、例えばハンクスの平衡塩溶液(HBSS)中に置き、ピンセットで細胞を押し出して脾臓リンパ球(B細胞)を得る。得られた脾臓リンパ球は、トリパンブルー等の染色液で染めて生細胞数をカウントし、ミエローマ細胞と融合させてハイブリドーマとする。
細胞融合に用いられるミエローマ細胞は特に限定されず、公知のものを使用できる。例えば、P3-X63.Ag8 (X63)、P3-X63.Ag8.U1 (P3U1)、P3/NS I/1-Ag4-1(NSI)、Sp2/0-Ag14(Sp2/0)等を挙げることができる。ミエローマ細胞の選択にあたっては、抗体産生細胞との適合性を適宜考慮する。
【0055】
細胞融合は、血清を含まないDMEM、RPMI-1640培地などの動物細胞培養用培地中で、1×106〜1×107個/mlの抗体産生細胞と2×105〜2×106個/mlのミエローマ細胞とを混合し(抗体産生細胞とミエローマ細胞との細胞比5:1が好ましい)、細胞融合促進剤存在のもとで融合反応を行う。
細胞の融合方法は、センダイウイルス法、ポリエチレングリコール法、プロトプラスト法等、当該分野で公知の方法を任意に選択して用いることができるが、特にポリエチレングリコール法が、細胞毒性が比較的少なく、融合操作も簡単であるという理由から好ましい。細胞融合促進剤としてのポリエチレングリコールは、平均分子量1000〜6000ダルトンのものを使用することができる。なお、抗体を大量に作り出したい場合は、ビニルピリジン誘導体で刺激した抗体産生細胞と骨髄腫細胞を細胞融合させたハイブリドーマを用いることが好ましい。
【0056】
得られたハイブリドーマは、常法に従い、HAT培地(ヒポキサンチン、アミノプテリン、およびチミジン含有培地)中で適当な期間培養し、ハイブリドーマの選択を行う。次いで、目的とする抗体産生ハイブリドーマのスクリーニングを行った後、ハイブリドーマのクローニングを行う。
スクリーニング法としては、公知の抗体検出方法を用いることができ、例えば、ELISA法、ラジオイムノアッセイ(以下「RIA」という)法、プラーク法、凝集反応法等を用いることができる。また、クローニング法としては、当該分野で公知の方法を用いることができ、例えば、限界希釈法、軟寒天法およびFACS法等を用いることができる。得られたハイブリドーマは、適当な培養液中で培養するか、あるいはハイブリドーマと適合性のある、例えばマウス腹腔内に投与する。こうして得られる培養液中または腹水中から、塩析、イオン交換クロマトグラフィー、ゲル濾過、アフィニティークロマトグラフィー等により、所望のモノクローナル抗体を単離精製することができる。
【0057】
また、上記した抗体の断片及びV領域の一本鎖抗体も本発明の範囲内である。抗体の断片としては、前述したポリクローナル抗体又はモノクローナル抗体の一部分の領域を意味し、具体的にはF(ab')2、Fab'、Fab、Fv(variable fragment of antibody)、sFv、dsFv(disulphide stabilized Fv)あるいはdAb(single domain antibody)等が挙げられる。ここで、「F(ab')2」及び「Fab'」とは、イムノグロブリン(モノクローナル抗体)を、それぞれ蛋白分解酵素であるペプシン、パパイン等で処理することにより製造され、ヒンジ領域中の2本のH鎖間に存在するジスルフィド結合の前後で消化されて生成される抗体フラグメントを意味する。例えば、IgGをパパインで処理すると、ヒンジ領域中の2本のH鎖間に存在するジスルフィド結合の上流で切断されてVL(L鎖可変領域)とCL(L鎖定常領域)からなるL鎖、及びVH(H鎖可変領域)とCHγ1(H鎖定常領域中のγ1領域)とからなるH鎖フラグメントがC末端領域でジスルフィド結合により結合した相同な2つの抗体フラグメントを製造することができる。これら2つの相同な抗体フラグメントを各々Fab'という。またIgGをペプシンで処理すると、ヒンジ領域中の2本のH鎖間に存在するジスルフィド結合の下流で切断されて前記2つのFab'がヒンジ領域でつながったものよりやや大きい抗体フラグメントを製造することができる。この抗体フラグメントをF(ab')2という。一本鎖抗体は、VL とVHをリンカーでつないだ構造を持つ。
【0058】
本発明の高親和性抗体はヒト型化抗体やヒト抗体でもよい。これらの抗体は、免疫系をヒトのものと入れ換えた哺乳動物を用いて、該哺乳動物を免疫して、通常のモノクローナル抗体と同様に直接ヒト抗体を作製することができる。
ヒト型化抗体を作製する場合は、マウス抗体の可変領域から相補性決定領域(complementarity determining region;CDR)をヒト可変領域に移植して、フレームワーク領域(FR)はヒト由来のものを、CDRはマウス由来のものからなる再構成した可変領域を作製する。
次に、これらのヒト型化された再構成ヒト可変領域をヒト定常領域に連結する。最終的に再構成されたヒト型化抗体のヒト以外のアミノ酸配列に由来する部分は、CDR及び極く一部のFRのみである。CDRは超可変アミノ酸配列により構成されており、これらは種特異的配列を示さないため、マウスCDRを有するヒト型化抗体を使用することが可能である。ヒト型化抗体の作製法は、当分野において周知である。
【0059】
ヒト抗体は、一般にV領域の抗原結合部位、すなわち超過変領域(Hyper Variable region)についてはその特異性と結合親和性が問題となるが、構造的にどの動物で作製してもかまわない(マウス、ラット等)。一方、V領域のそのほかの部分や定常領域の構造はヒトの抗体と同じ構造をしていることが望ましい。ヒトに共通の遺伝子配列については遺伝子工学的手法によって作成する方法が確立されている。
本発明の抗体のアイソタイプは特に限定されず、例えば、IgG(IgG1、IgG2、IgG3、IgG4)、IgM、IgA(IgA1、IgA2)、IgDまたはIgEの任意のアイソタイプを有することができる。
【0060】
4.高親和性抗体の利用
本発明の高親和性抗体は、疾患の診断、治療又は予防のための薬剤として有用である。
【0061】
(1) 疾患の診断
本発明の抗体を用いた各種疾患の診断方法は、各種疾患の疑いのある被験者から採取した検体、例えば血清等と本発明の抗体とを抗原抗体反応によって結合させ、結合した抗体量により検体中の目的とする抗原の量を検出することにより行う。抗体量の検出は、公知の免疫学的測定法に従って行えばよく、例えば、免疫沈降法、免疫凝集法、標識免疫測定法、免疫比ろう法、免疫比濁法等を用いることができる。特に標識免疫測定法が簡便かつ高感度という点で好ましい。標識免疫測定法では、検体中の抗体価は標識抗体を用いて直接検出した標識量で表すほか、既知濃度あるいは既知抗体価の抗体を標準液として用いて相対的に表してもよい。すなわち、標準液と検体を同測定系にて同時に測定し、標準液の値を基準にして検体中の抗体価を相対的に表すことができる。
【0062】
標識免疫測定法としては、公知の測定法、例えば、ELISA法、RIA法、蛍光免疫測定法、化学発光免疫測定法等を任意に利用することができる。用いる標識物質は、上記測定法に応じて、酵素、放射性同位体、蛍光化合物、および化学発光化合物等を適宜選択すればよい。前記酵素としては、例えば、ペルオキシダーゼ、アルカリホスファターゼ、酸性ホスファターゼ、グルコースオキシダーゼ等を挙げることができる。上記標識物質はアビジン−ビオチン複合体を用いることにより、標識物質の検出感度を向上させることも可能である。また、放射性同位体としては、主に125Iが、蛍光化合物としては、フルオレセインイソチオシアネート(FITC)やテトラメチルローダミンイソチオシアネート(TRITC)等が挙げられる。化学発光化合物としては、ロフィン、ルミノール、ルシゲニン等が挙げられる。上記標識物質による抗体の標識は、常法に従って行うことができる。以下、標識抗体を用いた標識免疫測定法について説明する。
【0063】
標識免疫測定法による、各種疾患を検出する方法としては、公知の非競合反応系あるいは競合反応系を用いて行うことができる。非競合反応系においては、固相が必要である(固相法)。競合反応系においては、必ずしも固相を必要としない(液相法)が、固相を用いた方が、測定操作が簡便になるため好ましい。固相の材質としては、例えば、ポリスチレン、ナイロン、ガラス、シリコンラバー、セルロース等が挙げられ、固相の形状としては、球状、ウェル状、チューブ状、シート状等が挙げられるが、これらに限定されず、標識免疫測定法に用いられる公知のものを任意に用いることができる。
【0064】
非競合反応系の場合、測定操作は、検体または本発明の抗体を固相化した後、本発明の抗体または検体と反応させ、次いであらかじめ標識しておいた抗免疫グロブリン抗体(二次抗体)を加えて固相化した検体と反応している抗体と反応させる。この二次抗体の標識により、検体に結合した抗体量を検出することができる。検出された標識化二次抗体の量は、検体中の目的とする抗原の量と正相関するので、これにより検体中の目的とする抗原の量を求めることができる。
競合反応系では、一定量の抗体に対して、検体と一定量の目的とする抗原を競合的に結合させる。例えば、検体を固相化した後に、あらかじめ目的とする抗原を添加し反応させた本発明の抗体と反応させる。次に、固相化された検体と反応した抗体を、あらかじめ標識しておいた抗免疫グロブリン抗体(二次抗体)と反応させ、標識物質によって抗体の量を検出することができる。検出される標識量は、添加された目的とする抗原の量と逆相関する。そのほかの競合反応系としては、本発明の抗体を固相化して、これに検体と反応させた後、あらかじめ標識しておいた目的とする抗原を反応させる。検出される標識量は、抗体と結合した検体中のGANP蛋白質量と逆相関する。
【0065】
前記した抗原または抗体の固相化法としては、物理的吸着法、共有結合法、イオン結合法、架橋法等、公知の方法を使用できる。特に、物理的吸着法が簡便という点で好ましい。また、抗免疫グロブリン抗体(二次抗体)としては、例えば、抗IgG抗体、抗IgM抗体等を用いることができる。これらの抗体は、抗体分子をそのまま使用してもよいし、あるいは抗体を酵素処理して得られる抗原結合部位を含む抗体フラグメントであるFab、Fab'、F(ab')2を使用してもよい。さらに、標識した抗免疫グロブリン抗体の代わりに、抗体分子に特異的な親和性をもつ物質、例えばIgGに特異的な親和性をもつプロテインA等を標識して使用してもよい。
【0066】
前記標識免疫測定法の好適な例として、酵素を標識とした免疫測定法、ELISA法を挙げることができる。ELISA法は、例えば、96穴プレートに検体またはその希釈液を入れて、4℃〜室温で一晩、または37℃で1〜3時間程度静置して検出すべきGANP蛋白質を吸着させて固相化する。次に、本発明の抗体を反応させ、次いであらかじめ酵素を結合させた抗免疫グロブリン抗体(二次抗体)を反応させる。最後に酵素と反応する適当な発色性の基質(例えば、酵素がホスファターゼの場合はp-ニトロフェニルリン酸等)を加え、この発色によって抗体を検出する。
また、本発明の高親和性抗体を利用することにより、各種疾患の治療薬の薬効評価を行うことができる。本発明の高親和性抗体を利用した薬効評価方法は、各種疾患患者あるいは各種疾患モデル動物に対して薬剤を投与後、これら生体中のウイルス等の抗原の量を本発明の抗体を用いて検出し、その量を比較することにより、生体中の抗原の量を通して各種疾患の治療薬としての薬効を評価することができる。
【0067】
本発明の高親和性抗体は、各種疾患診断用キットの形態で提供することができる。該キットは、本発明の診断方法や本発明の薬効評価方法に使用することができる。本発明のキットは以下の(a)及び(b)から選ばれる少なくとも一つ以上を含む。
(a)本発明の抗体またはその標識物
(b)前項(a)記載の抗体またはその標識物を固定した固相化試薬
ここで、抗体の標識物とは、酵素、放射性同位体、蛍光化合物、または化学発光化合物によって標識されたものを意味する。
また本発明のキットにおける抗体、もしくはこれらの標識物を固定する固相の材質としては、ポリスチレン、ナイロン、ガラス、シリコンラバー、セルロース等が挙げられ、固相の形状としては、球状、ウェル状、チューブ状、シート状等が挙げられるが、これらに限定されない。該固相化試薬の代わりに、固相と固相化に必要な固相化試薬を添付したものでもよい。固相化試薬として、例えば物理的吸着による固相化の場合は、50mM炭酸塩緩衝液(pH 9.6)、10 mMトリス-塩酸緩衝液(pH8.5、100mM塩化ナトリウム含有)、PBS等のコーティング液と、さらに必要に応じてコーティング液に0.5%のゼラチン等を含有させたブロッキング液が挙げられる。
【0068】
また、本発明のキットにおける抗体は、PBS等に溶解させた状態、あるいはゲルに結合させた状態(以下、「吸収用ゲル」と略す)であってもよい。前記吸収用ゲルはさらに適量を、バッチ法による吸収処理用に0.5〜2ml程度のマイクロ遠沈チューブに予めパッケージングされた状態であってもよく、あるいはカラム法による吸収処理用にカラム容量が0.1 〜5 mlのミニカラムに予め充填された状態であってもよい。
本発明のキットは、上記した構成要素のほか、本発明の検出を実施するための他の試薬、例えば標識物が酵素標識物の場合は、酵素基質(発色性基質等)、酵素基質溶解液、酵素反応停止液、あるいは検体用希釈液等を含んでいてもよい。前記検体用希釈液としては、例えばPBS(生理的リン酸緩衝液、pH7.4)、137mM塩化ナトリウムおよび3mM塩化カリウムを含むpH7.4かつ20mMのトリス-塩酸緩衝液(以下、「TBS」と略す)、0.05%Tween20、0.1〜1%のBSAを含有させたPBS、あるいはTBS等を挙げることができる。該検体用希釈液は、検体希釈以外、例えば抗体の希釈等に用いてもよい。
【0069】
(2) 疾患の治療又は予防用医薬組成物
本発明の高親和性抗体は、疾患の病原となる抗原の活性を中和させる作用を有するものであれば、疾患の治療又は予防のための医薬組成物として有用である。本発明の医薬組成物は、本発明の高親和性抗体またはその断片を有効成分として含み、さらに薬学的に許容される担体を含む医薬組成物の形態で提供することが好ましい。
ここで「薬学的に許容され得る担体」とは、賦形剤、希釈剤、増量剤、崩壊剤、安定剤、保存剤、緩衝剤、乳化剤、芳香剤、着色剤、甘味剤、粘稠剤、矯味剤、溶解補助剤あるいはその他の添加剤等が挙げられる。そのような担体の一つ以上を用いることにより、錠剤、丸剤、散剤、顆粒剤、注射剤、液剤、カプセル剤、トローチ剤、エリキシル剤、懸濁剤、乳剤あるいはシロップ剤等の形態の医薬組成物を調製することができる。これらの医薬組成物は、経口あるいは非経口的に投与することができる。非経口投与のためのその他の形態としては、一つまたはそれ以上の活性物質を含み、常法により処方される外用液剤、腸溶内投与のための坐剤およびペッサリーなどが含まれる。
【0070】
本発明の薬剤の投与量は、患者の年齢、性別、体重及び症状、治療効果、投与方法、処理時間、あるいは該薬剤に含有される活性成分である高親和性抗体の種類などにより異なるが、通常成人一人当たり、一回につき10μgから1000mg、好ましくは10μgから100mgの範囲で投与することができるが、この範囲に限定されるものではない。
【0071】
例えば、注射剤の場合には、例えば生理食塩水あるいは市販の注射用蒸留水等の薬学的に許容される担体中に0.1μg抗体/ml担体〜10mg抗体/ml担体の濃度となるように溶解または懸濁することにより製造することができる。このようにして製造された注射剤は、処置を必要とするヒト患者に対し、1回の投与において1kg体重あたり、1μg〜100mgの割合で、好ましくは50μg〜50mgの割合で、1日あたり1回〜数回投与することができる。投与の形態としては、静脈内注射、皮下注射、皮内注射、筋肉内注射あるいは腹腔内注射などが挙げられるが、好ましくは静脈内注射である。また、注射剤は、場合により、非水性の希釈剤(例えばプロピレングリコール、ポリエチレングリコール、オリーブ油のような植物油、エタノールのようなアルコール類など)、懸濁剤あるいは乳濁剤として調製することもできる。そのような注射剤の無菌化は、バクテリア保留フィルターを通す濾過滅菌、殺菌剤の配合または照射により行うことができる。注射剤は、用時調製の形態として製造することができる。即ち、凍結乾燥法などによって無菌の固体組成物とし、使用前に無菌の注射用蒸留水または他の溶媒に溶解して使用することができる。
【0072】
5.本発明の応用
本発明者らは、B細胞腫瘍株にGANPの過剰発現を誘導した上で解析を行った結果、B細胞腫瘍株はGANP遺伝子導入によって、飛躍的なV領域遺伝子の体細胞突然変異誘導効果を有することを示した。この効果は、GANPのRNAプライマーゼ活性に必要な502番目のセリンのリン酸化が起こらないような変異遺伝子を用いた場合には見られないことから、V領域遺伝子の体細胞突然変異の飛躍的な誘導には、RNAプライマーゼ活性が必要であることを示している。この結果は、臨床的な補助免疫賦活剤として、GANPが特異的抗体産生の増強効果を有することを示すものである。
【0073】
ベクターとしてレトロウイルスベクターを使用し、CD40、BAFFなどのTNFファミリー分子を介する刺激をGANPと併用することも、臨床的な補助免疫賦活作用にとって効果的である。また、この遺伝子導入を骨髄細胞レベルで行うことによって、T細胞における高親和性結合の誘導も期待できる。エイズ、C型肝炎ウイルス、成人T細胞白血病、狂牛病などの高親和性抗体が得られない場合や、あるいは得られたとしてもすぐに抗原の変異が起こるために十分に高親和性抗体の産生が持続できない場合には、この遺伝子導入は優れた効果を発揮すると期待できる。
【0074】
本発明のGANP遺伝子過剰発現哺乳動物は、生物学研究試薬、臨床検査試薬作製に有効なモノクローナル抗体の開発に有効である。例えば、特定のシグナル伝達分子に対するモノクローナル抗体を機能ドメインや機能モチーフに特異的にそして結合力の高い高親和性抗体を簡便に作製することは非常に活用される範囲が広い。多くの抗体はそれほど多くのスクリーニングをかけないため、ウエスタン解析と免疫沈降に用いることができない場合がある。この場合、本発明のトランスジェニック哺乳動物を用いれば、比較的少ないクローンの抗体から高親和性抗体産生細胞を短時間で選別することができ、経費、時間、労力の削減する効果は大きい。特にリン酸化抗体、遺伝子変異部分に対する特異抗体の作製は診断薬、あるいは抗体を用いた薬物の選択的注入法に応用できる。また遺伝子の配列やヌクレオチド部分に選択的に結合する高親和性抗体の産生も可能となる。
【0075】
無機物、炭水化物、化学合成物など、任意の物質の立体構造の一部は、抗原モチーフとして認識される。従来には高親和性抗体は得られていないが、自己免疫疾患マウスとの交配で作製されるマウスは、あらゆる抗原に対して高親和性抗体を得るために有効である。この方法で結合力が10-11Mオーダーの高親和性抗体ができる可能性があり、ELISA法の技術開発を導入することにより、微量物質の検出を簡便に行う技術を開発することが可能である。
また、本発明によれば、RNAプライマーゼ不活性型GANP遺伝子を含む、アレルギー疾患又は自己免疫疾患のための遺伝子治療剤を提供することも可能である。「RNAプライマーゼ不活性型GANP遺伝子」とは、RNAプライマーゼドメイン欠損、又はRNAプライマーゼドメインが変異した遺伝子を意味し、502番目のセリン残基の変異を含む近傍の遺伝子の変異によってGANP分子の構造および機能的な変化を生じた遺伝子のことを意味する。
【0076】
本発明の遺伝子治療剤は、RNAプライマーゼ不活性型GANP遺伝子を含む組み換えベクターを、遺伝子治療剤に用いる基剤と一緒に配合することにより製造することができる。組み換えベクターの構築の際に用いるベクターとしては、ウイルスベクターとしてレトロウイルスベクター、アデノウイルスベクター、アデノ随伴ウイルスベクター、バキュロウイルスベクター、ワクシニアウイルスベクターなどが挙げられ、あるいは動物発現用プラスミドを使用することもできる。ベクターは好ましくはウイルスベクターである。RNAプライマーゼ不活性型GANP遺伝子をウイルスベクターに組み込んだ場合は、組換え体DNAを含有するウイルス粒子を調製し、遺伝子治療剤に用いる基剤と一緒に配合することにより遺伝子治療剤を製造することができる。
【0077】
遺伝子治療剤に用いる基剤としては、通常注射剤に用いる基剤を使用することができ、例えば、蒸留水、塩化ナトリウム又は塩化ナトリウムと無機塩との混合物等の塩溶液、マンニトール、ラクトース、デキストラン、グルコース等の溶液、グリシン、アルギニン等のアミノ酸溶液、有機酸溶液又は塩溶液とグルコース溶液との混合溶液等が挙げられる。あるいはまた、当業者に既知の常法に従って、これらの基剤に浸透圧調整剤、pH調整剤、植物油、もしくは界面活性剤等の助剤を用いて、溶液、懸濁液、分散液として注射剤を調製することもできる。これらの注射剤は、粉末化、凍結乾燥等の操作により用時溶解用製剤として調製することもできる。
本発明の遺伝子治療剤の投与形態としては、通常の静脈内、動脈内等の全身投与でもよいし、局所注射又は経口投与等の局所投与を行ってもよい。本発明の遺伝子治療剤の投与量は、年齢、性別、症状、投与経路、投与回数、剤型によって異なるが、一般に、成人では一日当たり組み換え遺伝子の重量として1μg/kgから1000mg/kg程度の範囲であり、好ましくは10μg/kgから100mg/kg程度の範囲である。投与回数は特に限定されない。
【0078】
実施例
以下の実施例により本発明をさらに具体的に説明するが、本発明は実施例によって限定されるものではない。
【実施例1】
【0079】
自己免疫疾患モデル動物におけるGANPの発現とその機能
(材料及び方法)
【0080】
1.動物
NZB、NZW、B/WF1、MRL/lpr、及びBXSB マウスはJapan SLC Co.から購入した。
C57BL/6 及びBALB/cマウスはCharles River Japanから購入した。NODマウスは大阪大学大学院の宮崎博士から供与された。
【0081】
2.抗体及び試薬
マウスB220(RA3-6B2)、マウスIgM (AM/3) 及びマウスIgD (CS/15)に対するラットモノクローナル抗体はハイブリドーマの培養上清から精製し、D-ビオチン-N-ヒドロキシスクシンイミドエステル(Roche diagnostics, Branchburg, NJ)で標識した。ビオチン標識ラット抗マウスSyndecan-1及び抗マウスCD5モノクローナル抗体は購入した(BD PharMingen, San Diego, CA)。ビオチン標識ピーナッツアグルチニン(PNA)はVector Laboratories (Burlingame, CA)から購入した。
【0082】
3.免疫
トリニトロフェニル-キーホールリンペットヘモシアニン(Trinitrophenyl keyhole limpet hemocyanin (TNP-KLH)及びTNP-FicollはBiosearch Technologies (Novato, CA)から購入した。完全フロイントアジュバンドに乳化した100μgのTNP-KLHまたはPBS中の25μgのTNP-Ficollをマウスの腹腔内に注入した。14日後、リンパ器官を取得し、免疫組織分析用にOCT 化合物とともに凍結した。
【0083】
4.免疫組織分析
6μmの凍結切片をアセトンで5分間固定し、PBS中の3% BSAで15分間ブロッキングし、ラット抗マウスGANPモノクローナル抗体(42-23) [Kuwahara K., 他、2000, Blood 95: 2321-2328]またはラット抗-pSer502 GANPモノクローナル抗体 (PG/103) [Kuwahara K.,他、2001, Proc. Natl. Acad. Sci. USA 98: 10279-10283]と一緒に1時間インキュベートした。切片をのせたスライドグラスをPBSで数回洗浄し、アルカリホスファターゼ (ALP)-結合ヤギ抗ラットIgG 抗体(ICN Pharmaceuticals, Costa Mesa, CA)と一緒にインキュベートした。発色はVector Blue kit (Vector)を用いて行った。二重染色のために、反応はビオチン標識抗体と西洋ワサビペルオキシダーゼ(HRP)-結合ストレプトアビジン(Kirkegaard & Perry Laboratories, Gaithersburg, MD)を組み合わせて行った。3-3'-ジアミノベンジジンテトラヒドロクロライド(DAB; 同仁化学)による発色後、切片をPBS中1%グルタルアルデヒドで1分間固定した。Aquatex (Merck, Darmstadt, Germany)をマウンティングのために用いた。インビボで増殖活性のある細胞を検出するために、ブロモデオキシウリジン (BrdU) (Sigma Chemicals Co., St. Louis, MO; 1 mg/マウス)を屠殺する2時間前に静脈内に注入した。DNA合成を行う細胞を抗BrdUモノクローナル抗体 (BD PharMingen)とALP-結合ヤギ抗マウスIg 抗体(Sigma)とを組み合わせて染色しVector Red (Vector)により発色させて検出した。PAS染色は既報の通り行った [Jiang Y.他、1997, J. Immunol. 158: 992-997]。
【0084】
5.結果
(1) MRL/lpr マウスのリンパ節におけるGANPhi細胞の出現
GANP発現は自己免疫傾向の高活性B細胞で高発現している。高レベルのGANPを発現するリンパ細胞(GANPhi 細胞)は、MRL/lprマウスの末梢リンパ節において非免疫状態において自発的に出現する。
抗GANPモノクローナル抗体およびALP結合抗ラットIg抗体を用いて、自己免疫疾患モデルMRL/lpr、NZBおよび正常C57BL/6雌マウス由来の膝窩リンパ節について免疫組織化学分析を行った。
結果を図1に示す。Vector Blue(ALP基質)で染色されたGANPhi細胞は7週目にMRL/lprマウスのリンパ節で観察されたのに対し、同年齢のNZBマウスでは観察されず、40週目に出現した(図1)。正常C57BL/6マウスでは、極少数のGANPhi細胞が全期間を通じて観察された。
【0085】
自己免疫疾患モデルマウスは、C57BL/6 マウスと比較してリンパ細胞の増加は顕著であり、非免疫条件下ではGANPhi細胞は示されない(図1)。そのようなGANPhi細胞の出現を、加齢の間少しずつ自己免疫状態を引き起こすNZBマウスのリンパ節で調べた。若いNZBマウス(7週齢)は、膝窩リンパ節にGANPhi 細胞を有さないが、加齢したNZB (40 週齢)は多数のGANPhi 細胞を有した。
GANP RNAプライマーゼ活性はB細胞の活性化と分化において重要な役割を担っている可能性がある。そこで、NZBマウスで抗pSer502 モノクローナル抗体を用いて、RNAプライマーゼ活性の重要なリン酸化部位であるSer502のリン酸化状態を比較した。
NZBマウスのリンパ節中のGANPおよびpSer502 GANPの発現を比較した。pSer502 GANPは抗pSer502 GANP(PG/103)モノクローナル抗体(青)で検出し、全切片をビオチン標識抗B220モノクローナル抗体で染色した後、HRP結合ストレプトアビジンとDAB(茶色)を組み合わせて検出した。2回の独立した実験から代表的データを示した(図2)。
【0086】
図2において、下段の図(グラフ)は、加齢の間に濾胞外領域のGANPhi(黒の棒)およびpSer502 GANPhi(斜線の棒)の細胞数を示す。
GANPの発現は8週目で顕著であり、GANPhi 細胞が32週までの全期間を通じて検出された (図2;上図)。対照的に、pSer502-陽性細胞は8週目に最大であったが、その後、陽性細胞は顕著に減少した(図2;真中の図)。 ピーク年齢に基づく顕微鏡観察での反応性細胞数を図に示す(図2;下図)。これらの結果から、GANP発現は最初にRNAプライマーゼ活性を伴うが、この活性は長期間に渡っては調節されていないことが分かる。
【0087】
(2) 自己免疫傾向マウスの脾臓の赤脾髄におけるGANPhi細胞の自発的出現
自己免疫傾向NZBマウスの膝窩リンパ節で検出されたGANPhi細胞が非免疫状況下の脾臓に出現するかどうかを調べた。
免疫染色は前記(1)の操作(図2)と同様に行った。3回の独立した実験からの代表的データを示した(図3)。
GANPhi細胞は4週目に脾臓に出現し、細胞数は12週目に最大値に達したが、24週後に消失した(図3;上図)。pSer502 GANPの発現も8週目と12週目に検出された (図3;真中の図)。赤脾髄の相対細胞数と比較した結果、脾臓に出現したGANPhi細胞は 12週後には末梢リンパ節に移動していることが分かる。GANPhi の増加は、自己免疫疾患の発症に先行する自己抗体の産生量に比例している (図2及び図3; Theofilopoulos A.N.,他、1985, Adv. Immunol. 37: 269-390)。
【0088】
GANPhi細胞の出現は自己免疫傾向マウスにおけるB細胞の異常と関連している可能性があるので、非免疫条件下で各種の自己免疫傾向マウス(8週齢)におけるGANPhi細胞の出現を調べた。
結果を図4に示す。GANPhi 細胞はMRL/lpr、NZB及びB/WF1の赤脾髄で顕著に出現した。
GANPhi細胞の数はSLE-モデルマウスのBXSB 及びNODの脾臓にはそれほど増加しなかったが、対照のBALB/cマウス(図4) 及びC57BL/6マウス (図1)と比較すれば増加していた。脾臓の切片は、GC様構造としてPNA+ B細胞の連想又は未熟の会合を示した。GC様領域でのGANPの発現は、正常C57BL/6マウス及びBALB/cマウスに、T細胞依存性抗原(T cell-dependent Ags: TD-Ags)を免疫することによって作製したGCでのGANPの発現と比較して高くない。しかし、GANPhi 細胞は、自己免疫傾向マウスの赤脾髄領域で顕著に出現していた(図4)。
【0089】
さらに、GANPhi 細胞集団を、リンパ系細胞のマーカー分析によって解析した。
ビオチン標識抗B220モノクローナル抗体、ビオチン標識抗Syndecan-1モノクローナル抗体、ビオチン標識抗IgMモノクローナル抗体、及び抗IgG抗体を用いて、NZBマウスの脾臓切片について二重染色を行い、GANPhi 細胞を同定した。
結果を図5に示す。図5に示すパネルの左の列は、上から下に順にビオチン標識抗IgMモノクローナル抗体、抗IgG抗体、ビオチン標識抗B220モノクローナル抗体及びビオチン標識抗Syndecan-1モノクローナル抗体を用いたときの図である。中央の列は、上記それぞれの抗体を用いたのと同じ切片でのGANPの発現を示す。右の列は左の列と中央の列を重ね合わせた図である。右列の二重に染色された細胞は、GANPhi 細胞がB220-Syndecan1+IgM+であることを示す。GANP発現は、IgM、IgG及びB200の場合は赤色で示し、Syndecan-1の場合は緑色で示す。マーカーは、IgM、IgG及びB200は緑色で示し、Syndecan-1の場合は赤色で示す。
【0090】
GANPhi 細胞はB220-Syndecan-1+ の表現型を発現し、多量のIgM を細胞中に発現する(図5)。CR1、Thy-1、GL-7、CD23及びPNAについては陰性であり、これらの結果から、GANPhi 細胞がB系細胞の後期成熟段階、おそらく形質細胞であることが示される。このGANPhi 細胞が増殖性形質芽細胞であるかどうかを調べるために、NZBマウスにBrdU (1 mg/マウス)を投与(静脈内注入)し、インビボでBrdUを取り込ませるために2時間インキュベートした。その後、マウスから脾臓切片を調製した。
切片を抗GANPモノクローナル抗体(青)及び抗BrdUモノクローナル抗体(赤)で二重染色した。PAS染色を常法に従って行った。
結果を図6に示す。GCは胚中心を示す(左)。GANP単一陽性GANPhi 細胞は矢印で示し、PAS単一陽性細胞を矢頭で示す(中央)。
【0091】
また、切片をビオチン標識抗CD-5モノクローナル抗体で染色した。PALS領域は、リンパ節動脈周囲鞘を示す(右)。図6は、3回の独立した実験から代表的データを示した。
GANPhi 細胞はBrdU 取り込みについて陽性ではないことから(図6)、これらの細胞は増殖性ではなく、形質芽細胞段階より成熟していることが示唆された。
B-1細胞の異常な分化として、Mott細胞形成が自己免疫傾向マウスで観察される。Mott細胞は形質細胞の異常な形態であり、多量のIgM 分子が、PAS染色により細胞質内Russell小体として検出される粗面小胞体結合小胞に蓄積している [Jiang Y.,他、1997, J. Immunol. 158: 992-997]。GANPhi 細胞はPAS-染色で染色されず(図6)、これによりGANPhi 細胞をB-1 細胞由来形質細胞のMott細胞と区別することができる。脾臓GANPhi 集団はCD5発現が陰性であり(図6)、NZBマウス(12週)から得た腹膜細胞は GANPhi 細胞について陰性であったことから、B-1細胞は多量のGANPを発現していないことが示される。これらの結果から、GANPhi 細胞は自己免疫状態で高活性のB細胞に分類され、この集団がB-1細胞の起源とは異なる起源の系統であることが示唆される。
【0092】
(3) TD-Agでの免疫による正常マウスにおけるGANPhi細胞の誘導
二次リンパ器官におけるGANPhi形質細胞の出現が、自己免疫傾向マウスに限られているかどうかを調べた。
雌C57BL/6 マウス(7週齢)を、TNP-Ficoll (TI-2-Ag)又はTNP-KLH (TD-Ag)で腹腔内免疫し、14日後に脾臓を得た。TNP-Ficollで免疫したマウスは、ビオチン標識抗IgDモノクローナル抗体で対比染色した場合、GANPhi 細胞を赤脾髄領域で示さなかった(図7左)。TNP-KLHで免疫したマウスは 、GANPhi細胞の誘導を赤脾髄領域で示した(図7右)。図7において、GANPhi細胞は矢印で示す。WPは白脾髄領域を示す。
GANPhi 形質細胞集団は、数は非常に少ないが、TD-Agsによる免疫によって正常C57BL/6及びBALB/c マウスの脾臓においても誘導される(図7)。T細胞非依存性Ag (T cell-independent Ag: TI-Ag) による免疫はそのような細胞の誘導において効果は小さい。GANPhi 細胞集団はB220loIgMhiIgDloGL-7loPNAlo CD5loCD40lo と同様の表現型を示したが、Syndecan-1+を示した。
【0093】
これらの結果から、自己免疫傾向マウスにおけるGANPhi 形質細胞の生成は、TD-Agに対する免疫応答のために提供されるのと同様の刺激によって誘導されることが示される。GCで増殖と分化を経たAg駆動B細胞は、GANPを発現しながらより長い間、形質細胞段階として赤脾髄領域に局在化している可能性がある。
【実施例2】
【0094】
GANPの過剰発現
(方法)
1.Daudi細胞への安定なトランスフェクション
10μgの線状化したpCXN-2マウスGANP又はGANPS/A502 cDNAをDaudi細胞に、Gene Pulser II (Bio-Rad)を用いてエレクトロポレーションを行った。48時間後、G418 (Promega; 1 mg/ml)により選択を開始して、マウスGANPを安定に発現するDaudi細胞を得た。
【0095】
2.DaudiトランスフェクタントのIg VH 転写物の分析
全RNAを全細胞からTrizol (Invitrogen)を用いて抽出した。cDNAを既報の通り取得した(Kuwahara, K. et al., Blood 95, 2321-2328 (2000))。LVH3- CH1Cμ転写物を以下のプライマー及び反応液を用いて増幅した。増幅は、Pfu Turbo (Stratagene)を用いた。
【0096】
5'-LVH3 プライマー:
5'-CTATAACCATGGACCATGGACATACTTTGTTCC-3' (配列番号5)
3'-XbaI-CH1-Cμプライマー :
5'-TGCATGCATTCTAGAGTTGCCGTTGGGGTGCTGGAC-3' (配列番号6)
