
IGG抗体の安定な液体医薬製剤
【課題】抗体濃度が50mg/ml又はそれを超える、安定した液体抗体製剤を提供すること。
【解決手段】約pH5.5から約pH6.5のpH、約0.01〜0.1%のポリソルベート、製剤の等張性に寄与する浸透圧調節剤を有する約20〜60mMのコハク酸緩衝液又は30〜70mMのヒスチジン緩衝液に中に高濃度、例えば50mg/ml又はそれ以上の抗体を含む安定な液体医薬製剤による。この液体製剤は、冷蔵庫の温度(2〜8℃)で少なくても1年間、及び好ましくは2年間安定である。この液体製剤は皮下注射に適している。本発明はダクリズマブ、ヒトに適応させた抗IL−2受容体モノクローナル抗体;HAIL−12、ヒトに適応させた抗IL−12モノクローナル抗体;HuEP5C7、ヒトに適応させた抗Lセレクチンモノクローナル抗体;及びフリントズマブ、ヒトに適応させた抗γインターフェロンモノクローナル抗体によって例示される。
【解決手段】約pH5.5から約pH6.5のpH、約0.01〜0.1%のポリソルベート、製剤の等張性に寄与する浸透圧調節剤を有する約20〜60mMのコハク酸緩衝液又は30〜70mMのヒスチジン緩衝液に中に高濃度、例えば50mg/ml又はそれ以上の抗体を含む安定な液体医薬製剤による。この液体製剤は、冷蔵庫の温度(2〜8℃)で少なくても1年間、及び好ましくは2年間安定である。この液体製剤は皮下注射に適している。本発明はダクリズマブ、ヒトに適応させた抗IL−2受容体モノクローナル抗体;HAIL−12、ヒトに適応させた抗IL−12モノクローナル抗体;HuEP5C7、ヒトに適応させた抗Lセレクチンモノクローナル抗体;及びフリントズマブ、ヒトに適応させた抗γインターフェロンモノクローナル抗体によって例示される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、一般に抗体の医薬製剤の分野に関する。特に、本発明は、安定した、液体の高濃度の抗体製剤に関する。本発明は、ダクリズマブ、抗IL−2受容体抗体;HAIL−12、ヒト化抗IL−12モノクローナル抗体;及びHuEP5C7、ヒト化抗L選択モノクローナル抗体の安定な液体製剤によって例証される。
【背景技術】
【0002】
ヒトへの使用を目的とする多くのタンパク質製剤は、製剤の使用前におけるタンパク質に対する変性、凝集、及び変化を防ぐために安定剤が必要とされる。この不安定性は、可溶/不溶性の粒子の形成の点て顕著であり、タンパク質製剤が期限を越えて及び輸送の間貯蔵されるとき、増加することがある。タンパク質薬製剤の開発における主要な目標は、タンパク質の可溶性、安定性、及び生理活性を維持することである。
特に免疫グロブリンは、溶液中で凝集体及び粒子を形成する傾向があるという特徴を有していると認識されており、そうしたものとして、静脈注射又は皮下注射に使用する前にろ過が必要とされうる。タンパク質の凝集体及び粒子の形成は、非経口の免疫グロブリン製品の開発において、特に免疫グロブリンが高濃度で処方される場合に、長い間、問題であった。シナジス(商標)(メディミューン)は、組換えDNA技術によって作られたヒトに適応させたモノクローナルIgG1抗体であって、呼吸器合胞体ウイルス(RSV)のTタンパク質のA抗原サイトにおけるエピトープに対する抗体である。シナジス(商標)はヒト(90%)とマウス(10%)抗体配列の複合体である。シナジス(商標)は、注射のために滅菌水で再構成される無菌凍結乾燥製品として供給される。再構成されたシナジス(商標)は筋肉内注射でのみ投与される。再構成する際に、シナジス(商標)は、以下の賦形剤;47mMヒスチジン、3.0mMグリシン、5.6%マンニトール、及び活性成分、つまりIgG1抗体をバイアル当り100mgの濃度で含む。再構成されたシナジス(商標)は、再構成している6時間のうちに投与されるべきである。
【0003】
WO 89/11297は、1〜25mg/mlのIgGモノクローナル抗体の凍結乾燥された製剤、2〜10%マルトース、及びp3.0〜6.0の間のpHを有する酢酸ナトリウム、リン酸ナトリウム、又はクエン酸ナトリウム緩衝液を含む凍結乾燥されたモノクローナル抗体製剤を開示する。
WO 97/45140は、100mMクエン酸ナトリウム、0.05mM EDTA、pH6.0中におおよそ100mg/mlの濃度にされた抗CD4抗体の水性製剤を開示する。この出願は、抗体の濃縮後における濁度のわずかな上昇を開示し、濁度の上昇はタンパク質の凝集を表しているようである。この凝集を取り除くためには、ポリソルベート80の添加及び滅菌ろ過が必要になる。
WO 90/11091は、約5mg/mlのIgM、2.5〜5%(w/v)のヒト血清アルブミンを8〜20mMのリン酸緩衝液、270mMの塩化ナトリウム、pH6.8〜7.4中に含む注射可能な水性組成物を開示する。
【0004】
米国特許6,171,586号は、安定な水性医薬製剤であって、以前に凍結乾燥を受けていない治療有効量の抗体、約pH4.8〜約5.5の酢酸緩衝液、界面活性剤、及びポリオールを含む製剤を開示する。そこでは、製剤は等張化する量の塩化ナトリウムを欠如する。
米国特許出願公開US2001/0014326A1号は、前に凍結乾燥された抗体製剤で、5mg/mlの抗IgE抗体、5mMのヒスチジン、pH6.0、85mMスクロース、及び0.01%ポリソルベート20を含む製剤を開示する。
米国特許5,744,132号は、約5.6のpHを有する1〜1000μg/mlのIL−12抗体、2%スクロース、4.15%マンニトール、10mMのコハク酸ナトリウム、及び約0.02%のトゥィーン(商標)20を含む組成物を開示する。
米国特許6,267,958号は、20mMヒスチジン、pH6.0、340mMスクロース、0.04%ポリソルベート20、及び0.9%ベンジルアルコール中に100mg/ml rhuMab E25を再構成させた製剤を開示する。
【0005】
米国特許6,165,467号は、受入番号HB8307を有する融合細胞腫の細胞株により作られるヒトモノクローナル抗体組成物を安定化するための過程であって、7.2から7.4のpHを有するリン酸塩安定化緩衝溶液中でヒトモノクローナル抗体を透析することを含む過程を開示し、前記溶液は前記モノクローナル抗体1mgあたり1〜20mgのD−マンニトール、前記モノクローナル抗体1mgあたり0.005〜0.2ミリモルのグリシン、及び前記溶液のpHを安定化させるための多量のpH安定化リン酸塩を含む。
抗体濃度が50mg/ml又はそれを超える、安定した液体抗体製剤への需要があり;そういった製剤は、ヒトに対する静脈内、筋肉内、腹腔内、又は皮下注射を含む非経口投与に適している。
【発明の概要】
【0006】
発明の要約
本発明は、20〜60mMコハク酸緩衝液又は30〜70mMヒスチジン緩衝液(pHは約pH5.5から約pH6.5)、浸透圧調節剤、及び約0.01〜0.1%のポリソルベート中において高濃度の、例えば50mg/ml以上の、抗体を含む安定な液体医薬製剤に関する。本製剤は、抗体の物理的、化学的、及び生物的安定性を保っており、ヒト患者に対する投与を意図した免疫グロブリンが最終製品において凝集体及び粒子を形成することを防ぐ。本発明の好ましい抗体は、ダクリズマブ、ヒト化抗IL−2受容体モノクローナル抗体;HAIL−12、ヒト化抗IL−12モノクローナル抗体;HuEP5C7、ヒト化抗Lセレクチンモノクローナル抗体;及びフリントズマブ(Flintozumab)、ヒト化抗γインターフェロンモノクローナル抗体を含む。
液体の抗体製剤は、冷蔵庫の温度(2〜8℃)において少なくても1年間、好ましくは2年間は安定である。この液体製剤は、室温(23〜27℃)においても、少なくても6ヶ月は安定である。この液体製剤は皮下注射に適している。
【図面の簡単な説明】
【0007】
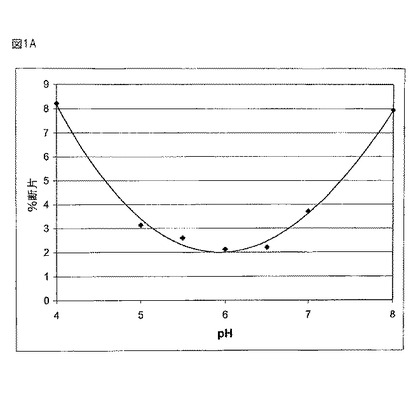
【図1A】図1Aは断片形成の割合(%)を、図1Bは凝集体の割合(%)を、様々なpHレベルで45℃においてサンプルを4週間インキュベーションした後、SEC−HPLCによって評価したものとして示す。
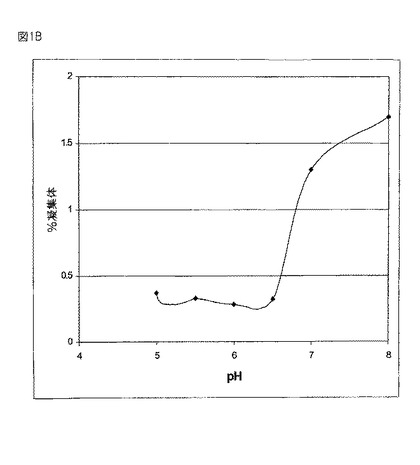
【図1B】図1Aは断片形成の割合(%)を、図1Bは凝集体の割合(%)を、様々なpHレベルで45℃においてサンプルを4週間インキュベーションした後、SEC−HPLCによって評価したものとして示す。
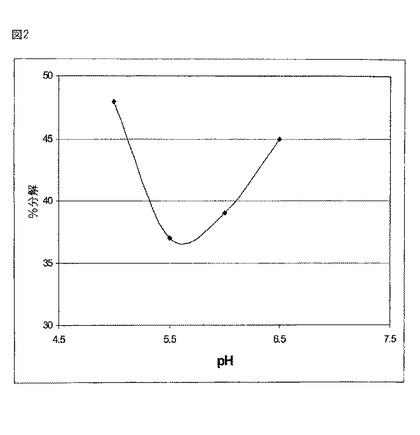
【図2】図2は、cIEFにより評価される分解であって、様々なpHレベルで45℃においてサンプルを4週間インキュベーションの後の分解の割合(%)を示す。
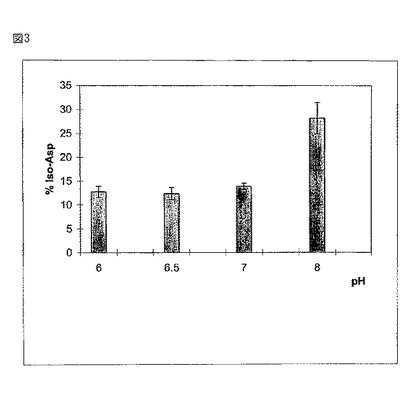
【図3】図3は、プロメガイソクオントキットにより評価されるイソアスパラギン酸の割合(%)であって、様々なpHレベルで45℃においてサンプルを4週間インキュベーションの後のイソアスパラギン酸の割合(%)を示す。
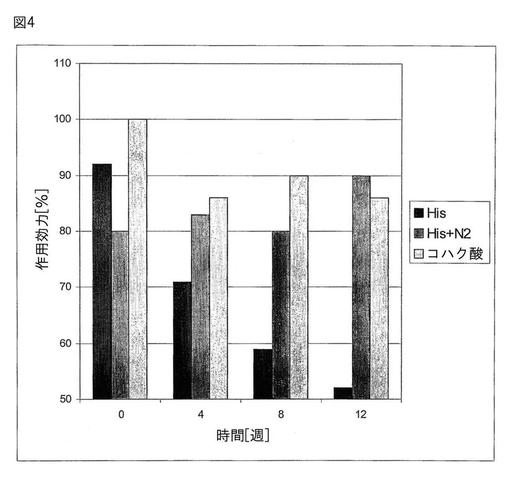
【図4】図4は、37℃でインキュベーション後における作用効力に対する種々の緩衝液の効果を時間に対して示す。
【発明を実施するための形態】
【0008】
発明の詳細な説明
I.定義
本明細書中で用いられる場合、「緩衝液」という用語は、溶液のpHを許容される範囲に維持する薬剤を含み、そしてクエン酸(ナトリウム)、ヒスチジン、リン酸(ナトリウム又はカリウム)、トリス(トリス(ヒドロキシメチル)アミノメタン)、ジエタノールアミンなどを含みうる。本発明の緩衝液は、約5.5から約6.5の範囲におけるpHを有し、好ましくは、約6.0のpHを有する。この範囲におけるpHを制御する緩衝液の例として、コハク酸(例えばコハク酸ナトリウム)、グルコン酸、ヒスチジン、クエン酸、リン酸、及び別の有機酸緩衝液が挙げられる。
【0009】
「医薬として許容される賦形剤(ビヒクル、添加剤)」は、適度に哺乳動物の対象に投与されうる不活性な物質であり、かつ使用される活性成分の有効な投与量を提供する。これらの物質は、抗体の物理的、化学的、生物的構造を安定化するために製剤に加えられる。この用語はまた、意図した投与様式に適した等張性の製剤に達するために必要とされうる添加剤のことも指す。
【0010】
「医薬製剤」という用語は、活性成分の生理活性が明確に有効であることを可能にするような形態であり、及び製剤が投与される対象に対し毒性を有する追加の成分を含まない製剤のことを指す。
【0011】
「安定な」製剤は、貯蔵のあいだに、その中にあるタンパク質が、その物理的安定性、化学的安定性、及び生理活性を本質的に十分に保持する製剤である。タンパク質の安定性を計測する様々な分析用の技術が当該技術分野で利用でき、Peptide and Protein Drug Delivery,247-301, Vincent Lee Ed., Marcel Dekker,Inc., New York, N.Y., Pubs.(1991)及び Jones, A. adv. Drug Delivery Rev. 10:29-90(1993)に総説される。安定性は、選択された温度で選択された期間計測される。
【0012】
「安定な」液体の抗体製剤は、冷蔵庫の温度で(2〜8℃)少なくとも12ヶ月間、好ましくは2年間、より好ましくは3年間、又は室温で(23〜27℃)少なくとも3ヶ月間、好ましくは6ヶ月間、及びより好ましくは1年間、有意な変化が認められない液体の抗体製剤である。安定性の基準は以下のとおりである:SEC−HPLCにより計測した場合に、抗体の単量体のうち10%以上、好ましくは5%以上が分解されない。視覚分析によると、この溶液は無色であるか、又は透明からわずかに乳白色を呈する。製剤の濃度、pH及び浸透圧は、+/−10%以上の変化を有しない。作用効力は対照の70%〜130%、好ましくは80%〜120%内である。10%以下の、好ましくは5%以下の分解(加水分解)しか観察されない。10%、好ましくは5%以下の凝集しか形成されない。
【0013】
色及び/又は透明度の視覚試験を行った場合、或いは紫外線の散乱、サイズ排除クロマトグラフィー(SEC-HPLC)、及び動的光散乱によって計測された場合に、もし凝集、沈殿、及び/又は変性の有意な上昇が見られないならば、抗体は、医薬製剤において「その物理的な安定性を保持する」といえる。さらに、タンパク質の高次構造は変化することはない。タンパク質の高次構造の変化は、タンパク質の3次構造を決定する蛍光分光法によって、及びタンパク質の2次構造を決定するFTIR分光法によって評価することができる。
【0014】
もし有意な化学変換が示されないなら、医薬製剤において抗体は「その化学的安定性を保持する」といえる。化学的安定性は、化学的に変換されたタンパク質の形態を検出し、定量することによって、評価することができる。タンパクの化学的構造をしばしば変換する分解過程としては、加水分解又は分解(例えばサイズ排除クロマトグラフィー及びSDS−PAGEの方法で評価される)、酸化(例えば、マススペクトロメトリー又はMALDI/TOF/MSと組み合わせたペプチドマッピングの方法により評価される)、アミド分解(例えばイオン交換クロマトグラフィー、キャピラリー等電点電気泳動、ペプチドマッピング、イソアスパラギン酸計測法といった方法で評価される)、及び異性化(イソアスパラギン酸の含量の計測、ペプチドマッピング等によって評価される)が挙げられる。
【0015】
所定の時間における抗体の生理活性が、医薬製剤が調製された際に示された既定の生理活性の範囲内であるなら、抗体は医薬製剤において「生理活性を保持する」といえる。抗体の生理活性は、例えば抗原に結合するELISAアッセイによって決定することができる。
【0016】
