
GPR119調節因子
Gタンパク質共役受容体GPR119の活性を調節する式(I)の化合物、および動物における、Gタンパク質共役受容体GPR119の調節と関連付けられる疾患の治療のためのその使用が本明細書に記載される。
【図1】

【図1】
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規な部類の環縮合ピロリジン、それらの化合物を含有する医薬組成物、およびGタンパク質共役受容体GPR119の活性を調節するためのその使用に関する。
【背景技術】
【0002】
糖尿病は、異常なグルコース恒常性の結果として高濃度の血中グルコースが存在する障害である。最も一般的な形態の糖尿病は、I型糖尿病(インスリン依存型糖尿病とも呼ばれる)およびII型糖尿病(インスリン非依存型糖尿病とも呼ばれる)である。すべての糖尿病症例のおよそ90%を占めるII型糖尿病は、微小血管合併症(たとえば、網膜症、神経障害、および腎障害を含める)、ならびに大血管合併症(たとえば、加速性のアテローム性動脈硬化症、冠動脈心疾患、および卒中を含める)をもたらす深刻な進行性疾患である。
【0003】
現在、糖尿病の治療法は存在しない。この疾患の標準的な治療は限られており、血中グルコース濃度をコントロールして、合併症を最小限に抑える、または遅らせることに集中している。現行の治療は、インスリン抵抗性(メトホルミンもしくはチアゾリジンジオン)、またはβ細胞からのインスリン放出(スルホニル尿素、エキサナチド(exanatide))のどちらかをターゲットとしている。β細胞の脱分極を介して働くスルホニル尿素および他の化合物は、循環グルコース濃度とは無関係にインスリン分泌を刺激するので、低血糖を促進する。認可されている薬物の1つであるエキサナチドは、高グルコースの存在下でインスリン分泌を刺激するが、経口による生物学的利用能を欠くので、注射しなければならない。ジペプチジルペプチダーゼIV阻害剤であるシタグリプチンは、インクレチンホルモンの血中濃度を上昇させる薬物であり、そのインクレチンホルモンは、インスリン分泌を増加させ、グルカゴン分泌を減少させることができ、あまり特徴付けられていない他の効果ももち得る。しかし、シタグリプチンおよび他のジペプチジルペプチダーゼIV阻害剤は、他のホルモンおよびペプチドの組織濃度にも影響を及ぼすことがあり、このより広範な影響による長期的な結果については、十分な調査がなされていない。
【0004】
II型糖尿病では、筋細胞、脂肪細胞、および肝細胞がインスリンに正常に反応しない。この状態(インスリン抵抗性)は、細胞のインスリン受容体の数の減少、細胞のシグナル伝達経路の破綻、またはこの両方に起因する可能性がある。最初のうち、β細胞は、インスリン産生量を増加させてインスリン抵抗性の埋合せをする。しかし結局、β細胞は、正常なグルコース濃度(正常血糖)を維持するのに十分なインスリンを産生できなくなり、II型糖尿病への進行を示す。
【0005】
II型糖尿病では、β細胞の機能不全と相まったインスリン抵抗性により空腹時高血糖が生じる。β細胞異常機能不全には2つの態様がある。すなわち、1)(非刺激性の低グルコース濃度で起こる)基礎インスリン放出の増加(これは、II型糖尿病だけでなく、肥満のインスリン抵抗性前糖尿病段階でも認められる)、および2)高血糖負荷に反応して、インスリン放出が、すでに上昇している基礎レベルを上回って増加しないこと(この現象は、前糖尿病段階では起こらず、正常血糖のインスリン抵抗性段階からII型糖尿病へと移行する前兆となる場合もある)。後者の態様を治療する現行の療法としては、内在性の貯蔵インスリンの放出を誘発するためのATP感受性β細胞カリウムチャネルの阻害剤、および外因性インスリンの投与が挙げられる。どちらも血中グルコース濃度の的確な正常化を実現するものでなく、また低血糖を惹起するリスクを伴う。
【発明の概要】
【発明が解決しようとする課題】
【0006】
したがって、グルコース依存的に機能する薬剤の発見に大きな関心が寄せられている。このように機能する生理学的なシグナル伝達経路は、腸管ペプチドのGLP−1およびGIPを含めて、よく知られている。これらのホルモンは、同種のGタンパク質共役受容体を介して信号を送って、膵臓β細胞におけるcAMPの産生を刺激する。cAMPが増加しても、空腹時または食事前の状態の間はインスリン放出が刺激されないようである。しかし、ATP感受性カリウムチャネル、電位感受性カリウムチャネル、および開口分泌機構を含めた、cAMPのいくつかの生化学的ターゲットは、食後のグルコース刺激によるインスリン分泌が顕著に強化されるように調節される。したがって、同様に機能する、GPR119を含めた新規なβ細胞GPCRのアゴニスト調節因子も、II型糖尿病患者において、内在性インスリンの放出を刺激し、グルコース濃度の正常化を促進するということになる。たとえばGLP−1が刺激された結果としてcAMPが増加すると、β細胞増殖が促進され、β細胞死が抑制され、したがって膵島質量が向上することもわかっている。β細胞質量に対するこのプラスの効果は、インスリンが十分に産生されないII型糖尿病において有益となるはずである。
【0007】
代謝性疾患が他の生理系に悪影響を及ぼすことはよく知られており、複合的な疾患状態(たとえば、「シンドロームX」におけるI型糖尿病、II型糖尿病、不十分な耐糖能、インスリン抵抗性、高血糖、高脂血症、高トリグリセリド血症、高コレステロール血症、異脂肪症、肥満、または心血管疾患)、または腎疾患や末梢神経障害などの、糖尿病に引き続いて生じる続発性疾患がしばしば併発される。したがって、糖尿病状態の新しい治療に対する必要性が存在する。
【課題を解決するための手段】
【0008】
本発明によれば、新規な部類のGPR119調節因子が発見された。これらの化合物は、以下に示す式I
【0009】
【化1】
[式中、
Xは、AまたはB
【0010】
【化2】
であり、
Yは、Oまたは結合であり、
R1は、−C(O)−O−R3または
【0011】
【化3】
であり、
R2は、水素、シアノ、C1〜C6アルキル、またはC3〜C6シクロアルキルであり、
R3は、C1〜C6アルキル、C3〜C6シクロアルキル、またはC1〜C6アルキル、C1〜C6アルコキシ、C1〜C6フルオロアルキル、ハロ、もしくはヒドロキシで置換されているC3〜C6シクロアルキルであり、但し、ハロ、C1〜C6アルコキシ、またはヒドロキシ基は、R1中のOに連結している炭素原子では結合しておらず、
R4は、C1〜C6ハロアルキル、C1〜C6アルキル、ハロ、シアノ、またはC3〜C6シクロアルキルであり、
R5は、水素、シアノ、ニトロ、C1〜C6フルオロアルキル、C1〜C6アルキル、C1〜C6アルコキシ、C1〜C6フルオロアルコキシ、またはC3〜C6シクロアルキルであり、
R6は、水素、C1〜C6アルキル、C1〜C6フルオロアルキル、C3〜C6シクロアルキル、またはC3〜C6シクロアルキル、C1〜C6アルコキシ、もしくはヒドロキシルで置換されているC1〜C6アルキルであり、但し、C1〜C6アルコキシ基またはヒドロキシル基は、ピラゾール窒素に連結している炭素には結合しておらず、
R7aおよびR7bは、それぞれ独立に、水素、フルオロ、またはC1〜C6アルキルであり、
R8a、R8b、R8cおよびR8dは、それぞれ独立に、水素、C1〜C6アルキル、C3〜C6シクロアルキル、またはヒドロキシもしくはC1〜C6アルコキシで置換されているC1〜C6アルキルであり、
またはR8aおよびR8bは、これらが結合している炭素と一緒になって、C3〜C6シクロアルキルを形成していてもよく、
またはR8cおよびR8dは、これらが結合している炭素と一緒になって、C3〜C6シクロアルキルを形成していてもよく、
またはR8aとR8cは一緒になって、完全飽和二炭素橋を形成していてもよく、但し、R8aおよびR8cは、これらが結合している環系の同じ平面上にある]
または薬学的に許容できるその塩によって表すことができる。
【0012】
式Iの化合物は、Gタンパク質共役受容体の活性を調節する。より詳細には、式Iの化合物は、GPR119を調節する。そのため、前記化合物は、糖尿病などの、GPR119の活性が疾患の病理または症状の一因となる疾患の治療に有用である。そのような状態の例として、高脂血症、I型糖尿病、II型糖尿病、特発性I型糖尿病(Ib型)、成人潜在性自己免疫性糖尿病(LADA)、早発性2型糖尿病(EOD)、若年性非定型糖尿病(YOAD)、若年発症成人型糖尿病(MODY)、栄養不良関連糖尿病、妊娠糖尿病、冠動脈心疾患、虚血発作、血管形成術後の再狭窄、末梢血管疾患、間欠性跛行、心筋梗塞(たとえば、壊死およびアポトーシス)、異脂肪症、食後脂肪血症、耐糖能障害(IGT)状態、空腹時血漿グルコース異常(impaired fasting plasma glucose)状態、代謝性アシドーシス、ケトーシス、関節炎、肥満、骨粗鬆症、高血圧、うっ血性心不全、左室肥大、末梢動脈疾患、糖尿病性網膜症、黄斑変性、白内障、糖尿病性腎症、糸球体硬化症、慢性腎不全、糖尿病性ニューロパシー、メタボリック症候群、シンドロームX、月経前症候群、冠動脈心疾患、狭心症、血栓症、アテローム性動脈硬化症、一過性脳虚血発作、卒中、血管再狭窄、高血糖、高インスリン血症、高脂血症、高トリグリセリド血症、インスリン抵抗性、グルコース代謝障害、耐糖能障害状態、空腹時血漿グルコースの異常状態、肥満、勃起機能不全、皮膚および結合組織の障害、足の潰瘍化および潰瘍性大腸炎、内皮障害、ならびに血管伸展性の障害が挙げられる。式Iの化合物は、アルツハイマー病、統合失調症、認知障害などの神経障害の治療に使用することもできる。式Iの化合物は、炎症性腸疾患、潰瘍性大腸炎、クローン病、過敏性腸症候群などの胃腸疾患においても有益となる。上述のように、式Iの化合物は、肥満患者、特に糖尿病に罹患している肥満患者において体重減少を刺激するのにも使用することができる。
【0013】
本発明の別の実施形態は、式Iの化合物を含有する医薬組成物を対象とする。そのような製剤は通常、少なくとも1種の薬学的に許容できる賦形剤と混和された式Iの化合物を含有する。そうした製剤はまた、少なくとも1種の追加の(本明細書に記載の)薬剤を含有してもよい。そのような薬剤の例として、(後述する)抗肥満薬および/または抗糖尿病薬が挙げられる。本発明の追加の態様は、糖尿病および本明細書に記載の関連状態を治療する医薬の調製における式Iの化合物の使用に関する。
【図面の簡単な説明】
【0014】
【発明を実施するための形態】
【0015】
本発明は、本発明の例示的な実施形態についての以下の詳細な記述およびその中に含まれる実施例を参照することにより、より一層容易に理解することができる。
【0016】
本発明は、当然様々に異なり得る、特定の合成的製造方法に限定されないことを理解されたい。本明細書で使用する専門用語は、特定の実施形態について述べるためのものにすぎず、限定するものではないことも理解されたい。複数形および単数形は、数を示す以外では、交換可能であるとして扱うべきである。
【0017】
本文書内の見出しは、読者が文書に手早く目を通せるようにするために利用されるにすぎない。それらの見出しは、本発明または特許請求の範囲をいかなる形でも限定しないものと解釈すべきである。
a.「ハロ」または「ハロゲン」とは、塩素、フッ素、ヨウ素、または臭素原子を指す。
b.「アルキル」とは、分枝状または直鎖状アルキル基、たとえば、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、ペンチルなどを指す。
c.「アルコキシ」とは、直鎖または分枝鎖アルコキシ基、たとえば、メトキシ、エトキシ、n−プロポキシ、イソプロポキシ、n−ブトキシ、イソブトキシ、ペントキシなどを指す。
d.「シクロアルキル」とは、完全に水素化され、単環として存在する、非芳香族の環を指す。そのような炭素環の例として、シクロプロピル、シクロブチル、シクロペンチル、およびシクロヘキシルが挙げられる。
e.「ハロアルキル」とは、1個または複数のハロ基で置換されている直鎖または分枝鎖アルキル基、たとえば、クロロメタン、フルオロメタン、ジクロロメタン、ジフルオロメタン、ジブロモメタン、トリクロロメタン、トリフルオロメタン、クロロフルオロメタン、1,1,1,2−テトラフルオロエタンなどを指す。
f.「フルオロアルキル」とは、1個または複数のフルオロ基で置換されている直鎖または分枝鎖アルキル基、たとえば、フルオロメタン、ジフルオロメタン、トリフルオロメタンなどを指す。
g.「ハロアルコキシ」とは、1個または複数のハロ基で置換されている直鎖または分枝鎖アルコキシ基、たとえば、クロロメトキシ、フルオロメトキシ、ジクロロメトキシ、ジフルオロメトキシ、ジブロモメトキシ、トリクロロメトキシ、トリフルオロメトキシ、クロロフルオロメトキシ、1,1,1,2−テトラフルオロエトキシなどを指す。
h.「治療有効量」とは、(i)特定の疾患、状態、または障害を治療もしくは予防する、(ii)特定の疾患、状態、または障害の1つまたは複数の症状を緩和し、寛解させ、もしくは除去する、または(iii)本明細書に記載の特定の疾患、状態、または障害の1つまたは複数の症状の発症を予防し、もしくは遅らせる、本発明の化合物の量を意味する。
i.「患者」とは、温血動物、たとえば、モルモット、マウス、ラット、アレチネズミ、ネコ、ウサギ、イヌ、サル、チンパンジー、およびヒトを指す。
j.「治療する」とは、化合物が、患者の疾患(もしくは状態)または疾患に関連する任意の組織損傷を軽減し、解消し、またはその進行を緩慢にし得ることを指す。
h.用語「調節された」、「調節すること」、または「調節する」とは、本明細書では、別段指摘しない限り、本発明の化合物を用いた、Gタンパク質共役受容体GPR119の活性化を指す。
i.「薬学的に許容できる」とは、物質または組成物が、製剤を構成する他の成分および/またはそれによる治療を受ける哺乳動物と化学的および/または毒物学的に適合性でなければならないことを示す。
j.「塩」とは、薬学的に許容できる塩、および化合物の調製などの工業的プロセスでの使用に適する塩を指すものとする。
k.「薬学的に許容できる塩」とは、化合物の実際の構造に応じて、薬学的に許容できる酸付加塩」または「薬学的に許容できる塩基付加塩」のどちらかを指すものとする。
l.「薬学的に許容できる酸付加塩」とは、式Iまたはその中間体のいずれかによって表される化合物の非毒性のいかなる有機酸または無機酸付加塩にも該当するものとする。適切な塩を形成する無機酸の実例として、塩化水素酸、臭化水素酸、硫酸、およびリン酸、ならびにオルトリン酸一水素ナトリウム(sodium monohydrogen orthophosphate)や硫酸水素カリウムなどの酸金属塩が挙げられる。適切な塩を形成する有機酸の実例として、モノ、ジ、およびトリカルボン酸が挙げられる。そのような酸の実例は、たとえば、酢酸、グリコール酸、乳酸、ピルビン酸、マロン酸、コハク酸、グルタル酸、フマル酸、リンゴ酸、酒石酸、クエン酸、アスコルビン酸、マレイン酸、ヒドロキシマレイン酸、安息香酸、ヒドロキシ−安息香酸、フェニル酢酸、ケイ皮酸、サリチル酸、2−フェノキシ安息香酸、p−トルエンスルホン酸、ならびにメタンスルホン酸や2−ヒドロキシエタンスルホン酸などのスルホン酸である。このような塩は、水和した形態または実質的に無水の形態のどちらで存在してもよい。一般に、こうした化合物の酸付加塩は、水および種々の親水性有機溶媒に可溶性である。
m.「薬学的に許容できる塩基付加塩」とは、式Iまたはその中間体のいずれかによって表される化合物の非毒性のいかなる有機または無機塩基付加塩にも該当するものとする。適切な塩を形成する塩基の実例として、水酸化ナトリウム、カリウム、カルシウム、マグネシウム、バリウムなどの、アルカリ金属またはアルカリ土類金属水酸化物;アンモニア;ならびにメチルアミン、ジメチルアミン、トリメチルアミン、ピコリンなどの、脂肪族、脂環式、または芳香族有機アミンが挙げられる。
n.「式Iの化合物」、「本発明の化合物」、および「化合物」は、本出願全体にわたり区別なく使用しており、同義語として扱うべきである。
「異性体」とは、以下で定義するような「立体異性体」および「幾何異性体」を意味する。
o.「立体異性体」とは、1つまたは複数のキラル中心を有する化合物を意味し、各中心は、RまたはS立体配置で存在し得る。立体異性体として、すべてのジアステレオ異性体、鏡像異性体、およびエピマーの形態、ならびにそのラセミ体および混合物が挙げられる。
p.「幾何異性体」とは、シス、トランス、アンチ、シン、エントゲーゲン(E)、およびツザンメン(Z)の形態、ならびにその混合物として存在し得る化合物を意味する。
【0018】
本発明の化合物は、不斉中心またはキラル中心を含んでおり、したがって、異なる立体異性体形態で存在する。別段指定しない限り、本発明の化合物のすべての立体異性体形態、ならびにラセミ混合物を含めたその混合物は、本発明の一部をなすものとする。加えて、本発明は、すべての幾何異性体および位置異性体も包含する。たとえば、本発明の化合物に二重結合または縮合環が組み込まれている場合、シスおよびトランス両方の形態ならびに混合物が本発明の範囲内に包含される。
【0019】
ジアステレオ異性体混合物は、その物理化学的差異に基づき、クロマトグラフィーおよび/または分別結晶、蒸留、昇華などの当技術分野でよく知られている方法によって、その個々のジアステレオ異性体に分離することができる。鏡像異性体は、鏡像異性体混合物を、光学活性のある適切な化合物(たとえば、キラルアルコールやMosherの酸塩化物などのキラル助剤)との反応によってジアステレオ異性体混合物に変換し、ジアステレオ異性体を分離し、個々のジアステレオ異性体を対応する純粋な鏡像異性体に変換(たとえば、加水分解)することにより分離できる。また、本発明の化合物の一部は、アトロプ異性体(たとえば、置換ビアリール)である場合もあり、本発明の一部とみなされる。鏡像異性体は、キラルHPLC(高圧液体クロマトグラフィー)カラムを使用して分離することもできる。
【0020】
本発明の中間体および化合物は、異なる互変異性体形態で存在し得ることも考えられ、そうしたすべての形態が本発明の範囲内に包含される。用語「互変異性体」または「互変異性体形態」とは、低いエネルギー障壁で相互変換可能な、異なるエネルギーの構造異性体を指す。たとえば、プロトン互変異性体(プロトトロピー互変異性体としても知られる)は、ケト−エノール異性体およびイミン−エナミン異性体などの、プロトンの移動による相互変換を伴う。プロトン互変異性体の具体的な例は、プロトンが2つの環窒素間を移動し得るイミダゾール部分である。原子価互変異性体は、結合電子の一部が再編成されることによる相互変換を伴う。一部の中間体(および/または中間体の混合物)の閉じた形態と開いた形態の平衡は、当業者に知られている、アルドースが関与する変旋光の過程を連想させる。
【0021】
加えて、本発明の化合物は、溶媒和していない形で存在しても、水、エタノールなどの薬学的に許容できる溶媒と溶媒和した形で存在してもよい。一般に、本発明の目的では、溶媒和した形態は、溶媒和していない形態と同等であるとみなす。化合物は、1種または複数の結晶状態、すなわち多形体で存在する場合もあり、または非晶質固体として存在する場合もある。そのようなすべての形態が特許請求の範囲に包含される。
【0022】
本発明は、1個または複数の原子が、原子質量または質量数が自然界で通常見られる原子質量または質量数とは異なる原子で置き換えられていること以外は、本明細書で列挙するものと同一である、同位体標識された本発明の化合物も包含する。本発明の化合物に組み込むことのできる同位体の例として、2H、3H、11C、13C、14C、13N、15N、15O、17O、18O、31P、32P、35S、18F、123I、125I、36Clなどの、それぞれ水素、炭素、窒素、酸素、リン、硫黄、フッ素、ヨウ素、および塩素の同位体が挙げられる。
【0023】
本発明の特定の同位体標識された化合物(たとえば、3Hおよび14Cで標識されたもの)は、化合物および/または基質の組織分布アッセイにおいて有用である。トリチウム化同位体(すなわち3H)および炭素14同位体(すなわち14C)は、調製しやすく検出可能であるので特に好ましい。さらに、ジュウテリウム(すなわち2H)などのより重い同位体で置換すると、代謝安定性がより高いために得られる特定の治療上の利点(たとえば、in vivo半減期の延長または投与必要量の減少)がもたらされる場合もあり、したがってある状況においては好ましいこともある。15O、13N、11C、18Fなどの陽電子放射同位体は、基質占有率を調べる陽電子放射断層撮影(PET)研究に有用である。同位体標識された本発明の化合物は、一般に、同位体標識されていない試薬の代わりに同位体標識された試薬を用いることにより、本明細書において以下でスキームおよび/または実施例の中で開示する手順と同様の手順に従って調製することができる。
【0024】
式Iの化合物のいくつかは、以下で図示するように、エーテル結合を介してピリミジン環に結合した3−オキサ−7−アザビシクロ[3.3.1]ノナン環を含んでいる。このアザビシクロ−ノナンは、幾何異性体として存在し、以下で図示するシンまたはアンチ異性体のいずれかとして存在し得る。
【0025】
【化4】
【0026】
式Iを有する化合物の一実施形態では、XはAであり、R1は−C(O)−O−R3である。
【0027】
式Iを有する化合物の別の実施形態では、R8a、R8b、R8cおよびR8dは、それぞれ水素であり、R3は、C1〜C3アルキルで置換されているC3〜C6シクロアルキルである。
【0028】
式Iを有する化合物の別の実施形態では、R7aおよびR7bは、それぞれ独立に、水素、フルオロ、またはC1〜C3アルキルである。
【0029】
式Iを有する化合物の別の実施形態では、R2は水素であり、R5はC1〜C6アルキルである。
【0030】
合成
本発明の化合物は、化学分野でよく知られている方法と類似した方法を包含する合成経路によって、特に本明細書に収められている記述に照らして、合成することができる。出発材料は、Aldrich Chemicals(ウィスコンシン州ミルウォーキー)などの市販品供給元から一般に入手可能であり、または当業者に知られている方法を使用して容易に調製される(たとえば、Louis F.FieserおよびMary Fieser、Reagents for Organic Synthesis、1〜19巻、Wiley、ニューヨーク(1967〜1999年版)、またはBeilsteins Handbuch der organischen Chemie、第4版、ベルリン、Springer−Verlag編(増刊を含める)(Beilsteinオンラインデータベースからも入手可能)に一般に記載されている方法によって調製される)。
【0031】
例示する目的で、以下で図示する反応スキームにより、本発明の化合物ならびに重要中間体を合成する潜在的経路を示す。個々の反応ステップのより詳細な説明については、以下の実施例の部を参照されたい。当業者なら、本発明の化合物の合成に他の合成経路を使用してもよいことがわかるであろう。詳細な出発材料および試薬をスキームの中で示し、以下で論じるが、他の出発材料および試薬で容易に置き換えて、様々な誘導体および/または反応条件を準備することができる。加えて、以下で述べる方法によって調製される化合物の多くは、当業者によく知られている従来の化学を使用し、この開示に照らしてさらに改変することができる。
【0032】
式Iの化合物は、当技術分野で同様に知られているエーテル生成の方法を使用して調製することができる。1)Hughes,D.L.、Organic Reactions 1992、42 335−656、米国ニュージャージー州ホーボーケン、2)Tikad,A.、Routier,S.、Akssira,M.、Leger,J.−M.l、Jarry,C.、Guillaumet,G.、Synlett 2006、12、1938〜42、および3)Loksha,Y.M.、Globisch,D.、Pedersen,E.B.、La Colla,P.、Collu,G.、Loddo,R.、J.Het.Chem.2008、45、1161〜6などの、そうした反応についてより詳細に記載している教本に目を向けられたい。
【0033】
すぐ下のスキームIは、式Iの化合物を組み立てるための代替方法を例示するものである。分子の中心部分は、置換されていてもよいピリミジン環である。式Iの化合物は、以下に示すように、ピリミジンとエーテル結合およびアミノ結合の両方を形成することにより生成される。この反応シーケンスをどの順序で実施するかは重要でないが、但し、R5がシアノまたはニトロである場合は例外である。そうした場合では、ステップI−BおよびI−Cを使用して式Iの化合物を組み立てる。
【0034】
【化5】
【0035】
反応スキームIの出発材料は、R2およびR5が通常、本明細書に記載する通り、最終生成物において所望されるものと同じ置換基によって表される、構造化合物I−1のジヒドキシ−ピリミジンである。このようなピリミジンの生成方法は、当技術分野で知られている。
【0036】
ステップI−Aの塩素化反応は、当技術分野で知られているとおりに実施する。構造I−1の化合物を、トリエチルアミン、N,N−ジメチルアニリン、N,N−ジイソプロピルエチルアミンなどの添加剤を加えまたは加えずに、過剰に使用されるか、またはトルエン、ベンゼン、キシレンなどの溶媒中で使用される、POCl3(オキシ塩化リン)などの塩素化試薬と反応させる(Matulenko,M.A.ら、Bioorg.Med.Chem.2007、15、1586〜1605)。この反応は、条件の選択に応じて、室温(約23℃)〜約140℃の範囲の温度で実施することができる。代替塩素化試薬は、PCl3(三塩化リン)、POCl3/PCl5(五塩化リン)、塩化チオニル、塩化オキサリル、またはホスゲンからなり、構造I−2のジクロロピリミジンを与えることができる。場合によっては、構造I−2のジクロロピリミジンは、市販品供給元から入手することもできる。任意選択により、構造I−2のジクロロピリミジンを反応液から単離および回収し、さらに当技術分野で知られているとおりに精製してもよい。別法として、以下で記載するステップI−Bにおいて未精製材料を使用してもよい。
【0037】
スキームIのステップI−Bでは、構造I−3のテトラヒドロピロロ[3,4−c]ピラゾールと構造I−2のジクロロピリミジン間にアミノ結合を形成する。構造I−3の縮合ピロリジンにおいて、R6、R8a、R8b、R8cおよびR8dは通常、本明細書に記載するとおり、最終生成物において所望されるものと同じ置換基によって表される。このようなテトラヒドロピロロ[3,4−c]ピラゾール誘導体は、文献で知られており、または当業者によく知られている様々な方法によって好都合に調製することができる(Heterocycles、2002、56、257〜264)。アミノ結合は、エタノール、プロパノール、イソプロパノール、ブタノールなどの極性プロトン性溶媒中にて、当量の構造I−2とI−3の化合物を、どの溶媒を使用するかに応じて約0〜120℃の範囲の温度で0.5〜24時間反応させることにより形成する。この反応に利用する典型的な条件は、108℃で1時間加熱を行う溶媒としてのイソプロパノールの使用である。別法として、トリエチルアミンやジエチルイソプロピルアミンなどのアミン塩基、または炭酸水素ナトリウム、炭酸カリウム、炭酸ナトリウムなどの無機塩基をこの反応に加えてもよい。上記アミンまたは無機塩基の1種を使用する場合では、溶媒をアセトニトリル、N,N−ジメチルホルムアミド(「DMF」)、テトラヒドロフラン(「THF」)、1,4−ジオキサンなどの極性非プロトン性溶媒(約0〜100℃で0.5〜24時間)に変更することができる。この反応に利用する典型的な条件として、アセトニトリル中にて室温で3時間のジエチルイソプロピルアミンの使用が挙げられる。また、水、メタノール、エタノール、プロパノールなどの単独または組合せの極性プロトン性溶媒中での塩酸の使用も、約0〜110℃の温度でのこの変換に使用することができる。典型的な条件は、エタノール中にて78℃で水を使用するものである。構造I−5の中間体は、反応液から単離および回収し、当技術分野で知られているとおりにさらに精製することができる。別法として、以下で記載するステップI−Cにおいて未精製材料を使用してもよい。
【0038】
スキームIのステップI−Cでは、構造I−5の中間体と構造I−4のアルコール間にエーテル結合を形成して、式Iの化合物を生成する。構造I−4のアルコールにおいて、Xは、A、BまたはCであり、R7aおよびR7bは、所望の最終生成物に見られるものと同じ置換基によって表される。R1によって表される置換基は、式Iの核を生成した後に操作することができる。そのような変形形態は、当業者によく知られており、本発明の一部とみなすべきである。ステップI−Cでは、DMF、THF、1,2−ジメトキシエタン、1,4−ジオキサン、N,N−ジメチルアセトアミド、ジメチルスルホキシド(「DMSO」)などの溶媒中にて、水素化ナトリウム;ナトリウムおよびカリウムtert−ブトキシド;ナトリウム、カリウム、およびリチウムビス(トリメチルシリル)アミド;ナトリウム、カリウム、およびリチウムtert−アミルオキシドなどの塩基の存在下、当量の反応物を反応させる。この変換の典型的な条件として、1,4−ジオキサン中にて約105℃で1時間のナトリウムビス(トリメチルシリル)アミドの使用が挙げられる。
【0039】
反応が完了した後、所望の式Iの化合物は、当技術分野で知られているとおりに回収し、単離することができる。化合物は、当技術分野で知られているように、蒸発、抽出などによって回収することができる。化合物は、場合により、クロマトグラフィー、再結晶、蒸留、または当技術分野で知られている他の技術によって精製してもよい。
【0040】
上で反応スキームIに示した代替合成では、構造I−2のジクロロ−ピリミジンを構造I−4のアルコールと最初に反応させて、構造I−6によって示される中間体を生成する。ステップI−Cでのように、構造I−4は、所望の最終生成物に応じて、XがA、BまたはCであるアルコールとなる。これらのヘテロ環において、R1およびR4は通常、最終生成物において所望されるものと同じ置換基で表されることとなり、またはR1は、式Iの核を生成した後に操作することができる。
【0041】
当量の構造I−2と構造I−4の化合物を極性非プロトン性溶媒および塩基の存在下で反応させて、ステップI−Dに示す構造I−6の中間体を生成する。適切な系として、0〜140℃の温度で、DMF、THF、1,2−ジメトキシエタン、1,4−ジオキサン、N,N−ジメチルアセトアミド、DMSOなどの溶媒中の、水素化ナトリウム、ナトリウムおよびカリウムtert−ブトキシド、ナトリウム、カリウム、およびリチウムビス(トリメチルシリル)アミド;ナトリウム、カリウム、およびリチウムtert−アミルオキシドなどの塩基が挙げられる。この変換の典型的な条件として、THF中にて約0℃〜室温で14時間のカリウムtert−ブトキシドの使用が挙げられる。構造I−6の中間体は、反応液から単離および回収し、当技術分野で知られているとおりにさらに精製することができる。別法として、以下で記載するステップI−Eにおいて未精製材料を使用してもよい。
【0042】
次いで、構造I−6の中間体を、上で予め記載した縮合テトラヒドロピロロ[3,4−c]ピラゾール誘導体I−3と反応させることにより、式Iの化合物を生成することができる。通常、塩基の存在下で、当量の構造I−3の縮合ピロリジンを式I−6のクロロ中間体と反応させる。適切な塩基は、DMF、THF、1,2−ジメトキシエタン、1,4−ジオキサン、N,N−ジメチルアセトアミド、もしくはDMSO、またはこれらの混合物などの溶媒中の、水素化ナトリウム、ナトリウムまたはカリウムtert−ブトキシド、ナトリウム、カリウム、またはリチウムビス(トリメチルシリル)アミド;およびナトリウム、カリウム、またはリチウムtert−アミルオキシドとすることができる。これらの反応は、使用する溶媒に応じて、約−10〜150℃の範囲の温度で実施することができる。通常、反応は、不活性雰囲気中にて約15分〜24時間の範囲の時間をかけて進行させる。適切な条件として、105℃で1時間、1,4−ジオキサン中の、ナトリウムビス(ジメチルシリル)アミドが挙げられる。
【0043】
別法として、この反応は、構造I−6の中間体と構造I−3のテトラヒドロピロロ[3,4−c]ピラゾール誘導体を、メタノール、エタノール、プロパノール、イソプロパノール、ブタノールなどの極性プロトン性溶媒中で0.5〜24時間加熱することにより実施してもよい。この変換の典型的な条件は、イソプロパノール中にて108℃で2時間加熱するものである。
【0044】
この反応は、式Iの化合物中に見られる重要な置換アミン結合の形成に遷移金属触媒を使用して実施することもできる。遷移金属触媒は、限定はしないが、トリフェニルホスフィン)パラジウム(Pd(PPh3)4)、塩化パラジウム(II)(PdCl2)、酢酸パラジウム(II)(Pd(OAc)2)、(トリス(ジベンジリデンアセトン)ジパラジウム(0)(Pd2(dba)3)、ヨウ化銅(I)(CuI)、酢酸銅(II)(Cu(OAc)2)、および銅(II)トリフルオロメタン(Cu(OTf)2)からなるものとすることができる。通常、これらの反応では塩基を利用する。パラジウム触媒と共に使用するのに適する塩基は、1,4−ジオキサン、THF、1,2−ジメトキシエタン、トルエンなどの適切な溶媒中のナトリウムtert−ブトキシド、カリウムtert−ブトキシド、カリウムtert−アミルオキシド、またはK3PO4とすることができる。銅触媒の使用について、適切な塩基は、DMF、DMSO、ジメチルアセトアミドなどの適切な溶媒中の炭酸ナトリウム、炭酸カリウム、炭酸セシウムなどのアルカリ塩基からなるものとすることができる。
【0045】
通常、配位子を加えると、アミン生成反応を促進することができる。パラジウムを触媒とする反応のための配位子として、限定はしないが、9,9−ジメチル−4,5−ビス(ジフェニルホスフィノ)キサンテン(キサントホス)、2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチル(BINAP)、1,1’−ビス(ジフェニルホスフィノ)フェロセン(DPPF)、2,8,9−トリイソブチル−2,5,8,9−テトラアザ−1−ホスファビシクロ[3.3.3]ウンデカン(P[N(i−Bu)CH2CH3]3N)、トリ−tert−ブチルホスフィン(tert−Bu3P)、(ビフェニル−2−イル)ビス(tert−ブチル)ホスフィン(JohnPhos)、Pd−PEPPSI(商標)−SIPr:(1,3−ビス(2,6−ジイソプロピルフェニル)−4,5−ジヒドロイミダゾール−2−イリデン)(3−クロロピリジル)パラジウム(II)ジクロリドを挙げることができる。銅を触媒とする反応に適する配位子として、限定はしないが、L−プロリン、N−メチルグリシン、ジエチルサリチルアミドを挙げることができる。式Iの化合物の生成に適する条件は、トルエン中にてPd2(dba)3をナトリウムtert−ブトキシドと共に120℃で12時間使用するものである。
【0046】
反応を完了した後、所望の式Iの化合物は、当技術分野で知られているとおりに回収および単離することができる。化合物は、当技術分野で知られているような蒸発、抽出などによって回収することができる。化合物は、クロマトグラフィー、再結晶、蒸留、または先行技術で知られている他の技術によって、場合により精製してもよい。
【0047】
また、当業者には容易に理解されるように、R1によって表される置換基の多くは、式Iの核を生成した後に操作することができる。そのような変形形態は、当業者によく知られており、本発明の一部とみなすべきである。
【0048】
【化6】
【0049】
スキームIIは、本発明の式IにおいてX=Bに対応する構造II−14およびII−15のアルコールの生成方法について記載したものである。R3、R6、R7aおよびR7bは通常、本明細書に記載するとおり、最終生成物で所望されるものと同じ置換基によって表される。構造II−7の化合物からの構造II−8の化合物の合成は、当技術分野で知られている。それらの変換(ステップII−A)は、文献で教示されており、J.Org.Chem.、1981、46、3196〜3204、JP2009096744、WC035303、J.Am.Chem.Soc.2008、130、5654〜5655、およびOrg.Lett.、2006、3、430〜436において例示されている。スキームIIのステップII−Bでは、メタノールのようなアルコール系溶媒中にて約0℃〜室温の範囲の温度で水素化ホウ素ナトリウムを使用するなどの、当技術分野で知られている標準のプロトコールを使用して、ケトンのカルボニル基を還元する。ステップII−D、すなわち、構造II−10からのベンジル保護基を除去してII−11を得るステップは、水素化分解によって実現することができる。この反応の典型的な条件として、水素、および5〜20%パラジウム担持炭素や10〜20%水酸化パラジウムなどのパラジウム触媒の利用が挙げられる。この反応の典型的な溶媒は、エタノール、メタノール、テトラヒドロフラン、または酢酸エチルである。
