
IAPアンタゴニストに対するヒトまたは非ヒト動物細胞の感受性を決定する方法
cFLIPは、Smacペプチド模倣体を含むIAPアンタゴニストを用いた処置の有効性に関するバイオマーカーとしての役割を果たす。
【発明の詳細な説明】
【技術分野】
【0001】
関連出願の相互参照
本出願は、2009年1月29日出願の米国特許仮出願第61/148,164号の恩典を主張し、同出願の全開示が参照により全体として本明細書に組み入れられる。
【0002】
発明の分野
本発明は、癌を含む増殖性障害を処置するためのSMAC模倣体ならびにその組成物および使用の分野にある。
【背景技術】
【0003】
発明の背景
アポトーシス抵抗性の発生は腫瘍形成の中核であり、腫瘍療法の主たる妨げとなる1。アポトーシス機構の再活性化を目的とした新規な治療方式は、熱心に研究されており、結果としてデスレセプターアゴニスト、Bcl-2アンタゴニスト、またはアポトーシス阻害タンパク質(IAP)阻害剤などの、アポトーシスシグナル伝達カスケード内の中心分子を標的とする多様な化合物が、その臨床的使用のため現在探究されている2〜4。
【0004】
TNF関連アポトーシス誘発リガンド(TRAIL)およびCD95Lは、広く研究されているデスリガンドであり、多数の研究がこれらのデスレセプターのシグナル伝達能力を検討してきた(総説については5、6参照)。特にTRAILは、腫瘍細胞の特異的除去を可能にする有望なリガンドと見なされている3,7。デスレセプターシグナル伝達経路は、細胞表面での受容体発現、細胞性FLICE抑制タンパク質(cFLIP)、X連鎖IAP(XIAP)、またはBcl-2ファミリータンパク質(例えばBcl-2、Bcl-X)などの抑制因子の発現を含む多レベルで制御される(総説については8参照)。最近の根拠は、デスレセプターのトリガーが、TNF経路に類似して、一次膜結合型複合体だけでなく二次的な独立シグナル伝達プラットフォームを誘導することを示している9〜11。これらの二次複合体の形成を導くメカニズム、ならびにデスレセプターによって活性化されるアポトーシス感受性および非アポトーシス性シグナルに対する二次複合体の重大な寄与は、詳細には解明されていない。外因性アポトーシスシグナル伝達経路の調節をさらに複雑化することには、過去10年間に壊死性および自己貪食性細胞死などのカスパーゼの活性化を必要としない追加型の細胞死が同定された12。これらは、単に細胞死経路が多様であることを表すのではなく、癌細胞に対する効率的な免疫応答に必要な多細胞生物内の免疫学的応答に特に関連する可能性がある。興味深いことに、細胞死の様式は、先に提案されたように細胞死を「免疫原性」対「不活動性」と定義することができよう13,14。
【0005】
過去10年間にわたる大規模な研究は、多数の腫瘍が腫瘍形成の間または初回処置までにアポトーシス抵抗性を有するか、または獲得するかのいずれかであること、および初期臨床研究においてデスレセプターアゴニストが単独ではまだ有望な結果を収めていないことを明らかにした15。IAPアンタゴニストは、XIAPのBIR2/BIR3ドメインに対するミトコンドリアタンパク質Smac/DIABLOのN末端IAP結合モチーフ(IBM)に応じてモデル化された合成化合物である16。デスリガンドに対するアポトーシス抵抗性に関するcIAPの役割は、あまりよく理解されていない17,18。しかしながらXIAP機能の干渉は、異種移植腫瘍モデルにおけるTRAILの治療有効性にとって重大である19。IAP阻害剤が、エフェクターであるカスパーゼへの結合からXIAPを移動させるだけではないことを数グループが最近になって独立して発見したことによって、癌治療のためのIAP阻害剤の使用が刺激された。むしろ、IAP阻害剤は、cIAP1およびcIAP2の迅速な自己ユビキチン化および欠如、NF-κB誘導、ならびに最終的にTNF介在性カスパーゼ8活性化および細胞死を導くTNFの自己分泌型産生もまた誘導する20〜24。この状況でcIAP1およびcIAP2は、カスパーゼ抑制因子というよりもむしろカスパーゼ調節因子であって、まだ詳細に探究されていない追加的な機能を有する見込みが最も高いことが明らかとなった。
【0006】
細胞性アポトーシス阻害タンパク質(cIAP)は、TNF介在性細胞死から保護するために必要である。CD95またはTRAIL-Rなどのプロトタイプのデスレセプターシグナル伝達に対する腫瘍細胞の感受性についてのcIAPの役割は、これまで詳細には研究されていない。
【0007】
細胞型FLICE様抑制因子cFLIPは、デスレセプターFas、DR4、およびDR5によって仲介されるアポトーシスの抑制因子であり、長鎖(cFLIPL)スプライス型および短鎖(cFLIPS)スプライス型として発現される。c-FLIPは、デスレセプターFas、DR4、およびDR5によって仲介されるアポトーシスの抑制因子であり、長鎖(c-FLIPL)および短鎖(c-FLIPS)スプライス型として発現される。cFLIPは、FAS介在性デスドメイン(FADD)およびカスパーゼ8と相互作用することによって、TNFレセプター遺伝子スーパーファミリーのメンバーにより仲介されるアポトーシスを阻害することができる。
【0008】
アポトーシス阻害タンパク質(IAP)は、カスパーゼ依存性アポトーシスを抑制する天然の細胞内タンパク質である。DIABLOとしても知られているSMACは、IAPと拮抗するように、すなわちIAPの活性を阻害するように機能する別の細胞内タンパク質である。健常細胞において、SMACおよびIAPは一緒に機能して健康な細胞を維持する。しかし、ある種の疾患状態、例えば癌および他の増殖性障害では、IAPは十分に拮抗されず、したがってアポトーシスを阻止し、そして異常増殖および異常生存を引き起こすかまたは増悪させる。
【0009】
IAPアンタゴニストとしても知られているSMAC模倣体は、SMACのN末端アミノ酸4個の構造およびIAPアンタゴニスト活性を模倣する合成小分子である(SMAC模倣体は、IAPアンタゴニストと呼ばれることもある)。増殖性障害を患う動物に投与した場合、SMAC模倣体はIAPと拮抗し、異常増殖細胞の間でアポトーシスの増加を引き起こす。
【0010】
SMACペプチド模倣体の例は、とりわけUS 7,517,906;US 7,309,792;US 7,419,975;US 2005/0234042;US 2005/0261203;US 2006/0014700;US 2006/0025347;US 2006/0052311;US 2006/0128632;US 2006/0167066;US 2007/0042428;US 2007/032437;US 2008/0132485;WO 2005/069888;WO 2005/069894;WO 2006/010118;WO 2006/122408;WO 2006/017295;WO 2006/133147;WO 2006/128455;WO 2006/091972;WO 2006/020060;WO 2006/014361;WO 2006/097791;WO 2005/094818;WO 2008/045905;WO 2008/016893;WO 2007/136921;WO 2007/021825;WO 2007/130626;WO 2007/106192;およびWO 2007/101347に開示されているものである。
【発明の概要】
【0011】
例示的な一態様では、本発明は、IAPアンタゴニスト、すなわちIAP阻害剤によるアポトーシス誘導に対する抵抗性に関するバイオマーカーである。具体的にはこの態様では、IAPアンタゴニストを用いた処置に対する抵抗性は、cFLIPの長鎖アイソフォーム、すなわちcFLIPLをアッセイすることによって決定される。cFLIPLを発現するヒトまたは非ヒト動物細胞は、IAPアンタゴニストに抵抗性の傾向がある。
【0012】
別の例示的な態様では、本発明は、IAPアンタゴニストによるアポトーシス誘導に対する感受性に関するバイオマーカーである。具体的にはこの態様では、IAPアンタゴニストを用いた処置に対する感受性または受容性は、cFLIPの短鎖アイソフォーム、すなわちcFLIPSをアッセイすることによって決定される。cFLIPSを発現するヒトまたは非ヒト動物細胞は、IAPアンタゴニストに感受性の傾向がある。
【0013】
さらに具体的で例示的な態様では、本発明は、TRAILレセプターアゴニスト、CD95レセプターアゴニスト、またはTNFaレセプターアゴニストと組み合わせたIAPアンタゴニストに対する細胞の感受性が決定される方法であって、例えば、TRAILレセプターアゴニストがTRAILであり、CD95レセプターアゴニストがCD95Lであり、かつTNFaレセプターがTNFαである方法を含む。
【0014】
特に例示的な態様では、ヒトまたは非ヒト動物細胞は、生検試料または細胞株由来である。細胞は、異常に増殖している任意の細胞、例えば腫瘍細胞または自己免疫障害における異常増殖細胞でありうる。
【0015】
特に例示的な態様では、細胞におけるcFLIPLまたはcFLIPS遺伝子の発現ポテンシャルは、
(a)細胞におけるcFLIPL mRNAまたはcFLIPS mRNAの存在を決定すること、
(b)細胞におけるcFLIPLまたはcFLIPSの存在を決定すること
によってアッセイされる。
【0016】
別の例示的な態様では、本発明は、
(a)IAPアンタゴニストを用いた処置に対する増殖細胞の感受性を、該細胞がcFLIPLまたはcFLIPSを発現できるかどうかを決定することによって決定する段階であって、それに従うと、cFLIPSを発現することができる細胞はIAPアンタゴニストに感受性である、段階、および
(b)細胞がcFLIPSを発現することができる場合に、IAPアンタゴニストで該細胞を処置する段階、あるいは
(c)細胞がcFLIPLを発現することができる場合に、IAPアンタゴニスト以外の剤もしくはIAPアンタゴニストに追加的な剤で該細胞を処置するか、またはIAPアンタゴニストの用量を増大させる段階
を含む、増殖性障害を患う患者を処置する方法である。
【0017】
本発明の方法は、ヒトまたは動物の身体に直接実施することができるが、そうする必要はない。むしろ、この方法は、生検などの試料を使用して実施することができる。したがって、別の例示的な態様では、本発明は、増殖性障害を患う患者を処置するために、かかる患者をIAPアンタゴニストで処置するかどうか、およびもし処置するならばどのくらいの用量で処置するかを決定するために、cFLIPLもしくはcFLIPSの存在、またはcFLIPLもしくはcFLIPSのmRNAの存在を検出する剤の使用を含む。本明細書下記にさらに説明するように、かかる剤は、例えば抗体またはヌクレオチドプローブでありうる。
【0018】
したがって他の例示的な態様における本発明は、本発明の方法を実施するためのキットであって、例えばcFLIPLもしくはcFLIPSの存在、またはcFLIPLもしくはcFLIPSのmRNAの存在を検出するための手段を含むキットも含み、該手段は、例えば、上記および本明細書下記などのcFLIPLもしくはcFLIPSの検出、またはcFLIPLもしくはcFLIPSのmRNAの検出に有用な剤である。
【図面の簡単な説明】
【0019】
【図1】IAP阻害剤は、自己分泌性TNF分泌とは独立して、SCCおよびHaCaTをデスリガンド(DL)介在性アポトーシスに対して感受性にすることを示す図である。A)HaCaT、MET1、またはA5RT3細胞を、100nM IAP阻害剤単独で、または10μg/ml TNFR2-Fcと組み合わせてのいずれかで30分間前処置し、次に3つ組のウェルにおいて表示した濃度のTRAIL(ng/ml)またはCD95L(U/ml)で刺激した。18〜24時間後に、クリスタルバイオレットアッセイによって細胞生存率を分析した。未刺激細胞は対照として用い、デスリガンド非依存性感受性との比較を可能にするために100%に設定した。4回の独立した実験のまとめを示し、エラーバーは平均の標準誤差(SEM)を示す。B〜E)IAP阻害剤は、CD95L介在性細胞死を増大させる。HaCaT細胞を、IAP阻害剤(100nM)単独で30分間予備刺激するか、あるいはCD95L(10U/ml)で4時間(B)、8時間(低二倍性分析;8時間)、またはカスパーゼもしくはPARP-1の切断のために(表示した時間)刺激/共刺激するかのいずれかとした。B)アネキシン-V-Cy5およびヨウ化プロピジウム(PI)で細胞を染色し、次にFACSによって分析した。C)細胞を8時間インキュベートし、続いてPIを含む低張緩衝液中に再懸濁し(材料および方法を参照されたい)、続いてFACS分析を行った。D)クローン形成アッセイのために、HaCaT細胞をIAP阻害剤(100nM)で30分間予備刺激し、続いてCD95L(2.5U/ml)で共刺激した。刺激の24時間後に、滅菌PBS中で数回洗浄後に培地を交換し、新しい培地を加え、細胞をさらに5または7日間培養し、続いてクリスタルバイオレット染色を行った。合計3回の独立した実験の代表的な一実験を示す。E)生化学分析のために、表示した時点についてTNF-R2-Fc(10μg/ml)の存在または非存在下の両方で、HaCaT細胞をIAP阻害剤(100nM)またはCD95L(2.5U/ml)の一方で、または両者を組み合わせてのいずれかで処置し、cIAP1、cIAP2、cFLIP、カスパーゼ8、PARP-1、FADD、およびRIP1の発現に関するウエスタンブロット分析を行った。βチューブリンは、内部負荷対照として用いた。代表的な2回の実験の一つを示す。
【図2】A)HaCaT、A5RT3、およびMET1細胞を表示時間にわたりIAPアンタゴニスト(100nM)で処置するか、またはTNF-R2-Fc(10μg/ml)で共刺激した。全ての細胞株におけるcIAP1およびcIAP2の発現ならびにMET1細胞におけるXIAPおよびカスパーゼ3の発現の十分な減少を、それぞれのタンパク質に特異的な吸収を有するウエスタンブロット分析によって確認した。βチューブリンを内部対照として用いた。4つの代表的な結果の一つを示す。B)様々な濃度のSMAC模倣体(6〜400nMのIAP)を、HaCaT、A5RT3、MET1およびSCC25細胞株に添加して細胞生存率を決定した。インキュベーションの終了時に、細胞をクリスタルバイオレットで染色し、生存率を決定した。C)クリスタルバイオレットで生存率を決定するために、Cと同様に細胞株をSMAC模倣体(6〜400nM)および10μg/ml TNF-R2-Fcと共に同時インキュベートした。D)HaCaTおよびMET1細胞を刺激しないか、またはIAPアンタゴニスト(100nM)で4時間刺激するかのいずれかとした。CD95/TRAIL-R1、およびTRAIL-R2の表面発現をデスレセプターに対するそれぞれの抗体で特異的に染色し、FACSによって視覚化した。二つの代表的な結果のうち一つを示す。
【図3】IAP阻害剤存在下のデスレセプター介在性細胞死は、カスパーゼ完全依存性でもなければ、カスパーゼ非依存性でもないことを示す。独特なカスパーゼ阻害剤zVAD-fmkによるカスパーゼ活性の阻害は、IAP阻害剤の存在下でHaCaT細胞のデスリガンド介在性細胞死を部分的に保護する。A)HaCaT細胞をzVAD-fmk(10μM;1時間)、ネクロスタチン(necrostatin)-1(50μM、1時間)、およびIAP阻害剤(100nM、30分間)で予備刺激または共刺激した。続いて細胞を表示濃度のTRAILまたはCD95Lで3つ組のウェルにおいて刺激した。18〜24時間のインキュベーション後に、材料および方法に示したように細胞生存率をクリスタルバイオレットアッセイによって分析した。7回の独立した実験についてSEMを示す。B)DNA凝縮の分析のために、HaCaT細胞をzVAD-fmk(10μM、1時間)またはIAP阻害剤(100nM、30分間)のいずれかで前処置した。続いて細胞をCD95L(5U/ml)でそれぞれ4時間または24時間刺激した。ヘキスト(Hoechst)-33342(5μg/ml)を37℃で15分間添加し、直後に透過(左)または蛍光(右)顕微鏡観察を行った。2回の独立した実験のうちの一つを代表として示す。
【図4】RIP1は、cIAP非存在下でデスリガンド介在性細胞死の重要な調節因子であることを示す図である。A)HaCaT、A5RT3、MET1、およびSCC25細胞の全細胞溶解物5μgのウエスタンブロットによって、FADD、cFLIP、カスパーゼ8、TRAF2、RIP1、cIAP1、cIAP2、およびXIAPの内因性タンパク質発現レベルを分析した。βチューブリンを、均等負荷の内部対照として用いた。三つの代表的な結果のうち一つを示す。B)zVAD-fmkによるカスパーゼ活性およびネクロスタチン-1によるRIP1キナーゼ活性の両方の阻害が、IAP阻害剤存在下でHaCaT細胞のデスリガンド介在性細胞死を完全に保護した。クリスタルバイオレットアッセイによってHaCaT細胞を3つ組のウェルにおいて、zVAD-fmk(10μM;1時間)、ネクロスタチン-1(50μM、1時間)およびIAP阻害剤(100nM、30分間)で別々に予備刺激し、続いてTRAIL(50ng/ml)またはCD95L(2,5U/ml)で刺激した。3回(TRAIL)または5回(CD95L)の独立した実験のSEMを示す。C)RIP1の安定的ノックダウンは、HaCaT細胞をデスリガンド誘導性細胞死から保護する。レトロウイルスによってハイパーランダム配列shRNA(HRS)またはRIP1特異的shRNAのいずれかをHaCaT細胞に形質導入し、ピューロマイシン(3μg/ml)で3日間選択した。RIP1のノックダウン効率は、RIP1に関するウエスタンブロット分析によって確認した。βチューブリンに対するAbを用いた膜の再プロービングを、タンパク質負荷の内部対照として用いる。D)C)に示したように形質導入されたHaCaT細胞を100nM IAP阻害剤およびTNF-R2-Fc(10μg/ml)で30分間予備刺激し、続いて表示濃度のTRAILまたはCD95Lで18〜24時間刺激し、クリスタルバイオレットアッセイによってアッセイした。3回(TRAIL)または4回(CD95L)の独立した実験のSEMを示す。E)C)に記載したように、形質導入されたHaCaTをIAP阻害剤(100nM)と共に30分間予備インキュベートし、続いてCD95L(0.5U/ml)で刺激した。24時間後に、培養液を除去し、細胞を無菌PBSで洗浄し、新しい培地を加えた。続いて細胞をさらに5日間培養し、続いてクリスタルバイオレット染色を行った。4回の代表的な独立した実験のうち一つを示す。
【図5】IAP阻害剤の存在下または非存在下でのリガンド誘導性レセプター結合型CD95複合体(DISC)または細胞内カスパーゼ8含有複合体(複合体II)の誘導を示す。A)CD95L-Fcで2時間刺激したMET1またはA5-RT3細胞からCD95 DISCを沈降させた。続いて、材料および方法に詳述したようにリガンドアフィニティー沈降を用いてCD95L DISC(左パネル)を沈降させた。溶解後のレセプター複合体の沈降(-)を、リガンドアフィニティー沈降(IP;+)と比べたときの内部特異性対照として用いた。続いて表示した分子について等量のDISC(CD95L-IP)またはカスパーゼ8相互作用タンパク質(複合体II)をウエスタンブロットによって分析した。等量の全細胞溶解物(TL)を同じゲルに負荷し、CD95L-IP、複合体II、およびTLの間でシグナル強度を比較可能にした。
【図6】cFLIPは、cIAP非存在下でデスリガンド介在性細胞死の重要な調節因子であることを示す。A)レトロウイルスによって、ハイパーランダム配列shRNA(HRS)またはcFLIP特異的shRNAのいずれかをA5-RT3細胞に形質導入し、ピューロマイシン(3μg/ml)で3日間選択した。cFLIPLおよびcFLIPSのノックダウン効率をウエスタンブロット分析によって確認した。RIP1、FADD、カスパーゼ8、およびβチューブリンに対するAbを用いた膜の再プロービングを、タンパク質負荷のための内部対照として用いる。3回の独立した実験の代表を示す。B)A)に示すように形質導入されたA5-RT3を100nM IAP阻害剤およびTNF-R2-Fc(10μg/ml)で30分間予備刺激し、続いて表示濃度のTRAIL(左パネル)またはCD95L(右パネル)で18〜24時間刺激し、クリスタルバイオレットアッセイによってアッセイした。C)カスパーゼ活性の阻害(zVAD-fmk;10μM)およびネクロスタチン-1(50μM)によるRIP1キナーゼ活性の阻害は、IAP阻害剤の存在下でA5-RT3細胞をデスリガンド介在性細胞死から完全に保護した。形質導入されたA5-RT3細胞を、3つ組のウェルにおいてzVAD-fmk(10μM;1時間)、ネクロスタチン-1(50μM、1時間)およびIAP阻害剤(100nM、30分間)で別々に予備刺激し、続いてCD95L(25U/ml)で刺激した。細胞の生存率をクリスタルバイオレットアッセイによって分析した。4回の独立した実験のSEMを示す。D)レトロウイルスによってHaCaT細胞にcFLIPLまたはcFLIPSまたは対照ベクターを形質導入した。cFLIPおよびカスパーゼ8について全細胞溶解物を分析した。βチューブリンは、タンパク質負荷のための内部対照として用いる。2回の追加的な独立した実験で同等の結果が得られた;E)D)に示されるような形質導入されたHaCaT細胞を、zVAD-fmk(10μM;1時間)、ネクロスタチン-1(50μM、1時間)、およびIAP阻害剤(100nM、30分間)または希釈液単独で予備刺激した。続いて3つ組のウェルにおいて細胞をCD95L(25U/ml)で刺激した。18〜24時間後に細胞生存率をクリスタルバイオレットアッセイによって分析した。7回の独立した実験のSEMを示す。F)クローン形成アッセイのために、形質導入されたHaCaT細胞をIAP阻害剤(100nM)で30分間予備刺激し、続いてCD95L(2,5U/ml)で共刺激した。刺激の24時間後に、滅菌PBSで数回洗浄してから培地を交換し、新しい培地を加え、細胞をさらに5または7日間培養し、続いてクリスタルバイオレット染色を行った。合計3回の独立した実験のうち代表的な一実験を示す。
【図7】cFLIPSではなくcFLIPLが複合体IIの形成を遮断することを示す。IAP阻害剤の存在下または非存在下でのリガンド誘導性レセプター結合型CD95複合体(DISC)または細胞内カスパーゼ8含有複合体(複合体II)の誘導。CD95L-Fcで2時間刺激したHaCaT細胞からCD95 DISCを沈降させた。続いて、材料および方法に詳述したようにリガンドアフィニティー沈降を用いてCD95L DISC(左パネル)を沈降させた。溶解後のレセプター複合体の沈降(-)を、リガンドアフィニティー沈降(IP;+)と比べたときの内部特異性対照として用いた。続いて、表示した分子について等量のDISC(CD95L-IP)またはカスパーゼ8-相互作用タンパク質(複合体II)をウエスタンブロットによって分析した。等量の全細胞溶解物(TL)を同じゲルに負荷して、IPとTLとの間のシグナル強度を比較できるようにした。
【図8】デスレセプター介在性細胞死の間のcIAPの役割を示す。cIAPは、完全長RIP1を有する質的に異なるDISCの形成を遮断する。このシグナル伝達プラットフォームは、カスパーゼ依存的と同様にカスパーゼ非依存的にも細胞死を誘導する。壊死性細胞死に重要な二次レセプター非依存性複合体IIもまた、開始因子カスパーゼ8およびカスパーゼ10を有する。カスパーゼ8によるRIP1切断は、複合体内のRIP1ダウンレギュレーションの仮説上の一メカニズムであることによって、RIP1依存的シグナル伝達を妨害する。または、RIP1は、ユビキチン化されるとDISCだけに動員される。
【図9】siRNAによるcFLIPのノックダウンは、抵抗性細胞をSmac模倣体およびTNFαの組み合わせに対して感受性にすることを示す。cFLIPのsiRNA介在性ノックダウンによってSmac模倣体、TNFαおよび両者の組み合わせに抵抗性の細胞株は感受性になる。A549およびIGROV-1細胞を24ウェルプレートに蒔き、一晩付着させた。翌日、細胞に100nMの対照siRNAまたはcFLIPターゲティングsiRNAのいずれかをトランスフェクトした。トランスフェクションの48時間後に、細胞を100ng/ml TNFα、100nM Smacペプチド模倣体または両者の組み合わせのいずれかで処理した。さらに24時間後に、細胞を回収し、FITCラベル化アネキシンVおよびヨウ化プロピジウムで染色した。アポトーシスをFACS分析によって定量した。
【発明を実施するための形態】
【0020】
発明の詳細な説明
添付の実験方法および結果の記載にさらに完全に説明および裏付けを行うように、本発明は、アポトーシス阻害タンパク質アンタゴニスト(IAPアンタゴニスト)を、単独、すなわち単独療法で、または他の抗増殖療法と組み合わせて、例えばTRAIL、CD95L、もしくはTNFα、またはそれらの関連アゴニストとの同時投与で用いた処置に対する細胞の感受性(または抵抗性)を予測する方法を提供する。別の述べ方をすると、本発明は、IAPアンタゴニストを用いた処置に対する特定の増殖性細胞障害の感受性または受容性を決定するアッセイ法に関する。細胞がIAPアンタゴニストに応答してアポトーシスを受ける場合、その細胞はIAPアンタゴニストに感受性である。本発明の方法は、どの細胞がアポトーシスを受けることによってIAPアンタゴニストに応答する見込みが高いかを予測するために有用である。これらの方法は、実験室または臨床状況のいずれかで使用することができる。
