
HIV・TATトランス活性化を抑制するための化合物
【要約書】
【課題】HIV・TATトランス活性化を抑制するための化合物を提供すること。
【解決手段】下記構造式:

の化合物であって、ここで、R1、R2、R3およびR4は夫々、HO−、CH3O−およびCH3(C=O)O−からなる群から選択される。但し、R1、R2、R3およびR4は夫々同時にHO−ではなく、各化合物は、クレオソートブッシュであるラレア・トリデンタータ(Larrea tridentata)の葉−花抽出物から単離されたものであり、1,4−ビス−(3,4−ジヒドロキシフェニル)−2,3−ジメチルブタン(ノルジヒドロカイアレ酸(nordihydroquaiaretic acid)、NDGA)の誘導体であり、加えて、これらのNDGAおよび各誘導体は、NDGAまたはその誘導体を細胞に投与することによって、該細胞において、HIVウイルスを含むレンチウイルスのTatトランス活性化を抑制するために使用することができる、化合物。
【課題】HIV・TATトランス活性化を抑制するための化合物を提供すること。
【解決手段】下記構造式:
の化合物であって、ここで、R1、R2、R3およびR4は夫々、HO−、CH3O−およびCH3(C=O)O−からなる群から選択される。但し、R1、R2、R3およびR4は夫々同時にHO−ではなく、各化合物は、クレオソートブッシュであるラレア・トリデンタータ(Larrea tridentata)の葉−花抽出物から単離されたものであり、1,4−ビス−(3,4−ジヒドロキシフェニル)−2,3−ジメチルブタン(ノルジヒドロカイアレ酸(nordihydroquaiaretic acid)、NDGA)の誘導体であり、加えて、これらのNDGAおよび各誘導体は、NDGAまたはその誘導体を細胞に投与することによって、該細胞において、HIVウイルスを含むレンチウイルスのTatトランス活性化を抑制するために使用することができる、化合物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、1,4−ビス−(3,4−ジヒドロキシフェニル)−2,3−ジメチルブタン(ノルジヒドロカイアレ酸(nordihydroquaiaretic acid)、NDGA)の誘導体の単離、精製および特徴付けに関する。この誘導体は、メキシコ・ハマビシまたはクレオソートブッシュ(creosolte bush; Larrea tridentata,Zygophyllaceae)の葉および花の抽出物から単離されたものであり、NDGAと共に、HIVウイルスを含むレンチウイルスにおけるTatトランス活性化を抑制するために用いることができる。
【背景技術】
【0002】
(関連技術の説明)
Tatはヒト免疫不全ウイルス(HIV)遺伝子発現のトランス活性化因子であり、HIV遺伝子発現のために必要な二以上のウイルス性調節因子(TatおよびRev)の一つである。Tatは、TAR・RNA要素に結合することによって作用し、長い末端繰り返し(LTR)プロモータからの転写を活性化する。
【0003】
Tatタンパクは転写の延長を安定化させるものであり、また転写の開始にも関与することが示されている。以前の研究によって、Tatは抗体依存性T細胞増殖の減少を媒介し、免疫応答の失敗に実質的に寄与することが示されている。Tatはまた、カポシ細胞増殖(Kaposi'scell growth)を直接刺激する。
【0004】
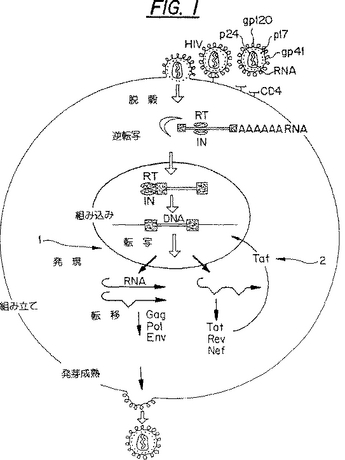
Tatは明らかな細胞同族体を有していないので、この強力な陽性調節因子は抗AIDS薬の開発のための魅力的な目標になっている(図1)。感染の新たなラウンドを妨害する現在入手可能なHIV逆転写阻害因子(AZT、DDI)または潜在的なプロテアーゼ阻害因子とは対照的に、組み込まれたプロウイルスDNAの、ウイルス遺伝子Tatで調節された発現を抑制する阻害因子は、該ウイルスを初期段階で停止させるであろう(非特許文献1)。
【0005】
真核宿主における転写レベルおよびポスト転写レベルでの遺伝子発現を制御する因子の解明を目的とした努力によって、最近、Tat機能の定量的な評価が可能になった(非特許文献2)。Tat調節されたトランス活性化(Tat−TRS)の阻害因子をスクリーニングするために、分泌された胎盤アルカリホスファターゼ(SEAP)リポータ遺伝子を、プラスミドpBC12/HIV/SEAPの中のHIV−1・LTRプロモータの制御下に置く。Tatにコードされる活性は、第二のプラスミド構築物pBC/CMV/t2によって供給される。これら二つのプラスミドを用いたCOS細胞の一時的な共感染によって、培地中へのアルカリホスファターゼの分泌が導かれ、この培地は単純な発色試験によって分析される(非特許文献3)。従って、このSEAP試験は、Tatトランス活性化の間接的な測定を提供する。阻害剤は、Tatタンパク(Tat−TRS)がHIV−1・LTRのトランス活性化を介してSEAP・mRNAを発現させるのを阻害することにより、SEAP活性の減少を生じさせるに違いない。
【非特許文献1】Hsu et al,Science 1991年 第254巻 p.1799-1820
【非特許文献2】Sim, Ann.N.Y.Acad.Sci 1990年 .第616巻 p.64-70
【非特許文献3】Berger et al.,Gene 1988年 第66巻 p.1-10
【発明の開示】
【課題を解決するための手段】
【0006】
本発明によって、以下が提供される:
1.下記構造式の化合物。
【0007】
ここで、R1、R2、R3およびR4は夫々、HO−、CH3O−およびCH3(C=O)O−からなる群から選択される。但し、R1、R2、R3およびR4は夫々同時にHO−ではない。
2.項目1に記載の化合物であって、R1、R2およびR3は夫々HO−、R4はCH3O−であり、下記の構造式を有する化合物。
【0008】
3.項目1に記載の化合物であって、R1、R2およびR4は夫々HO−、R3はCH3O−であり、下記の構造式を有する化合物。
【0009】
4.項目1に記載の化合物であって、R1およびR2は夫々HO−、R3はCH3(C=O)O−、R4はCH3O−であり、下記の構造式を有する化合物。
【0010】
5.項目1に記載の化合物であって、R1、R3およびR4は夫々CH3O−、R2はHO−であり、下記の構造式を有する化合物。
【0011】
6.項目1に記載の化合物であって、R1はHO−、R2、R4およびR3は夫々CH3O−であり、下記の構造式を有する化合物。
【0012】
7.項目1に記載の化合物であって、R1およびR3は夫々CH3O−、R2はHO−、R4はCH3(C=O)O−であり、下記の構造式を有する化合物。
【0013】
8.項目1に記載の化合物であって、R1およびR4は夫々CH3O−、R2はHO−、R3はCH3(C=O)O−であり、下記の構造式を有する化合物。
【0014】
9.項目1に記載の化合物であって、R1はHO−、R2およびR3は夫々CH3O−、R4はCH3(C=O)O−であり、下記の構造式を有する化合物。
【0015】
10.項目1に記載の化合物であって、R1はHO−、R2およびR4は夫々CH3O−、R3はCH3(C=O)O−であり、下記の構造式を有する化合物。
【0016】
11.細胞中のレンチウイルスのTatトランス活性化を抑制する方法であって:
a)ラレア・トリデンタータ(Larrea tridentata)からの抽出物を前記細胞に投与する工程であって、該抽出物は、図4に示すガスクロマトグラフィープロファイルを有する抽出物および図5に示すガスクロマトグラフィープロファイルを有する抽出物からなる群から選択される工程と;
b)該抽出物によって、前記細胞中のレンチウイルスのTatトランス活性化を抑制する工程とを具備した方法。
12.細胞中のレンチウイルスのTatトランス活性化を抑制する方法であって:
a)下記構造の化合物を前記細胞に投与する工程と;
【0017】
b)該化合物によって、前記細胞中のレンチウイルスのTatトランス活性化を抑制する工程とを具備した方法。
13.細胞中のレンチウイルスのTatトランス活性化を抑制する方法であって:
a)下記構造式の化合物を前記細胞に投与する工程と;
【0018】
ここで、R1、R2、R3およびR4は夫々、HO−、CH3O−およびCH3(C=O)O−からなる群から選択される。但し、R1、R2、R3およびR4は夫々同時にHO−ではない。
【0019】
b)該化合物によって、前記細胞中のレンチウイルスのTatトランス活性化を抑制する工程とを具備した方法。
14.項目13に記載の方法であって、R1、R2およびR3は夫々HO−、R4はCH3O−であり、前記化合物は下記の構造式を有する方法。
【0020】
15.項目13に記載の方法であって、R1、R2およびR4は夫々HO−、R3はCH3O−であり、前記化合物は下記の構造式を有する方法。
【0021】
16.項目13に記載の方法であって、R1およびR2は夫々HO−、R3はCH3(C=O)O−、R4はCH3O−であり、前記化合物は下記の構造式を有する方法。
【0022】
17.項目13に記載の方法であって、R1、R3およびR4は夫々CH3O−、R2はHO−であり、前記化合物は下記の構造式を有する方法。
【0023】
18.項目13に記載の方法であって、R1はHO−、R2、R4およびR3は夫々CH3O−であり、前記化合物は下記の構造式を有する方法。
【0024】
19.項目13に記載の方法であって、R1およびR3は夫々CH3O−、R2はHO−、R4はCH3(C=O)O−であり、前記化合物は下記の構造式を有する方法。
【0025】
20.項目13に記載の方法であって、R1およびR4は夫々CH3O−、R2はHO−、R3はCH3(C=O)O−であり、前記化合物は下記の構造式を有する方法。
【0026】
21.項目13に記載の方法であって、R1はHO−、R2およびR3は夫々CH3O−、R4はCH3(C=O)O−であり、前記化合物は下記の構造式を有する方法。
【0027】
22.項目13に記載の方法であって、R1はHO−、R2およびR4は夫々CH3O−、R3はCH3(C=O)O−であり、前記化合物は下記の構造式を有する方法。
【0028】
(発明の概要)
本件出願において、我々は、砂漠植物であるラレア・トリデンタータ(Larrea tridentata)のTat−TRS阻害活性を開示する。
【0029】
熱帯多雨林および砂漠の薬草植物から調製された、ウイルス疾患に対する伝統的な薬に用いられる幾つかの植物抽出物のうちで、メキシコ・ハマビシまたはクレオソートブッシュ(Larrea tridentata)の葉および花からの全抽出物のみが、Tat−TRS阻害活性を示した。この抽出物はまた、新しく開発された可溶性ホルマザン試験(soluble-formazan assay)によって評価されたように、該ウイルスに慢性的に感染したヒトリンパ芽球に対するHIV細胞変性効果を阻害する(Weislow et al.,JNCI81:577-586,1989)。
【0030】
本発明は、下記構造式の化合物を開示する。
【0031】
【化1】
【0032】
ここで、R1、R2、R3およびR4は夫々、HO−、CH3O−およびCH3(C=O)O−からなる群から選択される。但し、R1、R2、R3およびR4は夫々同時にHO−ではない。
【0033】
各化合物は、クレオソートブッシュであるラレア・トリデンタータ(Larrea tridentata)の葉−花抽出物から単離されたものであり、1,4−ビス−(3,4−ジヒドロキシフェニル)−2,3−ジメチルブタン(ノルジヒドロカイアレ酸(nordihydroquaiaretic acid)、NDGA)の誘導体である。
加えて、NDGAおよび各誘導体は、NDGAまたはその誘導体を細胞に投与することによって、該細胞において、HIVウイルスを含むレンチウイルスのTatトランス活性化を抑制するために使用することができる。
【発明を実施するための最良の形態】
【0034】
好ましい態様の詳細な説明
本発明は、1,4−ビス−(3,4−ジヒドロキシフェニル)−2,3−ジメチルブタン、即ち、ノルジヒドロカイアレ酸(NDGA)の誘導体の単離、精製および特徴付けに関する。以下の手順に従って、NDGAの各誘導体が単離され、精製され、特徴付けされた。
〈材料および方法〉
細胞ライン:SV40起源の複製を有するCOS−7細胞が、10%ウシ胎児血清(FCS)および抗生物質を補充したイスコーブ改変ダルベッコ培地(Isocove's Modified Dulbeccos Medium:IMDM)中で維持された。細胞は、37℃の95%O2/5%CO2湿潤インキュベータ中でインキュベートされた。