【0050】
最終生成物中にピリミジン置換基が所望される場合、エタノールやメタノールなどのプロトン性溶媒中、または1,4−ジオキサン、テトラヒドロフラン、N,N−ジメチルホルムアミドもしくはジメチルスルホキシドなどの極性非プロトン性溶媒中にて、炭酸セシウムやN,N−ジイソプロピルエチルアミンなどの塩基の存在下、化合物II−11を、構造II−12として示す適切に置換された2−クロロピリミジンに付加して、構造II−14を生成することができる。こうした反応は、およそ室温〜約110℃の範囲の温度で実施することができる。別法として、溶媒を使用せずN,N−ジイソプロピルエチルアミンなどの塩基の存在下、または化合物II−11を過剰に使用する場合では塩基または溶媒を使用せずに、構造II−11と構造II−12の化合物を一緒に加熱することもできる。
【0051】
最終生成物中にカルバメート置換基が所望される場合、ジクロロメタンまたはクロロホルム中にて、N,N−ジイソプロピルエチルアミン、トリエチルアミン、ピリジンなどの塩基の存在下、等価な量の構造II−13のアルキルハロホルメートを、構造II−11の化合物と反応させる。別法として、ジクロロメタン、クロロホルム、テトラヒドロフランなどの溶媒中にて、N,N−ジイソプロピルエチルアミン、ピリジン、2,6−ルチジン、トリエチルアミンなどのアミン塩基の存在下、二炭酸ジ−tert−ブチル(BOC無水物)や二炭酸ジイソプロピルなどの二炭酸ジアルキルを使用して、構造II−11の化合物から構造II−15の化合物を生成することもできる。加えて、R3=1−メチル−シクロプロピルまたは1−ジフルオロメチル−シクロプロピルであるとき、ジクロロメタン、ジクロロエタン、ジメトキシエタン、テトラヒドロフランのような溶媒中にて、トリエチルアミン、N,N−ジイソプロピルエチルアミンなどのような塩基の存在下、約0℃〜周囲温度程度の範囲の温度でカーボネートII−13’(WO09105717およびWO09005677を参照されたい)を使用して、カルバメート官能基を導入することができる。
【0052】
最終の構造II−14またはII−15は、当技術分野で知られているとおりに単離し、精製することができる。所望なら、この最終構造を分離ステップにかけて所望のシンまたはアンチ異性体を得ることもできる。
【0053】
別法として、ビス−アミノールエーテル誘導体II−9とケトンII−7間の二重マンニッヒ反応を介して、その後、ケトンカルボニルを還元し、官能基を操作してII−10型の構造を得ることにより、R7aおよびR7bの少なくとも一方が水素である式II−10の非対称構造に到達することができる。特定の例では、R9aは、構造II−10において示すベンジル基よりもα−メチル−ベンジル基とすることが好ましいことが認識される。適切なR9b基としては、メチルまたはエチルが挙げられる。式II−8の構造を得るための二重マンニッヒ反応の使用は、化学文献に発表されており(Tetrahedron 2005 61、5876〜5888;Org.Lett.2006 8、3399〜3401)、当業者によって実現することができる。同様に、容易に入手できる出発材料から出発し、シス−またはトランス−ジフルオロ−2,6シクロヘキサノンの生成に関する化学文献で知られているプロトコール(Tetrahedron 1970 26、2447)を使用して、R7aおよびR7bが両方ともフルオロである式II−10の構造に到達することもできる。
【0054】
スキームIIIでは、式IにおいてX=Aに対応する式III−19の化合物の調製について記載する。
【0055】
【化7】
【0056】
スキームIIIに示すように、R3が本明細書に記載のとおりであり、R7aおよびR7bの少なくとも一方が水素である式III−19の化合物は、市販されているN−tert−ブトキシカルボニル−4−ピペリドン(Aldrich)から出発して、または4−ピペリドンから、その後にカルバメート生成して調製することができる。式III−19の化合物は、ステップIII−Aによって示すように、式III−16またはIII−18の化合物のケトンカルボニルの還元による還元によって調製する。このための適切な条件として、エタノールなどのアルコール系溶媒とTHFの混合物中での水素化ホウ素ナトリウムの使用が挙げられる。R7aおよびR7bの少なくとも一方がフルオロである式III−19の化合物は、J.Org.Chem.2003 68、3232およびJ.Org.Chem.2002 67、8610に記載されているように、ケトンをエノール化してシリルエノールエーテルとしてトラップし、適切な求電子性フルオロ供給源と反応させることにより調製することができる。R7aおよびR7bの少なくとも一方がアルキル基である式III−18の化合物は、ハロゲン化アルキルやアルキルスルホネートなどの適切な求電子性アルキル基を使用して同様に調製することができる。加えて、R7aおよびR7bが両方ともフルオロなどのハロである式III−19の構造には、容易に入手できるN−tert−ブトキシカルボニル−4−ピペリドンから、化学文献で知られている同様のプロトコール(Tetrahedron、1970、26、2447)を使用して到達することができる。本発明の式IにおいてXがAであるとき、そのようなピペリジン化合物は、市販されているか、文献で知られているか、または市販(Aldrich)のN−tert−ブトキシカルボニル−4−ピペリドンもしくは適切にN保護された他のピペリドンから調製できることもまた認識される。
【0057】
tert−ブチルオキシカルボニル基(R3はtert−ブチルである)は、合成の多くの段階で、塩酸やトリフルオロ酢酸などの酸を使用して除去することができ、その結果生じる遊離アミンは、スキームIIのステップII−E’およびII−Eでそれぞれ記載した一般的な条件を使用して、別のカルバメートまたはピリミジンに変換できることが認識される。式III−19の化合物の調製は、WO2009014910にも記載されている。
【0058】
スキームIVでは、式IV−23の化合物の合成について記載する。
【0059】
【化8】
【0060】
スキームIVの式IV−23のテトラヒドロピロロ−ピラゾールは、式IV−21の化合物から、適切な式IV−22のヒドラジンとの付加/脱水環化(ステップIV−B)に続いて、tert−ブチルオキシカルボニル基を脱保護する(ステップIV−C)ことにより調製することができる。式IV−21の化合物は、スキームIVの式IV−20の化合物から、ホルミル化反応(ステップIV−A)によって調製することができる。スキームIVのステップIV−Aの適切な条件として、化合物IV−20をN,N−ジメチルホルムアミドジメチルアセタール(DMFDMA)の存在下で加熱することが挙げられる。スキームIVの式IV−20の化合物は、市販されており(Aldrich)、文献で知られており、または当業者により容易に調製することができる。
【0061】
R8aおよびR8bまたはR8cおよびR8dが、これらが結合している炭素と一緒になって、C3〜C6シクロアルキルを形成している例は、Journal of Organic Chemistry 2004 69、2755〜2759およびJournal of Organic Chemistry 1962 27、2901〜5において見られる。R8aとR8cは一緒になって、完全飽和二炭素橋を形成していてもよく、R8aおよびR8cが、これらが結合している環系の同じ平面上にある例としては、(1R,4S)−2−オキソ−7−アザビシクロ[2.2.1]ヘプタン−7−カルボン酸tert−ブチル(Brother Chemistry Co.から市販されている、CAS番号16513−98−2)が挙げられる。この化合物のラセミ体も、文献で知られている(Journal of Medicinal Chemistry 2003 46、921〜924;Journal of Organic Chemistry 1994 59、1771〜8、Bioorganic & Medicinal Chemistry Letters 2008 18、4651〜4654)。
【0062】
当業者には容易にわかるように、中間体の遠位官能基(たとえば、第一級または第二級アミン)を保護する必要がある場合もある。そのような保護の必要性は、その遠位官能基の性質および調製方法の条件に応じて異なってくる。適切なアミノ保護基(NH−Pg)として、アセチル、トリフルオロアセチル、t−ブトキシカルボニル(BOC)、ベンジルオキシカルボニル(CBZ)、および9−フルオレニルメチレンオキシカルボニル(Fmoc)が挙げられる。同様に、「ヒドロキシ保護基」とは、ヒドロキシ官能基を封鎖または保護する、ヒドロキシ基の置換基を指す。適切なヒドロキシル保護基(O−Pg)として、たとえば、アリル、アセチル、シリル、ベンジル、para−メトキシベンジル、トリチルなどが挙げられる。このような保護の必要性は、当業者によって容易に決定される。保護基およびその使用に関する総説については、T.W.Greene、Protective Groups in Organic Synthesis、John Wiley&Sons、ニューヨーク、1991を参照されたい。
【0063】
上述のとおり、本発明の化合物の一部は、薬学的に許容できるカチオンと塩を形成することができる。本発明の化合物の一部は、薬学的に許容できるアニオンと塩を形成することができる。そのような塩はすべて、本発明の範囲内にあり、従来の方法によって、たとえば、適宜、水性、非水性、または部分的に水性の媒質中にて、酸性実体と塩基性実体とを、通常は化学量論比で合わせるなどして調製することができる。塩は、適宜、濾過、非溶媒で沈殿させてからの濾過、溶媒の蒸発、または水溶液の場合では凍結乾燥によって回収する。化合物は、エタノール、ヘキサン、水/エタノール混合物などの適切な(1種または複数の)溶媒に溶解させるなどの、当技術分野で知られている手順に従って、結晶の形で得る。
【0064】
本発明は、原子質量または質量数が自然界で通常見られる原子質量または質量数と異なっている原子で1個または複数の原子が置き換えられていること以外は本明細書で列挙したものと同一である、同位体標識された本発明の化合物も包含する。本発明の化合物に組み込むことのできる同位体の例として、2H、3H、11C、13C、14C、13N、15N、15O、17O、18O、31P、32P、35S、18F、123I、125I、36Clなどの、それぞれ、水素、炭素、窒素、酸素、リン、硫黄、フッ素、ヨウ素、および塩素の同位体が挙げられる。
【0065】
特定の同位体標識された本発明の化合物(たとえば、3Hおよび14Cで標識された化合物)は、化合物および/または基質の組織分布アッセイにおいて有用である。トリチウム、14C、35S、および125Iが組み込まれている特定の同位体標識されたリガンドは、放射リガンド結合アッセイにおいて有用となり得るはずである。トリチウム化(すなわち3H)およびカーボン14(すなわち14C)同位体は、調製しやすく、検出性がよいので、特に好ましい。さらに、ジュウテリウム(すなわち2H)などのより重い同位体での置換は、代謝安定性がより高いために生じる特定の治療上の優位性(たとえば、in vivo半減期の延長または投与必要量の減少)をもたらす場合もあり、したがって、状況によっては好ましいこともある。15O、13N、11C、18Fなどの陽電子放射同位体は、受容体占有率を調べるための陽電子放射断層撮影(PET)研究に有用である。本発明の同位体標識された化合物は、一般に、同位体標識されていない試薬の代わりに同位体標識された試薬を用いて、スキームおよび/または後述の実施例で開示する手順と類似した手順に従って調製することができる。
【0066】
特定の本発明の化合物は、2種以上の結晶形で存在する場合もある(一般に「多形体」と呼ばれる)。多形体は、種々の条件下での結晶化によって、たとえば、結晶化の際に、異なる再結晶用溶媒もしくは溶媒混合物、異なる温度での結晶化、および/または超急速冷却から超緩速冷却の範囲の種々の冷却モードを使用して、調製することができる。多形体は、本発明の化合物を加熱または溶融した後、徐々にまたは急速に冷却して得ることもできる。多形体の存在は、固体プローブNMR分光法、IR分光法、示差走査熱量測定、粉末X線回折、または他のそのような技術によって判定することができる。
【0067】
医学的使用
本発明の化合物は、Gタンパク質共役受容体GPR119の活性を調節する。そのため、前記化合物は、糖尿病などの、GPR119の活性が疾患の病理または症状の一因となる疾患の予防および治療に有用である。したがって、本発明の別の態様は、個体において代謝性疾患および/または代謝関連障害を治療する方法であって、そのような治療が必要である個体に、治療有効量の本発明の化合物、前記化合物の塩、またはそのような化合物を含有する医薬組成物を投与することを含む方法を包含する。代謝性疾患および代謝関連障害は、限定はしないが、高脂血症、I型糖尿病、II型糖尿病、特発性I型糖尿病(Ib型)、成人潜在性自己免疫性糖尿病(LADA)、早発性2型糖尿病(EOD)、若年性非定型糖尿病(YOAD)、若年発症成人型糖尿病(MODY)、栄養不良関連糖尿病、妊娠糖尿病、冠動脈心疾患、虚血発作、血管形成術後の再狭窄、末梢血管疾患、間欠性跛行、心筋梗塞(たとえば、壊死およびアポトーシス)、異脂肪症、食後脂肪血症、耐糖能障害(IGT)状態、空腹時血漿グルコースの異常状態、代謝性アシドーシス、ケトーシス、関節炎、肥満、骨粗鬆症、高血圧、うっ血性心不全、左室肥大、末梢動脈疾患、糖尿病性網膜症、黄斑変性、白内障、糖尿病性腎症、糸球体硬化症、慢性腎不全、糖尿病性ニューロパシー、メタボリック症候群、シンドロームX、月経前症候群、冠動脈心疾患、狭心症、血栓症、アテローム性動脈硬化症、心筋梗塞、一過性脳虚血発作、卒中、血管再狭窄、高血糖、高インスリン血症、高脂血症、高トリグリセリド血症、インスリン抵抗性、グルコース代謝障害、耐糖能障害状態、空腹時血漿グルコースの異常状態、肥満、勃起機能不全、皮膚および結合組織の障害、足の潰瘍化、内皮障害、高アポBリポタンパク質血症(hyper apo B lipoproteinemia)、ならびに血管伸展性の障害から選択される。さらに、本発明の化合物は、アルツハイマー病、統合失調症、認知障害などの神経障害の治療に使用することもできる。本発明の化合物は、炎症性腸疾患、潰瘍性大腸炎、クローン病、過敏性腸症候群などの胃腸疾患においても有益となる。上述のように、本発明の化合物は、肥満患者、特に糖尿病に罹患している肥満患者において体重減少を刺激するのにも使用することができる。
【0068】
前述の内容によれば、本発明はさらに、その必要がある対象において、上述の疾患または障害のいずれかの症状を予防し、または寛解させる方法であって、治療有効量の本発明の化合物を対象に投与することを含む方法を提供する。本発明の別の態様は、糖尿病およびその関連合併症を治療する医薬の調製を包含する。
【0069】
上述の治療特性を示すためには、化合物は、Gタンパク質共役受容体GPR119の活性化を調節するのに十分な量で投与する必要がある。この量は、治療する特定の疾患/状態、患者の疾患/状態の重症度、患者、投与する特定の化合物、投与経路、および患者内の他の基礎病態の存在などに応じて様々となり得る。全身に投与するとき、化合物は通常、上で挙げた疾患または状態のいずれに対しても、約0.1mg/kg/日〜約100mg/kg/日の投与量範囲でその効果を示す。毎日の連続投与が望ましい場合もあり、上で概略を述べた状態に応じて異なってくる。
【0070】
本発明の化合物は、様々な経路によって投与することができる。本発明の化合物は、経口投与することができる。本発明の化合物は、非経口(すなわち、皮下、静脈内、筋肉内、腹腔内、またはくも膜下腔内)、直腸、または局所投与することもできる。
【0071】
共投与
本発明の化合物は、本明細書に記載の疾患、状態、および/または障害を治療するための他の薬剤と共に使用することもできる。したがって、本発明の化合物を他の薬剤と組み合わせて投与することを含む治療方法も提供する。本発明の化合物と組み合わせて使用することのできる適切な薬剤として、抗肥満薬(食欲抑制薬を含める)、抗糖尿病薬、抗高血糖薬(anti−hyperglycemic agent)、高脂血症治療薬、および降圧薬が挙げられる。
【0072】
適切な抗糖尿病薬として、アセチル−CoAカルボキシラーゼ2(ACC−2)阻害剤、ジアシルグリセロールO−アシルトランスフェラーゼ1(DGAT−1)阻害剤、ホスホジエステラーゼ(PDE)10阻害剤、スルホニル尿素(たとえば、アセトヘキサミド、クロルプロパミド、ジアビネース(diabinese)、グリベンクラミド、グリピジド、グリブリド、グリメピリド、グリクラジド、グリペンチド(glipentide)、グリキドン、グリソラミド、トラザミド、およびトルブタミド)、メグリチニド、α−アミラーゼ阻害剤(たとえば、テンダミスタット(tendamistat)、トレスタチン、およびAL−3688)、α−グルコシドヒドロラーゼ阻害剤(たとえば、アカルボース)、α−グルコシダーゼ阻害剤(たとえば、アジポシン、カミグリボース、エミグリテート、ミグリトール、ボグリボース、プラジミシンQ、およびサルボスタチン(salbostatin))、PPARγ作動薬(たとえば、バラグリタゾン、シグリタゾン、ダルグリタゾン、エングリタゾン、イサグリタゾン(isaglitazone)、ピオグリタゾン、ロシグリタゾン、およびトログリタゾン)、PPARα/γ作動薬(たとえば、CLX−0940、GW−1536、GW−1929、GW−2433、KRP−297、L−796449、LR−90、MK−0767、およびSB−219994)、ビグアナイド(たとえば、メトホルミン)、グルカゴン様ペプチド1(GLP−1)作動薬(たとえば、エキセンディン3およびエキセンディン4)、タンパク質チロシンホスファターゼ1B(PTP−1B)阻害剤(たとえば、トロダスケミン(trodusquemine)、ヒルチオサール抽出物(hyrtiosal extract)、およびZhang,S.ら、Drug Discovery Today、12(9/10)、373〜381(2007)で開示されている化合物)、SIRT−1阻害剤(たとえば、レセルバトロール(reservatrol))、ジペプチジルペプチダーゼIV(DPP−IV)阻害剤(たとえば、シタグリプチン、ビルダグリプチン、アログリプチン、およびサクサグリプチン)、インスリン分泌刺激物質、脂肪酸酸化阻害剤、A2拮抗薬、c−junアミノ末端キナーゼ(JNK)阻害剤、インスリン、インスリン模倣物、グリコーゲンホスホリラーゼ阻害剤、VPAC2受容体作動薬、およびSGLT2阻害剤(ナトリウム依存性グルコース輸送体阻害剤、たとえば、ダパグリフロジンなど)が挙げられる。好ましい抗糖尿病薬は、メトホルミンおよびDPP−IV阻害剤(たとえば、シタグリプチン、ビルダグリプチン、アログリプチン、およびサクサグリプチン)である。
【0073】
適切な抗肥満薬として、11β−ヒドロキシステロイドデヒドロゲナーゼ1(11β−HSD1型)阻害剤、ステアロイルCoAデサチュラーゼ1(SCD−1)阻害剤、MCR−4作動薬、コレシストキニンA(CCK−A)作動薬、モノアミン再取込み阻害剤(シブトラミンなど)、交感神経様作動薬、β3アドレナリン作動薬、ドーパミン作動薬(ブロモクリプチンなど)、メラノサイト刺激ホルモン類似体、5HT2c作動薬、メラニン濃縮ホルモン拮抗薬、レプチン(OBタンパク質)、レプチン類似体、レプチン作動薬、ガラニン拮抗薬、リパーゼ阻害剤(テトラヒドロリプスタチン、すなわちオルリスタットなど)、食欲抑制薬(ボンベシン作動薬など)、ニューロペプチドY拮抗薬(たとえば、NPY Y5拮抗薬、PYY3−36(その類似体を含める)、甲状腺模倣薬(thyromimetic agent)、デヒドロエピアンドロステロンまたはその類似体、糖質コルチコイド作動薬または拮抗薬、オレキシン拮抗薬、グルカゴン様ペプチド1作動薬、毛様体神経栄養因子(Regeneron Pharmaceuticals,Inc.、ニューヨーク州タリータウン、およびProcter&Gamble Company、オハイオ州シンシナティーから入手可能なAxokine(商標)など)、ヒトアグーチ関連タンパク質(AGRP)阻害剤、グレリン拮抗薬、ヒスタミン3拮抗薬または逆作動薬、ニューロメジンU作動薬、MTP/ApoB阻害剤(たとえば、ジルロタピド(dirlotapide)などの消化管選択的MTP阻害剤)、オピオイド拮抗薬、オレキシン拮抗薬などが挙げられる。
【0074】
本発明の組合せ態様で使用するのに好ましい抗肥満薬として、消化管選択的MTP阻害剤(たとえば、ジルロタピド(dirlotapide)、ミトラタピド(mitratapide)およびイミプリタピド(implitapide)、R56918(CAS番号403987)、およびCAS番号913541−47−6)、CCKa作動薬(たとえば、PCT公開第WO2005/116034号または米国公開第2005−0267100A1号に記載のN−ベンジル−2−[4−(1H−インドール−3−イルメチル)−5−オキソ−1−フェニル−4,5−ジヒドロ−2,3,6,10b−テトラアザ−ベンゾ[e]アズレン−6−イル]−N−イソプロピル−アセトアミド)、5HT2c作動薬(たとえば、ロルカセリン)、MCR4作動薬(たとえば、US6,818,658に記載の化合物)、リパーゼ阻害剤(たとえば、セチリスタット)、PYY3−36(本明細書では、「PYY3−36」は、ベグ化PYY3−36(たとえば、米国公開2006/0178501に記載のもの)などの類似体を包含する)、オピオイド拮抗薬(たとえば、ナルトレキソン)、オレオイル−エストロン(CAS番号180003−17−2)、オビネピチド(obinepitide)(TM30338)、プラムリンチド(Symlin(登録商標))、テソフェンシン(NS2330)、レプチン、リラグルチド、ブロモクリプチン、オルリスタット、エクセナチド(Byetta(登録商標))、AOD−9604(CAS番号221231−10−3)、およびシブトラミンが挙げられる。本発明の化合物および併用療法は、運動および賢明な食生活と合わせて投与することが好ましい。
【0075】
上で引用した米国特許および公開はすべて、参照により本明細書に援用する。
【0076】
医薬製剤
本発明は、少なくとも1種の薬学的に許容できる賦形剤と混和された治療有効量の化合物または薬学的に許容できるその塩を含む医薬組成物も提供する。医薬組成物は、経口、局所、または非経口の使用に適合させた形態の組成物を包含し、上述のような糖尿病および関連状態の治療に使用することができる。
【0077】
医薬組成物は、皮下、吸入、経口、局所、非経口などの、当技術分野で知られている任意の経路による投与用に製剤することができる。医薬組成物は、限定はしないが、錠剤、カプセル剤、粉末、顆粒、ロゼンジ、または経口もしくは滅菌非経口溶液もしくは懸濁液などの液体製剤を含めて、当技術分野で知られているどんな形態でもよい。
【0078】
経口投与用の錠剤およびカプセル剤は、単位用量体裁にすることができ、従来の賦形剤、たとえば、シロップ、アカシア、ゼラチン、ソルビトール、トラガカント、ポリビニルピロリドンなどの結合剤;ラクトース、糖、トウモロコシデンプン、リン酸カルシウム、ソルビトール、グリシンなどの充填剤;ステアリン酸マグネシウム、タルク、ポリエチレングリコール、シリカなどの打錠滑沢剤;バレイショデンプンなどの崩壊剤;またはラウリル硫酸ナトリウムなどの許容される湿潤剤を含有してよい。錠剤は、標準の薬務でよく知られている方法に従ってコーティングしてもよい。
【0079】
経口液体製剤は、たとえば、水性もしくは油性の懸濁液、溶液、乳濁液、シロップ、もしくはエリキシルの形にすることもでき、または使用前に水もしくは適切な他のビヒクルで再形成する乾燥製品としての体裁にすることもできる。このような液体製剤は、従来の添加剤、たとえば、ソルビトール、メチルセルロース、グルコースシロップ、ゼラチン、ヒドロキシエチルセルロース、カルボキシメチルセルロース、ステアリン酸アルミニウムゲル、水素添加食用脂などの懸濁化剤;レシチン、モノオレイン酸ソルビタン、アカシアなどの乳化剤;扁桃油、グリセリンのような油性エステル、プロピレングリコール、エチルアルコールなどの非水性ビヒクル(食用油を含めてもよい);p−ヒドロキシ安息香酸メチルもしくはプロピル、ソルビン酸などの保存剤、および所望なら、従来の着香剤または着色剤を含有してよい。
【0080】
非経口投与では、化合物および無菌ビヒクル(水が好ましい)を利用して、流動性の単位剤形を調製する。化合物は、使用するビヒクルおよび濃度に応じて、ビヒクルまたは適切な他の溶媒に懸濁または溶解させることができる。溶液の調製では、化合物を、注射用の水に溶解させ、濾過滅菌した後、適切なバイアルまたはアンプルに充填し、密封することができる。有利にするため、局所麻酔剤、保存剤、緩衝剤などの薬品をビヒクルに溶解させることができる。安定性を高めるために、組成物をバイアルに充填した後凍結させ、真空中で水を除去することができる。次いで、凍結乾燥した乾燥粉末をバイアルに密閉し、使用前に液体を再形成するための付属のバイアルの注射用水を支給することができる。非経口懸濁液は、化合物をビヒクルに溶解させる代わりに懸濁させること、および滅菌が濾過によって実現できないことを除き、実質上同じようにして調製する。無菌ビヒクルに懸濁させる前にエチレンオキシドにさらすことにより、化合物を滅菌することができる。組成物に界面活性剤または湿潤剤を組み込んで、化合物の均一な分布を促進すると有利である。
【0081】
組成物は、投与方法に応じて、たとえば約0.1重量%〜約99重量%の活性材料を含有してよい。組成物が投与量単位を構成する場合、各単位は、たとえば、約0.1〜900mg、より典型的な場合では1mg〜250mgの活性成分を含有することになる。
【0082】
本発明の化合物は、他の抗糖尿病薬による類推によって、ヒト医学または獣医学で使用するための好都合な任意の方法における投与用に製剤することができる。そのような方法は、当技術分野で知られており、上で概略を述べている。そうした製剤の調製に関するより詳細な論述については、University of the Sciences in PhiladelphiaによるRemington’s Pharmaceutical Sciences、第21版に目を向けられたい。
【0083】
以下の実施例によって、本発明の実施形態を例示する。しかし、本発明の実施形態は、実施例の詳細な項目を限定せず、このためそれらの他の変形形態が、当業者には知るところとなり、またはこの開示に照らして明白となることを理解されたい。
【0084】
(実施例)
別段指定しない限り、出発材料は一般に、Aldrich Chemicals Co.(ウィスコンシン州ミルウォーキー)、Lancaster Synthesis,Inc.(ニューハンプシャー州Windham)、Acros Organics(ニュージャージー州フェアローン)、Maybridge Chemical Company,Ltd.(英国コーンウォール)、Tyger Scientific(ニュージャージー州プリンストン)、AstraZeneca Pharmaceuticals(英国ロンドン)、Mallinckrodt Baker(ニュージャージー州フィリップスバーグ)、EMD(ニュージャージー州Gibbstown)などの市販品供給元から入手可能である。
【0085】
一般実験手順
プロトン分析について、NMRスペクトルは、Varian Unity(商標)400(DG400−5プローブ)または500(DG500−5プローブ)(共にVarian Inc.、カリフォルニア州パロアルトから入手可能)を用い、室温にてそれぞれ400MHzまたは500MHzで記録した。化学シフトは、内部標準としての残存溶媒を基準とした百万分率(δ)で表示する。ピーク形状は、次のとおりに表記する。すなわち、s:一重線、d:二重線、dd:二重二重線、t:三重線、q:四重線、m:多重線、bs:ブロード一重線、2s:2本の一重線。
【0086】
大気圧化学イオン化質量スペクトル(APCI)は、Waters(商標)分光計(Micromass ZMD、キャリヤーガス:窒素)(Waters Corp.、米国マサチューセッツ州ミルフォードから入手可能)を用い、0.3mL/分の流量で、50:50の水/アセトニトリル溶離液系を利用して取得した。エレクトロスプレーイオン化質量スペクトル(ES)は、Waters(商標)(Micromass ZQまたはZMD機器(キャリヤーガス:窒素)(Waters Corp.、米国マサチューセッツ州ミルフォード)の液体クロマトグラフィー質量分析計を用い、各溶媒に0.01%のギ酸を加えた95:5〜0:100の勾配の水アセトニトリル溶液を利用して取得した。これらの機器には、3.75分間1mL/分または1.95分間2mL/分の流量で、Varian Polaris 5 C18−A20×2.0mmカラム(Varian Inc.、カリフォルニア州パロアルト)を用いた。
【0087】
カラムクロマトグラフィーは、Flash 40 Biotage(商標)カラム(ISC,Inc.、コネティカット州シェルトン)またはBiotage(商標)SNAPカートリッジKPsilまたはRedisep Rfシリカ(Teledyne Isco Incより)のいずれかを用い、シリカゲルを使用して窒素圧下で実施した。分取HPLCは、フォトダイオードアレイ(Waters 2996)および質量分析計(Waters/Micromass ZQ)検出スキームを備えたWaters FractionLynxシステムを使用して実施した。分析HPLC作業は、フォトダイオードアレイ、単収束四極子質量検出および蒸発光散乱検出スキームを備えたWaters 2795 Alliance HPLCまたはWaters ACQUITY UPLCを用いて行った。
【0088】
真空中での濃縮とは、ロータリーエバポレーターを使用して、減圧下で溶媒を蒸発させることを指す。
【0089】
別段言及しない限り、化学反応は室温(摂氏約23度)で実施した。また、別段言及しない限り、化学反応は窒素雰囲気中で進めた。
【0090】
薬理学的データ
Gタンパク質共役受容体GPR119の本発明の化合物によるアゴニスト活性化によって調節される疾患を治療する本発明の実施は、後述する機能アッセイの1つまたは複数における活性によって証明することができる。供給元は括弧内に示す。
【0091】
in−vitro機能アッセイ
β−ラクタマーゼ:
GPR119アゴニストについてのアッセイでは、ヒトGPR119のアゴニスト活性化を、環状AMP反応要素(CRE)によってβ−ラクタマーゼ産生と合体させた、細胞を主体とした(hGPR119 HEK293−CRE β−ラクタマーゼ)レポーター構築物を利用する。そしてGPR119活性は、FRETを可能にするβ−ラクタマーゼ基質であるCCF4−AM(Live Blazer FRET−B/G Loadingキット、Invitrogenカタログ番号K1027)を利用して測定する。詳細には、hGPR119−HEK−CRE−β−ラクタマーゼ細胞(Invitrogen 2.5×107/mL)を液体窒素貯蔵庫から取り出し、プレーティング培地(ダルベッコ変法イーグル培地高グルコース(DMEM、Gibcoカタログ番号11995−065)、10%熱不活性化ウシ胎児血清(HIFBS、Sigmaカタログ番号F4135)、1×MEM非必須アミノ酸(Gibcoカタログ番号15630−080)、25mMのHEPES pH7.0(Gibcoカタログ番号15630−080)、200nMのクラブラン酸カリウム(Sigmaカタログ番号P3494)に希釈した。細胞プレーティング培地を使用して細胞濃度を調節し、この細胞懸濁液(12.5×104生細胞)50μLを、ポリ−d−リシンでコートされた黒色透明底384ウェルプレート(Greiner Bio−Oneカタログ番号781946)の各ウェルに加え、5%の二酸化炭素を含有する加湿した環境において37℃でインキュベートした。4時間後、プレーティング培地を除去し、40μLのアッセイ培地(アッセイ培地は、クラブラン酸カリウムおよびHIFBSを含有しないプレーティング培地である)と入れ替えた。次いで、試験対象の様々な濃度の各化合物を10uLの体積(最終DMSO≦0.5%)で加え、5%の二酸化炭素を含有する加湿した環境において細胞を37℃で16時間インキュベートした。プレートをインキュベーターから取り出し、約15分間かけて室温に平衡化させた。10uLの6×CCF4/AM作用色素溶液(Live Blazer FRET−B/G Loadingキット(Invitrogenカタログ番号K1027)に入っている説明書に従って調製)をウェル毎に加え、暗所にて室温で2時間インキュベートした。EnVision蛍光定量プレートリーダー(励起405nm、発光460nm/535nm)で蛍光を測定した。4パラメータロジスティック用量反応方程式を使用した曲線適合プログラムで分析を行ったアゴニスト反応曲線から、EC50を決定した。
【0092】
cAMP:
細胞中のcAMPレベルを測定するHTRF(均一時間分解蛍光)cAMP検出キット(cAMP dynamic 2アッセイキット Cis Bioカタログ番号62AM4PEC)を利用する細胞アッセイでも、GPR119アゴニスト活性を求めた。この方法は、細胞によって産生される自然なcAMPと、色素d2で標識したcAMPの競合イムノアッセイである。トレーサー結合を、クリプテートで標識したMab抗cAMPによって可視化する。特異的シグナル(すなわち、エネルギー移動)は、標準またはサンプル中のcAMPの濃度に反比例する。
【0093】
詳細には、hGPR119 HEK−CREβ−ラクタマーゼ細胞(Invitrogen 2.5×107/mL、上述のβ−ラクタマーゼアッセイで使用した同じ細胞系)を凍結保存から取り出し、増殖培地(ダルベッコ変法イーグル培地高グルコース(DMEM、Gibcoカタログ番号11995−065)、1%チャコールデキストラン処理ウシ胎児血清(CD血清、HyCloneカタログ番号SH30068.03)、1×MEM非必須アミノ酸(Gibcoカタログ番号15630−080)、および25mMのHEPES pH7.0(Gibcoカタログ番号15630−080))中に希釈する。細胞濃度を1.5×105細胞/mlに調整し、この懸濁液30mlをT−175フラスコに加え、5%二酸化炭素の加湿環境において37℃でインキュベートした。16時間(一晩経過)後、細胞をT−175フラスコから(フラスコの側面を叩いて)取り出し、800×gで遠心分離し、次いでアッセイ培地(1×HBSS+CaCl2+MgCl2(Gibcoカタログ番号14025−092)および25mMのHEPES pH7.0(Gibcoカタログ番号15630−080))に再懸濁した。アッセイ培地を用いて細胞濃度を6.25×105細胞/mlに調整し、この細胞懸濁液8μl(5000細胞)を、Greiner384ウェル白色小体積アッセイプレート(VWRカタログ番号82051−458)の各ウェルに加えた。
【0094】
試験対象の様々な濃度の各化合物を3−イソブチル−1−メチルキサンチン(IBMX、Sigmaカタログ番号I5879)を含有するアッセイ緩衝液に希釈し、2μLの体積でアッセイプレートウェルに加えた(最終IBMX濃度は400μMとし、最終DMSO濃度は0.58%とした)。室温で30分間のインキュベートに続いて、5μLの標識したd2cAMPおよび5μLの抗cAMP抗体(両方とも細胞溶解緩衝液に1:20希釈したもの、製造者のアッセイプロトコールに記載のとおり)をアッセイプレートの各ウェルに加えた。次いでプレートを室温でインキュベートし、60分後、Envision 2104マルチラベルプレートリーダーで、励起波長330nm、発光波長615nmおよび665nmを使用し、HTRFシグナルの変化を読み取った。生データを、cAMP検量線からの内挿によってnM cAMPに変換し(製造者のアッセイプロトコールに記載のとおり)、4パラメータロジスティック用量反応式を使用して曲線適合プログラムで分析したアゴニスト反応曲線から、EC50を決定した。
【0095】
GPR119の活性化によるcAMP反応は、ここで使用した特定の細胞系以外の細胞でも発生し得ることが認められている。
【0096】
β−アレスチン:
DiscoverX PathHunterβ−アレスチン細胞アッセイ技術およびそのU2OS hGPR119β−アレスチン細胞系(DiscoverXカタログ番号93−0356C3)を利用する細胞アッセイでも、GPR119アゴニスト活性を求めた。このアッセイでは、アゴニストによって誘発される、β−アレスチンと活性化型GPR119の相互作用を測定することにより、アゴニスト活性化を判定する。ProLinkと呼ばれる、小さい42アミノ酸の酵素断片を、GPR119のC末端に付加した。アレスチンを、EA(酵素アクセプター)と呼ばれるより大きい酵素断片に融合した。GPR119が活性化すると、アレスチンの結合が刺激され、2つの酵素断片の相補性が引き出される結果、基質を加水分解し、化学発光シグナルを発生させることのできる機能性β−ガラクトシダーゼ酵素が生成する。
