
HIV複製阻害剤
【課題】HIVの複製の仕組みや、LSD1の機能を研究するために有用な研究ツールとなりうる、HIV複製阻害剤を提供することを解決すべき課題とする。
【解決手段】下記一般式(1)(ただし、式中R1は水素又はフェニルアミド基を示し、R2は分枝を有することのあるアルキレン基を示し、シクロプロピルアミン置換基はメタ位又はパラ位に結合している)の化合物又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグからなることを特徴とするHIV複製阻害剤。

【解決手段】下記一般式(1)(ただし、式中R1は水素又はフェニルアミド基を示し、R2は分枝を有することのあるアルキレン基を示し、シクロプロピルアミン置換基はメタ位又はパラ位に結合している)の化合物又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグからなることを特徴とするHIV複製阻害剤。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、HIVのDNAの複製を阻害することのできるHIV複製阻害剤に関する。
【背景技術】
【0002】
ヒストンは、真核生物においてDNAを折りたたんでクロマチン構造を形成するタンパク質であり、遺伝子の発現に深くかかわっている。すなわち、ヒストンは様々な酵素の働きによって化学修飾され、これによりクロマチン構造が変化し、遺伝子の発現が制御されると考えられている。近年、こうしたエピジェネティックな遺伝子制御に関する様々な知見が発見されている。
【0003】
Lysine-specific demethylase(以下「LSD1」という)は、最初に発見されたヒストンの脱メチル化酵素であり、ヒストンH3の4番目のリシン残基のモノメチル化体及びジメチル化体(H3K4me1/2)の脱メチル化反応を触媒し、ホルムアルデヒドを副生する(非特許文献1参照)。また、LSD1は補酵素の一種であるフラビンアデニンジヌクレオチド(以下「FAD」という)と複合体をつくり、FADが酸化還元のメディエーターとなって、酸素によるリジン残基の酸化が進行する(下記反応式参照)。
【0004】
【化1】
【0005】
一方、LSD1は前立腺がんにおいて過剰発現し、アンドロゲン受容体(AR)と相互作用してAR依存性の転写を活性化することが明らかになっている。また、LSD1をノックダウンすると、アンドロゲンによって誘発された転写活性化が抑制され、細胞増殖が阻害されるということが報告されている(非特許文献2)。しかし、現在の段階ではLSD1が生体にどのような影響を与えるかなど、生物学的意義に関して不明な点が多い。また、LSD1とHIVの複製との関係についても、よく分かっていない。
【0006】
本発明のHIV複製阻害剤に用いられる、LSD1阻害剤としての下記化合物(1a)は、本発明者らがその合成法及びLSD1の阻害作用について既に発表している(非特許文献3)。また、本発明者らは、HIV−LTR近傍のメチル化状態に関する研究発表を行っている(非特許文献4)。
【0007】
【化2】
【先行技術文献】
【非特許文献】
【0008】
【非特許文献1】Biochemistry 2007, 46, 4408-4416
【非特許文献2】Nature 2005, 437, 436-439
【非特許文献3】Ueda et.al,J Am Chem Soc 2009, 131, 17536-17537
【非特許文献4】Kenichi Imai,Hiroaki Togami,Takashi Okamoto,J Biological Chemistryvol.285 N0.22 MAY 28 2010, 16538-16545
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明は、上記従来の問題に鑑みなされたものであり、HIVの複製の仕組みや、LSD1の機能を研究するために有用な研究ツールとなりうる、HIV複製阻害剤を提供することを解決すべき課題とする。
【課題を解決するための手段】
【0010】
本発明者らは、LSD1が遺伝子の転写活性に関係していることから、HIV複製とLSD1との関係について注目し、鋭意研究を行った。そして、LSD1阻害剤によってLSD1の酵素活性を抑制すると、HIVの複製が阻害されるという事実を見出し、本発明を完成するに至った。すなわち、本発明のHIV複製阻害剤は、LSD1阻害剤を有効成分として含有することを特徴とする。
【0011】
前記LSD1阻害剤としては、下記一般式(I)若しくは(II)(ただし、両式中R1は水素、置換基が結合していてもよいアルキル基、置換基が結合していてもよいフェニル基及び置換基が結合していてもよい複素環基のいずれかを示し、R2は分枝を有することがあり置換基が結合していてもよいアルキレン基を示し、R3は置換基が結合していてもよいアルキル基、置換基が結合していてもよいフェニル基、置換基が結合していてもよい複素環基及び置換基が結合していてもよいベンジル基のいずれかを示し、R4は置換基が結合していてもよいアルキル基、置換基が結合していてもよいフェニル基、置換基が結合していてもよい複素環基、置換基が結合していてもよいアルキルオキシ基、置換基が結合していてもよいフェニルオキシ基、置換基が結合していてもよいアルキルアミノ基及び置換基が結合していてもよいフェニルアミノ基のいずれかを示し、XはO、NH2、NHCO、CONH、S又はCH2を示す)のフェニルシクロプロピルアミン誘導体(I)及び(II)、又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグを用いることができる。
【化3】
【0012】
本発明者らの試験結果によれば、上記のフェニルシクロプロピルアミン誘導体(I)及び(II)は、LSD1の阻害活性が高く、MAO-AやMAO-Bなどの阻害作用は、LSD1阻害作用に比べて小さい。換言すれば、選択的にLSD1阻害を起こすことができる。そして、さらには、HIVの増殖を抑制する作用も有している。このため、LSD1とHIVの増殖との関係を調べる上で、MAO-AやMAO-Bなどの阻害作用の影響を避けることができる。このためLSD1とHIVの増殖との関係を調べる研究ツールとして、特に好ましい。
【0013】
なお、「プロドラッグ」とは、生体内で加水分解されてフェニルシクロプロピルアミン誘導体(I)又は(II)を再生する化合物をいい、例えばアミノ基の水素をアルカノイル基(アシル基)に置換した誘導体(すなわちアミド化した誘導体)、ヘミアミナールエーテル誘導体、アルコキシカルボニルオキシメチル基に置換した誘導体、N-オキシド誘導体等が挙げられる。
また、薬学上許容される塩としては、また、薬学上許容される塩としては、例えば塩酸塩、臭化水素酸塩、リン酸塩、硫酸塩、硝酸塩等の無機酸塩、ギ酸塩、酢酸塩、プロピオン酸塩、マレイン酸塩、フマル酸塩、コハク酸塩、乳酸塩、リンゴ酸塩、酒石酸塩、クエン酸塩、アスコルビン酸塩、マロン酸塩、シュウ酸塩、グリコール酸塩、フタル酸塩、ベンゼンスルホン酸塩等の有機酸塩が挙げられる。また、これらの塩を組み合わせて用いることもできる。
【0014】
上記フェニルシクロプロピルアミン誘導体(I)又は(II)の中でも、下記一般式(1)(ただし、式中R1は水素又はフェニルアミド基を示し、R2は分枝を有することのあるアルキレン基を示し、シクロプロピルアミン置換基はメタ位又はパラ位に結合している)の化合物又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグからなることを特徴とするフェニルシクロプロピルアミン誘導体が好ましい。
【化4】
【0015】
また、R2はエチレン基とすることが特に好ましい。さらに好ましいのは、下記構造式(1a)に示すフェニルアミド基を有するフェニルシクロプロピルアミン誘導体(1a)又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグである。本発明者らの試験結果によれば、この化合物は特にLSD1阻害活性が大きく、MAO阻害との選択性にも優れている。このため、LSD1とHIVの増殖との関係を調べる上で、MAO-AやMAO-Bなどの阻害作用の影響を避けることができ、LSD1とHIVの増殖との関係を調べる研究ツールとして、特に好ましい。
【0016】
【化5】
【0017】
また、前記LSD1阻害剤は、下記一般式(2)の化合物又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグでもよい。
【0018】
【化6】
【0019】
本発明者らは、この化合物(2)によっても、HIVの複製が阻害されることを見出している。なお、この化合物(2)はMAO阻害剤としても知られているが、MAO阻害作用を受けないLSD1阻害剤との比較を行うことにより、新たな知見を得ることができることから、LSD1とHIVの増殖との関係を調べる研究ツールとして有用である。
【発明を実施するための形態】
【0020】
以下、本発明を具体化した実施例について詳細に説明する。
【0021】
(実施例1)
実施例1では、(S)-トランス-N-3-[3-(2-アミノシクロプロピル) フェノキシ]-1-ベンジルカルバモイルプロピルベンズアミド塩酸塩(NCL-1)を合成した(下記化学式参照)。詳細は以下のとおりである。
【0022】
【化7】
【0023】
まず、NCL-1を合成するための一方の前駆体として、化合物(9)を以下の反応ルートにしたがって合成した。
【化8】
【0024】
以下に合成法の詳細を示す。
(ステップ1−1)
トランス-3- (3-ヒドロキシフェニル)アクリル酸メチルエステル(4)の合成
3- (3-ヒドロキシフェニル)アクリル酸(3)(25.0 g)をメタノール(82 mL)に溶解し、濃硫酸(2.0 mL)を加え、24時間還流した。反応液を濃縮し、残査を酢酸エチル(1000 mL)に溶解させ、水(500 mL)、飽和炭酸水素ナトリウム水溶液(500 mL)で洗浄した。有機層を飽和食塩水(500 mL)で洗浄して、無水硫酸ナトリウムで乾燥し、ろ過後、減圧濃縮し、化合物(4)(25.9 g, 収率96%)を白色固体として得た。化合物(4)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.60 (1H, d, J = 15.8 Hz), 7.21 (1H, t, J = 8.0 Hz), 7.04 (1H, d, J = 8.0 Hz), 6.98 (1H, t, J =1.8 Hz), 6.82 (1H,dd, J =8.0, 1.8Hz), 6.44 (1H, d, J =15.8 Hz), 3.77 (3H, s).
【0025】
(ステップ1−2)
トランス-3-(3メトキシメトキシフェニル)アクリル酸メチルエステル(5)の合成
ステップ1−1で得られたトランス-3- (3-ヒドロキシフェニル)アクリル酸メチルエステル(4)(25.8 g)をアセトン(220 mL)に溶解し、炭酸カリウム(40.0 g)を加え、室温で20分攪拌した。その溶液にメトキシメチルクロリド(11 mL)をゆっくりと加え、室温で12時間攪拌した。炭酸カリウムをろ過後、ろ液を濃縮し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(展開溶媒 n-ヘキサン:酢酸エチル=10 :1)で精製し、化合物(5)(27.2 g, 収率84%)を無色の油状物として得た。化合物(5)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.66 (1H, d, J = 16.2 Hz), 7.30 (1H, t, J = 8.0 Hz), 7.20 (1H, s), 7.17 (1H, d, J = 8.0 Hz), 7.07 (1H, dd, J = 8.0, 2.4 Hz), 6.43 (1H, d, J = 16.0 Hz), 5.19 (2H, s), 3.81 (3H, s), 3.49 (3H, s).
【0026】
(ステップ1−3)
トランス-2-(3-メトキシメトキシフェニル)シクロプロパンカルボン酸メチルエステル(6)の合成
水素化ナトリウム(60%)(6.32 g)とトリメチルスルホニウムヨージド(34.8 g)の混合物に、ジメチルスルホキシド(137 mL)を室温でゆっくりと滴下した。その反応液を室温で1時間攪拌した後、ステップ1−3で得られたトランス-3-(3メトキシメトキシフェニル)アクリル酸メチルエステル(5)(27.0 g)のジメチルスルホキシド(137 mL)溶液を滴下した。反応液を室温で4時間攪拌した後、10%クエン酸水溶液(500 mL)を加え、酢酸エチル(500 mL)で抽出した。有機層を飽和食塩水(500 mL)で洗浄して、無水硫酸ナトリウムで乾燥し、ろ過後、残渣をシリカゲルフラッシュカラムクロマトグラフィー(展開溶媒 n-ヘキサン:酢酸エチル=10 :1)で精製し、化合物(6)(6.70 g, 収率23%)を無色の油状物として得た。化合物(6)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.19 (1H, t, J = 8.0 Hz), 6.89 (1H, dd, J =8.0, 2.5 Hz), 6.78 (1H, t, J = 2.5 Hz), 6.74 (1H, d, J =8.0 Hz), 5.16 (2H, s), 3.71 (3H, s), 3.47 (3H, s), 2.51-2.49 (1H, m), 1.92-1.89 (1H, m), 1.61-1.57 (1H, m) 1.34-1.31 (1H, m).
【0027】
(ステップ1−4)
トランス-2-(3-メトキシメトキシフェニル)シクロプロパンカルボン酸(7)の合成
ステップ1−3で得られたトランス-2-(3-メトキシメトキシフェニル)シクロプロパンカルボン酸メチルエステル(6)(6.70 g)をメタノール(56 mL)に溶解させ、氷冷下、水酸化カリウム(16.0 g)のメタノール溶液(130 mL)を加え、反応液を室温で15時間攪拌した。反応液を濃縮し、残査を水(100 mL)に懸濁させ、ジクロロメタン(100 mL)で洗浄した。水層に2N塩酸を加えて酸性(pH 1)にし、ジクロロメタン(300 mL)で抽出した。有機層を無水硫酸ナトリウムで乾燥し、ろ過後、減圧濃縮し、化合物(7)(6.00 g, 収率95%)を無色の油状物として得た。化合物(7)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.20 (1H, t, J = 8.0 Hz), 6.90 (1H, dd, J = 8.0, 1.9 Hz), 6.79 (1H, t, J = 1.9 Hz), 6.75 (1H, d, J = 8.0 Hz), 5.16 (2H, s), 3.48 (3H, s), 2.60-2.56 (1H, m), 1.92-1.89 (1H, m), 1.65 (1H, quintet, J = 5.0 Hz)1.42-1.38 (1H, m).