【0097】
反応液組成:
【表1】
【0098】
反応条件:
94℃ 1 min
[94℃ 1 min; 62℃ 1 min ; 72℃ 1 min] ×35 サイクル
72℃ 10 min
4℃
【0099】
PCR産物をNcoI及びXbaIで消化し、ゲルで精製し、NcoI-XbaIで消化したプラスミドとライゲーションした。コンピテント細菌に形質転換後、QIAprepキット(QIAGEN)を用いて調製した少量のプラスミドDNAの塩基配列を自動シークエンサー(Applied Biosystems)により決定した。
【0100】
3.GANP-トランスジェニック(Tg)マウスの作製
導入遺伝子は、pLGベクターのXhoIサイトに5.6 kbのマウスGANP遺伝子を導入して作製した。このベクターはヒト免疫グロブリンイントロンエンハンサー領域(2kb EcoRIフラグメント)を持ち、B細胞での強力な発現を行う、特異的ベクターである。この遺伝子を直線化してマウスに遺伝子導入を行った。マウスGANP全長cDNAを含む線状化したpLG vector(Koike, M. et al. Int. Immunol. 7, 21-30 (1995))をC57BL/6 マウスの受精卵にマイクロインジェクションした。マウスの尾のゲノムDNAおよび以下のプライマー及び反応液を用いて導入遺伝子の存在についてスクリーニングした。
【0101】
1-5' プライマー:
5'-TCCCGCCTTCCAGCT GTGAC-3'(配列番号7)
1-3'プライマー:
5'-GTGCTGCTGTGTTATGTCCT-3' (配列番号8)
【0102】
反応液組成:
【表2】
【0103】
反応条件:
[98℃ 5 sec; 59℃ 5 sec ; 72℃ 10 sec] ×35 サイクル
4℃
【0104】
4.RT-PCR
全RNAは、脾臓又は脾臓B細胞からTrizol(Invitrogen)を用いて抽出し、RT-PCRは、2種のプライマー1-5'及び 1-3'を用いて行い、cDNAを合成した(Kuwahara, K. et al., Blood 95, 2321-2328 (2000))。GANP 転写物はアガロースゲル電気泳動により検出した。β-アクチン転写物は対照として用いた。
【0105】
5.結果
(1) Daudi細胞にマウスGANPを安定的に発現させたトランスフェクタントのV領域遺伝子の体細胞突然変異(SHM)
GANP遺伝子を、インビトロでSHMの分析に使用される各種ヒトBリンパ球細胞に導入した(Rogozin, I. B., et al., Nat. Immunol. 2, 530-536 (2001); Kuwahara, K. et al. Blood 95, 2321-2328 (2000); 及びDenepoux, S. et al., Immunity 6, 35-46 (1997))。多くのB細胞株にはトランスフェクションできなかったが、維持中はSHMを通常は生成しないAIDを発現するDaudi B細胞にはGANP遺伝子を導入できた。
このクローンは、野生型及び偽トランスフェクションした細胞と比較して高頻度のSHM (5 x 10-4/bp)をV領域で示した。
VH3-CH1Cμの断片をPCRで増幅してプラスミドにサブクローニングし、シークエンスした。
【0106】
体細胞変異の模式図を図8A〜Cに示す。縦線(「|」)はサイレントミューテーション(アミノ酸が変わらない)、もう一つの記号(縦線に●印を付したもの)にはアミノ酸が置換している変異を示す。Daudi/mockではほとんど変異が入っていないが、Daudi/GANP-14, 15, 17, 21の4クローンは、程度の差はあるが変異を多く認める。DNAプライマーゼ活性の制御に関係する502番目のセリンをアラニンに置換した変異体(GANP S/A)を導入したトランスフェクタントでは変異の入る効率が減少する。
SHMは、定常領域遺伝子には誘導されなかった(図8A〜C)。GANPのRNAプライマーゼ活性はS502でのリン酸化により調節され、これは特異的モノクローナル抗体により検出できる(Kuwahara, K. et al., Proc. Natl. Acad. Sci. USA 98, 10279-10283 (2001))。インビトロ及びインビボでのB細胞の刺激は共にS502のリン酸化を誘導するので(Kuwahara, K. et al., Proc. Natl. Acad. Sci. USA 98, 10279-10283 (2001))、このリン酸化がDaudi B細胞でのSHMの生成に関与するかどうかを調べた。
非リン酸化GANP変異体(GANP-S502A)を導入した場合、SHMは誘発されなかったことから(図8A)、S502のリン酸化が、GC-B細胞でのSHMの生成に重要であることが示唆された。
【0107】
(2) B細胞でGANPを過剰発現させたトランスジェニックマウス
免疫応答におけるGANPの関与を調べるために、ヒトIgエンハンサー及びプロモーターの制御下にマウスGANPを過剰発現させたGANP-トランスジェニック (Tg)マウスを作製した(図9A及びB)。GANP mRNAの発現の亢進は、RT-PCRで確認した。
このマウスは、B細胞でGANPの発現の増加を示し(図9C)、骨髄、脾臓及びリンパ節の細胞の表層マーカー分析において、B系細胞の通常の分化を示した。
SHMにおけるGANPのインビボでの役割を調べるために、TD-AgであるNP-CGの免疫後におけるVH186.2 領域について調べた。すなわち、GANP過剰発現トランスジェニック(Tg)マウスに50μgのNP-CGを2週間おきに3回免疫して、VH186.2をPCRで増幅し、体細胞突然変異を解析した。
【0108】
結果を図10に示す。変異の数はTgでやや増加しているが、高親和性を示す33番目のWがLに変わる変異(SHM)は、Tgで約3倍に増加していた。なお、CDRは相補鎖決定領域を示す。
VH186.2ローカスは、高親和性IgG(γ1λ1)NP-応答について特有のパターンのSHMを示す。NP-CGでの免疫後における全脾臓B細胞の配列分析により、野生型マウスと比較してGANP-Tg マウスではSHMの頻度が僅かに増加していることが示された(図10)。
この変異は、ハプテン特異的B細胞の親和性成熟に重要であることが以前に示されている(Allen, D. et al., EMBO J. 1995-2001 (1988))。
【実施例3】
【0109】
B細胞特異的GANP欠損マウス(B-GANP-/-マウス)の作製
(方法)
1.CD19-Cre/+GANP flox マウスの樹立
GANPゲノムDNAを用いて、エクソンIIの下流にネオマイシン耐性遺伝子(neo)を挿入することによってターゲッティングベクターを調製した。loxP部位は、neoの3’側の隣接領域とエクソンI及びIIの間のイントロンに導入した。
つまり、エクソンIIをloxP配列で挿んだfloxマウスを作製し、これとCD19-Creマウスとを交配させて、B細胞でGANPが欠損するマウスを樹立した(図11A及びB)。
直線化したターゲッティングベクターをTT2 ES 細胞(Yagi, T. et al. Anal. Biochem. 214, 70-76 (1993))にエレクトロポレーションによりトランスフェクションした。G418で選択後、ES コロニーを拾い上げ、プロテナーゼKと一緒にインキュベートした。相同組換え体を下記neo2プライマー及びCGK3'-2プライマーによりスクリーニングした。
【0110】
neo2プライマー:
5'-GCCTGCTTGCCGAATATCATGGTGGAAAAT-3' (配列番号9)
CGK3'-2プライマー
5'-GGCACCAAGCATGCACGGAGTACACAGA-3' (配列番号10)
【0111】
相同組み換えは、BamHIで消化したESクローンのDNAをプローブAを用いてサザンブロット分析することにより確認した。4kbのバンドを示す3個の陽性クローンを用いて、ICR胚盤胞へのマイクロインジェクションによりキメラ初代マウスを作製した。なお、B細胞でGANPの発現が見られないことは、サザンブロッティング、RT-PCRと細胞染色で確認した(図11C、D及びE)。
GANP flox/+マウスは少なくとも10回、C57BL/6マウスと戻し交配した。B細胞におけるGANP遺伝子を欠失させるために、GANP-floxed マウスとCD19-Cre ノックインマウス(Rickert, R. C., et al., Nucleic Acids Res. 25, 1317-1318 (1997))とを交配した。
【0112】
2.FACS分析
リンパ器官由来の単一細胞懸濁物を、各ビオチン標識モノクローナル抗体により氷上で1時間染色した。染色バッファーで洗浄後、細胞をFITC結合ストレプトアビジン (Amersham Bioscience)及びPE結合モノクローナル抗体で1時間インキュベートした。リンパ細胞を、CellQuestソフトウエアを用いて FACScan (Becton Dickinson)により分析した。
【0113】
3.B細胞の精製
脾臓細胞をCre-flox/+マウス及び B-GANP-/-マウス(7から8週齢) から単離し、0.15 M 塩化アンモニウム緩衝液で処理して赤血球を除いた。プラスチック皿上で37℃で30分間インキュベーションした後、未接着細胞をリンパ球として回収し、T細胞をDynabeads-anti-mouse Thy1.2 モノクローナル抗体(Dynal)を添付のプロトコールに従って使用して除去した。B細胞の純度(90%以上)をFITC結合抗B220モノクローナル抗体(BD Pharmingen)を用いた細胞表層染色により確認した。
【0114】
4.インビトロ増殖アッセイ
精製したB細胞を、10%熱不働化FCS(JRH Biosciences)、2 mMのL-グルタミン及び5×10-5 Mの2-メルカプトエタノールを含むRPMI-1640 培地(分裂促進剤を含むものと含まないもの)中において、96穴マイクロタイタープレートで2×105 細胞/ウエルで48時間インキュベートした。細胞を、0.2μCi/ウエルの[3H]-チミジン(ICN)で16時間パルスしてから回収し、取り込まれた放射活性をシンチレーションカウンターで測定した。
分裂促進剤は、アフィニティー精製したヤギ抗マウスμ鎖特異的抗体(10μg/ml)[F(ab')2] (ICN)、ラット抗マウスCD40モノクローナル抗体(LB429; 10μg/ml)及びLPS(Sigma; 10μg/ml)を用いた。
【0115】
5.抗原及び免疫
TNP-KLH、TNP-Ficoll及びニトロフェニル-ニワトリγグロブリン(NP-CG) (23:1)は、Biosearch Technologiesから購入した。50μgのTNP-KLH及びNP-CG (アルミニウムで沈殿)、又は25μgのTNP-Ficoll(PBSに溶解)をCre-flox/+マウス及びB-GANP-/-マウスの腹腔内に注入した。
【0116】
6.抗原特異的抗体産生の測定
抗原投与の10日後と14日後に、免疫したマウスから血清を回収した。5μg/ウエルのTNP-BSA (Biosearch Technology) をELISAプレートに被覆した。各ウエルをPBS中の3% BSAでブッロキングし、段階希釈した血清とインキュベートした。PBS-0.1% Tween 20で洗浄後、ウエルをビオチン結合isotype特異的モノクローナル抗体及びアルカリホスファターゼ(ALP)結合ストレプトアビジン(Southern Biotechnology)とともにインキュベートした。発色は基質の存在下で行った。
血清中のNP結合抗体の親和性を求めるために、NP2結合抗体のNP25結合抗体に対する割合を、被覆抗原にNP2-BSA(1分子あたり2個のNPがBSAに結合したもの)及びNP25-BSA(1分子あたり25個のNPがBSAに結合したもの)(Biosearch Technology)を用いたディファレンシャルELISAにより計算した。
【0117】
7.免疫組織化学
免疫したマウスからの8μmの脾臓切片をアセトンで軽く固定した。サンプルをPBS-Tween 20中の3% BSAでブロックし、抗IgDモノクローナル抗体及びALP-結合抗ラットIgG(ICN)抗体とともにインキュベートした。第一の発色はVector Blueキット(Vector)を用いて行った。第二の発色は、サンプルをビオチン結合ピーナッツアグルチニン(PNA)(Vecor)及び西洋わさびペルオキシダーゼ結合ストレプトアビジン(Kirkegaard & Perry)とともにインキュベートし、次に3,3'-ジアミノベンジジンテトラ塩酸塩(Dojindo)とともにインキュベートした。PBS中の1%グルタルアルデヒドを用いてサンプルを固定した後、Aquatex(Merck)によりマウンティングを行った。
【0118】
8.VH186.2 遺伝子の配列分析
NP-CGで免疫したCre-flox/+及びB-GANP-/-マウスからのNP-結合IgG1dullCD38low B細胞を、(4-ヒドロキシ-5-ヨード-3-ニトロフェニル)アセチル (NIP)を用いてFACS Vantage (Becton Dickinson Biosciences)で分画し、プロテナーゼKと一緒に37℃で一晩インキュベートした。ライセートを用いて、PCRを既報の通り(Takahashi, Y., et al. Immunity 14, 181-192 (2001))2回実施した。VH186.2 遺伝子DNAをpBluescriptにライゲーションし、自動シークエンサーにより配列を決定した。
【0119】
9.アポトーシス細胞の検出
Cre-flox/+及びB-GANP-/-マウスから精製したB細胞を40時間、種々の試薬で刺激した(Watanabe, N. et al. (1998) Scand. J. Immunol. 47, 541-547)。AICDには、24ウエルプレートに抗μ抗体(50μg/ml)を固定した。そのほかの場合には、精製したB細胞を種々の刺激物質で48時間刺激し、続いて抗Fasモノクローナル抗体(Jo2;BD Pharmingen)で4時間インキュベートした(Wang, J. et al. (1996) J. Exp. Med. 184, 831-838)。細胞を、プロピジウムアイオダイド(PI)溶液(50μg/ml PI, 0.1% Triton X-100, 0.1% クエン酸ナトリウム)を用い、室温で1時間インキュベートし、アポトーシス細胞(%)をFACScanでG1以下領域として計算した。なお、アポトーシス細胞は、トリパンブルーによる染色後、顕微鏡下での確認も行った。
【0120】
10.TUNELアッセイ
Cre-flox/+及びB-GANP-/-マウスをSRBC(ヒツジ赤血球細胞)で免役した後、それぞれの脾臓切片を調製し、PBS中の4%パラホルムアルデヒドで凍結切片を固定した。切片サンプルを、MEBSTAIN Apoptosis Kit II(MBL)で処理し、PIで対比染色した。TdT媒介dUTP-ビオチンニック-エンド標識(TUNEL)アッセイと共に行う実験用に、切片は抗IgG1 モノクローナル抗体(BD Pharmingen)及びAlexa546-結合ヤギ抗ラットIgG抗体(Molecular Probes)を用いた染色も行った。陽性シグナルを検出し、結果を蛍光顕微鏡(BX51; Olympus)により確認した。
【0121】
11.結果
(1) RNAプライマーゼGANPの役割
RNAプライマーゼGANPの役割を調べるために、Cre-loxP システムを用いてCD19+ B細胞にしてGANP遺伝子を欠失させた B-GANP-/-マウスを作製した(図11A及びB)。B-GANP-/-マウス遺伝子は、エクソンIIをほとんど欠損していた(図11C)。B-GANP-/-細胞はGANP mRNAを発現せず(図11D)、また免疫染色によればタンパク質をほとんど発現しなかった(図11E)。B-GANP-/-マウスは正常に成育し、骨髄、脾臓、胸腺、リンパ節で正常な数のリンパ細胞を示した。フローサイトメトリー分析では、B-GANP-/-は、骨髄、脾臓及びリンパ節の細胞上に、対照であるCre-flox/+マウスと同様の表面マーカープロフィールを示し(図12)、Cre-flox/+(対照)と差はなかった。
【0122】
B-GANP-/-のリンパ節では、sIgMlowsIgDhigh を発現する成熟B細胞(IgM+IgD+)の数が減少していた。B-GANP-/-マウス由来のB細胞は、インビトロで抗μ抗体、抗μ抗体+抗CD40モノクローナル抗体、またはリポ多糖による刺激後に通常の増殖応答を示した(図13:B-GANP-/-は白の棒、Cre-flox/+は黒の棒)。一方、B-GANP-/-マウス由来のB細胞は、抗CD40モノクローナル抗体(5, 10μg/mL)による刺激後に増殖活性が低下した(図13)。このことは、B-GANP-/-マウスのB細胞の増殖では、CD40/CD145相互作用への反応がわずかに損なわれていることを示す。血清Ig量はCre-flox/+マウスと同様であった(図14)。
【0123】
(2) B-GANP-/-マウスにおける抗原特異的抗体産生
TI-Ag及びTD-Agによる免疫後のB-GANP-/-マウスの免疫応答を調べた。TI抗原であるトリニトロフェニル(TNP)-Ficollを免疫して14日後に、抗TNP抗体価をELISAで測定した。その結果、(TNP)-FicollはB-GANP-/-マウス及びCre-flox/+マウスにおいて同様の応答を誘導し、特に差はなかった(図15)。
胚中心(GC)形成を調べてみると、TD-AgであるTNP-keyhole limpet hemocyanin (KLH)及びNP-CGなどのTD-Agsに応答して、変異体マウスはCre-flox/+マウスと比べて遅延したGC形成を示した。
【0124】
GC形成のピーク応答については、Cre-flox/+は、10日目にGC-B細胞のマーカーであるピーナッツアグルチニンで染色された大きな成熟したGCを示した(図16の矢印)。免疫後10日ではB-GANP-/-の方がGC形成がやや少なかった。しかし、14日後ではCre-flox/+に比べてB-GANP-/-の方が数が多くなっており、20日後でもGC形成が遷延していた(図16の矢印)。
B-GANP-/-マウスは14日目にGCの明白な形成を示したので、抗原特異的抗体応答を測定した(図17)。TD抗原であるTNP-KLHによる免疫では、B-GANP-/-マウスは10日目まで明確なGCを示さず、抗体価はほとんど差がなかったが、TNP-KLHによる免疫後の14日目には、B-GANP-/-でGCの段階的な増加と拡大を示した (図17)。変異体マウスはTNP-KLHに対してCre-flox/+マウスと同様の抗体応答を示した。
【0125】
(3) B-GANP-/-マウスにおける親和性成熟の障害
B-GANP-/-マウスにおけるGCの特徴(抗体応答が低親和性であること)をさらに調べるために、抗原特異的IgG1+ GC-B細胞をNP-CGによる免疫後に調べた。
異なる分子量を有するNPのハプテン/タンパク質コンジュゲートを用いたディファレンシャルELISAにより、マルチハプテンNP25-BSAコンジュゲートに対する応答と比較した。
B-GANP-/-マウスにおいて、NP2-BSAコンジュゲートに対する抗体応答は、NP-CG免疫後35日目において低親和性(13%)であった。この値は、Cre-flox/+マウスの値(42%)と比較して抗体反応は著しく減少していた(図18)。
【0126】
また、図19に示す通り、NP-特異的IgG1dullCD38low B細胞はB-GANP-/-マウスでは著しく減少した。すなわち、免疫後10日目において、Cre-flox/+マウスでは1,164細胞/106細胞であるのに対し、B-GANP-/-マウスでは88細胞/106細胞であった。また、14日目においては、Cre-flox/+が879細胞に対し、B-GANP-/-は83細胞であった。なお、この傾向は20日目も同様であった。
対照的に、IgG1highCD38high メモリーB細胞は減少しなかった。これらの結果は、GANP の無発現変異はIgG1highCD38low GC-B細胞段階でB細胞の分化に欠陥を生じさせていることを示す。
B-GANP-/-マウスにおける抗体の親和性成熟の減少を確認するため、NP-CGで免疫した後の脾臓B細胞のVH186.2領域の配列を調べた。
【0127】
SHMはB細胞分化のこの段階で生じるため、VH186.2 ローカスのSHMについて各種の精製したB細胞を調べた。なお、VH186.2 ローカスは、高親和性IgG (γ1λ1) NP-応答のために利用されている(Cumano, A. & Rajewsky, K. (1985) Eur. J. Immunol. 15, 512-520)。IgM ローカスはCre-flox/+と比較して差異がなかった。
次に、Cre-flox/+又はB-GANP-/-にNP-CGを免疫し、NP-結合IgG1弱陽性CD38弱陽性を示すGC-B細胞(Ag-結合IgG1 B細胞)についてソーティングした(図19)。また、ソーティング後の細胞からゲノムDNAを抽出し、VH186.2をPCRで増幅し、シークエンス解析を行い、VH186.2 の配列を比較した(図20A〜L)。図20A〜FはCre-flox/+のVH186.2 の配列を、図20G〜LはB-GANP-/-のVH186.2 の配列を比較したものである。
【0128】
B-GANP-/-マウスにおいて、IgV領域配列の全体の変異は14×10-3であり、Cre-flox/+マウス(21×10-3)と比較して変異の数が減少していた(図21)。さらに、W33からLへの高親和性型の変異(33番目のアミノ酸残基トリプトファンからリシンへの変異、C57BL/6 マウスで顕著に見られる)は13%(2/15 V領域)であり、変異型マウスでは、Cre-flox/+マウスの値(71%, 10/14 V領域)と比較してより顕著に減少し、親和性は1/3に低下した(図22)。
以上の結果より、GANPは抗体の親和性の成熟に必須であることが示された。
【0129】
(4) B-GANP-/-マウスB細胞におけるアポトーシスからの保護機能
B-GANP-/-マウスにおいて、高親和性抗体の産生が減少するのは、抗原刺激後のB細胞が不安定であるためであると考えられる。そこで、B細胞の感受性を調べるため、in vitroでのB細胞のアポトーシスを検討した。
正常なB細胞は、強力に架橋したB細胞抗原受容体により活性化誘導細胞死(AICD)が誘導され、AICDは、CD40の刺激によって阻止された。B-GANP-/- B細胞では、AICDの刺激に対する感受性は正常B細胞(対照)と同程度であったが、抗CD40を介するアポトーシスの抑制の程度については、Cre-flox/+対照B細胞よりも劣っていた(図23)。このことは、B-GANP-/-マウスは、GC形成期の間、抗原反応性B細胞の保護機能に欠けることを示す。
【0130】
GCでは、抗原刺激されたB細胞とCD40/CD154との相互作用によってFas/CD95の表面発現が誘導され、Fas誘導性アポトーシスに感受性を持つようになる。そこで、本発明者は、in vitroにおいてアポトーシスを誘導する抗CD95刺激に対するB-GANP-/-B細胞の感受性を測定した。
まず、脾臓B細胞を抗CD40モノクローナル抗体(LB429)、抗μ抗体+抗CD40モノクローナル抗体、IL-4+抗CD40モノクローナル抗体、及び抗μ抗体+IL-4+抗CD40モノクローナル抗体で48時間刺激し、次に抗CD95モノクローナル抗体を培地に添加した。
その結果、B-GANP-/-マウスのB細胞のアポトーシス応答は、Cre-flox/+マウスのB細胞の応答と同様であり、B-GANP-/-マウス及び Cre-flox/+マウスでは差がなかった(図24)。
【0131】
上記の通り、抗CD40(LB429)処理後に抗CD95で刺激しても、発現誘導は、B-GANP-/-マウス及び Cre-flox/+マウスでは差がなかった。このことは、B-GANP-/- マウスのB細胞においては、in vivoでGC-B細胞により通常受けるアポトーシス刺激に対して感受性を有することを示す。そこで、TUNELアッセイを、TD-AgとしてSRBCを用いて免疫した後の組織切片について行った。
その結果、TUNEL陽性細胞は、B-GANP-/-マウスのGC領域において増加しており、ほとんどの細胞は、IgG1を発現することも示された(図25、26)。これらの結果は、B-GANP-/-マウスのアポトーシス細胞は、ほとんどがGC-B細胞であることが示された(図25、26)
次に、種々の悪性リンパ腫細胞及び正常B細胞のCD40媒介性アポトーシス抑制に必要な分子であると認識されているBcl-2ファミリーメンバーのRNA発現を検討した。
【0132】
抗μ抗体+IL-4で刺激すると、Cre-flox/+ B細胞におけるbcl-2の転写の明らかな増加を誘導し、抗CD40 mAbはその発現をさらにアップレギュレートした(図27)。また、B-GANP-/- B細胞については、抗μ抗体によるbcl-2転写物と同様のアップレギュレーションを示したが、抗CD40 mAb(抗CD40 mAb単独又は抗μAb + IL-4 + 抗CD40 mAb)に対する応答は、Cre-flox/+のB細胞ほど高くはなかった(図27)。すなわち、アポトーシス抑制に関与するBcl-2ファミリーのRNA発現レベルは、抗CD40抗体で刺激したときのB-GANP-/-マウスB細胞において、対照よりも低下していた(図27)。
【0133】
変異B細胞中のbcl-XL、bax及びbadについては、Cre-flox/+ B細胞のレベルと同程度であった(図27)。
以上の結果は、GANPは、GC-B細胞においてCD-40を介したBcl-2の誘導に関する情報伝達を制御しており、in vivoでの高親和性BCR+ B細胞の生存に大きく寄与していることを示す。
【0134】
(5) まとめ
B-GANP-/-マウス及びGANP-Tg マウスの結果は、GANPがTD-Agによる免疫後における高親和性B細胞の生成に関与していることを実証している。GANPの役割としては、GC-B細胞でDNAポリメラーゼの効率的なリクルートと調節を仲介している可能性がある。V-領域SHMを有する GC-B細胞が高親和性BCRを取得すると、それらは選択されるべきであり、そしてさらにB細胞のV領域のSHM は抑制されて、インビボで高親和性抗体の産生が保証されるはずである。GC-B細胞でのAIDの発現はDNA変異を絶えず生成する可能性があるため、AID活性の調節がB細胞での高親和性BCRを維持するために必要であるかもしれない。B-GANP-/-マウスでの結果から、GANPがSHMプロセスに必要であることが示される。
【実施例4】
【0135】
GANPトランスジェニックマウスを用いた高親和性抗体の産生
1.ディファレンシャルELISAによる抗体価の比較
野生型(WT)マウスとGANPトランスジェニック(Tg)マウスの2匹ずつにNP-CG 100 μgを免疫し、28日後の血清を取り、ELISAを行った。20μg/mlのNP2-BSAとNP17-BSAをELISAプレートに4℃で1晩コーティングした。3% BSA/PBS-0.1% Tween20で1時間ブロックし、血清を1時間反応させた。PBS-0.1%Tween20で3回洗い、ビオチン標識抗マウスIgG1抗体(Southern Biotechnology)で1時間反応させた。PBS-0.1%Tween20で3回洗い、アルカリホスファターゼ標識ストレプトアビジン(Southern Biotechnology)で30分反応させた。PBS-0.1%Tween20で3回、TBSで1回洗い、p-Nitrophenyl phosphateを基質として発色させた。吸光度を405nmで測定した。
結果を図28に示す。図28の結果から、GANPトランスジェニックマウスを使用することにより高親和性抗体が産生されることが分かる。
【0136】
2.ELISA及びBiacoreを用いた抗原-抗体結合親和性解析
野生型(WT)マウスとGANPトランスジェニック(Tg)マウスに、免疫抗原としてNP-CGを免疫し、それぞれのマウスを用いて細胞融合を行い、得られた陽性ハイブリドーマの培養上清において、ELISA法及びBiacoreを用いて抗原と反応する抗体の結合曲線を得た。得られた結合曲線から、Tgマウスの有用性を導いた。
(1) 材料
(a) 動物
野生型(WT)マウスとGANPトランスジェニック(Tg)マウス。
(b) 抗体及び試薬
NP16-CG(一分子あたり16個のNPがCGニワトリイムノグロブリンに結合したもの)、NP2-BSA(一分子あたり2個のNPがBSA(牛血清アルブミン)に結合したもの)、NP17-BSA(一分子あたり17個のNPがBSAに結合したもの)、HRP標識抗マウスIgG、IgA及びIgMを用いた。
【0137】
(2) 方法及び結果
野生型(WT)マウスとGANPトランスジェニック(Tg)マウスの各5匹に、免疫抗原としてNP16-CGを、2週間をおきに3回免疫し、3回免疫後に採血し、抗血清を用いて抗体価の比較をしたところ、前記1項に記載の結果と同様にGANP-Tgマウスの有用性が確認された。
この中から、力価の高いマウスの脾細胞を用いて、P3U1ミエローマ細胞と細胞融合を実施し、GANP-Tgマウスの脾細胞数(6.0×107個)、WTの脾細胞数(4.8×107)に基づき、1×105/ウェルになるようにまきこみ、GANP-Tgマウス由来の細胞は600クローン、WTマウス由来の細胞は480クローンをHAT培地にて培養した。
HAT培養9日後の培養上清を用いて、NP2-BSA(1μg/mL)を固相化抗原としてELISA法を実施した。GANP-TgマウスおよびWTマウスの培養上清のそれぞれから、ELISA吸光度結果において高親和性抗体の産生を示す上位2.5%のクローンを選出し、HT培地にてクローニングを実施した。
【0138】
HT培養9日後の培養上清を用いて、NP2-BSA(1μg/mL)を固相化抗原としてELISA法を実施した結果、GANP-Tgは6クローン(クローン名:G2-6, G2-9, G2-12, G2-14, G2-15, G2-16)、WTは1クローン(クローン名:W2-7)のハイブリドーマを樹立した。
GANP-Tgマウス、WTマウスのそれぞれのクローンをRPMI培地にて培養し、以下の実験に用いるのに適した培養上清を1mLずつ確保した。この培養上清を用いて、以下の評価検討を実施した。
【0139】
(a) ELISA法
抗体価を評価するにあたり、抗原の性質の異なるもの、いわゆる、CG一分子あたりNP含有率の異なる物質を用いて、そのELISA反応性の比率をもって評価した。
本法は、NPの親和性を測るのに有用な測定法であり、簡便で多くの検査をすることができるので、一次スクリーニングとして適当で信頼できるものである。
【0140】
まず、NP2-BSA(1μg/mL)とNP17-BSA(1μg/mL)をそれぞれ固相化抗原として用い、4℃で一晩固相化した。固相化プレートをPBS-Tween20で洗浄した後、スキムミルクPBS-Tween20にてブロッキングを実施した。さらにプレートをPBS-Tween20で洗浄した後、GANP-Tgマウスの6クローン(クローン名:G2-6, G2-9, G2-12, G2-14, G2-15, G2-16)及びWTマウスの1クローン(クローン名:W2-7)のRPMI培養上清(原液〜256倍希釈液)を用いて、室温で1時間固相化抗原と反応させた。次に、プレートをPBS-Tween20で洗浄した後、HRP標識抗マウスIgG、IgA及びIgMを室温で1時間反応させた。さらに、プレートをPBS-Tween20で洗浄した後、オルトフェニレンジアミン(OPD)にて5分間発色し、2N硫酸を用いて反応を停止した。
吸光度は、ELISA READERを用いて、490nmにて測定した。
ELISAの結果を図29に示す。図29の結果よりGANP-Tgマウスを使用することにより、高親和性抗体が産生されることがわかる。
【0141】
(b) Biacoreを用いた高親和性解析
上記ELISAの結果で、一番親和性の高いと予想されるクローンについて、Biacoreを用いて物理化学的結合能を調べた。
Biacoreによる解析は、NP2-BSA(1μg/mL)をリガンドとしてBiacoreチップに結合させたものに、アナライト溶液として一番親和性の高いと思われるTg(G2-9)、一番低いと思われるTg(G2-15)、及びWT(W2-7)の3クローンのRPMI培養上清を用いて、それぞれの結合速度定数(k ass)、解離速度定数(k diss)、及び親和性の指標となる解離定数KD (KD = k diss/k ass)を算出した。KDが小さい程、親和性は高いと評価される。
【0142】
その結果として、G2-9(ELISA: 82.9% NP2/NP17比)のビアコアのパターンを図30に示す。図30に示す曲線(a)〜(e)は、抗体濃度がそれぞれ26.6、13.3、6.65、3.33及び1.66 nMのものである。上記結果から、結合速度定数(k ass)=1.48×105、解離速度定数(k diss) = 9.63×10-4、そして親和性の指標となる解離定数は、KD (KD = k diss/k ass)=6.50×10-9となった。
一方、ELISAの結果から比較的親和性の低いと予想されるG2-15(ELISA: 33.9% NP2/NP17比)のビアコアのパターンを図31に示す。図31に示す曲線(a)〜(e)は、抗体濃度がそれぞれ23.0、11.5、5.75、2.88及び1.44nMのものである。
【0143】
結合速度定数(k ass) = 5.33×104、解離速度定数(k diss) = 1.56×10-2、そして親和性の指標となる解離定数は、KD (KD = k diss/k ass)=2.92×10-7となった。この値は、ELISA法でも同等の親和性を示すW2-7(ELISA: 31.6% NP2/NP17比)の活性を持つKD値=1.67×10-7と近似していた。
以上をもって、GANPトランスジェニック(Tg)マウスを使用することにより、高親和性抗体が産生されることが明らかである。
【実施例5】
【0144】
GANPとMCM3の会合、細胞周期中の核/細胞質間の移動
1.概要
本実施例では、本発明者は部分切断型の変異体GANPを用いることによってGANPのMCM3結合領域を決定し、GANP特異的な領域における502番目のアミノ酸のリン酸化セリン(pSer502)に対する特異的なモノクローナル抗体を用いてNIH-3T3細胞におけるGANPの局在を特徴づけた。
プライマーゼとMCMとの結合は、一つの連動した機能であり、両者が結合した分子複合体はDNAを二重鎖から解く作用を有する。従って、GANP部分断片がMCMと結合すれば、当該GANP断片もプライマーゼ活性を発揮し、高親和性抗体の産生作用を有するものと考えられる。
そこで、GANP及びその部分断片、Map80及びMCM3の核/細胞質区画の局在を、cDNAトランスフェクションと細胞融合実験を用いて解析した。
得られたデータから、GANPはMCM3と結合することが示され、さらに、GANPの局在はMCM3の発現によって影響されることが示された。GANPはMap80の結合機序とは異なる結合機序によってMCM3と会合する。これらの結果は、MCM3に結合したGANPは、Map80/MCM3APの機能とは異なる独特の機能を介することを示す。
【0145】
2.材料および方法
2.1. 細胞および細胞培養
NIH-3T3、COS7、HelaおよびSwiss-3T3細胞を、10%熱不活化FCS(大日本製薬)、2 mM L-グルタミン(Biowhittaker)、100 μg/mlストレプトマイシン、100 U/mlペニシリン及び50 μM 2-メルカプトエタノールを補充したD-MEM培地(Invitrogen)中で、37℃で、5% CO2のもとで維持した(Takei, Y. et al., (1998) J. Biol. Chem. 273, 22177-22180、 Sakaguchi, N. et al., (1988) EMBO J. 7, 3457-3464、Kimura, H. et al., (1995) Nucl. Acids Res. 23, 2097-2104)。BAL17はRPMI-1640培地(Invitrogen)中で培養した。
