
H−1パルボウイルスに結合する抗体
完全または中空H−1パルボウイルスキャプシドに特異的に結合する抗体またはその抗原結合フラグメントについて記載する。当該抗体は、様々な診断および治療方法、例えば妊娠中のH−1パルボウイルス感染症の検出/療法に有用であるが、それはパルボウイルスが妊婦約400例中1例を罹患させ、胎児消失または胎児水腫を誘発する可能性があるためである。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、完全または中空H−1パルボウイルスキャプシドに特異的に結合する抗体またはその抗原結合フラグメントに関する。当該抗体は、様々な診断法および治療法、例えばH−1パルボウイルス感染症の検出/治療に有用である。
【背景技術】
【0002】
パルボウイルスは、ウイルスファミリーのパルボウイルス科(Parvoviridae)の1つの属名である。パルボウイルス属は、ヘルパーウイルスの非存在下で複製可能な多数の小さな正20面体ウイルスを含んでいる。パルボウイルスは長さ約5,000bpの一本鎖DNAを含有している。DNAの3’および5’末端には、各々1本のパリンドローム(回文)配列が存在する。このDNAは、2つのキャプシドタンパク質であるVP1およびVP2(図1)ならびに2つの調節非構造タンパク質であるNS−1およびNS−2をコードする。後者のタンパク質はリン酸化されており、核または細胞質および核両方の局在をそれぞれ示す。第3のより小さなキャプシドタンパク質であるVP3は、タンパク質分解的開裂によりVP2から引き出される。これら2つのキャプシドタンパク質VP1およびVP2は、VP2コーディング領域がVP1コーディング領域内に完全に含まれるように、重複するオープンリーディングフレームによってコードされる。自然感染では、キャプシドタンパク質は、選択的スプライシングに起因して1:10のVP1:VP2比で発現する。近年、VP1に対して特異的なキャプシドタンパク質の領域(N末端領域)は、細胞ホスホリパーゼAと共通するモチーフを含有しており、実際にin vitroでこの活性を発揮することが証明されている。パルボウイルスVP1が類似性を共有するカルシウム依存性の分泌型PLA2は、膜の透過による細胞溶解を包含するシグナリング経路に関与する。他方、NS1は、順にホスホリパーゼを経由して調節される、プロテインキナーゼCファミリーのメンバーによって調節されることが証明されている。
【0003】
パルボウイルスは、通常はそれらの自然宿主の集団によって良好に耐容され、その中で明白な病理学的徴候を示すことなく存続する。これは、先天性免疫による胎児および新生児の保護、ならびに成体動物においてはパルボウイルス複製が狭い範囲の標的増殖組織へ顕著に制限されることの両方に起因する。この宿主耐性は、特に齧歯類パルボウイルス、例えばマウス微小ウイルス(MVM)ならびにそれらの各天然宿主、つまりマウスおよびラット内のH−1ウイルスに関する。
【0004】
さらに、ヒトは、パルボウイルス、例えばH−1パルボウイルスに感染する可能性がある。実際、パルボウイルスは、一般的感染症であり、通常は小児における伝染性紅斑として現れる。この感染症は、通常は軽症であり、健常者においては自己限定性である。しかしパルボウイルスは、成人における反応性関節炎、および血液学的状態もしくは免疫抑制状態の成人における重症貧血もまた誘発することがある。さらに、パルボウイルスは、妊婦約400例中1例で発症し、胎児消失または胎児水腫を誘発することがある。そこで、特に妊婦におけるパルボウイルス感染症の同定は、モニタリングおよび考え得る治療のために重要である。残念なことに、パルボウイルス感染症の診断/療法のための現行法は改良を必要としており、例えば現時点では、集合H−1パルボウイルスに対する特異的抗体は入手することができない。
【発明の概要】
【発明が解決しようとする課題】
【0005】
そこで、本発明の基礎にある技術的課題は、パルボウイルス感染症またはそれに関連する疾患を診断および治療するための改良された手段を提供することである。
【課題を解決するための手段】
【0006】
前記技術的課題の解決策は、特許請求の範囲において特徴付けられた実施形態を提供することによって達成される。H−1パルボウイルスを用いてBalb/cマウスを免疫した後、本発明者らは、抗体産生B細胞を脾臓から単離し、Ag8骨髄腫細胞と融合させることができた。淘汰圧を用いた3ラウンドの選択後、H−1パルボウイルスに対するモノクローナル抗体を産生するハイブリドーマ細胞系BL−H1を単離することができた。
【0007】
そこで、本発明は、完全または中空H−1パルボウイルスキャプシドに特異的に結合する抗体またはその抗原結合フラグメントに関する。
【0008】
本明細書で使用する用語「抗体」は、完全抗体、または特異的結合に対して完全抗体と競合するその結合フラグメントを意味する。(抗原)結合フラグメントは、組換えDNA技術によって、または完全抗体の酵素的もしくは化学的開裂によって生成される。結合フラグメントには、Fab、Fab’、F(ab’)2、Fvおよび一本鎖抗体ならびに「ディアボディ(二重特異性抗体)」が含まれる。「二重特異性」もしくは「二機能性」以外の抗体は、その各々が同一である結合部位を有すると理解されている。
【0009】
本明細書で使用する用語「キャプシド」は、「キャプソマー」と呼ばれる、異なる、または同一のタンパク質サブユニットから形成されるウイルス核酸を取り囲むタンパク質の保護膜である。キャプシドは、ウイルスゲノムによってコードされるタンパク質から生成され、その形状は形態学的識別の根拠として役立つ。ウイルスによりコードされたタンパク質サブユニットは、自己集合してキャプシドを形成し、一般にウイルスゲノムの存在を必要とする。パルボウイルスキャプシドは、完全粒子の成熟の一部としてタンパク質VP1、VP2およびVP3から構成される。
【0010】
本明細書で使用する用語「完全キャプシド」は、ウイルス核酸、好ましくは生存ウイルスを含有するパルボウイルスキャプシドを意味する。用語「中空キャプシド」は、包埋された核酸を含まないキャプシドを意味する。
【0011】
好ましい実施形態では、本発明の抗体は、天然H−1パルボウイルスキャプシドには結合するが、変性キャプシド、例えば界面活性剤もしくは熱を使用することによって変性したキャプシドには結合しない抗体(またはその抗原結合フラグメント)である。
【0012】
より好ましい実施形態では、本発明の抗体(またはその抗原結合フラグメント)は、中和抗体である。本明細書で使用する用語「中和抗体」は、パルボウイルスの生物学的活性、例えば複製および/または感染性を低減させる、または無効にする抗体を意味する。
【0013】
いっそうより好ましい実施形態では、本発明の抗体は、モノクローナル抗体である。H−1パルボウイルスに特異的に結合するモノクローナル抗体は、培養中の連続細胞系による抗体分子の産生を提供する任意の技術を使用して調製することができる。これらの技術には、ハイブリドーマ技術、ヒトB細胞ハイブリドーマ技術およびEBVハイブリドーマ技術が含まれる(Kohler et al., Nature 256 (1985), 495−7)。全キャプシド、VP1、VP2、VP3またはそれらのフラグメントを使用して哺乳動物、例えばマウス、ラット、ウサギ、モルモット、サルまたはヒトを免疫し、ポリクローナル抗体を産生することができる。所望であれば、免疫原は、担体タンパク質、例えばウシ血清アルブミン、サイログロブリンおよびキーホール リンペット ヘモシアニンへ抱合させることができる。宿主種に依存して、様々なアジュバントを使用すると、免疫学的応答を増加させることができる。当該アジュバントには、フロイントアジュバント、無機質ゲル(例えば、水酸化アルミニウム)および界面活性剤(例えば、リゾレシチン、プルロニック(Pluronic、登録商標)ポリオール類、ポリアニオン類、ペプチド類、油乳濁剤、キーホール リンペット ヘモシアニンおよびジニトロフェノール)が含まれる。特にヒトにおいて使用されるアジュバントのうち、BCG(カルメット−ゲラン桿菌(Bacilli Calmette−Guerin))およびコリネバクテリウム パルヴム(Corynebacterium parvum)が特に有用である。
【0014】
一本鎖抗体を生成するために記載された技術は、H−1パルボウイルスキャプシドに特異的に結合する一本鎖抗体を産生するために当該技術分野において公知の方法を使用して適合させることができる。関連する特異性を備えるが別個のイディオタイプ組成の抗体は、ランダムコンビナトリアル免疫グロブリンライブラリーからのチェイン シャフリングによって生成することができる(Burton, PNAS USA 88 (1991), 11120−3)。一本鎖抗体は、さらにまたDNA増幅法、例えばPCRを使用して、鋳型としてハイブリドーマcDNAを用いて構築することもできる(Thirion et al., Eur.J.Cancer Prev. 5 (1996), 507−11)。一本鎖抗体は、単一特異性または二重特異性であってよく、および二価または四価であってよい。四価の二重特異性一本鎖抗体の構築は、例えば、Coloma & Morrison, Nat.Biotechnol. 15 (1997), 159−63に教示されている。二価の二重特異性一本鎖抗体の構築は、Mallender & Voss, J.Biol.Chem. X no9 (1994), 199−206に教示されている。
【0015】
最も好ましい実施形態では、本発明のモノクローナル抗体は、ブダペスト条約を遵守して、2009年11月25日に番号DSM ACC3030を付してDSMZに寄託されているハイブリドーマ細胞系BL−H1によって生成される。
【0016】
本発明はさらに、ブダペスト条約を遵守して、2009年11月25日に番号DSM ACC3030を付してDSMZに寄託されているハイブリドーマ細胞系BL−H1によって生成される抗体と同一エピトープに結合するモノクローナル抗体またはその抗原結合フラグメントを提供する。
【0017】
本明細書で使用する用語「エピトープ」には、抗体に特異的に結合することができる任意のタンパク質決定基が含まれる。エピトープ決定基は、通常は分子の化学的活性表面基、例えばアミノ酸もしくは糖側鎖からなり、通常は特異的三次元構造特性ならびに特異的電荷特性を有する。本発明の抗体は、解離定数が≦1μMである、好ましくは≦μM 100nMである、および最も好ましくは≦10nMである場合に抗原に特異的に結合すると言われている。典型的には、1つのエピトープを形成するために、少なくとも6、8、10または12連続アミノ酸が必要とされる。しかし、非連続アミノ酸を包含するエピトープは、より多くの、例えば少なくとも15、25または50アミノ酸を必要とすることがある。
【0018】
本発明はさらに、完全または中空H−1パルボウイルスキャプシドへの結合について上記で特徴付けた本発明の抗体と競合する、モノクローナル抗体またはその抗原結合フラグメントも提供する。
【0019】
好ましくは、抗体またはその抗原結合フラグメントは、検出可能な標識を持っている。抗体/フラグメントは、直接的または間接的に検出可能に、例えば、放射性同位体、蛍光化合物、生物発光化合物、化学発光化合物、金属キレート剤または酵素を用いて標識することができる。当業者であれば、抗体に結合させるための他の適切な標識を認識でき、またはルーチンの実験を使用して当該の標識を確認することができる。
【0020】
本発明はさらに、細胞系、すなわち本発明による抗体またはその抗原結合フラグメントを産生する細胞系に関する。好ましくは、この細胞系は、哺乳動物細胞系である。発現のための宿主として利用可能な哺乳動物細胞系は、当該技術分野において周知であり、DSMZまたはアメリカン培養細胞系統保存機関(ATCC)から入手可能な不死化細胞系が含まれ、CHO細胞、NSO細胞、HeLa細胞、BHK細胞、COS細胞、Hep細胞および多数の他の細胞系が含まれる。細菌、酵母、昆虫および植物を含む非哺乳動物細胞もまた組換え抗体を発現させるために使用できる。発現方法は、どの系が最高発現レベルを生成するのか、および所望の結合特性を備える抗体を産生するのかを決定することによって選択される。
【0021】