【0028】
(ステップ1−5)
トランス- 2-(3-メトキシメトキシフェニル)シクロプロピルtert-ブチルカルバメート(8)の合成
ステップ1−4で得られたトランス-2-(3-メトキシメトキシフェニル)シクロプロパンカルボン酸(7)(6.00 g)をシクロヘキサン(320 mL)に溶解させ、ジフェニルホスホリルアジド(6.7 mL)とトリエチルアミン(4.5 mL)をアルゴン雰囲気下、0 ℃で加えた。反応液を3時間還流後、tert-ブタノール(52 mL)を加え、さらに11時間還流した。反応液を濃縮後、残渣をシリカゲルフラッシュカラムクロマトグラフィー(展開溶媒 n-ヘキサン:酢酸エチル= 7 :1)で精製し、化合物(8)(3.70 g, 収率47%)を白色固体として得た。化合物(7)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.17 (1H, t, J = 8.0 Hz), 6.85 (1H, d, J = 9.2 Hz), 6.78 (1H, s), 6.76 (1H, s), 5.15 (2H, s),4.82 (1H, broad s), 3.47 (3H, s)2.72-2.76 (1H, m), 1.98-2.02 (1H, m), 1.45 (9H, s), 1.74-1.16 (2H, m).
【0029】
(ステップ1−6)
トランス-2-(3-ヒドロキシフェニル)シクロプロピルtert-ブチルカルバメート(9)の合成
ステップ1−5で得られたトランス-2-(3-メトキシメトキシフェニル)シクロプロピルtert-ブチルカルバメート(8)(3.70 g)をジクロロメタン(60 mL)に溶解させ、4N塩酸酢酸エチル溶液(68 mL)を加え、室温で4時間攪拌した。反応液を濃縮し、残渣を1,4-ジオキサン(33 mL) と水 (33 mL)に溶解させ、トリエチルアミン (20 mL) と Boc2O (4.6 mL)を加えた。反応液を室温で12時間攪拌後、10%クエン酸溶液(200 mL)に注ぎ、酢酸エチル(300 mL)で抽出した。有機層を飽和食塩水(300 mL)で洗浄して、無水硫酸ナトリウムで乾燥し、ろ過後、残渣をシリカゲルフラッシュカラムクロマトグラフィー(展開溶媒 n-ヘキサン:酢酸エチル= 5 :1)で精製し、化合物(9)(1.59 g, 収率35%)を無色の油状物として得た。化合物(9)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.10 (1H, d, J = 7.6 Hz), 6.67 (1H, d, J =8.0 Hz), 6.64 (1H, dd, J = 8.0, 2.4 Hz), 6.59 (1H, s), 5.28 (1H, broad s), 4.86 (1H, broad s), 2.69-2.72 (1H, m), 1.97-2.01 (1H, m), 1.46 (9H, s), 1.15-1.12 (2H, m).
【0030】
さらに、NCL-1を合成するための他方の前駆体として、化合物(13)を以下の反応ルートにしたがって合成した。
【化9】
【0031】
以下に合成法の詳細を示す。
(ステップ1−7)
(S)-1-ベンジルカルバモイル-3-ヒドロキシプロピルtert-ブチルカルバメート(11)の合成
N- tert-ブトキシカルボニル(S) -ホモセリン(10)(5.50 g)、ベンジルアミン(2.8 mL)、PyBOP(13.1 g)、トリエチルアミン(7.0 mL)をN,N-ジメチルホルムアミド(55 mL)に溶解させ、室温で10時間攪拌した。反応液を水(300 mL)で希釈し、クロロホルム(300 mL)で抽出した。有機層を飽和食塩水(300 mL)で洗浄して、無水硫酸ナトリウムで乾燥し、ろ過後、残渣をシリカゲルフラッシュカラムクロマトグラフィー(展開溶媒 n-ヘキサン:酢酸エチル= 1 :2)で精製し、化合物(11)(5.46 g, 収率70%)を白色固体として得た。化合物(11)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.35-7.32 (2H, m), 7.29-7.25 (3H, m), 6.71 (1H, broad s), 5.58 (1H, d, J = 7.3 Hz), 4.45 (2H, s), 4.35 (1H, s), 3.71 (2H, s), 3.27 (1H, broad s), 2.04-1.98 (1H, m), 1.72-1.85 (1H, m), 1.43 (9H, s)
【0032】
(ステップ1−8)
(S)-2-アミノ-N-ベンジル-4-ヒドロキシブタンアミド塩酸塩(12)の合成
ステップ1−7で得られた(S)-1-ベンジルカルバモイル-3-ヒドロキシプロピルtert-ブチルカルバメート(11)(5.40 g)をジクロロメタン(60 mL)に溶解させ、4N塩酸1,4-ジオキサン溶液(87 mL)を加え、室温で5時間攪拌した。反応液を濃縮し、化合物(4.27 g, 収率100%)を白色固体として得た。化合物(12)の1H NMRのデータを以下に示す。
δ:1H NMR (CD3OD, 500 MHz, δ; ppm)
7.44-7.41 (2H, m), 7.34-7.30 (3H, m), 7.27 (1H, broad s), 4.43 (2H, s), 3.94 (1H, d, J = 7.0 Hz) 2.03 (2H, s)
【0033】
(ステップ1−9)
(S)-N-(1-ベンジルカルバモイル-3-ヒドロキシプロピル)ベンズアミド(13)の合成
ステップ1−8で得られた(S)-2-アミノ-N-ベンジル-4-ヒドロキシブタンアミド塩酸塩(12)(118 mg)、安息香酸(60 mg)、PyBOP(303 mg)、トリエチルアミン(0.2 mL)をN,N-ジメチルホルムアミド(1 mL)に溶解させ、室温で20時間攪拌した。反応液を水(100 mL)で希釈し、ジクロロメタン(100 mL)で抽出した。有機層を飽和食塩水(100 mL)で洗浄して、無水硫酸ナトリウムで乾燥し、ろ過後、残渣をシリカゲルフラッシュカラムクロマトグラフィー(展開溶媒 n-ヘキサン:酢酸エチル=2:3)で精製し、化合物(13)(72 mg, 収率47%)を白色固体として得た。化合物(13)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.79 (2H, d, J = 7.3 Hz), 7.53 (1H, t, J = 7.3 Hz), 7.44 (2H, t, J = 8.0 Hz), 7.33-7.25 (5H, m), 7.06 (1H, d, J = 7.3 Hz), 6.89 (1H, broad s), 4.77 (1H, q, J = 6.4 Hz), 4.47-4.45 (2H, m), 4.32-4.28 (1H, m), 4.22-4.18 (1H, m), 2.28-2.19 (2H, m), 2.03 (3H, s).
【0034】
<カップリング反応>
ステップ1−1〜ステップ1−6で合成したカップリング前駆体である化合物(9)と、ステップ1−7〜ステップ1−9で合成したカップリング前駆体(13)とを用いて、化10に示す合成ルートに従い、光延反応を用いたカップリング反応を行った。
【化10】
【0035】
以下にカップリング反応の詳細を示す。
(ステップ1−10)
(S)-トランス-[2-[3-(3-ベンゾイルアミノ-3-ベンジルカルバモイルプロポキシ)フェニル] シクロプロピルtert-ブチルカルバメート(14)の合成
ステップ1−9で得られた(S)-N-(1-ベンジルカルバモイル-3-ヒドロキシプロピル)ベンズアミド(13)(200 mg)と、ステップ1−6で得られたトランス-2-(3-ヒドロキシフェニル)シクロプロピルtert-ブチルカルバメート(9)(230 mg)と、トリフェニルホスフィン(504 mg)とを無水テトラヒドロフラン(3 mL)に溶解させ、氷冷下、ジイソプロピルアゾジカルボキシレート(1mL)を加えた。反応液を室温で5時間攪拌後、濃縮し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(展開溶媒 n-ヘキサン:酢酸エチル= 1 :1)で精製し、化合物(14)(64 mg, 収率18%)を淡黄色固体として得た。化合物(14)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.82 (2H, d, J = 8.5 Hz), 7.52 (1H, t, J = 8.5 Hz), 7.46 (2H, t, J = 8.5 Hz), 7.26-7.21 (5H, m), 7.16 (1H, t, J = 8.0 Hz), 6.92 (1H, s), 6.75 (1H, d, J = 8.0 Hz), 6.63 (1H, d, J = 8.0 Hz), 6.60 (1H, broad s), 4.88 (1H, quartet, J = 6.0 Hz), 4.47 (2H, d, J = 6.0 Hz), 4.32-4.29 (1H, m), 4.13-4.10 (1H, m), 2.65-2.70 (1H, m), 2.43-2.37 (2H, m), 1.99-1.97 (1H, m), 1.45 (9H, s), 1.14-1.10 (2H, m).
【0036】
(ステップ1−11)
(S)-トランス-N-3-[3-(2-アミノシクロプロピル) フェノキシ]-1-ベンジルカルバモイルプロピルベンズアミド塩酸塩(NCL-1)の合成
ステップ1−10で得られた(S)-トランス-[2-[3-(3-ベンゾイルアミノ-3-ベンジルカルバモイルプロポキシ)フェニル] シクロプロピルtert-ブチルカルバメート(14)(70 mg)をジクロロメタン(1 mL)に溶解させ、4N塩酸酢酸エチル溶液(0.8 mL)を加え、室温で4時間攪拌した。反応液を濃縮し、残渣をクロロホルム-メタノールから再結晶し、実施例1の化合物(NCL-1)(22 mg, 収率36%)を淡黄色固体として得た。NCL-1の融点、1H NMR、13C NMR、MS (FAB)のデータを示す。
融点 134.8-139.1 ℃
1H NMR (CD3OD, 500 MHz, δ; ppm)
δ:7.84 (2H, d, J = 8.5 Hz), 7.54 (1H, t, J = 8.5 Hz), 7.46 (2H, t, J = 8.5 Hz), 7.26-7.17 (6H, m), 6.77 (1H, dd, J = 8.0, 1.5 Hz), 6.74-6.72 (1H, m), 6.67 (1H, d, J = 1.5 Hz), 4.47-4.37 (2H, m), 4.08 (2H, t, J = 6.0 Hz), 2.28-2.24 (2H, m), 1.35-1.33 (1H, m), 1.29-1.26 (1H, m)
13C NMR (CD3OD, 500 MHz, δ; ppm)
δ:173.93, 170.34, 160.52, 141.32, 139.79, 135.21, 132.96, 130.81, 129.55, 128.54, 128.46, 128.19, 120.00, 114.15, 114.05, 113.84, 65.92, 53.13, 44.15, 32.68, 31.96, 22.58, 13.84
MS (FAB) m/z = 444 (M+)
Anal. Calcd. for C27H30ClN3O3 3/2H2O: C, 63.96; H, 6.56; N, 8.29.
Found: C, 63.83; H, 6.29; N, 7.98. 3/2
【0037】
(実施例2)
実施例2では、(S)-トランス-N-3-[4-(2-アミノシクロプロピル)フェノキシ]-1-ベンジルカルバモイルプロピルベンズアミド塩酸塩(NCL-2)を合成した(下記化学式参照)。詳細は以下のとおりである。
【0038】
【化11】
【0039】
まず、NCL-2を合成するための一方の前駆体として、化合物(21)を以下の反応ルートにしたがって合成した。
【0040】
【化12】
【0041】
以下に合成法の詳細を示す。
(ステップ2−1)
トランス-3- (4-ヒドロキシフェニル)アクリル酸メチルエステル(16)の合成
実施例1のステップ1−1と同様の方法により、3-(4-ヒドロキシフェニル)アクリル酸(15)(20.0 g)を出発原料とし、化合物(16)(21.2 g, 収率99%)を白色固体として得た。化合物(16)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.64 (1H, d, J = 15.8 Hz), 7.43 (2H, d, J = 8.5 Hz), 6.84 (2H, d, J = 8.5 Hz), 6.31 (1H, d, J =15.8 Hz), 5.21 (1H,s), 3.80 (3H, s).
【0042】
(ステップ2−2)
トランス-3-(4-メトキシメトキシフェニル)アクリル酸メチルエステル(17)の合成
実施例1のステップ1−2と同様の方法により、ステップ2−1より得られたトランス-3- (4-ヒドロキシフェニル)アクリル酸メチルエステル(16)(10.0 g)より、化合物(17)(11.1 g, 収率89%)を無色油状物として得た。化合物(17)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.65 (1H, d, J = 6.2 Hz), 7.47 (2H, d, J = 8.8 Hz), 7.04 (2H, d, J = 8.8 Hz), 6.33 (1H, d, J = 6.2 Hz), 5.20 (2H, s), 3.80 (3H, s), 3.48 (3H, s).
【0043】
(ステップ2−3)
トランス-2-(4-メトキシメトキシフェニル)シクロプロパンカルボン酸メチルエステル(18)の合成
実施例1のステップ1−3と同様の方法により、ステップ2−2より得られたトランス-3-(4-メトキシメトキシフェニル)アクリル酸メチルエステル(15)(11.0 g)より、化合物(18)(1.68 g, 収率14%)を無色油状物として得た。化合物(18)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.03 (2H, d, J = 8.5 Hz), 6.95 (2H, d, J =8.5 Hz), 5.14 (2H, s), 3.71 (3H, s), 3.46 (3H, s), 2.51-2.47 (1H, m), 1.85-1.81 (1H, m), 1.58-1.54 (1H, m) 1.29-1.25 (1H, m).
【0044】
(ステップ2−4)
トランス-2-(4-メトキシメトキシフェニル)シクロプロパンカルボン酸(19)の合成
実施例1のステップ1−4と同様の方法により、ステップ2−3より得られたトランス-2-(4-メトキシメトキシフェニル)シクロプロパンカルボン酸メチルエステル(18)(1.68 g)より、化合物(19)(1.50 g, 収率95%)を白色固体として得た。化合物(19)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.05 (2H, d, J = 6.7 Hz), 6.94 (2H, d, J = 6.7 Hz), 5.12 (2H, s), 3.42 (3H, s), 2.43-2.39 (1H, m), 1.76-1.73 (1H, m), 1.49-1.46 (1H, m), 1.33-1.28 (1H, m).
【0045】
(ステップ2−5)
トランス- 2-(4-メトキシメトキシフェニル)シクロプロピルtert-ブチルカルバメート(20)の合成
実施例1のステップ1−5と同様の方法により、ステップ2−4より得られたトランス-2-(4-メトキシメトキシフェニル)シクロプロパンカルボン酸(19)(1.05 g)より、化合物(20)(788 mg, 収率56%)を淡黄色固体として得た。化合物(20)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.07 (2H, d, J = 8.2 Hz), 6.94 (2H, d, J = 8.2 Hz), 5.13 (2H, s), 4.89 (1H, broad s), 3.46 (3H, s), 2.64-2.69 (1H, m), 2.02-1.98 (1H, m), 1.46 (9H, s), 1.12-1.09 (2H, m).
