
HDAC阻害剤としてのFK228誘導体
一般式(I)または(I’)(式中、R1、R2、R3およびR4は、同じであるか異なり、アミノ酸側鎖部分を表し、ならびに各R6は、同じであるか異なり、水素またはC1−C4アルキルを表す)のFK228類似体である化合物、それらのアイソスターおよびそれらの医薬的に許容される塩は、HDACを阻害することが判明される。
【化40】

【化40】
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、HDACの阻害剤として作用する特定のデプシペプチドに関する。
【背景技術】
【0002】
ヒストンデアセチラーゼ(HDAC)は、アセチル化リシン残基の加水分解を触媒する亜鉛金属酵素である。ヒストンにおいて、これはリシンをそれらの正常なプロトン化状態に戻し、またこれはヌクレオソームにおいて緊密なDNAパッケージングを生じさせる真核性転写制御の包括的メカニズムである。加えて、可逆的リシンアセチル化は、非ヒストンタンパク質にとっての重要な調節プロセスである。従って、HDACを修飾することができる化合物は、重要な治療上の可能性を有する。
【発明の開示】
【発明が解決しようとする課題】
【0003】
2つの天然産物デプシペプチド、FK228およびスピルコスタチン(Spiruchostatin)Aには、HDAC阻害剤としての可能性があるとの報告がある。しかし、これらの天然産物を化学的に修飾してさらなる類似体を生じさせる可能性は、非常に限られている。
【課題を解決するための手段】
【0004】
驚くべきことに、今般、下に記載する一般式(I)および(I’)の化合物がHDACの阻害剤として作用することを発見した。従って本発明は、HDACの阻害剤として使用するための医薬品の製造における、式(I)もしくは(I’)
【0005】
【化15】
【0006】
(式中、R1、R2、R3およびR4は、同じであるか異なり、アミノ酸側鎖部分を表し、各R6は、同じであるか異なり、水素またはC1−C4アルキルを表し、ならびにPr1およびPr2は、同じであるか異なり、水素またはチオール保護基を表す)
のFK228類似体である化合物、そのアイソスターまたは医薬的に許容されるその塩の使用を提供する。
【0007】
さらに、本発明は、HDACの阻害剤として使用するための医薬品の製造における、上で定義したFK228類似体または医薬的に許容されるその塩の使用を提供する。
【発明を実施するための最良の形態】
【0008】
本明細書で用いられる場合、用語「アミノ酸側鎖部分」は、天然および非天然アミノ酸中に存在する任意のアミノ酸側鎖を指す。非天然アミノ酸由来のアミノ酸側鎖部分の例(括弧内にそれらが由来するアミノ酸を示す)は、−(CH2)2−C(O)−O−C(CH3)3(t−ブトキシカルボニルメチルアラニン)、−(CH2)4−NH−C(O)−O−C(CH3)3(Nε−(t−ブトキシカルボニル)−リシン)、−(CH2)3−NH−C(O)NH2(シトルリン)、−CH2−CH2OH(ホモセリン)および−(CH2)2−CH2NH2(オルニチン)である。特に、−(CH2)3−NH−C(O)NH2(シトルリン)、−CH2−CH2OH(ホモセリン)および−(CH2)2−CH2NH2(オルニチン)を挙げることができる。
【0009】
C1−C6アルキル基または部分は、線状である場合もあり、または分枝状である場合もある。一般に、それは、C1−C4アルキル基または部分、例えば、メチル、エチル、n−プロピル、i−プロピル、n−ブチル、s−ブチルおよびt−ブチルである。好ましい例としては、メチル、i−プロピルおよびt−ブチルが挙げられる。
【0010】
C2−C6アルケニル基または部分は、線状である場合もあり、または分枝状である場合もある。一般に、それは、C2−C4アルケニル基または部分である。アルケニルラジカルは、一または二不飽和であり、さらに好ましくは一不飽和であることが好ましい。例としては、ビニル、アリル、1−プロペニル、イソプロペニル、1−ブテニル、2−ブテニルおよび3−ブテニルが挙げられる。
【0011】
アルキレン基は、二価である前記アルキル基である。
【0012】
前記チオール保護基は、一般に、
(a)チオエーテルを形成してチオール基を保護する保護基、例えば、C1−C6アルコキシ(例えば、メトキシ)、C1−C6アシルオキシ(例えば、アセトキシ)、ヒドロキシおよびニトロにより所望により置換されているベンジル基、ピコリル、ピコリル−N−オキシド、アントリルメチル、ジフェニルメチル、フェニル、t−ブチル、アダマンチル、C1−C6アシルオキシメチル(例えば、ピバロイルオキシメチル、t−ブトキシカルボニルオキシメチル)、
(b)モノチオ、ジチオまたはアミノチオアセタールを形成してチオール基を保護する保護基、例えば、C1−C6アルコキシメチル(例えば、メトキシメチル、イソブトキシメチル)、テトラヒドロピラニル、ベンジルチオメチル、フェニルチオメチル、チアゾリジン、アセトアミドメチル、ベンズアミドメチル、
(c)チオエステルを形成してチオール基を保護する保護基、例えば、t−ブトキシカルボニル(BOC)、アセチルおよびその誘導体、ベンゾイルおよびその誘導体;または
(d)カルバミン酸チオエステルを形成してチオール基を保護する保護基、例えば、カルバモイル、フェニルカルバモイル、C1−C6アルキルカルバモイル(例えば、メチルカルバモイルおよびエチルカルバモイル)
である。
【0013】
一般に、Pr1およびPr2は、同じであるか異なり、各々、水素を表すか、チオエーテル、モノチオ、ジチオもしくはアミノチオアセタール、チオエステルまたはカルバミン酸チオエステルを形成してチオール基を保護する保護基を表す。好ましくは、Pr1およびPr2は、同じであるか異なり、各々、水素を表すか、C1−C6アルコキシ(例えば、メトキシ)、C1−C6アシルオキシ(例えば、アセトキシ)、ヒドロキシおよびニトロにより所望により置換されているベンジル基、ピコリル、ピコリル−N−オキシド、アントリルメチル、ジフェニルメチル、フェニル、t−ブチル、アダマンチル、C1−C6アシルオキシメチル(例えば、ピバロイルオキシメチル、t−ブトキシカルボニルオキシメチル)、C1−C6アルコキシメチル(例えば、メトキシメチル、イソブトキシメチル)、テトラヒドロピラニル、ベンジルチオメチル、フェニルチオメチル、チアゾリジン、アセトアミドメチル、ベンズアミドメチル、t−ブトキシカルボニル(BOC)、アセチルおよびその誘導体、ベンゾイルおよびその誘導体、カルバモイル、フェニルカルバモイルおよびC1−C6アルキルカルバモイル(例えば、メチルカルバモイルおよびエチルカルバモイル)から選択される保護基を表す。最も好ましくは、Pr1およびPr2は、水素である。
【0014】
1つの実施形態において、前記アミノ酸側鎖部分は、天然アミノ酸由来のものである。天然アミノ酸由来のアミノ酸側鎖部分の例(括弧内にそれらが由来するアミノ酸を示す)は、−H(グリシン)、−CH3(アラニン)、−CH(CH3)2(バリン)、−CH2CH(CH3)2(ロイシン)、−CH(CH3)CH2CH3(イソロイシン)、−(CH2)4NH2(リシン)、−(CH2)3NHC(=NH)NH2(アルギニン)、−CH2−(5−1H−イミダゾリル)(ヒスチジン)、−CH2CONH2(アスパラギン)、−CH2CH2CONH2(グルタミン)、−CH2COOH(アスパラギン酸)、−CH2CH2COOH(グルタミン酸)、−CH2−フェニル(フェニルアラニン)、−CH2−(4−OH−フェニル)(チロシン)、−CH2−(3−1H−インドリル)(トリプロファン)、−CH2SH(システイン)、−CH2CH2SCH3(メチオニン)、−CH2OH(セリン)、および−CH(OH)CH3(トレオニン)である。
【0015】
1つの実施形態において、各アミノ酸側鎖は、天然アミノ酸中に存在するアミノ酸側鎖部分であるか、−(CH2)2−C(O)−O−C(CH3)3(t−ブトキシカルボニルメチルアラニン)、−(CH2)4−NH−C(O)−O−C(CH3)3(Nε−(t−ブトキシカルボニル)−リシン)、−(CH2)3−NH−C(O)NH2(シトルリン)、−CH2−CH2OH(ホモセリン)または−(CH2)2−CH2NH2(オルニチン)である。
【0016】
本発明の好ましい実施形態において、各アミノ酸側鎖は、−H、−C1−C6アルキル、−C2−C6アルケニル、−L−O−C(O)−R’、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’、−L−Het−C(O)−Het−R’’および−L−Het−R’’から選択される部分であり、Lは、C1−C6アルキレン基であり、Aは、フェニルまたは5員から6員ヘテロアリール基であり、各R’は、同じであるか異なり、C1−C4アルキルを表し、各R’’は、同じであるか異なり、HまたはC1−C6アルキルを表し、各−Het−は、同じであるか異なり、ならびに−O−、−N(R’’’)−および−S−から選択されるヘテロ原子スペーサーであり、各R’’’は、同じであるか異なり、HまたはC1−C4アルキルを表す。
【0017】
基Aが、5員から6員ヘテロアリール基であるとき、それは、例えば、フラニル、チエニル、ピロリル、オキサゾリル、チアゾリル、イミダゾリル、ピラゾリル、イソオキサゾリル、イソチアゾリル、オキサジアゾリル、トリアゾリル、チアジアゾリル、ピリジル、ピリダジル、ピリミジニル、ピラジニル、トリアジニルであり得る。しかし、一般には、各A部分は、フェニルである。
【0018】
ヘテロ原子スペーサー基Hetは、一般に、−O−または−N(R’’’)−である。さらに一般的には、−O−または−N(H)−である。
【0019】
好ましくは、各アミノ酸側鎖は、−H、−C1−C6アルキル、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’および−L−N(R’’)−C(O)−O−R’’から選択される部分であり、L、AおよびR’’は、先に定義したとおりである。
【0020】
一般に、本発明の化合物のアミノ酸側鎖部分は、−(CH2)2−C(O)−O−C(CH3)3、−(CH2)4−NH−C(O)−O−C(CH3)3、−(CH2)2−C(O)OH、−CH2−C6H5、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2、−CH(CH3)CH2CH3、−(CH2)4NH2、CH2SH、−CH2CH2SCH3、−CH2OHおよび−CH(OH)CH3から選択される。さらに一般的には、アミノ酸側鎖部分は、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2、−CH(CH3)CH2CH3、−(CH2)4NH2、CH2SH、−CH2CH2SCH3、−CH2OHおよび−CH(OH)CH3から選択される。
【0021】
好ましくは、アミノ酸側鎖部分は、−H、−CH3、−(CH2)2−C(O)−O−C(CH3)3、−(CH2)4−NH−C(O)−O−C(CH3)3、−(CH2)2−C(O)OH、−CH2−C6H5、−(CH2)4NH2および−CH(CH3)2から選択される。本発明の1つの実施形態において、アミノ酸側鎖部分は、−H、−CH3および−CH(CH3)2から選択される。
【0022】
一般に、R1は、−H、−C1−C6アルキル、−C2−C6アルケニル、−L−O−C(O)−R’、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’、−L−Het−C(O)−Het−R’’および−L−Het−R’’から選択されるアミノ酸側鎖部分であり、L、R’、R’’、−Het−およびR’’’は、先に定義したとおりである。好ましくは、R1は、−H、−C1−C6アルキル、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’および−L−N(R’’)−C(O)−O−R’’から選択される部分であり、L、AおよびR’’は、先に定義したとおりである。さらに好ましくは、R1は、−Hおよび−C1−C6アルキルから選択される部分である。さらにいっそう好ましくは、R1は、−C1−C4アルキル、特にイソプロピルである。
【0023】
一般に、R2は、−H、−C1−C6アルキル、−C2−C6アルケニル、−L−O−C(O)−R’、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’、−L−Het−C(O)−Het−R’’および−L−Het−R’’から選択されるアミノ酸側鎖部分であり、L、R’、R’’、−Het−およびR’’’は、先に定義したとおりである。好ましくは、R2は、−H、−C1−C6アルキル、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’および−L−N(R’’)−C(O)−O−R’’から選択される部分であり、L、AおよびR’’は、先に定義したとおりである。さらに好ましくは、R2は、−Hおよび−C1−C4アルキルから選択される部分である。さらにいっそう好ましくは、R2は、−H、−CH3または−CH(CH3)2である。さらにより好ましくは、R2は−Hまたは−CH3である。
【0024】
一般に、R3は、−H、−C1−C6アルキル、−C2−C6アルケニル、−L−O−C(O)−R’、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’、−L−Het−C(O)−Het−R’’および−L−Het−R’’から選択されるアミノ酸側鎖部分であり、L、R’、R’’、−Het−およびR’’’は、先に定義したとおりである。好ましくは、R3は、−H、−C1−C6アルキル、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’および−L−N(R’’)−C(O)−O−R’’から選択される部分であり、L、AおよびR’’は、先に定義したとおりである。
【0025】
さらに好ましくは、R3は、−(CH2)2−C(O)−O−C(CH3)3、−(CH2)4−NH−C(O)−O−C(CH3)3、−(CH2)2−C(O)OH、−CH2−C6H5、−CH3、−CH(CH3)2または−(CH2)4NH2である。
【0026】
一般に、R4は、−H、−C1−C6アルキル、−C2−C6アルケニル、−L−O−C(O)−R’、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’、−L−Het−C(O)−Het−R’’および−L−Het−R’’から選択されるアミノ酸側鎖部分であり、L、R’、R’’、−Het−およびR’’’は、先に定義したとおりである。好ましくは、R4は、−H、−C1−C6アルキル、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’および−L−N(R’’)−C(O)−O−R’’から選択される部分であり、L、AおよびR’’は、先に定義したとおりである。さらに好ましくは、R4は、水素、−C1−C6アルキルまたは−C2−C6アルケニルである。さらにいっそう好ましくは、R4は、水素または−C1−C4アルキル、さらに好ましくは水素である。
【0027】
一般に、各R6は、同じであるか異なり、水素または−C1−C2アルキルである。好ましくは、各R6は、水素である。
【0028】
1つの実施形態において、本発明は、式(I)の化合物である、上で定義したとおりのFK228類似体、そのアイソスターまたは医薬的に許容されるその塩を提供する。
【0029】
本発明の好ましい化合物は、式中のR1が、−Hおよび−C1−C6アルキルから選択され、R2が、−Hおよび−C1−C4アルキルから選択され、R3が、−H、−C1−C6アルキル、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’および−L−N(R’’)−C(O)−O−R’’(この場合、L、AおよびR’’は、先に定義したとおりである)から選択され、R4が、−Hおよび−C1−C6アルキルから選択され、ならびにR6が、同じであるか異なり、水素または−C1−C2アルキルである、上で定義したFK228類似体、それらのアイソスターおよびそれらの医薬的に許容される塩である。
【0030】
本発明のさらに好ましい化合物は、(a)式中のR1が、−C1−C4アルキルであり、R2が、−Hおよび−CH3から選択され、R3が、−H、−C1−C6アルキル、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’および−L−N(R’’)−C(O)−O−R’’(この場合、L、AおよびR’’は、先に定義したとおりである)から選択され、R4が、−Hであり、各R6が、−Hである式(I)の化合物、それらのアイソスターおよびそれらの医薬的に許容される塩、ならびに(b)式中のR1が、−C1−C4アルキルであり、R2が、−Hおよび−CH3から選択され、R3が、−H、−C1−C6アルキル、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’および−L−N(R’’)−C(O)−O−R’’(この場合、L、AおよびR’’は、先に定義したとおりである)から選択され、R4が、−Hであり、R6が、−Hであり、Pr1およびPr2が、水素である式(I’)の化合物、それらのアイソスターおよびそれらの医薬的に許容される塩である。
【0031】
式(I)のさらに特に好ましい化合物は、式中の
R1が、−CH(CH3)2であり、R2が、−CH3であり、R3が、−CH3であり、R4が、水素であり、およびR6が、−Hである、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)2−C(O)−O−C(CH3)3であり、R4が、水素であり、およびR6が、−Hである、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)2−C(O)−OHであり、R4が、水素であり、およびR6が、−Hである、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)−C6H5であり、R4が、水素であり、およびR6が、−Hである、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−CH(CH3)2であり、R4が、水素であり、およびR6が、−Hである、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)4−NH−C(O)−O−C(CH3)3であり、R4が、水素であり、およびR6が、−Hである;または
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)4−NH2であり、R4が、水素であり、およびR6が、−Hである
化合物、およびそれらの医薬的に許容される塩である。
【0032】
式(I’)のさらに特に好ましい化合物は、式中の
R1が、−CH(CH3)2であり、R2が、−CH3であり、R3が、−CH3であり、R4が、水素であり、およびR6が、−Hであり、Pr1およびPr2が、水素である、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)2−C(O)−O−C(CH3)3であり、R4が、水素であり、およびR6が、−Hであり、Pr1およびPr2が、水素である、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)2−C(O)−OHであり、R4が、水素であり、およびR6が、−Hであり、Pr1およびPr2が、水素である、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)−C6H5であり、R4が、水素であり、およびR6が、−Hであり、Pr1およびPr2が、水素である、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−CH(CH3)2であり、R4が、水素であり、およびR6が、−Hであり、Pr1およびPr2が、水素である、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)4−NH−C(O)−O−C(CH3)3であり、R4が、水素であり、およびR6が、−Hであり、Pr1およびPr2が、水素である;または
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)4−NH2であり、R4が、水素であり、R6が、−Hであり、ならびにPr1およびPr2が、水素である
化合物、およびそれらの医薬的に許容される塩である。
【0033】
好ましいFK228類似体は、式(2)または(2’)である。
【0034】
【化16】
【0035】
別の好ましいFK228類似体は、式(3)または(3’)である。
【0036】
【化17】
【0037】
別の好ましいFK228類似体は、式(4)または(4’)である。
【0038】
【化18】
【0039】
別の好ましいFK228類似体は、式(5)または(5’)である。
【0040】
【化19】
【0041】
別の好ましいFK228類似体は、式(6)または(6’)である。
【0042】
【化20】
【0043】
別の好ましいFK228類似体は、式(7)または(7’)である。
【0044】
【化21】
【0045】
別の好ましいFK228類似体は、式(8)または(8’)である。
【0046】
【化22】
【0047】
別の好ましいFK228類似体は、式(9)または(9’)である。
【0048】
【化23】
【0049】
本発明の1つの実施形態において、FK228類似体は、式(II)または(II’)である。
【0050】
【化24】
【0051】
(式中、R1、R2、R3、R4、R6、Pr1およびPr2は、先に定義したとおりである)
本発明のもう1つの実施形態において、FK228類似体は、式(III)または(III’)である。
【0052】
【化25】
【0053】
(式中、R1、R2、R3、Pr1およびPr2は、先に定義したとおりである)
本発明のもう1つの実施形態において、FK228類似体は、式(IV)または(IV’)である。
【0054】
【化26】
【0055】
(式中、R1、R2、R3、Pr1およびPr2は、先に定義したとおりである)
本発明のもう1つの実施形態において、FK228類似体は、式(V)または(V’)である。
【0056】
【化27】
【0057】
(式中、R1、R2、R3、Pr1およびPr2は、先に定義したとおりである)
さらなる実施形態において、本発明は、HDACの阻害剤として使用するための医薬品の製造における、式(VI)
【0058】
【化28】
【0059】
(式中、R1、R2およびR3は、同じであるか異なり、アミノ酸側鎖部分を表し、ならびにR4は、水素、C1−C6アルキルまたはC2−C6アルケニルである)
の化合物、そのアイソスターまたは医薬的に許容されるその塩の使用を提供する。
【0060】
一般に、この実施形態において、R1は、天然アミノ酸由来のアミノ酸側鎖部分である。好ましくは、この実施形態において、R1は、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2、−CH(CH3)CH2CH3、−(CH2)4NH2、−CH2SH、−CH2CH2SCH3、−CH2OHまたは−CH(OH)CH3である。さらに好ましくは、R1は、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2または−CH(CH3)CH2CH3である。最も好ましくは、R1は、−CH(CH3)2である。
【0061】
一般に、この実施形態において、R2は、天然アミノ酸由来のアミノ酸側鎖部分である。好ましくは、R2は、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2、−CH(CH3)CH2CH3、−(CH2)4NH2、−CH2SH、−CH2CH2SCH3、−CH2OHまたは−CH(OH)CH3である。さらに好ましくは、R2は、−H、−CH3または−CH(CH3)2である。最も好ましくは、R2は、−Hである。
【0062】
一般に、この実施形態において、R3は、天然アミノ酸由来のアミノ酸側鎖部分である。好ましくは、R3は、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2、−CH(CH3)CH2CH3、−(CH2)4NH2、−CH2SH、−CH2CH2SCH3、−CH2OHまたは−CH(OH)CH3である。さらに好ましくは、R3は、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2または−CH(CH3)CH2CH3である。最も好ましくは、R3は、−CH3である。
【0063】
一般に、この実施形態において、R4は、水素またはC1−C6アルキルである。好ましくは、R4は、水素またはC1−C2アルキルである。さらに好ましくは、R4は、水素である。
【0064】
この実施形態において、本発明の好ましい化合物は、式中の
R1が、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2、−CH(CH3)CH2CH3、−(CH2)4NH2、−CH2SH、−CH2CH2SCH3、−CH2OHまたは−CH(OH)CH3であり、
R2が、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2、−CH(CH3)CH2CH3、−(CH2)4NH2、−CH2SH、−CH2CH2SCH3、−CH2OHまたは−CH(OH)CH3であり、
R3が、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2、−CH(CH3)CH2CH3、−(CH2)4NH2、−CH2SH、−CH2CH2SCH3、−CH2OHまたは−CH(OH)CH3であり;および
R4が、水素またはC1−C2アルキルである、
式(VI)の化合物である。
【0065】
この実施形態の好ましい態様において、本発明は、式中の
R1が、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2または−CH(CH3)CH2CH3であり、
R2が、−H、−CH3または−CH(CH3)2であり、
R3が、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2または−CH(CH3)CH2CH3であり;および
R4が、水素である、
式(VI)の化合物を提供する。
【0066】
この実施形態の特に好ましい化合物は、式(VI)中のR1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−CH3であり、およびR4が、水素である。
【0067】
この実施形態のさらに好ましい化合物は、式(III)の化合物である。
【0068】
【化29】
【0069】
(式中、R1、R2およびR3は、先に定義したとおりである)
この実施形態のさらに好ましい化合物は、式(IV)の化合物である。
【0070】
【化30】
【0071】
(式中、R1、R2およびR3は、先に定義したとおりである)
本発明の化合物は、新規であると考えられ、従って本発明は、式(I)の化合物、そのアイソスターまたは医薬的に許容されるその塩を提供する。さらに、本発明は、式(I)の化合物または医薬的に許容されるその塩を提供する。
【0072】
本発明は、ヒトまたは動物を治療する方法において使用するための、式(I)の化合物、そのアイソスターまたは医薬的に許容されるその塩も提供する。さらに本発明は、ヒトまたは動物を治療する方法において使用するための、式(I)の化合物または医薬的に許容されるその塩も提供する。
【0073】
本発明の化合物は、高い治療効果を示すので、特に有利である。また、そのテトラペプチドコアを容易に合成することができるので、有利である。
【0074】
本明細書で用いられる場合、医薬的に許容される塩は、医薬的に許容される酸または塩基との塩である。医薬的に許容される酸は、無機酸(例えば、塩酸、硫酸、リン酸、二リン酸、臭化水素酸または硝酸)と有機酸(例えば、クエン酸、フマル酸、マレイン酸、リンゴ酸、アスコルビン酸、コハク酸、酒石酸、安息香酸、酢酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸またはp−トルエンスルホン酸)の両方を含む。医薬的に許容される塩基としては、アルカリ金属(例えば、ナトリウムまたはカリウム)およびアルカリ土類金属(例えば、カルシウムまたはマグネシウム)水酸化物、ならびに有機塩基、例えば、アルキルアミン、アラルキルアミンまたは複素環式アミンが挙げられる。
【0075】
本明細書で用いられる場合、用語「アイソスター」は、ある原子または原子群と別の、概して類似している、原子または原子群の交換の結果として生じた化合物を指す。式(I)の化合物において、アイソステリック基を含有する部分は、好ましくは、−NH−CHR1−CO−、−NH−CHR2−CO−O−および−NH−CO−CHR3−NH−CO−である。式(I)の化合物において、アイソステリック基を含有する部分は、さらに好ましくは、−NH−CHR1−CO−および−NH−CHR2−CO−O−である。このようなアイソスターの例は、式中の部分−NH−が、−CH2−、−O−または−S−により置換されている、部分−CO−が、−CS−または−C(=NH)−により置換されている、および部分−O−が、−S−、CH2−または−NH−により置換されている、式(I)の化合物である。
【0076】
本発明の化合物は、式(I)に示すキラリティーを有する。しかし、基R1、R2およびR3の空間配置に起因して化合物内に追加のキラル中心が形成される場合がある。疑いを避けるために、本明細書に描かれている化学構造は、ラセミおよび非ラセミ混合物ならびに純粋なエナンチオマーおよび/またはジアステレオマーを含む、これらの追加のキラル中心を随伴する立体異性体配置のすべてを包含することを意図する。
【0077】
疑いを避けるために、本発明は、インビボで反応して本発明の化合物またはそのアイソスターもしくは医薬的に許容される塩を生じさせるプロドラッグも包含する。
【0078】
本発明の好ましい化合物は、光学異性体である。従って、例えば、キラル中心を1つだけ含有する式(I)の好ましい化合物としては、実質的に純粋な形態のR異性体、実質的に純粋な形態のSエナンチオマー、および過剰なRエナンチオマーまたは過剰なSエナンチオマーを含有するエナンチオマー混合物が挙げられる。
【0079】
本発明は、式(I)の化合物、そのアイソスターまたは医薬的に許容されるその塩および医薬的に許容される担体または希釈剤も提供する。
【0080】
前記医薬組成物は、一般に、85重量%以下の本発明の化合物を含有する。さらに一般的には、それは、50重量%以下の本発明の化合物を含有する。好ましい医薬組成物は、無菌であり、発熱物質を含まない。さらに、本発明により提供される医薬組成物は、実質的に純粋な光学異性体である本発明の化合物を、一般に含有する。好ましくは、本医薬組成物は、式(I)の化合物またはそのアイソスターの医薬的に許容される塩を含む。
【0081】
本FK228類似体は、従来の経路、例えば、以下の図式(この場合、基R1からR4は、上で定義したとおりである)を用いて調製することができる。
【0082】
【化31】
【0083】
段階(a)において、側鎖R2を有するアミノ酸エステルを、側鎖R1を有する第二のアミノ酸と縮合させて、ジペプチドを得る。段階(b)において、そのジペプチドを保護システイン誘導体と縮合させて、トリペプチドを得る。段階(c)において、そのトリペプチドをアミノ酸とカップリングさせて、保護テトラペプチドを生じさせる。段階(d)において、そのテトラペプチドのN末端を脱保護し、その遊離アミンを、β−ヒドロキシ酸誘導体(式中、R5は、一時的ブロッキング基であり、これを除去して、R5がHである化合物を生じさせることができ、およびXは、Yurek−George,A.;Habens,F.;Brimmell,M.;Packham,G.;Ganesan,A.J.Am.Chem.Soc.2004,126,1030−1031に報告されているようなキラル補助基である)とカップリングさせる。段階(e)においてそのエステル基を加水分解し、その後、段階(f)において環化し、(g)においてジスルフィド結合を形成させて、二環式デプシペプチド化合物(I)の合成を完了する。
【0084】
R6が水素以外である本発明の化合物は、R6が水素である本発明の対応する化合物もしくは中間体をアルキル化することにより、または適切に置換された出発材料を使用することにより、得ることができる。
【0085】
式(I’)の化合物は、ジスルフィド結合を開裂するように上記段階(g)の生成物を反応させることによって、得ることができる。一般に、このジスルフィド結合の開裂は、ジスルフィド結合を有するタンパク質の還元処理のために一般に使用されるチオール化合物、例えば、メルカプトエタノール、チオグリコール酸、2−メルカプトエチルアミン、ベンゼンチオール、パラチオクレゾールおよびジチオトレイトールを使用して達成される。好ましくは、メルカプトエタノールおよびジチオトレイトールを使用する。例えば透析またはゲル濾過により、過剰のチオール化合物を除去することができる。あるいは、例えば電解、テトラヒドロホウ酸ナトリウム、水素化アルミニウムリチウムまたは亜流酸塩を使用して、ジスルフィド結合を開裂することができる。
【0086】
Pr1および/またはPr2が水素以外である式(I’)の化合物は、Pr1および/またはPr2が水素である対応する化合物にチオール保護基を導入することによって、調製することができる。この態様において、この反応に用いられるチオール保護基の導入に適する薬剤は、その導入される保護基に依存して適切に決定される。例としては、その対応する保護基の塩化物(例えば、塩化ベンジル、塩化メトキシベンジル、塩化アセトキシベンジル、塩化ニトロベンジル、塩化ピコリル、塩化ピコリル−N−オキシド、塩化アントリルメチル、塩化イソブトキシメチル、塩化フェニルチオメチル)およびその対応する保護基のアルコール(例えば、ジフェニルメチルアルコール、アダマンチルアルコール、アセトアミドメチルアルコール、ベンズアミドメチルアルコール)、ジニトロフェニル、イソブチレン、ジメトキシメタン、ジヒドロピランならびにクロロギ酸t−ブチルが挙げられる。
【0087】
当業者には理解されるように、R1、R2、R3およびR4の1つが、官能基、例えば、−OH、−SH、−NH2または−COOHを有するときには、その導入後のもう1つの反応のためにその基を保護することが好ましい場合がある。このような場合、問題の基は、その導入後、別の段階で保護してもよいし、またはそれを導入する時点で既に保護されていてもよい。当業者は、これに関して使用することができる適当な保護基を知っていることであろう。
【0088】
このようにして得たFK228類似体を、適切な酸または塩基での処理により、塩化することができる。上記プロセスのうちのいずれかによって得られたラセミ混合物は、標準的な技術、例えばキラルクロマトグラフィーカラムでの溶離によって分解することができる。
【0089】
本発明の好ましい化合物は、スベロイルアニリドヒドロキサム酸(SAHA)と少なくとも等しいHDAC阻害活性を有する。従って、さらなる実施形態において、本発明は、SAHAにより示されるものに少なくとも等しいHDAC阻害活性を有する化合物を選択するためのプロセスであって、式(I)または(I’)の化合物を調製することを含み、該調製は以下を含む該プロセス:
(i)式(VI)の化合物と式(VII)の化合物を反応させ、
【0090】
【化32】
【0091】
(式中、R1およびR2は、上で定義したとおりであり、R7は、C1−C4アルキルまたはC2−C4アルケニルであり、ならびにYは、アミノ保護基である)
(ii)得られた中間体を脱保護し、それを式(VIII)の化合物と反応させ、
【0092】
【化33】
【0093】
(式中、Y’は、アミノ保護基であり、およびY’’は、水素または保護基である)
(iii)得られた中間体を脱保護し、それを式(IX)の化合物と反応させ、
【0094】
【化34】
【0095】
(式中、R3は、上で定義したとおりであり、およびY’’’は、アミノ酸保護基である)
(iv)得られた中間体を脱保護し、それを式(X)の化合物と反応させ、
【0096】
【化35】
【0097】
(式中、R4は、上で定義したとおりであり、R5は、水素またはヒドロキシ保護基であり、LGは、脱離基であり、およびY’’’’は、水素または保護基である)
(v)得られた中間体上のβ−ヒドロキシ基を所望により脱保護して、R5保護基を除去し、それをHで置換し、
(vi)得られた中間体を加水分解および環化すること、
(vii)得られた中間体を、所望により、ジスルフィド結合を形成させるように反応させ、そしてジスルフィド結合が形成される場合には、得られた化合物中のジスルフィド結合を所望により開裂し、得られた化合物がチオール基を含有する場合には、チオール保護基を所望により導入し;および
(viii)得られた化合物をスクリーニングして、HDAC阻害剤としてのその活性を測定する。
【0098】
一般に、段階(vi)において、エステル基の加水分解は、環化の前に行う。
【0099】
一般に、段階(vii)において、DTT(ジチオトレイトール)を使用して、ジスルフィド結合の開裂を行う。
【0100】
様々な実体を保護基Y、Y’、Y’’、Y’’’およびY’’’’に用いることができること、ならびにその好ましい実体が、それぞれの場合、存在する特定の基の性質に依存することは、当業者には理解されるであろう。
【0101】
基Y、Y’およびY’’’は、例えば、t−ブトキシカルボニル(Boc)または9−フルオレニルメトキシカルボニル(Fmoc)であり得る。一般に、それらはFmocである。
【0102】
基Y’’およびY’’’’は、例えば、トリチル(Trt)であり得る。
【0103】
当業者は、脱離基LGについての適する実体を知っている。例えば、それは、Yurek−George,A.ら(J.Am.Chem.Soc.2004,126,1030−1031)において説明されているような、そのN原子により結合されているチアゾリジンチオン基などのキラル補助基であり得る。あるいは、それは、−OH基であり得る。
【0104】
基R7は、一般に、C1−C4アルキルまたはC1−C4アルケニル基である。さらに一般的には、メチルまたはアリルである。
【0105】
様々なアッセイが、HDAC阻害についての試験に適すること、およびそれらを用いて、段階(vii)から得られた化合物の活性を測定し、既知HDAC阻害剤SAHAのものと比較できることは、当業者には理解される。従って、HDACに対する試験化合物のIC50を、例えば、あるインビトロアッセイで決定し、その同じアッセイ条件下でのSAHAのIC50と比較することができる。試験化合物が、SAHAのものに等しいか、それより低いIC50値を有する場合、それは、SAHAにより示されるものに少なくとも等しいHDAC阻害活性を有すると理解されよう。
【0106】
好ましい実施形態において、本発明は、段階(viii)におけるスクリーニング段階が、インビトロHDACアッセイである、上で定義したSAHAにより示されるものに少なくとも等しいHDAC阻害活性を有する化合物を選択するためのプロセスを提供する。一般に、前記アッセイは、様々な濃度の試験化合物およびSAHAを希釈Hela核抽出物と接触させて、Hela核抽出物に対するその試験化合物およびSAHAのIC50を決定することを含む。同じアッセイ条件下でのSAHAのIC50と等しいかそれより低い、Hela核抽出物に対して測定されたIC50値を有する試験化合物は、SAHAにより示されるものに少なくとも等しい阻害活性を有すると理解されよう。一般に、前記アッセイは、HDAC蛍光活性アッセイキット(英国のバイオモール(Biomol))を使用して行い、それらの試験化合物は、分析する前に還元する。前記アッセイ試験は、例えば、下の標題「活性アッセイ5」の下で説明するとおり行うことができる。
【0107】
もう1つの実施形態において、本発明は、SAHAにより示されるものに少なくとも等しいヒト癌細胞増殖阻害活性を有する化合物を選択するためのプロセスを提供し、このプロセスは、上で定義したように段階(i)から(vii)により式(I)または(I’)の化合物を調製すること、その後、(viii)得られた化合物をスクリーニングして、ヒト癌細胞増殖阻害剤としてのその活性を測定することを含む。
【0108】
様々なアッセイがヒト癌細胞増殖阻害についての試験に適し、それらを用いて、段階(vii)から得られた化合物の活性を測定し、SAHAのものと比較できることは、当業者には理解されるであろう。従って、ヒト癌細胞増殖に対する試験化合物のIC50を、例えば、あるインビトロアッセイで決定することができ、その同じアッセイ条件下でのSAHAのIC50と比較することができる。試験化合物が、SAHAの活性に等しいかそれより低いIC50値を有する場合、それは、SAHAにより示される活性に少なくとも等しい阻害活性を有すると理解されよう。一般に、この実施形態における段階(viii)は、様々な濃度の試験化合物およびSAHAを、MCF7乳癌細胞系、HUT78 T細胞白血病細胞系、A2780卵巣癌細胞系、PC3またはLNCAP前立腺癌細胞系と接触させて、その細胞系に対するその試験化合物およびSAHAのIC50を決定することを含む、インビトロアッセイを含む。同じアッセイ条件下でのSAHAのIC50に等しいかそれより低い、これらの細胞系のうちのいずれかに対して測定したIC50値を有する試験化合物は、SAHAの活性に少なくとも等しい阻害活性を有すると理解されよう。一般にこの実施形態において、前記アッセイは、商標シクワント(CyQuant)アッセイシステム(米国のモレキュラー・プローブス社(Molecular Probes,Inc.))を用いて行う。前記アッセイ試験は、例えば、下の標題「活性アッセイ6」および/または「活性アッセイ1」の下で説明するとおり行うことができる。
【0109】
もう1つの好ましい実施形態において、本発明は、SAHAにより示される活性に少なくとも等しい抗炎症活性を有する化合物を選択するためのプロセスを提供し、このプロセスは、上で定義したように段階(i)から(vii)により式(I)または(I’)の化合物を調製すること、その後、(viii)そのようにして得られた化合物をスクリーニングして、その抗炎症活性を測定することを含む。
【0110】
様々なアッセイが化合物の抗炎症活性の評価に適することは、当業者には理解されよう。SAHAを基準にした試験化合物の抗炎症活性は、SAHAを基準にして、末梢血単核細胞(PBMC)からのTNFαの生産の阻害に関する化合物の活性を測定することにより、判定することができる。従って、PBMCからのTNFαの生産を阻害する試験化合物の能力を、例えば、あるアッセイにおいて判定し、その同じアッセイ条件下でのSAHAの活性と比較することができる。試験化合物が、同じアッセイ条件下でのSAHAの活性に等しいかそれより高いTNFα生産に対するインビトロ阻害活性を有する場合、それは、SAHAにより示される活性に少なくとも等しい抗炎症活性を有すると理解されよう。一般に、段階(viii)は、登録商標クアンティキン(Quantikine)ヒト−αアッセイキット(英国、アビンドン(Abingdon,UK)のアール・アンド・ディー・システムズ(R&D systems))を使用して行う。前記アッセイ試験は、例えば、下の標題「活性アッセイ7」の下で説明するとおり行うことができる。
【0111】
この実施形態のもう1つの態様において、SAHAを基準にした試験化合物の抗炎症活性は、SAHAを基準にしてBalb/cマウスにおける炎症の阻害に関する化合物の活性を評価することにより判定することができる。試験化合物が、同じ試験条件下でのSAHAのものに等しいかそれより高いインビボ阻害活性を有する場合、それは、SAHAにより示される活性に少なくとも等しい抗炎症活性を有すると理解されよう。一般に、この実施形態における段階(viii)は、化学物質負荷(chemical challenge)により誘発されたBalb/cマウスにおける炎症の阻害に関する試験化合物およびSAHAのインビボ活性を評価することにより行う。一般に、前記化学物質負荷は、前記マウスへのオキサラゾンまたはアセトンの局所投与を含む。この実施形態において、調査中の化合物は、その化学物質負荷の前に適用される場合もあり、または後に適用される場合もある。このような方針に沿った抗炎症活性の評価は、例えば、下の標題「活性アッセイ8」の下で説明するとおり行うことができる。
【0112】
もう1つの好ましい実施形態において、本発明は、SAHAにより示される活性に少なくとも等しいMCF7細胞における主G2/M期停止または細胞死の誘導に関する活性を有する化合物を選択するためのプロセスを提供し、このプロセスは、上で定義したように段階(i)から(vii)により式(I)または(I’)の化合物を調製すること、その後、(viii)得られた化合物をスクリーニングして、SAHAを基準にしてMCF7細胞における主G2/M期停止または細胞死の誘導に関する活性を測定することを含む。スクリーニング段階(viii)は、例えば、下の標題「活性アッセイ3」の下で説明するとおり行うアッセイを含むことができる。
【0113】
上で説明したスクリーニングにおいて、試験化合物の好ましい形態は、そのスクリーニング段階の性質に依存する。従って、スクリーニング段階が、下の「活性アッセイ5」の下で説明するようなインビトロアッセイである場合、その試験化合物は、好ましくは、上で定義したとおりの式(I’)の化合物である。しかし、スクリーニング段階が、細胞系に合わない場合には、試験化合物は、好ましくは、式(I)の化合物である。
【0114】
本発明の化合物は、HDACの阻害剤であることが分かる。従って、本発明の化合物は、治療に有用である。
【0115】
本発明の化合物およびそれらを含む組成物は、様々な剤形で投与することができる。1つの実施形態において、本発明の化合物を含む医薬組成物は、経口、直腸内、非経口、鼻孔内もしくは経皮投与または吸入もしくは坐剤による投与に適する形式で調合することができる。典型的な投与経路は、非経口、鼻孔内もしくは経皮投与または吸入による投与である。
【0116】
本発明の化合物は、経口的に、例えば錠剤、トローチ、ロゼンジ、水性または油性懸濁剤、分散性粉末または顆粒として、投与することができる。本発明の好ましい医薬組成物は、経口投与に適する組成物、例えば、錠剤およびカプセルである。
【0117】
本発明の化合物は、皮下的であっても、静脈内的であっても、筋肉内的であっても、胸骨内的であっても、経皮的であっても、または注入法によっても、非経口的に投与することもできる。本化合物は、坐剤として投与することもできる。
【0118】
1つの好ましい投与経路は、吸入である。吸入薬の主な利点は、経口経路により摂取される多くの薬物と比較して、血液供給が豊富な領域へのそれらの直接送達である。例えば、肺胞は、膨大な表面積および豊富な血液供給を有するので、および初回通過代謝が回避されるので、吸収が非常に速い。
【0119】
従って、本発明の好ましい医薬組成物は、吸入に適するものを含む。本発明は、そのような医薬組成物が含有されている吸入装置も提供する。一般に、前記装置は、定量噴霧式吸入器(MDI)であり、この吸入器には、この吸入器から薬物を押出すための医薬的に許容される化学的噴射剤が含有されている。一般に、前記噴射剤は、フルオロカーボンである。
【0120】
さらに好ましい吸入装置は、ネブライザーを含む。ネブライザーは、加圧下の空気または酸素を用いて、鼻および口に装着する「マスク」を通して薬物の細かい液体ミストを送達することができる装置である。これらは、乳児、小児および実際に病気の全年齢の患者を含む、吸入器を使用することができない喘息患者を治療するために、多くの場合使用される。
【0121】
上記吸入装置は、例えば、噴射剤を用いずに本発明の化合物を送達することができる、回転式吸入器またはドライパウダー用吸入器であってもよい。
【0122】