「等張」という用語は、目的の製剤が基本的にヒト血液と同じ浸透圧を有するということを意味する。等張製剤は、通常約270〜328mOsmの浸透圧を有する。わずかに低張の浸透圧は250〜269mOsmであり、わずかに高張の浸透圧は328〜350mOsmである。浸透圧は、例えば蒸気圧又は氷冷型の浸透圧計を使って計測することができる。
【0017】
「浸透圧調節剤」は、医薬として許容される不活性な物質であって、製剤に等張性を与えるために製剤に加えられる物質である。本発明に適した浸透圧調節剤として、塩及びアミノ酸が挙げられる。
【0018】
II.分析方法
以下の判定基準は安定な医薬の抗体製剤を開発する際に重要である。抗体製剤は医薬として許容される賦形剤を含む。抗体がその物理的、化学的、及び生理活性を保持するように、抗体製剤が剤形される。製剤は、好ましくは、冷蔵庫の温度(2〜8℃)で少なくても1年間は安定であり、室温で(23〜27℃)で少なくても6ヶ月は安定である。
【0019】
製品の安定性を評価する分析方法としては、サイズ排除クロマトグラフィー(SEC−HPLC)、動的光散乱試験(DLS)、示唆走査熱量測定(DSC)、イソアスパラギン酸定量、作用効力、340nmのUV、及びUV分光法が挙げられる。SEC(J. Pharm.Scien., 83:1645-1650,(1994); Pharm. Res.,11:485(1994); J. Pharm. Bio. Anal., 15:1928(1997); J. Pharm. Bio. Anal., 14:1133-1140(1986))は、製品中の単量体の割合を測定し、可溶性の凝集体及び断片の量についての情報を与える。DSC(Pharm.Res., 15:200(1998); Pharm.Res., 9:109(1982))タンパク質の変性温度及びガラス転移温度についての情報を与える。DLS(American Lab., Nov.(1991))は、拡散係数の平均値を計測し、可溶及び不可溶な凝集体の量についての情報を与える。340nmにおけるUVは、340nmにおける散乱光の強度を計測し、可溶及び不溶性の凝集体の量についての情報を与える。UV分光法は、278nmにおける吸収を計測し、タンパク質濃度についての情報を与える。
【0020】
サンプルにおけるiso−Aspの含量は、イソクアントイソアスパラテートディテクションキット(プロメガ)を使って計測される。このキットは、標的タンパク質におけるイソアスパラギン酸残基の存在を特異的に検出する酵素、タンパク質イソアスパラギン酸メチル転移酵素 (PIMT)を使用する。PIMTは、S−アデノシル−L−メチオニン由来のメチル基をイソアスパラギン酸のα−カルボキシル位に転移することを触媒しており、その過程においてS−アデノシル−L−ホモシステイン(SAH)を形成する。SAHは比較的小さな分子量であり、キットに提供されるSAH HPLCの標準物質を用いることにより、逆相HPLCで通常単離及び定量される。
【0021】
抗体の効力又は生理活性は、その抗原に結合する能力によって測られうる。抗体のその抗原に対する特異的な結合は、当業者に周知であるいずれかの方法、例えばイムノアッセイ、例えばELISA(酵素結合免疫吸着検定法)によって定量される。
【0022】
III. 抗体の調製
本明細書中の発明は、抗体を含む安定した水性製剤に関する。当該技術分野で利用可能な抗体を作る技術と以下の記載においてより詳細に記述される例示の方法を使って、製剤内の抗体が調製される。
【0023】
抗体は、目的の抗原に対するものである。好ましくは、当該抗原は生物学的に重要なポリペプチドであり、哺乳動物に対する抗体の投与は疾患を予防又は治療することができる。しかしながら、非ポリペプチド抗原に対する抗体(例えば腫瘍に関連する糖脂質抗原;米国特許No.5,091,178号参照のこと。)にも関する。
【0024】
抗原がポリペプチドである場合、抗原は膜貫通分子(例えば受容体)又は成長因子などのリガンドであってもよい。例示の抗原としては、分子、例えばレニン;ヒト成長ホルモン及びウシ成長ホルモンを含む成長ホルモン;成長ホルモン放出因子;副甲状腺ホルモン;甲状腺刺激ホルモン;リポタンパク質;α−1−抗トリプシン;A鎖インスリン;B鎖インスリン;プロインスリン;卵胞刺激ホルモン;カルシトニン;黄体形成ホルモン;グルカゴン;凝血因子、例えば第VIII因子C、第IX因子、組織因子、及びフォンウィルブランド因子;抗凝血因子、例えばタンパク質C;心房性ナトリウム利尿因子;肺サーファクタント;プラスミノーゲン活性化因子、例えばウロキナーゼ又はヒト尿型若しくは組織型プラスミノーゲン活性化因子(t−PA);ボンベシン;トロンビン;造血性成長因子;腫瘍壊死因子−α及び−β;エンケファリン分解酵素;RANTES(regulated on activation nomally T-cell expressed and secreted);ヒトマクロファージ炎症タンパク質(MIP−1−α);血清アルブミン例えばヒト血清アルブミン;ミュラー管抑制因子;A鎖リラキシン;B鎖リラキシン;プロリラキシン:マウス性腺刺激ホルモン関連ペプチド;微生物タンパク質、例えばβラクタマーゼ;DNase;IgE;細胞傷害性Tリンパ球関連抗原(CTLA)、例えばCTLA−4;インヒビン;アクチビン;血管内皮増殖因子(VEGF);ホルモン又は成長因子の受容体;タンパク質A及びD;リウマチ因子;神経栄養性因子、例えばウシ由来神経栄養性因子(BDNF);ニューロトロフィン−3,−4,−5,又は−6(NT−3,NT−4,NT−5,又はNT−6) 又は神経成長因子、例えばNGF−β;血小板由来成長因子(PDGF);繊維芽細胞成長因子、例えばaFGF及びbFGF;上皮細胞成長因子(EGF);トランスフォーミング成長因子(TGF)、例えばTGF−α並びにTBF−β1、TBF−β2、TBF−β3、TBF−β4、及びTBF−β5を含むTGF−β;インスリン様成長因子−I及び−II(IGF−I及びIGF−II);des(1−3)−IGF−I(脳IGF−I)、インスリン様成長因子結合タンパク質;CDタンパク質、例えばCD3,CD4,CD8,CD19及びCD20;エリスロポエチン;骨誘導因子;免疫毒素;骨形成タンパク質(BMP);インターフェロン、例えばインターフェロン−α、−β、及び−γ;コロニー刺激因子(CSFs)、例えばM−CSF、GM−CSF、及びG−CSF;インターロイキン(ILs)、例えばIL1〜IL−12;インターロイキンIL−1〜IL−12の受容体:セレクチン、例えばL、E、及びP−セレクチン;スーパーオキシドジムスターゼ;T細胞受容体;膜表面タンパク質;腐食促進因子;ウイルス抗原、例えばAIDS外皮部位;輸送タンパク質;ホーミング受容体;アドレシン;調節タンパク質;インテグリン、例えばCD11a、CD11b、CD11c、CD18、ICAM、VLA−4及びVCAM;腫瘍関連抗原、例えばHER2、HER3又はHER4受容体;並びに上記リストのポリペプチドのいずれかの断片が挙げられる。
【0025】
組換え技術を用いる場合、抗体は細胞内に、細胞膜周辺腔において生産されるか、又は培養液に直接分泌されることもある。抗体が細胞内に産出される場合、最初のステップとして、粒子性細胞片、これらは宿主細胞又は溶解された細胞のいずれかである、は、例えば遠心又は限外ろ過により取り除かれる。抗体が培養液に分泌される場合、そういった発現システムの上清は最初に、商業的に利用できるタンパク質濃縮膜、例えばアミコン又はミリポアペリコン限外ろ過ユニットを使って濃縮される。プロテアーゼ阻害剤、例えばPMSFは、タンパク分解を阻害するため前述のステップいずれの中にも入れられるし、及び外来性の汚染菌の生育を防ぐため抗生物質が入れられる。
【0026】
細胞から調製された抗体組成物は、例えばヒドロキシルアパタイトクロマトグラフィー、ゲル電気泳動、透析、及びアフィニティークロマトグラフィーを用いて精製される。アフィニティークロマトグラフィーは好ましい精製技術である。Aタンパク質のアフィニティーリガンドとしての適合性は、種及び抗体内に存在する免疫グロブリンFcドメインのアイソタイプに依存する。Aタンパク質は、ヒトγ1、γ2、及びγ4重鎖(Lindmark et al.,J. Immunol. Meth.62:1-13(1983))に基づく抗体を精製するために使われうる。Gタンパク質は、全てのマウスアイソタイプ及びヒトγ3(Guss et al., EMBO J.5:1567-1575(1986))鎖に基づく抗体を精製するために推奨されている。アフィニティリガンドが結合するマトリクスはアガロースであることが最も多いが、別のマトリクスも用いることができる。機械的に安定なマトリックス、例えば、ガラス又はポリ(スチレンジビニル)ベンゼンの制御された細孔は、アガロースで達成されるより早い流れ、及び短い過程を可能にする。抗体がCH3ドメインを含む場合、バッカーボンドのABX(商標)樹脂(J.T.Baker, Phillipsburg, N.J.)が精製に有用である。タンパク質を精製する別の技術、例えばイオン交換カラムに基づく分画、エタノール沈殿、逆相HPLC、シリカ上でのクロマトグラフィー、ヘパリン セファロセット(商標)上でのクロマトグラフィー、陰イオン又は陽イオン交換樹脂(例えばポリアスパラギン酸カラム)上のクロマトグラフィー、等電点電気泳動、SDS−PAGE、及びリン酸アンモニウム沈殿が、回収される抗体に依存して利用される。
【0027】
本発明に含まれる好ましい抗体としてはダクリズマブ(USAN、United States Adopted Names)、ヒト化抗IL-2受容体抗体が挙げられる。ダクリズマブは、腎臓移植の後の組織拒絶を防ぐためにゼナパックス(商標)として現在売られており、経静脈で投与される。ダクリズマブは感染の治療にも有効であり、そのためには抗体の皮下投与が好ましい投与経路である。抗体の皮下投与のためには、高濃度の抗体が好ましい。ダクリズマブは、ヒト化組換え替えモノクローナル抗体、IgG1のサブクラスである。この分子は、2つの同一な重鎖及び2つの同一な軽鎖のサブユニットから構成される。ジスルフィド架橋が4つの鎖を結びつける。ダクリズマブ単量体は、分子量で大体150000ダルトンである。ダクリズマブは、活性化されたT細胞において発現するIL−2受容体のp55サブユニットに結合する。抗原標的は、CD25と命名される。ダクリズマブは、発酵流加培養法により重鎖及び軽鎖遺伝子を含むGS−NS0細胞株から生産される。バイオリアクターからの収集物を処理して、細胞及び細胞片を取り除き、そしてイオン交換及びゲルろ過クロマトグラフィー並びにいくつかの限外ろ過及びろ過技術を使って精製し、95%を超える単量体からなる薬を生産する。
【0028】
別の好ましい抗体は抗インターロイキン12(IL−12)抗体である。IL−12は、抗原提示細胞により合成されるサイトカインである。IL−12は、2つのサブユニット(p35及びp40)から構成されており、その両方が、機能的な活性のために存在しなければならない。機能的なIL−12は、IL−12p70とも呼ばれる。このサイトカインは、好ましくはヘルパーT細胞タイプ1(Th1)リンパ球及びナチュラルキラー細胞においてそれらの増殖速度を増加させることにより作用する。下流の影響の一つに、Th1細胞によるインターフェロンγ(IFNg)の分泌がある。これらの機能(増殖とIFNgの生産)の両方とも、容易に検定でき、サンプルにおけるIL−12の活性を検出するのに使われる。IL−12に対する特定の抗体は、上記の活性を中和すると示されてきた。Th1細胞は、様々な病気において中心的な役割を担っていることに結び付けられてきているので、中和特性を有する抗体は、潜在的な治療上の価値を持つ。16G2(ホフマン ラ ロシュ)は、IL−12p70に対するマウス抗体である。16G2は、機能アッセイ−つまりヒト抹消血由来の活性型T細胞(PBMC)の増殖阻害において、IL−12に対して近い化学量数で作用することが示されてきた。IL−12のp40の二量体は血漿中に存在し、p40サブユニットに対して産出された抗体は、与えられた量のIL−12の増殖能力を中和するために過度の量で使われる必要があるので、16G2がIL−12に対して近い化学量数で作用することは重要な性質である。16G2はプロテインデザインラボ(Fremont,CA)でヒト化され、HAIL−12(ヒト化抗IL−12、IgG1抗体)を生じる。
【0029】
別の好ましい抗体は、抗Lセレクチン抗体である。セレクチン、例えばL、E、及びP-セレクチンは、虚血及び再灌流の過程における組織の損傷に関連にすると分かってきた。好中球がこの関連に重要な役割を担う。セレクチンが好中球をリクルートするのに必要であると考えられる。L-セレクチンは、骨格筋及び肺において損傷を完全に発生するのに重要である。(Seekamp, et al., Am. J. `athol. 11:592-598(1994). Mulligan, et al., J. Immunol. 151:832-840(1994))。HuEp5C7(スマート 抗Lセレクチン)は、IgG2Fcミュータントを含み、ヒトE及びPセレクチン抗原と相互反応するヒト化抗-Lセレクチンモノクローナル抗体である。HuEP5C7は、プロテインデザインラボによって、様々な適応症、例えば喘息、脳卒中、傷害、及び自己免疫疾患に対して現在開発されている。
【0030】
