
HER−3に対して誘導された抗体及びその使用
【課題】HER−3に結合する結合タンパク質及び該タンパク質をコードするポリヌクレオチドを提供する。
【解決手段】抗ヒトHER−3抗体に由来する特定のアミノ酸配列およびそれをコードするDNA配列であって、該結合タンパク質を作製するための発現ベクター及び該ベクターを含む宿主細胞も提供される。さらに、本発明は、HER−3によって媒介されるシグナル伝達及び/又はそのリガンドであるヘレグリンに関連する疾病を診断及び治療するための組成物及び方法を提供する。
【解決手段】抗ヒトHER−3抗体に由来する特定のアミノ酸配列およびそれをコードするDNA配列であって、該結合タンパク質を作製するための発現ベクター及び該ベクターを含む宿主細胞も提供される。さらに、本発明は、HER−3によって媒介されるシグナル伝達及び/又はそのリガンドであるヘレグリンに関連する疾病を診断及び治療するための組成物及び方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、抗体及びその結合断片を含む、HER−3に結合する結合タンパク質及び該タンパク質をコードするポリヌクレオチドに関する。本発明の結合タンパク質を作製するための発現ベクター及び該ベクターを含む宿主細胞も提供される。さらに、本発明は、HER−3によって媒介されるシグナル伝達及び/又はそのリガンドであるヘレグリンに関連する疾病を診断及び治療するための組成物及び方法を提供する。
【背景技術】
【0002】
ヒト上皮増殖因子受容体3(HER−3、ErbB3としても知られる。)は、受容体タンパク質チロシンキナーゼであり、HER−1(EGFRとしても知られる。)、HER−2及びHER−4(Plowman et al., Proc.Natl.Acad.Sci.U.S.A.87(1990),4905−4909;Kraus et al., Proc.Natl.Acad.Sci.U.S.A.86(1989),9193−9197;and Kraus et al., Proc.Natl.Acad.Sci.U.S.A.90(1993),2900−2904)も含まれる、受容体タンパク質チロシンキナーゼの上皮成長因子受容体(EGFR)サブファミリーに属する。原型の上皮成長因子受容体と同様、膜貫通受容体であるHER−3は、細胞外リガンド結合ドメイン(ECD)、ECD内の二量体化ドメイン、膜貫通ドメイン、細胞内タンパク質チロシンキナーゼ(TKD)及びC末端リン酸化ドメインからなる。
【0003】
リガンドであるヘレグリン(HRG)は、HER−3の細胞外ドメインに結合し、他のヒト上皮増殖因子受容体(HER)ファミリーのメンバーとの二量体化及びその細胞内ドメインのトランスリン酸化を促進することによって、受容体媒介性シグナル伝達経路を活性化する。HERファミリーのメンバー間での二量体形成は、HER−3のシグナル伝達能力を拡張し、シグナルの多様化のみならず、シグナルの増幅のための手段でもある。例えば、HER−2/HER−3へテロ二量体は、HERファミリーメンバーの中で最も重要な有糸分裂促進シグナルの1つを誘導する。
【0004】
HER−3は、乳癌、胃腸癌及び膵臓癌など、癌の幾つかの種類において過剰発現されることが見出されている。興味深いことに、HER−2/HER−3の発現と非浸潤性段階から浸潤性段階への進行との間に相関が示されている(Alimandi et al., Oncogene 10,1813−1821;deFazio et al., Cancer 87, 487−498;Naidu et al., Br.J.Cancer 78, 1385−1390)。従って、HER−3によって媒介されるシグナル伝達を妨害する因子は望ましい。マウス又はキメラHER−3抗体が、US5968511、US5480968及びWO03013602などに報告されている。
【0005】
最近、HER−2に対するヒト化されたモノクローナル抗体であるHerceptin(R)は、HER−2によって媒介されるシグナル伝達を妨害し、ヒトにおいて、治療的に有効であることが示された(Fendly et al., Hybridoma 6, 359−370;Hudziak et al., Mol.Cell.Biol.9, 1165−1172;Stebbing et al., Cancer Treat.Rev.26, 287−290)。Herceptin(R)は、2つの異なる機序、すなわち、免疫系のエフェクター細胞の関与及び直接的な細胞傷害性アポトーシス誘導効果を通じて作用することが示されている。
【0006】
しかしながら、高度に増幅されたHER−2を有する患者のみが、Herceptin(R)治療に有意に応答するので、治療に適した患者の数を限定される。さらに、薬物に対する耐性の発生又は腫瘍細胞上のHER−2の発現若しくはエピトープ配列の変化のために、治療を適用することが可能な患者でさえ、抗体に対して非反応性となり、従って、その治療的な利点が喪失する場合があり得る。従って、HER−3などのHERファミリーのさらなるメンバーにアプローチする、標的をベースとする療法のためのさらなる薬物が必要とされている。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】米国特許第5968511号明細書
【特許文献2】米国特許第5480968号明細書
【特許文献3】国際公開第03013602号
【非特許文献】
【0008】
【非特許文献1】Plowman et al., Proc.Natl.Acad.Sci.U.S.A.87(1990),4905−4909
【非特許文献2】Kraus et al., Proc.Natl.Acad.Sci.U.S.A.86(1989),9193−9197
【非特許文献3】Kraus et al., Proc.Natl.Acad.Sci.U.S.A.90(1993),2900−2904
【非特許文献4】Alimandi et al., Oncogene 10,1813−1821
【非特許文献5】deFazio et al., Cancer 87, 487−498
【非特許文献6】Naidu et al., Br.J.Cancer 78, 1385−1390
【非特許文献7】Fendly et al., Hybridoma 6, 359−370
【非特許文献8】Hudziak et al., Mol.Cell.Biol.9, 1165−1172
【非特許文献9】Stebbing et al., Cancer Treat.Rev.26, 287−290
【発明の概要】
【0009】
本発明の第一の態様は、HER−3に結合する単離された結合タンパク質に関する。
【0010】
本発明の一実施形態において、本発明の単離された結合タンパク質は、(a)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH1;(b)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH2;並びに(c)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH3からなる群から選択されるCDRの少なくとも1つを含む重鎖アミノ酸配列、及び/又は(d)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL1;(e)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL2;並びに(f)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL3からなる群から選択されるCDRの少なくとも1つを含む軽鎖アミノ酸配列を含む。
【0011】
本発明の別の実施形態において、本発明の単離された結合タンパク質は、配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230からなる群から選択される重鎖アミノ酸配列、及び/又は配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232からなる群から選択される軽鎖アミノ酸配列を含む。
【0012】
本発明のさらに別の実施形態において、本発明の単離された結合タンパク質は、配列番号2及び4、6及び8、10及び12、14及び16、18及び20、22及び24、26及び28、30及び32、36及び38、42及び44、46及び48、50及び52、54及び56、60及び58、62及び64、66及び68、70及び72、74及び76、78及び82、80及び82、84及び86、88及び90、92及び94、96及び98、100及び102、104及び106、108及び110、112及び114、116及び118、122及び124、126及び128、130及び132、134及び136、138及び140、142及び144、146及び148、150及び152、154及び156、158及び160、162及び164、166及び168、170及び172、174及び176、178及び180、182及び184、186及び188、190及び192、194及び196、198及び200、202及び204、206及び208、210及び212、214及び216、218及び220、222及び224、226及び228、230及び232に示されている重鎖アミノ酸配列及び軽鎖アミノ酸配列、又は配列番号34、40、60、62若しくは120に示されている重鎖アミノ酸配列又は配列番号58若しくは64に示されている軽鎖アミノ酸配列を含む。
【0013】
本発明によれば、HER−3に結合することができる単離された結合タンパク質は、HER−3の細胞外部分中の少なくとも1つのエピトープと相互作用する。エピトープは、好ましくは、アミノ末端ドメインであるドメインL1(アミノ酸19から184)、システインに富む2つのドメインであるドメインS1(アミノ酸185から327)及びS2(アミノ酸500から632)、又はシステインに富む2つのドメインに隣接しているドメインL2(328から499)の中に位置する。エピトープは、L1及びS1の一部によって含まれるエピトープなど(但し、これに限定されない。)のドメインの組み合わせ中にも位置し得る。さらに好ましいのは、成熟HER−3、特に、成熟ヒトHER−3のアミノ酸残基1から160、161から358、359から575、1から358及び/又は359から604によって形成される三次元構造に結合する単離された結合タンパク質である。
【0014】
好ましくは、本発明の単離された結合タンパク質は、抗体様結合活性を有する足場タンパク質又は抗体、例えば、抗HER−3抗体である。特に、抗HER−3抗体は、U1−1抗体、U1−2抗体、U1−3抗体、U1−4抗体、U1−5抗体、U1−6抗体、U1−7抗体、U1−8抗体、U1−9抗体、U1−10抗体、U1−11抗体、U1−12抗体、U1−13抗体、U1−14抗体、U1−15抗体、U1−16抗体、U1−17抗体、U1−18抗体、U1−19抗体、U1−20抗体、U1−21抗体、U1−22抗体、U1−23抗体、U1−24抗体、U1−25抗体、U1−26抗体、U1−27抗体、U1−28抗体、U1−29抗体、U1−30抗体、U1−31抗体、U1−32抗体、U1−33抗体、U1−34抗体、U1−35抗体、U1−36抗体、U1−37抗体、U1−38抗体、U1−39抗体、U1−40抗体、U1−41抗体、U1−42抗体、U1−43抗体、U1−44抗体、U1−45抗体、U1−46抗体、U1−47抗体、U1−48抗体、U1−49抗体、U1−50抗体、U1−51抗体、U1−52抗体、U1−53抗体、U1−55.1抗体、U1−55抗体、U1−57.1抗体、U1−57抗体、U1−58抗体、U1−59抗体、U1−61.1抗体、U1−61抗体、U1−62又は前記抗体の1つの少なくとも1つの重鎖若しくは軽鎖を有する抗体からなる群から選択される。特に好ましくは、抗体U1−49(配列番号42/44)、U1−53(配列番号54/56)及びU1−59(配列番号70/72)又は前記抗体の1つの少なくとも1つの重鎖若しくは軽鎖を有する抗体である。
【0015】
さらに、本発明のさらなる実施形態は、標識基又はエフェクター基に連結された、単離された結合タンパク質を提供する。好ましくは、このような結合タンパク質は、過剰増殖性疾患、特に、乳癌、胃腸癌、膵臓癌、前立腺癌、卵巣癌、胃癌、子宮内膜癌、唾液腺癌、肺癌、腎臓癌、大腸癌、直腸結腸癌、甲状腺癌、膀胱癌、神経膠腫、悪性黒色腫、精巣癌、軟部組織肉腫、頭部及び頸部癌などの腫瘍疾患又はHER−3を発現若しくは過剰発現する他の癌並びに腫瘍転移の形成の治療に対して有用である。
【0016】
本発明の別の態様は、本発明の結合タンパク質をコードする単離された核酸分子、本発明の結合タンパク質をコードする核酸分子を有するベクター、並びに、このような核酸分子又はベクターで形質転換された宿主細胞、例えばCHO細胞、NS/0骨髄腫細胞を提供する。
【0017】
本発明のさらなる態様は、結合タンパク質を分泌する宿主細胞から前記結合タンパク質を調製することによって、本発明の結合タンパク質を作製する方法に関する。好ましくは、本発明の結合タンパク質は、結合タンパク質を分泌するハイブリドーマ細胞株又は本発明の結合タンパク質をコードする核酸分子で形質転換されたCHO若しくは他の細胞種から調製される。
【0018】
本発明の別の態様は、本発明の結合タンパク質をコードする1又は複数の核酸分子に関して遺伝子導入された動物、植物又は真菌の組織、産物又は分泌物から前記結合タンパク質を調製することによって、本発明の結合タンパク質を製造する方法に関する。好ましくは、本発明の結合タンパク質は、ウシ、ヒツジ、ウサギ、ニワトリ若しくは他の哺乳動物又は鳥類などのトランスジェニック動物、トウモロコシ、タバコ若しくは他の植物などのトランスジェニック植物又はアスペルギルス、ピチア又は他の真菌種などのトランスジェニック真菌の組織、産物又は分泌物から調製される。
【0019】
本発明の別の態様は、医薬として許容される担体、希釈剤及び/又は佐剤と混合された、本発明の少なくとも1つの結合タンパク質を活性因子として含む医薬組成物に関する。本発明の別の好ましい実施形態において、本発明の医薬組成物は、少なくとも1つの他の活性因子、例えば、少なくとも1つの抗新生物因子をさらに含有する。本発明のさらに別の態様は、医薬組成物の調製のための、医薬として許容される担体、希釈剤及び/又は佐剤と混合された、本発明の少なくとも1つの結合タンパク質、及び、場合によって使用される少なくとも1つの他の活性因子(例えば、少なくとも1つの抗新生物因子)の使用に関する。前記医薬組成物は、、過剰増殖性疾患、特に、乳癌、胃腸癌、膵臓癌、前立腺癌、卵巣癌、胃癌、子宮内膜癌、唾液腺癌、肺癌、腎臓癌、大腸癌、直腸結腸癌、甲状腺癌、膀胱癌、神経膠腫、悪性黒色腫又はHER−3を発現若しくは過剰発現する他の癌並びに腫瘍転移の形成を診断し、予防し、又は治療するのに適している。
【0020】
さらに、本発明は、さらなる態様において、HER−3の発現を伴う疾病又は症状を診断するための方法であり、試料を少なくとも1つの本発明の結合タンパク質と接触させること、及びHER−3の存在を検出することを含む、前記方法に関する。好ましい疾病又は症状には、上記過剰増殖性疾患が含まれる。
【0021】
本発明のさらに別の態様は、HER−3の発現を伴う疾病又は症状の予防又は治療を必要としている患者において、HER−3の発現を伴う疾病又は症状を予防又は治療する方法であり、少なくとも1つの本発明の結合タンパク質の有効量、及び、場合によって、少なくとも1つの他の活性因子(例えば、少なくとも1つの抗新生物因子)を前記患者に投与することを含む、前記方法である。好ましくは、患者は、哺乳動物患者、より好ましくは、ヒト患者である。HER−3の発現を伴う好ましい疾病又は症状は、上記過剰増殖性疾患である。
【0022】
本発明のさらなる態様は、HER−3の発現を伴う疾病又は症状を診断し、予防し、又は治療するためのキットであり、少なくとも1つの本発明の結合タンパク質及び/又は核酸分子及び/又はベクターを含む前記キットに関する。場合によって、本発明のキットは、少なくとも1つの他の活性因子、例えば、少なくとも1つの抗新生物因子をさらに含むことができる。好ましくは、HER−3の発現を伴う好ましい疾病又は症状は、上記過剰増殖性疾患である。
【図面の簡単な説明】
【0023】
【図1】図1は、ヒト癌細胞株の群におけるHER−3発現の程度を示しており、HER−3が様々なヒト癌内で発現されていることを示す。
【図2】図2は、HERファミリーの異なるメンバー又は空のベクターのみを安定に発現しているRat1細胞の何れかへのHER−3抗体結合のFACS分析の結果を示している。
【図3】図3は、HER3ドメインにマッピングされた抗体結合競合ビンを示している。
【図4】図4は、本発明の抗HER−3抗体を用いて実施された間接FACSScathcard抗体親和性分析の結果を示している。本分析は、本発明の抗HER−3抗体が、細胞表面上に発現されたHER−3に対して、高い親和性と強い結合定数を有することを示している。
【図5】図5は、本発明の抗HER−3抗体によって誘導された、HER−3の加速された飲食細胞運動を示している。
【図6a】図6aからeは、本発明の抗HER−3抗体を用いて実施されたリガンド競合アッセイの結果を示している。結果は、本発明の抗体が、内在性HER−3を発現する細胞への、[125I]−α−HRG/[125I]−β−HRGの結合を特異的に低下させることを示している。
【図6b】図6aからeは、本発明の抗HER−3抗体を用いて実施されたリガンド競合アッセイの結果を示している。結果は、本発明の抗体が、内在性HER−3を発現する細胞への、[125I]−α−HRG/[125I]−β−HRGの結合を特異的に低下させることを示している。
【図6c】図6aからeは、本発明の抗HER−3抗体を用いて実施されたリガンド競合アッセイの結果を示している。結果は、本発明の抗体が、内在性HER−3を発現する細胞への、[125I]−α−HRG/[125I]−β−HRGの結合を特異的に低下させることを示している。
【図6d】図6aからeは、本発明の抗HER−3抗体を用いて実施されたリガンド競合アッセイの結果を示している。結果は、本発明の抗体が、内在性HER−3を発現する細胞への、[125I]−α−HRG/[125I]−β−HRGの結合を特異的に低下させることを示している。
【図6e】図6aからeは、本発明の抗HER−3抗体を用いて実施されたリガンド競合アッセイの結果を示している。結果は、本発明の抗体が、内在性HER−3を発現する細胞への、[125I]−α−HRG/[125I]−β−HRGの結合を特異的に低下させることを示している。
【図7a】図7aは、本発明の抗HER−3抗体を用いて実施された、HER−3ホスホチロシンELISAの結果を示している。本発明の抗体は、増加された受容体チロシンリン酸化によって示されたように、β−HRGによって媒介されたHER−3の活性化を阻害することができた。
【図7b】本発明の抗HER−3抗体を用いて実施された、HER−3ホスホチロシンELISAの結果を示している。本発明の抗体は、増加された受容体チロシンリン酸化によって示されたように、β−HRGによって媒介されたHER−3の活性化を阻害することができた。さらに、図7bは、滴定された抗体を用いた本実験の代表的な結果を示している。
【図8】図8は、本発明の抗HER−3抗体を用いて実施された、p42/p4MAPキナーゼELISAの結果を示している。本発明の抗体は、増加されたMAP−キナーゼのリン酸化によって示されたように、β−HRGによって媒介されたp42/p44MAP−キナーゼの活性化を低下させることができた。
【図9】図9は、本発明の抗HER−3抗体を用いて実施された、ホスホ−AKTELISAの結果を示している。本発明の抗体は、AKTのリン酸化によって示されたように、β−HRGによって媒介されるAKTの活性化を低下することができた。
【図10】図10は、本発明のヒト抗HER−3抗体による、MCF7細胞の増殖の阻害を示している。本発明の抗体は、ヒト癌細胞中でのHRG誘導性細胞増殖を阻害する。
【図11】図11は、本発明のヒト抗HER−3抗体によって阻害された、MCF7細胞の遊出を示している。
【図12a】図12aからiは、本発明のヒトHER−3抗体による、足場非依存性細胞増殖の阻害を示している。
【図12b】図12aからiは、本発明のヒトHER−3抗体による、足場非依存性細胞増殖の阻害を示している。
【図12c】図12aからiは、本発明のヒトHER−3抗体による、足場非依存性細胞増殖の阻害を示している。
【図12d】図12aからiは、本発明のヒトHER−3抗体による、足場非依存性細胞増殖の阻害を示している。
【図12e】図12aからiは、本発明のヒトHER−3抗体による、足場非依存性細胞増殖の阻害を示している。
【図12f】図12aからiは、本発明のヒトHER−3抗体による、足場非依存性細胞増殖の阻害を示している。
【図12g】図12aからiは、本発明のヒトHER−3抗体による、足場非依存性細胞増殖の阻害を示している。
【図12h】図12aからiは、本発明のヒトHER−3抗体による、足場非依存性細胞増殖の阻害を示している。
【図12i】図12aからiは、本発明のヒトHER−3抗体による、足場非依存性細胞増殖の阻害を示している。
【図13】図13は、本発明のヒト抗HER−3抗体による、T47Dヒト乳癌細胞の異種移植片増殖の阻害を示している。
【図14】図14は、抗Her3(U1−59及びU1−53)又は抗EGFR(Erbitux)抗体の投与後における、マウス中のBxPC3ヒト膵臓癌細胞の低下を示している。
【図15】図15は、本発明のヒト抗HER−3抗体及び抗EGFR(Erbitux)抗体との組み合わせによる、BxPC3ヒト膵臓癌細胞の異種移植片増殖の低下を示している。
【図16】図16は、本発明の抗体が、nu/nuマウス中でのヒト悪性黒色腫(HT144)細胞の増殖を遅延させることを示す。
【図17】図17は、本発明のヒトHER−3抗体(U1−53、U1−59及びU1−7)による、HT−29ヒト大腸癌細胞の異種移植片増殖の低下を示している。
【図18】図18は、本発明のヒト抗HER−3抗体(U1−59、U1−53及びU1−7)抗体による、Calu−3ヒト肺癌細胞の異種移植片増殖の低下を示している。
【図19】図19は、本発明のヒト抗HER−3抗体(U1−7、U1−59及びU1−53)抗体による、BxPC−3ヒト膵臓癌細胞の異種移植片増殖の低下を示している。
【図20】図20は、本発明の抗体(U1−59)が、BxPC3ヒト膵臓癌異種移植片中でHER−3の抑制を引き起こすことを示す。
【発明を実施するための形態】
【0024】
本発明の第一の態様は、HER−3に結合する単離された結合タンパク質に関する。
【0025】
本発明の一実施形態において、本発明の単離された結合タンパク質は、(a)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH1;(b)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH2;並びに(c)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH3からなる群から選択されるCDRの少なくとも1つを含む重鎖アミノ酸配列、及び/又は(d)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL1;(e)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL2;並びに(f)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL3からなる群から選択されるCDRの少なくとも1つを含む軽鎖アミノ酸配列を含む。
【0026】
本発明の別の実施形態において、本発明の単離された結合タンパク質は、配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230からなる群から選択される重鎖アミノ酸配列、及び/又は配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232からなる群から選択される軽鎖アミノ酸配列を含む。
【0027】
本発明のさらに別の実施形態において、本発明の単離された結合タンパク質は、配列番号2及び4、6及び8、10及び12、14及び16、18及び20、22及び24、26及び28、30及び32、36及び38、42及び44、46及び48、50及び52、54及び56、60及び58、62及び64、66及び68、70及び72、74及び76、78及び82、80及び82、84及び86、88及び90、92及び94、96及び98、100及び102、104及び106、108及び110、112及び114、116及び118、122及び124、126及び128、130及び132、134及び136、138及び140、142及び144、146及び148、150及び152、154及び156、158及び160、162及び164、166及び168、170及び172、174及び176、178及び180、182及び184、186及び188、190及び192、194及び196、198及び200、202及び204、206及び208、210及び212、214及び216、218及び220、222及び224、226及び228、230及び232に示されている重鎖アミノ酸配列及び軽鎖アミノ酸配列、又は配列番号34、40、60、62若しくは120に示されている重鎖アミノ酸配列又は配列番号58若しくは64に示されている軽鎖アミノ酸配列を含む。
【0028】
本発明において、本発明の結合タンパク質のアミノ酸配列は、20の一般的なアミノ酸に限定されないことを理解すべきである(Immunology−A Synthesis(2nd Edition, E.S.Golub and D.R.Gren, Eds., Sinauer Associates, Sunderland, Mass.(1991)(参照により、本明細書に組み込まれる。)を参照されたい。)。例えば、アミノ酸には、20の一般的なアミノ酸の立体異性体(例えば、Dアミノ酸)、α,α−二置換されたアミノ酸、N−アルキルアミノ酸、乳酸及び他の非慣用アミノ酸などの非天然アミノ酸が含まれ得る。本発明の結合タンパク質に対する適切な成分でもあり得る非慣用アミノ酸の例には、4−ヒドロキシプロリン、γ−カルボキシグルタミン酸、ε−N,N,N−トリメチルリジン、ε−N−アセチルリジン、O−ホスホセリン、N−アセチルセリン、N−ホルミルメチオニン、3−メチルヒスチジン、5−ヒドロキシリジン、σ−N−メチルアルギニン並びに他の類似のアミノ酸及びイミノ酸(例えば、4−ヒドロキシプロリン)が含まれる。
【0029】
さらに、本発明において、配列番号1から232に示されているアミノ酸配列中の僅かな変動は、本発明によって包含されるものと想定されるが、但し、アミノ酸配列中の変動は、配列番号1から232中に示されている配列の少なくとも75%、より好ましくは少なくとも80%、90%、95%、及び最も好ましくは99%を維持する。変動は、フレームワーク領域内(すなわち、CDR外)、CDR内、又はフレームワーク領域及びCDR内に生じ得る。配列番号1から232に示されているアミノ酸配列中の好ましい変動、すなわち、少なくとも1つのアミノ酸の欠失、挿入及び/又は置換は、機能的ドメインの境界付近に生じる。構造的及び機能的ドメインは、ヌクレオチド及び/又はアミノ酸配列データを、公共の配列データベース又は独自の配列データベースと比較することによって同定することが可能である。公知の構造及び/又は機能の他の結合タンパク質中に生じる配列モチーフ又は予測されるタンパク質立体構造ドメインを同定するために、コンピュータ化された比較法を使用することが可能である。公知の三次元構造に折り畳まれるタンパク質配列を同定するための方法は公知である。例えばBowie et al., Science 253, 164(1991);Proteins, Structures and Molecular Principles(Creighton, Ed., W.H.Freeman and Company, New York(1984));Introduction to Protein Structure(C.Branden and J.Tooze, eds., Garland Publishing, New York, N.Y.(1991 ));及びThornton et al., Nature 354:105(1991)を参照されたい(これらは全て、参照により本明細書中に組み込まれる。)。従って、当業者であれば、本発明に従って構造及び機能的ドメインを定義するために使用され得る配列モチーフ及び構造的立体構造を認めることができる。
【0030】
配列番号1から174及び1から232に示されているアミノ酸配列の特に好ましい変動は、タンパク分解若しくは酸化に対して低下した感受性をもたらし、グリコシル化パターンを変化させ、又は結合親和性を変化させ、又は結合タンパク質の他の物理化学的若しくは機能的特性を付与若しくは修飾する変動である。特に、保存的アミノ酸置換が想定される。保存的な置換とは、それらの側鎖が関連するアミノ酸のファミリー内で生じる置換である。好ましいアミノ酸ファミリーは、以下のとおりである。酸性ファミリー=アスパギン酸、グルタミン酸;塩基性ファミリー=リジン、アルギニン、ヒスチジン;非極性ファミリー=アラニン、バリン、ロイシン、イソロイシン、プロリン、フェニルアラニン、メチオニン、トリプトファン;及び非帯電極性ファミリー=グリシン、アスパラギン、グルタミン、システイン、セリン、スレオニン、チロシン。より好ましいファミリーは、脂肪族ヒドロキシファミリー=セリン及びスレオニン;アミド含有ファミリー=アスパラギン及びグルタミン;脂肪族ファミリー=アラニン、バリン、ロイシン及びイソロイシン;並びに芳香族ファミリー=フェニルアラニン、トリプトファン及びチロシン。例えば、ロイシンのイソロイシン又はバリンとの単離された置換、アスパラギン酸のグルタミン酸との単離された置換、スレオニンのセリンとの単離された置換、又はあるアミノ酸の構造的に関連したアミノ酸との類似の置換は、特に置換がフレームワーク部位内のアミノ酸を含まなければ、生じた結合タンパク質の結合又は特性に対して多大な影響を有しない。しかしながら、全ての他の可能なアミノ酸置換も、本発明によって包含される。アミノ酸の変化が、機能的な結合タンパク質、すなわち、HER−3へ結合する結合タンパク質をもたらし、HERファミリーのメンバーのシグナル伝達を低下させるかどうかは、HER−3への結合に関するELISA若しくはFACSにおいて、又はインビトロ若しくはインビボでの機能的アッセイにおいて、得られた結合タンパク質の特異的活性をアッセイすることによって容易に決定することができる。
【0031】
本発明によれば、本発明の結合タンパク質は、HER−3の細胞外部分中の少なくとも1つのエピトープと相互作用する。エピトープは、好ましくは、アミノ末端ドメインであるドメインL1(アミノ酸19から184)、システインに富む2つのドメインであるドメインS1(アミノ酸185から327)及びS2(アミノ酸500から632)、システインに富む2つのドメインに隣接しているドメインL2(328から499)又はHER−3ドメインの組み合わせの中に位置する。エピトープは、L1及びS1の一部によって構成されるエピトープなど(但し、これに限定されない。)のドメインの組み合わせ中にも位置し得る。さらに、本発明の結合タンパク質は、HER−3へのその結合がHER−3によって媒介されるシグナル伝達を低下させることをさらに特徴とする。本発明に従って、HER−3によって媒介されるシグナル伝達の低下は、例えば、細胞表面からHER−3分子を少なくとも部分的に消失させるHER−3の下方制御によって、又は実質的に不活性な形態(すなわち、安定化されていない形態に比べて、より低いシグナル伝達を示す形態)の、細胞表面上のHER−3の安定化によって引き起こされ得る。あるいは、HER−3によって媒介されるシグナル伝達の低下は、HER−3への、リガンド又はHERファミリーの別のメンバーの結合、HER−2へのGRB2の結合若しくはSHCへのGRB2の結合に影響を与える(例えば、減少又は阻害する)ことによって、受容体チロシンリン酸化、AKTリン酸化、PYK2チロシンリン酸化若しくはERK2リン酸化を阻害することによって、又は腫瘍の浸潤性を減少させることによっても引き起こされ得る。あるいは、HER−3によって媒介されるシグナル伝達の低下は、他のHERファミリーのメンバーとのHER−3含有二量体の形成に影響を与える(例えば、減少又は阻害する)ことによっても引き起こされ得る。とりわけ、1つの例は、HER−3−EGFRタンパク質複合体の形成を減少又は阻害することであり得る。
【0032】
好ましくは、本発明の結合タンパク質は、抗体様結合活性を有する足場タンパク質又は抗体、すなわち、抗HER−3抗体である。
【0033】
本発明の文脈内で、本明細書において使用される「足場タンパク質」という用語は、アミノ酸の挿入、置換又は欠失が高度に許容される露出された表面を有するポリペプチド又はタンパク質を意味する。本発明に従って使用することができる足場タンパク質の例は、スタフィロコッカス・オーレウス(Staphylococcus aureus)から得られるプロテインA、ピエリス・ブラッシッカエ(Pieris brassicae)から得られるビリン結合タンパク質又はその他のリポカリン、アンキリン反復タンパク質及びヒトフィブロネクチンである(Binz and Pluckthun, Curr Opin Biotechnol.16, 459−69中に概説されている。)。足場タンパク質の操作は、安定に折り畳まれたタンパク質の構造的フレームワーク上に又は構造的フレームワーク中に親和性機能を移植し、又は組み込むこととみなすことができる。親和性機能とは、本発明に従うタンパク質結合親和性を意味する。足場は、結合特異性を付与するアミノ酸配列から構造的に分離可能である。一般に、このような人工的親和性試薬の開発に適していると思われるタンパク質は、推論によって、又は最も一般的には、HER3(精製されたタンパク質又は細胞表面上にディスプレイされたタンパク質の何れか)に対するパニングなどの、インビトロでディスプレイされた人工的足場ライブラリーの結合剤に対するコンビナトリアルタンパク質工学技術(これらの技術は、本分野において公知である(Skerra, J.Mol.Recog., 2000;Binz and Pluckthun, 2005))によって取得され得る。さらに、抗体様結合活性を有する足場タンパク質は、足場ドメインを含有するアクセプターポリペプチドに由来することができ、アクセプターポリペプチドを含有する足場ドメイン上にドナーポリペプチドの結合特異性を付与するために、アクセプターポリペプチドには、ドナーポリペプチドの結合ドメインを移植することができる。前記挿入された結合ドメインは、例えば、抗体、特に、抗HER−3抗体の相補性決定領域(CDR)であり得る。挿入は、例えば、当業者に周知の組み換え法の様々な形態による、ポリペプチド合成、コードするアミノ酸の核酸合成など、当業者に公知の様々な方法によって達成することが可能である。
【0034】
さらに、本明細書において使用される「抗体」又は「抗HER−3抗体」という用語は、モノクローナル抗体、ポリクローナル抗体、組換え抗体、ヒト化抗体(Jones et al., Nature 321(1986),522−525;Riechmann et al., Nature 332(1988), 323−329;and Presta, Curr.Op.Struct.Biol.2(1992),593−596)、キメラ抗体(Morrison et al.,Proc.Natl.Acad.Sci.U.S.A.81(1984),6851−6855)、少なくとも2つの抗体から形成された多重特異的抗体(例えば、二重特異的抗体)又はこれらの抗体断片を意味する。「抗体断片」という用語は、上記抗体のあらゆる一部、好ましくはそれらの抗原結合領域又は可変領域を含む。抗体断片の例には、Fab断片、Fab’断片、F(ab’)2断片、Fv断片、ダイアボディ(Hollinger et al., Proc.Natl.Acad.Sci.U.S.A.90(1993),6444−6448)、一本鎖抗体分子(Pluckthun in:The Pharmacology of Monoclonal Antibodies 113, Rosenburg and Moore, EDS, Springer Verlag,N.Y.(1994),269−315)及びHER−3への所望の結合能を示す限り他の断片が含まれる。
【0035】
さらに、本明細書において使用される「抗体」又は「抗HER−3抗体」という用語は、抗体の加工されたサブドメインを含有する抗体様分子又は天然に存在する抗体変種を含み得る。これらの抗体様分子は、ラクダ科の動物(camelid)などの天然の取得源から得られた(Muyldermans et al.,Reviews in Molecular Biotechnology 74,277−302)、又は、ヒト、ラクダ科の動物若しくはその他の種から得たライブラリーのインビトロディスプレーを通じて得られた(Holt et al.,Trends Biotechnol.,21,484−90)VHのみ又はVLのみのドメインなどの単一ドメイン抗体であり得る。
【0036】
本発明において、「Fv断片」は、完全な抗原認識及び結合部位を含有する最低の抗体断片である。これらの領域は、緊密に非共有会合した1つの重鎖可変ドメインと1つの軽鎖可変ドメインの二量体からなる。各可変ドメインの3つのCDRが、VH−VL二量体の表面上の抗原結合部位を規定するために相互作用するのは、この立体配置中である。一括して、6つのCDRが抗体への抗原結合特異性を付与する。しかしながら、単一の可変ドメイン(又は抗原に対して特異的な3つのCDRのみを含むFvの半分)でさえ、一般に、完全な結合部位より低い親和性であるが、抗原を認識及び結合する能力を有する。「Fab断片」は、軽鎖の定常ドメイン及び重鎖の第一の定常ドメイン(CH1)も含有する。「Fab断片」は、重鎖CH1ドメインのカルボキシ末端に、抗体ヒンジ領域由来の1つ又はそれ以上のシステインなど、数個の残基が付加されている点で、「Fab’断片」と異なる。「F(ab’)2断片」は、当初、ヒンジシステインをその間に有する「Fab’断片」の対として作製される。パパイン又はペプシン消化など、このような抗体断片を調製する方法は、当業者に公知である。
【0037】
本発明の好ましい実施形態において、本発明の抗HER−3抗体は、IgA−、IgD、IgE、IgG又はIgM型、好ましくは、IgG1、IgG2、IgG3、IgG4、IgM1及びIgM2型など(但し、これらに限定されない。)のIgG又はIgM型のものである。最も好ましい実施形態において、抗体は、IgG1、IgG2又はIgG4種である。
【0038】
本発明の別の好ましい実施形態において、本発明の抗HER−3抗体は、HER−3の細胞外ドメイン(ECD)に対して誘導された抗HER−3抗体である。
【0039】
ある観点において、例えば、治療候補物としてHER−3に対する抗体を作製することに関して、本発明の抗HER−3抗体は、補体を固定し、補体依存性細胞傷害(CDC)に関与できることが望ましい場合があり得る。マウスIgM、マウスIgG2a、マウスIgG2b、マウスIgG3、ヒトIgM、ヒトIgG1、ヒトIgG3及びヒトIgAなど(但し、これらに限定されない。)、補体の固定及び補体依存性細胞傷害(CDC)への関与が可能な抗体のイソタイプが多数存在する。作製される抗体は、このようなイソタイプを最初から有する必要はなく、むしろ、作製された抗体は、あらゆるイソタイプを有することができること、並びに、本分野において周知である慣用の分子生物学的技術を用いて、適切な発現ベクター中に分子クローニングされた定常領域遺伝子又はcDNAへ、分子クローニングされたV領域遺伝子又はcDNAを付加し、次いで、本分野で公知の技術を用いて宿主細胞中で抗体を発現させることによって抗体をイソタイプ交換できることが理解される。イソタイプ交換された抗体は、天然に存在する変種に比べて優れたCDCを有するように分子的に加工され(Idusogie et al., J Immunol.,166,2571−2575)、本分野で公知の技術を用いて、宿主細胞中で組換え的に発現されたFc領域も有し得る。このような技術には、とりわけ、直接的な組換え技術(例えば、U.S.Patent No.4,816,397を参照。)、細胞−細胞融合技術(例えば、U.S.Patent Nos.5,916,771及び6,207,418)の使用が含まれる。細胞−細胞融合技術では、何れかの所望のイソタイプを有する重鎖を保有する骨髄腫又は他の細胞株(CHOなど)が調製され、及び軽鎖を保有する別の骨髄腫又は他の細胞株(CHOなど)が調製される。その後、このような細胞を融合し、完全な状態の抗体を発現する細胞株を単離することができる。例として、(抗体の特異性及び抗体の親和性の幾つかを規定する)同じ可変領域を有しながら、ヒトIgM、ヒトIgG1又はヒトIgG3イソタイプを作製するために、HER−3抗原への所望の結合を有するヒト抗HER−3IgG4抗体を容易にイソタイプ交換させ得る。その後、このような分子は、補体を固定し、CDCへ関与することが可能であり得る。
【0040】
さらに、本発明の抗HER−3抗体は、単球及びナチュラルキラー(NK)細胞などのエフェクター細胞上のFc受容体へ結合することができ、抗体依存性細胞傷害(ADCC)に関与することも望ましい場合があり得る。マウスIgG2a、マウスIgG2b、マウスIgG3、ヒトIgG1及びヒトIgG3など(但し、これらに限定されない。)、上記のことが可能な抗体のイソタイプが多数存在する。作製される抗体は、このようなイソタイプを最初から有する必要はなく、むしろ、作製された抗体は、あらゆるイソタイプを有することができること、並びに、本分野において周知である慣用の分子生物学的技術を用いて、適切な発現ベクター中に分子クローニングされた定常領域遺伝子又はcDNAへ、分子クローニングされたV領域遺伝子又はcDNAを付加した後、本分野で公知の技術を用いて抗体を宿主細胞中で発現させることによって、抗体のイソタイプ交換を実施できることが理解される。イソタイプ交換された抗体は、天然に存在する変種に比べて優れたADCCを有するように分子的に加工され(Shields et al.J Biol Chem.,276,6591−6604)、本分野で公知の技術を用いて、宿主細胞中で組換え的に発現されたFc領域も有し得る。このような技術には、とりわけ、直接的な組換え技術(例えば、U.S.Patent No.4,816,397を参照。)、細胞−細胞融合技術(例えば、U.S.Patent Nos.5,916,771及び6,207,418を参照。)の使用が含まれる。細胞−細胞融合技術では、何れかの所望のイソタイプを有する重鎖を保有する骨髄腫又は他の細胞株(CHOなど)が調製され、及び軽鎖を保有する別の骨髄腫又は他の細胞株(CHOなど)が調製される。その後、このような細胞を融合し、完全な状態の抗体を発現する細胞株を単離することができる。例として、(抗体の特異性及び抗体の親和性の幾つかを規定する)同じ可変領域を有しながら、ヒトIgG1又はヒトIgG3イソタイプを作製するために、HER−3抗原への所望の結合を有するヒト抗HER−3IgG4抗体を容易にイソタイプ交換させ得る。次いで、このような分子は、エフェクター細胞上のFcγRへ結合し、ADCCに関与することが可能であり得る。
【0041】
さらに、本発明によれば、本発明の抗HER−3抗体は、完全なヒト抗体又はヒト化抗体であることが理解される。ヒト抗体は、異種抗体、例えば、マウス又はラットの可変及び/又は定常領域を有する抗体に伴う問題の幾つかを回避する。マウス又はラットに由来するタンパク質など、異種由来のタンパク質の存在は、患者による、抗体に対する免疫反応を惹起させることができ、続いて、抗体の迅速な除去、抗体の中和を通じた治療的有用性の喪失及び/又は重篤な、さらには生命を脅かすアレルギー反応をもたらし得る。
【0042】
