
GBSトキシン/CM101の精製方法
【課題】新規GBSトキシン/CM101の精製方法。
【解決手段】B群β−溶血性ストレプトコッカス(group B β-hemolytic Streptococcus(GBS))バクテリアからの多糖類の精製方法は、バクテリアの発酵ストックを疎水性相互作用クロマトグラフィー(HIC)樹脂と接触させることを含む。追加の段階は、フェノール/生理食塩水抽出及びイオン交換クロマトグラフィーを含むことができる。本法は、ひじょうに高い純度をもつ製品をもたらす。上記精製からの製品は、研究及び治療環境において高く有用である組成物を提供する。
【解決手段】B群β−溶血性ストレプトコッカス(group B β-hemolytic Streptococcus(GBS))バクテリアからの多糖類の精製方法は、バクテリアの発酵ストックを疎水性相互作用クロマトグラフィー(HIC)樹脂と接触させることを含む。追加の段階は、フェノール/生理食塩水抽出及びイオン交換クロマトグラフィーを含むことができる。本法は、ひじょうに高い純度をもつ製品をもたらす。上記精製からの製品は、研究及び治療環境において高く有用である組成物を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
緒 言
技術分野
本発明は、多糖類の改良精製方法に関する。
【背景技術】
【0002】
背 景
CM101、GBSトキシンは、B群β−溶血性ストレプトコッカス(group B β-hemolytic Streptococcus(GBS))バクテリアから単離された病原性分子である。新生児は、GBSに感染するようになることができ、この症状は、GBS肺炎又は“早期発症疾患(early-onset disease)"として知られており、そして敗血症、顆粒球減少症、呼吸器の苦痛(respiratory distress)、すなわち、肺高血圧及びタンパク質性肺浮腫を患うことができる(Hellerqvist.C.G. et al., Studies on group B β-hemolytic streptococcus I.Isolation and partial characterization of an extra-cellular toxim.,Pediar.Res., 12: 892-898 (1981))。
【0003】
GBSに晒される新生児に対する有害な効果にも拘らず、CM101は加齢したヒトにおいて毒性を生じさせることは知られていない。実際、このトキシンについての研究は、かなりの治療的適用を現した。米国特許第5,010,062号及びHellerqvist, C.G. et al., Early Results of a Phase I Trial of CM101 in Cancer Patieuts., Proceedings of the American Association of Cancer Research Annual Meeting (1995)を参照のこと。ここでは、CM101は腫瘍の血管形成を阻害するために使用されている。それ故、純粋なCM101を得ることは、研究目的及び治療目的のために、重要である。
【0004】
CM101は、約300,000ダルトンの分子量を有し、かつ、N−アセチル−ガラクトサミン、N−アセチル−グルコサミン、グルコース、ガラクトース、及びマンノース残基を含む複合多糖トキシンである。Nmr(核磁気共鳴)の結果は、アルジトール(alditol)残基も存在することができるということを示唆している。カルボン酸官能基、好ましくは、ガラクツロン酸も、上記分子の不可欠な部分であると信じられている。活性エピトープの繰り返しは、おそらく、標的内皮上のレセプターを架橋することにより、病理生理学的応答において重要な役害を演じているようである(Hellerqvist, C.G. et al., Early Results of a Phase I Trial of CM101 in Cancer Patients., Proceedings of the American Association of Cancer Research Annual Meeting (1995); DeVore, R.F., et al., A Phase IStudy of the Antineovascularization Drug CM101, J.Clin.Can.Res. , 3: 365-372 (1997))。
【0005】
米国特許第5,010,062号は、GBSトキシンの精製方法を提供する。しかしながら、教示された方法は、労働集約的であり、連続的なレベルの生物学的活性の損失を伴う多くの工程を必要とする。
【0006】
本分野において現在知られているCM101の精製は、化学的分析及び生物学的アッセイにより計測されるときほんの40%の純度である末端材料を提供する。他の60%は、植物と酵母の多糖類並びに内因性のバクテリアの多糖類を含む。この植物と酵母の汚染物質は、大部分、GBSバクテリアの最適な成長のために使用される商業的な培養基に対する添加物に起因する。上記内因的な汚染物質は、群及びタイプ(group and type)特異的な抗原を含むGBS多糖類を含む(Paoletti, L.C. et al., Neonatol mouse protection against infection with multiple group B streptococcal (GBS) serotypes by maternal immunization with a tetravalent GBS polysaccharide-tetanus toxid conjugate vaccine, Infect.Immun. 62(8):3236-43(1994);Michon, F., Multiantennary group-specific polysaccharide of Group B Streptococcus, Biochem., 27: 5341-51(1988))。この40%純度のCM101は、現在の臨床的なグレードを表している。それ故、高められた全体純度をもち、好ましくは、外因性の植物と酵母の多糖類及びGBS多糖類をもつ末端製品をもたらすCM101の精製方法についての必要性が存在する。
【0007】
さらに、本分野において知られている精製スキームは、環境的に有害な工程、例えば、フェノール:水抽出における多量のフェノールの使用を含む、フェノールは、周知の焼灼(caustic)材料である。
【0008】
それ故、本発明の目的は、(i)高純度材料を、(ii)最小数の工程を使用して、(iii)焼灼性又は毒性材料、例えば、フェノールの使用を最小化して、そして(iv)上記材料の収率を増加させて、もたらす精製方法を提供することである。
【発明の概要】
【0009】
本発明の要約
上記目的は、本明細書中に記載する発明により達成された。特に、GBSバクテリア培養基からCM101を精製するための疎水性相互作用クロマトグラフィー(HIC)を含む精製スキームは、95%を上廻る純度の製品をもたらす。
【0010】
本発明の1の側面は、GBSバクテリアからの多糖類トキシンの精製方法であってHIC樹脂の使用を含む方法である。本発明は、本明細書中に開示する方法により製造されたGBSバクテリアからの実質的に純粋な多糖類トキシン、及び実質的に純粋なトキシンと医薬として許容される担体を含む医薬組成物をも含む。本医薬組成物は、医学的症状をもつ患者を治療するために使用されることができる。例えば、腫瘍患者は、本発明に係る組成物により治療されることができる。
【図面の簡単な説明】
【0011】
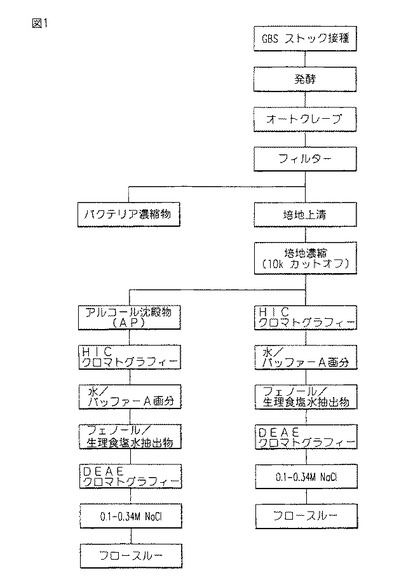
【図1】図1は、本発明のCM101精製スキームを示す。
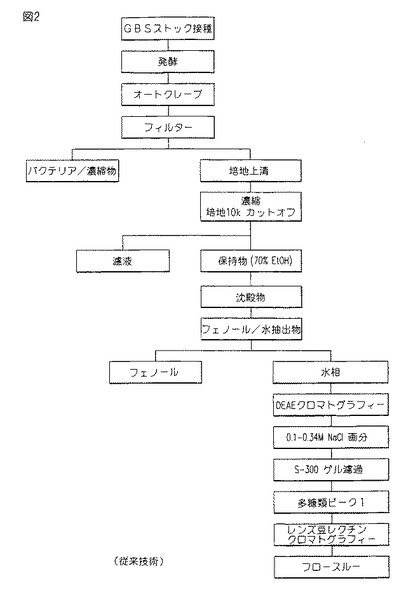
【図2】図2は、知られたCM101精製スキームを示す。
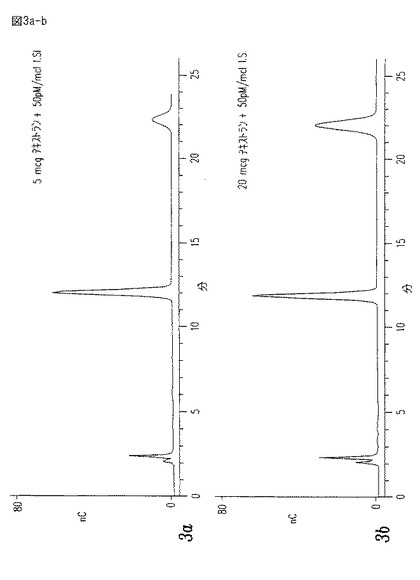
【図3a−b】一定内部標準として6−デオキシ・グルコースを含む、5,20、及び50μgのデキストラン(グルコース・ポリマー)についてのPAD検出器の投与量応答を示す、定量的加水分解標準曲線である。
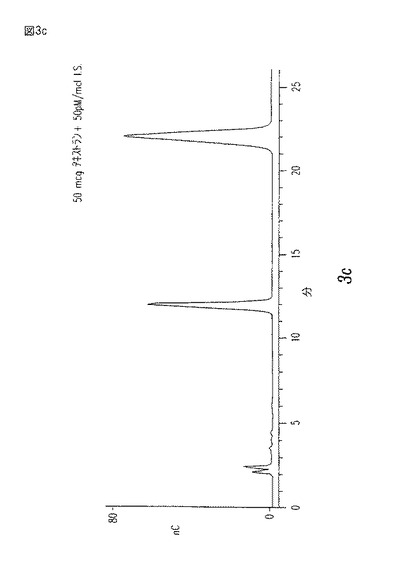
【図3c】一定内部標準として6−デオキシ・グルコースを含む、5,20、及び50μgのデキストラン(グルコース・ポリマー)についてのPAD検出器の投与量応答を示す、定量的加水分解標準曲線である。
【図4】図4は、標準糖サンプルの分離を示す。
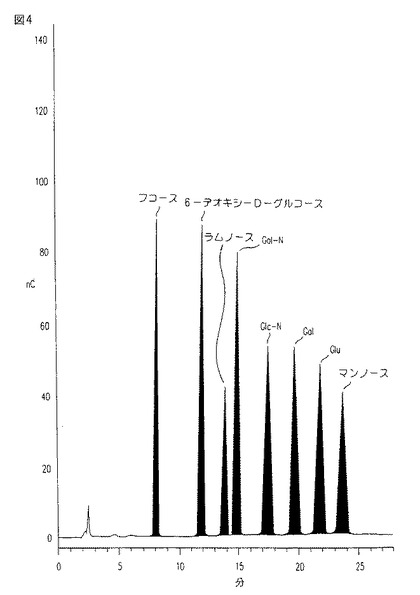
【図5a−b】図5a−bは、ブチル−セファロースHICカラム上での媒体濃縮物の溶出プロフィールである。図5aは、UV 206吸収において計測される。図5bは、UV 280吸収において計測される。
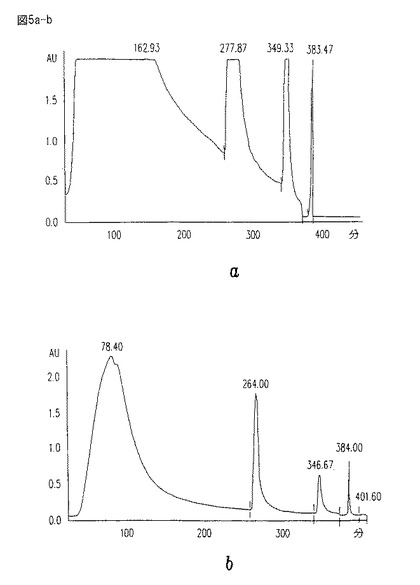
【図6a】図6aは、Millennium 2000 Diodo-Ray検出器(Waters, Millford, MA)上でのUV 203吸収においてモニターされ、そしてCM101(16分ピーク)を含むHIC−精製水−溶出画分のHPLCプロフィールである。
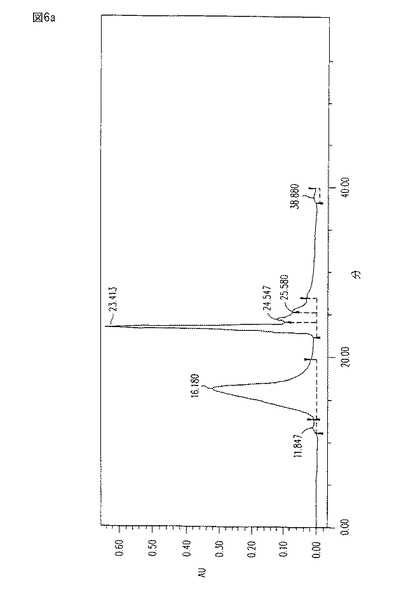
【図6b】図6bは、CM101含有(16分)ピークについての、260吸収(RNAとDNA)及び280吸収(チロシン含有タンパク質)の最小の存在を示し、そして図6aに対応するDiodo-Rayスペクトルである。
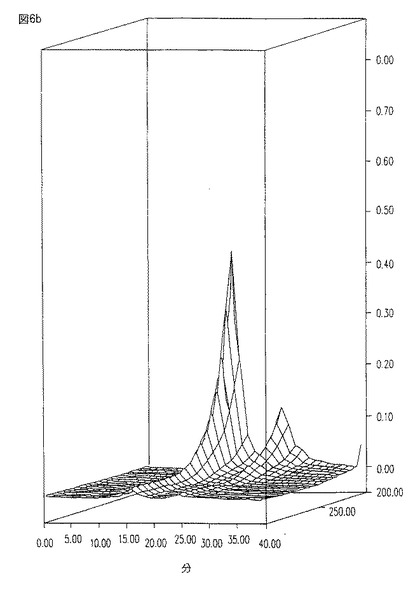
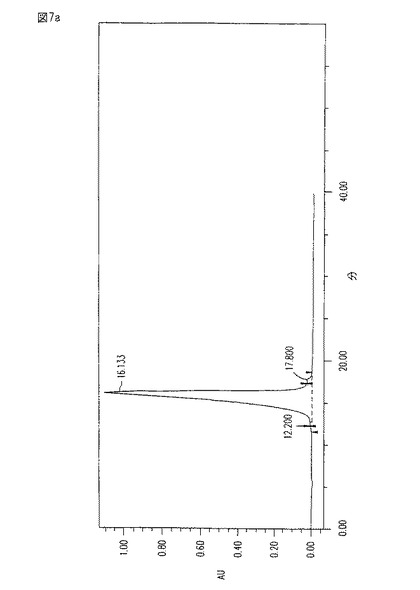
【図7a】図7aは、フェノール/生理食塩水抽出及びその後のDEAEクロマトグラフィーにさらに供された、図6aのHIC水−溶出ピークの純度を示す、203nmにおいてモニターされた溶出プロフィールである。
【図7b】図7bは、狭い対称ピークにより、及び260nm(RNA/DNA)と280nm(タンパク質)における吸収の欠如により、証明される、図7aのCM101−含有ピークの純度を示すDiode-Rayスペクトルである。
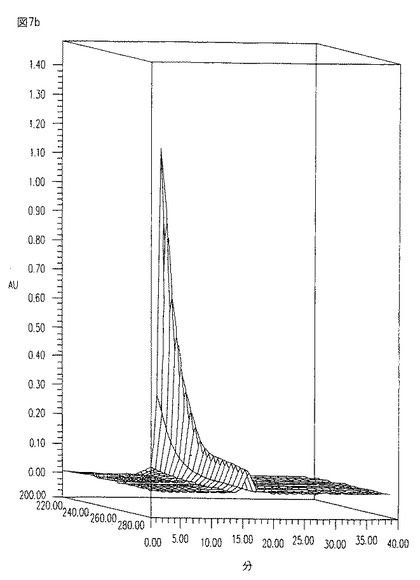
【図8】図8は、HICカラムから得られた画分のANA-1アッセイによるIL-6活性のプロフィール、より特に、100mlブチル・セファロース上を流れた10K5P6濃縮物から得られた画分のIL-6活性プロフィールである(FT=flow-through;1M=1Mホスフェート画分;0.25M=0.25Mホスフェート画分;H2O=水画分;EtoH=エタノール画分)。
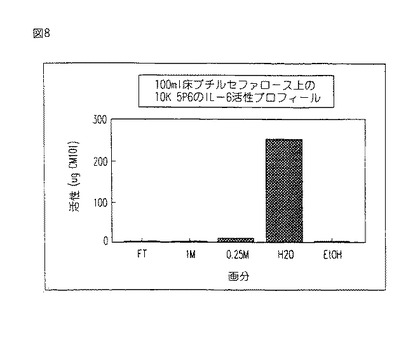
【図9】図9は、本発明に係る方法により精製されたCM101の糖分析を示す。
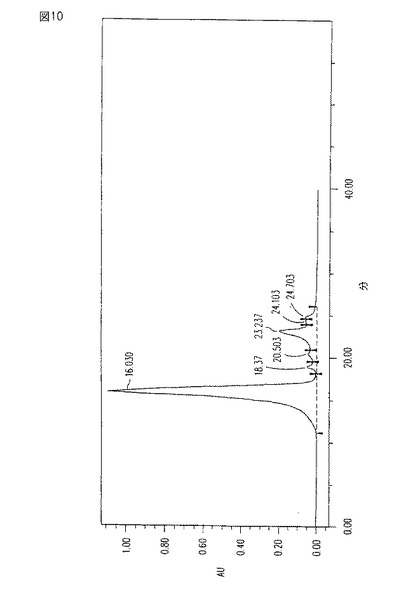
【図10】図10は、HICクロマトグラフィーにさらに供された最近の臨床グレードのCM101のHPLCプロフィールである。
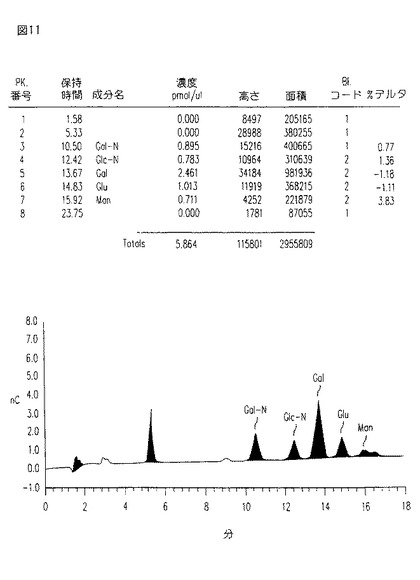
【図11】図11は、HIC及びHPLCによりさらに精製された最近の臨床グレードのCM101のサンプルの糖分析を示す。
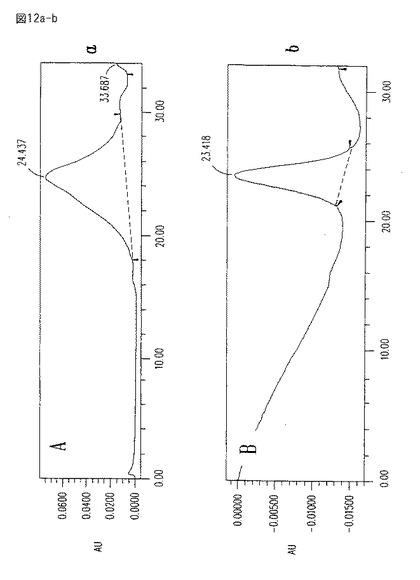
【図12a−b】図12aは、10mMホスフェート・バッファー、pH8.4を使用した知られた方法により精製されたCM101のHPLCプロフィールである。図12bは、図12aのHPLCプロフィールと同じ操作条件を使用して、本発明に係る方法により精製されたCM101のHPLCプロフィールである。
【発明を実施するための形態】
【0012】
特定の態様の説明
本明細書中に使用するとき、GBSトキシンは、天然又は溶解されたGBSバクテリアから単離され、又は溶解され、そして/又はオートクレーブにかけられたGBSバクテリアの培地上清から得られたいずれかの画分又は成分として定義され、そしてこれは、ヒツジのアッセイにおいて呼吸器苦痛の誘発(Hellerqvist, C.G. et al., Studies on group B β-hemolytic streptococcus I.Isolation and partial characterization of an extra-cellular toxin., Pediatr.Res., 12: 892-898 (1981))又は腫瘍組織試料のペルオキシダーゼ−アンチペルオキシダーゼ(peroxidase-antiperoxidase(PAP))アッセイにより証明されるような新生脈管構造への結合及び補体の活性化(Hellerqvist, C.G. et al., Anti-tumor effects of GBS toxin: a polysaccharide exotoxin from group B β-hemolytic streptococcus J.Canc Res.Clin.Oncol., 120: 63-70 (1993); 及びHellerqvist, C.G. et al., Early Results of a Phase I Trial of CM101 in Cancer Patients., Proceedings of the American Association of Cancer Research Annual Meeting (1995))により証明される生物学的活性を有する。