【0021】
本発明の方法は、様々な良性腫瘍または悪性腫瘍(癌)、良性増殖性疾患(例えば、乾癬、良性前立腺肥大、および再狭窄)、または自己免疫疾患(例えば、自己免疫性増殖性糸球体腎炎、リンパ増殖性自己免疫応答)を処置するためのIAPアンタゴニストの投与から利益を受け得る者を同定するために、例えば増殖性障害を患う患者をスクリーニングするために特に有用である。IAPアンタゴニストで潜在的に処置することができる癌には、非限定的に以下の一つまたは複数が挙げられる:肺腺癌、膵臓癌、結腸癌、卵巣癌、乳癌、中皮腫、末梢神経腫、膀胱癌、神経膠芽腫、黒色腫、副腎皮質癌、AIDS関連リンパ腫、肛門癌、膀胱癌、髄膜腫、神経膠腫、星状細胞腫、乳癌、子宮頸癌、慢性骨髄増殖性障害(例えば、慢性リンパ性白血病、慢性骨髄性白血病)、結腸癌、内分泌癌、子宮内膜癌、上衣腫、食道癌、ユーイング肉腫、頭蓋外胚細胞腫瘍、性腺外胚細胞腫瘍、肝外胆管癌、胆嚢癌、胃癌、消化管カルチノイド腫瘍、妊娠性トロホブラスト腫瘍、ヘアリー細胞白血病、ホジキンリンパ腫、非ホジキンリンパ腫、下咽頭癌、眼球内黒色腫、膵島細胞癌、カポジ肉腫、喉頭癌、白血病、急性リンパ芽球性白血病、急性骨髄性白血病、口唇癌、口腔癌、肝臓癌、男性乳癌、悪性中皮腫、髄芽腫、黒色腫、メルケル細胞癌、転移性扁平上皮性頸部癌(metastatic squamous neck cancer)、多発性骨髄腫および他の形質細胞腫瘍、菌状息肉腫およびセザリー症候群、骨髄異形成症候群、鼻咽頭癌、神経芽細胞腫、非小細胞肺癌、小細胞肺癌、中咽頭癌、骨肉腫および骨悪性線維性組織球腫を含む骨癌、卵巣上皮癌、卵巣胚細胞腫瘍、卵巣低悪性度腫瘍(ovarian low malignant potential tumor)、膵臓癌、副鼻腔癌、副甲状腺癌、陰茎癌、褐色細胞腫、下垂体腫瘍、前立腺癌、直腸癌、腎細胞癌、網膜芽細胞腫、横紋筋肉腫、唾液腺癌、皮膚癌、小腸癌、軟部組織肉腫、テント上原始神経外胚葉性腫瘍(supratentorial primitive neuroectodermal tumor)、松果体芽腫、精巣癌、胸腺腫、胸腺癌、甲状腺癌、腎盂尿管移行上皮癌、尿道癌、子宮肉腫、膣癌、外陰癌、ならびにウィルムス腫瘍および他の小児腎臓癌。
【0022】
本発明のいくつかの方法は、cFLIPLもしくはcFLIPS遺伝子の発現またはcFLIPLもしくはcFLIPS遺伝子発現ポテンシャルについて細胞をアッセイすることを伴う。cFLIPSアイソフォームを発現する細胞またはcFLIPSアイソフォームを発現するポテンシャルを有する細胞は、一つまたは複数のIAPアンタゴニストに感受性の傾向があり、すなわちIAPアンタゴニストで処置した場合にアポトーシスを受ける傾向がある。逆に、cFLIPLアイソフォームを発現する細胞またはcFLIPLアイソフォームを発現するポテンシャルを有する細胞は、一つまたは複数のIAPアンタゴニストに対して感受性が低い傾向があり、すなわち抵抗性である傾向がある。cFLIPLまたはcFLIPSの遺伝子発現は、当技術分野で公知の任意の手段によってアッセイすることができる。いくつかの態様では、遺伝子発現は、細胞のcFLIPLまたはcFLIPSタンパク質を検出することによってアッセイされる。ヒトcFLIPLおよびcFLIPSに関するアミノ酸配列は公知である(それぞれ配列番号:1および3)。cFLIPLまたはcFLIPSタンパク質(例えば分泌された、細胞内に含まれる、細胞表面に発現されたもの)は、例えば様々なイムノアッセイ(ELISA、ウエスタンブロット、フローサイトメトリー、ラジオイムノアッセイなど)を用いて検出することができる。cFLIPLおよびcFLIPS抗体は、入手可能である。例えば、抗cFLIPL抗体であるChemiconカタログ番号AB16963; Enzo Life Sciences カタログ番号ALX-804-127; Santa Cruz、カタログ番号SC7108、7111、8346、および7109を参照されたい。SまたはLアイソフォームに特異的でない抗体もまた、公的に入手可能である。当業者は、cFLIPおよびその各アイソフォームを同定するためにそのような抗体を使用する方法を知っている。例えば、細胞溶解物由来のタンパク質を、例えばSDS-PAGEによって単離し、次に抗cFLIP抗体を使用して、例えばELISAまたはウエスタンブロットで、例えばcFLIPSについて約25〜28KDであり、cFLIPLについて約55KDである分子量に基づき、cFLIPLまたはcFLIPSタンパク質を同定することができる。
【0023】
他の態様では、cFLIPL mRNAまたはcFLIPS mRNAを検出(例えばノーザンブロット、ドットブロット、RT-PCRなどによって)することにより遺伝子発現をアッセイする。ヒトcFLIPLおよびcFLIPSのDNA配列は公知である(それぞれ配列番号:2および4)。例えば、Goto et. al. J. Reproduction and Development, 2004, 50(5)549-555を参照されたい。公知の配列を使用してプローブを調製することができるし、また公知のアミノ酸配列に基づいて縮重プローブを作製することもできる。
【0024】
検出できる遺伝子発現レベルは、用いるアッセイに依存するであろうが、任意の検出可能なレベルのcFLIPLまたはcFLIPSのタンパク質またはmRNAを産生する細胞は、それぞれcFLIPLまたはcFLIPSアイソフォームを発現する細胞である。
【0025】
cFLIPLまたはcFLIPSの遺伝子発現について任意の細胞型をアッセイすることができる。細胞は、初代細胞(例えば患者から得られた生検の細胞)または細胞株由来でありうる。本発明は、ヒトまたは動物の身体での実施を必要としない。特に関心が持たれるのは、病的に増殖する細胞および疾患の症状を引き起こすかまたは誘導する細胞を含む、異常増殖細胞である。異常増殖細胞は、例えば癌、良性増殖性障害、および自己免疫性疾患で生ずる。
【0026】
cFLIP遺伝子を発現するように細胞を誘導することができ、方法は当業者に公知である;転写または翻訳レベルでのcFLIPLまたはcFLIPS発現の選択的誘導は、IAPに対する感受性または抵抗性に関して表現型を変化させることができる。
【0027】
cFLIPを発現している細胞は、SiRNAおよび当業者に公知の方法を用いてサイレンシングすることができ;cFLIPの選択的サイレンシングは、IAPに対する感受性に関する表現型の変化を招く。
【0028】
本発明のいくつかの態様は、細胞の、特に病的に増殖している細胞のアポトーシスを誘導することを含む。その方法は、インビトロまたはインビボで実施することができ、IAPアンタゴニストを用いた患者の処置を含むことができる。そのような処置は、単一のIAPアンタゴニストの投与、IAPアンタゴニストの組み合わせの投与、または一つまたは複数のIAPアンタゴニストおよび一つまたは複数の追加的な化学療法剤の投与を含みうる。多数の剤の投与は、同時または連続的でありうる。有用な化学療法剤には、非限定的に、アルキル化剤(例えばシクロホスファミド、メクロレタミン、クロラムブシル、メルファラン)、アントラサイクリン(例えばダウノルビシン、ドキソルビシン、エピルビシン、イダルビシン、ミトキサントロン、バルルビシン)、細胞骨格破壊剤(例えば、パクリタキセル、ドセタキセル)、エポチロン(例えば、エポチロンA、エポチロンB、エポチロンD)、トポイソメラーゼII阻害剤(例えば、エトポシド、テニポシド、タフルポシド)、ヌクレオチドアナログ前駆体アナログ(例えばアザシチジン、アザチオプリン、カペシタビン、シタラビン、ドキシフルリジン、フルオロウラシル、ゲムシタビン、メルカプトプリン、メトトレキサート、チオグアニン)、ペプチド系抗生物質(例えばブレオマイシン)、白金ベースの剤(例えばカルボプラチン、シスプラチン、オキサリプラチン)、レチノイド(例えば、オールトランスレチノイン酸)、ならびにビンカアルカロイドおよび誘導体(例えばビンブラスチン、ビンクリスチン、ビンデシン、ビノレルビン)が挙げられる。いくつかの態様では、化学療法剤には、フルダラビン、ドキソルビシン、パクリタキセル、ドセタキセル、カンプトテシン、エトポシド、トポテカン、イリノテカン、シスプラチン、カルボプラチン、オキサリプラチン、アムサクリン、ミトキサントロン、5-フルオロウラシル、またはゲムシタビンが挙げられる。
【0029】
IAPアンタゴニスト
本発明における使用のためのIAPアンタゴニストは、細胞性IAP(cIAP、例えばcIAP-1またはcIAP-2)またはX連鎖IAP(XIAP)などの一つまたは複数のIAPに結合してその活性を阻害する任意の分子である。いくつかの態様では、IAPアンタゴニストは、XIAP、cIAP-1、またはcIAP-2に優先的に結合する。いくつかの態様では、IAPアンタゴニストは、Smac(第二ミトコンドリア性カスパーゼ活性化因子)の模倣体であり、特定の態様では、Smac模倣体は、成熟SmacのN末端の4個のアミノ酸(Ala-Val-Pro-Ile)、またはより一般的にはAla-Val-Pro-Xaa(ここで、Xaaは、Phe、Tyr、Ile、またはVal、好ましくはPheまたはIleである)の模倣体またはペプチド模倣体である。
【0030】
本発明のいくつかの態様では、IAPアンタゴニストを含む薬学的組成物は、ヒトまたは動物対象に投与される。薬学的組成物は、典型的には薬学的に許容される担体または希釈剤を含み、全身、局所、または経口経路を含む経路によって従来方式で投与することができる。例えば、投与は、非限定的に、非経口、皮下、静脈内、筋肉内、腹腔内、経皮、経口、口内、膣内、または眼球経路、吸入による、デポー注射による、または植込みによるものでありうる。特定の投与様式は、適応症および投与される特定の化合物を含めたその他の要因に依存すると考えられる。投与されるべき化合物の量は、治療的に有効な量である。投与される用量は、処置される対象の性質、例えば処置される特定の患者、年齢、体重、健康状態、もしあれば同時に行う処置の種類に依存すると考えられる。処置の回数は、当業者(例えば臨床医)が容易に決定することができる。
【0031】
本発明のいくつかの態様には、cFLIP遺伝子発現の評価および分析を行うためのキットが含まれる。そのようなキットには、例えばcFLIPLまたはcFLIPSの配列決定の前のQiagen EPITECT(登録商標)Bisulfite Conversion Kit、抗体、プローブ、検出可能なマーカーなど、ならびに上記のIAPアンタゴニスト処置の評価および分析を行うために必要な試薬、ゲル、装置、分析ツールなどが含まれる。
【0032】
本発明には、IAPアンタゴニスト、キット、システムを市販するための方法、およびIAPアンタゴニストを使用した処置の成功見込みを決定するのに有用なバイオマーカーを使用するための方法が含まれる。一態様では、そのような方法、システムおよびキットの有効性に関するデータは、IAPアンタゴニストでのヒト臨床試験を行う承認を求めるための関係書類の一部として規制当局に提出されて、例えば、除外基準もしくは組み入れ基準が制定されるか、または臨床試験データの評価が容易になる。そのようなデータを規制当局に提出して、IAPアンタゴニストを使用した処置に関連するバイオマーカーを使用するための方法、システム、およびキットを市販するための承認申請を裏付けることができる。例えば、そのようなデータを、新医薬品承認申請(New Drug Approval Application)(NDA)の一部として米国食品医薬品局(United States Food and Drug Administration)(FDA)に提出することができる。
【0033】
本発明の様々な態様には、IAPアンタゴニストを用いた処置に応答してcFLIPLまたはcFLIPSを発現することができる細胞の応答性についての情報を提供すること、およびIAPアンタゴニストを含むそのような薬学的組成物に関心を持ちうる個人にこの情報を普及させることが含まれる。そのような個人には、増殖性障害を有する個人、そのような障害を処置する医療関係者、および薬剤を調剤する、または流通させる個人が含まれる。
【0034】
ヒト臨床試験の承認が得られた場合、前記の情報は、増殖性障害を示しているヒト対象への薬学的組成物の有効性を裏付けるデータ、ならびに投薬情報および細胞毒性データなどの他のデータと共に、IAPアンタゴニスト、およびIAPアンタゴニストを含む薬学的組成物を市販する承認を求めて規制当局に提出することのできる関係書類に添付することができる。
【0035】
態様には、承認が得られた後にIAPアンタゴニストまたはIAPアンタゴニストを含む薬学的組成物を市販するための方法も含まれる。そのような方法では、cFLIPLまたはcFLIPSを発現することができる細胞においてIAPアンタゴニストが有効である可能性があるという事実に関する情報を、例えば、医師、薬剤師、処方者、保険提供業者、流通業者、患者など、またはこれらの人々の組み合わせに普及させることができる。なお他の態様では、情報は、将来の患者および/または将来の処方者、および/または将来の流通業者に普及させることができる。
【0036】
情報は、非限定的に、消費者への直接広告、テレビ広告、ラジオ広告、新聞広告、印刷物による広告(例えば、パンフレット、チラシ、郵便はがき、手紙など)、ウェブサイトを経由したまたはウェブサイト上の広告(例えば、「バナー」アドオンウェブサイトを使用)、看板広告、ダイレクトメール、Eメール、伝聞、およびそれらの任意の組み合わせを含めた、当技術分野で公知の任意の方法により普及させることができる。
【0037】
他の態様では、データは、ユーザがアクセス可能なデータベースに保存することができる。データベースに保存されたデータには、例えばIAPアンタゴニストを使用した処置に関連するバイオマーカーを使用する方法、システム、およびキットを検査する間に生成されたデータを含むIAPアンタゴニストまたは薬学的組成物に関係する任意のデータ、IAPアンタゴニスト、薬学的組成物、方法、システムおよびキットの安全性および/または有効性に関する情報、投薬情報、該化合物を使用して処置することのできる障害の一覧、一つまたは複数の規制当局からの承認情報、流通業者の情報、処方情報、ならびにそれらの組み合わせが含まれうる。
【0038】
様々な態様にはまた、IAPアンタゴニストを使用した処置に関連するバイオマーカーを使用するための、IAPアンタゴニスト、薬学的組成物、方法、システムおよびキットを市販するためのシステムが含まれ、該システムには、IAPアンタゴニストを使用した処置に関連するバイオマーカーを使用するための、方法、システム、およびキットに関する情報、ならびに方法、システム、およびキットの有効性についてのデータを含む、上記のデータベースなどのデータベースが含まれる。そのような態様では、データベースに保管される情報は、例えば経営者、販売担当者、マーケティング担当者およびそれらの人々の組み合わせなどの選ばれた個人だけがアクセス可能でありうる。システムにはまた、データベースに保管された情報のサブセットが含まれてもよく、それは、例えば、医師、薬剤師、処方者、保険提供業者、患者、流通業者およびそれらの人々の組み合わせなどの選択された個人ではない任意の者でありうる、選択されていない個人に普及される。普及は、上記の当技術分野で公知の任意の普及方法によって行うことができる。
【0039】
データのサブセットには、データベースに保管された任意の情報が含まれてもよく、方法、システム、およびキットを市場性のあるものにすると考えられる情報、例えば、安全性および/または有効性のデータ、該化合物を使用して処置することができる障害の一覧、薬学的組成物中の薬剤、成分の一覧または活性剤の投与の潜在的副作用、一つまたは複数の規制当局からの承認情報、流通業者の情報、処方情報およびそれらの組み合わせが含まれうる。ある種の態様では、選択された個体は、データのサブセットに提供された情報を選択および/または承認することができる。
【0040】
上記態様のそれぞれにおいて、提供および/または普及された情報ならびにデータベースに保存されたデータには、さらに、別の抗自己免疫剤または抗増殖剤を含みうる併用療法用の組成物、方法、またはプロトコールが含まれうる。
【0041】
本開示に引用された全ての特許、特許出願、および参照は、参照により本明細書に明確に組み入れられる。上記開示は、全般的に本発明を説明する。さらに完全な理解は、以下の具体例を参照することによって得ることができ、それらの例は、例示のためだけに提供され、本発明の範囲を限定する意図はない。
【0042】
実験の方法およびデータを示す添付の項において本発明をさらに十分に説明する。
【0043】
1. デスリガンド(DL)介在性細胞死に対して感受性にするIAP阻害剤
最初に本発明者らは、記載されたIAP阻害剤である化合物A(IAP阻害剤)を用いて異なる角化細胞株および扁平上皮癌(SCC)細胞の感受性を特徴づけた22。細胞を100nMの本化合物と共に異なる時間の間、インキュベートしたとき、cIAP1、さらにcIAP2の急速分解が検出された(図2A)。cIAP1の欠如は24時間にわたり継続したが、cIAP2は、A5-RT3において、そしてそれよりも低度であるがHaCaT細胞において再発現された。MET1細胞では、TNF-R2-Fcを使用した自己分泌TNFシグナル伝達阻害とは無関係に、XIAPの30kDaのカスパーゼ切断断片が24時間後に検出されたが、HaCaTおよびA5-RT3(図4Aと比較)は、タンパク質レベルでXIAPを発現しない17,26。これらのデータは、IAPの欠如がcIAP1およびcIAP2の急速分解を誘導し、TNF非依存的にXIAPのカスパーゼ介在性切断を導くことを示した(図2A)。用量反応実験(6〜400nMのIAP阻害剤)から、HaCaT細胞に加えて、派生した転移細胞株A5-RT327が、最大400nMまでIAP阻害剤単独を用いた処置に大いに抵抗性であることが明らかとなった。対照的に、二つの遺伝的に異種のSCC細胞株(SCC25と同様にMET1)は、IAP阻害剤単独を用いた処置に部分的に感受性であった(20〜25%の細胞死;補足の図1B)。10μg/ml TNF-R2-Fcとの同時インキュベーションは、これらの細胞における細胞死を減少させたが、それは、TNFの産生および分泌の自己分泌ループがこれらの細胞株における細胞死を担うことを示している22。本発明者らは次に、これらの腫瘍細胞におけるデスリガンドの感受性に関するIAP阻害剤の影響を検討した。A5-RT3ではなく、HaCaTおよびMET1細胞は、TRAILまたはCD95L介在性細胞死に対してTNF依存的に劇的に感受性になった(図1A)。細胞死は、4時間後のアネキシン-Vの外部移行(図1B)またはデスリガンドと共に8時間インキュベーション後の低二倍性分析(図1C)によって決定されたようにアポトーシスによって起こった。さらに、IAP阻害剤とのインキュベーションは、クローン形成性生存を減少させた(図1D)。本発明者らが、TRAILまたはCD95Lで処置後のカスパーゼ活性化を調べたとき、本発明者らは、IAP阻害剤の存在下でそれぞれのデスリガンドを用いて刺激してから4時間以内に、開始因子であるカスパーゼ8およびエフェクターであるカスパーゼ3の活性化が増大し、それがエフェクターカスパーゼの標的であるPARPの切断増加を招くことを見出した(図1E)。独立した実験システムでこれらのデータを確認するために、本発明者らはcIAP1を欠如したMEFを使用した。IAP阻害剤を使用したヒト細胞でのデータに一致して、cIAP1ノックアウトMEFはCD95介在性細胞死に感受性となった。さらに、cIAP1 MEFへのcIAP1の誘導性再構成によって、デスリガンドに対する抵抗性が完全に回復した。まとめると、本発明者らのデータは、cIAPの細胞レベルがCD95LまたはTRAIL抵抗性の維持に重要であることを実証している。
【0044】
2. カスパーゼ完全依存性でもなければ、カスパーゼ非依存性でもなく、かつRIP1を必要とする、IAP阻害剤存在下のデスレセプター介在性細胞死
デスレセプター介在性アポトーシスは、DISC活性化カスパーゼ8によって開始される6,28。カスパーゼ活性化がIAP阻害剤存在下でデスレセプター介在性細胞死に重要であるかどうかを研究するために、本発明者らは、次に広域スペクトルカスパーゼ阻害剤zVAD-fmkの存在下で細胞死を研究した。多数のグループによって報告されたように、TRAILまたはCD95Lで細胞を24時間刺激したとき、zVAD-fmkは細胞死を完全に遮断した。しかし、IAP阻害剤存在下のTRAIL介在性またはCD95L介在性細胞死は、異なる濃度のデスリガンドでzVAD-fmkによって部分的に遮断されただけであった(図3A)。これらのデータは、カスパーゼ非依存型の細胞死が、IAP阻害剤の存在下でデスレセプターの刺激によって誘導されることを示唆した。これらの細胞における細胞死の形態学的特徴をさらに特徴づけるために、本発明者らは蛍光顕微鏡研究を行った。膜泡状突起、DNA凝縮および断片化を示している典型的なアポトーシス細胞の数増加は、CD95L処置から4時間以内ではカスパーゼ阻害剤の非存在下で処置された細胞のみに検出可能であった(図3B、左パネル)。これらの初期時点で、zVAD-fmkは、IAP阻害剤が存在する場合にもまた細胞形態またはDNA断片化を完全に保護した。しかしながら、zVAD-fmkは、処置の24時間後に細胞を細胞死から保護しなかったのに対して、zVAD-fmkは、IAPが存在する場合は完全な保護を達成した(図3B、右パネル)。これらのデータは、IAPが、デスレセプターの刺激によって誘導されるカスパーゼ非依存性遅延型細胞死から保護することができることを示した。以前の研究は、CD95がキナーゼRIP1を介してカスパーゼ非依存型細胞死を活性化することを明らかにしたが、RIP1の下流の標的は依然として未知である29〜31。IAPはRIP1のユビキチン化を妨害することができるので24、本発明者らは、次に本発明者らの実験システムにおけるRIP1の役割を検討した。興味深いことに、IAP阻害剤の感受性化効果に高度に抵抗性であることが証明されたA5-RT3細胞において、RIP1レベルは最低であった(図4A)。ネクロスタチン-1などのネクロスタチンは、RIP1のキナーゼ活性を特異的に遮断することが示された32。カスパーゼ阻害は、TRAILまたはCD95L介在性細胞死から部分的に保護しただけであったが、zVAD-fmk保護細胞へのネクロスタチン-1の添加は、cIAPの非存在下で生存率を完全に回復させた。対照的に、ネクロスタチン-1単独は、カスパーゼ阻害剤非存在下でデスリガンド介在性細胞死からの保護に無効であった(図4B)。これらのデータは、cIAPの欠如がカスパーゼおよびRIP1の組み合わせ阻害によってのみ遮断することができるデスレセプター介在性シグナルを明らかにしうるという事実をほのめかした。RIP1の役割にさらに直接的に取り組むために、本発明者らは、安定的なshRNA発現を使用して、RIP1レベルが減少した細胞株を作出した(図4C)。対照感染細胞は、IAP阻害剤によってTRAILまたはCD95L介在性細胞死に対して感受性にされたが、この感受性化は、生存率アッセイ(図4D)およびクローン形成アッセイ(図4E)によって決定されたように、RIP1抑制細胞では主として打ち消された。まとめると、これらのデータは、cIAPが抑制された場合は常に、CD95介在性細胞死およびTRAIL介在性細胞死が細胞死に不可欠なRIP1依存性シグナル伝達経路を利用することを示している。ノックダウンのデータをさらに裏付けるために、本発明者らは、RIP1ノックアウトMEFをそれらの野生型対照細胞と比較した。RIP1欠損細胞は、cIAPの非存在下でTRAILまたはCD95L介在性細胞死の誘導への感受性が低かった。まとめると、これらのデータは、RIP1がIAPの非存在下でデスレセプター介在性細胞死に決定的に関与することを実証している。
【0045】
3. DISCへのRIP1動員を負に調節し、レセプター非依存性複合体IIの形成増加を可能にする、IAP