【0046】
(ステップ2−6)
トランス-2-(4-ヒドロキシフェニル)シクロプロピルtert-ブチルカルバメート(21)の合成
実施例1のステップ1−6と同様の方法により、ステップ2−5より得られたトランス- 2-(4-メトキシメトキシフェニル)シクロプロピルtert-ブチルカルバメート(20)(476 mg)より、化合物(21)(300 mg, 収率74%)を淡黄色油状物として得た。化合物(21)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:6.94 (2H, d, J = 8.5 Hz), 6.72 (2H, d, J =8.5 Hz), 5.13 (1H, broad s), 4.84 (1H, broad s), 2.60-2.70 (1H, m), 1.98 (1H, m), 2.09-1.96 (1H, m), 1.10-1.05 (2H, m).
【0047】
NCL-2を合成するための他方の前駆体として、実施例1のステップ1−7〜ステップ1−9で合成した化合物(13)を用いた。
【0048】
<カップリング反応>
上記ステップ2−1〜ステップ2−6で合成したカップリング前駆体である化合物(21)と、化合物(13)とを用いて、光延反応によるたカップリング反応によって、実施例2の化合物NCL-2を合成した。
【0049】
【化13】
【0050】
以下に合成法の詳細を示す。
(ステップ2−7)
(S)-トランス-[2-[4-(3-ベンゾイルアミノ-3-ベンジルカルバモイルプロポキシ)フェニル] シクロプロピルtert-ブチルカルバメート(22)の合成
実施例1のステップ1−7と同様の方法により、ステップ2−6より得られたトランス-2-(4-ヒドロキシフェニル)シクロプロピルtert-ブチルカルバメート(21)(220 mg)と
実施例1のステップ1−7〜1−9で合成したカップリング前駆体(13)のカップリングを行い、化合物(22)(83 mg, 収率25%)を淡黄色固体として得た。化合物(22)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.79 (2H, d, J = 8.5 Hz), 7.53-7.49 (1H, m), 7.41 (2H, t, J = 8.5 Hz), 7.26-7.13 (5H, m), 7.04 (2H, d, J = 8.5 Hz), 6.71 (2H, d, J = 8.5 Hz), 4.94-4.89 (2H, m), 4.46-4.38 (2H, m), 4.25-4.21 (1H, m), 4.08-4.04 (1H, m), 2.55-2.59 (1H, m), 2.43-2.37 (2H, m), 1.95-2.01 (1H, m), 1.45 (9H, s), 1.08 (2H, t, J = 7.0 Hz).
【0051】
(ステップ2−8)
(S)-トランス-N-3-[4-(2-アミノシクロプロピル) フェノキシ]-1-ベンジルカルバモイルプロピルベンズアミド塩酸塩(NCL-2)の合成
実施例1のステップ1−11と同様の方法により、ステップ2−7より得られた、(S)-トランス-[2-[4-(3-ベンゾイルアミノ-3-ベンジルカルバモイルプロポキシ)フェニル] シクロプロピルtert-ブチルカルバメート(22)(83 mg)より、化合物(NCL-2)(18 mg, 収率25%)を淡黄色固体として得た。NCL-2の融点、1H NMR、13C NMR、MS (FAB)のデータを示す。
融点:141.2-144.5 ℃
1H NMR (CD3OD, 500 MHz, δ; ppm)
δ:7.83 (2H, d, J = 7.0 Hz), 7.54 (1H, t, J = 7.0 Hz), 7.45 (2H, t, J = 7.0 Hz), 7.27-7.19 (6H, m), 7.05 (2H, dd, J = 9.0 Hz), 6.84 (2H, d, J = 9.0 Hz), 4.40 (2H, d, J = 8.0 Hz), 4.06 (2H, t, J = 8.0 Hz), 2.74-2.72 (1H, m), 2.42-2.36 (1H, m), 2.29-2.21 (1H, m), 1.34-1.29 (1H, m), 1.26-1.22 (1H, m)
13C NMR (CD3OD, 500 MHz, δ; ppm)
δ:173.91, 170.32, 159.23, 139.80, 135.21, 132.93, 131.84, 129.55, 129.52, 128.62, 128.52, 128.48, 128.17, 115.89, 65.95, 53.13, 44.16, 32.70, 31.73, 21.96, 13.41
MS (FAB) m/z = 444 (M+);
Anal. Calcd For C27H30ClN3O3 2H2O: C, 62.84; H, 6.64; N, 8.14.
Found: C, 62.47; H, 6.08; N, 8.21.
【0052】
(実施例3)
実施例3では、トランス-4-[3-(2-アミノシクロプロピル)フェノキシ]-N-ベンジルブチルアミド塩酸塩(NCL-3)を合成した(下記化学式参照)。詳細は以下のとおりである。
【0053】
【化14】
【0054】
<カップリング前駆体>
実施例3では、NCL-3を合成するための前駆体として、実施例1(ステップ1−1〜1−6)で合成した、化合物(9)を用いた。また、NCL-3を合成するためのもう一方の前駆体として、化合物(24)を次の反応によって合成した。
【化15】
【0055】
(ステップ3−1)
N-ベンジル-4-クロロブチルアミド(24)の合成
ベンジルアミン(1.1 mL)とトリエチルアミン(4.5 mL)をジクロロメタン(26 mL)に溶解し、氷冷下、4-クロロブチリルクロリド(23)(1.5 mL)のジクロロメタン(15 mL)溶液を滴下した。0°Cで30分攪拌後、反応液を水(100 mL)に注ぎ、酢酸エチル(200 mL)で抽出した。有機層を飽和食塩水(100 mL)で洗浄して、無水硫酸ナトリウムで乾燥し、ろ過後、残渣をシリカゲルフラッシュカラムクロマトグラフィー(展開溶媒 n-ヘキサン:酢酸エチル= 3 :1)で精製し、化合物(24)(1.26 g, 収率59%)を淡黄色油状物として得た。化合物(24)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.36-7.26 (5H, m), 5.84 (1H, broad s), 4.44 (2H, d, J = 5.8 Hz), 3.62 (2H, t, J = 6.0 Hz), 2.40 (2H, t, J = 7.0 Hz), 2.14 (2H, quintet, J = 7.0 Hz).
【0056】
NCL-3を合成するための他方の前駆体として、実施例1のステップ1−1〜ステップ1−6で合成した化合物(9)を用いた。
【0057】
<カップリング反応>
上記のようにして合成した化合物(24)と化合物(9)とを光延反応によってカップリングさせて実施例3の化合物NCL-3を得た。
【化16】
【0058】
以下に合成法の詳細を示す。
(ステップ3−2)
トランス-2-[3-(3-ベンジルカルバモイルプロポキシ)フェニル]シクロプロピル tert-ブチルカルバメート(25)の合成
実施例1のステップ1−6で得られたトランス-2-(3-ヒドロキシフェニル)シクロプロピルtert-ブチルカルバメート(9)(400 mg)をアセトニトリル(7.5 mL)に溶解させ、炭酸セシウム(1.54 g)、ヨウ化ナトリウム(70 mg)を加えて、室温で15分間攪拌した。反応液にステップ3−1で得られたN-ベンジル-4-クロロブチルアミド(25)(900 mg)のアセトニトリル(10 mL)溶液を添加し、9時間加熱還流した。反応液をろ過し、不溶物を取り除き、ろ液を濃縮後、残渣をシリカゲルフラッシュカラムクロマトグラフィー(展開溶媒 n-ヘキサン:酢酸エチル= 1:1)で精製し、化合物(24)(140 mg, 収率21%)を淡黄色油状物として得た。化合物(25)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.30-7.24 (5H, m), 7.15 (1H, t, J = 7.0 Hz), 6.71 (1H, d, J = 7.0 Hz), 6.66 (1H, d, J = 7.0 Hz), 6.62 (1H, s), 5.85 (1H, broad s), 4.84 (1H, broad s), 4.44 (2H, d, J = 5.8 Hz), 3.99 (2H, t, J = 5.8 Hz), 2.67-2.75 (1H, m), 2.43 (2H, t, J = 7.2 Hz), 2.16-2.13 (2H, m), 2.01-1.96 (1H, m), 1.45 (9H, s), 1.12-1.15 (2H, m).
【0059】
(ステップ3−3)
トランス-4-[3-(2-アミノシクロプロピル)フェノキシ]-N-ベンジルブチルアミド塩酸塩(NCL-3)の合成
実施例1のステップ1−11と同様の方法により、ステップ3−2より得られた、トランス-2-[3-(3-ベンジルカルバモイルプロポキシ)フェニル]シクロプロピル tert-ブチルカルバメート(25)(140 mg)より、実施例3のフェニルシクロプロピルアミン誘導体である化合物(NCL-3)(82 mg, 収率69%)を白色固体として得た。NCL-3の融点、1H NMR、13C NMR、MS (ELC)のデータを示す。
融点:83.3-86.7 °C
1H NMR (CD3OD, 500 MHz, δ; ppm)
δ:7.26-7.17 (6H, m), 6.77 (1H, dd, J = 7.6, 2.5 Hz), 6.72 (1H, d, J = 7.6 Hz), 6.69 (1H, s), 4.36 (2H, s), 3.98 (2H, t, J = 6.4 Hz), 2.82-2.80 (1H, m), 2.43 (2H, t, J = 7.2 Hz), 2.32-2.30 (1H, m), 2.10-2.06 (2H, m), 1.39-1.35 (1H, m), 1.32-1.28 (1H, m)
13C NMR (CD3OD, 500 MHz, δ; ppm)
δ:175.37, 160.71, 141.30, 140.02, 130.78, 129.53, 128.52, 128.18, 119.68, 113.94 113.81, 68.06, 33.46, 31.96, 26.57, 22.62, 13.84
MS (ELC) m/z: 325 (M+)
Anal. Calcd For C20H25ClN2O2 2/3H2O: C, 64.42; H, 7.12; N, 7.51.
Found: C, 64.44; H, 6.87; N, 7.80.
【0060】
(実施例4)
実施例4では、トランス-4-[4-(2-アミノシクロプロピル)フェノキシ]-N-ベンジルブチルアミド塩酸塩(NCL-4)を合成した(下記化学式参照)。詳細は以下のとおりである。
【0061】
【化17】
【0062】
<カップリング前駆体>
実施例4では、カップリング前駆体は、実施例2におけるステップ2−1〜ステップ2−6で合成した、化合物(21)を用いた。また、もう一方のカップリング前駆体としては、実施例3におけるステップ3−1で合成した化合物(24)を用いた。
【0063】
<カップリング反応>
上記のようにして合成した化合物(24)と化合物(21)とを光延反応によってカップリングさせて実施例4の化合物NCL-4を得た。
【0064】
【化18】
【0065】
以下に合成法の詳細を示す。
(ステップ4−1)
トランス-2-[4-(3-ベンジルカルバモイルプロポキシ)フェニル]シクロプロピル tert-ブチルカルバメート(26)の合成
実施例3のステップ3−2と同様の方法により、化合物(21)(500 mg)と化合物(24)とをカップリングさせ、実施例4のフェニルシクロプロピルアミン誘導体である化合物(26)(NCL-3)(82 mg, 収率69%)を白色固体として得た。化合物(26)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.32-7.24 (5H, m), 7.05 (2H, d, J = 8.2 Hz), 6.75 (2H, d, J = 8.2 Hz), 5.82 (1H, broad s), 4.84 (1H, broad s), 4.44 (2H, d, J = 5.8 Hz), 3.97 (2H, t, J = 5.8 Hz), 2.60-2.70 (1H, m), 2.42 (2H, t, J = 7.2 Hz), 2.16-2.12 (2H, m), 1.97-2.04(1H, m), 1.46 (9H, s), 1.02-1.15 (2H, m).
【0066】
(ステップ4−2)
トランス-4-[4-(2-アミノシクロプロピル)フェノキシ]-N-ベンジルブチルアミド塩酸塩(NCL-4)の合成
実施例1のステップ1−11と同様の方法により、ステップ4−1より得られた、化合物(26)(140 mg)より、実施例4のフェニルシクロプロピルアミン誘導体である化合物(NCL-4)(82 mg, 収率69%)を白色固体として得た。NCL-4の融点、1H NMR、13C NMR、MS (ELC)のデータを示す。
融点:156.3-159.4 °C; 1H NMR (CD3OD, 500 MHz, δ; ppm)
δ:7.27-7.21 (5H, m), 7.06 (2H, d, J = 8.5 Hz), 6.83 (2H, d, J = 8.5 Hz), 4.35 (2H, s), 3.95 (2H, t, J = 6.4 Hz), 2.75-2.74 (1H, m), 2.42 (2H, t, J = 7.2 Hz), 2.30-2.27 (1H, m), 2.09-2.05 (2H, m), 1.35-1.32 (1H, m), 1.27-1.24 (1H, m)
δ:13C NMR (CD3OD, 500 MHz, δ; ppm) 175.34, 159.44, 140.06, 131.58, 129.51, 128.61, 128.53, 128.16, 115.81, 68.17, 44.11, 33.48, 31.73, 26.59, 21.96, 13.40
MS (ELC) m/z: 325 (M+)
Anal. Calcd For C20H25ClN2O2 H2O: C, 63.40; H, 7.18; N, 7.39.
Found: C, 63.50; H, 6.68; N, 7.44.
