
HEPG2細胞のコレステロール恒常性に関与するダイズ7Sグロブリンα’サブユニットの伸長領域のクローニング、酵母発現、精製および生物活性
コレステロールおよびトリグリセリドの恒常性のin vitroおよびin vivoモデルにおける制御において活性なα’鎖の切断形態(eα’)、ダイズ7Sグロブリンを酵母Pichia pastorisにおいてクローニングし、発現させた。組換えポリペプチドはN末端側から142のアミノ酸残基におよび、ダイズα’サブユニットのN末端伸長領域を含んでいた。従来の生化学的技術によりeα’ポリペプチドを精製し、標識LDLの取込みおよび分解をモニタリングすることにより、ヒト肝細胞癌細胞系(HepG2)においてLDL受容体の活性を調節するその潜在能力を評価した。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ダイズ7Sグロブリンの天然のα’サブユニットのN末端親水性フラグメントに対応する組換えα’フラグメント、ならびにその調製方法ならびにコレステロールおよびトリグリセリドの恒常性を制御するのに有用な活性成分としてそれを含有する組成物に関する。
【背景技術】
【0002】
(発明の背景)
高コレステロール血症患者の血中脂質レベル(lipidemic level)の制御における食事性のダイズタンパク質の役割は、広く受け入れられている論点である[1]。以前の研究[2〜4]においては、LDL受容体のアップレギュレーションにおけるダイズ7Sグロブリンの1つのサブユニットであるα’サブユニットの直接的な関与がin vitroおよびin vivo系において証明され、コレステロール恒常性を調節することができる生物活性(ポリ)ペプチドが、細胞酵素処理により産生される可能性が高いことが示唆されている。
【0003】
天然の7Sグロブリンは、異なる遺伝子によりコードされた、3つのランダムに分類されたポリペプチド鎖、α’、αおよびβサブユニット[5]からなる。成熟α’(受入番号P11827 UniProtKB/Swiss−Protデータベース)およびα鎖(受入番号P13916 UniProtKB/Swiss−Protデータベース)は、βサブユニット(受入番号P25974 UniProtKB/Swiss−Protデータベース)には存在していない約145のアミノ酸残基の伸長N末端領域を共有している。α’伸長領域の特有のアミノ酸配列に基づいて、このサブユニットは金属アフィニティークロマトグラフィーにより精製され、高コレステロール血症ラットに経口投与され、それにより、その血漿脂質低下特性および肝臓β−VLDL受容体のアップレギュレーションの両方を示すことが可能となった[4]。一方、α’サブユニットの分子量はおよそ71kDaである[6]ため、修飾されない限りin vivoで腸の障壁を越えるとは考えられない。
【0004】
このため、本発明者らの研究は、薬理学的効果に関与するα’サブユニットのアミノ酸配列(複数可)を探求することに向けられてきた。3つのサブユニットのコア領域は非常に類似のアミノ酸配列を有するため、生物活性は該伸長領域の1つまたは複数の(ポリ)ペプチドにあるはずであると考えられる。原理上、限定的ではあるが有意なα’およびα鎖の伸長領域間のアミノ酸の差異により、生物活性に関与するペプチドの数が制限されるであろう。
【0005】
これらの以前の記述から、最初に探求した戦略は、Croksoy(登録商標)70(高コレステロール血症患者の食事療法において通常利用されるイソフラボンを含まないダイズ濃縮物[7〜8])のin vitro消化(ペプシン/トリプシン)から得られたポリペプチド、および7Sダイズグロブリンサブユニット間で異なっていた特定のアミノ酸配列に対応する合成ペプチドの両方の、HepG2細胞におけるLDL受容体(LDL−R)調節に対する効果を試験することであった。これらの研究において得られた結果は、3000〜20000Daの範囲のMWを有するCroksoy(登録商標)70の酵素消化産物、ならびに7Sダイズグロブリン由来の小さい合成ペプチド(2271Da)(10−4Mの濃度で細胞に添加した)に曝されたHepG2細胞において顕著なLDL−Rアップレギュレーションを誘発できたことを指摘していた[9]。小さいペプチドを用いて得られた研究結果は、現在、依然として調査中であり、今までのところ結論が出ていない[10]。
【0006】
ダイズタンパク質のコレステロールおよびトリグリセリドを低下させる能力は確固たる論点である。ダイズタンパク質の食事は、高コレステロール血症患者を治療するための現在最も有力な食事性のツールであり、したがって、成人および非常に若い被験者の管理のユニークな機会を提供している。さらに、血漿コレステロールの低減が、コレステロール血症のベースラインの程度が高い患者においてより大きいことは明確に確立されている[14]。
【0007】
タンパク質自体が血中コレステロールを低減するという仮定は、食事における動物タンパク質から植物タンパク質への転換により、実験動物の肝臓[15]、ならびに高コレステロール血症患者の循環リンパ単球[16]においてLDL受容体系が活性化されることを示している実験的研究に起因していた。コレステロール低下効果に関与するダイズタンパク質成分を同定するために、LDL受容体の発現およびコレステロールの生合成/分解を調節する因子に対して非常に感受性であるヒト肝細胞癌細胞系を用いてin vitro研究を実施した。7Sダイズグロブリン由来の精製α’サブユニットがHepG2細胞においてLDL受容体をアップレギュレーションする[3]ことが分かり、この発見は、コレステロール給餌ラットにおいて確認された[4]。これらのデータは、観察された生物学的効果に該タンパク質部分が関与しているという仮定を支持するものであるが、ペプチドおよびアミノ酸は通常胃および/または腸のタンパク質分解酵素の作用により産生されるため、α’鎖のin vivoの生体内動態(biological fate)に対する議論が生じる可能性がある。しかし、抗酸化、抗増殖および抗炎症効果に起因する場合が多い関連する調節機能を果たすと主張されている動物および植物(ポリ)ペプチドの数が増大している[17]。ダイズに関する限り、ペプチドおよびBowman−Birk阻害物質などのさらに小さい小型のタンパク質を吸着することができ[18]、したがって、抗癌、抗炎症性、放射線防護効果を含めた多数の効果を引き出す[19]可能性が、実験的証拠により明確に示されている。遺伝子組換えダイズ(ポリ)ペプチドも、降圧効果などの生物学的応答を誘発することが示されてきた[20]。最近になって、7Sグロブリンβ鎖に由来するLDL−R転写を刺激するペプチド(FVVNATSN)が、Bacillus amyloliquefaciens由来のプロテアーゼにより、次いで、化学的合成により調製されたダイズ加水分解物から同定された[21]。この場合、100μMの濃度の該ペプチドに曝されたHepG2細胞においてLDL−R転写の増大(+148%)が検出された。11Sグロブリンから生じる他のペプチドは、類似の活性を発揮するがその活性はより低いことが示されてきた[21]。
【0008】
完全長タンパク質の生物学的特性を維持または改善さえする、より短いポリペプチドを入手可能にすることが望ましいであろう。
【先行技術文献】
【非特許文献】
【0009】
【非特許文献1】Sirtori CR. et al.,Curr Atheroscler Rep.2001;3:47−53
【非特許文献2】Lovati MR. et al.,J Nutr.1992;122:1971−8
【発明の概要】
【課題を解決するための手段】
【0010】
(発明の説明)
今般、ダイズ7SグロブリンのN末端側に対応するそのいわゆるα’伸長領域が有益な生物活性を有することが分かり、LDLの取込みおよび分解に対してフルサイズのα’鎖より効果的でさえあることが証明された。
【0011】
本発明は、したがって、以下eα’と呼ぶ前記α’伸長領域、ならびにダイズα’サブユニットのN末端伸長領域を含有する組換えポリペプチドのクローニング、酵母発現および精製によるその調製方法を提供する。
【0012】