別の好ましい抗体は、フリントズマブ、つまり抗γインターフェロン抗体である。フリントズマブは、プロテインデザインラボによって開発されたヒト化IgG1モノクローナル抗体であって、インターフェロンγ(IFN−g)、つまり炎症誘発性サイトカインによる自己免疫疾患の治療のための抗体である。IFN−gは主要組織適合性複合体(MHC)クラス1及び又はクラス2(HLA−DR)抗原の発現を促し、ナチュラルキラー細胞の細胞溶解活性を高め、マクロファージを活性化し、及び液性反応の特性である免疫グロブリンのアイソタイプを調節する。リンフォカインとして、IFN-gはまた、2型ヘルパーT細胞(Th2)の発生を抑制する一方で、1型ヘルパーT細胞(Th1)の発生を高める。Th1/Th2割合の異常は、様々な自己免疫疾患に結び付けられてきている。
【0031】
IV. 製剤の調製
目的の抗体が上記のように調製された後、抗体を含む医薬製剤が調製される。製剤開発のアプローチは以下の通りである:至適溶液pHを選び、緩衝液のタイプ及び濃度を選び、様々な液体の安定性を与える賦形剤の影響を評価し、及び選別した賦形剤の濃度を、I-至適実験デザイン(Statistics for Experimenters:An Introduction to Design, Date
Analysis, and Model Building, Box, George E.P. et al., John Wiley and Sons, Inc., 1978)を用いて最適化する。
【0032】
本発明の組成物は抗体凝集体及び粒子の形成を最小限にし、抗体がその生理活性を一定の期間保持することを保証する。組成物は、中性の又は少し酸性のpH(pH5.5〜6.5)、界面活性剤、及び浸透圧調節剤を有する緩衝液中に高い抗体濃度を含む医薬として許容される液体の製剤である。
【0033】
組成物中の抗体は50mg/ml又はそれを超える、好ましくは100mg/ml又はそれを超える高濃度である。本発明の好ましい組成物は、ダクリズマブ、つまりヒト化抗IL−2受容体抗体;HAIL−12、つまりヒト化抗IL−12抗体;HaEP5C7、つまりヒト化抗Lセレクチン抗体;及びフリントズマブ、ヒト化抗γインターフェロン抗体を含む。
【0034】
pH5.5〜6.5の緩衝液が組成物中で使われる。pH6.0〜6.5の緩衝液が好ましい。この範囲内でpHを制御する緩衝液の例として、コハク酸(例えばコハク酸ナトリウム)、グルコン酸、ヒスチジン、クエン酸、リン酸、及び別の有機酸緩衝液が挙げられる。コハク酸(pKa5.63)は皮下注射の好ましい緩衝液である。ヒスチジン(pK5.97)は、酸化に対する感受性のため好ましくないが、そういった酸化は、バイアルの上部をN2で置換すること又は抗酸化剤を添加することにより遅らせることができる。クエン酸及びリン酸緩衝液は、ずっと好ましくない。なぜなら、皮下注射されたとき有痛性の反応を起こすからである。好ましい緩衝液は約20〜60mMのコハク酸ナトリウムを含む。別の好ましい緩衝液は30〜70mMのN2で覆われたヒスチジン緩衝液である。
【0035】
界面活性剤もまた、抗体製剤に添加される。実例として、界面活性剤は、非イオン性界面活性剤、例えばポリソルベート(例えばポリソルベート20、80、例えばTween(商標)20、Tween(商標)80)又はポロクサマー類(例えばポロクサマー188)が挙げられる。剤形された抗体の凝集を減少し、及び/又は製剤中の粒子の形成を最小限にし、及び/又は吸着を減少させるように、ある量の界面活性剤が添加される。界面活性剤は、約0.005%〜約0.5%の量で、好ましくは約0.01%〜約0.1%の量で、より好ましくは0.01%〜約0.05%の量で、及び最も好ましくは約0.02%〜約0.04%の量で製剤内に存在しうる。
【0036】
製剤の等張性に貢献する浸透圧調節剤は、本組成物に加えられる。本発明にとって有用な浸透圧調節剤としては、塩及びアミノ酸が挙げられる。医薬として許容され、及び本発明に適している塩としては、塩化ナトリウム、コハク酸ナトリウム、硫酸ナトリウム、塩化カリウム、塩化マグネシウム、硫酸マグネシウム、及び塩化カルシウムが挙げられる。本発明の好ましい塩はNaCl及びMgCl2である。MgCl2はまた、タンパク質をアミド分解から守ることにより、抗体の安定性を改良しうる。NaClの好ましい濃度は、約75〜150mMである。MgCl2の好ましい濃度は、約1〜100mMである。医薬として許容され、及び本発明に適しているアミノ酸として、プロリン、アラニン、L−アルギニン、アスパラギン、L−アスパラギン酸、グリシン、セリン、リジン、及びヒスチジンが挙げられる。本発明に好ましいアミノ酸はプロリンである。好ましいプロリンの濃度はおよそ200mMである。
【0037】
タンパク質製剤を安定するために通常使われるEDTAもまた、製剤内に含まれうる。EDTAは、キレート剤として、金属に触媒されるスルフヒドリル基の酸化を阻害し、こうしてジスルフィド結合された凝集体の形成を減少させる。EDTAの好ましい濃度は、0.01〜0.2%である。
【0038】
例示の液体組成物は、抗体を約100mg/ml又はそれを超える量、約20〜60mMコハク酸ナトリウム(pH6)、約0.01〜0.1%ポリソルベート20又は80、及び約75〜150mMのNaClを含む製剤である。この製剤は、モノクローナル抗体の生理活性の安定性を保持し、ヒト患者に対して投与されるように意図される免疫グロブリンを、最終製品中における物理的、化学的、生物的分解から保護される。
【0039】
本発明の液体抗体製剤は、非経口投与、例えば静脈内、筋肉内、腹腔内、又は皮下注射に適しており;特に皮下注射に適している。
本発明は、次の実施例によってさらに記述される。実施例は実施例の中で記述される特定の手順の範囲に本発明を制限することとして解釈するべきではない。
【実施例】
【0040】
実施例1:pHの最適化
至適製剤のpH範囲を確認するため、及び主要の分解経路を確認するため、pH特性試験を行った。サンプル製剤は、3つの緩衝液:pH4.0若しくは5.0の50mM酢酸ナトリウム緩衝液、pH5.0、6.0、若しくは6.5の50mMヒスチジン緩衝液、又はpH7.0若しくは8.5の50mMリン酸ナトリウム緩衝液のうちの1つの緩衝液内に5.0mg/ml抗IL−2受容体抗体(ダクリズマブ)を含んだ。各製剤を5℃又は45℃で、100RPMの振蕩で4週間インキュベーションした。各サンプルの物理的又は化学的安定性を、0週目と4週目において、pH及び視覚分析、340nmのUV分光法、サイズ排除クロマトグラフィー(SEC−HPLC)、蛍光分光法、動的光散乱(DLS)、示唆走査熱量測定(DSC)、プロメガ イソクアント アッセイ、キャピラリー等電点電気泳動(cIEF)、SDS−PAGE(減少性、又は非減少性)、及び生理活性検定(ELISA)を含む分析方法によって評価した。
【0041】
図1Aにおいて、SECを用いることにより調べられた様々なpHレベルで回収された断片の割合により示されるように、45℃で4週間インキュベーションされた後のサンプルについて行われたSEC−HPLCにより、液体の製剤にとって切断が主要な分解経路であるということが示された。SECにより決定される断片の割合及び凝集体の割合(図1B)は、5.5から6.5の中間範囲のpH値において減少した。
【0042】
図2は、45℃でサンプルを4週間インキュベーションした後に、cIEFによって評価された際の、様々なpHレベルでの分解の割合を示す。最小の分解は約5.5のpH値において獲得される。
【0043】
図3は、45℃でサンプルを4週間インキュベーションした後に、プロメガのイソクアントキットによって評価された際の、様々なpHレベルで獲得されるイソアスパラギン酸の割合を示す。イソアスパラギン酸の形成(アミド分解)は、pH6.0及び6.5で最小であり、そしてpH8.0において急激に増加した。
【0044】
この実験の結果により、pH5.5〜6.5、及び好ましくはpH6.0〜6.5が最適pHであって、抗体の分解及び凝集を最小にするpHであるということが示される。
【0045】
実施例2:緩衝液の最適化
本実験において、各製剤は、5.0mg/mlダクリズマブ抗体を、50mMのコハク酸ナトリウム(pH6.0)内に;及び窒素ガス存在下、又は窒素ガス非存在下における50mMヒスチジン(pH6.0)内に含んだ。クエン酸ナトリウム緩衝液は、皮下注射された際に痛みの報告があるので、使用されなかった。生理活性(効力)を、0時間において、並びに4、8、及び12週間の37℃インキュベーションの後において、組換えヒトIL−2α受容体(IL−2sRα)抗原でコートされたマイクロプレート及びHRP標識されたヤギの抗ヒトIgGを使ってELISAによって計測した。
【0046】
図4は、37℃でのインキュベーション後の異なる緩衝液の効力に対する時間経過の影響を示す。抗体製剤の最も高い安定性が、pH6.0の50mMのコハク酸酸ナトリウム緩衝液において8週間をとおして達成された。ヒスチジンのみの緩衝液内における製剤は、緩衝液が酸化されるので即座に(8週以内に)その作用効力を失う。コハク酸ナトリウム又は酸化を防ぐために窒素ガスで満たされたヒスチジン緩衝液においては、製剤の作用効力は少なくとも12週の間、80%超を残した。
【0047】
実施例3:賦形剤の選別
対象
本試験は、50mg/mlダクリズマブ抗体の製剤のための様々な賦形剤を選別するため行われた。先に行われたpHの最適化試験(例1)から、製剤の安定性はpH6.0〜6.5のpH範囲において最大となった。それゆえ、本試験では、賦形剤は、二つの緩衝液;pH6.5の50mMリン酸緩衝液、とpH6.0の50mMコハク酸緩衝液において選別がされた。抗体の安定性を、濃度50mg/ml、5℃及び45℃で3週間、100RPMで攪拌された2つの緩衝液中でモニターした。試験される賦形剤として、界面活性剤(Tween80(商標)及びTween20(商標))、塩(NaCl及びMgCl2)、抗酸化剤(EDTA及びメチオニン)、アミノ酸(グリシン、リジン、セリン、及びプロリン)、及び混合溶媒(グリセロール及びエタノール)が挙げられる。様々な分析技術(透明度、pH、SEC−HPLC、UV−Vis、及びcIEF)が賦形剤を含んでいる製剤の性質決定のために使われた。
【0048】
サンプル調製
ダクリズマブ抗体は、67mMのリン酸ナトリウム製剤(Tween80を含まない)中に6.6mg/mlの濃度であった。この材料を、ペリコンII(ミリポア)で約30mg/mlまで濃縮し、続いて、2つの選択された緩衝液(50mMリン酸ナトリウムpH6.5、及び50mMコハク酸ナトリウムpH6.0)に、50mlのアミコンスターセル(ミリポア)を用いて緩衝液交換がされた。3回目及び最後の緩衝液交換ステップの間に、材料は最終濃度〜125mg/mlに濃縮された。最終的に抗体を0.8μm膜(ユニフロ)でろ過した。ろ過後のタンパク質濃度は、リン酸緩衝液サンプルについて約100mg/mlになるように、コハク酸緩衝液サンプルについて97mg/mlになるように決定された。
【0049】
賦形剤が選別されるところの賦形剤の標的濃度が、表1に示される。製剤は、必要とされる量の賦形剤を直接バイアルに量り入れることによって、又は賦形剤が濃縮されたストック溶液を調製することによって、調整された。賦形剤は、適切な緩衝溶液0.5mlに加えられ、pHは、1N HCL又は10%NaOHによって望ましい値に調節された。続いて、適切な緩衝液中の0.5mlの濃縮された抗体溶液(〜100mg/ml)が、標的濃度50mg/mlに達するように加えられる。この手順は、濃縮された賦形剤と直接接触することによるタンパク分解を防ぐために適用される。1mlの溶液は、2つのバイアルに0.5mlづつ分けて充填された。1つのバイアルは、最初のT=0の分析で使われ、その後2〜8℃の3週の時間点のため、2〜8℃で貯蔵された。別のバイアルを45℃100RPMで振蕩して3週間インキュベーションし、その期間の最後に分析した。
【0050】
【表1】

【0051】
分析方法
各2つの時間点において、様々な分析技術を用いてサンプルを分析した。溶液の透明度は、蛍光灯の下で黒い背景に対してサンプルバイアルを持つことによって、視覚的に試験した。溶液について不溶性の物質を検査し、色の変化を記録した。サイズ排除クロマトグラフィーを、ダイオードアレイ検出及び一連に結合される2つのトソハス(Tosohaas)カラムを有するパーキンエルマーのHPLCユニットを用いて行った。サンプルを、約5倍量の対応する緩衝液に約1mg/mlの濃度になるよう希釈し、そして100μlのサンプルをカラムに注入した。UV分光法により、パーキンエルマーラムダバイオ40分光光度計を用いてサンプル濃度を計測した。
【0052】
三週間の時点のサンプルを、バイオラッドCE(BioFocus3000)システムのキャピラリー等電点電気泳動により分析した。全てのサンプルを0.25mg/mlに水で希釈し、そして1:1希釈(終濃度0.125mg/ml)を、TEMED及び2つの内部のpIマーカー(8.4及び10.1)を含むファーマライト(pharmalyte)溶液を用いて行った。キャピラリーは、中性のコーティングを持つeCAP(ベックマン、長さ56cm、50umID)であった。
【0053】
コハク酸緩衝溶液中に賦形剤、つまりTween80、EDTA、NaCl及びMgCl2と供に剤形されたサンプルの効力は、5℃及び45℃で3週間のインキュベーション後に試験した。それは、KIT−225−K6細胞を含むバイオアッセイであった。
【0054】
結果
12の異なる賦形剤が2つの異なる緩衝液中でモニターされたので、T=0の時点で24のサンプルがあった。3週後の時点においては、48の被分析サンプルがあった(12の異なった賦形剤×2つの温度×2つの緩衝液=48)。実施したアッセイとして、UV−Vis、pH、透明度、SEC−HPLC、及びCIEFによる濃度決定が挙げられる。
(a)サンプル透明度
