
GPR49の使用による、神経変性疾患の治療
本発明は、神経生成(neurogenerative)疾患治療用の薬剤組成物を調製するためのGPR49相互作用分子の使用に関する。これによって、GPR49相互作用分子は、好ましくはGPR49の阻害剤であり、そして特に、該分子は、ガンマ−セクレターゼおよび/またはベータ−セクレターゼの活性を調節する能力を有する。さらに、本発明は、ガンマ−セクレターゼおよび/またはベータ−セクレターゼの調節剤を同定する方法であって:a.既定の試験化合物がGPR49相互作用分子であるかどうか決定することによって、GPR49相互作用分子を同定し、b.工程a)のGPR49相互作用分子がガンマ−セクレターゼおよび/またはベータ−セクレターゼの活性を調節可能であるかどうか決定する工程を含む、前記方法に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、GPR49タンパク質を含む、APPプロセシング経路のタンパク質複合体に関し、それとともに神経生成(neurogenerative)疾患の治療における、これらの複合体の阻害剤、並びにGPR49の阻害剤の使用に関する。
【背景技術】
【0002】
アルツハイマー病は、世界中で何百万という個体に影響を及ぼす慢性状態である。
アルツハイマー病患者の脳は、顕著な神経病理学的損傷の特徴的な病変、例えば初期の細胞内の神経原線維のもつれ(neurofibrillary tangle)(NFT)、および細胞外のアミロイド・リッチ老人斑を示す。これらの損傷は、CNSニューロン集団の大量の損失と関連し、そしてその進行には、ADと関連する臨床的認知症が伴う。アミロイド斑の主な構成要素は、多様な長さのアミロイド・ベータ(A−ベータ、AベータまたはAβ)ペプチドである。その変異体であるAβ1−42ペプチド(Aベータ−42)は、アミロイド形成の主な原因物質である。別の変異体はAβ1−40ペプチド(Aベータ−40)である。アミロイド・ベータは、前駆タンパク質、ベータ・アミロイド前駆タンパク質(ベータ−APPまたはAPP)のタンパク質分解産物である。APPは、I型膜貫通タンパク質であり、該タンパク質は、いくつかの異なる膜会合プロテアーゼによって、順次、切断される。APPの最初の切断は、2つのプロテアーゼ、アルファ−セクレターゼまたはベータ−セクレターゼの1つによって起こる。アルファ−セクレターゼは、メタロプロテアーゼであり、その活性は、タンパク質ADAM−10およびADAM−17の1つまたは組み合わせによって提供される可能性が最も高い。アルファ−セクレターゼによる切断は、アミロイド・ペプチド形成を妨げ、そしてしたがって、アミロイド非生成性と称される。対照的に、ベータ−セクレターゼによるAPPの切断は、アミロイド・ペプチド形成が続くための必要条件である。このセクレターゼは、BACE1(ベータ部位APP切断酵素)とも称され、アスパルチル・プロテアーゼ活性を含有するI型膜貫通タンパク質である(以下に詳細に記載される)。
【0003】
ベータ−セクレターゼ(BACE)活性は、細胞外ドメインでAPPを切断し、分泌可溶性APPbの分断(shedding)を生じ、そして99残基のC末端膜貫通断片(APP−C99)を生じる。Vassarら(Science 286, 735−741)は、APPのベータ−セクレターゼと推測される特性を有する膜貫通アスパラギン酸プロテアーゼをクローニングし、該プロテアーゼをBACE1と名づけた。BACE1ノックアウトマウス由来の脳および初代皮質培養物は、検出可能なベータ−セクレターゼ活性を示さず、そしてBACEノックアウトマウス由来の初代皮質培養物は、APPからはるかに少ないアミロイド−ベータしか産生しなかった。これによって、BACE1のパラログであるBACE2ではなく、BACE1がAPPの主なベータ−セクレターゼであることが示唆される。BACE1は、501アミノ酸(aa)のタンパク質であり、21aaのシグナルペプチドに続いて、aa22〜45に渡る、プロ配列ドメインを含有する。選択的スプライシング型、BACE−I−457およびBACE−I−476がある。成熟タンパク質の細胞外ドメインに続いて、予測される1つの膜貫通ドメインおよび24aaの短い細胞質ゾルC末端テールがある。BACE1は、1型膜貫通タンパク質であり、活性部位が膜の細胞外側にあると予測され、ここでベータ−セクレターゼがAPP、およびありうる他の未同定の基質を切断する。BACE1は、明らかに、APPをA−ベータにプロセシングするのに必要とされる重要な酵素であるが、最近の証拠によって、BACE1のさらなる潜在的基質および機能が示唆されている(J. Biol. Chem. 279, 10542−10550)。現在まで、制御機能または調節機能を持つBACE1相互作用タンパク質は、まったく記載されてきていない。
【0004】
BACE1切断によって生じるAPP断片であるAPP−C99は、ガンマ−セクレターゼ活性の基質であり、該活性は、膜平面内でAPP−C99を切断してA−ベータ・ペプチド(アミロイド生成性Aβ1−42ペプチドなど)、およびAPP細胞内ドメイン(AICD)と称されるC末端断片にする(Annu Rev Cell Dev Biol 19, 25−51)。ガンマ−セクレターゼ活性は、少なくとも4つの別個のサブユニットを持つ多タンパク質複合体内にある。発見された第一のサブユニットは、プレセニリンであった(Proc Natl Acad Sci USA 94, 8208−13)。ガンマ−セクレターゼ複合体の他の既知のタンパク質構成要素は、Pen−2、ニカストリンおよびAph−1a(Aph1a)である。
【0005】
アルツハイマー病の病因の根底にある分子事象の描写が近年、進歩しているにもかかわらず、これまでに疾患を緩和する療法は開発されてきていない。この目的に向け、業界は、BACE1を阻害するための適切なリード化合物を同定しようと苦闘している。さらに、ガンマ−セクレターゼのより多くの別の基質、最も著名には、Notchタンパク質が存在することが認められてきている。その結果、ガンマ−セクレターゼの阻害は、機構に基づく副作用を引き起こす可能性がある。現在、最も優れた薬剤(例えばアリセプト(登録商標)/ドネペジル)は、アセチルコリン・エステラーゼを阻害して、脳において、神経伝達物質アセチルコリンのレベル増加を生じることによって、認知機能の一時的な改善を達成することを試みる。これらの療法は、疾患のより後期の病期には適しておらず、根底にある疾患病変は治療せず、そして疾患進行を停止させることはない。
【0006】
したがって、アルツハイマー病の治療のための新規分子戦略を可能にする、新規ターゲットの同定に関して、満たされていない必要性がある。さらに、前記新規ターゲットをターゲティングすることによって、前述の分子プロセスを修飾する新規療法化合物に関する、強い必要性がある。
【発明の開示】
【課題を解決するための手段】
【0007】
第一の側面において、本発明は、神経生成疾患治療用の薬剤組成物を調製するための、「GPR49相互作用分子」の使用を提供する。
本発明の関連において、驚くべきことに、GPR49(ロイシン・リッチ・リピート含有Gタンパク質共役型受容体5(LGR5);FEX HG38、GPR67としても知られる)が、アルツハイマー病において、ガンマ−セクレターゼによるAPPの異常なプロセシングに関与する、異なるタンパク質複合体の一部を形成することが発見されている。特に、GPR49がAph1a複合体、Fe65L2複合体、APP−C99複合体およびBACE1複合体の一部であることが発見されている。これらの複合体は、TAP技術エントリーポイントとして用いられている、それぞれの重要なタンパク質化合物にちなんで命名されている(以下を参照されたい)。
【0008】
GPR49がこれらの複合体において重要な分子と同定されたことによって、神経変性(neurodegenerative)疾患の治療のため、GPR49と相互作用する分子の使用が可能になる。これは特に、GPR49に対して向けられるsiRNAが、Aベータ−42の生成および/または分泌の減弱を生じることを立証する実施例において示される。
【0009】
本発明の関連において、「GPR49相互作用分子」は、少なくとも一時的にGPR49に結合し、そして好ましくは、GPR49活性を調節し、そして特に阻害する、分子である。
【0010】
GPR49は、糖タンパク質ホルモン受容体スーパーファミリーの推定上のメンバーであり、このスーパーファミリーは、ある場合には糖タンパク質リガンドとの相互作用に重要であることが示されたロイシン・リッチ・リピートを含有する、巨大N末端細胞外ドメインによって特徴付けられる、異常なGタンパク質共役型受容体(GPCR)である。スーパーファミリーのメンバーを系統学的に分析すると、3つのサブファミリーに分かれると示唆され、第一のサブファミリーは、LH、FSH(卵胞刺激ホルモン)およびTSH(甲状腺刺激ホルモン)の受容体を特徴とし;第二のサブファミリーは、リラキシン受容体LGR7およびLGR8を含有する。GPR49/LGR5とともにLGR4およびLGR6(図4)は、FSH受容体とわずか〜35%の配列同一性しか示さない、オーファン受容体サブファミリーを構成する(Hsu SY, Liang SG, Hsueh AJ(1998)Characterization of two LGR genes homologous to gonadotropin and thyrotropin receptors with extracellular leucine−rich repeats and a G protein−coupled, seven−transmembrane region. Mol Endocrinol. 12(12):1830−45;McDonald T, Wang R, Bailey W, Xie G, Chen F, Caskey CT, Liu Q(1998)Identification and cloning of an orphan G protein−coupled receptor of the glycoprotein hormone receptor subfamily. Biochem Biophys Res Commun. 247(2):266−70)。
【0011】
GPR49は、骨格筋、胎盤および脊髄で顕著に発現され、そしてより低いレベルで、結腸、副腎および脳の多様な小領域で発現される(Hsu SYら(1998)、上記)。成体マウス脳では、GPR49転写物は、主に嗅球に限定されており、嗅球の神経細胞層に見られる(Hermey G, Methner A, Schaller HC, Hermans−Borgmeyer I(1999)Identification of a novel seven−transmembrane receptor with homology to glycoprotein receptors and its expression in the adult and developing mouse. Biochem Biophys Res Commun. 254(1):273−9)。成体におけるGPR49の機能はそれほど性質決定されていない。マウスにおいて、GPR49遺伝子をターゲットとして欠失させると、新生児致死が生じ、そしてこの欠失は舌小帯短縮症および胃腸膨張と関連する(Morita H, Mazerbourg S, Bouley DM, Luo CW, Kawamura K, Kuwabara Y, Baribault H, Tian H, Hsueh AJ(2004)Neonatal lethality of LGR5 null mice is associated with ankyloglossia and gastrointestinal distension. Mol Cell Biol. 24(22):9736−43)。
【0012】
GPR49は、最近、ベータ−カテニン突然変異を持つヒト肝細胞癌で過剰発現される遺伝子と同定された(Yamamoto Y, Sakamoto M, Fujii G, Tsuiji H, Kenetaka K, Asaka M, Hirohashi S(2003) Overexpression of orphan G−protein−coupled receptor, Gpr49, in human hepatocellular carcinomas with beta−catenin mutations. Hepatology. 37(3):528−33)。
【0013】
本発明にしたがって、表現「GPR49」は、図3に示すようなタンパク質だけでなく、機能的に活性なその誘導体、または機能的に活性なその断片、またはその相同体、または低ストリンジェンシー条件下で、前記タンパク質をコードする核酸にハイブリダイズする核酸にコードされる変異体もまた、意味する。好ましくは、これらの低ストリンジェンシー条件は、35%ホルムアミド、5xSSC、50mM Tris−HCl(pH7.5)、5mM EDTA、0.02% PVP、0.02% BSA、100μg/ml変性サケ精子DNA、および10%(重量/体積)デキストラン硫酸を含む緩衝液中、40℃で18〜20時間ハイブリダイゼーションし、2xSSC、25mM Tris−HCl(pH7.4)、5mM EDTA、および0.1% SDSからなる緩衝液中、55℃で1〜5時間洗浄し、そして2xSSC、25mM Tris−HCl(pH7.4)、5mM EDTA、および0.1% SDSからなる緩衝液中、60℃で1.5時間洗浄することを含む。
【0014】
本発明で挙げる他のタンパク質すべてにも同じことがあてはまる。したがって、既定のタンパク質または核酸の名称は、配列表に示すようなタンパク質または核酸を指すだけでなく、機能的に活性な誘導体、または機能的に活性なその断片、またはその相同体、または低ストリンジェンシー条件下で、好ましくは上述のような条件下で、前記タンパク質をコードする核酸にハイブリダイズする核酸にコードされる変異体もまた、指す。
【0015】
用語「機能的に活性な」は、本明細書において、このポリペプチド、すなわち断片または誘導体が関連する態様にしたがって、タンパク質の構造的機能、制御機能、または生化学的機能を有する、ポリペプチド、すなわち断片または誘導体を指す。
【0016】
本発明にしたがって、用語「活性」は、本明細書において、最も広い意味での分子の機能を指す。活性は、一般的に、限定されるわけではないが、分子の生物学的機能、生化学的機能、物理的機能または化学的機能を含む。活性は、例えば、酵素活性、他の分子と相互作用する能力、および他の分子の機能を活性化するか、促進するか、安定化するか、阻害するか、抑制するか、または不安定化する能力、安定性、特定の細胞内位置に局在する能力を含む。適用可能な場合、前記用語はまた、最も広い意味でのタンパク質複合体の機能にも関する。
【0017】
本発明にしたがって、用語「誘導体」または「構成要素タンパク質の類似体」または「変異体」は、本明細書において、好ましくは、限定されるわけではないが、構成要素タンパク質に実質的に相同な領域を含む分子を含み、こうした分子は、多様な態様において、同一サイズのアミノ酸配列に渡って、または当該技術分野に知られるコンピュータ相同性プログラムによって並列を行って並列される配列と比較した際、少なくとも30%、40%、50%、60%、70%、80%、90%、95%または99%同一であるか、あるいはこうした分子をコードする核酸は、ストリンジェントな条件、中程度にストリンジェントな条件、またはストリンジェントでない条件下で、構成要素タンパク質をコードする配列にハイブリダイズ可能である。これは、それぞれ、アミノ酸置換、欠失および付加によって、天然存在タンパク質を修飾した所産であり、その誘導体がなお天然存在タンパク質の生物学的機能を、必ずしも同じ度合いでなくてもよいが示す、タンパク質を意味する。例えば、本発明で提供するような、適切で利用可能なin vitroアッセイによって、こうしたタンパク質の生物学的機能を調べることも可能である。
【0018】
用語「断片」は、本明細書において、態様にしたがって、構成要素タンパク質の少なくとも10、20、30、40または50アミノ酸のポリペプチドを指す。特定の態様において、こうした断片は、35、100または200アミノ酸より長くはない。
【0019】
用語「遺伝子」は、本明細書において、別に言及しない限り、本発明のポリペプチドをコードするオープンリーディングフレームを含む核酸を指し、エクソンおよび場合によるイントロン配列両方を含む。
【0020】
用語「相同体」または「相同遺伝子産物」は、本明細書において、本明細書にさらに記載する複合体のタンパク質構成要素と同じ生物学的機能を実行する、別の種、好ましくは哺乳動物におけるタンパク質を意味する。こうした相同体はまた、「オルソログ遺伝子産物」とも称される。ヒトおよび哺乳動物または他の種からオルソログ遺伝子対を検出するためのアルゴリズムは、これらの生物の全ゲノムを用いる。まず、予測されるタンパク質の完全Smith−Waterman並列を用いて、対合ベストヒットを回収する。信頼性をさらに改善するため、キイロショウジョウバエ(Drosophila melanogaster)および線虫(C. elegans)タンパク質を含む対合ベストヒットを用いて、これらの対のクラスターを形成する。こうした分析は、例えば、Nature, 2001, 409:860−921に提供される。他の種の遺伝子に対する、本明細書に提供するタンパク質をコードする遺伝子の配列相同性に基づいて、慣用的技術を適用してそれぞれの遺伝子をクローニングし、そしてこうした遺伝子からタンパク質を発現させることによって、あるいは本明細書に提供する方法にしたがって、または当該技術分野に一般的に知られる他の適切な方法にしたがって、類似の複合体を単離することにより、他の種のタンパク質を単離することによって、本発明記載のタンパク質の相同体を単離することも可能である。
【0021】
本発明の好ましい態様において、「GPR49相互作用分子」はGPR49阻害剤である。
本発明にしたがって、用語「阻害剤」は、GPR49の活性を好ましくは阻害するかまたは減少させる、生化学的化合物または化学的化合物を指す。これは、例えば、対応する遺伝子の発現の抑制を介して起こることも可能である。RT−PCRまたはウェスタンブロット分析によって、遺伝子の発現を測定することも可能である。さらに、これは、例えばGPR49への結合による、活性の阻害を介して起こることも可能である。
【0022】
こうしたGPR49阻害剤の例は、GPR49に対して向けられる、特にGPR49の活性部位に対して向けられる、結合性タンパク質または結合性ペプチド、並びにGPR49遺伝子に対して向けられる核酸である。
【0023】
用語「GPR49に対する核酸」は、例えばGPR49遺伝子の発現またはGPR49の活性を阻害する、二本鎖または一本鎖DNAまたはRNA、あるいはその修飾物または誘導体を指し、そして限定なしに、アンチセンス核酸、アプタマー、siRNA(低分子干渉RNA)およびリボザイムを含む。
【0024】
好ましくは、阻害剤は、抗体、アンチセンス・オリゴヌクレオチド、siRNA、低分子量分子(LMW)、結合性ペプチド、アプタマー、リボザイムおよびペプチド模倣体(peptidomimetic)からなる群より選択される。
【0025】
これらの核酸を細胞に直接投与することも可能であるし、または外因性の導入された配列を転写させることによって、細胞内で産生することも可能である。
「アンチセンス」核酸は、本明細書において、何らかの配列相補性のため、構成要素タンパク質RNA(好ましくはmRNA)の配列特異的部分にハイブリダイズ可能な核酸を指す。アンチセンス核酸は、構成要素タンパク質mRNAのコード領域および/または非コード領域に相補的であることも可能である。複合体形成または活性を阻害する、こうしたアンチセンス核酸は、療法剤として有用性を有し、そして本明細書に記載するような障害の治療または予防に使用可能である。
【0026】
アンチセンス核酸は、少なくとも6ヌクレオチドのものであり、そして好ましくは、6〜約200ヌクレオチドの範囲のオリゴヌクレオチドである。特定の側面において、オリゴヌクレオチドは、少なくとも10ヌクレオチド、少なくとも15ヌクレオチド、少なくとも100ヌクレオチド、または少なくとも200ヌクレオチドである。
【0027】
核酸、例えばアンチセンス核酸またはsiRNAを、例えばホスホトリエステル法にしたがって、化学的に合成することも可能である(例えばUhlmann, E. & Peyman, A.(1990)Chemical Reviews, 90, 543−584を参照されたい)。アプタマーは、ポリペプチド、本明細書ではGPR49に、高い親和性で結合する核酸である。アプタマーは、異なる一本鎖RNA分子の巨大プールから、SELEXなどの選択法によって単離可能である(例えば、Jayasena(1999)Clin. Chem., 45, 1628−50;KlugおよびFamulok(1994)M. Mol. Biol. Rep., 20, 97−107;US 5,582,981を参照されたい)。アプタマーを、例えばL−リボヌクレオシドとして、鏡像型で合成し、そして選択することもまた可能である(Nolteら(1996)Nat. Biotechnol., 14, 1116−9;Klussmannら(1996)Nat. Biotechnol., 14, 1112−5)。この方式で単離された型は、天然存在リボヌクレアーゼによって分解されず、そしてしたがってより高い安定性を所持する利点を享受する。
【0028】
核酸は、エンドヌクレアーゼまたはエキソヌクレアーゼ、特に細胞で見出されることも可能なDNaseおよびRNaseによって分解されうる。したがって、分解に対して安定化させ、それによって長期間に渡って、細胞中で、核酸が高濃度で維持されることを保証するため、核酸を修飾することは好都合である(Beigelmanら(1995)Nucleic Acids Res. 23:3989−94;WO 95/11910;WO 98/37240;WO 97/29116)。典型的には、こうした安定化は、1以上のヌクレオチド間リン基を導入することによって、または1以上の非リン基をヌクレオチド間に導入することによって、得ることも可能である。
【0029】
適切なヌクレオチド間修飾は、UhlmannおよびPeyman(1990)、上記に集められている(Beigelmanら(1995)Nucleic Acids Res. 23:3989−94;WO 95/11910;WO 98/37240;WO 97/29116もまた参照されたい)。本発明にしたがった使用の1つで使用可能な、核酸における修飾ヌクレオチド間ホスフェート・ラジカルおよび/または非リン架橋は、例えば、メチル・ホスホネート、ホスホロチオエート、ホスホロアミデート、ホスホロジチオエートおよび/またはホスフェート・エステルを含有し、一方、非リン・ヌクレオチド間類似体(analogue)は、例えばシロキサン架橋、カーボネート架橋、カルボキシメチル・エステル、アセトアミデート(acetamidate)架橋および/またはチオエーテル架橋を含有する。この修飾が、本発明記載の使用の1つで使用可能である薬剤組成物の耐久性を改善するはずであることもまた意図される。一般的に、オリゴヌクレオチドを、塩基部分、糖部分、またはホスフェート主鎖で修飾することも可能である。
【0030】
オリゴヌクレオチドは、ペプチド、細胞膜を渡る輸送を促進する剤(例えば、Letsingerら, 1989, Proc. Natl. Acad. Sci. USA 86:6553−6556;Lemaitreら, 1987, Proc. Natl. Acad. Sci. USA 84:648−652;国際特許公報第WO 88/09810号)または血液−脳関門を渡る輸送を促進する剤(例えば国際特許公報第WO 89/10134号)、ハイブリダイゼーションに誘発される切断剤(例えば、Krolら, 1988, BioTechniques 6:958−976を参照されたい)、またはインターカレート剤(例えばZon, 1988, Pharm. Res. 5:539−549を参照されたい)などの、他の付随する基を含むことも可能である。
【0031】
詳細には、アンチセンス・オリゴヌクレオチドは、限定されるわけではないが、5−フルオロウラシル、5−ブロモウラシル、5−クロロウラシル、5−ヨードウラシル、ヒポキサンチン、キサンチン、4−アセチルシトシン、5−(カルボキシヒドロキシメチル)ウラシル、5−カルボキシメチルアミノメチル−2−チオ−ウリジン、5−カルボキシメチルアミノメチルウラシル、ジヒドロウラシル、D−ガラクトシルケオシン、イノシン、N6−イソペンテニルアデニン、1−メチルグアニン、1−メチルイノシン、2,2−ジメチルグアニン、2−メチルアデニン、2−メチルグアニン、3−メチルシトシン、5−メチルシトシン、N6−アデニン、7−メチルグアニン、5−メチルアミノメチルウラシル、5−メトキシアミノメチル−2−チオウラシル、D−マンノシルケオシン、5N−メトキシカルボキシメチルウラシル、5−メトキシウラシル、2−メチル−チオ−N6−イソペンテニルアデニン、ウラシル−5−オキシ酢酸(v)、ワイブトキソシン(wybutoxosine)、シュードウラシル、ケオシン、2−チオシトシン、5−メチル−2−チオウラシル、2−チオウラシル、4−チオウラシル、5−メチルウラシル、ウラシル−5−オキシ酢酸メチルエステル、ウラシル−5−オキシ酢酸(v)、5−メチル−2−チオウラシル、3−(3−アミノ−3−N−2−カルボキシプロピル)ウラシル、(acp3)w、および2,6−ジアミノプリンを含む群より選択される、少なくとも1つの修飾塩基部分を含むことも可能である。
【0032】
別の態様において、オリゴヌクレオチドは、限定されるわけではないが、アラビノース、2−フルオロアラビノース、キシルロース、およびヘキソースを含む群より選択される、少なくとも1つの修飾糖部分を含む。
【0033】
適切なアンチセンス核酸の使用が、例えば、ZhengおよびKemeny(1995)Clin. Exp. Immunol., 100, 380−2;NellenおよびLichtenstein(1993)Trends Biochem. Sci., 18, 419−23, Stein(1992)Leukemia, 6, 697−74またはYacyshyn, B.R.ら(1998)Gastroenterology, 114, 1142にさらに記載される。
【0034】
さらに別の態様において、オリゴヌクレオチドは、2−a−アノマー・オリゴヌクレオチドである。a−アノマー・オリゴヌクレオチド(2−a−アノマーまたはa−アノマー)は、通常のβ−ユニットと対照的に、鎖が互いに平行に走る相補RNAと特異的な二本鎖ハイブリッドを形成する(Gautierら, 1987, Nucl. Acids Res. 15:6625−6641)。
【0035】
オリゴヌクレオチドを別の分子、例えばペプチド、ハイブリダイゼーションが誘発する架橋剤、輸送剤、ハイブリダイゼーションが誘発する切断剤等にコンジュゲート化することも可能である。
【0036】
本発明全体で、当該技術分野に知られる標準法によって、例えば自動化DNA合成装置(Biosearch、Applied Biosystems等から商業的に入手可能なものなど)の使用によって、本発明のオリゴヌクレオチドを合成することも可能である。例えば、Steinら(1988, Nucl. Acids Res. 16:3209)の方法によって、ホスホロチオエート・オリゴヌクレオチドを合成することも可能であるし、調節孔ガラスポリマー支持体(Sarinら, 1988, Proc. Natl. Acad. Sci. USA 85:7448−7451)等の使用によって、メチルホスホネート・オリゴヌクレオチドを調製することも可能である。
【0037】
特定の態様において、アンチセンス・オリゴヌクレオチドは、触媒性RNAまたはリボザイムを含む(例えば、国際特許公報第WO 90/11364号;Sarverら, 1990, Science 247:1222−1225を参照されたい)。別の態様において、オリゴヌクレオチドは2’−O−メチルリボヌクレオチド(Inoueら, 1987, Nucl. Acids Res. 15:6131−6148)、またはキメラRNA−DNA類似体(Inoueら, 1987, FEBS Lett. 215:327−330)である。
【0038】
別の態様において、本発明のアンチセンス核酸は、外因性配列からの転写によって細胞内で産生される。例えば、細胞に取り込まれるように、ベクターをin vivoで導入することも可能であり、その細胞内でベクターまたはその一部が転写され、本発明のアンチセンス核酸(RNA)が産生される。こうしたベクターは、構成要素タンパク質をコードする配列を含有するであろう。こうしたベクターは、望ましいアンチセンスRNAを産生するように転写されることが可能である限り、エピソームにとどまることも可能であるし、または染色体に組み込まれることも可能である。当該技術分野で標準的な組換えDNA技術法によって、こうしたベクターを構築することも可能である。ベクターは、プラスミド・ベクター、ウイルス・ベクター、または哺乳動物細胞における複製および発現が可能であることが当該技術分野に知られる他のものであることも可能である。アンチセンスRNAをコードする配列の発現は、哺乳動物細胞、好ましくはヒト細胞において、作用することが当該技術分野に知られるプロモーターいずれによることも可能である。こうしたプロモーターは誘導性または恒常性であることも可能である。こうしたプロモーターには、限定されるわけではないが、SV40初期プロモーター領域(BernoistおよびChambon, 1981, Nature 290:304−310)、ラウス肉腫ウイルスの3’末端反復配列(long terminal repeat)に含有されるプロモーター(Yamamotoら, 1980, Cell 22:787−797)、ヘルペス・チミジンキナーゼ・プロモーター(Wagnerら, 1981, Proc. Natl. Acad. Sci. USA 78:1441−1445)、メタロチオネイン遺伝子の制御配列(Brinsterら, 1982, Nature 296:39−42)等が含まれる。
【0039】
本発明のアンチセンス核酸は、構成要素タンパク質遺伝子、好ましくはヒト遺伝子のRNA転写物の少なくとも一部に相補的な配列を含む。しかし、絶対相補性は好ましいが必要ではない。「RNAの少なくとも一部に相補的な」配列は、本明細書において、RNAとハイブリダイズ可能であるのに十分な相補性を有し、安定な二重鎖を形成する配列を意味し;したがって、二本鎖アンチセンス核酸の場合、二重鎖DNAの一本鎖を試験することも可能であるし、または三重鎖形成をアッセイすることも可能である。ハイブリダイズする能力は、相補性の度合いおよびアンチセンス核酸の長さの両方に依存するであろう。一般的に、ハイブリダイズする核酸がより長ければ、構成要素タンパク質RNAとの塩基ミスマッチをより多く含有して、そしてなお安定な二重鎖(または場合によっては三重鎖)を形成することが可能になる。当業者は、ハイブリダイズした複合体の融点を決定する標準法の使用によって、ミスマッチの容認しうる度合いを確認することも可能である。
【0040】
遺伝子発現、本明細書ではGPR49遺伝子発現を下方制御するかまたはスイッチオフするプロセスにおける、RNA干渉のためのツールとしてのsiRNAの産生および使用は、例えばElbashir, S.M.ら(2001)Genes Dev., 15, 188またはElbashir, S.M.ら(2001)Nature, 411, 494に記載される。好ましくは、siRNAは30ヌクレオチド未満の長さを示し、siRNAのセンス鎖の同一ストレッチは、好ましくは少なくとも19ヌクレオチドである。
【0041】
リボザイムもまた、核酸、本明細書ではGPR49遺伝子の翻訳を阻害するのに適したツールであり、これはリボザイムがmRNAに特異的に結合し、そして切断することが可能であるためである。これらは例えば、Amarzguiouiら(1998)Cell. Mol. Life Sci., 54, 1175−202;Vaishら(1998)Nucleic Acids Res., 26, 5237−42;Persidis(1997)Nat. Biotechnol., 15, 921−2またはCoutureおよびStinchcomb(1996)Trends Genet., 12, 510−5に記載される。
【0042】
薬学的に許容しうるキャリアー中に有効量の核酸を含む、本発明の薬剤組成物を、本発明のタンパク質複合体を発現するかまたは過剰発現する種類の疾患または障害を有する患者に投与することも可能である。
【0043】
特定の障害または状態の治療に有効であろう核酸の量は、障害または状態の性質に応じるであろうし、そして標準的臨床技術によって決定可能である。可能な場合、ヒトで試験しそして使用する前に、核酸細胞傷害性をin vitroで測定し、そして次いで有用な動物モデル系で測定することが望ましい。
【0044】
特定の態様において、核酸を含む薬剤組成物を、リポソーム、微小粒子、または微小カプセルを介して投与する。本発明の多様な態様において、こうした組成物を用いて、核酸の持続放出を達成することが有用である可能性もある。特定の態様において、同定可能な中枢神経系細胞種に特異的な抗体を介してターゲティングされるリポソームを利用することが望ましい可能性もある(Leonettiら, 1990, Proc. Natl. Acad. Sci. U.S.A. 87:2448−2451;Renneisenら, 1990, J. Biol. Chem. 265:16337−16342)。
【0045】
いわゆる「低分子量分子」(以下、「LMW」と称する)は、タンパク質、ペプチド、抗体または核酸ではなく、そして5000Da未満、好ましくは2000Da未満、より好ましくは1000Da未満、最も好ましくは500Da未満の分子量を示す、分子である。こうしたLMWは、ライブラリーから出発するハイスループット法において同定可能である。こうした方法は当該技術分野で知られ、そして以下に詳細に論じられる。
【0046】
用語「結合性タンパク質」または「結合性ペプチド」は、GPR49に結合し、そしてこれを阻害する種類のタンパク質またはペプチドを指し、そして限定なしに、GPR49に対して向けられるポリクローナル抗体またはモノクローナル抗体、抗体断片およびタンパク質骨格を含む。
【0047】
本発明にしたがって、用語、抗体または抗体断片はまた、組換え的に調製され、そして適切な場合、修飾された、抗体またはその抗原結合部分、例えばキメラ抗体、ヒト化抗体、多機能抗体、二重特異性またはオリゴ特異性(oligospecific)抗体、一本鎖抗体およびF(ab)またはF(ab)2断片(例えば、EP−B1−0 368 684、US 4,816,567、US 4,816,397、WO 88/01649、WO 93/06213またはWO 98/24884を参照されたい)を意味するとして理解され、好ましくはFAB発現ライブラリーの補助で産生される。