【0035】
プラスミド:Tat−感受性HIV LTRプロモータを有するプラスミドpBC12/HIV/SEAPであって、SEAPリポータ遺伝子を有するがTatがコ−ドする機能は有さないプラスミドを使用して、基底の活性のSEAPを発現した。pBC12/RSV/SEAPは、Tatがコ−ドする機能、すなわち誘導されたSEAPレベルを提供した。ラウス肉腫ウイルス由来の、Tat−非感受性の構成性LTRプロモ−タを有するpBC12/RSV/SEAPを、陽性の対照として利用した。プラスミドは全て、デュ−ク医療センタ−のブライアン・カレン(Bryan Cullen)博士より入手した。プラスミドpSEAP及びpBKCMV、並びにHIVLTR及びTat DNAは、クロンテック(Clontech)とストラタジ−ン(Stratagene)より、商業的に入手可能である。プラスミドの形質転換は、エ−ル(Yale)大学生物学科のバ−バラ・バックマン(Barbara Bachmann)博士から入手した大腸菌(E.coli)株MC1061で行った。大腸菌株MC1061はまた、クロンテックからも入手可能である。プラスミドDNAをキアゲン(Qiagen(登録商))・精製キット(キアゲン社)を利用して精製した。
【0036】
化学試薬:ジエタノ−ルアミン(#31589)及びp-ニトロフェニルリン酸塩(#71768)は、フルカ・バイオケミカより購入し、L-ホモアルギニン(#H−1007)はシグマ社より購入した。リポスペルミンDOGS(トランスフェクタム(登録商標)、#E123A、プロメガ社)をDNAの形質移入実験に使用した。
【0037】
植物性試験物質の準備:メキシコ・ハマビシの葉と花を、民族薬物学的要求に基づいて回収した。植物性物質は最初、クロロホルム:メタノ−ルを使用した連続的浸出により抽出した。抽出物を残渣にまで濃縮した。得られた全量176mgの粗抽出物を3mlのヘキサンで7回処理した。このステップにより、137mgのヘキサン不溶性(HI)物質と、31mgのヘキサン可溶性(HS)物質になった。これらの抽出物全てを、5.6%のH2SO4(v/v)中の2%CeSO4、硫酸セリウムチャ−リング(charring)を利用したSiO2TLCと、抗Tat-TRS活性のSEAPアッセイにより、ステップごとにモニタ−した。
【0038】
試験物質をSEAPアッセイ用に、カルシウム/マグネシウム・フリ−のPBSで調製して、10%DMSO溶液に溶解した。懸濁液を遠心して、ストック溶液(10mg/ml)を、マイレックス(Millex(登録商標))−GS22μmフィルタを(ミリポア社)利用してフィルタ−滅菌した。ストック溶液の適切な希釈液を、最終DMSO濃度がPBS中で0.2%になるように調製して、種々の濃度の試験物質を得た。
【0039】
向流クロマトグラフィ−(CCC)による、活性成分の差動的分画、及び精製:SEAPアッセイにおける、上記の分画の予備的結果に基づいて、CCCによる活性HI画分をさらに行った。これにより、二つの主要な活性画分(緑と、黄色の成分)が同定されるに至った。この見込みのある実験から、最も活性な画分が同定されたため、植物粉末を利用したフルスケ−ルの差動的分画を行い、大量の緑、及び黄色の画分を得る試みを行った。この分画は、101.4gの植物粉末を使って行なわれ、表1に概略が示されているように、ヘキサン処理から開始した。
【0040】
クロロホルム:水による分画後に得られた有機相(OG)由来の主要な成分に関する更なる分画は、イトとコンウェイにより記載されている、可変性交差軸プラネット遠心(versatile cross-axis planet centrifuge,CPC)を利用した、向流液−液分画クロマトグラフィ−により行った(Ito and Conway CRC Critical Reviews of Analytical Chemistry 17:65−,1986)。最適の溶媒系はヘキサン:EtOAc:MeOH:0.5%NaClが6:4:5:5の比率である、上相(有機層)が可動性相の混合物であった。5gの有機画分を23mlの二層混合物に溶解して、ル−プバルブを経由してコイル内へ導入した。800rpmで回転している間に、可動性相がコイルからくみ出された。約4ml/分の流速で、約32%の不変相が初期喪失した(68%が保持された)。可動性相が溶出物中に現れた後に、可動性相の画分を回収して、蒸発により乾燥させ、TLCでモニタ−して、5つのバッチ(前面溶媒(solvent front)、緑(Gr)、黄色(Ye)、赤、不変相)にプ−ルした。次にこれらの画分全てを、Tat-TRS活性に関してのSEAPアッセイによりモニタ−した。
【0041】
緑と黄色の画分の更なる精製を、イトーの多層コイル分離器−抽出器(Ito and Conway,CRC Critical Reviews of Analytical Chemistry 17:65-,1986)として知られる、周転円状(epicyclic)コイルプラネット遠心を利用したCCCにより行った。溶媒系は、ヘキサン:EtOH:MeOH:0.5%NaCl(7:3:5:5)であった。緑の画分200mgから、6.8mgの、Grと呼ばれる成分(すなわち元の植物粉末に基づくと約0.051%の全収量)が得られた。同様の実験を黄色の画分(Ye)でも行い、9.3mgの成分(Loと表される)が得られた。これらの精製済み成分(LoとGr)はそれぞれ複数の成分からなり、それぞれの「母」画分は、生物学的活性の試験と更なる特徴付けに供されるまで4℃で保存した。
【0042】
【数1】
【0043】
細胞培養、及びDNAの形質移入:COS細胞は、以前に記載された方法(Cullen Cell 46:973-982,1986)で維持した。DNAの形質移入は、リポスペルミン(トランスフェクタム、プロメガ社、#E123A)法(元はLoeffer and Behr,Methods in Enzymology 217:599-618,1993)を改変した方法を使用して行った。簡潔に言うと、DNA形質移入の前日に、直径17mmの平底面のリンブロ24(Linbro24(登録商標))プレ−トを、0.5mlの無菌の0.1%ゲラチン溶液で処理した。このプレ−トをフ−ド(hood)内で1時間保持した(特に言及しないかぎり、ステップは全てフ−ド内で行なわれた)。ゲラチン溶液を吸引して、該プレ−トを0.5mlのIMDM(10%のウシ胎児血清及び抗生物質で補充したもの(完全培地))で洗浄した。COS細胞を17mmのプレ−ト当たり、約1.5X105細胞でまいて、湿度95%/CO25%のインキュベ−タ−内で37℃でインキュベーションした。DNAの形質移入は、細胞の集密度が30−50%の時に行った。トランスフェクタム試薬(DOGS)のストック溶液は、製造業者推奨の方法にしたがって、蒸留水で作成した10%(v/v)のエタノ−ル溶液0.380ml当たり1mg(2.38mg/ml、または3.4mM)に調製した。
【0044】
形質移入用カクテルは、無菌のチュ−ブに調製した二つの溶液よりなる:
(a)溶液Aは、無菌の150mMNaCl溶液+プラスミドDNA(非選択遺伝子/選択遺伝子の比率が2:1)(ウェル当たり0.35μgのpBC12/HIV/SEAP+ウェル当たり0.175μgのpBC12/CMB/t2(Tat機能をコ−ド))
を含む;
(b)溶液Bは、等量の150mMNaClと、全量DNAの6倍であると決定されているトランスフェクタムを1容量を含む。
溶液AとBとを均一化してすぐに混合した。
【0045】
10分間反応を行わせた。その間に、増殖用培地をサブコンフルエントなCOS細胞より除去して、それぞれのウェルに300μl(完全IMDM100μlと無血清培地200μl)を加えた。形質移入カクテルを、等量でウェルに入れた。対照試料には無菌の150mMNaCl溶液のみを加えた。試料は全て、700μlの完全増殖培地を加えた後に、10から12時間インキュベーションした。5%DMSO/Ca-Mgフリ−PBS(非水溶性物質に対して)中に調製した試験化合物をすぐに、ウェルへ種々の濃度で加えた。薬剤未処理対照試料には、5%DMSO/PBS溶液のみを加えた(DMSOの最終濃度は0.2%)。次いで、試料全部を、30μlの培養上清をSEAPアッセイ用に除去した後に、更に48時間インキュベーションした。
【0046】
分泌アルカリホスファタ−ゼ(SEAP)アッセイ: 分泌アルカリホスファタ−ゼ分析は、最初に記載されたようにして行った(Berger et al.,Gene 66:1-10,1988)。簡潔に言うと、250μlのアリコットをCOS細胞の培養上清より除去して、65℃で5分間加熱して、選択的に内在性ホスファタ−ゼを不活化し(SEAPは熱にたいして安定である)、微量遠心管で2分間遠心した。100μlの2XSEAPアッセイ用緩衝液(1.0Mのジエタノ−ルアミンpH9.8、0.5mMMgCl2、10mML-ホモアルギニン)を100μlの試料のアリコットへ加えた。この溶液を混合して96穴の平底面の培養皿(Corning)へ移した。20μlの、予め暖めておいた基質溶液(1XSEAPアッセイ用緩衝液に溶解させた、120mMp-ニトロフェニルリン酸塩)をマルチピペッタ−で、反応混合液を含むそれぞれのウェルに分配した。反応物のA405を5分間隔で60分間、37℃で、EL3401マイクロプレ−トリ−ダ−(Biotek Instruments社)を使用して、各測定前に5秒間の自動振動を行って測定した。標準的なSEAP誘導のアッセイでの、吸光度の変化を時間に対してプロットした。薬剤スクリ−ニングアッセイでは、SEAP発現の阻害率を次のようにして30分時に計算した:
阻害率(%)=100−[(CT+−C+)X100]
ここで
C-:対照試料(DNAなし、薬剤なし);
CT-:対照試料(+DNA、薬剤なし);
C+:薬剤処理済み試料(DNAなし、+薬剤);
CT+:薬剤処理試料(+DNA、+薬剤)。
【0047】
形質移入技術の最適化: 種々の技術が、真核細胞のDNA形質転換に利用されている。これらの方法には、リン酸カルシウムやカチオン性ポリマ−によるDNA共沈殿法、化学的方法(洗剤、溶媒、酵素、両極性ポリマ−)、または物理的方法(熱的、浸透圧的若しくは電気ショック的、または粒子砲撃的なもの)によって細胞膜を弱らせる方法がある。これらの技術では、ある程度、効率と細胞毒性に関して、一定しないという難点がある。
【0048】
DNAの取り込みに関しての細胞の受入れ、すなわち無傷の細胞膜の通過に対して必要な条件は、「コンパクションとDNA電荷のマスキング」である(Loffler and Behr,Methods in Enzymology 217:599-618,1993)。新規のトランスフェクタム(transfectam;商標名)法では上記の条件をうまく満たしている。トランスフェクタム試薬(ジオクタデシルアミドグリシルスペルミン(dioctadecylamidoglycyl spermine))は、DNAに対して強い親和性(Kd=10-5−10-7M)を有する、+に帯電したスペルミン頭部を有した、カチオン性の合成リポポリアミンである。このスペルミン頭部はペプチド結合により、脂質部分に共有結合している。リポスペルミン分子は、DNAに結合して、それを脂質層にコ−ティングする。過剰なリポスペルミン存在下では、カチオン性脂質でコ−ティングされたプラスミドDNAの小胞が形成されて、該複合体の脂質部分が細胞膜と融合する。DNAのインタ−ナリゼ−ションはエンドサイトーシスにより起こると考えられている。
【0049】
トランスフェクタム−仲介性形質移入は、すでにある方法よりもより効率のよいものであることが示された(Barthel et al.,DNA and Cel Biology12(6):553-560,1993)。更に、トランスフェクタムは安定で、実質的に細胞無毒性の試薬である。しかしながら、特定のCOS細胞株における形質移入の最適化のための因子に関しての処理が必要である。これらの因子には、形質移入の期間、トランスフェクタム試薬のDNAに対する比率、DNA濃度、及びその他の希釈因子(例えばNaClの容量と強度)が含まれる。形質移入の条件の最適化を以下に示す。
【0050】
(a)形質移入の期間: 経時試験においてCOS細胞は、固定した濃度のプラスミドDNAとインキュベーションした。これらの試験は、種々の試験化合物によるSEAP発現試験のために、サブオプティマルなインキュベーション時間を選択することを目的とした。SEAP発現の経時誘導の結果は(非掲載)、SEAP発現は、漸進的な時間異存性上昇を示した。この誘導の開始は4時間未満でおきて、24時間で最大になった。10、12、15時間で見られた値の間には顕著な違いはなかった。よって、12−15時間の終末点を、以下の薬剤スクリ−ニングの全てにおける、SEAP発現の阻害に対して適切なサブオプティマル・インキュベーション期間として選択した。