この目的のために、α’鎖のN末端フラグメントの異種発現を試みた。酵母Pichia pastorisの分泌能力のある酵母細胞(secretion-competent yeast cell)においてこの目標を達成した。組換えポリペプチドを精製し、HepG2細胞においてその生物活性を評価した。この生物工学的手法の使用により、in vitro試験、さらにはin vivo実験において試験するのに十分な量の組換えポリペプチドを得ることができた。
【図面の簡単な説明】
【0013】
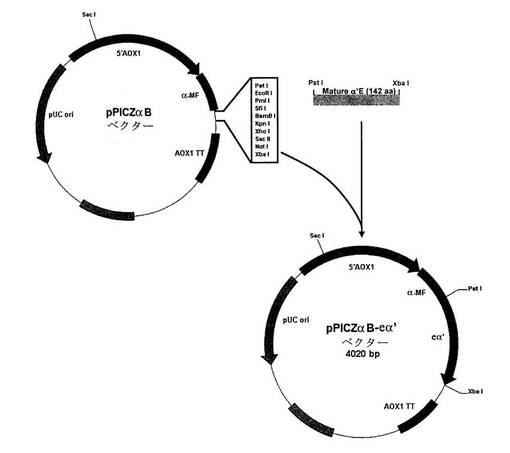
【図1】pPICZαB−eα’コンストラクトの概要である。eα’の発現は、AOX(アルコールオキシダーゼ)メタノール誘導性プロモーター(5'AOX1)により駆動され、α接合因子(α−MF)は組換えタンパク質の培地への分泌を促進し、AOX1 TTはAOX転写終結領域である。Sh ble遺伝子はゼオシンに対する耐性を付与し、pUC OriはE.coliにおける多数のプラスミドコピーの複製開始点である。他の略語は制限酵素の開裂位置を指し、bpは塩基対である。
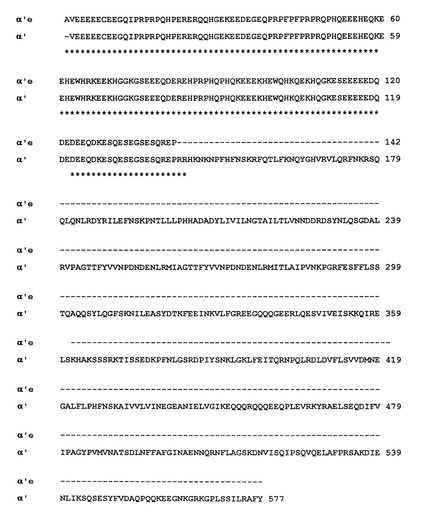
【図2】図2は、組換えポリペプチド(eα’)および野生型のダイズα’サブユニットの配列アラインメントである。星印は、2つの配列における同一のアミノ酸残基を示す。
【図2A】図2Aは組換えポリペプチド(eα)’の配列(配列番号1)である。
【図2B】図2Bは野生型のダイズα’サブユニットの配列(配列番号2)である。
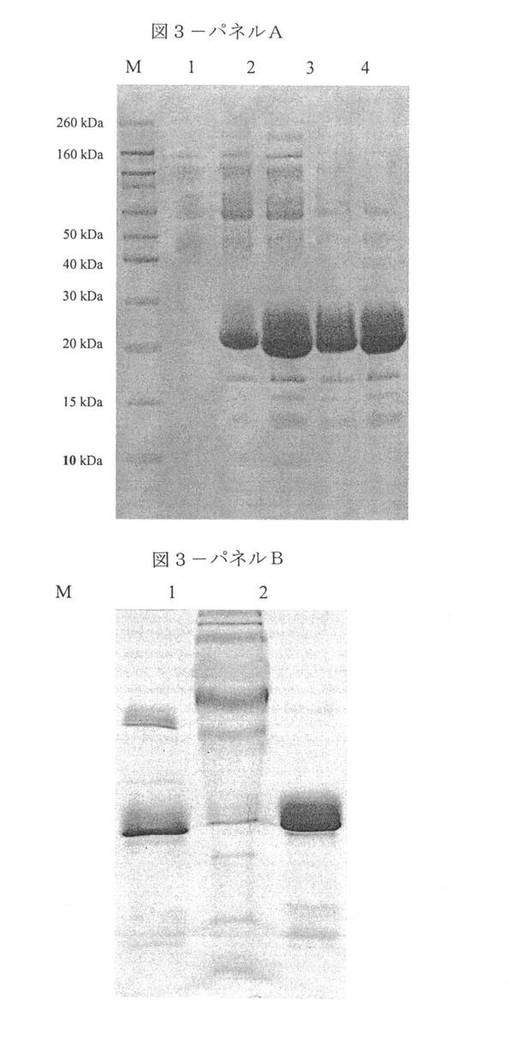
【図3】図3−パネルAは組換えPichia pastoris培養物の還元条件下でのSDS−PAGEである。レーンM:分子量マーカー。レーン1:メタノールによる誘導の前のTCA沈殿培養上清(TCA-Precipitated culture supernatant)(無細胞)。レーン2:メタノールによる誘導の1h後のTCA沈殿培養上清(無細胞)。レーン3:メタノールによる誘導の8h後のTCA沈殿培養上清(無細胞)。レーン4:メタノールによる誘導の19h後のTCA沈殿培養上清(無細胞)。レーン5:メタノールによる誘導の25h後のTCA沈殿培養上清(無細胞)。図3−パネルBはeα’精製ステップの還元条件下でのSDS−PAGEである。レーンM:分子量マーカー。レーン1:発酵ブロスの凍結乾燥粉末。レーン2:DEAE−セルロース 150mM NaCl溶出画分。レーン3:DEAE−セルロース 250mM NaCl溶出画分。
【発明を実施するための形態】
【0014】
発明の詳細な説明
本発明は、次に、以下の実験の項において詳細に説明する。
【0015】
材料および方法
酵母、細菌株および化学物質。酵母発現にPichia pastoris X33(WT)株(Invitrogen、San Diego、CA)を使用した。遺伝子操作に利用した細菌株はE.coli XL1−Blue(Invitrogen、San Diego、CA)であった。制限酵素Pst IおよびXba IをRoche(Indianapolis、IN)から、Sac IをFermentas(Ontario、Canada)から購入した。Taq DNAポリメラーゼはInvitrogen(San Diego、CA)から購入した。ゼオシンはInvivogen(San Diego、CA)から購入した。PCR用のオリゴヌクレオチドはPrimm(Milano、Italy)から得た。ペプトン、トリプトン、酵母エキスおよび寒天はBecton Dickinson and Company(Sparks、MD)から購入した。グルコースおよびソルビトールはSigma(St.Louis MO)から購入した。透析膜はSpectrum Laboratories(Rancho Dominguez、CA)から購入した。DEAE樹脂はWhatman(Maidstone、England)から購入した。NiNTA樹脂はQiagen(Hilden、Germany)から購入した。Symmetry C4 HPLCカラムはWaters(Milford、MA)から購入した。
【0016】
他の化学物質はSigma(St.Luis、MO)からまたはMerck(Darmstadt、Germany)からの試薬グレードであった。
【0017】
培地および増殖条件。P.pastoris X33株をYPD(酵母ペプトンデキストロース)完全培地(2%ペプトン、1%酵母エキス、2%グルコース)において培養した。YPD、寒天1.5%、100mg/mLゼオシンを含有するプレートにおいて、Mut+形質転換体を選択した。全ての酵母培養物を30℃で維持した。異種タンパク質検出のために、YPS(酵母ペプトンソルビトール)培地(2%ペプトン、1%酵母エキス、2%ソルビトール)において、Zeor−Mut+形質転換体を600nmで光学濃度5まで培養した。次いで、メタノールを1%の最終濃度まで添加した。ゼオシンを含有する低塩LB(Luria−Bertani)培地のプレート(1%トリプトン、0.5%酵母エキス、0.5%塩化ナトリウム、1.5%寒天、25mg/mLゼオシン)において全ての細菌形質転換体を選択した。全ての細菌培養物を37℃で維持した。
【0018】
eα’遺伝子発現ベクターの構築。関心対象(eα’)の配列を含む挿入物(tα’)を含有するPichia pastoris発現プラスミドDNA鋳型(expression Pichia pastoris plasmid DNA template)(pPICZαB)に対するPCRによりeα’遺伝子を増幅した。eα’遺伝子の5'末端でPstI制限部位を生成するようにオリゴヌクレオチド5'−GAAAAGATAGATTAAAGCTGCAGTGGAGGAAG−3'を設計した。この変異により、eα'のN末端位置においてアラニン残基挿入が生じた。eα’遺伝子の3'末端でXbaI制限部位を生成するように第2のオリゴヌクレオチド5'−CCCTTCTTATTCTTTCTAGATCATGGTTCTCTTTGAGACTC−3'を設計した。いずれのオリゴヌクレオチドもmQ滅菌水に溶解した。PCR反応混合物は、0.5mMプライマー、0.8mM dNTPs(Eppendorf、Hamburg、Germany)、30ng鋳型(pPICZαB/tα’)、2.5U Taq DNAポリメラーゼ、PCR緩衝液(最終組成:50mM KCl、1.5mM MgCl2、20mM Tris−Cl、pH8.4および最終容量が25mLとなるmQ滅菌水)からなっていた。以下の条件を使用して、Perkin Elmer Geneamp PCR System 2400サーモサイクラー(Perkin Elmer Corp.、Wellesley、MA)でPCR増幅を実施した。94℃で10minで開始;94℃で40sec、60℃で40sec、72℃で20secで30サイクル;最終伸長72℃で10minおよび4℃で維持。465bpのPCR産物を酵素で消化して423bpのフルサイズのコンストラクトとし、それをpPICZαBベクターにクローニングし、それにより、pPICZαB−eα’コンストラクトが生じ、それをXL1−Blue E.coli細胞において形質転換した。テトラサイクリンおよびゼオシンを含有する半流動LB培地上で陽性クローン(positives clones)を選択した。Primm(Milano、Italy)によりこれらのクローンの1つを配列決定して、pPICZαB−eα’コンストラクト配列において変異が発生しないようにした。制限酵素SacIを用いた消化により20μgのpPICZaB−eα’コンストラクトおよび30μgの発現ベクターpPICZαB(負の対照)を線状化し、次いで、精製した。
【0019】