当該細胞系によって産生された抗体は、当該技術分野において周知の方法によって精製することができる。例えば、抗体は、パルボウイルスキャプシドまたはパルボウイルスエンベロープタンパク質が結合しているカラムを通過させることによって親和性精製することができる。結合した抗体は、次に塩濃度の高い緩衝液を用いてカラムから溶出させることができる。
【0022】
最も好ましい細胞系は、ブダペスト条約を遵守して2009年11月25日に番号ACC3030を付してDSMZに寄託されたハイブリドーマ細胞系BL−H1である。
【0023】
本発明は、さらに本発明のモノクローナル抗体またはその抗原結合フラグメントをコードする核酸も提供する。本発明の抗体、例えば一本鎖抗体をコードする核酸を、手動もしくは自動ヌクレオチド合成を使用して構築し、標準組換えDNA法を用いて発現構築物にクローン化し、および細胞内へ導入して該コード配列を発現させることができる。または、抗体は、例えば繊維状ファージ テクノロジー(Verhaar et al., Int.J.Cancer 61 (1995), 497−501)を使用して直接的に産生することができる。
【0024】
本発明は、さらに本発明の抗体またはその抗原結合フラグメントを含む診断用組成物も提供する。当該組成物は、パルボウイルス感染症の診断に有用である可能性がある。さらに、当該組成物は、
(a)H−1パルボウイルス標本およびさらに感染した動物および患者(薬物動態学的ウイルス血症)における、例えばELISAによるH−1パルボウイルスの定量、
(b)細胞内でのH−1パルボウイルス集合および輸送の検出、
(c)実験動物、特にラットにおけるH−1パルボウイルスによる汚染の検出のために使用することができる。
【0025】
診断には、完全または中空H−1パルボウイルスキャプシドに特異的に結合する抗体を、免疫化学的測定法、例えばウェスタンブロット、ELISA、放射免疫測定法、免疫組織化学的測定法、免疫沈降法、または当該技術分野において公知の他の免疫化学的測定法において使用できる。競合的結合または免疫放射線測定法のための多数のプロトコールは、当該技術分野において周知である。当該免疫測定法は、典型的には免疫源と該免疫源に特異的に結合する抗体との間の錯体形成の測定を含んでいる。
【0026】
診断は、特定の免疫測定法には限定されず、同種および異種方法の両方を含むことが意図されている。実施可能な代表的な免疫測定法には、蛍光偏光免疫測定法(FPIA)、蛍光免疫測定法(FIA)、酵素免疫測定法(EIA)、比濁阻害免疫測定法(NIA)、酵素結合免疫吸着検定法(ELISA)および放射免疫測定法(RIA)が含まれる。指標成分、もしくは標識基は、対象抗体に付着させることができ、測定装置の利用可能性および適合する免疫測定法によって指示されることが多い方法の様々な使用のニーズを満たすよう選択される。上記に記載した様々な免疫測定法を実施する際に使用可能な一般的技術は、当業者には公知である。
【0027】
本発明は、さらに本発明の抗体またはその抗原結合フラグメントおよび薬学的に許容される担体を含む医薬組成物も提供する。抗体またはその抗原結合フラグメントは、例えばH−1パルボウイルス感染症またはそれに関連する疾患を治療するための、該抗体がウイルスを中和する能力(antidot)を使用する方法において使用することができる。
【0028】
療法には、抗体またはその抗原結合フラグメントは、有効量で、薬学的に許容される担体と組み合わされて存在する。「薬学的に許容される」は、有効成分の生物学的活性の有効性を妨害しない、およびそれが投与される患者にとって毒性ではない任意の担体を含むことが意図されている。適切な医薬担体の例は当該技術分野において周知であり、リン酸緩衝生理食塩水、水、乳濁液、例えば水中油型乳濁液、様々なタイプの湿潤剤、無菌溶液などが含まれる。当該担体は、従来の方法によって調製することができる。
【0029】
本発明による抗体またはその抗原結合フラグメントを、被験者に有効量で投与することができる。「有効量」は、感染症/疾患の経過および重症度に影響を及ぼし、当該病状の低減または緩解をもたらすために十分な抗体またはその抗原結合フラグメントの量を意味する。これらの感染症、疾患または障害を治療および/または予防するために有用な「有効量」は、当業者には公知の方法を使用して決定することができる(例えば、Fingl et al., The Pharmocological Basis of Therapeutics, Goodman and Gilman, eds. Macmillan Publishing Co., New York, pp.1−46(1975)を参照されたい)。
【0030】
最後に、本発明は、さらにH−1パルボウイルス感染症または当該感染症と関連する疾患の診断または療法に有用な本発明による抗体またはその抗原結合フラグメントを含むキットを提供する。好ましくは、本キットは、1つ以上の容器、例えばバイアル、試験管などを、その中にきっちりと密封して収容するために区分化されている輸送手段を含み、該容器手段の各々は該分析において使用される個別要素の1つを含んでいる。例えば、容器手段の1つは、検出可能に標識される、または検出可能に標識することができる本発明による抗体またはその抗原結合フラグメントを含むことができる。本キットは、さらに緩衝液を含有する容器および/またはレポーター分子(例えば、酵素もしくは蛍光標識)に結合したレポーター手段(例えば、ビオチン結合タンパク質、例えばアビジンもしくはストレプトアビジン)を含む容器も有することができる。
【図面の簡単な説明】
【0031】
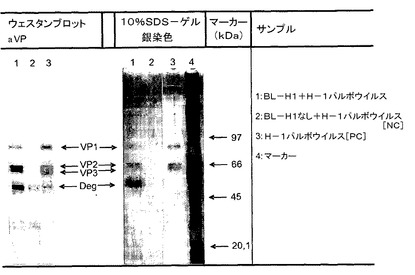
【図1】図1は、H−1パルボウイルスの免疫沈降反応を示す。 レーン1:BL−H1モノクローナル抗体を用いて免疫沈降させられたH−1パルボウイルス。レーン2:BL−H1モノクローナル抗体を伴わない、免疫沈降反応が生じないH−1パルボウイルスを意味する陰性コントロール。レーン3:陽性コントロール:免疫沈降反応方法を用いないSDS−ゲルおよびウェスタンブロット上のH−1パルボウイルス単独。レーン4:タンパク質マーカー。 PBS:リン酸緩衝生理食塩水。mAb:BL−H1。aVP:変性H−1パルボウイルスのウイルスタンパク質を認識するポリクローナル抗体。
【図2】図2は、ウェスタンドットブロットを示す。 ウェスタンドットブロットは、天然および変性H−1パルボウイルスを用いて生成された。BL−H1:H−1パルボウイルスに対するモノクローナル抗体。aVP:変性ウイルスタンパク質を認識するポリクローナル抗体(ドデシル硫酸ナトリウムポリアクリルアミドおよびジチオトレイトールを用いる)。天然:未処理H−1パルボウイルス。陰性コントロール:H−1パルボウイルスを含まない。
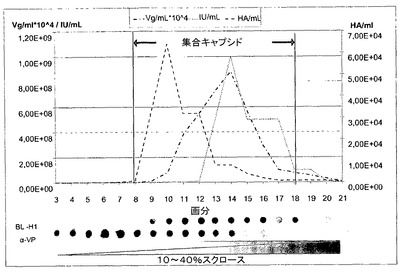
【図3】図3は、野生型H−1の分布およびBL−H1モノクローナル抗体を用いた検出を示す。 モノクローナル抗体BL−H1がキャプシド、モノ/オリゴマーキャプシドタンパク質または変性キャプシドタンパク質と反応するか決定するため、細胞抽出物は、pUC 19ΔHindIIIでのトランスフェクションの24時間後に、293T HEK細胞から調製した。細胞抽出物は、直線スクロース勾配上で分画させる。 BL−H1:H−1パルボウイルスに対するモノクローナル抗体。aVP:変性H−1パルボウイルスのウイルスタンパク質を認識するポリクローナル抗体。プラスミドpUC19ΔHindIIIは、完全H−1パルボウイルスゲノムを含有する。Vg:リアルタイムPCRによって分析されたウイルスゲノム含有粒子。IU:感染単位。HA:完全または中空H−1パルボウイルスの赤血球凝集反応。
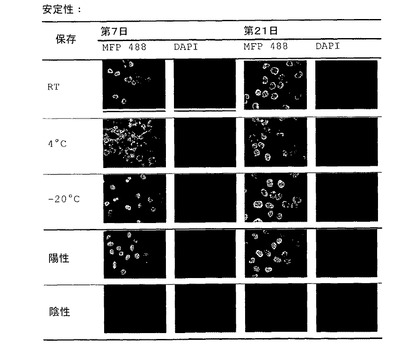
【図4】図4は、mab BL−H1の免疫蛍光試験(精製7および21日後のBL−H1活性の安定性)を示す。 精製後、BL−H1抗体を、各々室温、4℃および−20℃で保存する。活性は、感染したNB−324K細胞上でIF(免疫蛍光)によって21日後に試験する。陽性コントロールは、ポリクローナル抗体C8B10である。分析は、顕微鏡Axioskop 2 plusおよびカメラAxioCamMRc(倍率:40倍)によって実施する。
【図5】図5は、H−1パルボウイルスに感染したNB−324K細胞の中和を示す。 H−1パルボウイルスの中和は、野生型および組換えH−1パルボウイルスの何れかでの1のMOIで、550nm(吸光度550nm)で測定するMTT測定法によって示されている。0ng〜5000ngのB1−H1濃度を試験すると、3E5〜3E7のH−1パルボウイルス/μg(BL−H1)に対応した。
【図6】図6は、mab BL−H1を用いた野生型H−1 PVの中和を示す。 H−1パルボウイルスの中和は、1および10のMOIでのNB−324K細胞上でのプラークアッセイによって示されている。3E4または1E5のH−1パルボウイルスはいずれも1μgのBL−H1で中和された。 NC=陰性コントロール。PC=陽性コントロール。
【図7】図7は、組換えH−1 EGFP(緑色蛍光タンパク質)を示す。 1および10のMOIで緑色蛍光タンパク質(GFP)を含有する組換えH−1パルボウイルスは、感染したNB−324K細胞である0ng〜5000ngのB1−H1濃度で中和された。GFPは、40倍の倍率で分析する。 NB−324K細胞上でのBL−H1を用いた感染阻害作用は、一晩(o/n)のB1−H1プレインキュベーションおよびその後のH−1パルボウイルスによる感染によって所見されない。
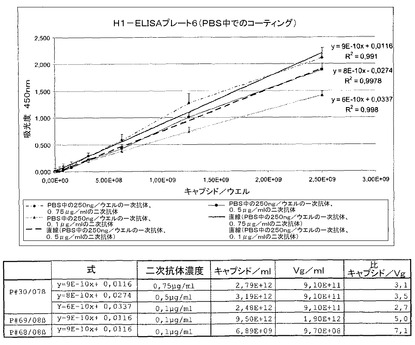
【図8】図8は、H−1 PV検出のためのELISAの開発を示す。 H−1パルボウイルスキャプシドは、標準曲線を評価するために分光光度計において450nmで測定する。現在もなお、3種の異なる濃度(0.75μg/ml、0.5μg/mlおよび0.1μg/mlの二次抗体)を使用する。
【0032】
以下、実施例により本発明を具体的に例示する。
【実施例1】
【0033】
一般的方法
(A)H−1パルボウイルスによるBalb/cマウスの免疫
マウスを以下のスケジュールに従って免疫した。
(1)注射1週間前
麻酔後、眼窩後穿刺により15μlの血液を採取した(陰性コントロール)。
【0034】
(2)免疫
麻酔:PBS中の100μlの0.2%塩酸キシラジン(Rompun(登録商標)、Bayer社)およびPBS中の体重10g当たり各10μgの塩酸ケタミン(Ketavalt、Parke−Davis社)の腹腔内[i.p.]注射。
【0035】
(3)1.H−1パルボウイルスの注射
40%のビジパーク(Visipaque、登録商標)−リンガー液中の100μlウイルス(=7×1010ウイルスゲノム含有粒子[Vg]=1.2×108PFU(プラーク形成単位))、
+50μlのPBS、
+150μlの不完全アジュバント、
を混合し、4つの相違する位置に各60μlを皮下[s.c.]注射。
【0036】
(4)2.初回注射の1カ月後のH−1パルボウイルスの注射
1.注射と同様の手順。
【0037】
(5)3. 初回注射の2カ月後のH−1パルボウイルスの注射
追加免疫:脾臓を摘出する1週間前
100μlのウイルス(7×1010Vg=1.2×108PFU)
+200μlのPBSの腹腔内[i.p.]注射。
【0038】
(B)ハイブリドーマの生成
ハイブリドーマは、以下のスケジュールに従って生成した。
【0039】
培地:
作業培地:RPMI 1640(Gibco社番号R8758)。
ハイブリドーマ培地:500mlのRPMI 1640(Gibco社番号R8758)
+5ml L−グルタミン200mM(Gibco社)。
+5ml Pen/Strep(ペニシリン10000U/ml、ストレプトマイシン10mg/ml(Gibco社))
+10mlの1M Hepes(pH7.2)(Sigma社)
+50mlのFCS(融合試験済み、30分間56℃で熱不活性化)。
選択培地:ハイブリドーマ培地+10mlのHAT50倍濃縮液(Gibco社)。
BM Condi−Med(選択用)10%。
【0040】
融合試薬:
PEG 1500(Roche社番号783641)。
【0041】
細胞:
Ag8/X63(B細胞腫瘍細胞)。
細胞は、融合の少なくとも1週間前に解凍し、ハイブリドーマ培地中で培養し、融合1日前に分割させた。細胞は、増殖期にあるはずである。
【0042】