【0051】
(b)DNA濃度: 形質移入に対して最適なDNA濃度は、トランスフェクタム試薬を使用した以前の研究に基づいて決定した(Loeffler and Behr,Methods in Enzymology,217:599-618,1993)。共形質移入に関しては、2:1(非選択遺伝子/選択遺伝子)の比率が最も適しているということが、他で報告されている(Hsuet al.,Science 254:1799-1802,1991)。リンブロ(登録商標)の24穴平底面の直径17mmプレ−トを使用して、非選択プラスミドpBC12/HIV/SEAPは、0.35μg/ウェルの濃度で、Tat機能をコ−ドしているpBC12/CMV/t2は、0.75μg/ウェルで使用した。
【0052】
(c)トランスフェクタムのDNAに対する比率と、イオン強度の決定: プラスミドDNAに対するトランスフェクタム(DOGS)の最適な比率、及び使用したNaClのイオン強度は、以前報告されている値を修飾したものであり(Loeffler and Behr,Methods in Enzymology,217:1799-1802,1993)、次のようにして決定した:
蒸留水で作成した10%(v/v)のエタノ−ル溶液0.400ml当たり、1mg(2.38mg/ml)の、オリジナルのトランスフェクトラムストック溶液からは、6倍量(μl)のストック溶液が、使用した各μgのDNAに対して必要であった。該溶液の最適のイオン強度は、適切な容量の150mMNaClにより提供され、以下の関係で決定される:
NaClの容量(μl)=トランスフェクタムの容量(μl)/0.6。
【0053】
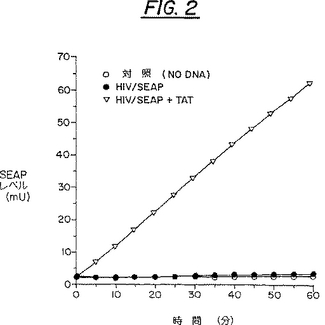
上記の条件の最適化後の、標準的アッセイにおけるTat-誘導SEAPレベルの結果は、図2に例示されている。簡単に言うと、COS細胞はイスコ−ブ改変ダルベッコ培地(IMDM)(10%ウシ胎仔血清(FCS)及び抗生物質で補充済み)中で維持した。3つ一組の細胞試料を、リンブロの24穴平底面、直径17mmプレ−トのウェル当たり、約1.5X105の密度でまいて、湿度95%/CO25%のインキュベ−タ中で、50%集密度が得られるまで37℃でインキュベーションした。サブコンフルエントな細胞をリポスペルミン法形質移入した(Loeffler and Behr,Methods in Enzymology,217:599-618,1993)。サブコンフルエントな細胞の培地を吸引して300μlの新鮮な最小培地(3%FCS補充済みIMDM)で置き換えた。COS細胞をpBC12/HIV/SEAPのみで(0.35μg/ウェル)、またはpBC12/CMV/t2(Tat機能をコ−ド、(0.175μg/ウェル))+pBC12/HIV/SEAP(0.35μg/ウェル)で、または緩衝液のみ(DNA対照試料なし)で形質移入した。プレ−トを12から15時間インキュベーションして、その後で700μlの完全培地(10%FCS補充済みIMDM)を加えた。次いで細胞を48時間インキュベーションし、その後で、COS細胞培養上清の250μlのアリコットをを除去して65℃で5分間加熱し、選択的に内在性ホスファタ−ゼを不活化した(SEAPは熱に安定である)。次いで試料を微量遠心管で2分間遠心した。100μlの2XSEAPアッセイ緩衝液(1.0Mジエタノ−ルアミンpH9.8;0.5mMMgCl2;10mML-ホモアルギニン)を、試料のアリコット100μlに加えた。この溶液を混合して、96穴の平底面培養皿(Corning社)に移した。マルチピペッタ−を使用して20μlの、予め暖めておいた基質溶液(1XSEAPアッセイ緩衝液に溶解した、120mMのp-ニトロフェニルリン酸塩)を、反応混合物が含まれているそれぞれのウェルに分配した。EL3401マイクロプレ−トリ−ダ−(Bio-tek Instrument社)を使用し、それぞれの測定ごとに5秒間の自動振動を行い、反応物のA405を60分間、37℃で5分間おきに測定した。吸光度を、以前に記載されているようにして、SEAPのmUに変換して(Berger et al.,Gene 66:1-10,1988)、時間に対してプロットした。
【0054】
これらの結果は、一時間後のSEAP誘導に関して、対照(DNAなし)のレベル、または非選択遺伝子(HIV/SEAP)のみの誘導と比較して、ほぼ65倍もの増加を示した。
【0055】
向流クロマトグラフィ−によるアッセイ先導性のメキシコ・ハマビシ抽出物活性組成物の単離: 上記したように、メキシコ・ハマビシ抽出物の、差動分画と、向流クロマトグラフィ−(CCC)による精製により、二つの主要な成分が単離された(表1)。Grと呼ばれる、6.8mgの成分はSiO2TLCの緑の画分より単離された。全回収率は、元の植物粉末に基づくと、約0.051%であった。成分Lo、9.3mgが、黄色い画分(Ye)より単離された。
【0056】
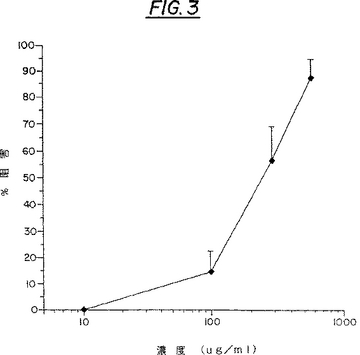
メキシコ・ハマビシの葉と花からの抽出物による、Tat-TRS活性の阻害:SEAPアッセイで試験した複数の植物抽出物のなかでは、メキシコ・ハマビシである、ラレア・トリデンタタ(Larrea tridentata)の葉と花からの抽出物のみが、HIVのTatタンパク質に関しての顕著な阻害活性を示した。メキシコ・ハマビシは、図3に示されているように、SEAP発現に関して、用量−反応性阻害を示した。簡単に言うと、上記したリポスペルミン法を使用して、3つ一組のCOS細胞の試料を、pBC12/HIV/SEAPとpCB12/CMV/t2(Tat機能をコ−ドしている)との2:1の比の混合物で形質移入した。形質移入後、細胞を12−15時間インキュベーションした。メキシコ・ハマビシの抽出物ストック溶液(10mg/ml)は、カルシウム/マグネシウム−フリ−PBSと10%DMSOで調製して、マイレックス−GS22μmフィルタ−(ミリポア)で滅菌した。適切な濃度のメキシコ・ハマビシ抽出物を、DMSOの最終濃度が0.2%になるようにして、形質移入細胞へ加えて、48時間インキュベーションした。SEAP分析用に250μlのアリコットをCOS細胞培養上清より除去して、65℃で5分間加熱して内在性ホスファタ−ゼを選択的に不活化し(SEAPは熱に対して安定)、微量遠心管で2分間遠心した。100μlの2XSEAPアッセイ用緩衝液(1.0Mジエタノ−ルアミンpH9.8,0.5mMMgCl2,10mML-ホモアルギニン)を100μlの試料のアリコットへ加えた。この溶液を混合して96穴の平底面培養皿(Corning)へ移した。20μlの予め暖めておいた基質溶液(1XSEAPアッセイ用緩衝液に溶解した、120mMp-ニトロフェニル-リン酸塩)を、反応物が含まれているそれぞれのウェルへ、マルチピペッタ−で分配した。反応物のA405を、60分間37℃で5分間ごとに、各測定前の5秒間自動振動付きEL340iマイクロプレ−トリ−ダ−(Bio-tekInstruments社)で測定した。SEAP発現の阻害率を以下のようにして、30分時に計算した:
阻害率(%)=100−[(CT+−C+)/(CT-−C-)X100]:
ここで、
C-:対照試料(DNAなし、薬剤なし)
CT-:対照試料(+DNA、薬剤なし)
C+:薬剤処理済み試料(DNAなし、+薬剤)
CT+:薬剤処理済み試料(+DNA、+薬剤)
図3で示されるように、この阻害は20μg/mlの濃度で始まり、600μg/mlで最大の阻害活性になった。この粗物質に対しての、推定のEC50(50%阻害率を示す濃度)は110μg/mlであった。活性成分の精製が進むにつれて、活性成分の活性度に上昇が見られ、これは元の全粗抽出物(21%)と比較して、有機相(OG)では3倍(68%)になった。
【0057】
HIVの細胞病変性効果の阻害: Tatのトランスアクティベ−ションを阻害する化合物は原理的に、HIVの複製を阻害するはずである。そのため国立癌研究所(NCI)において、可溶性フォルマザン(formazan)アッセイ(Weislow et al.,JNCI81:577-586,1989)を利用して、HIV-1の細胞病変性効果の阻害に関して、メキシコ・ハマビシの抽出物が試験された。おおまかに言うと次のとおりである。CEM-SS細胞(ATCC,Rockville,MD)をHIV-産生H9細胞と共培養する。ウイルスを宿主細胞CEM-SSに感染、複製させて、ほとんどのCEM-SS細胞を1週間で殺傷する。もし薬剤がHIVの産生を阻害するならば、CEM-SS細胞はHIV−誘導性の細胞死から保護されることになる。よってテトラゾリウム(XTT)試薬は、生細胞により代謝的に還元され、450nmで比色法により測定可能な有色のフォルマザン産物を生ずる。
【0058】
実際には、3つ一組のCEM-SS細胞(5000)を96穴マイクロタイタ−プレ−トにまいた。100μlの最終容量(カルシウム/マグネシウムフリ−PBS、5%DMSO)の、試験化合物を適切な濃度で加えた。5分後に500倍感染性のHIV-1を産生するH9細胞、または正常なH9細胞を、適切な濃度の薬剤を含むウェルに加えた。マイクロタイタ−プレ−トを、湿度95%/CO25%で、37℃で6日間インキュベーションして、その後にXTTとN-メチルフェナゾニウムメトサルフェ−ト(N-methylphenazolium methosulfate,PMS)の混合物50μlを加えた。プレ−トを再び4時間インキュベーションして発色させた(XTTフォルマザン産生)。プレ−トを密封し、その内容物を自動振動装置で混合し、試料のOD450をマイクロプレ−トリ−ダ−で決定した。それぞれの値は3回の決定の平均を表す。非感染細胞とHIV-1感染細胞との間の2つ一組の値の平均値には、試験化合物存在下において顕著な違いが見られなかった。対照的に、試験化合物存在下、または非存在下での、HIV-1感染細胞間には顕著な違い(p<0.05)が見られた。
【0059】
この実験の結果を表2にまとめた。成分Grの濃度0.75μg/mlで、平均して58%の対HIV保護(細胞生存度)が得られ、一方、薬剤フリ−の、HIV感染試料では生存度は15%であった。0.187μg/mlもの低い濃度において成分Loは、HIV細胞病変性効果に関しての更に強い阻害活性を示した。
細胞生存度は87%であって、これは、処理していない対照試料のもの(89%)と非常に近い値であり、薬剤フリ−のHIV感染試料の生存度14%と対照的であった。上記化合物は使用した濃度においては、細胞毒性がなかった。
【0060】
【表2】
【0061】
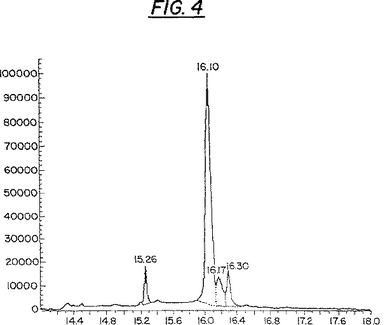
クレオソートブッシュ抽出物の活性成分の構造解明: 精製された植物活性成分の化学的な特徴付けは、主としてマススペクトル、並びにH−およびC−核磁気共鳴(NMR)によって行われた。成分Loは、四つの関連した化合物(L1,L2,L3およびL4)の混合物であることがわかった。該混合物の夫々のピークの解析および特徴付けは、マススペクトル分析器(MS)に取り付けられた、分析用の非破壊的キャピラリー架橋5%フェニルメチルシロキサン(HP−5)カラムを用いたガスクロマトグラフィー(GC)によって達成された。このGC研究によって、第一の化合物(L1)は当該混合物の6%に対応し;第二の化合物(L2)は当該混合物の76%に対応し(MW=316);また第三の化合物(L3)は当該混合物の9%に対応する(MW=358)ことが明らかになった。これらの化合物の溶出時間(分)は、各ピーク上に示されている(図4)。
【0062】
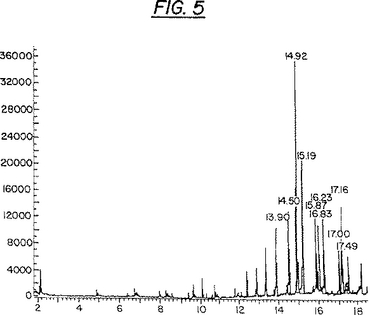
成分Grは15の化合物からなっている。該混合物の各ピークの解析および特徴付けは、マススペクトル分析器(MS)に取り付けられた、分析用の非破壊的キャピラリー架橋5%フェニルメチルシロキサン(HP−5)カラムを用いたガスクロマトグラフィー(GC)によって達成された。このGC研究により、15の化合物のうちの四つ(G1,G2,G3およびG4)はリガンドであり、構造的にはL化合物に関連していることが明らかになった。これらの化合物の溶出時間(分)は夫々のピーク上に示されている(図5)。
【0063】
これらの8種類の化合物(L1,L2,L3,L4,G1,G2,G3およびG4)の構造を以下に説明する。
【0064】