好ましくは、本発明の抗HER−3抗体は、U1−1抗体、U1−2抗体、U1−3抗体、U1−4抗体、U1−5抗体、U1−6抗体、U1−7抗体、U1−8抗体、U1−9抗体、U1−10抗体、U1−11抗体、U1−12抗体、U1−13抗体、U1−14抗体、U1−15抗体、U1−16抗体、U1−17抗体、U1−18抗体、U1−19抗体、U1−20抗体、U1−21抗体、U1−22抗体、U1−23抗体、U1−24抗体、U1−25抗体、U1−26抗体、U1−27抗体、U1−28抗体、U1−29抗体、U1−30抗体、U1−31抗体、U1−32抗体、U1−33抗体、U1−34抗体、U1−35抗体、U1−36抗体、U1−37抗体、U1−38抗体、U1−39抗体、U1−40抗体、U1−41抗体、U1−42抗体、U1−43抗体、U1−44抗体、U1−45抗体、U1−46抗体、U1−47抗体、U1−48抗体、U1−49抗体、U1−50抗体、U1−51抗体、U1−52抗体、U1−53抗体、U1−55.1抗体、U1−55抗体、U1−57.1抗体、U1−57抗体、U1−58抗体、U1−59抗体、U1−61.1抗体、U1−61抗体及びU1−62からなる群から選択される。
【0043】
本発明の好ましい実施形態において、本発明の結合タンパク質は、標識基に連結される。このような結合タンパク質は、特に、診断用途に適している。本明細書において使用される、「標識基」という用語は、検出可能なマーカー、例えば、放射性標識されたアミノ酸又は印を付けたアビジン(例えば、光学的方法又は比色分析法によって検出することができる、蛍光マーカー又は酵素活性に結合されたストレプトアビジン)によって検出することができるビオチン部分を表す。抗体などのポリペプチド及び糖タンパク質を標識するための様々な方法が本分野において公知であり、本発明を実施する際に使用され得る。適切な標識基の例には、以下の放射性同位体又は放射性核種(例えば、3H、14C、15N、35S、90Y、99Tc、111In、125I、131I)、蛍光基(例えば、FITC、ローダミン、ランタニドリン光体)、酵素基(例えば、西洋ワサビペルオキシダーゼ、β−ガラクトシダーゼ、ルシフェラーゼ、アルカリホスファターゼ)、化学発光基、ビオチン基又は二次レポーターによって認識される所定のポリペプチドエピトープ(例えば、ロイシンジッパー対配列、二次抗体に対する結合部位、金属結合ドメイン、エピトープタグ)が含まれるが、これらに限定されない。ある観点において、立体的障害の可能性を減らすために、標識基には、様々な長さのスペーサーアームを付着させることが望ましい場合があり得る。
【0044】
あるいは、本発明の別の好ましい実施形態において、本発明の結合タンパク質は、エフェクター基に連結され得る。このような結合タンパク質は、特に、治療用途に適している。本明細書において使用される「エフェクター基」という用語は、放射線同位体若しくは放射線核種、毒素、治療基又は本分野において公知の他のエフェクター基などの細胞毒性基を表す。適切なエフェクター基の例は、放射線同位体又は放射線核種(例えば、3H、14C、15N、35S、90Y、99Tc、111In、125I、131I)、カリチアマイシン、アウリスタチンなどのドラスタチン類縁体並びにゲルダナマイシン及びマイタンシン誘導体(DM1を含む。)などの化学療法剤である。ある観点において、立体的障害の可能性を減らすために、エフェクター基には、様々な長さのスペーサーアームを付着させることが望ましい場合があり得る。
【0045】
本発明の第二の態様は、結合タンパク質を分泌する宿主細胞から結合タンパク質を調製する工程を含む、本発明の単離された結合タンパク質を調製する方法に関する。本発明に従って使用され得る宿主細胞は、ハイブリドーマ;哺乳動物細胞(例えば、ハムスター、ウサギ、ラット、ブタ、マウス又はその他の動物細胞)、植物細胞、真菌細胞(例えば、サッカロミセス・セレビシアエ(Saccharomyces cerevisiae)、ピチア・パストリス(Pichia pastoris))などの真核細胞;イー・コリ(E.coli)などの原核細胞及び結合タンパク質の産生のために本分野で使用される他の細胞である。足場タンパク質又は抗体などの結合タンパク質を宿主細胞から調製及び単離するための様々な方法が本分野において公知であり、本発明を実施する際に使用され得る。さらに、パパイン又はペプシン消化、近代的なクローニング技術、一本鎖抗体分子を調製するための技術(Pluckthun in:The Pharmacology of Monoclonal Antibodies 113, Rosenburg and Moore, EDS, Springer Veriag, N.Y.(1994), 269−315)及びダイアボディ(Hollinger et al., Proc.Natl.Acad.Sci.U.S.A.90(1993),6444−6448)など、結合タンパク質断片(例えば、足場タンパク質断片又は抗体断片)を調製するための方法も当業者に公知であり、本発明を実施する際に使用し得る。
【0046】
本発明の好ましい実施形態において、本発明の結合タンパク質は、結合タンパク質を分泌するハイブリドーマから調製される。例えば、「Kohler et al., Nature 256(1975),495」を参照されたい。
【0047】
本発明のさらなる好ましい実施形態において、本発明の結合タンパク質は、宿主細胞中での結合タンパク質の発現を最適化及び/又は増幅し、前記宿主細胞からの結合タンパク質を単離することによって、組み換え的に調製される。この目的のために、結合タンパク質をコードするDNAで、又は結合タンパク質をコードするDNAを含有するベクターで宿主細胞を形質転換又は形質移入し、本発明の結合タンパク質を産生するのに適した条件下で培養する。例えば、U.S.Patent No.4,816,567を参照されたい。好ましい宿主細胞は、CHO細胞、NS/0骨髄腫細胞、ヒト胎児性腎臓293細胞、イー・コリ及びサッカロミセス・セレビシアエであり得る。
【0048】
抗体である結合タンパク質に関して、これらの抗体は、完全なヒト抗体を作製するように遺伝子操作された動物から、又はバクテリオファージ、酵母、リボソーム又はイー・コリ中に作製された抗体ディスプレイライブラリーから調製され得る。例えば、Clackson et al., Nature 352(1991), 624−628, Marks et al., J.Mol.Biol.222(1991), 581−597, Feldhaus and Siegel J Immunol Methods.290, 69−80, Groves and Osbourn, Expert Opin Biol Ther., 5, 125−135 and Jostock and Dubel, Comb Chem High Throughput Screen.8, 127−133を参照されたい。
【0049】
ヒト抗体は、マウス又はラットの可変及び/又は定常領域を有する抗体に伴う問題の幾つかを回避する。このようなマウス又はラット由来のタンパク質の存在は、抗体の急速な除去をもたらすことができ、又は、患者による、抗体に対する免疫応答の生成をもたらすことができる。マウス又はラット由来の抗体の使用を回避するために、げっ歯類、他の哺乳動物又は動物が完全なヒト抗体を産生するように、げっ歯類、他の哺乳動物又は動物中への機能的なヒト抗体遺伝子坐の導入を通じて、完全なヒト抗体を作製することが可能である。
【0050】
完全なヒト抗体を作製するための1つの方法は、ヒト重鎖遺伝子坐及びκ軽鎖遺伝子坐の245kb及び190kbサイズの生殖系列配置断片を含有するように操作されたマウスのXENOMOUSE(R)系統の使用を通じた方法である。マウスの他のXenoMouse系統は、ヒト重鎖遺伝子坐及びκ軽鎖遺伝子坐の980kb及び800kbサイズの生殖系列配置断片を含有する。マウスのさらに他のXenoMouse系統は、ヒト重鎖遺伝子坐及びκ軽鎖遺伝子坐の980kb及び800kbサイズの生殖系列配置断片に加えて、740kbのサイズの生殖系列構成された完全なヒトλ軽鎖遺伝子坐を含有する。「Mendez et al.Nature Genetics 15:146−156(1997)」及び「Green and Jakobovits J.Exp.Med.188:483−495(1998)」を参照されたい。XENOMOUSE(R)系統は、Abgenix,Inc.(Fremont,CA)から入手可能である。
【0051】
XENOMOUSE(R)マウスの作製は、1990年1月12日に出願されたU.S.Patent Application Serial Nos.07/466,008、1990年11月8日に出願された07/610,515、1992年7月24日に出願された07/919,297、1992年7月30日に出願された07/922,649、1993年3月15日に出願された08/031,801、1993年8月27日に出願された08/112,848、1994年4月28日に出願された08/234,145、1995年1月20日に出願された08/376,279、1995年4月27日に出願された08/430,938、1995年6月5日に出願された08/464,584、1995年6月5日に出願された08/464,582、1995年6月5日に出願された08/463,191、1995年6月5日に出願された08/462,837、1995年6月5日に出願された08/486,853、1995年6月5日に出願された08/486,857、1995年6月5日に出願された08/486,859、1995年6月5日に出願された08/462,513、1996年10月2日に出願された08/724,752及び1996年12月3日に出願された08/759,620、2002年11月20日に出願されたU.S.Patent Publication 2003/0217373並びにU.S.Patent Nos.6,833,268、6,162,963、6,150,584、6,114,598、6,075,181及び5,939,598並びにJapanese Patent Nos.3 068 180 B2、3 068 506 B2及び3 068 507 B2に、さらに論述及び描写されている。1996年6月12日に公開されたEuropean Patent No., EP 0 463 151 B1、1994年2月3日に公開されたInternational Patent Application No., WO 94/02602、1996年10月31日に公開されたInternational Patent Application No., WO 96/34096、1998年6月11日に公開されたWO98/24893、2000年12月21日公開されたWO00/76310も参照されたい。上記特許、出願及び参考文献の各々の開示の全体が、参照により、本明細書に組み込まれる。
【0052】
別のアプローチにおいて、GenPharm International,Inc.,などの他の者は、「ミニローカス」アプローチを使用してきた。ミニローカスアプローチにおいて、Ig遺伝子坐から得た片(各遺伝子)を含めることによって、外来Ig遺伝子坐が模倣される。従って、1つ又はそれ以上のVH遺伝子、1つ又はそれ以上のDH遺伝子、1つ又はそれ以上のJH遺伝子、μ定常領域及び第二の定常領域(好ましくは、γ定常領域)が、動物内への挿入のために、構築物の中に形成される。本アプローチは、Suraniらに対するU.S.Patent No.5,545,807及びそれぞれ、LonbergとKayに対するU.S.Patent Nos.5,545,806、5,625,825、5,625,126、5,633,425、5,661,016、5,770,429、5,789,650、5,814,318、5,877,397、5,874,299及び6,255,458、Krimpenfort及びBernsに対するU.S.Patent No.5,591,669及び6,023.010、Bernsらに対するU.S.Patent Nos.5,612,205、5,721,367及び5,789,215並びにChoi及びDunnに対するU.S.Patent No.5,643,763、並びに1990年8月29日に出願されたGenPharm International U.S.Patent Application Serial Nos.07/574,748、1990年8月31日に出願された07/575,962、1991年12月17日に出願された07/810,279、1992年3月18日に出願された07/853,408、1992年6月23日に出願された07/904,068、1992年12月16日に出願された07/990,860、1993年4月26日に出願された08/053,131、1993年7月22日に出願された08/096,762、1993年11月18日に出願された08/155,301、1993年12月3日に出願された08/161,739、1993年12月10日に出願された08/165,699、1994年3月9日に出願された08/209,741(これらの開示内容は、参照により、本明細書に組み込まれる。)に記載されている。European Patent No.0 546 073 B1、International Patent Application Nos.WO92/03918、WO92/22645、WO92/22647、WO92/22670、WO93/12227、WO94/00569、WO94/25585、WO96/14436、WO97/13852及びWO 98/24884並びにU.S.Patent No.5,981,175(これらの開示内容全体が、参照により、本明細書に組み込まれる。)も参照されたい。さらに、Taylor et al., 1992、Chen et al., 1993、Tuaillon et al., 1993、Choi et al., 1993、Lonberg et al.,(1994)、Taylor et al.,(1994)及びTuaillon et al.,(1995)、Fishwild et al.,(1996)(これらの開示内容全体が、参照により、本明細書に組み込まれる。)を参照されたい。
【0053】
Kirinは、ミクロセル融合を通じて、染色体の巨大片又は完全な染色体が導入されたマウスからのヒト抗体の作製も示した。European Patent Application Nos.773 288及び843 961(これらの開示内容全体が、参照により、本明細書に組み込まれる。)を参照されたい。さらに、KirinのTcマウスとMedarexのミニローカス(Humab)マウスの交雑から得られるKMTMマウスが作製されている。これらのマウスは、KirinマウスのHC導入染色体(transchromosome)とMedarexマウスのκ鎖導入遺伝子を有する(Ishida et al., Cloning Stem Cells,(2002) 4:91−102)。
【0054】
ヒト抗体は、インビトロ法によって得ることもできる。適切な例には、ファージディスプレイ(Cambridge Antibody Technology、Morphosys、Dyax、Biosite/Medarex、Xoma、Symphogen、Alexion(旧Proliferon)、Affimedによって市販されている。)、リボソームディスプレイ(Cambridge Antibody Technologyによって市販されている。)、酵母ディスプレイなどが含まれるが、これらに限定されない。
【0055】
抗体は、本明細書に記載されているように、以下に記載されているとおりにXENOMOUSE(R)技術を使用することによって調製された。その後、このようなマウスは、ヒト免疫グロブリン分子及び抗体を産生することができ、マウス免疫グロブリン分子及び抗体の産生を欠失している。これを達成するために使用された技術は、本明細書の背景技術中に開示されている特許、出願及び参考文献中に開示されている。しかしながら、とりわけ、マウス及びマウスから得られる抗体のトランスジェニック産生の好ましい実施形態は、1996年12月3日に出願されたU.S.Patent Application Serial No.08/759,620及び1998年6月11日に公開されたInternational Patent Application Nos.WO 98/24893及び2000年12月21日に公開されたWO00/76310(これらの開示内容は、参照により、本明細書に組み込まれる。)に開示されている。「Mendez et al.Nature Genetics 15:146−156(1997)」(本開示内容は、参照により、本明細書に組み込まれる。)も参照されたい。
【0056】
このような技術の使用を通じて、様々な抗原に対する完全なヒトモノクローナル抗体が作製されてきた。基本的には、マウスのXENOMOUSE(R)株を目的の抗原(例えば、HER−3)で免疫し、抗体を発現したマウスから(B細胞などの)リンパ系細胞を回収し、不死化ハイブリドーマ細胞株を調製するために、回収された細胞株を骨髄球型細胞株と融合する。目的の抗原に対して特異的な抗体を産生するハイブリドーマ細胞株を同定するために、これらのハイブリドーマ細胞株をスクリーニングし、選択する。本明細書には、HER−3に対して特異的な抗体を産生する複数のハイブリドーマ細胞株を作製するための方法が提供されている。さらに、本明細書において、このような抗体の重鎖及び軽鎖のヌクレオチド及びアミノ酸配列分析など、このような細胞株によって産生された抗体の性質決定が提供される。
【0057】
一般に、融合されたハイブリドーマによって産生される抗体は、完全なヒトκ軽鎖を有するヒトIgG1重鎖であった。本明細書に記載されている抗体は、ヒトIgG4重鎖及びIgG1重鎖を有する。抗体は、IgG2又はIgG3など、他のヒトイソタイプとすることもできる。抗体は、高い親和性を有し、典型的には、固相及び細胞ベースの技術によって測定された場合に、約10−6から約10−13M又はそれ以下のKDを有する。
【0058】
本発明の別の態様は、本発明の結合タンパク質をコードする単離された核酸分子に関する。本発明の文脈の中で、本明細書で使用される「単離された核酸分子」という用語は、ゲノム、cDNA若しくは合成起源又はこれらの幾つかの組み合わせのポリヌクレオチドを意味し、その起源に基づいて、「単離されたポリヌクレオチド」は、(1)「単離されたポリヌクレオチド」が本来その中に見出されるポリヌクレオチドの全部又は一部と会合していない、(2)本来連結されていないポリヌクレオチドに作用可能に連結されている、又は(3)より大きな配列の一部として本来存在しない。さらに本明細書において使用される「核酸分子」という用語は、少なくとも10塩基長のヌクレオチド(リボヌクレオチド若しくはデオキシヌクレオチドの何れか、又は修飾された若しくは置換された糖基を有するヌクレオチドなど、ヌクレオチドの何れかのタイプの修飾された形態)のポリマー形態を意味する。本用語は、DNAの一本鎖及び二本鎖形態も含む。
【0059】
本発明の一実施形態において、本発明の核酸分子は、調節配列へ作用可能に連結されている。本明細書において使用される「調節配列」という用語は、これらが連結されているコード配列の発現及びプロセッシングに影響を与えるのに必要なポリヌクレオチド配列を表す。このような調節配列の性質は、宿主生物に応じて異なる。原核生物では、このような調節配列には、一般に、プロモーター、リボソーム結合部位及び転写終結配列が含まれる。真核生物では、このような調節配列には、一般に、プロモーター及び転写終結配列が含まれる。本発明において、「調節配列」という用語は、最小限、その存在が発現及びプロセッシングに不可欠である全ての成分を含むものとし、その存在が有利である追加の成分、例えば、リーダー配列及び融合対配列も含むことが可能である。さらに、本明細書において使用される「作用可能に連結された」という用語は、所期の様式でそれらを機能させることができる関係にある、このように記載された成分の位置を表す。さらに、本発明によれば、コード配列に対して作用可能に連結された発現調節配列は、発現調節配列と適合的な条件下で、コード配列の発現が達成されるように連結されている。
【0060】
本発明のさらなる態様は、本発明の結合タンパク質をコードする核酸分子を含むベクターである。核酸分子は、調節配列に、作用可能に連結することができる。さらに、ベクターは、複製起点又は選択マーカー遺伝子をさらに含有し得る。本発明に従って使用され得るベクターの例は、例えば、プラスミド、コスミド、ファージ、ウイルスなどである。
【0061】
本発明の別の態様は、本発明の核酸分子又はベクターで形質転換された宿主細胞に関する。形質転換は、宿主細胞中にポリヌクレオチドを導入するためのあらゆる公知の方法によって、例えば、ウイルス中への(又はウイルスベクター中への)ポリヌクレオチドのパッケージング及びウイルス(又はベクター)での宿主細胞の形質導入、又は、U.S.Patent Nos.4,399,216、4,912,040、4,740,461及び4,959,455(これらの特許は、参照により、本明細書中に組み込まれる。)を例とする本分野で公知の形質移入操作によって実施され得る。特に、哺乳動物細胞中に異種のポリヌクレオチドを導入するための方法が本分野において周知であり、デキストランによって媒介される形質移入、リン酸カルシウム沈殿、ポリブレンによって媒介される形質移入、プロトプラスト融合、電気穿孔、リポソーム中へのポリヌクレオチドの封入及び核内へのDNAの直接的微少注入が含まれる。本発明に従って使用され得る宿主細胞の例は、哺乳動物細胞(例えば、ハムスター、ウサギ、ラット、ブタ、マウス又はその他の動物細胞)などのハイブリドーマ真核細胞、、植物細胞及び真菌細胞(例えば、トウモロコシ、タバコ、サッカロミセス・セレビシアエ(Saccharomyces cerevisiae)、ピチア・パストリス(Pichia pastoris));イー・コリ(E.coli)などの原核細胞及び抗体の産生のために本分野で使用される他の細胞である。特に、発現のための宿主として利用可能な哺乳動物細胞株が本分野において周知であり、チャイニーズハムスター卵巣(CHO)細胞、HeLa細胞、ベビーハムスター腎臓(BHK)細胞、サル腎臓細胞(COS)、ヒト肝細胞癌細胞(例えば、HepG2)及び多数の他の細胞株など(但し、これらに限定されない。)、アメリカン・タイプ・カルチャー・コレクション(ATCC)から入手可能な多くの不死化された細胞株が含まれる。
【0062】
本発明のさらに別の態様は、活性因子としての本発明の少なくとも1つの結合タンパク質並びに医薬として許容される担体、希釈剤及び/又は佐剤を含む医薬組成物である。本明細書において使用される「医薬組成物」という用語は、患者に適切に投与された場合に、所望の治療効果を誘導することができる化学的化合物又は組成物を表す(McGraw−Hill Dictionary of Chemical Terms(Parker, S., Ed., McGraw−Hill, San Francisco(1985)、参照により、本明細書に組み込まれる。)。本発明において、本発明の医薬組成物の効力は、HER−3への少なくとも1つの結合タンパク質の結合を基礎とする。好ましくは、この結合は、HER−3によって媒介されるシグナル伝達の低下をもたらす。
【0063】
さらに、本明細書において使用される場合、「担体」という用語には、使用される用量及び濃度で、担体に曝露されている細胞又は哺乳動物に対して無毒である担体、賦形剤又は安定化剤が含まれる。しばしば、生理的に許容される担体は、pH緩衝化された水溶液又はリポソーム(哺乳動物に薬物を送達するのに有用である、脂質、リン脂質及び/又は界面活性剤の様々な種類から構成される小胞)である。生理的に許容される担体の例には、ホスファート、シトラート及びその他の有機酸などの緩衝剤;アスコルビン酸などの抗酸化剤;低分子量(約10残基未満の)ポリペプチド;血清アルブミン、ゼラチン若しくは免疫グロブリンなどのタンパク質;ポリビニルピロリドンなどの親水性ポリマー;グリシン、グルタミン、アスパラギン、アルギニン若しくはリジンなどのアミノ酸;グルコース、マンノース又はデキストリンなどの単糖、二糖及び他の炭水化物;EDTAなどのキレート剤;マニトール若しくはソルビトールなどの糖アルコール;ナトリウムなどの塩形成対イオン;及び/又はTWEENTM、ポリエチレングリコール(PEG)及びPLURONICSTMなどの非イオン性界面活性剤が含まれる。
【0064】
本発明の一実施形態において、医薬組成物中に含有される本発明の少なくとも1つの結合タンパク質は、カルケアマイシン、アウリスタチン−PE、放射性同位体又はゲルダナマイシン及びマイタンシンなどの毒性を有する化学療法剤などのエフェクターに連結される。特に、これらの結合タンパク質抱合体は、除去のために、HER−3を発現している細胞(例えば、癌細胞)を標的とする上で有用である。
【0065】
さらに、例えば、放射性同位体への本発明の結合タンパク質の連結は、腫瘍治療に対して利点を与える。化学療法及び癌治療の他の形態とは異なり、放射線免疫療法又は放射性同位体−結合タンパク質の組み合わせの投与は、周囲の正常で、健康な組織に対する損傷を最小限に抑えながら、癌細胞を直接標的とする。この「魔法の弾丸」によって、患者は、今日利用可能な治療の他の形態より、放射線同位体のずっと少ない量で治療することができる。好ましい放射性同位体には、イットリウム90(90Y)、インジウム111(111In)、131I、99mTc、放射性銀−111、放射性銀−199及びビスマス213が含まれる。本発明の結合タンパク質への放射性同位体の連結は、例えば、慣用の二官能性キレートを用いて実施され得る。銀は一価であるので、放射性銀−111及び放射性銀−199の連結に関しては、硫黄をベースとするリンカーを使用し得る(Hazra et al., Cell Biophys.24−25, 1−7(1994))。銀放射性同位体の連結は、アスコルビン酸による免疫グロブリンの還元を伴い得る。さらに、チウキセタンは、イブリツモマブに結合されて、イブリツモマブ・チウキセタン(Zevalin)を形成するMX−DTPAリンカーキレート剤である(Witzig, T.E, Cancer Chemother.Pharmacol.48 Suppl 1 , 91−5(2001)。イブリツモマブ・チウキセタンは、インジウム111(111In)又は90Yなどの放射性同位体と反応して、それぞれ、111In−イブリツモマブ・チウキセタン及び90Y−イブリツモマブ・チウキセタンを形成することができる。
【0066】
さらに、本発明の結合タンパク質は、特に、癌を治療するのに使用される場合に、カリチアマイシン(Hamann et al., Bioconjug.Chem.13(1),40−6(2002)、ゲルダナマイシン(Mandler et al., J.Natl.Cancer Inst.,92(19),1549−51(2000))及びメイタンシン、例えば、メイタンシノイド薬DM1(Liu et al., Proc.Natl.Acad.Sci.U.S.A.93:8618−8623(1996))などの毒性を有する化学療法剤と連結され得る。酸性もしくは還元条件下で、又は特異的なプロテアーゼへの曝露時に薬物を放出する様々なリンカーを、本技術とともに使用され得る。本発明によれば、本発明の結合タンパク質は、本分野において記載されているとおりに連結され得る。
【0067】
アウリスタチン−PE、例えば、海洋性無殻軟体動物のペプチド構成成分であるドラスタチン10の構造的な修飾物である抗微小管因子である。アウリスタチン−PEは、抗腫瘍活性及び抗腫瘍血管活性を併有する(Otani et al., Jpn.J.Cancer Res.91(8), 837−44(2000))。例えば、膵臓癌細胞株において、アウリスタチン−PEは細胞増殖を阻害し、細胞周期の停止とアポトーシスを誘導する(Li et al., Int.J.Mol.Med.3(6),647−53(1999))。従って、アウリスタチン−PEの抗腫瘍活性と抗腫瘍血管活性を特定の腫瘍に対して特異的に標的化するために、アウリスタチン−PEは、本発明の結合タンパク質へ連結され得る。
【0068】
本発明の一実施形態において、医薬組成物は、少なくとも1つのさらなる活性因子を含む。本発明に従って使用され得るさらなる活性因子に対する例は、抗体又はEGFR、HER−2、HER−4、IGFR−1若しくはc−metなどの他の受容体タンパク質キナーゼの低分子量阻害剤、血管内皮因子(VEGF)などの受容体リガンド、ドキソルビシン、シス−プラチン若しくはカルボプラチンなどの細胞毒性因子、サイトカイン又は抗新生物因子である。現在、多くの抗新生物因子が当分野で公知である。一実施形態において、抗新生物因子は、抗体又は免疫調節タンパク質を含む(但し、これらに限定されない。)治療用タンパク質の群から選択される。別の実施形態において、抗新生物因子は、有糸分裂阻害剤、キナーゼ阻害剤、アルキル化剤、代謝抑制剤、インターカレート抗生物質、成長因子阻害剤、細胞周期阻害剤、酵素、トポイソメラーゼ阻害剤、ヒストンデアセチラーゼ阻害剤、抗生存因子、生物応答修飾物質、抗ホルモン、例えば、抗アンドロゲン及び抗血管新生因子からなる小分子阻害剤又は化学療法剤の群から選択される。抗新生物因子が放射線照射である場合には、治療は、体内密封小線源治療(brachytherapyBT)又は体外(体外光線照射療法:EBRT)源のいずれかを用いて治療を達成することができる。
【0069】
本発明の医薬組成物は、過剰増殖性疾患の診断、予防又は治療に特に適している。過剰増殖性疾患は、例えば、増加されたHERファミリーシグナル伝達と関連し得る。特に、疾患は、増加されたHER−3リン酸化及び/又はHER−3とHERファミリーの他のメンバーとの間での増加された複合体形成及び/又は増加されたPI3キナーゼ活性及び/又は増加されたc−jun末端キナーゼ活性及び/又AKT活性及び/又は増加されたERK2活性及び/又はPYK2活性と関連することができる。好ましくは、過剰増殖性疾患は、乳癌、胃腸癌、膵臓癌、前立腺癌、卵巣癌、胃癌、子宮内膜癌、唾液腺癌、肺癌、腎臓癌、大腸癌、直腸結腸癌、甲状腺癌、膀胱癌、神経膠腫、悪性黒色腫又はHER−3を発現若しくは過剰発現する他の癌及び腫瘍転移の形成からなる群から選択される。
【0070】
本発明において、本明細書で使用される「予防又は治療」という用語は、治療的な処置及び予防的若しくは防止的措置の両方を表し、必要とする患者は、標的とされる病的症状又は疾患を予防又は遅延(軽減)することである。予防又は治療を必要とする者には、疾患を既に有する者及び疾患を有する傾向がある者又は疾患が予防されるべき者が含まれる。予防又は治療を必要とする患者は、哺乳動物患者、すなわち、ヒト、イヌ、ネコ、ウシ、ウマ、ヒツジ、ブタ、ヤギ、ウサギなどの家畜及び農場動物、並びに動物園、スポーツ又はペット動物を含む哺乳動物として分類される全ての動物である。好ましくは、治療を必要とする患者は、ヒト患者である。
【0071】
本発明によれば、本発明の医薬組成物は、活性因子を、生理的に許容される担体、希釈剤及び/又は佐剤、並びに、改善された転送、送達、耐性などを付与するために、製剤中に一般的に取り込まれる、場合によって使用される他の因子と混合することによって製剤化され得る。本発明の医薬組成物は、例えば、凍結乾燥された製剤、水溶液、分散液又は錠剤、糖衣錠若しくはカプセルなどの固体調製物の形態で製剤化され得る。多数の適切な製剤が、全ての薬化学者に公知の処方中に見出すことができる。Pharmaceutical Sciences(18th ed, Mack Publishing Company, Easton, PA(1990))、特に、Block, LawrenceによるChapter 87中に見出すことができる。これらの製剤には、例えば、粉末、ペースト、軟膏、ゼリー、蝋、油、脂質、脂質(陽イオン性又は陰イオン性)含有小胞(LipofectinTMなど)、DNA抱合体、無水吸収ペースト、水中油及び油中水エマルジョン、エマルジョンカルボワックス(様々な分子量のポリエチレングリコール)、半固体ゲル及びカルボワックスを含有する半固体混合物が含まれる。製剤化によって、製剤中の活性因子が不活化されず、製剤が投与の経路と生理的に適合し、耐容される限り、先述の混合物の何れもが、本発明に係る治療及び療法において適切であり得る。薬化学者に周知の製剤、賦形剤及び担体に関するさらなる情報に関しては、Baldrick P., “Pharmaceutical excipient development:the need for preclinical guidance.”,Regul.Toxicol.Pharmacol.32(2),210−218(2000);Wang W., “Lyophilization and development of solid 5 protein pharmaceuticals.”,Int.J.Pharm.203(1−2),1−60(2000);Charman W.N., “Lipids, lipophilic drugs, and oral drug delivery−some emerging concepts.”, J.Pharm.Sci.89(8),967−978(2000);Powell et al., “Compendium of excipients for parenteral formulations”, PDA J.Pharm.Sci.Technol.52, 238−311(1998)及びこれらの中の引用文献も参照されたい。
【0072】
本発明の別の態様は、過剰増殖性疾患の診断、予防又は治療用医薬組成物の製造のための、医薬として許容される担体、希釈剤及び/又は佐剤と混合された、本発明の少なくとも1つの単離された結合タンパク質、及び、場合によって使用される少なくとも1つの他の活性因子(例えば、少なくとも1つの上記抗新生物因子)の使用に関する。好ましくは、医薬組成物は上記の医薬組成物であり、過剰増殖性疾患は上記過剰増殖性疾患である。
【0073】
本発明のさらに別の態様は、HER−3の発現と関連する疾病又は症状を診断するための方法であり、試料を本発明の結合タンパク質と接触させること、及び前記試料中のHER−3の存在を検出することを含む、前記方法に関する。試料は、腫瘍細胞、血液試料又は別の適切な試料など、HER−3の発現を示す細胞であり得る。本発明の好ましい実施形態において、HER−3の発現に関連する疾病又は症状は、上に定義されている過剰増殖性疾患である。
【0074】
本発明によれば、前記方法は、例えば、細胞中のHER−3抗原の検出のために、上記過剰増殖性疾患に罹患している患者中のHER−3抗原濃度の測定のために、又は患者中の前記過剰増殖性疾患の段階決定のために使用され得る。研究されている対象中の過剰増殖性疾患の進行の段階を決定するために、又は療法の経過に対する対象の応答を特徴付けるために、例えば、血液の試料を対象から採取し、試料中に存在するHER−3抗原の濃度を測定する。このようにして得られた濃度は、値が属する濃度範囲を同定するために使用される。このようにして同定された範囲は、診断された対象の様々な集団中で同定された進行の段階又は療法の段階と相関し、これにより、研究中の対象における段階を付与する。疾病の生検、例えば、癌、患者から得られた組織も、存在するHER−3抗原の量を評価するために使用され得る。疾病組織中に存在するHER−3抗原の量は、本発明のHER3抗体を用いて、免疫組織化学、ELISA又は抗体アレイによって評価され得る。診断的に興味深い他のパラメータは、二量体化の状態並びにHER3タンパク質の二量体化対並びにHER3タンパク質及びその対の活性化状態である。これらのパラメータを決定するためのタンパク質分析法は本分野において周知であり、特に、ウェスタンブロット及び免疫沈降技術、FACS分析、化学架橋、生物発光共鳴エネルギー転移(BRET)、蛍光共鳴エネルギー転移(FRET)など(例えば、Price et al, Methods in Molecular Biology, 218:255−268(2002))又はeTag技術(WO0503707,WO04091384,WO04011900)である。
【0075】
さらに、本発明は、別の態様において、HER−3の発現が関連する疾病又は症状の予防又は治療を必要としている患者において、HER−3の発現が関連する疾病又は症状を予防又は治療する方法であり、少なくとも1つの本発明の結合タンパク質の有効量を前記患者に投与することを含む、前記方法に関する。好ましくは、HER−3の発現を伴う好ましい疾病又は症状は、上に定義されている過剰増殖性疾患である。予防又は治療を必要とする患者は、哺乳動物患者、すなわち、ヒト、家畜及び農場動物、並びに動物園、スポーツ又はペット動物(イヌ、ネコ、ウシ、ウマ、ヒツジ、ブタ、ヤギ、ウサギなど)を含む哺乳動物として分類される全ての動物である。好ましくは、必要とする患者は、ヒト患者である。
【0076】
本発明の好ましい実施形態において、過剰増殖性疾患の予防又は治療を必要としている患者において、過剰増殖性疾患を予防又は治療する方法は、少なくとも1つの本発明の結合タンパク質の有効量、及び、さらに、少なくとも1つの他の活性因子(例えば、少なくとも1つの上記抗新生物因子)を前記患者に投与することを含む。好ましくは、本方法は、異常な細胞増殖、遊走又は浸潤を阻害するための方法である。
【0077】
例えば、上記製剤を介した、結合タンパク質治療薬の候補の古典的な投与様式の他に、新たに開発された投与様式が、本発明においても有用であり得る。例えば、手術の切開後に原発性脳腫瘍を治療するための131I標識されたモノクローナル抗体の局所投与が報告されている。さらに、モノクローナル抗体及びモノクローナル抗体の断片の直接的な定位的脳内注射も、臨床的に、及び前臨床的に研究されている。頚動脈内高浸透圧性灌流は、薬物が連結されたヒトモノクローナル抗体を、原発性脳悪性腫瘍に標的誘導するための実験的戦略である。
【0078】
治療されるべき症状の種類及び重度に応じて、例えば、1つ若しくはそれ以上の分割された投与によって、又は継続的注入によって、本発明の結合タンパク質を必要としている患者に、少なくとも1つの本発明の結合タンパク質約1μg/kgから15mg/kgを投与し得る。典型的な一日投薬量は、上述の要因に応じて、約1μg/kgから約100mg/kg又はそれ以上の範囲であり得る。数日又はそれ以上にわたる反復投与に関しては、治療されるべき症状に応じて、疾病症候の望ましい抑制が生じるまで、治療が持続される。
【0079】
投与された少なくとも1つの抗新生物因子の用量は、様々な要因に依存する。これらは、例えば、因子の性質、腫瘍の種類又は投与の経路である。本発明は、いかなる用量にも限定されないことが強調されるべきである。
【0080】
最後に、本発明は、さらなる態様において、HER−3によって媒介されるシグナル伝達と関連する疾病を診断し、予防し、又は治療するためのキットであり、少なくとも1つの本発明の結合タンパク質及び/又は核酸分子及び/又はベクターを含む前記キットに関する。さらに、本発明のキットは、少なくとも1つの他の活性因子、例えば、少なくとも1つの上記他の抗新生物因子をさらに含むことができる。
【0081】
さらに、以下の実施例及び添付の図面によって、本発明を説明するものとする。
【0082】
実施例
実施された実験及び達成された結果を含む以下の実施例は、例示の目的のために提供されているものに過ぎず、本発明を限定するものと解釈すべきでない。
【実施例1】
【0083】
HER−3抗原及び細胞株の調製
本研究では、組換えHER−3タンパク質を調製した。HER−3の配列(Genebank AccNr.NM 001982)に基づくプライマーを用いて、pcDNA3−HER−3(完全長ヒトHER−3を有する発現ベクター、C.Wallasch et al., EMBO J.14, 4267−4275)からのポリメラーゼ連鎖反応(PCR)によって、HER−3の細胞外ドメイン(ECD)cDNAをクローニングした。
【0084】
HER−3の増幅のために使用されたプライマーは、以下のとおりであった。
【0085】
【化1】

【0086】
【化2】
【0087】
BamH1及びXbaIでPCR産物を消化し、BamHI及びXbaIで消化されたpcDNA3(Invitrogen)中に連結した。CaPO4法を用いて、プラスミドをHEK293細胞中に形質移入した。Ni−NTAアフィニティークロマトグラフィーを用いて、採集された馴化培地からHER−3−HIS融合タンパク質を精製した。
【0088】
レトロウイルス遺伝子転送によって、Rat1HER−3細胞を作製した。要約すると、GP+E86細胞(3×105)を60mm培養皿上に播種し、リン酸カルシウム法を用いて、2μg/mLpIXSNベクター又はpIXSN−HER−3cDNA(C.Wallasch, PhD Thesis, Max−Planck lnsitute of Biochemistry, Martinsried, Germany)を形質移入した。24時間後、培地を新鮮な培地と交換し、GP+E86細胞を、4から8時間温置した。次いで、Polybren(4mg/mL;Aldrich)の存在下で、4から12時間、高力価のpLXSN又はpLXSN−HER−3、pウイルス(>1×106G418c.f.u./mL;m.o.i.10)を放出するGP+E86細胞の上清とともに、集密状態を下回るRat1細胞(2×105細胞/6cm皿)を温置した。培地を交換した後、G418でのRat1細胞の選択を開始した。通常、21日間の選択後に、安定なクローンを摘み取った。
【実施例2】
【0089】
ヒト癌細胞株中でのHER−3発現
例えば,HER−3のような受容体チロシンキナーゼは、良性過形成細胞増殖から悪性癌腫への移行など、過剰増殖性疾患の開始及び進行において重大な役割を果たしている。腫瘍細胞と正常組織との間でHER−3発現が異なるので、HER−3発現の分析は、本発明の結合タンパク質での治療が有益である患者の亜群を同定するための重要な要因である。従って、ヒト癌形成におけるHER−3の役割を解明するために、ヒト癌細胞株の集団において、HER−3発現を定量した。癌細胞株は、ATCCによって推奨されているとおりに増殖させた。詳しく述べると、PBS中の10mMEDTAを用いて105細胞を採集し、FACS緩衝液(PBS、3%FCS、0.4%アジ化物)で1回洗浄し、96ウェルの丸底プレート上に播種した。上清を除去するために、1000rpmで3分間、細胞を遠心し、次いで、α−HER−3抗体2D1D12(WO03013602)(3μg/ml)とともに再懸濁した。細胞懸濁液を氷上で1時間温置し、FACS緩衝液で2回洗浄し、二次抗体(100μL/ウェル)ロバ抗ヒトPE(Jackson)によって再懸濁し、FACS緩衝液中に1:50希釈した。氷上及び暗所で、細胞懸濁液を30分間温置し、FACS緩衝液を用いて2回洗浄し、分析した(FACS、Beckman Coulter)。図1は、分析の代表的な結果を示しており、様々なヒト癌内で、HER−3が発現されていることを示している。
【実施例3】
【0090】
免疫化及び力価測定
実施例1に記載されているとおりに調製されたHER−3ECDタンパク質及びC32細胞(ヒト悪性黒色腫;ATCC#CRL−1585)を抗原として使用した。XenoMouse(R)マウス(XenoMouse(R)系統:XMG1及びXMG4、Abgenix,Inc.Fremont,CA)を順次免疫化することによって、HER−3に対するモノクローナル抗体を産生させた。全ての注射に関して、足蹠経路を介して、XenoMouse(R)動物を免疫した。各注射の総容量は、50μL/マウス、25μL/足蹠であった。
【0091】
コホート番号1(10匹のXMG1マウス)に関して、初期免疫化は、マウス当り、TITERMAX GOLD(R)(Sigma, Oakville, ON)が1:1(v/v)で混合されたHER−3ECDタンパク質10μgを用いて行った。その後の5回の強化免疫は、発熱物質を含まないD−PBS中のalumゲル100μg(Sigma,Oakville,ON)と1:1(v/v)で混合されたHER−3ECDタンパク質10μgを用いて行った。6回目の強化免疫は、TITERMAX GOLD(R)と1:1(v/v)混合されたHER−3ECDタンパク質10μgからなった。7回目の注射は、alumゲル100μgと1:1v/v混合されたHER−3ECDタンパク質10μgからなった。最終強化免疫は、アジュバントなしの、無発熱物物質DPBS中のHER−3ECDタンパク質10μgを用いて行った。このプロトコールに関しては、第0日、4日、7日、11日、15日、20日、24日及び29日目にXenoMouse(R)マウスを免疫し、第33日目に融合を行った。4回目の強化免疫後に13日目に、6回目の強化免疫後に19日目に、後眼窩採血操作を通じて、2回の採血を行った。コホート番号2は存在しなかった。
