
GDF−8に対する中和抗体およびそれらの使用
【課題】筋肉および骨の変性障害の治療方法を提供すること。
【解決手段】成長分化因子8(GDF−8)に対する新規の抗体(特にヒト抗体)および抗体フラグメント(in vitroおよび/またはin vivoでGDF−8活性を阻害するものが含まれる)を開示する。筋肉もしくは骨の変性障害またはインスリン代謝障害の診断、防止、または治療方法も開示する。Myo29、Myo28、およびMyo22と呼ばれる新規のヒト抗GDF−8抗体およびこれ由来の抗体および抗原結合フラグメントも提供する。本発明の抗体は、多数の有用な特性を有する。
【解決手段】成長分化因子8(GDF−8)に対する新規の抗体(特にヒト抗体)および抗体フラグメント(in vitroおよび/またはin vivoでGDF−8活性を阻害するものが含まれる)を開示する。筋肉もしくは骨の変性障害またはインスリン代謝障害の診断、防止、または治療方法も開示する。Myo29、Myo28、およびMyo22と呼ばれる新規のヒト抗GDF−8抗体およびこれ由来の抗体および抗原結合フラグメントも提供する。本発明の抗体は、多数の有用な特性を有する。
【発明の詳細な説明】
【技術分野】
【0001】
本願は、2002年10月22日提出の米国特許仮出願番号60/419,964号(その全体が本明細書中で参考として援用される)の優先権を主張する。
【0002】
(技術分野)
技術分野は、成長分化因子8(GDF−8)に対する抗体(特にヒト抗体)および抗体フラグメント(詳細には、in vitroおよび/またはin vivoでGDF−8活性を阻害するもの)に関する。分野は、さらに、筋肉もしくは骨の変性障害またはインスリン代謝障害の診断、予防、または治療方法に関する。
【背景技術】
【0003】
(背景)
ミオスタチンとしても公知の成長分化因子8(GDF−8)は、分泌タンパク質であり、構造的に関連する成長因子の形質転換成長因子β(TGF−β)スーパーファミリー(その全てが生理学的に重要な成長調節特性および形態形成特性を有する)のメンバ−である(Kingsley et al.(1994)Genes Dev.,8:133−146;Hoodless et al.(1998)Curr.Topics Microbiol.Immunol.,228:235−272)。TGF−βと同様に、 ヒトGDF−8は、375アミノ酸長前駆体タンパク質として合成される。前駆体GDF−8タンパク質は、ホモ二量体を形成する。プロセシング時に、アミノ末端プロペプチドは、Arg−266で切断される。「潜伏期関連ペプチド(latency−associated peptide)」(LAP)として公知の切断されたプロペプチドは、ホモ二量体への非共有結合を保持し、それにより複合体を不活化することができる(Miyazono et al.(1988)J.Biol.Chem.,263:6407−6415;Wakefield et al.(1988)J.Biol.Chem.,263:7646−7654;Brown et al.(1990)Growth Factors,3:35−43;およびThies et al.(2001)Growth Factors,18:251−259)。成熟GDF−8とプロペプチドとの複合体は、一般に、「小潜在複合体(small−latent complex)」と呼ばれる(Gentry et al.(1990)Biochemistry,29:6851−6857;Derynck et al.(1995)Nature,316:701−705;およびMassague(1990)Ann.Rev.Cell Biol.,12:597−641)。成熟GDF−8に結合してその生物活性を阻害する他のタンパク質も公知である。このような阻害タンパク質には、フォリスタチンおよびフォリスタチン関連タンパク質が含まれる(Gamer et al.(1999)Dev.Biol.,208:222−232)。
【0004】
種々の種由来の推定アミノ酸配列のアラインメントにより、進化を通してGDF−8が高度に保存されていることが証明されている(McPherron et al.(1997)Proc.Nat.Acad.Sci.U.S.A.,94:12457−12461)。実際、ヒト、マウス、ラット、ブタ、およびニワトリのGDF−8配列はC末端領域が100%同一であり、ヒヒ、ウシ、およびヒツジでは3アミノ酸しか異ならない。ゼブラフィッシュGDF−8が最も異なるが、以前としてヒトと88%同一である。
【0005】
この保存の高さにより、GDF−8は不可欠な機能を有することが示唆される。GDF−8は、発達中および成体の骨格筋で高度に発現し、筋肉および骨形成における重要な生体プロセスの調節に関与することが見いだされた。たとえば、GDF−8ノックアウトトランスジェニックマウスは、骨格筋量の顕著な肥大および過形成(McPherron et al.(1997)Nature,387:83−90)および皮質骨構造の変化(Hamrick et al.(2000)Bone,27(3):343−349)によって特徴づけられる。骨格筋の類似の増大は、ウシのGDF−8の天然に存在する変異で明らかである(Ashmore et al.(1974)Growth,38:501−507;Swatland et al.(1994)J.Anim.Sci.,38:752−757;McPherron et al.(1997)Proc.Nat.Acad.Sci.U.S.A.,94:12457−12461;およびKambadur et al.(1997)Genome Res.,7:910−915)。研究により、HIV感染に関連する筋肉の消耗に伴ってGDF−8発現が増大することが示されている(Gonzalez−Cadavid et al.(1998)Proc.Nat.Acad.Sci.U.S.A.,95:14938−14943)。GDF−8はまた、筋肉特異的酵素(例えば、クレアチンキナーゼ)の産生および筋芽細胞の増殖(WO 00/43781)に関与する。その成長調節および形態形成特性に加えて、GDF−8は、多数の他の生理学的プロセス(2型糖尿病の発症におけるグルコースホメオスタシス、耐糖能障害、代謝症候群(例えば、X症候群)、外傷によるインスリン抵抗性(火傷または窒素不均衡など)、および脂肪組織障害(例えば、肥満症)が含まれる)にも関与すると考えられる(Kim et al.(2001)BBRC,281:902−906)。
【0006】
多数のヒトおよび動物の障害が、機能障害筋肉組織に関連する(例えば、筋ジストロフィー(デュシェンヌ型筋ジストロフィーが含まれる)、筋萎縮性側索硬化症(ALS)、筋萎縮症、組織萎縮症、脆弱性、鬱血性閉塞性肺疾患、サルコペニア、悪液質、ならびに他の疾患および病態に起因する筋消耗症候群)。今日まで、これらの障害を治療するための信頼がおける、又は有効な治療法はほとんどなかった。
【0007】
特に高齢者および/または閉経後の女性における骨量の低下に関連する病態(骨粗鬆症または変形性関節症が含まれる)も多数存在する。さらに、代謝性骨疾患および障害には、慢性糖質コルチコイド療法に起因する低骨量、若年性性腺不全、アンドロゲン抑制、ビタミンD欠損症、二次性副甲状腺機能亢進症、栄養失調、および拒食症が含まれる。これらの病態のための現在利用可能な治療法は、骨吸収の阻害によって作用する。これらの治療法に代わる新規の骨形成を促進する治療法が望ましい。
【0008】
したがって、特にヒトにおける筋肉量および/または強度および/または骨密度の全体的増加に寄与する新規の治療法を開発する必要がある。
【発明の概要】
【発明が解決しようとする課題】
【0009】
(要旨)
本発明の1つの目的は、筋肉および/または骨関連障害のための安全且つ有効な治療方法を提供することである。
【0010】
本発明の別の目的は、脊椎動物の筋肉量および/または骨の強度および/または密度を増大させる方法を提供することである。
【0011】
本発明のなおさらなる目的は、in vivoで安全且つ有効なGDF−8のインヒビターを提供することである。
【0012】
本発明のさらに別の目的は、高い特異性および親和性でGDF−8と結合するヒト抗体およびそのフラグメントを提供することである。
【課題を解決するための手段】
【0013】
したがって、筋肉および骨の変性障害の治療方法を提供する。方法はまた、正常な動物の筋肉量および骨密度の増加に有用である。Myo29、Myo28、およびMyo22と呼ばれる新規のヒト抗GDF−8抗体およびこれ由来の抗体および抗原結合フラグメントも提供する。本発明の抗体は、多数の有用な特性を有する。第1に、本抗体は、高親和性で成熟GDF−8と結合することができる。第2に、本開示の抗体は、証明するように、例えば、ActRIIB結合の阻害およびレポーター遺伝子アッセイによってin vitroおよびin vivoでGDF−8活性を阻害する。第3に、本開示の抗体は、骨格筋の量および骨密度の負の調節に関連するGDF−8活性を阻害することができる。
本発明は、例えば以下の項目を提供する。
(項目1)
実質的に配列番号n(nは2、4、6、8、10、12、14、16、18、20、22、24、26、28、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、または48である)に記載のアミノ酸配列を含む単離抗体において、前記抗体がGDF−8またはBMP−11と特異的に結合することができる、単離抗体。
(項目2)
配列番号n(nは、2、4、6、8、10、12、14、16、18、20、22、24、26、28、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、または48である)のアミノ酸配列を含む、項目1に記載の抗体。
(項目3)
前記抗体が、ATCC受託番号PTA−4741、PTA−4740、またはPTA−4739のE.coliによって発現するscFvフラグメントである、項目1に記載の抗体。
(項目4)
前記抗体が、配列番号54に記載のアミノ酸配列を含むタンパク質と特異的に結合することができる、項目1に記載の抗体。
(項目5)
互いに独立して少なくとも(a)配列番号54のN末端由来の第2のアミノ酸がメチオニンであるか、(b)N末端由来の第3のアミノ酸がセリンであるか、(c)N末端由来の第5のアミノ酸がイソロイシンである、項目4に記載の抗体。
(項目6)
前記抗体がヒトである、項目1に記載の抗体。
(項目7)
前記抗体がIgG1またはIgG4である、項目1に記載の抗体。
(項目8)
前記抗体のアミノ酸配列が、エフェクター機能を減少または変化させるように修飾された、項目1に記載の抗体。
(項目9)
前記アミノ酸配列が、配列番号53のアミノ酸117またはアミノ酸120に対応する残基で修飾された、項目8に記載の抗体。
(項目10)
前記抗体がIgG1λまたはIgG1κである、項目1に記載の抗体。
(項目11)
項目1に記載の抗体を含む、薬学的組成物。
(項目12)
有効用量の項目11に記載の薬学的組成物と投与する工程を含む、治療方法。
(項目13)
前記薬学的組成物を、筋肉障害、神経筋障害、および骨変性障害から選択される障害の治療または防止が必要な哺乳動物に投与する、項目12に記載の方法。
(項目14)
前記薬学的組成物を、筋ジストロフィー、デュシェンヌ型筋ジストロフィー、筋萎縮症、組織萎縮症、手根管症候群、鬱血性閉塞性肺疾患、サルコペニア、悪液質、筋消耗症候群、および筋萎縮性側索硬化症から選択される障害の治療または防止が必要な哺乳動物に投与する、項目12に記載の方法。
(項目15)
前記薬学的組成物をデュシェンヌ型筋ジストロフィーの治療または防止が必要な哺乳動物に投与する、項目12に記載の方法。
(項目16)
前記薬学的組成物を、肥満症および脂肪組織障害から選択される障害の治療または防止が必要な哺乳動物に投与する、項目12に記載の方法。
(項目17)
前記薬学的組成物を、X症候群、グルコース寛容減損、外傷誘導性インスリン抵抗性、および2型糖尿病から選択される障害の治療または防止が必要な哺乳動物に投与する、項目12に記載の方法。
(項目18)
前記薬学的組成物を、2型糖尿病の治療または防止が必要な哺乳動物に投与する、項目12に記載の方法。
(項目19)
前記薬学的組成物を、肥満症の治療または防止が必要な哺乳動物に投与する、項目12に記載の方法。
(項目20)
前記薬学的組成物を、損傷した筋肉の修復が必要な哺乳動物に投与する、項目12に記載の方法。
(項目21)
前記損傷した筋肉が心筋である、項目21に記載の方法。
(項目22)
前記損傷した筋肉が横隔膜である、項目21に記載の方法。
(項目23)
前記抗体を、1μg/kg〜150mg/kg、1μg/kg〜100mg/kg、1μg/kg〜50mg/kg、1μg/kg〜20mg/kg、1μg/kg〜10mg/kg、1μg/kg〜1mg/kg、10μg/kg〜1mg/kg、10μg/kg〜100μg/kg、100μg〜1mg/kg、および500μg/kg〜1mg/kgから選択される有効用量で投与する、項目12に記載の方法。
(項目24)
項目1に記載の抗体をコードする単離核酸。
(項目25)
項目24に記載の核酸を含む、発現ベクター。
(項目26)
項目25に記載のベクターを含む、宿主細胞。
(項目27)
前記宿主細胞が、ATCC受託番号PTA−4741、PTA−4740、またはPTA−4739のE.coliである、項目26に記載の宿主細胞。
(項目28)
前記核酸が、配列番号n(nは、1、3、5、7、9、11、13、15、17、19、21、23、25、27、または29である)のヌクレオチド配列を含む、項目24に記載の核酸。
(項目29)
GDF−8と特異的に反応する抗体の作製方法において、
(a)置換すべきCDR3を含むかCDR3コード領域を欠く可変ドメインをコードする核酸の出発レパートリーを得る工程と
(b)可変ドメインをコードする核酸の産物レパートリーを得るために前記レパートリー中のCDR3領域にドナ−核酸が挿入されるように前記レパートリーと実質的に配列番号n(nは31〜48の整数である)に記載のアミノ酸配列をコードするドナ−核酸とを組み合わせる工程と
(c)前記産物レパートリーの核酸を発現する工程と、
(d)GDF−8に特異的な特異的抗原結合フラグメントを選択する工程と、
(e)前記特異的抗原結合フラグメントまたは前記結合フラグメントをコードする核酸を回収する工程とを含む、方法。
(項目30)
項目29に記載の方法によって産生された抗体。
(項目31)
(a)項目1に記載の抗体およびGDF−8を含む第1の結合混合物を調製する工程と、
(b)該第1の混合物中の該抗体と該GDF−8との間の結合量を測定する工程と、
(c)抗体、GDF−8、試験化合物を含む第2の結合混合物を調製する工程と、
(d)該第2の混合物中の該抗体と該GDF−8との間の結合量を測定する工程とを含む、GDF−8のインヒビターの同定方法。
(項目32)
治療有効量の項目1に記載の抗体を哺乳動物に投与し、それにより筋肉の強度または量が増大する工程を含む、筋肉の強度または量を増大させる方法。
(項目33)
前記抗体が、GDF−8のActRIIBへの結合を阻害することができる、GDF−8に対する単離抗体。
(項目34)
実質的に配列番号n(nは、2、4、6、8、10、12、14、16、18、20、22、24、26、28、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、または48である)に記載のアミノ酸配列を含む、項目33に記載の抗体。
(項目35)
配列番号n(nは、2、4、6、8、10、12、14、16、18、20、22、24、26、28、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、または48である)に記載のアミノ酸配列を含む、項目33に記載の抗体。
(項目36)
治療有効量の項目33に記載の抗体を哺乳動物に投与し、それにより筋肉の強度が増大する工程を含む、筋肉の強度を増大させる方法。
(項目37)
前記抗体が、BMP−11と特異的に結合することができる、項目33に記載の抗体。
(項目38)
ATCC受託番号PTA−4741、PTA−4740、またはPTA−4739のE.coliを培養する工程と、抗体を回収する工程を含む、抗体の作製方法。
(項目39)
Myo29、Myo28、またはMyo22のsvFvをコードする核酸を免疫グロブリンのFc部分をコードする核酸と融合する工程と、細胞中で前記融合核酸を発現させる工程とをさらに含む、項目38に記載の方法。
(項目40)
生殖系列化(germlining)する工程を含む、項目39に記載の方法。
(項目41)
項目40に記載の方法を使用して作製された抗体。
(項目42)
配列番号54に記載のアミノ酸配列によって特徴づけられたエピトープと特異的に結合することができる抗体。
(項目43)
哺乳動物の筋肉、骨、またはグルコースホメオスタシスの少なくとも1つの障害の治療または防止のための薬物の調製のための項目1〜項目10、項目31、項目34、項目35、項目36、項目38、項目41、および項目42のいずれか1項に記載の抗体の使用。
(項目44)
前記哺乳動物がヒトである、項目43に記載の使用。
(項目45)
前記障害が神経筋障害である、項目43に記載の使用。
(項目46)
前記障害が、筋ジストロフィー、デュシェンヌ型筋ジストロフィー、筋萎縮症、組織萎縮症、手根管症候群、鬱血性閉塞性肺疾患、サルコペニア、悪液質、筋消耗症候群、または筋萎縮性側索硬化症である、項目43に記載の使用。
(項目47)
前記障害が肥満症または脂肪組織障害である、項目43に記載の使用。
(項目48)
前記障害がX症候群、グルコース寛容減損、外傷誘導性インスリン抵抗性、または2型糖尿病である、項目43に記載の使用。
(項目49)
(a)筋肉損傷の修復、(b)筋肉の量または強度の増大、および(c)哺乳動物におけるグルコース耐性の増大の少なくとも1つのための薬物の調製のための項目1〜項目10、項目31、項目34、項目35、項目36、項目38、項目41、および項目42のいずれか1項に記載の抗体の使用。
(項目50)
前記(a)の損傷した筋肉が、心筋または横隔膜である、項目49に記載の使用。
(項目51)
前記抗体を、1μg/kg〜150mg/kg、1μg/kg〜100mg/kg、1μg/kg〜50mg/kg、1μg/kg〜20mg/kg、1μg/kg〜10mg/kg、1μg/kg〜1mg/kg、10μg/kg〜1mg/kg、10μg/kg〜100μg/kg、100μg〜1mg/kg、および500μg/kg〜1mg/kgから選択される有効用量で投与する、項目46〜51のいずれか1項に記載の使用。
【0014】
本発明の一定の実施形態は、Myo29、Myo28、またはMyo22のFvフラグメントのVHおよび/またはVLドメインを含む。さらなる実施形態は、これらの任意のVHおよびVLドメインの1つまたは複数の相補性決定領域(CDR)を含む。他の実施形態は、Myo29、Myo28、またはMyo22のVHドメインのH3フラグメントを含む。
【0015】
他の態様は、本発明の抗体またはその抗原結合フラグメントを含む組成物およびGDF−8の阻害または中和方法(ヒトまたは動物の治療方法が含まれる)でのその使用を提供する。本発明の抗体を使用して、筋組織または骨密度の増加が望ましい病態を治療または予防することができる。例えば、本発明で開示の抗体を、損傷した筋肉(例えば、心筋、横隔膜など)を修復する治療法で使用することができる。疾患および障害の例には、筋ジストロフィー(デュシェンヌ型筋ジストロフィーが含まれる);筋萎縮性側索硬化症;筋萎縮症;組織萎縮症;脆弱性;管症候群;鬱血性閉塞性肺疾患;サルコペニア,悪液質、および他の筋肉消耗症候群などの筋障害および神経筋障害;脂肪組織障害(例えば、肥満症);2型糖尿病;耐糖能障害;代謝症候群(例えば、X症候群);外傷によるインスリン抵抗性(火傷または窒素不均衡など);および骨変性疾患(例えば、変形性関節症および骨粗鬆症)が含まれる。
【0016】
さらに、本発明で開示の抗体を、生体サンプル中のGDF−8またはそのフラグメントを定量的または定性的に検出するための診断ツ−ルとして使用することができる。GDF−8の存在または検出量は、上記に列挙した1つまたは複数の医学的症状に相関し得る。
【0017】
別の態様は、Myo29、Myo28、またはMyo22のFvフラグメント由来のVHまたはVLドメインをコードする配列を含む単離核酸を提供する。任意の本発明で開示のVHおよびVLドメイン由来の少なくとも1つのCDRをコードする配列を含む単離核酸も開示する。別の態様は、このような核酸を含む宿主細胞を提供する。
【0018】
さらに別の態様は、Myo29、Myo28、またはMyo22の新規のVHおよびVLドメインおよび/またはVHおよび/またはVLドメイン由来のドメインの全てまたは一部を含む機能的抗体の産生方法を提供する。
【0019】
本発明のさらなる目的を以下の説明に一部記載し、且つこれらの一部は説明から明らかであるか、本発明の実施によって得ることができる。本発明の種々の目的、態様、および利点は、添付の特許請求の範囲に特に指摘されている要素および組み合わせによって実現および達成される。
上記の一般的説明および以下の詳細な説明の両方は例示および説明のみを目的とし、特許請求の範囲のように本発明を制限しないと理解すべきである。
【図面の簡単な説明】
【0020】
【図1】図1は、ビオチン化GDF−8およびBMP−11がそれぞれ15ng/mlおよび40ng/mlのED50でActRIIB受容体と結合することを示す。
【図2】図2は、本発明のscFvフラグメントによるGDF−8のActRIIB受容体への結合の阻害を示す。例示するように、Myo29、Myo28、およびMyo22のscFvのIC50は、それぞれ2.4nM、1.7nM、および60nMである。
【図3A】図3Aおよび3Bは、Myo29の10ng/mlのビオチン化GDF−8またはBMP−11とのプレインキュベーションにより、ActRIIB結合アッセイにおいて0.2〜0.4nMのIC50でGDF−8またはBMP−11のActRIIBへの結合を阻害することを示す。
【図3B】図3Aおよび3Bは、Myo29の10ng/mlのビオチン化GDF−8またはBMP−11とのプレインキュベーションにより、ActRIIB結合アッセイにおいて0.2〜0.4nMのIC50でGDF−8またはBMP−11のActRIIBへの結合を阻害することを示す。
【図4A】図4Bおよび4Cは、Myo29を試験したpGL3(CAGA)12受容体遺伝子アッセイの結果を示す。図4Aは、ベースライン条件(すなわち、GDF−8、BMP−11、およびアクチビンによるレポーター遺伝子活性の誘導)を証明する。図4Bおよび4Cは、Myo29により用量反応様式にて15〜30ng/mlのIC50でGDF−8活性を減少し、BMP−11の生物活性を同一の程度に阻害することを示す。図4Dは、このアッセイではMyo29がアクチビン活性に影響を与えないことを例示する。
【図4B】図4Bおよび4Cは、Myo29を試験したpGL3(CAGA)12受容体遺伝子アッセイの結果を示す。図4Aは、ベースライン条件(すなわち、GDF−8、BMP−11、およびアクチビンによるレポーター遺伝子活性の誘導)を証明する。図4Bおよび4Cは、Myo29により用量反応様式にて15〜30ng/mlのIC50でGDF−8活性を減少し、BMP−11の生物活性を同一の程度に阻害することを示す。図4Dは、このアッセイではMyo29がアクチビン活性に影響を与えないことを例示する。
【図4C】図4Bおよび4Cは、Myo29を試験したpGL3(CAGA)12受容体遺伝子アッセイの結果を示す。図4Aは、ベースライン条件(すなわち、GDF−8、BMP−11、およびアクチビンによるレポーター遺伝子活性の誘導)を証明する。図4Bおよび4Cは、Myo29により用量反応様式にて15〜30ng/mlのIC50でGDF−8活性を減少し、BMP−11の生物活性を同一の程度に阻害することを示す。図4Dは、このアッセイではMyo29がアクチビン活性に影響を与えないことを例示する。
【図4D】図4Bおよび4Cは、Myo29を試験したpGL3(CAGA)12受容体遺伝子アッセイの結果を示す。図4Aは、ベースライン条件(すなわち、GDF−8、BMP−11、およびアクチビンによるレポーター遺伝子活性の誘導)を証明する。図4Bおよび4Cは、Myo29により用量反応様式にて15〜30ng/mlのIC50でGDF−8活性を減少し、BMP−11の生物活性を同一の程度に阻害することを示す。図4Dは、このアッセイではMyo29がアクチビン活性に影響を与えないことを例示する。
【図5】図5は、Myo22、Myo28、およびMyo29のエピトープマッピングの結果を示す。Myo29のエピトープを、成熟GDF−8のアミノ酸72からアミノ酸88までをマッピングし、Myo22については、アミノ酸1からアミノ酸44までをマッピングし、Myo28については、アミノ酸1からアミノ酸98までをマッピングした。
【図6】図6は、Myo29エピトープの置換分析の結果を示す。成熟GDF−8中の残基Lys−78、Pro−81、およびAsn−83は、Myo29のGDF−8への結合に重要なようである。
【図7】図7は、Myo29およびMyo28を使用して実施した免疫沈降実験の結果を示す。35S−メチオニン/システインで放射性標識したGDF−8を発現するCHO細胞由来の馴化培地を、Myo29またはMyo28での免疫沈降に供した。次いで、還元条件下でのSDS−PAGEによって免疫沈降物を分析した。ゲル上のバンドを、成熟GDF−8、GDF−8プロペプチド、および非プロセシングGDF−8と同定した。
【図8】図8は、Myo29の単回静脈内(IV)または腹腔内(IP)投与として1mg/kgの用量をC57B6/SCIDマウスに投与する薬物動態学研究の結果を示す。Myo29は、約1週間の長期末端半減期および約1ml/時間/kgの低クリアランスを示す。IP注射後に吸収された画分は約77%である。
【図9】図9は、種々の用量のMyo29(60、10、および1mg/kg)または賦形剤(PBS)で毎週処置した雄C57B6/SCIDマウスにおける大腿四頭筋量の比較を示す。10および60mg/kgの用量レベルで4週間のMyo29での処置により、筋肉量がそれぞれ19%および23%統計的に有意に増加する。
【図10A】図10Aおよび10Bは、種々の用量のMyo29(10、5、2.5、および1mg/kg)またはPBSで4週間毎週処置した雌CB17 SCIDマウスにおける腓腹筋量および大腿四頭筋量を示す。筋肉量は、賦形剤コントロールと比較してMyo29で処置したマウスで10%〜20%増加する。
【図10B】図10Aおよび10Bは、種々の用量のMyo29(10、5、2.5、および1mg/kg)またはPBSで4週間毎週処置した雌CB17 SCIDマウスにおける腓腹筋量および大腿四頭筋量を示す。筋肉量は、賦形剤コントロールと比較してMyo29で処置したマウスで10%〜20%増加する。
【図11A】図11Aおよび11Bは、種々の用量のMyo29(10、5、2.5、および1mg/kg)またはPBSで12週間毎週処置した雌CB17 SCIDマウスにおける腓腹筋量および大腿四頭筋量をそれぞれ示す。Myo29で処置したマウスの筋肉量は12%〜28%の範囲で増加する。
【図11B】図11Aおよび11Bは、種々の用量のMyo29(10、5、2.5、および1mg/kg)またはPBSで12週間毎週処置した雌CB17 SCIDマウスにおける腓腹筋量および大腿四頭筋量をそれぞれ示す。Myo29で処置したマウスの筋肉量は12%〜28%の範囲で増加する。
【図12】図12は、Myo29(10および5mg/kg)またはPBSで12週間毎週処置した雌CB17 SCIDマウスにおける握力計によって測定した前肢の筋肉の強度を示す。前肢強度は、5mg/kgおよび10mg/kgのMyo29で処置したマウスでそれぞれ17%および23%増加する。
【発明を実施するための形態】
【0021】
(詳細な説明)
(1.定義)