一般に、前記吸入装置は、スペーサーを含む。スペーサーは、個体が下気道に大量の薬物を直接吸入できるようにする装置であり、喉ではなく下気道に行くようにするためのものである。多数のスペーサーが、吸入器の末端に適合し、あるものについては、薬物のキャニスターがその装置に適合する。保留チャンバおよび逆止め弁を有するスペーサーは、薬物が気道に逃げるのを防止する。多くの人々、特に小児および老人には、定量噴霧式吸入器からパフを発射するために必要な動作と吸入を調整することが難しいことがある。これらの患者には、スペーサーの使用が特に推奨される。
【0123】
もう1つの好ましい投与経路は、鼻孔内投与である。鼻孔の高透過性組織は、薬物を非常に受け入れやすく、錠剤形の薬物よりはるかに迅速、且つ、効率的に薬物を吸収する。経鼻薬物送達は、注射より痛くなく、侵襲でなく、患者間にあまり不安を生じさせない。薬物は、錠剤形で送達される薬物より少ない用量で経鼻送達することができる。この方法による吸収は非常に迅速であり、初回通過代謝が回避され、従って、患者間でのばらつきが低減される。さらに、経鼻送達装置は、正確な計測用量で薬物を投与することができる。従って、本発明の医薬組成物は、一般に、鼻孔内投与に適する。さらに、本発明は、このような医薬組成物が含有されている鼻孔内用装置も提供する。
【0124】
さらに好ましい投与経路は、経皮投与である。従って本発明は、本発明の化合物または医薬的に許容されるその塩を含有する経皮パッチも提供する。舌下投与も好ましい。従って本発明は、本発明の化合物または医薬的に許容されるその塩を含む舌下錠も提供する。
【0125】
本発明の化合物は、一般に、医薬的に許容される担体または希釈剤と共に投与するために調合される。例えば、固体経口剤形は、活性化合物と共に、希釈剤、例えば、ラクトース、デキストロース、サッカロース、セルロース、トウモロコシデンプンまたは馬鈴薯デンプン;滑沢剤、例えば、シリカ、タルク、ステアリン酸、ステアリン酸マグネシウムもしくはステアリン酸カルシウム、および/またはポリエチレングリコール;結合剤、例えば、デンプン、アラビアゴム、ゼラチン、メチルセルロース、カルボキシメチルセルロースまたはポリビニルピロリドン;崩壊剤、例えば、デンプン、アルギン酸、アルジネートまたはデンプングリコール酸ナトリウム;飽和剤;染料;甘味剤;湿潤剤、例えば、レシチン、ポリソルベート、ラウリルスルフェート;および一般に、医薬調合物に使用される非毒性で薬理学的に不活性な物質を含有することがある。このような医薬製剤は、既知の方法で、例えば、混合、造粒、タブレット形成、糖衣またはフィルムコーティングプロセスによって、製造することができる。
【0126】
経口投与用の分散液は、シロップ、乳剤および懸濁剤であり得る。シロップは、担体として、例えば、サッカロースまたはサッカロースとグリセリンおよび/またはマンニトールおよび/またはソルビトールを含有することがある。
【0127】
懸濁剤および乳剤は、担体として、例えば、天然ゴム、寒天、アルギン酸ナトリウム、ペクチン、メチルセルロース、カルボキシメチルセルロース、またはポリビニルアルコールを含有することがある。筋肉内注射用の懸濁剤または溶液は、活性化合物と共に、医薬的に許容される担体、例えば、滅菌水、オリーブ油、オレイン酸エチル、グリコール、例えばプロピレングリコール、および所望される場合には、適量の塩酸リドカインを含有することがある。
【0128】
注射または注入用の溶液は、担体として、例えば滅菌水を含有することがあり、または好ましくは、滅菌等張食塩水溶液の形態であり得る。
【0129】
本発明の化合物は、HDACにより媒介される状態の治療または予防において、治療上、有用である。従って本発明は、HDACにより媒介される状態の治療または予防に使用するための医薬品の製造における、式(I)の化合物または医薬的に許容されるその塩の使用を提供する。HDACにより媒介される状態に罹患しているまたは罹患しやすい患者の治療方法も提供し、この方法は、式(I)の化合物、そのアイソスターまたは医薬的に許容されるその塩の有効量を前記患者に投与することを含む。
【0130】
1つの実施形態において、本発明の化合物は、HDACの別の既知阻害剤、例えばSAHAと併用することができる。この実施形態において、この併用製品は、同時に、別にまたは逐次的に使用するために各医薬品を含むように調合することができる。
【0131】
従って本発明は、(a)上で定義したとおりの本発明のFK228類似体またはそのアイソスターもしくは医薬的に許容される塩;および(b)同時に、別にまたは逐次的に使用するためのHDACの別の既知阻害剤、例えばSAHAを含む製品を提供する。
【0132】
従って本発明は、HDACの別の既知阻害剤、例えばSAHAとの併用投与で使用するための医薬品の製造において使用するための、上で定義したとおりの本発明のFK228類似体またはそのアイソスターもしくは医薬的に許容される塩の使用も提供する。
【0133】
当業者は、HDACの他の既知阻害剤を知っている。例えば、米国特許第20040266769号に適する例が与えられている。例としては、スピルコスタチンA、FR−901228、トリコスタチンAおよびSAHAが挙げられる。
【0134】
本発明の化合物は、癌の治療と予防の両方に使用することができ、ならびに単独療法または併用療法で使用することができる。併用療法において使用されるとき、本発明の化合物は、一般に、小化合物、放射線、抗体ベースの療法(例えば、ヘルセプチンおよびリツキシマブ)、抗癌剤接種、遺伝子療法、細胞療法、ホルモン療法またはサイトカイン療法と共に使用される。
【0135】
本発明の好ましい実施形態において、本発明の化合物は、癌の治療において別の化学療法薬または抗腫瘍薬と併用される。このような他の化学療法薬または抗腫瘍薬の例としては、ミトキサントロン、ビンカアルカロイド、例えばビンクリスチンおよびビンブラスチン、アントラサイクリン、抗生物質、例えばダウノルビシンおよびドキソルビシン、アルキル化剤、例えばクロラムブシルおよびメルファラン、タキサン、例えばパクリタキセル、抗葉酸剤、例えばメトトレキサートおよびトムデックス、エピポドフィロトキシン、例えばエトポシド、カンプトテシン、例えばイリノテカンおよびその活性代謝産物SN38ならびにDNAメチル化阻害剤、例えばWO 02/085400に開示されているDNAメチル化阻害剤が挙げられる。
【0136】
従って、癌を軽減させるために同時に、別にまたは逐次的に使用するための併用製剤として、本発明の化合物と別の化学療法薬または抗腫瘍薬を含有する製品を、本発明に従って提供する。別の化学療法薬または抗腫瘍薬との併用投与により癌を軽減するために使用する医薬品の製造における、上で定義したFK228類似体またはそのアイソスターまたは医薬的に許容されるその塩の使用も、本発明に従って提供する。本発明の化合物および前記他の薬剤は、任意の順序で投与することができる。これら両方の場合において、本発明の化合物および他の薬剤は、共に投与することができ、または別々に投与する場合には、医師により決定されるような任意の順序で投与することができる。
【0137】
HDACは、いくつかの異なる疾病の病理および/または総合的症状の一因であると考えられるので、阻害による被験者におけるHDAC活性の低減を用いて、これらの疾病に治療的に対処することができる。本発明のHDAC阻害剤を使用して治療することができる様々な疾病の例を本明細書で説明する。本明細書に開示するもの以外のさらなる疾病が、後に、様々な経路においてHDACが果す生物学的役割がさらに完全に理解されるにつれて、同定されることがあることに留意する。
【0138】
本発明のHDAC阻害剤を使用して治療することができる適応症の1つのセットは、望ましくないまたは無制御の細胞増殖を伴うものである。このような適応症としては、良性腫瘍、様々なタイプの癌、例えば原発腫瘍および腫瘍転移、再狭窄(例えば、冠動脈、頚動脈および脳の病変)、内皮細胞の異常刺激(アテローム硬化症)、手術に起因する体組織への傷害、異常創傷治癒、異常血管形成、組織の線維増多を生じさせる疾病、反復運動障害、高度な血管新生がない組織の疾患、ならびに臓器移植に関連した増殖反応が挙げられる。HDAC阻害剤のさらに具体的な適応症としては、前立腺癌、肺癌、急性白血病、多発性骨髄腫、膀胱癌、腎癌、乳癌、結腸直腸癌、神経芽腫および黒色腫が挙げられるが、これらに限定されない。
【0139】
1つの実施形態において、望ましくない、無制御の細胞増殖に関連した疾病を治療するための方法を提供する。この方法は、無制御細胞増殖に罹患している患者に、その無制御細胞増殖が低減されるような、本発明のHDAC阻害剤の治療有効量を投与することを含む。使用される阻害剤の特定の投薬量は、その疾病状態の重症度、投与経路、および担当医により決定され得る関連因子に依存する。一般に、許容される有効な日用量は、無制御細胞増殖を有効に遅速または排除するために十分な量である。
【0140】
本発明のHDAC阻害剤を他の薬剤と併用して、望ましくない無制御の細胞増殖を阻害することもできる。本発明のHDAC阻害剤と併用することができる他の抗細胞増殖剤の例としては、レチノイド酸およびその誘導体、2−メトキシエストラジオール、商標アンジオスタチン(ANGIOSTATIN)タンパク質、商標エンドスタチン(ENDOSTATIN)タンパク質、スラミン、スクアラミン、メタロプロテイナーゼ−Iの組織阻害剤、メタロプロテイナーゼ−2の組織阻害剤、プラスミノゲンアクチベータ阻害剤−1、プラスミノゲンアクチベータ阻害剤−2、軟骨由来阻害剤、パクリタキセル、血小板第4因子、硫酸プロタミン(クルペイン)、硫酸化キチン誘導体(ズワイガニの甲羅(queen crab shells)から調製されたもの)、硫酸化多糖類ペプチドグリカン複合体(sp−pg)、スタウロスポリン、例えばプロリン類似体((1−アゼチジン−2−カルボン酸(LACA)、シスヒドロキシプロリン、d,1−3,4−デヒドロプロリン、チアプロリン)を含めた基質代謝の調節剤、β−アミノプロピオニトリルフマレート、4−プロピル−5−(4−ピリジニル)−2(3H)−オキサゾロン;メトトレキセート、ミトキサントロン、ヘパリン、インターフェロン、2 マクログロブリン−血清、chimp−3、キモスタチン、β−シクロデキストリンテトラデカスルフェート、エポネマイシン;フマギリン、金チオリンゴ酸ナトリウム、d−ペニシラミン(CDPT)、β−1−アンチコラゲナーゼ−血清、α2−抗プラスミン、ビスアントレン、ロベンザリット二ナトリウム、n−(2−カルボキシフェニル−4−クロロアントロニル酸二ナトリウムすなわち「CCA」、サリドマイド;angostatic ステロイド、カルボキシアミノイミダゾール;メタロプロテイナーゼ阻害剤、例えばBB94が挙げられるが、これらに限定されない。使用することができる他の抗血管形成剤としては、抗体、好ましくはこれらの血管由来増殖因子:bFGF、aFGF、FGF−5、VEGFアイソフォーム、VEGF−C、HGF/SFおよびAng−1/Ang−2に対するモノクローナル抗体が挙げられる。Ferrara N.およびAlitalo,K.「血管由来増殖因子およびそれらの阻害剤の臨床適用(Clinical application of angiogenic growth factors and their inhibitors)」(1999)Nature Medicine 5:1359−1364。
【0141】
一般に、良性腫瘍における細胞は、それらの分化特徴を保持し、完全無制御様式では分裂しない。良性腫瘍は、通常、局在し、転移性ではない。本発明のHDAC阻害剤を使用して治療することができる良性腫瘍の具体的なタイプとしては、血管腫、肝細胞腺腫、海綿状血管腫、局在性結節性過形成、聴神経腫、神経線維腫、胆管腺腫、胆管嚢胞腺腫、線維腫、リンパ腫、平滑筋腫、中皮腫、奇形腫、粘液腫、肝小結節再生過形成、トラコーマおよび化膿性肉芽腫が挙げられる。
【0142】
悪性腫瘍の場合、細胞は未分化となり、身体の成長制御シグナルに応答せず、無制御様式で増す。悪性腫瘍は、侵襲性であり、また遠位部位に拡大する(転移する)ことができる。悪性腫瘍は、一般に、2つのカテゴリー:原発腫瘍および続発性腫瘍に分けられる。原発腫瘍は、それらが見出される組織から直接発生する。続発性腫瘍、すなわち転移は、身体の他の場所で起こったが、今は遠隔器官に拡大している腫瘍である。一般的な転移経路は、血管またはリンパ系により拡大する隣接構造への直接増殖、ならびに組織面および体腔(腹水、脳脊髄液など)に沿ってのトラッキングである。
【0143】
本発明のDHAC阻害剤を使用して治療することができる、原発または続発性いずれかの癌または悪性腫瘍の具体的なタイプとしては、白血病、乳癌、皮膚癌、骨癌、前立腺癌、肝癌、肺癌、脳癌、喉頭癌、胆嚢癌、膵臓癌、直腸癌、副甲状腺癌、甲状腺癌、副腎癌、神経組織癌、頭頚部癌、結腸癌、胃癌、気管支癌、腎癌、基底細胞癌、潰瘍性タイプと乳頭状タイプの両方の扁平上皮癌、転移性皮膚癌、骨肉腫、ユーイング肉腫、細網細胞肉腫、骨髄腫、巨細胞腫、小細胞肺癌、胆石、島細胞腫瘍、原発性脳腫瘍、急性および慢性リンパ球性および顆粒球性腫瘍、ヘアリーセル腫瘍、腺腫、過形成、髄様癌、褐色細胞腫、粘膜神経腫、腸神経節細胞種、過形成性角膜神経腫瘍、マルファン様体型腫瘍、ウィルムス腫瘍、精上皮腫、卵巣腫瘍、平滑筋腫、子宮頚部形成異常および上皮内癌、神経芽細胞種、網膜芽腫、軟部組織肉腫、悪性カルチノイド、局所的皮膚病変、菌状息肉腫、横紋筋肉腫、カポジ肉腫、骨原性および他の肉腫、悪性高カルシウム血症、腎細胞腫瘍、真性多血症、腺癌、多形性神経膠芽腫、白血病、リンパ腫、悪性黒色腫、類表皮癌、ならびに他の癌腫および肉腫が挙げられるが、これらに限定されない。
【0144】
本発明のHDAC阻害剤を使用して、手術中の体組織への傷害に起因する異常細胞増殖を治療することもできる。これらの傷害は、様々な手術手順、例えば関節手術、腸手術およびケロイド瘢痕化の結果として生じ得る。線維症性組織を生じさせる疾病としては、気腫が挙げられる。本発明を用いて治療することができる反復運動障害としては、手根管症候群が挙げられる。本発明を用いて治療することができる細胞増殖性障害の一例は、骨腫瘍である。
【0145】
本発明のHDAC阻害剤を使用して治療することができる、臓器移植に関連した増殖応答としては、潜在的臓器拒絶反応または関連併発症の一因となる増殖応答が挙げられる。具体的には、これらの増殖応答は、心臓、肺、肝臓、腎臓および他の体器官または器官系の移植中に発生し得る。
【0146】
本発明を用いて治療することができる異常血管形成としては、関節リウマチ、虚血性再潅流関連脳浮腫および傷害、皮質性虚血、卵巣過形成および血管過多、(多嚢胞性卵巣症候群)、子宮内膜症、乾癬、糖尿病性網膜症、ならびに他の眼球の血管由来の疾病、例えば未熟児網膜症(水晶体後線維形成性)、黄斑変性、角膜移植後拒絶反応、神経血管性緑内障およびオスター・ウェーバー症候群に随伴する異常血管形成が挙げられる。
【0147】
本発明に従って治療することができる無制御血管形成に関連した疾病の例としては、網膜/脈絡膜血管新生および角膜血管新生が挙げられるが、これらに限定されない。網膜/脈絡膜血管新生の例としては、ベスト病、近視、眼陥凹、ジュタルガルト病、パジェット病、静脈閉塞症、動脈閉塞症、鎌状赤血球貧血、サルコイド、梅毒、弾性線維性仮性黄色腫頚動脈apo構造疾患、慢性ブドウ膜炎/硝子体炎、マイコバクテリア感染症、ライム病、全身性エリテマトーデス、未熟児網膜症、イールズ病、糖尿病性網膜症、黄斑変性、ベーチェット病、網膜炎または脈絡膜炎に起因する感染、推定眼ヒストプラズマ症、扁平部炎、慢性網膜剥離、過粘稠度症候群、トキソプラズマ症、外傷およびレーザー後併発症、ルベオーシスに関連した疾病(隅角の血管新生)、ならびにすべての形態の増殖性硝子体網膜症を含む線維血管性組織または線維組織の異常増殖に起因する疾病が挙げられるが、これらに限定されない。角膜血管新生の例としては、流行性角結膜炎、ビタミンA欠乏症、コンタクトレンズ装着時間過剰(contact lens overwear)、アトピー性角膜炎、上方輪部角膜炎、翼状片乾性角膜炎、シューグレン病、酒さ性角膜炎、フクテリン症、糖尿病性網膜症、未熟児網膜症、角膜移植後拒絶反応、モーレン潰瘍、テリエン辺縁変性、辺縁表皮剥離(marginal keratolysis)、多発性動脈炎、ウェグナー病、強膜炎、類天疱瘡の放射状角膜切開、血管新生緑内障および水晶体後方線維増殖、梅毒、マイコバクテリア感染症、脂質変性、化学熱傷、細菌性潰瘍、真菌性潰瘍、単純疱疹感染症、帯状疱疹感染症、原虫感染症ならびにカポジ肉腫が挙げられるが、これらに限定されない。
【0148】
無制御の血管形成に関連した慢性炎症性疾患も、本発明のHDAC阻害剤を使用して治療することができる。慢性炎症は、炎症性細胞の流入を維持する毛細血管の芽の継続的形成に依存する。炎症細胞の流入および存在が肉芽を生じさせ、そうして慢性炎症状態を維持する。HDAC阻害剤を単独で使用または他の抗炎症薬と併用して血管形成を阻害することにより、肉芽形成を予防することができ、従って、疾病を軽減させることができる。慢性炎症性疾患の例としては、炎症性腸疾患、例えばクローン病および潰瘍性大腸炎、乾癬、サルコイドーシス、および関節リウマチが挙げられるが、これらに限定されない。
【0149】
炎症性腸疾患、例えば、クローン病および潰瘍性大腸炎は、胃腸管内の様々な部位における慢性炎症および血管形成を特徴とする。例えば、クローン病は、最も一般的には遠位回腸および結腸に影響及ぼす慢性経壁性炎症性疾患として発生するが、口から肛門および肛門周囲領域までの胃腸管のあらゆる部分においても発生し得る。クローン病の患者は、一般に、腹痛、発熱、食欲低下、体重減少および腹部膨満を伴う慢性下痢を有する。潰瘍性大腸炎は、結腸粘膜において発生する慢性で非特異的な炎症性および潰瘍性疾患でもあり、血性下痢の存在を特徴とする。一般に、これらの炎症性腸疾患は、炎症性細胞の柱体によって包囲された新たな毛細血管の芽を伴う、胃腸管全体にわたる慢性肉芽腫性炎症によって引き起こされる。これらの阻害剤による血管形成の阻害は、それらの芽の形成を阻害し、肉芽腫の形成を防止するはずである。炎症性腸疾患は、皮膚病変などの追加の腸性症状発現も示す。このような病変は、炎症および血管形成を特徴とし、胃腸管以外の多数の部位で発生し得る。本発明のHDAC阻害剤による血管形成の阻害は、炎症性細胞の流入を低減させ、病変形成を予防することができる。
【0150】
サルコイドーシスはもう1つの慢性炎症性疾患であり、これは多系統型肉芽腫性疾患として特徴付けられる。この疾病の肉芽腫は、身体のあらゆる場所で形成し得る。従って、それらの症状は、肉芽腫の部位、およびその疾病が活性であるかどうかに依存する。肉芽腫は、炎症性細胞の恒常的供給をもたらす血管形成性毛細血管芽によって作られる。血管形成を阻害する本発明のHDAC阻害剤の使用により、そのような肉芽腫形成を阻害することができる。乾癬も慢性および再発性炎症性疾患であり、これは、様々なサイズの丘疹およびプラークを特徴とする。これらの阻害剤を単独で使用または他の抗炎症薬と併用することによる治療は、それらの特徴的病変を維持するために必要な新たな血管の形成を防止し、患者の症状を軽減させるはずである。
【0151】
関節リウマチ(RA)も、末梢関節の非特異的炎症を特徴とする慢性炎症性疾患である。関節滑膜表層内の血管が、血管形成されると考えられている。新たな血管網の形成に加えて、内皮細胞が、パンヌス成長および軟骨破壊を招く因子および反応性酸素種を放出する。血管形成に関与する因子は、関節リウマチの慢性炎症状態に積極的に貢献し、前記状態の維持を助長することができる。本発明のHDAC阻害剤を単独で使用または他の抗RA剤と併用することによる治療は、慢性炎症を維持するために必要な新たな血管の形成を防止することができる。
【0152】
さらに、本発明の化合物は、心臓/血管系疾患、例えば、肥大、高血圧症、心筋梗塞、再潅流、虚血性心疾患、アンギナ、不整脈、高コレステロール血症、アテローム硬化症および卒中の治療において使用することができる。さらに、これらの化合物は、神経変性疾患/CNS障害、例えば、卒中、ハンチングトン病、筋萎縮性側索硬化症およびアルツハイマー病を含む急性および慢性神経疾患を治療するために使用することができる。
【0153】
本発明の化合物は、抗微生物剤、例えば抗菌剤として使用することもできる。従って本発明は、細菌感染症の治療において使用するための化合物も提供する。本発明の化合物は、ウイルス、細菌、真菌および寄生虫感染に対する抗感染症化合物として使用することができる。感染症の例としては、(プラスモディウム属(plasmodium)、クリプトスポリジウム・パルヴム(cryptosporidium parvum)、トキソプラズマ・ゴンヂ(toxoplasma gondii)、サルコシスティス・ニューロナ(sarcocystis neurona)およびエイメリア属(Eimeria)種を含む)原虫寄生虫感染症が挙げられる。
【0154】
本発明の化合物は、望ましくないまたは無制御の細胞増殖の治療に、好ましくは良性腫瘍/過形成および悪性腫瘍の治療に、さらに好ましくは悪性腫瘍の治療に、ならびに最も好ましくはCCL、乳癌およびT細胞リンパ腫の治療に、特に適する。
【0155】
本発明の好ましい実施形態において、本発明の化合物は、癌、心肥大症、慢性心不全、炎症状態、心血管疾患、ヘモグロビン異常症、サラセミア、鎌状赤血球症、CNS障害、自己免疫疾患、糖尿病、骨粗しょう症、MDS、良性前立腺肥大症、口腔白板症、遺伝的代謝性疾患、感染症、ルビンス−テイビ(Rubens−Taybi)、脆弱X症候群もしくはα−1アンチトリプシン欠損症を軽減させるために、または創傷治癒を加速させるために、または毛包を保護するために、または免疫抑制薬として使用される。
【0156】
一般に、前記炎症状態は、皮膚の炎症状態(例えば、乾癬、アクネおよび湿疹)、喘息、慢性閉塞性肺疾患(COPD)、関節リウマチ(RA)、炎症性腸疾患(IBD)、クローン病または大腸炎である。
【0157】
一般に、前記癌は、慢性リンパ球性白血病、乳癌、前立腺癌、卵巣癌、中皮腫またはT細胞リンパ腫である。
【0158】
一般に、前記心血管疾患は、高血圧症(hypertension)、心筋梗塞(MI)、虚血性心疾患(IHD)(再潅流)、狭心症、不整脈、高コレステロール血症、高脂血症、アテローム硬化症、卒中、心筋炎、うっ血性心不全、原発性および続発性ie拡張型(うっ血型)心筋症、肥大性心筋症、収縮性心筋症、末梢血管疾患、頻脈、高血圧(high blood pressure)または血栓症である。
【0159】
一般に、前記遺伝的代謝性疾患は、嚢胞性線維症(CF)、ペルオキソーム形成不全(peroxisome biogenesis disorder)または副腎性白質ジストロフィーである。
【0160】
一般に、本発明の化合物は、臓器移植後に免疫抑制薬として使用される。
【0161】
一般に、前記感染症は、ウイルス、細菌、真菌または寄生虫感染症、特に、黄色ブドウ球菌(S.aureus)、瘡プロピオニバクテリウム(P.acne)、カンジダ属またはアスペルギルス属による感染症である。
【0162】
一般に、前記CNS障害は、ハンチングトン病、アルツハイマー病、多発性硬化症または筋萎縮性側索硬化症である。
【0163】
好ましくは、この実施形態において、本発明の化合物は、癌、心肥大症、慢性心不全、炎症状態、心血管疾患、ヘモグロビン異常症、サラセミア、鎌状赤血球症、CNS障害、自己免疫疾患、糖尿病または骨粗しょう症を軽減するために使用されるか、免疫抑制薬として使用される。
【0164】
最も好ましくは、本発明の化合物は、慢性リンパ球性白血病、乳癌、前立腺癌、卵巣癌、中皮腫、T細胞リンパ腫、心肥大症、慢性心不全または皮膚の炎症状態、特に、乾癬、アクネもしくは湿疹を軽減するために使用される。
【0165】
本発明の化合物は、動物の治療、好ましくは哺乳動物の治療、さらに好ましくはヒトの治療に使用することができる。
【0166】
本発明の化合物は、適切な場合には、このような状態の発生率を低下させるために予防的に使用することができる。
【0167】
本発明の化合物の治療有効量を患者に投与する。典型的な用量は、その具体的な化合物の活性、治療すべき被験者の年齢、体重および状態、その疾病のタイプおよび重症度、ならびに投与頻度および経路に従って、体重のkg当たり約0.001から50mgである。好ましくは、日用量レベルは、5mgから2gである。
【0168】
以下の実施例は、本発明を説明するものである。しかし、これらは、いかなる点においても本発明を限定するものではない。これに関連して、実施例セクションにおいて用いている特定のアッセイは、HDACの阻害に関する活性の指標を提供するためだけに設計されたものであること理解することが重要である。所与の化合物のHDAC拮抗薬としての活性を判定するために利用できるアッセイは多数あり、従って、いずれか1つの特定のアッセイにおける否定的な結果に決定力があるわけではない。
【0169】
[実施例]
(E)−(1S,7R,10S,21R)−7−イソプロピル−21−メチル−2−オキサ−12,13−ジチア−5,8,20,23−テトラアザ−ビシクロ[8.7.6]トリコス−16−エン−3,6,9,19,22−ペンタオン(以後、化合物001と呼ぶ)は、下に示す図式を用いて調製した。
【0170】
【化36】
【0171】
[(R)−2−(9H−フルオレン−9−イルメトキシカルボニルアミノ)−3−メチル−ブチリルアミノ]−酢酸メチルエステル(2)の調製
CH2Cl2(50mL)中のFmoc保護D−バリン(2.70g、7.96mmol)の攪拌溶液に、EDAC・HCl(1.83g、9.55mmol)、HOBt(1.30g、9.55mmol)およびDIEA(3.8mL、27.86mmol)を添加した。室温で15分間攪拌した後、グリシンメチルエステル(1、1.0g、7.96mmol)を添加し、その反応混合物を4時間攪拌し、CH2Cl2(50mL)で希釈し、水(25mL)、10% HCl(25mL)、5% NaHCO3(25mL)および飽和NaCl(25mL)溶液で洗浄し、乾燥させ(Na2SO4)、濾過し、濃縮して、オフホワイトの固体を得、それをCH3CNから再結晶させて、2を白色の固体として得た(2.81g、86%):融点=148〜150℃;[α]22D−7.32(c 0.50、CHCl3);[α]22D−7.32(c 0.50、CHCl3);IR νmax 3287、1750、1690、1649、1535cm-1;1H NMR 400MHz 7.75(d,J=7.5Hz,2H)、7.57(d,J=7.0Hz,2H)、7.39(t,J=7.0Hz,2H)、7.28(m,2H)、6.54(s,1H)、5.44(s,1H)、7.43−4.38(m,2H)、4.21(t,J=7.0Hz,1H)、4.01−3.96(m,3H)、3.72(s,3H)、2.16(m,1H)、0.96(t,J=9.0Hz,6H);13C NMR 100MHz 171.7(C)、170.1(C)、156.6(C)、143.9(C)、141.4(C)、127.8(CH)、127.2(CH)、125.2(CH)、120.1(CH)、67.2(CH2)、60.4(CH)、52.5(CH)、47.3(CH3)、41.2(CH2)、31.2(CH)、19.3(CH3)、17.9(CH);MS m/z 432.9(M+Na)+、842.8(2M+Na)+。
【0172】
{(R)−2−[(S)−2−(9H−フルオレン−9−イルメトキシカルボニルアミノ)−3−(トリチルスルファニル)−プロピオニルアミノ]−3−メチル−ブチリルアミノ}−酢酸メチルエステル(3)の調製
室温でCH3CN(48.5mL)中の2(1.0g、2.44mmol)の攪拌溶液に、Et2NH(2.5mL)を添加した。3時間、室温で攪拌した後、その反応混合物をヘキサン(100mL)で希釈し、濃縮して、粗製アミンを無色の油として得た。CH3CN(25mL)中のFmoc−(STrt)−D−システイン(1.70g、2.9mmol)の攪拌溶液に、EDAC・HCl(561.0mg、2.93mmol)、HOBt(396mg、2.93mmol)およびDIEA(1.31mL、7.32mmol)を添加した。室温で15分間攪拌した後、その粗製アミンを添加し、その反応混合物を4時間攪拌し、溶媒を除去し、残留物をCH2Cl2(100mL)に溶解し、水(25mL)、10% HCl(25mL)、5% NaHCO3(25mL)および飽和NaCl(25mL)溶液で洗浄し、乾燥させ(Na2SO4)、濾過し、溶媒を除去して、オフホワイトの固体を得、それをCH3CNから再結晶させて、3を白色の固体として得た(1.46g、79%):[α]22D−2.35(c 0.50、CHCl3);IR νmax 3267、1646、1543cm-1;1H NMR 400MHz 7.76(t,J=7.0Hz,2H)、7.54(d,J=6.5Hz,2H)、7.39−7.21(m 19H)、6.87(s,1H)、6.23(d,J=8.0,2H)、5.04(d,J=7.0,1H)、4.37(d,J=7.0,2H)、4.29(dd,J=8.6,J=5.0,1H)、4.17(t,J=6.5,1H)、3.96(m,1H)、3.72(dd,,J=18.1,J=5.0,1H)、3.65(s,3H)、3.60(m,1H)、2.70(d,J=6.5,2H)、2.30(m,1H)、1.70(s,1H)、0.87(dd,J=13.0,J=7.0Hz,6H);13C NMR 100MHz 170.6(C)、170.5(C)、170.1(C)、156.2(C)、144.3(C)、143.8(C)、143.7(C)141.4(C)、129.6(CH)、128.4(CH)、128.0(CH)、127.8(CH)、127.2(CH)、125.1(CH)、120.2(CH)、67.6(CH)、67.2(CH2)、58.4(CH)、54.3(CH)、52.4(CH3)、47.1(CH)、40.9(CH2)、33.6(CH2)、30.0(CH)、19.4(CH3)、17.4(CH3);MS m/z 777.8(M+Na)+。C45H45N3O6Sについての分析計算値:71.50;H,6.00;N,5.56。実測値 C,71.42;H,5.99;N,5.55。
【0173】
((R)−2−{(S)−2−[(R)−2−(9H−フルオレン−9−イルメトキシカルボニルアミノ)−プロピオニルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(4)の調製
室温でCH3CN(30mL)中の3(400mg、0.53mmol)の攪拌溶液に、Et2NH(1.5mL)を添加した。3時間、室温で攪拌した後、その反応混合物をヘプタン(60mL)で希釈し、濃縮して、粗製アミンを無色の油として得た。CH3CN(25mL)中のFmoc−D−アラニン(198mg、0.636mmol)の攪拌溶液に、EDAC・HCl(122.0mg、0.634mmol)、HOBt(86mg、0.636mmol)およびDIEA(502μL、1.91mmol)を添加した。室温で15分間攪拌した後、その粗製アミンを添加し、その反応混合物を18時間攪拌し、溶媒を除去し、残留物をCH2Cl2(50mL)に溶解し、水(15mL)、10% HCl(15mL)、5% NaHCO3(15mL)および飽和NaCl(15mL)溶液で洗浄し、乾燥させ(Na2SO4)、濾過し、溶媒を除去して、オフホワイトの固体を得、それをCH3CNから再結晶させて、4を白色の固体として得た(670mg、81%):融点=195〜197℃;[α]22D +5.1(c 0.50、CHCl3);IR νmax 3267、3054、1744、1706、1635、1531.1cm-1;1H NMR 400MHz 7.75(d,J=7.5Hz,2H)、7.52(d,J=7.0Hz,2H)、7.39(m,7H)、7.26−7.15(m,13H)、6.79(s,1H)、6.62(s,1H)、5.47(s,1H)、4.43−4.28(m,4H)、4.13(m,1H)、4.01(m,1H)、3.88(s,2H)、3.63(s,3H)、2.81(m,1H)、2.52(m,1H)、2.26,(m,1H)、1.30(d,J=6.0Hz,3H)、0.94(d,J=6.0Hz,3H)、0.89(d,J=7.0Hz,3H);13C NMR 100MHz 172.7、171.1、170.2、156.1、144.4、143.9、143.8、141.4、129.6、128.3、127.9、127.2、127.1、125.1、120.1、58.5、52.7、52.3、50.8、47.2、41.0、33.5、30.4、19.3、19.0、17.7;MS m/z 849.2(M+Na)+、865.1(M+K)+。
【0174】
((R)−2−{(S)−2−[(R)−2−((E)−(S)−3−ヒドロキシ−7−トリチルスルファニル−ヘプト−4−エノイルアミノ)−プロピオニルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(6)の調製
【0175】
【化37】
【0176】
室温でCH3CN(10mL)中の4(137mg、0.166mmol)の攪拌溶液に、Et2NH(0.5mL)を添加した。5時間、室温で攪拌した後、その反応混合物をヘプタン(30mL)で希釈し、溶媒を除去し、CH2Cl2(10mL)を添加し、濾過し、濃縮して、粗製アミンを無色の油として得た。CH2Cl2(4mL)中のその粗製アミンの攪拌溶液に、0℃で5(84.0mg、0.149mmol、Yurek−George,A.;Habens,F.;Brimmell,M.;Packham,G.;Ganesan,A.J.Am.Chem.Soc.2004,126,1030−1031における手順に従って調製したもの)およびDMAP(2.2mg、0.0176mmol)を添加した。室温で8時間攪拌した後、溶媒を除去し、残留物をフラッシュクロマトグラフィー(溶離剤 30〜40% EtOAc/ヘキサン)によって精製して、6を白色のガラスとして得た(127mg、85%):融点=191〜193℃;[α]22D−18.0(c 0.50、CHCl3);IR νmax 3272、3064、1758、1692、1621cm-1;1H NMR 400MHz(5% CD3OD/CDCl3)7.40(m,12H)、7.25(m,12H)、7.20(m,6H)、6.96および6.88(不安定なNH,d,J=8.0Hz,1H)、5.49(dt,J=15.0Hz,6.5Hz,1H)、5.37(dd,J=15.5Hz,6.0Hz,1H)、4.32(m,2H)、4.22(d,J=6.0Hz,1H)、3.98(t,J=7.0Hz,1H)、3.92(d,J=18.1Hz,1H)、3.72(d,J=17.6Hz,1H)、3.67(s,3H)、2.64(dd,J=13.0Hz,7.5Hz,1H)、2.58(dd,J=13.0Hz,7.5Hz,1H)、2.56−2.18(m,9H)、2.11(q,J=6.5Hz,2H)、1.31(d,J=7.5Hz,3H)、0.91(t,J=7.0Hz,6H);13C NMR 100MHz(5% CD3OD/CDCl3)173.0、172.0、171.3、170.5、170.2、144.9、144.2、132.8、129.9、129.7、129.6、129.5、128.2、128.1、127.9、127.1、126.7、69.6、67.1、66.7、58.9、58.8、52.7、52.2、50.0、49.8、49.6、49.5、49.4、49.1、43.7、40.9、40.8、33.1、31.5、31.3、30.0、19.2、17.5;MS m/z 1050.4(M+2Na)+。
【0177】
((R)−2−{(S)−2−[(R)−2−((E)−(S)−3−ヒドロキシ−7−トリチルスルファニル−ヘプト−4−エノイルアミノ)−プロピオニルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸(7)の調製
0℃で4:1 THF/H2O(4.0mL)中の6(220mg、0.222mmol)の攪拌溶液に、LiOH(11mg、0.440mmol)を添加した。1時間攪拌した後、その反応混合物をH2O(30mL)で希釈し、2M KHSO4でpH4〜5に酸性化し、EtOAc(3×30mL)で抽出した。有機層を飽和NaCl(15mL)で洗浄し、乾燥させ(Na2SO4)、濾過し、濃縮して、白色の固体を得、それをエーテルと研和して、7を白色の固体として得(199.5mg、91%)、それをそのまま次の段階で直接使用した。融点=191〜193℃;[α]22D−18.5(c 0.50、CHCl3);IR νmax 3413、1711、1678、1630、1451cm-1;1H NMR 400MHz(5% CD3OD/CDCl3)7.30(m,12H)、7.16(m,12H)、7.13(m,6H)、5.43(dt,J=15.5Hz,6.0Hz,1H)、5.30(dd,J=15.5Hz,6.0Hz,1H)、4.27(m,2H)、4.21(m,1H)、3.99(m,1H)、3.75(s,2H)、3.77(m,br,6H)、2.55(dd,J=12.5Hz,6.5Hz,1H)、2.44(dd,J=12.5Hz,7.5Hz,1H)、2.21(m,2H)、2.12(m,3H)、2.02(m,2H)、1.23(d,J=7.0Hz,3H)、0.83(d,J=7.0Hz,3H)、0.80(d,J=7.0Hz,3H);13C NMR 100MHz(5% CD3OD/CDCl3)173.0、172.1、171.6、171.5、170.5、144.9、144.3、132.7、129.8、129.6、129.5、128.2、127.9、127.0、126.7、69.5、67.1、66.7、58.7、52.7、43.5、40.9、33.2、31.5、31.3、30.3、19.2、19.1、17.6;MS m/z 1013.2(M+Na)+。
【0178】
(6R,9S,12R,16S)−6−イソプロピル−12−メチル−16−((E)−4−トリチルスルファニル−ブト−1−エニル)−9−トリチルスルファニルメチル−1−オキサ−4,7,10,13−テトラアザ−シクロヘキサデカン−2,5,8,11,14−ペンタオン(8)の調製
0℃でCH2Cl2(5mL)中のヒドロキシル酸7(100mg、0.102mmol)の攪拌溶液に、CH2Cl2(0.5mL)中のDCC(28.2mg、0.137mmol)の溶液を1滴ずつ添加した。30分間、0℃で攪拌した後、CH2Cl2(51mL)中のDMAP(2.5mg、0.0213mmol)の溶液を添加し、その後、16時間、室温で攪拌した。固体沈殿物を濾過して除去し、その濾液をCH2Cl2(5mL)で洗浄した。有機層を水(10mL)、5% KHSO4(10mL)、5% NaHCO3(10mL)および飽和NaCl(10mL)溶液で洗浄し、乾燥させ(Na2SO4)、濾過し、濃縮した。残留物をフラッシュクロマトグラフィー(溶離剤 30〜50% EtOAc/ヘキサン)によって精製して、8を白色のガラスとして得た(37mg、38%)。
【0179】
(E)−(1S,7R,10S,21R)−7−イソプロピル−21−メチル−2−オキサ−12,13−ジチア−5,8,20,23−テトラアザ−ビシクロ[8.7.6]トリコス−16−エン−3,6,9,19,22−ペンタオン(化合物001)の調製
MeOH(50mL)中のI2(37.1mg、0.146mmol)の強力攪拌溶液に、MeOH(20mL)中の保護ジオール8(35mg、0.0365mmol)を5分かけて1滴ずつ添加した。さらに10分間攪拌した後、0.2M クエン酸緩衝液(4mL)中の0.2M アスコルベートを添加し、有機相を濃縮し、1:1 EtOAc/NaCl(20mL)を添加し、EtOAc(5×25mL)で抽出し、合わせた有機抽出物を乾燥させ(Na2SO4)、濾過し、溶媒を除去した。残留物をフラッシュクロマトグラフィー(溶離剤 1〜6% MeOH/CHCl3)によって精製して、化合物001を白色の固体として得た(11.8mg、67%):[α]22D−98.0(c 0.50、CHCl3);IR νmax 3300、2477、1734、1659、1526、1446cm-1;1H NMR 400MHz 7.41(d,J=6.0Hz,1H)、7.23(d,J=8.5Hz,1H)、6.37(s,1H)、5.94(dt,J=16.5,7.5Hz,1H)、5.78−5.71(m,1H)、4.86(m,1H)、4.26(d,J=7.5Hz,1H)、4.17(d,J=14.0Hz,1H)、4.08(d,J=14.0Hz,1H)、3.44(dd,J=14.5Hz,10.0Hz,1H)、3.22(d,J=10.0Hz,1H)、2.88−2.56(m,8H)、1.49(d,J=7.5Hz,3H)、0.98(d,J=6.5Hz,3H)、0.92(d,J=6.5Hz,3H);13C NMR 100MHz 173.2、171.9、170.9、170.1、168.3、129.8、70.2、64.4、55.9、51.7、41.7、38.2、37.9、35.7、31.0、26.9、20.2、19.7、15.4;HRMS m/z 509.1495(M+Na)+ 予測値 509.1499。
【0180】
式(I)によって表される類似体の合成のための一般手順。
【0181】
段階(a)において、側鎖R2を有するアミノ酸エステルを、側鎖R1を有する第二のアミノ酸と縮合させて、ジペプチドを得る。段階(b)において、そのジペプチドを保護システイン誘導体と縮合させて、トリペプチドを得る。段階(c)において、そのトリペプチドをアミノ酸とカップリングさせて、保護テトラペプチドを生じさせる。段階(d)において、そのテトラペプチドのN末端を脱保護し、その遊離アミンをβ−ヒドロキシ酸誘導体とカップリングさせる。本発明の好ましい実施形態では、R4およびR5は、Hであり、Xは、Yurek−George,A.;Habens,F.;Brimmell,M.;Packham,G.;Ganesan,A.J.Am.Chem.Soc.2004,126,1030−1031において報告されているようなキラル補助基である。段階(e)においてそのエステル基を加水分解し、その後、段階(f)において環化し、(g)においてジスルフィド結合を形成して、二環式デプシペプチド化合物(I)の合成を完了する。
【0182】
【化38】
【0183】
一般図式による類似体9A〜Fの合成。
【0184】
【化39】
【0185】
[(R)−2−(9H−フルオレン−9−イルメトキシカルボニルアミノ)−3−メチル−ブチリルアミノ]−酢酸メチルエステル(2A)の調製。
【0186】
CH2Cl2(50mL)中のFmoc保護D−バリン(2.70g、7.96mmol)の攪拌溶液に、EDAC・HCl(1.83g、9.55mmol)、HOBt(1.30g、9.55mmol)およびDIEA(3.8mL、27.86mmol)を添加した。室温で15分間攪拌した後、グリシンメチルエステル(1A、1.0g、7.96mmol)を添加し、その反応混合物を4時間攪拌し、CH2Cl2(50mL)で希釈し、水(25mL)、10% HCl(25mL)、5% NaHCO3(25mL)および飽和NaCl(25mL)溶液で洗浄し、乾燥させ(Na2SO4)、濾過し、濃縮して、オフホワイトの固体を得、それをCH3CNから再結晶させて、2Aを白色の固体として得た(2.81g、86%):融点=148〜150℃;[α]22D−7.32(c 0.50、CHCl3);IR νmax 3287、1750、1690、1649、1535cm-1;1H NMR(400MHz,CDCl3)δ7.75(d,J=7.5Hz,2H)、7.57(d,J=7.0Hz,2H)、7.39(t,J=7.0Hz,2H)、7.28(m,2H)、6.54(s,1H)、5.44(s,1H)、4.43−4.38(m,2H)、4.21(t,J=7.0Hz,1H)、4.01−3.96(m,3H)、3.72(s,3H)、2.16(m,1H)、0.96(t,J=9.0Hz,6H);13C NMR(100MHz,CDCl3)δ171.7(C)、170.1(C)、156.6(C)、143.9(C)、141.4(C)、127.8(CH)、127.2(CH)、125.2(CH)、120.1(CH)、67.2(CH2)、60.4(CH)、52.5(CH)、47.3(CH3)、41.2 (CH2)、31.2(CH)、19.3(CH3)、17.9(CH);MS m/z 842.8(30%,[2M+Na]+)、432.9(100%,[M+Na]+)。
【0187】
{(R)−2−[(S)−2−(9H−フルオレン−9−イルメトキシカルボニルアミノ)−3−(トリチルスルファニル)−プロピオニルアミノ]−3−メチル−ブチリルアミノ}−酢酸メチルエステル(3A)の調製。
【0188】
室温でCH3CN(48.5mL)中の2A(1.0g、2.44mmol)の攪拌溶液に、Et2NH(2.5mL)を添加した。3時間、室温で攪拌した後、その反応混合物をヘキサン(100mL)で希釈し、濃縮して、粗製アミンを無色の油として得た。CH3CN(25mL)中のFmoc−(STrt)−D−システイン(1.70g、2.9mmol)の攪拌溶液に、EDAC・HCl(561.0mg、2.93mmol)、HOBt(396mg、2.93mmol)およびDIEA(1.31mL、7.32mmol)を添加した。室温で15分間攪拌した後、その粗製アミンを添加し、その反応混合物を4時間攪拌し、溶媒を除去し、残留物をCH2Cl2(100mL)に溶解し、水(25mL)、10% HCl(25mL)、5% NaHCO3(25mL)および飽和NaCl(25mL)溶液で洗浄し、乾燥させ(Na2SO4)、濾過し、溶媒を除去して、オフホワイトの固体を得、それをCH3CNから再結晶させて、3Aを白色の固体として得た(1.46g、79%):[α]22D−2.35(c 0.50、CHCl3);IR νmax 3267、1646、1543cm-1;1H NMR 400MHz(400MHz,CDCl3)δ7.76(t,J=7.0Hz,2H)、7.54(d,J=6.5Hz,2H)、7.39−7.21(m 19H)、6.87(s,1H)、6.23(d,J=8.0,2H)、5.04(d,J=7.0,1H)、4.37(d,J=7.0,2H)、4.29(dd,J=8.6,J=5.0,1H)、4.17(t,J=6.5,1H)、3.96(m,1H)、3.72(dd,,J=18.1,5.0,1H)、3.65(s,3H)、3.60(m,1H)、2.70(d,J=6.5,2H)、2.30(m,1H)、1.70(s,1H)、0.87(dd,J=13.0,J=7.0Hz,6H);13C NMR(100MHz,CDCl3)δ170.6(C)、170.5(C)、170.1(C)、156.2(C)、144.3(C)、143.8(C)、143.7(C)141.4(C)、129.6(CH)、128.4(CH)、128.0(CH)、127.8(CH)、127.2(CH)、125.1(CH)、120.2(CH)、67.6(CH)、67.2(CH2)、58.4(CH)、54.3(CH)、52.4(CH3)、47.1(CH)、40.9(CH2)、33.6(CH2)、30.0(CH)、19.4(CH3)、17.4(CH3);LRMS m/z 777.8(100%,[M+Na]+)。C45H45N3O6Sについての分析計算値:71.50;H,6.00;N,5.56。実測値 C,71.42;H,5.99;N,5.55。
【0189】
((R)−2−{(S)−2−[(R)−2−(9H−フルオレン−9−イルメトキシカルボニルアミノ)−プロピオニルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(4A)の調製。
【0190】