【0097】
詳細には、U2OS hGPR119β−アレスチン細胞(DiscoverX 1×107/ml)を凍結保存から取り出し、増殖培地(最小必須培地(MEM、Gibcoカタログ番号11095−080)、10%熱不活化ウシ胎児血清(HIFBS、Sigmaカタログ番号F4135−100)、100mMのピルビン酸ナトリウム(Sigmaカタログ番号S8636)、500μg/mLのG418(Sigmaカタログ番号G8168)、および250μg/mLのハイグロマイシンB(Invitrogenカタログ番号10687−010)中に希釈する。細胞濃度を1.66×105細胞/mlに調整し、この懸濁液30mlをT−175フラスコに加え、5%二酸化炭素の加湿環境において37℃でインキュベートした。24時間後、酵素不使用の細胞解離緩衝液(Gibcoカタログ番号13151−014)を用いて細胞をT−175フラスコから取り出し、800×gで遠心分離し、次いでプレーティング培地(Opti−MEM I(Invitrogen/BRLカタログ番号31985−070)および2%チャコールデキストラン処理ウシ胎児血清(CD血清、HyCloneカタログ番号SH30068.03))に再懸濁した。プレーティング培地を用いて細胞濃度を2.5×105細胞/mlに調整し、この細胞懸濁液10μL(2500細胞)を、Greiner 384ウェル白色小体積アッセイプレート(VWRカタログ番号82051−458)の各ウェルに加え、プレートを5%二酸化炭素の加湿環境において37℃でインキュベートした。
【0098】
16時間(一晩経過)後、インキュベーターからアッセイプレートを取り出し、試験対象の様々な濃度の各化合物(アッセイ緩衝液(1×HBSS+CaCl2+MgCl2(Gibcoカタログ番号14025−092)、20mMのHEPES pH7.0(Gibcoカタログ番号15630−080)、および0.1%のBSA(Sigmaカタログ番号A9576)中に希釈したもの)を2.5μLの体積でアッセイプレートウェルに加えた(最終DMSO濃度を0.5%とした)。5%二酸化炭素の加湿環境において37℃で90分インキュベートした後、7.5μLのGalacton Starβ−ガラクトシダーゼ基質(PathHunter Detection Kit(DiscoveRxカタログ番号93−0001)、製造者のアッセイプロトコールに記載のとおりに調製)をアッセイプレートの各ウェルに加えた。プレートを室温でインキュベートし、60分後、Envision 2104マルチラベルプレートリーダーを用い、ウェルあたり0.1秒で発光の変化を読み取った。4−パラメータロジスティック用量反応式を使用して曲線適合プログラムで分析したアゴニスト反応曲線から、EC50を決定した。
【0099】
BacMamを使用したGPR119の発現およびGPR119結合アッセイ
鋳型としてのpIRES−puro−hGPR119および以下のプライマーを使用するポリメラーゼ連鎖反応(PCR)(Pfu Turbo Mater Mix、Stratagene、カリフォルニア州ラホーヤ)によって、野生型ヒトGPR119(図1)を増幅した。
hGPR119 BamH1、上流
5’−TAAATTGGATCCACCATGGAATCATCTTTCTCATTTGGAG−3’
(5’末端にBamHI部位を挿入する)
hGPR119 EcoRI、下流
5’−TAAATTGAATTCTTATCAGCCATCAAACTCTGAGC−3’
(3’末端にEcoRI部位を挿入する)
【0100】
製造者のプロトコールに従い、増幅産物を精製し(Qiaquick Kit、Qiagen、カリフォルニア州バレンシア)、BamH1およびEcoRI(New England BioLabs、マサチューセッツ州イプスウィッチ)で消化した。ベクターpFB−VSVG−CMV−ポリ(図2)をBamHIおよびEcoRI(New England BioLabs、マサチューセッツ州イプスウィッチ)で消化した。消化したDNAを1%アガロースゲルでの電気泳動によって分離し、断片をゲルから切り出し、精製した(Qiaquick Kit、Qiagen、カリフォルニア州バレンシア)。ベクターと遺伝子断片を連結し(Rapid Ligase Kit、Roche、カリフォルニア州プレザントン)、OneShot DH5αT1R細胞(Invitrogen、カリフォルニア州カールスバッド)に形質転換した。8つのアンピシリン耐性コロニー(「クローン1〜8」)をミニプレップ(Qiagen Miniprep Kit、Qiagen、カリフォルニア州バレンシア)用に成長させ、配列決定して、同一性および正しい挿入の方向を確認した。
【0101】
製造者のプロトコールに従い、pFB−VSVG−CMV−ポリ−hGPR119構築物(クローン#1)をOneShot DH10Bac細胞(Invitrogen、カリフォルニア州カールスバッド)に形質転換した。8つの陽性(すなわち白色)のコロニーを画線し直して、「陽性」であると確認し、引き続いてバクミド単離に向けて成長させた。組換え型hGPR119バクミドは、Qiagen Miniprep Kit(Qiagen、カリフォルニア州バレンシア)の緩衝液を使用する変法アルカリ溶解手順によって単離した。簡潔に述べると、ペレット化した細胞を緩衝液P1に溶解させ、緩衝液P2中で中和し、緩衝液N3で沈殿させた。沈殿を遠心分離(17,900×gで10分間)によってペレット化し、上清をイソプロパノールと合わせてDNAを沈殿させた。DNAを遠心分離(17,900×gで30分間)によってペレット化し、70%エタノールで1回洗浄し、50μLの緩衝液EB(Tris−HCL、pH8.5)に再懸濁した。市販のプライマー(M13F、M13R、Invitrogen、カリフォルニア州カールスバッド)を用いたポリメラーゼ連鎖反応(PCR)を使用して、バクミド中にhGPR119挿入断片が存在することを確認した。
【0102】
hGPR119組換え型バキュロウイルスの生成
P0ウイルスストックの作製
Sf900II培地(Invitrogen、カリフォルニア州カールスバッド)で増殖させた、懸濁液に適合させたSf9細胞に、製造者のプロトコール(Cellfectin、Invitrogen、カリフォルニア州カールスバッド)に従って10μLのhGPR119バクミドDNAをトランスフェクトした。5日間インキュベートした後、条件培地(すなわち「P0」ウイルスストック)を遠心分離し、0.22μmフィルター(Steriflip、Millipore、マサチューセッツ州ビルリカ)で濾過した。
【0103】
凍結ウイルス(BIIC)ストックの作製
長期間のウイルス貯蔵および作業(すなわち「P1」)ウイルスストック生成に備えて、凍結BIIC(バキュロウイルスを感染させた昆虫細胞)ストックを以下のとおりに作製した。懸濁液に適合させたSf9細胞をSf900II培地(Invitrogen、カリフォルニア州カールスバッド)で増殖させ、hGPR119 P0ウイルスストックで感染させた。24時間増殖させた後、感染した細胞を穏やかに遠心分離し(約100×g)、凍結用培地(10%のDMSO、1%のアルブミンを含有するSf900II培地)に再懸濁して、最終密度を1×107細胞/mLとし、標準の凍結用プロトコールに従って、1mLずつの一定分量にして凍結させた。
【0104】
作業(「P1」)ウイルスストックの作製
Sf900II培地(Invitrogen、カリフォルニア州カールスバッド)で増殖させた、懸濁液に適合させたSf9細胞を、解凍したhGPR119 BIICストックの1:100希釈物で感染させ、数日間インキュベートした(振盪しながら27℃)。細胞の生存度が70%に到達したとき、条件培地を遠心分離によって回収し、ELISA(BaculoElisa Kit、Clontech、カリフォルニア州マウンテンヴュー)によってウイルス力価を決定した。
【0105】
懸濁液に適合させたHEK 293FT細胞におけるhGPR119の過剰発現
振盪フラスコにおいて、HEK293FT細胞(Invitrogen、カリフォルニア州カールスバッド)を、50μg/mLのネオマイシンおよび10mMのHEPESを補充した293Freestyle培地(Invitrogen)で増殖させた(37C、8%二酸化炭素、振盪)。細胞を穏やかに遠心分離し(約500×g、10分)、ペレットを、18%ウシ胎児血清(Sigma Aldrich)を補充したダルベッコPBS(Mg++/−Ca++なし)とP1ウイルスの混合物に再懸濁して、感染多重度(MOI)が10になり、最終細胞密度が1.3×106/mL(合計体積2.5リットル)になるようにした。細胞を5リットルのWave Bioreactor Wavebag(Wave Technologies、マサチューセッツ州)に移し、27℃で4時間インキュベートし(17振動(rock)/分、乗せ台角度7°)、インキュベート期間の終わりに、30mMの酪酸ナトリウム(Sigma Aldrich)を補充した等体積(2.5リットル)の293Freestyle培地を加え(最終濃度=15mM)、細胞を20時間増殖させた(37℃、8%CO2[0.2リットル/分}、25振動(rock)/分、乗せ台角度7°)。細胞を遠心分離(3,000×g、10分)によって収集し、DPBS(Ca++/Mg++なし)で1回洗浄し、0.25Mのスクロース、25mMのHEPES、0.5mMのEDTA、pH7.4に再懸濁し、−80℃で凍結させた。
【0106】
放射リガンド結合アッセイに向けた膜調製
凍結した細胞を氷上で解凍し、4℃で10分間700×g(1400rpm)で遠心分離した。細胞ペレットを20mLのリン酸緩衝溶液に再懸濁し、1400rpmで10分間遠心分離した。次いで細胞ペレットをホモジナイズ緩衝液(10mMのHEPES(Gibco #15630)、pH7.5、1mMのEDTA(BioSolutions、#BIO260−15)、1mMのEGTA(Sigma、#E−4378)、0.01mg/mlのベンズアミジン(Sigma #B6506)、0.01mg/mlのバシトラシン(Sigma #B0125)、0.005mg/mlのロイペプチン(Sigma #L8511)、0.005mg/mlのアプロチニン(Sigma #A1153))に再懸濁し、氷上で10分間インキュベートした。次いで細胞を、タイトフィット型ガラス製Dounceホモジナイザーで15回の穏やかなストロークで溶解させた。ホモジネートを4℃で10分間1000×g(2200rpm)で遠心分離した。上清を氷上の新たな遠心管に移した。細胞ペレットをホモジナイズ緩衝液に再懸濁し、4℃で10分間1000×g(2200rpm)で再び遠心分離し、その後上清を取り出し、ペレットをホモジナイズ緩衝液に再懸濁した。この過程を3回目も繰り返し、その後上清を合わせ、Benzonase(Novagen #71206)およびMgCl2(Fluka #63020)を加えて最終濃度をそれぞれ1U/mlおよび6mMとし、氷上で1時間インキュベートした。次いで溶液を4℃で20分間25,000×g(15000rpm)で遠心分離し、上清を廃棄し、ペレットを新鮮なホモジナイズ緩衝液(BenzonaseおよびMgCl2なし)に再懸濁した。25,000×gでの遠心分離ステップを繰り返した後、最終膜ペレットをホモジナイズ緩衝液に再懸濁し、−80℃で凍結させた。Pierce BCAタンパク質アッセイキット(Pierce試薬A #23223およびB #23224)を使用して、タンパク質濃度を決定した。
【0107】
[3H]Cmpd Aの合成および精製
【0108】
【化9】
化合物A(「Cmpd A」、4−(1−(4−(メチルスルホニル)フェニル)−3a,7a−ジヒドロ−1H−ピラゾロ[3,4−d]ピリミジン−4−イルオキシ)ピペリジン−1−カルボン酸イソプロピル、上で示したとおり)(4mg、0.009mmol)を0.5mLのジクロロメタンに溶解させ、得られる溶液を(1,5−シクロオクタジエン)(ピリジン)(トリシクロヘキシルホスフィン)−イリジウム(I)ヘキサフルオロホスフェート(J.Organometal.Chem.1979、168、183)(5mg、0.006mmol)で処理した。反応容器を密封し、溶液をトリチウムガス雰囲気中で17時間撹拌した。減圧下で反応溶媒を除去し、得られる残渣をエタノールに溶解させた。未精製の[3H]Cmpd Aの精製を、以下の条件を使用する分取HPLCによって実施した。
カラム:Atlantis、4.6×150mm、5μm
移動相A:水/アセトニトリル/ギ酸(98/2/0.1)
移動相B:アセトニトリル
勾配: 時間 %B
0.00 30.0
1.00 30.0
13.00 80.0
分析時間:16分
ポストタイム:5分
流量:1.5mL/分
注入体積:20〜50μL
注入溶媒:DMSO
検出:210nmおよび245nmでのUV
【0109】
精製した[3H]Cmpd Aの比活性は、質量分析によって、70Ci/mmolと決定した。
【0110】
GPR119放射リガンド結合アッセイ
試験化合物を100%DMSO(J.T.Baker #922401)に段階希釈した。各希釈液2μLを96ウェルプレートの適切なウェルに加えた(各濃度3通りにして)。未標識Cmpd Aを最終濃度10μMで使用して、非特異的結合を測定した。
【0111】
3H−Cmpd Aを結合緩衝液(50mMのTris−HCl、pH7.5(Sigma #T7443)、10mMのMgCl2(Fluka 63020)、1mMのEDTA(BioSolutions #BIO260−15)、0.15%のウシ血清アルブミン(Sigma #A7511)、0.01mg/mLのベンズアミジン(Sigma #B6506)、0.01mg/mLのバシトラシン(Sigma #B0125)、0.005mg/mLのロイペプチン(Sigma #L8511)、0.005mg/mLのアプロチニン(Sigma #A1153))中に希釈して濃度を60nMとし、96ウェルプレート(Nalge Nunc #267245)のすべてのウェルに100μLを加えた。
【0112】
GPR119を発現する膜を解凍し、結合緩衝液に希釈して最終濃度を20μg/各ウェル100μLとし、希釈した膜100μLを96ウェルプレートの各ウェルに加えた。
【0113】
プレートを振盪しながら室温(約25℃)で60分間インキュベートした。Packardハーベスターを使用した、0.3%ポリエチレンアミンに予浸したGF/Cフィルタープレート(Packard #6005174)での真空濾過によって、アッセイを終結させた。次いで、洗浄緩衝液(50mMのTris−HCl、pH7.5、4℃に保ったもの)を使用してフィルターを6回洗浄した。次いでフィルタープレートを室温で終夜風乾した。30μlのシンチレーション液(Ready Safe、Beckman Coulter #141349)を各ウェルに加え、プレートを密封し、Wallac Trilux MicroBetaプレートベースのシンチレーションカウンターを使用して、各フィルターに付随する放射能を測定した。
【0114】
3H−Cmpd AのKdは、一部位双曲線(Graph Pad Prism)に適合させる非線形回帰によるデータ解析を用いた飽和結合実験を実施して求めた。IC50は、専有の曲線適合プログラム(SIGHTS)および4パラメータロジスティック用量反応式を用いて分析した競合曲線から求めた。Ki値は、Cheng−Prusoff式を使用してIC50値から算出した。
【0115】
β−ラクタマーゼおよびβ−アレスチン機能アッセイについて、以下の結果が得られた。
【0116】
【表1】
【0117】
cAMPアッセイおよび結合アッセイについて、以下の結果が得られた。
【0118】
【表2】
【0119】
出発材料の調製
調製例1:3−フルオロ−4−ヒドロキシピペリジン−1−カルボン酸tert−ブチルの異性体(4および5)。実験の細目は以下のスキームAで詳述する。
【0120】
【化10】
【0121】
ステップA.4−[(トリメチルシリル)オキシ]−3,6−ジヒドロピリジン−1(2H)−カルボン酸tert−ブチル(2)
【0122】
【化11】
N−tert−ブトキシカルボニル−4−ピペリドン(30.0g、0.15mol)を無水N,N−ジメチルホルムアミド(300mL)に溶かした室温の溶液に、塩化トリメチルシリル(22.9mL、0.18mol)およびトリエチルアミン(50.4mL、0.36mol)を添加漏斗で連続的に加えた。得られる溶液を80℃で終夜加熱し、次いで室温に冷却した。反応混合物を水およびヘプタンで希釈した。層を分離し、水層をヘプタンで抽出した。ヘプタン層を合わせて水およびブラインで順次洗浄し、次いで硫酸マグネシウムで乾燥させた。混合物を濾過し、減圧下で濾液を濃縮して、粗生成物を黄色の油状物として得た。油状物を90:10のヘプタン/酢酸エチル中でシリカゲル充填物に通すことにより精製して、表題化合物を無色の油状物(33.6g、82%)として得た。1H NMR (400 MHz, 重水素化クロロホルム) δ 4.78 (br s, 1H), 3.86 (br s, 2H), 3.51
(t, 2H), 2.09 (br s, 2H), 1.45 (s, 9H), 0.18 (s, 9H).
【0123】
ステップB.3−フルオロ−4−オキソピペリジン−1−カルボン酸tert−ブチル(3)
【0124】
【化12】
4−[(トリメチルシリル)オキシ]−3,6−ジヒドロピリジン−1(2H)−カルボン酸tert−ブチル(28.8g、0.11mol)をアセトニトリル(300mL)に溶かした室温の撹拌した溶液に、Selectfluor(商標)(41.4g、0.12mol)を加えた。得られる淡黄色の懸濁液を室温で1.5時間撹拌した。飽和炭酸水素ナトリウム水溶液(300mL)および酢酸エチル(300mL)を加え、層を分離した。水層を酢酸エチルで2回抽出し、すべての有機層を合わせ、飽和炭酸水素ナトリウム水溶液およびブラインで順次洗浄し、次いで硫酸マグネシウムで乾燥させた。混合物を濾過し、減圧下で濾液を濃縮して、粗生成物を淡黄色の油状物として得た。この材料を、ヘプタン/酢酸エチル勾配(2:1〜1:1)を用いたシリカゲルでのカラムクロマトグラフィーを繰り返すことにより精製すると、表題化合物が白色の固体(15.5g、67%)として得られた。1H NMR (400 MHz, 重水素化クロロホルム): δ 4.88 (dd, 0.5 H), 4.77 (dd, 0.5H), 4.47
(br s, 1H), 4.17 (ddd, 1H), 3.25 (br s, 1H), 3.23 (ddd, 1H), 2.58 (m, 1H), 2.51
(m, 1H), 1.49 (s, 9H).
【0125】
ステップC.(R*)−3−(S)−フルオロ−4−(R)−ヒドロキシピペリジン−1−カルボン酸tert−ブチルの異性体(4および5)(ラセミ)
【0126】
【化13】
3−フルオロ−4−オキソピペリジン−1−カルボン酸tert−ブチル(15.5g、71.3mmol)をメタノール(150mL)に溶かした0℃の溶液に、水素化ホウ素ナトリウム(3.51g、93.7mmol)を加えた。得られる混合物を0℃で2時間撹拌し、次いで室温に温めた。飽和塩化アンモニウム水溶液(200mL)を加え、混合物を酢酸エチルで3回抽出した。抽出物を合わせてブラインで洗浄し、硫酸マグネシウムで乾燥させた。混合物を濾過し、減圧下で濾液を濃縮して、粗生成物混合物を得、これを、ヘプタン−酢酸エチル(3:2〜1:1)を溶離液とするシリカゲルでのカラムクロマトグラフィーによって精製して、第1の溶離生成物である(3,4−トランス)−3−フルオロ−4−ヒドロキシピペリジン−1−カルボン酸tert−ブチル(3.81g、24%)を淡黄色の油状物として得たが、油状物は静置すると凝固して白色の固体になった。1H NMR (400 MHz, 重水素化クロロホルム) δ 4.35 (ddd,
0.5 H), 4.18 (ddd, 0.5 H), 4.15 (br s, 1H), 3.89-3.74 (m, 2H), 2.97 (br s, 1H),
2.93 (ddd, 1H), 2.47 (s, 1H), 2.05-1.92 (m, 1H), 1.58-1.46 (m, 1H), 1.44 (s,
9H).
【0127】
次いで、第2の溶離化合物である(3,4−シス)−3−フルオロ−4−ヒドロキシ−ピペリジン−1−カルボン酸tert−ブチル(10.57g、68%)を白色の固体として単離した。1H NMR (400 MHz, 重水素化クロロホルム) δ 4.69 - 4.65
(m, 0.5H), 4.53-4.49 (m, 0.5H), 3.92 - 3.86 (m, 2H), 3.69 (br s, 1H), 3.39 (br
s, 1H), 3.16 (br s, 1H), 2.13 (s, 1H), 1.88 - 1.73 (m, 2H), 1.44 (s, 9H).
【0128】
ステップD.(3,4−シス)−3−フルオロ−4−ヒドロキシ−ピペリジン−1−カルボン酸tert−ブチルの鏡像異性体
ラセミ体の(3,4−シス)−3−フルオロ−4−ヒドロキシ−ピペリジン−1−カルボン酸tert−ブチルのサンプル1gを、Chiralpak AD−Hカラム(10×250mm)を利用し、それぞれ90:10の二酸化炭素とエタノールからなる移動相を10mL/分の流量で用いた分取高圧液体クロマトグラフィーによって、その鏡像異性体に精製した。分離をモニターする波長は、210nMとした。Chrialpak AD−H(4.6mm×25cm)カラムを使用し、それぞれ90:10の二酸化炭素とエタノールからなる定組成移動相を2.5mL/分の流量で用いた分析高圧クロマトグラフィーの使用によって、各鏡像異性体の純度分析値を決定した。ピークをモニターする波長は、210nmとした。以下の2種の異性体が得られた。
(3,4−シス)−3−フルオロ−4−ヒドロキシ−ピペリジン−1−カルボン酸tert−ブチル、鏡像異性体1(363mg):Rt=2.67分(100%ee)および(3,4−シス)−3−フルオロ−4−ヒドロキシ−ピペリジン−1−カルボン酸tert−ブチル、鏡像異性体2(403mg):Rt=2.99分(88%ee)。
【0129】
調製例2:−9−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸イソプロピル(シンおよびアンチ異性体の混合物)
【0130】
【化14】
【0131】
スキームBのステップA. 7−ベンジル−3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オン−塩酸塩(2)の合成:
テトラヒドロ−4H−ピラン−4−オン1(60.0g、0.60mol)、ベンジルアミン(63.4g、0.60mol)、および氷酢酸(35.9g、0.60mol)を無水メタノール(1.2L)に溶かした溶液を、パラホルムアルデヒド(39.6g、1.3mol)を無水メタノール(1.2L)に懸濁させた撹拌した懸濁液に、65℃で75分間かけて加えた。2回目の分のパラホルムアルデヒド(39.6g、1.3mol)を加え、混合物を65℃で1時間撹拌した。反応を水(1.2L)および1M水酸化カリウム水溶液(600mL)で失活させた。混合物を酢酸エチル(3L×3)で抽出した。有機層を合わせて硫酸ナトリウムで乾燥させ、濾過し、濾液を真空中で濃縮乾燥した。残渣をカラムクロマトグラフィー(石油エーテル/酢酸エチル=20:1〜2:1)によって精製して、褐色の油状物を得た。残渣を6M無水塩酸1,4ジオキサン溶液(500mL)で希釈し、混合物を30分間撹拌した。真空中で溶媒を除去し、アセトン(500mL)を加えた。得られる混合物を30分間音波処理して白色の沈殿物を生成させた。混合物を濾過し、固体をアセトンで洗浄し、次いで真空中で乾燥させて、所望の生成物を白色の固体(21g、13%)として得た。1H NMR (400 MHz, 酸化重水素) δ 7.43 - 7.42 (m, 5H), 4.66 (s, 2H), 3.95
- 3.90 (m, 4H), 3.54 - 3.47 (m, 4H); 1.96 (bs, 2H); LCMS (ES+): 232.0 (M + 1).
【0132】
スキームBのステップB. 7−ベンジル−3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オール(シンおよびアンチ異性体の混合物)(3)の合成:
7−ベンジル−3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オン塩酸塩(4.40g、16.9mmol)をエタノール(40mL)および無水テトラヒドロフラン(40mL)に懸濁させた。混合物を氷浴で冷却し、水素化ホウ素ナトリウム(1.5g、37.3mmol)を1回で加えた。混合物を4時間かけてゆっくりと室温に温めた。次いで反応液を真空中で濃縮して、エタノールおよびテトラヒドロフランの大部分を除去した。混合物をメチルtert−ブチルエーテルと1.0M水酸化ナトリウム水溶液とに分配した。溶液を30分間撹拌した後、2つの層を分離した。水層をメチルtert−ブチルエーテルで抽出した。有機抽出物を合わせ、ブラインで洗浄し、硫酸ナトリウムで乾燥させた。混合物を濾過し、真空中で濾液を濃縮して透明な油状物を得、これを静置すると部分的に凝固して油性の白色固体(3.71g、94%)になった。このシンおよびアンチの7−ベンジル−3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オール異性体の混合物を、それ以上精製せずに次のステップで使用した。LCMS (ES+): 234.1 (M+1).
【0133】
スキームBのステップC. 3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オール(シンおよびアンチ異性体の混合物)(4)の合成:
シンおよびアンチの7−ベンジル−3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オール異性体の出発混合物(3.71g、15.9mmol)をエタノール(120mL)に溶解させ、Pd(OH)2(450mg)を加えた。Parrシェーカーにおいて混合物を50psiの水素中で2.5時間振盪した。混合物をCelite(登録商標)で濾過し、収集した固体をメタノールで3回洗浄した。真空中で濾液を濃縮して油性の固体を得た。この油性の固体を酢酸エチルに溶解させ、ヘプタンを加えた。真空中で溶液を濃縮して、3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オールのシンおよびアンチ異性体混合物を白色の固体(2.08g、91%)として得た。この材料をそれ以上精製せずに次のステップで使用した。LCMS (ES+): 144.1 (M+1).
【0134】
スキームBのステップD.9−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸イソプロピル(シンおよびアンチ異性体の混合物)(5)の合成:
3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オールのシンおよびアンチ異性体混合物(2.08g、14.5mmol)およびN,Nジイソプロピルエチルアミン(2.80mL、16.0mmol)の0℃のジクロロメタン(15mL)溶液に、クロロギ酸イソプロピル(14.2mL、14.2mmol、1.0Mトルエン溶液)を滴下した。反応混合物を14時間かけて室温に温めた。次いで反応液を1M塩酸水溶液(50mL)で希釈し、水層を分離した。有機層を水(50mL)およびブライン(50mL)で順次洗浄し、硫酸ナトリウムで乾燥させた。混合物を濾過し、濾液を真空中で濃縮して無色の油状物を得た。この油状物を酢酸エチルに溶解させ、ヘプタンを加え、混合物を濃縮した。得られる油状物を真空中で乾燥させて、9−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸イソプロピルのシンおよびアンチ異性体混合物を透明な油状物(2.74g、82%)として得た。LCMS (ES+): 230.1 (M+1).
【0135】
ステップE.9−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボキン酸イソプロピルのシンおよびアンチ異性体の分離:
それぞれ65mL/分の流量の85:15の二酸化炭素およびメタノールを移動相とした、Chiralpak AD−Hカラム(21×250mm)を利用する分取高圧液体クロマトグラフィーによって、9−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸イソプロピルのシンおよびアンチ異性体混合物(5.04g、35.1mmol)を分離した。分離をモニターする波長は210nmとした。それぞれ2.5mL/分の流量の85:15の二酸化炭素およびメタノールを移動相とした、Chiralpak AD−H(4.6mm×25cm)カラムを用いた分析用高圧クロマトグラフィーを使用して、分析による各異性体の純度を決定した。ピークをモニターする波長は210nmとした。以下の2種の異性体が得られた。
9−シン−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸イソプロピル(6)(1.34g):透明な油状物であったが、静置すると凝固した。保持時間(Rt)=2.3分、1H NMR (400 MHz, 重水素化-DMSO): δ 5.12 (d, 1H, J=2.8Hz), 4.76 - 4.71
(m, 1H), 4.20 (d, 1H, J=13Hz), 4.16 (d, 1H, J=13Hz), 3.96 - 3.92 (m, 2H), 3.79
(d, 1H, J=3Hz), 3.55 (s, 1H), 3.52 (S, 1H), 3.08 (d, 1H, J=13Hz), 2.98 (d, 1H,
J=13Hz), 1.47 (m, 2H) 1.16 (d, 3H, J=3Hz), 1.15 (d, 3H, J=3Hz); LCMS (ES+):
230.2 (M+1).
9−アンチ−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸イソプロピル(7)(1.70g):琥珀色の油状物、Rt=3.08分、1H NMR (400 MHz, DMSO-d6): δ 5.11 (d, 1H, J=2.8Hz), 4.74 - 4.67 (m, 1H), 3.89 (d, 1H, J=13Hz),
3.84 - 3.78 (m, 2H, J=11Hz), 3.80 (d, 1H, J=6Hz), 3.78 (d, 1H, J=3Hz), 3.52 -
3.47 (m, 2H), 3.35 - 3.30 (m, 1H), 3.24 - 3.20 (m, 1H), 1.53 (s, 1H), 1.51 (s,
1H), 1.13 (d, 3H, J=1Hz), 1.16 (d, 3H, J=1Hz); LCMS (ES+): 230.2 (M+1)
【0136】
別法として、上記反応スキームAのステップAとBを以下で述べるように組み合わせて、7−ベンジル−3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オール(シンおよびアンチ異性体の混合物)を合成することもできる。
ベンジルアミン(21.35g、199.27mmol)、テトラヒドロ−4H−ピラン−4−オン(1)(19.95g、199.27mmol)、および酢酸(11.97g、199.27mmol)をメタノール(400mL)に溶解させた。混合物を還流加熱した。反応混合物に、ホルムアルデヒド水溶液(37%、32.34g、398.53mmol)およびメタノール(100mL)を、反応液を還流させたまま60分かけて加えた。反応液を室温に冷却した。次いで炭酸水素ナトリウム(16.74g、199.27mmol)を少量ずつ加えた。引き続いて、反応温度を25℃以下に保ちながら、水素化ホウ素ナトリウム(7.92g 209.23mmol)を少量ずつ加えた。混合物を周囲温度で30分間撹拌した。Celite(登録商標)(20g)を加えた後、水(100mL)および1N水酸化ナトリウム水溶液(100mL)を加えた。これを1時間撹拌した後、混合物を濾過し、フィルターケーキをメタノールおよび水(各20mL)で順次すすいだ。濾液を真空中で濃縮して、メタノールの大部分を除去した。得られる水性混合物を2−メチルテトラヒドロフラン(300mL)で抽出した。有機相をブライン溶液(100mL)で洗浄し、無水硫酸マグネシウムで乾燥させ、真空中で濃縮して、シンおよびアンチ−7−ベンジル−3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オール異性体の混合物を油状物として得たが、油状物は室温で静置すると凝固した(22.0g、47.3%)。
【0137】
調製例3:9−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸tert−ブチル(シンおよびアンチ異性体の混合物)
【0138】
【化15】
3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オール(シンおよびアンチ異性体の混合物、調製例2ステップCの生成物)(3.78g、26.4mmol)を水(30mL)およびテトラヒドロフラン(30mL)に溶かした0℃の溶液に、二炭酸ジ−tert−ブチル(5.76g、26.4mmol)のテトラヒドロフラン(20mL)溶液を滴下した。溶液を徐々に室温に温めながら約15時間撹拌した。反応液をジクロロメタンおよび水で希釈した。層を分離し、水層をジクロロメタンで抽出した。有機層を合わせ、硫酸ナトリウムで乾燥させた。混合物を濾過し、濾液を減圧下で濃縮すると、表題化合物が透明な油状物(6.55g)として現れ、これをそれ以上精製せずに使用した。
【0139】
調製例4:9−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸tert−ブチルのシンおよびアンチ異性体の分離
【0140】
【化16】
調製例3の9−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸tert−ブチル(5.04g、35.1mmol)のシンおよびアンチ異性体の混合物を、それぞれ85:15の二酸化炭素およびメタノールを移動相とし、流量を65mL/分としたChiralpak AD−Hカラム(21×250mm)を利用する分取高圧液体クロマトグラフィーによって分離した。分離をモニターする波長は210nmとした。各異性体の純度分析値は、それぞれ85:15の二酸化炭素およびメタノールを移動相とし、流量を2.5mL/分としたChiralpak AD−H(4.6mm×25cm)カラムを使用する分析高速クロマトグラフィーを使用して求めた。ピークをモニターする波長は210nmとした。以下の2種の異性体が得られた。
9−アンチ−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸tert−ブチル:(1.30g、100%de)、静置すると白色の固体に凝固した透明な油状物、保持時間(Rt)=3.15分。1H NMR (400 MHz, 重水素化クロロホルム) δ 1.44 (s, 9 H), 1.66 (d, J=16.79 Hz, 2 H), 1.84 (d, J=2.93 Hz, 1 H),
3.30 - 3.52 (m, 2 H), 3.64 (t, J=11.03 Hz, 2 H), 3.93 - 4.21 (m, 5 H).
9−シン−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸tert−ブチル:(1.64g、89%de)、静置すると白色の固体に凝固した透明な油状物、Rt=3.55分。1H NMR (400 MHz, 重水素化クロロホルム) δ 1.47 (s, 9 H), 1.64 (d, J=13.47 Hz, 2 H), 2.12 (d, J=3.32 Hz, 1 H),
2.92 - 3.22 (m, 2 H), 3.71 - 3.83 (m, 2 H), 3.99 (d, J=3.32 Hz, 1 H), 4.09 -
4.19 (m, 2 H), 4.32 (d, J=13.66 Hz, 1 H), 4.48 (d, J=13.66 Hz, 1 H).