【0067】
−評 価−
以上のようにして得られた実施例1〜実施例4の化合物(NCL-1〜NCL-4)について、LSD1阻害活性試験、モノアミンオキシダーゼ阻害活性試験及びHIV複製に対する阻害活性試験を行った。
【0068】
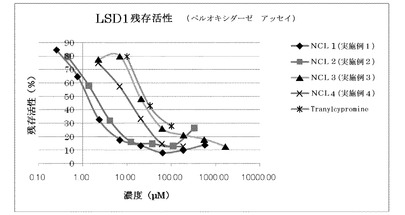
<LSD1阻害試験>
LSD1酵素は以下のようにして調製した。
全長のLSD1(1-851aa)のN末端にヒスチジン5残基を付加した組換えタンパク質をコードするプラスミドを調製し、このプラスミドで形質転換した組換え大腸菌を用いてLSD1を発現させた。その後、組換え大腸菌を超音波処理で溶解し、その可溶性画分をHisTrapクロマトグラフィーで精製し、LSD1酵素溶液を得た。LSD1の酵素活性は、LSD1の脱メチル化反応の際に生成する過酸化水素をペルオキシダーゼと試薬によって発色させ、吸光光度法で定量することにより測定した。すなわち、384ウエルマイクロタイタープレート内で、50mM Hepes-NaOH buffer(pH7.5)、0.1mM 4-アミノアンチピリン、1mM 3,5-ジクロロ-2-ヒドロキシベンゼンスルホン酸、20μM histone H3- lysine 4 dimethyl peptide、0.05μM LSD1、0.35μM horseradish peroxidaseからなる20μlの溶液を25℃で30分間、経時的に酵素反応を測定した。測定にはSpectra Max M2e(Molecular Devices社)を用いて、生成物の515nmでの吸光度を測定して求めた。また、阻害活性については、ジメチルスルホキシド(dimethyl sulfoxide)添加時の酵素活性を100%とし、フェニルシクロプロピルアミン誘導体の添加濃度を様々に変えて残存活性を測定し、50%の活性を阻害する濃度(IC50)を求めた。
【0069】
LSD1阻害試験の結果を図1に示す。この図に示すように、実施例1〜4の化合物(NCL-1〜4)は、いずれもtranylcypromineより高いLSD1阻害活性を示した。また、実施例1〜4のの化合物(NCL-1〜4)中でも、アミノ酸構造を有する実施例1のNCL-1及び実施例2のNCL-2では、極めて高いLSD1阻害活性を示した。
【0070】
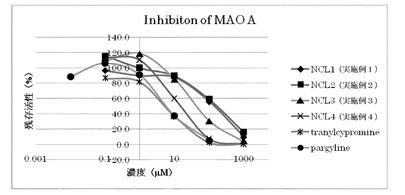
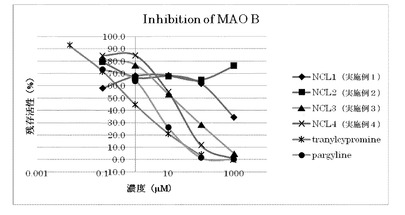
<モノアミンオキシダーゼ阻害活性測定試験>
モノアミンオキシダーゼA(MAO-A)及びモノアミンオキシダーゼB(MAO-B)阻害活性の測定を、Promega社のMAO-GloアッセイキットとSigma-Aldrich社から購入したMAO-AおよびMAO-Bを用いて以下のように行った。
12.5 μLの4倍MAO基質(最終濃度40 μM)、12.5 μLの4倍阻害剤溶液(最終濃度0.1〜1000 μM)、25 μLのMAO-A(最終濃度 9 unit/mL)あるいは25 μLのMAO-B(最終濃度2.3 unit/mL)を混合し、室温で1時間反応させた。この反応液に50 μLのルシフェリン検出試薬を添加し、室温で20分反応させ、蛍光プレートリーダーも用いて蛍光強度(蛍光測定波長:562 nm)を測定し、IC50値(酵素を50%阻害する阻害剤濃度)を求めた。
【0071】
MAO-Aに対する阻害活性の結果を図2に示す。この図に示すように、実施例1〜4の化合物(NCL-1〜4)は、いずれもtranylcypromineやpargylineより低いMAO A阻害活性を示した。その中でも、アミノ酸構造を有する実施例1のNCL-1及び実施例2のNCL-2では、特に阻害活性が低かった。
また、MAO-Bに対する阻害活性についても、図3に示すように、実施例1〜4の化合物(NCL-1〜4)は、いずれもtranylcypromineやpargyline(下記化学式参照)より低いMAO-B阻害活性を示し、その中でも、実施例1のNCL-1及び実施例2のNCL-2では、特に阻害活性が低かった。
【0072】
【化19】
【0073】
<HIV複製に対する阻害活性試験>
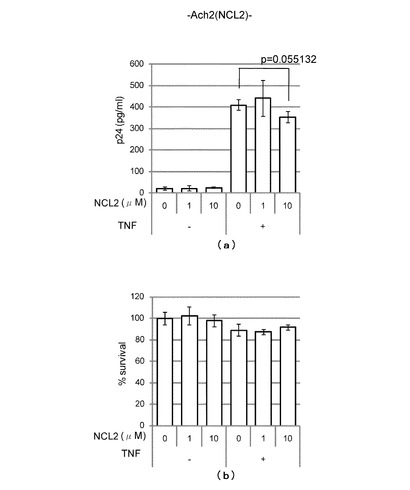
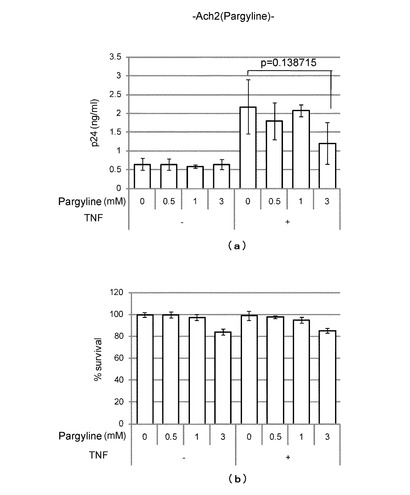
上記LSD1阻害試験において阻害活性を示したNCL-2及びpargylineについて、HIV複製に対する阻害活性試験を行った。詳細を以下に示す。
【0074】
(HIV複製に対するNCL-2の阻害活性)
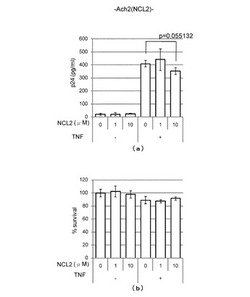
HIV潜伏感染細胞Ach2にNCL-2を0-10μM/Lとなるように加え、24時間後の細胞培養上清中のHIVのコア蛋白p24の濃度をp24antigen assay法により測定し、HIV複製の指標とした。
また、上記試験においてNCL-2とともにTNF (1μg/ml)を加えて潜伏感染細胞を刺激し、24時間後の細胞培養上清中のHIVのコア蛋白p24を測定し、HIV複製の指標とした。
【0075】
その結果、TNFを加えなかった場合には、図4(a)左側のグラフに示すように、NCL-2の添加量が0-10μM/Lの範囲において、HIVのコア蛋白p24はいずれも検出感度ぎりぎりで増減は認められず、HIV複製が阻害されることはなかった。
これに対して、TNFを加えた場合には、図4(a)右側のグラフに示すように、NCL-2の濃度の増加とともにHIVのコア蛋白p24が減少し(p値=0.055)、HIV複製が阻害されることが分かった。
また、上記HIV複製に対する阻害活性試験における細胞毒性効果をWST-1 assay法によって調べた結果、図4(b)に示すように、TNFの添加の有無にかかわらず、NCL-2の添加量が0-10μM/Lの範囲において、細胞毒性は20%を超えることはなかった。このことから、HIV複製の阻害が、細胞毒性によるものではないことが分かった。
【0076】
(HIV複製に対するPargylineの阻害活性)
HIV潜伏感染細胞Ach2にPargylineを0-3 mM/Lとなるように加え、24時間後の細胞培養上清中のHIVのコア蛋白p24の濃度をp24antigen assay法により測定し、HIV複製の指標とした。
また、上記試験においてPargylineとともにTNF (1μg/ml)を加えて潜伏感染細胞を刺激し、24時間後の細胞培養上清中のHIVのコア蛋白p24を測定し、HIV複製の指標とした。
【0077】
その結果、Pargylineを加えなかった場合には、図5(a)左側のグラフに示すように、Pargylineの添加量が0-3 mM/Lの範囲において、HIVのコア蛋白p24はいずれも検出感度ぎりぎりで増減は認められず、HIV複製が阻害されることはなかった。
これに対して、Pargylineを加えた場合には、図5(a)右側のグラフに示すように、Pargylineの濃度の増加とともにHIVのコア蛋白p24が減少し(p値=0.139)、HIV複製が阻害されることが分かった。
また、上記HIV複製に対する阻害活性試験における細胞毒性効果をWST-1 assay法によって調べた結果、図5(b)に示すように、TNFの添加の有無にかかわらず、Pargylineの添加量が0-3 mM/Lの範囲において、細胞毒性は20%を超えることはなかった。このことから、HIV複製の阻害が、細胞毒性によるものではないことが分かった。
【0078】
<HIV−LTR近傍のメチル化状態の解析>
本発明者らは、HIV−LTR近傍のメチル化状態について、以下に示す研究発表を既に行っている(非特許文献4)。
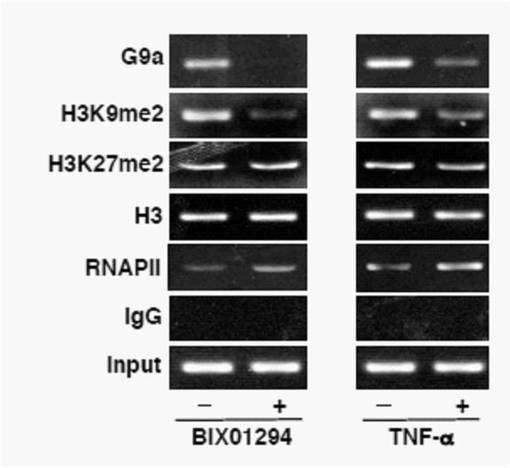
すなわち、HIV (AIDSウイルス)の転写を調節するLTRと呼ばれるプロウイルスDNAの「転写スイッチ」領域のメチル化状態とヒストンメチル化酵素G9aおよびRNA合成酵素II型(RNAPII)の結合状態を、ヒストンメチル化酵素G9a阻害剤BIX01294を加えた場合と加えない場合とについて、クロマチン免疫沈降(ChIP)アッセイで調べた。
【0079】
その結果、図6の左側のグラフが示すように、ヒストンメチル化酵素G9a阻害剤であるBIX阻害剤を加えない場合には、G9aはHIVプロウイルスLTRと結合していて、その部位のH3K9のジメチル化が十分に起こっている。これに対して、ヒストンメチル化酵素G9a阻害剤であるBIXを加えるとG9aは離脱し、しかもH3K9のジメチル化も著しく低下している。このことはG9aが離脱しただけでは説明ができず、ある種のヒストン脱メチル化酵素がG9aの代わりに新たに結合したことを示唆している。ただし、この事実のみからは、LSD1がHIV複製と関係しているか否かについては不明である。
【0080】
しかしながら、前述したように、NCL-2及びpargylineがHIV複製の阻害作用を示すこと、及び、NCL-2やpargylineがLSD1阻害剤作用を示すことを考え合わせれば、LSD1阻害剤作用を有する化合物はHIV複製の阻害作用を示すことを強く示唆しており、今回始めて明らかにされたことである。
【0081】
また、図6の右側のグラフは、潜伏感染HIVからの転写を促進する作用のあるTNFαを加えていない対照(左)と、TNFαを加えた場合(右)の、ChIPアッセイの結果を示す。このグラフから、TNFαを加えることによってもG9aの離脱は引き起こされるが、BIX01294を加えた場合の効果よりも小さいことが分かる。
【0082】
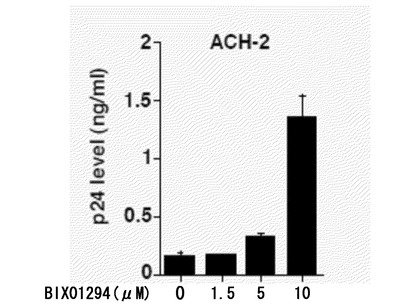
さらに、図7のグラフは、HIVが潜伏感染していて細胞からはほとんどウイルスの産生が起こっていないAch-2細胞(HIV感染ヒトT細胞株)にG9a阻害剤BIX01294を加えた場合の、細胞培養上清中のHIVのコア蛋白p24の濃度をp24antigen assay法により測定した結果である。この図から、G9a阻害剤BIX01294の添加量が多いほど、HIVのコア蛋白p24の量も多くなり、HIV複製が活性化されることが分かる。ただし、このことのみからは、LSD1とHIV複製との関係についての知見を得ることはできない。
【0083】
この発明は上記発明の実施の態様及び実施例の説明に何ら限定されるものではない。特許請求の範囲を逸脱せず、当業者が容易に想到できる範囲で種々の変形態様もこの発明に含まれる。
【図面の簡単な説明】
【0084】
【図1】LSD1に対する阻害活性を示す図である。
【図2】MAO-Aに対する阻害活性を示す図である。
【図3】MAO-Bに対する阻害活性を示す図である。
【図4】(a)は、NCL-2の添加量とHIVのコア蛋白p24の濃度との関係をTNF添加の場合及びTNF不添加の場合について調べたグラフである。また、(b)は、HIV複製に対する阻害活性試験における細胞毒性効果をWST-1 assay法によって調べたグラフである。
【図5】(a)は、Pargylineの添加量とHIVのコア蛋白p24の濃度との関係をTNF添加の場合及びTNF不添加の場合について調べたグラフである。また、(b)は、HIV複製に対する阻害活性試験における細胞毒性効果をWST-1 assay法によって調べたグラフである。
【図6】プロウイルスDNAの「転写スイッチ」領域のメチル化状態とヒストンメチル化酵素G9aおよびRNA合成酵素II型(RNAPII)の結合状態を、ヒストンメチル化酵素G9a阻害剤BIX01294を加えた場合と加えない場合とについて、クロマチン免疫沈降(ChIP)アッセイで調べた結果を示す図である。
【図7】Ach-2細胞にBIX01294を加えた場合の、細胞培養上清中のHIVのコア蛋白p24の濃度を示すグラフである。
【産業上の利用可能性】
【0085】
本発明のフェニルシクロプロピルアミン誘導体及びLSD1阻害剤は、LSD1の機能を調べるための生物学的ツールとして用いたり、抗がん剤として用いたりすることができると期待される。
【技術分野】
【0001】
本発明は、HIVのDNAの複製を阻害することのできるHIV複製阻害剤に関する。
【背景技術】
【0002】
ヒストンは、真核生物においてDNAを折りたたんでクロマチン構造を形成するタンパク質であり、遺伝子の発現に深くかかわっている。すなわち、ヒストンは様々な酵素の働きによって化学修飾され、これによりクロマチン構造が変化し、遺伝子の発現が制御されると考えられている。近年、こうしたエピジェネティックな遺伝子制御に関する様々な知見が発見されている。
【0003】
Lysine-specific demethylase(以下「LSD1」という)は、最初に発見されたヒストンの脱メチル化酵素であり、ヒストンH3の4番目のリシン残基のモノメチル化体及びジメチル化体(H3K4me1/2)の脱メチル化反応を触媒し、ホルムアルデヒドを副生する(非特許文献1参照)。また、LSD1は補酵素の一種であるフラビンアデニンジヌクレオチド(以下「FAD」という)と複合体をつくり、FADが酸化還元のメディエーターとなって、酸素によるリジン残基の酸化が進行する(下記反応式参照)。
【0004】
【化1】
【0005】
一方、LSD1は前立腺がんにおいて過剰発現し、アンドロゲン受容体(AR)と相互作用してAR依存性の転写を活性化することが明らかになっている。また、LSD1をノックダウンすると、アンドロゲンによって誘発された転写活性化が抑制され、細胞増殖が阻害されるということが報告されている(非特許文献2)。しかし、現在の段階ではLSD1が生体にどのような影響を与えるかなど、生物学的意義に関して不明な点が多い。また、LSD1とHIVの複製との関係についても、よく分かっていない。
【0006】
