
HER2に対する親和性を有する新規なポリペプチド
位置2におけるX1がM、I又はLであり、位置39におけるX2がS又はC(配列番号1)であるアミノ酸配列EX1RNAYWEIA LLPNLTNQQK RAFIRKLYDD PSQSSELLX2E AKKLNDSQを含むHER2結合ポリペプチドが開示されている。更に、キレート環境を含むそのようなペプチドが開示されている。また、キレート環境を含むペプチド及び放射性核種によって形成される、放射性標識されたポリペプチドが開示されている。更に、そのような放射性標識されたポリペプチドの投与と、それに続く医療用画像機器を使った身体の画像の取得を含む、HER2の過剰発現によって特徴付けられるがんを有するか、疑われる哺乳類対象の身体の、インビボ画像化の方法、そしてまたそのような癌の治療方法が開示されている。更に、HER2の過剰発現によって特徴付けられるがんの診断と治療における、そのような放射性標識されたポリペプチドの使用。また、ポリペプチドをコード化する核酸、核酸を含む発現ベクター及び発現ベクターを含む宿主細胞が開示されている。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ヒト上皮細胞成長因子受容体2(Human Epidermal Growth Factor Receptor
2)(以下、HER2と称す)に結合する新規なポリペプチドに関連する。本発明は、診断用薬及び/又は薬物としてのHER2結合ポリペプチドの使用、より詳しくは、HER2の過剰発現によって特徴付けられるがんの形態の診断及び/又は治療のための診断用薬及び/又は薬物としてのその使用に関する。
【背景技術】
【0002】
HER2とがんの疾病における役割
HER2がん原遺伝子は、HER2タンパク質又は受容体(非特許文献1)として知られている185kDa細胞表面受容体タンパク質の産生をコードする。この遺伝子は、また、しばしばneu、HER2/neu又はc-erbB-2と呼ばれる。neuは、最初はエチルニトロソ尿素で処理されていたラット中で発見され、そしてこの遺伝子の変異を示した(非特許文献2)。neuの変異バージョンは、構成的に活性な形態の受容体の産生をもたらし、低コピー数で細胞を形質転換できる強力ながん遺伝子を構成する(上記の非特許文献1を参照)。
【0003】
正常細胞は、組織特異性のパターンにおけるそれらの細胞膜上で小量のHER2タンパク質を発現する。HER2に対する一切の既知リガンドは解明されていない;しかしながら、HER2は、これらの受容体に対するリガンドと複合したHER1(上皮細胞成長因子受容体(EGFR))、HER3及びHER4とヘテロ二量体を形成することが示されている。そのようなヘテロ二量体形成は、活性化されたHER2受容体が増殖シグナルを細胞外から核へ伝達し、従って正常な細胞の成長と分裂の状況を制御することにつながる(非特許文献3)。
【0004】
腫瘍細胞では、DNA複製系でのエラーが、単一の染色体での遺伝子の複数のコピーの存在をもたらす可能性があり、それは遺伝子増幅として知られる現象である。HER2遺伝子の増幅は、この遺伝子の転写の増加をもたらす。これは、HER2mRNAレベルが上昇し、HER2タンパク質の随伴合成を増加し、これらの腫瘍細胞の表面でのHER2タンパク質の過剰発現をもたらす。この過剰発現は、隣の正常細胞に見られるものより10倍から100倍大きいHER2タンパク質レベルをもたらすことができる。このことが、ひいては細胞分裂の増加と付随的に高い細胞増殖の速度をもたらす。HER2遺伝子の増幅は、がん表現型への正常細胞の形質転換に関与している(上記の非特許文献1;上記の非特許文献3を参照)。
【0005】
HER2タンパク質の過剰発現は、HER2のホモダイマーの形成をもたらすと考えられ、それはひいては構成的に活性な受容体をもたらす(非特許文献4)。これらの条件下では、成長促進シグナルは、リガンドが無い状態で連続的に細胞に伝達され得る。その結果として、複数の細胞内シグナル伝達経路が活性化されるようになり、無秩序な細胞増殖、及び、ある場合には、がん遺伝子形質転換をもたらす(上記の非特許文献1を参照)。このように、増殖因子受容体によって仲介されるシグナル伝達機構は、細胞複製及び腫瘍増殖を抑制する重要な標的である。
【0006】
乳がんは、合衆国の女性の中で最も一般的な悪性腫瘍であり、2001年には19万2,200の新規症例が発生したと推定されている(非特許文献5)。全ての乳がん患者の約25%に、その増幅によるHER2遺伝子の過剰発現がある(非特許文献6)。HER2タンパク質のこの過剰発現は、エストロゲン受容体陰性状態を含む、いくつかの陰性の予後変数、高いS期分画、陽性結節状態、変異p53、及び高い核異型度と相関する(上記の非特許文献7を参照)。非特許文献6によれば、HER2遺伝子の増幅は、リンパ節陽性患者の短縮した無病生存及び短縮した全生存と強く相関することが分かった。
【0007】
このような理由で、乳がんの病因及び治療におけるHER2の役割への研究をさらに進めることは、重要な目標であったし、依然として然りである。
HER2と相互作用する分子の特定は、この取り組みの1部分を形成する。
【0008】
前臨床のインビトロの検討は、HER2活性の抑制が腫瘍細胞増殖に影響する可能性があったかどうかを調べている。対照モノクローナル抗体による投与に比較し、4D5、いくつかのマウス抗HER2モノクローナル抗体の1つ、によるHER2タンパク質を過剰発現しているSK−BR−3乳がん細胞の治療は、本当に腫瘍細胞増殖を抑制した。HER2を過剰発現するヒト乳がん及び卵巣がん(異種移植片)を有するマウスへの4D5の投与は、腫瘍の無い生存期間を延長した。同様の検討は、マウスにおけるヒト胃がん異種移植片で抗HER2ヒト化モノクローナル抗体の増殖抑制を実証した(非特許文献8)。
【0009】
腫瘍細胞表面に豊富に存在するHER2タンパク質を抗体で抑制する試みの中で、1つの療法は、ここ数年の間に市販されるようになった。このように、モノクローナル抗体4D5のヒト化変異体、又はトラスツズマブ(trastuzumab)は、Herceptin(登録商標)の商標名のもと、F Hoffman-La Roche 及び Genentechによってこの目的で市販されている。
【0010】
このように、HER2の過剰発現が乳がんについて記載されている。それは、また、とりわけ卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん(非特許文献9)及び食道におけるがん(非特許文献10)につながっている。
【0011】
HER2タンパク質の過剰発現によって特徴付けられるがんに対する抗体療法によって示される明らかな優位性にもかかわらず、種々のファクターには抗体の有効性(非特許文献11を参照)を減らす可能性があるという事実は残る。それらには以下のものがある:(1)大きい固形腫瘍又は脳のような生命の維持に必要な領域への抗体の制限的浸透;(2)血管透過性の減少により、標的部分への抗体の溢血の減少;(3)健常組織に対する交差反応性及び抗体の非特異的結合で、標的効果が低減する;(4)不均質な腫瘍取り込みが未治療区域をもたらす;(5)注入した抗体の代謝の増加で、治療効果が低減する;及び(6)HAMA及びヒト抗ヒト抗体の急速生成で、治療抗体を不活性化する。
【0012】
加えて、毒性作用は、がんのための治療抗体の開発の大きな障害である(非特許文献12、非特許文献13、非特許文献14)。健常組織との交差反応性は、非抱合型の(裸の)抗体に対してかなりの副作用を起こす可能性があり、その副作用が毒素又はラジオアイソトープを有する抗体の抱合で高められる可能性がある。免疫介在性合併症は、肺毒性作用、時折の中枢及び末梢神経系合併症、並びに肝及び腎機能の減少からの呼吸困難を含む。時として、HER2ターゲッティング抗体のトラスツズマブに関連している心毒性作用のような、予想外の毒性合併症が見られる(非特許文献15)。同位体抱合型抗体による放射免疫療法は、また、骨髄抑制を引き起こす可能性がある。
【0013】
現在使用されている抗がん抗体の最近の臨床的及び商業的成功にもかかわらず、このように相当な数の重要な疑問が、抗体の使用の将来に関して残っている。結果として、HER2に対して匹敵する親和性のある試薬の継続的な提供は、疾患の診断と治療におけるそのような分子の利用の提供と同様に、この分野での相当な関心事として残る。
【0014】
HER2結合のZ変異分子
ブドウ球菌プロンテインA(SPA)のドメインBから誘導された、タンパク質Zに関連する分子(非特許文献16)は、異なる相互作用ターゲットを使用している無作為化されたそのような分子のライブラリーから選択されている(例えば、特許文献1、非特許文献17、特許文献2、特許文献3、特許文献4、特許文献5を参照)。
【0015】
特許文献6には、相当な数のHER2と相互作用する能力のあるZの変形が開示されている。
【先行技術文献】
【特許文献】
【0016】
【特許文献1】国際公開公報第95/19374号
【特許文献2】国際公開公報第2005/000883号
【特許文献3】国際公開公報第2005/075507号
【特許文献4】国際公開公報第2006/092338号
【特許文献5】国際公開公報第2007/065635号
【特許文献6】国際公開公報第2005/003156号
【非特許文献】
【0017】
【非特許文献1】Hynes NE et al (1994), Biochim Biophys Acta1198: 165-184
【非特許文献2】Shih C et al (1981), Nature 290: 261-264)
【非特許文献3】Sundaresan S, et al (1999), Curr Oncol Rep1: 16-22)
【非特許文献4】Sliwkowski MX et al (1999), Semin Oncol, 26 (4 Suppl12): 60-70)
【非特許文献5】Greenlee R, et al (2001), CA Cancer J Clin 51: 15-36
【非特許文献6】Slamon DJ, et al (1989), Science 244: 707-712
【非特許文献7】Sjogren S, et al (1998), J Clin Oncol, 16(2): 462-469
【非特許文献8】Pietras RJ, et al (1994), Oncogene 9: 1829-1838
【非特許文献9】Holbro, et al, Annu. Rev. Pharmacol. Toxicol. 2004. 44: 195-217
【非特許文献10】Ekman, et al, Oncologist 2007 ; 12 : 1165-1177; see, in particular, pages 1170-1171
【非特許文献11】Reilly RM, et al (1995), Clin Pharmacokinet 28: 126-142
【非特許文献12】Carter P (2001), Nat Rev Cancer 1: 118-129
【非特許文献13】Goldenberg DM (2002), J Nucl Med 43: 693-713
【非特許文献14】Reichert JM (2002), Curr Opin Mol Ther 4: 110-118
【非特許文献15】Schneider JW, et al (2002), Semin Oncol 29 (3 suppl 11): 22-28
【非特許文献16】Nilson B, et al (1987), Protein Engineering 1: 107-133
【非特許文献17】Nord K, et al (1997), Nature Biotechnology 15: 772-777
【発明の概要】
【発明が解決しようとする課題】
【0018】
本発明の目的は、HER2への特異的結合によって特徴付けられるポリペプチドの提供を通して、HER2に対する親和性を有する試薬の継続的提供の要望を満たすことにある。
【0019】
本発明の関連する目的は、殆ど又は全く非特異的結合を示さない、HER2結合ポリペプチドである。
【0020】
本発明の別の目的は、容易に融合ポリペプチドの部分として使用できる、HER2結合ポリペプチドを提供することにある。
【0021】
別の目的は、既存の抗体試薬で経験する1つ又はそれ以上の公知の問題を解決する、HER2結合ポリペプチドの提供である。
【0022】
本発明の更なる目的は、治療用途での使用を可能にするHER2結合ポリペプチドを提供することである。
【0023】
本発明の更なる目的は、診断用途での使用を可能にするHER2結合ポリペプチドを提供することにである。
【0024】
関連する目的は、HER2タンパク質の過剰発現によって特徴付けられるがん疾患の臨床状況における、治療、阻害及び/又はターゲッティングの新規な形態を見出すことである。
【0025】
本発明の別の目的は、化学的ペプチド合成によって簡単に合成されるHER2結合ポリペプチドを提供することにある。
【0026】
関連する目的は、公知のHER2結合剤に対する改良された安定性を示す、HER2結合ポリペプチドを見出すことである。
【0027】
本発明の更に別の目的は、インビボで哺乳動物に使用する場合、低抗原性を示すHER2結合ポリペプチドを得ることである。
【0028】
本発明の別の目的は、哺乳動物への投与時に改善された生体内分布を有する、HER2結合ポリペプチドを提供することである。
【課題を解決するための手段】
【0029】
これらの及び他の目的は、添付の特許請求の範囲に記載されるように、本発明のさまざまの態様によって明らかになる。このように、第1の態様では、本発明は、以下の式:
EX1RNAYWEIALLPNLTNQQKRAFIRKLYDDPSQSSELLX2EAKKLNDSQ
式中、位置2におけるX1はM、I又はLであり、位置39におけるX2はS又はCである;
(配列番号1)
又は、ある場合にはより好ましくは
YAKEX1RNAYWEIALLPNLTNQQKRAFIRKLYDDPSQSSELLX2EAKKLNDSQ
式中、位置5におけるX1はM、I又はLであり、位置42におけるX2はS又はCである;
(配列番号2)
又は、ある場合にはより好ましくは
AEAKYAKEX1RNAYWEIALLPNLTNQQKRAFIRKLYDDPSQSSELLX2EAKK LNDSQ
式中、位置9におけるX1はM、I又はLであり、位置46におけるX2はS又はCである;
(配列番号3)
又は、ある場合にはより好ましくは
ESEKYAKEX1RNAYWEIALLPNLTNQQKRAFIRKLYDDPSQSSELLX2EAKK LNDSQ,
式中、位置9におけるX1はM、I又はLであり、位置46におけるX2は、S又はCである;
(配列番号4)
を含むアミノ酸配列を有するポリペプチドを提供する。
【発明を実施するための形態】
【0030】
これらに従って、本発明者は、配列番号配列番号1〜4の何れか1つを含むポリペプチドが、先に述べられたそれらのポリペプチドの標的HER2に結合する能力を保持しながら、例えば、特許文献6に開示されたHER2結合ポリペプチドに比べて、驚くべき優位性を示すことを見出した。本発明のポリペプチドの1つ又はそれ以上の実施態様によって示された、そのような優位性の非限定的な例は、以下のとおりである:
・本発明のポリペプチドは、アスパラギン、アルギニン、アスパラギン酸及びメチオニンのようなポリペプチド配列の化学合成において、低い収率及び成功率のような問題を引き起こす可能性があったアミノ酸残基を殆ど含まない。
・本発明のポリペプチドは、表面の疎水性を付与する、従って先に述べた、関連のHER2結合ポリペプチドより親水性の特性を持っているアミノ酸残基を殆ど含まない。このことは、低い溶解性及び凝集性に伴う問題が殆どないことを意味する。理論にこだわることを望むわけではないが、より親水性の特性が、肝胆道経路(肝臓を経由した排泄)からより望ましい腎臓経路(腎臓を経由した排泄)に向けて、投与時のポリペプチドの生体内分布を宿主にシフトするように作用することは、現在も同じように信じられている。
・本発明のポリペプチドは、メチオニン、アスパラギン及びジペプチドのアスパラギンープロリンのようなポリペプチドの安定性の問題に関連するアミノ酸残基を殆ど含まない。メチオニンは酸化を受けやすく、アスパラギンは脱アミド化を受けやすく、そしてアスパラギン−プロリン結合は分裂を受けやすく、それ故に、それらは最終製品の不均一性に寄与する。
・本発明のポリペプチドは、同様の配列構成において、VH3からの重鎖可変領域を含んでいる免疫グロブリンとの相互作用を高めることが分かっているアミノ酸残基を欠いている(Silverman G.J., Int. Rev. Immunol. 1992;9(1):57-78)。理論にこだわることを望むわけではないが、本発明のポリペプチド中のそのようなアミノ酸残基の置換が、同じものの投与時のポリペプチドの宿主に対する抗原性を減らすことは、今では信じられている。
【0031】
本発明のポリペプチドは、さまざまな適用におけるHER2に対する抗体の代替物としての応用を見出す。非限定的な例として、それらは、HER2過剰発現で特徴付けられるがんの治療に、細胞表面のHER2に結合することによって細胞シグナリングを抑制することに、インビボ及びインビトロの両方におけるHER2過剰発現で特徴付けられるがんの診断に、HER2を過剰発現している細胞に試薬のターゲッティングを行うことに、HER2の検出の組織化学的方法に、及び分離方法及び他の適用に有用である。本発明のポリペプチドは、試薬のHER2に対する親和性に頼るいかなる方法にも有用であることを立証することができる。このように、本ポリペプチドは、そのような方法における検出試薬、捕捉試薬又は分離試薬として、インビボ又はインビトロの診断用の診断薬として、又はそれ自体で治療薬として、又は他の治療薬若しくは診断用薬をHER2タンパク質にターゲットする方法として、使用することができる。インビトロで本発明のポリペプチドを用いる方法は、マイクロタイタープレート中で、プロテインアレイ中で、バイオセンサー表面上で、組織切片上で等のさまざまな型式で実施することができる。
【0032】
本発明の範囲を逸脱しないで、ポリペプチドを意図する特定用途に合わせるために、本発明にに記載のポリペプチドのさまざまな修飾及び/又は付加が実施されてもよい。そのような修飾及び付加は、以下により詳細に記載されており、同じポリペプチド鎖に含まれる更なるアミノ酸、又は本発明のポリペプチドに化学的に共役する若しくは別の様式で結合する標識及び/又は治療薬を含んでもよい。
【0033】
「HER2に対する結合親和性」、「HER2結合」等のような発現とは、例えば、Biacore(登録商標)機器(GE Healthcare)におけるような、表面プラズモン共鳴技術を用いて試験され得るポリペプチドの性質を意味する。HER2結合親和性は、HER2又はその断片、例えば、細胞外ドメイン又はその融合タンパク質が装置のセンサーチップ上に固定化され、検査されるポリペプチドを含む試料がそのチップ上を移動するような実験で試験することができる。或いはまた、検査されるポリペプチドが装置のセンサーチップ上に固定され、そしてHER2を含む試料、又はその断片、例えば、細胞外ドメインが、そのチップ上を通過する。次いで、当業者は、少なくともHER2に対するポリペプチドの結合親和性の少なくとも定性的測定を達成するために得られるセンサーグラム(sensorgrams)を解釈することができる。もし、例えば、相互作用の特定のKD値を確立する目的を持って定量的測定が求められる場合は、再び表面プラズモン共鳴方法を使用することが可能である。結合値は、例えば、Biacore(登録商標)2000装置(GE Healthcare)で定義することができる。HER2、又はその断片、例えば、細胞外ドメインは、装置のセンサーチップ上に固定され、親和性が決定されるべきポリペプチドの試料は連続希釈によって調製され、無作為の順序で注入される。次いで、KD値は、例えば、装置メーカーによって提供されるBIAevaluation 3.2ソフトウェアの1:1ラングミュア結合モデルを用いて、その結果から計算することができる。
【0034】
本発明は、また、上記のHER2結合ポリペプチドが、更なるアミノ酸残基がどちらかの末端に付加している、HER2結合ドメインとして存在するポリペプチドを包含する。これらの更なるアミノ酸残基は、ポリペプチドによりHER2の結合における役割を果たし得るが、同様に、例えば、ポリペプチドの産生、精製、安定化、カップリング又は検出の1つ又はそれ以上に関連する、他の目的をもよく果たし得る。そのような更なるアミノ酸残基は、化学カップリングのために追加される1つ又はそれ以上のアミノ酸残基を含んでもよい。この1つの例は、ポリペプチド鎖の中の一番最初の又は一番最後の位置、即ち、N又はC末端におけるシステイン残基の付加である。化学カップリングに使用されるシステイン残基は、また、タンパク質ドメイン上で、好ましくは標的結合に関与しない表面の部分の上で、別のアミノ酸の置換によって導入され得る。そのような更なるアミノ酸残基は、タグに固有の抗体との相互作用用のヘキサヒスチジル (His6)タグ、若しくは「myc」タグ又は「FLAG」タグのようなポリペプチドの精製又は検出のための「タグ」を含み得る。当業者は、他の選択肢を知っている。
【0035】
更なるアミノ酸残基を有するポリペプチドの具体的実施態様において、本発明は、下記のアミノ酸配列:
AEAKYAKEMRNAYWEIALLPNLTNQQKRAFIRKLYDDPSQSSELLSEAKK LNDSQAPKVDC
(配列番号5)
を含むHER2結合ポリペプチドを提供する。
【0036】
更なるアミノ酸残基を有するポリペプチドの別の具体的実施態様において、本発明は、下記のアミノ酸配列:
ESEKYAKEMR NAYWEIALLP NLTNQQKRAF IRKLYDDPSQ SSELLSEAKK LNDSQAPK
(配列番号6)
を含むHER2結合ポリペプチドを提供する。
【0037】
上で議論された「更なるアミノ酸残基」は、また、第1の、HER2結合ドメイン、又は別の結合機能、又は酵素機能、又は蛍光機能、又はそれらを混合したものと同じ結合機能のような、いかなる所望の機能を有する1つ又はそれ以上のポリペプチドドメインを構成してもよい。
【0038】
このように、本発明は、配列番号1〜6の何れか1つを含むポリペプチドの多量体を包含する。それは、例えば、がんの診断や治療のために又はHER2の精製の方法において、本発明のポリペプチドを使う場合、本発明の1つのポリペプチドで可能であるよりも更に強いHER2の結合を得ることに、関心が持たれるかもしれない。この場合、ポリペプチドの、二量体、三量体又は四量体のような、多量体の提供は、必要な結合力効果をもたらし得る。多量体は、本発明に記載の適する数のポリペプチドから成るり得る。本発明の多量体中の結合ポリペプチド「ユニット」は、公知の有機化学の方法を使って共有結合によって連結され得るし、又は、ポリペプチドの組換え発現のシステムにおいて1つ又はそれ以上の融合ポリペプチドとして表現されたり、又はいかなる他の方法ででも、直接に又はリンカー、例えば、アミノ酸リンカーを経由して連結され得る。
【0039】
さらに、配列番号1〜6の何れか1つを含むポリペプチドが、第1のドメイン、又は第1の部分を構成し、そして第2の部分及び更なる部分が結合性HER2以外の機能を有する「異種遺伝子型の」融合ポリペプチドも意図され、そして本発明の範囲に属する。融合ポリペプチドの第2の部分及び更なる1つ又は複数の部分は、HER2とは別のターゲット分子に対する親和性を有する結合ドメインを含み得る。その結果は、従って、少なくとも1つのHER2結合ドメイン及び該他の標的分子に対する親和性を有する少なくとも1つのドメインを有する融合ポリペプチドである。これは、治療剤又は捕捉、検出又は分離試薬として使用されるような、いくつかの生物工学的応用に使われ得る多特異的試薬を作成することを可能にする。少なくとも1つのポリペプチドドメインが配列番号1〜6のいずれか1つの配列を含むような、そのようなポリペプチドの多特異的多量体の製造は、いくつかのHER2結合性「ユニット」の多量体に対して上記のように行われ得る。第2の又は更なる1つの又は複数の部分は、「Zドメイン」又は「タンパク質Z」の変異体等の黄色ブドウ球菌(Staphylococcus aureus)のタンパク質A由来のドメインの変異体であり得る(Nilsson B et al (1987), Protein Engineering 1: 107-133を参照)、又は標的に対する結合親和性を有する無関係の、天然に存在する、又は組み換えのタンパク質(又は天然に存在する又は組み換えの蛋白の結合能力を保持しているその断片)を含む。ヒト血清アルブミンに対する親和性を有し、本発明のポリペプチドとの融合相手として使用され得るそのような結合タンパク質の例は、GA1、GA2又はGA3ドメインのような連鎖球菌菌株(Streptococcus strain)G148(Nygren P-A et al (1988), Mol Recogn 1: 69-74)のタンパク質Gからのアルブミン結合ドメインの1つである。プロテインGのGA3ドメインは、また、ABD、即ち、アルブミン結合ドメイン(albumin binding domain)で示される。配列番号1〜6のいずれか1つの配列を含むHER2結合ポリペプチドと連鎖球菌タンパク質Gのタンパク質結合ドメインの間の融合ポリペプチドは、このように本発明の範囲に属する。本発明のポリペプチドが診断薬、治療薬として、又は標的薬としてヒト被験者に投与される場合、血清アルブミンに結合する部分へのその融合は有益であることを証明することができる。その場合、単離時のHER2結合部分の半減期と比較して、そのような融合タンパク質のインビボでの半減期は延びる可能性が高い(この原理については、例えば、国際公開公報第91/01743号に記載されている)。同様に、国際公開公報第2005/097202号で明らかにされた原理に従って、融合ポリペプチドは、単離時のHER2結合部分より低い免疫原性を示すようである。
【0040】
融合ポリペプチドの創出に対する他の可能性が同様に意図される。このように、本発明の第1の態様に記載のポリペプチドは、第2の又は更なる部分等に共有結合的にカップリングすることができ、それは、標的結合に加えて、又は標的結合の代わりに、他の機能を示す。1つの例は、配列番号1〜6のいずれか1つの配列を含む1つ又はそれ以上のポリペプチドと、レポーター又はエフェクター部分として機能している酵素活性のポリペプチドとの間の融合である。融合タンパク質を形成する配列番号1〜6のいずれか1つの配列を含むポリペプチドにカップリングされ得るリポーター酵素の例は、当業者に知られており、そしてβ−ガラクトシダーゼ、アルカリホスファターゼ、西洋ワサビペルオキシダーゼ、カルボキシペプチダーゼのような酵素を含む。本発明の融合ポリペプチドの、第2の、及び更なる1つ又は複数の部分に対する他のオプションは、緑色蛍光タンパク質、赤色蛍光タンパク質、ルシフェラーゼ及びその変異体のような蛍光性ペプチドを含む。
【0041】
本発明の融合ポリペプチドの、第2の、及び更なる1つの又は複数の部分に対する他のオプションは、治療への適用のための部分等を含む。治療への適用においては、他の分子も、また、他の手段による本発明のポリペプチドに、共有結合で又は非共有結合でカップリングされ得る。非限定的な例は、エフェクター酵素(例えば、カルボキシペプチダーゼ)の方向に本発明のポリペプチドを用いるADEPT(抗体指向酵素プロドラッグ治療)(antibody-directed enzyme prodrug therapy)適用のための酵素;免疫系のエフェクター細胞及び他のコンポーネントの補充のためのタンパク質;IL−2、IL−12、TNFα、IP−10のようなサイトカイン;組織因子、フォン・ビルブラント因子のような凝固促進因子;リシンA、シュードモナス外毒素、カルケアマイシン、メイタンシノイドのような毒素;アウリスタチアナログ、ドキソルビシンのような毒性の小さな分子;を含む。
【0042】
本発明に記載のHER2結合ポリペプチドを取り込んでいる融合タンパク質の上記の記述に関して、第1の、第2の、更なる部分の命名は、一方ではHER2結合部分又は該部分と、他方では他の機能を発揮している部分との間を区別する明快な理由のためになされていることも注目すべきである。これらの命名は、融合タンパク質のポリペプチド鎖の中の、さまざまなドメインの実際の順序を示すよう意図されていない。従って、例えば、該第1の部分が、融合タンパク質のN末端に、中央に、又はC末端に制約なしに出現する可能性がある。
【0043】
本発明は、上記のHER2結合ポリペプチドが、例えば、ポリペプチドの検出の目的のために、少なくとも1つのフルオロフォア、ビオチン又は放射性同位元素のような標識基を備えているポリペプチドを含む。特に、本発明は、上記のHER2結合ポリペプチドの放射性キレート、及び放射性金属のような放射性核種から成る放射性標識ポリペプチドを含む。
【0044】
放射性核種の大部分は金属の性質を有し、そして金属は、一般的には、タンパク質及びペプチド中に存在する元素との安定な共有結合を形成することができない。このために、標的タンパク質を放射性金属で標識することが、キレート剤、キレートと呼ばれる、金属と非共有化合物を形成する、多座配位子を使って行われる。本発明のポリペプチドの実施態様において、放射性核種の取り込みは、キレート環境の提供によって可能になり、それによって放射性核種はポリペプチドに配位し、キレート化し又は錯体化され得る。
【0045】
本発明のポリペプチドの1つの具体的実施態様において、N3Sキレート剤として特徴付けられる四座配位子キレート環境を含む。N3Sという用語が意味するように、そのようなキレート剤の4つの付着している基は、3つの窒素原子及び1つの硫黄原子から形成される。N3Sキレート剤では、、N及びS原子は空間的に配置されて、放射性金属の錯体化又は付着のために適する「ポケット」を提供する。
【0046】
N3Sキレート剤は、ポリペプチドのアミノ酸配列中のアミノ酸残基を好適に選択して提供され得る。
【0047】
例えば、N3Sキレート環境は、本発明のポリペプチドのN末端に、メルカプトアセチ
ルのカップリングを通して供給され得る。この実施態様においては、メルカプトアセチルが必要なS原子を与え、一方、ペプチド骨格中の最初の3つのN原子が四座配位子キレート剤の3つのN原子を構成する。好ましくは、メルカプトアセチルが、最も多くのN末端アミノ酸配列である、配列番号4のアミノ酸配列を含むポリペプチドにカップリングされ、その結果、下記のアミノ酸配列:
maESEKYAKEX1RNAYWEIALLPNLTNQQKRAFIRKLYDDPSQSSELLX2EAKK LNDSQ、
式中、maはメルカプトアセチル、位置9のX1はM、I又はLであり、位置46のX2はS又はCである;
を含むポリペプチドになる。これの特に好ましい実施態様では、ポリペプチドが下記の配列:
maESEKYAKEMR NAYWEIALLP NLTNQQKRAF IRKLYDDPSQ SSELLSEAKK LNDSQAPK、
式中、maは、メルカプトアセチルである;
(配列番号7)
を有する。
【0048】
代わりの実施態様においては、3個のN基は、ポリペプチド鎖中の3つの連続したペプチド結合のN原子によって提供されることができ、その内、最後のものはシステイン残基であり、その側鎖にSH基を含む。システイン側鎖のS原子は、N3Sキレート剤におけるS原子を構成する。別の言葉で言えば、N3Sキレート剤がトリペプチド配列XXCの形で提供され、ここで、Xは、いかなるアミノ酸残基であってもよい。そのようなトリペプチド配列は、、1つ又はそれ以上の更なるアミノ酸として、最初に又は追加して、本発明のポリペプチドの中に含まれる。システイン残基が配列番号5のC末端に位置しているもの、又は追加のシステインが配列番号1〜4又は6〜7のいずれか1つのC末端に正しく位置していることが好ましい。システイン残基は、また、1つ又はそれ以上の他のアミノ酸残基がその後に続いてもよい。
【0049】
N3Sキレート剤として特徴付けられる四座配位子キレート環境を含み、3つの連続したペプチド結合のN末端及び窒素原子にカップリングしたメルカプトアセチルか、ポリペプチドのC末端で3つの連続したペプチド結合とシステイン残基の窒素原子のどちらかによって提供される本発明のポリペプチドは、HER2結合ポリペプチドの放射性キレート及び医用画像に適した放射性核種から成る放射性標識ポリペプチドを提供するために使用することができ、そして該放射性核種は99mTc (Engfeldt et al (2007) Eur. J. Nucl.