【0013】
実質的に純粋なGBSトキシンとは、GBSトキシンが40%を上廻る純度(例えば、少なくとも約40重量%の濃度で存在する)、好ましくは、少なくとも約60%の純度、より好ましくは、少なくとも約90%の純度、そして最も好ましくは少なくとも約95%の純度であるような調製物を意味する。
【0014】
本発明に係る方法における使用のためのGBS出発材料のための源は、最近感染し、又は新生児を感染させることができるB群β−溶血性ストレプトコッカス(Streptococcus)バクテリアの株を培養することにより得られることができる。このような株の単離物は、感染された乳児の血液から得られることができる。
【0015】
CM101の高い製造は、一般に、GBS最適成長及びCM101製造のための多糖類及びタンパク質の形態における高分子量材料を含有する、複合培地THBを用いた発酵を必要とする。
【0016】
発酵プロセスの間、バクテリアは、上記栄養から一定量のタンパク質、核酸、及びCM101以外の多糖類を製造する。上記発酵ブロス中のCM101の推定濃度は、0.1重量%未満である。
【0017】
本発明の精製方法は、知られた方法よりも効率的に内因性及び外因性汚染性タンパク質、核酸、及び多糖類のバルクを排除し、そして10〜50%の純度のCM101を含有する末端製品をもたらす、疎水性相互作用クロマトグラフィー(HIC)を使用する。GBS出発材料とHIC樹脂を接触させる唯一の工程において、これは、その出発材料からの100〜500倍の精製を表す。
【0018】
多糖類の精製のためのHIC樹脂の使用は驚ろくべきであり、そして新規である。なぜなら、HICカラムは、疎水性タンパク質の精製のためにデザインされており、そしてタンパク質及び脂質を含まない多糖類のために有用であるとは信じられていないからである。多糖類は、一般に、それらの多くのヒドロキシル基に因り親水性であると特徴付けられている。製造者により推奨され、そして当業者により使用される条件下でのHICカラムへの出発材料の適用は、それ故、タンパク質を保持し、そして多糖類が結合しないでカラムを通過することを許容する意図をもつであろう。
【0019】
驚ろくべき発見は、CM101が、高レベルの純度を達成するために本発明の精製スキームの使用を許容する疎水特性をもつということである。特に驚ろくべきことは、CM101がそれから単離されるところの上清中に存在するタンパク質及び多糖類の大部分よりも有意に大きな疎水特性をもつということである。上記タンパク質及び多糖類汚染物質の98%超が、HICカラムを通過する。HIC樹脂は一般にHIC−カラム中に使用されるけれども、この工程は、あるいは、いくつかの他のやり方で樹脂と出発材料とを接触させることにより行われることができる。例えば、GBS源及びその樹脂は、バッチ・プロセスで一緒に容器内に入れられ、そしてトキシン含有部分は、その後、例えば、遠心分離によりその樹脂から分離される。
【0020】
追加の精製工程は、従来の方法に比較して小容量(約1000倍減少)におけるフェノール/生理食塩水抽出、及びイオン交換カラムを含むことができる。これらの追加の精製工程は、95%超の純度をもつ末端製品に寄与する。
【0021】
HICは、それらの疎水性の性質に基づき、タンパク質、例えば膜タンパク質を分離するために使用される方法である。HIC樹脂は、この疎水性の基が、上記樹脂に接触している物質と相互作用しないように、支持体に一般に共有結合されている相互作用的疎水性基をもつ樹脂として定義される。疎水性基の例は、アルキル、アルコキシ、及びアリール基を含む。本発明に従って使用されるべき好ましいHIC樹脂は、2以上の炭素の脂肪族基、好ましくは2〜12炭素の範囲内のアルキル基、そしてより好ましくはノルマル又は分枝ブチル基が付着された支持体をもつ。フェニル基又は20炭素までのアルコキシ基も、好ましい相互作用性疎水性基である。相互作用性疎水性基は、好ましくは、セファロース(Sepharose)(Pharmacia)、アクリルアミド(Toso Haas, Montgomeryville, PA)、又はシリカにより支持される。HICカラムの使用のための標準的な手順に従えば、着目のタンパク質を含有する出発材料は、2Mまでの水性塩溶液中の上記カラムに適用され、そして次に結合したタンパク質は、溶出され、そして、展開(developing)バッファーのイオン強度を減少させることにより疎水性相互作用における減少を通じて、分離される。pH及び/又は温度における変更も、上記疎水性相互作用を変化させるために使用されることができる。
【0022】
B群ストレプトコッカス(Streptococcus)からのCM101精製は、GBSのバクテリア培養物を得ることを必要とする。バクテリア接種物は、2g/l以上のグルコース及びNa2HPO4を補うことにより修飾されたTodd Hewitt Broth (THB)中の対数増殖期後期までインキュベートされる。図1中に示すように、上記培養物はその後オートクレーブにかけられる。CM101は、オートクレーブ後、2〜15mg/lの濃度で、GBS発酵培養物の上清中に存在する。この培地は、約15g/lの他のバクテリア及び培地成分を含有する。従って、CM101は、上記上清中、上記成分の約0.01〜0.1%を構成する。オートクレーブ後、培地は濾過される。濾液は、好ましくは、10,000ダルトン(10kD)カット−オフ・フィルターを介して濃縮される。但し、50,000ダルトン(50kD)以下のカットオフをもつフィルターも使用されることができる。
【0023】
次に、CM101は、本発明に従って、図1中に示すように、リン酸カリウム又は他の塩、例えばリン酸ナトリウム、硫酸、塩化、又は酢酸ナトリウム又はカリウム中0.75〜2Mに調整された上清濃縮物又はその再構築されたアルコール沈殿物を、好ましくは同一モル濃度の同一塩中で平衡にされたHICカラムに、適用することにより、精製される。上記培地濃縮物、又は10kD〜50kDの出発材料が、アルコール沈殿及び再構築を伴わずに使用されるような方法が好ましい。なぜなら、培地濃縮物又は出発材料は、再構築されたアルコール沈殿物よりも高い収率のCM101を提供するからである。
【0024】
CM101含有出発材料は、HICカラムに適用され、そして0.75〜2Mにおける水性リン酸塩で洗浄される。0.75〜2M洗浄の後、このカラムを、さらに、0.5〜1M、そして次に0.25M塩で、好ましくはホスフェートで展開させる。好ましい態様においては、CM101は、10〜50%のCM101を含有する単一ピークとして水により上記カラムから溶出される。あるいは、水は、水中10%エタノール中の10mMホスフェート、pH6.8(バッファーA)により、その後水中20%エタノールにより、上記HICカラムから、CM101溶出物と置き替わる。CM101活性は、バッファーAと20%エタノール画分の両者中に回収される。バッファーAの使用は、HICカラムからCM101の全てを除去するために十分なものではない。従って、バッファーA洗浄の後に、追加の20%エタノール洗浄が続けられる。しかしながら、スケール−アップにおいては、エタノールは、環境ハザードを構成し、そして水ピーク又はバッファーA及び20%エタノール・ピーク画分のその後のフェノール/生理食塩水抽出は、凡そ等しい純度のCM101をもたらす。上記HIC手順は、10k濃縮物又は再構築されたアルコール沈殿物中に残存するタンパク質と培地多糖類の両者の98%以上を除去する。
【0025】
HICカラムからの濃縮されたCM101を、フェノール及び水性塩溶液、好ましくは0.05M生理食塩水中の抽出によりさらに精製されることができる。この追加の工程は、約95%の純度のCM101画分を提供する。
【0026】
HICカラムから溶出された、水又は、併合バッファーA及び20%エタノール画分は、水に対して透析され、そして凍結乾燥され、そして0.05M生理食塩水中で再構築されるか、又は濃縮後に生理食塩水に対して透析される。典型的には、フェノールが、上記材料に添加され、そしてその溶液は70〜80℃に急速加熱される。単一相が形成するとき、その溶液は4℃に冷却される。上記フェノール/生理食塩水抽出物の得られた生理食塩水相は、CM101を含み、そして次にカチオン交換カラム、例えば、DEAEに適用されることができる。
【0027】
DEAEカラム手順のために、DEAEカラムは、水中で平衡にされ、そして次に0.1M生理食塩水、0.05M NaOAc、pH7.4で洗浄され、そして0.34M NaClまでのステップ・グラジエントにより展開される。CM101の溶出はモニターされ、そしてANA-1アッセイを用いて定量される。HIC樹脂工程によるとき、イオン交換樹脂は、カラム以外の装置の使用を通じてトキシン含有材料と接触されることができる。次にCM101含有画分は、水に対して透析され、そして凍結乾燥される。フェノール/生理食塩水抽出及びイオン交換工程の後、CM101は95%を上廻る純度をもつ。
【0028】
上記樹脂接触工程から得られたカラム溶出物、又は材料は、ANA-1及び/又はドット・ブロット・アッセイを用いて生物学的活性についてアッセイされる。次に生物学的活性は、ヒツジ・アッセイを用いて確認される。表1は、出発材料の異なるバッチから得られ、そして上記HICカラムに適用されたAP及び10k材料のいくつかの分離を表す。変性タンパク質並びに培地多糖類及び他の材料の除去は同様である。
【0029】
好ましい精製の順序は、HIC工程、その後のフェノール/生理食塩水抽出、そしてその後のイオン交換工程を実施することであるけれども、精製は、他の順序において行われることもできる。
【0030】
図2は、CM101精製の知られた方法の1例である。特に、70%エタノール沈殿工程が使用され、その後、直ちに、フェノール/水抽出が行われる。この知られた精製方法の上記の初期段階において必要とされる多量のエタノール及びフェノールは、環境的に有害な実施を提供する。図2中に表された方法は、イオン交換カラム、ゲル濾過カラム、及びレンズ豆(lentil)レクチン・カラムをも要求する。
【0031】
従来の方法は、環境的に危険なものを含む、多くの工程を含む。一方において、本発明の方法は、有効であり、高純度及び2〜25倍の収率を与え、そして環境的に有害な材料の使用を最小化する。
【0032】
この環境的に危険なフェノール−水抽出工程は、従来使用されてきた手順に比較するとき、100倍減少される。さらに、例えば、ゲル透過手順による追加の精製が取り除かれる。従来方法のレンズ豆レクチン・クロマトグラフィー工程も取り除かれる。HICカラム、フェノール/生理食塩水抽出、及びイオン交換カラムの工程の末端製品は、約95%の純度をもち、それ故、他の処理は不要である。
【0033】
本発明の方法により精製されたCM101は、研究又治療目的をもって使用されることができる。本CM101は、医薬として許容される担体と併合されるとき、例えば、生理食塩水中で再構築され、そして患者に静脈内投与されるとき、特に有用である。精製されたCM101を投与するための他の剤形を使用することもできる。本発明の医薬組成物は、医薬として許容される担体との組合せにおいて本発明の実質的に純粋なGBSトキシンを含む。一般に、上記担体は、静脈内(IV)手段により投与されることができる組成物を形成するために、上記トキシンと容易に混合されるものであろう。従って、上記担体は、好ましくは、静脈内投与のためのその好適性を保証するために含有された他の医薬として許容される賦形剤をもつことができる、水である。得られた組成物は、滅菌され、そして許容される浸透圧特性をもつであろう。一般に、好適なIV配合物は、当業者に知られた標準的な技術に従って調製される。例えば、the Eighteenth Edition of Remington's Pharmaceutical Sciences, Mach Publishing Co. (1990) 中Salvatore J.Turco による“Intravenous Admixtures"と題する第85章(引用により本明細書中に取り込む)は、本発明に従って有用な、医薬として許容されるIV組成物を調製するための標準的な技術を提供する。
【0034】