P.pastorisゲノムにおけるpPICZaB−eα’の形質転換。1.5kVに設定したEppendorf(Hamburg、Germany)electroporator 2510装置で20μgの線状化pPICZαB−eα’コンストラクトおよび30μgの線状化pPICZαBを用いた電気穿孔法により、野生型(wt)の酵母細胞を形質転換した。100mg/mLのゼオシンを含有するYPDプレートでのプレーティングにより形質転換体を最初に選択した。本発明者らのコンストラクトの形質転換P.pastorisゲノムへの組込みを確認するために、Dneasy Plant Mini Kit[QIAGEN、Hilden、Germany]を使用してゲノムDNAを抽出した。5'AOX1(5'−GACTGGTTCCAATTGACAAGC−3')および3'AOX1プライマー(5'−GCAAATGGCATTCTGACATCC−3')を使用したPCRによりアルコールオキシダーゼプロモーター(AOX1)部位でのコンストラクトの挿入を確認するための鋳型としてゲノムDNAを使用した。PCR反応混合物は、0.5mMプライマー、0.25mM dNTPs、100ngのゲノム鋳型、2.5U Taq DNAポリメラーゼ、PCR緩衝液(最終組成は上記の通り)からなっていた。以下の条件を使用してPCR反応を実施した。94℃で10minで開始;94℃で40s、60℃で40s、72℃で20sで30サイクル;最終伸長72℃で10minおよび4℃で維持。
【0020】
形質転換体クローンの選択。振盪(180rpm)培養器中の30℃の50mLのYPS培地において、18のZeo+形質転換体およびpPICZαB発現ベクター(負の対照)を含有する1つの形質転換体をOD600=5まで増殖させた。1%の最終濃度までメタノールを添加することにより誘導期を誘発し、24時間延長した。SDS−PAGEによりeα’の発現について上清のアリコートを検査した。発酵槽規模(fermenter scale)での組換えタンパク質の生産のために、最高のeα’発現を有するクローン形質転換体を選択した。
【0021】
eα’の発現:発酵槽規模。大量生産のために、Invitrogenの「Pichia発酵プロセスガイドライン(Pichia Fermentation Process Guidelines)」(バージョンB、053002)に従って、14Lの発酵槽(Chemap、Switzerland)において選択したクローンを増殖させた。バッフルなし振盪フラスコ(unbaffled shake flask)中のYNB培地(KH2PO4 2.0g;(NH4)2SO4 10.0g;MgSO4 7H2O 1.0g;NaCl 0.2g;CaCl2 0.2g;グリセロール100% 10.0g;pH5.2までのKOH;1.0Lまでの蒸留水)で種菌培養物を調製する。250rpm、30℃で約24時間の培養後、1.0Lの種培養物を使用して、8.5Lの基礎塩培地(basal salt medium)(H3PO4 85% 227mL;CaSO4 2H2O 7.9g;K2SO4 155g;MgSO4 7H2O 127g;KOH 35g;グリセロール100% 340g;8.5Lまでの蒸留水;滅菌後にNH4OHを用いてpHを5.0に調節した)を用いて用意した発酵槽に接種する。前述の培養基のいずれにもビオチンおよびイノシトール(各1mg/L)を補充する。基礎塩培地にも修飾されたPTM1微量塩溶液(trace salt solution)(H2SO4 96% 2.5mL;CoC2O4°2H2O 243mg;CuSO4°5H2O 3.0g;KI 44.5mg;MnSO4°H2O 1.5g;Na2MoO4°2H2O 100mg;H3BO3 10mg;FeSO4°7H2O 32.5g;ZnCl2 10g;500mLまでの蒸留水)を補充する。
【0022】
微生物の増殖および異種タンパク質の発現は、3ステッププロセスを介して得られる。約24時間の最初の回分増殖期(炭素源としてグリセロール)の後に、約4時間の流加回分(fed-batch)期(制限炭素源(limiting C-source)としてグリセロール)が続く。これらの最初の2つの期の間、NH4OH 20%を添加することによりpHを5.0で維持する。一旦全てのグリセロールが消費されると、メタノールの供給を開始してeα’タンパク質発現を誘発し、NH4OH 20%を添加することによりpHを6.0にシフトする。メタノール流加回分期は約24時間にわたる。これらの2つの流加回分期の間、通気速度(vvm)は手動で徐々に増大させながら撹拌機速度(rpm)は電気的に微調節することにより、溶存酸素(DO%)を30%で一定に維持する(in maintained stable)。数時間毎に、DO%レベル(その値はメタノール供給の突然の停止後に急増を示すはずであり、逆に、その再開後に急速な減少を示すはずである)を追跡することにより炭素源制限条件をチェックする。数時間毎に、以下の分析のために無菌条件において発酵槽から試料を採取する。光学濃度(λ600nm)、細胞バイオマス%(湿量)、無菌性、顕微鏡観察、SDS−PAGE。
【0023】
eα’の精製:発酵ブロスの下流処理。約9.2Lの培養物を発酵槽から溢流させ、氷で冷却する。その後の操作は全て+4℃で実施する。3000xgで30minの遠心分離(Centrikon T−124、Kontron Instruments)により培養物全体を分離し、バイオマス(ペレット)を廃棄し、約7.5Lの上清をZetaplus 30SP(Cuno)での深層濾過(depth filtration)により清澄化し、引き続いて0.22μmフィルター(Millipak 100、Millipore)で精密濾過する。ポリエーテルスルホン膜、MWCO 10kDa(Omegaフィルター、Pall)での限外濾過を介して透明な濾液を濃縮する。約300mLの濃縮物を、3.0LのTris−HCl 10mM pH7.2に対してダイアフィルトレーションし、最終的に凍結乾燥する。この手順により、SDS−PAGEでかなり低い夾雑タンパク質の程度を示し、約25%p/pの総タンパク質含量(Bradfordタンパク質アッセイ、較正標準としてウシ血清アルブミン)を有する34.5gの凍結乾燥粉末を得た。
【0024】
eα’の精製:クロマトグラフィーによる精製
より高性能のゲル電気泳動のために、供給業者の手順に従ってNuPAGE(登録商標)プレキャストゲルシステム(Invitrogen)を使用した。2グラムの凍結乾燥粉末を150mL 50mM Tris−HCl、pH7.50に溶解し、同じ緩衝液を用いて平衡化したDEAE−セルロースカラム(6×10cm、Whatman、Maidstone、UK)に装填した。それぞれ150および250mM NaClを含有する同じ緩衝液を用いて、保持されたタンパク質の溶出を実施した。0.25M NaCl(300mL)を用いて溶出した画分が、最も多いeα’の含量を示した。凍結乾燥により該溶液を100mLまで濃縮し、次いで、6000〜8000Daの膜を用いてmilliQ限外濾過水により4℃で24時間透析し、次いで、凍結乾燥した。約370mgのタンパク質を得た。
【0025】
タンパク質の均一性を確認するために、約1mgのタンパク質をSymmetry C4(4.6×250mm)逆相カラムに装填した。緩衝液A(限外濾過水およびトリフルオロ酢酸0.1%)および緩衝液B(アセトニトリル100%+トリフルオロ酢酸0.1%)を使用した。
【0026】
電気泳動技術。参考文献11に従って、mini−Protean II細胞(Bio−Rad)を使用して、12%ポリアクリルアミドゲルで還元条件(2%β−メルカプトエタノール)下でのSDS−PAGEを実施した。クーマシーブルーを用いて該ゲルを染色した。
【0027】