融合:
調製:融合の1〜2日前
融合の1日前:Ag8細胞を分割させる(計18、増殖期にあるはずである)。
【0043】
融合日:
全培地および溶液を37℃に加温する。
【0044】
Ag8細胞の調製:
Ag8細胞を貯留し、7分間264×gで遠心し、30〜40mlの作業培地中に入れ、細胞を計数する(約1〜2×108細胞)。
【0045】
脾臓の調製:
麻酔後、マウスを致死させ、殺菌するために70%エタノール中に入れ、無菌ベースにピンで固定し、無菌ピンセットおよび剪刀を用いて脾臓を取り出し、それをシャーレ内の作業培地中へ入れ、心臓を穿刺して血液を採取し、1.5ml試験管中に収集する。リンパ節に注意を払う。
【0046】
脾臓細胞:
脾臓は剪刀を用いて数回切断し、無菌対物スライドを用いて脾臓を破砕し(粉砕)、脾嚢を取り出して作業培地を用いて洗浄し、脾嚢を廃棄し、脾細胞をパスツールピペットを用いてホモジナイズし、細胞懸濁液を50ml試験管内に収集し、嚢組織をセットし、上清を新しい50ml試験管内へ入れ、細胞を264×gで7分間遠心し、細胞を作業培地で1回洗浄し、細胞を264×gで7分間遠心し、ペレットを2〜5mlの作業培地中へ取り出し、細胞を計数する(5×107〜3×108細胞でなければならない)。
【0047】
脾細胞とAg8細胞との融合:
脾細胞:Ag8細胞=3:1を50ml試験管中で混合し、混合した細胞を264×gで7分間遠心する。各細胞ペレットは総計1×108細胞以下でなければならない。上清を廃棄し、試験管の底部を静かにタッピングすることによってペレットを崩壊させ、連続的に揺らしながら1分間にわたり1mlのピペットを使用して1mlのPEG 1500をペレットに加える。さらに2分間揺らし続け、3分間連続的に揺らすことによって慎重に10mlの選択培地を加える。さらに10mlの選択培地を加え、混合した細胞を120×gで7分間遠心し、上清を廃棄し、ペレットは試験管の底部を静かにタッピングすることによって崩壊させ、再び洗浄し(さらに10mlの選択培地を加え、混合した細胞を120×gで7分間遠心し、ペレットは試験管の底部を静かにタッピングすることによって崩壊させる)、100μlの選択培地を備える96ウエルプレートを調製する(各ウエルは細胞数2×105個/ウエル(=2×106個/ml、2×107個/10ml))。
【0048】
Ag8コントロール:
選択培地中に1E4細胞/ウエルを播種する(およそ24ウエル以下)。
【0049】
血液:
30分後(赤血球凝集反応)、遠心し(5000×g、10分間)、新しい試験管中に血清を採取する(陽性コントロール)。
【0050】
観察:
2〜7日後:HAT選択培地中のコントロールAg8細胞(死滅していなければならない)。
1週間後:融合した細胞を供給し、1ウエル当たり100μlを取り出し、廃棄し、100μlの新鮮HAT選択培地を供給する。
2週間後:スクリーニング(10%のBM CondiMedを備える培地)、免疫蛍光法、ウェスタンドットブロットを開始しなければならない。
【0051】
選択:
陽性スクリーニングのウエルを選択しなければならない。
1.選択:POSウエルを平板培養しなければならない。各96ウエルプレート、5細胞/ウエル、1細胞/ウエル。
2.選択:POSウエルを平板培養しなければならない。各96ウエルプレート、5細胞/ウエル、1細胞/ウエル。
3.選択:POSウエルを平板培養しなければならない。各96ウエルプレート、5細胞/ウエル、1細胞/ウエル。
【0052】
(C)H−1野生型キャプシドに対するモノクローナル抗体の産生
材料:
75cm2または150cm2の無菌フィルターを備えるGreiner Bio−one T−フラスコ。10%のFCS、1%のL−グルタミン(2.5mM)、1%のP/S、20mMのHEPESを備えるRPMI培地。BL−H1−ハイブリドーマ細胞。ピペット(5、10、20ml)。試験管(15本、50ml)。1lのボトル。水浴。そして遠心機。
【0053】
情報:
BL−H1の生成時間:約27時間。
懸濁細胞。
培地の交換:細胞を含む培地を遠心する:300×g、10分間。
B1−H1の採取:遠心:5000×g、5分間。
BL−H1細胞増殖は、グルコース濃度が高いほど良好である。
RPMIは2g/lのグルコースを含有し、3または4g/lへの増加は抗体産生を改良することができよう。
2×106個/mlまでの細胞密度が可能である。
【0054】
産生:
75cm2のフラスコには10ml、150cm2のフラスコには20mlの細胞懸濁液を入れる。
RPMI培地中でのBL−H1細胞の培養。開始時の細胞密度:2×105個/mlが理想的である。
細胞数1×105個/mlも可能であるが、細胞増殖は開始時にはより緩徐である。細胞数3×105個/mlも可能であるが、その場合、細胞を2または3日後に分割しなければならず、さもなければ生命力が低下する。
細胞を3日後(任意で4日後)に分割させる。
新鮮培地を備える15〜20%の馴化培地は、培地交換後、細胞生存性を増加させる。
【0055】
(D)親和性クロマトグラフィーを用いたモノクローナル抗体BL−H1の精製
材料:
AKTA Prime装置(GE Healthcare Europe社、フライブルク)。70%エタノール(H2O中)。20%エタノール(H2O中)。20mMリン酸ナトリウム緩衝液(pH7.0)(平衡バッファー=バッファーA)。1M Tris−HCl(pH9.0)(中和バッファー)。50ml試験管。ポリスチレン製丸底試験管。0.1Mクエン酸(pH4.0)(溶出バッファー=バッファーB)。HiTrap(商標)Protein A HP 5mlカラム。
【0056】
全緩衝液は、ボトルトップ フィルターを用いて濾過しなければならず、室温で10分間超音波を用いて100%脱気しなければならない(480W)。
【0057】
手順:
AKTA primeシステムの使用:最初に本システムを20%エタノールで洗浄する。試験管Aおよび試験管Bを20%エタノール中に入れ、「Run stored Method(保存されているメソッドを実行する)」を押し、プログラム1を選択する。その後、試験管にH2Oを入れ、再びプログラム1を選択する。ここで各試験管に平衡バッファーを満たし、全試験管を接続する。カラムを20%エタノール中に保管し、平衡バッファーを用いて直ちに洗浄しなければならない。プレッシャーリミットは、0.3MPaである。プロテインAカラムについての100%バッファーA(=0%バッファーB)の流速は、5ml/分である。代替のプロテインG:1ml/分(直列カラムの量とは無関係)。
【0058】
クロマトグラフィーは、メインメニューの「Manual Run(マニュアルで操作する)」で実行しなければならない。洗浄および装填のために、以下の条件を使用する。
メソッド ベースを設定する(ml)。
濃度%Bを設定する(0%)。
グラジエントを設定する(オフ)。
流速を設定する(A:1ml/分、B:5ml/分)。
フラクション ベースを設定する(ml)。
フラクション サイズを設定する(0)。
プレッシャーリミットを設定する(0.3MPa)。
バッファーバルブ ポジションを設定する(Pos 1)。
インジェクションバルブ ポジションを設定する(load)。
実行を開始する。
【0059】
クロマトグラフィーは、メインメニューの「Manual Run(マニュアル操作で作動する)」で実行しなければならない。カラムは、UVベースライン値が持続する限り、緩衝液を用いて洗浄しなければならない。カラムに充填する前に、マニュアル操作での作動で「auto zero(オートゼロ)」に入るが、これはUVベースライン値をゼロにリセットする。試験管Aを通して「抗体培地」を負荷する。このため、試験管Aを「抗体培地」内に入れ「break(中断する)」で保持する。さもないとシステム内に気泡が流入する。その後「continue(再開する)」を押す。最後に「break(中断する)」を押し、試験管Aに戻し、洗浄のために「continue(再開する)」を押す。UVベースライン値がほぼゼロである限り洗浄する。装填および洗浄中の素通り画分は追加ボトル内に収集しなければならない(カラムが過負荷になっている場合:これはUVシグナルの追加の増加を通して見ることができよう)。そこで、素通り画分は、二度目に使用することができよう。
【0060】
100%バッファーBへ目を向けると、他の全てのパラメーターは同一である。
メソッドベースを設定する(ml)。
濃度%Bを設定する(100%)。
グラジエントを設定する(オフ)。
流速を設定する(A:1ml/分、B:5ml/分)。
フラクション ベースを設定する(ml)。
フラクション サイズを設定する(0)。
プレッシャーリミットを設定する(0.3MPa)。
バッファーバルブ ポジションを設定する(Pos 1)。
インジェクションバルブ ポジションを設定する(load)。
実行を開始する。
【0061】
バッファーBがカラムに達すると、抗体の溶出が直ちに始まる。UVシグナルが強度に増加する。画分は、フラクションコレクターチューブを通して収集できよう。異なる抗体濃度のために、少なくとも2つの画分を収集することが推奨されている。
【0062】
画分を手作業で収集するため、「Manual Run(マニュアル操作で作動する)」の「set event mark(イベントマークを設定する)」を設定して開始時および終了時を見ることができよう。溶出された画分は、中和バッファーを用いて直ちにpH1にしなければならない。第1画分は今も少量のバッファーAを含有している可能性があるので、前もって中和バッファーの量を知ることは容易でない。不安定性を回避するために極めて迅速にpH紙を用いてチェックしなければならない。100%Puffer Bについての推奨値。
【0063】
【表1】

【0064】
ピークがベースライン値になった後、バッファーAへ切換える。
メソッド ベースを設定する(ml)。
濃度%Bを設定する(0%)。
グラジエントを設定する(オフ)。
流速を設定する(A:1ml/分、B:5ml/分)。
フラクション ベースを設定する(ml)。
フラクション サイズを設定する(0)。
プレッシャーリミットを設定する(0.3MPa)。
バッファーバルブ ポジションを設定する(Pos 1)。
インジェクションバルブ ポジションを設定する(load)。
実行を開始する。
【0065】
カラムおよびシステムの保管は20%エタノール中で行わなければならない。システム全体を洗浄しなければならない[プロトコールP−19−2Aを参照されたい]。
【0066】
(E)H1−キャプシド−ELISA
材料:
・フレキシブルプレート、96ウエル、U−ボトム、蓋なし、BD Falcon社。マイクロプレート用蓋、Greiner BioOne社。ウシ乳汁由来カゼイン、Sigma社。PBS+0.05%のTween(登録商標)。PBS(H2O中)。コーティング用:BL−H1画分E1、Tris−HClで中和した0.1Mグリシン緩衝液中の2mg/mlのIgG2A BL−H1(10mg/mlの全タンパク質、IgG2A+異種(foreighn)タンパク質)、4℃で保管。検出用:リン酸ナトリウム緩衝液中のBL−H1−HRP、およそ1mg/ml、−20℃で冷凍、試験管1本当たり10μl。標準物質:40%のビジパーク(商標)中のHV Pool green 1−2/1−3/3−3/4−2/4−3/5−2/6−2/7−2 1.5×1011Vg/ml、7.5×1011キャプシド/ml、4℃で保管。陽性コントロール:40%のビジパーク(商標)中のP#30/07β、9.1×1011Vg/ml(推定〜1×1013キャプシド/ml)、4℃で保管。光学接着カバー(Optical Adhesive Cover)(Applied Biosystems社)。TMB、ELISAのための超緩徐、液体物質システム、Sigma社、4℃で保管。停止液:室温で保管した1N H2SO4。Multiskan EXマイクロタイタープレート光度計、Thermo Scientific社。
【0067】
緩衝液:
洗浄バッファー:PBSに0.05%のTween(登録商標)を加える(ウェスタン(ドット)ブロット用のストック液を参照されたい)。
コーティング液:使用直前に調製する。E1をPBS(H2O中)中で2.5μg/mlへ希釈する、これは1:800を意味する。1プレート当たり10mlの希釈液。