さらに、CM101に有利に応答することが判明している医学的症状をもつ患者は、本発明の医薬組成物により治療されることができる。例えば、腫瘍をもつ患者は、本明細書中に教示される医薬組成物を静脈内投与することにより、有利に処置されることができる。米国特許第5,010,062号は、ヒトにおける特定の腫瘍の治療について討議しており、そしてそれを引用により本明細書中に取り込む。
【0035】
定量的及び定性的分析
HPLC分析
HICクロマトグラフィー後にサンプルから得られたCM101の純度及び量を、高圧液体クロマトグラフィー(HPLC)ゲル濾過分析により、確立する。ゲル濾過カラムは、典型的には、水中10%アセトニトリルにより平衡とされ、そして生物学的に活性なCM101が、それが含有された均質の狭いピークとして溶出される。あるいは、このカラムは、10mMホスフェート・バッファー、pH8.4中で展開され、それは、それがより含有されたピークをもたらす。酢酸アンモニウム(NH4OAc)バッファー、pH8.4は、10mMホスフェート・バッファーのさらなる代替物として使用されることができる。
【0036】
Hydragel 1000カラム(Waters, Millford, MA) 上での100μlの展開バッファー中にインジェクトされた、30,50、及び100μgの純粋なCM101標準を使用した典型的な検出器応答(UV 203吸収)は、それぞれ、26×106, 48×106、及び97×106の面積単位(area units)であり、それは、未知のサンプルの定量のための投与量応答曲線をもたらす。
【0037】
CM101の分子量も、ゲル濾過クロマトグラフィーにより計測されることができる。上記の非変性バッファー、例えばアセトニトリル、ホスフェート、又は酢酸アンモニウム・バッファーが、上記カラムを流すために使用される。CM101溶出は、異なる分子量をもつ標準デキストラン多糖類マーカーの溶出と比較される。CM101は、上記条件下、約300,000ダルトンの分子量をもつ。
【0038】
アミノ酸分析
定量的及び定性的な自動化されたアミノ酸分析は、標準的な商業的に入手可能な装置、例えば、Waters, Millford, MAから入手可能なPicotagを用いて、行われることができる。
【0039】
ANA-1 アッセイ
異なる発酵及び精製工程の生物学的活性をモニターするために、形質転換されたマクロファージ細胞系を使用するインビトロ・アッセイが使用されることができる。このアッセイは、CM101暴露に応答するマウス・マクロファージANA-1のIL-6生産を計測する。
【0040】
特に、CM101は、raf/myc形質転換ネズミ骨髄マクロファージ細胞系ANA-1が、IL-6生産によりインビトロにおいて応答することを誘発する。他のマクロファージ様細胞系及び新鮮末梢血液白血球も使用されることができる。
【0041】
ANA-1アッセイを実施するために、サンプルは、まず、(CM101活性の予想されたレベルに依存して)適当なレンジまで希釈され、そして4〜8つの濃縮物が1:4希釈においてテストされる。PBS中で再構築される臨床グレードのCM101を使用したCM101標準曲線が作製される。上記細胞が添加された後に2000ng/mlの最終濃度を与える4000ng/ml溶液を添加し、6つの連続した1:2希釈をもって、PBS中で調製する。例えば、2×106/mlの濃度における細胞を使用することができる。このアッセイの感度は、ANA-1細胞に200U/mlのネズミINF-γを添加することにより、高められる。最終培養物は、100U/ml IFN-γであった。
【0042】
培養物の入ったマイクロタイター・プレートは、一夜(16〜18時間)、37°、5% CO2/空気、加湿インキュベーター内に入れられなければならず、そしてその後、ELISA IL-6アッセイ(R.D.Systems, Minneapolis, MN)が続けられなければならない。特に、培養上清は、上記IL-6アッセイ・プレートに移され、そしてそのプレートは、IL-6アッセイが完結するまで、4℃で保たれる。
【0043】
ドット・ブロット・アッセイ
溶液又は生物学的液体中のCM101を定量的に検出するための他の迅速な手順は、段階的希釈においてポリビニリデン・ジフルオリド(PVDF)上でサンプルをブロットすることである。CM101の量は、CM101に対する蛍光タグ付マウス・モノクローナル抗体、又はCM101に対するマウス・モノクローナル抗体、その後の蛍光タグ付抗マウスIgGのいずれかを使用して、定量される。CM101抗原に対して向けられた抗体7A3は、この目的のために有用である。異なる画分中のCM101の定量は、連続的に希釈されたCM101標準の標準曲線に対する比較により確立される。
【0044】
ヒツジ肺動脈圧アッセイ
上記トキシンは、肺動脈圧の上昇及び肺血管透過性の上昇により顕示される、肺高血圧の上昇により、ヒツジ肺に影響を及ぼす。
ホスフェート・バッファー生理食塩水(PBS)中のCM101サンプルが、注入により子ヒツジに投与されることができ、そして肺動脈圧における変化は、15分間隔で記録される。圧力におけるこれらの変化は、CM101活性に相関する。(Hellerqvist.C.G. et al., Studies on group B β-hemolytic streptococcus I.Isolation and partial characterization of an extra-cellular toxin., Pediatr.Res., 15: 892-898 (1981))。
【0045】
糖分析
100μg量のサンプルを、5:70:25の比におけるトリフルオロ酢酸(TFA)、酢酸(HOAc)及び水の混合物中100℃において2時間加水分解する。この溶液を蒸発させ、そしてそのサンプルを、2:8の比におけるTFAと水の混合物中100℃で2時間さらに加水分解する。この方法は、サンプル中のグリコシド結合の全てを完全に加水分解する。アミノ糖上に元来存在するN−アセチル基も除去される。
【0046】
次にサンプルを、PAD(Pulsed Amperometric Detection)検出器を使用してDionex糖分析システム上で分析する。分離結果を図4に示す。
【0047】
上記サンプルの純度を、定量的及び定性的糖分析により確立する。この原理を図3a−cと図4に示す。HPLCにより定量された多糖類のサンプルに、加水分解され、かつ、分析された内部標準6−デオキシ−D−グルコースを補う。この章中に記載する方法は、テストされたレンジ内で線形投与量応答を与え、そして定性分析は、上記標準と未知の保持時間を比較することにより、達成される。
【実施例】
【0048】
実施例
実施例1:CM101のためのスケール・アップ精製
B群ストレプトコッカス(Streptococcus)血清型III単離物の通常在庫を、3,000ガロンの発酵槽と共に使用した。バクテリア培養物の25ml種母を、65リッターの発酵容量(liter working volume (lwv))をもつ80リッター容器のために使用し、これを次に、750lwv容器を接種するために使用し、そしてこれを次に、最終的に7500lwv(3,000ガロン)の発酵槽にもっていく。あるいは、この65lwvを、7500lwv発酵槽を直接的に接種するために使用することができる。
【0049】
上記培養を、オートクレーブすることにより対数増殖期後期に、終了させる。次に、バクテリアを、10,000×gにおける連続的遠心分離、その後の、0.45ミクロンのカセット濾過(Millipore Corporation, Bedford, MA)により除去する。
【0050】
次に、得られた培養上清を、10kD〜50kDのカット・オフ・カセット(Millipore)を使用したカセット濾過を使用して、500リッターまで、15倍濃縮する。次に、濃縮された材料、透析により、塩、好ましくは、リン酸ナトリウム、pH7.4(ローディング・バッファー)中2Mに調製する。
【0051】
次に、濃縮された上清を、BioPilotシステム(Pharmacia)を使用して60リッターのn−ブチル・セファロース・カラム(Pharmacia, Uppsala, Sweden)の使用を通して、疎水性相互作用クロマトグラフィーに供する。活性のflow-throughを伴わない、培質濃縮物中の生物学的CM101活性についてのn−ブチルSepharose樹脂の能力は、約80リッターの培地対1リッターの樹脂である。濃縮された上清が上記カラム上にロードされた後、このカラムを、上記ローディング・バッファー、その後、0.5〜1M、そして次に0.25Mホスフェート・バッファー、pH7.4で洗浄する。CM101含有画分を、約120リッター又は2カラム容量内の水で溶出し、そして10kD〜50kDの範囲内のカット−オフ・カセット内で2リッターに濃縮する。このカラムの溶出を、BioPilotにおいて事前に確立されたプログラムにより制御し、そして溶出物を、206及び280nmにおけるUV吸収、導電率、及びpHによりモニターする。
【0052】
CM101含有2リッター画分を、0.05M生理食塩水、pH7.0に対して透析し、そして次に75〜80℃の範囲に加熱し、そして0.2〜2リッターのフェノールを添加する。次に混合物を80℃に加熱し、そして5分間その温度で維持する。その後、この混合物を4℃に冷却する。この段階から得られた水相を、好ましくは、水中で平衡化されたDEAE Sephacel FFカラム(Pharmacia, Uppsala, Sweden)に適用する前に0.2容量のクロロホルムで2回抽出する。このカラムを、100mM生理食塩水、0.05M NaOAc、pH7.4で洗浄し、そして生理学的に活性な材料、CM101を、次に、NaClグラジエントで上記DEAEカラムから溶出する。この生物学的活性を、Il-6アッセイ及びHPLC分析により検出する。この手順を通して精製されたCM101の品質を、HPLC及び糖分析により並びにIl-6及びヒツジ・テストによる生物学的活性アッセイにより確立する。
【0053】
上記のスケール・アップされた精製スキームは、従来の手順において使用されたアルコール沈殿の、多量の、初期のフェノール−水抽出手順を回避するという利点を提供する。
【0054】
結 果
図5a−bは、2M K2HPO4、pH7.2中、ブチル−Sepharose HICカラム上の媒質濃縮物の溶出プロフィールを示す。さまざまなピークは、溶出グラジエントにおける、経時的、段階的な変化の結果である。図5aは、UV 206吸収において計測されたプロフィールを表し、これは、全有機材料についてのピーク画分を定量し、そして最後の狭いピーク(約383分)内のCM101を示す。図5bは、UV 280吸収において計測されたプロフィールを表し、これは、異る画分中のタンパク質の量を定量する。
【0055】
HICカラム段階を行うことにより、CM101は、上記カラムに結合するようにされるが、99.7%までのタンパク質及び98.5%までの中性及び電荷をもつ多糖類が、表1に示されるように、上記カラムを通過する。
【0056】
【表1】

【0057】
表1中、アルコール沈殿物(AP)としての異なる発酵ロット、AP1,AP2、及びAP6、及び10k濃縮物を、HICクロマトグラフィーに供し、そして水又はバッファーAのいずれかで溶出した。両プロセスは、外因性及び内因性タンパク質(UV 280)並びに多糖類及び一般有機物(UV 206)のほぼ同一の効率的な除去をもたらした。
【0058】
図6a−bは、CM101を含有し、そしてUV 203吸収をモニターされたHIC−精製水−溶出画分の、HPLCプロフィール、及びDiodo-Rayスペクトルを表す。これらの図は、CM101含有ピークについての206吸収(RNAとDNA)及び280吸収(タンパク質)を示す。
【0059】
HIC画分が、フェノール/生理食塩水抽出、及びイオン交換段階にさらに供された後、HIC水−溶出ピークの純度を、図7a−bに見られるように、さらに改良する。時刻0から約16分目における狭い対称ピーク、並びに260(RNA/DNA)及び280(タンパク質)における吸収の欠如に注目のこと。図6a−b及び7a−bにおいて示される溶出プロフィールについて、HPLCを、水中10%アセトニトリルを用いて行い、そしてその流速は、約0.3ml/分であった。
【0060】
これらの溶出プロフィール並びに生物学的活性は、アルコール沈殿物がHICカラムのための出発材料として使用されるときに得られるものと同様である。
【0061】
ANA-1細胞内でIL-6合成を誘発する10k出発材料からのHIC画分の能力を、図8中に示す。HICクロマトグラフィーは、ANA-1アッセイにより計測されるとき培地上清中の全生物学的活性のほぼ50%の回収をもたらした。CM101抗原の存在下で免疫反応性を示す同一材料のドット・プロット・アッセイを、ANA-1アッセイ結果を確認するために使用した。
【0062】
