
HIVプロテアーゼ阻害剤を含む組成物
本発明は、非晶質(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミド、またはその医薬上許容しうる塩または溶媒和物を含んでなる医薬組成物、その製造方法、HIVプロテアーゼ酵素の阻害におけるその使用、およびHIV感染した哺乳動物、例えばヒトを治療する医薬の製造におけるその使用に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、HIVプロテアーゼ阻害剤を含む医薬組成物に関する。
【背景技術】
【0002】
(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミド(別名「(R)−3−((2S,3S)−2−ヒドロキシ−3−{[1−(3−ヒドロキシ−2−メチル−フェニル)−メタノイル]−アミノ}−4−フェニル−ブタノイル)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド」、「(4R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチル−ベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド」、または「4−チアゾリジンカルボキサミド,3−[(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−1−オキソ−4−フェニルブチル]−5,5−ジメチル−N−2−プロペニル−,(4R)−」であり、以下、「化合物A」と称する)は、HIV感染した哺乳動物、例えばヒトの治療に使用することができるHIVプロテアーゼ酵素の阻害剤である。化合物Aおよびその製造は、2002年6月11日に出願された米国特許出願第10/166,979号;2003年12月4日に出願された同第60/527,477号、および2003年12月4日に出願された同第10/782,602号に開示されており、これらはすべて参照により本明細書に組み込まれている。結晶質化合物Aは水溶液中できわめて僅かの可溶性しかなく、非緩衝化水(pH8.3)、標準生理食塩水(pH8.2)および0.1N HCl(pH1.2)中では、水溶解度はそれぞれ約0.16mgA/mL、0.14mgA/mLおよび0.16mgA/mL(温度26℃)である。この低い水溶解度と低いin vivo浸透性が組み合わさると、結晶質化合物Aは経口生物学的利用能が低くなる。
【発明の開示】
【発明が解決しようとする課題】
【0003】
従って、剤形中の化合物Aの安定性を維持しながら、化合物Aの生物学的利用能を改善する必要がある。
【課題を解決するための手段】
【0004】
本発明は、非晶質化合物Aを含んでなる医薬組成物を提供する。非晶質化合物Aは、化合物Aの結晶質形態Iと比較して改善された溶解性を有し、経口的に哺乳動物、例えばヒトに投与した時に、結晶質形態と比較して改善された生物学的利用能が得られる。
【0005】
本発明の1つの態様は、非晶質化合物Aまたはその医薬上許容しうる塩または溶媒和物を提供する。
本発明の別の態様は、非晶質化合物Aまたはその医薬上許容しうる塩または溶媒和物を含んでなる医薬組成物を提供する。
【0006】
本発明のさらに別の態様において、存在する化合物Aの総量の少なくとも約5質量%が非晶質形態である、化合物Aまたはその医薬上許容しうる塩または溶媒和物を含んでなる医薬組成物が提供される。別法として、存在する化合物Aの総量の少なくとも約10質量%、または少なくとも約15質量%、または少なくとも約20質量%、または少なくとも約30質量%、または少なくとも約40質量%、または少なくとも約50質量%、または少なくとも約60質量%、または少なくとも約70質量%、または少なくとも約80質量%、または少なくとも約90質量%、または少なくとも約95質量%が非晶質形態である、化合物Aまたはその医薬上許容しうる塩または溶媒和物を含んでなる医薬組成物が提供される。
【0007】
一態様において、医薬組成物は、(1)非晶質化合物A、またはその医薬上許容しうる塩または溶媒和物、および(2)マトリックスを含んでなる。
【0008】
一実施態様において、医薬組成物は、(1)非晶質化合物A、および、(2)濃度増大ポリマーを含むマトリックスを含んでなる。濃度増大ポリマーは、使用環境中で溶解した化合物Aの濃度をさらに向上させる。結晶質化合物Aによって得られる高い溶解薬物濃度を持続させるには比較的少量のポリマーしか必要とされないことがわかった。非晶質化合物Aを使用環境に単独で投与する場合、化合物Aの溶解濃度は最初に高められており、時間とともに、結晶質形態Iによって得られる平衡濃度はより低い方へ減少する。しかしながら、濃度増大ポリマーを含んでなる組成物では、溶解濃度が最初に高められるだけでなく、生理学上妥当な時間にわたって溶解濃度が持続する。実際、組成物の約25質量%から約1質量%までの範囲の少量のポリマーのみで、結晶質形態Iの化合物A単独だけでなく、非晶質化合物A単独と比較しても化合物Aの溶解性能は実質的に改善される。
【0009】
さらに別の態様において、化合物Aまたはその医薬上許容しうる塩または溶媒和物およびマトリックスを含んでなり、その際、前記マトリックスは、イオン化できるセルロース系ポリマー、イオン化できないセルロース系ポリマー、または非セルロース系ポリマーの少なくとも1つからなる医薬組成物が提供される。
【0010】
なおさらなる態様において、少なくとも1つのイオン化可能なセルロース系ポリマーは、ヒドロキシプロピルメチルセルロースアセテートスクシネート、カルボキシメチルエチルセルロース、セルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースフタレート、メチルセルロースアセテートフタレート、セルロースアセテートトリメリテート、ヒドロキシプロピルセルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースアセテートフタレート、セルロースアセテートテレフタレートおよびセルロースアセテートイソフタレート、およびそれらの混合物の少なくとも1つから選ばれる。
【0011】
またさらに、少なくとも1つのイオン化できないセルロース系ポリマーが、ヒドロキシプロピルメチルセルロースアセテート、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、メチルセルロース、ヒドロキシエチルメチルセルロース、ヒドロキシエチルセルロースアセテートおよびヒドロキシエチルエチルセルロース、およびそれらの混合物から選ばれるこのような組成物が提供される。
【0012】
別の態様では、前記少なくとも1つの非セルロース系ポリマーが、カルボン酸官能化ポリメタクリレート、カルボン酸官能化ポリアクリレート、アミン官能化ポリアクリレート、アミン官能化ポリメタクリレート、タンパク質、カルボン酸官能化デンプン、ヒドロキシル、アルキルアシルオキシおよび環式アミドからなる群より選ばれる少なくとも1つの置換基を有するビニルポリマーおよびコポリマー、少なくとも1つの親水性のヒドロキシルを含む反復単位および少なくとも1つの疎水性のアルキル−またはアリールを含む反復単位のビニルコポリマー、加水分解されてない形態の反復単位の少なくとも一部を有するポリビニルアルコール、ポリビニルアルコールポリビニルアセテートコポリマー、ポリエチレングリコールポリプロピレングリコールコポリマー、ポリビニルピロリドン、ポリエチレンポリビニルアルコールコポリマー、ポリオキシエチレン−ポリオキシプロピレンブロックコポリマーおよびそれらの混合物から選ばれるこのような組成物が提供される。
【0013】
本発明の別の態様において、化合物Aまたはその医薬上許容しうる塩または溶媒和物およびマトリックスを含み、その際、存在する化合物Aの総量の少なくとも約5質量%が非晶質形態である医薬組成物が提供される。別の態様として、化合物A、またはその医薬上許容しうる塩または溶媒和物およびマトリックスを含み、その際、存在する化合物Aの総量の少なくとも約10質量%、または少なくとも約15質量%、または少なくとも質量%、または少なくとも約30質量%、または少なくとも約40質量%、または少なくとも約50質量%、または少なくとも約60質量%、または少なくとも70質量%または少なくとも約80質量%または少なくとも約90質量%、または少なくとも約95質量%が非晶質形態である医薬組成物が提供される。
【0014】
別の態様では、(1)化合物Aおよび、(2)マトリックスを含み、その際、化合物Aの少なくとも一部が非晶質である医薬組成物が提供される。
【0015】
別の実施態様において、医薬組成物は、化合物Aまたはその医薬上許容しうる塩または溶媒和物およびマトリックスを含む固体非晶質分散体を含んでなる。一態様において、固体非晶質分散体は、化合物Aまたはその医薬上許容しうる塩または溶媒和物少なくとも約30質量%を含む。別の態様において、固体非晶質分散体は、化合物Aまたはその医薬上許容しうる塩または溶媒和物の少なくとも約40質量%、少なくとも約50質量%、少なくとも約60質量%、少なくとも約70質量%、少なくとも約80質量%、少なくとも約90質量%、または少なくとも約95質量%を含む。
【0016】
別の実施態様において、本発明は、in vitro水性環境に投与した時に、(a)使用環境における化合物Aの最大溶解濃度が対照組成物によって得られるものの少なくとも約1.25倍である;および(b)使用環境へ導入した時間から使用環境へ導入後の約270分の間の少なくとも90分間のいずれかの期間について使用環境における化合物Aの濃度対時間曲線下の面積(AUC)が、対照組成物の少なくとも約1.25倍である:の少なくとも1つが得られる化合物Aを含んでなる医薬組成物を提供する。対照組成物は、本質的に等量の結晶質形態Iの化合物A単独からなる。別の実施態様において上記議論した使用環境は、本質的にpH6.5および290mOsm/kg、および37℃温度で、20mM Na2HPO4、47mM KH2PO4、87mM NaClおよび0.2mM KClからなり、その際、前記使用環境の総量は約1.8mLであり、使用する化合物Aの量は、化合物Aの全てが溶解した場合、化合物Aの全濃度が3000μg/mLとなるような量である。別の態様において、このような医薬組成物は、化合物Aの少なくとも一部が非晶質形態である化合物Aを含む。さらに別の態様において存在する化合物Aの総量の少なくとも約5質量%が非晶質形態であるこのような組成物が提供される。さらに別の態様において、存在する化合物Aの総量の少なくとも約10質量%、または少なくとも約15質量%、または少なくとも約20質量%、または少なくとも約30質量%、または少なくとも約40質量%、または少なくとも約50質量%、または少なくとも約60質量%、または少なくとも約70質量%、または少なくとも約80質量%、または少なくとも約90質量%、または少なくとも約95質量%が非晶質形態である、このような組成物が提供される。
【0017】
本発明の別の態様において、哺乳動物、例えばヒト中の化合物Aの血漿中濃度約0.001μM〜約5μMを達成する方法が提供され、前記方法は、化合物A単独またはその医薬上許容しうる塩または溶媒和物、またはマトリックスと組み合わせて含んでなる医薬組成物の十分な量を前記哺乳動物に投与することからなる。一実施態様において、組成物は非晶質化合物Aを含んでなる。別の実施態様において、組成物は、非晶質化合物Aおよび少なくとも1つのマトリックスを含んでなる。本発明の別の態様において、哺乳動物、例えばヒトにおける化合物Aの血漿濃度は、約0.01μM〜約2.5μM、または約0.02μM〜約1μM、または約0.025μM〜約1μM、または約0.05μM〜約1μMの範囲にある。
【0018】
本発明の別の態様において、哺乳動物、例えばヒト中の化合物Aの前記血漿中濃度が、前記投与後、少なくとも約6時間、または前記投与後、少なくとも約8、10、12、14、16、18、20、22または24時間維持されるこのような方法が提供される。
【0019】
本発明のさらなる態様において、約6〜約24時間、約0.001μMから約2.5μMの範囲で哺乳動物、例えばヒトの血漿中の化合物Aの平均血漿中濃度を達成する方法であって、前記哺乳動物に化合物A単独またはその医薬上許容しうる塩または溶媒和物、またはマトリックスと組み合わせて含んでなる医薬組成物の十分な量を投与することからなる方法が提供される。一実施態様において、組成物は、非晶質化合物Aを含んでなる。別の実施態様において、組成物は、非晶質化合物Aおよび少なくとも1つのマトリックスを含んでなる。本発明の別の態様において、哺乳動物における化合物Aの平均血漿中濃度は、約0.02μM〜約1μM、または約0.025μM〜約1μM、または約0.05μM〜約1μMの範囲にある。本発明の別の態様において、このような時間にわたるこのような平均血漿中濃度は、化合物A単独、またはその医薬上許容しうる塩または溶媒和物、またはマトリックスと組み合わせて含んでなる医薬組成物の用量を前記哺乳動物に投与することによって達成され、その際、化合物Aの前記用量は、前記組成物中で化合物A約300mgA〜約3600mgAの範囲にある。一実施態様において、このような組成物は、非晶質化合物Aを含んでなる。別の実施態様において、このような組成物は、非晶質化合物Aおよび少なくとも1つのマトリックスを含んでなる。「mgA」は、活性化合物Aのミリグラムを意味する。また、本発明は、化合物Aの用量が、約400mgA、600mgA、800mgA、1000mgA、1200mgA、1400mgA、1600mgA、1800mgA、2000mgA、2200mgA、2400mgA、2600mgA、2800mgA、3000mgA、3200mgAまたは3400mgAであるこのような方法を提供する。
【0020】
本発明のさらに別の態様において、非晶質化合物A単独、またはその医薬上許容しうる塩または溶媒和物またはマトリックスと組み合わせて含んでなる医薬組成物のHIV複製阻害量をHIV感染した哺乳動物に投与することからなる、HIV感染した哺乳動物、例えばヒトの治療方法が提供される。
【0021】
本発明のなおさらなる態様は、HIV複製阻害量の非晶質化合物Aまたは医薬上許容しうる塩またはその溶媒和物を、単独でまたはマトリックスと組み合わせて含んでなる医薬組成物をHIV感染した哺乳動物に投与することからなる、HIV感染した哺乳動物、例えばヒトにおけるAIDSまたはAIDS関連症候群の治療方法を提供する。
【0022】
また、本発明は、HIV複製阻害量の非晶質化合物A、またはその医薬上許容しうる塩または溶媒和物を、単独でまたはマトリックスと組み合わせて含んでなる医薬組成物をHIV感染した哺乳動物に投与することからなる、HIV感染した哺乳動物、例えばヒトにおけるHIVプロテアーゼ活性の阻害方法を提供する。
【0023】
本発明のなおさらなる態様において、HIV複製阻害量の非晶質化合物A、またはその医薬上許容しうる塩または溶媒和物を、単独でまたはマトリックス、およびヌクレオシドHIV逆転写酵素阻害剤、非ヌクレオシドHIV逆転写酵素阻害剤、HIVプロテアーゼ阻害剤、HIVインテグラーゼ阻害剤、HIV融合阻害剤、免疫モジュレーター、CCR5アンタゴニスト、および抗感染薬から選ばれる少なくとも1つのさらなる治療剤と組み合わせて含んでなる医薬組成物を、感染した哺乳動物に投与することからなる、感染した哺乳動物、例えばヒトにおけるHIVの治療方法を提供する。本発明の一態様において、非晶質化合物Aおよび少なくとも1つのさらなる治療剤は、同じ医薬組成物の一部として投与される。さらに別の態様において、非晶質化合物Aおよび少なくとも1つのさらなる治療剤は、同時にまたは順次投与される。
【0024】
また、本発明は、治療上有効量の非晶質化合物A単独、またはその医薬上許容しうる塩または溶媒和物、またはマトリックス、およびヌクレオシドHIV逆転写酵素阻害剤、非ヌクレオシドHIV逆転写酵素阻害剤、HIVプロテアーゼ阻害剤、HIVインテグラーゼ阻害剤、HIV融合阻害剤、免疫モジュレーター、CCR5アンタゴニスト、および抗感染薬から選ばれる少なくとも1つのさらなる治療剤と組み合わせて含んでなる医薬組成物を提供する。
【0025】
また、本発明は、マトリックスが、先に述べたようなイオン化できるセルロース系ポリマー、イオン化できないセルロース系ポリマーおよび非セルロース系ポリマーの少なくとも1つから選ばれるこのような方法を提供する。
【0026】
本発明のさらに別の態様において、医薬組成物の投与を1日1回、2回または3回実施する上述の方法を提供する。
【0027】
また、本発明は、感染した哺乳動物、例えばヒトにおいてHIV感染症を治療するための医薬の製造において非晶質化合物Aまたはその医薬上許容しうる塩または溶媒和物を単独でまたはマトリックスと組み合わせて使用する方法を提供する。さらに、本発明は、HIV感染した哺乳動物、例えばヒトにおいてAIDSまたはAIDS関連症候群を治療するための医薬の製造における、非晶質化合物Aまたはその医薬上許容しうる塩または溶媒和物を、単独でまたはマトリックスと組み合わせて使用する方法を提供する。
【0028】
本発明のさらに別の態様において、哺乳動物、例えばヒトの血漿中の化合物Aの平均血漿中濃度を、約0.001μM〜約2.5μMの範囲で約6〜約24時間達成する方法であって、チトクロームP450酵素の阻害剤および非晶質化合物A単独、またはその医薬上許容しうる塩または溶媒和物、またはマトリックスと組み合わせて含んでなる十分な量の医薬組成物を哺乳動物に投与することからなる方法が提供される。また、本発明は、チトクロームP450酵素が3A4アイソフォームであるこのような方法を提供する。また、チトクロームP450酵素の阻害剤がリトナビルまたはデラビルジンであるこのような方法を提供する。
【0029】
別の態様において、本発明は溶媒ベースのプロセスを用いた非晶質化合物Aの製造方法を提供する。
【0030】
また、本明細書では、(a) 少なくとも1つの溶媒を含んでなる噴霧溶液中に化合物を溶解し;そして(b) 前記噴霧溶液から前記少なくとも1つの溶媒を急速に蒸発させて前記化合物の非晶質形態を得る;ことからなる医薬組成物の製造方法であって、その際、前記化合物は、(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミドまたはその医薬上許容しうる塩または溶媒和物である、前記製造方法が提供される。
【0031】
別の態様において、噴霧溶液はさらにマトリックスを含むこのような方法が提供される。
【0032】
さらに本明細書において、マトリックスがイオン化できるセルロース系ポリマー、イオン化できないセルロース系ポリマーおよび非セルロース系ポリマーから選ばれる少なくとも1つのポリマーを含むこのような方法を提供する。
【0033】
また、本明細書において、前記少なくとも1つの溶媒が、メタノールおよび水とメタノールとの混合物から選ばれるこのような医薬組成物の製造方法が提供される。
【0034】
別の実施態様において、組成物は、化合物Aの化学安定性を改善するために、安定化剤を含む。安定化剤は、塩基または抗酸化剤であることができる。1つの好ましい実施態様において、組成物は固体非晶質分散体を含み、固体非晶質分散体はさらに安定化剤を含む。化合物Aの「化学安定性を改善する」とは、化合物Aが別の化学物質または化合物へ分解する速度を遅らせることを意味する。
【0035】
本発明のさらに別の態様において、非晶質化合物Aを含んでなる組成物は、化合物Aの分解を減らすためにパッケージされる。パッケージングは、化合物Aが湿度または酸素のいずれかまたは両方へ曝露されるのを制限することができる。
【0036】
本明細書に使用される用語「結晶質」は、三次元において広範囲の規則性を示す本発明の化合物の特定の固体形態を意味する。結晶質である物質は、当分野で知られている技術、例えば粉末X線回折(PXRD)結晶学、固体NMRまたは熱技術、例えば示差走査熱量測定法(DSC)によって特徴付けることができる。
【0037】
本明細書に使用される用語「非晶質」は、三次元において本質的に規則性を有しない本発明の化合物の特定の固体の形態を意味する。用語「非晶質」は、本質的に規則性を有しない物質だけでなく、いくらかの小規模の規則性を有しうるが、その規則性が三次元よりも少ないおよび/または短い距離しかない物質も含まれる。非晶質物質は、当分野で知られている技術、例えば粉末X線回折(PXRD)結晶学、固体NMRまたは熱技術、例えば示差走査熱量測定法(DSC)によって特徴付けることができる。
【0038】
本明細書に使用される用語「対照組成物」は、本質的に化合物Aの結晶質形態Iからなる組成物のことである。本明細書に使用される対照組成物は、本明細書に記載された使用環境において化合物Aの結晶質形態Iの溶解度に影響を与える他の化合物または成分を含まないことを理解すべきである。
【0039】
本明細書に使用される用語「化合物Aの結晶形態I」は、粉末X線回折(PXRD)、示差走査熱量測定法(DSC)、固体NMR(ssNMR)およびラマンIR分光法(ラマン)といった分析法を用いて当業者によって測定されうる化合物Aの非晶質形態を含まないことを特徴とする(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミドの結晶質形態を意味する。さらにまた、化合物Aの結晶質形態Iは、図I中の対照1として提供されたものと同様の粉末X線回折パターンを有することを特徴とする。「同様の」とは、当業者が、図Iの対照1のパターンと、化合物Aの結晶質形態を含む組成物の別の実験的に測定されたパターンとを比較して、パターンを得るために使用される特定の実験条件に左右されうる典型的な粉末X線回折パターンにおいて、強度および線の位置の知られている多様性を考慮して、それらが同じ多形性の形態であるということを意味する。
【0040】
本明細書に使用されるように、用語「化合物Aの少なくとも一部は非晶質形態である」は、組成物中の化合物Aの総量の少なくとも5質量%または少なくとも10質量%が非晶質形態であることを意味する。
【0041】
本明細書に使用される用語「等量」は、得られた組成物中に存在する、親化合物(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミドの理論上のモル数として測定された、化合物Aのモル量のことである。例えば、化合物Aの塩または溶媒和物を含んでなる組成物の得られた量について、化合物Aの結晶質形態Iの等量は、組成物中に存在する化合物Aの理論上のモル数を測定し、親化合物Aの同じ理論モル数が得られる化合物Aの結晶質形態Iの量を用いて算出される。
【0042】
本明細書に使用される用語「投与」、「投与する」、「用量」および「投薬」は、化合物が哺乳動物の血清または血漿に吸収されるように、化合物またはその医薬上許容しうる塩または溶媒和物、または化合物またはその医薬上許容しうる塩または溶媒和物を含んでなる医薬組成物を、哺乳動物に供給することである。
【0043】
本明細書に使用される用語「併用投与」または「併用投与する」は、第1の化合物および本発明の化合物またはその医薬上許容しうる塩または溶媒和物の組み合わせを単独でまたは医薬上許容しうる組成物の一部としていずれかで投与することである。このような併用投与は、第1の化合物および本発明の化合物が、同じ組成物の一部すなわち同じ一体的剤形の一部となるように実施することができる。また、併用投与には、第1の化合物および本発明の化合物を別々であるが、同じ治療処方計画の一部として投与することも含まれる。2つの成分を、別々に投与する場合、必ずしも本質的に同じ時間に投与する必要はないが、それが望ましい場合は可能である。従って、併用投与には、例えば、第1の化合物および本発明の化合物を、別々の用量または剤形として、しかし、同時に投与することが含まれる。また、併用投与には、異なる時間にいずれかのオーダーで別々の投与することが含まれる。
【0044】
「溶媒和物」は、明記された化合物の生物学的効果を保持しているこのような化合物の医薬上許容しうる溶媒和物の形態を意味するものとする。溶媒和物の例としては、限定されるわけではないが、本発明の化合物と水、イソプロパノール、エタノール、メタノール、ジメチルスルホキシド(DMSO)、酢酸エチル、酢酸、エタノールアミンまたはそれらの混合物との組み合わせが含まれる。「医薬上許容しうる塩」は、遊離酸の生物学的効果および薬理学的に許容しうるアニオンを含む明記された誘導体の塩基を保持しており、生物学的にまたはその他の点で好ましい塩を意味するものとする。医薬上許容しうる塩の例としては、限定されるわけではないが、酢酸塩、アクリル酸塩、ベンゼンスルホン酸塩、安息香酸塩(クロロ安息香酸塩、メチル安息香酸塩、ジニトロ安息香酸塩、ヒドロキシ安息香酸塩、およびメトキシ安息香酸塩)、炭酸水素塩、硫酸水素塩、亜硫酸水素塩、酒石酸水素塩、ホウ酸塩、臭化物、ブチン−1,4−ジオエート、カルシウムエデト酸塩、カムシル酸塩、炭酸塩、塩化物、カプロン酸塩、カプリル酸塩、クラブラン酸塩、クエン酸塩、デカン酸塩、二塩酸塩、リン酸二水素塩、エデト酸塩、エジスリ酸塩、エストール酸塩、エシル酸塩、琥珀酸エチル、ギ酸塩、フマル酸塩、グルセプト酸塩、グルコン酸塩、グルタミン酸塩、グリコール酸塩、グリコリルアルサニル酸塩、ヘプタン酸塩、ヘキシン−1,6−ジオエート、ヘキシルレソルシン酸塩、ヒドラバミン、臭化水素酸塩、塩酸塩、γ−ヒドロキシ酪酸塩、ヨウ化物、イソ酪酸塩、イソチオン酸塩、乳酸塩、ラクトビオン酸塩、ラウリン酸塩、リンゴ酸塩、マレイン酸塩、マロン酸塩、マンデル酸塩、メシル酸塩、メタリン酸塩、メタン−スルホン酸塩、メチル硫酸塩、リン酸一水素塩、粘液酸塩、ナプチル酸塩、ナフタレン−1−スルホネート、ナフタレン−2−スルホネート、硝酸塩、オレイン酸塩、シュウ酸塩、パモ酸塩(エンボン酸塩)、パルミチン酸塩、パントテン酸塩、フェニル酢酸塩、フェニル酪酸塩、フェニルプロピオン酸塩、フタル酸塩、リン酸塩/二リン酸塩、ポリガラクツロン酸塩、プロパンスルホン酸塩、プロピオン酸塩、プロピオル酸塩、ピロリン酸塩、ピロ硫酸塩、サリチル酸塩、ステアリン酸塩、塩基性酢酸塩、スベリン酸塩、琥珀酸塩、硫酸塩、スルホン酸塩、亜硫酸塩、タンニン酸塩、酒石酸、テオクル酸塩、トシル酸塩、トリエチオジド(triethiodode)、および吉草酸塩の塩が含まれる。
【0045】
本明細書に使用されるように、「使用環境」は、哺乳動物、例えば哺乳動物および特にヒトの消化管のin vivo環境、または試験溶液、例えばリン酸緩衝生理食塩水(PBS)またはModel Fasted Duodenal(MFD)溶液のin vitro環境のいずれかであることができる。濃度増大は、in vivo試験またはin vitro溶解試験のいずれかを通して測定することができる。本発明の組成物は、上記の試験環境で少なくとも1つにおいて濃度増大の基準を満たしている。
【0046】
本発明の前述および他の目的、特徴および利点は、本発明の以下の詳細な説明を考察することでより容易に理解される。
【0047】
〔実施態様の詳述〕
化合物Aは、(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミド(別名、「(R)−3−((2S,3S)−2−ヒドロキシ−3−{[1−(3−ヒドロキシ−2−メチル−フェニル)−メタノイル]−アミノ}−4−フェニル−ブタノイル)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド」、「(4R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチル−ベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド」、または「4−チアゾリジンカルボキサミド,3−[(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−1−オキソ−4−フェニルブチル]−5,5−ジメチル−N−2−プロペニル−,(4R)−」)である。化合物Aは、以下の構造を有する:
【化1】

それは、分子量512を有し、そして化合物Aの結晶質形態Iは176〜178℃の融点を有する。
【0048】
本明細書に使用されるように、用語「化合物」は、慣用的であり、上記の化学種を表し、そしてすべての医薬上許容しうる形態を含むことを理解すべきである。「医薬上許容しうる形態」は、立体異性体、立体異性体混合物、鏡像異性体、溶媒和物、水和物、同形体、多形体、仮像、中性の形態、塩の形態およびプロドラッグを含めたすべての医薬上許容しうる誘導体または変種を意味する。
【0049】
非晶質化合物A
一態様において、組成物は、非晶質化合物Aを含んでなる。「非晶質」とは、化合物Aが「結晶質」でないことを意味する。「結晶質」は、化合物Aが三次元において広範囲の規則性を示すことを意味する。従って、用語「非晶質」は、本質的に規則性を有しない物質だけでなく、いくつかの小規模の規則性を有しうるが、その規則性が三次元より低いおよび/または短い近距離しかない物質も含まれるものとする。非晶質物質は、当業者が、例えば粉末X線回折(PXRD)結晶学、固体NMR、ラマンIR分光法または熱技術、例えば示差走査熱量測定法(DSC)といったような当分野で知られている技術を用いて特徴付けることができる。本発明の組成物は、非晶質および結晶質化合物Aの両方を含むことができるが、具体的には本発明の組成物中に存在する化合物Aの総量の少なくとも約5質量%、または少なくとも約10質量%、または少なくとも約30質量%、または少なくとも約40質量%、または少なくとも約50質量%、または少なくとも約60質量%、または少なくとも約70質量%、または少なくとも約80質量%、または少なくとも約90質量%、または少なくとも約95質量%が、非晶質形態でありうることを意図する。
【0050】
非晶質形態化合物Aは、化合物Aの結晶質形態Iと比較して使用環境において溶解した化合物Aの濃度を改善することが見出された。典型的に、非晶質化合物Aでは、等量の化合物Aの結晶質形態Iからなる対照組成物と比較して使用環境で化合物Aのより高い最大薬物濃度(MDC)が得られる。本明細書において対照組成物として用いた化合物Aの結晶質形態Iは、適当な使用環境において化合物Aの溶解度に物質的に影響を及ぼす可溶化剤または他の成分を含まないことを理解しなければならない。化合物Aの非晶質形態は、対照組成物と比較して、水性使用環境における化合物Aの最大溶解濃度(MDC)を、少なくとも約1.25倍、または少なくとも2倍または少なくとも3倍まで高めることが好ましく、前記対照組成物は、本質的に化合物Aの結晶質形態Iからなる。さらに、また、化合物Aの非晶質形態は、等量の化合物Aの結晶質形態Iと比較して、使用環境において化合物Aの濃度対時間曲線下の溶解面積(AUC)を高めることもできる。AUCの計算は、医薬分野でよく知られた方法であり、例えば、Welling,“Pharmacokinetics Processes and Mathematics,”ACS Monograph 185 (1986)に記載されている。例えば、化合物Aの非晶質形態は、本質的に化合物Aの結晶質形態Iからなる対照組成物と比較して、水性使用環境中の化合物AのAUCを少なくとも約1.25倍、または少なくとも2倍または少なくとも3倍まで高める。例えば、37℃、pH6.5および290mOsm/kgのリン酸緩衝生理食塩水からなるin vitro使用環境で試験する場合、化合物Aの非晶質形態では、最大溶解薬物濃度(MDC)は、化合物Aの結晶質形態Iによって得られたものの10.3倍であり、そして使用環境に投与した後、最初の90分についての溶解薬物濃度対時間曲線下の面積(AUC90)は、化合物Aの結晶質形態Iによって得られたものの8.3倍であった。
【0051】
使用環境が動物の消化管である場合、溶解した薬物濃度は、当分野で知られているいずれかの慣用の方法によって測定することができる。1つの方法は、逆重畳積分方法である。この方法では、横座標(x軸)に沿った血液試料時間に対して縦座標(y軸)に沿って血清または血漿薬物濃度をプロットする。次いで、いずれかの慣用の分析、例えばWagner-NelsonまたはLoo-Riegelman分析を用いてデータを分析して消化管中での薬物放出速度を測定する。また、Welling,“Pharmacokinetics: Processes and Mathematics”(ACS Monograph 185, Amer. Chem. Soc., Washington, D.C., 1986)参照。このやり方でデータを処理して見かけのin vivo薬物放出プロファイルが得られる。別の方法は、患者に管を挿入しておよび定期的に消化管から直接、試料採取することである。
【0052】
また、水性使用環境においてAUCを改善するということは、非晶質化合物Aが、使用環境において、特に消化管中で溶解する化合物Aの濃度を高め、それによって血液に吸収される化合物Aの量を高めて化合物Aの高められた生物学的利用能が得られることを意味する。
【0053】
さらに別の態様において、非晶質化合物Aは、絶食状態のヒトまたは他の動物に経口的に投薬した時に、結晶質化合物Aと比較して血液中の溶解した化合物Aの改善された濃度を提供する。化合物Aの非晶質形態では、等量の化合物Aの結晶質形態Iから本質的になる対照組成物と比較して血液(血清または血漿)中で化合物Aのより高い最大薬物濃度(Cmax)が得られる。化合物Aの結晶質形態Iは、適当な使用環境において化合物Aの溶解度に物質的に影響を及ぼす可溶化剤または他の成分を含まないことを理解すべきである。例えば、化合物Aの非晶質形態では、適当な使用環境において本質的に化合物Aの結晶質形態Iからなる対照組成物によって得られる少なくとも約1.25倍または少なくとも約2倍、または少なくとも約3倍の血液中の化合物AのCmaxが得られる。
【0054】
さらに別の態様において、化合物Aの非晶質形態では、絶食状態のヒトまたは他の動物に経口的に投薬した時に、化合物Aの結晶質形態Iによって得られる少なくとも約1.25倍、または少なくとも約2倍、または少なくとも約3倍の血液(血清または血漿)中の化合物Aの濃度のAUCが得られる。また、このような組成物は、化合物Aの結晶質形態I対照の約1.25倍から約3倍までの相対生物学的利用能を有すると言えることに注目される。
【0055】
本発明の組成物は、単独で非晶質化合物Aを含むことができるし、またはさらに詳細に下に記載した賦形剤を含むことができる。
【0056】
化合物Aおよびマトリックスの固体非晶質分散体
別の実施態様において、医薬組成物は、化合物Aおよび1つまたはそれ以上の成分の固体非晶質分散体を含んでなり、これらの成分はひとまとめにして「マトリックス」と称する。「固体非晶質分散体」は、化合物Aの少なくとも一部が非晶質形態にあり、マトリックス中に分散されていることを意味する。好ましい実施態様において、マトリックスは、分散体において、非分散型の非晶質化合物A単独と比較して化合物Aの改善された物理安定性、改善された化学安定性、改善された濃度増大のいずれかまたはこれらのいずれかの組み合わせまたは3つ全てが得られるように選ばれる。「分散されてない化合物A」は、マトリックス中に分散されてない化合物Aを意味する。マトリックスは、単一の成分からなることもできるし、または2つまたはそれ以上の成分の混合物であってもよい。成分は、十分に混合して単一相または分子分散体を形成することができるし、または異なる組成を有する2つまたはそれ以上の異なる相として存在することができる。
【0057】
マトリックスの少なくとも一部は、生理学的に関連のあるpH(例えばpH1〜8)の水溶液中で水膨潤性、分散性または可溶性のいずれかである。マトリックスは、全体として室温で固体でなければならず、そして少なくとも約40℃の温度まで、好ましくは少なくとも約60℃の温度まで、そしてより好ましくは少なくとも約70℃の温度までは実質的に固体のままである。このため、マトリックスは、約40℃より上、好ましくは60℃より上、そしてより好ましくは約70℃より上の融点を有する成分を少なくとも1つまたはそれ以上含まなければならない。
【0058】
本発明の分散体中に存在する薬物の量に対するマトリックスの量は、マトリックスの特徴によって左右され、薬物対マトリックスの質量比は、約0.01〜約100で広く変化することができる(例えば薬物1質量%〜薬物99質量%)。化合物A対マトリックスの質量比は、約0.1〜約49(薬物約10質量%〜薬物約98質量%)の範囲であることが好ましい。
【0059】
マトリックスに使用する成分は、ポリマーまたは非ポリマーであってもよく、いくつかの成分の混合物を含むことができる。従って、マトリックスはポリマー成分の混合物、非ポリマー成分の混合物またはポリマーおよび非ポリマー成分の混合物を含むことができる。
【0060】
用語「ポリマー」は、慣用的に使用され、モノマーが一緒に結合してより大きい分子を形成してできた化合物を意味する。一般に、ポリマーマトリックス成分では、非ポリマーのマトリックス成分と比較して濃度増大が改善された分散体が得られる。ポリマー成分は中性であってもよいしまたはイオン化することができ、そしてセルロース系または非セルロース系であってもよい。マトリックスとして使用される典型的なポリマー成分としては、本明細書で下に記載された濃度増大ポリマー、ポリエチレングリコール、ポリオキシエチレングリコール、ポリエチレンオキシド、キサンタンガム、カラゲナン、キトサン、ポリデキストロース、デキストリンおよびデンプンが含まれる。また、この定義の中には高分子量タンパク質、例えばゼラチンおよびアルブミンが含まれる。マトリックスは後述する濃度増大ポリマーであることが好ましい。
【0061】
「非ポリマーの」は、成分がポリマーでないことを意味する。マトリックス成分として使用するための典型的な非ポリマー物質としては、限定されるわけではないが、アルコール、例えばステアリルアルコールおよびセチルアルコール、有機酸およびそれらの塩、例えばステアリン酸、クエン酸、フマル酸、酒石酸、リンゴ酸、およびその医薬上許容しうる塩;有機塩基、例えばグルコサミン、N−メチルグルカミン、トリス(ヒドロキシメチル)アミノメタンおよびドデシルアミン;塩、例えば塩化ナトリウム、塩化カリウム、塩化リチウム、塩化カルシウム、塩化マグネシウム、硫酸ナトリウム、硫酸カリウム、炭酸ナトリウムおよび硫酸マグネシウム;アミノ酸、例えばアラニンおよびグリシン;糖、例えばグルコース、シュクロース、キシリトール、フルクトース、ラクトース、トレハロース、マンニトール、ソルビトールおよびマルチトール;脂肪酸エステル、例えばグリセリル(モノ−および−)ステアレート、グリセリル(モノ−およびジ−)ベヘネート、トリグリセリド、ソルビタンモノステアレート、サッカロースモノステアレート、グリセリル(パルミチン酸ステアリン酸)エステル、水素化された綿実油、ポリオキシエチレンソルビタン脂肪酸エステル;ワックス、例えば微結晶ワックス、パラフィンワックス、蜜蝋、合成蝋、キャスターワックスおよびカルナバワックス;アルキルスルフェート、例えばラウリル硫酸ナトリウムおよびラウリル硫酸マグネシウム;およびリン脂質、例えばレシチン;およびそれらの混合物が含まれる。
【0062】
本発明の組成物において、固体非晶質分散体中に存在する化合物Aの少なくとも主要部分は、非晶質形態であることができる。本明細書に使用されるように、化合物Aの用語「主要部分」は、固体非晶質分散体中の化合物Aの少なくとも約60質量%が、結晶質形態ではなく非晶質形態であることを意味する。別法として、本発明の組成物は、実質的に非晶質である固体非晶質分散体中の化合物Aを含んでなることができる。本明細書に使用されるように、「実質的に非晶質」とは、結晶質形態中の化合物Aの量が約25質量%を超えないことを意味する。さらに、本発明の組成物は、「ほとんど完全に非晶質」である固体非晶質分散体中の化合物Aを含んでなり、結晶質形態の化合物Aの量が、存在する化合物Aの総量の約10質量%を超えないことを意味する。結晶質化合物Aの量は、粉末X線回折(PXRD)、走査型電子顕微鏡(SEM)分析、示差走査熱量測定法(DSC)、または他のいずれの標準定量的測定法を含めた分析技術を用いて当業者によって測定されうるが、これらに限定されない。
【0063】
化合物Aの非晶質形態は、ポリマー全体に均質に分散された薬物の固溶体としてまたはこれらの状態のいずれかの組み合わせまたはそれらの中間の状態として、固体非晶質分散体の中で、比較的純粋な非晶質薬物ドメインまたは領域中に存在することができる。
【0064】
濃度増大ポリマーを含んでなる組成物
別の実施態様において、医薬組成物は、化合物Aの非晶質形態および濃度増大ポリマーを含んでなる。濃度増大ポリマーは、適当な使用環境において溶解した薬物の濃度をさらに改善することができる。特に、本発明の組成物中に濃度増大ポリマーを含む利点は、このような濃度増大ポリマーを含まず、化合物Aを含んでなる組成物と比較して化合物AのAUCが改善されることである。本発明者らは、非晶質化合物Aでは、適当な使用環境中に溶解した場合、化合物Aの平衡濃度を超える化合物Aの初濃度が得られるが、しかしながら、溶解した化合物Aの濃度は、時間とともに実質的に減少することを見出した。理論によって拘束しようとするものではないが、本発明者らは、溶解した化合物Aを伴う使用環境に濃度増大ポリマーを添加すると、最初に高められた溶解薬物濃度が平衡濃度まで落ちる速度が遅くなると考えた。結果として、非晶質化合物Aおよび濃度増大ポリマーを含んでなる組成物では、化合物A単独で得られるよりも大きく改善された溶解曲線下の面積(「AUC」)が得られた。
【0065】
AUCを改善するということは、濃度増大ポリマーを含んでなる組成物では、使用環境中、特に哺乳動物、例えばヒトの消化(GI)管中に溶解したままの薬物の濃度を高めることによって化合物Aの高められた生物学的利用能を得ることができることを意味する。溶液中の化合物Aの濃度を改善することで哺乳動物においてより高い血中濃度が得られ、場合によっては、到達すべき有効レベルが可能であり、または別の場合には、より低い薬物用量レベルで到達すべき有効血中濃度を得ることができ、このため、投薬しなければならない薬物の量が減少し、血中濃度の変動が減り、そしてまた必要となるポリマーの量に応じて剤形のサイズが減少する。
【0066】
本発明の組成物に使用するのに適した濃度増大ポリマーは、有害なかたちで化合物Aと化学的に反応せず、医薬上許容しうるという点で不活性でなければならない。ポリマーは、中性またはイオン化可能であってもよく、pH約1〜8の範囲の少なくとも一部にわたって少なくとも約0.1mg/mlの水溶解度を有しなければならない。本発明の使用に適した濃度増大ポリマーは、セルロース系または非セルロース系であることができる。これらのうち、イオン化できるポリマーおよびセルロース系ポリマーが好ましく、イオン化できるセルロース系ポリマーがより好ましい。「セルロース系の」とは、糖類反復単位上のヒドロキシル基の少なくとも一部と化合物とが反応してエステルまたはエーテル置換基を形成することによって改良されたセルロースポリマーを意味する。
【0067】
ポリマーの好ましい種類としては、性質が「両親媒性」であるポリマーが含まれ、これはポリマーが疎水性および親水性部分を有することを意味する。疎水性部分は、脂肪族または芳香族炭化水素基のような基を含むことができる。親水性部分は、水素結合することができる、イオン化できるまたはイオン化できない基、例えばヒドロキシル、カルボン酸、エステル、アミンまたはアミドを含むことができる。
【0068】
本発明に使用するための適切なポリマーの1つの種類にイオン化できない(または中性の)非セルロース系ポリマーがある。典型的なポリマーとしては、ヒドロキシル、アルキルアシルオキシまたは環式アミドの置換基を有するビニルポリマーおよびコポリマー;加水分解されてない形態のそれらの反復単位(酢酸ビニル)の少なくとも一部を有するポリビニルアルコール;ポリビニルアルコールポリビニルアセテートコポリマー;ポリビニルピロリドン;ポリオキシエチレン−ポリオキシプロピレンコポリマー(別名ポロキサマー);およびポリエチレンポリビニルアルコールコポリマーが含まれる。典型的な非セルロース系の中性ポリマーとしては、ヒドロキシエチルメタクリレート、ポリビニルヒドロキシエチルエーテルおよびポリエチレングリコールが含まれる。
【0069】
中性の非セルロース系ポリマーの好ましい種類としては、ヒドロキシルを含む親水性の反復単位およびアルキルまたはアリールを含む疎水性の反復単位のビニルコポリマーが含まれる。このような中性のビニルコポリマーは、「両親媒性ヒドロキシル官能性ビニルコポリマー」と称する。「両親媒性ヒドロキシル官能性ビニルコポリマー」は、例外的であり、それらは、いずれも非イオン性であるが、驚くべきことに、低い溶解性の薬物用に分散体ポリマーとして使用する場合、使用する水性環境に投薬した時に高いレベルの薬物濃度増大をもたらす固体非晶質分散体が得られる。
【0070】
好ましいコポリマーは、一般構造:
【化2】
を有し、ここでAおよびBは、それぞれ「ヒドロキシルを含む親水性の」および「疎水性の」置換基を表し、そしてnおよびmは、それぞれポリマー分子当たりの親水性ビニル反復単位の平均数および疎水性ビニル反復単位の平均数を表す。コポリマーは、ブロックコポリマー、ランダムコポリマーであってもよいし、またはこれらの2の両極端の間のいずれかの構造を有することができる。ポリマーは、例えば約2,500〜約1,000,000ダルトンの分子量を有することができる。
【0071】
ヒドロキシルを含む親水性の反復単位(「A」)は、単にヒドロキシル(−OH)であってもよいし、またはそれに結合した1つまたはそれ以上のヒドロキシルを有する炭素1〜6個の短鎖アルキルのいずれかであってもよい。ヒドロキシル置換されたアルキルは、炭素−炭素またはエーテル結合を経てビニル骨格鎖に結合されていてもよい。従って、典型的な「A」構造としては、ヒドロキシルそれ自体に加えて、ヒドロキシメチル、ヒドロキシエチル、ヒドロキシプロピル、ヒドロキシメトキシ、ヒドロキシエトキシおよびヒドロキシプロポキシが含まれる。
【0072】
疎水性置換基(「B」)は、単に水素(−H)、この場合、疎水性反復単位は、エチレン;炭素−炭素結合を経て結合した炭素12個までのアルキルまたはアリール置換基、例えばメチル、エチルまたはフェニル;エーテル結合を経て結合した炭素12個までのアルキルまたはアリール置換基、例えばメトキシ、エトキシまたはフェノキシ;エステル結合を経て結合した炭素12個までのアルキルまたはアリール置換基、例えばアセテート、プロピオネート、ブチレートまたはベンゾエートであってもよい。本発明の両親媒性ヒドロキシル官能性ビニルコポリマーは、置換されたビニルコポリマーを製造するために使用するいずれかの慣用の方法によって合成することができる。ポリビニルアルコール/ポリビニルアセテートのようないくつかの置換されたビニルコポリマーは、よく知られており、商業的に入手可能である。
【0073】
このようなポリマーは、同一出願人による係属中の米国特許出願第10/175,132号にさらに詳細に開示されており、これは2001年6月22日に出願された仮出願第60/300,255号に対して優先権を主張し、参照により本明細書に組み込まれている。
【0074】
本発明に使用するのに適したポリマーの他の種類としては、イオン化できる非セルロース系ポリマーがある。典型的なポリマーとしては、カルボン酸官能化ビニルポリマー、例えばカルボン酸官能化ポリメタクリレートおよびカルボン酸官能化ポリアクリレート、例えばRohm Tech Inc., of Malden, Massachusettsによって製造されたEUDRAGITSR;アミン官能化ポリアクリレートおよびポリメタクリレート;タンパク質;およびカルボン酸官能化デンプン、例えばデンプングリコレートが含まれる。
【0075】
両親媒性非セルロース系ポリマーは、比較的親水性および比較的疎水性モノマーのコポリマーである。例としては、アクリレートおよびメタクリレートコポリマーおよびポリオキシエチレン−ポリオキシプロピレンコポリマーが含まれる。このようなコポリマーの典型的な商品名としてはEUDRAGITS(これはメタクリレートおよびアクリレートのコポリマーである)およびBASFによって供給されるPLURONICS(これはポリオキシエチレン−ポリオキ
シプロピレンコポリマーである)が含まれる。
【0076】
ポリマーの好ましい種類は、少なくとも1つのエステル−および/またはエーテル−結合した置換基を有するイオン化できるおよび中性のセルロース系ポリマーが含まれ、その際、ポリマーはそれぞれの置換基について少なくとも0.1の置換度を有する。本明細書に使用されるポリマー命名法において、エーテル結合した置換基は、エーテル基に結合した部分として「セルロース」の前に記載され;例えば「エチル安息香酸セルロース」は、エトキシ安息香酸置換基を有することに留意すべきである。同様に、エステル結合した置換基は、カルボキシレートとして「セルロース」の後に記載され;例えば「セルロースフ