室温でCH3CN/CH2Cl2(1:1,60mL)中の3A(900mg、1.19mmol)の攪拌溶液に、Et2NH(3mL)を添加した。3時間、室温で攪拌した後、その反応混合物をヘプタン(60mL)で希釈し、濃縮して、粗製アミンを無色の油として得た。CH2Cl2(30mL)中のFmoc−D−アラニン(529mg、1.7mmol)の攪拌溶液に、EDAC・HCl(326mg、1.7mmol)、HOBt(230mg、1.7mmol)およびDIEA(627μL、3.6mmol)を添加した。室温で10分間攪拌した後、CH2Cl2(20mL)中のその粗製アミンの溶液を添加し、その反応混合物を18時間攪拌した。その後、その反応物を水(15mL)、10% HCl(15mL)、5% NaHCO3(15mL)および飽和NaCl(15mL)溶液で洗浄し、乾燥させ(Na2SO4)、濾過し、溶媒を除去して、オフホワイトの固体を得、それをCH3CNから再結晶させて、4Aを白色の固体として得た(810mg、0.98mmol、82%):融点=195〜197℃;[α]22D +5.1(c 0.50、CHCl3);IR νmax 3267、3054、1744、1706、1635、1531.1cm-1;1H NMR(400MHz,CDCl3)δ7.75(d,J=7.5Hz,2H)、7.52(d,J=7.0Hz,2H)、7.39(m,7H)、7.26−7.15(m,13H)、6.79(s,1H)、6.62(s,1H)、5.47(s,1H)、4.43−4.28(m,4H)、4.13(m,1H)、4.01(m,1H)、3.88(s,2H)、3.63(s,3H)、2.81(m,1H)、2.52(m,1H)、2.26,(m,1H)、1.30(d,J=6.0Hz,3H)、0.94(d,J=6.0Hz,3H)、0.89(d,J=7.0Hz,3H);13C NMR(100MHz,CDCl3)δ172.7、171.1、170.2、156.1、144.4、143.9、143.8、141.4、129.6、128.3、127.9、127.2、127.1、125.1、120.1、58.5、52.7、52.3、50.8、47.2、41.0、33.5、30.4、19.3、19.0、17.7;LRMS m/z 849.2(100%,[M+Na]+)、865.1(20%,[M+K]+)。
【0191】
((R)−2−{(S)−2−[(R)−2−((E)−(S)−3−ヒドロキシ−7−トリチルスルファニル−ヘプト−4−エノイルアミノ)−プロピオニルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(6A)の調製。
【0192】
室温でCH2Cl2/CH3CN(3:2,150mL)中の4A(760mg、0.92mmol)の攪拌溶液に、Et2NH(7.5mL)を添加した。5時間、室温で攪拌した後、その反応混合物をヘプタン(60mL)で希釈し、溶媒を除去し、CH2Cl2(50mL)を添加し、濾過し、濃縮して、粗製アミンを無色の油として得た。CH2Cl2(30mL)中のその粗製アミンの攪拌溶液に、室温でCH2Cl2(5mL)中の5(672mg、1.20mmol、Yurek−George,A.;Habens,F.;Brimmell,M.;Packham,G.;Ganesan,A.J.Am.Chem.Soc.2004,126,1030−1031における手順に従って調製したもの)およびDMAP(15mg、0.12mmol)及びDMAP(15mg、0.12mmol)を添加した。室温で12時間攪拌した後、溶媒を除去し、残留物をフラッシュクロマトグラフィー(溶離剤 10〜100% EtOAc/CH2Cl2)によって精製して、6Aとして白色のガラスを得た(740mg、80%):融点=191〜193℃;[α]22D−18.0(c 0.50、CHCl3);IR νmax 3272、3064、1758、1692、1621cm-1;1H NMR(400MHz,5% CD3OD/CDCl3)δ7.40(m,12H)、7.25(m,12H)、7.20(m,6H)、6.96および6.88(不安定なNH,d,J=8.0Hz,1H)、5.49(dt,J=15.0Hz,6.5Hz,1H)、5.37(dd,J=15.5Hz,6.0Hz,1H)、4.32(m,2H)、4.22(d,J=6.0Hz,1H)、3.98(t,J=7.0Hz,1H)、3.92(d,J=18.1Hz,1H)、3.72(d,J=17.6Hz,1H)、3.67(s,3H)、2.64(dd,J=13.0Hz,7.5Hz,1H)、2.58(dd,J=13.0Hz,7.5Hz,1H)、2.56−2.18(m,9H)、2.11(q,J=6.5Hz,2H)、1.31(d,J=7.5Hz,3H)、0.91(t,J=7.0Hz,6H);13C NMR(100MHz,5% CD3OD/CDCl3)δ173.0、172.0、171.3、170.5、170.2、144.9、144.2、132.8、129.9、129.7、129.6、129.5、128.2、128.1、127.9、127.1、126.7、69.6、67.1、66.7、58.9、58.8、52.7、52.2、50.0、49.8、49.6、49.5、49.4、49.1、43.7、40.9、40.8、33.1、31.5、31.3、30.0、19.2、17.5;LRMS m/z 1050.4(100%,[M+Na]+)。
【0193】
((R)−2−{(S)−2−[(R)−2−((E)−(S)−3−ヒドロキシ−7−トリチルスルファニル−ヘプト−4−エノイルアミノ)−プロピオニルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸(7A)の調製
0℃でTHF(15mL)中の6A(700mg、0.70mmol)の攪拌溶液に、H2O(2.4mL)中のLiOH(25mg、1.05mmol)の溶液を添加した。1時間攪拌した後、その反応混合物をH2O(30mL)で希釈し、1M KHSO4でpH3〜4に酸性化し、EtOAc(3×30mL)で抽出した。有機層を飽和NaCl(15mL)で洗浄し、乾燥させ(Na2SO4)、濾過し、濃縮して、白色の固体を得、それをエーテルと研和して、7Aを白色の固体として得(693mg、99%)、それをそのまま次の段階で直接使用した。融点=191〜193℃;[α]22D−18.5(c 0.50、CHCl3);IR νmax 3413、1711、1678、1630、1451cm-1;1H NMR(400MHz,5% CD3OD/CDCl3)δ7.30(m,12H)、7.16(m,12H)、7.13(m,6H)、5.43(dt,J=15.5Hz,6.0Hz,1H)、5.30(dd,J=15.5Hz,6.0Hz,1H)、4.27(m,2H)、4.21(m,1H)、3.99(m,1H)、3.75(s,2H)、3.77(m,br,6H)、2.55(dd,J=12.5Hz,6.5Hz,1H)、2.44(dd,J=12.5Hz,7.5Hz,1H)、2.21(m,2H)、2.12(m,3H)、2.02(m,2H)、1.23(d,J=7.0Hz,3H)、0.83(d,J=7.0Hz,3H)、0.80(d,J=7.0Hz,3H);13C NMR(100MHz,5% CD3OD/CDCl3)δ173.0、172.1、171.6、171.5、170.5、144.9、144.3、132.7、129.8、129.6、129.5、128.2、127.9、127.0、126.7、69.5、67.1、66.7、58.7、52.7、43.5、40.9、33.2、31.5、31.3、30.3、19.2、19.1、17.6;LRMS m/z 1013.2(100%,[M+Na]+)。
【0194】
(6R,9S,12R,16S)−6−イソプロピル−12−メチル−16−((E)−4−トリチルスルファニル−ブト−1−エニル)−9−トリチルスルファニルメチル−1−オキサ−4,7,10,13−テトラアザ−シクロヘキサデカン−2,5,8,11,14−ペンタオン(8A)の調製。
【0195】
CH2Cl2(160mL)中の無水2−メチル−6−ニトロ安息香酸(MNBA)(289mg、0.84mmol)およびDMAP(205mg、1.68mmol)の溶液に、CH2Cl2/THF(2:1、600mL)中の酸7A(693mg、0.70mmol)の溶液を5時間かけて1滴ずつ添加した。さらに12時間後、1M HCl(150mL)を添加し、CH2Cl2(3×100mL)で抽出した。その後、合わせた有機相を飽和NaHCO3(150mL)、続いてブライン(80mL)で洗浄し、乾燥させ(MgSO4)、濾過し、濃縮した。残留物をフラッシュクロマトグラフィー(溶離剤 50〜100% EtOAc/ヘキサン)によって精製して、8Aを白色のガラスとして得た(478mg、0.49mmol、70%)。[α]22D−7.0(c 0.50、CHCl3);IR νmax 1735、1659、1526cm-1;1H NMR(400MHz,CDCl3)δ7.43−7.33(m,12H)、7.32−7.13(m,20H)、6.77(d,J=5.3Hz,1H)、6.54(d,J=7.3Hz,1H)、5.58(dt,J=15.1,6.8Hz,1H)、5.44−5.29(m,2H)、4.55(dd,J=16.8,9.0Hz,1H)、4.46(dd,J=9.5,4.3Hz,1H)、3.89(5重線,(dd,J=7.0Hz,1H)、3.77(dt,J=8.3,5.3Hz,1H)、3.43(dd,J=16.8,2.8Hz,1H)、3.03(dd,J=12.6,8.3Hz,1H)、2.61−2.49(m,3H)、2.18(t,J=7.5Hz,2H)、2.11−1.95(m,2H)、1.38(d,J=7.0Hz,3H)、0.94(d,J=6.8Hz,3H)、0.90(d,J=6.8Hz,3H);13C−NMR(100MHz,CDCl3)δ174.0(C)、170.9(C)、170.8(C)、169.8(C)、169.3(C)、144.9(C)、144.3(C)、133.2(CH)、129.7(CH)、129.5(CH)、128.3(CH)、128.0(CH)、127.2(CH)、126.8(CH)、72.3(CH)、67.3(C)、66.8(C)、58.3(CH)、55.9(CH)、50.5(CH)、41.7(2 x CH2)、32.5(CH2)、31.3(CH2)、31.2(CH2)、28.8(CH)、19.8(CH3)、19.8(CH3)、17.2(CH3)、16.7(CH3);LRMS(ES+)m/z 996(100%,[M+Na]+)、974(10%,[M+H]+)。
【0196】
(E)−(1S,7R,10S,21R)−7−イソプロピル−21−メチル−2−オキサ−12,13−ジチア−5,8,20,23−テトラアザ−ビシクロ[8.7.6]トリコス−16−エン−3,6,9,19,22−ペンタオン(9A)の調製
CH2Cl2/MeOH(9:1、1000mL)中のI2(1120mg、4.42mmol)の強力攪拌溶液に、CH2Cl2/MeOH(9:1、500mL)中の保護ジオール8A(430mg、0.44mmol)を30分かけて1滴ずつ添加した。さらに30分間攪拌した後、0.1M チオ硫酸ナトリウム(300mL)および飽和NaCl(100mL)を添加し、CH2Cl2(3×100mL)で抽出した。合わせた有機抽出物を乾燥させ(MgSO4)、濾過し、溶媒を除去した。残留物をフラッシュクロマトグラフィー(溶離剤 1〜6% MeOH/CH2Cl2)によって精製して、9A(205mg、0.42mmol、96%)を白色の固体として得た:[α]22D−98.0(c 0.50、CHCl3);IR νmax 3300、2477、1734、1659、1526、1446cm-1;1H NMR(400MHz,CDCl3)δ7.46(d,J=6.5Hz,1H)、7.33(d,J=5.3Hz,1H)、7.28(d,J=8.8Hz,1H)、6.60(d,J=3.8Hz,1H)、5,95(dtd,J=16.1,6.5,2.0Hz,1H)、5.80−5.70(m,2H)、4.85(ddd,J=9.8,8.5,3.8Hz,1H)、4.21(qd,J=7.3,3.8Hz,1H)、4.11(d,J=5.3Hz,2H)、3.44(dd,J=15.6,10.0Hz,1H)、3.20(dd,J=10.3,6.8Hz,1H)、3.01−2.95(m,2H)、2.92(dd,J=15.6,4.0Hz,1H)、2.85−2.58(m,5H)、1.49(d,J=7.5Hz,3H)、0.98(d,J=6.5Hz,3H)、0.92(d,J=6.5Hz,3H);13C NMR 100MHz 173.3(C)、171.6(C)、171.0(C)、169.4(C)、168.2(C)、130.4(CH)、130.3(CH)、69.8(CH)、64.9(CH)、54.5(CH)、52.0(CH)、42.4(2 x CH2)、38.7(CH2)、38.6(CH2)、32.7(CH2)、27.5(CH)、20.8(CH3)、20.1(CH3)、16.7(CH3);LRMS(ES+)m/z 995(50%,[2M+Na]+)、509(80%,[M+Na]+)、487(100%,[M+H]+);HRMS m/z 509.1495(M+Na)+ 予測値 509.1499。
【0197】
((R)−2−{(S)−2−[(R)−2−(9H−フルオレン−9−イルメトキシカルボニルアミノ)−3−フェニル−プロピオニルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(4B)の調製。
【0198】
室温でCH3CN/CH2Cl2(1:1、60mL)中の3A(836mg、1.11mmol)の攪拌溶液に、Et2NH(3mL)を添加した。室温で3時間攪拌した後、その反応混合物をヘプタン(60mL)で希釈し、濃縮して、粗製アミンを無色の油として得た。CH2Cl2(30mL)中のFmoc−D−フェニルアラニン(650mg、1.7mmol)の攪拌溶液に、EDAC・HCl(326mg、1.7mmol)、HOBt(230mg、1.7mmol)およびDIEA(627μL、3.6mmol)を添加した。室温で10分間攪拌した後、CH2Cl2(20mL)中のその粗製アミンの溶液を添加し、その反応混合物を12時間攪拌した。その後、その反応物を水(15mL)、10% HCl(15mL)、5% NaHCO3(15mL)および飽和NaCl(15mL)溶液で洗浄し、乾燥させ(Na2SO4)、濾過し、溶媒を除去して、オフホワイトの固体を得、それをCH3CNから再結晶させて、4Bを白色の固体として得た(800mg、0.89mmol、80%);IR νmax 3264、3053、1742、1701、1636、1532cm-1;1H−NMR(400MHz,CDCl3)δ7.75(d,J=7.5Hz,2H)、7.46(t,J=7.5Hz,2H)、7.41−7.31(m,8H)、7.30−7.07(m,16H)、7.01(br s,1H)、6.54(br s,2H)、5.29(br s,1H)、4.48−4.32(m,3H)、4.25−4.13(m,1H)、4.10(t,J=8.5Hz,1H)、3.99−3.76(m,3H)、3.65(s,3H)、3.10−2.88(m,2H)、2.83−2.69(m,1H)、2.49(dd,J=13.0,6.3Hz,1H)、2.36−2.22,(m,1H)、0.93(t,J=7.0Hz,6H);13C NMR 100MHz 171.2(C)、171.0(C)、170.2(C)、170.0(C)、156.2(C)、144.3(C)、143.9(C)、143.7(C)、141.4(C)、136.0(C)、129.5(CH)、129.3(CH)、129.0(CH)、128.3(CH)、127.9(CH)、127.4(CH)、127.2(CH)、127.1(CH)、125.2(CH)、120.1(CH)、67.3(CH2)、58.6(CH)、56.2(CH)、52.8(CH)、52.3(CH3)、47.2(CH)、41.0(CH2)、38.3(CH2)、33.5(CH2)、30.2(CH)、19.4(CH3)、17.7(CH3);LRMS(ES+)m/z 1828(30%,[2M+Na]+)、925(100%,[M+Na]+)。
【0199】
((R)−2−{(S)−2−[(R)−2−((E)−(S)−3−ヒドロキシ−7−トリチルスルファニル−ヘプト−4−エノイルアミノ)−3−フェニル−プロピオニルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(6B)の調製。
【0200】
室温でCHCl3/CH3CN(1:1、14mL)中の4B(245mg、0.28mmol)の攪拌溶液に、Et2NH(1mL)を添加した。室温で5時間攪拌した後、その反応混合物をヘプタン(10mL)で希釈し、溶媒を除去して、粗製アミンを無色の油として得た。CH2Cl2(10mL)中のその粗製アミンの攪拌溶液に、室温でCH2Cl2(5mL)中の5(193mg、0.34mmol)およびDMAP(4mg、0.034mmol)を添加した。室温で12時間攪拌した後、溶媒を除去し、残留物をフラッシュクロマトグラフィー(溶離剤 2〜4% MeOH/CH2Cl2)によって精製して、6B(190mg、64%)を白色の固体として得た;1H NMR(400MHz,CDCl3)δ7.46−7.05(m,37H)、6.85(br s,1H)、6.34(br s,1H)、5.44(dt,J=15.3,6.5Hz,1H)、5.29(dd,J=15.3,6.2Hz,1H)、4.74(br d,J=5.3Hz,1H)、4.37(t,J=7.0Hz,1H)、4.33−4.25(m,1H)、4.20−4.08(m,1H)、3.93−3.74(m,2H)、3.65(s,3H)、3.31(br s,1H)、3.06(dd,J=14.3,5.3Hz,1H)、2.96(dd,J=14.3,7.3Hz,1H)、2.59(dd,J=12.5,6.8Hz,1H)、2.33−2.08(m,5H)、2.04(q,J=7.0Hz,2H)、0.90(t,J=6.3Hz,6H);13C NMR(100MHz,CDCl3)δ171.9(C)、171.2(C)、171.1(C)、170.4(2 x C)、145.0(C)、144.3(C)、136.1(C)、132.5(CH)、130.1(CH)、129.7(CH)、129.5(CH)、129.3(CH)、128.8(CH)、128.2(CH)、128.0(CH)、127.3(CH)、127.1(CH)、126.7(CH)、69.8(CH)、67.1(C)、66.8(C)、58.9(CH)、54.4(CH)、52.6(CH)、52.3(CH3)、43.7(CH2)、41.0(CH2)、37.6(CH2)、33.8(CH2)、31.5(CH2)、31.4(CH2)、30.3(CH)、19.3(CH3)、17.9(CH3);LRMS(ES+)m/z 1104(100%,[M+Na]+)、1082(50%,[M+H]+)。
【0201】
(6R,9S,12R,16S)−12−ベンジル−6−イソプロピル−16−((E)−4−トリチルスルファニル−ブト−1−エニル)−9−トリチルスルファニルメチル−1−オキサ−4,7,10,13−テトラアザ−シクロヘキサデカン−2,5,8,11,14−ペンタオン(8B)の調製
0℃のTHF(10mL)中のメチルエステル6B(180mg、0.17mmol)の溶液に、H2O(1.5mL)中のLiOH(6mg、0.25)の溶液を添加した。1時間後、その反応を1M HCl(6mL)の添加により停止させた。CHCl3(50mL)を添加し、有機相を分離し、CHCl3(2×10mL)で抽出した。有機相をブライン(15mL)で洗浄し、乾燥させ(MgSO4)、濾過し、真空下で濃縮して、粗製酸7B(179mg、定量的)を白色の固体として得、それを次の段階で直ちに使用した:(ES-)m/z 1066(100%,[M−H]-)。
【0202】
CH2Cl2(40mL)中のMNBA(68mg、0.20mmol)およびDMAP(49mg、0.40mmol)の溶液に、CH2Cl2/THF(15:1、160mL)中の酸7B(179mg、0.17mmol)の溶液を3時間かけて1滴ずつ添加した(注意:先ず、THFに酸を溶解し、その後、CH2Cl2を添加する)。さらに14時間後、1M HCl(40mL)の添加により反応を停止させた。有機相を(CH2Cl2で抽出しながら)分離し、NaHCO3(30mL)、そしてブライン(20mL)で順次洗浄した。合わせた有機相を乾燥させ(MgSO4)、濾過し、真空下で濃縮して、黄色の油を得た。シリカゲルでのカラムクロマトグラフィー(30〜70% EtOAc/CH2Cl2)による精製によって、8B(108mg、0.10mmol、60%)を白色の固体として得た;1H−NMR(400MHz,CDCl3)δ7.42−7.08(m,35H)、7.05(d,J=7.3Hz,2H)、6.96(br s,1H)、6.54(br s,1H)、5.46(dt,J=15.3,6.5Hz,2H)、5.21(dd,J=15.3,6.6Hz,1H)、4.49(dd,J=13.5,4.3Hz,1H)、4.32(dd,J=16.8,9.0Hz,1H)、4.16−4.06(m,1H)、3.85(br q,J=6.0Hz,1H)、3.02−2.82(m,4H)、2.65−2.53(m,2H)、2.40(dd,J=14.6,3.3Hz,1H)、2.23(dd,J=14.6,11.3Hz,1H)、2.10(t,J=7.0Hz,2H)、1.97−1.84(m,2H)、0.98(d,J=7.0Hz,3H)、0.92(d,J=7.0Hz,3H);13C−NMR(100MHz,CDCl3)δ173.5(C)、170.8(C)、170.7(C)、169.9(2 x C)、144.9(C)、144.2(C)、136.3(C)、132.7(CH)、129.6(CH)、129.5(CH)、129.3(CH)、128.9(CH)、128.3(CH)、128.0(CH)、127.9(CH)、127.3(CH)、127.1(CH)、126.7(CH)、72.2(CH)、67.3(C)、66.7(C)、58.3(CH)、55.8(2 x CH)、42.0(CH2)、41.4(CH2)、36.7(CH2)、32.3(CH2)、31.3(CH2)、31.1(CH2)、28.7(CH)、19.8(CH3)、17.2(CH3);LRMS(ES+)m/z 1071(100%,[M+Na]+)、1066(10%,[M+NH4]+)、1049(10%,[M+H]+)。
【0203】
(E)−(1S,7R,10S,21R)−21−ベンジル−7−イソプロピル−2−オキサ−12,13−ジチア−5,8,20,23−テトラアザ−ビシクロ[8.7.6]トリコス−16−エン−3,6,9,19,22−ペンタオン(9B)の調製。
【0204】
CH2Cl2/MeOH(9:1、230mL)中のI2(254mg、1.0mmol)の強力攪拌溶液に、ビス−トリチル8B(105mg、0.10mmol)の溶液を30分かけて1滴ずつ添加した。さらに30分間後、チオ硫酸ナトリウム(0.05M、100mL)、続いてブライン(10mL)の添加により反応を停止させた。有機相を分離し、水性相をCH2Cl2(3×15mL)で抽出した。合わせた有機相を乾燥させ(MgSO4)、濾過し、真空下で濃縮して、白色の固体を得た。シリカゲルでのカラムクロマトグラフィー(1〜3.5% MeOH/CH2Cl2)による精製によって、二環式デプシペプチド9B(43mg、0.08mmol、75%)を白色の固体として得た:[α]25D−98(c 0.05、1:1 MeOH/CHCl3);IR νmax 3280、1732、1627、1538、1445cm-1;1H−NMR(400MHz,19:1 CD3OD/CDCl3)δ7.35−7.18(m,5H)、5.86−5.76(m,1H)、5.74−5.66(m,2H)、4.62(dd,J=11.3,4.0Hz,1H)、4.45(dd,J=9.3,5.3Hz,1H)、4.28(d,J=17.6Hz,1H)、3.77(d,J=17.6Hz,1H)、3.39(d,J=10.5Hz,1H)、3.27−3.17(m,2H)、3.11−2.99(m,3H)、2.97−2.83(m,2H)、2.81(dd,J=13.6,2.0Hz,1H)、2.70−2.53(m,3H)、0.96(d,J=6.5Hz,3H)、0.92(d,J=6.5Hz,3H);13C−NMR(100MHz,19:1 CD3OD/CDCl3)δ173.8(C)、173.2(C)、172.9(C)、171.3(C)、169.4(C)、137.7(CH)、131.3(CH)、131.3(CH)、129.7(CH)、129.6(CH)、128.0(CH)、71.9(CH)、65.9(CH)、58.3(CH)、58.1(CH)、42.6(CH2)、39.3(2 x CH2)、36.9(CH2)、36.8(CH2)、31.8(CH2)、28.1(CH)、20.6(CH3)、20.5(CH3);LRMS(ES+)m/z 580(50%,[2M+Na]+)、563(100%,[M+NH4]+)、563(10%,[M+H]+)。
【0205】
((R)−2−{(S)−2−[(R)−2−(9H−フルオレン−9−イルメトキシカルボニルアミノ)−3−メチル−ブチリルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(4C)の調製。
【0206】
室温でCH3CN/CH2Cl2(1:1、60mL)中の3A(836mg、1.11mmol)の攪拌溶液に、Et2NH(3mL)を添加した。室温で3時間攪拌した後、その反応混合物をヘプタン(60mL)で希釈し、濃縮して、粗製アミンを無色の油として得た。CH2Cl2(40mL)中のFmoc−D−バリン(577mg、1.7mmol)の攪拌溶液に、EDAC・HCl(326mg、1.7mmol)、HOBt(230mg、1.7mmol)およびDIEA(627μL、3.6mmol)を添加した。室温で10分間攪拌した後、CH2Cl2(20mL)中のその粗製アミンの溶液を添加し、その反応混合物を12時間攪拌した。その後、その反応物を水(15mL)、10% HCl(15mL)、5% NaHCO3(15mL)および飽和NaCl(15mL)溶液で洗浄し、乾燥させ(Na2SO4)、濾過し、溶媒を除去して、オフホワイトの固体を得、それをCH3CNから再結晶させて、4Cを白色の固体として得た(734mg、0.86mmol、78%);1H−NMR(400MHz,CDCl3)δ7.74(d,J=7.5Hz,2H)、7.53(t,J=7.5Hz,2H)、7.42−7.32(m,9H)、7.30−7.19(m,9H)、7.16(t,J=7.0Hz,2H)、6.80(d,J=8.0Hz,1H)、6.57(d,J=7.0Hz,1H)、5.52(d,J=8.0Hz,1H)、4.44(t,J=8.0Hz,1H)、4.38(dd,J=10.5,7.5Hz,1H)、4.25(dd,J=10.5,7.0Hz,1H)、4.17−3.95(m,3H)、3.87(dd,J=18.1,5.5Hz,1H)、3.78(dd,J=18.1,5.5Hz,1H)、3.63(s,3H)、2.71(dd,J=12.5,6.0Hz,1H)、2.53(dd,J=12.5,6.0Hz,1H)、2.21(sept,J=6.4Hz,1H)、2.07−1.93(m,1H)、0.93−0.83(m,12H);13C NMR 100MHz 171.5(C)、171.1(C)、170.2(2 x C)、156.6(C)、144.4(C)、144.0(C)、143.8(C)、141.4(C)、129.6(CH)、128.3(CH)、127.9(CH)、127.2(CH)、127.1(CH)、125.2(CH)、120.1(CH)、67.3(CH2)、67.2(C)、60.3(CH)、58.5(CH)、52.5(CH)、52.3(CH)、47.3(CH)、41.0(CH2)、33.8(CH2)、31.5(CH)、30.5(CH)、19.2(2 x CH3)、18.0(CH3)、17.8(CH3);LRMS(ES+)m/z 1832(10%,[2M+Na]+)、877.5(100%,[M+Na]+)。
【0207】
((R)−2−{(S)−2−[(R)−2−((E)−(S)−3−ヒドロキシ−7−トリチルスルファニル−ヘプト−4−エノイルアミノ)−3−メチル−ブチリルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(6C)の調製。
【0208】
室温でCHCl3/CH3CN(1:1、14mL)中の4C(235mg、0.28mmol)の攪拌溶液に、Et2NH(1mL)を添加した。室温で5時間攪拌した後、その反応混合物をヘプタン(10mL)で希釈し、溶媒を除去して、粗製アミンを無色の油として得た。CH2Cl2(10mL)中のその粗製アミンの攪拌溶液に、室温でCH2Cl2(5mL)中の5(193mg、0.34mmol)の溶液およびDMAP(4mg、0.034mmol)を添加した。室温で12時間攪拌した後、溶媒を除去し、残留物をフラッシュクロマトグラフィー(溶離剤 1〜3% MeOH/CH2Cl2)によって精製して、6C(230mg、81%)を白色の固体として得た;1H NMR(400MHz,CDCl3)δ7.42−7.38(m,12H)、7.30−7.14(m,20H)、6.74(br s,1H)、6.39(br s,1H)、5.49(dtd,J=15.3,6.5,0.8Hz,1H)、5.36(dd,J=15.3,6.3Hz,1H)、4.39−4.26(m,3H)、4.16−4.06(m,1H)、3.83(d,J=5.5Hz,2H)、3.65(s,3H)、3.33(br s,1H)、2.60(dd,J=12.8,7.3Hz,1H)、2.54(dd,J=12.8,6.8Hz,1H)、2.37(dd,J=14.0,3.0Hz,1H)、2.30−2.02(m,7H)、0.90(d,J=7.0Hz,3H)、0.89(d,J=7.0Hz,3H)、0.88(d,J=7.0Hz,3H)、0.85(d,J=7.0Hz,3H);13C NMR(100MHz,CDCl3)δ172.1(C)、171.4(C)、171.2(C)、170.5(C)、170.3(C)、145.0(C)、144.3(C)、132.6(CH)、130.2(CH)、129.7(CH)、129.5(CH)、128.3(CH)、128.0(CH)、127.1(CH)、126.8(CH)、69.8(CH)、67.1(C)、66.8(C)、59.0(CH)、58.9(CH)、52.5(CH)、52.3(CH3)、43.9(CH2)、41.0(CH2)、33.7(CH2)、31.5(2 x CH2)、30.4(CH)、30.2(CH2)、19.5(CH3)、19.3(CH3)、17.9(2 x CH3);LRMS(ES+)m/z 1056(100%,[M+Na]+)、1051(50%,[M+NH4]+)。
【0209】
(6R,9S,12R,16S)−6,12−ジイソプロピル−16−((E)−4−トリチルスルファニル−ブト−1−エニル)−9−トリチルスルファニルメチル−1−オキサ−4,7,10,13−テトラアザ−シクロヘキサデカン−2,5,8,11,14−ペンタオン(8C)の調製
0℃で、THF(12mL)中のメチルエステル6B(220mg、0.21mmol)の溶液に、H2O(2mL)中のLiOH(7.6mg、0.32)の溶液を添加した。1時間後、1M HCl(6mL)の添加により反応を停止させた。CHCl3(50mL)を添加し、有機相を分離し、CHCl3(2×15mL)で抽出した。有機相をブライン(10mL)で洗浄し、乾燥させ(MgSO4)、濾過し、真空下で濃縮して、粗製酸7C(217mg、定量的)を白色の固体として得、それを次の段階で直ちに使用した:(ES-)m/z 1017(100%,[M−H]-)。
【0210】
CH2Cl2(50mL)中のMNBA(90mg、0.26mmol)およびDMAP(62mg、0.51mmol)の溶液に、CH2Cl2/THF(15:1、200mL)中の酸7C(217mg、0.21mmol)の溶液を3時間かけて1滴ずつ添加した(注意:先ず、THFに酸を溶解し、その後、CH2Cl2を添加する)。さらに14時間後、1M HCl(40mL)の添加により反応を停止させた。有機相を(CH2Cl2で抽出しながら)分離し、NaHCO3(30mL)、そしてブライン(20mL)で順次洗浄した。合わせた有機相を乾燥させ(MgSO4)、濾過し、真空下で濃縮して、黄色の油を得た。シリカゲルでのカラムクロマトグラフィー(30〜70% EtOAc/CH2Cl2)による精製によって、8C(130mg、0.13mmol、62%)を白色の固体として得た;1H NMR(400MHz,CDCl3)δ7.88(br s,2H)、7.42−7.34(m,12H)、7.29−7.14(m,19H)、6.20(br d,J=7.0Hz,1H)、5.62−5.50(m,2H)、5.31(dd,J=15.3,6.4Hz,1H)、4.36(dd,J=17.3,8.3Hz,1H)、4.24(dd,J=8.5,4.5Hz,1H)、4.11(t,J=7.3Hz,1H)、3.65−3.56(m,1H)、3.42(d,J=14.5Hz,1H)、2.99(t,J=10.8Hz,1H)、2.64(dd,J=12.0,6.3Hz,1H)、2.50(dd,J=15.1,2.5Hz,1H)、2.35(dd,J=14.8,9.8Hz,1H)、2.17(t,J=7.5Hz,2H)、2.06−1.86(m,3H)、1.84−1.72(m,1H)、0.91(d,J=6.8Hz,3H)、0.88(d,J=6.3Hz,6H)、0.83(d,J=6.8Hz,3H);13C−NMR(100MHz,CDCl3)δ173.8(C)、172.2(C)、171.0(C)、169.5(C)、168.9(C)、145.0(C)、144.4(C)、132.7(CH)、129.7(CH)、128.2(CH)、128.1(CH)、128.0(CH)、127.0(CH)、126.8(CH)、72.2(CH)、67.3(C)、66.8(C)、58.8(CH)、58.7(CH)、58.4(CH)、42.1(CH2)、41.9(CH2)、32.0(CH2)、31.5(CH2)、31.3(CH)、31.2(CH2)、29.4(CH)、19.6(CH3)、19.4(CH3)18.5(CH3)、17.0(CH3);LRMS(ES+)m/z 1023(100%,[M+Na]+)、1018(60%,[M+NH4]+)。
【0211】
(E)−(1S,7R,10S,21R)−7,21−ジイソプロピル−2−オキサ−12,13−ジチア−5,8,20,23−テトラアザ−ビシクロ[8.7.6]トリコス−16−エン−3,6,9,19,22−ペンタオン(9C)の調製。
【0212】
CH2Cl2/MeOH(9:1、280mL)中のI2(305mg、1.2mmol)の強力攪拌溶液に、ビス−トリチル8C(120mg、0.12mmol)の溶液を30分かけて1滴ずつ添加した。さらに30分間後、チオ硫酸ナトリウム(0.05M、100mL)、続いてブライン(10mL)の添加により反応を停止させた。有機相を分離し、水性相をCH2Cl2(3×15mL)で抽出した。合わせた有機相を乾燥させ(MgSO4)、濾過し、真空下で濃縮して、白色の固体を得た。シリカゲルでのカラムクロマトグラフィー(1〜4% MeOH/CH2Cl2)による精製によって、二環式デプシペプチド9C(60mg、0.12mmol、97%)を白色の固体として得た:[α]25D−105.7(1:1 MeOH/CHCl3、c 0.15);1H NMR(400MHz,CD3OD)δ5.76−5.63(m,3H)、4.52(dd,J=11.3,4.7Hz,1H)、3.96(d,J=5.1Hz,1H)、3.74(d,J=17.5Hz,1H)、3.38(d,J=10.9Hz,1H)、3.19−3.11(m,2H)、3.09−2.96(m,3H)、2.90(dd,J=13.6,2.5Hz,1H)、2.71−2.62(m,2H)、2.58−2.44(m,1H)、2.31−2.18(m,1H)、1.11(d,J=7.0Hz,3H)、1.09(d,J=7.0Hz,3H)、1.01(d,J=6.6Hz,3H)、0.91(d,J=6.6Hz,3H);13C−NMR(100MHz,CD3OD)δ171.8(C)、171.7(C)、170.7(C)、170.4(C)、168.3(C)、130.0(CH)、129.6(CH)、69.8(CH)、64.6(CH)、61.9(CH)、54.8(CH)、42.2(CH2)、39.3(CH2)、37.5(CH2)、37.1(CH2)、32.1(CH2)、29.2(CH)、27.3(CH)、20.6(CH3)、20.0(CH3)、19.4(CH3)、19.0(CH3);LRMS(ES+)m/z 537(100%,[M+Na]+)、515(90%,[M+H]+)。
【0213】
((R)−2−{(S)−2−[(R)−6−t−ブトキシカルボニルアミノ−2−(9H−フルオレン−9−イルメトキシカルボニルアミノ)−ヘキサノイルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(4D)の調製。
【0214】
アルゴン下でトリペプチド3A(548mg、0.7mmol)をCH2Cl2/CH3CN(44mL、1:1)に溶解し、その後、攪拌しながらジエチルアミン(1.65mL、16mmol)を添加し、その反応混合物を室温で4.5時間攪拌した。その後、ヘキサン(100mL)をその反応混合物に添加し、溶媒を真空下で除去し、これを再びヘキサン(3×25mL)で繰り返した。その後、その粗製アミンを高真空下で40分間乾燥させた。その後、アルゴン下で攪拌しながら、CH3CN(16.5mL)中のPyBop(558mg、1.1mmol)およびFmoc−D−リシン(Boc)−OH(477mg、1mmol)の溶液にジイソプロピルエチルアミン(0.45mL、2.6mmol)を添加した。得られた3の脱保護アミンをCH2Cl2(19mL)に添加し、その反応混合物を室温で16時間攪拌した。その後、真空下で溶媒を除去し、形成した固体をシリカゲルでのカラムクロマトグラフィー(溶離剤 6:4 EtOAc/ヘキサン)によって精製して、4D(571mg、0.57mmol、81%)を白色の固体として得た:Rf 0.17 EtOAc/ヘキサン(6:4):[α]D26=+47(c 0.3、MeOH);IR 3294、1744、1646、1515、1445(m);1H−NMR(400MHz,CDCl3)δ7.74(d,J=8.0,2H)、7.56−7.51(m,2H)、7.42−7.34(m,8H)、7.30−7.17(m,11H)、7.02(br s,1H)、6.94(br s,1H)、6.59(d,J=8.03Hz,1H)、5.75(s,1H)、4.38−4.21(m,2H)、4.18(dd,J=8.5,6.0Hz,1H)、4.15−4.05(m,2H)、3.87(d,J=5.5Hz,3H)、3.55(s,3H)、3.03(br d,J=8.0Hz,2H)、2.76−2.67(m,1H)、2.66−2.58(m,1H)、2.20−2.10(m,1H)、1.93−1.84(m,2H)、1.82−1.72(m,1H)、1.70−1.59(m,1H)、1.43(s,9H)、1.50−1.30(m,3H)、0.89(d,J=7.02Hz,3H)、0.84(d,J=7.03Hz,3H)、13C NMR(100MHz,CDCl3)δ172.6(C)、171.4(C)、170.6(C)、170.6(C)、156.6(C)、156.3(CO)、144.4(C)、144.3(C)、143.9(C)、143.8(C)、141.4(C)、129.7(CH)、129.5(CH)、128.3(CH)、128.2(CH)、127.2(CH)、127.1(CH)、125.2(CH)、120.0(CH)、67.4(CH2)、47.2(CH3)、41.1(CH2)、40.0(CH2)、33.5(CH2)、31.9(CH2)、30.3(CH2)、29.8(CH)、28.6(CH3)、22.5(CH2)、19.4(CH3),17.9(CH3)、52.3(CH)、52.8(CH)、55.1(CH)、59.0(CH);LRMS(ES+)1007.0(100%,[M+Na]+)。
【0215】
((R)−2−{(S)−2−[(R)−6−t−ブトキシカルボニルアミノ−2−((S)−3−ヒドロキシ−7−トリチルスルファニル−ヘプト−4−エノイルアミノ)−ヘキサノイルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(6D)の調製。
【0216】
CH2Cl2(13.5mL)およびCH3CN(16.5mL)中のテトラペプチド4D(201mg、0.2mmol)の溶液に、アルゴン下で、トリエチルアミン(1mL、9.6mmol)を添加し、その反応混合物を5時間攪拌した。その後、真空下で溶媒を除去し、これをヘキサン(2×10mL)で繰り返した。その後、CH2Cl2(7mL)中のその粗製アミンの溶液にDMAP(4mg、0.03mmol)を添加し、続いてCH2Cl2(7mL)中の5(158mg、0.28mmol)の溶液を添加した。その後、その反応物を18時間攪拌した。その後、真空下で溶媒を除去し、形成した固体をシリカゲルでのカラムクロマトグラフィー(溶離剤 6:4〜7:3 EtOAc:ヘキサン)によって精製して、6D(136mg、0.12mmol、60%)を白色の固体として得た;融点85〜87℃;Rf 0.29 EtOAc/ヘキサン(8:2);[α]29D=+19(c 0.49、CH2Cl2);IR(薄膜)3289、1751、1687、1634、1521、1490、1443;1H−NMR(400MHz,CDCl3)δ7.42−7.35(m,15H)、7.29−7.16(m,17H)、6.95(d,J=7.5Hz,1H)、6.80(d,J=8.0Hz,1H)、5.48(dt,J=15.1,6.5Hz,1H,)、5.35(dd,J=15.1,6.0Hz,1H,)、4.65(s,1H)、4.46−4.35(m,2H)、4.21(dd,J=8.5,6.52Hz,1H)、4.04−3.96(m,1H)、3.93(d,J=5.5Hz,1H)、3.89−3.81(m,2H)、3.62(s,3H)、3.02(d,J=5.5Hz,2H)、2.60−2.45(m,2H)、2.35−2.00(m,9H)、1.85−1.74(m,1H)、1.66−1.54(m,1H)、1.40(s,9H)、1.47−1.25(m,2H)、0.88(d,J=7.0Hz,3H,)、0.84(d,J=7.0Hz,3H)、13C NMR(100MHz,CDCl3)δ172.3(C)、172.2(C)、171.5(C)、170.7(C)、170.5(C)、156.2(C)、145.0(C)、145.0(C)、144.3(C)、132.9(CH)、129.7(CH)、129.6(CH)、129.5(CH)、129.5(CH)、128.2(CH)、128.2(CH)、128.0(CH)、127.0(CH)、126.7(CH)、69.5(CH)、67.1(CH2)、66.7(CH2)、58.6(CH)、52.9(CH)、52.6(CH)、52.3(CH)、52.2(CH3)、44.2(CH2)、41.1(CH2)、40.1(CH2)、33.9(CH2)、31.4(CH2)、31.0(CH2)、31.0(CH2)、30.6(CH)、29.8(CH2)、28.6(CH3)、22.6(CH2)、19.3(CH3)、18.0(CH3)、MS(ES+)1185(100%,[M+Na]+)、1163(40%,[M+H+)。
【0217】
{4−[(6R,9S,12R)−6−イソプロピル−2,5,8,11,14−ペンタオキソ−16−((S)−4−トリチルスルファニル−ブト−1−エニル)−9−トリチルスルファニルメチル−1−オキサ−4,7,10,13−テトラアザ−シクロヘキサデク−12−イル]−ブチル}−カルバミン酸t−ブチルエステル(8D)の調製。
【0218】
0℃で、THF(1.9mL)中の6D(136mg、0.12mmol)の溶液に、水(0.5mL)中のLiOH(6.8mg、0.28mmol)の溶液を添加し、その反応混合物を55分間攪拌した。その後、pHが3〜4の間に低下するまでクエン酸を1滴ずつ添加し、その後、水(3.7mL)、続いてEtOAc(15mL)を添加した。層を分離し、生成物をEtOAc(2×10mL)で抽出した。その後、合わせた有機層を飽和ブライン(10mL)で洗浄し、Na2SO4で乾燥させ、溶媒を真空下で除去して、粗製酸7D(132mg、定量的)を白色の固体として得、それを次の段階で直ちに使用した:LRMS(ES-)1147(100%,[M−H]-)。
【0219】
その後、CH2Cl2(25mL)中のMNBA(47mg、0.14mmol)およびDMAP(33mg、0.27mmol)の溶液に、CH2Cl2(96mL)中の酸7D(132mg,0.11mmol)の溶液を10時間かけて1滴ずつ添加し、その反応混合物をさらに9.5時間、室温で攪拌した。その後、0.2% HCl(50mL)を添加し、形成した層を分離し、有機層を硫酸水素ナトリウム(30mL)および飽和ブライン(30mL)で洗浄し、MgSO4で乾燥させ、真空下で濃縮して、褐色の固体を得た。その後、これをEtOAc/ヘキサン(6:4)の溶離剤を用い、それを(3:1)に増加させるフラッシュカラムクロマトグラフィーによって精製して、生成物8D(52mg、0.05mmol、42%)を白色の固体として得た:Rf 0.07 EtOAc/ヘキサン(6:4);[α]25D=−76(c 0.1、CH2Cl2);IR(薄膜)3271 1739(m)、1683、1641、1536、1490、1444、1391;1H NMR(400MHz,CDCl3)δ7.54(s,1H)、7.44−7.36(m,12H)、7.30−7.17(m,19H)、6.80(d,J=8.5Hz,1H,)、6.47(d,J=8.5Hz,1H)、5.58(dt,J=15.1,7.0Hz,1H,)、5.47(td,J=6.5,2.5Hz,1H,)、5.39(dd,J=15.1,6.5Hz,1H)、4.68(s,1H)、4.29(q,J=7.0Hz,1H)、4.20(dd,J=17.1,7.5Hz,1H)、4.16(dd,J=9.0,6.0Hz,1H)、3.53(dd,J=17.1,5.5Hz,1H)、3.44(q,J=7.5Hz,1H)、3.00−2.90(m,2H)、2.66(d,J=7.5Hz,2H)、2.55−2.48(m,1H)、2.39(dd,J=15.1,8.0Hz,1H)、2.30−2.15(m,3H)、2.13−2.01(m,4H)、1.68−1.58(m,1H)、1.51(dd,J=13.5,7.0Hz,1H,)、1.40(s,9H)、1.45−1.34(m,2H)、1.36−1.28(m,2H)、1.31−1.23(m,2H)、0.89(d,J=6.5Hz,3H)、0.84(d,J=6.5Hz,3H,)、13C NMR(100MHz,CDCl3)δ173.1(C)、171.3(C)、171.3(C)、169.8(C)、168.9(C)、156.2(C)、145.0(C)、144.5(C)、132.6(CH)、129.7(CH)、128.2(CH)、128.0(CH)、127.0(CH)、126.8(CH)、79.1(CH)、67.5(C)、66.8(C)、58.7(CH)、53.1(CH)、53.0(CH)、42.0(CH2)、41.8(CH2)、40.2(CH2)、32.2(CH2)、31.5(CH2)、31.4(CH2)、31.2(CH2)、31.0(CH2)、29.7(CH)、28.6(CH3)、22.8(CH2)、19.5(CH3)、17.6(CH3);LRMS(ES+)1152(100%,[M+Na+])。
【0220】
[4−((7R,10S,14S,21R)−7−イソプロピル−3,6,9,19,22−ペンタオキソ−2−オキサ−12,13−ジチア−5,8,20,23−テトラアザ−ビシクロ[8.7.6]トリコス−16−エン−21−イル)−ブチル]−カルバミン酸t−ブチルエステル(9D)の調製。
【0221】
CH2Cl2/MeOH(9:1)(148mL)中のヨウ素(117mg、0.5mmol)の攪拌溶液に、CH2Cl2/MeOH(9:1)(77mL)中の8D(52mg、0.05mmol)の溶液を30分かけて1滴ずつ添加し、その後、その反応混合物をさらに30分間攪拌させておき、その後、チオ硫酸ナトリウム(128mL、0.02M)を添加した。層を分離し、その生成物をCH2Cl2(3×25mL)で抽出し、MgSO4で乾燥させ、真空下で溶媒を除去した。その後、シリカゲルでのカラムクロマトグラフィー(溶離剤 2:98〜8:92 MeOH/CH2Cl2)による精製を行い、それによって9D(3mg、0.005mmol、10%)を白色の固体として得た:Rf 0.05 MeOH/CH2Cl2(5:95);LRMS(ES+)666.8(100%,[M+Na]+)。
【0222】