HICクロマトグラフィー後の10k濃縮物から得られた異なる画分を、生物学的活性についてのヒツジ・モデルにおいてもテストした。CM101活性の量を、最近の臨床的CM101を使用した投与量応答曲線に基づいて決定する(1ユニットの活性は7.5μg/kgに対応する)。結果を表2中に示す。ここで、アルコール沈殿物(AP)と媒質濃縮物(10k)のHIC画分が比較される。
【0063】
【表2】
【0064】
本発明の方法により精製されるときのCM101の生物学的活性は、ヒツジにおける肺動脈圧アッセイを用いても計測され、そして例えば米国特許第5,010,062号中に教示されたような、古いプロセスにより精製されたCM101の活性と比較された。本発明に従って精製された材料は、上記の古いプロセス、すなわち、HIC樹脂と接触されていないプロセスにより、精製された材料よりも2〜3倍高い比活性を示した。
【0065】
本発明の方法の製品収率も、先に証明される。なぜなら、先に示す7520μg/l値に比較して、既知の方法は、約300μgのCM101/発酵容量1リッターを提供するからである。
【0066】
本発明の方法により得られた図7a−b中に示す精製されたCM101は、糖分析にも供された。糖の収量を図9中に示す。
【0067】
定量的には、本発明の方法により得られたCM101は95%を上廻る純度の炭水化物であり、そして先に提示されたような定量及び定性により、そして自動アミノ酸分析(Pico Tag, Waters, Millford,MA)により、確立された5%未満のタンパク質を含む。
【0068】
実施例2:最近の臨床グレード及び新規組成物の比較
本発明の方法により得られたCM101は、最近の臨床グレードのCM101よりも改善されている。特に、図7aと比較するとき、図10のHPLC溶出特性は、本発明に従って製造されたサンプル中の、より高い純度を示す。図7aは、いくつかのピークの代わりに、1つの狭い、かつ、対称のメイン・ピークを示す。
【0069】
HICカラムの有利な使用をさらに証明するために、そしてCM101として知られたトキシンの精製のさらなる証拠を提供するために、最近の臨床グレードのCM101をHICカラムに供し、そしてHPLC精製及び糖分析を行った。図1中の結果を図9に比較することができる。糖分析は、定量的、かつ、定性的に同様の末端製品を示し、そして本HICクロマトグラフィーが、生物学的に活性なCM101に関連しない糖を除去することを証明している。この結果を表3中にも示す。
【0070】
【表3】
【0071】
先に表す糖残基の表は、凡そのモル比を与える。本発明の方法に従って精製されたCM101の実際の残基(第1列)は、(0.2〜1マンノース):(2.5〜3.5ガラクトース):(0.5〜1グルコース):(1N−アセチル・グルコサミン):(0.5〜1N−アセチル・ガラクトサミン)のレンジ内にある。上記の数を、N−アセチル・グルコサミンに対して正規化する:従って、N−アセチル・グルコサミンを1に設定する。
【0072】
さらなる比較のために、図12a−bは、古いプロセス(図12a)により、そして本発明の方法に従って(図12b)製造されたCM101のHPLCプロフィールを示す。ゲル濾過カラム(Ultragel 100, Waters, Milford, MA)を、10mMホスフェート・バッファー、pH8.4中で展開する。図12b中に示すように、本発明の方法に従って精製されたCM101は、約5分間のレンジにわたり比較的狭いピークにおいて溶出する。この溶出時間は、0.3ml/分の流速をもって時刻0から約24分目である。これに対して、古いプロセスにより精製された材料は、約12分間にわたりブロードなピークにおいて溶出する。
【0073】
実施例3:SDS-PAGE/ウェスタン・ブロットによる CM101の分析
ドデシル硫酸ナトリウム−ポリアクリルアミド・ゲル電気泳動を4〜20%のグラジエント・ゲルを使用して行った。2%SDS、0.5M Tris-HCl、5%グリセロール、0.05%ブロモフェノール・ブルー、及び5% β−メルカプトエタノール pH0.8のバッファー中のCM101サンプルを、10分間55℃においてインキュベートし、そして4%スタッキング・ゲル中で、4〜20%SDSランニング・ゲルに適用した。このゲルを、90分間200ボルトにおいて走らせた。
【0074】
このゲルを、ウェスタン・ブロッティング・バッファー(25mM Tris、 192mMグリシン及び20%メタノール)中で展開し、そして2時間100ボルトにおいてブロットした。このゲルを、1時間室温においてPBS中で5%無脂肪乳でブロックし、そして結合バッファー(ホスフェート・バッファー生理食塩水、2%子ウシ胎児血清、及び 0.5%TWIN-20洗剤(BBT))で2回洗浄し、次に10μg/mlの7A3(CM101に対するモノクローナル抗体)で1時間インキュベートした。
【0075】
このゲルを、BBTで10分間4回インキュベートし、そして45分間、アルカリ・ホスファターゼ拘合抗−マウス抗体と共にインキュベートし、BBTで4回洗浄し、そして50分間Pierce Single-Step AP発色剤で、発色させた。
【0076】
SDS-PAGE/ウェスタン・ブロット分析は、CM101が上記条件下で分析されるとき、26,000ダルトンの成分又はそれらの多数体をもつということを示唆した。
【0077】
従って、本発明の方法は、危険な工程の困難性を最小化し、そしてすばらしい純度を提供する改良された精製法を提供する。さらに、本明細書中に教示された方法により製造される製品は、最近入手可能なCM101よりも改善されている。
【0078】
本明細書中に言及した全ての刊行物及び特許出願を、あたかも、個々の刊行物又は特許出願が特別に、かつ、個々に、引用により取り込まれることを示されるのと同程度で、引用により本明細書中に取り込む。
【0079】
本発明をこれまで十分に記載してきたが、多くの変更及び修正が添付請求の範囲の本質又は範囲から逸脱せずに行われることができるということは、当業者にとって明らかであろう。
【技術分野】
【0001】
緒 言
技術分野
本発明は、多糖類の改良精製方法に関する。
【背景技術】
【0002】
背 景
CM101、GBSトキシンは、B群β−溶血性ストレプトコッカス(group B β-hemolytic Streptococcus(GBS))バクテリアから単離された病原性分子である。新生児は、GBSに感染するようになることができ、この症状は、GBS肺炎又は“早期発症疾患(early-onset disease)"として知られており、そして敗血症、顆粒球減少症、呼吸器の苦痛(respiratory distress)、すなわち、肺高血圧及びタンパク質性肺浮腫を患うことができる(Hellerqvist.C.G. et al., Studies on group B β-hemolytic streptococcus I.Isolation and partial characterization of an extra-cellular toxim.,Pediar.Res., 12: 892-898 (1981))。
【0003】
GBSに晒される新生児に対する有害な効果にも拘らず、CM101は加齢したヒトにおいて毒性を生じさせることは知られていない。実際、このトキシンについての研究は、かなりの治療的適用を現した。米国特許第5,010,062号及びHellerqvist, C.G. et al., Early Results of a Phase I Trial of CM101 in Cancer Patieuts., Proceedings of the American Association of Cancer Research Annual Meeting (1995)を参照のこと。ここでは、CM101は腫瘍の血管形成を阻害するために使用されている。それ故、純粋なCM101を得ることは、研究目的及び治療目的のために、重要である。
【0004】
CM101は、約300,000ダルトンの分子量を有し、かつ、N−アセチル−ガラクトサミン、N−アセチル−グルコサミン、グルコース、ガラクトース、及びマンノース残基を含む複合多糖トキシンである。Nmr(核磁気共鳴)の結果は、アルジトール(alditol)残基も存在することができるということを示唆している。カルボン酸官能基、好ましくは、ガラクツロン酸も、上記分子の不可欠な部分であると信じられている。活性エピトープの繰り返しは、おそらく、標的内皮上のレセプターを架橋することにより、病理生理学的応答において重要な役害を演じているようである(Hellerqvist, C.G. et al., Early Results of a Phase I Trial of CM101 in Cancer Patients., Proceedings of the American Association of Cancer Research Annual Meeting (1995); DeVore, R.F., et al., A Phase IStudy of the Antineovascularization Drug CM101, J.Clin.Can.Res. , 3: 365-372 (1997))。
【0005】
米国特許第5,010,062号は、GBSトキシンの精製方法を提供する。しかしながら、教示された方法は、労働集約的であり、連続的なレベルの生物学的活性の損失を伴う多くの工程を必要とする。
【0006】
本分野において現在知られているCM101の精製は、化学的分析及び生物学的アッセイにより計測されるときほんの40%の純度である末端材料を提供する。他の60%は、植物と酵母の多糖類並びに内因性のバクテリアの多糖類を含む。この植物と酵母の汚染物質は、大部分、GBSバクテリアの最適な成長のために使用される商業的な培養基に対する添加物に起因する。上記内因的な汚染物質は、群及びタイプ(group and type)特異的な抗原を含むGBS多糖類を含む(Paoletti, L.C. et al., Neonatol mouse protection against infection with multiple group B streptococcal (GBS) serotypes by maternal immunization with a tetravalent GBS polysaccharide-tetanus toxid conjugate vaccine, Infect.Immun. 62(8):3236-43(1994);Michon, F., Multiantennary group-specific polysaccharide of Group B Streptococcus, Biochem., 27: 5341-51(1988))。この40%純度のCM101は、現在の臨床的なグレードを表している。それ故、高められた全体純度をもち、好ましくは、外因性の植物と酵母の多糖類及びGBS多糖類をもつ末端製品をもたらすCM101の精製方法についての必要性が存在する。
【0007】
さらに、本分野において知られている精製スキームは、環境的に有害な工程、例えば、フェノール:水抽出における多量のフェノールの使用を含む、フェノールは、周知の焼灼(caustic)材料である。
【0008】
それ故、本発明の目的は、(i)高純度材料を、(ii)最小数の工程を使用して、(iii)焼灼性又は毒性材料、例えば、フェノールの使用を最小化して、そして(iv)上記材料の収率を増加させて、もたらす精製方法を提供することである。
【発明の概要】
【0009】
本発明の要約
上記目的は、本明細書中に記載する発明により達成された。特に、GBSバクテリア培養基からCM101を精製するための疎水性相互作用クロマトグラフィー(HIC)を含む精製スキームは、95%を上廻る純度の製品をもたらす。
【0010】
本発明の1の側面は、GBSバクテリアからの多糖類トキシンの精製方法であってHIC樹脂の使用を含む方法である。本発明は、本明細書中に開示する方法により製造されたGBSバクテリアからの実質的に純粋な多糖類トキシン、及び実質的に純粋なトキシンと医薬として許容される担体を含む医薬組成物をも含む。本医薬組成物は、医学的症状をもつ患者を治療するために使用されることができる。例えば、腫瘍患者は、本発明に係る組成物により治療されることができる。
【図面の簡単な説明】
【0011】
【図1】図1は、本発明のCM101精製スキームを示す。
【図2】図2は、知られたCM101精製スキームを示す。
【図3a−b】一定内部標準として6−デオキシ・グルコースを含む、5,20、及び50μgのデキストラン(グルコース・ポリマー)についてのPAD検出器の投与量応答を示す、定量的加水分解標準曲線である。
【図3c】一定内部標準として6−デオキシ・グルコースを含む、5,20、及び50μgのデキストラン(グルコース・ポリマー)についてのPAD検出器の投与量応答を示す、定量的加水分解標準曲線である。
【図4】図4は、標準糖サンプルの分離を示す。