細胞培養物。株化ヒト肝細胞癌細胞系(HepG2)を米国培養細胞系統保存機関(American Type Culture Collection)(Rockville、MD)から得た。Eagleの最小必須培地(MEM)、ウシ胎仔血清、トリプシン−EDTA(1倍)、ペニシリン(105U/L)、ストレプトマイシン(100g/L)、トリシン緩衝液(1mmol/L、pH7.4)および非必須アミノ酸溶液(100倍)は、GIBCO(Madison、WI)から得た。ペトリ皿は、COSTAR(Cambridge、MA)から得た。フィルターはMillipore(Bedford、MA)から得た。タンパク質クーマシープラスタンパク質アッセイキット(Protein Coomassie Plus Protein Assay kit)はPierce(Rockford、IL、USA)から購入した。100mmol/L NaOH中の無担体の125ヨウ素はPerkin Elmer Life Sciences(Boston、MA)から得た。Sephadex G25カラム(PD10)はPharmacia Biotech(Uppsala、Sweden)から得た。LDHおよびMTTキットは、Sigma Diagnostics(Milano−Italy)から得た。全ての他の化学物質は、Merck(Darmstadt、Germany)からの分析用のものであった。直径90mmのペトリ皿において単層で細胞を増殖させ、95%空気、5%CO2の加湿雰囲気中で、10%ウシ胎仔血清(FCS)、非必須アミノ酸溶液(1%、v/v)、ペニシリン(105U/L)、ストレプトマイシン(0.1g/L)、トリシン緩衝液(20mmol/L、pH7.4)、NaHCO3(24mmol/L)およびピルビン酸ナトリウム(0.11g/L)を補充したMEMにおいて37℃で維持した。LDL受容体の調節を評価するように設計した実験のために、35mmのプラスチック皿に細胞を播種し(3〜5×105細胞)、コンフルエンスに達する直前に使用した。全ての細胞培養実験において、2〜3日毎に培地を交換した。細胞生存度を評価するために、基本的に参考文献9に記載されているように、メチルテトラゾリウム塩(MTT)アッセイにより、様々な濃度のeα’に曝された細胞由来の培養基を試験した。動態(LDH/LD)診断キット(Sigma Diagnostics)を使用して乳酸デヒドロゲナーゼ(LDH)活性を測定することにより細胞酵素の漏出を判定した。連続調製用超遠心分離(sequential preparative ultracentrifugation)[12]により、臨床的に健康な正常リポタンパク質のボランティアの血漿からLDL(1.019≦d≦1.063g/L)を単離した。Bilheimerらにより改善され[13]、以前に記載した[3]McFarlaneの方法に従ってリポタンパク質を標識した。濾過(Milliporeフィルター、孔径0.45μm)により125I−LDLを滅菌し、使用するまで4℃で貯蔵した。以前に記載した[9]ようにヒトリポタンパク質欠乏血清(LPDS)を調製した。
【0028】
125I−LDLの取込みおよび分解。5g/100g LPDSを補充したMEMにおいて細胞の単層を37℃で24h前培養して、表に列挙した様々な濃度のeα’または3.5μmol/Lのα’精製サブユニットまたは1.0μmol/Lのシンバスタチンの存在/不存在下でLDL受容体をアップレギュレーションした[2]。次いで、一定の濃度(7.5mg/L)の125I−LDLを該培地に添加し、培養を37℃でさらに5h継続した。以前に報告された[2]ように125I−LDLの特異的な取込み(結合+内部移行)および分解を評価した。
【0029】
統計的分析。様々な濃度のeα’を用いた細胞培養後のLDLにおける細胞の取込みおよび分解の差異を、ANOVAおよびその後のDunnettの検定により求めた。各値は平均値±SDとして表し、P値<0.05を統計的に有意であると見なした。
【0030】
結果
Pichia pastorisにおけるeα’の発現。Pichia pastoris細胞を形質転換するのに使用したプラスミドの構造を図1に示している。挿入物の配列およびα’サブユニットを有するそのアラインメントを図2に示している。材料および方法の項において述べたように、組換えポリペプチドと野生型のポリペプチドの間の唯一の差異はN末端第1アミノ酸残基にあり、それは、技術的な理由から、組換え鎖においてはアラニンであった。培養基のSDS−PAGE分析により判断したところ、最大の組換えポリペプチドの生産を示している(図示せず)クローンを大量生産のために選択した。図3−Aは、メタノールによる誘導の前(レーンn.1)および1〜8〜19および25時間後(レーンn.2、n.3、n.4、n.5)の酵母培養物の上清の電気泳動分析を示している。図3−Aが示しているように、約20kDaの明らかな分子量での明白なバンドが誘導の結果として生じている。
【0031】
eα’の精製。クロマトグラフィー法によりeα’の精製を達成した。各ステップから試料を採取し、SDS−PAGEにより分析した。同定したポリペプチドの均一性に対する精製ステップの効果を図3−Bに示している。培養基において酵母タンパク質による非常に低い程度の混入が既に達成されていたが、さらなるクロマトグラフィーによるステップにより主要な夾雑タンパク質を除去し、ほとんど均一な形態の組換えポリペプチドの回収を可能にした。このバンドのN末端配列分析により、この20kDaポリペプチドがeα’鎖に対応することが確認された。この試料の純度が細胞アッセイに適していると判断した。
【0032】
eα’の生物活性。正の対照としての精製α’サブユニット、およびその切断α’形態のHepG2細胞への添加により、以下の表において報告しているように、未処理の細胞と比較してLDL受容体媒介性の取込みおよび分解の有意な上昇がもたらされた。
【0033】
【表1】

【0034】
この結果は、LDL調節がeα’に用量依存的であり、最高濃度が正の対照、シンバスタチンの濃度と同様であったことを示していた。MTTおよびLDHアッセイにより測定したところ、いずれのeα’の濃度でも細胞毒性の証拠は見られなかった(図示せず)。
【0035】
したがって、本発明において、生物学的応答を誘導することができるアミノ酸配列がα’鎖のN末端伸長ドメインに存在することが分かった。さらに、本発明者らは、N末端親水性フラグメントが、強力な抗高脂血症薬であるシンバスタチンと同程度の濃度でその効果を発揮することを見出した。この効果は、少なくとも部分的に、本発明者らにより報告されている[3]ような、上記のフラグメントとチオレドキシン(保存活性部位配列Cys−Gly−Pro−Cysに酸化還元活性なジスルフィド−ジチオールを有する小さい多機能性タンパク質)の間のin vitro相互作用に起因し得る。この発見により、ダイズタンパク質を含有するコレステロールが豊富な食事を給餌したウサギにおいて観察された酸化第二銅により誘発されたLDL酸化の遅滞期が、同じ食事だがカゼインをタンパク質源として含有する食事を給餌したウサギにおいて見られたもの[22]より長い理由を説明し得る。
【0036】
得られたデータは、ダイズ7Sグロブリンα’鎖のN末端親水性フラグメントが、シンバスタチンについて報告されているのと同様に10μM未満の濃度でのin vitroモデルにおいて活性であることを初めて示しているため、特に興味深い。さらに、組換えタンパク質の使用により、明確な利益の欠如および潜在的な毒性が報告されてきた[23]イソフラボンを含めた他のタンパク質および非タンパク質のダイズ成分の関与が一切排除される。
【0037】
本発明は、脂質低下療法において単独でまたは薬物、すなわち、シンバスタチン、プラバスタチン、フルバスタチン、アトルバスタチン、ロバスタチンなどのスタチンと組み合わせて使用される、高脂血症および心血管疾患を含めた様々な疾患に対する有益な効果を有する機能性食品および組成物を提供する。
【0038】
本発明の組成物は、従来の賦形剤および方法を使用して調製する。本発明の組換えポリペプチドの投薬量は、患者の体重、年齢および性別などの複数の因子に依存し、該ポリペプチドの薬理学、薬物動態学および毒物学的特性に基づいて実務者により容易に決定される。しかし、一般に、前記投薬量は、約50〜約500mg、1日1〜3回の範囲である。
【0039】
参考文献
【0040】
【表2】
【技術分野】
【0001】
本発明は、ダイズ7Sグロブリンの天然のα’サブユニットのN末端親水性フラグメントに対応する組換えα’フラグメント、ならびにその調製方法ならびにコレステロールおよびトリグリセリドの恒常性を制御するのに有用な活性成分としてそれを含有する組成物に関する。
【背景技術】
【0002】
(発明の背景)