【0048】
古典的な抗体の代替物として、例えば、GPR49に対するタンパク質骨格、例えばリポカリンに基づくアンチカリン(Besteら(1999)Proc. Natl. Acad. Sci. USA, 96, 1898−1903)を使用することもまた可能である。選択されるハプテン、本明細書ではGPR49に結合する方式で、例えば「コンビナトリアル・タンパク質設計」アプローチによって、リポカリン、例えばレチノール結合性タンパク質またはビリン結合性タンパク質の天然リガンド結合部位を改変することも可能である(Skerra, 2000, Biochim. Biophys. Acta, 1482, 337−50)。他の既知のタンパク質骨格が、分子認識のための抗体の代替物であることが知られる(Skerra(2000)J. Mol. Recognit., 13, 167−187)。
【0049】
当業者に周知の方法にしたがって、例えば哺乳動物、例えばウサギを、適切な場合、例えばフロイントのアジュバントおよび/または水酸化アルミニウムゲルの存在下で、GPR49で免疫することによって、抗体または抗体断片を調製する方法が達成される(例えば、Diamond, B.A.ら(1981)The New England Journal of Medicine:1344−1349を参照されたい)。免疫学的反応の結果として、動物で形成されるポリクローナル抗体を、続いて、周知の方法を用いて血液から単離し、そして例えばカラム・クロマトグラフィーによって精製することも可能である。例えば、モノクローナル抗体をWinter & Milsteinの既知の方法(Winter, G. & Milstein, C.(1991)Nature, 349, 293−299)にしたがって、調製することも可能である。
【0050】
詳細には、適切な被験者を、免疫原としてのポリペプチドで免疫することによって、上述のように、ポリクローナル抗体を調製することも可能である。好ましいポリクローナル抗体組成物は、本発明の単数または複数のポリペプチドに対して向けられる抗体に関して選択されているものである。特に好ましいポリクローナル抗体調製物は、既定の単数または複数のポリペプチドに対して向けられる抗体のみを含有するものである。特に好ましい免疫原組成物は、他のヒト・タンパク質を含有しないもの、例えば本発明のポリペプチドの組換え発現のため、非ヒト宿主細胞を用いて作製した免疫原組成物である。こうした方式では、この免疫原に対して作製して生じた抗体組成物に認識されるのは、本発明の単数または複数のポリペプチドの一部として存在する、単数または複数のヒト・エピトープのみであろう。
【0051】
標準的技術、例えば固定ポリペプチドを用いた酵素連結免疫吸着アッセイ(ELISA)によって、一定時間に渡って、免疫した被験者における抗体力価を監視することも可能である。望ましい場合、哺乳動物から(例えば血液から)、抗体分子を単離し、そしてさらに、周知の技術、例えばプロテインAクロマトグラフィーによって、さらに精製してIgG分画を得ることも可能である。あるいは、本発明のタンパク質またはポリペプチドに特異的な抗体を、例えばアフィニティー・クロマトグラフィーによって、選択する(例えば部分的に精製する)かまたは精製することも可能である。例えば、組換え的に発現され、そして精製された(または部分的に精製された)本発明のタンパク質を、本明細書に記載するように産生し、そして例えばクロマトグラフィー・カラムなどの固体支持体に、共有的にまたは非共有的にカップリングする。次いで、該カラムを用いて、多数の異なるエピトープに対して向けられる抗体を含有する試料から、本発明のタンパク質に特異的な抗体をアフィニティー精製し、それによって実質的に精製された抗体組成物、すなわち混入抗体を実質的に含まないものを生成することも可能である。この関連において、実質的に精製された抗体組成物によって、抗体試料が、本発明の望ましいタンパク質またはポリペプチドに対するもの以外のエピトープに対して向けられる混入抗体を(乾燥重量にして)最大わずか30%しか含有せず、そして好ましくは(乾燥重量にして)試料の最大20%、さらにより好ましくは最大10%、そして最も好ましくは最大5%のみが混入抗体であることを意味する。精製抗体組成物は、組成物中の抗体の少なくとも99%が、本発明の望ましいタンパク質またはポリペプチドに対して向けられることを意味する。
【0052】
免疫後の適切な時点で、例えば特異的抗体力価が最も高いとき、抗体産生細胞を被験者から得て、そして該細胞を用いて、標準的技術、例えば元来、KohlerおよびMilstein, 1975, Nature 256:495−497に記載されたハイブリドーマ技術、ヒトB細胞ハイブリドーマ技術(Kozborら, 1983, Immunol. Today 4:72)、EBVハイブリドーマ技術(Coleら, 1985, Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, Inc., pp.77−96)またはトリオーマ技術によって、モノクローナル抗体を調製することも可能である。ハイブリドーマを産生するための技術は周知である(一般的に、Current Protocols in Immunology 1994, Coliganら(監修)John Wiley & Sons, Inc., ニューヨーク州ニューヨークを参照されたい)。本発明のモノクローナル抗体を産生するハイブリドーマ細胞は、例えば標準的ELISAアッセイを用いて、目的のポリペプチドに結合する抗体に関して、ハイブリドーマ培養上清をスクリーニングすることによって、検出される。
【0053】
モノクローナル抗体分泌ハイブリドーマを調製する代わりに、組換えコンビナトリアル免疫グロブリン・ライブラリー(例えば抗体ファージディスプレイ・ライブラリー)を、目的のポリペプチドでスクリーニングすることによって、本発明のポリペプチドに対して向けられるモノクローナル抗体を同定し、そして単離することも可能である。ファージディスプレイ・ライブラリーを生成し、そしてスクリーニングするためのキットが商業的に入手可能である(例えば、Pharmacia組換えファージ抗体系、カタログ番号27−9400−01;およびStratagene SurfZAPファージディスプレイ・キット、カタログ番号240612)。さらに、抗体ディスプレイ・ライブラリーを生成し、そしてスクリーニングする際に使用するのに特に受け入れられる方法および試薬の例は、例えば、米国特許第5,223,409号;PCT公報第WO 92/18619号;PCT公報第WO 91/17271号;PCT公報第WO 92/20791号;PCT公報第WO 92/15679号;PCT公報第WO 93/01288号;PCT公報第WO 92/01047号;PCT公報第WO 92/09690号;PCT公報第WO 90/02809号;Fuchsら, 1991, Bio/Technology 9:1370−1372;Hayら, 1992, Hum. Antibod. Hybridomas 3:81−85;Huseら, 1989, Science 246:1275−1281;Griffithsら, 1993, EMBO J. 12:725−734に見出すことも可能である。
【0054】
さらに、標準的組換えDNA技術を用いて作製可能である、ヒト部分および非ヒト部分両方を含む、キメラおよびヒト化モノクローナル抗体などの組換え抗体が、本発明の範囲内である。キメラ抗体は、異なる部分が異なる動物種に由来する分子、例えばネズミmAb由来の可変領域およびヒト免疫グロブリン定常領域を有するものである(例えば、本明細書に完全に援用される、Cabillyら、米国特許第4,816,567号;およびBossら、米国特許第4,816,397号を参照されたい)。ヒト化抗体は、非ヒト種由来の1以上の相補性決定領域(CDR)およびヒト免疫グロブリン分子由来のフレームワーク領域を有する、非ヒト種由来の抗体分子である(例えば、本明細書に完全に援用される、Queen、米国特許第5,585,089号を参照されたい)。こうしたキメラおよびヒト化モノクローナル抗体は、当該技術分野に知られる組換えDNA技術によって、例えば、PCT公報第WO 87/02671号;欧州特許出願184,187;欧州特許出願171,496;欧州特許出願173,494;PCT公報第WO 86/01533号;米国特許第4,816,567号;欧州特許出願125,023;Betterら, 1988, Science 240:1041−1043;Liuら, 1987, Proc. Natl. Acad. Sci. USA 84:3439−3443;Liuら, 1987, J. Immunol. 139:3521−3526;Sunら, 1987, Proc. Natl. Acad. Sci. USA 84:214−218;Nishimuraら, 1987, Canc. Res. 47:999−1005;Woodら, 1985, Nature 314:446−449;およびShawら, 1988, J. Natl. Cancer Inst. 80:1553−1559);Morrison, 1985, Science 229:1202−1207;Oiら, 1986, Bio/Techniques 4:214;米国特許第5,225,539号;Jonesら, 1986, Nature 321:552−525;Verhoeyanら, 1988, Science 239:1534;およびBeidlerら, 1988, J. Immunol. 141:4053−4060に記載される方法を用いて、産生可能である。
【0055】
完全ヒト抗体は、ヒト患者の療法的治療に特に望ましい。こうした抗体は、例えば、内因性免疫グロブリン重鎖および軽鎖遺伝子を発現不能であるが、ヒト重鎖および軽鎖遺伝子を発現可能である、トランスジェニックマウスを用いて、産生可能である。選択した抗原、例えば本発明のポリペプチドすべてまたは一部を用いて、通常の様式で該トランスジェニックマウスを免疫する。慣用的ハイブリドーマ技術を用いて、該抗原に対して向けられるモノクローナル抗体を得ることも可能である。該トランスジェニックマウスに宿するヒト免疫グロブリン導入遺伝子は、B細胞分化中に再編成し、そして続いてクラス・スイッチおよび体細胞突然変異を経る。したがって、こうした技術を用いて、療法的に有用なIgG、IgAおよびIgE抗体を産生することが可能である。ヒト抗体を産生するためのこの技術の概説には、LonbergおよびHuszar, 1995, Int. Rev. Immunol. 13:65−93を参照されたい。ヒト抗体およびヒト・モノクローナル抗体を産生するためのこの技術の詳細な考察、およびこうした抗体を産生するためのプロトコルには、例えば米国特許第5,625,126号;米国特許第5,633,425号;米国特許第5,569,825号;米国特許第5,661,016号;および米国特許第5,545,806号を参照されたい。さらに、Abgenix, Inc.(カリフォルニア州フリーモント)などの企業は、上述のものと類似の技術を用いて、選択した抗原に対して向けられるヒト抗体を提供するのに従事することも可能である。
【0056】
選択したエピトープを認識する完全ヒト抗体を、「ガイド選択」と称される技術を用いて、生成することも可能である。このアプローチでは、選択した非ヒト・モノクローナル抗体、例えばネズミ抗体を用いて、同一エピトープを認識する完全ヒト抗体の選択をガイドする(Jespersら, 1994, Bio/technology 12:899−903)。
【0057】
当該技術分野に知られる技術によって、複合体のイディオタイプを含有する抗体断片を生成することも可能である。例えば、こうした断片には、限定されるわけではないが、抗体分子のペプシン消化によって産生可能なF(ab’)2断片;F(ab’)2断片のジスルフィド架橋を還元することによって生成可能なFab’断片;パパインおよび還元剤で抗体分子を処理することによって生成可能なFab断片;およびFv断片が含まれる。
【0058】
抗体産生において、望ましい抗体に関するスクリーニングは、当該技術分野に知られる技術、例えばELISA(酵素連結免疫吸着アッセイ)によって達成可能である。複合体の特定のドメイン、またはその誘導体に特異的な抗体を選択するため、こうしたドメインを含有する複合体の断片、またはその誘導体に結合する産物に関して、生成されたハイブリドーマをアッセイすることも可能である。本発明の複合体、あるいはその誘導体または相同体に特異的に結合するが、複合体、あるいはその誘導体または相同体の個々のタンパク質に特異的に結合しない、抗体を選択するため、複合体に対する陽性結合および個々のタンパク質構成要素に対する結合の欠如に基づいて、選択することも可能である。
【0059】
既定の単数または複数のタンパク質の局在および/または定量に関連する、当該技術分野に知られる方法、例えばこれらのタンパク質を画像化するための方法、適切な生理学的試料中のそのレベルを測定する方法(イムノアッセイによる)、診断法などで、前述の抗体を使用することも可能である。これはまた、複合体の誘導体または相同体に関してもあてはまる。
【0060】
好ましい態様において、GPR49阻害剤は、配列:AACAGCAGTATGGACGACCTTを持つsiRNAである。
GPR49は、LH(黄体形成ホルモン)、リラキシン、IGF、FSH(卵胞刺激ホルモン)、およびTSHの受容体に類似性を示す。したがって、LH、リラキシン、IGF、FSH、およびTSHは、GPR49の生理学的リガンドでありうる。さらに、
WO−03 020 726における
【0061】
【化1】

【0062】
そしてWO−00 187 287における
【0063】
【化2】
【0064】
のLHおよびFSH受容体のアゴニストと同定されたチエノ[2,3−d]ピリミジンが、GPR49のアゴニストである可能性もある。
上に論じるように、GPR49は、ガンマ・セクレターゼ活性および/またはベータ・セクレターゼ活性の制御に関与するタンパク質複合体の一部である。したがって、好ましい態様において、GPR49相互作用分子または阻害剤は、タンパク質複合体、好ましくはAph1a複合体、Fe65L2複合体、APP−C99複合体、またはBACE1複合体の一部であるGPR49分子に作用する。
【0065】
前記タンパク質複合体は、ガンマ−セクレターゼ・サブユニットAph1a、ガンマ−セクレターゼ基質APP−C99(これに加えてそのアダプターFe65L2)およびベータ−セクレターゼ・タンパク質と相互作用するタンパク質の集合と同定されてきている。
【0066】
プレセニリン1および2(それぞれ、Psen1およびPsen2、PS1およびPS2とも称される)は、小胞体、ゴルジ体、および細胞表面にもまた局在する、内在性膜タンパク質である(Kovacs, Nat Med 2. 224)。これらは主に、NTFおよびCTF細胞内タンパク質分解断片のヘテロ二量体として見出される。プレセニリンを切断するプロテアーゼ(「プレセニリナーゼ」)は未知であり、プロセスが自己触媒性である可能性があり、またPS(自己)タンパク質分解の機能的な重要性は不明である。プレセニリンは、アミロイド前駆タンパク質(APP)(De Strooperら, Nature 391, 387)およびNotch受容体(De Strooperら, Nature 398, 518)のタンパク質分解プロセシングに関与する。さらに、プレセニリンは、細胞接着タンパク質、アルファおよびベータ−カテニン、N−カドヘリン、ならびにE−カドヘリン(Georgakopoulosら, Mol Cell 4, 893)およびアルマジロ・ファミリーの他のメンバー(Yuら, J Biol Chem 273, 16470)と会合する。プレセニリンによるAPPプロセシングは、APPを切断し、A−ベータ・ペプチドのC末端を生成する、ガンマ−セクレターゼに対する影響を通じる。PS1は、APPのC83およびC99プロセシングC末端断片(Xiaら, Proc Natl Acad Sci USA, 94, 8208)、ニカストリン(Yuら, Nature 407, 48)およびPen−2(Francisら, Dev Cell 3, 85)と会合する。
【0067】
プレセニリン・プロセシングには、Aph−1aなどのAph−1タンパク質(Francisら, Dev Cell 3, 85)が必要である。プレセニリンが、ガンマ−セクレターゼ活性を直接制御するかどうか、またはこれらがそれ自体、プロテアーゼ酵素であるのかどうかは明らかでない(KopanおよびGouate, Genes Dev 14, 2799)。ガンマ・セクレターゼ活性は、これらのタンパク質の多量体複合体を含むことも可能である(Yuら, Nature 407, 48)が、これらのタンパク質間の関係がどのようにセクレターゼ活性に影響を及ぼすかは知られていない。
【0068】
Aph−1およびPen−2は、線虫におけるプレセニリン増進剤(「pen」)に関するスクリーニングにおいて、最近クローニングされ、そしてAph−2(ニカストリン)と遺伝子相互作用することが示された。Aph−1の欠損は、Notchシグナル伝達およびニカストリン局在に影響を及ぼす。Aph−1およびPen−2は、Notch切断、ガンマ−セクレターゼ活性、およびプロセシングされたプレセニリンの集積に必要である。Francisらは、これらの遺伝子、Aph−1a、Aph−1bおよびPen−2の推定上のヒト・オルソログをクローニングし、そして最近、Leeらもまた、ヒトAph−1 cDNAをクローニングした。ガンマ−セクレターゼ複合体の正確な構成要素は知られていないが、これらの2つの新規タンパク質は、複合体の構成要素または付属因子である可能性もあり、そして一緒に、プレセニリンと、またはプレセニリン/ニカストリン複合体と直接相互作用することも可能である。したがって、ニカストリンは、活性ガンマ−セクレターゼ複合体のメンバーであり、そしてこの複合体に重要なのは、該タンパク質の完全グリコシル化型であるという最近の証拠がある。
【0069】
Fe65様2(Fe65L2)は、よく性質決定された細胞内APP相互作用アダプタータンパク質Fe65の相同体である(Duilio A, Faraonio R, Minopoli G, Zambrano N, Russo T(1998)Fe65L2: a new member of the Fe65 protein family interacting with the intracellular domain of the Alzheimer’s beta−amyloid precursor protein. Biochem J. 330(Pt 1):513−9)。該タンパク質は、脳で高発現され、そしてC末端PTBドメインを通じてAPPと相互作用する(Tanahashi H, Tabira T(1999)Molecular cloning of human Fe65L2 and its interaction with the Alzheimer’s beta−amyloid precursor protein. Neurosci Lett. 261(3):143−6)。Fe65L2は、核に転位置して、そして転写を制御することが知られており(Bruni P, Minopoli G, Brancaccio T, Napolitano M, Faraonio R, Zambrano N, Hansen U, Russo T(2002)Fe65, a ligand of the Alzheimer’s beta−amyloid precursor protein, blocks cell cycle progression by down−regulating thymidylate synthase expression. J. Biol. Chem. 277(38):35481−8)、該タンパク質が、APPの下流シグナル伝達機能に関与することが示唆される。in vitroでFe65L2を過剰発現すると、Aベータの分泌/生成が促進される(Tanahashi H, Tabira T(2002)Characterization of an amyloid precursor protein−binding protein Fe65L2 and its novel isoforms lacking phosphotyrosine−interaction domains. Biochem J. 367(Pt 3):687−95)。
【0070】
ベータ−セクレターゼ(BACE)活性は、細胞外ドメインでAPPを切断し、分泌可溶性APPbの分断を生じ、そして99残基のC末端膜貫通断片(APP−C99)を生じる。Vassarら(Science 286, 735−741)は、APPのベータ−セクレターゼと推測される特性を有する膜貫通アスパラギン酸プロテアーゼをクローニングし、該プロテアーゼをBACE1と名づけた。BACE1ノックアウトマウス由来の脳および初代皮質培養物は、検出可能なベータ−セクレターゼ活性を示さず、そしてBACEノックアウトマウス由来の初代皮質培養物は、APPからはるかに少ないアミロイド−ベータしか産生しなかった。これによって、BACE1のパラログであるBACE2ではなく、BACE1がAPPの主なベータ−セクレターゼであることが示唆される。BACE1は、501アミノ酸のタンパク質であり、21aaのシグナルペプチドに続いて、aa22〜45に渡る、プロタンパク質ドメインを含有する。選択的スプライシング型、BACE−I−457およびBACE−I−476がある。成熟タンパク質の管腔ドメインに続いて、予測される1つの膜貫通ドメインおよび24aaの短い細胞質ゾルC末端テールがある。BACE1は、1型膜貫通タンパク質であり、活性部位が膜の管腔側にあると予測され、ここでベータ−セクレターゼがAPP、およびありうる他の未同定の基質を切断する。BACE1は、明らかに、APPをA−ベータにプロセシングするのに必要とされる重要な酵素であるが、最近の証拠は、BACE1のさらなる潜在的基質および機能を示唆している(J. Biol. Chem. 279, 10542−10550)。現在まで、制御機能または調節機能を持つBACE1相互作用タンパク質は、まったく記載されてきていない。
【0071】
上に説明するように、驚くべきことに、本発明の関連において、GPR49が、特にベータ−セクレターゼおよび/またはガンマ−セクレターゼ活性によって、APPのタンパク質分解プロセシングを制御するタンパク質複合体の一部であることが発見された。したがって、好ましい態様において、阻害剤または相互作用分子は、ベータ−セクレターゼおよび/またはガンマ・セクレターゼの活性を調節する。
【0072】
本発明全体で、用語「ガンマ・セクレターゼおよび/またはベータ・セクレターゼの活性を調節する」は、酵素活性が直接または間接的に調節されることを含む。これは、GPR49調節剤がこれらの酵素のいずれかに直接結合可能であるか、あるいはより好ましくは、GPR49に影響を発揮して、次に例えばタンパク質−タンパク質相互作用によってまたはシグナル伝達によって、または小さい代謝産物を介して、これらの酵素のいずれかの活性を調節可能であることを意味する。
【0073】
本発明全体で、ベータ・セクレターゼ調節剤が、完全にまたは部分的に、ベータ・セクレターゼの活性を阻害することが好ましい。本発明全体で、GPR49調節剤の最も好ましい機能的結果は、Aベータ−42生成の減少である。
【0074】
本発明の関連において、「ガンマ・セクレターゼおよび/またはベータ・セクレターゼの活性を調節する」は、活性が減少して、より少ない産物が形成されるかまたは産物がまったく形成されず、最も好ましくはより少ないAベータ−42が形成されるかまたはAベータ−42がまったく形成されない(部分的または完全阻害)か、あるいはそれぞれの酵素が異なる産物(ガンマ−セクレターゼの場合、Aベータ−42の代わりにより短いアミノ酸配列の、例えばAベータ−38または他のAベータ・ペプチド)を産生するか、あるいは産物の相対量が異なる(ガンマ・セクレターゼの場合、例えばAベータ40対Aベータ−42の比が変化する、好ましくは増加する)ことを意味する。さらに、調節剤が、ガンマ・セクレターゼまたはベータ・セクレターゼいずれか、あるいは両酵素の活性を調節することが含まれる。
【0075】
ガンマ・セクレターゼ活性の調節剤に関しては、この調節剤が、ガンマ・セクレターゼ活性を阻害することが好ましい。しかし、Aベータ・ペプチド種の総量が変化しないが、Aベータ−42の代わりに、より多くのAベータ−38が産生される方式で、ガンマ・セクレターゼの活性がシフトすることもまた、好ましい。
【0076】
例えばAPPプロセシングを測定することによって、例えば、産生されるAベータ・ペプチド種のレベル、最も重要なことに、Aベータ−42のレベルを測定することによって、ガンマ・セクレターゼ活性を測定することも可能である(以下の実施例セクションを参照されたい)。
【0077】
BACE1活性を測定するため、ウェスタンブロッティングによって、アルファ−およびベータ−C末端APP断片間の比の変化を分析することも可能であり(Blaskoら, J Neural Transm 111, 523);BACE1活性アッセイのさらなる例には、限定されるわけではないが:ベータ−ガラクトシダーゼ・レポーター活性を再構成し、そして測定する、BACE1切断部位を含有する環状化酵素ドナー・ペプチドの使用(Naqviら, J Biomol Screen. 9, 398);消光蛍光ペプチド基質および蛍光測定の使用(Andrauら, J. Biol Chem 278, 25859);酵素(アルカリホスファターゼなど)を、BACE1認識配列を含有するアミノ酸ストレッチを介して、ゴルジ体常在タンパク質に連結する、組換えキメラタンパク質を利用する、細胞に基づくアッセイの使用(Ohら, Anal Biochem, 323, 7);蛍光共鳴エネルギー移動(FRET)に基づくアッセイ(Kennedyら, Anal Biochen 319, 49);酵母における細胞増殖選択系(Luthiら, Biochim Biophys Acta 1620, 167)が含まれる。
【0078】
好ましくは、神経変性疾患は、アルツハイマー病である。
本発明にしたがって、GPR49相互作用分子を用いて、薬剤組成物を調製する。
したがって、本発明は、有効量で被験者に投与可能な薬剤組成物を提供する。好ましい側面において、療法剤は、実質的に精製されている。被験者は、好ましくは、限定されるわけではないが、ウシ、ブタ、ウマ、ニワトリ、ネコ、イヌなどを含む動物であり、そして好ましくは哺乳動物であり、そして最も好ましくはヒトである。特定の態様において、非ヒト哺乳動物が被験者である。
【0079】
多様な搬送系が知られ、そしてこうした系を用いて、本発明の療法剤を投与することも可能であり、こうした系には、例えばリポソーム、微小粒子、および微小カプセル中の被包:療法剤を発現可能な組換え細胞の使用、受容体が仲介するエンドサイトーシスの使用(例えば、WuおよびWu, 1987, J. Biol. Chem. 262:4429−4432);レトロウイルスベクターまたは他のベクターの一部としての療法核酸の構築などがある。導入法には、限定されるわけではないが、皮内、筋内、腹腔内、静脈内、皮下、鼻内、硬膜外、および経口経路が含まれる。好適な経路いずれによって、例えば注入によって、ボーラス(bolus)注射によって、上皮または皮膚粘膜裏打ち(例えば口腔、直腸および腸粘膜など)を通じた吸収によって、化合物を投与することも可能であるし、そして他の生物学的活性剤と一緒に投与することも可能である。投与は全身性または局所であることも可能である。さらに、脳室内注射およびクモ膜下注射を含む、適切な経路いずれかによって、本発明の薬剤組成物を中枢神経系に導入することが望ましい可能性もあり;脳室内注射は、例えばOmmaya容器などの容器に取り付けた、脳室内カテーテルによって容易にすることも可能である。例えば吸入器または噴霧器を使用し、そしてエアロゾル化剤と配合することによって、肺投与もまた使用可能である。
【0080】
特定の態様において、治療の必要がある領域に、本発明の薬剤組成物を局所投与することが望ましい可能性もある。これは、例えば、そして限定なしに、手術中の局所注入によって、例えば手術後の創傷手当て(dressing)と組み合わせた局所適用によって、注射によって、カテーテルによって、座薬によって、あるいはシアラスティック(sialastic)膜などの膜またはファイバーを含む、多孔、無孔、またはゼラチン性物質である移植物によって、達成可能である。1つの態様において、悪性腫瘍または新生物組織もしくは新生物発生前組織の部位(または以前の部位)に直接注射することによって、投与することも可能である。
【0081】
別の態様において、療法剤を、小胞、特にリポソーム中で搬送することも可能である(Langer, 1990, Science 249:1527−1533;Treatら, 1989, :Liposomes in the Therapy of Infectious Disease and Cancer中, Lopez−BeresteinおよびFidler監修, Liss, ニューヨーク, pp.353−365;Lopez−Berestein, 同書, pp.317−327;一般的には同書を参照されたい)。
【0082】
さらに別の態様において、徐放系を介して、療法剤を搬送することも可能である。1つの態様において、ポンプを使用することも可能である(Langer、上記;Sefton, 1987, CRC Crit. Ref. Biomed. Eng. 14:201−240;Buchwaldら, 1980, Surgery 88:507−516;Saudekら, 1989, N. Engl. J. Med. 321:574−579)。別の態様において、ポリマー物質を用いることも可能である(Medical Applications of Controlled Release, LangerおよびWise監修, CRC Press, フロリダ州ボーカラートン, 1974;Controlled Drug Bioavailability, Drug Product Design and Performance, SmolenおよびBall監修, Wiley, ニューヨーク, 1984;RangerおよびPeppas, 1983, Macromol. Sci. Rev. Macromol. Chem. 23:61;Levyら, 1985, Science 228:190−192;Duringら, 1989, Ann. Neurol. 25:351−356;Howardら, 1989, J. Neurosurg. 71:858−863)。さらに別の態様において、療法ターゲット、すなわち脳に近接して、徐放系を配置し、こうして全身用量のうち、ごく少量しか必要としないことも可能である(例えばGoodson, 1984, :Medical Applications of Controlled Release中, 上記, Vol.2, pp.115−138)。他の徐放系が、Langer(1990, Science 249:1527−1533)による概説に論じられている。
【0083】
療法剤が、好ましくはタンパク質療法剤をコードする核酸である、特定の態様において、適切な核酸発現ベクターの一部として該核酸を構築し、そして細胞内に存在するように投与することによって、核酸をin vivoで投与して、コードされるタンパク質の発現を促進することも可能であり、これは、例えばレトロウイルスベクターの使用によって(米国特許第4,980,286号)、または直接注射によって、または微小粒子銃(例えば遺伝子銃;Biolistic、Dupont)の使用によって、または核酸を脂質、細胞表面受容体もしくはトランスフェクション剤でコーティングすることによって、または核に進入することが知られるホメオボックス様ペプチド(例えばJoliotら, 1991, Proc. Natl. Acad. Sci. USA 88:1864−1868)に連結した核酸を投与することによって、実行可能である。あるいは、核酸療法剤を細胞内に導入し、そして発現のため、宿主細胞DNA内に相同組換えによって取り込ませることも可能である。
【0084】
一般的に、本発明の薬剤組成物は、療法的有効量の療法剤、および薬学的に許容しうるキャリアーを含む。特定の態様において、用語「薬学的に許容しうる」は、動物、そしてより詳細にはヒトにおける使用のため、連邦政府または州政府の監督官庁に認可されたか、あるいは米国薬局方または他の一般的に認められる薬局方に列挙されていることを意味する。用語「キャリアー」は、療法剤を一緒に投与する、希釈剤、アジュバント、賦形剤、またはビヒクルを指す。こうした薬剤キャリアーは、無菌液体、例えば水および油であることも可能であり、石油、動物、植物または合成起源のものが含まれ、限定されるわけではないが、ピーナツ油、ダイズ油、ミネラルオイル、ゴマ油等が含まれる。薬剤組成物を経口投与する場合は、水が好ましいキャリアーである。薬剤組成物を静脈内投与する場合は、生理食塩水および水性デキストロースが好ましいキャリアーである。好ましくは、生理食塩水溶液、並びに水性デキストロースおよびグリセロール溶液が、注射可能溶液の液体キャリアーとして使用される。適切な薬剤賦形剤には、デンプン、グルコース、ラクトース、スクロース、ゼラチン、モルト、米、小麦粉、チョーク、シリカゲル、ステアリン酸ナトリウム、モノステアリン酸グリセロール、タルク、塩化ナトリウム、脱脂粉乳、グリセロール、プロピレン、グリコール、水、エタノール等が含まれる。組成物は、望ましい場合、少量の湿潤剤または乳化剤、あるいはpH緩衝剤もまた含有することも可能である。これらの組成物は、溶液、懸濁物、エマルジョン、錠剤、ピル、カプセル、粉末、持続放出配合物等の形を取ることも可能である。伝統的な結合剤およびキャリアー、例えばトリグリセリドを用いて、組成物を座薬として配合することも可能である。経口配合物は、薬剤等級のマンニトール、ラクトース、デンプン、ステアリン酸マグネシウム、サッカリン・ナトリウム、セルロース、炭酸マグネシウムなどの標準的キャリアーを含むことも可能である。適切な薬剤キャリアーの例は、E.W. Martinによる、“Remington’s Pharmaceutical Sciences”に記載される。こうした組成物は、患者に適切に投与する形を提供するように、適切な量のキャリアーと一緒に、療法的有効量の療法剤、好ましくは精製型のものを含有するであろう。配合物は、投与様式に適していなければならない。
【0085】