【図5a−b】図5a−bは、ブチル−セファロースHICカラム上での媒体濃縮物の溶出プロフィールである。図5aは、UV 206吸収において計測される。図5bは、UV 280吸収において計測される。
【図6a】図6aは、Millennium 2000 Diodo-Ray検出器(Waters, Millford, MA)上でのUV 203吸収においてモニターされ、そしてCM101(16分ピーク)を含むHIC−精製水−溶出画分のHPLCプロフィールである。
【図6b】図6bは、CM101含有(16分)ピークについての、260吸収(RNAとDNA)及び280吸収(チロシン含有タンパク質)の最小の存在を示し、そして図6aに対応するDiodo-Rayスペクトルである。
【図7a】図7aは、フェノール/生理食塩水抽出及びその後のDEAEクロマトグラフィーにさらに供された、図6aのHIC水−溶出ピークの純度を示す、203nmにおいてモニターされた溶出プロフィールである。
【図7b】図7bは、狭い対称ピークにより、及び260nm(RNA/DNA)と280nm(タンパク質)における吸収の欠如により、証明される、図7aのCM101−含有ピークの純度を示すDiode-Rayスペクトルである。
【図8】図8は、HICカラムから得られた画分のANA-1アッセイによるIL-6活性のプロフィール、より特に、100mlブチル・セファロース上を流れた10K5P6濃縮物から得られた画分のIL-6活性プロフィールである(FT=flow-through;1M=1Mホスフェート画分;0.25M=0.25Mホスフェート画分;H2O=水画分;EtoH=エタノール画分)。
【図9】図9は、本発明に係る方法により精製されたCM101の糖分析を示す。
【図10】図10は、HICクロマトグラフィーにさらに供された最近の臨床グレードのCM101のHPLCプロフィールである。
【図11】図11は、HIC及びHPLCによりさらに精製された最近の臨床グレードのCM101のサンプルの糖分析を示す。
【図12a−b】図12aは、10mMホスフェート・バッファー、pH8.4を使用した知られた方法により精製されたCM101のHPLCプロフィールである。図12bは、図12aのHPLCプロフィールと同じ操作条件を使用して、本発明に係る方法により精製されたCM101のHPLCプロフィールである。
【発明を実施するための形態】
【0012】
特定の態様の説明
本明細書中に使用するとき、GBSトキシンは、天然又は溶解されたGBSバクテリアから単離され、又は溶解され、そして/又はオートクレーブにかけられたGBSバクテリアの培地上清から得られたいずれかの画分又は成分として定義され、そしてこれは、ヒツジのアッセイにおいて呼吸器苦痛の誘発(Hellerqvist, C.G. et al., Studies on group B β-hemolytic streptococcus I.Isolation and partial characterization of an extra-cellular toxin., Pediatr.Res., 12: 892-898 (1981))又は腫瘍組織試料のペルオキシダーゼ−アンチペルオキシダーゼ(peroxidase-antiperoxidase(PAP))アッセイにより証明されるような新生脈管構造への結合及び補体の活性化(Hellerqvist, C.G. et al., Anti-tumor effects of GBS toxin: a polysaccharide exotoxin from group B β-hemolytic streptococcus J.Canc Res.Clin.Oncol., 120: 63-70 (1993); 及びHellerqvist, C.G. et al., Early Results of a Phase I Trial of CM101 in Cancer Patients., Proceedings of the American Association of Cancer Research Annual Meeting (1995))により証明される生物学的活性を有する。
【0013】
実質的に純粋なGBSトキシンとは、GBSトキシンが40%を上廻る純度(例えば、少なくとも約40重量%の濃度で存在する)、好ましくは、少なくとも約60%の純度、より好ましくは、少なくとも約90%の純度、そして最も好ましくは少なくとも約95%の純度であるような調製物を意味する。
【0014】
本発明に係る方法における使用のためのGBS出発材料のための源は、最近感染し、又は新生児を感染させることができるB群β−溶血性ストレプトコッカス(Streptococcus)バクテリアの株を培養することにより得られることができる。このような株の単離物は、感染された乳児の血液から得られることができる。
【0015】
CM101の高い製造は、一般に、GBS最適成長及びCM101製造のための多糖類及びタンパク質の形態における高分子量材料を含有する、複合培地THBを用いた発酵を必要とする。
【0016】
発酵プロセスの間、バクテリアは、上記栄養から一定量のタンパク質、核酸、及びCM101以外の多糖類を製造する。上記発酵ブロス中のCM101の推定濃度は、0.1重量%未満である。
【0017】
本発明の精製方法は、知られた方法よりも効率的に内因性及び外因性汚染性タンパク質、核酸、及び多糖類のバルクを排除し、そして10〜50%の純度のCM101を含有する末端製品をもたらす、疎水性相互作用クロマトグラフィー(HIC)を使用する。GBS出発材料とHIC樹脂を接触させる唯一の工程において、これは、その出発材料からの100〜500倍の精製を表す。
【0018】
多糖類の精製のためのHIC樹脂の使用は驚ろくべきであり、そして新規である。なぜなら、HICカラムは、疎水性タンパク質の精製のためにデザインされており、そしてタンパク質及び脂質を含まない多糖類のために有用であるとは信じられていないからである。多糖類は、一般に、それらの多くのヒドロキシル基に因り親水性であると特徴付けられている。製造者により推奨され、そして当業者により使用される条件下でのHICカラムへの出発材料の適用は、それ故、タンパク質を保持し、そして多糖類が結合しないでカラムを通過することを許容する意図をもつであろう。
【0019】
驚ろくべき発見は、CM101が、高レベルの純度を達成するために本発明の精製スキームの使用を許容する疎水特性をもつということである。特に驚ろくべきことは、CM101がそれから単離されるところの上清中に存在するタンパク質及び多糖類の大部分よりも有意に大きな疎水特性をもつということである。上記タンパク質及び多糖類汚染物質の98%超が、HICカラムを通過する。HIC樹脂は一般にHIC−カラム中に使用されるけれども、この工程は、あるいは、いくつかの他のやり方で樹脂と出発材料とを接触させることにより行われることができる。例えば、GBS源及びその樹脂は、バッチ・プロセスで一緒に容器内に入れられ、そしてトキシン含有部分は、その後、例えば、遠心分離によりその樹脂から分離される。
【0020】
追加の精製工程は、従来の方法に比較して小容量(約1000倍減少)におけるフェノール/生理食塩水抽出、及びイオン交換カラムを含むことができる。これらの追加の精製工程は、95%超の純度をもつ末端製品に寄与する。
【0021】
HICは、それらの疎水性の性質に基づき、タンパク質、例えば膜タンパク質を分離するために使用される方法である。HIC樹脂は、この疎水性の基が、上記樹脂に接触している物質と相互作用しないように、支持体に一般に共有結合されている相互作用的疎水性基をもつ樹脂として定義される。疎水性基の例は、アルキル、アルコキシ、及びアリール基を含む。本発明に従って使用されるべき好ましいHIC樹脂は、2以上の炭素の脂肪族基、好ましくは2〜12炭素の範囲内のアルキル基、そしてより好ましくはノルマル又は分枝ブチル基が付着された支持体をもつ。フェニル基又は20炭素までのアルコキシ基も、好ましい相互作用性疎水性基である。相互作用性疎水性基は、好ましくは、セファロース(Sepharose)(Pharmacia)、アクリルアミド(Toso Haas, Montgomeryville, PA)、又はシリカにより支持される。HICカラムの使用のための標準的な手順に従えば、着目のタンパク質を含有する出発材料は、2Mまでの水性塩溶液中の上記カラムに適用され、そして次に結合したタンパク質は、溶出され、そして、展開(developing)バッファーのイオン強度を減少させることにより疎水性相互作用における減少を通じて、分離される。pH及び/又は温度における変更も、上記疎水性相互作用を変化させるために使用されることができる。
【0022】
B群ストレプトコッカス(Streptococcus)からのCM101精製は、GBSのバクテリア培養物を得ることを必要とする。バクテリア接種物は、2g/l以上のグルコース及びNa2HPO4を補うことにより修飾されたTodd Hewitt Broth (THB)中の対数増殖期後期までインキュベートされる。図1中に示すように、上記培養物はその後オートクレーブにかけられる。CM101は、オートクレーブ後、2〜15mg/lの濃度で、GBS発酵培養物の上清中に存在する。この培地は、約15g/lの他のバクテリア及び培地成分を含有する。従って、CM101は、上記上清中、上記成分の約0.01〜0.1%を構成する。オートクレーブ後、培地は濾過される。濾液は、好ましくは、10,000ダルトン(10kD)カット−オフ・フィルターを介して濃縮される。但し、50,000ダルトン(50kD)以下のカットオフをもつフィルターも使用されることができる。
【0023】
次に、CM101は、本発明に従って、図1中に示すように、リン酸カリウム又は他の塩、例えばリン酸ナトリウム、硫酸、塩化、又は酢酸ナトリウム又はカリウム中0.75〜2Mに調整された上清濃縮物又はその再構築されたアルコール沈殿物を、好ましくは同一モル濃度の同一塩中で平衡にされたHICカラムに、適用することにより、精製される。上記培地濃縮物、又は10kD〜50kDの出発材料が、アルコール沈殿及び再構築を伴わずに使用されるような方法が好ましい。なぜなら、培地濃縮物又は出発材料は、再構築されたアルコール沈殿物よりも高い収率のCM101を提供するからである。
【0024】
CM101含有出発材料は、HICカラムに適用され、そして0.75〜2Mにおける水性リン酸塩で洗浄される。0.75〜2M洗浄の後、このカラムを、さらに、0.5〜1M、そして次に0.25M塩で、好ましくはホスフェートで展開させる。好ましい態様においては、CM101は、10〜50%のCM101を含有する単一ピークとして水により上記カラムから溶出される。あるいは、水は、水中10%エタノール中の10mMホスフェート、pH6.8(バッファーA)により、その後水中20%エタノールにより、上記HICカラムから、CM101溶出物と置き替わる。CM101活性は、バッファーAと20%エタノール画分の両者中に回収される。バッファーAの使用は、HICカラムからCM101の全てを除去するために十分なものではない。従って、バッファーA洗浄の後に、追加の20%エタノール洗浄が続けられる。しかしながら、スケール−アップにおいては、エタノールは、環境ハザードを構成し、そして水ピーク又はバッファーA及び20%エタノール・ピーク画分のその後のフェノール/生理食塩水抽出は、凡そ等しい純度のCM101をもたらす。上記HIC手順は、10k濃縮物又は再構築されたアルコール沈殿物中に残存するタンパク質と培地多糖類の両者の98%以上を除去する。
【0025】
HICカラムからの濃縮されたCM101を、フェノール及び水性塩溶液、好ましくは0.05M生理食塩水中の抽出によりさらに精製されることができる。この追加の工程は、約95%の純度のCM101画分を提供する。
【0026】
HICカラムから溶出された、水又は、併合バッファーA及び20%エタノール画分は、水に対して透析され、そして凍結乾燥され、そして0.05M生理食塩水中で再構築されるか、又は濃縮後に生理食塩水に対して透析される。典型的には、フェノールが、上記材料に添加され、そしてその溶液は70〜80℃に急速加熱される。単一相が形成するとき、その溶液は4℃に冷却される。上記フェノール/生理食塩水抽出物の得られた生理食塩水相は、CM101を含み、そして次にカチオン交換カラム、例えば、DEAEに適用されることができる。
【0027】
DEAEカラム手順のために、DEAEカラムは、水中で平衡にされ、そして次に0.1M生理食塩水、0.05M NaOAc、pH7.4で洗浄され、そして0.34M NaClまでのステップ・グラジエントにより展開される。CM101の溶出はモニターされ、そしてANA-1アッセイを用いて定量される。HIC樹脂工程によるとき、イオン交換樹脂は、カラム以外の装置の使用を通じてトキシン含有材料と接触されることができる。次にCM101含有画分は、水に対して透析され、そして凍結乾燥される。フェノール/生理食塩水抽出及びイオン交換工程の後、CM101は95%を上廻る純度をもつ。