【0092】
コホート番号3(10匹のXMG1マウス)及びコホート番号4(10匹のXMG4マウス)に関して、最初の注射は、マウス当り、TITERMAX GOLD(R)が1:1(v/v)で混合された無発熱物質Dulbecco’sPBS(DPBS)中の107個のC32細胞を用いて行った。次の4つの強化免疫は、マウス当り、Adju−Phos25μg及び10μgCpGと混合された無発熱物質DPBS中の107個のC32細胞を用いて行った。6回目の強化免疫は、マウス当り、TITERMAX GOLD(R)が1:1(v/v)で混合された無発熱物質DPBS中の107個のC32細胞を用いて行った。7回目、8回目、9回目の強化免疫は、マウス当り、Adju−Phos25μg及びCpG10μgと混合された無発熱物質DPBS中の107個のC32細胞を用いて行った。10回目から14回目の強化免疫は、マウス当り、Adju−Phos25μg及びCpG10μgと混合された無発熱物質DPBS中のHER−3ECDタンパク質5μgであった。最終強化免疫は、アジュバントなしの、無発熱物物質DPBS中のHER−3ECDタンパク質5μgからなった。コホート番号3及び4の両方で、このプロトコールに対して第0日、3日、7日、11日、14日、17日、21日、24日、28日、33日、35日、38日、42日及び45日目にXenoMouse(R)マウスを免疫し、第49日目に融合を行った。4回目の強化免疫後12日目に、6回目の強化免疫後19日目に、及び12回目の強化免疫後40日目に、後眼窩採血操作を通じて、3回の採血を行った。
【0093】
力価による、採集のための動物の選択
コホート番号1に関しては、免疫されたXenoMouse(R)マウスから得た血清中の抗HER−3抗体力価は、HER−3ECDタンパク質に対するELISAによって測定した。各XenoMouse(R)動物の特異的力価は、650nmでの光学密度から測定され、下表1に示されている。力価の値は、血清の最大希釈の逆数であり、ODの読み取りは、バックグラウンドの2倍である。従って、数値が高くなるほど、HER−3ECDに対する液性免疫応答は大きかった。
【0094】
【表1】
【0095】
コホート番号3及び4に関しては、免疫されたXenoMouse(R)マウスから得た血清中の抗HER−3抗体力価は、Rat1/HER−3細胞(抗原陽性細胞株)細胞及びRat1/pLSXN細胞(抗原陰性細胞株)を用いたFACSによって測定した。データは、血清試料の系列希釈による、細胞抗HER−3細胞染色の幾何平均(GeoMean)蛍光強度として表されている。
【0096】
【表2】
【0097】
【表3】
【実施例4】
【0098】
リンパ球の回収、B細胞の単離、融合及びハイブリドーマの作製
免疫されたマウスを屠殺し、各コホートからリンパ節を採集し、プールした。DMEM中で磨り潰すことによってリンパ球系細胞を解離させて、組織から細胞を放出させ、細胞をDMEM中に懸濁した。細胞を計数し、0.9mLDMEM/1億リンパ球を細胞ペレットに添加して、穏やかに、但し、完全に、細胞を再懸濁した。1億細胞当りCD90+磁気ビーズ100μLを用いて、4℃で15分間、磁気ビーズとともに細胞を温置することによって、細胞を標識した。最大108個の陽性細胞(又は最大2×109個の全細胞)を含有する磁気標識された細胞懸濁液をLS+カラムにかけ、DMEMでカラムを洗浄した。全ての流出物を、CD90陰性画分として集めた(これらの細胞の多くは、B細胞であると予想された。)。
【0099】
上で得た、洗浄され、濃縮されたB細胞と、ATCC(Cat.No.CRL1580)から購入された非分泌性骨髄腫P3X63Ag8.653細胞(Kearney et al, J.Immunol.123,1979,1548−1550)とを、1:1の比で混合することによって、融合を実施した。800gでの遠心によって、細胞混合物を穏やかに沈降させた。上清を完全に除去した後、最長2分間、プロナーゼ溶液2から4mL(CalBiochem、カタログ番号53702;PBS中0.5mg/ml)で細胞を処理した。次いで、酵素活性を停止させるために、FBS3から5mLを添加し、電気的細胞融合溶液ECFS(0.3Mショ糖、Sigma、カタログ番号S7903、0.1mM酢酸マグネシウム、Sigma、カタログ番号M2545、0.1mM酢酸カルシウム、Sigma、カタログ番号C4705)を用いて、40mLの総容積になるように懸濁液を調整した。遠心後、上清を除去し、40mLECFS中に細胞を再懸濁した。この洗浄工程を反復し、2×106細胞/mLの濃度になるように、再度、ECFS中に細胞を再懸濁した。
【0100】
融合生成装置モデルECM2001、Genetronic,Inc., San Diego, CAを用いて、電気的細胞融合を行った。使用した融合チャンバーのサイズは2.0mLであり、以下の機器設定を用いた。並列条件:電圧:50V、時間:50秒、膜破壊電圧:3000V、時間:30μ秒、融合後保持時間:3秒。
【0101】
ECF後、無菌条件下で、融合チャンバーから細胞懸濁液を慎重に取り出し、Lグルタミン、ペニシリン/ストレプトマイシン、OPI(オキサロアセタート、ピルバート、ウシインシュリン)(全てSigmaから入手)及びIL−6(Boehringer Mannheim)が補充された、Hybridoma Culture Medium(DMEM(JRH Biosciences)、15%FBS(Hyclone)の同じ容量を含有する無菌チューブ中に移した。37℃で15から30分間、細胞を温置し、次いで、400gで5分間遠心した。Hybridoma Selection Medium(0.5×HAが補充されたHybridoma Culture Medium(Sigma、カタログ番号A9666))の小容量中に、細胞を穏やかに再懸濁し、計5×106個B細胞/96ウェルプレート及び200μL/ウェルの最終播種を基礎として、さらなるHybridoma Selection Mediumを用いて、容量を適切に調整した。細胞を穏やかに混合し、96ウェルプレート中にピペットで添加し、増殖させた。第7日又は10日目に、培地の半分を除去し、細胞にHybridoma Selection Mediumを再度与えた。
【実施例5】
【0102】
ELISAによる候補抗体の選択
培養から14日後に、精製されたhisタグ化HER−3ECDを用いたELISAによって、HER−3特異的抗体に対して、コホート番号1(コホート1のマウスを、任意に融合番号1及び2に分割した。)から得られたハイブリドーマ上清の一次スクリーニングを行い、ELISAプレート上に固定化されたHER−3ECDへのヒトIgG結合を検出するために、ヤギ抗huIgGFc−HRP(Caltag Inc., Cat.No.H10507、使用した濃度は、1:2000希釈であった。)を用いたELISAによって、無関係なhisタグ化タンパク質に対するカウンタースクリーニングを行った。一次スクリーニングに基づく陽性ハイブリドーマ細胞増殖ウェルから得た古い培養上清を取り除き、新鮮なハイブリドーマ培地を用いて、HER−3陽性ハイブリドーマ細胞を懸濁し、24ウェルプレートに移した。培養から2日後には、これらの上清は、二次的確認スクリーニングを行える状態であった。HER−3特異的完全ヒトIgGκ抗体に対する二次的確認スクリーニングでは、ヒトγ鎖検出のためのヤギ抗huIgGFc−HRP(Caltag Inc.,Cat.No.H10507、使用した濃度は、1:2000希釈であった。)及びヒトκ軽鎖検出のためのヤギ抗hIgκ−HRP(Southern Biotechnology、カタログ番号2060−05)という検出用抗体の2つの組を用いたELISAによって、第一のスクリーニングにおいて陽性であったものをスクリーニングした。コホート番号1から生成された91の完全ヒトIgG/κHER−3特異的モノクローナル抗体が存在した。
【実施例6】
【0103】
FMAT/FACSによる候補抗体の選択
培養から14日後に、コホート番号3及び番号4から得られたハイブリドーマ上清(融合番号3及び番号4)を、FMATによって、HER−3−特異的モノクローナル抗体に関してスクリーニングした。一次スクリーニングにおいて、1:10最終希釈のハイブリドーマ上清を、ヒトHER−3を発現するRat1−Her3細胞及びCy5連結されたヤギF(ab’)2抗ヒトIgG、Fc特異的抗体400ng/mL(Jackson ImmunoResearch,カタログ番号109−176−098)とともに、室温で6時間温置した。細胞への抗体及び検出抗体複合体の結合を、FMAT(Applied Biosystems)によって測定した。細胞への抗体の非特異的結合は、親Rat1細胞への抗体の結合によって測定した。融合物3の一次スクリーニングから、HER−3−特異的抗体を産生する計420のハイブリドーマを選択した。同じFMATプロトコールを用いて、これらの拡張された培養物からの上清を再度検査し、これらのうちの262が、HER−3発現細胞へ特異的に結合することが確認された。融合物4の一次スクリーニングから、HER−3−特異的抗体を産生する計193のハイブリドーマを選択した。FACSによって、これらの拡張された培養物からの上清物を検査し、これらのうちの138が、HER−3発現細胞へ特異的に結合することを確認した。FACS確認アッセイでは、2%FBSを含有するPBS中において、1:2希釈で、1時間、40℃で、ハイブリドーマ上清とともに、Rat1−Xher3細胞と親Rat1細胞(陰性対照として)を温置した。PBSでの洗浄後、2.5μg/mLCy5連結されたヤギF(ab’)2抗ヒトIgG、Fc特異抗体(JIR#109−176−098)及び5μg/mLPE連結されたヤギF(ab’)2抗ヒトκ特異抗体(SB#2063−09)によって、細胞への抗体の結合を検出した。PBSでの洗浄によって、非結合抗体を除去した後、1:4の希釈で、cytofix(BD#51−2090KZ)によって、細胞を固定し、FACSCaliburによって分析した。
【実施例7】
【0104】
クローニングのためのハイブリドーマの選択
例えば、R&DBiosystemsから入手可能な精製された組換え細胞外ドメインを用いたELISAにおける、HER1(EGFR)、HER−2及びHER−4を上回るHER−3に対する特異性並びに異なるHERファミリーのメンバーを発現するヒト腫瘍細胞株のFACSベースの分析並びにバックグラウンドを上回るHER−3陽性細胞に対するFACS染色での平均蛍光強度の5倍超の増加に基づいて、ハイブリドーマクローニングのために、コホート1及び2から得た抗体を選択した。これらの基準に基づいて、限界希釈細胞播種によって、計23のハイブリドーマ株をクローニングのために選択した。
【0105】
HER−1(EGFR)、HER−2及びHER−4を上回るHER−3に対する特異性に加え、他の3つの基準に基づいて、ハイブリドーマクローニングのために、コホート3及び4から得た抗体を選択した。第一の基準は、HER−3のL2ドメイン内に含有されたエピトープを有する抗体に対するELISAスクリーニングであった(実施例「本発明における抗HER−3抗体の構造分析」を参照。)。
【0106】
第二の基準は、FACSベースのアッセイにおける、HER−3発現細胞へのビオチン化されたヘレグリンαの結合の中和であった。SKBR−3細胞を採集し、培地中で洗浄し、遠心によって沈降させ、培地中に再懸濁した。再懸濁された細胞を、96ウェルプレート中に分取した。細胞を沈降させるために、プレートを遠心した。排出ハイブリドーマ上清中の試験抗体を、25μL/ウェルで添加し、抗体を結合させるために、氷上で1時間温置した。10nMヘレグリンα(R&D Biosystems, Minneapolis, MN)溶液50μLを、5nMの最終濃度となるように、各ウェルに添加し、氷上で1.5時間温置した。150μLPBS中で細胞を洗浄し、遠心によって沈降させ、上清を除去した。10μg/mLで、ヤギ抗HRGαポリクローナル抗体50μL中に細胞を再懸濁し、45分間、氷上で温置した。PBS200μL中で細胞を洗浄し、遠心によって沈降させ、上清を除去した。10μg/mLの7AADを加えた5μg/mLのCy5標識されたウサギ抗ヤギポリクローナル抗体の溶液50μLを添加し、15分間、氷上で温置した。PBS200μL中で細胞を洗浄し、遠心によって沈降させ、上清を除去した。FACS緩衝液100μL中に細胞を再懸濁し、FACS中で読み取った。ヘレグリンαの結合を低下させた試験HER−3抗体は、最も低い蛍光強度を有するものであった。陽性対照として、マウスHER−3mAb(105.5)又はヒトIgG1HER−3mAB、U1−49の10,000ng/mLから16ng/mLまでの1:5系列希釈を使用した。陰性対照は、ヘレルギンαのみ、細胞のみ、ヤギ抗ヘレグリンαポリクローナル抗体のみ及びCy5標識されたウサギ抗ヤギポリクローナル抗体のみであった。
【0107】
第三の基準は、HER−3発現細胞株を用いたFACSにおける、親和性に対する相対順位付け及び/又はより高い相対平均蛍光強度であった。親和性に対する相対順位付けは、HER−3特異的抗体濃度を標準化し、以下のように、限界抗原ELISAから得られたデータに対してプロットすることによって行った。
【0108】
高抗原ELISAを用いた抗原特異的抗体濃度の標準化
ELISA法を用いて、抗原特異的抗体の濃度に対して上清を標準化した。平行して滴定された濃度既知のコホート1から得られた2つの抗HER−3ヒトIgG1抗体を用いて、標準曲線を作成し、コホート3及び4から得られた試験ハイブリドーマ上清中の抗原特異的抗体の量を標準と比較した。このようにして、各ハイブリドーマ培養中のヒトHER3IgG抗体の濃度を推定した。
【0109】
4℃で一晩温置しながら、50μL/ウェルで、Costar3368培地結合プレート上に、1×PBS/0.05%アジ化ナトリウム中の8μg/mLでニュートラビジンをコートすることによって、ニュートラビジンプレートを作製した。翌日、1×PBS/1%スキムミルクでプレートをブロックした。1×PBS/1%スキムミルク中の500ng/mLの光ビオチン化されたhisタグ化HER−3ECDを、室温で1時間温置することによって、ニュートラビジンプレートに結合させた。1×PBS/1%スキムミルク/0.05%アジ化物中に、1:31の出発希釈から1:7568の最終希釈まで、1:2.5系列希釈されたハイブリドーマ上清を、50μL/ウェルで添加し、次いで、室温で20時間温置した。未知の各々に対するODの読み取りが確実にアッセイの直線域内に得られるようにするために、系列希釈を使用した。次に、二次検出抗体である、1×PBX/1%スキムミルク中の400ng/mLのヤギ抗ヒトIgGFcHRPを、50μL/ウェルで添加した。室温で1時間後、再度、プレートを水で5回洗浄し、一成分TMB基質50μLを各ウェルに添加した。各ウェルに1M塩酸50μLを添加することによって、30分後に反応を停止させ、450nmの波長でプレートを読み取った。1000ng/mLから0.06ng/mLまで1:2で系列希釈された、コホート1から得られた2つのIgG1HER−3mAbから標準曲線を作成し、上記プロトコールを用いてELISA中で評価を行った。未知の各々に関しては、各試料中のヒトHER−3IgGの濃度を推定するために、アッセイの直線域中のODの読み取りを使用した。
【0110】
限定された抗原分析は、他の全ての抗原特異的抗体との比較において、B細胞培養上清中に調製された抗原特異的抗体の親和性を順位付けする方法である。抗原の極めて少量のコーティングの存在下では、最高の親和性の抗体のみが、平衡状態で、何れかの検出可能なレベルまで結合できるはずである。(例えば、2003年6月12日に公開された「IDENTIFICATION OF HIGH AFFINITY MOLECULES BY LIMITED DILUTION SCREENING」と題されたPCT Publication WO/03048730A2を参照されたい。)。この例では、コホート1から得られた2つのmAb(何れも、濃度既知及びKD既知)を、アッセイにおける基準として使用した。
【0111】
4℃で一晩温置しながら、50μL/ウェルで、Costar3368培地結合プレート上に、1×PBS/0.05%アジ化ナトリウム中の8μg/mLのニュートラビジンをコートすることによって、ニュートラビジンプレートを作製した。翌日、1×PBS/1%スキムミルクで、プレートをブロックした。室温で1時間、1×PBS/1%スキムミルク中、125、62.5、31.2、15.6及び7.8ng/mLという5つの濃度で、ビオチン化されたhisタグ化HER−3ECD(50μL/ウェル)を96ウェルニュートラビジンプレートに結合させた。各プレートを水で5回洗浄した。1×PBS/1%スキムミルク/0.05%アジ化物中に1:31希釈されたハイブリドーマ上清を、50μLウェルで添加した。震盪器上、室温での20時間の温置後、dH2Oで5回、プレートを再度洗浄した。次に、二次検出抗体である、1×PBS/1%スキムミルク中の400ng/mLのヤギ抗ヒトIgGFcHRP(西洋ワサビペルオキシダーゼ)を50μL/ウェルで添加した。室温で1時間後、dH2Oで、プレートを再度5回洗浄し、一成分TMB基質50μLを各ウェルに添加した。各ウェルに1M塩酸50μLを添加することによって、30分後に反応を停止させ、450nmの波長でプレートを読み取った。直線域内にOD値を与える抗原濃度から得られるOD読み取りを、データ解析のために使用した。
【0112】
特異的抗体濃度を比較的推定する(詳細については、上記参照)高抗原データを、限定された抗原ODに対してプロットすることによって、比較的より高い親和性抗体(例えば、結合された抗体が、限定された抗原アッセイでは、より高いODを有したが、上清中にIgGHER−3抗体のより低い量を有する。)を示した。
【0113】
アッセイのこれらの組において最も成績の優れていた33個の抗体に対するコホート3及び4から得たハイブリドーマは、限界希釈ハイブリドーマ播種によるクローニングに進んだ。
【0114】
あるいは、Rat1/pLXSN及びRat1/HER−3細胞のHER−3発現のFACS分析は、類似の結果を示した(内在性ラットエピトープとの交叉反応性なし)(図2)。
【0115】
詳しく述べると、PBS中の10mMEDTAを用いて1×105細胞を採集し、FACS緩衝液(PBS、3%FCS、0.4%アジ化物)で1回洗浄し、96ウェルの丸底プレート上に播種した。上清を除去するために、1000rpmで3分間、細胞を遠心し、次いで、特異的なHERファミリー抗体(3μg/ml)とともに再懸濁した。細胞懸濁液を氷上で45分間温置し、FACS緩衝液で2回洗浄し、二次抗体(100μL/ウェル)ロバ抗ヒトPE(Jackson Immunoresearch,PA)とともに再懸濁し、FACS緩衝液中に1:50希釈した。氷上及び暗所で、細胞懸濁液を30分間温置し、FACS緩衝液を用いて2回洗浄し、分析した(FACS、Beckman Coulter)。
【実施例8】
【0116】
本発明の抗HER−3抗体の構造的分析
以下の論述では、本発明に従って調製された抗体に関する構造情報が提供される。本発明に従って作製された抗体の構造を分析するために、特定のハイブリドーマから、重鎖及び軽鎖断片をコードする遺伝子を増幅した。配列決定は、以下のように行った。
【0117】
逆転写ポリメラーゼ連鎖反応(RT−PCR)を用いて、96ウェルプレート中の各ハイブリドーマクローンから、VH及びVL転写物を増幅した。Fast−Trackキット(Invitrogen)を用いて、約2×105個のハイブリドーマ細胞からポリ(A)+mRNAを単離した。各ハイブリドーマに対して、4つのPCR反応(軽鎖(κ(K)に対して2つ、及びγ重鎖(γ)に対して2つ)を実施した。増幅のために、QIAGENOneStep室温PCRキット(Qiagen、カタログ番号210212)を使用した。連結された室温PCR反応では、Cκ又はCγ遺伝子のCH1領域のコンセンサスに対応するアンチセンス配列特異的プライマーを使用し、室温酵素(Omniscript及びSensiscript)の混合物を用いてcDNAを合成した。50℃で1時間、逆転写を実施した後、高い特異性及び感受性のために、HotStarTaq DNA PolymeraseによるcDNAのPCR増幅を行った。各PCR反応は、5’センスプライマーの混合物を使用した。プライマー配列は、Vbaseウェブサイト(http://vbase.mrc−cpe.cam.ac.uk/)で入手可能なVH及びVKのリーダー配列に基づいた。
【0118】
94℃で15分間の初期ホットスタートでPCR反応を実行した後、94℃30秒間(変性)、60℃30秒間(アニーリング)及び72℃1分間(伸長)の40サイクルを行った。
【0119】
PCR産物を精製し、ABI PRISM BigDye terminator cycle sequencing ready reaction Kit(Perkin Elmer)を使用し、フォワード及びリバースPCRプライマーを用いて配列を直接決定した。Prism色素終結因子配列決定キット及びABI377配列決定機を用いて、両鎖を配列決定した。
【0120】
配列分析
HER3抗体のヒトV重及びVκcDNA配列の分析は、Abgenix社内ソフトウェア(5AS)を用いて、ヒト生殖系列V重及びVκ配列とHER−3配列を並置することによって行った。本ソフトウェアによって、V遺伝子、D遺伝子及びJ遺伝子並びに組換え連結におけるヌクレオチド挿入及び体細胞変異の使用が同定された。体細胞変異を同定するために、アミノ酸配列もコンピュータシミュレーションで作製された。市販の配列分析ソフトウェア並びにヒトV、D及びJ遺伝子の配列に関する公表された情報、例えばVbase(http://vbase.mrc−cpe.cam.ac.uk/)を用いて、同様の結果を得ることが可能である。
【0121】
mAbU1−59の分子クローニング
抗体U1−59を分泌するハイブリドーマ系列など、複数のハイブリドーマ系列を含有する組織培養ウェルから全RNAを抽出した。3’−C−γプライマーとともに、5’−リーダーVHファミリー特異的プライマーを用いて、重鎖可変領域を増幅した。VH4プライマーを用いて、大きなバンドが増幅され、他のバンドは見られなかった。ヒトγ1定常領域遺伝子とインフレームになるように、pCDNA発現ベクター中にVH4−34γ断片をクローニングした。
【0122】
3’μ定常領域プライマーとともに5’VHファミリー特異的プライマーを用いて、IgM重鎖可変領域を増幅した。VH2プライマーを用いて、主要なバンドが増幅され、他のバンドは見られなかった。ヒトμ定常領域遺伝子とインフレームになるように、pCDNA発現ベクター中にVH2−5μ断片をクローニングした。Vκ鎖を増幅し、配列を決定した。4つのκ鎖RT−PCR産物が同定された。産物を配列決定し、コンピュータシミュレーションでの翻訳を介した配列分析後に、産物の3つが読み取り枠を有するに過ぎなかった。(1)VK1A3−JK2、(2)VK1A20−JK3及び(3)B3−JK1としてのVκ遺伝子の使用に基づいて明確に同定されたオリゴクローン性U1−59ハイブリドーマから、これらの3つの機能的κ鎖をクローニングした。ヒトκ軽鎖定常領域遺伝子とインフレームになるように、pCDNA発現ベクター中に全てのVκをクローニングした。
【0123】
形質移入:
計6つの重鎖/κ軽鎖対に関して、一過性形質移入において、κ鎖の各々とともに、各重鎖を形質移入した。A20κ鎖とのγ鎖の形質移入は、乏しい抗体発現を与えたのに対して、A20κ鎖がμ鎖とともに同時形質移入された場合には、抗体は全く分泌又は検出されなかった。HER−3結合アッセイのために、計3つのIgGsup及び2つのIgMsupが利用可能であった。
【0124】
【表4】
【0125】
VH4−34及びB3κ鎖からなるIgG1mAbを用いたFACSにおいて、HER−3+細胞株への結合活性が検出された。他のVH/Vκの組み合わせは、HER−3+細胞株を用いたFACSにおいて、バックグラウンドを上回る蛍光シグナルを与えなかった。
【0126】
抗HER−3抗体の結合競合
HER−3への結合に関して競合するHER−3抗体のクラスターを評価するために、「Jia et al.J Immunol Methods.288, 91−98(2004)」に公表されているように、多重化された競合抗体ビニング(binning)を行った。コホート1由来の検査されたHER−3抗体は、結合に対する競合に基づいて、5つのビンにクラスター化された。
【0127】
【表5】
【0128】
抗HER−3抗体のエピトープ性質決定
本発明のヒト抗HER−3抗体のエピトープを性質決定した。まず、還元され、変性され、HER−3−Hisタグ化され、精製されたECDタンパク質のドットブロット分析が、検査された抗HER−3抗体(U1−59、U1−61、U1−41、U1−46、U1−53、U1−43、U1−44、U1−47、U1−52、U1−40、U1−49)による結合の不存在を示し、全てがジスルフィド結合の還元に対して感受性を有するエピトープを有することが実証され、全てが不連続なエピトープを有することが示唆された。次に、HER−3細胞外ドメインの以下の4つのドメインへの分割に基づいて、様々なヒト−ラットHER3キメラ分子を加工することによって、HER−3分子中の所定のドメインに抗体をマッピングした。
【0129】
1)L1(D1):非主要リガンド結合ドメイン、
2)S1(D2):第一のシステインリッチドメイン、
3)L2(D3):主要リガンド結合ドメイン、及び
4)S2(D4):第二のシステインリッチドメイン。
【0130】
RAT1−HER−3細胞から、ヒトHEr−3cDNAの細胞外ドメイン(ECR)を増幅した。ラット肝臓RNAからのRT−PCRによってラットHER−3cDNAを増幅し、配列決定によって確認した。ヒト及びラットHer3のECDを発現するcDNAを、V5−His融合タンパク質として、哺乳動物発現ベクター中にクローニングした。ヒトHER−3ECDから得られるドメインを、Mfe1、Bstx1及びDraIII内部制限部位を使用することによって、ラットHER−3ECDによって付与された足場中に取り替えた。この手段によって、様々なキメララット/ヒトHER−3ECDHIS融合タンパク質(アミノ酸1から160、161から358,359から575、1から358、359から604)を構築し、HEK293T細胞の一過性形質移入を介して発現させた。ヒトHER−3に対するラットポリクローナル抗体を用いて、構築物の発現を確認した。分泌されたキメラECDへの結合に関して、ヒトモノクローナル抗体をELISAにおいて検査した。
【0131】
抗体U1−59を含むヒト抗体の2つが、ラットHER−3と交叉反応した。結合ドメインを割り当てるために、HER3のL1−S1細胞外ドメインの発現をコードするプラスミドDNAが形質移入されたHEK293T細胞の上清から精製されたL1−S1−V5hisタグ化タンパク質からなるHER−3の末端切断形態に対して、これらのmAbを検査した。mAbU1−59は、ELISA中のL1−S1タンパク質に結合したので、そのエピトープがL1−S1中に存在することが示唆される。mAb2.5.1は、L1−S1タンパク質に結合しなかったので、そのエピトープがL2−S2中に存在することが示唆される。mAb−HER−3ECD複合体のチップ上タンパク質分解消化物とともに、飛行質量分析法のSELDI時間を用いて、抗体U1−59のさらなるマッピングを行った。
【0132】
SELDIを用いたU1−59のマッピング
mAb−HER−3ECD複合体のチップ上タンパク質分解消化物とともに、飛行質量分析法のSELDI時間を用いて、抗体U1−59のさらなるマッピングを行った。PS20タンパク質チップアレイに、プロテインAを共有結合させ、mAbU1−59を捕捉するために使用した。次いで、HER−3−His精製抗原とともに、PS20タンパク質チップとモノクローナル抗体の複合体を温置した。次に、Asp−Nの高濃度を用いて、抗体抗原複合体を消化した。チップを洗浄すると、チップ上の抗体に結合されたHER−3ペプチドのみが保持された。SELDIによってエピトープを決定し、断片の質量によってエピトープを同定した。同定された6814D断片は、HER−3hisECDの部分的消化物から生成される2つの可能な予想されたペプチドに対応する。重複するペプチドは何れも、ドメインS1にマッピングされる。HER−3欠失構築物への結合と、SELDIの結果を総合することによって、エピトープは、残基251から325にマッピングされた。
【0133】
本発明のヒト抗HER−3mAbによって認識されるHER−3の細胞外部分中における結合ドメインの位置が、表4に要約されている。エピトープドメインマッピングの結果は、抗体競合結合競合ビンから得られた結果と合致しており、HER−3への結合に関して互いに交叉競合した抗体は、HER−3上の同じドメインにマッピングされる(図3)。
【0134】
【表6】
XR=交叉反応性
【実施例9】
【0135】
抗体のカノニカルクラスの決定
Chothiaらは、各免疫グロブリン鎖の超可変領域に対する「カノニカルクラス」の観点で、抗体構造について記載している(J.Mol.Biol.,1987 Aug 20,196(4):901−17)。それらのアミノ酸配列とそれらの抗原結合部位の三次元構造との関係を決定するために、様々な免疫グロブリンのFab及びVL断片の原子構造を分析した。Chothiaらは、それらのパッキング、水素結合又は異常なφ、ψ若しくはω立体構造を採る能力を通じて、超可変領域の主鎖立体構造のために主として必要とされる残基は相対的に少ないことを見出した。これらの残基は、超可変領域内及び保存されたβシートフレームワーク中の部位に存在することが見出された。未知の構造を有する免疫グロブリンの配列を調べることによって、Chothiaらは、多くの免疫グロブリンが、公知の構造の1つとサイズが類似する超可変領域を有すること、さらに、観察された立体構造に必要とされる部位に同一の残基を含有することを示す。
【0136】
彼らの発見は、これらの超可変領域が公知の構造中のものに近い立体構造を有することを示唆した。超可変領域の5つに関しては、立体構造のレパートリーは、別個の構造クラスの比較的少数に限定されるようである。これらの一般的に存在する超可変領域の主鎖立体構造は、「カノニカル構造」と名付けられた。Chothiaら(Nature, 1989 Dec 21−28, 342(6252):877−83)及び他の者(Martin, et al.J.Mol.Biol., 1996 Nov 15, 263(5):800−15)によるさらなる研究は、抗体の6つの超可変領域のうち少なくとも5つに対して、主鎖立体構造の小さなレパートリーが存在することを確認した。
【0137】
それらのカノニカルクラスを決定するために、上記各抗体のCDRを分析した。既知のとおり、カノニカルクラスは、抗体軽鎖のCDR1、CDR2及びCDR3の他に、抗体重鎖のCDR1及びCDR2に対してのみ割り当てられている。下表は、分析の結果を要約している。カノニカルクラスのデータは、HCDR1−HCDR2−LCDR1−LCDR2−LCDR3の形態であり、「HCDR」は重鎖CDRを表し、「LCDR」は軽鎖CDRを表す。従って、例えば、1−3−2−1−5のカノニカルクラスは、カノニカルクラス1に属するHCDR1、カノニカルクラス3に属するHCDR2、カノニカルクラス2に属するLCDR1、カノニカルクラス1に属するLCDR2及びカノニカルクラス5に属するLCDR3を有する抗体を表す。
【0138】
各カノニカルクラスに対して定義されたアミノ酸と、抗体中のアミノ酸の70%又はそれ以上の同一性が存在する特定のカノニカルクラスに割り当てが為された。各抗体に対して定義されたアミノ酸は、例えば、上に引用されたChothiaらによる文献中に見出すことができる。表5及び表6は、HER−3抗体の各々に対するカノニカルクラスデータを報告する。70%未満の同一性が存在した場合には、各CDRの長さ及びデータの全体に基づいて、適切なカノニカルクラスの最良の推定が為されることを示すために、カノニカルクラスの割り当てにアスタリスク(「*」)が付されている。同じCDR長さを有する、合致したカノニカルクラスが存在しない場合には、「s18」(CDRが18のサイズであることを意味する。)のように、文字s及び数字で、カノニカルクラスの割り当てに印が付されている。重鎖又は軽鎖の1つに対して入手可能な配列データが存在しない場合には、カノニカルクラスは、「Z」でマークされる。
【0139】
【表7】
【0140】
表7は、クラス当りの抗体の数の分析である。左の欄に表記された特定のカノニカルクラスを有する抗体の数が、右の欄に示されている。1つの鎖配列データを欠如し、従って、カノニカル割り当てにおいて「Z」を有する4つのmAbは、このカウント中に含まれていない。
【0141】
最も一般的に見られる構造は、3−1−2−1−1である。重鎖及び軽鎖配列の両方を有する41のmAbのうち21が、この立体構造を有していた。
【0142】
【表8】
【実施例10】
【0143】
抗体親和性の測定
間接FACSScatchard分析によって、本発明の抗HER−3抗体の親和性測定を行った。従って、PBS中の10mMEDTAを用いて105個の目的の細胞又はSk−Br3細胞を採集し、FACS緩衝液(PBS、3%FCS、0.4%アジ化物)で1回洗浄し、96ウェルの丸底プレート上に播種した。上清を除去するために、1000rpmで3分間、細胞を遠心し、次いで、α―HER−3抗体(3μg/mL)又はFACS緩衝液中の20μg/mLヒトモノクローナル抗体から開始して、1:2の希釈工程で希釈された抗体希釈物(100μL/ウェル)とともに再懸濁した。細胞懸濁液を氷上で1時間温置し、FACS緩衝液で2回洗浄し、二次抗体(100μL/ウェル)ロバ抗ヒトPE(Jackson)によって再懸濁し、FACS緩衝液中に1:50希釈した。氷上及び暗所で、細胞懸濁液を30分間温置し、FACS緩衝液を用いて2回洗浄し、分析した(FACS、Beckman Coulter)。FACSScatchard分析に従って、各測定に対して蛍光平均を計算した。各蛍光平均からバックグラウンド染色(=第一の抗体なし)を差し引いた。x値=蛍光平均及びy値=蛍光平均/mAbの濃度(nM)であるScatchardプロットを作成した。KDは、線形方程式の1/mの絶対値と解した。図4は、本発明のU1−59抗体を用いた速度論分析を示している。以下の表8には、このようにして選択された本発明のある種の抗体に対する親和性測定が記載されている。
【0144】
【表9】
【実施例11】
【0145】
本発明の抗HER−3抗体は、HER−3受容体の飲食細胞運動を誘導する
HER−3は、HERファミリーによって媒介される細胞シグナル伝達の重要な門番としての役割を通じて、過剰増殖性疾患の開始及び進行に影響を与えることができる因子として同定されてきた。従って、HER−3が受容体の内部取り込みによって細胞表面/膜から効果的に除去されれば、細胞シグナル伝達が、従って、悪性腫瘍における細胞の形質転換及び/又は維持が、最終的に軽減又は抑圧され得る。
【0146】
本発明の抗HER−3抗体がHER−3の加速された飲食細胞運動を誘導できるかどうかを調べるために、本発明の抗HER−3抗体とともに細胞を0.5及び4時間温置した後に、細胞表面上のHER−3分子の相対量を比較した。24ウェル皿中の正常な増殖培地中に3×105個の細胞を播種し、一晩増殖させた。表記時間にわたって、37℃で、通常の増殖培地中の10μg/mL抗HER−3mAbとともに細胞を予め温置した。10mMEDTAを用いて細胞を剥離させ、洗浄緩衝液(PBS、3%FCS、0.04%アジ化物)中の10μg/mL抗HER−3mAbとともに、4℃で45分間温置した。洗浄緩衝液で細胞を2回洗浄し、1:100希釈されたロバ抗ヒトPE二次抗体(Jackson)とともに、4℃で45分間温置し、洗浄緩衝液で2回洗浄し、FACS(BeckmanCoulter、EXPO)によって分析した。
【0147】
図5に示されているデータは、抗HER−3抗体での細胞の処理が、受容体の内部取り込みをもたらすことを示している。データは、%内部取り込みとして示されており、対照処理された試料に対する、抗HER3処理された試料の平均蛍光強度の低下を表している。
【実施例12】
【0148】
本発明のヒト抗HER−3抗体による、ヒト癌細胞SKBr3へのリガンド結合の阻害
本発明の抗HER−3抗体が、細胞をベースとしたアッセイにおいて、HER−3へのリガンド結合を阻害する能力を定量するために、放射性リガンド競合実験を行った。従って、抗体の変動する濃度とともに、氷上で30分間温置された4×105個のSK−BR−3細胞を用いて、HER−3受容体結合アッセイを行った。各ウェルに、1.25nM[I125]−α−HRG[I125]−β−HRGを添加し、氷上で2時間、温置を継続した。プレートを5回洗浄し、風乾し、シンチレーションカウンターでカウントを計数した。図6aからeは、代表的な本発明の抗HER−3抗体を用いて実施されたこれらの実験の結果を示しており、本発明の抗体が、内在性HER−3を発現する細胞への、[125I]−α−HRG/[125I]−β−HRGの結合を特異的に低下させ得ることを示している。
【実施例13】
【0149】
本発明のヒト抗HER−3抗体による、リガンド誘導性HER−3リン酸化の阻害
本発明の抗体が、リガンドβ−HRGによって媒介されるHER−3の活性化を遮断することができるかどうかを調べるために、ELISA実験を行った。リガンドによって媒介されるHER−3の活性化は、増加した受容体チロシンリン酸化によって検出した。
【0150】
第1日:37℃で4時間、0.1M酢酸中の20μg/mLコラーゲンIで1×96ウェル皿をコートした。通常の増殖培地中に、2.5×105個の細胞を播種した。
【0151】
第2日:無血清培地100μL中で、24時間、細胞を飢餓状態にした。
【0152】
第3日:抗HER−3mAB10μg/mLとともに、37℃で1時間、細胞を予め温置し、次いで、β−HRG−EGFドメイン(R&D Systems)30ng/mLで、10分間処理した。溶媒を除去し、室温で1時間、PBS中の4%ホルムアルデヒド溶液で細胞を固定した。ホルムアルデヒド溶液を除去し、洗浄緩衝液(PBS/0.1%Tween20)で細胞を洗浄した。細胞緩衝液中の1%H2O2、0.1%NaN3で細胞を消光し、室温で20分間温置し、次いで、4℃で5時間、NET−ゼラチンでブロックした。4℃で一晩、一次抗体ホスホ−HER−3(Tyr1289)(ポリクローナルウサギ;Cell signaling #4791;1:300)を添加した。
【0153】
第4日:洗浄緩衝液で、プレートを3回洗浄した後、PBS中に1:3000希釈された抗ウサギ−PODとともに温置し、各ウェルに0.5%BSAを添加し、室温で1.5時間温置した。洗浄緩衝液で3回、PBSで1回、プレートを洗浄した。テトラメチルベンジジン(TMB、Calbiochem)を添加し、650nmでモニターした。250nMHCl100μLの添加によって反応を停止し、Vmaxプレートリーダー(Thermo LabSystems)を使用して、650nmの参照波長として、吸光度を450nmで読み取った。
【0154】
図7aは、本実験の代表的な結果を示し、減少した受容体チロシンのリン酸化によって示されたように、本発明の抗HER−3抗体が、リガンドによって媒介されるHER−3の活性化を低下させ得ることを示している。データは、対照抗体に対する、治療抗体によるパーセント低下として示されている。
【0155】
mAbU1−53が、リガンドによって媒介されるHER−3活性化を阻害する効力を検査するために、24時間、MCF−7細胞を飢餓状態とし、37℃で1時間、mAbU1−53とともに温置し、10分間、10nMHRG−βで刺激した。可溶化液を1B4(マウス抗HER−3mAb)ELISAプレートに移し、抗体4G10を用いて、HER−3のリン酸化を分析した。図7bに示されているように、HER−3のリン酸化は、0.14nMのIC50で、用量依存的な様式で、ほぼ完全に阻害された。
【実施例14】
【0156】
本発明のヒト抗HER−3抗体による、リガンド誘導性p42/p44MAPキナーゼリン酸化の阻害
本発明の抗体が、リガンドβ−HRGによって媒介されるp42/p44MAPキナーゼの活性化を遮断することができるかどうかを調べるために、次のELISA実験を行った。リガンドによって媒介されるHER−3の活性化は、増加したタンパク質(Thr202/Tyr204)チロシンリン酸化によって検出した。
【0157】
第1日:37℃で4時間、0.1M酢酸中の20μg/mLコラーゲンIで1×96ウェル皿をコートした。通常の増殖培地中に、3×105個の細胞を播種した。
【0158】
第2日:無血清培地100μL中で、24時間、細胞を飢餓状態にした。
【0159】
第3日:抗HER−3mAB5μg/mLとともに、37℃で1時間、細胞を予め温置し、次いで、β−HRG−EGFドメイン(R&D Systems)20ng/mLで、10分間処理した。溶媒を除去し、室温で1時間、PBS中の4%ホルムアルデヒド溶液で細胞を固定した。ホルムアルデヒド溶液を除去し、洗浄緩衝液(PBS/0.1%Tween20)で細胞を洗浄した。細胞緩衝液中の1%H2O2、0.1%NaN3で細胞を消光し、室温で20分間温置し、次いで、4℃で5時間、PBS/0.5%BSAでブロックした。4℃で一晩、一次抗体ホスホ−p44/p42MAPキナーゼ(Thr202/Tyr204)(ポリクローナルウサギ;Cell signaling #9101;1:300)を添加した。
【0160】
第4日:洗浄緩衝液で、プレートを3回洗浄した後、PBS中に1:5000希釈された抗ウサギ−HRPとともに温置し、各ウェルに0.5%BSAを添加し、室温で1.5時間温置した。洗浄緩衝液で3回、PBSで1回、プレートを洗浄した。テトラメチルベンジジン(TMB、Calbiochem)を添加し、650nmでモニターした。250nMHCl100μLの添加によって反応を停止し、Vmaxプレートリーダー(Thermo Lab Systems)を使用して、650nmの参照波長として、吸光度を450nmで読み取った。
【0161】
図8は、本実験の代表的な結果を示している。本発明の抗体は、減少されたリン酸化によって示されたように、リガンドによって媒介されたp42/p44MAP−キナーゼの活性化を低下させることができた。データは、対照抗体に対する、治療抗体によるパーセント低下として示されている。
【実施例15】
【0162】
本発明のヒト抗HER−3抗体による、β―HRG誘導性ホスホ−AKTリン酸化の阻害
以下のELISA実験において、本発明者らは、本発明の抗HER−3抗体が、リガンドβ−HRGによって媒介されるAKTキナーゼの活性化を遮断することができるかどうかを調べた。リガンドによって媒介されるAKTの活性化は、増加したタンパク質(Ser473)のリン酸化によって検出された。
【0163】
第1日:37℃で4時間、0.1M酢酸中の20μg/mLコラーゲンIで1×96ウェル皿をコートした。通常の増殖培地中に、3×105個の細胞を播種した。
【0164】
第2日:無血清培地100μL中で、24時間、細胞を飢餓状態にした。
【0165】
第3日:抗HER−3mAb5μg/mLとともに、37℃で1時間、細胞を予め温置し、次いで、β−HRG−EGFドメイン(R&D Systems)20ng/mLで10分間処理した。溶媒を除去し、室温で1時間、PBS中の4%ホルムアルデヒド溶液で細胞を固定した。ホルムアルデヒド溶液を除去し、洗浄緩衝液(PBS/0.1%Tween20)で細胞を洗浄した。細胞緩衝液中の1%H2O2、0.1%NaN3で細胞を消光し、室温で20分間温置し、次いで、4℃で5時間、PBS/0.5%BSAでブロックした。4℃で一晩、一次抗体ホスホ−Akt(Ser473)(ポリクローナルウサギ;Cell signaling #9217;1:1000)を添加した。