【0146】
2.2. リン酸化GANPおよびMCM3の細胞内局在化
NIH-3T3をPBS (pH 7.4)中で3.7%パラホルムアルデヒドで5分間固定し、0.2% Triton X-100を用いて透過性とした(Kimura, H. et al., (1994) EMBO J. 13, 4311-4320)。ラット抗pSer502 GANPモノクローナル抗体(Kuwahara, K. et al., (2001) Proc. Natl. Acad. Sci. USA 98, 10279-10283)及びウサギ抗MCM3抗体Kimura, H. et al., (1994) EMBO J. 13, 4311-4320を一次抗体として使用した。次に二次抗体として、GANPに対してはAlexa488結合ヤギ抗ラットIgG抗体(Molecular Probes)を使用し、MCM3に対してはAlexa546結合ヤギ抗ウサギIgG抗体(Molecular Probes)を使用して、ヨウ化TOTO-3 (Molecular Probes)で対比染色し、そして共焦点レーザー顕微鏡(FV500; オリンパス)によって観察した。
【0147】
2.3. 発現のためのcDNA構築物
pSRα-MCM3-HAは文献に記載されている(Kimura, H. et al., (1995) Nucl. Acids Res. 23, 2097-2104)。SV40 T-Agの3つの核局在化シグナル(NLS)を担持するpECFP-NucベクターはClontechより購入した。下記の組み合わせの3'及び5'プライマーを用いて得たPCR断片をpGEX-4T-1 (Amersham)に導入し、それらを用いて異なる形のマウスganp cDNAをグルタチオンS-トランスフェラーゼ(GST)との融合タンパク質として調製した。
【0148】
GANP1-5':5'-GGGGATCCATACCCGG TGAACCCCTT-3' (配列番号11)
GANP1-3':5'-GGGTCGACGCGCACAGACTTTCCCCTGA-3' (配列番号12)
GANP2-5':5'-GGGAATTCTCCCGCCTTCCAGCTGTGAC-3' (配列番号13)
GANP2-3':5'-GGGTCGACGTGCTGCTGTGTTATGTCCT-3' (配列番号14)
GANP3-5':5'-GGGAATTCCATGAGCT GAGACCCTCAGC-3' (配列番号15)
GANP3-3':5'-GGGTCGACTGAGGATGCAGGAGGCGGCT -3' (配列番号16)
GANP4-5':5'-GGGAATTCTACGTTGGAGAGAGCCTGGC-3' (配列番号17)
GANP4-3':5'-GGGTCGACCATGCTGTCATCTCCTGTGA-3' (配列番号18)
GANP5-5':5'-GGGAATTCGAGAA CCTGGCCAAGGGTCT-3' (配列番号19)
GANP5-3':5'-GGGTCGACGAAAAACCGACGGCTGA ACT-3' (配列番号20)
GANP6-5':5'-GGGAATTCAAGCCCTTCCAGCCTGCCCT-3' (配列番号21)
GANP6-3':5'-GGGTCGACCGAGGGAACGTGGTATTTTC-3' (配列番号22)
GANP7-5':5'-GGCCCGGGCC CGTGGGATGACATCATCA-3' (配列番号23)
GANP7-3':5'-GGCTCGAGCATGTCCACCATCTC CAGCA-3' (配列番号24)
【0149】
cDNA構築物は、PCR断片をpSVEGFP pA に導入することにより緑色蛍光タンパク質(GFP)をタグしたGanp変異体として調製した(Kuwata, N. et al., (1999) J. Immunol. 163, 6355-6359)。次に、これらの構築物をpCXN2哺乳動物発現ベクター中に導入した(Niwa, H. et al., (1991) Gene 108, 193-200)。プライマー配列は、Ganpをコードするように下記の通り設計した。
【0150】
Gp-gfp-5':
5'-GGGGATCCGAATTCCACCATGGCAGTCTTCAAACCGATA CC-3' (配列番号25)
Gp-gfp-3':
5'-GCAGGGGCTCCTCCTGATCT-3' (配列番号26)
Gsac-gfp-5':
5'-GGGGATC CGAATTCCACCATGTCCGAGGGCCTTGGTTCTTG-3' (配列番号27)
Gsac-gfp-3':
5'-CTGTCTT GTTTCTAAGCCGC-3' (配列番号28)
Gmap80-gfp-5':
5'-GGGGATCCGAATTCCACCATGGAGA ACCTGGCCAAGGGTCT-3' (配列番号29)
Gmap80-gfp-3':
5'-GAGGACTTGTAGATGTTTTCAC CATGG-3'(配列番号30)
【0151】
FLAGをタグしたGanp変異体については、以下のプライマーを用いてPCRにより得たcDNA断片をpCXN2に導入した。:
【0152】
FLAG-Gp-5':
5'-GGGAATTCCACCATGGATTACAAGGATGACGACGATAAGG CAGTCTTCAA CCGATACC-3' (配列番号31)
FLAG-Gp-3':
5'-GGGAATTCCTCCGGGTCTCCCTCAAGTA-3' (配列番号32)
FLAG-Gsac-5':
5'-GGGAATTCCACCATGGATTACAAGGATGACGACGATAAGTCCGAGGGCCTTGGTTCTTG-3' (配列番号33)
FLAGGsac-3':
5'-GGGAATTCGCTGTCTTGTTTCTAAGCCG-3' (配列番号34)
FLAG-Gmap-5':
5'-GGGAATTCCACCATGGATTACAAGGATGACGACGATAAGG AGAACCTGGCCAAGGGTCT-3' (配列番号35)
FLAG-Gmap-3':
5'-GGGAATTCTGAGGACTTG TAGATGTTTT-3' (配列番号36)
【0153】
内部欠失変異体GpΔNLS-GFP および I3変異体(MCMΔNLS-HA) は、文献に記述されているように調製した(Imai, Y. et al., (1991) Nucl. Acids Res. 19, 2785-2785)。全ての構築物は、ヌクレオチド配列の決定により配向が正しいこと、およびタグを付けた融合タンパク質としてコドンの読み枠が正しいことを確認し、品質を管理した。変異体RNA/DNAプライマーゼドメイン(PD) を有する発現ベクターは文献に記載されている(Ser502からAla [GpS502A]又はGlu [GpS502E]までのGp変異体)(Kuwahara, K. et al., (2001). Proc. Natl. Acad. Sci. USA 98, 10279-10283)。
【0154】
2.4. 導入した遺伝子産物の共焦点顕微鏡検査による検出
FuGENE 6 (Roche Diagnostics)を用いて、pCXN2-ganp-gfpおよび/またはpSRα-MCM3-HAによってNIH-3T3をトランスフェクトした。固定化の16時間前にレプトマイシンB (LMB;(Kudo, N. et al., (1999) Proc. Natl. Acad. Sci. USA 96, 9112-9117))を培養培地に添加した。共トランスフェクションの場合にはウサギ抗HA抗体 (Santa Cruz)およびAlexa546結合ヤギ抗ウサギIgG 抗体を用いて、そして単独トランスフェクションの場合には Alexa488結合ヤギ抗ウサギIgG 抗体 (Molecular Probes)を用いて、外因性MCM3タンパク質を染色した。核酸は、共トランスフェクション実験についてはヨウ化TOTO-3を用いて、単独トランスフェクション実験についてはヨウ化プロピジウム(PI; Sigma)を用いて対比染色した。
【0155】
2.5. GSTプルダウンアッセイ
GST融合タンパク質を文献に記述されているように精製した(Kuwahara, K. et al., (2000) Blood 95, 2321-2328)。グルタチオン-Sepharoseビーズ(Amersham)に固定した種々のGST融合タンパク質(5 μg)を、TNEバッファー(10 mM Tris-HCl [pH 7.8]、150 mM NaCl、1 mM EDTA、1% Nonidet P-40、10 μgアプロチニン、1 mMフェニルメチル-スルフォニルフルオリド[PMSF])を用いて調製したBAL17溶解物と共にインキュベートした。結合タンパク質を8% SDS-PAGEによって分離し、ニトロセルロースフィルターに転写し、ブロックした。次にウサギ抗マウスMCM3抗体(Kimura, H. et al., (1994) EMBO J. 13, 4311-4320)及びペルオキシダーゼ標識プロテインA (Amersham)と共に連続的にインキュベートし、最後にECL検出キット(Amersham)を用いてシグナルを可視化した。直接結合アッセイのためには、 [35S-メチオニン] (Amersham)を用いて、in vitro転写および翻訳結合システム(Novagen)を製造者の説明書に従って使用して放射性標識化MCM3を合成し、[35S]-標識MCM3をオートラジオグラフィーで検出した。
【0156】
2.6. 導入した遺伝子産物の免疫沈降およびウエスタンブロッティング
FuGENE 6を用いて、pCXN2-FLAG-ganpおよび/またはpSRα-MCM3-HAによってCOS7をトランスフェクトした。26時間後、TNEバッファーを用いて細胞を溶解し、得られた溶解物をプロテインA-Sepharose (Amersham)と組み合わせた抗HA抗体と共にインキュベートした。免疫沈降物を8% SDS-PAGEによって分離し、ニトロセルロースフィルターに転写し、ブロットをブロックし、マウス抗FLAG M2抗体(Stratagene)と共にインキュベートし、次にペルオキシダーゼ標識ヤギ抗マウスIgG (H+L)抗体(Zymed) と共にインキュベートした。Gp-GFPおよびその変異体の検出のためには、ブロッティングしたフィルターをウサギ抗GFP抗体(Santa Cruz)およびペルオキシダーゼ結合プロテインA(Zymed)により釣り上げた。
【0157】
2.7. ヘテロカリオンアッセイ
FuGENE 6を用いて、pSRα-MCM3-HAによってHela細胞をトランスフェクトした。20時間後、トランスフェクトしたHeLa細胞およびトランスフェクトしていないマウスSwiss-3T3細胞をトリプシン処理し、1:1の比で共に培養皿に播いた。24時間後、ポリエチレングリコール1500 (Roche diagnostics)を室温で2分間用いて細胞を融合させた(Schmidt-Zachmann, M.S. et al., (1993) Cell 74, 493-504)。培養皿を培地で4回洗浄した後、シクロヘキシミド含有培地を添加し(最終濃度20 μg/ml)、細胞をCO2インキュベーター中で 37℃で5時間インキュベートした。次に、250 mM HEPES-NaOH (pH 7.4)中で4%パラホルムアルデヒドを用いて細胞を20分間固定し、PBS中で0.5% Triton X-100を30分間用いて透過性とし、PBSで洗浄した。抗HA抗体(12CA5; Covence Research Products)およびCy3結合ロバ抗マウスIg抗体(Jackson)を用いて細胞を染色した。また、PBS中で100 ng/ml Hoechst 33342 (Sigma)を用いてDNAを20分間対比染色した。100x PlanNeofluar 位相差対物レンズ (NA 1.3) およびSpotII CCDを備えたZeiss Axioplanを用いて画像を集めた。
【0158】
3.結果および考察
3.1. GANPとMCM3の会合
これまでに、B細胞系統におけるGANPとMCM3の相互作用が、共免疫沈降によって実証されている(Kuwahara, K. et al., (2000) Blood 95, 2321-2328)。GANPのC末端領域は全Map80タンパク質と同一であるため、GANPは同一領域でMCM3と会合すると予想される。本発明者は、どの領域がMCM3と直接会合するのかを決定するため、図32に示す種々の末端切断型GANPを有するGST融合タンパク質を用いてプルダウンアッセイを実施した。GSTを図32に示される1から7及び5-7'と称した切断型GANPのN末端に融合した。図33の下のパネルにおいて、GANP5-7'と称するMap80領域は、以前に示されたように(Kimura, H. et al., (1994) EMBO J. 13, 4311-4320)、細胞抽出物からMCM3をプルダウンした。
驚くべきことに、GANPの部分断片であるGANP1およびGANP2もまたMCM3をプルダウンした(図33;上および下パネル)。
【0159】
次に、網状赤血球溶解物系により、in vitroで合成したMCM3を用いてこの結合を調べた(図34)。GST-GANP1 およびGST-GANP2 もまたin vitro翻訳カクテルから[35S]-MCM3 をプルダウンした。
GST単独を用いた陰性対照(最初のレーン)、又は、無関係なタンパク質と融合したGSTは、シグナルを示さなかった。そして、GST-GANP1との結合はMap80 領域 (GST-GANP5-7')との結合よりも強かった。この結合は、FLAGをタグした構築物を用いたDNAトランスフェクション実験により細胞においても確認された(図35)。
COS7細胞をpSRα-MCM3-HA又はpSRa-I3-HAと組み合わせて、pCXN2-FLAG-Ganp、pCXN2-FLAG-Gp及びpCXN2-FLAG-Gmap80によって共トランスフェクトした。抗HA抗体を用いた免疫沈降の後、抗FLAGモノクローナル抗体でウエスタンブロットを行った。FLAG標識タンパク質の予想サイズを、それぞれのレーンにおいて三角で示す。左及び右のパネルは、バンドの移動度(migration)が同様であるが、ECL検出の露光時間は左のパネルが1分、及び右のパネルは3分である(図35)。
【0160】
FLAG-Ganp、FLAG-Gp およびFLAG-Gmap80は野生型MCM3-HA (HAエピトープをタグしたMCM3)と結合した(図35; 左パネル)。FLAG-Gsac のみが結合しなかった。I3 変異体MCM3 (MCM3△NLS)との結合に関しては、FLAG-Gmap80 のみが陽性の結果を示した(図35;右パネル)。N末端NLSを担持するGp領域は、大量のMCM3を有する細胞において一貫してMCM3と会合している(図35;左パネル)。これらの結果は、GANPがGp領域を介してMCM3のNLS領域と会合していることを示している。
【0161】
本発明者はさらに、Gp領域のSer502のリン酸化の状態がMCM3との結合に影響するかどうかを検討した。プライマーゼ部位を欠損したGanp変異体(Ganp△PD-GFP)、及びGanpS502A又はGanpS502Eとして調製されるSer502におけるGanp変異体を、GFPと融合した(図36A)。細胞をpCXN2-Ganp-gfpとpSRα-MCM3-HAによって共トランスフェクトし、溶解液は抗HA抗体を用いた免疫沈降に用いた。
GFPシグナルは抗GFP抗体で検出され(図36B;上パネル)、このことは、GANPがMCMに結合したことを意味する。
Ganp-GFP変異体との共トランスフェクションも同様に行った。それぞれのタンパク質の予想位置を決定するために、溶解液をSDS-PAGE上で分離し、同じように抗GFP抗体でブロットした(図36B;下パネル)。
【0162】
非リン酸化性の変異体(GanpS502A-GFP)は、野生型Ganp-GFPまたはホスホセリンに良く似た変異体(GanpS502E-GFP)と同じくMCM3と結合した(図36B; 上パネル)。興味深いことに、Ganp△PD-GFP はMCM3-HAと共沈降しない (図36B; 上パネル)。
GANP分子全体としては、Map80領域の潜在的結合活性に関わりなく、MCM3との結合にはRNAプライマーゼ領域 (PD)が必要である。白抜き三角はGanp△PD-GFPの位置を示す。Ganp S502A-GFP およびGanp S502E-GFPのサイズと等しいGanp-GFPのサイズを黒三角で示す(図36B;下パネル)。これらの結果は、GANPのMCM3との結合はそのPD領域を介するが、Ser502 部位におけるリン酸化はこの結合に影響しないことを示している。
【0163】
末端切断型構築物を用いた実験によってGANPとMCM3との会合が広い領域にわたって示されたが、GANPのN末端の600個のアミノ酸領域が関与する全GANPとの結合にはMCM3のNLSが必要とされる。NLSを欠くMCM3変異体は、細胞中で全GANP分子と効果的に会合できなかった。Map80領域はNLS陰性MCM3と結合し、このことはGANPのMCM3との結合が主としてMap80との相互作用に必要とされる領域以外との結合であることを示している。Map80はMCM3インポート因子であると考えられているが、GANPはMCM3と共同して異なる役割を果たすのかもしれない。GANPは可能性のあるリン酸化部位を多数有し、そして細胞中に多くの会合成分をもつように思われる(Kuwahara, K. et al., (2000) Blood 95, 2321-2328)。したがって、その領域のリン酸化の状態がGANP/MCM3会合および細胞質と核コンパートメント間の輸送の調節に影響を及ぼす領域を特定することが必要であろう。
【0164】
3.2. トランスフェクションによって示されるMap80およびGanp変異体の細胞内局在化
GANPは2つの可能性のあるNLSを有する。1つはN末端プライマーゼ領域内にあり、もう1つはC末端Map80領域内にある。また、GANPは2つの核輸出シグナル(NES)様モチーフを有する。1つはSAC3相同領域とMap80領域との間にあり、もう1つはMap80領域内にある。
NIH-3T3細胞をpCXN2-Ganp-gfp又はpCXN2-Gmap80-gfpによってトランスフェクトし、48時間後に固定した。LMBを、固定の16時間前に添加した。核をPIで前染色し、共焦点顕微鏡を用いて画像を集積した。代表的な発現特性を図37に示す。異なる特性を持つ細胞数を計数し、パーセントとして表した(図37)。
【0165】
Ganp-GFP(GFPでタグしたほぼ全長GANP)は、核優性発現(NおよびN>C; 38%)又は細胞質優性発現(CおよびC>N; 31%)を示す細胞の割合に変動はあったが(合計500個の細胞から任意に計算された数の%)、細胞質および核コンパートメントの両方に存在することが見いだされた (図37; Ganp-GFP, LMB -)。対照的に、Gmap80-GFP は殆ど細胞質に見出され、発明者の分類による核優性発現は示さなかった (N>C, 0%; N=C, 35%; C and C>N, 65%)(図37; Gmap80-GFP, LMB -)。Ganp-GFPの局在化は、Gmap80-GFPの局在化とは異なる。
N末端NLSモチーフが機能性であるかどうかを確かめるため、RNA/DNAプライマーゼドメインおよびN末端NLSを担持する (ただしNES様モチーフは含まない) 5' 1-kb DNA 断片をGFPと融合させた (図38, Gp-GFP)。この Gp-GFP 産物は 核にのみ存在したが (NおよびN>C, 94%)(図38)、NLSを欠く変異体Gp-GFP (Gp△NLS-GFP; 図38に示すようにアミノ酸497から500が欠失している)は細胞質性であることを示し、N末端NLSが核局在化に関与していることが確認された。
【0166】
本発明者は、隣接するSer502 のアラニンへの突然変異(GpS502A-GFP; 非リン酸化型) 又はグルタミン酸への突然変異(GpS502E-GFP; ホスホセリンを模倣する型)がこの局在化に影響を及ぼすかどうかを検討した(図38)。そして、本発明者は、これらの突然変異がGpの局在化を変えないことを観察した。これは、N末端NLSがSer502 のリン酸化状態に関わりなく機能性であることを示唆している(図38)。対照的に、N末端NLSもC末端NLSも持たないGac-GFPは、殆どが細胞質中に存在するように思われる(N及びN>C, 0%; N=C, 3%; C及びC>N, 97%)(図38)。
これらの結果は、N末端NLSがGanpの核への移入に機能的な役割を有することを示唆している。しかし、NLSはGANP発現を核内に維持するほど強くはないのかもしれない。なぜなら、Ganp-GFPは細胞質中にも存在するからである(図37)。この問題をさらに検討するため、Crm1によって媒介される核への輸出を抑制するため、cDNAトランスフェクション後、細胞をレプトマイシンB (LMB)で処理した(Kudo, N. et al., (1999) Proc. Natl. Acad. Sci. USA 96, 9112-9117)。
【0167】
LMBで処理した細胞では、殆どのトランスフェクタントにおいてGanp-GFPは核に局在化した(図37)。優性な細胞質発現を示す細胞画分は31%から4% に減少し、対照的に、核において優性な発現を示す細胞が38%から81%に増大した。従って、Ganpの細胞質への移行はLMBによって抑制されるように思われる。
Gmap80-GFPの局在化もまたLMB処理後、劇的に変化した(図37)。すなわち、細胞質発現を示す細胞画分が65%から37%に減少し、優性な核発現を示す細胞画分が0%から41% に増大した。これらの所見は、COS7およびLtk- 細胞を含む他の細胞系においても再現され、核から細胞質へのGANP輸出がCrm1依存性経路によって調節されていることを示している。従って、GANP及びMap80は核と細胞質の間をシャトリング(往復)しているように思われ、そしてそれらの局在化は他の分子と共同している核移入および輸出機構の間のバランスに依存するようである。
【0168】
3.3. 共トランスフェクトした細胞におけるMCM3およびGANPの局在化
次に、本発明者はGANPの移行がMCM3発現と関連しているかどうかを検討した。哺乳動物のMCM3は、細胞周期中にクロマチンとの結合状態を変化させるが、間期の間を通して核にのみ存在する(Kimura, H. et al., (1994) EMBO J. 13, 4311-4320)。NIH-3T3細胞を、pSRα-MCM3-HA又はpSRα-I3-HAによってトランスフェクトし、固定し、抗HA抗体(Alexa488)で免疫標識し、PIで染色した。
トランスフェクトしたMCM3-HAは、典型的なNLSの存在と合致して(Kimura, H. et al., (1994) EMBO J. 13, 4311-4320、Takei, Y. et al., (1998) J. Biol. Chem. 273, 22177-22180)、核に局在化した(図39)。この核局在化はMCM3のNLSに依存した。なぜなら、このNLSを欠く変異体MCM3 (I3; MCM3△NLS-HA)は、細胞質にのみ発現されたからである (図39; 右パネル)。
【0169】
細胞を、pCXN2-Ganp-gfp又はpCXN2-Gmap80-gfp及びpSRα-MCM3-HAによって共トランスフェクトし、固定し、抗HA抗体(Alexa546)で免疫標識し、核をTOTO-3で前染色した(図40)。細胞数はパネルの下に示す(図40)。
興味深いことに、Ganp-GFP を用いて共トランスフェクションした場合、細胞の17%においてMCM3の細胞質局在化がもたらされた(図40, 白い矢印で示す)。Gmap80-GFPまたはGp-GFPを用いた共トランスフェクションの場合には、そのような結果はみられなかった。
MCM3へのGanpの効果が特異的であることを証明するために、核において出現するECFP-Nucの発現を、異なるganp-gfp構築物のトランスフェクション前後で調べた。Ganp-GFPでトランスフェクトした代表画像を示す(図41)。ECFP-Nucの核への局在化は、Ganp-GFP (図41)又はGmap80-GFP又はGp-GFPを用いた共トランスフェクションのいずれによっても影響されなかった。
【0170】
Ganp及びMCM3の共発現もまたGANP局在化の変更をもたらした。Ganp-GFP 単独(38%)及びGmap80-GFP単独(0%)のトランスフェクション(図37)と比較して、MCM3を用いた共トランスフェクションはGanp-GFP (74%)およびGmap80-GFP (64%) の核発現レベルを増大させた(図40)。MCM3は、GANPおよびMap80を核内に保持したが、Ganpの過剰発現のみがMCM3 の細胞質における出現を促進した(図40; Ganp-GFP発現によって17%)。他方、Gmap80 MCM3 の細胞質における出現を促進しなかった(Gmap80-GFP発現によって4%)。MCM3のNLSにおける突然変異(I3; MCM3△NLS-HA) (その結果、MCM3は細胞質に存在することになる)は、Ganp-GFPまたはGmap80-GFP の核への蓄積を引き起こさなかった(図42)。
野生型MCM3とは異なり、I3 変異体 (MCM3△NLS-HA) はGanp又はGpと会合しない(図35)という事実を考え合わせるならば、MCM3のNLSモチーフは、細胞においてGANPとの機能的会合および核と細胞質間のGANPの輸送に必要であることが示唆される。
【0171】
DNAトランスフェクション実験は、MCM3と共に導入された場合、Ganp-GFPが核コンパートメントに蓄積することを示し、核におけるGANP/MCM3複合体の形成を示唆した。MCM3 はCrm1によって認識されうる明らかな共通NES様モチーフを含まず、したがって核からのMCM3の輸出は、恐らく、NES様モチーフを有する他の結合分子または異なる輸出機構に依存するのであろう。GANP上の2つのNES様モチーフは、LMB感受性Crm1依存性輸出経路に関与しているように思われる(図37)。GANPによって担持される2つのNES様モチーフ(これらはCrm1によって認識されるのかもしれない)は、複合体の輸送に関与している可能性がある。
最近、GANP相同領域を担持する酵母SAC3が、あるタンパク質の核輸出に関与し、そして核膜孔複合体の成分と会合することが示された(Jones, A.L. et al., (2000) Proc. Natl. Acad. Sci. USA 97, 3224-3229)。GANPとの共発現は、MCM3の細胞質コンパートメントへの局在化を変化させた。
【0172】
細胞融合技法を用いて、MCM3の核-細胞質間シャトリング(往復)を調べた(Schmidt-Zachmann, M.S. et al., (1993) Cell 74, 493-504)。HeLa細胞を最初MCM3-HAでトランスフェクトし、次にトランスフェクトしていないマウスSwiss-3T3細胞と融合させた。タンパク質合成を抑制するためシクロヘキシミドの存在下で5時間インキュベートした後、細胞を固定し、MCM3-HA を免疫標識した。ヘキスト(Hoechst)染色によってヘテロカリオンを調べた。この染色は、「まだらの」ヘテロクロマチンを有するマウスの核(矢印)をヒトHeLa核と区別する。
図43に代表的なものを示すように、MCM3-HA はヘテロカリオン中にヒトおよびマウスの核の両方に見出された。融合されていないマウスの細胞は、そのような染色を示さない。このことは、MCM3-HAがHeLa核から細胞質へ輸出され、次にマウス核に移入されたことを示している。
【0173】
これらの結果から、MCM3-HAはシャトリングタンパク質であると結論される。なお、トランスジーン産物と同じような感度で内因性タンパク質の核から細胞質への移行を実証することはしばしば困難である(Kimura, H., Ohtomo, T.et al., (1996) Genes Cells 1, 977-993、Mizuno, T. et al., (1999) Mol. Cell. Biol. 19, 7886-7896)。本実施例に示す結果についても、そうであった。酵母のようなより原始的な細胞で達成されたとの同じような感度で、哺乳動物細胞における内因性MCMタンパク質の核から細胞質への移行を実証することもまた困難である。
しかしながら、本発明者の結果は、(実験はDNAトランスフェクション法によって行ったが)未操作細胞においてMCMタンパク質の核-細胞質間シャトリングが恐らく重要であることを示している。細胞周期の進行中におけるMCM複合体の核-細胞質間移動の明確な理解を容易にするには、さらなる成分の発見が必要であるかもしれない。
【0174】
3.4. 細胞周期の間のGANPの局在化
RNA/DNA プライマーゼドメインのGANPに特有のエピトープ(pSer502 GANP)に特異的なモノクローナル抗体を用いて、共焦点レーザー顕微鏡検査によってNIH-3T3細胞におけるGANPの局在化を調べた(Kuwahara, K. et al., (2001) Proc. Natl. Acad. Sci. USA 98, 10279-10283)。細胞周期の異なる時期にあるNIH-3T3細胞を、抗pSer502 GANP (Alexa488; 緑)及び抗MCM3 (Alexa546; 赤)抗体で免疫染色した。核をTOTO-3ヨウ化物(青)で前染色した。間期の間、上記モノクローナル抗体は核小体を除く核内のすべてで反応した(図44)。
抗MCM3抗体および核酸を染色するTOTO-3を用いた三重標識によって、有糸分裂の間のGANPの局在化を詳しく分析した。細胞が前中期から中期へ進むにつれ、GANPは濃縮クロマチンから解離されてくるように思われる(図44)。重ね合わせた画像の黄色いシグナルはGANPとMCM3との共局在化を示すが、若干の青い染色は前中期画像の中心部でGANPが単独でも見えることを表している。この段階では、GANP及びMCM3は濃縮された染色体と重なり合っていない。中期の細胞においては、GANPは紡錘体領域に検出される。そしてこのシグナルは後期に染色体が2つの娘細胞に分離する間に減少する。
【0175】
減数分裂後期においては、GANPの殆どは核が形成されるまで(終期)、細胞質コンパートメントに見出される。これらの結果は、GANPおよびMCM3の挙動が類似していて、これら2つは間期の間を通じて殆ど核に共局在化することを示している。このことは、これら2つの相互会合と一致している。しかし、有糸分裂の間に共焦点顕微鏡検査によって示されたように、GANPおよびMCM3は別々に存在することもできる(図44)。
第2型のRNA/DNAプライマーゼに関する核-細胞質間のGANPシャトリングの生物学的意味は、今後の解明を待たなければならない。それはDNA修復の最終段階におけるRNAプライマーの生成に関連するのかもしれない。正常細胞ではGANPの発現レベルは低いが、細胞が急速に増殖する胚中心においてはアップレギュレーションされる(Kuwahara, K.et al., (2000) Blood 95, 2321-2328 、Kuwahara, K. et al., (2001). Proc. Natl. Acad. Sci. USA 98, 10279-10283)。GANPはまた、速い細胞終期を有するある種の細胞においてより高レベルで発現される。このことは、MCM複合体との会合がDNA複製を刺激する可能性を示唆している(Kuwahara, K. et al., (2001). Proc. Natl. Acad. Sci. USA 98, 10279-10283)。RNA/DNAプライマーゼ活性、MCM3結合能、およびアセチルトランスフェラーゼドメイン(Takei, Y.et al., (2001) EMBO Rep. 2, 119-123を有するGANPの発現は、細胞周期進行の調節に関与しているのかもしれない。
【配列表フリーテキスト】
【0176】
配列番号5:プライマー
配列番号6:プライマー
配列番号7:プライマー
配列番号8:プライマー
配列番号9:プライマー
配列番号10:プライマー
配列番号11:プライマー
配列番号12:プライマー
配列番号13:プライマー
配列番号14:プライマー
配列番号15:プライマー
配列番号16:プライマー
配列番号17:プライマー
配列番号18:プライマー
配列番号19:プライマー
配列番号20:プライマー
配列番号21:プライマー
配列番号22:プライマー
配列番号23:プライマー
配列番号24:プライマー
配列番号25:プライマー
配列番号26:プライマー
配列番号27:プライマー
配列番号28:プライマー
配列番号29:プライマー
配列番号30:プライマー
配列番号31:プライマー
配列番号32:プライマー
配列番号33:プライマー
配列番号34:プライマー
配列番号35:プライマー
配列番号36:プライマー
【特許請求の範囲】
【請求項1】
GANP遺伝子が導入され、該遺伝子をB細胞で発現させたトランスジェニック非ヒト哺乳動物、又は、該遺伝子をB細胞で発現させたその子孫。
【請求項2】
GANP遺伝子をトランスフェクトしたES細胞から発生させ、該遺伝子をB細胞で発現させたものである、請求項1記載のトランスジェニック非ヒト哺乳動物又はその子孫。
【請求項3】
哺乳動物がマウスである請求項1又は2記載のトランスジェニック非ヒト哺乳動物又はその子孫。
【請求項4】
請求項1〜3のいずれか1項に記載のトランスジェニック非ヒト哺乳動物又はその子孫の一部。
【請求項5】
請求項1〜3のいずれか1項に記載のトランスジェニック非ヒト哺乳動物又はその子孫に抗原を投与し、得られる動物又は子孫から抗体を採取することを特徴とする高親和性抗体の製造方法により得られる、高親和性抗体又はその断片。
【請求項6】
ポリクローナル又はモノクローナル抗体である、請求項5記載の抗体又はその断片。
【請求項7】
請求項6記載の抗体又はその断片のV領域を含む、ヒト型化抗体若しくはヒト抗体又はそれらの断片。
【請求項8】
請求項5又は6記載の抗体又はその断片、及び請求項7記載のヒト型化抗体若しくはヒト抗体又はそれらの断片からなる群から選択される少なくとも1つを含有する医薬組成物。
【請求項9】
抗原を投与した請求項1〜3のいずれかに記載のトランスジェニック非ヒト哺乳動物又はその子孫から採取される、高親和性抗体産生細胞。
【請求項10】
高親和性抗体を産生するための、請求項1〜3のいずれか1項に記載のトランスジェニック非ヒト哺乳動物又はその子孫の使用。
【請求項1】
GANP遺伝子が導入され、該遺伝子をB細胞で発現させたトランスジェニック非ヒト哺乳動物、又は、該遺伝子をB細胞で発現させたその子孫。
【請求項2】
GANP遺伝子をトランスフェクトしたES細胞から発生させ、該遺伝子をB細胞で発現させたものである、請求項1記載のトランスジェニック非ヒト哺乳動物又はその子孫。
【請求項3】
哺乳動物がマウスである請求項1又は2記載のトランスジェニック非ヒト哺乳動物又はその子孫。
【請求項4】
請求項1〜3のいずれか1項に記載のトランスジェニック非ヒト哺乳動物又はその子孫の一部。
【請求項5】
請求項1〜3のいずれか1項に記載のトランスジェニック非ヒト哺乳動物又はその子孫に抗原を投与し、得られる動物又は子孫から抗体を採取することを特徴とする高親和性抗体の製造方法により得られる、高親和性抗体又はその断片。
【請求項6】
ポリクローナル又はモノクローナル抗体である、請求項5記載の抗体又はその断片。
【請求項7】
請求項6記載の抗体又はその断片のV領域を含む、ヒト型化抗体若しくはヒト抗体又はそれらの断片。
【請求項8】
請求項5又は6記載の抗体又はその断片、及び請求項7記載のヒト型化抗体若しくはヒト抗体又はそれらの断片からなる群から選択される少なくとも1つを含有する医薬組成物。
【請求項9】
抗原を投与した請求項1〜3のいずれかに記載のトランスジェニック非ヒト哺乳動物又はその子孫から採取される、高親和性抗体産生細胞。
【請求項10】
高親和性抗体を産生するための、請求項1〜3のいずれか1項に記載のトランスジェニック非ヒト哺乳動物又はその子孫の使用。
【図1】