(R)−2−((R)−2−{(R)−2−[(R)−2−((E)−(S)−3−ヒドロキシ−9,9,9−トリフェニル−ノン−4−エノイルアミノ)−プロピオニルアミノ]−5,5,5−トリフェニル−ペンタノイルアミノ}−3−メチル−ブチリルアミノ)−プロピオン酸メチルエステル(6E)の調製。
【0223】
CH2Cl2/THF(1:1、30mL)中のH2N−D−Ala−D−Cys(STrt)−D−Val−D−Ala−OMe(96mg、0.155mmol、米国、CA 92121−1510のバイオペピド社(Biopepide Co.)から購入したもの、純度約60%)の溶液に、CH2Cl2(5mL)中の5(87mg、0.155mmol)の溶液を添加し、続いてDMAP(2mg、0.02mmol)を添加した。18時間後、その反応混合物を真空下で濃縮した。シリカゲルでのカラムクロマトグラフィー(1〜3.5% MeOH/CH2Cl2)による精製によって、ビス−トリチル6E(55mg、0.054mmol、35%)を溶解度の低い白色の固体として得た:LRMS(5μm 粒径、3.0×50mm、1.25mL/分で5分間にわたって5%メタノール(0.1% HCOOH)/H2Oから100%への線形勾配で、XTerra MS C18カラムを使用する、HPLC ES+。その後、1.5mL/分で5分間、100%メタノール(0.1% HCOOH))室温=7.93分、m/z 1041.7(100%、[M+Na]+)、1019.7(30%、[M+H]+)。
【0224】
(3R,6R,9S,12R,16S)−6−イソプロピル−3,12−ジメチル−16−((E)−4−トリチルスルファニル−ブト−1−エニル)−9−トリチルスルファニルメチル−1−オキサ−4,7,10,13−テトラアザ−シクロヘキサデカン−2,5,8,11,14−ペンタオン(8E)の調製。
【0225】
0℃で、THF(5mL)中のメチルエステル6E(81mg、0.080mmol)の溶液に、H2O(0.8mL)中のLiOH(3mg、0.12)の溶液を添加した。1時間後、1M HCl(10mL)の添加により反応を停止させた。CHCl3(20mL)を添加し、有機相を分離し、CHCl3(2×10mL)で抽出した。有機相をブライン(15mL)で洗浄し、乾燥させ(MgSO4)、真空下で濃縮して、粗製酸7E(80mg、定量的)を白色の固体として得、それを次の段階で直ちに使用した:LRMS(ES-)m/z 1003(100%,[M−H]-)。
【0226】
CH2Cl2(20mL)中のMNBA(35mg、0.10mmol)およびDMAP(25mg、0.20mmol)の溶液に、CH2Cl2/THF(75:5、80mL)中の酸7E(80mg、0.080mmol)の溶液を3時間かけて1滴ずつ添加した。さらに14時間後、1M HCl(25mL)の添加により反応を停止させた。有機相を(CH2Cl2で抽出しながら)分離し、NaHCO3(30mL)そしてブライン(20mL)で順次洗浄した。合わせた有機相を乾燥させ(MgSO4)、真空下で濃縮して、黄色の油を得た。シリカゲルでのカラムクロマトグラフィー(70〜100% EtOAc/CH2Cl2)による精製によって、8E(34mg、0.035mmol、43%)を白色の固体として得た:1H−NMR(400MHz,CDCl3)δ7.49−7.08(m,33H)、6.31(br s,1H)、5.57(dt,J=15.3,6.5Hz,1H)、5.47(q,J=6.1Hz,1H)、5.36(dd,J=15.3,6.8Hz,1H)、4.45(5重線,J=7.0Hz,1H)、4.21(5重線,J=7.0Hz,1H)、3.87(t,J=7.3Hz,1H)、3.59(br s,1H)、2.92(dd,J=12.5,8.5Hz,1H)、2.55(dd,J=12.5,5.4Hz,1H)、2.51−2.36(m,2H)、2.17(t,J=7.3Hz,2H)、2.05−1.86(m,3H)、1.36(d,J=7.0Hz,3H)、1.33(d,J=7.0Hz,3H)、0.88(d,J=6.8Hz,3H)、0.83(d,J=7.0Hz 3H);13C−NMR(100MHz,CDCl3)δ173.6(C)、170.8(C)、170.4(C)、170.2(C)、169.7(C)、144.9(C)、144.4(C)、132.9(CH)、129.7(CH)、129.6(CH)、128.2(CH)、128.1(CH)、128.0(CH)、127.0(CH)、126.8(CH)、72.0(CH)、67.2(C)、66.8(C)、61.4(CH)、55.3(CH)、49.9(CH)、49.5(CH)、41.3(CH2)、32.4(CH2)、31.4(CH2)、31.3(CH2)、29.8(CH)、19.8(CH3)、18.4(CH3)、17.9(CH3)、17.8(CH3);LRMS(ES+)m/z 1009(100%,[M+Na]+)、987(10%,[M+H]+)。
【0227】
(E)−(1S,4R,7R,10S,21R)−7−イソプロピル−4,21−ジメチル−2−オキサ−12,13−ジチア−5,8,20,23−テトラアザ−ビシクロ[8.7.6]トリコス−16−エン−3,6,9,19,22−ペンタオン(9E)の調製。
【0228】
CH2Cl2/MeOH(9:1、100mL)中のI2(87mg、0.34mmol)の溶液に、ビス−トリチル8E(34mg、0.034mmol)の溶液を30分かけて1滴ずつ添加した。さらに30分間後、チオ硫酸ナトリウム(0.05M、25mL)、続いてブライン(10mL)の添加により反応を停止させた。有機相を分離し、水性相をCH2Cl2(3×15mL)で抽出した。合わせた有機相を乾燥させ(MgSO4)、濾過し、その後、真空下で濃縮することにより、白色の固体を得た。シリカゲルでのカラムクロマトグラフィー(3〜5% MeOH/CH2Cl2)による精製によって、環化デプシペプチド9E(10mg、0.02mmol、60%)を白色の固体として得た:[α]25D−51.8(c 0.45、MeOH);1H NMR(400MHz,CDCl3)δ7.52(d,J=6.5Hz,1H)、7.40(d,J=6.3Hz,1H)、6.94(d,J=9.5Hz,1H)、6.46(d,J=3.0Hz,1H)、6.23−6.10(m,1H)、5.80−5.72(m,2H)、5.06(ddd,J=10.0,6.8,3.7Hz,1H)、4.47(5重線,J=7.0Hz,1H)、4.20(qd,J=7.3,3.5Hz,1H)、3.81(dd,J=14.3,6.5Hz,1H)、2.96−2.77(m,3H)、2.76−2.64(m,5H)、2.61(dd,J=13.5,2.2Hz,1H)、1.49(d,J=7.0Hz,3H)、1.48(d,J=7.2Hz,3H)、0.95(d,J=6.5Hz,3H)、0.93(d,J=6.5Hz,3H);13C−NMR(100MHz,CD3OD)δ174.9(C)、173.9(C)、172.4(C)、172.2(C)、172.0(C)、131.9(CH)、131.1(CH)、71.8(CH)、67.2(CH)、56.3(CH)、53.0(CH)、50.7(CH)、40.4(CH2)、39.9(CH2)、39.3(CH2)、34.4(CH2)、28.7(CH)、20.7(CH3)、20.3(CH3)、17.7(CH3)、16.4(CH3);LRMS(ES+)m/z 1024(200%,[2M+Na]+)、523(50%,[M+Na]+)、501(100%,[M+H]+)。
【0229】
(R)−4−((E)−(S)−3−ヒドロキシ−7−トリチルスルファニル−ヘプト−4−エノイルアミノ)−4−{(S)−1−[(R)−1−(メトキシカルボニルメチル−カルバモイル)−2−メチル−プロピルカルバモイル]−2−トリチルスルファニル−エチルカルバモイル}−酪酸t−ブチルエステル(6F)の調製。
【0230】
CH2Cl2(40mL)中のH2N−D−Glu(OtBu)−D−Cys(STrt)−D−Val−Gly−OMe(145mg、0.2mmol、米国、CA 92121−1510のバイオペピド社(Biopepide Co.)から購入したもの、純度約60%)の溶液に、CH2Cl2(10mL)中の5(113mg、0.2mmol)の溶液を添加し、続いてDMAP(2.5mg、0.02mmol)を添加した。18時間後、その反応物を真空下で濃縮した。シリカゲルでのカラムクロマトグラフィー(1〜3% MeOH/CH2Cl2)による精製によって、6F(97mg、0.087mmol、44%)を白色の固体として得た:1H NMR(400MHz,CDCl3)δ7.46−7.33(m,12H)、7.32−7.13(m,20H)、7.07(br s,1H)、6.77(d,J=8.3Hz,1H)、5.46(dt,J=15.3,6.5Hz,1H)、5.34(dd,J=15.3,6.3Hz,1H)、4.40−4.23(m,3H)、4.11−3.86(m,2H)、3.79(dd,J=17.8,5.5Hz,1H)、3.70−3.58(m,1H)、3.64(s,3H)、3.06(br s,1H)、2.63(dd,J=12.8,7.5Hz,1H)、2.53(dd,J=12.8,5.8Hz,1H)、2.48−2.12(m,6H)、2.11−1.85(m,4H)、1.41(s,9H)、0.92(d,J=6.7Hz,3H)、0.91(d,J=6.7Hz,3H);13C−NMR(100MHz,CDCl3)δ173.7(C)、172.3(C)、171.6(C)、171.3(C)、170.4(C)、170.3(C)、145.0(C)、144.3(C)、133.6(CH)、130.3(CH)、129.7(CH)、129.5(CH)、128.2(CH)、128.0(CH)、127.1(CH)、126.7(CH)、81.5(C)、70.0(CH)、67.1(C)、66.7(C)、59.3(CH)、54.6(CH)、53.0(CH)、52.2(CH3)、44.1(CH2)、41.0(CH2)、33.3(CH2)、32.2(CH2)、31.4(CH2 x 2)、29.9(CH)、28.1(CH3)、26.1(CH2)、19.3(CH3)、17.7(CH3);LRMS(ES+)m/z 1142(100%,[M+Na]+)。
【0231】
3−[(6R,9S,12R,16S)−6−イソプロピル−2,5,8,11,14−ペンタオキソ−16−((E)−4−トリチルスルファニル−ブト−1−エニル)−9−トリチルスルファニルメチル−1−オキサ−4,7,10,13−テトラアザ−シクロヘキサデク−12−イル]−プロピオン酸t−ブチルエステル(8F)の調製。
【0232】
0℃で、THF(5mL)中のメチルエステル6F(98mg、0.088mmol)の溶液に、H2O(0.8mL)中のLiOH(3mg、0.13)の溶液を添加した。1時間後、1M HCl(15mL)の添加により反応を停止させた。CHCl3(20mL)を添加し、有機相を分離し、CHCl3(2×10mL)で抽出した。有機相をブライン(15mL)で洗浄し、乾燥させ(MgSO4)、真空下で濃縮して、粗製酸7F(96mg、定量的)を白色の固体として得、それを次の段階で直ちに使用した:(ES-)m/z 1103(100%,[M−H]-)。
【0233】
CH2Cl2(20mL)中のMNBA(36mg、0.10mmol)およびDMAP(25mg、0.21mmol)の溶液に、CH2Cl2/THF(75:2、77mL)中の酸7F(96mg、0.087mmol)の溶液を3時間かけて1滴ずつ添加した。さらに14時間後、1M HCl(25mL)の添加により反応を停止させた。有機相を(CH2Cl2で抽出しながら)分離し、NaHCO3(30mL)そしてブライン(20mL)で順次洗浄した。合わせた有機相を乾燥させ(MgSO4)、真空下で濃縮して、黄色の油を得た。シリカゲルでのカラムクロマトグラフィー(50〜70% EtOAc/CH2Cl2、その後、+0.1% MeOH)による精製によって、8F(53mg、0.049mmol、56%)を白色の固体として得た:1H NMR(400MHz,CDCl3)δ7.44−7.32(m,12H)、7.31−7.15(m,19H)、7.11−7.03(m,2H)、6.68(br s,1H)、5.58(dt,J=15.6,6.5Hz,1H)、5.43(ddd,J=9.5,7.0,3.0Hz,1H)、5.33(dd,J=15.6,7.0Hz,1H)、4.44(dd,J=17.0,9.6Hz,1H)、4.30(dd,J=9.0,4.5Hz,1H)、4.04(q,J=7.0Hz,1H)、3.59(m,1H)、3.50(dd,J=6.5,2.0Hz,1H)、3.07(t,J=11.0Hz,1H)、2.60−2.45(m,3H)、2.42−2.23(m,3H)、2.22−2.12(m,2H)、2.08−1.89(m,4H)、1.40(s,9H)、0.88(d,J=7.0Hz,3H)、0.83(d,J=7.0Hz,3H);13C−NMR(100MHz,CDCl3)δ173.4(C)、172.6(C)、171.1(C)、170.9(C)、170.0(C)、169.0(C)、145.0(C)、144.3(C)、133.2(CH)、129.7(CH)、129.6(CH)、128.3(CH)、128.0(CH)、127.1(CH)、126.8(CH)、81.3(C)、72.1(CH)、67.3(C)、66.8(C)、58.7(CH)、56.8(CH)、54.1(CH)、41.9(CH2)、41.3(CH2)、32.4(CH2)、31.8(CH2)、31.4(CH2)、31.2(CH2)、29.0(CH)、28.2(CH3)、25.9(CH2)、19.7(CH3)、17.2(CH3);LRMS(ES+)m/z 1109(100%,[M+Na]+)、1087(50%,[M+H]+)。
【0234】
3−((E)−(1S,7R,10S,21R)−7−イソプロピル−3,6,9,19,22−ペンタオキソ−2−オキサ−12,13−ジチア−5,8,20,23−テトラアザ−ビシクロ[8.7.6]トリコス−16−エン−21−イル)−プロピオン酸t−ブチルエステル(9F)の調製。
【0235】
CH2Cl2/MeOH(9:1、130mL)中のI2(117mg、0.46mmol)の溶液に、ビス−トリチル8F(50mg、0.046mmol)の溶液を30分かけて1滴ずつ添加した。さらに30分間後、チオ硫酸ナトリウム(0.05M、50mL)、続いてブライン(20mL)の添加により反応を停止させた。有機相を分離し、水性相をCH2Cl2(3×25mL)で抽出した。合わせた有機相を乾燥させ(MgSO4)、その後、真空下で濃縮することにより、白色の固体を得た。シリカゲルでのカラムクロマトグラフィー(1〜3% MeOH/CH2Cl2)による精製によって、二環式デプシペプチド9F(19mg、0.032mmol、69%)を白色の固体として得た:[α]25D−76.7(c 0.60、MeOH);1H NMR(400MHz,CDCl3)δ8.15(d,J=4.0Hz,1H)、7.35(d,J=6.5Hz,1H)、7.23(t,J=5.5Hz,1H)、7.11(d,J=9.0Hz,1H)、6.05(ddt,J=15.5,7.0,2.0Hz,1H)、5.84−5.73(m,2H)、4.97(td,J=8.0,4.5Hz,1H)、4.18−4.01(m,3H)、3.61(dd,J=15.1,8.0Hz,1H)、3.24(dd,J=10.5,6.5Hz,1H)、2.95−2.75(m,5H)、2.73−2.62(m,3H)、2.51(dd,J=13.6,2.0Hz,1H)、2.42(ddd,J=18.0,9.0,3.5Hz,1H)、2.19−2.04(m,2H)、1.83(s,9H)、0.98(d,J=6.5Hz,3H)、0.92(d,J=6.5Hz,3H);13C−NMR(100MHz,CDCl3)δ175.7(C)、171.8(C)、171.7(C)、170.9(C)、169.9(C)、168.2(C)、130.2(CH)、129.6(CH)、82.6(C)、69.4(CH)、65.4(CH)、57.6(CH)、53.8(CH)、42.4(CH2)、40.0(2 x CH2)、38.6(CH2)、34.1(CH2)、33.1(CH2)、28.2(CH3)、27.4(CH)、24.9(CH2)、20.6(CH3)、20.2(CH3);LRMS(ES+)m/z 623(90%,[M+Na]+)、601(100%,[M+H]+)。
【0236】
3−((E)−(1S,7R,10S,21R)−7−イソプロピル−3,6,9,19,22−ペンタオキソ−2−オキサ−12,13−ジチア−5,8,20,23−テトラアザ−ビシクロ[8.7.6]トリコス−16−エン−21−イル)−プロピオン酸(10F)の調製。
【0237】
CH2Cl2(1.5mL)中の9F(12mg、0.02mmol)およびEt3SiH(10μL、0.06mmol)の溶液に、TFA(150μL、2.0mmol)を添加した。6時間後、溶媒を除去し、残留物をシリカゲルでのカラムクロマトグラフィー(10% メタノール/CH2Cl2、その後、+1% AcOH)によって精製して、10F(5.6mg、0.01mmol、55%)を白色の固体として得た:[α]25D−63.4(c 0.25、MeOH);1H NMR(400MHz,CD3OD)δ7.5(1 x NHが観察された,d,J=7.5Hz,1H)、5.81(dddd,J=16.8,6.8,4.8,2.5Hz,1H)、5.75−5.67(m,2H)、4.62(ddd,J=11.0,8.0,5.5Hz,1H)、4.28(d,J=17.5Hz,1H)、4.15(dd,J=9.0,6.0Hz,1H)、3.78(d,J=17.5Hz,1H)、3.43(d,J=10.5Hz,1H)、3.20−3.10(m,2H)、3.07−3.00(m,2H)、2.94−2.81(m,1H)、2.82(dd,J=13.2,2.2Hz,1H)、2.73−2.50(m,4H)、2.38(q,J=7.0Hz,1H)、2.23−1.99(m,2H)、0.98(d,J=6.5Hz,3H)、0.92(d,J=6.5Hz,3H);13C−NMR(100MHz,CD3OD)δ176.5(C)、174.0(C)、173.5(C)、173.0(C)、171.6(C)、169.5(C)、131.4(CH)、131.3(CH)、72.1(CH)、66.0(CH)、58.5(CH)、57.0(CH)、42.7(CH2)、39.7(CH2)、39.4(CH2)、36.8(CH2)、31.8(CH2)、31.6(CH2)、28.2(CH3)、26.6(CH2)、20.6(CH3)、20.5(CH3);LRMS(ES-)m/z 543(100%,[M−H]-)。
【0238】
[結果]
[活性アッセイ1]
本発明者らは、MCF7ヒト乳癌細胞および正常ヒト皮膚線維芽細胞(NHDF)の増殖を阻害するスベロイルアニリドヒドロキサム酸(SAHA)、既知HDAC阻害剤、および化合物001(7頁の式(2)の化合物)の能力を比較した。
【0239】
方法;細胞増殖アッセイは、商標シクワント(CyQuant)アッセイシステム(米国のモレキュラー・プローブス社(Molecular Probes,Inc.))をその製造業者の説示に従って使用して行った。このシステムは、細胞の核酸に結合すると蛍光が増強されて、細胞数50から250,000の間の線形検出範囲での細胞数の決定を可能にする、有標緑色蛍光色素を利用する。エストロゲン受容体陽性MCF7乳癌細胞(ECAAC)およびNHDF細胞(英国のキャンブレックス(Cambrex))を96ウエルプレートに、1ウエル当たり100μLの細胞培養基中に細胞数1000の密度で、プレーティングした。化合物は、最低5時間後、細胞培養基中の系列希釈物で、2×最終濃度での100μL量で添加した。6日後、そのプレートを吸い取り紙の上で逆さにすることにより細胞培養基を除去し、細胞を200μLのPBSで1回、穏やかに洗浄した。直ちにプレートを−80℃で最低1時間冷凍し、その後、解凍した。上記製造業者の説示に従って作った色素を補足した200μLの1x シクワント細胞溶解緩衝液を各ウエルに直ちに添加し、室温で3〜5分間インキュベートした。その後、サイトフルーアII蛍光マルチウエルプレートリーダー(Cytofluor II Fluorescence Multiwell Plate Reader)およびサイトフルーアIIソフトウェアと励起用に480nmおよび最大発光用に520nmのフィルターを用いて、各ウエルについての蛍光を測定した。細胞増殖は、二重反復試験サンプルの平均値について、未処理細胞サンプル(=100%)を基準にしたパーセンテージとして判定した。増殖阻害曲線を用いて、IC50値を導出した。
【0240】
図1は、MCF7および正常ヒト皮膚線維芽細胞(NHDF)に対する化合物001およびSAHAの効果を示すものである。MCF7およびNHDFを指示濃度の化合物と共にインキュベートした。6日後、未処理細胞(=100%)を基準にした細胞増殖を、商標シクワント(CyQuant)アッセイを用いて判定した。溶媒対照としての等量のDMSOは、細胞増殖に対してまったく効果がなかった(示されていない)。データは、二重反復判定についての平均±標準偏差であり、複数の実験の代表値である。
【0241】
結果;細胞増殖は、これらの化合物により阻害された。重要なこととして、阻害剤間および細胞タイプ間に差があった。増殖阻害についての平均IC50(±SD)を複数の実験から計算した。それらを下の表1に示す。
【0242】
【表1】
【0243】
化合物001は、癌細胞増殖の強力な阻害剤であった。重要なこととして、化合物001は、現在II相試験中のSAHA(p=0.003、スチューデントt検定)より有意に強力であった。両方の化合物が、NHDFに対してMCF7での方が比較的有効であった。これは、悪性細胞に対する選択性の証拠となる。HUT78 T細胞白血病細胞を使用して行った(上で説明したように行った)同様の試験は、化合物001がこれらの細胞の増殖も阻害することを明示した(データは示さない)。
【0244】
[活性アッセイ2]
本発明者らは、HDAC媒介抑制に応答することが以前に証明されているSV40プロモーターを化合物001(7頁の式(2)の化合物)が活性化するかどうかを調査した。
【0245】
方法.本発明者らは、ルシフェラーゼ遺伝子の上流にクローニングされたSV40即時初期プロモーターが組み込まれたレポーター構築物を含有するMCF7細胞由来の安定なクローンを作製した。トランスファスト(Transfast)トランスフェクション試薬を使用して、pGL2−Basic(SV40/ルシフェラーゼレポーター)(英国のプロメガ(Promega))およびpcDNA3(ネオマイシン耐性遺伝子を発現するもの)をMCF7細胞にトランスフェクトした。24時間後、トリプシン処理により細胞を回収し、低密度でプレーティングした。翌日、G418をその増殖培地に添加した。〜21日後、個々の薬物耐性コロニーを単離し、培養した。本発明者らは、さらなる研究のためにHDAC阻害剤の添加後に高いルシフェラーゼ活性誘発レベルを明示したクローン(2.1.1細胞)を選択した。化合物001の効果を検査するために、2.1.1細胞を様々な濃度の化合物001または対照として等量のDMSOと共にインキュベートした。翌日、細胞を回収し、プロメガ(Promega)ルシフェラーゼアッセイシステムおよびトップカウント(パーキン・エルマー)(TopCount(Perkin−Elmer))を使用してルシフェラーゼ活性を検出した。
【0246】
図2は、HDAC応答性SV40プロモーターの活性に対する化合物001の効果を示すものである。SV40プロモーターが安定的に組み込まれたルシフェラーゼレポータープラスミドを含有するMCF7由来細胞を、指示濃度の化合物001、または溶媒対照としてのDMSOで処理した。ルシフェラーゼ活性は、約16時間後に判定した。データは、二重反復で行った単一の実験から導出したものであり、複数の実験の代表である。
【0247】
結果;この実験は、化合物001が、SV40即時初期プロモーターのHDAC媒介抑制を効果的に逆行させることを示す。
【0248】
[活性アッセイ3]
本発明者らは、MCF7細胞における細胞周期分布および生存に対する化合物001(7頁の式(2)の化合物)およびSAHAの効果を調査した。
【0249】
方法;図の説明文に詳述されているように、MCF7細胞を様々な時間、指示濃度の化合物と共にインキュベートするか、対照として未処理で放置した。細胞を回収し、以前に説明されている(Purohitら,Int J Ca 85,584−9 2000)ようなヨウ化プロピジウム染色法およびフローサイトメトリーを用いて細胞周期の異なる期における細胞の比率を判定した。全細胞を基準にして、<G0/G1含量(プレG0)を有する細胞の比率を、先ず計算した。細胞周期の異なる期(G0/G1、S、G2/M)にある細胞の比率は、その細胞周期内の全細胞(すなわち、<G0/G1含量を有する細胞を引いた全細胞)に対する比率として計算した。
【0250】
図3は、MCF7細胞における細胞周期分布および生存に対する化合物001およびSAHAの効果を示すものである。MCF7細胞を指示濃度(各々、増殖阻害についてのIC50値の10倍までに相当する)の化合物、または溶媒対照としてのDMSOと共に、24、48または72時間インキュベートした。細胞周期の異なる期にある細胞の比率は、フローサイトメトリーによって判定した。データは、1に正規化される未処理細胞にを基準にして、2つの別の実験から導出された平均値±SDを示すものである。
【0251】
結果;この図は、等価有効濃度で、化合物001およびSAHAが、MCF7細胞において主G2/M期停止および細胞死を誘導することを示している。
【0252】
[活性アッセイ4]
本発明者らは、MCF7乳癌細胞および心筋細胞におけるヒストンアセチル化に対する化合物001(7頁の式(2)の化合物)の効果を調査した。
【0253】
方法;心筋細胞を、Chembiochem.2005 Jan;6(1):162−70に記載されているように準備し、指示濃度の化合物001と共に24時間インキュベートした。MCF7細胞を指示濃度の化合物001と共に24時間インキュベートした。アップステート・バイオテック(Upstate Biotech)からの抗体を使用し、以前に説明されている(Brimmellら.Br J Cancer.1999 Nov;81(6):1042−51)ように、ウエスタンブロッティングによって、全ヒストンH4アセチル化、またはヒストンH4−K8もしくはヒストンH3−K9の特異的アセチル化の発現を分析した。
【0254】
図4は、ヒストンアセチル化に対する化合物001の効果を示すものである。(A)MCF7細胞を化合物001と共に24時間インキュベートした。ヒストンH4アセチル化を免疫ブロッティングによって測定した。(B)心筋細胞を指示化合物001と共に24時間インキュベートした。ヒストンH4アセチル化を免疫ブロッティングによって測定した。(C)MCF7細胞を化合物001と共に24時間インキュベートした。ヒストンH3−K9およびヒストンH4−K8アセチル化を免疫ブロッティングによって測定した。
【0255】
結果;この実験は、化合物001が、アセチル化ヒストンH4の蓄積ならびに特異的ヒストンH3−K9およびヒストンH4−K8アセチル化を誘導することを示している。本発明者らは、化合物001が、原発性慢性リンパ球性白血病細胞においてアセチル化ヒストンH4のレベルを増加させることも実証した(データは示さない)。
【0256】
[活性アッセイ5]
本発明者らは、インビトロHDAC活性を阻害する化合物の能力を分析した。インビトロHDACアッセイは、HDAC蛍光活性アッセイキット(英国のバイオモール(Biomol))をその製造業者の説示に従って使用して行った。化合物は、分析前に還元した。一晩、室温で、光から保護して、1mMの化合物をDMSO中30mMのDTTで還元した。その後、96ウエルプレートでの反応を準備した。各反応について、10μLの化合物(アッセイ用緩衝液中の必要濃度の5倍)を15μLの希釈Hela核抽出物(アッセイ用緩衝液中30倍)と混合した。各化合物についての系列希釈物を準備した。Hela抽出物のみを含有する反応物およびアッセイ用緩衝液のみを含む反応物も準備した。25μLの希釈した商標フルーア・ド・リス(Fluor de Lys)基質(アッセイ用緩衝液中100倍)を各反応物に添加し、その後、それらを37℃で1時間インキュベートした。50μLの商標フルーア・ド・リス(Fluor de Lys)顕色剤(アッセイ用緩衝液中20倍の希釈剤、加えて、100倍希釈したTSA)の添加により反応を停止させた。その後、反応物を室温で10分間インキュベートし、その後、サイトフルーアII蛍光マルチウエルプレートリーダーおよびサイトフルーアIIソフトウェアと励起用に360nMおよび発光用に460nMに設定したフィルターを用いて蛍光を測定した。インビトロHDAC活性の阻害は、二重反復試験サンプルの平均値について、Hela抽出物のみの反応物を基準にしたパーセンテージとして判定した。IC50値は、グラフパッド・プリズム(GraphPad Prism)ソフトウェアを使用して計算した。
【0257】
【表2】
【0258】
[活性アッセイ6]
本発明者らは、ヒト癌細胞のインビトロ増殖を阻害する化合物の能力を分析した。細胞増殖アッセイは、商標シクワント(CyQuant)アッセイシステム(米国のモレキュラー・プローブス社(Molecular Probes,Inc.))をその製造業者の説示に従って用いて行った。MCF7乳癌細胞、A2780卵巣癌細胞ならびにPC3およびLNCAP前立腺癌細胞を、96ウエルプレートに、ウエル当たり100μLの細胞培養基中、MCF7細胞については細胞数1000、またはA2780/PC3/LNCAP細胞については細胞数5000の密度でプレーティングした。化合物は、最低5時間後、細胞培養基中の系列希釈で、2×最終濃度での100μL量で添加した。4日後(A2780、PC3もしくはLNCAP細胞)または6日後(MCF7細胞)、プレートを吸い取り紙の上で逆さにすることにより細胞培養基を除去し、細胞を200μLのPBSで1回、穏やかに洗浄した。直ちにプレートを−80℃で最低1時間冷凍し、その後、解凍した。製造業者の説示に従って作った色素を補足した200μLの1x シクワント細胞溶解緩衝液を各ウエルに直ちに添加し、室温で3〜5分間インキュベートした。その後、サイトフルーアII蛍光マルチウエルプレートリーダーおよびサイトフルーアIIソフトウェアと励起用に480nmおよび最大発光用に520nmのフィルターを用いて、各ウエルについての蛍光を測定した。細胞増殖は、二重反復試験サンプルの平均値について、未処理細胞サンプル(=100%)を基準にしたパーセンテージとして判定した。増殖阻害曲線を用いて、IC50値を導出した。
【0259】
【表3】
【0260】
[活性アッセイ7]
本発明者らは、末梢血単核細胞(PBMC)からのTNFαの生産を阻害する化合物の能力を分析した。TNF−αイムノアッセイは、登録商標クアンティキン(Quantikine)ヒトTNF−αアッセイキット(英国、アビンドン(Abingdon,UK)のアール・アンド・ディー・システムズ(R&D systems))をその製造業者の説示に従って使用して行った。商標フィコール・パクー(Ficol paque)プラス(英国、アマシャム(Amersham,UK)のジーイー・ヘルスケア(GE Healthcare)を使用してヒト全血を分離し、PBMCを、24ウエルプレートに、ウエル当たり500μLの細胞培養基中2.5×106の密度でプレーティングした。1時間後、化合物を、6×最終濃度での100μL量で添加した。5時間後、リポ多糖類(LSP;英国、プール(Poole,UK)のシグマ(Sigma))を60×最終濃度の10μL量で添加し、プレートを混合し、37℃、5%CO2で一晩、放置した。プレートを遠心分離し、細胞上清を新たなプレートに移した。登録商標クアンティキン(Quantikine)アッセイ試薬および標準物質をその製造業者の説示に従って調製した。登録商標クアンティキン(Quantikine)アッセイプレートは、50μLのアッセイ希釈液RD1Fを各ウエルに添加することにより準備した。この後、200μLの標準物質または100μLの校正物質希釈液、加えて、100μLの細胞上清を添加し、これを2時間、室温(RT)でインキュベートした。洗浄緩衝液を使用してプレートを4回洗浄した。最終洗浄後、プレートを清浄なペーパータオルで軽くたたいてすべての残留緩衝液を除去した。200μLのTNF−αコンジュゲートを各ウエルに添加し、1時間、室温でインキュベートした。その後、プレートを前のとおり洗浄した。光から保護しながら等量の着色試薬AおよびBを添加することにより必要量の基質溶液を調製し、光から保護して、この混合物の200μLを各ウエルに添加し、20分間、室温でインキュベートした。50uLの停止溶液を基質溶液と同じオーダーで各ウエルに添加し、バイオラド(Biorad)680・96ウエルプレートリーダー(英国、ヘメルヘムステッド(Hemel Hempstead,UK)のバイオラド(Bio−Rad))を用い、570nmのλ補正で、450nmで、プレートを読み取った。TNF−αレベルは、nmでの吸収を用い、二重反復試験サンプルの平均値について判定し、校正ゼロ値をすべての結果から減算して、このアッセイにおける校正物質希釈液の添加を補正した。
【0261】
【表4】
【0262】
[活性アッセイ8]
本発明者らは、マウスにおいて炎症を阻害する化合物9A(7頁の式(2)の化合物)の能力を分析した。毛を刈った腹部の皮膚に5%(w/v)オキサゾロンを塗布することにより、Balb/cマウスを感作した。7日後、左耳に3%(w/v)オキサゾロンおよび右耳にアセトンを局所塗布することによりマウスの炎症を惹起(challenge)した。30分後、アセトンに溶解した試験化合物を耳(背側および腹側の面)に塗布した。24時間後、マウスを安楽死させ、耳の重量を測定した。阻害%は、次の式(Wは、平均重量である)。
【0263】
阻害%=1−((W刺激薬+試験材料−Wヒ゛ヒクル x 100)/(刺激薬ノミ−Wヒ゛ヒクル))
を用いて決定した。
【0264】
【表5】
【図面の簡単な説明】
【0265】
【図1】MCF7および正常ヒト皮膚線維芽細胞(NHDF)に対する化合物001およびSAHAの効果を示す。
【図2】HDAC応答性SV40プロモーターの活性に対する化合物001の効果を示す。
【図3】MCF7細胞における細胞周期分布および生存に対する化合物001およびSAHAの効果示す。
【図4】ヒストンアセチル化に対する化合物001の効果を示す。
【技術分野】
【0001】
本発明は、HDACの阻害剤として作用する特定のデプシペプチドに関する。
【背景技術】
【0002】
ヒストンデアセチラーゼ(HDAC)は、アセチル化リシン残基の加水分解を触媒する亜鉛金属酵素である。ヒストンにおいて、これはリシンをそれらの正常なプロトン化状態に戻し、またこれはヌクレオソームにおいて緊密なDNAパッケージングを生じさせる真核性転写制御の包括的メカニズムである。加えて、可逆的リシンアセチル化は、非ヒストンタンパク質にとっての重要な調節プロセスである。従って、HDACを修飾することができる化合物は、重要な治療上の可能性を有する。
【発明の開示】
【発明が解決しようとする課題】
【0003】
2つの天然産物デプシペプチド、FK228およびスピルコスタチン(Spiruchostatin)Aには、HDAC阻害剤としての可能性があるとの報告がある。しかし、これらの天然産物を化学的に修飾してさらなる類似体を生じさせる可能性は、非常に限られている。
【課題を解決するための手段】
【0004】
驚くべきことに、今般、下に記載する一般式(I)および(I’)の化合物がHDACの阻害剤として作用することを発見した。従って本発明は、HDACの阻害剤として使用するための医薬品の製造における、式(I)もしくは(I’)
【0005】
【化15】
【0006】
(式中、R1、R2、R3およびR4は、同じであるか異なり、アミノ酸側鎖部分を表し、各R6は、同じであるか異なり、水素またはC1−C4アルキルを表し、ならびにPr1およびPr2は、同じであるか異なり、水素またはチオール保護基を表す)
のFK228類似体である化合物、そのアイソスターまたは医薬的に許容されるその塩の使用を提供する。
【0007】
さらに、本発明は、HDACの阻害剤として使用するための医薬品の製造における、上で定義したFK228類似体または医薬的に許容されるその塩の使用を提供する。
【発明を実施するための最良の形態】
【0008】
本明細書で用いられる場合、用語「アミノ酸側鎖部分」は、天然および非天然アミノ酸中に存在する任意のアミノ酸側鎖を指す。非天然アミノ酸由来のアミノ酸側鎖部分の例(括弧内にそれらが由来するアミノ酸を示す)は、−(CH2)2−C(O)−O−C(CH3)3(t−ブトキシカルボニルメチルアラニン)、−(CH2)4−NH−C(O)−O−C(CH3)3(Nε−(t−ブトキシカルボニル)−リシン)、−(CH2)3−NH−C(O)NH2(シトルリン)、−CH2−CH2OH(ホモセリン)および−(CH2)2−CH2NH2(オルニチン)である。特に、−(CH2)3−NH−C(O)NH2(シトルリン)、−CH2−CH2OH(ホモセリン)および−(CH2)2−CH2NH2(オルニチン)を挙げることができる。
【0009】
C1−C6アルキル基または部分は、線状である場合もあり、または分枝状である場合もある。一般に、それは、C1−C4アルキル基または部分、例えば、メチル、エチル、n−プロピル、i−プロピル、n−ブチル、s−ブチルおよびt−ブチルである。好ましい例としては、メチル、i−プロピルおよびt−ブチルが挙げられる。
【0010】
C2−C6アルケニル基または部分は、線状である場合もあり、または分枝状である場合もある。一般に、それは、C2−C4アルケニル基または部分である。アルケニルラジカルは、一または二不飽和であり、さらに好ましくは一不飽和であることが好ましい。例としては、ビニル、アリル、1−プロペニル、イソプロペニル、1−ブテニル、2−ブテニルおよび3−ブテニルが挙げられる。
【0011】
アルキレン基は、二価である前記アルキル基である。
【0012】
前記チオール保護基は、一般に、
(a)チオエーテルを形成してチオール基を保護する保護基、例えば、C1−C6アルコキシ(例えば、メトキシ)、C1−C6アシルオキシ(例えば、アセトキシ)、ヒドロキシおよびニトロにより所望により置換されているベンジル基、ピコリル、ピコリル−N−オキシド、アントリルメチル、ジフェニルメチル、フェニル、t−ブチル、アダマンチル、C1−C6アシルオキシメチル(例えば、ピバロイルオキシメチル、t−ブトキシカルボニルオキシメチル)、
(b)モノチオ、ジチオまたはアミノチオアセタールを形成してチオール基を保護する保護基、例えば、C1−C6アルコキシメチル(例えば、メトキシメチル、イソブトキシメチル)、テトラヒドロピラニル、ベンジルチオメチル、フェニルチオメチル、チアゾリジン、アセトアミドメチル、ベンズアミドメチル、
(c)チオエステルを形成してチオール基を保護する保護基、例えば、t−ブトキシカルボニル(BOC)、アセチルおよびその誘導体、ベンゾイルおよびその誘導体;または
(d)カルバミン酸チオエステルを形成してチオール基を保護する保護基、例えば、カルバモイル、フェニルカルバモイル、C1−C6アルキルカルバモイル(例えば、メチルカルバモイルおよびエチルカルバモイル)
である。
【0013】
一般に、Pr1およびPr2は、同じであるか異なり、各々、水素を表すか、チオエーテル、モノチオ、ジチオもしくはアミノチオアセタール、チオエステルまたはカルバミン酸チオエステルを形成してチオール基を保護する保護基を表す。好ましくは、Pr1およびPr2は、同じであるか異なり、各々、水素を表すか、C1−C6アルコキシ(例えば、メトキシ)、C1−C6アシルオキシ(例えば、アセトキシ)、ヒドロキシおよびニトロにより所望により置換されているベンジル基、ピコリル、ピコリル−N−オキシド、アントリルメチル、ジフェニルメチル、フェニル、t−ブチル、アダマンチル、C1−C6アシルオキシメチル(例えば、ピバロイルオキシメチル、t−ブトキシカルボニルオキシメチル)、C1−C6アルコキシメチル(例えば、メトキシメチル、イソブトキシメチル)、テトラヒドロピラニル、ベンジルチオメチル、フェニルチオメチル、チアゾリジン、アセトアミドメチル、ベンズアミドメチル、t−ブトキシカルボニル(BOC)、アセチルおよびその誘導体、ベンゾイルおよびその誘導体、カルバモイル、フェニルカルバモイルおよびC1−C6アルキルカルバモイル(例えば、メチルカルバモイルおよびエチルカルバモイル)から選択される保護基を表す。最も好ましくは、Pr1およびPr2は、水素である。
【0014】
1つの実施形態において、前記アミノ酸側鎖部分は、天然アミノ酸由来のものである。天然アミノ酸由来のアミノ酸側鎖部分の例(括弧内にそれらが由来するアミノ酸を示す)は、−H(グリシン)、−CH3(アラニン)、−CH(CH3)2(バリン)、−CH2CH(CH3)2(ロイシン)、−CH(CH3)CH2CH3(イソロイシン)、−(CH2)4NH2(リシン)、−(CH2)3NHC(=NH)NH2(アルギニン)、−CH2−(5−1H−イミダゾリル)(ヒスチジン)、−CH2CONH2(アスパラギン)、−CH2CH2CONH2(グルタミン)、−CH2COOH(アスパラギン酸)、−CH2CH2COOH(グルタミン酸)、−CH2−フェニル(フェニルアラニン)、−CH2−(4−OH−フェニル)(チロシン)、−CH2−(3−1H−インドリル)(トリプロファン)、−CH2SH(システイン)、−CH2CH2SCH3(メチオニン)、−CH2OH(セリン)、および−CH(OH)CH3(トレオニン)である。
【0015】
1つの実施形態において、各アミノ酸側鎖は、天然アミノ酸中に存在するアミノ酸側鎖部分であるか、−(CH2)2−C(O)−O−C(CH3)3(t−ブトキシカルボニルメチルアラニン)、−(CH2)4−NH−C(O)−O−C(CH3)3(Nε−(t−ブトキシカルボニル)−リシン)、−(CH2)3−NH−C(O)NH2(シトルリン)、−CH2−CH2OH(ホモセリン)または−(CH2)2−CH2NH2(オルニチン)である。
【0016】
本発明の好ましい実施形態において、各アミノ酸側鎖は、−H、−C1−C6アルキル、−C2−C6アルケニル、−L−O−C(O)−R’、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’、−L−Het−C(O)−Het−R’’および−L−Het−R’’から選択される部分であり、Lは、C1−C6アルキレン基であり、Aは、フェニルまたは5員から6員ヘテロアリール基であり、各R’は、同じであるか異なり、C1−C4アルキルを表し、各R’’は、同じであるか異なり、HまたはC1−C6アルキルを表し、各−Het−は、同じであるか異なり、ならびに−O−、−N(R’’’)−および−S−から選択されるヘテロ原子スペーサーであり、各R’’’は、同じであるか異なり、HまたはC1−C4アルキルを表す。
【0017】
基Aが、5員から6員ヘテロアリール基であるとき、それは、例えば、フラニル、チエニル、ピロリル、オキサゾリル、チアゾリル、イミダゾリル、ピラゾリル、イソオキサゾリル、イソチアゾリル、オキサジアゾリル、トリアゾリル、チアジアゾリル、ピリジル、ピリダジル、ピリミジニル、ピラジニル、トリアジニルであり得る。しかし、一般には、各A部分は、フェニルである。
【0018】
ヘテロ原子スペーサー基Hetは、一般に、−O−または−N(R’’’)−である。さらに一般的には、−O−または−N(H)−である。
【0019】
好ましくは、各アミノ酸側鎖は、−H、−C1−C6アルキル、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’および−L−N(R’’)−C(O)−O−R’’から選択される部分であり、L、AおよびR’’は、先に定義したとおりである。
【0020】
一般に、本発明の化合物のアミノ酸側鎖部分は、−(CH2)2−C(O)−O−C(CH3)3、−(CH2)4−NH−C(O)−O−C(CH3)3、−(CH2)2−C(O)OH、−CH2−C6H5、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2、−CH(CH3)CH2CH3、−(CH2)4NH2、CH2SH、−CH2CH2SCH3、−CH2OHおよび−CH(OH)CH3から選択される。さらに一般的には、アミノ酸側鎖部分は、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2、−CH(CH3)CH2CH3、−(CH2)4NH2、CH2SH、−CH2CH2SCH3、−CH2OHおよび−CH(OH)CH3から選択される。
【0021】
好ましくは、アミノ酸側鎖部分は、−H、−CH3、−(CH2)2−C(O)−O−C(CH3)3、−(CH2)4−NH−C(O)−O−C(CH3)3、−(CH2)2−C(O)OH、−CH2−C6H5、−(CH2)4NH2および−CH(CH3)2から選択される。本発明の1つの実施形態において、アミノ酸側鎖部分は、−H、−CH3および−CH(CH3)2から選択される。
【0022】
一般に、R1は、−H、−C1−C6アルキル、−C2−C6アルケニル、−L−O−C(O)−R’、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’、−L−Het−C(O)−Het−R’’および−L−Het−R’’から選択されるアミノ酸側鎖部分であり、L、R’、R’’、−Het−およびR’’’は、先に定義したとおりである。好ましくは、R1は、−H、−C1−C6アルキル、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’および−L−N(R’’)−C(O)−O−R’’から選択される部分であり、L、AおよびR’’は、先に定義したとおりである。さらに好ましくは、R1は、−Hおよび−C1−C6アルキルから選択される部分である。さらにいっそう好ましくは、R1は、−C1−C4アルキル、特にイソプロピルである。
【0023】
一般に、R2は、−H、−C1−C6アルキル、−C2−C6アルケニル、−L−O−C(O)−R’、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’、−L−Het−C(O)−Het−R’’および−L−Het−R’’から選択されるアミノ酸側鎖部分であり、L、R’、R’’、−Het−およびR’’’は、先に定義したとおりである。好ましくは、R2は、−H、−C1−C6アルキル、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’および−L−N(R’’)−C(O)−O−R’’から選択される部分であり、L、AおよびR’’は、先に定義したとおりである。さらに好ましくは、R2は、−Hおよび−C1−C4アルキルから選択される部分である。さらにいっそう好ましくは、R2は、−H、−CH3または−CH(CH3)2である。さらにより好ましくは、R2は−Hまたは−CH3である。
【0024】
一般に、R3は、−H、−C1−C6アルキル、−C2−C6アルケニル、−L−O−C(O)−R’、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’、−L−Het−C(O)−Het−R’’および−L−Het−R’’から選択されるアミノ酸側鎖部分であり、L、R’、R’’、−Het−およびR’’’は、先に定義したとおりである。好ましくは、R3は、−H、−C1−C6アルキル、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’および−L−N(R’’)−C(O)−O−R’’から選択される部分であり、L、AおよびR’’は、先に定義したとおりである。
【0025】
さらに好ましくは、R3は、−(CH2)2−C(O)−O−C(CH3)3、−(CH2)4−NH−C(O)−O−C(CH3)3、−(CH2)2−C(O)OH、−CH2−C6H5、−CH3、−CH(CH3)2または−(CH2)4NH2である。
【0026】