好ましい態様において、日常的な方法にしたがって、ヒトへの静脈内投与に適応させた薬剤組成物として、組成物を配合する。典型的には、静脈内投与のための組成物は、無菌等張水性緩衝剤中の溶液である。必要な場合、組成物はまた、可溶化剤および注射部位での疼痛を和らげるリドカインなどの局所麻酔剤も含むことも可能である。一般的に、成分を別個に供給するか、または単位投薬型中で一緒に混合して供給し、例えば活性剤の量を示すアンプルまたはサシェ(sachette)などの密封容器中、凍結乾燥粉末または水不含濃縮物として供給する。組成物を注入によって投与しようとする場合、無菌薬剤等級の水または生理食塩水を含有する注入ビンを用いて、分配することも可能である。組成物を注射によって投与しようとする場合、投与前に、成分を混合可能であるように、注射用の無菌水または生理食塩水のアンプルを提供することも可能である。
【0086】
本発明の療法剤を中性型または塩型で配合することも可能である。薬学的に許容しうる塩には、塩酸、リン酸、酢酸、シュウ酸、酒石酸などに由来する遊離型のカルボキシル基とともに形成されるもの、イソプロピルアミン、トリエチルアミン、2−エチルアミノエタノール、ヒスチジン、プロカインなどに由来するものなどの遊離型のアミン基とともに形成されるもの、並びにナトリウム、カリウム、アンモニウム、カルシウム、および水酸化第二鉄などに由来するものが含まれる。
【0087】
特定の障害または状態の治療に有効であろう、本発明の療法剤の量は、障害または状態の性質に応じるであろうし、そして標準的臨床技術によって決定可能である。さらに、場合によって、in vitroアッセイを使用して、最適投薬量範囲を同定するのを補助することも可能である。配合物に使用しようとする正確な用量はまた、投与経路、および疾患または障害の重大性にも応じるであろうし、そして担当医の判断および各患者の状況にしたがって、決定すべきである。しかし、静脈内投与に適した投薬量範囲は、一般的に、キログラム体重あたり、活性化合物約20〜500マイクログラムである。鼻内投与に適した投薬量範囲は、一般的に、約0.01pg/kg体重〜1mg/kg体重である。有効用量は、in vitroまたは動物モデル試験系から得られる用量−反応曲線から推定可能である。
【0088】
座薬は、一般的に、重量にして0.5%〜10%の範囲の活性成分を含有し;経口配合物は、好ましくは、10%〜95%の活性成分を含有する。
本発明はまた、本発明の薬剤組成物の1以上の成分が充填された、1以上の容器を含む、薬剤パックまたはキットも提供する。場合によって、こうした容器(単数または複数)に付随して、薬剤または生物学的製品の製造、使用または販売を規制する政府機関によって規定された形で、政府機関による、ヒト投与のための製造、使用または販売の認可を示す情報を示すことも可能である。
【0089】
本発明のキットはまた、複合体機構の本質的な構成要素をコードする発現ベクターも含有することも可能であり、この構成要素は、発現された後、生物学的に活性な複合体を形成するため、再構成されることも可能である。こうしたキットは、好ましくはまた、必要な緩衝剤および試薬も含有する。場合によって、こうした容器(単数または複数)に付随して、キット使用のための説明書、並びに/あるいは薬剤または生物学的製品の製造、使用または販売を規制する政府機関によって規定された形で、政府機関による、ヒト投与のための製造、使用または販売の認可を示す情報を示すことも可能である。
【0090】
本発明はさらに、有効量のGPR49相互作用分子または阻害剤あるいは本発明の薬剤組成物を、神経変性疾患、好ましくはアルツハイマー病を患う被験者に投与する、治療法に関する。
【0091】
本発明のこの方法に関して、上記のすべての態様は、本発明の使用のため、あてはまる。
本発明はさらに、ガンマ−セクレターゼ調節剤および/またはベータ−セクレターゼ調節剤を同定する方法であって:
a.既定の試験化合物がGPR49相互作用分子であるかどうか決定することによって、GPR49相互作用分子を同定し、
b.工程a)のGPR49相互作用分子がガンマ−セクレターゼ活性またはベータ−セクレターゼ活性を調節可能であるかどうか決定する
工程を含む、前記方法に関する。
【0092】
本発明の好ましい態様において、工程a)において、試験化合物をGPR49と接触させ、そして試験化合物とGPR49の相互作用を決定する。好ましくは、候補分子がGPR49に結合するかどうかを測定する。
【0093】
本発明の好ましい態様において、GPR49活性を調節する、好ましくは阻害するかどうかを見出すために、工程a)で同定されたGPR49相互作用分子をまず、上記のようなGPR49活性試験に供し(実施例3も参照されたい)、そして次いで、工程b)に供する(Aベータ低下効果に関する試験)。
【0094】
本発明の方法を、好ましくは、ハイスループットアッセイの関連で行う。こうしたアッセイは、当業者に知られる。
スクリーニングしようとする試験分子または候補分子は、限定された数の明記される化合物の混合物として、または化合物ライブラリー、ペプチド・ライブラリー等として、提供されることも可能である。スクリーニングしようとする剤/分子はまた、複合体活性または形成を調節可能な、抗血清、アンチセンス核酸等のすべての形を含むことも可能である。スクリーニングのための典型的な候補分子およびライブラリーを以下に示す。
【0095】
ライブラリーのスクリーニングは、一般的に知られる多様な方法のいずれによって、達成することも可能である。例えばペプチド・ライブラリーのスクリーニングを開示する、以下の参考文献を参照されたい:ParmleyおよびSmith, 1989, Adv. Exp. Med. Biol. 251:215−218;ScottおよびSmith, 1990, Science 249:386−390;Fowlkesら, 1992, BioTechniques 13:422−427;Oldenburgら, 1992, Proc. Natl. Acad. Sci. USA 89:5393−5397;Yuら, 1994, Cell 76:933−945;Staudtら, 1988, Science 241:577−580;Bockら, 1992, Nature 355:564−566;Tuerkら, 1992, Proc. Natl. Acad. Sci. USA 89:6988−6992;Ellingtonら, 1992, Nature 355:850−852;すべてLadnerらに対する米国特許第5,096,815号、米国特許第5,223,409号、および米国特許第5,198,346号;RebarおよびPabo, 1993, Science 263:671−673;並びに国際特許公報第WO 94/18318号。
【0096】
特定の態様において、固相上に固定したGPR49とライブラリーメンバーを接触させ、そしてタンパク質(またはコード核酸もしくは誘導体)に結合するライブラリーメンバーを採取することによって、スクリーニングを行うことも可能である。こうしたスクリーニング法の例は、「パニング」技術と称され、例えば、ParmleyおよびSmith, 1988, Gene 73:305 318;Fowlkesら, 1992, BioTechniques 13:422−427;国際特許公報第WO 94/18318号;および上述の文献に引用される参考文献に記載される。
【0097】
特定の態様において、GPR49断片および/または類似体、特にペプチド模倣体を、GPR49と別のタンパク質、例えば表1に提供するタンパク質との複合体の形成(複合体の量または複合体の組成)または細胞中のGPR49活性の、細胞における複合体活性または形成を阻害する、競合的または非競合的阻害剤としての活性に関して、スクリーニングする。
【0098】
1つの態様において、試験しようとする剤の非存在下では複合体形成が起こる、水性または生理学的結合条件下で、複合体形成を調節する能力に関して剤をスクリーニングする、結合阻害アッセイを用いて、GPR49活性またはGPR49−タンパク質複合体形成を調節する(すなわちアンタゴナイズするかまたはアゴナイズする)剤をスクリーニングすることも可能である。本発明の複合体形成に干渉する剤を、複合体形成のアンタゴニストとして同定する。複合体形成を促進する剤を、複合体形成のアゴニストとして同定する。複合体形成を完全に遮断する剤を、複合体形成の阻害剤として同定する。
【0099】
スクリーニング法は、放射性リガンド(例えば125Iまたは3H)、磁気リガンド(例えばフォトビオチン・アセテートに共有結合した常磁性ビーズ)、蛍光リガンド(例えばフルオレセインまたはローダミン)、または酵素リガンド(例えばルシフェラーゼまたはβ−ガラクトシダーゼ)で、複合体の構成要素タンパク質を標識することを含むことも可能である。次いで、当該技術分野に知られる多くの技術の1つによって、溶液中で結合する反応剤を単離することも可能であり、こうした技術には、限定されるわけではないが、非標識結合パートナー(または第二の標識複合体部分上で用いるものと区別可能なマーカーで標識された結合パートナー)に対する抗血清を用いた、標識複合体部分の同時免疫沈降、免疫アフィニティー・クロマトグラフィー、サイズ排除クロマトグラフィー、および勾配密度遠心分離が含まれる。好ましい態様において、標識結合パートナーは、商業的に入手可能なフィルターに保持されない小さい断片またはペプチド模倣体である。結合したならば、次いで、標識種は、フィルターを通過不能となり、複合体形成の単純なアッセイが提供される。
【0100】
当該技術分野に一般的に知られる方法を用いて、複合体の構成要素メンバーの少なくとも1つを標識する。適切な標識法には、限定されるわけではないが、放射標識アミノ酸、例えば3H−ロイシンまたは35S−メチオニンの取り込みによる放射標識、クロラミンT法、Bolton−Hunter試薬などを用いた、125Iまたは131Iでの翻訳後ヨウ素化による放射標識、あるいはホスホリラーゼおよび無機放射標識リンを用いた32Pでの標識、フォトビオチン−アセテートでのビオチン標識、および太陽灯曝露などが含まれる。複合体メンバーの1つが、例えば以下に記載するように、固定されている場合、遊離種を標識する。相互作用種のいずれも固定されていない場合、各々を区別可能なマーカーで標識して、その後、両方の部分を単離して、より正確な定量化を提供し、そしてヘテロマー複合体からホモマー複合体の形成を区別可能であるようにすることも可能である。修飾相互作用因子の1つに結合する付属タンパク質を利用して、検出感度を改善し、複合体の安定性を増加させるなどの方法を提供する。
【0101】
典型的な結合条件は、限定ではないが、例えば、10〜250mM NaCl、5〜50mM Tris−HCl、pH5〜8、および0.5% Triton X−100または相互作用の特異性を改善する他の界面活性剤の水性塩溶液中である。金属キレート剤および/または二価陽イオンを添加して、結合を改善し、そして/またはタンパク質分解を減少させることも可能である。反応温度は、摂氏4、10、15、22、25、35、または42度を含むことも可能であり、そしてインキュベーション時間は、典型的には少なくとも15秒間であるが、結合平衡が起こることを可能にするには、より長い時間が好ましい。日常的なタンパク質結合アッセイを用いて、特定の複合体をアッセイして、再現可能な結合に最適な結合条件を決定することも可能である。
【0102】
用いる標識に特異的なアッセイ法、例えば放射能検出のための液体シンチレーション計測、酵素標識部分のための酵素活性などを用いた、複合体形成の定量化によって、複合体形成の物理的パラメーターを分析することも可能である。次いで、Scatchard分析、Hill分析、および当該技術分野に一般的に知られる他の方法(例えば、Proteins, Structures, and Molecular Principles, 第2版(1993)Creighton監修, W.H. Freeman and Company, ニューヨークを参照されたい)を利用して、反応結果を分析する。
【0103】
結合アッセイに対する第二の一般的なアプローチにおいて、結合種の1つをフィルター上、マイクロタイタープレートウェル中、試験管中、クロマトグラフィーマトリックスなどに、共有的または非共有的に固定する。当該技術分野に周知の方法いずれか、例えば限定されるわけではないが、KadonagaおよびTjian, 1986, Proc. Natl. Acad. Sci. USA 83:5889−5893の方法、すなわちCNBr−Sepharose 4B(Pharmacia)などの臭化シアン誘導体化支持体への連結を用いて、タンパク質を共有的に固定することも可能である。必要な場合、スペーサーを使用すると、支持体による立体障害を減少させることも可能である。支持体へのタンパク質の非共有結合には、限定されるわけではないが、荷電表面へのタンパク質の付着、特異的抗体との結合、関連しない第三の相互作用タンパク質への結合などが含まれる。
【0104】
いずれかの手段(例えば上述の手段)によって標識した複合体の別のメンバーと、複合体の1つのメンバー(またはその誘導体)の結合に関して競合する剤(細胞抽出物またはライブラリー・プールを含む)のアッセイを提供して、複合体形成の競合剤または増進剤に関してスクリーニングする。
【0105】
特定の態様において、他のタンパク質構成要素への試薬の非特異的結合、またはプラスチック、固定マトリックス等に試薬が吸収される損失を阻害する、ブロッキング剤をアッセイ混合物に含む。ブロッキング剤には、限定されるわけではないが、ウシ血清アルブミン、カゼイン、脱脂粉乳、デンハルト試薬、Ficoll、ポリビニルピロリジン、非イオン性界面活性剤(NP40、Triton X−100、Tween20、Tween80など)、イオン性界面活性剤(例えばSDS、LDSなど)、ポリエチレングリコールなどが含まれる。適切なブロッキング剤濃度によって、複合体形成が可能である。
【0106】
結合を行った後、上清中の未結合標識タンパク質を取り除き、そして結合した標識タンパク質いずれかを保持する固定タンパク質を徹底的に洗浄する。次いで、上述のように標識を検出する当該技術分野の標準法を用いて、結合標識の量を定量化する。
【0107】
別の特定の態様において、本明細書に提供するようなタンパク質複合体/タンパク質および/または抗体を固体キャリアーに付着させることによって、本明細書に提供するようなタンパク質複合体/タンパク質の調節剤に関するスクリーニングを行うことも可能である。
【0108】
異なる種類のタンパク質(抗体を含む)を含有するアレイの調製が当該技術分野に周知であり、そして当業者に明らかである(例えば、Ekinsら, 1989, J. Pharm. Biomed. Anal. 7:155−168;Mitchellら 2002, Nature Biotechnol. 20:225−229;Petricoinら, 2002, Lancet 359:572−577;Templinら, 2001, Trends Biotechnol. 20:160−166;WilsonおよびNock, 2001, Curr. Opin. Chem. Biol. 6:81−85;Leeら, 2002 Science 295:1702−1705;MacBeathおよびSchreiber, 2000, Science 289:1760;BlawasおよびReichert, 1998, Biomaterials 19:595;Kaneら, 1999, Biomaterials 20:2363;Chenら, 1997, Science 276:1425;Vaughamら, 1996, Nature Biotechnol. 14:309−314;Mahlerら, 1997, Immunotechnology 3:31−43;Robertsら, 1999, Curr. Opin. Chem. Biol. 3:268−273;Nordら, 1997, Nature Biotechnol. 15:772−777;Nordら, 2001, Eur. J. Biochem. 268:4269−4277;BrodyおよびGold, 2000, Rev. Mol. Biotechnol. 74:5−13;KarlstroemおよびNygren, 2001, Anal. Biochem. 295:22−30;Nelsonら, 2000, Electrophoresis 21:1155−1163;Honoreら, 2001, Expert Rev. Mol. Diagn. 3:265−274;Albala, 2001, Expert Rev. Mol. Diagn. 2:145−152, FigeysおよびPinto, 2001, Electrophoresis 2:208−216、並びにここに列挙する刊行物中の参考文献を参照されたい)。
【0109】
タンパク質またはタンパク質複合体を、当業者に明らかであろうような異なる手段によって、アレイに付着させることも可能である。精製工程後、または当業者に明らかであろうような別の適切な精製スキームによって、例えばTAP−タグ(WO/0009716およびRigautら, 1999, Nature Biotechnol. 10:1030−1032に記載されるようなもの)を介して、複合体をアレイに付加させることも可能である。
【0110】
場合によって、複合体のタンパク質を架橋して、複合体の安定性を増進させることも可能である。タンパク質を架橋する、異なる方法が、当該技術分野で周知である。架橋剤の反応性末端基には、限定されるわけではないが、−COOH、−SH、−NH2またはN−オキシ−スクシンアマートが含まれる。
【0111】
架橋剤のスペーサーは、架橋しようとする複合体のサイズに対して選択すべきである。いくつかのタンパク質しか含まない、小さいタンパク質複合体に関しては、反応混合物中、別個の複合体を架橋する可能性を減少させるため、比較的短いスペーサーが好ましい。より大きいタンパク質複合体に関しては、複合体内のタンパク質間の架橋を容易にするため、より大きいスペーサーのさらなる使用が好ましい。
【0112】
複合体をキャリアーに連結する前に、架橋の成功率をチェックすることが好ましい。
当業者に明らかであろうように、架橋の最適な率を、ケースバイケースで決定する必要がある。当該技術分野に周知の方法によって、これを達成することも可能であり、これらの方法のいくつかを以下に例示的に記載する。
【0113】
例えば、変性タンパク質ゲル上で、架橋されない複合体に対して、架橋された複合体を分析することによって、架橋が十分な率であることをチェックすることも可能である。
架橋が成功裡に行われたならば、複合体のタンパク質は、同一レーンに見られると予期され、一方、架橋されない複合体のタンパク質は、個々の特性にしたがって分離されると予期される。場合によって、質量分析および/またはエドマン分解などの当該技術分野に周知の方法を用いて、それぞれのバンド中のタンパク質をペプチド配列決定することによって、複合体のすべてのタンパク質の存在をさらにチェックすることも可能である。
【0114】
さらに、高すぎる架橋率もまた、回避しなければならない。架橋があまりにも大規模に起こった場合、個々のタンパク質複合体の架橋量が増加し、これが、アレイを用いた、潜在的な結合パートナーおよび/または調節剤等に関するスクリーニングと干渉する可能性があるであろう。
【0115】
当該技術分野に周知の方法によって、こうした構造の存在を決定することも可能であり、そしてこうした方法には、例えば、架橋されない複合体に対して、架橋された複合体を含有する溶液のゲルろ過プロフィールを比較する、ゲルろ過実験が含まれる。
【0116】
場合によって、いくつかが本明細書に例示的に提供される、当業者に明らかであろうような機能アッセイを行って、複合体の完全性をチェックすることも可能である。
あるいは、単数または複数のタンパク質を単一の融合タンパク質として発現させ、そして当業者に明らかであろうように、マトリックスにカップリングすることも可能である。
【0117】
場合によって、当業者に明らかな多様な方法によって、上に概略するような複合体またはタンパク質または抗体の付着をさらに監視することも可能である。これらには、限定されるわけではないが、表面プラズモン共鳴が含まれる(例えば、McDonnel, 2001, Curr. Opin. Chem. Biol. 5:572−577;Lee, 2001, Trends Biotechnol. 19:217−222;Weinbergerら, 2000, 1:395−416;Pearsonら, 2000, Ann. Clin. Biochem. 37:119−145;Velyら, 2000, Methods Mol. Biol. 121:313−321;Slepak, 2000, J. Mol Recognit. 13:20−26を参照されたい)。
【0118】
Aベータ−40およびAベータ−42ペプチドの産生を、ELISAによって測定するのに有用な典型的なアッセイには、限定されるわけではないが、Vassar Rら, 1999, Science, 286:735−41に記載されるものが含まれる。
【0119】
細胞株またはトランスジェニック動物におけるC末端APP断片の産生を、ウェスタンブロットによって測定するのに有用な典型的なアッセイには、限定されるわけではないが、Yan Rら, 1999, Nature, 402:533−7に記載されるものが含まれる。
【0120】
BACE1複合体の、細菌で発現されたAPP断片に対するベータ−セクレターゼまたはガンマ・セクレターゼのタンパク質分解活性を、in vitroで(例えばRNAi(siRNA)および/または相互作用タンパク質(単数または複数)をコードするプラスミドにより細胞中の1つまたはいくつかの相互作用タンパク質の発現を修飾することによって)測定するのに有用な典型的なアッセイには、限定されるわけではないが、Tian Gら, 2002, J Biol Chem, 277:31499−505に記載されるものが含まれる。
【0121】
BACE1複合体の、Gal4駆動レポーター遺伝子のトランス活性化を(例えばRNAi(siRNA)および/または相互作用タンパク質(単数または複数)をコードするプラスミドにより細胞中の1つまたはいくつかの相互作用タンパク質の発現を修飾することによって)測定するのに有用な典型的なアッセイには、限定されるわけではないが、Cao Xら, 2001, Science, 293:115−20に記載されるものが含まれる。
【0122】
本発明記載の相互作用分子または阻害剤である能力に関して、当該技術分野に知られるいかなる分子を試験することも可能である。GPR49複合体機構を発現する細胞に、候補分子を直接提供することも可能であるし、または候補タンパク質の場合、核酸が組換え的に発現されて候補タンパク質を産生する条件下で、コード核酸を提供することによって、候補タンパク質を提供することも可能である。
【0123】
本発明の方法は、複合体の量、活性、またはタンパク質構成要素組成を調節する分子、例えば阻害するか、アンタゴナイズするか、またはアゴナイズする分子に関して、化学的ライブラリーをスクリーニングするのによく適している。化学的ライブラリーは、ペプチド・ライブラリー、ペプチド模倣体ライブラリー、化学的合成ライブラリー、組換えライブラリー、例えばファージディスプレイ・ライブラリー、およびin vitro翻訳に基づくライブラリー、他の非ペプチド合成有機ライブラリーなどであることも可能である。
【0124】
典型的なライブラリーは、いくつかの供給源(ArQule、Tripos/PanLabs、ChemDesign、Pharmacopoeia)から商業的に入手可能である。いくつかの場合、コンビナトリアル戦略を用いて、メンバー化合物が付着した支持体上にライブラリーの各メンバーの同一性をコードし、こうして有効な調節剤である分子を直接、そして直ちに同定することを可能にする、これらの化学的ライブラリーを生成する。したがって、多くのコンビナトリアル・アプローチにおいて、化合物プレート上の位置は、その化合物の組成を特定する。また、1つの例において、単一プレート位置に、1〜20の化学薬品を有することも可能であり、この化学薬品を、目的の相互作用を含有するウェルに投与することによってスクリーニングすることも可能である。したがって調節が検出されたならば、調節活性に関して、相互作用対の徐々に小さいプールをアッセイすることも可能である。こうした方法によって、多くの候補分子をスクリーニングすることも可能である。
【0125】
使用に適した多くの多様性ライブラリーが当該技術分野に知られ、そしてこれを用いて、本発明にしたがって試験しようとする化合物を提供することも可能である。あるいは、標準法を用いて、ライブラリーを構築することも可能である。化学的(合成)ライブラリー、組換え発現ライブラリー、またはポリソームに基づくライブラリーが、使用可能なライブラリーの典型的な種類である。
【0126】
ライブラリーは、拘束性または半硬直性(semirigid)(ある程度の度合いの構造硬直性を有するもの)であることも可能であるし、あるいは直鎖または非拘束性であることも可能である。ライブラリーは、cDNAまたはゲノム発現ライブラリー、ランダムペプチド発現ライブラリーまたは化学的合成ランダムペプチド・ライブラリー、または非ペプチド・ライブラリーであることも可能である。発現ライブラリーを細胞に導入し、該細胞中でライブラリーの核酸を発現させて、コードされるタンパク質を産生し、アッセイを行う。
【0127】
1つの態様において、本発明で使用可能なペプチド・ライブラリーは、in vitroで化学的に合成されるライブラリーであることも可能である。こうしたライブラリーの例が、各ペプチドの第一の残基および第二の残基が個々にそして具体的に定義される遊離ヘキサペプチドの混合物を記載する、Houghtenら, 1991, Nature 354:84−86;コレクション中の各ビーズが該ビーズ上にアミノ酸残基の単一ランダム配列を固定するペプチド・ライブラリーを、固相スプリット合成スキームによって産生する、「1ビーズ、1ペプチド」アプローチを記載する、Lamら, 1991, Nature 354:82−84;スプリット合成およびT−バッグ合成法を記載する、Medynski, 1994, Bio/Technology 12:709−710;およびGallopら, 1994, J. Med. Chem. 37:1233−1251に提供される。単に他の例として、Ohlmeyerら, 1993, Proc. Natl. Acad. Sci. USA 90:10922−10926;Erbら, 1994, Proc. Natl. Acad. Sci. USA 91:11422−11426;Houghtenら, 1992, Biotechniques 13:412;Jayawickremeら, 1994, Proc. Natl. Acad. Sci. USA 91:1614−1618;またはSalmonら, 1993, Proc. Natl. Acad. Sci. USA 90:11708−11712の方法にしたがって、使用のためのコンビナトリアルライブラリーを調製することも可能である。PCT公報第WO 93/20242号、並びにBrennerおよびLerner, 1992, Proc. Natl. Acad. Sci. USA 89:5381−5383は、各化学ポリマーライブラリーメンバーに関してオリゴヌクレオチド識別子を含有する、「コードされるコンビナトリアル化学ライブラリー」を記載する。
【0128】
好ましい態様において、スクリーニングされるライブラリーは、ランダムペプチドが(例えばジスルフィド結合を有することによって)拘束されているランダムペプチド・ファージディスプレイ・ライブラリーである、生物学的発現ライブラリーである。
【0129】
さらに、より一般的には、構造的に拘束された有機多様性(例えば非ペプチド)ライブラリーもまた、使用可能である。例えば、ベンゾジアゼピン・ライブラリー(例えばBuninら, 1994, Proc. Natl. Acad. Sci. USA 91:4708−4712を参照されたい)を使用することも可能である。
【0130】
使用可能な、コンホメーション的に拘束されたライブラリーには、限定されるわけではないが、酸化環境中でジスルフィド結合によって架橋されて、シスチンを形成する、不変システイン残基、修飾されたペプチド(例えばフッ素、金属、同位体標識を取り込む、リン酸化されているなど)、1以上の非天然存在アミノ酸を含有するペプチド、非ペプチド構造、およびかなりの割合の−カルボキシグルタミン酸を含有するペプチド、を含有するものが含まれる。
【0131】
非ペプチド、例えばペプチド誘導体(例えば1以上の非天然存在アミノ酸を含有するもの)のライブラリーを用いることもまた可能である。これらの一例は、ペプトイド・ライブラリーである(Simonら, 1992, Proc. Natl. Acad. Sci. USA 89:9367−9371)。ペプトイドは、アルファ炭素に結合せず、主鎖アミノ窒素に結合する天然存在側鎖を有する、非天然アミノ酸のポリマーである。ペプトイドは、ヒト消化酵素によって容易には分解されないため、これらは好適に、薬剤使用に、より容易に適応可能である。ペプチド中のアミド官能性が過メチル化されて、化学的に変換されたコンビナトリアルライブラリーが生成される、使用可能なライブラリーの別の例が、Ostreshら, 1994, Proc. Natl. Acad. Sci. USA 91:11138−11142に記載されている。
【0132】
本発明にしたがってスクリーニング可能なペプチド・ライブラリーのメンバーは、20の天然存在アミノ酸を含有するものに限定されない。特に、化学的に合成されたライブラリーおよびポリソームに基づくライブラリーは、20の天然存在アミノ酸に加えたアミノ酸の使用を可能にする(ライブラリー産生に用いるアミノ酸の前駆体プールに含むことによる)。特定の態様において、ライブラリーメンバーは、1以上の非天然または非古典的アミノ酸または環状ペプチドを含有する。非古典的アミノ酸には、限定されるわけではないが、一般的なアミノ酸のD−異性体、−アミノイソ酪酸、4−アミノ酪酸、Abu、2−アミノ酪酸;−Abu、−Ahx、6−アミノヘキサン酸;Aib、2−アミノイソ酪酸;3−アミノプロピオン酸;オルニチン;ノルロイシン;ノルバリン、ヒドロキシプロリン、ザルコシン、シトルリン、シスチン酸、t−ブチルグリシン、t−ブチルアラニン、フェニルグリシン、シクロヘキシルアラニン、β−アラニン、デザイナーアミノ酸、例えばβ−メチルアミノ酸、C−メチルアミノ酸、N−メチルアミノ酸、フルオロ−アミノ酸およびアミノ酸類似体一般が含まれる。さらに、アミノ酸は、D(右旋性)またはL(左旋性)であることも可能である。
【0133】
特定の態様において、本発明の複合体の断片および/または類似体、またはそのタンパク質構成要素、特にペプチド模倣体を、複合体活性または形成の競合的または非競合的阻害剤としての活性に関して、スクリーニングする。
【0134】
本発明の別の態様において、コンビナトリアル化学を用いて、複合体の調節剤を同定することも可能である。コンビナトリアル化学は、その多くが構造的に類似でありうる、数十万の化合物を含有するライブラリーを生成することが可能である。ハイスループットスクリーニング・プログラムは、既知のターゲットに対する親和性に関して、これらの非常に大きいライブラリーをスクリーニングすることが可能である一方、より小規模であるが、最大化学的多様性を提供するライブラリーを得る、新規アプローチが開発されてきている(例えば、Matter, 1997, J. Med. Chem. 40:1219−1229を参照されたい)。
【0135】
明示される一団のタンパク質に対する結合親和性に関して、小分子の別個のライブラリーを試験するため、コンビナトリアル化学の1つの方法である、アフィニティー・フィンガープリンティングが、以前から用いられている。スクリーニングによって得られたフィンガープリントを用いて、目的の他のタンパク質または受容体(本発明において、本発明のタンパク質複合体およびそのタンパク質構成要素)に対する、個々のライブラリーメンバーの親和性を予測する。目的のタンパク質と反応することが知られる他の化合物から得たフィンガープリントと、フィンガープリントを比較して、ライブラリー化合物が同様に反応可能であるかどうかを予測する。例えば、複合体またはタンパク質構成要素との相互作用に関して、巨大ライブラリー中のすべてのリガンドを試験するのでなく、その活性を有することが知られる他の化合物と類似のフィンガープリントを有するリガンドのみを試験することも可能である(例えば、Kauvarら, 1995, Chem. Biol. 2:107−118;Kauvar, 1995, Affinity fingerprinting, Pharmaceutical Manufacturing International. 8:25−28;およびKauvar, Toxic−Chemical Detection by Pattern Recognition in New Frontiers in Agrochemical Immunoassay, Kurtz, StankerおよびSkerritt(監修), 1995, AOAC:ワシントンD.C., 305−312を参照されたい)。
【0136】
Kayら(1993, Gene 128:59−65)は、先の慣用的ライブラリーいずれのものより長い、完全にランダムな配列のペプチドをコードする、ペプチド・ライブラリーを構築する方法を開示した。Kayらに開示されるライブラリーは、長さ約20アミノ酸より長い、完全に合成のランダムペプチドをコードする。こうしたライブラリーを好適にスクリーニングして、複合体調節剤を同定することも可能である(米国特許第5,498,538号、1996年3月12日;およびPCT公報第WO 94/18318号、1994年8月18日もまた参照されたい)。
【0137】
ペプチド・ライブラリーの多様な種類の包括的な概説を、Gallopら, 1994, J. Med. Chem. 37:1233−1251に見出すことも可能である。
【0138】
好ましい態様において、GPR49と試験化合物の相互作用は、GPR49活性の阻害を生じる。
好ましい態様にしたがって、工程b)において、ガンマ−セクレターゼがAPPを切断する能力を測定する。これは上に示すように測定可能である。
【0139】
さらに、本発明はまた、神経変性疾患、好ましくはアルツハイマー病治療用の薬剤組成物を調製するための方法であって:
a)本発明の方法にしたがって、ガンマ−セクレターゼ調節剤および/またはベータ−セクレターゼ調節剤、好ましくは阻害剤を同定し、そして
b)ガンマ−セクレターゼおよび/またはベータ−セクレターゼの調節剤、好ましくは阻害剤を薬剤組成物に配合する
工程を含む、前記方法にも関する。
【0140】
ここでもまた、薬剤組成物に関して、上に示すようなすべての態様があてはまる。