タレート」は、ポリマーにエステル結合した各フタレート部分の1つカルボン酸と未反応の別のカルボン酸とを有する。
【0077】
また、「セルロースアセテートフタレート」(CAP)のようなポリマー名称は、エステル結合を経てセルロース系ポリマーのヒドロキシル基の有意な画分に結合したアセテートおよびフタレート基を有するセルロース系ポリマーのいずれかのファミリーのことであると留意すべきである。一般に、それぞれの置換基の置換度は、ポリマーの他の基準を満たせば、0.1〜2.9の範囲であることができる。「置換度」は、置換されたセルロース鎖上で糖類反復単位当たり3個の平均ヒドロキシル数に相当する。例えば、セルロース鎖のヒドロキシルの全てが置換されたフタレートである場合、フタレートの置換度は、3である。また、それぞれのポリマーファミリータイプには、ポリマーの性能を実質的に変えない比較的少量で加えられたさらなる置換基を有するセルロース系ポリマーが含まれる。このように、例えば、ポリマー名称「ヒドロキシプロピルメチルセルロースアセテートスクシネート」は、商業グレードL、MおよびHがあり、信越化学工業(東京,日本)から入手できる。
【0078】
両親媒性セルロースには、少なくとも1つの比較的疎水性の置換基を有するそれぞれの糖類反復単位上に存在する3個のヒドロキシル基のいずれかまたは全てで親セルロースポリマーが置換されたポリマーが含まれる。疎水性置換基は、十分に高いレベルまたは置換度に置換された場合、セルロース系ポリマーを本質的に水不溶性にすることができる本質的にすべての置換基であるといえる。疎水性置換基の例としては、エーテル結合したアルキル基、例えばメチル、エチル、プロピル、ブチル、等;またはエステル結合したアルキル基、例えばアセテート、プロピオネート、ブチレート、等;およびエーテルおよび/またはエステル結合したアリール基、例えばフェニル、ベンゾエートまたはフェニレートが含まれる。ポリマーの親水性領域は、非置換のヒドロキシルはそれ自体比較的親水性であるため、比較的非置換であるか、または親水性置換基で置換された領域のいずれかでありうる。親水性置換基としては、エーテル−またはエステル結合したイオン化できない基、例えばヒドロキシアルキル置換基ヒドロキシエチル、ヒドロキシプロピル、およびアルキルエーテル基、例えばエトキシエトキシまたはメトキシエトキシが含まれる。特に好ましい親水性置換基は、エーテル−またはエステル結合したイオン化できる基、例えばカルボン酸、チオカルボン酸、置換されたフェノキシ基、アミン、ホスフェートまたはスルホネートであるものである。
【0079】
セルロース系のポリマーの1つの種類には、中性ポリマーがあり、これはポリマーが水溶液中で実質的にイオン化できないことを意味する。このようなポリマーには、イオン化できない置換基が含まれており、それはエーテル結合またはエステル結合することができる。典型的なエーテル結合したイオン化できない置換基としては、アルキル基、例えばメチル、エチル、プロピル、ブチル、等;ヒドロキシアルキル基、例えばヒドロキシメチル、ヒドロキシエチル、ヒドロキシプロピル、等;およびアリール基、例えばフェニルが含まれる。典型的なエステル結合したイオン化できない置換基としては、アルキル基、例えばアセテート、プロピオネート、ブチレート、等;およびアリール基、例えばフェニレートが含まれる。しかしながら、アリール基が含まれる場合、生理学的に関連のあるpH1〜8のいずれかでポリマーが少なくともいくらか水溶解度を有するように、ポリマーは十分な量の親水性置換基を含む必要がありうる。
【0080】
ポリマーとして使用することができる典型的なイオン化できないポリマーとしてはヒドロキシプロピルメチルセルロースアセテート、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、メチルセルロース、ヒドロキシエチルメチルセルロース、ヒドロキシエチルセルロースアセテートおよびヒドロキシエチルエチルセルロースが含まれる。
【0081】
中性のセルロース系ポリマーの好ましいものは、両親媒性ものである。典型的なポリマーとしては、ヒドロキシプロピルメチルセルロースおよびヒドロキシプロピルセルロースアセテートが含まれ、その際、非置換のヒドロキシルまたはヒドロキシプロピル置換基と比較してメチルまたはアセテート置換基の数が比較的多いセルロース反復単位は、ポリマー上の他の反復単位と比較して疎水性部分を構成する。
【0082】
セルロース系ポリマーの好ましい種類としては、生理学的に関連のあるpHで少なくとも部分的にイオン化でき、そして少なくとも1つのイオン化できる置換基を含み、その置換基はエーテル結合またはエステル結合することができるポリマーが含まれる。典型的なエーテル結合したイオン化できる置換基としては、カルボン酸、例えば酢酸、プロピオン酸、安息香酸、サリチル酸、アルコキシ安息香酸、例えばエトキシ安息香酸またはプロポキシ安息香酸、アルコキシフタル酸の種々の異性体、例えばエトキシフタル酸およびエトキシイソフタル酸、アルコキシニコチン酸の種々の異性体、例えばエトキシニコチン酸、およびピコリン酸の種々の異性体、例えばエトキシピコリン酸、等;チオカルボン酸、例えばチオ酢酸;置換されたフェノキシ基、例えばヒドロキシフェノキシ、等;アミン、例えばアミノエトキシ、ジエチルアミノエトキシ、トリメチルアミノエトキシ、等;ホスフェート(例えばホスフェートエトキシ);およびスルホネート、例えばスルホネートエトキシが含まれる。典型的なエステル結合したイオン化できる置換基としては、カルボン酸、例えばスクシネート、シトレート、フタレート、テレフタレート、イソフタレート、トリメリテート、およびピリジンジカルボン酸の種々の異性体、等;チオカルボン酸、例えばチオスクシネート;置換されたフェノキシ基、例えばアミノサリチル酸;アミン、例えば天然または合成アミノ酸、例えばアラニンまたはフェニルアラニン;ホスフェート、例えばアセチルホスフェート;およびスルホネート、例えばアセチルスルホネートが含まれる。また、必要な水溶解度を有する芳香族置換されたポリマーについては、十分な親水基、例えばヒドロキシプロピルまたはカルボン酸官能基がポリマーに結合して、少なくともいずれかのイオン化できる基がイオン化されるpH値でポリマーを水溶解度にすることが望ましい。場合によっては、フタレートまたはトリメリテート置換基のような芳香族基はそれ自体イオン化できる。
【0083】
生理学的に関連のあるpHで少なくとも部分的にイオン化された典型的なセルロースのポリマーとしては、ヒドロキシプロピルメチルセルロースアセテートスクシネート、ヒドロキシプロピルメチルセルローススクシネート、ヒドロキシプロピルセルロースアセテートスクシネート、ヒドロキシエチルメチルセルローススクシネート、ヒドロキシエチルセルロースアセテートスクシネート、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシエチルメチルセルロースアセテートスクシネート、ヒドロキシエチルメチルセルロースアセテートフタレート、カルボキシエチルセルロース、カルボキシメチルセルロース、カルボキシメチルエチルセルロース、セルロースアセテートフタレート、メチルセルロースアセテートフタレート、エチルセルロースアセテートフタレート、ヒドロキシプロピルセルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースアセテートフタレート、ヒドロキシプロピルセルロースアセテートフタレートスクシネート、ヒドロキシプロピルメチルセルロースアセテートスクシネートフタレート、ヒドロキシプロピルメチルセルローススクシネートフタレート、セルロースプロピオネートフタレート、ヒドロキシプロピルセルロースブチレートフタレート、セルロースアセテートトリメリテート、メチルセルロースアセテートトリメリテート、エチルセルロースアセテートトリメリテート、ヒドロキシプロピルセルロースアセテートトリメリテート、ヒドロキシプロピルメチルセルロースアセテートトリメリテート、ヒドロキシプロピルセルロースアセテートトリメリテートスクシネート、セルロースプロピオネートトリメリテート、セルロースブチレートトリメリテート、セルロースアセテートテレフタレート、セルロースアセテートイソフタレート、セルロースアセテートピリジンジカルボキシレート、サリチル酸セルロースアセテート、ヒドロキシプロピルサリチル酸セルロースアセテート、エチル安息香酸セルロースアセテート、ヒドロキシプロピルエチル安息香酸セルロースアセテート、エチルフタル酸セルロースアセテート、エチルニコチン酸セルロースアセテート、およびエチルピコリン酸セルロースアセテートが含まれる。
【0084】
親水性および疎水性領域を有する両親媒性の定義を満たす典型的なイオン化できるセルロース系ポリマーとしては、セルロースアセテートフタレートおよびセルロースアセテートトリメリテートといったようなポリマーが含まれ、この場合、1つまたはそれ以上のアセテート置換基を有するセルロース反復単位は、アセテート置換基を有しないまたは1つまたはそれ以上のイオン化されたフタレートまたはトリメリテート置換基を有するものと比べて疎水性である。
【0085】
セルロース系のイオン化できるポリマーの特に望ましいサブセットは、カルボン酸官能性芳香族置換基およびアルキレート置換基の両方を有し、このため両親媒性であるものである。典型的なポリマーとしては、セルロースアセテートフタレート、メチルセルロースアセテートフタレート、エチルセルロースアセテートフタレート、ヒドロキシプロピルセルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートフタレート、ヒドロキシプロピルセルロースアセテートフタレートスクシネート、セルロースプロピオネートフタレート、ヒドロキシプロピルセルロースブチレートフタレート、セルロースアセテートトリメリテート、メチルセルロースアセテートトリメリテート、エチルセルロースアセテートトリメリテート、ヒドロキシプロピルセルロースアセテートトリメリテート、ヒドロキシプロピルメチルセルロースアセテートトリメリテート、ヒドロキシプロピルセルロースアセテートトリメリテートスクシネート、セルロースプロピオネートトリメリテート、セルロースブチレートトリメリテート、セルロースアセテートテレフタレート、セルロースアセテートイソフタレート、セルロースアセテートピリジンジカルボキシレート、サリチル酸セルロースアセテート、ヒドロキシプロピルサリチル酸セルロースアセテート、エチル安息香酸セルロースアセテート、ヒドロキシプロピルエチル安息香酸セルロースアセテート、エチルフタル酸セルロースアセテート、エチルニコチン酸セルロースアセテート、およびエチルピコリン酸セルロースアセテートが含まれる。
【0086】
セルロース系のイオン化できるポリマーの別の特に望ましいサブセットは、両親媒性であり非芳香族カルボキシレート置換基を有するものである。典型的なポリマーとしては、ヒドロキシプロピルメチルセルロースアセテートスクシネート、ヒドロキシプロピルメチルセルローススクシネート、ヒドロキシプロピルセルロースアセテートスクシネート、ヒドロキシエチルメチルセルロースアセテートスクシネート、ヒドロキシエチルメチルセルローススクシネート、ヒドロキシエチルセルロースアセテートスクシネート、およびカルボキシメチルエチルセルロースが含まれる。
【0087】
別の好ましい種類のポリマーは、中和された酸性ポリマーのからなる。「中和された酸性ポリマー」とは、「酸性部分」または「酸性置換基」の有意な画分が「中和された」;すなわち、それらの脱プロトン化された形態で存在するすべての酸性ポリマーを意味する。「酸性ポリマー」は、有意な数の酸性部分を有するすべてのポリマーを意味する。一般に、有意な数の酸性部分とは、ポリマー1グラム当たり約0.1ミリグラム当量またはそれ以上の酸性部分である。「酸性部分」としては、水と接触してまたは溶解して少なくとも水に水素カチオンを部分的に供与し、これによって水素イオン濃度を高めることができる十分に酸性であるすべての官能基が含まれる。この定義には、実際には、官能基がポリマーに共有結合した時に、約10未満のpKaを有するすべての官能基または「置換基」が含まれる。上記の説明に含まれる官能基の典型的な種類としては、カルボン酸、チオカルボン酸、ホスフェート、フェノール基およびスルホネートが含まれる。このような官能基は、例えばポリアクリル酸では、ポリマーの一次構造を構成することができるが、より一般には親ポリマーの骨格鎖に共有結合しており、このため「置換基」と称する。
【0088】
一塩基酸(例えばカルボン酸)で置換されたポリマーの「中和度」、αは、中和された;すなわち塩基によって脱プロトン化されたポリマー上で酸性部分の画分として定義される。典型的に、「中和された酸性ポリマー」とみなされる酸性ポリマーでは、αは、少なくとも約0.001(または0.1%)、好ましくは約0.01(1%)、そしてより好ましくは少なくとも0.1(10%)でなければならない。このような小さい中和度は許容され、それは中和度が少し増加するとばしばポリマーの有効pHが劇的に変化するためである。それにもかかわらず、より大きい中和度は、よりいっそう好ましい。従って、αは、少なくとも0.5(酸性部分の少なくとも50%が中和されたことを意味する)であることが好ましく、そしてαは少なくとも0.9(酸性部分の少なくとも90%が中和されたことを意味する)であることがより好ましい。
【0089】
中和された酸性ポリマーは、同一出願人による係属中の米国特許出願第10/175,566号にさらに詳細に記載されており、これは、2001年6月22日に出願された“Pharmaceutical Compositions of Drugs and Neutralized Acidic Polymers”と題する米国仮特許出願第60/300,256号の優先権を主張し、この関連した開示は、参照により組み込まれている。
【0090】
特定のポリマーは、本発明の組成物に使用するために適切であることを議論してきたが、このようなポリマーのブレンドも適切であるといえる。従って、用語「ポリマー」は、単一種類のポリマーに加えてポリマーのブレンドを含むものとする。
【0091】
組成物中に存在する濃度増大ポリマーの量は、濃度増大をもたらすのに十分であり、下により詳細に記載されている。一般に、薬物対ポリマーの比率は、約0.01(薬物1部対ポリマー100部)〜約100の範囲であることができる。
【0092】
非晶質化合物Aおよび濃度増大ポリマーは、なんらかの方法で合わせることができる。一実施態様において、組成物は非晶質化合物Aおよび濃度増大ポリマーの組み合わせからなる。別の実施態様において、組成物は、(1)化合物Aおよびマトリックスを含んでなる固体非晶質分散体および(2)濃度増大ポリマー:の組み合わせからなる。本明細書に使用される「組み合わせ」は、非晶質化合物Aまたは化合物Aおよびマトリックスおよび濃度増大ポリマーを含んでなる固体非晶質分散体が、互いに物理的に接触しているかまたは密接に接近しているが、物質的に混合する必要はないことを意味する。例えば、当分野で知られているように、組成物は、多層錠剤の形態をとることができ、そこでは1つまたはそれ以上の層が非晶質化合物Aを含み、そして1つまたはそれ以上の異なる層が濃度増大ポリマーを含む。さらに別の例としてコーティング錠剤を挙げることができ、そこでは、化合物Aまたは濃度増大ポリマーまたは両方が錠剤の核中に存在することができ、そしてコーティングは、非晶質化合物Aまたは濃度増大ポリマーまたは両方を含むことができる。別法として、組み合わせは、非晶質化合物Aおよび濃度増大ポリマーの両方が微粒子の形態で混合され、そしてそれぞれの粒子が、サイズに関係なく、それらがバルク中で示す個々の同じ物理特性を保持している、単純な乾燥した物理的混合物の形態をとることができる。
【0093】
非晶質化合物Aおよび濃度増大ポリマーの組み合わせは、V型ブレンダー、遊星型ミキサー、渦流ブレンダー(vortex blenders)、ミル、押出機、例えばツインスクリーン押出機(twin-screen extruders)および摩砕プロセスを用いて非晶質化合物A、1つまたはそれ以上の濃度増大ポリマー、および所望の剤形を形成するのに適当な他のいずれか賦形剤を含めた乾燥成分を混合するといったようななんらかの慣用の方法で形成することができる。成分は、ボールミルまたはローラーコンパクターのような機械的エネルギーを用いた造粒プロセスにおいて合わせることができる。また、成分は、高剪断造粒機または流動層造粒機中で湿式造粒法を用いて合わせることができ、その際、溶媒または湿潤剤を成分または濃度増大ポリマーに加え、溶媒中に溶解して造粒液として使用することができる。濃度増大ポリマーは、非晶質化合物Aを含む混合物から圧縮プロセスによって予め形成された錠剤にコーティングとして加えることができ、コーティングは、噴霧コーティングプロセスにおいて、例えばパンコーターまたは流動層コーターを用いて実施される。
【0094】
別法として、本発明の組成物は併用投与することができ、これは、非晶質薬物と濃度増大ポリマーとを別々であるが、同じ一般時間枠内に投与することができることを意味する。従って、非晶質薬物を、例えばそれ自体の剤形で投与し、これを別の剤形中にある濃度増大ポリマーとほぼ同じ時間に行う。別々に投与する場合、一般に、使用環境において2つが一緒に存在するように互いに60分の範囲内で、非晶質化合物Aおよび濃度増大ポリマーの両方を投与することが好ましい。同時に投与しない場合、濃度増大ポリマーを、化合物Aの非晶質形態の前に投与することが好ましい。
【0095】
別の実施態様において、化合物Aおよび濃度増大ポリマーを合わせて固体非晶質分散体を形成する。化合物Aおよび濃度増大ポリマーを含んでなる固体非晶質分散体は好ましく、それはこのような固体非晶質分散体では、しばしばin vitroおよびin vivo使用環境において溶解薬物の高い濃度を得ることができるためである。
【0096】
濃度増大ポリマーを含んでなる固体非晶質分散体は、化合物Aの用量および濃度増大ポリマーの有効性に応じて約99質量%までの化合物Aを含むことができる。非晶質分散体は、非常に高充填の化合物Aを含んで形成することができる。固体非晶質分散体が化合物Aおよび濃度増大ポリマーのみで構成される場合、固体非晶質分散体は、少なくとも約0.67の薬物対ポリマー質量の比率を有する。固体非晶質分散体は、化合物Aまたはその医薬上許容しうる塩または溶媒和物を少なくとも約30質量%、少なくとも約40質量%、少なくとも約50質量%、少なくとも約60質量%、少なくとも約70質量%、少なくとも約80質量%、少なくとも約90質量%、または少なくとも約95質量%含むことができる。マトリックス、例えばセルロース系ポリマーからなるマトリックス、例えばHPMCAS(ヒドロキシプロピルメチルセルロースアセテートスクシネート)、中に化合物Aを含んでなる固体非晶質分散体は、長期間貯蔵した後でも分散体が化合物Aの改善された溶解性能を提供し続けるという点で、長期間にわたって物理的に安定であるといえる。
【0097】
安定化剤
一実施態様において、組成物は、化合物Aの化学安定性を促進する安定化剤をさらに含む。安定化剤は、化合物Aが、例えば酸化分解プロセスを経て別の化学物質に分解する速度を低下させる。分解を防ぐために、本発明の組成物は、安定化剤を含むことができる。有用な安定化剤の1つの種類は、抗酸化剤である。分散体中に含まれうる典型的な抗酸化剤としては、ブチルヒドロキシトルエン(BHT)、ブチルヒドロキシアニソール(BHA)、プロピルガレート、ビタミンEおよびビタミンEスクシネートが含まれるが、これらに限定されない。このような抗酸化剤は、酸化を低下させるが、医薬上許容しうる量を超えない十分な量で存在することができる。典型的な量は、約0〜約10質量%の範囲である。
【0098】
有用に含まれる別の安定化剤は、塩基である。塩基の例としては、(限定されるわけではないが)水酸化物、例えば水酸化ナトリウム、水酸化カルシウム、水酸化アンモニウムおよびコリン水酸化物;炭酸水素塩、例えば(限定されるわけではないが)炭酸水素ナトリウム、炭酸水素カリウムおよび炭酸水素アンモニウム;炭酸塩、例えば(限定されるわけではないが)炭酸アンモニウム、炭酸カルシウムおよび炭酸ナトリウム;アミン、例えば(限定されるわけではないが)トリス(ヒドロキシメチル)アミノメタン、エタノールアミン、ジエタノールアミン、N−メチルグルカミン、グルコサミン、エチレンジアミン、N,N'−ジベンジルエチレンジアミン、N−ベンジル−2−フェネチルアミン、シクロヘキシルアミン、シクロペンチルアミン、ジエチルアミン、イソプロピルアミン、ジイソプロピルアミン、ドデシルアミンおよびトリエチルアミン;タンパク質、例えば(限定されるわけではないが)ゼラチン;アミノ酸、例えば(限定されるわけではないが)リシン、アルギニン、グアニン、グリシンおよびアデニン;ポリマーアミン、例えばポリアミノメタクリレート、例えばEudragit E;種々の酸の共役塩基、例えば(限定されるわけではないが)酢酸ナトリウム、酢酸カリウム、酢酸カルシウム、酢酸マグネシウム、酢酸アンモニウム、クエン酸カリウム、クエン酸カルシウム、クエン酸ナトリウム、クエン酸二ナトリウム、クエン酸三ナトリウム、安息香酸ナトリウム、安息香酸カリウム、安息香酸カルシウム、プロピオン酸ナトリウム、リン酸二ナトリウム、リン酸三ナトリウム、リン酸カルシウム、ナトリウムフェノキシド、硫酸ナトリウム、塩化アンモニウムおよび硫酸アンモニウム;EDTA(エチレンジアミン四酢酸)の塩、例えば(限定されるわけではないが)四ナトリウムEDTA;種々の酸性ポリマーの塩、例えばナトリウムスターチグリコレート、カルボキシルメチルセルロースナトリウムおよびナトリウムポリアクリル酸;およびN−メチルモルホリンが含まれる。好ましい塩基は、有機酸の共役塩基、例えば(限定されるわけではないが)酢酸ナトリウム、酢酸カリウム、酢酸カルシウム、酢酸マグネシウム、酢酸アンモニウム、枸櫞酸カリウム、クエン酸カルシウム、クエン酸ナトリウム、クエン酸二ナトリウム、クエン酸三ナトリウム、安息香酸ナトリウム、安息香酸カリウム、安息香酸カルシウムおよびプロピオン酸ナトリウムである。
【0099】
分散体ポリマーがHPMCASのような酸性である場合、固体非晶質分散体へ塩基を添加することは、特に好ましい。この実施態様では、塩基は、ポリマー上で酸性基のかなりの量を中和するのに十分な量で存在することができる。塩基は、酸性基の少なくとも約40%、酸性基の少なくとも約50%、または酸性基の少なくとも約90%を中和するのに十分な量で存在することができる。塩基は、酸性基よりも過剰に存在して本質的に酸性基の100%が中和されてもよい。
【0100】
酸性ポリマーの中和された形態が多価カチオン種、例えばCa2+、Mg2+、Al3+、Fe2+、Fe3+、またはジアミン、例えばエチレンジアミンを含む場合、カチオン種は、複数のポリマー鎖上で2つまたはそれ以上の中和された酸性部分と相互作用してポリマー鎖間でイオン架橋を生成することができる酸性ポリマーは、ポリマー1グラム当たり多価カチオン種のミリグラム当量数が、ポリマー1グラム当たり(ポリマーの)酸性部分のミリグラム当量数の少なくとも約5%、好ましくは少なくとも約10%である場合、「イオン架橋された」とみなすことができる。別法として、酸性ポリマーは、十分な多価カチオン種が存在して、中和された酸性ポリマーが、本質的に多価カチオン種を含んでいない同じポリマーよりも高いガラス転移温度(Tg)を有する場合、「イオン架橋された」とみなすことができる。このようなイオンによって架橋されたポリマーから製造され分散体中の易動度は、同じポリマーの酸性形態から形成された分散体と比較して特に低い。このようなイオン架橋されたポリマーは、塩基のカチオン性対イオンが二価であるいずれかの塩基を用いて酸性ポリマーを中和によって形成することができる。従って、水酸化マグネシウム、酢酸カルシウムまたはエチレンジアミンを酸性ポリマー、例えばセルロースのアセテートフタレートまたはヒドロキシプロピルメチルセルロースアセテートスクシネートに加えて、中和されてイオン架橋された酸性セルロース系ポリマーを形成することができる。このようなポリマーにおける低い易動度は、高いTg値またはより典型的にTg付近の熱容量増加規模における減少または場合によっては、分散体が示差熱分析を受けた時の明確なTgの欠如によって示すことができる。従って、ポリマーが本質的に完全に中和された場合、中和されたポリマーを示差熱分析にかける時には、Tgは明確ではない。このようなイオン架橋されたポリマーでは、非イオン的に架橋された中和された酸性ポリマーと比較して分散体中の化合物Aについて改善された物理的または化学物質安定性を得ることができる。
【0101】
非晶質化合物Aの製造
非晶質化合物Aおよび化合物Aの固体非晶質分散体は、当業者に知られているいずれかの慣用のプロセスに従って製造することができる。このようなプロセスとしては、機械的、熱的および溶媒プロセスがある。典型的な機械的プロセスは、ミル処理および押出;高温融合を含む溶融プロセス、溶媒変性された融合および溶融凝固プロセス;および非溶媒沈殿、噴霧コーティングおよび噴霧乾燥を含めた溶媒プロセスが含まれる。多くの場合、プロセスは、2つまたはそれ以上のプロセスタイプを組み合わせて分散体を形成することができる。例えば、押出プロセスを使用する場合、押出機を高められた温度で運転して機械的(剪断)および熱的(加熱する)手段を用いて分散体を形成することができる。典型的な方法の例は、以下の米国特許に開示されており、それらの適切な開示は、参照により本明細書に組み込まれている:米国特許第5,456,923号および同第5,939,099号、これらは押出プロセスによる分散体の形成を記載している;米国特許第5,340,591号および同第4,673,564号、これらはミル処理プロセスによる分散体の形成を記載している;および米国特許第5,707,646号および同第4,894,235号、これらは溶融凝固プロセスによる分散体の形成を記載している。
【0102】
一実施態様において、分散体は、熱プロセス、例えば押出プロセス、融合プロセス、または溶融凝固プロセスによって形成される。このような場合、マトリックスは、熱プロセスに使用するために適切となるように選ばれる。一般に、処理温度を化合物Aの熱分解を回避するためにできる限り低く維持することが望ましい。このように、マトリックス全体として、約200℃未満、より好ましくは約160℃未満、そして最も好ましくは約120℃未満の温度で流体になることが好ましい。
【0103】
熱プロセスのマトリックス成分として使用するのに適した典型的な物質としては、アルコール、例えばステアリルアルコールおよびセチルアルコール、有機酸およびその医薬上許容しうる塩、例えばステアリン酸、クエン酸およびリンゴ酸;糖、例えばグルコース、トレハロース、キシリトール、ソルビトールおよびマルチトール;脂肪酸エステル、例えばモノ、ジおよびトリ−グリセリド、グリセリルモノ、ジおよびトリステアレート、グリセリルモノ、ジおよびトリベヘネート、ソルビタンモノステアレート、サッカロースモノステアレート、グリセリル(パルミチン酸ステアリン酸)エステル、水素化された綿実油、ポリオキシエチレンソルビタン脂肪酸エステル;ワックス、例えば微結晶ワックス、パラフィンワックス、蜜蝋、合成蝋、キャスターワックスおよびカルナバワックス;アルキルスルフェート、例えばナトリウムラウリルスルフェート;およびポリマー、例えばポリエチレングリコール、ポリオキシエチレングリコール、ポリエチレンプロピレングリコールコポリマー、ポロキサマー、ポリエチレンオキシド、ポリビニルピロリジノン(別名ポリビニルピロリドンまたはポビドンまたはPVP)、ポリビニルアルコール、ポリエチレンビニルアルコールコポリマー、ポリビニルアルコールポリビニルアセテートコポリマー、カルボン酸官能化ポリメタクリレートおよびアミン官能化ポリメタクリレートが含まれる。熱処理によって形成される分散体中に単独で使用するのに適したものとして特定の物質について議論してきたが、物質のブレンドもまた適切であるといえる。例えば、水不溶性マトリックス成分、例えば微結晶ワックスを高い水溶性のマトリックス成分、例えばポロキサマーと混合して水分散性マトリックスを形成することができる。
【0104】
マトリックスは、加工温度を下げるために、マトリックスの1つ成分として可塑剤を含むことができる。典型的な可塑剤としては、鉱油、ペトロラタム、ラノリンアルコール、ポリエチレングリコール、ポリプロピレングリコール、ソルビトール、トリエタノールアミン、安息香酸ベンジル、セバシン酸ジブチル、ジエチルフタレート、グリセリルモノステアレート、トリアセチン、およびトリエチルシトレートが含まれる。使用する可塑剤の量は、別のマトリックス成分の融点および所望の処理温度に左右される。典型的に、可塑剤対マトリックスの比率は、0.01〜0.5、より典型的には0.05〜0.1である。また、水、アルコール、ケトン、等のような溶媒または膨潤剤を使用して処理温度を低下させ、組成物の処理性を改善することができる。
【0105】
1つの好ましい熱プロセスは、押出プロセスである。ここでは、化合物Aおよび1つまたはそれ以上のマトリックス成分を、可塑剤を添加してまたは添加せずに乾燥混合して、ブレンドを二軸スクリュー押出し装置に送る。二軸スクリュー押出し装置は、化合物Aまたはマトリックスの分解なしに分散体を形成するための熱および機械的エネルギー(例えば剪断)が十分であるように設計されている。処理温度は、マトリックス物質の融点に応じて約50℃〜約200℃で変化することができる。一般に、マトリックス成分の融点がより高いほど、処理温度はより高い。
【0106】
化合物Aがマトリックス中で高い溶解度を有する場合、分散体を形成するために必要な機械的エネルギーの量はより低い。このような場合、処理温度は、化合物Aの融解温度よりも低くてもよいが、化合物Aは融解したマトリックス中に溶解するため、少なくとも一部のマトリックス物質の融点よりは高い。
【0107】
化合物Aがマトリックス中で低い溶解度を有する場合、分散体を形成するためにより多量の機械的エネルギー量が必要となりうる。ここで、処理温度は、化合物Aおよび少なくともいくつかのマトリックス成分の融点よりも上である必要がありうる。融解した化合物Aをマトリックス成分と混合して分散体を形成するには、多量の機械的エネルギーが必要でありうる。典型的に、苛酷な条件に化合物Aを曝露するのを最小限にするために最も低い処理温度および満足な分散体を製造するための最も少ない量の機械的エネルギー(例えば剪断)を付与する押出機の設計を選択する。
【0108】
分散体を形成するための別の好ましい方法は「溶媒処理」であり、化合物Aの少なくとも一部および1つまたはそれ以上のマトリックス成分の少なくとも一部を共通の溶媒中に溶解することからなる。用語「溶媒」は広く使用され、溶媒の混合物が含まれる。ここで「共通の」は、化合物Aの少なくとも一部およびマトリックス物質を溶解する溶媒を意味し、これは化合物の混合物であってもよい。
【0109】
溶媒処理のためのマトリックス成分として使用するのに適した典型的な物質は、前に記載した濃度増大ポリマー;アルコール、例えばステアリルアルコールおよびセチルアルコール;有機酸およびそれらの医薬上許容しうる塩、例えばステアリン酸、クエン酸、フマル酸、酒石酸およびリンゴ酸;塩、例えば塩化ナトリウム、塩化カリウム、塩化リチウム、塩化カルシウム、塩化マグネシウム、硫酸ナトリウム、硫酸カリウム、炭酸ナトリウムおよび硫酸マグネシウム;アミノ酸、例えばアラニンおよびグリシン;糖、例えばグルコース、シュクロース、キシリトール、フルクトース、ラクトース、マンニトール、ソルビトールおよびマルチトール;脂肪酸エステル、例えばグリセリル(モノ−およびジ−)ステアレート、トリグリセリド、水素化された綿実油、ソルビタンモノステアレート、サッカロースモノステアレート、グリセリル(パルミチン酸ステアリン酸)エステル、ポリオキシエチレンソルビタン脂肪酸エステル;ワックス、例えば微結晶ワックス、パラフィンワックス、蜜蝋、合成蝋、キャスターワックスおよびカルナバワックス;アルキルスルフェート、例えばラウリル硫酸ナトリウムおよびラウリル硫酸マグネシウム;リン脂質、例えばレシチン;タンパク質、例えばゼラチンおよびアルブミン;およびポリマー例えばポリエチレングリコール、ポリオキシエチレングリコール、ポリエチレンオキシド、キサンタンガム、カラゲナン、キトサン、ポリデキストロース、デキストリンおよびデンプンが含まれる。溶媒処理によって形成される分散体に単独で使用するのに適したものとして特定の物質を議論してきたが、物質のブレンドも適切でありうる。
【0110】
化合物Aの少なくとも一部およびマトリックスを溶解した後、溶媒は、蒸発によってまたは非溶媒と混合することによって除去される。典型的なプロセスは、噴霧乾燥、噴霧コーティング(パン−コーティング、流動層コーティング、等)、ならびに化合物Aおよびマトリックスの溶液をCO2、ヘキサン、ヘプタン、適当なpHの水またはいくつかの他の非溶媒と急速に混合することによる沈殿である。溶媒を除去して実質的に均質である固体分散体を得ることが好ましい。この目的を達成するには、例えば、溶液を霧化して化合物Aおよびマトリックスを急速に凝固させるプロセスにおいて溶液から急速に溶媒を除去することが一般に望ましい。
【0111】
化合物Aおよびマトリックスが溶解した後、溶媒を、蒸発によってまたは非溶媒と混合することによって急速に除去する。典型的なプロセスは、噴霧乾燥、噴霧コーティング(パンコーティング、流動層コーティング、等)ならびにマトリックスおよび化合物Aの溶液をCO2、水またはいくつかの他の非溶媒と急速に混合することによる沈殿である。
【0112】
固体非晶質分散体の場合、分散体は相分離していてもよいし(これは、化合物Aおよびマトリックスが上記分散体内でそれぞれ別々のドメイン中にあることを意味する)、または相互に均質に分散して単一相を形成してもよいし、またはこれらのいずれかの組み合わせまたは中間のままのこれらの状態を形成してもよい。溶媒を除去して、実質的に均質の固体非晶質分散体を形成することが好ましい。このような分散体において、化合物Aおよびマトリックスは、相互にできるだけ均質に分散され、そしてマトリックス中に分散された化合物Aの固溶体とみなすことができ、その際、固体非晶質分散体は、熱力学的に安定であり、これはマトリックス中の化合物Aの濃度がその平衡値またはそれ未満であるか、またはマトリックス中の化合物Aの濃度がその平衡値より上にある過飽和固溶体であると考えることができる。
【0113】
溶媒は、噴霧乾燥によって除去することができる。用語「噴霧乾燥」は、慣用的に使用され、大まかに言えば、液滴から溶媒を蒸発させる強い推進力を有する噴霧乾燥装置中で液体の混合物を小さな液滴に分割(噴霧化)し、混合物から溶媒を急速に除去するプロセスのことである。噴霧乾燥プロセスおよび噴霧乾燥装置は、一般にPerry's Chemical Engineers' Handbook, pages 20-54 to 20-57 (Sixth Edition 1984)に記載されている。噴霧乾燥プロセスおよび装置のより詳細は、Marshall,“Atomization and Spray-Drying,”50 Chem. Eng. Prog. Monogr. Series 2 (1954)および Masters, Spray Drying Handbook (Fourth Edition 1985)によって概説されている。溶媒蒸発のための強い推進力は、一般に、噴霧乾燥装置中、乾燥する液滴の温度で溶媒の分圧を溶媒の蒸気圧より下に維持することによって得られる。これは、(1)噴霧乾燥装置中で圧力を部分的な真空(例えば0.01〜0.50気圧)に維持する;または(2)液滴を暖かい乾燥ガスと混合する;または(3)(1)および(2)の両方によって実施される。さらに、溶媒蒸発に必要な熱の少なくとも一部は、噴霧溶液を加熱することによって得ることができる。
【0114】
噴霧乾燥に適した溶媒は、化合物Aおよびマトリックスが相互に可溶性であるすべての化合物であるといえる。また、溶媒は揮発性であり、150℃またはそれ未満の沸点を有することが好ましい。さらに、溶媒は、比較的低い毒性を有し、そして固体非晶質分散体からThe International Committee on Harmonization(ICH)ガイドラインに従って許容されうるまで除去しなければならない。このレベルまで溶媒を除去するには、トレー乾燥のような後処理工程が必要となりうる。適切な溶媒としては、限定されるわけではないが、アルコール、例えばメタノール、エタノール、n−プロパノール、イソ−プロパノールおよびブタノール;ケトン、例えばアセトン、メチルエチルケトンおよびメチルイソ−ブチルケトン;エステル、例えば酢酸エチルおよびプロピル酢酸;および種々の他の溶媒、例えばアセトニトリル、塩化メチレン、トルエンおよび1,1,1−トリクロロエタンが含まれる。また、ジメチルアセトアミドまたはジメチルスルホキシドのようなより低い揮発性の溶媒を使用することもできる。また、溶媒の混合物、例えば50%メタノールおよび50%アセトンを使用することができ、ポリマーおよび化合物Aが、噴霧乾燥プロセスを実施できるくらい十分に可溶性ならば、水との混合物であってもよい。
【0115】
噴霧溶液に水を添加すると、生成した非晶質化合物Aの化学安定性を改善することができる。一般に、水は、濃度増大ポリマーおよび存在する他の溶媒に応じて溶媒の約30質量%までの量で存在することができる。一実施態様において、溶媒は約80/20(質量
/質量)の比率でメタノールおよび水を含む。
【0116】
生成した非晶質化合物Aの化学安定性を改善するための別の方法は、窒素のような不活性ガスを噴霧溶液に通してバブリングすることによって噴霧溶液を酸素パージすることである。
【0117】
固体非晶質分散体が塩基を含む場合、塩基が化合物Aを分解しないように注意しなければならない。化合物Aは、強塩基の存在下で加水分解を受けやすい。従って、酸性ポリマーを中和するために塩基を使用する場合、塩基が最初にポリマーと反応するように塩基および酸性ポリマーを最初に溶媒中で合わせることが好ましい。次いで、化合物Aを加えて噴霧溶液を形成する。
【0118】
さらに、塩基を噴霧溶液に加えて酸性ポリマーを中和する場合、噴霧溶液に水を加えて噴霧溶液中のポリマーに十分な溶解度を与える必要がある。噴霧溶液は、水を30質量%まで含むことができる。例えば、固体10質量%(化合物A90%、HPMCAS8%および酢酸カルシウム2%からなる)を有する噴霧溶液を得るには、噴霧溶液は、メタノール80%および水20%(質量)を含むことができる。
【0119】
噴霧溶液中の化合物Aおよびマトリックスの量は、噴霧溶液中のそれぞれの溶解度および生成する固体非晶質分散体中の化合物A対マトリックスの所望の比率に左右される。噴霧溶液は、溶解物質を少なくとも約1質量%、より好ましくは少なくとも約3質量%そしてさらにより好ましくは少なくとも約10質量%含むことが好ましい。化合物A対マトリックスの比率は、0.01〜100の範囲でありうる。化合物A対マトリックスの比率は、少なくとも約0.66〜約49、より好ましくは約3〜約19そしてさらにより好ましくは約5から約15であることが好ましい。
【0120】
溶媒を有する供給物は、様々な条件下で噴霧乾燥させることができるが、それでも許容しうる性質を有する非晶質薬物または固体非晶質分散体が得られる。例えば、種々のタイプのノズルを使用して噴霧溶液を霧化することができ、それによって小さな液滴の集まりとして噴霧乾燥室中に噴霧溶液が導入される。形成された液滴が、噴霧乾燥室壁に固着しないように、すなわち噴霧乾燥室壁をコーティングしないように十分に小さく十分に乾燥(溶媒蒸発により)すれば、本質的にあらゆるタイプのノズルを使用して溶液を噴霧することができる。
【0121】
最大液滴サイズは、噴霧乾燥器内でサイズ、形状およびフローパターンの関数として、広く変化するが、一般に、液滴は、ノズルから出る時に、直径約500μm未満でなければならない。固体非晶質分散体を形成するために使用することができるノズルのタイプの例としては、二流体ノズル、噴水型ノズル、フラットファン型ノズル、加圧ノズルおよびロータリーアトマイザーがある。好ましい実施態様において、加圧ノズルを使用することができ、これは、2003年1月24日に出願され、同一出願人による係属中の米国特許出願第10/351,568号に詳細に開示されており、2002年2月1日に出願された米国仮出願第60/353,986号に対して優先権を主張し、この開示は参照により本明細書に組み込まれている。
【0122】
噴霧溶液は、広範囲にわたる温度および流速で噴霧ノズルまたはノズルに供給することができる。一般に、噴霧溶液温度は、溶媒の凝固点のすぐ上から周囲圧力の沸点より約20℃上(溶液に加圧することによって)までそして場合によってはさらに高いいずれかの範囲であることができる。噴霧ノズルへの噴霧溶液の流速は、ノズルのタイプ、噴霧乾燥機のサイズおよび噴霧乾燥条件、例えば乾燥ガスの入口温度および流速に応じて広い範囲で変化することができる。一般に、噴霧乾燥プロセスにおける噴霧溶液から溶媒を蒸発させるエネルギーは、主として乾燥ガスから生じる。
【0123】
乾燥ガスは、原則として、本質的にすべてのガスであることができるが、安全性の理由のため、そして固体非晶質分散体中で化合物Aまたは他の物質の望ましくない酸化を最小限にするため、窒素のような不活性ガス、窒素の豊富な空気またはアルゴンが使用される。乾燥ガスは、典型的に約60℃〜約300℃、そして好ましくは約80℃〜約240℃の温度で乾燥室に導入される。噴霧乾燥プロセス中およびその後に化合物Aの化学分解を最小限にするために、化合物Aを高い温度に曝露するのを制限することが好ましい。例えば、噴霧溶液は化合物A、HPMCAS、メタノールおよび水を含み、NIRO PSD-1噴霧乾燥機を用いて噴霧する場合、入口ガス温度は150℃およびより好ましくは130℃またはそれ未満でありうる。
【0124】
液滴の比表面積が大きく、溶媒蒸発の推進力が大きいと、液滴の凝固時間が早くなる。凝固時間は、約20秒未満、好ましくは約10秒未満、そしてさらに好ましくは1秒未満でなければならない。この急速な凝固は、多くの場合、粒子が、薬物Aの豊富な相およびポリマーの豊富な相に分離しないで均一で均質な分散体を維持するのに重要である。好ましい実施態様において、噴霧乾燥器の高さおよび容積は、液滴が、噴霧乾燥機の内側面に衝突する前に乾燥するのに十分な時間が得られるように調整され、これは同一出願人による米国特許第6,763,607号に詳述されており、参照により本明細書に組み込まれている。上記したように、濃度および生物学的利用能における大きな増大を得るには多くの場合、できるだけ均質な分散体を得る必要がある。
【0125】
凝固した後、固体粉末は、典型的に約5〜60秒間、噴霧乾燥室中にとどまり、さらに固体粉末から溶媒を蒸発させる。固体分散体の最終的な溶媒含量は、乾燥機を出る時には低くなければならず、その理由は、これによって固体非晶質分散体中での化合物A分子の易動度が低減され、そのため安定性が改善されるためである。一般に、固体非晶質分散体の溶媒含量は、噴霧乾燥室から出る時には、10質量%未満、そして好ましくは2質量%未満でなければならない。
【0126】
形成した後、適切な乾燥プロセス、例えばトレー乾燥、真空乾燥、流動層乾燥、高周波乾燥、ベルト乾燥、回転乾燥および当分野で知られている他の乾燥プロセスを用いて固体非晶質分散体を乾燥させて残留溶媒を除去することができる。固体非晶質分散体は、熱い乾燥状態への曝露を最小限にする条件下で乾燥させることが好ましい。好ましくは、温度は50℃未満で、そしてより好ましくは40℃未満である。相対湿度は、相対湿度25%〜75%の間で維持するのが好ましい。好ましい二次的な乾燥方法としては、真空乾燥または周囲条件下のトレー乾燥が含まれる。乾燥の際に化学分解を最小限にするために、乾燥は窒素のような不活性ガス下で行うことができまたは真空下で行うことができる。
【0127】
固体非晶質分散体は、通常、小さな粒子の形態である。粒子の平均サイズは、直径500μm未満、直径100μm未満、直径50μm未満または直径25μm未満であることができる。固体非晶質分散体を噴霧乾燥によって形成する場合、生成した分散体は、このような小さな粒子の形態である。固体非晶質分散体を溶融凝固または押出プロセスのような別の方法によって形成する場合、生成した分散体を、ふるいにかけ、粉砕し、ミル処理し、または別のやり方で処理して多くの小さな粒子を得ることができる。
【0128】
処理を容易にするには、乾燥した粒子は、一定の密度およびサイズの特徴を有することになる。一実施態様において、生成した固体非晶質分散体粒子は、噴霧乾燥によって形成され、嵩比容約4cc/gまたはそれ未満、そしてより好ましくは約3.5cc/gまたはそれ未満を有することができる。粒子は、タップ比容約3cc/gまたはそれ未満、そしてより好ましくは約2cc/gまたはそれ未満を有することができる。粒子は、Hausner比約3またはそれ未満、そしてより好ましくは約2またはそれ未満を有する。粒子は平均粒子直径約150μmまで、そしてより好ましくは約1〜約25μmを有することができる。粒子は、スパン3またはそれ未満、そしてより好ましくは約2.5またはそれ未満を有することができる。本明細書に使用されるように、「スパン」は、
【数1】
として定義され、ここで、D10は、直径が等しいまたはより小さい粒子を含んでなる、総容積の10%を占める粒子の直径に対応する直径であり、D50は、直径が等しいまたはより小さい粒子を含んでなる、総容積の50%を占める粒子の直径に相当する直径であり、そしてD90は、直径が等しいまたはより小さい直径の粒子を含んでなる、総容積の90%を占める粒子の直径に対応する直径である。
【0129】
分散体を形成するのに適当であるとして前記されたプロセスは、非晶質化合物Aの形成に使用することができる。特に、非晶質化合物Aは、化合物Aを溶媒、例えばアセトンまたはメタノール中に溶解し、そしてマトリックス中の化合物Aの分散体を製造するために上記したのと同じ方法で噴霧乾燥することによって製造することができる。また、非晶質化合物Aは、例えば、結晶質化合物Aを溶融凝固装置に供給することによって製造することができ、これは、米国特許第5,183,493号または第5,549,917号に開示されており、融解した化合物Aの液滴を形成し、次いで、冷却ガスによって冷却し、直径約1〜約500μmそして好ましくは直径10〜300μmの範囲の化合物Aの非晶質粒子を形成する。
【0130】
固体非晶質分散体または非晶質薬物は、封入パッケージ中に保存して湿気(水蒸気)および/または酸素への曝露を減らすことができる。湿気または酸素との接触を減らすための典型的な方法としては、水蒸気および/または酸素に対して実質的に不透過性である保護パッキング、例えばホイルポーチまたはホイルブリスターパック、または組成物のパッケージング中に乾燥剤または酸素吸収剤、例えば酸素吸着剤パケットを封入することが含まれる。パッケージングは、湿気を減らすための乾燥剤、酸素吸収剤、または両方を含むことができる。典型的な乾燥剤としては、アルミノシリケート(Sorb-it(R),Sud-Chemieから入手可能)および無水硫酸カルシウムが含まれ、そして典型的な酸素吸収剤としては、米国特許第6,558,571号に開示された酸素吸収組成物およびMultisorb Technologies, Inc.から入手可能な商品名Fresh PaxTMの下で販売されているパケットおよび細片が含まれる。本発明は、(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミド、またはその医薬上許容しうる塩または溶媒和物を含んでなる医薬組成物を含む医薬パッケージであって、酸素吸収剤をさらに含んでなる前記パッケージを提供する。さらなる態様において、存在する酸素ガスの量は、前記パッケージ中に存在するガスの総量の約5体積%未満であるこのようなパッケージが提供される。さらなる態様において、存在する酸素ガスの量は、前記パッケージ中に存在するガスの総量の約2%未満、または約1%未満、または約0.5%未満、または約0.25%未満、または約0.1%未満、または約0.01%未満である。
【0131】
濃度増大
好ましい実施態様において、本発明の組成物は、有用な水性環境に投薬した時に、濃度増大が得られ、これは、少なくとも以下の状態の1つを満たすことを意味する。第1の状態は、使用環境中において本発明の組成物が、化合物Aの結晶質形態Iの等量からなる対照組成物と比較して化合物Aの最大溶解濃度(MDC)を高める状態である。すなわち、組成物がいったん使用環境に導入されたら、組成物は、対照組成物と比較して化合物Aの水性濃度を高める。対照組成物は、可溶化剤または化合物Aの結晶質形態Iの溶解度に物質的に影響を及ぼす他の成分を含まず、そして化合物Aは、対照組成物中で固体状態であることを理解すべきである。対照組成物は、化合物Aの慣用的に分散されてない結晶質形態I単独である。本発明の組成物では、水溶液中の化合物AのMDCが、対照組成物によって得られるものの少なくとも1.25倍または少なくとも2倍または少なくとも3倍であ