サンプルの外見を表2に示す。全てのサンプルは最初の時点T=0において両者の緩衝液中で透明であった。3週後の時点においては、リジンを含む1つを除いてリン酸緩衝液中のサンプル全てが5℃において透明であった。同じ緩衝液中で、45℃では、アミノ酸(グリシン、セリン、プロリン及びリジン)を含むサンプルでは透明に見えたが、いくつかの針様の浮遊物をバイアル中に有した。MgCl2のサンプルは、バイアルの底に明らかな結晶を有した。
コハク酸緩衝液中では、アミノ酸を含む製剤を除く全てのサンプルが5℃3週間のインキュベーションの後で透明であった。プロリン及びリジンを有するサンプルはもっともにごっていた。45℃においては、コハク酸緩衝液の全てのサンプルが3週後の時点においても透明であった。
【0055】
【表2】
【0056】
(b)SEC−HPLC
SEC−HPLCの結果を表3(A−C)に示した。表3Aは、本研究で調査された全てのサンプルにおける単量体野割合(%)を示す。全てのサンプルのT=0における単量体割合は99%以上であった。3週の時点における両緩衝液中における5℃のサンプルの単量体において、有意な変化は観測されなかった。しかしながら、45℃においては、全てのサンプルが単量体%の少しの低下(5%未満)が示された。リン酸緩衝液中に剤形されるサンプルでは、単量体%は94.08(メチオニン)から97.29(プロリン)の間であり、一方、コハク酸緩衝液で剤形されるサンプルでは、単量体割合は95.86%(メチオニン)から97.55(トゥィーン80)%の間であった。両方の緩衝液中で、メチオニン及びグリシンを含む製剤は、単量体%において最も有意な低下を示した。%単量体の減少は、主に断片の形成のせいであった。
【0057】
表3Bは本研究で調査された全てのサンプルにおいて凝集体形成割合(%)を掲載した。3週間の持続時間における凝集体の形成の増加は、両方の緩衝液の5℃における全てのサンプルで最小であることが明らかになった。45℃、3週間のインキュベーションの後、リン酸緩衝液中のサンプルは、0.40%(EDTA)から2.40%(グリシン)の範囲の凝集体割合(%)における増加を示した。コハク酸緩衝液中では、凝集体の形成はかなり低く;3週間のインキュベーションの後で0.7%(メチオニン)から1.09%(グリシン)の範囲であった。これらの結果を支持する仮説の一つは、もし凝集体の形成の原因が酸化であるなら、コハク酸緩衝液の金属をキレートする性質のため、凝集体の形成はコハク酸緩衝液中ではゆっくりになりうるということである。
【0058】
表3Cは、この研究で調査された全てのサンプルにおいて断片形成%を掲載する。最初の時間点においては、断片%は全てのサンプルにおいて0.2〜0.4%の範囲であった。5℃でインキュベートされた全てのサンプルで、断片%の増加は三週間の期間にわたってわずかなものであった。45℃においては、断片形成の割合の有意な増加が観測された。リン酸緩衝液中に剤形されるサンプルでは、断片%は4.74(メチオニン)から1.5%(プロリン、グリセロール、及びエタノール)の間であり、一方、コハク酸緩衝液中では、範囲は1.48%(トゥィーン80)から3.44(メチオニン)であった。一般的に、断片形成の増加は、アミノ酸を含む製剤で観察された。さらに、断片形成の割合は、リン酸緩衝液で高いようである。このことはNa−コハク酸緩衝液とNa−リン酸緩衝液との間におけるpHの違いに(それぞれpH6.0及びpH6.5)起因し、断片形成の第一の根拠として、塩基性に触媒される加水分解が示されうる。
【0059】
【表3】
【0060】
【表4】
【0061】
(c)キャピラリー電気泳動
本試験におけるすべてのサンプルは、バイオラッドシステムに基づくキャピラリー電気泳動(cIEF)によって分析された。ダクリズマブの典型的なcIEF特性は4つのピークを示す。典型的に、高温で時間効果が促進されたとき、主要なアイソフォームのピーク面積の減少に続いて別のアイソフォームのピーク面積が増加する。このことは、あるアイソフォームから別のアイソフォームへの変換を示唆する。分解割合(%)は、主要なアイソフォームのピーク面積における減少の割合によって計算される。
分解割合(%)=[T=0におけるピーク面積−45℃におけるピーク面積]×100%/[T=0におけるピーク面積]
【0062】
本発明者らの結果は、コハク酸緩衝液(pH6.0)中の同じサンプルと比較した場合に、45℃のサンプルがリン酸緩衝液(pH6.5)中でより分解されていることを示す。最良の電気泳動図は賦形剤、EDTA、NaCl、リジン、及びMgCl2で見られた。3週間後の45℃に対する5℃の分解%は、Tween80、Tween20、セリン及びプロリンを含むサンプルにおいてその電気泳動図がかなりつぶれていて、ピークが区別できなかったので、それらを含むサンプルでは計算できなかった。
【0063】
(d)作用効力
本試験の結果に基づくと、Na−コハク酸緩衝液は、Na−リン酸緩衝液とくらべてより有望であるようである。このように作用効力の評価は、Na−コハク酸緩衝液でのみ最も安定化する賦形剤についてされた。Na−コハク酸緩衝液はTween−80、EDTA、NaCl、及びMgCl2から成る製剤を含み、5℃及び45℃で3週間インキュベーションした。結果(表4)により、全ての製剤の作用効力が明細書の範囲内であり、強調される化学的及び物理的分解の過程はタンパク質の活性を有意には変えないということが示される。
【0064】
【表5】
【0065】
考察
本研究の結果に基づくと、pH6.5のNa−リン酸緩衝液と比較すると、pH6.0におけるNa−コハク酸緩衝液の方が製剤の安定性が高かった。このことは、pH6.5以上で促進される塩基性に触媒される加水分解が第一の原因であり、断片形態の割合の増加がもたらされる。このように、pH6.0におけるNa−コハク酸緩衝液は、今後の試験全てに選ばれる緩衝液である。両方の緩衝液中においてアミノ酸(グリシン、リジン、セリン、プロリン、及びメチオニン)はタンパク質の安定性に関して安定させる効果をもたないということを本試験の結果は明らかに示した。サンプルに関する明瞭なデータに示されたように、全てのアミノ酸を含む製剤は、45℃における不溶性の凝集体の形成を示した。
【0066】
MgCl2がタンパク質の減少から保護しうるという仮定に基づいて、賦形剤としてMgCl2が本研究において選択された。MgCl2がNa−リン酸緩衝液中で沈殿した一方で;Na−コハク酸緩衝液中では、cIEFデータに基づくとMgCl2はタンパク質を安定化する効果を有する。エタノールはまた、溶液の比誘電率を低めることで、タンパク質を脱アミド化に対し安定化させるかを試験するために、賦形剤として含まれた。しかしながら、その結果は上記仮説を支持することはない。最終的に、最も一般的にタンパク質製剤を安定するために使われるTween80、EDTA、及びNaCl、といった賦形剤は、どちらの緩衝液内においてもタンパク質を不安定化させる効果を全く与えないことを示した。
【0067】
更なる実験が、pH6.0のNa−コハク酸緩衝液中で行われ;賦形剤(MgCl2、Tween80、NaCl、及びEDTA)の影響がタンパク質の安定性において調べられた。その結果により、抗体を100mM NaClで100mg/mlに剤形すると、Tween80の最適濃度は0.02%〜0.03%の範囲内になることが示された。この結果により、塩の濃度の増加(100〜150mM)は製剤をさらに安定化しうることも示された。このように、NaClの濃度は、浸透圧を要求範囲に維持する一方で最大にするべきである。この結果により、Tween80及びNaClを含む製剤の安定性が、EDTAを0.35〜0.5%の範囲の濃度で加えることにより、高めることができることが示される。MgCl2を0〜50mMの範囲の濃度で添加することはまた、好ましい結果を与えうる。最も安定な剤形のための賦形剤の濃度は;150mM NaCl,0.05%Tween80、0.03%〜0.04%EDTA、及び60〜70mM MgCl2であるが、等張性の条件を提供しないため、この条件は実用的ではない。
【0068】
実施例4: コハク酸緩衝液中における2つのダクリズマブ抗体製剤の安定性データ
製剤1及び製剤2は実施例3に従って調製された。
製剤1:100mg/mlダクリズマブ抗体、30mMコハク酸ナトリウム(pH6.0)、100mM NaCl及び0.03%トゥィーン(商標)80である製剤
製剤2:製剤1と同じ製剤に0.05%EDTAを加えた製剤
製剤1及び製剤2の安定性の結果が、T=0、2週、4週、8週及び12週において、5、25、37℃での安定性の結果が以下に示される。(表5)
【0069】
【表6】
【0070】
【表7】
【0071】
実施例5: ヒスチジン緩衝液中における2つのダクリズマブ製剤の安定性データー
製剤3及び製剤4は実施例3に従って調製される。
製剤3:100mg/mlダクリズマブ抗体、50mMヒスチジン(pH6.0)、115mM NaCl、0.03%Tween(商標)80であり窒素置換を行った製剤。
製剤4:製剤3と同じ製剤に0.05%のEDTAを添加した製剤
製剤3及び製剤4のT=0、2週、4週、8週、及び12週における5、25、37℃での安定性の結果が以下に示される。(表6)
【0072】
【表8】
【0073】
【表9】
【0074】
実施例6: 室温で1年後におけるダクリズマブ製剤の安定性データ
100mg/mlダクリズマブを30mMコハク酸ナトリウム、pH6、100mM NaCl、及び0.03%トゥィーン(商標)80における液体の抗体製剤の安定性は、25℃で1年貯蔵した後に試験された。安定性の結果は、この製剤は少なくとも1年は25℃において安定であることを示す。(表7)
【0075】
【表10】
【0076】
実施例7: 5℃18ヶ月の期間後におけるダクリズマブ製剤の安定性データ
100mg/mlダクリズマブ、30mMコハク酸ナトリウム、pH6.0、100mM NaCl、及び0.03%トゥィーン(商標)80を含む液体の抗体製剤は、5℃(2〜8℃)でインキュベートされ、異なる時点において安定性が試験された。製剤が少なくとも18ヶ月間は冷蔵庫の温度で安定であることを安定性の結果は示す。
【0077】
【表11】
【0078】
実施例8: HAIL−12(ヒスチジン緩衝液)の安定性データ
HAIL−12(抗IL−12抗体、50mg/ml)を、50mMヒスチジン緩衝液、120mM塩化ナトリウム、0.03%トゥィーン80、pH6.0中に剤形した。進行中の安定性試験により、製剤が5℃で少なくとも9ヶ月間は安定であることが示される(表9)。
【0079】
【表12】
【0080】
実施例9: HAIL−12(コハク酸緩衝液)の安定性データ
HAIL−12(50及び100mg/ml)を、40mM Na−コハク酸緩衝液、100mM NaCl、及び0.03%トゥィーン80、pH6.0中に剤形した。進行中の安定性試験により、製剤が5℃、25、及び37℃で少なくとも12週間安定であることが示された(表10及び表11)。
【0081】
【表13】
【0082】
【表14】
【0083】
実施例11:HuEP5C7の安定性データ
HuEP5C7(抗Lセレクチン抗体、50及び100mg/ml)を、50mMヒスチジン緩衝液、125mM塩化ナトリウム、0.01%トゥィーン80、pH6.0中に剤刑した。進行中の安定性試験により、製剤は25℃及び45℃で3ヶ月間、並びに5℃で少なくても9ヶ月間安定であるということが示された。5℃における9ヶ月の安定性試験の結果を表12に示す。3ヶ月の促進された安定性試験を表13に示す。
【0084】
【表15】
【0085】
【表16】
本発明、並びに本発明を製造し、そして使用する方法及び過程は、発明が属する技術分野の当業者が、同発明を製造し、そして使用できるように十分、明瞭、簡潔、及び正確な用語で記述されている。実施例の記載は、本発明の好ましい態様を記述しており、そして特許請求の範囲において述べられるとき本発明の範囲から逸脱することなく変更がそこに加えられることは理解されるべきである。発明とみなされる対象の内容を特に明らかにし、明確に主張するために、以下の特許請求の範囲により本明細書が締めくくられる。
【技術分野】
【0001】
本発明は、一般に抗体の医薬製剤の分野に関する。特に、本発明は、安定した、液体の高濃度の抗体製剤に関する。本発明は、ダクリズマブ、抗IL−2受容体抗体;HAIL−12、ヒト化抗IL−12モノクローナル抗体;及びHuEP5C7、ヒト化抗L選択モノクローナル抗体の安定な液体製剤によって例証される。
【背景技術】
【0002】
ヒトへの使用を目的とする多くのタンパク質製剤は、製剤の使用前におけるタンパク質に対する変性、凝集、及び変化を防ぐために安定剤が必要とされる。この不安定性は、可溶/不溶性の粒子の形成の点て顕著であり、タンパク質製剤が期限を越えて及び輸送の間貯蔵されるとき、増加することがある。タンパク質薬製剤の開発における主要な目標は、タンパク質の可溶性、安定性、及び生理活性を維持することである。
特に免疫グロブリンは、溶液中で凝集体及び粒子を形成する傾向があるという特徴を有していると認識されており、そうしたものとして、静脈注射又は皮下注射に使用する前にろ過が必要とされうる。タンパク質の凝集体及び粒子の形成は、非経口の免疫グロブリン製品の開発において、特に免疫グロブリンが高濃度で処方される場合に、長い間、問題であった。シナジス(商標)(メディミューン)は、組換えDNA技術によって作られたヒトに適応させたモノクローナルIgG1抗体であって、呼吸器合胞体ウイルス(RSV)のTタンパク質のA抗原サイトにおけるエピトープに対する抗体である。シナジス(商標)はヒト(90%)とマウス(10%)抗体配列の複合体である。シナジス(商標)は、注射のために滅菌水で再構成される無菌凍結乾燥製品として供給される。再構成されたシナジス(商標)は筋肉内注射でのみ投与される。再構成する際に、シナジス(商標)は、以下の賦形剤;47mMヒスチジン、3.0mMグリシン、5.6%マンニトール、及び活性成分、つまりIgG1抗体をバイアル当り100mgの濃度で含む。再構成されたシナジス(商標)は、再構成している6時間のうちに投与されるべきである。