本明細書中で使用される、用語「抗体」は、免疫グロブリンまたはその一部をいい、その供給源、起源となる種、産生方法、および特徴と無関係に抗原結合部位を含む任意のポリペプチドを含む。非限定的な例として、用語「抗体」には、ヒト、オランウータン、マウス、ラット、ヤギ、ヒツジ、およびニワトリの抗体が含まれる。この用語には、ポリクローナル抗体、モノクローナル抗体、単一特異性抗体、多重特異性抗体、非特異性抗体、ヒト化抗体、単鎖抗体、キメラ抗体、合成抗体、組換え抗体、ハイブリッド抗体、変異抗体、およびCDRグラフティング抗体が含まれるが、これらに限定されない。本発明の目的のために、他で記載しない限り、Fab、F(ab’)2、Fv、scFv、Fd、dAbなどの抗体フラグメントおよび抗原結合機能を保持した他の抗体フラグメントも含まれる。
【0022】
抗体を、例えば、伝統的なハイブリドーマ技術(Kohler and Milstein(1975)Nature,256:495−499)、組換えDNA法(米国特許第4,816,567号)、または抗体ライブラリーを使用したファージディスプレイ技術(Clackson et al.(1991)Nature,352:624−628;Marks et al.(1991)J.Mol.Biol.,222:581−597)によって作製することができる。種々の他の抗体産生技術については、Antibodies:A Laboratory Manual,eds.Harlow et
al.,Cold Spring Harbor Laboratory,1988を参照のこと。
【0023】
用語「抗原結合ドメイン」は、抗原の一部または全部と特異的に結合するか相補的である領域を含む抗体分子の一部をいう。抗原が巨大な場合、抗体は抗原の特定の部分のみと結合することができる。「エピトープ」または「抗原決定基」は、抗体の抗原結合ドメインとの特異的相互作用を担う抗原分子の一部である。1つまたは複数の抗体可変ドメイン(例えば、いわゆるVHドメインからなるFd抗体フラグメント)によって抗原結合ドメインを得ることができる。抗原結合ドメインは、抗体軽鎖可変領域(VL)および抗体重鎖可変領域(VH)を含む。
【0024】
用語「レパートリー」は、発現した免疫グロブリンをコードする配列の全体または一部に由来するヌクレオチド(例えば、DNA)配列の遺伝的に多様な集団をいう。例えば、H鎖についてはV、D、およびJセグメント、L鎖についてはVおよびJセグメントのin vivo再編成によって配列を作製する。あるいは、in vitro刺激および再編成が起こる応答によって細胞株から配列を作製することができる。あるいは、例えば、非再編成VセグメントとDおよびJセグメントとの組み合わせ、ヌクレオチド合成、無作為化変異誘発、および米国特許第5,565,332号に開示の他の方法によって配列の一部または全部を得ることができる。
【0025】
用語「特異的相互作用」または「特異的に結合する」などは、2つの分子が生理学的条件下で比較的安定な複合体を形成することを意味する。この用語はまた、例えば、抗原結合ドメインが多数の抗原によって保有される特定のエピトープに特異的である場合に適用可能であり、この場合、抗原結合ドメインを保有する抗体はエピトープを保有する種々の抗原に結合することができる。したがって、抗体は、例えば、その両方が保有するエピトープに結合する限り、BMP−11およびGDF−8と特異的に結合することができる。
【0026】
高親和性および低〜中程度の能力によって特異的結合を特徴づける。非特異的結合は、通常、親和性が低く、中程度から高い能力を有する。典型的には、親和性定数Kaが106M−1より高い、好ましくは108M−1より高い場合、結合は特異的と見なされる。必要に応じて、結合条件の変化によって実質的に特異的結合に影響を与えることなく非特異的結合を減少させることができる。このような条件は当該分野で公知であり、当業者は日常的技術を使用して適切な条件を選択することができる。条件は、通常、抗体濃度、溶液のイオン強度、温度、結合時間、非関連分子(例えば、血清アルブミン、ミルクカゼイン)の濃度などに関して定義される。条件の例を、実施例4、7、および10に示す。
【0027】
句「実質的に記載の」は、関連するCDR、VH、またはVLドメインが記載の配列の特定の領域と同一であるか類似性が高いことを意味する。例えば、このような置換には、CDR(H1、H2、H3、L1、L2、またはL3)の配列中の任意の5個のアミノ酸のうちの1個または2個が含まれる。
【0028】
用語「TGF−βスーパーファミリー」は、構造的に関連した成長因子のファミリ−をいう。関連する成長因子のこのファミリ−は、当該分野で周知である(Kingsley
et al.(1994)Genes Dev.,8:133−146;Hoodless et al.(1998)Curr.Topics Microbiol.Immunol.,228:235−72)。TGF−βスーパーファミリーには、骨形態形成タンパク質(BMP)、アクチビン、インヒビン、ミューラー阻害物質、グリア由来の神経栄養因子、以前として増加している成長分化因子(GDF)(GDF−8(ミオスタチン)など)が含まれる。多数のこのようなタンパク質は、構造的および/または機能的にGDF−8と関連する。例えば、GDF−11としても公知のヒトBMP−11は、アミノ酸レベルでGDF−8と90%同一である(Gamer et al.(1999)Dev.Biol.208,222−232;Nakshima et al.(1999)Mech.Dev.,80:185−189)。
【0029】
用語「GDF−8」は、特異的な成長分化因子−8をいい、必要に応じて、GDF−8と構造的または機能的に関連する因子(例えば、BMP−11およびTGF−βスーパーファミリーに属する他の因子)をいう。この用語は、GDF−8の全長非プロセシング前駆体形態ならびに翻訳後切断に起因する成熟およびプロペプチド形態をいう。この用語はまた、本明細書中で考察された成熟GDF−8に関連する少なくともいくつかの生物活性を保持するGDF−8の任意のフラグメントおよび変異型(修飾された配列が含まれる)をいう。成熟ヒトGDF−8のアミノ酸配列を、配列番号49に提供する。本発明は、全ての脊椎動物種(ヒト、ウシ、ニワトリ、マウス、ラット、ブタ、ヒツジ、シチメンチョウ、ヒヒ、および魚類が含まれるが、これらに限定されない)由来のGDF−8に関する(配列情報については、例えば、McPherron et al.(1997)Proc.Nat.Acad.Sci.U.S.A.,94:12457−12461を参照のこと)。
【0030】
用語「成熟GDF−8」は、GDF−8前駆体タンパク質のカルボキシル末端ドメインから切断したタンパク質をいう。成熟GDF−8は、単量体、ホモ二量体として存在するか、GDF−8潜在複合体中に存在し得る。条件によって、成熟GDF−8は、任意または全てのこれらの異なる形態の間で平衡になり得る。その生物活性形態では、成熟GDF−8は、「活性GDF−8」ともいう。
【0031】
用語「GDF−8プロペプチド」は、GDF−8前駆体タンパク質のアミノ末端ドメインから切断されたポリペプチドをいう。GDF−8プロペプチドは、成熟GDF−8上のプロペプチド結合ドメインと結合することができる。
【0032】
用語「GDF−8潜在複合体」は、成熟GDF−8ホモ二量体とGDF−8プロペプチドとの間で形成されたタンパク質複合体をいう。2つのGDF−8プロペプチドがホモ二量体中の成熟GDF−8の2分子と会合して不活性四量体複合体を形成すると考えられている。潜在複合体は、1つまたは複数のGDF−8プロペプチドの代わりまたはそれに加えて他のGDF−8インヒビターを含み得る。
【0033】
用語「GDF−8活性」は、活性GDF−8タンパク質に関連する1つまたは複数の生理学的成長調節活性または形態形成活性をいう。例えば、活性GDF−8は、骨格筋量の負のレギュレーターである。活性GDF−8はまた、筋肉特異的酵素(例えば、クレアチンキナーゼ)の産生を調整し、筋芽細胞増殖を刺激し、前脂肪細胞の脂肪細胞への分化を調整することができる。in vivoおよびin vitroでのGDF−8活性の測定手順の例を、実施例2、3、6、および13に記載する。
【0034】
用語「GDF−8インヒビター」には、GDF−8の活性、発現、プロセシング、または分泌を阻害することができる任意の薬剤が含まれる。このようなインヒビターには、タンパク質、抗体、ペプチド、ペプチド模倣物、リボザイム、アンチセンスオリゴヌクレオチド、二本鎖RNA、およびGDF−8を特異的に阻害する他の小分子が含まれる。このようなインヒビターは、GDF−8の生物活性を「阻害」、「中和」、または「減少」すると言われる。
【0035】
用語「中和する」、「中和」、「阻害」、およびその同族語は、GDF−8インヒビターの非存在下でのGDF−8活性と比較したGDF−8インヒビターによるGDF−8活性の減少をいう。活性の減少は、好ましくは、少なくとも約10%、20%、30%、40%、50%、60%、70%、80%、90%、またはそれ以上である。
【0036】
用語「治療」は、本明細書中の用語「治療方法」と交換可能に使用され、治療上の処置および予防(prophylactic)/予防(preventative)手段の両方をいう。治療を必要とする者には、特定の内科的疾患を既に罹患している個体および最終的に障害を獲得し得る個体(すなわち、予防手段を必要とする個体)が含まれ得る。
【0037】
用語「単離された」は、その天然の環境を実質的に含まない分子をいう。例えば、単離タンパク質は、そのタンパク質が由来する細胞もしくは組織供給源由来の細胞物質または他のタンパク質を実質的に含まない。この用語は、単離タンパク質が治療組成物としての投与のために十分に純粋であるか、純度が少なくとも70%〜80%(w/w)、より好ましくは少なくとも80%〜90%(w/w)、さらにより好ましくは純度が90〜95%、最も好ましくは純度が少なくとも95%、96%、97%、98%、99%、または100%(w/w)である調製物をいう。
【0038】
用語「哺乳動物」は、哺乳動物として分類された任意の動物(ヒト、家畜、動物園の動物、競技用動物、またはペット動物(イヌ、ウマ、ネコ、ヒツジ、ブタ、ウシなど)が含まれる)をいう。
【0039】
用語「有効用量」または「有効量」は、患者の症状を改善するか所望の生物学的結果(例えば、骨格筋量および/または骨密度の増加)が得られる化合物の量をいう。このような量は、骨格筋量および骨密度の負の制御に関連するGDF−8活性の減少に十分なはずである。以下の節に記載のように有効量を決定することができる。
【0040】
(II.GDF−8に対する抗体および抗原結合フラグメント)
A.ヒト抗体Myo28、Myo29、およびMyo22
本開示は、GDF−8に対する新規の抗体およびその抗原結合フラグメントを提供する。このような抗体の非限定的な実施形態を、Myo29、Myo28、およびMyo22と呼ぶ。これらの例示的実施形態を、ヒトIgG1抗体の形態で提供する。
【0041】
本発明の抗体は、固有且つ有益な特徴を有する。第1に、これらの抗体は、高親和性で成熟GDF−8と結合することができる。第2に、本発明の抗体は、例えば、ActRIIB結合の阻害およびレポーター遺伝子アッセイによって証明されるように、in vitroおよびin vivoでGDF−8活性を阻害することができる。本発明の抗体はまた、例えば、ActRIIB結合の阻害およびレポーター遺伝子アッセイによって証明されるように、BMP−11に特異的に結合し、そして/またはその活性を阻害することができる。第3に、本開示の抗体は、骨格筋量および骨密度の負の制御に関連するGDF−8活性を阻害することができる。
【0042】
例示的実施形態では、本発明に開示の抗体は、GDF−8およびBMP−11の両方に特異的に結合することができる。抗体は他のタンパク質(例えば、TGF−βスーパーファミリーに属するもの(ミューラー阻害物質、グリア由来の神経栄養因子など)またはGDF−8以外の成長分化因子)とも反応することができることが意図される。一定の実施形態では、Myo29は、配列番号49のアミノ酸72〜88と同一の配列を含むタンパク質と反応する。さらなる実施形態では、Myo29は、配列Lys−Xaa1−Xaa2−Pro−Xaa3−Asn(配列番号54)(式中、Xaa1,Xaa2,およびXaa3はそれぞれ任意のアミノ酸である)を含むタンパク質と結合する。さらなる実施形態では、少なくとも1つの以下の条件を満たす:(1)Xaa1 = Met、(2)Xaa2=Ser、および(3)Xaa3 = Ile(全て互いに独立している)。他の実施形態では、Myo22は、成熟GDF−8配列中の最初の44個のN末端アミノ酸(配列番号49のアミノ酸1〜44)内のエピトープを認識する。
【0043】
当業者は、本発明の抗体を使用して、上記と異なるタンパク質を検出、測定、および阻害することができると認識する。一般に、本発明の抗体を、配列番号49に記載のGDF−8の成熟形態の配列中の少なくとも100個、80個、60個、40個、または20個の連続するアミノ酸の任意の配列と少なくとも約70%、80%、90%、95%、またはそれ以上同一である配列を含む任意のタンパク質とともに使用することができる。このようなタンパク質の非限定的な例には、本明細書中に記載の種々の種由来のGDF−8配列が含まれる。標準的なアラインメントアルゴリズム(例えば、Altschul et
al.(1990)J.Mol.Biol.,215:403−410に記載のBasic Local Alignment Tool(BLAST)、Needleman
et al.(1970)J.Mol.Biol.,48:444−453のアルゴリズム、またはMeyers et al.(1988)Comput.Appl.Biosci.,4:11−17のアルゴリズムなど)によって同一率を決定する。
【0044】
B.可変ドメイン
免疫グロブリンとしても公知のインタクトな抗体は、典型的には、それぞれ約25kDaの2つの軽鎖(L)およびそれぞれ約50kDaの2つの重鎖(H)から構成される四量体のグリコシル化タンパク質である。λおよびκと呼ばれる2つの軽鎖型が抗体中で見いだされる。重鎖の定常ドメインのアミノ酸配列に依存して、免疫グロブリンを以下の5つの主なクラス(A、D、E、G、およびM)に割り当てることができ、これらのいくつかをサブクラス(イソ型)(例えば、IgG1、IgG2、IgG3、IgG4、IgA1、およびIgA2)にさらに分類することができる。免疫グロブリンの異なるクラスのサブユニット構造および三次元高次構造は当該分野で周知である。抗体構造の概説については、Antibodies:A Laboratory Manual,Cold Spring Harbor Laboratory,eds.Harlow et al.,1988を参照のこと。簡単に述べれば、各軽鎖は、N末端可変(V)ドメイン(VL)および定常(C)ドメイン(CL)から構成される。各重鎖は、N末端Vドメイン、3つまたは4つのCドメイン、およびヒンジ領域から構成される。VHドメインに最も近いCHドメインを、CH1と命名する。VHおよびVLドメインは、超可変配列(相補性決定領域、CDR)の3つの領域のための足場を形成するフレームワーク領域(FR1、FR2、FR3、およびFR4)と呼ばれる比較的保存された配列の4つの領域からなる。CDRは、抗原との特異的相互作用を担うほとんどの残基を含む。CDRを、CDR1、CDR2、およびCDR3という。したがって、重鎖上のCDR構成要素を、H1、H2、およびH3といい、軽鎖上のCDR構成要素を、L1、L2、L3という。CDR3は、抗体結合部位内の分子多様性の最も巨大な供給源である。例えば、H3は、2アミノ酸残基ほどの短さであり得るか、26個より長くあり得る。最も小さな抗原結合フラグメントは、VHおよびVLドメインからなるFvである。Fabフラグメント(Fragment antigen binding)は、定常領域間のジスルフィド結合によって共有結合したVH−CH1およびVL−CLドメインからなる。宿主細胞中で同時発現した場合にFv中の非共有結合したVHドメインとVLドメインとが解離する傾向を克服するために、可動性があり且つ適切な長さのポリペプチドによりVHのC末端がVLのN末端と結合するかVLのC末端がVHのN末端と結合するいわゆる単鎖(sc)Fvフラグメント(scFv)を構築することができる。最も一般的に使用されているリンカーは、15残基の(Gly4Ser)3ペプチドであったが、他のリンカーも当該分野で公知である。
【0045】
可変領域をコードする複数の生殖系列遺伝子および種々の体細胞事象の使用によって抗体が多様化する。体細胞事象には、完全なVH領域を作製するための多様性(D)遺伝子セグメントおよび連結(J)遺伝子セグメントでの可変遺伝子セグメントの組換えならびに完全なVL領域を作製するための可変遺伝子セグメントおよび連結遺伝子セグメントの組換えが含まれる。組換えプロセス自体の不正確さにより、V(D)J連結点でアミノ酸が喪失または付加される。これらの多様性の機構は、抗原曝露前の発達中のB細胞で起こる。抗原刺激後、B細胞中の発現抗体遺伝子は体細胞変異を受ける。予測生殖系列遺伝子セグメント数、これらのセグメントのランダム組換え、およびランダムVH−VL対合に基づいて、1.6×107個までの異なる抗体を産生することができる(Fundamental Immunology,3rd ed.,ed.Paul,Raven Press,New York,NY,1993)。抗体多様性に寄与する他のプロセス(体細胞変異など)を考慮した場合、1×1010個以上の異なる抗体を作製することができると考えられる(Immunoglobulin Genes,2nd ed.,eds.Jonio et al.,Academic Press,San Diego,CA,1995)。抗体多様性の作製に関与する多数のプロセスにより、同一の抗原特異性を有する独立して誘導されたモノクローナル抗体が同一のアミノ酸配列を有する可能性は低い。
【0046】
したがって、本発明は、さらに、ヒト免疫グロブリン遺伝子ライブラリー由来の新規のCDRを提供する。本発明のCDRを保有するための構造は、一般に、天然に存在するVHおよびVLのCDRに対応する位置にCDRが存在する抗体重鎖配列もしくは軽鎖配列またはその実質的部分である。免疫グロブリン可変ドメインの構造および位置を、Sequences of Proteins of Immunological Interest,US Department of Health and Human Services,eds.Kabat et al.,1991に記載のように決定することができる。
【0047】
本発明に開示された抗体のDNAおよびアミノ酸(AA)配列、そのscFvフラグメント、VHおよびVLドメイン、ならびにCDRを配列表に記載し、表1に列挙する。便宜上、VHおよびVLドメイン内の各CDRの位置を表2に列挙する。VHおよびVLドメインを除く重鎖および軽鎖の配列は、Myo29、Myo28、およびMyo22で同一である。
【0048】
【表1】

【0049】
【表2】
本発明で開示された抗体は、抗体定常領域またはその一部をさらに含み得る。例えば、VLドメインのそのC末端を、抗体軽鎖定常ドメイン(ヒトCκ鎖またはCλ鎖が含まれ、好ましくはCλ鎖)に結合させることができる。同様に、VHドメインに基づいた特異的抗原結合フラグメントのそのC末端に、任意の抗体イソ型(例えば、IgG、IgA、IgE、およびIgM)および任意のイソ型サブクラス(特に、IgG1およびIgG4)由来の免疫グロブリン重鎖の全部または一部を結合させることができる。例示的実施形態では、抗体は、ヒトIgG1λのC末端フラグメントの重鎖および軽鎖を含む。軽鎖λのC末端フラグメントのDNA配列およびアミノ酸配列を、それぞれ配列番号50および配列番号51に記載する。IgG1重鎖のC末端フラグメントのDNA配列およびアミノ酸配列を、それぞれ配列番号52および配列番号53に記載する。
【0050】
本発明の一定の実施形態は、Myo29、Myo28、またはMyo22のFvフラグメントのVHおよび/またはVLドメインを含む。さらなる実施形態は、任意のこれらのVHおよびVLドメインの1つまたは複数の相補性決定領域(CDR)を含む。1つの実施形態は、Myo29、Myo28、またはMyo22のVHドメインのH3フラグメントを含む。一定の実施形態では、本発明のVHおよびVLドメインは、生殖系列化されている(すなわち、これらのドメインのフレームワーク領域(FR)が、ヒト生殖系列遺伝子産物のコンセンサスアミノ酸配列に適合するように従来の分子生物学技術を使用して変化している)。他の実施形態では、フレームワーク配列は、生殖系列と異なったままである。
【0051】
C.修飾抗体およびそのフラグメント
本発明のさらなる態様は、GDF−8に特異的な抗体抗原結合ドメインを得る方法を提供する。当業者は、本発明の抗体が表1に記載のVHおよびVLの特異的配列に限定されず、抗原結合能力を保持するこれらの配列の変異型も含まれることを認識する。このような変異型は、当該分野で公知の技術を使用して提供した配列に由来し得る。FRまたはCDRのいずれかのアミノ酸を、置換、欠失、または付加することができる。通常、抗体の安定性を改良して免疫原性を減少させるようにフレームワーク領域の変化をデザインする一方で、通常、抗体のその標的に対する親和性を増大させるようにCDRの変化をデザインする。このような親和性が増大する変化を、典型的に、CDR領域の変更および抗体の試験によって経験的に決定する。このような変更を、Antibody Engineering,2nd.ed.,ed.Borrebaeck,Oxford University Press,1995に記載の方法にしたがって行うことができる。
【0052】
本明細書中に記載のVHドメインのアミノ酸配列変異型であるVHドメインの作製方法は、本明細書中に開示のVHドメインのアミノ酸配列中の1つまたは複数のアミノ酸を付加、欠失、置換、または挿入する工程と、任意選択的にこのようにして得られたVHドメインと1つまたは複数のVLドメインと組み合わせる工程と、GDF−8への特異的結合についてVHドメインまたはVH/VL組み合わせを試験する工程と、任意選択的にこのような抗原結合ドメインがGDF−8活性を中和する能力を試験する工程とを含む。VLドメインは、実質的に本明細書中に記載のアミノ酸配列を有し得る。
【0053】
本明細書中に開示のVLドメインの1つまたは複数の配列変異型を1つまたは複数のVHドメインと組み合わせた類似の方法を使用することができる。
【0054】
本発明のさらなる態様は、GDF−8と特異的に反応する抗原結合フラグメントの調製方法を提供する。この方法は、
(a)置換すべきCDR3を含むかCDR3コード領域を欠くVHドメインをコードする核酸の出発レパートリーを得る工程と、
(b)VHドメインをコードする核酸の産物レパートリーを得るためにレパートリー中のCDR3領域にドナー核酸が挿入されるように前記レパートリーと実質的に本明細書中に記載のVHCDR3(すなわち、H3)のアミノ酸配列をコードするドナー核酸とを組み合わせる工程と、
(c)前記産物レパートリーの核酸を発現する工程と、
(d)GDF−8に特異的な特異的抗原結合フラグメントを選択する工程と、
(e)前記特異的抗原結合フラグメントまたはこれをコードする核酸を回収する工程とを含む。
【0055】
さらに、本発明のVLCDR3(すなわち、L3)を置換すべきCDR3を含むかCDR3コード領域を欠くVLドメインをコードする核酸レパートリーと組み合わせた類似の方法を使用することができる。
【0056】
本発明のコード配列CDR(例えば、CDR3)を、組換え(recombinant)DNAテクノロジ−を使用してCDR(例えば、CDR3)を欠く可変ドメインのレパートリーに移入することができる。例えば、Marks et al.(Bio/Technology(1992)10:779−783)は、可変ドメイン領域の5’末端に指向するか隣接するコンセンサスプライマーをヒトVH遺伝子の第3のフレームワーク領域に対するコンセンサスプライマーと組み合わせて使用してCDR3を欠くVH可変ドメインのレパートリーが得られる、抗体可変ドメインのレパートリーの産生方法を記載している。レパートリーを、特定の抗体のCDR3と組み合わせることができる。類似の技術を使用して、本発明のCDR3由来の配列を、CDR3を欠くVHまたはVLドメインのレパートリーで組み替え(shuffle)、組み替えられた完全なVHまたはVLドメインを同族VLまたはVHドメインと組み合わせて本発明の特異的抗原結合フラグメントを得ることができる。次いで、適切な抗原結合フラグメントを選択することができるように、レパートリーを、WO92/01047のファージディスプレイ系などの適切な宿主系にディスプレイすることができる。
【0057】
類似の組み替えまたは組替え技術は、Stemmer(Nature(1994)370:389−391)によっても開示されており、これはβ−ラクタマーゼ遺伝子に関する技術を記載しているが、このアプローチを抗体作製のために使用することができる。
【0058】
さらなる代替法は、全可変ドメイン内に変異を作製するための1つまたは複数の選択されたVHおよび/またはVL遺伝子のランダム変異誘発を使用した本発明のCDR由来の配列を保有する新規のVHまたはVL領域を作製することである。このような技術は、変異性PCRを使用したGram et al.(Proc.Nat.Acad.Sci.U.S.A.(1992)89:3576−3580)に記載されている。
【0059】
使用することができる別の方法は、VHまたはVL遺伝子のCDR領域に対する直接的変異誘発である。このような技術は、Barbas et al.(Proc.Nat.Acad.Sci.U.S.A.(1994)91:3809−3813)およびSchier et al.(J.Mol.Biol.(1996)263:551−567)に開示されている。
【0060】
同様に、1つまたは複数または3つ全てのCDRをVHまたはVLドメインのレパートリーにグラフティングし、その後GDF−8に特異的な特異的結合パートナーまたは結合フラグメントについてスクリーニングすることができる。
【0061】
免疫グロブリン可変ドメインの実質的部分は、少なくともCDR領域および選択的に本明細書中に記載のscFvフラグメント由来の介在フレームワーク領域を含む。この部分はまた、少なくとも約50%のFR1およびFR4のいずれかまたは両方(50%はFR1のC末端の50%であり、50%はFR4のN末端のである)を含む。可変ドメインの実質的部分のN末端またはC末端のさらなる残基は、通常は天然に存在する可変ドメイン領域に関連しない残基であり得る。例えば、組換えDNA技術によって作製された本発明の特異的抗原結合フラグメントの構築によって、導入されたリンカーによってコードされるN末端またはC末端残基が導入されて、クローニングまたは他の操作工程を容易にすることができる。他の操作工程は、本発明の可変ドメインを免疫グロブリン重鎖を含むさらなるタンパク質配列、(例えば、ダイアボディ(diabody)の産生における)他の可変ドメイン、または以下により詳細に記載したタンパク質標識に連結するためのリンカーの導入を含む。
【0062】
実施例に例示の実施形態はVHおよびVLドメインの「マッチング」対を含んでいるが、本発明は、VHまたはVLドメイン配列のいずれか(特に、VHドメイン)由来の1つの可変ドメインを含む結合フラグメントも含む。一本鎖特異的結合ドメインのいずれかの場合、これらのドメインを使用して、GDF−8に結合することができる2ドメイン特異的抗原結合ドメインを形成することができる相補性ドメインについてスクリーニングすることができる。HまたはL鎖クローンのいずれかを含む各コロニーを使用して他の鎖(LまたはH)をコードするクローンの完全なライブラリーに感染させ、WO92/01047に開示されるファージディスプレイ技術などにしたがって、得られた二鎖特異的抗原結合ドメインを選択する、WO92/01047に開示のいわゆる階層的二重組み合わせアプローチを使用したファージディスプレイスクリーニング法によってこれを行うことができる。この技術は、Marks et al.,supraにも開示されている。
【0063】
放射性核種、薬物、高分子、または他の薬剤を使用した化学的方法によって抗体を抱合することができるか、1つまたは複数の本発明のCDRを含む融合タンパク質として作製することができる。
【0064】
抗体融合タンパク質は、VH−VL対を含み、これらの鎖の1つ(通常VH)および別のタンパク質を1つのポリペプチド鎖として合成する。これらの産物型は、一般にさらなる機能的エレメント(小分子の活性部分)を有するかまたは抱合または融合高分子の主な分子構造の特徴を有するという点で抗体と異なる。