Med. Mol. Imaging 34(5):722-22; Engfeldt et al (2007) Eur. J. Nucl. Med. Mol. Imaging 34(11):1843-53)であり、又は治療に適する放射性核種を有しており、該放射性核種は186Re及び188Reから成るグループから選択される。いくつかの実施態様において、放射性核種は、キレート環境を介してHER2結合ポリペプチドと錯体を形成する。
【0050】
本発明のポリペプチドが2つ、3つ又はそれ以上のXXCトリペプチドを含む場合、それは、好ましくは放射性核種を錯体化するのに使われる末端のものである。ポリペプチドがこのような方法で放射性標識される場合、他のXXCトリペプチドは、好ましくは保護されている。
【0051】
本発明の放射性標識ポリペプチドの1つの特に好ましい実施態様は、アミノ酸配列:
maESEKYAKEMR NAYWEIALLP NLTNQQKRAF IRKLYDDPSQ SSELLSEAKK LNDSQAPK
式中、maはメルカプトアセチルである;
(配列番号7)
を有するHER2結合ポリペプチドの放射性キレート及び99mTcである。
【0052】
ポリペプチドのN末端にあるmaESE又はC末端にあるVDCを使用してキレート化した、99mTcの仮説の構造は、それぞれ以下のものである。
【化1】
【0053】
本発明のポリペプチドの代替実施態様では、ポリアミノポリカルボキシラート・キレート剤を使用して放射性核種を組み込む。好ましくはその場合、ポリペプチドは少なくとも1つのシステインを含み、最も好ましくはたった 1つのシステインを含む。システインは、本発明のポリペプチドに最初に存在していても、又は1つ又はそれ以上の更なるアミノ酸として後の段階で追加されてもよい。
【0054】
当業者は、また、ペプチド合成中ポリペプチドに部位特異的に結合できる、又は放射性標識のための公知の共役化学を使って組換え的に産生されるポリペプチドに共役させることができる、「裸の」Me、Me=O、O=Me=O、Me≡N、Me(CO)3のようなコア(core)、又はHYNIC−Me−co−リガンド・コア(ここで、MeはTc又はReのラジオアイソトープである)をキレートする能力がある、多くの他のキレート剤の予測を行うことができよう。好ましくは、そのようなキレート剤は親水性の特性を有する。そのようなキレート剤の良い概説は、Liu S and Edwards DS (1999) Chem Rev. 99(9):2235-68により提供されている。
【0055】
誰でもポリアミノポリカルボキシラート・キレート剤の2つのクラス:大環状及び非環式、を区別することができる。
【0056】
インジウム、ガリウム、イットリウム、ビスマス、放射性アクチニド及び放射性ランタニドのラジオアイソトープ用に最も一般的に使用される大環状キレート剤は、DOTA(1,4,7,10−テトラアザシクロドデカン−1,4,7,10−四酢酸)(以下を参照)のさまざまな誘導体である。本発明のポリペプチドとのキレート化剤として使用するために適した、いくつかのさまざまのDOTAベースの化合物は、例えばMacrocyclics Inc., USAから市販されており、例を以下に示す。
【化2】
【0057】
この図式では、AはDOTAであり、Bはアミノ反応性の4−イソチオシアナート−ベンジル−DOTA、CはDOTA−TFPエステル、そしてDはチオール反応性のマレイミド−モノ−アミドDOTAである。
【0058】
高い動力学的不活性、即ちDOTAからの金属の解離の遅速性は、放射性核種の安定結合に好都合である。しかし、会合遅速性のため、標識のためには高温が必要となる。この理由のために、60〜90℃への加熱に比較的敏感でないDOTA誘導体が、短いペプチドの標識に広く使用される。
【0059】
本発明におけるキレート剤として使用するための1つの好ましい誘導体は、1,4,7,10−テトラアザシクロドデカン−1,4,7−トリス酢酸−10−マレイミドエチルアセトアミドである。
【0060】
上に開示したように、本発明のポリペプチドは、例えばアミノ酸配列番号5の配列を含み得る。より格別な実施態様では、ポリペプチドは配列番号5を含み、そしてアミノ酸残基C61にカップリングしたテトラアザシクロ化合物を有する。特に好ましい実施態様では、テトラアザシクロ化合物が、残基C61のSH基の、テトラアザシクロ化合物のマレイミド部分との反応を通じてポリペプチドにカップリングされている、1,4,7,10−テトラアザシクロドデカン−1,4,7−トリス酢酸−10−マレイミドエチルアセトアミドである。本発明の1つの特に好ましい実施態様は、従って、1,4,7,10−テトラアザシクロ−ドデカン−1,4,7−トリス−酢酸−10−マレイミドエチルアセトアミドにカップリングしたアミノ酸配列:
AEAKYAKEMRNAYWEIALLPNLTNQQKRAFIRKLYDDPSQSSELLSEAKK LNDSQAPKVDC
(配列番号5)
を含むHER2結合ポリペプチドである。
【0061】
最も一般的に使用される非環状ポリアミノポリカルボキシラート・キレート剤は、以下の図式に示されるそれらのようなDTPA(ジエチレントリアミン五酢酸)のさまざまの誘導体であり、ここで、AはDTPAであり、Bはアミノ反応性誘導体イソチオシアナートベンジル−DTPAであり、Cはアミノ反応性誘導体半硬質の2−(パラ−イソチオシアナートベンジル)−6−メチル−DTPS(IB4M)であり、Dはアミノ反応性誘導体CHX−A”−DTPAである。
【化3】
【0062】
DTPAの骨格修飾した半硬質の変異体は、例えば、Zevalin(登録商標)の90Yでの標識化にとって適切な安定性を与えることが見出されている。非環式キレート剤が大環状のものより不活性の程度が弱く、従ってより不安定であるけれども、その標識化は周囲温度でも十分に迅速である。このために、それらは、加熱に耐えることができないモノクローナル抗体の標識化には好ましいかもしれない。ポリアミノポリカルボキシラート・キレート剤のターゲッティングタンパク質とペプチドに対するカップリング用の詳細なプロトコルは、Cooper及び共同研究者(Nat.Protoc.1: 314-7.2006)並びにSosabowski及びMather (Nat.Protoc1: 972-6(2006))によって公表されている。
【0063】
ポリアミノポリカルボキシレート・キレート剤にカップリングした本発明のポリペプチドは、キレート剤及び医用画像に適した放射性核種にカップリングしたHER2結合ポリペプチドの放射性キレートから成る放射性標識ポリペプチドを提供するために使用されてもよく、該放射性核種は、61Cu、64Cu、66Ga、67Ga、68Ga、110mIn、111In、44Sc及び86Yから成るグループから選択され、又は治療に適した放射性核種とのものでは、該放射性核種は、225Ac、212Bi、213Bi、67Cu、166Ho、177Lu、212Pb、149Pm、153Sm、227Th及び90Yから成るグループから選択され、ここで、放射性核種はキレート環境を介してHER2結合ポリペプチドと錯体を形成する。
【0064】
本発明の放射性標識ポリペプチドの1つの特に好ましい実施態様は、1,4,7,10−テトラアザシクロ−ドデカン−1,4,7−トリス−酢酸−10−マレイミドエチルアセト−アミド及び111Inにカップリングした、アミノ酸配列:
AEAKYAKEMRNAYWEIALLPNLTNQQKRAF IRKLYDD
PSQSSELLSEAKK LNDSQAPKVDC
(配列番号5)
を有する、HER2結合ポリペプチドの放射性キレートである。
【0065】
本発明の放射性標識ポリペプチドの1つの特に好ましい実施態様は、1,4,7,10−テトラアザシクロ−ドデカン−1,4,7−トリス−酢酸−10−マレイミドエチルアセト−アミド及び61Cu又は64Cuにカップリングした、アミノ酸配列:
AEAKYAKEMRNAYWEIALLPNLTNQQKRAF IRKLYDDPSQSSELLSEAKK LNDSQAPKVDC
(配列番号5)
を有する、HER2結合ポリペプチドの放射性キレートである。
【0066】
本発明の放射性標識ポリペプチドの1つの特に好ましい実施態様は、1,4,7,10−テトラアザシクロ−ドデカン−1,4,7−トリス−酢酸−10−マレイミドエチルアセト−アミド及び66Ga、67Ga又は68Gaにカップリングした、アミノ酸配列:
AEAKYAKEMRNAYWEIALLPNLTNQQKRAF IRKLYDDPSQSSELLSEAKK LNDSQAPKVDC
(配列番号5)
を有する、HER2結合ポリペプチドの放射性キレートである。
【0067】
本発明に記載の放射性標識ポリペプチドの1つの特に好ましい実施態様は、1,4,7,10−テトラアザシクロ−ドデカン−1,4,7−トリス−酢酸−10−マレイミドエチルアセト−アミド及び86Yにカップリングした、アミノ酸配列:
AEAKYAKEMRNAYWEIALLPNLTNQQKRAF IRKLYDDPSQSSELLSEAKK LNDSQAPKVDC
(配列番号5)
を有する、HER2結合ポリペプチドの放射性キレートである。
【0068】
本発明の放射性標識ポリペプチドは、また、前記に示すようなHER2結合性ポリペプチドの間接標識化によって得ることができる。好ましくは、ポリペプチドは少なくとも1つのシステインを含有し、最も好ましくはたった1つのシステインを含有する。システインは、本発明のポリペプチドに最初に存在していてもよいし、又は1つ又はそれ以上の更なるアミノ酸として後の段階で付加されてもよい。例えば18F、76Brによる非間接標識化のためには、さまざまのヨウ素同位体及び211At、中間体「リンカー分子」が標識化のために用いられる。そのようなリンカーは2つの官能性部分を含有し、1つは迅速で効率的な放射性標識化を与え、別のものは、タンパク質への、例えばアミン基への、又は好ましくはユニークシステインのチオール基への、迅速で効率的なカップリングを与える。例えば、マレイミド基は、チオール基と反応して安定なチオエーテル結合を形成する。この考えは、最初に「リンカー分子」を放射性標識と反応させ、続いてタンパク質のチオール基と反応させることである。
【0069】
いくつかの代替案が放射性ヨウ素化について徹底的に検討されており、そこではリンカー分子の放射標識化が、例えば活性化フェノール環又は適した脱離基を有する芳香環上で為されている。非標識リンカーの製造と、N−スクシンイミジル3−[*I]ヨードベンゾアートを使用する間接的放射性ヨウ素化の詳細なプロトコルは、Vaidyanathan et al.(Vaidyanathan G and Zalutsky MR(2006),Nat Protoc. 1(2): 707-13)によって提供されている。N−スクシンイミジルトリメチルスタンニルベンゾアートを使用する間接放射性ヨウ素化の例は、以下の図式に示される。
【化4】
【0070】
この図式では、リンカー分子は先ず酸性条件下で放射性ヨード化され、それからアルカリ条件下で遊離アミン(リジンのε−アミノ基のN末端)にカップリングされる。ベンゾアートのメタとパラヨード誘導体の両方は、文献に記載されている。
【0071】
リンカー分子は、、放射性標識(例えば、2−、3−又は4−の位置で)をリンクするアリール基を含んでもよい。但し、放射性標識が76Brである場合は、リンカー分子はフェノール性OH基を含まない。リンカー分子の非限定的な例は、本発明のポリペプチドにリンクするために使用できる複素環のイミドを含み、そして放射性標識は、N−[2−ベンズアミドエチル]マレイミド(malemide),4−マレイミドベンゾフェノン(BPMal)、スクシンイミジル4−(p−マレイミドフェニル)ブチラート(SMPB)、4−(4−N−マレイミドフェニル)酪酸ヒドラジド塩酸塩(MPBH)、及びマレイミドベンゾイル−N−ヒドロキシスクシンイミドエステル(MBS)を含む。
【0072】
N−[2−ベンズアミドエチル]マレイミドは、18Fと反応してN−[2−(4−(18F−フルオロベンズアミド)エチル]マレミドを形成し得る。同様にCai et al. (J. Nucl. Med.47:1172-80(2006))によって記載されているように、76Brと反応し得る。
【0073】
本発明は、また、前記HER2結合ポリペプチドの使用の別の態様、並びにポリペプチドがその結合特性故に有用である治療、診断及び検出のためのさまざまの方法、に関する。これらの使用及び方法の以下の説明で「HER2結合ポリペプチド」を言及する場合、この用語は、単独でHER2結合ポリペプチドを包含するが、しかしまた、例えば融合タンパク質中の部分としてHER2結合ポリペプチドを組み込む、及び/又は標識、キレート剤、治療薬及び/又は診断薬に共役されている、及び/又はタグとして又は他の目的で更なるアミノ酸残基と共に提供される、上記のこのポリペプチドに基づく全てのそれらの分子を包含するものとする。上で説明したように、そのような融合タンパク質、誘導体などは、本発明の一部を構成する。
【0074】
従って、1つのそのような態様において、本発明は、HER2の過剰発現によって特徴付けられるがんを有する又は疑われるヒトを含む、哺乳動物対象(subject)の体のインビボ画像化の方法を提供し、その方法は下記の工程を含む:
・医用画像に適した放射性核種を含む、前述の放射性標識ポリペプチドを哺乳類対象の体内に投与すること:及び
・医用画像機器を使用して対象の体の少なくとも一部分の画像、該画像は該体の内部の放射性核種の存在を指示する、を得ること。好ましくは、画像は、体への放射性標識ポリペプチドの投与の1〜72時間、又はある場合にはより好ましくは1〜24時間内に得られる。投与と画像取得の間の時間は、使用される放射性核種の半減期に依存する。画像を得る工程を2回、3回又はそれ以上繰り返すことが可能であり、それによって画像の系列が取得される。人が標的薬の経時的な体内分布を追跡しようとする場合、これは有用であり、薬物動態学的試験を可能にする。当業者には当然のことながら、そのような試験に必要な程度の時間分解能を達成するために、任意の数の画像を得ることができる。
【0075】
本発明の画像化方法の1つの実施態様において、本方法は、投与工程の前に、本発明の放射性標識ポリペプチドを製造する準備工程を含み、その工程は、第1の態様に記載のポリペプチドを医用画像に適した放射性核種と混合することを含む。
【0076】
本発明の画像化方法のより格別な実施態様において、本方法は、投与工程の前に、(i)アミノ酸配列が配列番号7から成るポリペプチド、の放射性標識ポリペプチドを、ii)99mTc、を用いて製造する準備工程を含み、その工程は、ポリペプチドを、適切な緩衝液中で、適切な還元剤、例えば塩化第一スズ又はフッ化第一スズの存在下で、99mTc−過テクネチウム酸と混合することを含む。これは、また、中間体の弱いキレート剤、例えば酒石酸若しくはクエン酸又はグルコン酸の存在下で実行され得る。
【0077】
本発明の画像化方法の別の格別な実施態様において、本方法は、投与工程の前に、(i)アミノ酸配列が1,4,7,10−テトラアザシクロドデカン−1,4,7−トリス酢酸−10−マレイミドエチルアセトアミドに共役した配列番号5から成るポリペプチド、の放射性標識ポリペプチドを、ii)111Inを用いて製造する準備工程を含み、その工程は、非溶解性コロイドの生成を防止する適切な緩衝液、例えば酢酸又はクエン酸緩衝液(しかし、これに限定されない)中、共役体を111Inと混合することを含む。
【0078】
本発明の画像化方法の別の格別な実施態様において、本方法は、投与工程の前に、(i)アミノ酸配列が1,4,7,10−テトラアザシクロドデカン−1,4,7−トリス酢酸−10−マレイミドエチルアセトアミドに共役した配列番号5から成るポリペプチド、の放射性標識ポリペプチドを、ii)61Cu又は64Cuを用いて製造する準備工程を含み、その工程は、非溶解性コロイドの生成を防止する適切な緩衝液、例えば、酢酸又はクエン酸緩衝液(しかし、これに限定されない)中、共役体を61Cu又は64Cuと混合することを含む。
【0079】
本発明の画像化方法の別の格別な実施態様において、本方法は、投与工程の前に、(i)アミノ酸配列が1,4,7,10−テトラアザシクロドデカン−1,4,7−トリス酢酸−10−マレイミドエチルアセトアミドに共役した配列番号5から成るポリペプチド、の放射性標識ポリペプチドを、ii)66Ga、67Ga又は68Gaを用いて製造する準備工程を含み、その工程は、非溶解性コロイドの生成を防止する適切な緩衝液、例えば酢酸又はクエン酸緩衝液(しかし、これに限定されない)中、共役体を66Ga、67Ga又は68Gaと混合することを含む。
【0080】
本発明のこの態様のいくつかの実施態様において、前記がんは、乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんから選択される。
【0081】
別の態様において、本発明は、診断に利用するための上記のような放射性標識ポリペプチドを提供する。「診断」という用語は、本明細書において、患者の病歴、試験及び臨床検査データのレビューの評価を通じて疾患の特質及び原因を同定したり、決定する行為又は方法を記述するために用いられる。
【0082】
放射性標識ポリペプチドは、頻発するHER2の過剰発現によって特徴付けられるがんの群に属しているがんの診断に用いることができる。「頻発する」という用語は、HER2の過剰発現が、特定のがんを有する全ての患者の少なくとも10%に存在していることを示すために、本明細書で用いられる。このように、放射性標識ポリペプチドは、患者がそのようながんを有するかどうかを決定するプロセスに使用することができる。また、患者の適する治療を決定するプロセスでも使用することができる。特に、HER2過剰発現の検出は、HER2標的の療法、例えばトラスツズマブ(Herceptin(登録商標))での治療に対する患者の層別化に有用であり得る。
【0083】
放射性標識ポリペプチドは、また、乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんより選択されるがんの診断及び/又は分子的特性化に、使用することができる。このような状況において、分子的特性化は、例えばいずれかのHER2の発現があるかどうかを決定し、例えば治療の前後での画像を取得することによって達成できる治療の前後でのHER2発現の量を決定し、例えば、手術目的のために、HER2発現の程度又は解剖学的含量(content)を決定するためのHER2発現の特性化に関する。
【0084】
また、本発明の別の態様において、ヒトを含む哺乳動物の対象物の体のインビボの画像化の診断薬の製造において、上記のような放射性標識ポリペプチドの使用が提供される。
【0085】
別の応用の態様において、本発明は、HER2の過剰発現によって特徴付けられるがんを有する、ヒトを含む哺乳類対象の治療方法であって、前記哺乳類対象物の身体に、治療に適する放射性核種を含む上記の放射性標識ポリペプチドを、治療的有効量で投与する工程を含む方法を提供する。
【0086】
この態様の1つの実施態様において、前記方法は、投与工程の前に、本発明の放射性標識ポリペプチドを製造する準備工程を含み、その工程は、以下を含み得る:
・適切なバッファー中で、第1の態様に記載のポリペプチドを治療に適する金属放射性核種と混合すること;又は
・適切な還元剤の存在下で、第1の態様に記載のポリペプチドを適切な緩衝液中で過レニウム酸塩と混合すること;これは、また、中間体の弱いキレート剤、例えば、酒石酸、又はクエン酸若しくはグルコン酸の存在下でも実行され得る;又は
・第1の態様に記載のポリペプチドをトリカルボニル錯体のようなレニウムの反応性中間体と混合すること;又は
・第1の態様に記載のポリペプチドを適切な緩衝液中で、付着ハロゲン原子を有する反応性中間体と混合すること。この混合は、アスコルピン酸又はゲンチジン酸のようなポリペプチドの放射線分解を抑制するために指定される物質の存在下で実行され得る。
【0087】
本発明のこの態様のいくつかの実施態様において、前記がんは、乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんから選択される。
【0088】
別の態様において、本発明は、HER2の過剰発現によって特徴付けられるがんの治療における使用のような、例えば、乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんから選択されるがんの治療における使用のような、治療における使用のための、上記のような放射性標識ポリペプチドを提供する。
【0089】
また、本発明の別の態様において、乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんから選択されるがんのような、HER2の過剰発現によって特徴付けられるがんの治療のための薬剤の製造における、上記の放射性標識ポリペプチドの使用が提供される。
【0090】
本発明の別の態様は、上記に示すようなポリペプチドをコードする配列を含む核酸分子に関する。
【0091】
本発明の更なる態様は、前記の態様の核酸分子を含む発現ベクター、及び核酸分子の発現を通して、本発明のポリペプチドの産生を可能にする他の核酸エレメントに関する。
【0092】
本発明の別の態様は、前記の態様の発現ベクターを含む宿主細胞に関する。
【0093】
本発明の後者の3つの態様は、本発明のポリペプチドの産生ツールであり、本明細書では発現されることになっているポリペプチドに関する情報を条件に、及びタンパク質の組み換え発現の当業者の最新の技術レベルを条件に、当業者はそれらを得ることができ、それらを必要以上の負担無く実用化できるであろう。
【0094】
しかしながら、本発明のポリペプチドは、また、植物及びトランスジェニック動物を含む、さまざまな原核生物の又は真核生物の宿主の中での化学的合成又は発現を含む、他の公知の手段によって産生し得る。
【0095】
さて、本発明を、それに従って実施される実験の説明を通して詳細に説明する。下記の実施例は、限定するものとして解釈されるべきではない。実施例では、添付の図面が参照される。
【0096】
Affibody(登録商標)分子の配列は、以下の表1に示される。
【図面の簡単な説明】
【0097】
【図1】A:7.8nM;B:1.9Nm及びC:0.49nMのAffibody(登録商標)分子IIを、固定化したHER2/Fcキメラ融合タンパク質を有するセンサーチップ上に注入した後に得られたBiacoreセンサーグラムを示す。C末端システインのSH基は、N−エチルマレイミド(NEM)で遮断された。
【図2】A:6nM;B:1.5Nm及びC:0.38nMのC末端のシステインにカップリングしたDOTAを有するAffibody(登録商標)分子II(配列番号5)を、固定化したHER2/Fcキメラ融合タンパク質を有するセンサーチップ上に注入した後に得られたBiacoreセンサーグラムを示す。
【図3】A:6nM;B:1.5Nm及びC:0.38nMのAffibody(登録商標)分子X(配列番号7)を、固定化したHER2/Fcキメラ融合タンパク質を有するセンサーチップ上に注入した後に得られたBiacoreセンサーグラムを示す。
【図4】BALB/c nu/nu マウスSKOV−3腫瘍異種移植片中の、DOTAと共に111In標識したAffibody(登録商標)分子IIの体内分布を示す。全てのマウスに、1μg(100kBq)の111In標識されたAffibody(登録商標)分子IIを注射した。注射後0.5、1.4及び24時間で、臓器を解剖して、秤量し、そしてその放射能の量をガンマ計数器で測定した。放射能の取り込みは、グラム組織当たり注射した活性のパーセント(%IA/g)として計算された。
【図5】図4で説明した体内分布の試験で測定された放射能の取込みの腫瘍対臓器比を示す。
【図6】SKOV−3腫瘍異種移植片中、DOTAと共に111In標識したAffibody(登録商標)分子IIで、標的化する腫瘍の特異性を示す。全てのマウスに、DOTAを有する1μg(100kBq)の111In標識したAffibody(登録商標)分子IIを注射した。動物の1つの群は、111In標識したAffibody(登録商標)分子の注射の50分前に過剰の非標識Affibody(登録商標)分子(保護基)で前処理された。臓器は、注射後4時間で解剖し、秤量し、そして放射能の量をガンマ計数器で測定した。放射能の取り込みは、グラム組織当たり注射した活性のパーセント(%IA/g)として計算された。
【図7】BALB/c nu/nu マウス中のSKOV−3異種移植片腫瘍を発現するHER2(左側の部分、30分及び4時間p.i.)、及びHER2陰性のA431異種移植片(右側部分、4時間p.i.)のガンマカメラ画像を示す。全てのマウスに、DOTAを有する3μg(4.5MBq)の111In標識したAffibody(登録商標)分子IIを注射した。K=腎、T=腫瘍。
【図8】注射後4時間のBALB/c nu/nu マウスSKOV−3腫瘍異種移植片中の99mTc−標識Affibody(登録商標)分子Xの体内分布を示す。全てのマウスに、1μg(65kBq)の99mTc標識Affibody(登録商標)分子Xを注射した。動物の1つの群は、99mTc標識Affibody(登録商標)分子の注射の50分前に過剰の非標識Affibody(登録商標)分子(ブロッキング群)で前処理された。臓器は注射後4時間で解剖し、秤量し、そしてその放射能の量をガンマ計数器で測定した。グラム組織当たり注射した活性のパーセント(%IA/g)として計算された。
【図9】図8に記載の生体内分布の試験で測定された、放射能取り込みの腫瘍対組織比を示す。
【図10】BALB/c nu/nu マウス中のSKOV−3異種移植片腫瘍を発現するHER2(左側の部分、1時間及び4時間p.i.)、及びHER2陰性のA431異種移植片(右側部分、4時間p.i.)のガンマカメラ画像を示す。全てのマウスに、DOTAを有する3μg(15MBq)の111In標識したAffibody(登録商標)分子IIを注射した。K=腎臓、T=腫瘍。
【図11A】アミノ酸18〜58の合成の段階でのAffibody(登録商標)分子Xについての分析用HPLC溶離プロファイルを示す。Affibody(登録商標)分子は、VydacTP218TP54カラムで分離された。流量:1ml/分;温度:35℃;検出:220nm;溶離液A:0.1%TFA、H2O中1%アセトニトリル;溶離液B:アセトニトリル中0.1%TFA。 位置22−23、41−42、45−46及び53−54に疑似プロリンを使用しているポリスチレン樹脂上で行なった合成。
【図11B】アミノ酸18〜58の合成の段階でのAffibody(登録商標)分子Xについての分析用HPLC溶離プロファイルを示す。Affibody(登録商標)分子は、VydacTP218TP54カラムで分離された。流量:1ml/分;温度:35℃;検出:220nm;溶離液A:0.1%TFA、H2O中1%アセトニトリル;溶離液B:アセトニトリル中0.1%TFA。 疑似プロリンのないポリスチレン樹脂上での標準ペプチドの合成。
【図12A】アミノ酸1〜58(A)及び10〜58(B)の合成の段階でのAffibody(登録商標)分子Xに対する分析用HPLC溶離プロファイルを示す。Affibody(登録商標)分子は、VydacTP218TP54カラムで分離された。流量:1ml/分;温度:35℃;検出:220nm;溶媒:0.1%TFA、H2O中1%アセトニトリル;溶媒:アセトニトリル中0.1%TFA。 位置22−23、41−42、45−46及び53−54に疑似プロリンを使用しているポリスチレン樹脂上で行なった合成。
【図12B】アミノ酸1〜58(A)及び10〜58(B)の合成の段階でのAffibody(登録商標)分子Xに対する分析用HPLC溶離プロファイルを示す。Affibody (登録商標)分子は、VydacTP218TP54カラムで分離された。流量:1ml/分;温度:35℃;検出:220nm;溶媒:0.1%TFA、H2O中1%アセトニトリル;溶媒:アセトニトリル中0.1%TFA。 疑似プロリンのないポリスチレン樹脂上での標準ペプチドの合成。
【図13A】表1に示されたいくつかのAffibody(登録商標)分子に対する疎水性/親水性プロットを示す。Affibody(登録商標)分子II(配列番号5)についてのプロット。図面の上部は、Kyte-Doolittleスケールを使用するプロットを示し、そして下部は、HPLCによって決定されたpH3.4での疎水性指標を示す。疎水性アミノ酸は正の値を有し、親水性アミノ酸は負の値を有する。7アミノ酸の移動ウインドウは、両方のプロットに使用された。
【図13B】表1に示されたいくつかのAffibody(登録商標)分子に対する疎水性/親水性プロットを示す。配列番号X(配列番号7)を有するAffibody (登録商標)分子についてのプロット。図面の上部は、Kyte-Doolittleスケールを使用するプロットを示し、そして下部は、HPLCによって決定されたpH3.4での疎水性指標を示す。疎水性アミノ酸は正の値を有し、親水性アミノ酸は負の値を有する。7アミノ酸の移動ウインドウは、両方のプロットに使用された。 Affibody(登録商標)分子X(配列番号7)のアミノ酸配列のみが、即ち、maのないAffibody(登録商標)分子Xが、プロットを作成するために使用された。
【図13C】表1に示されたいくつかのAffibody (登録商標)分子に対する疎水性/親水性プロットを示す。Affibody(登録商標)分子Z00342、対照としての変異のない親Affibody(登録商標)分子についてのプロット。図面の上部は、Kyte-Doolittleスケールを使用するプロットを示し、そして下部は、HPLCによって決定されたpH3.4での疎水性指標を示す。疎水性アミノ酸は正の値を有し、親水性アミノ酸は負の値を有する。7アミノ酸の移動ウインドウは、両方のプロットに使用された。
【図14】本発明に記載のAffibody(登録商標)分子II(黒線)及びAffibody(登録商標)分子Z00342(灰色の破線)についてのHPLC溶離プロファイルを示す。Affibody(登録商標)分子は、0.5ml/分の流量とTFA、水、アセトニトリル溶剤系を用いるZorbax 300SB C8 150×2.1mm、3.5のμmのカラム上で分離した。