本発明のHIV複製阻害剤に用いられる、LSD1阻害剤としての下記化合物(1a)は、本発明者らがその合成法及びLSD1の阻害作用について既に発表している(非特許文献3)。また、本発明者らは、HIV−LTR近傍のメチル化状態に関する研究発表を行っている(非特許文献4)。
【0007】
【化2】
【先行技術文献】
【非特許文献】
【0008】
【非特許文献1】Biochemistry 2007, 46, 4408-4416
【非特許文献2】Nature 2005, 437, 436-439
【非特許文献3】Ueda et.al,J Am Chem Soc 2009, 131, 17536-17537
【非特許文献4】Kenichi Imai,Hiroaki Togami,Takashi Okamoto,J Biological Chemistryvol.285 N0.22 MAY 28 2010, 16538-16545
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明は、上記従来の問題に鑑みなされたものであり、HIVの複製の仕組みや、LSD1の機能を研究するために有用な研究ツールとなりうる、HIV複製阻害剤を提供することを解決すべき課題とする。
【課題を解決するための手段】
【0010】
本発明者らは、LSD1が遺伝子の転写活性に関係していることから、HIV複製とLSD1との関係について注目し、鋭意研究を行った。そして、LSD1阻害剤によってLSD1の酵素活性を抑制すると、HIVの複製が阻害されるという事実を見出し、本発明を完成するに至った。すなわち、本発明のHIV複製阻害剤は、LSD1阻害剤を有効成分として含有することを特徴とする。
【0011】
前記LSD1阻害剤としては、下記一般式(I)若しくは(II)(ただし、両式中R1は水素、置換基が結合していてもよいアルキル基、置換基が結合していてもよいフェニル基及び置換基が結合していてもよい複素環基のいずれかを示し、R2は分枝を有することがあり置換基が結合していてもよいアルキレン基を示し、R3は置換基が結合していてもよいアルキル基、置換基が結合していてもよいフェニル基、置換基が結合していてもよい複素環基及び置換基が結合していてもよいベンジル基のいずれかを示し、R4は置換基が結合していてもよいアルキル基、置換基が結合していてもよいフェニル基、置換基が結合していてもよい複素環基、置換基が結合していてもよいアルキルオキシ基、置換基が結合していてもよいフェニルオキシ基、置換基が結合していてもよいアルキルアミノ基及び置換基が結合していてもよいフェニルアミノ基のいずれかを示し、XはO、NH2、NHCO、CONH、S又はCH2を示す)のフェニルシクロプロピルアミン誘導体(I)及び(II)、又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグを用いることができる。
【化3】
【0012】
本発明者らの試験結果によれば、上記のフェニルシクロプロピルアミン誘導体(I)及び(II)は、LSD1の阻害活性が高く、MAO-AやMAO-Bなどの阻害作用は、LSD1阻害作用に比べて小さい。換言すれば、選択的にLSD1阻害を起こすことができる。そして、さらには、HIVの増殖を抑制する作用も有している。このため、LSD1とHIVの増殖との関係を調べる上で、MAO-AやMAO-Bなどの阻害作用の影響を避けることができる。このためLSD1とHIVの増殖との関係を調べる研究ツールとして、特に好ましい。
【0013】
なお、「プロドラッグ」とは、生体内で加水分解されてフェニルシクロプロピルアミン誘導体(I)又は(II)を再生する化合物をいい、例えばアミノ基の水素をアルカノイル基(アシル基)に置換した誘導体(すなわちアミド化した誘導体)、ヘミアミナールエーテル誘導体、アルコキシカルボニルオキシメチル基に置換した誘導体、N-オキシド誘導体等が挙げられる。
また、薬学上許容される塩としては、また、薬学上許容される塩としては、例えば塩酸塩、臭化水素酸塩、リン酸塩、硫酸塩、硝酸塩等の無機酸塩、ギ酸塩、酢酸塩、プロピオン酸塩、マレイン酸塩、フマル酸塩、コハク酸塩、乳酸塩、リンゴ酸塩、酒石酸塩、クエン酸塩、アスコルビン酸塩、マロン酸塩、シュウ酸塩、グリコール酸塩、フタル酸塩、ベンゼンスルホン酸塩等の有機酸塩が挙げられる。また、これらの塩を組み合わせて用いることもできる。
【0014】
上記フェニルシクロプロピルアミン誘導体(I)又は(II)の中でも、下記一般式(1)(ただし、式中R1は水素又はフェニルアミド基を示し、R2は分枝を有することのあるアルキレン基を示し、シクロプロピルアミン置換基はメタ位又はパラ位に結合している)の化合物又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグからなることを特徴とするフェニルシクロプロピルアミン誘導体が好ましい。
【化4】
【0015】
また、R2はエチレン基とすることが特に好ましい。さらに好ましいのは、下記構造式(1a)に示すフェニルアミド基を有するフェニルシクロプロピルアミン誘導体(1a)又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグである。本発明者らの試験結果によれば、この化合物は特にLSD1阻害活性が大きく、MAO阻害との選択性にも優れている。このため、LSD1とHIVの増殖との関係を調べる上で、MAO-AやMAO-Bなどの阻害作用の影響を避けることができ、LSD1とHIVの増殖との関係を調べる研究ツールとして、特に好ましい。
【0016】
【化5】
【0017】
また、前記LSD1阻害剤は、下記一般式(2)の化合物又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグでもよい。
【0018】
【化6】
【0019】
本発明者らは、この化合物(2)によっても、HIVの複製が阻害されることを見出している。なお、この化合物(2)はMAO阻害剤としても知られているが、MAO阻害作用を受けないLSD1阻害剤との比較を行うことにより、新たな知見を得ることができることから、LSD1とHIVの増殖との関係を調べる研究ツールとして有用である。
【発明を実施するための形態】
【0020】
以下、本発明を具体化した実施例について詳細に説明する。
【0021】
(実施例1)
実施例1では、(S)-トランス-N-3-[3-(2-アミノシクロプロピル) フェノキシ]-1-ベンジルカルバモイルプロピルベンズアミド塩酸塩(NCL-1)を合成した(下記化学式参照)。詳細は以下のとおりである。
【0022】
【化7】
【0023】
まず、NCL-1を合成するための一方の前駆体として、化合物(9)を以下の反応ルートにしたがって合成した。
【化8】
【0024】
以下に合成法の詳細を示す。
(ステップ1−1)
トランス-3- (3-ヒドロキシフェニル)アクリル酸メチルエステル(4)の合成
3- (3-ヒドロキシフェニル)アクリル酸(3)(25.0 g)をメタノール(82 mL)に溶解し、濃硫酸(2.0 mL)を加え、24時間還流した。反応液を濃縮し、残査を酢酸エチル(1000 mL)に溶解させ、水(500 mL)、飽和炭酸水素ナトリウム水溶液(500 mL)で洗浄した。有機層を飽和食塩水(500 mL)で洗浄して、無水硫酸ナトリウムで乾燥し、ろ過後、減圧濃縮し、化合物(4)(25.9 g, 収率96%)を白色固体として得た。化合物(4)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.60 (1H, d, J = 15.8 Hz), 7.21 (1H, t, J = 8.0 Hz), 7.04 (1H, d, J = 8.0 Hz), 6.98 (1H, t, J =1.8 Hz), 6.82 (1H,dd, J =8.0, 1.8Hz), 6.44 (1H, d, J =15.8 Hz), 3.77 (3H, s).
【0025】
(ステップ1−2)
トランス-3-(3メトキシメトキシフェニル)アクリル酸メチルエステル(5)の合成
ステップ1−1で得られたトランス-3- (3-ヒドロキシフェニル)アクリル酸メチルエステル(4)(25.8 g)をアセトン(220 mL)に溶解し、炭酸カリウム(40.0 g)を加え、室温で20分攪拌した。その溶液にメトキシメチルクロリド(11 mL)をゆっくりと加え、室温で12時間攪拌した。炭酸カリウムをろ過後、ろ液を濃縮し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(展開溶媒 n-ヘキサン:酢酸エチル=10 :1)で精製し、化合物(5)(27.2 g, 収率84%)を無色の油状物として得た。化合物(5)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.66 (1H, d, J = 16.2 Hz), 7.30 (1H, t, J = 8.0 Hz), 7.20 (1H, s), 7.17 (1H, d, J = 8.0 Hz), 7.07 (1H, dd, J = 8.0, 2.4 Hz), 6.43 (1H, d, J = 16.0 Hz), 5.19 (2H, s), 3.81 (3H, s), 3.49 (3H, s).
【0026】
(ステップ1−3)
トランス-2-(3-メトキシメトキシフェニル)シクロプロパンカルボン酸メチルエステル(6)の合成
水素化ナトリウム(60%)(6.32 g)とトリメチルスルホニウムヨージド(34.8 g)の混合物に、ジメチルスルホキシド(137 mL)を室温でゆっくりと滴下した。その反応液を室温で1時間攪拌した後、ステップ1−3で得られたトランス-3-(3メトキシメトキシフェニル)アクリル酸メチルエステル(5)(27.0 g)のジメチルスルホキシド(137 mL)溶液を滴下した。反応液を室温で4時間攪拌した後、10%クエン酸水溶液(500 mL)を加え、酢酸エチル(500 mL)で抽出した。有機層を飽和食塩水(500 mL)で洗浄して、無水硫酸ナトリウムで乾燥し、ろ過後、残渣をシリカゲルフラッシュカラムクロマトグラフィー(展開溶媒 n-ヘキサン:酢酸エチル=10 :1)で精製し、化合物(6)(6.70 g, 収率23%)を無色の油状物として得た。化合物(6)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.19 (1H, t, J = 8.0 Hz), 6.89 (1H, dd, J =8.0, 2.5 Hz), 6.78 (1H, t, J = 2.5 Hz), 6.74 (1H, d, J =8.0 Hz), 5.16 (2H, s), 3.71 (3H, s), 3.47 (3H, s), 2.51-2.49 (1H, m), 1.92-1.89 (1H, m), 1.61-1.57 (1H, m) 1.34-1.31 (1H, m).
【0027】
(ステップ1−4)
トランス-2-(3-メトキシメトキシフェニル)シクロプロパンカルボン酸(7)の合成
ステップ1−3で得られたトランス-2-(3-メトキシメトキシフェニル)シクロプロパンカルボン酸メチルエステル(6)(6.70 g)をメタノール(56 mL)に溶解させ、氷冷下、水酸化カリウム(16.0 g)のメタノール溶液(130 mL)を加え、反応液を室温で15時間攪拌した。反応液を濃縮し、残査を水(100 mL)に懸濁させ、ジクロロメタン(100 mL)で洗浄した。水層に2N塩酸を加えて酸性(pH 1)にし、ジクロロメタン(300 mL)で抽出した。有機層を無水硫酸ナトリウムで乾燥し、ろ過後、減圧濃縮し、化合物(7)(6.00 g, 収率95%)を無色の油状物として得た。化合物(7)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.20 (1H, t, J = 8.0 Hz), 6.90 (1H, dd, J = 8.0, 1.9 Hz), 6.79 (1H, t, J = 1.9 Hz), 6.75 (1H, d, J = 8.0 Hz), 5.16 (2H, s), 3.48 (3H, s), 2.60-2.56 (1H, m), 1.92-1.89 (1H, m), 1.65 (1H, quintet, J = 5.0 Hz)1.42-1.38 (1H, m).
【0028】
(ステップ1−5)
トランス- 2-(3-メトキシメトキシフェニル)シクロプロピルtert-ブチルカルバメート(8)の合成
ステップ1−4で得られたトランス-2-(3-メトキシメトキシフェニル)シクロプロパンカルボン酸(7)(6.00 g)をシクロヘキサン(320 mL)に溶解させ、ジフェニルホスホリルアジド(6.7 mL)とトリエチルアミン(4.5 mL)をアルゴン雰囲気下、0 ℃で加えた。反応液を3時間還流後、tert-ブタノール(52 mL)を加え、さらに11時間還流した。反応液を濃縮後、残渣をシリカゲルフラッシュカラムクロマトグラフィー(展開溶媒 n-ヘキサン:酢酸エチル= 7 :1)で精製し、化合物(8)(3.70 g, 収率47%)を白色固体として得た。化合物(7)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.17 (1H, t, J = 8.0 Hz), 6.85 (1H, d, J = 9.2 Hz), 6.78 (1H, s), 6.76 (1H, s), 5.15 (2H, s),4.82 (1H, broad s), 3.47 (3H, s)2.72-2.76 (1H, m), 1.98-2.02 (1H, m), 1.45 (9H, s), 1.74-1.16 (2H, m).