高コレステロール血症患者の血中脂質レベル(lipidemic level)の制御における食事性のダイズタンパク質の役割は、広く受け入れられている論点である[1]。以前の研究[2〜4]においては、LDL受容体のアップレギュレーションにおけるダイズ7Sグロブリンの1つのサブユニットであるα’サブユニットの直接的な関与がin vitroおよびin vivo系において証明され、コレステロール恒常性を調節することができる生物活性(ポリ)ペプチドが、細胞酵素処理により産生される可能性が高いことが示唆されている。
【0003】
天然の7Sグロブリンは、異なる遺伝子によりコードされた、3つのランダムに分類されたポリペプチド鎖、α’、αおよびβサブユニット[5]からなる。成熟α’(受入番号P11827 UniProtKB/Swiss−Protデータベース)およびα鎖(受入番号P13916 UniProtKB/Swiss−Protデータベース)は、βサブユニット(受入番号P25974 UniProtKB/Swiss−Protデータベース)には存在していない約145のアミノ酸残基の伸長N末端領域を共有している。α’伸長領域の特有のアミノ酸配列に基づいて、このサブユニットは金属アフィニティークロマトグラフィーにより精製され、高コレステロール血症ラットに経口投与され、それにより、その血漿脂質低下特性および肝臓β−VLDL受容体のアップレギュレーションの両方を示すことが可能となった[4]。一方、α’サブユニットの分子量はおよそ71kDaである[6]ため、修飾されない限りin vivoで腸の障壁を越えるとは考えられない。
【0004】
このため、本発明者らの研究は、薬理学的効果に関与するα’サブユニットのアミノ酸配列(複数可)を探求することに向けられてきた。3つのサブユニットのコア領域は非常に類似のアミノ酸配列を有するため、生物活性は該伸長領域の1つまたは複数の(ポリ)ペプチドにあるはずであると考えられる。原理上、限定的ではあるが有意なα’およびα鎖の伸長領域間のアミノ酸の差異により、生物活性に関与するペプチドの数が制限されるであろう。
【0005】
これらの以前の記述から、最初に探求した戦略は、Croksoy(登録商標)70(高コレステロール血症患者の食事療法において通常利用されるイソフラボンを含まないダイズ濃縮物[7〜8])のin vitro消化(ペプシン/トリプシン)から得られたポリペプチド、および7Sダイズグロブリンサブユニット間で異なっていた特定のアミノ酸配列に対応する合成ペプチドの両方の、HepG2細胞におけるLDL受容体(LDL−R)調節に対する効果を試験することであった。これらの研究において得られた結果は、3000〜20000Daの範囲のMWを有するCroksoy(登録商標)70の酵素消化産物、ならびに7Sダイズグロブリン由来の小さい合成ペプチド(2271Da)(10−4Mの濃度で細胞に添加した)に曝されたHepG2細胞において顕著なLDL−Rアップレギュレーションを誘発できたことを指摘していた[9]。小さいペプチドを用いて得られた研究結果は、現在、依然として調査中であり、今までのところ結論が出ていない[10]。
【0006】
ダイズタンパク質のコレステロールおよびトリグリセリドを低下させる能力は確固たる論点である。ダイズタンパク質の食事は、高コレステロール血症患者を治療するための現在最も有力な食事性のツールであり、したがって、成人および非常に若い被験者の管理のユニークな機会を提供している。さらに、血漿コレステロールの低減が、コレステロール血症のベースラインの程度が高い患者においてより大きいことは明確に確立されている[14]。
【0007】
タンパク質自体が血中コレステロールを低減するという仮定は、食事における動物タンパク質から植物タンパク質への転換により、実験動物の肝臓[15]、ならびに高コレステロール血症患者の循環リンパ単球[16]においてLDL受容体系が活性化されることを示している実験的研究に起因していた。コレステロール低下効果に関与するダイズタンパク質成分を同定するために、LDL受容体の発現およびコレステロールの生合成/分解を調節する因子に対して非常に感受性であるヒト肝細胞癌細胞系を用いてin vitro研究を実施した。7Sダイズグロブリン由来の精製α’サブユニットがHepG2細胞においてLDL受容体をアップレギュレーションする[3]ことが分かり、この発見は、コレステロール給餌ラットにおいて確認された[4]。これらのデータは、観察された生物学的効果に該タンパク質部分が関与しているという仮定を支持するものであるが、ペプチドおよびアミノ酸は通常胃および/または腸のタンパク質分解酵素の作用により産生されるため、α’鎖のin vivoの生体内動態(biological fate)に対する議論が生じる可能性がある。しかし、抗酸化、抗増殖および抗炎症効果に起因する場合が多い関連する調節機能を果たすと主張されている動物および植物(ポリ)ペプチドの数が増大している[17]。ダイズに関する限り、ペプチドおよびBowman−Birk阻害物質などのさらに小さい小型のタンパク質を吸着することができ[18]、したがって、抗癌、抗炎症性、放射線防護効果を含めた多数の効果を引き出す[19]可能性が、実験的証拠により明確に示されている。遺伝子組換えダイズ(ポリ)ペプチドも、降圧効果などの生物学的応答を誘発することが示されてきた[20]。最近になって、7Sグロブリンβ鎖に由来するLDL−R転写を刺激するペプチド(FVVNATSN)が、Bacillus amyloliquefaciens由来のプロテアーゼにより、次いで、化学的合成により調製されたダイズ加水分解物から同定された[21]。この場合、100μMの濃度の該ペプチドに曝されたHepG2細胞においてLDL−R転写の増大(+148%)が検出された。11Sグロブリンから生じる他のペプチドは、類似の活性を発揮するがその活性はより低いことが示されてきた[21]。
【0008】
完全長タンパク質の生物学的特性を維持または改善さえする、より短いポリペプチドを入手可能にすることが望ましいであろう。
【先行技術文献】
【非特許文献】
【0009】
【非特許文献1】Sirtori CR. et al.,Curr Atheroscler Rep.2001;3:47−53
【非特許文献2】Lovati MR. et al.,J Nutr.1992;122:1971−8
【発明の概要】
【課題を解決するための手段】
【0010】
(発明の説明)
今般、ダイズ7SグロブリンのN末端側に対応するそのいわゆるα’伸長領域が有益な生物活性を有することが分かり、LDLの取込みおよび分解に対してフルサイズのα’鎖より効果的でさえあることが証明された。
【0011】
本発明は、したがって、以下eα’と呼ぶ前記α’伸長領域、ならびにダイズα’サブユニットのN末端伸長領域を含有する組換えポリペプチドのクローニング、酵母発現および精製によるその調製方法を提供する。
【0012】
この目的のために、α’鎖のN末端フラグメントの異種発現を試みた。酵母Pichia pastorisの分泌能力のある酵母細胞(secretion-competent yeast cell)においてこの目標を達成した。組換えポリペプチドを精製し、HepG2細胞においてその生物活性を評価した。この生物工学的手法の使用により、in vitro試験、さらにはin vivo実験において試験するのに十分な量の組換えポリペプチドを得ることができた。
【図面の簡単な説明】
【0013】
【図1】pPICZαB−eα’コンストラクトの概要である。eα’の発現は、AOX(アルコールオキシダーゼ)メタノール誘導性プロモーター(5'AOX1)により駆動され、α接合因子(α−MF)は組換えタンパク質の培地への分泌を促進し、AOX1 TTはAOX転写終結領域である。Sh ble遺伝子はゼオシンに対する耐性を付与し、pUC OriはE.coliにおける多数のプラスミドコピーの複製開始点である。他の略語は制限酵素の開裂位置を指し、bpは塩基対である。
【図2】図2は、組換えポリペプチド(eα’)および野生型のダイズα’サブユニットの配列アラインメントである。星印は、2つの配列における同一のアミノ酸残基を示す。
【図2A】図2Aは組換えポリペプチド(eα)’の配列(配列番号1)である。