【0166】
第4日:洗浄緩衝液で、プレートを3回洗浄した後、PBS中に1:5000希釈された抗ウサギ−HRPとともに温置し、各ウェルに0.5%BSAを添加し、室温で1.5時間温置した。洗浄緩衝液で3回、PBSで1回、プレートを洗浄した。テトラメチルベンジジン(TMB、Calbiochem)を添加し、650nmでモニターした。250nMHCl100μLの添加によって反応を停止し、Vmaxプレートリーダー(Thermo Lab Systems)を使用して、650nmの参照波長として、吸光度を450nmで読み取った。
【0167】
図9は、本実験の代表的な結果を示している。本発明の抗HER−3抗体は、減少されたリン酸化によって示されたように、β−HRGによって媒介されたAKTを低下させることができた。データは、対照抗体に対する、治療抗体によるパーセント低下として示されている。
【実施例16】
【0168】
本発明のヒト抗HER−3抗体による、α−HRG/β−HRG媒介性MCF7細胞の阻害
本発明の抗体がHGRによって刺激された細胞増殖を阻害する能力を決定するために、インビトロ実験を実施した。96ウェルプレート上のFCS含有培地中に、一晩、2000個のMCF7細胞を播種した。37℃で1時間、0.5%FCSを有する培地中で希釈された抗体とともに、細胞を4つ組みで予め温置した。抗体溶液にリガンドを直接添加することによって、30ng/mLのα−HRG又は20ng/mLβ−HRG(R&DSystems)で細胞を刺激した後、72時間、増殖させた。AlamarBlueTM(BIOSOURCE)を添加し、37℃で、暗所にて温置した。30分毎に、590nmでの吸光度を測定した。アラマール・ブルーの添加から90分後に、データを採取した。図10に示されている結果は、本発明の代表的な抗体がヒト癌細胞中でのHRG誘導性細胞増殖を阻害することを示している。データは、対照抗体に対する、治療抗体によるパーセント低下として示されている。
【実施例17】
【0169】
本発明のヒト抗HER−3抗体による、β―HRG誘導性MCF7細胞遊走の阻害
本発明の抗体が細胞遊走を遮断するかどうかを調べるために、遊出実験を行った。細胞懸濁液に抗体の表記量を添加し、37℃で45分間、両者を温置することによって、血清飢餓状態にされたMCF7細胞を予め温置した。次いで、コラーゲンIによって被覆されたtranswell(BD Falcon,8μm孔)の上部チャンバー中に、細胞懸濁液(50,000細胞)500μLを配置した。単独の、又はリガンドβ−HRG−EGFドメイン(R&DSystems)を含有する750μL(MEM、アミノ酸、ピルビン酸ナトリウム、ペニシリン−ストレプトマイシン、0.1%BSA、ウシ胎児血清なし)を、下部チャンバー中で使用した。37℃で8時間、細胞を遊走させ、DAPIで染色した。
【0170】
染色された核を手作業で計数した。対照抗体と比較した阻害として、%阻害を表した。
【0171】
図11は、本発明の代表的な抗HER−3抗体が、HRGによって誘導される細胞遊走を低下させることを示す実験の結果を示している。
【実施例18】
【0172】
コロニー形成アッセイ(軟寒天アッセイ)
本発明の抗HER−3抗体が足場非依存性細胞増殖を阻害する能力を調べるために、軟寒天アッセイを実施した。このような形質転換された細胞のみが軟寒天中で増殖することができるので、軟寒天コロニー形成アッセイは、形質転換された細胞にして検査を行うための標準的なインビトロアッセイである。
【0173】
IMDM培地(Gibco)中の10μg/mLの表記抗体とともに、30分間、750から2000個の細胞(細胞株による)を予め温置し、0.4%Dfico noble寒天中に再懸濁した。96ウェルプレート中に、4つ組みで、20%FCSを含有する0.75%アガロース下層上に細胞懸濁液を播種した。コロニーを14日間形成させた後、50μLMTT(PBS中、0.5mg/mL)で一晩染色した。図12aからiは、本発明の3つの代表的な抗体を用いて実施されたこれらの実験の結果を示している。これらの結果は、本発明の抗HER−3抗体が、MDA−MB316及びNCI−ADR乳癌細胞(図12a、b)、MKN−28胃癌(図12c)、HT144悪性黒色腫(図12d)、Skov3卵巣癌細胞(図12e)、PPC−1前立腺癌(図12f)、BX−PC3膵臓癌細胞(図12g)、A431類表皮癌細胞(図12h)及び肺癌細胞(図12i)の足場非依存性細胞増殖を低下させることを示す。Scanalyzer HTSカメラシステム(Lemnatec,Wuerselen)を用いて、コロニーを計数した。
【実施例19】
【0174】
ヒト抗HRE−3抗体は、ヌードマウス中でのヒト乳癌の増殖を阻害する
しばしば、ヒト異種移植腫瘍研究において、治療用抗体の抗腫瘍効果が評価される。これらの研究では、ヒト腫瘍は、免疫無防備状態のマウス中で、異種移植片として増殖し、治療効果は、腫瘍増殖阻害の程度によって測定される。本発明の抗HER−3抗体が、ヌードマウス中でのヒト乳癌細胞の腫瘍増殖を妨害するかどうかを測定するために、雌のNMRIヌード/ヌードマウス中に、5×106個のT47D細胞を移植した。腫瘍は、動物の背中上において、皮下で増殖された。腫瘍は、移植から8日後に、20mm3の平均容積に到達した時点で、治療を開始した。第一の治療前に、マウスを無作為化し、治療群にわたって、最初の腫瘍容積(平均、中央値及び標準偏差)の均一性を確保するために、統計学的検定を行った。治療は、50mg/kgの搭載容量から開始し、続いて、腹腔内注射によって、週に一回、25mg/kgを注射した。対照腕には、ドキソルビシン(医薬等級)を与えた。全ての動物に、0.5mg/kg/週のエストロゲンを腹腔内注射して補充した。
【0175】
処置群の詳細は、以下に掲載されている。
【0176】
【表10】
【0177】
中央値腫瘍容積のデータ(図13)は、本発明の抗HER−3抗体の投与が、腫瘍増殖の低下をもたらすことを示した。
【実施例20】
【0178】
ヒト抗HRE−3抗体は、SCIDマウス中でのヒト膵臓腫瘍の増殖を阻害する
他の固体腫瘍の種類中での抗HER3抗体の治療的な可能性を検査するために、ヒト膵臓腫瘍細胞株B×PC3に由来する確立された腫瘍を有するマウスにおいて、抗HER−3抗体、U1−53及びU1−59を検査した。ビヒクル対照、PBS又は確立された治療用抗体Erbituxの何れかで処理されたマウスの対照群として含めた。5×106個のBxPC3細胞を、Matrigelなしに、CB17SCiDマウス中の皮下に接種した。140mm2の平均容積を有する確立された腫瘍を有するマウスに、腹腔内注射を介して、U1−53、U1−59、Erbituxの50mg/kg又はPBSの等容量を与えた。その後、マウスに、研究期間中、毎週1回、25mg/kgの注射を与えた。
【0179】
この実験に対する結果は、図14に示されている。U1−53及びU1−59は、細胞分裂停止状態の様式でヒト膵臓腫瘍の増殖を低下させた。注目すべきことに、本実験では、U1−53及びU1−59は、腫瘍増殖の遅延に関して、EGFR標的抗体であるErbituxより有効であった。これらのデータは、基準治療剤と比較した、本発明の抗HER−3抗体の治療的効果を示した。
【実施例21】
【0180】
ヒト抗HER−3抗体を抗EGFR抗体と組み合わせることによって、抗腫瘍活性が増加する
標的化された抗体による過剰増殖性疾患の単独療法は、しばしば、一方で、薬物耐性の発生、他方で、抗原性の変化などの問題によって妨害される。例えば、標的化された抗原を発現していない腫瘍細胞又は標的化された抗原を喪失した腫瘍細胞は選択的な増殖という利点を有するので、長期の治療後に抗原性の喪失が、腫瘍細胞を治療抗体に対して非感受性とさせ得る。これらの問題は、腫瘍細胞上の異なる受容体を標的とする治療抗体又は別の抗新生物因子と組み合わせて本発明の抗体を使用することによって回避され得る。複数のシグナル伝達経路又は関連する経路において、但し、複数の介入工程で介入することも、治療的な利点を与え得る。これらの組み合わせ治療様式は、各々、異なる作用機序を介して作用する2つの抗癌剤を組み合わせるので、より有効である可能性がある。
【0181】
本発明の抗HER−3抗体U1−53及びU1−59の適切な組み合わせ因子としての実用可能性を示すために、本発明者らは、U1−53又はU1−59の何れかが、抗EGR特異的抗体Erbituxと組み合わされる療法と、U1−53又はU1−59の単独療法の投与を比較した。5×106個のBxPC3細胞を、Matrigelとともに、CB17SCIDマウス中の皮下に接種した。腫瘍容積が200mm3に達した後、各処置群にマウスを無作為に振り分けた。単一の薬剤として、又はErbituxと抗HER3抗体の組み合わせとして、又は2つの抗HER−3抗体のカクテルとして、U1−53、U1−59及びErbituxの腹腔内投与を毎週行った。全ての抗体は、50mg/kg/週の単回投薬用量で投薬した後、6週間、25mg/kgを毎週注射した。対照腕に、ゲムシタビン(120mg/kg)に隔週投与し、ヒトIgGを毎週プールし、又はビヒクル(PBS)注射を毎週プールした。治療計画は、以下に詳述されている。
【0182】
【表11】
【0183】
この実験に対する結果は、図15に示されている。単一の薬剤として投与された場合に、抗体U1−53及びU1−59は、しばしば標準的な抗膵臓癌化学療法として使用されるゲムシタビンと同じ程度まで、ヒト膵臓腫瘍の増殖を遅延させた。U1−53又はU1−59とのErbituxの同時投与は、U1−53、U1−59又はErbituxの何れかの単一薬剤投与を用いて観察されたものより、腫瘍増殖を有意により大きく低下させた。従って、別個の腫瘍抗原を標的とする適切な抗体と本発明の抗HER−3抗体を組み合わせることによって、有益な治療的応答を達成することが可能である。
【0184】
要約すると、本発明の抗HER−3抗体は、ヒト腫瘍に対して、インビボで強力な治療効果を有する。本発明の抗HER−3抗体は、抗腫瘍活性を増加させるために、他の抗新生物治療薬と効果的に組み合わせることが可能である。
【実施例22】
【0185】
ヒト抗HRE−3抗体は、nu/nuマウス中でのヒト悪性黒色腫瘍の増殖を阻害する
Her3を含む受容体のerbBファミリーのメンバーは、極めて様々な上皮癌中で異常に発現されており、多くのこれらの固形腫瘍の増殖及び生存において重要な役割を果たしていることが知られている。これらの腫瘍には、悪性黒色腫、頭部及び頸部扁平上皮細胞癌、非小細胞肺癌及び前立腺癌、神経膠腫、胃癌、乳癌、結腸直腸癌、膵臓癌、卵巣癌が含まれる。本発明の抗Her3抗体の抗癌活性が個別の腫瘍種(例えば、膵臓癌(実施例21参照))に限定されず、多くのHER−3依存性腫瘍に対する治療薬として使用することができることを確かめるために、本発明者らは、さらなる異種移植片研究においてU1−53及びU1−59を検査した。一例が図16に示されている。CB17SCIDマウス中に、5×105個のヒト悪性黒色腫細胞HT144を皮下注射した後、U1−53及びU1−59の50mg/kg、PBSの等しい容積又は200mg/kgのダカルバシン(DITC)を直ちにその後腹腔内注射した。その後、U1−53又はU1−59の25m/kgは、週一回、マウスに与えられたのに対して、200mg/kgで、2週毎に1回、DITCを与えた。
【0186】
各処置群から得られた中央値腫瘍容積が、図16に示されている。本発明の抗体の投与は、ビヒクル対照で処理された腫瘍と比べた場合に、ヒト悪性黒色腫の増殖の低下をもたらした。これらの結果は、本発明の抗体が、それらの治療的な潜在能力に限定されず、HER−3を発現している多様な癌を標的とすることを示している。
【実施例23】
【0187】
ヒト抗HRE−3抗体は、マウス中の大腸癌異種移植片の増殖を阻害する
10×106細胞/mLの最終濃度になるように、Matrigelの2:1の比率を用いて、HT−29ヒト大腸癌細胞を培地中に懸濁した。4から5週齢のCD1nu/nuマウスの右側腹部中に、細胞懸濁液0.2mLを皮下注射した。計95匹のマウスを使用した。
【0188】
マウスを、対照群及び処置群へ無作為に割り当てた。処置は、同じ日に開始した。治療の期間は、29日であった。研究が完了した時点で、処置を施してから3時間後に、群当り3つの腫瘍を集めた。腫瘍を迅速に凍結し、−80℃に保った。
【0189】
以下の治療プロトコールを実施した。
【0190】
対照群:非特異的ヒトIgG25mg/kg、週2回、腹腔内。
【0191】
処置群:抗体U1−53、25m/kg、週2回、腹腔内。
【0192】
処置群:抗体U1−7、25m/kg、週2回、腹腔内。
【0193】
処置群:抗体U1−59、25m/kg、週2回、腹腔内。
【0194】
処置群5−FU:5−フルオロウラシル、50mg/kg、9d×5、腹腔内。
【0195】
各群から得られた中央値腫瘍容積が、図17に示されている。本発明の抗体の投与は、非特異的ヒトIgG1で処理された腫瘍と比べた場合に、HT−29大腸癌腫瘍の増殖低下をもたらした。
【実施例24】
【0196】
ヒト抗HRE−3抗体は、マウス中の肺癌増殖を阻害する
5×106細胞/mLの最終濃度になるように、Matrigelの1:1の比率を用いて、Calu−3ヒト非小細胞肺癌細胞を培地中に懸濁した。9週齢の雌CB17scidマウスの右側腹部中に、細胞懸濁液0.05mLを皮下注射した。計60匹のマウスを使用した。
【0197】
マウスを、対照群及び処置群へ無作為に選択した。処置は、同じ日に開始した。治療の期間は、32日であった。
【0198】
以下の治療プロトコールを実施した。
【0199】
PBSビヒクル群
hG対照群:非特異的ヒトIgG:25mg/kg、週2回、腹腔内。
【0200】
処置群:抗体U1−53、25m/kg、週2回、腹腔内。
【0201】
処置群:抗体U1−7、25m/kg、週2回、腹腔内。
【0202】
処置群:抗体U1−59、25m/kg、週2回、腹腔内。
【0203】
各対照群及び処置群から得られた中央値腫瘍容積が、図18に示されている。本発明の抗体の投与は、PBSビヒクル対照又は非特異的ヒトIgGで処理された腫瘍と比べた場合に、ヒト非小肺癌異種移植片の増殖の低下をもたらした。
【実施例25】
【0204】
ヒト抗HRE−3抗体は、Balb/Cマウス中でのヒト膵臓腫瘍の増殖を阻害する
5×106細胞/mLの最終濃度になるように、Matrigelの2:1の比率を用いて、ヒト膵臓BxPC3腫瘍細胞を培地中に懸濁した。5から5週齢の雌BalbCnu/nuマウスの右側腹部中に、細胞懸濁液0.2mLを皮下注射した。計100匹のマウスを使用した。
【0205】
マウスを、対照群及び処置群へ無作為に割り当てた。処置は、同じ日に開始した。治療の期間は、27日であった。
【0206】
以下の治療プロトコールを実施した。
【0207】
hIgG対照群:非特異的ヒトIgG、25mg/kg、週2回、腹腔内。
【0208】
処置群:抗体U1−53、25m/kg、週2回、腹腔内。
【0209】
処置群:抗体U1−7、25m/kg、週2回、腹腔内。
【0210】
処置群:抗体U1−59、25m/kg、毎週、腹腔内。
【0211】
Gemzar処置群、ゲムシタビン、80m/kg、毎週、腹腔内。
【0212】
各対照群及び処置群から得られた中央値腫瘍容積が、図19に示されている。本発明の抗体の投与は、非特異的ヒトIgG又はGemzarで処理された腫瘍と比べた場合に、ヒト膵臓腫瘍の増殖低下をもたらした。
【0213】
ヒト膵臓腫瘍中でのHER−3の阻害は、薬力学的実験においても示すことができた。BxPC3腫瘍異種移植片は、上述のように増殖させた。IgG1対照抗体500μgで3匹のマウスを処理し、抗HER−3抗体U1−59の500μgで3匹のマウスを処理した。第1日及び第4日目にマウスを処理した後、HER−3リン酸化(pHER−3)の抗体依存性阻害を測定するために、第5日目に屠殺した。
【0214】
プロテアーゼ阻害剤を加えた標準的なRIPA緩衝液中で、腫瘍をホモゲナイズした。4から20%のTris−グリシンゲル上で50μgの透明な可溶化液を分離し、ニトロセルロース膜上に転写し、3%ウシ血清アルブミン(BSA)中にブロックした。抗HER−3抗体(抗体21D3、Cell Signaling technology)を用いて、イムノブロッティングを行った。抗アクチン抗体(ABa−2066、Sigma)を対照として使用した。
【0215】
増強された化学発光(Amersham Biosciences,Piscataway,NJ)によって、発現を検出した。Versadoc5000Imaging System(BioRad, Hercules, CA)を用いて、画像を捕捉した。
【0216】
結果は、図20に示されている。ヒト抗HER−3抗体U1−59の投与後、HER−3のリン酸化は、もはや検出できなかった。従って、本発明の抗体は、ヒト膵臓腫瘍細胞中でのHER−3の活性化を著しく低下させることができる。
【実施例26】
【0217】
診断剤としての本発明の抗HER−3抗体の使用
抗HER−3mAbは、悪性腫瘍疾患の診断において使用することが可能である。HER−3は、正常な組織と比べて極めて異なった様式で腫瘍細胞上に発現されており、従って、HER−3の発現分析は、固形腫瘍の一次診断、固形腫瘍の段階決定及び等級付け、増殖性疾患及び悪性新生物の予後診断基準の評価並びにHER−3陽性腫瘍を有する患者におけるリスク管理を補助する。
【0218】
A.試料中でのHER−3抗原の検出
試料中でのHER−3抗原の検出用酵素結合免疫吸着検定法(ELISA)が開発されている。本アッセイでは、96ウェルミクロタイタープレート又は384ウェルマイクロタイタープレートなどのマイクロタイタープレートのウェルに、HER−3抗原に対して誘導された第一の完全なヒトモノクローナル抗体を数時間吸着させる。固定化された抗体は、検査試料中に存在し得るHER−3抗原の何れかに対する捕捉抗体としての役割を果たす。ウェルを濯ぎ、分析物の非特異的吸着を防ぐために、ミルクタンパク質又はアルブミンなどのブロッキング剤で処理した。
【0219】
続いて、HER−3抗原を含有すると疑われる検査試料で、又はHER−3抗原の標準量を含有する溶液でウェルを処理する。例えば、このような試料は、病状を診断するものと考えられる循環HER−3抗原のレベルを有すると疑われる対象から得られる血清試料である。検査試料又は標準を濯いだ後、ビオチンの連結によって標識される本発明の第二の完全ヒトモノクローナル抗HER−3抗体でウェルを処理する。標識された抗HER−3抗体は、検出抗体としての役割を果たす。過剰な二次抗体を濯いで除去した後、アビジン連結された西洋ワサビペルオキシダーゼ(HRP)及び適切な色素産生性基質でウェルを処理する。検査試料中のHER−3抗原の濃度は、標準試料から作成された標準曲線との比較によって決定される。
【0220】
B.免疫組織化学(IHC)中のHER3抗原の検出
IHCによる組織切片中のHER3抗原を測定するために、まず、パラフィン包埋された組織を、2回、5分間、キシレン中でパラフィン除去し、次いで、100%エタノールで3分間、2回、95%エタノールで1分間、水和し、蒸留水中で濯いだ。エピトープ遮蔽の除去、酵素消化又はサポニンによって、ホルマリン固定及びパラフィン包埋によって遮蔽された抗原エピトープを露出させる。エピトープの遮蔽を除去するために、例えば、2NHCl溶液(pH1.0)のようなエピトープ修復溶液中で、20から40分間にわたって、蒸し器、水槽又は電子レンジ中で切片を加熱する。酵素消化の場合には、プロテイナーゼK、トリプシン、プロナーゼ、ペプシンなどの様々な酵素溶液中において、37℃で10から30分間、組織切片を温置する。
【0221】
エピトープ修復溶液又は過剰な酵素を濯いで除去した後、非特異的な相互作用を防ぐために、ブロッキング緩衝液で組織切片を処理する。室温で1時間、又は一晩、希釈緩衝液中において、適切な希釈で一次抗体を温置する。過剰の一次抗体を濯いで除去し、室温で10分間、ペルオキシダーゼブロッキング溶液中で切片を温置する。別の洗浄工程後、酵素に対する足場としての役割を果たし得る基で標識された二次抗体とともに組織切片を温置する。従って、例は、ストレプトアビジンが連結された西洋ワサビペルオキシダーゼによって認識されるビオチン標識された二次抗体である。前記抗体/酵素複合体の検出は、適切な色素産生性基質とともに温置することによって達成される。
【0222】
C.患者の血清中のHER−3抗原濃度の測定
ヒト血清中のHER−3レベルを定量するために、サンドイッチELISAを開発する。サンドイッチELISA中で使用された2つの完全なヒトモノクローナル抗HER−3抗体は、HER−3分子上の異なるドメインを認識し、結合に関して競合しない。例えば、実施例8を参照されたい。以下のように、ELISAを実施する。2μg/mLの濃度の、コーティング緩衝液(0.1MNaHCO3、pH9.6)中の捕捉抗HER−3抗体50μLを、ELISAプレート(Fisher)上に被覆した。4℃で一晩温置した後、ブロッキング緩衝液200μL(PBS中、0.5%BSA、0.1%Tween20、0.01%チメロサール)で、25℃で1時間、プレートを処理する。PBS中の0.05%Tween20を用いて、プレートを洗浄した(3回)(洗浄緩衝液、WB)。50%ヒト血清を含有するブロッキング緩衝液中に正常な血清又は患者の血清(Clinomics,Bioreclaimation)を希釈する。4℃で一晩、血清試料とともにプレートを温置し、洗浄緩衝液で洗浄し、次いで、25℃で1時間、ビオチン化された検出抗HER−3抗体100μL/ウェルとともに温置する。洗浄後、HRP−ストレプトアビジンとともにプレートを15分間温置し、前述のように洗浄し、次いで、発色させるために、H2O2中のo−フェニレンジアミン(Sigma発色溶液)100μL/ウェルで処理する。H2SO4(2M)の50μL/ウェルで反応を停止させ、492nmでELISAプレートリーダーを用いて分析する。4パラメータ曲線フィッティングプログラムを用いて、精製されたHER−3抗原の希釈と比較することによって、血清試料中のHER−3抗原の濃度を計算する。
【0223】
患者中の癌の段階決定
項目A、B及びCで記載及び論述した結果に基づき、本発明の使用を通じて、HER−3抗原の発現レベルを基礎として、対象中の癌の段階を決定することが可能である。癌の所定の種類に関して、疾病の進行における様々な段階であるとして、及び/又は癌の治療的処置における様々な点であるとして診断された対象から血液の試料を採取する。血液試料中に存在するHER−3抗原の濃度は、存在する抗原の量を特異的に測定する方法を用いて決定される。このような方法には、項目A及びBで記載されている方法などのELISA法が含まれる。進行又は治療の各段階に対して統計学的に有意な結果を与える試料の集団を用いて、各段階に関して特徴的であると考え得るHER−3抗原の濃度範囲が表記される。
【0224】
研究されている対象中の癌の進行の段階を決定するために、又は療法の経過に対する対象の応答を特徴付けるために、血液の試料を、対象から採取し、試料中に存在するHER−3抗原の濃度を測定する。このようにして得られた濃度は、値が属する濃度範囲を同定するために使用される。このようにして同定された範囲は、診断された対象の様々な集団において同定された進行の段階又は療法の段階と相関し、これにより、研究されている対象中の段階を決定する。
【実施例27】
【0225】
過剰増殖性疾患の治療又は予防のための、本発明の抗HER−3抗体及び抗体抱合体の使用
多くの固形腫瘍は、HERファミリーに媒介されるシグナル伝達によって駆動され、HER−1、HER−2及びHER−4間の複合体形成を通じて、HER−3は不可欠なパートナーであることが示されている。従って、HER−3によって媒介されるシグナル伝達の低下又は除去は、全ての他のHERファミリーのメンバーに影響を与え、細胞シグナル伝達を損なわせ、治療的介入の幅広い枠並びに標的化される他の因子、生物試薬及び細胞毒性因子との併用療法の可能性をもたらす。従って、本発明の抗HER−3抗体は、例えば、HER−3発現のような多数の因子に基づくある種の過剰増殖性疾患又はHER−3関連疾患の治療のために使用することが可能である。乳癌、胃腸癌、膵臓癌、前立腺癌、卵巣癌、胃癌、子宮内膜癌、唾液腺癌、肺癌、腎臓癌、大腸癌、直腸結腸癌、甲状腺癌、膀胱癌、神経膠腫、悪性黒色腫、HER−3を発現又は過剰発現する他の癌などの腫瘍の種類は、好ましい適応症に該当すると思われるが、適応症は、前記のリストに掲載されているものに限定されるものではない。さらに、患者の以下の群には、抗HER−3誘導性mAb治療が有益である。
【0226】
・抗HER−2mAb治療に対して耐性を有する患者
・抗HER−2mAb治療を用いた治療に対する適格性を欠く患者
・抗HER−1mAb又は小分子抗EGFR阻害剤に対して耐性を有する患者
・エルロチニブ又はゲフィチニブに対して耐性を示す非小細胞肺癌を有する患者
本発明の抗HER−3抗体は、単独療法として、又は、いわゆる「併用療法」において、1つ若しくはそれ以上の因子と組み合わせて使用される。前記併用療法は、本発明において前記した因子を含み得るが、これに限定されるものではない。抗HER3抗体及び他の因子との併用療法は、患者の生存、腫瘍進行までの時間を延長し、又は患者の生活の質を向上させ得る。プロトコール及び投与の設計は、治療の効果に対処するのみならず、標準的な療法(例えば、化学療法又は放射線療法など)の通常用量を低下させることができる。
【0227】
本発明の抗HER−3抗体を用いたヒトの治療
腫瘍を有するヒト患者における抗HER−3抗体治療のインビボでの効果を測定するために、本発明の抗HER−3抗体の有効量を、一定回数にわたって、このようなヒト患者に注射する。治療中の一定期間に、腫瘍が進行するかどうか、特に、腫瘍が増殖及び転移するかどうかを明らかにするために、ヒト患者をモニターする。
【0228】
本発明の抗HER−3抗体で治療された腫瘍患者は、現在の治療薬の標準で治療された腫瘍患者における腫瘍増殖及び転移のレベルと比べて、腫瘍増殖及び/又は転移のより低いレベルを有する。
【0229】
本発明の抗HER−3抗体抱合体を用いた治療
本発明の抗HER−3抗体抱合体のインビボでの効果を測定するために、本発明の抗HER−3抗体抱合体の有効量を、一定回数、腫瘍を呈するヒト患者又は動物に注射する。例えば、投与される抗HER−3抗体抱合体は、DM1−抗HER−3抗体抱合体、アウリスタチン−抗HER−3抗体抱合体又は放射性同位体−抗HER−3抗体抱合体である。治療中、定期的に、腫瘍が進行するかどうか、特に、腫瘍が増殖及び転移するかどうかを明らかにするために、ヒト患者又は動物をモニターする。
【0230】
腫瘍を呈し、例えば、DM1−抗HER−3抗体抱合体又は放射線同位体−抗HER−3抗体抱合体での治療を受けているヒト患者又は動物は、腫瘍を呈し、別の療法での治療を受けている対照患者又は動物と比べて、腫瘍増殖及び転移のより低いレベルを有する。動物中で使用され得る対照DM1−抗体には、本発明の抗HER−3抗体と同じイソタイプであるが、より具体的には、HER−3腫瘍体抗原への結合能を有しない抗体に連結されたDM1を含む抱合体が含まれる。動物検査において使用され得る対照放射性同位体−抗体には、本発明の抗HER−3抗体と同じイソタイプであるが、より具体的には、HER−3腫瘍抗原への結合能を有しない抗体に連結された放射性同位体を含む抱合体が含まれる。注意:対照抱合体は、ヒトに投与されない。
【0231】
総論
前記明細書は、当業者が本発明を実施可能であるのに十分であると考えられる。寄託された実施形態は、本発明のある種の目的の一つの例示であることが意図され、機能的に均等である全ての構築物が、本発明の範囲に属するので、本発明の範囲は、寄託された構築物による範囲に限定されない。本明細書中の寄託物は、本明細書中に含まれる記載が本発明の何れかの対象(本発明の最良の態様を含む。)を実施可能とするのに不十分であることを認めるものではなく、それが表す特定の例示に特許請求の範囲を限定するものと解釈すべきでない。
【0232】
先述の記載及び実施例は、本発明のある種の好ましい実施形態を詳述し、本発明者らによって想定される最良の態様を記載する。しかしながら、先述の記載が本文中でいかに詳しく記されていても、本発明は多くの様式で実施することができ、本発明は、添付の特許請求の範囲及びそのあらゆる均等物に従って解釈しなければならないことが理解される。
【0233】
さらに、別段の定義がなければ、本発明に関連して使用される科学及び技術用語は、当業者によって一般的に理解される意味を有するものとする。さらに、文脈上別段の必要がなければ、単数形の用語は複数を含むものとし、複数形の用語は単数を含むものとする。一般的に、本明細書に記載されている細胞及び組織培養、分子生物学並びにタンパク質及びオリゴヌクレオチド又はポリヌクレオチド化学及びハイブリダイゼーションに関連して使用される命名法及びこれらの技術は、周知のものであり、本分野において一般的に使用されている。組換えDNA、オリゴヌクレオチド合成及び組織培養及び形質転換のために、標準的な技術が使用されている(例えば、電気穿孔、リポフェクション)。酵素反応及び精製技術は、製造業者の説明書に従って、又は本分野で一般的に遂行されているように、又は本明細書に記載されているように実施される。一般に、先述の技術及び操作は、本分野で周知の慣用的な方法に従い、並びに本明細書を通じて引用及び論述されている様々な一般的参考文献及びより具体的な参考文献中に記載されているように、実施される。例えば、「Sambrook et al.Molecular Cloning:A Laboratory Manual(3rd ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.(2001))」(参照により、あらゆる目的のために、本明細書に組み込まれる。)を参照されたい。本明細書に記載されている分析化学、合成有機化学及び医薬品化学及び薬化学に関して使用される命名法、並びに本明細書に記載されている分析化学、合成有機化学及び医薬品化学及び薬化学の実験室操作及び技術は、周知のものであり、本分野で一般的に使用されているものである。化学合成、化学分析、薬学的な調製、調合、及び送達、及び患者の治療に関しては、標準的な技術が使用される。
【0234】
参照による組み込み
本願に引用されている全ての参考文献(特許、特許出願、論文、教科書(これらの中に引用されている参考文献を含む。)など)は、参照により、その全体が、本明細書に組み込まれる。
【0235】
【化3】
【0236】
【化4】
【0237】
【表12】
【技術分野】
【0001】
本発明は、抗体及びその結合断片を含む、HER−3に結合する結合タンパク質及び該タンパク質をコードするポリヌクレオチドに関する。本発明の結合タンパク質を作製するための発現ベクター及び該ベクターを含む宿主細胞も提供される。さらに、本発明は、HER−3によって媒介されるシグナル伝達及び/又はそのリガンドであるヘレグリンに関連する疾病を診断及び治療するための組成物及び方法を提供する。
【背景技術】
【0002】
ヒト上皮増殖因子受容体3(HER−3、ErbB3としても知られる。)は、受容体タンパク質チロシンキナーゼであり、HER−1(EGFRとしても知られる。)、HER−2及びHER−4(Plowman et al., Proc.Natl.Acad.Sci.U.S.A.87(1990),4905−4909;Kraus et al., Proc.Natl.Acad.Sci.U.S.A.86(1989),9193−9197;and Kraus et al., Proc.Natl.Acad.Sci.U.S.A.90(1993),2900−2904)も含まれる、受容体タンパク質チロシンキナーゼの上皮成長因子受容体(EGFR)サブファミリーに属する。原型の上皮成長因子受容体と同様、膜貫通受容体であるHER−3は、細胞外リガンド結合ドメイン(ECD)、ECD内の二量体化ドメイン、膜貫通ドメイン、細胞内タンパク質チロシンキナーゼ(TKD)及びC末端リン酸化ドメインからなる。
【0003】
リガンドであるヘレグリン(HRG)は、HER−3の細胞外ドメインに結合し、他のヒト上皮増殖因子受容体(HER)ファミリーのメンバーとの二量体化及びその細胞内ドメインのトランスリン酸化を促進することによって、受容体媒介性シグナル伝達経路を活性化する。HERファミリーのメンバー間での二量体形成は、HER−3のシグナル伝達能力を拡張し、シグナルの多様化のみならず、シグナルの増幅のための手段でもある。例えば、HER−2/HER−3へテロ二量体は、HERファミリーメンバーの中で最も重要な有糸分裂促進シグナルの1つを誘導する。
【0004】
HER−3は、乳癌、胃腸癌及び膵臓癌など、癌の幾つかの種類において過剰発現されることが見出されている。興味深いことに、HER−2/HER−3の発現と非浸潤性段階から浸潤性段階への進行との間に相関が示されている(Alimandi et al., Oncogene 10,1813−1821;deFazio et al., Cancer 87, 487−498;Naidu et al., Br.J.Cancer 78, 1385−1390)。従って、HER−3によって媒介されるシグナル伝達を妨害する因子は望ましい。マウス又はキメラHER−3抗体が、US5968511、US5480968及びWO03013602などに報告されている。
【0005】
最近、HER−2に対するヒト化されたモノクローナル抗体であるHerceptin(R)は、HER−2によって媒介されるシグナル伝達を妨害し、ヒトにおいて、治療的に有効であることが示された(Fendly et al., Hybridoma 6, 359−370;Hudziak et al., Mol.Cell.Biol.9, 1165−1172;Stebbing et al., Cancer Treat.Rev.26, 287−290)。Herceptin(R)は、2つの異なる機序、すなわち、免疫系のエフェクター細胞の関与及び直接的な細胞傷害性アポトーシス誘導効果を通じて作用することが示されている。
【0006】
しかしながら、高度に増幅されたHER−2を有する患者のみが、Herceptin(R)治療に有意に応答するので、治療に適した患者の数を限定される。さらに、薬物に対する耐性の発生又は腫瘍細胞上のHER−2の発現若しくはエピトープ配列の変化のために、治療を適用することが可能な患者でさえ、抗体に対して非反応性となり、従って、その治療的な利点が喪失する場合があり得る。従って、HER−3などのHERファミリーのさらなるメンバーにアプローチする、標的をベースとする療法のためのさらなる薬物が必要とされている。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】米国特許第5968511号明細書
【特許文献2】米国特許第5480968号明細書
【特許文献3】国際公開第03013602号
【非特許文献】
【0008】
【非特許文献1】Plowman et al., Proc.Natl.Acad.Sci.U.S.A.87(1990),4905−4909
【非特許文献2】Kraus et al., Proc.Natl.Acad.Sci.U.S.A.86(1989),9193−9197
【非特許文献3】Kraus et al., Proc.Natl.Acad.Sci.U.S.A.90(1993),2900−2904
【非特許文献4】Alimandi et al., Oncogene 10,1813−1821
【非特許文献5】deFazio et al., Cancer 87, 487−498
【非特許文献6】Naidu et al., Br.J.Cancer 78, 1385−1390
【非特許文献7】Fendly et al., Hybridoma 6, 359−370
【非特許文献8】Hudziak et al., Mol.Cell.Biol.9, 1165−1172
【非特許文献9】Stebbing et al., Cancer Treat.Rev.26, 287−290
【発明の概要】
【0009】
本発明の第一の態様は、HER−3に結合する単離された結合タンパク質に関する。
【0010】
本発明の一実施形態において、本発明の単離された結合タンパク質は、(a)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH1;(b)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH2;並びに(c)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH3からなる群から選択されるCDRの少なくとも1つを含む重鎖アミノ酸配列、及び/又は(d)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL1;(e)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL2;並びに(f)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL3からなる群から選択されるCDRの少なくとも1つを含む軽鎖アミノ酸配列を含む。
【0011】
本発明の別の実施形態において、本発明の単離された結合タンパク質は、配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230からなる群から選択される重鎖アミノ酸配列、及び/又は配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232からなる群から選択される軽鎖アミノ酸配列を含む。
【0012】
本発明のさらに別の実施形態において、本発明の単離された結合タンパク質は、配列番号2及び4、6及び8、10及び12、14及び16、18及び20、22及び24、26及び28、30及び32、36及び38、42及び44、46及び48、50及び52、54及び56、60及び58、62及び64、66及び68、70及び72、74及び76、78及び82、80及び82、84及び86、88及び90、92及び94、96及び98、100及び102、104及び106、108及び110、112及び114、116及び118、122及び124、126及び128、130及び132、134及び136、138及び140、142及び144、146及び148、150及び152、154及び156、158及び160、162及び164、166及び168、170及び172、174及び176、178及び180、182及び184、186及び188、190及び192、194及び196、198及び200、202及び204、206及び208、210及び212、214及び216、218及び220、222及び224、226及び228、230及び232に示されている重鎖アミノ酸配列及び軽鎖アミノ酸配列、又は配列番号34、40、60、62若しくは120に示されている重鎖アミノ酸配列又は配列番号58若しくは64に示されている軽鎖アミノ酸配列を含む。
【0013】
本発明によれば、HER−3に結合することができる単離された結合タンパク質は、HER−3の細胞外部分中の少なくとも1つのエピトープと相互作用する。エピトープは、好ましくは、アミノ末端ドメインであるドメインL1(アミノ酸19から184)、システインに富む2つのドメインであるドメインS1(アミノ酸185から327)及びS2(アミノ酸500から632)、又はシステインに富む2つのドメインに隣接しているドメインL2(328から499)の中に位置する。エピトープは、L1及びS1の一部によって含まれるエピトープなど(但し、これに限定されない。)のドメインの組み合わせ中にも位置し得る。さらに好ましいのは、成熟HER−3、特に、成熟ヒトHER−3のアミノ酸残基1から160、161から358、359から575、1から358及び/又は359から604によって形成される三次元構造に結合する単離された結合タンパク質である。
【0014】
好ましくは、本発明の単離された結合タンパク質は、抗体様結合活性を有する足場タンパク質又は抗体、例えば、抗HER−3抗体である。特に、抗HER−3抗体は、U1−1抗体、U1−2抗体、U1−3抗体、U1−4抗体、U1−5抗体、U1−6抗体、U1−7抗体、U1−8抗体、U1−9抗体、U1−10抗体、U1−11抗体、U1−12抗体、U1−13抗体、U1−14抗体、U1−15抗体、U1−16抗体、U1−17抗体、U1−18抗体、U1−19抗体、U1−20抗体、U1−21抗体、U1−22抗体、U1−23抗体、U1−24抗体、U1−25抗体、U1−26抗体、U1−27抗体、U1−28抗体、U1−29抗体、U1−30抗体、U1−31抗体、U1−32抗体、U1−33抗体、U1−34抗体、U1−35抗体、U1−36抗体、U1−37抗体、U1−38抗体、U1−39抗体、U1−40抗体、U1−41抗体、U1−42抗体、U1−43抗体、U1−44抗体、U1−45抗体、U1−46抗体、U1−47抗体、U1−48抗体、U1−49抗体、U1−50抗体、U1−51抗体、U1−52抗体、U1−53抗体、U1−55.1抗体、U1−55抗体、U1−57.1抗体、U1−57抗体、U1−58抗体、U1−59抗体、U1−61.1抗体、U1−61抗体、U1−62又は前記抗体の1つの少なくとも1つの重鎖若しくは軽鎖を有する抗体からなる群から選択される。特に好ましくは、抗体U1−49(配列番号42/44)、U1−53(配列番号54/56)及びU1−59(配列番号70/72)又は前記抗体の1つの少なくとも1つの重鎖若しくは軽鎖を有する抗体である。
【0015】
さらに、本発明のさらなる実施形態は、標識基又はエフェクター基に連結された、単離された結合タンパク質を提供する。好ましくは、このような結合タンパク質は、過剰増殖性疾患、特に、乳癌、胃腸癌、膵臓癌、前立腺癌、卵巣癌、胃癌、子宮内膜癌、唾液腺癌、肺癌、腎臓癌、大腸癌、直腸結腸癌、甲状腺癌、膀胱癌、神経膠腫、悪性黒色腫、精巣癌、軟部組織肉腫、頭部及び頸部癌などの腫瘍疾患又はHER−3を発現若しくは過剰発現する他の癌並びに腫瘍転移の形成の治療に対して有用である。
【0016】
本発明の別の態様は、本発明の結合タンパク質をコードする単離された核酸分子、本発明の結合タンパク質をコードする核酸分子を有するベクター、並びに、このような核酸分子又はベクターで形質転換された宿主細胞、例えばCHO細胞、NS/0骨髄腫細胞を提供する。
【0017】
本発明のさらなる態様は、結合タンパク質を分泌する宿主細胞から前記結合タンパク質を調製することによって、本発明の結合タンパク質を作製する方法に関する。好ましくは、本発明の結合タンパク質は、結合タンパク質を分泌するハイブリドーマ細胞株又は本発明の結合タンパク質をコードする核酸分子で形質転換されたCHO若しくは他の細胞種から調製される。
【0018】
本発明の別の態様は、本発明の結合タンパク質をコードする1又は複数の核酸分子に関して遺伝子導入された動物、植物又は真菌の組織、産物又は分泌物から前記結合タンパク質を調製することによって、本発明の結合タンパク質を製造する方法に関する。好ましくは、本発明の結合タンパク質は、ウシ、ヒツジ、ウサギ、ニワトリ若しくは他の哺乳動物又は鳥類などのトランスジェニック動物、トウモロコシ、タバコ若しくは他の植物などのトランスジェニック植物又はアスペルギルス、ピチア又は他の真菌種などのトランスジェニック真菌の組織、産物又は分泌物から調製される。
【0019】
本発明の別の態様は、医薬として許容される担体、希釈剤及び/又は佐剤と混合された、本発明の少なくとも1つの結合タンパク質を活性因子として含む医薬組成物に関する。本発明の別の好ましい実施形態において、本発明の医薬組成物は、少なくとも1つの他の活性因子、例えば、少なくとも1つの抗新生物因子をさらに含有する。本発明のさらに別の態様は、医薬組成物の調製のための、医薬として許容される担体、希釈剤及び/又は佐剤と混合された、本発明の少なくとも1つの結合タンパク質、及び、場合によって使用される少なくとも1つの他の活性因子(例えば、少なくとも1つの抗新生物因子)の使用に関する。前記医薬組成物は、、過剰増殖性疾患、特に、乳癌、胃腸癌、膵臓癌、前立腺癌、卵巣癌、胃癌、子宮内膜癌、唾液腺癌、肺癌、腎臓癌、大腸癌、直腸結腸癌、甲状腺癌、膀胱癌、神経膠腫、悪性黒色腫又はHER−3を発現若しくは過剰発現する他の癌並びに腫瘍転移の形成を診断し、予防し、又は治療するのに適している。