【実施例】
【0098】
〔実施例1〕
本発明のポリペプチドの製造
要約
この実施例では、選択されたHER2特異的Affibody(登録商標)分子を、下記のプロトコルに従って適切な細菌細胞中で大規模に発現され、精製された、pETシステム発現ベクターにサブクローニングした。対応する全長HER2特異的Affibody(登録商標)分子のアミノ酸配列は、Affibody(登録商標)分子I−XIとして表1に収載されている。
【0099】
Affibody(登録商標)分子のクローニング
Affibody(登録商標)分子I−VIIIをコード化する遺伝子を、テンプレートとして2つのオリゴヌクレオチド(一緒になってAffibody(登録商標)配列を包含している)を使用することによってPCR増幅した。各々の構築物について、28bpの相補的オーバーラップを備えた特定のテンプレート・オリゴヌクレオチド(各々、102長い)ペアを使用した。Affibody(登録商標)遺伝子を、NdeI部位及びATG開始コドンを含むフォワードプライマー(センス) (Affibody(登録商標)分子Iについては、配列をコードするHis6-タグもまたプライマーに含まれた)、並びにC末端アミノ酸についてのコドン、終止コドン及びNotI部位を有するリバースプライマー(アンチセンス)で増幅した。
【0100】
Affibody(登録商標)分子IX中のAffibody(登録商標)アルブミン結合ドメイン(ABD)遺伝子融合を、オーバーラップ伸長PCRによって生産した。リバースプライマーがABD遺伝子のリンカー配列(Cysコドンと共に)及び5’末端を含んでいたことを除いては、Affibody(登録商標)分子コード化フラグメントを、Affibody(登録商標)分子I−VIIIについて上述したように作成した。ABD遺伝子を、Affibody(登録商標)遺伝子の3’末端及びリンカーの配列及び終止コドンを有するリバースプライマー、及びNotI部位を含むフォワードプライマーで増幅した。続いて、25bpオーバーラップを有する2つのPCR産物を、Affibody(登録商標)遺伝子フォワードプライマー及びAffibody(登録商標)ABD遺伝子融合を作成するABD遺伝子リバースプライマーを用いて、最後のPCR増幅におけるテンプレートとして使用した。
【0101】
PCR産物を、制限酵素NdeI及びNotIで消化し、同じ酵素で先に消化したT7発現ベクターに連結した。
【0102】
【表1】
【0103】
Affibody(登録商標)分子の発現及び採取
得られた発現プラスミドを、エレクトロポレーションを使用して大腸菌BL21(DE3)細胞に形質転換した。細菌含有発現プラスミドを、カナマイシンを有する選択寒天から選び取り、マルチ発酵装置システム(System Greta, Belach Bioteknik AB, Sweden)を使用して、50mg/lのカナマイシンで補充された800mlのTSB+YE培地(30g/lトリプシン大豆ブロス+5g/l酵母エキス)の中で37℃で増殖させた。約1の光学密度(OD600nm)で、Affibody (登録商標)分子の発現を、0.5mmのIPTGの添加によって自動的に誘導した。誘導剤の添加の5時間後に、培養物を自動的に<12℃に冷却した。次いで、培養物を遠心分離(15900×gで20分)により採取し、5〜10gの細菌の細胞ペレットを採取し、冷凍庫(−20℃)で保管した。
【0104】
Affibody(登録商標)分子Iの精製
細胞ペレット(3g)を、7Mの尿素及び1,000ユニットのBenzonase(Merck)を含むpH8の緩衝液の30ml中に再懸濁させた。30分のインキュベーションの後、溶解物を遠心分離によって清澄化し、His6タグの付いたAffibody(登録商標)分子を含んでいる上清を、1.5mlの Superflow NiNTAカラム(Qiagen)にかけた。カラムを、7Mの尿素を含む20mlのpH6.3の緩衝液で洗浄し、8Mの尿素を含むpH4.5の緩衝液で溶離した。最後に、緩衝液を、PD-10カラム(GE Healthcare)を使用することによって、1×PBS(2.68mmKCl、137mmNaCl、1.47mmKH2PO4、8.1mmNa2HPO4、pH7.4)に交換した。
【0105】
Affibody(登録商標)分子II−VIIIの精製
細胞ペレット(10g)を、100mlの20mMのTris−HCl、pH7.5に再懸濁させ、2,000ユニットのBenzonase(Merck)を補充して、超音波処理によって破壊し、10分間75℃で加熱した。熱処理された溶解物を遠心分離によって清澄化し、そして20mMのTris−HClpH7.5であらかじめ平衡化した25mlQ−セファロースFF(GE Healtcare)を詰めたXK26カラムにかけた。カラムから流れ出た画分を集め、C末端システインを有するAffibody (登録商標)分子を、常温で1時間20mM DTTと共にインキュベートした。ACNを5%に加え、5%ACNと0.1%TFAを含有する水で予め平衡化した3mlのResource 15RPCカラム(GE Healtcare)にかけた。水中の5カラム容積(CV)の5%ACN及び1%TFAで洗浄した後、水中の20CVの5〜32%ACN及び0.1%TFAの直線勾配で、結合したタンパク質を溶離した。Affibody(登録商標)分子を含む画分をSDS−PAGE分析によって同定し、プールした。最後に、緩衝液を、HiPrep 26/10脱塩カラム(GE Healthcare)を使用することによって1×PBSに交換した。
【0106】
Affibody(登録商標)分子IXの精製
細胞ペレット(5g)を、50mlTST緩衝液(25mMTris−HCl、200mMのNaCl、1mMのEDTA、0.05%のTween20、pH8.0)に再懸濁させ、超音波処理によって破壊した。遠心分離による清澄化の後、上清を20mlHSAセファロース(CNBr活性セファロース4 Fast Flow, GE Healtcare上に固定化されたヒト血清アルブミン)を詰めたXK26カラム(GE Healtcare)にかけた。カラムを、10CVのTST緩衝液、次いで、5CVの5mMのNH4Ac、pH5.6によって洗浄した。結合したABDタグ付きAffibody(登録商標)分子を、0.5MのHAc、pH2.8で溶離した。最後に、緩衝液を、HiPrep26/10脱塩カラム(GE Healthcare)を使用することによって1×PBSに交換した。
【0107】
〔実施例2〕
インビトロの本発明のポリペプチドのHER2結合特性
要約
この実施例を形成している実験では、Biacore(登録商標)2000システムでの表面プラズモン共鳴(SPR)を使用して、本発明のポリペプチドの結合を分析した。HER2標的結合活性は、C末端のシステイン及びX(配列番号7)にカップリングしたAffibody(登録商標)分子II(配列番号5)(DOTAを有するII)について示される。
【0108】
バイオセンサー分析
C末端His6タグ(R &D System #1129-ER)でヒトIgG1(Pro100−Lys330)のFc領域に融合したErbB2(HER2、Met1−Thr652)の細胞外ドメインから成る、組み換えヒトHER2/Fc(ErbB2/Fc)キメラタンパク質を、アミンカップリング化学を使用するCM5センサーチップの表面上に(1,800のレゾナンスユニット(RU))固定化した。チップ上の1つの表面は、参照セルとして使用するために活性化され、不活化された。HBS−EP緩衝液(10mMのHEPES、150mMのNaCl、3mMのEDTA、0.005%の界面活性剤P−20、pH7.4)中希釈したAffibody (登録商標)分子を、被検体として使用した。
【0109】
結合速度論の解析に関しては、3つの被検体の濃度を、50μ/分の一定流量を用いてチップ上に2連で(dupulicate)注入した。会合相は5分で、続いてAffibody(登録商標)分子の遅速性を説明する長い解離相(30分)であった。解離平衡定数(KD)、会合速度定数(ka)及び解離速度定数(kd)は、BIAevaluation 4.1ソフトウェア(GE Healthcare)の質量移動(mass transfer)補正を有する1:1ラングミュア結合モデルを使って計算された。
【0110】
システインのSH基のブロッキング
システインのSH基をブロックし、それによって分子の二量化を防ぐために、単一のC末端のシステインを有するAffibody(登録商標)分子を、N−エチルマレイミド(NEM、Pierce No.23030)で処理した。Affibody (登録商標)分子II(配列番号5)を、1ml(2.1mg)のペプチドを10μlのTris−HCl、pH8.5及び40μlの0.5Mジチオトレイトール(DTT)で、室温で2時間処理することによって還元した。過剰のDDTは、カップリング緩衝液(100mMのリン酸ナトリウム、150mMのNaCl、pH7.0)で平衡化したNAP5カラム(GE Healthcare)上のゲル濾過によって除去した。0.5mlの還元タンパク質を有するロードカラムは、1mlのカップリング緩衝液でタンパク質を溶離する。
【0111】
遊離SH基のブロッキングは、タンパク質に対して20倍モル過剰のNEMを使って行われた:水(200mm)中15μlのNEM溶液を0.5mlの還元したタンパク質溶液(1mg/ml)に加え、そしてヘッド・オーバーヘッド・ミキサー(head-over-head mixer)上、室温で1時間インキュベートする。過剰のNEMは、前記のようにNAP5−カラム上でのゲル濾過によって、しかし平衡緩衝液としてPBSを用いて、除去した。
【0112】
C末端システインへのマレイミド−DOTAのカップリング
3mgのAffibody(登録商標)分子II(配列番号5)を、40℃で30分間20mMのDTTで還元した。過剰のDTTは、PD−10カラム上での0.2Mの酢酸アンモニウム、pH5.5への緩衝液交換によって除いた。カップリングは、3倍モル過剰のキレート剤、水中のマレイミド−モノ−アミドDOTA(Mal-DOTA, Macrocyclics No.B-272)溶液(1mg/ml)を用いて行った。混合物を、連続振盪しながら40℃で1時間インキュベートした。非複合化キレート剤からの精製は、セミ分離用RPCカラム、Zorbax 300SB C18、9.4×250mm、5μm上で行った。精製物質のカップリング程度は、Zorbax 300SB C8 150×2.1mm、3.5μmの分析用カラムの上でHPLC−MSによって分析した。マレイミド−DOTA共役Affibody(登録商標)分子のみが、本方法によって検出された。
【0113】
Affibody(登録商標)分子IIのHER2−結合相互作用を、図1のセンサーグラフに、C末端のシステインにカップリングしたDOTAを有するAffibody(登録商標)分子IIを図2に、及びAffibody(登録商標)分子X(配列番号7)のものを図3に示す。
【0114】
HER2標的とのAffibody(登録商標)分子の相互作用についての結合パラメーターを決定するために、動力学的試験を行なった。3つの異なる濃度のAffibody(登録商標)分子を、固定化HER2/Fcのキメラ融合タンパク質でBiacoreチップ表面に注入した。解離平衡定数(KD=kd/ka)、会合速度定数(ka)及び解離速度定数(kd)を表2に要約する。C末端システインへのDOTAカップリングは、NEMブロッキングAffibody(登録商標)分子IIに比べてKDを有意に変化させなかった。
【0115】
【表2】
【0116】
〔実施例3a〕
ポリアミノポリカルボキシラートキレート剤を有する本発明のポリペプチドのインビボのHER2結合特性
要約
この実施例を構成している実験では、本発明の放射標識ポリペプチドの結合を、インビボマウス異種移植片モデルで分析した。C末端のシステインにカップリングしたDOTAを有するAffibody(登録商標)分子IIを111Inで放射標識し、HER2を過剰発現するSKOV−3腫瘍異種移植片及び対照としてHER2陰性A431腫瘍異種移植片を担持するマウスに注射した。全動物中でのさまざまな臓器の放射性標識Affibody(登録商標)分子の分布及び局在を分析するために、体内分布及びガンマカメラ画像の試験を行なった。
【0117】
111Inラベル化
実施例No.2に記載したように、C末端のシステインにモノアミドDOTA(Mal−DOTA)をカップリングさせてAffibody (登録商標)分子を修飾した。得られた分子は「DOTAを有するAffibody(登録商標)分子II」と名付けられた。
【0118】
111Inで標識するために、DOTA(30μlのpH5.5の酢酸アンモニウム緩衝液)を有する30μgのAffibody(登録商標)分子IIを、40.5μlの111InCl3(標識化時15MBq)の保存液と混合した。反応混合液のpHは約5.0であった。反応混合物を、60℃で60分間インキュベートした。
【0119】
標識化効率は、シリカゲル含浸ガラス繊維シート(ITLC SGシート(Gelman Sciences社))上のインスタント薄層クロマトグラフィー(ITLC)により、0.02Mのクエン酸を溶離液として使用して分析した。ITLC片に沿った放射能の分布を可視化し、PhosphorImager(CycloneTM Storage Phosphor System, Packard)を使用して定量化した。放射性標識Affibody(登録商標)分子は、ITLCシートの元の場所に残り、一方、遊離の111Inは溶媒先端に移動した。
【0120】
放射化学的純度は99.8から99.9%であった、即ち、放射性標識化は殆ど定量的であった。放射性標識化Affibody(登録商標)分子の一切の更なる精製は必要でなかった。反応混合物は、PBSで1.4mlに希釈し、実験の前に凍結保存した(1〜2日)。
【0121】
腫瘍モデル
雌の非近交系のBALB/c nu/nu マウスに、1×107SKOV−3(ヒト卵巣腹水腺がん)細胞又は1×107a431(ヒト表皮がん)細胞を、右の後脚中に移植した。SKOV−3細胞はHER2を過剰発現し、A431細胞はHER2について陰性であった。
【0122】
腫瘍担持マウスにおける体内分布
体内分布の試験のため、異種移植片腫瘍(1×107個のSKOV−3細胞移植の30〜40日後)担持マウスを、4つの群に無作為に割り付けた。マウスに、100μlPBS中の1μg(100kBq)の111In標識Affibody(登録商標)分子を、尾静脈に注射した。全ての注射の忍容性は良好であった。動物の1つの群に、1.5ml(0.9mg)の非標識Affibody(登録商標)分子IIを皮下注射し(ここで、C末端Cysが実施例2に記述したようにNEMによってブロックされている)、次いで50分後に、1μg(100kBq)の111In標識Affibody(登録商標)分子を静脈注射した(ブロッキング実験)。
【0123】
致死量のKetalarの溶液及びRompun溶液の注射後、マウスを、注射後0.5、1、4及び24時間後 (h p.i)に心臓穿刺を介して放血により犠牲にした。ブロッキンググループからの動物は、注射後4時間で犠牲にした。臓器を解剖し、秤量し、そしてそれらの放射性含量をガンマ計数器で測定した。放射能の取り込みは、グラム組織当たり注射した活性のパーセントとして計算された(%IA/g)。
【0124】
画像化
ガンマカメラによる画像化のため、異種移植片腫瘍(1×107個のSKOV−3細胞移植の30〜40日後又は1×107個のa431細胞移植の10〜15日後)担持マウスを使用した。マウスに、100μlPBS中3μg(4.5MBq)の111In標識Affibody(登録商標)分子IIを、尾静脈に注射した。注射後0.5又は4時間で、動物を致死量のKetalar及びRompunの溶液により安楽死させた。MEGPコリメータを装着したMillenium GEガンマカメラを使用し、画像化を行った。Hermesソフトウェア(Nuclear Diagnostics, Stockholm, Sweden)を使用し、シンチグラフィーの結果を、視覚的に評価した。
【0125】
結果
DOTAを有する111In標識Affibody(登録商標)分子IIを使用した、体内分布の試験からの結果を図4に示す。全ての時点での放射能の取り込みは、腎臓は別として、全ての他の臓器中よりも腫瘍中で高かった。腫瘍の取り込みは、注射後0.5時間で既に13±2%IA/gであった、注射後1時間で17±2%IA/gに上昇し、注射後1時間で15±3%IA/gから注射後24時間で11±4%IA/gまでゆっくり減少した。放射能の高い取り込みが、また、腎臓にも見出されたが、腎臓は、Affibody(登録商標)分子のような小さいタンパク質のための主要な排泄経路であるので、それは、予期されたことでもある。注射後1時間で測定された186±24%IA/gを有する最も高い腎臓の取り込みは、注射後24時間で144±13%IA/gに減少した。このように、腎臓の取り込みは、N末端にカップリングしたDOTAを有するAffibody(登録商標)分子Z00342(ZHER2:107として国際公開公報第2005/003456号に開示され、しばしば、また、ZHER2:342とも呼ばれる)について測定された腎臓の取り込み、即ち注射後1時間で243±22%IA/g及び注射後24時間で232±34%IA/gより相当低かった(Orlova, A. et al. (2006), Cancer Research 67: 2178-2186)。加えて、DOTAを有する111In標識Affibody(登録商標)分子IIは、Orlova, et al. (2006) によって記述されたHER2特異的Affibody(登録商標)分子と比較して、全ての測定時点において全ての臓器でより低い取り込みであった。高い腫瘍の取り込みは、他の臓器での減少した取り込みと共に、先に記述されたAffibody(登録商標)分子について公表された比率を超える腫瘍対組織比をもたらす(図5)。例えば、腫瘍対血液比は、Orlova A. et al. (2006)によって記述されたAffibody(登録商標)分子に対する注射後1時間で8、注射後4時間で12及び注射後24時間で47と比較して、注射後1時間で16、注射後4時間で88及び注射後24時間で275であった。
【0126】
本発明に記載のAffibodyの(登録商標)分子を使用する腫瘍標的の特異性は、ブロッキング実験によって証明され、ここで過剰な非標識Affibody(登録商標)分子の腫瘍担持マウスへの事前注射は、DOTAを有する111In標識Affibody(登録商標)分子IIの腫瘍取り込みを86%だけ減少させたが、他の臓器における取り込みは減少させなかった(図6)。注射後30分及び4時間で撮られたガンマカメラ画像により、DOTAを有する111In標識Affibody(登録商標)分子IIの高い特異的な腫瘍ターゲッティングが確認された(図7)。SKOV−3腫瘍を注射後0.5時間で既に可視化し、注射後4時間で、バックグラウンド放射能を洗い落とした。腫瘍とは別に、明確な取り込みがまた腎臓でも見えた。HER2陰性A431異種移植片腫瘍を担持するマウスは、注射後4時間で腫瘍中には無視できる放射線の取り込みしかなかった。
【0127】
〔実施例3b〕
N3Sキレート剤を有する本発明に記載のポリペプチドのインビボのHER2結合特性
要約
この実施例を構成している実験では、本発明の放射性標識ポリペプチドの結合を、インビボで、マウス異種移植片モデルを用いて分析した。N末端メルカプトアセチル−グルタミル−セリル−グルタミル(ma−ESE)配列を含むAffibody(登録商標)分子X(配列番号7)を99mTcで放射性標識し、HER2を過剰発現するSKOV−3腫瘍異種移植片を担持するマウスに注射した。全動物中のさまざまな臓器への放射性標識Affibody(登録商標)分子の分布及び局在を分析するために、体内分布及びガンマカメラ画像の試験を行なった。
【0128】
99mTc標識
Affibody(登録商標)分子Xを、間接法を用いて99mTcで標識した。99mTcを用いた標識化については、下記成分を含むキットを製造した:1ml当たり50Mgのグルコン酸二水和物、1mgのEDTAのジナトリウム塩及び0.5mgの錫(II)クロリド二水和物。この溶液を1mlを含むアリコートに分割し、アリコートを凍結乾燥した。凍結乾燥標識キットを、使用の前に−20℃で貯蔵した。
【0129】
50μlPBS(2.68mMのKCl、137mMのNaCl、1.47mMのKH2PO4、8.1mMのNa2HPO4、pH7.4)を、50μgのABY−024溶液(脱イオン化した脱気水中2mg/ml)に追加した。ボルテックス撹拌後、混合物にテクネチウム標識キットを混ぜ、99mTc発生装置(UltratechneKow FM発生装置、Tyco Healthcare Nordic AB(Mallinckrodt Sweden AB)からの200μlの99mTcO4-含有溶離液を添加した。混合物を、90℃で60分間インキュベートした。
【0130】
各混合物の2つの小さい(0.8μl)アリコートを、2つのITLC・SG片に適用し、そして1つをPBS(収量を決定する)で溶離し、他方はピリジン:酢酸:水(5:3:1.5)(還元した加水分解されたテクネチウム(RHT)の存在を決定するため)で溶離した。この溶離液では、テクネチウムコロイドは元の場所に残ったが、放射性標識Affibody(登録商標)分子、過テクネチウム酸及び他の99mTcの錯体は、溶媒先端に移動した。ITLC片に沿った放射活性の分布を視覚化し、PhosphorImager (CycloneTM Storage Phosphor System, Packard)を使用して定量した。標識化は殆ど定量的であって、それはいかなる追加の精製も不要であった。
【0131】
放射性標識Affibody(登録商標)分子を、放射能濃度が100μl当たり65kBqであることを確実にするために、PBSで希釈した。Affibody(登録商標)分子の濃度を、100μl当たり1μgに調整した。
【0132】
腫瘍担持マウスにおける体内分布
雌の非近交系のBALB/c nu/nu マウスに、1×107SKOV−3(ヒト卵巣腹水腺がん)細胞を、右の後脚中に移植した。
【0133】
体内分布の試験については、異種移植片腫瘍(1×107個のSKOV−3細胞移植の40〜45日後)担持マウスを、4群に無作為に割り付けた。
【0134】
マウスに、100μlPBS中の1μg(65kBq)の111Tc標識Affibody(登録商標)分子を、尾静脈に注射した。全ての注射は良好な忍容性を示した。動物の1群に、100ml(0.5mg)の非標識His6-Z00342を皮下注射し、次いで50分後に、1μg(65kBq)の111Tc標識Affibody(登録商標)分子Xを静脈注射した(ブロッキング実験)。
【0135】
致死量のKetalarの溶液及びRompun溶液の注射後、マウスを、注射後4時間後に心臓穿刺を介して放血により犠牲にした。ブロッキンググループからの動物は、注射後4時間で犠牲にした。臓器を解剖し、秤量し、そしてそれらの放射能含量をガンマ計数器で測定した。放射能の取り込みは、グラム組織当たり注射した活性のパーセントとして計算された(%IA/g)。
【0136】
画像化
ガンマカメラによる画像化については、異種移植片腫瘍(1×107個のSKOV−3細胞移植の40日後又は1×107個のa431細胞移植の10〜15日後)担持マウスを使用した。
【0137】
マウスには、100μlPBS中の3μg(15MBq)の99mTc標識Affibody(登録商標)分子Xを尾静脈に注入した。注射後1又は4時間で、動物を致死量のKetalar及びRompunの溶液により安楽死させた。LEHRコリメータを装着したCam Siemensガンマカメラを使用し、画像化を行った。Hermesソフトウェア(Nuclear Diagnostics, Stockholm, Sweden)を使用し、シンチグラフィーの結果を、視覚的に評価した。
【0138】
結果
99mTc標識Affibody(登録商標)分子Xを使用した、体内分布の試験からの結果を図8に示す。注射後4時間での放射能の取り込みは、腎臓は別として、全ての他の臓器中よりも腫瘍中で高かった。腫瘍の取り込みは、注射後4時間で17±4%IA/gであった。放射能の高い取り込みが、また、腎臓にも見出されたが、腎臓がAffibody(登録商標)分子のよな小さいタンパク質のための主要な排泄経路であるので、それは予期されたことでもある。腎臓の取り込みは、注射後4時間で58±10%IA/gであった。このように、腎臓の取り込みは、上記実施例3aに記述されたAffibody(登録商標)分子及びN末端にカップリングしたDOTAを有するZ00342について測定された腎臓の取り込みより相当低かった(Orlova,A. et al. (2006), Cancer Research 67: 2178-2186)。また、腸(3.52%IA/g)及び肝臓(1.06%IA/g)での低い取り込みは、腎臓でのより高い取り込みと共に、99mTc標識Affibody(登録商標)分子Xが主に腎臓の経路によって浄化され、肝胆道の経路によって行われないことを示す。欧州特許第6123095号は、好ましいma−Xaa1−Xaa2−Xaa3キレート成分が1つ又は2つのSerを含み、残りの2つ又は1つのアミノ酸残基がGluであるものが、肝胆道系から腎臓クリアランス経路へのシフトをもたらすことを開示している。Affibody(登録商標)分子Xは、欧州特許第6123095号に開示された好ましいキレート剤部分の1つ、即ちma−Glu−Ser−Glu及びこれらのポリペプチドの親水性の増加をもたらす本発明の実施例1(表1)に記載のアミノ酸交換の両方を含む。このように、本明細書で説明する99mTc−標識Affibody (登録商標)分子Xは、高い腎臓クリアランスと低肝胆道系クリアランスを両立させるが、腎臓での再吸収を最小化した、ma−Xaa1−Xaa2−Xaa3キレート部分を含んでいるポリペプチドの追加的実施例を示す。
【0139】
全ての臓器における99mTc標識Affibody(登録商標)分子Xの取り込みは、実施例3aに記述された111In標識Affibody(登録商標)分子の取り込みと同様であった。
【0140】
上述の体内分布実験の腫瘍対組織比を図9に示す。再び高い腫瘍対組織比を得た。特に、99mTc標識Affibody(登録商標)分子Xの腫瘍対肝臓比は、注射後4時間で10の比率の実施例3aに記述されたDOTAを有する111In標識Affibody(登録商標)分子IIに比べて、17であった。腫瘍対血液比は、注射後4時間で42であった。
【0141】
Affibody (登録商標)分子Xを使用した腫瘍標的の特異性は、ブロッキング実験によって証明され、その場合、過剰な非標識Affibody(登録商標)分子の腫瘍担持マウスへの事前注射により、99mTc標識Affibody(登録商標)分子Xの腫瘍吸収を90%も減少させたが他の臓器での取り込みでは減少させなかった(図8)。
【0142】
注射後1時間及び4時間に撮られたガンマカメラ画像は、本発明に記載の99mTc標識Affibody(登録商標)分子の高い特異的な腫瘍ターゲッティングを確認した(図10)。SKOV−3腫瘍を注射後1時間で既に可視化し、注射後4時間で、バックグラウンド放射能が洗い落とした。腫瘍とは別に、明白な取り込みもまた腎臓に見える、しかしながら、実施例3aに記述されたDOTAを有する111In標識Affibody(登録商標)分子IIの腎臓の取り込みに比べて、この取り込みはかなり低かった。HER2陰性A431異種移植片腫瘍を担持するマウスは、注射後4時間で腫瘍中の放射能取り込みは無視できるものであった。
【0143】
〔実施例4〕
本発明のポリペプチド及びZ00342の化学的合成に関する比較実験
要約
この実施例を形成する実験では、本発明のポリペプチドの固相ペプチド合成が記述される。4つの位置、即ち[N23T]、[A42S]、[A46S]及び[A54S]で導入された変異は、簡略化された略号Fmoc−Xxx−Yyy−OHを有する疑似プロリン前駆体との代替合成戦略を用いることを可能にした。
上述の4つの位置で疑似プロリンを使うことによって、全体のAffibody(登録商標)分子X(配列番号7)(即ち、アミノ酸1〜58)を合成することを可能にしたが、一方、標準的な合成はペプチドを製造できなかった。
【0144】
新規なセリン又はトレオニン残基の導入は、また、イソアシルジペプチドの使用を可能にし、それはペプチド合成中凝集を減らすことによって合成効率を高めるための疑似プロリンに対する代替である(Sohma et al., Tetrahedron Lett.47:3013(2006))。いくつかのこのようなビルディングブロックは、Novabiochem of MerckBiosciences AGから入手することができる。
【0145】
理論的根拠
このAffibody(登録商標)分子のN末端へのDOTAのカップリングと同様にAffibody(登録商標)分子Z00342(ZHER2:107として国際公開第2005/003156号に開示され、ときどきZHER2:342とも呼ばれる)のペプチド合成は可能であり、文献(Orlova, A