ることが好ましい。場合によっては、本発明の組成物によって得られる化合物AのMDCは、対照組成物によって得られる平衡濃度の少なくとも5倍または少なくとも10倍である。
【0132】
第2の状態は、本発明の組成物が、ポリマーを有しない化合物Aの結晶質形態Iの等量を含んでなる対照組成物と比較して使用環境中で化合物Aの濃度対時間曲線より下の溶解面積(AUC)を増加させる状態である。AUCの計算は、医薬分野においてよく知られている方法であって、例えば、Welling,“Pharmacokinetics Processes and Mathematics,
”ACS Monograph 185 (1986)に記載されている。より詳しくは、使用環境において、本発明の組成物では、使用環境に導入後の約0〜約270分の間のいずれか90分の期間のAUCが、上記の対照組成物の少なくとも約1.25倍である。組成物によって得られるAUCは、対照組成物の少なくとも1.25倍または少なくとも約2倍または少なくとも約3倍であることが好ましい。本発明のいくつかの組成物では、上記対照組成物の少なくとも約5倍または少なくとも約10倍のAUC値を得ることができる。
【0133】
本発明の一実施態様において、組成物は、非晶質化合物Aおよび濃度増大ポリマーを含まない非晶質化合物Aを含んでなる第2の対照組成物と比較して組成物が濃度増大をもたらすのに十分な量の濃度増大ポリマーを含んでなる。これらの組成物において、ポリマーは水溶液中で化合物AのMDCまたはAUCの少なくとも1つを、第2の対照組成物と比較して少なくとも1.25倍または少なくとも約2倍または少なくとも約3倍高める。
【0134】
前述のように、「使用環境」とは、in vivo環境、例えば動物、特にヒトの消化管、または試験溶液のin vitro環境、例えばリン酸緩衝生理食塩水(PBS)溶液またはModel Fasted Duodenal(MFD)溶液のいずれかであることができる。本発明者らは、in vitro溶解試験は、in vivo性質の良好な予測因子であり、従って、組成物はin vitroおよびin vivo使用環境のいずれかまたは両方において濃度増大をもたらす場合、それらは本発明の範囲内にあることを見出した。
【0135】
本発明の組成物では、in vitro溶解試験において溶解した化合物Aの高められた濃度が得られる。in vitro溶出試験においてMFD溶液中またはPBS溶液中の高められた薬物濃度は、in vivo性能および生物学的利用能の良好な指標であることがわかった。適当なPBS溶液は、20mM Na2HPO4、47mM KH2PO4、87mM NaClおよび0.2mM KClを含み、NaOHでpH6.5に調整された水溶液である。適当なMFD溶液は、7.3mMナトリウムタウロコール酸および1.4mMの1−パルミトイル−2−オレイル−sn−グリセロ−3−ホスホコリンがさらに存在する同様のPBS溶液である。特に、本発明の方法によって形成された組成物はMFDまたはPBS溶液に加えて撹拌して溶解を促進することによって溶解試験することができる。
【0136】
水溶液中で高められた化合物Aの濃度を評価するin vitro試験は、(1)十分な量の対照組成物、典型的には分散されてない化合物A単独を、in vitro試験媒体、例えばMFDまたはPBS溶液に撹拌しながら加えて、化合物Aの平衡濃度にし;(2)別の試験において、全ての化合物Aが溶解した場合、化合物Aの理論濃度が、化合物Aの平衡濃度を少なくとも2倍または少なくとも10倍超えるように、十分な量の試験組成物(例えば、化合物Aおよびポリマーを含んでなる組成物)を同じ試験媒体中に撹拌しながら加え;そして(3)試験媒体中の試験組成物の測定したMDCおよび/または水性AUCを平衡濃度、および/または対照組成物の水性AUCと比較する:ことによって実施することができる。このような溶解試験を実施する際、使用した試験組成物または対照組成物の量は、化合物Aの全てが溶解した場合、化合物Aの濃度が平衡濃度の少なくとも2倍、少なくとも10倍、そしてもっとも好ましくは少なくとも100倍となるような量である。
【0137】
溶解した化合物Aの濃度は、典型的には、MDCを確認することができるように、試験媒体の試料を採取し、試験媒体中の化合物Aの濃度対時間をプロットして時間の関数として測定する。MDCは、試験の持続期間にわたって測定された溶解化合物Aの最大値である。水性AUCは、水性使用環境中へ組成物を導入した時間(時間がゼロの時)および使用環境に導入後270分(時間が270分の時)の間のいずれか90分の期間にわたって濃度対時間曲線を積分することによって算出される。典型的には、組成物が急速に、つまり約30分未満にそのMDCに到達した時、AUCを算出するために使用する時間的間隔は、時間0から時間90分までである。しかしながら、上記のいずれか90分の期間にわたる組成物のAUCが本発明の基準を満たす場合、形成された組成物は、本発明の範囲内であるとみなす。
【0138】
測定を誤る大きな薬物微粒子を取り除くため、試験溶液を濾過または遠心分離する。「溶解した薬物」は、典型的に0.45μmシリンジフィルタを通過する物質、またはそれとは別に遠心分離後の上澄み中に残る物質のいずれかである。濾過は、商標TITAN(R)の下でScientific Resourcesによって販売された13mm、0.45μmポリビニリジンジフルオリドシリンジフィルタを用いて実施することができる。遠心分離は、典型的にポリプロピレンマイクロ遠心分離管中、13,000Gで60秒間遠心分離することによって実施される。他の同様の濾過または遠心分離方法を使用することができ、有用な結果が得られる。例えば、別のタイプのミクロフィルターを使用すると、上記のフィルタで得るよりいくらか高いまたは低い値(±10〜40%)を得ることができるが、好ましい分散体の確認は依然として可能である。「溶解した薬物」のこの定義は、単一の溶媒和された薬物分子だけでなく、また、広範囲にわたる種類、例えばサブミクロンの寸法を有するポリマー/薬物集合体、例えば薬物凝集体、ポリマーおよび薬物の混合物の凝集体、ミセル、ポリマーミセル、コロイド粒子またはナノクリスタル、ポリマー/薬物複合体およびその他、明記された溶解試験において濾液または上澄み中に存在するこのような薬物を含む種類を包含することを認識すべきである。
【0139】
さらにまた、本発明の組成物は、絶食状態のヒトまたは他の哺乳動物に経口的に投薬した時に、等量の化合物の結晶質形態I単独からなる対照組成物によって得られるCmaxの少なくとも約1.25倍、または少なくとも約2倍、または少なくとも約3倍、または少なくとも約5倍、または少なくとも約10倍のCmaxが得られる。
【0140】
別法として、組成物では、絶食状態のヒトまたは他の哺乳動物に経口的に投薬した時に、血液(血清または血漿)中の化合物A濃度におけるAUCが、等量の化合物Aの結晶質形態Iからなる対照組成物を、追加ポリマーなしに単独で投薬した時に観察されるものの少なくとも約1.25倍、または少なくとも約2倍、または少なくとも約3倍、または少なくとも約5倍、または少なくとも約10倍得られる。このような組成物は、対照組成物の約1.25倍〜約10倍の相対的な生物学的利用能を有するといえることに留意する。
【0141】
組成物中の化合物Aの相対的な生物学的利用能は、このような測定を行うための慣用の方法を用いて動物またはヒトにおいてin vivo試験することができる。in vivo試験、例えば交差試験法は、化合物Aおよびマトリックスの組成物では、上記対照組成物と比較して高められた相対的な生物学的利用能が得られるかどうかを測定するのに使用することができる。in vivo交差試験法では、化合物Aおよびマトリックスの固体非晶質分散体の試験組成物を試験の被験者のグループの半分に投薬し、そして適当な休薬期間(例えば1週間)の後、同じ被験者に試験組成物として等量の化合物Aの結晶質形態Iからなる対照組成物(しかし、マトリックスは存在しない)を投薬する。グループのもう半分には、最初に対照組成物、続いて試験組成物を投薬した。相対的な生物学的利用能は、試験グループについて測定した血液(血清または血漿)中の濃度対時間曲線下の面積(AUC)を、対照組成物によって得られた血液中のAUCによって割ったものとして測定される。この試験/対照の比率は、それぞれの被験者について測定し、比率を本研究における全ての被験者で平均することが好ましい。AUCのin vivo測定は、横座標(x軸)に沿った時間に対して縦座標(y軸)に沿って化合物Aの血清または血漿中濃度をプロットすることによって行うことができる。投薬を促進するため、投薬ビヒクルを用いて用量を投与することができる。投薬ビヒクルは、好ましくは水であり、さらに試験または対照組成物を懸濁するための物質を含むことができるが、但し、これらの物質はin vivoで組成物を溶解せず、または化合物Aの溶解性を変化させない。
【0142】
剤形
組成物は、経口、鼻、直腸、膣、皮下、静脈内および肺を含めた様々な経路によって供給することができるが、これらに限定されない。一般に、経口経路が好ましい。
【0143】
また、組成物は、薬物を投与するための様々な剤形で使用することができる。典型的な剤形としては、ドライであるかまたは水もしくは他の液体を添加して再構成してペースト、スラリー、懸濁液または溶液を形成して経口的に摂取することができる散剤または顆粒剤;錠剤;カプセル剤;多粒剤;および丸剤である。種々の添加剤を本発明の組成物と共に混合、粉砕または造粒して上記の剤形に適した物質を形成することができる。
【0144】
本発明の組成物は、種々の形態で処方することができ、例えば液体ビヒクル中の粒子の懸濁液として供給される。このような懸濁液は、製造時に液体またはペーストとして処方することができ、またはドライ粉末として処方してあとで経口投与の前に、液体、典型的に水を加えることができる。懸濁液中に構成されるこのような粉末は、しばしばサシェまたは構成用経口粉末(OPC)製剤と称する。このような剤形は、いずれかの知られている方法によって処方および再構成することができる。最も単純なアプローチは、単に水を加えて撹拌することによって再構成される乾燥粉末として剤形を処方することである。別法として、剤形は、液体としておよび合わせて撹拌して経口懸濁液を形成する乾燥粉末として処方することができる。さらに別の実施態様において、剤形は、2種の粉末として処方することができ、これは、最初に1つの粉末に水を加え、溶液を形成し、これに第2の粉末を撹拌しながら合わせて懸濁液を形成することによって再構成される。
【0145】
さらに、本発明の組成物は、HIVウイルスによる感染症、AIDS、AIDS関連症候群(ARC)、またはHIVウイルスによる感染症と関連がある他のいずれかの疾患または状態にかかっている哺乳動物、例えばヒトを治療するためのさらなる薬剤(1種またはそれ以上)と組み合わせて投与することができる。本発明の組成物と組み合わせて使用することができる薬剤としては、HIVプロテアーゼ阻害剤として有用なもの、HIV逆転写酵素阻害剤、非ヌクレオシドHIV逆転写酵素阻害剤、HIVインテグラーゼの阻害剤、CCR5阻害剤、HIV融合阻害剤、免疫調節物質として有用な化合物、未知のメカニズムによってHIVウイルスを阻害する化合物、ヘルペスウイルスの治療に有用な化合物、抗感染薬として有用な化合物、および後述する他のものが含まれるが、これらに限定されない。
【0146】
本発明の組成物と組み合わせて使用することができるHIVプロテアーゼ阻害剤として有用な化合物としては、141W94(アンプレナビル)、CGP−73547、CGP−61755、DMP−450、ネルフィナビル、リトナビル、サキナビル(インビラーゼ)、ロピナビル、TMC−126、アタザナビル、パリナビル、GS−3333、KNI−413、KNI−272、LG−71350、CGP−61755、PD173606、PD177298、PD178390、PD178392、U−140690、ABT−378、DMP−450、AG−1776、MK−944、VX−478、インジナビル、チプラナビル、TMC−114、DPC−681、DPC−684、フォサンプレナビルカルシウム(レキシバ)、WO 03053435に開示されたベンゼンスルホンアミド誘導体、R−944、Ro−03−34649、VX−385、GS−224338、OPT−TL3、PL−100、SM−309515、AG−148、DG−35−VIII、DMP−850、GW−5950X、KNI−1039、L−756423、LB−71262、LP−130、RS−344、SE−063、UIC−94−003、Vb−19038、A−77003、BMS−182193、BMS−186318、SM−309515、JE−2147、GS−9005が含まれるが、これらに限定されない。
【0147】
本発明の組成物と組み合わせて使用することができるHIV逆転写酵素の阻害剤として有用な化合物には、アバカビル、FTC、GS−840、ラミブジン、アデホビルジピボキシル、ベータ−フルオロddA、ザルシタビン、ジダノシン、スタブジン、ジドブジン、テノホビル、アムドキソビル(amdoxovir)、SPD−754、SPD−756、ラシビール、ラベルセット(DPC−817)、MIV−210(FLG)、ベータ−L−Fd4C(ACH−126443)、MIV−310(アロブジン、FLT)、dOTC、DAPD、エンテカビル、GS−7340、エムトリシタビン、アロブジンが含まれるが、これらに限定されない。
【0148】
HIV逆転写酵素の非ヌクレオシドの阻害剤として有用な化合物には、エファビレンツ、HBY−097、ネビラピン、TMC−120(ダピビリン)、TMC−125、エトラビリン、デラビルジン、DPC−083、DPC−961、TMC−120、カプラビリン、GW−678248、GW−695634、カラノリド、およびWO 03062238に開示された三環式ピリミジノン誘導体が含まれるが、これらに限定されない。
【0149】
本発明の組成物と組み合わせて使用することができるCCR5阻害剤として有用な化合物には、TAK−779、SC−351125、SCH−D、UK−427857、PRO−140およびGW−873140(Ono−4128,AK−602)が含まれるが、これらに限定されない。
【0150】
本発明の組成物と組み合わせて使用することができるHIVインテグラーゼ酵素の阻害剤として有用な化合物には、GW−810781(WO 03062204に開示された1,5−ナフチリジン−3−カルボキサミド誘導体)、WO 03047564に開示された化合物、WO 03049690に開示された化合物、およびWO 03035076に開示された5−ヒドロキシピリミジン−4−カルボキサミド誘導体が含まれるが、これらに限定されない。
【0151】
本発明の組成物と組み合わせて使用することができるHIV治療のための融合阻害剤としては、エンフビルチド(T−20)、T−1249、AMD−3100、およびJP2003171381に開示された縮合三環式化合物が含まれるが、これらに限定されない。
【0152】
本発明の組成物と組み合わせて使用することができるHIVの有用な阻害剤である他の化合物としては、可溶性のCD4、TNX−355、PRO−542、BMS−806、フマル酸テノホビルジソプロキシル、およびJP2003119137に開示された化合物が含まれるが、これらに限定されない。
【0153】
本発明の組成物と組み合わせて使用することができる、HIV以外のウイルス感染症の治療または処置に有用な化合物としては、アシクロビル、ホミビルセン、ペンシクロビル、HPMPC、オキセタノシンG、AL−721、シドホビル、サイトメガロウィルス免疫グロビン、サイトベン、ホミブガンシクロビル(fomivganciclovir)、ファムシクロビル、フォスカーネットナトリウム、Isis2922、KNI−272、バラシクロビル、ビラゾールリバビリン、バルガンシクロビル、ME−609、PCL−016が含まれるが、これらに限定されない。
【0154】
免疫調節物質として作用し、そして本発明の組成物と組み合わせて使用することができる化合物としては、AD−439、AD−519、アルファインターフェロン、AS−101、ブロピリミン、アセマンナン、CL246,738、EL10、FP−21399、ガンマインターフェロン、顆粒球マクロファージコロニー刺激因子、IL−2、静脈内免疫グロブリン、IMREG−1、IMREG−2、イムチオールジエチルジチオカルバメート、アルファ−2−インターフェロン、メチオニン−エンケファリン、MTP−PE、顆粒球コロニー刺激因子、レムン(remune)、rCD4、組換え型可溶性のヒトCD4、インターフェロンアルファ−2、SK&F106528、可溶性T4イモペンチン、腫瘍壊死因子(TNF)、ツカレソール、組換え型ヒトインターフェロンベータ、およびインターフェロンアルファn−3が含まれるが、これらに限定されない。
【0155】
本発明の組成物と組み合わせて使用することができる抗感染薬としては、アトバクオン、アジスロマイシン、クラリスロマイシン、トリメトプリム、トロバフロキサシン、ピリメタミン、ダウノルビシン、プリマキンと共にクリンダマイシン、フルコナゾール、パスチル、オルニジル、エフロルニチンペンタミジン、リファブチン、スピラマイシン、イトラコナゾール−R51211、トリメトレキサート、ダウノルビシン、組換え型ヒトエリスロポエチン、組換え型ヒト成長ホルモン、メゲストロールアセテート、テストロン、および全ての腸内栄養剤が含まれるが、これらに限定されない。
【0156】
本発明の組成物と組み合わせて使用することができる抗真菌剤としては、アニデュラファンギン、C31G、カスポファンギン、DB−289、フルコナゾール、イトラコナゾール、ケトコナゾール、ミカファンギン、ポサコナゾールおよびボリコナゾールが含まれるが、これらに限定されない。
【0157】
本発明の組成物と組み合わせて使用することができる他の化合物としては、アクマンナン、アンサマイシン、LM 427、AR177、BMS−232623、BMS−234475、CI−1012、硫酸カードラン、硫酸デキストラン、STOCRINE EL10、ヒペリシン、ロブカビル、ノバプレン、ペプチドTオクターブペプチド配列(peptide T octabpeptide sequence)、トリナトリウムホスホノホルメート、プロブコール、およびRBC−CD4が含まれるが、これらに限定されない。
【0158】
さらに、本発明の組成物は、カポジ肉腫のような状態を治療するため抗増殖剤と組み合わせて使用することができる。このような薬剤としては、メタロマトリックスプロテアーゼの阻害剤、A−007、ビバシズマブ、BMS−275291、ハロフギノン、インターロイキン−12、リタキシマブ、パクリタキセル、ポルフィマーナトリウム、レビマスタットおよびCOL−3が含まれるが、これらに限定されない。
【0159】
さらなる薬剤(1種またはそれ以上)の特定の選択は、治療する哺乳動物の状態、治療する特定の状態、さらなる薬剤(1種またはそれ以上)の特徴、および哺乳動物を治療するために使用されるいずれかのさらなる化合物の特徴を含めた多くの因子に左右されるが、これらに限定されない。さらなる薬剤(1種またはそれ以上)の特定の選択は、当業者の知識の範囲内である。
【0160】
本発明の組成物は、HIVウイルスによる感染症、AIDS、AIDS関連症候群(ARC)、またはHIVウイルスの感染症と関連がある他のいずれかの疾患または状態にかかっている哺乳動物、例えばヒトを治療するため、上記のさらなる薬剤のいずれかと組み合わせて投与することができる。このような組み合わせは、本発明の組成物が上記のさらなる薬剤と同じ製剤中に存在するようにして哺乳動物に投与することができる。別法として、このような組み合わせは、さらなる薬剤が存在する製剤とは別の製剤中に本発明の組成物が存在するようにしてHIVウイルスによる感染症にかかっている哺乳動物に投与することができる。本発明の組成物をさらなる薬剤と別々に投与する場合、このような投与は、同時にまたは間に適当な期間を置いて順次行うことができる。さらなる薬剤(1種またはそれ以上)と同じ製剤中に本発明の組成物を含むべきかどうかの選択は、当業者の知識の範囲内である。
【0161】
さらに、本発明の組成物は、哺乳動物の本発明の化合物への曝露を高める効果を有するさらなる薬剤と組み合わせて哺乳動物、例えばヒトに投与することができる。用語「曝露」は、本明細書に使用されるように、ある期間にわたって測定された哺乳動物の血漿中の本発明の化合物の濃度に相当する。哺乳動物の化合物Aへの曝露は、本発明の組成物を適当な形態で哺乳動物に投与し、所定の時間に血漿試料を採取し、適当な分析技術、例えば液体クロマトグラフィまたは液体クロマトグラフィ/質量分析を用いて血漿中の化合物Aの量を計量することによって測定することができる。ある時間における血漿中の化合物Aの量を測定し、全てのサンプルからの濃度および時間データをプロットして曲線を得る。この曲線下の面積を算出して哺乳動物の化合物に対する曝露を得る。用語「曝露」、「曲線下の面積」および「濃度/時間曲線下の面積」は、同じ意味を有するものとし、全体を通して取り換えて使用することができる。
【0162】
化合物Aに対する哺乳動物の曝露を高めるために使用することができる薬剤の中には、チトクロームP450(CYP450)酵素の少なくとも1つのアイソフォームの阻害剤として作用することができるものがある。有益に阻害することができるCYP450のアイソフォームとしては、CYP1A2、CYP2D6、CYP2C9、CYP2C19およびCYP3A4が含まれるが、これらに限定されない。CYP3A4を阻害するために使用することができる適切な薬剤としては、リトナビルおよびデラビルジンが含まれるが、これらに限定されない。
【0163】
このような組み合わせは、化合物Aが上記のさらなる薬剤と同じ製剤中に存在するようにして哺乳動物に投与することができる。別法として、このような組み合わせは、化合物Aは、さらなる薬剤が存在する製剤とは別々の製剤中に存在するようにして投与することができる。化合物Aを、さらなる薬剤と別々に投与する場合、このような投与は、同時にまたは間に適当な期間をおいて順次行うことができる。さらなる薬剤(1種またはそれ以上)と同じ製剤中に本発明の組成物を含むべきかどうかの選択は、当業者の知識の範囲内である。
【0164】
本発明の他の特徴および実施態様は、以下の実施例から明らかとなり、これは本発明を説明するために記載したものであって、その意図する範囲を制限するものではない。
【実施例】
【0165】
実施例1
非晶質化合物Aを含む実施例1は、以下の通りに製造した。最初に、化合物A250.1mgおよびメタノール24.5gを含む噴霧溶液を形成した。Cole Parmer 74900シリーズの速度制御シリンジポンプを経て70mL/時の速度で溶液を「ミニ」噴霧乾燥装置にポンピングした。化合物Aの溶液を、窒素の加熱流れを用いて1SCFMの流速でSpraying Systems Co.の二流体ノズルModel No. SU1Aを通して霧化した。噴霧溶液を、直径11cmのステンレス鋼室に噴霧した。入口温度100℃で室に入った加熱されたガスは、出口温度22℃で排出された。生成した非晶質化合物Aを濾紙上に集め、真空下で乾燥させてデシケータ中に保存した。収量は約78%であった。
【0166】
実施例1の非晶質化合物Aが化合物Aの結晶質形態と比較して濃度増大をもたらすかどうかを測定するためにin vitro溶解試験を実施した。この試験では、全ての化合物Aが溶解した場合化合物Aの濃度が3000μg/mLとなるように、十分な量の物質を、マイクロ遠心分離試験管に加えた。試験は、同じものを二通り行った。管を、温度37℃の制御された室に置き、PBS 1.8mLをpH6.5および290mOsm/kgでそれぞれ個々の管に加えた。渦流ミキサーを用いて試料を約60秒間、急速に混合した。試料を、37℃、13,000Gで1分間遠心分離した。次いで、生成した上澄液から試料を採取し、メタノールで1:6(体積)に希釈してから高性能液体クロマトグラフィー(HPLC)で分析した。HPLC分析は、Phenomenex Luna C18カラムを用いて実施した。移動相は、H3PO4でpH3に調整された55%20mM KH2PO4および45%アセトニトリルからなる。UV吸光度は、210nmで測定した。各管の内容物を渦流ミキサーで混合し、次の試料を採取するまで、37℃で静かに放置した。試料は、4、10、20、40、90および1200分に集めた。結果を表1に示した。
【0167】
対照1
対照1は、化合物Aの結晶質形態I単独からなり、そして全ての化合物Aが溶解した場合、化合物Aの濃度が3000μg/mLとなるように十分な量の物質を加えた。
【表1】
【0168】
これらの試料で得られた薬物の濃度を用いて薬物の最大濃度(「MDC90」)、最初の90分間に濃度対時間曲線より下の面積(「AUC90」)、および1200分での化合物Aの濃度(「C1200」)を測定した。結果を表2に示した。
【表2】
【0169】
結果から、化合物Aの非晶質形態では、化合物Aの結晶質形態I単独と比較して濃度増大が得られることがわかった。化合物Aの非晶質形態では、化合物A.実施例2の結晶質形態Iによって得られたものの10.3倍のMDCおよび8.3倍のAUC90が得られた。
【0170】
実施例2
実施例2では、非晶質化合物Aおよび濃度増大ポリマーを合わせて製造した。化合物Aおよび濃度増大ポリマーヒドロキシプロピルメチルセルロースアセテートスクシネート(AQUOT−MG,信越化学工業,東京,日本から入手可能)の単純な物理的混合物は、実施例1で製造した非晶質化合物A5.4mgおよびHPMCAS0.6mgを遠心分離管に加えることによって製造した。渦流ミキサーを用いて乾燥粉末を1分間混合した。溶解試験は、実施例1に記載された通り実施した。結果を表3に示した。
【表3】
【0171】
これらの試料で得た薬物の濃度を用いて、薬物の最大濃度、MDC90、最初の90分間の濃度対時間曲線より下の面積(AUC90)および1200分での化合物A濃度、C1200を測定した。結果を表4に示した。実施例1および対照1についての比較を再び示した。
【表4】
【0172】
非晶質化合物Aおよび濃度増大ポリマーの両方を含む実施例2の物理的混合物では、結晶質形態Iの化合物A(対照1)単独と比較して改善された溶解性能が得られた。実施例2では、結晶質形態I化合物A単独によって得られたものの8.7倍のMDC90、および結晶質形態Iの化合物A単独によって得られたものの8.3倍のAUC90が得られた。
また、実施例2では、非晶質化合物A単独よりも長い時間、溶解薬物濃度が持続した。非晶質化合物A単独(実施例1)および濃度増大ポリマー(実施例2)を加えた非晶質化合物Aの性能は、最初の90分間では同様の性能を有していたが、濃度増大ポリマーを添加すると、より遅い時間で化合物Aの溶解性能が実質的に改善された。実施例2では、1200分での溶解した化合物Aの濃度(「C1200」)は、非晶質化合物A単独の7.8倍得られた。
【0173】
実施例3
実施例3、化合物A90質量%およびHPMCAS(AQUOT−MG,信越化学工業,東京,日本から入手可能)10質量%を含んでなる固体非晶質分散体を、以下の通りに製造した。最初に、化合物A300g、HPMCAS33.3gおよびメタノール3000gを含む噴霧溶液を以下の通り形成した。HPMCASおよびメタノールを容器中で合わせ、約2時間混合して、HPMCASを溶解させた。ポリマーの全量を加えた後、生成した混合物はわずかな曇りがあった。次に、化合物Aをこの混合物に直接加え、混合物をさらに2時間撹拌した。次いで、この混合物を、スクリーンサイズ250μmのフィルタに通すことによって濾過し、混合物からすべての大きな不溶性物質を除去し、このようにして噴霧溶液を形成した。
【0174】
高圧ポンプを用いて噴霧溶液を、加圧ノズル(Spraying Systems Pressure Nozzle and Body) (SK 78-21)を備えた噴霧乾燥機(Liquid-Feed Process Vesselを有するNiro type XP Portable Spray-Dryer(PSD−1))にポンピングした。PSD−1は、9インチの室延長部を備えていた。9インチの室延長部を噴霧乾燥機に付け加え、乾燥機の縦の長さを延ばした。長さを延ばして、乾燥機内の滞留時間を増加させ、これによって生成物を噴霧乾燥機の曲がった部分に到達する前に乾燥させることができる。また、噴霧乾燥機は、316SSサーキュラーディフューザープレートを備えており、1/16インチのドリル穴があって1%の開口領域を有する。この小さな開口領域は、乾燥ガスの流れが噴霧乾燥機内で生成物の再循環を最小限にすることを目的としている。ノズルは、操作中にディフューザープレートと同一平面上にした。Bran+Lubbe高圧ポンプを用いて液体をノズルに供給した。ポンプの後にパルセイション緩衝装置があり、ノズルでのパルセイションを最小限にする。噴霧溶液を、圧力200psigで約180g/分で噴霧乾燥機にポンピングした。乾燥ガス(例えば窒素)は、入口温度200℃で、ディフューザープレートを通して循環させた。蒸発した溶媒および乾燥ガスは、60℃の温度で、噴霧乾燥機から排出された。生成した固体非晶質分散体を、サイクロン中に集めた。
【0175】
上記の方法を用いて形成された固体非晶質分散体を、Gruenberg単路対流トレー乾燥機を用いて40℃で6時間作動させて後乾燥させた。次いで、乾燥後、分散体を周囲空気および湿気(20℃/RH50%)で8時間平衡状態にした。
【0176】
二次乾燥後の固体非晶質分散体の性質を表5に示した。
【表5】
【0177】
実施例4〜11
「ミニ」噴霧乾燥装置を用いて、化合物A対濃度増大ポリマーの種々の比率で、種々の濃度増大ポリマーを用いて化合物Aの固体非晶質分散体を製造した。表6は、各分散体中の化合物Aの濃度および使用した濃度増大ポリマーを記載している。
【0178】
【表6】
【0179】
ミニ噴霧乾燥機を用いて分散体を製造するために、化合物Aをポリマーと共に溶媒中で混合して噴霧溶液を形成した。各溶液を、Cole Parmer 74900シリーズの速度制御シリンジポンプ経由で「ミニ」噴霧乾燥装置中にポンピングした。窒素の加熱された流れ(100℃)を用いてSpraying Systems Co.二流体ノズル(model No. SU1A)を通して化合物A/ポリマー溶液を霧化した。噴霧溶液を、直径11cmのステンレス鋼室中に噴霧した。生成した固体非晶質分散体を、濾紙上に集めて、真空下で乾燥させてデシケータ中に保存した。各分散体を噴霧乾燥するのに使用した条件を表7に記載した。
【0180】
【表7】
【0181】
実施例12
溶解試験を実施して、実施例3〜11の固体非晶質分散体が化合物Aの濃度増大をもたらすことを示した。in vitro溶解試験は、実施例1のようにして実施した。これらの試験では、化合物Aの全てが溶解した場合、化合物Aの濃度が3000μg/mLとなるように十分な量の物質を加えた。結果を表8に示した。
【0182】
【表8】
【0183】
【表9】
【0184】
これらの試料で得られた化合物Aの濃度を用いて薬物の最大濃度(「MDC90」)および最初の90分間の濃度対時間曲線より下の面積(「AUC90」)を測定した。結果を表9に示した。比較のため、実施例1(非晶質化合物A)および対照1(結晶質形態Iの化合物A)の結果を示した。
【0185】
【表10】
【0186】
固体非晶質分散体では、化合物Aの結晶質形態I(対照1)単独のものよりも、そして非晶質化合物A(実施例1)単独よりも濃度増大が得られた。本発明の分散体のAUC90値は、結晶質対照の10.4〜14.6倍であり、そして非晶質化合物A(実施例1)単独の1.3〜1.8倍である。本発明の分散体のAUC1200値は、結晶質対照の9.7〜14.4倍であり、そして非晶質化合物A(実施例1)単独の5.5〜8.2倍である。これは、濃度増大ポリマーを添加すると非晶質化合物A単独と比較して高い溶解薬物濃度が長時間持続することを示している。
【0187】
実施例13
示差走査熱量測定法(DSC)を用いて実施例7の固体非晶質分散体を分析し、分散体中の化合物Aの非晶質の特徴を測定した。試料パンを周囲条件でクリンプし、ロボットアームを有するPerkin-Elmer Pyris 1 DSC炉に装填した。試料を約200℃まで、2℃/分で加熱した。試料のガラス転移温度は、DSCスキャンから測定した。比較のため同じ手法を用いて結晶質化合物A(対照1)および非晶質化合物A単独(実施例1)を分析した。結果を表10に示した。
【0188】
【表11】
【0189】
表10は、非晶質化合物A単独のガラス転移温度(Tg)(99.1℃)および結晶質薬物の鋭い融解ピーク(Tm)(178.1℃)を示す。固体非晶質分散体(実施例7)は、非晶質薬物と同様のTg(97.5℃)を示し、そして融解ピークは示さず、化合物Aの結晶質形態I単独のものとは異なる物理状態を示している。
【0190】
実施例14
実施例3は、Bruker AXS D8 Advance回折計を有する粉末X線回折を用いて試験し、分散体中の化合物Aの非晶質特徴を測定した。バックグラウンドシグナルが生じないようにカップの底面にSi(511)プレートを備えたLucite試料カップに試料(約100mg)を充填した。試料を、30rpmの速度で、ψ平面でスピンさせて結晶の配向効果を最小限にした。X線源(KCuα、λ=1.54Å)を電圧45kVおよび電流40mAで操作した。各試料についてのデータを、走査速度1.8秒/ステップおよび0.04°/ステップのステップサイズで、連続検出器走査モードで27分間にわたって集めた。ディフラクトグラムは、4°〜30°の2θ範囲にわたって集めた。比較のため同じ手法を用いて化合物Aの結晶質形態I(対照1)を分析した。結果を図1に示した。パターンを同じ図中で別々に見ることができるように、図1に示した各パターンのベースラインは、互いにシフトさせた。実施例3は、非晶質ハロのみを示す回折パターンを示したが、対照1は、結晶質薬物の特徴である鋭いピークを示すパターンを示した。これらのデータは、実施例3の固体非晶質分散体中の化合物Aが非晶質であり、結晶質でないことを示す。
【0191】
実施例15
これらの試験は、分散体のエージングを促進するため制御された温度および湿度条件下で貯蔵した後の固体非晶質分散体の物理的安定性を示す。実施例3および対照1のPXRDディフラクトグラムを、以下の温度および相対湿度(RH)条件:40℃/0%RH、40℃/25%RH、40℃/50%RHまたは40℃/75%RH下に6週間貯蔵する前および貯蔵した後に測定した。結果を図2に示した。
図2の結果では、6週間の加速エージング条件下で貯蔵した後、実施例3は非晶質ハロのみの回折パターンを示し、結晶質薬物の特徴である鋭いピークは示さなかった。結果は、実施例3の分散体がこれらの条件下で物理的に安定なことを示している。
【0192】
実施例16
さらに、これらの試験は、制御された温度および湿度条件下で貯蔵した後の化合物Aの固体非晶質分散体の物理的安定性を示した。実施例3の固体非晶質分散体の溶解性能は、以下の条件:30℃/60%RH、40℃/25%RHまたは40℃/75%RH下で12週間貯蔵する前および貯蔵した後に測定した。溶解試験は、実施例1に記載された通り実施した。結果を表11に示した。
【0193】
【表12】
【0194】
これらの試料で得られた化合物Aの濃度を用いて薬物の最大濃度(MDC90)および最初の90分間の濃度対時間曲線下の面積(AUC90)を測定した。結果を表12に示した。
【表13】
データから分かるように、分散体のAUC90は、加速エージング条件下で12週間貯蔵した後、比較的一定のままであった。これは、実施例3の分散体が、これらの条件下で物理的に安定であることを示している。
【0195】
実施例17
実施例17の固体非晶質分散体は、「ミニ」噴霧乾燥装置を用いて実施例4〜11に記載した通り形成した。噴霧溶液は、メタノール31g中の化合物A2500mg、HPMCAS(AQUOT−MG)247.5mgおよびBHT2.5mgからなる。噴霧溶液を1.3mL/分の速度で噴霧室にポンピングし、入口温度は80℃であった。
【0196】
実施例18
化合物A分散体を抗酸化剤ブチルヒドロキシトルエン(BHT)で処方するか、または酸素吸収パケットと共に貯蔵して化合物Aの化学安定性を改善した。
商品名Fresh PaxTM(Multisorb Technologyから入手可能)を有する酸素吸収剤と共に密閉した容器中の実施例3の分散体、実施例17の分散体および実施例3の分散体を、40℃/75%RHで保存した。HPLCを用いて10日、3週間または6週間後に化合物Aの分解生成物について試料を分析して試料中に存在する分解物の量を測定した。結果を表13に示した。
【0197】
【表14】
分散体は、BHTまたはO2吸収剤と共に貯蔵した試料では、化合物Aの分解が減少したことを示した。
【0198】
実施例19〜34
化合物A、濃度増大ポリマーHPMCASおよび安定化剤を含んでなる固体非晶質分散体を処方して化学安定性を改善した。実施例19〜33の固体非晶質分散体は、異なるタイプおよび量の塩基、異なる量の抗酸化剤BHT、または塩基および抗酸化剤の両方を用いて製造した。実施例19〜28および30〜33の固体非晶質分散体についてはポリマーの一部を中和するために塩基を用いた。実施例34は、比較のため安定化剤なしで製造した。
【0199】
実施例19〜22、24〜29、32および33の分散体は、「ミニ」噴霧乾燥装置を用いて実施例4〜11に記載された通り形成した。それぞれの製剤の組成物を表14に示した。噴霧溶液は、それぞれ20/80水/メタノール(質量/質量)の溶液中で10質量%の固体を含んだ。噴霧溶液を1.3mL/分の速度で、噴霧室にポンピングし、そして分散体の全てについて、入口温度は74℃であった。
【0200】
実施例23、30、31および34の分散体は、Niro PSD-1を用いて実施例3に記載した通り形成した。各製剤の組成は、表14に示した。噴霧溶液は、それぞれ20/80水/メタノール(質量/質量)の溶液中で10%の固体を含んだ。
【表15】
実施例19〜34の分散体を、ゆるやかにホイルで覆ったHPLCバイアル中、40℃および75%相対湿度(RH)でエージングした。
【0201】
6週後にHPLCを用いて分解生成物について試料を分析した。結果を表15に示した。
【表16】
結果から、塩基、抗酸化剤または両方を添加すると、HPMCASを含む固体非晶質分散体中の化合物Aの化学安定性が改善されることがわかった。
【0202】
実施例35
化合物AおよびHPMCASの物理混合物の化学安定性を、評価した。非晶質化合物Aは。実施例4〜11に記載された通り、ミニ噴霧乾燥機を用いて製造した。メタノール/水(8/2,体積/体積)中の固体1.5質量%を含む噴霧溶液を、入口温度70℃および出口温度、周囲条件で1.3mL/分の速度で噴霧した。
実施例3に記載された通りPSD−1噴霧乾燥機を用いてHPMCASを製造した。アセトン中に固体12質量%を含む噴霧溶液を、入口温度100℃および出口温度37℃で183〜215g/分の速度で噴霧した。
【0203】
化合物AおよびHPMCASを9:1の比率で一緒に混合し、化学安定性試験のため種々の温度、および相対湿度条件下で貯蔵した。結果は、下の表16の通りである。
【表17】
【0204】
実施例36
イヌを用いてin vivo溶解試験を実施し、本発明の非晶質分散体では化合物Aの濃度増大が得られることを示した。実施例3の固体非晶質分散体を6匹の絶食したビーグル犬のグループに投薬し、定期的に血液を採取し、血漿薬物濃度を測定することによって薬物放出はモニターした。用量を投与した時間から最後の試料までの濃度対時間曲線下の面積(
「AUC0-Tlast「ng*hr/mL)を表17に示した。化合物Aの結晶質形態I(対照1)および非晶質化合物A(実施例1)を、同様のプロトコールを用いて試験した。
【0205】
Methocel(R)(HPMC,USPグレード,4000cps,Dow Chemical Co.)0.5質量%を含む溶液の懸濁液として用量をイヌに投与した。水性薬物懸濁液の経口投与は、ポリエチレン管挿入部分を備えた経口強制飼養を用いて促進した。ポリエチレン管挿入部分を使用することで、管をすすぐためのさらなる水量を必要とすることなく、変位によって所望の用量体積を正確に供給した。用量は、25mgA/kg(「mgA」は、活性薬物のmgに相当する)であった。
【表18】
【0206】
上記の結果は、明らかに非晶質化合物A単独(実施例1)および化合物Aおよび濃度増大ポリマー(実施例3)の固体非晶質分散体の両方では、化合物A単独の結晶質形態Iよりも高い in vivo 薬物濃度が得られることを示している。実施例1の非晶質化合物Aについての相対的生物学的利用能(試験組成物のAUCを化合物Aの結晶質形態I単独のAUCによって割ったもの)は、化合物Aの結晶質形態I単独の11.7倍であったが、実施例3の固体非晶質分散体の相対的生物学的利用能は、化合物Aの結晶質形態I単独の44.7倍であった。
【0207】
実施例37
この実施例は、非常に少量の濃度増大ポリマーを非晶質化合物Aと合わせると、in vitro使用環境において溶解した化合物Aの濃度が持続することを示している。実施例37A、37Bおよび37Cは、実施例2の通り製造したが、しかし以下を除く:実施例37Aは、化合物A5.4mgおよびHPMCAS0.6mgであり;実施例37Bは、化合物A5.4mgおよびHPMCAS0.167mgであり;そして実施例37Cは、化合物A5.4mgおよびHPMCAS0.055mgであった。実施例37dは、非晶質化合物A単独からなる。溶解試験は、実施例2の通り実施し、結果は表18にまとめた。
【0208】
【表19】
【0209】
MDC360は、最初の360分の範囲内の最大溶解薬物濃度であり、そしてAUC360は、360分での溶解薬物濃度対時間曲線下の面積である。結果は、非常に少量のポリマーを非晶質化合物Aと合わせると非晶質化合物単独と比較して水性使用環境において溶解薬物濃度が持続することを示している。
【0210】
後述する実施例において、特に明記しない限り、以下の説明における全ての温度は、摂氏(℃)であり、そして特記しない限り、すべての部およびパーセンテージは質量による。
種々の出発物質および他の試薬は、商業上の供給業者、例えばAldrich Chemical CompanyまたはLancaster Synthesis Ltd.から購入し、そして特に明記しない限り、さらに精製することなく使用した。
【0211】
下に記載した反応は、窒素、アルゴンの正の圧力下でまたは乾燥管を用いて、周囲温度(特に明記しない限り)で、無水溶媒中で実施した。分析薄層クロマトグラフィは、ガラスに裏打ちされたシリカゲル60°F254プレート(Analtech(0.25mm))上で実施し、適当な溶媒比率(v/v)で溶離した。反応は、高圧液体クロマトグラフィ(HPLC)または薄層クロマトグラフィ(TLC)によって評価し、出発物質の消費量によって判断して終了した。TLCプレートをUV、リンモリブデン酸染色またはヨード染色によって視覚化した。1H−NMRスペクトルは、Bruker機器を用いて300MHzで操作して記録し、13CNMRスペクトルは、75MHzで記録した。NMRスペクトルは、参照標準としてクロロホルム(7.25ppmおよび77.00ppm)またはDMSO−d6((2.50ppmおよび39.52ppm))を使用してDMSO−d6またはCDCl3溶液(ppmで報告)として得た。必要に応じて、他のNMR溶媒を使用した。ピークの多重度を報告する場合、以下の略語を使用した:s=一重線、d=二重線、t=三重線、m=多重線、br=ブロード、dd=二重線の=二重線、dt=三重線の二重線。結合定数を記載する場合は、ヘルツで報告した。赤外スペクトルは、ニートオイルとして、KBrペレットとして、またはCDCl3溶液としてPerkin-Elmer FT−IR分光計上で記録し、波数(cm-1)で報告した。質量スペクトルは、LC/MSまたはAPCIを用いて得た。全ての融点は補正してない。全ての最終生成物は、(220nmおよび254nmの波長のHPLCによって)95%を超える純度を有した。
【0212】
化合物Aの製造
以下の実施例および製造において、「Et」はエチルを意味し、「Ac」はアセチルを意味し、「Me」はメチルを意味し、「Ph」はフェニルを意味し、(PhO)2POClはクロロジフェニルホスフェートを意味し、「HCl」は塩酸を意味し、「EtOAc」は酢酸エチルを意味し、「Na2CO3」は炭酸ナトリウムを意味し、「NaOH」は水酸化ナトリウムを意味し、「NaCl」は塩化ナトリウムを意味し、「NEt3」はトリエチルアミンを意味し、「THF」はテトラヒドロフランを意味し、「DIC」はジイソプロピルカルボジイミドを意味し、「HOBt」はヒドロキシベンゾトリアゾールを意味し、「H2O」は水を意味し、「NaHCO3」は炭酸水素ナトリウムを意味し、「K2CO3」は炭酸カリウムを意味し、「MeOH」はメタノールを意味し、「i−PrOAc」は酢酸イソプロピルを意味し、「MgSO4」は硫酸マグネシウムを意味し、「DMSO」はジメチルスルホキシドを意味し、「AcCl」はアセチルクロリドを意味し、「CH2Cl2」は塩化メチレンを意味し、「MTBE」はメチルt−ブチルエーテルを意味し、「DMF」はジメチルホルムアミドを意味し、「SOCl2」は塩化チオニルを意味し、「H3PO4」はリン酸を意味し、「CH3SO3H」はメタンスルホン酸を意味し、「Ac2O」は無水酢酸を意味し、「CH3CN」はアセトニトリルを意味し、そして「KOH」は水酸化カリウムを意味する。
【0213】
実施例38:(4R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−カルボン酸tert−ブチルエステルの製造
【化3】
【0214】
(4R)-5,5−ジメチル−チアゾリジン−3,4−ジカルボン酸3−tert−ブチルエステル(これは、Ikunaka, M. et al., Tetrahedron Asymm. 2002, 13, 1201; Mimoto, T. et al., J. Med. Chem. 1999, 42, 1789; and Mimoto, T. et al., European Patent Application 0574135A1 (1993)の方法に従って製造することができる。250g;0.957mol)をアルゴンパージした5Lフラスコに加え、そしてEtOAc(1.25L)中に溶解した。次いで、溶液を2℃に冷却し、(PhO)2POCl(208mL;1.00mol)をひとかたまりで加えた。滴下ロートを通してNEt3(280mL;2.01mol)を滴加し、次いで生成した懸濁液を0℃で撹拌した。7分後、アリルアミン(75.4mL;1.00mol)を滴加した。氷浴をはずし、懸濁液を室温に加温させた。半時間後、1N HCl(750mL;0.750mol)を加えた。すすぎ用にEtOAc(50mL)を用いて混合物を4L分液ロートへ移した。層を分離した。有機画分を7.2%水性Na2CO3(2×1.25L)で洗浄し、次いで3L蒸留フラスコへ移し、そしてEtOAc(400mL)で希釈した。溶液を共沸的に乾燥し、1気圧でEtOAcを蒸留することによって濃縮して体積800mLにした。25℃に冷却した後、生成した(4R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−カルボン酸tert−ブチルエステルの透明な黄色がかったEtOAc溶液を、次の工程に直接かけた。アリコートを取り出し、濃縮して白色結晶質固形物として(4R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−カルボン酸tert−ブチルエステルを得た:融点=94〜98℃,1HNMR(300MHz,CDCl3)δ6.12 (br s, 1H), 5.88 (app ddt, J = 10.2, 17.1, 5.6 Hz, 1H), 5.28 (app dq, J = 17.1, 1.5 Hz, 1H), 5.18 (app dd, J = 1.2, 10.2 Hz, 1H), 4.68 (s, 2H), 4.14 (br s, 1H), 3.95 (br t, J = 5.4 Hz, 2H), 1.62 (s, 3H), 1.49 (s, 9H), 1.46 (s, 3H); 13CNMR(75MHz,CDCl3)δ170.0, 154.0, 134.4, 116.9, 82.0, 73.3, 54.0, 48.7, 42.0, 30.6, 28.6, 24.6; MS(CI) m/z 301.1599(C14H25N2O3Sについての計算値301.1586,M+H+);C14H24N2O3Sについての元素分析 計算値:C,55.97;H,8.05;N,9.32;実測値:C,56.11;H,8.01;N,9.11.