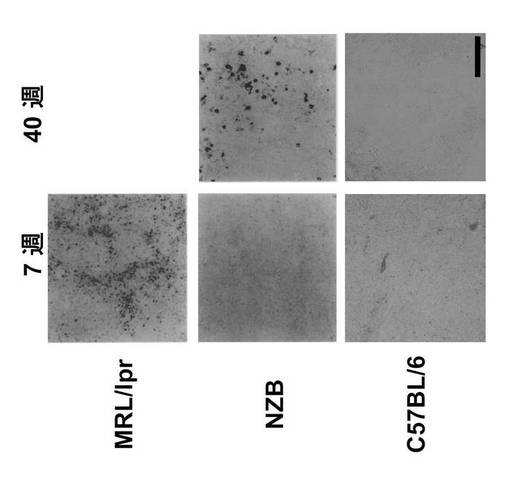
【図2】
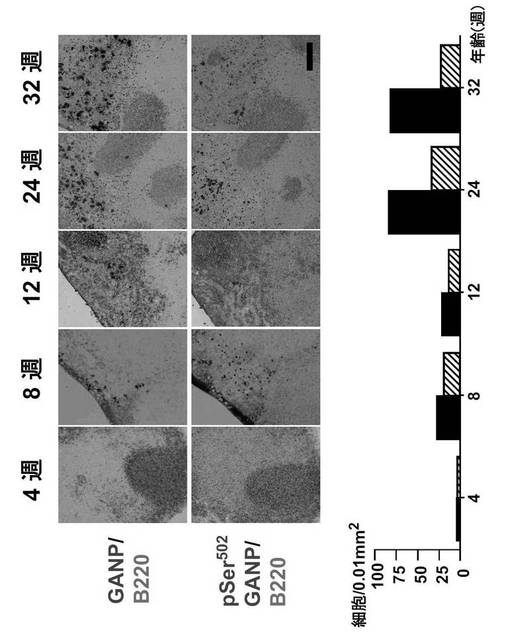
【図3】