cIAPがどのようにデスレセプター介在性細胞死を負に調節するかの分子メカニズムを特徴づけるために、本発明者らは、次に本発明者らの実験システムにおけるネイティブの細胞死誘導性シグナル伝達複合体(DISC)の成分および形成を特徴づけた。さらに、最近記載されたレセプター非依存性複合体IIの形成10をIAPの非存在下で検討した。本発明者らは、IAP阻害剤(MET1)の感受性化効果にどちらも応答性であった二つの細胞株におけるCD95L誘導複合体を分析することを選び、それを、IAPの欠如によって感受性化されなかった細胞(A5-RT3)と比較した。最初の実験は、IAP阻害剤がデスレセプターの発現に影響を与えなかったことを明らかにした(図2Dおよびデータは示さず)。本発明者らは、リガンドアフィニティー沈降物(DISC)およびカスパーゼ8免疫共沈物(複合体II)を各条件で同量の総細胞溶解物と比較した場合に、カスパーゼ8関連複合体(複合体IIと呼ぶ)中にCD95が非存在であることを最初に確認した(図5、TL)。HaCaT角化細胞33および他の細胞型34におけるTRAIL DISCに関する本発明者らの以前のデータに一致して、そしてデスリガンドに対する全体的な感受性に一致して(図1と比較)、本発明者らは、両細胞型におけるCD95 DISCへのcFLIP、カスパーゼ8、カスパーゼ10、FADD、およびRIP1の刺激依存性動員を検出した(図5、左パネル、レーン3〜4、7〜8)。著しいことに、カスパーゼ8のより高い発現にかかわらず、カスパーゼ8のDISC結合は、A5-RT3の方が弱く、これは、CD95についてのウエスタンブロットによって決定されたような、より低レベルのCD95表面発現によって潜在的に説明された(左および右パネル、CD95)。これら二つの細胞株のDISCを比較して、A5-RT3細胞は、cFLIPの類似の動員を示したが、RIP1、FADD、カスパーゼ8、およびカスパーゼ10の動員はA5-RT3の方が低下していた(図5、左パネルと比較、レーン1〜4;5〜8)。驚くことに、RIP1の動員は、cIAPの非存在下でMET1細胞と同様にA5-RT3細胞のCD95 DISCにおいて堅調に増加した(図5、左パネル、レーン3〜4)。対照的に、A5-RT3細胞においてRIP1の動員増加は検出可能であったが実質的にさらに弱まった(図5、左パネル、レーン7〜8)。そのうえ本発明者らは、MET1細胞ではなくA5-RT3細胞においてcIAP2の特異的刺激依存性動員を検出した(図5、左パネル、レーン3〜4、7〜8)。カスパーゼ8関連タンパク質(複合体II)の免疫共沈によってレセプター非依存性的なDISC成分の相互作用を調べて、本発明者らは、両細胞株におけるFADD、カスパーゼ10、cFLIP、およびRIP1とカスパーゼ8との刺激依存性相互作用を検出した。最初の結果から、カスパーゼ阻害剤の存在下でDISCと同様に複合体IIが強く安定化されたことが確認された(データは示さず)。したがって本発明者らは、刺激の間に、本発明者らの実験において検出可能なカスパーゼ依存性切断の程度を変化させうるzVAD-fmkの存在下でこれらの実験を行った。興味深いことに、RIP1、FADD、cFLIPLおよびcFLIPL p43の大きな増加がカスパーゼ8に関連して検出された(図5、右パネル、レーン19〜20、23〜24)。これらのデータは、cIAPの発現抑制時に、細胞質において複合体IIが漸増的に形成されることを示唆した(図5右パネル)。興味深いことに、本発明者らは、A5-RT3の複合体II中にcIAP1ではなくcIAP2を見出したが、これは、もしかするとMET1細胞においてcIAP2の発現レベルがより低いことから起こったのかもしれない(図4Aと比較)。まとめると、本発明者らのデータによって、cIAPの欠如がCD95 DISCへのRIP1動員を促進し、RIP1-FADD-カスパーゼ10およびカスパーゼ8を含有する複合体IIの形成増加を可能にすることが示唆された。さらに、IAP阻害剤の感受性化効果に抵抗性の細胞は、IAP阻害剤を用いた処置にかかわらず複合体II内にcIAP2が存在することを実証したが、これは、cIAP2がIAP阻害剤の存在下で(ゆえにcIAP1の欠如下で)細胞死経路のさらなる活性化を遮断するために十分でありうることを示唆している。
【0046】
4. IAPの非存在下でデスリガンド介在性細胞死への抵抗性に異なる寄与を果たす、cFLIPアイソフォーム
デスレセプター介在性細胞死に対するIAP阻害剤介在性感受性化に感受性であった細胞は、DISC内でカスパーゼ8を高効率で動員および切断した一方で、cFLIPの動員は同等であり、先に示唆されたように異なるcFLIPアイソフォームがDISCに高親和性であることを示した33。これに関連して、cFLIPがTRAILまたはCD95L細胞死抵抗性の重大な一決定因子であることが広く受け入れられている(総説35参照)。したがって本発明者らは、次にcFLIPがデスリガンドに対するIAP阻害剤介在性感受性化への抵抗性を担うかどうか試験した。このために本発明者らは、主として、XIAPを欠如するIAP阻害剤抵抗性SCC細胞株A5-RT3を選択し、cFLIPに対するshRNAの安定的発現によってcFLIPをダウンレギュレーションさせた。ウエスタンブロット分析によって、cFLIPLおよびcFLIPSの両方の効率的なダウンレギュレーションが確認された(図6A)。興味深いことに、A5-RT3細胞におけるcFLIP欠如は、IAP阻害剤存在下でのTRAIL介在性またはCD95L介在性細胞死の増加を招いた(図6B)。しかし、HaCaT細胞でのデータと対照的に、IAP欠如による感受性化は、完全にカスパーゼ依存性であった一方で、ネクロスタチン-1はこれらの細胞において無効であった(図6C)。これらのデータから、デスリガンド介在性細胞死に対する感受性の細胞型高特異性IAP調節が確認された。さらに重要なことには、本発明者らのデータは、RIP1およびcFLIPがこの調節に重大であること、ならびにA5-RT3では、cFLIPアイソフォームがIAP阻害剤介在性細胞死を遮断することができることを示した。cFLIPのシグナル伝達能に関して、異なるcFLIPアイソフォームの機能と同様にcFLIPLの切断断片の機能についていくつかの対立する結果がある(総説36参照)。デスレセプター介在性細胞死についてのIAP阻害剤介在性感受性化のメカニズム、さらに具体的にはcFLIPの影響に取り組むために、本発明者らは、異なるcFLIPアイソフォームを発現している安定的細胞株を生成した(図6D)。これらの実験について本発明者らは、内因性cFLIPが低レベルであること37およびタンパク質レベルでXIAPを欠如していることから、HaCaT角化細胞を選択した17。予期されたように、cFLIPLと同様にcFLIPSは、CD95LまたはTRAIL介在性細胞死(図6E、パネル2)、デスレセプター介在性アネキシン-Vの外部移行、およびDNAの低二倍性(データは示さず)を効率的に遮断した。興味深いことに、IAP阻害剤単独に対する感受性は、cFLIPLではなくcFLIPSを発現している細胞において堅調に増加した(図7E、パネル6)。興味深いことにcFLIPSは、IAP阻害剤存在下でデスレセプター介在性細胞死から保護することができず(図6E、パネル7)、その細胞死はzVAD-fmkによって保護されなかった(図6E、パネル8)。しかしながら、図3に示される本発明者らのデータに一致して、両cFLIPアイソフォームは、アポトーシス細胞死の初期特徴を遮断した(データは示さず)。しかしながら、cFLIPS発現HaCaTにおけるIAP阻害剤存在下のCD95L介在性細胞死は、ネクロスタチン-1によって24時間完全に保護され、それは、RIP1依存性シグナル伝達の寄与を示した(図6E、パネル9)。まとめると、本発明者らのデータは、RIP1動員を介してデスレセプターによって活性化されるカスパーゼ非依存型細胞死を、cFLIPSではなくcFLIPLが遮断できることを示唆している。
【0047】
5. 複合体IIへのRIP1のCD95誘導性動員に異なる影響を与える、cFLIPアイソフォーム
この現象の潜在的メカニズムを明らかにするために、本発明者らは次に、cFLIPLおよびcFLIPS発現HaCaT細胞においてCD95 DISCと同様に複合体IIを沈降させ、対照感染HaCaTの結果と比較した。初回の結果は、DISCと同様に複合体IIが、カスパーゼ阻害剤の存在下で堅調に安定化されることを確認した(データは示さず)。カスパーゼ阻害は、細胞死からの保護に十分でなかったことから、本発明者らは、zVAD-fmkが存在してもこれらのデスリガンド誘導性複合体における差は相変わらずモニタリング可能であるという仮説を立てた。したがって本発明者らは、zVAD-fmkの存在下で、異なるcFLIPアイソフォームを発現しているHaCaT細胞をIAP阻害剤の存在下または非存在下で、CD95Lで刺激した。A5-RT3およびMET-1について図5に示した本発明者らのデータに一致して、cIAPが欠如している場合に対照細胞のDISCにおいてRIP1の劇的な増加が検出された(図7、左パネル、レーン1〜4)。TRAIL DISC33についての本発明者らの先の報告に一致して、cFLIPLと同様にcFLIPSは、CD95 DISCへのRIP1動員を抑制した。リンパ腫細胞における先の報告と適合することに、cFLIPLは、カスパーゼ8 p43/41の動員増加を導いたが、一方でcFLIPSは、DISCにおけるカスパーゼ8切断を完全に遮断した38。複合体IIにおいて、カスパーゼ8と共に免疫共沈したRIP1、FADD、およびcFLIPL(プロ型およびp43)の実質的な増加があった(図7、右パネル、レーン1〜4)。対照的にcFLIPLは、複合体IIの形成を遮断した。興味深いことに本発明者らは、デスリガンド刺激の非存在下でcFLIPS発現細胞の複合体II形成を再現性よく検出したが(図7、右パネル、レーン6)、これは、これらの細胞においてcIAP非存在下で複合体IIが自然形成したことを示している。さらに、CD95Lに刺激された複合体IIの形成は、cFLIPL存在下の複合体IIよりも非常に強かったことから、cIAPがcFLIPS細胞中で非存在である場合はいつもCD95Lに応答した細胞死が増加することの説明を与えている。
【0048】
考察
本発明において、本発明者らは、IAP阻害の状況でのデスレセプター介在性細胞死のメカニズムを検討した。本発明者らは、IAP阻害剤が自己分泌性TNF阻害にほとんど依存せずにSCC細胞をDR介在性細胞死に対して劇的に感受性にすることを示す。それどころか、IAP阻害剤は、カスパーゼ8依存型およびRIP1依存型細胞死の両方を増加させる。本発明者らが驚いたことには、異なるcFLIPアイソフォームは、IAPの存在に応じて別個の阻害能を有する。cFLIPLおよびcFLIPSは、IAPの存在下でデスレセプター介在性アポトーシスを同様に阻害するが、cFLIPLはRIP1依存性細胞死に加えてカスパーゼ8依存性細胞死を遮断し、cFLIPSはカスパーゼ8依存性アポトーシスだけを妨害するが、RIP1依存性細胞死の保護には著しく非効率的であった。本発明者らのデータは、異なるcFLIPアイソフォームが、cIAPの非存在下でのみ明らかな別個のシグナル伝達能を有することを初めて示している。cIAPのこの機能は、腫瘍療法の障壁としてのアポトーシス抵抗性にとってのみかかわりがあるというわけではなく、細胞死の様式が最も重要であるウイルス感染または腫瘍免疫の際に関係しうる25。
【0049】
本発明は、TRAIL-R1、TRAIL-R2、およびCD95デスレセプターによって活性化されるシグナル伝達経路の理解についてのいくつかの重要な発見に寄与するものである。第一に、本発明者らは、−細胞表面上でデスレセプターの調節が顕著に非存在の状況で(データは示さず)− cIAPの欠如がTRAILまたはCD95L誘導性細胞死への劇的な感受性化を導くことを予想外に見い出している。本発明者らは、数分間以内にIAPの分解を誘導する薬理学的阻害剤(IAP阻害剤)で処置されたヒトSCC腫瘍細胞において、またはその代わりに異なるcIAPを欠如したMEF遺伝的モデルを使用してのいずれかで、これらの局面を研究した。本発明者らのデータは、TRAILまたはCD95Lシグナル伝達経路が、示唆されたTNFシグナル伝達自己分泌性ループにほとんど依存せずにcIAPによって顕著に調節されることを実証しているが、それは、デスレセプターシグナル伝達経路におけるcIAPの独立した機能を示している20〜24。重要なことには、使用された細胞の一部がXIAPをタンパク質レベルで完全に欠如していることから、本発明者らのデータは、XIAPの機能とは独立して作動する分子メカニズムを示唆している17。IAP阻害剤への感受性化もまた、XIAP MEFで得られ、そのことはさらに、CD95およびTRAIL-R介在性細胞死の調節のためのcIAPのこの新規な機能を裏付けている。
【0050】
本発明者らが、この現象の分子メカニズムを研究したとき、本発明者らは、キナーゼRIP1をデスレセプター介在性細胞死に果たすcIAPの負の調節の役割に重要であると確認した。RIP1は、デスレセプターによるNF-κB活性化におけるその関連性が何年にもわたり知られている39。しかしながら、RIP1が細胞死を遮断する能力は、間接であって、デスレセプター誘導性NF-κB活性化の欠如によって仲介されると見なされた。他のグループは、TNFまたはCD95刺激に応答するプログラム壊死を先に検討していた40。
【0051】
過剰発現研究により、FADDのDDが壊死誘導に必要である一方で、FADDのDEDはカスパーゼ活性化に必要とされることが示唆された41。さらにHollerらは、FADD欠損Jurkat T細胞におけるCD95誘導性壊死を示した29。したがってFADDは、デスレセプターのトリガー後にアポトーシス細胞死経路および壊死細胞死経路の両方に関与するが、FADDがこれら二つのシグナル伝達経路についての重要な分子スイッチに相当するかどうかは不明である。酸性の細胞外条件が少なくともTRAILに応答したRIP1依存性細胞死に有利に働くとはいえ、FADDおよびRIP1を発現している細胞における壊死性細胞死の理由ははっきりしないままであった42。今回、本発明者らのデータは、cIAPが壊死性細胞死経路を負に調節すること、およびRIP1がDISCレベルでCD95またはTRAIL-R誘導性細胞死に必要であることを示唆している。この結論は、RIP1ノックダウン、RIP1欠損MEF、およびcIAPの非存在下で大量のRIP1を含有する腫瘍細胞におけるネイティブCD95複合体の沈降を用いた本発明者らのデータに基づく。さらに、本発明者らのデータは、膜結合型CD95複合体(CD95 DISC)へのRIP1動員がcIAPによって劇的に減少するが、RIP1の合計細胞レベルは影響を受けないことを実証している。特異的RIP1キナーゼ阻害剤であるネクロスタチン-1によって、本発明者らは、RIP1キナーゼ活性の必要性をさらに研究できた32。本発明者らのデータは、カスパーゼが遮断されたときはいつも、RIP1キナーゼが重要なタンパク質となり、カスパーゼおよびRIP1キナーゼの二重阻害がCD95介在性またはTRAIL介在性細胞死の細胞生存率の回復を可能にすることを明らかに示している。本発明者らは、汎カスパーゼ阻害剤であるzVAD-fmkの存在下であってもDISC中でRIP1のDD含有断片が強く濃縮することを検出している(データは示さず)。不幸にも、これまでのところ入手可能な全ての抗体は、DDにおけるエピトープを認識し、このことがRIP1のキナーゼドメインを有するN末端断片の検出を妨げている。タグ付きタンパク質またはキナーゼドメインに対する抗体を使用した将来的な研究は、RIP1の切断が、a)細胞死を誘導するためにDDによる阻害からキナーゼ活性を解放することを導くか、またはb)機能的RIP1を全体的に除去する有効なメカニズムであるかどうかをさらに解明すると考えられる。
【0052】
本発明者らは、IAPの欠如がRIP1依存的にCD95LまたはTRAILへの劇的な感受性化を導くことを見出している。本発明者らは、もはやデスレセプターに結合していないカスパーゼ8-FADD-cFLIP-RIP1含有細胞質複合体(複合体II)を検出している。この複合体の形成は、cIAPによって阻害される。驚くことに、異なるアイソフォームのカスパーゼ8抑制因子cFLIPは、cIAPが非存在のときはいつも差次的な効果を有する。全てのcFLIPLアイソフォームは、cIAPの存在下で細胞死から保護するが、cFLIPSではなくcFLIPLだけがネイティブ複合体IIの形成を遮断することができ、cIAPの非存在下で効率的な細胞死を可能にする。したがって本発明者らのデータは、CD95またはTRAILデスレセプター下流の細胞死シグナル伝達に関連する重要な細胞内タンパク質複合体を同定している。
【0053】
本発明者らの研究の別の重要な発見は、少なくともFADD、カスパーゼ8、カスパーゼ10、RIP1、および異なるアイソフォームのcFLIPを含有するレセプター非依存性複合体(複合体II)の同定である。最近の報告は、CD95またはTRAIL刺激後にそのような複合体を同定している10,11。本発明者らの実験において、複合体IIの形成およびカスパーゼ8とRIP1との結合は、IAP阻害剤に非感受性の細胞では抑制される。顕著には、これらの細胞においてcIAP2は高発現され、IAP阻害後に動力学的に調べた場合、未処置細胞の定常状態レベルまで急速に再発現される(補足の図1A)。このパターンのcIAP2発現は、DISCと結合したcIAP2の検出に加えて、これらの細胞の複合体IIにおけるRIP1の検出減少と同時に起こったことから(図5と比較)、cIAP1およびcIAP2がIAP阻害剤による阻害に差次的な感受性を有するか、または全体で差次的に調節されうることを示唆している。自己分泌性TNF分泌がcIAP2のデノボ発現を導きうることを認知できる。これに関連して、本発明者らの細胞モデルにおいてcIAP1はIAP阻害剤によって長期的に抑制されるが、cIAP2は、TNF介在性細胞死の調節に不要であるが、NF-κBによって堅調に誘導される17。したがって、両方のcIAPは別個の役割を有する可能性があり、cIAP1はかなり構成的に発現されるcIAPである一方で、これらの高度に類似したcIAPの誘導型のcIAP2は、そのために別個の病態生理学的過程の間に差次的な機能を有する。
【0054】
細胞死に重要なDISCまたは複合体II関連シグナルの機能をさらに詳しく検討するために、本発明者らは、内因性カスパーゼ8ホモログであるcFLIPを使用して実験を行った。本発明者らは、IAP阻害剤に非感受性の細胞(例えば重複する用量反応曲線を有する)を、cFLIPアイソフォームの特異的ダウンレギュレーションによって感受性化することができることを実証している。重要なことには、これらの細胞は、zVAD-fmkによってまだ完全に保護されており、TNFシグナル伝達について示されたように、IAPがカスパーゼ依存性シグナル伝達経路も遮断することを示している43。DISC内のRIP1依存性細胞死を活性化するためのカスパーゼ活性化の関連性は何であろうか?本発明者らは、zVAD-fmkの非存在下および存在下でCD95複合体に動員されたRIP1の量を比較したところ、TRAIL DISCについての本発明者らの先の報告33に一致して、zVAD-fmkの存在下でネイティブCD95 DISC中の完全長RIP1の顕著な増加を見出した(データは示さず)。しかしzVAD-fmkは、DISCにおけるcFLIPLの切断に基づきカスパーゼ8のDISC関連活性を完全に遮断することができない33。加えてzVAD-fmkは、プロ型カスパーゼ8の酵素活性を遮断しない44。したがって、本発明者らの実験は、cFLIPがカスパーゼ依存性細胞死およびカスパーゼ非依存性細胞死に必要なDISCによって生成するシグナルと拮抗することを示している。これに一致して、本発明者らの過剰発現研究は、cFLIPが二重の役割を有することを示唆している:cIAPが存在するときはいつもcFLIPの全アイソフォームが同等の効率でデスレセプター介在性アポトーシスを遮断するのに対し、cIAPの非存在下では、cFLIPSではなくcFLIPLだけがデスレセプターによってトリガーされる細胞死を完全に遮断する。重要なことには、RIP1キナーゼ活性は、cFLIPS発現細胞において細胞をデスレセプター介在性細胞死から保護するために重要であり、本発明者らは、これらの細胞におけるFADDおよびRIP1のレベル増加と共に、複合体IIの自然形成および誘導形成の増加を検出している。これらのデータは、cFLIPSが、cFLIPLのカスパーゼ様ドメインによって負に調節される複合体IIの形成を遮断できないことを立証している。この断片は、cFLIPLおよびcFLIPSから生成することができる45ので、cFLIP p22はこの差次的効果の原因となりそうにない。主に過剰発現研究に基づくと、cFLIPLのカスパーゼ様ドメインは、TRAF、RIP1、またはその他などのタンパク質への結合を仲介し、内因性TRAF2はDISCによって生成したcFLIPL p43と相互作用することが報告された(総説については46参照)。そのうえTRAF2は、cIAPおよびRIP1の結合パートナーである。しかし本発明者らは、それぞれ本発明者らのDISCリガンドアフィニティー沈降または複合体IIの免疫共沈からTRAF2を検出することができなかった(データは示さず)。したがって、これらの追加的な相互作用タンパク質の役割をさらに詳細に解明するために、さらなる研究が必要である。
【0055】
いくつかの異なるSCC細胞株を使用した本発明者らのデータは、異なるDISC成分の化学量論比およびIAPによるそれらの修飾が、アポトーシス細胞死経路および壊死性細胞死経路の活性化に高度に関連することを立証している。CD95 DISCのネイティブリガンドアフィニティー沈降を用いて、本発明者らは、cIAPがDISCに動員されるRIP1の量を負に調節する一方で、RIP1の合計細胞レベルは不変であることを示している(図5、7を比較)。RIP1は、デスレセプター介在性カスパーゼ活性化の伝播(薬理学的カスパーゼ阻害剤によって研究されたように)がいったん遮断されたならば大いに関連性を増し、DISCレベルでカスパーゼ依存性細胞死およびカスパーゼ非依存性細胞死を並行活性化することを立証している。この概念を裏付けて、Hollerらは、FADD欠損Jurkat細胞を使用してFADDの非存在下でRIP1がDISCに動員されることを示した29。したがって本発明者らは、RIP1が、DISCでTRAILおよびCD95デスレセプターによって活性化され、cIAPによって負に調節される、FADD非依存性シグナル伝達経路の重要な成分を構成すると提案する。DISC関連カスパーゼは、DISCに利用可能なRIP1をダウンレギュレーションするように作用することができ、DISCにおけるRIP1のa)cIAP介在性ユビキチン化、またはb)カスパーゼ介在性切断が、DISC活性化RIP1依存性細胞死シグナル伝達経路の重大な負の調節メカニズムとなることを示唆している(図8と比較)。zVAD-fmkを使用した本発明者らの研究によって示されたように、FADD、RIP1、またはカスパーゼ8を欠損した細胞を使用した将来的な研究は、複合体IIの潜在的不安定化因子としてのカスパーゼ活性の必要性をさらに解明すると考えられる。さらに重要なことには、RIP1キナーゼの重要な標的を同定するであろう将来的な研究は、CD95またはTRAIL-Rによって活性化されるアポトーシス性細胞死経路および壊死性細胞死経路の間のクロストークを支配しているシグナル伝達メカニズムをさらに解明すると考えられる。
【0056】
cIAP1およびcIAP2は、もともとTRAF結合タンパク質として報告された47。さらに最近になって、RIP1がcIAPの直接のターゲットであること24,48、およびRIP1についての構成的E3ユビキチンリガーゼとしてのcIAPの機能が、デスレセプターの刺激に依存せずに作用しうることが示唆されている。特にTRAF2は、RIP1についてのリシン63(K63)ユビキチンリガーゼであり、K63-RIP1は、NF-κBの活性化に必要なシグナル伝達モジュールのさらなる集合を可能にする31。対照的にcIAPは、RIP1のK63およびリシン48(K48)のユビキチン化に関与すると示唆されている24。今回本発明者ら自身の実験は、cIAPの重大な一機能が、DISC全体へのRIP1の動員を遮断することであると示すか、またはその代わりにcIAPがDISC内のRIP1の急速分解に必要な可能性があることを示している。重要なことには、本発明者らの実験は、インビトロユビキチン化アッセイによって研究されたようなcIAPによるRIP1の構成的K48またはK63ユビキチン化のDISC関連(刺激依存性)強化を区別することができない24。本発明者らがDISC中のRIP1のユビキチン化パターンを比較したとき、本発明者らは、長期曝露でcIAP非存在下と比較した場合にcIAP存在下で高い分子量のRIP1種を検出している(図5-長期曝露と比較)。本発明者らのデータは、構成的にユビキチン化されたRIP1がDISCに動員されないこと、またはその代わりにDISC内のK48ユビキチン化によってユビキチン化RIP1をダウンレギュレーションすることを示唆している。最近記載されたように49、ユビキチン化特異性抗体を使用した将来的な研究は、異なるデスレセプター誘導性膜結合型複合体内でこれらの点に動力学的にさらに詳細に取り組むことができると考えられる。これに関連して、最近の報告は、Lys377でユビキチン化することのできないRIP1変異体を研究した。これらの著者は、細胞質複合体で細胞死を誘導してカスパーゼ8と相互作用するのが非ユビキチン化形態のRIP1であることを示した。対照的にユビキチン化形態のRIP1は、細胞死を誘導するのではなく、細胞死からの保護のために、おそらくNF-κB活性化によるLys377でのK63ユビキチン化を必要とする50。
【0057】
なぜIAPがダウンレギュレーションされたときにいつも、cFLIPSではなくcFLIPLが、細胞死を遮断することができるのか?本発明者らのデータは、cFLIPSではなくcFLIPLだけが、DISCまたは複合体II全体内のカスパーゼ8に関連したRIP1の効率的な形成(または維持)を抑制するという事実を立証している。図7から明らかなように、複合体IIは、FADD-RIP1-cFLIPS-カスパーゼ8を含有し、特にRIP1とカスパーゼ8との相互作用は、cFLIPSではなくcFLIPLによって抑制される。これらのデータは、cFLIPLのカスパーゼ様ドメインの重要な役割を指摘している。この機能は、DISC内で関連し、切断によってRIP1をダウンレギュレーションすることができよう。または、cFLIPアイソフォームは、先にリンパ系細胞において示唆されたように45、デスレセプター複合体に依存せずに作用することもできる。非ユビキチン化型RIP1は、DISCに依存せずにFADDに結合し、続いて複合体IIの形成および壊死性細胞死を導くことができると推測することができよう。それにもかかわらず本発明者らのデータは、cFLIPSが複合体IIを遮断する能力を有さないことをはっきりと示し、cIAP非存在下での異なるcFLIPアイソフォームの新規で差次的な機能を指摘している。