【0028】
上記樹脂接触工程から得られたカラム溶出物、又は材料は、ANA-1及び/又はドット・ブロット・アッセイを用いて生物学的活性についてアッセイされる。次に生物学的活性は、ヒツジ・アッセイを用いて確認される。表1は、出発材料の異なるバッチから得られ、そして上記HICカラムに適用されたAP及び10k材料のいくつかの分離を表す。変性タンパク質並びに培地多糖類及び他の材料の除去は同様である。
【0029】
好ましい精製の順序は、HIC工程、その後のフェノール/生理食塩水抽出、そしてその後のイオン交換工程を実施することであるけれども、精製は、他の順序において行われることもできる。
【0030】
図2は、CM101精製の知られた方法の1例である。特に、70%エタノール沈殿工程が使用され、その後、直ちに、フェノール/水抽出が行われる。この知られた精製方法の上記の初期段階において必要とされる多量のエタノール及びフェノールは、環境的に有害な実施を提供する。図2中に表された方法は、イオン交換カラム、ゲル濾過カラム、及びレンズ豆(lentil)レクチン・カラムをも要求する。
【0031】
従来の方法は、環境的に危険なものを含む、多くの工程を含む。一方において、本発明の方法は、有効であり、高純度及び2〜25倍の収率を与え、そして環境的に有害な材料の使用を最小化する。
【0032】
この環境的に危険なフェノール−水抽出工程は、従来使用されてきた手順に比較するとき、100倍減少される。さらに、例えば、ゲル透過手順による追加の精製が取り除かれる。従来方法のレンズ豆レクチン・クロマトグラフィー工程も取り除かれる。HICカラム、フェノール/生理食塩水抽出、及びイオン交換カラムの工程の末端製品は、約95%の純度をもち、それ故、他の処理は不要である。
【0033】
本発明の方法により精製されたCM101は、研究又治療目的をもって使用されることができる。本CM101は、医薬として許容される担体と併合されるとき、例えば、生理食塩水中で再構築され、そして患者に静脈内投与されるとき、特に有用である。精製されたCM101を投与するための他の剤形を使用することもできる。本発明の医薬組成物は、医薬として許容される担体との組合せにおいて本発明の実質的に純粋なGBSトキシンを含む。一般に、上記担体は、静脈内(IV)手段により投与されることができる組成物を形成するために、上記トキシンと容易に混合されるものであろう。従って、上記担体は、好ましくは、静脈内投与のためのその好適性を保証するために含有された他の医薬として許容される賦形剤をもつことができる、水である。得られた組成物は、滅菌され、そして許容される浸透圧特性をもつであろう。一般に、好適なIV配合物は、当業者に知られた標準的な技術に従って調製される。例えば、the Eighteenth Edition of Remington's Pharmaceutical Sciences, Mach Publishing Co. (1990) 中Salvatore J.Turco による“Intravenous Admixtures"と題する第85章(引用により本明細書中に取り込む)は、本発明に従って有用な、医薬として許容されるIV組成物を調製するための標準的な技術を提供する。
【0034】
さらに、CM101に有利に応答することが判明している医学的症状をもつ患者は、本発明の医薬組成物により治療されることができる。例えば、腫瘍をもつ患者は、本明細書中に教示される医薬組成物を静脈内投与することにより、有利に処置されることができる。米国特許第5,010,062号は、ヒトにおける特定の腫瘍の治療について討議しており、そしてそれを引用により本明細書中に取り込む。
【0035】
定量的及び定性的分析
HPLC分析
HICクロマトグラフィー後にサンプルから得られたCM101の純度及び量を、高圧液体クロマトグラフィー(HPLC)ゲル濾過分析により、確立する。ゲル濾過カラムは、典型的には、水中10%アセトニトリルにより平衡とされ、そして生物学的に活性なCM101が、それが含有された均質の狭いピークとして溶出される。あるいは、このカラムは、10mMホスフェート・バッファー、pH8.4中で展開され、それは、それがより含有されたピークをもたらす。酢酸アンモニウム(NH4OAc)バッファー、pH8.4は、10mMホスフェート・バッファーのさらなる代替物として使用されることができる。
【0036】
Hydragel 1000カラム(Waters, Millford, MA) 上での100μlの展開バッファー中にインジェクトされた、30,50、及び100μgの純粋なCM101標準を使用した典型的な検出器応答(UV 203吸収)は、それぞれ、26×106, 48×106、及び97×106の面積単位(area units)であり、それは、未知のサンプルの定量のための投与量応答曲線をもたらす。
【0037】
CM101の分子量も、ゲル濾過クロマトグラフィーにより計測されることができる。上記の非変性バッファー、例えばアセトニトリル、ホスフェート、又は酢酸アンモニウム・バッファーが、上記カラムを流すために使用される。CM101溶出は、異なる分子量をもつ標準デキストラン多糖類マーカーの溶出と比較される。CM101は、上記条件下、約300,000ダルトンの分子量をもつ。
【0038】
アミノ酸分析
定量的及び定性的な自動化されたアミノ酸分析は、標準的な商業的に入手可能な装置、例えば、Waters, Millford, MAから入手可能なPicotagを用いて、行われることができる。
【0039】
ANA-1 アッセイ
異なる発酵及び精製工程の生物学的活性をモニターするために、形質転換されたマクロファージ細胞系を使用するインビトロ・アッセイが使用されることができる。このアッセイは、CM101暴露に応答するマウス・マクロファージANA-1のIL-6生産を計測する。
【0040】
特に、CM101は、raf/myc形質転換ネズミ骨髄マクロファージ細胞系ANA-1が、IL-6生産によりインビトロにおいて応答することを誘発する。他のマクロファージ様細胞系及び新鮮末梢血液白血球も使用されることができる。
【0041】
ANA-1アッセイを実施するために、サンプルは、まず、(CM101活性の予想されたレベルに依存して)適当なレンジまで希釈され、そして4〜8つの濃縮物が1:4希釈においてテストされる。PBS中で再構築される臨床グレードのCM101を使用したCM101標準曲線が作製される。上記細胞が添加された後に2000ng/mlの最終濃度を与える4000ng/ml溶液を添加し、6つの連続した1:2希釈をもって、PBS中で調製する。例えば、2×106/mlの濃度における細胞を使用することができる。このアッセイの感度は、ANA-1細胞に200U/mlのネズミINF-γを添加することにより、高められる。最終培養物は、100U/ml IFN-γであった。
【0042】
培養物の入ったマイクロタイター・プレートは、一夜(16〜18時間)、37°、5% CO2/空気、加湿インキュベーター内に入れられなければならず、そしてその後、ELISA IL-6アッセイ(R.D.Systems, Minneapolis, MN)が続けられなければならない。特に、培養上清は、上記IL-6アッセイ・プレートに移され、そしてそのプレートは、IL-6アッセイが完結するまで、4℃で保たれる。
【0043】
ドット・ブロット・アッセイ
溶液又は生物学的液体中のCM101を定量的に検出するための他の迅速な手順は、段階的希釈においてポリビニリデン・ジフルオリド(PVDF)上でサンプルをブロットすることである。CM101の量は、CM101に対する蛍光タグ付マウス・モノクローナル抗体、又はCM101に対するマウス・モノクローナル抗体、その後の蛍光タグ付抗マウスIgGのいずれかを使用して、定量される。CM101抗原に対して向けられた抗体7A3は、この目的のために有用である。異なる画分中のCM101の定量は、連続的に希釈されたCM101標準の標準曲線に対する比較により確立される。
【0044】
ヒツジ肺動脈圧アッセイ
上記トキシンは、肺動脈圧の上昇及び肺血管透過性の上昇により顕示される、肺高血圧の上昇により、ヒツジ肺に影響を及ぼす。
ホスフェート・バッファー生理食塩水(PBS)中のCM101サンプルが、注入により子ヒツジに投与されることができ、そして肺動脈圧における変化は、15分間隔で記録される。圧力におけるこれらの変化は、CM101活性に相関する。(Hellerqvist.C.G. et al., Studies on group B β-hemolytic streptococcus I.Isolation and partial characterization of an extra-cellular toxin., Pediatr.Res., 15: 892-898 (1981))。
【0045】
糖分析
100μg量のサンプルを、5:70:25の比におけるトリフルオロ酢酸(TFA)、酢酸(HOAc)及び水の混合物中100℃において2時間加水分解する。この溶液を蒸発させ、そしてそのサンプルを、2:8の比におけるTFAと水の混合物中100℃で2時間さらに加水分解する。この方法は、サンプル中のグリコシド結合の全てを完全に加水分解する。アミノ糖上に元来存在するN−アセチル基も除去される。
【0046】
次にサンプルを、PAD(Pulsed Amperometric Detection)検出器を使用してDionex糖分析システム上で分析する。分離結果を図4に示す。
【0047】
上記サンプルの純度を、定量的及び定性的糖分析により確立する。この原理を図3a−cと図4に示す。HPLCにより定量された多糖類のサンプルに、加水分解され、かつ、分析された内部標準6−デオキシ−D−グルコースを補う。この章中に記載する方法は、テストされたレンジ内で線形投与量応答を与え、そして定性分析は、上記標準と未知の保持時間を比較することにより、達成される。
【実施例】
【0048】
実施例
実施例1:CM101のためのスケール・アップ精製
B群ストレプトコッカス(Streptococcus)血清型III単離物の通常在庫を、3,000ガロンの発酵槽と共に使用した。バクテリア培養物の25ml種母を、65リッターの発酵容量(liter working volume (lwv))をもつ80リッター容器のために使用し、これを次に、750lwv容器を接種するために使用し、そしてこれを次に、最終的に7500lwv(3,000ガロン)の発酵槽にもっていく。あるいは、この65lwvを、7500lwv発酵槽を直接的に接種するために使用することができる。
【0049】
上記培養を、オートクレーブすることにより対数増殖期後期に、終了させる。次に、バクテリアを、10,000×gにおける連続的遠心分離、その後の、0.45ミクロンのカセット濾過(Millipore Corporation, Bedford, MA)により除去する。
【0050】
次に、得られた培養上清を、10kD〜50kDのカット・オフ・カセット(Millipore)を使用したカセット濾過を使用して、500リッターまで、15倍濃縮する。次に、濃縮された材料、透析により、塩、好ましくは、リン酸ナトリウム、pH7.4(ローディング・バッファー)中2Mに調製する。
【0051】
次に、濃縮された上清を、BioPilotシステム(Pharmacia)を使用して60リッターのn−ブチル・セファロース・カラム(Pharmacia, Uppsala, Sweden)の使用を通して、疎水性相互作用クロマトグラフィーに供する。活性のflow-throughを伴わない、培質濃縮物中の生物学的CM101活性についてのn−ブチルSepharose樹脂の能力は、約80リッターの培地対1リッターの樹脂である。濃縮された上清が上記カラム上にロードされた後、このカラムを、上記ローディング・バッファー、その後、0.5〜1M、そして次に0.25Mホスフェート・バッファー、pH7.4で洗浄する。CM101含有画分を、約120リッター又は2カラム容量内の水で溶出し、そして10kD〜50kDの範囲内のカット−オフ・カセット内で2リッターに濃縮する。このカラムの溶出を、BioPilotにおいて事前に確立されたプログラムにより制御し、そして溶出物を、206及び280nmにおけるUV吸収、導電率、及びpHによりモニターする。
【0052】
CM101含有2リッター画分を、0.05M生理食塩水、pH7.0に対して透析し、そして次に75〜80℃の範囲に加熱し、そして0.2〜2リッターのフェノールを添加する。次に混合物を80℃に加熱し、そして5分間その温度で維持する。その後、この混合物を4℃に冷却する。この段階から得られた水相を、好ましくは、水中で平衡化されたDEAE Sephacel FFカラム(Pharmacia, Uppsala, Sweden)に適用する前に0.2容量のクロロホルムで2回抽出する。このカラムを、100mM生理食塩水、0.05M NaOAc、pH7.4で洗浄し、そして生理学的に活性な材料、CM101を、次に、NaClグラジエントで上記DEAEカラムから溶出する。この生物学的活性を、Il-6アッセイ及びHPLC分析により検出する。この手順を通して精製されたCM101の品質を、HPLC及び糖分析により並びにIl-6及びヒツジ・テストによる生物学的活性アッセイにより確立する。