et al.(2006),Cancer Research 67:2178-2186)に記述されている。しかしながら、合成後のペプチド収率における大きな変動が観察された。ペプチドを再現よく合成する上での困難は、一次アミノ酸配列と同様にペプチドの長さの両方に関連づけることができる。また、さらに保護されたアミノ酸側鎖の反応基を持った長いペプチドは、好ましくない二次構造、即ち固相ペプチド合成を妨げ得るβシートを生成し得る(Quibell, M. & Johnson, T., in Fmoc Solid Phase Peptide Synthesis-A Practical Approach, W.C. Chan, P.D. White Eds, Oxford University Press 2000, 115-135)。ペプチド合成中二次構造形成を防ぐ1つの方法は、疑似プロリンの使用である。もしアミノ酸Yyyがセリン、トレオニン又はシステインであれば、簡略化された略記Fmoc−Xxx−Yyy−OHを有する疑似プロリンが使用できる。これらの疑似プロリンは、骨格に結合した側鎖を有する閉環したプロリン状構造を示し、そして酸処理によって正常なアミノ酸の構造に変換できる(Haack, T., & Mutter, M. Tetrahedron Letters (1992), 33(12), 1589-92)。疑似プロリンは、位置Yyy中のセリン又はトレオニンと共に、位置Xxx(Arg,Cys,His,Met,Pro,Thrを除く)における14のアミノ酸について市販されている。
【0146】
親Affibody(登録商標)分子Z00342は、一次配列に一切のトレオニン及びシステインを持たない。セリンは、単に位置33、39及び41にのみ見出される。疑似プロリン前駆体は、わずかセリン41(Q40〜S41)に対してのみ入手できる。2つの他のセリンについては、入手可能な前駆体(R32〜S33及びP38〜S39)が一切ないので、位置Xxxにあるアミノ酸が疑似プロリンの使用を妨げる。
【0147】
本発明のポリペプチドに導入される変異は、ペプチド合成を促進することを目標とするが、それに限定されるものではない。特に、位置23、42、46及び54、即ち[N23T]、[A42S]、[A46S]及び[A54S]での変異は、SPPSで確認された問題の2つを解決する能力を有し得る:それらは、疑似プロリンの使用を可能にし、アミノ酸21〜26のまわりの臨界領域(critical region)はアスパラギンをスレオニンに置換することによって、位置23に変化する。
【0148】
固相ペプチド合成(SPPS)。
Affibody(登録商標)分子X(配列番号7)のアミノ酸配列は、完全に自動化ペプチド合成機でのFmoc−Lys(Boc)−Wangポリスチレン樹脂に組み立てられた。この樹脂は、Fmoc戦略を持ったペプチドの形成に非常に適している。57個のアミノ酸(適切な側鎖の保護で)は樹脂上でカップリングされた。最終工程の、S−トリチル保護メルカプト酢酸のカップリングは手動で行った。
【0149】
工程1:固相ペプチド合成
Fmoc−Lys(Boc)−Wangポリスチレン樹脂を、撹拌機を備えたSPPS反応器中に移した。次いで、合成は樹脂のFmoc脱保護から始め、続いて以下に示される概要に記載のFmoc−Pro−OHを有するカップリング手順を続けた。この工程の後、再度、Fmoc脱保護及びその後手順に従ってアミノ酸誘導体のカップリングを続けた。イソプロピルエーテル(IPE)での樹脂の最終洗浄の後、ペプチド樹脂を減圧下のデシケーターで乾燥した。
【0150】
標準的なFmocペプチド合成と、4つの位置で疑似プロリンを使用する合成の両方が行われた。標準的なペプチド合成については、Fmocアミノ酸しか使用されなかった。Fmocアミノ酸は別として代替ペプチド合成については、以下の疑似プロリンが使用された:位置22〜23でのFmoc−Leu−Thr−OH、位置41〜42でのFmoc−Ser−Ser−OH、位置45〜46でのFmoc−Leu−Ser−OH、及び位置53〜54でのFmoc−Asp−Ser−OH。
【0151】
Fmoc脱保護手順
N−α−Fmoc保護基の開裂を達成するために、樹脂を、また、NMP中の20%ピペリジンで処理した。次いで樹脂の洗浄をNMPで行った。
【0152】
カップリング手順
Glu1へのアミノ酸誘導体Pro57の自動化カップリング。最大3当量のFmoc−AA誘導体をNMP中に溶解した。カップリングのために、ジメチルホルムアミド(DMF)中の2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムテトラフルオロボラート(TBTU)及びNMP中のsym.−コリジン(2,4,6−トリメチルピリジン)を添加した。生成溶液を、樹脂上に注ぐ前に室温で混合した。NMPを溶媒として使用した。60℃で少なくとも15分のカップリング時間の後で、樹脂をNMPで洗浄した。
【0153】
各カップリング手順の後、試薬としてのDMF及び溶媒としてのジクロルエタン中の2−(1H−7−アザベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムテトラフルオロボラート(TATU)でのカップリングを自動的に繰り返し、引き続いて無水酢酸キャッピングを行った。
【0154】
工程2メルカプト酢酸のカップリング
アシル化は、5モル当量のアミノ酸、2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3,−テトラメチルウロニウムヘキサフルオロホスファート(HBTU)及び1−ヒドロキシベンゾトリアゾール(HOBt)及び10当量のN−エチルジイソプロピルアミン(DIEA、Lancaster Synthesis, Morecambe, England製)によって行なった。S−トリチルメルカプト酢酸は、AnaSpec Inc(San Jose, CA, USA)製であった。
【0155】
工程3:残りの保護基の開裂を含む樹脂からの開裂
ペプチド樹脂を、純水、エタンジチオール(EDT)及びトリイソプロピルシラン(TIS)の存在下にトリフルオロ酢酸(TFA)処理した。室温で約2時間の開裂後に、反応混合物を約0℃に冷却した。ヨウ化アンモニウム及びジメチルサルファイドを添加して、酸化メチオニンを還元した。約0℃で更に60分の開裂の後に、形成したヨウ素をアスコルビン酸で還元した。生成物を濾別した後、冷却しながらIPE中に沈殿させ、再度濾別し、IPEで洗浄し、減圧下で乾燥した。
【0156】
HPLCによる純度分析
58個のアミノ酸の長いペプチド及びいくつかの中間体の純度は、Vydac 218 TP54(5μm、250×4.6mm)カラムを使用し、0.1%のTFA及び1%のアセトニトリルを含有する水、0.1%のTFAを含有するアセトニトリル溶液をそれぞれ溶媒A及びBとして、逆相HPLCによって決定した。カラムオーブン温度は35℃に設定した。カラムを、30分で溶媒Bを15〜45%にするグラジエント、又は30分でBを20〜50%にするグラジエントのどちらかで溶離した。UV検出は220nmで行った。純度は、面積正規化によって計算した。
【0157】
結果
疑似プロリンを使用して又はしないで合成された、Affibody(登録商標)分子X(配列番号7)の収率及び純度は、分析用逆相クロマトグラフィーによって分析した。合成の進行をフォローするために、少量の合成樹脂を、いくつかのカップリング工程後に抜き取り、所望のペプチド中間体の存在、純度及び収率を分析した。図11は、41のアミノ酸の長さのペプチド中間体(アミノ酸18〜58)の分析を示す。ペプチド合成のこの段階では、正しい配列を有する1つの明確な及び主たるペプチドピーク(保持時間=15.33分、収率49%)が、疑似プロリンを使用して合成が行われたかどうかを同定した(図11A)。しかしながら、標準的なFmoc合成は、同じサイズを有する膨大な数の小さなペプチドピーク及び2つのメインピークをもたらしたが、低収率であった。この2つのピーク(保持時間=20.82分)の1つは、正しい配列(aa18〜58)を有するペプチド中間体として同定された(図11B)。全長ペプチド(アミノ酸1〜58)は、合成が疑似プロリンを使用して行われた場合に限り得られた。図12Aは、26%の最終的なペプチドの収率を有する単一の生成物ピークを示す。しかしながら、標準的なFmoc合成は、最終的なペプチド生成物を生成できなかた。標準的な合成からの49個のアミノ酸の長さの中間体(アミノ酸10〜58)の分析は、所望の中間体が検出できなかったこと及び合成は中止されたことを明らかにした(図12B)。
【0158】
〔実施例5〕
本発明のポリペプチド及びZ00342の親水性の比較試験
要約
この実施例を形成している実験では、2つの方法:1)タンパク質の疎水性/親水性プロット;及び2)逆相HPLC溶離プロフィール;を使用し、本発明のポリペプチドの親水性の増加が記述される。
【0159】
1)タンパク質の疎水性/親水性プロット
疎水性/親水性プロットは、タンパク質の配列に沿った極性及び無極性の残基の分布を表示するために作成される。高度に疎水性の領域、例えば膜貫通部分又は親水性の配列、即ちタンパク質の表面に曝されやすい領域を予測するために、これらのプロットは一般に使用される。
【0160】
プロットは、分析の目的に適合できる移動ウインドウ付きのタンパク質配列を走査することによって作成される。例えば、7アミノ酸の移動ウインドウは、推定表面露出領域を見つけるためのよい値であると示唆されている。各位置では、ウインドウ中のアミノ酸の平均の疎水性指数が算出される。この値は、ウインドウの中間点としてプロットされる。この実施例を構成している疎水性/疎水性プロットのため、以下の2つの疎水性スケールが使われた:1)Kyte-Doolittleスケールは、正の値を達成するためのハイドロパティック(Hydropathic)領域であった(Kyte J., Doolittle R.F. (1982) J. Mol. Biol. 157:105-132)、そして2)HPLCによって求められるpH3.4での疎水性指標(Cowan R., Whittaker R.G. (1990) Peptide Research 3:75-80)。プロットは、Vector NTI Suite 9.0.0の構成要素であるプログラムBioAnnotatorwo(Invitrogen)を使用して作成された。
【0161】
E.coliにおける発現のため及び放射性核種での標識化のために配列を最適化するために、10〜12個のアミノ酸のAffibody(登録商標)分子、即ち分子の標的結合活性に関与しないアミノ酸(1つのMet9は別として)は、可能な場合は、より親水性アミノ酸及び/又は全体的な抗原性を低減するアミノ酸に変更された。
【0162】
本発明のポリペプチドに対する疎水性/親水性プロットの2つの実施例は、図13A及びBに示される。比較のために、親Affibody(登録商標)分子Z00342用の図が、図13Cに示される。
【0163】
Affibody (登録商標)分子II(配列番号5)の配列では、以下のアミノ酸の変更がなされた:
・これらの変異の4つは、非極性疎水性アミノ酸を極性アミノ酸[F5Y]、[A42S]、[A46S]及び[A54S]に交換した;
・その2つは、極性アミノ酸を無極性疎水性アミノ酸[N3A]及び[N6A]に変更した;
・その1つは、未変化の極性アミノ酸を正に荷電した極性アミノ酸[S33K]に変更した;
・その1つは、極性アミノ酸を負に荷電したアミノ酸(N43E)に変更した;
・その1つは、極性アミノ酸をより小さい極性アミノ酸(N23T)に変更した;
・その1つは、非極性疎水性アミノ酸をより小さい疎水性アミノ酸(V1A)に変更した;
・その1つは、負に荷電したアミノ酸を別の負に荷電したアミノ酸(D2E)に変更した。
【0164】
親Affibody(登録商標)分子Z00342に比べて、図13AのAffibody(登録商標)分子II用のプロットは、αヘリックス2及び3及びC末端(アミノ酸24〜58の間)を構成しているAffibody(登録商標)分子の領域における親水性を高めた(図13C)。
【0165】
Affibody (登録商標)分子X(配列番号7)の配列では、Affibody(登録商標)分子IIに関しては同一のアミノ酸の変更が行われたが、最初の3つのアミノ酸は、変更した場合はより親水性のアミノ酸に変わった:
・これらの変異の1つは、非極性疎水性アミノ酸を負に荷電したアミノ酸(V1E)に交換した;
・その1つは、負に荷電したアミノ酸を極性アミノ酸(D2S)に変更した;
・その1つは、荷電していない極性アミノ酸を負に荷電したアミノ酸(N3E)に変更した。
【0166】
図13B中のAffibody(登録商標)(配列番号7)分子Xに対するプロットは、親Affibody(登録商標)分子Z00342と比較すると、αへリックス2及び3(アミノ酸24〜58の間)及び親水性N末端でも、同じ親水性の増加を示す(図13C)。
【0167】
2)逆相HPLC溶離プロフィール
逆相(RP)クロマトグラフィーによって効率的にタンパク質を分離することができる。タンパク質の親水性/疎水性の違いは、より親水性のタンパク質がより疎水性タンパク質よりも早く、即ち低濃度のアセトニトリル溶離液で溶離するRPカラム、例えばC8又はC18カラム上で分析することができる。Affibody (登録商標)分子II(配列番号5)及び親Affibody(登録商標)分子Z00342の溶離を比較するために、以下の条件を使用した:HPLC機器:アジレント1100(Agilent);カラム:Zorbax 300SB C8 150×2.1mm、3.5μm(Agilent);温度:30℃;ロード:20μlの2.2mg/mlを有するタンパク質溶液;流量0.5ml/分;溶媒A:0.5%のトリフルオロ酢酸(TFA)水溶液;溶媒B:90%アセトニトリル+0.5%TFAの水溶液;グラジエント;0〜2分は10%のB、2〜17分は10〜70%のB、17〜18分は70〜100%のB、18〜21分は100%のB、21〜22分は100〜10%のB、22〜25分は10%のB。分析結果を図14に示す。保持時間におけるシフトは、親Affibody(登録商標)分子Z00342と比較して、Affibody(登録商標)分子IIの親水性の増加を示している。上述の全ての変異を有するこのAffibody(登録商標)分子は、親Affibody(登録商標)分子Z00342での9.860と比較すると、9.614分の保持時間で溶離する。
【0168】
実施例6
−本発明のポリペプチド及びZ00342の抗原性(IgG結合)の比較試験
要約
本発明に記載のHER2特異的ポリペプチドの1つの望ましい性質は、免疫グロブリン(Ig)との低い相互作用の可能性を示すことを意味する、低抗原性プロファイルを持つことである。この実施例では、インビトロで抗原性(IgG結合)を測定する方法が記述されており、行われたアミノ酸の変更による影響を評価するために、C末端システインに共役したAffibody(登録商標)分子I、II、DOTAを有するII及び内部のシステイン(位置42)上で共役したDOTAを有するXIを、標準的なHER2特異的Affibody(登録商標)分子Z00342と比較した。使用されたアッセイは、インビトロの抗原性(IVA)ELISAと呼ばれている。
【0169】
インビトロの抗原性(IVA)ELISA
IVA ELISAでは、96ウェルのプレートを、2μg/mlの標準コーティング試験として選ばれたHER2特異的Affibody(登録商標)分子Z00342でコーティングした。また、プレートを、2μg/mlのHER2特異的Affibody(登録商標)分子I、II、DOTAを有するII及びDOTAを有するXIでコーティングした。50μlのコーティング溶液をウェル毎に添加し、そして半面積のプレートを+4oCで一夜インキュベートした。ELISAプレートを水道水で2回洗浄し、100μlのブロッキング溶液(0.5%のカゼインを有するリン酸緩衝食塩液(PBS)、pH7.4)を各ウェルに添加した。プレートを、その後室温(RT)で1時間インキュベートして空にし、続いて、1から開始して100倍希釈の2倍希釈系列で、50μl/ウェルのさまざまな霊長類の血清を加えた。プレートを室温で1時間インキュベートし、その後PBS+0.05%のTween(PBS−T)で5回洗浄した。検出には、ヤギ抗ヒトIg−HRP抗体を、1/5000に希釈して使用した(Southern Biotechnology)。プレートを、室温で50分間インキュベートし、その後PBS−Tを使用して5回洗浄工程にかけた。発色については、50μlのImmunoPureTMB基質(Pierce)を添加し、プレートを室温で12分間暗所でインキュベーションした。停止液(2MのH2SO4)を加えて呈色反応を止め、吸光度を450nmで測定した。
【0170】
IVA値の計算
上記に示すように、続いてIVA・ELISAを行った。得られた滴定曲線を、XY非線形回帰式を使用してグラフにプロットし、OD0.3での希釈値を求めた。OD0.3での標準希釈液値を100に設定し、それを基準に関連付けることによって、各サンプルのIVA値を算出した。従って、100以下のIVA値は、標準的なZ00342と比較して、その特別のHER2特異的ポリペプチドについては、より低い程度の霊長類Ig結合を反映している。4つの異なるHER2特異的ポリペプチド、即ち、Affibody(登録商標)分子I、II、DOTAを有するII及びXIを有するDOTAについて、インビトロ抗原性を評価した。標準的なタンパク質と比較して、これらのポリペプチドは低いインビトロの抗原性プロファイルを有することを示した。結論は、行われたアミノ酸変更は、免疫グロブリンとの相互作用の可能性の減少をもたらしたということである。
【0171】
【表3】
【技術分野】
【0001】
本発明は、ヒト上皮細胞成長因子受容体2(Human Epidermal Growth Factor Receptor
2)(以下、HER2と称す)に結合する新規なポリペプチドに関連する。本発明は、診断用薬及び/又は薬物としてのHER2結合ポリペプチドの使用、より詳しくは、HER2の過剰発現によって特徴付けられるがんの形態の診断及び/又は治療のための診断用薬及び/又は薬物としてのその使用に関する。
【背景技術】
【0002】
HER2とがんの疾病における役割
HER2がん原遺伝子は、HER2タンパク質又は受容体(非特許文献1)として知られている185kDa細胞表面受容体タンパク質の産生をコードする。この遺伝子は、また、しばしばneu、HER2/neu又はc-erbB-2と呼ばれる。neuは、最初はエチルニトロソ尿素で処理されていたラット中で発見され、そしてこの遺伝子の変異を示した(非特許文献2)。neuの変異バージョンは、構成的に活性な形態の受容体の産生をもたらし、低コピー数で細胞を形質転換できる強力ながん遺伝子を構成する(上記の非特許文献1を参照)。
【0003】
正常細胞は、組織特異性のパターンにおけるそれらの細胞膜上で小量のHER2タンパク質を発現する。HER2に対する一切の既知リガンドは解明されていない;しかしながら、HER2は、これらの受容体に対するリガンドと複合したHER1(上皮細胞成長因子受容体(EGFR))、HER3及びHER4とヘテロ二量体を形成することが示されている。そのようなヘテロ二量体形成は、活性化されたHER2受容体が増殖シグナルを細胞外から核へ伝達し、従って正常な細胞の成長と分裂の状況を制御することにつながる(非特許文献3)。
【0004】
腫瘍細胞では、DNA複製系でのエラーが、単一の染色体での遺伝子の複数のコピーの存在をもたらす可能性があり、それは遺伝子増幅として知られる現象である。HER2遺伝子の増幅は、この遺伝子の転写の増加をもたらす。これは、HER2mRNAレベルが上昇し、HER2タンパク質の随伴合成を増加し、これらの腫瘍細胞の表面でのHER2タンパク質の過剰発現をもたらす。この過剰発現は、隣の正常細胞に見られるものより10倍から100倍大きいHER2タンパク質レベルをもたらすことができる。このことが、ひいては細胞分裂の増加と付随的に高い細胞増殖の速度をもたらす。HER2遺伝子の増幅は、がん表現型への正常細胞の形質転換に関与している(上記の非特許文献1;上記の非特許文献3を参照)。
【0005】
HER2タンパク質の過剰発現は、HER2のホモダイマーの形成をもたらすと考えられ、それはひいては構成的に活性な受容体をもたらす(非特許文献4)。これらの条件下では、成長促進シグナルは、リガンドが無い状態で連続的に細胞に伝達され得る。その結果として、複数の細胞内シグナル伝達経路が活性化されるようになり、無秩序な細胞増殖、及び、ある場合には、がん遺伝子形質転換をもたらす(上記の非特許文献1を参照)。このように、増殖因子受容体によって仲介されるシグナル伝達機構は、細胞複製及び腫瘍増殖を抑制する重要な標的である。
【0006】
乳がんは、合衆国の女性の中で最も一般的な悪性腫瘍であり、2001年には19万2,200の新規症例が発生したと推定されている(非特許文献5)。全ての乳がん患者の約25%に、その増幅によるHER2遺伝子の過剰発現がある(非特許文献6)。HER2タンパク質のこの過剰発現は、エストロゲン受容体陰性状態を含む、いくつかの陰性の予後変数、高いS期分画、陽性結節状態、変異p53、及び高い核異型度と相関する(上記の非特許文献7を参照)。非特許文献6によれば、HER2遺伝子の増幅は、リンパ節陽性患者の短縮した無病生存及び短縮した全生存と強く相関することが分かった。
【0007】
このような理由で、乳がんの病因及び治療におけるHER2の役割への研究をさらに進めることは、重要な目標であったし、依然として然りである。
HER2と相互作用する分子の特定は、この取り組みの1部分を形成する。
【0008】
前臨床のインビトロの検討は、HER2活性の抑制が腫瘍細胞増殖に影響する可能性があったかどうかを調べている。対照モノクローナル抗体による投与に比較し、4D5、いくつかのマウス抗HER2モノクローナル抗体の1つ、によるHER2タンパク質を過剰発現しているSK−BR−3乳がん細胞の治療は、本当に腫瘍細胞増殖を抑制した。HER2を過剰発現するヒト乳がん及び卵巣がん(異種移植片)を有するマウスへの4D5の投与は、腫瘍の無い生存期間を延長した。同様の検討は、マウスにおけるヒト胃がん異種移植片で抗HER2ヒト化モノクローナル抗体の増殖抑制を実証した(非特許文献8)。
【0009】
腫瘍細胞表面に豊富に存在するHER2タンパク質を抗体で抑制する試みの中で、1つの療法は、ここ数年の間に市販されるようになった。このように、モノクローナル抗体4D5のヒト化変異体、又はトラスツズマブ(trastuzumab)は、Herceptin(登録商標)の商標名のもと、F Hoffman-La Roche 及び Genentechによってこの目的で市販されている。
【0010】
このように、HER2の過剰発現が乳がんについて記載されている。それは、また、とりわけ卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん(非特許文献9)及び食道におけるがん(非特許文献10)につながっている。
【0011】
HER2タンパク質の過剰発現によって特徴付けられるがんに対する抗体療法によって示される明らかな優位性にもかかわらず、種々のファクターには抗体の有効性(非特許文献11を参照)を減らす可能性があるという事実は残る。それらには以下のものがある:(1)大きい固形腫瘍又は脳のような生命の維持に必要な領域への抗体の制限的浸透;(2)血管透過性の減少により、標的部分への抗体の溢血の減少;(3)健常組織に対する交差反応性及び抗体の非特異的結合で、標的効果が低減する;(4)不均質な腫瘍取り込みが未治療区域をもたらす;(5)注入した抗体の代謝の増加で、治療効果が低減する;及び(6)HAMA及びヒト抗ヒト抗体の急速生成で、治療抗体を不活性化する。
【0012】
加えて、毒性作用は、がんのための治療抗体の開発の大きな障害である(非特許文献12、非特許文献13、非特許文献14)。健常組織との交差反応性は、非抱合型の(裸の)抗体に対してかなりの副作用を起こす可能性があり、その副作用が毒素又はラジオアイソトープを有する抗体の抱合で高められる可能性がある。免疫介在性合併症は、肺毒性作用、時折の中枢及び末梢神経系合併症、並びに肝及び腎機能の減少からの呼吸困難を含む。時として、HER2ターゲッティング抗体のトラスツズマブに関連している心毒性作用のような、予想外の毒性合併症が見られる(非特許文献15)。同位体抱合型抗体による放射免疫療法は、また、骨髄抑制を引き起こす可能性がある。
【0013】
現在使用されている抗がん抗体の最近の臨床的及び商業的成功にもかかわらず、このように相当な数の重要な疑問が、抗体の使用の将来に関して残っている。結果として、HER2に対して匹敵する親和性のある試薬の継続的な提供は、疾患の診断と治療におけるそのような分子の利用の提供と同様に、この分野での相当な関心事として残る。
【0014】
HER2結合のZ変異分子
ブドウ球菌プロンテインA(SPA)のドメインBから誘導された、タンパク質Zに関連する分子(非特許文献16)は、異なる相互作用ターゲットを使用している無作為化されたそのような分子のライブラリーから選択されている(例えば、特許文献1、非特許文献17、特許文献2、特許文献3、特許文献4、特許文献5を参照)。
【0015】
特許文献6には、相当な数のHER2と相互作用する能力のあるZの変形が開示されている。
【先行技術文献】
【特許文献】
【0016】
【特許文献1】国際公開公報第95/19374号
【特許文献2】国際公開公報第2005/000883号
【特許文献3】国際公開公報第2005/075507号
【特許文献4】国際公開公報第2006/092338号
【特許文献5】国際公開公報第2007/065635号
【特許文献6】国際公開公報第2005/003156号
【非特許文献】
【0017】
【非特許文献1】Hynes NE et al (1994), Biochim Biophys Acta1198: 165-184
【非特許文献2】Shih C et al (1981), Nature 290: 261-264)
【非特許文献3】Sundaresan S, et al (1999), Curr Oncol Rep1: 16-22)
【非特許文献4】Sliwkowski MX et al (1999), Semin Oncol, 26 (4 Suppl12): 60-70)
【非特許文献5】Greenlee R, et al (2001), CA Cancer J Clin 51: 15-36
【非特許文献6】Slamon DJ, et al (1989), Science 244: 707-712
【非特許文献7】Sjogren S, et al (1998), J Clin Oncol, 16(2): 462-469
【非特許文献8】Pietras RJ, et al (1994), Oncogene 9: 1829-1838
【非特許文献9】Holbro, et al, Annu. Rev. Pharmacol. Toxicol. 2004. 44: 195-217
【非特許文献10】Ekman, et al, Oncologist 2007 ; 12 : 1165-1177; see, in particular, pages 1170-1171
【非特許文献11】Reilly RM, et al (1995), Clin Pharmacokinet 28: 126-142
【非特許文献12】Carter P (2001), Nat Rev Cancer 1: 118-129
【非特許文献13】Goldenberg DM (2002), J Nucl Med 43: 693-713
【非特許文献14】Reichert JM (2002), Curr Opin Mol Ther 4: 110-118
【非特許文献15】Schneider JW, et al (2002), Semin Oncol 29 (3 suppl 11): 22-28
【非特許文献16】Nilson B, et al (1987), Protein Engineering 1: 107-133
【非特許文献17】Nord K, et al (1997), Nature Biotechnology 15: 772-777
【発明の概要】
【発明が解決しようとする課題】
【0018】
本発明の目的は、HER2への特異的結合によって特徴付けられるポリペプチドの提供を通して、HER2に対する親和性を有する試薬の継続的提供の要望を満たすことにある。
【0019】
本発明の関連する目的は、殆ど又は全く非特異的結合を示さない、HER2結合ポリペプチドである。
【0020】
本発明の別の目的は、容易に融合ポリペプチドの部分として使用できる、HER2結合ポリペプチドを提供することにある。
【0021】
別の目的は、既存の抗体試薬で経験する1つ又はそれ以上の公知の問題を解決する、HER2結合ポリペプチドの提供である。
【0022】
本発明の更なる目的は、治療用途での使用を可能にするHER2結合ポリペプチドを提供することである。
【0023】
本発明の更なる目的は、診断用途での使用を可能にするHER2結合ポリペプチドを提供することにである。
【0024】
関連する目的は、HER2タンパク質の過剰発現によって特徴付けられるがん疾患の臨床状況における、治療、阻害及び/又はターゲッティングの新規な形態を見出すことである。
【0025】
本発明の別の目的は、化学的ペプチド合成によって簡単に合成されるHER2結合ポリペプチドを提供することにある。
【0026】
関連する目的は、公知のHER2結合剤に対する改良された安定性を示す、HER2結合ポリペプチドを見出すことである。
【0027】
本発明の更に別の目的は、インビボで哺乳動物に使用する場合、低抗原性を示すHER2結合ポリペプチドを得ることである。
【0028】
本発明の別の目的は、哺乳動物への投与時に改善された生体内分布を有する、HER2結合ポリペプチドを提供することである。
【課題を解決するための手段】
【0029】
これらの及び他の目的は、添付の特許請求の範囲に記載されるように、本発明のさまざまの態様によって明らかになる。このように、第1の態様では、本発明は、以下の式:
EX1RNAYWEIALLPNLTNQQKRAFIRKLYDDPSQSSELLX2EAKKLNDSQ
式中、位置2におけるX1はM、I又はLであり、位置39におけるX2はS又はCである;
(配列番号1)
又は、ある場合にはより好ましくは
YAKEX1RNAYWEIALLPNLTNQQKRAFIRKLYDDPSQSSELLX2EAKKLNDSQ
式中、位置5におけるX1はM、I又はLであり、位置42におけるX2はS又はCである;
(配列番号2)
又は、ある場合にはより好ましくは