ブロッキングバッファー(BB):PBS中の2mg/mlカゼイン+0.05%のTween(登録商標)。常に新しく調製する。
400mlのPBS+0.05%のTween(登録商標)を〜50℃に(マイクロウェーブで40〜50秒間)加熱し、それをビーカー内へ注入し、必要量のカゼインを加える。30分間加熱しながら、全てが溶解するまでおよそ50℃で1時間まで攪拌する。使用のためには、水浴中で37℃へ冷却する。1プレート当たり100mlで十分である。
検出:使用直前に調製する。BL−H1−HRP(二次抗体)をブロッキングバッファー中で0.1g/mlへ希釈する、これは1:10,000を意味する。1プレート当たり10mlの希釈液。
標準物質:HV pool greenを2.5×109〜3.91×107キャプシド/ウエルの標準物質へ希釈する。2つの標準物質列については、600μlの2.50×109(douplex)キャプシド/ウエル標準物質をストック標準物質からブロッキングバッファー中での1:30(2.5×1010キャプシド/ml)希釈によって調製する。300μlのブロッキングバッファーを6本の1.5ml試験管へ加え、1.25E+09、6.25E+08、3.13E0+8、1.56E+08、7.81E+07、3.91E+07キャプシド/ウエルと表示する。300μlの各標準物質を次の試験管に加え、各移行間にボルテックスミキサーにかけて連続希釈を実施する。PBSは、ブランクとして機能する(ブロッキングバッファーも使用することができる)。
陽性コントロール:BB中1:700に希釈すると、およそ4.2×109キャプシド/ウエル、各々最初は2.9×1012キャプシド/ウエル。1プレートに付きコントロールを含む少なくとも4ウエルを実施する。P#30/07βについての陰性コントロールは、40%のビジパーク(商標)をBB中1:700に希釈する。
【0068】
実行:
第0日
午後:フレキシブルプレートを100μl/ウエルのコーティング液で一晩4℃にてコーティングする。マイクロプレート用の蓋を用いてプレートを閉じる。この作業は、ウエルの液体を蓋に押し出さないように慎重に実施する。
【0069】
第1日
または、プレートを午前中、室温で2時間インキュベートすることでコーティングすることができる。
1.ブロッキングバッファーを上述したように調製する。コーティング液は、残っているコーティング液を全て除去するために吸収紙上で反転させて吸い取ることによってプレートから除去する。200μl/ウエルのブロッキングバッファーを加え、プレートを蓋で閉じ、37℃で1時間インキュベートする。
2.プレートのブロッキング中、上述したように標準物質および陽性コントロールを調製する。サンプル希釈のためにVg/ml力価によってキャプシド数/mlを推定する。より広い範囲をカバーするためにほぼ推定値の数種の希釈液を作製する。バッファーを用いてサンプルマトリックスの陰性コントロールを調製する(サンプルの希釈に注意を払う!)。
3.ウエルを吸引し、200μl/ウエルの洗浄バッファーを用いて2回洗浄する。最終洗浄後、プレートを反転させて、残っている緩衝液を全て取り除くために吸水性のペーパータオルで吸い取る。
4.ここから、プレートを用いる全ての工程は無菌フード下で行う。100μlの標準物質、陽性コントロール、各陰性コントロールまたはサンプルを各ウエルに加える。プレートを蓋で閉じる。37℃で1時間インキュベートする。
5.陽性コントロール、各陰性コントロールまたはサンプルを、異なるサンプル毎にチップを交換しながらピペットで取り除く。200μl/ウエルの洗浄バッファーを用いて3回洗浄する。最終洗浄後、プレートの表面をペーパータオルで乾燥させる。廃棄物をビーカー内に収集する。
6.100μl/ウエルの希釈BL−H1−HRPを各ウエルに加える。プレートを蓋で閉じ、37℃で1時間インキュベートする。
7.検出抗体を12チャンネルピペットを用いて取り出す。200μl/ウエルの洗浄バッファーを用いて4回洗浄する。最終洗浄後、プレートの表面をペーパータオルで乾燥させる。できる限り多くの液体を取り除く努力をする。廃棄物を無菌フード下でビーカー内に収集する(12チャンネルピペットを使用することができる)。
8.依然として無菌フード下で、100μl/ウエルのTMB溶液を各ウエルに加え、室温の暗所でおよそ15分間インキュベートする。およそ10分後に反応強度が申し分ないかチェックする。
9.100μl/ウエルの停止液を各ウエルに加えることによって反応を停止させる。プレートを光学接着カバーで密封する。
10.反応を停止させるため30分以内に450nmで吸光度を読み取る。Multiskan EXマイクロタイタープレート光度計を使用する。結果の選択的補正のために、プレートを550nmおよび595nmで追加して読み取る。
【0070】
追加の勧告:
ブロッキングは、一晩より長い時間実施してはならない。インキュベーション時間が長くなると凝集体の形成などが生じる。ウイルスおよび抗体の全希釈液は、使用直前に調製しなければならない。ウエルは、結合した抗体の消失を回避するため慎重に洗浄しなければならない。さらに、各列には同一時間工程でTMBを加えることが重要である。反応を停止させるため、2つの列間の変動を最小限に抑えるため、同一時間工程を使用しなければならない。残っている液体をピペットで取り除く場合は、結合した反応物質の消失を回避するために、ウエルの底部に極めて軽く触れるだけでこすってはならない。37℃でのプレートのインキュベーションは、ポリスチレンが弱熱導体であるため、周辺効果を誘発することがある。このため、分析の実施中にはブロッキングバッファーを37℃に維持することが重要である。これにより、周辺効果を下方レベルへ抑えることができる。標識された二次抗体は、使用後に再冷凍してはならない。再冷凍は、シグナルの低強度を引き起こすことがある。カゼインを含有するブロッキングバッファーは、−20℃で冷凍すれば少なくとも4週間保管することができる。この希釈手順は、間違えると結果が不正確になるので、慎重に実施しなければならない。多数回の希釈を次々に実施しなければ、極めて正確に実施することができる。
【実施例2】
【0071】
H−1キャプシドに対するBL−H1モノクローナル抗体の特性付け
BL−H1抗体がH−1パルボウイルスを免疫沈降させる能力を評価するため、抗体およびパルボウイルスを一緒に37℃で一晩インキュベートした。免疫複合体をプロテインAセファロースによって沈降させ、ウェスタンブロッティングによって変性ウイルスタンパク質に対する抗体を使用して分析した。これは、BL−H1抗体が集合H−1パルボウイルスキャプシドを認識することを証明している(図1)。
【0072】
この同定を特性付けるために、天然または変性H−1パルボウイルス何れかをニトロセルロース膜へ移した(図2)。BL−H1抗体は天然粒子しか認識しないが、ポリクローナル抗体は変性キャプシドを認識する。
【0073】
BL−H1抗体がキャプシドと、または天然ウイルスタンパク質とも反応するか決定するために、キャプシドタンパク質発現プラスミドでトランスフェクトした293T細胞の抽出物を調製し、直線スクロース勾配上で分画させた(図3)。BL−H1は、中空および完全H−1パルボウイルス粒子に対応する画分8〜18の間でピークを示す。完全または中空キャプシドは、赤血球凝集を介して実証される(画分8〜17)。完全キャプシドは、ウイルスゲノム含有粒子(画分10〜17)および感染単位を介した活性粒子(画分12〜18)によって決定される。これとは対照的に、非集合ウイルスタンパク質は、ウイルスタンパク質に対する抗体での画分3〜14において検出された。最後に、BL−H1と非変性ならびに変性キャプシドタンパク質との間に反応性は見られない。BL−H1は、集合H−1パルボウイルスキャプシド上に存在する立体構造エピトープを特異的に認識する。全てまとめると、これは、BL−H1が集合粒子によってのみ表示される構造的エピトープを認識することを証明している。
【0074】
図4は、感染したNB−324K細胞におけるH−1産生の免疫蛍光法によるBL−H1を用いた検出を示している。BL−H1抗体は、室温、4℃および−20℃で保管した場合、少なくとも3週間安定性である。
【0075】
図5、6および7では、H−1パルボウイルスのBL−H1中和能力が証明されている。
【0076】
MTT測定法は、H−1パルボウイルスとBL−H1抗体との正しい比率を見つけるために実施した。1E5までのH−1パルボウイルスを中和するには、BL−H1は1μgで十分である(図5)。
【0077】
プラークアッセイによって決定すると、1E5 H−1パルボウイルス当たり1μgのBL−H1を用いて95%の中和を達成することができた(図6)。この中和能力は、さらに緑色蛍光タンパク質を発現する組み換えH−1パルボウイルスを用いても証明されている(図7、上方パネル)。BL−H1自体が細胞レポーターと相互作用することによって間接的に作用するのではないことを証明するため、NB−324K細胞をBL−H1抗体とともに一晩プレインキュベートし、その後H−1パルボウイルスにより感染させた(図7、下方パネル)。
【実施例3】
【0078】
H−1−キャプシドのELISA
BL−H1を用いたH−1パルボウイルスの定量をELISAによって評価した。最初の結果は、キャプシドとウイルスゲノム含有粒子との比率が約5であることを示している(図8、下方部分)。使用した二次抗体の3種の濃度(0.1μg/ml〜0.75μg/ml)は、回収率における有意差を示さなかった(図8、上方部分)。
【技術分野】
【0001】
本発明は、完全または中空H−1パルボウイルスキャプシドに特異的に結合する抗体またはその抗原結合フラグメントに関する。当該抗体は、様々な診断法および治療法、例えばH−1パルボウイルス感染症の検出/治療に有用である。
【背景技術】
【0002】
パルボウイルスは、ウイルスファミリーのパルボウイルス科(Parvoviridae)の1つの属名である。パルボウイルス属は、ヘルパーウイルスの非存在下で複製可能な多数の小さな正20面体ウイルスを含んでいる。パルボウイルスは長さ約5,000bpの一本鎖DNAを含有している。DNAの3’および5’末端には、各々1本のパリンドローム(回文)配列が存在する。このDNAは、2つのキャプシドタンパク質であるVP1およびVP2(図1)ならびに2つの調節非構造タンパク質であるNS−1およびNS−2をコードする。後者のタンパク質はリン酸化されており、核または細胞質および核両方の局在をそれぞれ示す。第3のより小さなキャプシドタンパク質であるVP3は、タンパク質分解的開裂によりVP2から引き出される。これら2つのキャプシドタンパク質VP1およびVP2は、VP2コーディング領域がVP1コーディング領域内に完全に含まれるように、重複するオープンリーディングフレームによってコードされる。自然感染では、キャプシドタンパク質は、選択的スプライシングに起因して1:10のVP1:VP2比で発現する。近年、VP1に対して特異的なキャプシドタンパク質の領域(N末端領域)は、細胞ホスホリパーゼAと共通するモチーフを含有しており、実際にin vitroでこの活性を発揮することが証明されている。パルボウイルスVP1が類似性を共有するカルシウム依存性の分泌型PLA2は、膜の透過による細胞溶解を包含するシグナリング経路に関与する。他方、NS1は、順にホスホリパーゼを経由して調節される、プロテインキナーゼCファミリーのメンバーによって調節されることが証明されている。
【0003】
パルボウイルスは、通常はそれらの自然宿主の集団によって良好に耐容され、その中で明白な病理学的徴候を示すことなく存続する。これは、先天性免疫による胎児および新生児の保護、ならびに成体動物においてはパルボウイルス複製が狭い範囲の標的増殖組織へ顕著に制限されることの両方に起因する。この宿主耐性は、特に齧歯類パルボウイルス、例えばマウス微小ウイルス(MVM)ならびにそれらの各天然宿主、つまりマウスおよびラット内のH−1ウイルスに関する。
【0004】
さらに、ヒトは、パルボウイルス、例えばH−1パルボウイルスに感染する可能性がある。実際、パルボウイルスは、一般的感染症であり、通常は小児における伝染性紅斑として現れる。この感染症は、通常は軽症であり、健常者においては自己限定性である。しかしパルボウイルスは、成人における反応性関節炎、および血液学的状態もしくは免疫抑制状態の成人における重症貧血もまた誘発することがある。さらに、パルボウイルスは、妊婦約400例中1例で発症し、胎児消失または胎児水腫を誘発することがある。そこで、特に妊婦におけるパルボウイルス感染症の同定は、モニタリングおよび考え得る治療のために重要である。残念なことに、パルボウイルス感染症の診断/療法のための現行法は改良を必要としており、例えば現時点では、集合H−1パルボウイルスに対する特異的抗体は入手することができない。