【0215】
実施例39:(4R)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミドの製造
【化4】
【0216】
メタンスルホン酸(155mL;2.39mol)を3Lフラスコ中の(4R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−カルボン酸tert−ブチルエステルのEtOAc溶液に滴加した。室温で一夜撹拌した後、溶液を7℃に冷却し、H2O(400mL)を中に注いだ。混合物を4L分液ロート[すすぎ用にH2O(30mL)を用いた]へ移し、層を分離した。有機画分をH2O(190mL)で抽出した。合わせたH2O抽出物を5Lフラスコへ移して8℃に冷却した。3N NaOH(〜1.05L)を用いてpHを0.4から9.3に調整した。2−メチルテトラヒドロフラン(1.55L)を注ぎ、続いてNaCl(150g)を添加した。氷浴をはずし、混合物を室温に加温させた。3N NaOH(〜1mL)を用いてpHを9.0に再調整した。すすぎ用に2−メチルテトラヒドロフラン(50mL)を用いて混合物を4L分液ロートへ移し、層を分離した。水性相を2−メチルテトラヒドロフラン(950mL)で抽出した。すすぎ用に2−メチルテトラヒドロフラン(200mL)を用いて、セライトを通して有機抽出物を5L蒸留フラスコ中に直接、真空濾過した。1気圧で2−メチルテトラヒドロフランを蒸留させることによって溶液を共沸的に乾燥させて濃縮し、体積1.2Lにした。計量したアリコートを濃縮し、秤量したところ(4R)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド161gが溶液中に存在することがわかった[(4R)−5,5−ジメチル−チアゾリジン−3,4−ジカルボン酸3−tert−ブチルエステルから84%]。次いで、この溶液を次の工程に直接かけた。上記を濃縮したアリコートから結晶質固形物として(4R)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミドを得た:融点=45〜47℃,1HNMR(300MHz,CDCl3)δ6.73 (br s, 1H), 5.87 (app ddt, J = 10.2, 17.1, 5.7 Hz, 1H), 5.17- 5.27 (m, 2H), 4.27 (AB q, JAB = 9.7 Hz, Δv=22.5 Hz, 2H), 2.94 (app tt, J = 1.5, 5.8 Hz, 2H), 3.51 (s, 1H), 1.74 (s, 3H), 1.38 (s, 3H); 13CNMR(75MHz,CDCl3)δ169.7, 134.4, 116.9, 74.8, 57.2, 51.6, 41.9, 29.1, 27.3; MS(CI)m/z 201.1063(C9H17N2OSについての計算値201.1062,M+H+);C9H16N2OSについての元素分析 計算値:C,53.97;H,8.05;N,13.99;実測値:C,53.93;H,8.09;N,14.07.
【0217】
実施例40:(2S,3S)−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−2ヒドロキシ−4−フェニル−酪酸の製造
【化5】
【0218】
(2S,3S)−3−アミノ−2−ヒドロキシ−4−フェニル−酪酸(これは、Pedrosa et al., Tetrahedron Asymm. 2001, 12, 347; M. Shibasaki et al., Tetrahedron Lett. 1994, 35, 6123;および Ikunaka, M. et al. Tetrahedron Asymm. 2002, 13, 1201の方法に従って製造することができる;185g;948mmol)を5Lフラスコに加え、THF(695mL)中で懸濁した。H2O(695mL)、続いてNEt3(277mL;1990mmol)を注いだ。45分間撹拌した後、溶液を6℃に冷却した。次いで、THF(350mL)中の酢酸3−クロロカルボニル−2−メチルフェニルエステル(201g;948mmol)の溶液を滴加した。半時間後、6N HCl(〜170mL)を用いてpHを8.7から2.5に調整した。固体NaCl(46g)を加え、次いで氷浴をはずし、混合物を激しく撹拌しながら室温に加温させた。移すために1:1 THF/H2O(50mL)を用いて混合物を4L分液ロートへ移し、次いで低い方の水性相を除去した。有機画分を5L蒸留フラスコへ移し、次いで新たなTHF(2.5L)で希釈した。1気圧でTHFを蒸留させることによって溶液を共沸的に乾燥させて濃縮し、体積1.3Lにした。共沸的な乾燥を完了するため、新たなTHF(2.0L)を加え、1気圧での蒸留によって溶液を濃縮して1.85Lにし、次いで55℃に保った。滴下ロートを通してn−ヘプタン(230mL)を滴加し、次いで溶液に直ちに結晶種を入れた。結晶化が始まった後、さらなるn−ヘプタン(95mL)を滴加した。生成した結晶スラリーを7分間激しく撹拌した。次いで、さらなるn−ヘプタン(1.52L)をゆるやかな流れとして加えた。次いで、結晶スラリーをゆっくりと室温に冷却させて一夜撹拌した。懸濁液を真空濾過し、次いで、濾過ケークを1:1THF/n−ヘプタン(700mL)で洗浄した。真空オーブン中45〜50℃で乾燥させた後、〜7mol%Et3N・HClが混入した結晶質固体として(2S,3S)−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−2−ヒドロキシ−4−フェニル−酪酸324g(92%)を得た:融点=189〜191℃,1HNMR(300MHz,DMSO−d6)δ12.65 (br s, 1H), 3.80 (d, J = 9.7 Hz, 1H), 7.16- 7.30 (m, 6H), 7.07 (dd, J = 1.1, 8.0 Hz, 1H), 7.00 (dd, J = 1.1, 7.5 Hz), 4.40- 4.52 (m, 1H), 4.09 (d, J = 6.0 Hz, 1H), 2.92 (app dd, J = 2.9, 13.9 Hz, 1H), 2.76 (app dd, J = 11.4, 13.9 Hz, 1H), 2.29 (s, 3H), 1.80 (s, 3H); 13CNMR(75MHz,DMSO−d6)δ174.4, 169.3, 168.1, 149.5, 139.7, 139.4, 129.5, 128.3, 127.9, 126.5, 126.3, 124.8, 123.3, 73.2, 53.5, 35.4, 20.8, 12.6; MS(CI)m/z 372.1464(C20H22NO6についての計算値372.1447,M+H+);C20H21NO6・0.07Et3N・HClについての元素分析 計算値:C,64.34;H,5.86;N,3.95;Cl,0.70;実測値:C,64.27;H,5.79;N,3.96;Cl;0.86.
【0219】
実施例41:酢酸3−{(1S,2S)−3−[(4R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−イル]−1−ベンジル−2−ヒドロキシ−3−オキソ−プロピルカルバモイル}−2−メチルフェニルエステルの製造
【化6】
【0220】
(2S,3S)−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−2−ヒドロキシ−4−フェニル−酪酸(271g;731mmol)を、2−メチルテトラヒドロフラン中の(4R)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド(161g;804mmol)溶液(合計1.20Lの溶液)を含む5Lフラスコに加え、すすぎ用に2−メチルテトラヒドロフラン(500mL)を用いた。すすぎ用に2−メチルテトラヒドロフラン(50mL)を用いてHOBt・H2O(32.6g;241mmol)を加えた。白色懸濁液を室温で10分間を撹拌した。ジイソプロピルカルボジイミド(119mL;760mmol)を3つの部分(40mL+40mL+39mL)にわけて30分間隔で加えた。最後にDICを加えて1時間後、セライト(100g)を加え、懸濁液を室温で3時間撹拌した。混合物を真空濾過し、2−メチルテトラヒドロフラン(400mL)を用いて固形物をすすぎ、生成した濾過ケーキを洗浄した。濾液を4L分液ロートへ移し、すすぎ用に2−メチルテトラヒドロフラン(50mL)を用いた。溶液を1N HCl(1.25L)、次いでNaHCO3(27g)、NaCl(134g)およびH2O(1.25L)の水溶液で洗浄した。生成した有機相を3L蒸留フラスコへ移し、次いで、1気圧で2−メチルテトラヒドロフランを蒸留させることよって溶液を体積1.12Lに減らした。次いで、溶液を2−メチルテトラヒドロフラン(230mL)で希釈し、総体積1.35Lをした。溶液を23℃にさました後、粗酢酸3−{(1S,2S)−3−[(4R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−イル]−1−ベンジル−2−ヒドロキシ−3−オキソ−プロピルカルバモイル}−2−メチルフェニルエステルの溶液を次の工程に直接かけた。
【0221】
実施例42:(4R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチルベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミドの製造
【化7】
【0222】
MeOH(330mL)およびK2CO3(66.9g;484mmol)を、3Lフラスコ中の粗酢酸3−{(1S,2S)−3−[(4R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−イル]−1−ベンジル−2−ヒドロキシ−3−オキソ−プロピルカルバモイル}−2−メチルフェニルエステル(理論量:405g;731mmol)の2−メチルテトラヒドロフラン溶液に室温で順次加えた。2時間半後、さらなるK2CO3(20g;144mmol)を加えた。3時間後、固形物をすすいで濾過ケーキを洗浄するために4:1の2−メチルテトラヒドロフラン/MeOH(330mL)を用いてセライトのパッド上で反応混合物を真空濾過した。すすぎ用に4:1の2−メチルテトラヒドロフラン/MeOH(80mL)を用いて濾液を6L分液ロートへ移した。溶液をi−PrOAc(1.66L)で希釈し、次いで、H2O(1.60L)中のNaCl(83.0g)の溶液で洗浄した。有機画分を0.5NHCl(1.66L)、次いで飽和水性NaCl溶液(400mL)で洗浄した。生成した有機画分を4Lエルレンマイヤーフラスコへ移し、MgSO4(120g)を加えた。10分間撹拌した後、分液ロートおよびエルレンマイヤーフラスコをすすぎ、MgSO4を洗浄するために2:1のi−PrOAc/2−メチルテトラヒドロフラン(600mL)を用いて混合物を5L蒸留フラスコ中に直接、真空濾過した。最小ポット体積を〜2.50Lに維持しながらi−PrOAcを5つの部分(合計3.60L用いた)に分けて添加し、同時に1気圧で蒸留することによって2−メチルテトラヒドロフランを置き換えた。生成した結晶化混合物を75℃にさまし、この温度で30分間保持した。懸濁液を室温に一夜ゆっくりとさました。結晶を移して洗浄するためにi−PrOAc(600mL)を用いて懸濁液を真空濾過した。真空オーブン中40℃で乾燥後、結晶質(4R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチル−ベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド204g((2S,3S)−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−2−ヒドロキシ−4−フェニル−酪酸から54%)を得た。この物質を後述するように再結晶した。
【0223】
実施例43:(4R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチルベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミドの再結晶
【化8】
【0224】
(4R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチル−ベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド(193g,378mmol)を5Lフラスコに加え、次いでEtOAc(1.28L)中で懸濁した。懸濁液を76℃に加熱した後、MeOH(68mL)を加え、次いで内部温度を70℃に下げた。内部の温度を70℃に維持しながら、n−ヘプタン(810mL)を溶液に滴加した。n−ヘプタンの添加を完了した後、生成した結晶の懸濁液は、70℃で30分間保ち、次いで一夜室温にゆっくりとさました。懸濁液を真空濾過し、1.6:1のEtOAc/n−ヘプタン(500mL)を用いて結晶を移して洗浄した。次いで真空オーブン中45℃で結晶を乾燥させて、白色結晶質固形物として精製された(4R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチルベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド162g(回収率84%)を得た:融点=173〜175℃、1HNMR(300MHz,DMSO−d6)は、〜10:1の回転異性体の混合物を示した、主要な回転異性体の共鳴δ9.35 (s, 1H), 8.04- 8.15 (m, 2H), 7.13- 7.38 (m, 5H), 6.96 (t, J = 7.7 Hz, 1H), 6.79 (d, J = 7.2 Hz, 1H), 6.55 (d, J = 7.5 Hz, 1H), 5.71-5.87 (m, 1H), 5.45 (br d, J = 6.2 Hz, 1H), 4.98- 5.27 (m, 4H), 4.38- 4.52 (m, 3H), 3.58- 3.86 (m, 2H), 2.68- 2.90 (m, 2H), 1.84 (s, 3H), 1.52 (s, 3H), 1.37 (s, 3H) [特徴的な微量回転異性体の共鳴δ9.36 (s), 8.21 (d, J = 10.5 Hz), 7.82 (5, J = 5.8 Hz), 4.89 (s), 4.78 (AB q, JAB = 9.8 Hz, Δv = 27.1 Hz), 4.17- 4.24 (m), 2.93-3.01 (m), 1.87 (s), 1.41 (s)]; 13CNMR(75MHz,DMSO−d6)は、〜10:1の回転異性体の混合物を示した。主要な回転異性体の共鳴δ170.4, 169.5, 168.2, 155.7, 139.6, 139.4, 135.5, 135.4, 129.9, 128.2, 126.2, 126.1, 121.9, 117.8, 115.6, 72.4, 72.1, 53.1, 51.4, 48.2, 41.3, 34.2, 30.5, 25.0, 12.6[特徴的な微量回転異性体の共鳴δ171.4, 169.7, 168.6, 139.0, 129.5, 128.4, 70.6, 54.2, 49.1, 41.5, 31.4, 24.8]; MS(CI)m/z 512.2224(C27H34N3O5Sについての計算値512.2219,M+H+),C27H33N3O5Sについての元素分析 計算値:C,63.38;H,6.50;N,8.22;実測値:C,63.19;H,6.52;N,8.10.
【0225】
実施例44:(R)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド塩酸塩の製造;
【化9】
【0226】
(R)−5,5−ジメチル−チアゾリジン−3,4−ジカルボン酸3−tert−ブチルエステル(105kg,402mol)および酢酸エチル(690L)の溶液をジフェニルクロロホスフェート(113kg,422mol)で処理してから0℃に冷却した。温度を5℃に維持しながらNEt3(85.5kg,844mol)を加え、次いで混合物をこの温度で2時間保持した。混合物を0℃に冷却し、次いで温度を5℃に維持しながらアリルアミン(24.1kg,422mol)を加えた。混合物を20℃に加温し、次いで水性HCl10質量%(310L)でクエンチした。層を分離した後、有機画分を水性Na2CO38.6質量%(710L)で洗浄した。層を分離した後、水性画分を酢酸エチル(315L)で抽出した。AG-074278を含む合わせた酢酸エチル抽出物を、最小ポット体積を約315Lに維持しながら1気圧で共沸蒸留することによって乾燥させた。生成した(R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−カルボン酸tert−ブチルエステルの懸濁液を5℃に冷却した。酢酸エチル(263L)中の無水HCl(36.8kg,1008mol)の溶液13質量%を5℃に冷却し、次いで温度を15℃に維持しながら(R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−カルボン酸tert−ブチルエステル懸濁液に加えた。生成した懸濁液を20℃で19時間保持し、次いで冷却し、5℃で2時間保持した。次いで、すすぎ用に冷酢酸エチルを用いて懸濁液を濾過した。湿ったケークを真空下45℃で乾燥させて白色固形物として(R)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド塩酸塩90.5kg(95.2%)を得た:1HNMR(300MHz,DMSO−d6)δ8.94 (app t, J = 5.5 Hz, 1H), 5.82 (ddt, J = 10.4, 17.2, 5.2 Hz, 1H), 5.19- 5.25 (m, 1H), 5.10- 5.14 (m, 1H), 4.38 (AB q, JAB = 9.8 Hz, Δv = 14.5 Hz, 2H), 4.08 (s, 1H), 3.72- 3.91 (m, 2H), 1.58 (s, 3H), 1.32 (s, 3H); 13CNMR(75MHz,DMSO−d6)δ161.7, 132.2, 114.0, 67.9, 51.4, 43.5, 39.3, 25.3, 24.3; MS(CI)m/z 201.1070(C9H17N2OSについての計算値201.1062,M+H+);C9H17ClN2OSについての元素分析 計算値:C,45.65;H,7.24;N,11.83;Cl,14.97;実測値:C,45.41;H,7.33;N,11.69;Cl,15.22.
【0227】
実施例45:(2S,3S)−2−アセトキシ−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−4−フェニル−酪酸の製造
【化10】
【0228】
(2S,3S)−3−アミノ−2−ヒドロキシ−4−フェニル−酪酸(110kg,563mol)、NaCl(195kg)およびTHF(413L)の混合物に、周囲温度でNEt3(120kg,1183mol)およびH2O(414L)を投入した。生成した混合物を0℃に冷却した。酢酸3−クロロカルボニル−2−メチルフェニルエステル(120kg,563mol)を別の反応器に加えてからTHF(185L)中に溶解した。生成した酢酸3−クロロカルボニル−2−メチルフェニルエステルの溶液を10℃に冷却し、次いで添加中の温度を<10℃に維持しながら(2S,3S)−3−アミノ−2−ヒドロキシ−4−フェニル−酪酸混合物に加えた。生成した二相混合物を5℃で1時間撹拌し、次いで濃HCl(62kg)を用いてpH2.5〜3.0に調整した。次いで、混合物を25℃に加温させ、層を分離した。(2S,3S)−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−2−ヒドロキシ−4−フェニル−酪酸を含む生成したTHF画分を1気圧で蒸留することによって部分的に濃縮した。次いで最小ポット体積を1500Lに維持しながら、1気圧で蒸留することによってTHFを酢酸エチルで置き換えた。生成した溶液を25℃に冷却し、次いで無水酢酸(74.8kg,733mol)およびメタンスルホン酸(10.8kg,112mol)を投入した。混合物を70℃で約3時間加熱した。混合物を25℃にさまし、次いで温度を20℃に維持しながらH2O(1320L)で急冷した。水性層を除去した後、有機画分に酢酸エチル(658L)およびH2O(563L)を投入した。撹拌した後、水性相を除去した。有機画分を13質量%水性NaCl(2×650L)で二回洗浄した。有機画分を部分的に濃縮し、減圧蒸留(70〜140mmHg)によって乾燥させて体積約1500Lにした。生成した溶液を40℃に加熱し、次いで温度を40℃に維持しながらn−ヘプタン(1042L)を投入した。(2S,3S)−2−アセトキシ−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−4−フェニル−酪酸(0.1kg)を用いて溶液に結晶種を入れ、次いでさらなるn−ヘプタン(437L)をゆっくりと加えた。結晶化混合物を40℃で1時間維持した。温度を40℃に維持しながら、さらなるn−ヘプタン(175L)を加えた。結晶質懸濁液をさまし、25℃で1時間、次いで0℃で2時間保持した。すすぎ用にn−ヘプタンを用いて懸濁液を濾過した。湿ったケークを真空下55℃で乾燥させて白色固体として(2S,3S)−2−アセトキシ−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−4−フェニル−酪酸174kg(74.5%)を得た:融点=152〜154℃;1HNMR(300MHz,CDCl3)δ7.21- 7.35 (m, 5H), 7.13 (app t, J = 7.9 Hz, 1H), 7.01 (app d, J = 8.1 Hz, 1H), 6.94 (app d, J = 7.2 Hz, 1H), 5.99 (d, J = 9.0 Hz, 1H), 5.33 (d, J = 4.1 Hz, 1H), 4.96- 5.07 (m, 1H), 3.07 (dd, J = 5.5, 14.6 Hz, 1H), 2.90 (dd, J = 10.0, 14.5 Hz, 1H), 2.30 (s, 3H), 2.18 (s, 3H), 1.96 (s, 3H); 13CNMR(125MHz,CDCl3)δ170.4, 170.2, 169.6, 169.5, 149.5, 137.81, 136.5, 129.2, 128.6, 128.4, 127.0, 126.6, 124.5, 123.7, 73.1, 50.9, 35.9, 20.6, 20.5, 12.4; C22H23NO7についての元素分析 計算値:C,63.92;H,5.61;N,3.39;実測値:C,64.22;H,5.68;N,3.33;MS(CI)m/z 414.1572(C22H24NO7についての計算値414.1553,M+H+).
【0229】
実施例46:(4R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチル−ベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミドの製造
【化11】
【0230】
(2S,3S)−2−アセトキシ−3−(3−アセトキシ−2−メチルベンゾイルアミノ)
−4−フェニル−酪酸(140kg,339mol)、CH3CN(560L)およびピリジン(64.3kg,813mol)の溶液を15℃に冷却した。温度を15℃に維持しながら、SOCl2(44.3kg,373mol)を投入した。混合物を15℃で1時間保持した。別の
反応器に(R)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド塩酸塩(96.6kg,408mol)、CH3CN(254L)およびピリジン(29.5kg,373mol)を投入し、次いで15℃に冷却した。温度を15℃に維持しながら、(2S,3S)−2−アセトキシ−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−4−フェニル酪酸塩化物溶液を(R)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド溶液を加えた。混合物を15℃で6時間保持した。0℃の冷却ジャケットを用いて別の反応器にKOH(167kg,2709mol)およびメタノール(280L)を投入した。生成したKOH/メタノール溶液を5℃に冷却した。温度を10℃に維持しながら、粗酢酸3−{(1S,2S)−2−アセトキシ−3−[(R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−イル]−1−ベンジル−3−オキソ−プロピルカルバモイル}−2−メチルフェニルエステル混合物を、KOH/メタノール溶液に加えた。添加を完了した後、混合物を25℃で3時間保持した。混合物にH2O(840L)および酢酸エチル(840L)を投入し、次いで温度を20℃に維持しながら、濃HCl(85kg)を用いてpH5〜6.5に酸性化した。生成した層を分離した。水性NaHCO36.8質量%(770L)、水性HCl/NaCl溶液(H2O:875L;濃HCl:207kg;NaCl:56kg)、水性NaHCO38.5質量%(322L)、次いで水性NaCl3.8質量%(728L)を用いて有機画分を順次洗浄した。生成した有機画分を、1気圧で蒸留することによって部分的に濃縮した。蒸留を続け、ポット温度を〜70℃に維持することによって溶媒を酢酸エチルと交換した。ポット体積が約840Lのままであるようにして、酢酸エチルを加えた。次いで、溶液を20℃に冷まし、結晶化が観察されるまでこの温度で保持した。n−ヘプタン(280L)を加え、懸濁液を15℃で4時間撹拌した。すすぎ用に冷2.4:1(v/v)酢酸エチル/n−ヘプタンを用いて結晶をすすいだ。湿ったケークを真空下、45℃で乾燥させて粗(R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチルベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミドを得た。脱色および再結晶は、以下の通りに実施した:粗(R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチルベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド、ADP炭素(21kg)、Supercel(3kg)および酢酸エチル(780L)の混合物を70℃に加熱した。CH3OH(40L)を混合物に加えた。混合物を濾過し、生成した透明な濾液を1気圧で加熱して還流させ、蒸留を開始した。CH3OHを以下のようにして置き換えた:ポット体積を約840Lおよび70℃で維持しながら酢酸エチル(388L)を投入した。温度を70℃に維持しながら溶液にn−ヘプタン(316L)をゆっくりと投入した。次いで、混合物を20℃に冷却し、この温度で4時間保持した。結晶は濾過し、すすぎ用に冷2.1:1(v/v)酢酸エチル/n−ヘプタンを用いて濾過した。湿ったケークを真空下、45℃で乾燥させて白色結晶質固形物として(4R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチルベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド103kg(59.6%)を得た:融点=173〜175℃,1HNMR(300MHz,DMSO−d6)は、〜10:1回転異性体の混合物を示した。主要な回転異性体の共鳴δ9.35 (s, 1H), 8.04- 8.15 (m, 2H), 7.13- 7.38 (m, 5H), 6.96 (t, J = 7.7 Hz, 1H), 6.79 (d, J = 7.2 Hz, 1H), 6.55 (d, J = 7.5 Hz, 1H), 5.71- 5.87 (m, 1H), 5.45 (br d, J = 6.2 Hz, 1H), 4.98- 5.27 (m, 4H), 4.38- 4.52 (m, 3H), 3.58- 3.86 (m, 2H), 2.68- 2.90 (m, 2H), 1.84 (s, 3H), 1.52 (s, 3H), 1.37 (s, 3H) [特徴的な微量回転異性体の共鳴δ9.36 (s), 8.21 (d, J = 10.5 Hz), 7.82 (5, J = 5.8 Hz), 4.89 (s), 4.78 (AB q, JAB = 9.8 Hz, Δv = 27.1 Hz), 4.17- 4.24 (m), 2.93- 3.01 (m), 1.87 (s), 1.41 (s)];13CNMR(75MHz,DMSO−d6)は、〜10:1回転異性体の混合物を示した。主要な回転異性体の共鳴δ170.4, 169.5, 168.2, 155.7, 139.6, 139.4, 135.5, 135.4, 129.9, 128.2, 126.2, 126.1, 121.9, 117.8, 115.6, 72.4, 72.1, 53.1, 51.4, 48.2, 41.3, 34.2, 30.5, 25.0, 12.6[特徴的な微量回転異性体の共鳴δ171.4, 169.7, 168.6, 139.0, 129.5, 128.4, 70.6, 54.2, 49.1, 41.5, 31.4, 24.8];MS(CI)m/z 512.2224(C27H34N3O5Sについての計算値512.2219,M+H+)、C27H33N3O5Sについての元素分析 計算値:C,63.38;H,6.50;N,8.22;実測値:C,63.19;H,6.52;N,8.10.