AEAKYAKEX1RNAYWEIALLPNLTNQQKRAFIRKLYDDPSQSSELLX2EAKK LNDSQ
式中、位置9におけるX1はM、I又はLであり、位置46におけるX2はS又はCである;
(配列番号3)
又は、ある場合にはより好ましくは
ESEKYAKEX1RNAYWEIALLPNLTNQQKRAFIRKLYDDPSQSSELLX2EAKK LNDSQ,
式中、位置9におけるX1はM、I又はLであり、位置46におけるX2は、S又はCである;
(配列番号4)
を含むアミノ酸配列を有するポリペプチドを提供する。
【発明を実施するための形態】
【0030】
これらに従って、本発明者は、配列番号配列番号1〜4の何れか1つを含むポリペプチドが、先に述べられたそれらのポリペプチドの標的HER2に結合する能力を保持しながら、例えば、特許文献6に開示されたHER2結合ポリペプチドに比べて、驚くべき優位性を示すことを見出した。本発明のポリペプチドの1つ又はそれ以上の実施態様によって示された、そのような優位性の非限定的な例は、以下のとおりである:
・本発明のポリペプチドは、アスパラギン、アルギニン、アスパラギン酸及びメチオニンのようなポリペプチド配列の化学合成において、低い収率及び成功率のような問題を引き起こす可能性があったアミノ酸残基を殆ど含まない。
・本発明のポリペプチドは、表面の疎水性を付与する、従って先に述べた、関連のHER2結合ポリペプチドより親水性の特性を持っているアミノ酸残基を殆ど含まない。このことは、低い溶解性及び凝集性に伴う問題が殆どないことを意味する。理論にこだわることを望むわけではないが、より親水性の特性が、肝胆道経路(肝臓を経由した排泄)からより望ましい腎臓経路(腎臓を経由した排泄)に向けて、投与時のポリペプチドの生体内分布を宿主にシフトするように作用することは、現在も同じように信じられている。
・本発明のポリペプチドは、メチオニン、アスパラギン及びジペプチドのアスパラギンープロリンのようなポリペプチドの安定性の問題に関連するアミノ酸残基を殆ど含まない。メチオニンは酸化を受けやすく、アスパラギンは脱アミド化を受けやすく、そしてアスパラギン−プロリン結合は分裂を受けやすく、それ故に、それらは最終製品の不均一性に寄与する。
・本発明のポリペプチドは、同様の配列構成において、VH3からの重鎖可変領域を含んでいる免疫グロブリンとの相互作用を高めることが分かっているアミノ酸残基を欠いている(Silverman G.J., Int. Rev. Immunol. 1992;9(1):57-78)。理論にこだわることを望むわけではないが、本発明のポリペプチド中のそのようなアミノ酸残基の置換が、同じものの投与時のポリペプチドの宿主に対する抗原性を減らすことは、今では信じられている。
【0031】
本発明のポリペプチドは、さまざまな適用におけるHER2に対する抗体の代替物としての応用を見出す。非限定的な例として、それらは、HER2過剰発現で特徴付けられるがんの治療に、細胞表面のHER2に結合することによって細胞シグナリングを抑制することに、インビボ及びインビトロの両方におけるHER2過剰発現で特徴付けられるがんの診断に、HER2を過剰発現している細胞に試薬のターゲッティングを行うことに、HER2の検出の組織化学的方法に、及び分離方法及び他の適用に有用である。本発明のポリペプチドは、試薬のHER2に対する親和性に頼るいかなる方法にも有用であることを立証することができる。このように、本ポリペプチドは、そのような方法における検出試薬、捕捉試薬又は分離試薬として、インビボ又はインビトロの診断用の診断薬として、又はそれ自体で治療薬として、又は他の治療薬若しくは診断用薬をHER2タンパク質にターゲットする方法として、使用することができる。インビトロで本発明のポリペプチドを用いる方法は、マイクロタイタープレート中で、プロテインアレイ中で、バイオセンサー表面上で、組織切片上で等のさまざまな型式で実施することができる。
【0032】
本発明の範囲を逸脱しないで、ポリペプチドを意図する特定用途に合わせるために、本発明にに記載のポリペプチドのさまざまな修飾及び/又は付加が実施されてもよい。そのような修飾及び付加は、以下により詳細に記載されており、同じポリペプチド鎖に含まれる更なるアミノ酸、又は本発明のポリペプチドに化学的に共役する若しくは別の様式で結合する標識及び/又は治療薬を含んでもよい。
【0033】
「HER2に対する結合親和性」、「HER2結合」等のような発現とは、例えば、Biacore(登録商標)機器(GE Healthcare)におけるような、表面プラズモン共鳴技術を用いて試験され得るポリペプチドの性質を意味する。HER2結合親和性は、HER2又はその断片、例えば、細胞外ドメイン又はその融合タンパク質が装置のセンサーチップ上に固定化され、検査されるポリペプチドを含む試料がそのチップ上を移動するような実験で試験することができる。或いはまた、検査されるポリペプチドが装置のセンサーチップ上に固定され、そしてHER2を含む試料、又はその断片、例えば、細胞外ドメインが、そのチップ上を通過する。次いで、当業者は、少なくともHER2に対するポリペプチドの結合親和性の少なくとも定性的測定を達成するために得られるセンサーグラム(sensorgrams)を解釈することができる。もし、例えば、相互作用の特定のKD値を確立する目的を持って定量的測定が求められる場合は、再び表面プラズモン共鳴方法を使用することが可能である。結合値は、例えば、Biacore(登録商標)2000装置(GE Healthcare)で定義することができる。HER2、又はその断片、例えば、細胞外ドメインは、装置のセンサーチップ上に固定され、親和性が決定されるべきポリペプチドの試料は連続希釈によって調製され、無作為の順序で注入される。次いで、KD値は、例えば、装置メーカーによって提供されるBIAevaluation 3.2ソフトウェアの1:1ラングミュア結合モデルを用いて、その結果から計算することができる。
【0034】
本発明は、また、上記のHER2結合ポリペプチドが、更なるアミノ酸残基がどちらかの末端に付加している、HER2結合ドメインとして存在するポリペプチドを包含する。これらの更なるアミノ酸残基は、ポリペプチドによりHER2の結合における役割を果たし得るが、同様に、例えば、ポリペプチドの産生、精製、安定化、カップリング又は検出の1つ又はそれ以上に関連する、他の目的をもよく果たし得る。そのような更なるアミノ酸残基は、化学カップリングのために追加される1つ又はそれ以上のアミノ酸残基を含んでもよい。この1つの例は、ポリペプチド鎖の中の一番最初の又は一番最後の位置、即ち、N又はC末端におけるシステイン残基の付加である。化学カップリングに使用されるシステイン残基は、また、タンパク質ドメイン上で、好ましくは標的結合に関与しない表面の部分の上で、別のアミノ酸の置換によって導入され得る。そのような更なるアミノ酸残基は、タグに固有の抗体との相互作用用のヘキサヒスチジル (His6)タグ、若しくは「myc」タグ又は「FLAG」タグのようなポリペプチドの精製又は検出のための「タグ」を含み得る。当業者は、他の選択肢を知っている。
【0035】
更なるアミノ酸残基を有するポリペプチドの具体的実施態様において、本発明は、下記のアミノ酸配列:
AEAKYAKEMRNAYWEIALLPNLTNQQKRAFIRKLYDDPSQSSELLSEAKK LNDSQAPKVDC
(配列番号5)
を含むHER2結合ポリペプチドを提供する。
【0036】
更なるアミノ酸残基を有するポリペプチドの別の具体的実施態様において、本発明は、下記のアミノ酸配列:
ESEKYAKEMR NAYWEIALLP NLTNQQKRAF IRKLYDDPSQ SSELLSEAKK LNDSQAPK
(配列番号6)
を含むHER2結合ポリペプチドを提供する。
【0037】
上で議論された「更なるアミノ酸残基」は、また、第1の、HER2結合ドメイン、又は別の結合機能、又は酵素機能、又は蛍光機能、又はそれらを混合したものと同じ結合機能のような、いかなる所望の機能を有する1つ又はそれ以上のポリペプチドドメインを構成してもよい。
【0038】
このように、本発明は、配列番号1〜6の何れか1つを含むポリペプチドの多量体を包含する。それは、例えば、がんの診断や治療のために又はHER2の精製の方法において、本発明のポリペプチドを使う場合、本発明の1つのポリペプチドで可能であるよりも更に強いHER2の結合を得ることに、関心が持たれるかもしれない。この場合、ポリペプチドの、二量体、三量体又は四量体のような、多量体の提供は、必要な結合力効果をもたらし得る。多量体は、本発明に記載の適する数のポリペプチドから成るり得る。本発明の多量体中の結合ポリペプチド「ユニット」は、公知の有機化学の方法を使って共有結合によって連結され得るし、又は、ポリペプチドの組換え発現のシステムにおいて1つ又はそれ以上の融合ポリペプチドとして表現されたり、又はいかなる他の方法ででも、直接に又はリンカー、例えば、アミノ酸リンカーを経由して連結され得る。
【0039】
さらに、配列番号1〜6の何れか1つを含むポリペプチドが、第1のドメイン、又は第1の部分を構成し、そして第2の部分及び更なる部分が結合性HER2以外の機能を有する「異種遺伝子型の」融合ポリペプチドも意図され、そして本発明の範囲に属する。融合ポリペプチドの第2の部分及び更なる1つ又は複数の部分は、HER2とは別のターゲット分子に対する親和性を有する結合ドメインを含み得る。その結果は、従って、少なくとも1つのHER2結合ドメイン及び該他の標的分子に対する親和性を有する少なくとも1つのドメインを有する融合ポリペプチドである。これは、治療剤又は捕捉、検出又は分離試薬として使用されるような、いくつかの生物工学的応用に使われ得る多特異的試薬を作成することを可能にする。少なくとも1つのポリペプチドドメインが配列番号1〜6のいずれか1つの配列を含むような、そのようなポリペプチドの多特異的多量体の製造は、いくつかのHER2結合性「ユニット」の多量体に対して上記のように行われ得る。第2の又は更なる1つの又は複数の部分は、「Zドメイン」又は「タンパク質Z」の変異体等の黄色ブドウ球菌(Staphylococcus aureus)のタンパク質A由来のドメインの変異体であり得る(Nilsson B et al (1987), Protein Engineering 1: 107-133を参照)、又は標的に対する結合親和性を有する無関係の、天然に存在する、又は組み換えのタンパク質(又は天然に存在する又は組み換えの蛋白の結合能力を保持しているその断片)を含む。ヒト血清アルブミンに対する親和性を有し、本発明のポリペプチドとの融合相手として使用され得るそのような結合タンパク質の例は、GA1、GA2又はGA3ドメインのような連鎖球菌菌株(Streptococcus strain)G148(Nygren P-A et al (1988), Mol Recogn 1: 69-74)のタンパク質Gからのアルブミン結合ドメインの1つである。プロテインGのGA3ドメインは、また、ABD、即ち、アルブミン結合ドメイン(albumin binding domain)で示される。配列番号1〜6のいずれか1つの配列を含むHER2結合ポリペプチドと連鎖球菌タンパク質Gのタンパク質結合ドメインの間の融合ポリペプチドは、このように本発明の範囲に属する。本発明のポリペプチドが診断薬、治療薬として、又は標的薬としてヒト被験者に投与される場合、血清アルブミンに結合する部分へのその融合は有益であることを証明することができる。その場合、単離時のHER2結合部分の半減期と比較して、そのような融合タンパク質のインビボでの半減期は延びる可能性が高い(この原理については、例えば、国際公開公報第91/01743号に記載されている)。同様に、国際公開公報第2005/097202号で明らかにされた原理に従って、融合ポリペプチドは、単離時のHER2結合部分より低い免疫原性を示すようである。
【0040】
融合ポリペプチドの創出に対する他の可能性が同様に意図される。このように、本発明の第1の態様に記載のポリペプチドは、第2の又は更なる部分等に共有結合的にカップリングすることができ、それは、標的結合に加えて、又は標的結合の代わりに、他の機能を示す。1つの例は、配列番号1〜6のいずれか1つの配列を含む1つ又はそれ以上のポリペプチドと、レポーター又はエフェクター部分として機能している酵素活性のポリペプチドとの間の融合である。融合タンパク質を形成する配列番号1〜6のいずれか1つの配列を含むポリペプチドにカップリングされ得るリポーター酵素の例は、当業者に知られており、そしてβ−ガラクトシダーゼ、アルカリホスファターゼ、西洋ワサビペルオキシダーゼ、カルボキシペプチダーゼのような酵素を含む。本発明の融合ポリペプチドの、第2の、及び更なる1つ又は複数の部分に対する他のオプションは、緑色蛍光タンパク質、赤色蛍光タンパク質、ルシフェラーゼ及びその変異体のような蛍光性ペプチドを含む。
【0041】
本発明の融合ポリペプチドの、第2の、及び更なる1つの又は複数の部分に対する他のオプションは、治療への適用のための部分等を含む。治療への適用においては、他の分子も、また、他の手段による本発明のポリペプチドに、共有結合で又は非共有結合でカップリングされ得る。非限定的な例は、エフェクター酵素(例えば、カルボキシペプチダーゼ)の方向に本発明のポリペプチドを用いるADEPT(抗体指向酵素プロドラッグ治療)(antibody-directed enzyme prodrug therapy)適用のための酵素;免疫系のエフェクター細胞及び他のコンポーネントの補充のためのタンパク質;IL−2、IL−12、TNFα、IP−10のようなサイトカイン;組織因子、フォン・ビルブラント因子のような凝固促進因子;リシンA、シュードモナス外毒素、カルケアマイシン、メイタンシノイドのような毒素;アウリスタチアナログ、ドキソルビシンのような毒性の小さな分子;を含む。
【0042】
本発明に記載のHER2結合ポリペプチドを取り込んでいる融合タンパク質の上記の記述に関して、第1の、第2の、更なる部分の命名は、一方ではHER2結合部分又は該部分と、他方では他の機能を発揮している部分との間を区別する明快な理由のためになされていることも注目すべきである。これらの命名は、融合タンパク質のポリペプチド鎖の中の、さまざまなドメインの実際の順序を示すよう意図されていない。従って、例えば、該第1の部分が、融合タンパク質のN末端に、中央に、又はC末端に制約なしに出現する可能性がある。
【0043】
本発明は、上記のHER2結合ポリペプチドが、例えば、ポリペプチドの検出の目的のために、少なくとも1つのフルオロフォア、ビオチン又は放射性同位元素のような標識基を備えているポリペプチドを含む。特に、本発明は、上記のHER2結合ポリペプチドの放射性キレート、及び放射性金属のような放射性核種から成る放射性標識ポリペプチドを含む。
【0044】
放射性核種の大部分は金属の性質を有し、そして金属は、一般的には、タンパク質及びペプチド中に存在する元素との安定な共有結合を形成することができない。このために、標的タンパク質を放射性金属で標識することが、キレート剤、キレートと呼ばれる、金属と非共有化合物を形成する、多座配位子を使って行われる。本発明のポリペプチドの実施態様において、放射性核種の取り込みは、キレート環境の提供によって可能になり、それによって放射性核種はポリペプチドに配位し、キレート化し又は錯体化され得る。
【0045】
本発明のポリペプチドの1つの具体的実施態様において、N3Sキレート剤として特徴付けられる四座配位子キレート環境を含む。N3Sという用語が意味するように、そのようなキレート剤の4つの付着している基は、3つの窒素原子及び1つの硫黄原子から形成される。N3Sキレート剤では、、N及びS原子は空間的に配置されて、放射性金属の錯体化又は付着のために適する「ポケット」を提供する。
【0046】
N3Sキレート剤は、ポリペプチドのアミノ酸配列中のアミノ酸残基を好適に選択して提供され得る。
【0047】
例えば、N3Sキレート環境は、本発明のポリペプチドのN末端に、メルカプトアセチ
ルのカップリングを通して供給され得る。この実施態様においては、メルカプトアセチルが必要なS原子を与え、一方、ペプチド骨格中の最初の3つのN原子が四座配位子キレート剤の3つのN原子を構成する。好ましくは、メルカプトアセチルが、最も多くのN末端アミノ酸配列である、配列番号4のアミノ酸配列を含むポリペプチドにカップリングされ、その結果、下記のアミノ酸配列:
maESEKYAKEX1RNAYWEIALLPNLTNQQKRAFIRKLYDDPSQSSELLX2EAKK LNDSQ、
式中、maはメルカプトアセチル、位置9のX1はM、I又はLであり、位置46のX2はS又はCである;
を含むポリペプチドになる。これの特に好ましい実施態様では、ポリペプチドが下記の配列:
maESEKYAKEMR NAYWEIALLP NLTNQQKRAF IRKLYDDPSQ SSELLSEAKK LNDSQAPK、
式中、maは、メルカプトアセチルである;
(配列番号7)
を有する。
【0048】
代わりの実施態様においては、3個のN基は、ポリペプチド鎖中の3つの連続したペプチド結合のN原子によって提供されることができ、その内、最後のものはシステイン残基であり、その側鎖にSH基を含む。システイン側鎖のS原子は、N3Sキレート剤におけるS原子を構成する。別の言葉で言えば、N3Sキレート剤がトリペプチド配列XXCの形で提供され、ここで、Xは、いかなるアミノ酸残基であってもよい。そのようなトリペプチド配列は、、1つ又はそれ以上の更なるアミノ酸として、最初に又は追加して、本発明のポリペプチドの中に含まれる。システイン残基が配列番号5のC末端に位置しているもの、又は追加のシステインが配列番号1〜4又は6〜7のいずれか1つのC末端に正しく位置していることが好ましい。システイン残基は、また、1つ又はそれ以上の他のアミノ酸残基がその後に続いてもよい。
【0049】
N3Sキレート剤として特徴付けられる四座配位子キレート環境を含み、3つの連続したペプチド結合のN末端及び窒素原子にカップリングしたメルカプトアセチルか、ポリペプチドのC末端で3つの連続したペプチド結合とシステイン残基の窒素原子のどちらかによって提供される本発明のポリペプチドは、HER2結合ポリペプチドの放射性キレート及び医用画像に適した放射性核種から成る放射性標識ポリペプチドを提供するために使用することができ、そして該放射性核種は99mTc (Engfeldt et al (2007) Eur. J. Nucl.
Med. Mol. Imaging 34(5):722-22; Engfeldt et al (2007) Eur. J. Nucl. Med. Mol. Imaging 34(11):1843-53)であり、又は治療に適する放射性核種を有しており、該放射性核種は186Re及び188Reから成るグループから選択される。いくつかの実施態様において、放射性核種は、キレート環境を介してHER2結合ポリペプチドと錯体を形成する。
【0050】
本発明のポリペプチドが2つ、3つ又はそれ以上のXXCトリペプチドを含む場合、それは、好ましくは放射性核種を錯体化するのに使われる末端のものである。ポリペプチドがこのような方法で放射性標識される場合、他のXXCトリペプチドは、好ましくは保護されている。
【0051】
本発明の放射性標識ポリペプチドの1つの特に好ましい実施態様は、アミノ酸配列:
maESEKYAKEMR NAYWEIALLP NLTNQQKRAF IRKLYDDPSQ SSELLSEAKK LNDSQAPK
式中、maはメルカプトアセチルである;
(配列番号7)
を有するHER2結合ポリペプチドの放射性キレート及び99mTcである。
【0052】
ポリペプチドのN末端にあるmaESE又はC末端にあるVDCを使用してキレート化した、99mTcの仮説の構造は、それぞれ以下のものである。
【化1】
【0053】
本発明のポリペプチドの代替実施態様では、ポリアミノポリカルボキシラート・キレート剤を使用して放射性核種を組み込む。好ましくはその場合、ポリペプチドは少なくとも1つのシステインを含み、最も好ましくはたった 1つのシステインを含む。システインは、本発明のポリペプチドに最初に存在していても、又は1つ又はそれ以上の更なるアミノ酸として後の段階で追加されてもよい。
【0054】
当業者は、また、ペプチド合成中ポリペプチドに部位特異的に結合できる、又は放射性標識のための公知の共役化学を使って組換え的に産生されるポリペプチドに共役させることができる、「裸の」Me、Me=O、O=Me=O、Me≡N、Me(CO)3のようなコア(core)、又はHYNIC−Me−co−リガンド・コア(ここで、MeはTc又はReのラジオアイソトープである)をキレートする能力がある、多くの他のキレート剤の予測を行うことができよう。好ましくは、そのようなキレート剤は親水性の特性を有する。そのようなキレート剤の良い概説は、Liu S and Edwards DS (1999) Chem Rev. 99(9):2235-68により提供されている。
【0055】
誰でもポリアミノポリカルボキシラート・キレート剤の2つのクラス:大環状及び非環式、を区別することができる。
【0056】
インジウム、ガリウム、イットリウム、ビスマス、放射性アクチニド及び放射性ランタニドのラジオアイソトープ用に最も一般的に使用される大環状キレート剤は、DOTA(1,4,7,10−テトラアザシクロドデカン−1,4,7,10−四酢酸)(以下を参照)のさまざまな誘導体である。本発明のポリペプチドとのキレート化剤として使用するために適した、いくつかのさまざまのDOTAベースの化合物は、例えばMacrocyclics Inc., USAから市販されており、例を以下に示す。
【化2】
【0057】
この図式では、AはDOTAであり、Bはアミノ反応性の4−イソチオシアナート−ベンジル−DOTA、CはDOTA−TFPエステル、そしてDはチオール反応性のマレイミド−モノ−アミドDOTAである。
【0058】
高い動力学的不活性、即ちDOTAからの金属の解離の遅速性は、放射性核種の安定結合に好都合である。しかし、会合遅速性のため、標識のためには高温が必要となる。この理由のために、60〜90℃への加熱に比較的敏感でないDOTA誘導体が、短いペプチドの標識に広く使用される。
【0059】
本発明におけるキレート剤として使用するための1つの好ましい誘導体は、1,4,7,10−テトラアザシクロドデカン−1,4,7−トリス酢酸−10−マレイミドエチルアセトアミドである。
【0060】
上に開示したように、本発明のポリペプチドは、例えばアミノ酸配列番号5の配列を含み得る。より格別な実施態様では、ポリペプチドは配列番号5を含み、そしてアミノ酸残基C61にカップリングしたテトラアザシクロ化合物を有する。特に好ましい実施態様では、テトラアザシクロ化合物が、残基C61のSH基の、テトラアザシクロ化合物のマレイミド部分との反応を通じてポリペプチドにカップリングされている、1,4,7,10−テトラアザシクロドデカン−1,4,7−トリス酢酸−10−マレイミドエチルアセトアミドである。本発明の1つの特に好ましい実施態様は、従って、1,4,7,10−テトラアザシクロ−ドデカン−1,4,7−トリス−酢酸−10−マレイミドエチルアセトアミドにカップリングしたアミノ酸配列:
AEAKYAKEMRNAYWEIALLPNLTNQQKRAFIRKLYDDPSQSSELLSEAKK LNDSQAPKVDC
(配列番号5)
を含むHER2結合ポリペプチドである。
【0061】
最も一般的に使用される非環状ポリアミノポリカルボキシラート・キレート剤は、以下の図式に示されるそれらのようなDTPA(ジエチレントリアミン五酢酸)のさまざまの誘導体であり、ここで、AはDTPAであり、Bはアミノ反応性誘導体イソチオシアナートベンジル−DTPAであり、Cはアミノ反応性誘導体半硬質の2−(パラ−イソチオシアナートベンジル)−6−メチル−DTPS(IB4M)であり、Dはアミノ反応性誘導体CHX−A”−DTPAである。
【化3】
【0062】
DTPAの骨格修飾した半硬質の変異体は、例えば、Zevalin(登録商標)の90Yでの標識化にとって適切な安定性を与えることが見出されている。非環式キレート剤が大環状のものより不活性の程度が弱く、従ってより不安定であるけれども、その標識化は周囲温度でも十分に迅速である。このために、それらは、加熱に耐えることができないモノクローナル抗体の標識化には好ましいかもしれない。ポリアミノポリカルボキシラート・キレート剤のターゲッティングタンパク質とペプチドに対するカップリング用の詳細なプロトコルは、Cooper及び共同研究者(Nat.Protoc.1: 314-7.2006)並びにSosabowski及びMather (Nat.Protoc1: 972-6(2006))によって公表されている。
【0063】
ポリアミノポリカルボキシレート・キレート剤にカップリングした本発明のポリペプチドは、キレート剤及び医用画像に適した放射性核種にカップリングしたHER2結合ポリペプチドの放射性キレートから成る放射性標識ポリペプチドを提供するために使用されてもよく、該放射性核種は、61Cu、64Cu、66Ga、67Ga、68Ga、110mIn、111In、44Sc及び86Yから成るグループから選択され、又は治療に適した放射性核種とのものでは、該放射性核種は、225Ac、212Bi、213Bi、67Cu、166Ho、177Lu、212Pb、149Pm、153Sm、227Th及び90Yから成るグループから選択され、ここで、放射性核種はキレート環境を介してHER2結合ポリペプチドと錯体を形成する。
【0064】
本発明の放射性標識ポリペプチドの1つの特に好ましい実施態様は、1,4,7,10−テトラアザシクロ−ドデカン−1,4,7−トリス−酢酸−10−マレイミドエチルアセト−アミド及び111Inにカップリングした、アミノ酸配列:
AEAKYAKEMRNAYWEIALLPNLTNQQKRAF IRKLYDD
PSQSSELLSEAKK LNDSQAPKVDC
(配列番号5)
を有する、HER2結合ポリペプチドの放射性キレートである。
【0065】
本発明の放射性標識ポリペプチドの1つの特に好ましい実施態様は、1,4,7,10−テトラアザシクロ−ドデカン−1,4,7−トリス−酢酸−10−マレイミドエチルアセト−アミド及び61Cu又は64Cuにカップリングした、アミノ酸配列:
AEAKYAKEMRNAYWEIALLPNLTNQQKRAF IRKLYDDPSQSSELLSEAKK LNDSQAPKVDC
(配列番号5)
を有する、HER2結合ポリペプチドの放射性キレートである。
【0066】
本発明の放射性標識ポリペプチドの1つの特に好ましい実施態様は、1,4,7,10−テトラアザシクロ−ドデカン−1,4,7−トリス−酢酸−10−マレイミドエチルアセト−アミド及び66Ga、67Ga又は68Gaにカップリングした、アミノ酸配列:
AEAKYAKEMRNAYWEIALLPNLTNQQKRAF IRKLYDDPSQSSELLSEAKK LNDSQAPKVDC
(配列番号5)
を有する、HER2結合ポリペプチドの放射性キレートである。
【0067】
本発明に記載の放射性標識ポリペプチドの1つの特に好ましい実施態様は、1,4,7,10−テトラアザシクロ−ドデカン−1,4,7−トリス−酢酸−10−マレイミドエチルアセト−アミド及び86Yにカップリングした、アミノ酸配列:
AEAKYAKEMRNAYWEIALLPNLTNQQKRAF IRKLYDDPSQSSELLSEAKK LNDSQAPKVDC
(配列番号5)
を有する、HER2結合ポリペプチドの放射性キレートである。
【0068】
本発明の放射性標識ポリペプチドは、また、前記に示すようなHER2結合性ポリペプチドの間接標識化によって得ることができる。好ましくは、ポリペプチドは少なくとも1つのシステインを含有し、最も好ましくはたった1つのシステインを含有する。システインは、本発明のポリペプチドに最初に存在していてもよいし、又は1つ又はそれ以上の更なるアミノ酸として後の段階で付加されてもよい。例えば18F、76Brによる非間接標識化のためには、さまざまのヨウ素同位体及び211At、中間体「リンカー分子」が標識化のために用いられる。そのようなリンカーは2つの官能性部分を含有し、1つは迅速で効率的な放射性標識化を与え、別のものは、タンパク質への、例えばアミン基への、又は好ましくはユニークシステインのチオール基への、迅速で効率的なカップリングを与える。例えば、マレイミド基は、チオール基と反応して安定なチオエーテル結合を形成する。この考えは、最初に「リンカー分子」を放射性標識と反応させ、続いてタンパク質のチオール基と反応させることである。
【0069】
いくつかの代替案が放射性ヨウ素化について徹底的に検討されており、そこではリンカー分子の放射標識化が、例えば活性化フェノール環又は適した脱離基を有する芳香環上で為されている。非標識リンカーの製造と、N−スクシンイミジル3−[*I]ヨードベンゾアートを使用する間接的放射性ヨウ素化の詳細なプロトコルは、Vaidyanathan et al.(Vaidyanathan G and Zalutsky MR(2006),Nat Protoc. 1(2): 707-13)によって提供されている。N−スクシンイミジルトリメチルスタンニルベンゾアートを使用する間接放射性ヨウ素化の例は、以下の図式に示される。
【化4】
【0070】
この図式では、リンカー分子は先ず酸性条件下で放射性ヨード化され、それからアルカリ条件下で遊離アミン(リジンのε−アミノ基のN末端)にカップリングされる。ベンゾアートのメタとパラヨード誘導体の両方は、文献に記載されている。
【0071】
リンカー分子は、、放射性標識(例えば、2−、3−又は4−の位置で)をリンクするアリール基を含んでもよい。但し、放射性標識が76Brである場合は、リンカー分子はフェノール性OH基を含まない。リンカー分子の非限定的な例は、本発明のポリペプチドにリンクするために使用できる複素環のイミドを含み、そして放射性標識は、N−[2−ベンズアミドエチル]マレイミド(malemide),4−マレイミドベンゾフェノン(BPMal)、スクシンイミジル4−(p−マレイミドフェニル)ブチラート(SMPB)、4−(4−N−マレイミドフェニル)酪酸ヒドラジド塩酸塩(MPBH)、及びマレイミドベンゾイル−N−ヒドロキシスクシンイミドエステル(MBS)を含む。