一般に、R4は、−H、−C1−C6アルキル、−C2−C6アルケニル、−L−O−C(O)−R’、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’、−L−Het−C(O)−Het−R’’および−L−Het−R’’から選択されるアミノ酸側鎖部分であり、L、R’、R’’、−Het−およびR’’’は、先に定義したとおりである。好ましくは、R4は、−H、−C1−C6アルキル、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’および−L−N(R’’)−C(O)−O−R’’から選択される部分であり、L、AおよびR’’は、先に定義したとおりである。さらに好ましくは、R4は、水素、−C1−C6アルキルまたは−C2−C6アルケニルである。さらにいっそう好ましくは、R4は、水素または−C1−C4アルキル、さらに好ましくは水素である。
【0027】
一般に、各R6は、同じであるか異なり、水素または−C1−C2アルキルである。好ましくは、各R6は、水素である。
【0028】
1つの実施形態において、本発明は、式(I)の化合物である、上で定義したとおりのFK228類似体、そのアイソスターまたは医薬的に許容されるその塩を提供する。
【0029】
本発明の好ましい化合物は、式中のR1が、−Hおよび−C1−C6アルキルから選択され、R2が、−Hおよび−C1−C4アルキルから選択され、R3が、−H、−C1−C6アルキル、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’および−L−N(R’’)−C(O)−O−R’’(この場合、L、AおよびR’’は、先に定義したとおりである)から選択され、R4が、−Hおよび−C1−C6アルキルから選択され、ならびにR6が、同じであるか異なり、水素または−C1−C2アルキルである、上で定義したFK228類似体、それらのアイソスターおよびそれらの医薬的に許容される塩である。
【0030】
本発明のさらに好ましい化合物は、(a)式中のR1が、−C1−C4アルキルであり、R2が、−Hおよび−CH3から選択され、R3が、−H、−C1−C6アルキル、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’および−L−N(R’’)−C(O)−O−R’’(この場合、L、AおよびR’’は、先に定義したとおりである)から選択され、R4が、−Hであり、各R6が、−Hである式(I)の化合物、それらのアイソスターおよびそれらの医薬的に許容される塩、ならびに(b)式中のR1が、−C1−C4アルキルであり、R2が、−Hおよび−CH3から選択され、R3が、−H、−C1−C6アルキル、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’および−L−N(R’’)−C(O)−O−R’’(この場合、L、AおよびR’’は、先に定義したとおりである)から選択され、R4が、−Hであり、R6が、−Hであり、Pr1およびPr2が、水素である式(I’)の化合物、それらのアイソスターおよびそれらの医薬的に許容される塩である。
【0031】
式(I)のさらに特に好ましい化合物は、式中の
R1が、−CH(CH3)2であり、R2が、−CH3であり、R3が、−CH3であり、R4が、水素であり、およびR6が、−Hである、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)2−C(O)−O−C(CH3)3であり、R4が、水素であり、およびR6が、−Hである、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)2−C(O)−OHであり、R4が、水素であり、およびR6が、−Hである、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)−C6H5であり、R4が、水素であり、およびR6が、−Hである、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−CH(CH3)2であり、R4が、水素であり、およびR6が、−Hである、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)4−NH−C(O)−O−C(CH3)3であり、R4が、水素であり、およびR6が、−Hである;または
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)4−NH2であり、R4が、水素であり、およびR6が、−Hである
化合物、およびそれらの医薬的に許容される塩である。
【0032】
式(I’)のさらに特に好ましい化合物は、式中の
R1が、−CH(CH3)2であり、R2が、−CH3であり、R3が、−CH3であり、R4が、水素であり、およびR6が、−Hであり、Pr1およびPr2が、水素である、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)2−C(O)−O−C(CH3)3であり、R4が、水素であり、およびR6が、−Hであり、Pr1およびPr2が、水素である、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)2−C(O)−OHであり、R4が、水素であり、およびR6が、−Hであり、Pr1およびPr2が、水素である、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)−C6H5であり、R4が、水素であり、およびR6が、−Hであり、Pr1およびPr2が、水素である、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−CH(CH3)2であり、R4が、水素であり、およびR6が、−Hであり、Pr1およびPr2が、水素である、
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)4−NH−C(O)−O−C(CH3)3であり、R4が、水素であり、およびR6が、−Hであり、Pr1およびPr2が、水素である;または
R1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−(CH2)4−NH2であり、R4が、水素であり、R6が、−Hであり、ならびにPr1およびPr2が、水素である
化合物、およびそれらの医薬的に許容される塩である。
【0033】
好ましいFK228類似体は、式(2)または(2’)である。
【0034】
【化16】
【0035】
別の好ましいFK228類似体は、式(3)または(3’)である。
【0036】
【化17】
【0037】
別の好ましいFK228類似体は、式(4)または(4’)である。
【0038】
【化18】
【0039】
別の好ましいFK228類似体は、式(5)または(5’)である。
【0040】
【化19】
【0041】
別の好ましいFK228類似体は、式(6)または(6’)である。
【0042】
【化20】
【0043】
別の好ましいFK228類似体は、式(7)または(7’)である。
【0044】
【化21】
【0045】
別の好ましいFK228類似体は、式(8)または(8’)である。
【0046】
【化22】
【0047】
別の好ましいFK228類似体は、式(9)または(9’)である。
【0048】
【化23】
【0049】
本発明の1つの実施形態において、FK228類似体は、式(II)または(II’)である。
【0050】
【化24】
【0051】
(式中、R1、R2、R3、R4、R6、Pr1およびPr2は、先に定義したとおりである)
本発明のもう1つの実施形態において、FK228類似体は、式(III)または(III’)である。
【0052】
【化25】
【0053】
(式中、R1、R2、R3、Pr1およびPr2は、先に定義したとおりである)
本発明のもう1つの実施形態において、FK228類似体は、式(IV)または(IV’)である。
【0054】
【化26】
【0055】
(式中、R1、R2、R3、Pr1およびPr2は、先に定義したとおりである)
本発明のもう1つの実施形態において、FK228類似体は、式(V)または(V’)である。
【0056】
【化27】
【0057】
(式中、R1、R2、R3、Pr1およびPr2は、先に定義したとおりである)
さらなる実施形態において、本発明は、HDACの阻害剤として使用するための医薬品の製造における、式(VI)
【0058】
【化28】
【0059】
(式中、R1、R2およびR3は、同じであるか異なり、アミノ酸側鎖部分を表し、ならびにR4は、水素、C1−C6アルキルまたはC2−C6アルケニルである)
の化合物、そのアイソスターまたは医薬的に許容されるその塩の使用を提供する。
【0060】
一般に、この実施形態において、R1は、天然アミノ酸由来のアミノ酸側鎖部分である。好ましくは、この実施形態において、R1は、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2、−CH(CH3)CH2CH3、−(CH2)4NH2、−CH2SH、−CH2CH2SCH3、−CH2OHまたは−CH(OH)CH3である。さらに好ましくは、R1は、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2または−CH(CH3)CH2CH3である。最も好ましくは、R1は、−CH(CH3)2である。
【0061】
一般に、この実施形態において、R2は、天然アミノ酸由来のアミノ酸側鎖部分である。好ましくは、R2は、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2、−CH(CH3)CH2CH3、−(CH2)4NH2、−CH2SH、−CH2CH2SCH3、−CH2OHまたは−CH(OH)CH3である。さらに好ましくは、R2は、−H、−CH3または−CH(CH3)2である。最も好ましくは、R2は、−Hである。
【0062】
一般に、この実施形態において、R3は、天然アミノ酸由来のアミノ酸側鎖部分である。好ましくは、R3は、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2、−CH(CH3)CH2CH3、−(CH2)4NH2、−CH2SH、−CH2CH2SCH3、−CH2OHまたは−CH(OH)CH3である。さらに好ましくは、R3は、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2または−CH(CH3)CH2CH3である。最も好ましくは、R3は、−CH3である。
【0063】
一般に、この実施形態において、R4は、水素またはC1−C6アルキルである。好ましくは、R4は、水素またはC1−C2アルキルである。さらに好ましくは、R4は、水素である。
【0064】
この実施形態において、本発明の好ましい化合物は、式中の
R1が、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2、−CH(CH3)CH2CH3、−(CH2)4NH2、−CH2SH、−CH2CH2SCH3、−CH2OHまたは−CH(OH)CH3であり、
R2が、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2、−CH(CH3)CH2CH3、−(CH2)4NH2、−CH2SH、−CH2CH2SCH3、−CH2OHまたは−CH(OH)CH3であり、
R3が、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2、−CH(CH3)CH2CH3、−(CH2)4NH2、−CH2SH、−CH2CH2SCH3、−CH2OHまたは−CH(OH)CH3であり;および
R4が、水素またはC1−C2アルキルである、
式(VI)の化合物である。
【0065】
この実施形態の好ましい態様において、本発明は、式中の
R1が、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2または−CH(CH3)CH2CH3であり、
R2が、−H、−CH3または−CH(CH3)2であり、
R3が、−H、−CH3、−CH(CH3)2、−CH2CH(CH3)2または−CH(CH3)CH2CH3であり;および
R4が、水素である、
式(VI)の化合物を提供する。
【0066】
この実施形態の特に好ましい化合物は、式(VI)中のR1が、−CH(CH3)2であり、R2が、−Hであり、R3が、−CH3であり、およびR4が、水素である。
【0067】
この実施形態のさらに好ましい化合物は、式(III)の化合物である。
【0068】
【化29】
【0069】
(式中、R1、R2およびR3は、先に定義したとおりである)
この実施形態のさらに好ましい化合物は、式(IV)の化合物である。
【0070】
【化30】
【0071】
(式中、R1、R2およびR3は、先に定義したとおりである)
本発明の化合物は、新規であると考えられ、従って本発明は、式(I)の化合物、そのアイソスターまたは医薬的に許容されるその塩を提供する。さらに、本発明は、式(I)の化合物または医薬的に許容されるその塩を提供する。
【0072】
本発明は、ヒトまたは動物を治療する方法において使用するための、式(I)の化合物、そのアイソスターまたは医薬的に許容されるその塩も提供する。さらに本発明は、ヒトまたは動物を治療する方法において使用するための、式(I)の化合物または医薬的に許容されるその塩も提供する。
【0073】
本発明の化合物は、高い治療効果を示すので、特に有利である。また、そのテトラペプチドコアを容易に合成することができるので、有利である。
【0074】
本明細書で用いられる場合、医薬的に許容される塩は、医薬的に許容される酸または塩基との塩である。医薬的に許容される酸は、無機酸(例えば、塩酸、硫酸、リン酸、二リン酸、臭化水素酸または硝酸)と有機酸(例えば、クエン酸、フマル酸、マレイン酸、リンゴ酸、アスコルビン酸、コハク酸、酒石酸、安息香酸、酢酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸またはp−トルエンスルホン酸)の両方を含む。医薬的に許容される塩基としては、アルカリ金属(例えば、ナトリウムまたはカリウム)およびアルカリ土類金属(例えば、カルシウムまたはマグネシウム)水酸化物、ならびに有機塩基、例えば、アルキルアミン、アラルキルアミンまたは複素環式アミンが挙げられる。
【0075】
本明細書で用いられる場合、用語「アイソスター」は、ある原子または原子群と別の、概して類似している、原子または原子群の交換の結果として生じた化合物を指す。式(I)の化合物において、アイソステリック基を含有する部分は、好ましくは、−NH−CHR1−CO−、−NH−CHR2−CO−O−および−NH−CO−CHR3−NH−CO−である。式(I)の化合物において、アイソステリック基を含有する部分は、さらに好ましくは、−NH−CHR1−CO−および−NH−CHR2−CO−O−である。このようなアイソスターの例は、式中の部分−NH−が、−CH2−、−O−または−S−により置換されている、部分−CO−が、−CS−または−C(=NH)−により置換されている、および部分−O−が、−S−、CH2−または−NH−により置換されている、式(I)の化合物である。
【0076】
本発明の化合物は、式(I)に示すキラリティーを有する。しかし、基R1、R2およびR3の空間配置に起因して化合物内に追加のキラル中心が形成される場合がある。疑いを避けるために、本明細書に描かれている化学構造は、ラセミおよび非ラセミ混合物ならびに純粋なエナンチオマーおよび/またはジアステレオマーを含む、これらの追加のキラル中心を随伴する立体異性体配置のすべてを包含することを意図する。
【0077】
疑いを避けるために、本発明は、インビボで反応して本発明の化合物またはそのアイソスターもしくは医薬的に許容される塩を生じさせるプロドラッグも包含する。
【0078】
本発明の好ましい化合物は、光学異性体である。従って、例えば、キラル中心を1つだけ含有する式(I)の好ましい化合物としては、実質的に純粋な形態のR異性体、実質的に純粋な形態のSエナンチオマー、および過剰なRエナンチオマーまたは過剰なSエナンチオマーを含有するエナンチオマー混合物が挙げられる。
【0079】
本発明は、式(I)の化合物、そのアイソスターまたは医薬的に許容されるその塩および医薬的に許容される担体または希釈剤も提供する。
【0080】
前記医薬組成物は、一般に、85重量%以下の本発明の化合物を含有する。さらに一般的には、それは、50重量%以下の本発明の化合物を含有する。好ましい医薬組成物は、無菌であり、発熱物質を含まない。さらに、本発明により提供される医薬組成物は、実質的に純粋な光学異性体である本発明の化合物を、一般に含有する。好ましくは、本医薬組成物は、式(I)の化合物またはそのアイソスターの医薬的に許容される塩を含む。
【0081】
本FK228類似体は、従来の経路、例えば、以下の図式(この場合、基R1からR4は、上で定義したとおりである)を用いて調製することができる。
【0082】
【化31】
【0083】
段階(a)において、側鎖R2を有するアミノ酸エステルを、側鎖R1を有する第二のアミノ酸と縮合させて、ジペプチドを得る。段階(b)において、そのジペプチドを保護システイン誘導体と縮合させて、トリペプチドを得る。段階(c)において、そのトリペプチドをアミノ酸とカップリングさせて、保護テトラペプチドを生じさせる。段階(d)において、そのテトラペプチドのN末端を脱保護し、その遊離アミンを、β−ヒドロキシ酸誘導体(式中、R5は、一時的ブロッキング基であり、これを除去して、R5がHである化合物を生じさせることができ、およびXは、Yurek−George,A.;Habens,F.;Brimmell,M.;Packham,G.;Ganesan,A.J.Am.Chem.Soc.2004,126,1030−1031に報告されているようなキラル補助基である)とカップリングさせる。段階(e)においてそのエステル基を加水分解し、その後、段階(f)において環化し、(g)においてジスルフィド結合を形成させて、二環式デプシペプチド化合物(I)の合成を完了する。
【0084】
R6が水素以外である本発明の化合物は、R6が水素である本発明の対応する化合物もしくは中間体をアルキル化することにより、または適切に置換された出発材料を使用することにより、得ることができる。
【0085】
式(I’)の化合物は、ジスルフィド結合を開裂するように上記段階(g)の生成物を反応させることによって、得ることができる。一般に、このジスルフィド結合の開裂は、ジスルフィド結合を有するタンパク質の還元処理のために一般に使用されるチオール化合物、例えば、メルカプトエタノール、チオグリコール酸、2−メルカプトエチルアミン、ベンゼンチオール、パラチオクレゾールおよびジチオトレイトールを使用して達成される。好ましくは、メルカプトエタノールおよびジチオトレイトールを使用する。例えば透析またはゲル濾過により、過剰のチオール化合物を除去することができる。あるいは、例えば電解、テトラヒドロホウ酸ナトリウム、水素化アルミニウムリチウムまたは亜流酸塩を使用して、ジスルフィド結合を開裂することができる。
【0086】
Pr1および/またはPr2が水素以外である式(I’)の化合物は、Pr1および/またはPr2が水素である対応する化合物にチオール保護基を導入することによって、調製することができる。この態様において、この反応に用いられるチオール保護基の導入に適する薬剤は、その導入される保護基に依存して適切に決定される。例としては、その対応する保護基の塩化物(例えば、塩化ベンジル、塩化メトキシベンジル、塩化アセトキシベンジル、塩化ニトロベンジル、塩化ピコリル、塩化ピコリル−N−オキシド、塩化アントリルメチル、塩化イソブトキシメチル、塩化フェニルチオメチル)およびその対応する保護基のアルコール(例えば、ジフェニルメチルアルコール、アダマンチルアルコール、アセトアミドメチルアルコール、ベンズアミドメチルアルコール)、ジニトロフェニル、イソブチレン、ジメトキシメタン、ジヒドロピランならびにクロロギ酸t−ブチルが挙げられる。
【0087】
当業者には理解されるように、R1、R2、R3およびR4の1つが、官能基、例えば、−OH、−SH、−NH2または−COOHを有するときには、その導入後のもう1つの反応のためにその基を保護することが好ましい場合がある。このような場合、問題の基は、その導入後、別の段階で保護してもよいし、またはそれを導入する時点で既に保護されていてもよい。当業者は、これに関して使用することができる適当な保護基を知っていることであろう。
【0088】
このようにして得たFK228類似体を、適切な酸または塩基での処理により、塩化することができる。上記プロセスのうちのいずれかによって得られたラセミ混合物は、標準的な技術、例えばキラルクロマトグラフィーカラムでの溶離によって分解することができる。
【0089】
本発明の好ましい化合物は、スベロイルアニリドヒドロキサム酸(SAHA)と少なくとも等しいHDAC阻害活性を有する。従って、さらなる実施形態において、本発明は、SAHAにより示されるものに少なくとも等しいHDAC阻害活性を有する化合物を選択するためのプロセスであって、式(I)または(I’)の化合物を調製することを含み、該調製は以下を含む該プロセス:
(i)式(VI)の化合物と式(VII)の化合物を反応させ、
【0090】
【化32】
【0091】
(式中、R1およびR2は、上で定義したとおりであり、R7は、C1−C4アルキルまたはC2−C4アルケニルであり、ならびにYは、アミノ保護基である)
(ii)得られた中間体を脱保護し、それを式(VIII)の化合物と反応させ、
【0092】
【化33】
【0093】
(式中、Y’は、アミノ保護基であり、およびY’’は、水素または保護基である)
(iii)得られた中間体を脱保護し、それを式(IX)の化合物と反応させ、
【0094】
【化34】
【0095】
(式中、R3は、上で定義したとおりであり、およびY’’’は、アミノ酸保護基である)
(iv)得られた中間体を脱保護し、それを式(X)の化合物と反応させ、
【0096】
【化35】
【0097】
(式中、R4は、上で定義したとおりであり、R5は、水素またはヒドロキシ保護基であり、LGは、脱離基であり、およびY’’’’は、水素または保護基である)
(v)得られた中間体上のβ−ヒドロキシ基を所望により脱保護して、R5保護基を除去し、それをHで置換し、
(vi)得られた中間体を加水分解および環化すること、
(vii)得られた中間体を、所望により、ジスルフィド結合を形成させるように反応させ、そしてジスルフィド結合が形成される場合には、得られた化合物中のジスルフィド結合を所望により開裂し、得られた化合物がチオール基を含有する場合には、チオール保護基を所望により導入し;および
(viii)得られた化合物をスクリーニングして、HDAC阻害剤としてのその活性を測定する。
【0098】
一般に、段階(vi)において、エステル基の加水分解は、環化の前に行う。
【0099】
一般に、段階(vii)において、DTT(ジチオトレイトール)を使用して、ジスルフィド結合の開裂を行う。
【0100】
様々な実体を保護基Y、Y’、Y’’、Y’’’およびY’’’’に用いることができること、ならびにその好ましい実体が、それぞれの場合、存在する特定の基の性質に依存することは、当業者には理解されるであろう。
【0101】
基Y、Y’およびY’’’は、例えば、t−ブトキシカルボニル(Boc)または9−フルオレニルメトキシカルボニル(Fmoc)であり得る。一般に、それらはFmocである。
【0102】
基Y’’およびY’’’’は、例えば、トリチル(Trt)であり得る。
【0103】
当業者は、脱離基LGについての適する実体を知っている。例えば、それは、Yurek−George,A.ら(J.Am.Chem.Soc.2004,126,1030−1031)において説明されているような、そのN原子により結合されているチアゾリジンチオン基などのキラル補助基であり得る。あるいは、それは、−OH基であり得る。
【0104】
基R7は、一般に、C1−C4アルキルまたはC1−C4アルケニル基である。さらに一般的には、メチルまたはアリルである。
【0105】
様々なアッセイが、HDAC阻害についての試験に適すること、およびそれらを用いて、段階(vii)から得られた化合物の活性を測定し、既知HDAC阻害剤SAHAのものと比較できることは、当業者には理解される。従って、HDACに対する試験化合物のIC50を、例えば、あるインビトロアッセイで決定し、その同じアッセイ条件下でのSAHAのIC50と比較することができる。試験化合物が、SAHAのものに等しいか、それより低いIC50値を有する場合、それは、SAHAにより示されるものに少なくとも等しいHDAC阻害活性を有すると理解されよう。
【0106】
好ましい実施形態において、本発明は、段階(viii)におけるスクリーニング段階が、インビトロHDACアッセイである、上で定義したSAHAにより示されるものに少なくとも等しいHDAC阻害活性を有する化合物を選択するためのプロセスを提供する。一般に、前記アッセイは、様々な濃度の試験化合物およびSAHAを希釈Hela核抽出物と接触させて、Hela核抽出物に対するその試験化合物およびSAHAのIC50を決定することを含む。同じアッセイ条件下でのSAHAのIC50と等しいかそれより低い、Hela核抽出物に対して測定されたIC50値を有する試験化合物は、SAHAにより示されるものに少なくとも等しい阻害活性を有すると理解されよう。一般に、前記アッセイは、HDAC蛍光活性アッセイキット(英国のバイオモール(Biomol))を使用して行い、それらの試験化合物は、分析する前に還元する。前記アッセイ試験は、例えば、下の標題「活性アッセイ5」の下で説明するとおり行うことができる。
【0107】
もう1つの実施形態において、本発明は、SAHAにより示されるものに少なくとも等しいヒト癌細胞増殖阻害活性を有する化合物を選択するためのプロセスを提供し、このプロセスは、上で定義したように段階(i)から(vii)により式(I)または(I’)の化合物を調製すること、その後、(viii)得られた化合物をスクリーニングして、ヒト癌細胞増殖阻害剤としてのその活性を測定することを含む。
【0108】
様々なアッセイがヒト癌細胞増殖阻害についての試験に適し、それらを用いて、段階(vii)から得られた化合物の活性を測定し、SAHAのものと比較できることは、当業者には理解されるであろう。従って、ヒト癌細胞増殖に対する試験化合物のIC50を、例えば、あるインビトロアッセイで決定することができ、その同じアッセイ条件下でのSAHAのIC50と比較することができる。試験化合物が、SAHAの活性に等しいかそれより低いIC50値を有する場合、それは、SAHAにより示される活性に少なくとも等しい阻害活性を有すると理解されよう。一般に、この実施形態における段階(viii)は、様々な濃度の試験化合物およびSAHAを、MCF7乳癌細胞系、HUT78 T細胞白血病細胞系、A2780卵巣癌細胞系、PC3またはLNCAP前立腺癌細胞系と接触させて、その細胞系に対するその試験化合物およびSAHAのIC50を決定することを含む、インビトロアッセイを含む。同じアッセイ条件下でのSAHAのIC50に等しいかそれより低い、これらの細胞系のうちのいずれかに対して測定したIC50値を有する試験化合物は、SAHAの活性に少なくとも等しい阻害活性を有すると理解されよう。一般にこの実施形態において、前記アッセイは、商標シクワント(CyQuant)アッセイシステム(米国のモレキュラー・プローブス社(Molecular Probes,Inc.))を用いて行う。前記アッセイ試験は、例えば、下の標題「活性アッセイ6」および/または「活性アッセイ1」の下で説明するとおり行うことができる。
【0109】
もう1つの好ましい実施形態において、本発明は、SAHAにより示される活性に少なくとも等しい抗炎症活性を有する化合物を選択するためのプロセスを提供し、このプロセスは、上で定義したように段階(i)から(vii)により式(I)または(I’)の化合物を調製すること、その後、(viii)そのようにして得られた化合物をスクリーニングして、その抗炎症活性を測定することを含む。
【0110】
様々なアッセイが化合物の抗炎症活性の評価に適することは、当業者には理解されよう。SAHAを基準にした試験化合物の抗炎症活性は、SAHAを基準にして、末梢血単核細胞(PBMC)からのTNFαの生産の阻害に関する化合物の活性を測定することにより、判定することができる。従って、PBMCからのTNFαの生産を阻害する試験化合物の能力を、例えば、あるアッセイにおいて判定し、その同じアッセイ条件下でのSAHAの活性と比較することができる。試験化合物が、同じアッセイ条件下でのSAHAの活性に等しいかそれより高いTNFα生産に対するインビトロ阻害活性を有する場合、それは、SAHAにより示される活性に少なくとも等しい抗炎症活性を有すると理解されよう。一般に、段階(viii)は、登録商標クアンティキン(Quantikine)ヒト−αアッセイキット(英国、アビンドン(Abingdon,UK)のアール・アンド・ディー・システムズ(R&D systems))を使用して行う。前記アッセイ試験は、例えば、下の標題「活性アッセイ7」の下で説明するとおり行うことができる。
【0111】
この実施形態のもう1つの態様において、SAHAを基準にした試験化合物の抗炎症活性は、SAHAを基準にしてBalb/cマウスにおける炎症の阻害に関する化合物の活性を評価することにより判定することができる。試験化合物が、同じ試験条件下でのSAHAのものに等しいかそれより高いインビボ阻害活性を有する場合、それは、SAHAにより示される活性に少なくとも等しい抗炎症活性を有すると理解されよう。一般に、この実施形態における段階(viii)は、化学物質負荷(chemical challenge)により誘発されたBalb/cマウスにおける炎症の阻害に関する試験化合物およびSAHAのインビボ活性を評価することにより行う。一般に、前記化学物質負荷は、前記マウスへのオキサラゾンまたはアセトンの局所投与を含む。この実施形態において、調査中の化合物は、その化学物質負荷の前に適用される場合もあり、または後に適用される場合もある。このような方針に沿った抗炎症活性の評価は、例えば、下の標題「活性アッセイ8」の下で説明するとおり行うことができる。
【0112】
もう1つの好ましい実施形態において、本発明は、SAHAにより示される活性に少なくとも等しいMCF7細胞における主G2/M期停止または細胞死の誘導に関する活性を有する化合物を選択するためのプロセスを提供し、このプロセスは、上で定義したように段階(i)から(vii)により式(I)または(I’)の化合物を調製すること、その後、(viii)得られた化合物をスクリーニングして、SAHAを基準にしてMCF7細胞における主G2/M期停止または細胞死の誘導に関する活性を測定することを含む。スクリーニング段階(viii)は、例えば、下の標題「活性アッセイ3」の下で説明するとおり行うアッセイを含むことができる。
【0113】
上で説明したスクリーニングにおいて、試験化合物の好ましい形態は、そのスクリーニング段階の性質に依存する。従って、スクリーニング段階が、下の「活性アッセイ5」の下で説明するようなインビトロアッセイである場合、その試験化合物は、好ましくは、上で定義したとおりの式(I’)の化合物である。しかし、スクリーニング段階が、細胞系に合わない場合には、試験化合物は、好ましくは、式(I)の化合物である。
【0114】
本発明の化合物は、HDACの阻害剤であることが分かる。従って、本発明の化合物は、治療に有用である。
【0115】
本発明の化合物およびそれらを含む組成物は、様々な剤形で投与することができる。1つの実施形態において、本発明の化合物を含む医薬組成物は、経口、直腸内、非経口、鼻孔内もしくは経皮投与または吸入もしくは坐剤による投与に適する形式で調合することができる。典型的な投与経路は、非経口、鼻孔内もしくは経皮投与または吸入による投与である。
【0116】
本発明の化合物は、経口的に、例えば錠剤、トローチ、ロゼンジ、水性または油性懸濁剤、分散性粉末または顆粒として、投与することができる。本発明の好ましい医薬組成物は、経口投与に適する組成物、例えば、錠剤およびカプセルである。
【0117】
本発明の化合物は、皮下的であっても、静脈内的であっても、筋肉内的であっても、胸骨内的であっても、経皮的であっても、または注入法によっても、非経口的に投与することもできる。本化合物は、坐剤として投与することもできる。
【0118】
1つの好ましい投与経路は、吸入である。吸入薬の主な利点は、経口経路により摂取される多くの薬物と比較して、血液供給が豊富な領域へのそれらの直接送達である。例えば、肺胞は、膨大な表面積および豊富な血液供給を有するので、および初回通過代謝が回避されるので、吸収が非常に速い。
【0119】
従って、本発明の好ましい医薬組成物は、吸入に適するものを含む。本発明は、そのような医薬組成物が含有されている吸入装置も提供する。一般に、前記装置は、定量噴霧式吸入器(MDI)であり、この吸入器には、この吸入器から薬物を押出すための医薬的に許容される化学的噴射剤が含有されている。一般に、前記噴射剤は、フルオロカーボンである。
【0120】
さらに好ましい吸入装置は、ネブライザーを含む。ネブライザーは、加圧下の空気または酸素を用いて、鼻および口に装着する「マスク」を通して薬物の細かい液体ミストを送達することができる装置である。これらは、乳児、小児および実際に病気の全年齢の患者を含む、吸入器を使用することができない喘息患者を治療するために、多くの場合使用される。
【0121】
上記吸入装置は、例えば、噴射剤を用いずに本発明の化合物を送達することができる、回転式吸入器またはドライパウダー用吸入器であってもよい。
【0122】
一般に、前記吸入装置は、スペーサーを含む。スペーサーは、個体が下気道に大量の薬物を直接吸入できるようにする装置であり、喉ではなく下気道に行くようにするためのものである。多数のスペーサーが、吸入器の末端に適合し、あるものについては、薬物のキャニスターがその装置に適合する。保留チャンバおよび逆止め弁を有するスペーサーは、薬物が気道に逃げるのを防止する。多くの人々、特に小児および老人には、定量噴霧式吸入器からパフを発射するために必要な動作と吸入を調整することが難しいことがある。これらの患者には、スペーサーの使用が特に推奨される。
【0123】
もう1つの好ましい投与経路は、鼻孔内投与である。鼻孔の高透過性組織は、薬物を非常に受け入れやすく、錠剤形の薬物よりはるかに迅速、且つ、効率的に薬物を吸収する。経鼻薬物送達は、注射より痛くなく、侵襲でなく、患者間にあまり不安を生じさせない。薬物は、錠剤形で送達される薬物より少ない用量で経鼻送達することができる。この方法による吸収は非常に迅速であり、初回通過代謝が回避され、従って、患者間でのばらつきが低減される。さらに、経鼻送達装置は、正確な計測用量で薬物を投与することができる。従って、本発明の医薬組成物は、一般に、鼻孔内投与に適する。さらに、本発明は、このような医薬組成物が含有されている鼻孔内用装置も提供する。
【0124】
さらに好ましい投与経路は、経皮投与である。従って本発明は、本発明の化合物または医薬的に許容されるその塩を含有する経皮パッチも提供する。舌下投与も好ましい。従って本発明は、本発明の化合物または医薬的に許容されるその塩を含む舌下錠も提供する。
【0125】
本発明の化合物は、一般に、医薬的に許容される担体または希釈剤と共に投与するために調合される。例えば、固体経口剤形は、活性化合物と共に、希釈剤、例えば、ラクトース、デキストロース、サッカロース、セルロース、トウモロコシデンプンまたは馬鈴薯デンプン;滑沢剤、例えば、シリカ、タルク、ステアリン酸、ステアリン酸マグネシウムもしくはステアリン酸カルシウム、および/またはポリエチレングリコール;結合剤、例えば、デンプン、アラビアゴム、ゼラチン、メチルセルロース、カルボキシメチルセルロースまたはポリビニルピロリドン;崩壊剤、例えば、デンプン、アルギン酸、アルジネートまたはデンプングリコール酸ナトリウム;飽和剤;染料;甘味剤;湿潤剤、例えば、レシチン、ポリソルベート、ラウリルスルフェート;および一般に、医薬調合物に使用される非毒性で薬理学的に不活性な物質を含有することがある。このような医薬製剤は、既知の方法で、例えば、混合、造粒、タブレット形成、糖衣またはフィルムコーティングプロセスによって、製造することができる。
【0126】
経口投与用の分散液は、シロップ、乳剤および懸濁剤であり得る。シロップは、担体として、例えば、サッカロースまたはサッカロースとグリセリンおよび/またはマンニトールおよび/またはソルビトールを含有することがある。
【0127】
懸濁剤および乳剤は、担体として、例えば、天然ゴム、寒天、アルギン酸ナトリウム、ペクチン、メチルセルロース、カルボキシメチルセルロース、またはポリビニルアルコールを含有することがある。筋肉内注射用の懸濁剤または溶液は、活性化合物と共に、医薬的に許容される担体、例えば、滅菌水、オリーブ油、オレイン酸エチル、グリコール、例えばプロピレングリコール、および所望される場合には、適量の塩酸リドカインを含有することがある。
【0128】
注射または注入用の溶液は、担体として、例えば滅菌水を含有することがあり、または好ましくは、滅菌等張食塩水溶液の形態であり得る。
【0129】
本発明の化合物は、HDACにより媒介される状態の治療または予防において、治療上、有用である。従って本発明は、HDACにより媒介される状態の治療または予防に使用するための医薬品の製造における、式(I)の化合物または医薬的に許容されるその塩の使用を提供する。HDACにより媒介される状態に罹患しているまたは罹患しやすい患者の治療方法も提供し、この方法は、式(I)の化合物、そのアイソスターまたは医薬的に許容されるその塩の有効量を前記患者に投与することを含む。
【0130】
1つの実施形態において、本発明の化合物は、HDACの別の既知阻害剤、例えばSAHAと併用することができる。この実施形態において、この併用製品は、同時に、別にまたは逐次的に使用するために各医薬品を含むように調合することができる。
【0131】
従って本発明は、(a)上で定義したとおりの本発明のFK228類似体またはそのアイソスターもしくは医薬的に許容される塩;および(b)同時に、別にまたは逐次的に使用するためのHDACの別の既知阻害剤、例えばSAHAを含む製品を提供する。
【0132】
従って本発明は、HDACの別の既知阻害剤、例えばSAHAとの併用投与で使用するための医薬品の製造において使用するための、上で定義したとおりの本発明のFK228類似体またはそのアイソスターもしくは医薬的に許容される塩の使用も提供する。
【0133】
当業者は、HDACの他の既知阻害剤を知っている。例えば、米国特許第20040266769号に適する例が与えられている。例としては、スピルコスタチンA、FR−901228、トリコスタチンAおよびSAHAが挙げられる。
【0134】
本発明の化合物は、癌の治療と予防の両方に使用することができ、ならびに単独療法または併用療法で使用することができる。併用療法において使用されるとき、本発明の化合物は、一般に、小化合物、放射線、抗体ベースの療法(例えば、ヘルセプチンおよびリツキシマブ)、抗癌剤接種、遺伝子療法、細胞療法、ホルモン療法またはサイトカイン療法と共に使用される。
【0135】
本発明の好ましい実施形態において、本発明の化合物は、癌の治療において別の化学療法薬または抗腫瘍薬と併用される。このような他の化学療法薬または抗腫瘍薬の例としては、ミトキサントロン、ビンカアルカロイド、例えばビンクリスチンおよびビンブラスチン、アントラサイクリン、抗生物質、例えばダウノルビシンおよびドキソルビシン、アルキル化剤、例えばクロラムブシルおよびメルファラン、タキサン、例えばパクリタキセル、抗葉酸剤、例えばメトトレキサートおよびトムデックス、エピポドフィロトキシン、例えばエトポシド、カンプトテシン、例えばイリノテカンおよびその活性代謝産物SN38ならびにDNAメチル化阻害剤、例えばWO 02/085400に開示されているDNAメチル化阻害剤が挙げられる。
【0136】
従って、癌を軽減させるために同時に、別にまたは逐次的に使用するための併用製剤として、本発明の化合物と別の化学療法薬または抗腫瘍薬を含有する製品を、本発明に従って提供する。別の化学療法薬または抗腫瘍薬との併用投与により癌を軽減するために使用する医薬品の製造における、上で定義したFK228類似体またはそのアイソスターまたは医薬的に許容されるその塩の使用も、本発明に従って提供する。本発明の化合物および前記他の薬剤は、任意の順序で投与することができる。これら両方の場合において、本発明の化合物および他の薬剤は、共に投与することができ、または別々に投与する場合には、医師により決定されるような任意の順序で投与することができる。
【0137】
HDACは、いくつかの異なる疾病の病理および/または総合的症状の一因であると考えられるので、阻害による被験者におけるHDAC活性の低減を用いて、これらの疾病に治療的に対処することができる。本発明のHDAC阻害剤を使用して治療することができる様々な疾病の例を本明細書で説明する。本明細書に開示するもの以外のさらなる疾病が、後に、様々な経路においてHDACが果す生物学的役割がさらに完全に理解されるにつれて、同定されることがあることに留意する。
【0138】
本発明のHDAC阻害剤を使用して治療することができる適応症の1つのセットは、望ましくないまたは無制御の細胞増殖を伴うものである。このような適応症としては、良性腫瘍、様々なタイプの癌、例えば原発腫瘍および腫瘍転移、再狭窄(例えば、冠動脈、頚動脈および脳の病変)、内皮細胞の異常刺激(アテローム硬化症)、手術に起因する体組織への傷害、異常創傷治癒、異常血管形成、組織の線維増多を生じさせる疾病、反復運動障害、高度な血管新生がない組織の疾患、ならびに臓器移植に関連した増殖反応が挙げられる。HDAC阻害剤のさらに具体的な適応症としては、前立腺癌、肺癌、急性白血病、多発性骨髄腫、膀胱癌、腎癌、乳癌、結腸直腸癌、神経芽腫および黒色腫が挙げられるが、これらに限定されない。
【0139】
1つの実施形態において、望ましくない、無制御の細胞増殖に関連した疾病を治療するための方法を提供する。この方法は、無制御細胞増殖に罹患している患者に、その無制御細胞増殖が低減されるような、本発明のHDAC阻害剤の治療有効量を投与することを含む。使用される阻害剤の特定の投薬量は、その疾病状態の重症度、投与経路、および担当医により決定され得る関連因子に依存する。一般に、許容される有効な日用量は、無制御細胞増殖を有効に遅速または排除するために十分な量である。
【0140】
本発明のHDAC阻害剤を他の薬剤と併用して、望ましくない無制御の細胞増殖を阻害することもできる。本発明のHDAC阻害剤と併用することができる他の抗細胞増殖剤の例としては、レチノイド酸およびその誘導体、2−メトキシエストラジオール、商標アンジオスタチン(ANGIOSTATIN)タンパク質、商標エンドスタチン(ENDOSTATIN)タンパク質、スラミン、スクアラミン、メタロプロテイナーゼ−Iの組織阻害剤、メタロプロテイナーゼ−2の組織阻害剤、プラスミノゲンアクチベータ阻害剤−1、プラスミノゲンアクチベータ阻害剤−2、軟骨由来阻害剤、パクリタキセル、血小板第4因子、硫酸プロタミン(クルペイン)、硫酸化キチン誘導体(ズワイガニの甲羅(queen crab shells)から調製されたもの)、硫酸化多糖類ペプチドグリカン複合体(sp−pg)、スタウロスポリン、例えばプロリン類似体((1−アゼチジン−2−カルボン酸(LACA)、シスヒドロキシプロリン、d,1−3,4−デヒドロプロリン、チアプロリン)を含めた基質代謝の調節剤、β−アミノプロピオニトリルフマレート、4−プロピル−5−(4−ピリジニル)−2(3H)−オキサゾロン;メトトレキセート、ミトキサントロン、ヘパリン、インターフェロン、2 マクログロブリン−血清、chimp−3、キモスタチン、β−シクロデキストリンテトラデカスルフェート、エポネマイシン;フマギリン、金チオリンゴ酸ナトリウム、d−ペニシラミン(CDPT)、β−1−アンチコラゲナーゼ−血清、α2−抗プラスミン、ビスアントレン、ロベンザリット二ナトリウム、n−(2−カルボキシフェニル−4−クロロアントロニル酸二ナトリウムすなわち「CCA」、サリドマイド;angostatic ステロイド、カルボキシアミノイミダゾール;メタロプロテイナーゼ阻害剤、例えばBB94が挙げられるが、これらに限定されない。使用することができる他の抗血管形成剤としては、抗体、好ましくはこれらの血管由来増殖因子:bFGF、aFGF、FGF−5、VEGFアイソフォーム、VEGF−C、HGF/SFおよびAng−1/Ang−2に対するモノクローナル抗体が挙げられる。Ferrara N.およびAlitalo,K.「血管由来増殖因子およびそれらの阻害剤の臨床適用(Clinical application of angiogenic growth factors and their inhibitors)」(1999)Nature Medicine 5:1359−1364。
【0141】
一般に、良性腫瘍における細胞は、それらの分化特徴を保持し、完全無制御様式では分裂しない。良性腫瘍は、通常、局在し、転移性ではない。本発明のHDAC阻害剤を使用して治療することができる良性腫瘍の具体的なタイプとしては、血管腫、肝細胞腺腫、海綿状血管腫、局在性結節性過形成、聴神経腫、神経線維腫、胆管腺腫、胆管嚢胞腺腫、線維腫、リンパ腫、平滑筋腫、中皮腫、奇形腫、粘液腫、肝小結節再生過形成、トラコーマおよび化膿性肉芽腫が挙げられる。
【0142】
悪性腫瘍の場合、細胞は未分化となり、身体の成長制御シグナルに応答せず、無制御様式で増す。悪性腫瘍は、侵襲性であり、また遠位部位に拡大する(転移する)ことができる。悪性腫瘍は、一般に、2つのカテゴリー:原発腫瘍および続発性腫瘍に分けられる。原発腫瘍は、それらが見出される組織から直接発生する。続発性腫瘍、すなわち転移は、身体の他の場所で起こったが、今は遠隔器官に拡大している腫瘍である。一般的な転移経路は、血管またはリンパ系により拡大する隣接構造への直接増殖、ならびに組織面および体腔(腹水、脳脊髄液など)に沿ってのトラッキングである。
【0143】
本発明のDHAC阻害剤を使用して治療することができる、原発または続発性いずれかの癌または悪性腫瘍の具体的なタイプとしては、白血病、乳癌、皮膚癌、骨癌、前立腺癌、肝癌、肺癌、脳癌、喉頭癌、胆嚢癌、膵臓癌、直腸癌、副甲状腺癌、甲状腺癌、副腎癌、神経組織癌、頭頚部癌、結腸癌、胃癌、気管支癌、腎癌、基底細胞癌、潰瘍性タイプと乳頭状タイプの両方の扁平上皮癌、転移性皮膚癌、骨肉腫、ユーイング肉腫、細網細胞肉腫、骨髄腫、巨細胞腫、小細胞肺癌、胆石、島細胞腫瘍、原発性脳腫瘍、急性および慢性リンパ球性および顆粒球性腫瘍、ヘアリーセル腫瘍、腺腫、過形成、髄様癌、褐色細胞腫、粘膜神経腫、腸神経節細胞種、過形成性角膜神経腫瘍、マルファン様体型腫瘍、ウィルムス腫瘍、精上皮腫、卵巣腫瘍、平滑筋腫、子宮頚部形成異常および上皮内癌、神経芽細胞種、網膜芽腫、軟部組織肉腫、悪性カルチノイド、局所的皮膚病変、菌状息肉腫、横紋筋肉腫、カポジ肉腫、骨原性および他の肉腫、悪性高カルシウム血症、腎細胞腫瘍、真性多血症、腺癌、多形性神経膠芽腫、白血病、リンパ腫、悪性黒色腫、類表皮癌、ならびに他の癌腫および肉腫が挙げられるが、これらに限定されない。
【0144】
本発明のHDAC阻害剤を使用して、手術中の体組織への傷害に起因する異常細胞増殖を治療することもできる。これらの傷害は、様々な手術手順、例えば関節手術、腸手術およびケロイド瘢痕化の結果として生じ得る。線維症性組織を生じさせる疾病としては、気腫が挙げられる。本発明を用いて治療することができる反復運動障害としては、手根管症候群が挙げられる。本発明を用いて治療することができる細胞増殖性障害の一例は、骨腫瘍である。
【0145】
本発明のHDAC阻害剤を使用して治療することができる、臓器移植に関連した増殖応答としては、潜在的臓器拒絶反応または関連併発症の一因となる増殖応答が挙げられる。具体的には、これらの増殖応答は、心臓、肺、肝臓、腎臓および他の体器官または器官系の移植中に発生し得る。
【0146】