【0003】
WO 89/11297は、1〜25mg/mlのIgGモノクローナル抗体の凍結乾燥された製剤、2〜10%マルトース、及びp3.0〜6.0の間のpHを有する酢酸ナトリウム、リン酸ナトリウム、又はクエン酸ナトリウム緩衝液を含む凍結乾燥されたモノクローナル抗体製剤を開示する。
WO 97/45140は、100mMクエン酸ナトリウム、0.05mM EDTA、pH6.0中におおよそ100mg/mlの濃度にされた抗CD4抗体の水性製剤を開示する。この出願は、抗体の濃縮後における濁度のわずかな上昇を開示し、濁度の上昇はタンパク質の凝集を表しているようである。この凝集を取り除くためには、ポリソルベート80の添加及び滅菌ろ過が必要になる。
WO 90/11091は、約5mg/mlのIgM、2.5〜5%(w/v)のヒト血清アルブミンを8〜20mMのリン酸緩衝液、270mMの塩化ナトリウム、pH6.8〜7.4中に含む注射可能な水性組成物を開示する。
【0004】
米国特許6,171,586号は、安定な水性医薬製剤であって、以前に凍結乾燥を受けていない治療有効量の抗体、約pH4.8〜約5.5の酢酸緩衝液、界面活性剤、及びポリオールを含む製剤を開示する。そこでは、製剤は等張化する量の塩化ナトリウムを欠如する。
米国特許出願公開US2001/0014326A1号は、前に凍結乾燥された抗体製剤で、5mg/mlの抗IgE抗体、5mMのヒスチジン、pH6.0、85mMスクロース、及び0.01%ポリソルベート20を含む製剤を開示する。
米国特許5,744,132号は、約5.6のpHを有する1〜1000μg/mlのIL−12抗体、2%スクロース、4.15%マンニトール、10mMのコハク酸ナトリウム、及び約0.02%のトゥィーン(商標)20を含む組成物を開示する。
米国特許6,267,958号は、20mMヒスチジン、pH6.0、340mMスクロース、0.04%ポリソルベート20、及び0.9%ベンジルアルコール中に100mg/ml rhuMab E25を再構成させた製剤を開示する。
【0005】
米国特許6,165,467号は、受入番号HB8307を有する融合細胞腫の細胞株により作られるヒトモノクローナル抗体組成物を安定化するための過程であって、7.2から7.4のpHを有するリン酸塩安定化緩衝溶液中でヒトモノクローナル抗体を透析することを含む過程を開示し、前記溶液は前記モノクローナル抗体1mgあたり1〜20mgのD−マンニトール、前記モノクローナル抗体1mgあたり0.005〜0.2ミリモルのグリシン、及び前記溶液のpHを安定化させるための多量のpH安定化リン酸塩を含む。
抗体濃度が50mg/ml又はそれを超える、安定した液体抗体製剤への需要があり;そういった製剤は、ヒトに対する静脈内、筋肉内、腹腔内、又は皮下注射を含む非経口投与に適している。
【発明の概要】
【0006】
発明の要約
本発明は、20〜60mMコハク酸緩衝液又は30〜70mMヒスチジン緩衝液(pHは約pH5.5から約pH6.5)、浸透圧調節剤、及び約0.01〜0.1%のポリソルベート中において高濃度の、例えば50mg/ml以上の、抗体を含む安定な液体医薬製剤に関する。本製剤は、抗体の物理的、化学的、及び生物的安定性を保っており、ヒト患者に対する投与を意図した免疫グロブリンが最終製品において凝集体及び粒子を形成することを防ぐ。本発明の好ましい抗体は、ダクリズマブ、ヒト化抗IL−2受容体モノクローナル抗体;HAIL−12、ヒト化抗IL−12モノクローナル抗体;HuEP5C7、ヒト化抗Lセレクチンモノクローナル抗体;及びフリントズマブ(Flintozumab)、ヒト化抗γインターフェロンモノクローナル抗体を含む。
液体の抗体製剤は、冷蔵庫の温度(2〜8℃)において少なくても1年間、好ましくは2年間は安定である。この液体製剤は、室温(23〜27℃)においても、少なくても6ヶ月は安定である。この液体製剤は皮下注射に適している。
【図面の簡単な説明】
【0007】
【図1A】図1Aは断片形成の割合(%)を、図1Bは凝集体の割合(%)を、様々なpHレベルで45℃においてサンプルを4週間インキュベーションした後、SEC−HPLCによって評価したものとして示す。
【図1B】図1Aは断片形成の割合(%)を、図1Bは凝集体の割合(%)を、様々なpHレベルで45℃においてサンプルを4週間インキュベーションした後、SEC−HPLCによって評価したものとして示す。
【図2】図2は、cIEFにより評価される分解であって、様々なpHレベルで45℃においてサンプルを4週間インキュベーションの後の分解の割合(%)を示す。
【図3】図3は、プロメガイソクオントキットにより評価されるイソアスパラギン酸の割合(%)であって、様々なpHレベルで45℃においてサンプルを4週間インキュベーションの後のイソアスパラギン酸の割合(%)を示す。
【図4】図4は、37℃でインキュベーション後における作用効力に対する種々の緩衝液の効果を時間に対して示す。
【発明を実施するための形態】
【0008】
発明の詳細な説明
I.定義
本明細書中で用いられる場合、「緩衝液」という用語は、溶液のpHを許容される範囲に維持する薬剤を含み、そしてクエン酸(ナトリウム)、ヒスチジン、リン酸(ナトリウム又はカリウム)、トリス(トリス(ヒドロキシメチル)アミノメタン)、ジエタノールアミンなどを含みうる。本発明の緩衝液は、約5.5から約6.5の範囲におけるpHを有し、好ましくは、約6.0のpHを有する。この範囲におけるpHを制御する緩衝液の例として、コハク酸(例えばコハク酸ナトリウム)、グルコン酸、ヒスチジン、クエン酸、リン酸、及び別の有機酸緩衝液が挙げられる。
【0009】
「医薬として許容される賦形剤(ビヒクル、添加剤)」は、適度に哺乳動物の対象に投与されうる不活性な物質であり、かつ使用される活性成分の有効な投与量を提供する。これらの物質は、抗体の物理的、化学的、生物的構造を安定化するために製剤に加えられる。この用語はまた、意図した投与様式に適した等張性の製剤に達するために必要とされうる添加剤のことも指す。
【0010】
「医薬製剤」という用語は、活性成分の生理活性が明確に有効であることを可能にするような形態であり、及び製剤が投与される対象に対し毒性を有する追加の成分を含まない製剤のことを指す。
【0011】
「安定な」製剤は、貯蔵のあいだに、その中にあるタンパク質が、その物理的安定性、化学的安定性、及び生理活性を本質的に十分に保持する製剤である。タンパク質の安定性を計測する様々な分析用の技術が当該技術分野で利用でき、Peptide and Protein Drug Delivery,247-301, Vincent Lee Ed., Marcel Dekker,Inc., New York, N.Y., Pubs.(1991)及び Jones, A. adv. Drug Delivery Rev. 10:29-90(1993)に総説される。安定性は、選択された温度で選択された期間計測される。
【0012】
「安定な」液体の抗体製剤は、冷蔵庫の温度で(2〜8℃)少なくとも12ヶ月間、好ましくは2年間、より好ましくは3年間、又は室温で(23〜27℃)少なくとも3ヶ月間、好ましくは6ヶ月間、及びより好ましくは1年間、有意な変化が認められない液体の抗体製剤である。安定性の基準は以下のとおりである:SEC−HPLCにより計測した場合に、抗体の単量体のうち10%以上、好ましくは5%以上が分解されない。視覚分析によると、この溶液は無色であるか、又は透明からわずかに乳白色を呈する。製剤の濃度、pH及び浸透圧は、+/−10%以上の変化を有しない。作用効力は対照の70%〜130%、好ましくは80%〜120%内である。10%以下の、好ましくは5%以下の分解(加水分解)しか観察されない。10%、好ましくは5%以下の凝集しか形成されない。
【0013】
色及び/又は透明度の視覚試験を行った場合、或いは紫外線の散乱、サイズ排除クロマトグラフィー(SEC-HPLC)、及び動的光散乱によって計測された場合に、もし凝集、沈殿、及び/又は変性の有意な上昇が見られないならば、抗体は、医薬製剤において「その物理的な安定性を保持する」といえる。さらに、タンパク質の高次構造は変化することはない。タンパク質の高次構造の変化は、タンパク質の3次構造を決定する蛍光分光法によって、及びタンパク質の2次構造を決定するFTIR分光法によって評価することができる。
【0014】
もし有意な化学変換が示されないなら、医薬製剤において抗体は「その化学的安定性を保持する」といえる。化学的安定性は、化学的に変換されたタンパク質の形態を検出し、定量することによって、評価することができる。タンパクの化学的構造をしばしば変換する分解過程としては、加水分解又は分解(例えばサイズ排除クロマトグラフィー及びSDS−PAGEの方法で評価される)、酸化(例えば、マススペクトロメトリー又はMALDI/TOF/MSと組み合わせたペプチドマッピングの方法により評価される)、アミド分解(例えばイオン交換クロマトグラフィー、キャピラリー等電点電気泳動、ペプチドマッピング、イソアスパラギン酸計測法といった方法で評価される)、及び異性化(イソアスパラギン酸の含量の計測、ペプチドマッピング等によって評価される)が挙げられる。
【0015】
所定の時間における抗体の生理活性が、医薬製剤が調製された際に示された既定の生理活性の範囲内であるなら、抗体は医薬製剤において「生理活性を保持する」といえる。抗体の生理活性は、例えば抗原に結合するELISAアッセイによって決定することができる。
【0016】
「等張」という用語は、目的の製剤が基本的にヒト血液と同じ浸透圧を有するということを意味する。等張製剤は、通常約270〜328mOsmの浸透圧を有する。わずかに低張の浸透圧は250〜269mOsmであり、わずかに高張の浸透圧は328〜350mOsmである。浸透圧は、例えば蒸気圧又は氷冷型の浸透圧計を使って計測することができる。
【0017】
「浸透圧調節剤」は、医薬として許容される不活性な物質であって、製剤に等張性を与えるために製剤に加えられる物質である。本発明に適した浸透圧調節剤として、塩及びアミノ酸が挙げられる。
【0018】
II.分析方法
以下の判定基準は安定な医薬の抗体製剤を開発する際に重要である。抗体製剤は医薬として許容される賦形剤を含む。抗体がその物理的、化学的、及び生理活性を保持するように、抗体製剤が剤形される。製剤は、好ましくは、冷蔵庫の温度(2〜8℃)で少なくても1年間は安定であり、室温で(23〜27℃)で少なくても6ヶ月は安定である。
【0019】
製品の安定性を評価する分析方法としては、サイズ排除クロマトグラフィー(SEC−HPLC)、動的光散乱試験(DLS)、示唆走査熱量測定(DSC)、イソアスパラギン酸定量、作用効力、340nmのUV、及びUV分光法が挙げられる。SEC(J. Pharm.Scien., 83:1645-1650,(1994); Pharm. Res.,11:485(1994); J. Pharm. Bio. Anal., 15:1928(1997); J. Pharm. Bio. Anal., 14:1133-1140(1986))は、製品中の単量体の割合を測定し、可溶性の凝集体及び断片の量についての情報を与える。DSC(Pharm.Res., 15:200(1998); Pharm.Res., 9:109(1982))タンパク質の変性温度及びガラス転移温度についての情報を与える。DLS(American Lab., Nov.(1991))は、拡散係数の平均値を計測し、可溶及び不可溶な凝集体の量についての情報を与える。340nmにおけるUVは、340nmにおける散乱光の強度を計測し、可溶及び不溶性の凝集体の量についての情報を与える。UV分光法は、278nmにおける吸収を計測し、タンパク質濃度についての情報を与える。
【0020】
サンプルにおけるiso−Aspの含量は、イソクアントイソアスパラテートディテクションキット(プロメガ)を使って計測される。このキットは、標的タンパク質におけるイソアスパラギン酸残基の存在を特異的に検出する酵素、タンパク質イソアスパラギン酸メチル転移酵素 (PIMT)を使用する。PIMTは、S−アデノシル−L−メチオニン由来のメチル基をイソアスパラギン酸のα−カルボキシル位に転移することを触媒しており、その過程においてS−アデノシル−L−ホモシステイン(SAH)を形成する。SAHは比較的小さな分子量であり、キットに提供されるSAH HPLCの標準物質を用いることにより、逆相HPLCで通常単離及び定量される。
【0021】
抗体の効力又は生理活性は、その抗原に結合する能力によって測られうる。抗体のその抗原に対する特異的な結合は、当業者に周知であるいずれかの方法、例えばイムノアッセイ、例えばELISA(酵素結合免疫吸着検定法)によって定量される。
【0022】
III. 抗体の調製
本明細書中の発明は、抗体を含む安定した水性製剤に関する。当該技術分野で利用可能な抗体を作る技術と以下の記載においてより詳細に記述される例示の方法を使って、製剤内の抗体が調製される。
【0023】
抗体は、目的の抗原に対するものである。好ましくは、当該抗原は生物学的に重要なポリペプチドであり、哺乳動物に対する抗体の投与は疾患を予防又は治療することができる。しかしながら、非ポリペプチド抗原に対する抗体(例えば腫瘍に関連する糖脂質抗原;米国特許No.5,091,178号参照のこと。)にも関する。
【0024】