【0053】
上記のスケール・アップされた精製スキームは、従来の手順において使用されたアルコール沈殿の、多量の、初期のフェノール−水抽出手順を回避するという利点を提供する。
【0054】
結 果
図5a−bは、2M K2HPO4、pH7.2中、ブチル−Sepharose HICカラム上の媒質濃縮物の溶出プロフィールを示す。さまざまなピークは、溶出グラジエントにおける、経時的、段階的な変化の結果である。図5aは、UV 206吸収において計測されたプロフィールを表し、これは、全有機材料についてのピーク画分を定量し、そして最後の狭いピーク(約383分)内のCM101を示す。図5bは、UV 280吸収において計測されたプロフィールを表し、これは、異る画分中のタンパク質の量を定量する。
【0055】
HICカラム段階を行うことにより、CM101は、上記カラムに結合するようにされるが、99.7%までのタンパク質及び98.5%までの中性及び電荷をもつ多糖類が、表1に示されるように、上記カラムを通過する。
【0056】
【表1】
【0057】
表1中、アルコール沈殿物(AP)としての異なる発酵ロット、AP1,AP2、及びAP6、及び10k濃縮物を、HICクロマトグラフィーに供し、そして水又はバッファーAのいずれかで溶出した。両プロセスは、外因性及び内因性タンパク質(UV 280)並びに多糖類及び一般有機物(UV 206)のほぼ同一の効率的な除去をもたらした。
【0058】
図6a−bは、CM101を含有し、そしてUV 203吸収をモニターされたHIC−精製水−溶出画分の、HPLCプロフィール、及びDiodo-Rayスペクトルを表す。これらの図は、CM101含有ピークについての206吸収(RNAとDNA)及び280吸収(タンパク質)を示す。
【0059】
HIC画分が、フェノール/生理食塩水抽出、及びイオン交換段階にさらに供された後、HIC水−溶出ピークの純度を、図7a−bに見られるように、さらに改良する。時刻0から約16分目における狭い対称ピーク、並びに260(RNA/DNA)及び280(タンパク質)における吸収の欠如に注目のこと。図6a−b及び7a−bにおいて示される溶出プロフィールについて、HPLCを、水中10%アセトニトリルを用いて行い、そしてその流速は、約0.3ml/分であった。
【0060】
これらの溶出プロフィール並びに生物学的活性は、アルコール沈殿物がHICカラムのための出発材料として使用されるときに得られるものと同様である。
【0061】
ANA-1細胞内でIL-6合成を誘発する10k出発材料からのHIC画分の能力を、図8中に示す。HICクロマトグラフィーは、ANA-1アッセイにより計測されるとき培地上清中の全生物学的活性のほぼ50%の回収をもたらした。CM101抗原の存在下で免疫反応性を示す同一材料のドット・プロット・アッセイを、ANA-1アッセイ結果を確認するために使用した。
【0062】
HICクロマトグラフィー後の10k濃縮物から得られた異なる画分を、生物学的活性についてのヒツジ・モデルにおいてもテストした。CM101活性の量を、最近の臨床的CM101を使用した投与量応答曲線に基づいて決定する(1ユニットの活性は7.5μg/kgに対応する)。結果を表2中に示す。ここで、アルコール沈殿物(AP)と媒質濃縮物(10k)のHIC画分が比較される。
【0063】
【表2】
【0064】
本発明の方法により精製されるときのCM101の生物学的活性は、ヒツジにおける肺動脈圧アッセイを用いても計測され、そして例えば米国特許第5,010,062号中に教示されたような、古いプロセスにより精製されたCM101の活性と比較された。本発明に従って精製された材料は、上記の古いプロセス、すなわち、HIC樹脂と接触されていないプロセスにより、精製された材料よりも2〜3倍高い比活性を示した。
【0065】
本発明の方法の製品収率も、先に証明される。なぜなら、先に示す7520μg/l値に比較して、既知の方法は、約300μgのCM101/発酵容量1リッターを提供するからである。
【0066】
本発明の方法により得られた図7a−b中に示す精製されたCM101は、糖分析にも供された。糖の収量を図9中に示す。
【0067】
定量的には、本発明の方法により得られたCM101は95%を上廻る純度の炭水化物であり、そして先に提示されたような定量及び定性により、そして自動アミノ酸分析(Pico Tag, Waters, Millford,MA)により、確立された5%未満のタンパク質を含む。
【0068】
実施例2:最近の臨床グレード及び新規組成物の比較
本発明の方法により得られたCM101は、最近の臨床グレードのCM101よりも改善されている。特に、図7aと比較するとき、図10のHPLC溶出特性は、本発明に従って製造されたサンプル中の、より高い純度を示す。図7aは、いくつかのピークの代わりに、1つの狭い、かつ、対称のメイン・ピークを示す。
【0069】
HICカラムの有利な使用をさらに証明するために、そしてCM101として知られたトキシンの精製のさらなる証拠を提供するために、最近の臨床グレードのCM101をHICカラムに供し、そしてHPLC精製及び糖分析を行った。図1中の結果を図9に比較することができる。糖分析は、定量的、かつ、定性的に同様の末端製品を示し、そして本HICクロマトグラフィーが、生物学的に活性なCM101に関連しない糖を除去することを証明している。この結果を表3中にも示す。
【0070】
【表3】
【0071】
先に表す糖残基の表は、凡そのモル比を与える。本発明の方法に従って精製されたCM101の実際の残基(第1列)は、(0.2〜1マンノース):(2.5〜3.5ガラクトース):(0.5〜1グルコース):(1N−アセチル・グルコサミン):(0.5〜1N−アセチル・ガラクトサミン)のレンジ内にある。上記の数を、N−アセチル・グルコサミンに対して正規化する:従って、N−アセチル・グルコサミンを1に設定する。
【0072】
さらなる比較のために、図12a−bは、古いプロセス(図12a)により、そして本発明の方法に従って(図12b)製造されたCM101のHPLCプロフィールを示す。ゲル濾過カラム(Ultragel 100, Waters, Milford, MA)を、10mMホスフェート・バッファー、pH8.4中で展開する。図12b中に示すように、本発明の方法に従って精製されたCM101は、約5分間のレンジにわたり比較的狭いピークにおいて溶出する。この溶出時間は、0.3ml/分の流速をもって時刻0から約24分目である。これに対して、古いプロセスにより精製された材料は、約12分間にわたりブロードなピークにおいて溶出する。
【0073】
実施例3:SDS-PAGE/ウェスタン・ブロットによる CM101の分析
ドデシル硫酸ナトリウム−ポリアクリルアミド・ゲル電気泳動を4〜20%のグラジエント・ゲルを使用して行った。2%SDS、0.5M Tris-HCl、5%グリセロール、0.05%ブロモフェノール・ブルー、及び5% β−メルカプトエタノール pH0.8のバッファー中のCM101サンプルを、10分間55℃においてインキュベートし、そして4%スタッキング・ゲル中で、4〜20%SDSランニング・ゲルに適用した。このゲルを、90分間200ボルトにおいて走らせた。
【0074】
このゲルを、ウェスタン・ブロッティング・バッファー(25mM Tris、 192mMグリシン及び20%メタノール)中で展開し、そして2時間100ボルトにおいてブロットした。このゲルを、1時間室温においてPBS中で5%無脂肪乳でブロックし、そして結合バッファー(ホスフェート・バッファー生理食塩水、2%子ウシ胎児血清、及び 0.5%TWIN-20洗剤(BBT))で2回洗浄し、次に10μg/mlの7A3(CM101に対するモノクローナル抗体)で1時間インキュベートした。
【0075】
このゲルを、BBTで10分間4回インキュベートし、そして45分間、アルカリ・ホスファターゼ拘合抗−マウス抗体と共にインキュベートし、BBTで4回洗浄し、そして50分間Pierce Single-Step AP発色剤で、発色させた。
【0076】
SDS-PAGE/ウェスタン・ブロット分析は、CM101が上記条件下で分析されるとき、26,000ダルトンの成分又はそれらの多数体をもつということを示唆した。
【0077】
従って、本発明の方法は、危険な工程の困難性を最小化し、そしてすばらしい純度を提供する改良された精製法を提供する。さらに、本明細書中に教示された方法により製造される製品は、最近入手可能なCM101よりも改善されている。
【0078】
本明細書中に言及した全ての刊行物及び特許出願を、あたかも、個々の刊行物又は特許出願が特別に、かつ、個々に、引用により取り込まれることを示されるのと同程度で、引用により本明細書中に取り込む。
【0079】
本発明をこれまで十分に記載してきたが、多くの変更及び修正が添付請求の範囲の本質又は範囲から逸脱せずに行われることができるということは、当業者にとって明らかであろう。
【特許請求の範囲】
【請求項1】
GBSバクテリアからの多糖類トキシンの精製方法であって、上記トキシンを含有する水性混合物をHIC樹脂と接触させることを含む、前記方法。
【請求項2】
前記HIC樹脂接触工程が、上記HIC樹脂からの上記トキシンの分離をさらに含む、請求項1に記載の方法。
【請求項3】
前記トキシンがCM101である、請求項1に記載の方法。
【請求項4】
前記HIC樹脂接触工程から得られる上記トキシンの純度が約100〜500倍に高められる、請求項1に記載の方法。
【請求項5】
前記HIC樹脂接触工程から得られるトキシンが40%を上廻る純度である、請求項1に記載の方法。
【請求項6】
前記HIC樹脂接触工程から得られるトキシンが少なくとも約60%の純度である、請求項5に記載の方法。
【請求項7】
前記HIC樹脂接触工程から得られるトキシンが少なくとも約90%純度である、請求項6に記載の方法。
【請求項8】
前記HIC樹脂接触工程から得られるトキシンが少なくとも約95%純度である、請求項1に記載の方法。
【請求項9】
前記HIC樹脂が、アルキル、アルコキシ、及びアリール基から成る群から選ばれた相互作用性基をもつ樹脂を含む、請求項1に記載の方法。
【請求項10】
前記相互作用性基が、2〜12の範囲内の炭素をもつアルキル基である、請求項9に記載の方法。
【請求項11】
前記相互作用性基がブチル基である、請求項10に記載の方法。
【請求項12】
前記相互作用性基がノルマル・ブチル基である、請求項11に記載の方法。
【請求項13】
前記相互作用性基がフェニル基である、請求項9に記載の方法。
【請求項14】
水性フェノール混合物により前記トキシンを抽出することをさらに含む、請求項1に記載の方法。
【請求項15】
前記トキシンをイオン交換樹脂と接触させることをさらに含む、請求項1に記載の方法。
【請求項16】
前記HIC樹脂接触工程の後に、水性フェノール混合物により前記トキシンを抽出し、そして前記トキシンをイオン交換樹脂と接触させることをさらに含む、請求項1に記載の方法。
【請求項17】
請求項1に記載の方法により精製されたGBSバクテリアからのトキシン。
【請求項18】
前記トキシンがCM101である、請求項17に記載のトキシン。
【請求項19】
前記トキシンが少なくとも約60%の純度である、請求項18に記載のトキシン。
【請求項20】
前記トキシンが少なくとも約90%の純度である、請求項19に記載のトキシン。
【請求項21】
前記トキシンが少なくとも約95%の純度である、請求項20に記載のトキシン。
【請求項22】
(0.2〜1マンノース):(2.5〜3.5ガラクトース):(0.5〜1グルコース):(1N−アセチル・グルコサミン):(0.5〜1N−アセチル・ガラクトサミン)のモル比で、マンノース、ガラクトース、グルコース、N−アセチル・グルコサミン、及びN−アセチル・ガラクトサミンの糖残基をさらに含む、請求項18に記載のトキシン。
【請求項23】
(1マンノース):(3ガラクトース):(1グルコース):(1N−アセチル・グルコサミン):(1N−アセチル・ガラクトサミン)のモル比で、マンノース、ガラクトース、グルコース、N−アセチル・グルコサミン、及びN−アセチル・ガラクトサミンが存在する、請求項22に記載のトキシン。
【請求項24】
治療方法における使用のための、請求項18に記載のトキシン。
【請求項25】
非変性条件におけるゲル濾過クロマトグラフィーにより計測されるとき約300,000ダルトンの分子量をもつ、請求項18に記載のトキシン。
【請求項26】