【0072】
N−[2−ベンズアミドエチル]マレイミドは、18Fと反応してN−[2−(4−(18F−フルオロベンズアミド)エチル]マレミドを形成し得る。同様にCai et al. (J. Nucl. Med.47:1172-80(2006))によって記載されているように、76Brと反応し得る。
【0073】
本発明は、また、前記HER2結合ポリペプチドの使用の別の態様、並びにポリペプチドがその結合特性故に有用である治療、診断及び検出のためのさまざまの方法、に関する。これらの使用及び方法の以下の説明で「HER2結合ポリペプチド」を言及する場合、この用語は、単独でHER2結合ポリペプチドを包含するが、しかしまた、例えば融合タンパク質中の部分としてHER2結合ポリペプチドを組み込む、及び/又は標識、キレート剤、治療薬及び/又は診断薬に共役されている、及び/又はタグとして又は他の目的で更なるアミノ酸残基と共に提供される、上記のこのポリペプチドに基づく全てのそれらの分子を包含するものとする。上で説明したように、そのような融合タンパク質、誘導体などは、本発明の一部を構成する。
【0074】
従って、1つのそのような態様において、本発明は、HER2の過剰発現によって特徴付けられるがんを有する又は疑われるヒトを含む、哺乳動物対象(subject)の体のインビボ画像化の方法を提供し、その方法は下記の工程を含む:
・医用画像に適した放射性核種を含む、前述の放射性標識ポリペプチドを哺乳類対象の体内に投与すること:及び
・医用画像機器を使用して対象の体の少なくとも一部分の画像、該画像は該体の内部の放射性核種の存在を指示する、を得ること。好ましくは、画像は、体への放射性標識ポリペプチドの投与の1〜72時間、又はある場合にはより好ましくは1〜24時間内に得られる。投与と画像取得の間の時間は、使用される放射性核種の半減期に依存する。画像を得る工程を2回、3回又はそれ以上繰り返すことが可能であり、それによって画像の系列が取得される。人が標的薬の経時的な体内分布を追跡しようとする場合、これは有用であり、薬物動態学的試験を可能にする。当業者には当然のことながら、そのような試験に必要な程度の時間分解能を達成するために、任意の数の画像を得ることができる。
【0075】
本発明の画像化方法の1つの実施態様において、本方法は、投与工程の前に、本発明の放射性標識ポリペプチドを製造する準備工程を含み、その工程は、第1の態様に記載のポリペプチドを医用画像に適した放射性核種と混合することを含む。
【0076】
本発明の画像化方法のより格別な実施態様において、本方法は、投与工程の前に、(i)アミノ酸配列が配列番号7から成るポリペプチド、の放射性標識ポリペプチドを、ii)99mTc、を用いて製造する準備工程を含み、その工程は、ポリペプチドを、適切な緩衝液中で、適切な還元剤、例えば塩化第一スズ又はフッ化第一スズの存在下で、99mTc−過テクネチウム酸と混合することを含む。これは、また、中間体の弱いキレート剤、例えば酒石酸若しくはクエン酸又はグルコン酸の存在下で実行され得る。
【0077】
本発明の画像化方法の別の格別な実施態様において、本方法は、投与工程の前に、(i)アミノ酸配列が1,4,7,10−テトラアザシクロドデカン−1,4,7−トリス酢酸−10−マレイミドエチルアセトアミドに共役した配列番号5から成るポリペプチド、の放射性標識ポリペプチドを、ii)111Inを用いて製造する準備工程を含み、その工程は、非溶解性コロイドの生成を防止する適切な緩衝液、例えば酢酸又はクエン酸緩衝液(しかし、これに限定されない)中、共役体を111Inと混合することを含む。
【0078】
本発明の画像化方法の別の格別な実施態様において、本方法は、投与工程の前に、(i)アミノ酸配列が1,4,7,10−テトラアザシクロドデカン−1,4,7−トリス酢酸−10−マレイミドエチルアセトアミドに共役した配列番号5から成るポリペプチド、の放射性標識ポリペプチドを、ii)61Cu又は64Cuを用いて製造する準備工程を含み、その工程は、非溶解性コロイドの生成を防止する適切な緩衝液、例えば、酢酸又はクエン酸緩衝液(しかし、これに限定されない)中、共役体を61Cu又は64Cuと混合することを含む。
【0079】
本発明の画像化方法の別の格別な実施態様において、本方法は、投与工程の前に、(i)アミノ酸配列が1,4,7,10−テトラアザシクロドデカン−1,4,7−トリス酢酸−10−マレイミドエチルアセトアミドに共役した配列番号5から成るポリペプチド、の放射性標識ポリペプチドを、ii)66Ga、67Ga又は68Gaを用いて製造する準備工程を含み、その工程は、非溶解性コロイドの生成を防止する適切な緩衝液、例えば酢酸又はクエン酸緩衝液(しかし、これに限定されない)中、共役体を66Ga、67Ga又は68Gaと混合することを含む。
【0080】
本発明のこの態様のいくつかの実施態様において、前記がんは、乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんから選択される。
【0081】
別の態様において、本発明は、診断に利用するための上記のような放射性標識ポリペプチドを提供する。「診断」という用語は、本明細書において、患者の病歴、試験及び臨床検査データのレビューの評価を通じて疾患の特質及び原因を同定したり、決定する行為又は方法を記述するために用いられる。
【0082】
放射性標識ポリペプチドは、頻発するHER2の過剰発現によって特徴付けられるがんの群に属しているがんの診断に用いることができる。「頻発する」という用語は、HER2の過剰発現が、特定のがんを有する全ての患者の少なくとも10%に存在していることを示すために、本明細書で用いられる。このように、放射性標識ポリペプチドは、患者がそのようながんを有するかどうかを決定するプロセスに使用することができる。また、患者の適する治療を決定するプロセスでも使用することができる。特に、HER2過剰発現の検出は、HER2標的の療法、例えばトラスツズマブ(Herceptin(登録商標))での治療に対する患者の層別化に有用であり得る。
【0083】
放射性標識ポリペプチドは、また、乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんより選択されるがんの診断及び/又は分子的特性化に、使用することができる。このような状況において、分子的特性化は、例えばいずれかのHER2の発現があるかどうかを決定し、例えば治療の前後での画像を取得することによって達成できる治療の前後でのHER2発現の量を決定し、例えば、手術目的のために、HER2発現の程度又は解剖学的含量(content)を決定するためのHER2発現の特性化に関する。
【0084】
また、本発明の別の態様において、ヒトを含む哺乳動物の対象物の体のインビボの画像化の診断薬の製造において、上記のような放射性標識ポリペプチドの使用が提供される。
【0085】
別の応用の態様において、本発明は、HER2の過剰発現によって特徴付けられるがんを有する、ヒトを含む哺乳類対象の治療方法であって、前記哺乳類対象物の身体に、治療に適する放射性核種を含む上記の放射性標識ポリペプチドを、治療的有効量で投与する工程を含む方法を提供する。
【0086】
この態様の1つの実施態様において、前記方法は、投与工程の前に、本発明の放射性標識ポリペプチドを製造する準備工程を含み、その工程は、以下を含み得る:
・適切なバッファー中で、第1の態様に記載のポリペプチドを治療に適する金属放射性核種と混合すること;又は
・適切な還元剤の存在下で、第1の態様に記載のポリペプチドを適切な緩衝液中で過レニウム酸塩と混合すること;これは、また、中間体の弱いキレート剤、例えば、酒石酸、又はクエン酸若しくはグルコン酸の存在下でも実行され得る;又は
・第1の態様に記載のポリペプチドをトリカルボニル錯体のようなレニウムの反応性中間体と混合すること;又は
・第1の態様に記載のポリペプチドを適切な緩衝液中で、付着ハロゲン原子を有する反応性中間体と混合すること。この混合は、アスコルピン酸又はゲンチジン酸のようなポリペプチドの放射線分解を抑制するために指定される物質の存在下で実行され得る。
【0087】
本発明のこの態様のいくつかの実施態様において、前記がんは、乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんから選択される。
【0088】
別の態様において、本発明は、HER2の過剰発現によって特徴付けられるがんの治療における使用のような、例えば、乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんから選択されるがんの治療における使用のような、治療における使用のための、上記のような放射性標識ポリペプチドを提供する。
【0089】
また、本発明の別の態様において、乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんから選択されるがんのような、HER2の過剰発現によって特徴付けられるがんの治療のための薬剤の製造における、上記の放射性標識ポリペプチドの使用が提供される。
【0090】
本発明の別の態様は、上記に示すようなポリペプチドをコードする配列を含む核酸分子に関する。
【0091】
本発明の更なる態様は、前記の態様の核酸分子を含む発現ベクター、及び核酸分子の発現を通して、本発明のポリペプチドの産生を可能にする他の核酸エレメントに関する。
【0092】
本発明の別の態様は、前記の態様の発現ベクターを含む宿主細胞に関する。
【0093】
本発明の後者の3つの態様は、本発明のポリペプチドの産生ツールであり、本明細書では発現されることになっているポリペプチドに関する情報を条件に、及びタンパク質の組み換え発現の当業者の最新の技術レベルを条件に、当業者はそれらを得ることができ、それらを必要以上の負担無く実用化できるであろう。
【0094】
しかしながら、本発明のポリペプチドは、また、植物及びトランスジェニック動物を含む、さまざまな原核生物の又は真核生物の宿主の中での化学的合成又は発現を含む、他の公知の手段によって産生し得る。
【0095】
さて、本発明を、それに従って実施される実験の説明を通して詳細に説明する。下記の実施例は、限定するものとして解釈されるべきではない。実施例では、添付の図面が参照される。
【0096】
Affibody(登録商標)分子の配列は、以下の表1に示される。
【図面の簡単な説明】
【0097】
【図1】A:7.8nM;B:1.9Nm及びC:0.49nMのAffibody(登録商標)分子IIを、固定化したHER2/Fcキメラ融合タンパク質を有するセンサーチップ上に注入した後に得られたBiacoreセンサーグラムを示す。C末端システインのSH基は、N−エチルマレイミド(NEM)で遮断された。
【図2】A:6nM;B:1.5Nm及びC:0.38nMのC末端のシステインにカップリングしたDOTAを有するAffibody(登録商標)分子II(配列番号5)を、固定化したHER2/Fcキメラ融合タンパク質を有するセンサーチップ上に注入した後に得られたBiacoreセンサーグラムを示す。
【図3】A:6nM;B:1.5Nm及びC:0.38nMのAffibody(登録商標)分子X(配列番号7)を、固定化したHER2/Fcキメラ融合タンパク質を有するセンサーチップ上に注入した後に得られたBiacoreセンサーグラムを示す。
【図4】BALB/c nu/nu マウスSKOV−3腫瘍異種移植片中の、DOTAと共に111In標識したAffibody(登録商標)分子IIの体内分布を示す。全てのマウスに、1μg(100kBq)の111In標識されたAffibody(登録商標)分子IIを注射した。注射後0.5、1.4及び24時間で、臓器を解剖して、秤量し、そしてその放射能の量をガンマ計数器で測定した。放射能の取り込みは、グラム組織当たり注射した活性のパーセント(%IA/g)として計算された。
【図5】図4で説明した体内分布の試験で測定された放射能の取込みの腫瘍対臓器比を示す。
【図6】SKOV−3腫瘍異種移植片中、DOTAと共に111In標識したAffibody(登録商標)分子IIで、標的化する腫瘍の特異性を示す。全てのマウスに、DOTAを有する1μg(100kBq)の111In標識したAffibody(登録商標)分子IIを注射した。動物の1つの群は、111In標識したAffibody(登録商標)分子の注射の50分前に過剰の非標識Affibody(登録商標)分子(保護基)で前処理された。臓器は、注射後4時間で解剖し、秤量し、そして放射能の量をガンマ計数器で測定した。放射能の取り込みは、グラム組織当たり注射した活性のパーセント(%IA/g)として計算された。
【図7】BALB/c nu/nu マウス中のSKOV−3異種移植片腫瘍を発現するHER2(左側の部分、30分及び4時間p.i.)、及びHER2陰性のA431異種移植片(右側部分、4時間p.i.)のガンマカメラ画像を示す。全てのマウスに、DOTAを有する3μg(4.5MBq)の111In標識したAffibody(登録商標)分子IIを注射した。K=腎、T=腫瘍。
【図8】注射後4時間のBALB/c nu/nu マウスSKOV−3腫瘍異種移植片中の99mTc−標識Affibody(登録商標)分子Xの体内分布を示す。全てのマウスに、1μg(65kBq)の99mTc標識Affibody(登録商標)分子Xを注射した。動物の1つの群は、99mTc標識Affibody(登録商標)分子の注射の50分前に過剰の非標識Affibody(登録商標)分子(ブロッキング群)で前処理された。臓器は注射後4時間で解剖し、秤量し、そしてその放射能の量をガンマ計数器で測定した。グラム組織当たり注射した活性のパーセント(%IA/g)として計算された。
【図9】図8に記載の生体内分布の試験で測定された、放射能取り込みの腫瘍対組織比を示す。
【図10】BALB/c nu/nu マウス中のSKOV−3異種移植片腫瘍を発現するHER2(左側の部分、1時間及び4時間p.i.)、及びHER2陰性のA431異種移植片(右側部分、4時間p.i.)のガンマカメラ画像を示す。全てのマウスに、DOTAを有する3μg(15MBq)の111In標識したAffibody(登録商標)分子IIを注射した。K=腎臓、T=腫瘍。
【図11A】アミノ酸18〜58の合成の段階でのAffibody(登録商標)分子Xについての分析用HPLC溶離プロファイルを示す。Affibody(登録商標)分子は、VydacTP218TP54カラムで分離された。流量:1ml/分;温度:35℃;検出:220nm;溶離液A:0.1%TFA、H2O中1%アセトニトリル;溶離液B:アセトニトリル中0.1%TFA。 位置22−23、41−42、45−46及び53−54に疑似プロリンを使用しているポリスチレン樹脂上で行なった合成。
【図11B】アミノ酸18〜58の合成の段階でのAffibody(登録商標)分子Xについての分析用HPLC溶離プロファイルを示す。Affibody(登録商標)分子は、VydacTP218TP54カラムで分離された。流量:1ml/分;温度:35℃;検出:220nm;溶離液A:0.1%TFA、H2O中1%アセトニトリル;溶離液B:アセトニトリル中0.1%TFA。 疑似プロリンのないポリスチレン樹脂上での標準ペプチドの合成。
【図12A】アミノ酸1〜58(A)及び10〜58(B)の合成の段階でのAffibody(登録商標)分子Xに対する分析用HPLC溶離プロファイルを示す。Affibody(登録商標)分子は、VydacTP218TP54カラムで分離された。流量:1ml/分;温度:35℃;検出:220nm;溶媒:0.1%TFA、H2O中1%アセトニトリル;溶媒:アセトニトリル中0.1%TFA。 位置22−23、41−42、45−46及び53−54に疑似プロリンを使用しているポリスチレン樹脂上で行なった合成。
【図12B】アミノ酸1〜58(A)及び10〜58(B)の合成の段階でのAffibody(登録商標)分子Xに対する分析用HPLC溶離プロファイルを示す。Affibody (登録商標)分子は、VydacTP218TP54カラムで分離された。流量:1ml/分;温度:35℃;検出:220nm;溶媒:0.1%TFA、H2O中1%アセトニトリル;溶媒:アセトニトリル中0.1%TFA。 疑似プロリンのないポリスチレン樹脂上での標準ペプチドの合成。
【図13A】表1に示されたいくつかのAffibody(登録商標)分子に対する疎水性/親水性プロットを示す。Affibody(登録商標)分子II(配列番号5)についてのプロット。図面の上部は、Kyte-Doolittleスケールを使用するプロットを示し、そして下部は、HPLCによって決定されたpH3.4での疎水性指標を示す。疎水性アミノ酸は正の値を有し、親水性アミノ酸は負の値を有する。7アミノ酸の移動ウインドウは、両方のプロットに使用された。
【図13B】表1に示されたいくつかのAffibody(登録商標)分子に対する疎水性/親水性プロットを示す。配列番号X(配列番号7)を有するAffibody (登録商標)分子についてのプロット。図面の上部は、Kyte-Doolittleスケールを使用するプロットを示し、そして下部は、HPLCによって決定されたpH3.4での疎水性指標を示す。疎水性アミノ酸は正の値を有し、親水性アミノ酸は負の値を有する。7アミノ酸の移動ウインドウは、両方のプロットに使用された。 Affibody(登録商標)分子X(配列番号7)のアミノ酸配列のみが、即ち、maのないAffibody(登録商標)分子Xが、プロットを作成するために使用された。
【図13C】表1に示されたいくつかのAffibody (登録商標)分子に対する疎水性/親水性プロットを示す。Affibody(登録商標)分子Z00342、対照としての変異のない親Affibody(登録商標)分子についてのプロット。図面の上部は、Kyte-Doolittleスケールを使用するプロットを示し、そして下部は、HPLCによって決定されたpH3.4での疎水性指標を示す。疎水性アミノ酸は正の値を有し、親水性アミノ酸は負の値を有する。7アミノ酸の移動ウインドウは、両方のプロットに使用された。
【図14】本発明に記載のAffibody(登録商標)分子II(黒線)及びAffibody(登録商標)分子Z00342(灰色の破線)についてのHPLC溶離プロファイルを示す。Affibody(登録商標)分子は、0.5ml/分の流量とTFA、水、アセトニトリル溶剤系を用いるZorbax 300SB C8 150×2.1mm、3.5のμmのカラム上で分離した。
【実施例】
【0098】
〔実施例1〕
本発明のポリペプチドの製造
要約
この実施例では、選択されたHER2特異的Affibody(登録商標)分子を、下記のプロトコルに従って適切な細菌細胞中で大規模に発現され、精製された、pETシステム発現ベクターにサブクローニングした。対応する全長HER2特異的Affibody(登録商標)分子のアミノ酸配列は、Affibody(登録商標)分子I−XIとして表1に収載されている。
【0099】
Affibody(登録商標)分子のクローニング
Affibody(登録商標)分子I−VIIIをコード化する遺伝子を、テンプレートとして2つのオリゴヌクレオチド(一緒になってAffibody(登録商標)配列を包含している)を使用することによってPCR増幅した。各々の構築物について、28bpの相補的オーバーラップを備えた特定のテンプレート・オリゴヌクレオチド(各々、102長い)ペアを使用した。Affibody(登録商標)遺伝子を、NdeI部位及びATG開始コドンを含むフォワードプライマー(センス) (Affibody(登録商標)分子Iについては、配列をコードするHis6-タグもまたプライマーに含まれた)、並びにC末端アミノ酸についてのコドン、終止コドン及びNotI部位を有するリバースプライマー(アンチセンス)で増幅した。
【0100】
Affibody(登録商標)分子IX中のAffibody(登録商標)アルブミン結合ドメイン(ABD)遺伝子融合を、オーバーラップ伸長PCRによって生産した。リバースプライマーがABD遺伝子のリンカー配列(Cysコドンと共に)及び5’末端を含んでいたことを除いては、Affibody(登録商標)分子コード化フラグメントを、Affibody(登録商標)分子I−VIIIについて上述したように作成した。ABD遺伝子を、Affibody(登録商標)遺伝子の3’末端及びリンカーの配列及び終止コドンを有するリバースプライマー、及びNotI部位を含むフォワードプライマーで増幅した。続いて、25bpオーバーラップを有する2つのPCR産物を、Affibody(登録商標)遺伝子フォワードプライマー及びAffibody(登録商標)ABD遺伝子融合を作成するABD遺伝子リバースプライマーを用いて、最後のPCR増幅におけるテンプレートとして使用した。
【0101】
PCR産物を、制限酵素NdeI及びNotIで消化し、同じ酵素で先に消化したT7発現ベクターに連結した。
【0102】
【表1】
【0103】
Affibody(登録商標)分子の発現及び採取
得られた発現プラスミドを、エレクトロポレーションを使用して大腸菌BL21(DE3)細胞に形質転換した。細菌含有発現プラスミドを、カナマイシンを有する選択寒天から選び取り、マルチ発酵装置システム(System Greta, Belach Bioteknik AB, Sweden)を使用して、50mg/lのカナマイシンで補充された800mlのTSB+YE培地(30g/lトリプシン大豆ブロス+5g/l酵母エキス)の中で37℃で増殖させた。約1の光学密度(OD600nm)で、Affibody (登録商標)分子の発現を、0.5mmのIPTGの添加によって自動的に誘導した。誘導剤の添加の5時間後に、培養物を自動的に<12℃に冷却した。次いで、培養物を遠心分離(15900×gで20分)により採取し、5〜10gの細菌の細胞ペレットを採取し、冷凍庫(−20℃)で保管した。
【0104】
Affibody(登録商標)分子Iの精製
細胞ペレット(3g)を、7Mの尿素及び1,000ユニットのBenzonase(Merck)を含むpH8の緩衝液の30ml中に再懸濁させた。30分のインキュベーションの後、溶解物を遠心分離によって清澄化し、His6タグの付いたAffibody(登録商標)分子を含んでいる上清を、1.5mlの Superflow NiNTAカラム(Qiagen)にかけた。カラムを、7Mの尿素を含む20mlのpH6.3の緩衝液で洗浄し、8Mの尿素を含むpH4.5の緩衝液で溶離した。最後に、緩衝液を、PD-10カラム(GE Healthcare)を使用することによって、1×PBS(2.68mmKCl、137mmNaCl、1.47mmKH2PO4、8.1mmNa2HPO4、pH7.4)に交換した。
【0105】
Affibody(登録商標)分子II−VIIIの精製
細胞ペレット(10g)を、100mlの20mMのTris−HCl、pH7.5に再懸濁させ、2,000ユニットのBenzonase(Merck)を補充して、超音波処理によって破壊し、10分間75℃で加熱した。熱処理された溶解物を遠心分離によって清澄化し、そして20mMのTris−HClpH7.5であらかじめ平衡化した25mlQ−セファロースFF(GE Healtcare)を詰めたXK26カラムにかけた。カラムから流れ出た画分を集め、C末端システインを有するAffibody (登録商標)分子を、常温で1時間20mM DTTと共にインキュベートした。ACNを5%に加え、5%ACNと0.1%TFAを含有する水で予め平衡化した3mlのResource 15RPCカラム(GE Healtcare)にかけた。水中の5カラム容積(CV)の5%ACN及び1%TFAで洗浄した後、水中の20CVの5〜32%ACN及び0.1%TFAの直線勾配で、結合したタンパク質を溶離した。Affibody(登録商標)分子を含む画分をSDS−PAGE分析によって同定し、プールした。最後に、緩衝液を、HiPrep 26/10脱塩カラム(GE Healthcare)を使用することによって1×PBSに交換した。
【0106】
Affibody(登録商標)分子IXの精製
細胞ペレット(5g)を、50mlTST緩衝液(25mMTris−HCl、200mMのNaCl、1mMのEDTA、0.05%のTween20、pH8.0)に再懸濁させ、超音波処理によって破壊した。遠心分離による清澄化の後、上清を20mlHSAセファロース(CNBr活性セファロース4 Fast Flow, GE Healtcare上に固定化されたヒト血清アルブミン)を詰めたXK26カラム(GE Healtcare)にかけた。カラムを、10CVのTST緩衝液、次いで、5CVの5mMのNH4Ac、pH5.6によって洗浄した。結合したABDタグ付きAffibody(登録商標)分子を、0.5MのHAc、pH2.8で溶離した。最後に、緩衝液を、HiPrep26/10脱塩カラム(GE Healthcare)を使用することによって1×PBSに交換した。
【0107】
〔実施例2〕
インビトロの本発明のポリペプチドのHER2結合特性
要約
この実施例を形成している実験では、Biacore(登録商標)2000システムでの表面プラズモン共鳴(SPR)を使用して、本発明のポリペプチドの結合を分析した。HER2標的結合活性は、C末端のシステイン及びX(配列番号7)にカップリングしたAffibody(登録商標)分子II(配列番号5)(DOTAを有するII)について示される。
【0108】
バイオセンサー分析
C末端His6タグ(R &D System #1129-ER)でヒトIgG1(Pro100−Lys330)のFc領域に融合したErbB2(HER2、Met1−Thr652)の細胞外ドメインから成る、組み換えヒトHER2/Fc(ErbB2/Fc)キメラタンパク質を、アミンカップリング化学を使用するCM5センサーチップの表面上に(1,800のレゾナンスユニット(RU))固定化した。チップ上の1つの表面は、参照セルとして使用するために活性化され、不活化された。HBS−EP緩衝液(10mMのHEPES、150mMのNaCl、3mMのEDTA、0.005%の界面活性剤P−20、pH7.4)中希釈したAffibody (登録商標)分子を、被検体として使用した。
【0109】
結合速度論の解析に関しては、3つの被検体の濃度を、50μ/分の一定流量を用いてチップ上に2連で(dupulicate)注入した。会合相は5分で、続いてAffibody(登録商標)分子の遅速性を説明する長い解離相(30分)であった。解離平衡定数(KD)、会合速度定数(ka)及び解離速度定数(kd)は、BIAevaluation 4.1ソフトウェア(GE Healthcare)の質量移動(mass transfer)補正を有する1:1ラングミュア結合モデルを使って計算された。
【0110】
システインのSH基のブロッキング
システインのSH基をブロックし、それによって分子の二量化を防ぐために、単一のC末端のシステインを有するAffibody(登録商標)分子を、N−エチルマレイミド(NEM、Pierce No.23030)で処理した。Affibody (登録商標)分子II(配列番号5)を、1ml(2.1mg)のペプチドを10μlのTris−HCl、pH8.5及び40μlの0.5Mジチオトレイトール(DTT)で、室温で2時間処理することによって還元した。過剰のDDTは、カップリング緩衝液(100mMのリン酸ナトリウム、150mMのNaCl、pH7.0)で平衡化したNAP5カラム(GE Healthcare)上のゲル濾過によって除去した。0.5mlの還元タンパク質を有するロードカラムは、1mlのカップリング緩衝液でタンパク質を溶離する。
【0111】
遊離SH基のブロッキングは、タンパク質に対して20倍モル過剰のNEMを使って行われた:水(200mm)中15μlのNEM溶液を0.5mlの還元したタンパク質溶液(1mg/ml)に加え、そしてヘッド・オーバーヘッド・ミキサー(head-over-head mixer)上、室温で1時間インキュベートする。過剰のNEMは、前記のようにNAP5−カラム上でのゲル濾過によって、しかし平衡緩衝液としてPBSを用いて、除去した。
【0112】
C末端システインへのマレイミド−DOTAのカップリング
3mgのAffibody(登録商標)分子II(配列番号5)を、40℃で30分間20mMのDTTで還元した。過剰のDTTは、PD−10カラム上での0.2Mの酢酸アンモニウム、pH5.5への緩衝液交換によって除いた。カップリングは、3倍モル過剰のキレート剤、水中のマレイミド−モノ−アミドDOTA(Mal-DOTA, Macrocyclics No.B-272)溶液(1mg/ml)を用いて行った。混合物を、連続振盪しながら40℃で1時間インキュベートした。非複合化キレート剤からの精製は、セミ分離用RPCカラム、Zorbax 300SB C18、9.4×250mm、5μm上で行った。精製物質のカップリング程度は、Zorbax 300SB C8 150×2.1mm、3.5μmの分析用カラムの上でHPLC−MSによって分析した。マレイミド−DOTA共役Affibody(登録商標)分子のみが、本方法によって検出された。
【0113】
Affibody(登録商標)分子IIのHER2−結合相互作用を、図1のセンサーグラフに、C末端のシステインにカップリングしたDOTAを有するAffibody(登録商標)分子IIを図2に、及びAffibody(登録商標)分子X(配列番号7)のものを図3に示す。
【0114】
HER2標的とのAffibody(登録商標)分子の相互作用についての結合パラメーターを決定するために、動力学的試験を行なった。3つの異なる濃度のAffibody(登録商標)分子を、固定化HER2/Fcのキメラ融合タンパク質でBiacoreチップ表面に注入した。解離平衡定数(KD=kd/ka)、会合速度定数(ka)及び解離速度定数(kd)を表2に要約する。C末端システインへのDOTAカップリングは、NEMブロッキングAffibody(登録商標)分子IIに比べてKDを有意に変化させなかった。
【0115】