【0231】
実施例47:(2S,3S)−3−アミノ−2−ヒドロキシ−4−フェニル−酪酸;塩酸塩の製造
【化12】
【0232】
(2S,3S)−3−tert−ブトキシカルボニルアミノ−2−ヒドロキシ−4−フェニル−酪酸(163g,551mmol)およびCH2Cl2(2.0L)の懸濁液中にHClガス(51g,1.4mol)を0℃でバブリングした。生成したオフホワイト懸濁液を周囲温度に加温させて一夜撹拌した。濃縮したアリコートの1HNMR分析は、生成物への転化率約95%を示した。懸濁液を0℃に冷却し、懸濁液にさらなるHClガス(46g,1.3mol)をバブリングした。周囲温度に加温させた後、懸濁液を一夜撹拌した。懸濁液を真空濾過し、固形物をCH2Cl2(200mL)ですすぎ、次いで、固形物を真空オーブン中、45℃で24時間乾燥させて白色固形物として(2S,3S)−3−アミノ−2−ヒドロキシ−4−フェニル−酪酸;塩酸塩129g(100%)を得た:1HNMR(300MHz,DMSO−d6)δ13.05 (br s, 1H), 8.25 (br s, 3H), 7.22-7.34 (m, 5H), 4.41 (d, J = 2.6 Hz, 1H), 3.66 (br s, 1H), 2.84 (ABXのAB部分, JAX = 11.0 Hz, JBX = 2.8 Hz, Δv = 19.6 Hz, 2H); 13CNMR(75MHz,DMSO−d6)d 172.4, 136.6, 129.8, 128.7, 127.1, 69.6, 55.0, 33.6;MS(CI)m/z196.0979(C10H14NO3についての計算値196.0974,M−Cl-)
【0233】
実施例48:(2S,3S)−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−2−
ヒドロキシ−4−フェニル−酪酸の製造
【化13】
【0234】
NEt3(186mL,1.34mol)を、(2S,3S)−3−アミノ−2−ヒドロキシ−4−フェニル−酪酸;塩酸塩(100g,432mmol)、H2O(320mL)およびテトラヒドロフラン(320mL)の懸濁液に加えた。懸濁液を4℃に冷却し、酢酸3−クロロカルボニル−2−メチルフェニルエステル(93.6g,440mmol)およびTHF(160mL)の溶液を滴加した。生成した溶液を周囲温度に加温させて1時間撹拌した。溶液を10℃に冷却し、6N HCl(87mL)を用いてpHを2.0に調整した。NaCl(25g)およびテトラヒドロフラン(200mL)を加え、混合物を周囲温度に加温させた。相を分離し、テトラヒドロフラン画分をMgSO4で乾燥させて濾過した。ロータリーエバポレーターを用いて濾液を濃縮して体積330mLにしてからテトラヒドロフラン(230mL)で希釈した。n−ヘプタン(1.2L)をゆっくりと加え、生成した固形物の白色懸濁液を周囲温度で一夜撹拌した。懸濁液を真空濾過し、固形物をn−ヘプタン(2×500mL)ですすぎ、固形物を真空オーブン中45℃で24時間乾燥させ、〜7.7mol%Et3N・HClが混入した白色固形物として(2S,3S)−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−2−ヒドロキシ−4−フェニル−酪酸150g(93.6%)を得た:融点=189〜191℃,1HNMR(300MHz,DMSO−d6)δ12.65 (br s, 1H), 3.80 (d, J = 9.7 Hz, 1H), 7.16- 7.30 (m, 6H), 7.07 (dd, J = 1.1, 8.0 Hz, 1H), 7.00 (dd, J = 1.1, 7.5 Hz), 4.40- 4.52 (m, 1H), 4.09 (d, J = 6.0 Hz, 1H), 2.92 (app dd, J = 2.9, 13.9 Hz, 1H), 2.76 (app dd, J = 11.4, 13.9 Hz, 1H), 2.29 (s, 3H), 1.80 (s, 3H); 13CNMR(75MHz,DMSO−d6)δ174.4, 169.3, 168.1, 149.5, 139.7, 139.4, 129.5, 128.3, 127.9, 126.5, 126.3, 124.8, 123.3, 73.2, 53.5, 35.4, 20.8, 12.6; MS(CI)m/z 372.1464(C20H22NO6についての計算値372.1447,M+H+)
【図面の簡単な説明】
【0235】
【図1】実施例14の化合物Aの結晶質形態Iおよび本発明の固体非晶質分散体の粉末X線回折をプロットし、そして固体非晶質分散体中の化合物Aが結晶質でないことを示している。
【図2】異なる条件で貯蔵した後の、実施例15の化合物Aの結晶質形態Iおよび本発明の固体非晶質分散体の粉末X線回折をプロットし、そして固体非晶質分散体が物理的に安定であることを示す。
【技術分野】
【0001】
本発明は、HIVプロテアーゼ阻害剤を含む医薬組成物に関する。
【背景技術】
【0002】
(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミド(別名「(R)−3−((2S,3S)−2−ヒドロキシ−3−{[1−(3−ヒドロキシ−2−メチル−フェニル)−メタノイル]−アミノ}−4−フェニル−ブタノイル)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド」、「(4R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチル−ベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド」、または「4−チアゾリジンカルボキサミド,3−[(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−1−オキソ−4−フェニルブチル]−5,5−ジメチル−N−2−プロペニル−,(4R)−」であり、以下、「化合物A」と称する)は、HIV感染した哺乳動物、例えばヒトの治療に使用することができるHIVプロテアーゼ酵素の阻害剤である。化合物Aおよびその製造は、2002年6月11日に出願された米国特許出願第10/166,979号;2003年12月4日に出願された同第60/527,477号、および2003年12月4日に出願された同第10/782,602号に開示されており、これらはすべて参照により本明細書に組み込まれている。結晶質化合物Aは水溶液中できわめて僅かの可溶性しかなく、非緩衝化水(pH8.3)、標準生理食塩水(pH8.2)および0.1N HCl(pH1.2)中では、水溶解度はそれぞれ約0.16mgA/mL、0.14mgA/mLおよび0.16mgA/mL(温度26℃)である。この低い水溶解度と低いin vivo浸透性が組み合わさると、結晶質化合物Aは経口生物学的利用能が低くなる。
【発明の開示】
【発明が解決しようとする課題】
【0003】
従って、剤形中の化合物Aの安定性を維持しながら、化合物Aの生物学的利用能を改善する必要がある。
【課題を解決するための手段】
【0004】
本発明は、非晶質化合物Aを含んでなる医薬組成物を提供する。非晶質化合物Aは、化合物Aの結晶質形態Iと比較して改善された溶解性を有し、経口的に哺乳動物、例えばヒトに投与した時に、結晶質形態と比較して改善された生物学的利用能が得られる。
【0005】
本発明の1つの態様は、非晶質化合物Aまたはその医薬上許容しうる塩または溶媒和物を提供する。
本発明の別の態様は、非晶質化合物Aまたはその医薬上許容しうる塩または溶媒和物を含んでなる医薬組成物を提供する。
【0006】
本発明のさらに別の態様において、存在する化合物Aの総量の少なくとも約5質量%が非晶質形態である、化合物Aまたはその医薬上許容しうる塩または溶媒和物を含んでなる医薬組成物が提供される。別法として、存在する化合物Aの総量の少なくとも約10質量%、または少なくとも約15質量%、または少なくとも約20質量%、または少なくとも約30質量%、または少なくとも約40質量%、または少なくとも約50質量%、または少なくとも約60質量%、または少なくとも約70質量%、または少なくとも約80質量%、または少なくとも約90質量%、または少なくとも約95質量%が非晶質形態である、化合物Aまたはその医薬上許容しうる塩または溶媒和物を含んでなる医薬組成物が提供される。
【0007】
一態様において、医薬組成物は、(1)非晶質化合物A、またはその医薬上許容しうる塩または溶媒和物、および(2)マトリックスを含んでなる。
【0008】
一実施態様において、医薬組成物は、(1)非晶質化合物A、および、(2)濃度増大ポリマーを含むマトリックスを含んでなる。濃度増大ポリマーは、使用環境中で溶解した化合物Aの濃度をさらに向上させる。結晶質化合物Aによって得られる高い溶解薬物濃度を持続させるには比較的少量のポリマーしか必要とされないことがわかった。非晶質化合物Aを使用環境に単独で投与する場合、化合物Aの溶解濃度は最初に高められており、時間とともに、結晶質形態Iによって得られる平衡濃度はより低い方へ減少する。しかしながら、濃度増大ポリマーを含んでなる組成物では、溶解濃度が最初に高められるだけでなく、生理学上妥当な時間にわたって溶解濃度が持続する。実際、組成物の約25質量%から約1質量%までの範囲の少量のポリマーのみで、結晶質形態Iの化合物A単独だけでなく、非晶質化合物A単独と比較しても化合物Aの溶解性能は実質的に改善される。
【0009】
さらに別の態様において、化合物Aまたはその医薬上許容しうる塩または溶媒和物およびマトリックスを含んでなり、その際、前記マトリックスは、イオン化できるセルロース系ポリマー、イオン化できないセルロース系ポリマー、または非セルロース系ポリマーの少なくとも1つからなる医薬組成物が提供される。
【0010】
なおさらなる態様において、少なくとも1つのイオン化可能なセルロース系ポリマーは、ヒドロキシプロピルメチルセルロースアセテートスクシネート、カルボキシメチルエチルセルロース、セルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースフタレート、メチルセルロースアセテートフタレート、セルロースアセテートトリメリテート、ヒドロキシプロピルセルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースアセテートフタレート、セルロースアセテートテレフタレートおよびセルロースアセテートイソフタレート、およびそれらの混合物の少なくとも1つから選ばれる。
【0011】
またさらに、少なくとも1つのイオン化できないセルロース系ポリマーが、ヒドロキシプロピルメチルセルロースアセテート、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、メチルセルロース、ヒドロキシエチルメチルセルロース、ヒドロキシエチルセルロースアセテートおよびヒドロキシエチルエチルセルロース、およびそれらの混合物から選ばれるこのような組成物が提供される。
【0012】
別の態様では、前記少なくとも1つの非セルロース系ポリマーが、カルボン酸官能化ポリメタクリレート、カルボン酸官能化ポリアクリレート、アミン官能化ポリアクリレート、アミン官能化ポリメタクリレート、タンパク質、カルボン酸官能化デンプン、ヒドロキシル、アルキルアシルオキシおよび環式アミドからなる群より選ばれる少なくとも1つの置換基を有するビニルポリマーおよびコポリマー、少なくとも1つの親水性のヒドロキシルを含む反復単位および少なくとも1つの疎水性のアルキル−またはアリールを含む反復単位のビニルコポリマー、加水分解されてない形態の反復単位の少なくとも一部を有するポリビニルアルコール、ポリビニルアルコールポリビニルアセテートコポリマー、ポリエチレングリコールポリプロピレングリコールコポリマー、ポリビニルピロリドン、ポリエチレンポリビニルアルコールコポリマー、ポリオキシエチレン−ポリオキシプロピレンブロックコポリマーおよびそれらの混合物から選ばれるこのような組成物が提供される。
【0013】
本発明の別の態様において、化合物Aまたはその医薬上許容しうる塩または溶媒和物およびマトリックスを含み、その際、存在する化合物Aの総量の少なくとも約5質量%が非晶質形態である医薬組成物が提供される。別の態様として、化合物A、またはその医薬上許容しうる塩または溶媒和物およびマトリックスを含み、その際、存在する化合物Aの総量の少なくとも約10質量%、または少なくとも約15質量%、または少なくとも質量%、または少なくとも約30質量%、または少なくとも約40質量%、または少なくとも約50質量%、または少なくとも約60質量%、または少なくとも70質量%または少なくとも約80質量%または少なくとも約90質量%、または少なくとも約95質量%が非晶質形態である医薬組成物が提供される。
【0014】
別の態様では、(1)化合物Aおよび、(2)マトリックスを含み、その際、化合物Aの少なくとも一部が非晶質である医薬組成物が提供される。
【0015】
別の実施態様において、医薬組成物は、化合物Aまたはその医薬上許容しうる塩または溶媒和物およびマトリックスを含む固体非晶質分散体を含んでなる。一態様において、固体非晶質分散体は、化合物Aまたはその医薬上許容しうる塩または溶媒和物少なくとも約30質量%を含む。別の態様において、固体非晶質分散体は、化合物Aまたはその医薬上許容しうる塩または溶媒和物の少なくとも約40質量%、少なくとも約50質量%、少なくとも約60質量%、少なくとも約70質量%、少なくとも約80質量%、少なくとも約90質量%、または少なくとも約95質量%を含む。
【0016】
別の実施態様において、本発明は、in vitro水性環境に投与した時に、(a)使用環境における化合物Aの最大溶解濃度が対照組成物によって得られるものの少なくとも約1.25倍である;および(b)使用環境へ導入した時間から使用環境へ導入後の約270分の間の少なくとも90分間のいずれかの期間について使用環境における化合物Aの濃度対時間曲線下の面積(AUC)が、対照組成物の少なくとも約1.25倍である:の少なくとも1つが得られる化合物Aを含んでなる医薬組成物を提供する。対照組成物は、本質的に等量の結晶質形態Iの化合物A単独からなる。別の実施態様において上記議論した使用環境は、本質的にpH6.5および290mOsm/kg、および37℃温度で、20mM Na2HPO4、47mM KH2PO4、87mM NaClおよび0.2mM KClからなり、その際、前記使用環境の総量は約1.8mLであり、使用する化合物Aの量は、化合物Aの全てが溶解した場合、化合物Aの全濃度が3000μg/mLとなるような量である。別の態様において、このような医薬組成物は、化合物Aの少なくとも一部が非晶質形態である化合物Aを含む。さらに別の態様において存在する化合物Aの総量の少なくとも約5質量%が非晶質形態であるこのような組成物が提供される。さらに別の態様において、存在する化合物Aの総量の少なくとも約10質量%、または少なくとも約15質量%、または少なくとも約20質量%、または少なくとも約30質量%、または少なくとも約40質量%、または少なくとも約50質量%、または少なくとも約60質量%、または少なくとも約70質量%、または少なくとも約80質量%、または少なくとも約90質量%、または少なくとも約95質量%が非晶質形態である、このような組成物が提供される。
【0017】
本発明の別の態様において、哺乳動物、例えばヒト中の化合物Aの血漿中濃度約0.001μM〜約5μMを達成する方法が提供され、前記方法は、化合物A単独またはその医薬上許容しうる塩または溶媒和物、またはマトリックスと組み合わせて含んでなる医薬組成物の十分な量を前記哺乳動物に投与することからなる。一実施態様において、組成物は非晶質化合物Aを含んでなる。別の実施態様において、組成物は、非晶質化合物Aおよび少なくとも1つのマトリックスを含んでなる。本発明の別の態様において、哺乳動物、例えばヒトにおける化合物Aの血漿濃度は、約0.01μM〜約2.5μM、または約0.02μM〜約1μM、または約0.025μM〜約1μM、または約0.05μM〜約1μMの範囲にある。
【0018】
本発明の別の態様において、哺乳動物、例えばヒト中の化合物Aの前記血漿中濃度が、前記投与後、少なくとも約6時間、または前記投与後、少なくとも約8、10、12、14、16、18、20、22または24時間維持されるこのような方法が提供される。
【0019】
本発明のさらなる態様において、約6〜約24時間、約0.001μMから約2.5μMの範囲で哺乳動物、例えばヒトの血漿中の化合物Aの平均血漿中濃度を達成する方法であって、前記哺乳動物に化合物A単独またはその医薬上許容しうる塩または溶媒和物、またはマトリックスと組み合わせて含んでなる医薬組成物の十分な量を投与することからなる方法が提供される。一実施態様において、組成物は、非晶質化合物Aを含んでなる。別の実施態様において、組成物は、非晶質化合物Aおよび少なくとも1つのマトリックスを含んでなる。本発明の別の態様において、哺乳動物における化合物Aの平均血漿中濃度は、約0.02μM〜約1μM、または約0.025μM〜約1μM、または約0.05μM〜約1μMの範囲にある。本発明の別の態様において、このような時間にわたるこのような平均血漿中濃度は、化合物A単独、またはその医薬上許容しうる塩または溶媒和物、またはマトリックスと組み合わせて含んでなる医薬組成物の用量を前記哺乳動物に投与することによって達成され、その際、化合物Aの前記用量は、前記組成物中で化合物A約300mgA〜約3600mgAの範囲にある。一実施態様において、このような組成物は、非晶質化合物Aを含んでなる。別の実施態様において、このような組成物は、非晶質化合物Aおよび少なくとも1つのマトリックスを含んでなる。「mgA」は、活性化合物Aのミリグラムを意味する。また、本発明は、化合物Aの用量が、約400mgA、600mgA、800mgA、1000mgA、1200mgA、1400mgA、1600mgA、1800mgA、2000mgA、2200mgA、2400mgA、2600mgA、2800mgA、3000mgA、3200mgAまたは3400mgAであるこのような方法を提供する。
【0020】
本発明のさらに別の態様において、非晶質化合物A単独、またはその医薬上許容しうる塩または溶媒和物またはマトリックスと組み合わせて含んでなる医薬組成物のHIV複製阻害量をHIV感染した哺乳動物に投与することからなる、HIV感染した哺乳動物、例えばヒトの治療方法が提供される。
【0021】
本発明のなおさらなる態様は、HIV複製阻害量の非晶質化合物Aまたは医薬上許容しうる塩またはその溶媒和物を、単独でまたはマトリックスと組み合わせて含んでなる医薬組成物をHIV感染した哺乳動物に投与することからなる、HIV感染した哺乳動物、例えばヒトにおけるAIDSまたはAIDS関連症候群の治療方法を提供する。
【0022】
また、本発明は、HIV複製阻害量の非晶質化合物A、またはその医薬上許容しうる塩または溶媒和物を、単独でまたはマトリックスと組み合わせて含んでなる医薬組成物をHIV感染した哺乳動物に投与することからなる、HIV感染した哺乳動物、例えばヒトにおけるHIVプロテアーゼ活性の阻害方法を提供する。
【0023】
本発明のなおさらなる態様において、HIV複製阻害量の非晶質化合物A、またはその医薬上許容しうる塩または溶媒和物を、単独でまたはマトリックス、およびヌクレオシドHIV逆転写酵素阻害剤、非ヌクレオシドHIV逆転写酵素阻害剤、HIVプロテアーゼ阻害剤、HIVインテグラーゼ阻害剤、HIV融合阻害剤、免疫モジュレーター、CCR5アンタゴニスト、および抗感染薬から選ばれる少なくとも1つのさらなる治療剤と組み合わせて含んでなる医薬組成物を、感染した哺乳動物に投与することからなる、感染した哺乳動物、例えばヒトにおけるHIVの治療方法を提供する。本発明の一態様において、非晶質化合物Aおよび少なくとも1つのさらなる治療剤は、同じ医薬組成物の一部として投与される。さらに別の態様において、非晶質化合物Aおよび少なくとも1つのさらなる治療剤は、同時にまたは順次投与される。
【0024】
また、本発明は、治療上有効量の非晶質化合物A単独、またはその医薬上許容しうる塩または溶媒和物、またはマトリックス、およびヌクレオシドHIV逆転写酵素阻害剤、非ヌクレオシドHIV逆転写酵素阻害剤、HIVプロテアーゼ阻害剤、HIVインテグラーゼ阻害剤、HIV融合阻害剤、免疫モジュレーター、CCR5アンタゴニスト、および抗感染薬から選ばれる少なくとも1つのさらなる治療剤と組み合わせて含んでなる医薬組成物を提供する。
【0025】
また、本発明は、マトリックスが、先に述べたようなイオン化できるセルロース系ポリマー、イオン化できないセルロース系ポリマーおよび非セルロース系ポリマーの少なくとも1つから選ばれるこのような方法を提供する。
【0026】
本発明のさらに別の態様において、医薬組成物の投与を1日1回、2回または3回実施する上述の方法を提供する。
【0027】
また、本発明は、感染した哺乳動物、例えばヒトにおいてHIV感染症を治療するための医薬の製造において非晶質化合物Aまたはその医薬上許容しうる塩または溶媒和物を単独でまたはマトリックスと組み合わせて使用する方法を提供する。さらに、本発明は、HIV感染した哺乳動物、例えばヒトにおいてAIDSまたはAIDS関連症候群を治療するための医薬の製造における、非晶質化合物Aまたはその医薬上許容しうる塩または溶媒和物を、単独でまたはマトリックスと組み合わせて使用する方法を提供する。
【0028】
本発明のさらに別の態様において、哺乳動物、例えばヒトの血漿中の化合物Aの平均血漿中濃度を、約0.001μM〜約2.5μMの範囲で約6〜約24時間達成する方法であって、チトクロームP450酵素の阻害剤および非晶質化合物A単独、またはその医薬上許容しうる塩または溶媒和物、またはマトリックスと組み合わせて含んでなる十分な量の医薬組成物を哺乳動物に投与することからなる方法が提供される。また、本発明は、チトクロームP450酵素が3A4アイソフォームであるこのような方法を提供する。また、チトクロームP450酵素の阻害剤がリトナビルまたはデラビルジンであるこのような方法を提供する。
【0029】
別の態様において、本発明は溶媒ベースのプロセスを用いた非晶質化合物Aの製造方法を提供する。
【0030】
また、本明細書では、(a) 少なくとも1つの溶媒を含んでなる噴霧溶液中に化合物を溶解し;そして(b) 前記噴霧溶液から前記少なくとも1つの溶媒を急速に蒸発させて前記化合物の非晶質形態を得る;ことからなる医薬組成物の製造方法であって、その際、前記化合物は、(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミドまたはその医薬上許容しうる塩または溶媒和物である、前記製造方法が提供される。
【0031】
別の態様において、噴霧溶液はさらにマトリックスを含むこのような方法が提供される。
【0032】
さらに本明細書において、マトリックスがイオン化できるセルロース系ポリマー、イオン化できないセルロース系ポリマーおよび非セルロース系ポリマーから選ばれる少なくとも1つのポリマーを含むこのような方法を提供する。
【0033】
また、本明細書において、前記少なくとも1つの溶媒が、メタノールおよび水とメタノールとの混合物から選ばれるこのような医薬組成物の製造方法が提供される。
【0034】
別の実施態様において、組成物は、化合物Aの化学安定性を改善するために、安定化剤を含む。安定化剤は、塩基または抗酸化剤であることができる。1つの好ましい実施態様において、組成物は固体非晶質分散体を含み、固体非晶質分散体はさらに安定化剤を含む。化合物Aの「化学安定性を改善する」とは、化合物Aが別の化学物質または化合物へ分解する速度を遅らせることを意味する。
【0035】
本発明のさらに別の態様において、非晶質化合物Aを含んでなる組成物は、化合物Aの分解を減らすためにパッケージされる。パッケージングは、化合物Aが湿度または酸素のいずれかまたは両方へ曝露されるのを制限することができる。
【0036】
本明細書に使用される用語「結晶質」は、三次元において広範囲の規則性を示す本発明の化合物の特定の固体形態を意味する。結晶質である物質は、当分野で知られている技術、例えば粉末X線回折(PXRD)結晶学、固体NMRまたは熱技術、例えば示差走査熱量測定法(DSC)によって特徴付けることができる。
【0037】
本明細書に使用される用語「非晶質」は、三次元において本質的に規則性を有しない本発明の化合物の特定の固体の形態を意味する。用語「非晶質」は、本質的に規則性を有しない物質だけでなく、いくらかの小規模の規則性を有しうるが、その規則性が三次元よりも少ないおよび/または短い距離しかない物質も含まれる。非晶質物質は、当分野で知られている技術、例えば粉末X線回折(PXRD)結晶学、固体NMRまたは熱技術、例えば示差走査熱量測定法(DSC)によって特徴付けることができる。
【0038】
本明細書に使用される用語「対照組成物」は、本質的に化合物Aの結晶質形態Iからなる組成物のことである。本明細書に使用される対照組成物は、本明細書に記載された使用環境において化合物Aの結晶質形態Iの溶解度に影響を与える他の化合物または成分を含まないことを理解すべきである。
【0039】
本明細書に使用される用語「化合物Aの結晶形態I」は、粉末X線回折(PXRD)、示差走査熱量測定法(DSC)、固体NMR(ssNMR)およびラマンIR分光法(ラマン)といった分析法を用いて当業者によって測定されうる化合物Aの非晶質形態を含まないことを特徴とする(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミドの結晶質形態を意味する。さらにまた、化合物Aの結晶質形態Iは、図I中の対照1として提供されたものと同様の粉末X線回折パターンを有することを特徴とする。「同様の」とは、当業者が、図Iの対照1のパターンと、化合物Aの結晶質形態を含む組成物の別の実験的に測定されたパターンとを比較して、パターンを得るために使用される特定の実験条件に左右されうる典型的な粉末X線回折パターンにおいて、強度および線の位置の知られている多様性を考慮して、それらが同じ多形性の形態であるということを意味する。
【0040】
本明細書に使用されるように、用語「化合物Aの少なくとも一部は非晶質形態である」は、組成物中の化合物Aの総量の少なくとも5質量%または少なくとも10質量%が非晶質形態であることを意味する。
【0041】
本明細書に使用される用語「等量」は、得られた組成物中に存在する、親化合物(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミドの理論上のモル数として測定された、化合物Aのモル量のことである。例えば、化合物Aの塩または溶媒和物を含んでなる組成物の得られた量について、化合物Aの結晶質形態Iの等量は、組成物中に存在する化合物Aの理論上のモル数を測定し、親化合物Aの同じ理論モル数が得られる化合物Aの結晶質形態Iの量を用いて算出される。
【0042】
本明細書に使用される用語「投与」、「投与する」、「用量」および「投薬」は、化合物が哺乳動物の血清または血漿に吸収されるように、化合物またはその医薬上許容しうる塩または溶媒和物、または化合物またはその医薬上許容しうる塩または溶媒和物を含んでなる医薬組成物を、哺乳動物に供給することである。
【0043】
本明細書に使用される用語「併用投与」または「併用投与する」は、第1の化合物および本発明の化合物またはその医薬上許容しうる塩または溶媒和物の組み合わせを単独でまたは医薬上許容しうる組成物の一部としていずれかで投与することである。このような併用投与は、第1の化合物および本発明の化合物が、同じ組成物の一部すなわち同じ一体的剤形の一部となるように実施することができる。また、併用投与には、第1の化合物および本発明の化合物を別々であるが、同じ治療処方計画の一部として投与することも含まれる。2つの成分を、別々に投与する場合、必ずしも本質的に同じ時間に投与する必要はないが、それが望ましい場合は可能である。従って、併用投与には、例えば、第1の化合物および本発明の化合物を、別々の用量または剤形として、しかし、同時に投与することが含まれる。また、併用投与には、異なる時間にいずれかのオーダーで別々の投与することが含まれる。
【0044】
「溶媒和物」は、明記された化合物の生物学的効果を保持しているこのような化合物の医薬上許容しうる溶媒和物の形態を意味するものとする。溶媒和物の例としては、限定されるわけではないが、本発明の化合物と水、イソプロパノール、エタノール、メタノール、ジメチルスルホキシド(DMSO)、酢酸エチル、酢酸、エタノールアミンまたはそれらの混合物との組み合わせが含まれる。「医薬上許容しうる塩」は、遊離酸の生物学的効果および薬理学的に許容しうるアニオンを含む明記された誘導体の塩基を保持しており、生物学的にまたはその他の点で好ましい塩を意味するものとする。医薬上許容しうる塩の例としては、限定されるわけではないが、酢酸塩、アクリル酸塩、ベンゼンスルホン酸塩、安息香酸塩(クロロ安息香酸塩、メチル安息香酸塩、ジニトロ安息香酸塩、ヒドロキシ安息香酸塩、およびメトキシ安息香酸塩)、炭酸水素塩、硫酸水素塩、亜硫酸水素塩、酒石酸水素塩、ホウ酸塩、臭化物、ブチン−1,4−ジオエート、カルシウムエデト酸塩、カムシル酸塩、炭酸塩、塩化物、カプロン酸塩、カプリル酸塩、クラブラン酸塩、クエン酸塩、デカン酸塩、二塩酸塩、リン酸二水素塩、エデト酸塩、エジスリ酸塩、エストール酸塩、エシル酸塩、琥珀酸エチル、ギ酸塩、フマル酸塩、グルセプト酸塩、グルコン酸塩、グルタミン酸塩、グリコール酸塩、グリコリルアルサニル酸塩、ヘプタン酸塩、ヘキシン−1,6−ジオエート、ヘキシルレソルシン酸塩、ヒドラバミン、臭化水素酸塩、塩酸塩、γ−ヒドロキシ酪酸塩、ヨウ化物、イソ酪酸塩、イソチオン酸塩、乳酸塩、ラクトビオン酸塩、ラウリン酸塩、リンゴ酸塩、マレイン酸塩、マロン酸塩、マンデル酸塩、メシル酸塩、メタリン酸塩、メタン−スルホン酸塩、メチル硫酸塩、リン酸一水素塩、粘液酸塩、ナプチル酸塩、ナフタレン−1−スルホネート、ナフタレン−2−スルホネート、硝酸塩、オレイン酸塩、シュウ酸塩、パモ酸塩(エンボン酸塩)、パルミチン酸塩、パントテン酸塩、フェニル酢酸塩、フェニル酪酸塩、フェニルプロピオン酸塩、フタル酸塩、リン酸塩/二リン酸塩、ポリガラクツロン酸塩、プロパンスルホン酸塩、プロピオン酸塩、プロピオル酸塩、ピロリン酸塩、ピロ硫酸塩、サリチル酸塩、ステアリン酸塩、塩基性酢酸塩、スベリン酸塩、琥珀酸塩、硫酸塩、スルホン酸塩、亜硫酸塩、タンニン酸塩、酒石酸、テオクル酸塩、トシル酸塩、トリエチオジド(triethiodode)、および吉草酸塩の塩が含まれる。
【0045】
本明細書に使用されるように、「使用環境」は、哺乳動物、例えば哺乳動物および特にヒトの消化管のin vivo環境、または試験溶液、例えばリン酸緩衝生理食塩水(PBS)またはModel Fasted Duodenal(MFD)溶液のin vitro環境のいずれかであることができる。濃度増大は、in vivo試験またはin vitro溶解試験のいずれかを通して測定することができる。本発明の組成物は、上記の試験環境で少なくとも1つにおいて濃度増大の基準を満たしている。
【0046】
本発明の前述および他の目的、特徴および利点は、本発明の以下の詳細な説明を考察することでより容易に理解される。
【0047】
〔実施態様の詳述〕
化合物Aは、(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミド(別名、「(R)−3−((2S,3S)−2−ヒドロキシ−3−{[1−(3−ヒドロキシ−2−メチル−フェニル)−メタノイル]−アミノ}−4−フェニル−ブタノイル)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド」、「(4R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチル−ベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド」、または「4−チアゾリジンカルボキサミド,3−[(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−1−オキソ−4−フェニルブチル]−5,5−ジメチル−N−2−プロペニル−,(4R)−」)である。化合物Aは、以下の構造を有する:
【化1】
それは、分子量512を有し、そして化合物Aの結晶質形態Iは176〜178℃の融点を有する。
【0048】
本明細書に使用されるように、用語「化合物」は、慣用的であり、上記の化学種を表し、そしてすべての医薬上許容しうる形態を含むことを理解すべきである。「医薬上許容しうる形態」は、立体異性体、立体異性体混合物、鏡像異性体、溶媒和物、水和物、同形体、多形体、仮像、中性の形態、塩の形態およびプロドラッグを含めたすべての医薬上許容しうる誘導体または変種を意味する。
【0049】
非晶質化合物A
一態様において、組成物は、非晶質化合物Aを含んでなる。「非晶質」とは、化合物Aが「結晶質」でないことを意味する。「結晶質」は、化合物Aが三次元において広範囲の規則性を示すことを意味する。従って、用語「非晶質」は、本質的に規則性を有しない物質だけでなく、いくつかの小規模の規則性を有しうるが、その規則性が三次元より低いおよび/または短い近距離しかない物質も含まれるものとする。非晶質物質は、当業者が、例えば粉末X線回折(PXRD)結晶学、固体NMR、ラマンIR分光法または熱技術、例えば示差走査熱量測定法(DSC)といったような当分野で知られている技術を用いて特徴付けることができる。本発明の組成物は、非晶質および結晶質化合物Aの両方を含むことができるが、具体的には本発明の組成物中に存在する化合物Aの総量の少なくとも約5質量%、または少なくとも約10質量%、または少なくとも約30質量%、または少なくとも約40質量%、または少なくとも約50質量%、または少なくとも約60質量%、または少なくとも約70質量%、または少なくとも約80質量%、または少なくとも約90質量%、または少なくとも約95質量%が、非晶質形態でありうることを意図する。
【0050】
非晶質形態化合物Aは、化合物Aの結晶質形態Iと比較して使用環境において溶解した化合物Aの濃度を改善することが見出された。典型的に、非晶質化合物Aでは、等量の化合物Aの結晶質形態Iからなる対照組成物と比較して使用環境で化合物Aのより高い最大薬物濃度(MDC)が得られる。本明細書において対照組成物として用いた化合物Aの結晶質形態Iは、適当な使用環境において化合物Aの溶解度に物質的に影響を及ぼす可溶化剤または他の成分を含まないことを理解しなければならない。化合物Aの非晶質形態は、対照組成物と比較して、水性使用環境における化合物Aの最大溶解濃度(MDC)を、少なくとも約1.25倍、または少なくとも2倍または少なくとも3倍まで高めることが好ましく、前記対照組成物は、本質的に化合物Aの結晶質形態Iからなる。さらに、また、化合物Aの非晶質形態は、等量の化合物Aの結晶質形態Iと比較して、使用環境において化合物Aの濃度対時間曲線下の溶解面積(AUC)を高めることもできる。AUCの計算は、医薬分野でよく知られた方法であり、例えば、Welling,“Pharmacokinetics Processes and Mathematics,”ACS Monograph 185 (1986)に記載されている。例えば、化合物Aの非晶質形態は、本質的に化合物Aの結晶質形態Iからなる対照組成物と比較して、水性使用環境中の化合物AのAUCを少なくとも約1.25倍、または少なくとも2倍または少なくとも3倍まで高める。例えば、37℃、pH6.5および290mOsm/kgのリン酸緩衝生理食塩水からなるin vitro使用環境で試験する場合、化合物Aの非晶質形態では、最大溶解薬物濃度(MDC)は、化合物Aの結晶質形態Iによって得られたものの10.3倍であり、そして使用環境に投与した後、最初の90分についての溶解薬物濃度対時間曲線下の面積(AUC90)は、化合物Aの結晶質形態Iによって得られたものの8.3倍であった。
【0051】
使用環境が動物の消化管である場合、溶解した薬物濃度は、当分野で知られているいずれかの慣用の方法によって測定することができる。1つの方法は、逆重畳積分方法である。この方法では、横座標(x軸)に沿った血液試料時間に対して縦座標(y軸)に沿って血清または血漿薬物濃度をプロットする。次いで、いずれかの慣用の分析、例えばWagner-NelsonまたはLoo-Riegelman分析を用いてデータを分析して消化管中での薬物放出速度を測定する。また、Welling,“Pharmacokinetics: Processes and Mathematics”(ACS Monograph 185, Amer. Chem. Soc., Washington, D.C., 1986)参照。このやり方でデータを処理して見かけのin vivo薬物放出プロファイルが得られる。別の方法は、患者に管を挿入しておよび定期的に消化管から直接、試料採取することである。
【0052】
また、水性使用環境においてAUCを改善するということは、非晶質化合物Aが、使用環境において、特に消化管中で溶解する化合物Aの濃度を高め、それによって血液に吸収される化合物Aの量を高めて化合物Aの高められた生物学的利用能が得られることを意味する。
【0053】
さらに別の態様において、非晶質化合物Aは、絶食状態のヒトまたは他の動物に経口的に投薬した時に、結晶質化合物Aと比較して血液中の溶解した化合物Aの改善された濃度を提供する。化合物Aの非晶質形態では、等量の化合物Aの結晶質形態Iから本質的になる対照組成物と比較して血液(血清または血漿)中で化合物Aのより高い最大薬物濃度(Cmax)が得られる。化合物Aの結晶質形態Iは、適当な使用環境において化合物Aの溶解度に物質的に影響を及ぼす可溶化剤または他の成分を含まないことを理解すべきである。例えば、化合物Aの非晶質形態では、適当な使用環境において本質的に化合物Aの結晶質形態Iからなる対照組成物によって得られる少なくとも約1.25倍または少なくとも約2倍、または少なくとも約3倍の血液中の化合物AのCmaxが得られる。
【0054】
さらに別の態様において、化合物Aの非晶質形態では、絶食状態のヒトまたは他の動物に経口的に投薬した時に、化合物Aの結晶質形態Iによって得られる少なくとも約1.25倍、または少なくとも約2倍、または少なくとも約3倍の血液(血清または血漿)中の化合物Aの濃度のAUCが得られる。また、このような組成物は、化合物Aの結晶質形態I対照の約1.25倍から約3倍までの相対生物学的利用能を有すると言えることに注目される。
【0055】
本発明の組成物は、単独で非晶質化合物Aを含むことができるし、またはさらに詳細に下に記載した賦形剤を含むことができる。
【0056】
化合物Aおよびマトリックスの固体非晶質分散体
別の実施態様において、医薬組成物は、化合物Aおよび1つまたはそれ以上の成分の固体非晶質分散体を含んでなり、これらの成分はひとまとめにして「マトリックス」と称する。「固体非晶質分散体」は、化合物Aの少なくとも一部が非晶質形態にあり、マトリックス中に分散されていることを意味する。好ましい実施態様において、マトリックスは、分散体において、非分散型の非晶質化合物A単独と比較して化合物Aの改善された物理安定性、改善された化学安定性、改善された濃度増大のいずれかまたはこれらのいずれかの組み合わせまたは3つ全てが得られるように選ばれる。「分散されてない化合物A」は、マトリックス中に分散されてない化合物Aを意味する。マトリックスは、単一の成分からなることもできるし、または2つまたはそれ以上の成分の混合物であってもよい。成分は、十分に混合して単一相または分子分散体を形成することができるし、または異なる組成を有する2つまたはそれ以上の異なる相として存在することができる。
【0057】
マトリックスの少なくとも一部は、生理学的に関連のあるpH(例えばpH1〜8)の水溶液中で水膨潤性、分散性または可溶性のいずれかである。マトリックスは、全体として室温で固体でなければならず、そして少なくとも約40℃の温度まで、好ましくは少なくとも約60℃の温度まで、そしてより好ましくは少なくとも約70℃の温度までは実質的に固体のままである。このため、マトリックスは、約40℃より上、好ましくは60℃より上、そしてより好ましくは約70℃より上の融点を有する成分を少なくとも1つまたはそれ以上含まなければならない。
【0058】
本発明の分散体中に存在する薬物の量に対するマトリックスの量は、マトリックスの特徴によって左右され、薬物対マトリックスの質量比は、約0.01〜約100で広く変化することができる(例えば薬物1質量%〜薬物99質量%)。化合物A対マトリックスの質量比は、約0.1〜約49(薬物約10質量%〜薬物約98質量%)の範囲であることが好ましい。
【0059】
マトリックスに使用する成分は、ポリマーまたは非ポリマーであってもよく、いくつかの成分の混合物を含むことができる。従って、マトリックスはポリマー成分の混合物、非ポリマー成分の混合物またはポリマーおよび非ポリマー成分の混合物を含むことができる。
【0060】
用語「ポリマー」は、慣用的に使用され、モノマーが一緒に結合してより大きい分子を形成してできた化合物を意味する。一般に、ポリマーマトリックス成分では、非ポリマーのマトリックス成分と比較して濃度増大が改善された分散体が得られる。ポリマー成分は中性であってもよいしまたはイオン化することができ、そしてセルロース系または非セルロース系であってもよい。マトリックスとして使用される典型的なポリマー成分としては、本明細書で下に記載された濃度増大ポリマー、ポリエチレングリコール、ポリオキシエチレングリコール、ポリエチレンオキシド、キサンタンガム、カラゲナン、キトサン、ポリデキストロース、デキストリンおよびデンプンが含まれる。また、この定義の中には高分子量タンパク質、例えばゼラチンおよびアルブミンが含まれる。マトリックスは後述する濃度増大ポリマーであることが好ましい。
【0061】
「非ポリマーの」は、成分がポリマーでないことを意味する。マトリックス成分として使用するための典型的な非ポリマー物質としては、限定されるわけではないが、アルコール、例えばステアリルアルコールおよびセチルアルコール、有機酸およびそれらの塩、例えばステアリン酸、クエン酸、フマル酸、酒石酸、リンゴ酸、およびその医薬上許容しうる塩;有機塩基、例えばグルコサミン、N−メチルグルカミン、トリス(ヒドロキシメチル)アミノメタンおよびドデシルアミン;塩、例えば塩化ナトリウム、塩化カリウム、塩化リチウム、塩化カルシウム、塩化マグネシウム、硫酸ナトリウム、硫酸カリウム、炭酸ナトリウムおよび硫酸マグネシウム;アミノ酸、例えばアラニンおよびグリシン;糖、例えばグルコース、シュクロース、キシリトール、フルクトース、ラクトース、トレハロース、マンニトール、ソルビトールおよびマルチトール;脂肪酸エステル、例えばグリセリル(モノ−および−)ステアレート、グリセリル(モノ−およびジ−)ベヘネート、トリグリセリド、ソルビタンモノステアレート、サッカロースモノステアレート、グリセリル(パルミチン酸ステアリン酸)エステル、水素化された綿実油、ポリオキシエチレンソルビタン脂肪酸エステル;ワックス、例えば微結晶ワックス、パラフィンワックス、蜜蝋、合成蝋、キャスターワックスおよびカルナバワックス;アルキルスルフェート、例えばラウリル硫酸ナトリウムおよびラウリル硫酸マグネシウム;およびリン脂質、例えばレシチン;およびそれらの混合物が含まれる。
【0062】
本発明の組成物において、固体非晶質分散体中に存在する化合物Aの少なくとも主要部分は、非晶質形態であることができる。本明細書に使用されるように、化合物Aの用語「主要部分」は、固体非晶質分散体中の化合物Aの少なくとも約60質量%が、結晶質形態ではなく非晶質形態であることを意味する。別法として、本発明の組成物は、実質的に非晶質である固体非晶質分散体中の化合物Aを含んでなることができる。本明細書に使用されるように、「実質的に非晶質」とは、結晶質形態中の化合物Aの量が約25質量%を超えないことを意味する。さらに、本発明の組成物は、「ほとんど完全に非晶質」である固体非晶質分散体中の化合物Aを含んでなり、結晶質形態の化合物Aの量が、存在する化合物Aの総量の約10質量%を超えないことを意味する。結晶質化合物Aの量は、粉末X線回折(PXRD)、走査型電子顕微鏡(SEM)分析、示差走査熱量測定法(DSC)、または他のいずれの標準定量的測定法を含めた分析技術を用いて当業者によって測定されうるが、これらに限定されない。
【0063】
化合物Aの非晶質形態は、ポリマー全体に均質に分散された薬物の固溶体としてまたはこれらの状態のいずれかの組み合わせまたはそれらの中間の状態として、固体非晶質分散体の中で、比較的純粋な非晶質薬物ドメインまたは領域中に存在することができる。
【0064】
濃度増大ポリマーを含んでなる組成物
別の実施態様において、医薬組成物は、化合物Aの非晶質形態および濃度増大ポリマーを含んでなる。濃度増大ポリマーは、適当な使用環境において溶解した薬物の濃度をさらに改善することができる。特に、本発明の組成物中に濃度増大ポリマーを含む利点は、このような濃度増大ポリマーを含まず、化合物Aを含んでなる組成物と比較して化合物AのAUCが改善されることである。本発明者らは、非晶質化合物Aでは、適当な使用環境中に溶解した場合、化合物Aの平衡濃度を超える化合物Aの初濃度が得られるが、しかしながら、溶解した化合物Aの濃度は、時間とともに実質的に減少することを見出した。理論によって拘束しようとするものではないが、本発明者らは、溶解した化合物Aを伴う使用環境に濃度増大ポリマーを添加すると、最初に高められた溶解薬物濃度が平衡濃度まで落ちる速度が遅くなると考えた。結果として、非晶質化合物Aおよび濃度増大ポリマーを含んでなる組成物では、化合物A単独で得られるよりも大きく改善された溶解曲線下の面積(「AUC」)が得られた。
【0065】
AUCを改善するということは、濃度増大ポリマーを含んでなる組成物では、使用環境中、特に哺乳動物、例えばヒトの消化(GI)管中に溶解したままの薬物の濃度を高めることによって化合物Aの高められた生物学的利用能を得ることができることを意味する。溶液中の化合物Aの濃度を改善することで哺乳動物においてより高い血中濃度が得られ、場合によっては、到達すべき有効レベルが可能であり、または別の場合には、より低い薬物用量レベルで到達すべき有効血中濃度を得ることができ、このため、投薬しなければならない薬物の量が減少し、血中濃度の変動が減り、そしてまた必要となるポリマーの量に応じて剤形のサイズが減少する。
【0066】
本発明の組成物に使用するのに適した濃度増大ポリマーは、有害なかたちで化合物Aと化学的に反応せず、医薬上許容しうるという点で不活性でなければならない。ポリマーは、中性またはイオン化可能であってもよく、pH約1〜8の範囲の少なくとも一部にわたって少なくとも約0.1mg/mlの水溶解度を有しなければならない。本発明の使用に適した濃度増大ポリマーは、セルロース系または非セルロース系であることができる。これらのうち、イオン化できるポリマーおよびセルロース系ポリマーが好ましく、イオン化できるセルロース系ポリマーがより好ましい。「セルロース系の」とは、糖類反復単位上のヒドロキシル基の少なくとも一部と化合物とが反応してエステルまたはエーテル置換基を形成することによって改良されたセルロースポリマーを意味する。
【0067】
ポリマーの好ましい種類としては、性質が「両親媒性」であるポリマーが含まれ、これはポリマーが疎水性および親水性部分を有することを意味する。疎水性部分は、脂肪族または芳香族炭化水素基のような基を含むことができる。親水性部分は、水素結合することができる、イオン化できるまたはイオン化できない基、例えばヒドロキシル、カルボン酸、エステル、アミンまたはアミドを含むことができる。
【0068】
本発明に使用するための適切なポリマーの1つの種類にイオン化できない(または中性の)非セルロース系ポリマーがある。典型的なポリマーとしては、ヒドロキシル、アルキルアシルオキシまたは環式アミドの置換基を有するビニルポリマーおよびコポリマー;加水分解されてない形態のそれらの反復単位(酢酸ビニル)の少なくとも一部を有するポリビニルアルコール;ポリビニルアルコールポリビニルアセテートコポリマー;ポリビニルピロリドン;ポリオキシエチレン−ポリオキシプロピレンコポリマー(別名ポロキサマー);およびポリエチレンポリビニルアルコールコポリマーが含まれる。典型的な非セルロース系の中性ポリマーとしては、ヒドロキシエチルメタクリレート、ポリビニルヒドロキシエチルエーテルおよびポリエチレングリコールが含まれる。
【0069】
中性の非セルロース系ポリマーの好ましい種類としては、ヒドロキシルを含む親水性の反復単位およびアルキルまたはアリールを含む疎水性の反復単位のビニルコポリマーが含まれる。このような中性のビニルコポリマーは、「両親媒性ヒドロキシル官能性ビニルコポリマー」と称する。「両親媒性ヒドロキシル官能性ビニルコポリマー」は、例外的であり、それらは、いずれも非イオン性であるが、驚くべきことに、低い溶解性の薬物用に分散体ポリマーとして使用する場合、使用する水性環境に投薬した時に高いレベルの薬物濃度増大をもたらす固体非晶質分散体が得られる。
【0070】
好ましいコポリマーは、一般構造:
【化2】
を有し、ここでAおよびBは、それぞれ「ヒドロキシルを含む親水性の」および「疎水性の」置換基を表し、そしてnおよびmは、それぞれポリマー分子当たりの親水性ビニル反復単位の平均数および疎水性ビニル反復単位の平均数を表す。コポリマーは、ブロックコポリマー、ランダムコポリマーであってもよいし、またはこれらの2の両極端の間のいずれかの構造を有することができる。ポリマーは、例えば約2,500〜約1,000,000ダルトンの分子量を有することができる。
【0071】
ヒドロキシルを含む親水性の反復単位(「A」)は、単にヒドロキシル(−OH)であってもよいし、またはそれに結合した1つまたはそれ以上のヒドロキシルを有する炭素1〜6個の短鎖アルキルのいずれかであってもよい。ヒドロキシル置換されたアルキルは、炭素−炭素またはエーテル結合を経てビニル骨格鎖に結合されていてもよい。従って、典型的な「A」構造としては、ヒドロキシルそれ自体に加えて、ヒドロキシメチル、ヒドロキシエチル、ヒドロキシプロピル、ヒドロキシメトキシ、ヒドロキシエトキシおよびヒドロキシプロポキシが含まれる。