抗原がポリペプチドである場合、抗原は膜貫通分子(例えば受容体)又は成長因子などのリガンドであってもよい。例示の抗原としては、分子、例えばレニン;ヒト成長ホルモン及びウシ成長ホルモンを含む成長ホルモン;成長ホルモン放出因子;副甲状腺ホルモン;甲状腺刺激ホルモン;リポタンパク質;α−1−抗トリプシン;A鎖インスリン;B鎖インスリン;プロインスリン;卵胞刺激ホルモン;カルシトニン;黄体形成ホルモン;グルカゴン;凝血因子、例えば第VIII因子C、第IX因子、組織因子、及びフォンウィルブランド因子;抗凝血因子、例えばタンパク質C;心房性ナトリウム利尿因子;肺サーファクタント;プラスミノーゲン活性化因子、例えばウロキナーゼ又はヒト尿型若しくは組織型プラスミノーゲン活性化因子(t−PA);ボンベシン;トロンビン;造血性成長因子;腫瘍壊死因子−α及び−β;エンケファリン分解酵素;RANTES(regulated on activation nomally T-cell expressed and secreted);ヒトマクロファージ炎症タンパク質(MIP−1−α);血清アルブミン例えばヒト血清アルブミン;ミュラー管抑制因子;A鎖リラキシン;B鎖リラキシン;プロリラキシン:マウス性腺刺激ホルモン関連ペプチド;微生物タンパク質、例えばβラクタマーゼ;DNase;IgE;細胞傷害性Tリンパ球関連抗原(CTLA)、例えばCTLA−4;インヒビン;アクチビン;血管内皮増殖因子(VEGF);ホルモン又は成長因子の受容体;タンパク質A及びD;リウマチ因子;神経栄養性因子、例えばウシ由来神経栄養性因子(BDNF);ニューロトロフィン−3,−4,−5,又は−6(NT−3,NT−4,NT−5,又はNT−6) 又は神経成長因子、例えばNGF−β;血小板由来成長因子(PDGF);繊維芽細胞成長因子、例えばaFGF及びbFGF;上皮細胞成長因子(EGF);トランスフォーミング成長因子(TGF)、例えばTGF−α並びにTBF−β1、TBF−β2、TBF−β3、TBF−β4、及びTBF−β5を含むTGF−β;インスリン様成長因子−I及び−II(IGF−I及びIGF−II);des(1−3)−IGF−I(脳IGF−I)、インスリン様成長因子結合タンパク質;CDタンパク質、例えばCD3,CD4,CD8,CD19及びCD20;エリスロポエチン;骨誘導因子;免疫毒素;骨形成タンパク質(BMP);インターフェロン、例えばインターフェロン−α、−β、及び−γ;コロニー刺激因子(CSFs)、例えばM−CSF、GM−CSF、及びG−CSF;インターロイキン(ILs)、例えばIL1〜IL−12;インターロイキンIL−1〜IL−12の受容体:セレクチン、例えばL、E、及びP−セレクチン;スーパーオキシドジムスターゼ;T細胞受容体;膜表面タンパク質;腐食促進因子;ウイルス抗原、例えばAIDS外皮部位;輸送タンパク質;ホーミング受容体;アドレシン;調節タンパク質;インテグリン、例えばCD11a、CD11b、CD11c、CD18、ICAM、VLA−4及びVCAM;腫瘍関連抗原、例えばHER2、HER3又はHER4受容体;並びに上記リストのポリペプチドのいずれかの断片が挙げられる。
【0025】
組換え技術を用いる場合、抗体は細胞内に、細胞膜周辺腔において生産されるか、又は培養液に直接分泌されることもある。抗体が細胞内に産出される場合、最初のステップとして、粒子性細胞片、これらは宿主細胞又は溶解された細胞のいずれかである、は、例えば遠心又は限外ろ過により取り除かれる。抗体が培養液に分泌される場合、そういった発現システムの上清は最初に、商業的に利用できるタンパク質濃縮膜、例えばアミコン又はミリポアペリコン限外ろ過ユニットを使って濃縮される。プロテアーゼ阻害剤、例えばPMSFは、タンパク分解を阻害するため前述のステップいずれの中にも入れられるし、及び外来性の汚染菌の生育を防ぐため抗生物質が入れられる。
【0026】
細胞から調製された抗体組成物は、例えばヒドロキシルアパタイトクロマトグラフィー、ゲル電気泳動、透析、及びアフィニティークロマトグラフィーを用いて精製される。アフィニティークロマトグラフィーは好ましい精製技術である。Aタンパク質のアフィニティーリガンドとしての適合性は、種及び抗体内に存在する免疫グロブリンFcドメインのアイソタイプに依存する。Aタンパク質は、ヒトγ1、γ2、及びγ4重鎖(Lindmark et al.,J. Immunol. Meth.62:1-13(1983))に基づく抗体を精製するために使われうる。Gタンパク質は、全てのマウスアイソタイプ及びヒトγ3(Guss et al., EMBO J.5:1567-1575(1986))鎖に基づく抗体を精製するために推奨されている。アフィニティリガンドが結合するマトリクスはアガロースであることが最も多いが、別のマトリクスも用いることができる。機械的に安定なマトリックス、例えば、ガラス又はポリ(スチレンジビニル)ベンゼンの制御された細孔は、アガロースで達成されるより早い流れ、及び短い過程を可能にする。抗体がCH3ドメインを含む場合、バッカーボンドのABX(商標)樹脂(J.T.Baker, Phillipsburg, N.J.)が精製に有用である。タンパク質を精製する別の技術、例えばイオン交換カラムに基づく分画、エタノール沈殿、逆相HPLC、シリカ上でのクロマトグラフィー、ヘパリン セファロセット(商標)上でのクロマトグラフィー、陰イオン又は陽イオン交換樹脂(例えばポリアスパラギン酸カラム)上のクロマトグラフィー、等電点電気泳動、SDS−PAGE、及びリン酸アンモニウム沈殿が、回収される抗体に依存して利用される。
【0027】
本発明に含まれる好ましい抗体としてはダクリズマブ(USAN、United States Adopted Names)、ヒト化抗IL-2受容体抗体が挙げられる。ダクリズマブは、腎臓移植の後の組織拒絶を防ぐためにゼナパックス(商標)として現在売られており、経静脈で投与される。ダクリズマブは感染の治療にも有効であり、そのためには抗体の皮下投与が好ましい投与経路である。抗体の皮下投与のためには、高濃度の抗体が好ましい。ダクリズマブは、ヒト化組換え替えモノクローナル抗体、IgG1のサブクラスである。この分子は、2つの同一な重鎖及び2つの同一な軽鎖のサブユニットから構成される。ジスルフィド架橋が4つの鎖を結びつける。ダクリズマブ単量体は、分子量で大体150000ダルトンである。ダクリズマブは、活性化されたT細胞において発現するIL−2受容体のp55サブユニットに結合する。抗原標的は、CD25と命名される。ダクリズマブは、発酵流加培養法により重鎖及び軽鎖遺伝子を含むGS−NS0細胞株から生産される。バイオリアクターからの収集物を処理して、細胞及び細胞片を取り除き、そしてイオン交換及びゲルろ過クロマトグラフィー並びにいくつかの限外ろ過及びろ過技術を使って精製し、95%を超える単量体からなる薬を生産する。
【0028】
別の好ましい抗体は抗インターロイキン12(IL−12)抗体である。IL−12は、抗原提示細胞により合成されるサイトカインである。IL−12は、2つのサブユニット(p35及びp40)から構成されており、その両方が、機能的な活性のために存在しなければならない。機能的なIL−12は、IL−12p70とも呼ばれる。このサイトカインは、好ましくはヘルパーT細胞タイプ1(Th1)リンパ球及びナチュラルキラー細胞においてそれらの増殖速度を増加させることにより作用する。下流の影響の一つに、Th1細胞によるインターフェロンγ(IFNg)の分泌がある。これらの機能(増殖とIFNgの生産)の両方とも、容易に検定でき、サンプルにおけるIL−12の活性を検出するのに使われる。IL−12に対する特定の抗体は、上記の活性を中和すると示されてきた。Th1細胞は、様々な病気において中心的な役割を担っていることに結び付けられてきているので、中和特性を有する抗体は、潜在的な治療上の価値を持つ。16G2(ホフマン ラ ロシュ)は、IL−12p70に対するマウス抗体である。16G2は、機能アッセイ−つまりヒト抹消血由来の活性型T細胞(PBMC)の増殖阻害において、IL−12に対して近い化学量数で作用することが示されてきた。IL−12のp40の二量体は血漿中に存在し、p40サブユニットに対して産出された抗体は、与えられた量のIL−12の増殖能力を中和するために過度の量で使われる必要があるので、16G2がIL−12に対して近い化学量数で作用することは重要な性質である。16G2はプロテインデザインラボ(Fremont,CA)でヒト化され、HAIL−12(ヒト化抗IL−12、IgG1抗体)を生じる。
【0029】
別の好ましい抗体は、抗Lセレクチン抗体である。セレクチン、例えばL、E、及びP-セレクチンは、虚血及び再灌流の過程における組織の損傷に関連にすると分かってきた。好中球がこの関連に重要な役割を担う。セレクチンが好中球をリクルートするのに必要であると考えられる。L-セレクチンは、骨格筋及び肺において損傷を完全に発生するのに重要である。(Seekamp, et al., Am. J. `athol. 11:592-598(1994). Mulligan, et al., J. Immunol. 151:832-840(1994))。HuEp5C7(スマート 抗Lセレクチン)は、IgG2Fcミュータントを含み、ヒトE及びPセレクチン抗原と相互反応するヒト化抗-Lセレクチンモノクローナル抗体である。HuEP5C7は、プロテインデザインラボによって、様々な適応症、例えば喘息、脳卒中、傷害、及び自己免疫疾患に対して現在開発されている。
【0030】
別の好ましい抗体は、フリントズマブ、つまり抗γインターフェロン抗体である。フリントズマブは、プロテインデザインラボによって開発されたヒト化IgG1モノクローナル抗体であって、インターフェロンγ(IFN−g)、つまり炎症誘発性サイトカインによる自己免疫疾患の治療のための抗体である。IFN−gは主要組織適合性複合体(MHC)クラス1及び又はクラス2(HLA−DR)抗原の発現を促し、ナチュラルキラー細胞の細胞溶解活性を高め、マクロファージを活性化し、及び液性反応の特性である免疫グロブリンのアイソタイプを調節する。リンフォカインとして、IFN-gはまた、2型ヘルパーT細胞(Th2)の発生を抑制する一方で、1型ヘルパーT細胞(Th1)の発生を高める。Th1/Th2割合の異常は、様々な自己免疫疾患に結び付けられてきている。
【0031】
IV. 製剤の調製
目的の抗体が上記のように調製された後、抗体を含む医薬製剤が調製される。製剤開発のアプローチは以下の通りである:至適溶液pHを選び、緩衝液のタイプ及び濃度を選び、様々な液体の安定性を与える賦形剤の影響を評価し、及び選別した賦形剤の濃度を、I-至適実験デザイン(Statistics for Experimenters:An Introduction to Design, Date
Analysis, and Model Building, Box, George E.P. et al., John Wiley and Sons, Inc., 1978)を用いて最適化する。
【0032】
本発明の組成物は抗体凝集体及び粒子の形成を最小限にし、抗体がその生理活性を一定の期間保持することを保証する。組成物は、中性の又は少し酸性のpH(pH5.5〜6.5)、界面活性剤、及び浸透圧調節剤を有する緩衝液中に高い抗体濃度を含む医薬として許容される液体の製剤である。
【0033】
組成物中の抗体は50mg/ml又はそれを超える、好ましくは100mg/ml又はそれを超える高濃度である。本発明の好ましい組成物は、ダクリズマブ、つまりヒト化抗IL−2受容体抗体;HAIL−12、つまりヒト化抗IL−12抗体;HaEP5C7、つまりヒト化抗Lセレクチン抗体;及びフリントズマブ、ヒト化抗γインターフェロン抗体を含む。
【0034】
pH5.5〜6.5の緩衝液が組成物中で使われる。pH6.0〜6.5の緩衝液が好ましい。この範囲内でpHを制御する緩衝液の例として、コハク酸(例えばコハク酸ナトリウム)、グルコン酸、ヒスチジン、クエン酸、リン酸、及び別の有機酸緩衝液が挙げられる。コハク酸(pKa5.63)は皮下注射の好ましい緩衝液である。ヒスチジン(pK5.97)は、酸化に対する感受性のため好ましくないが、そういった酸化は、バイアルの上部をN2で置換すること又は抗酸化剤を添加することにより遅らせることができる。クエン酸及びリン酸緩衝液は、ずっと好ましくない。なぜなら、皮下注射されたとき有痛性の反応を起こすからである。好ましい緩衝液は約20〜60mMのコハク酸ナトリウムを含む。別の好ましい緩衝液は30〜70mMのN2で覆われたヒスチジン緩衝液である。
【0035】
界面活性剤もまた、抗体製剤に添加される。実例として、界面活性剤は、非イオン性界面活性剤、例えばポリソルベート(例えばポリソルベート20、80、例えばTween(商標)20、Tween(商標)80)又はポロクサマー類(例えばポロクサマー188)が挙げられる。剤形された抗体の凝集を減少し、及び/又は製剤中の粒子の形成を最小限にし、及び/又は吸着を減少させるように、ある量の界面活性剤が添加される。界面活性剤は、約0.005%〜約0.5%の量で、好ましくは約0.01%〜約0.1%の量で、より好ましくは0.01%〜約0.05%の量で、及び最も好ましくは約0.02%〜約0.04%の量で製剤内に存在しうる。
【0036】
製剤の等張性に貢献する浸透圧調節剤は、本組成物に加えられる。本発明にとって有用な浸透圧調節剤としては、塩及びアミノ酸が挙げられる。医薬として許容され、及び本発明に適している塩としては、塩化ナトリウム、コハク酸ナトリウム、硫酸ナトリウム、塩化カリウム、塩化マグネシウム、硫酸マグネシウム、及び塩化カルシウムが挙げられる。本発明の好ましい塩はNaCl及びMgCl2である。MgCl2はまた、タンパク質をアミド分解から守ることにより、抗体の安定性を改良しうる。NaClの好ましい濃度は、約75〜150mMである。MgCl2の好ましい濃度は、約1〜100mMである。医薬として許容され、及び本発明に適しているアミノ酸として、プロリン、アラニン、L−アルギニン、アスパラギン、L−アスパラギン酸、グリシン、セリン、リジン、及びヒスチジンが挙げられる。本発明に好ましいアミノ酸はプロリンである。好ましいプロリンの濃度はおよそ200mMである。