【0065】
上記概説のアミノ酸配列の変化に加えて、抗体を、グリコシル化するか、ペグ化するか、アルブミンもしくは非タンパク質性ポリマーに結合させることができる。例えば、米国特許第4,640,835号;同第4,496,689号;同第4,301,144号;同第4,670,417号;同第4,791,192号;または同第4,179,337号に記載の様式で、本発明で開示の抗体を、例えばポリエチレングリコール、ポリプロピレングリコール、又はポリオキシアルキレンなどの種々の非タンパク質性ポリマーの1つに結合させることができる。例えば、循環半減期を増大させるために、ポリマーへの共有結合による抱合によって、抗体を化学修飾する。典型的なポリマーおよびポリマーのペプチドへの結合方法は、米国特許第4,766,106号;同第4,179,337号;同第4,495,285号;および同第4,609,546号にも示されている。
【0066】
他の実施形態では、グリコシル化パターンが変化するように(すなわち、元のまたは天然のグリコシル化パターンと異なる)抗体を修飾することができる。本明細書中で使用される、「変化した」は、1つまたは複数の炭水化物部分の欠失および/または元の抗体への1つまたは複数のグリコシル化部位の付加を意味する。グリコシル化部位のコンセンサス配列を含めるためのアミノ酸配列の変化によって達成された本発明で開示の抗体へのグリコシル化部位の付加は当該分野で周知である。抗体上の炭水化物部分の数の別の増加方法は、抗体のアミノ酸残基へのグリコシドの化学的または酵素的結合によるものである。これらの方法は、WO87/05330およびAplin and Wriston(1981)CRC Crit.Rev.Biochem.,22:259−306に記載されている。Hakimuddin et al.(1987)Arch.Biochem.Biophys.,259:52;Edge et al.(1981)Anal.Biochem.,118:131;およびThotakura et al.(1987)Meth.Enzymol.,138:350に記載のように、抗体上に存在する任意の炭水化物部分の除去を化学的または酵素的に行うことができる。
【0067】
本発明の抗体を、検出可能な標識または機能的標識でタグ化することもできる。検出可能な標識には、当該分野で公知の従来の化学的性質を使用して本発明の抗体に結合することができる131Iまたは99Tcなどの放射性標識が含まれる。標識には、西洋ワサビペルオキシダーゼまたはアルカリホスファタ−ゼなどの酵素標識も含まれる。標識には、さらに、特異的同族検出可能部分(例えば、標識ビオチン)への結合を介して検出することができるビオチンなどの化学的部分が含まれる。
【0068】
CDR配列が配列番号n(nは、2、4、6、8、10、12、14、16、18、20、22、24、26、28、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、また48である)に記載のCDR配列と非本質的にのみ異なる抗体は、本発明の範囲内に含まれる。非実質的相違には、CDR配列中の任意の5個のアミノ酸のうちの1個または2個の置換などのアミノ酸の小さな変化が含まれる。典型的には、アミノ酸を、類似の電荷、疎水性、または立体化学的性質を有する関連アミノ酸と置換する。このような置換は、当業者の範囲内である。CDR中と異なり、抗体の結合特性に悪影響を与えることなく構造フレームワーク領域(FR)中のより実質的な変化を行うことができる。FRの変化には、非ヒト由来のフレームワークのヒト化または抗原接触または結合部位の安定化に重要な一定のフレームワーク残基の操作(例えば、定常領域のクラスまたはサブクラスの変化、Fc受容体結合などのエフェクター機能を変化させることができる特異的アミノ酸残基の変化(Lund et al.(1991)J.Immun.147:2657−2662およびMorgan et al.(1995)Immunology 86:319−324)、または定常領域が由来する種の変化)が含まれるが、これらに限定されない。抗体は、重鎖のCH2領域にエフェクター機能が低下または変化する(例えば、Fc受容体結合および補体活性化)変異を有し得る。例えば、抗体は、米国特許第5,624,821号および同第5,648,260号などに記載の変異を有し得る。例えば、IgG1またはIgG2重鎖では、IgG1のFc部分を示す配列番号53のアミノ酸残基117および120にこのような変異を作製することができる(これらの残基は、IgG1またはIgG2の全長配列中のアミノ酸234および237に相当する)。抗体は、免疫グロブリンの2つの重鎖の間のジスルフィド結合を安定化する変異(Angal et al.(1993)Mol.Immunol.30:105−108に開示のIgG4のヒンジ領域の変異など)も有し得る。
【0069】
D.核酸、クローニング系、および発現系
本発明は、さらに、本発明の抗体または結合フラグメントをコードする単離核酸を提供する。本発明の核酸は、DNAまたはRNAを含むことができ、且つ全体または部分的に合成であり得る。本明細書中に記載のヌクレオチド配列の基準は、特定の配列を有するDNA分子を含み、且つ、他で特記しない限り、TがUに置換された特定の配列を有するRNA分子を含む。
【0070】
本発明の核酸は、本明細書中に記載の本発明のCDRまたはVHもしくはVLドメインのコード配列を含む。
【0071】
本発明はまた、上記の、少なくとも1つの本発明の核酸を含むプラスミド、ベクター、転写もしくは発現カセットの形態の構築物を提供する。
【0072】
本発明はまた、上記の1つまたは複数の構築物を含む宿主細胞を提供する。コードされた産物の産生方法と同様に、本明細書中に提供する任意のCDR(H1、H2、H3、L1、L2、またはL3)、VHもしくはVLドメイン、または特異的抗原結合フラグメントをコードする核酸は、本発明の態様を形成する。この方法は、コード核酸からの発現を含む。核酸を含む組換え宿主細胞の適切な条件下での培養によって発現させることができる。発現による産生後、VHもしくはVLドメイン、または特異的抗原結合フラグメントを、任意の適切な技術を使用して単離および/または精製し、必要に応じて使用することができる。
【0073】
本発明の特異的抗原結合フラグメント、VHおよび/またはVLドメイン、ならびにコード核酸分子およびベクターを、例えば、その天然の環境から単離および/または精製するか、実質的に純粋もしくは均一な形態で提供するか、核酸の場合、必要な機能を有するポリペプチドをコードする配列以外の元の核酸または遺伝子を含まないか実質的に含まずに提供することができる。
【0074】
種々の異なる宿主細胞におけるポリペプチドのクローニング系および発現系が周知である。適切な宿主細胞には、細菌系、哺乳動物細胞系、ならびに酵母およびバキュロウイルス系が含まれる。異種ポリペプチド発現のために当該分野で利用可能な哺乳動物細胞株には、チャイニーズハムスター卵巣細胞、HeLa細胞、ベビーハムスター腎臓細胞、NS0マウス黒色腫細胞、および多くの他の細胞が含まれる。一般的な細菌宿主は、E.coliである。抗体産生に適切な細胞については、Gene Expression Systems,eds.Fernandez et al.,Academic Press,1999を参照のこと。本発明に適合する任意の細胞を本発明で開示の抗体の産生に使用することができる。
【0075】
適切な調節配列(プロモーター配列、ターミネーター配列、ポリアデニル化配列、エンハンサー配列、マーカー遺伝子、および必要に応じて他の配列が含まれる)を含む適切なベクターを選択または構築することができる。必要に応じて、ベクターは、プラスミドベクターまたはウイルスベクター(例えば、ファージまたはファージミド)であり得る。さらなる詳細については、例えば、Molecular Cloning:a Laboratory Manual:2nd ed.,Sambrook et al.,Cold Spring Harbor Laboratory Press,1989を参照のこと。例えば、核酸構築物の調製、変異誘発、配列決定、細胞へのDNAの導入および遺伝子発現、ならびにタンパク質分析における核酸操作のための多数の公知の技術およびプロトコールは、Current Protocols in Molecular Biology,2nd ed.,Ausubel et al.eds.,JohnWiley & Sons,1992に詳細に記載されている。
【0076】
したがって、本発明のさらなる態様は、本明細書中に開示の核酸を含む宿主細胞を提供する。なおさらなる態様は、このような核酸を宿主細胞に導入する工程を含む方法を提供する。導入は、任意の利用可能な技術を使用することができる。真核細胞のための適切な技術には、リン酸カルシウムトランスフェクション、DEAE−デキストラン、エレクトロポレーション、リポソ−ム媒介トランスフェクション、およびレトロウイルスまたは他のウイルス(例えば、ワクシニアまたは昆虫細胞についてはバキュロウイルス)を使用した形質導入が含まれ得る。細菌細胞のための適切な技術には、塩化カルシウム形質転換、エレクトロポレーション、およびバクテリオファージを使用したトランスフェクションが含まれ得る。
【0077】
移入後に、例えば、遺伝子発現条件下での宿主細胞の培養によって核酸から発現させることができる。
【0078】
E.微生物寄託
非生殖系列化scFvのMyo29、Myo28、またはMyo22をコードするファージミドベクターpCANTAB6でそれぞれ形質転換したE.coli培養物を、2002年10月2日にAmerican Tissue Culture Collection(ATCC)にそれぞれ受託番号PTA−4741、PTA−4740、およびPTA−4739で寄託した。寄託機関の住所は、10801 University Blvd,Manassas,VA 20110,U.S.A.である。
【0079】
(II.疾患の治療方法および他の用途)
本発明の抗体は、ヒトまたは動物の種々の内科的疾患の予防、診断、または治療に有用である。抗体を使用して、GDF−8または関連タンパク質に関連する1つまたは複数の活性を阻害または減少させることができる。より好ましくは、抗体は、抗体に結合していないGDF−8と比較して1つまたは複数のGDF−8活性を阻害または減少させることができる。一定の実施形態では、1つまたは複数の本発明に開示の抗体と結合した場合、GDF−8活性は、1つまたは複数の本発明に開示の抗体に結合していない成熟GDF−8タンパク質と比較して、少なくとも50%、好ましくは少なくとも60、62、64、66、68、70、72、72、76、78、80、82、84、86、または88%、より好ましくは少なくとも90、91、92、93、または94%、さらにより好ましくは少なくとも95%〜100%阻害される。GDF−8活性の阻害を、Thies et
al.(Growth Factors(2001)18:251−259)に記載され、且つ実施例2および9に例示のpGL3(CAGA)12レポーター遺伝子アッセイ(RGA)または実施例3および6に例示のActRIIB受容体アッセイで測定することができる。
【0080】
本発明に開示の抗体によって診断、治療、または予防される内科的疾患は、筋肉または神経筋障害;肥満症などの脂肪組織障害;2型糖尿病;耐糖能障害;代謝症候群(例えば、X症候群);火傷または窒素不均衡などの外傷によるインスリン抵抗性;または骨粗鬆症などの骨変性疾患である。
【0081】
本発明に開示の抗体によって診断、治療、または予防される他の内科的疾患は、骨の喪失に関連する障害(骨粗鬆症(特に高齢者および/または閉経後の女性)、糖質コルチコイド誘導性骨粗鬆症、オステオペニア、変形性関節症、および骨粗鬆症関連骨折が含まれる)である。他の標的代謝性骨の疾患または障害には、慢性糖質コルチコイド療法に起因する低骨量、若年性性腺不全、アンドロゲン抑制、ビタミンD欠損症、二次性副甲状腺機能亢進症、栄養失調、および拒食症が含まれる。好ましくは、抗体を使用して、哺乳動物、特にヒトにおけるこのような内科的疾患を予防、診断、または治療する。
【0082】
本発明の抗体または抗体組成物を、治療有効量で投与する。一般に、治療有効量は、被験体の年齢、病態、および性別ならびに被験体の病状の重症度によって変化し得る。医師が投薬量を決定し、必要に応じて認められた治療効果に適合するように調整することができる。このような化合物の毒性および治療効率を、例えば、LD50(集団の50%に致命的な用量)およびED50(集団の50%の治療有効量)の決定のための細胞培養または実験動物における標準的な薬学的手順によって決定することができる。有毒と治療効果との間の用量の比が治療指数であり、LD50/ED50比として示すことができる。高い治療指数を示す抗体が好ましい。
【0083】
細胞培養アッセイおよび動物研究から得られたデータを、ヒトで使用するための投薬量範囲の処方において使用することができる。このような化合物の投薬量は、毒性がほとんどないか全くないED50を含む循環濃度範囲内であることが好ましい。投薬量は、使用した投薬形態および使用した投与経路によってこの範囲内で変化させることができる。本発明で使用した任意の抗体について、最初に細胞培養アッセイから治療有効用量を評価することができる。細胞培養において決定したIC50(すなわち、最大の症状の阻害の半分を達成する試験抗体の濃度)を含む循環血漿濃度範囲を達成するために、動物モデルにおける投与量を処方することができる。例えば、高速液体クロマトグラフィによって血漿レベルを測定することができる。任意の特定の投薬量の効果を、適切なバイオアッセイによってモニタリングすることができる。適切なバイオアッセイの例には、DNA複製アッセイ、転写ベースのアッセイ、GDF−8タンパク質/受容体結合アッセイ、クレアチンキナーゼアッセイ、前脂肪細胞の分化に基づいたアッセイ、脂肪細胞におけるグルコース取り込みに基づいたアッセイ、および免疫学的アッセイが含まれる。
【0084】
一般に、抗体またはその結合フラグメントが、1μg/kg〜150mg/kg、1μg/kg〜100mg/kg、1μg/kg〜50mg/kg、1μg/kg〜20mg/kg、1μg/kg〜10mg/kg、1μg/kg〜1mg/kg、10μg/kg〜1mg/kg、10μg/kg〜100μg/kg、100μg〜1mg/kg、および500μg/kg〜1mg/kgの用量で投与されるように組成物を投与する。好ましくは、投与後最も長時間循環抗体レベルを最大にするために、抗体をボーラス投与として投与する。ボーラス投与後、連続的注入を使用することもできる。
【0085】
本発明に開示の抗体を使用した上記病状の治療、診断、または予防方法を、TGF−βスーパーファミリー中の他のタンパク質で使用することもできる。多数のこれらのタンパク質は、BMP−11などのGDF−8の構造と類似している。したがって、別の実施形態は、BMP−11またはアクチビンを阻害することができる抗体の単独または他のTGF−βインヒビター(GDF−8に対する中和抗体など)との組み合わせの被験体への投与による上記障害の治療方法を提供する。本発明の抗体を使用して、BMP−11と関連するか媒介される疾患または病態を治療することもできる。例えば、米国特許第5,639,638号および同第6,437,111号を参照のこと。
【0086】
本発明の抗体を使用して、in vivoまたはin vitroでBMP−11およびGDF−8などのTGF−βスーパーファミリーに属するタンパク質の存在を検出することができる。これらのタンパク質の存在またはレベルと病状との相関により、当業者は関連する病状を診断することができる。本発明に開示の抗体によって診断することができる病状を上に記載する。
【0087】
このような検出方法は当該分野で周知であり、ELISA、ラジオイムノアッセイ、免疫ブロット、ウェスタンブロット、免疫蛍光、免疫沈降、および他の類似の技術が含まれる。タンパク質(例えば、GDF−8)を検出するための1つまたは複数のこれらの技術を組み込んだ診断キット中に抗体をさらに提供することができる。このようなキットは、他の構成要素、輸送容器、説明書、またはタンパク質の検出およびキットの使用を補助するための他の材料を含むことができる。
【0088】
抗体が診断を目的とする場合、例えば、リガンド基(ビオチンなど)または検出可能なマーカー基(蛍光基、放射性同位体、または酵素など)で修飾することが望ましい。所望ならば、従来技術を使用して抗体(ポリクローナル抗体またはモノクローナル抗体のいずれか)を標識することができる。適切な標識には、フルオロフォア、ケモフォア、放射性原子、高電子密度試薬、酵素、および特異的結合パートナーを有するリガンドが含まれる。酵素は、典型的には、その活性によって検出される。例えば、西洋ワサビペルオキシダーゼを、テトラメチルベンジジン(TMB)を分光光度計で定量可能な青色色素に変換する能力によって検出することができる。他の適切な標識には、ビオチンおよびアビジンまたはストレプトアビジン、IgGおよびタンパク質A、ならびに当該分野で公知の多数の受容体−リガンド結合物が含まれ得る。他の順列および可能性は当業者に容易に明らかであり、本発明の範囲内で等価物と見なされる。
【0089】
本発明のなおさらなる態様は、筋肉および骨の障害の治療で有用な治療薬の同定方法を提供する。適切なスクリーニングアッセイ(例えば、ELISAベースのアッセイ)は当該分野で公知である。このようなスクリーニングアッセイでは、第1の結合混合物を、本発明の抗体とリガンド(例えば、GDF−8、BMP−11、アクチビン)との組み合わせによって形成し、第1の結合混合物中のリガンドと抗体との間の結合量(M0)を測定する。第2の結合混合物も抗体と、リガンドと、スクリーニングすべき化合物または薬剤との組み合わせによって形成し、第2の結合混合物中のリガンドと抗体との間の結合量(M1)を測定する。次いで、例えば、M1/M0比の計算によって第1および第2の結合混合物の結合量を比較する。第1の結合混合物と比較して第2の結合混合物で結合の減少が認められる場合、化合物または薬剤はGDF−8活性を阻害することができると見なされる。結合混合物の処方および至適化は当業者のレベルの範囲内であり、このような結合混合物はまた、結合の強化または至適化に必要な緩衝液および塩を含むことができ、本発明のスクリーニングアッセイにさらなるコントロールアッセイが含まれ得る。
【0090】
したがって、少なくとも約10%(すなわち、M1/M0<0.9)、好ましくは約30%を超えて抗体−リガンド結合が減少することが見いだされた化合物を同定することができ、所望ならば、ActRIIB結合アッセイ(実施例2)ならびに実施例13、15、および16に記載の他の細胞ベースのアッセイおよびin vivoアッセイなどの他のアッセイでGDF−8活性を阻害する能力について二次的にスクリーニングすることができる。
【0091】
(III.薬学的組成物および投与方法)
本発明は、本発明に開示の抗体を含む組成物を提供する。このような組成物は、薬学的使用および患者への投与に適切であり得る。組成物は、典型的には、1つまたは複数の本発明の抗体および薬学的に許容可能な賦形剤を含む。本明細書中で使用される、句「薬学的に許容可能な賦形剤」には、薬学的投与に適合する任意および全ての溶媒、分散媒、コーティング、抗生物質および抗真菌薬、ならびに等張化剤および吸収遅延剤などが含まれる。薬学的に活性な物質のためのこのような溶剤および薬剤の使用は当該分野で周知である。組成物は、補足的、付加的、または強化された治療機能を提供する他の活性化合物も含み得る。薬学的組成物を、投与説明書とともにコンテナ、パック、またディスペンサーに含めることもできる。
【0092】
本発明の薬学的組成物を、その意図する投与経路に適合するように処方する。投与方法は、当業者に公知である。局所的または経口投与することができるか、粘膜を通過することができる組成物を得ることも可能である。投与は、例えば、静脈内、腹腔内、筋肉内、腔内、皮下、または経皮であり得る。
【0093】
皮内または皮下投与で使用される溶液または懸濁液には、典型的には、1つまたは複数の以下の構成要素が含まれる:注射用の水、生理食塩水、不揮発油、ポリエチレングリコール、グリセリン、プロピレングリコール、または他の合成溶媒などの滅菌希釈剤;ベンジルアルコールまたはメチルパラベンなどの抗菌薬;アスコルビン酸または亜硫酸水素ナトリウムなどの抗酸化剤;エチレンジアミン四酢酸などのキレート化剤;酢酸、クエン酸、またはリン酸などの緩衝液;および塩化ナトリウムまたはデキストロースなどの等張化剤。塩酸または水酸化ナトリウムなどの酸または塩基でpHを調整することができる。このような調製物を、ガラス製またはプラスチック製のアンプル、使い捨てシリンジ、または複数回投与用バイアルに同封することができる。
【0094】
注射に適切な薬学的組成物には、滅菌水溶液または分散液および滅菌注射用溶液または分散液の即時調製のための滅菌粉末が含まれる。静脈内投与に適切なキャリアには、生理食塩水、静菌水、Cremophor(商標)EL(BASF,Parsippany,NJ)、またはリン酸緩衝化生理食塩水(PBS)が含まれる。全ての場合において、組成物は滅菌されていなければならず、容易にシリンジで使用できる範囲の流動物であるべきである。製造および保存条件下で安定でなければならず、細菌および真菌などの微生物の汚染作用に対して保存されるべきである。キャリアは、例えば、水、エタノール、ポリオ−ル(例えば、グリセロール、プロピレングリコール、および液体ポリエチレングリコールなど)、およびその適切な混合物を含む溶媒または分散媒であり得る。例えば、レシチンなどのコーティングの使用、分散液の場合の必要な粒子サイズの維持、および界面活性剤の使用によって適切な流動性を維持することができる。種々の抗菌薬および抗真菌薬(例えば、パラベン、クロロブタノ−ル、フェノール、アスコルビン酸、およびチメロサールなど)によって微生物作用を防止することができる。多くの場合、組成物中に等張化剤(例えば、糖、マンニトールなどのポリアルコール、ソルビトール、および塩化ナトリウム)を含むことが好ましい。注射用組成物は、組成物中に吸収を遅延させる薬剤(例えば、モノステアリン酸アルミニウムおよびゼラチン)を含めることによって長期間吸収され得る。
【0095】
経口組成物には、一般に、不活性希釈剤または食用キャリアが含まれる。これらをゼラチンカプセルに封入するか、打錠することができる。治療のための経口投与の目的で、抗体を賦形剤に組み込み、錠剤またはカプセル形態で使用することができる。薬学的に適合する結合剤および/またはアジュバント材料を、組成物の一部として含めることができる。錠剤、丸薬、およびカプセルなどは、任意の以下の成分または類似の性質の化合物を含み得る:微結晶性セルロース、トラガカントガム、またはゼラチンなどの結合剤;デンプンまたはラクトースなどの賦形剤;アルギン酸、Primogel(商標)、またはコーンスターチなどの崩壊薬;ステアリン酸マグネシウムまたはSterotes(商標)などの潤滑剤;コロイド状二酸化ケイ素などの流動促進剤;スクロースまたはサッカリンなどの甘味料;またはペパーミント、サリチル酸メチル、またはオレンジフレーバーなどの矯味矯臭薬。
【0096】
吸入による投与のために、適切な高圧ガス(例えば、二酸化炭素などのガス)を含む加圧コンテナもしくディスペンサーまたはネブライザーからエアゾールスプレーの形態で抗体を送達させる。
【0097】
経粘膜または経皮手段によって全身投与を行うこともできる。例えば、Fc部分を含む抗体の場合、組成物は、FcRn受容体媒介経路を介して粘膜(例えば、腸、口腔、または肺)を通過することができる(米国特許第6,030,613号)。例えば、ロゼンジ、鼻腔用スプレー、吸入器、または坐剤の使用によって経粘膜投与を行うことができる。経皮投与のために、活性化合物を、当該分野で公知の軟膏(ointment)、軟膏(salve)、ゲル、またはクリームに処方する。経粘膜投与または経皮投与のために、処方物に通過すべきバリアに適切な浸透剤を使用する。このような浸透剤は一般に当該分野で公知であり、例えば、界面活性剤、胆汁酸塩、およびフシジン酸誘導体が含まれる。
【0098】
本発明に開示の抗体を、徐放性処方物などの身体からの急速な排除から化合物を保護するキャリア(移植片およびマイクロカプセル化した送達システムが含まれる)を使用して調製することができる。エチレンビニルアセテート、ポリ酸無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、およびポリ乳酸などの生分解性生体適合性ポリマーを使用することができる。このような処方物の調製方法は、当業者に自明である。本発明に開示の抗体を含むリポソーム懸濁液を、薬学的に許容可能なキャリアとして使用することもできる。例えば、米国特許第4,522,811号に記載の当業者に公知の方法にしたがって、これらを調製することができる。
【0099】
投与を容易にするためおよび投薬量の均一性のために、単位投薬形態で経口または非経口組成物を処方することが有利であり得る。本明細書中で使用される、「単位投薬形態」は、処置すべき被験体のための単一投薬量として適切な物理的に個別の単位をいい、必要な薬学的キャリアと共同して所望の治療効果が得られるように所定量の活性化合物を含む単位を計算した。本発明の単位投薬形態の仕様は、活性化合物の固有の特徴および達成すべき特定の治療効果、および個体治療のためのこのような活性化合物の処方分野が本来有する制限について言及しているか、これらに直接依存する。
【0100】
以下の実施例は、本発明の例示的実施形態を提供し、本発明を決して限定しない。当業者は、多数の他の実施形態が本発明の範囲内に含まれることを認識する。
【0101】
本願で引用された全ての引用文献、特許、および公開された特許出願の内容全体が本明細書中で参考として援用される。
【実施例】
【0102】
(実施例1:GDF−8の精製)
組換えヒトGDF−8タンパク質(成熟GDF−8およびGDF−8プロペプチド)を発現する選択細胞株由来の馴化培地をpH6.5に酸性化し、80×50mmPOROS(商標)SP陽イオン交換カラムと縦に並べた80×50mmPOROS(商標)HQ陰イオン交換カラムにアプライした(PerSeptive Biosystems,Foster City,CA)。通過物(flow through)をpH5.0に調整し、75×20mmPOROS(商標)SP陽イオン交換カラム(PerSeptive
Biosystems)にアプライし、NaCl勾配を使用して溶離した。SDS−PAGEによって確認したGDF−8潜在複合体を含む画分をプールし、トリフルオロ酢酸(TFA)でpH2〜3に酸性化し、粘度を低下させるために0.1%TFAで200mlにした。次いで、プールを、60×21.2mmのガードカラム(Phenomenex,Torrance,CA)を取りつけた250×21.2mmのC5カラム(Phenomenex)にアプライし、TFA/アセトニトリル勾配を使用して溶離し、GDF−8プロペプチドから成熟GDF−8を分離した。アセトニトリルを除去するために成熟GDF−8を含むプール画分を凍結乾燥によって濃縮し、20mlの0.1%TFAを添加した。次いで、サンプルを、分離を補助するために60℃に加熱した250×10mmのC5カラム(Phenomenex)にアプライした。もはやさらに分離することができなくなるまでこれを繰り返した。次いで、成熟GDF−8を含む画分をプールし、40%アセトニトリルで増量し(bring up)、60×21.2のガードカラムを取りつけた600×21.2のBioSep(商標)S−3000サイズ排除カラム(Phenomenex)にアプライした。精製成熟GDF−8を含む画分をプールし、その後の実験での使用のために濃縮した。
【0103】
SDS−PAGEにおいて、精製成熟GDF−8は、非還元条件下で25kDaおよび還元条件下で13kDaの広いバンドとして移動した。McPherron et al.(Proc.Nat.Acad.Sci.U.S.A.(1997)94:12457−12461)によってマウスGDF−8について類似のSDS−PAGEプロフィ−ルが報告されており、成熟タンパク質の二量体特性を反映する。活性成熟BMP−11二量体を、類似の様式で組換えヒトBMP−11を発現する細胞株由来の馴化培地から精製した。