L1はC18H22O4の組成を有しており、以前に知られている化学物質、即ち、1,4−ビス−(3,4−ジヒドロキシフェニル)−2,3−ジメチルブタン(ノルジヒドロカイアレ酸、NDGA、メルクインデックス、第10版、#6534)として同定されている。L1の構造式は下記の通りである。
【0065】
【化2】
【0066】
L2はC19H24O4の組成を有しており、3−O−メチル−NDGA、または1−(3,4−ジヒドロキシフェニル)−4−(3−メトキシ−4−ヒドロキシフェニル)−2,3−ジメチルブタンとして同定されている。3−O−メチル−NDGAの構造式は下記の通りである。
【0067】
【化3】
【0068】
L3もまたC19H24O4の組成を有しており、4−O−メチル−NDGA、または1−(3,4−ジヒドロキシフェニル)−4−(3−ヒドロキシ−4−メトキシフェニル)−2,3−ジメチルブタンとして同定されている。4−O−メチル−NDGAはまた、Malachi4:5-6またはMal4としても知られている。4−O−メチル−NDGAの構造式は下記の通りである。
【0069】
【化4】
【0070】
L4はC21H26O5の組成を有しており、3−O−メチル−4−O−アセチル−NDGA、または1−(3,4−ジヒドロキシフェニル)−4−(3−メトキシ−4−アセトキシフェニル)−2,3−ジメチルブタンとして同定されている。3−O−メチル−4−O−アセチル−NDGAの構造式は下記の通りである。
【0071】
【化5】
【0072】
G1は分子量344およびC21H22O4の組成を有しており、3,3´,4−トリ−O−メチル−NDGA、または1−(3−ヒドロキシ−4−メトキシフェニル)−4−(3,4−ジメトキシフェニル)−2,3−ジメチルブタンとして同定されている。3,3´,4−トリ−O−メチル−NDGAは下記の構造式を有している。
【0073】
【化6】
【0074】
G2は分子量344およびC21H22O4の組成を有しており、3,4,4´−トリ−O−メチル−NDGA、または1−(3−メトキシ−4−ヒドロキシフェニル)−4−(3,4−ジメトキシフェニル)−2,3−ジメチルブタンとして同定されている。3,4,4´−トリ−O−メチル−NDGAは下記の構造式を有している。
【0075】
【化7】
【0076】
G3およびG4は夫々、分子量374およびC22H28O5の組成を有している。G3は、3´,4−ジ−O−メチル−3−O−アセチル−NDGA(G3a)、または3,3´−ジ−O−メチル−4−O−アセチル−NDGA(G3b)の何れかである。3´,4−ジ−O−メチル−3−O−アセチル−NDGAはまた、1−(3−メトキシ−4−ヒドロキシフェニル)−4−(3−アセトキシ−4−メトキシフェニル)−2,3−ジメチルブタンとしても知られており、下記の構造式を有している。
G3a:
【0077】
【化8】
【0078】
3,3´−ジ−O−メチル−4−O−アセチル−NDGAはまた、1−(3−メトキシ−4−ヒドロキシフェニル)−4−(3−メトキシ−4−アセトキシフェニル)−2,3−ジメチルブタンとしても知られており、下記の構造式を有している。
G3b:
【0079】
【化9】
【0080】
同様にして、G4は、4,4´−ジ−O−メチル−3−O−アセチル−NDGA(G4a)、または3,4´−ジ−O−メチル−4−O−アセチル−NDGA(G4b)の何れかである。4,4´−ジ−O−メチル−3−O−アセチル−NDGAはまた、1−(3−ヒドロキシ−4−メトキシフェニル)−4−(3−アセトキシ−4−メトキシフェニル)−2,3−ジメチルブタンとしても知られており、下記の構造式を有している。
G4a:
【0081】
【化10】
【0082】
3,4´−ジ−O−メチル−4−O−アセチル−NDGAはまた、1−(3−ヒドロキシ−4−メトキシフェニル)−4−(3−メトキシ−4−アセトキシフェニル)−2,3−ジメチルブタンとしても知られており、下記の構造式を有している。
G4b:
【0083】
【化11】
【0084】
上記で述べた単離および精製の手順に加えて、開示された夫々のNDGA誘導体は、イケヤ等の方法(Ikeya et.al.,Chem.Pharm.Bull.27(7): 1583-1588,1979)に従って、NDGAのメチル化および/またはアシル化による化学合成によって製造してもよい。
【0085】
成分Loおよび成分Loからの二つの純粋な化合物(L2およびL3)の大規模精製: 植物材料バッチの大規模CCC分画を開始して、大量の成分Loを生じさせた。合計100gの植物粉末を、まず700mlのヘキサンで5回処理した。ヘキサン可溶物(1.17g)を廃棄した。ヘキサン不溶物(HI画分)を乾燥し、これを800mlのクロロホルム:メタノールで連続的に浸出することにより3回抽出した。これによって20gの全抽出物(Tex)を得、これを先のバッチ式微分抽出(batch differential extraction)で得たHI画分7.6gと混合した。こうして得られた組成植物材料の27.6gの全体を、10gおよび17.6gの二つのバッチに分割した。これらのバッチは、最初は、比率が6:4:5:5のヘキサン:EtOAc:MeOH:0.5%NaClの溶媒系を、上相(有機相)を可動相として用いることにより、大容量の多用途交差軸CPC(versatile Cross-Axis CPC;Shinomiya et al.,J.Chromatogr.644:215-229,1993)上で別々に運転された。各画分はTLCパターンに従って独自にプールされた。二つのCCC操作から四つの主要な画分が同定され、画分緑(Gr)(1.12g)、画分Lo(2.87g)、画分End(1.78g)および最後に静止相SP(20.84g)と表示された。画分Loの全体の2.87gは、大モデルのトリプレットCPC中において、水相を移動相に用いたヘキサン:EtOAc:MeOH:H2O(比率は7:3:5:5)によって更に分画された。溶出の順序およびTLCパターンに従って、LYI(0.375g);LYII(0.113g);LYIII(0.280g);およびLYIV(2.
80g)と表示された四種類の画分が同定された。これらの画分は、10μg/mlの濃度で抗Tat−TRS活性について試験された。この試験結果に基づいて、LYIが更なる精製のために選択された。
【0086】
Lo成分のLYI画分からの純粋なL2化合物およびL3化合物の単離: 親水性のより小さい画分LYIが、先に改良された条件、即ち、トリプレットCPCと、ヘキサン:CHCl3:MeOH:10mMNaClの溶媒系(比率は1:4:4:2)との条件を用いた更なる精製のために選択された。これによって、NMRおよびマススペクトルで試験したときに、148mgの均一なL3および109.3mgの純粋なL2の調製品が得られた。L3(Malachi4:5-6)およびL2の構造は上記で述べた通りである。
【0087】
化合物L2およびL3は、以前に同定された化学物質である1,4−ビス−(3,4−ジヒドロキシフェニル)−2,3−ジメチルブタン(ノルジヒドロカイアレ酸、NDGA、メルクインデックス、第10版、#6534)の誘導体である。NDGAの構造式は、既述したL1の構造式と同じであり、下記の通りである。
【0088】
【化12】
【0089】
NDGAおよびその誘導体の抗HIV活性(Tatに調節されたHIVトランス活性化の阻害)は、以前は知られていなかった。NDGAおよび誘導体Malachi4の抗HIVトランス活性化能の比較が図6に示されている。簡単にいえば、サブコンフルーエントなCOS細胞の複製サンプルを、上記のリポスペルミン法を用いて、プラスミドpBC12/HIV/SEAPおよびpBC12/CMB/t2(Tat機能をコードする)で共トランスフェクトした。次いで、12時間〜15時間、細胞をインキュベートした。試験サンプルは、先ず10%DMSO/カルシウム−マグネシウムを含まないPBS中で可溶化され、最終DMSO濃度0.2%の適切な濃度で、形質転換された細胞に添加された。サンプルは48時間インキュベートされ、その後、250μlのアリコートをCOS細胞培養上清から取り出し、図3に示すような標準的な試験でSEAPを分析した。SEAP発現のパーセント阻害は、30分の時点で次のようにして計算された。
【0090】
%阻害=100−[(CT+−C+)/(CT-−C-)×100]
但し、
C- : 対照サンプル(DNAなし、薬物なし)
CT-: 対照サンプル(+DNA、薬物なし)
C+ : 薬物処理したサンプル(DNAなし、+薬物)
CT+: 薬物処理したサンプル(+DNA、+薬物)
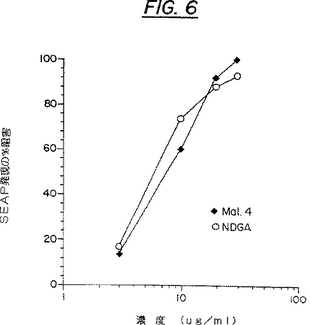
各点は、二回の測定の平均を表している。Mal4およびNDGAのEC50Sの間に顕著な相違はみられず、夫々8μg/ml(25μM)および6μg/ml(20μM)であった。このEC50Sは、Tat調節されたHIVトランス活性化が未処理対照細胞のそれの50%にまで減少するような、その化合物の阻害濃度として定義される。
【0091】
表3において、Mal4およびNDGAによる、HIVプロモータ活性のトランス活性化阻害を比較した。
【0092】
【表3】
【0093】
化合物NDGAおよびMal4は、図6に記載されたようにして試験された。対照サンプルは4回繰り返して実行された。パーセント阻害は30分後に測定去れ、OD405値は、次の通りであった。
【0094】
C-: 対照サンプル(DNAなし、薬物なし):0.091
CT-: 対照サンプル(+DNA、薬物なし):0.805
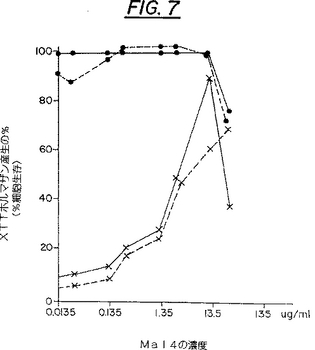
Mal4処理されたCEM−SS細胞の培養物における生存細胞の測定としての、XTTホルマザン産生の定量が図7に示されている。EC50は、感染した培養物中でのXTTホルマザン産生を、非感染の非処理培養細胞中におけるそれの50%にまで増加させる(保護する)、Mal4の濃度(例えば13.4μM)を表している。IC50は、非感染培養物中でのXTTホルマザン産生を、非感染の非処理対照細胞中におけるそれの50%にまで減少させる、Mal4の阻害濃度または有毒濃度(例えば325μM、見積り)を表している。非処理の感染された細胞におけるXTTホルマザンのレベルは、非処理かつ非感染の対照細胞におけるレベルの9%であった。HIV−1細胞変成効果の可溶性−ホルマザン試験は、文献(Weislow et al.,JNCI81:577-586,1989)に記載の方法に従って行われた。
【0095】
現在のところ最も実際的で且つ好ましいと思われる態様に関連して本発明を説明してきたが、本発明はこの開示された態様に限定されるものではなく、添付の請求の範囲の精神および範囲内に含まれる種々の変形および均等な構成をもカバーするものとして理解されるべきである。
【0096】
従って、請求の範囲に定義された本発明の新規な側面を逸脱することなく、NDGA誘導体およびTatトランス活性の抑制方法における変形を行い得ることが理解されるべきである。
【図面の簡単な説明】
【0097】
【図1】図1は、HIVのライフサイクルと、Tat−TRS阻害剤を含む潜在的治療剤の異なった作用部位を図示的に示している。基本的な転写ステップは1で示されており、ウイルス調節タンパク依存性のトランス活性化ステップは2で示されている。
【図2】図2は、標準的なSEAP試験における分泌されたアルカリホスファターゼ(SEAP)発現の誘導を示している。
【図3】図3は、分泌されたアルカリホスファターゼ(SEAP)試験における、クレオソートブッシュ全抽出物によるTat−TRS活性の阻害を示している。
【図4】図4は、分析用の非破壊性キャピラリー架橋5%フェニルメチルシロキサン(HP−5)カラムを用いたガスクロマトグラフィー(GC)による、成分Lo中の植物由来のHIV・Tat阻害剤の分析を示している。成分Loは、四つの成分の混合物である。溶出の時間(分)は各ピークについて出現する。
【図5】図5は、分析用の非破壊的キャピラリー架橋5%フェニルメチルシロキサン(analytical non-destructive capillarycross-linked 5% phenylmethylsiloxane)(HP−5)カラムを用いたガスクロマトグラフィー(GC)による、成分Gr中の植物由来HIV・Tat阻害剤の分析を示している。成分Grは複雑な混合物である。溶出の時間(分)は各ピークについて出現する。
【図6】図6は、分泌されたアルカリホスファターゼ(SEAP)試験における、植物由来の単一化合物であるMalachi4:5-6(Mal 4)およびNDGAによる、Tat誘導SEAP発現の阻害を示している。