【図4】
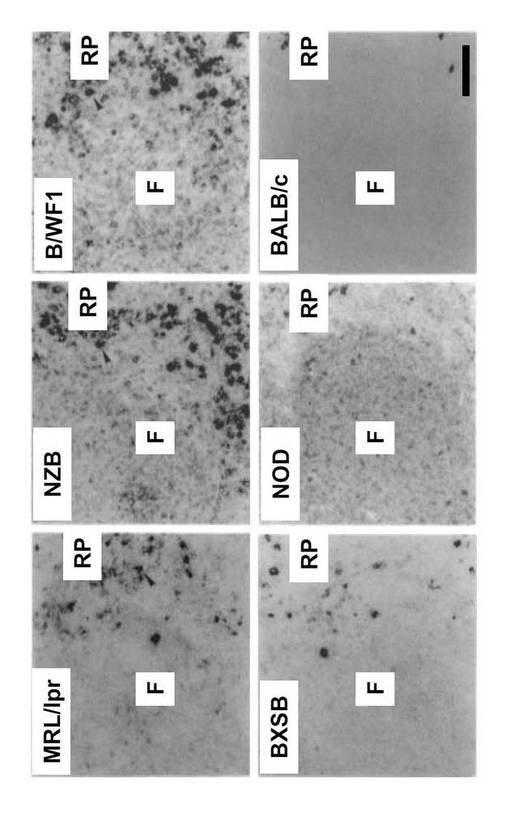
【図5】
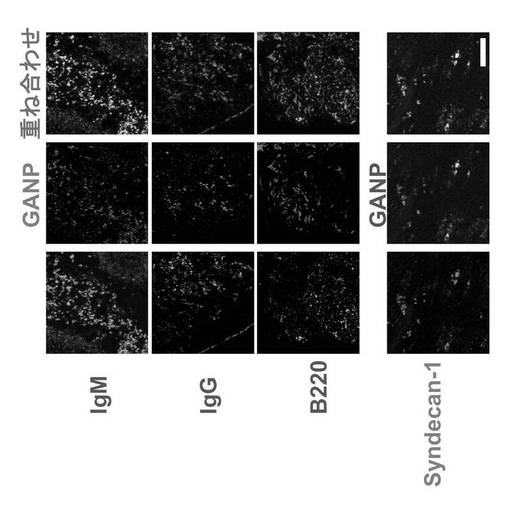
【図6】
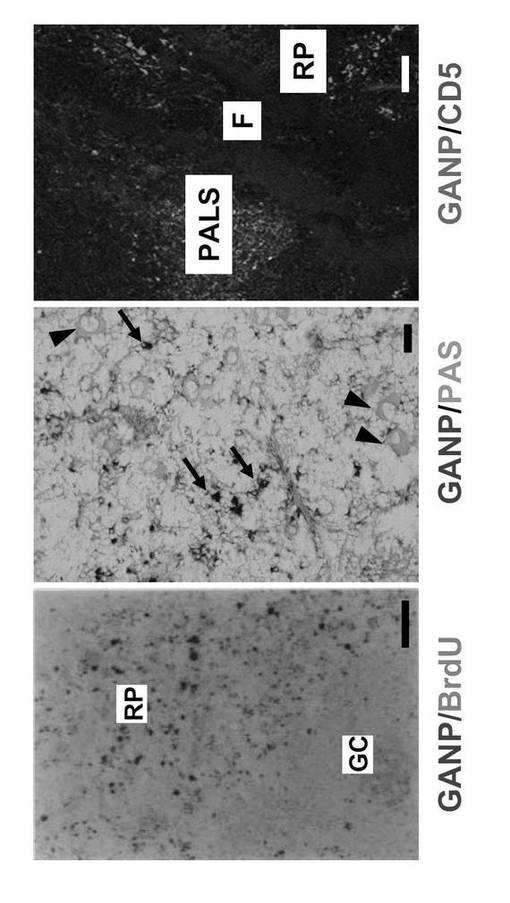
【図7】
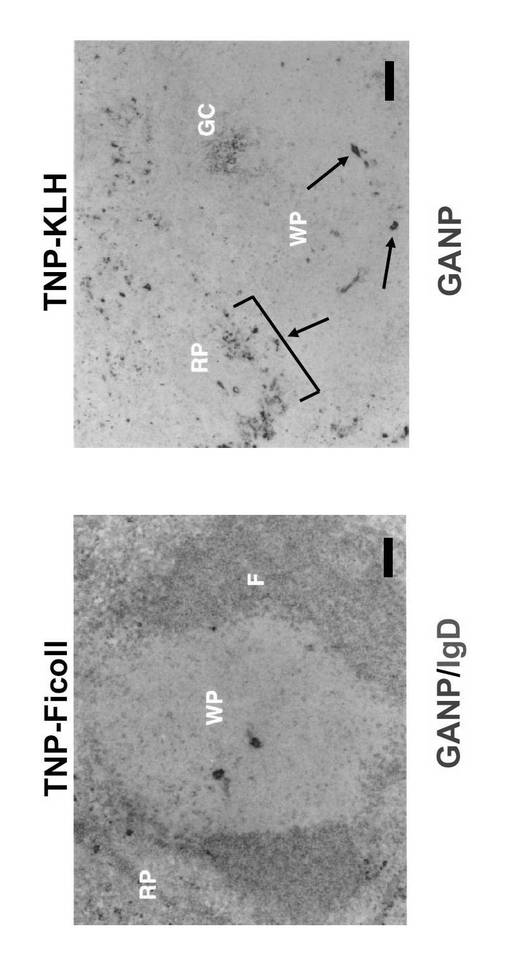
【図8A】