約0.3ml/分の流速で10%アセトニトリル・バッファー中でHPLCにより分析されるとき、時刻0から約16分目に203nmの吸収において計測される狭い対称ピークにおいて、前記トキシンが溶出されることを特徴とする、請求項18に記載のトキシン。
【請求項27】
10mMホスフェート・バッファー、pH8.4中HPLCにより分析されるとき、時刻0から約24分目に203nmの吸収において計測される狭い対称ピークにおいて、前記トキシンが溶出されることを特徴とする、請求項18に記載のトキシン。
【請求項28】
ヒツジにおける上昇した肺動脈圧についてのアッセイにより計測されるとき、HIC樹脂と接触していないGBSトキシンの比活性よりも約2〜3倍大きな比活性をもつ、請求項18に記載のトキシン。
【請求項29】
(1)GBSバクテリア・トキシンの源をHICカラムに適用し、
(2)上記HICカラムを溶出させて、前記トキシンを含むHIC溶出物を得て、
(3)フェノールと水性溶液の組合せ物を用いて上記HIC溶出物を抽出して、上記トキシンを含む水相を作り、
(4)上記水相をイオン交換カラムを適用し、そして
(5)上記イオン交換カラムを溶出させて、イオン交換溶出物を得る、
ことを含む、GBSバクテリアからの多糖類トキシンの精製方法であって、ここで、前記イオン交換溶出物が実質的に純粋なトキシンを含む、前記方法。
【請求項30】
前記の実質的に純粋なトキシンがCM101である、請求項29に記載の方法。
【請求項31】
前記の実質的に純粋なトキシンが少なくとも約60%の純度である、請求項30に記載の方法。
【請求項32】
前記の実質的に純粋なトキシンが少なくとも約95%の純度である、請求項31に記載の方法。
【請求項33】
前記の実質的に純粋なGBSトキシンから本質的に成る組成物。
【請求項34】
前記の実質的に純粋なGBSトキシンが CM101である、請求項33に記載の組成物。
【請求項35】
前記トキシンが少なくとも約60%の純度である、請求項34に記載の組成物。
【請求項36】
前記トキシンが少なくとも約90%の純度である、請求項35に記載の組成物。
【請求項37】
前記トキシンが少なくとも約95%の純度である、請求項36に記載の組成物。
【請求項38】
前記トキシンが、(0.2〜1マンノース):(2.5〜3.5ガラクトース):(0.5〜1グルコース):(1N−アセチル・グルコサミン):(0.5〜1N−アセチル・ガラクトサミン)のモル比で、マンノース、ガラクトース、グルコース、N−アセチル・グルコサミン、及びN−アセチル・ガラクトサミンの糖残基をさらに含む、請求項34に記載の組成物。
【請求項39】
(1マンノース):(3ガラクトース):(1グルコース):(1N−アセチル・グルコサミン):(1N−アセチル・ガラクトサミン)のモル比で、マンノース、ガラクトース、グルコース、N−アセチル・グルコサミン、及びN−アセチル・ガラクトサミンの糖残が存在する、請求項38に記載のトキシン。
【請求項40】
前記トキシンが、非変性条件下、ゲル濾過クロマトグラフィーにより計測されるとき約300,000ダルトンの分子量をもつ、請求項34に記載の組成物。
【請求項41】
約0.3ml/分の流速で10%アセトニトリル・バッファー中HPLCにより分析されるとき、時刻0から約16分目に203nm吸収において計測される狭い対称ピークにおいて、前記トキシンが溶出される、請求項34に記載の組成物。
【請求項42】
10mMホスフェート・バッファー、pH8.4中HPLCにより分析されるとき、時刻0から約24分目に203nm吸収において計測される狭い対称ピークにおいて、前記トキシンが溶出される、請求項34に記載の組成物。
【請求項43】
実質的に純粋なCM101と医薬として許容される担体を含んで成る医薬組成物。
【請求項44】
前記組成物が少なくとも約60%の純度である、請求項43に記載の医薬組成物。
【請求項45】
患者に請求項43に記載の医薬組成物を投与することを含む、医学的症状をもつ患者の治療方法。
【請求項46】
患者に請求項43に記載の医薬組成物を投与することを含む、腫瘍をもつ患者の治療方法。
【請求項1】
GBSバクテリアからの多糖類トキシンの精製方法であって、上記トキシンを含有する水性混合物をHIC樹脂と接触させることを含む、前記方法。
【請求項2】
前記HIC樹脂接触工程が、上記HIC樹脂からの上記トキシンの分離をさらに含む、請求項1に記載の方法。
【請求項3】
前記トキシンがCM101である、請求項1に記載の方法。
【請求項4】
前記HIC樹脂接触工程から得られる上記トキシンの純度が約100〜500倍に高められる、請求項1に記載の方法。
【請求項5】
前記HIC樹脂接触工程から得られるトキシンが40%を上廻る純度である、請求項1に記載の方法。
【請求項6】
前記HIC樹脂接触工程から得られるトキシンが少なくとも約60%の純度である、請求項5に記載の方法。
【請求項7】
前記HIC樹脂接触工程から得られるトキシンが少なくとも約90%純度である、請求項6に記載の方法。
【請求項8】
前記HIC樹脂接触工程から得られるトキシンが少なくとも約95%純度である、請求項1に記載の方法。
【請求項9】
前記HIC樹脂が、アルキル、アルコキシ、及びアリール基から成る群から選ばれた相互作用性基をもつ樹脂を含む、請求項1に記載の方法。
【請求項10】
前記相互作用性基が、2〜12の範囲内の炭素をもつアルキル基である、請求項9に記載の方法。
【請求項11】
前記相互作用性基がブチル基である、請求項10に記載の方法。
【請求項12】
前記相互作用性基がノルマル・ブチル基である、請求項11に記載の方法。
【請求項13】
前記相互作用性基がフェニル基である、請求項9に記載の方法。
【請求項14】
水性フェノール混合物により前記トキシンを抽出することをさらに含む、請求項1に記載の方法。
【請求項15】
前記トキシンをイオン交換樹脂と接触させることをさらに含む、請求項1に記載の方法。
【請求項16】
前記HIC樹脂接触工程の後に、水性フェノール混合物により前記トキシンを抽出し、そして前記トキシンをイオン交換樹脂と接触させることをさらに含む、請求項1に記載の方法。
【請求項17】
請求項1に記載の方法により精製されたGBSバクテリアからのトキシン。
【請求項18】
前記トキシンがCM101である、請求項17に記載のトキシン。
【請求項19】
前記トキシンが少なくとも約60%の純度である、請求項18に記載のトキシン。
【請求項20】
前記トキシンが少なくとも約90%の純度である、請求項19に記載のトキシン。
【請求項21】
前記トキシンが少なくとも約95%の純度である、請求項20に記載のトキシン。
【請求項22】
(0.2〜1マンノース):(2.5〜3.5ガラクトース):(0.5〜1グルコース):(1N−アセチル・グルコサミン):(0.5〜1N−アセチル・ガラクトサミン)のモル比で、マンノース、ガラクトース、グルコース、N−アセチル・グルコサミン、及びN−アセチル・ガラクトサミンの糖残基をさらに含む、請求項18に記載のトキシン。
【請求項23】
(1マンノース):(3ガラクトース):(1グルコース):(1N−アセチル・グルコサミン):(1N−アセチル・ガラクトサミン)のモル比で、マンノース、ガラクトース、グルコース、N−アセチル・グルコサミン、及びN−アセチル・ガラクトサミンが存在する、請求項22に記載のトキシン。
【請求項24】
治療方法における使用のための、請求項18に記載のトキシン。
【請求項25】
非変性条件におけるゲル濾過クロマトグラフィーにより計測されるとき約300,000ダルトンの分子量をもつ、請求項18に記載のトキシン。
【請求項26】
約0.3ml/分の流速で10%アセトニトリル・バッファー中でHPLCにより分析されるとき、時刻0から約16分目に203nmの吸収において計測される狭い対称ピークにおいて、前記トキシンが溶出されることを特徴とする、請求項18に記載のトキシン。
【請求項27】
10mMホスフェート・バッファー、pH8.4中HPLCにより分析されるとき、時刻0から約24分目に203nmの吸収において計測される狭い対称ピークにおいて、前記トキシンが溶出されることを特徴とする、請求項18に記載のトキシン。
【請求項28】
ヒツジにおける上昇した肺動脈圧についてのアッセイにより計測されるとき、HIC樹脂と接触していないGBSトキシンの比活性よりも約2〜3倍大きな比活性をもつ、請求項18に記載のトキシン。
【請求項29】
(1)GBSバクテリア・トキシンの源をHICカラムに適用し、
(2)上記HICカラムを溶出させて、前記トキシンを含むHIC溶出物を得て、
(3)フェノールと水性溶液の組合せ物を用いて上記HIC溶出物を抽出して、上記トキシンを含む水相を作り、
(4)上記水相をイオン交換カラムを適用し、そして
(5)上記イオン交換カラムを溶出させて、イオン交換溶出物を得る、
ことを含む、GBSバクテリアからの多糖類トキシンの精製方法であって、ここで、前記イオン交換溶出物が実質的に純粋なトキシンを含む、前記方法。
【請求項30】
前記の実質的に純粋なトキシンがCM101である、請求項29に記載の方法。
【請求項31】
前記の実質的に純粋なトキシンが少なくとも約60%の純度である、請求項30に記載の方法。
【請求項32】
前記の実質的に純粋なトキシンが少なくとも約95%の純度である、請求項31に記載の方法。
【請求項33】
前記の実質的に純粋なGBSトキシンから本質的に成る組成物。
【請求項34】
前記の実質的に純粋なGBSトキシンが CM101である、請求項33に記載の組成物。
【請求項35】
前記トキシンが少なくとも約60%の純度である、請求項34に記載の組成物。
【請求項36】
前記トキシンが少なくとも約90%の純度である、請求項35に記載の組成物。
【請求項37】
前記トキシンが少なくとも約95%の純度である、請求項36に記載の組成物。
【請求項38】
前記トキシンが、(0.2〜1マンノース):(2.5〜3.5ガラクトース):(0.5〜1グルコース):(1N−アセチル・グルコサミン):(0.5〜1N−アセチル・ガラクトサミン)のモル比で、マンノース、ガラクトース、グルコース、N−アセチル・グルコサミン、及びN−アセチル・ガラクトサミンの糖残基をさらに含む、請求項34に記載の組成物。
【請求項39】
(1マンノース):(3ガラクトース):(1グルコース):(1N−アセチル・グルコサミン):(1N−アセチル・ガラクトサミン)のモル比で、マンノース、ガラクトース、グルコース、N−アセチル・グルコサミン、及びN−アセチル・ガラクトサミンの糖残が存在する、請求項38に記載のトキシン。
【請求項40】
前記トキシンが、非変性条件下、ゲル濾過クロマトグラフィーにより計測されるとき約300,000ダルトンの分子量をもつ、請求項34に記載の組成物。
【請求項41】
約0.3ml/分の流速で10%アセトニトリル・バッファー中HPLCにより分析されるとき、時刻0から約16分目に203nm吸収において計測される狭い対称ピークにおいて、前記トキシンが溶出される、請求項34に記載の組成物。
【請求項42】
10mMホスフェート・バッファー、pH8.4中HPLCにより分析されるとき、時刻0から約24分目に203nm吸収において計測される狭い対称ピークにおいて、前記トキシンが溶出される、請求項34に記載の組成物。
【請求項43】
実質的に純粋なCM101と医薬として許容される担体を含んで成る医薬組成物。
【請求項44】
前記組成物が少なくとも約60%の純度である、請求項43に記載の医薬組成物。
【請求項45】
患者に請求項43に記載の医薬組成物を投与することを含む、医学的症状をもつ患者の治療方法。
【請求項46】
患者に請求項43に記載の医薬組成物を投与することを含む、腫瘍をもつ患者の治療方法。
【図1】

【図2】
【図3a−b】
【図3c】
【図4】
【図5a−b】
【図6a】
【図6b】
【図7a】
【図7b】
【図8】
【図9】
【図10】
【図11】
【図12a−b】
【図2】
【図3a−b】
【図3c】
【図4】
【図5a−b】
【図6a】
【図6b】
【図7a】
【図7b】
【図8】
【図9】
【図10】
【図11】
【図12a−b】
【公開番号】特開2009−173945(P2009−173945A)
【公開日】平成21年8月6日(2009.8.6)
【国際特許分類】
【出願番号】特願2009−106600(P2009−106600)
【出願日】平成21年4月24日(2009.4.24)
【分割の表示】特願平10−516772の分割
【原出願日】平成9年9月30日(1997.9.30)
【出願人】(500167881)バンダービルト ユニバーシティ (11)
【Fターム(参考)】
【公開日】平成21年8月6日(2009.8.6)
【国際特許分類】
【出願日】平成21年4月24日(2009.4.24)
【分割の表示】特願平10−516772の分割
【原出願日】平成9年9月30日(1997.9.30)
【出願人】(500167881)バンダービルト ユニバーシティ (11)
【Fターム(参考)】
[ Back to top ]