【図2B】図2Bは野生型のダイズα’サブユニットの配列(配列番号2)である。
【図3】図3−パネルAは組換えPichia pastoris培養物の還元条件下でのSDS−PAGEである。レーンM:分子量マーカー。レーン1:メタノールによる誘導の前のTCA沈殿培養上清(TCA-Precipitated culture supernatant)(無細胞)。レーン2:メタノールによる誘導の1h後のTCA沈殿培養上清(無細胞)。レーン3:メタノールによる誘導の8h後のTCA沈殿培養上清(無細胞)。レーン4:メタノールによる誘導の19h後のTCA沈殿培養上清(無細胞)。レーン5:メタノールによる誘導の25h後のTCA沈殿培養上清(無細胞)。図3−パネルBはeα’精製ステップの還元条件下でのSDS−PAGEである。レーンM:分子量マーカー。レーン1:発酵ブロスの凍結乾燥粉末。レーン2:DEAE−セルロース 150mM NaCl溶出画分。レーン3:DEAE−セルロース 250mM NaCl溶出画分。
【発明を実施するための形態】
【0014】
発明の詳細な説明
本発明は、次に、以下の実験の項において詳細に説明する。
【0015】
材料および方法
酵母、細菌株および化学物質。酵母発現にPichia pastoris X33(WT)株(Invitrogen、San Diego、CA)を使用した。遺伝子操作に利用した細菌株はE.coli XL1−Blue(Invitrogen、San Diego、CA)であった。制限酵素Pst IおよびXba IをRoche(Indianapolis、IN)から、Sac IをFermentas(Ontario、Canada)から購入した。Taq DNAポリメラーゼはInvitrogen(San Diego、CA)から購入した。ゼオシンはInvivogen(San Diego、CA)から購入した。PCR用のオリゴヌクレオチドはPrimm(Milano、Italy)から得た。ペプトン、トリプトン、酵母エキスおよび寒天はBecton Dickinson and Company(Sparks、MD)から購入した。グルコースおよびソルビトールはSigma(St.Louis MO)から購入した。透析膜はSpectrum Laboratories(Rancho Dominguez、CA)から購入した。DEAE樹脂はWhatman(Maidstone、England)から購入した。NiNTA樹脂はQiagen(Hilden、Germany)から購入した。Symmetry C4 HPLCカラムはWaters(Milford、MA)から購入した。
【0016】
他の化学物質はSigma(St.Luis、MO)からまたはMerck(Darmstadt、Germany)からの試薬グレードであった。
【0017】
培地および増殖条件。P.pastoris X33株をYPD(酵母ペプトンデキストロース)完全培地(2%ペプトン、1%酵母エキス、2%グルコース)において培養した。YPD、寒天1.5%、100mg/mLゼオシンを含有するプレートにおいて、Mut+形質転換体を選択した。全ての酵母培養物を30℃で維持した。異種タンパク質検出のために、YPS(酵母ペプトンソルビトール)培地(2%ペプトン、1%酵母エキス、2%ソルビトール)において、Zeor−Mut+形質転換体を600nmで光学濃度5まで培養した。次いで、メタノールを1%の最終濃度まで添加した。ゼオシンを含有する低塩LB(Luria−Bertani)培地のプレート(1%トリプトン、0.5%酵母エキス、0.5%塩化ナトリウム、1.5%寒天、25mg/mLゼオシン)において全ての細菌形質転換体を選択した。全ての細菌培養物を37℃で維持した。
【0018】
eα’遺伝子発現ベクターの構築。関心対象(eα’)の配列を含む挿入物(tα’)を含有するPichia pastoris発現プラスミドDNA鋳型(expression Pichia pastoris plasmid DNA template)(pPICZαB)に対するPCRによりeα’遺伝子を増幅した。eα’遺伝子の5'末端でPstI制限部位を生成するようにオリゴヌクレオチド5'−GAAAAGATAGATTAAAGCTGCAGTGGAGGAAG−3'を設計した。この変異により、eα'のN末端位置においてアラニン残基挿入が生じた。eα’遺伝子の3'末端でXbaI制限部位を生成するように第2のオリゴヌクレオチド5'−CCCTTCTTATTCTTTCTAGATCATGGTTCTCTTTGAGACTC−3'を設計した。いずれのオリゴヌクレオチドもmQ滅菌水に溶解した。PCR反応混合物は、0.5mMプライマー、0.8mM dNTPs(Eppendorf、Hamburg、Germany)、30ng鋳型(pPICZαB/tα’)、2.5U Taq DNAポリメラーゼ、PCR緩衝液(最終組成:50mM KCl、1.5mM MgCl2、20mM Tris−Cl、pH8.4および最終容量が25mLとなるmQ滅菌水)からなっていた。以下の条件を使用して、Perkin Elmer Geneamp PCR System 2400サーモサイクラー(Perkin Elmer Corp.、Wellesley、MA)でPCR増幅を実施した。94℃で10minで開始;94℃で40sec、60℃で40sec、72℃で20secで30サイクル;最終伸長72℃で10minおよび4℃で維持。465bpのPCR産物を酵素で消化して423bpのフルサイズのコンストラクトとし、それをpPICZαBベクターにクローニングし、それにより、pPICZαB−eα’コンストラクトが生じ、それをXL1−Blue E.coli細胞において形質転換した。テトラサイクリンおよびゼオシンを含有する半流動LB培地上で陽性クローン(positives clones)を選択した。Primm(Milano、Italy)によりこれらのクローンの1つを配列決定して、pPICZαB−eα’コンストラクト配列において変異が発生しないようにした。制限酵素SacIを用いた消化により20μgのpPICZaB−eα’コンストラクトおよび30μgの発現ベクターpPICZαB(負の対照)を線状化し、次いで、精製した。
【0019】
P.pastorisゲノムにおけるpPICZaB−eα’の形質転換。1.5kVに設定したEppendorf(Hamburg、Germany)electroporator 2510装置で20μgの線状化pPICZαB−eα’コンストラクトおよび30μgの線状化pPICZαBを用いた電気穿孔法により、野生型(wt)の酵母細胞を形質転換した。100mg/mLのゼオシンを含有するYPDプレートでのプレーティングにより形質転換体を最初に選択した。本発明者らのコンストラクトの形質転換P.pastorisゲノムへの組込みを確認するために、Dneasy Plant Mini Kit[QIAGEN、Hilden、Germany]を使用してゲノムDNAを抽出した。5'AOX1(5'−GACTGGTTCCAATTGACAAGC−3')および3'AOX1プライマー(5'−GCAAATGGCATTCTGACATCC−3')を使用したPCRによりアルコールオキシダーゼプロモーター(AOX1)部位でのコンストラクトの挿入を確認するための鋳型としてゲノムDNAを使用した。PCR反応混合物は、0.5mMプライマー、0.25mM dNTPs、100ngのゲノム鋳型、2.5U Taq DNAポリメラーゼ、PCR緩衝液(最終組成は上記の通り)からなっていた。以下の条件を使用してPCR反応を実施した。94℃で10minで開始;94℃で40s、60℃で40s、72℃で20sで30サイクル;最終伸長72℃で10minおよび4℃で維持。
【0020】
形質転換体クローンの選択。振盪(180rpm)培養器中の30℃の50mLのYPS培地において、18のZeo+形質転換体およびpPICZαB発現ベクター(負の対照)を含有する1つの形質転換体をOD600=5まで増殖させた。1%の最終濃度までメタノールを添加することにより誘導期を誘発し、24時間延長した。SDS−PAGEによりeα’の発現について上清のアリコートを検査した。発酵槽規模(fermenter scale)での組換えタンパク質の生産のために、最高のeα’発現を有するクローン形質転換体を選択した。