好ましい態様において、本発明のこの方法は、同定された分子と、上に説明したような薬学的に許容しうるキャリアーを混合する工程をさらに含む。
【0141】
本発明はまた、上に定義するようなGPR49阻害剤を含む、薬剤組成物にも関する。
さらに、本発明はまた、薬剤組成物調製のための上記方法によって得られうる、薬剤組成物にも関する。
【0142】
本発明はまた、アルツハイマー病などの神経変性疾患および関連する神経変性障害の治療用の、本発明の薬剤組成物にも関する。
本発明はまた、神経変性疾患、好ましくはアルツハイマー病を治療するかまたは予防する方法であって、こうした治療または予防の必要がある被験者に、本発明の薬剤組成物の療法的有効量を投与することを含む、前記方法にも関する。
【0143】
本発明のこの方法に関して、上記のすべての態様は、本発明の使用のため、やはりあてはまる。
本発明はまた、ベータ−セクレターゼおよび/またはガンマ−セクレターゼ活性をin vitroで調節する、好ましくは阻害するための、GPR49相互作用分子の使用にも関する。例えば、GPR49相互作用分子によって、細胞培養において、ベータ−セクレターゼおよび/またはガンマ−セクレターゼの活性を調節する、好ましくは阻害することが、本発明内に含まれる。上述のような、GPR49相互作用分子に関するすべての態様もまた、本発明のこの使用にあてはまる。
【0144】
以下の実施例は、本発明の主題をより詳細に記載するであろう。
【実施例】
【0145】
(実施例1)
それぞれ、EP 1 105 508 B1およびRigautら, 1999, Nature Biotechnol. 17:1030−1032に、より十分に記載されるTAP技術を用いて、そしてタンパク質精製のため、以下にさらに記載するように、さらに適応させた。以下にさらに記載するように、質量分析を用いて、タンパク質を同定した。
【0146】
GPR49は、TAP技術エントリーポイントAph1a、APP−C99、BACE1およびFe65L2で、タンパク質複合体のメンバーと同定された(図1)。
パート1:TAPタグ化ベイト(bait)の構築
RT−PCRによって、完全ORFをコードするcDNAを得た。RNeasyミニキット(Qiagen)を用いて、適切な細胞株から総RNAを調製した。遺伝子特異的プライマーを用い、長鎖テンプレート用SUPERSCRIPT One−Step RT−PCRキット(Life Technologies)を用いて、cDNA合成およびPCR両方を行った。35〜40周期増幅した後、予期されるサイズのPCR産物をMinElute PCR精製キット(Qiagen)を用いてゲル精製し、そして必要な場合、さらなる増幅に用いた。低存在量のRNAを、ゲル精製前に、入れ子(nested)PCRによって増幅した。NotI制限部位をPCRプライマーに付着させて、レトロウイルスベクターpIE94−N/C−TAPに、増幅されたcDNAをサブクローニングすることを可能にし、それによって、TAPタグとのN末端融合体またはC末端融合体を生成した(Rigautら, 1999, Nature Biotechnol. 17:1030−1032)。以下のベイト/エントリーポイントに関しては、N末端タグ化を選択した:プレセニリン1、プレセニリン2、Aph−1a、Aph−1b、Pen−2、APP、タウ、Fe65、カルセニリン。以下のベイト/エントリーポイントに関しては、C末端タグ化を選択した:ニカストリン、Aph−1a、Aph−1b、BACE1 D215N、APP、APP695SW、APP−C99、Fe65、Fe65L2、X11ベータ。
【0147】
制限消化、DNA配列決定、およびTNT T7迅速カップリング転写/翻訳系(Promega inc.)を用いたin vitro翻訳によって、クローンを分析した。検出のため、TAPタグのプロテインA部分を用いたウェスタンブロッティングによって、タンパク質の存在を証明した。簡潔には、標準的SDS−PAGEによってタンパク質を分離した後、Pharmacia BiotechのMultiphorIIブロッティング装置を用いて、ニトロセルロース膜(PROTRAN、Schleicher&Schuell)に半乾性トランスファーした。トランスファー緩衝液は、48mM Tris、39mMグリシン、10%メタノールおよび0.0375%ドデシル硫酸ナトリウムからなった。10%脱脂粉乳および0.1% Tweeen20を補ったリン酸緩衝生理食塩水(PBS)中でブロッキングした後、トランスファーしたタンパク質を、ブロッキング溶液中で希釈したペルオキシダーゼ−抗−ペルオキシダーゼ可溶性複合体(Sigma)で探査した(probed)。徹底的に洗浄した後、高感度化学発光(ECL;Amersham Pharmacia Biotech)によって、免疫反応性タンパク質を視覚化した。
【0148】
パート2:ウイルスの調製および感染
ベクターとして、MoMLVに基づく組換えウイルスを用いた。
調製は、以下のとおり行った:
2.1.ウイルスの調製
293gp細胞を100%集密(confluency)に増殖させた。これらを、ポリ−L−リジンプレート(PBS中、1:5希釈したポリーL−リジン[0.01%ストック溶液、Sigma P−4832]を、プレート上に少なくとも10分間放置した)上、1:5にスプリットした。第2日、63マイクログラムのレトロウイルスベクターDNAと一緒に、適切なエンベロープ・タンパク質をコードするプラスミドDNA 13マイクログラムを、293gp細胞にトランスフェクションした(Somiaら, 1999, Proc. Natl. Acad. Sci. USA 96:12667−12672;Somiaら 2000, J. Virol. 74:4420−4424)。第3日、15cmディッシュあたり、15mlのDMEM+10% FBSと培地を交換した。第4日、ウイルスを含有する培地(上清)を採取した(トランスフェクション後の培地交換の24時間後)。第二の収集を計画している場合、DMEM 10% FBSをプレートに添加し、そしてプレートをさらに24時間インキュベーションした。すべての収集を以下のように行った:0.45マイクロメートル・フィルター(Corning GmbH、酢酸セルロース、431155)を通じて、上清をろ過した。フィルターをコニカル・ポリアロマー遠心管(Beckman、358126)に入れ、これをSW28ローター(Beckman)のバケットに入れた。ろ過した上清をSW28ローター中、19400rpm、摂氏21度で2時間、超遠心した。上清を廃棄した。ウイルスを含有するペレットを、エアロゾル安全チップを用い、上下に100回ピペッティングすることによって、少量(例えば300マイクロリットル)のハンクス平衡塩溶液(Gibco BRL、14025−092)に再懸濁した。ウイルスを以下のようにトランスフェクションに用いた。
【0149】
2.2.感染
感染させる細胞を1日前に6ウェルプレートの1つのウェルに蒔いた。感染4時間前に、細胞上の古い培地を新鮮な培地と交換した。最小限の体積のみを添加して、細胞が完全に覆われるようにした(例えば700マイクロリットル)。感染中、細胞は活発に分裂していた。
【0150】
細胞およびその増殖条件の説明を、以下にさらに提供する(「3.細胞株」)。
濃縮ウイルスに、ポリブレン(臭化ヘキサジメトリン;Sigma、H9268)を添加して、最終濃度8マイクログラム/mlを達成した(これは、濃縮レトロウイルス300マイクロリットルあたり、1ミリグラム/mlポリブレンストックの2.4マイクロリットルと同等である)。ウイルスをポリブレン中、室温で1時間インキュベーションした。感染のため、ウイルス/ポリブレン混合物を細胞に添加して、そして摂氏37度で数時間、適切なCO2濃度でインキュベーションした(例えば1日または1晩)。感染後、感染細胞上の培地を新鮮な培地と交換した。集密になったら、通常どおりに細胞を継代した。細胞は染色体に組み込まれたレトロウイルスを含有し、そして目的の遺伝子を安定に発現する。
【0151】
2.3.細胞株
発現のため、SKN−BE2細胞を用いた。SKN−BE2細胞(アメリカン・タイプ・カルチャー・コレクション第CRL−2271号)を95% OptiMEM+5%鉄補充ウシ血清中で増殖させた。
【0152】
パート3:TAPタグ化タンパク質の発現パターンのチェック
イムノブロット分析によって、そして/または免疫蛍光によって、TAPタグ化タンパク質の発現パターンをチェックした。免疫蛍光分析を、TAPタグ化タンパク質の種類に応じて、No.1またはNo.2にしたがって行った。イムノブロット分析を、No.3にしたがって行った。
【0153】
3.1.原形質膜およびER結合タンパク質に関する、固定哺乳動物細胞の間接的免疫蛍光染色のプロトコル
ポリリジンでコーティングした8ウェルチャンバー・スライド上、FCS培地中で、細胞を50%集密まで増殖させた。次いで、リン酸緩衝生理食塩水(PBS)溶液(0.14Mホスフェート、0.1M NaCl pH7.4)中で希釈した4%パラホルムアルデヒド中で、細胞の固定を行った。ウェルあたり300マイクロリットル中、室温で30分間、細胞をインキュベーションした。PBS中の0.1Mグリシン中で、室温で2x20分間、反応停止を行った。0.3%サポニン+PBS中の1%ウシ血清アルブミン(BSA)で、室温で少なくとも1時間、ブロッキングを行った。ブロッキング溶液中、+4℃で1晩、一次抗体のインキュベーションを行った。ケースバイケースで、抗体の適切な希釈を決定した。0.3%サポニンを含有するPBS中、室温で2x20分間、細胞を洗浄した。ブロッキング溶液中で、二次抗体のインキュベーションを行う。Alexa594にカップリングしたヤギ抗ウサギを1:1000に希釈した(Molecular Probes)。Alexa488にカップリングしたヤギ抗マウスを1:1000に希釈した(Molecular Probes)。DAPIを用いてDNAを標識した。F−アクチンを標識するのにファロイジンを用いた場合、該薬剤を1:500に希釈し、そして二次抗体とインキュベーションした。次いで、細胞を再び、PBS中、室温で2x20分間洗浄した。過剰な緩衝液を取り除き、そしてアンチ・ブリーチング剤(Vectashield、Vector Laboratories)を含有する培地中で細胞をマウントした。
【0154】
3.2.非原形質膜結合タンパク質に関する、固定哺乳動物細胞の間接的免疫蛍光染色のためのプロトコル:
ポリリジンでコーティングした8ウェルチャンバー・スライド上、FCS培地中で、細胞を50%集密まで増殖させた。リン酸緩衝生理食塩水(PBS)溶液(0.14Mホスフェート、0.1M NaCl pH7.4)中で希釈した4%パラホルムアルデヒド中、室温(RT)で30分間、ウェルあたり300マイクロリットルで、細胞の固定を行った。PBS中の0.1Mグリシン中で、室温で2x20分間、反応停止を行った。PBS中、0.5% Triton X−100で、室温で10分間、細胞の透過化を行った。次いで、0.3%サポニン+PBS中の1%ウシ血清アルブミン(BSA)で、RTで少なくとも1時間、ブロッキングを行った(ブロッキング溶液)。ブロッキング溶液中、+4℃で1晩、一次抗体のインキュベーションを行った。ケースバイケースで、抗体の適切な希釈を決定しなければならない。0.3%サポニンを含有するPBS中、RTで2x20分間、細胞を洗浄した。ブロッキング溶液中で、二次抗体のインキュベーションを行った。Alexa594にカップリングしたヤギ抗ウサギを1:1000に希釈し(Molecular Probes)、Alexa488にカップリングしたヤギ抗マウスを1:1000に希釈した(Molecular Probes)。DAPIを用いてDNAを標識した。F−アクチンを標識するのにファロイジンを用いた場合、該薬剤を1:500に希釈し、そして二次抗体とインキュベーションした。細胞を、PBS中、RTで2x20分間洗浄した。過剰な緩衝液を取り除き、そしてアンチ・ブリーチング剤(Vectashield、Vector Laboratories)を含有する培地中で細胞をマウントした。
【0155】
3.3.イムノブロット分析
TAPタグ化タンパク質の発現レベルを分析するため、細胞ペレット(6ウェルプレート由来)を60μlのDNase I緩衝液(5%グリセロール、100mM NaCl、0.8% NP−40(IGEPAL)、5mM硫酸マグネシウム、100μg/ml DNアーゼI(Roche Diagnositics)、50mM Tris、pH7.5、プロテアーゼ阻害剤カクテル)中、氷上で15分間溶解した。各試料を2つのアリコットにスプリットした。最初の半分を13,000rpmで5分間遠心分離して、上清中にNP−40抽出可能物質を得た;次の半分(全物質)を注意深くすりつぶした(triturated)。各50μgのNP−40抽出可能物質および全物質を、DTT含有試料緩衝液と、シェーカー上、50℃で30分間混合し、そしてSDSポリアクリルアミドゲル電気泳動によって、プレキャスト4〜12% Bis−Trisゲル(Invitrogen)上で分離した。次いで、不連続緩衝剤系で、半乾法を用いて、タンパク質をニトロセルロースにトランスファーした。簡潔には、ゲルおよびニトロセルロース膜を、陽極緩衝液(3層の緩衝液A1(0.3M Tris−HCl)および3層の緩衝液A2(0.03M Tris−HCl))または陰極緩衝液(3層の0.03M Tris−HCl、pH9.4、0.1% SDS、40mM □−アミノカプロン酸)のいずれかに浸したろ紙の間に積んだ。600mAで25分間、同時に2つのゲルのエレクトロトランスファーを行った。トランスファー効率の対照とするため、Ponceau S溶液で、トランスファーしたタンパク質を1分間視覚化して、そして次いで、水中で脱染色した。TBST(0.05% Tween−20を含有するTBS)中の5%脱脂粉乳で、室温で30分間、膜をブロッキングした。続いて、HRPカップリングPAP抗体(5%ミルク/TBST中、1:5000に希釈)と室温で1時間インキュベーションし、TBST中、10分間、3回洗浄した。ブロット膜を最後に、化学発光基質(ECL、Roche Diagnostics)に2分間浸し、そしてX線フィルムに曝露するか、またはイメージング・ステーション上で分析した。
【0156】
パート4 精製またはタンパク質複合体
タンパク質複合体精製を、TAPタグ化タンパク質の細胞内局在に適応させ、そして以下に記載するように行った。
【0157】
4.1.細胞質タンパク質の溶解物調製
約1x109の接着細胞(平均)を細胞スクレーパーで採取し、そして氷冷PBS中で3回洗浄した(3分間、550g)。収集した細胞を液体窒素中で凍結するか、または直ちにさらにプロセシングした。細胞溶解のため、細胞ペレットを10mlのCZ溶解緩衝液(50mM Tris−HCl、pH7.4;5%グリセロール;0.2% IGEPAL;1.5mM MgCl2;100mM NaCl;25mM NaF;1mM Na3VO4;1mM DTT;緩衝液25mlあたり1タブレットのEDTA不含プロテアーゼ阻害剤カクテル(CompleteTM、Roche)を含有する)に再懸濁し、そして加圧型細胞破砕装置中、きつくはまったペッスルを10回ストロークすることによって、ホモジナイズした。氷上で30分間、溶解物をインキュベーションし、そして20,000gで10分間回転させた。上清を、100,000gで1時間のさらなる超遠心工程に供した。上清を回収し、そして液体窒素中で迅速に凍結させるか、または直ちにさらにプロセシングした。
【0158】
4.2.膜タンパク質のための溶解物調製
約1x109の接着細胞(平均)を細胞スクレーパーで採取し、そして氷冷PBS中で3回洗浄した(3分間、550g)。収集した細胞を液体窒素中で凍結するか、または直ちにさらにプロセシングした。細胞溶解のため、細胞ペレットを10mlの膜溶解緩衝液(50mM Tris、pH7.4;7.5%グリセロール;1mM EDTA;150mM NaCl;25mM NaF;1mM Na3VO4;1mM DTT;緩衝液25mlあたり1タブレットのEDTA不含プロテアーゼ阻害剤カクテル(CompleteTM、Roche)を含有する)に再懸濁し、そして加圧型細胞破砕装置中、きつくはまったペッスルを10回ストロークすることによって、ホモジナイズした。溶解物を750gで10分間回転させ、上清を回収し、そして100,000gで1時間の超遠心工程に供した。0.8%のn−ドデシル−D−マルトシドを含有する7.5mlの膜溶解緩衝液に膜ペレットを再懸濁し、そして休まず攪拌しながら、4℃で1時間インキュベーションした。100,000gで1時間の別の超遠心工程に試料を供し、そして可溶化された物質を液体窒素中で迅速に凍結させるか、または直ちにさらにプロセシングした。
【0159】
4.3.核タンパク質のための溶解物調製
約1x109の接着細胞(平均)を細胞スクレーパーで採取し、そして氷冷PBS中で3回洗浄した(3分間、550g)。収集した細胞を液体窒素中で凍結するか、または直ちにさらにプロセシングした。細胞溶解のため、細胞ペレットを10mlの低張溶解緩衝液(10mM Tris、pH7.4;1.5mM MgCl2;10mM KCl;25mM NaF;1mM Na3VO4;1mM DTT;緩衝液25mlあたり1タブレットのEDTA不含プロテアーゼ阻害剤カクテル(CompleteTM、Roche)を含有する)に再懸濁し、そして加圧型細胞破砕装置中、きつくはまったペッスルを10回ストロークすることによって、ホモジナイズした。溶解物を2,000gで10分間回転させ、そして生じた上清(S1)を氷上で保存した。核ペレット(P1)を5mlの核溶解緩衝液(50mM Tris、pH7.4;1.5mM MgCl2;20%グリセロール;420mM NaCl;25mM NaF;1mM Na3VO4;1mM DTT;緩衝液25mlあたり1タブレットのEDTA不含プロテアーゼ阻害剤カクテル(CompleteTM、Roche)を含有する)に再懸濁し、そして氷上で30分間インキュベーションした。試料をS1と合わせ、7mlの希釈緩衝液(110mM Tris、pH7.4;0.7% NP40;1.5mM MgCl2;25mM NaF;1mM Na3VO4;1mM DTT)でさらに希釈し、氷上で10分間インキュベーションし、そして100,000gで1時間遠心分離した。最終上清(S2)を液体窒素中で迅速に凍結させた。
【0160】
4.4.タンデムアフィニティー精製
凍結した溶解物を37℃の水槽で迅速に融解し、そして100,000gで20分間回転させた。上清を回収し、そして0.2mlの定着した(settled)ウサギIgG−アガロースビーズ(Sigma)と、休みなく攪拌しながら4℃で2時間インキュベーションした。固定タンパク質複合体を10mlのCZ溶解緩衝液(50mlの緩衝液あたり1タブレットのCompleteTM(Roche)を含有する)で洗浄し、そしてさらに5mlのTEV切断緩衝液(10mM Tris、pH7.4;100mM NaCl;0.1% IGEPAL;0.5mM EDTA;1mM DTT)で洗浄した。150μlのTEV切断緩衝液中、5μlのTEVプロテアーゼ(GibcoBRL、カタログ番号10127−017)と、16℃で1時間インキュベーションすることによって、タンパク質複合体を溶出させた。溶出液を回収し、そして0.2ml CBP結合緩衝液(10mM Tris、pH7.4;100mM NaCl;0.1% IGEPAL;2mM MgAc;2mMイミダゾール;1mM DTT;4mM CaCl2)中、0.2mlの定着したカルモジュリン親和性ビーズ(Stratagene)と合わせ、その後、休みなく攪拌しながら4℃で1時間インキュベーションした。10mlのCBP洗浄緩衝液(10mM Tris、pH7.4;100mM NaCl;0.1% IGEPAL;1mM MgAc;1mMイミダゾール;1mM DTT;2mM CaCl2)で固定タンパク質複合体を洗浄し、そして600μl CBP溶出緩衝液(10mM Tris、pH8.0;5mM EGTA)の添加によって、37℃で5分間溶出した。溶出物を、シリコン処理試験管に回収し、そして凍結乾燥した。残ったカルモジュリン樹脂を50μl 4x Laemmli試料緩衝液中で5分間煮沸した。試料緩衝液を単離し、凍結乾燥分画と合わせ、そしてNuPAGE勾配ゲル(Invitrogen、4〜12%、1.5mm、10ウェル)上に装填した。
【0161】
パート5 質量分析によるタンパク質同定
5.1.質量分析前のタンパク質消化
本質的に、Shevchenkoら, 1996, Anal. Chem. 68:850−858に記載される方法にしたがって、ゲル中で、ゲル分離したタンパク質を還元し、アルキル化し、そして消化した。簡潔には、ゲル分離したタンパク質を、清潔なメスを用いて、ゲルから切り出し、10mM DTT(5mM重炭酸アンモニウム中、54℃、45分間)を用いて還元し、そして続いて、55mMヨードアセトアミド(5mM重炭酸アンモニウム中)で、暗所中、室温でアルキル化した(30分間)。還元し、そしてアルキル化したタンパク質を、5mM重炭酸アンモニウム中、12.5ng/μlのプロテアーゼ濃度のブタ・トリプシン(Promega)を用いて、ゲル中で消化した。消化を37℃で4時間進行させ、そして続いて、5μlの5%ギ酸を用いて、反応を停止した。
【0162】
5.2.質量分析による分析前の試料調製
20μlの1% TFAを用いて、ゲル・プラグを2回抽出し、そして酸性化した消化上清とともにプールした。真空遠心分離機中で試料を乾燥させ、そして13μlの1% TFA中に再懸濁した。
【0163】
5.3.質量分析データ収集
四極子TOF(QTOF2、QTOF Ultima、QTOF Micro、MicromassまたはQSTAR Pulsar、Sciex)またはイオン捕捉(LCQ Deca XP)質量分析計いずれかに直接カップリングしたナノLC系(CapLC、WatersまたはUltimate、Dionex)に、ペプチド試料を注入した。水性溶媒および有機溶媒の勾配を用いて、LC系上でペプチドを分離した(以下を参照されたい)。溶媒Aは0.5%ギ酸中の5%アセトニトリルであり、そして溶媒Bは、0.5%ギ酸中の70%アセトニトリルであった。
【0164】
【表1】
【0165】
LC系から溶出したペプチドを、質量分析計内で、部分的に配列決定した。
5.4.タンパク質同定
LC−MS/MS実験で得たペプチド質量および断片化データを用いて、NCBI(NCBInr、dbEST、並びにヒトおよびマウスゲノムに関して)および欧州バイオインフォマティクス研究所(EBI、ヒト、マウス、キイロショウジョウバエおよび線虫プロテオーム・データベースに関して)で、維持され、そして定期的に更新される、fasta形式のタンパク質およびヌクレオチド配列データベースにクエリーを行った。測定したペプチド質量および断片化データを、ソフトウェア・ツール、Mascot(Matrix Science;Perkinsら, 1999, Electrophoresis 20:3551−3567)を用いてデータベース中のエントリーから計算した同じデータと相関させることによって、タンパク質を同定した。検索規準は、分析にどの質量分析計を用いたかに応じて、多様であった。
【0166】
(実施例2)
siRNAが仲介するGPR49のノックダウンのAβ1−42レベルに対する影響
結果:
我々は、APPプロセシングの既知のエフェクターであるBACE1およびニカストリンに対して向けられるsiRNA同様、GPR49をターゲットとするsiRNAが、Aβ1−42分泌の有意な減弱を引き起こし、一方、Luc3 siRNAは影響しないことに注目し(図2A)、GPR49が、APPのプロセシング/分泌を制御する際に機能的な役割を果たすことを立証した。
【0167】
我々は、GPR49 siRNAが、実際に、GPR49の発現に干渉することを確認した(図2B)。
APPプロセシングのエフェクターとしてのGPR49の機能的検証のため、RNAi遺伝子発現摂動(perturbation)戦略を使用した:GPR49に対して向けられるsiRNA、あるいはAPPプロセシングの既知のエフェクターであるBACE1またはニカストリンに対して向けられるsiRNA、あるいは関連しないLuc3に対して向けられるsiRNAを、ヒトAPP695を発現するSK−N−BE2神経芽細胞にトランスフェクションした。ヒトGPR49に対するsiRNAは、Dharmacon Research Inc.によって合成された。
【0168】
GPR49に対して用いるsiRNA配列は:AACAGCAGTATGGACGACCTTであった。
製造者の指示にしたがって、LipofectAMINE 2000(Invitrogen)を用いて、SK−N−BE2細胞のトランスフェクションを行った。簡潔には、96ウェルあたり最終体積85μl中、1.0x104細胞の密度で、トランスフェクション12〜16時間前に、細胞を植え付けた。25nMのsiRNAを8μlのOpti−MEM緩衝液(Gibco)および60ngのキャリアーDNAと混合し、そして混合物を細胞に添加する前に、室温で20分間インキュベーションした。トランスフェクション16〜48時間後、培地を、それぞれ血清を含むまたは含まない、100μlまたは200μlの増殖培地と交換した。トランスフェクション72時間後、Aβ1−42 ELISA(Innogenetics)のため、100μlの上清を採取した。製造者の指示にしたがって、アッセイを行った。
【0169】
定量的RT−PCRによって、選択したsiRNAのノックダウン効率を評価した。簡潔には、6ウェルあたり、5x105 SKNBE2細胞を蒔き、そして翌日、25nM siRNAをトランスフェクションした。トランスフェクション36時間後、細胞を採取し、そして標準法を用いて、総RNAを調製し、そして逆転写した。製造者の指示にしたがって、GPR49の相対発現レベルを決定するため、等量のcDNAおよびGPR49特異的プライマーを利用した。すべての値を、ヒト参照RNA(Stratagene)に対して標準化した。
【0170】
(実施例3)
GPR49活性の決定
GPR49に誘発されるシグナル伝達カスケードは、現在、未知である。しかし、他のファミリーメンバーである、ヒト糖ホルモン受容体、並びに系統学的に遠い、線虫における推定上のLGRオルソログ(Kudo M, Chen T, Nakabayashi K, Hsu SY, Hsueh AJ.(2000)The nematode leucine−rich repeat−containing, G protein−coupled receptor(LGR) protein homologous to vertebrate gonadotropin and thyrotropin receptors is constitutively active in mammalian cells. Mol Endocrinol. 14(2):272−84)は、アデニル酸シクラーゼ依存機構を通じて、シグナル伝達することが示されてきている。後者は、哺乳動物細胞において、異種発現されると、細胞の環状アデノシン一リン酸(cAMP)レベルの恒常的増加を引き起こす。
【0171】
GPR49がcAMP経路にカップリングしていることを確認するため、公知のものとして利用可能ないくつかのアッセイを、使用することも可能である。例えば、Bresnickら(Bresnick JN, Skynner HA, Chapman KL, Jack AD, Zamiara E, Negulescu P, Beaumont K, Patel S, McAllister G(2003)Identification of signal transduction pathways used by orphan g protein−coupled receptors. Assay Drug Dev Technol. 1(2):239−49)は、ベータ−ラクタマーゼ・レポーター構築物を用いて、オーファンGPRCによって用いられるシグナル伝達経路を同定した。
【0172】
次いで、GPR49発現細胞で観察されるが、GPR49欠損細胞では観察されない、細胞のcAMP上昇を誘発する能力に基づいて、GPR49調節剤を同定する。cAMPの直接測定は、例えば、cAMP応答要素(CRE)−ルシフェラーゼ・レポーター細胞株を用いた、ハイスループット形式で実行可能である(Gabriel D, Vernier M, Pfeifer MJ, Dasen B, Tenaillon L, Bouhelal R(2003)High throughput screening technologies for direct cyclic AMP measurement. Assay Drug Dev Technol. 1(2):291−303)。cAMP上昇またはcAMP依存性シグナル伝達の他の測定法が、当業者に明らかである。以前、オーファンGPCRの小分子調節剤を同定した前例の概説には、Howard AD, McAllister G, Feighner SD, Liu Q, Nargund RP, Van der Ploeg LH, Patchett AA(2001)Orphan G−protein−coupled receptors and natural ligand discovery. Trends Pharmacol Sci. 22(3):132−40を参照されたい。
【0173】
(実施例4)
GPR49調節剤による、Aβ1−42生成/分泌の調節
ヒトAPP695を安定に過剰発現するSKNBE2細胞(SKNBE2/APP695)(または別の適切な細胞株)またはベータ−/ガンマ−セクレターゼ切断反応速度が増進した適切な突然変異体を増殖培地中で蒔き、そして翌朝、4時間、血清枯渇させた。次いで、血清不含培地中で希釈した、GPR49調節剤、好ましくは阻害剤を添加し、そして適切な期間、インキュベーションした。細胞上清を収集し、そしてELISA(INNOGENETICS N.V.、BelgiumのInnotest β−アミロイド(1−42))によって、Aβ1−42のレベルを決定した。
【0174】
本発明は図面に、より詳細に記載される。
【図面の簡単な説明】
【0175】
【図1】図1:GPR49が相互作用することが見出されたTAPエントリーポイント(白)の略図。
【図2】図2:siRNAが仲介するGPR49発現のノックダウンは、Aβ1−42の生成/分泌を減弱する。
【0176】
図2A:BACE1、ニカストリン、GPR49またはLuc3に対して向けられるSiRNAを、APP695を過剰発現するSK−N−BE2神経芽腫細胞にトランスフェクションした。トランスフェクション48時間後、増殖培地を取り除き、そして細胞を血清不含培地中で1晩インキュベーションした。上清を収集し、そしてAβ1−42レベルをELISA(INNOGENETICS N.V.、BelgiumのInnotest β−アミロイド(1−42))によって決定した。少なくとも3回の独立実験を2つ組で行った。代表的な例を示す。
【0177】
図2B:GPR49に対して向けられるsiRNAは、定量的RT−PCR分析で評価した際、GPR49 mRNAを特異的に減少させたが、関連しないLuc3に対して向けられるsiRNAは、該mRNAを減少させなかった。各siRNAを示す2つのバーは、2つの独立実験に相当する。
【図3】図3:1文字コードで示す、ヒトGPR(LGR5;ロイシン・リッチ・リピート含有Gタンパク質共役型受容体5)のアミノ酸配列。
【図4−1】図4:ヒトLGR4、LGR5/GPR49およびLGR6の複数配列並列。
【図4−2】図4:ヒトLGR4、LGR5/GPR49およびLGR6の複数配列並列。
【技術分野】
【0001】
本発明は、GPR49タンパク質を含む、APPプロセシング経路のタンパク質複合体に関し、それとともに神経生成(neurogenerative)疾患の治療における、これらの複合体の阻害剤、並びにGPR49の阻害剤の使用に関する。
【背景技術】
【0002】
アルツハイマー病は、世界中で何百万という個体に影響を及ぼす慢性状態である。
アルツハイマー病患者の脳は、顕著な神経病理学的損傷の特徴的な病変、例えば初期の細胞内の神経原線維のもつれ(neurofibrillary tangle)(NFT)、および細胞外のアミロイド・リッチ老人斑を示す。これらの損傷は、CNSニューロン集団の大量の損失と関連し、そしてその進行には、ADと関連する臨床的認知症が伴う。アミロイド斑の主な構成要素は、多様な長さのアミロイド・ベータ(A−ベータ、AベータまたはAβ)ペプチドである。その変異体であるAβ1−42ペプチド(Aベータ−42)は、アミロイド形成の主な原因物質である。別の変異体はAβ1−40ペプチド(Aベータ−40)である。アミロイド・ベータは、前駆タンパク質、ベータ・アミロイド前駆タンパク質(ベータ−APPまたはAPP)のタンパク質分解産物である。APPは、I型膜貫通タンパク質であり、該タンパク質は、いくつかの異なる膜会合プロテアーゼによって、順次、切断される。APPの最初の切断は、2つのプロテアーゼ、アルファ−セクレターゼまたはベータ−セクレターゼの1つによって起こる。アルファ−セクレターゼは、メタロプロテアーゼであり、その活性は、タンパク質ADAM−10およびADAM−17の1つまたは組み合わせによって提供される可能性が最も高い。アルファ−セクレターゼによる切断は、アミロイド・ペプチド形成を妨げ、そしてしたがって、アミロイド非生成性と称される。対照的に、ベータ−セクレターゼによるAPPの切断は、アミロイド・ペプチド形成が続くための必要条件である。このセクレターゼは、BACE1(ベータ部位APP切断酵素)とも称され、アスパルチル・プロテアーゼ活性を含有するI型膜貫通タンパク質である(以下に詳細に記載される)。
【0003】
ベータ−セクレターゼ(BACE)活性は、細胞外ドメインでAPPを切断し、分泌可溶性APPbの分断(shedding)を生じ、そして99残基のC末端膜貫通断片(APP−C99)を生じる。Vassarら(Science 286, 735−741)は、APPのベータ−セクレターゼと推測される特性を有する膜貫通アスパラギン酸プロテアーゼをクローニングし、該プロテアーゼをBACE1と名づけた。BACE1ノックアウトマウス由来の脳および初代皮質培養物は、検出可能なベータ−セクレターゼ活性を示さず、そしてBACEノックアウトマウス由来の初代皮質培養物は、APPからはるかに少ないアミロイド−ベータしか産生しなかった。これによって、BACE1のパラログであるBACE2ではなく、BACE1がAPPの主なベータ−セクレターゼであることが示唆される。BACE1は、501アミノ酸(aa)のタンパク質であり、21aaのシグナルペプチドに続いて、aa22〜45に渡る、プロ配列ドメインを含有する。選択的スプライシング型、BACE−I−457およびBACE−I−476がある。成熟タンパク質の細胞外ドメインに続いて、予測される1つの膜貫通ドメインおよび24aaの短い細胞質ゾルC末端テールがある。BACE1は、1型膜貫通タンパク質であり、活性部位が膜の細胞外側にあると予測され、ここでベータ−セクレターゼがAPP、およびありうる他の未同定の基質を切断する。BACE1は、明らかに、APPをA−ベータにプロセシングするのに必要とされる重要な酵素であるが、最近の証拠によって、BACE1のさらなる潜在的基質および機能が示唆されている(J. Biol. Chem. 279, 10542−10550)。現在まで、制御機能または調節機能を持つBACE1相互作用タンパク質は、まったく記載されてきていない。