【0037】
タンパク質製剤を安定するために通常使われるEDTAもまた、製剤内に含まれうる。EDTAは、キレート剤として、金属に触媒されるスルフヒドリル基の酸化を阻害し、こうしてジスルフィド結合された凝集体の形成を減少させる。EDTAの好ましい濃度は、0.01〜0.2%である。
【0038】
例示の液体組成物は、抗体を約100mg/ml又はそれを超える量、約20〜60mMコハク酸ナトリウム(pH6)、約0.01〜0.1%ポリソルベート20又は80、及び約75〜150mMのNaClを含む製剤である。この製剤は、モノクローナル抗体の生理活性の安定性を保持し、ヒト患者に対して投与されるように意図される免疫グロブリンを、最終製品中における物理的、化学的、生物的分解から保護される。
【0039】
本発明の液体抗体製剤は、非経口投与、例えば静脈内、筋肉内、腹腔内、又は皮下注射に適しており;特に皮下注射に適している。
本発明は、次の実施例によってさらに記述される。実施例は実施例の中で記述される特定の手順の範囲に本発明を制限することとして解釈するべきではない。
【実施例】
【0040】
実施例1:pHの最適化
至適製剤のpH範囲を確認するため、及び主要の分解経路を確認するため、pH特性試験を行った。サンプル製剤は、3つの緩衝液:pH4.0若しくは5.0の50mM酢酸ナトリウム緩衝液、pH5.0、6.0、若しくは6.5の50mMヒスチジン緩衝液、又はpH7.0若しくは8.5の50mMリン酸ナトリウム緩衝液のうちの1つの緩衝液内に5.0mg/ml抗IL−2受容体抗体(ダクリズマブ)を含んだ。各製剤を5℃又は45℃で、100RPMの振蕩で4週間インキュベーションした。各サンプルの物理的又は化学的安定性を、0週目と4週目において、pH及び視覚分析、340nmのUV分光法、サイズ排除クロマトグラフィー(SEC−HPLC)、蛍光分光法、動的光散乱(DLS)、示唆走査熱量測定(DSC)、プロメガ イソクアント アッセイ、キャピラリー等電点電気泳動(cIEF)、SDS−PAGE(減少性、又は非減少性)、及び生理活性検定(ELISA)を含む分析方法によって評価した。
【0041】
図1Aにおいて、SECを用いることにより調べられた様々なpHレベルで回収された断片の割合により示されるように、45℃で4週間インキュベーションされた後のサンプルについて行われたSEC−HPLCにより、液体の製剤にとって切断が主要な分解経路であるということが示された。SECにより決定される断片の割合及び凝集体の割合(図1B)は、5.5から6.5の中間範囲のpH値において減少した。
【0042】
図2は、45℃でサンプルを4週間インキュベーションした後に、cIEFによって評価された際の、様々なpHレベルでの分解の割合を示す。最小の分解は約5.5のpH値において獲得される。
【0043】
図3は、45℃でサンプルを4週間インキュベーションした後に、プロメガのイソクアントキットによって評価された際の、様々なpHレベルで獲得されるイソアスパラギン酸の割合を示す。イソアスパラギン酸の形成(アミド分解)は、pH6.0及び6.5で最小であり、そしてpH8.0において急激に増加した。
【0044】
この実験の結果により、pH5.5〜6.5、及び好ましくはpH6.0〜6.5が最適pHであって、抗体の分解及び凝集を最小にするpHであるということが示される。
【0045】
実施例2:緩衝液の最適化
本実験において、各製剤は、5.0mg/mlダクリズマブ抗体を、50mMのコハク酸ナトリウム(pH6.0)内に;及び窒素ガス存在下、又は窒素ガス非存在下における50mMヒスチジン(pH6.0)内に含んだ。クエン酸ナトリウム緩衝液は、皮下注射された際に痛みの報告があるので、使用されなかった。生理活性(効力)を、0時間において、並びに4、8、及び12週間の37℃インキュベーションの後において、組換えヒトIL−2α受容体(IL−2sRα)抗原でコートされたマイクロプレート及びHRP標識されたヤギの抗ヒトIgGを使ってELISAによって計測した。
【0046】
図4は、37℃でのインキュベーション後の異なる緩衝液の効力に対する時間経過の影響を示す。抗体製剤の最も高い安定性が、pH6.0の50mMのコハク酸酸ナトリウム緩衝液において8週間をとおして達成された。ヒスチジンのみの緩衝液内における製剤は、緩衝液が酸化されるので即座に(8週以内に)その作用効力を失う。コハク酸ナトリウム又は酸化を防ぐために窒素ガスで満たされたヒスチジン緩衝液においては、製剤の作用効力は少なくとも12週の間、80%超を残した。
【0047】
実施例3:賦形剤の選別
対象
本試験は、50mg/mlダクリズマブ抗体の製剤のための様々な賦形剤を選別するため行われた。先に行われたpHの最適化試験(例1)から、製剤の安定性はpH6.0〜6.5のpH範囲において最大となった。それゆえ、本試験では、賦形剤は、二つの緩衝液;pH6.5の50mMリン酸緩衝液、とpH6.0の50mMコハク酸緩衝液において選別がされた。抗体の安定性を、濃度50mg/ml、5℃及び45℃で3週間、100RPMで攪拌された2つの緩衝液中でモニターした。試験される賦形剤として、界面活性剤(Tween80(商標)及びTween20(商標))、塩(NaCl及びMgCl2)、抗酸化剤(EDTA及びメチオニン)、アミノ酸(グリシン、リジン、セリン、及びプロリン)、及び混合溶媒(グリセロール及びエタノール)が挙げられる。様々な分析技術(透明度、pH、SEC−HPLC、UV−Vis、及びcIEF)が賦形剤を含んでいる製剤の性質決定のために使われた。
【0048】
サンプル調製
ダクリズマブ抗体は、67mMのリン酸ナトリウム製剤(Tween80を含まない)中に6.6mg/mlの濃度であった。この材料を、ペリコンII(ミリポア)で約30mg/mlまで濃縮し、続いて、2つの選択された緩衝液(50mMリン酸ナトリウムpH6.5、及び50mMコハク酸ナトリウムpH6.0)に、50mlのアミコンスターセル(ミリポア)を用いて緩衝液交換がされた。3回目及び最後の緩衝液交換ステップの間に、材料は最終濃度〜125mg/mlに濃縮された。最終的に抗体を0.8μm膜(ユニフロ)でろ過した。ろ過後のタンパク質濃度は、リン酸緩衝液サンプルについて約100mg/mlになるように、コハク酸緩衝液サンプルについて97mg/mlになるように決定された。
【0049】
賦形剤が選別されるところの賦形剤の標的濃度が、表1に示される。製剤は、必要とされる量の賦形剤を直接バイアルに量り入れることによって、又は賦形剤が濃縮されたストック溶液を調製することによって、調整された。賦形剤は、適切な緩衝溶液0.5mlに加えられ、pHは、1N HCL又は10%NaOHによって望ましい値に調節された。続いて、適切な緩衝液中の0.5mlの濃縮された抗体溶液(〜100mg/ml)が、標的濃度50mg/mlに達するように加えられる。この手順は、濃縮された賦形剤と直接接触することによるタンパク分解を防ぐために適用される。1mlの溶液は、2つのバイアルに0.5mlづつ分けて充填された。1つのバイアルは、最初のT=0の分析で使われ、その後2〜8℃の3週の時間点のため、2〜8℃で貯蔵された。別のバイアルを45℃100RPMで振蕩して3週間インキュベーションし、その期間の最後に分析した。
【0050】
【表1】
【0051】
分析方法
各2つの時間点において、様々な分析技術を用いてサンプルを分析した。溶液の透明度は、蛍光灯の下で黒い背景に対してサンプルバイアルを持つことによって、視覚的に試験した。溶液について不溶性の物質を検査し、色の変化を記録した。サイズ排除クロマトグラフィーを、ダイオードアレイ検出及び一連に結合される2つのトソハス(Tosohaas)カラムを有するパーキンエルマーのHPLCユニットを用いて行った。サンプルを、約5倍量の対応する緩衝液に約1mg/mlの濃度になるよう希釈し、そして100μlのサンプルをカラムに注入した。UV分光法により、パーキンエルマーラムダバイオ40分光光度計を用いてサンプル濃度を計測した。
【0052】
三週間の時点のサンプルを、バイオラッドCE(BioFocus3000)システムのキャピラリー等電点電気泳動により分析した。全てのサンプルを0.25mg/mlに水で希釈し、そして1:1希釈(終濃度0.125mg/ml)を、TEMED及び2つの内部のpIマーカー(8.4及び10.1)を含むファーマライト(pharmalyte)溶液を用いて行った。キャピラリーは、中性のコーティングを持つeCAP(ベックマン、長さ56cm、50umID)であった。
【0053】
コハク酸緩衝溶液中に賦形剤、つまりTween80、EDTA、NaCl及びMgCl2と供に剤形されたサンプルの効力は、5℃及び45℃で3週間のインキュベーション後に試験した。それは、KIT−225−K6細胞を含むバイオアッセイであった。
【0054】
結果
12の異なる賦形剤が2つの異なる緩衝液中でモニターされたので、T=0の時点で24のサンプルがあった。3週後の時点においては、48の被分析サンプルがあった(12の異なった賦形剤×2つの温度×2つの緩衝液=48)。実施したアッセイとして、UV−Vis、pH、透明度、SEC−HPLC、及びCIEFによる濃度決定が挙げられる。
(a)サンプル透明度
サンプルの外見を表2に示す。全てのサンプルは最初の時点T=0において両者の緩衝液中で透明であった。3週後の時点においては、リジンを含む1つを除いてリン酸緩衝液中のサンプル全てが5℃において透明であった。同じ緩衝液中で、45℃では、アミノ酸(グリシン、セリン、プロリン及びリジン)を含むサンプルでは透明に見えたが、いくつかの針様の浮遊物をバイアル中に有した。MgCl2のサンプルは、バイアルの底に明らかな結晶を有した。
コハク酸緩衝液中では、アミノ酸を含む製剤を除く全てのサンプルが5℃3週間のインキュベーションの後で透明であった。プロリン及びリジンを有するサンプルはもっともにごっていた。45℃においては、コハク酸緩衝液の全てのサンプルが3週後の時点においても透明であった。
【0055】
【表2】
【0056】
(b)SEC−HPLC
SEC−HPLCの結果を表3(A−C)に示した。表3Aは、本研究で調査された全てのサンプルにおける単量体野割合(%)を示す。全てのサンプルのT=0における単量体割合は99%以上であった。3週の時点における両緩衝液中における5℃のサンプルの単量体において、有意な変化は観測されなかった。しかしながら、45℃においては、全てのサンプルが単量体%の少しの低下(5%未満)が示された。リン酸緩衝液中に剤形されるサンプルでは、単量体%は94.08(メチオニン)から97.29(プロリン)の間であり、一方、コハク酸緩衝液で剤形されるサンプルでは、単量体割合は95.86%(メチオニン)から97.55(トゥィーン80)%の間であった。両方の緩衝液中で、メチオニン及びグリシンを含む製剤は、単量体%において最も有意な低下を示した。%単量体の減少は、主に断片の形成のせいであった。
【0057】
表3Bは本研究で調査された全てのサンプルにおいて凝集体形成割合(%)を掲載した。3週間の持続時間における凝集体の形成の増加は、両方の緩衝液の5℃における全てのサンプルで最小であることが明らかになった。45℃、3週間のインキュベーションの後、リン酸緩衝液中のサンプルは、0.40%(EDTA)から2.40%(グリシン)の範囲の凝集体割合(%)における増加を示した。コハク酸緩衝液中では、凝集体の形成はかなり低く;3週間のインキュベーションの後で0.7%(メチオニン)から1.09%(グリシン)の範囲であった。これらの結果を支持する仮説の一つは、もし凝集体の形成の原因が酸化であるなら、コハク酸緩衝液の金属をキレートする性質のため、凝集体の形成はコハク酸緩衝液中ではゆっくりになりうるということである。
【0058】
表3Cは、この研究で調査された全てのサンプルにおいて断片形成%を掲載する。最初の時間点においては、断片%は全てのサンプルにおいて0.2〜0.4%の範囲であった。5℃でインキュベートされた全てのサンプルで、断片%の増加は三週間の期間にわたってわずかなものであった。45℃においては、断片形成の割合の有意な増加が観測された。リン酸緩衝液中に剤形されるサンプルでは、断片%は4.74(メチオニン)から1.5%(プロリン、グリセロール、及びエタノール)の間であり、一方、コハク酸緩衝液中では、範囲は1.48%(トゥィーン80)から3.44(メチオニン)であった。一般的に、断片形成の増加は、アミノ酸を含む製剤で観察された。さらに、断片形成の割合は、リン酸緩衝液で高いようである。このことはNa−コハク酸緩衝液とNa−リン酸緩衝液との間におけるpHの違いに(それぞれpH6.0及びpH6.5)起因し、断片形成の第一の根拠として、塩基性に触媒される加水分解が示されうる。
【0059】
【表3】
【0060】
【表4】
【0061】
(c)キャピラリー電気泳動
本試験におけるすべてのサンプルは、バイオラッドシステムに基づくキャピラリー電気泳動(cIEF)によって分析された。ダクリズマブの典型的なcIEF特性は4つのピークを示す。典型的に、高温で時間効果が促進されたとき、主要なアイソフォームのピーク面積の減少に続いて別のアイソフォームのピーク面積が増加する。このことは、あるアイソフォームから別のアイソフォームへの変換を示唆する。分解割合(%)は、主要なアイソフォームのピーク面積における減少の割合によって計算される。
分解割合(%)=[T=0におけるピーク面積−45℃におけるピーク面積]×100%/[T=0におけるピーク面積]