【発明の概要】
【発明が解決しようとする課題】
【0005】
そこで、本発明の基礎にある技術的課題は、パルボウイルス感染症またはそれに関連する疾患を診断および治療するための改良された手段を提供することである。
【課題を解決するための手段】
【0006】
前記技術的課題の解決策は、特許請求の範囲において特徴付けられた実施形態を提供することによって達成される。H−1パルボウイルスを用いてBalb/cマウスを免疫した後、本発明者らは、抗体産生B細胞を脾臓から単離し、Ag8骨髄腫細胞と融合させることができた。淘汰圧を用いた3ラウンドの選択後、H−1パルボウイルスに対するモノクローナル抗体を産生するハイブリドーマ細胞系BL−H1を単離することができた。
【0007】
そこで、本発明は、完全または中空H−1パルボウイルスキャプシドに特異的に結合する抗体またはその抗原結合フラグメントに関する。
【0008】
本明細書で使用する用語「抗体」は、完全抗体、または特異的結合に対して完全抗体と競合するその結合フラグメントを意味する。(抗原)結合フラグメントは、組換えDNA技術によって、または完全抗体の酵素的もしくは化学的開裂によって生成される。結合フラグメントには、Fab、Fab’、F(ab’)2、Fvおよび一本鎖抗体ならびに「ディアボディ(二重特異性抗体)」が含まれる。「二重特異性」もしくは「二機能性」以外の抗体は、その各々が同一である結合部位を有すると理解されている。
【0009】
本明細書で使用する用語「キャプシド」は、「キャプソマー」と呼ばれる、異なる、または同一のタンパク質サブユニットから形成されるウイルス核酸を取り囲むタンパク質の保護膜である。キャプシドは、ウイルスゲノムによってコードされるタンパク質から生成され、その形状は形態学的識別の根拠として役立つ。ウイルスによりコードされたタンパク質サブユニットは、自己集合してキャプシドを形成し、一般にウイルスゲノムの存在を必要とする。パルボウイルスキャプシドは、完全粒子の成熟の一部としてタンパク質VP1、VP2およびVP3から構成される。
【0010】
本明細書で使用する用語「完全キャプシド」は、ウイルス核酸、好ましくは生存ウイルスを含有するパルボウイルスキャプシドを意味する。用語「中空キャプシド」は、包埋された核酸を含まないキャプシドを意味する。
【0011】
好ましい実施形態では、本発明の抗体は、天然H−1パルボウイルスキャプシドには結合するが、変性キャプシド、例えば界面活性剤もしくは熱を使用することによって変性したキャプシドには結合しない抗体(またはその抗原結合フラグメント)である。
【0012】
より好ましい実施形態では、本発明の抗体(またはその抗原結合フラグメント)は、中和抗体である。本明細書で使用する用語「中和抗体」は、パルボウイルスの生物学的活性、例えば複製および/または感染性を低減させる、または無効にする抗体を意味する。
【0013】
いっそうより好ましい実施形態では、本発明の抗体は、モノクローナル抗体である。H−1パルボウイルスに特異的に結合するモノクローナル抗体は、培養中の連続細胞系による抗体分子の産生を提供する任意の技術を使用して調製することができる。これらの技術には、ハイブリドーマ技術、ヒトB細胞ハイブリドーマ技術およびEBVハイブリドーマ技術が含まれる(Kohler et al., Nature 256 (1985), 495−7)。全キャプシド、VP1、VP2、VP3またはそれらのフラグメントを使用して哺乳動物、例えばマウス、ラット、ウサギ、モルモット、サルまたはヒトを免疫し、ポリクローナル抗体を産生することができる。所望であれば、免疫原は、担体タンパク質、例えばウシ血清アルブミン、サイログロブリンおよびキーホール リンペット ヘモシアニンへ抱合させることができる。宿主種に依存して、様々なアジュバントを使用すると、免疫学的応答を増加させることができる。当該アジュバントには、フロイントアジュバント、無機質ゲル(例えば、水酸化アルミニウム)および界面活性剤(例えば、リゾレシチン、プルロニック(Pluronic、登録商標)ポリオール類、ポリアニオン類、ペプチド類、油乳濁剤、キーホール リンペット ヘモシアニンおよびジニトロフェノール)が含まれる。特にヒトにおいて使用されるアジュバントのうち、BCG(カルメット−ゲラン桿菌(Bacilli Calmette−Guerin))およびコリネバクテリウム パルヴム(Corynebacterium parvum)が特に有用である。
【0014】
一本鎖抗体を生成するために記載された技術は、H−1パルボウイルスキャプシドに特異的に結合する一本鎖抗体を産生するために当該技術分野において公知の方法を使用して適合させることができる。関連する特異性を備えるが別個のイディオタイプ組成の抗体は、ランダムコンビナトリアル免疫グロブリンライブラリーからのチェイン シャフリングによって生成することができる(Burton, PNAS USA 88 (1991), 11120−3)。一本鎖抗体は、さらにまたDNA増幅法、例えばPCRを使用して、鋳型としてハイブリドーマcDNAを用いて構築することもできる(Thirion et al., Eur.J.Cancer Prev. 5 (1996), 507−11)。一本鎖抗体は、単一特異性または二重特異性であってよく、および二価または四価であってよい。四価の二重特異性一本鎖抗体の構築は、例えば、Coloma & Morrison, Nat.Biotechnol. 15 (1997), 159−63に教示されている。二価の二重特異性一本鎖抗体の構築は、Mallender & Voss, J.Biol.Chem. X no9 (1994), 199−206に教示されている。
【0015】
最も好ましい実施形態では、本発明のモノクローナル抗体は、ブダペスト条約を遵守して、2009年11月25日に番号DSM ACC3030を付してDSMZに寄託されているハイブリドーマ細胞系BL−H1によって生成される。
【0016】
本発明はさらに、ブダペスト条約を遵守して、2009年11月25日に番号DSM ACC3030を付してDSMZに寄託されているハイブリドーマ細胞系BL−H1によって生成される抗体と同一エピトープに結合するモノクローナル抗体またはその抗原結合フラグメントを提供する。
【0017】
本明細書で使用する用語「エピトープ」には、抗体に特異的に結合することができる任意のタンパク質決定基が含まれる。エピトープ決定基は、通常は分子の化学的活性表面基、例えばアミノ酸もしくは糖側鎖からなり、通常は特異的三次元構造特性ならびに特異的電荷特性を有する。本発明の抗体は、解離定数が≦1μMである、好ましくは≦μM 100nMである、および最も好ましくは≦10nMである場合に抗原に特異的に結合すると言われている。典型的には、1つのエピトープを形成するために、少なくとも6、8、10または12連続アミノ酸が必要とされる。しかし、非連続アミノ酸を包含するエピトープは、より多くの、例えば少なくとも15、25または50アミノ酸を必要とすることがある。
【0018】
本発明はさらに、完全または中空H−1パルボウイルスキャプシドへの結合について上記で特徴付けた本発明の抗体と競合する、モノクローナル抗体またはその抗原結合フラグメントも提供する。
【0019】
好ましくは、抗体またはその抗原結合フラグメントは、検出可能な標識を持っている。抗体/フラグメントは、直接的または間接的に検出可能に、例えば、放射性同位体、蛍光化合物、生物発光化合物、化学発光化合物、金属キレート剤または酵素を用いて標識することができる。当業者であれば、抗体に結合させるための他の適切な標識を認識でき、またはルーチンの実験を使用して当該の標識を確認することができる。
【0020】
本発明はさらに、細胞系、すなわち本発明による抗体またはその抗原結合フラグメントを産生する細胞系に関する。好ましくは、この細胞系は、哺乳動物細胞系である。発現のための宿主として利用可能な哺乳動物細胞系は、当該技術分野において周知であり、DSMZまたはアメリカン培養細胞系統保存機関(ATCC)から入手可能な不死化細胞系が含まれ、CHO細胞、NSO細胞、HeLa細胞、BHK細胞、COS細胞、Hep細胞および多数の他の細胞系が含まれる。細菌、酵母、昆虫および植物を含む非哺乳動物細胞もまた組換え抗体を発現させるために使用できる。発現方法は、どの系が最高発現レベルを生成するのか、および所望の結合特性を備える抗体を産生するのかを決定することによって選択される。
【0021】
当該細胞系によって産生された抗体は、当該技術分野において周知の方法によって精製することができる。例えば、抗体は、パルボウイルスキャプシドまたはパルボウイルスエンベロープタンパク質が結合しているカラムを通過させることによって親和性精製することができる。結合した抗体は、次に塩濃度の高い緩衝液を用いてカラムから溶出させることができる。
【0022】
最も好ましい細胞系は、ブダペスト条約を遵守して2009年11月25日に番号ACC3030を付してDSMZに寄託されたハイブリドーマ細胞系BL−H1である。
【0023】
本発明は、さらに本発明のモノクローナル抗体またはその抗原結合フラグメントをコードする核酸も提供する。本発明の抗体、例えば一本鎖抗体をコードする核酸を、手動もしくは自動ヌクレオチド合成を使用して構築し、標準組換えDNA法を用いて発現構築物にクローン化し、および細胞内へ導入して該コード配列を発現させることができる。または、抗体は、例えば繊維状ファージ テクノロジー(Verhaar et al., Int.J.Cancer 61 (1995), 497−501)を使用して直接的に産生することができる。
【0024】
本発明は、さらに本発明の抗体またはその抗原結合フラグメントを含む診断用組成物も提供する。当該組成物は、パルボウイルス感染症の診断に有用である可能性がある。さらに、当該組成物は、
(a)H−1パルボウイルス標本およびさらに感染した動物および患者(薬物動態学的ウイルス血症)における、例えばELISAによるH−1パルボウイルスの定量、
(b)細胞内でのH−1パルボウイルス集合および輸送の検出、
(c)実験動物、特にラットにおけるH−1パルボウイルスによる汚染の検出のために使用することができる。
【0025】
診断には、完全または中空H−1パルボウイルスキャプシドに特異的に結合する抗体を、免疫化学的測定法、例えばウェスタンブロット、ELISA、放射免疫測定法、免疫組織化学的測定法、免疫沈降法、または当該技術分野において公知の他の免疫化学的測定法において使用できる。競合的結合または免疫放射線測定法のための多数のプロトコールは、当該技術分野において周知である。当該免疫測定法は、典型的には免疫源と該免疫源に特異的に結合する抗体との間の錯体形成の測定を含んでいる。
【0026】
診断は、特定の免疫測定法には限定されず、同種および異種方法の両方を含むことが意図されている。実施可能な代表的な免疫測定法には、蛍光偏光免疫測定法(FPIA)、蛍光免疫測定法(FIA)、酵素免疫測定法(EIA)、比濁阻害免疫測定法(NIA)、酵素結合免疫吸着検定法(ELISA)および放射免疫測定法(RIA)が含まれる。指標成分、もしくは標識基は、対象抗体に付着させることができ、測定装置の利用可能性および適合する免疫測定法によって指示されることが多い方法の様々な使用のニーズを満たすよう選択される。上記に記載した様々な免疫測定法を実施する際に使用可能な一般的技術は、当業者には公知である。
【0027】
本発明は、さらに本発明の抗体またはその抗原結合フラグメントおよび薬学的に許容される担体を含む医薬組成物も提供する。抗体またはその抗原結合フラグメントは、例えばH−1パルボウイルス感染症またはそれに関連する疾患を治療するための、該抗体がウイルスを中和する能力(antidot)を使用する方法において使用することができる。
【0028】