【表2】
【0116】
〔実施例3a〕
ポリアミノポリカルボキシラートキレート剤を有する本発明のポリペプチドのインビボのHER2結合特性
要約
この実施例を構成している実験では、本発明の放射標識ポリペプチドの結合を、インビボマウス異種移植片モデルで分析した。C末端のシステインにカップリングしたDOTAを有するAffibody(登録商標)分子IIを111Inで放射標識し、HER2を過剰発現するSKOV−3腫瘍異種移植片及び対照としてHER2陰性A431腫瘍異種移植片を担持するマウスに注射した。全動物中でのさまざまな臓器の放射性標識Affibody(登録商標)分子の分布及び局在を分析するために、体内分布及びガンマカメラ画像の試験を行なった。
【0117】
111Inラベル化
実施例No.2に記載したように、C末端のシステインにモノアミドDOTA(Mal−DOTA)をカップリングさせてAffibody (登録商標)分子を修飾した。得られた分子は「DOTAを有するAffibody(登録商標)分子II」と名付けられた。
【0118】
111Inで標識するために、DOTA(30μlのpH5.5の酢酸アンモニウム緩衝液)を有する30μgのAffibody(登録商標)分子IIを、40.5μlの111InCl3(標識化時15MBq)の保存液と混合した。反応混合液のpHは約5.0であった。反応混合物を、60℃で60分間インキュベートした。
【0119】
標識化効率は、シリカゲル含浸ガラス繊維シート(ITLC SGシート(Gelman Sciences社))上のインスタント薄層クロマトグラフィー(ITLC)により、0.02Mのクエン酸を溶離液として使用して分析した。ITLC片に沿った放射能の分布を可視化し、PhosphorImager(CycloneTM Storage Phosphor System, Packard)を使用して定量化した。放射性標識Affibody(登録商標)分子は、ITLCシートの元の場所に残り、一方、遊離の111Inは溶媒先端に移動した。
【0120】
放射化学的純度は99.8から99.9%であった、即ち、放射性標識化は殆ど定量的であった。放射性標識化Affibody(登録商標)分子の一切の更なる精製は必要でなかった。反応混合物は、PBSで1.4mlに希釈し、実験の前に凍結保存した(1〜2日)。
【0121】
腫瘍モデル
雌の非近交系のBALB/c nu/nu マウスに、1×107SKOV−3(ヒト卵巣腹水腺がん)細胞又は1×107a431(ヒト表皮がん)細胞を、右の後脚中に移植した。SKOV−3細胞はHER2を過剰発現し、A431細胞はHER2について陰性であった。
【0122】
腫瘍担持マウスにおける体内分布
体内分布の試験のため、異種移植片腫瘍(1×107個のSKOV−3細胞移植の30〜40日後)担持マウスを、4つの群に無作為に割り付けた。マウスに、100μlPBS中の1μg(100kBq)の111In標識Affibody(登録商標)分子を、尾静脈に注射した。全ての注射の忍容性は良好であった。動物の1つの群に、1.5ml(0.9mg)の非標識Affibody(登録商標)分子IIを皮下注射し(ここで、C末端Cysが実施例2に記述したようにNEMによってブロックされている)、次いで50分後に、1μg(100kBq)の111In標識Affibody(登録商標)分子を静脈注射した(ブロッキング実験)。
【0123】
致死量のKetalarの溶液及びRompun溶液の注射後、マウスを、注射後0.5、1、4及び24時間後 (h p.i)に心臓穿刺を介して放血により犠牲にした。ブロッキンググループからの動物は、注射後4時間で犠牲にした。臓器を解剖し、秤量し、そしてそれらの放射性含量をガンマ計数器で測定した。放射能の取り込みは、グラム組織当たり注射した活性のパーセントとして計算された(%IA/g)。
【0124】
画像化
ガンマカメラによる画像化のため、異種移植片腫瘍(1×107個のSKOV−3細胞移植の30〜40日後又は1×107個のa431細胞移植の10〜15日後)担持マウスを使用した。マウスに、100μlPBS中3μg(4.5MBq)の111In標識Affibody(登録商標)分子IIを、尾静脈に注射した。注射後0.5又は4時間で、動物を致死量のKetalar及びRompunの溶液により安楽死させた。MEGPコリメータを装着したMillenium GEガンマカメラを使用し、画像化を行った。Hermesソフトウェア(Nuclear Diagnostics, Stockholm, Sweden)を使用し、シンチグラフィーの結果を、視覚的に評価した。
【0125】
結果
DOTAを有する111In標識Affibody(登録商標)分子IIを使用した、体内分布の試験からの結果を図4に示す。全ての時点での放射能の取り込みは、腎臓は別として、全ての他の臓器中よりも腫瘍中で高かった。腫瘍の取り込みは、注射後0.5時間で既に13±2%IA/gであった、注射後1時間で17±2%IA/gに上昇し、注射後1時間で15±3%IA/gから注射後24時間で11±4%IA/gまでゆっくり減少した。放射能の高い取り込みが、また、腎臓にも見出されたが、腎臓は、Affibody(登録商標)分子のような小さいタンパク質のための主要な排泄経路であるので、それは、予期されたことでもある。注射後1時間で測定された186±24%IA/gを有する最も高い腎臓の取り込みは、注射後24時間で144±13%IA/gに減少した。このように、腎臓の取り込みは、N末端にカップリングしたDOTAを有するAffibody(登録商標)分子Z00342(ZHER2:107として国際公開公報第2005/003456号に開示され、しばしば、また、ZHER2:342とも呼ばれる)について測定された腎臓の取り込み、即ち注射後1時間で243±22%IA/g及び注射後24時間で232±34%IA/gより相当低かった(Orlova, A. et al. (2006), Cancer Research 67: 2178-2186)。加えて、DOTAを有する111In標識Affibody(登録商標)分子IIは、Orlova, et al. (2006) によって記述されたHER2特異的Affibody(登録商標)分子と比較して、全ての測定時点において全ての臓器でより低い取り込みであった。高い腫瘍の取り込みは、他の臓器での減少した取り込みと共に、先に記述されたAffibody(登録商標)分子について公表された比率を超える腫瘍対組織比をもたらす(図5)。例えば、腫瘍対血液比は、Orlova A. et al. (2006)によって記述されたAffibody(登録商標)分子に対する注射後1時間で8、注射後4時間で12及び注射後24時間で47と比較して、注射後1時間で16、注射後4時間で88及び注射後24時間で275であった。
【0126】
本発明に記載のAffibodyの(登録商標)分子を使用する腫瘍標的の特異性は、ブロッキング実験によって証明され、ここで過剰な非標識Affibody(登録商標)分子の腫瘍担持マウスへの事前注射は、DOTAを有する111In標識Affibody(登録商標)分子IIの腫瘍取り込みを86%だけ減少させたが、他の臓器における取り込みは減少させなかった(図6)。注射後30分及び4時間で撮られたガンマカメラ画像により、DOTAを有する111In標識Affibody(登録商標)分子IIの高い特異的な腫瘍ターゲッティングが確認された(図7)。SKOV−3腫瘍を注射後0.5時間で既に可視化し、注射後4時間で、バックグラウンド放射能を洗い落とした。腫瘍とは別に、明確な取り込みがまた腎臓でも見えた。HER2陰性A431異種移植片腫瘍を担持するマウスは、注射後4時間で腫瘍中には無視できる放射線の取り込みしかなかった。
【0127】
〔実施例3b〕
N3Sキレート剤を有する本発明に記載のポリペプチドのインビボのHER2結合特性
要約
この実施例を構成している実験では、本発明の放射性標識ポリペプチドの結合を、インビボで、マウス異種移植片モデルを用いて分析した。N末端メルカプトアセチル−グルタミル−セリル−グルタミル(ma−ESE)配列を含むAffibody(登録商標)分子X(配列番号7)を99mTcで放射性標識し、HER2を過剰発現するSKOV−3腫瘍異種移植片を担持するマウスに注射した。全動物中のさまざまな臓器への放射性標識Affibody(登録商標)分子の分布及び局在を分析するために、体内分布及びガンマカメラ画像の試験を行なった。
【0128】
99mTc標識
Affibody(登録商標)分子Xを、間接法を用いて99mTcで標識した。99mTcを用いた標識化については、下記成分を含むキットを製造した:1ml当たり50Mgのグルコン酸二水和物、1mgのEDTAのジナトリウム塩及び0.5mgの錫(II)クロリド二水和物。この溶液を1mlを含むアリコートに分割し、アリコートを凍結乾燥した。凍結乾燥標識キットを、使用の前に−20℃で貯蔵した。
【0129】
50μlPBS(2.68mMのKCl、137mMのNaCl、1.47mMのKH2PO4、8.1mMのNa2HPO4、pH7.4)を、50μgのABY−024溶液(脱イオン化した脱気水中2mg/ml)に追加した。ボルテックス撹拌後、混合物にテクネチウム標識キットを混ぜ、99mTc発生装置(UltratechneKow FM発生装置、Tyco Healthcare Nordic AB(Mallinckrodt Sweden AB)からの200μlの99mTcO4-含有溶離液を添加した。混合物を、90℃で60分間インキュベートした。
【0130】
各混合物の2つの小さい(0.8μl)アリコートを、2つのITLC・SG片に適用し、そして1つをPBS(収量を決定する)で溶離し、他方はピリジン:酢酸:水(5:3:1.5)(還元した加水分解されたテクネチウム(RHT)の存在を決定するため)で溶離した。この溶離液では、テクネチウムコロイドは元の場所に残ったが、放射性標識Affibody(登録商標)分子、過テクネチウム酸及び他の99mTcの錯体は、溶媒先端に移動した。ITLC片に沿った放射活性の分布を視覚化し、PhosphorImager (CycloneTM Storage Phosphor System, Packard)を使用して定量した。標識化は殆ど定量的であって、それはいかなる追加の精製も不要であった。
【0131】
放射性標識Affibody(登録商標)分子を、放射能濃度が100μl当たり65kBqであることを確実にするために、PBSで希釈した。Affibody(登録商標)分子の濃度を、100μl当たり1μgに調整した。
【0132】
腫瘍担持マウスにおける体内分布
雌の非近交系のBALB/c nu/nu マウスに、1×107SKOV−3(ヒト卵巣腹水腺がん)細胞を、右の後脚中に移植した。
【0133】
体内分布の試験については、異種移植片腫瘍(1×107個のSKOV−3細胞移植の40〜45日後)担持マウスを、4群に無作為に割り付けた。
【0134】
マウスに、100μlPBS中の1μg(65kBq)の111Tc標識Affibody(登録商標)分子を、尾静脈に注射した。全ての注射は良好な忍容性を示した。動物の1群に、100ml(0.5mg)の非標識His6-Z00342を皮下注射し、次いで50分後に、1μg(65kBq)の111Tc標識Affibody(登録商標)分子Xを静脈注射した(ブロッキング実験)。
【0135】
致死量のKetalarの溶液及びRompun溶液の注射後、マウスを、注射後4時間後に心臓穿刺を介して放血により犠牲にした。ブロッキンググループからの動物は、注射後4時間で犠牲にした。臓器を解剖し、秤量し、そしてそれらの放射能含量をガンマ計数器で測定した。放射能の取り込みは、グラム組織当たり注射した活性のパーセントとして計算された(%IA/g)。
【0136】
画像化
ガンマカメラによる画像化については、異種移植片腫瘍(1×107個のSKOV−3細胞移植の40日後又は1×107個のa431細胞移植の10〜15日後)担持マウスを使用した。
【0137】
マウスには、100μlPBS中の3μg(15MBq)の99mTc標識Affibody(登録商標)分子Xを尾静脈に注入した。注射後1又は4時間で、動物を致死量のKetalar及びRompunの溶液により安楽死させた。LEHRコリメータを装着したCam Siemensガンマカメラを使用し、画像化を行った。Hermesソフトウェア(Nuclear Diagnostics, Stockholm, Sweden)を使用し、シンチグラフィーの結果を、視覚的に評価した。
【0138】
結果
99mTc標識Affibody(登録商標)分子Xを使用した、体内分布の試験からの結果を図8に示す。注射後4時間での放射能の取り込みは、腎臓は別として、全ての他の臓器中よりも腫瘍中で高かった。腫瘍の取り込みは、注射後4時間で17±4%IA/gであった。放射能の高い取り込みが、また、腎臓にも見出されたが、腎臓がAffibody(登録商標)分子のよな小さいタンパク質のための主要な排泄経路であるので、それは予期されたことでもある。腎臓の取り込みは、注射後4時間で58±10%IA/gであった。このように、腎臓の取り込みは、上記実施例3aに記述されたAffibody(登録商標)分子及びN末端にカップリングしたDOTAを有するZ00342について測定された腎臓の取り込みより相当低かった(Orlova,A. et al. (2006), Cancer Research 67: 2178-2186)。また、腸(3.52%IA/g)及び肝臓(1.06%IA/g)での低い取り込みは、腎臓でのより高い取り込みと共に、99mTc標識Affibody(登録商標)分子Xが主に腎臓の経路によって浄化され、肝胆道の経路によって行われないことを示す。欧州特許第6123095号は、好ましいma−Xaa1−Xaa2−Xaa3キレート成分が1つ又は2つのSerを含み、残りの2つ又は1つのアミノ酸残基がGluであるものが、肝胆道系から腎臓クリアランス経路へのシフトをもたらすことを開示している。Affibody(登録商標)分子Xは、欧州特許第6123095号に開示された好ましいキレート剤部分の1つ、即ちma−Glu−Ser−Glu及びこれらのポリペプチドの親水性の増加をもたらす本発明の実施例1(表1)に記載のアミノ酸交換の両方を含む。このように、本明細書で説明する99mTc−標識Affibody (登録商標)分子Xは、高い腎臓クリアランスと低肝胆道系クリアランスを両立させるが、腎臓での再吸収を最小化した、ma−Xaa1−Xaa2−Xaa3キレート部分を含んでいるポリペプチドの追加的実施例を示す。
【0139】
全ての臓器における99mTc標識Affibody(登録商標)分子Xの取り込みは、実施例3aに記述された111In標識Affibody(登録商標)分子の取り込みと同様であった。
【0140】
上述の体内分布実験の腫瘍対組織比を図9に示す。再び高い腫瘍対組織比を得た。特に、99mTc標識Affibody(登録商標)分子Xの腫瘍対肝臓比は、注射後4時間で10の比率の実施例3aに記述されたDOTAを有する111In標識Affibody(登録商標)分子IIに比べて、17であった。腫瘍対血液比は、注射後4時間で42であった。
【0141】
Affibody (登録商標)分子Xを使用した腫瘍標的の特異性は、ブロッキング実験によって証明され、その場合、過剰な非標識Affibody(登録商標)分子の腫瘍担持マウスへの事前注射により、99mTc標識Affibody(登録商標)分子Xの腫瘍吸収を90%も減少させたが他の臓器での取り込みでは減少させなかった(図8)。
【0142】
注射後1時間及び4時間に撮られたガンマカメラ画像は、本発明に記載の99mTc標識Affibody(登録商標)分子の高い特異的な腫瘍ターゲッティングを確認した(図10)。SKOV−3腫瘍を注射後1時間で既に可視化し、注射後4時間で、バックグラウンド放射能が洗い落とした。腫瘍とは別に、明白な取り込みもまた腎臓に見える、しかしながら、実施例3aに記述されたDOTAを有する111In標識Affibody(登録商標)分子IIの腎臓の取り込みに比べて、この取り込みはかなり低かった。HER2陰性A431異種移植片腫瘍を担持するマウスは、注射後4時間で腫瘍中の放射能取り込みは無視できるものであった。
【0143】
〔実施例4〕
本発明のポリペプチド及びZ00342の化学的合成に関する比較実験
要約
この実施例を形成する実験では、本発明のポリペプチドの固相ペプチド合成が記述される。4つの位置、即ち[N23T]、[A42S]、[A46S]及び[A54S]で導入された変異は、簡略化された略号Fmoc−Xxx−Yyy−OHを有する疑似プロリン前駆体との代替合成戦略を用いることを可能にした。
上述の4つの位置で疑似プロリンを使うことによって、全体のAffibody(登録商標)分子X(配列番号7)(即ち、アミノ酸1〜58)を合成することを可能にしたが、一方、標準的な合成はペプチドを製造できなかった。
【0144】
新規なセリン又はトレオニン残基の導入は、また、イソアシルジペプチドの使用を可能にし、それはペプチド合成中凝集を減らすことによって合成効率を高めるための疑似プロリンに対する代替である(Sohma et al., Tetrahedron Lett.47:3013(2006))。いくつかのこのようなビルディングブロックは、Novabiochem of MerckBiosciences AGから入手することができる。
【0145】
理論的根拠
このAffibody(登録商標)分子のN末端へのDOTAのカップリングと同様にAffibody(登録商標)分子Z00342(ZHER2:107として国際公開第2005/003156号に開示され、ときどきZHER2:342とも呼ばれる)のペプチド合成は可能であり、文献(Orlova, A
et al.(2006),Cancer Research 67:2178-2186)に記述されている。しかしながら、合成後のペプチド収率における大きな変動が観察された。ペプチドを再現よく合成する上での困難は、一次アミノ酸配列と同様にペプチドの長さの両方に関連づけることができる。また、さらに保護されたアミノ酸側鎖の反応基を持った長いペプチドは、好ましくない二次構造、即ち固相ペプチド合成を妨げ得るβシートを生成し得る(Quibell, M. & Johnson, T., in Fmoc Solid Phase Peptide Synthesis-A Practical Approach, W.C. Chan, P.D. White Eds, Oxford University Press 2000, 115-135)。ペプチド合成中二次構造形成を防ぐ1つの方法は、疑似プロリンの使用である。もしアミノ酸Yyyがセリン、トレオニン又はシステインであれば、簡略化された略記Fmoc−Xxx−Yyy−OHを有する疑似プロリンが使用できる。これらの疑似プロリンは、骨格に結合した側鎖を有する閉環したプロリン状構造を示し、そして酸処理によって正常なアミノ酸の構造に変換できる(Haack, T., & Mutter, M. Tetrahedron Letters (1992), 33(12), 1589-92)。疑似プロリンは、位置Yyy中のセリン又はトレオニンと共に、位置Xxx(Arg,Cys,His,Met,Pro,Thrを除く)における14のアミノ酸について市販されている。
【0146】
親Affibody(登録商標)分子Z00342は、一次配列に一切のトレオニン及びシステインを持たない。セリンは、単に位置33、39及び41にのみ見出される。疑似プロリン前駆体は、わずかセリン41(Q40〜S41)に対してのみ入手できる。2つの他のセリンについては、入手可能な前駆体(R32〜S33及びP38〜S39)が一切ないので、位置Xxxにあるアミノ酸が疑似プロリンの使用を妨げる。
【0147】
本発明のポリペプチドに導入される変異は、ペプチド合成を促進することを目標とするが、それに限定されるものではない。特に、位置23、42、46及び54、即ち[N23T]、[A42S]、[A46S]及び[A54S]での変異は、SPPSで確認された問題の2つを解決する能力を有し得る:それらは、疑似プロリンの使用を可能にし、アミノ酸21〜26のまわりの臨界領域(critical region)はアスパラギンをスレオニンに置換することによって、位置23に変化する。
【0148】
固相ペプチド合成(SPPS)。
Affibody(登録商標)分子X(配列番号7)のアミノ酸配列は、完全に自動化ペプチド合成機でのFmoc−Lys(Boc)−Wangポリスチレン樹脂に組み立てられた。この樹脂は、Fmoc戦略を持ったペプチドの形成に非常に適している。57個のアミノ酸(適切な側鎖の保護で)は樹脂上でカップリングされた。最終工程の、S−トリチル保護メルカプト酢酸のカップリングは手動で行った。
【0149】
工程1:固相ペプチド合成
Fmoc−Lys(Boc)−Wangポリスチレン樹脂を、撹拌機を備えたSPPS反応器中に移した。次いで、合成は樹脂のFmoc脱保護から始め、続いて以下に示される概要に記載のFmoc−Pro−OHを有するカップリング手順を続けた。この工程の後、再度、Fmoc脱保護及びその後手順に従ってアミノ酸誘導体のカップリングを続けた。イソプロピルエーテル(IPE)での樹脂の最終洗浄の後、ペプチド樹脂を減圧下のデシケーターで乾燥した。
【0150】
標準的なFmocペプチド合成と、4つの位置で疑似プロリンを使用する合成の両方が行われた。標準的なペプチド合成については、Fmocアミノ酸しか使用されなかった。Fmocアミノ酸は別として代替ペプチド合成については、以下の疑似プロリンが使用された:位置22〜23でのFmoc−Leu−Thr−OH、位置41〜42でのFmoc−Ser−Ser−OH、位置45〜46でのFmoc−Leu−Ser−OH、及び位置53〜54でのFmoc−Asp−Ser−OH。
【0151】
Fmoc脱保護手順
N−α−Fmoc保護基の開裂を達成するために、樹脂を、また、NMP中の20%ピペリジンで処理した。次いで樹脂の洗浄をNMPで行った。
【0152】
カップリング手順
Glu1へのアミノ酸誘導体Pro57の自動化カップリング。最大3当量のFmoc−AA誘導体をNMP中に溶解した。カップリングのために、ジメチルホルムアミド(DMF)中の2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムテトラフルオロボラート(TBTU)及びNMP中のsym.−コリジン(2,4,6−トリメチルピリジン)を添加した。生成溶液を、樹脂上に注ぐ前に室温で混合した。NMPを溶媒として使用した。60℃で少なくとも15分のカップリング時間の後で、樹脂をNMPで洗浄した。
【0153】
各カップリング手順の後、試薬としてのDMF及び溶媒としてのジクロルエタン中の2−(1H−7−アザベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムテトラフルオロボラート(TATU)でのカップリングを自動的に繰り返し、引き続いて無水酢酸キャッピングを行った。
【0154】
工程2メルカプト酢酸のカップリング
アシル化は、5モル当量のアミノ酸、2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3,−テトラメチルウロニウムヘキサフルオロホスファート(HBTU)及び1−ヒドロキシベンゾトリアゾール(HOBt)及び10当量のN−エチルジイソプロピルアミン(DIEA、Lancaster Synthesis, Morecambe, England製)によって行なった。S−トリチルメルカプト酢酸は、AnaSpec Inc(San Jose, CA, USA)製であった。
【0155】
工程3:残りの保護基の開裂を含む樹脂からの開裂
ペプチド樹脂を、純水、エタンジチオール(EDT)及びトリイソプロピルシラン(TIS)の存在下にトリフルオロ酢酸(TFA)処理した。室温で約2時間の開裂後に、反応混合物を約0℃に冷却した。ヨウ化アンモニウム及びジメチルサルファイドを添加して、酸化メチオニンを還元した。約0℃で更に60分の開裂の後に、形成したヨウ素をアスコルビン酸で還元した。生成物を濾別した後、冷却しながらIPE中に沈殿させ、再度濾別し、IPEで洗浄し、減圧下で乾燥した。
【0156】
HPLCによる純度分析
58個のアミノ酸の長いペプチド及びいくつかの中間体の純度は、Vydac 218 TP54(5μm、250×4.6mm)カラムを使用し、0.1%のTFA及び1%のアセトニトリルを含有する水、0.1%のTFAを含有するアセトニトリル溶液をそれぞれ溶媒A及びBとして、逆相HPLCによって決定した。カラムオーブン温度は35℃に設定した。カラムを、30分で溶媒Bを15〜45%にするグラジエント、又は30分でBを20〜50%にするグラジエントのどちらかで溶離した。UV検出は220nmで行った。純度は、面積正規化によって計算した。
【0157】
結果
疑似プロリンを使用して又はしないで合成された、Affibody(登録商標)分子X(配列番号7)の収率及び純度は、分析用逆相クロマトグラフィーによって分析した。合成の進行をフォローするために、少量の合成樹脂を、いくつかのカップリング工程後に抜き取り、所望のペプチド中間体の存在、純度及び収率を分析した。図11は、41のアミノ酸の長さのペプチド中間体(アミノ酸18〜58)の分析を示す。ペプチド合成のこの段階では、正しい配列を有する1つの明確な及び主たるペプチドピーク(保持時間=15.33分、収率49%)が、疑似プロリンを使用して合成が行われたかどうかを同定した(図11A)。しかしながら、標準的なFmoc合成は、同じサイズを有する膨大な数の小さなペプチドピーク及び2つのメインピークをもたらしたが、低収率であった。この2つのピーク(保持時間=20.82分)の1つは、正しい配列(aa18〜58)を有するペプチド中間体として同定された(図11B)。全長ペプチド(アミノ酸1〜58)は、合成が疑似プロリンを使用して行われた場合に限り得られた。図12Aは、26%の最終的なペプチドの収率を有する単一の生成物ピークを示す。しかしながら、標準的なFmoc合成は、最終的なペプチド生成物を生成できなかた。標準的な合成からの49個のアミノ酸の長さの中間体(アミノ酸10〜58)の分析は、所望の中間体が検出できなかったこと及び合成は中止されたことを明らかにした(図12B)。
【0158】
〔実施例5〕
本発明のポリペプチド及びZ00342の親水性の比較試験
要約
この実施例を形成している実験では、2つの方法:1)タンパク質の疎水性/親水性プロット;及び2)逆相HPLC溶離プロフィール;を使用し、本発明のポリペプチドの親水性の増加が記述される。
【0159】
1)タンパク質の疎水性/親水性プロット
疎水性/親水性プロットは、タンパク質の配列に沿った極性及び無極性の残基の分布を表示するために作成される。高度に疎水性の領域、例えば膜貫通部分又は親水性の配列、即ちタンパク質の表面に曝されやすい領域を予測するために、これらのプロットは一般に使用される。
【0160】
プロットは、分析の目的に適合できる移動ウインドウ付きのタンパク質配列を走査することによって作成される。例えば、7アミノ酸の移動ウインドウは、推定表面露出領域を見つけるためのよい値であると示唆されている。各位置では、ウインドウ中のアミノ酸の平均の疎水性指数が算出される。この値は、ウインドウの中間点としてプロットされる。この実施例を構成している疎水性/疎水性プロットのため、以下の2つの疎水性スケールが使われた:1)Kyte-Doolittleスケールは、正の値を達成するためのハイドロパティック(Hydropathic)領域であった(Kyte J., Doolittle R.F. (1982) J. Mol. Biol. 157:105-132)、そして2)HPLCによって求められるpH3.4での疎水性指標(Cowan R., Whittaker R.G. (1990) Peptide Research 3:75-80)。プロットは、Vector NTI Suite 9.0.0の構成要素であるプログラムBioAnnotatorwo(Invitrogen)を使用して作成された。
【0161】
E.coliにおける発現のため及び放射性核種での標識化のために配列を最適化するために、10〜12個のアミノ酸のAffibody(登録商標)分子、即ち分子の標的結合活性に関与しないアミノ酸(1つのMet9は別として)は、可能な場合は、より親水性アミノ酸及び/又は全体的な抗原性を低減するアミノ酸に変更された。
【0162】
本発明のポリペプチドに対する疎水性/親水性プロットの2つの実施例は、図13A及びBに示される。比較のために、親Affibody(登録商標)分子Z00342用の図が、図13Cに示される。
【0163】
Affibody (登録商標)分子II(配列番号5)の配列では、以下のアミノ酸の変更がなされた:
・これらの変異の4つは、非極性疎水性アミノ酸を極性アミノ酸[F5Y]、[A42S]、[A46S]及び[A54S]に交換した;
・その2つは、極性アミノ酸を無極性疎水性アミノ酸[N3A]及び[N6A]に変更した;
・その1つは、未変化の極性アミノ酸を正に荷電した極性アミノ酸[S33K]に変更した;
・その1つは、極性アミノ酸を負に荷電したアミノ酸(N43E)に変更した;
・その1つは、極性アミノ酸をより小さい極性アミノ酸(N23T)に変更した;
・その1つは、非極性疎水性アミノ酸をより小さい疎水性アミノ酸(V1A)に変更した;
・その1つは、負に荷電したアミノ酸を別の負に荷電したアミノ酸(D2E)に変更した。
【0164】
親Affibody(登録商標)分子Z00342に比べて、図13AのAffibody(登録商標)分子II用のプロットは、αヘリックス2及び3及びC末端(アミノ酸24〜58の間)を構成しているAffibody(登録商標)分子の領域における親水性を高めた(図13C)。
【0165】
Affibody (登録商標)分子X(配列番号7)の配列では、Affibody(登録商標)分子IIに関しては同一のアミノ酸の変更が行われたが、最初の3つのアミノ酸は、変更した場合はより親水性のアミノ酸に変わった:
・これらの変異の1つは、非極性疎水性アミノ酸を負に荷電したアミノ酸(V1E)に交換した;