【0020】
さらに、本発明は、さらなる態様において、HER−3の発現を伴う疾病又は症状を診断するための方法であり、試料を少なくとも1つの本発明の結合タンパク質と接触させること、及びHER−3の存在を検出することを含む、前記方法に関する。好ましい疾病又は症状には、上記過剰増殖性疾患が含まれる。
【0021】
本発明のさらに別の態様は、HER−3の発現を伴う疾病又は症状の予防又は治療を必要としている患者において、HER−3の発現を伴う疾病又は症状を予防又は治療する方法であり、少なくとも1つの本発明の結合タンパク質の有効量、及び、場合によって、少なくとも1つの他の活性因子(例えば、少なくとも1つの抗新生物因子)を前記患者に投与することを含む、前記方法である。好ましくは、患者は、哺乳動物患者、より好ましくは、ヒト患者である。HER−3の発現を伴う好ましい疾病又は症状は、上記過剰増殖性疾患である。
【0022】
本発明のさらなる態様は、HER−3の発現を伴う疾病又は症状を診断し、予防し、又は治療するためのキットであり、少なくとも1つの本発明の結合タンパク質及び/又は核酸分子及び/又はベクターを含む前記キットに関する。場合によって、本発明のキットは、少なくとも1つの他の活性因子、例えば、少なくとも1つの抗新生物因子をさらに含むことができる。好ましくは、HER−3の発現を伴う好ましい疾病又は症状は、上記過剰増殖性疾患である。
【図面の簡単な説明】
【0023】
【図1】図1は、ヒト癌細胞株の群におけるHER−3発現の程度を示しており、HER−3が様々なヒト癌内で発現されていることを示す。
【図2】図2は、HERファミリーの異なるメンバー又は空のベクターのみを安定に発現しているRat1細胞の何れかへのHER−3抗体結合のFACS分析の結果を示している。
【図3】図3は、HER3ドメインにマッピングされた抗体結合競合ビンを示している。
【図4】図4は、本発明の抗HER−3抗体を用いて実施された間接FACSScathcard抗体親和性分析の結果を示している。本分析は、本発明の抗HER−3抗体が、細胞表面上に発現されたHER−3に対して、高い親和性と強い結合定数を有することを示している。
【図5】図5は、本発明の抗HER−3抗体によって誘導された、HER−3の加速された飲食細胞運動を示している。
【図6a】図6aからeは、本発明の抗HER−3抗体を用いて実施されたリガンド競合アッセイの結果を示している。結果は、本発明の抗体が、内在性HER−3を発現する細胞への、[125I]−α−HRG/[125I]−β−HRGの結合を特異的に低下させることを示している。
【図6b】図6aからeは、本発明の抗HER−3抗体を用いて実施されたリガンド競合アッセイの結果を示している。結果は、本発明の抗体が、内在性HER−3を発現する細胞への、[125I]−α−HRG/[125I]−β−HRGの結合を特異的に低下させることを示している。
【図6c】図6aからeは、本発明の抗HER−3抗体を用いて実施されたリガンド競合アッセイの結果を示している。結果は、本発明の抗体が、内在性HER−3を発現する細胞への、[125I]−α−HRG/[125I]−β−HRGの結合を特異的に低下させることを示している。
【図6d】図6aからeは、本発明の抗HER−3抗体を用いて実施されたリガンド競合アッセイの結果を示している。結果は、本発明の抗体が、内在性HER−3を発現する細胞への、[125I]−α−HRG/[125I]−β−HRGの結合を特異的に低下させることを示している。
【図6e】図6aからeは、本発明の抗HER−3抗体を用いて実施されたリガンド競合アッセイの結果を示している。結果は、本発明の抗体が、内在性HER−3を発現する細胞への、[125I]−α−HRG/[125I]−β−HRGの結合を特異的に低下させることを示している。
【図7a】図7aは、本発明の抗HER−3抗体を用いて実施された、HER−3ホスホチロシンELISAの結果を示している。本発明の抗体は、増加された受容体チロシンリン酸化によって示されたように、β−HRGによって媒介されたHER−3の活性化を阻害することができた。
【図7b】本発明の抗HER−3抗体を用いて実施された、HER−3ホスホチロシンELISAの結果を示している。本発明の抗体は、増加された受容体チロシンリン酸化によって示されたように、β−HRGによって媒介されたHER−3の活性化を阻害することができた。さらに、図7bは、滴定された抗体を用いた本実験の代表的な結果を示している。
【図8】図8は、本発明の抗HER−3抗体を用いて実施された、p42/p4MAPキナーゼELISAの結果を示している。本発明の抗体は、増加されたMAP−キナーゼのリン酸化によって示されたように、β−HRGによって媒介されたp42/p44MAP−キナーゼの活性化を低下させることができた。
【図9】図9は、本発明の抗HER−3抗体を用いて実施された、ホスホ−AKTELISAの結果を示している。本発明の抗体は、AKTのリン酸化によって示されたように、β−HRGによって媒介されるAKTの活性化を低下することができた。
【図10】図10は、本発明のヒト抗HER−3抗体による、MCF7細胞の増殖の阻害を示している。本発明の抗体は、ヒト癌細胞中でのHRG誘導性細胞増殖を阻害する。
【図11】図11は、本発明のヒト抗HER−3抗体によって阻害された、MCF7細胞の遊出を示している。
【図12a】図12aからiは、本発明のヒトHER−3抗体による、足場非依存性細胞増殖の阻害を示している。
【図12b】図12aからiは、本発明のヒトHER−3抗体による、足場非依存性細胞増殖の阻害を示している。
【図12c】図12aからiは、本発明のヒトHER−3抗体による、足場非依存性細胞増殖の阻害を示している。
【図12d】図12aからiは、本発明のヒトHER−3抗体による、足場非依存性細胞増殖の阻害を示している。
【図12e】図12aからiは、本発明のヒトHER−3抗体による、足場非依存性細胞増殖の阻害を示している。
【図12f】図12aからiは、本発明のヒトHER−3抗体による、足場非依存性細胞増殖の阻害を示している。
【図12g】図12aからiは、本発明のヒトHER−3抗体による、足場非依存性細胞増殖の阻害を示している。
【図12h】図12aからiは、本発明のヒトHER−3抗体による、足場非依存性細胞増殖の阻害を示している。
【図12i】図12aからiは、本発明のヒトHER−3抗体による、足場非依存性細胞増殖の阻害を示している。
【図13】図13は、本発明のヒト抗HER−3抗体による、T47Dヒト乳癌細胞の異種移植片増殖の阻害を示している。
【図14】図14は、抗Her3(U1−59及びU1−53)又は抗EGFR(Erbitux)抗体の投与後における、マウス中のBxPC3ヒト膵臓癌細胞の低下を示している。
【図15】図15は、本発明のヒト抗HER−3抗体及び抗EGFR(Erbitux)抗体との組み合わせによる、BxPC3ヒト膵臓癌細胞の異種移植片増殖の低下を示している。
【図16】図16は、本発明の抗体が、nu/nuマウス中でのヒト悪性黒色腫(HT144)細胞の増殖を遅延させることを示す。
【図17】図17は、本発明のヒトHER−3抗体(U1−53、U1−59及びU1−7)による、HT−29ヒト大腸癌細胞の異種移植片増殖の低下を示している。
【図18】図18は、本発明のヒト抗HER−3抗体(U1−59、U1−53及びU1−7)抗体による、Calu−3ヒト肺癌細胞の異種移植片増殖の低下を示している。
【図19】図19は、本発明のヒト抗HER−3抗体(U1−7、U1−59及びU1−53)抗体による、BxPC−3ヒト膵臓癌細胞の異種移植片増殖の低下を示している。
【図20】図20は、本発明の抗体(U1−59)が、BxPC3ヒト膵臓癌異種移植片中でHER−3の抑制を引き起こすことを示す。
【発明を実施するための形態】
【0024】
本発明の第一の態様は、HER−3に結合する単離された結合タンパク質に関する。
【0025】
本発明の一実施形態において、本発明の単離された結合タンパク質は、(a)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH1;(b)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH2;並びに(c)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH3からなる群から選択されるCDRの少なくとも1つを含む重鎖アミノ酸配列、及び/又は(d)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL1;(e)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL2;並びに(f)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL3からなる群から選択されるCDRの少なくとも1つを含む軽鎖アミノ酸配列を含む。
【0026】
本発明の別の実施形態において、本発明の単離された結合タンパク質は、配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230からなる群から選択される重鎖アミノ酸配列、及び/又は配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232からなる群から選択される軽鎖アミノ酸配列を含む。
【0027】
本発明のさらに別の実施形態において、本発明の単離された結合タンパク質は、配列番号2及び4、6及び8、10及び12、14及び16、18及び20、22及び24、26及び28、30及び32、36及び38、42及び44、46及び48、50及び52、54及び56、60及び58、62及び64、66及び68、70及び72、74及び76、78及び82、80及び82、84及び86、88及び90、92及び94、96及び98、100及び102、104及び106、108及び110、112及び114、116及び118、122及び124、126及び128、130及び132、134及び136、138及び140、142及び144、146及び148、150及び152、154及び156、158及び160、162及び164、166及び168、170及び172、174及び176、178及び180、182及び184、186及び188、190及び192、194及び196、198及び200、202及び204、206及び208、210及び212、214及び216、218及び220、222及び224、226及び228、230及び232に示されている重鎖アミノ酸配列及び軽鎖アミノ酸配列、又は配列番号34、40、60、62若しくは120に示されている重鎖アミノ酸配列又は配列番号58若しくは64に示されている軽鎖アミノ酸配列を含む。
【0028】
本発明において、本発明の結合タンパク質のアミノ酸配列は、20の一般的なアミノ酸に限定されないことを理解すべきである(Immunology−A Synthesis(2nd Edition, E.S.Golub and D.R.Gren, Eds., Sinauer Associates, Sunderland, Mass.(1991)(参照により、本明細書に組み込まれる。)を参照されたい。)。例えば、アミノ酸には、20の一般的なアミノ酸の立体異性体(例えば、Dアミノ酸)、α,α−二置換されたアミノ酸、N−アルキルアミノ酸、乳酸及び他の非慣用アミノ酸などの非天然アミノ酸が含まれ得る。本発明の結合タンパク質に対する適切な成分でもあり得る非慣用アミノ酸の例には、4−ヒドロキシプロリン、γ−カルボキシグルタミン酸、ε−N,N,N−トリメチルリジン、ε−N−アセチルリジン、O−ホスホセリン、N−アセチルセリン、N−ホルミルメチオニン、3−メチルヒスチジン、5−ヒドロキシリジン、σ−N−メチルアルギニン並びに他の類似のアミノ酸及びイミノ酸(例えば、4−ヒドロキシプロリン)が含まれる。
【0029】
さらに、本発明において、配列番号1から232に示されているアミノ酸配列中の僅かな変動は、本発明によって包含されるものと想定されるが、但し、アミノ酸配列中の変動は、配列番号1から232中に示されている配列の少なくとも75%、より好ましくは少なくとも80%、90%、95%、及び最も好ましくは99%を維持する。変動は、フレームワーク領域内(すなわち、CDR外)、CDR内、又はフレームワーク領域及びCDR内に生じ得る。配列番号1から232に示されているアミノ酸配列中の好ましい変動、すなわち、少なくとも1つのアミノ酸の欠失、挿入及び/又は置換は、機能的ドメインの境界付近に生じる。構造的及び機能的ドメインは、ヌクレオチド及び/又はアミノ酸配列データを、公共の配列データベース又は独自の配列データベースと比較することによって同定することが可能である。公知の構造及び/又は機能の他の結合タンパク質中に生じる配列モチーフ又は予測されるタンパク質立体構造ドメインを同定するために、コンピュータ化された比較法を使用することが可能である。公知の三次元構造に折り畳まれるタンパク質配列を同定するための方法は公知である。例えばBowie et al., Science 253, 164(1991);Proteins, Structures and Molecular Principles(Creighton, Ed., W.H.Freeman and Company, New York(1984));Introduction to Protein Structure(C.Branden and J.Tooze, eds., Garland Publishing, New York, N.Y.(1991 ));及びThornton et al., Nature 354:105(1991)を参照されたい(これらは全て、参照により本明細書中に組み込まれる。)。従って、当業者であれば、本発明に従って構造及び機能的ドメインを定義するために使用され得る配列モチーフ及び構造的立体構造を認めることができる。
【0030】
配列番号1から174及び1から232に示されているアミノ酸配列の特に好ましい変動は、タンパク分解若しくは酸化に対して低下した感受性をもたらし、グリコシル化パターンを変化させ、又は結合親和性を変化させ、又は結合タンパク質の他の物理化学的若しくは機能的特性を付与若しくは修飾する変動である。特に、保存的アミノ酸置換が想定される。保存的な置換とは、それらの側鎖が関連するアミノ酸のファミリー内で生じる置換である。好ましいアミノ酸ファミリーは、以下のとおりである。酸性ファミリー=アスパギン酸、グルタミン酸;塩基性ファミリー=リジン、アルギニン、ヒスチジン;非極性ファミリー=アラニン、バリン、ロイシン、イソロイシン、プロリン、フェニルアラニン、メチオニン、トリプトファン;及び非帯電極性ファミリー=グリシン、アスパラギン、グルタミン、システイン、セリン、スレオニン、チロシン。より好ましいファミリーは、脂肪族ヒドロキシファミリー=セリン及びスレオニン;アミド含有ファミリー=アスパラギン及びグルタミン;脂肪族ファミリー=アラニン、バリン、ロイシン及びイソロイシン;並びに芳香族ファミリー=フェニルアラニン、トリプトファン及びチロシン。例えば、ロイシンのイソロイシン又はバリンとの単離された置換、アスパラギン酸のグルタミン酸との単離された置換、スレオニンのセリンとの単離された置換、又はあるアミノ酸の構造的に関連したアミノ酸との類似の置換は、特に置換がフレームワーク部位内のアミノ酸を含まなければ、生じた結合タンパク質の結合又は特性に対して多大な影響を有しない。しかしながら、全ての他の可能なアミノ酸置換も、本発明によって包含される。アミノ酸の変化が、機能的な結合タンパク質、すなわち、HER−3へ結合する結合タンパク質をもたらし、HERファミリーのメンバーのシグナル伝達を低下させるかどうかは、HER−3への結合に関するELISA若しくはFACSにおいて、又はインビトロ若しくはインビボでの機能的アッセイにおいて、得られた結合タンパク質の特異的活性をアッセイすることによって容易に決定することができる。
【0031】
本発明によれば、本発明の結合タンパク質は、HER−3の細胞外部分中の少なくとも1つのエピトープと相互作用する。エピトープは、好ましくは、アミノ末端ドメインであるドメインL1(アミノ酸19から184)、システインに富む2つのドメインであるドメインS1(アミノ酸185から327)及びS2(アミノ酸500から632)、システインに富む2つのドメインに隣接しているドメインL2(328から499)又はHER−3ドメインの組み合わせの中に位置する。エピトープは、L1及びS1の一部によって構成されるエピトープなど(但し、これに限定されない。)のドメインの組み合わせ中にも位置し得る。さらに、本発明の結合タンパク質は、HER−3へのその結合がHER−3によって媒介されるシグナル伝達を低下させることをさらに特徴とする。本発明に従って、HER−3によって媒介されるシグナル伝達の低下は、例えば、細胞表面からHER−3分子を少なくとも部分的に消失させるHER−3の下方制御によって、又は実質的に不活性な形態(すなわち、安定化されていない形態に比べて、より低いシグナル伝達を示す形態)の、細胞表面上のHER−3の安定化によって引き起こされ得る。あるいは、HER−3によって媒介されるシグナル伝達の低下は、HER−3への、リガンド又はHERファミリーの別のメンバーの結合、HER−2へのGRB2の結合若しくはSHCへのGRB2の結合に影響を与える(例えば、減少又は阻害する)ことによって、受容体チロシンリン酸化、AKTリン酸化、PYK2チロシンリン酸化若しくはERK2リン酸化を阻害することによって、又は腫瘍の浸潤性を減少させることによっても引き起こされ得る。あるいは、HER−3によって媒介されるシグナル伝達の低下は、他のHERファミリーのメンバーとのHER−3含有二量体の形成に影響を与える(例えば、減少又は阻害する)ことによっても引き起こされ得る。とりわけ、1つの例は、HER−3−EGFRタンパク質複合体の形成を減少又は阻害することであり得る。
【0032】
好ましくは、本発明の結合タンパク質は、抗体様結合活性を有する足場タンパク質又は抗体、すなわち、抗HER−3抗体である。
【0033】
本発明の文脈内で、本明細書において使用される「足場タンパク質」という用語は、アミノ酸の挿入、置換又は欠失が高度に許容される露出された表面を有するポリペプチド又はタンパク質を意味する。本発明に従って使用することができる足場タンパク質の例は、スタフィロコッカス・オーレウス(Staphylococcus aureus)から得られるプロテインA、ピエリス・ブラッシッカエ(Pieris brassicae)から得られるビリン結合タンパク質又はその他のリポカリン、アンキリン反復タンパク質及びヒトフィブロネクチンである(Binz and Pluckthun, Curr Opin Biotechnol.16, 459−69中に概説されている。)。足場タンパク質の操作は、安定に折り畳まれたタンパク質の構造的フレームワーク上に又は構造的フレームワーク中に親和性機能を移植し、又は組み込むこととみなすことができる。親和性機能とは、本発明に従うタンパク質結合親和性を意味する。足場は、結合特異性を付与するアミノ酸配列から構造的に分離可能である。一般に、このような人工的親和性試薬の開発に適していると思われるタンパク質は、推論によって、又は最も一般的には、HER3(精製されたタンパク質又は細胞表面上にディスプレイされたタンパク質の何れか)に対するパニングなどの、インビトロでディスプレイされた人工的足場ライブラリーの結合剤に対するコンビナトリアルタンパク質工学技術(これらの技術は、本分野において公知である(Skerra, J.Mol.Recog., 2000;Binz and Pluckthun, 2005))によって取得され得る。さらに、抗体様結合活性を有する足場タンパク質は、足場ドメインを含有するアクセプターポリペプチドに由来することができ、アクセプターポリペプチドを含有する足場ドメイン上にドナーポリペプチドの結合特異性を付与するために、アクセプターポリペプチドには、ドナーポリペプチドの結合ドメインを移植することができる。前記挿入された結合ドメインは、例えば、抗体、特に、抗HER−3抗体の相補性決定領域(CDR)であり得る。挿入は、例えば、当業者に周知の組み換え法の様々な形態による、ポリペプチド合成、コードするアミノ酸の核酸合成など、当業者に公知の様々な方法によって達成することが可能である。
【0034】
さらに、本明細書において使用される「抗体」又は「抗HER−3抗体」という用語は、モノクローナル抗体、ポリクローナル抗体、組換え抗体、ヒト化抗体(Jones et al., Nature 321(1986),522−525;Riechmann et al., Nature 332(1988), 323−329;and Presta, Curr.Op.Struct.Biol.2(1992),593−596)、キメラ抗体(Morrison et al.,Proc.Natl.Acad.Sci.U.S.A.81(1984),6851−6855)、少なくとも2つの抗体から形成された多重特異的抗体(例えば、二重特異的抗体)又はこれらの抗体断片を意味する。「抗体断片」という用語は、上記抗体のあらゆる一部、好ましくはそれらの抗原結合領域又は可変領域を含む。抗体断片の例には、Fab断片、Fab’断片、F(ab’)2断片、Fv断片、ダイアボディ(Hollinger et al., Proc.Natl.Acad.Sci.U.S.A.90(1993),6444−6448)、一本鎖抗体分子(Pluckthun in:The Pharmacology of Monoclonal Antibodies 113, Rosenburg and Moore, EDS, Springer Verlag,N.Y.(1994),269−315)及びHER−3への所望の結合能を示す限り他の断片が含まれる。
【0035】
さらに、本明細書において使用される「抗体」又は「抗HER−3抗体」という用語は、抗体の加工されたサブドメインを含有する抗体様分子又は天然に存在する抗体変種を含み得る。これらの抗体様分子は、ラクダ科の動物(camelid)などの天然の取得源から得られた(Muyldermans et al.,Reviews in Molecular Biotechnology 74,277−302)、又は、ヒト、ラクダ科の動物若しくはその他の種から得たライブラリーのインビトロディスプレーを通じて得られた(Holt et al.,Trends Biotechnol.,21,484−90)VHのみ又はVLのみのドメインなどの単一ドメイン抗体であり得る。
【0036】
本発明において、「Fv断片」は、完全な抗原認識及び結合部位を含有する最低の抗体断片である。これらの領域は、緊密に非共有会合した1つの重鎖可変ドメインと1つの軽鎖可変ドメインの二量体からなる。各可変ドメインの3つのCDRが、VH−VL二量体の表面上の抗原結合部位を規定するために相互作用するのは、この立体配置中である。一括して、6つのCDRが抗体への抗原結合特異性を付与する。しかしながら、単一の可変ドメイン(又は抗原に対して特異的な3つのCDRのみを含むFvの半分)でさえ、一般に、完全な結合部位より低い親和性であるが、抗原を認識及び結合する能力を有する。「Fab断片」は、軽鎖の定常ドメイン及び重鎖の第一の定常ドメイン(CH1)も含有する。「Fab断片」は、重鎖CH1ドメインのカルボキシ末端に、抗体ヒンジ領域由来の1つ又はそれ以上のシステインなど、数個の残基が付加されている点で、「Fab’断片」と異なる。「F(ab’)2断片」は、当初、ヒンジシステインをその間に有する「Fab’断片」の対として作製される。パパイン又はペプシン消化など、このような抗体断片を調製する方法は、当業者に公知である。
【0037】
本発明の好ましい実施形態において、本発明の抗HER−3抗体は、IgA−、IgD、IgE、IgG又はIgM型、好ましくは、IgG1、IgG2、IgG3、IgG4、IgM1及びIgM2型など(但し、これらに限定されない。)のIgG又はIgM型のものである。最も好ましい実施形態において、抗体は、IgG1、IgG2又はIgG4種である。
【0038】
本発明の別の好ましい実施形態において、本発明の抗HER−3抗体は、HER−3の細胞外ドメイン(ECD)に対して誘導された抗HER−3抗体である。
【0039】
ある観点において、例えば、治療候補物としてHER−3に対する抗体を作製することに関して、本発明の抗HER−3抗体は、補体を固定し、補体依存性細胞傷害(CDC)に関与できることが望ましい場合があり得る。マウスIgM、マウスIgG2a、マウスIgG2b、マウスIgG3、ヒトIgM、ヒトIgG1、ヒトIgG3及びヒトIgAなど(但し、これらに限定されない。)、補体の固定及び補体依存性細胞傷害(CDC)への関与が可能な抗体のイソタイプが多数存在する。作製される抗体は、このようなイソタイプを最初から有する必要はなく、むしろ、作製された抗体は、あらゆるイソタイプを有することができること、並びに、本分野において周知である慣用の分子生物学的技術を用いて、適切な発現ベクター中に分子クローニングされた定常領域遺伝子又はcDNAへ、分子クローニングされたV領域遺伝子又はcDNAを付加し、次いで、本分野で公知の技術を用いて宿主細胞中で抗体を発現させることによって抗体をイソタイプ交換できることが理解される。イソタイプ交換された抗体は、天然に存在する変種に比べて優れたCDCを有するように分子的に加工され(Idusogie et al., J Immunol.,166,2571−2575)、本分野で公知の技術を用いて、宿主細胞中で組換え的に発現されたFc領域も有し得る。このような技術には、とりわけ、直接的な組換え技術(例えば、U.S.Patent No.4,816,397を参照。)、細胞−細胞融合技術(例えば、U.S.Patent Nos.5,916,771及び6,207,418)の使用が含まれる。細胞−細胞融合技術では、何れかの所望のイソタイプを有する重鎖を保有する骨髄腫又は他の細胞株(CHOなど)が調製され、及び軽鎖を保有する別の骨髄腫又は他の細胞株(CHOなど)が調製される。その後、このような細胞を融合し、完全な状態の抗体を発現する細胞株を単離することができる。例として、(抗体の特異性及び抗体の親和性の幾つかを規定する)同じ可変領域を有しながら、ヒトIgM、ヒトIgG1又はヒトIgG3イソタイプを作製するために、HER−3抗原への所望の結合を有するヒト抗HER−3IgG4抗体を容易にイソタイプ交換させ得る。その後、このような分子は、補体を固定し、CDCへ関与することが可能であり得る。
【0040】
さらに、本発明の抗HER−3抗体は、単球及びナチュラルキラー(NK)細胞などのエフェクター細胞上のFc受容体へ結合することができ、抗体依存性細胞傷害(ADCC)に関与することも望ましい場合があり得る。マウスIgG2a、マウスIgG2b、マウスIgG3、ヒトIgG1及びヒトIgG3など(但し、これらに限定されない。)、上記のことが可能な抗体のイソタイプが多数存在する。作製される抗体は、このようなイソタイプを最初から有する必要はなく、むしろ、作製された抗体は、あらゆるイソタイプを有することができること、並びに、本分野において周知である慣用の分子生物学的技術を用いて、適切な発現ベクター中に分子クローニングされた定常領域遺伝子又はcDNAへ、分子クローニングされたV領域遺伝子又はcDNAを付加した後、本分野で公知の技術を用いて抗体を宿主細胞中で発現させることによって、抗体のイソタイプ交換を実施できることが理解される。イソタイプ交換された抗体は、天然に存在する変種に比べて優れたADCCを有するように分子的に加工され(Shields et al.J Biol Chem.,276,6591−6604)、本分野で公知の技術を用いて、宿主細胞中で組換え的に発現されたFc領域も有し得る。このような技術には、とりわけ、直接的な組換え技術(例えば、U.S.Patent No.4,816,397を参照。)、細胞−細胞融合技術(例えば、U.S.Patent Nos.5,916,771及び6,207,418を参照。)の使用が含まれる。細胞−細胞融合技術では、何れかの所望のイソタイプを有する重鎖を保有する骨髄腫又は他の細胞株(CHOなど)が調製され、及び軽鎖を保有する別の骨髄腫又は他の細胞株(CHOなど)が調製される。その後、このような細胞を融合し、完全な状態の抗体を発現する細胞株を単離することができる。例として、(抗体の特異性及び抗体の親和性の幾つかを規定する)同じ可変領域を有しながら、ヒトIgG1又はヒトIgG3イソタイプを作製するために、HER−3抗原への所望の結合を有するヒト抗HER−3IgG4抗体を容易にイソタイプ交換させ得る。次いで、このような分子は、エフェクター細胞上のFcγRへ結合し、ADCCに関与することが可能であり得る。
【0041】
さらに、本発明によれば、本発明の抗HER−3抗体は、完全なヒト抗体又はヒト化抗体であることが理解される。ヒト抗体は、異種抗体、例えば、マウス又はラットの可変及び/又は定常領域を有する抗体に伴う問題の幾つかを回避する。マウス又はラットに由来するタンパク質など、異種由来のタンパク質の存在は、患者による、抗体に対する免疫反応を惹起させることができ、続いて、抗体の迅速な除去、抗体の中和を通じた治療的有用性の喪失及び/又は重篤な、さらには生命を脅かすアレルギー反応をもたらし得る。
【0042】
好ましくは、本発明の抗HER−3抗体は、U1−1抗体、U1−2抗体、U1−3抗体、U1−4抗体、U1−5抗体、U1−6抗体、U1−7抗体、U1−8抗体、U1−9抗体、U1−10抗体、U1−11抗体、U1−12抗体、U1−13抗体、U1−14抗体、U1−15抗体、U1−16抗体、U1−17抗体、U1−18抗体、U1−19抗体、U1−20抗体、U1−21抗体、U1−22抗体、U1−23抗体、U1−24抗体、U1−25抗体、U1−26抗体、U1−27抗体、U1−28抗体、U1−29抗体、U1−30抗体、U1−31抗体、U1−32抗体、U1−33抗体、U1−34抗体、U1−35抗体、U1−36抗体、U1−37抗体、U1−38抗体、U1−39抗体、U1−40抗体、U1−41抗体、U1−42抗体、U1−43抗体、U1−44抗体、U1−45抗体、U1−46抗体、U1−47抗体、U1−48抗体、U1−49抗体、U1−50抗体、U1−51抗体、U1−52抗体、U1−53抗体、U1−55.1抗体、U1−55抗体、U1−57.1抗体、U1−57抗体、U1−58抗体、U1−59抗体、U1−61.1抗体、U1−61抗体及びU1−62からなる群から選択される。
【0043】
本発明の好ましい実施形態において、本発明の結合タンパク質は、標識基に連結される。このような結合タンパク質は、特に、診断用途に適している。本明細書において使用される、「標識基」という用語は、検出可能なマーカー、例えば、放射性標識されたアミノ酸又は印を付けたアビジン(例えば、光学的方法又は比色分析法によって検出することができる、蛍光マーカー又は酵素活性に結合されたストレプトアビジン)によって検出することができるビオチン部分を表す。抗体などのポリペプチド及び糖タンパク質を標識するための様々な方法が本分野において公知であり、本発明を実施する際に使用され得る。適切な標識基の例には、以下の放射性同位体又は放射性核種(例えば、3H、14C、15N、35S、90Y、99Tc、111In、125I、131I)、蛍光基(例えば、FITC、ローダミン、ランタニドリン光体)、酵素基(例えば、西洋ワサビペルオキシダーゼ、β−ガラクトシダーゼ、ルシフェラーゼ、アルカリホスファターゼ)、化学発光基、ビオチン基又は二次レポーターによって認識される所定のポリペプチドエピトープ(例えば、ロイシンジッパー対配列、二次抗体に対する結合部位、金属結合ドメイン、エピトープタグ)が含まれるが、これらに限定されない。ある観点において、立体的障害の可能性を減らすために、標識基には、様々な長さのスペーサーアームを付着させることが望ましい場合があり得る。
【0044】
あるいは、本発明の別の好ましい実施形態において、本発明の結合タンパク質は、エフェクター基に連結され得る。このような結合タンパク質は、特に、治療用途に適している。本明細書において使用される「エフェクター基」という用語は、放射線同位体若しくは放射線核種、毒素、治療基又は本分野において公知の他のエフェクター基などの細胞毒性基を表す。適切なエフェクター基の例は、放射線同位体又は放射線核種(例えば、3H、14C、15N、35S、90Y、99Tc、111In、125I、131I)、カリチアマイシン、アウリスタチンなどのドラスタチン類縁体並びにゲルダナマイシン及びマイタンシン誘導体(DM1を含む。)などの化学療法剤である。ある観点において、立体的障害の可能性を減らすために、エフェクター基には、様々な長さのスペーサーアームを付着させることが望ましい場合があり得る。
【0045】
本発明の第二の態様は、結合タンパク質を分泌する宿主細胞から結合タンパク質を調製する工程を含む、本発明の単離された結合タンパク質を調製する方法に関する。本発明に従って使用され得る宿主細胞は、ハイブリドーマ;哺乳動物細胞(例えば、ハムスター、ウサギ、ラット、ブタ、マウス又はその他の動物細胞)、植物細胞、真菌細胞(例えば、サッカロミセス・セレビシアエ(Saccharomyces cerevisiae)、ピチア・パストリス(Pichia pastoris))などの真核細胞;イー・コリ(E.coli)などの原核細胞及び結合タンパク質の産生のために本分野で使用される他の細胞である。足場タンパク質又は抗体などの結合タンパク質を宿主細胞から調製及び単離するための様々な方法が本分野において公知であり、本発明を実施する際に使用され得る。さらに、パパイン又はペプシン消化、近代的なクローニング技術、一本鎖抗体分子を調製するための技術(Pluckthun in:The Pharmacology of Monoclonal Antibodies 113, Rosenburg and Moore, EDS, Springer Veriag, N.Y.(1994), 269−315)及びダイアボディ(Hollinger et al., Proc.Natl.Acad.Sci.U.S.A.90(1993),6444−6448)など、結合タンパク質断片(例えば、足場タンパク質断片又は抗体断片)を調製するための方法も当業者に公知であり、本発明を実施する際に使用し得る。
【0046】
本発明の好ましい実施形態において、本発明の結合タンパク質は、結合タンパク質を分泌するハイブリドーマから調製される。例えば、「Kohler et al., Nature 256(1975),495」を参照されたい。
【0047】
本発明のさらなる好ましい実施形態において、本発明の結合タンパク質は、宿主細胞中での結合タンパク質の発現を最適化及び/又は増幅し、前記宿主細胞からの結合タンパク質を単離することによって、組み換え的に調製される。この目的のために、結合タンパク質をコードするDNAで、又は結合タンパク質をコードするDNAを含有するベクターで宿主細胞を形質転換又は形質移入し、本発明の結合タンパク質を産生するのに適した条件下で培養する。例えば、U.S.Patent No.4,816,567を参照されたい。好ましい宿主細胞は、CHO細胞、NS/0骨髄腫細胞、ヒト胎児性腎臓293細胞、イー・コリ及びサッカロミセス・セレビシアエであり得る。
【0048】
抗体である結合タンパク質に関して、これらの抗体は、完全なヒト抗体を作製するように遺伝子操作された動物から、又はバクテリオファージ、酵母、リボソーム又はイー・コリ中に作製された抗体ディスプレイライブラリーから調製され得る。例えば、Clackson et al., Nature 352(1991), 624−628, Marks et al., J.Mol.Biol.222(1991), 581−597, Feldhaus and Siegel J Immunol Methods.290, 69−80, Groves and Osbourn, Expert Opin Biol Ther., 5, 125−135 and Jostock and Dubel, Comb Chem High Throughput Screen.8, 127−133を参照されたい。
【0049】
ヒト抗体は、マウス又はラットの可変及び/又は定常領域を有する抗体に伴う問題の幾つかを回避する。このようなマウス又はラット由来のタンパク質の存在は、抗体の急速な除去をもたらすことができ、又は、患者による、抗体に対する免疫応答の生成をもたらすことができる。マウス又はラット由来の抗体の使用を回避するために、げっ歯類、他の哺乳動物又は動物が完全なヒト抗体を産生するように、げっ歯類、他の哺乳動物又は動物中への機能的なヒト抗体遺伝子坐の導入を通じて、完全なヒト抗体を作製することが可能である。
【0050】
完全なヒト抗体を作製するための1つの方法は、ヒト重鎖遺伝子坐及びκ軽鎖遺伝子坐の245kb及び190kbサイズの生殖系列配置断片を含有するように操作されたマウスのXENOMOUSE(R)系統の使用を通じた方法である。マウスの他のXenoMouse系統は、ヒト重鎖遺伝子坐及びκ軽鎖遺伝子坐の980kb及び800kbサイズの生殖系列配置断片を含有する。マウスのさらに他のXenoMouse系統は、ヒト重鎖遺伝子坐及びκ軽鎖遺伝子坐の980kb及び800kbサイズの生殖系列配置断片に加えて、740kbのサイズの生殖系列構成された完全なヒトλ軽鎖遺伝子坐を含有する。「Mendez et al.Nature Genetics 15:146−156(1997)」及び「Green and Jakobovits J.Exp.Med.188:483−495(1998)」を参照されたい。XENOMOUSE(R)系統は、Abgenix,Inc.(Fremont,CA)から入手可能である。
【0051】
XENOMOUSE(R)マウスの作製は、1990年1月12日に出願されたU.S.Patent Application Serial Nos.07/466,008、1990年11月8日に出願された07/610,515、1992年7月24日に出願された07/919,297、1992年7月30日に出願された07/922,649、1993年3月15日に出願された08/031,801、1993年8月27日に出願された08/112,848、1994年4月28日に出願された08/234,145、1995年1月20日に出願された08/376,279、1995年4月27日に出願された08/430,938、1995年6月5日に出願された08/464,584、1995年6月5日に出願された08/464,582、1995年6月5日に出願された08/463,191、1995年6月5日に出願された08/462,837、1995年6月5日に出願された08/486,853、1995年6月5日に出願された08/486,857、1995年6月5日に出願された08/486,859、1995年6月5日に出願された08/462,513、1996年10月2日に出願された08/724,752及び1996年12月3日に出願された08/759,620、2002年11月20日に出願されたU.S.Patent Publication 2003/0217373並びにU.S.Patent Nos.6,833,268、6,162,963、6,150,584、6,114,598、6,075,181及び5,939,598並びにJapanese Patent Nos.3 068 180 B2、3 068 506 B2及び3 068 507 B2に、さらに論述及び描写されている。1996年6月12日に公開されたEuropean Patent No., EP 0 463 151 B1、1994年2月3日に公開されたInternational Patent Application No., WO 94/02602、1996年10月31日に公開されたInternational Patent Application No., WO 96/34096、1998年6月11日に公開されたWO98/24893、2000年12月21日公開されたWO00/76310も参照されたい。上記特許、出願及び参考文献の各々の開示の全体が、参照により、本明細書に組み込まれる。
【0052】
別のアプローチにおいて、GenPharm International,Inc.,などの他の者は、「ミニローカス」アプローチを使用してきた。ミニローカスアプローチにおいて、Ig遺伝子坐から得た片(各遺伝子)を含めることによって、外来Ig遺伝子坐が模倣される。従って、1つ又はそれ以上のVH遺伝子、1つ又はそれ以上のDH遺伝子、1つ又はそれ以上のJH遺伝子、μ定常領域及び第二の定常領域(好ましくは、γ定常領域)が、動物内への挿入のために、構築物の中に形成される。本アプローチは、Suraniらに対するU.S.Patent No.5,545,807及びそれぞれ、LonbergとKayに対するU.S.Patent Nos.5,545,806、5,625,825、5,625,126、5,633,425、5,661,016、5,770,429、5,789,650、5,814,318、5,877,397、5,874,299及び6,255,458、Krimpenfort及びBernsに対するU.S.Patent No.5,591,669及び6,023.010、Bernsらに対するU.S.Patent Nos.5,612,205、5,721,367及び5,789,215並びにChoi及びDunnに対するU.S.Patent No.5,643,763、並びに1990年8月29日に出願されたGenPharm International U.S.Patent Application Serial Nos.07/574,748、1990年8月31日に出願された07/575,962、1991年12月17日に出願された07/810,279、1992年3月18日に出願された07/853,408、1992年6月23日に出願された07/904,068、1992年12月16日に出願された07/990,860、1993年4月26日に出願された08/053,131、1993年7月22日に出願された08/096,762、1993年11月18日に出願された08/155,301、1993年12月3日に出願された08/161,739、1993年12月10日に出願された08/165,699、1994年3月9日に出願された08/209,741(これらの開示内容は、参照により、本明細書に組み込まれる。)に記載されている。European Patent No.0 546 073 B1、International Patent Application Nos.WO92/03918、WO92/22645、WO92/22647、WO92/22670、WO93/12227、WO94/00569、WO94/25585、WO96/14436、WO97/13852及びWO 98/24884並びにU.S.Patent No.5,981,175(これらの開示内容全体が、参照により、本明細書に組み込まれる。)も参照されたい。さらに、Taylor et al., 1992、Chen et al., 1993、Tuaillon et al., 1993、Choi et al., 1993、Lonberg et al.,(1994)、Taylor et al.,(1994)及びTuaillon et al.,(1995)、Fishwild et al.,(1996)(これらの開示内容全体が、参照により、本明細書に組み込まれる。)を参照されたい。
【0053】
Kirinは、ミクロセル融合を通じて、染色体の巨大片又は完全な染色体が導入されたマウスからのヒト抗体の作製も示した。European Patent Application Nos.773 288及び843 961(これらの開示内容全体が、参照により、本明細書に組み込まれる。)を参照されたい。さらに、KirinのTcマウスとMedarexのミニローカス(Humab)マウスの交雑から得られるKMTMマウスが作製されている。これらのマウスは、KirinマウスのHC導入染色体(transchromosome)とMedarexマウスのκ鎖導入遺伝子を有する(Ishida et al., Cloning Stem Cells,(2002) 4:91−102)。