【0058】
デスレセプターシグナル伝達におけるRIP1の生理学的意味は何でありうるか?T細胞レセプターの刺激後に、細胞死がT細胞区画においてカスパーゼ8非依存的に進行することが最近実証された。この形態の細胞死は、決定的にRIP1を必要とした51。まとめると、これらのデータは、RIP1がカスパーゼ8の非存在下でT細胞刺激後にT細胞を排除するために重要でありうること、およびカスパーゼ8欠損マウスにおいて記載されたいくつかの細胞の増殖欠損52が、活性カスパーゼ8によるRIP1の負の調節の欠如によって引き起こされるおそれがあることを立証している。このRIP1分解は細胞質カスパーゼ8-FADD-RIP1-複合体II内で起こると推測したくなる。最近のデータは、カスパーゼ8の自己プロセシング欠如がカスパーゼ8の非アポトーシス機能を妨害しない一方で、アポトーシスが損なわれることを示しており53、レセプター非依存性複合体IIでのカスパーゼ8の潜在的シャペロン機能を立証している。本発明者らのデータはさらに、これらの観察に付け加え、デスレセプター刺激が主にRIP1依存性シグナルとカスパーゼ8依存性シグナルの両方を活性化するものの、cIAPが重大な負の調節因子に相当することを示している。RIP1は、いくつかの生理学的および病態生理学的に関連する状況でカスパーゼ8に依存せずに作用しうる。カスパーゼ8およびRIP1の両方を欠損した条件付きマウスを調べる将来的な研究は、デスレセプターおよび/またはTCR介在性細胞死経路に関するこれら二つの分子が免疫系に果たす生理学的役割をさらに詳細に明らかにすると考えられる。いくつかの腫瘍実体はIAPを高発現する16。本発明者らのデータに基づき、cIAPは、壊死性の、したがって「免疫学的に強い(loud)」形態の細胞死に関連する可能性のあるRIP1介在性細胞死を逸脱させるために役立つ可能性がありうる。したがって、cIAPは、デスレセプター介在性壊死性細胞死を回避することにより効率的な抗腫瘍免疫反応を回避する重要な役割を果たすことができる16。さらに最近になって、カスパーゼ8が細胞形質転換54および転移55を負に調節することが示された。加えてNF-□Bは、いくつかの細胞株において腫瘍促進転写因子である。したがってcIAPは、腫瘍形成の間にRIP1の死滅誘導性機能をNF-□B誘導性機能に変化させる重要な役割もまた果たしうる。これに関連して、IAPを過剰発現しない腫瘍が、複合体II介在性細胞死または免疫活性化を回避するためにRIP1の追加的な欠如を必要とするかどうかを決定することは興味深いと考えられる。
【実施例】
【0059】
材料および方法
材料
ウエスタンブロット分析のために以下の抗体(Ab)を使用した:カスパーゼ8に対するAb(C-15;親切にもP.H. Krammerによって提供された、C-20; Santa Cruz, Delaware Avenue, California)、cFLIPに対するAb(NF-6; Alexis, San Diego, California)、FADDおよびRIP1に対するAb(Transduction Laboratories, San Diego, California)、CPP32に対するAb(親切にもH. Mehmetによって提供された、Merck Frost)、カスパーゼ10に対するAb(MLB)、PARP-1に対するAb(クローンC-2-10、Biomol)、cIAP1およびcIAP2に対するラットAb56、ならびにSigma製(Saint Louise, Missouri, USA)のβチューブリンに対するAb(クローン2.1)。βアクチンAbは、適切な供給元からのものであった。His-FLAG-TRAIL(HF-TRAIL)は、最近記載されたように産生させた17。Fc-CD95Lの発現のために、本発明者らは、最近公表された構築物を使用した57(親切にもP. Schneider, Epalinges, Switzerlandによって提供された)。1ユニットのFc-CD95Lを、Fc-CD95L上清の原液を1:500希釈したものと規定し、最近記載されたように58、1ユニット/mlのFc-CD95L上清は、A375黒色腫細胞の50パーセントを死滅させるために十分であった(LD50)。リガンド介在性細胞死は、それぞれ可溶性TRAIL-R2-Fcタンパク質またはCD95-Fcタンパク質のいずれかの添加によって完全に遮断された。ホースラディッシュペルオキシダーゼ(HRP)結合ヤギ抗ウサギ、ヤギ抗ラットIgG、ヤギ抗マウスIgG AbならびにHRP結合ヤギ抗マウスIgG1、IgG2a、IgG2b、およびIgG1κは、Southern Biotechnology Associates(Birmingham, AL)から得た。表面レセプターの発現のFACScan分析用mAbであるTRAIL-R1(HS101)、TRAIL-R2(HS201)は、先に記載されたように使用したが26、これらはAlexis(San Diego, California)から入手可能である。CD95 Ab(Apo-1 IgG1およびIgG3a)は、親切にもP. H. Krammer(German Cancer Research Center, Heidelberg, Germany)によって提供された。Cy5結合アネキシンVは、Pharmingen(Hamburg, Germany)から購入した。IAP阻害剤(化合物A)は、Tetralogic Pharmaceuticals(Malvern, Pennsylvania, USA)によって提供された。化合物Aは、参照により全体として本明細書に組み入れられるUS20060194741に例示されており、化合物Aは、以下の構造を有する。

【0060】
細胞培養
自然形質転換角化細胞株HaCaTおよび派生した転移性クローンA5-RT327は、親切にもDr. Petra Boukamp(DKFZ, Heidelberg)によって提供された。MET1細胞59は、I. Leigh, Skin Tumor Laboratory, Cancer Research UK, London, UK)によって提供された。細胞株を記載のように27,60,61正確に培養した。
【0061】
レトロウイルス感染
HaCaT細胞の感染のために、cFLIPLまたはcFLIPSのcDNA挿入部を有するpCFG5-IEGZレトロウイルスベクターを、先に報告されたように使用した62,62。簡潔には、リン酸カルシウム沈殿によってアンホトロピック産生細胞株ΦNXに10μgのレトロウイルスベクターをトランスフェクトした。産生細胞をHaCaT培地(10%FCS含有DMEM)と共に一晩インキュベーションすることによって、ウイルス粒子を含有する細胞培養上清を生成させた。濾過(45μmのフィルター、Schleicher & Schuell, Dassel, Germany)後に、1μg/mlポリブレンの存在下で24時間前に6ウェルプレートに蒔いておいたHaCaT細胞に培養上清を添加した。30℃で3時間遠心分離後に、ウイルス粒子含有上清を新鮮培地と交換した。ポリクローナル細胞に関して10〜14日後に感染培養物全体のゼオシン選択、GFP発現についてのFACS分析(常に>90%、データは示さず)およびウエスタンブロット分析を行い、それぞれの分子の異所性発現を確認した。空のレトロウイルスベクターを対照として用いた。初回の特徴づけの後に2〜6回継代した一定分量の細胞を、その後の全研究のための実験に使用した。
【0062】
siRNAの安定的発現
本発明者らは、最近公表されたように58cFLIPに対するsiRNAの安定的発現を利用した。RIP1 siRNAと同様にNCBIデータベースのどの遺伝子とも一致しないハイパーランダム配列(HRS)63を使用した。構築物の生成のために、RIP1ターゲティング配列を有するcDNA 64塩基長オリゴマー(nt開始位置+193:要望に応じて完全配列が入手可能)を、HindIIIおよびBglII制限部位を使用してpSuper.retroレトロウイルスベクター(pRS)にクローニングした。結果として生じたベクターまたはヒトゲノムに見られないものを有する対照ベクターを、上記に概説したように正確にアンホトロピック産生細胞株にトランスフェクトした。次に、レトロウイルス含有上清を使用して、A5RT3およびMET1細胞にそれぞれHRS shRNAまたはcFLIP shRNAを感染させた。HaCaT細胞にHRSおよびRIP1 shRNAを感染させ、さらなる分析のためにピューロマイシン耐性感染培養物全体を得るために感染細胞をピューロマイシン(1μg/ml; Sigma, Taufkirchen, Germany)で3日間選択した。それぞれの対照構築物を、内部対照として用いた。GFP発現のFACS分析(常に>90%、データは示さず)およびウエスタンブロット分析をポリクローナル細胞について行って、それぞれの分子の異所性発現を確認した。抗生物質選択の後に2〜6回継代した一定分量の細胞を、細胞毒性アッセイおよび生化学的特徴付けのために使用した。
【0063】
FACScan分析
TRAILレセプター(TRAIL-R1およびTRAIL-R2)およびCD95の表面染色のために、細胞をトリプシン処理し、4×105個の細胞をTRAIL-R1 TRAIL-R2、CD95に対するモノクローナルAb、またはアイソタイプが一致する対照IgGと共に60分間インキュベートし、続いて記載のように33ビオチン化ヤギ抗マウス二次Ab(BD Pharmingen)およびCy5-フィコエリトリン標識ストレプトアビジン(Caltag, Burlingame, CA)と共にインキュベーションした。全ての実験について、2×104個の細胞をFACScan(Becton Dickinson & Co, San Jose, CA)によって分析した。
【0064】
ウエスタンブロット分析
記載のように17,58細胞溶解液を調製し、総細胞タンパク質5μgをSDS-PAGEによって4〜12%勾配のゲルで分離し(Invitrogen, Karlsruhe, Germany)、続いてニトロセルロースまたはPVDFメンブランに転写させた。メンブランのブロッキングおよび一次Abおよび適切な二次Abとのインキュベーションは、本質的に先に記載されたように行った33,62。ECL検出キットでバンドを視覚化した(Amersham, Freiburg, Germany)。
【0065】
細胞毒性アッセイ
記載のように37、96ウェルプレート中で一条件について3つ組のウェルを使用して表示濃度のTRAILまたはCD95Lで刺激した20〜24時間後に、付着した生存細胞のクリスタルバイオレット染色を行った。対照培養液の吸光度(OD)を100%に基準化し、それを刺激された細胞と比較した。統計解析のために、細胞株および刺激条件毎に3〜7回の独立した実験について平均の標準誤差(SEM)を決定した。
【0066】
低二倍性分析
先に行われたように33低二倍体(subdiploid)DNA含量を分析した。簡潔には、細胞を表示の試薬で8時間刺激した。次に、細胞を剥離させ、冷PBSで洗浄し、緩衝液N(クエン酸ナトリウム0.1%(w/v)、Triton X100 0.1%(v/v)、PI 50μg/ml)に再懸濁した。細胞を4℃で36〜48時間暗条件に保ち、次に二倍性をFACScan分析によって測定した。
【0067】
免疫蛍光顕微鏡観察
核形態の検出のために、それぞれの細胞の5×104個を12ウェルプレートの各ウェルに蒔いた。接着させるために24時間インキュベーションした後に、図の凡例に示したように細胞を4または24時間刺激した。続いて、細胞をヘキスト33342(5μg/ml)と共に37℃で15分間インキュベートし、直後に適切な装置(Leica)を使用して位相差または蛍光顕微鏡観察を行った。デジタル画像は適切なソフトウェアを使用して全く同様に処理した。
【0068】
アネキシンVの外部移行
ホスファチジルセリンの外部移行を検出するために、図の凡例に示したように細胞を刺激した。細胞を4時間インキュベーションした後に、トリプシン処理した細胞を1×アネキシン-V結合緩衝液(10mM Hepes、pH7.4、140mM NaCl、2,5mM CaCl2)に再懸濁し、続いて製造業者(Pharmingen)に厳密にしたがって2〜4×105個の細胞をCy5結合アネキシン-Vで染色し、その後室温の暗条件で15分間対比染色(ヨウ化プロピジウム;10μg/ml)を行った。全ての実験について、2×104個の細胞をFACScan(Becton Dickinson & Co, San Jose, CA)によって分析した。
【0069】
コロニー形成アッセイ
コロニー形成アッセイのために、1×104個の親HaCaT細胞に加えてレトロウイルスを形質導入されたHaCaT細胞(HRS、shRNA RIP1、pCFG5-IEGZレトロウイルスベクターおよびcFLIPLまたはcFLIPS)を24ウェルプレートの各ウェルに蒔いた。24時間インキュベーション後に接着細胞をIAP阻害剤(100nM、30分間)、zVAD-fmk(10μM、1時間)、ネクロスタチン-1(50μM、1時間)で別々に、または全ての化合物を組み合わせてのいずれかで予備刺激し、続いてCD95Lと共に24時間共刺激した。その時に培地を除去し、細胞を滅菌PBSで2回洗浄し、完全培地を添加した。細胞を3、5、または7日間培養し、続いて生存細胞のコロニーを上記のようにクリスタルバイオレットによって染色した。
【0070】
レセプター複合体のリガンドアフィニティー沈降
CD95L DISCを沈降させるために、各条件について5×106個の細胞を使用した。細胞を培地で37℃で1回洗浄し、続いて10μM zVAD-fmkと共におよび表示のように100nMのIAP阻害剤と共に37℃で1時間予備インキュベートした。続いて細胞を250ユニット/mlのCD95L-Fcで2時間、または未刺激対照についてはリガンドの非存在下で処理した。単層を氷冷PBSで4回洗浄することによってレセプター複合体の形成を停止した。2mlの溶解緩衝液(30mM Tris-HCl、pH7.5、21℃、120mM NaCl、10%グリセロール、1%Triton X-100、Complete(登録商標)プロテアーゼ阻害剤カクテル(Roche Molecular Diagnostics, Mannheim, Germany))の添加によって細胞を氷上で溶解させた。氷上で30分間溶解させた後に、溶解液を20,000×gで2回、それぞれ5分間および30分間遠心分離して細胞の破片を除去した。これらの透明溶解液の小画分を使用してそれぞれのタンパク質の投入量を確認した。CD95レセプターおよび動員された刺激依存性タンパク質を沈降させるために、未刺激細胞および刺激細胞から調製した溶解液にApo-1 IgG3抗体(親切にもP. H. Krammerによって提供された)を添加して、レセプター相互作用タンパク質を沈降させた。リガンドアフィニティー沈降またはカスパーゼ8免疫沈降のいずれかによって沈降したレセプターのレベルを、全ての実験においてCD95についてのウエスタンブロットによって比較した(図5、7を比較)。40μlプロテインGビーズ(Roche, Mannheim, Germany)を反復振盪装置上で4℃で16〜24時間使用して、溶解液からレセプター複合体を沈降させた。リガンドアフィニティー沈降物を溶解緩衝液で4回洗浄してから、標準還元試料緩衝液を添加して95℃で沸騰させることによって、乾燥ビーズからタンパク質複合体を溶出させた。続いて、タンパク質を4〜12%NuPAGE勾配ゲル(Invitrogen, Karlsruhe, Germany)を用いたSDS-PAGEによって分離してから、ウエスタンブロット分析によってDISC成分を検出した。
【0071】
複合体IIのカスパーゼ8免疫沈降
CD95 DISCの沈降後に、残りの溶解液を20,000×gで5分間、2回遠心分離した。続いて1μgのカスパーゼ8抗体(C-20、Santa Cruz)を全ての溶解液に添加した。40μlのプロテインGビーズ(Roche, Mannheim, Germany)と共に反復振盪装置上で、4℃で16〜24時間同時インキュベーションすることによって、溶解液からカスパーゼ8含有複合体を沈降させた。リガンドアフィニティー沈降物を溶解緩衝液で4回洗浄してから、標準還元試料緩衝液を添加して95℃で沸騰させることによって、乾燥ビーズからタンパク質複合体を溶出させた。続いて、タンパク質を4〜12%NuPAGE勾配ゲル(Invitrogen, Karlsruhe, Germany)でSDS-PAGEによって分離してから、ウエスタンブロット分析によってカスパーゼ8相互作用タンパク質を検出した。残りのレセプター結合型DISC複合体を排除するために、CD95の存在について全てのカスパーゼ8相互作用複合体を分析した(図5、7を比較)。
【0072】
cFLIPのノックダウン
細胞株を24ウェルプレートにウェル1個あたり35,000個の細胞密度で蒔いた。翌日、細胞株に、100nMの対照siRNAまたはcFLIPの発現をサイレンシングするように設計されたsiRNAをトランスフェクトした(LおよびSアイソフォーム)。トランスフェクションの48時間後に細胞を100ng/ml TNFα、100nM Smac模倣体、または両者の組み合わせで処理した(ここで使用したSmac模倣体は、化合物A以外の化合物であった。)。追加的に24時間インキュベーションした後に、全ての細胞を回収し、FITC標識アネキシンVおよびヨウ化プロピジウムで染色した。フローサイトメトリーによって染色を分析した。
【0073】
対照siRNAがトランスフェクトされた細胞の処理は、どの条件でもアポトーシスの増加を招かなかった。対照的に、cFLIP特異的siRNAがトランスフェクトされた細胞の処理は、Smac模倣体単独に対する感受性の幾分の増加に加えて、Smac模倣体およびTNFαの組み合わせに対する有意な感受性化を招いた。TNFα単独に対する有意な感受性化は検出されなかった(図9)。
【0074】
これらのノックダウンデータは、cFLIPがSmac模倣体およびTNFαの組み合わせに対する抵抗性の細胞性伝達物質であることを示している。Smac模倣体ならびにSmac模倣体およびTNFαの組み合わせに完全に抵抗性の細胞株は、cFLIPのsiRNA介在性ノックダウンによる処置に感受性になることができる。臨床状況では、Smac模倣体化合物を用いた処置に対する抵抗性を予測して患者の選択を助けるために、腫瘍内のcFLIPレベルを使用することができる。
【0075】
参考文献
【技術分野】
【0001】
関連出願の相互参照
本出願は、2009年1月29日出願の米国特許仮出願第61/148,164号の恩典を主張し、同出願の全開示が参照により全体として本明細書に組み入れられる。
【0002】
発明の分野
本発明は、癌を含む増殖性障害を処置するためのSMAC模倣体ならびにその組成物および使用の分野にある。
【背景技術】
【0003】
発明の背景
アポトーシス抵抗性の発生は腫瘍形成の中核であり、腫瘍療法の主たる妨げとなる1。アポトーシス機構の再活性化を目的とした新規な治療方式は、熱心に研究されており、結果としてデスレセプターアゴニスト、Bcl-2アンタゴニスト、またはアポトーシス阻害タンパク質(IAP)阻害剤などの、アポトーシスシグナル伝達カスケード内の中心分子を標的とする多様な化合物が、その臨床的使用のため現在探究されている2〜4。
【0004】
TNF関連アポトーシス誘発リガンド(TRAIL)およびCD95Lは、広く研究されているデスリガンドであり、多数の研究がこれらのデスレセプターのシグナル伝達能力を検討してきた(総説については5、6参照)。特にTRAILは、腫瘍細胞の特異的除去を可能にする有望なリガンドと見なされている3,7。デスレセプターシグナル伝達経路は、細胞表面での受容体発現、細胞性FLICE抑制タンパク質(cFLIP)、X連鎖IAP(XIAP)、またはBcl-2ファミリータンパク質(例えばBcl-2、Bcl-X)などの抑制因子の発現を含む多レベルで制御される(総説については8参照)。最近の根拠は、デスレセプターのトリガーが、TNF経路に類似して、一次膜結合型複合体だけでなく二次的な独立シグナル伝達プラットフォームを誘導することを示している9〜11。これらの二次複合体の形成を導くメカニズム、ならびにデスレセプターによって活性化されるアポトーシス感受性および非アポトーシス性シグナルに対する二次複合体の重大な寄与は、詳細には解明されていない。外因性アポトーシスシグナル伝達経路の調節をさらに複雑化することには、過去10年間に壊死性および自己貪食性細胞死などのカスパーゼの活性化を必要としない追加型の細胞死が同定された12。これらは、単に細胞死経路が多様であることを表すのではなく、癌細胞に対する効率的な免疫応答に必要な多細胞生物内の免疫学的応答に特に関連する可能性がある。興味深いことに、細胞死の様式は、先に提案されたように細胞死を「免疫原性」対「不活動性」と定義することができよう13,14。
【0005】
過去10年間にわたる大規模な研究は、多数の腫瘍が腫瘍形成の間または初回処置までにアポトーシス抵抗性を有するか、または獲得するかのいずれかであること、および初期臨床研究においてデスレセプターアゴニストが単独ではまだ有望な結果を収めていないことを明らかにした15。IAPアンタゴニストは、XIAPのBIR2/BIR3ドメインに対するミトコンドリアタンパク質Smac/DIABLOのN末端IAP結合モチーフ(IBM)に応じてモデル化された合成化合物である16。デスリガンドに対するアポトーシス抵抗性に関するcIAPの役割は、あまりよく理解されていない17,18。しかしながらXIAP機能の干渉は、異種移植腫瘍モデルにおけるTRAILの治療有効性にとって重大である19。IAP阻害剤が、エフェクターであるカスパーゼへの結合からXIAPを移動させるだけではないことを数グループが最近になって独立して発見したことによって、癌治療のためのIAP阻害剤の使用が刺激された。むしろ、IAP阻害剤は、cIAP1およびcIAP2の迅速な自己ユビキチン化および欠如、NF-κB誘導、ならびに最終的にTNF介在性カスパーゼ8活性化および細胞死を導くTNFの自己分泌型産生もまた誘導する20〜24。この状況でcIAP1およびcIAP2は、カスパーゼ抑制因子というよりもむしろカスパーゼ調節因子であって、まだ詳細に探究されていない追加的な機能を有する見込みが最も高いことが明らかとなった。
【0006】
細胞性アポトーシス阻害タンパク質(cIAP)は、TNF介在性細胞死から保護するために必要である。CD95またはTRAIL-Rなどのプロトタイプのデスレセプターシグナル伝達に対する腫瘍細胞の感受性についてのcIAPの役割は、これまで詳細には研究されていない。
【0007】
細胞型FLICE様抑制因子cFLIPは、デスレセプターFas、DR4、およびDR5によって仲介されるアポトーシスの抑制因子であり、長鎖(cFLIPL)スプライス型および短鎖(cFLIPS)スプライス型として発現される。c-FLIPは、デスレセプターFas、DR4、およびDR5によって仲介されるアポトーシスの抑制因子であり、長鎖(c-FLIPL)および短鎖(c-FLIPS)スプライス型として発現される。cFLIPは、FAS介在性デスドメイン(FADD)およびカスパーゼ8と相互作用することによって、TNFレセプター遺伝子スーパーファミリーのメンバーにより仲介されるアポトーシスを阻害することができる。
【0008】
アポトーシス阻害タンパク質(IAP)は、カスパーゼ依存性アポトーシスを抑制する天然の細胞内タンパク質である。DIABLOとしても知られているSMACは、IAPと拮抗するように、すなわちIAPの活性を阻害するように機能する別の細胞内タンパク質である。健常細胞において、SMACおよびIAPは一緒に機能して健康な細胞を維持する。しかし、ある種の疾患状態、例えば癌および他の増殖性障害では、IAPは十分に拮抗されず、したがってアポトーシスを阻止し、そして異常増殖および異常生存を引き起こすかまたは増悪させる。
【0009】
IAPアンタゴニストとしても知られているSMAC模倣体は、SMACのN末端アミノ酸4個の構造およびIAPアンタゴニスト活性を模倣する合成小分子である(SMAC模倣体は、IAPアンタゴニストと呼ばれることもある)。増殖性障害を患う動物に投与した場合、SMAC模倣体はIAPと拮抗し、異常増殖細胞の間でアポトーシスの増加を引き起こす。
【0010】
SMACペプチド模倣体の例は、とりわけUS 7,517,906;US 7,309,792;US 7,419,975;US 2005/0234042;US 2005/0261203;US 2006/0014700;US 2006/0025347;US 2006/0052311;US 2006/0128632;US 2006/0167066;US 2007/0042428;US 2007/032437;US 2008/0132485;WO 2005/069888;WO 2005/069894;WO 2006/010118;WO 2006/122408;WO 2006/017295;WO 2006/133147;WO 2006/128455;WO 2006/091972;WO 2006/020060;WO 2006/014361;WO 2006/097791;WO 2005/094818;WO 2008/045905;WO 2008/016893;WO 2007/136921;WO 2007/021825;WO 2007/130626;WO 2007/106192;およびWO 2007/101347に開示されているものである。