・その1つは、負に荷電したアミノ酸を極性アミノ酸(D2S)に変更した;
・その1つは、荷電していない極性アミノ酸を負に荷電したアミノ酸(N3E)に変更した。
【0166】
図13B中のAffibody(登録商標)(配列番号7)分子Xに対するプロットは、親Affibody(登録商標)分子Z00342と比較すると、αへリックス2及び3(アミノ酸24〜58の間)及び親水性N末端でも、同じ親水性の増加を示す(図13C)。
【0167】
2)逆相HPLC溶離プロフィール
逆相(RP)クロマトグラフィーによって効率的にタンパク質を分離することができる。タンパク質の親水性/疎水性の違いは、より親水性のタンパク質がより疎水性タンパク質よりも早く、即ち低濃度のアセトニトリル溶離液で溶離するRPカラム、例えばC8又はC18カラム上で分析することができる。Affibody (登録商標)分子II(配列番号5)及び親Affibody(登録商標)分子Z00342の溶離を比較するために、以下の条件を使用した:HPLC機器:アジレント1100(Agilent);カラム:Zorbax 300SB C8 150×2.1mm、3.5μm(Agilent);温度:30℃;ロード:20μlの2.2mg/mlを有するタンパク質溶液;流量0.5ml/分;溶媒A:0.5%のトリフルオロ酢酸(TFA)水溶液;溶媒B:90%アセトニトリル+0.5%TFAの水溶液;グラジエント;0〜2分は10%のB、2〜17分は10〜70%のB、17〜18分は70〜100%のB、18〜21分は100%のB、21〜22分は100〜10%のB、22〜25分は10%のB。分析結果を図14に示す。保持時間におけるシフトは、親Affibody(登録商標)分子Z00342と比較して、Affibody(登録商標)分子IIの親水性の増加を示している。上述の全ての変異を有するこのAffibody(登録商標)分子は、親Affibody(登録商標)分子Z00342での9.860と比較すると、9.614分の保持時間で溶離する。
【0168】
実施例6
−本発明のポリペプチド及びZ00342の抗原性(IgG結合)の比較試験
要約
本発明に記載のHER2特異的ポリペプチドの1つの望ましい性質は、免疫グロブリン(Ig)との低い相互作用の可能性を示すことを意味する、低抗原性プロファイルを持つことである。この実施例では、インビトロで抗原性(IgG結合)を測定する方法が記述されており、行われたアミノ酸の変更による影響を評価するために、C末端システインに共役したAffibody(登録商標)分子I、II、DOTAを有するII及び内部のシステイン(位置42)上で共役したDOTAを有するXIを、標準的なHER2特異的Affibody(登録商標)分子Z00342と比較した。使用されたアッセイは、インビトロの抗原性(IVA)ELISAと呼ばれている。
【0169】
インビトロの抗原性(IVA)ELISA
IVA ELISAでは、96ウェルのプレートを、2μg/mlの標準コーティング試験として選ばれたHER2特異的Affibody(登録商標)分子Z00342でコーティングした。また、プレートを、2μg/mlのHER2特異的Affibody(登録商標)分子I、II、DOTAを有するII及びDOTAを有するXIでコーティングした。50μlのコーティング溶液をウェル毎に添加し、そして半面積のプレートを+4oCで一夜インキュベートした。ELISAプレートを水道水で2回洗浄し、100μlのブロッキング溶液(0.5%のカゼインを有するリン酸緩衝食塩液(PBS)、pH7.4)を各ウェルに添加した。プレートを、その後室温(RT)で1時間インキュベートして空にし、続いて、1から開始して100倍希釈の2倍希釈系列で、50μl/ウェルのさまざまな霊長類の血清を加えた。プレートを室温で1時間インキュベートし、その後PBS+0.05%のTween(PBS−T)で5回洗浄した。検出には、ヤギ抗ヒトIg−HRP抗体を、1/5000に希釈して使用した(Southern Biotechnology)。プレートを、室温で50分間インキュベートし、その後PBS−Tを使用して5回洗浄工程にかけた。発色については、50μlのImmunoPureTMB基質(Pierce)を添加し、プレートを室温で12分間暗所でインキュベーションした。停止液(2MのH2SO4)を加えて呈色反応を止め、吸光度を450nmで測定した。
【0170】
IVA値の計算
上記に示すように、続いてIVA・ELISAを行った。得られた滴定曲線を、XY非線形回帰式を使用してグラフにプロットし、OD0.3での希釈値を求めた。OD0.3での標準希釈液値を100に設定し、それを基準に関連付けることによって、各サンプルのIVA値を算出した。従って、100以下のIVA値は、標準的なZ00342と比較して、その特別のHER2特異的ポリペプチドについては、より低い程度の霊長類Ig結合を反映している。4つの異なるHER2特異的ポリペプチド、即ち、Affibody(登録商標)分子I、II、DOTAを有するII及びXIを有するDOTAについて、インビトロ抗原性を評価した。標準的なタンパク質と比較して、これらのポリペプチドは低いインビトロの抗原性プロファイルを有することを示した。結論は、行われたアミノ酸変更は、免疫グロブリンとの相互作用の可能性の減少をもたらしたということである。
【0171】
【表3】
【特許請求の範囲】
【請求項1】
位置2におけるX1がM、I又はLであり、位置39におけるX2がS又はCである、アミノ酸配列EX1RNAYWEIA LLPNLTNQQK RAFIRKLYDD PSQSSELLX2E AKKLNDSQ(配列番号1)を含むHER2結合ポリペプチド。
【請求項2】
位置5におけるX1がM、I又はLであり、位置42におけるX2がS又はCである、アミノ酸配列YAKEX1RNAYW EIALLPNLTN QQKRAFIRKL YDDPSQSSEL LX2EAKKLNDS Q(配列番号2)を含む、請求項1に記載のHER2結合ポリペプチド。
【請求項3】
位置9におけるX1がM、I又はLであり、位置46におけるX2がS又はCである、アミノ酸配列AEAKYAKEX1R NAYWEIALLP NLTNQQKRAF IRKLYDDPSQ SSELLX2EAKK LNDSQ(配列番号3)を含む、請求項1又は2に記載のHER2結合ポリペプチド。
【請求項4】
位置9におけるX1はM、I又はLであり、位置46におけるX2はS又はCである、アミノ酸配列ESEKYAKEX1R NAYWEIALLP NLTNQQKRAF IRKLYDDPSQ SSELLX2EAKK LNDSQ(配列番号4)を含む、請求項1又は2に記載のHER2結合ポリペプチド。
【請求項5】
アミノ酸配列AEAKYAKEMR NAYWEIALLP NLTNQQKRAF IRKLYDDPSQ SSELLSEAKK LNDSQAPKVD C(配列番号5)を含む、請求項3に記載のHER2結合ポリペプチド。
【請求項6】
アミノ酸配列ESEKYAKEMR NAYWEIALLP NLTNQQKRAF IRKLYDDPSQ SSELLSEAKK LNDSQAPK(配列番号6)を含む、請求項4に記載のHER2結合ポリペプチド。
【請求項7】
そのN末端にカップリングしたメルカプトアセチルを更に含む、請求項1〜6のいずれか1項に記載のHER2結合ポリペプチドであって、N3Sキレート剤モチーフによって構成されるキレート環境が、メルカプトアセチルのSH基と一緒になってN末端からの最初の3つの連続したペプチド結合の窒素原子によって与えられるHER2結合ポリペプチド。
【請求項8】
N末端からの最初の3つの連続したペプチド結合が、残基ESEのアミノ基によって与えられる、請求項7に記載のHER2結合ポリペプチド。
【請求項9】
maがメルカプトアセチルである、配列maESEKYAKEMR NAYWEIALLP NLTNQQKRAF IRKLYDDPSQ SSELLSEAKK LNDSQAPK(配列番号7)を含む、請求項6〜8のいずれか1項に記載のHER2結合ポリペプチド。
【請求項10】
配列中に既に存在するか、又は更なるアミノ酸残基として存在するか、又はHER2結合に関与しない表面に露出したアミノ酸残基を置換することによって存在するいずれかのシステイン残基を含む、請求項1〜6のいずれか1項に記載のHER2結合ポリペプチド。
【請求項11】
請求項10に記載のHER2結合ポリペプチドであって、システイン残基が配列番号5のC末端に位置するものか又は更なるシステインが配列番号1〜4若しくは6〜7のいずれか1つのC末端に位置しており、ここでシステイン残基には、場合により1つ又はそれ以上の他のアミノ酸残基が続き、そしてここでN3Sキレート剤モチーフによって構成されるキレート環境が、システイン残基のSH基と一緒になって、システインによって構成されるアミノ酸残基及び2つの先行するアミノ酸残基の広がりの中の3つの連続したペプチド結合の窒素原子によって与えられる、HER2結合ポリペプチド。
【請求項12】
チオール基を介して、ポリペプチドにカップリングしたポリアミノポリカルボキシラートのキレート剤によって与えられるキレート環境を含む、請求項10に記載のHER2結合ポリペプチド。
【請求項13】
ポリアミノポリカルボキシラートのキレート剤が、1,4,7,10−テトラアザシクロドデカン−1,4,7,10−四酢酸又はその誘導体である、請求項12に記載のHER2結合ポリペプチド。
【請求項14】
1,4,7,10−テトラアザシクロドデカン−1,4,7,10−四酢酸誘導体が、1,4,7,10−テトラアザシクロドデカン−1,4,7−トリス酢酸−10−マレイミドエチルアセトアミドである、請求項13に記載のHER2結合ポリペプチド。
【請求項15】
1,4,7,10−テトラアザシクロドデカン−1,4,7−トリス酢酸−10−マレイミドエチルアセトアミドにカップリングした、配列番号5から成るHER2結合ポリペプチド。
【請求項16】
ポリアミノポリカルボキシラートのキレート剤がジエチレントリアミンペンタ酢酸又はその誘導体である、請求項12に記載のHER2結合ポリペプチド。
【請求項17】
請求項7〜9及び11のいずれか1項に記載のHER2結合ポリペプチド、及び医用画像に適した放射性核種の放射性キレートから成る放射性標識化ポリペプチドであって、該放射性核種が99mTcであるか、又は治療に適した放射性核種とのものであって、該放射性核種が86Re及び188Reから成るグループから選択される、放射性標識ポリペプチド。
【請求項18】
請求項9に記載のHER2結合ポリペプチド及び99mTcの放射性キレートから成る、放射性標識ポリペプチド。
【請求項19】
医用画像に適した放射性核種を有する請求項12〜16のいずれか1項に記載のHER2結合ポリペプチドの放射性キレートから成る放射性標識ポリペプチドであって、該放射性核種が61Cu、64Cu、66Ga、67Ga、68Ga、110mIn、111In、44Sc及び86Yから成るグループから選択され、又は治療に適した放射性核種とのものであって、該放射性核種が225Ac、212Bi、213Bi、67Cu、166Ho、177Lu、212Pb、149Pm、153Sm、227Th及び90Yから成るグループから選択され、ここで、放射性核種は、キレート環境を介してHER2結合ポリペプチドと錯体を形成する、放射性標識ポリペプチド。
【請求項20】
請求項15に記載のHER2結合ポリペプチド及び111Inの放射性キレートから成る、放射性標識ポリペプチド。
【請求項21】
211At、76Br、18F及びヨウ素同位体から成るグループから選択される医用画像及び/又は治療に適した放射性核種に、リンカー分子を介して結合した、請求項10に記載のHER2結合ポリペプチドから成る放射性標識ポリペプチド。
【請求項22】
HER2の過剰発現によって特徴付けられるがんを有する又は疑われる、ヒトを含む哺乳類対象の身体のインビボ画像化の方法であって、以下の工程:
・放射性核種が画像化に適する請求項17〜21のいずれか1項に記載の放射性標識ポリペプチドを、哺乳類対象の体内に投与すること;及び
・放射性標識ポリペプチドの投与の1〜72時間以内に、医用画像機器を使用して、対象の体の少なくとも1部分の1つ又はそれ以上の画像、ここで、該画像は体の内部の放射性核種の存在を指示する、を取得すること;
を含む、上記方法。
【請求項23】
投与工程前に、請求項10〜16のいずれか1項に記載のポリペプチドを適した放射性核種と混合するこを含む、放射性核種が画像化に適する請求項17〜21のいずれか1項に記載の放射性標識ポリペプチドを製造する工程を含む、請求項22に記載の方法。
【請求項24】
投与工程前に、請求項9に記載のポリペプチドを99mTcと混合することを含む、請求項18に記載の放射性標識ポリペプチドを製造する工程を含む、請求項22に記載の方法。
【請求項25】
投与工程前に、請求項15に記載のポリペプチドを111Inと混合することを含む、請求項20に記載の放射性標識ポリペプチドを製造する工程を含む、請求項22に記載の方法。
【請求項26】
がんが、乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんから選択される、請求項22〜25のいずれか1項に記載の方法。
【請求項27】
放射性核種が診断に用いるための画像化に適する、請求項17〜21のいずれか1項に記載の放射性標識化ポリペプチド。
【請求項28】
放射性核種が、HER2の頻発する過剰発現によって特徴付けられるがんのグループに属するがんの診断に用いるための画像化に適する、請求項17〜21のいずれか1項に記載の放射性標識ポリペプチド。
【請求項29】
放射性核種が、乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんから選択されるがんの診断及び/又は分子的特性化に用いるための画像化に適する、請求項17〜21のいずれか1項に記載の放射性標識ポリペプチド。
【請求項30】
放射性核種が画像化に適する請求項17〜21のいずれか1項に記載の放射性標識ポリペプチドの、ヒトを含む哺乳動物対象の体のインビボ画像化のための診断薬の製造における使用。
【請求項31】
HER2の過剰発現によって特徴付けられるがんを有する、ヒトを含む哺乳動物対象の治療方法であって、放射性核種が治療に適する、請求項17、19又は21に記載の放射性標識ポリペプチドの治療有効量を対象の身体内に投与する工程を含む方法。
【請求項32】
投与工程前に、放射性核種が治療に適する請求項17、19又は21に記載の放射性標識ポリペプチドを製造する工程であって、請求項10〜16のいずれか1項に記載のポリペプチドを適した放射性核種と混合することを含む工程を含む、請求項31に記載の方法。
【請求項33】
がんが、乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんから選択される、請求項31又は32に記載の方法。
【請求項34】
治療に使用するための、放射性核種が治療に適する請求項17、19又は21に記載の放射性標識化ポリペプチド。
【請求項35】
HER2の過剰発現によって特徴付けられるがんの治療に使用するための、放射性核種が治療に適する請求項17、19又は21に記載の放射性標識ポリペプチド。
【請求項36】
乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんより選択されるがんの治療に使用するための、放射性核種が治療に適する請求項17、19又は21に記載の放射性標識ポリペプチド。
【請求項37】
HER2の過剰発現によって特徴付けられるがんの治療のための薬剤の製造のための、放射性核種が治療に適する請求項17、19又は21に記載の放射性標識ポリペプチドの使用。
【請求項38】
乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんから選ばれたがんの治療用の薬剤を調製するための、放射性核種が治療に適する請求項17、19又は21に記載の放射性標識ポリペプチドの使用。
【請求項39】
請求項1〜6又は10のいずれか1項に記載のポリペプチドをコードする核酸。
【請求項40】
請求項39に記載の核酸を含む発現ベクター。
【請求項41】
請求項39に記載の発現ベクターを含む宿主細胞。
【請求項1】
位置2におけるX1がM、I又はLであり、位置39におけるX2がS又はCである、アミノ酸配列EX1RNAYWEIA LLPNLTNQQK RAFIRKLYDD PSQSSELLX2E AKKLNDSQ(配列番号1)を含むHER2結合ポリペプチド。
【請求項2】
位置5におけるX1がM、I又はLであり、位置42におけるX2がS又はCである、アミノ酸配列YAKEX1RNAYW EIALLPNLTN QQKRAFIRKL YDDPSQSSEL LX2EAKKLNDS Q(配列番号2)を含む、請求項1に記載のHER2結合ポリペプチド。
【請求項3】
位置9におけるX1がM、I又はLであり、位置46におけるX2がS又はCである、アミノ酸配列AEAKYAKEX1R NAYWEIALLP NLTNQQKRAF IRKLYDDPSQ SSELLX2EAKK LNDSQ(配列番号3)を含む、請求項1又は2に記載のHER2結合ポリペプチド。
【請求項4】
位置9におけるX1はM、I又はLであり、位置46におけるX2はS又はCである、アミノ酸配列ESEKYAKEX1R NAYWEIALLP NLTNQQKRAF IRKLYDDPSQ SSELLX2EAKK LNDSQ(配列番号4)を含む、請求項1又は2に記載のHER2結合ポリペプチド。
【請求項5】
アミノ酸配列AEAKYAKEMR NAYWEIALLP NLTNQQKRAF IRKLYDDPSQ SSELLSEAKK LNDSQAPKVD C(配列番号5)を含む、請求項3に記載のHER2結合ポリペプチド。
【請求項6】
アミノ酸配列ESEKYAKEMR NAYWEIALLP NLTNQQKRAF IRKLYDDPSQ SSELLSEAKK LNDSQAPK(配列番号6)を含む、請求項4に記載のHER2結合ポリペプチド。
【請求項7】
そのN末端にカップリングしたメルカプトアセチルを更に含む、請求項1〜6のいずれか1項に記載のHER2結合ポリペプチドであって、N3Sキレート剤モチーフによって構成されるキレート環境が、メルカプトアセチルのSH基と一緒になってN末端からの最初の3つの連続したペプチド結合の窒素原子によって与えられるHER2結合ポリペプチド。
【請求項8】
N末端からの最初の3つの連続したペプチド結合が、残基ESEのアミノ基によって与えられる、請求項7に記載のHER2結合ポリペプチド。
【請求項9】
maがメルカプトアセチルである、配列maESEKYAKEMR NAYWEIALLP NLTNQQKRAF IRKLYDDPSQ SSELLSEAKK LNDSQAPK(配列番号7)を含む、請求項6〜8のいずれか1項に記載のHER2結合ポリペプチド。
【請求項10】
配列中に既に存在するか、又は更なるアミノ酸残基として存在するか、又はHER2結合に関与しない表面に露出したアミノ酸残基を置換することによって存在するいずれかのシステイン残基を含む、請求項1〜6のいずれか1項に記載のHER2結合ポリペプチド。
【請求項11】
請求項10に記載のHER2結合ポリペプチドであって、システイン残基が配列番号5のC末端に位置するものか又は更なるシステインが配列番号1〜4若しくは6〜7のいずれか1つのC末端に位置しており、ここでシステイン残基には、場合により1つ又はそれ以上の他のアミノ酸残基が続き、そしてここでN3Sキレート剤モチーフによって構成されるキレート環境が、システイン残基のSH基と一緒になって、システインによって構成されるアミノ酸残基及び2つの先行するアミノ酸残基の広がりの中の3つの連続したペプチド結合の窒素原子によって与えられる、HER2結合ポリペプチド。
【請求項12】
チオール基を介して、ポリペプチドにカップリングしたポリアミノポリカルボキシラートのキレート剤によって与えられるキレート環境を含む、請求項10に記載のHER2結合ポリペプチド。
【請求項13】
ポリアミノポリカルボキシラートのキレート剤が、1,4,7,10−テトラアザシクロドデカン−1,4,7,10−四酢酸又はその誘導体である、請求項12に記載のHER2結合ポリペプチド。
【請求項14】
1,4,7,10−テトラアザシクロドデカン−1,4,7,10−四酢酸誘導体が、1,4,7,10−テトラアザシクロドデカン−1,4,7−トリス酢酸−10−マレイミドエチルアセトアミドである、請求項13に記載のHER2結合ポリペプチド。
【請求項15】
1,4,7,10−テトラアザシクロドデカン−1,4,7−トリス酢酸−10−マレイミドエチルアセトアミドにカップリングした、配列番号5から成るHER2結合ポリペプチド。
【請求項16】
ポリアミノポリカルボキシラートのキレート剤がジエチレントリアミンペンタ酢酸又はその誘導体である、請求項12に記載のHER2結合ポリペプチド。
【請求項17】
請求項7〜9及び11のいずれか1項に記載のHER2結合ポリペプチド、及び医用画像に適した放射性核種の放射性キレートから成る放射性標識化ポリペプチドであって、該放射性核種が99mTcであるか、又は治療に適した放射性核種とのものであって、該放射性核種が86Re及び188Reから成るグループから選択される、放射性標識ポリペプチド。
【請求項18】
請求項9に記載のHER2結合ポリペプチド及び99mTcの放射性キレートから成る、放射性標識ポリペプチド。
【請求項19】
医用画像に適した放射性核種を有する請求項12〜16のいずれか1項に記載のHER2結合ポリペプチドの放射性キレートから成る放射性標識ポリペプチドであって、該放射性核種が61Cu、64Cu、66Ga、67Ga、68Ga、110mIn、111In、44Sc及び86Yから成るグループから選択され、又は治療に適した放射性核種とのものであって、該放射性核種が225Ac、212Bi、213Bi、67Cu、166Ho、177Lu、212Pb、149Pm、153Sm、227Th及び90Yから成るグループから選択され、ここで、放射性核種は、キレート環境を介してHER2結合ポリペプチドと錯体を形成する、放射性標識ポリペプチド。
【請求項20】
請求項15に記載のHER2結合ポリペプチド及び111Inの放射性キレートから成る、放射性標識ポリペプチド。
【請求項21】
211At、76Br、18F及びヨウ素同位体から成るグループから選択される医用画像及び/又は治療に適した放射性核種に、リンカー分子を介して結合した、請求項10に記載のHER2結合ポリペプチドから成る放射性標識ポリペプチド。
【請求項22】
HER2の過剰発現によって特徴付けられるがんを有する又は疑われる、ヒトを含む哺乳類対象の身体のインビボ画像化の方法であって、以下の工程:
・放射性核種が画像化に適する請求項17〜21のいずれか1項に記載の放射性標識ポリペプチドを、哺乳類対象の体内に投与すること;及び
・放射性標識ポリペプチドの投与の1〜72時間以内に、医用画像機器を使用して、対象の体の少なくとも1部分の1つ又はそれ以上の画像、ここで、該画像は体の内部の放射性核種の存在を指示する、を取得すること;
を含む、上記方法。
【請求項23】
投与工程前に、請求項10〜16のいずれか1項に記載のポリペプチドを適した放射性核種と混合するこを含む、放射性核種が画像化に適する請求項17〜21のいずれか1項に記載の放射性標識ポリペプチドを製造する工程を含む、請求項22に記載の方法。
【請求項24】
投与工程前に、請求項9に記載のポリペプチドを99mTcと混合することを含む、請求項18に記載の放射性標識ポリペプチドを製造する工程を含む、請求項22に記載の方法。
【請求項25】
投与工程前に、請求項15に記載のポリペプチドを111Inと混合することを含む、請求項20に記載の放射性標識ポリペプチドを製造する工程を含む、請求項22に記載の方法。
【請求項26】
がんが、乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんから選択される、請求項22〜25のいずれか1項に記載の方法。
【請求項27】
放射性核種が診断に用いるための画像化に適する、請求項17〜21のいずれか1項に記載の放射性標識化ポリペプチド。
【請求項28】
放射性核種が、HER2の頻発する過剰発現によって特徴付けられるがんのグループに属するがんの診断に用いるための画像化に適する、請求項17〜21のいずれか1項に記載の放射性標識ポリペプチド。
【請求項29】
放射性核種が、乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんから選択されるがんの診断及び/又は分子的特性化に用いるための画像化に適する、請求項17〜21のいずれか1項に記載の放射性標識ポリペプチド。
【請求項30】
放射性核種が画像化に適する請求項17〜21のいずれか1項に記載の放射性標識ポリペプチドの、ヒトを含む哺乳動物対象の体のインビボ画像化のための診断薬の製造における使用。
【請求項31】
HER2の過剰発現によって特徴付けられるがんを有する、ヒトを含む哺乳動物対象の治療方法であって、放射性核種が治療に適する、請求項17、19又は21に記載の放射性標識ポリペプチドの治療有効量を対象の身体内に投与する工程を含む方法。
【請求項32】
投与工程前に、放射性核種が治療に適する請求項17、19又は21に記載の放射性標識ポリペプチドを製造する工程であって、請求項10〜16のいずれか1項に記載のポリペプチドを適した放射性核種と混合することを含む工程を含む、請求項31に記載の方法。
【請求項33】
がんが、乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんから選択される、請求項31又は32に記載の方法。
【請求項34】
治療に使用するための、放射性核種が治療に適する請求項17、19又は21に記載の放射性標識化ポリペプチド。
【請求項35】
HER2の過剰発現によって特徴付けられるがんの治療に使用するための、放射性核種が治療に適する請求項17、19又は21に記載の放射性標識ポリペプチド。
【請求項36】
乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんより選択されるがんの治療に使用するための、放射性核種が治療に適する請求項17、19又は21に記載の放射性標識ポリペプチド。
【請求項37】
HER2の過剰発現によって特徴付けられるがんの治療のための薬剤の製造のための、放射性核種が治療に適する請求項17、19又は21に記載の放射性標識ポリペプチドの使用。
【請求項38】
乳がん、卵巣がん、胃がん、膀胱がん、唾液腺がん、肺がん及び食道におけるがんから選ばれたがんの治療用の薬剤を調製するための、放射性核種が治療に適する請求項17、19又は21に記載の放射性標識ポリペプチドの使用。
【請求項39】
請求項1〜6又は10のいずれか1項に記載のポリペプチドをコードする核酸。
【請求項40】
請求項39に記載の核酸を含む発現ベクター。
【請求項41】
請求項39に記載の発現ベクターを含む宿主細胞。
【図1】

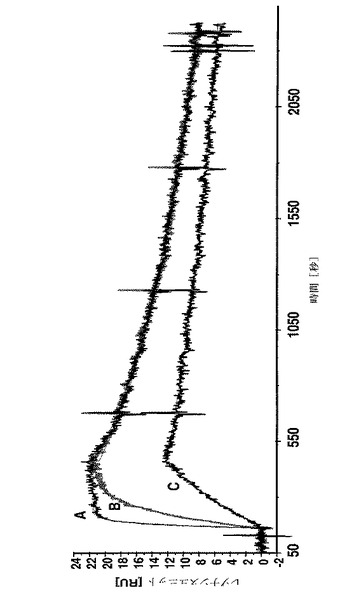
【図2】
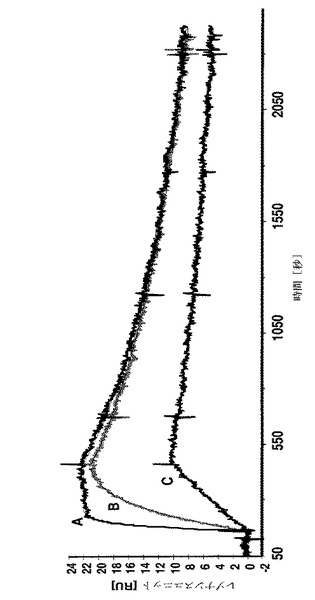
【図3】

【図4】
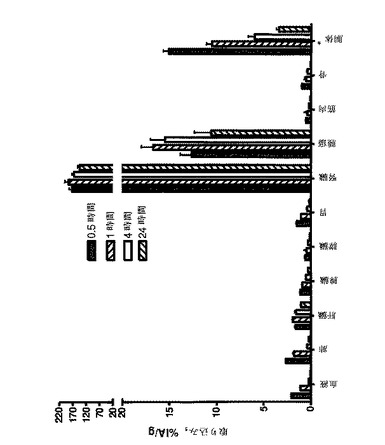
【図5】
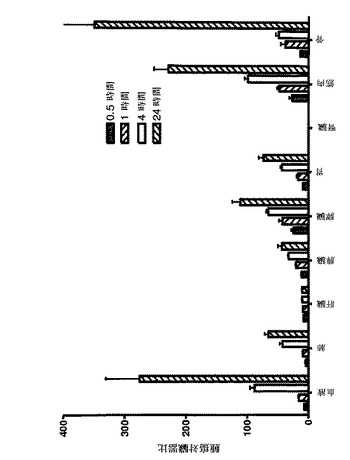
【図6】
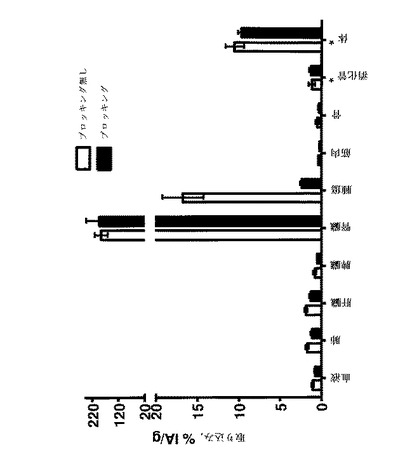
【図7】
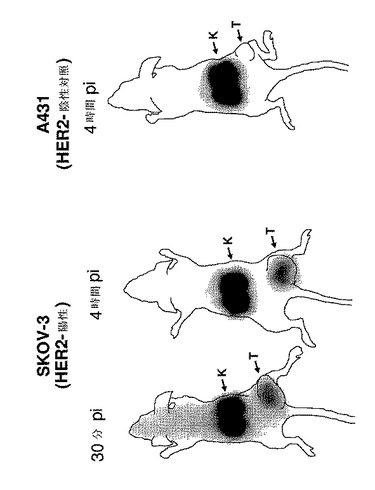
【図8】
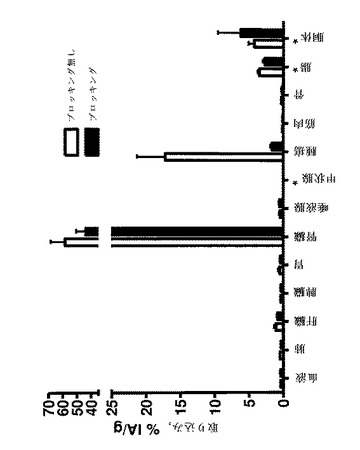
【図9】
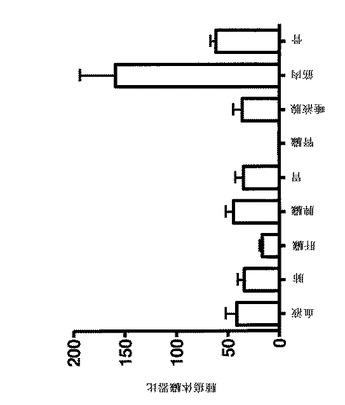
【図10】
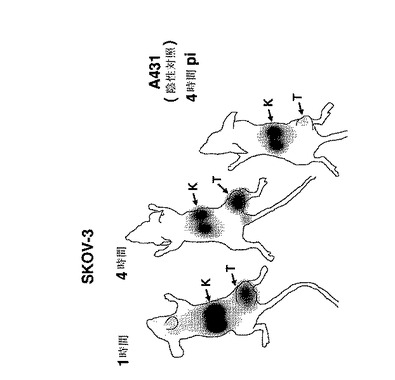
【図11A】
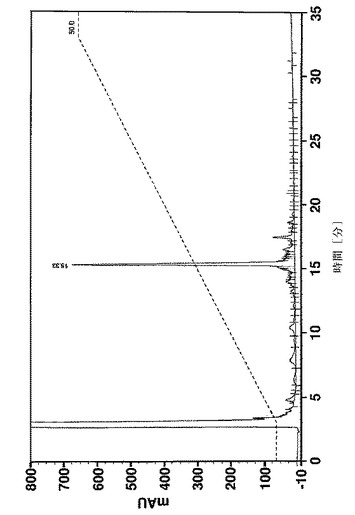
【図11B】
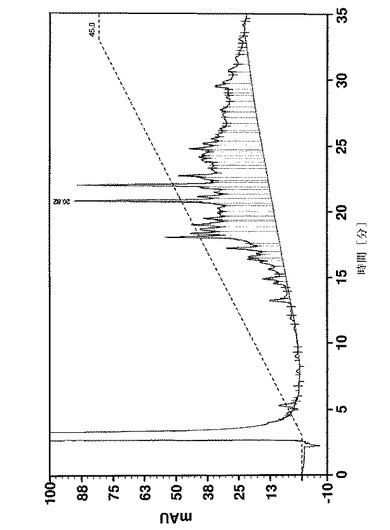
【図12A】
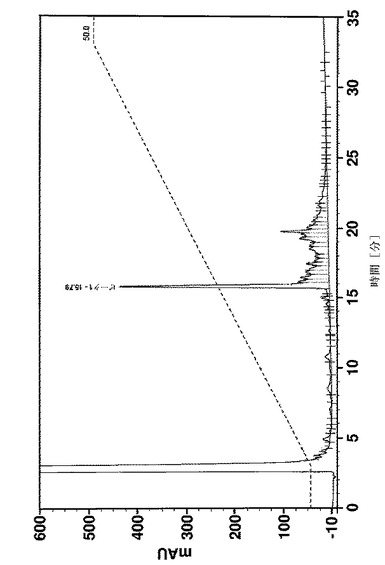
【図12B】
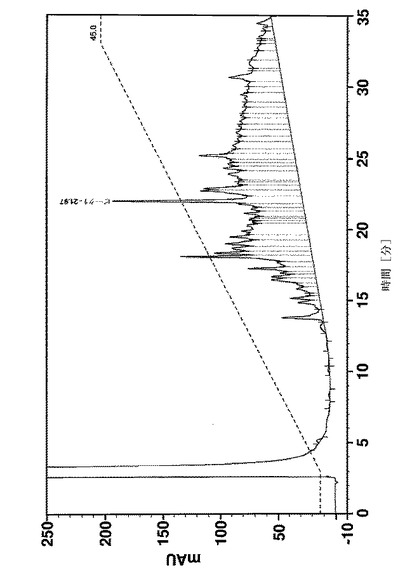
【図13A】
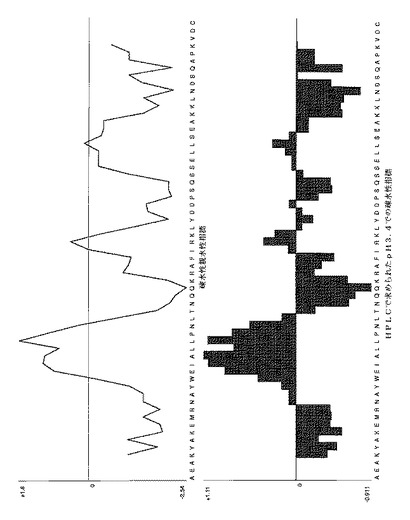
【図13B】
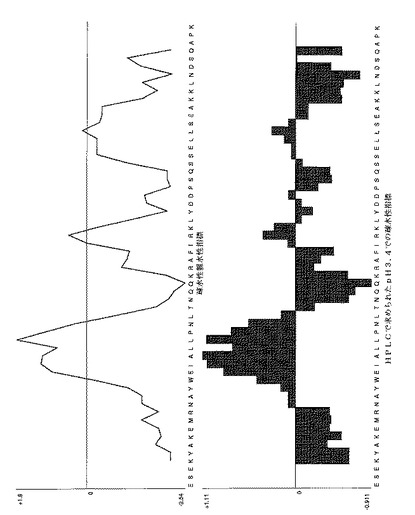
【図13C】
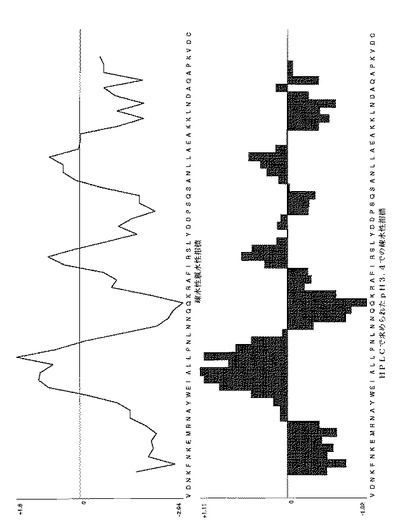
【図14】
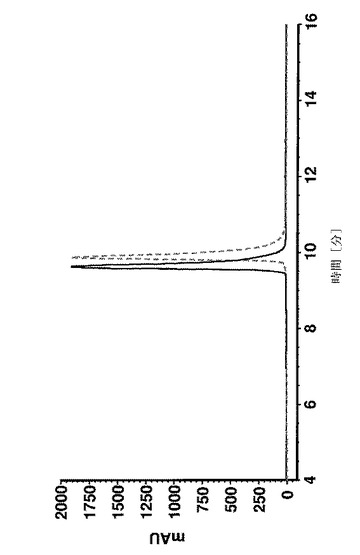
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11A】
【図11B】
【図12A】
【図12B】
【図13A】
【図13B】
【図13C】
【図14】
【公表番号】特表2011−507497(P2011−507497A)
【公表日】平成23年3月10日(2011.3.10)
【国際特許分類】
【出願番号】特願2010−538788(P2010−538788)
【出願日】平成20年12月22日(2008.12.22)
【国際出願番号】PCT/EP2008/068167
【国際公開番号】WO2009/080810
【国際公開日】平成21年7月2日(2009.7.2)
【出願人】(307015286)アフィボディ・アーベー (9)
【Fターム(参考)】
【公表日】平成23年3月10日(2011.3.10)
【国際特許分類】
【出願日】平成20年12月22日(2008.12.22)
【国際出願番号】PCT/EP2008/068167
【国際公開番号】WO2009/080810
【国際公開日】平成21年7月2日(2009.7.2)
【出願人】(307015286)アフィボディ・アーベー (9)
【Fターム(参考)】
[ Back to top ]