【0004】
BACE1切断によって生じるAPP断片であるAPP−C99は、ガンマ−セクレターゼ活性の基質であり、該活性は、膜平面内でAPP−C99を切断してA−ベータ・ペプチド(アミロイド生成性Aβ1−42ペプチドなど)、およびAPP細胞内ドメイン(AICD)と称されるC末端断片にする(Annu Rev Cell Dev Biol 19, 25−51)。ガンマ−セクレターゼ活性は、少なくとも4つの別個のサブユニットを持つ多タンパク質複合体内にある。発見された第一のサブユニットは、プレセニリンであった(Proc Natl Acad Sci USA 94, 8208−13)。ガンマ−セクレターゼ複合体の他の既知のタンパク質構成要素は、Pen−2、ニカストリンおよびAph−1a(Aph1a)である。
【0005】
アルツハイマー病の病因の根底にある分子事象の描写が近年、進歩しているにもかかわらず、これまでに疾患を緩和する療法は開発されてきていない。この目的に向け、業界は、BACE1を阻害するための適切なリード化合物を同定しようと苦闘している。さらに、ガンマ−セクレターゼのより多くの別の基質、最も著名には、Notchタンパク質が存在することが認められてきている。その結果、ガンマ−セクレターゼの阻害は、機構に基づく副作用を引き起こす可能性がある。現在、最も優れた薬剤(例えばアリセプト(登録商標)/ドネペジル)は、アセチルコリン・エステラーゼを阻害して、脳において、神経伝達物質アセチルコリンのレベル増加を生じることによって、認知機能の一時的な改善を達成することを試みる。これらの療法は、疾患のより後期の病期には適しておらず、根底にある疾患病変は治療せず、そして疾患進行を停止させることはない。
【0006】
したがって、アルツハイマー病の治療のための新規分子戦略を可能にする、新規ターゲットの同定に関して、満たされていない必要性がある。さらに、前記新規ターゲットをターゲティングすることによって、前述の分子プロセスを修飾する新規療法化合物に関する、強い必要性がある。
【発明の開示】
【課題を解決するための手段】
【0007】
第一の側面において、本発明は、神経生成疾患治療用の薬剤組成物を調製するための、「GPR49相互作用分子」の使用を提供する。
本発明の関連において、驚くべきことに、GPR49(ロイシン・リッチ・リピート含有Gタンパク質共役型受容体5(LGR5);FEX HG38、GPR67としても知られる)が、アルツハイマー病において、ガンマ−セクレターゼによるAPPの異常なプロセシングに関与する、異なるタンパク質複合体の一部を形成することが発見されている。特に、GPR49がAph1a複合体、Fe65L2複合体、APP−C99複合体およびBACE1複合体の一部であることが発見されている。これらの複合体は、TAP技術エントリーポイントとして用いられている、それぞれの重要なタンパク質化合物にちなんで命名されている(以下を参照されたい)。
【0008】
GPR49がこれらの複合体において重要な分子と同定されたことによって、神経変性(neurodegenerative)疾患の治療のため、GPR49と相互作用する分子の使用が可能になる。これは特に、GPR49に対して向けられるsiRNAが、Aベータ−42の生成および/または分泌の減弱を生じることを立証する実施例において示される。
【0009】
本発明の関連において、「GPR49相互作用分子」は、少なくとも一時的にGPR49に結合し、そして好ましくは、GPR49活性を調節し、そして特に阻害する、分子である。
【0010】
GPR49は、糖タンパク質ホルモン受容体スーパーファミリーの推定上のメンバーであり、このスーパーファミリーは、ある場合には糖タンパク質リガンドとの相互作用に重要であることが示されたロイシン・リッチ・リピートを含有する、巨大N末端細胞外ドメインによって特徴付けられる、異常なGタンパク質共役型受容体(GPCR)である。スーパーファミリーのメンバーを系統学的に分析すると、3つのサブファミリーに分かれると示唆され、第一のサブファミリーは、LH、FSH(卵胞刺激ホルモン)およびTSH(甲状腺刺激ホルモン)の受容体を特徴とし;第二のサブファミリーは、リラキシン受容体LGR7およびLGR8を含有する。GPR49/LGR5とともにLGR4およびLGR6(図4)は、FSH受容体とわずか〜35%の配列同一性しか示さない、オーファン受容体サブファミリーを構成する(Hsu SY, Liang SG, Hsueh AJ(1998)Characterization of two LGR genes homologous to gonadotropin and thyrotropin receptors with extracellular leucine−rich repeats and a G protein−coupled, seven−transmembrane region. Mol Endocrinol. 12(12):1830−45;McDonald T, Wang R, Bailey W, Xie G, Chen F, Caskey CT, Liu Q(1998)Identification and cloning of an orphan G protein−coupled receptor of the glycoprotein hormone receptor subfamily. Biochem Biophys Res Commun. 247(2):266−70)。
【0011】
GPR49は、骨格筋、胎盤および脊髄で顕著に発現され、そしてより低いレベルで、結腸、副腎および脳の多様な小領域で発現される(Hsu SYら(1998)、上記)。成体マウス脳では、GPR49転写物は、主に嗅球に限定されており、嗅球の神経細胞層に見られる(Hermey G, Methner A, Schaller HC, Hermans−Borgmeyer I(1999)Identification of a novel seven−transmembrane receptor with homology to glycoprotein receptors and its expression in the adult and developing mouse. Biochem Biophys Res Commun. 254(1):273−9)。成体におけるGPR49の機能はそれほど性質決定されていない。マウスにおいて、GPR49遺伝子をターゲットとして欠失させると、新生児致死が生じ、そしてこの欠失は舌小帯短縮症および胃腸膨張と関連する(Morita H, Mazerbourg S, Bouley DM, Luo CW, Kawamura K, Kuwabara Y, Baribault H, Tian H, Hsueh AJ(2004)Neonatal lethality of LGR5 null mice is associated with ankyloglossia and gastrointestinal distension. Mol Cell Biol. 24(22):9736−43)。
【0012】
GPR49は、最近、ベータ−カテニン突然変異を持つヒト肝細胞癌で過剰発現される遺伝子と同定された(Yamamoto Y, Sakamoto M, Fujii G, Tsuiji H, Kenetaka K, Asaka M, Hirohashi S(2003) Overexpression of orphan G−protein−coupled receptor, Gpr49, in human hepatocellular carcinomas with beta−catenin mutations. Hepatology. 37(3):528−33)。
【0013】
本発明にしたがって、表現「GPR49」は、図3に示すようなタンパク質だけでなく、機能的に活性なその誘導体、または機能的に活性なその断片、またはその相同体、または低ストリンジェンシー条件下で、前記タンパク質をコードする核酸にハイブリダイズする核酸にコードされる変異体もまた、意味する。好ましくは、これらの低ストリンジェンシー条件は、35%ホルムアミド、5xSSC、50mM Tris−HCl(pH7.5)、5mM EDTA、0.02% PVP、0.02% BSA、100μg/ml変性サケ精子DNA、および10%(重量/体積)デキストラン硫酸を含む緩衝液中、40℃で18〜20時間ハイブリダイゼーションし、2xSSC、25mM Tris−HCl(pH7.4)、5mM EDTA、および0.1% SDSからなる緩衝液中、55℃で1〜5時間洗浄し、そして2xSSC、25mM Tris−HCl(pH7.4)、5mM EDTA、および0.1% SDSからなる緩衝液中、60℃で1.5時間洗浄することを含む。
【0014】
本発明で挙げる他のタンパク質すべてにも同じことがあてはまる。したがって、既定のタンパク質または核酸の名称は、配列表に示すようなタンパク質または核酸を指すだけでなく、機能的に活性な誘導体、または機能的に活性なその断片、またはその相同体、または低ストリンジェンシー条件下で、好ましくは上述のような条件下で、前記タンパク質をコードする核酸にハイブリダイズする核酸にコードされる変異体もまた、指す。
【0015】
用語「機能的に活性な」は、本明細書において、このポリペプチド、すなわち断片または誘導体が関連する態様にしたがって、タンパク質の構造的機能、制御機能、または生化学的機能を有する、ポリペプチド、すなわち断片または誘導体を指す。
【0016】
本発明にしたがって、用語「活性」は、本明細書において、最も広い意味での分子の機能を指す。活性は、一般的に、限定されるわけではないが、分子の生物学的機能、生化学的機能、物理的機能または化学的機能を含む。活性は、例えば、酵素活性、他の分子と相互作用する能力、および他の分子の機能を活性化するか、促進するか、安定化するか、阻害するか、抑制するか、または不安定化する能力、安定性、特定の細胞内位置に局在する能力を含む。適用可能な場合、前記用語はまた、最も広い意味でのタンパク質複合体の機能にも関する。
【0017】
本発明にしたがって、用語「誘導体」または「構成要素タンパク質の類似体」または「変異体」は、本明細書において、好ましくは、限定されるわけではないが、構成要素タンパク質に実質的に相同な領域を含む分子を含み、こうした分子は、多様な態様において、同一サイズのアミノ酸配列に渡って、または当該技術分野に知られるコンピュータ相同性プログラムによって並列を行って並列される配列と比較した際、少なくとも30%、40%、50%、60%、70%、80%、90%、95%または99%同一であるか、あるいはこうした分子をコードする核酸は、ストリンジェントな条件、中程度にストリンジェントな条件、またはストリンジェントでない条件下で、構成要素タンパク質をコードする配列にハイブリダイズ可能である。これは、それぞれ、アミノ酸置換、欠失および付加によって、天然存在タンパク質を修飾した所産であり、その誘導体がなお天然存在タンパク質の生物学的機能を、必ずしも同じ度合いでなくてもよいが示す、タンパク質を意味する。例えば、本発明で提供するような、適切で利用可能なin vitroアッセイによって、こうしたタンパク質の生物学的機能を調べることも可能である。
【0018】
用語「断片」は、本明細書において、態様にしたがって、構成要素タンパク質の少なくとも10、20、30、40または50アミノ酸のポリペプチドを指す。特定の態様において、こうした断片は、35、100または200アミノ酸より長くはない。
【0019】
用語「遺伝子」は、本明細書において、別に言及しない限り、本発明のポリペプチドをコードするオープンリーディングフレームを含む核酸を指し、エクソンおよび場合によるイントロン配列両方を含む。
【0020】
用語「相同体」または「相同遺伝子産物」は、本明細書において、本明細書にさらに記載する複合体のタンパク質構成要素と同じ生物学的機能を実行する、別の種、好ましくは哺乳動物におけるタンパク質を意味する。こうした相同体はまた、「オルソログ遺伝子産物」とも称される。ヒトおよび哺乳動物または他の種からオルソログ遺伝子対を検出するためのアルゴリズムは、これらの生物の全ゲノムを用いる。まず、予測されるタンパク質の完全Smith−Waterman並列を用いて、対合ベストヒットを回収する。信頼性をさらに改善するため、キイロショウジョウバエ(Drosophila melanogaster)および線虫(C. elegans)タンパク質を含む対合ベストヒットを用いて、これらの対のクラスターを形成する。こうした分析は、例えば、Nature, 2001, 409:860−921に提供される。他の種の遺伝子に対する、本明細書に提供するタンパク質をコードする遺伝子の配列相同性に基づいて、慣用的技術を適用してそれぞれの遺伝子をクローニングし、そしてこうした遺伝子からタンパク質を発現させることによって、あるいは本明細書に提供する方法にしたがって、または当該技術分野に一般的に知られる他の適切な方法にしたがって、類似の複合体を単離することにより、他の種のタンパク質を単離することによって、本発明記載のタンパク質の相同体を単離することも可能である。
【0021】
本発明の好ましい態様において、「GPR49相互作用分子」はGPR49阻害剤である。
本発明にしたがって、用語「阻害剤」は、GPR49の活性を好ましくは阻害するかまたは減少させる、生化学的化合物または化学的化合物を指す。これは、例えば、対応する遺伝子の発現の抑制を介して起こることも可能である。RT−PCRまたはウェスタンブロット分析によって、遺伝子の発現を測定することも可能である。さらに、これは、例えばGPR49への結合による、活性の阻害を介して起こることも可能である。
【0022】
こうしたGPR49阻害剤の例は、GPR49に対して向けられる、特にGPR49の活性部位に対して向けられる、結合性タンパク質または結合性ペプチド、並びにGPR49遺伝子に対して向けられる核酸である。
【0023】
用語「GPR49に対する核酸」は、例えばGPR49遺伝子の発現またはGPR49の活性を阻害する、二本鎖または一本鎖DNAまたはRNA、あるいはその修飾物または誘導体を指し、そして限定なしに、アンチセンス核酸、アプタマー、siRNA(低分子干渉RNA)およびリボザイムを含む。
【0024】
好ましくは、阻害剤は、抗体、アンチセンス・オリゴヌクレオチド、siRNA、低分子量分子(LMW)、結合性ペプチド、アプタマー、リボザイムおよびペプチド模倣体(peptidomimetic)からなる群より選択される。
【0025】
これらの核酸を細胞に直接投与することも可能であるし、または外因性の導入された配列を転写させることによって、細胞内で産生することも可能である。
「アンチセンス」核酸は、本明細書において、何らかの配列相補性のため、構成要素タンパク質RNA(好ましくはmRNA)の配列特異的部分にハイブリダイズ可能な核酸を指す。アンチセンス核酸は、構成要素タンパク質mRNAのコード領域および/または非コード領域に相補的であることも可能である。複合体形成または活性を阻害する、こうしたアンチセンス核酸は、療法剤として有用性を有し、そして本明細書に記載するような障害の治療または予防に使用可能である。
【0026】
アンチセンス核酸は、少なくとも6ヌクレオチドのものであり、そして好ましくは、6〜約200ヌクレオチドの範囲のオリゴヌクレオチドである。特定の側面において、オリゴヌクレオチドは、少なくとも10ヌクレオチド、少なくとも15ヌクレオチド、少なくとも100ヌクレオチド、または少なくとも200ヌクレオチドである。
【0027】
核酸、例えばアンチセンス核酸またはsiRNAを、例えばホスホトリエステル法にしたがって、化学的に合成することも可能である(例えばUhlmann, E. & Peyman, A.(1990)Chemical Reviews, 90, 543−584を参照されたい)。アプタマーは、ポリペプチド、本明細書ではGPR49に、高い親和性で結合する核酸である。アプタマーは、異なる一本鎖RNA分子の巨大プールから、SELEXなどの選択法によって単離可能である(例えば、Jayasena(1999)Clin. Chem., 45, 1628−50;KlugおよびFamulok(1994)M. Mol. Biol. Rep., 20, 97−107;US 5,582,981を参照されたい)。アプタマーを、例えばL−リボヌクレオシドとして、鏡像型で合成し、そして選択することもまた可能である(Nolteら(1996)Nat. Biotechnol., 14, 1116−9;Klussmannら(1996)Nat. Biotechnol., 14, 1112−5)。この方式で単離された型は、天然存在リボヌクレアーゼによって分解されず、そしてしたがってより高い安定性を所持する利点を享受する。
【0028】
核酸は、エンドヌクレアーゼまたはエキソヌクレアーゼ、特に細胞で見出されることも可能なDNaseおよびRNaseによって分解されうる。したがって、分解に対して安定化させ、それによって長期間に渡って、細胞中で、核酸が高濃度で維持されることを保証するため、核酸を修飾することは好都合である(Beigelmanら(1995)Nucleic Acids Res. 23:3989−94;WO 95/11910;WO 98/37240;WO 97/29116)。典型的には、こうした安定化は、1以上のヌクレオチド間リン基を導入することによって、または1以上の非リン基をヌクレオチド間に導入することによって、得ることも可能である。
【0029】
適切なヌクレオチド間修飾は、UhlmannおよびPeyman(1990)、上記に集められている(Beigelmanら(1995)Nucleic Acids Res. 23:3989−94;WO 95/11910;WO 98/37240;WO 97/29116もまた参照されたい)。本発明にしたがった使用の1つで使用可能な、核酸における修飾ヌクレオチド間ホスフェート・ラジカルおよび/または非リン架橋は、例えば、メチル・ホスホネート、ホスホロチオエート、ホスホロアミデート、ホスホロジチオエートおよび/またはホスフェート・エステルを含有し、一方、非リン・ヌクレオチド間類似体(analogue)は、例えばシロキサン架橋、カーボネート架橋、カルボキシメチル・エステル、アセトアミデート(acetamidate)架橋および/またはチオエーテル架橋を含有する。この修飾が、本発明記載の使用の1つで使用可能である薬剤組成物の耐久性を改善するはずであることもまた意図される。一般的に、オリゴヌクレオチドを、塩基部分、糖部分、またはホスフェート主鎖で修飾することも可能である。
【0030】
オリゴヌクレオチドは、ペプチド、細胞膜を渡る輸送を促進する剤(例えば、Letsingerら, 1989, Proc. Natl. Acad. Sci. USA 86:6553−6556;Lemaitreら, 1987, Proc. Natl. Acad. Sci. USA 84:648−652;国際特許公報第WO 88/09810号)または血液−脳関門を渡る輸送を促進する剤(例えば国際特許公報第WO 89/10134号)、ハイブリダイゼーションに誘発される切断剤(例えば、Krolら, 1988, BioTechniques 6:958−976を参照されたい)、またはインターカレート剤(例えばZon, 1988, Pharm. Res. 5:539−549を参照されたい)などの、他の付随する基を含むことも可能である。
【0031】
詳細には、アンチセンス・オリゴヌクレオチドは、限定されるわけではないが、5−フルオロウラシル、5−ブロモウラシル、5−クロロウラシル、5−ヨードウラシル、ヒポキサンチン、キサンチン、4−アセチルシトシン、5−(カルボキシヒドロキシメチル)ウラシル、5−カルボキシメチルアミノメチル−2−チオ−ウリジン、5−カルボキシメチルアミノメチルウラシル、ジヒドロウラシル、D−ガラクトシルケオシン、イノシン、N6−イソペンテニルアデニン、1−メチルグアニン、1−メチルイノシン、2,2−ジメチルグアニン、2−メチルアデニン、2−メチルグアニン、3−メチルシトシン、5−メチルシトシン、N6−アデニン、7−メチルグアニン、5−メチルアミノメチルウラシル、5−メトキシアミノメチル−2−チオウラシル、D−マンノシルケオシン、5N−メトキシカルボキシメチルウラシル、5−メトキシウラシル、2−メチル−チオ−N6−イソペンテニルアデニン、ウラシル−5−オキシ酢酸(v)、ワイブトキソシン(wybutoxosine)、シュードウラシル、ケオシン、2−チオシトシン、5−メチル−2−チオウラシル、2−チオウラシル、4−チオウラシル、5−メチルウラシル、ウラシル−5−オキシ酢酸メチルエステル、ウラシル−5−オキシ酢酸(v)、5−メチル−2−チオウラシル、3−(3−アミノ−3−N−2−カルボキシプロピル)ウラシル、(acp3)w、および2,6−ジアミノプリンを含む群より選択される、少なくとも1つの修飾塩基部分を含むことも可能である。
【0032】
別の態様において、オリゴヌクレオチドは、限定されるわけではないが、アラビノース、2−フルオロアラビノース、キシルロース、およびヘキソースを含む群より選択される、少なくとも1つの修飾糖部分を含む。
【0033】
適切なアンチセンス核酸の使用が、例えば、ZhengおよびKemeny(1995)Clin. Exp. Immunol., 100, 380−2;NellenおよびLichtenstein(1993)Trends Biochem. Sci., 18, 419−23, Stein(1992)Leukemia, 6, 697−74またはYacyshyn, B.R.ら(1998)Gastroenterology, 114, 1142にさらに記載される。
【0034】
さらに別の態様において、オリゴヌクレオチドは、2−a−アノマー・オリゴヌクレオチドである。a−アノマー・オリゴヌクレオチド(2−a−アノマーまたはa−アノマー)は、通常のβ−ユニットと対照的に、鎖が互いに平行に走る相補RNAと特異的な二本鎖ハイブリッドを形成する(Gautierら, 1987, Nucl. Acids Res. 15:6625−6641)。
【0035】
オリゴヌクレオチドを別の分子、例えばペプチド、ハイブリダイゼーションが誘発する架橋剤、輸送剤、ハイブリダイゼーションが誘発する切断剤等にコンジュゲート化することも可能である。
【0036】
本発明全体で、当該技術分野に知られる標準法によって、例えば自動化DNA合成装置(Biosearch、Applied Biosystems等から商業的に入手可能なものなど)の使用によって、本発明のオリゴヌクレオチドを合成することも可能である。例えば、Steinら(1988, Nucl. Acids Res. 16:3209)の方法によって、ホスホロチオエート・オリゴヌクレオチドを合成することも可能であるし、調節孔ガラスポリマー支持体(Sarinら, 1988, Proc. Natl. Acad. Sci. USA 85:7448−7451)等の使用によって、メチルホスホネート・オリゴヌクレオチドを調製することも可能である。
【0037】
特定の態様において、アンチセンス・オリゴヌクレオチドは、触媒性RNAまたはリボザイムを含む(例えば、国際特許公報第WO 90/11364号;Sarverら, 1990, Science 247:1222−1225を参照されたい)。別の態様において、オリゴヌクレオチドは2’−O−メチルリボヌクレオチド(Inoueら, 1987, Nucl. Acids Res. 15:6131−6148)、またはキメラRNA−DNA類似体(Inoueら, 1987, FEBS Lett. 215:327−330)である。
【0038】
別の態様において、本発明のアンチセンス核酸は、外因性配列からの転写によって細胞内で産生される。例えば、細胞に取り込まれるように、ベクターをin vivoで導入することも可能であり、その細胞内でベクターまたはその一部が転写され、本発明のアンチセンス核酸(RNA)が産生される。こうしたベクターは、構成要素タンパク質をコードする配列を含有するであろう。こうしたベクターは、望ましいアンチセンスRNAを産生するように転写されることが可能である限り、エピソームにとどまることも可能であるし、または染色体に組み込まれることも可能である。当該技術分野で標準的な組換えDNA技術法によって、こうしたベクターを構築することも可能である。ベクターは、プラスミド・ベクター、ウイルス・ベクター、または哺乳動物細胞における複製および発現が可能であることが当該技術分野に知られる他のものであることも可能である。アンチセンスRNAをコードする配列の発現は、哺乳動物細胞、好ましくはヒト細胞において、作用することが当該技術分野に知られるプロモーターいずれによることも可能である。こうしたプロモーターは誘導性または恒常性であることも可能である。こうしたプロモーターには、限定されるわけではないが、SV40初期プロモーター領域(BernoistおよびChambon, 1981, Nature 290:304−310)、ラウス肉腫ウイルスの3’末端反復配列(long terminal repeat)に含有されるプロモーター(Yamamotoら, 1980, Cell 22:787−797)、ヘルペス・チミジンキナーゼ・プロモーター(Wagnerら, 1981, Proc. Natl. Acad. Sci. USA 78:1441−1445)、メタロチオネイン遺伝子の制御配列(Brinsterら, 1982, Nature 296:39−42)等が含まれる。
【0039】
本発明のアンチセンス核酸は、構成要素タンパク質遺伝子、好ましくはヒト遺伝子のRNA転写物の少なくとも一部に相補的な配列を含む。しかし、絶対相補性は好ましいが必要ではない。「RNAの少なくとも一部に相補的な」配列は、本明細書において、RNAとハイブリダイズ可能であるのに十分な相補性を有し、安定な二重鎖を形成する配列を意味し;したがって、二本鎖アンチセンス核酸の場合、二重鎖DNAの一本鎖を試験することも可能であるし、または三重鎖形成をアッセイすることも可能である。ハイブリダイズする能力は、相補性の度合いおよびアンチセンス核酸の長さの両方に依存するであろう。一般的に、ハイブリダイズする核酸がより長ければ、構成要素タンパク質RNAとの塩基ミスマッチをより多く含有して、そしてなお安定な二重鎖(または場合によっては三重鎖)を形成することが可能になる。当業者は、ハイブリダイズした複合体の融点を決定する標準法の使用によって、ミスマッチの容認しうる度合いを確認することも可能である。
【0040】
遺伝子発現、本明細書ではGPR49遺伝子発現を下方制御するかまたはスイッチオフするプロセスにおける、RNA干渉のためのツールとしてのsiRNAの産生および使用は、例えばElbashir, S.M.ら(2001)Genes Dev., 15, 188またはElbashir, S.M.ら(2001)Nature, 411, 494に記載される。好ましくは、siRNAは30ヌクレオチド未満の長さを示し、siRNAのセンス鎖の同一ストレッチは、好ましくは少なくとも19ヌクレオチドである。
【0041】
リボザイムもまた、核酸、本明細書ではGPR49遺伝子の翻訳を阻害するのに適したツールであり、これはリボザイムがmRNAに特異的に結合し、そして切断することが可能であるためである。これらは例えば、Amarzguiouiら(1998)Cell. Mol. Life Sci., 54, 1175−202;Vaishら(1998)Nucleic Acids Res., 26, 5237−42;Persidis(1997)Nat. Biotechnol., 15, 921−2またはCoutureおよびStinchcomb(1996)Trends Genet., 12, 510−5に記載される。
【0042】
薬学的に許容しうるキャリアー中に有効量の核酸を含む、本発明の薬剤組成物を、本発明のタンパク質複合体を発現するかまたは過剰発現する種類の疾患または障害を有する患者に投与することも可能である。
【0043】
特定の障害または状態の治療に有効であろう核酸の量は、障害または状態の性質に応じるであろうし、そして標準的臨床技術によって決定可能である。可能な場合、ヒトで試験しそして使用する前に、核酸細胞傷害性をin vitroで測定し、そして次いで有用な動物モデル系で測定することが望ましい。
【0044】
特定の態様において、核酸を含む薬剤組成物を、リポソーム、微小粒子、または微小カプセルを介して投与する。本発明の多様な態様において、こうした組成物を用いて、核酸の持続放出を達成することが有用である可能性もある。特定の態様において、同定可能な中枢神経系細胞種に特異的な抗体を介してターゲティングされるリポソームを利用することが望ましい可能性もある(Leonettiら, 1990, Proc. Natl. Acad. Sci. U.S.A. 87:2448−2451;Renneisenら, 1990, J. Biol. Chem. 265:16337−16342)。
【0045】
いわゆる「低分子量分子」(以下、「LMW」と称する)は、タンパク質、ペプチド、抗体または核酸ではなく、そして5000Da未満、好ましくは2000Da未満、より好ましくは1000Da未満、最も好ましくは500Da未満の分子量を示す、分子である。こうしたLMWは、ライブラリーから出発するハイスループット法において同定可能である。こうした方法は当該技術分野で知られ、そして以下に詳細に論じられる。
【0046】
用語「結合性タンパク質」または「結合性ペプチド」は、GPR49に結合し、そしてこれを阻害する種類のタンパク質またはペプチドを指し、そして限定なしに、GPR49に対して向けられるポリクローナル抗体またはモノクローナル抗体、抗体断片およびタンパク質骨格を含む。
【0047】
本発明にしたがって、用語、抗体または抗体断片はまた、組換え的に調製され、そして適切な場合、修飾された、抗体またはその抗原結合部分、例えばキメラ抗体、ヒト化抗体、多機能抗体、二重特異性またはオリゴ特異性(oligospecific)抗体、一本鎖抗体およびF(ab)またはF(ab)2断片(例えば、EP−B1−0 368 684、US 4,816,567、US 4,816,397、WO 88/01649、WO 93/06213またはWO 98/24884を参照されたい)を意味するとして理解され、好ましくはFAB発現ライブラリーの補助で産生される。
【0048】
古典的な抗体の代替物として、例えば、GPR49に対するタンパク質骨格、例えばリポカリンに基づくアンチカリン(Besteら(1999)Proc. Natl. Acad. Sci. USA, 96, 1898−1903)を使用することもまた可能である。選択されるハプテン、本明細書ではGPR49に結合する方式で、例えば「コンビナトリアル・タンパク質設計」アプローチによって、リポカリン、例えばレチノール結合性タンパク質またはビリン結合性タンパク質の天然リガンド結合部位を改変することも可能である(Skerra, 2000, Biochim. Biophys. Acta, 1482, 337−50)。他の既知のタンパク質骨格が、分子認識のための抗体の代替物であることが知られる(Skerra(2000)J. Mol. Recognit., 13, 167−187)。
【0049】
当業者に周知の方法にしたがって、例えば哺乳動物、例えばウサギを、適切な場合、例えばフロイントのアジュバントおよび/または水酸化アルミニウムゲルの存在下で、GPR49で免疫することによって、抗体または抗体断片を調製する方法が達成される(例えば、Diamond, B.A.ら(1981)The New England Journal of Medicine:1344−1349を参照されたい)。免疫学的反応の結果として、動物で形成されるポリクローナル抗体を、続いて、周知の方法を用いて血液から単離し、そして例えばカラム・クロマトグラフィーによって精製することも可能である。例えば、モノクローナル抗体をWinter & Milsteinの既知の方法(Winter, G. & Milstein, C.(1991)Nature, 349, 293−299)にしたがって、調製することも可能である。
【0050】
詳細には、適切な被験者を、免疫原としてのポリペプチドで免疫することによって、上述のように、ポリクローナル抗体を調製することも可能である。好ましいポリクローナル抗体組成物は、本発明の単数または複数のポリペプチドに対して向けられる抗体に関して選択されているものである。特に好ましいポリクローナル抗体調製物は、既定の単数または複数のポリペプチドに対して向けられる抗体のみを含有するものである。特に好ましい免疫原組成物は、他のヒト・タンパク質を含有しないもの、例えば本発明のポリペプチドの組換え発現のため、非ヒト宿主細胞を用いて作製した免疫原組成物である。こうした方式では、この免疫原に対して作製して生じた抗体組成物に認識されるのは、本発明の単数または複数のポリペプチドの一部として存在する、単数または複数のヒト・エピトープのみであろう。
【0051】
標準的技術、例えば固定ポリペプチドを用いた酵素連結免疫吸着アッセイ(ELISA)によって、一定時間に渡って、免疫した被験者における抗体力価を監視することも可能である。望ましい場合、哺乳動物から(例えば血液から)、抗体分子を単離し、そしてさらに、周知の技術、例えばプロテインAクロマトグラフィーによって、さらに精製してIgG分画を得ることも可能である。あるいは、本発明のタンパク質またはポリペプチドに特異的な抗体を、例えばアフィニティー・クロマトグラフィーによって、選択する(例えば部分的に精製する)かまたは精製することも可能である。例えば、組換え的に発現され、そして精製された(または部分的に精製された)本発明のタンパク質を、本明細書に記載するように産生し、そして例えばクロマトグラフィー・カラムなどの固体支持体に、共有的にまたは非共有的にカップリングする。次いで、該カラムを用いて、多数の異なるエピトープに対して向けられる抗体を含有する試料から、本発明のタンパク質に特異的な抗体をアフィニティー精製し、それによって実質的に精製された抗体組成物、すなわち混入抗体を実質的に含まないものを生成することも可能である。この関連において、実質的に精製された抗体組成物によって、抗体試料が、本発明の望ましいタンパク質またはポリペプチドに対するもの以外のエピトープに対して向けられる混入抗体を(乾燥重量にして)最大わずか30%しか含有せず、そして好ましくは(乾燥重量にして)試料の最大20%、さらにより好ましくは最大10%、そして最も好ましくは最大5%のみが混入抗体であることを意味する。精製抗体組成物は、組成物中の抗体の少なくとも99%が、本発明の望ましいタンパク質またはポリペプチドに対して向けられることを意味する。