【0141】
調製例5:4−[(6−クロロ−ピリミジン−4−イル)オキシ]ピペリジン−1−カルボン酸イソプロピル
【0142】
【化17】
4−ヒドロキシピペリジン−1−カルボン酸イソプロピル(553mg、2.95mmol)を無水テトラヒドロフラン(20mL)に溶かした溶液に、カリウムtert−ブトキシド(0.450mL、4.00mmol)を0℃で加えた。反応混合物を65℃で10分間撹拌した。上記混合物に4,6−ジクロロピリミジン(0.400g、2.68mmol)を加えた。次いで、得られる溶液を65℃で1時間撹拌した。混合物を周囲温度に冷却し、水(100mL)で失活させ、酢酸エチル(100mL×3)で抽出した。有機層を合わせてブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧下で濃縮した。残渣を、シリカゲルでのカラムクロマトグラフィー(石油エーテル:酢酸エチル=20:1)によって精製して、生成物を白色の固体(350mg、44%)として得た。
【0143】
調製例6:4−[(6−クロロ−5−メチルピリミジン−4−イル)オキシ]ピペリジン−1−カルボン酸イソプロピル
【0144】
【化18】
4−ヒドロキシピペリジン−1−カルボン酸イソプロピル(482mg、2.68mmol)の無水テトラヒドロフラン(15mL)溶液に、0℃でカリウムtert−ブトキシド(0.41g、3.6mmol)を加えた。反応混合物を65℃で10分間撹拌した。上記混合物に4,6−ジクロロ−5−メチルピリミジン(0.40g、2.4mmol)を加えた。次いで、得られる溶液を65℃で1時間撹拌した。混合物を周囲温度に冷却し、水(100mL)で失活させ、酢酸エチル(100mL×3)で抽出した。有機抽出物を合わせてブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過し、減圧下で濾液を濃縮した。残渣をシリカゲルでのカラムクロマトグラフィー(石油エーテル:酢酸エチル=20:1)によって精製して、生成物を白色の固体(680mg、80%)として得た。
【0145】
調製例7:4−クロロ−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−5−カルボニトリル
【0146】
【化19】
4,6−ジクロロピリミジン−5−カルボニトリル(174mg、1.00mmol)および1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール(Heterocycles 2002 56 257〜264およびUS2007232676)(123mg、1.00mmol)を無水ジクロロメタン(5mL)に溶かした溶液に、室温でN,N−ジイソプロピルエチルアミン(0.50mL、3.5mmol)を加えた。反応混合物を室温で2時間撹拌した。水(50mL)を加え、得られる混合物をジクロロメタン(50mL×3)で抽出した。有機層を合わせてブライン(100mL)で洗浄し、硫酸ナトリウムで乾燥させ、濾過し、減圧下で濾液を濃縮した。残渣を分取薄層クロマトグラフィーによって精製して、生成物を白色の固体(150mg、58%)として得た。
【0147】
調製例8:4−[(6−クロロ−5−メチルピリミジン−4−イル)オキシ]ピペリジン−1−カルボン酸tert−ブチル
【0148】
【化20】
20mLのBiotage(商標)マイクロ波対応管を窒素パージし、4,6−ジクロロ−5−メチルピリミジン(0.600g、2.98mmol)および4−ヒドロキシピペリジン−1−カルボン酸tert−ブチル(534mg、3.28mmol)を投入した。1,4−ジオキサン(14.9mL)を加え、混合物を100℃に加熱した。混合物にナトリウムビス(トリメチルシリル)アミド(3.58mL、3.58mmol、1.0Mテトラヒドロフラン溶液)を10分かけて滴下した。混合物を60分間撹拌し、次いで室温で12時間撹拌した。水で反応を失活させ、水層を酢酸エチルで抽出した(3回)。有機抽出物を合わせて硫酸ナトリウムで乾燥させ、濾過し、濾液を真空中で濃縮した。未精製材料をシリカゲルクロマトグラフィー(40gのSiO2カラム、0〜50%の酢酸エチルヘプタン溶液の勾配)によって精製して、表題化合物(842mg、86%)を得た。
【0149】
調製例9:5−(6−クロロ−5−メチルピリミジン−4−イル)−1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール
【0150】
【化21】
1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール二塩酸塩(2.00g、10.2mmol)および4,6−ジクロロ−5−メチルピリミジン(1.66g、10.2mmol)を室温でテトラヒドロフラン(51mL)に懸濁させた。これにトリエチルアミン(4.41mL、31.6mmol)を加えると、混合物が濁り、フラスコの壁面に褐色の固体が固着した。この混合物を室温で4時間撹拌し、次いでさらに19時間50℃に加熱した。反応混合物を室温に冷却し、水(100mL)で希釈した。この混合物を酢酸エチル(3×100mL)で抽出した。有機抽出物をプールし、ブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過した。真空中で濾液を乾燥状態まで減少させて、表題化合物を淡褐色の固体(1.95g、78%)として得、これをそれ以上精製せずに次のステップで使用した。
1H NMR
(500 MHz, 重水素化クロロホルム) δ 2.54
(s, 3 H) 3.88 (s, 3 H) 4.90 (見かけd, J=3.66 Hz, 4 H) 7.28
(s, 1 H) 8.29 (s, 1 H).
【実施例】
【0151】
(実施例1)
4−{[6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸イソプロピル
【0152】
【化22】
調製例5の4−[(6−クロロピリミジン−4−イル)オキシ]ピペリジン−1−カルボン酸イソプロピル(0.200g、0.667mmol)のN−メチルピロリジノン(5mL)溶液に、周囲温度で、1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール(82mg、0.67mmol)、次いで炭酸セシウム(1.08g、3.33mmol)を加えた。反応混合物を3時間150℃に加熱した。反応混合物を周囲温度に冷却した。水(50mL)を加え、次いで、得られる混合物をジクロロメタン(100mL、3回)で抽出した。有機層を合わせてブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過し、次いで濃縮して残渣を得、これを、63%のアセトニトリル(改質剤として0.05%の水酸化アンモニウム)水(改質剤として0.05%の水酸化アンモニウム)溶液からなる移動相を溶離液とするXBridge C18カラム150×30mmでの分取逆相HPLCによって精製して、生成物を白色の固体(35mg、14%)として得た。1H NMR (400 MHz, 重水素化クロロホルム): δ 8.16 (s, 1H), 7.14 (s, 1H), 5.51 (s,
1H), 5.09-5.13 (m, 1H), 4.74-4.80 (m, 1H), 4.48-4.60 (s, 2H), 4.19-4.48 (s,
2H), 3.70 (s, 3H), 3.63-3.66 (m, 2H), 3.13-3.19 (m, 2H), 1.80-1.83 (m, 2H),
1.55-1.57 (m, 2H), 1.09 (d, J=6.4 Hz, 6H); LCMS (ES+): 387.3 (M+H).
【0153】
(実施例2)
4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸イソプロピル
【0154】
【化23】
調製例6の4−[(6−クロロ−5−メチルピリミジン−4−イル)オキシ]ピペリジン−1−カルボン酸イソプロピル(0.020g、0.056mmol)のN−メチルピロリジノン(0.56mL)溶液に、1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール(0.010g、0.056mmol)に続いて炭酸セシウム(91mg、0.28mmol)を加えた。混合物を3時間150℃に加熱した。反応液を水で希釈し、水層をジクロロメタンで3回抽出した。有機抽出物を合わせて硫酸ナトリウムで乾燥させ、濾過し、濾液を真空中で濃縮した。未精製材料を、水アセトニトリル溶液(0.03%の水酸化アンモニウム改質剤)の勾配で溶離するWaters XBridge C18 19×100mm、0.005mmカラムでの分取HPLCによって精製して、生成物(8.3mg、13%)を得た。分析LCMS:保持時間0.97分(Atlantis C18 4.6×50mm、5μmカラム、1.8分かけて95%の水/アセトニトリルから5%の水/アセトニトリルへの線形勾配、5%の水/アセトニトリルで2.0分まで保持、0.05%のトリフルオロ酢酸改質剤、流量1.3mL/分)、LCMS (ES+): 401.5 (M+H).
【0155】
(実施例3)
4−{[5−シアノ−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸イソプロピル
【0156】
【化24】
4−ヒドロキシピペリジン−1−カルボン酸イソプロピル(77mg、0.63mmol)の無水テトラヒドロフラン(4mL)溶液に、周囲温度でナトリウムビス(トリメチルシリル)アミド(1.0M無水テトラヒドロフラン溶液、0.63mL、0.63mmol)を加えた。混合物を周囲温度で2時間撹拌した。上記混合物に、室温で、調製例7の4−クロロ−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−5−カルボニトリル(65mg、0.25mmol)の無水テトラヒドロフラン(2mL)溶液を加えた。得られる混合物を70℃で1時間撹拌した。反応混合物を飽和塩化アンモニウム水溶液(50mL)で失活させ、酢酸エチル(100mL、3回)で抽出した。有機抽出物を合わせてブライン(100mL)で洗浄し、硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧下で濃縮した。残渣を、66%のアセトニトリル(改質剤としての0.05%の水酸化アンモニウム)水(改質剤としての0.05%の水酸化アンモニウム)溶液からなる移動相で溶離するXBridge C18カラム150×30mmでの分取逆相HPLCによって精製して、生成物を白色の固体(25mg、24%)として得た。1H NMR (400 MHz, 重水素化クロロホルム): δ 8.23 (s, 1H), 7.24 (s, 1H), 5.33-5.36
(m, 1H), 4.83-4.89 (m, 5H), 3.80 (s, 3H), 3.64-3.70 (m, 2H), 3.38-3.41 (m, 2H),
1.80-1.91 (m, 2H), 1.75-1.79 (m, 2H), 1.19-1.23 (d, J=6.4 Hz, 6H): LCMS (ES+):
434.4 (M+Na).
【0157】
(実施例4)
4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチル
【0158】
【化25】
調製例8の4−[(6−クロロ−5−メチルピリミジン−4−イル)オキシ]ピペリジン−1−カルボン酸tert−ブチル(0.400g、1.22mmol)を含有するN−メチルピロリジノン(4.07mL)に、1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール(287mg、1.46mmol)に続いて炭酸セシウム(1.99g、6.10mmol)を加えた。混合物を1時間150℃に加熱した。反応を水で失活させ、水層を酢酸エチルで3回抽出した。有機層を合わせて硫酸ナトリウムで乾燥させ、濾過し、真空中で濃縮した。未精製材料をシリカゲルクロマトグラフィー(0〜100%の酢酸エチルヘプタン溶液の勾配)によって精製して、所望の生成物(77mg、15%)を得た。
【0159】
(実施例5)
4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピル
【0160】
【化26】
4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチル(実施例4)(77mg、0.19mmol)に、ジクロロメタン(1.5mL)に続いてトリフルオロ酢酸(1.50mL)を加えた。混合物を周囲温度で12時間撹拌した。反応混合物を真空中で濃縮し、残りのトリフルオロ酢酸を減圧下でトルエン共沸混合物によって除去した。
【0161】
未精製の1−メチル−5−[5−メチル−6−(ピペリジン−4−イルオキシ)ピリミジン−4−イル]−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾールを含有するジクロロメタン(1.8mL)に、炭酸1−メチルシクロプロピル4−ニトロフェニル(WO09105717)(87mg、0.37mmol、約10%の炭酸1−イソプロピル4−ニトロフェニルが混入したもの)に続いてトリエチルアミン(0.256mL、1.84mmol)を加えた。混合物が濃い黄色になった。反応液を室温で12時間撹拌した。未精製材料をシリカゲルクロマトグラフィー(0〜100%の酢酸エチルヘプタン溶液の勾配)によって精製して、約10%の4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸イソプロピルが混入している所望の生成物(76mg、33%)を得た。1H NMR (400 MHz, 重水素化クロロホルム): δ 8.17 (s, 1H), 7.23 (s, 1H), 5.30-5.22
(m, 1H), 4.86-4.83 (m, 2H), 4.82-4.79 (m, 2H), 3.83 (s, 3H), 3.77-3.55 (m, 2H),
3.45-3.28 (m, 2H), 2.25 (s, 3H), 2.00-1.85 (m, 2H), 1.79-1.65 (m, 2H), 1.54 (s,
3H), 0.88-0.82 (m, 2H), 0.63-0.58 (m, 2H); LCMS (ES+): 413.5 (M+H).
【0162】
(実施例6)
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチル(ラセミ体)
【0163】
【化27】
(3,4−シス)−3−フルオロ−4−ヒドロキシピペリジン−1−カルボン酸tert−ブチル(1.67g、7.62mmol)と調製例9の5−(6−クロロ−5−メチルピリミジン−4−イル)−1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール(900mg、3.60mmol)の混合物を1,4−ジオキサン(20mL)に溶解させ、105℃に加熱した。10分間加熱した後、すべての材料が溶液になっており、混合物にナトリウムビス(トリメチルシリル)アミド(4.3mL、4.3mmol、1Mトルエン溶液)を速やかに加えると、濁った黄色の混合物となり、次いでこれを105℃で2時間撹拌した。次いで反応液を室温に冷却し、水と飽和炭酸水素ナトリウム水溶液の等体積混合物を加えて失活させた。混合物を酢酸エチル(3×15mL)で抽出した。有機抽出物を合わせてブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過した。濾液を真空中で濃縮して黄色の残渣を得、これを、60〜100%の酢酸エチルヘプタン溶液を溶離液とするシリカゲルでのカラムクロマトグラフィーによって精製した。表題化合物と出発5−(6−クロロ−5−メチルピリミジン−4−イル)−1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾールの混合物を白色の固体(1.20g)として単離し、それ以上精製せずに後続の反応で使用した。
【0164】
同じ条件下で進めた別個の反応の1回分の未精製(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチルをHPLCによって精製した。未精製サンプル(9.5mg)をジメチルスルホキシド(1mL)に溶解させ、80%の水/アセトニトリル(0.03%の水酸化アンモニウム改質剤)から8.5分で0%の水/アセトニトリルへの線形勾配、続いて0%の水/アセトニトリルで1.5分間、流量:25mL/分で溶離するWaters XBridge C18 19×100mm、0.005mmカラムでの分取逆相HPLCによって精製した。そうして表題化合物(5mg)が得られた。分析LCMS:保持時間2.81分(Waters XBridge C18 4.6×50mm、0.005mmカラム、4.0分かけて90%の水/アセトニトリルから5%の水/アセトニトリルへの線形勾配、続いて5%の水/アセトニトリルで1分間、0.03%の水酸化アンモニウム改質剤、流量:2.0mL/分)、LCMS (ES+) 433.2 (M+1).
【0165】
(実施例7)
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピル(ラセミ体)
【0166】
【化28】
実施例6の未精製(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチル(1.20g)をジクロロメタン(12mL)に溶解させ、この溶液にトリフルオロ酢酸(5mL)を加えた。反応液を室温で1時間撹拌した。真空中で溶媒を除去し、残渣を水(50mL)および1N塩酸水溶液(10mL)に溶解させた。混合物をジクロロメタン(10×30mL)で抽出した。次いで、1N水酸化ナトリウム水溶液(20mL)を加えて水層をpH12とし、ジクロロメタン(40mL)で3回抽出した。有機抽出物を合わせてブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過した。濾液を減圧下で濃縮して、5−(6−{[(3,4−シス)−3−フルオロピペリジン−4−イル]オキシ}−5−メチルピリミジン−4−イル)−1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール(0.72g、2ステップで60%)を白色の固体として得、これをそれ以上精製せずに使用した。
1H NMR
(500 MHz, 重水素化クロロホルム) δ 1.84 -
2.08 (m, 2 H) 2.33 (s, 3 H) 2.69 - 2.84 (m, 1 H) 2.83 - 3.01 (m, 1 H) 3.16 (d,
J=13.66 Hz, 1 H) 3.27 - 3.44 (m, 1 H) 3.86 (s, 3 H) 4.78-4.91 (m, 1 H) 4.86 (d,
J=1.95 Hz, 2 H) 4.88 (d, J=1.95 Hz, 2 H) 5.21 - 5.32 (m, 1 H) 7.26 (s, 1 H)
8.18 (s, 1 H); LCMS (ES+) 333.4 (M+1).
【0167】
5−(6−{[(3,4−シス)−3−フルオロピペリジン−4−イル]オキシ}−5−メチルピリミジン−4−イル)−1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール(717mg、2.16mmol)および炭酸1−メチルシクロプロピル4−ニトロフェニル(NMR積分によると約10%の炭酸イソプロピル4−ニトロフェニルが混入したもの)(620mg、2.59mmol)をジクロロメタン(11mL)に溶かした溶液に、トリエチルアミン(0.60mL、4.31mmol)を加え、反応混合物を室温で15時間撹拌した。次いで反応混合物をさらに4時間還流加熱し、室温に冷却し、1N水酸化ナトリウム水溶液(30mL)で希釈し、ジクロロメタン(30mL)で3回抽出した。有機抽出物を合わせて、1N水酸化ナトリウム水溶液/ブライン溶液(10mL)の2:1混合物で2回洗浄し、硫酸ナトリウムで乾燥させ、濾過した。真空中で濾液を減圧乾固させ、黄色の泡沫を得た。70〜100%の酢酸エチルヘプタン溶液を溶離液としながらシリカゲルで精製すると、表題化合物が白色の固体(0.84g、90%、純度91%)として得られた。サンプルは、0.91ppmでのシクロプロピルメチレンおよび1.28ppmでのイソプロピルメチルシグナルのNMR積分によって求めたところ、それぞれ10:1の比で、(3,4−トランス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸イソプロピルが混入していた。
1H NMR
(500 MHz, 重水素化クロロホルム) δ 0.62 -
0.67 (m, 2 H) 0.87 - 0.94 (m, 2 H) 1.57 (s, 3 H) 1.88 (br. s., 1 H) 2.10 (br.
s., 1 H) 2.32 (s, 3 H) 3.04 - 3.23 (m, 1 H) 3.23 - 3.49 (m, 1 H) 3.86 (s, 3 H)
3.99 - 4.34 (m, 2 H) 4.66 - 5.04 (m, 5 H) 5.32 (m, 1 H) 7.26 (s, 1 H) 8.17 (s,
1H); LCMS (ES+) 431.4 (M+1).
【0168】
また、in vitroでの生物学的な特徴付けのために、同じ条件下で進めた別個の反応の1回分の未精製(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピルをHPLCによって精製した。未精製材料(52mg)をジメチルスルホキシド(1mL)に溶解させ、80%の水/アセトニトリル(0.03%の水酸化アンモニウム改質剤)から8.5分で0%の水/アセトニトリルへの線形勾配、流量:25mL/分で溶離するWaters XBridge C18 19×100mm、0.005mmカラムでの分取逆相HPLCによって精製した。分析LCMS:保持時間2.59分(Waters XBridge C18 4.6×50mm、0.005mmカラム、4.0分かけて90%の水/アセトニトリルから5%の水/アセトニトリルへの線形勾配、0.03%の水酸化アンモニウム改質剤、流量:2.0mL/分)、LCMS (ES+) 431.2 (M+1)。そうして表題化合物が得られた(22mg、55%)。このサンプルの純度は、使用した炭酸シクロプロピルメチルが不純であったために、90%であると推定された。
【0169】
(実施例8)
(3,4−トランス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチル(ラセミ体)
【0170】
【化29】
セプタムでふたをしたバイアルにおいて、調製例1の(3,4−トランス)−3−フルオロ−4−ヒドロキシピペリジン−1−カルボン酸tert−ブチル(66mg、0.30mmol)と調製例9の5−(6−クロロ−5−メチルピリミジン−4−イル)−1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール(50mg、0.20mmol)の混合物を1,4−ジオキサン(1.5mL)に溶解させ、105℃に加熱した。5分後、溶液に、ナトリウムビス(トリメチルシリル)アミド(0.32mL、0.32mmol、1Mトルエン溶液)を速やかに加え、琥珀色から濃緑色へと色を変化させた。105℃で10分間加熱した後、反応混合物が濁った褐色の混合物になり、加熱をさらに2時間続けた。反応混合物を室温に冷却し、水と飽和炭酸水素ナトリウム水溶液の等体積混合物で失活させ、混合物を酢酸エチル(10mL)で3回抽出した。有機抽出物をプールし、ブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過した。濾液を真空中で濃縮して残渣を得、これをジメチルスルホキシド(1mL)に溶解させ、80%の水/アセトニトリル(0.03%の水酸化アンモニウム改質剤)から8.5分で0%の水/アセトニトリルへの線形勾配、流量:25mL/分で溶離するWaters XBridge C18 19×100mm、0.005mmカラムでの分取逆相HPLCによって精製して、表題化合物(9.2mg、11%)を得た。分析LCMS:保持時間3.21分(Waters Atlantis C18 4.6×50mm、0.005mmカラム、4.0分かけて90%の水/アセトニトリルから5%の水/アセトニトリルへの線形勾配、0.05%のトリフルオロ酢酸改質剤、流量:2.0mL/分)、LCMS (ES+) 433.2 (M+1).
【0171】
(実施例9)
(9−アンチ)−9−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸tert−ブチル
【0172】
【化30】
セプタムでふたをしたバイアルにおいて、(9−アンチ)−9−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸tert−ブチル(73mg、0.30mmol)と調製例9の5−(6−クロロ−5−メチルピリミジン−4−イル)−1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール(50mg、0.20mmol)の混合物を1,4−ジオキサン(1.5mL)に溶解させ、105℃で5分間加熱した。混合物に、ナトリウムビス(トリメチルシリル)アミド(0.32ml、0.32mmol、1Mトルエン溶液)を速やかに加え、琥珀色の溶液を濃緑色に変色させた。105℃で10分間加熱した後、反応混合物が濁った褐色の混合物になり、加熱をさらに2時間続けた。反応液を室温に冷却し、水と飽和炭酸水素ナトリウム水溶液の等体積混合物を加えて失活させ、酢酸エチル(15mL)で3回抽出した。有機抽出物をプールし、ブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過した。濾液を真空中で濃縮乾燥して、橙褐色の泡沫を得た。この材料のサンプルをジメチルスルホキシド(1mL)に溶解させ、80%の水/アセトニトリル(0.03%の水酸化アンモニウム改質剤)から8.5分で0%の水/アセトニトリルへの線形勾配、流量:25mL/分で溶離するWaters XBridge C18 19×100mm、0.005mmカラムでの分取逆相HPLCによって精製して、表題化合物(10.2mg、11%)を得た。分析LCMS:保持時間2.53分(Waters XBridge C18 4.6×50mm、0.005mmカラム、4.0かけて90%の水/アセトニトリルから5%の水/アセトニトリルへの線形勾配、0.03%の水酸化アンモニウム改質剤、流量:2.0mL/分)、LCMS (ES+) 457.2 (M+1)。材料の残りを、50〜100%の酢酸エチルを溶離液とするシリカゲルクロマトグラフィーによって精製して、表題化合物を淡黄色の固体(33mg、36%)として得た。1H NMR (500 MHz, 重水素化クロロホルム) δ 1.49 (s, 9 H) 1.92 - 2.12 (m, 2 H) 2.35
(s, 3 H) 3.37 (d, J=13.42 Hz, 1 H) 3.47 (d, J=13.42 Hz, 1 H) 3.74 - 3.96 (m, 5
H) 4.07 - 4.23 (m, 3 H) 4.29 (d, J=13.42 Hz, 1 H) 4.70 - 5.02 (m, 4 H) 5.37 (t,
J=3.42 Hz, 1 H) 7.27 (s, 1 H) 8.19 (s, 1 H); LCMS (ES+) 457.5 (M+1).
【0173】
(実施例10)
(9−アンチ)−9−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸1−メチルシクロプロピル
【0174】
【化31】
(9−アンチ)−9−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸tert−ブチル(実施例9)(32mg、0.07mmol)をジクロロメタン(1mL)に溶解させ、トリフルオロ酢酸(0.2mL)で処理し、室温で2時間撹拌した。得られる反応混合物を真空中で濃縮すると、黄色の残渣が残った。残渣をジクロロメタン(1mL)に溶解させ、トリエチルアミン(0.1mL)で処理した後、1H NMR積分によって求めたところ約10%の炭酸イソプロピル4−ニトロフェニルが混入している炭酸1−メチルシクロプロピル4−ニトロフェニル(20mg、0.08mmol)を加え、反応液を室温で24時間撹拌した。反応混合物をジクロロメタン(5mL)で希釈し、溶液に1N水酸化ナトリウム水溶液(10mL)を加えた。ジクロロメタン層を除去し、水層をジクロロメタン(10mL)で2回抽出した。有機抽出物をプールし、1N水酸化ナトリウム水溶液と飽和ブライン(20mL)の1:1溶液で洗浄し、硫酸ナトリウムで乾燥させ、濾過した。濾液を真空中で濃縮乾燥して淡黄色の泡沫を得、これを、酢酸エチルを溶離液とするシリカゲルクロマトグラフィーによって精製した。そうして、表題化合物と(9−アンチ)−9−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸イソプロピルのそれぞれ13:1の比(0.79ppmでのシクロプロピルメチレンおよび1.18ppmでのイソプロピルメチルシグナルの1H NMR積分によって求めたもの)の混合物が、白色の固体(27.4mg、86%、純度93%)として得られた。1H NMR (500 MHz, 重水素化ジメチルスルホキシド) δ 0.53 - 0.63 (m, 2 H) 0.75 - 0.83 (m, 2
H) 1.46 (s, 3 H) 1.93 (d, 2 H) 2.32 (s, 3 H) 3.23 (d, J=13.17 Hz, 1 H) 3.33 (m,
1 H) 3.64 - 3.75 (m, 2 H) 3.79 (s, 3 H) 3.87 - 4.02 (m, 3 H) 4.12 (d, J=13.17
Hz, 1 H) 4.79 (s, 2 H) 4.90 (s, 2 H) 5.29 (t, J=3.29 Hz, 1 H) 7.24 (s, 1 H)
8.15 (s, 1 H); LCMS (ES+) 455.4 (M+1).
【0175】
(実施例11および12)
キラルな(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピルの鏡像異性体(個々の鏡像異性体の絶対立体化学は不明)
【0176】
【化32】
実施例7にあるように調製したラセミ体の(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピルの約700mgのサンプルを、Chiralpak AD−Hカラム(21×250mm)を利用し、それぞれ70:30の二酸化炭素とメタノールからなる移動相を65mL/分の流量で用いたキラル分取高圧液体クロマトグラフィーによって、その鏡像異性体に精製した。分離をモニターする波長は、210nmとした。Chrialpak AD−H(4.6mm×25cm)カラムを使用し、それぞれ70:30の二酸化炭素とメタノールからなる移動相を2.5mL/分の流量で用いた分析高圧クロマトグラフィーの使用によって、各鏡像異性体の純度分析値を決定した。ピークをモニターする波長は、210nmとした。以下の2種の異性体が得られた。
【0177】
(実施例11)
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピル(鏡像異性体2、絶対立体化学不明)(266mg):Rt=6.54分(100%)は、丸底フラスコにおいて60%の酢酸エチル/ヘプタン混合物(10mL)中に入れ、室温で20時間スラリー化した。混合物を濾過し、固体を60%の酢酸エチル/ヘプタン混合物(3mL)で2回すすぎ、窒素流中で乾燥させた。固体を真空中でさらに乾燥させると、完全に結晶質の白色の固体(199mg)が得られた。LCMS (ES+) 431.3 (M+1)
【0178】
(実施例12)
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピル(鏡像異性体1、絶対立体化学不明)(305mg):Rt=5.63分(100%ee、対応するカルバミン酸イソプロピルを約8%含有する)。この材料を、Chiralcel OD−Hカラム(21×250mm)を利用し、それぞれ75:25の二酸化炭素とメタノールからなる移動相を65mL/分の流量で用いたキラル分取高圧液体クロマトグラフィーで精製し直して、対応するカルバミン酸イソプロピル不純物を除去した。分離をモニターする波長は、210nmとした。Rt=6.2分(100%ee)。
【0179】
(実施例13)
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−イソプロピル(ラセミ体)
【0180】
【化33】
表題化合物は、クロロギ酸イソプロピルを使用したことを除き、実施例7に記載のとおりに調製した。1H NMR (500 MHz, 重水素化クロロホルム) δ 1.27 (d, J=6.34 Hz, 6 H) 1.83 - 1.95 (m,