【図8B】
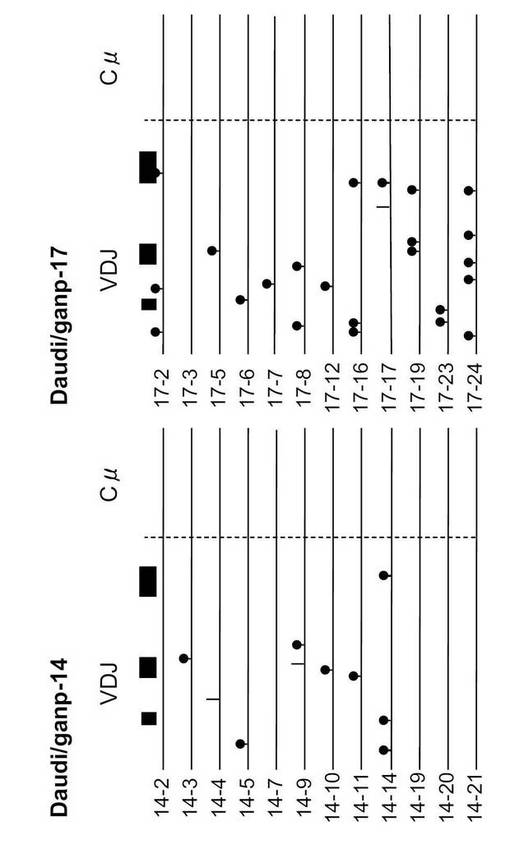
【図8C】
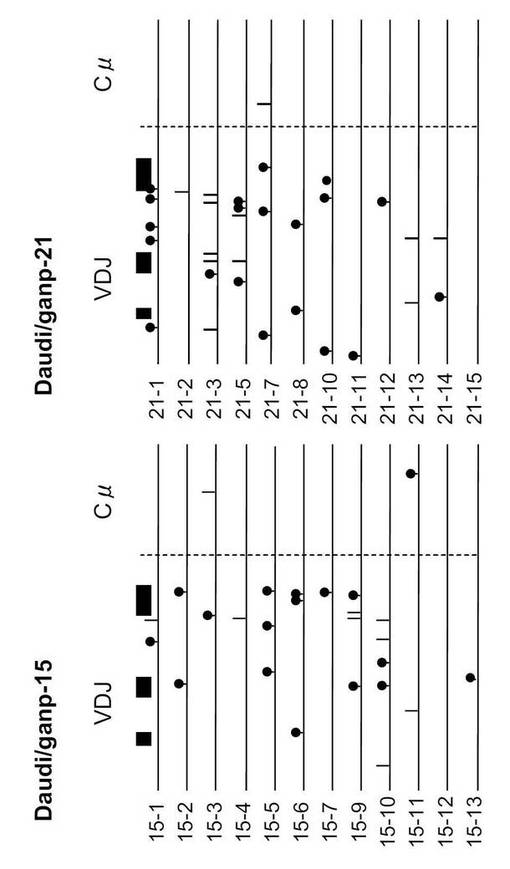
【図9A】
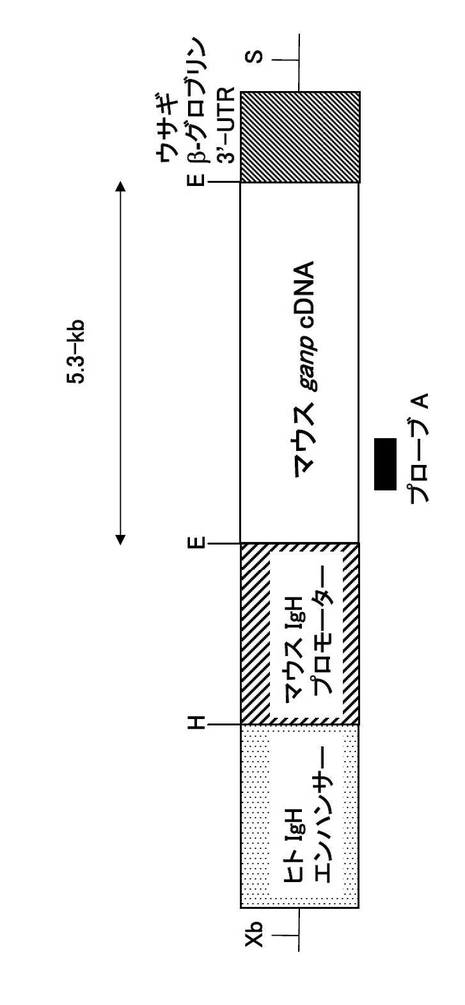
【図9B】
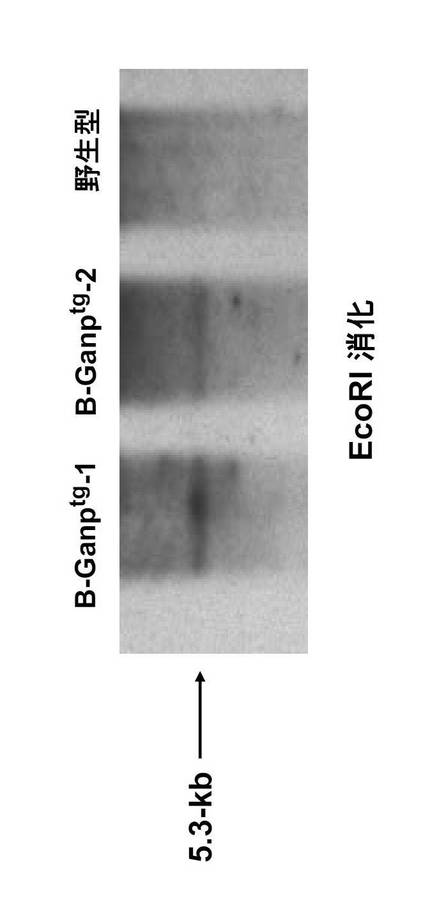
【図9C】
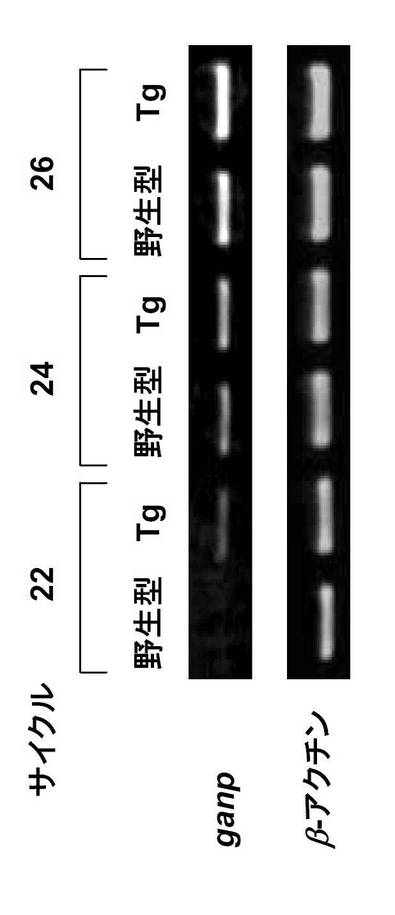
【図10】