【0054】
ヒト抗体は、インビトロ法によって得ることもできる。適切な例には、ファージディスプレイ(Cambridge Antibody Technology、Morphosys、Dyax、Biosite/Medarex、Xoma、Symphogen、Alexion(旧Proliferon)、Affimedによって市販されている。)、リボソームディスプレイ(Cambridge Antibody Technologyによって市販されている。)、酵母ディスプレイなどが含まれるが、これらに限定されない。
【0055】
抗体は、本明細書に記載されているように、以下に記載されているとおりにXENOMOUSE(R)技術を使用することによって調製された。その後、このようなマウスは、ヒト免疫グロブリン分子及び抗体を産生することができ、マウス免疫グロブリン分子及び抗体の産生を欠失している。これを達成するために使用された技術は、本明細書の背景技術中に開示されている特許、出願及び参考文献中に開示されている。しかしながら、とりわけ、マウス及びマウスから得られる抗体のトランスジェニック産生の好ましい実施形態は、1996年12月3日に出願されたU.S.Patent Application Serial No.08/759,620及び1998年6月11日に公開されたInternational Patent Application Nos.WO 98/24893及び2000年12月21日に公開されたWO00/76310(これらの開示内容は、参照により、本明細書に組み込まれる。)に開示されている。「Mendez et al.Nature Genetics 15:146−156(1997)」(本開示内容は、参照により、本明細書に組み込まれる。)も参照されたい。
【0056】
このような技術の使用を通じて、様々な抗原に対する完全なヒトモノクローナル抗体が作製されてきた。基本的には、マウスのXENOMOUSE(R)株を目的の抗原(例えば、HER−3)で免疫し、抗体を発現したマウスから(B細胞などの)リンパ系細胞を回収し、不死化ハイブリドーマ細胞株を調製するために、回収された細胞株を骨髄球型細胞株と融合する。目的の抗原に対して特異的な抗体を産生するハイブリドーマ細胞株を同定するために、これらのハイブリドーマ細胞株をスクリーニングし、選択する。本明細書には、HER−3に対して特異的な抗体を産生する複数のハイブリドーマ細胞株を作製するための方法が提供されている。さらに、本明細書において、このような抗体の重鎖及び軽鎖のヌクレオチド及びアミノ酸配列分析など、このような細胞株によって産生された抗体の性質決定が提供される。
【0057】
一般に、融合されたハイブリドーマによって産生される抗体は、完全なヒトκ軽鎖を有するヒトIgG1重鎖であった。本明細書に記載されている抗体は、ヒトIgG4重鎖及びIgG1重鎖を有する。抗体は、IgG2又はIgG3など、他のヒトイソタイプとすることもできる。抗体は、高い親和性を有し、典型的には、固相及び細胞ベースの技術によって測定された場合に、約10−6から約10−13M又はそれ以下のKDを有する。
【0058】
本発明の別の態様は、本発明の結合タンパク質をコードする単離された核酸分子に関する。本発明の文脈の中で、本明細書で使用される「単離された核酸分子」という用語は、ゲノム、cDNA若しくは合成起源又はこれらの幾つかの組み合わせのポリヌクレオチドを意味し、その起源に基づいて、「単離されたポリヌクレオチド」は、(1)「単離されたポリヌクレオチド」が本来その中に見出されるポリヌクレオチドの全部又は一部と会合していない、(2)本来連結されていないポリヌクレオチドに作用可能に連結されている、又は(3)より大きな配列の一部として本来存在しない。さらに本明細書において使用される「核酸分子」という用語は、少なくとも10塩基長のヌクレオチド(リボヌクレオチド若しくはデオキシヌクレオチドの何れか、又は修飾された若しくは置換された糖基を有するヌクレオチドなど、ヌクレオチドの何れかのタイプの修飾された形態)のポリマー形態を意味する。本用語は、DNAの一本鎖及び二本鎖形態も含む。
【0059】
本発明の一実施形態において、本発明の核酸分子は、調節配列へ作用可能に連結されている。本明細書において使用される「調節配列」という用語は、これらが連結されているコード配列の発現及びプロセッシングに影響を与えるのに必要なポリヌクレオチド配列を表す。このような調節配列の性質は、宿主生物に応じて異なる。原核生物では、このような調節配列には、一般に、プロモーター、リボソーム結合部位及び転写終結配列が含まれる。真核生物では、このような調節配列には、一般に、プロモーター及び転写終結配列が含まれる。本発明において、「調節配列」という用語は、最小限、その存在が発現及びプロセッシングに不可欠である全ての成分を含むものとし、その存在が有利である追加の成分、例えば、リーダー配列及び融合対配列も含むことが可能である。さらに、本明細書において使用される「作用可能に連結された」という用語は、所期の様式でそれらを機能させることができる関係にある、このように記載された成分の位置を表す。さらに、本発明によれば、コード配列に対して作用可能に連結された発現調節配列は、発現調節配列と適合的な条件下で、コード配列の発現が達成されるように連結されている。
【0060】
本発明のさらなる態様は、本発明の結合タンパク質をコードする核酸分子を含むベクターである。核酸分子は、調節配列に、作用可能に連結することができる。さらに、ベクターは、複製起点又は選択マーカー遺伝子をさらに含有し得る。本発明に従って使用され得るベクターの例は、例えば、プラスミド、コスミド、ファージ、ウイルスなどである。
【0061】
本発明の別の態様は、本発明の核酸分子又はベクターで形質転換された宿主細胞に関する。形質転換は、宿主細胞中にポリヌクレオチドを導入するためのあらゆる公知の方法によって、例えば、ウイルス中への(又はウイルスベクター中への)ポリヌクレオチドのパッケージング及びウイルス(又はベクター)での宿主細胞の形質導入、又は、U.S.Patent Nos.4,399,216、4,912,040、4,740,461及び4,959,455(これらの特許は、参照により、本明細書中に組み込まれる。)を例とする本分野で公知の形質移入操作によって実施され得る。特に、哺乳動物細胞中に異種のポリヌクレオチドを導入するための方法が本分野において周知であり、デキストランによって媒介される形質移入、リン酸カルシウム沈殿、ポリブレンによって媒介される形質移入、プロトプラスト融合、電気穿孔、リポソーム中へのポリヌクレオチドの封入及び核内へのDNAの直接的微少注入が含まれる。本発明に従って使用され得る宿主細胞の例は、哺乳動物細胞(例えば、ハムスター、ウサギ、ラット、ブタ、マウス又はその他の動物細胞)などのハイブリドーマ真核細胞、、植物細胞及び真菌細胞(例えば、トウモロコシ、タバコ、サッカロミセス・セレビシアエ(Saccharomyces cerevisiae)、ピチア・パストリス(Pichia pastoris));イー・コリ(E.coli)などの原核細胞及び抗体の産生のために本分野で使用される他の細胞である。特に、発現のための宿主として利用可能な哺乳動物細胞株が本分野において周知であり、チャイニーズハムスター卵巣(CHO)細胞、HeLa細胞、ベビーハムスター腎臓(BHK)細胞、サル腎臓細胞(COS)、ヒト肝細胞癌細胞(例えば、HepG2)及び多数の他の細胞株など(但し、これらに限定されない。)、アメリカン・タイプ・カルチャー・コレクション(ATCC)から入手可能な多くの不死化された細胞株が含まれる。
【0062】
本発明のさらに別の態様は、活性因子としての本発明の少なくとも1つの結合タンパク質並びに医薬として許容される担体、希釈剤及び/又は佐剤を含む医薬組成物である。本明細書において使用される「医薬組成物」という用語は、患者に適切に投与された場合に、所望の治療効果を誘導することができる化学的化合物又は組成物を表す(McGraw−Hill Dictionary of Chemical Terms(Parker, S., Ed., McGraw−Hill, San Francisco(1985)、参照により、本明細書に組み込まれる。)。本発明において、本発明の医薬組成物の効力は、HER−3への少なくとも1つの結合タンパク質の結合を基礎とする。好ましくは、この結合は、HER−3によって媒介されるシグナル伝達の低下をもたらす。
【0063】
さらに、本明細書において使用される場合、「担体」という用語には、使用される用量及び濃度で、担体に曝露されている細胞又は哺乳動物に対して無毒である担体、賦形剤又は安定化剤が含まれる。しばしば、生理的に許容される担体は、pH緩衝化された水溶液又はリポソーム(哺乳動物に薬物を送達するのに有用である、脂質、リン脂質及び/又は界面活性剤の様々な種類から構成される小胞)である。生理的に許容される担体の例には、ホスファート、シトラート及びその他の有機酸などの緩衝剤;アスコルビン酸などの抗酸化剤;低分子量(約10残基未満の)ポリペプチド;血清アルブミン、ゼラチン若しくは免疫グロブリンなどのタンパク質;ポリビニルピロリドンなどの親水性ポリマー;グリシン、グルタミン、アスパラギン、アルギニン若しくはリジンなどのアミノ酸;グルコース、マンノース又はデキストリンなどの単糖、二糖及び他の炭水化物;EDTAなどのキレート剤;マニトール若しくはソルビトールなどの糖アルコール;ナトリウムなどの塩形成対イオン;及び/又はTWEENTM、ポリエチレングリコール(PEG)及びPLURONICSTMなどの非イオン性界面活性剤が含まれる。
【0064】
本発明の一実施形態において、医薬組成物中に含有される本発明の少なくとも1つの結合タンパク質は、カルケアマイシン、アウリスタチン−PE、放射性同位体又はゲルダナマイシン及びマイタンシンなどの毒性を有する化学療法剤などのエフェクターに連結される。特に、これらの結合タンパク質抱合体は、除去のために、HER−3を発現している細胞(例えば、癌細胞)を標的とする上で有用である。
【0065】
さらに、例えば、放射性同位体への本発明の結合タンパク質の連結は、腫瘍治療に対して利点を与える。化学療法及び癌治療の他の形態とは異なり、放射線免疫療法又は放射性同位体−結合タンパク質の組み合わせの投与は、周囲の正常で、健康な組織に対する損傷を最小限に抑えながら、癌細胞を直接標的とする。この「魔法の弾丸」によって、患者は、今日利用可能な治療の他の形態より、放射線同位体のずっと少ない量で治療することができる。好ましい放射性同位体には、イットリウム90(90Y)、インジウム111(111In)、131I、99mTc、放射性銀−111、放射性銀−199及びビスマス213が含まれる。本発明の結合タンパク質への放射性同位体の連結は、例えば、慣用の二官能性キレートを用いて実施され得る。銀は一価であるので、放射性銀−111及び放射性銀−199の連結に関しては、硫黄をベースとするリンカーを使用し得る(Hazra et al., Cell Biophys.24−25, 1−7(1994))。銀放射性同位体の連結は、アスコルビン酸による免疫グロブリンの還元を伴い得る。さらに、チウキセタンは、イブリツモマブに結合されて、イブリツモマブ・チウキセタン(Zevalin)を形成するMX−DTPAリンカーキレート剤である(Witzig, T.E, Cancer Chemother.Pharmacol.48 Suppl 1 , 91−5(2001)。イブリツモマブ・チウキセタンは、インジウム111(111In)又は90Yなどの放射性同位体と反応して、それぞれ、111In−イブリツモマブ・チウキセタン及び90Y−イブリツモマブ・チウキセタンを形成することができる。
【0066】
さらに、本発明の結合タンパク質は、特に、癌を治療するのに使用される場合に、カリチアマイシン(Hamann et al., Bioconjug.Chem.13(1),40−6(2002)、ゲルダナマイシン(Mandler et al., J.Natl.Cancer Inst.,92(19),1549−51(2000))及びメイタンシン、例えば、メイタンシノイド薬DM1(Liu et al., Proc.Natl.Acad.Sci.U.S.A.93:8618−8623(1996))などの毒性を有する化学療法剤と連結され得る。酸性もしくは還元条件下で、又は特異的なプロテアーゼへの曝露時に薬物を放出する様々なリンカーを、本技術とともに使用され得る。本発明によれば、本発明の結合タンパク質は、本分野において記載されているとおりに連結され得る。
【0067】
アウリスタチン−PE、例えば、海洋性無殻軟体動物のペプチド構成成分であるドラスタチン10の構造的な修飾物である抗微小管因子である。アウリスタチン−PEは、抗腫瘍活性及び抗腫瘍血管活性を併有する(Otani et al., Jpn.J.Cancer Res.91(8), 837−44(2000))。例えば、膵臓癌細胞株において、アウリスタチン−PEは細胞増殖を阻害し、細胞周期の停止とアポトーシスを誘導する(Li et al., Int.J.Mol.Med.3(6),647−53(1999))。従って、アウリスタチン−PEの抗腫瘍活性と抗腫瘍血管活性を特定の腫瘍に対して特異的に標的化するために、アウリスタチン−PEは、本発明の結合タンパク質へ連結され得る。
【0068】
本発明の一実施形態において、医薬組成物は、少なくとも1つのさらなる活性因子を含む。本発明に従って使用され得るさらなる活性因子に対する例は、抗体又はEGFR、HER−2、HER−4、IGFR−1若しくはc−metなどの他の受容体タンパク質キナーゼの低分子量阻害剤、血管内皮因子(VEGF)などの受容体リガンド、ドキソルビシン、シス−プラチン若しくはカルボプラチンなどの細胞毒性因子、サイトカイン又は抗新生物因子である。現在、多くの抗新生物因子が当分野で公知である。一実施形態において、抗新生物因子は、抗体又は免疫調節タンパク質を含む(但し、これらに限定されない。)治療用タンパク質の群から選択される。別の実施形態において、抗新生物因子は、有糸分裂阻害剤、キナーゼ阻害剤、アルキル化剤、代謝抑制剤、インターカレート抗生物質、成長因子阻害剤、細胞周期阻害剤、酵素、トポイソメラーゼ阻害剤、ヒストンデアセチラーゼ阻害剤、抗生存因子、生物応答修飾物質、抗ホルモン、例えば、抗アンドロゲン及び抗血管新生因子からなる小分子阻害剤又は化学療法剤の群から選択される。抗新生物因子が放射線照射である場合には、治療は、体内密封小線源治療(brachytherapyBT)又は体外(体外光線照射療法:EBRT)源のいずれかを用いて治療を達成することができる。
【0069】
本発明の医薬組成物は、過剰増殖性疾患の診断、予防又は治療に特に適している。過剰増殖性疾患は、例えば、増加されたHERファミリーシグナル伝達と関連し得る。特に、疾患は、増加されたHER−3リン酸化及び/又はHER−3とHERファミリーの他のメンバーとの間での増加された複合体形成及び/又は増加されたPI3キナーゼ活性及び/又は増加されたc−jun末端キナーゼ活性及び/又AKT活性及び/又は増加されたERK2活性及び/又はPYK2活性と関連することができる。好ましくは、過剰増殖性疾患は、乳癌、胃腸癌、膵臓癌、前立腺癌、卵巣癌、胃癌、子宮内膜癌、唾液腺癌、肺癌、腎臓癌、大腸癌、直腸結腸癌、甲状腺癌、膀胱癌、神経膠腫、悪性黒色腫又はHER−3を発現若しくは過剰発現する他の癌及び腫瘍転移の形成からなる群から選択される。
【0070】
本発明において、本明細書で使用される「予防又は治療」という用語は、治療的な処置及び予防的若しくは防止的措置の両方を表し、必要とする患者は、標的とされる病的症状又は疾患を予防又は遅延(軽減)することである。予防又は治療を必要とする者には、疾患を既に有する者及び疾患を有する傾向がある者又は疾患が予防されるべき者が含まれる。予防又は治療を必要とする患者は、哺乳動物患者、すなわち、ヒト、イヌ、ネコ、ウシ、ウマ、ヒツジ、ブタ、ヤギ、ウサギなどの家畜及び農場動物、並びに動物園、スポーツ又はペット動物を含む哺乳動物として分類される全ての動物である。好ましくは、治療を必要とする患者は、ヒト患者である。
【0071】
本発明によれば、本発明の医薬組成物は、活性因子を、生理的に許容される担体、希釈剤及び/又は佐剤、並びに、改善された転送、送達、耐性などを付与するために、製剤中に一般的に取り込まれる、場合によって使用される他の因子と混合することによって製剤化され得る。本発明の医薬組成物は、例えば、凍結乾燥された製剤、水溶液、分散液又は錠剤、糖衣錠若しくはカプセルなどの固体調製物の形態で製剤化され得る。多数の適切な製剤が、全ての薬化学者に公知の処方中に見出すことができる。Pharmaceutical Sciences(18th ed, Mack Publishing Company, Easton, PA(1990))、特に、Block, LawrenceによるChapter 87中に見出すことができる。これらの製剤には、例えば、粉末、ペースト、軟膏、ゼリー、蝋、油、脂質、脂質(陽イオン性又は陰イオン性)含有小胞(LipofectinTMなど)、DNA抱合体、無水吸収ペースト、水中油及び油中水エマルジョン、エマルジョンカルボワックス(様々な分子量のポリエチレングリコール)、半固体ゲル及びカルボワックスを含有する半固体混合物が含まれる。製剤化によって、製剤中の活性因子が不活化されず、製剤が投与の経路と生理的に適合し、耐容される限り、先述の混合物の何れもが、本発明に係る治療及び療法において適切であり得る。薬化学者に周知の製剤、賦形剤及び担体に関するさらなる情報に関しては、Baldrick P., “Pharmaceutical excipient development:the need for preclinical guidance.”,Regul.Toxicol.Pharmacol.32(2),210−218(2000);Wang W., “Lyophilization and development of solid 5 protein pharmaceuticals.”,Int.J.Pharm.203(1−2),1−60(2000);Charman W.N., “Lipids, lipophilic drugs, and oral drug delivery−some emerging concepts.”, J.Pharm.Sci.89(8),967−978(2000);Powell et al., “Compendium of excipients for parenteral formulations”, PDA J.Pharm.Sci.Technol.52, 238−311(1998)及びこれらの中の引用文献も参照されたい。
【0072】
本発明の別の態様は、過剰増殖性疾患の診断、予防又は治療用医薬組成物の製造のための、医薬として許容される担体、希釈剤及び/又は佐剤と混合された、本発明の少なくとも1つの単離された結合タンパク質、及び、場合によって使用される少なくとも1つの他の活性因子(例えば、少なくとも1つの上記抗新生物因子)の使用に関する。好ましくは、医薬組成物は上記の医薬組成物であり、過剰増殖性疾患は上記過剰増殖性疾患である。
【0073】
本発明のさらに別の態様は、HER−3の発現と関連する疾病又は症状を診断するための方法であり、試料を本発明の結合タンパク質と接触させること、及び前記試料中のHER−3の存在を検出することを含む、前記方法に関する。試料は、腫瘍細胞、血液試料又は別の適切な試料など、HER−3の発現を示す細胞であり得る。本発明の好ましい実施形態において、HER−3の発現に関連する疾病又は症状は、上に定義されている過剰増殖性疾患である。
【0074】
本発明によれば、前記方法は、例えば、細胞中のHER−3抗原の検出のために、上記過剰増殖性疾患に罹患している患者中のHER−3抗原濃度の測定のために、又は患者中の前記過剰増殖性疾患の段階決定のために使用され得る。研究されている対象中の過剰増殖性疾患の進行の段階を決定するために、又は療法の経過に対する対象の応答を特徴付けるために、例えば、血液の試料を対象から採取し、試料中に存在するHER−3抗原の濃度を測定する。このようにして得られた濃度は、値が属する濃度範囲を同定するために使用される。このようにして同定された範囲は、診断された対象の様々な集団中で同定された進行の段階又は療法の段階と相関し、これにより、研究中の対象における段階を付与する。疾病の生検、例えば、癌、患者から得られた組織も、存在するHER−3抗原の量を評価するために使用され得る。疾病組織中に存在するHER−3抗原の量は、本発明のHER3抗体を用いて、免疫組織化学、ELISA又は抗体アレイによって評価され得る。診断的に興味深い他のパラメータは、二量体化の状態並びにHER3タンパク質の二量体化対並びにHER3タンパク質及びその対の活性化状態である。これらのパラメータを決定するためのタンパク質分析法は本分野において周知であり、特に、ウェスタンブロット及び免疫沈降技術、FACS分析、化学架橋、生物発光共鳴エネルギー転移(BRET)、蛍光共鳴エネルギー転移(FRET)など(例えば、Price et al, Methods in Molecular Biology, 218:255−268(2002))又はeTag技術(WO0503707,WO04091384,WO04011900)である。
【0075】
さらに、本発明は、別の態様において、HER−3の発現が関連する疾病又は症状の予防又は治療を必要としている患者において、HER−3の発現が関連する疾病又は症状を予防又は治療する方法であり、少なくとも1つの本発明の結合タンパク質の有効量を前記患者に投与することを含む、前記方法に関する。好ましくは、HER−3の発現を伴う好ましい疾病又は症状は、上に定義されている過剰増殖性疾患である。予防又は治療を必要とする患者は、哺乳動物患者、すなわち、ヒト、家畜及び農場動物、並びに動物園、スポーツ又はペット動物(イヌ、ネコ、ウシ、ウマ、ヒツジ、ブタ、ヤギ、ウサギなど)を含む哺乳動物として分類される全ての動物である。好ましくは、必要とする患者は、ヒト患者である。
【0076】
本発明の好ましい実施形態において、過剰増殖性疾患の予防又は治療を必要としている患者において、過剰増殖性疾患を予防又は治療する方法は、少なくとも1つの本発明の結合タンパク質の有効量、及び、さらに、少なくとも1つの他の活性因子(例えば、少なくとも1つの上記抗新生物因子)を前記患者に投与することを含む。好ましくは、本方法は、異常な細胞増殖、遊走又は浸潤を阻害するための方法である。
【0077】
例えば、上記製剤を介した、結合タンパク質治療薬の候補の古典的な投与様式の他に、新たに開発された投与様式が、本発明においても有用であり得る。例えば、手術の切開後に原発性脳腫瘍を治療するための131I標識されたモノクローナル抗体の局所投与が報告されている。さらに、モノクローナル抗体及びモノクローナル抗体の断片の直接的な定位的脳内注射も、臨床的に、及び前臨床的に研究されている。頚動脈内高浸透圧性灌流は、薬物が連結されたヒトモノクローナル抗体を、原発性脳悪性腫瘍に標的誘導するための実験的戦略である。
【0078】
治療されるべき症状の種類及び重度に応じて、例えば、1つ若しくはそれ以上の分割された投与によって、又は継続的注入によって、本発明の結合タンパク質を必要としている患者に、少なくとも1つの本発明の結合タンパク質約1μg/kgから15mg/kgを投与し得る。典型的な一日投薬量は、上述の要因に応じて、約1μg/kgから約100mg/kg又はそれ以上の範囲であり得る。数日又はそれ以上にわたる反復投与に関しては、治療されるべき症状に応じて、疾病症候の望ましい抑制が生じるまで、治療が持続される。
【0079】
投与された少なくとも1つの抗新生物因子の用量は、様々な要因に依存する。これらは、例えば、因子の性質、腫瘍の種類又は投与の経路である。本発明は、いかなる用量にも限定されないことが強調されるべきである。
【0080】
最後に、本発明は、さらなる態様において、HER−3によって媒介されるシグナル伝達と関連する疾病を診断し、予防し、又は治療するためのキットであり、少なくとも1つの本発明の結合タンパク質及び/又は核酸分子及び/又はベクターを含む前記キットに関する。さらに、本発明のキットは、少なくとも1つの他の活性因子、例えば、少なくとも1つの上記他の抗新生物因子をさらに含むことができる。
【0081】
さらに、以下の実施例及び添付の図面によって、本発明を説明するものとする。
【0082】
実施例
実施された実験及び達成された結果を含む以下の実施例は、例示の目的のために提供されているものに過ぎず、本発明を限定するものと解釈すべきでない。
【実施例1】
【0083】
HER−3抗原及び細胞株の調製
本研究では、組換えHER−3タンパク質を調製した。HER−3の配列(Genebank AccNr.NM 001982)に基づくプライマーを用いて、pcDNA3−HER−3(完全長ヒトHER−3を有する発現ベクター、C.Wallasch et al., EMBO J.14, 4267−4275)からのポリメラーゼ連鎖反応(PCR)によって、HER−3の細胞外ドメイン(ECD)cDNAをクローニングした。
【0084】
HER−3の増幅のために使用されたプライマーは、以下のとおりであった。
【0085】
【化1】
【0086】
【化2】
【0087】
BamH1及びXbaIでPCR産物を消化し、BamHI及びXbaIで消化されたpcDNA3(Invitrogen)中に連結した。CaPO4法を用いて、プラスミドをHEK293細胞中に形質移入した。Ni−NTAアフィニティークロマトグラフィーを用いて、採集された馴化培地からHER−3−HIS融合タンパク質を精製した。
【0088】
レトロウイルス遺伝子転送によって、Rat1HER−3細胞を作製した。要約すると、GP+E86細胞(3×105)を60mm培養皿上に播種し、リン酸カルシウム法を用いて、2μg/mLpIXSNベクター又はpIXSN−HER−3cDNA(C.Wallasch, PhD Thesis, Max−Planck lnsitute of Biochemistry, Martinsried, Germany)を形質移入した。24時間後、培地を新鮮な培地と交換し、GP+E86細胞を、4から8時間温置した。次いで、Polybren(4mg/mL;Aldrich)の存在下で、4から12時間、高力価のpLXSN又はpLXSN−HER−3、pウイルス(>1×106G418c.f.u./mL;m.o.i.10)を放出するGP+E86細胞の上清とともに、集密状態を下回るRat1細胞(2×105細胞/6cm皿)を温置した。培地を交換した後、G418でのRat1細胞の選択を開始した。通常、21日間の選択後に、安定なクローンを摘み取った。
【実施例2】
【0089】
ヒト癌細胞株中でのHER−3発現
例えば,HER−3のような受容体チロシンキナーゼは、良性過形成細胞増殖から悪性癌腫への移行など、過剰増殖性疾患の開始及び進行において重大な役割を果たしている。腫瘍細胞と正常組織との間でHER−3発現が異なるので、HER−3発現の分析は、本発明の結合タンパク質での治療が有益である患者の亜群を同定するための重要な要因である。従って、ヒト癌形成におけるHER−3の役割を解明するために、ヒト癌細胞株の集団において、HER−3発現を定量した。癌細胞株は、ATCCによって推奨されているとおりに増殖させた。詳しく述べると、PBS中の10mMEDTAを用いて105細胞を採集し、FACS緩衝液(PBS、3%FCS、0.4%アジ化物)で1回洗浄し、96ウェルの丸底プレート上に播種した。上清を除去するために、1000rpmで3分間、細胞を遠心し、次いで、α−HER−3抗体2D1D12(WO03013602)(3μg/ml)とともに再懸濁した。細胞懸濁液を氷上で1時間温置し、FACS緩衝液で2回洗浄し、二次抗体(100μL/ウェル)ロバ抗ヒトPE(Jackson)によって再懸濁し、FACS緩衝液中に1:50希釈した。氷上及び暗所で、細胞懸濁液を30分間温置し、FACS緩衝液を用いて2回洗浄し、分析した(FACS、Beckman Coulter)。図1は、分析の代表的な結果を示しており、様々なヒト癌内で、HER−3が発現されていることを示している。
【実施例3】
【0090】
免疫化及び力価測定
実施例1に記載されているとおりに調製されたHER−3ECDタンパク質及びC32細胞(ヒト悪性黒色腫;ATCC#CRL−1585)を抗原として使用した。XenoMouse(R)マウス(XenoMouse(R)系統:XMG1及びXMG4、Abgenix,Inc.Fremont,CA)を順次免疫化することによって、HER−3に対するモノクローナル抗体を産生させた。全ての注射に関して、足蹠経路を介して、XenoMouse(R)動物を免疫した。各注射の総容量は、50μL/マウス、25μL/足蹠であった。
【0091】
コホート番号1(10匹のXMG1マウス)に関して、初期免疫化は、マウス当り、TITERMAX GOLD(R)(Sigma, Oakville, ON)が1:1(v/v)で混合されたHER−3ECDタンパク質10μgを用いて行った。その後の5回の強化免疫は、発熱物質を含まないD−PBS中のalumゲル100μg(Sigma,Oakville,ON)と1:1(v/v)で混合されたHER−3ECDタンパク質10μgを用いて行った。6回目の強化免疫は、TITERMAX GOLD(R)と1:1(v/v)混合されたHER−3ECDタンパク質10μgからなった。7回目の注射は、alumゲル100μgと1:1v/v混合されたHER−3ECDタンパク質10μgからなった。最終強化免疫は、アジュバントなしの、無発熱物物質DPBS中のHER−3ECDタンパク質10μgを用いて行った。このプロトコールに関しては、第0日、4日、7日、11日、15日、20日、24日及び29日目にXenoMouse(R)マウスを免疫し、第33日目に融合を行った。4回目の強化免疫後に13日目に、6回目の強化免疫後に19日目に、後眼窩採血操作を通じて、2回の採血を行った。コホート番号2は存在しなかった。
【0092】
コホート番号3(10匹のXMG1マウス)及びコホート番号4(10匹のXMG4マウス)に関して、最初の注射は、マウス当り、TITERMAX GOLD(R)が1:1(v/v)で混合された無発熱物質Dulbecco’sPBS(DPBS)中の107個のC32細胞を用いて行った。次の4つの強化免疫は、マウス当り、Adju−Phos25μg及び10μgCpGと混合された無発熱物質DPBS中の107個のC32細胞を用いて行った。6回目の強化免疫は、マウス当り、TITERMAX GOLD(R)が1:1(v/v)で混合された無発熱物質DPBS中の107個のC32細胞を用いて行った。7回目、8回目、9回目の強化免疫は、マウス当り、Adju−Phos25μg及びCpG10μgと混合された無発熱物質DPBS中の107個のC32細胞を用いて行った。10回目から14回目の強化免疫は、マウス当り、Adju−Phos25μg及びCpG10μgと混合された無発熱物質DPBS中のHER−3ECDタンパク質5μgであった。最終強化免疫は、アジュバントなしの、無発熱物物質DPBS中のHER−3ECDタンパク質5μgからなった。コホート番号3及び4の両方で、このプロトコールに対して第0日、3日、7日、11日、14日、17日、21日、24日、28日、33日、35日、38日、42日及び45日目にXenoMouse(R)マウスを免疫し、第49日目に融合を行った。4回目の強化免疫後12日目に、6回目の強化免疫後19日目に、及び12回目の強化免疫後40日目に、後眼窩採血操作を通じて、3回の採血を行った。
【0093】
力価による、採集のための動物の選択
コホート番号1に関しては、免疫されたXenoMouse(R)マウスから得た血清中の抗HER−3抗体力価は、HER−3ECDタンパク質に対するELISAによって測定した。各XenoMouse(R)動物の特異的力価は、650nmでの光学密度から測定され、下表1に示されている。力価の値は、血清の最大希釈の逆数であり、ODの読み取りは、バックグラウンドの2倍である。従って、数値が高くなるほど、HER−3ECDに対する液性免疫応答は大きかった。
【0094】
【表1】
【0095】
コホート番号3及び4に関しては、免疫されたXenoMouse(R)マウスから得た血清中の抗HER−3抗体力価は、Rat1/HER−3細胞(抗原陽性細胞株)細胞及びRat1/pLSXN細胞(抗原陰性細胞株)を用いたFACSによって測定した。データは、血清試料の系列希釈による、細胞抗HER−3細胞染色の幾何平均(GeoMean)蛍光強度として表されている。
【0096】
【表2】
【0097】
【表3】
【実施例4】
【0098】
リンパ球の回収、B細胞の単離、融合及びハイブリドーマの作製
免疫されたマウスを屠殺し、各コホートからリンパ節を採集し、プールした。DMEM中で磨り潰すことによってリンパ球系細胞を解離させて、組織から細胞を放出させ、細胞をDMEM中に懸濁した。細胞を計数し、0.9mLDMEM/1億リンパ球を細胞ペレットに添加して、穏やかに、但し、完全に、細胞を再懸濁した。1億細胞当りCD90+磁気ビーズ100μLを用いて、4℃で15分間、磁気ビーズとともに細胞を温置することによって、細胞を標識した。最大108個の陽性細胞(又は最大2×109個の全細胞)を含有する磁気標識された細胞懸濁液をLS+カラムにかけ、DMEMでカラムを洗浄した。全ての流出物を、CD90陰性画分として集めた(これらの細胞の多くは、B細胞であると予想された。)。
【0099】
上で得た、洗浄され、濃縮されたB細胞と、ATCC(Cat.No.CRL1580)から購入された非分泌性骨髄腫P3X63Ag8.653細胞(Kearney et al, J.Immunol.123,1979,1548−1550)とを、1:1の比で混合することによって、融合を実施した。800gでの遠心によって、細胞混合物を穏やかに沈降させた。上清を完全に除去した後、最長2分間、プロナーゼ溶液2から4mL(CalBiochem、カタログ番号53702;PBS中0.5mg/ml)で細胞を処理した。次いで、酵素活性を停止させるために、FBS3から5mLを添加し、電気的細胞融合溶液ECFS(0.3Mショ糖、Sigma、カタログ番号S7903、0.1mM酢酸マグネシウム、Sigma、カタログ番号M2545、0.1mM酢酸カルシウム、Sigma、カタログ番号C4705)を用いて、40mLの総容積になるように懸濁液を調整した。遠心後、上清を除去し、40mLECFS中に細胞を再懸濁した。この洗浄工程を反復し、2×106細胞/mLの濃度になるように、再度、ECFS中に細胞を再懸濁した。
【0100】
融合生成装置モデルECM2001、Genetronic,Inc., San Diego, CAを用いて、電気的細胞融合を行った。使用した融合チャンバーのサイズは2.0mLであり、以下の機器設定を用いた。並列条件:電圧:50V、時間:50秒、膜破壊電圧:3000V、時間:30μ秒、融合後保持時間:3秒。
【0101】
ECF後、無菌条件下で、融合チャンバーから細胞懸濁液を慎重に取り出し、Lグルタミン、ペニシリン/ストレプトマイシン、OPI(オキサロアセタート、ピルバート、ウシインシュリン)(全てSigmaから入手)及びIL−6(Boehringer Mannheim)が補充された、Hybridoma Culture Medium(DMEM(JRH Biosciences)、15%FBS(Hyclone)の同じ容量を含有する無菌チューブ中に移した。37℃で15から30分間、細胞を温置し、次いで、400gで5分間遠心した。Hybridoma Selection Medium(0.5×HAが補充されたHybridoma Culture Medium(Sigma、カタログ番号A9666))の小容量中に、細胞を穏やかに再懸濁し、計5×106個B細胞/96ウェルプレート及び200μL/ウェルの最終播種を基礎として、さらなるHybridoma Selection Mediumを用いて、容量を適切に調整した。細胞を穏やかに混合し、96ウェルプレート中にピペットで添加し、増殖させた。第7日又は10日目に、培地の半分を除去し、細胞にHybridoma Selection Mediumを再度与えた。
【実施例5】
【0102】
ELISAによる候補抗体の選択
培養から14日後に、精製されたhisタグ化HER−3ECDを用いたELISAによって、HER−3特異的抗体に対して、コホート番号1(コホート1のマウスを、任意に融合番号1及び2に分割した。)から得られたハイブリドーマ上清の一次スクリーニングを行い、ELISAプレート上に固定化されたHER−3ECDへのヒトIgG結合を検出するために、ヤギ抗huIgGFc−HRP(Caltag Inc., Cat.No.H10507、使用した濃度は、1:2000希釈であった。)を用いたELISAによって、無関係なhisタグ化タンパク質に対するカウンタースクリーニングを行った。一次スクリーニングに基づく陽性ハイブリドーマ細胞増殖ウェルから得た古い培養上清を取り除き、新鮮なハイブリドーマ培地を用いて、HER−3陽性ハイブリドーマ細胞を懸濁し、24ウェルプレートに移した。培養から2日後には、これらの上清は、二次的確認スクリーニングを行える状態であった。HER−3特異的完全ヒトIgGκ抗体に対する二次的確認スクリーニングでは、ヒトγ鎖検出のためのヤギ抗huIgGFc−HRP(Caltag Inc.,Cat.No.H10507、使用した濃度は、1:2000希釈であった。)及びヒトκ軽鎖検出のためのヤギ抗hIgκ−HRP(Southern Biotechnology、カタログ番号2060−05)という検出用抗体の2つの組を用いたELISAによって、第一のスクリーニングにおいて陽性であったものをスクリーニングした。コホート番号1から生成された91の完全ヒトIgG/κHER−3特異的モノクローナル抗体が存在した。
【実施例6】
【0103】
FMAT/FACSによる候補抗体の選択
培養から14日後に、コホート番号3及び番号4から得られたハイブリドーマ上清(融合番号3及び番号4)を、FMATによって、HER−3−特異的モノクローナル抗体に関してスクリーニングした。一次スクリーニングにおいて、1:10最終希釈のハイブリドーマ上清を、ヒトHER−3を発現するRat1−Her3細胞及びCy5連結されたヤギF(ab’)2抗ヒトIgG、Fc特異的抗体400ng/mL(Jackson ImmunoResearch,カタログ番号109−176−098)とともに、室温で6時間温置した。細胞への抗体及び検出抗体複合体の結合を、FMAT(Applied Biosystems)によって測定した。細胞への抗体の非特異的結合は、親Rat1細胞への抗体の結合によって測定した。融合物3の一次スクリーニングから、HER−3−特異的抗体を産生する計420のハイブリドーマを選択した。同じFMATプロトコールを用いて、これらの拡張された培養物からの上清を再度検査し、これらのうちの262が、HER−3発現細胞へ特異的に結合することが確認された。融合物4の一次スクリーニングから、HER−3−特異的抗体を産生する計193のハイブリドーマを選択した。FACSによって、これらの拡張された培養物からの上清物を検査し、これらのうちの138が、HER−3発現細胞へ特異的に結合することを確認した。FACS確認アッセイでは、2%FBSを含有するPBS中において、1:2希釈で、1時間、40℃で、ハイブリドーマ上清とともに、Rat1−Xher3細胞と親Rat1細胞(陰性対照として)を温置した。PBSでの洗浄後、2.5μg/mLCy5連結されたヤギF(ab’)2抗ヒトIgG、Fc特異抗体(JIR#109−176−098)及び5μg/mLPE連結されたヤギF(ab’)2抗ヒトκ特異抗体(SB#2063−09)によって、細胞への抗体の結合を検出した。PBSでの洗浄によって、非結合抗体を除去した後、1:4の希釈で、cytofix(BD#51−2090KZ)によって、細胞を固定し、FACSCaliburによって分析した。
【実施例7】
【0104】
クローニングのためのハイブリドーマの選択
例えば、R&DBiosystemsから入手可能な精製された組換え細胞外ドメインを用いたELISAにおける、HER1(EGFR)、HER−2及びHER−4を上回るHER−3に対する特異性並びに異なるHERファミリーのメンバーを発現するヒト腫瘍細胞株のFACSベースの分析並びにバックグラウンドを上回るHER−3陽性細胞に対するFACS染色での平均蛍光強度の5倍超の増加に基づいて、ハイブリドーマクローニングのために、コホート1及び2から得た抗体を選択した。これらの基準に基づいて、限界希釈細胞播種によって、計23のハイブリドーマ株をクローニングのために選択した。
【0105】
HER−1(EGFR)、HER−2及びHER−4を上回るHER−3に対する特異性に加え、他の3つの基準に基づいて、ハイブリドーマクローニングのために、コホート3及び4から得た抗体を選択した。第一の基準は、HER−3のL2ドメイン内に含有されたエピトープを有する抗体に対するELISAスクリーニングであった(実施例「本発明における抗HER−3抗体の構造分析」を参照。)。
【0106】
第二の基準は、FACSベースのアッセイにおける、HER−3発現細胞へのビオチン化されたヘレグリンαの結合の中和であった。SKBR−3細胞を採集し、培地中で洗浄し、遠心によって沈降させ、培地中に再懸濁した。再懸濁された細胞を、96ウェルプレート中に分取した。細胞を沈降させるために、プレートを遠心した。排出ハイブリドーマ上清中の試験抗体を、25μL/ウェルで添加し、抗体を結合させるために、氷上で1時間温置した。10nMヘレグリンα(R&D Biosystems, Minneapolis, MN)溶液50μLを、5nMの最終濃度となるように、各ウェルに添加し、氷上で1.5時間温置した。150μLPBS中で細胞を洗浄し、遠心によって沈降させ、上清を除去した。10μg/mLで、ヤギ抗HRGαポリクローナル抗体50μL中に細胞を再懸濁し、45分間、氷上で温置した。PBS200μL中で細胞を洗浄し、遠心によって沈降させ、上清を除去した。10μg/mLの7AADを加えた5μg/mLのCy5標識されたウサギ抗ヤギポリクローナル抗体の溶液50μLを添加し、15分間、氷上で温置した。PBS200μL中で細胞を洗浄し、遠心によって沈降させ、上清を除去した。FACS緩衝液100μL中に細胞を再懸濁し、FACS中で読み取った。ヘレグリンαの結合を低下させた試験HER−3抗体は、最も低い蛍光強度を有するものであった。陽性対照として、マウスHER−3mAb(105.5)又はヒトIgG1HER−3mAB、U1−49の10,000ng/mLから16ng/mLまでの1:5系列希釈を使用した。陰性対照は、ヘレルギンαのみ、細胞のみ、ヤギ抗ヘレグリンαポリクローナル抗体のみ及びCy5標識されたウサギ抗ヤギポリクローナル抗体のみであった。
【0107】
第三の基準は、HER−3発現細胞株を用いたFACSにおける、親和性に対する相対順位付け及び/又はより高い相対平均蛍光強度であった。親和性に対する相対順位付けは、HER−3特異的抗体濃度を標準化し、以下のように、限界抗原ELISAから得られたデータに対してプロットすることによって行った。
【0108】
高抗原ELISAを用いた抗原特異的抗体濃度の標準化
ELISA法を用いて、抗原特異的抗体の濃度に対して上清を標準化した。平行して滴定された濃度既知のコホート1から得られた2つの抗HER−3ヒトIgG1抗体を用いて、標準曲線を作成し、コホート3及び4から得られた試験ハイブリドーマ上清中の抗原特異的抗体の量を標準と比較した。このようにして、各ハイブリドーマ培養中のヒトHER3IgG抗体の濃度を推定した。
【0109】