【発明の概要】
【0011】
例示的な一態様では、本発明は、IAPアンタゴニスト、すなわちIAP阻害剤によるアポトーシス誘導に対する抵抗性に関するバイオマーカーである。具体的にはこの態様では、IAPアンタゴニストを用いた処置に対する抵抗性は、cFLIPの長鎖アイソフォーム、すなわちcFLIPLをアッセイすることによって決定される。cFLIPLを発現するヒトまたは非ヒト動物細胞は、IAPアンタゴニストに抵抗性の傾向がある。
【0012】
別の例示的な態様では、本発明は、IAPアンタゴニストによるアポトーシス誘導に対する感受性に関するバイオマーカーである。具体的にはこの態様では、IAPアンタゴニストを用いた処置に対する感受性または受容性は、cFLIPの短鎖アイソフォーム、すなわちcFLIPSをアッセイすることによって決定される。cFLIPSを発現するヒトまたは非ヒト動物細胞は、IAPアンタゴニストに感受性の傾向がある。
【0013】
さらに具体的で例示的な態様では、本発明は、TRAILレセプターアゴニスト、CD95レセプターアゴニスト、またはTNFaレセプターアゴニストと組み合わせたIAPアンタゴニストに対する細胞の感受性が決定される方法であって、例えば、TRAILレセプターアゴニストがTRAILであり、CD95レセプターアゴニストがCD95Lであり、かつTNFaレセプターがTNFαである方法を含む。
【0014】
特に例示的な態様では、ヒトまたは非ヒト動物細胞は、生検試料または細胞株由来である。細胞は、異常に増殖している任意の細胞、例えば腫瘍細胞または自己免疫障害における異常増殖細胞でありうる。
【0015】
特に例示的な態様では、細胞におけるcFLIPLまたはcFLIPS遺伝子の発現ポテンシャルは、
(a)細胞におけるcFLIPL mRNAまたはcFLIPS mRNAの存在を決定すること、
(b)細胞におけるcFLIPLまたはcFLIPSの存在を決定すること
によってアッセイされる。
【0016】
別の例示的な態様では、本発明は、
(a)IAPアンタゴニストを用いた処置に対する増殖細胞の感受性を、該細胞がcFLIPLまたはcFLIPSを発現できるかどうかを決定することによって決定する段階であって、それに従うと、cFLIPSを発現することができる細胞はIAPアンタゴニストに感受性である、段階、および
(b)細胞がcFLIPSを発現することができる場合に、IAPアンタゴニストで該細胞を処置する段階、あるいは
(c)細胞がcFLIPLを発現することができる場合に、IAPアンタゴニスト以外の剤もしくはIAPアンタゴニストに追加的な剤で該細胞を処置するか、またはIAPアンタゴニストの用量を増大させる段階
を含む、増殖性障害を患う患者を処置する方法である。
【0017】
本発明の方法は、ヒトまたは動物の身体に直接実施することができるが、そうする必要はない。むしろ、この方法は、生検などの試料を使用して実施することができる。したがって、別の例示的な態様では、本発明は、増殖性障害を患う患者を処置するために、かかる患者をIAPアンタゴニストで処置するかどうか、およびもし処置するならばどのくらいの用量で処置するかを決定するために、cFLIPLもしくはcFLIPSの存在、またはcFLIPLもしくはcFLIPSのmRNAの存在を検出する剤の使用を含む。本明細書下記にさらに説明するように、かかる剤は、例えば抗体またはヌクレオチドプローブでありうる。
【0018】
したがって他の例示的な態様における本発明は、本発明の方法を実施するためのキットであって、例えばcFLIPLもしくはcFLIPSの存在、またはcFLIPLもしくはcFLIPSのmRNAの存在を検出するための手段を含むキットも含み、該手段は、例えば、上記および本明細書下記などのcFLIPLもしくはcFLIPSの検出、またはcFLIPLもしくはcFLIPSのmRNAの検出に有用な剤である。
【図面の簡単な説明】
【0019】
【図1】IAP阻害剤は、自己分泌性TNF分泌とは独立して、SCCおよびHaCaTをデスリガンド(DL)介在性アポトーシスに対して感受性にすることを示す図である。A)HaCaT、MET1、またはA5RT3細胞を、100nM IAP阻害剤単独で、または10μg/ml TNFR2-Fcと組み合わせてのいずれかで30分間前処置し、次に3つ組のウェルにおいて表示した濃度のTRAIL(ng/ml)またはCD95L(U/ml)で刺激した。18〜24時間後に、クリスタルバイオレットアッセイによって細胞生存率を分析した。未刺激細胞は対照として用い、デスリガンド非依存性感受性との比較を可能にするために100%に設定した。4回の独立した実験のまとめを示し、エラーバーは平均の標準誤差(SEM)を示す。B〜E)IAP阻害剤は、CD95L介在性細胞死を増大させる。HaCaT細胞を、IAP阻害剤(100nM)単独で30分間予備刺激するか、あるいはCD95L(10U/ml)で4時間(B)、8時間(低二倍性分析;8時間)、またはカスパーゼもしくはPARP-1の切断のために(表示した時間)刺激/共刺激するかのいずれかとした。B)アネキシン-V-Cy5およびヨウ化プロピジウム(PI)で細胞を染色し、次にFACSによって分析した。C)細胞を8時間インキュベートし、続いてPIを含む低張緩衝液中に再懸濁し(材料および方法を参照されたい)、続いてFACS分析を行った。D)クローン形成アッセイのために、HaCaT細胞をIAP阻害剤(100nM)で30分間予備刺激し、続いてCD95L(2.5U/ml)で共刺激した。刺激の24時間後に、滅菌PBS中で数回洗浄後に培地を交換し、新しい培地を加え、細胞をさらに5または7日間培養し、続いてクリスタルバイオレット染色を行った。合計3回の独立した実験の代表的な一実験を示す。E)生化学分析のために、表示した時点についてTNF-R2-Fc(10μg/ml)の存在または非存在下の両方で、HaCaT細胞をIAP阻害剤(100nM)またはCD95L(2.5U/ml)の一方で、または両者を組み合わせてのいずれかで処置し、cIAP1、cIAP2、cFLIP、カスパーゼ8、PARP-1、FADD、およびRIP1の発現に関するウエスタンブロット分析を行った。βチューブリンは、内部負荷対照として用いた。代表的な2回の実験の一つを示す。
【図2】A)HaCaT、A5RT3、およびMET1細胞を表示時間にわたりIAPアンタゴニスト(100nM)で処置するか、またはTNF-R2-Fc(10μg/ml)で共刺激した。全ての細胞株におけるcIAP1およびcIAP2の発現ならびにMET1細胞におけるXIAPおよびカスパーゼ3の発現の十分な減少を、それぞれのタンパク質に特異的な吸収を有するウエスタンブロット分析によって確認した。βチューブリンを内部対照として用いた。4つの代表的な結果の一つを示す。B)様々な濃度のSMAC模倣体(6〜400nMのIAP)を、HaCaT、A5RT3、MET1およびSCC25細胞株に添加して細胞生存率を決定した。インキュベーションの終了時に、細胞をクリスタルバイオレットで染色し、生存率を決定した。C)クリスタルバイオレットで生存率を決定するために、Cと同様に細胞株をSMAC模倣体(6〜400nM)および10μg/ml TNF-R2-Fcと共に同時インキュベートした。D)HaCaTおよびMET1細胞を刺激しないか、またはIAPアンタゴニスト(100nM)で4時間刺激するかのいずれかとした。CD95/TRAIL-R1、およびTRAIL-R2の表面発現をデスレセプターに対するそれぞれの抗体で特異的に染色し、FACSによって視覚化した。二つの代表的な結果のうち一つを示す。
【図3】IAP阻害剤存在下のデスレセプター介在性細胞死は、カスパーゼ完全依存性でもなければ、カスパーゼ非依存性でもないことを示す。独特なカスパーゼ阻害剤zVAD-fmkによるカスパーゼ活性の阻害は、IAP阻害剤の存在下でHaCaT細胞のデスリガンド介在性細胞死を部分的に保護する。A)HaCaT細胞をzVAD-fmk(10μM;1時間)、ネクロスタチン(necrostatin)-1(50μM、1時間)、およびIAP阻害剤(100nM、30分間)で予備刺激または共刺激した。続いて細胞を表示濃度のTRAILまたはCD95Lで3つ組のウェルにおいて刺激した。18〜24時間のインキュベーション後に、材料および方法に示したように細胞生存率をクリスタルバイオレットアッセイによって分析した。7回の独立した実験についてSEMを示す。B)DNA凝縮の分析のために、HaCaT細胞をzVAD-fmk(10μM、1時間)またはIAP阻害剤(100nM、30分間)のいずれかで前処置した。続いて細胞をCD95L(5U/ml)でそれぞれ4時間または24時間刺激した。ヘキスト(Hoechst)-33342(5μg/ml)を37℃で15分間添加し、直後に透過(左)または蛍光(右)顕微鏡観察を行った。2回の独立した実験のうちの一つを代表として示す。
【図4】RIP1は、cIAP非存在下でデスリガンド介在性細胞死の重要な調節因子であることを示す図である。A)HaCaT、A5RT3、MET1、およびSCC25細胞の全細胞溶解物5μgのウエスタンブロットによって、FADD、cFLIP、カスパーゼ8、TRAF2、RIP1、cIAP1、cIAP2、およびXIAPの内因性タンパク質発現レベルを分析した。βチューブリンを、均等負荷の内部対照として用いた。三つの代表的な結果のうち一つを示す。B)zVAD-fmkによるカスパーゼ活性およびネクロスタチン-1によるRIP1キナーゼ活性の両方の阻害が、IAP阻害剤存在下でHaCaT細胞のデスリガンド介在性細胞死を完全に保護した。クリスタルバイオレットアッセイによってHaCaT細胞を3つ組のウェルにおいて、zVAD-fmk(10μM;1時間)、ネクロスタチン-1(50μM、1時間)およびIAP阻害剤(100nM、30分間)で別々に予備刺激し、続いてTRAIL(50ng/ml)またはCD95L(2,5U/ml)で刺激した。3回(TRAIL)または5回(CD95L)の独立した実験のSEMを示す。C)RIP1の安定的ノックダウンは、HaCaT細胞をデスリガンド誘導性細胞死から保護する。レトロウイルスによってハイパーランダム配列shRNA(HRS)またはRIP1特異的shRNAのいずれかをHaCaT細胞に形質導入し、ピューロマイシン(3μg/ml)で3日間選択した。RIP1のノックダウン効率は、RIP1に関するウエスタンブロット分析によって確認した。βチューブリンに対するAbを用いた膜の再プロービングを、タンパク質負荷の内部対照として用いる。D)C)に示したように形質導入されたHaCaT細胞を100nM IAP阻害剤およびTNF-R2-Fc(10μg/ml)で30分間予備刺激し、続いて表示濃度のTRAILまたはCD95Lで18〜24時間刺激し、クリスタルバイオレットアッセイによってアッセイした。3回(TRAIL)または4回(CD95L)の独立した実験のSEMを示す。E)C)に記載したように、形質導入されたHaCaTをIAP阻害剤(100nM)と共に30分間予備インキュベートし、続いてCD95L(0.5U/ml)で刺激した。24時間後に、培養液を除去し、細胞を無菌PBSで洗浄し、新しい培地を加えた。続いて細胞をさらに5日間培養し、続いてクリスタルバイオレット染色を行った。4回の代表的な独立した実験のうち一つを示す。
【図5】IAP阻害剤の存在下または非存在下でのリガンド誘導性レセプター結合型CD95複合体(DISC)または細胞内カスパーゼ8含有複合体(複合体II)の誘導を示す。A)CD95L-Fcで2時間刺激したMET1またはA5-RT3細胞からCD95 DISCを沈降させた。続いて、材料および方法に詳述したようにリガンドアフィニティー沈降を用いてCD95L DISC(左パネル)を沈降させた。溶解後のレセプター複合体の沈降(-)を、リガンドアフィニティー沈降(IP;+)と比べたときの内部特異性対照として用いた。続いて表示した分子について等量のDISC(CD95L-IP)またはカスパーゼ8相互作用タンパク質(複合体II)をウエスタンブロットによって分析した。等量の全細胞溶解物(TL)を同じゲルに負荷し、CD95L-IP、複合体II、およびTLの間でシグナル強度を比較可能にした。
【図6】cFLIPは、cIAP非存在下でデスリガンド介在性細胞死の重要な調節因子であることを示す。A)レトロウイルスによって、ハイパーランダム配列shRNA(HRS)またはcFLIP特異的shRNAのいずれかをA5-RT3細胞に形質導入し、ピューロマイシン(3μg/ml)で3日間選択した。cFLIPLおよびcFLIPSのノックダウン効率をウエスタンブロット分析によって確認した。RIP1、FADD、カスパーゼ8、およびβチューブリンに対するAbを用いた膜の再プロービングを、タンパク質負荷のための内部対照として用いる。3回の独立した実験の代表を示す。B)A)に示すように形質導入されたA5-RT3を100nM IAP阻害剤およびTNF-R2-Fc(10μg/ml)で30分間予備刺激し、続いて表示濃度のTRAIL(左パネル)またはCD95L(右パネル)で18〜24時間刺激し、クリスタルバイオレットアッセイによってアッセイした。C)カスパーゼ活性の阻害(zVAD-fmk;10μM)およびネクロスタチン-1(50μM)によるRIP1キナーゼ活性の阻害は、IAP阻害剤の存在下でA5-RT3細胞をデスリガンド介在性細胞死から完全に保護した。形質導入されたA5-RT3細胞を、3つ組のウェルにおいてzVAD-fmk(10μM;1時間)、ネクロスタチン-1(50μM、1時間)およびIAP阻害剤(100nM、30分間)で別々に予備刺激し、続いてCD95L(25U/ml)で刺激した。細胞の生存率をクリスタルバイオレットアッセイによって分析した。4回の独立した実験のSEMを示す。D)レトロウイルスによってHaCaT細胞にcFLIPLまたはcFLIPSまたは対照ベクターを形質導入した。cFLIPおよびカスパーゼ8について全細胞溶解物を分析した。βチューブリンは、タンパク質負荷のための内部対照として用いる。2回の追加的な独立した実験で同等の結果が得られた;E)D)に示されるような形質導入されたHaCaT細胞を、zVAD-fmk(10μM;1時間)、ネクロスタチン-1(50μM、1時間)、およびIAP阻害剤(100nM、30分間)または希釈液単独で予備刺激した。続いて3つ組のウェルにおいて細胞をCD95L(25U/ml)で刺激した。18〜24時間後に細胞生存率をクリスタルバイオレットアッセイによって分析した。7回の独立した実験のSEMを示す。F)クローン形成アッセイのために、形質導入されたHaCaT細胞をIAP阻害剤(100nM)で30分間予備刺激し、続いてCD95L(2,5U/ml)で共刺激した。刺激の24時間後に、滅菌PBSで数回洗浄してから培地を交換し、新しい培地を加え、細胞をさらに5または7日間培養し、続いてクリスタルバイオレット染色を行った。合計3回の独立した実験のうち代表的な一実験を示す。
【図7】cFLIPSではなくcFLIPLが複合体IIの形成を遮断することを示す。IAP阻害剤の存在下または非存在下でのリガンド誘導性レセプター結合型CD95複合体(DISC)または細胞内カスパーゼ8含有複合体(複合体II)の誘導。CD95L-Fcで2時間刺激したHaCaT細胞からCD95 DISCを沈降させた。続いて、材料および方法に詳述したようにリガンドアフィニティー沈降を用いてCD95L DISC(左パネル)を沈降させた。溶解後のレセプター複合体の沈降(-)を、リガンドアフィニティー沈降(IP;+)と比べたときの内部特異性対照として用いた。続いて、表示した分子について等量のDISC(CD95L-IP)またはカスパーゼ8-相互作用タンパク質(複合体II)をウエスタンブロットによって分析した。等量の全細胞溶解物(TL)を同じゲルに負荷して、IPとTLとの間のシグナル強度を比較できるようにした。
【図8】デスレセプター介在性細胞死の間のcIAPの役割を示す。cIAPは、完全長RIP1を有する質的に異なるDISCの形成を遮断する。このシグナル伝達プラットフォームは、カスパーゼ依存的と同様にカスパーゼ非依存的にも細胞死を誘導する。壊死性細胞死に重要な二次レセプター非依存性複合体IIもまた、開始因子カスパーゼ8およびカスパーゼ10を有する。カスパーゼ8によるRIP1切断は、複合体内のRIP1ダウンレギュレーションの仮説上の一メカニズムであることによって、RIP1依存的シグナル伝達を妨害する。または、RIP1は、ユビキチン化されるとDISCだけに動員される。
【図9】siRNAによるcFLIPのノックダウンは、抵抗性細胞をSmac模倣体およびTNFαの組み合わせに対して感受性にすることを示す。cFLIPのsiRNA介在性ノックダウンによってSmac模倣体、TNFαおよび両者の組み合わせに抵抗性の細胞株は感受性になる。A549およびIGROV-1細胞を24ウェルプレートに蒔き、一晩付着させた。翌日、細胞に100nMの対照siRNAまたはcFLIPターゲティングsiRNAのいずれかをトランスフェクトした。トランスフェクションの48時間後に、細胞を100ng/ml TNFα、100nM Smacペプチド模倣体または両者の組み合わせのいずれかで処理した。さらに24時間後に、細胞を回収し、FITCラベル化アネキシンVおよびヨウ化プロピジウムで染色した。アポトーシスをFACS分析によって定量した。
【発明を実施するための形態】
【0020】
発明の詳細な説明
添付の実験方法および結果の記載にさらに完全に説明および裏付けを行うように、本発明は、アポトーシス阻害タンパク質アンタゴニスト(IAPアンタゴニスト)を、単独、すなわち単独療法で、または他の抗増殖療法と組み合わせて、例えばTRAIL、CD95L、もしくはTNFα、またはそれらの関連アゴニストとの同時投与で用いた処置に対する細胞の感受性(または抵抗性)を予測する方法を提供する。別の述べ方をすると、本発明は、IAPアンタゴニストを用いた処置に対する特定の増殖性細胞障害の感受性または受容性を決定するアッセイ法に関する。細胞がIAPアンタゴニストに応答してアポトーシスを受ける場合、その細胞はIAPアンタゴニストに感受性である。本発明の方法は、どの細胞がアポトーシスを受けることによってIAPアンタゴニストに応答する見込みが高いかを予測するために有用である。これらの方法は、実験室または臨床状況のいずれかで使用することができる。
【0021】
本発明の方法は、様々な良性腫瘍または悪性腫瘍(癌)、良性増殖性疾患(例えば、乾癬、良性前立腺肥大、および再狭窄)、または自己免疫疾患(例えば、自己免疫性増殖性糸球体腎炎、リンパ増殖性自己免疫応答)を処置するためのIAPアンタゴニストの投与から利益を受け得る者を同定するために、例えば増殖性障害を患う患者をスクリーニングするために特に有用である。IAPアンタゴニストで潜在的に処置することができる癌には、非限定的に以下の一つまたは複数が挙げられる:肺腺癌、膵臓癌、結腸癌、卵巣癌、乳癌、中皮腫、末梢神経腫、膀胱癌、神経膠芽腫、黒色腫、副腎皮質癌、AIDS関連リンパ腫、肛門癌、膀胱癌、髄膜腫、神経膠腫、星状細胞腫、乳癌、子宮頸癌、慢性骨髄増殖性障害(例えば、慢性リンパ性白血病、慢性骨髄性白血病)、結腸癌、内分泌癌、子宮内膜癌、上衣腫、食道癌、ユーイング肉腫、頭蓋外胚細胞腫瘍、性腺外胚細胞腫瘍、肝外胆管癌、胆嚢癌、胃癌、消化管カルチノイド腫瘍、妊娠性トロホブラスト腫瘍、ヘアリー細胞白血病、ホジキンリンパ腫、非ホジキンリンパ腫、下咽頭癌、眼球内黒色腫、膵島細胞癌、カポジ肉腫、喉頭癌、白血病、急性リンパ芽球性白血病、急性骨髄性白血病、口唇癌、口腔癌、肝臓癌、男性乳癌、悪性中皮腫、髄芽腫、黒色腫、メルケル細胞癌、転移性扁平上皮性頸部癌(metastatic squamous neck cancer)、多発性骨髄腫および他の形質細胞腫瘍、菌状息肉腫およびセザリー症候群、骨髄異形成症候群、鼻咽頭癌、神経芽細胞腫、非小細胞肺癌、小細胞肺癌、中咽頭癌、骨肉腫および骨悪性線維性組織球腫を含む骨癌、卵巣上皮癌、卵巣胚細胞腫瘍、卵巣低悪性度腫瘍(ovarian low malignant potential tumor)、膵臓癌、副鼻腔癌、副甲状腺癌、陰茎癌、褐色細胞腫、下垂体腫瘍、前立腺癌、直腸癌、腎細胞癌、網膜芽細胞腫、横紋筋肉腫、唾液腺癌、皮膚癌、小腸癌、軟部組織肉腫、テント上原始神経外胚葉性腫瘍(supratentorial primitive neuroectodermal tumor)、松果体芽腫、精巣癌、胸腺腫、胸腺癌、甲状腺癌、腎盂尿管移行上皮癌、尿道癌、子宮肉腫、膣癌、外陰癌、ならびにウィルムス腫瘍および他の小児腎臓癌。
【0022】
本発明のいくつかの方法は、cFLIPLもしくはcFLIPS遺伝子の発現またはcFLIPLもしくはcFLIPS遺伝子発現ポテンシャルについて細胞をアッセイすることを伴う。cFLIPSアイソフォームを発現する細胞またはcFLIPSアイソフォームを発現するポテンシャルを有する細胞は、一つまたは複数のIAPアンタゴニストに感受性の傾向があり、すなわちIAPアンタゴニストで処置した場合にアポトーシスを受ける傾向がある。逆に、cFLIPLアイソフォームを発現する細胞またはcFLIPLアイソフォームを発現するポテンシャルを有する細胞は、一つまたは複数のIAPアンタゴニストに対して感受性が低い傾向があり、すなわち抵抗性である傾向がある。cFLIPLまたはcFLIPSの遺伝子発現は、当技術分野で公知の任意の手段によってアッセイすることができる。いくつかの態様では、遺伝子発現は、細胞のcFLIPLまたはcFLIPSタンパク質を検出することによってアッセイされる。ヒトcFLIPLおよびcFLIPSに関するアミノ酸配列は公知である(それぞれ配列番号:1および3)。cFLIPLまたはcFLIPSタンパク質(例えば分泌された、細胞内に含まれる、細胞表面に発現されたもの)は、例えば様々なイムノアッセイ(ELISA、ウエスタンブロット、フローサイトメトリー、ラジオイムノアッセイなど)を用いて検出することができる。cFLIPLおよびcFLIPS抗体は、入手可能である。例えば、抗cFLIPL抗体であるChemiconカタログ番号AB16963; Enzo Life Sciences カタログ番号ALX-804-127; Santa Cruz、カタログ番号SC7108、7111、8346、および7109を参照されたい。SまたはLアイソフォームに特異的でない抗体もまた、公的に入手可能である。当業者は、cFLIPおよびその各アイソフォームを同定するためにそのような抗体を使用する方法を知っている。例えば、細胞溶解物由来のタンパク質を、例えばSDS-PAGEによって単離し、次に抗cFLIP抗体を使用して、例えばELISAまたはウエスタンブロットで、例えばcFLIPSについて約25〜28KDであり、cFLIPLについて約55KDである分子量に基づき、cFLIPLまたはcFLIPSタンパク質を同定することができる。
【0023】
他の態様では、cFLIPL mRNAまたはcFLIPS mRNAを検出(例えばノーザンブロット、ドットブロット、RT-PCRなどによって)することにより遺伝子発現をアッセイする。ヒトcFLIPLおよびcFLIPSのDNA配列は公知である(それぞれ配列番号:2および4)。例えば、Goto et. al. J. Reproduction and Development, 2004, 50(5)549-555を参照されたい。公知の配列を使用してプローブを調製することができるし、また公知のアミノ酸配列に基づいて縮重プローブを作製することもできる。
【0024】
検出できる遺伝子発現レベルは、用いるアッセイに依存するであろうが、任意の検出可能なレベルのcFLIPLまたはcFLIPSのタンパク質またはmRNAを産生する細胞は、それぞれcFLIPLまたはcFLIPSアイソフォームを発現する細胞である。
【0025】
cFLIPLまたはcFLIPSの遺伝子発現について任意の細胞型をアッセイすることができる。細胞は、初代細胞(例えば患者から得られた生検の細胞)または細胞株由来でありうる。本発明は、ヒトまたは動物の身体での実施を必要としない。特に関心が持たれるのは、病的に増殖する細胞および疾患の症状を引き起こすかまたは誘導する細胞を含む、異常増殖細胞である。異常増殖細胞は、例えば癌、良性増殖性障害、および自己免疫性疾患で生ずる。
【0026】
cFLIP遺伝子を発現するように細胞を誘導することができ、方法は当業者に公知である;転写または翻訳レベルでのcFLIPLまたはcFLIPS発現の選択的誘導は、IAPに対する感受性または抵抗性に関して表現型を変化させることができる。
【0027】
cFLIPを発現している細胞は、SiRNAおよび当業者に公知の方法を用いてサイレンシングすることができ;cFLIPの選択的サイレンシングは、IAPに対する感受性に関する表現型の変化を招く。
【0028】