【図10−1】

【図10−2】

【図10−3】

【図10−4】

【図10−5】

【図11A】
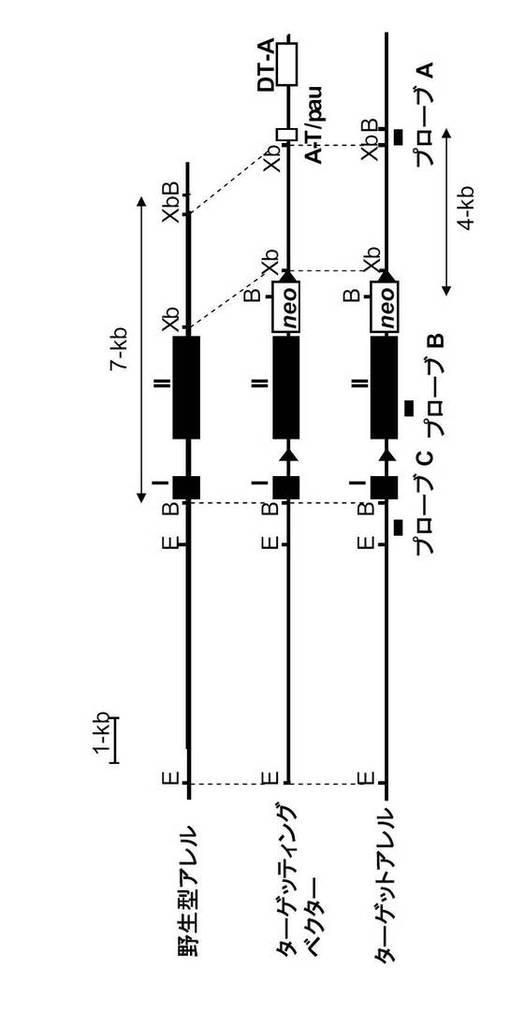
【図11B】
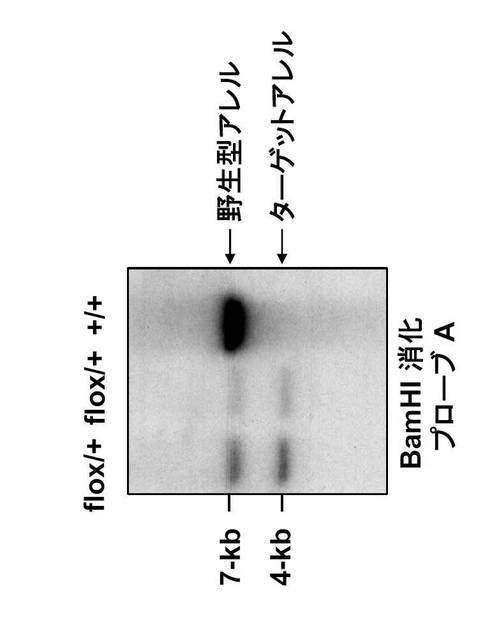
【図11C】
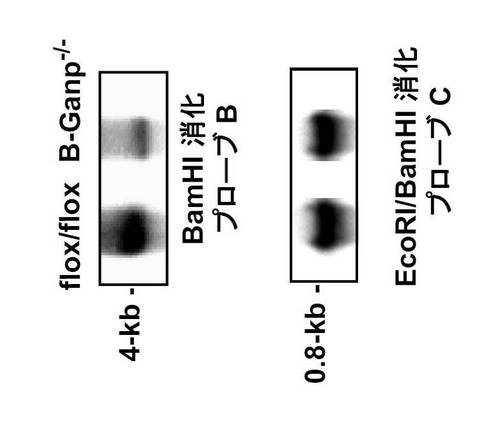
【図11D】
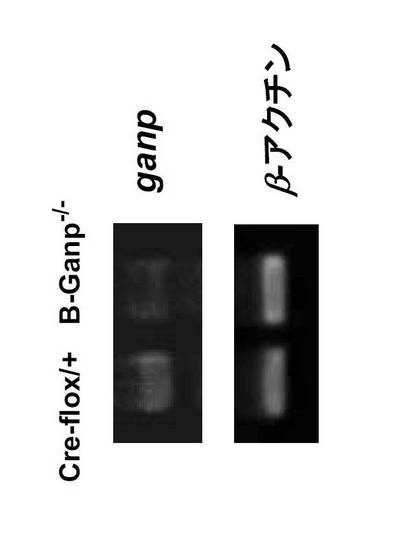
【図11E】
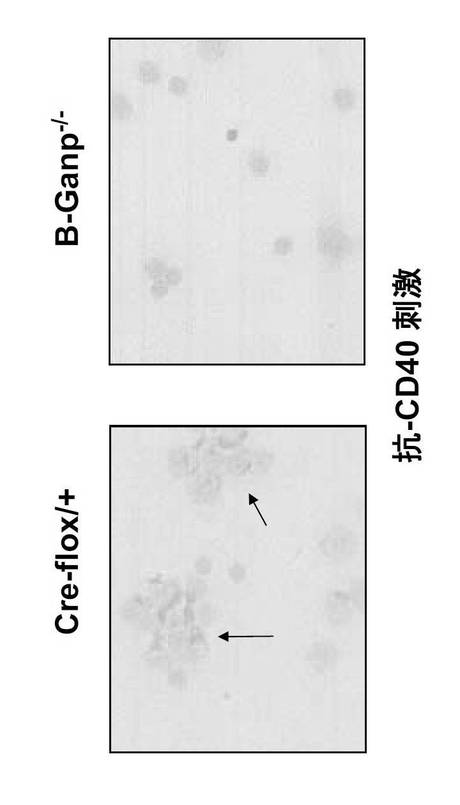
【図12】
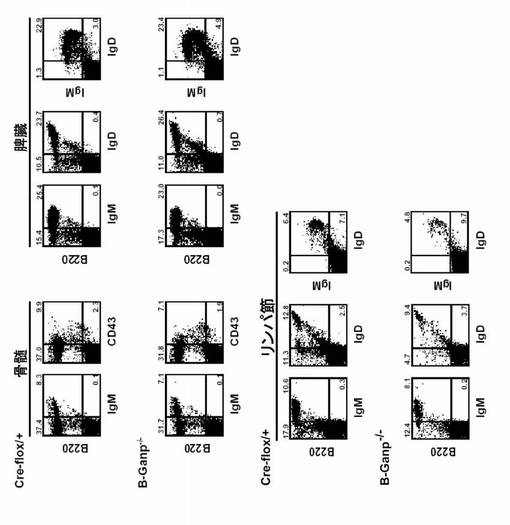
【図13】
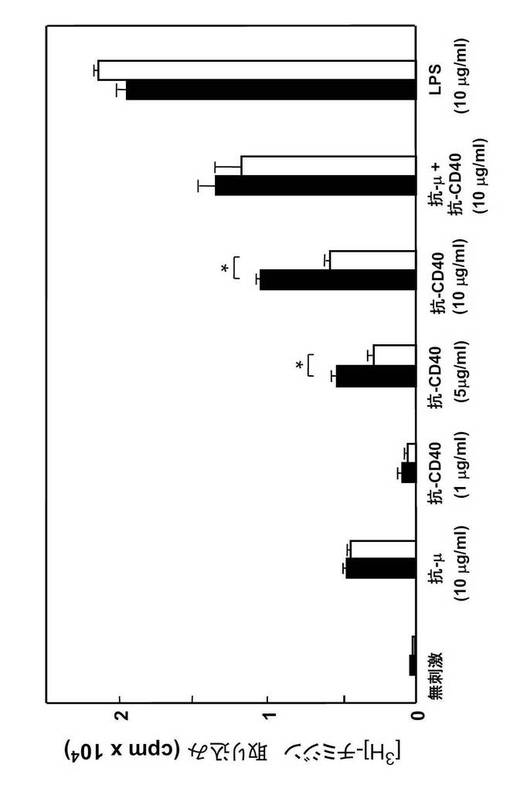
【図14】
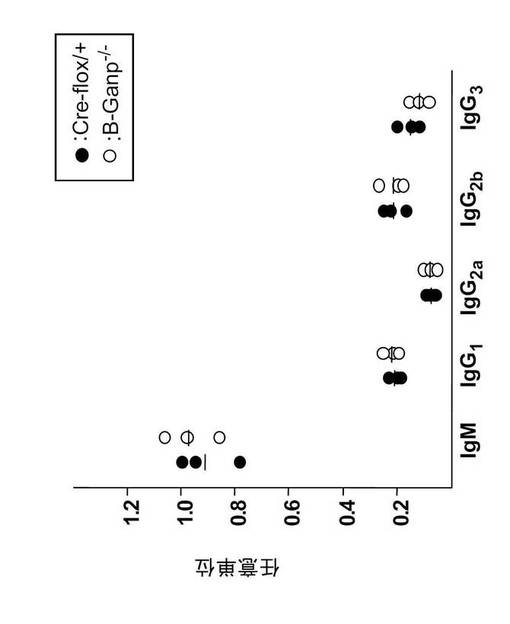
【図15】
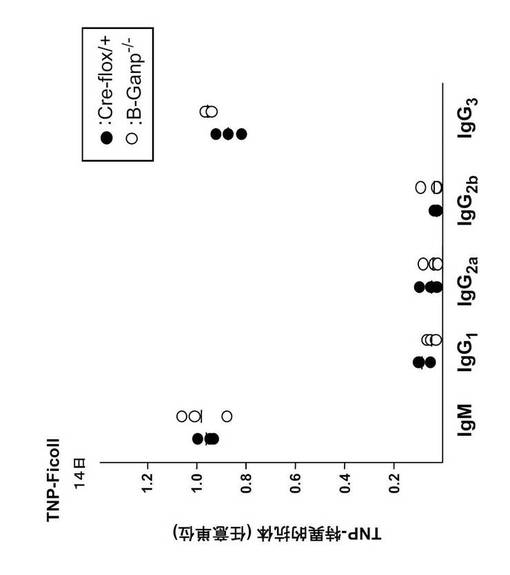
【図16】
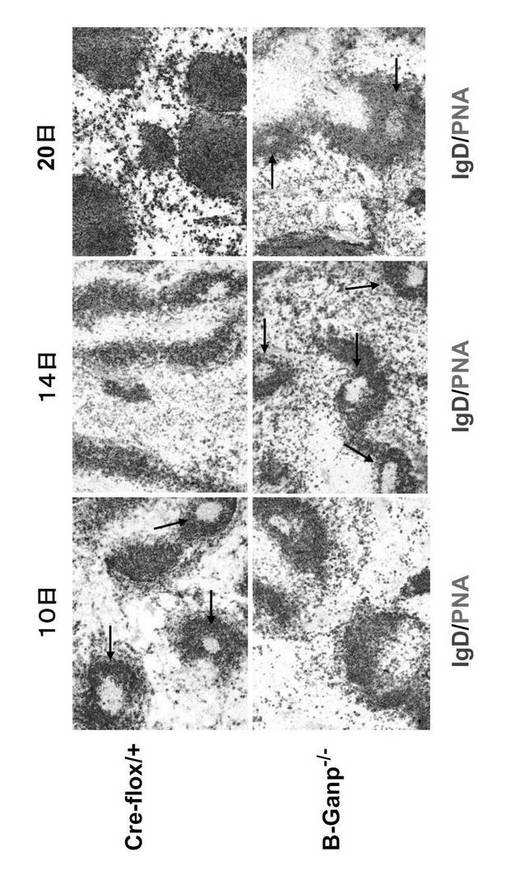
【図17】
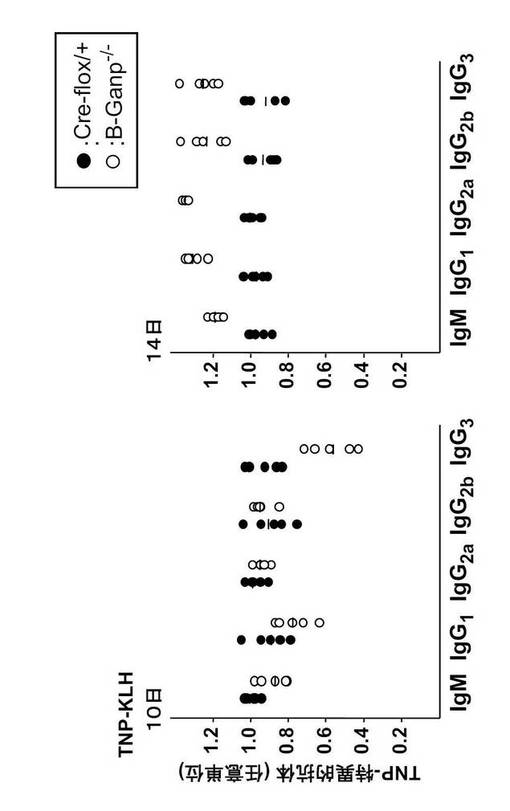
【図18】
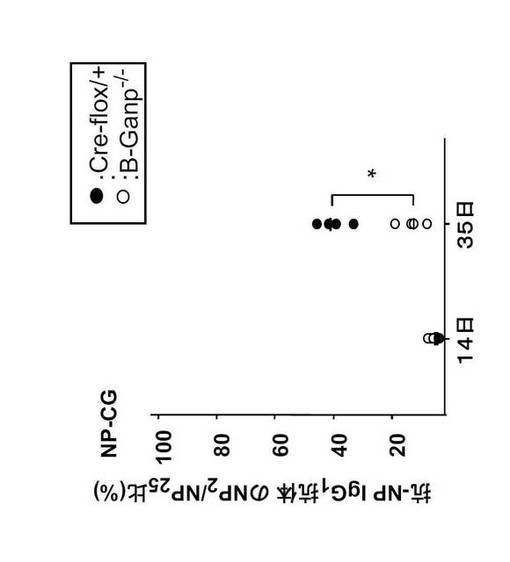
【図19】
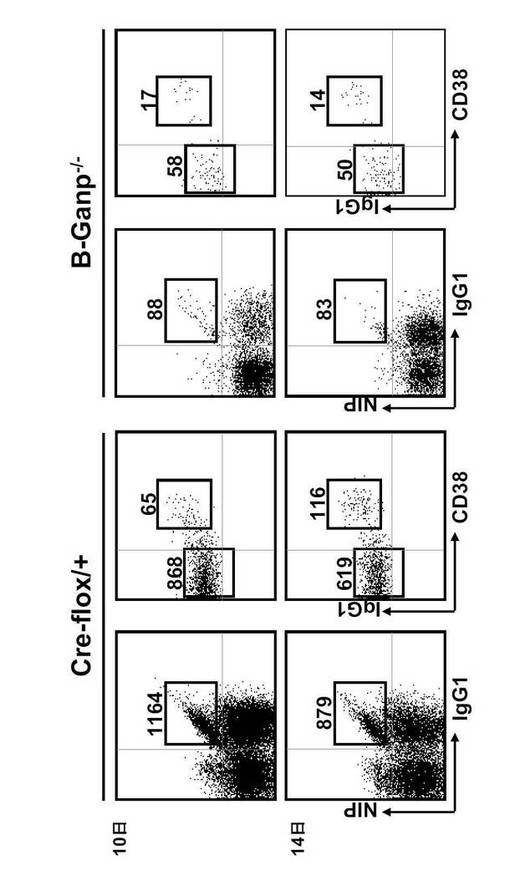
【図20A】
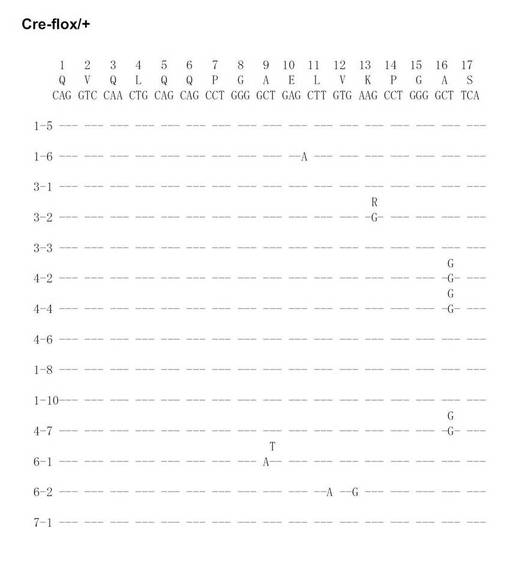
【図20B】
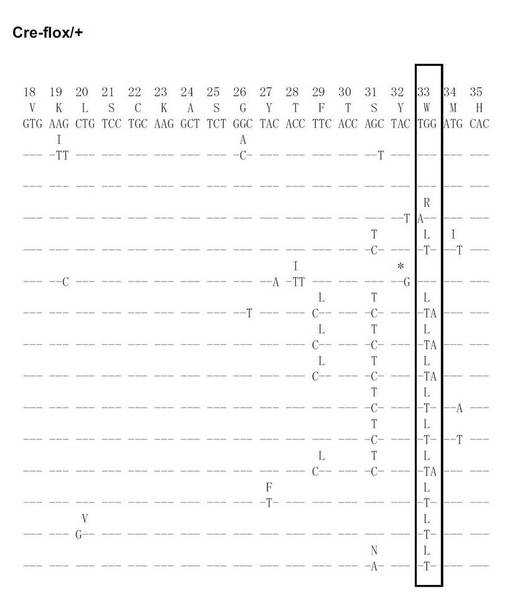
【図20C】

【図20D】
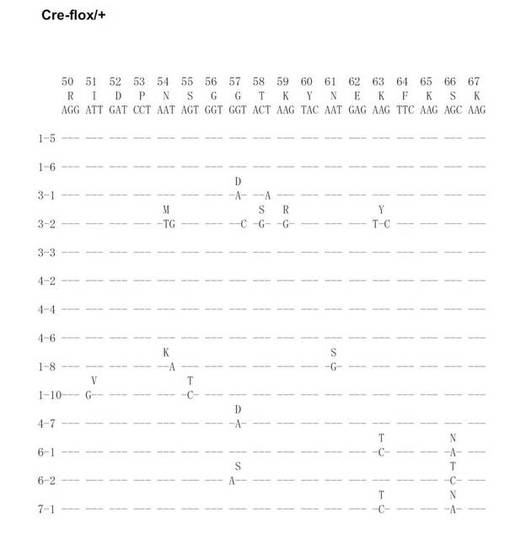
【図20E】
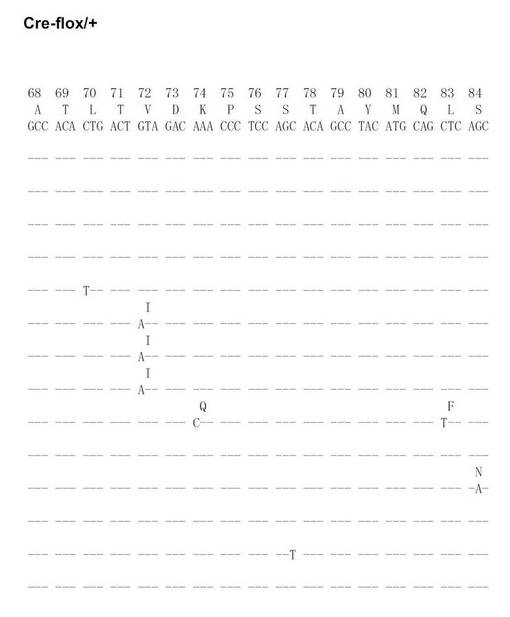
【図20F】

【図20G】

【図20H】
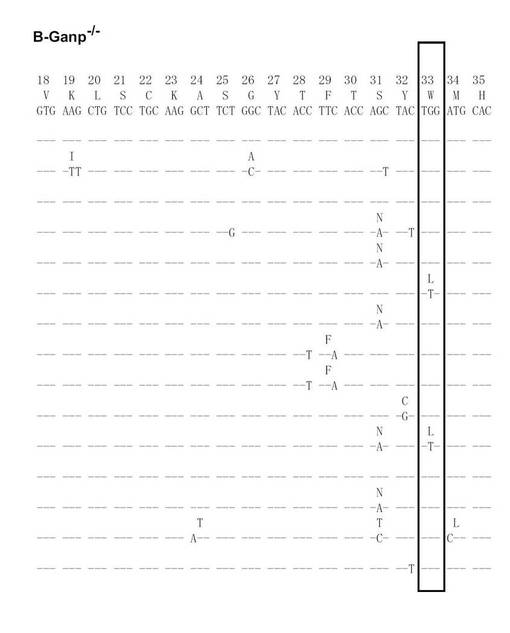
【図20I】

【図20J】
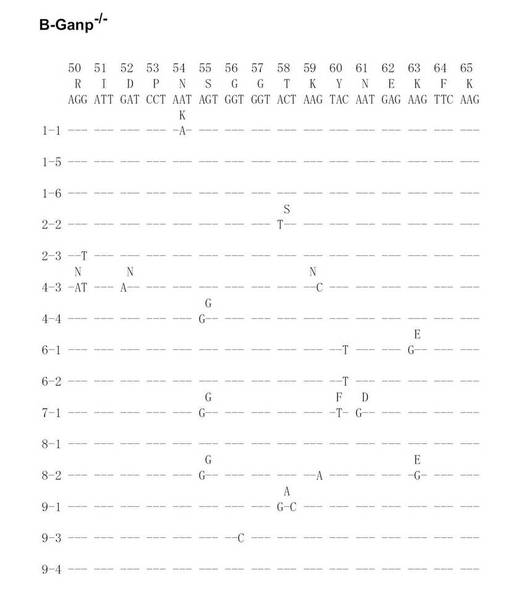
【図20K】

【図20L】

【図21】
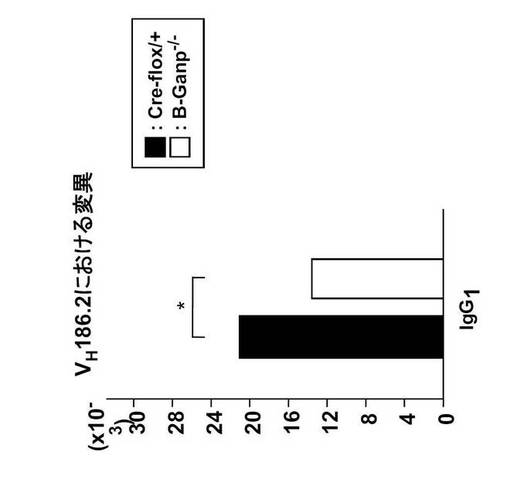
【図22】

【図23】
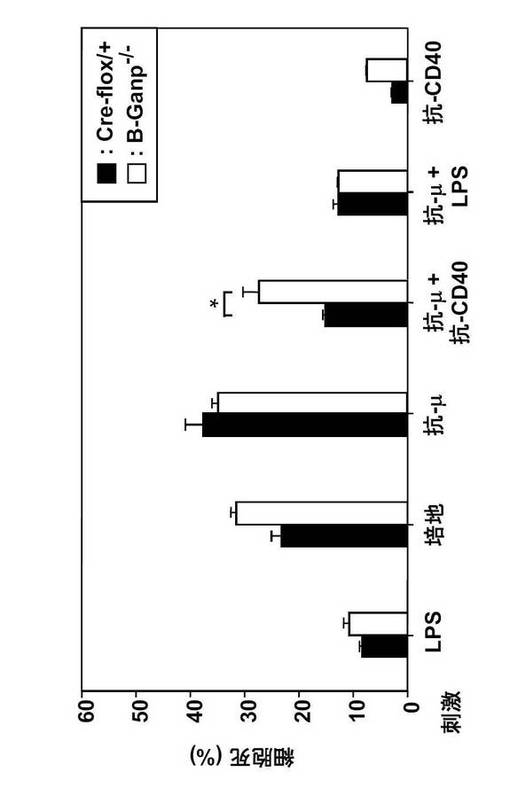
【図24】
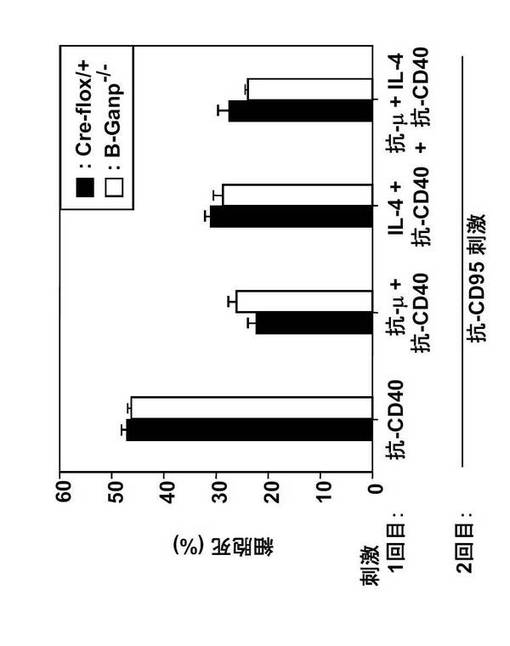
【図25】