4℃で一晩温置しながら、50μL/ウェルで、Costar3368培地結合プレート上に、1×PBS/0.05%アジ化ナトリウム中の8μg/mLでニュートラビジンをコートすることによって、ニュートラビジンプレートを作製した。翌日、1×PBS/1%スキムミルクでプレートをブロックした。1×PBS/1%スキムミルク中の500ng/mLの光ビオチン化されたhisタグ化HER−3ECDを、室温で1時間温置することによって、ニュートラビジンプレートに結合させた。1×PBS/1%スキムミルク/0.05%アジ化物中に、1:31の出発希釈から1:7568の最終希釈まで、1:2.5系列希釈されたハイブリドーマ上清を、50μL/ウェルで添加し、次いで、室温で20時間温置した。未知の各々に対するODの読み取りが確実にアッセイの直線域内に得られるようにするために、系列希釈を使用した。次に、二次検出抗体である、1×PBX/1%スキムミルク中の400ng/mLのヤギ抗ヒトIgGFcHRPを、50μL/ウェルで添加した。室温で1時間後、再度、プレートを水で5回洗浄し、一成分TMB基質50μLを各ウェルに添加した。各ウェルに1M塩酸50μLを添加することによって、30分後に反応を停止させ、450nmの波長でプレートを読み取った。1000ng/mLから0.06ng/mLまで1:2で系列希釈された、コホート1から得られた2つのIgG1HER−3mAbから標準曲線を作成し、上記プロトコールを用いてELISA中で評価を行った。未知の各々に関しては、各試料中のヒトHER−3IgGの濃度を推定するために、アッセイの直線域中のODの読み取りを使用した。
【0110】
限定された抗原分析は、他の全ての抗原特異的抗体との比較において、B細胞培養上清中に調製された抗原特異的抗体の親和性を順位付けする方法である。抗原の極めて少量のコーティングの存在下では、最高の親和性の抗体のみが、平衡状態で、何れかの検出可能なレベルまで結合できるはずである。(例えば、2003年6月12日に公開された「IDENTIFICATION OF HIGH AFFINITY MOLECULES BY LIMITED DILUTION SCREENING」と題されたPCT Publication WO/03048730A2を参照されたい。)。この例では、コホート1から得られた2つのmAb(何れも、濃度既知及びKD既知)を、アッセイにおける基準として使用した。
【0111】
4℃で一晩温置しながら、50μL/ウェルで、Costar3368培地結合プレート上に、1×PBS/0.05%アジ化ナトリウム中の8μg/mLのニュートラビジンをコートすることによって、ニュートラビジンプレートを作製した。翌日、1×PBS/1%スキムミルクで、プレートをブロックした。室温で1時間、1×PBS/1%スキムミルク中、125、62.5、31.2、15.6及び7.8ng/mLという5つの濃度で、ビオチン化されたhisタグ化HER−3ECD(50μL/ウェル)を96ウェルニュートラビジンプレートに結合させた。各プレートを水で5回洗浄した。1×PBS/1%スキムミルク/0.05%アジ化物中に1:31希釈されたハイブリドーマ上清を、50μLウェルで添加した。震盪器上、室温での20時間の温置後、dH2Oで5回、プレートを再度洗浄した。次に、二次検出抗体である、1×PBS/1%スキムミルク中の400ng/mLのヤギ抗ヒトIgGFcHRP(西洋ワサビペルオキシダーゼ)を50μL/ウェルで添加した。室温で1時間後、dH2Oで、プレートを再度5回洗浄し、一成分TMB基質50μLを各ウェルに添加した。各ウェルに1M塩酸50μLを添加することによって、30分後に反応を停止させ、450nmの波長でプレートを読み取った。直線域内にOD値を与える抗原濃度から得られるOD読み取りを、データ解析のために使用した。
【0112】
特異的抗体濃度を比較的推定する(詳細については、上記参照)高抗原データを、限定された抗原ODに対してプロットすることによって、比較的より高い親和性抗体(例えば、結合された抗体が、限定された抗原アッセイでは、より高いODを有したが、上清中にIgGHER−3抗体のより低い量を有する。)を示した。
【0113】
アッセイのこれらの組において最も成績の優れていた33個の抗体に対するコホート3及び4から得たハイブリドーマは、限界希釈ハイブリドーマ播種によるクローニングに進んだ。
【0114】
あるいは、Rat1/pLXSN及びRat1/HER−3細胞のHER−3発現のFACS分析は、類似の結果を示した(内在性ラットエピトープとの交叉反応性なし)(図2)。
【0115】
詳しく述べると、PBS中の10mMEDTAを用いて1×105細胞を採集し、FACS緩衝液(PBS、3%FCS、0.4%アジ化物)で1回洗浄し、96ウェルの丸底プレート上に播種した。上清を除去するために、1000rpmで3分間、細胞を遠心し、次いで、特異的なHERファミリー抗体(3μg/ml)とともに再懸濁した。細胞懸濁液を氷上で45分間温置し、FACS緩衝液で2回洗浄し、二次抗体(100μL/ウェル)ロバ抗ヒトPE(Jackson Immunoresearch,PA)とともに再懸濁し、FACS緩衝液中に1:50希釈した。氷上及び暗所で、細胞懸濁液を30分間温置し、FACS緩衝液を用いて2回洗浄し、分析した(FACS、Beckman Coulter)。
【実施例8】
【0116】
本発明の抗HER−3抗体の構造的分析
以下の論述では、本発明に従って調製された抗体に関する構造情報が提供される。本発明に従って作製された抗体の構造を分析するために、特定のハイブリドーマから、重鎖及び軽鎖断片をコードする遺伝子を増幅した。配列決定は、以下のように行った。
【0117】
逆転写ポリメラーゼ連鎖反応(RT−PCR)を用いて、96ウェルプレート中の各ハイブリドーマクローンから、VH及びVL転写物を増幅した。Fast−Trackキット(Invitrogen)を用いて、約2×105個のハイブリドーマ細胞からポリ(A)+mRNAを単離した。各ハイブリドーマに対して、4つのPCR反応(軽鎖(κ(K)に対して2つ、及びγ重鎖(γ)に対して2つ)を実施した。増幅のために、QIAGENOneStep室温PCRキット(Qiagen、カタログ番号210212)を使用した。連結された室温PCR反応では、Cκ又はCγ遺伝子のCH1領域のコンセンサスに対応するアンチセンス配列特異的プライマーを使用し、室温酵素(Omniscript及びSensiscript)の混合物を用いてcDNAを合成した。50℃で1時間、逆転写を実施した後、高い特異性及び感受性のために、HotStarTaq DNA PolymeraseによるcDNAのPCR増幅を行った。各PCR反応は、5’センスプライマーの混合物を使用した。プライマー配列は、Vbaseウェブサイト(http://vbase.mrc−cpe.cam.ac.uk/)で入手可能なVH及びVKのリーダー配列に基づいた。
【0118】
94℃で15分間の初期ホットスタートでPCR反応を実行した後、94℃30秒間(変性)、60℃30秒間(アニーリング)及び72℃1分間(伸長)の40サイクルを行った。
【0119】
PCR産物を精製し、ABI PRISM BigDye terminator cycle sequencing ready reaction Kit(Perkin Elmer)を使用し、フォワード及びリバースPCRプライマーを用いて配列を直接決定した。Prism色素終結因子配列決定キット及びABI377配列決定機を用いて、両鎖を配列決定した。
【0120】
配列分析
HER3抗体のヒトV重及びVκcDNA配列の分析は、Abgenix社内ソフトウェア(5AS)を用いて、ヒト生殖系列V重及びVκ配列とHER−3配列を並置することによって行った。本ソフトウェアによって、V遺伝子、D遺伝子及びJ遺伝子並びに組換え連結におけるヌクレオチド挿入及び体細胞変異の使用が同定された。体細胞変異を同定するために、アミノ酸配列もコンピュータシミュレーションで作製された。市販の配列分析ソフトウェア並びにヒトV、D及びJ遺伝子の配列に関する公表された情報、例えばVbase(http://vbase.mrc−cpe.cam.ac.uk/)を用いて、同様の結果を得ることが可能である。
【0121】
mAbU1−59の分子クローニング
抗体U1−59を分泌するハイブリドーマ系列など、複数のハイブリドーマ系列を含有する組織培養ウェルから全RNAを抽出した。3’−C−γプライマーとともに、5’−リーダーVHファミリー特異的プライマーを用いて、重鎖可変領域を増幅した。VH4プライマーを用いて、大きなバンドが増幅され、他のバンドは見られなかった。ヒトγ1定常領域遺伝子とインフレームになるように、pCDNA発現ベクター中にVH4−34γ断片をクローニングした。
【0122】
3’μ定常領域プライマーとともに5’VHファミリー特異的プライマーを用いて、IgM重鎖可変領域を増幅した。VH2プライマーを用いて、主要なバンドが増幅され、他のバンドは見られなかった。ヒトμ定常領域遺伝子とインフレームになるように、pCDNA発現ベクター中にVH2−5μ断片をクローニングした。Vκ鎖を増幅し、配列を決定した。4つのκ鎖RT−PCR産物が同定された。産物を配列決定し、コンピュータシミュレーションでの翻訳を介した配列分析後に、産物の3つが読み取り枠を有するに過ぎなかった。(1)VK1A3−JK2、(2)VK1A20−JK3及び(3)B3−JK1としてのVκ遺伝子の使用に基づいて明確に同定されたオリゴクローン性U1−59ハイブリドーマから、これらの3つの機能的κ鎖をクローニングした。ヒトκ軽鎖定常領域遺伝子とインフレームになるように、pCDNA発現ベクター中に全てのVκをクローニングした。
【0123】
形質移入:
計6つの重鎖/κ軽鎖対に関して、一過性形質移入において、κ鎖の各々とともに、各重鎖を形質移入した。A20κ鎖とのγ鎖の形質移入は、乏しい抗体発現を与えたのに対して、A20κ鎖がμ鎖とともに同時形質移入された場合には、抗体は全く分泌又は検出されなかった。HER−3結合アッセイのために、計3つのIgGsup及び2つのIgMsupが利用可能であった。
【0124】
【表4】
【0125】
VH4−34及びB3κ鎖からなるIgG1mAbを用いたFACSにおいて、HER−3+細胞株への結合活性が検出された。他のVH/Vκの組み合わせは、HER−3+細胞株を用いたFACSにおいて、バックグラウンドを上回る蛍光シグナルを与えなかった。
【0126】
抗HER−3抗体の結合競合
HER−3への結合に関して競合するHER−3抗体のクラスターを評価するために、「Jia et al.J Immunol Methods.288, 91−98(2004)」に公表されているように、多重化された競合抗体ビニング(binning)を行った。コホート1由来の検査されたHER−3抗体は、結合に対する競合に基づいて、5つのビンにクラスター化された。
【0127】
【表5】
【0128】
抗HER−3抗体のエピトープ性質決定
本発明のヒト抗HER−3抗体のエピトープを性質決定した。まず、還元され、変性され、HER−3−Hisタグ化され、精製されたECDタンパク質のドットブロット分析が、検査された抗HER−3抗体(U1−59、U1−61、U1−41、U1−46、U1−53、U1−43、U1−44、U1−47、U1−52、U1−40、U1−49)による結合の不存在を示し、全てがジスルフィド結合の還元に対して感受性を有するエピトープを有することが実証され、全てが不連続なエピトープを有することが示唆された。次に、HER−3細胞外ドメインの以下の4つのドメインへの分割に基づいて、様々なヒト−ラットHER3キメラ分子を加工することによって、HER−3分子中の所定のドメインに抗体をマッピングした。
【0129】
1)L1(D1):非主要リガンド結合ドメイン、
2)S1(D2):第一のシステインリッチドメイン、
3)L2(D3):主要リガンド結合ドメイン、及び
4)S2(D4):第二のシステインリッチドメイン。
【0130】
RAT1−HER−3細胞から、ヒトHEr−3cDNAの細胞外ドメイン(ECR)を増幅した。ラット肝臓RNAからのRT−PCRによってラットHER−3cDNAを増幅し、配列決定によって確認した。ヒト及びラットHer3のECDを発現するcDNAを、V5−His融合タンパク質として、哺乳動物発現ベクター中にクローニングした。ヒトHER−3ECDから得られるドメインを、Mfe1、Bstx1及びDraIII内部制限部位を使用することによって、ラットHER−3ECDによって付与された足場中に取り替えた。この手段によって、様々なキメララット/ヒトHER−3ECDHIS融合タンパク質(アミノ酸1から160、161から358,359から575、1から358、359から604)を構築し、HEK293T細胞の一過性形質移入を介して発現させた。ヒトHER−3に対するラットポリクローナル抗体を用いて、構築物の発現を確認した。分泌されたキメラECDへの結合に関して、ヒトモノクローナル抗体をELISAにおいて検査した。
【0131】
抗体U1−59を含むヒト抗体の2つが、ラットHER−3と交叉反応した。結合ドメインを割り当てるために、HER3のL1−S1細胞外ドメインの発現をコードするプラスミドDNAが形質移入されたHEK293T細胞の上清から精製されたL1−S1−V5hisタグ化タンパク質からなるHER−3の末端切断形態に対して、これらのmAbを検査した。mAbU1−59は、ELISA中のL1−S1タンパク質に結合したので、そのエピトープがL1−S1中に存在することが示唆される。mAb2.5.1は、L1−S1タンパク質に結合しなかったので、そのエピトープがL2−S2中に存在することが示唆される。mAb−HER−3ECD複合体のチップ上タンパク質分解消化物とともに、飛行質量分析法のSELDI時間を用いて、抗体U1−59のさらなるマッピングを行った。
【0132】
SELDIを用いたU1−59のマッピング
mAb−HER−3ECD複合体のチップ上タンパク質分解消化物とともに、飛行質量分析法のSELDI時間を用いて、抗体U1−59のさらなるマッピングを行った。PS20タンパク質チップアレイに、プロテインAを共有結合させ、mAbU1−59を捕捉するために使用した。次いで、HER−3−His精製抗原とともに、PS20タンパク質チップとモノクローナル抗体の複合体を温置した。次に、Asp−Nの高濃度を用いて、抗体抗原複合体を消化した。チップを洗浄すると、チップ上の抗体に結合されたHER−3ペプチドのみが保持された。SELDIによってエピトープを決定し、断片の質量によってエピトープを同定した。同定された6814D断片は、HER−3hisECDの部分的消化物から生成される2つの可能な予想されたペプチドに対応する。重複するペプチドは何れも、ドメインS1にマッピングされる。HER−3欠失構築物への結合と、SELDIの結果を総合することによって、エピトープは、残基251から325にマッピングされた。
【0133】
本発明のヒト抗HER−3mAbによって認識されるHER−3の細胞外部分中における結合ドメインの位置が、表4に要約されている。エピトープドメインマッピングの結果は、抗体競合結合競合ビンから得られた結果と合致しており、HER−3への結合に関して互いに交叉競合した抗体は、HER−3上の同じドメインにマッピングされる(図3)。
【0134】
【表6】
XR=交叉反応性
【実施例9】
【0135】
抗体のカノニカルクラスの決定
Chothiaらは、各免疫グロブリン鎖の超可変領域に対する「カノニカルクラス」の観点で、抗体構造について記載している(J.Mol.Biol.,1987 Aug 20,196(4):901−17)。それらのアミノ酸配列とそれらの抗原結合部位の三次元構造との関係を決定するために、様々な免疫グロブリンのFab及びVL断片の原子構造を分析した。Chothiaらは、それらのパッキング、水素結合又は異常なφ、ψ若しくはω立体構造を採る能力を通じて、超可変領域の主鎖立体構造のために主として必要とされる残基は相対的に少ないことを見出した。これらの残基は、超可変領域内及び保存されたβシートフレームワーク中の部位に存在することが見出された。未知の構造を有する免疫グロブリンの配列を調べることによって、Chothiaらは、多くの免疫グロブリンが、公知の構造の1つとサイズが類似する超可変領域を有すること、さらに、観察された立体構造に必要とされる部位に同一の残基を含有することを示す。
【0136】
彼らの発見は、これらの超可変領域が公知の構造中のものに近い立体構造を有することを示唆した。超可変領域の5つに関しては、立体構造のレパートリーは、別個の構造クラスの比較的少数に限定されるようである。これらの一般的に存在する超可変領域の主鎖立体構造は、「カノニカル構造」と名付けられた。Chothiaら(Nature, 1989 Dec 21−28, 342(6252):877−83)及び他の者(Martin, et al.J.Mol.Biol., 1996 Nov 15, 263(5):800−15)によるさらなる研究は、抗体の6つの超可変領域のうち少なくとも5つに対して、主鎖立体構造の小さなレパートリーが存在することを確認した。
【0137】
それらのカノニカルクラスを決定するために、上記各抗体のCDRを分析した。既知のとおり、カノニカルクラスは、抗体軽鎖のCDR1、CDR2及びCDR3の他に、抗体重鎖のCDR1及びCDR2に対してのみ割り当てられている。下表は、分析の結果を要約している。カノニカルクラスのデータは、HCDR1−HCDR2−LCDR1−LCDR2−LCDR3の形態であり、「HCDR」は重鎖CDRを表し、「LCDR」は軽鎖CDRを表す。従って、例えば、1−3−2−1−5のカノニカルクラスは、カノニカルクラス1に属するHCDR1、カノニカルクラス3に属するHCDR2、カノニカルクラス2に属するLCDR1、カノニカルクラス1に属するLCDR2及びカノニカルクラス5に属するLCDR3を有する抗体を表す。
【0138】
各カノニカルクラスに対して定義されたアミノ酸と、抗体中のアミノ酸の70%又はそれ以上の同一性が存在する特定のカノニカルクラスに割り当てが為された。各抗体に対して定義されたアミノ酸は、例えば、上に引用されたChothiaらによる文献中に見出すことができる。表5及び表6は、HER−3抗体の各々に対するカノニカルクラスデータを報告する。70%未満の同一性が存在した場合には、各CDRの長さ及びデータの全体に基づいて、適切なカノニカルクラスの最良の推定が為されることを示すために、カノニカルクラスの割り当てにアスタリスク(「*」)が付されている。同じCDR長さを有する、合致したカノニカルクラスが存在しない場合には、「s18」(CDRが18のサイズであることを意味する。)のように、文字s及び数字で、カノニカルクラスの割り当てに印が付されている。重鎖又は軽鎖の1つに対して入手可能な配列データが存在しない場合には、カノニカルクラスは、「Z」でマークされる。
【0139】
【表7】
【0140】
表7は、クラス当りの抗体の数の分析である。左の欄に表記された特定のカノニカルクラスを有する抗体の数が、右の欄に示されている。1つの鎖配列データを欠如し、従って、カノニカル割り当てにおいて「Z」を有する4つのmAbは、このカウント中に含まれていない。
【0141】
最も一般的に見られる構造は、3−1−2−1−1である。重鎖及び軽鎖配列の両方を有する41のmAbのうち21が、この立体構造を有していた。
【0142】
【表8】
【実施例10】
【0143】
抗体親和性の測定
間接FACSScatchard分析によって、本発明の抗HER−3抗体の親和性測定を行った。従って、PBS中の10mMEDTAを用いて105個の目的の細胞又はSk−Br3細胞を採集し、FACS緩衝液(PBS、3%FCS、0.4%アジ化物)で1回洗浄し、96ウェルの丸底プレート上に播種した。上清を除去するために、1000rpmで3分間、細胞を遠心し、次いで、α―HER−3抗体(3μg/mL)又はFACS緩衝液中の20μg/mLヒトモノクローナル抗体から開始して、1:2の希釈工程で希釈された抗体希釈物(100μL/ウェル)とともに再懸濁した。細胞懸濁液を氷上で1時間温置し、FACS緩衝液で2回洗浄し、二次抗体(100μL/ウェル)ロバ抗ヒトPE(Jackson)によって再懸濁し、FACS緩衝液中に1:50希釈した。氷上及び暗所で、細胞懸濁液を30分間温置し、FACS緩衝液を用いて2回洗浄し、分析した(FACS、Beckman Coulter)。FACSScatchard分析に従って、各測定に対して蛍光平均を計算した。各蛍光平均からバックグラウンド染色(=第一の抗体なし)を差し引いた。x値=蛍光平均及びy値=蛍光平均/mAbの濃度(nM)であるScatchardプロットを作成した。KDは、線形方程式の1/mの絶対値と解した。図4は、本発明のU1−59抗体を用いた速度論分析を示している。以下の表8には、このようにして選択された本発明のある種の抗体に対する親和性測定が記載されている。
【0144】
【表9】
【実施例11】
【0145】
本発明の抗HER−3抗体は、HER−3受容体の飲食細胞運動を誘導する
HER−3は、HERファミリーによって媒介される細胞シグナル伝達の重要な門番としての役割を通じて、過剰増殖性疾患の開始及び進行に影響を与えることができる因子として同定されてきた。従って、HER−3が受容体の内部取り込みによって細胞表面/膜から効果的に除去されれば、細胞シグナル伝達が、従って、悪性腫瘍における細胞の形質転換及び/又は維持が、最終的に軽減又は抑圧され得る。
【0146】
本発明の抗HER−3抗体がHER−3の加速された飲食細胞運動を誘導できるかどうかを調べるために、本発明の抗HER−3抗体とともに細胞を0.5及び4時間温置した後に、細胞表面上のHER−3分子の相対量を比較した。24ウェル皿中の正常な増殖培地中に3×105個の細胞を播種し、一晩増殖させた。表記時間にわたって、37℃で、通常の増殖培地中の10μg/mL抗HER−3mAbとともに細胞を予め温置した。10mMEDTAを用いて細胞を剥離させ、洗浄緩衝液(PBS、3%FCS、0.04%アジ化物)中の10μg/mL抗HER−3mAbとともに、4℃で45分間温置した。洗浄緩衝液で細胞を2回洗浄し、1:100希釈されたロバ抗ヒトPE二次抗体(Jackson)とともに、4℃で45分間温置し、洗浄緩衝液で2回洗浄し、FACS(BeckmanCoulter、EXPO)によって分析した。
【0147】
図5に示されているデータは、抗HER−3抗体での細胞の処理が、受容体の内部取り込みをもたらすことを示している。データは、%内部取り込みとして示されており、対照処理された試料に対する、抗HER3処理された試料の平均蛍光強度の低下を表している。
【実施例12】
【0148】
本発明のヒト抗HER−3抗体による、ヒト癌細胞SKBr3へのリガンド結合の阻害
本発明の抗HER−3抗体が、細胞をベースとしたアッセイにおいて、HER−3へのリガンド結合を阻害する能力を定量するために、放射性リガンド競合実験を行った。従って、抗体の変動する濃度とともに、氷上で30分間温置された4×105個のSK−BR−3細胞を用いて、HER−3受容体結合アッセイを行った。各ウェルに、1.25nM[I125]−α−HRG[I125]−β−HRGを添加し、氷上で2時間、温置を継続した。プレートを5回洗浄し、風乾し、シンチレーションカウンターでカウントを計数した。図6aからeは、代表的な本発明の抗HER−3抗体を用いて実施されたこれらの実験の結果を示しており、本発明の抗体が、内在性HER−3を発現する細胞への、[125I]−α−HRG/[125I]−β−HRGの結合を特異的に低下させ得ることを示している。
【実施例13】
【0149】
本発明のヒト抗HER−3抗体による、リガンド誘導性HER−3リン酸化の阻害
本発明の抗体が、リガンドβ−HRGによって媒介されるHER−3の活性化を遮断することができるかどうかを調べるために、ELISA実験を行った。リガンドによって媒介されるHER−3の活性化は、増加した受容体チロシンリン酸化によって検出した。
【0150】
第1日:37℃で4時間、0.1M酢酸中の20μg/mLコラーゲンIで1×96ウェル皿をコートした。通常の増殖培地中に、2.5×105個の細胞を播種した。
【0151】
第2日:無血清培地100μL中で、24時間、細胞を飢餓状態にした。
【0152】
第3日:抗HER−3mAB10μg/mLとともに、37℃で1時間、細胞を予め温置し、次いで、β−HRG−EGFドメイン(R&D Systems)30ng/mLで、10分間処理した。溶媒を除去し、室温で1時間、PBS中の4%ホルムアルデヒド溶液で細胞を固定した。ホルムアルデヒド溶液を除去し、洗浄緩衝液(PBS/0.1%Tween20)で細胞を洗浄した。細胞緩衝液中の1%H2O2、0.1%NaN3で細胞を消光し、室温で20分間温置し、次いで、4℃で5時間、NET−ゼラチンでブロックした。4℃で一晩、一次抗体ホスホ−HER−3(Tyr1289)(ポリクローナルウサギ;Cell signaling #4791;1:300)を添加した。
【0153】
第4日:洗浄緩衝液で、プレートを3回洗浄した後、PBS中に1:3000希釈された抗ウサギ−PODとともに温置し、各ウェルに0.5%BSAを添加し、室温で1.5時間温置した。洗浄緩衝液で3回、PBSで1回、プレートを洗浄した。テトラメチルベンジジン(TMB、Calbiochem)を添加し、650nmでモニターした。250nMHCl100μLの添加によって反応を停止し、Vmaxプレートリーダー(Thermo LabSystems)を使用して、650nmの参照波長として、吸光度を450nmで読み取った。
【0154】
図7aは、本実験の代表的な結果を示し、減少した受容体チロシンのリン酸化によって示されたように、本発明の抗HER−3抗体が、リガンドによって媒介されるHER−3の活性化を低下させ得ることを示している。データは、対照抗体に対する、治療抗体によるパーセント低下として示されている。
【0155】
mAbU1−53が、リガンドによって媒介されるHER−3活性化を阻害する効力を検査するために、24時間、MCF−7細胞を飢餓状態とし、37℃で1時間、mAbU1−53とともに温置し、10分間、10nMHRG−βで刺激した。可溶化液を1B4(マウス抗HER−3mAb)ELISAプレートに移し、抗体4G10を用いて、HER−3のリン酸化を分析した。図7bに示されているように、HER−3のリン酸化は、0.14nMのIC50で、用量依存的な様式で、ほぼ完全に阻害された。
【実施例14】
【0156】
本発明のヒト抗HER−3抗体による、リガンド誘導性p42/p44MAPキナーゼリン酸化の阻害
本発明の抗体が、リガンドβ−HRGによって媒介されるp42/p44MAPキナーゼの活性化を遮断することができるかどうかを調べるために、次のELISA実験を行った。リガンドによって媒介されるHER−3の活性化は、増加したタンパク質(Thr202/Tyr204)チロシンリン酸化によって検出した。
【0157】
第1日:37℃で4時間、0.1M酢酸中の20μg/mLコラーゲンIで1×96ウェル皿をコートした。通常の増殖培地中に、3×105個の細胞を播種した。
【0158】
第2日:無血清培地100μL中で、24時間、細胞を飢餓状態にした。
【0159】
第3日:抗HER−3mAB5μg/mLとともに、37℃で1時間、細胞を予め温置し、次いで、β−HRG−EGFドメイン(R&D Systems)20ng/mLで、10分間処理した。溶媒を除去し、室温で1時間、PBS中の4%ホルムアルデヒド溶液で細胞を固定した。ホルムアルデヒド溶液を除去し、洗浄緩衝液(PBS/0.1%Tween20)で細胞を洗浄した。細胞緩衝液中の1%H2O2、0.1%NaN3で細胞を消光し、室温で20分間温置し、次いで、4℃で5時間、PBS/0.5%BSAでブロックした。4℃で一晩、一次抗体ホスホ−p44/p42MAPキナーゼ(Thr202/Tyr204)(ポリクローナルウサギ;Cell signaling #9101;1:300)を添加した。
【0160】
第4日:洗浄緩衝液で、プレートを3回洗浄した後、PBS中に1:5000希釈された抗ウサギ−HRPとともに温置し、各ウェルに0.5%BSAを添加し、室温で1.5時間温置した。洗浄緩衝液で3回、PBSで1回、プレートを洗浄した。テトラメチルベンジジン(TMB、Calbiochem)を添加し、650nmでモニターした。250nMHCl100μLの添加によって反応を停止し、Vmaxプレートリーダー(Thermo Lab Systems)を使用して、650nmの参照波長として、吸光度を450nmで読み取った。
【0161】
図8は、本実験の代表的な結果を示している。本発明の抗体は、減少されたリン酸化によって示されたように、リガンドによって媒介されたp42/p44MAP−キナーゼの活性化を低下させることができた。データは、対照抗体に対する、治療抗体によるパーセント低下として示されている。
【実施例15】
【0162】
本発明のヒト抗HER−3抗体による、β―HRG誘導性ホスホ−AKTリン酸化の阻害
以下のELISA実験において、本発明者らは、本発明の抗HER−3抗体が、リガンドβ−HRGによって媒介されるAKTキナーゼの活性化を遮断することができるかどうかを調べた。リガンドによって媒介されるAKTの活性化は、増加したタンパク質(Ser473)のリン酸化によって検出された。
【0163】
第1日:37℃で4時間、0.1M酢酸中の20μg/mLコラーゲンIで1×96ウェル皿をコートした。通常の増殖培地中に、3×105個の細胞を播種した。
【0164】
第2日:無血清培地100μL中で、24時間、細胞を飢餓状態にした。
【0165】
第3日:抗HER−3mAb5μg/mLとともに、37℃で1時間、細胞を予め温置し、次いで、β−HRG−EGFドメイン(R&D Systems)20ng/mLで10分間処理した。溶媒を除去し、室温で1時間、PBS中の4%ホルムアルデヒド溶液で細胞を固定した。ホルムアルデヒド溶液を除去し、洗浄緩衝液(PBS/0.1%Tween20)で細胞を洗浄した。細胞緩衝液中の1%H2O2、0.1%NaN3で細胞を消光し、室温で20分間温置し、次いで、4℃で5時間、PBS/0.5%BSAでブロックした。4℃で一晩、一次抗体ホスホ−Akt(Ser473)(ポリクローナルウサギ;Cell signaling #9217;1:1000)を添加した。
【0166】
第4日:洗浄緩衝液で、プレートを3回洗浄した後、PBS中に1:5000希釈された抗ウサギ−HRPとともに温置し、各ウェルに0.5%BSAを添加し、室温で1.5時間温置した。洗浄緩衝液で3回、PBSで1回、プレートを洗浄した。テトラメチルベンジジン(TMB、Calbiochem)を添加し、650nmでモニターした。250nMHCl100μLの添加によって反応を停止し、Vmaxプレートリーダー(Thermo Lab Systems)を使用して、650nmの参照波長として、吸光度を450nmで読み取った。
【0167】
図9は、本実験の代表的な結果を示している。本発明の抗HER−3抗体は、減少されたリン酸化によって示されたように、β−HRGによって媒介されたAKTを低下させることができた。データは、対照抗体に対する、治療抗体によるパーセント低下として示されている。
【実施例16】
【0168】
本発明のヒト抗HER−3抗体による、α−HRG/β−HRG媒介性MCF7細胞の阻害
本発明の抗体がHGRによって刺激された細胞増殖を阻害する能力を決定するために、インビトロ実験を実施した。96ウェルプレート上のFCS含有培地中に、一晩、2000個のMCF7細胞を播種した。37℃で1時間、0.5%FCSを有する培地中で希釈された抗体とともに、細胞を4つ組みで予め温置した。抗体溶液にリガンドを直接添加することによって、30ng/mLのα−HRG又は20ng/mLβ−HRG(R&DSystems)で細胞を刺激した後、72時間、増殖させた。AlamarBlueTM(BIOSOURCE)を添加し、37℃で、暗所にて温置した。30分毎に、590nmでの吸光度を測定した。アラマール・ブルーの添加から90分後に、データを採取した。図10に示されている結果は、本発明の代表的な抗体がヒト癌細胞中でのHRG誘導性細胞増殖を阻害することを示している。データは、対照抗体に対する、治療抗体によるパーセント低下として示されている。
【実施例17】
【0169】
本発明のヒト抗HER−3抗体による、β―HRG誘導性MCF7細胞遊走の阻害
本発明の抗体が細胞遊走を遮断するかどうかを調べるために、遊出実験を行った。細胞懸濁液に抗体の表記量を添加し、37℃で45分間、両者を温置することによって、血清飢餓状態にされたMCF7細胞を予め温置した。次いで、コラーゲンIによって被覆されたtranswell(BD Falcon,8μm孔)の上部チャンバー中に、細胞懸濁液(50,000細胞)500μLを配置した。単独の、又はリガンドβ−HRG−EGFドメイン(R&DSystems)を含有する750μL(MEM、アミノ酸、ピルビン酸ナトリウム、ペニシリン−ストレプトマイシン、0.1%BSA、ウシ胎児血清なし)を、下部チャンバー中で使用した。37℃で8時間、細胞を遊走させ、DAPIで染色した。
【0170】
染色された核を手作業で計数した。対照抗体と比較した阻害として、%阻害を表した。
【0171】
図11は、本発明の代表的な抗HER−3抗体が、HRGによって誘導される細胞遊走を低下させることを示す実験の結果を示している。
【実施例18】
【0172】
コロニー形成アッセイ(軟寒天アッセイ)
本発明の抗HER−3抗体が足場非依存性細胞増殖を阻害する能力を調べるために、軟寒天アッセイを実施した。このような形質転換された細胞のみが軟寒天中で増殖することができるので、軟寒天コロニー形成アッセイは、形質転換された細胞にして検査を行うための標準的なインビトロアッセイである。
【0173】
IMDM培地(Gibco)中の10μg/mLの表記抗体とともに、30分間、750から2000個の細胞(細胞株による)を予め温置し、0.4%Dfico noble寒天中に再懸濁した。96ウェルプレート中に、4つ組みで、20%FCSを含有する0.75%アガロース下層上に細胞懸濁液を播種した。コロニーを14日間形成させた後、50μLMTT(PBS中、0.5mg/mL)で一晩染色した。図12aからiは、本発明の3つの代表的な抗体を用いて実施されたこれらの実験の結果を示している。これらの結果は、本発明の抗HER−3抗体が、MDA−MB316及びNCI−ADR乳癌細胞(図12a、b)、MKN−28胃癌(図12c)、HT144悪性黒色腫(図12d)、Skov3卵巣癌細胞(図12e)、PPC−1前立腺癌(図12f)、BX−PC3膵臓癌細胞(図12g)、A431類表皮癌細胞(図12h)及び肺癌細胞(図12i)の足場非依存性細胞増殖を低下させることを示す。Scanalyzer HTSカメラシステム(Lemnatec,Wuerselen)を用いて、コロニーを計数した。
【実施例19】
【0174】
ヒト抗HRE−3抗体は、ヌードマウス中でのヒト乳癌の増殖を阻害する
しばしば、ヒト異種移植腫瘍研究において、治療用抗体の抗腫瘍効果が評価される。これらの研究では、ヒト腫瘍は、免疫無防備状態のマウス中で、異種移植片として増殖し、治療効果は、腫瘍増殖阻害の程度によって測定される。本発明の抗HER−3抗体が、ヌードマウス中でのヒト乳癌細胞の腫瘍増殖を妨害するかどうかを測定するために、雌のNMRIヌード/ヌードマウス中に、5×106個のT47D細胞を移植した。腫瘍は、動物の背中上において、皮下で増殖された。腫瘍は、移植から8日後に、20mm3の平均容積に到達した時点で、治療を開始した。第一の治療前に、マウスを無作為化し、治療群にわたって、最初の腫瘍容積(平均、中央値及び標準偏差)の均一性を確保するために、統計学的検定を行った。治療は、50mg/kgの搭載容量から開始し、続いて、腹腔内注射によって、週に一回、25mg/kgを注射した。対照腕には、ドキソルビシン(医薬等級)を与えた。全ての動物に、0.5mg/kg/週のエストロゲンを腹腔内注射して補充した。
【0175】
処置群の詳細は、以下に掲載されている。
【0176】
【表10】
【0177】
中央値腫瘍容積のデータ(図13)は、本発明の抗HER−3抗体の投与が、腫瘍増殖の低下をもたらすことを示した。
【実施例20】
【0178】
ヒト抗HRE−3抗体は、SCIDマウス中でのヒト膵臓腫瘍の増殖を阻害する
他の固体腫瘍の種類中での抗HER3抗体の治療的な可能性を検査するために、ヒト膵臓腫瘍細胞株B×PC3に由来する確立された腫瘍を有するマウスにおいて、抗HER−3抗体、U1−53及びU1−59を検査した。ビヒクル対照、PBS又は確立された治療用抗体Erbituxの何れかで処理されたマウスの対照群として含めた。5×106個のBxPC3細胞を、Matrigelなしに、CB17SCiDマウス中の皮下に接種した。140mm2の平均容積を有する確立された腫瘍を有するマウスに、腹腔内注射を介して、U1−53、U1−59、Erbituxの50mg/kg又はPBSの等容量を与えた。その後、マウスに、研究期間中、毎週1回、25mg/kgの注射を与えた。
【0179】
この実験に対する結果は、図14に示されている。U1−53及びU1−59は、細胞分裂停止状態の様式でヒト膵臓腫瘍の増殖を低下させた。注目すべきことに、本実験では、U1−53及びU1−59は、腫瘍増殖の遅延に関して、EGFR標的抗体であるErbituxより有効であった。これらのデータは、基準治療剤と比較した、本発明の抗HER−3抗体の治療的効果を示した。
【実施例21】
【0180】
ヒト抗HER−3抗体を抗EGFR抗体と組み合わせることによって、抗腫瘍活性が増加する
標的化された抗体による過剰増殖性疾患の単独療法は、しばしば、一方で、薬物耐性の発生、他方で、抗原性の変化などの問題によって妨害される。例えば、標的化された抗原を発現していない腫瘍細胞又は標的化された抗原を喪失した腫瘍細胞は選択的な増殖という利点を有するので、長期の治療後に抗原性の喪失が、腫瘍細胞を治療抗体に対して非感受性とさせ得る。これらの問題は、腫瘍細胞上の異なる受容体を標的とする治療抗体又は別の抗新生物因子と組み合わせて本発明の抗体を使用することによって回避され得る。複数のシグナル伝達経路又は関連する経路において、但し、複数の介入工程で介入することも、治療的な利点を与え得る。これらの組み合わせ治療様式は、各々、異なる作用機序を介して作用する2つの抗癌剤を組み合わせるので、より有効である可能性がある。
【0181】
本発明の抗HER−3抗体U1−53及びU1−59の適切な組み合わせ因子としての実用可能性を示すために、本発明者らは、U1−53又はU1−59の何れかが、抗EGR特異的抗体Erbituxと組み合わされる療法と、U1−53又はU1−59の単独療法の投与を比較した。5×106個のBxPC3細胞を、Matrigelとともに、CB17SCIDマウス中の皮下に接種した。腫瘍容積が200mm3に達した後、各処置群にマウスを無作為に振り分けた。単一の薬剤として、又はErbituxと抗HER3抗体の組み合わせとして、又は2つの抗HER−3抗体のカクテルとして、U1−53、U1−59及びErbituxの腹腔内投与を毎週行った。全ての抗体は、50mg/kg/週の単回投薬用量で投薬した後、6週間、25mg/kgを毎週注射した。対照腕に、ゲムシタビン(120mg/kg)に隔週投与し、ヒトIgGを毎週プールし、又はビヒクル(PBS)注射を毎週プールした。治療計画は、以下に詳述されている。
【0182】
【表11】
【0183】
この実験に対する結果は、図15に示されている。単一の薬剤として投与された場合に、抗体U1−53及びU1−59は、しばしば標準的な抗膵臓癌化学療法として使用されるゲムシタビンと同じ程度まで、ヒト膵臓腫瘍の増殖を遅延させた。U1−53又はU1−59とのErbituxの同時投与は、U1−53、U1−59又はErbituxの何れかの単一薬剤投与を用いて観察されたものより、腫瘍増殖を有意により大きく低下させた。従って、別個の腫瘍抗原を標的とする適切な抗体と本発明の抗HER−3抗体を組み合わせることによって、有益な治療的応答を達成することが可能である。
【0184】
要約すると、本発明の抗HER−3抗体は、ヒト腫瘍に対して、インビボで強力な治療効果を有する。本発明の抗HER−3抗体は、抗腫瘍活性を増加させるために、他の抗新生物治療薬と効果的に組み合わせることが可能である。
【実施例22】
【0185】
ヒト抗HRE−3抗体は、nu/nuマウス中でのヒト悪性黒色腫瘍の増殖を阻害する
Her3を含む受容体のerbBファミリーのメンバーは、極めて様々な上皮癌中で異常に発現されており、多くのこれらの固形腫瘍の増殖及び生存において重要な役割を果たしていることが知られている。これらの腫瘍には、悪性黒色腫、頭部及び頸部扁平上皮細胞癌、非小細胞肺癌及び前立腺癌、神経膠腫、胃癌、乳癌、結腸直腸癌、膵臓癌、卵巣癌が含まれる。本発明の抗Her3抗体の抗癌活性が個別の腫瘍種(例えば、膵臓癌(実施例21参照))に限定されず、多くのHER−3依存性腫瘍に対する治療薬として使用することができることを確かめるために、本発明者らは、さらなる異種移植片研究においてU1−53及びU1−59を検査した。一例が図16に示されている。CB17SCIDマウス中に、5×105個のヒト悪性黒色腫細胞HT144を皮下注射した後、U1−53及びU1−59の50mg/kg、PBSの等しい容積又は200mg/kgのダカルバシン(DITC)を直ちにその後腹腔内注射した。その後、U1−53又はU1−59の25m/kgは、週一回、マウスに与えられたのに対して、200mg/kgで、2週毎に1回、DITCを与えた。
【0186】
各処置群から得られた中央値腫瘍容積が、図16に示されている。本発明の抗体の投与は、ビヒクル対照で処理された腫瘍と比べた場合に、ヒト悪性黒色腫の増殖の低下をもたらした。これらの結果は、本発明の抗体が、それらの治療的な潜在能力に限定されず、HER−3を発現している多様な癌を標的とすることを示している。
【実施例23】
【0187】
ヒト抗HRE−3抗体は、マウス中の大腸癌異種移植片の増殖を阻害する
10×106細胞/mLの最終濃度になるように、Matrigelの2:1の比率を用いて、HT−29ヒト大腸癌細胞を培地中に懸濁した。4から5週齢のCD1nu/nuマウスの右側腹部中に、細胞懸濁液0.2mLを皮下注射した。計95匹のマウスを使用した。
【0188】
マウスを、対照群及び処置群へ無作為に割り当てた。処置は、同じ日に開始した。治療の期間は、29日であった。研究が完了した時点で、処置を施してから3時間後に、群当り3つの腫瘍を集めた。腫瘍を迅速に凍結し、−80℃に保った。
【0189】
以下の治療プロトコールを実施した。
【0190】
対照群:非特異的ヒトIgG25mg/kg、週2回、腹腔内。
【0191】
処置群:抗体U1−53、25m/kg、週2回、腹腔内。
【0192】
処置群:抗体U1−7、25m/kg、週2回、腹腔内。
【0193】
処置群:抗体U1−59、25m/kg、週2回、腹腔内。
【0194】
処置群5−FU:5−フルオロウラシル、50mg/kg、9d×5、腹腔内。
【0195】
各群から得られた中央値腫瘍容積が、図17に示されている。本発明の抗体の投与は、非特異的ヒトIgG1で処理された腫瘍と比べた場合に、HT−29大腸癌腫瘍の増殖低下をもたらした。
【実施例24】
【0196】
ヒト抗HRE−3抗体は、マウス中の肺癌増殖を阻害する
5×106細胞/mLの最終濃度になるように、Matrigelの1:1の比率を用いて、Calu−3ヒト非小細胞肺癌細胞を培地中に懸濁した。9週齢の雌CB17scidマウスの右側腹部中に、細胞懸濁液0.05mLを皮下注射した。計60匹のマウスを使用した。
【0197】
マウスを、対照群及び処置群へ無作為に選択した。処置は、同じ日に開始した。治療の期間は、32日であった。
【0198】
以下の治療プロトコールを実施した。
【0199】
PBSビヒクル群
hG対照群:非特異的ヒトIgG:25mg/kg、週2回、腹腔内。
【0200】
処置群:抗体U1−53、25m/kg、週2回、腹腔内。
【0201】
処置群:抗体U1−7、25m/kg、週2回、腹腔内。
【0202】
処置群:抗体U1−59、25m/kg、週2回、腹腔内。
【0203】
各対照群及び処置群から得られた中央値腫瘍容積が、図18に示されている。本発明の抗体の投与は、PBSビヒクル対照又は非特異的ヒトIgGで処理された腫瘍と比べた場合に、ヒト非小肺癌異種移植片の増殖の低下をもたらした。
【実施例25】
【0204】
ヒト抗HRE−3抗体は、Balb/Cマウス中でのヒト膵臓腫瘍の増殖を阻害する
5×106細胞/mLの最終濃度になるように、Matrigelの2:1の比率を用いて、ヒト膵臓BxPC3腫瘍細胞を培地中に懸濁した。5から5週齢の雌BalbCnu/nuマウスの右側腹部中に、細胞懸濁液0.2mLを皮下注射した。計100匹のマウスを使用した。
【0205】
マウスを、対照群及び処置群へ無作為に割り当てた。処置は、同じ日に開始した。治療の期間は、27日であった。