療法には、抗体またはその抗原結合フラグメントは、有効量で、薬学的に許容される担体と組み合わされて存在する。「薬学的に許容される」は、有効成分の生物学的活性の有効性を妨害しない、およびそれが投与される患者にとって毒性ではない任意の担体を含むことが意図されている。適切な医薬担体の例は当該技術分野において周知であり、リン酸緩衝生理食塩水、水、乳濁液、例えば水中油型乳濁液、様々なタイプの湿潤剤、無菌溶液などが含まれる。当該担体は、従来の方法によって調製することができる。
【0029】
本発明による抗体またはその抗原結合フラグメントを、被験者に有効量で投与することができる。「有効量」は、感染症/疾患の経過および重症度に影響を及ぼし、当該病状の低減または緩解をもたらすために十分な抗体またはその抗原結合フラグメントの量を意味する。これらの感染症、疾患または障害を治療および/または予防するために有用な「有効量」は、当業者には公知の方法を使用して決定することができる(例えば、Fingl et al., The Pharmocological Basis of Therapeutics, Goodman and Gilman, eds. Macmillan Publishing Co., New York, pp.1−46(1975)を参照されたい)。
【0030】
最後に、本発明は、さらにH−1パルボウイルス感染症または当該感染症と関連する疾患の診断または療法に有用な本発明による抗体またはその抗原結合フラグメントを含むキットを提供する。好ましくは、本キットは、1つ以上の容器、例えばバイアル、試験管などを、その中にきっちりと密封して収容するために区分化されている輸送手段を含み、該容器手段の各々は該分析において使用される個別要素の1つを含んでいる。例えば、容器手段の1つは、検出可能に標識される、または検出可能に標識することができる本発明による抗体またはその抗原結合フラグメントを含むことができる。本キットは、さらに緩衝液を含有する容器および/またはレポーター分子(例えば、酵素もしくは蛍光標識)に結合したレポーター手段(例えば、ビオチン結合タンパク質、例えばアビジンもしくはストレプトアビジン)を含む容器も有することができる。
【図面の簡単な説明】
【0031】
【図1】図1は、H−1パルボウイルスの免疫沈降反応を示す。 レーン1:BL−H1モノクローナル抗体を用いて免疫沈降させられたH−1パルボウイルス。レーン2:BL−H1モノクローナル抗体を伴わない、免疫沈降反応が生じないH−1パルボウイルスを意味する陰性コントロール。レーン3:陽性コントロール:免疫沈降反応方法を用いないSDS−ゲルおよびウェスタンブロット上のH−1パルボウイルス単独。レーン4:タンパク質マーカー。 PBS:リン酸緩衝生理食塩水。mAb:BL−H1。aVP:変性H−1パルボウイルスのウイルスタンパク質を認識するポリクローナル抗体。
【図2】図2は、ウェスタンドットブロットを示す。 ウェスタンドットブロットは、天然および変性H−1パルボウイルスを用いて生成された。BL−H1:H−1パルボウイルスに対するモノクローナル抗体。aVP:変性ウイルスタンパク質を認識するポリクローナル抗体(ドデシル硫酸ナトリウムポリアクリルアミドおよびジチオトレイトールを用いる)。天然:未処理H−1パルボウイルス。陰性コントロール:H−1パルボウイルスを含まない。
【図3】図3は、野生型H−1の分布およびBL−H1モノクローナル抗体を用いた検出を示す。 モノクローナル抗体BL−H1がキャプシド、モノ/オリゴマーキャプシドタンパク質または変性キャプシドタンパク質と反応するか決定するため、細胞抽出物は、pUC 19ΔHindIIIでのトランスフェクションの24時間後に、293T HEK細胞から調製した。細胞抽出物は、直線スクロース勾配上で分画させる。 BL−H1:H−1パルボウイルスに対するモノクローナル抗体。aVP:変性H−1パルボウイルスのウイルスタンパク質を認識するポリクローナル抗体。プラスミドpUC19ΔHindIIIは、完全H−1パルボウイルスゲノムを含有する。Vg:リアルタイムPCRによって分析されたウイルスゲノム含有粒子。IU:感染単位。HA:完全または中空H−1パルボウイルスの赤血球凝集反応。
【図4】図4は、mab BL−H1の免疫蛍光試験(精製7および21日後のBL−H1活性の安定性)を示す。 精製後、BL−H1抗体を、各々室温、4℃および−20℃で保存する。活性は、感染したNB−324K細胞上でIF(免疫蛍光)によって21日後に試験する。陽性コントロールは、ポリクローナル抗体C8B10である。分析は、顕微鏡Axioskop 2 plusおよびカメラAxioCamMRc(倍率:40倍)によって実施する。
【図5】図5は、H−1パルボウイルスに感染したNB−324K細胞の中和を示す。 H−1パルボウイルスの中和は、野生型および組換えH−1パルボウイルスの何れかでの1のMOIで、550nm(吸光度550nm)で測定するMTT測定法によって示されている。0ng〜5000ngのB1−H1濃度を試験すると、3E5〜3E7のH−1パルボウイルス/μg(BL−H1)に対応した。
【図6】図6は、mab BL−H1を用いた野生型H−1 PVの中和を示す。 H−1パルボウイルスの中和は、1および10のMOIでのNB−324K細胞上でのプラークアッセイによって示されている。3E4または1E5のH−1パルボウイルスはいずれも1μgのBL−H1で中和された。 NC=陰性コントロール。PC=陽性コントロール。
【図7】図7は、組換えH−1 EGFP(緑色蛍光タンパク質)を示す。 1および10のMOIで緑色蛍光タンパク質(GFP)を含有する組換えH−1パルボウイルスは、感染したNB−324K細胞である0ng〜5000ngのB1−H1濃度で中和された。GFPは、40倍の倍率で分析する。 NB−324K細胞上でのBL−H1を用いた感染阻害作用は、一晩(o/n)のB1−H1プレインキュベーションおよびその後のH−1パルボウイルスによる感染によって所見されない。
【図8】図8は、H−1 PV検出のためのELISAの開発を示す。 H−1パルボウイルスキャプシドは、標準曲線を評価するために分光光度計において450nmで測定する。現在もなお、3種の異なる濃度(0.75μg/ml、0.5μg/mlおよび0.1μg/mlの二次抗体)を使用する。
【0032】
以下、実施例により本発明を具体的に例示する。
【実施例1】
【0033】
一般的方法
(A)H−1パルボウイルスによるBalb/cマウスの免疫
マウスを以下のスケジュールに従って免疫した。
(1)注射1週間前
麻酔後、眼窩後穿刺により15μlの血液を採取した(陰性コントロール)。
【0034】
(2)免疫
麻酔:PBS中の100μlの0.2%塩酸キシラジン(Rompun(登録商標)、Bayer社)およびPBS中の体重10g当たり各10μgの塩酸ケタミン(Ketavalt、Parke−Davis社)の腹腔内[i.p.]注射。
【0035】
(3)1.H−1パルボウイルスの注射
40%のビジパーク(Visipaque、登録商標)−リンガー液中の100μlウイルス(=7×1010ウイルスゲノム含有粒子[Vg]=1.2×108PFU(プラーク形成単位))、
+50μlのPBS、
+150μlの不完全アジュバント、
を混合し、4つの相違する位置に各60μlを皮下[s.c.]注射。
【0036】
(4)2.初回注射の1カ月後のH−1パルボウイルスの注射
1.注射と同様の手順。
【0037】
(5)3. 初回注射の2カ月後のH−1パルボウイルスの注射
追加免疫:脾臓を摘出する1週間前
100μlのウイルス(7×1010Vg=1.2×108PFU)
+200μlのPBSの腹腔内[i.p.]注射。
【0038】
(B)ハイブリドーマの生成
ハイブリドーマは、以下のスケジュールに従って生成した。
【0039】
培地:
作業培地:RPMI 1640(Gibco社番号R8758)。
ハイブリドーマ培地:500mlのRPMI 1640(Gibco社番号R8758)
+5ml L−グルタミン200mM(Gibco社)。
+5ml Pen/Strep(ペニシリン10000U/ml、ストレプトマイシン10mg/ml(Gibco社))
+10mlの1M Hepes(pH7.2)(Sigma社)
+50mlのFCS(融合試験済み、30分間56℃で熱不活性化)。
選択培地:ハイブリドーマ培地+10mlのHAT50倍濃縮液(Gibco社)。
BM Condi−Med(選択用)10%。
【0040】
融合試薬:
PEG 1500(Roche社番号783641)。
【0041】
細胞:
Ag8/X63(B細胞腫瘍細胞)。
細胞は、融合の少なくとも1週間前に解凍し、ハイブリドーマ培地中で培養し、融合1日前に分割させた。細胞は、増殖期にあるはずである。
【0042】
融合:
調製:融合の1〜2日前
融合の1日前:Ag8細胞を分割させる(計18、増殖期にあるはずである)。
【0043】
融合日:
全培地および溶液を37℃に加温する。
【0044】
Ag8細胞の調製:
Ag8細胞を貯留し、7分間264×gで遠心し、30〜40mlの作業培地中に入れ、細胞を計数する(約1〜2×108細胞)。
【0045】
脾臓の調製:
麻酔後、マウスを致死させ、殺菌するために70%エタノール中に入れ、無菌ベースにピンで固定し、無菌ピンセットおよび剪刀を用いて脾臓を取り出し、それをシャーレ内の作業培地中へ入れ、心臓を穿刺して血液を採取し、1.5ml試験管中に収集する。リンパ節に注意を払う。
【0046】
脾臓細胞:
脾臓は剪刀を用いて数回切断し、無菌対物スライドを用いて脾臓を破砕し(粉砕)、脾嚢を取り出して作業培地を用いて洗浄し、脾嚢を廃棄し、脾細胞をパスツールピペットを用いてホモジナイズし、細胞懸濁液を50ml試験管内に収集し、嚢組織をセットし、上清を新しい50ml試験管内へ入れ、細胞を264×gで7分間遠心し、細胞を作業培地で1回洗浄し、細胞を264×gで7分間遠心し、ペレットを2〜5mlの作業培地中へ取り出し、細胞を計数する(5×107〜3×108細胞でなければならない)。
【0047】
脾細胞とAg8細胞との融合:
脾細胞:Ag8細胞=3:1を50ml試験管中で混合し、混合した細胞を264×gで7分間遠心する。各細胞ペレットは総計1×108細胞以下でなければならない。上清を廃棄し、試験管の底部を静かにタッピングすることによってペレットを崩壊させ、連続的に揺らしながら1分間にわたり1mlのピペットを使用して1mlのPEG 1500をペレットに加える。さらに2分間揺らし続け、3分間連続的に揺らすことによって慎重に10mlの選択培地を加える。さらに10mlの選択培地を加え、混合した細胞を120×gで7分間遠心し、上清を廃棄し、ペレットは試験管の底部を静かにタッピングすることによって崩壊させ、再び洗浄し(さらに10mlの選択培地を加え、混合した細胞を120×gで7分間遠心し、ペレットは試験管の底部を静かにタッピングすることによって崩壊させる)、100μlの選択培地を備える96ウエルプレートを調製する(各ウエルは細胞数2×105個/ウエル(=2×106個/ml、2×107個/10ml))。
【0048】
Ag8コントロール:
選択培地中に1E4細胞/ウエルを播種する(およそ24ウエル以下)。
【0049】
血液:
30分後(赤血球凝集反応)、遠心し(5000×g、10分間)、新しい試験管中に血清を採取する(陽性コントロール)。
【0050】
観察:
2〜7日後:HAT選択培地中のコントロールAg8細胞(死滅していなければならない)。
1週間後:融合した細胞を供給し、1ウエル当たり100μlを取り出し、廃棄し、100μlの新鮮HAT選択培地を供給する。
2週間後:スクリーニング(10%のBM CondiMedを備える培地)、免疫蛍光法、ウェスタンドットブロットを開始しなければならない。
【0051】
選択:
陽性スクリーニングのウエルを選択しなければならない。
1.選択:POSウエルを平板培養しなければならない。各96ウエルプレート、5細胞/ウエル、1細胞/ウエル。
2.選択:POSウエルを平板培養しなければならない。各96ウエルプレート、5細胞/ウエル、1細胞/ウエル。
3.選択:POSウエルを平板培養しなければならない。各96ウエルプレート、5細胞/ウエル、1細胞/ウエル。
【0052】
(C)H−1野生型キャプシドに対するモノクローナル抗体の産生
材料:
75cm2または150cm2の無菌フィルターを備えるGreiner Bio−one T−フラスコ。10%のFCS、1%のL−グルタミン(2.5mM)、1%のP/S、20mMのHEPESを備えるRPMI培地。BL−H1−ハイブリドーマ細胞。ピペット(5、10、20ml)。試験管(15本、50ml)。1lのボトル。水浴。そして遠心機。