【0052】
免疫後の適切な時点で、例えば特異的抗体力価が最も高いとき、抗体産生細胞を被験者から得て、そして該細胞を用いて、標準的技術、例えば元来、KohlerおよびMilstein, 1975, Nature 256:495−497に記載されたハイブリドーマ技術、ヒトB細胞ハイブリドーマ技術(Kozborら, 1983, Immunol. Today 4:72)、EBVハイブリドーマ技術(Coleら, 1985, Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, Inc., pp.77−96)またはトリオーマ技術によって、モノクローナル抗体を調製することも可能である。ハイブリドーマを産生するための技術は周知である(一般的に、Current Protocols in Immunology 1994, Coliganら(監修)John Wiley & Sons, Inc., ニューヨーク州ニューヨークを参照されたい)。本発明のモノクローナル抗体を産生するハイブリドーマ細胞は、例えば標準的ELISAアッセイを用いて、目的のポリペプチドに結合する抗体に関して、ハイブリドーマ培養上清をスクリーニングすることによって、検出される。
【0053】
モノクローナル抗体分泌ハイブリドーマを調製する代わりに、組換えコンビナトリアル免疫グロブリン・ライブラリー(例えば抗体ファージディスプレイ・ライブラリー)を、目的のポリペプチドでスクリーニングすることによって、本発明のポリペプチドに対して向けられるモノクローナル抗体を同定し、そして単離することも可能である。ファージディスプレイ・ライブラリーを生成し、そしてスクリーニングするためのキットが商業的に入手可能である(例えば、Pharmacia組換えファージ抗体系、カタログ番号27−9400−01;およびStratagene SurfZAPファージディスプレイ・キット、カタログ番号240612)。さらに、抗体ディスプレイ・ライブラリーを生成し、そしてスクリーニングする際に使用するのに特に受け入れられる方法および試薬の例は、例えば、米国特許第5,223,409号;PCT公報第WO 92/18619号;PCT公報第WO 91/17271号;PCT公報第WO 92/20791号;PCT公報第WO 92/15679号;PCT公報第WO 93/01288号;PCT公報第WO 92/01047号;PCT公報第WO 92/09690号;PCT公報第WO 90/02809号;Fuchsら, 1991, Bio/Technology 9:1370−1372;Hayら, 1992, Hum. Antibod. Hybridomas 3:81−85;Huseら, 1989, Science 246:1275−1281;Griffithsら, 1993, EMBO J. 12:725−734に見出すことも可能である。
【0054】
さらに、標準的組換えDNA技術を用いて作製可能である、ヒト部分および非ヒト部分両方を含む、キメラおよびヒト化モノクローナル抗体などの組換え抗体が、本発明の範囲内である。キメラ抗体は、異なる部分が異なる動物種に由来する分子、例えばネズミmAb由来の可変領域およびヒト免疫グロブリン定常領域を有するものである(例えば、本明細書に完全に援用される、Cabillyら、米国特許第4,816,567号;およびBossら、米国特許第4,816,397号を参照されたい)。ヒト化抗体は、非ヒト種由来の1以上の相補性決定領域(CDR)およびヒト免疫グロブリン分子由来のフレームワーク領域を有する、非ヒト種由来の抗体分子である(例えば、本明細書に完全に援用される、Queen、米国特許第5,585,089号を参照されたい)。こうしたキメラおよびヒト化モノクローナル抗体は、当該技術分野に知られる組換えDNA技術によって、例えば、PCT公報第WO 87/02671号;欧州特許出願184,187;欧州特許出願171,496;欧州特許出願173,494;PCT公報第WO 86/01533号;米国特許第4,816,567号;欧州特許出願125,023;Betterら, 1988, Science 240:1041−1043;Liuら, 1987, Proc. Natl. Acad. Sci. USA 84:3439−3443;Liuら, 1987, J. Immunol. 139:3521−3526;Sunら, 1987, Proc. Natl. Acad. Sci. USA 84:214−218;Nishimuraら, 1987, Canc. Res. 47:999−1005;Woodら, 1985, Nature 314:446−449;およびShawら, 1988, J. Natl. Cancer Inst. 80:1553−1559);Morrison, 1985, Science 229:1202−1207;Oiら, 1986, Bio/Techniques 4:214;米国特許第5,225,539号;Jonesら, 1986, Nature 321:552−525;Verhoeyanら, 1988, Science 239:1534;およびBeidlerら, 1988, J. Immunol. 141:4053−4060に記載される方法を用いて、産生可能である。
【0055】
完全ヒト抗体は、ヒト患者の療法的治療に特に望ましい。こうした抗体は、例えば、内因性免疫グロブリン重鎖および軽鎖遺伝子を発現不能であるが、ヒト重鎖および軽鎖遺伝子を発現可能である、トランスジェニックマウスを用いて、産生可能である。選択した抗原、例えば本発明のポリペプチドすべてまたは一部を用いて、通常の様式で該トランスジェニックマウスを免疫する。慣用的ハイブリドーマ技術を用いて、該抗原に対して向けられるモノクローナル抗体を得ることも可能である。該トランスジェニックマウスに宿するヒト免疫グロブリン導入遺伝子は、B細胞分化中に再編成し、そして続いてクラス・スイッチおよび体細胞突然変異を経る。したがって、こうした技術を用いて、療法的に有用なIgG、IgAおよびIgE抗体を産生することが可能である。ヒト抗体を産生するためのこの技術の概説には、LonbergおよびHuszar, 1995, Int. Rev. Immunol. 13:65−93を参照されたい。ヒト抗体およびヒト・モノクローナル抗体を産生するためのこの技術の詳細な考察、およびこうした抗体を産生するためのプロトコルには、例えば米国特許第5,625,126号;米国特許第5,633,425号;米国特許第5,569,825号;米国特許第5,661,016号;および米国特許第5,545,806号を参照されたい。さらに、Abgenix, Inc.(カリフォルニア州フリーモント)などの企業は、上述のものと類似の技術を用いて、選択した抗原に対して向けられるヒト抗体を提供するのに従事することも可能である。
【0056】
選択したエピトープを認識する完全ヒト抗体を、「ガイド選択」と称される技術を用いて、生成することも可能である。このアプローチでは、選択した非ヒト・モノクローナル抗体、例えばネズミ抗体を用いて、同一エピトープを認識する完全ヒト抗体の選択をガイドする(Jespersら, 1994, Bio/technology 12:899−903)。
【0057】
当該技術分野に知られる技術によって、複合体のイディオタイプを含有する抗体断片を生成することも可能である。例えば、こうした断片には、限定されるわけではないが、抗体分子のペプシン消化によって産生可能なF(ab’)2断片;F(ab’)2断片のジスルフィド架橋を還元することによって生成可能なFab’断片;パパインおよび還元剤で抗体分子を処理することによって生成可能なFab断片;およびFv断片が含まれる。
【0058】
抗体産生において、望ましい抗体に関するスクリーニングは、当該技術分野に知られる技術、例えばELISA(酵素連結免疫吸着アッセイ)によって達成可能である。複合体の特定のドメイン、またはその誘導体に特異的な抗体を選択するため、こうしたドメインを含有する複合体の断片、またはその誘導体に結合する産物に関して、生成されたハイブリドーマをアッセイすることも可能である。本発明の複合体、あるいはその誘導体または相同体に特異的に結合するが、複合体、あるいはその誘導体または相同体の個々のタンパク質に特異的に結合しない、抗体を選択するため、複合体に対する陽性結合および個々のタンパク質構成要素に対する結合の欠如に基づいて、選択することも可能である。
【0059】
既定の単数または複数のタンパク質の局在および/または定量に関連する、当該技術分野に知られる方法、例えばこれらのタンパク質を画像化するための方法、適切な生理学的試料中のそのレベルを測定する方法(イムノアッセイによる)、診断法などで、前述の抗体を使用することも可能である。これはまた、複合体の誘導体または相同体に関してもあてはまる。
【0060】
好ましい態様において、GPR49阻害剤は、配列:AACAGCAGTATGGACGACCTTを持つsiRNAである。
GPR49は、LH(黄体形成ホルモン)、リラキシン、IGF、FSH(卵胞刺激ホルモン)、およびTSHの受容体に類似性を示す。したがって、LH、リラキシン、IGF、FSH、およびTSHは、GPR49の生理学的リガンドでありうる。さらに、
WO−03 020 726における
【0061】
【化1】
【0062】
そしてWO−00 187 287における
【0063】
【化2】
【0064】
のLHおよびFSH受容体のアゴニストと同定されたチエノ[2,3−d]ピリミジンが、GPR49のアゴニストである可能性もある。
上に論じるように、GPR49は、ガンマ・セクレターゼ活性および/またはベータ・セクレターゼ活性の制御に関与するタンパク質複合体の一部である。したがって、好ましい態様において、GPR49相互作用分子または阻害剤は、タンパク質複合体、好ましくはAph1a複合体、Fe65L2複合体、APP−C99複合体、またはBACE1複合体の一部であるGPR49分子に作用する。
【0065】
前記タンパク質複合体は、ガンマ−セクレターゼ・サブユニットAph1a、ガンマ−セクレターゼ基質APP−C99(これに加えてそのアダプターFe65L2)およびベータ−セクレターゼ・タンパク質と相互作用するタンパク質の集合と同定されてきている。
【0066】
プレセニリン1および2(それぞれ、Psen1およびPsen2、PS1およびPS2とも称される)は、小胞体、ゴルジ体、および細胞表面にもまた局在する、内在性膜タンパク質である(Kovacs, Nat Med 2. 224)。これらは主に、NTFおよびCTF細胞内タンパク質分解断片のヘテロ二量体として見出される。プレセニリンを切断するプロテアーゼ(「プレセニリナーゼ」)は未知であり、プロセスが自己触媒性である可能性があり、またPS(自己)タンパク質分解の機能的な重要性は不明である。プレセニリンは、アミロイド前駆タンパク質(APP)(De Strooperら, Nature 391, 387)およびNotch受容体(De Strooperら, Nature 398, 518)のタンパク質分解プロセシングに関与する。さらに、プレセニリンは、細胞接着タンパク質、アルファおよびベータ−カテニン、N−カドヘリン、ならびにE−カドヘリン(Georgakopoulosら, Mol Cell 4, 893)およびアルマジロ・ファミリーの他のメンバー(Yuら, J Biol Chem 273, 16470)と会合する。プレセニリンによるAPPプロセシングは、APPを切断し、A−ベータ・ペプチドのC末端を生成する、ガンマ−セクレターゼに対する影響を通じる。PS1は、APPのC83およびC99プロセシングC末端断片(Xiaら, Proc Natl Acad Sci USA, 94, 8208)、ニカストリン(Yuら, Nature 407, 48)およびPen−2(Francisら, Dev Cell 3, 85)と会合する。
【0067】
プレセニリン・プロセシングには、Aph−1aなどのAph−1タンパク質(Francisら, Dev Cell 3, 85)が必要である。プレセニリンが、ガンマ−セクレターゼ活性を直接制御するかどうか、またはこれらがそれ自体、プロテアーゼ酵素であるのかどうかは明らかでない(KopanおよびGouate, Genes Dev 14, 2799)。ガンマ・セクレターゼ活性は、これらのタンパク質の多量体複合体を含むことも可能である(Yuら, Nature 407, 48)が、これらのタンパク質間の関係がどのようにセクレターゼ活性に影響を及ぼすかは知られていない。
【0068】
Aph−1およびPen−2は、線虫におけるプレセニリン増進剤(「pen」)に関するスクリーニングにおいて、最近クローニングされ、そしてAph−2(ニカストリン)と遺伝子相互作用することが示された。Aph−1の欠損は、Notchシグナル伝達およびニカストリン局在に影響を及ぼす。Aph−1およびPen−2は、Notch切断、ガンマ−セクレターゼ活性、およびプロセシングされたプレセニリンの集積に必要である。Francisらは、これらの遺伝子、Aph−1a、Aph−1bおよびPen−2の推定上のヒト・オルソログをクローニングし、そして最近、Leeらもまた、ヒトAph−1 cDNAをクローニングした。ガンマ−セクレターゼ複合体の正確な構成要素は知られていないが、これらの2つの新規タンパク質は、複合体の構成要素または付属因子である可能性もあり、そして一緒に、プレセニリンと、またはプレセニリン/ニカストリン複合体と直接相互作用することも可能である。したがって、ニカストリンは、活性ガンマ−セクレターゼ複合体のメンバーであり、そしてこの複合体に重要なのは、該タンパク質の完全グリコシル化型であるという最近の証拠がある。
【0069】
Fe65様2(Fe65L2)は、よく性質決定された細胞内APP相互作用アダプタータンパク質Fe65の相同体である(Duilio A, Faraonio R, Minopoli G, Zambrano N, Russo T(1998)Fe65L2: a new member of the Fe65 protein family interacting with the intracellular domain of the Alzheimer’s beta−amyloid precursor protein. Biochem J. 330(Pt 1):513−9)。該タンパク質は、脳で高発現され、そしてC末端PTBドメインを通じてAPPと相互作用する(Tanahashi H, Tabira T(1999)Molecular cloning of human Fe65L2 and its interaction with the Alzheimer’s beta−amyloid precursor protein. Neurosci Lett. 261(3):143−6)。Fe65L2は、核に転位置して、そして転写を制御することが知られており(Bruni P, Minopoli G, Brancaccio T, Napolitano M, Faraonio R, Zambrano N, Hansen U, Russo T(2002)Fe65, a ligand of the Alzheimer’s beta−amyloid precursor protein, blocks cell cycle progression by down−regulating thymidylate synthase expression. J. Biol. Chem. 277(38):35481−8)、該タンパク質が、APPの下流シグナル伝達機能に関与することが示唆される。in vitroでFe65L2を過剰発現すると、Aベータの分泌/生成が促進される(Tanahashi H, Tabira T(2002)Characterization of an amyloid precursor protein−binding protein Fe65L2 and its novel isoforms lacking phosphotyrosine−interaction domains. Biochem J. 367(Pt 3):687−95)。
【0070】
ベータ−セクレターゼ(BACE)活性は、細胞外ドメインでAPPを切断し、分泌可溶性APPbの分断を生じ、そして99残基のC末端膜貫通断片(APP−C99)を生じる。Vassarら(Science 286, 735−741)は、APPのベータ−セクレターゼと推測される特性を有する膜貫通アスパラギン酸プロテアーゼをクローニングし、該プロテアーゼをBACE1と名づけた。BACE1ノックアウトマウス由来の脳および初代皮質培養物は、検出可能なベータ−セクレターゼ活性を示さず、そしてBACEノックアウトマウス由来の初代皮質培養物は、APPからはるかに少ないアミロイド−ベータしか産生しなかった。これによって、BACE1のパラログであるBACE2ではなく、BACE1がAPPの主なベータ−セクレターゼであることが示唆される。BACE1は、501アミノ酸のタンパク質であり、21aaのシグナルペプチドに続いて、aa22〜45に渡る、プロタンパク質ドメインを含有する。選択的スプライシング型、BACE−I−457およびBACE−I−476がある。成熟タンパク質の管腔ドメインに続いて、予測される1つの膜貫通ドメインおよび24aaの短い細胞質ゾルC末端テールがある。BACE1は、1型膜貫通タンパク質であり、活性部位が膜の管腔側にあると予測され、ここでベータ−セクレターゼがAPP、およびありうる他の未同定の基質を切断する。BACE1は、明らかに、APPをA−ベータにプロセシングするのに必要とされる重要な酵素であるが、最近の証拠は、BACE1のさらなる潜在的基質および機能を示唆している(J. Biol. Chem. 279, 10542−10550)。現在まで、制御機能または調節機能を持つBACE1相互作用タンパク質は、まったく記載されてきていない。
【0071】
上に説明するように、驚くべきことに、本発明の関連において、GPR49が、特にベータ−セクレターゼおよび/またはガンマ−セクレターゼ活性によって、APPのタンパク質分解プロセシングを制御するタンパク質複合体の一部であることが発見された。したがって、好ましい態様において、阻害剤または相互作用分子は、ベータ−セクレターゼおよび/またはガンマ・セクレターゼの活性を調節する。
【0072】
本発明全体で、用語「ガンマ・セクレターゼおよび/またはベータ・セクレターゼの活性を調節する」は、酵素活性が直接または間接的に調節されることを含む。これは、GPR49調節剤がこれらの酵素のいずれかに直接結合可能であるか、あるいはより好ましくは、GPR49に影響を発揮して、次に例えばタンパク質−タンパク質相互作用によってまたはシグナル伝達によって、または小さい代謝産物を介して、これらの酵素のいずれかの活性を調節可能であることを意味する。
【0073】
本発明全体で、ベータ・セクレターゼ調節剤が、完全にまたは部分的に、ベータ・セクレターゼの活性を阻害することが好ましい。本発明全体で、GPR49調節剤の最も好ましい機能的結果は、Aベータ−42生成の減少である。
【0074】
本発明の関連において、「ガンマ・セクレターゼおよび/またはベータ・セクレターゼの活性を調節する」は、活性が減少して、より少ない産物が形成されるかまたは産物がまったく形成されず、最も好ましくはより少ないAベータ−42が形成されるかまたはAベータ−42がまったく形成されない(部分的または完全阻害)か、あるいはそれぞれの酵素が異なる産物(ガンマ−セクレターゼの場合、Aベータ−42の代わりにより短いアミノ酸配列の、例えばAベータ−38または他のAベータ・ペプチド)を産生するか、あるいは産物の相対量が異なる(ガンマ・セクレターゼの場合、例えばAベータ40対Aベータ−42の比が変化する、好ましくは増加する)ことを意味する。さらに、調節剤が、ガンマ・セクレターゼまたはベータ・セクレターゼいずれか、あるいは両酵素の活性を調節することが含まれる。
【0075】
ガンマ・セクレターゼ活性の調節剤に関しては、この調節剤が、ガンマ・セクレターゼ活性を阻害することが好ましい。しかし、Aベータ・ペプチド種の総量が変化しないが、Aベータ−42の代わりに、より多くのAベータ−38が産生される方式で、ガンマ・セクレターゼの活性がシフトすることもまた、好ましい。
【0076】
例えばAPPプロセシングを測定することによって、例えば、産生されるAベータ・ペプチド種のレベル、最も重要なことに、Aベータ−42のレベルを測定することによって、ガンマ・セクレターゼ活性を測定することも可能である(以下の実施例セクションを参照されたい)。
【0077】
BACE1活性を測定するため、ウェスタンブロッティングによって、アルファ−およびベータ−C末端APP断片間の比の変化を分析することも可能であり(Blaskoら, J Neural Transm 111, 523);BACE1活性アッセイのさらなる例には、限定されるわけではないが:ベータ−ガラクトシダーゼ・レポーター活性を再構成し、そして測定する、BACE1切断部位を含有する環状化酵素ドナー・ペプチドの使用(Naqviら, J Biomol Screen. 9, 398);消光蛍光ペプチド基質および蛍光測定の使用(Andrauら, J. Biol Chem 278, 25859);酵素(アルカリホスファターゼなど)を、BACE1認識配列を含有するアミノ酸ストレッチを介して、ゴルジ体常在タンパク質に連結する、組換えキメラタンパク質を利用する、細胞に基づくアッセイの使用(Ohら, Anal Biochem, 323, 7);蛍光共鳴エネルギー移動(FRET)に基づくアッセイ(Kennedyら, Anal Biochen 319, 49);酵母における細胞増殖選択系(Luthiら, Biochim Biophys Acta 1620, 167)が含まれる。
【0078】
好ましくは、神経変性疾患は、アルツハイマー病である。
本発明にしたがって、GPR49相互作用分子を用いて、薬剤組成物を調製する。
したがって、本発明は、有効量で被験者に投与可能な薬剤組成物を提供する。好ましい側面において、療法剤は、実質的に精製されている。被験者は、好ましくは、限定されるわけではないが、ウシ、ブタ、ウマ、ニワトリ、ネコ、イヌなどを含む動物であり、そして好ましくは哺乳動物であり、そして最も好ましくはヒトである。特定の態様において、非ヒト哺乳動物が被験者である。
【0079】
多様な搬送系が知られ、そしてこうした系を用いて、本発明の療法剤を投与することも可能であり、こうした系には、例えばリポソーム、微小粒子、および微小カプセル中の被包:療法剤を発現可能な組換え細胞の使用、受容体が仲介するエンドサイトーシスの使用(例えば、WuおよびWu, 1987, J. Biol. Chem. 262:4429−4432);レトロウイルスベクターまたは他のベクターの一部としての療法核酸の構築などがある。導入法には、限定されるわけではないが、皮内、筋内、腹腔内、静脈内、皮下、鼻内、硬膜外、および経口経路が含まれる。好適な経路いずれによって、例えば注入によって、ボーラス(bolus)注射によって、上皮または皮膚粘膜裏打ち(例えば口腔、直腸および腸粘膜など)を通じた吸収によって、化合物を投与することも可能であるし、そして他の生物学的活性剤と一緒に投与することも可能である。投与は全身性または局所であることも可能である。さらに、脳室内注射およびクモ膜下注射を含む、適切な経路いずれかによって、本発明の薬剤組成物を中枢神経系に導入することが望ましい可能性もあり;脳室内注射は、例えばOmmaya容器などの容器に取り付けた、脳室内カテーテルによって容易にすることも可能である。例えば吸入器または噴霧器を使用し、そしてエアロゾル化剤と配合することによって、肺投与もまた使用可能である。
【0080】
特定の態様において、治療の必要がある領域に、本発明の薬剤組成物を局所投与することが望ましい可能性もある。これは、例えば、そして限定なしに、手術中の局所注入によって、例えば手術後の創傷手当て(dressing)と組み合わせた局所適用によって、注射によって、カテーテルによって、座薬によって、あるいはシアラスティック(sialastic)膜などの膜またはファイバーを含む、多孔、無孔、またはゼラチン性物質である移植物によって、達成可能である。1つの態様において、悪性腫瘍または新生物組織もしくは新生物発生前組織の部位(または以前の部位)に直接注射することによって、投与することも可能である。
【0081】
別の態様において、療法剤を、小胞、特にリポソーム中で搬送することも可能である(Langer, 1990, Science 249:1527−1533;Treatら, 1989, :Liposomes in the Therapy of Infectious Disease and Cancer中, Lopez−BeresteinおよびFidler監修, Liss, ニューヨーク, pp.353−365;Lopez−Berestein, 同書, pp.317−327;一般的には同書を参照されたい)。
【0082】
さらに別の態様において、徐放系を介して、療法剤を搬送することも可能である。1つの態様において、ポンプを使用することも可能である(Langer、上記;Sefton, 1987, CRC Crit. Ref. Biomed. Eng. 14:201−240;Buchwaldら, 1980, Surgery 88:507−516;Saudekら, 1989, N. Engl. J. Med. 321:574−579)。別の態様において、ポリマー物質を用いることも可能である(Medical Applications of Controlled Release, LangerおよびWise監修, CRC Press, フロリダ州ボーカラートン, 1974;Controlled Drug Bioavailability, Drug Product Design and Performance, SmolenおよびBall監修, Wiley, ニューヨーク, 1984;RangerおよびPeppas, 1983, Macromol. Sci. Rev. Macromol. Chem. 23:61;Levyら, 1985, Science 228:190−192;Duringら, 1989, Ann. Neurol. 25:351−356;Howardら, 1989, J. Neurosurg. 71:858−863)。さらに別の態様において、療法ターゲット、すなわち脳に近接して、徐放系を配置し、こうして全身用量のうち、ごく少量しか必要としないことも可能である(例えばGoodson, 1984, :Medical Applications of Controlled Release中, 上記, Vol.2, pp.115−138)。他の徐放系が、Langer(1990, Science 249:1527−1533)による概説に論じられている。
【0083】
療法剤が、好ましくはタンパク質療法剤をコードする核酸である、特定の態様において、適切な核酸発現ベクターの一部として該核酸を構築し、そして細胞内に存在するように投与することによって、核酸をin vivoで投与して、コードされるタンパク質の発現を促進することも可能であり、これは、例えばレトロウイルスベクターの使用によって(米国特許第4,980,286号)、または直接注射によって、または微小粒子銃(例えば遺伝子銃;Biolistic、Dupont)の使用によって、または核酸を脂質、細胞表面受容体もしくはトランスフェクション剤でコーティングすることによって、または核に進入することが知られるホメオボックス様ペプチド(例えばJoliotら, 1991, Proc. Natl. Acad. Sci. USA 88:1864−1868)に連結した核酸を投与することによって、実行可能である。あるいは、核酸療法剤を細胞内に導入し、そして発現のため、宿主細胞DNA内に相同組換えによって取り込ませることも可能である。
【0084】
一般的に、本発明の薬剤組成物は、療法的有効量の療法剤、および薬学的に許容しうるキャリアーを含む。特定の態様において、用語「薬学的に許容しうる」は、動物、そしてより詳細にはヒトにおける使用のため、連邦政府または州政府の監督官庁に認可されたか、あるいは米国薬局方または他の一般的に認められる薬局方に列挙されていることを意味する。用語「キャリアー」は、療法剤を一緒に投与する、希釈剤、アジュバント、賦形剤、またはビヒクルを指す。こうした薬剤キャリアーは、無菌液体、例えば水および油であることも可能であり、石油、動物、植物または合成起源のものが含まれ、限定されるわけではないが、ピーナツ油、ダイズ油、ミネラルオイル、ゴマ油等が含まれる。薬剤組成物を経口投与する場合は、水が好ましいキャリアーである。薬剤組成物を静脈内投与する場合は、生理食塩水および水性デキストロースが好ましいキャリアーである。好ましくは、生理食塩水溶液、並びに水性デキストロースおよびグリセロール溶液が、注射可能溶液の液体キャリアーとして使用される。適切な薬剤賦形剤には、デンプン、グルコース、ラクトース、スクロース、ゼラチン、モルト、米、小麦粉、チョーク、シリカゲル、ステアリン酸ナトリウム、モノステアリン酸グリセロール、タルク、塩化ナトリウム、脱脂粉乳、グリセロール、プロピレン、グリコール、水、エタノール等が含まれる。組成物は、望ましい場合、少量の湿潤剤または乳化剤、あるいはpH緩衝剤もまた含有することも可能である。これらの組成物は、溶液、懸濁物、エマルジョン、錠剤、ピル、カプセル、粉末、持続放出配合物等の形を取ることも可能である。伝統的な結合剤およびキャリアー、例えばトリグリセリドを用いて、組成物を座薬として配合することも可能である。経口配合物は、薬剤等級のマンニトール、ラクトース、デンプン、ステアリン酸マグネシウム、サッカリン・ナトリウム、セルロース、炭酸マグネシウムなどの標準的キャリアーを含むことも可能である。適切な薬剤キャリアーの例は、E.W. Martinによる、“Remington’s Pharmaceutical Sciences”に記載される。こうした組成物は、患者に適切に投与する形を提供するように、適切な量のキャリアーと一緒に、療法的有効量の療法剤、好ましくは精製型のものを含有するであろう。配合物は、投与様式に適していなければならない。
【0085】
好ましい態様において、日常的な方法にしたがって、ヒトへの静脈内投与に適応させた薬剤組成物として、組成物を配合する。典型的には、静脈内投与のための組成物は、無菌等張水性緩衝剤中の溶液である。必要な場合、組成物はまた、可溶化剤および注射部位での疼痛を和らげるリドカインなどの局所麻酔剤も含むことも可能である。一般的に、成分を別個に供給するか、または単位投薬型中で一緒に混合して供給し、例えば活性剤の量を示すアンプルまたはサシェ(sachette)などの密封容器中、凍結乾燥粉末または水不含濃縮物として供給する。組成物を注入によって投与しようとする場合、無菌薬剤等級の水または生理食塩水を含有する注入ビンを用いて、分配することも可能である。組成物を注射によって投与しようとする場合、投与前に、成分を混合可能であるように、注射用の無菌水または生理食塩水のアンプルを提供することも可能である。
【0086】
本発明の療法剤を中性型または塩型で配合することも可能である。薬学的に許容しうる塩には、塩酸、リン酸、酢酸、シュウ酸、酒石酸などに由来する遊離型のカルボキシル基とともに形成されるもの、イソプロピルアミン、トリエチルアミン、2−エチルアミノエタノール、ヒスチジン、プロカインなどに由来するものなどの遊離型のアミン基とともに形成されるもの、並びにナトリウム、カリウム、アンモニウム、カルシウム、および水酸化第二鉄などに由来するものが含まれる。
【0087】
特定の障害または状態の治療に有効であろう、本発明の療法剤の量は、障害または状態の性質に応じるであろうし、そして標準的臨床技術によって決定可能である。さらに、場合によって、in vitroアッセイを使用して、最適投薬量範囲を同定するのを補助することも可能である。配合物に使用しようとする正確な用量はまた、投与経路、および疾患または障害の重大性にも応じるであろうし、そして担当医の判断および各患者の状況にしたがって、決定すべきである。しかし、静脈内投与に適した投薬量範囲は、一般的に、キログラム体重あたり、活性化合物約20〜500マイクログラムである。鼻内投与に適した投薬量範囲は、一般的に、約0.01pg/kg体重〜1mg/kg体重である。有効用量は、in vitroまたは動物モデル試験系から得られる用量−反応曲線から推定可能である。
【0088】
座薬は、一般的に、重量にして0.5%〜10%の範囲の活性成分を含有し;経口配合物は、好ましくは、10%〜95%の活性成分を含有する。
本発明はまた、本発明の薬剤組成物の1以上の成分が充填された、1以上の容器を含む、薬剤パックまたはキットも提供する。場合によって、こうした容器(単数または複数)に付随して、薬剤または生物学的製品の製造、使用または販売を規制する政府機関によって規定された形で、政府機関による、ヒト投与のための製造、使用または販売の認可を示す情報を示すことも可能である。
【0089】
本発明のキットはまた、複合体機構の本質的な構成要素をコードする発現ベクターも含有することも可能であり、この構成要素は、発現された後、生物学的に活性な複合体を形成するため、再構成されることも可能である。こうしたキットは、好ましくはまた、必要な緩衝剤および試薬も含有する。場合によって、こうした容器(単数または複数)に付随して、キット使用のための説明書、並びに/あるいは薬剤または生物学的製品の製造、使用または販売を規制する政府機関によって規定された形で、政府機関による、ヒト投与のための製造、使用または販売の認可を示す情報を示すことも可能である。
【0090】
本発明はさらに、有効量のGPR49相互作用分子または阻害剤あるいは本発明の薬剤組成物を、神経変性疾患、好ましくはアルツハイマー病を患う被験者に投与する、治療法に関する。
【0091】
本発明のこの方法に関して、上記のすべての態様は、本発明の使用のため、あてはまる。
本発明はさらに、ガンマ−セクレターゼ調節剤および/またはベータ−セクレターゼ調節剤を同定する方法であって:
a.既定の試験化合物がGPR49相互作用分子であるかどうか決定することによって、GPR49相互作用分子を同定し、
b.工程a)のGPR49相互作用分子がガンマ−セクレターゼ活性またはベータ−セクレターゼ活性を調節可能であるかどうか決定する
工程を含む、前記方法に関する。
【0092】
本発明の好ましい態様において、工程a)において、試験化合物をGPR49と接触させ、そして試験化合物とGPR49の相互作用を決定する。好ましくは、候補分子がGPR49に結合するかどうかを測定する。