【0206】
以下の治療プロトコールを実施した。
【0207】
hIgG対照群:非特異的ヒトIgG、25mg/kg、週2回、腹腔内。
【0208】
処置群:抗体U1−53、25m/kg、週2回、腹腔内。
【0209】
処置群:抗体U1−7、25m/kg、週2回、腹腔内。
【0210】
処置群:抗体U1−59、25m/kg、毎週、腹腔内。
【0211】
Gemzar処置群、ゲムシタビン、80m/kg、毎週、腹腔内。
【0212】
各対照群及び処置群から得られた中央値腫瘍容積が、図19に示されている。本発明の抗体の投与は、非特異的ヒトIgG又はGemzarで処理された腫瘍と比べた場合に、ヒト膵臓腫瘍の増殖低下をもたらした。
【0213】
ヒト膵臓腫瘍中でのHER−3の阻害は、薬力学的実験においても示すことができた。BxPC3腫瘍異種移植片は、上述のように増殖させた。IgG1対照抗体500μgで3匹のマウスを処理し、抗HER−3抗体U1−59の500μgで3匹のマウスを処理した。第1日及び第4日目にマウスを処理した後、HER−3リン酸化(pHER−3)の抗体依存性阻害を測定するために、第5日目に屠殺した。
【0214】
プロテアーゼ阻害剤を加えた標準的なRIPA緩衝液中で、腫瘍をホモゲナイズした。4から20%のTris−グリシンゲル上で50μgの透明な可溶化液を分離し、ニトロセルロース膜上に転写し、3%ウシ血清アルブミン(BSA)中にブロックした。抗HER−3抗体(抗体21D3、Cell Signaling technology)を用いて、イムノブロッティングを行った。抗アクチン抗体(ABa−2066、Sigma)を対照として使用した。
【0215】
増強された化学発光(Amersham Biosciences,Piscataway,NJ)によって、発現を検出した。Versadoc5000Imaging System(BioRad, Hercules, CA)を用いて、画像を捕捉した。
【0216】
結果は、図20に示されている。ヒト抗HER−3抗体U1−59の投与後、HER−3のリン酸化は、もはや検出できなかった。従って、本発明の抗体は、ヒト膵臓腫瘍細胞中でのHER−3の活性化を著しく低下させることができる。
【実施例26】
【0217】
診断剤としての本発明の抗HER−3抗体の使用
抗HER−3mAbは、悪性腫瘍疾患の診断において使用することが可能である。HER−3は、正常な組織と比べて極めて異なった様式で腫瘍細胞上に発現されており、従って、HER−3の発現分析は、固形腫瘍の一次診断、固形腫瘍の段階決定及び等級付け、増殖性疾患及び悪性新生物の予後診断基準の評価並びにHER−3陽性腫瘍を有する患者におけるリスク管理を補助する。
【0218】
A.試料中でのHER−3抗原の検出
試料中でのHER−3抗原の検出用酵素結合免疫吸着検定法(ELISA)が開発されている。本アッセイでは、96ウェルミクロタイタープレート又は384ウェルマイクロタイタープレートなどのマイクロタイタープレートのウェルに、HER−3抗原に対して誘導された第一の完全なヒトモノクローナル抗体を数時間吸着させる。固定化された抗体は、検査試料中に存在し得るHER−3抗原の何れかに対する捕捉抗体としての役割を果たす。ウェルを濯ぎ、分析物の非特異的吸着を防ぐために、ミルクタンパク質又はアルブミンなどのブロッキング剤で処理した。
【0219】
続いて、HER−3抗原を含有すると疑われる検査試料で、又はHER−3抗原の標準量を含有する溶液でウェルを処理する。例えば、このような試料は、病状を診断するものと考えられる循環HER−3抗原のレベルを有すると疑われる対象から得られる血清試料である。検査試料又は標準を濯いだ後、ビオチンの連結によって標識される本発明の第二の完全ヒトモノクローナル抗HER−3抗体でウェルを処理する。標識された抗HER−3抗体は、検出抗体としての役割を果たす。過剰な二次抗体を濯いで除去した後、アビジン連結された西洋ワサビペルオキシダーゼ(HRP)及び適切な色素産生性基質でウェルを処理する。検査試料中のHER−3抗原の濃度は、標準試料から作成された標準曲線との比較によって決定される。
【0220】
B.免疫組織化学(IHC)中のHER3抗原の検出
IHCによる組織切片中のHER3抗原を測定するために、まず、パラフィン包埋された組織を、2回、5分間、キシレン中でパラフィン除去し、次いで、100%エタノールで3分間、2回、95%エタノールで1分間、水和し、蒸留水中で濯いだ。エピトープ遮蔽の除去、酵素消化又はサポニンによって、ホルマリン固定及びパラフィン包埋によって遮蔽された抗原エピトープを露出させる。エピトープの遮蔽を除去するために、例えば、2NHCl溶液(pH1.0)のようなエピトープ修復溶液中で、20から40分間にわたって、蒸し器、水槽又は電子レンジ中で切片を加熱する。酵素消化の場合には、プロテイナーゼK、トリプシン、プロナーゼ、ペプシンなどの様々な酵素溶液中において、37℃で10から30分間、組織切片を温置する。
【0221】
エピトープ修復溶液又は過剰な酵素を濯いで除去した後、非特異的な相互作用を防ぐために、ブロッキング緩衝液で組織切片を処理する。室温で1時間、又は一晩、希釈緩衝液中において、適切な希釈で一次抗体を温置する。過剰の一次抗体を濯いで除去し、室温で10分間、ペルオキシダーゼブロッキング溶液中で切片を温置する。別の洗浄工程後、酵素に対する足場としての役割を果たし得る基で標識された二次抗体とともに組織切片を温置する。従って、例は、ストレプトアビジンが連結された西洋ワサビペルオキシダーゼによって認識されるビオチン標識された二次抗体である。前記抗体/酵素複合体の検出は、適切な色素産生性基質とともに温置することによって達成される。
【0222】
C.患者の血清中のHER−3抗原濃度の測定
ヒト血清中のHER−3レベルを定量するために、サンドイッチELISAを開発する。サンドイッチELISA中で使用された2つの完全なヒトモノクローナル抗HER−3抗体は、HER−3分子上の異なるドメインを認識し、結合に関して競合しない。例えば、実施例8を参照されたい。以下のように、ELISAを実施する。2μg/mLの濃度の、コーティング緩衝液(0.1MNaHCO3、pH9.6)中の捕捉抗HER−3抗体50μLを、ELISAプレート(Fisher)上に被覆した。4℃で一晩温置した後、ブロッキング緩衝液200μL(PBS中、0.5%BSA、0.1%Tween20、0.01%チメロサール)で、25℃で1時間、プレートを処理する。PBS中の0.05%Tween20を用いて、プレートを洗浄した(3回)(洗浄緩衝液、WB)。50%ヒト血清を含有するブロッキング緩衝液中に正常な血清又は患者の血清(Clinomics,Bioreclaimation)を希釈する。4℃で一晩、血清試料とともにプレートを温置し、洗浄緩衝液で洗浄し、次いで、25℃で1時間、ビオチン化された検出抗HER−3抗体100μL/ウェルとともに温置する。洗浄後、HRP−ストレプトアビジンとともにプレートを15分間温置し、前述のように洗浄し、次いで、発色させるために、H2O2中のo−フェニレンジアミン(Sigma発色溶液)100μL/ウェルで処理する。H2SO4(2M)の50μL/ウェルで反応を停止させ、492nmでELISAプレートリーダーを用いて分析する。4パラメータ曲線フィッティングプログラムを用いて、精製されたHER−3抗原の希釈と比較することによって、血清試料中のHER−3抗原の濃度を計算する。
【0223】
患者中の癌の段階決定
項目A、B及びCで記載及び論述した結果に基づき、本発明の使用を通じて、HER−3抗原の発現レベルを基礎として、対象中の癌の段階を決定することが可能である。癌の所定の種類に関して、疾病の進行における様々な段階であるとして、及び/又は癌の治療的処置における様々な点であるとして診断された対象から血液の試料を採取する。血液試料中に存在するHER−3抗原の濃度は、存在する抗原の量を特異的に測定する方法を用いて決定される。このような方法には、項目A及びBで記載されている方法などのELISA法が含まれる。進行又は治療の各段階に対して統計学的に有意な結果を与える試料の集団を用いて、各段階に関して特徴的であると考え得るHER−3抗原の濃度範囲が表記される。
【0224】
研究されている対象中の癌の進行の段階を決定するために、又は療法の経過に対する対象の応答を特徴付けるために、血液の試料を、対象から採取し、試料中に存在するHER−3抗原の濃度を測定する。このようにして得られた濃度は、値が属する濃度範囲を同定するために使用される。このようにして同定された範囲は、診断された対象の様々な集団において同定された進行の段階又は療法の段階と相関し、これにより、研究されている対象中の段階を決定する。
【実施例27】
【0225】
過剰増殖性疾患の治療又は予防のための、本発明の抗HER−3抗体及び抗体抱合体の使用
多くの固形腫瘍は、HERファミリーに媒介されるシグナル伝達によって駆動され、HER−1、HER−2及びHER−4間の複合体形成を通じて、HER−3は不可欠なパートナーであることが示されている。従って、HER−3によって媒介されるシグナル伝達の低下又は除去は、全ての他のHERファミリーのメンバーに影響を与え、細胞シグナル伝達を損なわせ、治療的介入の幅広い枠並びに標的化される他の因子、生物試薬及び細胞毒性因子との併用療法の可能性をもたらす。従って、本発明の抗HER−3抗体は、例えば、HER−3発現のような多数の因子に基づくある種の過剰増殖性疾患又はHER−3関連疾患の治療のために使用することが可能である。乳癌、胃腸癌、膵臓癌、前立腺癌、卵巣癌、胃癌、子宮内膜癌、唾液腺癌、肺癌、腎臓癌、大腸癌、直腸結腸癌、甲状腺癌、膀胱癌、神経膠腫、悪性黒色腫、HER−3を発現又は過剰発現する他の癌などの腫瘍の種類は、好ましい適応症に該当すると思われるが、適応症は、前記のリストに掲載されているものに限定されるものではない。さらに、患者の以下の群には、抗HER−3誘導性mAb治療が有益である。
【0226】
・抗HER−2mAb治療に対して耐性を有する患者
・抗HER−2mAb治療を用いた治療に対する適格性を欠く患者
・抗HER−1mAb又は小分子抗EGFR阻害剤に対して耐性を有する患者
・エルロチニブ又はゲフィチニブに対して耐性を示す非小細胞肺癌を有する患者
本発明の抗HER−3抗体は、単独療法として、又は、いわゆる「併用療法」において、1つ若しくはそれ以上の因子と組み合わせて使用される。前記併用療法は、本発明において前記した因子を含み得るが、これに限定されるものではない。抗HER3抗体及び他の因子との併用療法は、患者の生存、腫瘍進行までの時間を延長し、又は患者の生活の質を向上させ得る。プロトコール及び投与の設計は、治療の効果に対処するのみならず、標準的な療法(例えば、化学療法又は放射線療法など)の通常用量を低下させることができる。
【0227】
本発明の抗HER−3抗体を用いたヒトの治療
腫瘍を有するヒト患者における抗HER−3抗体治療のインビボでの効果を測定するために、本発明の抗HER−3抗体の有効量を、一定回数にわたって、このようなヒト患者に注射する。治療中の一定期間に、腫瘍が進行するかどうか、特に、腫瘍が増殖及び転移するかどうかを明らかにするために、ヒト患者をモニターする。
【0228】
本発明の抗HER−3抗体で治療された腫瘍患者は、現在の治療薬の標準で治療された腫瘍患者における腫瘍増殖及び転移のレベルと比べて、腫瘍増殖及び/又は転移のより低いレベルを有する。
【0229】
本発明の抗HER−3抗体抱合体を用いた治療
本発明の抗HER−3抗体抱合体のインビボでの効果を測定するために、本発明の抗HER−3抗体抱合体の有効量を、一定回数、腫瘍を呈するヒト患者又は動物に注射する。例えば、投与される抗HER−3抗体抱合体は、DM1−抗HER−3抗体抱合体、アウリスタチン−抗HER−3抗体抱合体又は放射性同位体−抗HER−3抗体抱合体である。治療中、定期的に、腫瘍が進行するかどうか、特に、腫瘍が増殖及び転移するかどうかを明らかにするために、ヒト患者又は動物をモニターする。
【0230】
腫瘍を呈し、例えば、DM1−抗HER−3抗体抱合体又は放射線同位体−抗HER−3抗体抱合体での治療を受けているヒト患者又は動物は、腫瘍を呈し、別の療法での治療を受けている対照患者又は動物と比べて、腫瘍増殖及び転移のより低いレベルを有する。動物中で使用され得る対照DM1−抗体には、本発明の抗HER−3抗体と同じイソタイプであるが、より具体的には、HER−3腫瘍体抗原への結合能を有しない抗体に連結されたDM1を含む抱合体が含まれる。動物検査において使用され得る対照放射性同位体−抗体には、本発明の抗HER−3抗体と同じイソタイプであるが、より具体的には、HER−3腫瘍抗原への結合能を有しない抗体に連結された放射性同位体を含む抱合体が含まれる。注意:対照抱合体は、ヒトに投与されない。
【0231】
総論
前記明細書は、当業者が本発明を実施可能であるのに十分であると考えられる。寄託された実施形態は、本発明のある種の目的の一つの例示であることが意図され、機能的に均等である全ての構築物が、本発明の範囲に属するので、本発明の範囲は、寄託された構築物による範囲に限定されない。本明細書中の寄託物は、本明細書中に含まれる記載が本発明の何れかの対象(本発明の最良の態様を含む。)を実施可能とするのに不十分であることを認めるものではなく、それが表す特定の例示に特許請求の範囲を限定するものと解釈すべきでない。
【0232】
先述の記載及び実施例は、本発明のある種の好ましい実施形態を詳述し、本発明者らによって想定される最良の態様を記載する。しかしながら、先述の記載が本文中でいかに詳しく記されていても、本発明は多くの様式で実施することができ、本発明は、添付の特許請求の範囲及びそのあらゆる均等物に従って解釈しなければならないことが理解される。
【0233】
さらに、別段の定義がなければ、本発明に関連して使用される科学及び技術用語は、当業者によって一般的に理解される意味を有するものとする。さらに、文脈上別段の必要がなければ、単数形の用語は複数を含むものとし、複数形の用語は単数を含むものとする。一般的に、本明細書に記載されている細胞及び組織培養、分子生物学並びにタンパク質及びオリゴヌクレオチド又はポリヌクレオチド化学及びハイブリダイゼーションに関連して使用される命名法及びこれらの技術は、周知のものであり、本分野において一般的に使用されている。組換えDNA、オリゴヌクレオチド合成及び組織培養及び形質転換のために、標準的な技術が使用されている(例えば、電気穿孔、リポフェクション)。酵素反応及び精製技術は、製造業者の説明書に従って、又は本分野で一般的に遂行されているように、又は本明細書に記載されているように実施される。一般に、先述の技術及び操作は、本分野で周知の慣用的な方法に従い、並びに本明細書を通じて引用及び論述されている様々な一般的参考文献及びより具体的な参考文献中に記載されているように、実施される。例えば、「Sambrook et al.Molecular Cloning:A Laboratory Manual(3rd ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.(2001))」(参照により、あらゆる目的のために、本明細書に組み込まれる。)を参照されたい。本明細書に記載されている分析化学、合成有機化学及び医薬品化学及び薬化学に関して使用される命名法、並びに本明細書に記載されている分析化学、合成有機化学及び医薬品化学及び薬化学の実験室操作及び技術は、周知のものであり、本分野で一般的に使用されているものである。化学合成、化学分析、薬学的な調製、調合、及び送達、及び患者の治療に関しては、標準的な技術が使用される。
【0234】
参照による組み込み
本願に引用されている全ての参考文献(特許、特許出願、論文、教科書(これらの中に引用されている参考文献を含む。)など)は、参照により、その全体が、本明細書に組み込まれる。
【0235】
【化3】
【0236】
【化4】
【0237】
【表12】
【特許請求の範囲】
【請求項1】
HER−3に結合する単離された結合タンパク質であり、
(a)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDHR1;(b)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH2;並びに(c)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH3からなる群から選択されるCDRの少なくとも1つを含む重鎖アミノ酸配列を含む、前記タンパク質。
【請求項2】
HER−3に結合する単離された結合タンパク質であり、
(d)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL1;(e)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL2;並びに(f)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL3からなる群から選択されるCDRの少なくとも1つを含む軽鎖アミノ酸配列を含む、前記タンパク質。
【請求項3】
請求項1又は2に記載の単離された結合タンパク質であり、前記結合タンパク質が、
(a)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDHR1;(b)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH2;並びに(c)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH3からなる群から選択されるCDRの少なくとも1つを含む重鎖アミノ酸配列、並びに
(d)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL1;(e)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL2;並びに(f)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL3からなる群から選択されるCDRの少なくとも1つを含む軽鎖アミノ酸配列を含む、前記タンパク質。
【請求項4】
前記結合タンパク質が、配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230の何れかのCDRH1、CDRH2及びCDRH3を含む重鎖アミノ酸配列、又は配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232の何れかのCDRL1、CDRL2及びCDRL3を含む軽鎖アミノ酸配列を含む、請求項1又は2に記載の単離された結合タンパク質。
【請求項5】
前記結合タンパク質が、配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230の何れかのCDRH1、CDRH2及びCDRH3を含む重鎖アミノ酸配列、並びに配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232の何れかのCDRL1、CDRL2及びCDRL3を含む軽鎖アミノ酸配列を含む、請求項3に記載の単離された結合タンパク質。
【請求項6】
配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230からなる群から選択される重鎖アミノ酸配列を含む、請求項1に記載の単離された結合タンパク質。
【請求項7】
配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232からなる群から選択される軽鎖アミノ酸配列を含む、請求項2に記載の単離された結合タンパク質。
【請求項8】
配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230からなる群から選択される重鎖アミノ酸配列、並びに
配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232からなる群から選択される軽鎖アミノ酸配列を含む、請求項1に記載の単離された結合タンパク質。
【請求項9】
配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230の何れかのCDRH3を含む、請求項1に記載の単離された結合タンパク質。
【請求項10】
配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232の何れかの軽鎖アミノ酸配列を含む、請求項9に記載の単離された結合タンパク質。
【請求項11】
配列番号42の重鎖アミノ酸配列及び配列番号44の軽鎖アミノ酸配列を含む、請求項1から9の何れか一項に記載の単離された結合タンパク質。
【請求項12】
配列番号54の重鎖アミノ酸配列及び配列番号56の軽鎖アミノ酸配列を含む、請求項1から9の何れか一項に記載の単離された結合タンパク質。
【請求項13】
配列番号70の重鎖アミノ酸配列及び配列番号72の軽鎖アミノ酸配列を含む、請求項1から9の何れか一項に記載の単離された結合タンパク質。
【請求項14】
成熟HER−3、特に、成熟ヒトHER−3のアミノ酸残基1から160、161から358、359から575、1から358、359から604によって形成される三次元構造に結合する単離された結合タンパク質。
【請求項15】
結合タンパク質が、HER−3の細胞外ドメインに対して誘導される、請求項1から14の何れかに記載の単離された結合タンパク質。
【請求項16】
HER−3への結合タンパク質の結合が、HER−3によって媒介されるシグナル伝達を低下させる、請求項1から15の何れかに記載の単離された結合タンパク質。
【請求項17】
HER−3への結合タンパク質の結合が、HER−3のリン酸化を低下させる、請求項1から15の何れか一項に記載の単離された結合タンパク質。
【請求項18】
HER−3への結合タンパク質の結合が、細胞増殖を低下させる、請求項1から15の何れか一項に記載の単離された結合タンパク質。
【請求項19】
HER−3への結合タンパク質の結合が、細胞の遊走を低下させる、請求項1から15の何れか一項に記載の単離された結合タンパク質。
【請求項20】
HER−3への結合タンパク質の結合が、HER−3の下方制御を増加させる、請求項1から15の何れか一項に記載の単離された結合タンパク質。
【請求項21】
抗体である、請求項1から20の何れか一項に記載の単離された結合タンパク質。
【請求項22】
抗体が、モノクローナル抗体、ポリクローナル抗体、組換え抗体、ヒト化抗体、ヒト抗体、キメラ抗体、多重特異的抗体又はこれらの抗体断片である、請求項21に記載の単離された結合タンパク質。
【請求項23】
抗体断片が、Fab断片、Fab’断片、F(ab’)2断片、Fv断片、ダイアボディ又は一本鎖抗体分子である、請求項22に記載の単離された結合タンパク質。
【請求項24】
前記単離された結合タンパク質が、IgG1、IgG2、IgG3又はIgG4型である、請求項1から23の何れか一項に記載の単離された結合タンパク質。
【請求項25】
結合タンパク質が、標識基に連結されている、請求項1から24の何れか一項に記載の単離された結合タンパク質。
【請求項26】
標識基が、放射性同位体若しくは放射性核種、蛍光基、酵素基、化学発光基、ビオチニル基又は所定のポリペプチドエピトープである、請求項25に記載の単離された結合タンパク質。
【請求項27】
結合タンパク質が、エフェクター基に連結されている、請求項1から24の何れか一項に記載の単離された結合タンパク質。
【請求項28】
エフェクター基が、放射性同位体若しくは放射性核種、毒素又は治療基若しくは化学療法基である、請求項27に記載の単離された結合タンパク質。
【請求項29】
治療基若しくは化学療法基が、カリチアマイシン、アウリスタチン−PE、ゲルダナマイシン、マイタンシン及びこれらの誘導体からなる群から選択される、請求項28に記載の単離された結合タンパク質。
【請求項30】
請求項1から29の何れか1項に記載の結合タンパク質をコードする単離された核酸分子。
【請求項31】
核酸分子が、調節配列に作用可能に連結されている、請求項30に記載の単離された核酸分子。
【請求項32】
請求項30に記載の核酸分子を含むベクター。
【請求項33】
請求項31に記載の核酸分子を含むベクター。
【請求項34】
請求項32に記載のベクターで形質転換された宿主細胞。
【請求項35】
請求項33に記載のベクターで形質転換された宿主細胞。
【請求項36】
宿主細胞から前記結合タンパク質を単離する工程を含む、請求項1から29の何れか一項に記載の単離された結合タンパク質を調製する方法。
【請求項37】
宿主細胞が、哺乳動物細胞、植物細胞、真菌細胞又は原核細胞である、請求項36に記載の方法。
【請求項38】
請求項1か29の何れか一項に記載の少なくとも1つの単離された結合タンパク質を活性因子として含み、及び医薬として許容される、担体、希釈剤又は佐剤を含む医薬組成物。
【請求項39】
治療的使用のための請求項37又は38に記載の組成物。
【請求項40】
診断的使用のための請求項37又は38に記載の組成物。
【請求項41】
請求項38又は39に記載の医薬組成物の医薬有効量を、HER−3と関連する疾病の治療又は予防を必要としている対象に投与することを含む、患者中のHER−3に関連する疾病を治療又は予防する方法。
【請求項42】
疾病が過剰増殖性疾患である、請求項41に記載の方法。
【請求項43】
前記過剰増殖性疾患が、乳癌、胃腸癌、膵臓癌、前立腺癌、卵巣癌、胃癌、子宮内膜癌、唾液腺癌、肺癌、腎臓癌、大腸癌、直腸結腸癌、甲状腺癌、膀胱癌、神経膠腫、悪性黒色腫、精巣癌、軟部組織肉腫、頭部及び頸部癌、HER−3を発現又は過剰発現する他の癌並びに腫瘍転移の形成からなる群から選択される、請求項42に記載の方法。
【請求項44】
前記過剰増殖性疾患が、増加したHER−3のリン酸化、増加したHER−2/HER−3のヘテロ二量体化又はPI3−キナーゼ、c−jun末端キナーゼ、AKT、ERK2及び/又はPYK2の増加した活性を伴う、請求項42又は43に記載の方法。
【請求項45】
HER−3に関連する疾病を診断する方法であり、
(a)請求項1から26の何れか一項に記載の結合タンパク質のHER−3への結合を可能とするのに適した条件下で、請求項1から26の何れか一項に記載の結合タンパク質と試料を接触させること、及び
(c)HER−3への前記結合タンパク質の結合を同定すること、
を含む、前記方法。
【請求項46】
疾病が過剰増殖性疾患である、請求項45に記載の方法。
【請求項47】
前記過剰増殖性疾患が、乳癌、胃腸癌、膵臓癌、前立腺癌、卵巣癌、胃癌、子宮内膜癌、唾液腺癌、肺癌、腎臓癌、大腸癌、直腸結腸癌、甲状腺癌、膀胱癌、神経膠腫、悪性黒色腫、HER−3を発現又は過剰発現する他の癌、精巣癌、軟部組織肉腫、頭部及び頸部癌並びに腫瘍転移の形成からなる群から選択される、請求項46に記載の方法。
【請求項48】
前記過剰増殖性疾患が、増加したHER−3のリン酸化、増加したHER−2/HER−3のヘテロ二量体化又はPI3−キナーゼ、c−jun末端キナーゼ、AKT、ERK2及び/又はPYK2の増加した活性を伴う、請求項46又は47に記載の方法。
【請求項49】
請求項1から28の何れか一項に記載の単離された結合タンパク質を含むキット。
【請求項50】
さらなる治療剤を含む、請求項49に記載のキット。
【請求項51】
さらなる治療剤が抗新生物剤である、請求項50に記載のキット。
【請求項52】
抗新生物剤が、抗腫瘍抗体又は化学療法剤である、請求項51に記載のキット。
【請求項1】
HER−3に結合する単離された結合タンパク質であり、
(a)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDHR1;(b)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH2;並びに(c)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH3からなる群から選択されるCDRの少なくとも1つを含む重鎖アミノ酸配列を含む、前記タンパク質。
【請求項2】
HER−3に結合する単離された結合タンパク質であり、
(d)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL1;(e)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL2;並びに(f)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL3からなる群から選択されるCDRの少なくとも1つを含む軽鎖アミノ酸配列を含む、前記タンパク質。
【請求項3】
請求項1又は2に記載の単離された結合タンパク質であり、前記結合タンパク質が、
(a)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDHR1;(b)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH2;並びに(c)配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230に示されているCDRH3からなる群から選択されるCDRの少なくとも1つを含む重鎖アミノ酸配列、並びに
(d)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL1;(e)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL2;並びに(f)配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232に示されているCDRL3からなる群から選択されるCDRの少なくとも1つを含む軽鎖アミノ酸配列を含む、前記タンパク質。
【請求項4】
前記結合タンパク質が、配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230の何れかのCDRH1、CDRH2及びCDRH3を含む重鎖アミノ酸配列、又は配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232の何れかのCDRL1、CDRL2及びCDRL3を含む軽鎖アミノ酸配列を含む、請求項1又は2に記載の単離された結合タンパク質。
【請求項5】
前記結合タンパク質が、配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230の何れかのCDRH1、CDRH2及びCDRH3を含む重鎖アミノ酸配列、並びに配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232の何れかのCDRL1、CDRL2及びCDRL3を含む軽鎖アミノ酸配列を含む、請求項3に記載の単離された結合タンパク質。
【請求項6】
配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230からなる群から選択される重鎖アミノ酸配列を含む、請求項1に記載の単離された結合タンパク質。
【請求項7】
配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232からなる群から選択される軽鎖アミノ酸配列を含む、請求項2に記載の単離された結合タンパク質。
【請求項8】
配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230からなる群から選択される重鎖アミノ酸配列、並びに
配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232からなる群から選択される軽鎖アミノ酸配列を含む、請求項1に記載の単離された結合タンパク質。
【請求項9】
配列番号2、6、10、14、18、22、26、30、34、36、40、42、46、50、54、60、62、66、70、74、78、80、84、88、92、96、100、104、108、112、116、120、122、126、130、134、138、142、146、150、154、158、162、166、170、174、178、182、186、190、194、198、202、206、210、214、218、222、226及び230の何れかのCDRH3を含む、請求項1に記載の単離された結合タンパク質。
【請求項10】
配列番号4、8、12、16、20、24、28、32、38、44、48、52、56、58、64、68、72、76、82、86、90、94、98、102、106、110、114、118、124、128、132、136、140、144、148、152、156、160、164、168、172、176、180、184、188、192、196、200、204、208、212、216、220、224、228及び232の何れかの軽鎖アミノ酸配列を含む、請求項9に記載の単離された結合タンパク質。
【請求項11】
配列番号42の重鎖アミノ酸配列及び配列番号44の軽鎖アミノ酸配列を含む、請求項1から9の何れか一項に記載の単離された結合タンパク質。
【請求項12】
配列番号54の重鎖アミノ酸配列及び配列番号56の軽鎖アミノ酸配列を含む、請求項1から9の何れか一項に記載の単離された結合タンパク質。
【請求項13】
配列番号70の重鎖アミノ酸配列及び配列番号72の軽鎖アミノ酸配列を含む、請求項1から9の何れか一項に記載の単離された結合タンパク質。
【請求項14】
成熟HER−3、特に、成熟ヒトHER−3のアミノ酸残基1から160、161から358、359から575、1から358、359から604によって形成される三次元構造に結合する単離された結合タンパク質。
【請求項15】
結合タンパク質が、HER−3の細胞外ドメインに対して誘導される、請求項1から14の何れかに記載の単離された結合タンパク質。
【請求項16】
HER−3への結合タンパク質の結合が、HER−3によって媒介されるシグナル伝達を低下させる、請求項1から15の何れかに記載の単離された結合タンパク質。
【請求項17】
HER−3への結合タンパク質の結合が、HER−3のリン酸化を低下させる、請求項1から15の何れか一項に記載の単離された結合タンパク質。
【請求項18】
HER−3への結合タンパク質の結合が、細胞増殖を低下させる、請求項1から15の何れか一項に記載の単離された結合タンパク質。
【請求項19】
HER−3への結合タンパク質の結合が、細胞の遊走を低下させる、請求項1から15の何れか一項に記載の単離された結合タンパク質。
【請求項20】
HER−3への結合タンパク質の結合が、HER−3の下方制御を増加させる、請求項1から15の何れか一項に記載の単離された結合タンパク質。
【請求項21】
抗体である、請求項1から20の何れか一項に記載の単離された結合タンパク質。
【請求項22】
抗体が、モノクローナル抗体、ポリクローナル抗体、組換え抗体、ヒト化抗体、ヒト抗体、キメラ抗体、多重特異的抗体又はこれらの抗体断片である、請求項21に記載の単離された結合タンパク質。
【請求項23】
抗体断片が、Fab断片、Fab’断片、F(ab’)2断片、Fv断片、ダイアボディ又は一本鎖抗体分子である、請求項22に記載の単離された結合タンパク質。
【請求項24】
前記単離された結合タンパク質が、IgG1、IgG2、IgG3又はIgG4型である、請求項1から23の何れか一項に記載の単離された結合タンパク質。
【請求項25】
結合タンパク質が、標識基に連結されている、請求項1から24の何れか一項に記載の単離された結合タンパク質。
【請求項26】
標識基が、放射性同位体若しくは放射性核種、蛍光基、酵素基、化学発光基、ビオチニル基又は所定のポリペプチドエピトープである、請求項25に記載の単離された結合タンパク質。
【請求項27】
結合タンパク質が、エフェクター基に連結されている、請求項1から24の何れか一項に記載の単離された結合タンパク質。
【請求項28】
エフェクター基が、放射性同位体若しくは放射性核種、毒素又は治療基若しくは化学療法基である、請求項27に記載の単離された結合タンパク質。
【請求項29】
治療基若しくは化学療法基が、カリチアマイシン、アウリスタチン−PE、ゲルダナマイシン、マイタンシン及びこれらの誘導体からなる群から選択される、請求項28に記載の単離された結合タンパク質。
【請求項30】
請求項1から29の何れか1項に記載の結合タンパク質をコードする単離された核酸分子。
【請求項31】
核酸分子が、調節配列に作用可能に連結されている、請求項30に記載の単離された核酸分子。
【請求項32】
請求項30に記載の核酸分子を含むベクター。
【請求項33】
請求項31に記載の核酸分子を含むベクター。
【請求項34】
請求項32に記載のベクターで形質転換された宿主細胞。
【請求項35】
請求項33に記載のベクターで形質転換された宿主細胞。
【請求項36】
宿主細胞から前記結合タンパク質を単離する工程を含む、請求項1から29の何れか一項に記載の単離された結合タンパク質を調製する方法。
【請求項37】
宿主細胞が、哺乳動物細胞、植物細胞、真菌細胞又は原核細胞である、請求項36に記載の方法。
【請求項38】
請求項1か29の何れか一項に記載の少なくとも1つの単離された結合タンパク質を活性因子として含み、及び医薬として許容される、担体、希釈剤又は佐剤を含む医薬組成物。
【請求項39】
治療的使用のための請求項37又は38に記載の組成物。
【請求項40】
診断的使用のための請求項37又は38に記載の組成物。
【請求項41】
請求項38又は39に記載の医薬組成物の医薬有効量を、HER−3と関連する疾病の治療又は予防を必要としている対象に投与することを含む、患者中のHER−3に関連する疾病を治療又は予防する方法。
【請求項42】
疾病が過剰増殖性疾患である、請求項41に記載の方法。
【請求項43】
前記過剰増殖性疾患が、乳癌、胃腸癌、膵臓癌、前立腺癌、卵巣癌、胃癌、子宮内膜癌、唾液腺癌、肺癌、腎臓癌、大腸癌、直腸結腸癌、甲状腺癌、膀胱癌、神経膠腫、悪性黒色腫、精巣癌、軟部組織肉腫、頭部及び頸部癌、HER−3を発現又は過剰発現する他の癌並びに腫瘍転移の形成からなる群から選択される、請求項42に記載の方法。
【請求項44】
前記過剰増殖性疾患が、増加したHER−3のリン酸化、増加したHER−2/HER−3のヘテロ二量体化又はPI3−キナーゼ、c−jun末端キナーゼ、AKT、ERK2及び/又はPYK2の増加した活性を伴う、請求項42又は43に記載の方法。
【請求項45】
HER−3に関連する疾病を診断する方法であり、
(a)請求項1から26の何れか一項に記載の結合タンパク質のHER−3への結合を可能とするのに適した条件下で、請求項1から26の何れか一項に記載の結合タンパク質と試料を接触させること、及び
(c)HER−3への前記結合タンパク質の結合を同定すること、
を含む、前記方法。
【請求項46】
疾病が過剰増殖性疾患である、請求項45に記載の方法。
【請求項47】
前記過剰増殖性疾患が、乳癌、胃腸癌、膵臓癌、前立腺癌、卵巣癌、胃癌、子宮内膜癌、唾液腺癌、肺癌、腎臓癌、大腸癌、直腸結腸癌、甲状腺癌、膀胱癌、神経膠腫、悪性黒色腫、HER−3を発現又は過剰発現する他の癌、精巣癌、軟部組織肉腫、頭部及び頸部癌並びに腫瘍転移の形成からなる群から選択される、請求項46に記載の方法。
【請求項48】
前記過剰増殖性疾患が、増加したHER−3のリン酸化、増加したHER−2/HER−3のヘテロ二量体化又はPI3−キナーゼ、c−jun末端キナーゼ、AKT、ERK2及び/又はPYK2の増加した活性を伴う、請求項46又は47に記載の方法。
【請求項49】
請求項1から28の何れか一項に記載の単離された結合タンパク質を含むキット。
【請求項50】
さらなる治療剤を含む、請求項49に記載のキット。
【請求項51】
さらなる治療剤が抗新生物剤である、請求項50に記載のキット。
【請求項52】
抗新生物剤が、抗腫瘍抗体又は化学療法剤である、請求項51に記載のキット。
【図1】


【図2】

【図3】

【図4】

【図5】
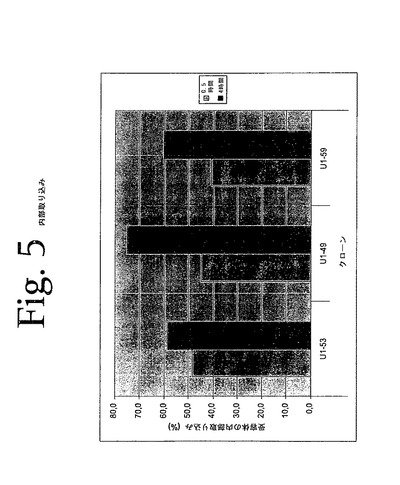
【図6a】

【図6b】
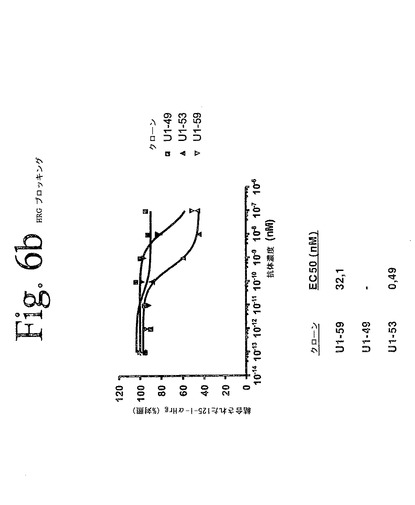
【図6c】

【図6d】

【図6e】
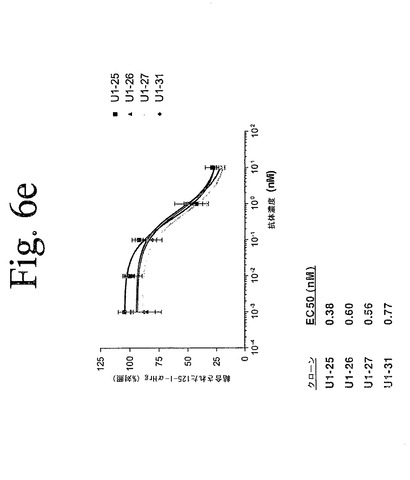
【図7a】
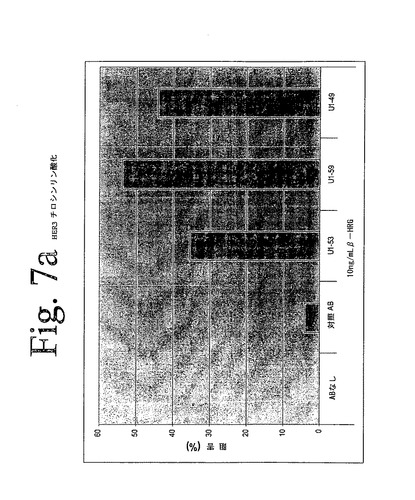
【図7b】
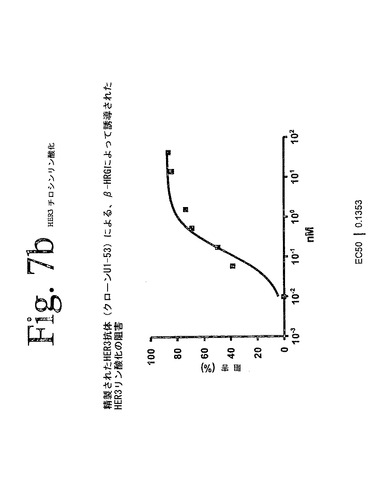
【図8】
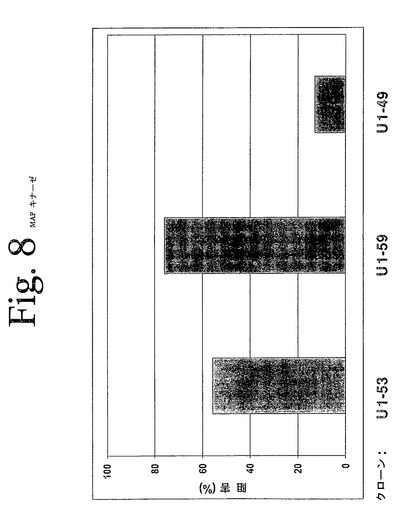
【図9】
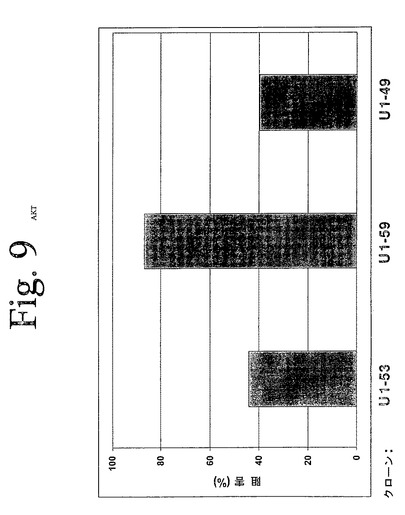
【図10】
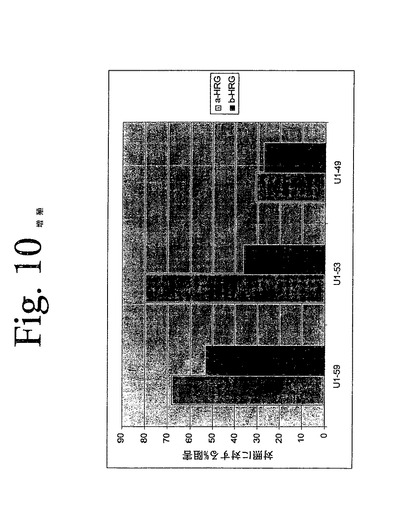
【図11】
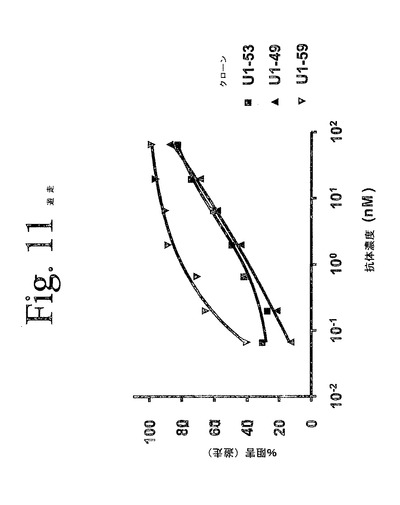
【図12a】
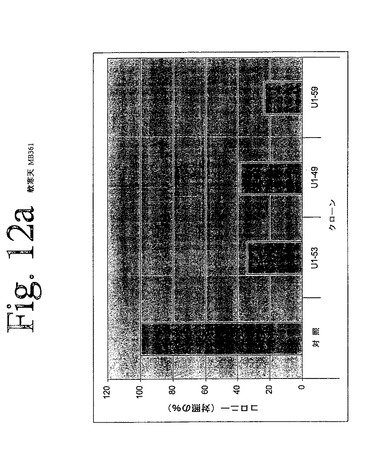
【図12b】

【図12c】
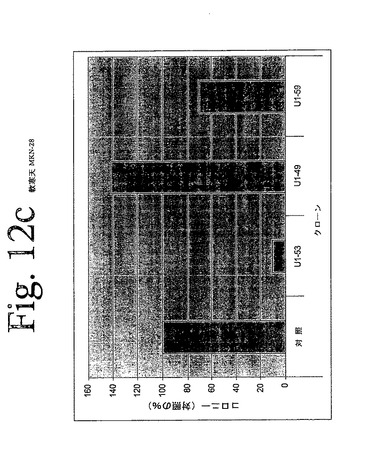
【図12d】
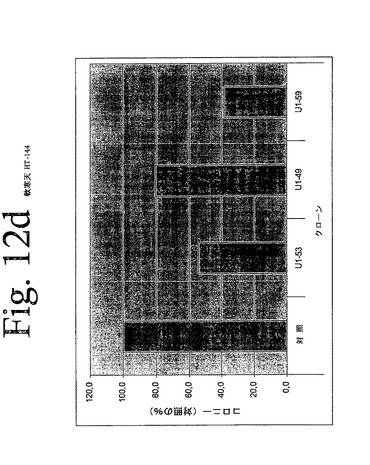
【図12e】
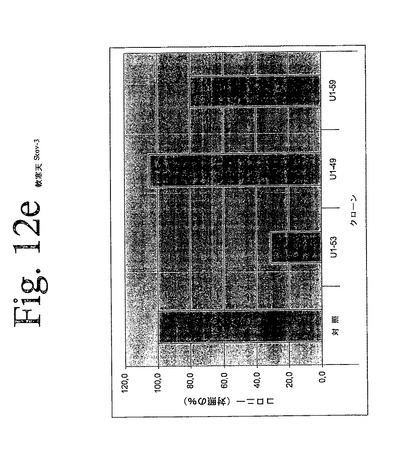
【図12f】

【図12g】
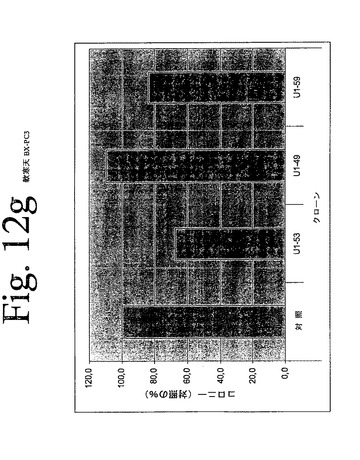
【図12h】
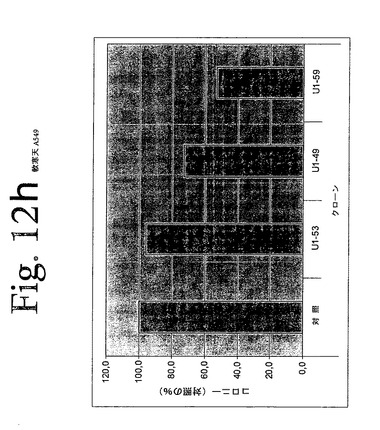
【図12i】
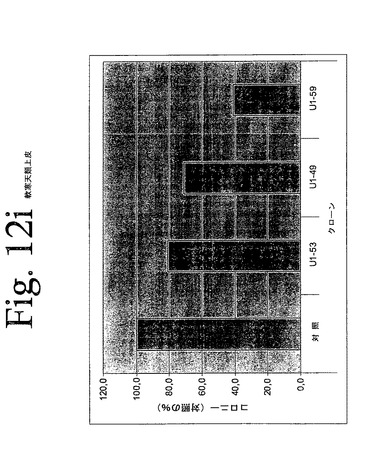
【図13】
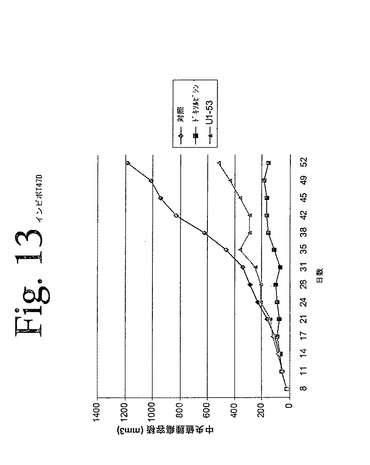
【図14】

【図15】

【図16】

【図17】
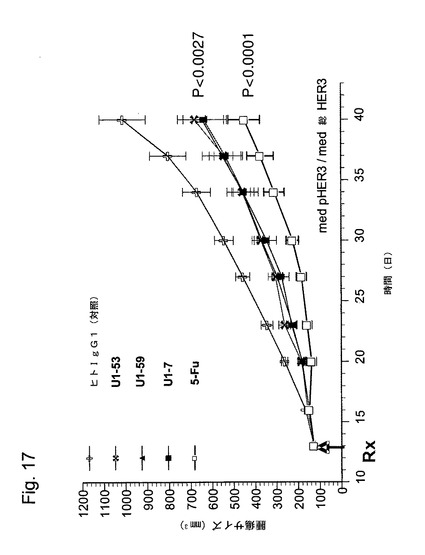
【図18】
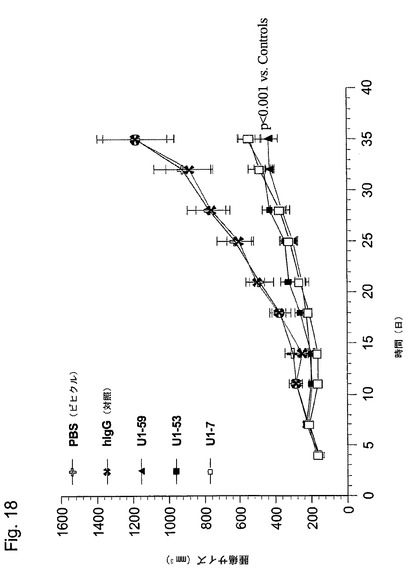
【図19】
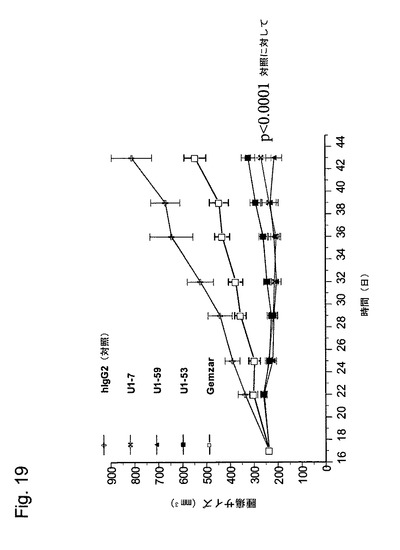
【図20】
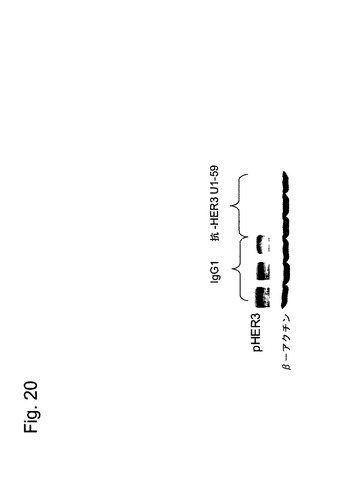
【図2】
【図3】
【図4】
【図5】
【図6a】
【図6b】
【図6c】
【図6d】
【図6e】
【図7a】
【図7b】
【図8】
【図9】
【図10】
【図11】
【図12a】
【図12b】
【図12c】
【図12d】
【図12e】
【図12f】
【図12g】
【図12h】
【図12i】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【公開番号】特開2013−56884(P2013−56884A)
【公開日】平成25年3月28日(2013.3.28)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−220833(P2012−220833)
【出願日】平成24年10月2日(2012.10.2)
【分割の表示】特願2008−547904(P2008−547904)の分割
【原出願日】平成18年12月29日(2006.12.29)
【出願人】(507107992)ウー3・フアルマ・ゲー・エム・ベー・ハー (5)
【氏名又は名称原語表記】U3 PHARMA
【出願人】(500049716)アムジエン・インコーポレーテツド (242)
【Fターム(参考)】
【公開日】平成25年3月28日(2013.3.28)
【国際特許分類】
【出願番号】特願2012−220833(P2012−220833)
【出願日】平成24年10月2日(2012.10.2)
【分割の表示】特願2008−547904(P2008−547904)の分割
【原出願日】平成18年12月29日(2006.12.29)
【出願人】(507107992)ウー3・フアルマ・ゲー・エム・ベー・ハー (5)
【氏名又は名称原語表記】U3 PHARMA
【出願人】(500049716)アムジエン・インコーポレーテツド (242)
【Fターム(参考)】
[ Back to top ]