【0053】
情報:
BL−H1の生成時間:約27時間。
懸濁細胞。
培地の交換:細胞を含む培地を遠心する:300×g、10分間。
B1−H1の採取:遠心:5000×g、5分間。
BL−H1細胞増殖は、グルコース濃度が高いほど良好である。
RPMIは2g/lのグルコースを含有し、3または4g/lへの増加は抗体産生を改良することができよう。
2×106個/mlまでの細胞密度が可能である。
【0054】
産生:
75cm2のフラスコには10ml、150cm2のフラスコには20mlの細胞懸濁液を入れる。
RPMI培地中でのBL−H1細胞の培養。開始時の細胞密度:2×105個/mlが理想的である。
細胞数1×105個/mlも可能であるが、細胞増殖は開始時にはより緩徐である。細胞数3×105個/mlも可能であるが、その場合、細胞を2または3日後に分割しなければならず、さもなければ生命力が低下する。
細胞を3日後(任意で4日後)に分割させる。
新鮮培地を備える15〜20%の馴化培地は、培地交換後、細胞生存性を増加させる。
【0055】
(D)親和性クロマトグラフィーを用いたモノクローナル抗体BL−H1の精製
材料:
AKTA Prime装置(GE Healthcare Europe社、フライブルク)。70%エタノール(H2O中)。20%エタノール(H2O中)。20mMリン酸ナトリウム緩衝液(pH7.0)(平衡バッファー=バッファーA)。1M Tris−HCl(pH9.0)(中和バッファー)。50ml試験管。ポリスチレン製丸底試験管。0.1Mクエン酸(pH4.0)(溶出バッファー=バッファーB)。HiTrap(商標)Protein A HP 5mlカラム。
【0056】
全緩衝液は、ボトルトップ フィルターを用いて濾過しなければならず、室温で10分間超音波を用いて100%脱気しなければならない(480W)。
【0057】
手順:
AKTA primeシステムの使用:最初に本システムを20%エタノールで洗浄する。試験管Aおよび試験管Bを20%エタノール中に入れ、「Run stored Method(保存されているメソッドを実行する)」を押し、プログラム1を選択する。その後、試験管にH2Oを入れ、再びプログラム1を選択する。ここで各試験管に平衡バッファーを満たし、全試験管を接続する。カラムを20%エタノール中に保管し、平衡バッファーを用いて直ちに洗浄しなければならない。プレッシャーリミットは、0.3MPaである。プロテインAカラムについての100%バッファーA(=0%バッファーB)の流速は、5ml/分である。代替のプロテインG:1ml/分(直列カラムの量とは無関係)。
【0058】
クロマトグラフィーは、メインメニューの「Manual Run(マニュアルで操作する)」で実行しなければならない。洗浄および装填のために、以下の条件を使用する。
メソッド ベースを設定する(ml)。
濃度%Bを設定する(0%)。
グラジエントを設定する(オフ)。
流速を設定する(A:1ml/分、B:5ml/分)。
フラクション ベースを設定する(ml)。
フラクション サイズを設定する(0)。
プレッシャーリミットを設定する(0.3MPa)。
バッファーバルブ ポジションを設定する(Pos 1)。
インジェクションバルブ ポジションを設定する(load)。
実行を開始する。
【0059】
クロマトグラフィーは、メインメニューの「Manual Run(マニュアル操作で作動する)」で実行しなければならない。カラムは、UVベースライン値が持続する限り、緩衝液を用いて洗浄しなければならない。カラムに充填する前に、マニュアル操作での作動で「auto zero(オートゼロ)」に入るが、これはUVベースライン値をゼロにリセットする。試験管Aを通して「抗体培地」を負荷する。このため、試験管Aを「抗体培地」内に入れ「break(中断する)」で保持する。さもないとシステム内に気泡が流入する。その後「continue(再開する)」を押す。最後に「break(中断する)」を押し、試験管Aに戻し、洗浄のために「continue(再開する)」を押す。UVベースライン値がほぼゼロである限り洗浄する。装填および洗浄中の素通り画分は追加ボトル内に収集しなければならない(カラムが過負荷になっている場合:これはUVシグナルの追加の増加を通して見ることができよう)。そこで、素通り画分は、二度目に使用することができよう。
【0060】
100%バッファーBへ目を向けると、他の全てのパラメーターは同一である。
メソッドベースを設定する(ml)。
濃度%Bを設定する(100%)。
グラジエントを設定する(オフ)。
流速を設定する(A:1ml/分、B:5ml/分)。
フラクション ベースを設定する(ml)。
フラクション サイズを設定する(0)。
プレッシャーリミットを設定する(0.3MPa)。
バッファーバルブ ポジションを設定する(Pos 1)。
インジェクションバルブ ポジションを設定する(load)。
実行を開始する。
【0061】
バッファーBがカラムに達すると、抗体の溶出が直ちに始まる。UVシグナルが強度に増加する。画分は、フラクションコレクターチューブを通して収集できよう。異なる抗体濃度のために、少なくとも2つの画分を収集することが推奨されている。
【0062】
画分を手作業で収集するため、「Manual Run(マニュアル操作で作動する)」の「set event mark(イベントマークを設定する)」を設定して開始時および終了時を見ることができよう。溶出された画分は、中和バッファーを用いて直ちにpH1にしなければならない。第1画分は今も少量のバッファーAを含有している可能性があるので、前もって中和バッファーの量を知ることは容易でない。不安定性を回避するために極めて迅速にpH紙を用いてチェックしなければならない。100%Puffer Bについての推奨値。
【0063】
【表1】
【0064】
ピークがベースライン値になった後、バッファーAへ切換える。
メソッド ベースを設定する(ml)。
濃度%Bを設定する(0%)。
グラジエントを設定する(オフ)。
流速を設定する(A:1ml/分、B:5ml/分)。
フラクション ベースを設定する(ml)。
フラクション サイズを設定する(0)。
プレッシャーリミットを設定する(0.3MPa)。
バッファーバルブ ポジションを設定する(Pos 1)。
インジェクションバルブ ポジションを設定する(load)。
実行を開始する。
【0065】
カラムおよびシステムの保管は20%エタノール中で行わなければならない。システム全体を洗浄しなければならない[プロトコールP−19−2Aを参照されたい]。
【0066】
(E)H1−キャプシド−ELISA
材料:
・フレキシブルプレート、96ウエル、U−ボトム、蓋なし、BD Falcon社。マイクロプレート用蓋、Greiner BioOne社。ウシ乳汁由来カゼイン、Sigma社。PBS+0.05%のTween(登録商標)。PBS(H2O中)。コーティング用:BL−H1画分E1、Tris−HClで中和した0.1Mグリシン緩衝液中の2mg/mlのIgG2A BL−H1(10mg/mlの全タンパク質、IgG2A+異種(foreighn)タンパク質)、4℃で保管。検出用:リン酸ナトリウム緩衝液中のBL−H1−HRP、およそ1mg/ml、−20℃で冷凍、試験管1本当たり10μl。標準物質:40%のビジパーク(商標)中のHV Pool green 1−2/1−3/3−3/4−2/4−3/5−2/6−2/7−2 1.5×1011Vg/ml、7.5×1011キャプシド/ml、4℃で保管。陽性コントロール:40%のビジパーク(商標)中のP#30/07β、9.1×1011Vg/ml(推定〜1×1013キャプシド/ml)、4℃で保管。光学接着カバー(Optical Adhesive Cover)(Applied Biosystems社)。TMB、ELISAのための超緩徐、液体物質システム、Sigma社、4℃で保管。停止液:室温で保管した1N H2SO4。Multiskan EXマイクロタイタープレート光度計、Thermo Scientific社。
【0067】
緩衝液:
洗浄バッファー:PBSに0.05%のTween(登録商標)を加える(ウェスタン(ドット)ブロット用のストック液を参照されたい)。
コーティング液:使用直前に調製する。E1をPBS(H2O中)中で2.5μg/mlへ希釈する、これは1:800を意味する。1プレート当たり10mlの希釈液。
ブロッキングバッファー(BB):PBS中の2mg/mlカゼイン+0.05%のTween(登録商標)。常に新しく調製する。
400mlのPBS+0.05%のTween(登録商標)を〜50℃に(マイクロウェーブで40〜50秒間)加熱し、それをビーカー内へ注入し、必要量のカゼインを加える。30分間加熱しながら、全てが溶解するまでおよそ50℃で1時間まで攪拌する。使用のためには、水浴中で37℃へ冷却する。1プレート当たり100mlで十分である。
検出:使用直前に調製する。BL−H1−HRP(二次抗体)をブロッキングバッファー中で0.1g/mlへ希釈する、これは1:10,000を意味する。1プレート当たり10mlの希釈液。
標準物質:HV pool greenを2.5×109〜3.91×107キャプシド/ウエルの標準物質へ希釈する。2つの標準物質列については、600μlの2.50×109(douplex)キャプシド/ウエル標準物質をストック標準物質からブロッキングバッファー中での1:30(2.5×1010キャプシド/ml)希釈によって調製する。300μlのブロッキングバッファーを6本の1.5ml試験管へ加え、1.25E+09、6.25E+08、3.13E0+8、1.56E+08、7.81E+07、3.91E+07キャプシド/ウエルと表示する。300μlの各標準物質を次の試験管に加え、各移行間にボルテックスミキサーにかけて連続希釈を実施する。PBSは、ブランクとして機能する(ブロッキングバッファーも使用することができる)。
陽性コントロール:BB中1:700に希釈すると、およそ4.2×109キャプシド/ウエル、各々最初は2.9×1012キャプシド/ウエル。1プレートに付きコントロールを含む少なくとも4ウエルを実施する。P#30/07βについての陰性コントロールは、40%のビジパーク(商標)をBB中1:700に希釈する。
【0068】
実行:
第0日
午後:フレキシブルプレートを100μl/ウエルのコーティング液で一晩4℃にてコーティングする。マイクロプレート用の蓋を用いてプレートを閉じる。この作業は、ウエルの液体を蓋に押し出さないように慎重に実施する。
【0069】
第1日
または、プレートを午前中、室温で2時間インキュベートすることでコーティングすることができる。
1.ブロッキングバッファーを上述したように調製する。コーティング液は、残っているコーティング液を全て除去するために吸収紙上で反転させて吸い取ることによってプレートから除去する。200μl/ウエルのブロッキングバッファーを加え、プレートを蓋で閉じ、37℃で1時間インキュベートする。
2.プレートのブロッキング中、上述したように標準物質および陽性コントロールを調製する。サンプル希釈のためにVg/ml力価によってキャプシド数/mlを推定する。より広い範囲をカバーするためにほぼ推定値の数種の希釈液を作製する。バッファーを用いてサンプルマトリックスの陰性コントロールを調製する(サンプルの希釈に注意を払う!)。
3.ウエルを吸引し、200μl/ウエルの洗浄バッファーを用いて2回洗浄する。最終洗浄後、プレートを反転させて、残っている緩衝液を全て取り除くために吸水性のペーパータオルで吸い取る。
4.ここから、プレートを用いる全ての工程は無菌フード下で行う。100μlの標準物質、陽性コントロール、各陰性コントロールまたはサンプルを各ウエルに加える。プレートを蓋で閉じる。37℃で1時間インキュベートする。
5.陽性コントロール、各陰性コントロールまたはサンプルを、異なるサンプル毎にチップを交換しながらピペットで取り除く。200μl/ウエルの洗浄バッファーを用いて3回洗浄する。最終洗浄後、プレートの表面をペーパータオルで乾燥させる。廃棄物をビーカー内に収集する。
6.100μl/ウエルの希釈BL−H1−HRPを各ウエルに加える。プレートを蓋で閉じ、37℃で1時間インキュベートする。