【0093】
本発明の好ましい態様において、GPR49活性を調節する、好ましくは阻害するかどうかを見出すために、工程a)で同定されたGPR49相互作用分子をまず、上記のようなGPR49活性試験に供し(実施例3も参照されたい)、そして次いで、工程b)に供する(Aベータ低下効果に関する試験)。
【0094】
本発明の方法を、好ましくは、ハイスループットアッセイの関連で行う。こうしたアッセイは、当業者に知られる。
スクリーニングしようとする試験分子または候補分子は、限定された数の明記される化合物の混合物として、または化合物ライブラリー、ペプチド・ライブラリー等として、提供されることも可能である。スクリーニングしようとする剤/分子はまた、複合体活性または形成を調節可能な、抗血清、アンチセンス核酸等のすべての形を含むことも可能である。スクリーニングのための典型的な候補分子およびライブラリーを以下に示す。
【0095】
ライブラリーのスクリーニングは、一般的に知られる多様な方法のいずれによって、達成することも可能である。例えばペプチド・ライブラリーのスクリーニングを開示する、以下の参考文献を参照されたい:ParmleyおよびSmith, 1989, Adv. Exp. Med. Biol. 251:215−218;ScottおよびSmith, 1990, Science 249:386−390;Fowlkesら, 1992, BioTechniques 13:422−427;Oldenburgら, 1992, Proc. Natl. Acad. Sci. USA 89:5393−5397;Yuら, 1994, Cell 76:933−945;Staudtら, 1988, Science 241:577−580;Bockら, 1992, Nature 355:564−566;Tuerkら, 1992, Proc. Natl. Acad. Sci. USA 89:6988−6992;Ellingtonら, 1992, Nature 355:850−852;すべてLadnerらに対する米国特許第5,096,815号、米国特許第5,223,409号、および米国特許第5,198,346号;RebarおよびPabo, 1993, Science 263:671−673;並びに国際特許公報第WO 94/18318号。
【0096】
特定の態様において、固相上に固定したGPR49とライブラリーメンバーを接触させ、そしてタンパク質(またはコード核酸もしくは誘導体)に結合するライブラリーメンバーを採取することによって、スクリーニングを行うことも可能である。こうしたスクリーニング法の例は、「パニング」技術と称され、例えば、ParmleyおよびSmith, 1988, Gene 73:305 318;Fowlkesら, 1992, BioTechniques 13:422−427;国際特許公報第WO 94/18318号;および上述の文献に引用される参考文献に記載される。
【0097】
特定の態様において、GPR49断片および/または類似体、特にペプチド模倣体を、GPR49と別のタンパク質、例えば表1に提供するタンパク質との複合体の形成(複合体の量または複合体の組成)または細胞中のGPR49活性の、細胞における複合体活性または形成を阻害する、競合的または非競合的阻害剤としての活性に関して、スクリーニングする。
【0098】
1つの態様において、試験しようとする剤の非存在下では複合体形成が起こる、水性または生理学的結合条件下で、複合体形成を調節する能力に関して剤をスクリーニングする、結合阻害アッセイを用いて、GPR49活性またはGPR49−タンパク質複合体形成を調節する(すなわちアンタゴナイズするかまたはアゴナイズする)剤をスクリーニングすることも可能である。本発明の複合体形成に干渉する剤を、複合体形成のアンタゴニストとして同定する。複合体形成を促進する剤を、複合体形成のアゴニストとして同定する。複合体形成を完全に遮断する剤を、複合体形成の阻害剤として同定する。
【0099】
スクリーニング法は、放射性リガンド(例えば125Iまたは3H)、磁気リガンド(例えばフォトビオチン・アセテートに共有結合した常磁性ビーズ)、蛍光リガンド(例えばフルオレセインまたはローダミン)、または酵素リガンド(例えばルシフェラーゼまたはβ−ガラクトシダーゼ)で、複合体の構成要素タンパク質を標識することを含むことも可能である。次いで、当該技術分野に知られる多くの技術の1つによって、溶液中で結合する反応剤を単離することも可能であり、こうした技術には、限定されるわけではないが、非標識結合パートナー(または第二の標識複合体部分上で用いるものと区別可能なマーカーで標識された結合パートナー)に対する抗血清を用いた、標識複合体部分の同時免疫沈降、免疫アフィニティー・クロマトグラフィー、サイズ排除クロマトグラフィー、および勾配密度遠心分離が含まれる。好ましい態様において、標識結合パートナーは、商業的に入手可能なフィルターに保持されない小さい断片またはペプチド模倣体である。結合したならば、次いで、標識種は、フィルターを通過不能となり、複合体形成の単純なアッセイが提供される。
【0100】
当該技術分野に一般的に知られる方法を用いて、複合体の構成要素メンバーの少なくとも1つを標識する。適切な標識法には、限定されるわけではないが、放射標識アミノ酸、例えば3H−ロイシンまたは35S−メチオニンの取り込みによる放射標識、クロラミンT法、Bolton−Hunter試薬などを用いた、125Iまたは131Iでの翻訳後ヨウ素化による放射標識、あるいはホスホリラーゼおよび無機放射標識リンを用いた32Pでの標識、フォトビオチン−アセテートでのビオチン標識、および太陽灯曝露などが含まれる。複合体メンバーの1つが、例えば以下に記載するように、固定されている場合、遊離種を標識する。相互作用種のいずれも固定されていない場合、各々を区別可能なマーカーで標識して、その後、両方の部分を単離して、より正確な定量化を提供し、そしてヘテロマー複合体からホモマー複合体の形成を区別可能であるようにすることも可能である。修飾相互作用因子の1つに結合する付属タンパク質を利用して、検出感度を改善し、複合体の安定性を増加させるなどの方法を提供する。
【0101】
典型的な結合条件は、限定ではないが、例えば、10〜250mM NaCl、5〜50mM Tris−HCl、pH5〜8、および0.5% Triton X−100または相互作用の特異性を改善する他の界面活性剤の水性塩溶液中である。金属キレート剤および/または二価陽イオンを添加して、結合を改善し、そして/またはタンパク質分解を減少させることも可能である。反応温度は、摂氏4、10、15、22、25、35、または42度を含むことも可能であり、そしてインキュベーション時間は、典型的には少なくとも15秒間であるが、結合平衡が起こることを可能にするには、より長い時間が好ましい。日常的なタンパク質結合アッセイを用いて、特定の複合体をアッセイして、再現可能な結合に最適な結合条件を決定することも可能である。
【0102】
用いる標識に特異的なアッセイ法、例えば放射能検出のための液体シンチレーション計測、酵素標識部分のための酵素活性などを用いた、複合体形成の定量化によって、複合体形成の物理的パラメーターを分析することも可能である。次いで、Scatchard分析、Hill分析、および当該技術分野に一般的に知られる他の方法(例えば、Proteins, Structures, and Molecular Principles, 第2版(1993)Creighton監修, W.H. Freeman and Company, ニューヨークを参照されたい)を利用して、反応結果を分析する。
【0103】
結合アッセイに対する第二の一般的なアプローチにおいて、結合種の1つをフィルター上、マイクロタイタープレートウェル中、試験管中、クロマトグラフィーマトリックスなどに、共有的または非共有的に固定する。当該技術分野に周知の方法いずれか、例えば限定されるわけではないが、KadonagaおよびTjian, 1986, Proc. Natl. Acad. Sci. USA 83:5889−5893の方法、すなわちCNBr−Sepharose 4B(Pharmacia)などの臭化シアン誘導体化支持体への連結を用いて、タンパク質を共有的に固定することも可能である。必要な場合、スペーサーを使用すると、支持体による立体障害を減少させることも可能である。支持体へのタンパク質の非共有結合には、限定されるわけではないが、荷電表面へのタンパク質の付着、特異的抗体との結合、関連しない第三の相互作用タンパク質への結合などが含まれる。
【0104】
いずれかの手段(例えば上述の手段)によって標識した複合体の別のメンバーと、複合体の1つのメンバー(またはその誘導体)の結合に関して競合する剤(細胞抽出物またはライブラリー・プールを含む)のアッセイを提供して、複合体形成の競合剤または増進剤に関してスクリーニングする。
【0105】
特定の態様において、他のタンパク質構成要素への試薬の非特異的結合、またはプラスチック、固定マトリックス等に試薬が吸収される損失を阻害する、ブロッキング剤をアッセイ混合物に含む。ブロッキング剤には、限定されるわけではないが、ウシ血清アルブミン、カゼイン、脱脂粉乳、デンハルト試薬、Ficoll、ポリビニルピロリジン、非イオン性界面活性剤(NP40、Triton X−100、Tween20、Tween80など)、イオン性界面活性剤(例えばSDS、LDSなど)、ポリエチレングリコールなどが含まれる。適切なブロッキング剤濃度によって、複合体形成が可能である。
【0106】
結合を行った後、上清中の未結合標識タンパク質を取り除き、そして結合した標識タンパク質いずれかを保持する固定タンパク質を徹底的に洗浄する。次いで、上述のように標識を検出する当該技術分野の標準法を用いて、結合標識の量を定量化する。
【0107】
別の特定の態様において、本明細書に提供するようなタンパク質複合体/タンパク質および/または抗体を固体キャリアーに付着させることによって、本明細書に提供するようなタンパク質複合体/タンパク質の調節剤に関するスクリーニングを行うことも可能である。
【0108】
異なる種類のタンパク質(抗体を含む)を含有するアレイの調製が当該技術分野に周知であり、そして当業者に明らかである(例えば、Ekinsら, 1989, J. Pharm. Biomed. Anal. 7:155−168;Mitchellら 2002, Nature Biotechnol. 20:225−229;Petricoinら, 2002, Lancet 359:572−577;Templinら, 2001, Trends Biotechnol. 20:160−166;WilsonおよびNock, 2001, Curr. Opin. Chem. Biol. 6:81−85;Leeら, 2002 Science 295:1702−1705;MacBeathおよびSchreiber, 2000, Science 289:1760;BlawasおよびReichert, 1998, Biomaterials 19:595;Kaneら, 1999, Biomaterials 20:2363;Chenら, 1997, Science 276:1425;Vaughamら, 1996, Nature Biotechnol. 14:309−314;Mahlerら, 1997, Immunotechnology 3:31−43;Robertsら, 1999, Curr. Opin. Chem. Biol. 3:268−273;Nordら, 1997, Nature Biotechnol. 15:772−777;Nordら, 2001, Eur. J. Biochem. 268:4269−4277;BrodyおよびGold, 2000, Rev. Mol. Biotechnol. 74:5−13;KarlstroemおよびNygren, 2001, Anal. Biochem. 295:22−30;Nelsonら, 2000, Electrophoresis 21:1155−1163;Honoreら, 2001, Expert Rev. Mol. Diagn. 3:265−274;Albala, 2001, Expert Rev. Mol. Diagn. 2:145−152, FigeysおよびPinto, 2001, Electrophoresis 2:208−216、並びにここに列挙する刊行物中の参考文献を参照されたい)。
【0109】
タンパク質またはタンパク質複合体を、当業者に明らかであろうような異なる手段によって、アレイに付着させることも可能である。精製工程後、または当業者に明らかであろうような別の適切な精製スキームによって、例えばTAP−タグ(WO/0009716およびRigautら, 1999, Nature Biotechnol. 10:1030−1032に記載されるようなもの)を介して、複合体をアレイに付加させることも可能である。
【0110】
場合によって、複合体のタンパク質を架橋して、複合体の安定性を増進させることも可能である。タンパク質を架橋する、異なる方法が、当該技術分野で周知である。架橋剤の反応性末端基には、限定されるわけではないが、−COOH、−SH、−NH2またはN−オキシ−スクシンアマートが含まれる。
【0111】
架橋剤のスペーサーは、架橋しようとする複合体のサイズに対して選択すべきである。いくつかのタンパク質しか含まない、小さいタンパク質複合体に関しては、反応混合物中、別個の複合体を架橋する可能性を減少させるため、比較的短いスペーサーが好ましい。より大きいタンパク質複合体に関しては、複合体内のタンパク質間の架橋を容易にするため、より大きいスペーサーのさらなる使用が好ましい。
【0112】
複合体をキャリアーに連結する前に、架橋の成功率をチェックすることが好ましい。
当業者に明らかであろうように、架橋の最適な率を、ケースバイケースで決定する必要がある。当該技術分野に周知の方法によって、これを達成することも可能であり、これらの方法のいくつかを以下に例示的に記載する。
【0113】
例えば、変性タンパク質ゲル上で、架橋されない複合体に対して、架橋された複合体を分析することによって、架橋が十分な率であることをチェックすることも可能である。
架橋が成功裡に行われたならば、複合体のタンパク質は、同一レーンに見られると予期され、一方、架橋されない複合体のタンパク質は、個々の特性にしたがって分離されると予期される。場合によって、質量分析および/またはエドマン分解などの当該技術分野に周知の方法を用いて、それぞれのバンド中のタンパク質をペプチド配列決定することによって、複合体のすべてのタンパク質の存在をさらにチェックすることも可能である。
【0114】
さらに、高すぎる架橋率もまた、回避しなければならない。架橋があまりにも大規模に起こった場合、個々のタンパク質複合体の架橋量が増加し、これが、アレイを用いた、潜在的な結合パートナーおよび/または調節剤等に関するスクリーニングと干渉する可能性があるであろう。
【0115】
当該技術分野に周知の方法によって、こうした構造の存在を決定することも可能であり、そしてこうした方法には、例えば、架橋されない複合体に対して、架橋された複合体を含有する溶液のゲルろ過プロフィールを比較する、ゲルろ過実験が含まれる。
【0116】
場合によって、いくつかが本明細書に例示的に提供される、当業者に明らかであろうような機能アッセイを行って、複合体の完全性をチェックすることも可能である。
あるいは、単数または複数のタンパク質を単一の融合タンパク質として発現させ、そして当業者に明らかであろうように、マトリックスにカップリングすることも可能である。
【0117】
場合によって、当業者に明らかな多様な方法によって、上に概略するような複合体またはタンパク質または抗体の付着をさらに監視することも可能である。これらには、限定されるわけではないが、表面プラズモン共鳴が含まれる(例えば、McDonnel, 2001, Curr. Opin. Chem. Biol. 5:572−577;Lee, 2001, Trends Biotechnol. 19:217−222;Weinbergerら, 2000, 1:395−416;Pearsonら, 2000, Ann. Clin. Biochem. 37:119−145;Velyら, 2000, Methods Mol. Biol. 121:313−321;Slepak, 2000, J. Mol Recognit. 13:20−26を参照されたい)。
【0118】
Aベータ−40およびAベータ−42ペプチドの産生を、ELISAによって測定するのに有用な典型的なアッセイには、限定されるわけではないが、Vassar Rら, 1999, Science, 286:735−41に記載されるものが含まれる。
【0119】
細胞株またはトランスジェニック動物におけるC末端APP断片の産生を、ウェスタンブロットによって測定するのに有用な典型的なアッセイには、限定されるわけではないが、Yan Rら, 1999, Nature, 402:533−7に記載されるものが含まれる。
【0120】
BACE1複合体の、細菌で発現されたAPP断片に対するベータ−セクレターゼまたはガンマ・セクレターゼのタンパク質分解活性を、in vitroで(例えばRNAi(siRNA)および/または相互作用タンパク質(単数または複数)をコードするプラスミドにより細胞中の1つまたはいくつかの相互作用タンパク質の発現を修飾することによって)測定するのに有用な典型的なアッセイには、限定されるわけではないが、Tian Gら, 2002, J Biol Chem, 277:31499−505に記載されるものが含まれる。
【0121】
BACE1複合体の、Gal4駆動レポーター遺伝子のトランス活性化を(例えばRNAi(siRNA)および/または相互作用タンパク質(単数または複数)をコードするプラスミドにより細胞中の1つまたはいくつかの相互作用タンパク質の発現を修飾することによって)測定するのに有用な典型的なアッセイには、限定されるわけではないが、Cao Xら, 2001, Science, 293:115−20に記載されるものが含まれる。
【0122】
本発明記載の相互作用分子または阻害剤である能力に関して、当該技術分野に知られるいかなる分子を試験することも可能である。GPR49複合体機構を発現する細胞に、候補分子を直接提供することも可能であるし、または候補タンパク質の場合、核酸が組換え的に発現されて候補タンパク質を産生する条件下で、コード核酸を提供することによって、候補タンパク質を提供することも可能である。
【0123】
本発明の方法は、複合体の量、活性、またはタンパク質構成要素組成を調節する分子、例えば阻害するか、アンタゴナイズするか、またはアゴナイズする分子に関して、化学的ライブラリーをスクリーニングするのによく適している。化学的ライブラリーは、ペプチド・ライブラリー、ペプチド模倣体ライブラリー、化学的合成ライブラリー、組換えライブラリー、例えばファージディスプレイ・ライブラリー、およびin vitro翻訳に基づくライブラリー、他の非ペプチド合成有機ライブラリーなどであることも可能である。
【0124】
典型的なライブラリーは、いくつかの供給源(ArQule、Tripos/PanLabs、ChemDesign、Pharmacopoeia)から商業的に入手可能である。いくつかの場合、コンビナトリアル戦略を用いて、メンバー化合物が付着した支持体上にライブラリーの各メンバーの同一性をコードし、こうして有効な調節剤である分子を直接、そして直ちに同定することを可能にする、これらの化学的ライブラリーを生成する。したがって、多くのコンビナトリアル・アプローチにおいて、化合物プレート上の位置は、その化合物の組成を特定する。また、1つの例において、単一プレート位置に、1〜20の化学薬品を有することも可能であり、この化学薬品を、目的の相互作用を含有するウェルに投与することによってスクリーニングすることも可能である。したがって調節が検出されたならば、調節活性に関して、相互作用対の徐々に小さいプールをアッセイすることも可能である。こうした方法によって、多くの候補分子をスクリーニングすることも可能である。
【0125】
使用に適した多くの多様性ライブラリーが当該技術分野に知られ、そしてこれを用いて、本発明にしたがって試験しようとする化合物を提供することも可能である。あるいは、標準法を用いて、ライブラリーを構築することも可能である。化学的(合成)ライブラリー、組換え発現ライブラリー、またはポリソームに基づくライブラリーが、使用可能なライブラリーの典型的な種類である。
【0126】
ライブラリーは、拘束性または半硬直性(semirigid)(ある程度の度合いの構造硬直性を有するもの)であることも可能であるし、あるいは直鎖または非拘束性であることも可能である。ライブラリーは、cDNAまたはゲノム発現ライブラリー、ランダムペプチド発現ライブラリーまたは化学的合成ランダムペプチド・ライブラリー、または非ペプチド・ライブラリーであることも可能である。発現ライブラリーを細胞に導入し、該細胞中でライブラリーの核酸を発現させて、コードされるタンパク質を産生し、アッセイを行う。
【0127】
1つの態様において、本発明で使用可能なペプチド・ライブラリーは、in vitroで化学的に合成されるライブラリーであることも可能である。こうしたライブラリーの例が、各ペプチドの第一の残基および第二の残基が個々にそして具体的に定義される遊離ヘキサペプチドの混合物を記載する、Houghtenら, 1991, Nature 354:84−86;コレクション中の各ビーズが該ビーズ上にアミノ酸残基の単一ランダム配列を固定するペプチド・ライブラリーを、固相スプリット合成スキームによって産生する、「1ビーズ、1ペプチド」アプローチを記載する、Lamら, 1991, Nature 354:82−84;スプリット合成およびT−バッグ合成法を記載する、Medynski, 1994, Bio/Technology 12:709−710;およびGallopら, 1994, J. Med. Chem. 37:1233−1251に提供される。単に他の例として、Ohlmeyerら, 1993, Proc. Natl. Acad. Sci. USA 90:10922−10926;Erbら, 1994, Proc. Natl. Acad. Sci. USA 91:11422−11426;Houghtenら, 1992, Biotechniques 13:412;Jayawickremeら, 1994, Proc. Natl. Acad. Sci. USA 91:1614−1618;またはSalmonら, 1993, Proc. Natl. Acad. Sci. USA 90:11708−11712の方法にしたがって、使用のためのコンビナトリアルライブラリーを調製することも可能である。PCT公報第WO 93/20242号、並びにBrennerおよびLerner, 1992, Proc. Natl. Acad. Sci. USA 89:5381−5383は、各化学ポリマーライブラリーメンバーに関してオリゴヌクレオチド識別子を含有する、「コードされるコンビナトリアル化学ライブラリー」を記載する。
【0128】
好ましい態様において、スクリーニングされるライブラリーは、ランダムペプチドが(例えばジスルフィド結合を有することによって)拘束されているランダムペプチド・ファージディスプレイ・ライブラリーである、生物学的発現ライブラリーである。
【0129】
さらに、より一般的には、構造的に拘束された有機多様性(例えば非ペプチド)ライブラリーもまた、使用可能である。例えば、ベンゾジアゼピン・ライブラリー(例えばBuninら, 1994, Proc. Natl. Acad. Sci. USA 91:4708−4712を参照されたい)を使用することも可能である。
【0130】
使用可能な、コンホメーション的に拘束されたライブラリーには、限定されるわけではないが、酸化環境中でジスルフィド結合によって架橋されて、シスチンを形成する、不変システイン残基、修飾されたペプチド(例えばフッ素、金属、同位体標識を取り込む、リン酸化されているなど)、1以上の非天然存在アミノ酸を含有するペプチド、非ペプチド構造、およびかなりの割合の−カルボキシグルタミン酸を含有するペプチド、を含有するものが含まれる。
【0131】
非ペプチド、例えばペプチド誘導体(例えば1以上の非天然存在アミノ酸を含有するもの)のライブラリーを用いることもまた可能である。これらの一例は、ペプトイド・ライブラリーである(Simonら, 1992, Proc. Natl. Acad. Sci. USA 89:9367−9371)。ペプトイドは、アルファ炭素に結合せず、主鎖アミノ窒素に結合する天然存在側鎖を有する、非天然アミノ酸のポリマーである。ペプトイドは、ヒト消化酵素によって容易には分解されないため、これらは好適に、薬剤使用に、より容易に適応可能である。ペプチド中のアミド官能性が過メチル化されて、化学的に変換されたコンビナトリアルライブラリーが生成される、使用可能なライブラリーの別の例が、Ostreshら, 1994, Proc. Natl. Acad. Sci. USA 91:11138−11142に記載されている。
【0132】
本発明にしたがってスクリーニング可能なペプチド・ライブラリーのメンバーは、20の天然存在アミノ酸を含有するものに限定されない。特に、化学的に合成されたライブラリーおよびポリソームに基づくライブラリーは、20の天然存在アミノ酸に加えたアミノ酸の使用を可能にする(ライブラリー産生に用いるアミノ酸の前駆体プールに含むことによる)。特定の態様において、ライブラリーメンバーは、1以上の非天然または非古典的アミノ酸または環状ペプチドを含有する。非古典的アミノ酸には、限定されるわけではないが、一般的なアミノ酸のD−異性体、−アミノイソ酪酸、4−アミノ酪酸、Abu、2−アミノ酪酸;−Abu、−Ahx、6−アミノヘキサン酸;Aib、2−アミノイソ酪酸;3−アミノプロピオン酸;オルニチン;ノルロイシン;ノルバリン、ヒドロキシプロリン、ザルコシン、シトルリン、シスチン酸、t−ブチルグリシン、t−ブチルアラニン、フェニルグリシン、シクロヘキシルアラニン、β−アラニン、デザイナーアミノ酸、例えばβ−メチルアミノ酸、C−メチルアミノ酸、N−メチルアミノ酸、フルオロ−アミノ酸およびアミノ酸類似体一般が含まれる。さらに、アミノ酸は、D(右旋性)またはL(左旋性)であることも可能である。
【0133】
特定の態様において、本発明の複合体の断片および/または類似体、またはそのタンパク質構成要素、特にペプチド模倣体を、複合体活性または形成の競合的または非競合的阻害剤としての活性に関して、スクリーニングする。
【0134】
本発明の別の態様において、コンビナトリアル化学を用いて、複合体の調節剤を同定することも可能である。コンビナトリアル化学は、その多くが構造的に類似でありうる、数十万の化合物を含有するライブラリーを生成することが可能である。ハイスループットスクリーニング・プログラムは、既知のターゲットに対する親和性に関して、これらの非常に大きいライブラリーをスクリーニングすることが可能である一方、より小規模であるが、最大化学的多様性を提供するライブラリーを得る、新規アプローチが開発されてきている(例えば、Matter, 1997, J. Med. Chem. 40:1219−1229を参照されたい)。
【0135】
明示される一団のタンパク質に対する結合親和性に関して、小分子の別個のライブラリーを試験するため、コンビナトリアル化学の1つの方法である、アフィニティー・フィンガープリンティングが、以前から用いられている。スクリーニングによって得られたフィンガープリントを用いて、目的の他のタンパク質または受容体(本発明において、本発明のタンパク質複合体およびそのタンパク質構成要素)に対する、個々のライブラリーメンバーの親和性を予測する。目的のタンパク質と反応することが知られる他の化合物から得たフィンガープリントと、フィンガープリントを比較して、ライブラリー化合物が同様に反応可能であるかどうかを予測する。例えば、複合体またはタンパク質構成要素との相互作用に関して、巨大ライブラリー中のすべてのリガンドを試験するのでなく、その活性を有することが知られる他の化合物と類似のフィンガープリントを有するリガンドのみを試験することも可能である(例えば、Kauvarら, 1995, Chem. Biol. 2:107−118;Kauvar, 1995, Affinity fingerprinting, Pharmaceutical Manufacturing International. 8:25−28;およびKauvar, Toxic−Chemical Detection by Pattern Recognition in New Frontiers in Agrochemical Immunoassay, Kurtz, StankerおよびSkerritt(監修), 1995, AOAC:ワシントンD.C., 305−312を参照されたい)。
【0136】
Kayら(1993, Gene 128:59−65)は、先の慣用的ライブラリーいずれのものより長い、完全にランダムな配列のペプチドをコードする、ペプチド・ライブラリーを構築する方法を開示した。Kayらに開示されるライブラリーは、長さ約20アミノ酸より長い、完全に合成のランダムペプチドをコードする。こうしたライブラリーを好適にスクリーニングして、複合体調節剤を同定することも可能である(米国特許第5,498,538号、1996年3月12日;およびPCT公報第WO 94/18318号、1994年8月18日もまた参照されたい)。
【0137】
ペプチド・ライブラリーの多様な種類の包括的な概説を、Gallopら, 1994, J. Med. Chem. 37:1233−1251に見出すことも可能である。
【0138】
好ましい態様において、GPR49と試験化合物の相互作用は、GPR49活性の阻害を生じる。
好ましい態様にしたがって、工程b)において、ガンマ−セクレターゼがAPPを切断する能力を測定する。これは上に示すように測定可能である。
【0139】
さらに、本発明はまた、神経変性疾患、好ましくはアルツハイマー病治療用の薬剤組成物を調製するための方法であって:
a)本発明の方法にしたがって、ガンマ−セクレターゼ調節剤および/またはベータ−セクレターゼ調節剤、好ましくは阻害剤を同定し、そして
b)ガンマ−セクレターゼおよび/またはベータ−セクレターゼの調節剤、好ましくは阻害剤を薬剤組成物に配合する
工程を含む、前記方法にも関する。
【0140】
ここでもまた、薬剤組成物に関して、上に示すようなすべての態様があてはまる。
好ましい態様において、本発明のこの方法は、同定された分子と、上に説明したような薬学的に許容しうるキャリアーを混合する工程をさらに含む。
【0141】
本発明はまた、上に定義するようなGPR49阻害剤を含む、薬剤組成物にも関する。
さらに、本発明はまた、薬剤組成物調製のための上記方法によって得られうる、薬剤組成物にも関する。
【0142】
本発明はまた、アルツハイマー病などの神経変性疾患および関連する神経変性障害の治療用の、本発明の薬剤組成物にも関する。
本発明はまた、神経変性疾患、好ましくはアルツハイマー病を治療するかまたは予防する方法であって、こうした治療または予防の必要がある被験者に、本発明の薬剤組成物の療法的有効量を投与することを含む、前記方法にも関する。
【0143】
本発明のこの方法に関して、上記のすべての態様は、本発明の使用のため、やはりあてはまる。
本発明はまた、ベータ−セクレターゼおよび/またはガンマ−セクレターゼ活性をin vitroで調節する、好ましくは阻害するための、GPR49相互作用分子の使用にも関する。例えば、GPR49相互作用分子によって、細胞培養において、ベータ−セクレターゼおよび/またはガンマ−セクレターゼの活性を調節する、好ましくは阻害することが、本発明内に含まれる。上述のような、GPR49相互作用分子に関するすべての態様もまた、本発明のこの使用にあてはまる。
【0144】
以下の実施例は、本発明の主題をより詳細に記載するであろう。
【実施例】
【0145】
(実施例1)
それぞれ、EP 1 105 508 B1およびRigautら, 1999, Nature Biotechnol. 17:1030−1032に、より十分に記載されるTAP技術を用いて、そしてタンパク質精製のため、以下にさらに記載するように、さらに適応させた。以下にさらに記載するように、質量分析を用いて、タンパク質を同定した。
【0146】
GPR49は、TAP技術エントリーポイントAph1a、APP−C99、BACE1およびFe65L2で、タンパク質複合体のメンバーと同定された(図1)。
パート1:TAPタグ化ベイト(bait)の構築
RT−PCRによって、完全ORFをコードするcDNAを得た。RNeasyミニキット(Qiagen)を用いて、適切な細胞株から総RNAを調製した。遺伝子特異的プライマーを用い、長鎖テンプレート用SUPERSCRIPT One−Step RT−PCRキット(Life Technologies)を用いて、cDNA合成およびPCR両方を行った。35〜40周期増幅した後、予期されるサイズのPCR産物をMinElute PCR精製キット(Qiagen)を用いてゲル精製し、そして必要な場合、さらなる増幅に用いた。低存在量のRNAを、ゲル精製前に、入れ子(nested)PCRによって増幅した。NotI制限部位をPCRプライマーに付着させて、レトロウイルスベクターpIE94−N/C−TAPに、増幅されたcDNAをサブクローニングすることを可能にし、それによって、TAPタグとのN末端融合体またはC末端融合体を生成した(Rigautら, 1999, Nature Biotechnol. 17:1030−1032)。以下のベイト/エントリーポイントに関しては、N末端タグ化を選択した:プレセニリン1、プレセニリン2、Aph−1a、Aph−1b、Pen−2、APP、タウ、Fe65、カルセニリン。以下のベイト/エントリーポイントに関しては、C末端タグ化を選択した:ニカストリン、Aph−1a、Aph−1b、BACE1 D215N、APP、APP695SW、APP−C99、Fe65、Fe65L2、X11ベータ。
【0147】
制限消化、DNA配列決定、およびTNT T7迅速カップリング転写/翻訳系(Promega inc.)を用いたin vitro翻訳によって、クローンを分析した。検出のため、TAPタグのプロテインA部分を用いたウェスタンブロッティングによって、タンパク質の存在を証明した。簡潔には、標準的SDS−PAGEによってタンパク質を分離した後、Pharmacia BiotechのMultiphorIIブロッティング装置を用いて、ニトロセルロース膜(PROTRAN、Schleicher&Schuell)に半乾性トランスファーした。トランスファー緩衝液は、48mM Tris、39mMグリシン、10%メタノールおよび0.0375%ドデシル硫酸ナトリウムからなった。10%脱脂粉乳および0.1% Tweeen20を補ったリン酸緩衝生理食塩水(PBS)中でブロッキングした後、トランスファーしたタンパク質を、ブロッキング溶液中で希釈したペルオキシダーゼ−抗−ペルオキシダーゼ可溶性複合体(Sigma)で探査した(probed)。徹底的に洗浄した後、高感度化学発光(ECL;Amersham Pharmacia Biotech)によって、免疫反応性タンパク質を視覚化した。