【0104】
活性成熟BMP−11を、組換えヒトGDF−8プロペプチド/成熟BMP−11キメラタンパク質を発現する細胞株由来の馴化培地から精製した。馴化培地を、50mM Tris(pH8.0)、1M NaClを含む10mlのTALON(商標)カラム(Clonetech,Palo Alto,CA)に1ml/分でロードした。結合したタンパク質を、50mM Tris(pH8.0)、1M NaCl、500mMイミダゾールで溶離した。GDF−8プロペプチド/BMP−11潜在複合体を含むプール画分を、10%TFAでpH3に酸性化した。次いで、プールを、成熟BMP−11およびGDF−8プロペプチドのより良好な分離のために60℃に加熱した250×4.6mmのJupiter C4カラム(Phenomenex,Torrance,CA)にアプライし、TFA/アセトニトリル勾配を使用して溶離した。成熟BMP−11を含むプール画分を、凍結乾燥によって濃縮した。SDS−PAGE、精製成熟BMP−11は、非還元条件下で25kDaおよび還元条件下で12kDaに移動した。
【0105】
(実施例2:精製組換えヒトGDF−8の生物活性)
GDF−8の活性を証明するために、ルシフェラーゼを発現するレポーターベクターpGL3(CAGA)12を使用してレポーター遺伝子アッセイ(RGA)を開発した。CAGA配列は、TGF−β誘導性遺伝子PAI−1のプロモーター内のTGF−β応答配列であることが以前に報告されていた(Denner et al.(1998)EMBO J.,17:3091−3100)。
【0106】
基本ルシフェラーゼレポータープラスミドpGL3(Promega,Madison,WI)を使用して、12個のCAGAボックスを含むレポーターベクターを作製した。アデノウイルス主要後期プロモーター(−35/+10)由来のTATAボックスおよび転写開始部位を、BglIIとHindIII部位との間に挿入した。CAGAボックスAGCCAGACAの12回反復を含むオリゴヌクレオチドをアニーリングし、XhoI部位にクローン化した。FuGENE(商標)6トランスフェクション試薬(Boehringer Manheim,Germany)を使用して、ヒト横紋筋肉腫細胞株A204(ATCC HTB−82)をpGL3(CAGA)12で一過性にトランスフェクトした。トランスフェクション後、2mMグルタミン、100U/mlストレプトマイシン、100μg/mlペニシリン、および10%ウシ胎児血清を補足したMcCoyの5A培地を含む48ウェルプレートに細胞を16時間培養した。次いで、細胞を、McCoyの5A培地中の10ng/mlのGDF−8の存在下又は非存在下で、グルタミン、ストレプトマイシン、ペニシリン、および1mg/mlのウシ血清アルブミンで、37℃にて6時間処理した。ルシフェラーゼアッセイ系(Promega)を使用して、処置した細胞中のルシフェラーゼを定量した。
【0107】
図4Aは、GDF−8は、10ng/mlのED50でレポーター構築物を最大で10倍活性化し、精製組換えGDF−8が生物学的に活性であることを示す。BMP−11およびアクチビンは類似の生物学的応答を誘発した。
【0108】
(実施例3:ActRIIB結合アッセイにおける精製GDF−8の結合特性)
GDF−8潜在複合体を、1モルのGDF−8複合体に対して20モルのEZ結合スルホ−NHS−ビオチン(Pierce,Rockford,Illinois,Cat.No.21217)の比で氷上で2時間ビオチン化した。0.5%TFAを使用したpHの低下によって反応を停止させ、複合体をC4 Jupiter 250×4.6mmカラム(Phenomenex)におけるクロマトグラフィに供してGDF−8プロペプチドから成熟GDF−8を分離した。TFA/CH3CN勾配を使用して溶離したビオチン化成熟GDF−8画分をプールし、濃縮し、MicroBCA(商標)タンパク質アッセイ試薬キット(Pierce,Rockford,IL,Cat.No.23235)によって定量した。
【0109】
上記と同一の様式で、BMP−11潜在複合体からビオチン化成熟BMP−11を調製した。1μg/mlの組換えActRIIB−Fcキメラ(R&D Systems,Minneapolis,MN,Cat.No.339−RB/CF)を含む0.2M炭酸ナトリウム緩衝液を、4℃で一晩96ウェルの平底アッセイプレート(Costar,NY,Cat.No.3590)にコートした。次いで、プレートを1mg/mlのウシ血清アルブミンでブロッキングし、標準的なELISAプロトコールにしたがって洗浄した。種々の濃度の100μアリコートのビオチン化GDF−8またはBMP−11を、ブロッキングしたELISAプレートに添加し、1時間インキュベートし、洗浄し、ストレプトアビジン−西洋ワサビペルオキシダーゼ(SA−HRP,BD PharMingen,San Diego,CA,Cat.No.13047E)およびその後のTMB(KPL,Gaithersburg,MD,Cat.No.50−76−04)の添加によってGDF−8またはBMP−11の結合量を検出した。Molecular Devicesマイクロプレートリーダーにおいて450nMで比色測定を行った。
【0110】
図1に示すように、ビオチン化GDF−8およびBMP−11はActRIIB(ED50が15および40ng/mlである推定GDF−8II型受容体)にそれぞれ結合し、ActRIIB結合アッセイがin vitro結合アッセイでGDF−8およびBMP−11に感受性を示すことを示す。
【0111】
(実施例4:scFvライブラリーにおけるGDF−8のパニングによるMyo22の単離)
記載の1.38×1010個のライブラリー(Vaughan et al.(1996)Nature Biotech.,14:309−314)の拡大バ−ジョンであるscFvファージミドライブラリーを使用して、GDF−8に特異的な抗体を選択した。可溶性GDF−8タンパク質(10μg/mlを含む50mM炭酸ナトリウム緩衝液(pH9.6))でマイクロタイタープレートのウェルを4℃で一晩コートした。ウェルをPBSで洗浄し、MPBS(3%Marvel(商標)脱脂粉乳を含むPBS)にて37℃で2時間ブロッキングした。精製したファージ(1012形質導入単位(tu))を含む100lの3%MPBSをブロッキングしたウェルに添加し、室温で1時間インキュベートした。ウェルをPBST(0.1%v/vのTween(商標)20を含むPBS)で10回洗浄し、次いでPBSで10回洗浄した。結合したファージ粒子を、100μlの100mMトリエチルアミンにて室温で10分間溶離し、その直後に50μlの1M Tris−HCl(pH7.4)で中和した。溶離したファージを使用して、指数関数的に成長した10mlのE.coli TG1に感染させた。感染細胞を、2TYブロス中で静置にて37℃で30分間およびその後曝気しながら37℃で30分間成長させ、2TYAGプレート上に画線し、30℃で一晩インキュベートした。コロニーをプレートから10mのl2TYブロスに掻き取り、−70℃での保存のために15%グリセロールを添加した。
【0112】
第1ラウンドのパニング選択由来のグリセロールストック培養物にヘルパーファージを重感染させ、レスキューして第2のパニングラウンドのためのscFv抗体発現ファージ粒子を得た。このようにして、全部で3ラウンドのパニングを行った。
【0113】
(実施例5:scFvライブラリーからのMyo28およびMyo29の選択)
ビオチン化GDF−8タンパク質(bioGDF−8)を使用して、可溶性選択(soluble selections)を行った。1μg/mlの濃度でbioGDF−8を使用した。実施例4に記載するように、scFvライブラリーを使用した。精製scFvファージ(1012tu)を100μlの3%MPBSで30分間ブロッキングし、ビオチン化抗体を添加し、室温で1時間インキュベートした。ファージ/抗原を、1mlの3%MPBS中にて37℃で1時間ブロッキングした50μlのDynal(商標)M280ストレプトアビジン磁性ビーズに添加し、室温でさらに15分間インキュベートした。磁性ラックを使用してビーズを補足し、0.1%(v/v)Tween(商標)20を含む1mlの3%MPBSで4回洗浄し、PBSで3回洗浄した。最後のPBSでの洗浄後、ビーズを100μlのPBS中で再懸濁し、5mlの指数関数的に成長したE.coli TG−1細胞を感染させるのに使用した。細胞およびファージを、37℃で1時間(静置で30分間および250rpmで震盪しながら30分間)インキュベートし、2TYAGプレートに広げた。プレートを30℃で一晩インキュベートし、翌日にコロニーを視覚化した。産生されたコロニーをプレートから掻き取り、上記のようにファージをレスキューした。上記のように第2ラウンドの可溶性選択を行った。
【0114】
(実施例6:ActRIIB受容体阻害アッセイおよびスクリーニング)
実施例4および5に記載のようにして得た産生コロニーを、100μlの2TYAGを含む96ウェルプレートに採取した。指数関数的に成長した培養物への1mMのIPTGの添加および30℃で一晩のインキュベーションによってscFv産生を誘導した。本質的に実施例3に記載のように、粗scFv含有培養物上清を、bioGDF−8のActRIIBへの結合を阻害する能力についてスクリーニングした。bioGDF−8の結合をユウロピウム標識ストレプトアビジンで検出し、時間分解蛍光アッセイ(TRF)でDELFIA(商標)試薬キット(PerkinElmer Life Sciences,Boston,MA)を使用するという点でアッセイをわずかに修正した。無関係のクローンを超える結合シグナルの阻害を示す正のクローンを採取し、活性を確認するためにアッセイした。
【0115】
受容体阻害スクリーニングから同定された正のクローン由来の精製scFvを、上記の阻害アッセイで試験した。アッセイにおけるIC50値によって測定したクローンの能力を確立するために、scFv濃度の滴定を使用した。実験結果を図2に示す。これらのアッセイで決定したところ、Myo29、Myo28、およびMyo22のscFvについてのIC50は、それぞれ2.4nM、1.7nM、および60nMである。したがって、これらの抗体は、GDF−8活性の強力なインヒビターである。
【0116】
(実施例7:ファージELISAによる特異性の特徴づけ)
抗体の特異性を決定するために、GDF−8および無関係のタンパク質に対するActRIIBスクリーニング由来の正のクローンについてファージELISAを行った。ファージミドを含む各E.coliコロニーを、ウェルあたり100μlの2TYAG培地を含む96ウェルプレートに接種した。M13K07ヘルパーファージを、10の感染多重度(moi)の指数関数的に成長した培養物に添加し、プレートを37℃でさらに1時間インキュベートした。プレートを、ベンチトップ遠心分離機にて2000rpmで10分間遠心分離した。上清を除去し、細胞ペレットを100μlの2TYAKに再懸濁し、震盪しながら30℃で一晩インキュベートした。翌日、プレートを2000rpmで10分間遠心分離し、各ウェル由来の100μlのファージ含有上清を新鮮な96ウェルプレートに移した。ファージサンプルを、ELISA前に最終濃度3%のMPBSにて室温で1時間ブロッキングした。
【0117】
1μg/mlのGDF−8または無関係のタンパク質を、96ウェルマイクロタイタープレートに4℃で一晩コートした。コーティング後、ウェルから溶液を取り出し、プレートを3%MPBSにて室温で1時間ブロッキングした。プレートをPBSでリンスし、各ウェルに50μlのプレブロッキングファージを添加した。プレートを室温で1時間インキュベートし、PBSTで3回洗浄し、その後PBSで3回洗浄した。各ウェルに、50μlの5,000倍希釈の抗M13−HRP抱合体(Pharmacia)を添加し、プレートを室温で1時間インキュベートした。各プレートを、PBSTで3回洗浄し、その後PBSで3回洗浄した。50μlのTMB基質を各ウェルに添加し、発色するまでインキュベートした。25μlの0.5M H2SO4の添加によって反応を停止させた。作製されたシグナルを、マイクロタイタープレートリーダーを使用した450nmの吸光度の読み取りによって測定した。GDF−8の特異的結合を確認した。
【0118】
(実施例8:scFvの配列決定、IgGへの変換、および生殖系列化)
中和scFv E.coliクローンを2TYAGプレートに画線し、30℃で一晩インキュベートした。scFvクローン由来のVHおよびVL領域を増幅するために、これらのプレート由来の三連のコロニーをpCANTAB6ベクター配列オリゴを使用して配列決定した。Myo29、Myo28、およびMyo22のIgGの作製に使用したscFvフラグメントのDNA配列を、それぞれ配列番号13、配列番号7、および配列番号1に示す。
【0119】
PCRおよびクローン特異的プライマーを使用して、scFvクローン由来の重鎖および軽鎖のV領域を増幅した。適切な制限酵素でPCR産物を消化し、ヒトIgG1重鎖定常ドメイン(VHドメインについて)を含むベクターまたは必要に応じてヒトλ軽鎖定常ドメイン(VLドメインについて)を含むベクターにサブクローン化した。各E.coliコロニー由来のプラスミドDNAの配列決定によってプラスミドへのV領域ドメインの正確な挿入を確認した。標準的技術によってE.coli培養物からプラスミドを調製し、標準的な技術を使用して重鎖および軽鎖構築物をCOS細胞に同時トランスフェクトした。プロテインAセファロ−ス(Pharmacia,Peapack,NJ)を使用して分泌IgGを精製し、緩衝液をPBSに交換した。
【0120】
scFvクローンの配列データを使用して、各クローンの重鎖および軽鎖の最も近い生殖系列配列を同定した。適切な変異誘発プライマーを使用した標準的な部位特異的変異誘発技術を使用して適切に変異させた。配列分析によってscFv配列の変異を確認した。Myo28およびMyo29の生殖系列化scFvならびにVHおよびVLドメイン配列を、配列番号19および配列番号25にそれぞれ示す。
【0121】
(実施例9:抗体の生物活性)
図3Aは、実施例3に記載のActRIIB結合アッセイにおいてMyo29の10ng/mlのビオチン化GDF−8とのプレインキュベーションにより、0.2〜0.4nMのIC50にてActRIIBへのGDF−8結合が阻害されることを示す。同様に図3Bでは、Myo29は同一のIC50でのActRIIBへのビオチン化BMP−11結合を阻害した。
【0122】
in vitroバイオアッセイにおいてMyo29はGDF−8活性も遮断した。例として、GDF−8をMyo29と室温で1時間プレインキュベートした場合、本質的に実施例2に記載のように実施したRGAアッセイで決定したところ、GDF−8の生物活性が減少した。図4Cは、Myo29の存在下におけるGDF−8のED50(20ng/ml)でのpGL3(CAGA)12レポーター活性の誘導を示す。Myo29は、15〜30ng/mlのIC50(0.1〜0.2nM)にて用量応答様式でGDF−8誘導を減少させた。Myo29は、同程度にBMP−11の生物活性も阻害した(図4B)。対照的に、本アッセイのアクチビン活性は、GDF−8およびBMP−11と比較してMyo29に影響を受けず(図4D)、これはおそらくGDF−8とアクチビンとの間の相同性が相対的に低いためである。
【0123】
RGAおよびActRIIB結合アッセイにおいてMyo22およびMyo28も試験した。両抗体は、GDF−8およびBMP−11活性を遮断する。Myo28のIC50は、例えば、0.2〜0.35nMである。
【0124】
(実施例10:Myo22、Myo28、およびMyo29のエピトープのマッピング)
抗体の正確なエピトープをマッピングするために、配列番号49に記載の成熟GDF−8の全配列を示す48の重複する13残基のペプチドを、スポット合成技術を使用してセルロースペーパー上で直接合成した(Molina et al.(1996)Peptide Research,9:151−155;Frank et al.(1992)Tetrahedron,48:9217−9232)。ペプチドの重複は11アミノ酸であった。このアレイでは、システインの存在によって生じる化学的な複雑さを減少させるために、システイン残基をセリンに置換した。ポリエチレングリコールで修飾したセルロースメンブレンおよびFmoc保護アミノ酸を、Abimed(Lagenfeld,Germany)から購入した。以前に記載のように、β−アラニンスペーサーのカップリングによってメンブレン上でアレイを定義し、ペプチドを標準的なDIC(ジイソプロピルカルボジイミド)/HOBt(ヒドロキシベンゾトリアゾール)カップリング化学を使用して合成した(Molina et al.(1996)Peptide Research,9:151−155;Frank et al.(1992)Tetrahedron,48:9217−9232)。
【0125】
活性化アミノ酸を、Abimed ASP 222ロボットを用いてスポットした。洗浄および脱保護工程を手動で行い、最終合成サイクル後にペプチドのN末端をアセチル化した。ペプチド合成後、メンブレンをメタノールで10分間洗浄し、ブロッカー(TBST(0.1%(v/v)Tween(商標)20を含むTris緩衝化生理食塩水)および1%(w/v)カゼイン)で10分間洗浄した。次いで、メンブレンを、2.5μg/mlの抗GDF−8抗体を含むブロッカーと穏やかに撹拌しながら1時間インキュベートした。ブロッカーでの10分間で3回の洗浄後、メンブレンをHRP標識二次抗体(0.25μg/mlを含むブロッカー)と30分間インキュベートした。次いで、メンブレンをブロッカーにて10分間ずつ3回およびTBSTにて10分間ずつ2回洗浄した。結合した抗体をSuperSignal(商標)West試薬(Pierce)およびデジタルカメラ(Alphananotech Fluoromager)を使用して視覚化した。結果を図5に示す。特に、図5で認められるように、Myo29のエピトープは、成熟GDF−8のアミノ酸72と88との間にマッピングされた。それに対して、Myo22は、成熟GDF−8配列の最初の44個のN末端アミノ酸(配列番号49のアミノ酸1〜44)内のエピトープを認識する。最後に、Myo28のエピトープは、成熟GDF−8の最初の98個のN末端アミノ酸内に存在する残基を含む。
【0126】
Myo29エピトープをさらに特徴づけるために、スポット合成を使用して削除および置換分析を行った。置換分析では、ペプチドの各残基を個別にシステイン以外の20種の天然アミノ酸にそれぞれ置換した。上記のように、合成および結合アッセイを行った。結果を図6に示し、最初の行、最初の2つの列、および最後の3つの列は、野生型ペプチドコントロールを示す。結果は、Lys−78、Pro−81、およびAsn−83を個別に別のアミノ酸に変異させた場合、ペプチドに対するMyo29の結合親和性を有意に減少させることを示している。したがって、Myo29は、Lys−Xaa1−Xaa2−Pro−Xaa3−Asn(配列番号54)(Xaa1、Xaa2、およびXaa3はそれぞれ任意のアミノ酸のいずれかであるか、互いに独立して、Xaa1=Met、Xaa2=Ser、およびXaa3=Ile)を含む配列を認識する。
【0127】
(実施例11:GDF−8の免疫沈降)
成熟GDF−8およびGDF−8複合体へのMyo29およびMyo28の結合を評価するために、免疫沈降研究を行った。GDF−8を発現するCHO細胞を、35S−メチオニンおよび35S−システインで標識した。GDF−8タンパク質(成熟GDF−8および潜在複合体)を含む100μlのこれらの細胞由来の馴化培地を、20μg/mlのMyo29またはMyo28と4℃で1時間インキュベートした。プロテインA−セファロ−ス(商標)を添加し、4℃で一晩インキュベートした。免疫沈降物を回収し、還元サンプル緩衝液中に再懸濁し、SDS−PAGEで分析した。ゲルを固定し、オ−トラジオグラフィ増強溶液で増強し、乾燥させ、オ−トラジグラム(autorad)を現像した。図7は、Myo29およびMyo28は成熟GDF−8、GDF−8潜在複合体、および非プロセシングGDF−8を免疫沈殿させることができることを示す。ウェスタンブロッティングによって決定したところ、両抗体は、非還元条件下でGDF−8二量体と結合する。
【0128】
(実施例12:薬物動態学)
単回静脈内(IV)または腹腔内(IP)投与後のC57B6/SCIDマウスにおける1mg/kgの用量でのMyo29の薬物動態学(PK)を評価した。上記用量での非標識および125I標識Myo29の混合物を動物に投与し、血清中の125I放射能および注射用量の比活性に基づいて血清濃度を決定した。図8は、IVまたはIPのいずれかで投与したMyo29の時間に体する血清濃度のプロットを示す。
【0129】
Myo29は、約1週間の長期末端半減期および約1ml/時間/kgの低クリアランスを示した。初期分布体積は、約83ml/kgであった。外見上の分布体積は、約227ml/kgであった。Myo29は、注射後約6時間でピーク濃度に達した。IP注射後に吸収した画分は、約77%であった。
【0130】
(実施例13:筋肉量および強度に対するMyo29のin vivo効果)
Myo29がin vivoでGDF−8活性を遮断するかどうかを決定するために、成体SCIDマウスにおいてMyo29を試験した。SCIDマウスは、重症複合型免疫不全を罹患しているので、Myo29などのヒト抗体の注射後に免疫反応を起こさない。筋肉量は、Myo29で処置したマウスのGDF−8活性の指標として使用した。
【0131】
8週齢の雄C57B6 SCIDマウスを秤量し、体重に関して均一に8つの群に分配した。Myo29を含むPBS緩衝液を、種々の用量(60、10、および1mg/kg)で毎週マウスに腹腔内注射した。最初の週は二重用量を投与した。賦形剤(PBS)で処置した、あるいは処置を行わなかったマウスをコントロールとして用いた。処置を4週間継続した。処置後に腓腹筋および大腿四頭筋を解剖および秤量することによって筋肉量を評価した。処置から4週間後、Myo29で処置した全ての群で筋肉量が10%〜23%の範囲で増加し、より高用量で処置した群は有意なレベルに達した(図9、p<0.01)。
【0132】
別の実験では、雌CB17 SCIDマウスを、種々の用量(10、5、2.5、および1mg/kg)のMyo29で4週間または12週間毎週処置した。さらに、Myo29での4週間の処置により、腓腹筋および大腿四頭筋の重量が10%〜20%の範囲で増加した(図10Aおよび10B)。より長い処置(12週間)により、筋肉量がより増加し(12%〜28%)、Myo29で処置した全ての群は統計的に有意なレベルに達した(図11Aおよび11B)。
【0133】
筋肉量の増加により筋肉がより強くなるかどうかを決定するために、前肢の筋肉の強度を、握力計(model 1027 csx,Columbus Instruments,Columbus,OH)で測定した。処置から12週間後、前肢強度は、賦形剤コントロールと比較して5mg/kgまたは10mg/kgのMyo29で処置したマウスでそれぞれ17%および23%高かった(p<0.01、図12)。この研究結果は、Myo29がin vivoでGDF−8活性を阻害して筋肉量および筋肉の強度を有意に増加させることを証明する。
【0134】
(実施例14:代謝疾患の治療)
GDF−8のインヒビター(例えば、阻害抗体など)は、2型糖尿病、耐糖能障害、代謝症候群(例えば、X症候群)、外傷によるインスリン抵抗性(例えば、火傷または窒素不均衡)、および脂肪組織障害(例えば、肥満症)などの代謝障害の治療に有用である。本発明の抗GDF−8抗体を使用して、疾患を発症している被験体または確立された代謝疾患を有する被験体を治療する。
【0135】
代謝障害(例えば、2型糖尿病および/肥満症)の治療のための抗GDF−8抗体の有効性を、肥満症、インスリン抵抗性、および2型糖尿病の確立されたマウスモデル(ob/ob、db/db、および致死性黄色症変異を有する株が含まれる)を使用して確認する。インスリン抵抗性を、一定のマウス株(C57BL/6Jが含まれる)の高脂肪または高カロリー飼料によって誘導することもできる。ヒトと同様に、これらのげっ歯類は、インスリン抵抗性、高インスリン血症、異常脂質血症、および高血糖症を発症するグルコースホメオスタシスの悪化を発症する。結果の評価は、血清中のグルコース、インスリン、および脂質の測定に基づく。インスリン抵抗性試験およびグルコース負荷試験によってインスリン感度の改良を測定することができる。より感度の高い技術には、血糖コントロールおよびインスリン感度の改善を評価するためのオイグリセミック高インスリン血症クランプの使用が含まれる。さらに、クランプ技術により、改良された血糖コントロールにおける主なグルコース処理組織(筋肉、脂肪、および肝臓)の役割が定量的に評価可能である。
【0136】
1つの研究では、1週間から6ヶ月間Myo29(IP注射)などの抗GDF−8抗体または賦形剤での処置を行う。治療プロトコールは、異なる用量の試験および治療計画(例えば、毎日、毎週、2週間毎の注射)によって変化し得る。抗GDF−8抗体で処置したマウスは、プラシーボ処置を受けたマウスと比較して、グルコース取り込みが増加し、解糖およびグリコーゲン合成が増加し、血清中の遊離脂肪酸およびトリグリセリドが低下すると予想される。
【0137】
また、GDF−8に対する阻害抗体を使用して、疾患の重症度および/または症状を予防および/または軽減する。抗GDF−8抗体を1日1回の頻度および1ヶ月に1回の頻度の皮下注射として投与することが予想される。治療継続期間は、1ヶ月から数年までの範囲であり得る。
【0138】
ヒトにおける抗GDF−8の臨床効果を試験するために、2型糖尿病を罹患しているかリスクのある被験体を同定し、無作為に治療群に分けた。治療群は、プラシーボ群および抗体(異なる用量)を投与された1〜3つの群を含む。個体のグルコース代謝の変化を評価するために1ヶ月〜3年間予測的に追跡する。処置を受けた個体は改善すると予想される。
【0139】
唯一の活性化合物として、又は別の化合物または組成物と組み合わせて、抗体を投与する。唯一の活性化合物として、又は別の化合物または組成物と組み合わせて投与した場合、投薬量は、好ましくは、疾患の症状の重症度および進行に依存して約1μg/kg〜20mg/kgである。適切な有効用量を、治療を行う医師によって以下の範囲から選択される:1μg/kg〜20mg/kg、1μg/kg〜10mg/kg、1μg/kg〜1mg/kg、10μg/kg〜1mg/kg、10μg/kg〜100μg/kg、100μg/kg〜1mg/kg、および500μg/kg〜1mg/kg。治療計画の例および結果を表3にまとめる。
【0140】
【表3】
本明細書中で引用された引用文献(その全体が本明細書中で参考として援用される)の教示に照らして、明細書がほぼ完全に理解される。本明細書中の実施形態は、本発明の実施形態の例示であり、本発明の範囲を制限すると解釈すべきではない。当業者は、多数の他の実施形態が特許請求の範囲に含まれ、本明細書および実施例は例示のみを目的とし、本発明の真の範囲および精神を以下の特許請求の範囲に示すことが意図されることを認識する。
【技術分野】
【0001】
本願は、2002年10月22日提出の米国特許仮出願番号60/419,964号(その全体が本明細書中で参考として援用される)の優先権を主張する。
【0002】
(技術分野)
技術分野は、成長分化因子8(GDF−8)に対する抗体(特にヒト抗体)および抗体フラグメント(詳細には、in vitroおよび/またはin vivoでGDF−8活性を阻害するもの)に関する。分野は、さらに、筋肉もしくは骨の変性障害またはインスリン代謝障害の診断、予防、または治療方法に関する。
【背景技術】
【0003】
(背景)