【0029】
(ステップ1−6)
トランス-2-(3-ヒドロキシフェニル)シクロプロピルtert-ブチルカルバメート(9)の合成
ステップ1−5で得られたトランス-2-(3-メトキシメトキシフェニル)シクロプロピルtert-ブチルカルバメート(8)(3.70 g)をジクロロメタン(60 mL)に溶解させ、4N塩酸酢酸エチル溶液(68 mL)を加え、室温で4時間攪拌した。反応液を濃縮し、残渣を1,4-ジオキサン(33 mL) と水 (33 mL)に溶解させ、トリエチルアミン (20 mL) と Boc2O (4.6 mL)を加えた。反応液を室温で12時間攪拌後、10%クエン酸溶液(200 mL)に注ぎ、酢酸エチル(300 mL)で抽出した。有機層を飽和食塩水(300 mL)で洗浄して、無水硫酸ナトリウムで乾燥し、ろ過後、残渣をシリカゲルフラッシュカラムクロマトグラフィー(展開溶媒 n-ヘキサン:酢酸エチル= 5 :1)で精製し、化合物(9)(1.59 g, 収率35%)を無色の油状物として得た。化合物(9)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.10 (1H, d, J = 7.6 Hz), 6.67 (1H, d, J =8.0 Hz), 6.64 (1H, dd, J = 8.0, 2.4 Hz), 6.59 (1H, s), 5.28 (1H, broad s), 4.86 (1H, broad s), 2.69-2.72 (1H, m), 1.97-2.01 (1H, m), 1.46 (9H, s), 1.15-1.12 (2H, m).
【0030】
さらに、NCL-1を合成するための他方の前駆体として、化合物(13)を以下の反応ルートにしたがって合成した。
【化9】
【0031】
以下に合成法の詳細を示す。
(ステップ1−7)
(S)-1-ベンジルカルバモイル-3-ヒドロキシプロピルtert-ブチルカルバメート(11)の合成
N- tert-ブトキシカルボニル(S) -ホモセリン(10)(5.50 g)、ベンジルアミン(2.8 mL)、PyBOP(13.1 g)、トリエチルアミン(7.0 mL)をN,N-ジメチルホルムアミド(55 mL)に溶解させ、室温で10時間攪拌した。反応液を水(300 mL)で希釈し、クロロホルム(300 mL)で抽出した。有機層を飽和食塩水(300 mL)で洗浄して、無水硫酸ナトリウムで乾燥し、ろ過後、残渣をシリカゲルフラッシュカラムクロマトグラフィー(展開溶媒 n-ヘキサン:酢酸エチル= 1 :2)で精製し、化合物(11)(5.46 g, 収率70%)を白色固体として得た。化合物(11)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.35-7.32 (2H, m), 7.29-7.25 (3H, m), 6.71 (1H, broad s), 5.58 (1H, d, J = 7.3 Hz), 4.45 (2H, s), 4.35 (1H, s), 3.71 (2H, s), 3.27 (1H, broad s), 2.04-1.98 (1H, m), 1.72-1.85 (1H, m), 1.43 (9H, s)
【0032】
(ステップ1−8)
(S)-2-アミノ-N-ベンジル-4-ヒドロキシブタンアミド塩酸塩(12)の合成
ステップ1−7で得られた(S)-1-ベンジルカルバモイル-3-ヒドロキシプロピルtert-ブチルカルバメート(11)(5.40 g)をジクロロメタン(60 mL)に溶解させ、4N塩酸1,4-ジオキサン溶液(87 mL)を加え、室温で5時間攪拌した。反応液を濃縮し、化合物(4.27 g, 収率100%)を白色固体として得た。化合物(12)の1H NMRのデータを以下に示す。
δ:1H NMR (CD3OD, 500 MHz, δ; ppm)
7.44-7.41 (2H, m), 7.34-7.30 (3H, m), 7.27 (1H, broad s), 4.43 (2H, s), 3.94 (1H, d, J = 7.0 Hz) 2.03 (2H, s)
【0033】
(ステップ1−9)
(S)-N-(1-ベンジルカルバモイル-3-ヒドロキシプロピル)ベンズアミド(13)の合成
ステップ1−8で得られた(S)-2-アミノ-N-ベンジル-4-ヒドロキシブタンアミド塩酸塩(12)(118 mg)、安息香酸(60 mg)、PyBOP(303 mg)、トリエチルアミン(0.2 mL)をN,N-ジメチルホルムアミド(1 mL)に溶解させ、室温で20時間攪拌した。反応液を水(100 mL)で希釈し、ジクロロメタン(100 mL)で抽出した。有機層を飽和食塩水(100 mL)で洗浄して、無水硫酸ナトリウムで乾燥し、ろ過後、残渣をシリカゲルフラッシュカラムクロマトグラフィー(展開溶媒 n-ヘキサン:酢酸エチル=2:3)で精製し、化合物(13)(72 mg, 収率47%)を白色固体として得た。化合物(13)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.79 (2H, d, J = 7.3 Hz), 7.53 (1H, t, J = 7.3 Hz), 7.44 (2H, t, J = 8.0 Hz), 7.33-7.25 (5H, m), 7.06 (1H, d, J = 7.3 Hz), 6.89 (1H, broad s), 4.77 (1H, q, J = 6.4 Hz), 4.47-4.45 (2H, m), 4.32-4.28 (1H, m), 4.22-4.18 (1H, m), 2.28-2.19 (2H, m), 2.03 (3H, s).
【0034】
<カップリング反応>
ステップ1−1〜ステップ1−6で合成したカップリング前駆体である化合物(9)と、ステップ1−7〜ステップ1−9で合成したカップリング前駆体(13)とを用いて、化10に示す合成ルートに従い、光延反応を用いたカップリング反応を行った。
【化10】
【0035】
以下にカップリング反応の詳細を示す。
(ステップ1−10)
(S)-トランス-[2-[3-(3-ベンゾイルアミノ-3-ベンジルカルバモイルプロポキシ)フェニル] シクロプロピルtert-ブチルカルバメート(14)の合成
ステップ1−9で得られた(S)-N-(1-ベンジルカルバモイル-3-ヒドロキシプロピル)ベンズアミド(13)(200 mg)と、ステップ1−6で得られたトランス-2-(3-ヒドロキシフェニル)シクロプロピルtert-ブチルカルバメート(9)(230 mg)と、トリフェニルホスフィン(504 mg)とを無水テトラヒドロフラン(3 mL)に溶解させ、氷冷下、ジイソプロピルアゾジカルボキシレート(1mL)を加えた。反応液を室温で5時間攪拌後、濃縮し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(展開溶媒 n-ヘキサン:酢酸エチル= 1 :1)で精製し、化合物(14)(64 mg, 収率18%)を淡黄色固体として得た。化合物(14)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.82 (2H, d, J = 8.5 Hz), 7.52 (1H, t, J = 8.5 Hz), 7.46 (2H, t, J = 8.5 Hz), 7.26-7.21 (5H, m), 7.16 (1H, t, J = 8.0 Hz), 6.92 (1H, s), 6.75 (1H, d, J = 8.0 Hz), 6.63 (1H, d, J = 8.0 Hz), 6.60 (1H, broad s), 4.88 (1H, quartet, J = 6.0 Hz), 4.47 (2H, d, J = 6.0 Hz), 4.32-4.29 (1H, m), 4.13-4.10 (1H, m), 2.65-2.70 (1H, m), 2.43-2.37 (2H, m), 1.99-1.97 (1H, m), 1.45 (9H, s), 1.14-1.10 (2H, m).
【0036】
(ステップ1−11)
(S)-トランス-N-3-[3-(2-アミノシクロプロピル) フェノキシ]-1-ベンジルカルバモイルプロピルベンズアミド塩酸塩(NCL-1)の合成
ステップ1−10で得られた(S)-トランス-[2-[3-(3-ベンゾイルアミノ-3-ベンジルカルバモイルプロポキシ)フェニル] シクロプロピルtert-ブチルカルバメート(14)(70 mg)をジクロロメタン(1 mL)に溶解させ、4N塩酸酢酸エチル溶液(0.8 mL)を加え、室温で4時間攪拌した。反応液を濃縮し、残渣をクロロホルム-メタノールから再結晶し、実施例1の化合物(NCL-1)(22 mg, 収率36%)を淡黄色固体として得た。NCL-1の融点、1H NMR、13C NMR、MS (FAB)のデータを示す。
融点 134.8-139.1 ℃
1H NMR (CD3OD, 500 MHz, δ; ppm)
δ:7.84 (2H, d, J = 8.5 Hz), 7.54 (1H, t, J = 8.5 Hz), 7.46 (2H, t, J = 8.5 Hz), 7.26-7.17 (6H, m), 6.77 (1H, dd, J = 8.0, 1.5 Hz), 6.74-6.72 (1H, m), 6.67 (1H, d, J = 1.5 Hz), 4.47-4.37 (2H, m), 4.08 (2H, t, J = 6.0 Hz), 2.28-2.24 (2H, m), 1.35-1.33 (1H, m), 1.29-1.26 (1H, m)
13C NMR (CD3OD, 500 MHz, δ; ppm)
δ:173.93, 170.34, 160.52, 141.32, 139.79, 135.21, 132.96, 130.81, 129.55, 128.54, 128.46, 128.19, 120.00, 114.15, 114.05, 113.84, 65.92, 53.13, 44.15, 32.68, 31.96, 22.58, 13.84
MS (FAB) m/z = 444 (M+)
Anal. Calcd. for C27H30ClN3O3 3/2H2O: C, 63.96; H, 6.56; N, 8.29.
Found: C, 63.83; H, 6.29; N, 7.98. 3/2
【0037】
(実施例2)
実施例2では、(S)-トランス-N-3-[4-(2-アミノシクロプロピル)フェノキシ]-1-ベンジルカルバモイルプロピルベンズアミド塩酸塩(NCL-2)を合成した(下記化学式参照)。詳細は以下のとおりである。
【0038】
【化11】
【0039】
まず、NCL-2を合成するための一方の前駆体として、化合物(21)を以下の反応ルートにしたがって合成した。
【0040】
【化12】
【0041】
以下に合成法の詳細を示す。
(ステップ2−1)
トランス-3- (4-ヒドロキシフェニル)アクリル酸メチルエステル(16)の合成
実施例1のステップ1−1と同様の方法により、3-(4-ヒドロキシフェニル)アクリル酸(15)(20.0 g)を出発原料とし、化合物(16)(21.2 g, 収率99%)を白色固体として得た。化合物(16)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.64 (1H, d, J = 15.8 Hz), 7.43 (2H, d, J = 8.5 Hz), 6.84 (2H, d, J = 8.5 Hz), 6.31 (1H, d, J =15.8 Hz), 5.21 (1H,s), 3.80 (3H, s).
【0042】
(ステップ2−2)
トランス-3-(4-メトキシメトキシフェニル)アクリル酸メチルエステル(17)の合成
実施例1のステップ1−2と同様の方法により、ステップ2−1より得られたトランス-3- (4-ヒドロキシフェニル)アクリル酸メチルエステル(16)(10.0 g)より、化合物(17)(11.1 g, 収率89%)を無色油状物として得た。化合物(17)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.65 (1H, d, J = 6.2 Hz), 7.47 (2H, d, J = 8.8 Hz), 7.04 (2H, d, J = 8.8 Hz), 6.33 (1H, d, J = 6.2 Hz), 5.20 (2H, s), 3.80 (3H, s), 3.48 (3H, s).
【0043】
(ステップ2−3)
トランス-2-(4-メトキシメトキシフェニル)シクロプロパンカルボン酸メチルエステル(18)の合成
実施例1のステップ1−3と同様の方法により、ステップ2−2より得られたトランス-3-(4-メトキシメトキシフェニル)アクリル酸メチルエステル(15)(11.0 g)より、化合物(18)(1.68 g, 収率14%)を無色油状物として得た。化合物(18)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.03 (2H, d, J = 8.5 Hz), 6.95 (2H, d, J =8.5 Hz), 5.14 (2H, s), 3.71 (3H, s), 3.46 (3H, s), 2.51-2.47 (1H, m), 1.85-1.81 (1H, m), 1.58-1.54 (1H, m) 1.29-1.25 (1H, m).
【0044】
(ステップ2−4)
トランス-2-(4-メトキシメトキシフェニル)シクロプロパンカルボン酸(19)の合成
実施例1のステップ1−4と同様の方法により、ステップ2−3より得られたトランス-2-(4-メトキシメトキシフェニル)シクロプロパンカルボン酸メチルエステル(18)(1.68 g)より、化合物(19)(1.50 g, 収率95%)を白色固体として得た。化合物(19)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.05 (2H, d, J = 6.7 Hz), 6.94 (2H, d, J = 6.7 Hz), 5.12 (2H, s), 3.42 (3H, s), 2.43-2.39 (1H, m), 1.76-1.73 (1H, m), 1.49-1.46 (1H, m), 1.33-1.28 (1H, m).
【0045】
(ステップ2−5)
トランス- 2-(4-メトキシメトキシフェニル)シクロプロピルtert-ブチルカルバメート(20)の合成
実施例1のステップ1−5と同様の方法により、ステップ2−4より得られたトランス-2-(4-メトキシメトキシフェニル)シクロプロパンカルボン酸(19)(1.05 g)より、化合物(20)(788 mg, 収率56%)を淡黄色固体として得た。化合物(20)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.07 (2H, d, J = 8.2 Hz), 6.94 (2H, d, J = 8.2 Hz), 5.13 (2H, s), 4.89 (1H, broad s), 3.46 (3H, s), 2.64-2.69 (1H, m), 2.02-1.98 (1H, m), 1.46 (9H, s), 1.12-1.09 (2H, m).
【0046】
(ステップ2−6)
トランス-2-(4-ヒドロキシフェニル)シクロプロピルtert-ブチルカルバメート(21)の合成
実施例1のステップ1−6と同様の方法により、ステップ2−5より得られたトランス- 2-(4-メトキシメトキシフェニル)シクロプロピルtert-ブチルカルバメート(20)(476 mg)より、化合物(21)(300 mg, 収率74%)を淡黄色油状物として得た。化合物(21)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:6.94 (2H, d, J = 8.5 Hz), 6.72 (2H, d, J =8.5 Hz), 5.13 (1H, broad s), 4.84 (1H, broad s), 2.60-2.70 (1H, m), 1.98 (1H, m), 2.09-1.96 (1H, m), 1.10-1.05 (2H, m).