本発明を用いて治療することができる異常血管形成としては、関節リウマチ、虚血性再潅流関連脳浮腫および傷害、皮質性虚血、卵巣過形成および血管過多、(多嚢胞性卵巣症候群)、子宮内膜症、乾癬、糖尿病性網膜症、ならびに他の眼球の血管由来の疾病、例えば未熟児網膜症(水晶体後線維形成性)、黄斑変性、角膜移植後拒絶反応、神経血管性緑内障およびオスター・ウェーバー症候群に随伴する異常血管形成が挙げられる。
【0147】
本発明に従って治療することができる無制御血管形成に関連した疾病の例としては、網膜/脈絡膜血管新生および角膜血管新生が挙げられるが、これらに限定されない。網膜/脈絡膜血管新生の例としては、ベスト病、近視、眼陥凹、ジュタルガルト病、パジェット病、静脈閉塞症、動脈閉塞症、鎌状赤血球貧血、サルコイド、梅毒、弾性線維性仮性黄色腫頚動脈apo構造疾患、慢性ブドウ膜炎/硝子体炎、マイコバクテリア感染症、ライム病、全身性エリテマトーデス、未熟児網膜症、イールズ病、糖尿病性網膜症、黄斑変性、ベーチェット病、網膜炎または脈絡膜炎に起因する感染、推定眼ヒストプラズマ症、扁平部炎、慢性網膜剥離、過粘稠度症候群、トキソプラズマ症、外傷およびレーザー後併発症、ルベオーシスに関連した疾病(隅角の血管新生)、ならびにすべての形態の増殖性硝子体網膜症を含む線維血管性組織または線維組織の異常増殖に起因する疾病が挙げられるが、これらに限定されない。角膜血管新生の例としては、流行性角結膜炎、ビタミンA欠乏症、コンタクトレンズ装着時間過剰(contact lens overwear)、アトピー性角膜炎、上方輪部角膜炎、翼状片乾性角膜炎、シューグレン病、酒さ性角膜炎、フクテリン症、糖尿病性網膜症、未熟児網膜症、角膜移植後拒絶反応、モーレン潰瘍、テリエン辺縁変性、辺縁表皮剥離(marginal keratolysis)、多発性動脈炎、ウェグナー病、強膜炎、類天疱瘡の放射状角膜切開、血管新生緑内障および水晶体後方線維増殖、梅毒、マイコバクテリア感染症、脂質変性、化学熱傷、細菌性潰瘍、真菌性潰瘍、単純疱疹感染症、帯状疱疹感染症、原虫感染症ならびにカポジ肉腫が挙げられるが、これらに限定されない。
【0148】
無制御の血管形成に関連した慢性炎症性疾患も、本発明のHDAC阻害剤を使用して治療することができる。慢性炎症は、炎症性細胞の流入を維持する毛細血管の芽の継続的形成に依存する。炎症細胞の流入および存在が肉芽を生じさせ、そうして慢性炎症状態を維持する。HDAC阻害剤を単独で使用または他の抗炎症薬と併用して血管形成を阻害することにより、肉芽形成を予防することができ、従って、疾病を軽減させることができる。慢性炎症性疾患の例としては、炎症性腸疾患、例えばクローン病および潰瘍性大腸炎、乾癬、サルコイドーシス、および関節リウマチが挙げられるが、これらに限定されない。
【0149】
炎症性腸疾患、例えば、クローン病および潰瘍性大腸炎は、胃腸管内の様々な部位における慢性炎症および血管形成を特徴とする。例えば、クローン病は、最も一般的には遠位回腸および結腸に影響及ぼす慢性経壁性炎症性疾患として発生するが、口から肛門および肛門周囲領域までの胃腸管のあらゆる部分においても発生し得る。クローン病の患者は、一般に、腹痛、発熱、食欲低下、体重減少および腹部膨満を伴う慢性下痢を有する。潰瘍性大腸炎は、結腸粘膜において発生する慢性で非特異的な炎症性および潰瘍性疾患でもあり、血性下痢の存在を特徴とする。一般に、これらの炎症性腸疾患は、炎症性細胞の柱体によって包囲された新たな毛細血管の芽を伴う、胃腸管全体にわたる慢性肉芽腫性炎症によって引き起こされる。これらの阻害剤による血管形成の阻害は、それらの芽の形成を阻害し、肉芽腫の形成を防止するはずである。炎症性腸疾患は、皮膚病変などの追加の腸性症状発現も示す。このような病変は、炎症および血管形成を特徴とし、胃腸管以外の多数の部位で発生し得る。本発明のHDAC阻害剤による血管形成の阻害は、炎症性細胞の流入を低減させ、病変形成を予防することができる。
【0150】
サルコイドーシスはもう1つの慢性炎症性疾患であり、これは多系統型肉芽腫性疾患として特徴付けられる。この疾病の肉芽腫は、身体のあらゆる場所で形成し得る。従って、それらの症状は、肉芽腫の部位、およびその疾病が活性であるかどうかに依存する。肉芽腫は、炎症性細胞の恒常的供給をもたらす血管形成性毛細血管芽によって作られる。血管形成を阻害する本発明のHDAC阻害剤の使用により、そのような肉芽腫形成を阻害することができる。乾癬も慢性および再発性炎症性疾患であり、これは、様々なサイズの丘疹およびプラークを特徴とする。これらの阻害剤を単独で使用または他の抗炎症薬と併用することによる治療は、それらの特徴的病変を維持するために必要な新たな血管の形成を防止し、患者の症状を軽減させるはずである。
【0151】
関節リウマチ(RA)も、末梢関節の非特異的炎症を特徴とする慢性炎症性疾患である。関節滑膜表層内の血管が、血管形成されると考えられている。新たな血管網の形成に加えて、内皮細胞が、パンヌス成長および軟骨破壊を招く因子および反応性酸素種を放出する。血管形成に関与する因子は、関節リウマチの慢性炎症状態に積極的に貢献し、前記状態の維持を助長することができる。本発明のHDAC阻害剤を単独で使用または他の抗RA剤と併用することによる治療は、慢性炎症を維持するために必要な新たな血管の形成を防止することができる。
【0152】
さらに、本発明の化合物は、心臓/血管系疾患、例えば、肥大、高血圧症、心筋梗塞、再潅流、虚血性心疾患、アンギナ、不整脈、高コレステロール血症、アテローム硬化症および卒中の治療において使用することができる。さらに、これらの化合物は、神経変性疾患/CNS障害、例えば、卒中、ハンチングトン病、筋萎縮性側索硬化症およびアルツハイマー病を含む急性および慢性神経疾患を治療するために使用することができる。
【0153】
本発明の化合物は、抗微生物剤、例えば抗菌剤として使用することもできる。従って本発明は、細菌感染症の治療において使用するための化合物も提供する。本発明の化合物は、ウイルス、細菌、真菌および寄生虫感染に対する抗感染症化合物として使用することができる。感染症の例としては、(プラスモディウム属(plasmodium)、クリプトスポリジウム・パルヴム(cryptosporidium parvum)、トキソプラズマ・ゴンヂ(toxoplasma gondii)、サルコシスティス・ニューロナ(sarcocystis neurona)およびエイメリア属(Eimeria)種を含む)原虫寄生虫感染症が挙げられる。
【0154】
本発明の化合物は、望ましくないまたは無制御の細胞増殖の治療に、好ましくは良性腫瘍/過形成および悪性腫瘍の治療に、さらに好ましくは悪性腫瘍の治療に、ならびに最も好ましくはCCL、乳癌およびT細胞リンパ腫の治療に、特に適する。
【0155】
本発明の好ましい実施形態において、本発明の化合物は、癌、心肥大症、慢性心不全、炎症状態、心血管疾患、ヘモグロビン異常症、サラセミア、鎌状赤血球症、CNS障害、自己免疫疾患、糖尿病、骨粗しょう症、MDS、良性前立腺肥大症、口腔白板症、遺伝的代謝性疾患、感染症、ルビンス−テイビ(Rubens−Taybi)、脆弱X症候群もしくはα−1アンチトリプシン欠損症を軽減させるために、または創傷治癒を加速させるために、または毛包を保護するために、または免疫抑制薬として使用される。
【0156】
一般に、前記炎症状態は、皮膚の炎症状態(例えば、乾癬、アクネおよび湿疹)、喘息、慢性閉塞性肺疾患(COPD)、関節リウマチ(RA)、炎症性腸疾患(IBD)、クローン病または大腸炎である。
【0157】
一般に、前記癌は、慢性リンパ球性白血病、乳癌、前立腺癌、卵巣癌、中皮腫またはT細胞リンパ腫である。
【0158】
一般に、前記心血管疾患は、高血圧症(hypertension)、心筋梗塞(MI)、虚血性心疾患(IHD)(再潅流)、狭心症、不整脈、高コレステロール血症、高脂血症、アテローム硬化症、卒中、心筋炎、うっ血性心不全、原発性および続発性ie拡張型(うっ血型)心筋症、肥大性心筋症、収縮性心筋症、末梢血管疾患、頻脈、高血圧(high blood pressure)または血栓症である。
【0159】
一般に、前記遺伝的代謝性疾患は、嚢胞性線維症(CF)、ペルオキソーム形成不全(peroxisome biogenesis disorder)または副腎性白質ジストロフィーである。
【0160】
一般に、本発明の化合物は、臓器移植後に免疫抑制薬として使用される。
【0161】
一般に、前記感染症は、ウイルス、細菌、真菌または寄生虫感染症、特に、黄色ブドウ球菌(S.aureus)、瘡プロピオニバクテリウム(P.acne)、カンジダ属またはアスペルギルス属による感染症である。
【0162】
一般に、前記CNS障害は、ハンチングトン病、アルツハイマー病、多発性硬化症または筋萎縮性側索硬化症である。
【0163】
好ましくは、この実施形態において、本発明の化合物は、癌、心肥大症、慢性心不全、炎症状態、心血管疾患、ヘモグロビン異常症、サラセミア、鎌状赤血球症、CNS障害、自己免疫疾患、糖尿病または骨粗しょう症を軽減するために使用されるか、免疫抑制薬として使用される。
【0164】
最も好ましくは、本発明の化合物は、慢性リンパ球性白血病、乳癌、前立腺癌、卵巣癌、中皮腫、T細胞リンパ腫、心肥大症、慢性心不全または皮膚の炎症状態、特に、乾癬、アクネもしくは湿疹を軽減するために使用される。
【0165】
本発明の化合物は、動物の治療、好ましくは哺乳動物の治療、さらに好ましくはヒトの治療に使用することができる。
【0166】
本発明の化合物は、適切な場合には、このような状態の発生率を低下させるために予防的に使用することができる。
【0167】
本発明の化合物の治療有効量を患者に投与する。典型的な用量は、その具体的な化合物の活性、治療すべき被験者の年齢、体重および状態、その疾病のタイプおよび重症度、ならびに投与頻度および経路に従って、体重のkg当たり約0.001から50mgである。好ましくは、日用量レベルは、5mgから2gである。
【0168】
以下の実施例は、本発明を説明するものである。しかし、これらは、いかなる点においても本発明を限定するものではない。これに関連して、実施例セクションにおいて用いている特定のアッセイは、HDACの阻害に関する活性の指標を提供するためだけに設計されたものであること理解することが重要である。所与の化合物のHDAC拮抗薬としての活性を判定するために利用できるアッセイは多数あり、従って、いずれか1つの特定のアッセイにおける否定的な結果に決定力があるわけではない。
【0169】
[実施例]
(E)−(1S,7R,10S,21R)−7−イソプロピル−21−メチル−2−オキサ−12,13−ジチア−5,8,20,23−テトラアザ−ビシクロ[8.7.6]トリコス−16−エン−3,6,9,19,22−ペンタオン(以後、化合物001と呼ぶ)は、下に示す図式を用いて調製した。
【0170】
【化36】
【0171】
[(R)−2−(9H−フルオレン−9−イルメトキシカルボニルアミノ)−3−メチル−ブチリルアミノ]−酢酸メチルエステル(2)の調製
CH2Cl2(50mL)中のFmoc保護D−バリン(2.70g、7.96mmol)の攪拌溶液に、EDAC・HCl(1.83g、9.55mmol)、HOBt(1.30g、9.55mmol)およびDIEA(3.8mL、27.86mmol)を添加した。室温で15分間攪拌した後、グリシンメチルエステル(1、1.0g、7.96mmol)を添加し、その反応混合物を4時間攪拌し、CH2Cl2(50mL)で希釈し、水(25mL)、10% HCl(25mL)、5% NaHCO3(25mL)および飽和NaCl(25mL)溶液で洗浄し、乾燥させ(Na2SO4)、濾過し、濃縮して、オフホワイトの固体を得、それをCH3CNから再結晶させて、2を白色の固体として得た(2.81g、86%):融点=148〜150℃;[α]22D−7.32(c 0.50、CHCl3);[α]22D−7.32(c 0.50、CHCl3);IR νmax 3287、1750、1690、1649、1535cm-1;1H NMR 400MHz 7.75(d,J=7.5Hz,2H)、7.57(d,J=7.0Hz,2H)、7.39(t,J=7.0Hz,2H)、7.28(m,2H)、6.54(s,1H)、5.44(s,1H)、7.43−4.38(m,2H)、4.21(t,J=7.0Hz,1H)、4.01−3.96(m,3H)、3.72(s,3H)、2.16(m,1H)、0.96(t,J=9.0Hz,6H);13C NMR 100MHz 171.7(C)、170.1(C)、156.6(C)、143.9(C)、141.4(C)、127.8(CH)、127.2(CH)、125.2(CH)、120.1(CH)、67.2(CH2)、60.4(CH)、52.5(CH)、47.3(CH3)、41.2(CH2)、31.2(CH)、19.3(CH3)、17.9(CH);MS m/z 432.9(M+Na)+、842.8(2M+Na)+。
【0172】
{(R)−2−[(S)−2−(9H−フルオレン−9−イルメトキシカルボニルアミノ)−3−(トリチルスルファニル)−プロピオニルアミノ]−3−メチル−ブチリルアミノ}−酢酸メチルエステル(3)の調製
室温でCH3CN(48.5mL)中の2(1.0g、2.44mmol)の攪拌溶液に、Et2NH(2.5mL)を添加した。3時間、室温で攪拌した後、その反応混合物をヘキサン(100mL)で希釈し、濃縮して、粗製アミンを無色の油として得た。CH3CN(25mL)中のFmoc−(STrt)−D−システイン(1.70g、2.9mmol)の攪拌溶液に、EDAC・HCl(561.0mg、2.93mmol)、HOBt(396mg、2.93mmol)およびDIEA(1.31mL、7.32mmol)を添加した。室温で15分間攪拌した後、その粗製アミンを添加し、その反応混合物を4時間攪拌し、溶媒を除去し、残留物をCH2Cl2(100mL)に溶解し、水(25mL)、10% HCl(25mL)、5% NaHCO3(25mL)および飽和NaCl(25mL)溶液で洗浄し、乾燥させ(Na2SO4)、濾過し、溶媒を除去して、オフホワイトの固体を得、それをCH3CNから再結晶させて、3を白色の固体として得た(1.46g、79%):[α]22D−2.35(c 0.50、CHCl3);IR νmax 3267、1646、1543cm-1;1H NMR 400MHz 7.76(t,J=7.0Hz,2H)、7.54(d,J=6.5Hz,2H)、7.39−7.21(m 19H)、6.87(s,1H)、6.23(d,J=8.0,2H)、5.04(d,J=7.0,1H)、4.37(d,J=7.0,2H)、4.29(dd,J=8.6,J=5.0,1H)、4.17(t,J=6.5,1H)、3.96(m,1H)、3.72(dd,,J=18.1,J=5.0,1H)、3.65(s,3H)、3.60(m,1H)、2.70(d,J=6.5,2H)、2.30(m,1H)、1.70(s,1H)、0.87(dd,J=13.0,J=7.0Hz,6H);13C NMR 100MHz 170.6(C)、170.5(C)、170.1(C)、156.2(C)、144.3(C)、143.8(C)、143.7(C)141.4(C)、129.6(CH)、128.4(CH)、128.0(CH)、127.8(CH)、127.2(CH)、125.1(CH)、120.2(CH)、67.6(CH)、67.2(CH2)、58.4(CH)、54.3(CH)、52.4(CH3)、47.1(CH)、40.9(CH2)、33.6(CH2)、30.0(CH)、19.4(CH3)、17.4(CH3);MS m/z 777.8(M+Na)+。C45H45N3O6Sについての分析計算値:71.50;H,6.00;N,5.56。実測値 C,71.42;H,5.99;N,5.55。
【0173】
((R)−2−{(S)−2−[(R)−2−(9H−フルオレン−9−イルメトキシカルボニルアミノ)−プロピオニルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(4)の調製
室温でCH3CN(30mL)中の3(400mg、0.53mmol)の攪拌溶液に、Et2NH(1.5mL)を添加した。3時間、室温で攪拌した後、その反応混合物をヘプタン(60mL)で希釈し、濃縮して、粗製アミンを無色の油として得た。CH3CN(25mL)中のFmoc−D−アラニン(198mg、0.636mmol)の攪拌溶液に、EDAC・HCl(122.0mg、0.634mmol)、HOBt(86mg、0.636mmol)およびDIEA(502μL、1.91mmol)を添加した。室温で15分間攪拌した後、その粗製アミンを添加し、その反応混合物を18時間攪拌し、溶媒を除去し、残留物をCH2Cl2(50mL)に溶解し、水(15mL)、10% HCl(15mL)、5% NaHCO3(15mL)および飽和NaCl(15mL)溶液で洗浄し、乾燥させ(Na2SO4)、濾過し、溶媒を除去して、オフホワイトの固体を得、それをCH3CNから再結晶させて、4を白色の固体として得た(670mg、81%):融点=195〜197℃;[α]22D +5.1(c 0.50、CHCl3);IR νmax 3267、3054、1744、1706、1635、1531.1cm-1;1H NMR 400MHz 7.75(d,J=7.5Hz,2H)、7.52(d,J=7.0Hz,2H)、7.39(m,7H)、7.26−7.15(m,13H)、6.79(s,1H)、6.62(s,1H)、5.47(s,1H)、4.43−4.28(m,4H)、4.13(m,1H)、4.01(m,1H)、3.88(s,2H)、3.63(s,3H)、2.81(m,1H)、2.52(m,1H)、2.26,(m,1H)、1.30(d,J=6.0Hz,3H)、0.94(d,J=6.0Hz,3H)、0.89(d,J=7.0Hz,3H);13C NMR 100MHz 172.7、171.1、170.2、156.1、144.4、143.9、143.8、141.4、129.6、128.3、127.9、127.2、127.1、125.1、120.1、58.5、52.7、52.3、50.8、47.2、41.0、33.5、30.4、19.3、19.0、17.7;MS m/z 849.2(M+Na)+、865.1(M+K)+。
【0174】
((R)−2−{(S)−2−[(R)−2−((E)−(S)−3−ヒドロキシ−7−トリチルスルファニル−ヘプト−4−エノイルアミノ)−プロピオニルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(6)の調製
【0175】
【化37】
【0176】
室温でCH3CN(10mL)中の4(137mg、0.166mmol)の攪拌溶液に、Et2NH(0.5mL)を添加した。5時間、室温で攪拌した後、その反応混合物をヘプタン(30mL)で希釈し、溶媒を除去し、CH2Cl2(10mL)を添加し、濾過し、濃縮して、粗製アミンを無色の油として得た。CH2Cl2(4mL)中のその粗製アミンの攪拌溶液に、0℃で5(84.0mg、0.149mmol、Yurek−George,A.;Habens,F.;Brimmell,M.;Packham,G.;Ganesan,A.J.Am.Chem.Soc.2004,126,1030−1031における手順に従って調製したもの)およびDMAP(2.2mg、0.0176mmol)を添加した。室温で8時間攪拌した後、溶媒を除去し、残留物をフラッシュクロマトグラフィー(溶離剤 30〜40% EtOAc/ヘキサン)によって精製して、6を白色のガラスとして得た(127mg、85%):融点=191〜193℃;[α]22D−18.0(c 0.50、CHCl3);IR νmax 3272、3064、1758、1692、1621cm-1;1H NMR 400MHz(5% CD3OD/CDCl3)7.40(m,12H)、7.25(m,12H)、7.20(m,6H)、6.96および6.88(不安定なNH,d,J=8.0Hz,1H)、5.49(dt,J=15.0Hz,6.5Hz,1H)、5.37(dd,J=15.5Hz,6.0Hz,1H)、4.32(m,2H)、4.22(d,J=6.0Hz,1H)、3.98(t,J=7.0Hz,1H)、3.92(d,J=18.1Hz,1H)、3.72(d,J=17.6Hz,1H)、3.67(s,3H)、2.64(dd,J=13.0Hz,7.5Hz,1H)、2.58(dd,J=13.0Hz,7.5Hz,1H)、2.56−2.18(m,9H)、2.11(q,J=6.5Hz,2H)、1.31(d,J=7.5Hz,3H)、0.91(t,J=7.0Hz,6H);13C NMR 100MHz(5% CD3OD/CDCl3)173.0、172.0、171.3、170.5、170.2、144.9、144.2、132.8、129.9、129.7、129.6、129.5、128.2、128.1、127.9、127.1、126.7、69.6、67.1、66.7、58.9、58.8、52.7、52.2、50.0、49.8、49.6、49.5、49.4、49.1、43.7、40.9、40.8、33.1、31.5、31.3、30.0、19.2、17.5;MS m/z 1050.4(M+2Na)+。
【0177】
((R)−2−{(S)−2−[(R)−2−((E)−(S)−3−ヒドロキシ−7−トリチルスルファニル−ヘプト−4−エノイルアミノ)−プロピオニルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸(7)の調製
0℃で4:1 THF/H2O(4.0mL)中の6(220mg、0.222mmol)の攪拌溶液に、LiOH(11mg、0.440mmol)を添加した。1時間攪拌した後、その反応混合物をH2O(30mL)で希釈し、2M KHSO4でpH4〜5に酸性化し、EtOAc(3×30mL)で抽出した。有機層を飽和NaCl(15mL)で洗浄し、乾燥させ(Na2SO4)、濾過し、濃縮して、白色の固体を得、それをエーテルと研和して、7を白色の固体として得(199.5mg、91%)、それをそのまま次の段階で直接使用した。融点=191〜193℃;[α]22D−18.5(c 0.50、CHCl3);IR νmax 3413、1711、1678、1630、1451cm-1;1H NMR 400MHz(5% CD3OD/CDCl3)7.30(m,12H)、7.16(m,12H)、7.13(m,6H)、5.43(dt,J=15.5Hz,6.0Hz,1H)、5.30(dd,J=15.5Hz,6.0Hz,1H)、4.27(m,2H)、4.21(m,1H)、3.99(m,1H)、3.75(s,2H)、3.77(m,br,6H)、2.55(dd,J=12.5Hz,6.5Hz,1H)、2.44(dd,J=12.5Hz,7.5Hz,1H)、2.21(m,2H)、2.12(m,3H)、2.02(m,2H)、1.23(d,J=7.0Hz,3H)、0.83(d,J=7.0Hz,3H)、0.80(d,J=7.0Hz,3H);13C NMR 100MHz(5% CD3OD/CDCl3)173.0、172.1、171.6、171.5、170.5、144.9、144.3、132.7、129.8、129.6、129.5、128.2、127.9、127.0、126.7、69.5、67.1、66.7、58.7、52.7、43.5、40.9、33.2、31.5、31.3、30.3、19.2、19.1、17.6;MS m/z 1013.2(M+Na)+。
【0178】
(6R,9S,12R,16S)−6−イソプロピル−12−メチル−16−((E)−4−トリチルスルファニル−ブト−1−エニル)−9−トリチルスルファニルメチル−1−オキサ−4,7,10,13−テトラアザ−シクロヘキサデカン−2,5,8,11,14−ペンタオン(8)の調製
0℃でCH2Cl2(5mL)中のヒドロキシル酸7(100mg、0.102mmol)の攪拌溶液に、CH2Cl2(0.5mL)中のDCC(28.2mg、0.137mmol)の溶液を1滴ずつ添加した。30分間、0℃で攪拌した後、CH2Cl2(51mL)中のDMAP(2.5mg、0.0213mmol)の溶液を添加し、その後、16時間、室温で攪拌した。固体沈殿物を濾過して除去し、その濾液をCH2Cl2(5mL)で洗浄した。有機層を水(10mL)、5% KHSO4(10mL)、5% NaHCO3(10mL)および飽和NaCl(10mL)溶液で洗浄し、乾燥させ(Na2SO4)、濾過し、濃縮した。残留物をフラッシュクロマトグラフィー(溶離剤 30〜50% EtOAc/ヘキサン)によって精製して、8を白色のガラスとして得た(37mg、38%)。
【0179】
(E)−(1S,7R,10S,21R)−7−イソプロピル−21−メチル−2−オキサ−12,13−ジチア−5,8,20,23−テトラアザ−ビシクロ[8.7.6]トリコス−16−エン−3,6,9,19,22−ペンタオン(化合物001)の調製
MeOH(50mL)中のI2(37.1mg、0.146mmol)の強力攪拌溶液に、MeOH(20mL)中の保護ジオール8(35mg、0.0365mmol)を5分かけて1滴ずつ添加した。さらに10分間攪拌した後、0.2M クエン酸緩衝液(4mL)中の0.2M アスコルベートを添加し、有機相を濃縮し、1:1 EtOAc/NaCl(20mL)を添加し、EtOAc(5×25mL)で抽出し、合わせた有機抽出物を乾燥させ(Na2SO4)、濾過し、溶媒を除去した。残留物をフラッシュクロマトグラフィー(溶離剤 1〜6% MeOH/CHCl3)によって精製して、化合物001を白色の固体として得た(11.8mg、67%):[α]22D−98.0(c 0.50、CHCl3);IR νmax 3300、2477、1734、1659、1526、1446cm-1;1H NMR 400MHz 7.41(d,J=6.0Hz,1H)、7.23(d,J=8.5Hz,1H)、6.37(s,1H)、5.94(dt,J=16.5,7.5Hz,1H)、5.78−5.71(m,1H)、4.86(m,1H)、4.26(d,J=7.5Hz,1H)、4.17(d,J=14.0Hz,1H)、4.08(d,J=14.0Hz,1H)、3.44(dd,J=14.5Hz,10.0Hz,1H)、3.22(d,J=10.0Hz,1H)、2.88−2.56(m,8H)、1.49(d,J=7.5Hz,3H)、0.98(d,J=6.5Hz,3H)、0.92(d,J=6.5Hz,3H);13C NMR 100MHz 173.2、171.9、170.9、170.1、168.3、129.8、70.2、64.4、55.9、51.7、41.7、38.2、37.9、35.7、31.0、26.9、20.2、19.7、15.4;HRMS m/z 509.1495(M+Na)+ 予測値 509.1499。
【0180】
式(I)によって表される類似体の合成のための一般手順。
【0181】
段階(a)において、側鎖R2を有するアミノ酸エステルを、側鎖R1を有する第二のアミノ酸と縮合させて、ジペプチドを得る。段階(b)において、そのジペプチドを保護システイン誘導体と縮合させて、トリペプチドを得る。段階(c)において、そのトリペプチドをアミノ酸とカップリングさせて、保護テトラペプチドを生じさせる。段階(d)において、そのテトラペプチドのN末端を脱保護し、その遊離アミンをβ−ヒドロキシ酸誘導体とカップリングさせる。本発明の好ましい実施形態では、R4およびR5は、Hであり、Xは、Yurek−George,A.;Habens,F.;Brimmell,M.;Packham,G.;Ganesan,A.J.Am.Chem.Soc.2004,126,1030−1031において報告されているようなキラル補助基である。段階(e)においてそのエステル基を加水分解し、その後、段階(f)において環化し、(g)においてジスルフィド結合を形成して、二環式デプシペプチド化合物(I)の合成を完了する。
【0182】
【化38】
【0183】
一般図式による類似体9A〜Fの合成。
【0184】
【化39】
【0185】
[(R)−2−(9H−フルオレン−9−イルメトキシカルボニルアミノ)−3−メチル−ブチリルアミノ]−酢酸メチルエステル(2A)の調製。
【0186】
CH2Cl2(50mL)中のFmoc保護D−バリン(2.70g、7.96mmol)の攪拌溶液に、EDAC・HCl(1.83g、9.55mmol)、HOBt(1.30g、9.55mmol)およびDIEA(3.8mL、27.86mmol)を添加した。室温で15分間攪拌した後、グリシンメチルエステル(1A、1.0g、7.96mmol)を添加し、その反応混合物を4時間攪拌し、CH2Cl2(50mL)で希釈し、水(25mL)、10% HCl(25mL)、5% NaHCO3(25mL)および飽和NaCl(25mL)溶液で洗浄し、乾燥させ(Na2SO4)、濾過し、濃縮して、オフホワイトの固体を得、それをCH3CNから再結晶させて、2Aを白色の固体として得た(2.81g、86%):融点=148〜150℃;[α]22D−7.32(c 0.50、CHCl3);IR νmax 3287、1750、1690、1649、1535cm-1;1H NMR(400MHz,CDCl3)δ7.75(d,J=7.5Hz,2H)、7.57(d,J=7.0Hz,2H)、7.39(t,J=7.0Hz,2H)、7.28(m,2H)、6.54(s,1H)、5.44(s,1H)、4.43−4.38(m,2H)、4.21(t,J=7.0Hz,1H)、4.01−3.96(m,3H)、3.72(s,3H)、2.16(m,1H)、0.96(t,J=9.0Hz,6H);13C NMR(100MHz,CDCl3)δ171.7(C)、170.1(C)、156.6(C)、143.9(C)、141.4(C)、127.8(CH)、127.2(CH)、125.2(CH)、120.1(CH)、67.2(CH2)、60.4(CH)、52.5(CH)、47.3(CH3)、41.2 (CH2)、31.2(CH)、19.3(CH3)、17.9(CH);MS m/z 842.8(30%,[2M+Na]+)、432.9(100%,[M+Na]+)。
【0187】
{(R)−2−[(S)−2−(9H−フルオレン−9−イルメトキシカルボニルアミノ)−3−(トリチルスルファニル)−プロピオニルアミノ]−3−メチル−ブチリルアミノ}−酢酸メチルエステル(3A)の調製。
【0188】
室温でCH3CN(48.5mL)中の2A(1.0g、2.44mmol)の攪拌溶液に、Et2NH(2.5mL)を添加した。3時間、室温で攪拌した後、その反応混合物をヘキサン(100mL)で希釈し、濃縮して、粗製アミンを無色の油として得た。CH3CN(25mL)中のFmoc−(STrt)−D−システイン(1.70g、2.9mmol)の攪拌溶液に、EDAC・HCl(561.0mg、2.93mmol)、HOBt(396mg、2.93mmol)およびDIEA(1.31mL、7.32mmol)を添加した。室温で15分間攪拌した後、その粗製アミンを添加し、その反応混合物を4時間攪拌し、溶媒を除去し、残留物をCH2Cl2(100mL)に溶解し、水(25mL)、10% HCl(25mL)、5% NaHCO3(25mL)および飽和NaCl(25mL)溶液で洗浄し、乾燥させ(Na2SO4)、濾過し、溶媒を除去して、オフホワイトの固体を得、それをCH3CNから再結晶させて、3Aを白色の固体として得た(1.46g、79%):[α]22D−2.35(c 0.50、CHCl3);IR νmax 3267、1646、1543cm-1;1H NMR 400MHz(400MHz,CDCl3)δ7.76(t,J=7.0Hz,2H)、7.54(d,J=6.5Hz,2H)、7.39−7.21(m 19H)、6.87(s,1H)、6.23(d,J=8.0,2H)、5.04(d,J=7.0,1H)、4.37(d,J=7.0,2H)、4.29(dd,J=8.6,J=5.0,1H)、4.17(t,J=6.5,1H)、3.96(m,1H)、3.72(dd,,J=18.1,5.0,1H)、3.65(s,3H)、3.60(m,1H)、2.70(d,J=6.5,2H)、2.30(m,1H)、1.70(s,1H)、0.87(dd,J=13.0,J=7.0Hz,6H);13C NMR(100MHz,CDCl3)δ170.6(C)、170.5(C)、170.1(C)、156.2(C)、144.3(C)、143.8(C)、143.7(C)141.4(C)、129.6(CH)、128.4(CH)、128.0(CH)、127.8(CH)、127.2(CH)、125.1(CH)、120.2(CH)、67.6(CH)、67.2(CH2)、58.4(CH)、54.3(CH)、52.4(CH3)、47.1(CH)、40.9(CH2)、33.6(CH2)、30.0(CH)、19.4(CH3)、17.4(CH3);LRMS m/z 777.8(100%,[M+Na]+)。C45H45N3O6Sについての分析計算値:71.50;H,6.00;N,5.56。実測値 C,71.42;H,5.99;N,5.55。
【0189】
((R)−2−{(S)−2−[(R)−2−(9H−フルオレン−9−イルメトキシカルボニルアミノ)−プロピオニルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(4A)の調製。
【0190】
室温でCH3CN/CH2Cl2(1:1,60mL)中の3A(900mg、1.19mmol)の攪拌溶液に、Et2NH(3mL)を添加した。3時間、室温で攪拌した後、その反応混合物をヘプタン(60mL)で希釈し、濃縮して、粗製アミンを無色の油として得た。CH2Cl2(30mL)中のFmoc−D−アラニン(529mg、1.7mmol)の攪拌溶液に、EDAC・HCl(326mg、1.7mmol)、HOBt(230mg、1.7mmol)およびDIEA(627μL、3.6mmol)を添加した。室温で10分間攪拌した後、CH2Cl2(20mL)中のその粗製アミンの溶液を添加し、その反応混合物を18時間攪拌した。その後、その反応物を水(15mL)、10% HCl(15mL)、5% NaHCO3(15mL)および飽和NaCl(15mL)溶液で洗浄し、乾燥させ(Na2SO4)、濾過し、溶媒を除去して、オフホワイトの固体を得、それをCH3CNから再結晶させて、4Aを白色の固体として得た(810mg、0.98mmol、82%):融点=195〜197℃;[α]22D +5.1(c 0.50、CHCl3);IR νmax 3267、3054、1744、1706、1635、1531.1cm-1;1H NMR(400MHz,CDCl3)δ7.75(d,J=7.5Hz,2H)、7.52(d,J=7.0Hz,2H)、7.39(m,7H)、7.26−7.15(m,13H)、6.79(s,1H)、6.62(s,1H)、5.47(s,1H)、4.43−4.28(m,4H)、4.13(m,1H)、4.01(m,1H)、3.88(s,2H)、3.63(s,3H)、2.81(m,1H)、2.52(m,1H)、2.26,(m,1H)、1.30(d,J=6.0Hz,3H)、0.94(d,J=6.0Hz,3H)、0.89(d,J=7.0Hz,3H);13C NMR(100MHz,CDCl3)δ172.7、171.1、170.2、156.1、144.4、143.9、143.8、141.4、129.6、128.3、127.9、127.2、127.1、125.1、120.1、58.5、52.7、52.3、50.8、47.2、41.0、33.5、30.4、19.3、19.0、17.7;LRMS m/z 849.2(100%,[M+Na]+)、865.1(20%,[M+K]+)。
【0191】
((R)−2−{(S)−2−[(R)−2−((E)−(S)−3−ヒドロキシ−7−トリチルスルファニル−ヘプト−4−エノイルアミノ)−プロピオニルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(6A)の調製。
【0192】
室温でCH2Cl2/CH3CN(3:2,150mL)中の4A(760mg、0.92mmol)の攪拌溶液に、Et2NH(7.5mL)を添加した。5時間、室温で攪拌した後、その反応混合物をヘプタン(60mL)で希釈し、溶媒を除去し、CH2Cl2(50mL)を添加し、濾過し、濃縮して、粗製アミンを無色の油として得た。CH2Cl2(30mL)中のその粗製アミンの攪拌溶液に、室温でCH2Cl2(5mL)中の5(672mg、1.20mmol、Yurek−George,A.;Habens,F.;Brimmell,M.;Packham,G.;Ganesan,A.J.Am.Chem.Soc.2004,126,1030−1031における手順に従って調製したもの)およびDMAP(15mg、0.12mmol)及びDMAP(15mg、0.12mmol)を添加した。室温で12時間攪拌した後、溶媒を除去し、残留物をフラッシュクロマトグラフィー(溶離剤 10〜100% EtOAc/CH2Cl2)によって精製して、6Aとして白色のガラスを得た(740mg、80%):融点=191〜193℃;[α]22D−18.0(c 0.50、CHCl3);IR νmax 3272、3064、1758、1692、1621cm-1;1H NMR(400MHz,5% CD3OD/CDCl3)δ7.40(m,12H)、7.25(m,12H)、7.20(m,6H)、6.96および6.88(不安定なNH,d,J=8.0Hz,1H)、5.49(dt,J=15.0Hz,6.5Hz,1H)、5.37(dd,J=15.5Hz,6.0Hz,1H)、4.32(m,2H)、4.22(d,J=6.0Hz,1H)、3.98(t,J=7.0Hz,1H)、3.92(d,J=18.1Hz,1H)、3.72(d,J=17.6Hz,1H)、3.67(s,3H)、2.64(dd,J=13.0Hz,7.5Hz,1H)、2.58(dd,J=13.0Hz,7.5Hz,1H)、2.56−2.18(m,9H)、2.11(q,J=6.5Hz,2H)、1.31(d,J=7.5Hz,3H)、0.91(t,J=7.0Hz,6H);13C NMR(100MHz,5% CD3OD/CDCl3)δ173.0、172.0、171.3、170.5、170.2、144.9、144.2、132.8、129.9、129.7、129.6、129.5、128.2、128.1、127.9、127.1、126.7、69.6、67.1、66.7、58.9、58.8、52.7、52.2、50.0、49.8、49.6、49.5、49.4、49.1、43.7、40.9、40.8、33.1、31.5、31.3、30.0、19.2、17.5;LRMS m/z 1050.4(100%,[M+Na]+)。
【0193】
((R)−2−{(S)−2−[(R)−2−((E)−(S)−3−ヒドロキシ−7−トリチルスルファニル−ヘプト−4−エノイルアミノ)−プロピオニルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸(7A)の調製
0℃でTHF(15mL)中の6A(700mg、0.70mmol)の攪拌溶液に、H2O(2.4mL)中のLiOH(25mg、1.05mmol)の溶液を添加した。1時間攪拌した後、その反応混合物をH2O(30mL)で希釈し、1M KHSO4でpH3〜4に酸性化し、EtOAc(3×30mL)で抽出した。有機層を飽和NaCl(15mL)で洗浄し、乾燥させ(Na2SO4)、濾過し、濃縮して、白色の固体を得、それをエーテルと研和して、7Aを白色の固体として得(693mg、99%)、それをそのまま次の段階で直接使用した。融点=191〜193℃;[α]22D−18.5(c 0.50、CHCl3);IR νmax 3413、1711、1678、1630、1451cm-1;1H NMR(400MHz,5% CD3OD/CDCl3)δ7.30(m,12H)、7.16(m,12H)、7.13(m,6H)、5.43(dt,J=15.5Hz,6.0Hz,1H)、5.30(dd,J=15.5Hz,6.0Hz,1H)、4.27(m,2H)、4.21(m,1H)、3.99(m,1H)、3.75(s,2H)、3.77(m,br,6H)、2.55(dd,J=12.5Hz,6.5Hz,1H)、2.44(dd,J=12.5Hz,7.5Hz,1H)、2.21(m,2H)、2.12(m,3H)、2.02(m,2H)、1.23(d,J=7.0Hz,3H)、0.83(d,J=7.0Hz,3H)、0.80(d,J=7.0Hz,3H);13C NMR(100MHz,5% CD3OD/CDCl3)δ173.0、172.1、171.6、171.5、170.5、144.9、144.3、132.7、129.8、129.6、129.5、128.2、127.9、127.0、126.7、69.5、67.1、66.7、58.7、52.7、43.5、40.9、33.2、31.5、31.3、30.3、19.2、19.1、17.6;LRMS m/z 1013.2(100%,[M+Na]+)。
【0194】
(6R,9S,12R,16S)−6−イソプロピル−12−メチル−16−((E)−4−トリチルスルファニル−ブト−1−エニル)−9−トリチルスルファニルメチル−1−オキサ−4,7,10,13−テトラアザ−シクロヘキサデカン−2,5,8,11,14−ペンタオン(8A)の調製。
【0195】
CH2Cl2(160mL)中の無水2−メチル−6−ニトロ安息香酸(MNBA)(289mg、0.84mmol)およびDMAP(205mg、1.68mmol)の溶液に、CH2Cl2/THF(2:1、600mL)中の酸7A(693mg、0.70mmol)の溶液を5時間かけて1滴ずつ添加した。さらに12時間後、1M HCl(150mL)を添加し、CH2Cl2(3×100mL)で抽出した。その後、合わせた有機相を飽和NaHCO3(150mL)、続いてブライン(80mL)で洗浄し、乾燥させ(MgSO4)、濾過し、濃縮した。残留物をフラッシュクロマトグラフィー(溶離剤 50〜100% EtOAc/ヘキサン)によって精製して、8Aを白色のガラスとして得た(478mg、0.49mmol、70%)。[α]22D−7.0(c 0.50、CHCl3);IR νmax 1735、1659、1526cm-1;1H NMR(400MHz,CDCl3)δ7.43−7.33(m,12H)、7.32−7.13(m,20H)、6.77(d,J=5.3Hz,1H)、6.54(d,J=7.3Hz,1H)、5.58(dt,J=15.1,6.8Hz,1H)、5.44−5.29(m,2H)、4.55(dd,J=16.8,9.0Hz,1H)、4.46(dd,J=9.5,4.3Hz,1H)、3.89(5重線,(dd,J=7.0Hz,1H)、3.77(dt,J=8.3,5.3Hz,1H)、3.43(dd,J=16.8,2.8Hz,1H)、3.03(dd,J=12.6,8.3Hz,1H)、2.61−2.49(m,3H)、2.18(t,J=7.5Hz,2H)、2.11−1.95(m,2H)、1.38(d,J=7.0Hz,3H)、0.94(d,J=6.8Hz,3H)、0.90(d,J=6.8Hz,3H);13C−NMR(100MHz,CDCl3)δ174.0(C)、170.9(C)、170.8(C)、169.8(C)、169.3(C)、144.9(C)、144.3(C)、133.2(CH)、129.7(CH)、129.5(CH)、128.3(CH)、128.0(CH)、127.2(CH)、126.8(CH)、72.3(CH)、67.3(C)、66.8(C)、58.3(CH)、55.9(CH)、50.5(CH)、41.7(2 x CH2)、32.5(CH2)、31.3(CH2)、31.2(CH2)、28.8(CH)、19.8(CH3)、19.8(CH3)、17.2(CH3)、16.7(CH3);LRMS(ES+)m/z 996(100%,[M+Na]+)、974(10%,[M+H]+)。
【0196】
(E)−(1S,7R,10S,21R)−7−イソプロピル−21−メチル−2−オキサ−12,13−ジチア−5,8,20,23−テトラアザ−ビシクロ[8.7.6]トリコス−16−エン−3,6,9,19,22−ペンタオン(9A)の調製
CH2Cl2/MeOH(9:1、1000mL)中のI2(1120mg、4.42mmol)の強力攪拌溶液に、CH2Cl2/MeOH(9:1、500mL)中の保護ジオール8A(430mg、0.44mmol)を30分かけて1滴ずつ添加した。さらに30分間攪拌した後、0.1M チオ硫酸ナトリウム(300mL)および飽和NaCl(100mL)を添加し、CH2Cl2(3×100mL)で抽出した。合わせた有機抽出物を乾燥させ(MgSO4)、濾過し、溶媒を除去した。残留物をフラッシュクロマトグラフィー(溶離剤 1〜6% MeOH/CH2Cl2)によって精製して、9A(205mg、0.42mmol、96%)を白色の固体として得た:[α]22D−98.0(c 0.50、CHCl3);IR νmax 3300、2477、1734、1659、1526、1446cm-1;1H NMR(400MHz,CDCl3)δ7.46(d,J=6.5Hz,1H)、7.33(d,J=5.3Hz,1H)、7.28(d,J=8.8Hz,1H)、6.60(d,J=3.8Hz,1H)、5,95(dtd,J=16.1,6.5,2.0Hz,1H)、5.80−5.70(m,2H)、4.85(ddd,J=9.8,8.5,3.8Hz,1H)、4.21(qd,J=7.3,3.8Hz,1H)、4.11(d,J=5.3Hz,2H)、3.44(dd,J=15.6,10.0Hz,1H)、3.20(dd,J=10.3,6.8Hz,1H)、3.01−2.95(m,2H)、2.92(dd,J=15.6,4.0Hz,1H)、2.85−2.58(m,5H)、1.49(d,J=7.5Hz,3H)、0.98(d,J=6.5Hz,3H)、0.92(d,J=6.5Hz,3H);13C NMR 100MHz 173.3(C)、171.6(C)、171.0(C)、169.4(C)、168.2(C)、130.4(CH)、130.3(CH)、69.8(CH)、64.9(CH)、54.5(CH)、52.0(CH)、42.4(2 x CH2)、38.7(CH2)、38.6(CH2)、32.7(CH2)、27.5(CH)、20.8(CH3)、20.1(CH3)、16.7(CH3);LRMS(ES+)m/z 995(50%,[2M+Na]+)、509(80%,[M+Na]+)、487(100%,[M+H]+);HRMS m/z 509.1495(M+Na)+ 予測値 509.1499。
【0197】
((R)−2−{(S)−2−[(R)−2−(9H−フルオレン−9−イルメトキシカルボニルアミノ)−3−フェニル−プロピオニルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(4B)の調製。
【0198】
室温でCH3CN/CH2Cl2(1:1、60mL)中の3A(836mg、1.11mmol)の攪拌溶液に、Et2NH(3mL)を添加した。室温で3時間攪拌した後、その反応混合物をヘプタン(60mL)で希釈し、濃縮して、粗製アミンを無色の油として得た。CH2Cl2(30mL)中のFmoc−D−フェニルアラニン(650mg、1.7mmol)の攪拌溶液に、EDAC・HCl(326mg、1.7mmol)、HOBt(230mg、1.7mmol)およびDIEA(627μL、3.6mmol)を添加した。室温で10分間攪拌した後、CH2Cl2(20mL)中のその粗製アミンの溶液を添加し、その反応混合物を12時間攪拌した。その後、その反応物を水(15mL)、10% HCl(15mL)、5% NaHCO3(15mL)および飽和NaCl(15mL)溶液で洗浄し、乾燥させ(Na2SO4)、濾過し、溶媒を除去して、オフホワイトの固体を得、それをCH3CNから再結晶させて、4Bを白色の固体として得た(800mg、0.89mmol、80%);IR νmax 3264、3053、1742、1701、1636、1532cm-1;1H−NMR(400MHz,CDCl3)δ7.75(d,J=7.5Hz,2H)、7.46(t,J=7.5Hz,2H)、7.41−7.31(m,8H)、7.30−7.07(m,16H)、7.01(br s,1H)、6.54(br s,2H)、5.29(br s,1H)、4.48−4.32(m,3H)、4.25−4.13(m,1H)、4.10(t,J=8.5Hz,1H)、3.99−3.76(m,3H)、3.65(s,3H)、3.10−2.88(m,2H)、2.83−2.69(m,1H)、2.49(dd,J=13.0,6.3Hz,1H)、2.36−2.22,(m,1H)、0.93(t,J=7.0Hz,6H);13C NMR 100MHz 171.2(C)、171.0(C)、170.2(C)、170.0(C)、156.2(C)、144.3(C)、143.9(C)、143.7(C)、141.4(C)、136.0(C)、129.5(CH)、129.3(CH)、129.0(CH)、128.3(CH)、127.9(CH)、127.4(CH)、127.2(CH)、127.1(CH)、125.2(CH)、120.1(CH)、67.3(CH2)、58.6(CH)、56.2(CH)、52.8(CH)、52.3(CH3)、47.2(CH)、41.0(CH2)、38.3(CH2)、33.5(CH2)、30.2(CH)、19.4(CH3)、17.7(CH3);LRMS(ES+)m/z 1828(30%,[2M+Na]+)、925(100%,[M+Na]+)。