ミオスタチンとしても公知の成長分化因子8(GDF−8)は、分泌タンパク質であり、構造的に関連する成長因子の形質転換成長因子β(TGF−β)スーパーファミリー(その全てが生理学的に重要な成長調節特性および形態形成特性を有する)のメンバ−である(Kingsley et al.(1994)Genes Dev.,8:133−146;Hoodless et al.(1998)Curr.Topics Microbiol.Immunol.,228:235−272)。TGF−βと同様に、 ヒトGDF−8は、375アミノ酸長前駆体タンパク質として合成される。前駆体GDF−8タンパク質は、ホモ二量体を形成する。プロセシング時に、アミノ末端プロペプチドは、Arg−266で切断される。「潜伏期関連ペプチド(latency−associated peptide)」(LAP)として公知の切断されたプロペプチドは、ホモ二量体への非共有結合を保持し、それにより複合体を不活化することができる(Miyazono et al.(1988)J.Biol.Chem.,263:6407−6415;Wakefield et al.(1988)J.Biol.Chem.,263:7646−7654;Brown et al.(1990)Growth Factors,3:35−43;およびThies et al.(2001)Growth Factors,18:251−259)。成熟GDF−8とプロペプチドとの複合体は、一般に、「小潜在複合体(small−latent complex)」と呼ばれる(Gentry et al.(1990)Biochemistry,29:6851−6857;Derynck et al.(1995)Nature,316:701−705;およびMassague(1990)Ann.Rev.Cell Biol.,12:597−641)。成熟GDF−8に結合してその生物活性を阻害する他のタンパク質も公知である。このような阻害タンパク質には、フォリスタチンおよびフォリスタチン関連タンパク質が含まれる(Gamer et al.(1999)Dev.Biol.,208:222−232)。
【0004】
種々の種由来の推定アミノ酸配列のアラインメントにより、進化を通してGDF−8が高度に保存されていることが証明されている(McPherron et al.(1997)Proc.Nat.Acad.Sci.U.S.A.,94:12457−12461)。実際、ヒト、マウス、ラット、ブタ、およびニワトリのGDF−8配列はC末端領域が100%同一であり、ヒヒ、ウシ、およびヒツジでは3アミノ酸しか異ならない。ゼブラフィッシュGDF−8が最も異なるが、以前としてヒトと88%同一である。
【0005】
この保存の高さにより、GDF−8は不可欠な機能を有することが示唆される。GDF−8は、発達中および成体の骨格筋で高度に発現し、筋肉および骨形成における重要な生体プロセスの調節に関与することが見いだされた。たとえば、GDF−8ノックアウトトランスジェニックマウスは、骨格筋量の顕著な肥大および過形成(McPherron et al.(1997)Nature,387:83−90)および皮質骨構造の変化(Hamrick et al.(2000)Bone,27(3):343−349)によって特徴づけられる。骨格筋の類似の増大は、ウシのGDF−8の天然に存在する変異で明らかである(Ashmore et al.(1974)Growth,38:501−507;Swatland et al.(1994)J.Anim.Sci.,38:752−757;McPherron et al.(1997)Proc.Nat.Acad.Sci.U.S.A.,94:12457−12461;およびKambadur et al.(1997)Genome Res.,7:910−915)。研究により、HIV感染に関連する筋肉の消耗に伴ってGDF−8発現が増大することが示されている(Gonzalez−Cadavid et al.(1998)Proc.Nat.Acad.Sci.U.S.A.,95:14938−14943)。GDF−8はまた、筋肉特異的酵素(例えば、クレアチンキナーゼ)の産生および筋芽細胞の増殖(WO 00/43781)に関与する。その成長調節および形態形成特性に加えて、GDF−8は、多数の他の生理学的プロセス(2型糖尿病の発症におけるグルコースホメオスタシス、耐糖能障害、代謝症候群(例えば、X症候群)、外傷によるインスリン抵抗性(火傷または窒素不均衡など)、および脂肪組織障害(例えば、肥満症)が含まれる)にも関与すると考えられる(Kim et al.(2001)BBRC,281:902−906)。
【0006】
多数のヒトおよび動物の障害が、機能障害筋肉組織に関連する(例えば、筋ジストロフィー(デュシェンヌ型筋ジストロフィーが含まれる)、筋萎縮性側索硬化症(ALS)、筋萎縮症、組織萎縮症、脆弱性、鬱血性閉塞性肺疾患、サルコペニア、悪液質、ならびに他の疾患および病態に起因する筋消耗症候群)。今日まで、これらの障害を治療するための信頼がおける、又は有効な治療法はほとんどなかった。
【0007】
特に高齢者および/または閉経後の女性における骨量の低下に関連する病態(骨粗鬆症または変形性関節症が含まれる)も多数存在する。さらに、代謝性骨疾患および障害には、慢性糖質コルチコイド療法に起因する低骨量、若年性性腺不全、アンドロゲン抑制、ビタミンD欠損症、二次性副甲状腺機能亢進症、栄養失調、および拒食症が含まれる。これらの病態のための現在利用可能な治療法は、骨吸収の阻害によって作用する。これらの治療法に代わる新規の骨形成を促進する治療法が望ましい。
【0008】
したがって、特にヒトにおける筋肉量および/または強度および/または骨密度の全体的増加に寄与する新規の治療法を開発する必要がある。
【発明の概要】
【発明が解決しようとする課題】
【0009】
(要旨)
本発明の1つの目的は、筋肉および/または骨関連障害のための安全且つ有効な治療方法を提供することである。
【0010】
本発明の別の目的は、脊椎動物の筋肉量および/または骨の強度および/または密度を増大させる方法を提供することである。
【0011】
本発明のなおさらなる目的は、in vivoで安全且つ有効なGDF−8のインヒビターを提供することである。
【0012】
本発明のさらに別の目的は、高い特異性および親和性でGDF−8と結合するヒト抗体およびそのフラグメントを提供することである。
【課題を解決するための手段】
【0013】
したがって、筋肉および骨の変性障害の治療方法を提供する。方法はまた、正常な動物の筋肉量および骨密度の増加に有用である。Myo29、Myo28、およびMyo22と呼ばれる新規のヒト抗GDF−8抗体およびこれ由来の抗体および抗原結合フラグメントも提供する。本発明の抗体は、多数の有用な特性を有する。第1に、本抗体は、高親和性で成熟GDF−8と結合することができる。第2に、本開示の抗体は、証明するように、例えば、ActRIIB結合の阻害およびレポーター遺伝子アッセイによってin vitroおよびin vivoでGDF−8活性を阻害する。第3に、本開示の抗体は、骨格筋の量および骨密度の負の調節に関連するGDF−8活性を阻害することができる。
本発明は、例えば以下の項目を提供する。
(項目1)
実質的に配列番号n(nは2、4、6、8、10、12、14、16、18、20、22、24、26、28、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、または48である)に記載のアミノ酸配列を含む単離抗体において、前記抗体がGDF−8またはBMP−11と特異的に結合することができる、単離抗体。
(項目2)
配列番号n(nは、2、4、6、8、10、12、14、16、18、20、22、24、26、28、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、または48である)のアミノ酸配列を含む、項目1に記載の抗体。
(項目3)
前記抗体が、ATCC受託番号PTA−4741、PTA−4740、またはPTA−4739のE.coliによって発現するscFvフラグメントである、項目1に記載の抗体。
(項目4)
前記抗体が、配列番号54に記載のアミノ酸配列を含むタンパク質と特異的に結合することができる、項目1に記載の抗体。
(項目5)
互いに独立して少なくとも(a)配列番号54のN末端由来の第2のアミノ酸がメチオニンであるか、(b)N末端由来の第3のアミノ酸がセリンであるか、(c)N末端由来の第5のアミノ酸がイソロイシンである、項目4に記載の抗体。
(項目6)
前記抗体がヒトである、項目1に記載の抗体。
(項目7)
前記抗体がIgG1またはIgG4である、項目1に記載の抗体。
(項目8)
前記抗体のアミノ酸配列が、エフェクター機能を減少または変化させるように修飾された、項目1に記載の抗体。
(項目9)
前記アミノ酸配列が、配列番号53のアミノ酸117またはアミノ酸120に対応する残基で修飾された、項目8に記載の抗体。
(項目10)
前記抗体がIgG1λまたはIgG1κである、項目1に記載の抗体。
(項目11)
項目1に記載の抗体を含む、薬学的組成物。
(項目12)
有効用量の項目11に記載の薬学的組成物と投与する工程を含む、治療方法。
(項目13)
前記薬学的組成物を、筋肉障害、神経筋障害、および骨変性障害から選択される障害の治療または防止が必要な哺乳動物に投与する、項目12に記載の方法。
(項目14)
前記薬学的組成物を、筋ジストロフィー、デュシェンヌ型筋ジストロフィー、筋萎縮症、組織萎縮症、手根管症候群、鬱血性閉塞性肺疾患、サルコペニア、悪液質、筋消耗症候群、および筋萎縮性側索硬化症から選択される障害の治療または防止が必要な哺乳動物に投与する、項目12に記載の方法。
(項目15)
前記薬学的組成物をデュシェンヌ型筋ジストロフィーの治療または防止が必要な哺乳動物に投与する、項目12に記載の方法。
(項目16)
前記薬学的組成物を、肥満症および脂肪組織障害から選択される障害の治療または防止が必要な哺乳動物に投与する、項目12に記載の方法。
(項目17)
前記薬学的組成物を、X症候群、グルコース寛容減損、外傷誘導性インスリン抵抗性、および2型糖尿病から選択される障害の治療または防止が必要な哺乳動物に投与する、項目12に記載の方法。
(項目18)
前記薬学的組成物を、2型糖尿病の治療または防止が必要な哺乳動物に投与する、項目12に記載の方法。
(項目19)
前記薬学的組成物を、肥満症の治療または防止が必要な哺乳動物に投与する、項目12に記載の方法。
(項目20)
前記薬学的組成物を、損傷した筋肉の修復が必要な哺乳動物に投与する、項目12に記載の方法。
(項目21)
前記損傷した筋肉が心筋である、項目21に記載の方法。
(項目22)
前記損傷した筋肉が横隔膜である、項目21に記載の方法。
(項目23)
前記抗体を、1μg/kg〜150mg/kg、1μg/kg〜100mg/kg、1μg/kg〜50mg/kg、1μg/kg〜20mg/kg、1μg/kg〜10mg/kg、1μg/kg〜1mg/kg、10μg/kg〜1mg/kg、10μg/kg〜100μg/kg、100μg〜1mg/kg、および500μg/kg〜1mg/kgから選択される有効用量で投与する、項目12に記載の方法。
(項目24)
項目1に記載の抗体をコードする単離核酸。
(項目25)
項目24に記載の核酸を含む、発現ベクター。
(項目26)
項目25に記載のベクターを含む、宿主細胞。
(項目27)
前記宿主細胞が、ATCC受託番号PTA−4741、PTA−4740、またはPTA−4739のE.coliである、項目26に記載の宿主細胞。
(項目28)
前記核酸が、配列番号n(nは、1、3、5、7、9、11、13、15、17、19、21、23、25、27、または29である)のヌクレオチド配列を含む、項目24に記載の核酸。
(項目29)
GDF−8と特異的に反応する抗体の作製方法において、
(a)置換すべきCDR3を含むかCDR3コード領域を欠く可変ドメインをコードする核酸の出発レパートリーを得る工程と
(b)可変ドメインをコードする核酸の産物レパートリーを得るために前記レパートリー中のCDR3領域にドナ−核酸が挿入されるように前記レパートリーと実質的に配列番号n(nは31〜48の整数である)に記載のアミノ酸配列をコードするドナ−核酸とを組み合わせる工程と
(c)前記産物レパートリーの核酸を発現する工程と、
(d)GDF−8に特異的な特異的抗原結合フラグメントを選択する工程と、
(e)前記特異的抗原結合フラグメントまたは前記結合フラグメントをコードする核酸を回収する工程とを含む、方法。
(項目30)
項目29に記載の方法によって産生された抗体。
(項目31)
(a)項目1に記載の抗体およびGDF−8を含む第1の結合混合物を調製する工程と、
(b)該第1の混合物中の該抗体と該GDF−8との間の結合量を測定する工程と、
(c)抗体、GDF−8、試験化合物を含む第2の結合混合物を調製する工程と、
(d)該第2の混合物中の該抗体と該GDF−8との間の結合量を測定する工程とを含む、GDF−8のインヒビターの同定方法。
(項目32)
治療有効量の項目1に記載の抗体を哺乳動物に投与し、それにより筋肉の強度または量が増大する工程を含む、筋肉の強度または量を増大させる方法。
(項目33)
前記抗体が、GDF−8のActRIIBへの結合を阻害することができる、GDF−8に対する単離抗体。
(項目34)
実質的に配列番号n(nは、2、4、6、8、10、12、14、16、18、20、22、24、26、28、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、または48である)に記載のアミノ酸配列を含む、項目33に記載の抗体。
(項目35)
配列番号n(nは、2、4、6、8、10、12、14、16、18、20、22、24、26、28、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、または48である)に記載のアミノ酸配列を含む、項目33に記載の抗体。
(項目36)
治療有効量の項目33に記載の抗体を哺乳動物に投与し、それにより筋肉の強度が増大する工程を含む、筋肉の強度を増大させる方法。
(項目37)
前記抗体が、BMP−11と特異的に結合することができる、項目33に記載の抗体。
(項目38)
ATCC受託番号PTA−4741、PTA−4740、またはPTA−4739のE.coliを培養する工程と、抗体を回収する工程を含む、抗体の作製方法。
(項目39)
Myo29、Myo28、またはMyo22のsvFvをコードする核酸を免疫グロブリンのFc部分をコードする核酸と融合する工程と、細胞中で前記融合核酸を発現させる工程とをさらに含む、項目38に記載の方法。
(項目40)
生殖系列化(germlining)する工程を含む、項目39に記載の方法。
(項目41)
項目40に記載の方法を使用して作製された抗体。
(項目42)
配列番号54に記載のアミノ酸配列によって特徴づけられたエピトープと特異的に結合することができる抗体。
(項目43)
哺乳動物の筋肉、骨、またはグルコースホメオスタシスの少なくとも1つの障害の治療または防止のための薬物の調製のための項目1〜項目10、項目31、項目34、項目35、項目36、項目38、項目41、および項目42のいずれか1項に記載の抗体の使用。
(項目44)
前記哺乳動物がヒトである、項目43に記載の使用。
(項目45)
前記障害が神経筋障害である、項目43に記載の使用。
(項目46)
前記障害が、筋ジストロフィー、デュシェンヌ型筋ジストロフィー、筋萎縮症、組織萎縮症、手根管症候群、鬱血性閉塞性肺疾患、サルコペニア、悪液質、筋消耗症候群、または筋萎縮性側索硬化症である、項目43に記載の使用。
(項目47)
前記障害が肥満症または脂肪組織障害である、項目43に記載の使用。
(項目48)
前記障害がX症候群、グルコース寛容減損、外傷誘導性インスリン抵抗性、または2型糖尿病である、項目43に記載の使用。
(項目49)
(a)筋肉損傷の修復、(b)筋肉の量または強度の増大、および(c)哺乳動物におけるグルコース耐性の増大の少なくとも1つのための薬物の調製のための項目1〜項目10、項目31、項目34、項目35、項目36、項目38、項目41、および項目42のいずれか1項に記載の抗体の使用。
(項目50)
前記(a)の損傷した筋肉が、心筋または横隔膜である、項目49に記載の使用。
(項目51)
前記抗体を、1μg/kg〜150mg/kg、1μg/kg〜100mg/kg、1μg/kg〜50mg/kg、1μg/kg〜20mg/kg、1μg/kg〜10mg/kg、1μg/kg〜1mg/kg、10μg/kg〜1mg/kg、10μg/kg〜100μg/kg、100μg〜1mg/kg、および500μg/kg〜1mg/kgから選択される有効用量で投与する、項目46〜51のいずれか1項に記載の使用。
【0014】
本発明の一定の実施形態は、Myo29、Myo28、またはMyo22のFvフラグメントのVHおよび/またはVLドメインを含む。さらなる実施形態は、これらの任意のVHおよびVLドメインの1つまたは複数の相補性決定領域(CDR)を含む。他の実施形態は、Myo29、Myo28、またはMyo22のVHドメインのH3フラグメントを含む。
【0015】
他の態様は、本発明の抗体またはその抗原結合フラグメントを含む組成物およびGDF−8の阻害または中和方法(ヒトまたは動物の治療方法が含まれる)でのその使用を提供する。本発明の抗体を使用して、筋組織または骨密度の増加が望ましい病態を治療または予防することができる。例えば、本発明で開示の抗体を、損傷した筋肉(例えば、心筋、横隔膜など)を修復する治療法で使用することができる。疾患および障害の例には、筋ジストロフィー(デュシェンヌ型筋ジストロフィーが含まれる);筋萎縮性側索硬化症;筋萎縮症;組織萎縮症;脆弱性;管症候群;鬱血性閉塞性肺疾患;サルコペニア,悪液質、および他の筋肉消耗症候群などの筋障害および神経筋障害;脂肪組織障害(例えば、肥満症);2型糖尿病;耐糖能障害;代謝症候群(例えば、X症候群);外傷によるインスリン抵抗性(火傷または窒素不均衡など);および骨変性疾患(例えば、変形性関節症および骨粗鬆症)が含まれる。
【0016】
さらに、本発明で開示の抗体を、生体サンプル中のGDF−8またはそのフラグメントを定量的または定性的に検出するための診断ツ−ルとして使用することができる。GDF−8の存在または検出量は、上記に列挙した1つまたは複数の医学的症状に相関し得る。
【0017】
別の態様は、Myo29、Myo28、またはMyo22のFvフラグメント由来のVHまたはVLドメインをコードする配列を含む単離核酸を提供する。任意の本発明で開示のVHおよびVLドメイン由来の少なくとも1つのCDRをコードする配列を含む単離核酸も開示する。別の態様は、このような核酸を含む宿主細胞を提供する。
【0018】
さらに別の態様は、Myo29、Myo28、またはMyo22の新規のVHおよびVLドメインおよび/またはVHおよび/またはVLドメイン由来のドメインの全てまたは一部を含む機能的抗体の産生方法を提供する。
【0019】
本発明のさらなる目的を以下の説明に一部記載し、且つこれらの一部は説明から明らかであるか、本発明の実施によって得ることができる。本発明の種々の目的、態様、および利点は、添付の特許請求の範囲に特に指摘されている要素および組み合わせによって実現および達成される。
上記の一般的説明および以下の詳細な説明の両方は例示および説明のみを目的とし、特許請求の範囲のように本発明を制限しないと理解すべきである。
【図面の簡単な説明】
【0020】
【図1】図1は、ビオチン化GDF−8およびBMP−11がそれぞれ15ng/mlおよび40ng/mlのED50でActRIIB受容体と結合することを示す。
【図2】図2は、本発明のscFvフラグメントによるGDF−8のActRIIB受容体への結合の阻害を示す。例示するように、Myo29、Myo28、およびMyo22のscFvのIC50は、それぞれ2.4nM、1.7nM、および60nMである。
【図3A】図3Aおよび3Bは、Myo29の10ng/mlのビオチン化GDF−8またはBMP−11とのプレインキュベーションにより、ActRIIB結合アッセイにおいて0.2〜0.4nMのIC50でGDF−8またはBMP−11のActRIIBへの結合を阻害することを示す。
【図3B】図3Aおよび3Bは、Myo29の10ng/mlのビオチン化GDF−8またはBMP−11とのプレインキュベーションにより、ActRIIB結合アッセイにおいて0.2〜0.4nMのIC50でGDF−8またはBMP−11のActRIIBへの結合を阻害することを示す。
【図4A】図4Bおよび4Cは、Myo29を試験したpGL3(CAGA)12受容体遺伝子アッセイの結果を示す。図4Aは、ベースライン条件(すなわち、GDF−8、BMP−11、およびアクチビンによるレポーター遺伝子活性の誘導)を証明する。図4Bおよび4Cは、Myo29により用量反応様式にて15〜30ng/mlのIC50でGDF−8活性を減少し、BMP−11の生物活性を同一の程度に阻害することを示す。図4Dは、このアッセイではMyo29がアクチビン活性に影響を与えないことを例示する。
【図4B】図4Bおよび4Cは、Myo29を試験したpGL3(CAGA)12受容体遺伝子アッセイの結果を示す。図4Aは、ベースライン条件(すなわち、GDF−8、BMP−11、およびアクチビンによるレポーター遺伝子活性の誘導)を証明する。図4Bおよび4Cは、Myo29により用量反応様式にて15〜30ng/mlのIC50でGDF−8活性を減少し、BMP−11の生物活性を同一の程度に阻害することを示す。図4Dは、このアッセイではMyo29がアクチビン活性に影響を与えないことを例示する。
【図4C】図4Bおよび4Cは、Myo29を試験したpGL3(CAGA)12受容体遺伝子アッセイの結果を示す。図4Aは、ベースライン条件(すなわち、GDF−8、BMP−11、およびアクチビンによるレポーター遺伝子活性の誘導)を証明する。図4Bおよび4Cは、Myo29により用量反応様式にて15〜30ng/mlのIC50でGDF−8活性を減少し、BMP−11の生物活性を同一の程度に阻害することを示す。図4Dは、このアッセイではMyo29がアクチビン活性に影響を与えないことを例示する。
【図4D】図4Bおよび4Cは、Myo29を試験したpGL3(CAGA)12受容体遺伝子アッセイの結果を示す。図4Aは、ベースライン条件(すなわち、GDF−8、BMP−11、およびアクチビンによるレポーター遺伝子活性の誘導)を証明する。図4Bおよび4Cは、Myo29により用量反応様式にて15〜30ng/mlのIC50でGDF−8活性を減少し、BMP−11の生物活性を同一の程度に阻害することを示す。図4Dは、このアッセイではMyo29がアクチビン活性に影響を与えないことを例示する。
【図5】図5は、Myo22、Myo28、およびMyo29のエピトープマッピングの結果を示す。Myo29のエピトープを、成熟GDF−8のアミノ酸72からアミノ酸88までをマッピングし、Myo22については、アミノ酸1からアミノ酸44までをマッピングし、Myo28については、アミノ酸1からアミノ酸98までをマッピングした。
【図6】図6は、Myo29エピトープの置換分析の結果を示す。成熟GDF−8中の残基Lys−78、Pro−81、およびAsn−83は、Myo29のGDF−8への結合に重要なようである。
【図7】図7は、Myo29およびMyo28を使用して実施した免疫沈降実験の結果を示す。35S−メチオニン/システインで放射性標識したGDF−8を発現するCHO細胞由来の馴化培地を、Myo29またはMyo28での免疫沈降に供した。次いで、還元条件下でのSDS−PAGEによって免疫沈降物を分析した。ゲル上のバンドを、成熟GDF−8、GDF−8プロペプチド、および非プロセシングGDF−8と同定した。
【図8】図8は、Myo29の単回静脈内(IV)または腹腔内(IP)投与として1mg/kgの用量をC57B6/SCIDマウスに投与する薬物動態学研究の結果を示す。Myo29は、約1週間の長期末端半減期および約1ml/時間/kgの低クリアランスを示す。IP注射後に吸収された画分は約77%である。
【図9】図9は、種々の用量のMyo29(60、10、および1mg/kg)または賦形剤(PBS)で毎週処置した雄C57B6/SCIDマウスにおける大腿四頭筋量の比較を示す。10および60mg/kgの用量レベルで4週間のMyo29での処置により、筋肉量がそれぞれ19%および23%統計的に有意に増加する。
【図10A】図10Aおよび10Bは、種々の用量のMyo29(10、5、2.5、および1mg/kg)またはPBSで4週間毎週処置した雌CB17 SCIDマウスにおける腓腹筋量および大腿四頭筋量を示す。筋肉量は、賦形剤コントロールと比較してMyo29で処置したマウスで10%〜20%増加する。
【図10B】図10Aおよび10Bは、種々の用量のMyo29(10、5、2.5、および1mg/kg)またはPBSで4週間毎週処置した雌CB17 SCIDマウスにおける腓腹筋量および大腿四頭筋量を示す。筋肉量は、賦形剤コントロールと比較してMyo29で処置したマウスで10%〜20%増加する。
【図11A】図11Aおよび11Bは、種々の用量のMyo29(10、5、2.5、および1mg/kg)またはPBSで12週間毎週処置した雌CB17 SCIDマウスにおける腓腹筋量および大腿四頭筋量をそれぞれ示す。Myo29で処置したマウスの筋肉量は12%〜28%の範囲で増加する。
【図11B】図11Aおよび11Bは、種々の用量のMyo29(10、5、2.5、および1mg/kg)またはPBSで12週間毎週処置した雌CB17 SCIDマウスにおける腓腹筋量および大腿四頭筋量をそれぞれ示す。Myo29で処置したマウスの筋肉量は12%〜28%の範囲で増加する。
【図12】図12は、Myo29(10および5mg/kg)またはPBSで12週間毎週処置した雌CB17 SCIDマウスにおける握力計によって測定した前肢の筋肉の強度を示す。前肢強度は、5mg/kgおよび10mg/kgのMyo29で処置したマウスでそれぞれ17%および23%増加する。
【発明を実施するための形態】
【0021】
(詳細な説明)
(1.定義)
本明細書中で使用される、用語「抗体」は、免疫グロブリンまたはその一部をいい、その供給源、起源となる種、産生方法、および特徴と無関係に抗原結合部位を含む任意のポリペプチドを含む。非限定的な例として、用語「抗体」には、ヒト、オランウータン、マウス、ラット、ヤギ、ヒツジ、およびニワトリの抗体が含まれる。この用語には、ポリクローナル抗体、モノクローナル抗体、単一特異性抗体、多重特異性抗体、非特異性抗体、ヒト化抗体、単鎖抗体、キメラ抗体、合成抗体、組換え抗体、ハイブリッド抗体、変異抗体、およびCDRグラフティング抗体が含まれるが、これらに限定されない。本発明の目的のために、他で記載しない限り、Fab、F(ab’)2、Fv、scFv、Fd、dAbなどの抗体フラグメントおよび抗原結合機能を保持した他の抗体フラグメントも含まれる。
【0022】
抗体を、例えば、伝統的なハイブリドーマ技術(Kohler and Milstein(1975)Nature,256:495−499)、組換えDNA法(米国特許第4,816,567号)、または抗体ライブラリーを使用したファージディスプレイ技術(Clackson et al.(1991)Nature,352:624−628;Marks et al.(1991)J.Mol.Biol.,222:581−597)によって作製することができる。種々の他の抗体産生技術については、Antibodies:A Laboratory Manual,eds.Harlow et
al.,Cold Spring Harbor Laboratory,1988を参照のこと。
【0023】
用語「抗原結合ドメイン」は、抗原の一部または全部と特異的に結合するか相補的である領域を含む抗体分子の一部をいう。抗原が巨大な場合、抗体は抗原の特定の部分のみと結合することができる。「エピトープ」または「抗原決定基」は、抗体の抗原結合ドメインとの特異的相互作用を担う抗原分子の一部である。1つまたは複数の抗体可変ドメイン(例えば、いわゆるVHドメインからなるFd抗体フラグメント)によって抗原結合ドメインを得ることができる。抗原結合ドメインは、抗体軽鎖可変領域(VL)および抗体重鎖可変領域(VH)を含む。
【0024】
用語「レパートリー」は、発現した免疫グロブリンをコードする配列の全体または一部に由来するヌクレオチド(例えば、DNA)配列の遺伝的に多様な集団をいう。例えば、H鎖についてはV、D、およびJセグメント、L鎖についてはVおよびJセグメントのin vivo再編成によって配列を作製する。あるいは、in vitro刺激および再編成が起こる応答によって細胞株から配列を作製することができる。あるいは、例えば、非再編成VセグメントとDおよびJセグメントとの組み合わせ、ヌクレオチド合成、無作為化変異誘発、および米国特許第5,565,332号に開示の他の方法によって配列の一部または全部を得ることができる。