【0021】
eα’の発現:発酵槽規模。大量生産のために、Invitrogenの「Pichia発酵プロセスガイドライン(Pichia Fermentation Process Guidelines)」(バージョンB、053002)に従って、14Lの発酵槽(Chemap、Switzerland)において選択したクローンを増殖させた。バッフルなし振盪フラスコ(unbaffled shake flask)中のYNB培地(KH2PO4 2.0g;(NH4)2SO4 10.0g;MgSO4 7H2O 1.0g;NaCl 0.2g;CaCl2 0.2g;グリセロール100% 10.0g;pH5.2までのKOH;1.0Lまでの蒸留水)で種菌培養物を調製する。250rpm、30℃で約24時間の培養後、1.0Lの種培養物を使用して、8.5Lの基礎塩培地(basal salt medium)(H3PO4 85% 227mL;CaSO4 2H2O 7.9g;K2SO4 155g;MgSO4 7H2O 127g;KOH 35g;グリセロール100% 340g;8.5Lまでの蒸留水;滅菌後にNH4OHを用いてpHを5.0に調節した)を用いて用意した発酵槽に接種する。前述の培養基のいずれにもビオチンおよびイノシトール(各1mg/L)を補充する。基礎塩培地にも修飾されたPTM1微量塩溶液(trace salt solution)(H2SO4 96% 2.5mL;CoC2O4°2H2O 243mg;CuSO4°5H2O 3.0g;KI 44.5mg;MnSO4°H2O 1.5g;Na2MoO4°2H2O 100mg;H3BO3 10mg;FeSO4°7H2O 32.5g;ZnCl2 10g;500mLまでの蒸留水)を補充する。
【0022】
微生物の増殖および異種タンパク質の発現は、3ステッププロセスを介して得られる。約24時間の最初の回分増殖期(炭素源としてグリセロール)の後に、約4時間の流加回分(fed-batch)期(制限炭素源(limiting C-source)としてグリセロール)が続く。これらの最初の2つの期の間、NH4OH 20%を添加することによりpHを5.0で維持する。一旦全てのグリセロールが消費されると、メタノールの供給を開始してeα’タンパク質発現を誘発し、NH4OH 20%を添加することによりpHを6.0にシフトする。メタノール流加回分期は約24時間にわたる。これらの2つの流加回分期の間、通気速度(vvm)は手動で徐々に増大させながら撹拌機速度(rpm)は電気的に微調節することにより、溶存酸素(DO%)を30%で一定に維持する(in maintained stable)。数時間毎に、DO%レベル(その値はメタノール供給の突然の停止後に急増を示すはずであり、逆に、その再開後に急速な減少を示すはずである)を追跡することにより炭素源制限条件をチェックする。数時間毎に、以下の分析のために無菌条件において発酵槽から試料を採取する。光学濃度(λ600nm)、細胞バイオマス%(湿量)、無菌性、顕微鏡観察、SDS−PAGE。
【0023】
eα’の精製:発酵ブロスの下流処理。約9.2Lの培養物を発酵槽から溢流させ、氷で冷却する。その後の操作は全て+4℃で実施する。3000xgで30minの遠心分離(Centrikon T−124、Kontron Instruments)により培養物全体を分離し、バイオマス(ペレット)を廃棄し、約7.5Lの上清をZetaplus 30SP(Cuno)での深層濾過(depth filtration)により清澄化し、引き続いて0.22μmフィルター(Millipak 100、Millipore)で精密濾過する。ポリエーテルスルホン膜、MWCO 10kDa(Omegaフィルター、Pall)での限外濾過を介して透明な濾液を濃縮する。約300mLの濃縮物を、3.0LのTris−HCl 10mM pH7.2に対してダイアフィルトレーションし、最終的に凍結乾燥する。この手順により、SDS−PAGEでかなり低い夾雑タンパク質の程度を示し、約25%p/pの総タンパク質含量(Bradfordタンパク質アッセイ、較正標準としてウシ血清アルブミン)を有する34.5gの凍結乾燥粉末を得た。
【0024】
eα’の精製:クロマトグラフィーによる精製
より高性能のゲル電気泳動のために、供給業者の手順に従ってNuPAGE(登録商標)プレキャストゲルシステム(Invitrogen)を使用した。2グラムの凍結乾燥粉末を150mL 50mM Tris−HCl、pH7.50に溶解し、同じ緩衝液を用いて平衡化したDEAE−セルロースカラム(6×10cm、Whatman、Maidstone、UK)に装填した。それぞれ150および250mM NaClを含有する同じ緩衝液を用いて、保持されたタンパク質の溶出を実施した。0.25M NaCl(300mL)を用いて溶出した画分が、最も多いeα’の含量を示した。凍結乾燥により該溶液を100mLまで濃縮し、次いで、6000〜8000Daの膜を用いてmilliQ限外濾過水により4℃で24時間透析し、次いで、凍結乾燥した。約370mgのタンパク質を得た。
【0025】
タンパク質の均一性を確認するために、約1mgのタンパク質をSymmetry C4(4.6×250mm)逆相カラムに装填した。緩衝液A(限外濾過水およびトリフルオロ酢酸0.1%)および緩衝液B(アセトニトリル100%+トリフルオロ酢酸0.1%)を使用した。
【0026】
電気泳動技術。参考文献11に従って、mini−Protean II細胞(Bio−Rad)を使用して、12%ポリアクリルアミドゲルで還元条件(2%β−メルカプトエタノール)下でのSDS−PAGEを実施した。クーマシーブルーを用いて該ゲルを染色した。
【0027】
細胞培養物。株化ヒト肝細胞癌細胞系(HepG2)を米国培養細胞系統保存機関(American Type Culture Collection)(Rockville、MD)から得た。Eagleの最小必須培地(MEM)、ウシ胎仔血清、トリプシン−EDTA(1倍)、ペニシリン(105U/L)、ストレプトマイシン(100g/L)、トリシン緩衝液(1mmol/L、pH7.4)および非必須アミノ酸溶液(100倍)は、GIBCO(Madison、WI)から得た。ペトリ皿は、COSTAR(Cambridge、MA)から得た。フィルターはMillipore(Bedford、MA)から得た。タンパク質クーマシープラスタンパク質アッセイキット(Protein Coomassie Plus Protein Assay kit)はPierce(Rockford、IL、USA)から購入した。100mmol/L NaOH中の無担体の125ヨウ素はPerkin Elmer Life Sciences(Boston、MA)から得た。Sephadex G25カラム(PD10)はPharmacia Biotech(Uppsala、Sweden)から得た。LDHおよびMTTキットは、Sigma Diagnostics(Milano−Italy)から得た。全ての他の化学物質は、Merck(Darmstadt、Germany)からの分析用のものであった。直径90mmのペトリ皿において単層で細胞を増殖させ、95%空気、5%CO2の加湿雰囲気中で、10%ウシ胎仔血清(FCS)、非必須アミノ酸溶液(1%、v/v)、ペニシリン(105U/L)、ストレプトマイシン(0.1g/L)、トリシン緩衝液(20mmol/L、pH7.4)、NaHCO3(24mmol/L)およびピルビン酸ナトリウム(0.11g/L)を補充したMEMにおいて37℃で維持した。LDL受容体の調節を評価するように設計した実験のために、35mmのプラスチック皿に細胞を播種し(3〜5×105細胞)、コンフルエンスに達する直前に使用した。全ての細胞培養実験において、2〜3日毎に培地を交換した。細胞生存度を評価するために、基本的に参考文献9に記載されているように、メチルテトラゾリウム塩(MTT)アッセイにより、様々な濃度のeα’に曝された細胞由来の培養基を試験した。動態(LDH/LD)診断キット(Sigma Diagnostics)を使用して乳酸デヒドロゲナーゼ(LDH)活性を測定することにより細胞酵素の漏出を判定した。連続調製用超遠心分離(sequential preparative ultracentrifugation)[12]により、臨床的に健康な正常リポタンパク質のボランティアの血漿からLDL(1.019≦d≦1.063g/L)を単離した。Bilheimerらにより改善され[13]、以前に記載した[3]McFarlaneの方法に従ってリポタンパク質を標識した。濾過(Milliporeフィルター、孔径0.45μm)により125I−LDLを滅菌し、使用するまで4℃で貯蔵した。以前に記載した[9]ようにヒトリポタンパク質欠乏血清(LPDS)を調製した。