【0148】
パート2:ウイルスの調製および感染
ベクターとして、MoMLVに基づく組換えウイルスを用いた。
調製は、以下のとおり行った:
2.1.ウイルスの調製
293gp細胞を100%集密(confluency)に増殖させた。これらを、ポリ−L−リジンプレート(PBS中、1:5希釈したポリーL−リジン[0.01%ストック溶液、Sigma P−4832]を、プレート上に少なくとも10分間放置した)上、1:5にスプリットした。第2日、63マイクログラムのレトロウイルスベクターDNAと一緒に、適切なエンベロープ・タンパク質をコードするプラスミドDNA 13マイクログラムを、293gp細胞にトランスフェクションした(Somiaら, 1999, Proc. Natl. Acad. Sci. USA 96:12667−12672;Somiaら 2000, J. Virol. 74:4420−4424)。第3日、15cmディッシュあたり、15mlのDMEM+10% FBSと培地を交換した。第4日、ウイルスを含有する培地(上清)を採取した(トランスフェクション後の培地交換の24時間後)。第二の収集を計画している場合、DMEM 10% FBSをプレートに添加し、そしてプレートをさらに24時間インキュベーションした。すべての収集を以下のように行った:0.45マイクロメートル・フィルター(Corning GmbH、酢酸セルロース、431155)を通じて、上清をろ過した。フィルターをコニカル・ポリアロマー遠心管(Beckman、358126)に入れ、これをSW28ローター(Beckman)のバケットに入れた。ろ過した上清をSW28ローター中、19400rpm、摂氏21度で2時間、超遠心した。上清を廃棄した。ウイルスを含有するペレットを、エアロゾル安全チップを用い、上下に100回ピペッティングすることによって、少量(例えば300マイクロリットル)のハンクス平衡塩溶液(Gibco BRL、14025−092)に再懸濁した。ウイルスを以下のようにトランスフェクションに用いた。
【0149】
2.2.感染
感染させる細胞を1日前に6ウェルプレートの1つのウェルに蒔いた。感染4時間前に、細胞上の古い培地を新鮮な培地と交換した。最小限の体積のみを添加して、細胞が完全に覆われるようにした(例えば700マイクロリットル)。感染中、細胞は活発に分裂していた。
【0150】
細胞およびその増殖条件の説明を、以下にさらに提供する(「3.細胞株」)。
濃縮ウイルスに、ポリブレン(臭化ヘキサジメトリン;Sigma、H9268)を添加して、最終濃度8マイクログラム/mlを達成した(これは、濃縮レトロウイルス300マイクロリットルあたり、1ミリグラム/mlポリブレンストックの2.4マイクロリットルと同等である)。ウイルスをポリブレン中、室温で1時間インキュベーションした。感染のため、ウイルス/ポリブレン混合物を細胞に添加して、そして摂氏37度で数時間、適切なCO2濃度でインキュベーションした(例えば1日または1晩)。感染後、感染細胞上の培地を新鮮な培地と交換した。集密になったら、通常どおりに細胞を継代した。細胞は染色体に組み込まれたレトロウイルスを含有し、そして目的の遺伝子を安定に発現する。
【0151】
2.3.細胞株
発現のため、SKN−BE2細胞を用いた。SKN−BE2細胞(アメリカン・タイプ・カルチャー・コレクション第CRL−2271号)を95% OptiMEM+5%鉄補充ウシ血清中で増殖させた。
【0152】
パート3:TAPタグ化タンパク質の発現パターンのチェック
イムノブロット分析によって、そして/または免疫蛍光によって、TAPタグ化タンパク質の発現パターンをチェックした。免疫蛍光分析を、TAPタグ化タンパク質の種類に応じて、No.1またはNo.2にしたがって行った。イムノブロット分析を、No.3にしたがって行った。
【0153】
3.1.原形質膜およびER結合タンパク質に関する、固定哺乳動物細胞の間接的免疫蛍光染色のプロトコル
ポリリジンでコーティングした8ウェルチャンバー・スライド上、FCS培地中で、細胞を50%集密まで増殖させた。次いで、リン酸緩衝生理食塩水(PBS)溶液(0.14Mホスフェート、0.1M NaCl pH7.4)中で希釈した4%パラホルムアルデヒド中で、細胞の固定を行った。ウェルあたり300マイクロリットル中、室温で30分間、細胞をインキュベーションした。PBS中の0.1Mグリシン中で、室温で2x20分間、反応停止を行った。0.3%サポニン+PBS中の1%ウシ血清アルブミン(BSA)で、室温で少なくとも1時間、ブロッキングを行った。ブロッキング溶液中、+4℃で1晩、一次抗体のインキュベーションを行った。ケースバイケースで、抗体の適切な希釈を決定した。0.3%サポニンを含有するPBS中、室温で2x20分間、細胞を洗浄した。ブロッキング溶液中で、二次抗体のインキュベーションを行う。Alexa594にカップリングしたヤギ抗ウサギを1:1000に希釈した(Molecular Probes)。Alexa488にカップリングしたヤギ抗マウスを1:1000に希釈した(Molecular Probes)。DAPIを用いてDNAを標識した。F−アクチンを標識するのにファロイジンを用いた場合、該薬剤を1:500に希釈し、そして二次抗体とインキュベーションした。次いで、細胞を再び、PBS中、室温で2x20分間洗浄した。過剰な緩衝液を取り除き、そしてアンチ・ブリーチング剤(Vectashield、Vector Laboratories)を含有する培地中で細胞をマウントした。
【0154】
3.2.非原形質膜結合タンパク質に関する、固定哺乳動物細胞の間接的免疫蛍光染色のためのプロトコル:
ポリリジンでコーティングした8ウェルチャンバー・スライド上、FCS培地中で、細胞を50%集密まで増殖させた。リン酸緩衝生理食塩水(PBS)溶液(0.14Mホスフェート、0.1M NaCl pH7.4)中で希釈した4%パラホルムアルデヒド中、室温(RT)で30分間、ウェルあたり300マイクロリットルで、細胞の固定を行った。PBS中の0.1Mグリシン中で、室温で2x20分間、反応停止を行った。PBS中、0.5% Triton X−100で、室温で10分間、細胞の透過化を行った。次いで、0.3%サポニン+PBS中の1%ウシ血清アルブミン(BSA)で、RTで少なくとも1時間、ブロッキングを行った(ブロッキング溶液)。ブロッキング溶液中、+4℃で1晩、一次抗体のインキュベーションを行った。ケースバイケースで、抗体の適切な希釈を決定しなければならない。0.3%サポニンを含有するPBS中、RTで2x20分間、細胞を洗浄した。ブロッキング溶液中で、二次抗体のインキュベーションを行った。Alexa594にカップリングしたヤギ抗ウサギを1:1000に希釈し(Molecular Probes)、Alexa488にカップリングしたヤギ抗マウスを1:1000に希釈した(Molecular Probes)。DAPIを用いてDNAを標識した。F−アクチンを標識するのにファロイジンを用いた場合、該薬剤を1:500に希釈し、そして二次抗体とインキュベーションした。細胞を、PBS中、RTで2x20分間洗浄した。過剰な緩衝液を取り除き、そしてアンチ・ブリーチング剤(Vectashield、Vector Laboratories)を含有する培地中で細胞をマウントした。
【0155】
3.3.イムノブロット分析
TAPタグ化タンパク質の発現レベルを分析するため、細胞ペレット(6ウェルプレート由来)を60μlのDNase I緩衝液(5%グリセロール、100mM NaCl、0.8% NP−40(IGEPAL)、5mM硫酸マグネシウム、100μg/ml DNアーゼI(Roche Diagnositics)、50mM Tris、pH7.5、プロテアーゼ阻害剤カクテル)中、氷上で15分間溶解した。各試料を2つのアリコットにスプリットした。最初の半分を13,000rpmで5分間遠心分離して、上清中にNP−40抽出可能物質を得た;次の半分(全物質)を注意深くすりつぶした(triturated)。各50μgのNP−40抽出可能物質および全物質を、DTT含有試料緩衝液と、シェーカー上、50℃で30分間混合し、そしてSDSポリアクリルアミドゲル電気泳動によって、プレキャスト4〜12% Bis−Trisゲル(Invitrogen)上で分離した。次いで、不連続緩衝剤系で、半乾法を用いて、タンパク質をニトロセルロースにトランスファーした。簡潔には、ゲルおよびニトロセルロース膜を、陽極緩衝液(3層の緩衝液A1(0.3M Tris−HCl)および3層の緩衝液A2(0.03M Tris−HCl))または陰極緩衝液(3層の0.03M Tris−HCl、pH9.4、0.1% SDS、40mM □−アミノカプロン酸)のいずれかに浸したろ紙の間に積んだ。600mAで25分間、同時に2つのゲルのエレクトロトランスファーを行った。トランスファー効率の対照とするため、Ponceau S溶液で、トランスファーしたタンパク質を1分間視覚化して、そして次いで、水中で脱染色した。TBST(0.05% Tween−20を含有するTBS)中の5%脱脂粉乳で、室温で30分間、膜をブロッキングした。続いて、HRPカップリングPAP抗体(5%ミルク/TBST中、1:5000に希釈)と室温で1時間インキュベーションし、TBST中、10分間、3回洗浄した。ブロット膜を最後に、化学発光基質(ECL、Roche Diagnostics)に2分間浸し、そしてX線フィルムに曝露するか、またはイメージング・ステーション上で分析した。
【0156】
パート4 精製またはタンパク質複合体
タンパク質複合体精製を、TAPタグ化タンパク質の細胞内局在に適応させ、そして以下に記載するように行った。
【0157】
4.1.細胞質タンパク質の溶解物調製
約1x109の接着細胞(平均)を細胞スクレーパーで採取し、そして氷冷PBS中で3回洗浄した(3分間、550g)。収集した細胞を液体窒素中で凍結するか、または直ちにさらにプロセシングした。細胞溶解のため、細胞ペレットを10mlのCZ溶解緩衝液(50mM Tris−HCl、pH7.4;5%グリセロール;0.2% IGEPAL;1.5mM MgCl2;100mM NaCl;25mM NaF;1mM Na3VO4;1mM DTT;緩衝液25mlあたり1タブレットのEDTA不含プロテアーゼ阻害剤カクテル(CompleteTM、Roche)を含有する)に再懸濁し、そして加圧型細胞破砕装置中、きつくはまったペッスルを10回ストロークすることによって、ホモジナイズした。氷上で30分間、溶解物をインキュベーションし、そして20,000gで10分間回転させた。上清を、100,000gで1時間のさらなる超遠心工程に供した。上清を回収し、そして液体窒素中で迅速に凍結させるか、または直ちにさらにプロセシングした。
【0158】
4.2.膜タンパク質のための溶解物調製
約1x109の接着細胞(平均)を細胞スクレーパーで採取し、そして氷冷PBS中で3回洗浄した(3分間、550g)。収集した細胞を液体窒素中で凍結するか、または直ちにさらにプロセシングした。細胞溶解のため、細胞ペレットを10mlの膜溶解緩衝液(50mM Tris、pH7.4;7.5%グリセロール;1mM EDTA;150mM NaCl;25mM NaF;1mM Na3VO4;1mM DTT;緩衝液25mlあたり1タブレットのEDTA不含プロテアーゼ阻害剤カクテル(CompleteTM、Roche)を含有する)に再懸濁し、そして加圧型細胞破砕装置中、きつくはまったペッスルを10回ストロークすることによって、ホモジナイズした。溶解物を750gで10分間回転させ、上清を回収し、そして100,000gで1時間の超遠心工程に供した。0.8%のn−ドデシル−D−マルトシドを含有する7.5mlの膜溶解緩衝液に膜ペレットを再懸濁し、そして休まず攪拌しながら、4℃で1時間インキュベーションした。100,000gで1時間の別の超遠心工程に試料を供し、そして可溶化された物質を液体窒素中で迅速に凍結させるか、または直ちにさらにプロセシングした。
【0159】
4.3.核タンパク質のための溶解物調製
約1x109の接着細胞(平均)を細胞スクレーパーで採取し、そして氷冷PBS中で3回洗浄した(3分間、550g)。収集した細胞を液体窒素中で凍結するか、または直ちにさらにプロセシングした。細胞溶解のため、細胞ペレットを10mlの低張溶解緩衝液(10mM Tris、pH7.4;1.5mM MgCl2;10mM KCl;25mM NaF;1mM Na3VO4;1mM DTT;緩衝液25mlあたり1タブレットのEDTA不含プロテアーゼ阻害剤カクテル(CompleteTM、Roche)を含有する)に再懸濁し、そして加圧型細胞破砕装置中、きつくはまったペッスルを10回ストロークすることによって、ホモジナイズした。溶解物を2,000gで10分間回転させ、そして生じた上清(S1)を氷上で保存した。核ペレット(P1)を5mlの核溶解緩衝液(50mM Tris、pH7.4;1.5mM MgCl2;20%グリセロール;420mM NaCl;25mM NaF;1mM Na3VO4;1mM DTT;緩衝液25mlあたり1タブレットのEDTA不含プロテアーゼ阻害剤カクテル(CompleteTM、Roche)を含有する)に再懸濁し、そして氷上で30分間インキュベーションした。試料をS1と合わせ、7mlの希釈緩衝液(110mM Tris、pH7.4;0.7% NP40;1.5mM MgCl2;25mM NaF;1mM Na3VO4;1mM DTT)でさらに希釈し、氷上で10分間インキュベーションし、そして100,000gで1時間遠心分離した。最終上清(S2)を液体窒素中で迅速に凍結させた。
【0160】
4.4.タンデムアフィニティー精製
凍結した溶解物を37℃の水槽で迅速に融解し、そして100,000gで20分間回転させた。上清を回収し、そして0.2mlの定着した(settled)ウサギIgG−アガロースビーズ(Sigma)と、休みなく攪拌しながら4℃で2時間インキュベーションした。固定タンパク質複合体を10mlのCZ溶解緩衝液(50mlの緩衝液あたり1タブレットのCompleteTM(Roche)を含有する)で洗浄し、そしてさらに5mlのTEV切断緩衝液(10mM Tris、pH7.4;100mM NaCl;0.1% IGEPAL;0.5mM EDTA;1mM DTT)で洗浄した。150μlのTEV切断緩衝液中、5μlのTEVプロテアーゼ(GibcoBRL、カタログ番号10127−017)と、16℃で1時間インキュベーションすることによって、タンパク質複合体を溶出させた。溶出液を回収し、そして0.2ml CBP結合緩衝液(10mM Tris、pH7.4;100mM NaCl;0.1% IGEPAL;2mM MgAc;2mMイミダゾール;1mM DTT;4mM CaCl2)中、0.2mlの定着したカルモジュリン親和性ビーズ(Stratagene)と合わせ、その後、休みなく攪拌しながら4℃で1時間インキュベーションした。10mlのCBP洗浄緩衝液(10mM Tris、pH7.4;100mM NaCl;0.1% IGEPAL;1mM MgAc;1mMイミダゾール;1mM DTT;2mM CaCl2)で固定タンパク質複合体を洗浄し、そして600μl CBP溶出緩衝液(10mM Tris、pH8.0;5mM EGTA)の添加によって、37℃で5分間溶出した。溶出物を、シリコン処理試験管に回収し、そして凍結乾燥した。残ったカルモジュリン樹脂を50μl 4x Laemmli試料緩衝液中で5分間煮沸した。試料緩衝液を単離し、凍結乾燥分画と合わせ、そしてNuPAGE勾配ゲル(Invitrogen、4〜12%、1.5mm、10ウェル)上に装填した。
【0161】
パート5 質量分析によるタンパク質同定
5.1.質量分析前のタンパク質消化
本質的に、Shevchenkoら, 1996, Anal. Chem. 68:850−858に記載される方法にしたがって、ゲル中で、ゲル分離したタンパク質を還元し、アルキル化し、そして消化した。簡潔には、ゲル分離したタンパク質を、清潔なメスを用いて、ゲルから切り出し、10mM DTT(5mM重炭酸アンモニウム中、54℃、45分間)を用いて還元し、そして続いて、55mMヨードアセトアミド(5mM重炭酸アンモニウム中)で、暗所中、室温でアルキル化した(30分間)。還元し、そしてアルキル化したタンパク質を、5mM重炭酸アンモニウム中、12.5ng/μlのプロテアーゼ濃度のブタ・トリプシン(Promega)を用いて、ゲル中で消化した。消化を37℃で4時間進行させ、そして続いて、5μlの5%ギ酸を用いて、反応を停止した。
【0162】
5.2.質量分析による分析前の試料調製
20μlの1% TFAを用いて、ゲル・プラグを2回抽出し、そして酸性化した消化上清とともにプールした。真空遠心分離機中で試料を乾燥させ、そして13μlの1% TFA中に再懸濁した。
【0163】
5.3.質量分析データ収集
四極子TOF(QTOF2、QTOF Ultima、QTOF Micro、MicromassまたはQSTAR Pulsar、Sciex)またはイオン捕捉(LCQ Deca XP)質量分析計いずれかに直接カップリングしたナノLC系(CapLC、WatersまたはUltimate、Dionex)に、ペプチド試料を注入した。水性溶媒および有機溶媒の勾配を用いて、LC系上でペプチドを分離した(以下を参照されたい)。溶媒Aは0.5%ギ酸中の5%アセトニトリルであり、そして溶媒Bは、0.5%ギ酸中の70%アセトニトリルであった。
【0164】
【表1】
【0165】
LC系から溶出したペプチドを、質量分析計内で、部分的に配列決定した。
5.4.タンパク質同定
LC−MS/MS実験で得たペプチド質量および断片化データを用いて、NCBI(NCBInr、dbEST、並びにヒトおよびマウスゲノムに関して)および欧州バイオインフォマティクス研究所(EBI、ヒト、マウス、キイロショウジョウバエおよび線虫プロテオーム・データベースに関して)で、維持され、そして定期的に更新される、fasta形式のタンパク質およびヌクレオチド配列データベースにクエリーを行った。測定したペプチド質量および断片化データを、ソフトウェア・ツール、Mascot(Matrix Science;Perkinsら, 1999, Electrophoresis 20:3551−3567)を用いてデータベース中のエントリーから計算した同じデータと相関させることによって、タンパク質を同定した。検索規準は、分析にどの質量分析計を用いたかに応じて、多様であった。
【0166】
(実施例2)
siRNAが仲介するGPR49のノックダウンのAβ1−42レベルに対する影響
結果:
我々は、APPプロセシングの既知のエフェクターであるBACE1およびニカストリンに対して向けられるsiRNA同様、GPR49をターゲットとするsiRNAが、Aβ1−42分泌の有意な減弱を引き起こし、一方、Luc3 siRNAは影響しないことに注目し(図2A)、GPR49が、APPのプロセシング/分泌を制御する際に機能的な役割を果たすことを立証した。
【0167】
我々は、GPR49 siRNAが、実際に、GPR49の発現に干渉することを確認した(図2B)。
APPプロセシングのエフェクターとしてのGPR49の機能的検証のため、RNAi遺伝子発現摂動(perturbation)戦略を使用した:GPR49に対して向けられるsiRNA、あるいはAPPプロセシングの既知のエフェクターであるBACE1またはニカストリンに対して向けられるsiRNA、あるいは関連しないLuc3に対して向けられるsiRNAを、ヒトAPP695を発現するSK−N−BE2神経芽細胞にトランスフェクションした。ヒトGPR49に対するsiRNAは、Dharmacon Research Inc.によって合成された。
【0168】
GPR49に対して用いるsiRNA配列は:AACAGCAGTATGGACGACCTTであった。
製造者の指示にしたがって、LipofectAMINE 2000(Invitrogen)を用いて、SK−N−BE2細胞のトランスフェクションを行った。簡潔には、96ウェルあたり最終体積85μl中、1.0x104細胞の密度で、トランスフェクション12〜16時間前に、細胞を植え付けた。25nMのsiRNAを8μlのOpti−MEM緩衝液(Gibco)および60ngのキャリアーDNAと混合し、そして混合物を細胞に添加する前に、室温で20分間インキュベーションした。トランスフェクション16〜48時間後、培地を、それぞれ血清を含むまたは含まない、100μlまたは200μlの増殖培地と交換した。トランスフェクション72時間後、Aβ1−42 ELISA(Innogenetics)のため、100μlの上清を採取した。製造者の指示にしたがって、アッセイを行った。
【0169】
定量的RT−PCRによって、選択したsiRNAのノックダウン効率を評価した。簡潔には、6ウェルあたり、5x105 SKNBE2細胞を蒔き、そして翌日、25nM siRNAをトランスフェクションした。トランスフェクション36時間後、細胞を採取し、そして標準法を用いて、総RNAを調製し、そして逆転写した。製造者の指示にしたがって、GPR49の相対発現レベルを決定するため、等量のcDNAおよびGPR49特異的プライマーを利用した。すべての値を、ヒト参照RNA(Stratagene)に対して標準化した。
【0170】
(実施例3)
GPR49活性の決定
GPR49に誘発されるシグナル伝達カスケードは、現在、未知である。しかし、他のファミリーメンバーである、ヒト糖ホルモン受容体、並びに系統学的に遠い、線虫における推定上のLGRオルソログ(Kudo M, Chen T, Nakabayashi K, Hsu SY, Hsueh AJ.(2000)The nematode leucine−rich repeat−containing, G protein−coupled receptor(LGR) protein homologous to vertebrate gonadotropin and thyrotropin receptors is constitutively active in mammalian cells. Mol Endocrinol. 14(2):272−84)は、アデニル酸シクラーゼ依存機構を通じて、シグナル伝達することが示されてきている。後者は、哺乳動物細胞において、異種発現されると、細胞の環状アデノシン一リン酸(cAMP)レベルの恒常的増加を引き起こす。
【0171】
GPR49がcAMP経路にカップリングしていることを確認するため、公知のものとして利用可能ないくつかのアッセイを、使用することも可能である。例えば、Bresnickら(Bresnick JN, Skynner HA, Chapman KL, Jack AD, Zamiara E, Negulescu P, Beaumont K, Patel S, McAllister G(2003)Identification of signal transduction pathways used by orphan g protein−coupled receptors. Assay Drug Dev Technol. 1(2):239−49)は、ベータ−ラクタマーゼ・レポーター構築物を用いて、オーファンGPRCによって用いられるシグナル伝達経路を同定した。
【0172】
次いで、GPR49発現細胞で観察されるが、GPR49欠損細胞では観察されない、細胞のcAMP上昇を誘発する能力に基づいて、GPR49調節剤を同定する。cAMPの直接測定は、例えば、cAMP応答要素(CRE)−ルシフェラーゼ・レポーター細胞株を用いた、ハイスループット形式で実行可能である(Gabriel D, Vernier M, Pfeifer MJ, Dasen B, Tenaillon L, Bouhelal R(2003)High throughput screening technologies for direct cyclic AMP measurement. Assay Drug Dev Technol. 1(2):291−303)。cAMP上昇またはcAMP依存性シグナル伝達の他の測定法が、当業者に明らかである。以前、オーファンGPCRの小分子調節剤を同定した前例の概説には、Howard AD, McAllister G, Feighner SD, Liu Q, Nargund RP, Van der Ploeg LH, Patchett AA(2001)Orphan G−protein−coupled receptors and natural ligand discovery. Trends Pharmacol Sci. 22(3):132−40を参照されたい。
【0173】
(実施例4)
GPR49調節剤による、Aβ1−42生成/分泌の調節
ヒトAPP695を安定に過剰発現するSKNBE2細胞(SKNBE2/APP695)(または別の適切な細胞株)またはベータ−/ガンマ−セクレターゼ切断反応速度が増進した適切な突然変異体を増殖培地中で蒔き、そして翌朝、4時間、血清枯渇させた。次いで、血清不含培地中で希釈した、GPR49調節剤、好ましくは阻害剤を添加し、そして適切な期間、インキュベーションした。細胞上清を収集し、そしてELISA(INNOGENETICS N.V.、BelgiumのInnotest β−アミロイド(1−42))によって、Aβ1−42のレベルを決定した。
【0174】
本発明は図面に、より詳細に記載される。
【図面の簡単な説明】
【0175】
【図1】図1:GPR49が相互作用することが見出されたTAPエントリーポイント(白)の略図。
【図2】図2:siRNAが仲介するGPR49発現のノックダウンは、Aβ1−42の生成/分泌を減弱する。
【0176】
図2A:BACE1、ニカストリン、GPR49またはLuc3に対して向けられるSiRNAを、APP695を過剰発現するSK−N−BE2神経芽腫細胞にトランスフェクションした。トランスフェクション48時間後、増殖培地を取り除き、そして細胞を血清不含培地中で1晩インキュベーションした。上清を収集し、そしてAβ1−42レベルをELISA(INNOGENETICS N.V.、BelgiumのInnotest β−アミロイド(1−42))によって決定した。少なくとも3回の独立実験を2つ組で行った。代表的な例を示す。
【0177】
図2B:GPR49に対して向けられるsiRNAは、定量的RT−PCR分析で評価した際、GPR49 mRNAを特異的に減少させたが、関連しないLuc3に対して向けられるsiRNAは、該mRNAを減少させなかった。各siRNAを示す2つのバーは、2つの独立実験に相当する。
【図3】図3:1文字コードで示す、ヒトGPR(LGR5;ロイシン・リッチ・リピート含有Gタンパク質共役型受容体5)のアミノ酸配列。
【図4−1】図4:ヒトLGR4、LGR5/GPR49およびLGR6の複数配列並列。
【図4−2】図4:ヒトLGR4、LGR5/GPR49およびLGR6の複数配列並列。
【特許請求の範囲】
【請求項1】
神経生成(neurogenerative)疾患治療用の薬剤組成物を調製するための、GPR49相互作用分子の使用。
【請求項2】
GPR49相互作用分子がGPR49阻害剤である、請求項1の使用。
【請求項3】
阻害剤が、抗体、アンチセンス・オリゴヌクレオチド、siRNA、低分子量分子(LMW)、結合性ペプチド、アプタマー、リボザイムおよびペプチド模倣体(peptidomimetic)からなる群より選択される、請求項2の使用。
【請求項4】
GPR49が細胞内タンパク質複合体の一部である、請求項1〜3のいずれかの使用。
【請求項5】
相互作用分子または阻害剤が、ガンマ−セクレターゼおよび/またはベータ−セクレターゼの活性を調節する、請求項1〜4のいずれかの使用。
【請求項6】
神経変性(neurodegenerative)疾患がアルツハイマー病である、請求項1〜5のいずれかの使用。
【請求項7】
ガンマ−セクレターゼおよび/またはベータ−セクレターゼの調節剤を同定する方法であって:
a.既定の試験化合物がGPR49相互作用分子であるかどうか決定することによって、GPR49相互作用分子を同定し、
b.工程a)のGPR49相互作用分子がガンマ−セクレターゼおよび/またはベータ−セクレターゼの活性を調節可能であるかどうか決定する
工程を含む、前記方法。
【請求項8】
工程a)において、試験化合物をGPR49と接触させ、そして試験化合物とGPR49の相互作用を決定する、請求項7の方法。
【請求項9】
GPR49と試験化合物の相互作用がGPR49活性の阻害を生じる、請求項8の方法。
【請求項10】
工程b)において、ガンマ−セクレターゼおよび/またはベータ−セクレターゼがAPPを切断する能力を測定する、好ましくはAベータ42を産生する能力を測定する、請求項7〜9のいずれかの方法。
【請求項11】
神経変性疾患治療用の薬剤組成物を調製するための方法であって:
a.請求項7〜10にしたがって、ガンマ−セクレターゼおよび/またはベータ−セクレターゼの調節剤を同定し、そして
b.ガンマ−セクレターゼおよび/またはベータ−セクレターゼの調節剤を薬剤組成物に配合する
工程を含む、前記方法。
【請求項12】
同定された分子と薬学的に許容しうるキャリアーを混合する工程をさらに含む、請求項11の方法。
【請求項13】
請求項1〜5のいずれかに定義されるようなGPR49阻害剤を含む、薬剤組成物。
【請求項14】
請求項11または12のいずれかに記載の方法によって得られうる、薬剤組成物。
【請求項15】
アルツハイマー病などの神経変性疾患および関連する神経変性障害の治療用の、請求項13または14のいずれかに記載の薬剤組成物。
【請求項16】
神経変性疾患、好ましくはアルツハイマー病を治療するかまたは予防する方法であって、こうした治療または予防の必要がある被験者に、請求項13〜15のいずれかの薬剤組成物の療法的有効量を投与する、前記方法。
【請求項17】
ベータ−セクレターゼおよび/またはガンマ−セクレターゼの活性をin vitroで調節するための、GPR49相互作用分子の使用。
【請求項1】
神経生成(neurogenerative)疾患治療用の薬剤組成物を調製するための、GPR49相互作用分子の使用。
【請求項2】
GPR49相互作用分子がGPR49阻害剤である、請求項1の使用。
【請求項3】
阻害剤が、抗体、アンチセンス・オリゴヌクレオチド、siRNA、低分子量分子(LMW)、結合性ペプチド、アプタマー、リボザイムおよびペプチド模倣体(peptidomimetic)からなる群より選択される、請求項2の使用。
【請求項4】
GPR49が細胞内タンパク質複合体の一部である、請求項1〜3のいずれかの使用。
【請求項5】
相互作用分子または阻害剤が、ガンマ−セクレターゼおよび/またはベータ−セクレターゼの活性を調節する、請求項1〜4のいずれかの使用。
【請求項6】
神経変性(neurodegenerative)疾患がアルツハイマー病である、請求項1〜5のいずれかの使用。
【請求項7】
ガンマ−セクレターゼおよび/またはベータ−セクレターゼの調節剤を同定する方法であって:
a.既定の試験化合物がGPR49相互作用分子であるかどうか決定することによって、GPR49相互作用分子を同定し、
b.工程a)のGPR49相互作用分子がガンマ−セクレターゼおよび/またはベータ−セクレターゼの活性を調節可能であるかどうか決定する
工程を含む、前記方法。
【請求項8】
工程a)において、試験化合物をGPR49と接触させ、そして試験化合物とGPR49の相互作用を決定する、請求項7の方法。
【請求項9】
GPR49と試験化合物の相互作用がGPR49活性の阻害を生じる、請求項8の方法。
【請求項10】
工程b)において、ガンマ−セクレターゼおよび/またはベータ−セクレターゼがAPPを切断する能力を測定する、好ましくはAベータ42を産生する能力を測定する、請求項7〜9のいずれかの方法。
【請求項11】
神経変性疾患治療用の薬剤組成物を調製するための方法であって:
a.請求項7〜10にしたがって、ガンマ−セクレターゼおよび/またはベータ−セクレターゼの調節剤を同定し、そして
b.ガンマ−セクレターゼおよび/またはベータ−セクレターゼの調節剤を薬剤組成物に配合する
工程を含む、前記方法。
【請求項12】
同定された分子と薬学的に許容しうるキャリアーを混合する工程をさらに含む、請求項11の方法。
【請求項13】
請求項1〜5のいずれかに定義されるようなGPR49阻害剤を含む、薬剤組成物。
【請求項14】
請求項11または12のいずれかに記載の方法によって得られうる、薬剤組成物。
【請求項15】
アルツハイマー病などの神経変性疾患および関連する神経変性障害の治療用の、請求項13または14のいずれかに記載の薬剤組成物。
【請求項16】
神経変性疾患、好ましくはアルツハイマー病を治療するかまたは予防する方法であって、こうした治療または予防の必要がある被験者に、請求項13〜15のいずれかの薬剤組成物の療法的有効量を投与する、前記方法。
【請求項17】
ベータ−セクレターゼおよび/またはガンマ−セクレターゼの活性をin vitroで調節するための、GPR49相互作用分子の使用。
【図1】

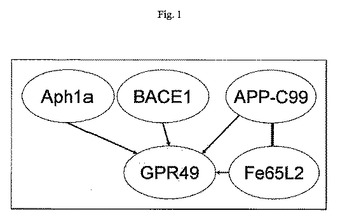
【図2】
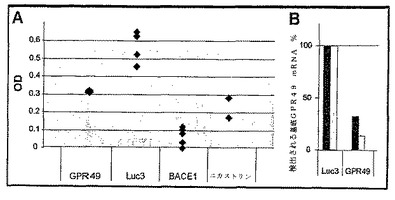
【図3】

【図4−1】
【図4−2】
【図2】
【図3】
【図4−1】
【図4−2】
【公表番号】特表2007−525491(P2007−525491A)
【公表日】平成19年9月6日(2007.9.6)
【国際特許分類】
【出願番号】特願2006−549895(P2006−549895)
【出願日】平成16年11月29日(2004.11.29)
【国際出願番号】PCT/EP2004/013539
【国際公開番号】WO2005/074980
【国際公開日】平成17年8月18日(2005.8.18)
【出願人】(505022105)セルゾーム・アクチェンゲゼルシャフト (4)
【氏名又は名称原語表記】CELLZOME AG
【Fターム(参考)】
【公表日】平成19年9月6日(2007.9.6)
【国際特許分類】
【出願日】平成16年11月29日(2004.11.29)
【国際出願番号】PCT/EP2004/013539
【国際公開番号】WO2005/074980
【国際公開日】平成17年8月18日(2005.8.18)
【出願人】(505022105)セルゾーム・アクチェンゲゼルシャフト (4)
【氏名又は名称原語表記】CELLZOME AG
【Fターム(参考)】
[ Back to top ]