本発明のいくつかの態様は、細胞の、特に病的に増殖している細胞のアポトーシスを誘導することを含む。その方法は、インビトロまたはインビボで実施することができ、IAPアンタゴニストを用いた患者の処置を含むことができる。そのような処置は、単一のIAPアンタゴニストの投与、IAPアンタゴニストの組み合わせの投与、または一つまたは複数のIAPアンタゴニストおよび一つまたは複数の追加的な化学療法剤の投与を含みうる。多数の剤の投与は、同時または連続的でありうる。有用な化学療法剤には、非限定的に、アルキル化剤(例えばシクロホスファミド、メクロレタミン、クロラムブシル、メルファラン)、アントラサイクリン(例えばダウノルビシン、ドキソルビシン、エピルビシン、イダルビシン、ミトキサントロン、バルルビシン)、細胞骨格破壊剤(例えば、パクリタキセル、ドセタキセル)、エポチロン(例えば、エポチロンA、エポチロンB、エポチロンD)、トポイソメラーゼII阻害剤(例えば、エトポシド、テニポシド、タフルポシド)、ヌクレオチドアナログ前駆体アナログ(例えばアザシチジン、アザチオプリン、カペシタビン、シタラビン、ドキシフルリジン、フルオロウラシル、ゲムシタビン、メルカプトプリン、メトトレキサート、チオグアニン)、ペプチド系抗生物質(例えばブレオマイシン)、白金ベースの剤(例えばカルボプラチン、シスプラチン、オキサリプラチン)、レチノイド(例えば、オールトランスレチノイン酸)、ならびにビンカアルカロイドおよび誘導体(例えばビンブラスチン、ビンクリスチン、ビンデシン、ビノレルビン)が挙げられる。いくつかの態様では、化学療法剤には、フルダラビン、ドキソルビシン、パクリタキセル、ドセタキセル、カンプトテシン、エトポシド、トポテカン、イリノテカン、シスプラチン、カルボプラチン、オキサリプラチン、アムサクリン、ミトキサントロン、5-フルオロウラシル、またはゲムシタビンが挙げられる。
【0029】
IAPアンタゴニスト
本発明における使用のためのIAPアンタゴニストは、細胞性IAP(cIAP、例えばcIAP-1またはcIAP-2)またはX連鎖IAP(XIAP)などの一つまたは複数のIAPに結合してその活性を阻害する任意の分子である。いくつかの態様では、IAPアンタゴニストは、XIAP、cIAP-1、またはcIAP-2に優先的に結合する。いくつかの態様では、IAPアンタゴニストは、Smac(第二ミトコンドリア性カスパーゼ活性化因子)の模倣体であり、特定の態様では、Smac模倣体は、成熟SmacのN末端の4個のアミノ酸(Ala-Val-Pro-Ile)、またはより一般的にはAla-Val-Pro-Xaa(ここで、Xaaは、Phe、Tyr、Ile、またはVal、好ましくはPheまたはIleである)の模倣体またはペプチド模倣体である。
【0030】
本発明のいくつかの態様では、IAPアンタゴニストを含む薬学的組成物は、ヒトまたは動物対象に投与される。薬学的組成物は、典型的には薬学的に許容される担体または希釈剤を含み、全身、局所、または経口経路を含む経路によって従来方式で投与することができる。例えば、投与は、非限定的に、非経口、皮下、静脈内、筋肉内、腹腔内、経皮、経口、口内、膣内、または眼球経路、吸入による、デポー注射による、または植込みによるものでありうる。特定の投与様式は、適応症および投与される特定の化合物を含めたその他の要因に依存すると考えられる。投与されるべき化合物の量は、治療的に有効な量である。投与される用量は、処置される対象の性質、例えば処置される特定の患者、年齢、体重、健康状態、もしあれば同時に行う処置の種類に依存すると考えられる。処置の回数は、当業者(例えば臨床医)が容易に決定することができる。
【0031】
本発明のいくつかの態様には、cFLIP遺伝子発現の評価および分析を行うためのキットが含まれる。そのようなキットには、例えばcFLIPLまたはcFLIPSの配列決定の前のQiagen EPITECT(登録商標)Bisulfite Conversion Kit、抗体、プローブ、検出可能なマーカーなど、ならびに上記のIAPアンタゴニスト処置の評価および分析を行うために必要な試薬、ゲル、装置、分析ツールなどが含まれる。
【0032】
本発明には、IAPアンタゴニスト、キット、システムを市販するための方法、およびIAPアンタゴニストを使用した処置の成功見込みを決定するのに有用なバイオマーカーを使用するための方法が含まれる。一態様では、そのような方法、システムおよびキットの有効性に関するデータは、IAPアンタゴニストでのヒト臨床試験を行う承認を求めるための関係書類の一部として規制当局に提出されて、例えば、除外基準もしくは組み入れ基準が制定されるか、または臨床試験データの評価が容易になる。そのようなデータを規制当局に提出して、IAPアンタゴニストを使用した処置に関連するバイオマーカーを使用するための方法、システム、およびキットを市販するための承認申請を裏付けることができる。例えば、そのようなデータを、新医薬品承認申請(New Drug Approval Application)(NDA)の一部として米国食品医薬品局(United States Food and Drug Administration)(FDA)に提出することができる。
【0033】
本発明の様々な態様には、IAPアンタゴニストを用いた処置に応答してcFLIPLまたはcFLIPSを発現することができる細胞の応答性についての情報を提供すること、およびIAPアンタゴニストを含むそのような薬学的組成物に関心を持ちうる個人にこの情報を普及させることが含まれる。そのような個人には、増殖性障害を有する個人、そのような障害を処置する医療関係者、および薬剤を調剤する、または流通させる個人が含まれる。
【0034】
ヒト臨床試験の承認が得られた場合、前記の情報は、増殖性障害を示しているヒト対象への薬学的組成物の有効性を裏付けるデータ、ならびに投薬情報および細胞毒性データなどの他のデータと共に、IAPアンタゴニスト、およびIAPアンタゴニストを含む薬学的組成物を市販する承認を求めて規制当局に提出することのできる関係書類に添付することができる。
【0035】
態様には、承認が得られた後にIAPアンタゴニストまたはIAPアンタゴニストを含む薬学的組成物を市販するための方法も含まれる。そのような方法では、cFLIPLまたはcFLIPSを発現することができる細胞においてIAPアンタゴニストが有効である可能性があるという事実に関する情報を、例えば、医師、薬剤師、処方者、保険提供業者、流通業者、患者など、またはこれらの人々の組み合わせに普及させることができる。なお他の態様では、情報は、将来の患者および/または将来の処方者、および/または将来の流通業者に普及させることができる。
【0036】
情報は、非限定的に、消費者への直接広告、テレビ広告、ラジオ広告、新聞広告、印刷物による広告(例えば、パンフレット、チラシ、郵便はがき、手紙など)、ウェブサイトを経由したまたはウェブサイト上の広告(例えば、「バナー」アドオンウェブサイトを使用)、看板広告、ダイレクトメール、Eメール、伝聞、およびそれらの任意の組み合わせを含めた、当技術分野で公知の任意の方法により普及させることができる。
【0037】
他の態様では、データは、ユーザがアクセス可能なデータベースに保存することができる。データベースに保存されたデータには、例えばIAPアンタゴニストを使用した処置に関連するバイオマーカーを使用する方法、システム、およびキットを検査する間に生成されたデータを含むIAPアンタゴニストまたは薬学的組成物に関係する任意のデータ、IAPアンタゴニスト、薬学的組成物、方法、システムおよびキットの安全性および/または有効性に関する情報、投薬情報、該化合物を使用して処置することのできる障害の一覧、一つまたは複数の規制当局からの承認情報、流通業者の情報、処方情報、ならびにそれらの組み合わせが含まれうる。
【0038】
様々な態様にはまた、IAPアンタゴニストを使用した処置に関連するバイオマーカーを使用するための、IAPアンタゴニスト、薬学的組成物、方法、システムおよびキットを市販するためのシステムが含まれ、該システムには、IAPアンタゴニストを使用した処置に関連するバイオマーカーを使用するための、方法、システム、およびキットに関する情報、ならびに方法、システム、およびキットの有効性についてのデータを含む、上記のデータベースなどのデータベースが含まれる。そのような態様では、データベースに保管される情報は、例えば経営者、販売担当者、マーケティング担当者およびそれらの人々の組み合わせなどの選ばれた個人だけがアクセス可能でありうる。システムにはまた、データベースに保管された情報のサブセットが含まれてもよく、それは、例えば、医師、薬剤師、処方者、保険提供業者、患者、流通業者およびそれらの人々の組み合わせなどの選択された個人ではない任意の者でありうる、選択されていない個人に普及される。普及は、上記の当技術分野で公知の任意の普及方法によって行うことができる。
【0039】
データのサブセットには、データベースに保管された任意の情報が含まれてもよく、方法、システム、およびキットを市場性のあるものにすると考えられる情報、例えば、安全性および/または有効性のデータ、該化合物を使用して処置することができる障害の一覧、薬学的組成物中の薬剤、成分の一覧または活性剤の投与の潜在的副作用、一つまたは複数の規制当局からの承認情報、流通業者の情報、処方情報およびそれらの組み合わせが含まれうる。ある種の態様では、選択された個体は、データのサブセットに提供された情報を選択および/または承認することができる。
【0040】
上記態様のそれぞれにおいて、提供および/または普及された情報ならびにデータベースに保存されたデータには、さらに、別の抗自己免疫剤または抗増殖剤を含みうる併用療法用の組成物、方法、またはプロトコールが含まれうる。
【0041】
本開示に引用された全ての特許、特許出願、および参照は、参照により本明細書に明確に組み入れられる。上記開示は、全般的に本発明を説明する。さらに完全な理解は、以下の具体例を参照することによって得ることができ、それらの例は、例示のためだけに提供され、本発明の範囲を限定する意図はない。
【0042】
実験の方法およびデータを示す添付の項において本発明をさらに十分に説明する。
【0043】
1. デスリガンド(DL)介在性細胞死に対して感受性にするIAP阻害剤
最初に本発明者らは、記載されたIAP阻害剤である化合物A(IAP阻害剤)を用いて異なる角化細胞株および扁平上皮癌(SCC)細胞の感受性を特徴づけた22。細胞を100nMの本化合物と共に異なる時間の間、インキュベートしたとき、cIAP1、さらにcIAP2の急速分解が検出された(図2A)。cIAP1の欠如は24時間にわたり継続したが、cIAP2は、A5-RT3において、そしてそれよりも低度であるがHaCaT細胞において再発現された。MET1細胞では、TNF-R2-Fcを使用した自己分泌TNFシグナル伝達阻害とは無関係に、XIAPの30kDaのカスパーゼ切断断片が24時間後に検出されたが、HaCaTおよびA5-RT3(図4Aと比較)は、タンパク質レベルでXIAPを発現しない17,26。これらのデータは、IAPの欠如がcIAP1およびcIAP2の急速分解を誘導し、TNF非依存的にXIAPのカスパーゼ介在性切断を導くことを示した(図2A)。用量反応実験(6〜400nMのIAP阻害剤)から、HaCaT細胞に加えて、派生した転移細胞株A5-RT327が、最大400nMまでIAP阻害剤単独を用いた処置に大いに抵抗性であることが明らかとなった。対照的に、二つの遺伝的に異種のSCC細胞株(SCC25と同様にMET1)は、IAP阻害剤単独を用いた処置に部分的に感受性であった(20〜25%の細胞死;補足の図1B)。10μg/ml TNF-R2-Fcとの同時インキュベーションは、これらの細胞における細胞死を減少させたが、それは、TNFの産生および分泌の自己分泌ループがこれらの細胞株における細胞死を担うことを示している22。本発明者らは次に、これらの腫瘍細胞におけるデスリガンドの感受性に関するIAP阻害剤の影響を検討した。A5-RT3ではなく、HaCaTおよびMET1細胞は、TRAILまたはCD95L介在性細胞死に対してTNF依存的に劇的に感受性になった(図1A)。細胞死は、4時間後のアネキシン-Vの外部移行(図1B)またはデスリガンドと共に8時間インキュベーション後の低二倍性分析(図1C)によって決定されたようにアポトーシスによって起こった。さらに、IAP阻害剤とのインキュベーションは、クローン形成性生存を減少させた(図1D)。本発明者らが、TRAILまたはCD95Lで処置後のカスパーゼ活性化を調べたとき、本発明者らは、IAP阻害剤の存在下でそれぞれのデスリガンドを用いて刺激してから4時間以内に、開始因子であるカスパーゼ8およびエフェクターであるカスパーゼ3の活性化が増大し、それがエフェクターカスパーゼの標的であるPARPの切断増加を招くことを見出した(図1E)。独立した実験システムでこれらのデータを確認するために、本発明者らはcIAP1を欠如したMEFを使用した。IAP阻害剤を使用したヒト細胞でのデータに一致して、cIAP1ノックアウトMEFはCD95介在性細胞死に感受性となった。さらに、cIAP1 MEFへのcIAP1の誘導性再構成によって、デスリガンドに対する抵抗性が完全に回復した。まとめると、本発明者らのデータは、cIAPの細胞レベルがCD95LまたはTRAIL抵抗性の維持に重要であることを実証している。
【0044】
2. カスパーゼ完全依存性でもなければ、カスパーゼ非依存性でもなく、かつRIP1を必要とする、IAP阻害剤存在下のデスレセプター介在性細胞死
デスレセプター介在性アポトーシスは、DISC活性化カスパーゼ8によって開始される6,28。カスパーゼ活性化がIAP阻害剤存在下でデスレセプター介在性細胞死に重要であるかどうかを研究するために、本発明者らは、次に広域スペクトルカスパーゼ阻害剤zVAD-fmkの存在下で細胞死を研究した。多数のグループによって報告されたように、TRAILまたはCD95Lで細胞を24時間刺激したとき、zVAD-fmkは細胞死を完全に遮断した。しかし、IAP阻害剤存在下のTRAIL介在性またはCD95L介在性細胞死は、異なる濃度のデスリガンドでzVAD-fmkによって部分的に遮断されただけであった(図3A)。これらのデータは、カスパーゼ非依存型の細胞死が、IAP阻害剤の存在下でデスレセプターの刺激によって誘導されることを示唆した。これらの細胞における細胞死の形態学的特徴をさらに特徴づけるために、本発明者らは蛍光顕微鏡研究を行った。膜泡状突起、DNA凝縮および断片化を示している典型的なアポトーシス細胞の数増加は、CD95L処置から4時間以内ではカスパーゼ阻害剤の非存在下で処置された細胞のみに検出可能であった(図3B、左パネル)。これらの初期時点で、zVAD-fmkは、IAP阻害剤が存在する場合にもまた細胞形態またはDNA断片化を完全に保護した。しかしながら、zVAD-fmkは、処置の24時間後に細胞を細胞死から保護しなかったのに対して、zVAD-fmkは、IAPが存在する場合は完全な保護を達成した(図3B、右パネル)。これらのデータは、IAPが、デスレセプターの刺激によって誘導されるカスパーゼ非依存性遅延型細胞死から保護することができることを示した。以前の研究は、CD95がキナーゼRIP1を介してカスパーゼ非依存型細胞死を活性化することを明らかにしたが、RIP1の下流の標的は依然として未知である29〜31。IAPはRIP1のユビキチン化を妨害することができるので24、本発明者らは、次に本発明者らの実験システムにおけるRIP1の役割を検討した。興味深いことに、IAP阻害剤の感受性化効果に高度に抵抗性であることが証明されたA5-RT3細胞において、RIP1レベルは最低であった(図4A)。ネクロスタチン-1などのネクロスタチンは、RIP1のキナーゼ活性を特異的に遮断することが示された32。カスパーゼ阻害は、TRAILまたはCD95L介在性細胞死から部分的に保護しただけであったが、zVAD-fmk保護細胞へのネクロスタチン-1の添加は、cIAPの非存在下で生存率を完全に回復させた。対照的に、ネクロスタチン-1単独は、カスパーゼ阻害剤非存在下でデスリガンド介在性細胞死からの保護に無効であった(図4B)。これらのデータは、cIAPの欠如がカスパーゼおよびRIP1の組み合わせ阻害によってのみ遮断することができるデスレセプター介在性シグナルを明らかにしうるという事実をほのめかした。RIP1の役割にさらに直接的に取り組むために、本発明者らは、安定的なshRNA発現を使用して、RIP1レベルが減少した細胞株を作出した(図4C)。対照感染細胞は、IAP阻害剤によってTRAILまたはCD95L介在性細胞死に対して感受性にされたが、この感受性化は、生存率アッセイ(図4D)およびクローン形成アッセイ(図4E)によって決定されたように、RIP1抑制細胞では主として打ち消された。まとめると、これらのデータは、cIAPが抑制された場合は常に、CD95介在性細胞死およびTRAIL介在性細胞死が細胞死に不可欠なRIP1依存性シグナル伝達経路を利用することを示している。ノックダウンのデータをさらに裏付けるために、本発明者らは、RIP1ノックアウトMEFをそれらの野生型対照細胞と比較した。RIP1欠損細胞は、cIAPの非存在下でTRAILまたはCD95L介在性細胞死の誘導への感受性が低かった。まとめると、これらのデータは、RIP1がIAPの非存在下でデスレセプター介在性細胞死に決定的に関与することを実証している。
【0045】
3. DISCへのRIP1動員を負に調節し、レセプター非依存性複合体IIの形成増加を可能にする、IAP
cIAPがどのようにデスレセプター介在性細胞死を負に調節するかの分子メカニズムを特徴づけるために、本発明者らは、次に本発明者らの実験システムにおけるネイティブの細胞死誘導性シグナル伝達複合体(DISC)の成分および形成を特徴づけた。さらに、最近記載されたレセプター非依存性複合体IIの形成10をIAPの非存在下で検討した。本発明者らは、IAP阻害剤(MET1)の感受性化効果にどちらも応答性であった二つの細胞株におけるCD95L誘導複合体を分析することを選び、それを、IAPの欠如によって感受性化されなかった細胞(A5-RT3)と比較した。最初の実験は、IAP阻害剤がデスレセプターの発現に影響を与えなかったことを明らかにした(図2Dおよびデータは示さず)。本発明者らは、リガンドアフィニティー沈降物(DISC)およびカスパーゼ8免疫共沈物(複合体II)を各条件で同量の総細胞溶解物と比較した場合に、カスパーゼ8関連複合体(複合体IIと呼ぶ)中にCD95が非存在であることを最初に確認した(図5、TL)。HaCaT角化細胞33および他の細胞型34におけるTRAIL DISCに関する本発明者らの以前のデータに一致して、そしてデスリガンドに対する全体的な感受性に一致して(図1と比較)、本発明者らは、両細胞型におけるCD95 DISCへのcFLIP、カスパーゼ8、カスパーゼ10、FADD、およびRIP1の刺激依存性動員を検出した(図5、左パネル、レーン3〜4、7〜8)。著しいことに、カスパーゼ8のより高い発現にかかわらず、カスパーゼ8のDISC結合は、A5-RT3の方が弱く、これは、CD95についてのウエスタンブロットによって決定されたような、より低レベルのCD95表面発現によって潜在的に説明された(左および右パネル、CD95)。これら二つの細胞株のDISCを比較して、A5-RT3細胞は、cFLIPの類似の動員を示したが、RIP1、FADD、カスパーゼ8、およびカスパーゼ10の動員はA5-RT3の方が低下していた(図5、左パネルと比較、レーン1〜4;5〜8)。驚くことに、RIP1の動員は、cIAPの非存在下でMET1細胞と同様にA5-RT3細胞のCD95 DISCにおいて堅調に増加した(図5、左パネル、レーン3〜4)。対照的に、A5-RT3細胞においてRIP1の動員増加は検出可能であったが実質的にさらに弱まった(図5、左パネル、レーン7〜8)。そのうえ本発明者らは、MET1細胞ではなくA5-RT3細胞においてcIAP2の特異的刺激依存性動員を検出した(図5、左パネル、レーン3〜4、7〜8)。カスパーゼ8関連タンパク質(複合体II)の免疫共沈によってレセプター非依存性的なDISC成分の相互作用を調べて、本発明者らは、両細胞株におけるFADD、カスパーゼ10、cFLIP、およびRIP1とカスパーゼ8との刺激依存性相互作用を検出した。最初の結果から、カスパーゼ阻害剤の存在下でDISCと同様に複合体IIが強く安定化されたことが確認された(データは示さず)。したがって本発明者らは、刺激の間に、本発明者らの実験において検出可能なカスパーゼ依存性切断の程度を変化させうるzVAD-fmkの存在下でこれらの実験を行った。興味深いことに、RIP1、FADD、cFLIPLおよびcFLIPL p43の大きな増加がカスパーゼ8に関連して検出された(図5、右パネル、レーン19〜20、23〜24)。これらのデータは、cIAPの発現抑制時に、細胞質において複合体IIが漸増的に形成されることを示唆した(図5右パネル)。興味深いことに、本発明者らは、A5-RT3の複合体II中にcIAP1ではなくcIAP2を見出したが、これは、もしかするとMET1細胞においてcIAP2の発現レベルがより低いことから起こったのかもしれない(図4Aと比較)。まとめると、本発明者らのデータによって、cIAPの欠如がCD95 DISCへのRIP1動員を促進し、RIP1-FADD-カスパーゼ10およびカスパーゼ8を含有する複合体IIの形成増加を可能にすることが示唆された。さらに、IAP阻害剤の感受性化効果に抵抗性の細胞は、IAP阻害剤を用いた処置にかかわらず複合体II内にcIAP2が存在することを実証したが、これは、cIAP2がIAP阻害剤の存在下で(ゆえにcIAP1の欠如下で)細胞死経路のさらなる活性化を遮断するために十分でありうることを示唆している。
【0046】
4. IAPの非存在下でデスリガンド介在性細胞死への抵抗性に異なる寄与を果たす、cFLIPアイソフォーム
デスレセプター介在性細胞死に対するIAP阻害剤介在性感受性化に感受性であった細胞は、DISC内でカスパーゼ8を高効率で動員および切断した一方で、cFLIPの動員は同等であり、先に示唆されたように異なるcFLIPアイソフォームがDISCに高親和性であることを示した33。これに関連して、cFLIPがTRAILまたはCD95L細胞死抵抗性の重大な一決定因子であることが広く受け入れられている(総説35参照)。したがって本発明者らは、次にcFLIPがデスリガンドに対するIAP阻害剤介在性感受性化への抵抗性を担うかどうか試験した。このために本発明者らは、主として、XIAPを欠如するIAP阻害剤抵抗性SCC細胞株A5-RT3を選択し、cFLIPに対するshRNAの安定的発現によってcFLIPをダウンレギュレーションさせた。ウエスタンブロット分析によって、cFLIPLおよびcFLIPSの両方の効率的なダウンレギュレーションが確認された(図6A)。興味深いことに、A5-RT3細胞におけるcFLIP欠如は、IAP阻害剤存在下でのTRAIL介在性またはCD95L介在性細胞死の増加を招いた(図6B)。しかし、HaCaT細胞でのデータと対照的に、IAP欠如による感受性化は、完全にカスパーゼ依存性であった一方で、ネクロスタチン-1はこれらの細胞において無効であった(図6C)。これらのデータから、デスリガンド介在性細胞死に対する感受性の細胞型高特異性IAP調節が確認された。さらに重要なことには、本発明者らのデータは、RIP1およびcFLIPがこの調節に重大であること、ならびにA5-RT3では、cFLIPアイソフォームがIAP阻害剤介在性細胞死を遮断することができることを示した。cFLIPのシグナル伝達能に関して、異なるcFLIPアイソフォームの機能と同様にcFLIPLの切断断片の機能についていくつかの対立する結果がある(総説36参照)。デスレセプター介在性細胞死についてのIAP阻害剤介在性感受性化のメカニズム、さらに具体的にはcFLIPの影響に取り組むために、本発明者らは、異なるcFLIPアイソフォームを発現している安定的細胞株を生成した(図6D)。これらの実験について本発明者らは、内因性cFLIPが低レベルであること37およびタンパク質レベルでXIAPを欠如していることから、HaCaT角化細胞を選択した17。予期されたように、cFLIPLと同様にcFLIPSは、CD95LまたはTRAIL介在性細胞死(図6E、パネル2)、デスレセプター介在性アネキシン-Vの外部移行、およびDNAの低二倍性(データは示さず)を効率的に遮断した。興味深いことに、IAP阻害剤単独に対する感受性は、cFLIPLではなくcFLIPSを発現している細胞において堅調に増加した(図7E、パネル6)。興味深いことにcFLIPSは、IAP阻害剤存在下でデスレセプター介在性細胞死から保護することができず(図6E、パネル7)、その細胞死はzVAD-fmkによって保護されなかった(図6E、パネル8)。しかしながら、図3に示される本発明者らのデータに一致して、両cFLIPアイソフォームは、アポトーシス細胞死の初期特徴を遮断した(データは示さず)。しかしながら、cFLIPS発現HaCaTにおけるIAP阻害剤存在下のCD95L介在性細胞死は、ネクロスタチン-1によって24時間完全に保護され、それは、RIP1依存性シグナル伝達の寄与を示した(図6E、パネル9)。まとめると、本発明者らのデータは、RIP1動員を介してデスレセプターによって活性化されるカスパーゼ非依存型細胞死を、cFLIPSではなくcFLIPLが遮断できることを示唆している。
【0047】
5. 複合体IIへのRIP1のCD95誘導性動員に異なる影響を与える、cFLIPアイソフォーム