1 H) 2.07 - 2.18 (m, 1 H) 2.32 (s, 3 H) 3.20 (br. s., 1 H) 3.29 - 3.50 (m, 1 H)
3.86 (s, 3 H) 3.90 - 4.11 (m, 1 H) 4.24 (br. s., 1 H) 4.79 - 4.93 (m, 1 H)4.84
- 4.90 (ほぼd, 4 H) 4.93 - 5.01 (m, 1 H) 5.28 - 5.43 (m,
1 H) 7.27 (s, 1 H) 8.18 (s, 1 H); LCMS (ES+) 419.4 (M+1).
【0181】
本出願では終始、様々な刊行物を参照文献として引用している。それらの刊行物の開示は、その全体が、あらゆる目的で参照により本出願に援用される。
【0182】
本発明の範囲および精神から逸脱することなく、本発明に様々な変更および変化を添えてよいことは、当業者に明白となろう。本発明の他の実施形態は、本明細書を検討し、本明細書で開示する本発明を実践する中で、当業者に明らかとなろう。本明細書および実施例は、例示的なものにすぎないとみなし、本発明の真の範囲および精神は、以下の特許請求の範囲によって示すものとする。
【技術分野】
【0001】
本発明は、新規な部類の環縮合ピロリジン、それらの化合物を含有する医薬組成物、およびGタンパク質共役受容体GPR119の活性を調節するためのその使用に関する。
【背景技術】
【0002】
糖尿病は、異常なグルコース恒常性の結果として高濃度の血中グルコースが存在する障害である。最も一般的な形態の糖尿病は、I型糖尿病(インスリン依存型糖尿病とも呼ばれる)およびII型糖尿病(インスリン非依存型糖尿病とも呼ばれる)である。すべての糖尿病症例のおよそ90%を占めるII型糖尿病は、微小血管合併症(たとえば、網膜症、神経障害、および腎障害を含める)、ならびに大血管合併症(たとえば、加速性のアテローム性動脈硬化症、冠動脈心疾患、および卒中を含める)をもたらす深刻な進行性疾患である。
【0003】
現在、糖尿病の治療法は存在しない。この疾患の標準的な治療は限られており、血中グルコース濃度をコントロールして、合併症を最小限に抑える、または遅らせることに集中している。現行の治療は、インスリン抵抗性(メトホルミンもしくはチアゾリジンジオン)、またはβ細胞からのインスリン放出(スルホニル尿素、エキサナチド(exanatide))のどちらかをターゲットとしている。β細胞の脱分極を介して働くスルホニル尿素および他の化合物は、循環グルコース濃度とは無関係にインスリン分泌を刺激するので、低血糖を促進する。認可されている薬物の1つであるエキサナチドは、高グルコースの存在下でインスリン分泌を刺激するが、経口による生物学的利用能を欠くので、注射しなければならない。ジペプチジルペプチダーゼIV阻害剤であるシタグリプチンは、インクレチンホルモンの血中濃度を上昇させる薬物であり、そのインクレチンホルモンは、インスリン分泌を増加させ、グルカゴン分泌を減少させることができ、あまり特徴付けられていない他の効果ももち得る。しかし、シタグリプチンおよび他のジペプチジルペプチダーゼIV阻害剤は、他のホルモンおよびペプチドの組織濃度にも影響を及ぼすことがあり、このより広範な影響による長期的な結果については、十分な調査がなされていない。
【0004】
II型糖尿病では、筋細胞、脂肪細胞、および肝細胞がインスリンに正常に反応しない。この状態(インスリン抵抗性)は、細胞のインスリン受容体の数の減少、細胞のシグナル伝達経路の破綻、またはこの両方に起因する可能性がある。最初のうち、β細胞は、インスリン産生量を増加させてインスリン抵抗性の埋合せをする。しかし結局、β細胞は、正常なグルコース濃度(正常血糖)を維持するのに十分なインスリンを産生できなくなり、II型糖尿病への進行を示す。
【0005】
II型糖尿病では、β細胞の機能不全と相まったインスリン抵抗性により空腹時高血糖が生じる。β細胞異常機能不全には2つの態様がある。すなわち、1)(非刺激性の低グルコース濃度で起こる)基礎インスリン放出の増加(これは、II型糖尿病だけでなく、肥満のインスリン抵抗性前糖尿病段階でも認められる)、および2)高血糖負荷に反応して、インスリン放出が、すでに上昇している基礎レベルを上回って増加しないこと(この現象は、前糖尿病段階では起こらず、正常血糖のインスリン抵抗性段階からII型糖尿病へと移行する前兆となる場合もある)。後者の態様を治療する現行の療法としては、内在性の貯蔵インスリンの放出を誘発するためのATP感受性β細胞カリウムチャネルの阻害剤、および外因性インスリンの投与が挙げられる。どちらも血中グルコース濃度の的確な正常化を実現するものでなく、また低血糖を惹起するリスクを伴う。
【発明の概要】
【発明が解決しようとする課題】
【0006】
したがって、グルコース依存的に機能する薬剤の発見に大きな関心が寄せられている。このように機能する生理学的なシグナル伝達経路は、腸管ペプチドのGLP−1およびGIPを含めて、よく知られている。これらのホルモンは、同種のGタンパク質共役受容体を介して信号を送って、膵臓β細胞におけるcAMPの産生を刺激する。cAMPが増加しても、空腹時または食事前の状態の間はインスリン放出が刺激されないようである。しかし、ATP感受性カリウムチャネル、電位感受性カリウムチャネル、および開口分泌機構を含めた、cAMPのいくつかの生化学的ターゲットは、食後のグルコース刺激によるインスリン分泌が顕著に強化されるように調節される。したがって、同様に機能する、GPR119を含めた新規なβ細胞GPCRのアゴニスト調節因子も、II型糖尿病患者において、内在性インスリンの放出を刺激し、グルコース濃度の正常化を促進するということになる。たとえばGLP−1が刺激された結果としてcAMPが増加すると、β細胞増殖が促進され、β細胞死が抑制され、したがって膵島質量が向上することもわかっている。β細胞質量に対するこのプラスの効果は、インスリンが十分に産生されないII型糖尿病において有益となるはずである。
【0007】
代謝性疾患が他の生理系に悪影響を及ぼすことはよく知られており、複合的な疾患状態(たとえば、「シンドロームX」におけるI型糖尿病、II型糖尿病、不十分な耐糖能、インスリン抵抗性、高血糖、高脂血症、高トリグリセリド血症、高コレステロール血症、異脂肪症、肥満、または心血管疾患)、または腎疾患や末梢神経障害などの、糖尿病に引き続いて生じる続発性疾患がしばしば併発される。したがって、糖尿病状態の新しい治療に対する必要性が存在する。
【課題を解決するための手段】
【0008】
本発明によれば、新規な部類のGPR119調節因子が発見された。これらの化合物は、以下に示す式I
【0009】
【化1】
[式中、
Xは、AまたはB
【0010】
【化2】
であり、
Yは、Oまたは結合であり、
R1は、−C(O)−O−R3または
【0011】
【化3】
であり、
R2は、水素、シアノ、C1〜C6アルキル、またはC3〜C6シクロアルキルであり、
R3は、C1〜C6アルキル、C3〜C6シクロアルキル、またはC1〜C6アルキル、C1〜C6アルコキシ、C1〜C6フルオロアルキル、ハロ、もしくはヒドロキシで置換されているC3〜C6シクロアルキルであり、但し、ハロ、C1〜C6アルコキシ、またはヒドロキシ基は、R1中のOに連結している炭素原子では結合しておらず、
R4は、C1〜C6ハロアルキル、C1〜C6アルキル、ハロ、シアノ、またはC3〜C6シクロアルキルであり、
R5は、水素、シアノ、ニトロ、C1〜C6フルオロアルキル、C1〜C6アルキル、C1〜C6アルコキシ、C1〜C6フルオロアルコキシ、またはC3〜C6シクロアルキルであり、
R6は、水素、C1〜C6アルキル、C1〜C6フルオロアルキル、C3〜C6シクロアルキル、またはC3〜C6シクロアルキル、C1〜C6アルコキシ、もしくはヒドロキシルで置換されているC1〜C6アルキルであり、但し、C1〜C6アルコキシ基またはヒドロキシル基は、ピラゾール窒素に連結している炭素には結合しておらず、
R7aおよびR7bは、それぞれ独立に、水素、フルオロ、またはC1〜C6アルキルであり、
R8a、R8b、R8cおよびR8dは、それぞれ独立に、水素、C1〜C6アルキル、C3〜C6シクロアルキル、またはヒドロキシもしくはC1〜C6アルコキシで置換されているC1〜C6アルキルであり、
またはR8aおよびR8bは、これらが結合している炭素と一緒になって、C3〜C6シクロアルキルを形成していてもよく、
またはR8cおよびR8dは、これらが結合している炭素と一緒になって、C3〜C6シクロアルキルを形成していてもよく、
またはR8aとR8cは一緒になって、完全飽和二炭素橋を形成していてもよく、但し、R8aおよびR8cは、これらが結合している環系の同じ平面上にある]
または薬学的に許容できるその塩によって表すことができる。
【0012】
式Iの化合物は、Gタンパク質共役受容体の活性を調節する。より詳細には、式Iの化合物は、GPR119を調節する。そのため、前記化合物は、糖尿病などの、GPR119の活性が疾患の病理または症状の一因となる疾患の治療に有用である。そのような状態の例として、高脂血症、I型糖尿病、II型糖尿病、特発性I型糖尿病(Ib型)、成人潜在性自己免疫性糖尿病(LADA)、早発性2型糖尿病(EOD)、若年性非定型糖尿病(YOAD)、若年発症成人型糖尿病(MODY)、栄養不良関連糖尿病、妊娠糖尿病、冠動脈心疾患、虚血発作、血管形成術後の再狭窄、末梢血管疾患、間欠性跛行、心筋梗塞(たとえば、壊死およびアポトーシス)、異脂肪症、食後脂肪血症、耐糖能障害(IGT)状態、空腹時血漿グルコース異常(impaired fasting plasma glucose)状態、代謝性アシドーシス、ケトーシス、関節炎、肥満、骨粗鬆症、高血圧、うっ血性心不全、左室肥大、末梢動脈疾患、糖尿病性網膜症、黄斑変性、白内障、糖尿病性腎症、糸球体硬化症、慢性腎不全、糖尿病性ニューロパシー、メタボリック症候群、シンドロームX、月経前症候群、冠動脈心疾患、狭心症、血栓症、アテローム性動脈硬化症、一過性脳虚血発作、卒中、血管再狭窄、高血糖、高インスリン血症、高脂血症、高トリグリセリド血症、インスリン抵抗性、グルコース代謝障害、耐糖能障害状態、空腹時血漿グルコースの異常状態、肥満、勃起機能不全、皮膚および結合組織の障害、足の潰瘍化および潰瘍性大腸炎、内皮障害、ならびに血管伸展性の障害が挙げられる。式Iの化合物は、アルツハイマー病、統合失調症、認知障害などの神経障害の治療に使用することもできる。式Iの化合物は、炎症性腸疾患、潰瘍性大腸炎、クローン病、過敏性腸症候群などの胃腸疾患においても有益となる。上述のように、式Iの化合物は、肥満患者、特に糖尿病に罹患している肥満患者において体重減少を刺激するのにも使用することができる。
【0013】
本発明の別の実施形態は、式Iの化合物を含有する医薬組成物を対象とする。そのような製剤は通常、少なくとも1種の薬学的に許容できる賦形剤と混和された式Iの化合物を含有する。そうした製剤はまた、少なくとも1種の追加の(本明細書に記載の)薬剤を含有してもよい。そのような薬剤の例として、(後述する)抗肥満薬および/または抗糖尿病薬が挙げられる。本発明の追加の態様は、糖尿病および本明細書に記載の関連状態を治療する医薬の調製における式Iの化合物の使用に関する。
【図面の簡単な説明】
【0014】
【発明を実施するための形態】
【0015】
本発明は、本発明の例示的な実施形態についての以下の詳細な記述およびその中に含まれる実施例を参照することにより、より一層容易に理解することができる。
【0016】
本発明は、当然様々に異なり得る、特定の合成的製造方法に限定されないことを理解されたい。本明細書で使用する専門用語は、特定の実施形態について述べるためのものにすぎず、限定するものではないことも理解されたい。複数形および単数形は、数を示す以外では、交換可能であるとして扱うべきである。
【0017】
本文書内の見出しは、読者が文書に手早く目を通せるようにするために利用されるにすぎない。それらの見出しは、本発明または特許請求の範囲をいかなる形でも限定しないものと解釈すべきである。
a.「ハロ」または「ハロゲン」とは、塩素、フッ素、ヨウ素、または臭素原子を指す。
b.「アルキル」とは、分枝状または直鎖状アルキル基、たとえば、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、ペンチルなどを指す。
c.「アルコキシ」とは、直鎖または分枝鎖アルコキシ基、たとえば、メトキシ、エトキシ、n−プロポキシ、イソプロポキシ、n−ブトキシ、イソブトキシ、ペントキシなどを指す。
d.「シクロアルキル」とは、完全に水素化され、単環として存在する、非芳香族の環を指す。そのような炭素環の例として、シクロプロピル、シクロブチル、シクロペンチル、およびシクロヘキシルが挙げられる。
e.「ハロアルキル」とは、1個または複数のハロ基で置換されている直鎖または分枝鎖アルキル基、たとえば、クロロメタン、フルオロメタン、ジクロロメタン、ジフルオロメタン、ジブロモメタン、トリクロロメタン、トリフルオロメタン、クロロフルオロメタン、1,1,1,2−テトラフルオロエタンなどを指す。
f.「フルオロアルキル」とは、1個または複数のフルオロ基で置換されている直鎖または分枝鎖アルキル基、たとえば、フルオロメタン、ジフルオロメタン、トリフルオロメタンなどを指す。
g.「ハロアルコキシ」とは、1個または複数のハロ基で置換されている直鎖または分枝鎖アルコキシ基、たとえば、クロロメトキシ、フルオロメトキシ、ジクロロメトキシ、ジフルオロメトキシ、ジブロモメトキシ、トリクロロメトキシ、トリフルオロメトキシ、クロロフルオロメトキシ、1,1,1,2−テトラフルオロエトキシなどを指す。
h.「治療有効量」とは、(i)特定の疾患、状態、または障害を治療もしくは予防する、(ii)特定の疾患、状態、または障害の1つまたは複数の症状を緩和し、寛解させ、もしくは除去する、または(iii)本明細書に記載の特定の疾患、状態、または障害の1つまたは複数の症状の発症を予防し、もしくは遅らせる、本発明の化合物の量を意味する。
i.「患者」とは、温血動物、たとえば、モルモット、マウス、ラット、アレチネズミ、ネコ、ウサギ、イヌ、サル、チンパンジー、およびヒトを指す。
j.「治療する」とは、化合物が、患者の疾患(もしくは状態)または疾患に関連する任意の組織損傷を軽減し、解消し、またはその進行を緩慢にし得ることを指す。
h.用語「調節された」、「調節すること」、または「調節する」とは、本明細書では、別段指摘しない限り、本発明の化合物を用いた、Gタンパク質共役受容体GPR119の活性化を指す。
i.「薬学的に許容できる」とは、物質または組成物が、製剤を構成する他の成分および/またはそれによる治療を受ける哺乳動物と化学的および/または毒物学的に適合性でなければならないことを示す。
j.「塩」とは、薬学的に許容できる塩、および化合物の調製などの工業的プロセスでの使用に適する塩を指すものとする。
k.「薬学的に許容できる塩」とは、化合物の実際の構造に応じて、薬学的に許容できる酸付加塩」または「薬学的に許容できる塩基付加塩」のどちらかを指すものとする。
l.「薬学的に許容できる酸付加塩」とは、式Iまたはその中間体のいずれかによって表される化合物の非毒性のいかなる有機酸または無機酸付加塩にも該当するものとする。適切な塩を形成する無機酸の実例として、塩化水素酸、臭化水素酸、硫酸、およびリン酸、ならびにオルトリン酸一水素ナトリウム(sodium monohydrogen orthophosphate)や硫酸水素カリウムなどの酸金属塩が挙げられる。適切な塩を形成する有機酸の実例として、モノ、ジ、およびトリカルボン酸が挙げられる。そのような酸の実例は、たとえば、酢酸、グリコール酸、乳酸、ピルビン酸、マロン酸、コハク酸、グルタル酸、フマル酸、リンゴ酸、酒石酸、クエン酸、アスコルビン酸、マレイン酸、ヒドロキシマレイン酸、安息香酸、ヒドロキシ−安息香酸、フェニル酢酸、ケイ皮酸、サリチル酸、2−フェノキシ安息香酸、p−トルエンスルホン酸、ならびにメタンスルホン酸や2−ヒドロキシエタンスルホン酸などのスルホン酸である。このような塩は、水和した形態または実質的に無水の形態のどちらで存在してもよい。一般に、こうした化合物の酸付加塩は、水および種々の親水性有機溶媒に可溶性である。
m.「薬学的に許容できる塩基付加塩」とは、式Iまたはその中間体のいずれかによって表される化合物の非毒性のいかなる有機または無機塩基付加塩にも該当するものとする。適切な塩を形成する塩基の実例として、水酸化ナトリウム、カリウム、カルシウム、マグネシウム、バリウムなどの、アルカリ金属またはアルカリ土類金属水酸化物;アンモニア;ならびにメチルアミン、ジメチルアミン、トリメチルアミン、ピコリンなどの、脂肪族、脂環式、または芳香族有機アミンが挙げられる。
n.「式Iの化合物」、「本発明の化合物」、および「化合物」は、本出願全体にわたり区別なく使用しており、同義語として扱うべきである。
「異性体」とは、以下で定義するような「立体異性体」および「幾何異性体」を意味する。
o.「立体異性体」とは、1つまたは複数のキラル中心を有する化合物を意味し、各中心は、RまたはS立体配置で存在し得る。立体異性体として、すべてのジアステレオ異性体、鏡像異性体、およびエピマーの形態、ならびにそのラセミ体および混合物が挙げられる。
p.「幾何異性体」とは、シス、トランス、アンチ、シン、エントゲーゲン(E)、およびツザンメン(Z)の形態、ならびにその混合物として存在し得る化合物を意味する。
【0018】
本発明の化合物は、不斉中心またはキラル中心を含んでおり、したがって、異なる立体異性体形態で存在する。別段指定しない限り、本発明の化合物のすべての立体異性体形態、ならびにラセミ混合物を含めたその混合物は、本発明の一部をなすものとする。加えて、本発明は、すべての幾何異性体および位置異性体も包含する。たとえば、本発明の化合物に二重結合または縮合環が組み込まれている場合、シスおよびトランス両方の形態ならびに混合物が本発明の範囲内に包含される。
【0019】
ジアステレオ異性体混合物は、その物理化学的差異に基づき、クロマトグラフィーおよび/または分別結晶、蒸留、昇華などの当技術分野でよく知られている方法によって、その個々のジアステレオ異性体に分離することができる。鏡像異性体は、鏡像異性体混合物を、光学活性のある適切な化合物(たとえば、キラルアルコールやMosherの酸塩化物などのキラル助剤)との反応によってジアステレオ異性体混合物に変換し、ジアステレオ異性体を分離し、個々のジアステレオ異性体を対応する純粋な鏡像異性体に変換(たとえば、加水分解)することにより分離できる。また、本発明の化合物の一部は、アトロプ異性体(たとえば、置換ビアリール)である場合もあり、本発明の一部とみなされる。鏡像異性体は、キラルHPLC(高圧液体クロマトグラフィー)カラムを使用して分離することもできる。
【0020】
本発明の中間体および化合物は、異なる互変異性体形態で存在し得ることも考えられ、そうしたすべての形態が本発明の範囲内に包含される。用語「互変異性体」または「互変異性体形態」とは、低いエネルギー障壁で相互変換可能な、異なるエネルギーの構造異性体を指す。たとえば、プロトン互変異性体(プロトトロピー互変異性体としても知られる)は、ケト−エノール異性体およびイミン−エナミン異性体などの、プロトンの移動による相互変換を伴う。プロトン互変異性体の具体的な例は、プロトンが2つの環窒素間を移動し得るイミダゾール部分である。原子価互変異性体は、結合電子の一部が再編成されることによる相互変換を伴う。一部の中間体(および/または中間体の混合物)の閉じた形態と開いた形態の平衡は、当業者に知られている、アルドースが関与する変旋光の過程を連想させる。
【0021】
加えて、本発明の化合物は、溶媒和していない形で存在しても、水、エタノールなどの薬学的に許容できる溶媒と溶媒和した形で存在してもよい。一般に、本発明の目的では、溶媒和した形態は、溶媒和していない形態と同等であるとみなす。化合物は、1種または複数の結晶状態、すなわち多形体で存在する場合もあり、または非晶質固体として存在する場合もある。そのようなすべての形態が特許請求の範囲に包含される。
【0022】
本発明は、1個または複数の原子が、原子質量または質量数が自然界で通常見られる原子質量または質量数とは異なる原子で置き換えられていること以外は、本明細書で列挙するものと同一である、同位体標識された本発明の化合物も包含する。本発明の化合物に組み込むことのできる同位体の例として、2H、3H、11C、13C、14C、13N、15N、15O、17O、18O、31P、32P、35S、18F、123I、125I、36Clなどの、それぞれ水素、炭素、窒素、酸素、リン、硫黄、フッ素、ヨウ素、および塩素の同位体が挙げられる。
【0023】
本発明の特定の同位体標識された化合物(たとえば、3Hおよび14Cで標識されたもの)は、化合物および/または基質の組織分布アッセイにおいて有用である。トリチウム化同位体(すなわち3H)および炭素14同位体(すなわち14C)は、調製しやすく検出可能であるので特に好ましい。さらに、ジュウテリウム(すなわち2H)などのより重い同位体で置換すると、代謝安定性がより高いために得られる特定の治療上の利点(たとえば、in vivo半減期の延長または投与必要量の減少)がもたらされる場合もあり、したがってある状況においては好ましいこともある。15O、13N、11C、18Fなどの陽電子放射同位体は、基質占有率を調べる陽電子放射断層撮影(PET)研究に有用である。同位体標識された本発明の化合物は、一般に、同位体標識されていない試薬の代わりに同位体標識された試薬を用いることにより、本明細書において以下でスキームおよび/または実施例の中で開示する手順と同様の手順に従って調製することができる。
【0024】
式Iの化合物のいくつかは、以下で図示するように、エーテル結合を介してピリミジン環に結合した3−オキサ−7−アザビシクロ[3.3.1]ノナン環を含んでいる。このアザビシクロ−ノナンは、幾何異性体として存在し、以下で図示するシンまたはアンチ異性体のいずれかとして存在し得る。
【0025】
【化4】
【0026】
式Iを有する化合物の一実施形態では、XはAであり、R1は−C(O)−O−R3である。
【0027】
式Iを有する化合物の別の実施形態では、R8a、R8b、R8cおよびR8dは、それぞれ水素であり、R3は、C1〜C3アルキルで置換されているC3〜C6シクロアルキルである。
【0028】
式Iを有する化合物の別の実施形態では、R7aおよびR7bは、それぞれ独立に、水素、フルオロ、またはC1〜C3アルキルである。
【0029】
式Iを有する化合物の別の実施形態では、R2は水素であり、R5はC1〜C6アルキルである。
【0030】
合成
本発明の化合物は、化学分野でよく知られている方法と類似した方法を包含する合成経路によって、特に本明細書に収められている記述に照らして、合成することができる。出発材料は、Aldrich Chemicals(ウィスコンシン州ミルウォーキー)などの市販品供給元から一般に入手可能であり、または当業者に知られている方法を使用して容易に調製される(たとえば、Louis F.FieserおよびMary Fieser、Reagents for Organic Synthesis、1〜19巻、Wiley、ニューヨーク(1967〜1999年版)、またはBeilsteins Handbuch der organischen Chemie、第4版、ベルリン、Springer−Verlag編(増刊を含める)(Beilsteinオンラインデータベースからも入手可能)に一般に記載されている方法によって調製される)。
【0031】
例示する目的で、以下で図示する反応スキームにより、本発明の化合物ならびに重要中間体を合成する潜在的経路を示す。個々の反応ステップのより詳細な説明については、以下の実施例の部を参照されたい。当業者なら、本発明の化合物の合成に他の合成経路を使用してもよいことがわかるであろう。詳細な出発材料および試薬をスキームの中で示し、以下で論じるが、他の出発材料および試薬で容易に置き換えて、様々な誘導体および/または反応条件を準備することができる。加えて、以下で述べる方法によって調製される化合物の多くは、当業者によく知られている従来の化学を使用し、この開示に照らしてさらに改変することができる。
【0032】
式Iの化合物は、当技術分野で同様に知られているエーテル生成の方法を使用して調製することができる。1)Hughes,D.L.、Organic Reactions 1992、42 335−656、米国ニュージャージー州ホーボーケン、2)Tikad,A.、Routier,S.、Akssira,M.、Leger,J.−M.l、Jarry,C.、Guillaumet,G.、Synlett 2006、12、1938〜42、および3)Loksha,Y.M.、Globisch,D.、Pedersen,E.B.、La Colla,P.、Collu,G.、Loddo,R.、J.Het.Chem.2008、45、1161〜6などの、そうした反応についてより詳細に記載している教本に目を向けられたい。
【0033】
すぐ下のスキームIは、式Iの化合物を組み立てるための代替方法を例示するものである。分子の中心部分は、置換されていてもよいピリミジン環である。式Iの化合物は、以下に示すように、ピリミジンとエーテル結合およびアミノ結合の両方を形成することにより生成される。この反応シーケンスをどの順序で実施するかは重要でないが、但し、R5がシアノまたはニトロである場合は例外である。そうした場合では、ステップI−BおよびI−Cを使用して式Iの化合物を組み立てる。
【0034】
【化5】
【0035】
反応スキームIの出発材料は、R2およびR5が通常、本明細書に記載する通り、最終生成物において所望されるものと同じ置換基によって表される、構造化合物I−1のジヒドキシ−ピリミジンである。このようなピリミジンの生成方法は、当技術分野で知られている。
【0036】
ステップI−Aの塩素化反応は、当技術分野で知られているとおりに実施する。構造I−1の化合物を、トリエチルアミン、N,N−ジメチルアニリン、N,N−ジイソプロピルエチルアミンなどの添加剤を加えまたは加えずに、過剰に使用されるか、またはトルエン、ベンゼン、キシレンなどの溶媒中で使用される、POCl3(オキシ塩化リン)などの塩素化試薬と反応させる(Matulenko,M.A.ら、Bioorg.Med.Chem.2007、15、1586〜1605)。この反応は、条件の選択に応じて、室温(約23℃)〜約140℃の範囲の温度で実施することができる。代替塩素化試薬は、PCl3(三塩化リン)、POCl3/PCl5(五塩化リン)、塩化チオニル、塩化オキサリル、またはホスゲンからなり、構造I−2のジクロロピリミジンを与えることができる。場合によっては、構造I−2のジクロロピリミジンは、市販品供給元から入手することもできる。任意選択により、構造I−2のジクロロピリミジンを反応液から単離および回収し、さらに当技術分野で知られているとおりに精製してもよい。別法として、以下で記載するステップI−Bにおいて未精製材料を使用してもよい。
【0037】
スキームIのステップI−Bでは、構造I−3のテトラヒドロピロロ[3,4−c]ピラゾールと構造I−2のジクロロピリミジン間にアミノ結合を形成する。構造I−3の縮合ピロリジンにおいて、R6、R8a、R8b、R8cおよびR8dは通常、本明細書に記載するとおり、最終生成物において所望されるものと同じ置換基によって表される。このようなテトラヒドロピロロ[3,4−c]ピラゾール誘導体は、文献で知られており、または当業者によく知られている様々な方法によって好都合に調製することができる(Heterocycles、2002、56、257〜264)。アミノ結合は、エタノール、プロパノール、イソプロパノール、ブタノールなどの極性プロトン性溶媒中にて、当量の構造I−2とI−3の化合物を、どの溶媒を使用するかに応じて約0〜120℃の範囲の温度で0.5〜24時間反応させることにより形成する。この反応に利用する典型的な条件は、108℃で1時間加熱を行う溶媒としてのイソプロパノールの使用である。別法として、トリエチルアミンやジエチルイソプロピルアミンなどのアミン塩基、または炭酸水素ナトリウム、炭酸カリウム、炭酸ナトリウムなどの無機塩基をこの反応に加えてもよい。上記アミンまたは無機塩基の1種を使用する場合では、溶媒をアセトニトリル、N,N−ジメチルホルムアミド(「DMF」)、テトラヒドロフラン(「THF」)、1,4−ジオキサンなどの極性非プロトン性溶媒(約0〜100℃で0.5〜24時間)に変更することができる。この反応に利用する典型的な条件として、アセトニトリル中にて室温で3時間のジエチルイソプロピルアミンの使用が挙げられる。また、水、メタノール、エタノール、プロパノールなどの単独または組合せの極性プロトン性溶媒中での塩酸の使用も、約0〜110℃の温度でのこの変換に使用することができる。典型的な条件は、エタノール中にて78℃で水を使用するものである。構造I−5の中間体は、反応液から単離および回収し、当技術分野で知られているとおりにさらに精製することができる。別法として、以下で記載するステップI−Cにおいて未精製材料を使用してもよい。
【0038】
スキームIのステップI−Cでは、構造I−5の中間体と構造I−4のアルコール間にエーテル結合を形成して、式Iの化合物を生成する。構造I−4のアルコールにおいて、Xは、A、BまたはCであり、R7aおよびR7bは、所望の最終生成物に見られるものと同じ置換基によって表される。R1によって表される置換基は、式Iの核を生成した後に操作することができる。そのような変形形態は、当業者によく知られており、本発明の一部とみなすべきである。ステップI−Cでは、DMF、THF、1,2−ジメトキシエタン、1,4−ジオキサン、N,N−ジメチルアセトアミド、ジメチルスルホキシド(「DMSO」)などの溶媒中にて、水素化ナトリウム;ナトリウムおよびカリウムtert−ブトキシド;ナトリウム、カリウム、およびリチウムビス(トリメチルシリル)アミド;ナトリウム、カリウム、およびリチウムtert−アミルオキシドなどの塩基の存在下、当量の反応物を反応させる。この変換の典型的な条件として、1,4−ジオキサン中にて約105℃で1時間のナトリウムビス(トリメチルシリル)アミドの使用が挙げられる。
【0039】
反応が完了した後、所望の式Iの化合物は、当技術分野で知られているとおりに回収し、単離することができる。化合物は、当技術分野で知られているように、蒸発、抽出などによって回収することができる。化合物は、場合により、クロマトグラフィー、再結晶、蒸留、または当技術分野で知られている他の技術によって精製してもよい。
【0040】
上で反応スキームIに示した代替合成では、構造I−2のジクロロ−ピリミジンを構造I−4のアルコールと最初に反応させて、構造I−6によって示される中間体を生成する。ステップI−Cでのように、構造I−4は、所望の最終生成物に応じて、XがA、BまたはCであるアルコールとなる。これらのヘテロ環において、R1およびR4は通常、最終生成物において所望されるものと同じ置換基で表されることとなり、またはR1は、式Iの核を生成した後に操作することができる。
【0041】
当量の構造I−2と構造I−4の化合物を極性非プロトン性溶媒および塩基の存在下で反応させて、ステップI−Dに示す構造I−6の中間体を生成する。適切な系として、0〜140℃の温度で、DMF、THF、1,2−ジメトキシエタン、1,4−ジオキサン、N,N−ジメチルアセトアミド、DMSOなどの溶媒中の、水素化ナトリウム、ナトリウムおよびカリウムtert−ブトキシド、ナトリウム、カリウム、およびリチウムビス(トリメチルシリル)アミド;ナトリウム、カリウム、およびリチウムtert−アミルオキシドなどの塩基が挙げられる。この変換の典型的な条件として、THF中にて約0℃〜室温で14時間のカリウムtert−ブトキシドの使用が挙げられる。構造I−6の中間体は、反応液から単離および回収し、当技術分野で知られているとおりにさらに精製することができる。別法として、以下で記載するステップI−Eにおいて未精製材料を使用してもよい。
【0042】
次いで、構造I−6の中間体を、上で予め記載した縮合テトラヒドロピロロ[3,4−c]ピラゾール誘導体I−3と反応させることにより、式Iの化合物を生成することができる。通常、塩基の存在下で、当量の構造I−3の縮合ピロリジンを式I−6のクロロ中間体と反応させる。適切な塩基は、DMF、THF、1,2−ジメトキシエタン、1,4−ジオキサン、N,N−ジメチルアセトアミド、もしくはDMSO、またはこれらの混合物などの溶媒中の、水素化ナトリウム、ナトリウムまたはカリウムtert−ブトキシド、ナトリウム、カリウム、またはリチウムビス(トリメチルシリル)アミド;およびナトリウム、カリウム、またはリチウムtert−アミルオキシドとすることができる。これらの反応は、使用する溶媒に応じて、約−10〜150℃の範囲の温度で実施することができる。通常、反応は、不活性雰囲気中にて約15分〜24時間の範囲の時間をかけて進行させる。適切な条件として、105℃で1時間、1,4−ジオキサン中の、ナトリウムビス(ジメチルシリル)アミドが挙げられる。
【0043】
別法として、この反応は、構造I−6の中間体と構造I−3のテトラヒドロピロロ[3,4−c]ピラゾール誘導体を、メタノール、エタノール、プロパノール、イソプロパノール、ブタノールなどの極性プロトン性溶媒中で0.5〜24時間加熱することにより実施してもよい。この変換の典型的な条件は、イソプロパノール中にて108℃で2時間加熱するものである。
【0044】
この反応は、式Iの化合物中に見られる重要な置換アミン結合の形成に遷移金属触媒を使用して実施することもできる。遷移金属触媒は、限定はしないが、トリフェニルホスフィン)パラジウム(Pd(PPh3)4)、塩化パラジウム(II)(PdCl2)、酢酸パラジウム(II)(Pd(OAc)2)、(トリス(ジベンジリデンアセトン)ジパラジウム(0)(Pd2(dba)3)、ヨウ化銅(I)(CuI)、酢酸銅(II)(Cu(OAc)2)、および銅(II)トリフルオロメタン(Cu(OTf)2)からなるものとすることができる。通常、これらの反応では塩基を利用する。パラジウム触媒と共に使用するのに適する塩基は、1,4−ジオキサン、THF、1,2−ジメトキシエタン、トルエンなどの適切な溶媒中のナトリウムtert−ブトキシド、カリウムtert−ブトキシド、カリウムtert−アミルオキシド、またはK3PO4とすることができる。銅触媒の使用について、適切な塩基は、DMF、DMSO、ジメチルアセトアミドなどの適切な溶媒中の炭酸ナトリウム、炭酸カリウム、炭酸セシウムなどのアルカリ塩基からなるものとすることができる。
【0045】
通常、配位子を加えると、アミン生成反応を促進することができる。パラジウムを触媒とする反応のための配位子として、限定はしないが、9,9−ジメチル−4,5−ビス(ジフェニルホスフィノ)キサンテン(キサントホス)、2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチル(BINAP)、1,1’−ビス(ジフェニルホスフィノ)フェロセン(DPPF)、2,8,9−トリイソブチル−2,5,8,9−テトラアザ−1−ホスファビシクロ[3.3.3]ウンデカン(P[N(i−Bu)CH2CH3]3N)、トリ−tert−ブチルホスフィン(tert−Bu3P)、(ビフェニル−2−イル)ビス(tert−ブチル)ホスフィン(JohnPhos)、Pd−PEPPSI(商標)−SIPr:(1,3−ビス(2,6−ジイソプロピルフェニル)−4,5−ジヒドロイミダゾール−2−イリデン)(3−クロロピリジル)パラジウム(II)ジクロリドを挙げることができる。銅を触媒とする反応に適する配位子として、限定はしないが、L−プロリン、N−メチルグリシン、ジエチルサリチルアミドを挙げることができる。式Iの化合物の生成に適する条件は、トルエン中にてPd2(dba)3をナトリウムtert−ブトキシドと共に120℃で12時間使用するものである。
【0046】
反応を完了した後、所望の式Iの化合物は、当技術分野で知られているとおりに回収および単離することができる。化合物は、当技術分野で知られているような蒸発、抽出などによって回収することができる。化合物は、クロマトグラフィー、再結晶、蒸留、または先行技術で知られている他の技術によって、場合により精製してもよい。
【0047】
また、当業者には容易に理解されるように、R1によって表される置換基の多くは、式Iの核を生成した後に操作することができる。そのような変形形態は、当業者によく知られており、本発明の一部とみなすべきである。
【0048】
【化6】
【0049】
スキームIIは、本発明の式IにおいてX=Bに対応する構造II−14およびII−15のアルコールの生成方法について記載したものである。R3、R6、R7aおよびR7bは通常、本明細書に記載するとおり、最終生成物で所望されるものと同じ置換基によって表される。構造II−7の化合物からの構造II−8の化合物の合成は、当技術分野で知られている。それらの変換(ステップII−A)は、文献で教示されており、J.Org.Chem.、1981、46、3196〜3204、JP2009096744、WC035303、J.Am.Chem.Soc.2008、130、5654〜5655、およびOrg.Lett.、2006、3、430〜436において例示されている。スキームIIのステップII−Bでは、メタノールのようなアルコール系溶媒中にて約0℃〜室温の範囲の温度で水素化ホウ素ナトリウムを使用するなどの、当技術分野で知られている標準のプロトコールを使用して、ケトンのカルボニル基を還元する。ステップII−D、すなわち、構造II−10からのベンジル保護基を除去してII−11を得るステップは、水素化分解によって実現することができる。この反応の典型的な条件として、水素、および5〜20%パラジウム担持炭素や10〜20%水酸化パラジウムなどのパラジウム触媒の利用が挙げられる。この反応の典型的な溶媒は、エタノール、メタノール、テトラヒドロフラン、または酢酸エチルである。
【0050】
最終生成物中にピリミジン置換基が所望される場合、エタノールやメタノールなどのプロトン性溶媒中、または1,4−ジオキサン、テトラヒドロフラン、N,N−ジメチルホルムアミドもしくはジメチルスルホキシドなどの極性非プロトン性溶媒中にて、炭酸セシウムやN,N−ジイソプロピルエチルアミンなどの塩基の存在下、化合物II−11を、構造II−12として示す適切に置換された2−クロロピリミジンに付加して、構造II−14を生成することができる。こうした反応は、およそ室温〜約110℃の範囲の温度で実施することができる。別法として、溶媒を使用せずN,N−ジイソプロピルエチルアミンなどの塩基の存在下、または化合物II−11を過剰に使用する場合では塩基または溶媒を使用せずに、構造II−11と構造II−12の化合物を一緒に加熱することもできる。
【0051】
最終生成物中にカルバメート置換基が所望される場合、ジクロロメタンまたはクロロホルム中にて、N,N−ジイソプロピルエチルアミン、トリエチルアミン、ピリジンなどの塩基の存在下、等価な量の構造II−13のアルキルハロホルメートを、構造II−11の化合物と反応させる。別法として、ジクロロメタン、クロロホルム、テトラヒドロフランなどの溶媒中にて、N,N−ジイソプロピルエチルアミン、ピリジン、2,6−ルチジン、トリエチルアミンなどのアミン塩基の存在下、二炭酸ジ−tert−ブチル(BOC無水物)や二炭酸ジイソプロピルなどの二炭酸ジアルキルを使用して、構造II−11の化合物から構造II−15の化合物を生成することもできる。加えて、R3=1−メチル−シクロプロピルまたは1−ジフルオロメチル−シクロプロピルであるとき、ジクロロメタン、ジクロロエタン、ジメトキシエタン、テトラヒドロフランのような溶媒中にて、トリエチルアミン、N,N−ジイソプロピルエチルアミンなどのような塩基の存在下、約0℃〜周囲温度程度の範囲の温度でカーボネートII−13’(WO09105717およびWO09005677を参照されたい)を使用して、カルバメート官能基を導入することができる。
【0052】
最終の構造II−14またはII−15は、当技術分野で知られているとおりに単離し、精製することができる。所望なら、この最終構造を分離ステップにかけて所望のシンまたはアンチ異性体を得ることもできる。
【0053】
別法として、ビス−アミノールエーテル誘導体II−9とケトンII−7間の二重マンニッヒ反応を介して、その後、ケトンカルボニルを還元し、官能基を操作してII−10型の構造を得ることにより、R7aおよびR7bの少なくとも一方が水素である式II−10の非対称構造に到達することができる。特定の例では、R9aは、構造II−10において示すベンジル基よりもα−メチル−ベンジル基とすることが好ましいことが認識される。適切なR9b基としては、メチルまたはエチルが挙げられる。式II−8の構造を得るための二重マンニッヒ反応の使用は、化学文献に発表されており(Tetrahedron 2005 61、5876〜5888;Org.Lett.2006 8、3399〜3401)、当業者によって実現することができる。同様に、容易に入手できる出発材料から出発し、シス−またはトランス−ジフルオロ−2,6シクロヘキサノンの生成に関する化学文献で知られているプロトコール(Tetrahedron 1970 26、2447)を使用して、R7aおよびR7bが両方ともフルオロである式II−10の構造に到達することもできる。
【0054】
スキームIIIでは、式IにおいてX=Aに対応する式III−19の化合物の調製について記載する。
【0055】
【化7】
【0056】
スキームIIIに示すように、R3が本明細書に記載のとおりであり、R7aおよびR7bの少なくとも一方が水素である式III−19の化合物は、市販されているN−tert−ブトキシカルボニル−4−ピペリドン(Aldrich)から出発して、または4−ピペリドンから、その後にカルバメート生成して調製することができる。式III−19の化合物は、ステップIII−Aによって示すように、式III−16またはIII−18の化合物のケトンカルボニルの還元による還元によって調製する。このための適切な条件として、エタノールなどのアルコール系溶媒とTHFの混合物中での水素化ホウ素ナトリウムの使用が挙げられる。R7aおよびR7bの少なくとも一方がフルオロである式III−19の化合物は、J.Org.Chem.2003 68、3232およびJ.Org.Chem.2002 67、8610に記載されているように、ケトンをエノール化してシリルエノールエーテルとしてトラップし、適切な求電子性フルオロ供給源と反応させることにより調製することができる。R7aおよびR7bの少なくとも一方がアルキル基である式III−18の化合物は、ハロゲン化アルキルやアルキルスルホネートなどの適切な求電子性アルキル基を使用して同様に調製することができる。加えて、R7aおよびR7bが両方ともフルオロなどのハロである式III−19の構造には、容易に入手できるN−tert−ブトキシカルボニル−4−ピペリドンから、化学文献で知られている同様のプロトコール(Tetrahedron、1970、26、2447)を使用して到達することができる。本発明の式IにおいてXがAであるとき、そのようなピペリジン化合物は、市販されているか、文献で知られているか、または市販(Aldrich)のN−tert−ブトキシカルボニル−4−ピペリドンもしくは適切にN保護された他のピペリドンから調製できることもまた認識される。
【0057】
tert−ブチルオキシカルボニル基(R3はtert−ブチルである)は、合成の多くの段階で、塩酸やトリフルオロ酢酸などの酸を使用して除去することができ、その結果生じる遊離アミンは、スキームIIのステップII−E’およびII−Eでそれぞれ記載した一般的な条件を使用して、別のカルバメートまたはピリミジンに変換できることが認識される。式III−19の化合物の調製は、WO2009014910にも記載されている。
【0058】
スキームIVでは、式IV−23の化合物の合成について記載する。
【0059】
【化8】
【0060】
スキームIVの式IV−23のテトラヒドロピロロ−ピラゾールは、式IV−21の化合物から、適切な式IV−22のヒドラジンとの付加/脱水環化(ステップIV−B)に続いて、tert−ブチルオキシカルボニル基を脱保護する(ステップIV−C)ことにより調製することができる。式IV−21の化合物は、スキームIVの式IV−20の化合物から、ホルミル化反応(ステップIV−A)によって調製することができる。スキームIVのステップIV−Aの適切な条件として、化合物IV−20をN,N−ジメチルホルムアミドジメチルアセタール(DMFDMA)の存在下で加熱することが挙げられる。スキームIVの式IV−20の化合物は、市販されており(Aldrich)、文献で知られており、または当業者により容易に調製することができる。
【0061】
R8aおよびR8bまたはR8cおよびR8dが、これらが結合している炭素と一緒になって、C3〜C6シクロアルキルを形成している例は、Journal of Organic Chemistry 2004 69、2755〜2759およびJournal of Organic Chemistry 1962 27、2901〜5において見られる。R8aとR8cは一緒になって、完全飽和二炭素橋を形成していてもよく、R8aおよびR8cが、これらが結合している環系の同じ平面上にある例としては、(1R,4S)−2−オキソ−7−アザビシクロ[2.2.1]ヘプタン−7−カルボン酸tert−ブチル(Brother Chemistry Co.から市販されている、CAS番号16513−98−2)が挙げられる。この化合物のラセミ体も、文献で知られている(Journal of Medicinal Chemistry 2003 46、921〜924;Journal of Organic Chemistry 1994 59、1771〜8、Bioorganic & Medicinal Chemistry Letters 2008 18、4651〜4654)。
【0062】