【0025】
用語「特異的相互作用」または「特異的に結合する」などは、2つの分子が生理学的条件下で比較的安定な複合体を形成することを意味する。この用語はまた、例えば、抗原結合ドメインが多数の抗原によって保有される特定のエピトープに特異的である場合に適用可能であり、この場合、抗原結合ドメインを保有する抗体はエピトープを保有する種々の抗原に結合することができる。したがって、抗体は、例えば、その両方が保有するエピトープに結合する限り、BMP−11およびGDF−8と特異的に結合することができる。
【0026】
高親和性および低〜中程度の能力によって特異的結合を特徴づける。非特異的結合は、通常、親和性が低く、中程度から高い能力を有する。典型的には、親和性定数Kaが106M−1より高い、好ましくは108M−1より高い場合、結合は特異的と見なされる。必要に応じて、結合条件の変化によって実質的に特異的結合に影響を与えることなく非特異的結合を減少させることができる。このような条件は当該分野で公知であり、当業者は日常的技術を使用して適切な条件を選択することができる。条件は、通常、抗体濃度、溶液のイオン強度、温度、結合時間、非関連分子(例えば、血清アルブミン、ミルクカゼイン)の濃度などに関して定義される。条件の例を、実施例4、7、および10に示す。
【0027】
句「実質的に記載の」は、関連するCDR、VH、またはVLドメインが記載の配列の特定の領域と同一であるか類似性が高いことを意味する。例えば、このような置換には、CDR(H1、H2、H3、L1、L2、またはL3)の配列中の任意の5個のアミノ酸のうちの1個または2個が含まれる。
【0028】
用語「TGF−βスーパーファミリー」は、構造的に関連した成長因子のファミリ−をいう。関連する成長因子のこのファミリ−は、当該分野で周知である(Kingsley
et al.(1994)Genes Dev.,8:133−146;Hoodless et al.(1998)Curr.Topics Microbiol.Immunol.,228:235−72)。TGF−βスーパーファミリーには、骨形態形成タンパク質(BMP)、アクチビン、インヒビン、ミューラー阻害物質、グリア由来の神経栄養因子、以前として増加している成長分化因子(GDF)(GDF−8(ミオスタチン)など)が含まれる。多数のこのようなタンパク質は、構造的および/または機能的にGDF−8と関連する。例えば、GDF−11としても公知のヒトBMP−11は、アミノ酸レベルでGDF−8と90%同一である(Gamer et al.(1999)Dev.Biol.208,222−232;Nakshima et al.(1999)Mech.Dev.,80:185−189)。
【0029】
用語「GDF−8」は、特異的な成長分化因子−8をいい、必要に応じて、GDF−8と構造的または機能的に関連する因子(例えば、BMP−11およびTGF−βスーパーファミリーに属する他の因子)をいう。この用語は、GDF−8の全長非プロセシング前駆体形態ならびに翻訳後切断に起因する成熟およびプロペプチド形態をいう。この用語はまた、本明細書中で考察された成熟GDF−8に関連する少なくともいくつかの生物活性を保持するGDF−8の任意のフラグメントおよび変異型(修飾された配列が含まれる)をいう。成熟ヒトGDF−8のアミノ酸配列を、配列番号49に提供する。本発明は、全ての脊椎動物種(ヒト、ウシ、ニワトリ、マウス、ラット、ブタ、ヒツジ、シチメンチョウ、ヒヒ、および魚類が含まれるが、これらに限定されない)由来のGDF−8に関する(配列情報については、例えば、McPherron et al.(1997)Proc.Nat.Acad.Sci.U.S.A.,94:12457−12461を参照のこと)。
【0030】
用語「成熟GDF−8」は、GDF−8前駆体タンパク質のカルボキシル末端ドメインから切断したタンパク質をいう。成熟GDF−8は、単量体、ホモ二量体として存在するか、GDF−8潜在複合体中に存在し得る。条件によって、成熟GDF−8は、任意または全てのこれらの異なる形態の間で平衡になり得る。その生物活性形態では、成熟GDF−8は、「活性GDF−8」ともいう。
【0031】
用語「GDF−8プロペプチド」は、GDF−8前駆体タンパク質のアミノ末端ドメインから切断されたポリペプチドをいう。GDF−8プロペプチドは、成熟GDF−8上のプロペプチド結合ドメインと結合することができる。
【0032】
用語「GDF−8潜在複合体」は、成熟GDF−8ホモ二量体とGDF−8プロペプチドとの間で形成されたタンパク質複合体をいう。2つのGDF−8プロペプチドがホモ二量体中の成熟GDF−8の2分子と会合して不活性四量体複合体を形成すると考えられている。潜在複合体は、1つまたは複数のGDF−8プロペプチドの代わりまたはそれに加えて他のGDF−8インヒビターを含み得る。
【0033】
用語「GDF−8活性」は、活性GDF−8タンパク質に関連する1つまたは複数の生理学的成長調節活性または形態形成活性をいう。例えば、活性GDF−8は、骨格筋量の負のレギュレーターである。活性GDF−8はまた、筋肉特異的酵素(例えば、クレアチンキナーゼ)の産生を調整し、筋芽細胞増殖を刺激し、前脂肪細胞の脂肪細胞への分化を調整することができる。in vivoおよびin vitroでのGDF−8活性の測定手順の例を、実施例2、3、6、および13に記載する。
【0034】
用語「GDF−8インヒビター」には、GDF−8の活性、発現、プロセシング、または分泌を阻害することができる任意の薬剤が含まれる。このようなインヒビターには、タンパク質、抗体、ペプチド、ペプチド模倣物、リボザイム、アンチセンスオリゴヌクレオチド、二本鎖RNA、およびGDF−8を特異的に阻害する他の小分子が含まれる。このようなインヒビターは、GDF−8の生物活性を「阻害」、「中和」、または「減少」すると言われる。
【0035】
用語「中和する」、「中和」、「阻害」、およびその同族語は、GDF−8インヒビターの非存在下でのGDF−8活性と比較したGDF−8インヒビターによるGDF−8活性の減少をいう。活性の減少は、好ましくは、少なくとも約10%、20%、30%、40%、50%、60%、70%、80%、90%、またはそれ以上である。
【0036】
用語「治療」は、本明細書中の用語「治療方法」と交換可能に使用され、治療上の処置および予防(prophylactic)/予防(preventative)手段の両方をいう。治療を必要とする者には、特定の内科的疾患を既に罹患している個体および最終的に障害を獲得し得る個体(すなわち、予防手段を必要とする個体)が含まれ得る。
【0037】
用語「単離された」は、その天然の環境を実質的に含まない分子をいう。例えば、単離タンパク質は、そのタンパク質が由来する細胞もしくは組織供給源由来の細胞物質または他のタンパク質を実質的に含まない。この用語は、単離タンパク質が治療組成物としての投与のために十分に純粋であるか、純度が少なくとも70%〜80%(w/w)、より好ましくは少なくとも80%〜90%(w/w)、さらにより好ましくは純度が90〜95%、最も好ましくは純度が少なくとも95%、96%、97%、98%、99%、または100%(w/w)である調製物をいう。
【0038】
用語「哺乳動物」は、哺乳動物として分類された任意の動物(ヒト、家畜、動物園の動物、競技用動物、またはペット動物(イヌ、ウマ、ネコ、ヒツジ、ブタ、ウシなど)が含まれる)をいう。
【0039】
用語「有効用量」または「有効量」は、患者の症状を改善するか所望の生物学的結果(例えば、骨格筋量および/または骨密度の増加)が得られる化合物の量をいう。このような量は、骨格筋量および骨密度の負の制御に関連するGDF−8活性の減少に十分なはずである。以下の節に記載のように有効量を決定することができる。
【0040】
(II.GDF−8に対する抗体および抗原結合フラグメント)
A.ヒト抗体Myo28、Myo29、およびMyo22
本開示は、GDF−8に対する新規の抗体およびその抗原結合フラグメントを提供する。このような抗体の非限定的な実施形態を、Myo29、Myo28、およびMyo22と呼ぶ。これらの例示的実施形態を、ヒトIgG1抗体の形態で提供する。
【0041】
本発明の抗体は、固有且つ有益な特徴を有する。第1に、これらの抗体は、高親和性で成熟GDF−8と結合することができる。第2に、本発明の抗体は、例えば、ActRIIB結合の阻害およびレポーター遺伝子アッセイによって証明されるように、in vitroおよびin vivoでGDF−8活性を阻害することができる。本発明の抗体はまた、例えば、ActRIIB結合の阻害およびレポーター遺伝子アッセイによって証明されるように、BMP−11に特異的に結合し、そして/またはその活性を阻害することができる。第3に、本開示の抗体は、骨格筋量および骨密度の負の制御に関連するGDF−8活性を阻害することができる。
【0042】
例示的実施形態では、本発明に開示の抗体は、GDF−8およびBMP−11の両方に特異的に結合することができる。抗体は他のタンパク質(例えば、TGF−βスーパーファミリーに属するもの(ミューラー阻害物質、グリア由来の神経栄養因子など)またはGDF−8以外の成長分化因子)とも反応することができることが意図される。一定の実施形態では、Myo29は、配列番号49のアミノ酸72〜88と同一の配列を含むタンパク質と反応する。さらなる実施形態では、Myo29は、配列Lys−Xaa1−Xaa2−Pro−Xaa3−Asn(配列番号54)(式中、Xaa1,Xaa2,およびXaa3はそれぞれ任意のアミノ酸である)を含むタンパク質と結合する。さらなる実施形態では、少なくとも1つの以下の条件を満たす:(1)Xaa1 = Met、(2)Xaa2=Ser、および(3)Xaa3 = Ile(全て互いに独立している)。他の実施形態では、Myo22は、成熟GDF−8配列中の最初の44個のN末端アミノ酸(配列番号49のアミノ酸1〜44)内のエピトープを認識する。
【0043】
当業者は、本発明の抗体を使用して、上記と異なるタンパク質を検出、測定、および阻害することができると認識する。一般に、本発明の抗体を、配列番号49に記載のGDF−8の成熟形態の配列中の少なくとも100個、80個、60個、40個、または20個の連続するアミノ酸の任意の配列と少なくとも約70%、80%、90%、95%、またはそれ以上同一である配列を含む任意のタンパク質とともに使用することができる。このようなタンパク質の非限定的な例には、本明細書中に記載の種々の種由来のGDF−8配列が含まれる。標準的なアラインメントアルゴリズム(例えば、Altschul et
al.(1990)J.Mol.Biol.,215:403−410に記載のBasic Local Alignment Tool(BLAST)、Needleman
et al.(1970)J.Mol.Biol.,48:444−453のアルゴリズム、またはMeyers et al.(1988)Comput.Appl.Biosci.,4:11−17のアルゴリズムなど)によって同一率を決定する。
【0044】
B.可変ドメイン
免疫グロブリンとしても公知のインタクトな抗体は、典型的には、それぞれ約25kDaの2つの軽鎖(L)およびそれぞれ約50kDaの2つの重鎖(H)から構成される四量体のグリコシル化タンパク質である。λおよびκと呼ばれる2つの軽鎖型が抗体中で見いだされる。重鎖の定常ドメインのアミノ酸配列に依存して、免疫グロブリンを以下の5つの主なクラス(A、D、E、G、およびM)に割り当てることができ、これらのいくつかをサブクラス(イソ型)(例えば、IgG1、IgG2、IgG3、IgG4、IgA1、およびIgA2)にさらに分類することができる。免疫グロブリンの異なるクラスのサブユニット構造および三次元高次構造は当該分野で周知である。抗体構造の概説については、Antibodies:A Laboratory Manual,Cold Spring Harbor Laboratory,eds.Harlow et al.,1988を参照のこと。簡単に述べれば、各軽鎖は、N末端可変(V)ドメイン(VL)および定常(C)ドメイン(CL)から構成される。各重鎖は、N末端Vドメイン、3つまたは4つのCドメイン、およびヒンジ領域から構成される。VHドメインに最も近いCHドメインを、CH1と命名する。VHおよびVLドメインは、超可変配列(相補性決定領域、CDR)の3つの領域のための足場を形成するフレームワーク領域(FR1、FR2、FR3、およびFR4)と呼ばれる比較的保存された配列の4つの領域からなる。CDRは、抗原との特異的相互作用を担うほとんどの残基を含む。CDRを、CDR1、CDR2、およびCDR3という。したがって、重鎖上のCDR構成要素を、H1、H2、およびH3といい、軽鎖上のCDR構成要素を、L1、L2、L3という。CDR3は、抗体結合部位内の分子多様性の最も巨大な供給源である。例えば、H3は、2アミノ酸残基ほどの短さであり得るか、26個より長くあり得る。最も小さな抗原結合フラグメントは、VHおよびVLドメインからなるFvである。Fabフラグメント(Fragment antigen binding)は、定常領域間のジスルフィド結合によって共有結合したVH−CH1およびVL−CLドメインからなる。宿主細胞中で同時発現した場合にFv中の非共有結合したVHドメインとVLドメインとが解離する傾向を克服するために、可動性があり且つ適切な長さのポリペプチドによりVHのC末端がVLのN末端と結合するかVLのC末端がVHのN末端と結合するいわゆる単鎖(sc)Fvフラグメント(scFv)を構築することができる。最も一般的に使用されているリンカーは、15残基の(Gly4Ser)3ペプチドであったが、他のリンカーも当該分野で公知である。
【0045】
可変領域をコードする複数の生殖系列遺伝子および種々の体細胞事象の使用によって抗体が多様化する。体細胞事象には、完全なVH領域を作製するための多様性(D)遺伝子セグメントおよび連結(J)遺伝子セグメントでの可変遺伝子セグメントの組換えならびに完全なVL領域を作製するための可変遺伝子セグメントおよび連結遺伝子セグメントの組換えが含まれる。組換えプロセス自体の不正確さにより、V(D)J連結点でアミノ酸が喪失または付加される。これらの多様性の機構は、抗原曝露前の発達中のB細胞で起こる。抗原刺激後、B細胞中の発現抗体遺伝子は体細胞変異を受ける。予測生殖系列遺伝子セグメント数、これらのセグメントのランダム組換え、およびランダムVH−VL対合に基づいて、1.6×107個までの異なる抗体を産生することができる(Fundamental Immunology,3rd ed.,ed.Paul,Raven Press,New York,NY,1993)。抗体多様性に寄与する他のプロセス(体細胞変異など)を考慮した場合、1×1010個以上の異なる抗体を作製することができると考えられる(Immunoglobulin Genes,2nd ed.,eds.Jonio et al.,Academic Press,San Diego,CA,1995)。抗体多様性の作製に関与する多数のプロセスにより、同一の抗原特異性を有する独立して誘導されたモノクローナル抗体が同一のアミノ酸配列を有する可能性は低い。
【0046】
したがって、本発明は、さらに、ヒト免疫グロブリン遺伝子ライブラリー由来の新規のCDRを提供する。本発明のCDRを保有するための構造は、一般に、天然に存在するVHおよびVLのCDRに対応する位置にCDRが存在する抗体重鎖配列もしくは軽鎖配列またはその実質的部分である。免疫グロブリン可変ドメインの構造および位置を、Sequences of Proteins of Immunological Interest,US Department of Health and Human Services,eds.Kabat et al.,1991に記載のように決定することができる。
【0047】
本発明に開示された抗体のDNAおよびアミノ酸(AA)配列、そのscFvフラグメント、VHおよびVLドメイン、ならびにCDRを配列表に記載し、表1に列挙する。便宜上、VHおよびVLドメイン内の各CDRの位置を表2に列挙する。VHおよびVLドメインを除く重鎖および軽鎖の配列は、Myo29、Myo28、およびMyo22で同一である。
【0048】
【表1】
【0049】
【表2】
本発明で開示された抗体は、抗体定常領域またはその一部をさらに含み得る。例えば、VLドメインのそのC末端を、抗体軽鎖定常ドメイン(ヒトCκ鎖またはCλ鎖が含まれ、好ましくはCλ鎖)に結合させることができる。同様に、VHドメインに基づいた特異的抗原結合フラグメントのそのC末端に、任意の抗体イソ型(例えば、IgG、IgA、IgE、およびIgM)および任意のイソ型サブクラス(特に、IgG1およびIgG4)由来の免疫グロブリン重鎖の全部または一部を結合させることができる。例示的実施形態では、抗体は、ヒトIgG1λのC末端フラグメントの重鎖および軽鎖を含む。軽鎖λのC末端フラグメントのDNA配列およびアミノ酸配列を、それぞれ配列番号50および配列番号51に記載する。IgG1重鎖のC末端フラグメントのDNA配列およびアミノ酸配列を、それぞれ配列番号52および配列番号53に記載する。
【0050】
本発明の一定の実施形態は、Myo29、Myo28、またはMyo22のFvフラグメントのVHおよび/またはVLドメインを含む。さらなる実施形態は、任意のこれらのVHおよびVLドメインの1つまたは複数の相補性決定領域(CDR)を含む。1つの実施形態は、Myo29、Myo28、またはMyo22のVHドメインのH3フラグメントを含む。一定の実施形態では、本発明のVHおよびVLドメインは、生殖系列化されている(すなわち、これらのドメインのフレームワーク領域(FR)が、ヒト生殖系列遺伝子産物のコンセンサスアミノ酸配列に適合するように従来の分子生物学技術を使用して変化している)。他の実施形態では、フレームワーク配列は、生殖系列と異なったままである。
【0051】
C.修飾抗体およびそのフラグメント
本発明のさらなる態様は、GDF−8に特異的な抗体抗原結合ドメインを得る方法を提供する。当業者は、本発明の抗体が表1に記載のVHおよびVLの特異的配列に限定されず、抗原結合能力を保持するこれらの配列の変異型も含まれることを認識する。このような変異型は、当該分野で公知の技術を使用して提供した配列に由来し得る。FRまたはCDRのいずれかのアミノ酸を、置換、欠失、または付加することができる。通常、抗体の安定性を改良して免疫原性を減少させるようにフレームワーク領域の変化をデザインする一方で、通常、抗体のその標的に対する親和性を増大させるようにCDRの変化をデザインする。このような親和性が増大する変化を、典型的に、CDR領域の変更および抗体の試験によって経験的に決定する。このような変更を、Antibody Engineering,2nd.ed.,ed.Borrebaeck,Oxford University Press,1995に記載の方法にしたがって行うことができる。
【0052】
本明細書中に記載のVHドメインのアミノ酸配列変異型であるVHドメインの作製方法は、本明細書中に開示のVHドメインのアミノ酸配列中の1つまたは複数のアミノ酸を付加、欠失、置換、または挿入する工程と、任意選択的にこのようにして得られたVHドメインと1つまたは複数のVLドメインと組み合わせる工程と、GDF−8への特異的結合についてVHドメインまたはVH/VL組み合わせを試験する工程と、任意選択的にこのような抗原結合ドメインがGDF−8活性を中和する能力を試験する工程とを含む。VLドメインは、実質的に本明細書中に記載のアミノ酸配列を有し得る。
【0053】
本明細書中に開示のVLドメインの1つまたは複数の配列変異型を1つまたは複数のVHドメインと組み合わせた類似の方法を使用することができる。
【0054】
本発明のさらなる態様は、GDF−8と特異的に反応する抗原結合フラグメントの調製方法を提供する。この方法は、
(a)置換すべきCDR3を含むかCDR3コード領域を欠くVHドメインをコードする核酸の出発レパートリーを得る工程と、
(b)VHドメインをコードする核酸の産物レパートリーを得るためにレパートリー中のCDR3領域にドナー核酸が挿入されるように前記レパートリーと実質的に本明細書中に記載のVHCDR3(すなわち、H3)のアミノ酸配列をコードするドナー核酸とを組み合わせる工程と、
(c)前記産物レパートリーの核酸を発現する工程と、
(d)GDF−8に特異的な特異的抗原結合フラグメントを選択する工程と、
(e)前記特異的抗原結合フラグメントまたはこれをコードする核酸を回収する工程とを含む。
【0055】
さらに、本発明のVLCDR3(すなわち、L3)を置換すべきCDR3を含むかCDR3コード領域を欠くVLドメインをコードする核酸レパートリーと組み合わせた類似の方法を使用することができる。
【0056】
本発明のコード配列CDR(例えば、CDR3)を、組換え(recombinant)DNAテクノロジ−を使用してCDR(例えば、CDR3)を欠く可変ドメインのレパートリーに移入することができる。例えば、Marks et al.(Bio/Technology(1992)10:779−783)は、可変ドメイン領域の5’末端に指向するか隣接するコンセンサスプライマーをヒトVH遺伝子の第3のフレームワーク領域に対するコンセンサスプライマーと組み合わせて使用してCDR3を欠くVH可変ドメインのレパートリーが得られる、抗体可変ドメインのレパートリーの産生方法を記載している。レパートリーを、特定の抗体のCDR3と組み合わせることができる。類似の技術を使用して、本発明のCDR3由来の配列を、CDR3を欠くVHまたはVLドメインのレパートリーで組み替え(shuffle)、組み替えられた完全なVHまたはVLドメインを同族VLまたはVHドメインと組み合わせて本発明の特異的抗原結合フラグメントを得ることができる。次いで、適切な抗原結合フラグメントを選択することができるように、レパートリーを、WO92/01047のファージディスプレイ系などの適切な宿主系にディスプレイすることができる。
【0057】
類似の組み替えまたは組替え技術は、Stemmer(Nature(1994)370:389−391)によっても開示されており、これはβ−ラクタマーゼ遺伝子に関する技術を記載しているが、このアプローチを抗体作製のために使用することができる。
【0058】
さらなる代替法は、全可変ドメイン内に変異を作製するための1つまたは複数の選択されたVHおよび/またはVL遺伝子のランダム変異誘発を使用した本発明のCDR由来の配列を保有する新規のVHまたはVL領域を作製することである。このような技術は、変異性PCRを使用したGram et al.(Proc.Nat.Acad.Sci.U.S.A.(1992)89:3576−3580)に記載されている。
【0059】
使用することができる別の方法は、VHまたはVL遺伝子のCDR領域に対する直接的変異誘発である。このような技術は、Barbas et al.(Proc.Nat.Acad.Sci.U.S.A.(1994)91:3809−3813)およびSchier et al.(J.Mol.Biol.(1996)263:551−567)に開示されている。
【0060】
同様に、1つまたは複数または3つ全てのCDRをVHまたはVLドメインのレパートリーにグラフティングし、その後GDF−8に特異的な特異的結合パートナーまたは結合フラグメントについてスクリーニングすることができる。
【0061】
免疫グロブリン可変ドメインの実質的部分は、少なくともCDR領域および選択的に本明細書中に記載のscFvフラグメント由来の介在フレームワーク領域を含む。この部分はまた、少なくとも約50%のFR1およびFR4のいずれかまたは両方(50%はFR1のC末端の50%であり、50%はFR4のN末端のである)を含む。可変ドメインの実質的部分のN末端またはC末端のさらなる残基は、通常は天然に存在する可変ドメイン領域に関連しない残基であり得る。例えば、組換えDNA技術によって作製された本発明の特異的抗原結合フラグメントの構築によって、導入されたリンカーによってコードされるN末端またはC末端残基が導入されて、クローニングまたは他の操作工程を容易にすることができる。他の操作工程は、本発明の可変ドメインを免疫グロブリン重鎖を含むさらなるタンパク質配列、(例えば、ダイアボディ(diabody)の産生における)他の可変ドメイン、または以下により詳細に記載したタンパク質標識に連結するためのリンカーの導入を含む。
【0062】
実施例に例示の実施形態はVHおよびVLドメインの「マッチング」対を含んでいるが、本発明は、VHまたはVLドメイン配列のいずれか(特に、VHドメイン)由来の1つの可変ドメインを含む結合フラグメントも含む。一本鎖特異的結合ドメインのいずれかの場合、これらのドメインを使用して、GDF−8に結合することができる2ドメイン特異的抗原結合ドメインを形成することができる相補性ドメインについてスクリーニングすることができる。HまたはL鎖クローンのいずれかを含む各コロニーを使用して他の鎖(LまたはH)をコードするクローンの完全なライブラリーに感染させ、WO92/01047に開示されるファージディスプレイ技術などにしたがって、得られた二鎖特異的抗原結合ドメインを選択する、WO92/01047に開示のいわゆる階層的二重組み合わせアプローチを使用したファージディスプレイスクリーニング法によってこれを行うことができる。この技術は、Marks et al.,supraにも開示されている。
【0063】
放射性核種、薬物、高分子、または他の薬剤を使用した化学的方法によって抗体を抱合することができるか、1つまたは複数の本発明のCDRを含む融合タンパク質として作製することができる。
【0064】
抗体融合タンパク質は、VH−VL対を含み、これらの鎖の1つ(通常VH)および別のタンパク質を1つのポリペプチド鎖として合成する。これらの産物型は、一般にさらなる機能的エレメント(小分子の活性部分)を有するかまたは抱合または融合高分子の主な分子構造の特徴を有するという点で抗体と異なる。
【0065】
上記概説のアミノ酸配列の変化に加えて、抗体を、グリコシル化するか、ペグ化するか、アルブミンもしくは非タンパク質性ポリマーに結合させることができる。例えば、米国特許第4,640,835号;同第4,496,689号;同第4,301,144号;同第4,670,417号;同第4,791,192号;または同第4,179,337号に記載の様式で、本発明で開示の抗体を、例えばポリエチレングリコール、ポリプロピレングリコール、又はポリオキシアルキレンなどの種々の非タンパク質性ポリマーの1つに結合させることができる。例えば、循環半減期を増大させるために、ポリマーへの共有結合による抱合によって、抗体を化学修飾する。典型的なポリマーおよびポリマーのペプチドへの結合方法は、米国特許第4,766,106号;同第4,179,337号;同第4,495,285号;および同第4,609,546号にも示されている。
【0066】
他の実施形態では、グリコシル化パターンが変化するように(すなわち、元のまたは天然のグリコシル化パターンと異なる)抗体を修飾することができる。本明細書中で使用される、「変化した」は、1つまたは複数の炭水化物部分の欠失および/または元の抗体への1つまたは複数のグリコシル化部位の付加を意味する。グリコシル化部位のコンセンサス配列を含めるためのアミノ酸配列の変化によって達成された本発明で開示の抗体へのグリコシル化部位の付加は当該分野で周知である。抗体上の炭水化物部分の数の別の増加方法は、抗体のアミノ酸残基へのグリコシドの化学的または酵素的結合によるものである。これらの方法は、WO87/05330およびAplin and Wriston(1981)CRC Crit.Rev.Biochem.,22:259−306に記載されている。Hakimuddin et al.(1987)Arch.Biochem.Biophys.,259:52;Edge et al.(1981)Anal.Biochem.,118:131;およびThotakura et al.(1987)Meth.Enzymol.,138:350に記載のように、抗体上に存在する任意の炭水化物部分の除去を化学的または酵素的に行うことができる。