【0072】
疎水性置換基(「B」)は、単に水素(−H)、この場合、疎水性反復単位は、エチレン;炭素−炭素結合を経て結合した炭素12個までのアルキルまたはアリール置換基、例えばメチル、エチルまたはフェニル;エーテル結合を経て結合した炭素12個までのアルキルまたはアリール置換基、例えばメトキシ、エトキシまたはフェノキシ;エステル結合を経て結合した炭素12個までのアルキルまたはアリール置換基、例えばアセテート、プロピオネート、ブチレートまたはベンゾエートであってもよい。本発明の両親媒性ヒドロキシル官能性ビニルコポリマーは、置換されたビニルコポリマーを製造するために使用するいずれかの慣用の方法によって合成することができる。ポリビニルアルコール/ポリビニルアセテートのようないくつかの置換されたビニルコポリマーは、よく知られており、商業的に入手可能である。
【0073】
このようなポリマーは、同一出願人による係属中の米国特許出願第10/175,132号にさらに詳細に開示されており、これは2001年6月22日に出願された仮出願第60/300,255号に対して優先権を主張し、参照により本明細書に組み込まれている。
【0074】
本発明に使用するのに適したポリマーの他の種類としては、イオン化できる非セルロース系ポリマーがある。典型的なポリマーとしては、カルボン酸官能化ビニルポリマー、例えばカルボン酸官能化ポリメタクリレートおよびカルボン酸官能化ポリアクリレート、例えばRohm Tech Inc., of Malden, Massachusettsによって製造されたEUDRAGITSR;アミン官能化ポリアクリレートおよびポリメタクリレート;タンパク質;およびカルボン酸官能化デンプン、例えばデンプングリコレートが含まれる。
【0075】
両親媒性非セルロース系ポリマーは、比較的親水性および比較的疎水性モノマーのコポリマーである。例としては、アクリレートおよびメタクリレートコポリマーおよびポリオキシエチレン−ポリオキシプロピレンコポリマーが含まれる。このようなコポリマーの典型的な商品名としてはEUDRAGITS(これはメタクリレートおよびアクリレートのコポリマーである)およびBASFによって供給されるPLURONICS(これはポリオキシエチレン−ポリオキ
シプロピレンコポリマーである)が含まれる。
【0076】
ポリマーの好ましい種類は、少なくとも1つのエステル−および/またはエーテル−結合した置換基を有するイオン化できるおよび中性のセルロース系ポリマーが含まれ、その際、ポリマーはそれぞれの置換基について少なくとも0.1の置換度を有する。本明細書に使用されるポリマー命名法において、エーテル結合した置換基は、エーテル基に結合した部分として「セルロース」の前に記載され;例えば「エチル安息香酸セルロース」は、エトキシ安息香酸置換基を有することに留意すべきである。同様に、エステル結合した置換基は、カルボキシレートとして「セルロース」の後に記載され;例えば「セルロースフ
タレート」は、ポリマーにエステル結合した各フタレート部分の1つカルボン酸と未反応の別のカルボン酸とを有する。
【0077】
また、「セルロースアセテートフタレート」(CAP)のようなポリマー名称は、エステル結合を経てセルロース系ポリマーのヒドロキシル基の有意な画分に結合したアセテートおよびフタレート基を有するセルロース系ポリマーのいずれかのファミリーのことであると留意すべきである。一般に、それぞれの置換基の置換度は、ポリマーの他の基準を満たせば、0.1〜2.9の範囲であることができる。「置換度」は、置換されたセルロース鎖上で糖類反復単位当たり3個の平均ヒドロキシル数に相当する。例えば、セルロース鎖のヒドロキシルの全てが置換されたフタレートである場合、フタレートの置換度は、3である。また、それぞれのポリマーファミリータイプには、ポリマーの性能を実質的に変えない比較的少量で加えられたさらなる置換基を有するセルロース系ポリマーが含まれる。このように、例えば、ポリマー名称「ヒドロキシプロピルメチルセルロースアセテートスクシネート」は、商業グレードL、MおよびHがあり、信越化学工業(東京,日本)から入手できる。
【0078】
両親媒性セルロースには、少なくとも1つの比較的疎水性の置換基を有するそれぞれの糖類反復単位上に存在する3個のヒドロキシル基のいずれかまたは全てで親セルロースポリマーが置換されたポリマーが含まれる。疎水性置換基は、十分に高いレベルまたは置換度に置換された場合、セルロース系ポリマーを本質的に水不溶性にすることができる本質的にすべての置換基であるといえる。疎水性置換基の例としては、エーテル結合したアルキル基、例えばメチル、エチル、プロピル、ブチル、等;またはエステル結合したアルキル基、例えばアセテート、プロピオネート、ブチレート、等;およびエーテルおよび/またはエステル結合したアリール基、例えばフェニル、ベンゾエートまたはフェニレートが含まれる。ポリマーの親水性領域は、非置換のヒドロキシルはそれ自体比較的親水性であるため、比較的非置換であるか、または親水性置換基で置換された領域のいずれかでありうる。親水性置換基としては、エーテル−またはエステル結合したイオン化できない基、例えばヒドロキシアルキル置換基ヒドロキシエチル、ヒドロキシプロピル、およびアルキルエーテル基、例えばエトキシエトキシまたはメトキシエトキシが含まれる。特に好ましい親水性置換基は、エーテル−またはエステル結合したイオン化できる基、例えばカルボン酸、チオカルボン酸、置換されたフェノキシ基、アミン、ホスフェートまたはスルホネートであるものである。
【0079】
セルロース系のポリマーの1つの種類には、中性ポリマーがあり、これはポリマーが水溶液中で実質的にイオン化できないことを意味する。このようなポリマーには、イオン化できない置換基が含まれており、それはエーテル結合またはエステル結合することができる。典型的なエーテル結合したイオン化できない置換基としては、アルキル基、例えばメチル、エチル、プロピル、ブチル、等;ヒドロキシアルキル基、例えばヒドロキシメチル、ヒドロキシエチル、ヒドロキシプロピル、等;およびアリール基、例えばフェニルが含まれる。典型的なエステル結合したイオン化できない置換基としては、アルキル基、例えばアセテート、プロピオネート、ブチレート、等;およびアリール基、例えばフェニレートが含まれる。しかしながら、アリール基が含まれる場合、生理学的に関連のあるpH1〜8のいずれかでポリマーが少なくともいくらか水溶解度を有するように、ポリマーは十分な量の親水性置換基を含む必要がありうる。
【0080】
ポリマーとして使用することができる典型的なイオン化できないポリマーとしてはヒドロキシプロピルメチルセルロースアセテート、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、メチルセルロース、ヒドロキシエチルメチルセルロース、ヒドロキシエチルセルロースアセテートおよびヒドロキシエチルエチルセルロースが含まれる。
【0081】
中性のセルロース系ポリマーの好ましいものは、両親媒性ものである。典型的なポリマーとしては、ヒドロキシプロピルメチルセルロースおよびヒドロキシプロピルセルロースアセテートが含まれ、その際、非置換のヒドロキシルまたはヒドロキシプロピル置換基と比較してメチルまたはアセテート置換基の数が比較的多いセルロース反復単位は、ポリマー上の他の反復単位と比較して疎水性部分を構成する。
【0082】
セルロース系ポリマーの好ましい種類としては、生理学的に関連のあるpHで少なくとも部分的にイオン化でき、そして少なくとも1つのイオン化できる置換基を含み、その置換基はエーテル結合またはエステル結合することができるポリマーが含まれる。典型的なエーテル結合したイオン化できる置換基としては、カルボン酸、例えば酢酸、プロピオン酸、安息香酸、サリチル酸、アルコキシ安息香酸、例えばエトキシ安息香酸またはプロポキシ安息香酸、アルコキシフタル酸の種々の異性体、例えばエトキシフタル酸およびエトキシイソフタル酸、アルコキシニコチン酸の種々の異性体、例えばエトキシニコチン酸、およびピコリン酸の種々の異性体、例えばエトキシピコリン酸、等;チオカルボン酸、例えばチオ酢酸;置換されたフェノキシ基、例えばヒドロキシフェノキシ、等;アミン、例えばアミノエトキシ、ジエチルアミノエトキシ、トリメチルアミノエトキシ、等;ホスフェート(例えばホスフェートエトキシ);およびスルホネート、例えばスルホネートエトキシが含まれる。典型的なエステル結合したイオン化できる置換基としては、カルボン酸、例えばスクシネート、シトレート、フタレート、テレフタレート、イソフタレート、トリメリテート、およびピリジンジカルボン酸の種々の異性体、等;チオカルボン酸、例えばチオスクシネート;置換されたフェノキシ基、例えばアミノサリチル酸;アミン、例えば天然または合成アミノ酸、例えばアラニンまたはフェニルアラニン;ホスフェート、例えばアセチルホスフェート;およびスルホネート、例えばアセチルスルホネートが含まれる。また、必要な水溶解度を有する芳香族置換されたポリマーについては、十分な親水基、例えばヒドロキシプロピルまたはカルボン酸官能基がポリマーに結合して、少なくともいずれかのイオン化できる基がイオン化されるpH値でポリマーを水溶解度にすることが望ましい。場合によっては、フタレートまたはトリメリテート置換基のような芳香族基はそれ自体イオン化できる。
【0083】
生理学的に関連のあるpHで少なくとも部分的にイオン化された典型的なセルロースのポリマーとしては、ヒドロキシプロピルメチルセルロースアセテートスクシネート、ヒドロキシプロピルメチルセルローススクシネート、ヒドロキシプロピルセルロースアセテートスクシネート、ヒドロキシエチルメチルセルローススクシネート、ヒドロキシエチルセルロースアセテートスクシネート、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシエチルメチルセルロースアセテートスクシネート、ヒドロキシエチルメチルセルロースアセテートフタレート、カルボキシエチルセルロース、カルボキシメチルセルロース、カルボキシメチルエチルセルロース、セルロースアセテートフタレート、メチルセルロースアセテートフタレート、エチルセルロースアセテートフタレート、ヒドロキシプロピルセルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースアセテートフタレート、ヒドロキシプロピルセルロースアセテートフタレートスクシネート、ヒドロキシプロピルメチルセルロースアセテートスクシネートフタレート、ヒドロキシプロピルメチルセルローススクシネートフタレート、セルロースプロピオネートフタレート、ヒドロキシプロピルセルロースブチレートフタレート、セルロースアセテートトリメリテート、メチルセルロースアセテートトリメリテート、エチルセルロースアセテートトリメリテート、ヒドロキシプロピルセルロースアセテートトリメリテート、ヒドロキシプロピルメチルセルロースアセテートトリメリテート、ヒドロキシプロピルセルロースアセテートトリメリテートスクシネート、セルロースプロピオネートトリメリテート、セルロースブチレートトリメリテート、セルロースアセテートテレフタレート、セルロースアセテートイソフタレート、セルロースアセテートピリジンジカルボキシレート、サリチル酸セルロースアセテート、ヒドロキシプロピルサリチル酸セルロースアセテート、エチル安息香酸セルロースアセテート、ヒドロキシプロピルエチル安息香酸セルロースアセテート、エチルフタル酸セルロースアセテート、エチルニコチン酸セルロースアセテート、およびエチルピコリン酸セルロースアセテートが含まれる。
【0084】
親水性および疎水性領域を有する両親媒性の定義を満たす典型的なイオン化できるセルロース系ポリマーとしては、セルロースアセテートフタレートおよびセルロースアセテートトリメリテートといったようなポリマーが含まれ、この場合、1つまたはそれ以上のアセテート置換基を有するセルロース反復単位は、アセテート置換基を有しないまたは1つまたはそれ以上のイオン化されたフタレートまたはトリメリテート置換基を有するものと比べて疎水性である。
【0085】
セルロース系のイオン化できるポリマーの特に望ましいサブセットは、カルボン酸官能性芳香族置換基およびアルキレート置換基の両方を有し、このため両親媒性であるものである。典型的なポリマーとしては、セルロースアセテートフタレート、メチルセルロースアセテートフタレート、エチルセルロースアセテートフタレート、ヒドロキシプロピルセルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートフタレート、ヒドロキシプロピルセルロースアセテートフタレートスクシネート、セルロースプロピオネートフタレート、ヒドロキシプロピルセルロースブチレートフタレート、セルロースアセテートトリメリテート、メチルセルロースアセテートトリメリテート、エチルセルロースアセテートトリメリテート、ヒドロキシプロピルセルロースアセテートトリメリテート、ヒドロキシプロピルメチルセルロースアセテートトリメリテート、ヒドロキシプロピルセルロースアセテートトリメリテートスクシネート、セルロースプロピオネートトリメリテート、セルロースブチレートトリメリテート、セルロースアセテートテレフタレート、セルロースアセテートイソフタレート、セルロースアセテートピリジンジカルボキシレート、サリチル酸セルロースアセテート、ヒドロキシプロピルサリチル酸セルロースアセテート、エチル安息香酸セルロースアセテート、ヒドロキシプロピルエチル安息香酸セルロースアセテート、エチルフタル酸セルロースアセテート、エチルニコチン酸セルロースアセテート、およびエチルピコリン酸セルロースアセテートが含まれる。
【0086】
セルロース系のイオン化できるポリマーの別の特に望ましいサブセットは、両親媒性であり非芳香族カルボキシレート置換基を有するものである。典型的なポリマーとしては、ヒドロキシプロピルメチルセルロースアセテートスクシネート、ヒドロキシプロピルメチルセルローススクシネート、ヒドロキシプロピルセルロースアセテートスクシネート、ヒドロキシエチルメチルセルロースアセテートスクシネート、ヒドロキシエチルメチルセルローススクシネート、ヒドロキシエチルセルロースアセテートスクシネート、およびカルボキシメチルエチルセルロースが含まれる。
【0087】
別の好ましい種類のポリマーは、中和された酸性ポリマーのからなる。「中和された酸性ポリマー」とは、「酸性部分」または「酸性置換基」の有意な画分が「中和された」;すなわち、それらの脱プロトン化された形態で存在するすべての酸性ポリマーを意味する。「酸性ポリマー」は、有意な数の酸性部分を有するすべてのポリマーを意味する。一般に、有意な数の酸性部分とは、ポリマー1グラム当たり約0.1ミリグラム当量またはそれ以上の酸性部分である。「酸性部分」としては、水と接触してまたは溶解して少なくとも水に水素カチオンを部分的に供与し、これによって水素イオン濃度を高めることができる十分に酸性であるすべての官能基が含まれる。この定義には、実際には、官能基がポリマーに共有結合した時に、約10未満のpKaを有するすべての官能基または「置換基」が含まれる。上記の説明に含まれる官能基の典型的な種類としては、カルボン酸、チオカルボン酸、ホスフェート、フェノール基およびスルホネートが含まれる。このような官能基は、例えばポリアクリル酸では、ポリマーの一次構造を構成することができるが、より一般には親ポリマーの骨格鎖に共有結合しており、このため「置換基」と称する。
【0088】
一塩基酸(例えばカルボン酸)で置換されたポリマーの「中和度」、αは、中和された;すなわち塩基によって脱プロトン化されたポリマー上で酸性部分の画分として定義される。典型的に、「中和された酸性ポリマー」とみなされる酸性ポリマーでは、αは、少なくとも約0.001(または0.1%)、好ましくは約0.01(1%)、そしてより好ましくは少なくとも0.1(10%)でなければならない。このような小さい中和度は許容され、それは中和度が少し増加するとばしばポリマーの有効pHが劇的に変化するためである。それにもかかわらず、より大きい中和度は、よりいっそう好ましい。従って、αは、少なくとも0.5(酸性部分の少なくとも50%が中和されたことを意味する)であることが好ましく、そしてαは少なくとも0.9(酸性部分の少なくとも90%が中和されたことを意味する)であることがより好ましい。
【0089】
中和された酸性ポリマーは、同一出願人による係属中の米国特許出願第10/175,566号にさらに詳細に記載されており、これは、2001年6月22日に出願された“Pharmaceutical Compositions of Drugs and Neutralized Acidic Polymers”と題する米国仮特許出願第60/300,256号の優先権を主張し、この関連した開示は、参照により組み込まれている。
【0090】
特定のポリマーは、本発明の組成物に使用するために適切であることを議論してきたが、このようなポリマーのブレンドも適切であるといえる。従って、用語「ポリマー」は、単一種類のポリマーに加えてポリマーのブレンドを含むものとする。
【0091】
組成物中に存在する濃度増大ポリマーの量は、濃度増大をもたらすのに十分であり、下により詳細に記載されている。一般に、薬物対ポリマーの比率は、約0.01(薬物1部対ポリマー100部)〜約100の範囲であることができる。
【0092】
非晶質化合物Aおよび濃度増大ポリマーは、なんらかの方法で合わせることができる。一実施態様において、組成物は非晶質化合物Aおよび濃度増大ポリマーの組み合わせからなる。別の実施態様において、組成物は、(1)化合物Aおよびマトリックスを含んでなる固体非晶質分散体および(2)濃度増大ポリマー:の組み合わせからなる。本明細書に使用される「組み合わせ」は、非晶質化合物Aまたは化合物Aおよびマトリックスおよび濃度増大ポリマーを含んでなる固体非晶質分散体が、互いに物理的に接触しているかまたは密接に接近しているが、物質的に混合する必要はないことを意味する。例えば、当分野で知られているように、組成物は、多層錠剤の形態をとることができ、そこでは1つまたはそれ以上の層が非晶質化合物Aを含み、そして1つまたはそれ以上の異なる層が濃度増大ポリマーを含む。さらに別の例としてコーティング錠剤を挙げることができ、そこでは、化合物Aまたは濃度増大ポリマーまたは両方が錠剤の核中に存在することができ、そしてコーティングは、非晶質化合物Aまたは濃度増大ポリマーまたは両方を含むことができる。別法として、組み合わせは、非晶質化合物Aおよび濃度増大ポリマーの両方が微粒子の形態で混合され、そしてそれぞれの粒子が、サイズに関係なく、それらがバルク中で示す個々の同じ物理特性を保持している、単純な乾燥した物理的混合物の形態をとることができる。
【0093】
非晶質化合物Aおよび濃度増大ポリマーの組み合わせは、V型ブレンダー、遊星型ミキサー、渦流ブレンダー(vortex blenders)、ミル、押出機、例えばツインスクリーン押出機(twin-screen extruders)および摩砕プロセスを用いて非晶質化合物A、1つまたはそれ以上の濃度増大ポリマー、および所望の剤形を形成するのに適当な他のいずれか賦形剤を含めた乾燥成分を混合するといったようななんらかの慣用の方法で形成することができる。成分は、ボールミルまたはローラーコンパクターのような機械的エネルギーを用いた造粒プロセスにおいて合わせることができる。また、成分は、高剪断造粒機または流動層造粒機中で湿式造粒法を用いて合わせることができ、その際、溶媒または湿潤剤を成分または濃度増大ポリマーに加え、溶媒中に溶解して造粒液として使用することができる。濃度増大ポリマーは、非晶質化合物Aを含む混合物から圧縮プロセスによって予め形成された錠剤にコーティングとして加えることができ、コーティングは、噴霧コーティングプロセスにおいて、例えばパンコーターまたは流動層コーターを用いて実施される。
【0094】
別法として、本発明の組成物は併用投与することができ、これは、非晶質薬物と濃度増大ポリマーとを別々であるが、同じ一般時間枠内に投与することができることを意味する。従って、非晶質薬物を、例えばそれ自体の剤形で投与し、これを別の剤形中にある濃度増大ポリマーとほぼ同じ時間に行う。別々に投与する場合、一般に、使用環境において2つが一緒に存在するように互いに60分の範囲内で、非晶質化合物Aおよび濃度増大ポリマーの両方を投与することが好ましい。同時に投与しない場合、濃度増大ポリマーを、化合物Aの非晶質形態の前に投与することが好ましい。
【0095】
別の実施態様において、化合物Aおよび濃度増大ポリマーを合わせて固体非晶質分散体を形成する。化合物Aおよび濃度増大ポリマーを含んでなる固体非晶質分散体は好ましく、それはこのような固体非晶質分散体では、しばしばin vitroおよびin vivo使用環境において溶解薬物の高い濃度を得ることができるためである。
【0096】
濃度増大ポリマーを含んでなる固体非晶質分散体は、化合物Aの用量および濃度増大ポリマーの有効性に応じて約99質量%までの化合物Aを含むことができる。非晶質分散体は、非常に高充填の化合物Aを含んで形成することができる。固体非晶質分散体が化合物Aおよび濃度増大ポリマーのみで構成される場合、固体非晶質分散体は、少なくとも約0.67の薬物対ポリマー質量の比率を有する。固体非晶質分散体は、化合物Aまたはその医薬上許容しうる塩または溶媒和物を少なくとも約30質量%、少なくとも約40質量%、少なくとも約50質量%、少なくとも約60質量%、少なくとも約70質量%、少なくとも約80質量%、少なくとも約90質量%、または少なくとも約95質量%含むことができる。マトリックス、例えばセルロース系ポリマーからなるマトリックス、例えばHPMCAS(ヒドロキシプロピルメチルセルロースアセテートスクシネート)、中に化合物Aを含んでなる固体非晶質分散体は、長期間貯蔵した後でも分散体が化合物Aの改善された溶解性能を提供し続けるという点で、長期間にわたって物理的に安定であるといえる。
【0097】
安定化剤
一実施態様において、組成物は、化合物Aの化学安定性を促進する安定化剤をさらに含む。安定化剤は、化合物Aが、例えば酸化分解プロセスを経て別の化学物質に分解する速度を低下させる。分解を防ぐために、本発明の組成物は、安定化剤を含むことができる。有用な安定化剤の1つの種類は、抗酸化剤である。分散体中に含まれうる典型的な抗酸化剤としては、ブチルヒドロキシトルエン(BHT)、ブチルヒドロキシアニソール(BHA)、プロピルガレート、ビタミンEおよびビタミンEスクシネートが含まれるが、これらに限定されない。このような抗酸化剤は、酸化を低下させるが、医薬上許容しうる量を超えない十分な量で存在することができる。典型的な量は、約0〜約10質量%の範囲である。
【0098】
有用に含まれる別の安定化剤は、塩基である。塩基の例としては、(限定されるわけではないが)水酸化物、例えば水酸化ナトリウム、水酸化カルシウム、水酸化アンモニウムおよびコリン水酸化物;炭酸水素塩、例えば(限定されるわけではないが)炭酸水素ナトリウム、炭酸水素カリウムおよび炭酸水素アンモニウム;炭酸塩、例えば(限定されるわけではないが)炭酸アンモニウム、炭酸カルシウムおよび炭酸ナトリウム;アミン、例えば(限定されるわけではないが)トリス(ヒドロキシメチル)アミノメタン、エタノールアミン、ジエタノールアミン、N−メチルグルカミン、グルコサミン、エチレンジアミン、N,N'−ジベンジルエチレンジアミン、N−ベンジル−2−フェネチルアミン、シクロヘキシルアミン、シクロペンチルアミン、ジエチルアミン、イソプロピルアミン、ジイソプロピルアミン、ドデシルアミンおよびトリエチルアミン;タンパク質、例えば(限定されるわけではないが)ゼラチン;アミノ酸、例えば(限定されるわけではないが)リシン、アルギニン、グアニン、グリシンおよびアデニン;ポリマーアミン、例えばポリアミノメタクリレート、例えばEudragit E;種々の酸の共役塩基、例えば(限定されるわけではないが)酢酸ナトリウム、酢酸カリウム、酢酸カルシウム、酢酸マグネシウム、酢酸アンモニウム、クエン酸カリウム、クエン酸カルシウム、クエン酸ナトリウム、クエン酸二ナトリウム、クエン酸三ナトリウム、安息香酸ナトリウム、安息香酸カリウム、安息香酸カルシウム、プロピオン酸ナトリウム、リン酸二ナトリウム、リン酸三ナトリウム、リン酸カルシウム、ナトリウムフェノキシド、硫酸ナトリウム、塩化アンモニウムおよび硫酸アンモニウム;EDTA(エチレンジアミン四酢酸)の塩、例えば(限定されるわけではないが)四ナトリウムEDTA;種々の酸性ポリマーの塩、例えばナトリウムスターチグリコレート、カルボキシルメチルセルロースナトリウムおよびナトリウムポリアクリル酸;およびN−メチルモルホリンが含まれる。好ましい塩基は、有機酸の共役塩基、例えば(限定されるわけではないが)酢酸ナトリウム、酢酸カリウム、酢酸カルシウム、酢酸マグネシウム、酢酸アンモニウム、枸櫞酸カリウム、クエン酸カルシウム、クエン酸ナトリウム、クエン酸二ナトリウム、クエン酸三ナトリウム、安息香酸ナトリウム、安息香酸カリウム、安息香酸カルシウムおよびプロピオン酸ナトリウムである。
【0099】
分散体ポリマーがHPMCASのような酸性である場合、固体非晶質分散体へ塩基を添加することは、特に好ましい。この実施態様では、塩基は、ポリマー上で酸性基のかなりの量を中和するのに十分な量で存在することができる。塩基は、酸性基の少なくとも約40%、酸性基の少なくとも約50%、または酸性基の少なくとも約90%を中和するのに十分な量で存在することができる。塩基は、酸性基よりも過剰に存在して本質的に酸性基の100%が中和されてもよい。
【0100】
酸性ポリマーの中和された形態が多価カチオン種、例えばCa2+、Mg2+、Al3+、Fe2+、Fe3+、またはジアミン、例えばエチレンジアミンを含む場合、カチオン種は、複数のポリマー鎖上で2つまたはそれ以上の中和された酸性部分と相互作用してポリマー鎖間でイオン架橋を生成することができる酸性ポリマーは、ポリマー1グラム当たり多価カチオン種のミリグラム当量数が、ポリマー1グラム当たり(ポリマーの)酸性部分のミリグラム当量数の少なくとも約5%、好ましくは少なくとも約10%である場合、「イオン架橋された」とみなすことができる。別法として、酸性ポリマーは、十分な多価カチオン種が存在して、中和された酸性ポリマーが、本質的に多価カチオン種を含んでいない同じポリマーよりも高いガラス転移温度(Tg)を有する場合、「イオン架橋された」とみなすことができる。このようなイオンによって架橋されたポリマーから製造され分散体中の易動度は、同じポリマーの酸性形態から形成された分散体と比較して特に低い。このようなイオン架橋されたポリマーは、塩基のカチオン性対イオンが二価であるいずれかの塩基を用いて酸性ポリマーを中和によって形成することができる。従って、水酸化マグネシウム、酢酸カルシウムまたはエチレンジアミンを酸性ポリマー、例えばセルロースのアセテートフタレートまたはヒドロキシプロピルメチルセルロースアセテートスクシネートに加えて、中和されてイオン架橋された酸性セルロース系ポリマーを形成することができる。このようなポリマーにおける低い易動度は、高いTg値またはより典型的にTg付近の熱容量増加規模における減少または場合によっては、分散体が示差熱分析を受けた時の明確なTgの欠如によって示すことができる。従って、ポリマーが本質的に完全に中和された場合、中和されたポリマーを示差熱分析にかける時には、Tgは明確ではない。このようなイオン架橋されたポリマーでは、非イオン的に架橋された中和された酸性ポリマーと比較して分散体中の化合物Aについて改善された物理的または化学物質安定性を得ることができる。
【0101】
非晶質化合物Aの製造
非晶質化合物Aおよび化合物Aの固体非晶質分散体は、当業者に知られているいずれかの慣用のプロセスに従って製造することができる。このようなプロセスとしては、機械的、熱的および溶媒プロセスがある。典型的な機械的プロセスは、ミル処理および押出;高温融合を含む溶融プロセス、溶媒変性された融合および溶融凝固プロセス;および非溶媒沈殿、噴霧コーティングおよび噴霧乾燥を含めた溶媒プロセスが含まれる。多くの場合、プロセスは、2つまたはそれ以上のプロセスタイプを組み合わせて分散体を形成することができる。例えば、押出プロセスを使用する場合、押出機を高められた温度で運転して機械的(剪断)および熱的(加熱する)手段を用いて分散体を形成することができる。典型的な方法の例は、以下の米国特許に開示されており、それらの適切な開示は、参照により本明細書に組み込まれている:米国特許第5,456,923号および同第5,939,099号、これらは押出プロセスによる分散体の形成を記載している;米国特許第5,340,591号および同第4,673,564号、これらはミル処理プロセスによる分散体の形成を記載している;および米国特許第5,707,646号および同第4,894,235号、これらは溶融凝固プロセスによる分散体の形成を記載している。
【0102】
一実施態様において、分散体は、熱プロセス、例えば押出プロセス、融合プロセス、または溶融凝固プロセスによって形成される。このような場合、マトリックスは、熱プロセスに使用するために適切となるように選ばれる。一般に、処理温度を化合物Aの熱分解を回避するためにできる限り低く維持することが望ましい。このように、マトリックス全体として、約200℃未満、より好ましくは約160℃未満、そして最も好ましくは約120℃未満の温度で流体になることが好ましい。
【0103】
熱プロセスのマトリックス成分として使用するのに適した典型的な物質としては、アルコール、例えばステアリルアルコールおよびセチルアルコール、有機酸およびその医薬上許容しうる塩、例えばステアリン酸、クエン酸およびリンゴ酸;糖、例えばグルコース、トレハロース、キシリトール、ソルビトールおよびマルチトール;脂肪酸エステル、例えばモノ、ジおよびトリ−グリセリド、グリセリルモノ、ジおよびトリステアレート、グリセリルモノ、ジおよびトリベヘネート、ソルビタンモノステアレート、サッカロースモノステアレート、グリセリル(パルミチン酸ステアリン酸)エステル、水素化された綿実油、ポリオキシエチレンソルビタン脂肪酸エステル;ワックス、例えば微結晶ワックス、パラフィンワックス、蜜蝋、合成蝋、キャスターワックスおよびカルナバワックス;アルキルスルフェート、例えばナトリウムラウリルスルフェート;およびポリマー、例えばポリエチレングリコール、ポリオキシエチレングリコール、ポリエチレンプロピレングリコールコポリマー、ポロキサマー、ポリエチレンオキシド、ポリビニルピロリジノン(別名ポリビニルピロリドンまたはポビドンまたはPVP)、ポリビニルアルコール、ポリエチレンビニルアルコールコポリマー、ポリビニルアルコールポリビニルアセテートコポリマー、カルボン酸官能化ポリメタクリレートおよびアミン官能化ポリメタクリレートが含まれる。熱処理によって形成される分散体中に単独で使用するのに適したものとして特定の物質について議論してきたが、物質のブレンドもまた適切であるといえる。例えば、水不溶性マトリックス成分、例えば微結晶ワックスを高い水溶性のマトリックス成分、例えばポロキサマーと混合して水分散性マトリックスを形成することができる。
【0104】
マトリックスは、加工温度を下げるために、マトリックスの1つ成分として可塑剤を含むことができる。典型的な可塑剤としては、鉱油、ペトロラタム、ラノリンアルコール、ポリエチレングリコール、ポリプロピレングリコール、ソルビトール、トリエタノールアミン、安息香酸ベンジル、セバシン酸ジブチル、ジエチルフタレート、グリセリルモノステアレート、トリアセチン、およびトリエチルシトレートが含まれる。使用する可塑剤の量は、別のマトリックス成分の融点および所望の処理温度に左右される。典型的に、可塑剤対マトリックスの比率は、0.01〜0.5、より典型的には0.05〜0.1である。また、水、アルコール、ケトン、等のような溶媒または膨潤剤を使用して処理温度を低下させ、組成物の処理性を改善することができる。
【0105】
1つの好ましい熱プロセスは、押出プロセスである。ここでは、化合物Aおよび1つまたはそれ以上のマトリックス成分を、可塑剤を添加してまたは添加せずに乾燥混合して、ブレンドを二軸スクリュー押出し装置に送る。二軸スクリュー押出し装置は、化合物Aまたはマトリックスの分解なしに分散体を形成するための熱および機械的エネルギー(例えば剪断)が十分であるように設計されている。処理温度は、マトリックス物質の融点に応じて約50℃〜約200℃で変化することができる。一般に、マトリックス成分の融点がより高いほど、処理温度はより高い。
【0106】
化合物Aがマトリックス中で高い溶解度を有する場合、分散体を形成するために必要な機械的エネルギーの量はより低い。このような場合、処理温度は、化合物Aの融解温度よりも低くてもよいが、化合物Aは融解したマトリックス中に溶解するため、少なくとも一部のマトリックス物質の融点よりは高い。
【0107】
化合物Aがマトリックス中で低い溶解度を有する場合、分散体を形成するためにより多量の機械的エネルギー量が必要となりうる。ここで、処理温度は、化合物Aおよび少なくともいくつかのマトリックス成分の融点よりも上である必要がありうる。融解した化合物Aをマトリックス成分と混合して分散体を形成するには、多量の機械的エネルギーが必要でありうる。典型的に、苛酷な条件に化合物Aを曝露するのを最小限にするために最も低い処理温度および満足な分散体を製造するための最も少ない量の機械的エネルギー(例えば剪断)を付与する押出機の設計を選択する。
【0108】
分散体を形成するための別の好ましい方法は「溶媒処理」であり、化合物Aの少なくとも一部および1つまたはそれ以上のマトリックス成分の少なくとも一部を共通の溶媒中に溶解することからなる。用語「溶媒」は広く使用され、溶媒の混合物が含まれる。ここで「共通の」は、化合物Aの少なくとも一部およびマトリックス物質を溶解する溶媒を意味し、これは化合物の混合物であってもよい。
【0109】
溶媒処理のためのマトリックス成分として使用するのに適した典型的な物質は、前に記載した濃度増大ポリマー;アルコール、例えばステアリルアルコールおよびセチルアルコール;有機酸およびそれらの医薬上許容しうる塩、例えばステアリン酸、クエン酸、フマル酸、酒石酸およびリンゴ酸;塩、例えば塩化ナトリウム、塩化カリウム、塩化リチウム、塩化カルシウム、塩化マグネシウム、硫酸ナトリウム、硫酸カリウム、炭酸ナトリウムおよび硫酸マグネシウム;アミノ酸、例えばアラニンおよびグリシン;糖、例えばグルコース、シュクロース、キシリトール、フルクトース、ラクトース、マンニトール、ソルビトールおよびマルチトール;脂肪酸エステル、例えばグリセリル(モノ−およびジ−)ステアレート、トリグリセリド、水素化された綿実油、ソルビタンモノステアレート、サッカロースモノステアレート、グリセリル(パルミチン酸ステアリン酸)エステル、ポリオキシエチレンソルビタン脂肪酸エステル;ワックス、例えば微結晶ワックス、パラフィンワックス、蜜蝋、合成蝋、キャスターワックスおよびカルナバワックス;アルキルスルフェート、例えばラウリル硫酸ナトリウムおよびラウリル硫酸マグネシウム;リン脂質、例えばレシチン;タンパク質、例えばゼラチンおよびアルブミン;およびポリマー例えばポリエチレングリコール、ポリオキシエチレングリコール、ポリエチレンオキシド、キサンタンガム、カラゲナン、キトサン、ポリデキストロース、デキストリンおよびデンプンが含まれる。溶媒処理によって形成される分散体に単独で使用するのに適したものとして特定の物質を議論してきたが、物質のブレンドも適切でありうる。
【0110】
化合物Aの少なくとも一部およびマトリックスを溶解した後、溶媒は、蒸発によってまたは非溶媒と混合することによって除去される。典型的なプロセスは、噴霧乾燥、噴霧コーティング(パン−コーティング、流動層コーティング、等)、ならびに化合物Aおよびマトリックスの溶液をCO2、ヘキサン、ヘプタン、適当なpHの水またはいくつかの他の非溶媒と急速に混合することによる沈殿である。溶媒を除去して実質的に均質である固体分散体を得ることが好ましい。この目的を達成するには、例えば、溶液を霧化して化合物Aおよびマトリックスを急速に凝固させるプロセスにおいて溶液から急速に溶媒を除去することが一般に望ましい。
【0111】
化合物Aおよびマトリックスが溶解した後、溶媒を、蒸発によってまたは非溶媒と混合することによって急速に除去する。典型的なプロセスは、噴霧乾燥、噴霧コーティング(パンコーティング、流動層コーティング、等)ならびにマトリックスおよび化合物Aの溶液をCO2、水またはいくつかの他の非溶媒と急速に混合することによる沈殿である。
【0112】
固体非晶質分散体の場合、分散体は相分離していてもよいし(これは、化合物Aおよびマトリックスが上記分散体内でそれぞれ別々のドメイン中にあることを意味する)、または相互に均質に分散して単一相を形成してもよいし、またはこれらのいずれかの組み合わせまたは中間のままのこれらの状態を形成してもよい。溶媒を除去して、実質的に均質の固体非晶質分散体を形成することが好ましい。このような分散体において、化合物Aおよびマトリックスは、相互にできるだけ均質に分散され、そしてマトリックス中に分散された化合物Aの固溶体とみなすことができ、その際、固体非晶質分散体は、熱力学的に安定であり、これはマトリックス中の化合物Aの濃度がその平衡値またはそれ未満であるか、またはマトリックス中の化合物Aの濃度がその平衡値より上にある過飽和固溶体であると考えることができる。
【0113】
溶媒は、噴霧乾燥によって除去することができる。用語「噴霧乾燥」は、慣用的に使用され、大まかに言えば、液滴から溶媒を蒸発させる強い推進力を有する噴霧乾燥装置中で液体の混合物を小さな液滴に分割(噴霧化)し、混合物から溶媒を急速に除去するプロセスのことである。噴霧乾燥プロセスおよび噴霧乾燥装置は、一般にPerry's Chemical Engineers' Handbook, pages 20-54 to 20-57 (Sixth Edition 1984)に記載されている。噴霧乾燥プロセスおよび装置のより詳細は、Marshall,“Atomization and Spray-Drying,”50 Chem. Eng. Prog. Monogr. Series 2 (1954)および Masters, Spray Drying Handbook (Fourth Edition 1985)によって概説されている。溶媒蒸発のための強い推進力は、一般に、噴霧乾燥装置中、乾燥する液滴の温度で溶媒の分圧を溶媒の蒸気圧より下に維持することによって得られる。これは、(1)噴霧乾燥装置中で圧力を部分的な真空(例えば0.01〜0.50気圧)に維持する;または(2)液滴を暖かい乾燥ガスと混合する;または(3)(1)および(2)の両方によって実施される。さらに、溶媒蒸発に必要な熱の少なくとも一部は、噴霧溶液を加熱することによって得ることができる。
【0114】
噴霧乾燥に適した溶媒は、化合物Aおよびマトリックスが相互に可溶性であるすべての化合物であるといえる。また、溶媒は揮発性であり、150℃またはそれ未満の沸点を有することが好ましい。さらに、溶媒は、比較的低い毒性を有し、そして固体非晶質分散体からThe International Committee on Harmonization(ICH)ガイドラインに従って許容されうるまで除去しなければならない。このレベルまで溶媒を除去するには、トレー乾燥のような後処理工程が必要となりうる。適切な溶媒としては、限定されるわけではないが、アルコール、例えばメタノール、エタノール、n−プロパノール、イソ−プロパノールおよびブタノール;ケトン、例えばアセトン、メチルエチルケトンおよびメチルイソ−ブチルケトン;エステル、例えば酢酸エチルおよびプロピル酢酸;および種々の他の溶媒、例えばアセトニトリル、塩化メチレン、トルエンおよび1,1,1−トリクロロエタンが含まれる。また、ジメチルアセトアミドまたはジメチルスルホキシドのようなより低い揮発性の溶媒を使用することもできる。また、溶媒の混合物、例えば50%メタノールおよび50%アセトンを使用することができ、ポリマーおよび化合物Aが、噴霧乾燥プロセスを実施できるくらい十分に可溶性ならば、水との混合物であってもよい。
【0115】
噴霧溶液に水を添加すると、生成した非晶質化合物Aの化学安定性を改善することができる。一般に、水は、濃度増大ポリマーおよび存在する他の溶媒に応じて溶媒の約30質量%までの量で存在することができる。一実施態様において、溶媒は約80/20(質量
/質量)の比率でメタノールおよび水を含む。
【0116】
生成した非晶質化合物Aの化学安定性を改善するための別の方法は、窒素のような不活性ガスを噴霧溶液に通してバブリングすることによって噴霧溶液を酸素パージすることである。
【0117】
固体非晶質分散体が塩基を含む場合、塩基が化合物Aを分解しないように注意しなければならない。化合物Aは、強塩基の存在下で加水分解を受けやすい。従って、酸性ポリマーを中和するために塩基を使用する場合、塩基が最初にポリマーと反応するように塩基および酸性ポリマーを最初に溶媒中で合わせることが好ましい。次いで、化合物Aを加えて噴霧溶液を形成する。
【0118】
さらに、塩基を噴霧溶液に加えて酸性ポリマーを中和する場合、噴霧溶液に水を加えて噴霧溶液中のポリマーに十分な溶解度を与える必要がある。噴霧溶液は、水を30質量%まで含むことができる。例えば、固体10質量%(化合物A90%、HPMCAS8%および酢酸カルシウム2%からなる)を有する噴霧溶液を得るには、噴霧溶液は、メタノール80%および水20%(質量)を含むことができる。
【0119】
噴霧溶液中の化合物Aおよびマトリックスの量は、噴霧溶液中のそれぞれの溶解度および生成する固体非晶質分散体中の化合物A対マトリックスの所望の比率に左右される。噴霧溶液は、溶解物質を少なくとも約1質量%、より好ましくは少なくとも約3質量%そしてさらにより好ましくは少なくとも約10質量%含むことが好ましい。化合物A対マトリックスの比率は、0.01〜100の範囲でありうる。化合物A対マトリックスの比率は、少なくとも約0.66〜約49、より好ましくは約3〜約19そしてさらにより好ましくは約5から約15であることが好ましい。
【0120】
溶媒を有する供給物は、様々な条件下で噴霧乾燥させることができるが、それでも許容しうる性質を有する非晶質薬物または固体非晶質分散体が得られる。例えば、種々のタイプのノズルを使用して噴霧溶液を霧化することができ、それによって小さな液滴の集まりとして噴霧乾燥室中に噴霧溶液が導入される。形成された液滴が、噴霧乾燥室壁に固着しないように、すなわち噴霧乾燥室壁をコーティングしないように十分に小さく十分に乾燥(溶媒蒸発により)すれば、本質的にあらゆるタイプのノズルを使用して溶液を噴霧することができる。
【0121】
最大液滴サイズは、噴霧乾燥器内でサイズ、形状およびフローパターンの関数として、広く変化するが、一般に、液滴は、ノズルから出る時に、直径約500μm未満でなければならない。固体非晶質分散体を形成するために使用することができるノズルのタイプの例としては、二流体ノズル、噴水型ノズル、フラットファン型ノズル、加圧ノズルおよびロータリーアトマイザーがある。好ましい実施態様において、加圧ノズルを使用することができ、これは、2003年1月24日に出願され、同一出願人による係属中の米国特許出願第10/351,568号に詳細に開示されており、2002年2月1日に出願された米国仮出願第60/353,986号に対して優先権を主張し、この開示は参照により本明細書に組み込まれている。
【0122】
噴霧溶液は、広範囲にわたる温度および流速で噴霧ノズルまたはノズルに供給することができる。一般に、噴霧溶液温度は、溶媒の凝固点のすぐ上から周囲圧力の沸点より約20℃上(溶液に加圧することによって)までそして場合によってはさらに高いいずれかの範囲であることができる。噴霧ノズルへの噴霧溶液の流速は、ノズルのタイプ、噴霧乾燥機のサイズおよび噴霧乾燥条件、例えば乾燥ガスの入口温度および流速に応じて広い範囲で変化することができる。一般に、噴霧乾燥プロセスにおける噴霧溶液から溶媒を蒸発させるエネルギーは、主として乾燥ガスから生じる。
【0123】
乾燥ガスは、原則として、本質的にすべてのガスであることができるが、安全性の理由のため、そして固体非晶質分散体中で化合物Aまたは他の物質の望ましくない酸化を最小限にするため、窒素のような不活性ガス、窒素の豊富な空気またはアルゴンが使用される。乾燥ガスは、典型的に約60℃〜約300℃、そして好ましくは約80℃〜約240℃の温度で乾燥室に導入される。噴霧乾燥プロセス中およびその後に化合物Aの化学分解を最小限にするために、化合物Aを高い温度に曝露するのを制限することが好ましい。例えば、噴霧溶液は化合物A、HPMCAS、メタノールおよび水を含み、NIRO PSD-1噴霧乾燥機を用いて噴霧する場合、入口ガス温度は150℃およびより好ましくは130℃またはそれ未満でありうる。
【0124】
液滴の比表面積が大きく、溶媒蒸発の推進力が大きいと、液滴の凝固時間が早くなる。凝固時間は、約20秒未満、好ましくは約10秒未満、そしてさらに好ましくは1秒未満でなければならない。この急速な凝固は、多くの場合、粒子が、薬物Aの豊富な相およびポリマーの豊富な相に分離しないで均一で均質な分散体を維持するのに重要である。好ましい実施態様において、噴霧乾燥器の高さおよび容積は、液滴が、噴霧乾燥機の内側面に衝突する前に乾燥するのに十分な時間が得られるように調整され、これは同一出願人による米国特許第6,763,607号に詳述されており、参照により本明細書に組み込まれている。上記したように、濃度および生物学的利用能における大きな増大を得るには多くの場合、できるだけ均質な分散体を得る必要がある。
【0125】
凝固した後、固体粉末は、典型的に約5〜60秒間、噴霧乾燥室中にとどまり、さらに固体粉末から溶媒を蒸発させる。固体分散体の最終的な溶媒含量は、乾燥機を出る時には低くなければならず、その理由は、これによって固体非晶質分散体中での化合物A分子の易動度が低減され、そのため安定性が改善されるためである。一般に、固体非晶質分散体の溶媒含量は、噴霧乾燥室から出る時には、10質量%未満、そして好ましくは2質量%未満でなければならない。
【0126】
形成した後、適切な乾燥プロセス、例えばトレー乾燥、真空乾燥、流動層乾燥、高周波乾燥、ベルト乾燥、回転乾燥および当分野で知られている他の乾燥プロセスを用いて固体非晶質分散体を乾燥させて残留溶媒を除去することができる。固体非晶質分散体は、熱い乾燥状態への曝露を最小限にする条件下で乾燥させることが好ましい。好ましくは、温度は50℃未満で、そしてより好ましくは40℃未満である。相対湿度は、相対湿度25%〜75%の間で維持するのが好ましい。好ましい二次的な乾燥方法としては、真空乾燥または周囲条件下のトレー乾燥が含まれる。乾燥の際に化学分解を最小限にするために、乾燥は窒素のような不活性ガス下で行うことができまたは真空下で行うことができる。
【0127】
固体非晶質分散体は、通常、小さな粒子の形態である。粒子の平均サイズは、直径500μm未満、直径100μm未満、直径50μm未満または直径25μm未満であることができる。固体非晶質分散体を噴霧乾燥によって形成する場合、生成した分散体は、このような小さな粒子の形態である。固体非晶質分散体を溶融凝固または押出プロセスのような別の方法によって形成する場合、生成した分散体を、ふるいにかけ、粉砕し、ミル処理し、または別のやり方で処理して多くの小さな粒子を得ることができる。
【0128】
処理を容易にするには、乾燥した粒子は、一定の密度およびサイズの特徴を有することになる。一実施態様において、生成した固体非晶質分散体粒子は、噴霧乾燥によって形成され、嵩比容約4cc/gまたはそれ未満、そしてより好ましくは約3.5cc/gまたはそれ未満を有することができる。粒子は、タップ比容約3cc/gまたはそれ未満、そしてより好ましくは約2cc/gまたはそれ未満を有することができる。粒子は、Hausner比約3またはそれ未満、そしてより好ましくは約2またはそれ未満を有する。粒子は平均粒子直径約150μmまで、そしてより好ましくは約1〜約25μmを有することができる。粒子は、スパン3またはそれ未満、そしてより好ましくは約2.5またはそれ未満を有することができる。本明細書に使用されるように、「スパン」は、
【数1】
として定義され、ここで、D10は、直径が等しいまたはより小さい粒子を含んでなる、総容積の10%を占める粒子の直径に対応する直径であり、D50は、直径が等しいまたはより小さい粒子を含んでなる、総容積の50%を占める粒子の直径に相当する直径であり、そしてD90は、直径が等しいまたはより小さい直径の粒子を含んでなる、総容積の90%を占める粒子の直径に対応する直径である。
【0129】
分散体を形成するのに適当であるとして前記されたプロセスは、非晶質化合物Aの形成に使用することができる。特に、非晶質化合物Aは、化合物Aを溶媒、例えばアセトンまたはメタノール中に溶解し、そしてマトリックス中の化合物Aの分散体を製造するために上記したのと同じ方法で噴霧乾燥することによって製造することができる。また、非晶質化合物Aは、例えば、結晶質化合物Aを溶融凝固装置に供給することによって製造することができ、これは、米国特許第5,183,493号または第5,549,917号に開示されており、融解した化合物Aの液滴を形成し、次いで、冷却ガスによって冷却し、直径約1〜約500μmそして好ましくは直径10〜300μmの範囲の化合物Aの非晶質粒子を形成する。
【0130】
固体非晶質分散体または非晶質薬物は、封入パッケージ中に保存して湿気(水蒸気)および/または酸素への曝露を減らすことができる。湿気または酸素との接触を減らすための典型的な方法としては、水蒸気および/または酸素に対して実質的に不透過性である保護パッキング、例えばホイルポーチまたはホイルブリスターパック、または組成物のパッケージング中に乾燥剤または酸素吸収剤、例えば酸素吸着剤パケットを封入することが含まれる。パッケージングは、湿気を減らすための乾燥剤、酸素吸収剤、または両方を含むことができる。典型的な乾燥剤としては、アルミノシリケート(Sorb-it(R),Sud-Chemieから入手可能)および無水硫酸カルシウムが含まれ、そして典型的な酸素吸収剤としては、米国特許第6,558,571号に開示された酸素吸収組成物およびMultisorb Technologies, Inc.から入手可能な商品名Fresh PaxTMの下で販売されているパケットおよび細片が含まれる。本発明は、(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミド、またはその医薬上許容しうる塩または溶媒和物を含んでなる医薬組成物を含む医薬パッケージであって、酸素吸収剤をさらに含んでなる前記パッケージを提供する。さらなる態様において、存在する酸素ガスの量は、前記パッケージ中に存在するガスの総量の約5体積%未満であるこのようなパッケージが提供される。さらなる態様において、存在する酸素ガスの量は、前記パッケージ中に存在するガスの総量の約2%未満、または約1%未満、または約0.5%未満、または約0.25%未満、または約0.1%未満、または約0.01%未満である。