当業者には容易にわかるように、中間体の遠位官能基(たとえば、第一級または第二級アミン)を保護する必要がある場合もある。そのような保護の必要性は、その遠位官能基の性質および調製方法の条件に応じて異なってくる。適切なアミノ保護基(NH−Pg)として、アセチル、トリフルオロアセチル、t−ブトキシカルボニル(BOC)、ベンジルオキシカルボニル(CBZ)、および9−フルオレニルメチレンオキシカルボニル(Fmoc)が挙げられる。同様に、「ヒドロキシ保護基」とは、ヒドロキシ官能基を封鎖または保護する、ヒドロキシ基の置換基を指す。適切なヒドロキシル保護基(O−Pg)として、たとえば、アリル、アセチル、シリル、ベンジル、para−メトキシベンジル、トリチルなどが挙げられる。このような保護の必要性は、当業者によって容易に決定される。保護基およびその使用に関する総説については、T.W.Greene、Protective Groups in Organic Synthesis、John Wiley&Sons、ニューヨーク、1991を参照されたい。
【0063】
上述のとおり、本発明の化合物の一部は、薬学的に許容できるカチオンと塩を形成することができる。本発明の化合物の一部は、薬学的に許容できるアニオンと塩を形成することができる。そのような塩はすべて、本発明の範囲内にあり、従来の方法によって、たとえば、適宜、水性、非水性、または部分的に水性の媒質中にて、酸性実体と塩基性実体とを、通常は化学量論比で合わせるなどして調製することができる。塩は、適宜、濾過、非溶媒で沈殿させてからの濾過、溶媒の蒸発、または水溶液の場合では凍結乾燥によって回収する。化合物は、エタノール、ヘキサン、水/エタノール混合物などの適切な(1種または複数の)溶媒に溶解させるなどの、当技術分野で知られている手順に従って、結晶の形で得る。
【0064】
本発明は、原子質量または質量数が自然界で通常見られる原子質量または質量数と異なっている原子で1個または複数の原子が置き換えられていること以外は本明細書で列挙したものと同一である、同位体標識された本発明の化合物も包含する。本発明の化合物に組み込むことのできる同位体の例として、2H、3H、11C、13C、14C、13N、15N、15O、17O、18O、31P、32P、35S、18F、123I、125I、36Clなどの、それぞれ、水素、炭素、窒素、酸素、リン、硫黄、フッ素、ヨウ素、および塩素の同位体が挙げられる。
【0065】
特定の同位体標識された本発明の化合物(たとえば、3Hおよび14Cで標識された化合物)は、化合物および/または基質の組織分布アッセイにおいて有用である。トリチウム、14C、35S、および125Iが組み込まれている特定の同位体標識されたリガンドは、放射リガンド結合アッセイにおいて有用となり得るはずである。トリチウム化(すなわち3H)およびカーボン14(すなわち14C)同位体は、調製しやすく、検出性がよいので、特に好ましい。さらに、ジュウテリウム(すなわち2H)などのより重い同位体での置換は、代謝安定性がより高いために生じる特定の治療上の優位性(たとえば、in vivo半減期の延長または投与必要量の減少)をもたらす場合もあり、したがって、状況によっては好ましいこともある。15O、13N、11C、18Fなどの陽電子放射同位体は、受容体占有率を調べるための陽電子放射断層撮影(PET)研究に有用である。本発明の同位体標識された化合物は、一般に、同位体標識されていない試薬の代わりに同位体標識された試薬を用いて、スキームおよび/または後述の実施例で開示する手順と類似した手順に従って調製することができる。
【0066】
特定の本発明の化合物は、2種以上の結晶形で存在する場合もある(一般に「多形体」と呼ばれる)。多形体は、種々の条件下での結晶化によって、たとえば、結晶化の際に、異なる再結晶用溶媒もしくは溶媒混合物、異なる温度での結晶化、および/または超急速冷却から超緩速冷却の範囲の種々の冷却モードを使用して、調製することができる。多形体は、本発明の化合物を加熱または溶融した後、徐々にまたは急速に冷却して得ることもできる。多形体の存在は、固体プローブNMR分光法、IR分光法、示差走査熱量測定、粉末X線回折、または他のそのような技術によって判定することができる。
【0067】
医学的使用
本発明の化合物は、Gタンパク質共役受容体GPR119の活性を調節する。そのため、前記化合物は、糖尿病などの、GPR119の活性が疾患の病理または症状の一因となる疾患の予防および治療に有用である。したがって、本発明の別の態様は、個体において代謝性疾患および/または代謝関連障害を治療する方法であって、そのような治療が必要である個体に、治療有効量の本発明の化合物、前記化合物の塩、またはそのような化合物を含有する医薬組成物を投与することを含む方法を包含する。代謝性疾患および代謝関連障害は、限定はしないが、高脂血症、I型糖尿病、II型糖尿病、特発性I型糖尿病(Ib型)、成人潜在性自己免疫性糖尿病(LADA)、早発性2型糖尿病(EOD)、若年性非定型糖尿病(YOAD)、若年発症成人型糖尿病(MODY)、栄養不良関連糖尿病、妊娠糖尿病、冠動脈心疾患、虚血発作、血管形成術後の再狭窄、末梢血管疾患、間欠性跛行、心筋梗塞(たとえば、壊死およびアポトーシス)、異脂肪症、食後脂肪血症、耐糖能障害(IGT)状態、空腹時血漿グルコースの異常状態、代謝性アシドーシス、ケトーシス、関節炎、肥満、骨粗鬆症、高血圧、うっ血性心不全、左室肥大、末梢動脈疾患、糖尿病性網膜症、黄斑変性、白内障、糖尿病性腎症、糸球体硬化症、慢性腎不全、糖尿病性ニューロパシー、メタボリック症候群、シンドロームX、月経前症候群、冠動脈心疾患、狭心症、血栓症、アテローム性動脈硬化症、心筋梗塞、一過性脳虚血発作、卒中、血管再狭窄、高血糖、高インスリン血症、高脂血症、高トリグリセリド血症、インスリン抵抗性、グルコース代謝障害、耐糖能障害状態、空腹時血漿グルコースの異常状態、肥満、勃起機能不全、皮膚および結合組織の障害、足の潰瘍化、内皮障害、高アポBリポタンパク質血症(hyper apo B lipoproteinemia)、ならびに血管伸展性の障害から選択される。さらに、本発明の化合物は、アルツハイマー病、統合失調症、認知障害などの神経障害の治療に使用することもできる。本発明の化合物は、炎症性腸疾患、潰瘍性大腸炎、クローン病、過敏性腸症候群などの胃腸疾患においても有益となる。上述のように、本発明の化合物は、肥満患者、特に糖尿病に罹患している肥満患者において体重減少を刺激するのにも使用することができる。
【0068】
前述の内容によれば、本発明はさらに、その必要がある対象において、上述の疾患または障害のいずれかの症状を予防し、または寛解させる方法であって、治療有効量の本発明の化合物を対象に投与することを含む方法を提供する。本発明の別の態様は、糖尿病およびその関連合併症を治療する医薬の調製を包含する。
【0069】
上述の治療特性を示すためには、化合物は、Gタンパク質共役受容体GPR119の活性化を調節するのに十分な量で投与する必要がある。この量は、治療する特定の疾患/状態、患者の疾患/状態の重症度、患者、投与する特定の化合物、投与経路、および患者内の他の基礎病態の存在などに応じて様々となり得る。全身に投与するとき、化合物は通常、上で挙げた疾患または状態のいずれに対しても、約0.1mg/kg/日〜約100mg/kg/日の投与量範囲でその効果を示す。毎日の連続投与が望ましい場合もあり、上で概略を述べた状態に応じて異なってくる。
【0070】
本発明の化合物は、様々な経路によって投与することができる。本発明の化合物は、経口投与することができる。本発明の化合物は、非経口(すなわち、皮下、静脈内、筋肉内、腹腔内、またはくも膜下腔内)、直腸、または局所投与することもできる。
【0071】
共投与
本発明の化合物は、本明細書に記載の疾患、状態、および/または障害を治療するための他の薬剤と共に使用することもできる。したがって、本発明の化合物を他の薬剤と組み合わせて投与することを含む治療方法も提供する。本発明の化合物と組み合わせて使用することのできる適切な薬剤として、抗肥満薬(食欲抑制薬を含める)、抗糖尿病薬、抗高血糖薬(anti−hyperglycemic agent)、高脂血症治療薬、および降圧薬が挙げられる。
【0072】
適切な抗糖尿病薬として、アセチル−CoAカルボキシラーゼ2(ACC−2)阻害剤、ジアシルグリセロールO−アシルトランスフェラーゼ1(DGAT−1)阻害剤、ホスホジエステラーゼ(PDE)10阻害剤、スルホニル尿素(たとえば、アセトヘキサミド、クロルプロパミド、ジアビネース(diabinese)、グリベンクラミド、グリピジド、グリブリド、グリメピリド、グリクラジド、グリペンチド(glipentide)、グリキドン、グリソラミド、トラザミド、およびトルブタミド)、メグリチニド、α−アミラーゼ阻害剤(たとえば、テンダミスタット(tendamistat)、トレスタチン、およびAL−3688)、α−グルコシドヒドロラーゼ阻害剤(たとえば、アカルボース)、α−グルコシダーゼ阻害剤(たとえば、アジポシン、カミグリボース、エミグリテート、ミグリトール、ボグリボース、プラジミシンQ、およびサルボスタチン(salbostatin))、PPARγ作動薬(たとえば、バラグリタゾン、シグリタゾン、ダルグリタゾン、エングリタゾン、イサグリタゾン(isaglitazone)、ピオグリタゾン、ロシグリタゾン、およびトログリタゾン)、PPARα/γ作動薬(たとえば、CLX−0940、GW−1536、GW−1929、GW−2433、KRP−297、L−796449、LR−90、MK−0767、およびSB−219994)、ビグアナイド(たとえば、メトホルミン)、グルカゴン様ペプチド1(GLP−1)作動薬(たとえば、エキセンディン3およびエキセンディン4)、タンパク質チロシンホスファターゼ1B(PTP−1B)阻害剤(たとえば、トロダスケミン(trodusquemine)、ヒルチオサール抽出物(hyrtiosal extract)、およびZhang,S.ら、Drug Discovery Today、12(9/10)、373〜381(2007)で開示されている化合物)、SIRT−1阻害剤(たとえば、レセルバトロール(reservatrol))、ジペプチジルペプチダーゼIV(DPP−IV)阻害剤(たとえば、シタグリプチン、ビルダグリプチン、アログリプチン、およびサクサグリプチン)、インスリン分泌刺激物質、脂肪酸酸化阻害剤、A2拮抗薬、c−junアミノ末端キナーゼ(JNK)阻害剤、インスリン、インスリン模倣物、グリコーゲンホスホリラーゼ阻害剤、VPAC2受容体作動薬、およびSGLT2阻害剤(ナトリウム依存性グルコース輸送体阻害剤、たとえば、ダパグリフロジンなど)が挙げられる。好ましい抗糖尿病薬は、メトホルミンおよびDPP−IV阻害剤(たとえば、シタグリプチン、ビルダグリプチン、アログリプチン、およびサクサグリプチン)である。
【0073】
適切な抗肥満薬として、11β−ヒドロキシステロイドデヒドロゲナーゼ1(11β−HSD1型)阻害剤、ステアロイルCoAデサチュラーゼ1(SCD−1)阻害剤、MCR−4作動薬、コレシストキニンA(CCK−A)作動薬、モノアミン再取込み阻害剤(シブトラミンなど)、交感神経様作動薬、β3アドレナリン作動薬、ドーパミン作動薬(ブロモクリプチンなど)、メラノサイト刺激ホルモン類似体、5HT2c作動薬、メラニン濃縮ホルモン拮抗薬、レプチン(OBタンパク質)、レプチン類似体、レプチン作動薬、ガラニン拮抗薬、リパーゼ阻害剤(テトラヒドロリプスタチン、すなわちオルリスタットなど)、食欲抑制薬(ボンベシン作動薬など)、ニューロペプチドY拮抗薬(たとえば、NPY Y5拮抗薬、PYY3−36(その類似体を含める)、甲状腺模倣薬(thyromimetic agent)、デヒドロエピアンドロステロンまたはその類似体、糖質コルチコイド作動薬または拮抗薬、オレキシン拮抗薬、グルカゴン様ペプチド1作動薬、毛様体神経栄養因子(Regeneron Pharmaceuticals,Inc.、ニューヨーク州タリータウン、およびProcter&Gamble Company、オハイオ州シンシナティーから入手可能なAxokine(商標)など)、ヒトアグーチ関連タンパク質(AGRP)阻害剤、グレリン拮抗薬、ヒスタミン3拮抗薬または逆作動薬、ニューロメジンU作動薬、MTP/ApoB阻害剤(たとえば、ジルロタピド(dirlotapide)などの消化管選択的MTP阻害剤)、オピオイド拮抗薬、オレキシン拮抗薬などが挙げられる。
【0074】
本発明の組合せ態様で使用するのに好ましい抗肥満薬として、消化管選択的MTP阻害剤(たとえば、ジルロタピド(dirlotapide)、ミトラタピド(mitratapide)およびイミプリタピド(implitapide)、R56918(CAS番号403987)、およびCAS番号913541−47−6)、CCKa作動薬(たとえば、PCT公開第WO2005/116034号または米国公開第2005−0267100A1号に記載のN−ベンジル−2−[4−(1H−インドール−3−イルメチル)−5−オキソ−1−フェニル−4,5−ジヒドロ−2,3,6,10b−テトラアザ−ベンゾ[e]アズレン−6−イル]−N−イソプロピル−アセトアミド)、5HT2c作動薬(たとえば、ロルカセリン)、MCR4作動薬(たとえば、US6,818,658に記載の化合物)、リパーゼ阻害剤(たとえば、セチリスタット)、PYY3−36(本明細書では、「PYY3−36」は、ベグ化PYY3−36(たとえば、米国公開2006/0178501に記載のもの)などの類似体を包含する)、オピオイド拮抗薬(たとえば、ナルトレキソン)、オレオイル−エストロン(CAS番号180003−17−2)、オビネピチド(obinepitide)(TM30338)、プラムリンチド(Symlin(登録商標))、テソフェンシン(NS2330)、レプチン、リラグルチド、ブロモクリプチン、オルリスタット、エクセナチド(Byetta(登録商標))、AOD−9604(CAS番号221231−10−3)、およびシブトラミンが挙げられる。本発明の化合物および併用療法は、運動および賢明な食生活と合わせて投与することが好ましい。
【0075】
上で引用した米国特許および公開はすべて、参照により本明細書に援用する。
【0076】
医薬製剤
本発明は、少なくとも1種の薬学的に許容できる賦形剤と混和された治療有効量の化合物または薬学的に許容できるその塩を含む医薬組成物も提供する。医薬組成物は、経口、局所、または非経口の使用に適合させた形態の組成物を包含し、上述のような糖尿病および関連状態の治療に使用することができる。
【0077】
医薬組成物は、皮下、吸入、経口、局所、非経口などの、当技術分野で知られている任意の経路による投与用に製剤することができる。医薬組成物は、限定はしないが、錠剤、カプセル剤、粉末、顆粒、ロゼンジ、または経口もしくは滅菌非経口溶液もしくは懸濁液などの液体製剤を含めて、当技術分野で知られているどんな形態でもよい。
【0078】
経口投与用の錠剤およびカプセル剤は、単位用量体裁にすることができ、従来の賦形剤、たとえば、シロップ、アカシア、ゼラチン、ソルビトール、トラガカント、ポリビニルピロリドンなどの結合剤;ラクトース、糖、トウモロコシデンプン、リン酸カルシウム、ソルビトール、グリシンなどの充填剤;ステアリン酸マグネシウム、タルク、ポリエチレングリコール、シリカなどの打錠滑沢剤;バレイショデンプンなどの崩壊剤;またはラウリル硫酸ナトリウムなどの許容される湿潤剤を含有してよい。錠剤は、標準の薬務でよく知られている方法に従ってコーティングしてもよい。
【0079】
経口液体製剤は、たとえば、水性もしくは油性の懸濁液、溶液、乳濁液、シロップ、もしくはエリキシルの形にすることもでき、または使用前に水もしくは適切な他のビヒクルで再形成する乾燥製品としての体裁にすることもできる。このような液体製剤は、従来の添加剤、たとえば、ソルビトール、メチルセルロース、グルコースシロップ、ゼラチン、ヒドロキシエチルセルロース、カルボキシメチルセルロース、ステアリン酸アルミニウムゲル、水素添加食用脂などの懸濁化剤;レシチン、モノオレイン酸ソルビタン、アカシアなどの乳化剤;扁桃油、グリセリンのような油性エステル、プロピレングリコール、エチルアルコールなどの非水性ビヒクル(食用油を含めてもよい);p−ヒドロキシ安息香酸メチルもしくはプロピル、ソルビン酸などの保存剤、および所望なら、従来の着香剤または着色剤を含有してよい。
【0080】
非経口投与では、化合物および無菌ビヒクル(水が好ましい)を利用して、流動性の単位剤形を調製する。化合物は、使用するビヒクルおよび濃度に応じて、ビヒクルまたは適切な他の溶媒に懸濁または溶解させることができる。溶液の調製では、化合物を、注射用の水に溶解させ、濾過滅菌した後、適切なバイアルまたはアンプルに充填し、密封することができる。有利にするため、局所麻酔剤、保存剤、緩衝剤などの薬品をビヒクルに溶解させることができる。安定性を高めるために、組成物をバイアルに充填した後凍結させ、真空中で水を除去することができる。次いで、凍結乾燥した乾燥粉末をバイアルに密閉し、使用前に液体を再形成するための付属のバイアルの注射用水を支給することができる。非経口懸濁液は、化合物をビヒクルに溶解させる代わりに懸濁させること、および滅菌が濾過によって実現できないことを除き、実質上同じようにして調製する。無菌ビヒクルに懸濁させる前にエチレンオキシドにさらすことにより、化合物を滅菌することができる。組成物に界面活性剤または湿潤剤を組み込んで、化合物の均一な分布を促進すると有利である。
【0081】
組成物は、投与方法に応じて、たとえば約0.1重量%〜約99重量%の活性材料を含有してよい。組成物が投与量単位を構成する場合、各単位は、たとえば、約0.1〜900mg、より典型的な場合では1mg〜250mgの活性成分を含有することになる。
【0082】
本発明の化合物は、他の抗糖尿病薬による類推によって、ヒト医学または獣医学で使用するための好都合な任意の方法における投与用に製剤することができる。そのような方法は、当技術分野で知られており、上で概略を述べている。そうした製剤の調製に関するより詳細な論述については、University of the Sciences in PhiladelphiaによるRemington’s Pharmaceutical Sciences、第21版に目を向けられたい。
【0083】
以下の実施例によって、本発明の実施形態を例示する。しかし、本発明の実施形態は、実施例の詳細な項目を限定せず、このためそれらの他の変形形態が、当業者には知るところとなり、またはこの開示に照らして明白となることを理解されたい。
【0084】
(実施例)
別段指定しない限り、出発材料は一般に、Aldrich Chemicals Co.(ウィスコンシン州ミルウォーキー)、Lancaster Synthesis,Inc.(ニューハンプシャー州Windham)、Acros Organics(ニュージャージー州フェアローン)、Maybridge Chemical Company,Ltd.(英国コーンウォール)、Tyger Scientific(ニュージャージー州プリンストン)、AstraZeneca Pharmaceuticals(英国ロンドン)、Mallinckrodt Baker(ニュージャージー州フィリップスバーグ)、EMD(ニュージャージー州Gibbstown)などの市販品供給元から入手可能である。
【0085】
一般実験手順
プロトン分析について、NMRスペクトルは、Varian Unity(商標)400(DG400−5プローブ)または500(DG500−5プローブ)(共にVarian Inc.、カリフォルニア州パロアルトから入手可能)を用い、室温にてそれぞれ400MHzまたは500MHzで記録した。化学シフトは、内部標準としての残存溶媒を基準とした百万分率(δ)で表示する。ピーク形状は、次のとおりに表記する。すなわち、s:一重線、d:二重線、dd:二重二重線、t:三重線、q:四重線、m:多重線、bs:ブロード一重線、2s:2本の一重線。
【0086】
大気圧化学イオン化質量スペクトル(APCI)は、Waters(商標)分光計(Micromass ZMD、キャリヤーガス:窒素)(Waters Corp.、米国マサチューセッツ州ミルフォードから入手可能)を用い、0.3mL/分の流量で、50:50の水/アセトニトリル溶離液系を利用して取得した。エレクトロスプレーイオン化質量スペクトル(ES)は、Waters(商標)(Micromass ZQまたはZMD機器(キャリヤーガス:窒素)(Waters Corp.、米国マサチューセッツ州ミルフォード)の液体クロマトグラフィー質量分析計を用い、各溶媒に0.01%のギ酸を加えた95:5〜0:100の勾配の水アセトニトリル溶液を利用して取得した。これらの機器には、3.75分間1mL/分または1.95分間2mL/分の流量で、Varian Polaris 5 C18−A20×2.0mmカラム(Varian Inc.、カリフォルニア州パロアルト)を用いた。
【0087】
カラムクロマトグラフィーは、Flash 40 Biotage(商標)カラム(ISC,Inc.、コネティカット州シェルトン)またはBiotage(商標)SNAPカートリッジKPsilまたはRedisep Rfシリカ(Teledyne Isco Incより)のいずれかを用い、シリカゲルを使用して窒素圧下で実施した。分取HPLCは、フォトダイオードアレイ(Waters 2996)および質量分析計(Waters/Micromass ZQ)検出スキームを備えたWaters FractionLynxシステムを使用して実施した。分析HPLC作業は、フォトダイオードアレイ、単収束四極子質量検出および蒸発光散乱検出スキームを備えたWaters 2795 Alliance HPLCまたはWaters ACQUITY UPLCを用いて行った。
【0088】
真空中での濃縮とは、ロータリーエバポレーターを使用して、減圧下で溶媒を蒸発させることを指す。
【0089】
別段言及しない限り、化学反応は室温(摂氏約23度)で実施した。また、別段言及しない限り、化学反応は窒素雰囲気中で進めた。
【0090】
薬理学的データ
Gタンパク質共役受容体GPR119の本発明の化合物によるアゴニスト活性化によって調節される疾患を治療する本発明の実施は、後述する機能アッセイの1つまたは複数における活性によって証明することができる。供給元は括弧内に示す。
【0091】
in−vitro機能アッセイ
β−ラクタマーゼ:
GPR119アゴニストについてのアッセイでは、ヒトGPR119のアゴニスト活性化を、環状AMP反応要素(CRE)によってβ−ラクタマーゼ産生と合体させた、細胞を主体とした(hGPR119 HEK293−CRE β−ラクタマーゼ)レポーター構築物を利用する。そしてGPR119活性は、FRETを可能にするβ−ラクタマーゼ基質であるCCF4−AM(Live Blazer FRET−B/G Loadingキット、Invitrogenカタログ番号K1027)を利用して測定する。詳細には、hGPR119−HEK−CRE−β−ラクタマーゼ細胞(Invitrogen 2.5×107/mL)を液体窒素貯蔵庫から取り出し、プレーティング培地(ダルベッコ変法イーグル培地高グルコース(DMEM、Gibcoカタログ番号11995−065)、10%熱不活性化ウシ胎児血清(HIFBS、Sigmaカタログ番号F4135)、1×MEM非必須アミノ酸(Gibcoカタログ番号15630−080)、25mMのHEPES pH7.0(Gibcoカタログ番号15630−080)、200nMのクラブラン酸カリウム(Sigmaカタログ番号P3494)に希釈した。細胞プレーティング培地を使用して細胞濃度を調節し、この細胞懸濁液(12.5×104生細胞)50μLを、ポリ−d−リシンでコートされた黒色透明底384ウェルプレート(Greiner Bio−Oneカタログ番号781946)の各ウェルに加え、5%の二酸化炭素を含有する加湿した環境において37℃でインキュベートした。4時間後、プレーティング培地を除去し、40μLのアッセイ培地(アッセイ培地は、クラブラン酸カリウムおよびHIFBSを含有しないプレーティング培地である)と入れ替えた。次いで、試験対象の様々な濃度の各化合物を10uLの体積(最終DMSO≦0.5%)で加え、5%の二酸化炭素を含有する加湿した環境において細胞を37℃で16時間インキュベートした。プレートをインキュベーターから取り出し、約15分間かけて室温に平衡化させた。10uLの6×CCF4/AM作用色素溶液(Live Blazer FRET−B/G Loadingキット(Invitrogenカタログ番号K1027)に入っている説明書に従って調製)をウェル毎に加え、暗所にて室温で2時間インキュベートした。EnVision蛍光定量プレートリーダー(励起405nm、発光460nm/535nm)で蛍光を測定した。4パラメータロジスティック用量反応方程式を使用した曲線適合プログラムで分析を行ったアゴニスト反応曲線から、EC50を決定した。
【0092】
cAMP:
細胞中のcAMPレベルを測定するHTRF(均一時間分解蛍光)cAMP検出キット(cAMP dynamic 2アッセイキット Cis Bioカタログ番号62AM4PEC)を利用する細胞アッセイでも、GPR119アゴニスト活性を求めた。この方法は、細胞によって産生される自然なcAMPと、色素d2で標識したcAMPの競合イムノアッセイである。トレーサー結合を、クリプテートで標識したMab抗cAMPによって可視化する。特異的シグナル(すなわち、エネルギー移動)は、標準またはサンプル中のcAMPの濃度に反比例する。
【0093】
詳細には、hGPR119 HEK−CREβ−ラクタマーゼ細胞(Invitrogen 2.5×107/mL、上述のβ−ラクタマーゼアッセイで使用した同じ細胞系)を凍結保存から取り出し、増殖培地(ダルベッコ変法イーグル培地高グルコース(DMEM、Gibcoカタログ番号11995−065)、1%チャコールデキストラン処理ウシ胎児血清(CD血清、HyCloneカタログ番号SH30068.03)、1×MEM非必須アミノ酸(Gibcoカタログ番号15630−080)、および25mMのHEPES pH7.0(Gibcoカタログ番号15630−080))中に希釈する。細胞濃度を1.5×105細胞/mlに調整し、この懸濁液30mlをT−175フラスコに加え、5%二酸化炭素の加湿環境において37℃でインキュベートした。16時間(一晩経過)後、細胞をT−175フラスコから(フラスコの側面を叩いて)取り出し、800×gで遠心分離し、次いでアッセイ培地(1×HBSS+CaCl2+MgCl2(Gibcoカタログ番号14025−092)および25mMのHEPES pH7.0(Gibcoカタログ番号15630−080))に再懸濁した。アッセイ培地を用いて細胞濃度を6.25×105細胞/mlに調整し、この細胞懸濁液8μl(5000細胞)を、Greiner384ウェル白色小体積アッセイプレート(VWRカタログ番号82051−458)の各ウェルに加えた。
【0094】
試験対象の様々な濃度の各化合物を3−イソブチル−1−メチルキサンチン(IBMX、Sigmaカタログ番号I5879)を含有するアッセイ緩衝液に希釈し、2μLの体積でアッセイプレートウェルに加えた(最終IBMX濃度は400μMとし、最終DMSO濃度は0.58%とした)。室温で30分間のインキュベートに続いて、5μLの標識したd2cAMPおよび5μLの抗cAMP抗体(両方とも細胞溶解緩衝液に1:20希釈したもの、製造者のアッセイプロトコールに記載のとおり)をアッセイプレートの各ウェルに加えた。次いでプレートを室温でインキュベートし、60分後、Envision 2104マルチラベルプレートリーダーで、励起波長330nm、発光波長615nmおよび665nmを使用し、HTRFシグナルの変化を読み取った。生データを、cAMP検量線からの内挿によってnM cAMPに変換し(製造者のアッセイプロトコールに記載のとおり)、4パラメータロジスティック用量反応式を使用して曲線適合プログラムで分析したアゴニスト反応曲線から、EC50を決定した。
【0095】
GPR119の活性化によるcAMP反応は、ここで使用した特定の細胞系以外の細胞でも発生し得ることが認められている。
【0096】
β−アレスチン:
DiscoverX PathHunterβ−アレスチン細胞アッセイ技術およびそのU2OS hGPR119β−アレスチン細胞系(DiscoverXカタログ番号93−0356C3)を利用する細胞アッセイでも、GPR119アゴニスト活性を求めた。このアッセイでは、アゴニストによって誘発される、β−アレスチンと活性化型GPR119の相互作用を測定することにより、アゴニスト活性化を判定する。ProLinkと呼ばれる、小さい42アミノ酸の酵素断片を、GPR119のC末端に付加した。アレスチンを、EA(酵素アクセプター)と呼ばれるより大きい酵素断片に融合した。GPR119が活性化すると、アレスチンの結合が刺激され、2つの酵素断片の相補性が引き出される結果、基質を加水分解し、化学発光シグナルを発生させることのできる機能性β−ガラクトシダーゼ酵素が生成する。
【0097】
詳細には、U2OS hGPR119β−アレスチン細胞(DiscoverX 1×107/ml)を凍結保存から取り出し、増殖培地(最小必須培地(MEM、Gibcoカタログ番号11095−080)、10%熱不活化ウシ胎児血清(HIFBS、Sigmaカタログ番号F4135−100)、100mMのピルビン酸ナトリウム(Sigmaカタログ番号S8636)、500μg/mLのG418(Sigmaカタログ番号G8168)、および250μg/mLのハイグロマイシンB(Invitrogenカタログ番号10687−010)中に希釈する。細胞濃度を1.66×105細胞/mlに調整し、この懸濁液30mlをT−175フラスコに加え、5%二酸化炭素の加湿環境において37℃でインキュベートした。24時間後、酵素不使用の細胞解離緩衝液(Gibcoカタログ番号13151−014)を用いて細胞をT−175フラスコから取り出し、800×gで遠心分離し、次いでプレーティング培地(Opti−MEM I(Invitrogen/BRLカタログ番号31985−070)および2%チャコールデキストラン処理ウシ胎児血清(CD血清、HyCloneカタログ番号SH30068.03))に再懸濁した。プレーティング培地を用いて細胞濃度を2.5×105細胞/mlに調整し、この細胞懸濁液10μL(2500細胞)を、Greiner 384ウェル白色小体積アッセイプレート(VWRカタログ番号82051−458)の各ウェルに加え、プレートを5%二酸化炭素の加湿環境において37℃でインキュベートした。
【0098】
16時間(一晩経過)後、インキュベーターからアッセイプレートを取り出し、試験対象の様々な濃度の各化合物(アッセイ緩衝液(1×HBSS+CaCl2+MgCl2(Gibcoカタログ番号14025−092)、20mMのHEPES pH7.0(Gibcoカタログ番号15630−080)、および0.1%のBSA(Sigmaカタログ番号A9576)中に希釈したもの)を2.5μLの体積でアッセイプレートウェルに加えた(最終DMSO濃度を0.5%とした)。5%二酸化炭素の加湿環境において37℃で90分インキュベートした後、7.5μLのGalacton Starβ−ガラクトシダーゼ基質(PathHunter Detection Kit(DiscoveRxカタログ番号93−0001)、製造者のアッセイプロトコールに記載のとおりに調製)をアッセイプレートの各ウェルに加えた。プレートを室温でインキュベートし、60分後、Envision 2104マルチラベルプレートリーダーを用い、ウェルあたり0.1秒で発光の変化を読み取った。4−パラメータロジスティック用量反応式を使用して曲線適合プログラムで分析したアゴニスト反応曲線から、EC50を決定した。
【0099】
BacMamを使用したGPR119の発現およびGPR119結合アッセイ
鋳型としてのpIRES−puro−hGPR119および以下のプライマーを使用するポリメラーゼ連鎖反応(PCR)(Pfu Turbo Mater Mix、Stratagene、カリフォルニア州ラホーヤ)によって、野生型ヒトGPR119(図1)を増幅した。
hGPR119 BamH1、上流
5’−TAAATTGGATCCACCATGGAATCATCTTTCTCATTTGGAG−3’
(5’末端にBamHI部位を挿入する)
hGPR119 EcoRI、下流
5’−TAAATTGAATTCTTATCAGCCATCAAACTCTGAGC−3’
(3’末端にEcoRI部位を挿入する)
【0100】
製造者のプロトコールに従い、増幅産物を精製し(Qiaquick Kit、Qiagen、カリフォルニア州バレンシア)、BamH1およびEcoRI(New England BioLabs、マサチューセッツ州イプスウィッチ)で消化した。ベクターpFB−VSVG−CMV−ポリ(図2)をBamHIおよびEcoRI(New England BioLabs、マサチューセッツ州イプスウィッチ)で消化した。消化したDNAを1%アガロースゲルでの電気泳動によって分離し、断片をゲルから切り出し、精製した(Qiaquick Kit、Qiagen、カリフォルニア州バレンシア)。ベクターと遺伝子断片を連結し(Rapid Ligase Kit、Roche、カリフォルニア州プレザントン)、OneShot DH5αT1R細胞(Invitrogen、カリフォルニア州カールスバッド)に形質転換した。8つのアンピシリン耐性コロニー(「クローン1〜8」)をミニプレップ(Qiagen Miniprep Kit、Qiagen、カリフォルニア州バレンシア)用に成長させ、配列決定して、同一性および正しい挿入の方向を確認した。
【0101】
製造者のプロトコールに従い、pFB−VSVG−CMV−ポリ−hGPR119構築物(クローン#1)をOneShot DH10Bac細胞(Invitrogen、カリフォルニア州カールスバッド)に形質転換した。8つの陽性(すなわち白色)のコロニーを画線し直して、「陽性」であると確認し、引き続いてバクミド単離に向けて成長させた。組換え型hGPR119バクミドは、Qiagen Miniprep Kit(Qiagen、カリフォルニア州バレンシア)の緩衝液を使用する変法アルカリ溶解手順によって単離した。簡潔に述べると、ペレット化した細胞を緩衝液P1に溶解させ、緩衝液P2中で中和し、緩衝液N3で沈殿させた。沈殿を遠心分離(17,900×gで10分間)によってペレット化し、上清をイソプロパノールと合わせてDNAを沈殿させた。DNAを遠心分離(17,900×gで30分間)によってペレット化し、70%エタノールで1回洗浄し、50μLの緩衝液EB(Tris−HCL、pH8.5)に再懸濁した。市販のプライマー(M13F、M13R、Invitrogen、カリフォルニア州カールスバッド)を用いたポリメラーゼ連鎖反応(PCR)を使用して、バクミド中にhGPR119挿入断片が存在することを確認した。
【0102】
hGPR119組換え型バキュロウイルスの生成
P0ウイルスストックの作製
Sf900II培地(Invitrogen、カリフォルニア州カールスバッド)で増殖させた、懸濁液に適合させたSf9細胞に、製造者のプロトコール(Cellfectin、Invitrogen、カリフォルニア州カールスバッド)に従って10μLのhGPR119バクミドDNAをトランスフェクトした。5日間インキュベートした後、条件培地(すなわち「P0」ウイルスストック)を遠心分離し、0.22μmフィルター(Steriflip、Millipore、マサチューセッツ州ビルリカ)で濾過した。
【0103】
凍結ウイルス(BIIC)ストックの作製
長期間のウイルス貯蔵および作業(すなわち「P1」)ウイルスストック生成に備えて、凍結BIIC(バキュロウイルスを感染させた昆虫細胞)ストックを以下のとおりに作製した。懸濁液に適合させたSf9細胞をSf900II培地(Invitrogen、カリフォルニア州カールスバッド)で増殖させ、hGPR119 P0ウイルスストックで感染させた。24時間増殖させた後、感染した細胞を穏やかに遠心分離し(約100×g)、凍結用培地(10%のDMSO、1%のアルブミンを含有するSf900II培地)に再懸濁して、最終密度を1×107細胞/mLとし、標準の凍結用プロトコールに従って、1mLずつの一定分量にして凍結させた。
【0104】
作業(「P1」)ウイルスストックの作製
Sf900II培地(Invitrogen、カリフォルニア州カールスバッド)で増殖させた、懸濁液に適合させたSf9細胞を、解凍したhGPR119 BIICストックの1:100希釈物で感染させ、数日間インキュベートした(振盪しながら27℃)。細胞の生存度が70%に到達したとき、条件培地を遠心分離によって回収し、ELISA(BaculoElisa Kit、Clontech、カリフォルニア州マウンテンヴュー)によってウイルス力価を決定した。
【0105】
懸濁液に適合させたHEK 293FT細胞におけるhGPR119の過剰発現
振盪フラスコにおいて、HEK293FT細胞(Invitrogen、カリフォルニア州カールスバッド)を、50μg/mLのネオマイシンおよび10mMのHEPESを補充した293Freestyle培地(Invitrogen)で増殖させた(37C、8%二酸化炭素、振盪)。細胞を穏やかに遠心分離し(約500×g、10分)、ペレットを、18%ウシ胎児血清(Sigma Aldrich)を補充したダルベッコPBS(Mg++/−Ca++なし)とP1ウイルスの混合物に再懸濁して、感染多重度(MOI)が10になり、最終細胞密度が1.3×106/mL(合計体積2.5リットル)になるようにした。細胞を5リットルのWave Bioreactor Wavebag(Wave Technologies、マサチューセッツ州)に移し、27℃で4時間インキュベートし(17振動(rock)/分、乗せ台角度7°)、インキュベート期間の終わりに、30mMの酪酸ナトリウム(Sigma Aldrich)を補充した等体積(2.5リットル)の293Freestyle培地を加え(最終濃度=15mM)、細胞を20時間増殖させた(37℃、8%CO2[0.2リットル/分}、25振動(rock)/分、乗せ台角度7°)。細胞を遠心分離(3,000×g、10分)によって収集し、DPBS(Ca++/Mg++なし)で1回洗浄し、0.25Mのスクロース、25mMのHEPES、0.5mMのEDTA、pH7.4に再懸濁し、−80℃で凍結させた。
【0106】
放射リガンド結合アッセイに向けた膜調製
凍結した細胞を氷上で解凍し、4℃で10分間700×g(1400rpm)で遠心分離した。細胞ペレットを20mLのリン酸緩衝溶液に再懸濁し、1400rpmで10分間遠心分離した。次いで細胞ペレットをホモジナイズ緩衝液(10mMのHEPES(Gibco #15630)、pH7.5、1mMのEDTA(BioSolutions、#BIO260−15)、1mMのEGTA(Sigma、#E−4378)、0.01mg/mlのベンズアミジン(Sigma #B6506)、0.01mg/mlのバシトラシン(Sigma #B0125)、0.005mg/mlのロイペプチン(Sigma #L8511)、0.005mg/mlのアプロチニン(Sigma #A1153))に再懸濁し、氷上で10分間インキュベートした。次いで細胞を、タイトフィット型ガラス製Dounceホモジナイザーで15回の穏やかなストロークで溶解させた。ホモジネートを4℃で10分間1000×g(2200rpm)で遠心分離した。上清を氷上の新たな遠心管に移した。細胞ペレットをホモジナイズ緩衝液に再懸濁し、4℃で10分間1000×g(2200rpm)で再び遠心分離し、その後上清を取り出し、ペレットをホモジナイズ緩衝液に再懸濁した。この過程を3回目も繰り返し、その後上清を合わせ、Benzonase(Novagen #71206)およびMgCl2(Fluka #63020)を加えて最終濃度をそれぞれ1U/mlおよび6mMとし、氷上で1時間インキュベートした。次いで溶液を4℃で20分間25,000×g(15000rpm)で遠心分離し、上清を廃棄し、ペレットを新鮮なホモジナイズ緩衝液(BenzonaseおよびMgCl2なし)に再懸濁した。25,000×gでの遠心分離ステップを繰り返した後、最終膜ペレットをホモジナイズ緩衝液に再懸濁し、−80℃で凍結させた。Pierce BCAタンパク質アッセイキット(Pierce試薬A #23223およびB #23224)を使用して、タンパク質濃度を決定した。
【0107】
[3H]Cmpd Aの合成および精製
【0108】
【化9】
化合物A(「Cmpd A」、4−(1−(4−(メチルスルホニル)フェニル)−3a,7a−ジヒドロ−1H−ピラゾロ[3,4−d]ピリミジン−4−イルオキシ)ピペリジン−1−カルボン酸イソプロピル、上で示したとおり)(4mg、0.009mmol)を0.5mLのジクロロメタンに溶解させ、得られる溶液を(1,5−シクロオクタジエン)(ピリジン)(トリシクロヘキシルホスフィン)−イリジウム(I)ヘキサフルオロホスフェート(J.Organometal.Chem.1979、168、183)(5mg、0.006mmol)で処理した。反応容器を密封し、溶液をトリチウムガス雰囲気中で17時間撹拌した。減圧下で反応溶媒を除去し、得られる残渣をエタノールに溶解させた。未精製の[3H]Cmpd Aの精製を、以下の条件を使用する分取HPLCによって実施した。
カラム:Atlantis、4.6×150mm、5μm
移動相A:水/アセトニトリル/ギ酸(98/2/0.1)
移動相B:アセトニトリル
勾配: 時間 %B
0.00 30.0
1.00 30.0
13.00 80.0
分析時間:16分
ポストタイム:5分
流量:1.5mL/分
注入体積:20〜50μL
注入溶媒:DMSO
検出:210nmおよび245nmでのUV
【0109】
精製した[3H]Cmpd Aの比活性は、質量分析によって、70Ci/mmolと決定した。
【0110】
GPR119放射リガンド結合アッセイ
試験化合物を100%DMSO(J.T.Baker #922401)に段階希釈した。各希釈液2μLを96ウェルプレートの適切なウェルに加えた(各濃度3通りにして)。未標識Cmpd Aを最終濃度10μMで使用して、非特異的結合を測定した。
【0111】
3H−Cmpd Aを結合緩衝液(50mMのTris−HCl、pH7.5(Sigma #T7443)、10mMのMgCl2(Fluka 63020)、1mMのEDTA(BioSolutions #BIO260−15)、0.15%のウシ血清アルブミン(Sigma #A7511)、0.01mg/mLのベンズアミジン(Sigma #B6506)、0.01mg/mLのバシトラシン(Sigma #B0125)、0.005mg/mLのロイペプチン(Sigma #L8511)、0.005mg/mLのアプロチニン(Sigma #A1153))中に希釈して濃度を60nMとし、96ウェルプレート(Nalge Nunc #267245)のすべてのウェルに100μLを加えた。
【0112】
GPR119を発現する膜を解凍し、結合緩衝液に希釈して最終濃度を20μg/各ウェル100μLとし、希釈した膜100μLを96ウェルプレートの各ウェルに加えた。
【0113】
プレートを振盪しながら室温(約25℃)で60分間インキュベートした。Packardハーベスターを使用した、0.3%ポリエチレンアミンに予浸したGF/Cフィルタープレート(Packard #6005174)での真空濾過によって、アッセイを終結させた。次いで、洗浄緩衝液(50mMのTris−HCl、pH7.5、4℃に保ったもの)を使用してフィルターを6回洗浄した。次いでフィルタープレートを室温で終夜風乾した。30μlのシンチレーション液(Ready Safe、Beckman Coulter #141349)を各ウェルに加え、プレートを密封し、Wallac Trilux MicroBetaプレートベースのシンチレーションカウンターを使用して、各フィルターに付随する放射能を測定した。
【0114】
3H−Cmpd AのKdは、一部位双曲線(Graph Pad Prism)に適合させる非線形回帰によるデータ解析を用いた飽和結合実験を実施して求めた。IC50は、専有の曲線適合プログラム(SIGHTS)および4パラメータロジスティック用量反応式を用いて分析した競合曲線から求めた。Ki値は、Cheng−Prusoff式を使用してIC50値から算出した。
【0115】
β−ラクタマーゼおよびβ−アレスチン機能アッセイについて、以下の結果が得られた。
【0116】
【表1】
【0117】
cAMPアッセイおよび結合アッセイについて、以下の結果が得られた。
【0118】
【表2】
【0119】
出発材料の調製
調製例1:3−フルオロ−4−ヒドロキシピペリジン−1−カルボン酸tert−ブチルの異性体(4および5)。実験の細目は以下のスキームAで詳述する。
【0120】
【化10】
【0121】
ステップA.4−[(トリメチルシリル)オキシ]−3,6−ジヒドロピリジン−1(2H)−カルボン酸tert−ブチル(2)
【0122】
【化11】
N−tert−ブトキシカルボニル−4−ピペリドン(30.0g、0.15mol)を無水N,N−ジメチルホルムアミド(300mL)に溶かした室温の溶液に、塩化トリメチルシリル(22.9mL、0.18mol)およびトリエチルアミン(50.4mL、0.36mol)を添加漏斗で連続的に加えた。得られる溶液を80℃で終夜加熱し、次いで室温に冷却した。反応混合物を水およびヘプタンで希釈した。層を分離し、水層をヘプタンで抽出した。ヘプタン層を合わせて水およびブラインで順次洗浄し、次いで硫酸マグネシウムで乾燥させた。混合物を濾過し、減圧下で濾液を濃縮して、粗生成物を黄色の油状物として得た。油状物を90:10のヘプタン/酢酸エチル中でシリカゲル充填物に通すことにより精製して、表題化合物を無色の油状物(33.6g、82%)として得た。1H NMR (400 MHz, 重水素化クロロホルム) δ 4.78 (br s, 1H), 3.86 (br s, 2H), 3.51
(t, 2H), 2.09 (br s, 2H), 1.45 (s, 9H), 0.18 (s, 9H).