【0062】
本発明者らの結果は、コハク酸緩衝液(pH6.0)中の同じサンプルと比較した場合に、45℃のサンプルがリン酸緩衝液(pH6.5)中でより分解されていることを示す。最良の電気泳動図は賦形剤、EDTA、NaCl、リジン、及びMgCl2で見られた。3週間後の45℃に対する5℃の分解%は、Tween80、Tween20、セリン及びプロリンを含むサンプルにおいてその電気泳動図がかなりつぶれていて、ピークが区別できなかったので、それらを含むサンプルでは計算できなかった。
【0063】
(d)作用効力
本試験の結果に基づくと、Na−コハク酸緩衝液は、Na−リン酸緩衝液とくらべてより有望であるようである。このように作用効力の評価は、Na−コハク酸緩衝液でのみ最も安定化する賦形剤についてされた。Na−コハク酸緩衝液はTween−80、EDTA、NaCl、及びMgCl2から成る製剤を含み、5℃及び45℃で3週間インキュベーションした。結果(表4)により、全ての製剤の作用効力が明細書の範囲内であり、強調される化学的及び物理的分解の過程はタンパク質の活性を有意には変えないということが示される。
【0064】
【表5】
【0065】
考察
本研究の結果に基づくと、pH6.5のNa−リン酸緩衝液と比較すると、pH6.0におけるNa−コハク酸緩衝液の方が製剤の安定性が高かった。このことは、pH6.5以上で促進される塩基性に触媒される加水分解が第一の原因であり、断片形態の割合の増加がもたらされる。このように、pH6.0におけるNa−コハク酸緩衝液は、今後の試験全てに選ばれる緩衝液である。両方の緩衝液中においてアミノ酸(グリシン、リジン、セリン、プロリン、及びメチオニン)はタンパク質の安定性に関して安定させる効果をもたないということを本試験の結果は明らかに示した。サンプルに関する明瞭なデータに示されたように、全てのアミノ酸を含む製剤は、45℃における不溶性の凝集体の形成を示した。
【0066】
MgCl2がタンパク質の減少から保護しうるという仮定に基づいて、賦形剤としてMgCl2が本研究において選択された。MgCl2がNa−リン酸緩衝液中で沈殿した一方で;Na−コハク酸緩衝液中では、cIEFデータに基づくとMgCl2はタンパク質を安定化する効果を有する。エタノールはまた、溶液の比誘電率を低めることで、タンパク質を脱アミド化に対し安定化させるかを試験するために、賦形剤として含まれた。しかしながら、その結果は上記仮説を支持することはない。最終的に、最も一般的にタンパク質製剤を安定するために使われるTween80、EDTA、及びNaCl、といった賦形剤は、どちらの緩衝液内においてもタンパク質を不安定化させる効果を全く与えないことを示した。
【0067】
更なる実験が、pH6.0のNa−コハク酸緩衝液中で行われ;賦形剤(MgCl2、Tween80、NaCl、及びEDTA)の影響がタンパク質の安定性において調べられた。その結果により、抗体を100mM NaClで100mg/mlに剤形すると、Tween80の最適濃度は0.02%〜0.03%の範囲内になることが示された。この結果により、塩の濃度の増加(100〜150mM)は製剤をさらに安定化しうることも示された。このように、NaClの濃度は、浸透圧を要求範囲に維持する一方で最大にするべきである。この結果により、Tween80及びNaClを含む製剤の安定性が、EDTAを0.35〜0.5%の範囲の濃度で加えることにより、高めることができることが示される。MgCl2を0〜50mMの範囲の濃度で添加することはまた、好ましい結果を与えうる。最も安定な剤形のための賦形剤の濃度は;150mM NaCl,0.05%Tween80、0.03%〜0.04%EDTA、及び60〜70mM MgCl2であるが、等張性の条件を提供しないため、この条件は実用的ではない。
【0068】
実施例4: コハク酸緩衝液中における2つのダクリズマブ抗体製剤の安定性データ
製剤1及び製剤2は実施例3に従って調製された。
製剤1:100mg/mlダクリズマブ抗体、30mMコハク酸ナトリウム(pH6.0)、100mM NaCl及び0.03%トゥィーン(商標)80である製剤
製剤2:製剤1と同じ製剤に0.05%EDTAを加えた製剤
製剤1及び製剤2の安定性の結果が、T=0、2週、4週、8週及び12週において、5、25、37℃での安定性の結果が以下に示される。(表5)
【0069】
【表6】
【0070】
【表7】
【0071】
実施例5: ヒスチジン緩衝液中における2つのダクリズマブ製剤の安定性データー
製剤3及び製剤4は実施例3に従って調製される。
製剤3:100mg/mlダクリズマブ抗体、50mMヒスチジン(pH6.0)、115mM NaCl、0.03%Tween(商標)80であり窒素置換を行った製剤。
製剤4:製剤3と同じ製剤に0.05%のEDTAを添加した製剤
製剤3及び製剤4のT=0、2週、4週、8週、及び12週における5、25、37℃での安定性の結果が以下に示される。(表6)
【0072】
【表8】
【0073】
【表9】
【0074】
実施例6: 室温で1年後におけるダクリズマブ製剤の安定性データ
100mg/mlダクリズマブを30mMコハク酸ナトリウム、pH6、100mM NaCl、及び0.03%トゥィーン(商標)80における液体の抗体製剤の安定性は、25℃で1年貯蔵した後に試験された。安定性の結果は、この製剤は少なくとも1年は25℃において安定であることを示す。(表7)
【0075】
【表10】
【0076】
実施例7: 5℃18ヶ月の期間後におけるダクリズマブ製剤の安定性データ
100mg/mlダクリズマブ、30mMコハク酸ナトリウム、pH6.0、100mM NaCl、及び0.03%トゥィーン(商標)80を含む液体の抗体製剤は、5℃(2〜8℃)でインキュベートされ、異なる時点において安定性が試験された。製剤が少なくとも18ヶ月間は冷蔵庫の温度で安定であることを安定性の結果は示す。
【0077】
【表11】
【0078】
実施例8: HAIL−12(ヒスチジン緩衝液)の安定性データ
HAIL−12(抗IL−12抗体、50mg/ml)を、50mMヒスチジン緩衝液、120mM塩化ナトリウム、0.03%トゥィーン80、pH6.0中に剤形した。進行中の安定性試験により、製剤が5℃で少なくとも9ヶ月間は安定であることが示される(表9)。
【0079】
【表12】
【0080】
実施例9: HAIL−12(コハク酸緩衝液)の安定性データ
HAIL−12(50及び100mg/ml)を、40mM Na−コハク酸緩衝液、100mM NaCl、及び0.03%トゥィーン80、pH6.0中に剤形した。進行中の安定性試験により、製剤が5℃、25、及び37℃で少なくとも12週間安定であることが示された(表10及び表11)。
【0081】
【表13】
【0082】
【表14】
【0083】
実施例11:HuEP5C7の安定性データ
HuEP5C7(抗Lセレクチン抗体、50及び100mg/ml)を、50mMヒスチジン緩衝液、125mM塩化ナトリウム、0.01%トゥィーン80、pH6.0中に剤刑した。進行中の安定性試験により、製剤は25℃及び45℃で3ヶ月間、並びに5℃で少なくても9ヶ月間安定であるということが示された。5℃における9ヶ月の安定性試験の結果を表12に示す。3ヶ月の促進された安定性試験を表13に示す。
【0084】
【表15】
【0085】
【表16】
本発明、並びに本発明を製造し、そして使用する方法及び過程は、発明が属する技術分野の当業者が、同発明を製造し、そして使用できるように十分、明瞭、簡潔、及び正確な用語で記述されている。実施例の記載は、本発明の好ましい態様を記述しており、そして特許請求の範囲において述べられるとき本発明の範囲から逸脱することなく変更がそこに加えられることは理解されるべきである。発明とみなされる対象の内容を特に明らかにし、明確に主張するために、以下の特許請求の範囲により本明細書が締めくくられる。
【特許請求の範囲】
【請求項1】
以下の:約pH5.5から約pH6.5までのpHを有する約20〜60mMコハク酸緩衝液、約0.01%〜0.1%のポリソルベート、製剤の等張性に寄与する浸透圧調節剤、及び50mg/ml超の抗体を含む安定な液体医薬製剤。
【請求項2】
以下の:約pH5.5から約pH6.5までのpHを有する約30〜70mMヒスチジン緩衝液、約0.01%〜0.1%のポリソルベート、製剤の等張性に寄与する浸透圧調節剤、及び50mg/ml超の抗体を含む安定な液体医薬製剤。
【請求項3】
前記抗体濃度が100mg/ml超である、請求項1又は2に記載の液体医薬製剤。
【請求項4】
前記浸透圧調節剤がNaCl又はMgCl2である、請求項1又は2に記載の安定な液体医薬製剤。
【請求項5】
前記NaClが75〜150mMである、請求項4に記載の安定な液体医薬製剤。
【請求項6】
前記MgCl2が1〜100mMである、請求項4に記載の安定な液体医薬製剤。
【請求項7】
前記pHがpH約6.0から6.5である、請求項1又は2に記載の安定な液体医薬製剤。
【請求項8】
前記ポリソルベートが約0.02〜0.04%の濃度である、請求項1又は2に記載の安定な液体医薬製剤。
【請求項9】
0.01〜0.5%のEDTAを更に含む、請求項1又は2に記載の安定な液体医薬製剤。
【請求項10】
前記製剤が約2〜8℃で少なくとも1年間は安定である、請求項1又は2に記載の安定な液体医薬製剤。
【請求項11】
前記製剤が約23〜27℃で少なくとも6ヶ月間は安定である、請求項1又は2に記載の安定な液体医薬製剤。
【請求項12】
以下の:約pH5.5から約pH6.5のpHを有する約20〜60mMコハク酸緩衝液、約0.01%〜0.05%のポリソルベート、約75〜150mMの塩化ナトリウム、並びにダクリズマブ、フリントズマブ、HAIL−12、及びHuEP5C7から成る群から選ばれる50mg/ml超の抗体を含む液体医薬製剤。
【請求項13】
以下の:約pH5.5から約pH6.5のpHを有する約30〜70mMヒスチジン緩衝液、約0.01%〜0.05%のポリソルベート、約75〜150mMの塩化ナトリウム、並びにダクリズマブ、フリントズマブ、HAIL−12、及びHuEP5C7から成る群から選ばれる50mg/ml超の抗体を含む液体医薬製剤。
【請求項1】
以下の:約pH5.5から約pH6.5までのpHを有する約20〜60mMコハク酸緩衝液、約0.01%〜0.1%のポリソルベート、製剤の等張性に寄与する浸透圧調節剤、及び50mg/ml超の抗体を含む安定な液体医薬製剤。
【請求項2】
以下の:約pH5.5から約pH6.5までのpHを有する約30〜70mMヒスチジン緩衝液、約0.01%〜0.1%のポリソルベート、製剤の等張性に寄与する浸透圧調節剤、及び50mg/ml超の抗体を含む安定な液体医薬製剤。
【請求項3】
前記抗体濃度が100mg/ml超である、請求項1又は2に記載の液体医薬製剤。
【請求項4】
前記浸透圧調節剤がNaCl又はMgCl2である、請求項1又は2に記載の安定な液体医薬製剤。
【請求項5】
前記NaClが75〜150mMである、請求項4に記載の安定な液体医薬製剤。
【請求項6】
前記MgCl2が1〜100mMである、請求項4に記載の安定な液体医薬製剤。
【請求項7】
前記pHがpH約6.0から6.5である、請求項1又は2に記載の安定な液体医薬製剤。
【請求項8】
前記ポリソルベートが約0.02〜0.04%の濃度である、請求項1又は2に記載の安定な液体医薬製剤。
【請求項9】
0.01〜0.5%のEDTAを更に含む、請求項1又は2に記載の安定な液体医薬製剤。
【請求項10】
前記製剤が約2〜8℃で少なくとも1年間は安定である、請求項1又は2に記載の安定な液体医薬製剤。
【請求項11】
前記製剤が約23〜27℃で少なくとも6ヶ月間は安定である、請求項1又は2に記載の安定な液体医薬製剤。
【請求項12】
以下の:約pH5.5から約pH6.5のpHを有する約20〜60mMコハク酸緩衝液、約0.01%〜0.05%のポリソルベート、約75〜150mMの塩化ナトリウム、並びにダクリズマブ、フリントズマブ、HAIL−12、及びHuEP5C7から成る群から選ばれる50mg/ml超の抗体を含む液体医薬製剤。
【請求項13】
以下の:約pH5.5から約pH6.5のpHを有する約30〜70mMヒスチジン緩衝液、約0.01%〜0.05%のポリソルベート、約75〜150mMの塩化ナトリウム、並びにダクリズマブ、フリントズマブ、HAIL−12、及びHuEP5C7から成る群から選ばれる50mg/ml超の抗体を含む液体医薬製剤。
【図1A】

【図1B】
【図2】
【図3】
【図4】
【図1B】
【図2】
【図3】
【図4】
【公開番号】特開2011−68675(P2011−68675A)
【公開日】平成23年4月7日(2011.4.7)
【国際特許分類】
【外国語出願】
【出願番号】特願2010−276222(P2010−276222)
【出願日】平成22年12月10日(2010.12.10)
【分割の表示】特願2003−541777(P2003−541777)の分割
【原出願日】平成14年11月8日(2002.11.8)
【出願人】(509189086)アボット バイオセラピューティクス コーポレイション (11)
【Fターム(参考)】
【公開日】平成23年4月7日(2011.4.7)
【国際特許分類】
【出願番号】特願2010−276222(P2010−276222)
【出願日】平成22年12月10日(2010.12.10)
【分割の表示】特願2003−541777(P2003−541777)の分割
【原出願日】平成14年11月8日(2002.11.8)
【出願人】(509189086)アボット バイオセラピューティクス コーポレイション (11)
【Fターム(参考)】
[ Back to top ]