【0199】
((R)−2−{(S)−2−[(R)−2−((E)−(S)−3−ヒドロキシ−7−トリチルスルファニル−ヘプト−4−エノイルアミノ)−3−フェニル−プロピオニルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(6B)の調製。
【0200】
室温でCHCl3/CH3CN(1:1、14mL)中の4B(245mg、0.28mmol)の攪拌溶液に、Et2NH(1mL)を添加した。室温で5時間攪拌した後、その反応混合物をヘプタン(10mL)で希釈し、溶媒を除去して、粗製アミンを無色の油として得た。CH2Cl2(10mL)中のその粗製アミンの攪拌溶液に、室温でCH2Cl2(5mL)中の5(193mg、0.34mmol)およびDMAP(4mg、0.034mmol)を添加した。室温で12時間攪拌した後、溶媒を除去し、残留物をフラッシュクロマトグラフィー(溶離剤 2〜4% MeOH/CH2Cl2)によって精製して、6B(190mg、64%)を白色の固体として得た;1H NMR(400MHz,CDCl3)δ7.46−7.05(m,37H)、6.85(br s,1H)、6.34(br s,1H)、5.44(dt,J=15.3,6.5Hz,1H)、5.29(dd,J=15.3,6.2Hz,1H)、4.74(br d,J=5.3Hz,1H)、4.37(t,J=7.0Hz,1H)、4.33−4.25(m,1H)、4.20−4.08(m,1H)、3.93−3.74(m,2H)、3.65(s,3H)、3.31(br s,1H)、3.06(dd,J=14.3,5.3Hz,1H)、2.96(dd,J=14.3,7.3Hz,1H)、2.59(dd,J=12.5,6.8Hz,1H)、2.33−2.08(m,5H)、2.04(q,J=7.0Hz,2H)、0.90(t,J=6.3Hz,6H);13C NMR(100MHz,CDCl3)δ171.9(C)、171.2(C)、171.1(C)、170.4(2 x C)、145.0(C)、144.3(C)、136.1(C)、132.5(CH)、130.1(CH)、129.7(CH)、129.5(CH)、129.3(CH)、128.8(CH)、128.2(CH)、128.0(CH)、127.3(CH)、127.1(CH)、126.7(CH)、69.8(CH)、67.1(C)、66.8(C)、58.9(CH)、54.4(CH)、52.6(CH)、52.3(CH3)、43.7(CH2)、41.0(CH2)、37.6(CH2)、33.8(CH2)、31.5(CH2)、31.4(CH2)、30.3(CH)、19.3(CH3)、17.9(CH3);LRMS(ES+)m/z 1104(100%,[M+Na]+)、1082(50%,[M+H]+)。
【0201】
(6R,9S,12R,16S)−12−ベンジル−6−イソプロピル−16−((E)−4−トリチルスルファニル−ブト−1−エニル)−9−トリチルスルファニルメチル−1−オキサ−4,7,10,13−テトラアザ−シクロヘキサデカン−2,5,8,11,14−ペンタオン(8B)の調製
0℃のTHF(10mL)中のメチルエステル6B(180mg、0.17mmol)の溶液に、H2O(1.5mL)中のLiOH(6mg、0.25)の溶液を添加した。1時間後、その反応を1M HCl(6mL)の添加により停止させた。CHCl3(50mL)を添加し、有機相を分離し、CHCl3(2×10mL)で抽出した。有機相をブライン(15mL)で洗浄し、乾燥させ(MgSO4)、濾過し、真空下で濃縮して、粗製酸7B(179mg、定量的)を白色の固体として得、それを次の段階で直ちに使用した:(ES-)m/z 1066(100%,[M−H]-)。
【0202】
CH2Cl2(40mL)中のMNBA(68mg、0.20mmol)およびDMAP(49mg、0.40mmol)の溶液に、CH2Cl2/THF(15:1、160mL)中の酸7B(179mg、0.17mmol)の溶液を3時間かけて1滴ずつ添加した(注意:先ず、THFに酸を溶解し、その後、CH2Cl2を添加する)。さらに14時間後、1M HCl(40mL)の添加により反応を停止させた。有機相を(CH2Cl2で抽出しながら)分離し、NaHCO3(30mL)、そしてブライン(20mL)で順次洗浄した。合わせた有機相を乾燥させ(MgSO4)、濾過し、真空下で濃縮して、黄色の油を得た。シリカゲルでのカラムクロマトグラフィー(30〜70% EtOAc/CH2Cl2)による精製によって、8B(108mg、0.10mmol、60%)を白色の固体として得た;1H−NMR(400MHz,CDCl3)δ7.42−7.08(m,35H)、7.05(d,J=7.3Hz,2H)、6.96(br s,1H)、6.54(br s,1H)、5.46(dt,J=15.3,6.5Hz,2H)、5.21(dd,J=15.3,6.6Hz,1H)、4.49(dd,J=13.5,4.3Hz,1H)、4.32(dd,J=16.8,9.0Hz,1H)、4.16−4.06(m,1H)、3.85(br q,J=6.0Hz,1H)、3.02−2.82(m,4H)、2.65−2.53(m,2H)、2.40(dd,J=14.6,3.3Hz,1H)、2.23(dd,J=14.6,11.3Hz,1H)、2.10(t,J=7.0Hz,2H)、1.97−1.84(m,2H)、0.98(d,J=7.0Hz,3H)、0.92(d,J=7.0Hz,3H);13C−NMR(100MHz,CDCl3)δ173.5(C)、170.8(C)、170.7(C)、169.9(2 x C)、144.9(C)、144.2(C)、136.3(C)、132.7(CH)、129.6(CH)、129.5(CH)、129.3(CH)、128.9(CH)、128.3(CH)、128.0(CH)、127.9(CH)、127.3(CH)、127.1(CH)、126.7(CH)、72.2(CH)、67.3(C)、66.7(C)、58.3(CH)、55.8(2 x CH)、42.0(CH2)、41.4(CH2)、36.7(CH2)、32.3(CH2)、31.3(CH2)、31.1(CH2)、28.7(CH)、19.8(CH3)、17.2(CH3);LRMS(ES+)m/z 1071(100%,[M+Na]+)、1066(10%,[M+NH4]+)、1049(10%,[M+H]+)。
【0203】
(E)−(1S,7R,10S,21R)−21−ベンジル−7−イソプロピル−2−オキサ−12,13−ジチア−5,8,20,23−テトラアザ−ビシクロ[8.7.6]トリコス−16−エン−3,6,9,19,22−ペンタオン(9B)の調製。
【0204】
CH2Cl2/MeOH(9:1、230mL)中のI2(254mg、1.0mmol)の強力攪拌溶液に、ビス−トリチル8B(105mg、0.10mmol)の溶液を30分かけて1滴ずつ添加した。さらに30分間後、チオ硫酸ナトリウム(0.05M、100mL)、続いてブライン(10mL)の添加により反応を停止させた。有機相を分離し、水性相をCH2Cl2(3×15mL)で抽出した。合わせた有機相を乾燥させ(MgSO4)、濾過し、真空下で濃縮して、白色の固体を得た。シリカゲルでのカラムクロマトグラフィー(1〜3.5% MeOH/CH2Cl2)による精製によって、二環式デプシペプチド9B(43mg、0.08mmol、75%)を白色の固体として得た:[α]25D−98(c 0.05、1:1 MeOH/CHCl3);IR νmax 3280、1732、1627、1538、1445cm-1;1H−NMR(400MHz,19:1 CD3OD/CDCl3)δ7.35−7.18(m,5H)、5.86−5.76(m,1H)、5.74−5.66(m,2H)、4.62(dd,J=11.3,4.0Hz,1H)、4.45(dd,J=9.3,5.3Hz,1H)、4.28(d,J=17.6Hz,1H)、3.77(d,J=17.6Hz,1H)、3.39(d,J=10.5Hz,1H)、3.27−3.17(m,2H)、3.11−2.99(m,3H)、2.97−2.83(m,2H)、2.81(dd,J=13.6,2.0Hz,1H)、2.70−2.53(m,3H)、0.96(d,J=6.5Hz,3H)、0.92(d,J=6.5Hz,3H);13C−NMR(100MHz,19:1 CD3OD/CDCl3)δ173.8(C)、173.2(C)、172.9(C)、171.3(C)、169.4(C)、137.7(CH)、131.3(CH)、131.3(CH)、129.7(CH)、129.6(CH)、128.0(CH)、71.9(CH)、65.9(CH)、58.3(CH)、58.1(CH)、42.6(CH2)、39.3(2 x CH2)、36.9(CH2)、36.8(CH2)、31.8(CH2)、28.1(CH)、20.6(CH3)、20.5(CH3);LRMS(ES+)m/z 580(50%,[2M+Na]+)、563(100%,[M+NH4]+)、563(10%,[M+H]+)。
【0205】
((R)−2−{(S)−2−[(R)−2−(9H−フルオレン−9−イルメトキシカルボニルアミノ)−3−メチル−ブチリルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(4C)の調製。
【0206】
室温でCH3CN/CH2Cl2(1:1、60mL)中の3A(836mg、1.11mmol)の攪拌溶液に、Et2NH(3mL)を添加した。室温で3時間攪拌した後、その反応混合物をヘプタン(60mL)で希釈し、濃縮して、粗製アミンを無色の油として得た。CH2Cl2(40mL)中のFmoc−D−バリン(577mg、1.7mmol)の攪拌溶液に、EDAC・HCl(326mg、1.7mmol)、HOBt(230mg、1.7mmol)およびDIEA(627μL、3.6mmol)を添加した。室温で10分間攪拌した後、CH2Cl2(20mL)中のその粗製アミンの溶液を添加し、その反応混合物を12時間攪拌した。その後、その反応物を水(15mL)、10% HCl(15mL)、5% NaHCO3(15mL)および飽和NaCl(15mL)溶液で洗浄し、乾燥させ(Na2SO4)、濾過し、溶媒を除去して、オフホワイトの固体を得、それをCH3CNから再結晶させて、4Cを白色の固体として得た(734mg、0.86mmol、78%);1H−NMR(400MHz,CDCl3)δ7.74(d,J=7.5Hz,2H)、7.53(t,J=7.5Hz,2H)、7.42−7.32(m,9H)、7.30−7.19(m,9H)、7.16(t,J=7.0Hz,2H)、6.80(d,J=8.0Hz,1H)、6.57(d,J=7.0Hz,1H)、5.52(d,J=8.0Hz,1H)、4.44(t,J=8.0Hz,1H)、4.38(dd,J=10.5,7.5Hz,1H)、4.25(dd,J=10.5,7.0Hz,1H)、4.17−3.95(m,3H)、3.87(dd,J=18.1,5.5Hz,1H)、3.78(dd,J=18.1,5.5Hz,1H)、3.63(s,3H)、2.71(dd,J=12.5,6.0Hz,1H)、2.53(dd,J=12.5,6.0Hz,1H)、2.21(sept,J=6.4Hz,1H)、2.07−1.93(m,1H)、0.93−0.83(m,12H);13C NMR 100MHz 171.5(C)、171.1(C)、170.2(2 x C)、156.6(C)、144.4(C)、144.0(C)、143.8(C)、141.4(C)、129.6(CH)、128.3(CH)、127.9(CH)、127.2(CH)、127.1(CH)、125.2(CH)、120.1(CH)、67.3(CH2)、67.2(C)、60.3(CH)、58.5(CH)、52.5(CH)、52.3(CH)、47.3(CH)、41.0(CH2)、33.8(CH2)、31.5(CH)、30.5(CH)、19.2(2 x CH3)、18.0(CH3)、17.8(CH3);LRMS(ES+)m/z 1832(10%,[2M+Na]+)、877.5(100%,[M+Na]+)。
【0207】
((R)−2−{(S)−2−[(R)−2−((E)−(S)−3−ヒドロキシ−7−トリチルスルファニル−ヘプト−4−エノイルアミノ)−3−メチル−ブチリルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(6C)の調製。
【0208】
室温でCHCl3/CH3CN(1:1、14mL)中の4C(235mg、0.28mmol)の攪拌溶液に、Et2NH(1mL)を添加した。室温で5時間攪拌した後、その反応混合物をヘプタン(10mL)で希釈し、溶媒を除去して、粗製アミンを無色の油として得た。CH2Cl2(10mL)中のその粗製アミンの攪拌溶液に、室温でCH2Cl2(5mL)中の5(193mg、0.34mmol)の溶液およびDMAP(4mg、0.034mmol)を添加した。室温で12時間攪拌した後、溶媒を除去し、残留物をフラッシュクロマトグラフィー(溶離剤 1〜3% MeOH/CH2Cl2)によって精製して、6C(230mg、81%)を白色の固体として得た;1H NMR(400MHz,CDCl3)δ7.42−7.38(m,12H)、7.30−7.14(m,20H)、6.74(br s,1H)、6.39(br s,1H)、5.49(dtd,J=15.3,6.5,0.8Hz,1H)、5.36(dd,J=15.3,6.3Hz,1H)、4.39−4.26(m,3H)、4.16−4.06(m,1H)、3.83(d,J=5.5Hz,2H)、3.65(s,3H)、3.33(br s,1H)、2.60(dd,J=12.8,7.3Hz,1H)、2.54(dd,J=12.8,6.8Hz,1H)、2.37(dd,J=14.0,3.0Hz,1H)、2.30−2.02(m,7H)、0.90(d,J=7.0Hz,3H)、0.89(d,J=7.0Hz,3H)、0.88(d,J=7.0Hz,3H)、0.85(d,J=7.0Hz,3H);13C NMR(100MHz,CDCl3)δ172.1(C)、171.4(C)、171.2(C)、170.5(C)、170.3(C)、145.0(C)、144.3(C)、132.6(CH)、130.2(CH)、129.7(CH)、129.5(CH)、128.3(CH)、128.0(CH)、127.1(CH)、126.8(CH)、69.8(CH)、67.1(C)、66.8(C)、59.0(CH)、58.9(CH)、52.5(CH)、52.3(CH3)、43.9(CH2)、41.0(CH2)、33.7(CH2)、31.5(2 x CH2)、30.4(CH)、30.2(CH2)、19.5(CH3)、19.3(CH3)、17.9(2 x CH3);LRMS(ES+)m/z 1056(100%,[M+Na]+)、1051(50%,[M+NH4]+)。
【0209】
(6R,9S,12R,16S)−6,12−ジイソプロピル−16−((E)−4−トリチルスルファニル−ブト−1−エニル)−9−トリチルスルファニルメチル−1−オキサ−4,7,10,13−テトラアザ−シクロヘキサデカン−2,5,8,11,14−ペンタオン(8C)の調製
0℃で、THF(12mL)中のメチルエステル6B(220mg、0.21mmol)の溶液に、H2O(2mL)中のLiOH(7.6mg、0.32)の溶液を添加した。1時間後、1M HCl(6mL)の添加により反応を停止させた。CHCl3(50mL)を添加し、有機相を分離し、CHCl3(2×15mL)で抽出した。有機相をブライン(10mL)で洗浄し、乾燥させ(MgSO4)、濾過し、真空下で濃縮して、粗製酸7C(217mg、定量的)を白色の固体として得、それを次の段階で直ちに使用した:(ES-)m/z 1017(100%,[M−H]-)。
【0210】
CH2Cl2(50mL)中のMNBA(90mg、0.26mmol)およびDMAP(62mg、0.51mmol)の溶液に、CH2Cl2/THF(15:1、200mL)中の酸7C(217mg、0.21mmol)の溶液を3時間かけて1滴ずつ添加した(注意:先ず、THFに酸を溶解し、その後、CH2Cl2を添加する)。さらに14時間後、1M HCl(40mL)の添加により反応を停止させた。有機相を(CH2Cl2で抽出しながら)分離し、NaHCO3(30mL)、そしてブライン(20mL)で順次洗浄した。合わせた有機相を乾燥させ(MgSO4)、濾過し、真空下で濃縮して、黄色の油を得た。シリカゲルでのカラムクロマトグラフィー(30〜70% EtOAc/CH2Cl2)による精製によって、8C(130mg、0.13mmol、62%)を白色の固体として得た;1H NMR(400MHz,CDCl3)δ7.88(br s,2H)、7.42−7.34(m,12H)、7.29−7.14(m,19H)、6.20(br d,J=7.0Hz,1H)、5.62−5.50(m,2H)、5.31(dd,J=15.3,6.4Hz,1H)、4.36(dd,J=17.3,8.3Hz,1H)、4.24(dd,J=8.5,4.5Hz,1H)、4.11(t,J=7.3Hz,1H)、3.65−3.56(m,1H)、3.42(d,J=14.5Hz,1H)、2.99(t,J=10.8Hz,1H)、2.64(dd,J=12.0,6.3Hz,1H)、2.50(dd,J=15.1,2.5Hz,1H)、2.35(dd,J=14.8,9.8Hz,1H)、2.17(t,J=7.5Hz,2H)、2.06−1.86(m,3H)、1.84−1.72(m,1H)、0.91(d,J=6.8Hz,3H)、0.88(d,J=6.3Hz,6H)、0.83(d,J=6.8Hz,3H);13C−NMR(100MHz,CDCl3)δ173.8(C)、172.2(C)、171.0(C)、169.5(C)、168.9(C)、145.0(C)、144.4(C)、132.7(CH)、129.7(CH)、128.2(CH)、128.1(CH)、128.0(CH)、127.0(CH)、126.8(CH)、72.2(CH)、67.3(C)、66.8(C)、58.8(CH)、58.7(CH)、58.4(CH)、42.1(CH2)、41.9(CH2)、32.0(CH2)、31.5(CH2)、31.3(CH)、31.2(CH2)、29.4(CH)、19.6(CH3)、19.4(CH3)18.5(CH3)、17.0(CH3);LRMS(ES+)m/z 1023(100%,[M+Na]+)、1018(60%,[M+NH4]+)。
【0211】
(E)−(1S,7R,10S,21R)−7,21−ジイソプロピル−2−オキサ−12,13−ジチア−5,8,20,23−テトラアザ−ビシクロ[8.7.6]トリコス−16−エン−3,6,9,19,22−ペンタオン(9C)の調製。
【0212】
CH2Cl2/MeOH(9:1、280mL)中のI2(305mg、1.2mmol)の強力攪拌溶液に、ビス−トリチル8C(120mg、0.12mmol)の溶液を30分かけて1滴ずつ添加した。さらに30分間後、チオ硫酸ナトリウム(0.05M、100mL)、続いてブライン(10mL)の添加により反応を停止させた。有機相を分離し、水性相をCH2Cl2(3×15mL)で抽出した。合わせた有機相を乾燥させ(MgSO4)、濾過し、真空下で濃縮して、白色の固体を得た。シリカゲルでのカラムクロマトグラフィー(1〜4% MeOH/CH2Cl2)による精製によって、二環式デプシペプチド9C(60mg、0.12mmol、97%)を白色の固体として得た:[α]25D−105.7(1:1 MeOH/CHCl3、c 0.15);1H NMR(400MHz,CD3OD)δ5.76−5.63(m,3H)、4.52(dd,J=11.3,4.7Hz,1H)、3.96(d,J=5.1Hz,1H)、3.74(d,J=17.5Hz,1H)、3.38(d,J=10.9Hz,1H)、3.19−3.11(m,2H)、3.09−2.96(m,3H)、2.90(dd,J=13.6,2.5Hz,1H)、2.71−2.62(m,2H)、2.58−2.44(m,1H)、2.31−2.18(m,1H)、1.11(d,J=7.0Hz,3H)、1.09(d,J=7.0Hz,3H)、1.01(d,J=6.6Hz,3H)、0.91(d,J=6.6Hz,3H);13C−NMR(100MHz,CD3OD)δ171.8(C)、171.7(C)、170.7(C)、170.4(C)、168.3(C)、130.0(CH)、129.6(CH)、69.8(CH)、64.6(CH)、61.9(CH)、54.8(CH)、42.2(CH2)、39.3(CH2)、37.5(CH2)、37.1(CH2)、32.1(CH2)、29.2(CH)、27.3(CH)、20.6(CH3)、20.0(CH3)、19.4(CH3)、19.0(CH3);LRMS(ES+)m/z 537(100%,[M+Na]+)、515(90%,[M+H]+)。
【0213】
((R)−2−{(S)−2−[(R)−6−t−ブトキシカルボニルアミノ−2−(9H−フルオレン−9−イルメトキシカルボニルアミノ)−ヘキサノイルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(4D)の調製。
【0214】
アルゴン下でトリペプチド3A(548mg、0.7mmol)をCH2Cl2/CH3CN(44mL、1:1)に溶解し、その後、攪拌しながらジエチルアミン(1.65mL、16mmol)を添加し、その反応混合物を室温で4.5時間攪拌した。その後、ヘキサン(100mL)をその反応混合物に添加し、溶媒を真空下で除去し、これを再びヘキサン(3×25mL)で繰り返した。その後、その粗製アミンを高真空下で40分間乾燥させた。その後、アルゴン下で攪拌しながら、CH3CN(16.5mL)中のPyBop(558mg、1.1mmol)およびFmoc−D−リシン(Boc)−OH(477mg、1mmol)の溶液にジイソプロピルエチルアミン(0.45mL、2.6mmol)を添加した。得られた3の脱保護アミンをCH2Cl2(19mL)に添加し、その反応混合物を室温で16時間攪拌した。その後、真空下で溶媒を除去し、形成した固体をシリカゲルでのカラムクロマトグラフィー(溶離剤 6:4 EtOAc/ヘキサン)によって精製して、4D(571mg、0.57mmol、81%)を白色の固体として得た:Rf 0.17 EtOAc/ヘキサン(6:4):[α]D26=+47(c 0.3、MeOH);IR 3294、1744、1646、1515、1445(m);1H−NMR(400MHz,CDCl3)δ7.74(d,J=8.0,2H)、7.56−7.51(m,2H)、7.42−7.34(m,8H)、7.30−7.17(m,11H)、7.02(br s,1H)、6.94(br s,1H)、6.59(d,J=8.03Hz,1H)、5.75(s,1H)、4.38−4.21(m,2H)、4.18(dd,J=8.5,6.0Hz,1H)、4.15−4.05(m,2H)、3.87(d,J=5.5Hz,3H)、3.55(s,3H)、3.03(br d,J=8.0Hz,2H)、2.76−2.67(m,1H)、2.66−2.58(m,1H)、2.20−2.10(m,1H)、1.93−1.84(m,2H)、1.82−1.72(m,1H)、1.70−1.59(m,1H)、1.43(s,9H)、1.50−1.30(m,3H)、0.89(d,J=7.02Hz,3H)、0.84(d,J=7.03Hz,3H)、13C NMR(100MHz,CDCl3)δ172.6(C)、171.4(C)、170.6(C)、170.6(C)、156.6(C)、156.3(CO)、144.4(C)、144.3(C)、143.9(C)、143.8(C)、141.4(C)、129.7(CH)、129.5(CH)、128.3(CH)、128.2(CH)、127.2(CH)、127.1(CH)、125.2(CH)、120.0(CH)、67.4(CH2)、47.2(CH3)、41.1(CH2)、40.0(CH2)、33.5(CH2)、31.9(CH2)、30.3(CH2)、29.8(CH)、28.6(CH3)、22.5(CH2)、19.4(CH3),17.9(CH3)、52.3(CH)、52.8(CH)、55.1(CH)、59.0(CH);LRMS(ES+)1007.0(100%,[M+Na]+)。
【0215】
((R)−2−{(S)−2−[(R)−6−t−ブトキシカルボニルアミノ−2−((S)−3−ヒドロキシ−7−トリチルスルファニル−ヘプト−4−エノイルアミノ)−ヘキサノイルアミノ]−3−トリチルスルファニル−プロピオニルアミノ}−3−メチル−ブチリルアミノ)−酢酸メチルエステル(6D)の調製。
【0216】
CH2Cl2(13.5mL)およびCH3CN(16.5mL)中のテトラペプチド4D(201mg、0.2mmol)の溶液に、アルゴン下で、トリエチルアミン(1mL、9.6mmol)を添加し、その反応混合物を5時間攪拌した。その後、真空下で溶媒を除去し、これをヘキサン(2×10mL)で繰り返した。その後、CH2Cl2(7mL)中のその粗製アミンの溶液にDMAP(4mg、0.03mmol)を添加し、続いてCH2Cl2(7mL)中の5(158mg、0.28mmol)の溶液を添加した。その後、その反応物を18時間攪拌した。その後、真空下で溶媒を除去し、形成した固体をシリカゲルでのカラムクロマトグラフィー(溶離剤 6:4〜7:3 EtOAc:ヘキサン)によって精製して、6D(136mg、0.12mmol、60%)を白色の固体として得た;融点85〜87℃;Rf 0.29 EtOAc/ヘキサン(8:2);[α]29D=+19(c 0.49、CH2Cl2);IR(薄膜)3289、1751、1687、1634、1521、1490、1443;1H−NMR(400MHz,CDCl3)δ7.42−7.35(m,15H)、7.29−7.16(m,17H)、6.95(d,J=7.5Hz,1H)、6.80(d,J=8.0Hz,1H)、5.48(dt,J=15.1,6.5Hz,1H,)、5.35(dd,J=15.1,6.0Hz,1H,)、4.65(s,1H)、4.46−4.35(m,2H)、4.21(dd,J=8.5,6.52Hz,1H)、4.04−3.96(m,1H)、3.93(d,J=5.5Hz,1H)、3.89−3.81(m,2H)、3.62(s,3H)、3.02(d,J=5.5Hz,2H)、2.60−2.45(m,2H)、2.35−2.00(m,9H)、1.85−1.74(m,1H)、1.66−1.54(m,1H)、1.40(s,9H)、1.47−1.25(m,2H)、0.88(d,J=7.0Hz,3H,)、0.84(d,J=7.0Hz,3H)、13C NMR(100MHz,CDCl3)δ172.3(C)、172.2(C)、171.5(C)、170.7(C)、170.5(C)、156.2(C)、145.0(C)、145.0(C)、144.3(C)、132.9(CH)、129.7(CH)、129.6(CH)、129.5(CH)、129.5(CH)、128.2(CH)、128.2(CH)、128.0(CH)、127.0(CH)、126.7(CH)、69.5(CH)、67.1(CH2)、66.7(CH2)、58.6(CH)、52.9(CH)、52.6(CH)、52.3(CH)、52.2(CH3)、44.2(CH2)、41.1(CH2)、40.1(CH2)、33.9(CH2)、31.4(CH2)、31.0(CH2)、31.0(CH2)、30.6(CH)、29.8(CH2)、28.6(CH3)、22.6(CH2)、19.3(CH3)、18.0(CH3)、MS(ES+)1185(100%,[M+Na]+)、1163(40%,[M+H+)。
【0217】
{4−[(6R,9S,12R)−6−イソプロピル−2,5,8,11,14−ペンタオキソ−16−((S)−4−トリチルスルファニル−ブト−1−エニル)−9−トリチルスルファニルメチル−1−オキサ−4,7,10,13−テトラアザ−シクロヘキサデク−12−イル]−ブチル}−カルバミン酸t−ブチルエステル(8D)の調製。
【0218】
0℃で、THF(1.9mL)中の6D(136mg、0.12mmol)の溶液に、水(0.5mL)中のLiOH(6.8mg、0.28mmol)の溶液を添加し、その反応混合物を55分間攪拌した。その後、pHが3〜4の間に低下するまでクエン酸を1滴ずつ添加し、その後、水(3.7mL)、続いてEtOAc(15mL)を添加した。層を分離し、生成物をEtOAc(2×10mL)で抽出した。その後、合わせた有機層を飽和ブライン(10mL)で洗浄し、Na2SO4で乾燥させ、溶媒を真空下で除去して、粗製酸7D(132mg、定量的)を白色の固体として得、それを次の段階で直ちに使用した:LRMS(ES-)1147(100%,[M−H]-)。
【0219】
その後、CH2Cl2(25mL)中のMNBA(47mg、0.14mmol)およびDMAP(33mg、0.27mmol)の溶液に、CH2Cl2(96mL)中の酸7D(132mg,0.11mmol)の溶液を10時間かけて1滴ずつ添加し、その反応混合物をさらに9.5時間、室温で攪拌した。その後、0.2% HCl(50mL)を添加し、形成した層を分離し、有機層を硫酸水素ナトリウム(30mL)および飽和ブライン(30mL)で洗浄し、MgSO4で乾燥させ、真空下で濃縮して、褐色の固体を得た。その後、これをEtOAc/ヘキサン(6:4)の溶離剤を用い、それを(3:1)に増加させるフラッシュカラムクロマトグラフィーによって精製して、生成物8D(52mg、0.05mmol、42%)を白色の固体として得た:Rf 0.07 EtOAc/ヘキサン(6:4);[α]25D=−76(c 0.1、CH2Cl2);IR(薄膜)3271 1739(m)、1683、1641、1536、1490、1444、1391;1H NMR(400MHz,CDCl3)δ7.54(s,1H)、7.44−7.36(m,12H)、7.30−7.17(m,19H)、6.80(d,J=8.5Hz,1H,)、6.47(d,J=8.5Hz,1H)、5.58(dt,J=15.1,7.0Hz,1H,)、5.47(td,J=6.5,2.5Hz,1H,)、5.39(dd,J=15.1,6.5Hz,1H)、4.68(s,1H)、4.29(q,J=7.0Hz,1H)、4.20(dd,J=17.1,7.5Hz,1H)、4.16(dd,J=9.0,6.0Hz,1H)、3.53(dd,J=17.1,5.5Hz,1H)、3.44(q,J=7.5Hz,1H)、3.00−2.90(m,2H)、2.66(d,J=7.5Hz,2H)、2.55−2.48(m,1H)、2.39(dd,J=15.1,8.0Hz,1H)、2.30−2.15(m,3H)、2.13−2.01(m,4H)、1.68−1.58(m,1H)、1.51(dd,J=13.5,7.0Hz,1H,)、1.40(s,9H)、1.45−1.34(m,2H)、1.36−1.28(m,2H)、1.31−1.23(m,2H)、0.89(d,J=6.5Hz,3H)、0.84(d,J=6.5Hz,3H,)、13C NMR(100MHz,CDCl3)δ173.1(C)、171.3(C)、171.3(C)、169.8(C)、168.9(C)、156.2(C)、145.0(C)、144.5(C)、132.6(CH)、129.7(CH)、128.2(CH)、128.0(CH)、127.0(CH)、126.8(CH)、79.1(CH)、67.5(C)、66.8(C)、58.7(CH)、53.1(CH)、53.0(CH)、42.0(CH2)、41.8(CH2)、40.2(CH2)、32.2(CH2)、31.5(CH2)、31.4(CH2)、31.2(CH2)、31.0(CH2)、29.7(CH)、28.6(CH3)、22.8(CH2)、19.5(CH3)、17.6(CH3);LRMS(ES+)1152(100%,[M+Na+])。
【0220】
[4−((7R,10S,14S,21R)−7−イソプロピル−3,6,9,19,22−ペンタオキソ−2−オキサ−12,13−ジチア−5,8,20,23−テトラアザ−ビシクロ[8.7.6]トリコス−16−エン−21−イル)−ブチル]−カルバミン酸t−ブチルエステル(9D)の調製。
【0221】
CH2Cl2/MeOH(9:1)(148mL)中のヨウ素(117mg、0.5mmol)の攪拌溶液に、CH2Cl2/MeOH(9:1)(77mL)中の8D(52mg、0.05mmol)の溶液を30分かけて1滴ずつ添加し、その後、その反応混合物をさらに30分間攪拌させておき、その後、チオ硫酸ナトリウム(128mL、0.02M)を添加した。層を分離し、その生成物をCH2Cl2(3×25mL)で抽出し、MgSO4で乾燥させ、真空下で溶媒を除去した。その後、シリカゲルでのカラムクロマトグラフィー(溶離剤 2:98〜8:92 MeOH/CH2Cl2)による精製を行い、それによって9D(3mg、0.005mmol、10%)を白色の固体として得た:Rf 0.05 MeOH/CH2Cl2(5:95);LRMS(ES+)666.8(100%,[M+Na]+)。
【0222】
(R)−2−((R)−2−{(R)−2−[(R)−2−((E)−(S)−3−ヒドロキシ−9,9,9−トリフェニル−ノン−4−エノイルアミノ)−プロピオニルアミノ]−5,5,5−トリフェニル−ペンタノイルアミノ}−3−メチル−ブチリルアミノ)−プロピオン酸メチルエステル(6E)の調製。
【0223】
CH2Cl2/THF(1:1、30mL)中のH2N−D−Ala−D−Cys(STrt)−D−Val−D−Ala−OMe(96mg、0.155mmol、米国、CA 92121−1510のバイオペピド社(Biopepide Co.)から購入したもの、純度約60%)の溶液に、CH2Cl2(5mL)中の5(87mg、0.155mmol)の溶液を添加し、続いてDMAP(2mg、0.02mmol)を添加した。18時間後、その反応混合物を真空下で濃縮した。シリカゲルでのカラムクロマトグラフィー(1〜3.5% MeOH/CH2Cl2)による精製によって、ビス−トリチル6E(55mg、0.054mmol、35%)を溶解度の低い白色の固体として得た:LRMS(5μm 粒径、3.0×50mm、1.25mL/分で5分間にわたって5%メタノール(0.1% HCOOH)/H2Oから100%への線形勾配で、XTerra MS C18カラムを使用する、HPLC ES+。その後、1.5mL/分で5分間、100%メタノール(0.1% HCOOH))室温=7.93分、m/z 1041.7(100%、[M+Na]+)、1019.7(30%、[M+H]+)。
【0224】
(3R,6R,9S,12R,16S)−6−イソプロピル−3,12−ジメチル−16−((E)−4−トリチルスルファニル−ブト−1−エニル)−9−トリチルスルファニルメチル−1−オキサ−4,7,10,13−テトラアザ−シクロヘキサデカン−2,5,8,11,14−ペンタオン(8E)の調製。
【0225】
0℃で、THF(5mL)中のメチルエステル6E(81mg、0.080mmol)の溶液に、H2O(0.8mL)中のLiOH(3mg、0.12)の溶液を添加した。1時間後、1M HCl(10mL)の添加により反応を停止させた。CHCl3(20mL)を添加し、有機相を分離し、CHCl3(2×10mL)で抽出した。有機相をブライン(15mL)で洗浄し、乾燥させ(MgSO4)、真空下で濃縮して、粗製酸7E(80mg、定量的)を白色の固体として得、それを次の段階で直ちに使用した:LRMS(ES-)m/z 1003(100%,[M−H]-)。
【0226】
CH2Cl2(20mL)中のMNBA(35mg、0.10mmol)およびDMAP(25mg、0.20mmol)の溶液に、CH2Cl2/THF(75:5、80mL)中の酸7E(80mg、0.080mmol)の溶液を3時間かけて1滴ずつ添加した。さらに14時間後、1M HCl(25mL)の添加により反応を停止させた。有機相を(CH2Cl2で抽出しながら)分離し、NaHCO3(30mL)そしてブライン(20mL)で順次洗浄した。合わせた有機相を乾燥させ(MgSO4)、真空下で濃縮して、黄色の油を得た。シリカゲルでのカラムクロマトグラフィー(70〜100% EtOAc/CH2Cl2)による精製によって、8E(34mg、0.035mmol、43%)を白色の固体として得た:1H−NMR(400MHz,CDCl3)δ7.49−7.08(m,33H)、6.31(br s,1H)、5.57(dt,J=15.3,6.5Hz,1H)、5.47(q,J=6.1Hz,1H)、5.36(dd,J=15.3,6.8Hz,1H)、4.45(5重線,J=7.0Hz,1H)、4.21(5重線,J=7.0Hz,1H)、3.87(t,J=7.3Hz,1H)、3.59(br s,1H)、2.92(dd,J=12.5,8.5Hz,1H)、2.55(dd,J=12.5,5.4Hz,1H)、2.51−2.36(m,2H)、2.17(t,J=7.3Hz,2H)、2.05−1.86(m,3H)、1.36(d,J=7.0Hz,3H)、1.33(d,J=7.0Hz,3H)、0.88(d,J=6.8Hz,3H)、0.83(d,J=7.0Hz 3H);13C−NMR(100MHz,CDCl3)δ173.6(C)、170.8(C)、170.4(C)、170.2(C)、169.7(C)、144.9(C)、144.4(C)、132.9(CH)、129.7(CH)、129.6(CH)、128.2(CH)、128.1(CH)、128.0(CH)、127.0(CH)、126.8(CH)、72.0(CH)、67.2(C)、66.8(C)、61.4(CH)、55.3(CH)、49.9(CH)、49.5(CH)、41.3(CH2)、32.4(CH2)、31.4(CH2)、31.3(CH2)、29.8(CH)、19.8(CH3)、18.4(CH3)、17.9(CH3)、17.8(CH3);LRMS(ES+)m/z 1009(100%,[M+Na]+)、987(10%,[M+H]+)。
【0227】
(E)−(1S,4R,7R,10S,21R)−7−イソプロピル−4,21−ジメチル−2−オキサ−12,13−ジチア−5,8,20,23−テトラアザ−ビシクロ[8.7.6]トリコス−16−エン−3,6,9,19,22−ペンタオン(9E)の調製。
【0228】
CH2Cl2/MeOH(9:1、100mL)中のI2(87mg、0.34mmol)の溶液に、ビス−トリチル8E(34mg、0.034mmol)の溶液を30分かけて1滴ずつ添加した。さらに30分間後、チオ硫酸ナトリウム(0.05M、25mL)、続いてブライン(10mL)の添加により反応を停止させた。有機相を分離し、水性相をCH2Cl2(3×15mL)で抽出した。合わせた有機相を乾燥させ(MgSO4)、濾過し、その後、真空下で濃縮することにより、白色の固体を得た。シリカゲルでのカラムクロマトグラフィー(3〜5% MeOH/CH2Cl2)による精製によって、環化デプシペプチド9E(10mg、0.02mmol、60%)を白色の固体として得た:[α]25D−51.8(c 0.45、MeOH);1H NMR(400MHz,CDCl3)δ7.52(d,J=6.5Hz,1H)、7.40(d,J=6.3Hz,1H)、6.94(d,J=9.5Hz,1H)、6.46(d,J=3.0Hz,1H)、6.23−6.10(m,1H)、5.80−5.72(m,2H)、5.06(ddd,J=10.0,6.8,3.7Hz,1H)、4.47(5重線,J=7.0Hz,1H)、4.20(qd,J=7.3,3.5Hz,1H)、3.81(dd,J=14.3,6.5Hz,1H)、2.96−2.77(m,3H)、2.76−2.64(m,5H)、2.61(dd,J=13.5,2.2Hz,1H)、1.49(d,J=7.0Hz,3H)、1.48(d,J=7.2Hz,3H)、0.95(d,J=6.5Hz,3H)、0.93(d,J=6.5Hz,3H);13C−NMR(100MHz,CD3OD)δ174.9(C)、173.9(C)、172.4(C)、172.2(C)、172.0(C)、131.9(CH)、131.1(CH)、71.8(CH)、67.2(CH)、56.3(CH)、53.0(CH)、50.7(CH)、40.4(CH2)、39.9(CH2)、39.3(CH2)、34.4(CH2)、28.7(CH)、20.7(CH3)、20.3(CH3)、17.7(CH3)、16.4(CH3);LRMS(ES+)m/z 1024(200%,[2M+Na]+)、523(50%,[M+Na]+)、501(100%,[M+H]+)。
【0229】
(R)−4−((E)−(S)−3−ヒドロキシ−7−トリチルスルファニル−ヘプト−4−エノイルアミノ)−4−{(S)−1−[(R)−1−(メトキシカルボニルメチル−カルバモイル)−2−メチル−プロピルカルバモイル]−2−トリチルスルファニル−エチルカルバモイル}−酪酸t−ブチルエステル(6F)の調製。
【0230】
CH2Cl2(40mL)中のH2N−D−Glu(OtBu)−D−Cys(STrt)−D−Val−Gly−OMe(145mg、0.2mmol、米国、CA 92121−1510のバイオペピド社(Biopepide Co.)から購入したもの、純度約60%)の溶液に、CH2Cl2(10mL)中の5(113mg、0.2mmol)の溶液を添加し、続いてDMAP(2.5mg、0.02mmol)を添加した。18時間後、その反応物を真空下で濃縮した。シリカゲルでのカラムクロマトグラフィー(1〜3% MeOH/CH2Cl2)による精製によって、6F(97mg、0.087mmol、44%)を白色の固体として得た:1H NMR(400MHz,CDCl3)δ7.46−7.33(m,12H)、7.32−7.13(m,20H)、7.07(br s,1H)、6.77(d,J=8.3Hz,1H)、5.46(dt,J=15.3,6.5Hz,1H)、5.34(dd,J=15.3,6.3Hz,1H)、4.40−4.23(m,3H)、4.11−3.86(m,2H)、3.79(dd,J=17.8,5.5Hz,1H)、3.70−3.58(m,1H)、3.64(s,3H)、3.06(br s,1H)、2.63(dd,J=12.8,7.5Hz,1H)、2.53(dd,J=12.8,5.8Hz,1H)、2.48−2.12(m,6H)、2.11−1.85(m,4H)、1.41(s,9H)、0.92(d,J=6.7Hz,3H)、0.91(d,J=6.7Hz,3H);13C−NMR(100MHz,CDCl3)δ173.7(C)、172.3(C)、171.6(C)、171.3(C)、170.4(C)、170.3(C)、145.0(C)、144.3(C)、133.6(CH)、130.3(CH)、129.7(CH)、129.5(CH)、128.2(CH)、128.0(CH)、127.1(CH)、126.7(CH)、81.5(C)、70.0(CH)、67.1(C)、66.7(C)、59.3(CH)、54.6(CH)、53.0(CH)、52.2(CH3)、44.1(CH2)、41.0(CH2)、33.3(CH2)、32.2(CH2)、31.4(CH2 x 2)、29.9(CH)、28.1(CH3)、26.1(CH2)、19.3(CH3)、17.7(CH3);LRMS(ES+)m/z 1142(100%,[M+Na]+)。
【0231】
3−[(6R,9S,12R,16S)−6−イソプロピル−2,5,8,11,14−ペンタオキソ−16−((E)−4−トリチルスルファニル−ブト−1−エニル)−9−トリチルスルファニルメチル−1−オキサ−4,7,10,13−テトラアザ−シクロヘキサデク−12−イル]−プロピオン酸t−ブチルエステル(8F)の調製。
【0232】
0℃で、THF(5mL)中のメチルエステル6F(98mg、0.088mmol)の溶液に、H2O(0.8mL)中のLiOH(3mg、0.13)の溶液を添加した。1時間後、1M HCl(15mL)の添加により反応を停止させた。CHCl3(20mL)を添加し、有機相を分離し、CHCl3(2×10mL)で抽出した。有機相をブライン(15mL)で洗浄し、乾燥させ(MgSO4)、真空下で濃縮して、粗製酸7F(96mg、定量的)を白色の固体として得、それを次の段階で直ちに使用した:(ES-)m/z 1103(100%,[M−H]-)。
【0233】
CH2Cl2(20mL)中のMNBA(36mg、0.10mmol)およびDMAP(25mg、0.21mmol)の溶液に、CH2Cl2/THF(75:2、77mL)中の酸7F(96mg、0.087mmol)の溶液を3時間かけて1滴ずつ添加した。さらに14時間後、1M HCl(25mL)の添加により反応を停止させた。有機相を(CH2Cl2で抽出しながら)分離し、NaHCO3(30mL)そしてブライン(20mL)で順次洗浄した。合わせた有機相を乾燥させ(MgSO4)、真空下で濃縮して、黄色の油を得た。シリカゲルでのカラムクロマトグラフィー(50〜70% EtOAc/CH2Cl2、その後、+0.1% MeOH)による精製によって、8F(53mg、0.049mmol、56%)を白色の固体として得た:1H NMR(400MHz,CDCl3)δ7.44−7.32(m,12H)、7.31−7.15(m,19H)、7.11−7.03(m,2H)、6.68(br s,1H)、5.58(dt,J=15.6,6.5Hz,1H)、5.43(ddd,J=9.5,7.0,3.0Hz,1H)、5.33(dd,J=15.6,7.0Hz,1H)、4.44(dd,J=17.0,9.6Hz,1H)、4.30(dd,J=9.0,4.5Hz,1H)、4.04(q,J=7.0Hz,1H)、3.59(m,1H)、3.50(dd,J=6.5,2.0Hz,1H)、3.07(t,J=11.0Hz,1H)、2.60−2.45(m,3H)、2.42−2.23(m,3H)、2.22−2.12(m,2H)、2.08−1.89(m,4H)、1.40(s,9H)、0.88(d,J=7.0Hz,3H)、0.83(d,J=7.0Hz,3H);13C−NMR(100MHz,CDCl3)δ173.4(C)、172.6(C)、171.1(C)、170.9(C)、170.0(C)、169.0(C)、145.0(C)、144.3(C)、133.2(CH)、129.7(CH)、129.6(CH)、128.3(CH)、128.0(CH)、127.1(CH)、126.8(CH)、81.3(C)、72.1(CH)、67.3(C)、66.8(C)、58.7(CH)、56.8(CH)、54.1(CH)、41.9(CH2)、41.3(CH2)、32.4(CH2)、31.8(CH2)、31.4(CH2)、31.2(CH2)、29.0(CH)、28.2(CH3)、25.9(CH2)、19.7(CH3)、17.2(CH3);LRMS(ES+)m/z 1109(100%,[M+Na]+)、1087(50%,[M+H]+)。
【0234】
3−((E)−(1S,7R,10S,21R)−7−イソプロピル−3,6,9,19,22−ペンタオキソ−2−オキサ−12,13−ジチア−5,8,20,23−テトラアザ−ビシクロ[8.7.6]トリコス−16−エン−21−イル)−プロピオン酸t−ブチルエステル(9F)の調製。
【0235】