7.検出抗体を12チャンネルピペットを用いて取り出す。200μl/ウエルの洗浄バッファーを用いて4回洗浄する。最終洗浄後、プレートの表面をペーパータオルで乾燥させる。できる限り多くの液体を取り除く努力をする。廃棄物を無菌フード下でビーカー内に収集する(12チャンネルピペットを使用することができる)。
8.依然として無菌フード下で、100μl/ウエルのTMB溶液を各ウエルに加え、室温の暗所でおよそ15分間インキュベートする。およそ10分後に反応強度が申し分ないかチェックする。
9.100μl/ウエルの停止液を各ウエルに加えることによって反応を停止させる。プレートを光学接着カバーで密封する。
10.反応を停止させるため30分以内に450nmで吸光度を読み取る。Multiskan EXマイクロタイタープレート光度計を使用する。結果の選択的補正のために、プレートを550nmおよび595nmで追加して読み取る。
【0070】
追加の勧告:
ブロッキングは、一晩より長い時間実施してはならない。インキュベーション時間が長くなると凝集体の形成などが生じる。ウイルスおよび抗体の全希釈液は、使用直前に調製しなければならない。ウエルは、結合した抗体の消失を回避するため慎重に洗浄しなければならない。さらに、各列には同一時間工程でTMBを加えることが重要である。反応を停止させるため、2つの列間の変動を最小限に抑えるため、同一時間工程を使用しなければならない。残っている液体をピペットで取り除く場合は、結合した反応物質の消失を回避するために、ウエルの底部に極めて軽く触れるだけでこすってはならない。37℃でのプレートのインキュベーションは、ポリスチレンが弱熱導体であるため、周辺効果を誘発することがある。このため、分析の実施中にはブロッキングバッファーを37℃に維持することが重要である。これにより、周辺効果を下方レベルへ抑えることができる。標識された二次抗体は、使用後に再冷凍してはならない。再冷凍は、シグナルの低強度を引き起こすことがある。カゼインを含有するブロッキングバッファーは、−20℃で冷凍すれば少なくとも4週間保管することができる。この希釈手順は、間違えると結果が不正確になるので、慎重に実施しなければならない。多数回の希釈を次々に実施しなければ、極めて正確に実施することができる。
【実施例2】
【0071】
H−1キャプシドに対するBL−H1モノクローナル抗体の特性付け
BL−H1抗体がH−1パルボウイルスを免疫沈降させる能力を評価するため、抗体およびパルボウイルスを一緒に37℃で一晩インキュベートした。免疫複合体をプロテインAセファロースによって沈降させ、ウェスタンブロッティングによって変性ウイルスタンパク質に対する抗体を使用して分析した。これは、BL−H1抗体が集合H−1パルボウイルスキャプシドを認識することを証明している(図1)。
【0072】
この同定を特性付けるために、天然または変性H−1パルボウイルス何れかをニトロセルロース膜へ移した(図2)。BL−H1抗体は天然粒子しか認識しないが、ポリクローナル抗体は変性キャプシドを認識する。
【0073】
BL−H1抗体がキャプシドと、または天然ウイルスタンパク質とも反応するか決定するために、キャプシドタンパク質発現プラスミドでトランスフェクトした293T細胞の抽出物を調製し、直線スクロース勾配上で分画させた(図3)。BL−H1は、中空および完全H−1パルボウイルス粒子に対応する画分8〜18の間でピークを示す。完全または中空キャプシドは、赤血球凝集を介して実証される(画分8〜17)。完全キャプシドは、ウイルスゲノム含有粒子(画分10〜17)および感染単位を介した活性粒子(画分12〜18)によって決定される。これとは対照的に、非集合ウイルスタンパク質は、ウイルスタンパク質に対する抗体での画分3〜14において検出された。最後に、BL−H1と非変性ならびに変性キャプシドタンパク質との間に反応性は見られない。BL−H1は、集合H−1パルボウイルスキャプシド上に存在する立体構造エピトープを特異的に認識する。全てまとめると、これは、BL−H1が集合粒子によってのみ表示される構造的エピトープを認識することを証明している。
【0074】
図4は、感染したNB−324K細胞におけるH−1産生の免疫蛍光法によるBL−H1を用いた検出を示している。BL−H1抗体は、室温、4℃および−20℃で保管した場合、少なくとも3週間安定性である。
【0075】
図5、6および7では、H−1パルボウイルスのBL−H1中和能力が証明されている。
【0076】
MTT測定法は、H−1パルボウイルスとBL−H1抗体との正しい比率を見つけるために実施した。1E5までのH−1パルボウイルスを中和するには、BL−H1は1μgで十分である(図5)。
【0077】
プラークアッセイによって決定すると、1E5 H−1パルボウイルス当たり1μgのBL−H1を用いて95%の中和を達成することができた(図6)。この中和能力は、さらに緑色蛍光タンパク質を発現する組み換えH−1パルボウイルスを用いても証明されている(図7、上方パネル)。BL−H1自体が細胞レポーターと相互作用することによって間接的に作用するのではないことを証明するため、NB−324K細胞をBL−H1抗体とともに一晩プレインキュベートし、その後H−1パルボウイルスにより感染させた(図7、下方パネル)。
【実施例3】
【0078】
H−1−キャプシドのELISA
BL−H1を用いたH−1パルボウイルスの定量をELISAによって評価した。最初の結果は、キャプシドとウイルスゲノム含有粒子との比率が約5であることを示している(図8、下方部分)。使用した二次抗体の3種の濃度(0.1μg/ml〜0.75μg/ml)は、回収率における有意差を示さなかった(図8、上方部分)。
【特許請求の範囲】
【請求項1】
完全または中空H−1パルボウイルスキャプシドに特異的に結合する抗体またはその抗原結合フラグメントであって、天然H−1パルボウイルスキャプシドにのみ結合して単離キャプシドタンパク質には結合しない抗体またはその抗原結合フラグメント。
【請求項2】
中和抗体である請求項1に記載の抗体またはその抗原結合フラグメント。
【請求項3】
モノクローナル抗体である請求項1または2に記載の抗体。
【請求項4】
DSMZにおいて番号ACC3030で寄託されたハイブリドーマ細胞系BL−H1によって産生される請求項3に記載のモノクローナル抗体。
【請求項5】
請求項1〜4の何れか一項に記載の前記抗体と同一エピトープに結合する請求項1に記載のモノクローナル抗体またはその抗原結合フラグメント。
【請求項6】
完全または中空H−1パルボウイルスキャプシドへの結合について請求項1〜4の何れか一項に記載の前記抗体と競合する請求項1に記載のモノクローナル抗体またはその抗原結合フラグメント。
【請求項7】
検出可能な標識を有する請求項1〜6の何れか一項に記載の抗体またはその抗原結合フラグメント。
【請求項8】
請求項1〜7の何れか一項に記載の抗体またはその抗原結合フラグメントを産生する細胞系。
【請求項9】
DSMZにおいて番号ACC3030で寄託されたハイブリドーマ細胞系BL−H1である請求項8に記載の細胞系。
【請求項10】
請求項3〜6の何れか一項に記載のモノクローナル抗体またはその抗原結合フラグメントをコードする核酸。
【請求項11】
請求項1〜7の何れか一項に記載の抗体またはその抗原結合フラグメントを含む診断用組成物。
【請求項12】
パルボウイルス感染症を診断するための請求項1〜7の何れか一項に記載の抗体またはその抗原結合フラグメントの使用。
【請求項13】
請求項1〜7の何れか一項に記載の抗体またはその抗原結合フラグメントおよび薬学的に許容される担体を含む医薬組成物。
【請求項14】
H−1パルボウイルス感染症または前記感染症によって誘発される疾患を治療するための方法において使用するための請求項1〜7の何れか一項に記載の抗体またはその抗原結合フラグメント。
【請求項15】
パルボウイルス感染症または前記感染症によって誘発される疾患を治療するための医薬組成物を調製するための請求項1〜7の何れか一項に記載の抗体またはその抗原結合フラグメントの使用。
【請求項16】
請求項1〜7の何れか一項に記載の抗体またはその抗原結合フラグメントを含むH−1パルボウイルス感染症または前記感染症によって誘発される疾患の診断または治療のために有用なキット。
【請求項17】
少なくとも1つの緩衝液をさらに含む請求項16に記載のキット。
【請求項1】
完全または中空H−1パルボウイルスキャプシドに特異的に結合する抗体またはその抗原結合フラグメントであって、天然H−1パルボウイルスキャプシドにのみ結合して単離キャプシドタンパク質には結合しない抗体またはその抗原結合フラグメント。
【請求項2】
中和抗体である請求項1に記載の抗体またはその抗原結合フラグメント。
【請求項3】
モノクローナル抗体である請求項1または2に記載の抗体。
【請求項4】
DSMZにおいて番号ACC3030で寄託されたハイブリドーマ細胞系BL−H1によって産生される請求項3に記載のモノクローナル抗体。
【請求項5】
請求項1〜4の何れか一項に記載の前記抗体と同一エピトープに結合する請求項1に記載のモノクローナル抗体またはその抗原結合フラグメント。
【請求項6】
完全または中空H−1パルボウイルスキャプシドへの結合について請求項1〜4の何れか一項に記載の前記抗体と競合する請求項1に記載のモノクローナル抗体またはその抗原結合フラグメント。
【請求項7】
検出可能な標識を有する請求項1〜6の何れか一項に記載の抗体またはその抗原結合フラグメント。
【請求項8】
請求項1〜7の何れか一項に記載の抗体またはその抗原結合フラグメントを産生する細胞系。
【請求項9】
DSMZにおいて番号ACC3030で寄託されたハイブリドーマ細胞系BL−H1である請求項8に記載の細胞系。
【請求項10】
請求項3〜6の何れか一項に記載のモノクローナル抗体またはその抗原結合フラグメントをコードする核酸。
【請求項11】
請求項1〜7の何れか一項に記載の抗体またはその抗原結合フラグメントを含む診断用組成物。
【請求項12】
パルボウイルス感染症を診断するための請求項1〜7の何れか一項に記載の抗体またはその抗原結合フラグメントの使用。
【請求項13】
請求項1〜7の何れか一項に記載の抗体またはその抗原結合フラグメントおよび薬学的に許容される担体を含む医薬組成物。
【請求項14】
H−1パルボウイルス感染症または前記感染症によって誘発される疾患を治療するための方法において使用するための請求項1〜7の何れか一項に記載の抗体またはその抗原結合フラグメント。
【請求項15】
パルボウイルス感染症または前記感染症によって誘発される疾患を治療するための医薬組成物を調製するための請求項1〜7の何れか一項に記載の抗体またはその抗原結合フラグメントの使用。
【請求項16】
請求項1〜7の何れか一項に記載の抗体またはその抗原結合フラグメントを含むH−1パルボウイルス感染症または前記感染症によって誘発される疾患の診断または治療のために有用なキット。
【請求項17】
少なくとも1つの緩衝液をさらに含む請求項16に記載のキット。
【図1】

【図2】

【図3】
【図4】
【図5】

【図6】

【図7】

【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2013−513358(P2013−513358A)
【公表日】平成25年4月22日(2013.4.22)
【国際特許分類】
【出願番号】特願2012−542386(P2012−542386)
【出願日】平成22年12月1日(2010.12.1)
【国際出願番号】PCT/EP2010/007301
【国際公開番号】WO2011/069614
【国際公開日】平成23年6月16日(2011.6.16)
【出願人】(512151403)
【氏名又は名称原語表記】DEUTSCHES KREBSFORSCHUNGSZENTRUM
【Fターム(参考)】
【公表日】平成25年4月22日(2013.4.22)
【国際特許分類】
【出願日】平成22年12月1日(2010.12.1)
【国際出願番号】PCT/EP2010/007301
【国際公開番号】WO2011/069614
【国際公開日】平成23年6月16日(2011.6.16)
【出願人】(512151403)
【氏名又は名称原語表記】DEUTSCHES KREBSFORSCHUNGSZENTRUM
【Fターム(参考)】
[ Back to top ]