この現象の潜在的メカニズムを明らかにするために、本発明者らは次に、cFLIPLおよびcFLIPS発現HaCaT細胞においてCD95 DISCと同様に複合体IIを沈降させ、対照感染HaCaTの結果と比較した。初回の結果は、DISCと同様に複合体IIが、カスパーゼ阻害剤の存在下で堅調に安定化されることを確認した(データは示さず)。カスパーゼ阻害は、細胞死からの保護に十分でなかったことから、本発明者らは、zVAD-fmkが存在してもこれらのデスリガンド誘導性複合体における差は相変わらずモニタリング可能であるという仮説を立てた。したがって本発明者らは、zVAD-fmkの存在下で、異なるcFLIPアイソフォームを発現しているHaCaT細胞をIAP阻害剤の存在下または非存在下で、CD95Lで刺激した。A5-RT3およびMET-1について図5に示した本発明者らのデータに一致して、cIAPが欠如している場合に対照細胞のDISCにおいてRIP1の劇的な増加が検出された(図7、左パネル、レーン1〜4)。TRAIL DISC33についての本発明者らの先の報告に一致して、cFLIPLと同様にcFLIPSは、CD95 DISCへのRIP1動員を抑制した。リンパ腫細胞における先の報告と適合することに、cFLIPLは、カスパーゼ8 p43/41の動員増加を導いたが、一方でcFLIPSは、DISCにおけるカスパーゼ8切断を完全に遮断した38。複合体IIにおいて、カスパーゼ8と共に免疫共沈したRIP1、FADD、およびcFLIPL(プロ型およびp43)の実質的な増加があった(図7、右パネル、レーン1〜4)。対照的にcFLIPLは、複合体IIの形成を遮断した。興味深いことに本発明者らは、デスリガンド刺激の非存在下でcFLIPS発現細胞の複合体II形成を再現性よく検出したが(図7、右パネル、レーン6)、これは、これらの細胞においてcIAP非存在下で複合体IIが自然形成したことを示している。さらに、CD95Lに刺激された複合体IIの形成は、cFLIPL存在下の複合体IIよりも非常に強かったことから、cIAPがcFLIPS細胞中で非存在である場合はいつもCD95Lに応答した細胞死が増加することの説明を与えている。
【0048】
考察
本発明において、本発明者らは、IAP阻害の状況でのデスレセプター介在性細胞死のメカニズムを検討した。本発明者らは、IAP阻害剤が自己分泌性TNF阻害にほとんど依存せずにSCC細胞をDR介在性細胞死に対して劇的に感受性にすることを示す。それどころか、IAP阻害剤は、カスパーゼ8依存型およびRIP1依存型細胞死の両方を増加させる。本発明者らが驚いたことには、異なるcFLIPアイソフォームは、IAPの存在に応じて別個の阻害能を有する。cFLIPLおよびcFLIPSは、IAPの存在下でデスレセプター介在性アポトーシスを同様に阻害するが、cFLIPLはRIP1依存性細胞死に加えてカスパーゼ8依存性細胞死を遮断し、cFLIPSはカスパーゼ8依存性アポトーシスだけを妨害するが、RIP1依存性細胞死の保護には著しく非効率的であった。本発明者らのデータは、異なるcFLIPアイソフォームが、cIAPの非存在下でのみ明らかな別個のシグナル伝達能を有することを初めて示している。cIAPのこの機能は、腫瘍療法の障壁としてのアポトーシス抵抗性にとってのみかかわりがあるというわけではなく、細胞死の様式が最も重要であるウイルス感染または腫瘍免疫の際に関係しうる25。
【0049】
本発明は、TRAIL-R1、TRAIL-R2、およびCD95デスレセプターによって活性化されるシグナル伝達経路の理解についてのいくつかの重要な発見に寄与するものである。第一に、本発明者らは、−細胞表面上でデスレセプターの調節が顕著に非存在の状況で(データは示さず)− cIAPの欠如がTRAILまたはCD95L誘導性細胞死への劇的な感受性化を導くことを予想外に見い出している。本発明者らは、数分間以内にIAPの分解を誘導する薬理学的阻害剤(IAP阻害剤)で処置されたヒトSCC腫瘍細胞において、またはその代わりに異なるcIAPを欠如したMEF遺伝的モデルを使用してのいずれかで、これらの局面を研究した。本発明者らのデータは、TRAILまたはCD95Lシグナル伝達経路が、示唆されたTNFシグナル伝達自己分泌性ループにほとんど依存せずにcIAPによって顕著に調節されることを実証しているが、それは、デスレセプターシグナル伝達経路におけるcIAPの独立した機能を示している20〜24。重要なことには、使用された細胞の一部がXIAPをタンパク質レベルで完全に欠如していることから、本発明者らのデータは、XIAPの機能とは独立して作動する分子メカニズムを示唆している17。IAP阻害剤への感受性化もまた、XIAP MEFで得られ、そのことはさらに、CD95およびTRAIL-R介在性細胞死の調節のためのcIAPのこの新規な機能を裏付けている。
【0050】
本発明者らが、この現象の分子メカニズムを研究したとき、本発明者らは、キナーゼRIP1をデスレセプター介在性細胞死に果たすcIAPの負の調節の役割に重要であると確認した。RIP1は、デスレセプターによるNF-κB活性化におけるその関連性が何年にもわたり知られている39。しかしながら、RIP1が細胞死を遮断する能力は、間接であって、デスレセプター誘導性NF-κB活性化の欠如によって仲介されると見なされた。他のグループは、TNFまたはCD95刺激に応答するプログラム壊死を先に検討していた40。
【0051】
過剰発現研究により、FADDのDDが壊死誘導に必要である一方で、FADDのDEDはカスパーゼ活性化に必要とされることが示唆された41。さらにHollerらは、FADD欠損Jurkat T細胞におけるCD95誘導性壊死を示した29。したがってFADDは、デスレセプターのトリガー後にアポトーシス細胞死経路および壊死細胞死経路の両方に関与するが、FADDがこれら二つのシグナル伝達経路についての重要な分子スイッチに相当するかどうかは不明である。酸性の細胞外条件が少なくともTRAILに応答したRIP1依存性細胞死に有利に働くとはいえ、FADDおよびRIP1を発現している細胞における壊死性細胞死の理由ははっきりしないままであった42。今回、本発明者らのデータは、cIAPが壊死性細胞死経路を負に調節すること、およびRIP1がDISCレベルでCD95またはTRAIL-R誘導性細胞死に必要であることを示唆している。この結論は、RIP1ノックダウン、RIP1欠損MEF、およびcIAPの非存在下で大量のRIP1を含有する腫瘍細胞におけるネイティブCD95複合体の沈降を用いた本発明者らのデータに基づく。さらに、本発明者らのデータは、膜結合型CD95複合体(CD95 DISC)へのRIP1動員がcIAPによって劇的に減少するが、RIP1の合計細胞レベルは影響を受けないことを実証している。特異的RIP1キナーゼ阻害剤であるネクロスタチン-1によって、本発明者らは、RIP1キナーゼ活性の必要性をさらに研究できた32。本発明者らのデータは、カスパーゼが遮断されたときはいつも、RIP1キナーゼが重要なタンパク質となり、カスパーゼおよびRIP1キナーゼの二重阻害がCD95介在性またはTRAIL介在性細胞死の細胞生存率の回復を可能にすることを明らかに示している。本発明者らは、汎カスパーゼ阻害剤であるzVAD-fmkの存在下であってもDISC中でRIP1のDD含有断片が強く濃縮することを検出している(データは示さず)。不幸にも、これまでのところ入手可能な全ての抗体は、DDにおけるエピトープを認識し、このことがRIP1のキナーゼドメインを有するN末端断片の検出を妨げている。タグ付きタンパク質またはキナーゼドメインに対する抗体を使用した将来的な研究は、RIP1の切断が、a)細胞死を誘導するためにDDによる阻害からキナーゼ活性を解放することを導くか、またはb)機能的RIP1を全体的に除去する有効なメカニズムであるかどうかをさらに解明すると考えられる。
【0052】
本発明者らは、IAPの欠如がRIP1依存的にCD95LまたはTRAILへの劇的な感受性化を導くことを見出している。本発明者らは、もはやデスレセプターに結合していないカスパーゼ8-FADD-cFLIP-RIP1含有細胞質複合体(複合体II)を検出している。この複合体の形成は、cIAPによって阻害される。驚くことに、異なるアイソフォームのカスパーゼ8抑制因子cFLIPは、cIAPが非存在のときはいつも差次的な効果を有する。全てのcFLIPLアイソフォームは、cIAPの存在下で細胞死から保護するが、cFLIPSではなくcFLIPLだけがネイティブ複合体IIの形成を遮断することができ、cIAPの非存在下で効率的な細胞死を可能にする。したがって本発明者らのデータは、CD95またはTRAILデスレセプター下流の細胞死シグナル伝達に関連する重要な細胞内タンパク質複合体を同定している。
【0053】
本発明者らの研究の別の重要な発見は、少なくともFADD、カスパーゼ8、カスパーゼ10、RIP1、および異なるアイソフォームのcFLIPを含有するレセプター非依存性複合体(複合体II)の同定である。最近の報告は、CD95またはTRAIL刺激後にそのような複合体を同定している10,11。本発明者らの実験において、複合体IIの形成およびカスパーゼ8とRIP1との結合は、IAP阻害剤に非感受性の細胞では抑制される。顕著には、これらの細胞においてcIAP2は高発現され、IAP阻害後に動力学的に調べた場合、未処置細胞の定常状態レベルまで急速に再発現される(補足の図1A)。このパターンのcIAP2発現は、DISCと結合したcIAP2の検出に加えて、これらの細胞の複合体IIにおけるRIP1の検出減少と同時に起こったことから(図5と比較)、cIAP1およびcIAP2がIAP阻害剤による阻害に差次的な感受性を有するか、または全体で差次的に調節されうることを示唆している。自己分泌性TNF分泌がcIAP2のデノボ発現を導きうることを認知できる。これに関連して、本発明者らの細胞モデルにおいてcIAP1はIAP阻害剤によって長期的に抑制されるが、cIAP2は、TNF介在性細胞死の調節に不要であるが、NF-κBによって堅調に誘導される17。したがって、両方のcIAPは別個の役割を有する可能性があり、cIAP1はかなり構成的に発現されるcIAPである一方で、これらの高度に類似したcIAPの誘導型のcIAP2は、そのために別個の病態生理学的過程の間に差次的な機能を有する。
【0054】
細胞死に重要なDISCまたは複合体II関連シグナルの機能をさらに詳しく検討するために、本発明者らは、内因性カスパーゼ8ホモログであるcFLIPを使用して実験を行った。本発明者らは、IAP阻害剤に非感受性の細胞(例えば重複する用量反応曲線を有する)を、cFLIPアイソフォームの特異的ダウンレギュレーションによって感受性化することができることを実証している。重要なことには、これらの細胞は、zVAD-fmkによってまだ完全に保護されており、TNFシグナル伝達について示されたように、IAPがカスパーゼ依存性シグナル伝達経路も遮断することを示している43。DISC内のRIP1依存性細胞死を活性化するためのカスパーゼ活性化の関連性は何であろうか?本発明者らは、zVAD-fmkの非存在下および存在下でCD95複合体に動員されたRIP1の量を比較したところ、TRAIL DISCについての本発明者らの先の報告33に一致して、zVAD-fmkの存在下でネイティブCD95 DISC中の完全長RIP1の顕著な増加を見出した(データは示さず)。しかしzVAD-fmkは、DISCにおけるcFLIPLの切断に基づきカスパーゼ8のDISC関連活性を完全に遮断することができない33。加えてzVAD-fmkは、プロ型カスパーゼ8の酵素活性を遮断しない44。したがって、本発明者らの実験は、cFLIPがカスパーゼ依存性細胞死およびカスパーゼ非依存性細胞死に必要なDISCによって生成するシグナルと拮抗することを示している。これに一致して、本発明者らの過剰発現研究は、cFLIPが二重の役割を有することを示唆している:cIAPが存在するときはいつもcFLIPの全アイソフォームが同等の効率でデスレセプター介在性アポトーシスを遮断するのに対し、cIAPの非存在下では、cFLIPSではなくcFLIPLだけがデスレセプターによってトリガーされる細胞死を完全に遮断する。重要なことには、RIP1キナーゼ活性は、cFLIPS発現細胞において細胞をデスレセプター介在性細胞死から保護するために重要であり、本発明者らは、これらの細胞におけるFADDおよびRIP1のレベル増加と共に、複合体IIの自然形成および誘導形成の増加を検出している。これらのデータは、cFLIPSが、cFLIPLのカスパーゼ様ドメインによって負に調節される複合体IIの形成を遮断できないことを立証している。この断片は、cFLIPLおよびcFLIPSから生成することができる45ので、cFLIP p22はこの差次的効果の原因となりそうにない。主に過剰発現研究に基づくと、cFLIPLのカスパーゼ様ドメインは、TRAF、RIP1、またはその他などのタンパク質への結合を仲介し、内因性TRAF2はDISCによって生成したcFLIPL p43と相互作用することが報告された(総説については46参照)。そのうえTRAF2は、cIAPおよびRIP1の結合パートナーである。しかし本発明者らは、それぞれ本発明者らのDISCリガンドアフィニティー沈降または複合体IIの免疫共沈からTRAF2を検出することができなかった(データは示さず)。したがって、これらの追加的な相互作用タンパク質の役割をさらに詳細に解明するために、さらなる研究が必要である。
【0055】
いくつかの異なるSCC細胞株を使用した本発明者らのデータは、異なるDISC成分の化学量論比およびIAPによるそれらの修飾が、アポトーシス細胞死経路および壊死性細胞死経路の活性化に高度に関連することを立証している。CD95 DISCのネイティブリガンドアフィニティー沈降を用いて、本発明者らは、cIAPがDISCに動員されるRIP1の量を負に調節する一方で、RIP1の合計細胞レベルは不変であることを示している(図5、7を比較)。RIP1は、デスレセプター介在性カスパーゼ活性化の伝播(薬理学的カスパーゼ阻害剤によって研究されたように)がいったん遮断されたならば大いに関連性を増し、DISCレベルでカスパーゼ依存性細胞死およびカスパーゼ非依存性細胞死を並行活性化することを立証している。この概念を裏付けて、Hollerらは、FADD欠損Jurkat細胞を使用してFADDの非存在下でRIP1がDISCに動員されることを示した29。したがって本発明者らは、RIP1が、DISCでTRAILおよびCD95デスレセプターによって活性化され、cIAPによって負に調節される、FADD非依存性シグナル伝達経路の重要な成分を構成すると提案する。DISC関連カスパーゼは、DISCに利用可能なRIP1をダウンレギュレーションするように作用することができ、DISCにおけるRIP1のa)cIAP介在性ユビキチン化、またはb)カスパーゼ介在性切断が、DISC活性化RIP1依存性細胞死シグナル伝達経路の重大な負の調節メカニズムとなることを示唆している(図8と比較)。zVAD-fmkを使用した本発明者らの研究によって示されたように、FADD、RIP1、またはカスパーゼ8を欠損した細胞を使用した将来的な研究は、複合体IIの潜在的不安定化因子としてのカスパーゼ活性の必要性をさらに解明すると考えられる。さらに重要なことには、RIP1キナーゼの重要な標的を同定するであろう将来的な研究は、CD95またはTRAIL-Rによって活性化されるアポトーシス性細胞死経路および壊死性細胞死経路の間のクロストークを支配しているシグナル伝達メカニズムをさらに解明すると考えられる。
【0056】
cIAP1およびcIAP2は、もともとTRAF結合タンパク質として報告された47。さらに最近になって、RIP1がcIAPの直接のターゲットであること24,48、およびRIP1についての構成的E3ユビキチンリガーゼとしてのcIAPの機能が、デスレセプターの刺激に依存せずに作用しうることが示唆されている。特にTRAF2は、RIP1についてのリシン63(K63)ユビキチンリガーゼであり、K63-RIP1は、NF-κBの活性化に必要なシグナル伝達モジュールのさらなる集合を可能にする31。対照的にcIAPは、RIP1のK63およびリシン48(K48)のユビキチン化に関与すると示唆されている24。今回本発明者ら自身の実験は、cIAPの重大な一機能が、DISC全体へのRIP1の動員を遮断することであると示すか、またはその代わりにcIAPがDISC内のRIP1の急速分解に必要な可能性があることを示している。重要なことには、本発明者らの実験は、インビトロユビキチン化アッセイによって研究されたようなcIAPによるRIP1の構成的K48またはK63ユビキチン化のDISC関連(刺激依存性)強化を区別することができない24。本発明者らがDISC中のRIP1のユビキチン化パターンを比較したとき、本発明者らは、長期曝露でcIAP非存在下と比較した場合にcIAP存在下で高い分子量のRIP1種を検出している(図5-長期曝露と比較)。本発明者らのデータは、構成的にユビキチン化されたRIP1がDISCに動員されないこと、またはその代わりにDISC内のK48ユビキチン化によってユビキチン化RIP1をダウンレギュレーションすることを示唆している。最近記載されたように49、ユビキチン化特異性抗体を使用した将来的な研究は、異なるデスレセプター誘導性膜結合型複合体内でこれらの点に動力学的にさらに詳細に取り組むことができると考えられる。これに関連して、最近の報告は、Lys377でユビキチン化することのできないRIP1変異体を研究した。これらの著者は、細胞質複合体で細胞死を誘導してカスパーゼ8と相互作用するのが非ユビキチン化形態のRIP1であることを示した。対照的にユビキチン化形態のRIP1は、細胞死を誘導するのではなく、細胞死からの保護のために、おそらくNF-κB活性化によるLys377でのK63ユビキチン化を必要とする50。
【0057】
なぜIAPがダウンレギュレーションされたときにいつも、cFLIPSではなくcFLIPLが、細胞死を遮断することができるのか?本発明者らのデータは、cFLIPSではなくcFLIPLだけが、DISCまたは複合体II全体内のカスパーゼ8に関連したRIP1の効率的な形成(または維持)を抑制するという事実を立証している。図7から明らかなように、複合体IIは、FADD-RIP1-cFLIPS-カスパーゼ8を含有し、特にRIP1とカスパーゼ8との相互作用は、cFLIPSではなくcFLIPLによって抑制される。これらのデータは、cFLIPLのカスパーゼ様ドメインの重要な役割を指摘している。この機能は、DISC内で関連し、切断によってRIP1をダウンレギュレーションすることができよう。または、cFLIPアイソフォームは、先にリンパ系細胞において示唆されたように45、デスレセプター複合体に依存せずに作用することもできる。非ユビキチン化型RIP1は、DISCに依存せずにFADDに結合し、続いて複合体IIの形成および壊死性細胞死を導くことができると推測することができよう。それにもかかわらず本発明者らのデータは、cFLIPSが複合体IIを遮断する能力を有さないことをはっきりと示し、cIAP非存在下での異なるcFLIPアイソフォームの新規で差次的な機能を指摘している。
【0058】
デスレセプターシグナル伝達におけるRIP1の生理学的意味は何でありうるか?T細胞レセプターの刺激後に、細胞死がT細胞区画においてカスパーゼ8非依存的に進行することが最近実証された。この形態の細胞死は、決定的にRIP1を必要とした51。まとめると、これらのデータは、RIP1がカスパーゼ8の非存在下でT細胞刺激後にT細胞を排除するために重要でありうること、およびカスパーゼ8欠損マウスにおいて記載されたいくつかの細胞の増殖欠損52が、活性カスパーゼ8によるRIP1の負の調節の欠如によって引き起こされるおそれがあることを立証している。このRIP1分解は細胞質カスパーゼ8-FADD-RIP1-複合体II内で起こると推測したくなる。最近のデータは、カスパーゼ8の自己プロセシング欠如がカスパーゼ8の非アポトーシス機能を妨害しない一方で、アポトーシスが損なわれることを示しており53、レセプター非依存性複合体IIでのカスパーゼ8の潜在的シャペロン機能を立証している。本発明者らのデータはさらに、これらの観察に付け加え、デスレセプター刺激が主にRIP1依存性シグナルとカスパーゼ8依存性シグナルの両方を活性化するものの、cIAPが重大な負の調節因子に相当することを示している。RIP1は、いくつかの生理学的および病態生理学的に関連する状況でカスパーゼ8に依存せずに作用しうる。カスパーゼ8およびRIP1の両方を欠損した条件付きマウスを調べる将来的な研究は、デスレセプターおよび/またはTCR介在性細胞死経路に関するこれら二つの分子が免疫系に果たす生理学的役割をさらに詳細に明らかにすると考えられる。いくつかの腫瘍実体はIAPを高発現する16。本発明者らのデータに基づき、cIAPは、壊死性の、したがって「免疫学的に強い(loud)」形態の細胞死に関連する可能性のあるRIP1介在性細胞死を逸脱させるために役立つ可能性がありうる。したがって、cIAPは、デスレセプター介在性壊死性細胞死を回避することにより効率的な抗腫瘍免疫反応を回避する重要な役割を果たすことができる16。さらに最近になって、カスパーゼ8が細胞形質転換54および転移55を負に調節することが示された。加えてNF-□Bは、いくつかの細胞株において腫瘍促進転写因子である。したがってcIAPは、腫瘍形成の間にRIP1の死滅誘導性機能をNF-□B誘導性機能に変化させる重要な役割もまた果たしうる。これに関連して、IAPを過剰発現しない腫瘍が、複合体II介在性細胞死または免疫活性化を回避するためにRIP1の追加的な欠如を必要とするかどうかを決定することは興味深いと考えられる。
【実施例】
【0059】
材料および方法
材料
ウエスタンブロット分析のために以下の抗体(Ab)を使用した:カスパーゼ8に対するAb(C-15;親切にもP.H. Krammerによって提供された、C-20; Santa Cruz, Delaware Avenue, California)、cFLIPに対するAb(NF-6; Alexis, San Diego, California)、FADDおよびRIP1に対するAb(Transduction Laboratories, San Diego, California)、CPP32に対するAb(親切にもH. Mehmetによって提供された、Merck Frost)、カスパーゼ10に対するAb(MLB)、PARP-1に対するAb(クローンC-2-10、Biomol)、cIAP1およびcIAP2に対するラットAb56、ならびにSigma製(Saint Louise, Missouri, USA)のβチューブリンに対するAb(クローン2.1)。βアクチンAbは、適切な供給元からのものであった。His-FLAG-TRAIL(HF-TRAIL)は、最近記載されたように産生させた17。Fc-CD95Lの発現のために、本発明者らは、最近公表された構築物を使用した57(親切にもP. Schneider, Epalinges, Switzerlandによって提供された)。1ユニットのFc-CD95Lを、Fc-CD95L上清の原液を1:500希釈したものと規定し、最近記載されたように58、1ユニット/mlのFc-CD95L上清は、A375黒色腫細胞の50パーセントを死滅させるために十分であった(LD50)。リガンド介在性細胞死は、それぞれ可溶性TRAIL-R2-Fcタンパク質またはCD95-Fcタンパク質のいずれかの添加によって完全に遮断された。ホースラディッシュペルオキシダーゼ(HRP)結合ヤギ抗ウサギ、ヤギ抗ラットIgG、ヤギ抗マウスIgG AbならびにHRP結合ヤギ抗マウスIgG1、IgG2a、IgG2b、およびIgG1κは、Southern Biotechnology Associates(Birmingham, AL)から得た。表面レセプターの発現のFACScan分析用mAbであるTRAIL-R1(HS101)、TRAIL-R2(HS201)は、先に記載されたように使用したが26、これらはAlexis(San Diego, California)から入手可能である。CD95 Ab(Apo-1 IgG1およびIgG3a)は、親切にもP. H. Krammer(German Cancer Research Center, Heidelberg, Germany)によって提供された。Cy5結合アネキシンVは、Pharmingen(Hamburg, Germany)から購入した。IAP阻害剤(化合物A)は、Tetralogic Pharmaceuticals(Malvern, Pennsylvania, USA)によって提供された。化合物Aは、参照により全体として本明細書に組み入れられるUS20060194741に例示されており、化合物Aは、以下の構造を有する。
【0060】
細胞培養
自然形質転換角化細胞株HaCaTおよび派生した転移性クローンA5-RT327は、親切にもDr. Petra Boukamp(DKFZ, Heidelberg)によって提供された。MET1細胞59は、I. Leigh, Skin Tumor Laboratory, Cancer Research UK, London, UK)によって提供された。細胞株を記載のように27,60,61正確に培養した。
【0061】
レトロウイルス感染