CH2Cl2/MeOH(9:1、130mL)中のI2(117mg、0.46mmol)の溶液に、ビス−トリチル8F(50mg、0.046mmol)の溶液を30分かけて1滴ずつ添加した。さらに30分間後、チオ硫酸ナトリウム(0.05M、50mL)、続いてブライン(20mL)の添加により反応を停止させた。有機相を分離し、水性相をCH2Cl2(3×25mL)で抽出した。合わせた有機相を乾燥させ(MgSO4)、その後、真空下で濃縮することにより、白色の固体を得た。シリカゲルでのカラムクロマトグラフィー(1〜3% MeOH/CH2Cl2)による精製によって、二環式デプシペプチド9F(19mg、0.032mmol、69%)を白色の固体として得た:[α]25D−76.7(c 0.60、MeOH);1H NMR(400MHz,CDCl3)δ8.15(d,J=4.0Hz,1H)、7.35(d,J=6.5Hz,1H)、7.23(t,J=5.5Hz,1H)、7.11(d,J=9.0Hz,1H)、6.05(ddt,J=15.5,7.0,2.0Hz,1H)、5.84−5.73(m,2H)、4.97(td,J=8.0,4.5Hz,1H)、4.18−4.01(m,3H)、3.61(dd,J=15.1,8.0Hz,1H)、3.24(dd,J=10.5,6.5Hz,1H)、2.95−2.75(m,5H)、2.73−2.62(m,3H)、2.51(dd,J=13.6,2.0Hz,1H)、2.42(ddd,J=18.0,9.0,3.5Hz,1H)、2.19−2.04(m,2H)、1.83(s,9H)、0.98(d,J=6.5Hz,3H)、0.92(d,J=6.5Hz,3H);13C−NMR(100MHz,CDCl3)δ175.7(C)、171.8(C)、171.7(C)、170.9(C)、169.9(C)、168.2(C)、130.2(CH)、129.6(CH)、82.6(C)、69.4(CH)、65.4(CH)、57.6(CH)、53.8(CH)、42.4(CH2)、40.0(2 x CH2)、38.6(CH2)、34.1(CH2)、33.1(CH2)、28.2(CH3)、27.4(CH)、24.9(CH2)、20.6(CH3)、20.2(CH3);LRMS(ES+)m/z 623(90%,[M+Na]+)、601(100%,[M+H]+)。
【0236】
3−((E)−(1S,7R,10S,21R)−7−イソプロピル−3,6,9,19,22−ペンタオキソ−2−オキサ−12,13−ジチア−5,8,20,23−テトラアザ−ビシクロ[8.7.6]トリコス−16−エン−21−イル)−プロピオン酸(10F)の調製。
【0237】
CH2Cl2(1.5mL)中の9F(12mg、0.02mmol)およびEt3SiH(10μL、0.06mmol)の溶液に、TFA(150μL、2.0mmol)を添加した。6時間後、溶媒を除去し、残留物をシリカゲルでのカラムクロマトグラフィー(10% メタノール/CH2Cl2、その後、+1% AcOH)によって精製して、10F(5.6mg、0.01mmol、55%)を白色の固体として得た:[α]25D−63.4(c 0.25、MeOH);1H NMR(400MHz,CD3OD)δ7.5(1 x NHが観察された,d,J=7.5Hz,1H)、5.81(dddd,J=16.8,6.8,4.8,2.5Hz,1H)、5.75−5.67(m,2H)、4.62(ddd,J=11.0,8.0,5.5Hz,1H)、4.28(d,J=17.5Hz,1H)、4.15(dd,J=9.0,6.0Hz,1H)、3.78(d,J=17.5Hz,1H)、3.43(d,J=10.5Hz,1H)、3.20−3.10(m,2H)、3.07−3.00(m,2H)、2.94−2.81(m,1H)、2.82(dd,J=13.2,2.2Hz,1H)、2.73−2.50(m,4H)、2.38(q,J=7.0Hz,1H)、2.23−1.99(m,2H)、0.98(d,J=6.5Hz,3H)、0.92(d,J=6.5Hz,3H);13C−NMR(100MHz,CD3OD)δ176.5(C)、174.0(C)、173.5(C)、173.0(C)、171.6(C)、169.5(C)、131.4(CH)、131.3(CH)、72.1(CH)、66.0(CH)、58.5(CH)、57.0(CH)、42.7(CH2)、39.7(CH2)、39.4(CH2)、36.8(CH2)、31.8(CH2)、31.6(CH2)、28.2(CH3)、26.6(CH2)、20.6(CH3)、20.5(CH3);LRMS(ES-)m/z 543(100%,[M−H]-)。
【0238】
[結果]
[活性アッセイ1]
本発明者らは、MCF7ヒト乳癌細胞および正常ヒト皮膚線維芽細胞(NHDF)の増殖を阻害するスベロイルアニリドヒドロキサム酸(SAHA)、既知HDAC阻害剤、および化合物001(7頁の式(2)の化合物)の能力を比較した。
【0239】
方法;細胞増殖アッセイは、商標シクワント(CyQuant)アッセイシステム(米国のモレキュラー・プローブス社(Molecular Probes,Inc.))をその製造業者の説示に従って使用して行った。このシステムは、細胞の核酸に結合すると蛍光が増強されて、細胞数50から250,000の間の線形検出範囲での細胞数の決定を可能にする、有標緑色蛍光色素を利用する。エストロゲン受容体陽性MCF7乳癌細胞(ECAAC)およびNHDF細胞(英国のキャンブレックス(Cambrex))を96ウエルプレートに、1ウエル当たり100μLの細胞培養基中に細胞数1000の密度で、プレーティングした。化合物は、最低5時間後、細胞培養基中の系列希釈物で、2×最終濃度での100μL量で添加した。6日後、そのプレートを吸い取り紙の上で逆さにすることにより細胞培養基を除去し、細胞を200μLのPBSで1回、穏やかに洗浄した。直ちにプレートを−80℃で最低1時間冷凍し、その後、解凍した。上記製造業者の説示に従って作った色素を補足した200μLの1x シクワント細胞溶解緩衝液を各ウエルに直ちに添加し、室温で3〜5分間インキュベートした。その後、サイトフルーアII蛍光マルチウエルプレートリーダー(Cytofluor II Fluorescence Multiwell Plate Reader)およびサイトフルーアIIソフトウェアと励起用に480nmおよび最大発光用に520nmのフィルターを用いて、各ウエルについての蛍光を測定した。細胞増殖は、二重反復試験サンプルの平均値について、未処理細胞サンプル(=100%)を基準にしたパーセンテージとして判定した。増殖阻害曲線を用いて、IC50値を導出した。
【0240】
図1は、MCF7および正常ヒト皮膚線維芽細胞(NHDF)に対する化合物001およびSAHAの効果を示すものである。MCF7およびNHDFを指示濃度の化合物と共にインキュベートした。6日後、未処理細胞(=100%)を基準にした細胞増殖を、商標シクワント(CyQuant)アッセイを用いて判定した。溶媒対照としての等量のDMSOは、細胞増殖に対してまったく効果がなかった(示されていない)。データは、二重反復判定についての平均±標準偏差であり、複数の実験の代表値である。
【0241】
結果;細胞増殖は、これらの化合物により阻害された。重要なこととして、阻害剤間および細胞タイプ間に差があった。増殖阻害についての平均IC50(±SD)を複数の実験から計算した。それらを下の表1に示す。
【0242】
【表1】
【0243】
化合物001は、癌細胞増殖の強力な阻害剤であった。重要なこととして、化合物001は、現在II相試験中のSAHA(p=0.003、スチューデントt検定)より有意に強力であった。両方の化合物が、NHDFに対してMCF7での方が比較的有効であった。これは、悪性細胞に対する選択性の証拠となる。HUT78 T細胞白血病細胞を使用して行った(上で説明したように行った)同様の試験は、化合物001がこれらの細胞の増殖も阻害することを明示した(データは示さない)。
【0244】
[活性アッセイ2]
本発明者らは、HDAC媒介抑制に応答することが以前に証明されているSV40プロモーターを化合物001(7頁の式(2)の化合物)が活性化するかどうかを調査した。
【0245】
方法.本発明者らは、ルシフェラーゼ遺伝子の上流にクローニングされたSV40即時初期プロモーターが組み込まれたレポーター構築物を含有するMCF7細胞由来の安定なクローンを作製した。トランスファスト(Transfast)トランスフェクション試薬を使用して、pGL2−Basic(SV40/ルシフェラーゼレポーター)(英国のプロメガ(Promega))およびpcDNA3(ネオマイシン耐性遺伝子を発現するもの)をMCF7細胞にトランスフェクトした。24時間後、トリプシン処理により細胞を回収し、低密度でプレーティングした。翌日、G418をその増殖培地に添加した。〜21日後、個々の薬物耐性コロニーを単離し、培養した。本発明者らは、さらなる研究のためにHDAC阻害剤の添加後に高いルシフェラーゼ活性誘発レベルを明示したクローン(2.1.1細胞)を選択した。化合物001の効果を検査するために、2.1.1細胞を様々な濃度の化合物001または対照として等量のDMSOと共にインキュベートした。翌日、細胞を回収し、プロメガ(Promega)ルシフェラーゼアッセイシステムおよびトップカウント(パーキン・エルマー)(TopCount(Perkin−Elmer))を使用してルシフェラーゼ活性を検出した。
【0246】
図2は、HDAC応答性SV40プロモーターの活性に対する化合物001の効果を示すものである。SV40プロモーターが安定的に組み込まれたルシフェラーゼレポータープラスミドを含有するMCF7由来細胞を、指示濃度の化合物001、または溶媒対照としてのDMSOで処理した。ルシフェラーゼ活性は、約16時間後に判定した。データは、二重反復で行った単一の実験から導出したものであり、複数の実験の代表である。
【0247】
結果;この実験は、化合物001が、SV40即時初期プロモーターのHDAC媒介抑制を効果的に逆行させることを示す。
【0248】
[活性アッセイ3]
本発明者らは、MCF7細胞における細胞周期分布および生存に対する化合物001(7頁の式(2)の化合物)およびSAHAの効果を調査した。
【0249】
方法;図の説明文に詳述されているように、MCF7細胞を様々な時間、指示濃度の化合物と共にインキュベートするか、対照として未処理で放置した。細胞を回収し、以前に説明されている(Purohitら,Int J Ca 85,584−9 2000)ようなヨウ化プロピジウム染色法およびフローサイトメトリーを用いて細胞周期の異なる期における細胞の比率を判定した。全細胞を基準にして、<G0/G1含量(プレG0)を有する細胞の比率を、先ず計算した。細胞周期の異なる期(G0/G1、S、G2/M)にある細胞の比率は、その細胞周期内の全細胞(すなわち、<G0/G1含量を有する細胞を引いた全細胞)に対する比率として計算した。
【0250】
図3は、MCF7細胞における細胞周期分布および生存に対する化合物001およびSAHAの効果を示すものである。MCF7細胞を指示濃度(各々、増殖阻害についてのIC50値の10倍までに相当する)の化合物、または溶媒対照としてのDMSOと共に、24、48または72時間インキュベートした。細胞周期の異なる期にある細胞の比率は、フローサイトメトリーによって判定した。データは、1に正規化される未処理細胞にを基準にして、2つの別の実験から導出された平均値±SDを示すものである。
【0251】
結果;この図は、等価有効濃度で、化合物001およびSAHAが、MCF7細胞において主G2/M期停止および細胞死を誘導することを示している。
【0252】
[活性アッセイ4]
本発明者らは、MCF7乳癌細胞および心筋細胞におけるヒストンアセチル化に対する化合物001(7頁の式(2)の化合物)の効果を調査した。
【0253】
方法;心筋細胞を、Chembiochem.2005 Jan;6(1):162−70に記載されているように準備し、指示濃度の化合物001と共に24時間インキュベートした。MCF7細胞を指示濃度の化合物001と共に24時間インキュベートした。アップステート・バイオテック(Upstate Biotech)からの抗体を使用し、以前に説明されている(Brimmellら.Br J Cancer.1999 Nov;81(6):1042−51)ように、ウエスタンブロッティングによって、全ヒストンH4アセチル化、またはヒストンH4−K8もしくはヒストンH3−K9の特異的アセチル化の発現を分析した。
【0254】
図4は、ヒストンアセチル化に対する化合物001の効果を示すものである。(A)MCF7細胞を化合物001と共に24時間インキュベートした。ヒストンH4アセチル化を免疫ブロッティングによって測定した。(B)心筋細胞を指示化合物001と共に24時間インキュベートした。ヒストンH4アセチル化を免疫ブロッティングによって測定した。(C)MCF7細胞を化合物001と共に24時間インキュベートした。ヒストンH3−K9およびヒストンH4−K8アセチル化を免疫ブロッティングによって測定した。
【0255】
結果;この実験は、化合物001が、アセチル化ヒストンH4の蓄積ならびに特異的ヒストンH3−K9およびヒストンH4−K8アセチル化を誘導することを示している。本発明者らは、化合物001が、原発性慢性リンパ球性白血病細胞においてアセチル化ヒストンH4のレベルを増加させることも実証した(データは示さない)。
【0256】
[活性アッセイ5]
本発明者らは、インビトロHDAC活性を阻害する化合物の能力を分析した。インビトロHDACアッセイは、HDAC蛍光活性アッセイキット(英国のバイオモール(Biomol))をその製造業者の説示に従って使用して行った。化合物は、分析前に還元した。一晩、室温で、光から保護して、1mMの化合物をDMSO中30mMのDTTで還元した。その後、96ウエルプレートでの反応を準備した。各反応について、10μLの化合物(アッセイ用緩衝液中の必要濃度の5倍)を15μLの希釈Hela核抽出物(アッセイ用緩衝液中30倍)と混合した。各化合物についての系列希釈物を準備した。Hela抽出物のみを含有する反応物およびアッセイ用緩衝液のみを含む反応物も準備した。25μLの希釈した商標フルーア・ド・リス(Fluor de Lys)基質(アッセイ用緩衝液中100倍)を各反応物に添加し、その後、それらを37℃で1時間インキュベートした。50μLの商標フルーア・ド・リス(Fluor de Lys)顕色剤(アッセイ用緩衝液中20倍の希釈剤、加えて、100倍希釈したTSA)の添加により反応を停止させた。その後、反応物を室温で10分間インキュベートし、その後、サイトフルーアII蛍光マルチウエルプレートリーダーおよびサイトフルーアIIソフトウェアと励起用に360nMおよび発光用に460nMに設定したフィルターを用いて蛍光を測定した。インビトロHDAC活性の阻害は、二重反復試験サンプルの平均値について、Hela抽出物のみの反応物を基準にしたパーセンテージとして判定した。IC50値は、グラフパッド・プリズム(GraphPad Prism)ソフトウェアを使用して計算した。
【0257】
【表2】
【0258】
[活性アッセイ6]
本発明者らは、ヒト癌細胞のインビトロ増殖を阻害する化合物の能力を分析した。細胞増殖アッセイは、商標シクワント(CyQuant)アッセイシステム(米国のモレキュラー・プローブス社(Molecular Probes,Inc.))をその製造業者の説示に従って用いて行った。MCF7乳癌細胞、A2780卵巣癌細胞ならびにPC3およびLNCAP前立腺癌細胞を、96ウエルプレートに、ウエル当たり100μLの細胞培養基中、MCF7細胞については細胞数1000、またはA2780/PC3/LNCAP細胞については細胞数5000の密度でプレーティングした。化合物は、最低5時間後、細胞培養基中の系列希釈で、2×最終濃度での100μL量で添加した。4日後(A2780、PC3もしくはLNCAP細胞)または6日後(MCF7細胞)、プレートを吸い取り紙の上で逆さにすることにより細胞培養基を除去し、細胞を200μLのPBSで1回、穏やかに洗浄した。直ちにプレートを−80℃で最低1時間冷凍し、その後、解凍した。製造業者の説示に従って作った色素を補足した200μLの1x シクワント細胞溶解緩衝液を各ウエルに直ちに添加し、室温で3〜5分間インキュベートした。その後、サイトフルーアII蛍光マルチウエルプレートリーダーおよびサイトフルーアIIソフトウェアと励起用に480nmおよび最大発光用に520nmのフィルターを用いて、各ウエルについての蛍光を測定した。細胞増殖は、二重反復試験サンプルの平均値について、未処理細胞サンプル(=100%)を基準にしたパーセンテージとして判定した。増殖阻害曲線を用いて、IC50値を導出した。
【0259】
【表3】
【0260】
[活性アッセイ7]
本発明者らは、末梢血単核細胞(PBMC)からのTNFαの生産を阻害する化合物の能力を分析した。TNF−αイムノアッセイは、登録商標クアンティキン(Quantikine)ヒトTNF−αアッセイキット(英国、アビンドン(Abingdon,UK)のアール・アンド・ディー・システムズ(R&D systems))をその製造業者の説示に従って使用して行った。商標フィコール・パクー(Ficol paque)プラス(英国、アマシャム(Amersham,UK)のジーイー・ヘルスケア(GE Healthcare)を使用してヒト全血を分離し、PBMCを、24ウエルプレートに、ウエル当たり500μLの細胞培養基中2.5×106の密度でプレーティングした。1時間後、化合物を、6×最終濃度での100μL量で添加した。5時間後、リポ多糖類(LSP;英国、プール(Poole,UK)のシグマ(Sigma))を60×最終濃度の10μL量で添加し、プレートを混合し、37℃、5%CO2で一晩、放置した。プレートを遠心分離し、細胞上清を新たなプレートに移した。登録商標クアンティキン(Quantikine)アッセイ試薬および標準物質をその製造業者の説示に従って調製した。登録商標クアンティキン(Quantikine)アッセイプレートは、50μLのアッセイ希釈液RD1Fを各ウエルに添加することにより準備した。この後、200μLの標準物質または100μLの校正物質希釈液、加えて、100μLの細胞上清を添加し、これを2時間、室温(RT)でインキュベートした。洗浄緩衝液を使用してプレートを4回洗浄した。最終洗浄後、プレートを清浄なペーパータオルで軽くたたいてすべての残留緩衝液を除去した。200μLのTNF−αコンジュゲートを各ウエルに添加し、1時間、室温でインキュベートした。その後、プレートを前のとおり洗浄した。光から保護しながら等量の着色試薬AおよびBを添加することにより必要量の基質溶液を調製し、光から保護して、この混合物の200μLを各ウエルに添加し、20分間、室温でインキュベートした。50uLの停止溶液を基質溶液と同じオーダーで各ウエルに添加し、バイオラド(Biorad)680・96ウエルプレートリーダー(英国、ヘメルヘムステッド(Hemel Hempstead,UK)のバイオラド(Bio−Rad))を用い、570nmのλ補正で、450nmで、プレートを読み取った。TNF−αレベルは、nmでの吸収を用い、二重反復試験サンプルの平均値について判定し、校正ゼロ値をすべての結果から減算して、このアッセイにおける校正物質希釈液の添加を補正した。
【0261】
【表4】
【0262】
[活性アッセイ8]
本発明者らは、マウスにおいて炎症を阻害する化合物9A(7頁の式(2)の化合物)の能力を分析した。毛を刈った腹部の皮膚に5%(w/v)オキサゾロンを塗布することにより、Balb/cマウスを感作した。7日後、左耳に3%(w/v)オキサゾロンおよび右耳にアセトンを局所塗布することによりマウスの炎症を惹起(challenge)した。30分後、アセトンに溶解した試験化合物を耳(背側および腹側の面)に塗布した。24時間後、マウスを安楽死させ、耳の重量を測定した。阻害%は、次の式(Wは、平均重量である)。
【0263】
阻害%=1−((W刺激薬+試験材料−Wヒ゛ヒクル x 100)/(刺激薬ノミ−Wヒ゛ヒクル))
を用いて決定した。
【0264】
【表5】
【図面の簡単な説明】
【0265】
【図1】MCF7および正常ヒト皮膚線維芽細胞(NHDF)に対する化合物001およびSAHAの効果を示す。
【図2】HDAC応答性SV40プロモーターの活性に対する化合物001の効果を示す。
【図3】MCF7細胞における細胞周期分布および生存に対する化合物001およびSAHAの効果示す。
【図4】ヒストンアセチル化に対する化合物001の効果を示す。
【特許請求の範囲】
【請求項1】
HDACにより媒介される状態の治療および予防に使用するための医薬品の製造における、式(I)もしくは(I’)
【化1】
(式中、R1、R2、R3およびR4は、同じであるか異なり、アミノ酸側鎖部分を表し、各R6は、同じであるか異なり、水素またはC1−C4アルキルを表し、ならびにPr1およびPr2は、同じであるか異なり、水素またはチオール保護基を表す)のFK228類似体である化合物、そのアイソスターまたは医薬的に許容されるその塩の使用。
【請求項2】
HDACにより媒介される状態の治療および予防に使用するための医薬品の製造における、請求項1に記載の式(I)もしくは(I’)の化合物であるFK228類似体、または医薬的に許容されるその塩の使用。
【請求項3】
各アミノ酸側鎖が、−H、−C1−C6アルキル、−C2−C6アルケニル、−L−O−C(O)−R’、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’、−L−Het−C(O)−Het−R’’および−L−Het−R’’から選択される部分であり、Lが、C1−C6アルキレン基であり、Aが、フェニルまたは5員から6員ヘテロアリール基であり、各R’が、同じであるか異なり、C1−C4アルキルを表し、各R’’が、同じであるか異なり、HまたはC1−C6アルキルを表し、各−Het−が、同じであるか異なり、−O−、−N(R’’’)−および−S−から選択されるヘテロ原子スペーサーであり、各R’’’が、同じであるか異なり、HまたはC1−C4アルキルを表す、先行する請求項のいずれか一項に記載の使用。
【請求項4】
Aが、フェニルである、請求項3に記載の使用。
【請求項5】
−Het−が、−O−または−NR’’’である、請求項3または請求項4に記載の使用。
【請求項6】
R1が、−Hおよび−C1−C6アルキルから選択される部分である、請求項3、請求項4および請求項5のいずれか一項に記載の使用。
【請求項7】
R2が、−Hおよび−C1−C4アルキルから選択される部分である、請求項3から請求項6のいずれか一項に記載の使用。
【請求項8】
R3が、−H、−C1−C6アルキル、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’および−L−N(R’’)−C(O)−O−R’’から選択される部分である、請求項3から請求項7のいずれか一項に記載の使用。
【請求項9】
R4が、−Hおよび−C1−C4アルキルから選択される部分である、請求項3から請求項8のいずれか一項に記載の使用。
【請求項10】
R6が、−Hである、請求項3から請求項9のいずれか一項に記載の使用。
【請求項11】
Pr1およびPr2が、同じであるか異なり、水素および保護基から選択され、前記保護基が、C1−C6アルコキシ、C1−C6アシルオキシ、ヒドロキシおよびニトロで所望により置換されているベンジル基、ピコリル、ピコリル−N−オキシド、アントリルメチル、ジフェニルメチル、フェニル、t−ブチル、アダマンチル、C1−C6アシルオキシメチル、C1−C6アルコキシメチル、テトラヒドロピラニル、ベンジルチオメチル、フェニルチオメチル、チアゾリジン、アセトアミドメチル、ベンズアミドメチル、t−ブトキシカルボニル(BOC)、アセチルおよびその誘導体、ベンゾイルおよびその誘導体、カルバモイル、フェニルカルバモイルおよびC1−C6アルキルカルバモイルから選択される、請求項3から請求項10のいずれか一項に記載の使用。
【請求項12】
Pr1およびPr2が、水素である、請求項3から請求項11のいずれか一項に記載の使用。
【請求項13】
各アミノ酸側鎖が、天然アミノ酸中に存在するアミノ酸側鎖部分であるか、−(CH2)2−C(O)−O−C(CH3)3、−(CH2)4−NH−C(O)−O−C(CH3)3、−(CH2)3−NH−C(O)NH2、−CH2−CH2OHまたは−(CH2)2−CH2NH2である、請求項1または請求項2に記載の使用。
【請求項14】
前記化合物が、式(2)、(2’)、(3)、(3’)、(4)、(4’)、(5)、(5’)、(6)、(6’)、(7)、(7’)、(8)、(8’)、(9)または(9’)の化合物である、請求項1に記載の使用。
【化2】
【請求項15】
前記FK228類似体が、式(I)の化合物、そのアイソスターまたは医薬的に許容されるその塩である、先行する請求項のいずれか一項に記載の使用。
【請求項16】
前記医薬品が、癌、心肥大症、慢性心不全、炎症状態、心血管疾患、ヘモグロビン異常症、サラセミア、鎌状赤血球症、CNS障害、自己免疫疾患、糖尿病、骨粗しょう症、MDS、良性前立腺肥大症、口腔白板症、遺伝的代謝性疾患、感染症、ルビンス−テイビ(Rubens−Taybi)、脆弱X症候群、またはアルファ−1アンチトリプシン欠損症の軽減のために使用されるか、創傷治癒の加速または毛包を保護するために使用されるか、免疫抑制薬として使用される、先行する請求項のいずれか一項に記載の使用。
【請求項17】
前記医薬品が、慢性リンパ球性白血病、乳癌、前立腺癌、卵巣癌、中皮腫、T細胞リンパ腫、心肥大症、慢性心不全または皮膚炎症状態、特に乾癬、アクネまたは湿疹を軽減させるための使用である、請求項16に記載の使用。
【請求項18】
ヒトまたは動物を治療する方法に使用するための、請求項1から請求項15のいずれか一項に記載のFK228類似体、そのアイソスターまたは医薬的に許容されるその塩。
【請求項19】
請求項1から請求項15のいずれか一項に記載のFK228類似体、そのアイソスターまたは医薬的に許容されるその塩。
【請求項20】
請求項1から請求項15のいずれか一項に記載のFK228類似体、そのアイソスターまたは医薬的に許容されるその塩と医薬的に許容される担体または希釈剤とを含む医薬組成物。
【請求項21】
経口、直腸内、非経口、鼻孔内もしくは経皮投与または吸入もしくは坐剤による投与に適する形態である、請求項20に記載の医薬組成物。
【請求項22】
錠剤、カプセル、トローチ、ロゼンジ、水性もしくは油性懸濁剤、分散性粉末もしくは顆粒または舌下錠の形態である、請求項21に記載の医薬組成物。
【請求項23】
HDACにより媒介される、請求項1、請求項16または請求項17に記載の状態に罹患しているまたは罹患しやすい患者を治療する方法であって、請求項1から請求項15のいずれか一項に記載の化合物、そのアイソスターまたは医薬的に許容されるその塩の有効量を前記患者に投与することを含む方法。
【請求項24】
スベロイルアニリドヒドロキサム酸(SAHA)と少なくとも等しいHDAC阻害活性を有する化合物を選択するプロセスであって、該プロセスは請求項1から請求項15のいずれか一項に記載の式(I)または(I’)の化合物を調製することを含み、該調製は以下を含む該プロセス:
(i)式(VI)の化合物と式(VII)の化合物を反応させ、
【化3】
(式中、R1およびR2は、先行する請求項のいずれか一項に記載のとおりであり、R7は、C1−C4アルキルまたはC2−C4アルケニルであり、ならびにYは、アミノ保護基である)
(ii)得られた中間体を脱保護した後、式(VIII)の化合物と反応させ、
【化4】
(式中、Y’は、アミノ保護基であり、Y’’は、水素または保護基である)
(iii)得られた中間体を脱保護した後、式(IX)の化合物と反応させ、
【化5】
(式中、R3は、先行する請求項のいずれか一項に記載のとおりであり、Y’’’は、アミノ保護基である)
(iv)得られた中間体を脱保護した後、式(X)の化合物と反応させ、
【化6】
(式中、R4は、先行する請求項のいずれか一項に記載のとおりであり、R5は、水素またはヒドロキシ保護基であり、LGは、脱離基であり、Y’’’’は、水素または保護基である)
(v)得られた中間体のβ−ヒドロキシ基を所望により脱保護して、R5保護基を除去し、Hで置換し、
(vi)得られた中間体を加水分解および環化し、
(vii)得られた中間体を、所望により、ジスルフィド結合を形成させるように反応させ、ジスルフィド結合が形成された場合には、得られた化合物中のジスルフィド結合を所望により切断し、得られた化合物がチオール基を含有する場合には、チオール保護基を所望により導入し;および
(viii)得られた化合物をスクリーニングして、HDAC阻害剤としての活性を測定すること。
【請求項25】
段階(viii)におけるスクリーニング段階が、インビトロHDACアッセイであって、様々な濃度で、試験化合物およびSAHAを希釈Hela核抽出物と接触させて、Hela核抽出物に対する試験化合物およびSAHAのIC50を決定することを含む、請求項24に記載のプロセス。
【請求項26】
SAHAと少なくとも等しいヒト癌細胞増殖阻害活性を有する化合物を選択するプロセスであって、請求項1から請求項15のいずれか一項に記載の式(I)または(I’)の化合物を調製することを含み、該調製が以下を含む該プロセス:
(i)式(VI)の化合物と式(VII)の化合物を反応させ、
【化7】
(式中、R1およびR2は、先行する請求項のいずれか一項に記載のとおりであり、R7は、C1−C4アルキルまたはC2−C4アルケニルであり、ならびにYは、アミノ保護基である)
(ii)得られた中間体を脱保護した後、式(VIII)の化合物と反応させ、
【化8】
(式中、Y’は、アミノ保護基であり、Y’’は、水素または保護基である)
(iii)得られた中間体を脱保護した後、式(IX)の化合物と反応させ、
【化9】
(式中、R3は、先行する請求項のいずれか一項に記載のとおりであり、Y’’’は、アミノ保護基である)
(iv)得られた中間体を脱保護した後、式(X)の化合物と反応させ、
【化10】
(式中、R4は、先行する請求項のいずれか一項に記載のとおりであり、R5は、水素またはヒドロキシ保護基であり、LGは、脱離基であり、Y’’’’は、水素または保護基である)
(v)得られた中間体のβ−ヒドロキシ基を所望により脱保護して、R5保護基を除去し、Hで置換し、
(vi)得られた中間体を加水分解および環化し、
(vii)得られた中間体を、所望により、ジスルフィド結合を形成させるように反応させ、ジスルフィド結合が形成された場合には、得られた化合物中のジスルフィド結合を所望により切断し、得られた化合物がチオール基を含有する場合には、チオール保護基を所望により導入し;および
(viii)得られた化合物をスクリーニングして、ヒト癌細胞増殖阻害剤としての活性を測定すること。
【請求項27】
段階(viii)におけるスクリーニング段階がインビトロアッセイであって、様々な濃度で試験化合物およびSAHAを、MCF7乳癌細胞系、HUT78T細胞白血病細胞系、A2780卵巣癌細胞系、PC3またはLNCAP前立腺癌細胞系と接触させて、該細胞系に対する該試験化合物およびSAHAのIC50を決定することを含む、請求項26に記載のプロセス。
【請求項28】
SAHAと少なくとも等しい抗炎症活性を有する化合物を選択するためのプロセスであって、該プロセスが請求項1から請求項15のいずれか一項に記載の式(I)または(I’)の化合物を調製することを含み、該調製が以下を含む該プロセス:
(i)式(VI)の化合物と式(VII)の化合物を反応させ、
【化11】
(式中、R1およびR2は、先行する請求項のいずれか一項に記載のとおりであり、R7は、C1−C4アルキルまたはC2−C4アルケニルであり、ならびにYは、アミノ保護基である)
(ii)得られた中間体を脱保護した後、式(VIII)の化合物と反応させ、
【化12】
(式中、Y’は、アミノ保護基であり、Y’’は、水素または保護基である)
(iii)得られた中間体を脱保護した後、式(IX)の化合物と反応させ、
【化13】
(式中、R3は、先行する請求項のいずれか一項に記載のとおりであり、Y’’’は、アミノ保護基である)
(iv)得られた中間体を脱保護した後、式(X)の化合物と反応させ、
【化14】
(式中、R4は、先行する請求項のいずれか一項に記載のとおりであり、R5は、水素またはヒドロキシ保護基であり、LGは、脱離基であり、Y’’’’は、水素または保護基である)
(v)得られた中間体のβ−ヒドロキシ基を所望により脱保護して、R5保護基を除去し、Hで置換し、
(vi)得られた中間体を加水分解および環化し、
(vii)得られた中間体を、所望により、ジスルフィド結合を形成させるように反応させ、ジスルフィド結合が形成された場合には、得られた化合物中のジスルフィド結合を所望により切断し、得られた化合物がチオール基を含有する場合には、チオール保護基を所望により導入し;および
(viii)得られた化合物をスクリーニングして、抗炎症活性を測定すること。
【請求項29】
段階(viii)におけるスクリーニング段階が、化合物の、SAHAを基準にして末梢血単核細胞(PBMC)からのTNFαの生産の阻害活性を測定することを含むアッセイである、請求項28に記載のプロセス。
【請求項30】
(a)請求項1から請求項15のいずれか一項に記載のFK228類似体、そのアイソスターまたは医薬的に許容されるその塩と、(b)請求項1、請求項16または請求項17に記載のHDACにより媒介される状態の治療または予防に同時に、別々にまたは逐次的に使用するためのHDACの別の阻害剤とを含有する製品。
【請求項31】
(a)請求項1から請求項15のいずれか一項に記載のFK228類似体、そのアイソスターまたは医薬的に許容されるその塩と、(b)癌の治療または予防に同時に、別々にまたは逐次的に使用するための別の化学療法薬または抗腫瘍薬とを含有する製品。
【請求項1】
HDACにより媒介される状態の治療および予防に使用するための医薬品の製造における、式(I)もしくは(I’)
【化1】
(式中、R1、R2、R3およびR4は、同じであるか異なり、アミノ酸側鎖部分を表し、各R6は、同じであるか異なり、水素またはC1−C4アルキルを表し、ならびにPr1およびPr2は、同じであるか異なり、水素またはチオール保護基を表す)のFK228類似体である化合物、そのアイソスターまたは医薬的に許容されるその塩の使用。
【請求項2】
HDACにより媒介される状態の治療および予防に使用するための医薬品の製造における、請求項1に記載の式(I)もしくは(I’)の化合物であるFK228類似体、または医薬的に許容されるその塩の使用。
【請求項3】
各アミノ酸側鎖が、−H、−C1−C6アルキル、−C2−C6アルケニル、−L−O−C(O)−R’、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’、−L−Het−C(O)−Het−R’’および−L−Het−R’’から選択される部分であり、Lが、C1−C6アルキレン基であり、Aが、フェニルまたは5員から6員ヘテロアリール基であり、各R’が、同じであるか異なり、C1−C4アルキルを表し、各R’’が、同じであるか異なり、HまたはC1−C6アルキルを表し、各−Het−が、同じであるか異なり、−O−、−N(R’’’)−および−S−から選択されるヘテロ原子スペーサーであり、各R’’’が、同じであるか異なり、HまたはC1−C4アルキルを表す、先行する請求項のいずれか一項に記載の使用。
【請求項4】
Aが、フェニルである、請求項3に記載の使用。
【請求項5】
−Het−が、−O−または−NR’’’である、請求項3または請求項4に記載の使用。
【請求項6】
R1が、−Hおよび−C1−C6アルキルから選択される部分である、請求項3、請求項4および請求項5のいずれか一項に記載の使用。
【請求項7】
R2が、−Hおよび−C1−C4アルキルから選択される部分である、請求項3から請求項6のいずれか一項に記載の使用。
【請求項8】
R3が、−H、−C1−C6アルキル、−L−C(O)−O−R’’、−L−A、−L−NR’’R’’および−L−N(R’’)−C(O)−O−R’’から選択される部分である、請求項3から請求項7のいずれか一項に記載の使用。
【請求項9】
R4が、−Hおよび−C1−C4アルキルから選択される部分である、請求項3から請求項8のいずれか一項に記載の使用。
【請求項10】
R6が、−Hである、請求項3から請求項9のいずれか一項に記載の使用。
【請求項11】
Pr1およびPr2が、同じであるか異なり、水素および保護基から選択され、前記保護基が、C1−C6アルコキシ、C1−C6アシルオキシ、ヒドロキシおよびニトロで所望により置換されているベンジル基、ピコリル、ピコリル−N−オキシド、アントリルメチル、ジフェニルメチル、フェニル、t−ブチル、アダマンチル、C1−C6アシルオキシメチル、C1−C6アルコキシメチル、テトラヒドロピラニル、ベンジルチオメチル、フェニルチオメチル、チアゾリジン、アセトアミドメチル、ベンズアミドメチル、t−ブトキシカルボニル(BOC)、アセチルおよびその誘導体、ベンゾイルおよびその誘導体、カルバモイル、フェニルカルバモイルおよびC1−C6アルキルカルバモイルから選択される、請求項3から請求項10のいずれか一項に記載の使用。
【請求項12】
Pr1およびPr2が、水素である、請求項3から請求項11のいずれか一項に記載の使用。
【請求項13】
各アミノ酸側鎖が、天然アミノ酸中に存在するアミノ酸側鎖部分であるか、−(CH2)2−C(O)−O−C(CH3)3、−(CH2)4−NH−C(O)−O−C(CH3)3、−(CH2)3−NH−C(O)NH2、−CH2−CH2OHまたは−(CH2)2−CH2NH2である、請求項1または請求項2に記載の使用。
【請求項14】
前記化合物が、式(2)、(2’)、(3)、(3’)、(4)、(4’)、(5)、(5’)、(6)、(6’)、(7)、(7’)、(8)、(8’)、(9)または(9’)の化合物である、請求項1に記載の使用。
【化2】
【請求項15】
前記FK228類似体が、式(I)の化合物、そのアイソスターまたは医薬的に許容されるその塩である、先行する請求項のいずれか一項に記載の使用。
【請求項16】
前記医薬品が、癌、心肥大症、慢性心不全、炎症状態、心血管疾患、ヘモグロビン異常症、サラセミア、鎌状赤血球症、CNS障害、自己免疫疾患、糖尿病、骨粗しょう症、MDS、良性前立腺肥大症、口腔白板症、遺伝的代謝性疾患、感染症、ルビンス−テイビ(Rubens−Taybi)、脆弱X症候群、またはアルファ−1アンチトリプシン欠損症の軽減のために使用されるか、創傷治癒の加速または毛包を保護するために使用されるか、免疫抑制薬として使用される、先行する請求項のいずれか一項に記載の使用。
【請求項17】
前記医薬品が、慢性リンパ球性白血病、乳癌、前立腺癌、卵巣癌、中皮腫、T細胞リンパ腫、心肥大症、慢性心不全または皮膚炎症状態、特に乾癬、アクネまたは湿疹を軽減させるための使用である、請求項16に記載の使用。
【請求項18】
ヒトまたは動物を治療する方法に使用するための、請求項1から請求項15のいずれか一項に記載のFK228類似体、そのアイソスターまたは医薬的に許容されるその塩。
【請求項19】
請求項1から請求項15のいずれか一項に記載のFK228類似体、そのアイソスターまたは医薬的に許容されるその塩。
【請求項20】
請求項1から請求項15のいずれか一項に記載のFK228類似体、そのアイソスターまたは医薬的に許容されるその塩と医薬的に許容される担体または希釈剤とを含む医薬組成物。
【請求項21】
経口、直腸内、非経口、鼻孔内もしくは経皮投与または吸入もしくは坐剤による投与に適する形態である、請求項20に記載の医薬組成物。
【請求項22】
錠剤、カプセル、トローチ、ロゼンジ、水性もしくは油性懸濁剤、分散性粉末もしくは顆粒または舌下錠の形態である、請求項21に記載の医薬組成物。
【請求項23】
HDACにより媒介される、請求項1、請求項16または請求項17に記載の状態に罹患しているまたは罹患しやすい患者を治療する方法であって、請求項1から請求項15のいずれか一項に記載の化合物、そのアイソスターまたは医薬的に許容されるその塩の有効量を前記患者に投与することを含む方法。
【請求項24】
スベロイルアニリドヒドロキサム酸(SAHA)と少なくとも等しいHDAC阻害活性を有する化合物を選択するプロセスであって、該プロセスは請求項1から請求項15のいずれか一項に記載の式(I)または(I’)の化合物を調製することを含み、該調製は以下を含む該プロセス:
(i)式(VI)の化合物と式(VII)の化合物を反応させ、
【化3】
(式中、R1およびR2は、先行する請求項のいずれか一項に記載のとおりであり、R7は、C1−C4アルキルまたはC2−C4アルケニルであり、ならびにYは、アミノ保護基である)
(ii)得られた中間体を脱保護した後、式(VIII)の化合物と反応させ、
【化4】
(式中、Y’は、アミノ保護基であり、Y’’は、水素または保護基である)
(iii)得られた中間体を脱保護した後、式(IX)の化合物と反応させ、
【化5】
(式中、R3は、先行する請求項のいずれか一項に記載のとおりであり、Y’’’は、アミノ保護基である)
(iv)得られた中間体を脱保護した後、式(X)の化合物と反応させ、
【化6】
(式中、R4は、先行する請求項のいずれか一項に記載のとおりであり、R5は、水素またはヒドロキシ保護基であり、LGは、脱離基であり、Y’’’’は、水素または保護基である)
(v)得られた中間体のβ−ヒドロキシ基を所望により脱保護して、R5保護基を除去し、Hで置換し、
(vi)得られた中間体を加水分解および環化し、
(vii)得られた中間体を、所望により、ジスルフィド結合を形成させるように反応させ、ジスルフィド結合が形成された場合には、得られた化合物中のジスルフィド結合を所望により切断し、得られた化合物がチオール基を含有する場合には、チオール保護基を所望により導入し;および
(viii)得られた化合物をスクリーニングして、HDAC阻害剤としての活性を測定すること。
【請求項25】
段階(viii)におけるスクリーニング段階が、インビトロHDACアッセイであって、様々な濃度で、試験化合物およびSAHAを希釈Hela核抽出物と接触させて、Hela核抽出物に対する試験化合物およびSAHAのIC50を決定することを含む、請求項24に記載のプロセス。
【請求項26】
SAHAと少なくとも等しいヒト癌細胞増殖阻害活性を有する化合物を選択するプロセスであって、請求項1から請求項15のいずれか一項に記載の式(I)または(I’)の化合物を調製することを含み、該調製が以下を含む該プロセス:
(i)式(VI)の化合物と式(VII)の化合物を反応させ、
【化7】
(式中、R1およびR2は、先行する請求項のいずれか一項に記載のとおりであり、R7は、C1−C4アルキルまたはC2−C4アルケニルであり、ならびにYは、アミノ保護基である)
(ii)得られた中間体を脱保護した後、式(VIII)の化合物と反応させ、
【化8】
(式中、Y’は、アミノ保護基であり、Y’’は、水素または保護基である)
(iii)得られた中間体を脱保護した後、式(IX)の化合物と反応させ、
【化9】
(式中、R3は、先行する請求項のいずれか一項に記載のとおりであり、Y’’’は、アミノ保護基である)
(iv)得られた中間体を脱保護した後、式(X)の化合物と反応させ、
【化10】
(式中、R4は、先行する請求項のいずれか一項に記載のとおりであり、R5は、水素またはヒドロキシ保護基であり、LGは、脱離基であり、Y’’’’は、水素または保護基である)
(v)得られた中間体のβ−ヒドロキシ基を所望により脱保護して、R5保護基を除去し、Hで置換し、
(vi)得られた中間体を加水分解および環化し、
(vii)得られた中間体を、所望により、ジスルフィド結合を形成させるように反応させ、ジスルフィド結合が形成された場合には、得られた化合物中のジスルフィド結合を所望により切断し、得られた化合物がチオール基を含有する場合には、チオール保護基を所望により導入し;および
(viii)得られた化合物をスクリーニングして、ヒト癌細胞増殖阻害剤としての活性を測定すること。
【請求項27】
段階(viii)におけるスクリーニング段階がインビトロアッセイであって、様々な濃度で試験化合物およびSAHAを、MCF7乳癌細胞系、HUT78T細胞白血病細胞系、A2780卵巣癌細胞系、PC3またはLNCAP前立腺癌細胞系と接触させて、該細胞系に対する該試験化合物およびSAHAのIC50を決定することを含む、請求項26に記載のプロセス。
【請求項28】
SAHAと少なくとも等しい抗炎症活性を有する化合物を選択するためのプロセスであって、該プロセスが請求項1から請求項15のいずれか一項に記載の式(I)または(I’)の化合物を調製することを含み、該調製が以下を含む該プロセス:
(i)式(VI)の化合物と式(VII)の化合物を反応させ、
【化11】
(式中、R1およびR2は、先行する請求項のいずれか一項に記載のとおりであり、R7は、C1−C4アルキルまたはC2−C4アルケニルであり、ならびにYは、アミノ保護基である)
(ii)得られた中間体を脱保護した後、式(VIII)の化合物と反応させ、
【化12】
(式中、Y’は、アミノ保護基であり、Y’’は、水素または保護基である)
(iii)得られた中間体を脱保護した後、式(IX)の化合物と反応させ、
【化13】
(式中、R3は、先行する請求項のいずれか一項に記載のとおりであり、Y’’’は、アミノ保護基である)
(iv)得られた中間体を脱保護した後、式(X)の化合物と反応させ、
【化14】
(式中、R4は、先行する請求項のいずれか一項に記載のとおりであり、R5は、水素またはヒドロキシ保護基であり、LGは、脱離基であり、Y’’’’は、水素または保護基である)
(v)得られた中間体のβ−ヒドロキシ基を所望により脱保護して、R5保護基を除去し、Hで置換し、
(vi)得られた中間体を加水分解および環化し、
(vii)得られた中間体を、所望により、ジスルフィド結合を形成させるように反応させ、ジスルフィド結合が形成された場合には、得られた化合物中のジスルフィド結合を所望により切断し、得られた化合物がチオール基を含有する場合には、チオール保護基を所望により導入し;および
(viii)得られた化合物をスクリーニングして、抗炎症活性を測定すること。
【請求項29】
段階(viii)におけるスクリーニング段階が、化合物の、SAHAを基準にして末梢血単核細胞(PBMC)からのTNFαの生産の阻害活性を測定することを含むアッセイである、請求項28に記載のプロセス。
【請求項30】
(a)請求項1から請求項15のいずれか一項に記載のFK228類似体、そのアイソスターまたは医薬的に許容されるその塩と、(b)請求項1、請求項16または請求項17に記載のHDACにより媒介される状態の治療または予防に同時に、別々にまたは逐次的に使用するためのHDACの別の阻害剤とを含有する製品。
【請求項31】
(a)請求項1から請求項15のいずれか一項に記載のFK228類似体、そのアイソスターまたは医薬的に許容されるその塩と、(b)癌の治療または予防に同時に、別々にまたは逐次的に使用するための別の化学療法薬または抗腫瘍薬とを含有する製品。
【図1】

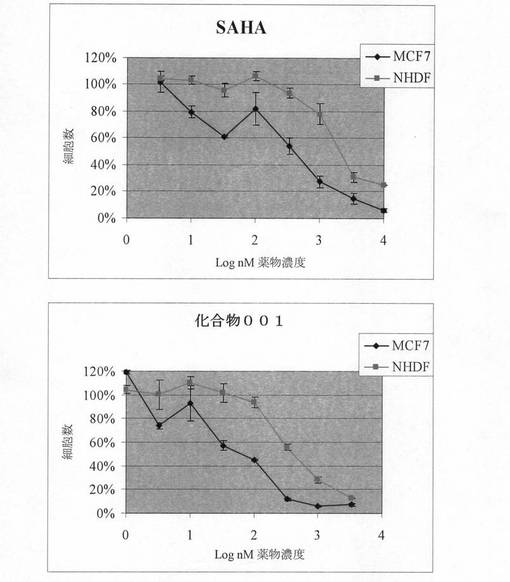
【図2】
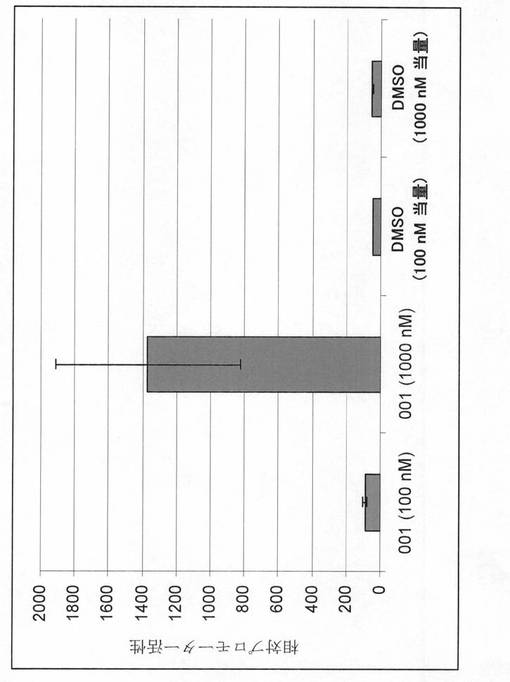
【図3】
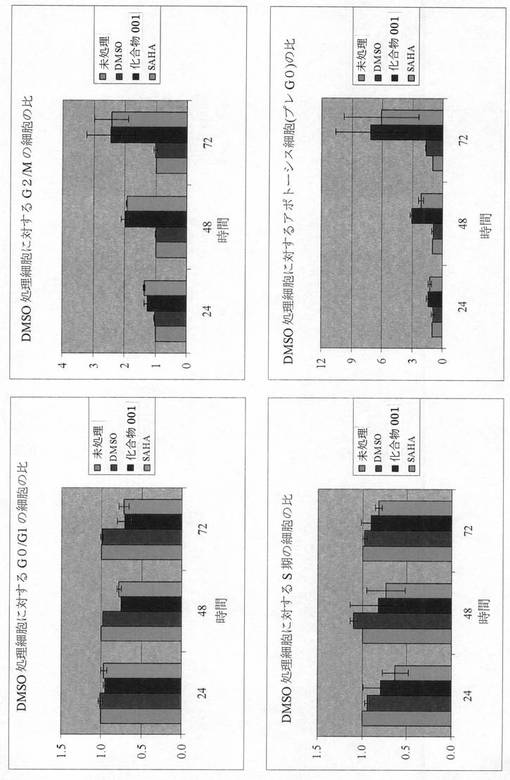
【図4】
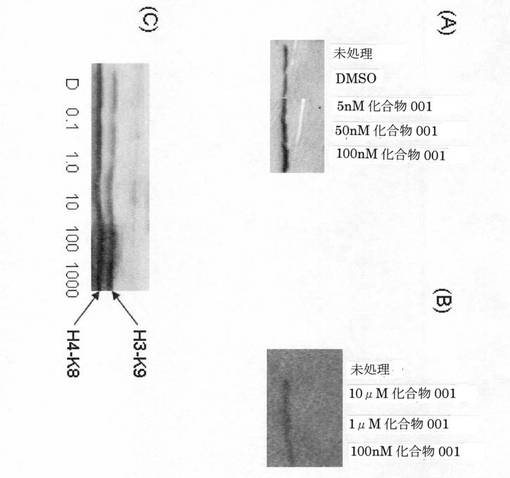
【図2】
【図3】
【図4】
【公表番号】特表2008−542347(P2008−542347A)
【公表日】平成20年11月27日(2008.11.27)
【国際特許分類】
【出願番号】特願2008−514194(P2008−514194)
【出願日】平成18年6月2日(2006.6.2)
【国際出願番号】PCT/GB2006/002022
【国際公開番号】WO2006/129105
【国際公開日】平成18年12月7日(2006.12.7)
【出願人】(500125788)ユニバーシティ、オブ、サウサンプトン (12)
【氏名又は名称原語表記】UNIVERSITY OF SOUTHAMPTON
【Fターム(参考)】
【公表日】平成20年11月27日(2008.11.27)
【国際特許分類】
【出願日】平成18年6月2日(2006.6.2)
【国際出願番号】PCT/GB2006/002022
【国際公開番号】WO2006/129105
【国際公開日】平成18年12月7日(2006.12.7)
【出願人】(500125788)ユニバーシティ、オブ、サウサンプトン (12)
【氏名又は名称原語表記】UNIVERSITY OF SOUTHAMPTON
【Fターム(参考)】
[ Back to top ]