【0123】
ステップB.3−フルオロ−4−オキソピペリジン−1−カルボン酸tert−ブチル(3)
【0124】
【化12】
4−[(トリメチルシリル)オキシ]−3,6−ジヒドロピリジン−1(2H)−カルボン酸tert−ブチル(28.8g、0.11mol)をアセトニトリル(300mL)に溶かした室温の撹拌した溶液に、Selectfluor(商標)(41.4g、0.12mol)を加えた。得られる淡黄色の懸濁液を室温で1.5時間撹拌した。飽和炭酸水素ナトリウム水溶液(300mL)および酢酸エチル(300mL)を加え、層を分離した。水層を酢酸エチルで2回抽出し、すべての有機層を合わせ、飽和炭酸水素ナトリウム水溶液およびブラインで順次洗浄し、次いで硫酸マグネシウムで乾燥させた。混合物を濾過し、減圧下で濾液を濃縮して、粗生成物を淡黄色の油状物として得た。この材料を、ヘプタン/酢酸エチル勾配(2:1〜1:1)を用いたシリカゲルでのカラムクロマトグラフィーを繰り返すことにより精製すると、表題化合物が白色の固体(15.5g、67%)として得られた。1H NMR (400 MHz, 重水素化クロロホルム): δ 4.88 (dd, 0.5 H), 4.77 (dd, 0.5H), 4.47
(br s, 1H), 4.17 (ddd, 1H), 3.25 (br s, 1H), 3.23 (ddd, 1H), 2.58 (m, 1H), 2.51
(m, 1H), 1.49 (s, 9H).
【0125】
ステップC.(R*)−3−(S)−フルオロ−4−(R)−ヒドロキシピペリジン−1−カルボン酸tert−ブチルの異性体(4および5)(ラセミ)
【0126】
【化13】
3−フルオロ−4−オキソピペリジン−1−カルボン酸tert−ブチル(15.5g、71.3mmol)をメタノール(150mL)に溶かした0℃の溶液に、水素化ホウ素ナトリウム(3.51g、93.7mmol)を加えた。得られる混合物を0℃で2時間撹拌し、次いで室温に温めた。飽和塩化アンモニウム水溶液(200mL)を加え、混合物を酢酸エチルで3回抽出した。抽出物を合わせてブラインで洗浄し、硫酸マグネシウムで乾燥させた。混合物を濾過し、減圧下で濾液を濃縮して、粗生成物混合物を得、これを、ヘプタン−酢酸エチル(3:2〜1:1)を溶離液とするシリカゲルでのカラムクロマトグラフィーによって精製して、第1の溶離生成物である(3,4−トランス)−3−フルオロ−4−ヒドロキシピペリジン−1−カルボン酸tert−ブチル(3.81g、24%)を淡黄色の油状物として得たが、油状物は静置すると凝固して白色の固体になった。1H NMR (400 MHz, 重水素化クロロホルム) δ 4.35 (ddd,
0.5 H), 4.18 (ddd, 0.5 H), 4.15 (br s, 1H), 3.89-3.74 (m, 2H), 2.97 (br s, 1H),
2.93 (ddd, 1H), 2.47 (s, 1H), 2.05-1.92 (m, 1H), 1.58-1.46 (m, 1H), 1.44 (s,
9H).
【0127】
次いで、第2の溶離化合物である(3,4−シス)−3−フルオロ−4−ヒドロキシ−ピペリジン−1−カルボン酸tert−ブチル(10.57g、68%)を白色の固体として単離した。1H NMR (400 MHz, 重水素化クロロホルム) δ 4.69 - 4.65
(m, 0.5H), 4.53-4.49 (m, 0.5H), 3.92 - 3.86 (m, 2H), 3.69 (br s, 1H), 3.39 (br
s, 1H), 3.16 (br s, 1H), 2.13 (s, 1H), 1.88 - 1.73 (m, 2H), 1.44 (s, 9H).
【0128】
ステップD.(3,4−シス)−3−フルオロ−4−ヒドロキシ−ピペリジン−1−カルボン酸tert−ブチルの鏡像異性体
ラセミ体の(3,4−シス)−3−フルオロ−4−ヒドロキシ−ピペリジン−1−カルボン酸tert−ブチルのサンプル1gを、Chiralpak AD−Hカラム(10×250mm)を利用し、それぞれ90:10の二酸化炭素とエタノールからなる移動相を10mL/分の流量で用いた分取高圧液体クロマトグラフィーによって、その鏡像異性体に精製した。分離をモニターする波長は、210nMとした。Chrialpak AD−H(4.6mm×25cm)カラムを使用し、それぞれ90:10の二酸化炭素とエタノールからなる定組成移動相を2.5mL/分の流量で用いた分析高圧クロマトグラフィーの使用によって、各鏡像異性体の純度分析値を決定した。ピークをモニターする波長は、210nmとした。以下の2種の異性体が得られた。
(3,4−シス)−3−フルオロ−4−ヒドロキシ−ピペリジン−1−カルボン酸tert−ブチル、鏡像異性体1(363mg):Rt=2.67分(100%ee)および(3,4−シス)−3−フルオロ−4−ヒドロキシ−ピペリジン−1−カルボン酸tert−ブチル、鏡像異性体2(403mg):Rt=2.99分(88%ee)。
【0129】
調製例2:−9−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸イソプロピル(シンおよびアンチ異性体の混合物)
【0130】
【化14】
【0131】
スキームBのステップA. 7−ベンジル−3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オン−塩酸塩(2)の合成:
テトラヒドロ−4H−ピラン−4−オン1(60.0g、0.60mol)、ベンジルアミン(63.4g、0.60mol)、および氷酢酸(35.9g、0.60mol)を無水メタノール(1.2L)に溶かした溶液を、パラホルムアルデヒド(39.6g、1.3mol)を無水メタノール(1.2L)に懸濁させた撹拌した懸濁液に、65℃で75分間かけて加えた。2回目の分のパラホルムアルデヒド(39.6g、1.3mol)を加え、混合物を65℃で1時間撹拌した。反応を水(1.2L)および1M水酸化カリウム水溶液(600mL)で失活させた。混合物を酢酸エチル(3L×3)で抽出した。有機層を合わせて硫酸ナトリウムで乾燥させ、濾過し、濾液を真空中で濃縮乾燥した。残渣をカラムクロマトグラフィー(石油エーテル/酢酸エチル=20:1〜2:1)によって精製して、褐色の油状物を得た。残渣を6M無水塩酸1,4ジオキサン溶液(500mL)で希釈し、混合物を30分間撹拌した。真空中で溶媒を除去し、アセトン(500mL)を加えた。得られる混合物を30分間音波処理して白色の沈殿物を生成させた。混合物を濾過し、固体をアセトンで洗浄し、次いで真空中で乾燥させて、所望の生成物を白色の固体(21g、13%)として得た。1H NMR (400 MHz, 酸化重水素) δ 7.43 - 7.42 (m, 5H), 4.66 (s, 2H), 3.95
- 3.90 (m, 4H), 3.54 - 3.47 (m, 4H); 1.96 (bs, 2H); LCMS (ES+): 232.0 (M + 1).
【0132】
スキームBのステップB. 7−ベンジル−3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オール(シンおよびアンチ異性体の混合物)(3)の合成:
7−ベンジル−3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オン塩酸塩(4.40g、16.9mmol)をエタノール(40mL)および無水テトラヒドロフラン(40mL)に懸濁させた。混合物を氷浴で冷却し、水素化ホウ素ナトリウム(1.5g、37.3mmol)を1回で加えた。混合物を4時間かけてゆっくりと室温に温めた。次いで反応液を真空中で濃縮して、エタノールおよびテトラヒドロフランの大部分を除去した。混合物をメチルtert−ブチルエーテルと1.0M水酸化ナトリウム水溶液とに分配した。溶液を30分間撹拌した後、2つの層を分離した。水層をメチルtert−ブチルエーテルで抽出した。有機抽出物を合わせ、ブラインで洗浄し、硫酸ナトリウムで乾燥させた。混合物を濾過し、真空中で濾液を濃縮して透明な油状物を得、これを静置すると部分的に凝固して油性の白色固体(3.71g、94%)になった。このシンおよびアンチの7−ベンジル−3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オール異性体の混合物を、それ以上精製せずに次のステップで使用した。LCMS (ES+): 234.1 (M+1).
【0133】
スキームBのステップC. 3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オール(シンおよびアンチ異性体の混合物)(4)の合成:
シンおよびアンチの7−ベンジル−3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オール異性体の出発混合物(3.71g、15.9mmol)をエタノール(120mL)に溶解させ、Pd(OH)2(450mg)を加えた。Parrシェーカーにおいて混合物を50psiの水素中で2.5時間振盪した。混合物をCelite(登録商標)で濾過し、収集した固体をメタノールで3回洗浄した。真空中で濾液を濃縮して油性の固体を得た。この油性の固体を酢酸エチルに溶解させ、ヘプタンを加えた。真空中で溶液を濃縮して、3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オールのシンおよびアンチ異性体混合物を白色の固体(2.08g、91%)として得た。この材料をそれ以上精製せずに次のステップで使用した。LCMS (ES+): 144.1 (M+1).
【0134】
スキームBのステップD.9−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸イソプロピル(シンおよびアンチ異性体の混合物)(5)の合成:
3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オールのシンおよびアンチ異性体混合物(2.08g、14.5mmol)およびN,Nジイソプロピルエチルアミン(2.80mL、16.0mmol)の0℃のジクロロメタン(15mL)溶液に、クロロギ酸イソプロピル(14.2mL、14.2mmol、1.0Mトルエン溶液)を滴下した。反応混合物を14時間かけて室温に温めた。次いで反応液を1M塩酸水溶液(50mL)で希釈し、水層を分離した。有機層を水(50mL)およびブライン(50mL)で順次洗浄し、硫酸ナトリウムで乾燥させた。混合物を濾過し、濾液を真空中で濃縮して無色の油状物を得た。この油状物を酢酸エチルに溶解させ、ヘプタンを加え、混合物を濃縮した。得られる油状物を真空中で乾燥させて、9−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸イソプロピルのシンおよびアンチ異性体混合物を透明な油状物(2.74g、82%)として得た。LCMS (ES+): 230.1 (M+1).
【0135】
ステップE.9−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボキン酸イソプロピルのシンおよびアンチ異性体の分離:
それぞれ65mL/分の流量の85:15の二酸化炭素およびメタノールを移動相とした、Chiralpak AD−Hカラム(21×250mm)を利用する分取高圧液体クロマトグラフィーによって、9−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸イソプロピルのシンおよびアンチ異性体混合物(5.04g、35.1mmol)を分離した。分離をモニターする波長は210nmとした。それぞれ2.5mL/分の流量の85:15の二酸化炭素およびメタノールを移動相とした、Chiralpak AD−H(4.6mm×25cm)カラムを用いた分析用高圧クロマトグラフィーを使用して、分析による各異性体の純度を決定した。ピークをモニターする波長は210nmとした。以下の2種の異性体が得られた。
9−シン−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸イソプロピル(6)(1.34g):透明な油状物であったが、静置すると凝固した。保持時間(Rt)=2.3分、1H NMR (400 MHz, 重水素化-DMSO): δ 5.12 (d, 1H, J=2.8Hz), 4.76 - 4.71
(m, 1H), 4.20 (d, 1H, J=13Hz), 4.16 (d, 1H, J=13Hz), 3.96 - 3.92 (m, 2H), 3.79
(d, 1H, J=3Hz), 3.55 (s, 1H), 3.52 (S, 1H), 3.08 (d, 1H, J=13Hz), 2.98 (d, 1H,
J=13Hz), 1.47 (m, 2H) 1.16 (d, 3H, J=3Hz), 1.15 (d, 3H, J=3Hz); LCMS (ES+):
230.2 (M+1).
9−アンチ−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸イソプロピル(7)(1.70g):琥珀色の油状物、Rt=3.08分、1H NMR (400 MHz, DMSO-d6): δ 5.11 (d, 1H, J=2.8Hz), 4.74 - 4.67 (m, 1H), 3.89 (d, 1H, J=13Hz),
3.84 - 3.78 (m, 2H, J=11Hz), 3.80 (d, 1H, J=6Hz), 3.78 (d, 1H, J=3Hz), 3.52 -
3.47 (m, 2H), 3.35 - 3.30 (m, 1H), 3.24 - 3.20 (m, 1H), 1.53 (s, 1H), 1.51 (s,
1H), 1.13 (d, 3H, J=1Hz), 1.16 (d, 3H, J=1Hz); LCMS (ES+): 230.2 (M+1)
【0136】
別法として、上記反応スキームAのステップAとBを以下で述べるように組み合わせて、7−ベンジル−3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オール(シンおよびアンチ異性体の混合物)を合成することもできる。
ベンジルアミン(21.35g、199.27mmol)、テトラヒドロ−4H−ピラン−4−オン(1)(19.95g、199.27mmol)、および酢酸(11.97g、199.27mmol)をメタノール(400mL)に溶解させた。混合物を還流加熱した。反応混合物に、ホルムアルデヒド水溶液(37%、32.34g、398.53mmol)およびメタノール(100mL)を、反応液を還流させたまま60分かけて加えた。反応液を室温に冷却した。次いで炭酸水素ナトリウム(16.74g、199.27mmol)を少量ずつ加えた。引き続いて、反応温度を25℃以下に保ちながら、水素化ホウ素ナトリウム(7.92g 209.23mmol)を少量ずつ加えた。混合物を周囲温度で30分間撹拌した。Celite(登録商標)(20g)を加えた後、水(100mL)および1N水酸化ナトリウム水溶液(100mL)を加えた。これを1時間撹拌した後、混合物を濾過し、フィルターケーキをメタノールおよび水(各20mL)で順次すすいだ。濾液を真空中で濃縮して、メタノールの大部分を除去した。得られる水性混合物を2−メチルテトラヒドロフラン(300mL)で抽出した。有機相をブライン溶液(100mL)で洗浄し、無水硫酸マグネシウムで乾燥させ、真空中で濃縮して、シンおよびアンチ−7−ベンジル−3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オール異性体の混合物を油状物として得たが、油状物は室温で静置すると凝固した(22.0g、47.3%)。
【0137】
調製例3:9−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸tert−ブチル(シンおよびアンチ異性体の混合物)
【0138】
【化15】
3−オキサ−7−アザビシクロ[3.3.1]ノナン−9−オール(シンおよびアンチ異性体の混合物、調製例2ステップCの生成物)(3.78g、26.4mmol)を水(30mL)およびテトラヒドロフラン(30mL)に溶かした0℃の溶液に、二炭酸ジ−tert−ブチル(5.76g、26.4mmol)のテトラヒドロフラン(20mL)溶液を滴下した。溶液を徐々に室温に温めながら約15時間撹拌した。反応液をジクロロメタンおよび水で希釈した。層を分離し、水層をジクロロメタンで抽出した。有機層を合わせ、硫酸ナトリウムで乾燥させた。混合物を濾過し、濾液を減圧下で濃縮すると、表題化合物が透明な油状物(6.55g)として現れ、これをそれ以上精製せずに使用した。
【0139】
調製例4:9−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸tert−ブチルのシンおよびアンチ異性体の分離
【0140】
【化16】
調製例3の9−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸tert−ブチル(5.04g、35.1mmol)のシンおよびアンチ異性体の混合物を、それぞれ85:15の二酸化炭素およびメタノールを移動相とし、流量を65mL/分としたChiralpak AD−Hカラム(21×250mm)を利用する分取高圧液体クロマトグラフィーによって分離した。分離をモニターする波長は210nmとした。各異性体の純度分析値は、それぞれ85:15の二酸化炭素およびメタノールを移動相とし、流量を2.5mL/分としたChiralpak AD−H(4.6mm×25cm)カラムを使用する分析高速クロマトグラフィーを使用して求めた。ピークをモニターする波長は210nmとした。以下の2種の異性体が得られた。
9−アンチ−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸tert−ブチル:(1.30g、100%de)、静置すると白色の固体に凝固した透明な油状物、保持時間(Rt)=3.15分。1H NMR (400 MHz, 重水素化クロロホルム) δ 1.44 (s, 9 H), 1.66 (d, J=16.79 Hz, 2 H), 1.84 (d, J=2.93 Hz, 1 H),
3.30 - 3.52 (m, 2 H), 3.64 (t, J=11.03 Hz, 2 H), 3.93 - 4.21 (m, 5 H).
9−シン−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸tert−ブチル:(1.64g、89%de)、静置すると白色の固体に凝固した透明な油状物、Rt=3.55分。1H NMR (400 MHz, 重水素化クロロホルム) δ 1.47 (s, 9 H), 1.64 (d, J=13.47 Hz, 2 H), 2.12 (d, J=3.32 Hz, 1 H),
2.92 - 3.22 (m, 2 H), 3.71 - 3.83 (m, 2 H), 3.99 (d, J=3.32 Hz, 1 H), 4.09 -
4.19 (m, 2 H), 4.32 (d, J=13.66 Hz, 1 H), 4.48 (d, J=13.66 Hz, 1 H).
【0141】
調製例5:4−[(6−クロロ−ピリミジン−4−イル)オキシ]ピペリジン−1−カルボン酸イソプロピル
【0142】
【化17】
4−ヒドロキシピペリジン−1−カルボン酸イソプロピル(553mg、2.95mmol)を無水テトラヒドロフラン(20mL)に溶かした溶液に、カリウムtert−ブトキシド(0.450mL、4.00mmol)を0℃で加えた。反応混合物を65℃で10分間撹拌した。上記混合物に4,6−ジクロロピリミジン(0.400g、2.68mmol)を加えた。次いで、得られる溶液を65℃で1時間撹拌した。混合物を周囲温度に冷却し、水(100mL)で失活させ、酢酸エチル(100mL×3)で抽出した。有機層を合わせてブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧下で濃縮した。残渣を、シリカゲルでのカラムクロマトグラフィー(石油エーテル:酢酸エチル=20:1)によって精製して、生成物を白色の固体(350mg、44%)として得た。
【0143】
調製例6:4−[(6−クロロ−5−メチルピリミジン−4−イル)オキシ]ピペリジン−1−カルボン酸イソプロピル
【0144】
【化18】
4−ヒドロキシピペリジン−1−カルボン酸イソプロピル(482mg、2.68mmol)の無水テトラヒドロフラン(15mL)溶液に、0℃でカリウムtert−ブトキシド(0.41g、3.6mmol)を加えた。反応混合物を65℃で10分間撹拌した。上記混合物に4,6−ジクロロ−5−メチルピリミジン(0.40g、2.4mmol)を加えた。次いで、得られる溶液を65℃で1時間撹拌した。混合物を周囲温度に冷却し、水(100mL)で失活させ、酢酸エチル(100mL×3)で抽出した。有機抽出物を合わせてブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過し、減圧下で濾液を濃縮した。残渣をシリカゲルでのカラムクロマトグラフィー(石油エーテル:酢酸エチル=20:1)によって精製して、生成物を白色の固体(680mg、80%)として得た。
【0145】
調製例7:4−クロロ−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−5−カルボニトリル
【0146】
【化19】
4,6−ジクロロピリミジン−5−カルボニトリル(174mg、1.00mmol)および1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール(Heterocycles 2002 56 257〜264およびUS2007232676)(123mg、1.00mmol)を無水ジクロロメタン(5mL)に溶かした溶液に、室温でN,N−ジイソプロピルエチルアミン(0.50mL、3.5mmol)を加えた。反応混合物を室温で2時間撹拌した。水(50mL)を加え、得られる混合物をジクロロメタン(50mL×3)で抽出した。有機層を合わせてブライン(100mL)で洗浄し、硫酸ナトリウムで乾燥させ、濾過し、減圧下で濾液を濃縮した。残渣を分取薄層クロマトグラフィーによって精製して、生成物を白色の固体(150mg、58%)として得た。
【0147】
調製例8:4−[(6−クロロ−5−メチルピリミジン−4−イル)オキシ]ピペリジン−1−カルボン酸tert−ブチル
【0148】
【化20】
20mLのBiotage(商標)マイクロ波対応管を窒素パージし、4,6−ジクロロ−5−メチルピリミジン(0.600g、2.98mmol)および4−ヒドロキシピペリジン−1−カルボン酸tert−ブチル(534mg、3.28mmol)を投入した。1,4−ジオキサン(14.9mL)を加え、混合物を100℃に加熱した。混合物にナトリウムビス(トリメチルシリル)アミド(3.58mL、3.58mmol、1.0Mテトラヒドロフラン溶液)を10分かけて滴下した。混合物を60分間撹拌し、次いで室温で12時間撹拌した。水で反応を失活させ、水層を酢酸エチルで抽出した(3回)。有機抽出物を合わせて硫酸ナトリウムで乾燥させ、濾過し、濾液を真空中で濃縮した。未精製材料をシリカゲルクロマトグラフィー(40gのSiO2カラム、0〜50%の酢酸エチルヘプタン溶液の勾配)によって精製して、表題化合物(842mg、86%)を得た。
【0149】
調製例9:5−(6−クロロ−5−メチルピリミジン−4−イル)−1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール
【0150】
【化21】
1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール二塩酸塩(2.00g、10.2mmol)および4,6−ジクロロ−5−メチルピリミジン(1.66g、10.2mmol)を室温でテトラヒドロフラン(51mL)に懸濁させた。これにトリエチルアミン(4.41mL、31.6mmol)を加えると、混合物が濁り、フラスコの壁面に褐色の固体が固着した。この混合物を室温で4時間撹拌し、次いでさらに19時間50℃に加熱した。反応混合物を室温に冷却し、水(100mL)で希釈した。この混合物を酢酸エチル(3×100mL)で抽出した。有機抽出物をプールし、ブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過した。真空中で濾液を乾燥状態まで減少させて、表題化合物を淡褐色の固体(1.95g、78%)として得、これをそれ以上精製せずに次のステップで使用した。
1H NMR
(500 MHz, 重水素化クロロホルム) δ 2.54
(s, 3 H) 3.88 (s, 3 H) 4.90 (見かけd, J=3.66 Hz, 4 H) 7.28
(s, 1 H) 8.29 (s, 1 H).
【実施例】
【0151】
(実施例1)
4−{[6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸イソプロピル
【0152】
【化22】
調製例5の4−[(6−クロロピリミジン−4−イル)オキシ]ピペリジン−1−カルボン酸イソプロピル(0.200g、0.667mmol)のN−メチルピロリジノン(5mL)溶液に、周囲温度で、1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール(82mg、0.67mmol)、次いで炭酸セシウム(1.08g、3.33mmol)を加えた。反応混合物を3時間150℃に加熱した。反応混合物を周囲温度に冷却した。水(50mL)を加え、次いで、得られる混合物をジクロロメタン(100mL、3回)で抽出した。有機層を合わせてブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過し、次いで濃縮して残渣を得、これを、63%のアセトニトリル(改質剤として0.05%の水酸化アンモニウム)水(改質剤として0.05%の水酸化アンモニウム)溶液からなる移動相を溶離液とするXBridge C18カラム150×30mmでの分取逆相HPLCによって精製して、生成物を白色の固体(35mg、14%)として得た。1H NMR (400 MHz, 重水素化クロロホルム): δ 8.16 (s, 1H), 7.14 (s, 1H), 5.51 (s,
1H), 5.09-5.13 (m, 1H), 4.74-4.80 (m, 1H), 4.48-4.60 (s, 2H), 4.19-4.48 (s,
2H), 3.70 (s, 3H), 3.63-3.66 (m, 2H), 3.13-3.19 (m, 2H), 1.80-1.83 (m, 2H),
1.55-1.57 (m, 2H), 1.09 (d, J=6.4 Hz, 6H); LCMS (ES+): 387.3 (M+H).
【0153】
(実施例2)
4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸イソプロピル
【0154】
【化23】
調製例6の4−[(6−クロロ−5−メチルピリミジン−4−イル)オキシ]ピペリジン−1−カルボン酸イソプロピル(0.020g、0.056mmol)のN−メチルピロリジノン(0.56mL)溶液に、1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール(0.010g、0.056mmol)に続いて炭酸セシウム(91mg、0.28mmol)を加えた。混合物を3時間150℃に加熱した。反応液を水で希釈し、水層をジクロロメタンで3回抽出した。有機抽出物を合わせて硫酸ナトリウムで乾燥させ、濾過し、濾液を真空中で濃縮した。未精製材料を、水アセトニトリル溶液(0.03%の水酸化アンモニウム改質剤)の勾配で溶離するWaters XBridge C18 19×100mm、0.005mmカラムでの分取HPLCによって精製して、生成物(8.3mg、13%)を得た。分析LCMS:保持時間0.97分(Atlantis C18 4.6×50mm、5μmカラム、1.8分かけて95%の水/アセトニトリルから5%の水/アセトニトリルへの線形勾配、5%の水/アセトニトリルで2.0分まで保持、0.05%のトリフルオロ酢酸改質剤、流量1.3mL/分)、LCMS (ES+): 401.5 (M+H).
【0155】
(実施例3)
4−{[5−シアノ−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸イソプロピル
【0156】
【化24】
4−ヒドロキシピペリジン−1−カルボン酸イソプロピル(77mg、0.63mmol)の無水テトラヒドロフラン(4mL)溶液に、周囲温度でナトリウムビス(トリメチルシリル)アミド(1.0M無水テトラヒドロフラン溶液、0.63mL、0.63mmol)を加えた。混合物を周囲温度で2時間撹拌した。上記混合物に、室温で、調製例7の4−クロロ−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−5−カルボニトリル(65mg、0.25mmol)の無水テトラヒドロフラン(2mL)溶液を加えた。得られる混合物を70℃で1時間撹拌した。反応混合物を飽和塩化アンモニウム水溶液(50mL)で失活させ、酢酸エチル(100mL、3回)で抽出した。有機抽出物を合わせてブライン(100mL)で洗浄し、硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧下で濃縮した。残渣を、66%のアセトニトリル(改質剤としての0.05%の水酸化アンモニウム)水(改質剤としての0.05%の水酸化アンモニウム)溶液からなる移動相で溶離するXBridge C18カラム150×30mmでの分取逆相HPLCによって精製して、生成物を白色の固体(25mg、24%)として得た。1H NMR (400 MHz, 重水素化クロロホルム): δ 8.23 (s, 1H), 7.24 (s, 1H), 5.33-5.36
(m, 1H), 4.83-4.89 (m, 5H), 3.80 (s, 3H), 3.64-3.70 (m, 2H), 3.38-3.41 (m, 2H),
1.80-1.91 (m, 2H), 1.75-1.79 (m, 2H), 1.19-1.23 (d, J=6.4 Hz, 6H): LCMS (ES+):
434.4 (M+Na).
【0157】
(実施例4)
4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチル
【0158】
【化25】
調製例8の4−[(6−クロロ−5−メチルピリミジン−4−イル)オキシ]ピペリジン−1−カルボン酸tert−ブチル(0.400g、1.22mmol)を含有するN−メチルピロリジノン(4.07mL)に、1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール(287mg、1.46mmol)に続いて炭酸セシウム(1.99g、6.10mmol)を加えた。混合物を1時間150℃に加熱した。反応を水で失活させ、水層を酢酸エチルで3回抽出した。有機層を合わせて硫酸ナトリウムで乾燥させ、濾過し、真空中で濃縮した。未精製材料をシリカゲルクロマトグラフィー(0〜100%の酢酸エチルヘプタン溶液の勾配)によって精製して、所望の生成物(77mg、15%)を得た。
【0159】
(実施例5)
4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピル
【0160】
【化26】
4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチル(実施例4)(77mg、0.19mmol)に、ジクロロメタン(1.5mL)に続いてトリフルオロ酢酸(1.50mL)を加えた。混合物を周囲温度で12時間撹拌した。反応混合物を真空中で濃縮し、残りのトリフルオロ酢酸を減圧下でトルエン共沸混合物によって除去した。
【0161】
未精製の1−メチル−5−[5−メチル−6−(ピペリジン−4−イルオキシ)ピリミジン−4−イル]−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾールを含有するジクロロメタン(1.8mL)に、炭酸1−メチルシクロプロピル4−ニトロフェニル(WO09105717)(87mg、0.37mmol、約10%の炭酸1−イソプロピル4−ニトロフェニルが混入したもの)に続いてトリエチルアミン(0.256mL、1.84mmol)を加えた。混合物が濃い黄色になった。反応液を室温で12時間撹拌した。未精製材料をシリカゲルクロマトグラフィー(0〜100%の酢酸エチルヘプタン溶液の勾配)によって精製して、約10%の4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸イソプロピルが混入している所望の生成物(76mg、33%)を得た。1H NMR (400 MHz, 重水素化クロロホルム): δ 8.17 (s, 1H), 7.23 (s, 1H), 5.30-5.22
(m, 1H), 4.86-4.83 (m, 2H), 4.82-4.79 (m, 2H), 3.83 (s, 3H), 3.77-3.55 (m, 2H),
3.45-3.28 (m, 2H), 2.25 (s, 3H), 2.00-1.85 (m, 2H), 1.79-1.65 (m, 2H), 1.54 (s,
3H), 0.88-0.82 (m, 2H), 0.63-0.58 (m, 2H); LCMS (ES+): 413.5 (M+H).