HaCaT細胞の感染のために、cFLIPLまたはcFLIPSのcDNA挿入部を有するpCFG5-IEGZレトロウイルスベクターを、先に報告されたように使用した62,62。簡潔には、リン酸カルシウム沈殿によってアンホトロピック産生細胞株ΦNXに10μgのレトロウイルスベクターをトランスフェクトした。産生細胞をHaCaT培地(10%FCS含有DMEM)と共に一晩インキュベーションすることによって、ウイルス粒子を含有する細胞培養上清を生成させた。濾過(45μmのフィルター、Schleicher & Schuell, Dassel, Germany)後に、1μg/mlポリブレンの存在下で24時間前に6ウェルプレートに蒔いておいたHaCaT細胞に培養上清を添加した。30℃で3時間遠心分離後に、ウイルス粒子含有上清を新鮮培地と交換した。ポリクローナル細胞に関して10〜14日後に感染培養物全体のゼオシン選択、GFP発現についてのFACS分析(常に>90%、データは示さず)およびウエスタンブロット分析を行い、それぞれの分子の異所性発現を確認した。空のレトロウイルスベクターを対照として用いた。初回の特徴づけの後に2〜6回継代した一定分量の細胞を、その後の全研究のための実験に使用した。
【0062】
siRNAの安定的発現
本発明者らは、最近公表されたように58cFLIPに対するsiRNAの安定的発現を利用した。RIP1 siRNAと同様にNCBIデータベースのどの遺伝子とも一致しないハイパーランダム配列(HRS)63を使用した。構築物の生成のために、RIP1ターゲティング配列を有するcDNA 64塩基長オリゴマー(nt開始位置+193:要望に応じて完全配列が入手可能)を、HindIIIおよびBglII制限部位を使用してpSuper.retroレトロウイルスベクター(pRS)にクローニングした。結果として生じたベクターまたはヒトゲノムに見られないものを有する対照ベクターを、上記に概説したように正確にアンホトロピック産生細胞株にトランスフェクトした。次に、レトロウイルス含有上清を使用して、A5RT3およびMET1細胞にそれぞれHRS shRNAまたはcFLIP shRNAを感染させた。HaCaT細胞にHRSおよびRIP1 shRNAを感染させ、さらなる分析のためにピューロマイシン耐性感染培養物全体を得るために感染細胞をピューロマイシン(1μg/ml; Sigma, Taufkirchen, Germany)で3日間選択した。それぞれの対照構築物を、内部対照として用いた。GFP発現のFACS分析(常に>90%、データは示さず)およびウエスタンブロット分析をポリクローナル細胞について行って、それぞれの分子の異所性発現を確認した。抗生物質選択の後に2〜6回継代した一定分量の細胞を、細胞毒性アッセイおよび生化学的特徴付けのために使用した。
【0063】
FACScan分析
TRAILレセプター(TRAIL-R1およびTRAIL-R2)およびCD95の表面染色のために、細胞をトリプシン処理し、4×105個の細胞をTRAIL-R1 TRAIL-R2、CD95に対するモノクローナルAb、またはアイソタイプが一致する対照IgGと共に60分間インキュベートし、続いて記載のように33ビオチン化ヤギ抗マウス二次Ab(BD Pharmingen)およびCy5-フィコエリトリン標識ストレプトアビジン(Caltag, Burlingame, CA)と共にインキュベーションした。全ての実験について、2×104個の細胞をFACScan(Becton Dickinson & Co, San Jose, CA)によって分析した。
【0064】
ウエスタンブロット分析
記載のように17,58細胞溶解液を調製し、総細胞タンパク質5μgをSDS-PAGEによって4〜12%勾配のゲルで分離し(Invitrogen, Karlsruhe, Germany)、続いてニトロセルロースまたはPVDFメンブランに転写させた。メンブランのブロッキングおよび一次Abおよび適切な二次Abとのインキュベーションは、本質的に先に記載されたように行った33,62。ECL検出キットでバンドを視覚化した(Amersham, Freiburg, Germany)。
【0065】
細胞毒性アッセイ
記載のように37、96ウェルプレート中で一条件について3つ組のウェルを使用して表示濃度のTRAILまたはCD95Lで刺激した20〜24時間後に、付着した生存細胞のクリスタルバイオレット染色を行った。対照培養液の吸光度(OD)を100%に基準化し、それを刺激された細胞と比較した。統計解析のために、細胞株および刺激条件毎に3〜7回の独立した実験について平均の標準誤差(SEM)を決定した。
【0066】
低二倍性分析
先に行われたように33低二倍体(subdiploid)DNA含量を分析した。簡潔には、細胞を表示の試薬で8時間刺激した。次に、細胞を剥離させ、冷PBSで洗浄し、緩衝液N(クエン酸ナトリウム0.1%(w/v)、Triton X100 0.1%(v/v)、PI 50μg/ml)に再懸濁した。細胞を4℃で36〜48時間暗条件に保ち、次に二倍性をFACScan分析によって測定した。
【0067】
免疫蛍光顕微鏡観察
核形態の検出のために、それぞれの細胞の5×104個を12ウェルプレートの各ウェルに蒔いた。接着させるために24時間インキュベーションした後に、図の凡例に示したように細胞を4または24時間刺激した。続いて、細胞をヘキスト33342(5μg/ml)と共に37℃で15分間インキュベートし、直後に適切な装置(Leica)を使用して位相差または蛍光顕微鏡観察を行った。デジタル画像は適切なソフトウェアを使用して全く同様に処理した。
【0068】
アネキシンVの外部移行
ホスファチジルセリンの外部移行を検出するために、図の凡例に示したように細胞を刺激した。細胞を4時間インキュベーションした後に、トリプシン処理した細胞を1×アネキシン-V結合緩衝液(10mM Hepes、pH7.4、140mM NaCl、2,5mM CaCl2)に再懸濁し、続いて製造業者(Pharmingen)に厳密にしたがって2〜4×105個の細胞をCy5結合アネキシン-Vで染色し、その後室温の暗条件で15分間対比染色(ヨウ化プロピジウム;10μg/ml)を行った。全ての実験について、2×104個の細胞をFACScan(Becton Dickinson & Co, San Jose, CA)によって分析した。
【0069】
コロニー形成アッセイ
コロニー形成アッセイのために、1×104個の親HaCaT細胞に加えてレトロウイルスを形質導入されたHaCaT細胞(HRS、shRNA RIP1、pCFG5-IEGZレトロウイルスベクターおよびcFLIPLまたはcFLIPS)を24ウェルプレートの各ウェルに蒔いた。24時間インキュベーション後に接着細胞をIAP阻害剤(100nM、30分間)、zVAD-fmk(10μM、1時間)、ネクロスタチン-1(50μM、1時間)で別々に、または全ての化合物を組み合わせてのいずれかで予備刺激し、続いてCD95Lと共に24時間共刺激した。その時に培地を除去し、細胞を滅菌PBSで2回洗浄し、完全培地を添加した。細胞を3、5、または7日間培養し、続いて生存細胞のコロニーを上記のようにクリスタルバイオレットによって染色した。
【0070】
レセプター複合体のリガンドアフィニティー沈降
CD95L DISCを沈降させるために、各条件について5×106個の細胞を使用した。細胞を培地で37℃で1回洗浄し、続いて10μM zVAD-fmkと共におよび表示のように100nMのIAP阻害剤と共に37℃で1時間予備インキュベートした。続いて細胞を250ユニット/mlのCD95L-Fcで2時間、または未刺激対照についてはリガンドの非存在下で処理した。単層を氷冷PBSで4回洗浄することによってレセプター複合体の形成を停止した。2mlの溶解緩衝液(30mM Tris-HCl、pH7.5、21℃、120mM NaCl、10%グリセロール、1%Triton X-100、Complete(登録商標)プロテアーゼ阻害剤カクテル(Roche Molecular Diagnostics, Mannheim, Germany))の添加によって細胞を氷上で溶解させた。氷上で30分間溶解させた後に、溶解液を20,000×gで2回、それぞれ5分間および30分間遠心分離して細胞の破片を除去した。これらの透明溶解液の小画分を使用してそれぞれのタンパク質の投入量を確認した。CD95レセプターおよび動員された刺激依存性タンパク質を沈降させるために、未刺激細胞および刺激細胞から調製した溶解液にApo-1 IgG3抗体(親切にもP. H. Krammerによって提供された)を添加して、レセプター相互作用タンパク質を沈降させた。リガンドアフィニティー沈降またはカスパーゼ8免疫沈降のいずれかによって沈降したレセプターのレベルを、全ての実験においてCD95についてのウエスタンブロットによって比較した(図5、7を比較)。40μlプロテインGビーズ(Roche, Mannheim, Germany)を反復振盪装置上で4℃で16〜24時間使用して、溶解液からレセプター複合体を沈降させた。リガンドアフィニティー沈降物を溶解緩衝液で4回洗浄してから、標準還元試料緩衝液を添加して95℃で沸騰させることによって、乾燥ビーズからタンパク質複合体を溶出させた。続いて、タンパク質を4〜12%NuPAGE勾配ゲル(Invitrogen, Karlsruhe, Germany)を用いたSDS-PAGEによって分離してから、ウエスタンブロット分析によってDISC成分を検出した。
【0071】
複合体IIのカスパーゼ8免疫沈降
CD95 DISCの沈降後に、残りの溶解液を20,000×gで5分間、2回遠心分離した。続いて1μgのカスパーゼ8抗体(C-20、Santa Cruz)を全ての溶解液に添加した。40μlのプロテインGビーズ(Roche, Mannheim, Germany)と共に反復振盪装置上で、4℃で16〜24時間同時インキュベーションすることによって、溶解液からカスパーゼ8含有複合体を沈降させた。リガンドアフィニティー沈降物を溶解緩衝液で4回洗浄してから、標準還元試料緩衝液を添加して95℃で沸騰させることによって、乾燥ビーズからタンパク質複合体を溶出させた。続いて、タンパク質を4〜12%NuPAGE勾配ゲル(Invitrogen, Karlsruhe, Germany)でSDS-PAGEによって分離してから、ウエスタンブロット分析によってカスパーゼ8相互作用タンパク質を検出した。残りのレセプター結合型DISC複合体を排除するために、CD95の存在について全てのカスパーゼ8相互作用複合体を分析した(図5、7を比較)。
【0072】
cFLIPのノックダウン
細胞株を24ウェルプレートにウェル1個あたり35,000個の細胞密度で蒔いた。翌日、細胞株に、100nMの対照siRNAまたはcFLIPの発現をサイレンシングするように設計されたsiRNAをトランスフェクトした(LおよびSアイソフォーム)。トランスフェクションの48時間後に細胞を100ng/ml TNFα、100nM Smac模倣体、または両者の組み合わせで処理した(ここで使用したSmac模倣体は、化合物A以外の化合物であった。)。追加的に24時間インキュベーションした後に、全ての細胞を回収し、FITC標識アネキシンVおよびヨウ化プロピジウムで染色した。フローサイトメトリーによって染色を分析した。
【0073】
対照siRNAがトランスフェクトされた細胞の処理は、どの条件でもアポトーシスの増加を招かなかった。対照的に、cFLIP特異的siRNAがトランスフェクトされた細胞の処理は、Smac模倣体単独に対する感受性の幾分の増加に加えて、Smac模倣体およびTNFαの組み合わせに対する有意な感受性化を招いた。TNFα単独に対する有意な感受性化は検出されなかった(図9)。
【0074】
これらのノックダウンデータは、cFLIPがSmac模倣体およびTNFαの組み合わせに対する抵抗性の細胞性伝達物質であることを示している。Smac模倣体ならびにSmac模倣体およびTNFαの組み合わせに完全に抵抗性の細胞株は、cFLIPのsiRNA介在性ノックダウンによる処置に感受性になることができる。臨床状況では、Smac模倣体化合物を用いた処置に対する抵抗性を予測して患者の選択を助けるために、腫瘍内のcFLIPレベルを使用することができる。
【0075】
参考文献
【特許請求の範囲】
【請求項1】
以下の段階を含む、IAPアンタゴニストに対するヒトまたは非ヒト動物細胞の感受性を決定する方法:
該細胞がcFLIPLを発現することができるかどうかを決定する段階であって、それに従うと、cFLIPLを発現することができる細胞はIAPアンタゴニストに抵抗性である、段階。
【請求項2】
TRAILレセプターアゴニスト、CD95レセプターアゴニスト、またはTNFαレセプターアゴニストと組み合わせたIAPアンタゴニストに対する細胞の感受性が決定される、請求項1記載の方法。
【請求項3】
TRAILレセプターゴニストがTRAILであり、CD95レセプターアゴニストがCD95L(FasL)であり、かつTNFαレセプターがTNFαである、請求項2記載の方法。
【請求項4】
細胞が生検試料由来である、請求項1、2、または3記載の方法。
【請求項5】
cFLIPL遺伝子の発現ポテンシャルが、
(a)細胞中のcFLIPL mRNAの存在を決定すること、および
(b)細胞中のcFLIPLの存在を決定すること
からなる群より選択される方法によってアッセイされる、請求項1、2、3、または4記載の方法。
【請求項6】
細胞が細胞株を含む、請求項1、2、3、4、または5記載の方法。
【請求項7】
細胞が、腫瘍細胞および自己免疫障害における異常増殖細胞からなる群より選択される、請求項1、2、3、4、または5記載の方法。
【請求項8】
以下の段階を含む、IAPアンタゴニストに対するヒトまたは非ヒト動物細胞の感受性を決定する方法:
該細胞がcFLIPSを発現することができるかどうかを決定する段階であって、それに従うと、cFLIPSを発現することができる細胞はIAPアンタゴニストに感受性である、段階。
【請求項9】
TRAILレセプターアゴニスト、CD95レセプターアゴニスト、またはTNFαレセプターアゴニストと組み合わせたIAPアンタゴニストに対する細胞の感受性が決定される、請求項8記載の方法。
【請求項10】
TRAILレセプターアゴニストがTRAILであり、CD95レセプターアゴニストがCD95Lであり、かつTNFαレセプターがTNFαである、請求項9記載の方法。
【請求項11】
細胞が生検試料由来である、請求項8、9、または10記載の方法。
【請求項12】
cFLIPS遺伝子の発現ポテンシャルが、
(a)細胞中のcFLIPS mRNAの存在を決定すること、および
(b)細胞中のcFLIPSの存在を決定すること
からなる群より選択される方法によってアッセイされる、請求項8、9、10、または11記載の方法。
【請求項13】
細胞が細胞株を含む、請求項8、9、10、11、または12記載の方法。
【請求項14】
細胞が、腫瘍細胞および自己免疫障害における異常増殖細胞からなる群より選択される、請求項8、9、10、11、または12記載の方法。
【請求項15】
以下の段階を含む、増殖性障害を患う患者を処置する方法:
(a)IAPアンタゴニストを用いた処置に対する一部または全ての増殖細胞の感受性を、該細胞がcFLIPLまたはcFLIPSを発現することができるかどうかを決定することによって決定する段階であって、それに従うと、cFLIPSを発現することができる細胞はIAPアンタゴニストに感受性である、段階、および
(b)該細胞がcFLIPSを発現することができる場合に、該細胞をIAPアンタゴニストで処置する段階、または
(c)該細胞がcFLIPLを発現することができる場合に、該細胞をIAPアンタゴニスト以外の剤またはIAPアンタゴニストに追加的な剤で処置する段階。
【請求項16】
TRAILレセプターアゴニスト、CD95レセプターアゴニスト、またはTNFαレセプターアゴニストと組み合わせたIAPアンタゴニストに対する細胞の感受性が決定される、請求項15記載の方法。
【請求項17】
TRAILレセプターアゴニストがTRAILであり、CD95レセプターアゴニストがCD95L(FasL)であり、かつTNFαレセプターがTNFαである、請求項16記載の方法。
【請求項18】
細胞が生検試料由来である、請求項15、16、または17記載の方法。
【請求項19】
cFLIPL遺伝子の発現ポテンシャルが、
(a)細胞中のcFLIPL mRNAの存在を決定すること、および
(b)細胞中のcFLIPLの存在を決定すること
からなる群より選択される方法によってアッセイされる、請求項15、16、17、または18記載の方法。
【請求項20】
細胞が細胞株を含む、請求項15、16、17、18、または19記載の方法。
【請求項21】
細胞が、腫瘍細胞および自己免疫障害における異常増殖細胞からなる群より選択される、請求項15、16、17、18、または19記載の方法。
【請求項22】
以下の段階を含む、アポトーシスを誘導する方法:
(a)cFLIPSの発現を決定するために細胞集団の細胞をアッセイする段階、および
(b)cFLIPSタンパク質の発現が検出された場合に、該細胞集団をIAPアンタゴニストと接触させる段階。
【請求項23】
以下の段階を含む、IAPアンタゴニストを用いた処置から利益を得る可能性のある患者を求めて癌患者をスクリーニングする方法:
(a)患者から得られた異常増殖細胞を、cFLIPS遺伝子/タンパク質発現についてアッセイする段階、および
(b)該増殖細胞がcFLIPSを発現することができる場合のそれらの患者を、IAPアンタゴニストを用いた処置のために選択する段階。
【請求項24】
以下の段階を含む、IAPアンタゴニストの臨床試験に組み入れるために増殖性障害を患う患者を選択する方法:
(a)各患者から得られた異常増殖細胞を、cFLIPS遺伝子/タンパク質の発現についてアッセイする段階、および
(b)該増殖細胞がcFLIPSを発現することができる場合のそれらの患者を、臨床試験に組み入れる段階。
【請求項1】
以下の段階を含む、IAPアンタゴニストに対するヒトまたは非ヒト動物細胞の感受性を決定する方法:
該細胞がcFLIPLを発現することができるかどうかを決定する段階であって、それに従うと、cFLIPLを発現することができる細胞はIAPアンタゴニストに抵抗性である、段階。
【請求項2】
TRAILレセプターアゴニスト、CD95レセプターアゴニスト、またはTNFαレセプターアゴニストと組み合わせたIAPアンタゴニストに対する細胞の感受性が決定される、請求項1記載の方法。
【請求項3】
TRAILレセプターゴニストがTRAILであり、CD95レセプターアゴニストがCD95L(FasL)であり、かつTNFαレセプターがTNFαである、請求項2記載の方法。
【請求項4】
細胞が生検試料由来である、請求項1、2、または3記載の方法。
【請求項5】
cFLIPL遺伝子の発現ポテンシャルが、
(a)細胞中のcFLIPL mRNAの存在を決定すること、および
(b)細胞中のcFLIPLの存在を決定すること
からなる群より選択される方法によってアッセイされる、請求項1、2、3、または4記載の方法。
【請求項6】
細胞が細胞株を含む、請求項1、2、3、4、または5記載の方法。
【請求項7】
細胞が、腫瘍細胞および自己免疫障害における異常増殖細胞からなる群より選択される、請求項1、2、3、4、または5記載の方法。
【請求項8】
以下の段階を含む、IAPアンタゴニストに対するヒトまたは非ヒト動物細胞の感受性を決定する方法:
該細胞がcFLIPSを発現することができるかどうかを決定する段階であって、それに従うと、cFLIPSを発現することができる細胞はIAPアンタゴニストに感受性である、段階。
【請求項9】
TRAILレセプターアゴニスト、CD95レセプターアゴニスト、またはTNFαレセプターアゴニストと組み合わせたIAPアンタゴニストに対する細胞の感受性が決定される、請求項8記載の方法。
【請求項10】
TRAILレセプターアゴニストがTRAILであり、CD95レセプターアゴニストがCD95Lであり、かつTNFαレセプターがTNFαである、請求項9記載の方法。
【請求項11】
細胞が生検試料由来である、請求項8、9、または10記載の方法。
【請求項12】
cFLIPS遺伝子の発現ポテンシャルが、
(a)細胞中のcFLIPS mRNAの存在を決定すること、および
(b)細胞中のcFLIPSの存在を決定すること
からなる群より選択される方法によってアッセイされる、請求項8、9、10、または11記載の方法。
【請求項13】
細胞が細胞株を含む、請求項8、9、10、11、または12記載の方法。
【請求項14】
細胞が、腫瘍細胞および自己免疫障害における異常増殖細胞からなる群より選択される、請求項8、9、10、11、または12記載の方法。
【請求項15】
以下の段階を含む、増殖性障害を患う患者を処置する方法:
(a)IAPアンタゴニストを用いた処置に対する一部または全ての増殖細胞の感受性を、該細胞がcFLIPLまたはcFLIPSを発現することができるかどうかを決定することによって決定する段階であって、それに従うと、cFLIPSを発現することができる細胞はIAPアンタゴニストに感受性である、段階、および
(b)該細胞がcFLIPSを発現することができる場合に、該細胞をIAPアンタゴニストで処置する段階、または
(c)該細胞がcFLIPLを発現することができる場合に、該細胞をIAPアンタゴニスト以外の剤またはIAPアンタゴニストに追加的な剤で処置する段階。
【請求項16】
TRAILレセプターアゴニスト、CD95レセプターアゴニスト、またはTNFαレセプターアゴニストと組み合わせたIAPアンタゴニストに対する細胞の感受性が決定される、請求項15記載の方法。
【請求項17】
TRAILレセプターアゴニストがTRAILであり、CD95レセプターアゴニストがCD95L(FasL)であり、かつTNFαレセプターがTNFαである、請求項16記載の方法。
【請求項18】
細胞が生検試料由来である、請求項15、16、または17記載の方法。
【請求項19】
cFLIPL遺伝子の発現ポテンシャルが、
(a)細胞中のcFLIPL mRNAの存在を決定すること、および
(b)細胞中のcFLIPLの存在を決定すること
からなる群より選択される方法によってアッセイされる、請求項15、16、17、または18記載の方法。
【請求項20】
細胞が細胞株を含む、請求項15、16、17、18、または19記載の方法。
【請求項21】
細胞が、腫瘍細胞および自己免疫障害における異常増殖細胞からなる群より選択される、請求項15、16、17、18、または19記載の方法。
【請求項22】
以下の段階を含む、アポトーシスを誘導する方法:
(a)cFLIPSの発現を決定するために細胞集団の細胞をアッセイする段階、および
(b)cFLIPSタンパク質の発現が検出された場合に、該細胞集団をIAPアンタゴニストと接触させる段階。
【請求項23】
以下の段階を含む、IAPアンタゴニストを用いた処置から利益を得る可能性のある患者を求めて癌患者をスクリーニングする方法:
(a)患者から得られた異常増殖細胞を、cFLIPS遺伝子/タンパク質発現についてアッセイする段階、および
(b)該増殖細胞がcFLIPSを発現することができる場合のそれらの患者を、IAPアンタゴニストを用いた処置のために選択する段階。
【請求項24】
以下の段階を含む、IAPアンタゴニストの臨床試験に組み入れるために増殖性障害を患う患者を選択する方法:
(a)各患者から得られた異常増殖細胞を、cFLIPS遺伝子/タンパク質の発現についてアッセイする段階、および
(b)該増殖細胞がcFLIPSを発現することができる場合のそれらの患者を、臨床試験に組み入れる段階。
【図1】

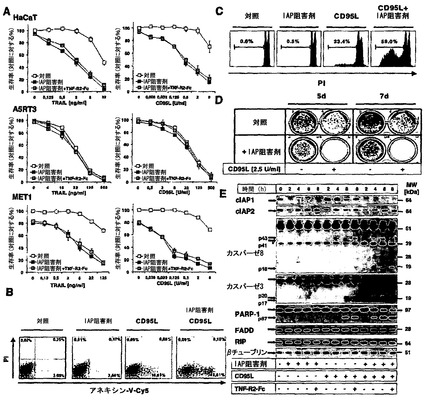
【図2】
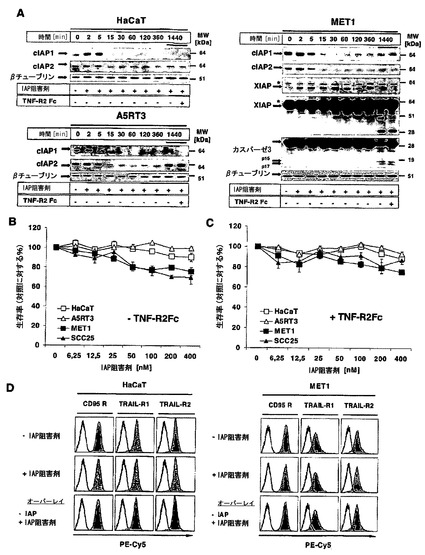
【図3】
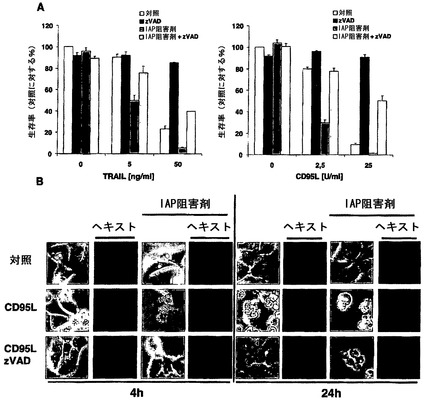
【図4】
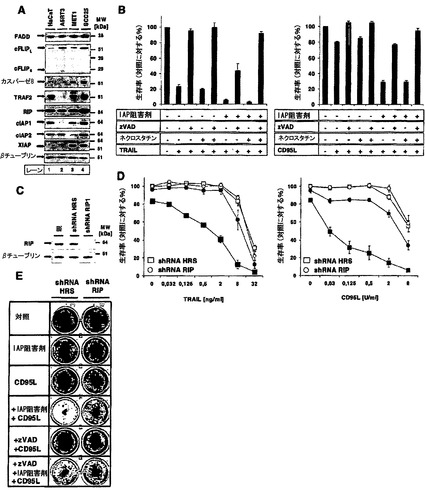
【図5】
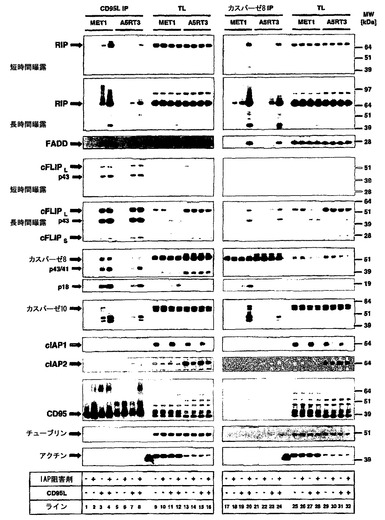
【図6】
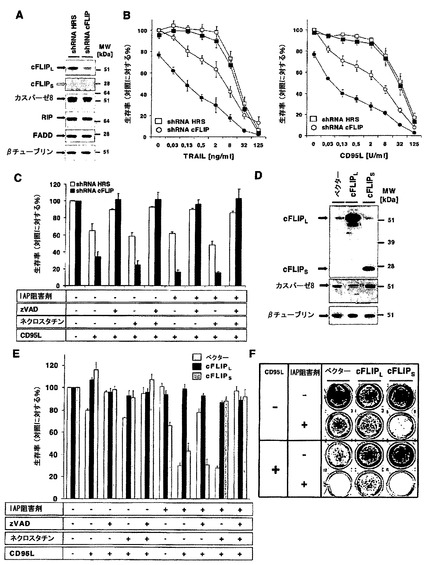
【図7】
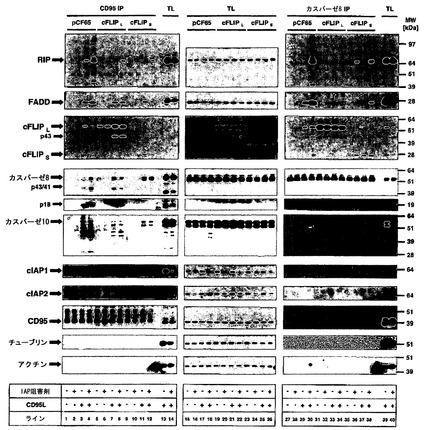
【図8】
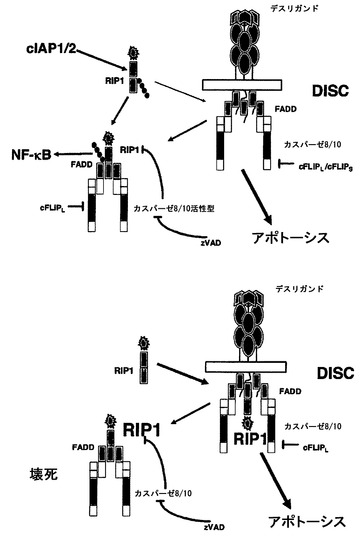
【図9】
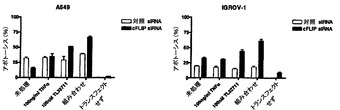
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公表番号】特表2012−516149(P2012−516149A)
【公表日】平成24年7月19日(2012.7.19)
【国際特許分類】
【出願番号】特願2011−546992(P2011−546992)
【出願日】平成22年1月29日(2010.1.29)
【国際出願番号】PCT/IB2010/000165
【国際公開番号】WO2010/086722
【国際公開日】平成22年8月5日(2010.8.5)
【出願人】(511183456)オットー−フォン−ゲーリケ−ウニヴェルジテート マグデブルク (1)
【Fターム(参考)】
【公表日】平成24年7月19日(2012.7.19)
【国際特許分類】
【出願日】平成22年1月29日(2010.1.29)
【国際出願番号】PCT/IB2010/000165
【国際公開番号】WO2010/086722
【国際公開日】平成22年8月5日(2010.8.5)
【出願人】(511183456)オットー−フォン−ゲーリケ−ウニヴェルジテート マグデブルク (1)
【Fターム(参考)】
[ Back to top ]