【図7】図7は、二つの別々の実験における、HIVに感染し若しくは感染しないCEM−SS細胞のMal4処理培養における生存細胞の測定としての、XTTホルマザン産生の定量を示している。Mal4処理されていない感染細胞についてのXTTホルマザン産生パーセントは、実験1については8.9%であり、実験2については7.5%である。実験1では、非感染細胞は●・・・・・・●で表されており、感染細胞は×…×で表されている。実験2では、非感染細胞は●・・・・・・●で表されており、感染細胞は×…×で表されている。EC50は4.25μg/mlまたは13.4μMであることが分かった。IC50は100μg/mlまたは325μMと見積もられた。
【技術分野】
【0001】
本発明は、1,4−ビス−(3,4−ジヒドロキシフェニル)−2,3−ジメチルブタン(ノルジヒドロカイアレ酸(nordihydroquaiaretic acid)、NDGA)の誘導体の単離、精製および特徴付けに関する。この誘導体は、メキシコ・ハマビシまたはクレオソートブッシュ(creosolte bush; Larrea tridentata,Zygophyllaceae)の葉および花の抽出物から単離されたものであり、NDGAと共に、HIVウイルスを含むレンチウイルスにおけるTatトランス活性化を抑制するために用いることができる。
【背景技術】
【0002】
(関連技術の説明)
Tatはヒト免疫不全ウイルス(HIV)遺伝子発現のトランス活性化因子であり、HIV遺伝子発現のために必要な二以上のウイルス性調節因子(TatおよびRev)の一つである。Tatは、TAR・RNA要素に結合することによって作用し、長い末端繰り返し(LTR)プロモータからの転写を活性化する。
【0003】
Tatタンパクは転写の延長を安定化させるものであり、また転写の開始にも関与することが示されている。以前の研究によって、Tatは抗体依存性T細胞増殖の減少を媒介し、免疫応答の失敗に実質的に寄与することが示されている。Tatはまた、カポシ細胞増殖(Kaposi'scell growth)を直接刺激する。
【0004】
Tatは明らかな細胞同族体を有していないので、この強力な陽性調節因子は抗AIDS薬の開発のための魅力的な目標になっている(図1)。感染の新たなラウンドを妨害する現在入手可能なHIV逆転写阻害因子(AZT、DDI)または潜在的なプロテアーゼ阻害因子とは対照的に、組み込まれたプロウイルスDNAの、ウイルス遺伝子Tatで調節された発現を抑制する阻害因子は、該ウイルスを初期段階で停止させるであろう(非特許文献1)。
【0005】
真核宿主における転写レベルおよびポスト転写レベルでの遺伝子発現を制御する因子の解明を目的とした努力によって、最近、Tat機能の定量的な評価が可能になった(非特許文献2)。Tat調節されたトランス活性化(Tat−TRS)の阻害因子をスクリーニングするために、分泌された胎盤アルカリホスファターゼ(SEAP)リポータ遺伝子を、プラスミドpBC12/HIV/SEAPの中のHIV−1・LTRプロモータの制御下に置く。Tatにコードされる活性は、第二のプラスミド構築物pBC/CMV/t2によって供給される。これら二つのプラスミドを用いたCOS細胞の一時的な共感染によって、培地中へのアルカリホスファターゼの分泌が導かれ、この培地は単純な発色試験によって分析される(非特許文献3)。従って、このSEAP試験は、Tatトランス活性化の間接的な測定を提供する。阻害剤は、Tatタンパク(Tat−TRS)がHIV−1・LTRのトランス活性化を介してSEAP・mRNAを発現させるのを阻害することにより、SEAP活性の減少を生じさせるに違いない。
【非特許文献1】Hsu et al,Science 1991年 第254巻 p.1799-1820
【非特許文献2】Sim, Ann.N.Y.Acad.Sci 1990年 .第616巻 p.64-70
【非特許文献3】Berger et al.,Gene 1988年 第66巻 p.1-10
【発明の開示】
【課題を解決するための手段】
【0006】
本発明によって、以下が提供される:
1.下記構造式の化合物。
【0007】
ここで、R1、R2、R3およびR4は夫々、HO−、CH3O−およびCH3(C=O)O−からなる群から選択される。但し、R1、R2、R3およびR4は夫々同時にHO−ではない。
2.項目1に記載の化合物であって、R1、R2およびR3は夫々HO−、R4はCH3O−であり、下記の構造式を有する化合物。
【0008】
3.項目1に記載の化合物であって、R1、R2およびR4は夫々HO−、R3はCH3O−であり、下記の構造式を有する化合物。
【0009】
4.項目1に記載の化合物であって、R1およびR2は夫々HO−、R3はCH3(C=O)O−、R4はCH3O−であり、下記の構造式を有する化合物。
【0010】
5.項目1に記載の化合物であって、R1、R3およびR4は夫々CH3O−、R2はHO−であり、下記の構造式を有する化合物。
【0011】
6.項目1に記載の化合物であって、R1はHO−、R2、R4およびR3は夫々CH3O−であり、下記の構造式を有する化合物。
【0012】
7.項目1に記載の化合物であって、R1およびR3は夫々CH3O−、R2はHO−、R4はCH3(C=O)O−であり、下記の構造式を有する化合物。
【0013】
8.項目1に記載の化合物であって、R1およびR4は夫々CH3O−、R2はHO−、R3はCH3(C=O)O−であり、下記の構造式を有する化合物。
【0014】
9.項目1に記載の化合物であって、R1はHO−、R2およびR3は夫々CH3O−、R4はCH3(C=O)O−であり、下記の構造式を有する化合物。
【0015】
10.項目1に記載の化合物であって、R1はHO−、R2およびR4は夫々CH3O−、R3はCH3(C=O)O−であり、下記の構造式を有する化合物。
【0016】
11.細胞中のレンチウイルスのTatトランス活性化を抑制する方法であって:
a)ラレア・トリデンタータ(Larrea tridentata)からの抽出物を前記細胞に投与する工程であって、該抽出物は、図4に示すガスクロマトグラフィープロファイルを有する抽出物および図5に示すガスクロマトグラフィープロファイルを有する抽出物からなる群から選択される工程と;
b)該抽出物によって、前記細胞中のレンチウイルスのTatトランス活性化を抑制する工程とを具備した方法。
12.細胞中のレンチウイルスのTatトランス活性化を抑制する方法であって:
a)下記構造の化合物を前記細胞に投与する工程と;
【0017】
b)該化合物によって、前記細胞中のレンチウイルスのTatトランス活性化を抑制する工程とを具備した方法。
13.細胞中のレンチウイルスのTatトランス活性化を抑制する方法であって:
a)下記構造式の化合物を前記細胞に投与する工程と;
【0018】
ここで、R1、R2、R3およびR4は夫々、HO−、CH3O−およびCH3(C=O)O−からなる群から選択される。但し、R1、R2、R3およびR4は夫々同時にHO−ではない。
【0019】
b)該化合物によって、前記細胞中のレンチウイルスのTatトランス活性化を抑制する工程とを具備した方法。
14.項目13に記載の方法であって、R1、R2およびR3は夫々HO−、R4はCH3O−であり、前記化合物は下記の構造式を有する方法。
【0020】
15.項目13に記載の方法であって、R1、R2およびR4は夫々HO−、R3はCH3O−であり、前記化合物は下記の構造式を有する方法。
【0021】
16.項目13に記載の方法であって、R1およびR2は夫々HO−、R3はCH3(C=O)O−、R4はCH3O−であり、前記化合物は下記の構造式を有する方法。
【0022】
17.項目13に記載の方法であって、R1、R3およびR4は夫々CH3O−、R2はHO−であり、前記化合物は下記の構造式を有する方法。
【0023】
18.項目13に記載の方法であって、R1はHO−、R2、R4およびR3は夫々CH3O−であり、前記化合物は下記の構造式を有する方法。
【0024】
19.項目13に記載の方法であって、R1およびR3は夫々CH3O−、R2はHO−、R4はCH3(C=O)O−であり、前記化合物は下記の構造式を有する方法。
【0025】
20.項目13に記載の方法であって、R1およびR4は夫々CH3O−、R2はHO−、R3はCH3(C=O)O−であり、前記化合物は下記の構造式を有する方法。
【0026】
21.項目13に記載の方法であって、R1はHO−、R2およびR3は夫々CH3O−、R4はCH3(C=O)O−であり、前記化合物は下記の構造式を有する方法。
【0027】
22.項目13に記載の方法であって、R1はHO−、R2およびR4は夫々CH3O−、R3はCH3(C=O)O−であり、前記化合物は下記の構造式を有する方法。
【0028】
(発明の概要)
本件出願において、我々は、砂漠植物であるラレア・トリデンタータ(Larrea tridentata)のTat−TRS阻害活性を開示する。
【0029】
熱帯多雨林および砂漠の薬草植物から調製された、ウイルス疾患に対する伝統的な薬に用いられる幾つかの植物抽出物のうちで、メキシコ・ハマビシまたはクレオソートブッシュ(Larrea tridentata)の葉および花からの全抽出物のみが、Tat−TRS阻害活性を示した。この抽出物はまた、新しく開発された可溶性ホルマザン試験(soluble-formazan assay)によって評価されたように、該ウイルスに慢性的に感染したヒトリンパ芽球に対するHIV細胞変性効果を阻害する(Weislow et al.,JNCI81:577-586,1989)。
【0030】
本発明は、下記構造式の化合物を開示する。
【0031】
【化1】
【0032】
ここで、R1、R2、R3およびR4は夫々、HO−、CH3O−およびCH3(C=O)O−からなる群から選択される。但し、R1、R2、R3およびR4は夫々同時にHO−ではない。
【0033】
各化合物は、クレオソートブッシュであるラレア・トリデンタータ(Larrea tridentata)の葉−花抽出物から単離されたものであり、1,4−ビス−(3,4−ジヒドロキシフェニル)−2,3−ジメチルブタン(ノルジヒドロカイアレ酸(nordihydroquaiaretic acid)、NDGA)の誘導体である。
加えて、NDGAおよび各誘導体は、NDGAまたはその誘導体を細胞に投与することによって、該細胞において、HIVウイルスを含むレンチウイルスのTatトランス活性化を抑制するために使用することができる。
【発明を実施するための最良の形態】
【0034】
好ましい態様の詳細な説明
本発明は、1,4−ビス−(3,4−ジヒドロキシフェニル)−2,3−ジメチルブタン、即ち、ノルジヒドロカイアレ酸(NDGA)の誘導体の単離、精製および特徴付けに関する。以下の手順に従って、NDGAの各誘導体が単離され、精製され、特徴付けされた。
〈材料および方法〉
細胞ライン:SV40起源の複製を有するCOS−7細胞が、10%ウシ胎児血清(FCS)および抗生物質を補充したイスコーブ改変ダルベッコ培地(Isocove's Modified Dulbeccos Medium:IMDM)中で維持された。細胞は、37℃の95%O2/5%CO2湿潤インキュベータ中でインキュベートされた。
【0035】
プラスミド:Tat−感受性HIV LTRプロモータを有するプラスミドpBC12/HIV/SEAPであって、SEAPリポータ遺伝子を有するがTatがコ−ドする機能は有さないプラスミドを使用して、基底の活性のSEAPを発現した。pBC12/RSV/SEAPは、Tatがコ−ドする機能、すなわち誘導されたSEAPレベルを提供した。ラウス肉腫ウイルス由来の、Tat−非感受性の構成性LTRプロモ−タを有するpBC12/RSV/SEAPを、陽性の対照として利用した。プラスミドは全て、デュ−ク医療センタ−のブライアン・カレン(Bryan Cullen)博士より入手した。プラスミドpSEAP及びpBKCMV、並びにHIVLTR及びTat DNAは、クロンテック(Clontech)とストラタジ−ン(Stratagene)より、商業的に入手可能である。プラスミドの形質転換は、エ−ル(Yale)大学生物学科のバ−バラ・バックマン(Barbara Bachmann)博士から入手した大腸菌(E.coli)株MC1061で行った。大腸菌株MC1061はまた、クロンテックからも入手可能である。プラスミドDNAをキアゲン(Qiagen(登録商))・精製キット(キアゲン社)を利用して精製した。
【0036】