【0067】
本発明の抗体を、検出可能な標識または機能的標識でタグ化することもできる。検出可能な標識には、当該分野で公知の従来の化学的性質を使用して本発明の抗体に結合することができる131Iまたは99Tcなどの放射性標識が含まれる。標識には、西洋ワサビペルオキシダーゼまたはアルカリホスファタ−ゼなどの酵素標識も含まれる。標識には、さらに、特異的同族検出可能部分(例えば、標識ビオチン)への結合を介して検出することができるビオチンなどの化学的部分が含まれる。
【0068】
CDR配列が配列番号n(nは、2、4、6、8、10、12、14、16、18、20、22、24、26、28、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、また48である)に記載のCDR配列と非本質的にのみ異なる抗体は、本発明の範囲内に含まれる。非実質的相違には、CDR配列中の任意の5個のアミノ酸のうちの1個または2個の置換などのアミノ酸の小さな変化が含まれる。典型的には、アミノ酸を、類似の電荷、疎水性、または立体化学的性質を有する関連アミノ酸と置換する。このような置換は、当業者の範囲内である。CDR中と異なり、抗体の結合特性に悪影響を与えることなく構造フレームワーク領域(FR)中のより実質的な変化を行うことができる。FRの変化には、非ヒト由来のフレームワークのヒト化または抗原接触または結合部位の安定化に重要な一定のフレームワーク残基の操作(例えば、定常領域のクラスまたはサブクラスの変化、Fc受容体結合などのエフェクター機能を変化させることができる特異的アミノ酸残基の変化(Lund et al.(1991)J.Immun.147:2657−2662およびMorgan et al.(1995)Immunology 86:319−324)、または定常領域が由来する種の変化)が含まれるが、これらに限定されない。抗体は、重鎖のCH2領域にエフェクター機能が低下または変化する(例えば、Fc受容体結合および補体活性化)変異を有し得る。例えば、抗体は、米国特許第5,624,821号および同第5,648,260号などに記載の変異を有し得る。例えば、IgG1またはIgG2重鎖では、IgG1のFc部分を示す配列番号53のアミノ酸残基117および120にこのような変異を作製することができる(これらの残基は、IgG1またはIgG2の全長配列中のアミノ酸234および237に相当する)。抗体は、免疫グロブリンの2つの重鎖の間のジスルフィド結合を安定化する変異(Angal et al.(1993)Mol.Immunol.30:105−108に開示のIgG4のヒンジ領域の変異など)も有し得る。
【0069】
D.核酸、クローニング系、および発現系
本発明は、さらに、本発明の抗体または結合フラグメントをコードする単離核酸を提供する。本発明の核酸は、DNAまたはRNAを含むことができ、且つ全体または部分的に合成であり得る。本明細書中に記載のヌクレオチド配列の基準は、特定の配列を有するDNA分子を含み、且つ、他で特記しない限り、TがUに置換された特定の配列を有するRNA分子を含む。
【0070】
本発明の核酸は、本明細書中に記載の本発明のCDRまたはVHもしくはVLドメインのコード配列を含む。
【0071】
本発明はまた、上記の、少なくとも1つの本発明の核酸を含むプラスミド、ベクター、転写もしくは発現カセットの形態の構築物を提供する。
【0072】
本発明はまた、上記の1つまたは複数の構築物を含む宿主細胞を提供する。コードされた産物の産生方法と同様に、本明細書中に提供する任意のCDR(H1、H2、H3、L1、L2、またはL3)、VHもしくはVLドメイン、または特異的抗原結合フラグメントをコードする核酸は、本発明の態様を形成する。この方法は、コード核酸からの発現を含む。核酸を含む組換え宿主細胞の適切な条件下での培養によって発現させることができる。発現による産生後、VHもしくはVLドメイン、または特異的抗原結合フラグメントを、任意の適切な技術を使用して単離および/または精製し、必要に応じて使用することができる。
【0073】
本発明の特異的抗原結合フラグメント、VHおよび/またはVLドメイン、ならびにコード核酸分子およびベクターを、例えば、その天然の環境から単離および/または精製するか、実質的に純粋もしくは均一な形態で提供するか、核酸の場合、必要な機能を有するポリペプチドをコードする配列以外の元の核酸または遺伝子を含まないか実質的に含まずに提供することができる。
【0074】
種々の異なる宿主細胞におけるポリペプチドのクローニング系および発現系が周知である。適切な宿主細胞には、細菌系、哺乳動物細胞系、ならびに酵母およびバキュロウイルス系が含まれる。異種ポリペプチド発現のために当該分野で利用可能な哺乳動物細胞株には、チャイニーズハムスター卵巣細胞、HeLa細胞、ベビーハムスター腎臓細胞、NS0マウス黒色腫細胞、および多くの他の細胞が含まれる。一般的な細菌宿主は、E.coliである。抗体産生に適切な細胞については、Gene Expression Systems,eds.Fernandez et al.,Academic Press,1999を参照のこと。本発明に適合する任意の細胞を本発明で開示の抗体の産生に使用することができる。
【0075】
適切な調節配列(プロモーター配列、ターミネーター配列、ポリアデニル化配列、エンハンサー配列、マーカー遺伝子、および必要に応じて他の配列が含まれる)を含む適切なベクターを選択または構築することができる。必要に応じて、ベクターは、プラスミドベクターまたはウイルスベクター(例えば、ファージまたはファージミド)であり得る。さらなる詳細については、例えば、Molecular Cloning:a Laboratory Manual:2nd ed.,Sambrook et al.,Cold Spring Harbor Laboratory Press,1989を参照のこと。例えば、核酸構築物の調製、変異誘発、配列決定、細胞へのDNAの導入および遺伝子発現、ならびにタンパク質分析における核酸操作のための多数の公知の技術およびプロトコールは、Current Protocols in Molecular Biology,2nd ed.,Ausubel et al.eds.,JohnWiley & Sons,1992に詳細に記載されている。
【0076】
したがって、本発明のさらなる態様は、本明細書中に開示の核酸を含む宿主細胞を提供する。なおさらなる態様は、このような核酸を宿主細胞に導入する工程を含む方法を提供する。導入は、任意の利用可能な技術を使用することができる。真核細胞のための適切な技術には、リン酸カルシウムトランスフェクション、DEAE−デキストラン、エレクトロポレーション、リポソ−ム媒介トランスフェクション、およびレトロウイルスまたは他のウイルス(例えば、ワクシニアまたは昆虫細胞についてはバキュロウイルス)を使用した形質導入が含まれ得る。細菌細胞のための適切な技術には、塩化カルシウム形質転換、エレクトロポレーション、およびバクテリオファージを使用したトランスフェクションが含まれ得る。
【0077】
移入後に、例えば、遺伝子発現条件下での宿主細胞の培養によって核酸から発現させることができる。
【0078】
E.微生物寄託
非生殖系列化scFvのMyo29、Myo28、またはMyo22をコードするファージミドベクターpCANTAB6でそれぞれ形質転換したE.coli培養物を、2002年10月2日にAmerican Tissue Culture Collection(ATCC)にそれぞれ受託番号PTA−4741、PTA−4740、およびPTA−4739で寄託した。寄託機関の住所は、10801 University Blvd,Manassas,VA 20110,U.S.A.である。
【0079】
(II.疾患の治療方法および他の用途)
本発明の抗体は、ヒトまたは動物の種々の内科的疾患の予防、診断、または治療に有用である。抗体を使用して、GDF−8または関連タンパク質に関連する1つまたは複数の活性を阻害または減少させることができる。より好ましくは、抗体は、抗体に結合していないGDF−8と比較して1つまたは複数のGDF−8活性を阻害または減少させることができる。一定の実施形態では、1つまたは複数の本発明に開示の抗体と結合した場合、GDF−8活性は、1つまたは複数の本発明に開示の抗体に結合していない成熟GDF−8タンパク質と比較して、少なくとも50%、好ましくは少なくとも60、62、64、66、68、70、72、72、76、78、80、82、84、86、または88%、より好ましくは少なくとも90、91、92、93、または94%、さらにより好ましくは少なくとも95%〜100%阻害される。GDF−8活性の阻害を、Thies et
al.(Growth Factors(2001)18:251−259)に記載され、且つ実施例2および9に例示のpGL3(CAGA)12レポーター遺伝子アッセイ(RGA)または実施例3および6に例示のActRIIB受容体アッセイで測定することができる。
【0080】
本発明に開示の抗体によって診断、治療、または予防される内科的疾患は、筋肉または神経筋障害;肥満症などの脂肪組織障害;2型糖尿病;耐糖能障害;代謝症候群(例えば、X症候群);火傷または窒素不均衡などの外傷によるインスリン抵抗性;または骨粗鬆症などの骨変性疾患である。
【0081】
本発明に開示の抗体によって診断、治療、または予防される他の内科的疾患は、骨の喪失に関連する障害(骨粗鬆症(特に高齢者および/または閉経後の女性)、糖質コルチコイド誘導性骨粗鬆症、オステオペニア、変形性関節症、および骨粗鬆症関連骨折が含まれる)である。他の標的代謝性骨の疾患または障害には、慢性糖質コルチコイド療法に起因する低骨量、若年性性腺不全、アンドロゲン抑制、ビタミンD欠損症、二次性副甲状腺機能亢進症、栄養失調、および拒食症が含まれる。好ましくは、抗体を使用して、哺乳動物、特にヒトにおけるこのような内科的疾患を予防、診断、または治療する。
【0082】
本発明の抗体または抗体組成物を、治療有効量で投与する。一般に、治療有効量は、被験体の年齢、病態、および性別ならびに被験体の病状の重症度によって変化し得る。医師が投薬量を決定し、必要に応じて認められた治療効果に適合するように調整することができる。このような化合物の毒性および治療効率を、例えば、LD50(集団の50%に致命的な用量)およびED50(集団の50%の治療有効量)の決定のための細胞培養または実験動物における標準的な薬学的手順によって決定することができる。有毒と治療効果との間の用量の比が治療指数であり、LD50/ED50比として示すことができる。高い治療指数を示す抗体が好ましい。
【0083】
細胞培養アッセイおよび動物研究から得られたデータを、ヒトで使用するための投薬量範囲の処方において使用することができる。このような化合物の投薬量は、毒性がほとんどないか全くないED50を含む循環濃度範囲内であることが好ましい。投薬量は、使用した投薬形態および使用した投与経路によってこの範囲内で変化させることができる。本発明で使用した任意の抗体について、最初に細胞培養アッセイから治療有効用量を評価することができる。細胞培養において決定したIC50(すなわち、最大の症状の阻害の半分を達成する試験抗体の濃度)を含む循環血漿濃度範囲を達成するために、動物モデルにおける投与量を処方することができる。例えば、高速液体クロマトグラフィによって血漿レベルを測定することができる。任意の特定の投薬量の効果を、適切なバイオアッセイによってモニタリングすることができる。適切なバイオアッセイの例には、DNA複製アッセイ、転写ベースのアッセイ、GDF−8タンパク質/受容体結合アッセイ、クレアチンキナーゼアッセイ、前脂肪細胞の分化に基づいたアッセイ、脂肪細胞におけるグルコース取り込みに基づいたアッセイ、および免疫学的アッセイが含まれる。
【0084】
一般に、抗体またはその結合フラグメントが、1μg/kg〜150mg/kg、1μg/kg〜100mg/kg、1μg/kg〜50mg/kg、1μg/kg〜20mg/kg、1μg/kg〜10mg/kg、1μg/kg〜1mg/kg、10μg/kg〜1mg/kg、10μg/kg〜100μg/kg、100μg〜1mg/kg、および500μg/kg〜1mg/kgの用量で投与されるように組成物を投与する。好ましくは、投与後最も長時間循環抗体レベルを最大にするために、抗体をボーラス投与として投与する。ボーラス投与後、連続的注入を使用することもできる。
【0085】
本発明に開示の抗体を使用した上記病状の治療、診断、または予防方法を、TGF−βスーパーファミリー中の他のタンパク質で使用することもできる。多数のこれらのタンパク質は、BMP−11などのGDF−8の構造と類似している。したがって、別の実施形態は、BMP−11またはアクチビンを阻害することができる抗体の単独または他のTGF−βインヒビター(GDF−8に対する中和抗体など)との組み合わせの被験体への投与による上記障害の治療方法を提供する。本発明の抗体を使用して、BMP−11と関連するか媒介される疾患または病態を治療することもできる。例えば、米国特許第5,639,638号および同第6,437,111号を参照のこと。
【0086】
本発明の抗体を使用して、in vivoまたはin vitroでBMP−11およびGDF−8などのTGF−βスーパーファミリーに属するタンパク質の存在を検出することができる。これらのタンパク質の存在またはレベルと病状との相関により、当業者は関連する病状を診断することができる。本発明に開示の抗体によって診断することができる病状を上に記載する。
【0087】
このような検出方法は当該分野で周知であり、ELISA、ラジオイムノアッセイ、免疫ブロット、ウェスタンブロット、免疫蛍光、免疫沈降、および他の類似の技術が含まれる。タンパク質(例えば、GDF−8)を検出するための1つまたは複数のこれらの技術を組み込んだ診断キット中に抗体をさらに提供することができる。このようなキットは、他の構成要素、輸送容器、説明書、またはタンパク質の検出およびキットの使用を補助するための他の材料を含むことができる。
【0088】
抗体が診断を目的とする場合、例えば、リガンド基(ビオチンなど)または検出可能なマーカー基(蛍光基、放射性同位体、または酵素など)で修飾することが望ましい。所望ならば、従来技術を使用して抗体(ポリクローナル抗体またはモノクローナル抗体のいずれか)を標識することができる。適切な標識には、フルオロフォア、ケモフォア、放射性原子、高電子密度試薬、酵素、および特異的結合パートナーを有するリガンドが含まれる。酵素は、典型的には、その活性によって検出される。例えば、西洋ワサビペルオキシダーゼを、テトラメチルベンジジン(TMB)を分光光度計で定量可能な青色色素に変換する能力によって検出することができる。他の適切な標識には、ビオチンおよびアビジンまたはストレプトアビジン、IgGおよびタンパク質A、ならびに当該分野で公知の多数の受容体−リガンド結合物が含まれ得る。他の順列および可能性は当業者に容易に明らかであり、本発明の範囲内で等価物と見なされる。
【0089】
本発明のなおさらなる態様は、筋肉および骨の障害の治療で有用な治療薬の同定方法を提供する。適切なスクリーニングアッセイ(例えば、ELISAベースのアッセイ)は当該分野で公知である。このようなスクリーニングアッセイでは、第1の結合混合物を、本発明の抗体とリガンド(例えば、GDF−8、BMP−11、アクチビン)との組み合わせによって形成し、第1の結合混合物中のリガンドと抗体との間の結合量(M0)を測定する。第2の結合混合物も抗体と、リガンドと、スクリーニングすべき化合物または薬剤との組み合わせによって形成し、第2の結合混合物中のリガンドと抗体との間の結合量(M1)を測定する。次いで、例えば、M1/M0比の計算によって第1および第2の結合混合物の結合量を比較する。第1の結合混合物と比較して第2の結合混合物で結合の減少が認められる場合、化合物または薬剤はGDF−8活性を阻害することができると見なされる。結合混合物の処方および至適化は当業者のレベルの範囲内であり、このような結合混合物はまた、結合の強化または至適化に必要な緩衝液および塩を含むことができ、本発明のスクリーニングアッセイにさらなるコントロールアッセイが含まれ得る。
【0090】
したがって、少なくとも約10%(すなわち、M1/M0<0.9)、好ましくは約30%を超えて抗体−リガンド結合が減少することが見いだされた化合物を同定することができ、所望ならば、ActRIIB結合アッセイ(実施例2)ならびに実施例13、15、および16に記載の他の細胞ベースのアッセイおよびin vivoアッセイなどの他のアッセイでGDF−8活性を阻害する能力について二次的にスクリーニングすることができる。
【0091】
(III.薬学的組成物および投与方法)
本発明は、本発明に開示の抗体を含む組成物を提供する。このような組成物は、薬学的使用および患者への投与に適切であり得る。組成物は、典型的には、1つまたは複数の本発明の抗体および薬学的に許容可能な賦形剤を含む。本明細書中で使用される、句「薬学的に許容可能な賦形剤」には、薬学的投与に適合する任意および全ての溶媒、分散媒、コーティング、抗生物質および抗真菌薬、ならびに等張化剤および吸収遅延剤などが含まれる。薬学的に活性な物質のためのこのような溶剤および薬剤の使用は当該分野で周知である。組成物は、補足的、付加的、または強化された治療機能を提供する他の活性化合物も含み得る。薬学的組成物を、投与説明書とともにコンテナ、パック、またディスペンサーに含めることもできる。
【0092】
本発明の薬学的組成物を、その意図する投与経路に適合するように処方する。投与方法は、当業者に公知である。局所的または経口投与することができるか、粘膜を通過することができる組成物を得ることも可能である。投与は、例えば、静脈内、腹腔内、筋肉内、腔内、皮下、または経皮であり得る。
【0093】
皮内または皮下投与で使用される溶液または懸濁液には、典型的には、1つまたは複数の以下の構成要素が含まれる:注射用の水、生理食塩水、不揮発油、ポリエチレングリコール、グリセリン、プロピレングリコール、または他の合成溶媒などの滅菌希釈剤;ベンジルアルコールまたはメチルパラベンなどの抗菌薬;アスコルビン酸または亜硫酸水素ナトリウムなどの抗酸化剤;エチレンジアミン四酢酸などのキレート化剤;酢酸、クエン酸、またはリン酸などの緩衝液;および塩化ナトリウムまたはデキストロースなどの等張化剤。塩酸または水酸化ナトリウムなどの酸または塩基でpHを調整することができる。このような調製物を、ガラス製またはプラスチック製のアンプル、使い捨てシリンジ、または複数回投与用バイアルに同封することができる。
【0094】
注射に適切な薬学的組成物には、滅菌水溶液または分散液および滅菌注射用溶液または分散液の即時調製のための滅菌粉末が含まれる。静脈内投与に適切なキャリアには、生理食塩水、静菌水、Cremophor(商標)EL(BASF,Parsippany,NJ)、またはリン酸緩衝化生理食塩水(PBS)が含まれる。全ての場合において、組成物は滅菌されていなければならず、容易にシリンジで使用できる範囲の流動物であるべきである。製造および保存条件下で安定でなければならず、細菌および真菌などの微生物の汚染作用に対して保存されるべきである。キャリアは、例えば、水、エタノール、ポリオ−ル(例えば、グリセロール、プロピレングリコール、および液体ポリエチレングリコールなど)、およびその適切な混合物を含む溶媒または分散媒であり得る。例えば、レシチンなどのコーティングの使用、分散液の場合の必要な粒子サイズの維持、および界面活性剤の使用によって適切な流動性を維持することができる。種々の抗菌薬および抗真菌薬(例えば、パラベン、クロロブタノ−ル、フェノール、アスコルビン酸、およびチメロサールなど)によって微生物作用を防止することができる。多くの場合、組成物中に等張化剤(例えば、糖、マンニトールなどのポリアルコール、ソルビトール、および塩化ナトリウム)を含むことが好ましい。注射用組成物は、組成物中に吸収を遅延させる薬剤(例えば、モノステアリン酸アルミニウムおよびゼラチン)を含めることによって長期間吸収され得る。
【0095】
経口組成物には、一般に、不活性希釈剤または食用キャリアが含まれる。これらをゼラチンカプセルに封入するか、打錠することができる。治療のための経口投与の目的で、抗体を賦形剤に組み込み、錠剤またはカプセル形態で使用することができる。薬学的に適合する結合剤および/またはアジュバント材料を、組成物の一部として含めることができる。錠剤、丸薬、およびカプセルなどは、任意の以下の成分または類似の性質の化合物を含み得る:微結晶性セルロース、トラガカントガム、またはゼラチンなどの結合剤;デンプンまたはラクトースなどの賦形剤;アルギン酸、Primogel(商標)、またはコーンスターチなどの崩壊薬;ステアリン酸マグネシウムまたはSterotes(商標)などの潤滑剤;コロイド状二酸化ケイ素などの流動促進剤;スクロースまたはサッカリンなどの甘味料;またはペパーミント、サリチル酸メチル、またはオレンジフレーバーなどの矯味矯臭薬。
【0096】
吸入による投与のために、適切な高圧ガス(例えば、二酸化炭素などのガス)を含む加圧コンテナもしくディスペンサーまたはネブライザーからエアゾールスプレーの形態で抗体を送達させる。
【0097】
経粘膜または経皮手段によって全身投与を行うこともできる。例えば、Fc部分を含む抗体の場合、組成物は、FcRn受容体媒介経路を介して粘膜(例えば、腸、口腔、または肺)を通過することができる(米国特許第6,030,613号)。例えば、ロゼンジ、鼻腔用スプレー、吸入器、または坐剤の使用によって経粘膜投与を行うことができる。経皮投与のために、活性化合物を、当該分野で公知の軟膏(ointment)、軟膏(salve)、ゲル、またはクリームに処方する。経粘膜投与または経皮投与のために、処方物に通過すべきバリアに適切な浸透剤を使用する。このような浸透剤は一般に当該分野で公知であり、例えば、界面活性剤、胆汁酸塩、およびフシジン酸誘導体が含まれる。
【0098】
本発明に開示の抗体を、徐放性処方物などの身体からの急速な排除から化合物を保護するキャリア(移植片およびマイクロカプセル化した送達システムが含まれる)を使用して調製することができる。エチレンビニルアセテート、ポリ酸無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、およびポリ乳酸などの生分解性生体適合性ポリマーを使用することができる。このような処方物の調製方法は、当業者に自明である。本発明に開示の抗体を含むリポソーム懸濁液を、薬学的に許容可能なキャリアとして使用することもできる。例えば、米国特許第4,522,811号に記載の当業者に公知の方法にしたがって、これらを調製することができる。
【0099】
投与を容易にするためおよび投薬量の均一性のために、単位投薬形態で経口または非経口組成物を処方することが有利であり得る。本明細書中で使用される、「単位投薬形態」は、処置すべき被験体のための単一投薬量として適切な物理的に個別の単位をいい、必要な薬学的キャリアと共同して所望の治療効果が得られるように所定量の活性化合物を含む単位を計算した。本発明の単位投薬形態の仕様は、活性化合物の固有の特徴および達成すべき特定の治療効果、および個体治療のためのこのような活性化合物の処方分野が本来有する制限について言及しているか、これらに直接依存する。
【0100】
以下の実施例は、本発明の例示的実施形態を提供し、本発明を決して限定しない。当業者は、多数の他の実施形態が本発明の範囲内に含まれることを認識する。
【0101】
本願で引用された全ての引用文献、特許、および公開された特許出願の内容全体が本明細書中で参考として援用される。
【実施例】
【0102】
(実施例1:GDF−8の精製)
組換えヒトGDF−8タンパク質(成熟GDF−8およびGDF−8プロペプチド)を発現する選択細胞株由来の馴化培地をpH6.5に酸性化し、80×50mmPOROS(商標)SP陽イオン交換カラムと縦に並べた80×50mmPOROS(商標)HQ陰イオン交換カラムにアプライした(PerSeptive Biosystems,Foster City,CA)。通過物(flow through)をpH5.0に調整し、75×20mmPOROS(商標)SP陽イオン交換カラム(PerSeptive
Biosystems)にアプライし、NaCl勾配を使用して溶離した。SDS−PAGEによって確認したGDF−8潜在複合体を含む画分をプールし、トリフルオロ酢酸(TFA)でpH2〜3に酸性化し、粘度を低下させるために0.1%TFAで200mlにした。次いで、プールを、60×21.2mmのガードカラム(Phenomenex,Torrance,CA)を取りつけた250×21.2mmのC5カラム(Phenomenex)にアプライし、TFA/アセトニトリル勾配を使用して溶離し、GDF−8プロペプチドから成熟GDF−8を分離した。アセトニトリルを除去するために成熟GDF−8を含むプール画分を凍結乾燥によって濃縮し、20mlの0.1%TFAを添加した。次いで、サンプルを、分離を補助するために60℃に加熱した250×10mmのC5カラム(Phenomenex)にアプライした。もはやさらに分離することができなくなるまでこれを繰り返した。次いで、成熟GDF−8を含む画分をプールし、40%アセトニトリルで増量し(bring up)、60×21.2のガードカラムを取りつけた600×21.2のBioSep(商標)S−3000サイズ排除カラム(Phenomenex)にアプライした。精製成熟GDF−8を含む画分をプールし、その後の実験での使用のために濃縮した。
【0103】
SDS−PAGEにおいて、精製成熟GDF−8は、非還元条件下で25kDaおよび還元条件下で13kDaの広いバンドとして移動した。McPherron et al.(Proc.Nat.Acad.Sci.U.S.A.(1997)94:12457−12461)によってマウスGDF−8について類似のSDS−PAGEプロフィ−ルが報告されており、成熟タンパク質の二量体特性を反映する。活性成熟BMP−11二量体を、類似の様式で組換えヒトBMP−11を発現する細胞株由来の馴化培地から精製した。
【0104】
活性成熟BMP−11を、組換えヒトGDF−8プロペプチド/成熟BMP−11キメラタンパク質を発現する細胞株由来の馴化培地から精製した。馴化培地を、50mM Tris(pH8.0)、1M NaClを含む10mlのTALON(商標)カラム(Clonetech,Palo Alto,CA)に1ml/分でロードした。結合したタンパク質を、50mM Tris(pH8.0)、1M NaCl、500mMイミダゾールで溶離した。GDF−8プロペプチド/BMP−11潜在複合体を含むプール画分を、10%TFAでpH3に酸性化した。次いで、プールを、成熟BMP−11およびGDF−8プロペプチドのより良好な分離のために60℃に加熱した250×4.6mmのJupiter C4カラム(Phenomenex,Torrance,CA)にアプライし、TFA/アセトニトリル勾配を使用して溶離した。成熟BMP−11を含むプール画分を、凍結乾燥によって濃縮した。SDS−PAGE、精製成熟BMP−11は、非還元条件下で25kDaおよび還元条件下で12kDaに移動した。
【0105】
(実施例2:精製組換えヒトGDF−8の生物活性)
GDF−8の活性を証明するために、ルシフェラーゼを発現するレポーターベクターpGL3(CAGA)12を使用してレポーター遺伝子アッセイ(RGA)を開発した。CAGA配列は、TGF−β誘導性遺伝子PAI−1のプロモーター内のTGF−β応答配列であることが以前に報告されていた(Denner et al.(1998)EMBO J.,17:3091−3100)。
【0106】