【0131】
濃度増大
好ましい実施態様において、本発明の組成物は、有用な水性環境に投薬した時に、濃度増大が得られ、これは、少なくとも以下の状態の1つを満たすことを意味する。第1の状態は、使用環境中において本発明の組成物が、化合物Aの結晶質形態Iの等量からなる対照組成物と比較して化合物Aの最大溶解濃度(MDC)を高める状態である。すなわち、組成物がいったん使用環境に導入されたら、組成物は、対照組成物と比較して化合物Aの水性濃度を高める。対照組成物は、可溶化剤または化合物Aの結晶質形態Iの溶解度に物質的に影響を及ぼす他の成分を含まず、そして化合物Aは、対照組成物中で固体状態であることを理解すべきである。対照組成物は、化合物Aの慣用的に分散されてない結晶質形態I単独である。本発明の組成物では、水溶液中の化合物AのMDCが、対照組成物によって得られるものの少なくとも1.25倍または少なくとも2倍または少なくとも3倍であ
ることが好ましい。場合によっては、本発明の組成物によって得られる化合物AのMDCは、対照組成物によって得られる平衡濃度の少なくとも5倍または少なくとも10倍である。
【0132】
第2の状態は、本発明の組成物が、ポリマーを有しない化合物Aの結晶質形態Iの等量を含んでなる対照組成物と比較して使用環境中で化合物Aの濃度対時間曲線より下の溶解面積(AUC)を増加させる状態である。AUCの計算は、医薬分野においてよく知られている方法であって、例えば、Welling,“Pharmacokinetics Processes and Mathematics,
”ACS Monograph 185 (1986)に記載されている。より詳しくは、使用環境において、本発明の組成物では、使用環境に導入後の約0〜約270分の間のいずれか90分の期間のAUCが、上記の対照組成物の少なくとも約1.25倍である。組成物によって得られるAUCは、対照組成物の少なくとも1.25倍または少なくとも約2倍または少なくとも約3倍であることが好ましい。本発明のいくつかの組成物では、上記対照組成物の少なくとも約5倍または少なくとも約10倍のAUC値を得ることができる。
【0133】
本発明の一実施態様において、組成物は、非晶質化合物Aおよび濃度増大ポリマーを含まない非晶質化合物Aを含んでなる第2の対照組成物と比較して組成物が濃度増大をもたらすのに十分な量の濃度増大ポリマーを含んでなる。これらの組成物において、ポリマーは水溶液中で化合物AのMDCまたはAUCの少なくとも1つを、第2の対照組成物と比較して少なくとも1.25倍または少なくとも約2倍または少なくとも約3倍高める。
【0134】
前述のように、「使用環境」とは、in vivo環境、例えば動物、特にヒトの消化管、または試験溶液のin vitro環境、例えばリン酸緩衝生理食塩水(PBS)溶液またはModel Fasted Duodenal(MFD)溶液のいずれかであることができる。本発明者らは、in vitro溶解試験は、in vivo性質の良好な予測因子であり、従って、組成物はin vitroおよびin vivo使用環境のいずれかまたは両方において濃度増大をもたらす場合、それらは本発明の範囲内にあることを見出した。
【0135】
本発明の組成物では、in vitro溶解試験において溶解した化合物Aの高められた濃度が得られる。in vitro溶出試験においてMFD溶液中またはPBS溶液中の高められた薬物濃度は、in vivo性能および生物学的利用能の良好な指標であることがわかった。適当なPBS溶液は、20mM Na2HPO4、47mM KH2PO4、87mM NaClおよび0.2mM KClを含み、NaOHでpH6.5に調整された水溶液である。適当なMFD溶液は、7.3mMナトリウムタウロコール酸および1.4mMの1−パルミトイル−2−オレイル−sn−グリセロ−3−ホスホコリンがさらに存在する同様のPBS溶液である。特に、本発明の方法によって形成された組成物はMFDまたはPBS溶液に加えて撹拌して溶解を促進することによって溶解試験することができる。
【0136】
水溶液中で高められた化合物Aの濃度を評価するin vitro試験は、(1)十分な量の対照組成物、典型的には分散されてない化合物A単独を、in vitro試験媒体、例えばMFDまたはPBS溶液に撹拌しながら加えて、化合物Aの平衡濃度にし;(2)別の試験において、全ての化合物Aが溶解した場合、化合物Aの理論濃度が、化合物Aの平衡濃度を少なくとも2倍または少なくとも10倍超えるように、十分な量の試験組成物(例えば、化合物Aおよびポリマーを含んでなる組成物)を同じ試験媒体中に撹拌しながら加え;そして(3)試験媒体中の試験組成物の測定したMDCおよび/または水性AUCを平衡濃度、および/または対照組成物の水性AUCと比較する:ことによって実施することができる。このような溶解試験を実施する際、使用した試験組成物または対照組成物の量は、化合物Aの全てが溶解した場合、化合物Aの濃度が平衡濃度の少なくとも2倍、少なくとも10倍、そしてもっとも好ましくは少なくとも100倍となるような量である。
【0137】
溶解した化合物Aの濃度は、典型的には、MDCを確認することができるように、試験媒体の試料を採取し、試験媒体中の化合物Aの濃度対時間をプロットして時間の関数として測定する。MDCは、試験の持続期間にわたって測定された溶解化合物Aの最大値である。水性AUCは、水性使用環境中へ組成物を導入した時間(時間がゼロの時)および使用環境に導入後270分(時間が270分の時)の間のいずれか90分の期間にわたって濃度対時間曲線を積分することによって算出される。典型的には、組成物が急速に、つまり約30分未満にそのMDCに到達した時、AUCを算出するために使用する時間的間隔は、時間0から時間90分までである。しかしながら、上記のいずれか90分の期間にわたる組成物のAUCが本発明の基準を満たす場合、形成された組成物は、本発明の範囲内であるとみなす。
【0138】
測定を誤る大きな薬物微粒子を取り除くため、試験溶液を濾過または遠心分離する。「溶解した薬物」は、典型的に0.45μmシリンジフィルタを通過する物質、またはそれとは別に遠心分離後の上澄み中に残る物質のいずれかである。濾過は、商標TITAN(R)の下でScientific Resourcesによって販売された13mm、0.45μmポリビニリジンジフルオリドシリンジフィルタを用いて実施することができる。遠心分離は、典型的にポリプロピレンマイクロ遠心分離管中、13,000Gで60秒間遠心分離することによって実施される。他の同様の濾過または遠心分離方法を使用することができ、有用な結果が得られる。例えば、別のタイプのミクロフィルターを使用すると、上記のフィルタで得るよりいくらか高いまたは低い値(±10〜40%)を得ることができるが、好ましい分散体の確認は依然として可能である。「溶解した薬物」のこの定義は、単一の溶媒和された薬物分子だけでなく、また、広範囲にわたる種類、例えばサブミクロンの寸法を有するポリマー/薬物集合体、例えば薬物凝集体、ポリマーおよび薬物の混合物の凝集体、ミセル、ポリマーミセル、コロイド粒子またはナノクリスタル、ポリマー/薬物複合体およびその他、明記された溶解試験において濾液または上澄み中に存在するこのような薬物を含む種類を包含することを認識すべきである。
【0139】
さらにまた、本発明の組成物は、絶食状態のヒトまたは他の哺乳動物に経口的に投薬した時に、等量の化合物の結晶質形態I単独からなる対照組成物によって得られるCmaxの少なくとも約1.25倍、または少なくとも約2倍、または少なくとも約3倍、または少なくとも約5倍、または少なくとも約10倍のCmaxが得られる。
【0140】
別法として、組成物では、絶食状態のヒトまたは他の哺乳動物に経口的に投薬した時に、血液(血清または血漿)中の化合物A濃度におけるAUCが、等量の化合物Aの結晶質形態Iからなる対照組成物を、追加ポリマーなしに単独で投薬した時に観察されるものの少なくとも約1.25倍、または少なくとも約2倍、または少なくとも約3倍、または少なくとも約5倍、または少なくとも約10倍得られる。このような組成物は、対照組成物の約1.25倍〜約10倍の相対的な生物学的利用能を有するといえることに留意する。
【0141】
組成物中の化合物Aの相対的な生物学的利用能は、このような測定を行うための慣用の方法を用いて動物またはヒトにおいてin vivo試験することができる。in vivo試験、例えば交差試験法は、化合物Aおよびマトリックスの組成物では、上記対照組成物と比較して高められた相対的な生物学的利用能が得られるかどうかを測定するのに使用することができる。in vivo交差試験法では、化合物Aおよびマトリックスの固体非晶質分散体の試験組成物を試験の被験者のグループの半分に投薬し、そして適当な休薬期間(例えば1週間)の後、同じ被験者に試験組成物として等量の化合物Aの結晶質形態Iからなる対照組成物(しかし、マトリックスは存在しない)を投薬する。グループのもう半分には、最初に対照組成物、続いて試験組成物を投薬した。相対的な生物学的利用能は、試験グループについて測定した血液(血清または血漿)中の濃度対時間曲線下の面積(AUC)を、対照組成物によって得られた血液中のAUCによって割ったものとして測定される。この試験/対照の比率は、それぞれの被験者について測定し、比率を本研究における全ての被験者で平均することが好ましい。AUCのin vivo測定は、横座標(x軸)に沿った時間に対して縦座標(y軸)に沿って化合物Aの血清または血漿中濃度をプロットすることによって行うことができる。投薬を促進するため、投薬ビヒクルを用いて用量を投与することができる。投薬ビヒクルは、好ましくは水であり、さらに試験または対照組成物を懸濁するための物質を含むことができるが、但し、これらの物質はin vivoで組成物を溶解せず、または化合物Aの溶解性を変化させない。
【0142】
剤形
組成物は、経口、鼻、直腸、膣、皮下、静脈内および肺を含めた様々な経路によって供給することができるが、これらに限定されない。一般に、経口経路が好ましい。
【0143】
また、組成物は、薬物を投与するための様々な剤形で使用することができる。典型的な剤形としては、ドライであるかまたは水もしくは他の液体を添加して再構成してペースト、スラリー、懸濁液または溶液を形成して経口的に摂取することができる散剤または顆粒剤;錠剤;カプセル剤;多粒剤;および丸剤である。種々の添加剤を本発明の組成物と共に混合、粉砕または造粒して上記の剤形に適した物質を形成することができる。
【0144】
本発明の組成物は、種々の形態で処方することができ、例えば液体ビヒクル中の粒子の懸濁液として供給される。このような懸濁液は、製造時に液体またはペーストとして処方することができ、またはドライ粉末として処方してあとで経口投与の前に、液体、典型的に水を加えることができる。懸濁液中に構成されるこのような粉末は、しばしばサシェまたは構成用経口粉末(OPC)製剤と称する。このような剤形は、いずれかの知られている方法によって処方および再構成することができる。最も単純なアプローチは、単に水を加えて撹拌することによって再構成される乾燥粉末として剤形を処方することである。別法として、剤形は、液体としておよび合わせて撹拌して経口懸濁液を形成する乾燥粉末として処方することができる。さらに別の実施態様において、剤形は、2種の粉末として処方することができ、これは、最初に1つの粉末に水を加え、溶液を形成し、これに第2の粉末を撹拌しながら合わせて懸濁液を形成することによって再構成される。
【0145】
さらに、本発明の組成物は、HIVウイルスによる感染症、AIDS、AIDS関連症候群(ARC)、またはHIVウイルスによる感染症と関連がある他のいずれかの疾患または状態にかかっている哺乳動物、例えばヒトを治療するためのさらなる薬剤(1種またはそれ以上)と組み合わせて投与することができる。本発明の組成物と組み合わせて使用することができる薬剤としては、HIVプロテアーゼ阻害剤として有用なもの、HIV逆転写酵素阻害剤、非ヌクレオシドHIV逆転写酵素阻害剤、HIVインテグラーゼの阻害剤、CCR5阻害剤、HIV融合阻害剤、免疫調節物質として有用な化合物、未知のメカニズムによってHIVウイルスを阻害する化合物、ヘルペスウイルスの治療に有用な化合物、抗感染薬として有用な化合物、および後述する他のものが含まれるが、これらに限定されない。
【0146】
本発明の組成物と組み合わせて使用することができるHIVプロテアーゼ阻害剤として有用な化合物としては、141W94(アンプレナビル)、CGP−73547、CGP−61755、DMP−450、ネルフィナビル、リトナビル、サキナビル(インビラーゼ)、ロピナビル、TMC−126、アタザナビル、パリナビル、GS−3333、KNI−413、KNI−272、LG−71350、CGP−61755、PD173606、PD177298、PD178390、PD178392、U−140690、ABT−378、DMP−450、AG−1776、MK−944、VX−478、インジナビル、チプラナビル、TMC−114、DPC−681、DPC−684、フォサンプレナビルカルシウム(レキシバ)、WO 03053435に開示されたベンゼンスルホンアミド誘導体、R−944、Ro−03−34649、VX−385、GS−224338、OPT−TL3、PL−100、SM−309515、AG−148、DG−35−VIII、DMP−850、GW−5950X、KNI−1039、L−756423、LB−71262、LP−130、RS−344、SE−063、UIC−94−003、Vb−19038、A−77003、BMS−182193、BMS−186318、SM−309515、JE−2147、GS−9005が含まれるが、これらに限定されない。
【0147】
本発明の組成物と組み合わせて使用することができるHIV逆転写酵素の阻害剤として有用な化合物には、アバカビル、FTC、GS−840、ラミブジン、アデホビルジピボキシル、ベータ−フルオロddA、ザルシタビン、ジダノシン、スタブジン、ジドブジン、テノホビル、アムドキソビル(amdoxovir)、SPD−754、SPD−756、ラシビール、ラベルセット(DPC−817)、MIV−210(FLG)、ベータ−L−Fd4C(ACH−126443)、MIV−310(アロブジン、FLT)、dOTC、DAPD、エンテカビル、GS−7340、エムトリシタビン、アロブジンが含まれるが、これらに限定されない。
【0148】
HIV逆転写酵素の非ヌクレオシドの阻害剤として有用な化合物には、エファビレンツ、HBY−097、ネビラピン、TMC−120(ダピビリン)、TMC−125、エトラビリン、デラビルジン、DPC−083、DPC−961、TMC−120、カプラビリン、GW−678248、GW−695634、カラノリド、およびWO 03062238に開示された三環式ピリミジノン誘導体が含まれるが、これらに限定されない。
【0149】
本発明の組成物と組み合わせて使用することができるCCR5阻害剤として有用な化合物には、TAK−779、SC−351125、SCH−D、UK−427857、PRO−140およびGW−873140(Ono−4128,AK−602)が含まれるが、これらに限定されない。
【0150】
本発明の組成物と組み合わせて使用することができるHIVインテグラーゼ酵素の阻害剤として有用な化合物には、GW−810781(WO 03062204に開示された1,5−ナフチリジン−3−カルボキサミド誘導体)、WO 03047564に開示された化合物、WO 03049690に開示された化合物、およびWO 03035076に開示された5−ヒドロキシピリミジン−4−カルボキサミド誘導体が含まれるが、これらに限定されない。
【0151】
本発明の組成物と組み合わせて使用することができるHIV治療のための融合阻害剤としては、エンフビルチド(T−20)、T−1249、AMD−3100、およびJP2003171381に開示された縮合三環式化合物が含まれるが、これらに限定されない。
【0152】
本発明の組成物と組み合わせて使用することができるHIVの有用な阻害剤である他の化合物としては、可溶性のCD4、TNX−355、PRO−542、BMS−806、フマル酸テノホビルジソプロキシル、およびJP2003119137に開示された化合物が含まれるが、これらに限定されない。
【0153】
本発明の組成物と組み合わせて使用することができる、HIV以外のウイルス感染症の治療または処置に有用な化合物としては、アシクロビル、ホミビルセン、ペンシクロビル、HPMPC、オキセタノシンG、AL−721、シドホビル、サイトメガロウィルス免疫グロビン、サイトベン、ホミブガンシクロビル(fomivganciclovir)、ファムシクロビル、フォスカーネットナトリウム、Isis2922、KNI−272、バラシクロビル、ビラゾールリバビリン、バルガンシクロビル、ME−609、PCL−016が含まれるが、これらに限定されない。
【0154】
免疫調節物質として作用し、そして本発明の組成物と組み合わせて使用することができる化合物としては、AD−439、AD−519、アルファインターフェロン、AS−101、ブロピリミン、アセマンナン、CL246,738、EL10、FP−21399、ガンマインターフェロン、顆粒球マクロファージコロニー刺激因子、IL−2、静脈内免疫グロブリン、IMREG−1、IMREG−2、イムチオールジエチルジチオカルバメート、アルファ−2−インターフェロン、メチオニン−エンケファリン、MTP−PE、顆粒球コロニー刺激因子、レムン(remune)、rCD4、組換え型可溶性のヒトCD4、インターフェロンアルファ−2、SK&F106528、可溶性T4イモペンチン、腫瘍壊死因子(TNF)、ツカレソール、組換え型ヒトインターフェロンベータ、およびインターフェロンアルファn−3が含まれるが、これらに限定されない。
【0155】
本発明の組成物と組み合わせて使用することができる抗感染薬としては、アトバクオン、アジスロマイシン、クラリスロマイシン、トリメトプリム、トロバフロキサシン、ピリメタミン、ダウノルビシン、プリマキンと共にクリンダマイシン、フルコナゾール、パスチル、オルニジル、エフロルニチンペンタミジン、リファブチン、スピラマイシン、イトラコナゾール−R51211、トリメトレキサート、ダウノルビシン、組換え型ヒトエリスロポエチン、組換え型ヒト成長ホルモン、メゲストロールアセテート、テストロン、および全ての腸内栄養剤が含まれるが、これらに限定されない。
【0156】
本発明の組成物と組み合わせて使用することができる抗真菌剤としては、アニデュラファンギン、C31G、カスポファンギン、DB−289、フルコナゾール、イトラコナゾール、ケトコナゾール、ミカファンギン、ポサコナゾールおよびボリコナゾールが含まれるが、これらに限定されない。
【0157】
本発明の組成物と組み合わせて使用することができる他の化合物としては、アクマンナン、アンサマイシン、LM 427、AR177、BMS−232623、BMS−234475、CI−1012、硫酸カードラン、硫酸デキストラン、STOCRINE EL10、ヒペリシン、ロブカビル、ノバプレン、ペプチドTオクターブペプチド配列(peptide T octabpeptide sequence)、トリナトリウムホスホノホルメート、プロブコール、およびRBC−CD4が含まれるが、これらに限定されない。
【0158】
さらに、本発明の組成物は、カポジ肉腫のような状態を治療するため抗増殖剤と組み合わせて使用することができる。このような薬剤としては、メタロマトリックスプロテアーゼの阻害剤、A−007、ビバシズマブ、BMS−275291、ハロフギノン、インターロイキン−12、リタキシマブ、パクリタキセル、ポルフィマーナトリウム、レビマスタットおよびCOL−3が含まれるが、これらに限定されない。
【0159】
さらなる薬剤(1種またはそれ以上)の特定の選択は、治療する哺乳動物の状態、治療する特定の状態、さらなる薬剤(1種またはそれ以上)の特徴、および哺乳動物を治療するために使用されるいずれかのさらなる化合物の特徴を含めた多くの因子に左右されるが、これらに限定されない。さらなる薬剤(1種またはそれ以上)の特定の選択は、当業者の知識の範囲内である。
【0160】
本発明の組成物は、HIVウイルスによる感染症、AIDS、AIDS関連症候群(ARC)、またはHIVウイルスの感染症と関連がある他のいずれかの疾患または状態にかかっている哺乳動物、例えばヒトを治療するため、上記のさらなる薬剤のいずれかと組み合わせて投与することができる。このような組み合わせは、本発明の組成物が上記のさらなる薬剤と同じ製剤中に存在するようにして哺乳動物に投与することができる。別法として、このような組み合わせは、さらなる薬剤が存在する製剤とは別の製剤中に本発明の組成物が存在するようにしてHIVウイルスによる感染症にかかっている哺乳動物に投与することができる。本発明の組成物をさらなる薬剤と別々に投与する場合、このような投与は、同時にまたは間に適当な期間を置いて順次行うことができる。さらなる薬剤(1種またはそれ以上)と同じ製剤中に本発明の組成物を含むべきかどうかの選択は、当業者の知識の範囲内である。
【0161】
さらに、本発明の組成物は、哺乳動物の本発明の化合物への曝露を高める効果を有するさらなる薬剤と組み合わせて哺乳動物、例えばヒトに投与することができる。用語「曝露」は、本明細書に使用されるように、ある期間にわたって測定された哺乳動物の血漿中の本発明の化合物の濃度に相当する。哺乳動物の化合物Aへの曝露は、本発明の組成物を適当な形態で哺乳動物に投与し、所定の時間に血漿試料を採取し、適当な分析技術、例えば液体クロマトグラフィまたは液体クロマトグラフィ/質量分析を用いて血漿中の化合物Aの量を計量することによって測定することができる。ある時間における血漿中の化合物Aの量を測定し、全てのサンプルからの濃度および時間データをプロットして曲線を得る。この曲線下の面積を算出して哺乳動物の化合物に対する曝露を得る。用語「曝露」、「曲線下の面積」および「濃度/時間曲線下の面積」は、同じ意味を有するものとし、全体を通して取り換えて使用することができる。
【0162】
化合物Aに対する哺乳動物の曝露を高めるために使用することができる薬剤の中には、チトクロームP450(CYP450)酵素の少なくとも1つのアイソフォームの阻害剤として作用することができるものがある。有益に阻害することができるCYP450のアイソフォームとしては、CYP1A2、CYP2D6、CYP2C9、CYP2C19およびCYP3A4が含まれるが、これらに限定されない。CYP3A4を阻害するために使用することができる適切な薬剤としては、リトナビルおよびデラビルジンが含まれるが、これらに限定されない。
【0163】
このような組み合わせは、化合物Aが上記のさらなる薬剤と同じ製剤中に存在するようにして哺乳動物に投与することができる。別法として、このような組み合わせは、化合物Aは、さらなる薬剤が存在する製剤とは別々の製剤中に存在するようにして投与することができる。化合物Aを、さらなる薬剤と別々に投与する場合、このような投与は、同時にまたは間に適当な期間をおいて順次行うことができる。さらなる薬剤(1種またはそれ以上)と同じ製剤中に本発明の組成物を含むべきかどうかの選択は、当業者の知識の範囲内である。
【0164】
本発明の他の特徴および実施態様は、以下の実施例から明らかとなり、これは本発明を説明するために記載したものであって、その意図する範囲を制限するものではない。
【実施例】
【0165】
実施例1
非晶質化合物Aを含む実施例1は、以下の通りに製造した。最初に、化合物A250.1mgおよびメタノール24.5gを含む噴霧溶液を形成した。Cole Parmer 74900シリーズの速度制御シリンジポンプを経て70mL/時の速度で溶液を「ミニ」噴霧乾燥装置にポンピングした。化合物Aの溶液を、窒素の加熱流れを用いて1SCFMの流速でSpraying Systems Co.の二流体ノズルModel No. SU1Aを通して霧化した。噴霧溶液を、直径11cmのステンレス鋼室に噴霧した。入口温度100℃で室に入った加熱されたガスは、出口温度22℃で排出された。生成した非晶質化合物Aを濾紙上に集め、真空下で乾燥させてデシケータ中に保存した。収量は約78%であった。
【0166】
実施例1の非晶質化合物Aが化合物Aの結晶質形態と比較して濃度増大をもたらすかどうかを測定するためにin vitro溶解試験を実施した。この試験では、全ての化合物Aが溶解した場合化合物Aの濃度が3000μg/mLとなるように、十分な量の物質を、マイクロ遠心分離試験管に加えた。試験は、同じものを二通り行った。管を、温度37℃の制御された室に置き、PBS 1.8mLをpH6.5および290mOsm/kgでそれぞれ個々の管に加えた。渦流ミキサーを用いて試料を約60秒間、急速に混合した。試料を、37℃、13,000Gで1分間遠心分離した。次いで、生成した上澄液から試料を採取し、メタノールで1:6(体積)に希釈してから高性能液体クロマトグラフィー(HPLC)で分析した。HPLC分析は、Phenomenex Luna C18カラムを用いて実施した。移動相は、H3PO4でpH3に調整された55%20mM KH2PO4および45%アセトニトリルからなる。UV吸光度は、210nmで測定した。各管の内容物を渦流ミキサーで混合し、次の試料を採取するまで、37℃で静かに放置した。試料は、4、10、20、40、90および1200分に集めた。結果を表1に示した。
【0167】
対照1
対照1は、化合物Aの結晶質形態I単独からなり、そして全ての化合物Aが溶解した場合、化合物Aの濃度が3000μg/mLとなるように十分な量の物質を加えた。
【表1】
【0168】
これらの試料で得られた薬物の濃度を用いて薬物の最大濃度(「MDC90」)、最初の90分間に濃度対時間曲線より下の面積(「AUC90」)、および1200分での化合物Aの濃度(「C1200」)を測定した。結果を表2に示した。
【表2】
【0169】
結果から、化合物Aの非晶質形態では、化合物Aの結晶質形態I単独と比較して濃度増大が得られることがわかった。化合物Aの非晶質形態では、化合物A.実施例2の結晶質形態Iによって得られたものの10.3倍のMDCおよび8.3倍のAUC90が得られた。
【0170】
実施例2
実施例2では、非晶質化合物Aおよび濃度増大ポリマーを合わせて製造した。化合物Aおよび濃度増大ポリマーヒドロキシプロピルメチルセルロースアセテートスクシネート(AQUOT−MG,信越化学工業,東京,日本から入手可能)の単純な物理的混合物は、実施例1で製造した非晶質化合物A5.4mgおよびHPMCAS0.6mgを遠心分離管に加えることによって製造した。渦流ミキサーを用いて乾燥粉末を1分間混合した。溶解試験は、実施例1に記載された通り実施した。結果を表3に示した。
【表3】
【0171】
これらの試料で得た薬物の濃度を用いて、薬物の最大濃度、MDC90、最初の90分間の濃度対時間曲線より下の面積(AUC90)および1200分での化合物A濃度、C1200を測定した。結果を表4に示した。実施例1および対照1についての比較を再び示した。
【表4】
【0172】
非晶質化合物Aおよび濃度増大ポリマーの両方を含む実施例2の物理的混合物では、結晶質形態Iの化合物A(対照1)単独と比較して改善された溶解性能が得られた。実施例2では、結晶質形態I化合物A単独によって得られたものの8.7倍のMDC90、および結晶質形態Iの化合物A単独によって得られたものの8.3倍のAUC90が得られた。
また、実施例2では、非晶質化合物A単独よりも長い時間、溶解薬物濃度が持続した。非晶質化合物A単独(実施例1)および濃度増大ポリマー(実施例2)を加えた非晶質化合物Aの性能は、最初の90分間では同様の性能を有していたが、濃度増大ポリマーを添加すると、より遅い時間で化合物Aの溶解性能が実質的に改善された。実施例2では、1200分での溶解した化合物Aの濃度(「C1200」)は、非晶質化合物A単独の7.8倍得られた。
【0173】
実施例3
実施例3、化合物A90質量%およびHPMCAS(AQUOT−MG,信越化学工業,東京,日本から入手可能)10質量%を含んでなる固体非晶質分散体を、以下の通りに製造した。最初に、化合物A300g、HPMCAS33.3gおよびメタノール3000gを含む噴霧溶液を以下の通り形成した。HPMCASおよびメタノールを容器中で合わせ、約2時間混合して、HPMCASを溶解させた。ポリマーの全量を加えた後、生成した混合物はわずかな曇りがあった。次に、化合物Aをこの混合物に直接加え、混合物をさらに2時間撹拌した。次いで、この混合物を、スクリーンサイズ250μmのフィルタに通すことによって濾過し、混合物からすべての大きな不溶性物質を除去し、このようにして噴霧溶液を形成した。
【0174】
高圧ポンプを用いて噴霧溶液を、加圧ノズル(Spraying Systems Pressure Nozzle and Body) (SK 78-21)を備えた噴霧乾燥機(Liquid-Feed Process Vesselを有するNiro type XP Portable Spray-Dryer(PSD−1))にポンピングした。PSD−1は、9インチの室延長部を備えていた。9インチの室延長部を噴霧乾燥機に付け加え、乾燥機の縦の長さを延ばした。長さを延ばして、乾燥機内の滞留時間を増加させ、これによって生成物を噴霧乾燥機の曲がった部分に到達する前に乾燥させることができる。また、噴霧乾燥機は、316SSサーキュラーディフューザープレートを備えており、1/16インチのドリル穴があって1%の開口領域を有する。この小さな開口領域は、乾燥ガスの流れが噴霧乾燥機内で生成物の再循環を最小限にすることを目的としている。ノズルは、操作中にディフューザープレートと同一平面上にした。Bran+Lubbe高圧ポンプを用いて液体をノズルに供給した。ポンプの後にパルセイション緩衝装置があり、ノズルでのパルセイションを最小限にする。噴霧溶液を、圧力200psigで約180g/分で噴霧乾燥機にポンピングした。乾燥ガス(例えば窒素)は、入口温度200℃で、ディフューザープレートを通して循環させた。蒸発した溶媒および乾燥ガスは、60℃の温度で、噴霧乾燥機から排出された。生成した固体非晶質分散体を、サイクロン中に集めた。
【0175】
上記の方法を用いて形成された固体非晶質分散体を、Gruenberg単路対流トレー乾燥機を用いて40℃で6時間作動させて後乾燥させた。次いで、乾燥後、分散体を周囲空気および湿気(20℃/RH50%)で8時間平衡状態にした。
【0176】
二次乾燥後の固体非晶質分散体の性質を表5に示した。
【表5】
【0177】
実施例4〜11
「ミニ」噴霧乾燥装置を用いて、化合物A対濃度増大ポリマーの種々の比率で、種々の濃度増大ポリマーを用いて化合物Aの固体非晶質分散体を製造した。表6は、各分散体中の化合物Aの濃度および使用した濃度増大ポリマーを記載している。
【0178】
【表6】
【0179】
ミニ噴霧乾燥機を用いて分散体を製造するために、化合物Aをポリマーと共に溶媒中で混合して噴霧溶液を形成した。各溶液を、Cole Parmer 74900シリーズの速度制御シリンジポンプ経由で「ミニ」噴霧乾燥装置中にポンピングした。窒素の加熱された流れ(100℃)を用いてSpraying Systems Co.二流体ノズル(model No. SU1A)を通して化合物A/ポリマー溶液を霧化した。噴霧溶液を、直径11cmのステンレス鋼室中に噴霧した。生成した固体非晶質分散体を、濾紙上に集めて、真空下で乾燥させてデシケータ中に保存した。各分散体を噴霧乾燥するのに使用した条件を表7に記載した。
【0180】
【表7】
【0181】
実施例12
溶解試験を実施して、実施例3〜11の固体非晶質分散体が化合物Aの濃度増大をもたらすことを示した。in vitro溶解試験は、実施例1のようにして実施した。これらの試験では、化合物Aの全てが溶解した場合、化合物Aの濃度が3000μg/mLとなるように十分な量の物質を加えた。結果を表8に示した。
【0182】
【表8】
【0183】
【表9】
【0184】
これらの試料で得られた化合物Aの濃度を用いて薬物の最大濃度(「MDC90」)および最初の90分間の濃度対時間曲線より下の面積(「AUC90」)を測定した。結果を表9に示した。比較のため、実施例1(非晶質化合物A)および対照1(結晶質形態Iの化合物A)の結果を示した。
【0185】
【表10】
【0186】
固体非晶質分散体では、化合物Aの結晶質形態I(対照1)単独のものよりも、そして非晶質化合物A(実施例1)単独よりも濃度増大が得られた。本発明の分散体のAUC90値は、結晶質対照の10.4〜14.6倍であり、そして非晶質化合物A(実施例1)単独の1.3〜1.8倍である。本発明の分散体のAUC1200値は、結晶質対照の9.7〜14.4倍であり、そして非晶質化合物A(実施例1)単独の5.5〜8.2倍である。これは、濃度増大ポリマーを添加すると非晶質化合物A単独と比較して高い溶解薬物濃度が長時間持続することを示している。
【0187】
実施例13
示差走査熱量測定法(DSC)を用いて実施例7の固体非晶質分散体を分析し、分散体中の化合物Aの非晶質の特徴を測定した。試料パンを周囲条件でクリンプし、ロボットアームを有するPerkin-Elmer Pyris 1 DSC炉に装填した。試料を約200℃まで、2℃/分で加熱した。試料のガラス転移温度は、DSCスキャンから測定した。比較のため同じ手法を用いて結晶質化合物A(対照1)および非晶質化合物A単独(実施例1)を分析した。結果を表10に示した。
【0188】
【表11】
【0189】
表10は、非晶質化合物A単独のガラス転移温度(Tg)(99.1℃)および結晶質薬物の鋭い融解ピーク(Tm)(178.1℃)を示す。固体非晶質分散体(実施例7)は、非晶質薬物と同様のTg(97.5℃)を示し、そして融解ピークは示さず、化合物Aの結晶質形態I単独のものとは異なる物理状態を示している。
【0190】
実施例14
実施例3は、Bruker AXS D8 Advance回折計を有する粉末X線回折を用いて試験し、分散体中の化合物Aの非晶質特徴を測定した。バックグラウンドシグナルが生じないようにカップの底面にSi(511)プレートを備えたLucite試料カップに試料(約100mg)を充填した。試料を、30rpmの速度で、ψ平面でスピンさせて結晶の配向効果を最小限にした。X線源(KCuα、λ=1.54Å)を電圧45kVおよび電流40mAで操作した。各試料についてのデータを、走査速度1.8秒/ステップおよび0.04°/ステップのステップサイズで、連続検出器走査モードで27分間にわたって集めた。ディフラクトグラムは、4°〜30°の2θ範囲にわたって集めた。比較のため同じ手法を用いて化合物Aの結晶質形態I(対照1)を分析した。結果を図1に示した。パターンを同じ図中で別々に見ることができるように、図1に示した各パターンのベースラインは、互いにシフトさせた。実施例3は、非晶質ハロのみを示す回折パターンを示したが、対照1は、結晶質薬物の特徴である鋭いピークを示すパターンを示した。これらのデータは、実施例3の固体非晶質分散体中の化合物Aが非晶質であり、結晶質でないことを示す。
【0191】
実施例15
これらの試験は、分散体のエージングを促進するため制御された温度および湿度条件下で貯蔵した後の固体非晶質分散体の物理的安定性を示す。実施例3および対照1のPXRDディフラクトグラムを、以下の温度および相対湿度(RH)条件:40℃/0%RH、40℃/25%RH、40℃/50%RHまたは40℃/75%RH下に6週間貯蔵する前および貯蔵した後に測定した。結果を図2に示した。
図2の結果では、6週間の加速エージング条件下で貯蔵した後、実施例3は非晶質ハロのみの回折パターンを示し、結晶質薬物の特徴である鋭いピークは示さなかった。結果は、実施例3の分散体がこれらの条件下で物理的に安定なことを示している。
【0192】
実施例16
さらに、これらの試験は、制御された温度および湿度条件下で貯蔵した後の化合物Aの固体非晶質分散体の物理的安定性を示した。実施例3の固体非晶質分散体の溶解性能は、以下の条件:30℃/60%RH、40℃/25%RHまたは40℃/75%RH下で12週間貯蔵する前および貯蔵した後に測定した。溶解試験は、実施例1に記載された通り実施した。結果を表11に示した。
【0193】
【表12】
【0194】
これらの試料で得られた化合物Aの濃度を用いて薬物の最大濃度(MDC90)および最初の90分間の濃度対時間曲線下の面積(AUC90)を測定した。結果を表12に示した。
【表13】
データから分かるように、分散体のAUC90は、加速エージング条件下で12週間貯蔵した後、比較的一定のままであった。これは、実施例3の分散体が、これらの条件下で物理的に安定であることを示している。
【0195】
実施例17
実施例17の固体非晶質分散体は、「ミニ」噴霧乾燥装置を用いて実施例4〜11に記載した通り形成した。噴霧溶液は、メタノール31g中の化合物A2500mg、HPMCAS(AQUOT−MG)247.5mgおよびBHT2.5mgからなる。噴霧溶液を1.3mL/分の速度で噴霧室にポンピングし、入口温度は80℃であった。
【0196】
実施例18
化合物A分散体を抗酸化剤ブチルヒドロキシトルエン(BHT)で処方するか、または酸素吸収パケットと共に貯蔵して化合物Aの化学安定性を改善した。
商品名Fresh PaxTM(Multisorb Technologyから入手可能)を有する酸素吸収剤と共に密閉した容器中の実施例3の分散体、実施例17の分散体および実施例3の分散体を、40℃/75%RHで保存した。HPLCを用いて10日、3週間または6週間後に化合物Aの分解生成物について試料を分析して試料中に存在する分解物の量を測定した。結果を表13に示した。
【0197】
【表14】
分散体は、BHTまたはO2吸収剤と共に貯蔵した試料では、化合物Aの分解が減少したことを示した。
【0198】
実施例19〜34
化合物A、濃度増大ポリマーHPMCASおよび安定化剤を含んでなる固体非晶質分散体を処方して化学安定性を改善した。実施例19〜33の固体非晶質分散体は、異なるタイプおよび量の塩基、異なる量の抗酸化剤BHT、または塩基および抗酸化剤の両方を用いて製造した。実施例19〜28および30〜33の固体非晶質分散体についてはポリマーの一部を中和するために塩基を用いた。実施例34は、比較のため安定化剤なしで製造した。
【0199】
実施例19〜22、24〜29、32および33の分散体は、「ミニ」噴霧乾燥装置を用いて実施例4〜11に記載された通り形成した。それぞれの製剤の組成物を表14に示した。噴霧溶液は、それぞれ20/80水/メタノール(質量/質量)の溶液中で10質量%の固体を含んだ。噴霧溶液を1.3mL/分の速度で、噴霧室にポンピングし、そして分散体の全てについて、入口温度は74℃であった。
【0200】
実施例23、30、31および34の分散体は、Niro PSD-1を用いて実施例3に記載した通り形成した。各製剤の組成は、表14に示した。噴霧溶液は、それぞれ20/80水/メタノール(質量/質量)の溶液中で10%の固体を含んだ。
【表15】
実施例19〜34の分散体を、ゆるやかにホイルで覆ったHPLCバイアル中、40℃および75%相対湿度(RH)でエージングした。
【0201】
6週後にHPLCを用いて分解生成物について試料を分析した。結果を表15に示した。
【表16】
結果から、塩基、抗酸化剤または両方を添加すると、HPMCASを含む固体非晶質分散体中の化合物Aの化学安定性が改善されることがわかった。
【0202】
実施例35
化合物AおよびHPMCASの物理混合物の化学安定性を、評価した。非晶質化合物Aは。実施例4〜11に記載された通り、ミニ噴霧乾燥機を用いて製造した。メタノール/水(8/2,体積/体積)中の固体1.5質量%を含む噴霧溶液を、入口温度70℃および出口温度、周囲条件で1.3mL/分の速度で噴霧した。
実施例3に記載された通りPSD−1噴霧乾燥機を用いてHPMCASを製造した。アセトン中に固体12質量%を含む噴霧溶液を、入口温度100℃および出口温度37℃で183〜215g/分の速度で噴霧した。
【0203】
化合物AおよびHPMCASを9:1の比率で一緒に混合し、化学安定性試験のため種々の温度、および相対湿度条件下で貯蔵した。結果は、下の表16の通りである。
【表17】
【0204】
実施例36
イヌを用いてin vivo溶解試験を実施し、本発明の非晶質分散体では化合物Aの濃度増大が得られることを示した。実施例3の固体非晶質分散体を6匹の絶食したビーグル犬のグループに投薬し、定期的に血液を採取し、血漿薬物濃度を測定することによって薬物放出はモニターした。用量を投与した時間から最後の試料までの濃度対時間曲線下の面積(
「AUC0-Tlast「ng*hr/mL)を表17に示した。化合物Aの結晶質形態I(対照1)および非晶質化合物A(実施例1)を、同様のプロトコールを用いて試験した。
【0205】
Methocel(R)(HPMC,USPグレード,4000cps,Dow Chemical Co.)0.5質量%を含む溶液の懸濁液として用量をイヌに投与した。水性薬物懸濁液の経口投与は、ポリエチレン管挿入部分を備えた経口強制飼養を用いて促進した。ポリエチレン管挿入部分を使用することで、管をすすぐためのさらなる水量を必要とすることなく、変位によって所望の用量体積を正確に供給した。用量は、25mgA/kg(「mgA」は、活性薬物のmgに相当する)であった。
【表18】
【0206】
上記の結果は、明らかに非晶質化合物A単独(実施例1)および化合物Aおよび濃度増大ポリマー(実施例3)の固体非晶質分散体の両方では、化合物A単独の結晶質形態Iよりも高い in vivo 薬物濃度が得られることを示している。実施例1の非晶質化合物Aについての相対的生物学的利用能(試験組成物のAUCを化合物Aの結晶質形態I単独のAUCによって割ったもの)は、化合物Aの結晶質形態I単独の11.7倍であったが、実施例3の固体非晶質分散体の相対的生物学的利用能は、化合物Aの結晶質形態I単独の44.7倍であった。
【0207】
実施例37
この実施例は、非常に少量の濃度増大ポリマーを非晶質化合物Aと合わせると、in vitro使用環境において溶解した化合物Aの濃度が持続することを示している。実施例37A、37Bおよび37Cは、実施例2の通り製造したが、しかし以下を除く:実施例37Aは、化合物A5.4mgおよびHPMCAS0.6mgであり;実施例37Bは、化合物A5.4mgおよびHPMCAS0.167mgであり;そして実施例37Cは、化合物A5.4mgおよびHPMCAS0.055mgであった。実施例37dは、非晶質化合物A単独からなる。溶解試験は、実施例2の通り実施し、結果は表18にまとめた。
【0208】
【表19】
【0209】
MDC360は、最初の360分の範囲内の最大溶解薬物濃度であり、そしてAUC360は、360分での溶解薬物濃度対時間曲線下の面積である。結果は、非常に少量のポリマーを非晶質化合物Aと合わせると非晶質化合物単独と比較して水性使用環境において溶解薬物濃度が持続することを示している。
【0210】
後述する実施例において、特に明記しない限り、以下の説明における全ての温度は、摂氏(℃)であり、そして特記しない限り、すべての部およびパーセンテージは質量による。
種々の出発物質および他の試薬は、商業上の供給業者、例えばAldrich Chemical CompanyまたはLancaster Synthesis Ltd.から購入し、そして特に明記しない限り、さらに精製することなく使用した。
【0211】
下に記載した反応は、窒素、アルゴンの正の圧力下でまたは乾燥管を用いて、周囲温度(特に明記しない限り)で、無水溶媒中で実施した。分析薄層クロマトグラフィは、ガラスに裏打ちされたシリカゲル60°F254プレート(Analtech(0.25mm))上で実施し、適当な溶媒比率(v/v)で溶離した。反応は、高圧液体クロマトグラフィ(HPLC)または薄層クロマトグラフィ(TLC)によって評価し、出発物質の消費量によって判断して終了した。TLCプレートをUV、リンモリブデン酸染色またはヨード染色によって視覚化した。1H−NMRスペクトルは、Bruker機器を用いて300MHzで操作して記録し、13CNMRスペクトルは、75MHzで記録した。NMRスペクトルは、参照標準としてクロロホルム(7.25ppmおよび77.00ppm)またはDMSO−d6((2.50ppmおよび39.52ppm))を使用してDMSO−d6またはCDCl3溶液(ppmで報告)として得た。必要に応じて、他のNMR溶媒を使用した。ピークの多重度を報告する場合、以下の略語を使用した:s=一重線、d=二重線、t=三重線、m=多重線、br=ブロード、dd=二重線の=二重線、dt=三重線の二重線。結合定数を記載する場合は、ヘルツで報告した。赤外スペクトルは、ニートオイルとして、KBrペレットとして、またはCDCl3溶液としてPerkin-Elmer FT−IR分光計上で記録し、波数(cm-1)で報告した。質量スペクトルは、LC/MSまたはAPCIを用いて得た。全ての融点は補正してない。全ての最終生成物は、(220nmおよび254nmの波長のHPLCによって)95%を超える純度を有した。
【0212】
化合物Aの製造
以下の実施例および製造において、「Et」はエチルを意味し、「Ac」はアセチルを意味し、「Me」はメチルを意味し、「Ph」はフェニルを意味し、(PhO)2POClはクロロジフェニルホスフェートを意味し、「HCl」は塩酸を意味し、「EtOAc」は酢酸エチルを意味し、「Na2CO3」は炭酸ナトリウムを意味し、「NaOH」は水酸化ナトリウムを意味し、「NaCl」は塩化ナトリウムを意味し、「NEt3」はトリエチルアミンを意味し、「THF」はテトラヒドロフランを意味し、「DIC」はジイソプロピルカルボジイミドを意味し、「HOBt」はヒドロキシベンゾトリアゾールを意味し、「H2O」は水を意味し、「NaHCO3」は炭酸水素ナトリウムを意味し、「K2CO3」は炭酸カリウムを意味し、「MeOH」はメタノールを意味し、「i−PrOAc」は酢酸イソプロピルを意味し、「MgSO4」は硫酸マグネシウムを意味し、「DMSO」はジメチルスルホキシドを意味し、「AcCl」はアセチルクロリドを意味し、「CH2Cl2」は塩化メチレンを意味し、「MTBE」はメチルt−ブチルエーテルを意味し、「DMF」はジメチルホルムアミドを意味し、「SOCl2」は塩化チオニルを意味し、「H3PO4」はリン酸を意味し、「CH3SO3H」はメタンスルホン酸を意味し、「Ac2O」は無水酢酸を意味し、「CH3CN」はアセトニトリルを意味し、そして「KOH」は水酸化カリウムを意味する。
【0213】
実施例38:(4R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−カルボン酸tert−ブチルエステルの製造
【化3】
【0214】
(4R)-5,5−ジメチル−チアゾリジン−3,4−ジカルボン酸3−tert−ブチルエステル(これは、Ikunaka, M. et al., Tetrahedron Asymm. 2002, 13, 1201; Mimoto, T. et al., J. Med. Chem. 1999, 42, 1789; and Mimoto, T. et al., European Patent Application 0574135A1 (1993)の方法に従って製造することができる。250g;0.957mol)をアルゴンパージした5Lフラスコに加え、そしてEtOAc(1.25L)中に溶解した。次いで、溶液を2℃に冷却し、(PhO)2POCl(208mL;1.00mol)をひとかたまりで加えた。滴下ロートを通してNEt3(280mL;2.01mol)を滴加し、次いで生成した懸濁液を0℃で撹拌した。7分後、アリルアミン(75.4mL;1.00mol)を滴加した。氷浴をはずし、懸濁液を室温に加温させた。半時間後、1N HCl(750mL;0.750mol)を加えた。すすぎ用にEtOAc(50mL)を用いて混合物を4L分液ロートへ移した。層を分離した。有機画分を7.2%水性Na2CO3(2×1.25L)で洗浄し、次いで3L蒸留フラスコへ移し、そしてEtOAc(400mL)で希釈した。溶液を共沸的に乾燥し、1気圧でEtOAcを蒸留することによって濃縮して体積800mLにした。25℃に冷却した後、生成した(4R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−カルボン酸tert−ブチルエステルの透明な黄色がかったEtOAc溶液を、次の工程に直接かけた。アリコートを取り出し、濃縮して白色結晶質固形物として(4R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−カルボン酸tert−ブチルエステルを得た:融点=94〜98℃,1HNMR(300MHz,CDCl3)δ6.12 (br s, 1H), 5.88 (app ddt, J = 10.2, 17.1, 5.6 Hz, 1H), 5.28 (app dq, J = 17.1, 1.5 Hz, 1H), 5.18 (app dd, J = 1.2, 10.2 Hz, 1H), 4.68 (s, 2H), 4.14 (br s, 1H), 3.95 (br t, J = 5.4 Hz, 2H), 1.62 (s, 3H), 1.49 (s, 9H), 1.46 (s, 3H); 13CNMR(75MHz,CDCl3)δ170.0, 154.0, 134.4, 116.9, 82.0, 73.3, 54.0, 48.7, 42.0, 30.6, 28.6, 24.6; MS(CI) m/z 301.1599(C14H25N2O3Sについての計算値301.1586,M+H+);C14H24N2O3Sについての元素分析 計算値:C,55.97;H,8.05;N,9.32;実測値:C,56.11;H,8.01;N,9.11.