化学試薬:ジエタノ−ルアミン(#31589)及びp-ニトロフェニルリン酸塩(#71768)は、フルカ・バイオケミカより購入し、L-ホモアルギニン(#H−1007)はシグマ社より購入した。リポスペルミンDOGS(トランスフェクタム(登録商標)、#E123A、プロメガ社)をDNAの形質移入実験に使用した。
【0037】
植物性試験物質の準備:メキシコ・ハマビシの葉と花を、民族薬物学的要求に基づいて回収した。植物性物質は最初、クロロホルム:メタノ−ルを使用した連続的浸出により抽出した。抽出物を残渣にまで濃縮した。得られた全量176mgの粗抽出物を3mlのヘキサンで7回処理した。このステップにより、137mgのヘキサン不溶性(HI)物質と、31mgのヘキサン可溶性(HS)物質になった。これらの抽出物全てを、5.6%のH2SO4(v/v)中の2%CeSO4、硫酸セリウムチャ−リング(charring)を利用したSiO2TLCと、抗Tat-TRS活性のSEAPアッセイにより、ステップごとにモニタ−した。
【0038】
試験物質をSEAPアッセイ用に、カルシウム/マグネシウム・フリ−のPBSで調製して、10%DMSO溶液に溶解した。懸濁液を遠心して、ストック溶液(10mg/ml)を、マイレックス(Millex(登録商標))−GS22μmフィルタを(ミリポア社)利用してフィルタ−滅菌した。ストック溶液の適切な希釈液を、最終DMSO濃度がPBS中で0.2%になるように調製して、種々の濃度の試験物質を得た。
【0039】
向流クロマトグラフィ−(CCC)による、活性成分の差動的分画、及び精製:SEAPアッセイにおける、上記の分画の予備的結果に基づいて、CCCによる活性HI画分をさらに行った。これにより、二つの主要な活性画分(緑と、黄色の成分)が同定されるに至った。この見込みのある実験から、最も活性な画分が同定されたため、植物粉末を利用したフルスケ−ルの差動的分画を行い、大量の緑、及び黄色の画分を得る試みを行った。この分画は、101.4gの植物粉末を使って行なわれ、表1に概略が示されているように、ヘキサン処理から開始した。
【0040】
クロロホルム:水による分画後に得られた有機相(OG)由来の主要な成分に関する更なる分画は、イトとコンウェイにより記載されている、可変性交差軸プラネット遠心(versatile cross-axis planet centrifuge,CPC)を利用した、向流液−液分画クロマトグラフィ−により行った(Ito and Conway CRC Critical Reviews of Analytical Chemistry 17:65−,1986)。最適の溶媒系はヘキサン:EtOAc:MeOH:0.5%NaClが6:4:5:5の比率である、上相(有機層)が可動性相の混合物であった。5gの有機画分を23mlの二層混合物に溶解して、ル−プバルブを経由してコイル内へ導入した。800rpmで回転している間に、可動性相がコイルからくみ出された。約4ml/分の流速で、約32%の不変相が初期喪失した(68%が保持された)。可動性相が溶出物中に現れた後に、可動性相の画分を回収して、蒸発により乾燥させ、TLCでモニタ−して、5つのバッチ(前面溶媒(solvent front)、緑(Gr)、黄色(Ye)、赤、不変相)にプ−ルした。次にこれらの画分全てを、Tat-TRS活性に関してのSEAPアッセイによりモニタ−した。
【0041】
緑と黄色の画分の更なる精製を、イトーの多層コイル分離器−抽出器(Ito and Conway,CRC Critical Reviews of Analytical Chemistry 17:65-,1986)として知られる、周転円状(epicyclic)コイルプラネット遠心を利用したCCCにより行った。溶媒系は、ヘキサン:EtOH:MeOH:0.5%NaCl(7:3:5:5)であった。緑の画分200mgから、6.8mgの、Grと呼ばれる成分(すなわち元の植物粉末に基づくと約0.051%の全収量)が得られた。同様の実験を黄色の画分(Ye)でも行い、9.3mgの成分(Loと表される)が得られた。これらの精製済み成分(LoとGr)はそれぞれ複数の成分からなり、それぞれの「母」画分は、生物学的活性の試験と更なる特徴付けに供されるまで4℃で保存した。
【0042】
【数1】
【0043】
細胞培養、及びDNAの形質移入:COS細胞は、以前に記載された方法(Cullen Cell 46:973-982,1986)で維持した。DNAの形質移入は、リポスペルミン(トランスフェクタム、プロメガ社、#E123A)法(元はLoeffer and Behr,Methods in Enzymology 217:599-618,1993)を改変した方法を使用して行った。簡潔に言うと、DNA形質移入の前日に、直径17mmの平底面のリンブロ24(Linbro24(登録商標))プレ−トを、0.5mlの無菌の0.1%ゲラチン溶液で処理した。このプレ−トをフ−ド(hood)内で1時間保持した(特に言及しないかぎり、ステップは全てフ−ド内で行なわれた)。ゲラチン溶液を吸引して、該プレ−トを0.5mlのIMDM(10%のウシ胎児血清及び抗生物質で補充したもの(完全培地))で洗浄した。COS細胞を17mmのプレ−ト当たり、約1.5X105細胞でまいて、湿度95%/CO25%のインキュベ−タ−内で37℃でインキュベーションした。DNAの形質移入は、細胞の集密度が30−50%の時に行った。トランスフェクタム試薬(DOGS)のストック溶液は、製造業者推奨の方法にしたがって、蒸留水で作成した10%(v/v)のエタノ−ル溶液0.380ml当たり1mg(2.38mg/ml、または3.4mM)に調製した。
【0044】
形質移入用カクテルは、無菌のチュ−ブに調製した二つの溶液よりなる:
(a)溶液Aは、無菌の150mMNaCl溶液+プラスミドDNA(非選択遺伝子/選択遺伝子の比率が2:1)(ウェル当たり0.35μgのpBC12/HIV/SEAP+ウェル当たり0.175μgのpBC12/CMB/t2(Tat機能をコ−ド))
を含む;
(b)溶液Bは、等量の150mMNaClと、全量DNAの6倍であると決定されているトランスフェクタムを1容量を含む。
溶液AとBとを均一化してすぐに混合した。
【0045】
10分間反応を行わせた。その間に、増殖用培地をサブコンフルエントなCOS細胞より除去して、それぞれのウェルに300μl(完全IMDM100μlと無血清培地200μl)を加えた。形質移入カクテルを、等量でウェルに入れた。対照試料には無菌の150mMNaCl溶液のみを加えた。試料は全て、700μlの完全増殖培地を加えた後に、10から12時間インキュベーションした。5%DMSO/Ca-Mgフリ−PBS(非水溶性物質に対して)中に調製した試験化合物をすぐに、ウェルへ種々の濃度で加えた。薬剤未処理対照試料には、5%DMSO/PBS溶液のみを加えた(DMSOの最終濃度は0.2%)。次いで、試料全部を、30μlの培養上清をSEAPアッセイ用に除去した後に、更に48時間インキュベーションした。
【0046】
分泌アルカリホスファタ−ゼ(SEAP)アッセイ: 分泌アルカリホスファタ−ゼ分析は、最初に記載されたようにして行った(Berger et al.,Gene 66:1-10,1988)。簡潔に言うと、250μlのアリコットをCOS細胞の培養上清より除去して、65℃で5分間加熱して、選択的に内在性ホスファタ−ゼを不活化し(SEAPは熱にたいして安定である)、微量遠心管で2分間遠心した。100μlの2XSEAPアッセイ用緩衝液(1.0Mのジエタノ−ルアミンpH9.8、0.5mMMgCl2、10mML-ホモアルギニン)を100μlの試料のアリコットへ加えた。この溶液を混合して96穴の平底面の培養皿(Corning)へ移した。20μlの、予め暖めておいた基質溶液(1XSEAPアッセイ用緩衝液に溶解させた、120mMp-ニトロフェニルリン酸塩)をマルチピペッタ−で、反応混合液を含むそれぞれのウェルに分配した。反応物のA405を5分間隔で60分間、37℃で、EL3401マイクロプレ−トリ−ダ−(Biotek Instruments社)を使用して、各測定前に5秒間の自動振動を行って測定した。標準的なSEAP誘導のアッセイでの、吸光度の変化を時間に対してプロットした。薬剤スクリ−ニングアッセイでは、SEAP発現の阻害率を次のようにして30分時に計算した:
阻害率(%)=100−[(CT+−C+)X100]
ここで
C-:対照試料(DNAなし、薬剤なし);
CT-:対照試料(+DNA、薬剤なし);
C+:薬剤処理済み試料(DNAなし、+薬剤);
CT+:薬剤処理試料(+DNA、+薬剤)。
【0047】
形質移入技術の最適化: 種々の技術が、真核細胞のDNA形質転換に利用されている。これらの方法には、リン酸カルシウムやカチオン性ポリマ−によるDNA共沈殿法、化学的方法(洗剤、溶媒、酵素、両極性ポリマ−)、または物理的方法(熱的、浸透圧的若しくは電気ショック的、または粒子砲撃的なもの)によって細胞膜を弱らせる方法がある。これらの技術では、ある程度、効率と細胞毒性に関して、一定しないという難点がある。
【0048】
DNAの取り込みに関しての細胞の受入れ、すなわち無傷の細胞膜の通過に対して必要な条件は、「コンパクションとDNA電荷のマスキング」である(Loffler and Behr,Methods in Enzymology 217:599-618,1993)。新規のトランスフェクタム(transfectam;商標名)法では上記の条件をうまく満たしている。トランスフェクタム試薬(ジオクタデシルアミドグリシルスペルミン(dioctadecylamidoglycyl spermine))は、DNAに対して強い親和性(Kd=10-5−10-7M)を有する、+に帯電したスペルミン頭部を有した、カチオン性の合成リポポリアミンである。このスペルミン頭部はペプチド結合により、脂質部分に共有結合している。リポスペルミン分子は、DNAに結合して、それを脂質層にコ−ティングする。過剰なリポスペルミン存在下では、カチオン性脂質でコ−ティングされたプラスミドDNAの小胞が形成されて、該複合体の脂質部分が細胞膜と融合する。DNAのインタ−ナリゼ−ションはエンドサイトーシスにより起こると考えられている。
【0049】
トランスフェクタム−仲介性形質移入は、すでにある方法よりもより効率のよいものであることが示された(Barthel et al.,DNA and Cel Biology12(6):553-560,1993)。更に、トランスフェクタムは安定で、実質的に細胞無毒性の試薬である。しかしながら、特定のCOS細胞株における形質移入の最適化のための因子に関しての処理が必要である。これらの因子には、形質移入の期間、トランスフェクタム試薬のDNAに対する比率、DNA濃度、及びその他の希釈因子(例えばNaClの容量と強度)が含まれる。形質移入の条件の最適化を以下に示す。
【0050】
(a)形質移入の期間: 経時試験においてCOS細胞は、固定した濃度のプラスミドDNAとインキュベーションした。これらの試験は、種々の試験化合物によるSEAP発現試験のために、サブオプティマルなインキュベーション時間を選択することを目的とした。SEAP発現の経時誘導の結果は(非掲載)、SEAP発現は、漸進的な時間異存性上昇を示した。この誘導の開始は4時間未満でおきて、24時間で最大になった。10、12、15時間で見られた値の間には顕著な違いはなかった。よって、12−15時間の終末点を、以下の薬剤スクリ−ニングの全てにおける、SEAP発現の阻害に対して適切なサブオプティマル・インキュベーション期間として選択した。
【0051】
(b)DNA濃度: 形質移入に対して最適なDNA濃度は、トランスフェクタム試薬を使用した以前の研究に基づいて決定した(Loeffler and Behr,Methods in Enzymology,217:599-618,1993)。共形質移入に関しては、2:1(非選択遺伝子/選択遺伝子)の比率が最も適しているということが、他で報告されている(Hsuet al.,Science 254:1799-1802,1991)。リンブロ(登録商標)の24穴平底面の直径17mmプレ−トを使用して、非選択プラスミドpBC12/HIV/SEAPは、0.35μg/ウェルの濃度で、Tat機能をコ−ドしているpBC12/CMV/t2は、0.75μg/ウェルで使用した。
【0052】
(c)トランスフェクタムのDNAに対する比率と、イオン強度の決定: プラスミドDNAに対するトランスフェクタム(DOGS)の最適な比率、及び使用したNaClのイオン強度は、以前報告されている値を修飾したものであり(Loeffler and Behr,Methods in Enzymology,217:1799-1802,1993)、次のようにして決定した:
蒸留水で作成した10%(v/v)のエタノ−ル溶液0.400ml当たり、1mg(2.38mg/ml)の、オリジナルのトランスフェクトラムストック溶液からは、6倍量(μl)のストック溶液が、使用した各μgのDNAに対して必要であった。該溶液の最適のイオン強度は、適切な容量の150mMNaClにより提供され、以下の関係で決定される:
NaClの容量(μl)=トランスフェクタムの容量(μl)/0.6。
【0053】
上記の条件の最適化後の、標準的アッセイにおけるTat-誘導SEAPレベルの結果は、図2に例示されている。簡単に言うと、COS細胞はイスコ−ブ改変ダルベッコ培地(IMDM)(10%ウシ胎仔血清(FCS)及び抗生物質で補充済み)中で維持した。3つ一組の細胞試料を、リンブロの24穴平底面、直径17mmプレ−トのウェル当たり、約1.5X105の密度でまいて、湿度95%/CO25%のインキュベ−タ中で、50%集密度が得られるまで37℃でインキュベーションした。サブコンフルエントな細胞をリポスペルミン法形質移入した(Loeffler and Behr,Methods in Enzymology,217:599-618,1993)。サブコンフルエントな細胞の培地を吸引して300μlの新鮮な最小培地(3%FCS補充済みIMDM)で置き換えた。COS細胞をpBC12/HIV/SEAPのみで(0.35μg/ウェル)、またはpBC12/CMV/t2(Tat機能をコ−ド、(0.175μg/ウェル))+pBC12/HIV/SEAP(0.35μg/ウェル)で、または緩衝液のみ(DNA対照試料なし)で形質移入した。プレ−トを12から15時間インキュベーションして、その後で700μlの完全培地(10%FCS補充済みIMDM)を加えた。次いで細胞を48時間インキュベーションし、その後で、COS細胞培養上清の250μlのアリコットをを除去して65℃で5分間加熱し、選択的に内在性ホスファタ−ゼを不活化した(SEAPは熱に安定である)。次いで試料を微量遠心管で2分間遠心した。100μlの2XSEAPアッセイ緩衝液(1.0Mジエタノ−ルアミンpH9.8;0.5mMMgCl2;10mML-ホモアルギニン)を、試料のアリコット100μlに加えた。この溶液を混合して、96穴の平底面培養皿(Corning社)に移した。マルチピペッタ−を使用して20μlの、予め暖めておいた基質溶液(1XSEAPアッセイ緩衝液に溶解した、120mMのp-ニトロフェニルリン酸塩)を、反応混合物が含まれているそれぞれのウェルに分配した。EL3401マイクロプレ−トリ−ダ−(Bio-tek Instrument社)を使用し、それぞれの測定ごとに5秒間の自動振動を行い、反応物のA405を60分間、37℃で5分間おきに測定した。吸光度を、以前に記載されているようにして、SEAPのmUに変換して(Berger et al.,Gene 66:1-10,1988)、時間に対してプロットした。
【0054】
これらの結果は、一時間後のSEAP誘導に関して、対照(DNAなし)のレベル、または非選択遺伝子(HIV/SEAP)のみの誘導と比較して、ほぼ65倍もの増加を示した。
【0055】
向流クロマトグラフィ−によるアッセイ先導性のメキシコ・ハマビシ抽出物活性組成物の単離: 上記したように、メキシコ・ハマビシ抽出物の、差動分画と、向流クロマトグラフィ−(CCC)による精製により、二つの主要な成分が単離された(表1)。Grと呼ばれる、6.8mgの成分はSiO2TLCの緑の画分より単離された。全回収率は、元の植物粉末に基づくと、約0.051%であった。成分Lo、9.3mgが、黄色い画分(Ye)より単離された。
【0056】
メキシコ・ハマビシの葉と花からの抽出物による、Tat-TRS活性の阻害:SEAPアッセイで試験した複数の植物抽出物のなかでは、メキシコ・ハマビシである、ラレア・トリデンタタ(Larrea tridentata)の葉と花からの抽出物のみが、HIVのTatタンパク質に関しての顕著な阻害活性を示した。メキシコ・ハマビシは、図3に示されているように、SEAP発現に関して、用量−反応性阻害を示した。簡単に言うと、上記したリポスペルミン法を使用して、3つ一組のCOS細胞の試料を、pBC12/HIV/SEAPとpCB12/CMV/t2(Tat機能をコ−ドしている)との2:1の比の混合物で形質移入した。形質移入後、細胞を12−15時間インキュベーションした。メキシコ・ハマビシの抽出物ストック溶液(10mg/ml)は、カルシウム/マグネシウム−フリ−PBSと10%DMSOで調製して、マイレックス−GS22μmフィルタ−(ミリポア)で滅菌した。適切な濃度のメキシコ・ハマビシ抽出物を、DMSOの最終濃度が0.2%になるようにして、形質移入細胞へ加えて、48時間インキュベーションした。SEAP分析用に250μlのアリコットをCOS細胞培養上清より除去して、65℃で5分間加熱して内在性ホスファタ−ゼを選択的に不活化し(SEAPは熱に対して安定)、微量遠心管で2分間遠心した。100μlの2XSEAPアッセイ用緩衝液(1.0Mジエタノ−ルアミンpH9.8,0.5mMMgCl2,10mML-ホモアルギニン)を100μlの試料のアリコットへ加えた。この溶液を混合して96穴の平底面培養皿(Corning)へ移した。20μlの予め暖めておいた基質溶液(1XSEAPアッセイ用緩衝液に溶解した、120mMp-ニトロフェニル-リン酸塩)を、反応物が含まれているそれぞれのウェルへ、マルチピペッタ−で分配した。反応物のA405を、60分間37℃で5分間ごとに、各測定前の5秒間自動振動付きEL340iマイクロプレ−トリ−ダ−(Bio-tekInstruments社)で測定した。SEAP発現の阻害率を以下のようにして、30分時に計算した:
阻害率(%)=100−[(CT+−C+)/(CT-−C-)X100]:
ここで、
C-:対照試料(DNAなし、薬剤なし)
CT-:対照試料(+DNA、薬剤なし)
C+:薬剤処理済み試料(DNAなし、+薬剤)
CT+:薬剤処理済み試料(+DNA、+薬剤)
図3で示されるように、この阻害は20μg/mlの濃度で始まり、600μg/mlで最大の阻害活性になった。この粗物質に対しての、推定のEC50(50%阻害率を示す濃度)は110μg/mlであった。活性成分の精製が進むにつれて、活性成分の活性度に上昇が見られ、これは元の全粗抽出物(21%)と比較して、有機相(OG)では3倍(68%)になった。
【0057】
HIVの細胞病変性効果の阻害: Tatのトランスアクティベ−ションを阻害する化合物は原理的に、HIVの複製を阻害するはずである。そのため国立癌研究所(NCI)において、可溶性フォルマザン(formazan)アッセイ(Weislow et al.,JNCI81:577-586,1989)を利用して、HIV-1の細胞病変性効果の阻害に関して、メキシコ・ハマビシの抽出物が試験された。おおまかに言うと次のとおりである。CEM-SS細胞(ATCC,Rockville,MD)をHIV-産生H9細胞と共培養する。ウイルスを宿主細胞CEM-SSに感染、複製させて、ほとんどのCEM-SS細胞を1週間で殺傷する。もし薬剤がHIVの産生を阻害するならば、CEM-SS細胞はHIV−誘導性の細胞死から保護されることになる。よってテトラゾリウム(XTT)試薬は、生細胞により代謝的に還元され、450nmで比色法により測定可能な有色のフォルマザン産物を生ずる。
【0058】
実際には、3つ一組のCEM-SS細胞(5000)を96穴マイクロタイタ−プレ−トにまいた。100μlの最終容量(カルシウム/マグネシウムフリ−PBS、5%DMSO)の、試験化合物を適切な濃度で加えた。5分後に500倍感染性のHIV-1を産生するH9細胞、または正常なH9細胞を、適切な濃度の薬剤を含むウェルに加えた。マイクロタイタ−プレ−トを、湿度95%/CO25%で、37℃で6日間インキュベーションして、その後にXTTとN-メチルフェナゾニウムメトサルフェ−ト(N-methylphenazolium methosulfate,PMS)の混合物50μlを加えた。プレ−トを再び4時間インキュベーションして発色させた(XTTフォルマザン産生)。プレ−トを密封し、その内容物を自動振動装置で混合し、試料のOD450をマイクロプレ−トリ−ダ−で決定した。それぞれの値は3回の決定の平均を表す。非感染細胞とHIV-1感染細胞との間の2つ一組の値の平均値には、試験化合物存在下において顕著な違いが見られなかった。対照的に、試験化合物存在下、または非存在下での、HIV-1感染細胞間には顕著な違い(p<0.05)が見られた。
【0059】
この実験の結果を表2にまとめた。成分Grの濃度0.75μg/mlで、平均して58%の対HIV保護(細胞生存度)が得られ、一方、薬剤フリ−の、HIV感染試料では生存度は15%であった。0.187μg/mlもの低い濃度において成分Loは、HIV細胞病変性効果に関しての更に強い阻害活性を示した。
細胞生存度は87%であって、これは、処理していない対照試料のもの(89%)と非常に近い値であり、薬剤フリ−のHIV感染試料の生存度14%と対照的であった。上記化合物は使用した濃度においては、細胞毒性がなかった。
【0060】
【表2】
【0061】
クレオソートブッシュ抽出物の活性成分の構造解明: 精製された植物活性成分の化学的な特徴付けは、主としてマススペクトル、並びにH−およびC−核磁気共鳴(NMR)によって行われた。成分Loは、四つの関連した化合物(L1,L2,L3およびL4)の混合物であることがわかった。該混合物の夫々のピークの解析および特徴付けは、マススペクトル分析器(MS)に取り付けられた、分析用の非破壊的キャピラリー架橋5%フェニルメチルシロキサン(HP−5)カラムを用いたガスクロマトグラフィー(GC)によって達成された。このGC研究によって、第一の化合物(L1)は当該混合物の6%に対応し;第二の化合物(L2)は当該混合物の76%に対応し(MW=316);また第三の化合物(L3)は当該混合物の9%に対応する(MW=358)ことが明らかになった。これらの化合物の溶出時間(分)は、各ピーク上に示されている(図4)。
【0062】
成分Grは15の化合物からなっている。該混合物の各ピークの解析および特徴付けは、マススペクトル分析器(MS)に取り付けられた、分析用の非破壊的キャピラリー架橋5%フェニルメチルシロキサン(HP−5)カラムを用いたガスクロマトグラフィー(GC)によって達成された。このGC研究により、15の化合物のうちの四つ(G1,G2,G3およびG4)はリガンドであり、構造的にはL化合物に関連していることが明らかになった。これらの化合物の溶出時間(分)は夫々のピーク上に示されている(図5)。
【0063】
これらの8種類の化合物(L1,L2,L3,L4,G1,G2,G3およびG4)の構造を以下に説明する。
【0064】
L1はC18H22O4の組成を有しており、以前に知られている化学物質、即ち、1,4−ビス−(3,4−ジヒドロキシフェニル)−2,3−ジメチルブタン(ノルジヒドロカイアレ酸、NDGA、メルクインデックス、第10版、#6534)として同定されている。L1の構造式は下記の通りである。
【0065】
【化2】
【0066】
L2はC19H24O4の組成を有しており、3−O−メチル−NDGA、または1−(3,4−ジヒドロキシフェニル)−4−(3−メトキシ−4−ヒドロキシフェニル)−2,3−ジメチルブタンとして同定されている。3−O−メチル−NDGAの構造式は下記の通りである。
【0067】
【化3】
【0068】
L3もまたC19H24O4の組成を有しており、4−O−メチル−NDGA、または1−(3,4−ジヒドロキシフェニル)−4−(3−ヒドロキシ−4−メトキシフェニル)−2,3−ジメチルブタンとして同定されている。4−O−メチル−NDGAはまた、Malachi4:5-6またはMal4としても知られている。4−O−メチル−NDGAの構造式は下記の通りである。
【0069】
【化4】
【0070】
L4はC21H26O5の組成を有しており、3−O−メチル−4−O−アセチル−NDGA、または1−(3,4−ジヒドロキシフェニル)−4−(3−メトキシ−4−アセトキシフェニル)−2,3−ジメチルブタンとして同定されている。3−O−メチル−4−O−アセチル−NDGAの構造式は下記の通りである。
【0071】
【化5】
【0072】
G1は分子量344およびC21H22O4の組成を有しており、3,3´,4−トリ−O−メチル−NDGA、または1−(3−ヒドロキシ−4−メトキシフェニル)−4−(3,4−ジメトキシフェニル)−2,3−ジメチルブタンとして同定されている。3,3´,4−トリ−O−メチル−NDGAは下記の構造式を有している。