【0047】
NCL-2を合成するための他方の前駆体として、実施例1のステップ1−7〜ステップ1−9で合成した化合物(13)を用いた。
【0048】
<カップリング反応>
上記ステップ2−1〜ステップ2−6で合成したカップリング前駆体である化合物(21)と、化合物(13)とを用いて、光延反応によるたカップリング反応によって、実施例2の化合物NCL-2を合成した。
【0049】
【化13】
【0050】
以下に合成法の詳細を示す。
(ステップ2−7)
(S)-トランス-[2-[4-(3-ベンゾイルアミノ-3-ベンジルカルバモイルプロポキシ)フェニル] シクロプロピルtert-ブチルカルバメート(22)の合成
実施例1のステップ1−7と同様の方法により、ステップ2−6より得られたトランス-2-(4-ヒドロキシフェニル)シクロプロピルtert-ブチルカルバメート(21)(220 mg)と
実施例1のステップ1−7〜1−9で合成したカップリング前駆体(13)のカップリングを行い、化合物(22)(83 mg, 収率25%)を淡黄色固体として得た。化合物(22)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.79 (2H, d, J = 8.5 Hz), 7.53-7.49 (1H, m), 7.41 (2H, t, J = 8.5 Hz), 7.26-7.13 (5H, m), 7.04 (2H, d, J = 8.5 Hz), 6.71 (2H, d, J = 8.5 Hz), 4.94-4.89 (2H, m), 4.46-4.38 (2H, m), 4.25-4.21 (1H, m), 4.08-4.04 (1H, m), 2.55-2.59 (1H, m), 2.43-2.37 (2H, m), 1.95-2.01 (1H, m), 1.45 (9H, s), 1.08 (2H, t, J = 7.0 Hz).
【0051】
(ステップ2−8)
(S)-トランス-N-3-[4-(2-アミノシクロプロピル) フェノキシ]-1-ベンジルカルバモイルプロピルベンズアミド塩酸塩(NCL-2)の合成
実施例1のステップ1−11と同様の方法により、ステップ2−7より得られた、(S)-トランス-[2-[4-(3-ベンゾイルアミノ-3-ベンジルカルバモイルプロポキシ)フェニル] シクロプロピルtert-ブチルカルバメート(22)(83 mg)より、化合物(NCL-2)(18 mg, 収率25%)を淡黄色固体として得た。NCL-2の融点、1H NMR、13C NMR、MS (FAB)のデータを示す。
融点:141.2-144.5 ℃
1H NMR (CD3OD, 500 MHz, δ; ppm)
δ:7.83 (2H, d, J = 7.0 Hz), 7.54 (1H, t, J = 7.0 Hz), 7.45 (2H, t, J = 7.0 Hz), 7.27-7.19 (6H, m), 7.05 (2H, dd, J = 9.0 Hz), 6.84 (2H, d, J = 9.0 Hz), 4.40 (2H, d, J = 8.0 Hz), 4.06 (2H, t, J = 8.0 Hz), 2.74-2.72 (1H, m), 2.42-2.36 (1H, m), 2.29-2.21 (1H, m), 1.34-1.29 (1H, m), 1.26-1.22 (1H, m)
13C NMR (CD3OD, 500 MHz, δ; ppm)
δ:173.91, 170.32, 159.23, 139.80, 135.21, 132.93, 131.84, 129.55, 129.52, 128.62, 128.52, 128.48, 128.17, 115.89, 65.95, 53.13, 44.16, 32.70, 31.73, 21.96, 13.41
MS (FAB) m/z = 444 (M+);
Anal. Calcd For C27H30ClN3O3 2H2O: C, 62.84; H, 6.64; N, 8.14.
Found: C, 62.47; H, 6.08; N, 8.21.
【0052】
(実施例3)
実施例3では、トランス-4-[3-(2-アミノシクロプロピル)フェノキシ]-N-ベンジルブチルアミド塩酸塩(NCL-3)を合成した(下記化学式参照)。詳細は以下のとおりである。
【0053】
【化14】
【0054】
<カップリング前駆体>
実施例3では、NCL-3を合成するための前駆体として、実施例1(ステップ1−1〜1−6)で合成した、化合物(9)を用いた。また、NCL-3を合成するためのもう一方の前駆体として、化合物(24)を次の反応によって合成した。
【化15】
【0055】
(ステップ3−1)
N-ベンジル-4-クロロブチルアミド(24)の合成
ベンジルアミン(1.1 mL)とトリエチルアミン(4.5 mL)をジクロロメタン(26 mL)に溶解し、氷冷下、4-クロロブチリルクロリド(23)(1.5 mL)のジクロロメタン(15 mL)溶液を滴下した。0°Cで30分攪拌後、反応液を水(100 mL)に注ぎ、酢酸エチル(200 mL)で抽出した。有機層を飽和食塩水(100 mL)で洗浄して、無水硫酸ナトリウムで乾燥し、ろ過後、残渣をシリカゲルフラッシュカラムクロマトグラフィー(展開溶媒 n-ヘキサン:酢酸エチル= 3 :1)で精製し、化合物(24)(1.26 g, 収率59%)を淡黄色油状物として得た。化合物(24)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.36-7.26 (5H, m), 5.84 (1H, broad s), 4.44 (2H, d, J = 5.8 Hz), 3.62 (2H, t, J = 6.0 Hz), 2.40 (2H, t, J = 7.0 Hz), 2.14 (2H, quintet, J = 7.0 Hz).
【0056】
NCL-3を合成するための他方の前駆体として、実施例1のステップ1−1〜ステップ1−6で合成した化合物(9)を用いた。
【0057】
<カップリング反応>
上記のようにして合成した化合物(24)と化合物(9)とを光延反応によってカップリングさせて実施例3の化合物NCL-3を得た。
【化16】
【0058】
以下に合成法の詳細を示す。
(ステップ3−2)
トランス-2-[3-(3-ベンジルカルバモイルプロポキシ)フェニル]シクロプロピル tert-ブチルカルバメート(25)の合成
実施例1のステップ1−6で得られたトランス-2-(3-ヒドロキシフェニル)シクロプロピルtert-ブチルカルバメート(9)(400 mg)をアセトニトリル(7.5 mL)に溶解させ、炭酸セシウム(1.54 g)、ヨウ化ナトリウム(70 mg)を加えて、室温で15分間攪拌した。反応液にステップ3−1で得られたN-ベンジル-4-クロロブチルアミド(25)(900 mg)のアセトニトリル(10 mL)溶液を添加し、9時間加熱還流した。反応液をろ過し、不溶物を取り除き、ろ液を濃縮後、残渣をシリカゲルフラッシュカラムクロマトグラフィー(展開溶媒 n-ヘキサン:酢酸エチル= 1:1)で精製し、化合物(24)(140 mg, 収率21%)を淡黄色油状物として得た。化合物(25)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.30-7.24 (5H, m), 7.15 (1H, t, J = 7.0 Hz), 6.71 (1H, d, J = 7.0 Hz), 6.66 (1H, d, J = 7.0 Hz), 6.62 (1H, s), 5.85 (1H, broad s), 4.84 (1H, broad s), 4.44 (2H, d, J = 5.8 Hz), 3.99 (2H, t, J = 5.8 Hz), 2.67-2.75 (1H, m), 2.43 (2H, t, J = 7.2 Hz), 2.16-2.13 (2H, m), 2.01-1.96 (1H, m), 1.45 (9H, s), 1.12-1.15 (2H, m).
【0059】
(ステップ3−3)
トランス-4-[3-(2-アミノシクロプロピル)フェノキシ]-N-ベンジルブチルアミド塩酸塩(NCL-3)の合成
実施例1のステップ1−11と同様の方法により、ステップ3−2より得られた、トランス-2-[3-(3-ベンジルカルバモイルプロポキシ)フェニル]シクロプロピル tert-ブチルカルバメート(25)(140 mg)より、実施例3のフェニルシクロプロピルアミン誘導体である化合物(NCL-3)(82 mg, 収率69%)を白色固体として得た。NCL-3の融点、1H NMR、13C NMR、MS (ELC)のデータを示す。
融点:83.3-86.7 °C
1H NMR (CD3OD, 500 MHz, δ; ppm)
δ:7.26-7.17 (6H, m), 6.77 (1H, dd, J = 7.6, 2.5 Hz), 6.72 (1H, d, J = 7.6 Hz), 6.69 (1H, s), 4.36 (2H, s), 3.98 (2H, t, J = 6.4 Hz), 2.82-2.80 (1H, m), 2.43 (2H, t, J = 7.2 Hz), 2.32-2.30 (1H, m), 2.10-2.06 (2H, m), 1.39-1.35 (1H, m), 1.32-1.28 (1H, m)
13C NMR (CD3OD, 500 MHz, δ; ppm)
δ:175.37, 160.71, 141.30, 140.02, 130.78, 129.53, 128.52, 128.18, 119.68, 113.94 113.81, 68.06, 33.46, 31.96, 26.57, 22.62, 13.84
MS (ELC) m/z: 325 (M+)
Anal. Calcd For C20H25ClN2O2 2/3H2O: C, 64.42; H, 7.12; N, 7.51.
Found: C, 64.44; H, 6.87; N, 7.80.
【0060】
(実施例4)
実施例4では、トランス-4-[4-(2-アミノシクロプロピル)フェノキシ]-N-ベンジルブチルアミド塩酸塩(NCL-4)を合成した(下記化学式参照)。詳細は以下のとおりである。
【0061】
【化17】
【0062】
<カップリング前駆体>
実施例4では、カップリング前駆体は、実施例2におけるステップ2−1〜ステップ2−6で合成した、化合物(21)を用いた。また、もう一方のカップリング前駆体としては、実施例3におけるステップ3−1で合成した化合物(24)を用いた。
【0063】
<カップリング反応>
上記のようにして合成した化合物(24)と化合物(21)とを光延反応によってカップリングさせて実施例4の化合物NCL-4を得た。
【0064】
【化18】
【0065】
以下に合成法の詳細を示す。
(ステップ4−1)
トランス-2-[4-(3-ベンジルカルバモイルプロポキシ)フェニル]シクロプロピル tert-ブチルカルバメート(26)の合成
実施例3のステップ3−2と同様の方法により、化合物(21)(500 mg)と化合物(24)とをカップリングさせ、実施例4のフェニルシクロプロピルアミン誘導体である化合物(26)(NCL-3)(82 mg, 収率69%)を白色固体として得た。化合物(26)の1H NMRのデータを以下に示す。
1H NMR (CDCl3, 500 MHz, δ; ppm)
δ:7.32-7.24 (5H, m), 7.05 (2H, d, J = 8.2 Hz), 6.75 (2H, d, J = 8.2 Hz), 5.82 (1H, broad s), 4.84 (1H, broad s), 4.44 (2H, d, J = 5.8 Hz), 3.97 (2H, t, J = 5.8 Hz), 2.60-2.70 (1H, m), 2.42 (2H, t, J = 7.2 Hz), 2.16-2.12 (2H, m), 1.97-2.04(1H, m), 1.46 (9H, s), 1.02-1.15 (2H, m).
【0066】
(ステップ4−2)
トランス-4-[4-(2-アミノシクロプロピル)フェノキシ]-N-ベンジルブチルアミド塩酸塩(NCL-4)の合成
実施例1のステップ1−11と同様の方法により、ステップ4−1より得られた、化合物(26)(140 mg)より、実施例4のフェニルシクロプロピルアミン誘導体である化合物(NCL-4)(82 mg, 収率69%)を白色固体として得た。NCL-4の融点、1H NMR、13C NMR、MS (ELC)のデータを示す。
融点:156.3-159.4 °C; 1H NMR (CD3OD, 500 MHz, δ; ppm)
δ:7.27-7.21 (5H, m), 7.06 (2H, d, J = 8.5 Hz), 6.83 (2H, d, J = 8.5 Hz), 4.35 (2H, s), 3.95 (2H, t, J = 6.4 Hz), 2.75-2.74 (1H, m), 2.42 (2H, t, J = 7.2 Hz), 2.30-2.27 (1H, m), 2.09-2.05 (2H, m), 1.35-1.32 (1H, m), 1.27-1.24 (1H, m)
δ:13C NMR (CD3OD, 500 MHz, δ; ppm) 175.34, 159.44, 140.06, 131.58, 129.51, 128.61, 128.53, 128.16, 115.81, 68.17, 44.11, 33.48, 31.73, 26.59, 21.96, 13.40
MS (ELC) m/z: 325 (M+)
Anal. Calcd For C20H25ClN2O2 H2O: C, 63.40; H, 7.18; N, 7.39.
Found: C, 63.50; H, 6.68; N, 7.44.