基本ルシフェラーゼレポータープラスミドpGL3(Promega,Madison,WI)を使用して、12個のCAGAボックスを含むレポーターベクターを作製した。アデノウイルス主要後期プロモーター(−35/+10)由来のTATAボックスおよび転写開始部位を、BglIIとHindIII部位との間に挿入した。CAGAボックスAGCCAGACAの12回反復を含むオリゴヌクレオチドをアニーリングし、XhoI部位にクローン化した。FuGENE(商標)6トランスフェクション試薬(Boehringer Manheim,Germany)を使用して、ヒト横紋筋肉腫細胞株A204(ATCC HTB−82)をpGL3(CAGA)12で一過性にトランスフェクトした。トランスフェクション後、2mMグルタミン、100U/mlストレプトマイシン、100μg/mlペニシリン、および10%ウシ胎児血清を補足したMcCoyの5A培地を含む48ウェルプレートに細胞を16時間培養した。次いで、細胞を、McCoyの5A培地中の10ng/mlのGDF−8の存在下又は非存在下で、グルタミン、ストレプトマイシン、ペニシリン、および1mg/mlのウシ血清アルブミンで、37℃にて6時間処理した。ルシフェラーゼアッセイ系(Promega)を使用して、処置した細胞中のルシフェラーゼを定量した。
【0107】
図4Aは、GDF−8は、10ng/mlのED50でレポーター構築物を最大で10倍活性化し、精製組換えGDF−8が生物学的に活性であることを示す。BMP−11およびアクチビンは類似の生物学的応答を誘発した。
【0108】
(実施例3:ActRIIB結合アッセイにおける精製GDF−8の結合特性)
GDF−8潜在複合体を、1モルのGDF−8複合体に対して20モルのEZ結合スルホ−NHS−ビオチン(Pierce,Rockford,Illinois,Cat.No.21217)の比で氷上で2時間ビオチン化した。0.5%TFAを使用したpHの低下によって反応を停止させ、複合体をC4 Jupiter 250×4.6mmカラム(Phenomenex)におけるクロマトグラフィに供してGDF−8プロペプチドから成熟GDF−8を分離した。TFA/CH3CN勾配を使用して溶離したビオチン化成熟GDF−8画分をプールし、濃縮し、MicroBCA(商標)タンパク質アッセイ試薬キット(Pierce,Rockford,IL,Cat.No.23235)によって定量した。
【0109】
上記と同一の様式で、BMP−11潜在複合体からビオチン化成熟BMP−11を調製した。1μg/mlの組換えActRIIB−Fcキメラ(R&D Systems,Minneapolis,MN,Cat.No.339−RB/CF)を含む0.2M炭酸ナトリウム緩衝液を、4℃で一晩96ウェルの平底アッセイプレート(Costar,NY,Cat.No.3590)にコートした。次いで、プレートを1mg/mlのウシ血清アルブミンでブロッキングし、標準的なELISAプロトコールにしたがって洗浄した。種々の濃度の100μアリコートのビオチン化GDF−8またはBMP−11を、ブロッキングしたELISAプレートに添加し、1時間インキュベートし、洗浄し、ストレプトアビジン−西洋ワサビペルオキシダーゼ(SA−HRP,BD PharMingen,San Diego,CA,Cat.No.13047E)およびその後のTMB(KPL,Gaithersburg,MD,Cat.No.50−76−04)の添加によってGDF−8またはBMP−11の結合量を検出した。Molecular Devicesマイクロプレートリーダーにおいて450nMで比色測定を行った。
【0110】
図1に示すように、ビオチン化GDF−8およびBMP−11はActRIIB(ED50が15および40ng/mlである推定GDF−8II型受容体)にそれぞれ結合し、ActRIIB結合アッセイがin vitro結合アッセイでGDF−8およびBMP−11に感受性を示すことを示す。
【0111】
(実施例4:scFvライブラリーにおけるGDF−8のパニングによるMyo22の単離)
記載の1.38×1010個のライブラリー(Vaughan et al.(1996)Nature Biotech.,14:309−314)の拡大バ−ジョンであるscFvファージミドライブラリーを使用して、GDF−8に特異的な抗体を選択した。可溶性GDF−8タンパク質(10μg/mlを含む50mM炭酸ナトリウム緩衝液(pH9.6))でマイクロタイタープレートのウェルを4℃で一晩コートした。ウェルをPBSで洗浄し、MPBS(3%Marvel(商標)脱脂粉乳を含むPBS)にて37℃で2時間ブロッキングした。精製したファージ(1012形質導入単位(tu))を含む100lの3%MPBSをブロッキングしたウェルに添加し、室温で1時間インキュベートした。ウェルをPBST(0.1%v/vのTween(商標)20を含むPBS)で10回洗浄し、次いでPBSで10回洗浄した。結合したファージ粒子を、100μlの100mMトリエチルアミンにて室温で10分間溶離し、その直後に50μlの1M Tris−HCl(pH7.4)で中和した。溶離したファージを使用して、指数関数的に成長した10mlのE.coli TG1に感染させた。感染細胞を、2TYブロス中で静置にて37℃で30分間およびその後曝気しながら37℃で30分間成長させ、2TYAGプレート上に画線し、30℃で一晩インキュベートした。コロニーをプレートから10mのl2TYブロスに掻き取り、−70℃での保存のために15%グリセロールを添加した。
【0112】
第1ラウンドのパニング選択由来のグリセロールストック培養物にヘルパーファージを重感染させ、レスキューして第2のパニングラウンドのためのscFv抗体発現ファージ粒子を得た。このようにして、全部で3ラウンドのパニングを行った。
【0113】
(実施例5:scFvライブラリーからのMyo28およびMyo29の選択)
ビオチン化GDF−8タンパク質(bioGDF−8)を使用して、可溶性選択(soluble selections)を行った。1μg/mlの濃度でbioGDF−8を使用した。実施例4に記載するように、scFvライブラリーを使用した。精製scFvファージ(1012tu)を100μlの3%MPBSで30分間ブロッキングし、ビオチン化抗体を添加し、室温で1時間インキュベートした。ファージ/抗原を、1mlの3%MPBS中にて37℃で1時間ブロッキングした50μlのDynal(商標)M280ストレプトアビジン磁性ビーズに添加し、室温でさらに15分間インキュベートした。磁性ラックを使用してビーズを補足し、0.1%(v/v)Tween(商標)20を含む1mlの3%MPBSで4回洗浄し、PBSで3回洗浄した。最後のPBSでの洗浄後、ビーズを100μlのPBS中で再懸濁し、5mlの指数関数的に成長したE.coli TG−1細胞を感染させるのに使用した。細胞およびファージを、37℃で1時間(静置で30分間および250rpmで震盪しながら30分間)インキュベートし、2TYAGプレートに広げた。プレートを30℃で一晩インキュベートし、翌日にコロニーを視覚化した。産生されたコロニーをプレートから掻き取り、上記のようにファージをレスキューした。上記のように第2ラウンドの可溶性選択を行った。
【0114】
(実施例6:ActRIIB受容体阻害アッセイおよびスクリーニング)
実施例4および5に記載のようにして得た産生コロニーを、100μlの2TYAGを含む96ウェルプレートに採取した。指数関数的に成長した培養物への1mMのIPTGの添加および30℃で一晩のインキュベーションによってscFv産生を誘導した。本質的に実施例3に記載のように、粗scFv含有培養物上清を、bioGDF−8のActRIIBへの結合を阻害する能力についてスクリーニングした。bioGDF−8の結合をユウロピウム標識ストレプトアビジンで検出し、時間分解蛍光アッセイ(TRF)でDELFIA(商標)試薬キット(PerkinElmer Life Sciences,Boston,MA)を使用するという点でアッセイをわずかに修正した。無関係のクローンを超える結合シグナルの阻害を示す正のクローンを採取し、活性を確認するためにアッセイした。
【0115】
受容体阻害スクリーニングから同定された正のクローン由来の精製scFvを、上記の阻害アッセイで試験した。アッセイにおけるIC50値によって測定したクローンの能力を確立するために、scFv濃度の滴定を使用した。実験結果を図2に示す。これらのアッセイで決定したところ、Myo29、Myo28、およびMyo22のscFvについてのIC50は、それぞれ2.4nM、1.7nM、および60nMである。したがって、これらの抗体は、GDF−8活性の強力なインヒビターである。
【0116】
(実施例7:ファージELISAによる特異性の特徴づけ)
抗体の特異性を決定するために、GDF−8および無関係のタンパク質に対するActRIIBスクリーニング由来の正のクローンについてファージELISAを行った。ファージミドを含む各E.coliコロニーを、ウェルあたり100μlの2TYAG培地を含む96ウェルプレートに接種した。M13K07ヘルパーファージを、10の感染多重度(moi)の指数関数的に成長した培養物に添加し、プレートを37℃でさらに1時間インキュベートした。プレートを、ベンチトップ遠心分離機にて2000rpmで10分間遠心分離した。上清を除去し、細胞ペレットを100μlの2TYAKに再懸濁し、震盪しながら30℃で一晩インキュベートした。翌日、プレートを2000rpmで10分間遠心分離し、各ウェル由来の100μlのファージ含有上清を新鮮な96ウェルプレートに移した。ファージサンプルを、ELISA前に最終濃度3%のMPBSにて室温で1時間ブロッキングした。
【0117】
1μg/mlのGDF−8または無関係のタンパク質を、96ウェルマイクロタイタープレートに4℃で一晩コートした。コーティング後、ウェルから溶液を取り出し、プレートを3%MPBSにて室温で1時間ブロッキングした。プレートをPBSでリンスし、各ウェルに50μlのプレブロッキングファージを添加した。プレートを室温で1時間インキュベートし、PBSTで3回洗浄し、その後PBSで3回洗浄した。各ウェルに、50μlの5,000倍希釈の抗M13−HRP抱合体(Pharmacia)を添加し、プレートを室温で1時間インキュベートした。各プレートを、PBSTで3回洗浄し、その後PBSで3回洗浄した。50μlのTMB基質を各ウェルに添加し、発色するまでインキュベートした。25μlの0.5M H2SO4の添加によって反応を停止させた。作製されたシグナルを、マイクロタイタープレートリーダーを使用した450nmの吸光度の読み取りによって測定した。GDF−8の特異的結合を確認した。
【0118】
(実施例8:scFvの配列決定、IgGへの変換、および生殖系列化)
中和scFv E.coliクローンを2TYAGプレートに画線し、30℃で一晩インキュベートした。scFvクローン由来のVHおよびVL領域を増幅するために、これらのプレート由来の三連のコロニーをpCANTAB6ベクター配列オリゴを使用して配列決定した。Myo29、Myo28、およびMyo22のIgGの作製に使用したscFvフラグメントのDNA配列を、それぞれ配列番号13、配列番号7、および配列番号1に示す。
【0119】
PCRおよびクローン特異的プライマーを使用して、scFvクローン由来の重鎖および軽鎖のV領域を増幅した。適切な制限酵素でPCR産物を消化し、ヒトIgG1重鎖定常ドメイン(VHドメインについて)を含むベクターまたは必要に応じてヒトλ軽鎖定常ドメイン(VLドメインについて)を含むベクターにサブクローン化した。各E.coliコロニー由来のプラスミドDNAの配列決定によってプラスミドへのV領域ドメインの正確な挿入を確認した。標準的技術によってE.coli培養物からプラスミドを調製し、標準的な技術を使用して重鎖および軽鎖構築物をCOS細胞に同時トランスフェクトした。プロテインAセファロ−ス(Pharmacia,Peapack,NJ)を使用して分泌IgGを精製し、緩衝液をPBSに交換した。
【0120】
scFvクローンの配列データを使用して、各クローンの重鎖および軽鎖の最も近い生殖系列配列を同定した。適切な変異誘発プライマーを使用した標準的な部位特異的変異誘発技術を使用して適切に変異させた。配列分析によってscFv配列の変異を確認した。Myo28およびMyo29の生殖系列化scFvならびにVHおよびVLドメイン配列を、配列番号19および配列番号25にそれぞれ示す。
【0121】
(実施例9:抗体の生物活性)
図3Aは、実施例3に記載のActRIIB結合アッセイにおいてMyo29の10ng/mlのビオチン化GDF−8とのプレインキュベーションにより、0.2〜0.4nMのIC50にてActRIIBへのGDF−8結合が阻害されることを示す。同様に図3Bでは、Myo29は同一のIC50でのActRIIBへのビオチン化BMP−11結合を阻害した。
【0122】
in vitroバイオアッセイにおいてMyo29はGDF−8活性も遮断した。例として、GDF−8をMyo29と室温で1時間プレインキュベートした場合、本質的に実施例2に記載のように実施したRGAアッセイで決定したところ、GDF−8の生物活性が減少した。図4Cは、Myo29の存在下におけるGDF−8のED50(20ng/ml)でのpGL3(CAGA)12レポーター活性の誘導を示す。Myo29は、15〜30ng/mlのIC50(0.1〜0.2nM)にて用量応答様式でGDF−8誘導を減少させた。Myo29は、同程度にBMP−11の生物活性も阻害した(図4B)。対照的に、本アッセイのアクチビン活性は、GDF−8およびBMP−11と比較してMyo29に影響を受けず(図4D)、これはおそらくGDF−8とアクチビンとの間の相同性が相対的に低いためである。
【0123】
RGAおよびActRIIB結合アッセイにおいてMyo22およびMyo28も試験した。両抗体は、GDF−8およびBMP−11活性を遮断する。Myo28のIC50は、例えば、0.2〜0.35nMである。
【0124】
(実施例10:Myo22、Myo28、およびMyo29のエピトープのマッピング)
抗体の正確なエピトープをマッピングするために、配列番号49に記載の成熟GDF−8の全配列を示す48の重複する13残基のペプチドを、スポット合成技術を使用してセルロースペーパー上で直接合成した(Molina et al.(1996)Peptide Research,9:151−155;Frank et al.(1992)Tetrahedron,48:9217−9232)。ペプチドの重複は11アミノ酸であった。このアレイでは、システインの存在によって生じる化学的な複雑さを減少させるために、システイン残基をセリンに置換した。ポリエチレングリコールで修飾したセルロースメンブレンおよびFmoc保護アミノ酸を、Abimed(Lagenfeld,Germany)から購入した。以前に記載のように、β−アラニンスペーサーのカップリングによってメンブレン上でアレイを定義し、ペプチドを標準的なDIC(ジイソプロピルカルボジイミド)/HOBt(ヒドロキシベンゾトリアゾール)カップリング化学を使用して合成した(Molina et al.(1996)Peptide Research,9:151−155;Frank et al.(1992)Tetrahedron,48:9217−9232)。
【0125】
活性化アミノ酸を、Abimed ASP 222ロボットを用いてスポットした。洗浄および脱保護工程を手動で行い、最終合成サイクル後にペプチドのN末端をアセチル化した。ペプチド合成後、メンブレンをメタノールで10分間洗浄し、ブロッカー(TBST(0.1%(v/v)Tween(商標)20を含むTris緩衝化生理食塩水)および1%(w/v)カゼイン)で10分間洗浄した。次いで、メンブレンを、2.5μg/mlの抗GDF−8抗体を含むブロッカーと穏やかに撹拌しながら1時間インキュベートした。ブロッカーでの10分間で3回の洗浄後、メンブレンをHRP標識二次抗体(0.25μg/mlを含むブロッカー)と30分間インキュベートした。次いで、メンブレンをブロッカーにて10分間ずつ3回およびTBSTにて10分間ずつ2回洗浄した。結合した抗体をSuperSignal(商標)West試薬(Pierce)およびデジタルカメラ(Alphananotech Fluoromager)を使用して視覚化した。結果を図5に示す。特に、図5で認められるように、Myo29のエピトープは、成熟GDF−8のアミノ酸72と88との間にマッピングされた。それに対して、Myo22は、成熟GDF−8配列の最初の44個のN末端アミノ酸(配列番号49のアミノ酸1〜44)内のエピトープを認識する。最後に、Myo28のエピトープは、成熟GDF−8の最初の98個のN末端アミノ酸内に存在する残基を含む。
【0126】
Myo29エピトープをさらに特徴づけるために、スポット合成を使用して削除および置換分析を行った。置換分析では、ペプチドの各残基を個別にシステイン以外の20種の天然アミノ酸にそれぞれ置換した。上記のように、合成および結合アッセイを行った。結果を図6に示し、最初の行、最初の2つの列、および最後の3つの列は、野生型ペプチドコントロールを示す。結果は、Lys−78、Pro−81、およびAsn−83を個別に別のアミノ酸に変異させた場合、ペプチドに対するMyo29の結合親和性を有意に減少させることを示している。したがって、Myo29は、Lys−Xaa1−Xaa2−Pro−Xaa3−Asn(配列番号54)(Xaa1、Xaa2、およびXaa3はそれぞれ任意のアミノ酸のいずれかであるか、互いに独立して、Xaa1=Met、Xaa2=Ser、およびXaa3=Ile)を含む配列を認識する。
【0127】
(実施例11:GDF−8の免疫沈降)
成熟GDF−8およびGDF−8複合体へのMyo29およびMyo28の結合を評価するために、免疫沈降研究を行った。GDF−8を発現するCHO細胞を、35S−メチオニンおよび35S−システインで標識した。GDF−8タンパク質(成熟GDF−8および潜在複合体)を含む100μlのこれらの細胞由来の馴化培地を、20μg/mlのMyo29またはMyo28と4℃で1時間インキュベートした。プロテインA−セファロ−ス(商標)を添加し、4℃で一晩インキュベートした。免疫沈降物を回収し、還元サンプル緩衝液中に再懸濁し、SDS−PAGEで分析した。ゲルを固定し、オ−トラジオグラフィ増強溶液で増強し、乾燥させ、オ−トラジグラム(autorad)を現像した。図7は、Myo29およびMyo28は成熟GDF−8、GDF−8潜在複合体、および非プロセシングGDF−8を免疫沈殿させることができることを示す。ウェスタンブロッティングによって決定したところ、両抗体は、非還元条件下でGDF−8二量体と結合する。
【0128】
(実施例12:薬物動態学)
単回静脈内(IV)または腹腔内(IP)投与後のC57B6/SCIDマウスにおける1mg/kgの用量でのMyo29の薬物動態学(PK)を評価した。上記用量での非標識および125I標識Myo29の混合物を動物に投与し、血清中の125I放射能および注射用量の比活性に基づいて血清濃度を決定した。図8は、IVまたはIPのいずれかで投与したMyo29の時間に体する血清濃度のプロットを示す。
【0129】
Myo29は、約1週間の長期末端半減期および約1ml/時間/kgの低クリアランスを示した。初期分布体積は、約83ml/kgであった。外見上の分布体積は、約227ml/kgであった。Myo29は、注射後約6時間でピーク濃度に達した。IP注射後に吸収した画分は、約77%であった。
【0130】
(実施例13:筋肉量および強度に対するMyo29のin vivo効果)
Myo29がin vivoでGDF−8活性を遮断するかどうかを決定するために、成体SCIDマウスにおいてMyo29を試験した。SCIDマウスは、重症複合型免疫不全を罹患しているので、Myo29などのヒト抗体の注射後に免疫反応を起こさない。筋肉量は、Myo29で処置したマウスのGDF−8活性の指標として使用した。
【0131】
8週齢の雄C57B6 SCIDマウスを秤量し、体重に関して均一に8つの群に分配した。Myo29を含むPBS緩衝液を、種々の用量(60、10、および1mg/kg)で毎週マウスに腹腔内注射した。最初の週は二重用量を投与した。賦形剤(PBS)で処置した、あるいは処置を行わなかったマウスをコントロールとして用いた。処置を4週間継続した。処置後に腓腹筋および大腿四頭筋を解剖および秤量することによって筋肉量を評価した。処置から4週間後、Myo29で処置した全ての群で筋肉量が10%〜23%の範囲で増加し、より高用量で処置した群は有意なレベルに達した(図9、p<0.01)。
【0132】
別の実験では、雌CB17 SCIDマウスを、種々の用量(10、5、2.5、および1mg/kg)のMyo29で4週間または12週間毎週処置した。さらに、Myo29での4週間の処置により、腓腹筋および大腿四頭筋の重量が10%〜20%の範囲で増加した(図10Aおよび10B)。より長い処置(12週間)により、筋肉量がより増加し(12%〜28%)、Myo29で処置した全ての群は統計的に有意なレベルに達した(図11Aおよび11B)。
【0133】
筋肉量の増加により筋肉がより強くなるかどうかを決定するために、前肢の筋肉の強度を、握力計(model 1027 csx,Columbus Instruments,Columbus,OH)で測定した。処置から12週間後、前肢強度は、賦形剤コントロールと比較して5mg/kgまたは10mg/kgのMyo29で処置したマウスでそれぞれ17%および23%高かった(p<0.01、図12)。この研究結果は、Myo29がin vivoでGDF−8活性を阻害して筋肉量および筋肉の強度を有意に増加させることを証明する。
【0134】
(実施例14:代謝疾患の治療)
GDF−8のインヒビター(例えば、阻害抗体など)は、2型糖尿病、耐糖能障害、代謝症候群(例えば、X症候群)、外傷によるインスリン抵抗性(例えば、火傷または窒素不均衡)、および脂肪組織障害(例えば、肥満症)などの代謝障害の治療に有用である。本発明の抗GDF−8抗体を使用して、疾患を発症している被験体または確立された代謝疾患を有する被験体を治療する。
【0135】
代謝障害(例えば、2型糖尿病および/肥満症)の治療のための抗GDF−8抗体の有効性を、肥満症、インスリン抵抗性、および2型糖尿病の確立されたマウスモデル(ob/ob、db/db、および致死性黄色症変異を有する株が含まれる)を使用して確認する。インスリン抵抗性を、一定のマウス株(C57BL/6Jが含まれる)の高脂肪または高カロリー飼料によって誘導することもできる。ヒトと同様に、これらのげっ歯類は、インスリン抵抗性、高インスリン血症、異常脂質血症、および高血糖症を発症するグルコースホメオスタシスの悪化を発症する。結果の評価は、血清中のグルコース、インスリン、および脂質の測定に基づく。インスリン抵抗性試験およびグルコース負荷試験によってインスリン感度の改良を測定することができる。より感度の高い技術には、血糖コントロールおよびインスリン感度の改善を評価するためのオイグリセミック高インスリン血症クランプの使用が含まれる。さらに、クランプ技術により、改良された血糖コントロールにおける主なグルコース処理組織(筋肉、脂肪、および肝臓)の役割が定量的に評価可能である。
【0136】
1つの研究では、1週間から6ヶ月間Myo29(IP注射)などの抗GDF−8抗体または賦形剤での処置を行う。治療プロトコールは、異なる用量の試験および治療計画(例えば、毎日、毎週、2週間毎の注射)によって変化し得る。抗GDF−8抗体で処置したマウスは、プラシーボ処置を受けたマウスと比較して、グルコース取り込みが増加し、解糖およびグリコーゲン合成が増加し、血清中の遊離脂肪酸およびトリグリセリドが低下すると予想される。
【0137】
また、GDF−8に対する阻害抗体を使用して、疾患の重症度および/または症状を予防および/または軽減する。抗GDF−8抗体を1日1回の頻度および1ヶ月に1回の頻度の皮下注射として投与することが予想される。治療継続期間は、1ヶ月から数年までの範囲であり得る。
【0138】
ヒトにおける抗GDF−8の臨床効果を試験するために、2型糖尿病を罹患しているかリスクのある被験体を同定し、無作為に治療群に分けた。治療群は、プラシーボ群および抗体(異なる用量)を投与された1〜3つの群を含む。個体のグルコース代謝の変化を評価するために1ヶ月〜3年間予測的に追跡する。処置を受けた個体は改善すると予想される。
【0139】
唯一の活性化合物として、又は別の化合物または組成物と組み合わせて、抗体を投与する。唯一の活性化合物として、又は別の化合物または組成物と組み合わせて投与した場合、投薬量は、好ましくは、疾患の症状の重症度および進行に依存して約1μg/kg〜20mg/kgである。適切な有効用量を、治療を行う医師によって以下の範囲から選択される:1μg/kg〜20mg/kg、1μg/kg〜10mg/kg、1μg/kg〜1mg/kg、10μg/kg〜1mg/kg、10μg/kg〜100μg/kg、100μg/kg〜1mg/kg、および500μg/kg〜1mg/kg。治療計画の例および結果を表3にまとめる。
【0140】
【表3】
本明細書中で引用された引用文献(その全体が本明細書中で参考として援用される)の教示に照らして、明細書がほぼ完全に理解される。本明細書中の実施形態は、本発明の実施形態の例示であり、本発明の範囲を制限すると解釈すべきではない。当業者は、多数の他の実施形態が特許請求の範囲に含まれ、本明細書および実施例は例示のみを目的とし、本発明の真の範囲および精神を以下の特許請求の範囲に示すことが意図されることを認識する。
【特許請求の範囲】
【請求項1】
明細書に記載の発明。
【請求項1】
明細書に記載の発明。
【図1】

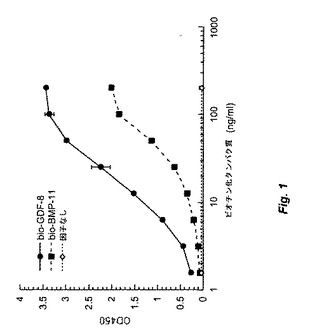
【図2】
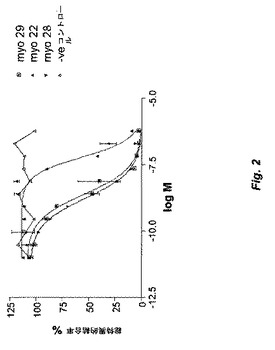
【図3A】
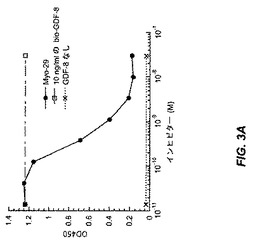
【図3B】
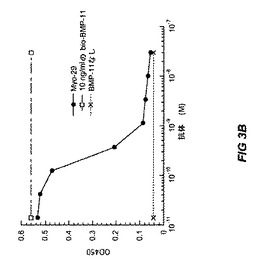
【図4A】
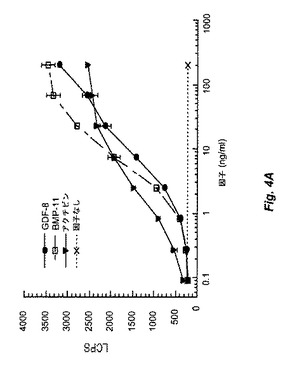
【図4B】
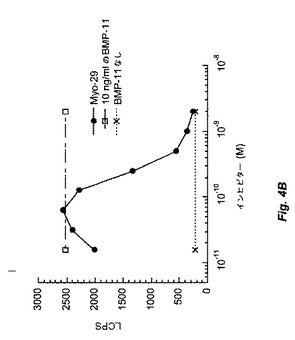
【図4C】
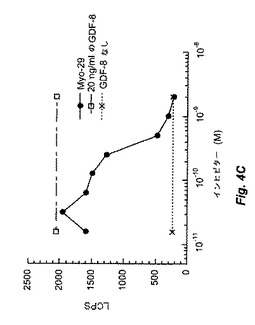
【図4D】
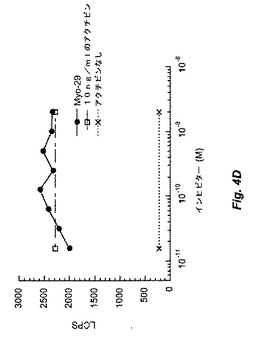
【図5】
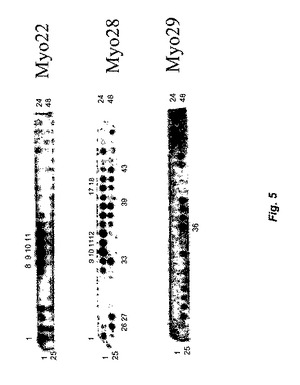
【図6】

【図7】
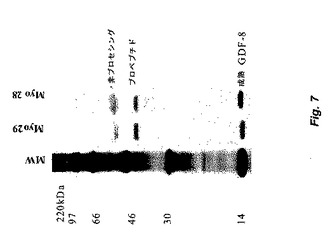
【図8】
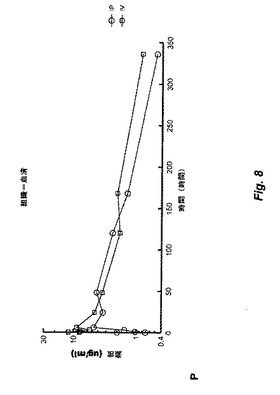
【図9】
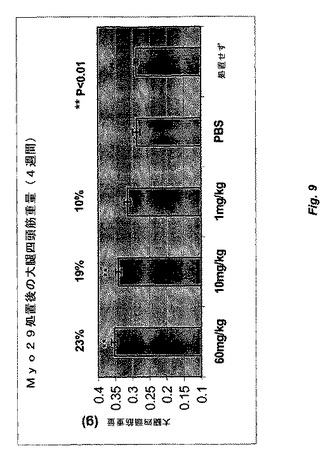
【図10A】
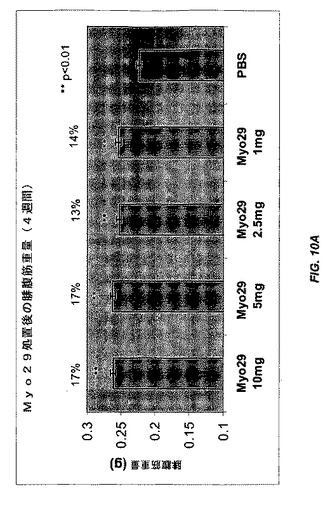
【図10B】
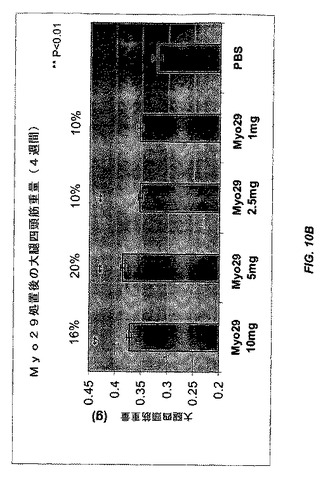
【図11A】
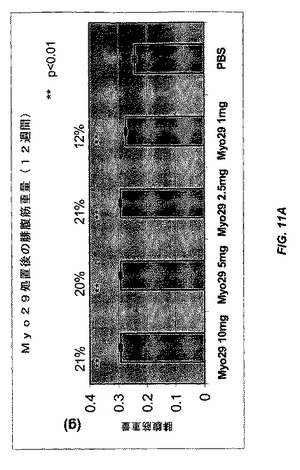
【図11B】
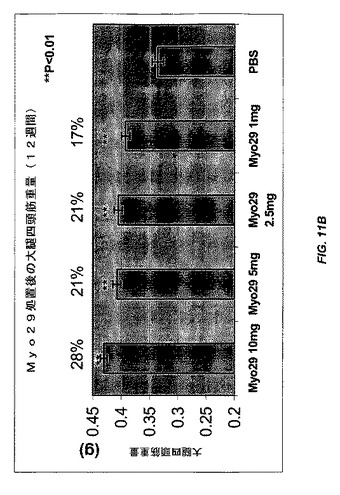
【図12】
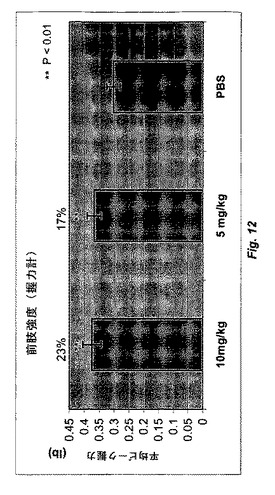
【図2】
【図3A】
【図3B】
【図4A】
【図4B】
【図4C】
【図4D】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10A】
【図10B】
【図11A】
【図11B】
【図12】
【公開番号】特開2010−148513(P2010−148513A)
【公開日】平成22年7月8日(2010.7.8)
【国際特許分類】
【出願番号】特願2010−19767(P2010−19767)
【出願日】平成22年1月29日(2010.1.29)
【分割の表示】特願2004−546306(P2004−546306)の分割
【原出願日】平成15年10月22日(2003.10.22)
【出願人】(591011502)ワイス エルエルシー (573)
【出願人】(505148324)メッドイミューン・リミテッド (8)
【Fターム(参考)】
【公開日】平成22年7月8日(2010.7.8)
【国際特許分類】
【出願日】平成22年1月29日(2010.1.29)
【分割の表示】特願2004−546306(P2004−546306)の分割
【原出願日】平成15年10月22日(2003.10.22)
【出願人】(591011502)ワイス エルエルシー (573)
【出願人】(505148324)メッドイミューン・リミテッド (8)
【Fターム(参考)】
[ Back to top ]