【0073】
【化6】
【0074】
G2は分子量344およびC21H22O4の組成を有しており、3,4,4´−トリ−O−メチル−NDGA、または1−(3−メトキシ−4−ヒドロキシフェニル)−4−(3,4−ジメトキシフェニル)−2,3−ジメチルブタンとして同定されている。3,4,4´−トリ−O−メチル−NDGAは下記の構造式を有している。
【0075】
【化7】
【0076】
G3およびG4は夫々、分子量374およびC22H28O5の組成を有している。G3は、3´,4−ジ−O−メチル−3−O−アセチル−NDGA(G3a)、または3,3´−ジ−O−メチル−4−O−アセチル−NDGA(G3b)の何れかである。3´,4−ジ−O−メチル−3−O−アセチル−NDGAはまた、1−(3−メトキシ−4−ヒドロキシフェニル)−4−(3−アセトキシ−4−メトキシフェニル)−2,3−ジメチルブタンとしても知られており、下記の構造式を有している。
G3a:
【0077】
【化8】
【0078】
3,3´−ジ−O−メチル−4−O−アセチル−NDGAはまた、1−(3−メトキシ−4−ヒドロキシフェニル)−4−(3−メトキシ−4−アセトキシフェニル)−2,3−ジメチルブタンとしても知られており、下記の構造式を有している。
G3b:
【0079】
【化9】
【0080】
同様にして、G4は、4,4´−ジ−O−メチル−3−O−アセチル−NDGA(G4a)、または3,4´−ジ−O−メチル−4−O−アセチル−NDGA(G4b)の何れかである。4,4´−ジ−O−メチル−3−O−アセチル−NDGAはまた、1−(3−ヒドロキシ−4−メトキシフェニル)−4−(3−アセトキシ−4−メトキシフェニル)−2,3−ジメチルブタンとしても知られており、下記の構造式を有している。
G4a:
【0081】
【化10】
【0082】
3,4´−ジ−O−メチル−4−O−アセチル−NDGAはまた、1−(3−ヒドロキシ−4−メトキシフェニル)−4−(3−メトキシ−4−アセトキシフェニル)−2,3−ジメチルブタンとしても知られており、下記の構造式を有している。
G4b:
【0083】
【化11】
【0084】
上記で述べた単離および精製の手順に加えて、開示された夫々のNDGA誘導体は、イケヤ等の方法(Ikeya et.al.,Chem.Pharm.Bull.27(7): 1583-1588,1979)に従って、NDGAのメチル化および/またはアシル化による化学合成によって製造してもよい。
【0085】
成分Loおよび成分Loからの二つの純粋な化合物(L2およびL3)の大規模精製: 植物材料バッチの大規模CCC分画を開始して、大量の成分Loを生じさせた。合計100gの植物粉末を、まず700mlのヘキサンで5回処理した。ヘキサン可溶物(1.17g)を廃棄した。ヘキサン不溶物(HI画分)を乾燥し、これを800mlのクロロホルム:メタノールで連続的に浸出することにより3回抽出した。これによって20gの全抽出物(Tex)を得、これを先のバッチ式微分抽出(batch differential extraction)で得たHI画分7.6gと混合した。こうして得られた組成植物材料の27.6gの全体を、10gおよび17.6gの二つのバッチに分割した。これらのバッチは、最初は、比率が6:4:5:5のヘキサン:EtOAc:MeOH:0.5%NaClの溶媒系を、上相(有機相)を可動相として用いることにより、大容量の多用途交差軸CPC(versatile Cross-Axis CPC;Shinomiya et al.,J.Chromatogr.644:215-229,1993)上で別々に運転された。各画分はTLCパターンに従って独自にプールされた。二つのCCC操作から四つの主要な画分が同定され、画分緑(Gr)(1.12g)、画分Lo(2.87g)、画分End(1.78g)および最後に静止相SP(20.84g)と表示された。画分Loの全体の2.87gは、大モデルのトリプレットCPC中において、水相を移動相に用いたヘキサン:EtOAc:MeOH:H2O(比率は7:3:5:5)によって更に分画された。溶出の順序およびTLCパターンに従って、LYI(0.375g);LYII(0.113g);LYIII(0.280g);およびLYIV(2.
80g)と表示された四種類の画分が同定された。これらの画分は、10μg/mlの濃度で抗Tat−TRS活性について試験された。この試験結果に基づいて、LYIが更なる精製のために選択された。
【0086】
Lo成分のLYI画分からの純粋なL2化合物およびL3化合物の単離: 親水性のより小さい画分LYIが、先に改良された条件、即ち、トリプレットCPCと、ヘキサン:CHCl3:MeOH:10mMNaClの溶媒系(比率は1:4:4:2)との条件を用いた更なる精製のために選択された。これによって、NMRおよびマススペクトルで試験したときに、148mgの均一なL3および109.3mgの純粋なL2の調製品が得られた。L3(Malachi4:5-6)およびL2の構造は上記で述べた通りである。
【0087】
化合物L2およびL3は、以前に同定された化学物質である1,4−ビス−(3,4−ジヒドロキシフェニル)−2,3−ジメチルブタン(ノルジヒドロカイアレ酸、NDGA、メルクインデックス、第10版、#6534)の誘導体である。NDGAの構造式は、既述したL1の構造式と同じであり、下記の通りである。
【0088】
【化12】
【0089】
NDGAおよびその誘導体の抗HIV活性(Tatに調節されたHIVトランス活性化の阻害)は、以前は知られていなかった。NDGAおよび誘導体Malachi4の抗HIVトランス活性化能の比較が図6に示されている。簡単にいえば、サブコンフルーエントなCOS細胞の複製サンプルを、上記のリポスペルミン法を用いて、プラスミドpBC12/HIV/SEAPおよびpBC12/CMB/t2(Tat機能をコードする)で共トランスフェクトした。次いで、12時間〜15時間、細胞をインキュベートした。試験サンプルは、先ず10%DMSO/カルシウム−マグネシウムを含まないPBS中で可溶化され、最終DMSO濃度0.2%の適切な濃度で、形質転換された細胞に添加された。サンプルは48時間インキュベートされ、その後、250μlのアリコートをCOS細胞培養上清から取り出し、図3に示すような標準的な試験でSEAPを分析した。SEAP発現のパーセント阻害は、30分の時点で次のようにして計算された。
【0090】
%阻害=100−[(CT+−C+)/(CT-−C-)×100]
但し、
C- : 対照サンプル(DNAなし、薬物なし)
CT-: 対照サンプル(+DNA、薬物なし)
C+ : 薬物処理したサンプル(DNAなし、+薬物)
CT+: 薬物処理したサンプル(+DNA、+薬物)
各点は、二回の測定の平均を表している。Mal4およびNDGAのEC50Sの間に顕著な相違はみられず、夫々8μg/ml(25μM)および6μg/ml(20μM)であった。このEC50Sは、Tat調節されたHIVトランス活性化が未処理対照細胞のそれの50%にまで減少するような、その化合物の阻害濃度として定義される。
【0091】
表3において、Mal4およびNDGAによる、HIVプロモータ活性のトランス活性化阻害を比較した。
【0092】
【表3】
【0093】
化合物NDGAおよびMal4は、図6に記載されたようにして試験された。対照サンプルは4回繰り返して実行された。パーセント阻害は30分後に測定去れ、OD405値は、次の通りであった。
【0094】
C-: 対照サンプル(DNAなし、薬物なし):0.091
CT-: 対照サンプル(+DNA、薬物なし):0.805
Mal4処理されたCEM−SS細胞の培養物における生存細胞の測定としての、XTTホルマザン産生の定量が図7に示されている。EC50は、感染した培養物中でのXTTホルマザン産生を、非感染の非処理培養細胞中におけるそれの50%にまで増加させる(保護する)、Mal4の濃度(例えば13.4μM)を表している。IC50は、非感染培養物中でのXTTホルマザン産生を、非感染の非処理対照細胞中におけるそれの50%にまで減少させる、Mal4の阻害濃度または有毒濃度(例えば325μM、見積り)を表している。非処理の感染された細胞におけるXTTホルマザンのレベルは、非処理かつ非感染の対照細胞におけるレベルの9%であった。HIV−1細胞変成効果の可溶性−ホルマザン試験は、文献(Weislow et al.,JNCI81:577-586,1989)に記載の方法に従って行われた。
【0095】
現在のところ最も実際的で且つ好ましいと思われる態様に関連して本発明を説明してきたが、本発明はこの開示された態様に限定されるものではなく、添付の請求の範囲の精神および範囲内に含まれる種々の変形および均等な構成をもカバーするものとして理解されるべきである。
【0096】
従って、請求の範囲に定義された本発明の新規な側面を逸脱することなく、NDGA誘導体およびTatトランス活性の抑制方法における変形を行い得ることが理解されるべきである。
【図面の簡単な説明】
【0097】
【図1】図1は、HIVのライフサイクルと、Tat−TRS阻害剤を含む潜在的治療剤の異なった作用部位を図示的に示している。基本的な転写ステップは1で示されており、ウイルス調節タンパク依存性のトランス活性化ステップは2で示されている。
【図2】図2は、標準的なSEAP試験における分泌されたアルカリホスファターゼ(SEAP)発現の誘導を示している。
【図3】図3は、分泌されたアルカリホスファターゼ(SEAP)試験における、クレオソートブッシュ全抽出物によるTat−TRS活性の阻害を示している。
【図4】図4は、分析用の非破壊性キャピラリー架橋5%フェニルメチルシロキサン(HP−5)カラムを用いたガスクロマトグラフィー(GC)による、成分Lo中の植物由来のHIV・Tat阻害剤の分析を示している。成分Loは、四つの成分の混合物である。溶出の時間(分)は各ピークについて出現する。
【図5】図5は、分析用の非破壊的キャピラリー架橋5%フェニルメチルシロキサン(analytical non-destructive capillarycross-linked 5% phenylmethylsiloxane)(HP−5)カラムを用いたガスクロマトグラフィー(GC)による、成分Gr中の植物由来HIV・Tat阻害剤の分析を示している。成分Grは複雑な混合物である。溶出の時間(分)は各ピークについて出現する。
【図6】図6は、分泌されたアルカリホスファターゼ(SEAP)試験における、植物由来の単一化合物であるMalachi4:5-6(Mal 4)およびNDGAによる、Tat誘導SEAP発現の阻害を示している。
【図7】図7は、二つの別々の実験における、HIVに感染し若しくは感染しないCEM−SS細胞のMal4処理培養における生存細胞の測定としての、XTTホルマザン産生の定量を示している。Mal4処理されていない感染細胞についてのXTTホルマザン産生パーセントは、実験1については8.9%であり、実験2については7.5%である。実験1では、非感染細胞は●・・・・・・●で表されており、感染細胞は×…×で表されている。実験2では、非感染細胞は●・・・・・・●で表されており、感染細胞は×…×で表されている。EC50は4.25μg/mlまたは13.4μMであることが分かった。IC50は100μg/mlまたは325μMと見積もられた。
【特許請求の範囲】
【請求項1】
HIV・TATトランス活性化を抑制するための化合物。
【請求項1】
HIV・TATトランス活性化を抑制するための化合物。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2006−273869(P2006−273869A)
【公開日】平成18年10月12日(2006.10.12)
【国際特許分類】
【出願番号】特願2006−184646(P2006−184646)
【出願日】平成18年7月4日(2006.7.4)
【分割の表示】特願平8−511844の分割
【原出願日】平成7年9月22日(1995.9.22)
【出願人】(398076227)ザ・ジョンズ・ホプキンス・ユニバーシティー (35)
【Fターム(参考)】
【公開日】平成18年10月12日(2006.10.12)
【国際特許分類】
【出願日】平成18年7月4日(2006.7.4)
【分割の表示】特願平8−511844の分割
【原出願日】平成7年9月22日(1995.9.22)
【出願人】(398076227)ザ・ジョンズ・ホプキンス・ユニバーシティー (35)
【Fターム(参考)】
[ Back to top ]