【図26】
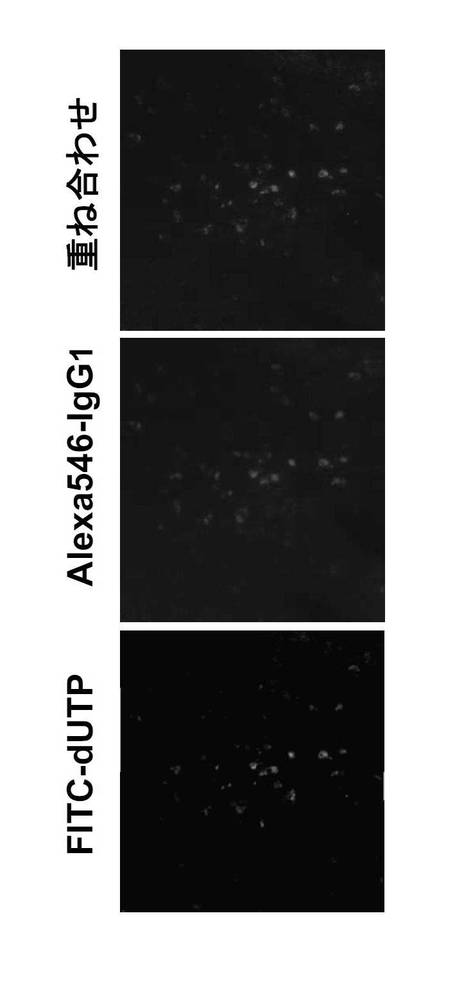
【図27】
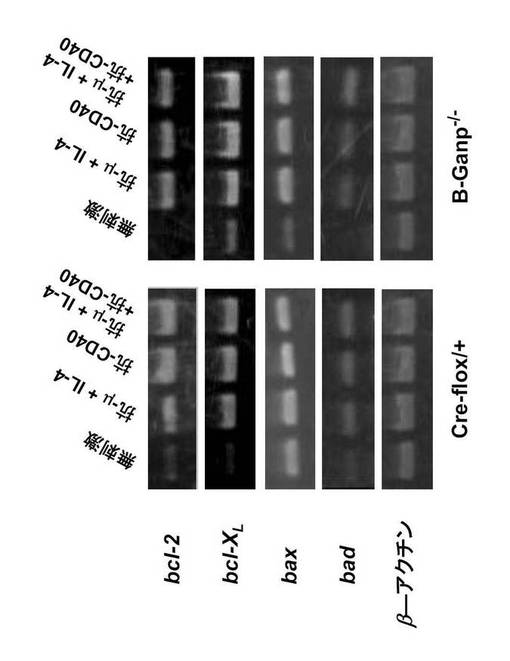
【図28】
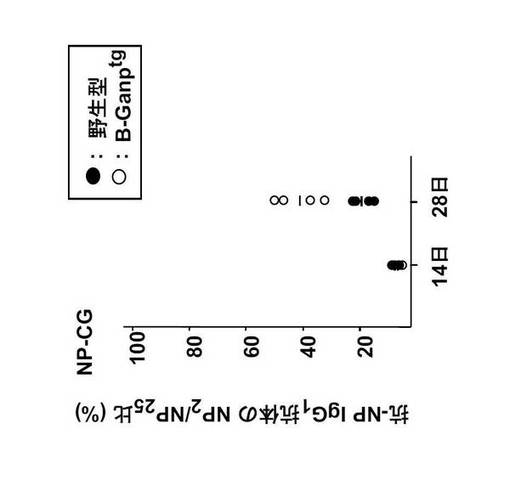
【図29】

【図30】
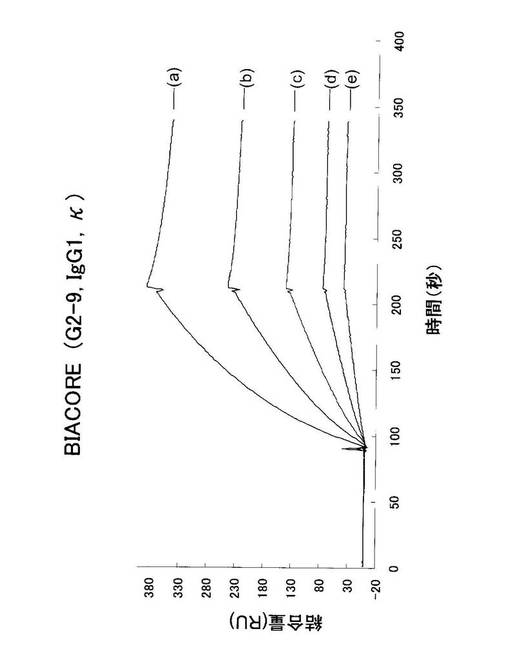
【図31】
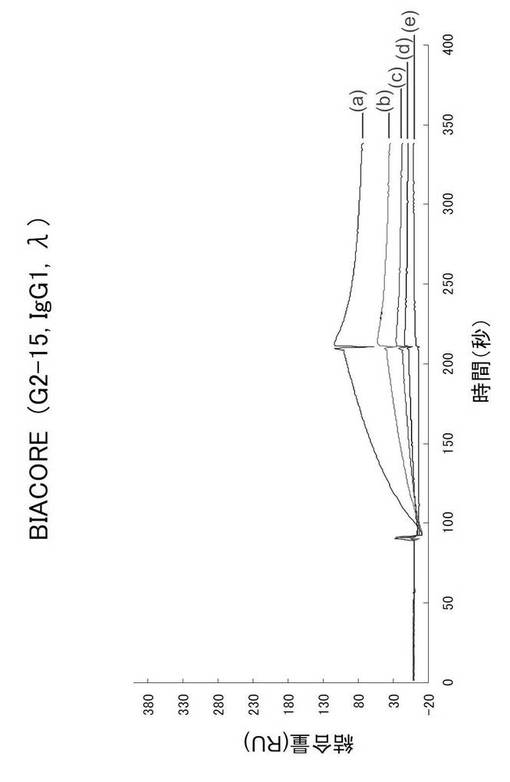
【図32】
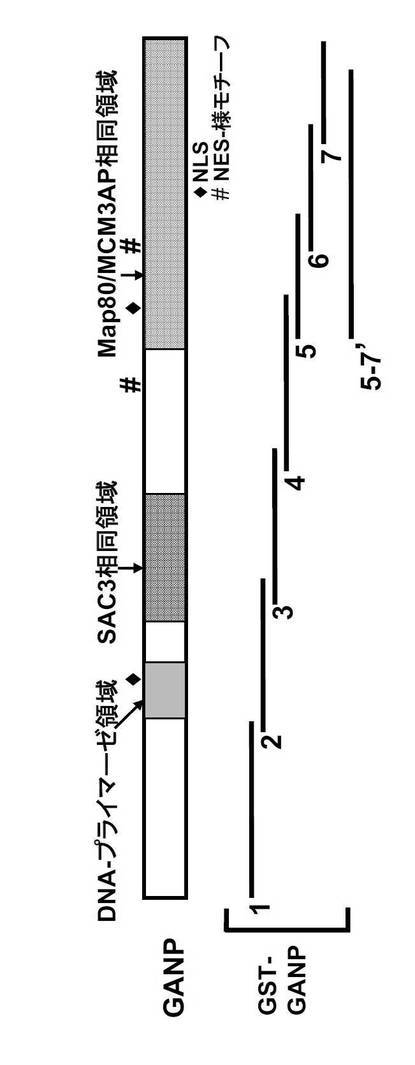
【図33】
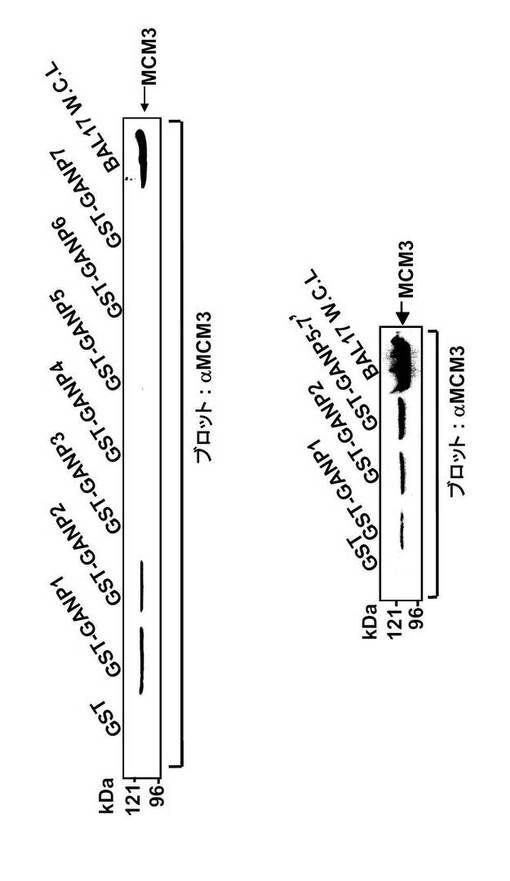
【図34】
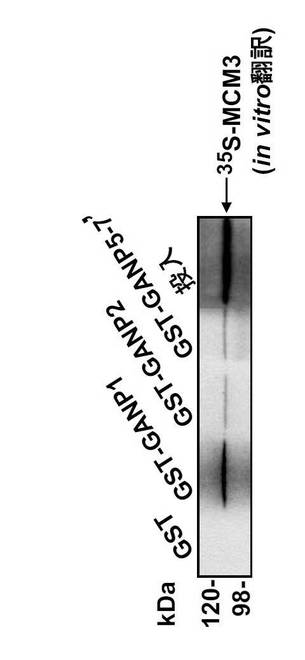
【図35】
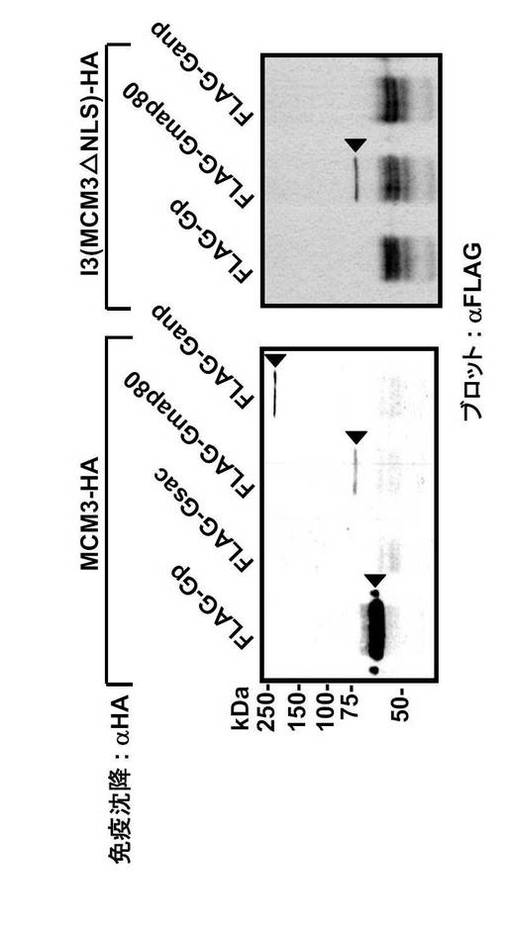
【図36A】
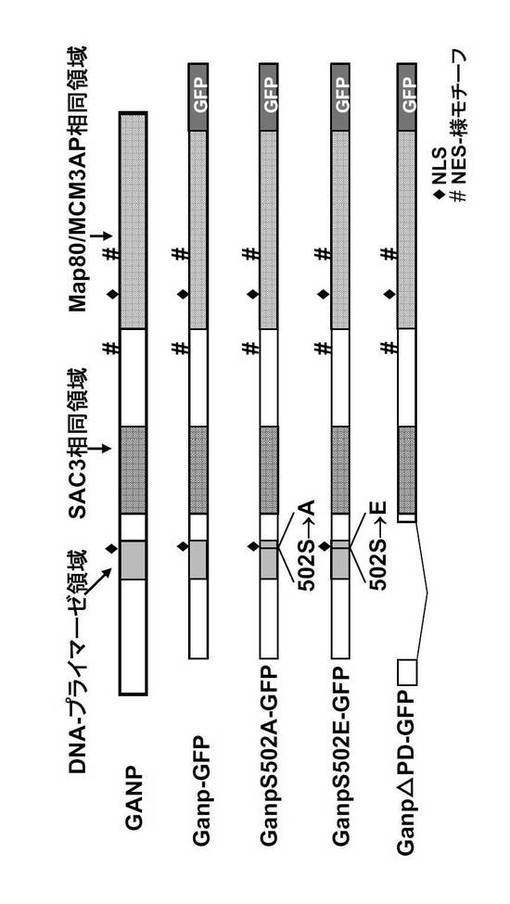
【図36B】
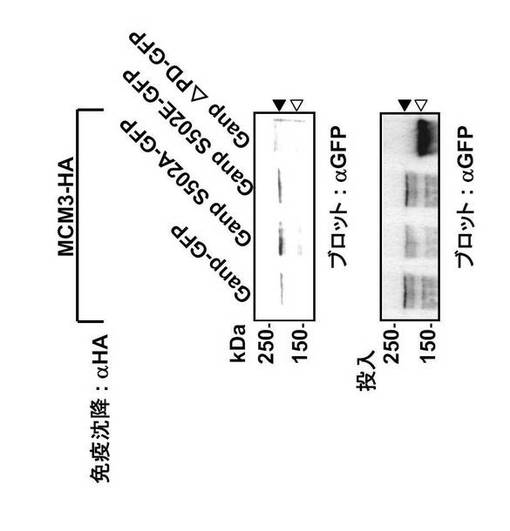
【図37】

【図38】
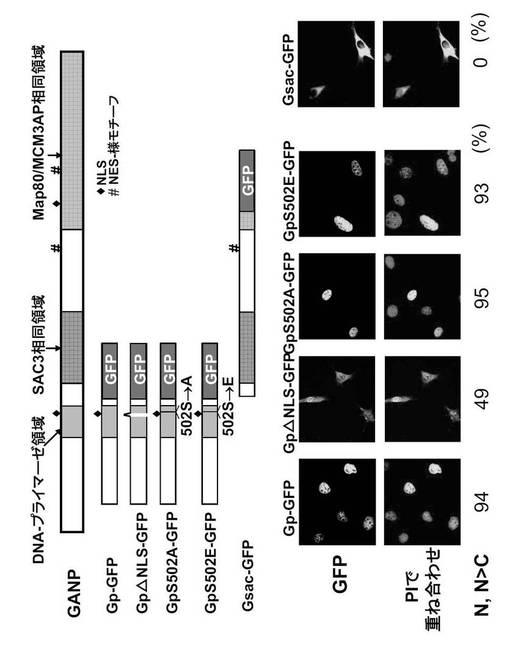
【図39】
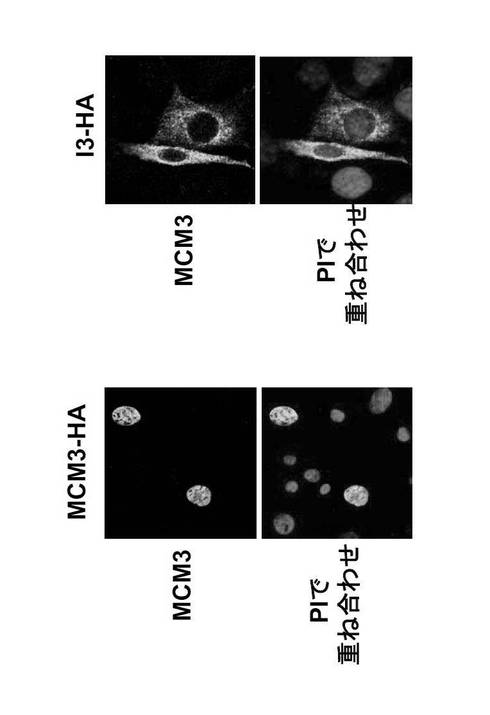
【図40】
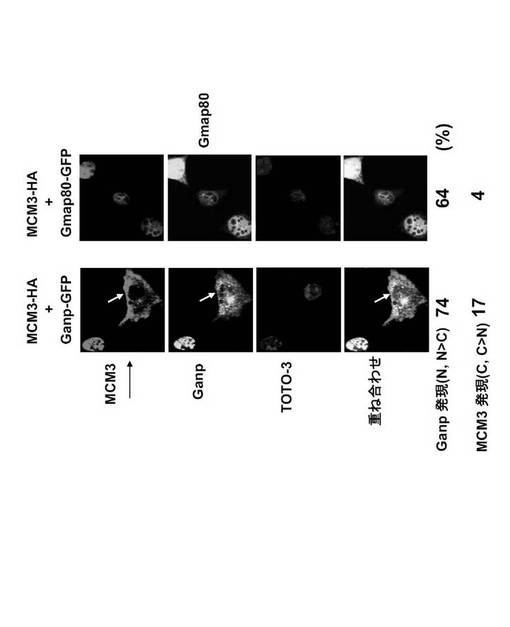
【図41】
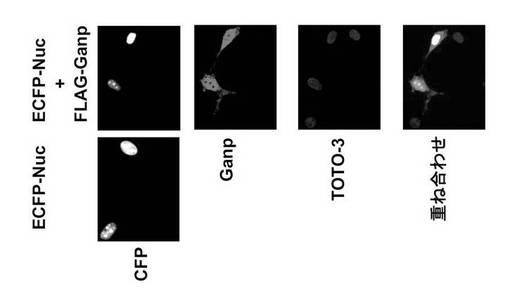
【図42】
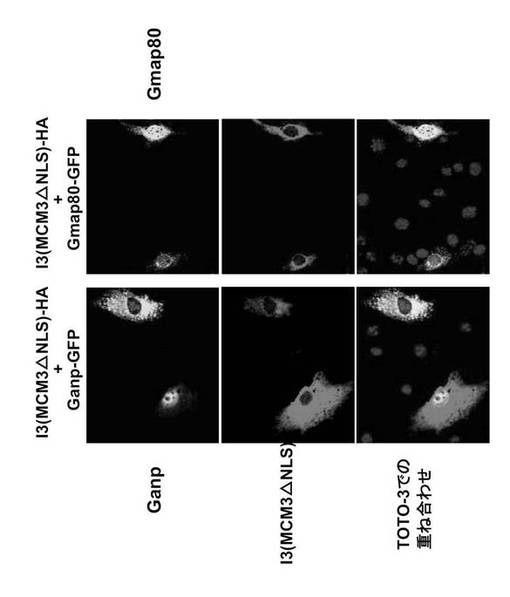
【図43】
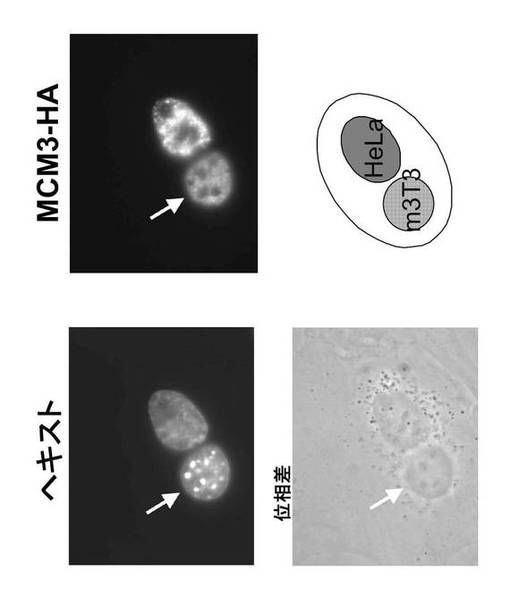
【図44】
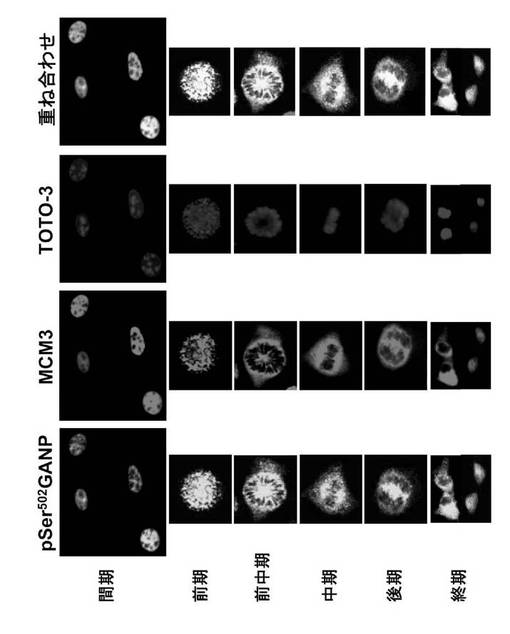
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8A】
【図8B】
【図8C】
【図9A】
【図9B】
【図9C】
【図10】
【図10−1】
【図10−2】
【図10−3】
【図10−4】
【図10−5】
【図11A】
【図11B】
【図11C】
【図11D】
【図11E】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20A】
【図20B】
【図20C】
【図20D】
【図20E】
【図20F】
【図20G】
【図20H】
【図20I】
【図20J】
【図20K】
【図20L】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36A】
【図36B】
【図37】
【図38】
【図39】
【図40】
【図41】
【図42】
【図43】
【図44】
【公開番号】特開2010−162026(P2010−162026A)
【公開日】平成22年7月29日(2010.7.29)
【国際特許分類】
【出願番号】特願2010−28977(P2010−28977)
【出願日】平成22年2月12日(2010.2.12)
【分割の表示】特願2004−549646(P2004−549646)の分割
【原出願日】平成15年11月7日(2003.11.7)
【出願人】(504112920)株式会社イムノキック (2)
【Fターム(参考)】
【公開日】平成22年7月29日(2010.7.29)
【国際特許分類】
【出願日】平成22年2月12日(2010.2.12)
【分割の表示】特願2004−549646(P2004−549646)の分割
【原出願日】平成15年11月7日(2003.11.7)
【出願人】(504112920)株式会社イムノキック (2)
【Fターム(参考)】
[ Back to top ]