【0028】
125I−LDLの取込みおよび分解。5g/100g LPDSを補充したMEMにおいて細胞の単層を37℃で24h前培養して、表に列挙した様々な濃度のeα’または3.5μmol/Lのα’精製サブユニットまたは1.0μmol/Lのシンバスタチンの存在/不存在下でLDL受容体をアップレギュレーションした[2]。次いで、一定の濃度(7.5mg/L)の125I−LDLを該培地に添加し、培養を37℃でさらに5h継続した。以前に報告された[2]ように125I−LDLの特異的な取込み(結合+内部移行)および分解を評価した。
【0029】
統計的分析。様々な濃度のeα’を用いた細胞培養後のLDLにおける細胞の取込みおよび分解の差異を、ANOVAおよびその後のDunnettの検定により求めた。各値は平均値±SDとして表し、P値<0.05を統計的に有意であると見なした。
【0030】
結果
Pichia pastorisにおけるeα’の発現。Pichia pastoris細胞を形質転換するのに使用したプラスミドの構造を図1に示している。挿入物の配列およびα’サブユニットを有するそのアラインメントを図2に示している。材料および方法の項において述べたように、組換えポリペプチドと野生型のポリペプチドの間の唯一の差異はN末端第1アミノ酸残基にあり、それは、技術的な理由から、組換え鎖においてはアラニンであった。培養基のSDS−PAGE分析により判断したところ、最大の組換えポリペプチドの生産を示している(図示せず)クローンを大量生産のために選択した。図3−Aは、メタノールによる誘導の前(レーンn.1)および1〜8〜19および25時間後(レーンn.2、n.3、n.4、n.5)の酵母培養物の上清の電気泳動分析を示している。図3−Aが示しているように、約20kDaの明らかな分子量での明白なバンドが誘導の結果として生じている。
【0031】
eα’の精製。クロマトグラフィー法によりeα’の精製を達成した。各ステップから試料を採取し、SDS−PAGEにより分析した。同定したポリペプチドの均一性に対する精製ステップの効果を図3−Bに示している。培養基において酵母タンパク質による非常に低い程度の混入が既に達成されていたが、さらなるクロマトグラフィーによるステップにより主要な夾雑タンパク質を除去し、ほとんど均一な形態の組換えポリペプチドの回収を可能にした。このバンドのN末端配列分析により、この20kDaポリペプチドがeα’鎖に対応することが確認された。この試料の純度が細胞アッセイに適していると判断した。
【0032】
eα’の生物活性。正の対照としての精製α’サブユニット、およびその切断α’形態のHepG2細胞への添加により、以下の表において報告しているように、未処理の細胞と比較してLDL受容体媒介性の取込みおよび分解の有意な上昇がもたらされた。
【0033】
【表1】
【0034】
この結果は、LDL調節がeα’に用量依存的であり、最高濃度が正の対照、シンバスタチンの濃度と同様であったことを示していた。MTTおよびLDHアッセイにより測定したところ、いずれのeα’の濃度でも細胞毒性の証拠は見られなかった(図示せず)。
【0035】
したがって、本発明において、生物学的応答を誘導することができるアミノ酸配列がα’鎖のN末端伸長ドメインに存在することが分かった。さらに、本発明者らは、N末端親水性フラグメントが、強力な抗高脂血症薬であるシンバスタチンと同程度の濃度でその効果を発揮することを見出した。この効果は、少なくとも部分的に、本発明者らにより報告されている[3]ような、上記のフラグメントとチオレドキシン(保存活性部位配列Cys−Gly−Pro−Cysに酸化還元活性なジスルフィド−ジチオールを有する小さい多機能性タンパク質)の間のin vitro相互作用に起因し得る。この発見により、ダイズタンパク質を含有するコレステロールが豊富な食事を給餌したウサギにおいて観察された酸化第二銅により誘発されたLDL酸化の遅滞期が、同じ食事だがカゼインをタンパク質源として含有する食事を給餌したウサギにおいて見られたもの[22]より長い理由を説明し得る。
【0036】
得られたデータは、ダイズ7Sグロブリンα’鎖のN末端親水性フラグメントが、シンバスタチンについて報告されているのと同様に10μM未満の濃度でのin vitroモデルにおいて活性であることを初めて示しているため、特に興味深い。さらに、組換えタンパク質の使用により、明確な利益の欠如および潜在的な毒性が報告されてきた[23]イソフラボンを含めた他のタンパク質および非タンパク質のダイズ成分の関与が一切排除される。
【0037】
本発明は、脂質低下療法において単独でまたは薬物、すなわち、シンバスタチン、プラバスタチン、フルバスタチン、アトルバスタチン、ロバスタチンなどのスタチンと組み合わせて使用される、高脂血症および心血管疾患を含めた様々な疾患に対する有益な効果を有する機能性食品および組成物を提供する。
【0038】
本発明の組成物は、従来の賦形剤および方法を使用して調製する。本発明の組換えポリペプチドの投薬量は、患者の体重、年齢および性別などの複数の因子に依存し、該ポリペプチドの薬理学、薬物動態学および毒物学的特性に基づいて実務者により容易に決定される。しかし、一般に、前記投薬量は、約50〜約500mg、1日1〜3回の範囲である。
【0039】
参考文献
【0040】
【表2】
【特許請求の範囲】
【請求項1】
配列番号1を有するN末端親水性フラグメントダイズ7Sグロブリンα’サブユニット。
【請求項2】
組換えポリペプチドのクローニング、Pichia pastorisにおける発現および精製を含む、請求項1に記載のダイズ7SグロブリンのN末端親水性フラグメントの調製方法。
【請求項3】
好適な担体との混合剤中に活性成分として請求項1に記載のダイズ7SグロブリンのN末端親水性フラグメントを含む組成物。
【請求項1】
配列番号1を有するN末端親水性フラグメントダイズ7Sグロブリンα’サブユニット。
【請求項2】
組換えポリペプチドのクローニング、Pichia pastorisにおける発現および精製を含む、請求項1に記載のダイズ7SグロブリンのN末端親水性フラグメントの調製方法。
【請求項3】
好適な担体との混合剤中に活性成分として請求項1に記載のダイズ7SグロブリンのN末端親水性フラグメントを含む組成物。
【図2】

【図2A】

【図2B】

【図1】
【図3】
【図2A】
【図2B】
【図1】
【図3】
【公表番号】特表2012−530090(P2012−530090A)
【公表日】平成24年11月29日(2012.11.29)
【国際特許分類】
【出願番号】特願2012−515392(P2012−515392)
【出願日】平成22年6月16日(2010.6.16)
【国際出願番号】PCT/EP2010/003627
【国際公開番号】WO2010/145820
【国際公開日】平成22年12月23日(2010.12.23)
【出願人】(591092198)インデナ エッセ ピ ア (52)
【Fターム(参考)】
【公表日】平成24年11月29日(2012.11.29)
【国際特許分類】
【出願日】平成22年6月16日(2010.6.16)
【国際出願番号】PCT/EP2010/003627
【国際公開番号】WO2010/145820
【国際公開日】平成22年12月23日(2010.12.23)
【出願人】(591092198)インデナ エッセ ピ ア (52)
【Fターム(参考)】
[ Back to top ]