【0162】
(実施例6)
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチル(ラセミ体)
【0163】
【化27】
(3,4−シス)−3−フルオロ−4−ヒドロキシピペリジン−1−カルボン酸tert−ブチル(1.67g、7.62mmol)と調製例9の5−(6−クロロ−5−メチルピリミジン−4−イル)−1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール(900mg、3.60mmol)の混合物を1,4−ジオキサン(20mL)に溶解させ、105℃に加熱した。10分間加熱した後、すべての材料が溶液になっており、混合物にナトリウムビス(トリメチルシリル)アミド(4.3mL、4.3mmol、1Mトルエン溶液)を速やかに加えると、濁った黄色の混合物となり、次いでこれを105℃で2時間撹拌した。次いで反応液を室温に冷却し、水と飽和炭酸水素ナトリウム水溶液の等体積混合物を加えて失活させた。混合物を酢酸エチル(3×15mL)で抽出した。有機抽出物を合わせてブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過した。濾液を真空中で濃縮して黄色の残渣を得、これを、60〜100%の酢酸エチルヘプタン溶液を溶離液とするシリカゲルでのカラムクロマトグラフィーによって精製した。表題化合物と出発5−(6−クロロ−5−メチルピリミジン−4−イル)−1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾールの混合物を白色の固体(1.20g)として単離し、それ以上精製せずに後続の反応で使用した。
【0164】
同じ条件下で進めた別個の反応の1回分の未精製(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチルをHPLCによって精製した。未精製サンプル(9.5mg)をジメチルスルホキシド(1mL)に溶解させ、80%の水/アセトニトリル(0.03%の水酸化アンモニウム改質剤)から8.5分で0%の水/アセトニトリルへの線形勾配、続いて0%の水/アセトニトリルで1.5分間、流量:25mL/分で溶離するWaters XBridge C18 19×100mm、0.005mmカラムでの分取逆相HPLCによって精製した。そうして表題化合物(5mg)が得られた。分析LCMS:保持時間2.81分(Waters XBridge C18 4.6×50mm、0.005mmカラム、4.0分かけて90%の水/アセトニトリルから5%の水/アセトニトリルへの線形勾配、続いて5%の水/アセトニトリルで1分間、0.03%の水酸化アンモニウム改質剤、流量:2.0mL/分)、LCMS (ES+) 433.2 (M+1).
【0165】
(実施例7)
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピル(ラセミ体)
【0166】
【化28】
実施例6の未精製(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチル(1.20g)をジクロロメタン(12mL)に溶解させ、この溶液にトリフルオロ酢酸(5mL)を加えた。反応液を室温で1時間撹拌した。真空中で溶媒を除去し、残渣を水(50mL)および1N塩酸水溶液(10mL)に溶解させた。混合物をジクロロメタン(10×30mL)で抽出した。次いで、1N水酸化ナトリウム水溶液(20mL)を加えて水層をpH12とし、ジクロロメタン(40mL)で3回抽出した。有機抽出物を合わせてブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過した。濾液を減圧下で濃縮して、5−(6−{[(3,4−シス)−3−フルオロピペリジン−4−イル]オキシ}−5−メチルピリミジン−4−イル)−1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール(0.72g、2ステップで60%)を白色の固体として得、これをそれ以上精製せずに使用した。
1H NMR
(500 MHz, 重水素化クロロホルム) δ 1.84 -
2.08 (m, 2 H) 2.33 (s, 3 H) 2.69 - 2.84 (m, 1 H) 2.83 - 3.01 (m, 1 H) 3.16 (d,
J=13.66 Hz, 1 H) 3.27 - 3.44 (m, 1 H) 3.86 (s, 3 H) 4.78-4.91 (m, 1 H) 4.86 (d,
J=1.95 Hz, 2 H) 4.88 (d, J=1.95 Hz, 2 H) 5.21 - 5.32 (m, 1 H) 7.26 (s, 1 H)
8.18 (s, 1 H); LCMS (ES+) 333.4 (M+1).
【0167】
5−(6−{[(3,4−シス)−3−フルオロピペリジン−4−イル]オキシ}−5−メチルピリミジン−4−イル)−1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール(717mg、2.16mmol)および炭酸1−メチルシクロプロピル4−ニトロフェニル(NMR積分によると約10%の炭酸イソプロピル4−ニトロフェニルが混入したもの)(620mg、2.59mmol)をジクロロメタン(11mL)に溶かした溶液に、トリエチルアミン(0.60mL、4.31mmol)を加え、反応混合物を室温で15時間撹拌した。次いで反応混合物をさらに4時間還流加熱し、室温に冷却し、1N水酸化ナトリウム水溶液(30mL)で希釈し、ジクロロメタン(30mL)で3回抽出した。有機抽出物を合わせて、1N水酸化ナトリウム水溶液/ブライン溶液(10mL)の2:1混合物で2回洗浄し、硫酸ナトリウムで乾燥させ、濾過した。真空中で濾液を減圧乾固させ、黄色の泡沫を得た。70〜100%の酢酸エチルヘプタン溶液を溶離液としながらシリカゲルで精製すると、表題化合物が白色の固体(0.84g、90%、純度91%)として得られた。サンプルは、0.91ppmでのシクロプロピルメチレンおよび1.28ppmでのイソプロピルメチルシグナルのNMR積分によって求めたところ、それぞれ10:1の比で、(3,4−トランス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸イソプロピルが混入していた。
1H NMR
(500 MHz, 重水素化クロロホルム) δ 0.62 -
0.67 (m, 2 H) 0.87 - 0.94 (m, 2 H) 1.57 (s, 3 H) 1.88 (br. s., 1 H) 2.10 (br.
s., 1 H) 2.32 (s, 3 H) 3.04 - 3.23 (m, 1 H) 3.23 - 3.49 (m, 1 H) 3.86 (s, 3 H)
3.99 - 4.34 (m, 2 H) 4.66 - 5.04 (m, 5 H) 5.32 (m, 1 H) 7.26 (s, 1 H) 8.17 (s,
1H); LCMS (ES+) 431.4 (M+1).
【0168】
また、in vitroでの生物学的な特徴付けのために、同じ条件下で進めた別個の反応の1回分の未精製(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピルをHPLCによって精製した。未精製材料(52mg)をジメチルスルホキシド(1mL)に溶解させ、80%の水/アセトニトリル(0.03%の水酸化アンモニウム改質剤)から8.5分で0%の水/アセトニトリルへの線形勾配、流量:25mL/分で溶離するWaters XBridge C18 19×100mm、0.005mmカラムでの分取逆相HPLCによって精製した。分析LCMS:保持時間2.59分(Waters XBridge C18 4.6×50mm、0.005mmカラム、4.0分かけて90%の水/アセトニトリルから5%の水/アセトニトリルへの線形勾配、0.03%の水酸化アンモニウム改質剤、流量:2.0mL/分)、LCMS (ES+) 431.2 (M+1)。そうして表題化合物が得られた(22mg、55%)。このサンプルの純度は、使用した炭酸シクロプロピルメチルが不純であったために、90%であると推定された。
【0169】
(実施例8)
(3,4−トランス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチル(ラセミ体)
【0170】
【化29】
セプタムでふたをしたバイアルにおいて、調製例1の(3,4−トランス)−3−フルオロ−4−ヒドロキシピペリジン−1−カルボン酸tert−ブチル(66mg、0.30mmol)と調製例9の5−(6−クロロ−5−メチルピリミジン−4−イル)−1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール(50mg、0.20mmol)の混合物を1,4−ジオキサン(1.5mL)に溶解させ、105℃に加熱した。5分後、溶液に、ナトリウムビス(トリメチルシリル)アミド(0.32mL、0.32mmol、1Mトルエン溶液)を速やかに加え、琥珀色から濃緑色へと色を変化させた。105℃で10分間加熱した後、反応混合物が濁った褐色の混合物になり、加熱をさらに2時間続けた。反応混合物を室温に冷却し、水と飽和炭酸水素ナトリウム水溶液の等体積混合物で失活させ、混合物を酢酸エチル(10mL)で3回抽出した。有機抽出物をプールし、ブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過した。濾液を真空中で濃縮して残渣を得、これをジメチルスルホキシド(1mL)に溶解させ、80%の水/アセトニトリル(0.03%の水酸化アンモニウム改質剤)から8.5分で0%の水/アセトニトリルへの線形勾配、流量:25mL/分で溶離するWaters XBridge C18 19×100mm、0.005mmカラムでの分取逆相HPLCによって精製して、表題化合物(9.2mg、11%)を得た。分析LCMS:保持時間3.21分(Waters Atlantis C18 4.6×50mm、0.005mmカラム、4.0分かけて90%の水/アセトニトリルから5%の水/アセトニトリルへの線形勾配、0.05%のトリフルオロ酢酸改質剤、流量:2.0mL/分)、LCMS (ES+) 433.2 (M+1).
【0171】
(実施例9)
(9−アンチ)−9−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸tert−ブチル
【0172】
【化30】
セプタムでふたをしたバイアルにおいて、(9−アンチ)−9−ヒドロキシ−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸tert−ブチル(73mg、0.30mmol)と調製例9の5−(6−クロロ−5−メチルピリミジン−4−イル)−1−メチル−1,4,5,6−テトラヒドロピロロ[3,4−c]ピラゾール(50mg、0.20mmol)の混合物を1,4−ジオキサン(1.5mL)に溶解させ、105℃で5分間加熱した。混合物に、ナトリウムビス(トリメチルシリル)アミド(0.32ml、0.32mmol、1Mトルエン溶液)を速やかに加え、琥珀色の溶液を濃緑色に変色させた。105℃で10分間加熱した後、反応混合物が濁った褐色の混合物になり、加熱をさらに2時間続けた。反応液を室温に冷却し、水と飽和炭酸水素ナトリウム水溶液の等体積混合物を加えて失活させ、酢酸エチル(15mL)で3回抽出した。有機抽出物をプールし、ブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過した。濾液を真空中で濃縮乾燥して、橙褐色の泡沫を得た。この材料のサンプルをジメチルスルホキシド(1mL)に溶解させ、80%の水/アセトニトリル(0.03%の水酸化アンモニウム改質剤)から8.5分で0%の水/アセトニトリルへの線形勾配、流量:25mL/分で溶離するWaters XBridge C18 19×100mm、0.005mmカラムでの分取逆相HPLCによって精製して、表題化合物(10.2mg、11%)を得た。分析LCMS:保持時間2.53分(Waters XBridge C18 4.6×50mm、0.005mmカラム、4.0かけて90%の水/アセトニトリルから5%の水/アセトニトリルへの線形勾配、0.03%の水酸化アンモニウム改質剤、流量:2.0mL/分)、LCMS (ES+) 457.2 (M+1)。材料の残りを、50〜100%の酢酸エチルを溶離液とするシリカゲルクロマトグラフィーによって精製して、表題化合物を淡黄色の固体(33mg、36%)として得た。1H NMR (500 MHz, 重水素化クロロホルム) δ 1.49 (s, 9 H) 1.92 - 2.12 (m, 2 H) 2.35
(s, 3 H) 3.37 (d, J=13.42 Hz, 1 H) 3.47 (d, J=13.42 Hz, 1 H) 3.74 - 3.96 (m, 5
H) 4.07 - 4.23 (m, 3 H) 4.29 (d, J=13.42 Hz, 1 H) 4.70 - 5.02 (m, 4 H) 5.37 (t,
J=3.42 Hz, 1 H) 7.27 (s, 1 H) 8.19 (s, 1 H); LCMS (ES+) 457.5 (M+1).
【0173】
(実施例10)
(9−アンチ)−9−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸1−メチルシクロプロピル
【0174】
【化31】
(9−アンチ)−9−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸tert−ブチル(実施例9)(32mg、0.07mmol)をジクロロメタン(1mL)に溶解させ、トリフルオロ酢酸(0.2mL)で処理し、室温で2時間撹拌した。得られる反応混合物を真空中で濃縮すると、黄色の残渣が残った。残渣をジクロロメタン(1mL)に溶解させ、トリエチルアミン(0.1mL)で処理した後、1H NMR積分によって求めたところ約10%の炭酸イソプロピル4−ニトロフェニルが混入している炭酸1−メチルシクロプロピル4−ニトロフェニル(20mg、0.08mmol)を加え、反応液を室温で24時間撹拌した。反応混合物をジクロロメタン(5mL)で希釈し、溶液に1N水酸化ナトリウム水溶液(10mL)を加えた。ジクロロメタン層を除去し、水層をジクロロメタン(10mL)で2回抽出した。有機抽出物をプールし、1N水酸化ナトリウム水溶液と飽和ブライン(20mL)の1:1溶液で洗浄し、硫酸ナトリウムで乾燥させ、濾過した。濾液を真空中で濃縮乾燥して淡黄色の泡沫を得、これを、酢酸エチルを溶離液とするシリカゲルクロマトグラフィーによって精製した。そうして、表題化合物と(9−アンチ)−9−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸イソプロピルのそれぞれ13:1の比(0.79ppmでのシクロプロピルメチレンおよび1.18ppmでのイソプロピルメチルシグナルの1H NMR積分によって求めたもの)の混合物が、白色の固体(27.4mg、86%、純度93%)として得られた。1H NMR (500 MHz, 重水素化ジメチルスルホキシド) δ 0.53 - 0.63 (m, 2 H) 0.75 - 0.83 (m, 2
H) 1.46 (s, 3 H) 1.93 (d, 2 H) 2.32 (s, 3 H) 3.23 (d, J=13.17 Hz, 1 H) 3.33 (m,
1 H) 3.64 - 3.75 (m, 2 H) 3.79 (s, 3 H) 3.87 - 4.02 (m, 3 H) 4.12 (d, J=13.17
Hz, 1 H) 4.79 (s, 2 H) 4.90 (s, 2 H) 5.29 (t, J=3.29 Hz, 1 H) 7.24 (s, 1 H)
8.15 (s, 1 H); LCMS (ES+) 455.4 (M+1).
【0175】
(実施例11および12)
キラルな(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピルの鏡像異性体(個々の鏡像異性体の絶対立体化学は不明)
【0176】
【化32】
実施例7にあるように調製したラセミ体の(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピルの約700mgのサンプルを、Chiralpak AD−Hカラム(21×250mm)を利用し、それぞれ70:30の二酸化炭素とメタノールからなる移動相を65mL/分の流量で用いたキラル分取高圧液体クロマトグラフィーによって、その鏡像異性体に精製した。分離をモニターする波長は、210nmとした。Chrialpak AD−H(4.6mm×25cm)カラムを使用し、それぞれ70:30の二酸化炭素とメタノールからなる移動相を2.5mL/分の流量で用いた分析高圧クロマトグラフィーの使用によって、各鏡像異性体の純度分析値を決定した。ピークをモニターする波長は、210nmとした。以下の2種の異性体が得られた。
【0177】
(実施例11)
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピル(鏡像異性体2、絶対立体化学不明)(266mg):Rt=6.54分(100%)は、丸底フラスコにおいて60%の酢酸エチル/ヘプタン混合物(10mL)中に入れ、室温で20時間スラリー化した。混合物を濾過し、固体を60%の酢酸エチル/ヘプタン混合物(3mL)で2回すすぎ、窒素流中で乾燥させた。固体を真空中でさらに乾燥させると、完全に結晶質の白色の固体(199mg)が得られた。LCMS (ES+) 431.3 (M+1)
【0178】
(実施例12)
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピル(鏡像異性体1、絶対立体化学不明)(305mg):Rt=5.63分(100%ee、対応するカルバミン酸イソプロピルを約8%含有する)。この材料を、Chiralcel OD−Hカラム(21×250mm)を利用し、それぞれ75:25の二酸化炭素とメタノールからなる移動相を65mL/分の流量で用いたキラル分取高圧液体クロマトグラフィーで精製し直して、対応するカルバミン酸イソプロピル不純物を除去した。分離をモニターする波長は、210nmとした。Rt=6.2分(100%ee)。
【0179】
(実施例13)
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−イソプロピル(ラセミ体)
【0180】
【化33】
表題化合物は、クロロギ酸イソプロピルを使用したことを除き、実施例7に記載のとおりに調製した。1H NMR (500 MHz, 重水素化クロロホルム) δ 1.27 (d, J=6.34 Hz, 6 H) 1.83 - 1.95 (m,
1 H) 2.07 - 2.18 (m, 1 H) 2.32 (s, 3 H) 3.20 (br. s., 1 H) 3.29 - 3.50 (m, 1 H)
3.86 (s, 3 H) 3.90 - 4.11 (m, 1 H) 4.24 (br. s., 1 H) 4.79 - 4.93 (m, 1 H)4.84
- 4.90 (ほぼd, 4 H) 4.93 - 5.01 (m, 1 H) 5.28 - 5.43 (m,
1 H) 7.27 (s, 1 H) 8.18 (s, 1 H); LCMS (ES+) 419.4 (M+1).
【0181】
本出願では終始、様々な刊行物を参照文献として引用している。それらの刊行物の開示は、その全体が、あらゆる目的で参照により本出願に援用される。
【0182】
本発明の範囲および精神から逸脱することなく、本発明に様々な変更および変化を添えてよいことは、当業者に明白となろう。本発明の他の実施形態は、本明細書を検討し、本明細書で開示する本発明を実践する中で、当業者に明らかとなろう。本明細書および実施例は、例示的なものにすぎないとみなし、本発明の真の範囲および精神は、以下の特許請求の範囲によって示すものとする。
【特許請求の範囲】
【請求項1】
式Iを有する化合物
【化1】
[式中、
Xは、AまたはB
【化2】
であり、
Yは、Oまたは結合であり、
R1は、−C(O)−O−R3または
【化3】
であり、
R2は、水素、シアノ、C1〜C6アルキル、またはC3〜C6シクロアルキルであり、
R3は、C1〜C6アルキル、C3〜C6シクロアルキル、またはC1〜C6アルキル、C1〜C6アルコキシ、C1〜C6フルオロアルキル、ハロ、もしくはヒドロキシで置換されているC3〜C6シクロアルキルであり、但し、ハロ、C1〜C6アルコキシ、またはヒドロキシ基は、R1中のOに連結している炭素原子では結合しておらず、
R4は、C1〜C6ハロアルキル、C1〜C6アルキル、ハロ、シアノ、またはC3〜C6シクロアルキルであり、
R5は、水素、シアノ、ニトロ、C1〜C6フルオロアルキル、C1〜C6アルキル、C1〜C6アルコキシ、C1〜C6フルオロアルコキシ、またはC3〜C6シクロアルキルであり、
R6は、水素、C1〜C6アルキル、C1〜C6フルオロアルキル、C3〜C6シクロアルキル、またはC3〜C6シクロアルキル、C1〜C6アルコキシ、もしくはヒドロキシルで置換されているC1〜C6アルキルであり、但し、C1〜C6アルコキシ基またはヒドロキシル基は、ピラゾール窒素に連結している炭素には結合しておらず、
R7aおよびR7bは、それぞれ独立に、水素、フルオロ、またはC1〜C6アルキルであり、
R8a、R8b、R8cおよびR8dは、それぞれ独立に、水素、C1〜C6アルキル、C3〜C6シクロアルキル、またはヒドロキシもしくはC1〜C6アルコキシで置換されているC1〜C6アルキルであり、
またはR8aおよびR8bは、これらが結合している炭素と一緒になって、C3〜C6シクロアルキルを形成していてもよく、
またはR8cおよびR8dは、これらが結合している炭素と一緒になって、C3〜C6シクロアルキルを形成していてもよく、
またはR8aとR8cは一緒になって、完全飽和二炭素橋を形成していてもよく、但し、R8aおよびR8cは、これらが結合している環系の同じ平面上にある]
または薬学的に許容できるその塩。
【請求項2】
XがAであり、R1が−C(O)−O−R3である、請求項1に記載の化合物。
【請求項3】
R8a、R8b、R8cおよびR8dが、それぞれ水素であり、R3が、C1〜C3アルキルで置換されているC3〜C6シクロアルキルである、請求項1または2に記載の化合物。
【請求項4】
R7aおよびR7bが、それぞれ独立に、水素、フルオロ、またはC1〜C3アルキルである、請求項1、2または3に記載の化合物。
【請求項5】
R2が水素であり、R5がC1〜C6アルキルである、請求項1から4のいずれかに記載の化合物。
【請求項6】
以下の化合物、すなわち、
4−{[6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸イソプロピル;
4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸イソプロピル;
4−{[5−シアノ−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸イソプロピル;
4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチル;
4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピル;
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチル;
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピル;
(3,4−トランス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチル;
(9−アンチ)−9−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸tert−ブチル;
(9−アンチ)−9−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸1−メチルシクロプロピル;
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピルの鏡像異性体1;
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピルの鏡像異性体2;
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−イソプロピル;
または薬学的に許容できるこれらの塩。
【請求項7】
以下の化合物、すなわち、
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチル;
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピル;
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピルの鏡像異性体1;
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピルの鏡像異性体2;
または薬学的に許容できるこれらの塩。
【請求項8】
少なくとも1種の薬学的に許容できる賦形剤と混和された、治療有効量で存在する請求項1から7のいずれかに記載の化合物を含む医薬組成物。
【請求項9】
抗肥満薬および抗糖尿病薬からなる群から選択される少なくとも1種の追加の薬剤をさらに含む、請求項8に記載の組成物。
【請求項10】
前記抗肥満薬が、ジルロタピド、ミトラタピド、イミプリタピド、R56918(CAS番号403987)、CAS番号913541−47−6、ロルカセリン、セチリスタット、PYY3−36、ナルトレキソン、オレオイル−エストロン、オビネピチド、プラムリンチド、テソフェンシン、レプチン、リラグルチド、ブロモクリプチン、オルリスタット、エクセナチド、AOD−9604(CAS番号221231−10−3)、およびシブトラミンからなる群から選択される、請求項9に記載の組成物。
【請求項11】
前記抗糖尿病薬が、メトホルミン、アセトヘキサミド、クロルプロパミド、ジアビネース、グリベンクラミド、グリピジド、グリブリド、グリメピリド、グリクラジド、グリペンチド、グリキドン、グリソラミド、トラザミド、トルブタミド、テンダミスタット、トレスタチン、アカルボース、アジポシン、カミグリボース、エミグリテート、ミグリトール、ボグリボース、プラジミシンQ、サルボスタチン、バラグリタゾン、シグリタゾン、ダルグリタゾン、エングリタゾン、イサグリタゾン、ピオグリタゾン、ロシグリタゾン、トログリタゾン、エキセンディン3、エキセンディン4、トロダスケミン、レセルバトロール、ヒルチオサール抽出物、シタグリプチン、ビルダグリプチン、アログリプチン、およびサクサグリプチンからなる群から選択される、請求項9に記載の組成物。
【請求項12】
治療有効量の請求項1から7のいずれかに記載の化合物の、その必要がある患者への投与を含む、糖尿病の治療方法。
【請求項13】
治療有効量の請求項1から7のいずれか一項に記載の化合物を患者に投与するステップを含む、代謝性または代謝関連の疾患、状態、または障害の治療方法。
【請求項14】
高脂血症、I型糖尿病、II型糖尿病、特発性I型糖尿病(Ib型)、成人潜在性自己免疫性糖尿病(LADA)、早発性2型糖尿病(EOD)、若年性非定型糖尿病(YOAD)、若年発症成人型糖尿病(MODY)、栄養不良関連糖尿病、妊娠糖尿病、冠動脈心疾患、虚血発作、血管形成術後の再狭窄、末梢血管疾患、間欠性跛行、心筋梗塞(たとえば、壊死およびアポトーシス)、異脂肪症、食後脂肪血症、耐糖能障害(IGT)状態、空腹時血漿グルコース異常状態、代謝性アシドーシス、ケトーシス、関節炎、肥満、骨粗鬆症、高血圧、うっ血性心不全、左室肥大、末梢動脈疾患、糖尿病性網膜症、黄斑変性、白内障、糖尿病性腎症、糸球体硬化症、慢性腎不全、糖尿病性ニューロパシー、メタボリック症候群、シンドロームX、月経前症候群、冠動脈心疾患、狭心症、血栓症、アテローム性動脈硬化症、心筋梗塞、一過性脳虚血発作、卒中、血管再狭窄、高血糖、高インスリン血症、高脂血症、高トリグリセリド血症、インスリン抵抗性、グルコース代謝障害、耐糖能障害状態、空腹時血漿グルコース異常状態、肥満、勃起機能不全、皮膚および結合組織の障害、足の潰瘍化および潰瘍性大腸炎、内皮障害および血管伸展性の障害、高アポBリポタンパク質血症、アルツハイマー病、統合失調症、認知障害、炎症性腸疾患、潰瘍性大腸炎、クローン病、ならびに過敏性腸症候群からなる群から選択される疾患、状態または障害の治療方法であって、治療有効量の請求項1から7のいずれかに記載の化合物の投与を含む方法。
【請求項15】
代謝性または代謝関連の疾患、状態、または障害の治療方法であって、そのような治療の必要がある患者に、
(i)請求項8に記載の第一の組成物、および
(ii)抗肥満薬および抗糖尿病薬からなる群から選択される少なくとも1種の追加の薬剤と少なくとも1種の薬学的に許容できる賦形剤とを含む第二の組成物
を含む2種の別個の医薬組成物を投与するステップを含む方法。
【請求項16】
前記第一の組成物と前記第二の組成物が同時に投与される、請求項15に記載の方法。
【請求項17】
前記第一の組成物と前記第二の組成物が逐次的に、任意の順序で投与される、請求項15に記載の方法。
【請求項18】
Gタンパク質共役受容体GPR119の活性を調節する疾患、状態、または障害を治療するための医薬の製造における、請求項1から7に記載の化合物の使用。
【請求項19】
糖尿病または前記糖尿病に付随する病的状態を治療するための医薬の調製における、請求項1から7のいずれかに記載の化合物の使用。
【請求項1】
式Iを有する化合物
【化1】
[式中、
Xは、AまたはB
【化2】
であり、
Yは、Oまたは結合であり、
R1は、−C(O)−O−R3または
【化3】
であり、
R2は、水素、シアノ、C1〜C6アルキル、またはC3〜C6シクロアルキルであり、
R3は、C1〜C6アルキル、C3〜C6シクロアルキル、またはC1〜C6アルキル、C1〜C6アルコキシ、C1〜C6フルオロアルキル、ハロ、もしくはヒドロキシで置換されているC3〜C6シクロアルキルであり、但し、ハロ、C1〜C6アルコキシ、またはヒドロキシ基は、R1中のOに連結している炭素原子では結合しておらず、
R4は、C1〜C6ハロアルキル、C1〜C6アルキル、ハロ、シアノ、またはC3〜C6シクロアルキルであり、
R5は、水素、シアノ、ニトロ、C1〜C6フルオロアルキル、C1〜C6アルキル、C1〜C6アルコキシ、C1〜C6フルオロアルコキシ、またはC3〜C6シクロアルキルであり、
R6は、水素、C1〜C6アルキル、C1〜C6フルオロアルキル、C3〜C6シクロアルキル、またはC3〜C6シクロアルキル、C1〜C6アルコキシ、もしくはヒドロキシルで置換されているC1〜C6アルキルであり、但し、C1〜C6アルコキシ基またはヒドロキシル基は、ピラゾール窒素に連結している炭素には結合しておらず、
R7aおよびR7bは、それぞれ独立に、水素、フルオロ、またはC1〜C6アルキルであり、
R8a、R8b、R8cおよびR8dは、それぞれ独立に、水素、C1〜C6アルキル、C3〜C6シクロアルキル、またはヒドロキシもしくはC1〜C6アルコキシで置換されているC1〜C6アルキルであり、
またはR8aおよびR8bは、これらが結合している炭素と一緒になって、C3〜C6シクロアルキルを形成していてもよく、
またはR8cおよびR8dは、これらが結合している炭素と一緒になって、C3〜C6シクロアルキルを形成していてもよく、
またはR8aとR8cは一緒になって、完全飽和二炭素橋を形成していてもよく、但し、R8aおよびR8cは、これらが結合している環系の同じ平面上にある]
または薬学的に許容できるその塩。
【請求項2】
XがAであり、R1が−C(O)−O−R3である、請求項1に記載の化合物。
【請求項3】
R8a、R8b、R8cおよびR8dが、それぞれ水素であり、R3が、C1〜C3アルキルで置換されているC3〜C6シクロアルキルである、請求項1または2に記載の化合物。
【請求項4】
R7aおよびR7bが、それぞれ独立に、水素、フルオロ、またはC1〜C3アルキルである、請求項1、2または3に記載の化合物。
【請求項5】
R2が水素であり、R5がC1〜C6アルキルである、請求項1から4のいずれかに記載の化合物。
【請求項6】
以下の化合物、すなわち、
4−{[6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸イソプロピル;
4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸イソプロピル;
4−{[5−シアノ−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸イソプロピル;
4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチル;
4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピル;
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチル;
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピル;
(3,4−トランス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチル;
(9−アンチ)−9−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸tert−ブチル;
(9−アンチ)−9−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}−3−オキサ−7−アザビシクロ[3.3.1]ノナン−7−カルボン酸1−メチルシクロプロピル;
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピルの鏡像異性体1;
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピルの鏡像異性体2;
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−イソプロピル;
または薬学的に許容できるこれらの塩。
【請求項7】
以下の化合物、すなわち、
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸tert−ブチル;
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピル;
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピルの鏡像異性体1;
(3,4−シス)−3−フルオロ−4−{[5−メチル−6−(1−メチル−4,6−ジヒドロピロロ[3,4−c]ピラゾール−5(1H)−イル)ピリミジン−4−イル]オキシ}ピペリジン−1−カルボン酸1−メチルシクロプロピルの鏡像異性体2;
または薬学的に許容できるこれらの塩。
【請求項8】
少なくとも1種の薬学的に許容できる賦形剤と混和された、治療有効量で存在する請求項1から7のいずれかに記載の化合物を含む医薬組成物。
【請求項9】
抗肥満薬および抗糖尿病薬からなる群から選択される少なくとも1種の追加の薬剤をさらに含む、請求項8に記載の組成物。
【請求項10】
前記抗肥満薬が、ジルロタピド、ミトラタピド、イミプリタピド、R56918(CAS番号403987)、CAS番号913541−47−6、ロルカセリン、セチリスタット、PYY3−36、ナルトレキソン、オレオイル−エストロン、オビネピチド、プラムリンチド、テソフェンシン、レプチン、リラグルチド、ブロモクリプチン、オルリスタット、エクセナチド、AOD−9604(CAS番号221231−10−3)、およびシブトラミンからなる群から選択される、請求項9に記載の組成物。
【請求項11】
前記抗糖尿病薬が、メトホルミン、アセトヘキサミド、クロルプロパミド、ジアビネース、グリベンクラミド、グリピジド、グリブリド、グリメピリド、グリクラジド、グリペンチド、グリキドン、グリソラミド、トラザミド、トルブタミド、テンダミスタット、トレスタチン、アカルボース、アジポシン、カミグリボース、エミグリテート、ミグリトール、ボグリボース、プラジミシンQ、サルボスタチン、バラグリタゾン、シグリタゾン、ダルグリタゾン、エングリタゾン、イサグリタゾン、ピオグリタゾン、ロシグリタゾン、トログリタゾン、エキセンディン3、エキセンディン4、トロダスケミン、レセルバトロール、ヒルチオサール抽出物、シタグリプチン、ビルダグリプチン、アログリプチン、およびサクサグリプチンからなる群から選択される、請求項9に記載の組成物。
【請求項12】
治療有効量の請求項1から7のいずれかに記載の化合物の、その必要がある患者への投与を含む、糖尿病の治療方法。
【請求項13】
治療有効量の請求項1から7のいずれか一項に記載の化合物を患者に投与するステップを含む、代謝性または代謝関連の疾患、状態、または障害の治療方法。
【請求項14】
高脂血症、I型糖尿病、II型糖尿病、特発性I型糖尿病(Ib型)、成人潜在性自己免疫性糖尿病(LADA)、早発性2型糖尿病(EOD)、若年性非定型糖尿病(YOAD)、若年発症成人型糖尿病(MODY)、栄養不良関連糖尿病、妊娠糖尿病、冠動脈心疾患、虚血発作、血管形成術後の再狭窄、末梢血管疾患、間欠性跛行、心筋梗塞(たとえば、壊死およびアポトーシス)、異脂肪症、食後脂肪血症、耐糖能障害(IGT)状態、空腹時血漿グルコース異常状態、代謝性アシドーシス、ケトーシス、関節炎、肥満、骨粗鬆症、高血圧、うっ血性心不全、左室肥大、末梢動脈疾患、糖尿病性網膜症、黄斑変性、白内障、糖尿病性腎症、糸球体硬化症、慢性腎不全、糖尿病性ニューロパシー、メタボリック症候群、シンドロームX、月経前症候群、冠動脈心疾患、狭心症、血栓症、アテローム性動脈硬化症、心筋梗塞、一過性脳虚血発作、卒中、血管再狭窄、高血糖、高インスリン血症、高脂血症、高トリグリセリド血症、インスリン抵抗性、グルコース代謝障害、耐糖能障害状態、空腹時血漿グルコース異常状態、肥満、勃起機能不全、皮膚および結合組織の障害、足の潰瘍化および潰瘍性大腸炎、内皮障害および血管伸展性の障害、高アポBリポタンパク質血症、アルツハイマー病、統合失調症、認知障害、炎症性腸疾患、潰瘍性大腸炎、クローン病、ならびに過敏性腸症候群からなる群から選択される疾患、状態または障害の治療方法であって、治療有効量の請求項1から7のいずれかに記載の化合物の投与を含む方法。
【請求項15】
代謝性または代謝関連の疾患、状態、または障害の治療方法であって、そのような治療の必要がある患者に、
(i)請求項8に記載の第一の組成物、および
(ii)抗肥満薬および抗糖尿病薬からなる群から選択される少なくとも1種の追加の薬剤と少なくとも1種の薬学的に許容できる賦形剤とを含む第二の組成物
を含む2種の別個の医薬組成物を投与するステップを含む方法。
【請求項16】
前記第一の組成物と前記第二の組成物が同時に投与される、請求項15に記載の方法。
【請求項17】
前記第一の組成物と前記第二の組成物が逐次的に、任意の順序で投与される、請求項15に記載の方法。
【請求項18】
Gタンパク質共役受容体GPR119の活性を調節する疾患、状態、または障害を治療するための医薬の製造における、請求項1から7に記載の化合物の使用。
【請求項19】
糖尿病または前記糖尿病に付随する病的状態を治療するための医薬の調製における、請求項1から7のいずれかに記載の化合物の使用。
【図1】


【図2】
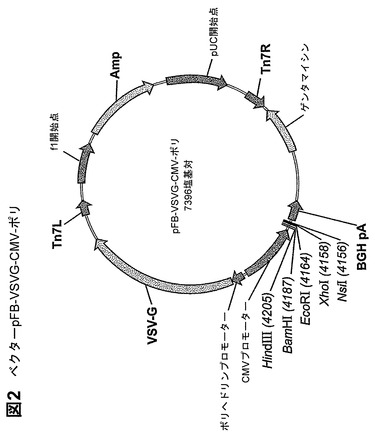
【図2】
【公表番号】特表2013−505290(P2013−505290A)
【公表日】平成25年2月14日(2013.2.14)
【国際特許分類】
【出願番号】特願2012−530363(P2012−530363)
【出願日】平成22年7月22日(2010.7.22)
【国際出願番号】PCT/IB2010/053347
【国際公開番号】WO2011/036576
【国際公開日】平成23年3月31日(2011.3.31)
【出願人】(593141953)ファイザー・インク (302)
【Fターム(参考)】
【公表日】平成25年2月14日(2013.2.14)
【国際特許分類】
【出願日】平成22年7月22日(2010.7.22)
【国際出願番号】PCT/IB2010/053347
【国際公開番号】WO2011/036576
【国際公開日】平成23年3月31日(2011.3.31)
【出願人】(593141953)ファイザー・インク (302)
【Fターム(参考)】
[ Back to top ]