【0067】
−評 価−
以上のようにして得られた実施例1〜実施例4の化合物(NCL-1〜NCL-4)について、LSD1阻害活性試験、モノアミンオキシダーゼ阻害活性試験及びHIV複製に対する阻害活性試験を行った。
【0068】
<LSD1阻害試験>
LSD1酵素は以下のようにして調製した。
全長のLSD1(1-851aa)のN末端にヒスチジン5残基を付加した組換えタンパク質をコードするプラスミドを調製し、このプラスミドで形質転換した組換え大腸菌を用いてLSD1を発現させた。その後、組換え大腸菌を超音波処理で溶解し、その可溶性画分をHisTrapクロマトグラフィーで精製し、LSD1酵素溶液を得た。LSD1の酵素活性は、LSD1の脱メチル化反応の際に生成する過酸化水素をペルオキシダーゼと試薬によって発色させ、吸光光度法で定量することにより測定した。すなわち、384ウエルマイクロタイタープレート内で、50mM Hepes-NaOH buffer(pH7.5)、0.1mM 4-アミノアンチピリン、1mM 3,5-ジクロロ-2-ヒドロキシベンゼンスルホン酸、20μM histone H3- lysine 4 dimethyl peptide、0.05μM LSD1、0.35μM horseradish peroxidaseからなる20μlの溶液を25℃で30分間、経時的に酵素反応を測定した。測定にはSpectra Max M2e(Molecular Devices社)を用いて、生成物の515nmでの吸光度を測定して求めた。また、阻害活性については、ジメチルスルホキシド(dimethyl sulfoxide)添加時の酵素活性を100%とし、フェニルシクロプロピルアミン誘導体の添加濃度を様々に変えて残存活性を測定し、50%の活性を阻害する濃度(IC50)を求めた。
【0069】
LSD1阻害試験の結果を図1に示す。この図に示すように、実施例1〜4の化合物(NCL-1〜4)は、いずれもtranylcypromineより高いLSD1阻害活性を示した。また、実施例1〜4のの化合物(NCL-1〜4)中でも、アミノ酸構造を有する実施例1のNCL-1及び実施例2のNCL-2では、極めて高いLSD1阻害活性を示した。
【0070】
<モノアミンオキシダーゼ阻害活性測定試験>
モノアミンオキシダーゼA(MAO-A)及びモノアミンオキシダーゼB(MAO-B)阻害活性の測定を、Promega社のMAO-GloアッセイキットとSigma-Aldrich社から購入したMAO-AおよびMAO-Bを用いて以下のように行った。
12.5 μLの4倍MAO基質(最終濃度40 μM)、12.5 μLの4倍阻害剤溶液(最終濃度0.1〜1000 μM)、25 μLのMAO-A(最終濃度 9 unit/mL)あるいは25 μLのMAO-B(最終濃度2.3 unit/mL)を混合し、室温で1時間反応させた。この反応液に50 μLのルシフェリン検出試薬を添加し、室温で20分反応させ、蛍光プレートリーダーも用いて蛍光強度(蛍光測定波長:562 nm)を測定し、IC50値(酵素を50%阻害する阻害剤濃度)を求めた。
【0071】
MAO-Aに対する阻害活性の結果を図2に示す。この図に示すように、実施例1〜4の化合物(NCL-1〜4)は、いずれもtranylcypromineやpargylineより低いMAO A阻害活性を示した。その中でも、アミノ酸構造を有する実施例1のNCL-1及び実施例2のNCL-2では、特に阻害活性が低かった。
また、MAO-Bに対する阻害活性についても、図3に示すように、実施例1〜4の化合物(NCL-1〜4)は、いずれもtranylcypromineやpargyline(下記化学式参照)より低いMAO-B阻害活性を示し、その中でも、実施例1のNCL-1及び実施例2のNCL-2では、特に阻害活性が低かった。
【0072】
【化19】
【0073】
<HIV複製に対する阻害活性試験>
上記LSD1阻害試験において阻害活性を示したNCL-2及びpargylineについて、HIV複製に対する阻害活性試験を行った。詳細を以下に示す。
【0074】
(HIV複製に対するNCL-2の阻害活性)
HIV潜伏感染細胞Ach2にNCL-2を0-10μM/Lとなるように加え、24時間後の細胞培養上清中のHIVのコア蛋白p24の濃度をp24antigen assay法により測定し、HIV複製の指標とした。
また、上記試験においてNCL-2とともにTNF (1μg/ml)を加えて潜伏感染細胞を刺激し、24時間後の細胞培養上清中のHIVのコア蛋白p24を測定し、HIV複製の指標とした。
【0075】
その結果、TNFを加えなかった場合には、図4(a)左側のグラフに示すように、NCL-2の添加量が0-10μM/Lの範囲において、HIVのコア蛋白p24はいずれも検出感度ぎりぎりで増減は認められず、HIV複製が阻害されることはなかった。
これに対して、TNFを加えた場合には、図4(a)右側のグラフに示すように、NCL-2の濃度の増加とともにHIVのコア蛋白p24が減少し(p値=0.055)、HIV複製が阻害されることが分かった。
また、上記HIV複製に対する阻害活性試験における細胞毒性効果をWST-1 assay法によって調べた結果、図4(b)に示すように、TNFの添加の有無にかかわらず、NCL-2の添加量が0-10μM/Lの範囲において、細胞毒性は20%を超えることはなかった。このことから、HIV複製の阻害が、細胞毒性によるものではないことが分かった。
【0076】
(HIV複製に対するPargylineの阻害活性)
HIV潜伏感染細胞Ach2にPargylineを0-3 mM/Lとなるように加え、24時間後の細胞培養上清中のHIVのコア蛋白p24の濃度をp24antigen assay法により測定し、HIV複製の指標とした。
また、上記試験においてPargylineとともにTNF (1μg/ml)を加えて潜伏感染細胞を刺激し、24時間後の細胞培養上清中のHIVのコア蛋白p24を測定し、HIV複製の指標とした。
【0077】
その結果、Pargylineを加えなかった場合には、図5(a)左側のグラフに示すように、Pargylineの添加量が0-3 mM/Lの範囲において、HIVのコア蛋白p24はいずれも検出感度ぎりぎりで増減は認められず、HIV複製が阻害されることはなかった。
これに対して、Pargylineを加えた場合には、図5(a)右側のグラフに示すように、Pargylineの濃度の増加とともにHIVのコア蛋白p24が減少し(p値=0.139)、HIV複製が阻害されることが分かった。
また、上記HIV複製に対する阻害活性試験における細胞毒性効果をWST-1 assay法によって調べた結果、図5(b)に示すように、TNFの添加の有無にかかわらず、Pargylineの添加量が0-3 mM/Lの範囲において、細胞毒性は20%を超えることはなかった。このことから、HIV複製の阻害が、細胞毒性によるものではないことが分かった。
【0078】
<HIV−LTR近傍のメチル化状態の解析>
本発明者らは、HIV−LTR近傍のメチル化状態について、以下に示す研究発表を既に行っている(非特許文献4)。
すなわち、HIV (AIDSウイルス)の転写を調節するLTRと呼ばれるプロウイルスDNAの「転写スイッチ」領域のメチル化状態とヒストンメチル化酵素G9aおよびRNA合成酵素II型(RNAPII)の結合状態を、ヒストンメチル化酵素G9a阻害剤BIX01294を加えた場合と加えない場合とについて、クロマチン免疫沈降(ChIP)アッセイで調べた。
【0079】
その結果、図6の左側のグラフが示すように、ヒストンメチル化酵素G9a阻害剤であるBIX阻害剤を加えない場合には、G9aはHIVプロウイルスLTRと結合していて、その部位のH3K9のジメチル化が十分に起こっている。これに対して、ヒストンメチル化酵素G9a阻害剤であるBIXを加えるとG9aは離脱し、しかもH3K9のジメチル化も著しく低下している。このことはG9aが離脱しただけでは説明ができず、ある種のヒストン脱メチル化酵素がG9aの代わりに新たに結合したことを示唆している。ただし、この事実のみからは、LSD1がHIV複製と関係しているか否かについては不明である。
【0080】
しかしながら、前述したように、NCL-2及びpargylineがHIV複製の阻害作用を示すこと、及び、NCL-2やpargylineがLSD1阻害剤作用を示すことを考え合わせれば、LSD1阻害剤作用を有する化合物はHIV複製の阻害作用を示すことを強く示唆しており、今回始めて明らかにされたことである。
【0081】
また、図6の右側のグラフは、潜伏感染HIVからの転写を促進する作用のあるTNFαを加えていない対照(左)と、TNFαを加えた場合(右)の、ChIPアッセイの結果を示す。このグラフから、TNFαを加えることによってもG9aの離脱は引き起こされるが、BIX01294を加えた場合の効果よりも小さいことが分かる。
【0082】
さらに、図7のグラフは、HIVが潜伏感染していて細胞からはほとんどウイルスの産生が起こっていないAch-2細胞(HIV感染ヒトT細胞株)にG9a阻害剤BIX01294を加えた場合の、細胞培養上清中のHIVのコア蛋白p24の濃度をp24antigen assay法により測定した結果である。この図から、G9a阻害剤BIX01294の添加量が多いほど、HIVのコア蛋白p24の量も多くなり、HIV複製が活性化されることが分かる。ただし、このことのみからは、LSD1とHIV複製との関係についての知見を得ることはできない。
【0083】
この発明は上記発明の実施の態様及び実施例の説明に何ら限定されるものではない。特許請求の範囲を逸脱せず、当業者が容易に想到できる範囲で種々の変形態様もこの発明に含まれる。
【図面の簡単な説明】
【0084】
【図1】LSD1に対する阻害活性を示す図である。
【図2】MAO-Aに対する阻害活性を示す図である。
【図3】MAO-Bに対する阻害活性を示す図である。
【図4】(a)は、NCL-2の添加量とHIVのコア蛋白p24の濃度との関係をTNF添加の場合及びTNF不添加の場合について調べたグラフである。また、(b)は、HIV複製に対する阻害活性試験における細胞毒性効果をWST-1 assay法によって調べたグラフである。
【図5】(a)は、Pargylineの添加量とHIVのコア蛋白p24の濃度との関係をTNF添加の場合及びTNF不添加の場合について調べたグラフである。また、(b)は、HIV複製に対する阻害活性試験における細胞毒性効果をWST-1 assay法によって調べたグラフである。
【図6】プロウイルスDNAの「転写スイッチ」領域のメチル化状態とヒストンメチル化酵素G9aおよびRNA合成酵素II型(RNAPII)の結合状態を、ヒストンメチル化酵素G9a阻害剤BIX01294を加えた場合と加えない場合とについて、クロマチン免疫沈降(ChIP)アッセイで調べた結果を示す図である。
【図7】Ach-2細胞にBIX01294を加えた場合の、細胞培養上清中のHIVのコア蛋白p24の濃度を示すグラフである。
【産業上の利用可能性】
【0085】
本発明のフェニルシクロプロピルアミン誘導体及びLSD1阻害剤は、LSD1の機能を調べるための生物学的ツールとして用いたり、抗がん剤として用いたりすることができると期待される。
【特許請求の範囲】
【請求項1】
LSD1阻害剤を有効成分として含有することを特徴とするHIV複製阻害剤。
【請求項2】
前記LSD1阻害剤は、下記一般式(I)若しくは(II)(ただし、両式中R1は水素、置換基が結合していてもよいアルキル基、置換基が結合していてもよいフェニル基及び置換基が結合していてもよい複素環基のいずれかを示し、R2は分枝を有することがあり置換基が結合していてもよいアルキレン基を示し、R3は置換基が結合していてもよいアルキル基、置換基が結合していてもよいフェニル基、置換基が結合していてもよい複素環基及び置換基が結合していてもよいベンジル基のいずれかを示し、R4は置換基が結合していてもよいアルキル基、置換基が結合していてもよいフェニル基、置換基が結合していてもよい複素環基、置換基が結合していてもよいアルキルオキシ基、置換基が結合していてもよいフェニルオキシ基、置換基が結合していてもよいアルキルアミノ基及び置換基が結合していてもよいフェニルアミノ基のいずれかを示し、XはO、NH2、NHCO、CONH、S又はCH2を示す)の化合物又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグからなることを特徴とするHIV複製阻害剤。
【化1】
【請求項3】
前記LSD1阻害剤は、下記一般式(1)(ただし、式中R1は水素又はフェニルアミド基を示し、R2は分枝を有することのあるアルキレン基を示し、シクロプロピルアミン置換基はメタ位又はパラ位に結合している)の化合物又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグからなることを特徴とする請求項1又は2記載のHIV複製阻害剤。
【化2】
【請求項4】
R2はエチレン基であることを特徴とする請求項3記載のHIV複製阻害剤。
【請求項5】
R1はフェニルアミド基であることを特徴とする請求項3又は4に記載のHIV複製阻害剤。
【請求項6】
前記LSD1阻害剤は、下記一般式(2)の化合物又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグからなることを特徴とするHIV複製阻害剤。
【化3】
【請求項1】
LSD1阻害剤を有効成分として含有することを特徴とするHIV複製阻害剤。
【請求項2】
前記LSD1阻害剤は、下記一般式(I)若しくは(II)(ただし、両式中R1は水素、置換基が結合していてもよいアルキル基、置換基が結合していてもよいフェニル基及び置換基が結合していてもよい複素環基のいずれかを示し、R2は分枝を有することがあり置換基が結合していてもよいアルキレン基を示し、R3は置換基が結合していてもよいアルキル基、置換基が結合していてもよいフェニル基、置換基が結合していてもよい複素環基及び置換基が結合していてもよいベンジル基のいずれかを示し、R4は置換基が結合していてもよいアルキル基、置換基が結合していてもよいフェニル基、置換基が結合していてもよい複素環基、置換基が結合していてもよいアルキルオキシ基、置換基が結合していてもよいフェニルオキシ基、置換基が結合していてもよいアルキルアミノ基及び置換基が結合していてもよいフェニルアミノ基のいずれかを示し、XはO、NH2、NHCO、CONH、S又はCH2を示す)の化合物又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグからなることを特徴とするHIV複製阻害剤。
【化1】
【請求項3】
前記LSD1阻害剤は、下記一般式(1)(ただし、式中R1は水素又はフェニルアミド基を示し、R2は分枝を有することのあるアルキレン基を示し、シクロプロピルアミン置換基はメタ位又はパラ位に結合している)の化合物又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグからなることを特徴とする請求項1又は2記載のHIV複製阻害剤。
【化2】
【請求項4】
R2はエチレン基であることを特徴とする請求項3記載のHIV複製阻害剤。
【請求項5】
R1はフェニルアミド基であることを特徴とする請求項3又は4に記載のHIV複製阻害剤。
【請求項6】
前記LSD1阻害剤は、下記一般式(2)の化合物又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグからなることを特徴とするHIV複製阻害剤。
【化3】
【図1】

【図2】
【図3】
【図4】
【図5】
【図7】
【図6】
【図2】
【図3】
【図4】
【図5】
【図7】
【図6】
【公開番号】特開2012−36124(P2012−36124A)
【公開日】平成24年2月23日(2012.2.23)
【国際特許分類】
【出願番号】特願2010−177176(P2010−177176)
【出願日】平成22年8月6日(2010.8.6)
【出願人】(506218664)公立大学法人名古屋市立大学 (48)
【Fターム(参考)】
【公開日】平成24年2月23日(2012.2.23)
【国際特許分類】
【出願日】平成22年8月6日(2010.8.6)
【出願人】(506218664)公立大学法人名古屋市立大学 (48)
【Fターム(参考)】
[ Back to top ]