【0215】
実施例39:(4R)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミドの製造
【化4】
【0216】
メタンスルホン酸(155mL;2.39mol)を3Lフラスコ中の(4R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−カルボン酸tert−ブチルエステルのEtOAc溶液に滴加した。室温で一夜撹拌した後、溶液を7℃に冷却し、H2O(400mL)を中に注いだ。混合物を4L分液ロート[すすぎ用にH2O(30mL)を用いた]へ移し、層を分離した。有機画分をH2O(190mL)で抽出した。合わせたH2O抽出物を5Lフラスコへ移して8℃に冷却した。3N NaOH(〜1.05L)を用いてpHを0.4から9.3に調整した。2−メチルテトラヒドロフラン(1.55L)を注ぎ、続いてNaCl(150g)を添加した。氷浴をはずし、混合物を室温に加温させた。3N NaOH(〜1mL)を用いてpHを9.0に再調整した。すすぎ用に2−メチルテトラヒドロフラン(50mL)を用いて混合物を4L分液ロートへ移し、層を分離した。水性相を2−メチルテトラヒドロフラン(950mL)で抽出した。すすぎ用に2−メチルテトラヒドロフラン(200mL)を用いて、セライトを通して有機抽出物を5L蒸留フラスコ中に直接、真空濾過した。1気圧で2−メチルテトラヒドロフランを蒸留させることによって溶液を共沸的に乾燥させて濃縮し、体積1.2Lにした。計量したアリコートを濃縮し、秤量したところ(4R)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド161gが溶液中に存在することがわかった[(4R)−5,5−ジメチル−チアゾリジン−3,4−ジカルボン酸3−tert−ブチルエステルから84%]。次いで、この溶液を次の工程に直接かけた。上記を濃縮したアリコートから結晶質固形物として(4R)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミドを得た:融点=45〜47℃,1HNMR(300MHz,CDCl3)δ6.73 (br s, 1H), 5.87 (app ddt, J = 10.2, 17.1, 5.7 Hz, 1H), 5.17- 5.27 (m, 2H), 4.27 (AB q, JAB = 9.7 Hz, Δv=22.5 Hz, 2H), 2.94 (app tt, J = 1.5, 5.8 Hz, 2H), 3.51 (s, 1H), 1.74 (s, 3H), 1.38 (s, 3H); 13CNMR(75MHz,CDCl3)δ169.7, 134.4, 116.9, 74.8, 57.2, 51.6, 41.9, 29.1, 27.3; MS(CI)m/z 201.1063(C9H17N2OSについての計算値201.1062,M+H+);C9H16N2OSについての元素分析 計算値:C,53.97;H,8.05;N,13.99;実測値:C,53.93;H,8.09;N,14.07.
【0217】
実施例40:(2S,3S)−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−2ヒドロキシ−4−フェニル−酪酸の製造
【化5】
【0218】
(2S,3S)−3−アミノ−2−ヒドロキシ−4−フェニル−酪酸(これは、Pedrosa et al., Tetrahedron Asymm. 2001, 12, 347; M. Shibasaki et al., Tetrahedron Lett. 1994, 35, 6123;および Ikunaka, M. et al. Tetrahedron Asymm. 2002, 13, 1201の方法に従って製造することができる;185g;948mmol)を5Lフラスコに加え、THF(695mL)中で懸濁した。H2O(695mL)、続いてNEt3(277mL;1990mmol)を注いだ。45分間撹拌した後、溶液を6℃に冷却した。次いで、THF(350mL)中の酢酸3−クロロカルボニル−2−メチルフェニルエステル(201g;948mmol)の溶液を滴加した。半時間後、6N HCl(〜170mL)を用いてpHを8.7から2.5に調整した。固体NaCl(46g)を加え、次いで氷浴をはずし、混合物を激しく撹拌しながら室温に加温させた。移すために1:1 THF/H2O(50mL)を用いて混合物を4L分液ロートへ移し、次いで低い方の水性相を除去した。有機画分を5L蒸留フラスコへ移し、次いで新たなTHF(2.5L)で希釈した。1気圧でTHFを蒸留させることによって溶液を共沸的に乾燥させて濃縮し、体積1.3Lにした。共沸的な乾燥を完了するため、新たなTHF(2.0L)を加え、1気圧での蒸留によって溶液を濃縮して1.85Lにし、次いで55℃に保った。滴下ロートを通してn−ヘプタン(230mL)を滴加し、次いで溶液に直ちに結晶種を入れた。結晶化が始まった後、さらなるn−ヘプタン(95mL)を滴加した。生成した結晶スラリーを7分間激しく撹拌した。次いで、さらなるn−ヘプタン(1.52L)をゆるやかな流れとして加えた。次いで、結晶スラリーをゆっくりと室温に冷却させて一夜撹拌した。懸濁液を真空濾過し、次いで、濾過ケークを1:1THF/n−ヘプタン(700mL)で洗浄した。真空オーブン中45〜50℃で乾燥させた後、〜7mol%Et3N・HClが混入した結晶質固体として(2S,3S)−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−2−ヒドロキシ−4−フェニル−酪酸324g(92%)を得た:融点=189〜191℃,1HNMR(300MHz,DMSO−d6)δ12.65 (br s, 1H), 3.80 (d, J = 9.7 Hz, 1H), 7.16- 7.30 (m, 6H), 7.07 (dd, J = 1.1, 8.0 Hz, 1H), 7.00 (dd, J = 1.1, 7.5 Hz), 4.40- 4.52 (m, 1H), 4.09 (d, J = 6.0 Hz, 1H), 2.92 (app dd, J = 2.9, 13.9 Hz, 1H), 2.76 (app dd, J = 11.4, 13.9 Hz, 1H), 2.29 (s, 3H), 1.80 (s, 3H); 13CNMR(75MHz,DMSO−d6)δ174.4, 169.3, 168.1, 149.5, 139.7, 139.4, 129.5, 128.3, 127.9, 126.5, 126.3, 124.8, 123.3, 73.2, 53.5, 35.4, 20.8, 12.6; MS(CI)m/z 372.1464(C20H22NO6についての計算値372.1447,M+H+);C20H21NO6・0.07Et3N・HClについての元素分析 計算値:C,64.34;H,5.86;N,3.95;Cl,0.70;実測値:C,64.27;H,5.79;N,3.96;Cl;0.86.
【0219】
実施例41:酢酸3−{(1S,2S)−3−[(4R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−イル]−1−ベンジル−2−ヒドロキシ−3−オキソ−プロピルカルバモイル}−2−メチルフェニルエステルの製造
【化6】
【0220】
(2S,3S)−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−2−ヒドロキシ−4−フェニル−酪酸(271g;731mmol)を、2−メチルテトラヒドロフラン中の(4R)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド(161g;804mmol)溶液(合計1.20Lの溶液)を含む5Lフラスコに加え、すすぎ用に2−メチルテトラヒドロフラン(500mL)を用いた。すすぎ用に2−メチルテトラヒドロフラン(50mL)を用いてHOBt・H2O(32.6g;241mmol)を加えた。白色懸濁液を室温で10分間を撹拌した。ジイソプロピルカルボジイミド(119mL;760mmol)を3つの部分(40mL+40mL+39mL)にわけて30分間隔で加えた。最後にDICを加えて1時間後、セライト(100g)を加え、懸濁液を室温で3時間撹拌した。混合物を真空濾過し、2−メチルテトラヒドロフラン(400mL)を用いて固形物をすすぎ、生成した濾過ケーキを洗浄した。濾液を4L分液ロートへ移し、すすぎ用に2−メチルテトラヒドロフラン(50mL)を用いた。溶液を1N HCl(1.25L)、次いでNaHCO3(27g)、NaCl(134g)およびH2O(1.25L)の水溶液で洗浄した。生成した有機相を3L蒸留フラスコへ移し、次いで、1気圧で2−メチルテトラヒドロフランを蒸留させることよって溶液を体積1.12Lに減らした。次いで、溶液を2−メチルテトラヒドロフラン(230mL)で希釈し、総体積1.35Lをした。溶液を23℃にさました後、粗酢酸3−{(1S,2S)−3−[(4R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−イル]−1−ベンジル−2−ヒドロキシ−3−オキソ−プロピルカルバモイル}−2−メチルフェニルエステルの溶液を次の工程に直接かけた。
【0221】
実施例42:(4R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチルベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミドの製造
【化7】
【0222】
MeOH(330mL)およびK2CO3(66.9g;484mmol)を、3Lフラスコ中の粗酢酸3−{(1S,2S)−3−[(4R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−イル]−1−ベンジル−2−ヒドロキシ−3−オキソ−プロピルカルバモイル}−2−メチルフェニルエステル(理論量:405g;731mmol)の2−メチルテトラヒドロフラン溶液に室温で順次加えた。2時間半後、さらなるK2CO3(20g;144mmol)を加えた。3時間後、固形物をすすいで濾過ケーキを洗浄するために4:1の2−メチルテトラヒドロフラン/MeOH(330mL)を用いてセライトのパッド上で反応混合物を真空濾過した。すすぎ用に4:1の2−メチルテトラヒドロフラン/MeOH(80mL)を用いて濾液を6L分液ロートへ移した。溶液をi−PrOAc(1.66L)で希釈し、次いで、H2O(1.60L)中のNaCl(83.0g)の溶液で洗浄した。有機画分を0.5NHCl(1.66L)、次いで飽和水性NaCl溶液(400mL)で洗浄した。生成した有機画分を4Lエルレンマイヤーフラスコへ移し、MgSO4(120g)を加えた。10分間撹拌した後、分液ロートおよびエルレンマイヤーフラスコをすすぎ、MgSO4を洗浄するために2:1のi−PrOAc/2−メチルテトラヒドロフラン(600mL)を用いて混合物を5L蒸留フラスコ中に直接、真空濾過した。最小ポット体積を〜2.50Lに維持しながらi−PrOAcを5つの部分(合計3.60L用いた)に分けて添加し、同時に1気圧で蒸留することによって2−メチルテトラヒドロフランを置き換えた。生成した結晶化混合物を75℃にさまし、この温度で30分間保持した。懸濁液を室温に一夜ゆっくりとさました。結晶を移して洗浄するためにi−PrOAc(600mL)を用いて懸濁液を真空濾過した。真空オーブン中40℃で乾燥後、結晶質(4R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチル−ベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド204g((2S,3S)−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−2−ヒドロキシ−4−フェニル−酪酸から54%)を得た。この物質を後述するように再結晶した。
【0223】
実施例43:(4R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチルベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミドの再結晶
【化8】
【0224】
(4R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチル−ベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド(193g,378mmol)を5Lフラスコに加え、次いでEtOAc(1.28L)中で懸濁した。懸濁液を76℃に加熱した後、MeOH(68mL)を加え、次いで内部温度を70℃に下げた。内部の温度を70℃に維持しながら、n−ヘプタン(810mL)を溶液に滴加した。n−ヘプタンの添加を完了した後、生成した結晶の懸濁液は、70℃で30分間保ち、次いで一夜室温にゆっくりとさました。懸濁液を真空濾過し、1.6:1のEtOAc/n−ヘプタン(500mL)を用いて結晶を移して洗浄した。次いで真空オーブン中45℃で結晶を乾燥させて、白色結晶質固形物として精製された(4R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチルベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド162g(回収率84%)を得た:融点=173〜175℃、1HNMR(300MHz,DMSO−d6)は、〜10:1の回転異性体の混合物を示した、主要な回転異性体の共鳴δ9.35 (s, 1H), 8.04- 8.15 (m, 2H), 7.13- 7.38 (m, 5H), 6.96 (t, J = 7.7 Hz, 1H), 6.79 (d, J = 7.2 Hz, 1H), 6.55 (d, J = 7.5 Hz, 1H), 5.71-5.87 (m, 1H), 5.45 (br d, J = 6.2 Hz, 1H), 4.98- 5.27 (m, 4H), 4.38- 4.52 (m, 3H), 3.58- 3.86 (m, 2H), 2.68- 2.90 (m, 2H), 1.84 (s, 3H), 1.52 (s, 3H), 1.37 (s, 3H) [特徴的な微量回転異性体の共鳴δ9.36 (s), 8.21 (d, J = 10.5 Hz), 7.82 (5, J = 5.8 Hz), 4.89 (s), 4.78 (AB q, JAB = 9.8 Hz, Δv = 27.1 Hz), 4.17- 4.24 (m), 2.93-3.01 (m), 1.87 (s), 1.41 (s)]; 13CNMR(75MHz,DMSO−d6)は、〜10:1の回転異性体の混合物を示した。主要な回転異性体の共鳴δ170.4, 169.5, 168.2, 155.7, 139.6, 139.4, 135.5, 135.4, 129.9, 128.2, 126.2, 126.1, 121.9, 117.8, 115.6, 72.4, 72.1, 53.1, 51.4, 48.2, 41.3, 34.2, 30.5, 25.0, 12.6[特徴的な微量回転異性体の共鳴δ171.4, 169.7, 168.6, 139.0, 129.5, 128.4, 70.6, 54.2, 49.1, 41.5, 31.4, 24.8]; MS(CI)m/z 512.2224(C27H34N3O5Sについての計算値512.2219,M+H+),C27H33N3O5Sについての元素分析 計算値:C,63.38;H,6.50;N,8.22;実測値:C,63.19;H,6.52;N,8.10.
【0225】
実施例44:(R)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド塩酸塩の製造;
【化9】
【0226】
(R)−5,5−ジメチル−チアゾリジン−3,4−ジカルボン酸3−tert−ブチルエステル(105kg,402mol)および酢酸エチル(690L)の溶液をジフェニルクロロホスフェート(113kg,422mol)で処理してから0℃に冷却した。温度を5℃に維持しながらNEt3(85.5kg,844mol)を加え、次いで混合物をこの温度で2時間保持した。混合物を0℃に冷却し、次いで温度を5℃に維持しながらアリルアミン(24.1kg,422mol)を加えた。混合物を20℃に加温し、次いで水性HCl10質量%(310L)でクエンチした。層を分離した後、有機画分を水性Na2CO38.6質量%(710L)で洗浄した。層を分離した後、水性画分を酢酸エチル(315L)で抽出した。AG-074278を含む合わせた酢酸エチル抽出物を、最小ポット体積を約315Lに維持しながら1気圧で共沸蒸留することによって乾燥させた。生成した(R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−カルボン酸tert−ブチルエステルの懸濁液を5℃に冷却した。酢酸エチル(263L)中の無水HCl(36.8kg,1008mol)の溶液13質量%を5℃に冷却し、次いで温度を15℃に維持しながら(R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−カルボン酸tert−ブチルエステル懸濁液に加えた。生成した懸濁液を20℃で19時間保持し、次いで冷却し、5℃で2時間保持した。次いで、すすぎ用に冷酢酸エチルを用いて懸濁液を濾過した。湿ったケークを真空下45℃で乾燥させて白色固形物として(R)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド塩酸塩90.5kg(95.2%)を得た:1HNMR(300MHz,DMSO−d6)δ8.94 (app t, J = 5.5 Hz, 1H), 5.82 (ddt, J = 10.4, 17.2, 5.2 Hz, 1H), 5.19- 5.25 (m, 1H), 5.10- 5.14 (m, 1H), 4.38 (AB q, JAB = 9.8 Hz, Δv = 14.5 Hz, 2H), 4.08 (s, 1H), 3.72- 3.91 (m, 2H), 1.58 (s, 3H), 1.32 (s, 3H); 13CNMR(75MHz,DMSO−d6)δ161.7, 132.2, 114.0, 67.9, 51.4, 43.5, 39.3, 25.3, 24.3; MS(CI)m/z 201.1070(C9H17N2OSについての計算値201.1062,M+H+);C9H17ClN2OSについての元素分析 計算値:C,45.65;H,7.24;N,11.83;Cl,14.97;実測値:C,45.41;H,7.33;N,11.69;Cl,15.22.
【0227】
実施例45:(2S,3S)−2−アセトキシ−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−4−フェニル−酪酸の製造
【化10】
【0228】
(2S,3S)−3−アミノ−2−ヒドロキシ−4−フェニル−酪酸(110kg,563mol)、NaCl(195kg)およびTHF(413L)の混合物に、周囲温度でNEt3(120kg,1183mol)およびH2O(414L)を投入した。生成した混合物を0℃に冷却した。酢酸3−クロロカルボニル−2−メチルフェニルエステル(120kg,563mol)を別の反応器に加えてからTHF(185L)中に溶解した。生成した酢酸3−クロロカルボニル−2−メチルフェニルエステルの溶液を10℃に冷却し、次いで添加中の温度を<10℃に維持しながら(2S,3S)−3−アミノ−2−ヒドロキシ−4−フェニル−酪酸混合物に加えた。生成した二相混合物を5℃で1時間撹拌し、次いで濃HCl(62kg)を用いてpH2.5〜3.0に調整した。次いで、混合物を25℃に加温させ、層を分離した。(2S,3S)−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−2−ヒドロキシ−4−フェニル−酪酸を含む生成したTHF画分を1気圧で蒸留することによって部分的に濃縮した。次いで最小ポット体積を1500Lに維持しながら、1気圧で蒸留することによってTHFを酢酸エチルで置き換えた。生成した溶液を25℃に冷却し、次いで無水酢酸(74.8kg,733mol)およびメタンスルホン酸(10.8kg,112mol)を投入した。混合物を70℃で約3時間加熱した。混合物を25℃にさまし、次いで温度を20℃に維持しながらH2O(1320L)で急冷した。水性層を除去した後、有機画分に酢酸エチル(658L)およびH2O(563L)を投入した。撹拌した後、水性相を除去した。有機画分を13質量%水性NaCl(2×650L)で二回洗浄した。有機画分を部分的に濃縮し、減圧蒸留(70〜140mmHg)によって乾燥させて体積約1500Lにした。生成した溶液を40℃に加熱し、次いで温度を40℃に維持しながらn−ヘプタン(1042L)を投入した。(2S,3S)−2−アセトキシ−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−4−フェニル−酪酸(0.1kg)を用いて溶液に結晶種を入れ、次いでさらなるn−ヘプタン(437L)をゆっくりと加えた。結晶化混合物を40℃で1時間維持した。温度を40℃に維持しながら、さらなるn−ヘプタン(175L)を加えた。結晶質懸濁液をさまし、25℃で1時間、次いで0℃で2時間保持した。すすぎ用にn−ヘプタンを用いて懸濁液を濾過した。湿ったケークを真空下55℃で乾燥させて白色固体として(2S,3S)−2−アセトキシ−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−4−フェニル−酪酸174kg(74.5%)を得た:融点=152〜154℃;1HNMR(300MHz,CDCl3)δ7.21- 7.35 (m, 5H), 7.13 (app t, J = 7.9 Hz, 1H), 7.01 (app d, J = 8.1 Hz, 1H), 6.94 (app d, J = 7.2 Hz, 1H), 5.99 (d, J = 9.0 Hz, 1H), 5.33 (d, J = 4.1 Hz, 1H), 4.96- 5.07 (m, 1H), 3.07 (dd, J = 5.5, 14.6 Hz, 1H), 2.90 (dd, J = 10.0, 14.5 Hz, 1H), 2.30 (s, 3H), 2.18 (s, 3H), 1.96 (s, 3H); 13CNMR(125MHz,CDCl3)δ170.4, 170.2, 169.6, 169.5, 149.5, 137.81, 136.5, 129.2, 128.6, 128.4, 127.0, 126.6, 124.5, 123.7, 73.1, 50.9, 35.9, 20.6, 20.5, 12.4; C22H23NO7についての元素分析 計算値:C,63.92;H,5.61;N,3.39;実測値:C,64.22;H,5.68;N,3.33;MS(CI)m/z 414.1572(C22H24NO7についての計算値414.1553,M+H+).
【0229】
実施例46:(4R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチル−ベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミドの製造
【化11】
【0230】
(2S,3S)−2−アセトキシ−3−(3−アセトキシ−2−メチルベンゾイルアミノ)
−4−フェニル−酪酸(140kg,339mol)、CH3CN(560L)およびピリジン(64.3kg,813mol)の溶液を15℃に冷却した。温度を15℃に維持しながら、SOCl2(44.3kg,373mol)を投入した。混合物を15℃で1時間保持した。別の
反応器に(R)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド塩酸塩(96.6kg,408mol)、CH3CN(254L)およびピリジン(29.5kg,373mol)を投入し、次いで15℃に冷却した。温度を15℃に維持しながら、(2S,3S)−2−アセトキシ−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−4−フェニル酪酸塩化物溶液を(R)−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド溶液を加えた。混合物を15℃で6時間保持した。0℃の冷却ジャケットを用いて別の反応器にKOH(167kg,2709mol)およびメタノール(280L)を投入した。生成したKOH/メタノール溶液を5℃に冷却した。温度を10℃に維持しながら、粗酢酸3−{(1S,2S)−2−アセトキシ−3−[(R)−4−アリルカルバモイル−5,5−ジメチル−チアゾリジン−3−イル]−1−ベンジル−3−オキソ−プロピルカルバモイル}−2−メチルフェニルエステル混合物を、KOH/メタノール溶液に加えた。添加を完了した後、混合物を25℃で3時間保持した。混合物にH2O(840L)および酢酸エチル(840L)を投入し、次いで温度を20℃に維持しながら、濃HCl(85kg)を用いてpH5〜6.5に酸性化した。生成した層を分離した。水性NaHCO36.8質量%(770L)、水性HCl/NaCl溶液(H2O:875L;濃HCl:207kg;NaCl:56kg)、水性NaHCO38.5質量%(322L)、次いで水性NaCl3.8質量%(728L)を用いて有機画分を順次洗浄した。生成した有機画分を、1気圧で蒸留することによって部分的に濃縮した。蒸留を続け、ポット温度を〜70℃に維持することによって溶媒を酢酸エチルと交換した。ポット体積が約840Lのままであるようにして、酢酸エチルを加えた。次いで、溶液を20℃に冷まし、結晶化が観察されるまでこの温度で保持した。n−ヘプタン(280L)を加え、懸濁液を15℃で4時間撹拌した。すすぎ用に冷2.4:1(v/v)酢酸エチル/n−ヘプタンを用いて結晶をすすいだ。湿ったケークを真空下、45℃で乾燥させて粗(R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチルベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミドを得た。脱色および再結晶は、以下の通りに実施した:粗(R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチルベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド、ADP炭素(21kg)、Supercel(3kg)および酢酸エチル(780L)の混合物を70℃に加熱した。CH3OH(40L)を混合物に加えた。混合物を濾過し、生成した透明な濾液を1気圧で加熱して還流させ、蒸留を開始した。CH3OHを以下のようにして置き換えた:ポット体積を約840Lおよび70℃で維持しながら酢酸エチル(388L)を投入した。温度を70℃に維持しながら溶液にn−ヘプタン(316L)をゆっくりと投入した。次いで、混合物を20℃に冷却し、この温度で4時間保持した。結晶は濾過し、すすぎ用に冷2.1:1(v/v)酢酸エチル/n−ヘプタンを用いて濾過した。湿ったケークを真空下、45℃で乾燥させて白色結晶質固形物として(4R)−3−[(2S,3S)−2−ヒドロキシ−3−(3−ヒドロキシ−2−メチルベンゾイルアミノ)−4−フェニル−ブチリル]−5,5−ジメチル−チアゾリジン−4−カルボン酸アリルアミド103kg(59.6%)を得た:融点=173〜175℃,1HNMR(300MHz,DMSO−d6)は、〜10:1回転異性体の混合物を示した。主要な回転異性体の共鳴δ9.35 (s, 1H), 8.04- 8.15 (m, 2H), 7.13- 7.38 (m, 5H), 6.96 (t, J = 7.7 Hz, 1H), 6.79 (d, J = 7.2 Hz, 1H), 6.55 (d, J = 7.5 Hz, 1H), 5.71- 5.87 (m, 1H), 5.45 (br d, J = 6.2 Hz, 1H), 4.98- 5.27 (m, 4H), 4.38- 4.52 (m, 3H), 3.58- 3.86 (m, 2H), 2.68- 2.90 (m, 2H), 1.84 (s, 3H), 1.52 (s, 3H), 1.37 (s, 3H) [特徴的な微量回転異性体の共鳴δ9.36 (s), 8.21 (d, J = 10.5 Hz), 7.82 (5, J = 5.8 Hz), 4.89 (s), 4.78 (AB q, JAB = 9.8 Hz, Δv = 27.1 Hz), 4.17- 4.24 (m), 2.93- 3.01 (m), 1.87 (s), 1.41 (s)];13CNMR(75MHz,DMSO−d6)は、〜10:1回転異性体の混合物を示した。主要な回転異性体の共鳴δ170.4, 169.5, 168.2, 155.7, 139.6, 139.4, 135.5, 135.4, 129.9, 128.2, 126.2, 126.1, 121.9, 117.8, 115.6, 72.4, 72.1, 53.1, 51.4, 48.2, 41.3, 34.2, 30.5, 25.0, 12.6[特徴的な微量回転異性体の共鳴δ171.4, 169.7, 168.6, 139.0, 129.5, 128.4, 70.6, 54.2, 49.1, 41.5, 31.4, 24.8];MS(CI)m/z 512.2224(C27H34N3O5Sについての計算値512.2219,M+H+)、C27H33N3O5Sについての元素分析 計算値:C,63.38;H,6.50;N,8.22;実測値:C,63.19;H,6.52;N,8.10.
【0231】
実施例47:(2S,3S)−3−アミノ−2−ヒドロキシ−4−フェニル−酪酸;塩酸塩の製造
【化12】
【0232】
(2S,3S)−3−tert−ブトキシカルボニルアミノ−2−ヒドロキシ−4−フェニル−酪酸(163g,551mmol)およびCH2Cl2(2.0L)の懸濁液中にHClガス(51g,1.4mol)を0℃でバブリングした。生成したオフホワイト懸濁液を周囲温度に加温させて一夜撹拌した。濃縮したアリコートの1HNMR分析は、生成物への転化率約95%を示した。懸濁液を0℃に冷却し、懸濁液にさらなるHClガス(46g,1.3mol)をバブリングした。周囲温度に加温させた後、懸濁液を一夜撹拌した。懸濁液を真空濾過し、固形物をCH2Cl2(200mL)ですすぎ、次いで、固形物を真空オーブン中、45℃で24時間乾燥させて白色固形物として(2S,3S)−3−アミノ−2−ヒドロキシ−4−フェニル−酪酸;塩酸塩129g(100%)を得た:1HNMR(300MHz,DMSO−d6)δ13.05 (br s, 1H), 8.25 (br s, 3H), 7.22-7.34 (m, 5H), 4.41 (d, J = 2.6 Hz, 1H), 3.66 (br s, 1H), 2.84 (ABXのAB部分, JAX = 11.0 Hz, JBX = 2.8 Hz, Δv = 19.6 Hz, 2H); 13CNMR(75MHz,DMSO−d6)d 172.4, 136.6, 129.8, 128.7, 127.1, 69.6, 55.0, 33.6;MS(CI)m/z196.0979(C10H14NO3についての計算値196.0974,M−Cl-)
【0233】
実施例48:(2S,3S)−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−2−
ヒドロキシ−4−フェニル−酪酸の製造
【化13】
【0234】
NEt3(186mL,1.34mol)を、(2S,3S)−3−アミノ−2−ヒドロキシ−4−フェニル−酪酸;塩酸塩(100g,432mmol)、H2O(320mL)およびテトラヒドロフラン(320mL)の懸濁液に加えた。懸濁液を4℃に冷却し、酢酸3−クロロカルボニル−2−メチルフェニルエステル(93.6g,440mmol)およびTHF(160mL)の溶液を滴加した。生成した溶液を周囲温度に加温させて1時間撹拌した。溶液を10℃に冷却し、6N HCl(87mL)を用いてpHを2.0に調整した。NaCl(25g)およびテトラヒドロフラン(200mL)を加え、混合物を周囲温度に加温させた。相を分離し、テトラヒドロフラン画分をMgSO4で乾燥させて濾過した。ロータリーエバポレーターを用いて濾液を濃縮して体積330mLにしてからテトラヒドロフラン(230mL)で希釈した。n−ヘプタン(1.2L)をゆっくりと加え、生成した固形物の白色懸濁液を周囲温度で一夜撹拌した。懸濁液を真空濾過し、固形物をn−ヘプタン(2×500mL)ですすぎ、固形物を真空オーブン中45℃で24時間乾燥させ、〜7.7mol%Et3N・HClが混入した白色固形物として(2S,3S)−3−(3−アセトキシ−2−メチルベンゾイルアミノ)−2−ヒドロキシ−4−フェニル−酪酸150g(93.6%)を得た:融点=189〜191℃,1HNMR(300MHz,DMSO−d6)δ12.65 (br s, 1H), 3.80 (d, J = 9.7 Hz, 1H), 7.16- 7.30 (m, 6H), 7.07 (dd, J = 1.1, 8.0 Hz, 1H), 7.00 (dd, J = 1.1, 7.5 Hz), 4.40- 4.52 (m, 1H), 4.09 (d, J = 6.0 Hz, 1H), 2.92 (app dd, J = 2.9, 13.9 Hz, 1H), 2.76 (app dd, J = 11.4, 13.9 Hz, 1H), 2.29 (s, 3H), 1.80 (s, 3H); 13CNMR(75MHz,DMSO−d6)δ174.4, 169.3, 168.1, 149.5, 139.7, 139.4, 129.5, 128.3, 127.9, 126.5, 126.3, 124.8, 123.3, 73.2, 53.5, 35.4, 20.8, 12.6; MS(CI)m/z 372.1464(C20H22NO6についての計算値372.1447,M+H+)
【図面の簡単な説明】
【0235】
【図1】実施例14の化合物Aの結晶質形態Iおよび本発明の固体非晶質分散体の粉末X線回折をプロットし、そして固体非晶質分散体中の化合物Aが結晶質でないことを示している。
【図2】異なる条件で貯蔵した後の、実施例15の化合物Aの結晶質形態Iおよび本発明の固体非晶質分散体の粉末X線回折をプロットし、そして固体非晶質分散体が物理的に安定であることを示す。
【特許請求の範囲】
【請求項1】
非晶質(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミドまたはその医薬上許容しうる塩もしくは溶媒和物。
【請求項2】
非晶質(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミドまたはその医薬上許容しうる塩もしくは溶媒和物を含む医薬組成物。
【請求項3】
存在する(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミドの全量の少なくとも5質量%が非晶質形態である、請求項2に記載の医薬組成物。
【請求項4】
マトリックスをさらに含む、請求項2または3に記載の医薬組成物。
【請求項5】
マトリックスは、少なくとも1つのイオン化できるセルロース系ポリマーから選ばれる、請求項4に記載の医薬組成物。
【請求項6】
少なくとも1つのイオン化できるセルロース系ポリマーは、ヒドロキシプロピルメチルセルロースアセテートスクシネート、カルボキシメチルエチルセルロース、セルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースフタレート、メチルセルロースアセテートフタレート、セルロースアセテートトリメリテート、ヒドロキシプロピルセルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースアセテートフタレート、セルロースアセテートテレフタレートおよびセルロースアセテートイソフタレート、およびそれらの混合物から選ばれる、請求項5に記載の医薬組成物。
【請求項7】
組成物は、固体非晶質分散体の形態である、請求項4〜6のいずれか1項に記載の医薬組成物。
【請求項8】
固体非晶質分散体は、 (4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミド、またはその医薬上許容しうる塩もしくは溶媒和物の少なくとも40質量%を含む、請求項7に記載の医薬組成物。
【請求項9】
水性使用環境に投与した時に、
(a) 該使用環境中で対照組成物によって得られる少なくとも1.25倍である(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミドの最大溶解濃度;および
(b) 該使用環境へ導入した時間と使用環境に導入後約270分との間の少なくとも90分のいずれかの期間について該使用環境対時間曲線下面積(AUC)において該対照組成物の少なくとも1.25倍である(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリ−4−カルボキサミドの濃度;
の少なくとも1つが得られ、ここにおいて、該対照組成物は、等量の結晶質形態Iの(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミド単独から本質的になる、(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミド、またはその医薬上許容しうる塩もしくは溶媒和物を含む医薬組成物。
【請求項10】
水性使用環境は、本質的に
(a) 20mM Na2HPO4;
(b) 47mM KH2PO4;
(c) 87mM NaCl;
(d) 0.2mM KCl;
(e) pH6.5;
(f) 290mOsm/kg;および
(e) 温度37℃;
からなり、ここにおいて、該使用環境の総量は1.8mLであり、そして使用する(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミドの量は、全て溶解した場合に(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミドの全濃度が3000μg/mLである、請求項9に記載の医薬組成物。
【請求項11】
(a) 溶媒を含む噴霧溶液中に化合物を溶解し;そして
(b) 噴霧溶液から溶媒を急速に蒸発させて該化合物の非晶質形態を得る;
ことからなる医薬組成物の製造方法であって、ここにおいて、該化合物は、(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミド、またはその医薬上許容しうる塩もしくは溶媒和物である、上記製造方法。
【請求項12】
噴霧溶液はマトリックスをさらに含む、請求項11に記載の方法。
【請求項13】
マトリックスは、少なくとも1つのイオン化できるセルロース系ポリマーから選ばれる、請求項12に記載の方法。
【請求項14】
マトリックスは、ヒドロキシプロピルメチルセルロースアセテート、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、メチルセルロース、ヒドロキシエチルメチルセルロース、ヒドロキシエチルセルロースアセテート、ヒドロキシエチルエチルセルロース、ヒドロキシプロピルメチルセルロースアセテートスクシネート、カルボキシメチルエチルセルロース、セルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートフタレート、メチルセルロースアセテートフタレート、セルロースアセテートトリメリテート、ヒドロキシプロピルセルロースアセテートフタレート、セルロースアセテートテレフタレートおよびセルロースアセテートイソフタレートからなる群より選ばれる、請求項13に記載の方法。
【請求項15】
HIV感染した哺乳動物を治療する医薬の製造における、請求項2〜10のいずれか1項に記載の医薬組成物の使用。
【請求項1】
非晶質(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミドまたはその医薬上許容しうる塩もしくは溶媒和物。
【請求項2】
非晶質(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミドまたはその医薬上許容しうる塩もしくは溶媒和物を含む医薬組成物。
【請求項3】
存在する(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミドの全量の少なくとも5質量%が非晶質形態である、請求項2に記載の医薬組成物。
【請求項4】
マトリックスをさらに含む、請求項2または3に記載の医薬組成物。
【請求項5】
マトリックスは、少なくとも1つのイオン化できるセルロース系ポリマーから選ばれる、請求項4に記載の医薬組成物。
【請求項6】
少なくとも1つのイオン化できるセルロース系ポリマーは、ヒドロキシプロピルメチルセルロースアセテートスクシネート、カルボキシメチルエチルセルロース、セルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースフタレート、メチルセルロースアセテートフタレート、セルロースアセテートトリメリテート、ヒドロキシプロピルセルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースアセテートフタレート、セルロースアセテートテレフタレートおよびセルロースアセテートイソフタレート、およびそれらの混合物から選ばれる、請求項5に記載の医薬組成物。
【請求項7】
組成物は、固体非晶質分散体の形態である、請求項4〜6のいずれか1項に記載の医薬組成物。
【請求項8】
固体非晶質分散体は、 (4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミド、またはその医薬上許容しうる塩もしくは溶媒和物の少なくとも40質量%を含む、請求項7に記載の医薬組成物。
【請求項9】
水性使用環境に投与した時に、
(a) 該使用環境中で対照組成物によって得られる少なくとも1.25倍である(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミドの最大溶解濃度;および
(b) 該使用環境へ導入した時間と使用環境に導入後約270分との間の少なくとも90分のいずれかの期間について該使用環境対時間曲線下面積(AUC)において該対照組成物の少なくとも1.25倍である(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリ−4−カルボキサミドの濃度;
の少なくとも1つが得られ、ここにおいて、該対照組成物は、等量の結晶質形態Iの(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミド単独から本質的になる、(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミド、またはその医薬上許容しうる塩もしくは溶媒和物を含む医薬組成物。
【請求項10】
水性使用環境は、本質的に
(a) 20mM Na2HPO4;
(b) 47mM KH2PO4;
(c) 87mM NaCl;
(d) 0.2mM KCl;
(e) pH6.5;
(f) 290mOsm/kg;および
(e) 温度37℃;
からなり、ここにおいて、該使用環境の総量は1.8mLであり、そして使用する(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミドの量は、全て溶解した場合に(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミドの全濃度が3000μg/mLである、請求項9に記載の医薬組成物。
【請求項11】
(a) 溶媒を含む噴霧溶液中に化合物を溶解し;そして
(b) 噴霧溶液から溶媒を急速に蒸発させて該化合物の非晶質形態を得る;
ことからなる医薬組成物の製造方法であって、ここにおいて、該化合物は、(4R)−N−アリル−3−{(2S,3S)−2−ヒドロキシ−3−[(3−ヒドロキシ−2−メチルベンゾイル)アミノ]−4−フェニルブタノイル}−5,5−ジメチル−1,3−チアゾリジン−4−カルボキサミド、またはその医薬上許容しうる塩もしくは溶媒和物である、上記製造方法。
【請求項12】
噴霧溶液はマトリックスをさらに含む、請求項11に記載の方法。
【請求項13】
マトリックスは、少なくとも1つのイオン化できるセルロース系ポリマーから選ばれる、請求項12に記載の方法。
【請求項14】
マトリックスは、ヒドロキシプロピルメチルセルロースアセテート、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、メチルセルロース、ヒドロキシエチルメチルセルロース、ヒドロキシエチルセルロースアセテート、ヒドロキシエチルエチルセルロース、ヒドロキシプロピルメチルセルロースアセテートスクシネート、カルボキシメチルエチルセルロース、セルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートフタレート、メチルセルロースアセテートフタレート、セルロースアセテートトリメリテート、ヒドロキシプロピルセルロースアセテートフタレート、セルロースアセテートテレフタレートおよびセルロースアセテートイソフタレートからなる群より選ばれる、請求項13に記載の方法。
【請求項15】
HIV感染した哺乳動物を治療する医薬の製造における、請求項2〜10のいずれか1項に記載の医薬組成物の使用。
【図1】


【図2】
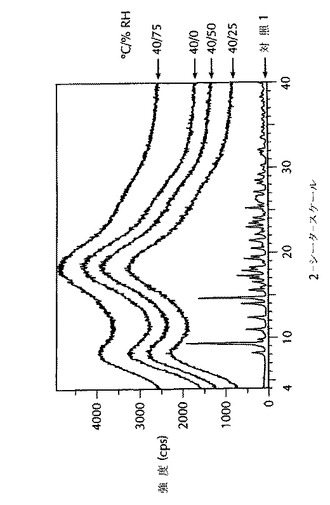
【図2】
【公表番号】特表2007−513937(P2007−513937A)
【公表日】平成19年5月31日(2007.5.31)
【国際特許分類】
【出願番号】特願2006−543642(P2006−543642)
【出願日】平成16年11月26日(2004.11.26)
【国際出願番号】PCT/IB2004/003921
【国際公開番号】WO2005/056542
【国際公開日】平成17年6月23日(2005.6.23)
【出願人】(503053011)ファイザー インコーポレイテッド (26)
【Fターム(参考)】
【公表日】平成19年5月31日(2007.5.31)
【国際特許分類】
【出願日】平成16年11月26日(2004.11.26)
【国際出願番号】PCT/IB2004/003921
【国際公開番号】WO2005/056542
【国際公開日】平成17年6月23日(2005.6.23)
【出願人】(503053011)ファイザー インコーポレイテッド (26)
【Fターム(参考)】
[ Back to top ]